CN101010339B - Human glucagon-like-peptide-1 modulators and their use in treatment of diabetes and related conditions - Google Patents
Human glucagon-like-peptide-1 modulators and their use in treatment of diabetes and related conditions Download PDFInfo
- Publication number
- CN101010339B CN101010339B CN2005800295435A CN200580029543A CN101010339B CN 101010339 B CN101010339 B CN 101010339B CN 2005800295435 A CN2005800295435 A CN 2005800295435A CN 200580029543 A CN200580029543 A CN 200580029543A CN 101010339 B CN101010339 B CN 101010339B
- Authority
- CN
- China
- Prior art keywords
- peptide
- group
- fmoc
- solution
- methyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- 0 *[C@@](C(*)=O)NP Chemical compound *[C@@](C(*)=O)NP 0.000 description 17
- QVIAMKXOQGCYCV-UHFFFAOYSA-N CC(C)CCCN Chemical compound CC(C)CCCN QVIAMKXOQGCYCV-UHFFFAOYSA-N 0.000 description 1
- HTHZMKQJDHBHNV-UHFFFAOYSA-N Cc1ccccc1-c(nc1)ccc1C#C Chemical compound Cc1ccccc1-c(nc1)ccc1C#C HTHZMKQJDHBHNV-UHFFFAOYSA-N 0.000 description 1
- OYTWDQHYIUDLIP-UHFFFAOYSA-N O=C(N1CCC1)SS Chemical compound O=C(N1CCC1)SS OYTWDQHYIUDLIP-UHFFFAOYSA-N 0.000 description 1
Images



Landscapes
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Peptides Or Proteins (AREA)
Abstract
The present invention provides novel human glucagon-like peptide-1 (GLP-1)-receptor modulators that have biological activity similar or superior to native GLP-1 peptide and thus are useful for the treatment or prevention of diseases or disorders associated with GLP activity. Further, the present invention provides novel, chemically modified peptides that not only stimulate insulin secretion in type II diabetics, but also produce other beneficial insulinotropic responses. These synthetic peptide GLP-1 receptor modulators exhibit increased stability to proteolytic cleavage making them ideal therapeutic candidates for oral or parenteral administration. The peptides of this invention show desirable pharmacokinetic properties and desirable potency in efficacy models of diabetes.
Description
This application claims priority from U.S. provisional patent application serial No. 60/585,358, filed on 7/2/2004 and U.S. provisional patent application serial No. 60/684,805, filed on 26/5/2005, each of which is incorporated herein by reference in its entirety.
Technical Field
The present invention provides novel human glucagon-like peptide-1 (GLP-1) peptide receptor modulators, agonists or partial agonists that exhibit superior biological properties of the native peptide GLP-1 and that exhibit increased stability to proteolytic cleavage when compared to the GLP-1 native sequence, and are therefore useful for ameliorating diabetic conditions.
Background
GLP-1 is an important gastrointestinal hormone that has a regulatory function on glucose metabolism and gastrointestinal secretion and metabolism. Human GLP-1 is a 30 amino acid peptide derived from preproglucagon, which is synthesized, for example, in L-cells in the terminal ileum, in the pancreas and in the brain. The process of processing preproglucagon to obtain GLP-1(7-36) amide and GLP-2 occurs mainly in L-cells and brainstem. GLP-1 is normally secreted in response to ingestion of food, and carbohydrates and lipids, in particular, stimulate GLP-1 secretion. GLP-1 has been determined to be a very potent and potent stimulator of glucose-dependent insulin release and has a reduced risk of causing hypoglycemia. GLP-1 lowers plasma glucagon concentration, slows gastric emptying, stimulates insulin biosynthesis and increases insulin sensitivity (Nauck, 1997, Horm. Metab. Res.47: 1253-one 1258). GLP-1 also increases the ability of pancreatic beta cells to sense and respond to glucose in patients with impaired glucose tolerance (Byrne, Eur.J.Clin.Invest., 28: 72-78, 1998). The insulinotropic effect of GLP-1 in humans increases the rate of glucose metabolism, partly due to increased insulin levels and partly due to increased insulin sensitivity (D' Alessio, Eur. J. Clin. invest., 28: 72-78, 1994.) inhibition of glucagon release is believed to be an additional mechanism that helps to improve glucose homeostasis, which is observed after GLP-1 treatment of type II diabetics (Nauck, M.A., et al, Diabetologia 36: 741-. The pharmacological properties of GLP-1 as illustrated above make it a highly desirable therapeutic agent for the treatment of type II diabetes.
In addition, recent studies have shown that infusion of slightly supraphysiological doses of GLP-1 significantly increases satiety and reduces food intake in normal subjects (Flint, A., Raben, A., Astrup, A. and Hoist, J.J., J.Clin.invest 101: 515-. The retention of said effects on food intake and satiety in obese subjects has also been reported (Naslund, e., barkling, b., King, n., Gutniak, m., Blundell, j.e., Holst, j.j., Rossner s., and Hellstrom, p.m., int.j.obes.relat.metab.d., 23: 304 + 311, 1999).
In the above cited studies, it is also suspected that a significant effect of GLP-1 on gastric emptying occurs. Gastric emptying leads to glucose excursions after feeding. GLP-1 has been shown to stimulate the expression of the transcription factor islet-duodenum homeobox-1 (IDX-1) in addition to insulin secretion, while stimulating B-cell neogenesis, and thus may be an effective therapeutic and/or prophylactic agent for diabetes (Stoffers, D.A., Kieffer, T.J.Hussain, M.A., Drucker, D.J., Bonner-Weir, S.A., Habener, J.F., and Egan, J.M.diabetes, 40: 741-748, 2000). GLP-1 has also been shown to inhibit gastric acid secretion (Wettergren, A., Schjoldager, B., Mortens, P.E., Myhre, J., Christiansen, J., Hoist, J.J., dig.Dis.Sci., 38: 665-.
It has recently been reported that GLP-1 has many other extra-pancreatic effects which can, for example, lead to cardioprotection, neuroprotection and induction of cognition and memory (Ahren, B., Horm. Metab. Res. 36: 842-845, 2004). Thus, GLP-1 has also been proposed for the treatment of heart failure (Nikolaidis, L.A. et al, Circulation 110: 955-.
GLP-1 is an incretin, e.g., an enterohormone that increases meal-induced insulin secretion (Holst, J.J., curr.Med.chem., 6: 1005-1017, 1999). It is the product of the glucagon gene, which encodes preproglucan. This gene is expressed not only in A cells of the pancreas but also in endocrine L-cells of the intestinal mucosa. Pro-glucagon is a peptide (protein) containing 160 amino acids. Further processing of preproglucagon leads to the production of: a) glucagon, b) an N-terminal, presumably inactive fragment, and C) a large C-terminal fragment commonly referred to as the "major pro-glucagon fragment". The fragment is considered biologically inactive. Although this fragment is present not only in the pancreas but also in the L-cells of the intestine, it is only in the intestine that the breakdown products of the "major pro-glucagon fragment" are observed to produce two highly homologous peptides commonly referred to as GLP-1 and GLP-2. Both peptides have important biological activities. Thus, the amino acid sequence of GLP-1 present in L-cells is identical to the 78-107 portion of pro-glucagon.
Currently, there has been a significant problem with treatments involving the use of GLP-1 type molecules, because the serum half-life of this peptide is rather short. For example, GLP-1(7-37) has a serum half-life of less than 5 minutes. Thus, there is an urgent need for GLP-1 receptor modulators, agonists or antagonists that are biologically active and have prolonged pharmacokinetic profiles. The present invention addresses this and other needs.
Thus, the present invention provides novel peptides that are GLP-1 receptor modulators, agonists or partial agonists that exhibit biological properties similar to or superior to the native peptide GLP-1 and are therefore useful for ameliorating diabetes and related conditions.
Summary of The Invention
In one aspect, the invention relates to an isolated polypeptide comprising a polypeptide having a sequence of formula I:
Xaa1-Xaa2-Xaa3-Xaa4-Xaa5-Xaa6-Xaa7-Xaa8-Xaa9-Xaa10-Xaa11
I
wherein,
Xaa1is a naturally or non-naturally occurring imidazole-containing amino acid; wherein one or more carbon atoms of the amino acid are optionally substituted with one or more alkyl groups; wherein the amino acid optionally has a free amino group, which group is optionally substituted with alkyl, acyl, benzoyl, L-lactyl, alkyloxycarbonyl, aryloxycarbonyl, arylalkyloxycarbonyl, heterocyclyloxycarbonyl, heteroarylalkyloxycarbonyl, alkylcarbamoyl, arylcarbamoyl, arylalkylcarbamoyl, heterocyclylsulfonyl, alkylsulfonyl, arylsulfonyl, arylalkylsulfonyl, heteroarylalkylsulfonyl or heteroarylsulfonyl; and wherein when said free amino group is absent, Xaa1A des-amino (des-amino) acid which is histidine wherein one or more carbon atoms of said amino acid is optionally substituted with one or more alkyl groups;
Xaa2an amino acid that is naturally or non-naturally occurring and selected from the group consisting of D-alanine, α -amino-isobutyric acid (Aib), N-methyl-D-alanine, N-ethyl-D-alanine, 2-methyl-azetidine-2-carboxylic acid, α -methyl- (L) -proline, 2-methylpiperidine-2-carboxylic acid and isovaline;
Xaa3an amino acid that is naturally or non-naturally occurring and has (1) a carboxylic acid-containing amino acid side chain or (2) an imidazole side chain, and wherein one or more carbon atoms of the amino acid are optionally substituted with one or more alkyl groups;
Xaa4is glycine;
Xaa5an amino acid which is naturally or non-naturally occurring and is selected from the group consisting of (L) -threonine and (L) -norvaline; and wherein one or more carbon atoms of the amino acid are optionally substituted with one or more alkyl groups;
Xaa6an alpha carbon with two side chains that is a naturally or non-naturally occurring amino acid and which has a di-substituted; wherein at least one of the two side chains has an aromatic ring and at least one of the two chains has an alkyl group; and wherein one or more carbon atoms of the amino acid are optionally substituted with one or more alkyl groups or one or more halogen groups.
Xaa7An amino acid that is naturally or non-naturally occurring and has an amino acid side chain substituted with a hydroxyl group; and wherein one or more carbon atoms of the amino acid are optionally substituted with one or more alkyl groups;
Xaa8an amino acid that is naturally or non-naturally occurring and selected from L-serine and L-histidine; and wherein one or more carbon atoms of the amino acid are optionally substituted with one or more alkyl groups;
Xaa9an amino acid that is naturally or non-naturally occurring and has an amino acid side chain comprising a carboxylic acid; and wherein one or more carbon atoms of said amino acid are optionally substituted with one or more alkyl groupsGroup substitution;
Xaa10is a naturally or non-naturally occurring amino acid of formula II:
formula II
Wherein R is4Selected from hydrogen, alkyl and halogen;
wherein R is3And R6Each is independently selected from hydrogen, halogen, methyl, ethyl, alkyl, hydroxy, methoxy, and alkoxy;
wherein the phenyl ring proximal to the β -carbon of the amino acid is additionally optionally substituted with alkyl or halogen; and
wherein the phenyl ring distal to the β -carbon of the amino acid is additionally optionally substituted with halogen, methyl, ethyl, alkyl, hydroxy, methoxy, and alkoxy;
Xaa11is a naturally or non-naturally occurring amino acid of formula IVa:
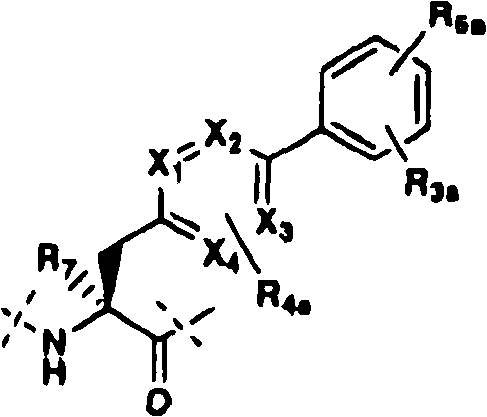
formula IVa
Wherein the C-terminal carbonyl carbon of the amino acid is attached to nitrogen to form a carboxamide (NH)2);
Wherein R is4aSelected from hydrogen, alkyl and halogen;
wherein R is3aAnd R6aEach is independently selected from hydrogen, halogen, methyl, ethyl, alkyl, hydroxy, methoxy, and alkoxy;
wherein R is7Selected from hydrogen, methyl, and ethyl; and
wherein X1、X2、X3And X4Each is C or N, and provided that X1、X2、X3And X4At least one of which is N;
wherein the phenyl ring proximal to the β -carbon of the amino acid is additionally optionally substituted with alkyl or halogen; and
wherein the phenyl ring distal to the β -carbon of the amino acid is additionally optionally substituted with halogen, methyl, ethyl, alkyl, hydroxy, methoxy, and alkoxy.
Further, Xaa3May be histidine, wherein said histidine is optionally substituted with one or more alkyl groups. Xaa3May be L-aspartic acid or L-glutamic acid, wherein each of the L-aspartic acid or L-glutamic acid is optionally substituted with one or more alkyl groups.
Xaa6May be alpha-methyl-phenylalanine, alpha-methyl-2-fluorophenylalanine or alpha-methyl-2, 6-difluorophenylalanine, wherein each of the alpha-methyl-phenylalanine, alpha-methyl-2-fluorophenylalanine or alpha-methyl-2, 6-difluorophenylalanine is optionally substituted with one or more alkyl groups.
Xaa7May be L-threonine, wherein the threonine is optionally substituted with one or more alkyl groups.
Xaa9May be L-aspartic acid or L-glutamic acid, wherein each of the L-aspartic acid or L-glutamic acid is optionally substituted with one or more alkyl groups.
Xaa1May be L-histidine having a terminal amino group optionally substituted with alkyl, dialkyl, acyl, benzoyl, L-lactyl, alkyloxycarbonyl, aryloxycarbonyl, aralkyloxycarbonyl, heterocyclyloxycarbonyl, heteroarylalkyloxycarbonyl, alkylcarbamoyl, arylcarbamoyl, aralkylcarbamoyl, heterocyclylsulfonyl, alkylsulfonyl, arylsulfonylAcyl, arylalkyl sulfonyl, heteroarylalkyl sulfonyl or heteroarylsulfonyl.
Xaa1Can be selected from L-N-methyl-His, L-alpha-methyl-His, des-amino-His, 3- (1H-imidazol-4-yl) -2-methylpropionyl, and (S) -3- (1H-imidazol-4-yl) -2-hydroxypropionyl (L-beta-imidazole lactyl).
Xaa2May be selected from alpha-amino-isobutyric acid (Aib), D-alanine, N-methyl-D-alanine, alpha-methyl- (L) -proline, 2-methyl-azetidine-2-carboxylic acid and 2-methylpiperidine-2-carboxylic acid.
Xaa4May be glycine.
Xaa5Can be selected from L-Thr and L-Nva.
Xaa6Can be selected from L-alpha-Me-Phe, L-alpha-Me-2-fluoro-Phe and L-alpha-Me-2, 6-difluoro-Phe.
Xaa7May be L-Thr.
Xaa8Can be selected from L-Ser and L-His.
Xaa9May be L-Asp.
Xaa10Can be selected from 4-phenyl-phenylalanine, 4- [ (4 '-methoxy-2' -ethyl) phenyl]Phenylalanine, 4- [ (4 '-ethoxy-2' -ethyl) phenyl]Phenylalanine, 4- [ (4 '-methoxy-2' -methyl) phenyl]Phenylalanine, 4- [ (4 '-ethoxy-2' -methyl) phenyl]Phenylalanine, 4- (2 '-ethylphenyl) phenylalanine, 4- (2' -methylphenyl) phenylalanine, 4- [ (3 ', 5' -dimethyl) phenyl]Phenylalanine and 4- [ (3 ', 4' -dimethoxy) phenyl]Phenylalanine, and mixtures thereof.
Xaa11Can be selected from 4-phenyl-3-pyridylalanine, 4- (2 ' -methylphenyl) -3-pyridylalanine, 4- (2 ' -fluorophenyl) -3-pyridylalanine, 4- (2 ' -chlorophenyl) -3-pyridylalanine, 4- [ (3 ', 5 ' -dimethyl) phenyl]-3-pyridylalanine, 4- (4' -trifluoromethylphenyl) -3-pyridylalanine4- (3 '-methoxyphenyl) -3-pyridylalanine, 4- (3' -methylphenyl) -3-pyridylalanine, 4- (2 '-methylphenyl) -3, 5-pyrimidylalanine and 4- (2' -ethylphenyl) -3-pyridylalanine;
wherein Xaa11To the nitrogen to form the carboxamide (NH)2) (ii) a And
wherein R is7Selected from hydrogen and methyl.
In another aspect, the isolated polypeptide can be a polypeptide of formula VI:

formula VI
Wherein:
Xaa2is an amino acid selected from D-Ala, N-methyl-D-Ala, α -methyl-L-Pro, 2-methyl-azetidine-2-carboxylic acid, 2-methylpiperidine-2-carboxylic acid and α -aminoisobutyric acid (Aib);
x and Y are each independently selected from hydrogen and fluorine;
Xaa8is an amino acid selected from L-Ser and L-His;
R3selected from hydrogen, methyl and ethyl;
R6selected from hydrogen, hydroxy, methoxy and ethoxy;
R3aselected from hydrogen, fluoro, methyl and ethyl;
R6aselected from hydrogen, methyl and methoxy; and
R7selected from hydrogen and methyl.
Further, Xaa2Can be ammonia selected from N-methyl-D-Ala, alpha-methyl-L-Pro and alpha-aminoisobutyric acid (Aib)An amino acid;
x may be fluorine;
y may be hydrogen;
Xaa8may be an amino acid selected from L-Ser and L-His;
R3may be ethyl;
R6may be methoxy;
R3amay be selected from methyl and ethyl;
R6amay be hydrogen;
R7may be hydrogen.
In another aspect, the isolated polypeptide can be a polypeptide of formula VII:
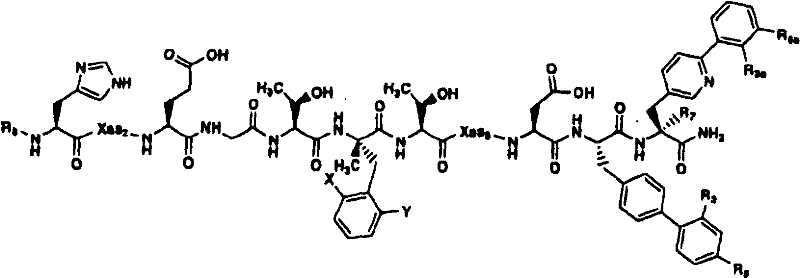
formula VII
Wherein:
R8selected from methyl, ethyl,

Xaa2Is an amino acid selected from the group consisting of D-Ala, N-methyl-D-Ala, α -methyl-L-Pro, α -aminoisobutyric acid (Aib), 2-methyl-azetidine-2-carboxylic acid and 2-methylpiperidine-2-carboxylic acid;
x and Y are each independently selected from hydrogen and fluorine;
Xaa8is an amino acid selected from L-Ser and L-His;
R3selected from hydrogen, methyl and ethyl;
R6selected from hydrogen, hydroxy, methoxy and ethoxy;
R3aselected from hydrogen, fluoro, methyl and ethyl;
R6aselected from hydrogen, methyl and methoxy; and
R7selected from hydrogen and methyl.
Further, R8Can be selected from methyl and
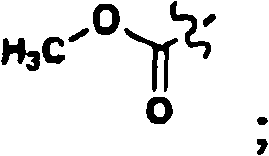
Xaa2may be an amino acid selected from N-methyl-D-Ala, alpha-methyl-L-Pro and aminoisobutyric acid (Aib);
x may be fluorine;
y may be hydrogen;
Xaa8may be an amino acid selected from L-Ser and L-His;
R3may be ethyl;
R6may be methoxy;
R3amay be selected from methyl and ethyl;
R6amay be hydrogen;
R7may be selected from hydrogen and methyl.
In another aspect, the isolated polypeptide can be a polypeptide of formula VIII:
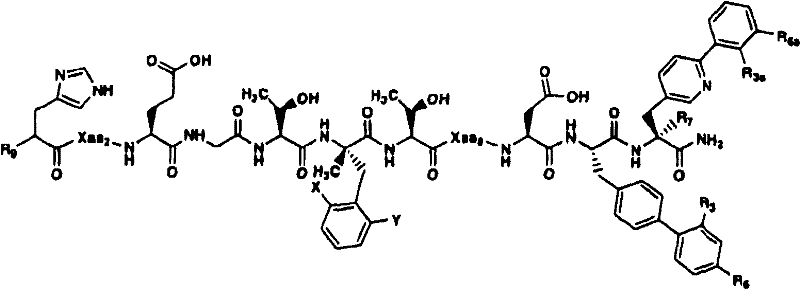
of the formula VIII
Wherein:
R9selected from hydrogen, methyl and alkyl;
Xaa2is an amino acid selected from the group consisting of D-Ala, N-methyl-D-Ala, α -methyl-L-Pro, α -aminoisobutyric acid (Aib), 2-methyl-azetidine-2-carboxylic acid and 2-methylpiperidine-2-carboxylic acid;
x and Y are each independently selected from hydrogen and fluorine;
Xaa8is an amino acid selected from L-Ser and L-His;
R3selected from hydrogen, methyl and ethyl;
R6selected from hydrogen, hydroxy, methoxy and ethoxy;
R3aselected from hydrogen, fluoro, methyl and ethyl;
R6aselected from hydrogen, methyl and methoxy;
R7selected from hydrogen and methyl.
Further, R9May be selected from hydrogen and methyl;
Xaa2may be an amino acid selected from N-methyl-D-Ala, alpha-methyl-L-Pro and alpha-aminoisobutyric acid (Aib);
x may be fluorine;
y may be hydrogen;
Xaa8may be an amino acid selected from L-Ser and L-His;
R3may be ethyl;
R6may be methoxy;
R3amay be selected from methyl and ethyl;
R6amay be hydrogen;
R7may be hydrogen.
In another aspect, the isolated polypeptide may be a compound selected from the following table:
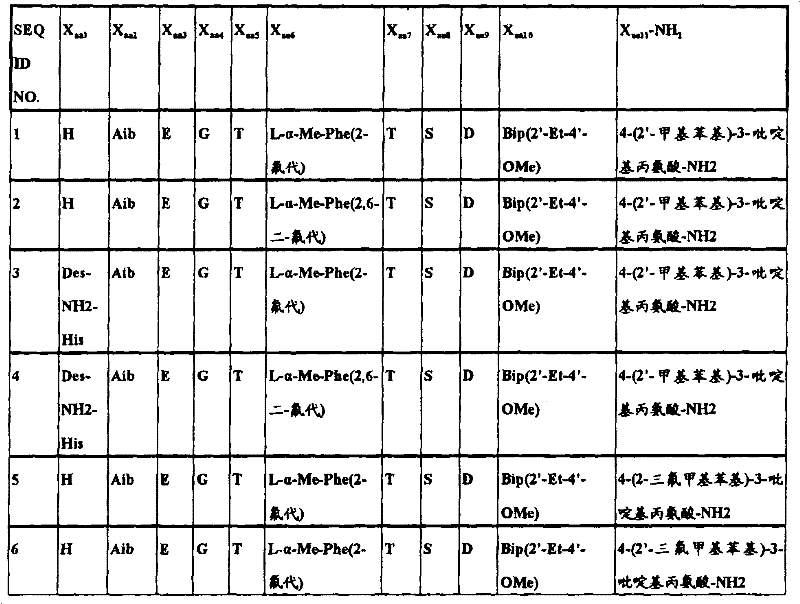
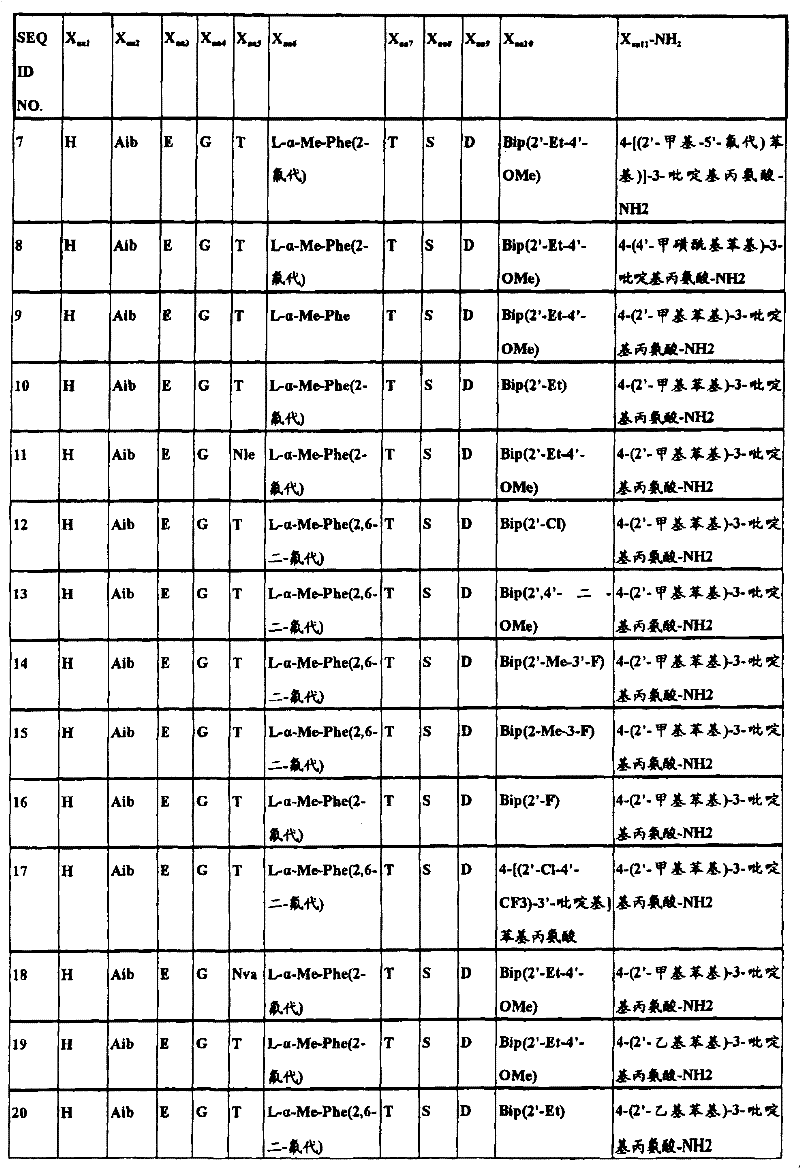
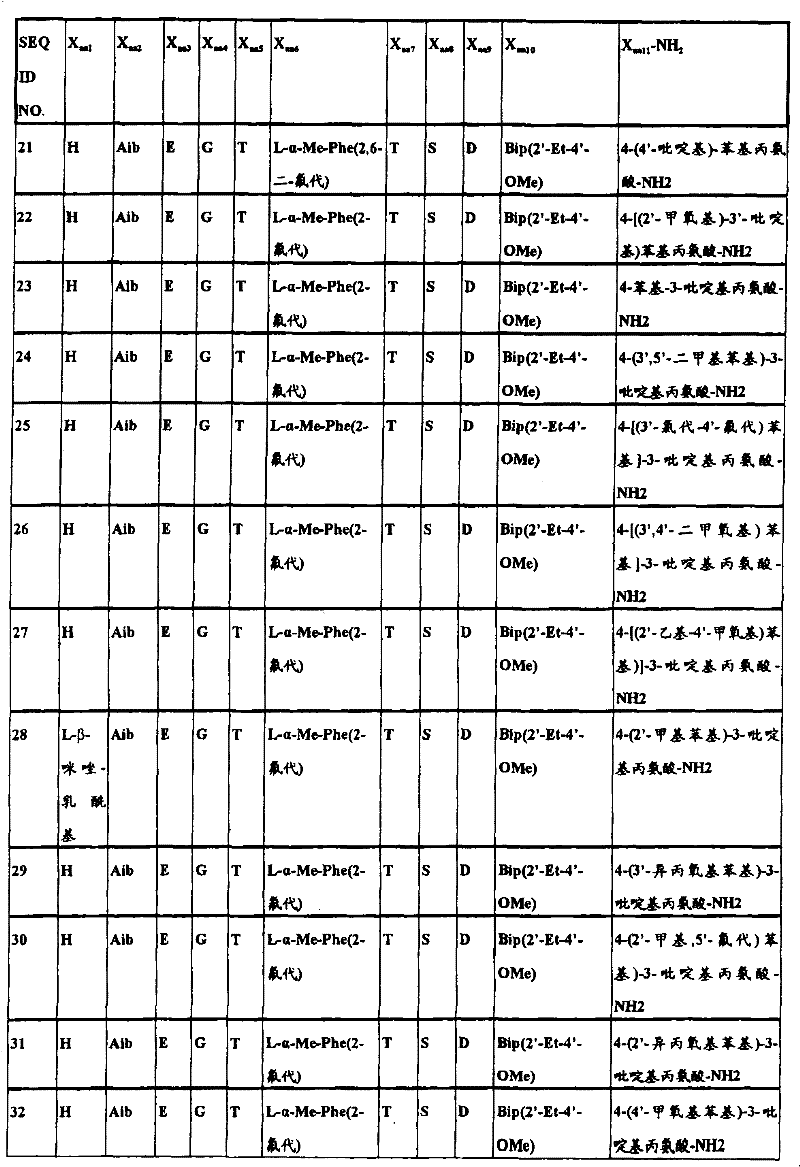

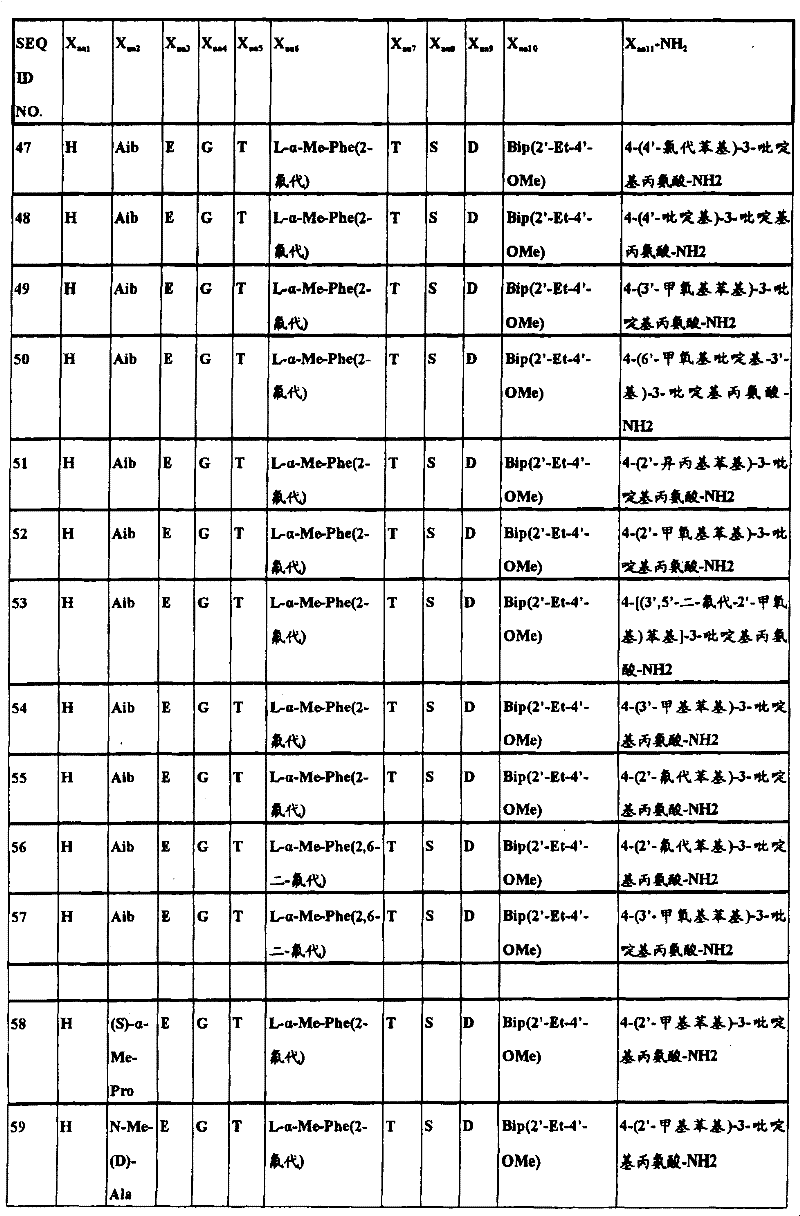
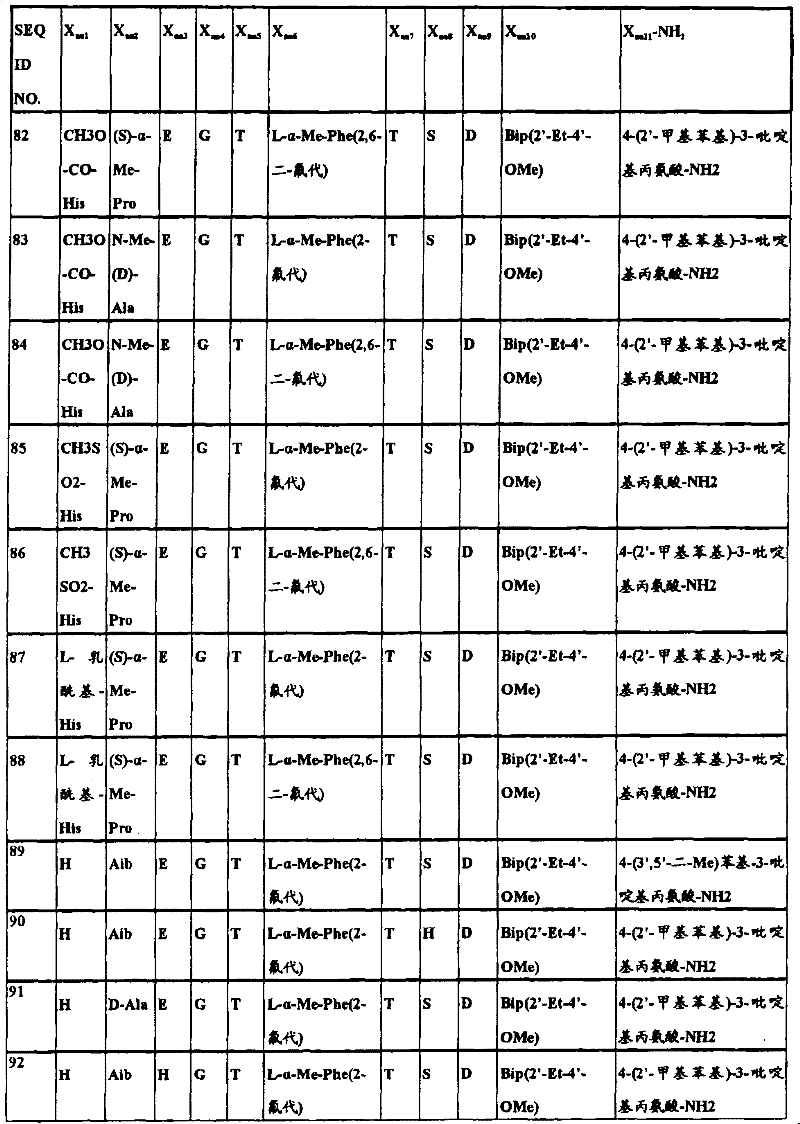
in preferred embodiments of the subject matter described and claimed herein, the polypeptide is selected from the group consisting of seq id NO's: 1.2, 4,9, 10, 19, 20, 23, 38, 43, 46, 49, 57, 58, 59, 61, 62, 63, 65, 69, 70, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83, 84, and 90.
In another aspect, the isolated polypeptide is:

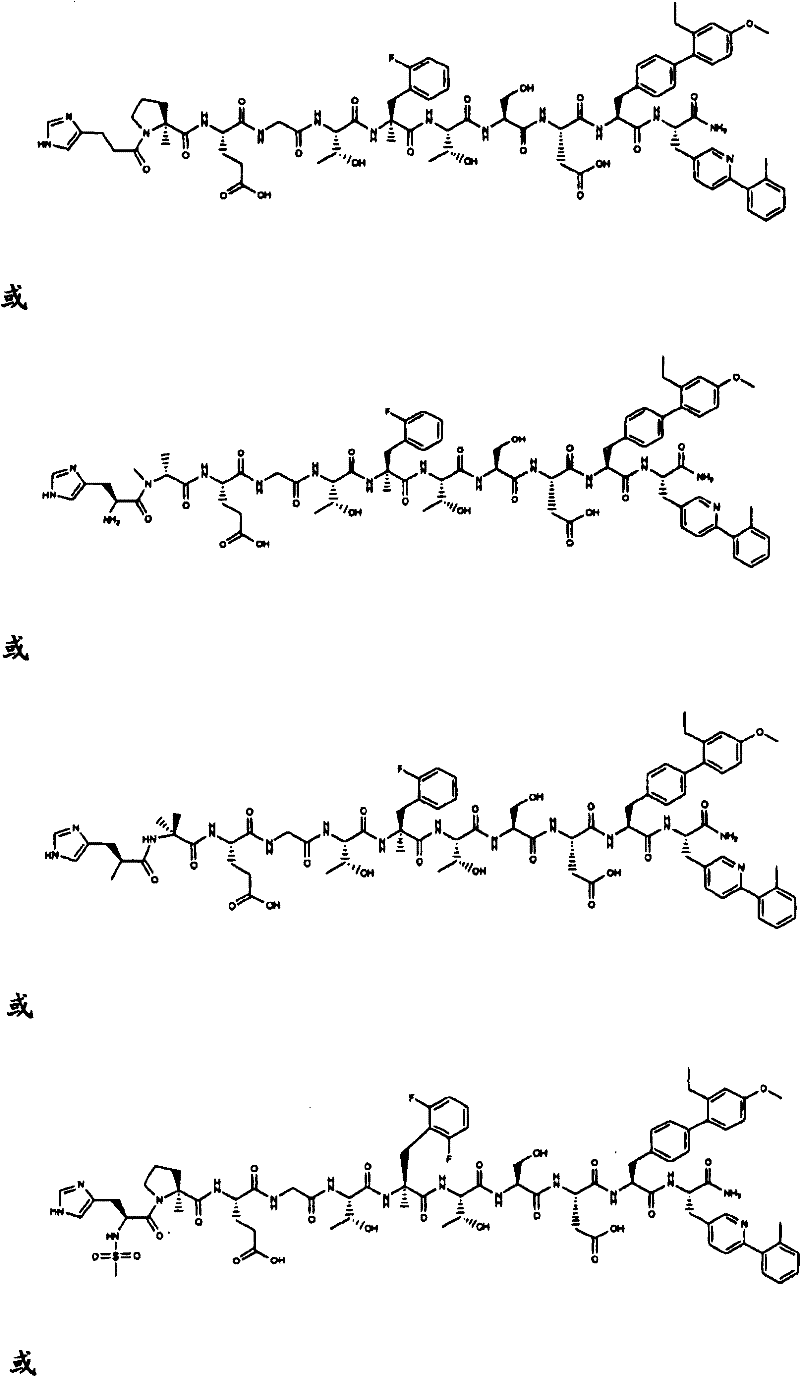

in another aspect, the isolated polypeptide is:
in another aspect, the isolated polypeptide is:
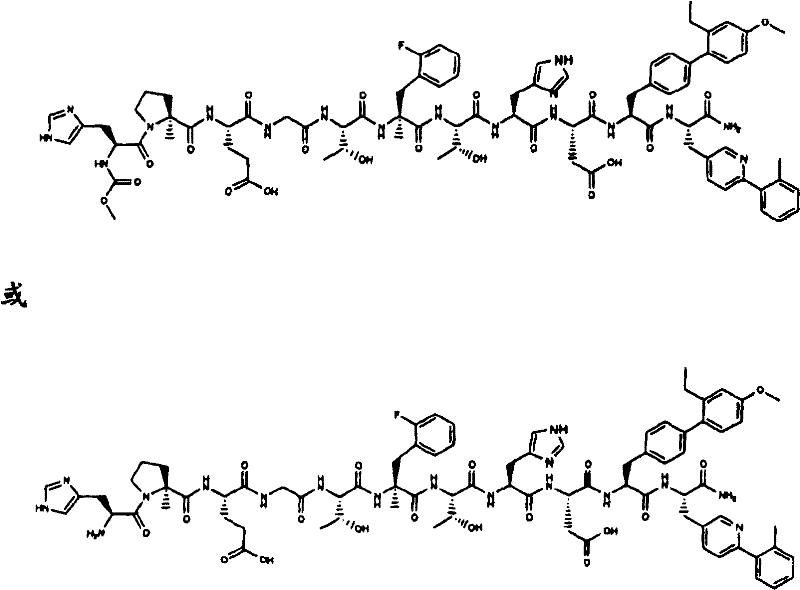
in another aspect, the invention relates to a compound of formula VIa:

formula VIa
Wherein P is hydrogen, fluorenylmethyloxycarbonyl (Fmoc) or tert-butyloxycarbonyl (t-Boc); wherein R is3aSelected from methyl, ethyl and fluoro; wherein R is10Selected from OH and NH2(ii) a And wherein R7Selected from hydrogen and methyl.
In another aspect, the invention relates to a compound of formula VIIa:

formula VIIa
Wherein P is hydrogen, fluorenylmethyloxycarbonyl (Fmoc) or tert-butyloxycarbonyl (t-Boc); wherein R is6aIs methoxy; wherein R is10Selected from OH and NH2(ii) a And wherein R7Selected from hydrogen and methyl.
In another aspect, the invention relates to a pharmaceutical composition comprising an isolated polypeptide described herein and a pharmaceutically acceptable carrier therefor.
In another aspect, the invention relates to a pharmaceutical composition comprising an isolated polypeptide described herein and at least one therapeutic agent; wherein the therapeutic agent is selected from antidiabetic agents, antiobesity agents, antihypertensive agents, antiatherosclerotic agents and lipid-lowering agents.
The antidiabetic agent may be selected from biguanides, sulfonylureas, glucosidase inhibitors, PPAR γ agonists, PPAR α/γ dual agonists, aP2 inhibitors, DPP4 inhibitors, insulin sensitizers, glucagon-like peptide-1 (GLP-1) analogs, insulin, and meglitinides.
The antidiabetic agent may be selected from the group consisting of metformin, glyburide, glimepiride, glipizide, chlorpropamide, gliclazide, acarbose, miglitol, pioglitazone, troglitazone, rosiglitazone, moglicazar, insulin, Gl-262570, iglitazone, JTT-501, NN-2344, L895645, YM-440, R-119702, AJ9677, repaglinide, nateglinide, KAD1129, AR-HO39242, GW-409, KRP297, AC2993, LY315902, NVP-DPP-728A, and saxagliptin.
The anti-obesity agent may be selected from beta 3 adrenergic agonists, lipase inhibitors, serotonin (and dopamine) reuptake inhibitors, thyroid receptor beta compounds, CB-1 antagonists, NPY-Y2 or NPY-Y4 receptor agonists, and anorectic agents.
The anti-obesity agent may be selected from orlistat, ATL-962, AJ9677, L750355, CP331648, sibutramine, topiramate, axokine (axokine), dexamphetamine, phentermine, phenylpropanolamine rimonabant (SR141716A), PYY (3-36), Pancreatic Polypeptide (PP), and mazindol.
The lipid lowering agent may be selected from MTP inhibitors, cholesteryl ester transfer protein, HMG CoA reductase inhibitors, squalene synthetase inhibitors, fibric acid (fibric acid) derivatives, upregulators of LDL receptor activity, lipoxygenase inhibitors and ACAT inhibitors.
The lipid lowering agent may be selected from pravastatin, lovastatin, simvastatin, atorvastatin, cerivastatin, fluvastatin, nivastatin (nisvastatin), visastatin, fenofibrate, gemfibrozil, clofibrate, avasimibe (avasimibe), TS-962, MD-700, CP-529414 and LY 295427.
In another aspect, the invention relates to a method of treating or delaying the progression or onset of diabetes, diabetic retinopathy, diabetic neuropathy, diabetic nephropathy, wound healing, insulin resistance, hyperglycemia, hyperinsulinemia, syndrome X, diabetic complications, elevated blood levels of free fatty acids or glycerol, hyperlipidemia, obesity, hypertriglyceridemia, atherosclerosis, or hypertension, comprising administering to a mammal in need of treatment a therapeutically effective amount of an isolated polypeptide described herein.
The method may further comprise the simultaneous or sequential administration of a therapeutically effective amount of one or more therapeutic agents selected from the group consisting of antidiabetic agents, antiobesity agents, antihypertensive agents, antiatherosclerotic agents and lipid-lowering agents.
In another aspect, the invention relates to a method of treating or delaying the progression or onset of diabetes, diabetic retinopathy, diabetic neuropathy, diabetic nephropathy, wound healing, insulin resistance, hyperglycemia, hyperinsulinemia, syndrome X, diabetic complications, elevated blood levels of free fatty acids or glycerol, hyperlipidemia, obesity, hypertriglyceridemia, atherosclerosis, or hypertension, comprising administering a therapeutically effective amount of a pharmaceutical composition described herein to a mammal in need of treatment.
In another aspect, the invention relates to a method of administering a polypeptide described herein, comprising parenteral administration of a formulation comprising a polypeptide described herein.
In another aspect, the invention relates to a method of administering a polypeptide described herein, comprising non-parenteral administration of a formulation comprising a polypeptide described herein.
The parenteral administration may be selected from Intravenous (IV) bolus, IV infusion, subcutaneous, intramuscular, intranasal, buccal, pulmonary, and ocular delivery.
The subcutaneous administration may include the use of an immediate release formulation or a sustained release formulation.
The intramuscular administration may include the use of an immediate release formulation or a sustained release formulation.
The formulation may further comprise pharmaceutically acceptable excipients selected from solvents and co-solvents, solubilizers, emulsifiers, thickeners, chelating agents, antioxidants, reducing agents, antimicrobial preservatives, buffers and pH adjusting agents, bulking agents, protectants and tonicity adjusting agents, and special additives.
The formulation may further comprise an encapsulated delivery system.
Brief description of the drawings
FIG. 1 illustrates the effect of subcutaneous injection of Compound I on plasma glucose in an intraperitoneal glucose tolerance test (ipGTT) in obese ob/ob mice.
FIG. 2 illustrates the effect of subcutaneous injection of Compound I on plasma insulin in ipGTT in ob/ob mice.
FIG. 3 illustrates the subcutaneous injection of the amino acid sequence of SEQ ID NO: 1 on plasma glucose.
Figure 4 illustrates the subcutaneous injection of SEQ ID NO: 58 on plasma glucose.
Detailed Description
The present invention provides novel human glucagon-like peptide-1 (GLP-1) peptide receptor modulators, agonists or partial agonists that exhibit the superior biological properties of the native peptide GLP-1 and that exhibit increased stability to proteolytic cleavage when compared to the GLP-1 native sequence, and are therefore useful for ameliorating diabetic conditions.
The synthetic isolated peptides of the invention and described herein are capable of modulating the GLP-1 receptor, desirably as agonists or partial agonists of the GLP-1 receptor. These synthetic peptides exhibit superior in vivo potency and pharmacokinetic properties relative to GLP-1, including lowering postprandial plasma glucose and simultaneously increasing plasma insulin levels, thus making them ideal candidates for therapeutic agents for subcutaneous, pulmonary, nasal, buccal or sustained release administration.
The invention includes, for example, an isolated polypeptide comprising a polypeptide having a sequence of formula I:
Xaa1-Xaa2-Xaa3-Xaa4-Xaa5-Xaa6-Xaa7-Xaa8-Xaa9-Xaa10-Xaa11
I
wherein,
Xaa1is a naturally or non-naturally occurring imidazole-containing amino acid; wherein one or more carbon atoms of the amino acid are optionally substituted with one or more alkyl groups; wherein the amino acid optionally has a free amino group, which group is optionally substituted with alkyl, acyl, benzoyl, L-lactyl, alkyloxycarbonyl, aryloxycarbonyl, arylalkyloxycarbonyl, heterocyclyloxycarbonyl, heteroarylalkyloxycarbonyl, alkylcarbamoyl, arylcarbamoyl, arylalkylcarbamoyl, heterocyclosulfonyl, alkylsulfonyl, arylsulfonyl, arylalkylsulfonyl, heteroarylalkylsulfonyl, or heteroarylsulfonyl; and wherein when said free amino group is absent, Xaa1A des-amino acid which is histidine wherein one or more carbon atoms of said amino acid is optionally substituted with one or more alkyl groups;
Xaa2is a naturally or non-naturally occurring amino acid selected from the group consisting of D-alanine, alpha-amino-isobutyric acid (Aib), N-methyl-D-alanine, N-ethyl-D-alanine, 2-methyl-azetidine-2-carboxylic acid, alpha-methyl- (L) -proline, 2-methylpiperidine-2-carboxylic acid and isovaline;
Xaa3an amino acid that is naturally or non-naturally occurring and has (1) a carboxylic acid-containing amino acid side chain or (2) an imidazole side chain, and wherein one or more carbon atoms of the amino acid are optionally substituted with one or more alkyl groups;
Xaa4is glycine;
Xaa5is a naturally or non-naturally occurring amino acid selected from the group consisting of (L) -threonine and (L) -norvaline; and wherein one or more carbon atoms of said amino acidOptionally substituted with one or more alkyl groups;
Xaa6an alpha carbon with two side chains that is a naturally or non-naturally occurring amino acid and which has a di-substituted; wherein at least one of the two side chains has an aromatic ring and at least one of the two chains has an alkyl group; and wherein one or more carbon atoms of the amino acid are optionally substituted with one or more alkyl groups or one or more halogen groups.
Xaa7Is a naturally or non-naturally occurring amino acid having an amino acid side chain substituted with a hydroxyl group; and wherein one or more carbon atoms of the amino acid are optionally substituted with one or more alkyl groups;
Xaa8an amino acid that is naturally or non-naturally occurring and selected from L-serine and L-histidine; and wherein one or more carbon atoms of the amino acid are optionally substituted with one or more alkyl groups;
Xaa9is a naturally or non-naturally occurring amino acid having an amino acid side chain comprising a carboxylic acid; and wherein one or more carbon atoms of the amino acid are optionally substituted with one or more alkyl groups;
Xaa10is a naturally or non-naturally occurring amino acid of formula II:

formula II
Wherein R is4Selected from hydrogen, alkyl and halogen;
wherein R is3And R6Each is independently selected from hydrogen, halogen, methyl, ethyl, alkyl, hydroxy, methoxy, and alkoxy;
wherein the phenyl ring proximal to the β -carbon of the amino acid is additionally optionally substituted with hydrogen, alkyl, or halogen; and
wherein the phenyl ring distal to the β -carbon of the amino acid is additionally optionally substituted with hydrogen, halogen, methyl, ethyl, alkyl, hydroxy, methoxy, and alkoxy;
Xaa11is a naturally or non-naturally occurring amino acid of formula IVa:
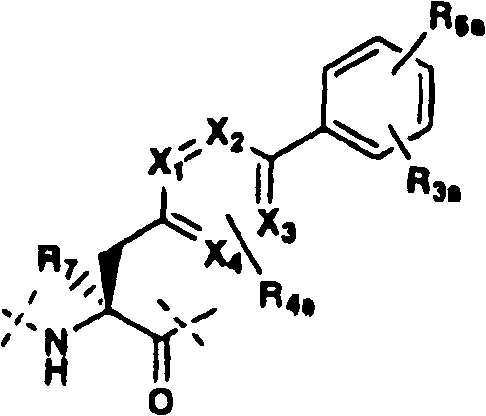
formula IVa
Wherein the C-terminal carbonyl carbon of the amino acid is attached to nitrogen to form a carboxamide (NH)2);
Wherein R is4aSelected from hydrogen, alkyl and halogen;
wherein R is3aAnd R6aEach is independently selected from hydrogen, halogen, methyl, ethyl, alkyl, hydroxy, methoxy, and alkoxy;
wherein R is7Selected from hydrogen, methyl, and ethyl; and
wherein X1、X2、X3And X4Each is C or N, and provided that X1、X2、X3And X4At least one of which is N;
wherein the phenyl ring proximal to the β -carbon of the amino acid is additionally optionally substituted with hydrogen, alkyl, or halogen; and
wherein the phenyl ring distal to the β -carbon of the amino acid is additionally optionally substituted with hydrogen, halogen, methyl, ethyl, alkyl, hydroxy, methoxy, and alkoxy.
Unless otherwise defined in a specific context, the definitions provided herein apply without limitation to the terms used throughout this specification.
Amino acids known to those skilled in the art of amino acid and peptide chemistry include compounds represented by the following general structure:

l-or S-alpha-amino acid D-or R-alpha-amino acid
(if R ═ H)
Wherein R and R' are as discussed herein. Unless otherwise indicated, the term "amino acid" used herein alone or as part of another group includes, but is not limited to, amino groups and carboxyl groups attached to the same carbon called the "α" carbon, where R and/or R' may be a natural or non-natural side chain, including hydrogen. The absolute "S" configuration at the "alpha" carbon is often referred to as the "L" or "native" configuration. In the case where both the "R" and the "R substituent" are hydrogen, the amino acid is glycine and is not chiral.
Unless otherwise indicated, the term "amino-alcohol" used herein alone or as part of another group includes, but is not limited to, natural or unnatural amino acids and wherein the carboxyl group is replaced (reduced) to a carbinol such as valinol, glycinol, alaninol, arylalaninol, heteroarylalaninol.
The term "alkyl" as used herein alone or as part of another group includes, but is not limited to, straight and branched chain hydrocarbons containing from 1 to 40 carbons in the normal chain, preferably from 1 to 20 carbons, more preferably from 1 to 8 carbons, such as methyl, ethyl, propyl, isopropyl, butyl, tert-butyl, isobutyl, pentyl, hexyl, isohexyl, heptyl, 4-dimethylpentyl, octyl, 2, 4-trimethylpentyl, nonyl, decyl, undecyl, dodecyl and various branched chain isomers thereof,and so on. Further, an alkyl group as defined herein may be optionally substituted on any available carbon atom with one or more functional groups typically attached to the chain such as, but not limited to, alkyl, aryl, alkenyl, alkynyl, hydroxy, aralkyl, cycloalkyl, cycloalkylalkyl, alkyloxy, arylalkyloxy, heteroaryloxy, heteroarylalkyloxy, alkanoyl, halogen, hydroxy, thio, nitro, cyano, carboxy, carbonyl (═ O), carboxamide (carboxamido), amino, alkylamino, dialkylamino, amido, alkylamino, arylamido, heteroarylamido, azido, guanidino, amidino, phosphonic (phosphonic), and the like, to form an alkyl group such as trifluoromethyl, 3-hydroxyhexyl, 2-carboxypropyl, 2-fluoroethyl, carboxymethyl, cyanobutyl, and the like, Phosphinic acid (phosphinic), sulfonic acid, sulfonamide, haloaryl, CF3、OCF2、OCF3Aryloxy, heteroaryl, cycloalkylalkyloxyalkyl, cycloheteroalkyl, and the like.
Unless otherwise specified, the term "alkenyl" used herein alone or as part of another group includes, but is not limited to, straight and branched chain hydrocarbons containing from 2 to 40 carbons in the normal chain and having one or more double bonds, preferably from 2 to 20 carbons and having one to three double bonds, more preferably from 2 to 8 carbons and having one to two double bonds, any of which carbons may be optionally substituted as described above for "alkyl".
Unless otherwise specified, the term "alkynyl" used herein alone or as part of another group includes, but is not limited to, straight and branched chain hydrocarbons containing from 2 to 40 carbons in the normal chain and having one or more triple bonds, preferably from 2 to 20 carbons and having one to three triple bonds, more preferably from 2 to 8 carbons and having one to two triple bonds, any of which may be optionally substituted as described above for "alkyl".
Unless otherwise indicated, the term "cycloalkyl" as used herein alone or as part of another group includes, but is not limited to, saturated or partially unsaturated (containing 1 or 2 double bonds) cyclic hydrocarbon groups containing 1 to 3 rings (appended or fused), including monocyclic alkyl, bicyclic alkyl, and tricyclic alkyl groups containing a total of 3 to 20 carbons forming the ring, preferably 4 to 7 carbons forming each ring; which may be fused to 1 aromatic ring such as those described for aryl, including cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl, cyclodecyl, cyclododecyl, cyclohexenyl,
any of which groups may optionally be substituted by one or more groups selected from hydrogen, halogen, haloalkyl, alkyl, haloalkyl, alkoxy, haloalkoxy, alkenyl, trifluoromethyl, trifluoromethoxy, alkynyl, cycloalkylalkyl, fluorenyl, heterocycloalkyl, heterocycloalkylalkyl, aryl, heteroaryl, aralkyl, aryloxy, aryloxyalkyl, arylalkyloxy, arylthio, arylazo, heteroarylalkyl, heteroarylalkenyl, heteroarylheteroaryl, heteroaryloxy, hydroxy, nitro, oxo, cyano, carboxy, carbonyl, through any available carbon atomsCarboxamido, amino, substituted amino where the amino includes 1 or 2 substituents (which are alkyl, aryl, or any other aryl compound mentioned in the definition), amido, azido, guanidino, amidino, phosphonate, phosphinate, sulfonate, sulfonamide, thiol, alkylthio, arylthio, heteroarylthio, arylthioalkyl, alkoxyarylthio, alkylcarbonyl, arylcarbonyl, alkylaminocarbonyl, arylaminocarbonyl, alkoxycarbonyl, aminocarbonyl, alkylcarbonyloxy, arylcarbonyloxy, alkylcarbonylamino, arylcarbonylamino, arylsulfinyl, arylsulfinylalkyl, arylsulfonylamino, or arylsulfonylaminocarbonyl, or the group of any alkyl substituent listed above.
The term "aryl" as used herein alone or as part of another group refers to, but is not limited to, monocyclic and bicyclic aromatic groups containing 6 to 10 carbons in the ring moiety (e.g., phenyl or naphthyl) and may optionally include one to three other rings fused to an "aryl" (e.g., aryl, cycloalkyl, heteroaryl or heterocycloalkyl rings) and may optionally be substituted with one or more substituents selected from hydrogen, alkyl, halo, haloalkyl, alkoxy, haloalkoxy, alkenyl, trifluoromethyl, trifluoromethoxy, alkynyl, cycloalkylalkyl, fluorenyl, heterocycloalkyl, heterocycloalkylalkyl, aryl, heteroaryl, aralkyl, aryloxy, aryloxyalkyl, arylalkyloxy, arylthio, arylazo, heteroarylalkyl, heteroarylalkenyl, heteroaryloxy, heteroarylalkyloxy, heteroarylalkyl, aryl, heteroaryl, and heteroaryl, Heteroarylalkyloxyalkyl, hydroxy, nitro, oxo, cyano, amino, substituted amino wherein the amino comprises 1 or 2 substituents (which are alkyl, cycloalkyl, heterocycloalkyl, heteroaryl, or aryl or any other aryl compound mentioned in the definitions), thiol, alkylthio, arylthio, heteroarylthio, arylthioalkyl, alkoxyarylthio, alkylcarbonyl, arylcarbonyl, alkylaminocarbonyl, cycloalkylaminocarbonyl, arylaminocarbonyl, heteroarylaminocarbonyl, heteroarylalkylaminocarbonyl, alkoxycarbonyl, aminocarbonyl, alkylcarbonyloxy, arylcarbonyloxy, alkylcarbonylamino, arylcarbonylamino, arylsulfinyl, arylsulfinylalkyl, arylsulfonylamino or arylsulfonylaminocarbonyl or any of the alkyl substituents listed above.
The term "aralkyl" as used herein alone or as part of another group refers to, but is not limited to, an alkyl group as defined above having an aryl substituent, such as benzyl, phenethyl, or naphthylpropyl, wherein the aryl and/or alkyl groups may be optionally substituted as defined above.
The terms "alkoxy", "aryloxy", "heteroaryloxy", "arylalkyloxy" or "heteroarylalkyloxy", used herein alone or as part of another group, include, but are not limited to, alkyl or aryl groups, as defined above, attached through an oxygen atom.
As used herein, the term "heterocycle or" heterocyclyl "means, but is not limited to, an unsubstituted or substituted stable 4-, 5-, 6-, or 7-membered monocyclic ring system which may be saturated or unsaturated and which consists of carbon atoms and one to four members selected from nitrogen, sulfur, oxygen, and/or SO2The heteroatoms of the group, wherein the nitrogen and sulfur heteroatoms may optionally be oxidized, and the nitrogen heteroatoms may optionally be quaternized. The heterocyclic ring may be attached at any heteroatom or carbon atom that results in the formation of a stable structure. Examples of such heterocyclic groups include, but are not limited to, tetrahydrofuranyl, tetrahydrothienyl, pyrrolidinyl, piperidinyl, piperazinyl, oxopyrrolidinyl, oxopiperazinyl, oxopiperidinyl, anda diazolyl group. The heterocyclic group may be optionally substituted with one or more functional groups such as those described for "alkyl" or "aryl".
The term "heterocycloalkyl" as used herein alone or as part of another group refers to, but is not limited to, an alkyl group as defined above having a heterocycloalkyl substituent, wherein the "heterocycle" and/or alkyl group may be optionally substituted as defined above.
As used herein, the term "heteroaryl" refers to, but is not limited to, a 5-, 6-, or 7-membered ring containing one or more members selected from nitrogen, sulfur, oxygen, and/or SO2Aromatic heterocycles of heteroatoms of the group. Such a ring may be fused to another aryl or heteroaryl ring and includes possible N-oxides; examples of such heteroaryl groups include, but are not limited to, furan, pyrrole, thiophene, pyridine, pyrimidine, pyrazine, pyridazine, isozyme Azole,Oxazoles, imidazoles, and the like. The heteroaryl group may be optionally substituted with one or more functional groups typically attached to the chain, such as those described for "alkyl" or "aryl".
Azole,Oxazoles, imidazoles, and the like. The heteroaryl group may be optionally substituted with one or more functional groups typically attached to the chain, such as those described for "alkyl" or "aryl".
The term "heteroarylalkyl" as used herein alone or as part of another group refers to, but is not limited to, an alkyl group as defined above having a heteroaryl substituent, wherein the heteroaryl and/or alkyl group may be optionally substituted as defined above.
The term "alkoxycarbonyl", as used herein alone or as part of another group, refers to, but is not limited to, an alkyl group as defined above attached to the oxygen of the-oc (o) -group, e.g., CH3OC(O)-、CH3CH2OC (O) -or CH2(OH)CH2OC(O)-。
The term "aryloxycarbonyl" as used herein alone or as part of another group, refers to, but is not limited to, an aryl group as defined above attached to the oxygen of the-oc (o) -group.
The term "arylalkyloxycarbonyl" as used herein alone or as part of another group refers to, but is not limited to, an arylalkyl group as defined above attached to the oxygen of the-oc (o) -group.
The term "heterocyclyloxycarbonyl" as used herein alone or as part of another group refers to, but is not limited to, a heterocyclyl group as defined above attached to the oxygen of the-oc (o) -group through any carbon atom of the heterocyclyl group.
The term "heterocyclyloxycarbonyl" as used herein alone or as part of another group refers to, but is not limited to, a heterocyclyl group as defined above attached to the oxygen of the-oc (o) -group through any carbon atom of the heterocyclyl group.
The term "heteroarylalkyloxycarbonyl," as used herein alone or as part of another group, refers to, but is not limited to, a heteroarylalkyl group, as defined above, attached to the oxygen of the-oc (o) -group through any carbon atom of the heterocyclyl group.
The term "alkylcarbamoyl", as used herein alone or as part of another group, refers to, but is not limited to, an alkyl group as defined above, e.g., CH, attached to the nitrogen of a-NC (O) -group3NHC(O)-、CH3CH2NHC (O) -or (CH)3)2Nhc (o) -, and wherein when 2 alkyl groups are present, said alkyl groups may optionally be linked to form a 4,5, 6 or 7 membered ring, for example,

the term "arylalkylcarbamoyl" as used herein alone or as part of another group refers to, but is not limited to, an arylalkyl group, as defined above, attached to the nitrogen of the-nc (o) -group.
The term "heterocyclylcarbamoyl" as used herein, alone or as part of another group, refers to, but is not limited to, a heterocyclyl group, as defined above, attached to the nitrogen of a-nc (o) -group.
The term "alkylsulfonyl" as used herein alone or as part of another group means but is not limited to a linkage to-S (O)2Alkyl radicals, as defined above, of sulfur of the radical, e.g. CH3S(O)2-、CH3CH2S(O)2-or (CH)3)2CH2S(O)2-。
The term "arylsulfonyl," used herein alone or as part of another group, refers to but is not limited to a linkage to-S (O)2The aryl radical as defined above of the sulphur of the radical.
The term "arylalkyl sulfonyl", used herein, alone or as part of another group, means, but is not limited to, a linkage to-S (O)2An aralkyl radical as defined above for the sulphur of the radical.
The term "heteroarylsulfonyl", as used herein, alone or as part of another group, means, but is not limited to, a linkage to-S (O)2-the heteroaryl group of the sulfur of the group as defined above.
The term "heteroarylalkylsulfonyl" as used herein alone or as part of another group means but is not limited to a linkage to-S (O)2-the heteroarylalkyl radical as defined above of the sulfur of the radical.
The term "receptor modulator" refers to a compound that acts on the GLP-1 receptor to alter its ability to modulate downstream signaling events. Examples of receptor modulators include agonists, antagonists, partial agonists, inverse agonists, allosteric antagonists, and allosteric potentiators as defined in standard Pharmacological textbooks (e.g., e.m.ross and t.p.kenakin Goodman and Gilman's the Pharmacological Basis of Therapeutics, 10th edition (2001) McGraw Hill, Chapter 2, pp.31-43).
Those skilled in the art will readily understand the meanings of these terms as provided in the present application and in the art.
The term "diabetes and related diseases or conditions" refers to, but is not limited to, type II diabetes, type I diabetes, glucose intolerance, obesity, hyperglycemia, syndrome X, metabolic dysfunction syndrome, diabetic complications, and hyperinsulinemia.
The term "lipid modulating agent" or "lipid lowering agent" as used herein refers to, but is not limited to, agents that lower LDL and/or raise HDL and/or lower triglycerides and/or lower total cholesterol and/or other known mechanisms for the therapeutic treatment of lipid disorders.
Administration of the therapeutic agents of the present invention includes, but is not limited to, administration of a therapeutically effective amount of the agents of the present invention. As used herein, the term "therapeutically effective amount" refers to, but is not limited to, the amount of a therapeutic agent that treats or prevents a condition that can be treated by administration of a composition of the present invention. The amount is an amount sufficient to exhibit a measurable therapeutic or prophylactic or ameliorating effect. The effect may include, for example, but is not limited to, the treatment or prevention of the conditions listed herein. The precise effective amount for a subject will depend upon the subject's individual size and health, the nature and extent of the condition to be treated, the recommendations of the treating physician, and the selected treatment or combination of treatments for administration. Therefore, it is not useful to specify an exact effective amount in advance.
The relative effect of the peptides of the invention in vivo was evaluated in a glucose tolerance test in ob/ob mice, as described in example 22 below. The peptides of the invention show superior potency and superior pharmacokinetics (measured by subcutaneous injection in dogs, as described in example 25) in the glucose-lowering potency model relative to the peptides exemplified by compound I from WO 2003/033671, WO 2003/033671, which is incorporated herein by reference in its entirety, as shown in tables 1 and 2:
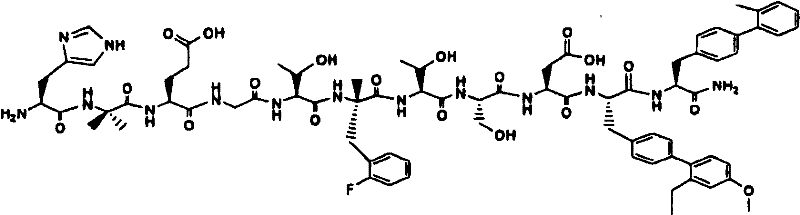
compound I
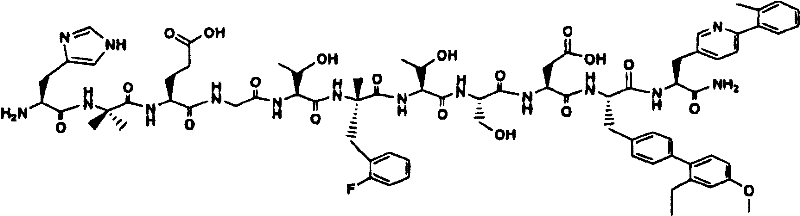
Compound II
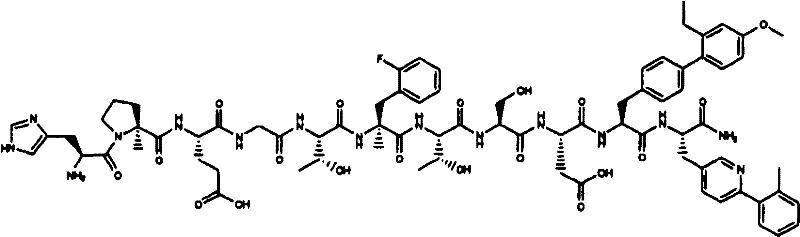
Compound III
Compound 139
TABLE 1
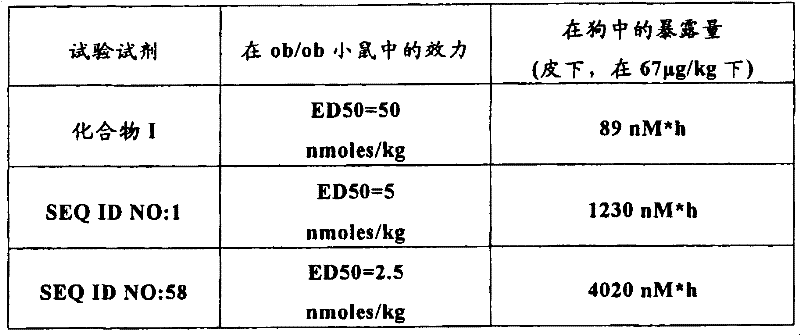
TABLE 2
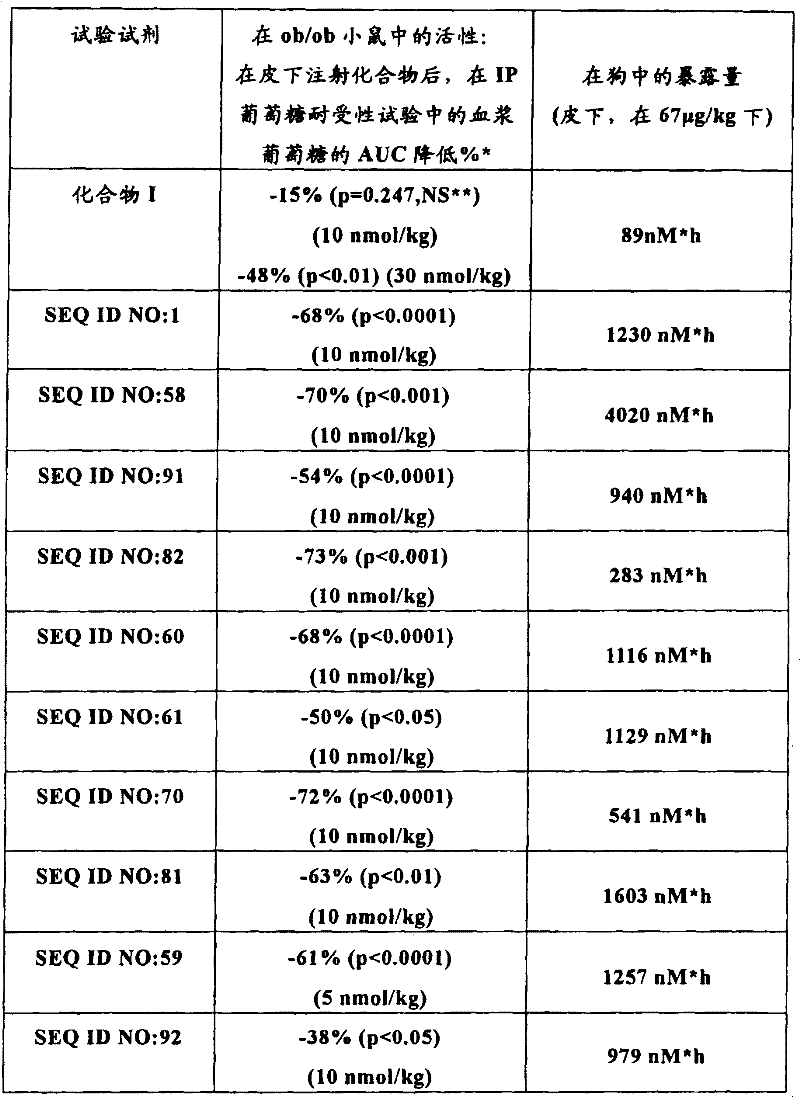
AUC is the area under the curve. AUC values were calculated using fasting blood glucose values as baseline for each individual animal. Percent change in AUC was calculated relative to the AUC of vehicle treated groups in the same study. The p-values given are determined by analysis of variance (ANOVA) with vehicle-treated groups followed by Fisher's post-hoc testing, with no statistically significant difference in NS.
The Peptides and analogs thereof described herein can be prepared by chemical synthesis using various solid phase techniques, such as those described in g.barany and r.b.merrifield, "The Peptides: analysis, Synthesis, Biology "; volume 2- "Special Methods in PeptideSeynthesis, Part A", pp.3-284, E.Gross and J.Meienhofer, eds., Academic Press, New York, 1980; and J.M.Stewart and J.Young, "Solid-Phase Peptide Synthesis" 2ndEd., Pierce Chemical co., Rockford, IL, 1984.
The desired strategy for use in The present invention is based on an Fmoc (9-fluorenylmethyloxycarbonyl) Group for temporary protection of The alpha-Amino Group in combination with a tert-butyl Group for temporary protection of The Amino acid side chain (see, e.g., E.Atherton and R.C.Shepard, "The fluorinyl methyl Amino Protecting Group", in "The peptides: Analysis, Synthesis, Biology"; Volume 9- "specialty methods Peptide Synthesis, Part C", pp.1-38, S.Unfriend and J.Meienhofer, eds., Academic Press, Sandiego, 1987.
The peptides of the invention can be synthesized in a stepwise manner starting from the C-terminus of the peptide on an insoluble polymer support (also referred to as a "resin"). The synthesis is initiated by attaching the C-terminal amino acid of the peptide to the resin via formation of an amide or ester bond. This allows the final release of the resulting peptide in the form of a C-terminal amide or carboxylic acid, respectively. Alternatively, if a C-terminal amino alcohol is present, the C-terminal residue as described herein may be attached to a 2-methoxy-4-alkoxybenzyl alcohol resin (SASRIN)TMBachem Bioscience, Inc., King of Prussia, PA) and, after assembly of the peptide sequence is complete, LiBH is used4The THF solution of (a) released the resulting peptide alcohol (see j.m. stewart and j.d. young, supra, page 92).
For the C-terminal amino acid used in the synthesis, as well as all other amino acids, it is necessary that their alpha-amino group and side chain functional groups (if present) be differentially protected, so that the alpha-amino protecting group can be selectively removed during the synthesis. The coupling of the amino acid is carried out by activating its carboxyl group in the form of an activated ester and reacting it with the non-blocked alpha-amino group of the N-terminal amino acid attached to the resin. The sequence of deprotection and coupling of the alpha-amino group is repeated until the entire peptide sequence is assembled. The peptide is then released from the resin and the side chain functionalities are deprotected simultaneously, typically in the presence of a suitable scavenger to limit side reactions. The resulting peptide was finally purified by reverse phase HPLC.
The synthesis of the peptidyl-resin required as precursor of the final peptide uses a commercially available cross-linked polystyrene polymer resin (Novabiochem, San Diego, Calif.; applied biosystems, Foster City, Calif.). Preferred solid supports for use in the present invention are: 4- (2 ', 4' -dimethoxyphenyl-Fmoc-aminomethyl) -phenoxyacetyl-p-methylbenzhydrylamine resin (Rink amide MBHA resin), 9-Fmoc-amino-xanthen-3-yloxy-Merrifield resin (Sieber amide resin), 4- (9-Fmoc) aminomethyl-3, 5-dimethoxyphenoxy) valeryl-aminomethyl-Merrifield resin (PAL resin) for C-terminal carboxamides. The first and subsequent coupling of amino acids can be accomplished using HOBT or HOAT active esters prepared from DIC/HOBT, HBTU/HOBT, BOP, PyBOP or from DIC/HOAT, HATU/HOAT, respectively. Preferred solid supports for use in the present invention are: 2-chlorotrityl chloride resin and 9-Fmoc-amino-xanthen-3-yloxy-Merrifield resin for protected peptide fragments (Sieber amide resin). The loading of the first amino acid on the 2-chlorotrityl chloride resin is best achieved by reacting the Fmoc-protected amino acid with the resin in dichloromethane and DIEA. If necessary, a small amount of DMF may be added to facilitate the dissolution of the amino acid.
The synthesis of the 11-mer peptide analogs described herein can be performed by using a peptide synthesizer such as the Advanced Chemtech multiple peptide synthesizer (MPS396) or the Applied Biosystems inc. If the MPS396 is used, up to 96 peptides are synthesized simultaneously. If the ABI 433A synthesizer was used, the individual peptides were synthesized sequentially. In both cases, the stepwise solid phase peptide synthesis was performed using the Fmoc/t-butyl protection strategy described herein.
Will be present at position-X in one of two waysaa11And position-Xaa10Is incorporated into the peptide chain. In the first method, Boc-or Fmoc-protected unnatural amino acids are prepared in solution using appropriate organic synthesis methods. The resulting derivative is then used in the stepwise synthesis of the peptide. Alternatively, the resin is built directly using synthetic organic chemistry methodsDesired unnatural amino acids. When a non-natural non-commercially available amino acid is desired for use at position Xaa6Or at any other XaaThe desired Fmoc-protected unnatural amino acid is synthesized in solution while positionally incorporated. This derivative is then used in stepwise solid phase peptide synthesis.
The Fmoc amino acid derivatives shown below are desirable for use in the present invention.
Examples of orthogonally protected amino acids for use in solid phase synthesis

Examples of protected amino acids for use in solid phase synthesis

Any standard method can be used to cleave and deprotect the peptidyl-resin precursor for its corresponding peptide (see, e.g., d.s.king et al,Int.J.Peptide Protein Res.36, 1990, 255-266). A desirable method for use in the present invention is to use TFA as a scavenger in the presence of water and TIS. Typically, the peptidyl-resin is stirred in TFA/water/TIS (94: 3, v: v; 1mL/100mg peptidyl-resin) at room temperature for 2-6 hours. The used resin was then filtered off and the TFA solution was concentrated or dried under reduced pressure. The resulting crude peptide was precipitated and Et2O washes or redissolved directly into DMSO or 50% aqueous acetic acid for purification by preparative HPLC.
The peptide can be obtained in the desired purity by purification using preparative HPLC, for example, on a Waters Model 4000 or Shimadzu Model LC-8A liquid chromatograph. The solution of the crude peptide was injected into a column of YMC S5ODS (20X 100mm) and eluted with a linear gradient of aqueous MeCN, both buffered with 0.1% TFA, using a flow rate of 14-20mL/min and the effluent monitored using UV absorbance at 220 nm. The structure of the purified peptide can be confirmed by electrospray mass spectrometry.
The following abbreviations are used in the examples and elsewhere herein:
Ph-phenyl-THF-tetrahydrofuran
Bn ═ benzyl TFA ═ trifluoroacetic acid
i-Bu ═ isobutyl TFE ═ alpha, alpha-trifluoroethanol
i-Pr ═ isopropyl Et2NH ═ diethylamine
Me-methyl-NMM-N-methylmorpholine
Et-ethyl NMP-N-methylpyrrolidone
Pr ═ n-propyl DCM ═ dichloromethane
Bu ═ n-butyl n-BuLi ═ n-butyl lithium
TMS-trimethylsilyl Pd/C-palladium/carbon
TIS-triisopropylsilane PtO2Platinum oxide (II)
Et2O-diethyl ether TEA-triethylamine
HOAc or AcOH acetic acid min
h or hr-hour
MeCN or CH3CN is acetonitrile L is liter
mL or mL-mL
DMF ═ N, N-dimethylformamide ═ L ═ microliter
g is g ═ g
EtOAc ═ ethyl acetate mg ═ mg
mol RT or RT at room temperature
mmol-millimole sat or sat'd-saturated
meq. aq. milliequivalents aqueous
mp is melting point
Bip ═ biphenylalanine
LiBH4Lithium borohydride
NBS ═ N-bromosuccinimide
BOP reagent ═ benzotriazole-1-yloxy-tris-dimethylamino- Hexafluorophosphate (Castro's reagent)
Hexafluorophosphate (Castro's reagent)
PyBOP reagent ═ benzotriazol-1-yloxy-tripyrrolidinyl Hexafluorophosphates
Hexafluorophosphates
HBTU-2- (1H-benzotriazol-1-yl) -1, 1,3, 3-tetramethylureaHexafluorophosphates
HATU ═ O- (7-azabenzotriazol-1-yl) -1, 1,3, 3-tetramethylurea Hexafluorophosphates
Hexafluorophosphates
HCTU 2- (6-chloro-1-H-benzotriazol-1-yl) -1, 1,3, 3-tetramethylurea Hexafluorophosphates
Hexafluorophosphates
DMAP ═ 4- (dimethylamino) pyridine
DIEA is diisopropylethylamine
EDAC ═ 3-ethyl-3' - (dimethylamino) propyl-carbodiimide hydrochloride (or 1- [ (3- (dimethyl) amino) propyl ]) -3-ethylcarbodiimide hydrochloride)
Fmoc or FMOC ═ fluorenylmethoxycarbonyl
Boc or BOC ═ tert-butoxycarbonyl
Cbz ═ benzyloxycarbonyl or benzyl ester group or benzyloxycarbonyl group
HOBT or HOBT. H2O-1-hydroxybenzotriazole hydrate
Cl-HOBt ═ 6-chloro-benzotriazole
HOAT ═ 1-hydroxy-7-azabenzotriazole
TLC (thin layer chromatography)
HPLC ═ high performance liquid chromatography
LC/MS-HPLC/Mass Spectrometry
MS or Mass Spec ═ Mass spectrometry
NMR (nuclear magnetic resonance)
Sc or SC ═ subcutaneous
IP or IP-intraperitoneal
GTT ═ glucose tolerance test
NBS ═ N-bromosuccinimide
General procedure for the synthesis of amino acids of formula IVa.
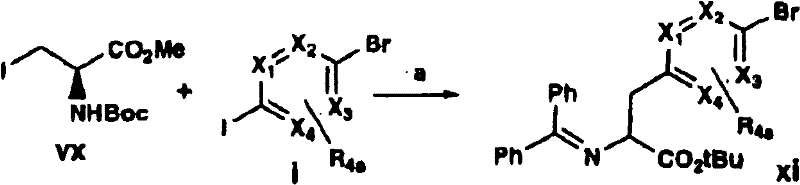
The protected amino acids of formula IVa can be prepared in several ways. For example (scheme A), iodine can be brominated-heterocycle i (where X is a ligand of formula I) via palladium-mediated catalysis according to standard literature procedures3N) with boronic acids to provide aryl heterocyclic bromides ii which are reacted by lithiation (lithiation) and with an acylating agent such as dimethylformamide to give aldehydes iii. The aldehyde is reduced to the alcohol iv with sodium borohydride or similar reagent and the corresponding bromide v is prepared by continuously refluxing iv in 48% hydrobromic acid. T-butyl 2- (benzhydrylideneamino) acetate is alkylated with v using a chiral catalyst according to the O' Donnell method (Tetrahedron Letters 398775(1998)) to give chiral esters vi which, after deprotection with a strong non-aqueous acid, are treated with FmocCl to give Fmoc t-butyl esters vii which are predominantly in one chiral form. Vii is recrystallized from common organic solvents to provide viii in enantiomeric excess (enantiomeric excess) > 95%. Removing the ester using a strong non-aqueous acid to provide the compound of formula IVa.
Alternatively, compounds of formula IVa can be prepared by subjecting methyl heterocycle ix to free radical induced bromination (scheme B) to give bromomethyl heterocycle x. The chiral ester xiii with high enantiomeric excess is produced by alkylation of x using the O' Donnell method described above and similar recrystallization. Boronic acid couplings are carried out as described in scheme a to yield compounds of formula IVa.
Flow chart A
a)R3R6C6H3B(OH)2,Pd(Ph3P)4Toluene/10% Na2CO3b) s-BuLi, DMF/toluene c) NaBH4MeOH d) 48% HBr, reflux e) PhC ═ NCH2CO2tBu, chiral catalyst, 2-tert-butylimino-2-diethylamino-1, 3-dimethyl-perhydro-1, 3, 2-diazaphosphorine/THFf) i.15% citric acid ii.FmocCl Na2CO3/THF-H2Og) recrystallization of h) TFA
Flow chart B
a)NBS,AIBN/CCl4b)PhC=NCH2CO2tBu, chiral catalyst, 2-tert-butylimino-2-diethylamino-1, 3-dimethyl-perhydro-1, 3, 2-diazaphosphorine/THFc) i.15% citric acid ii.FmocCl Na2CO3/THF-H2O d) recrystallizing e) R3R6H3B(OH)2,Pd(Ph3P)4Toluene-10% Na2CO3f)TFA
Compound ix can be prepared from hydroxy heterocyclic xiv by treatment with phosphoryl bromide (scheme C).
Flow chart C
An alternative synthesis for intermediate ix is to use xv, 3-iodo-alanine methyl ester, and i for zinc-copper coupling (scheme D).
Flow chart D
a)Zn-Cu(Ph3P)2PdCl2Benzene, DMA
Arylpyrimidinylmethyl bromide xxiii (X)2、X3=N,X1、X4=CR4a) Can be prepared from aryl nitriles xv (scheme E).
Flow chart E

Hydroxypyrimidines xvi are prepared from xv by treating the nitrile with hydroxylamine hydrochloride. The pyrimidine xvii results from the hydrogenation of xvi. Xvii is condensed with an enolmethylmalonate to yield a pyrimidine xix, which is chlorinated with phosphorus oxychloride to yield xx. Dehalogenation via catalytic hydrogenation to give xxi, followed by reduction with DiBAl to give alcohol xxii. Treatment of the alcohol with phosphoryl bromide to form the labile bromide xxiii, which must be used immediately as shown in scheme a to give the protected amino acid vi.
Prepared by the method of Kapadia, J.org.chem.661903(2001) Preparation of Compounds of formula IVa (R) by oxazolidine xxiv7Me) (scheme F). Thus, xxiv is alkylated at v using potassium hexamethyldisilazide (potassium hexamethyldisilazide) or other strong base to give xxv. Xxv is subjected to a strong acid hydrolysis, after which the amine is protected (with FmocCl or FmocOSu or the like), thus obtaining a compound of the type of formula IVa.
Preparation of Compounds of formula IVa (R) by oxazolidine xxiv7Me) (scheme F). Thus, xxiv is alkylated at v using potassium hexamethyldisilazide (potassium hexamethyldisilazide) or other strong base to give xxv. Xxv is subjected to a strong acid hydrolysis, after which the amine is protected (with FmocCl or FmocOSu or the like), thus obtaining a compound of the type of formula IVa.
Flow chart F
Amino acids are known to those skilled in the art of peptide chemistry to exist as both D and L isomers, and the present invention includes the use of one isomer or a mixture of isomers of an amino acid for incorporation into the synthesis of the peptides described herein.
Example 1
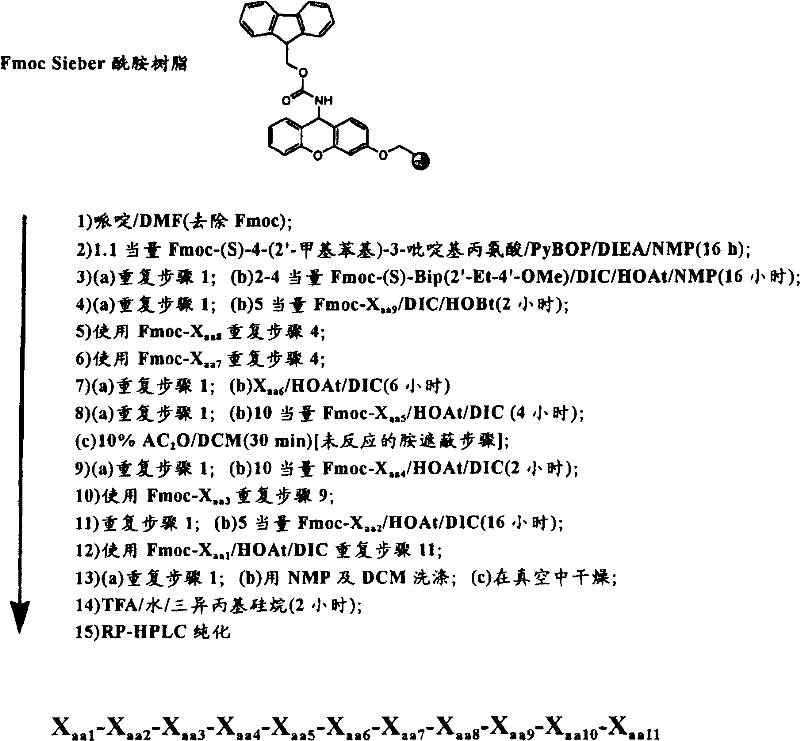
Simultaneous solid phase peptide synthesis of 11-mer peptides
Prepared in batch mode at position X using the following manual procedureaa10And Xaa11A dipeptide based resin containing amino acids followed by continuous peptide chain extension using an automated simultaneous synthesis protocol on an MPS-396 peptide synthesizer. The synthesis of the N- α -Fmoc-protected biphenylalanine or phenyl-heteroaryl-alanine derivatives used in the manual coupling is described in the general experimental procedure described above, and as described in examples 10-16 and examples 21-22.
An amount of 9-Fmoc-aminoxanthen-3-yloxy-Merrifield resin (Sieber amide resin; loading: 0.5 to 0.7mmol/g) sufficient to synthesize several 11-mer analogs was swollen by washing with DMF (4X 10mL/g, 5 min). The Fmoc group was then removed by treatment twice with 20% piperidine in DMF (10mL/g) for 5 and 15 minutes, respectively. The resin was washed with DMF (4X 10mL/g) and NMP (4X 10 mL/g). A solution of 0.5M Fmoc-L-4- (2' -methylphenyl) -3-pyridylalanine-OH (HCl salt) (1.1 eq), (or any other amino acid of formula IVa), PyBOP (1.1 eq) and DIEA (3.3 eq) in NMP was added to the resin. The resin is then shaken or vortexed for 16-24 hours. The completion of the coupling was monitored using the qualitative ninhydrin test. The resin was drained, washed with NMP (3X 10mL/g) and DMF (3X 10mL/g), and treated with 10% acetic anhydride in DCM (10mL/g) for 90 minutes. After DCM washing (4X 10mL/g), a second manual coupling cycle using DIC/HOAt mediated was then performed, starting with the removal of the Fmoc group with 20% piperidine in DMF and the Fmoc-protected biphenylalanine analogue as shown in formula II was used in the coupling step. The required dipeptidyl-Sieber amide resin protected by Fmoc is prepared by the synthetic process.
This dipeptidyl-resin, required for the synthesis of the designed analogue set, was then used in the automated MPS synthesis for up to 96 peptides per round of operation in the following manner. The dipeptidyl-resin was loaded into the 96-well reactor of the Advance ChemTech MPS396 synthesizer as a suspension in dichloromethane/DMF (60: 40) in an amount equivalent to 0.01-0.025mmol (20-50mg) of resin per reactor well. The reactor was placed on the instrument and drained. The wells were then washed with DMF (0.5-1.0mL, 3 × 2min) and several automated coupling cycles required to assemble the respective peptide sequences were performed as determined from a pre-programmed sequence synthesis table.
A detailed step-by-step synthetic scheme for a typical 0.025 mmol/well simultaneous synthesis of 96 compounds is described below. This procedure is suitable for simultaneous synthesis of sets of 12 to 96 analogs per single round of operation. A general synthetic scheme is depicted in scheme 1.
Prior to starting the synthesis, the following reagent solutions were prepared and placed on the instrument as needed: 1.5M (15%) piperidine in DMF, 0.5M DIEA in NMP, 0.36M DIC in NMP, 1M (10%) acetic anhydride in DMF. The desired Fmoc protected amino acid was prepared as a 0.36M solution in 0.36M HOAt/NMP and placed in position in the 32-position amino acid scaffold.
The Fmoc-protected dipeptidyl-resin prepared above was deprotected by treatment with 20% piperidine in DMF (1.0 mL; 1X 5 min; 1X 15 min). The resin was then washed with NMP (8X 1.0 mL).
The coupling of the next amino acid, typically Fmoc-Asp (OtBu) -OH or another Fmoc-amino acid appropriately orthogonally protected if required, was performed by manually adding a solution of the appropriate Fmoc-amino acid (0.075mmol, 3.0 equivalents), HCTU (0.075mmol, 3.0 equivalents) and DIEA (0.15mmol, 6.0 equivalents) in NMP (1mL) to all wells. The coupling was allowed to proceed for 3 hours. After draining the reactor with nitrogen pressure (3-5psi), the wells were washed with NMP (4X 1.0 mL).
The next coupling cycle begins with the removal of the Fmoc group as described above and involves the coupling of Fmoc-Ser (tBu) -OH or a different Fmoc-amino acid as required for the sequence substitution required at that position. The coupling was carried out in the same manner as described for Fmoc-Asp (OtBu) -OH. The next coupling step is carried out in the same way to incorporate Fmoc-Thr (tBu) -OH or any other selected Fmoc-amino acid in the desired position of this sequence.
The next Fmoc-amino acid (e.g., Fmoc- α -methyl-Phe-OH or analog thereof) is coupled as follows: after Fmoc deprotection in the usual way, Fmoc-amino acids (1-5 equiv.), HOAt (1-5 equiv.), and DIC (1-5 equiv.) were added manually as a solution in NMP (1.0mL) and the coupling was allowed to proceed for 16-24 hours. In this case the coupling is not repeated. After a conventional post-coupling wash, the peptidyl-resin is masked with acetic anhydride as described herein.
The next coupling step involves Fmoc-Thr (tBu) -OH or a substituted analogue with sequence substitutions at this position as required. The coupling was performed as described for the original Fmoc-Asp- (OtBu) -OH and MPS coupling of analogues thereof, except that 10 equivalents of Fmoc-Thr- (OtBu) -OH or substituted analogue were used and the coupling was allowed to proceed for 16 hours and the coupling reagent used was DIC/HOAt in NMP. After a conventional post-coupling wash, the peptidyl-resin was masked with 10% acetic anhydride in DCM (1 × 1mL × 60 mins.).
The same coupling procedure described for the coupling of Fmoc-Asp (OtBu) -OH was repeated for the coupling of the next three amino acid residues. To complete the sequence assembly of the desired 11-mer peptide analogs, Fmoc-His (Trt) -OH was coupled as described for the Fmoc-Thr (tBu) -OH residues in the above paragraphs. For coupling of the desired commercial and non-commercial unnatural amino acids at a sequence position, a similar pair as above was used at position 6 (X)aa6) The novel amino acids of (a) describes a single coupling scheme for those.
Finally, the Fmoc group was removed as described above with 20% piperidine in DMF and the peptidyl-resin was washed with DMF (4 × 1.0mL) and DCM (4 × 1.0 mL). They were then dried on the reactor block by applying nitrogen (5psi) at constant pressure for 10-15 min.
a. Cleavage/deprotection.
The desired peptides were cleaved/deprotected from their respective peptidyl-resins by treatment with TFA cleavage cocktail as follows. A TFA/DCM/triisopropylsilane solution (70: 28: 2) (1.0mL) was added to each well in the reactor block, which was then vortexed for 10 mins. This operation was repeated two more times and the TFA solution in the well was collected by positive pressure into a pre-prepared equilibrium vial located within a matched 96-vial stack on the bottom of the reactor. The vial was capped and gently vortexed for an additional 90 minutes. The vial was uncapped and placed in a SpeedVacTM(Savant) to a volume of about 0.2 mL. The crude peptide was then precipitated by adding diisopropyl ether (3mL) and vortexing it briefly. The pellet was pelleted by centrifugation and the supernatant decanted. The vial was placed in a SpeedVacTM(Savant) to give the crude peptide, generally in > 100% yield(20-40 mgs). The crude peptide was directly dissolved in 2mL of 0.6% ammonium hydroxide for purification by preparative HPLC as follows.
b. Preparative HPLC purification of the crude peptide.
Preparative HPLC was performed on a Waters Model 4000 or Shimadzu Model LC-8A liquid chromatograph. Each crude peptide solution was injected onto a column of YMC S5ODS (20X 100mm) and eluted with a linear gradient of aqueous MeCN, both buffered with 0.1% TFA. A typical gradient used was from 20% to 50% 0.1% TFA/MeCN in 0.1% TFA/water during 15min and a flow rate of 14mL/min and effluent UV detection at 220 nm. After 10-11min, generally, the eluted desired product separates well from impurities and is typically collected as a single fraction of 10-15mL on a fraction collector. The desired peptides were obtained as amorphous white powders by freeze-drying their HPLC fractions.
c. HPLC analysis of the purified peptide.
After purification by preparative HPLC as described above, each peptide was analyzed by analytical RP-HPLC on a Shimadzu LC-10AD or LC-10AT analytical HPLC system consisting of an SCL-10A system controller, SIL-10A auto-injector, SPD10AV or SPD-M6A UV/VIS detector, or SPD-M10A diode array detector. Elution was performed using a YMC ODS S3 (4.6X 50mm) column and with one of the following gradients: use 10-70% B in solution A during 8min, 2.5mL/min (method A); use 5-80% B in solution a during 8min, 2.5mL/min (method B); use 5-70% B in a solution during 8min, 2.5mL/min (method C); use 25-75% B in a solution during 8min, 2.5mL/min (method D); use 20-75% B in a solution during 8min, 2.5mL/min (method E); use 15-70% B in a solution during 8min, 2.5mL/min (method F); use 10-90% B in a solution during 8min, 2.5mL/min (method G); use 20-65% B in a solution during 8min, 2.5mL/min (method H); use 5-90% B in a solution during 8min, 2.0mL/min (method I); use 5-90% B in a solution during 8min, 2.5mL/min (method J); use 20-80% B in a solution during 8min, 2.5mL/min (method K); use 10-100% B in a solution during 8min, 2.5mL/min (method L); use 10-75% B in a solution during 8min, 2.5mL/min (method M). Mobile phase A: 0.1% TFA/water; mobile phase B: 0.1% TFA/acetonitrile. The purity is generally > 90%.
d. Characterization was by mass spectrometry.
Each peptide was characterized by electrospray mass spectrometry (ES-MS) in either flow injection or LC/MS mode. Finnigan SSQ7000 single quadrupole mass spectrometer (ThermoFinnigan, San Jose, Calif.) was used in all analyses in positive and negative ion electrospray mode. Full scan data was obtained at a scan time of 1.0 second over a mass range of 300 to 2200 amu. The quadrupole operates at unit resolution. For flow injection analysis, the mass spectrometer was connected to a Waters 616HPLC pump (Waters corp., MiIford, MA) and equipped with an HTS PAL autosampler (CTC analytical, Zwingen, Switzerland). The sample was injected into a mobile phase containing 50: 50 water: acetonitrile with 0.1% ammonium hydroxide. The flow rate for the assay was 0.42mL/min and the injection volume was 6 μ Ι. Thermoseparation Constameric 3500 liquid chromatograph (Thermoseparation products, San Jose, Calif.) and HTS PAL autosampler were used for LC/MS analysis. Use of Luna C18Chromatography was carried out on a 5 micron column, 2X 30mm (Phenomenex, Torrance, Calif.). The flow rate for the analysis was 1.0mL/min and the column effluent was split such that the flow rate into the electrospray interface was 400 μ l/min. a linear gradient of a solution from 0% to 100% B was run during 4 minutes, with mobile phase a being 98: 2 water: acetonitrile with 10mM ammonium acetate and mobile phase B being 10: 90 water: acetonitrile with 10mM ammonium acetate. The UV response was monitored at 220 nm. The sample was dissolved in 200. mu.l 50: 50H2MeCN (0.05% TFA). The injection volume was 5 μ l.
In all cases, the molecular weight determined by the experiment was within 0.5 daltons of the calculated monoisotopic molecular weight.
Example 2
A. General procedure for the synthesis of N-acylated 11-mer peptide analogs (scheme 2).
As shown in scheme 2, the synthesis of the N-acylated 11-mer peptide analog begins with the protected 11-mer peptidyl-resin intermediate (1) (0.015mmol) prepared in the manner described herein. The Fmoc group was removed using the methods described herein, and the resulting resin intermediate 2 was coupled with the relevant Fmoc-protected amino acid or carboxylic acid using the coupling scheme described in the general methods described herein. In the case where the appropriate anhydride is available, the N-acylation is carried out using a solution of 5 equivalents of the anhydride in NMP. The resulting N-acylated 11-mer analog (3) was cleaved/deprotected and purified by preparative HPLC as described generally herein.
The flow chart is as follows: synthesis of residue # 1 substituted/derivatized 11-mer peptide analogs

B. General procedure for the synthesis of N-carbamate derivatives of 11-mer peptide analogs
The method is carried out.
The synthesis of the N-carbamate derivative of the 11-mer peptide analog can begin with the protected 11-mer peptidyl-resin intermediate (1) (0.015mmol) prepared in the manner described herein. The Fmoc group is removed using the methods described herein and the resulting resin intermediate 2 is reacted with the relevant chloroformate in the presence of a suitable base such as a tertiary amine, or with a di-carbonate or activated carbonate such as p-nitrophenyl or phenyl carbonate or hydroxy-succinimidyl carbonate.
C. General procedure for the synthesis of N-urea derivatives of 11-mer peptide analogs.
The synthesis of the N-urea derivative of the 11-mer peptide analog can begin with the protected 11-mer peptidyl-resin intermediate (1) (0.025mmol) prepared in the manner described herein. The Fmoc group is removed using the methods described herein and the resulting resin intermediate 2 is reacted with a corresponding resin as described, for example, in k.burgess et al,J.Am.Chem.Soc.1997, 119, 1556 and 1564; alternatively, the resin intermediate 2 may be reacted with the relevant carbamoyl chloride. Similarly, an N-urea derivative of a 10-polymeric peptide analogue can be prepared starting from a protected 10-polymeric peptidyl-resin intermediate, removing the Fmoc and reacting the resulting peptidyl-resin intermediate with the relevant isocyanate or carbamoyl chloride.
D. General procedure for the synthesis of N-sulfonamides of 11-mer peptide analogs.
The synthesis of the N-sulfonamide of the 11-mer peptide analogs can begin with the protected 11-mer peptidyl-resin intermediate (1) (0.025mmol) prepared in the manner described herein. The Fmoc group was removed using the methods described herein, and the resulting resin intermediate 2 was reacted with the relevant sulfonyl chloride. Similarly, the N-sulfonamide of 10-polymeric peptide analogs can be prepared starting from a protected 10-polymeric peptidyl-resin intermediate, removing the Fmoc and reacting the resulting peptidyl-resin intermediate with the relevant sulfonyl chloride.
E. General procedure for the synthesis of N-sulfonylurea derivatives of 11-mer peptide analogs.
The synthesis of the N-sulfonylurea derivative of the 11-mer peptide analog can start from the protected 11-mer peptidyl-resin intermediate (1) (0.025mmol) prepared in the manner described herein. Using the method described herein toRemoving the Fmoc group and allowing the resulting resin intermediate 2 to react with the corresponding sulfonamide chloride R4R5N-SO2Cl to give a sulfonylurea intermediate (see, for example, P.Davern et al, J.chem.Soc., Perkin Trans.2, 1994(2), 381-387). Similarly, N-sulfonylurea derivatives of 10-polymeric peptide analogues can be prepared starting from a protected 10-polymeric peptidyl-resin intermediate, removing Fmoc and reacting the resulting peptidyl-resin intermediate with the associated sulfonamide chloride R4R5N-SO2-Cl.
Example 3
11-mer peptides Using an Applied Biosystems Model 433A peptide synthesizer
Solid phase synthesis of analogs
The following is a general description of solid phase synthesis of a typical 11-mer peptide analog using an upgraded Applied Biosystems Model 433A peptide synthesizer. Upgraded hardware and software of the synthesizer is able to monitor the Fmoc deprotection step conductively with coupled feedback control. The scheme allows for a synthesis scale in the range of 0.05 to 1.0 mmol.
The incorporation of the two unnatural C-terminal amino acids involved in the simultaneous synthesis of 11-mer analogs is described above. This Fmoc-protected dipeptidyl resin was used in this ABI synthesis. The Fmoc-protected dipeptidyl-resin (0.1mmol) was placed in an appropriately sized container on the instrument, washed 6 times with NMP and deprotected using two treatments with 22% piperidine/NMP (2 and 8min each). One or two additional monitored deprotection steps are performed until the conditions for the monitoring option are met (< 10% difference between the last two conductivity-based deprotected peaks). The total deprotection time is 10-12 min. The deprotected dipeptidyl-resin was washed 6 times with NMP and then coupled with the next amino acid. The process is illustrated by the example for the next step.
Thus, Fmoc-Asp (OtBu) -OH was then coupled using the following method: Fmoc-Asp (OtBu) -OH was dissolved in 2mL of NMP and activated by the subsequent addition of 0.45MHBTU/HOBt in DMF (2.2mL) and 2M DIEA/NMP (1 mL). Then, the solution of the activated Fmoc-protected amino acid is transferred to the reaction vessel and the coupling is allowed to proceed for 30 to 60min depending on the feedback from the deprotection step. The resin was then washed 6 times with NMP and another 8 deprotection/coupling cycles were performed as described above to complete the assembly of the desired sequence. The Fmoc-amino acids used in sequence were: Fmoc-Ser (tBu) -OH, Fmoc-Thr (tBu) -OH, Fmoc- α -methyl-Phe (2-fluoro) -OH or analogs thereof, Fmoc-Thr (tBu) -OH, Fmoc-Gly-OH, Fmoc-Glu (OtBu) -OH, Fmoc-Aib-OH, and Fmoc-His (Trt) -OH. Finally, the Fmoc group was removed with 22% piperidine in NMP as described above and the peptidyl-resin was washed 6 times with NMP and DCM and then dried in vacuo.
Alternatively, a modified coupling scheme was used in which the Fmoc protected amino acid (0.26mmol) was activated by the subsequent addition of 0.5M HOAt in DMF (0.52mL) and DIC (40 μ L), transferred manually to the reaction vessel and allowed to couple for 14-18 hours.
A. Cleavage/deprotection
The desired peptide was cleaved/deprotected from its corresponding peptidyl-resin by treatment with TFA/water/triisopropylsilane (96: 2) (3.0mL) for 2 hours. The resin was filtered off, rinsed with TFA (1.0mL), and the combined TFA filtrate was added to 35mL Et2And (4) in O. The resulting precipitate was collected by centrifugation and finally dried, yielding 232mg of crude peptide product as a white solid. It was purified using preparative HPLC as described herein. The gradient used was from 15% to 45% 0.1% TFA/MeCN in 0.1% TFA/water over 40min. Fractions containing pure product were pooled and freeze-dried to give 28.4mg (18% recovery) of pure product.
Example 4
position-X of formulae II and IVa
aa10
And position-X
aa11
Biphenylalanine and phenyl-
Synthesis of heteroaryl-alanine analogs.
For position-X thereinaa10And position-Xaa11The residues at (a) are those represented by substituted amino acid analogs represented by formulas II and IVa, i.e., biphenylalanine analogs (Bip analogs) or phenyl-heteroaryl-alanine analogs, which are incorporated into the peptide chain in one of two ways.
A. The method A comprises the following steps: solid phase Suzuki condensation.
In method a, solid phase Suzu ki condensation is carried out to produce the desired modified biphenylalanine or phenyl-heteroaryl-alanine residue in a manner suitable for subsequent solid phase peptide synthesis to yield the target peptide. When in position-X in the target peptideaa11When the amino acid at (a) is represented by a modified biphenylalanine or phenyl-heteroaryl-alanine residue, it is prepared as shown in scheme 3. After removal of the Boc α -amine protecting group, chain extension is continued using a multiplex peptide synthesis method as described in the preceding paragraph to give the desired 11-mer peptide or derivative thereof. When the modified biphenylalanine analog is at position-X of the target peptideaa10In this case, the desired amino acid is prepared on a solid support using a suitable dipeptide precursor as shown in scheme 4.
The resulting dipeptidyl fragment containing the desired modified biphenylalanine derivative is then used to perform the synthesis of the target 11-mer peptide or derivative thereof. When position is-Xaa10And position-Xaa11Both require novel biphenylalanine or phenyl-heteroaromaticsWhen the amino acid residue is a trans-alanine residue, the solid phase Suzuki reaction is performed twice in this order as shown in the scheme 5 (below).
1.Prepared in position-X aa11 Synphase comprising an amino acid of formula IVa TM General procedure for Lantern (Suzuki coupling).
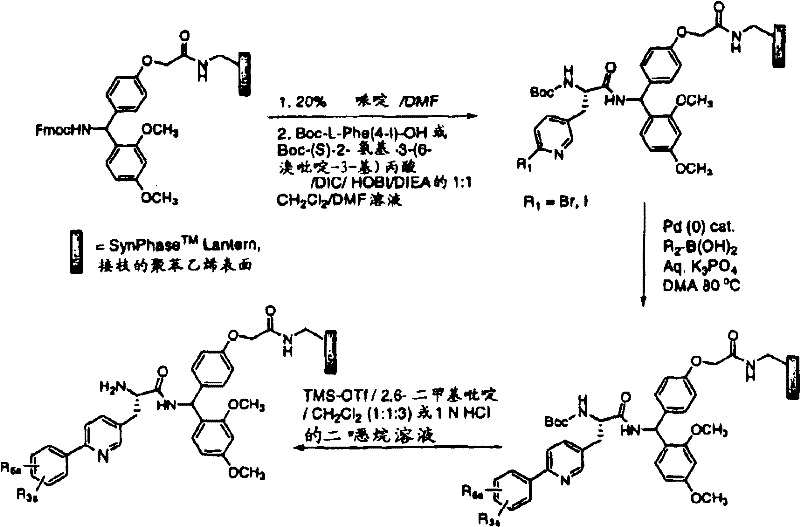
R2-B(OH)2Ariaryl-or heteroaryl-boronic acids
R3、R4、R6、R3aAnd R6aRepresented by the side chains described in formulae II and IVa
a.General procedure A.
N linked either directly via Knorr bond (Boc-amino acid-resin) or via amino acid-Knorr bond (Boc-dipeptide-resin)α-Boc- (S) -2-amino-3- (6-bromopyridin-3-yl) propionic acid residue or NαSynphase derivatized with-Boc-L-4-iodophenylalanine residueTMLantern (A-series (0.075 mmole/lanter) or D-series (0.035 mmole/lanter), from Mimotopes) was placed in a 13X 100mm glass culture tube with a screw cap. (the following procedure was used for the D-series lanters. similar proportions of reactants were used for reactions involving the a-series lanters.) aryl-or heteroaryl-boronic acids (0.140mmole, 4 equivalents) were dissolved in 0.30ml of N, N-dimethylacetamide. The resulting solution was added to the lanters in the 13X 100mm glass culture tubes.
Potassium phosphate (0.280mmole, 8 eq., 0.14ml of a 2M aqueous solution) was added to the aryl-or heteroaryl-boronic acid solution, followed by 0.10ml of a solution of N, N-dimethylacetamide containing 4.0mg of tetrakis (triphenylphosphine) palladium (0) catalyst (about 10 mole%, 0.0035 mmol). The resulting mixture was blanketed with nitrogen and the reaction vessel lid was screwed and then held at 80 ℃ for 17-20 hours while being placed on an orbital shaker. The lanters were transferred to a filtration unit and then washed with 3 × 1ml N, N-dimethylacetamide and 3 × 1ml dichloromethane (minimum 3 min/wash cycle per lantern), followed by cleavage of the Boc group (see general procedure below).
b.General procedure B.
The reaction was carried out as in general procedure a, except that a different catalyst was used. For this step, dichlorobis (triphenylphosphine) palladium (II) was used as the catalyst. For the D-series lantern grade reactions, approximately 10 mol% (0.0035mol) of catalyst was used.
2.Step for cleavage of the Boc group
a.Method A.
(the following procedure applies to D-series lanters, 0.035 mmol/lanter. an analogous and appropriate ranking procedure was applied to A-series lanters, 0.075 mmol/lanter.) Boc protected lanters prepared as described in general procedure A or B were treated with 0.5ml of a reagent solution consisting of trimethylsilyl trifluoromethanesulfonate, 2, 6-lutidine and dichloromethane (1: 3 by volume). After 2 treatments with this reagent with gentle agitation for 1 hour each, the resin was washed with 4X 1.0ml of dichloromethane, 3X 1.0ml of N, N-dimethylformamide and 3X 1.0ml of dichloromethane. The lanters were then subjected to the next acylation (coupling reaction) in the peptide synthesis procedure.
b.Method B.
Boc protected lanters prepared as described in general procedure A or B were reacted with 0.5ml of 1N HCl in anhydrous 1, 4-bis at room temperature with gentle agitation The alkane solution was treated for 1 hour. Mixing said lanterns 2X 1.0
The alkane solution was treated for 1 hour. Mixing said lanterns 2X 1.0ml 1, 4-bis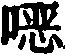 The alkanes, 2X 1.0ml of a 10% N, N-diisopropylethylamine in N, N-dimethylacetamide (v: v), 3X 1.0ml of N, N-dimethylacetamide and 3X 1.0ml of dichloromethane were washed to provide free amino-lanters which were then used in the next acylation (coupling) step.
The alkanes, 2X 1.0ml of a 10% N, N-diisopropylethylamine in N, N-dimethylacetamide (v: v), 3X 1.0ml of N, N-dimethylacetamide and 3X 1.0ml of dichloromethane were washed to provide free amino-lanters which were then used in the next acylation (coupling) step.
Example 6
Prepared in position-X
aa10
General of lantern comprising a modified biphenylalanine residue
And (5) carrying out the following steps.
The general steps described above for Suzuki coupling (A and B) to couple from Synphase to Synphase were used as shown in scheme 4TMAmino acid of Lantern (at position-X)aa11At) to obtain the desired at-position-Xaa10A dipeptidyl lantern comprising a modified Phe.
Flow chart 4
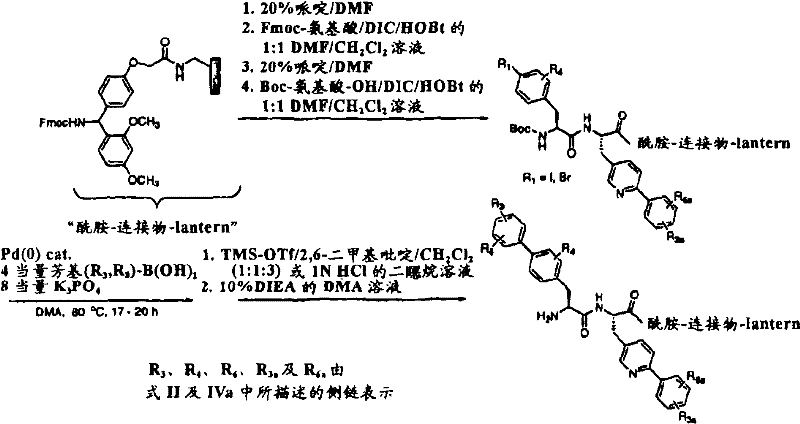
Example 7
Prepared in position-X
aa11
and-X
aa11
Comprising amino acids of formulae II and IVa
General procedure for lanters.
As shown in the following flow chart 5Illustratively, the above-described method for position-X is usedaa11Step of modified analogs (scheme 1) and two successive Suzuki coupling steps to make the linker at position-Xaa10and-Xaa11Dipeptidyl lanters in which both modified phenylalanine and phenyl-heteroarylalanine residues are contained.
Example 8
For acylation/elongation of SynPhase
TM
General procedure for peptides on Lanterns.
a.And (3) carrying out Fmoc deprotection.
The D-series SynPhaseTMLantern (0.035mmol/Lantern loading) was added to 0.5ml of 8: 2N, N-dimethylformamide/piperidine (vol: vol). Gentle agitation was performed. After 1h, the lantern was washed with 3 × 1.0ml of N, N-dimethylformamide and 3 × 1.0ml of dichloromethane and allowed to soak for at least 3 min/wash.
b.Procedure for acylation/amino acid coupling (scheme 6).
The side chain and the alpha-amine protected amino acid (0.105mmol) were dissolved in 0.5ml 1: 1N, N-dimethylformamide/dichloromethane. To the solution were added N-hydroxybenzotriazole (0.105mmol), N-diisopropylethylamine (0.315mmol) and N, N' -diisopropylcarbodiimide (0.105 mmol). The amino acid solution was allowed to stand for 10 minutes, after which the D-series lantern (0.035mmol/lantern) containing the α -amine deprotected peptide was added to the solution. The sample vial was capped and gently agitated for 16-20 h. The lantern was then washed with 3X 1.0ml N, N-dimethylformamide and 3X 1.0ml dichloromethane and allowed to soak for 3-5 min/wash cycle.
Flow chart 5
Flow chart 6
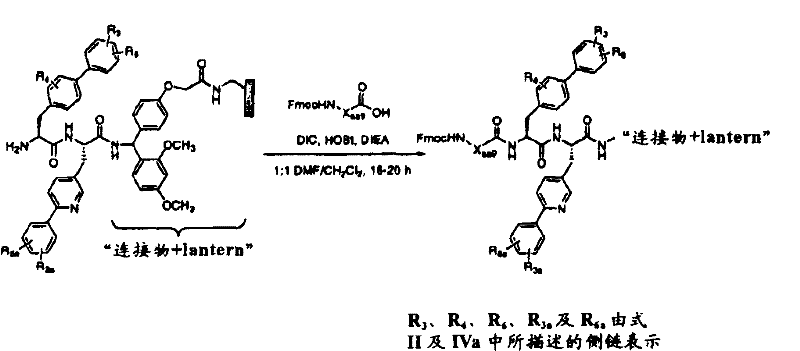
Example 9
General procedure for the preparation of peptides via fragment condensation.
In method A, as described in example 7, a solid phase Suzuki condensation is carried out to bring about the-X positionaa10and-Xaa11To prepare the desired amino acids represented by formula II and formula IVa. Cleaving the dipeptide from the support and simultaneously (step a) or subsequently (step B) removing the N-terminal alpha-amine protecting group. The dipeptide was then coupled to a 9 amino acid peptide with a fully protected side chain (see below). The side chain is subsequently deprotected and purified to give the desired 11-mer peptide product.
A.The method A comprises the following steps: solution phase fragment condensation.
In method A, solid phase Suzuki condensation and acylation (as described in example 7) are performed to prepare a conjugate to SynphaseTMThe desired dipeptide of Lantern, and the N-terminal alpha-amine is either Boc protected or Fmoc protected. Cleaving the dipeptide from the Lantern vector under acidic conditions. In the case of Boc protected N-terminal alpha-amines, the acidic cleavage simultaneously provides deprotection of the alpha-amines and their purification or direct introduction into the fragment coupling sequence as shown in scheme 7.
As shown in scheme 8, a dipeptide comprising an Fmoc-protected N-terminal alpha-amine is cleaved under acidic conditions and the N-terminal alpha-amine is deprotected in solution. These dipeptides were purified and then fed into the fragment-coupling sequence.
1.For the removal of dipeptides from Synphase TM Step of cleavage in Lantern.
a.Step A (Boc protected dipeptide; see scheme 7).
Subjecting the D-series SynPhase toTMLantern was placed into a 1 dram (dram) glass sample vial. A1: 1 trifluoroacetic acid/dichloromethane solution (0.5ml) was added to the sample vial. The sample vial was capped and gently agitated on an orbital shaker (100rpm) for 2 h. The lysis solution was transferred to a new sample bottle and an additional 0.5ml of 1: 1 trifluoroacetic acid/dichloromethane was added to the lantern. The sample vial was capped again and gently agitated on an orbital shaker (100rpm) for 2 h. The second lysis solution was added to the first solution, and the Cantern was then rinsed with dichloromethane. The rinsing solution was added to the lysis solution, and the solvent was evaporated to obtain the dipeptide as the trifluoroacetate salt of the α -amine.
Flow chart 7
b.Step B (Fmoc-protected dipeptide; scheme 8).
The Fmoc-protected dipeptide was removed from the SynPhase as described above in step ATMCleaved in Lantern. The lanters were rinsed with dichloromethane and the solvent was evaporated from the combined rinse/cleavage solution. To the resulting residue (in a 1 dram sample bottle) was added 0.40ml of 8: 2 dimethylformamide/piperidine (vol: vol). The sample vial was capped and allowed to react for 45 min. Evaporating the remaining solvent and purifying with a C-18 column and CH3CN/H2HPLC of O/TFA solvent System the resultingThe product was purified to give (after evaporation of the solvent) the dipeptide as the trifluoroacetate salt of the alpha-amine.
Flow chart 8

2.Procedure for solid phase Synthesis of C-terminal Carboxylic acids of side chain protected 9-mer peptides (flow) Fig. 9).
A solution of Fmoc- (L) -Ser (tBu) -OH (5 equiv.), 0.5M HOAt/DMF (5 equiv.), and DIC (5 equiv.) in NMP (5mL) was vortexed with (L) -Asp (OtBu) -2-chlorotrityl resin (3.0g, 2.16mmol) at room temperature for 18 h. After several washes with NMP, the Fmoc group was removed by treatment with 1.5M piperidine/DMF twice (5min and 10 min). These coupling and deprotection steps were repeated seven times to assemble the desired sequence, except that 1.1 and 1.5 equivalents of Fmoc- α -Me-Phe (2-R-6-R ") -OH and Boc- (L) -his (trt) -OH were used for their coupling, and HATU/HOAt and DIEA (4 equivalents) were used for coupling Fmoc-thr (tbu) -OH to (S) - α -Me-Phe (2-R-6-R") -peptidyl-resin, respectively.
After assembly was complete, the peptidyl-resin was washed with DCM and the protected 9-mer peptide C-terminal carboxylic acid was released from the resin by treatment with DCM/AcOH/TFE (8: 1, v: v) for 1 hour at room temperature. The resin was filtered off and the filtrate was evaporated to dryness, redissolved in AcCN/water (2: 1) and freeze-dried twice to give 2.777g of 81% pure product, which was used in the subsequent fragment coupling step without further purification.
Flow chart 9

3.Step for solution phase fragment coupling reaction.
These reactions were performed as single compounds in 1 dram sample vials, and as parallel arrays of compounds in 2ml96 well plates. The following description (shown in scheme 10) applies to the single compound case, but is entirely similar to the reaction carried out in a 96-well plate.
The TFA salt of the dipeptide (0.01mmol) was dissolved in 0.25ml THF containing 0.5% N, N-diisopropylethylamine in a 1.5ml glass sample vial. Macroporous carbonate resin (MP-carbonate, 0.03mmol, Argonaut Technologies) was added to the sample vial. The sample vial was capped and agitated for 2h at room temperature. The solution was filtered and excess solvent was removed by evaporation.
A solution of 0.15ml of 9: 1 chloroform/N, N-dimethylformamide containing the side chain protected 9-mer peptide C-terminal carboxylic acid (0.008mmol) and N-hydroxybenzotriazole (HOBt, 0.008mmol) was added to a sample vial containing the dipeptide amine. Diisopropylcarbodiimide (DIC, 0.08mmol) was added as a solution in 0.05ml of 9: 1 chloroform/N, N-dimethylformamide. The sample vial was capped and the reaction was stirred on an orbital shaker at room temperature for 16 h. The residual solvent was evaporated from the sample bottle.
Deprotection of the 11-mer peptide side chain and N-terminal a-amine with 0.40mL 97.5: 2.5 trifluoroacetic acid/triisopropylsilane (TFA/TIS) for 1h residual solvent was evaporated and CH3CN/H2HPLC with O/TFA solvent system purified the 11-mer peptide product and driven the collection of the effluent by detecting the desired product mass.
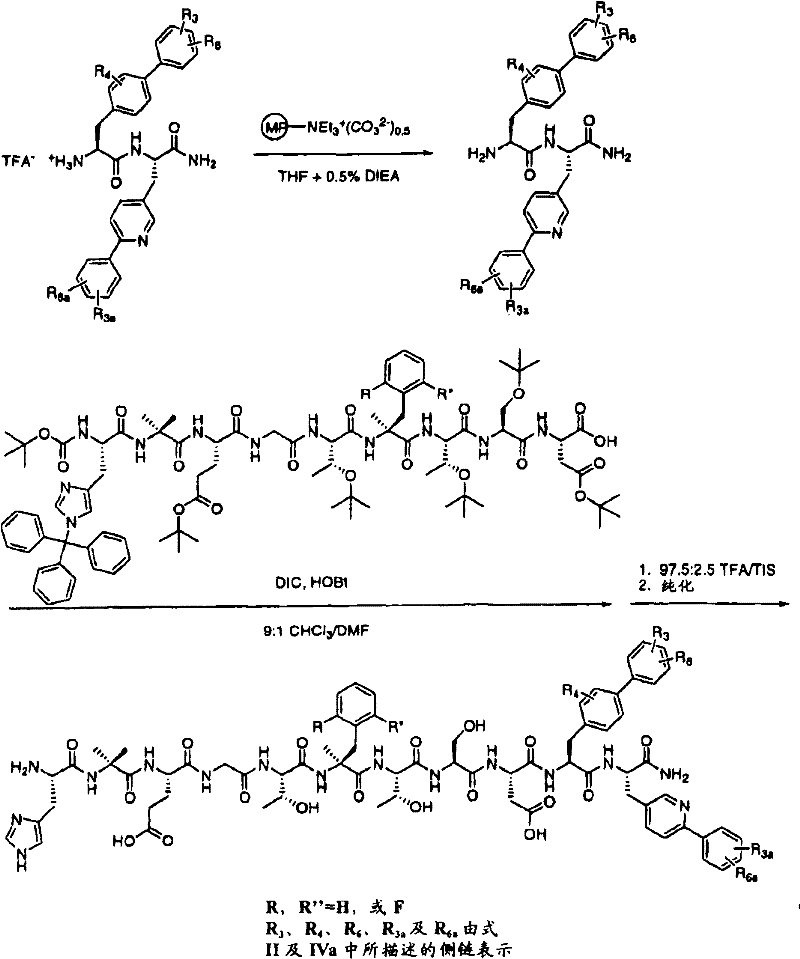
B.The method B comprises the following steps: synthesis of the formulae II and IVa in solution using the Suzuki coupling method The Fmoc-amino acid analog of (1).
The following examples illustrate the synthesis of several Fmoc-amino acid analogs of formula II and IVa, which were then used for solid phase synthesis of 11-mer and other peptide analogs as described in example 1.
Example 10
Fmoc- (S) -2 ' -ethyl-4 ' -methoxy-biphenylalanine [ Fmoc- (S) -Bip (2 ' -Et-4-)
OMe)]And (4) synthesizing.
Scheme 11 below depicts the synthesis of Fmoc- (S) -2 '-ethyl-4' -methoxy-biphenylalanine.
Flow chart 11
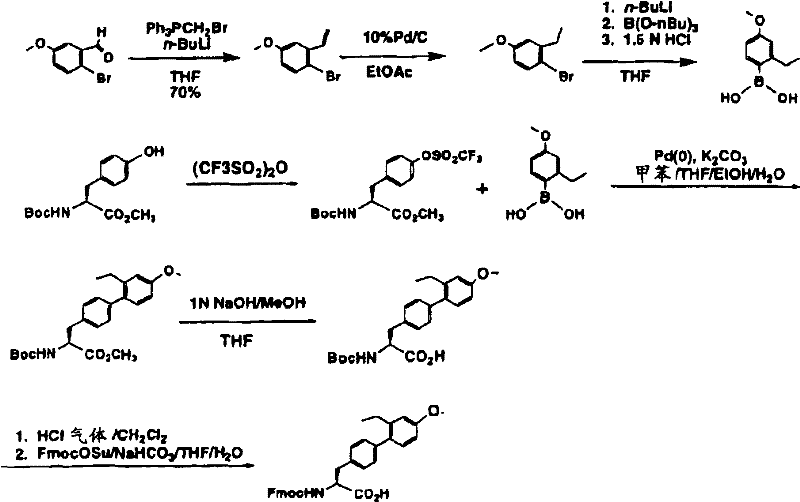
1.Boc-L-tyrosine-O-triflate.
A solution of 47.74mg (169.5mmol, 2 equivalents) of trifluoromethanesulfonic anhydride in DCM (100ml) was slowly added over a period of 30 minutes to a solution maintained at-40 deg.C in N2Next, 25g (85mmol) of Boc-L-tyrosine methyl ester and 36.25g (339mmol, 4 eq) of 2, 6-lutidine in dry DCM (200 mL). The solution was stirred at-40 ℃ for an additional 2 hours. HPLC analysis indicated the reaction was complete. The reaction was quenched by the addition of 20mL of water. The layers were separated and the organic layer was washed with 3X 200mL 1N HCl, 200mL saturated Na2CO3, 200mL water and 200mL brine. The organic layer was dried over magnesium sulfate, filtered and dried in vacuo to give the crude product as a red oil. It was subjected to silica gel flash chromatography (300g silica gel, 0 to 50% gradient of EtOAc in hexanes). Will compriseFractions of the product were concentrated in vacuo to afford the desired compound as a white solid (27g, 75% yield).
2.2-ethyl-4-methoxy-phenylboronic acid.
a.Method A.
Bromination of methyl triphenyl The suspension (199.5g, 0.465mol) in dry THF (800ml) was purged (purge) for 10min, then cooled to 10 deg.C and n-butyllithium (169ml, 0.465mol, 2.75M solution) was added slowly over 30min and stirred for 1h. A solution of 2-bromo-methoxybenzaldehyde (100g, 0.465mol) in anhydrous THF (300ml) was added slowly over a period of 30 min. After the addition, the reaction mixture was stirred for 1 hour. Petroleum ether (2L) was added and the reaction mixture was stirred for an additional 30 min. The reaction mixture was filtered on a silica gel filter pad. The filter pad was washed with diethyl ether. The combined organic washes were concentrated below 30 ℃ and the crude product was purified by 60-120 silica gel chromatography using 100% petroleum ether as eluent. Yield: 92g, 90% in the form of pale yellow liquid.
The suspension (199.5g, 0.465mol) in dry THF (800ml) was purged (purge) for 10min, then cooled to 10 deg.C and n-butyllithium (169ml, 0.465mol, 2.75M solution) was added slowly over 30min and stirred for 1h. A solution of 2-bromo-methoxybenzaldehyde (100g, 0.465mol) in anhydrous THF (300ml) was added slowly over a period of 30 min. After the addition, the reaction mixture was stirred for 1 hour. Petroleum ether (2L) was added and the reaction mixture was stirred for an additional 30 min. The reaction mixture was filtered on a silica gel filter pad. The filter pad was washed with diethyl ether. The combined organic washes were concentrated below 30 ℃ and the crude product was purified by 60-120 silica gel chromatography using 100% petroleum ether as eluent. Yield: 92g, 90% in the form of pale yellow liquid.
A solution of 2, 2' -bipyridine (24.3g, 0.15mol) and 2-bromo-5-methoxystyrene (65g, 0.31mol) in ethyl acetate (650ml) was cooled to 0 ℃. The solution was purged and then 10% palladium on carbon (16.25g, 25%) was added under a stream of nitrogen. The reaction mixture was stirred under hydrogen for 3 days under a pressure of 2kg in a Parr shaker. The progress of the reaction was monitored by HPLC. The reaction mixture was filtered through Celite and the filtrate was washed with 5% potassium hydrogen sulfate solution, dried over sodium sulfate and concentrated below 30 ℃. Yield: 60g, 91% in light yellow liquid.
A solution of 4-bromo-3-ethylanisole (94g, 0.437mol) in THF (900ml) was cooled to-78 deg.C and n-butyllithium (249ml, 0.55mol) was added dropwise at the same temperature. Stirring was continued for 1 hour at-78 ℃. Trin-butyl borate (177ml, 0.655mol) was added slowly at-78 ℃. The cooling bath was removed and the reaction mixture was allowed to warm to 0 ℃ and quenched with 1.5N hydrochloric acid at 0 ℃. The organic layer was separated. The aqueous layer was extracted with ethyl acetate and the combined organic layers were washed with brine and concentrated. The resulting residue was stirred in petroleum ether for 30 min. The resulting solid was filtered and dried under vacuum. Yield: 65g, 82% as white solid.
b.Method B (see flow chart 12).
Methyl iodide (290g, 2.05mol) was added to 3-ethylphenol (50g, 0.4mol, 98% purity, Fluka) and K2CO3(283g, 2.05mol) in a mixture of anhydrous acetone (500 ml). The reaction mixture was transferred to an autoclave and refluxed at 70 ℃ overnight. The reaction mixture was filtered through a pad of celite. The filter pad was washed with acetone and the combined filtrate and washings were concentrated. The product was dissolved in DCM, filtered and evaporated to dryness. Yield: 50g, 90% in brown liquid form.
A solution of 3-ethyl anisole (50g, 0.3676mol) and N-bromosuccinimide (72g, 0.4mol) in acetonitrile (1L) was stirred at room temperature in the dark for 8 hours. The reaction mixture was concentrated below 40 ℃ and the residue obtained was redissolved in CCl4Neutralized and filtered. The filtrate was concentrated and the product was purified by fractional distillation. Yield: 35g, 43% in the form of pale yellow liquid. The 4-bromo-3-ethyl anisole was converted to the corresponding boronic acid as described in method a.
For the purpose of upscaling the conversion of 4-bromo-3-ethylanisole to 2-ethyl-4-methoxy-boronic acid can be achieved using Grignard processes. The process comprises forming the Grignard reagent by reacting 4-bromo-3-ethylanisole with Mg (1.1 equivalents) in THF, followed by reacting the resulting Grignard intermediate with tri-n-butyl or trimethyl borate as described in method a.
Flow chart 12
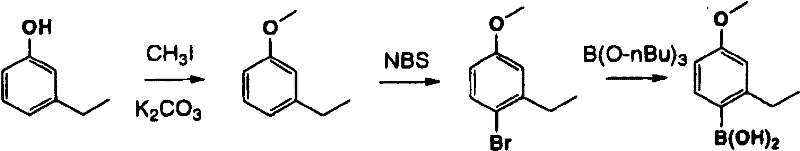
3.Fmoc- (S) -2 '-ethyl-4' -methoxy-biphenylalanine.
A solution of Boc-L-tyrosine-O-triflate (81g, 0.19mol) in dry toluene (600ml) was purged with nitrogen for 10 min. Adding K2CO3(36g, 0.26mol) in water (200ml) 2-ethyl-4-methoxy-benzeneboronic acid (36g, 0.2mol) was then added and the reaction mixture was purged with nitrogen for 10 min. Adding Pd (PPh)3)4(16.18g, 0.014mol), ethanol (200ml) and THF (400ml) the reaction mixture was then heated to 100 ℃ for 4 hours with stirring. The reaction mixture was concentrated under vacuum and the residue was dissolved in DCM (1.0L). The organic layer was washed with 10% sodium hydroxide solution, 15% citric acid solution, dried over sodium sulfate and concentrated. The crude product was purified by column chromatography on 60-120-mesh silica gel eluting with 10% ethyl acetate in petroleum ether. Yield: 50g, 65%, yellow liquid.
To a mixture of methyl ester of Boc- (S) -2 '-ethyl-4' -methoxy-biphenylalanine (60g, 0.146mol) in THF (450ml) and methanol (85ml) was added a solution of sodium hydroxide (24g, 0.58mol) in water (85 ml). The reaction mixture was stirred at room temperature overnight, concentrated and the residue was dissolved in water (100ml) and washed with diethyl ether. The aqueous layer was acidified to pH 1 with 20% citric acid and extracted with ethyl acetate. The extract was washed with brine, dried over sodium sulfate and evaporated to dryness. Yield: 55g, 94% in the form of a colorless liquid.
Boc- (S) -2 '-ethyl-4' -methoxy-biphenylalanine (55g, 0.138mol) was dissolved in anhydrous DCM (1 liter) and purged with dry HCl gas at room temperature for 6 h. The solid product obtained was filtered and dried under vacuum. Yield: 46g, 100%. To a solution of the free amino acid hydrochloride (30g, 0.089mol) in THF (700ml) was added NaHCO3(29g, 0.358mol) in water (240 ml). Fmoc-OSu (30g,0.089mol) was added in portions during 30 min. The reaction mixture was stirred at room temperature overnight. The THF was removed under vacuum and water (2.0L) was added. The clear solution was extracted with ether to remove any impurities. The aqueous solution was acidified to pH 1 and extracted with ethyl acetate. The organic layer was washed with water and brine and evaporated to dryness. Yield: 37g, 80%.
Example 11
(2S) -2- (((9H-fluoren-9-yl) methoxy) carbonylamino) -3- (6-o-tolylpyridin-3-yl) propionic acid
Hydrochloride [ Fmoc- (S) -4- (2' -methylphenyl) -3-pyridylalanine hydrochloride]And (4) synthesizing.
The following scheme 13 describes the synthesis of (2S) -2- (((9H-fluoren-9-yl) methoxy) carbonylamino) -3- (6-o-tolylpyridin-3-yl) propionic acid hydrochloride:
flow chart 13

1.5-bromo-2-o-tolylpyridine.
To a slurry of 910mg (3.21mmol) of 5-bromo-2-iodopyridine and 436mg (3.21mmol, 1.0 equiv.) of 2-o-tolylboronic acid in 8mL of toluene and 3.2mL of 2M aqueous sodium carbonate solution purged with argon and evacuated was added 36mg (0.032mmol, 0.01 equiv.) of tetrakis (triphenylphosphine) palladium. The reaction mixture was again purged with argon and evacuated twice and then refluxed under argon for 15 h. The reaction was cooled and partitioned between water and EtOAc. The layers were separated and the aqueous layer was extracted once more with EtOAc. The organic extracts were combined, dried over magnesium sulfate, filtered, concentrated and dried in vacuo to give the crude product as an orange oil. Chromatography on silica gel (7: 3 CH)2Cl2Hexane) to yield 666mg yellowThe title compound was obtained as an oil in 84% yield.
2.6-o-tolyl nicotinaldehyde.
To a stirred solution of 125mg of the above compound (0.50mmol) in THF (2.0mL) at-74 deg.C under argon, during 5min, was added 220. mu.L of a solution of nBuLi in hexane (2.5M, 0.55mmol, 1.1 equiv) without the temperature rising above-71 deg.C. A light green solution formed which turned dark green after 30 min. After 45min, 49.4 μ L (0.61mmol, 1.2 equiv.) of DMF was added and the reaction was allowed to warm to-40 ℃. After 14h, a bright orange solution formed. The reaction was quenched with 10% citric acid and the mixture was stirred rapidly at room temperature for 20 min. The resulting bright yellow solution was extracted twice with EtOAc. The organic extracts were combined and MgSO4Dried, filtered and concentrated to give a yellow oil. The crude mixture thus obtained was purified by silica gel chromatography (2.5X 10cm column) using ethyl acetate/dichloromethane (1: 24) as eluent to give a white solid, mp82-84 ℃, 90.3mg, 91% yield.
3.(6-o-tolylpyridin-3-yl) methanol.
To a solution of 1.070g (5.43mmol) 6-o-tolyl nicotinaldehyde in 19mL ethanol at a temperature of 0-5 deg.C was added 287mg (7.5mmol, 1.4 equiv.) of sodium borohydride. After 2h, the reaction mixture was quenched with saturated sodium bicarbonate solution and after 30min, it was partitioned between dichloromethane and brine. The organic extracts were dried over magnesium sulfate and concentrated to give the indicated product as a colourless oil, 1.08g, 100% yield.
4.5- (bromomethyl) -2-o-tolylpyridine hydrobromide.
A solution of 4.49g (22.5mmol) (6-o-tolylpyridin-3-yl) methanol in 75mL 48% hydrobromic acid was heated to reflux for 64 h. The reaction mixture was partially cooled and excess hydrobromic acid was removed by vacuum distillation (110 ℃ and 2 torr) until a brown solid residue remained in the flask. Distillation was performed using a large KOH pellet trap placed between the distillation apparatus and the vacuum pump. The solid residue was slurried in ether, filtered and dried under a stream of nitrogen to give 7.38g of product in 95% yield.
5.(2S) -2- (Diphenylmethyleneamino) -3- (6-o-tolylpyridin-3-yl) propionic acid tert-butyl ester And (3) an ester.
To a stirred amount of 800mg (2.33mmol) of 5- (bromomethyl) -2-O-tolylpyridine hydrobromide, 689mg (2.33mmol, 1.0 eq) of tert-butyl 2- (diphenylmethyleneamino) acetate and 141mg (0.233mmol, 0.1 eq) of O-allyl-N- (9-anthrylmethyl) cinchonidine under argon at-78 ℃ during 5min To a mixture of bromide in 14mL of dichloromethane was added 1.687mL (5.83mmol, 2.5 equivalents) of 2-tert-butylimino-2-diethylamino-1, 3-dimethyl-perhydro-1, 3, 2-diazaphosphorine. The reaction mixture was stirred at-78 ℃ for 10h and then allowed to warm in situ to room temperature. The mixture was directly purified by silica gel chromatography (5X 10cm column) using ethyl acetate/dichloromethane (1: 4) as eluent to give a brown oil, 1.10g, 100% yield.
To a mixture of bromide in 14mL of dichloromethane was added 1.687mL (5.83mmol, 2.5 equivalents) of 2-tert-butylimino-2-diethylamino-1, 3-dimethyl-perhydro-1, 3, 2-diazaphosphorine. The reaction mixture was stirred at-78 ℃ for 10h and then allowed to warm in situ to room temperature. The mixture was directly purified by silica gel chromatography (5X 10cm column) using ethyl acetate/dichloromethane (1: 4) as eluent to give a brown oil, 1.10g, 100% yield.
6.(2S) -2- (((9H-fluoren-9-yl) methoxy) carbonylamino) -3- (6-o-tolylpyridine-3- Yl) propionic acid tert-butyl ester.
To a stirred solution of 1.10g (2.33mmol) of tert-butyl (2S) -2- (diphenylmethyleneamino) -3- (6-o-tolylpyridin-3-yl) propionate in 9mL of THF at room temperature under argon was added 2.795g (14.54mmol, 6.5 equivalents) of citric acid in 9mL of water. After 20h, the reaction mixture was diluted with water (5mL) and washed twice with diethyl ether (10 mL). The aqueous phase was then adjusted to pH 9 with solid sodium carbonate and then extracted twice with dichloromethane.
The dichloromethane extracts were combined, dried over sodium sulfate and concentrated. The resulting oil was dissolved in 10mL of THF and treated successively with 7.2mL of 10% sodium carbonate solution and 703mg (2.56mmol, 1.1 eq.) of 9-fluorenylmethyloxycarbonyl chloride at room temperature. After 14h, the reaction mixture was extracted twice with dichloromethane, dried over sodium sulfate, filtered, concentrated and purified by silica gel chromatography (2.5 × 10cm column) using ethyl acetate/dichloromethane (1: 19) as eluent to give a colorless oil, 1.082g, 91% yield. Recrystallization from 20mL of 7: 1 hexane/dichloromethane provided a white solid, 287 mg. The mother liquor was concentrated to give an amorphous white solid, the title compound, 779mg, 63% yield. Chiral HPLC analysis (4.6X 250mm AD column with 38: 1 heptane: methanol: ethanol as eluent at a flow rate of 1mL/min) indicated 98% ee.
(2S) -2- (((9H-fluoren-9-yl) methoxy) carbonylamino) -3- (6-o-tolylpyridine-3-
Yl) propionic acid hydrochloride.
A solution of 1.75g (3.19mmol) of tert-butyl (2S) -2- (((9H-fluoren-9-yl) methoxy) carbonylamino) -3- (6-o-tolylpyridin-3-yl) propionate in TFA (5.0mL) was stirred at room temperature for two hours and a calcium chloride-filled drying tube was used to exclude air during this time. The reaction mixture was concentrated in vacuo at below 40 ℃ and the resulting orange oil was dissolved in 10mL of diethyl ether to which was added 5mL of 1M HCl/diethyl ether solution. The resulting white solid was filtered and washed with diethyl ether to give the desired compound as a white powder, 1.65g, 100% yield.
Example 12
(2S) -2- (((9H-fluoren-9-yl) methoxy) carbonylamino) -4- (6-bromopyridin-3-yl) propionic acid
And (3) synthesizing hydrochloride.
The following scheme 14 describes the synthesis of 3- (((9H-fluoren-9-yl) methoxy) carbonylamino) -3- (6-bromopyridin-3-yl) propionic acid hydrochloride:
flow chart 14

1.2-bromo-5- (bromomethyl) pyridine.
To a stirred slurry of 10.320g (60.0mmol) of 5-methyl-2-bromopyridine and 5.339g (30.0mmol, 0.5 equiv.) of recrystallized N-bromosuccinimide in 150mL of carbon tetrachloride was added 200mg of AIBN. The reaction mixture was purged twice with argon and evacuated and then refluxed under argon. After 90min, the reaction mixture was cooled to room temperature, filtered and the filtrate was concentrated to give a yellow oil. Proton NMR indicated that the mixture contained 53% (mol) unreacted 5-methyl-2-bromopyridine, 43% of the title product, and 4% 2-bromo-5- (dibromomethyl) pyridine. The mixture was used without further purification in the following step.
2.(S) -2- (((9H-fluoren-9-yl) methoxy) carbonylamino) -3- (6-bromopyridin-3-yl) propane And (3) tert-butyl ester.
Stirred 2-bromo-5- (bromomethyl) pyridine (nominally 26.4mmol), 7.798(26.4mmol, 1.0 eq) tert-butyl 2- (diphenylmethyleneamino) acetate and 1.60g (2.64mmol, 0.1 eq) O-allyl-N- (9-anthrylmethyl) cinchonidine at-78 deg.C under argon over a period of 5minTo a mixture of bromide in 100mL of dichloromethane was added 11.46mL (39.6mmol, 1.5 equivalents) of 2-t-butylimino-2-diethylamino-1, 3-dimethyl-perhydro-1, 3, 2-diazaphosphorine. The reaction mixture was stirred at-78 ℃ for 7h and then allowed to warm in situ to room temperature. The reaction mixture was then concentrated, redissolved in 75mL THF and treated with 75mL aqueous citric acid (22 g). After stirring vigorously for 7h, the mixture was extracted twice with ether (75 mL). The organic extracts were combined and washed once with water (25 mL). Combining the aqueous extractsAnd the pH was adjusted to 8 with solid sodium carbonate. The aqueous solution was used for the next reaction without further treatment.
3.(S) -2- (((9H-fluoren-9-yl) methoxy) carbonylamino) -3- (6-bromopyridin-3-yl) propane And (3) tert-butyl ester.
The above aqueous solution was added to a solution of 7.545g (27.5mmol, 1.04 eq.) of 9-fluorenylmethyloxycarbonyl chloride in 75mL of THF at room temperature. After 14h, the reaction mixture was extracted twice with ethyl acetate, dried over magnesium sulfate, filtered, concentrated and purified by chromatography on silica gel (12 × 25cm column) using ethyl acetate/dichloromethane (1: 24) as eluent to give a colorless oil, 7.25g, 91% yield. It was recrystallized from 120mL of 5: 1 hexane/dichloromethane to give a small amount of white solid, which was filtered off. The mother liquor was concentrated to give the title compound as an amorphous white solid, 4.96g, 62% yield. Chiral HPLC analysis (4.6X 250mm AD column, 38: 1 heptane: methanol: ethanol as eluent, flow rate 1mL/min) indicated 97.2% ee.
4.2- (((9H-fluoren-9-yl) methoxy) carbonylamino) -3- (6-bromopyridin-3-yl) propanoic acid salt An acid salt.
A solution of 1.02g (1.95mmol) of tert-butyl (2S) -2- (((9H-fluoren-9-yl) methoxy) carbonylamino) -3- (6-bromopyridin-3-yl) propionate in TFA (3.0mL) in a drying tube filled with calcium chloride with exclusion of air was stirred at room temperature for two hours. The reaction mixture was concentrated in vacuo at below 35 ℃ and the resulting orange oil was dissolved in 3mL of dichloromethane to which was added 6mL of 1M HCl/ether solution. The resulting white solid was filtered and washed with diethyl ether to give the title compound as a white powder, 845mg, 86% yield.
Example 13
(2S) -2- (((9H-fluoren-9-yl) methoxy) carbonylamino) -3- (6- (2-ethylphenyl) piperidine-3-
Yl) propionic acid hydrochloride [ Fmoc- (S) -4- (2' -ethylphenyl) -3-pyridylalanine]And (4) synthesizing.
The following scheme 15 describes the synthesis of (2S) -2- (((9H-fluoren-9-yl) methoxy) carbonylamino) -3- (6- (2-ethylphenyl) pyridin-3-yl) propionic acid hydrochloride:
flow chart 15
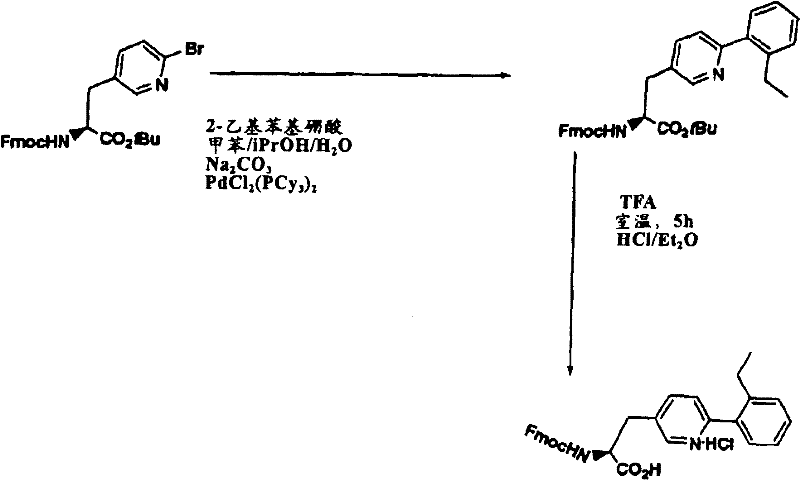
1.((S) -2- (((9H-fluoren-9-yl) methoxy) carbonylamino) -3- (6- (2-ethylphenyl) pyridine -3-yl) propionic acid tert-butyl ester.
To a stirred slurry of 1.75g (3.35mmol) of tert-butyl (S) -2- (((9H-fluoren-9-yl) methoxy) carbonylamino) -3- (6-bromo-pyridin-3-yl) propanoate and 1.005g (6.70mmol, 2 equivalents) of 2-ethylphenylboronic acid in 50mL of 1: 1 isopropanol/toluene was added 25.0mL of a 2M aqueous solution of sodium carbonate. The reaction mixture was purged twice with argon and evacuated then 124mg (0.167mmol, 0.05 eq) bis (tricyclohexylphosphine) palladium (II) chloride was added and then the mixture was again purged with argon and evacuated. The rapidly stirred mixture was heated at 80 ℃ under argon. After 20h, the reaction mixture was cooled to room temperature and partially concentrated to remove the isopropanol. The residue was partitioned between ethyl acetate and water and the aqueous phase was re-extracted with ethyl acetate. The organic extracts were combined, dried over magnesium sulfate, filtered and concentrated to give a brown oil. Purification by chromatography on silica gel (5X 15cm column) using ethyl acetate/dichloromethane (1: 9) as eluent gave the desired compound as a colourless oil, 1.25g, 77% yield.
2.(2S)2- (((9H-fluoren-9-yl) methoxy) carbonylamino) -3- (6- (2-ethylphenyl) pyridine -3-yl) propionic acid hydrochloride.
A solution of 1.53g (2.79mmol) of tert-butyl (2S) -2- (((9H-fluoren-9-yl) methoxy) carbonylamino) -4- (6- (2-ethylphenyl) pyridin-3-yl) propanoate in TFA (5.0mL) was stirred at room temperature for two hours with a drying tube filled with calcium chloride to exclude air. The reaction mixture was concentrated in vacuo at below 35 ℃ and the resulting orange oil was dissolved in ether to which was added 6mL of 1M HCl/ether solution. The resulting white solid was filtered and washed with diethyl ether to give the desired product as a white powder, 1.38g, 93% yield.
Example 14
(2S)2- (((9H-fluoren-9-yl) methoxy) carbonylamino) -3- (6- (2-ethyl-4-methoxy) benzene
Yl) pyridin-3-yl) propanoic acid hydrochloride [ Fmoc- (S) -4- [ (2 '-ethyl-4' -methoxy) phenyl)]-3-pyridine
Pyridylalanine]And (4) synthesizing.
The following scheme 16 describes the synthesis of (2S) -2- (((9H-fluoren-9-yl) methoxy) carbonylamino) -3- (6- (2-ethyl-4-methoxy) phenyl) pyridin-3-yl) propionic acid hydrochloride:
flow chart 16
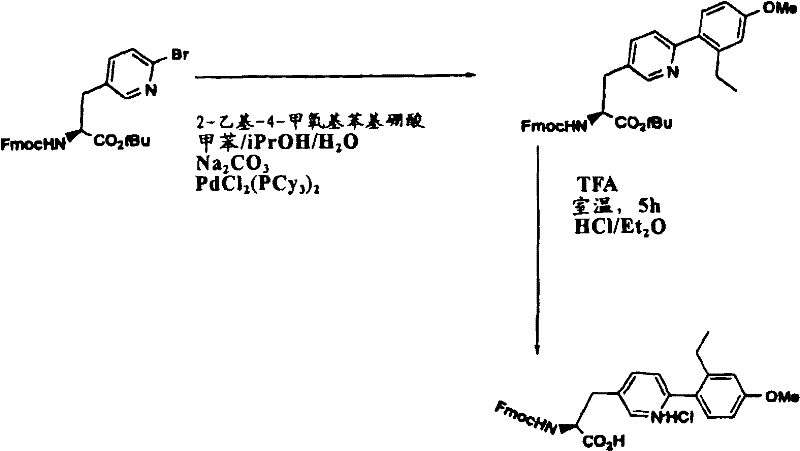
1.(S) -2- (((9H-fluoren-9-yl) methoxy) carbonylamino) -3- (6- (2-ethyl-4-methoxy) Phenyl) pyridin-3-yl) propionic acid tert-butyl ester:
To a stirred slurry of 613mg (1.17mmol) of tert-butyl (S) -2- (((9H-fluoren-9-yl) methoxy) carbonylamino) -3- (6-bromo-pyridin-3-yl) propanoate and 422mg (2.34mmol, 2 equiv.) of 2-ethylphenylboronic acid in 20mL of 1: 1 isopropanol/toluene was added 10.0mL of 2M aqueous sodium carbonate solution. The reaction mixture was purged twice with argon and evacuated then 43.2mg (0.059mmol, 0.05 eq) bis (tricyclohexylphosphine) palladium (II) chloride was added and then the mixture was again purged with argon and evacuated. The rapidly stirred mixture was heated at 80 ℃ under argon. After 9h, the reaction mixture was cooled to room temperature and partially concentrated to remove the isopropanol. The residue was partitioned between ethyl acetate and water and the aqueous phase was extracted again with ethyl acetate. The organic extracts were combined, dried over magnesium sulfate, filtered and concentrated to give a brown oil. Purification by chromatography on silica gel (5X 15cm column) using ethyl acetate/dichloromethane (3: 17) as eluent gave the desired compound as a colourless oil, 401mg, 59% yield.
2.(2S)2- (((9H-fluoren-9-yl) methoxy) carbonylamino) -3- (6- (2-ethyl-4-methoxy) Phenyl) pyridin-3-yl) propanoic acid hydrochloride: a solution of 401mg (0.69mmol) of tert-butyl (2S) -2- (((9H-fluoren-9-yl) methoxy) carbonylamino) -3- (6- (2-ethyl-4-methoxyphenyl) pyridin-3-yl) propanoate in TFA (2.0mL) in a drying tube filled with calcium chloride to exclude air was stirred at room temperature for two hours. The reaction mixture was concentrated in vacuo at below 30 ℃ and the resulting orange oil was dissolved in ether to which was added 2mL of 1M HCl/ether solution. The resulting white solid was filtered and washed with diethyl ether to give the desired product as a white powder, 336mg, 84% yield.
Example 15
(S) -2- (((9H-fluoren-9-yl) methoxy) carbonylamino) -3- (6- (2-methylphenyl) pyridin-3-yl)
Tert-butyl propionate [ Fmoc- (S) -4- (2' -methylphenyl) -3-pyridylalanine tert-butyl ester]To another one of
A synthetic method is provided.
Scheme 17 below depicts an alternative synthesis of tert-butyl (S) -2- (((9H-fluoren-9-yl) methoxy) carbonylamino) -3- (6- (2-methylphenyl) pyridin-3-yl) propionate:
flow chart 17

1.(S) -2- (((9H-fluoren-9-yl) methoxy) carbonylamino) -3- (6- (2-methylphenyl) pyridine- 3-yl) propionic acid tert-butyl ester: to a stirred slurry of 1.75g (3.35mmol) of tert-butyl (S) -2- (((9H-fluoren-9-yl) methoxy) carbonylamino) -3- (6-bromo-pyridin-3-yl) propanoate with 913mg (6.70mmol, 2 equivalents) of 2-methylphenylboronic acid in 50mL of 1: 1 isopropanol/toluene was added 25.0mL of 2M aqueous sodium carbonate solution. The reaction mixture was purged twice with argon and evacuated then 124mg (0.167mmol, 0.05 eq) bis (tricyclohexylphosphine) palladium (II) chloride was added and then the mixture was again purged with argon and evacuated.
The rapidly stirred mixture was heated at 80 ℃ under argon. After 20h, the reaction mixture was cooled to room temperature and partially concentrated to remove the isopropanol. The residue was partitioned between ethyl acetate and water and the aqueous phase was extracted again with ethyl acetate. The organic extracts were combined, dried over magnesium sulfate, filtered and concentrated to give a brown oil. Purification by chromatography on silica gel (5X 15cm column) using ethyl acetate/dichloromethane (1: 9) as eluent gave the desired compound as a colourless oil, 1.81g, 90% yield.
Example 16
Scheme 18 below depicts a general synthesis for analogs of (2S) -2- (((9H-fluoren-9-yl) methoxy) carbonylamino) -3- (6-phenyl) pyridin-3-yl) propanoate.
Flow chart 18
1.(2S) -2- (((9H-fluoren-9-yl) methoxy) carbonylamino) -3- [ (6- (3-chloro-4-fluoro) phenyl) Pyridin-3-yl)]Tert-butyl propionate.
Into a round bottom flask was added 300mg of Fmoc-L-bromo-3-pyridylalanine (0.573mmol), 200mg of 3-chloro-4-fluorophenylboronic acid (1.145mmol, 2 equiv.), 1.145mL of 2M sodium carbonate solution (2.29mmol, 4 equiv.), 5mL of toluene, 5mL of isopropanol, and 42mg of PdCl2(PCy)3)2(0.0573mmol, 0.1 equiv.). The reaction solution was purged with argon, after which the temperature was allowed to reach 80 ℃ for 5 hours. The reaction was cooled to room temperature and diluted with 50ml of etoac. The solution was washed with water (30mL) and brine (20mL), dried over magnesium sulfate, filtered and concentrated. The crude oil was chromatographed on silica gel (12gm silica, 0-40% EtOAc/hexanes gradient) to give 245mg of the desired compound as an oil (75% yield).
2.(2S) -2- (((9H-fluoren-9-yl) methoxy) carbonylamino) -3- (6- [ (3-chloro-4-fluoro) phenyl) Pyridin-3-yl) propionic acid.
To a solution of tert-butyl (2S) -2- (((9H-fluoren-9-yl) methoxy) carbonylamino) -3- (6- [ (3-chloro-4-fluoro) phenyl) pyridin-3-yl) propanoate (240mg, 0.429mmol) in 3mL dichloromethane was added TFA (3 mL). The reaction was stirred at room temperature for 5 hours. The solvent was evaporated to dryness and the residue was subjected to preparative HPLC (methanol-water gradient, 0.1% TFA). Concentration of the fractions containing the product gave 200mg (93% yield) of the desired compound as the TFA salt.
Example 17
SEQ ID NO: 1 Synthesis of peptides
Prepared as described in example 1 and including as said Xaa11(S) -4- (2' -methylphenyl) -3-pyridylalanine of amino acid and X as saidaa10(S) - (2 '-ethyl-4' -methoxy) biphenylalanine of amino acids. Then applied as in example 1Described for amino acid Xaa1-Xaa9To accomplish peptide chain extension. The resulting peptidyl-resin was dried and treated with 2mL TFA/TIS/water (96: 2) for 1.5 hours. The resin was filtered off and washed with TFA (1X 1 ml). The combined filtrates were added to diethyl ether (30mL), vortexed slightly and then held at-15 ℃ for 1 hour. The precipitated solid was collected by centrifugation and dried in a fast vacuum apparatus (speed-vac). The crude product was purified by preparative HPLC as follows: the crude peptide was dissolved in 1mL of 0.1M sodium bicarbonate, 2mL of water, and 1mL of acetonitrile. The peptide was loaded onto a YMC column (SH-343-10P), 250X 20mm I.D., containing ODS-A10 μm packing material. The column was equipped with a guard column, YMC (G-340-10P), 50X 20mm I.D., containing ODS 10 μm packing material. The peptide was eluted with a gradient of 0.1% TFA/MeCN in 0.1% TFA/water, 20% to 45% over a 50 minute period at a flow rate of 15 ml/min. The appropriate fractions collected were pooled and lyophilized to give the peptide 98.6% pure and an HPLC retention time of 14.4 min under the following conditions: gradient, from 10% to 70% solvent B in solvent A at 1mL/min over 20 min. Solvent A: aqueous 0.1% TFA, solvent B: 0.1% TFA in acetonitrile. Column: YMC ODS-A100X 4.6mm, particle size 3 μm, pore size 12 nm. Mass spectrum: ESI (M + H)+1528.9 and (M +2H)/2 765.3.
SEQ ID NO: synthesis of 58 peptides
Mixing the above Xaa4-Xaa11Peptidyl-resin sample (0.067mmole) with Fmoc-L-Glu (OtBu) -OH (5 eq) -residue Xaa3And 0.5MHOAt in DMF (5 equiv) which had been vortexed for 5 minutes, and DIC (5 equiv) vortexed for 18 hours. The resin was drained and washed with DMF (4X 3 mL).
As previously for residue Xaa3The described procedure deprotects the resin-bound peptide (0.034mmole) and reacts with Fmoc- [ (S) - α -Me-Pro]-OH (5 equivalents) coupling to give said resin bound Fmoc- [ Xaa2-Xaa11]-a peptide.
According to the use for the disabledRadical Xaa2The procedure described deprotected the resin (0.017mmole) and coupled with Boc-L-His (Trt) -OH (5 eq.). The desired peptide was cleaved/deprotected from its corresponding peptidyl-resin by treatment with TFA/water/triisopropylsilane (94: 3) (5.0mL) for 3 hours. The resin was filtered off, rinsed with TFA (1.0mL), and the combined TFA filtrates were evaporated to give 39mg of crude peptide product as an oily solid. It was purified by preparative HPLC using a gradient from 5% to 65% in 0.1% TFA/AcCN in 0.1% TFA/water over 20 min. The fractions containing pure product were pooled and lyophilized to give 5.4mg (18.9% recovery) of 92% pureSEQ ID NO: 58 of a polypeptide(ii) a HPLC retention time 5.65min under the following conditions: a gradient from 5 to 80% 0.1% TFA/MeCN in 0.1% TFA/water over a 10min period at a flow rate of 2.5 mL/min; column: YMC S5ODS (4.6X 50 mm); ESI: (M + H)+=1554.8amu。
SEQ ID NO: 59 Synthesis of peptides
Said Fmoc- [ X ] to be described in the previous synthetic methodsaa3-Xaa11]peptidyl-Sieber resin sample (0.015mmole) with Fmoc- [ N-methyl- (D) -Ala]OH (5 equiv.) and 0.5M HOAt (5 equiv.) in DMF for 5 minutes after pre-vortexing and DIC (5 equiv.) vortexing for 4 hours. The resin was drained and washed with DMF (4X 3 mL). The Fmoc group was removed by treatment with 20% piperidine in DMF (3mL) for 5 and 15 minutes. The resin was washed with DMF (8X 3mL) and then coupled with Boc-L-His (Trt) -OH (5 equivalents) as described in the previous synthetic methods. The desired peptide was cleaved/deprotected from its corresponding peptidyl-resin by treatment with TFA/water/triisopropylsilane (94: 3) (5.0mL) for 3 hours. The resin was filtered off, rinsed with TFA (1.0mL), and the combined TFA filtrate was evaporated. The resulting oily solid was dissolved (1: 1) in acetonitrile/water (2mL) and purified by preparative HPLC using a gradient of 0.1% TFA/MeCN in 0.1% TFA/water from 5% to 65% over 20 min. The fractions containing pure product were pooled and lyophilized to give 5.2mg (18.5% recovery) of 99% pureSEQ ID NO: 59 Polypeptides(ii) a In thatHPLC retention time 5.65min under the following conditions: a gradient from 5 to 80% 0.1% TFA/MeCN in 0.1% TFA/water over a 10min period at a flow rate of 2.5 mL/min; column: YMCS5ODS (4.6X 50 mm); ESI: (M + H)+=1528.9amu。
SEQ ID NO: synthesis of 60 peptide
Fmoc deprotected [ X ] prepared as previously described was subjected to the procedure described in example 3aa10-Xaa11]Samples of-dipeptidyl-Sieber resin (0.05mmol) FastMoc Using an applied biosystems 433A peptide synthesizerTMThe scheme was run for another 9 coupling cycles. The Fmoc-protected dipeptidyl-resin (0.05mmol) was placed in an appropriately sized container on the instrument, washed 6 times with NMP and deprotected using two treatments with 20% piperidine/NMP (2 and 8min each). An additional monitored deprotection step is performed until the monitoring option is satisfactory. The total deprotection time is 10-12 min. The deprotected dipeptidyl-resin was washed 6 times with NMP and then coupled with the next amino acid. The procedure is illustrated by the example used in the next step.
Fmoc-L-Asp (OtBu) -OH was then coupled using the following method: Fmoc-L-Asp (OtBu) -OH (1mmol, 20 equivalents) was dissolved in 2mL of NMP and activated by the sequential addition of 0.45M HBTU/HOBt in DMF (2.2mL) and 2M DIEA/NMP (1 mL). The activated Fmoc-protected amino acid solution was then transferred to the reaction vessel and the coupling was allowed to proceed for 30 to 60min depending on the feedback from the deprotection step. The resin was then washed 6 times with NMP and the coupling procedure was repeated. To accomplish the required Xaa4-Xaa11Assembly of the sequences an additional 5 deprotection/coupling cycles were performed as described above. The Fmoc-amino acids subsequently coupled were: fmoc- (L) -His (Trt) -OH, Fmoc- (L) -Thr (tBu) -OH, Fmoc- (S) -2-fluoro-alpha-Me-Phe-OH, Fmoc- (L) -Thr (tBu) -OH and Fmoc-Gly-OH. Finally, the peptidyl-resin was washed 6 times with NMP and DCM. Fmoc-protected dipeptidyl-resin (0.025mmole) in N, N-dimethylformamideA/dichloromethane (55: 45) slurry was added to an ACT 396 multiple peptide synthesizer. The resin was washed 2 times with DMF and deprotected as described in example 1 using treatment twice with 1.5M piperidine/DMF. Fmoc-L-Glu (OtBu) -OH (4.0 equiv.) was activated by sequential addition of 0.5M HOAt in DMF (4.0 equiv.) and DIC (4.0 equiv.), then manually transferred to the reaction vessel and allowed to couple for 2 hours. The resin was rinsed with NMP (4X 0.5mL) with vortexing for 1min. Fmoc- [ (S) - α -Me-Pro was coupled as follows after deprotection of the Fmoc group as described for the previous coupling]-OH: fmoc- [ (S) - α -Me-Pro was purified by sequential addition of 0.5M HOAt in DMF (2.4 equiv.) diluted with NMP (0.12mL) and DIC (2.4 equiv.)]-OH (2.4 equiv.) activation. The solution was manually transferred to the reaction vessel and allowed to couple for 18 hours. The resin was rinsed with NMP. After deprotection of the Fmoc group, Fmoc- (L) -his (trt) -OH was coupled by manual addition of a solution of the amino acid (4 eq) diluted with NMP (0.2mL) in 0.5M HOAt in DMF (4 eq) and DIC (4 eq) to the reaction vessel. The coupling reaction was allowed to couple for 18 hours. The resin was rinsed with NMP. The Fmoc group was removed as described for the previous coupling. TFA cleavage/deprotection of the peptide was performed as described in example 1. It was purified by preparative HPLC using a gradient from 10% to 60% in 0.1% TFA/MeCN in 0.1% TFA/water over 20 min. The fractions containing pure product were pooled and freeze-dried to give 21.7mg (42% recovery) 94% pureSEQ ID NO: 60 Polypeptides(ii) a HPLC retention time 4.88min under the following conditions: a gradient from 5 to 80% 0.1% TFA/MeCN in 0.1% TFA/water over a 10min period at a flow rate of 2.5 mL/min; column: YMC S5ODS (4.6X 50 mm); ESI: (M + H)+=1604.9amu。
SEQ ID NO: 73 Synthesis of peptide
Fmoc deprotected [ x ] described in previous synthetic methodsaa2-Xaa11]peptidyl-Sieber resin sample (0.017mmole) with des-amino-His (Trt) -OH (5 eq.) and HATU (5 eq.)) A solution of 0.5HOAt in DMF (5 equivalents) and a solution of 2M DIEA in NMP (5 equivalents) were vortexed for 18 hours. The resin was drained and washed with DMF (6X 2mL) and DCM (3X 2 mL). The desired peptide was cleaved/deprotected from its corresponding peptidyl-resin by treatment with TFA/water/triisopropylsilane (94: 3) (5.0mL) for 3 hours. The resin was filtered off, rinsed with TFA (1.0mL), and the combined TFA filtrate was evaporated. The resulting oily solid (32mg) was dissolved (1: 1) in acetonitrile/water (2mL) and purified by preparative HPLC using a gradient of 0.1% TFA/MeCN in 0.1% TFA/water from 5% to 65% over 20 min. The fractions containing pure product were pooled and freeze-dried to give 7.4mg (24.6% recovery) of 99% pureSEQ ID NO:73(ii) a HPLC retention time 6.01min under the following conditions: a gradient from 5 to 80% 0.1% TFA/MeCN in 0.1% TFA/water over a 10min period at a flow rate of 2.5 mL/min; column: YMC S5ODS (4.6X 50 mm); ESI: (M + H)+=1539.8amu.
SEQ ID NO: synthesis of 61 peptide
Fmoc deprotected [ X ] prepared as described previously is prepared as described in example 3aa10-Xaa11]Samples of-dipeptidyl-Sieber resin (0.05mmol) FastMoc Using an Applied Biosystems 433A peptide synthesizerTMThe scheme was run for another 9 coupling cycles. The Fmoc-protected dipeptidyl-resin (0.05mmol) was placed in an appropriately sized container on the instrument, washed 6 times with NMP and deprotected using two treatments with 20% piperidine/NMP (2 and 8min each). An additional monitored deprotection step is performed until the monitoring option is satisfactory. The total deprotection time is 10-12 min. The deprotected dipeptidyl-resin was washed 6 times with NMP and then coupled with the next amino acid. The process is illustrated by the example for the next step.
Fmoc-L-Asp (OtBu) -OH was then coupled using the following method: Fmoc-L-Asp (OtBu) -OH (1mmol, 20 equivalents) was dissolved in 2mL of NMP and purified by the subsequent addition of 0.45MHBTU/HOBt in DMF (2.2mL) and 2M DIEA/NMP (1mL) were activated. The activated Fmoc-protected amino acid solution was then transferred to the reaction vessel and the coupling was allowed to proceed for 30 to 60min depending on the feedback from the deprotection step. The resin was then washed 6 times with NMP and the coupling procedure was repeated. To accomplish the desired Xaa4-Xaa11Assembly of the sequences an additional 5 deprotection/coupling cycles were performed as described above. The Fmoc-amino acids subsequently coupled were: fmoc- (L) -His (Trt) -OH, Fmoc- (L) -Thr (tBu) -OH, Fmoc- (S) -2-fluoro-alpha-Me-Phe-OH, Fmoc- (L) -Thr (tBu) -OH and Fmoc-Gly-OH. Finally, the peptidyl-resin was washed 6 times with NMP and DCM. The Fmoc-protected dipeptidyl-resin (0.025mmole) was added to an ACT 396 multiple peptide synthesizer in a N, N-dimethylformamide/dichloromethane (55: 45) slurry. The resin was washed 2 times with DMF and deprotected by treatment twice with 1.5M piperidine/DMF as described in example 1. Fmoc-L-Glu (OtBu) -OH (4.0 equiv.) was activated by sequential addition of 0.5M HOAt in DMF (4.0 equiv.) and DIC (4.0 equiv.), then manually transferred to the reaction vessel and allowed to couple for 2 hours. The resin was rinsed with NMP (4X 0.5mL) with vortexing for 1min. Fmoc- [ (S) - α -Me-Pro was coupled as follows after deprotection of the Fmoc group as described for the previous coupling]-OH: fmoc- [ (S) - α -Me-Pro was purified by sequential addition of 0.5M HOAt in DMF (2.4 equiv.) diluted with NMP (0.12mL) and DIC (2.4 equiv.)]OH activation (2.4 equiv.). The solution was manually transferred to the reaction vessel and allowed to couple for 18 hours. The resin was rinsed with NMP. After deprotection of the Fmoc group, Fmoc- (L) -his (trt) -OH was coupled by manually adding a solution of the amino acid (4 eq) diluted with NMP (0.2mL) in 0.5M HOAt in DMF (4 eq) and DIC (4 eq) to the reaction vessel. The coupling reaction was allowed to couple for 18 hours. The resin was rinsed with NMP. The Fmoc group was removed as described for the previous coupling. TFA cleavage/deprotection of the peptide was performed as described in example 1. Preparative by gradient from 10% to 60% over 20min using 0.1% TFA/MeCN in 0.1% TFA/waterIt was purified by HPLC. The fractions containing pure product were pooled and freeze-dried to give 21.7mg (42% recovery) 91% pureSEQ ID NO: 61 PolypeptidesAs determined by HPLC; the retention time was 20.8 minutes using the following conditions: gradient, from 10% to 60% solvent B in solvent a at 1mL/min during 25 minutes. Solvent A: aqueous 0.1% TFA, solvent B: 0.1% TFA in acetonitrile. Column: YMC ODS-A150X 4.6mm, particle size 3 μm, pore size 12 nm. Mass spectrum: ESI (M + H)+1568.9 and (M +2H)/2 785.2.
Example 18
(R, S) -3- (1- (2, 4-dinitrophenyl) -imidazol-4-yl) -2-methylpropanoic acid [ alpha-methyl-beta- [1- (2, 4-)
Dinitrophenyl) -imidazol-4-yl]Propionic acid]The synthesis of [ 3- (1H-imidazol-4-yl) -2-methyl
Propionic acid is abbreviated as Imp, see "amino acid abbreviations and structures" below "]
1-tosyl-4 (5) -hydroxymethylimidazole
The following procedures were taken from Agr.biol.chem., 38(5), 1097-2CO3(8.4g., 0.08mole) to a solution in water (40mL) was added 4- (hydroxymethyl) imidazole hydrochloride (2.7g, 0.02 mole). After complete dissolution, a solution of p-toluenesulfonyl chloride (4.58g, 0.024mole) in ethyl acetate (30mL) was added dropwise over a period of 5 minutes. The reaction mixture was allowed to stir for 5 hours. The layers were separated and more ethyl acetate (20mLs) was added. The organic phase was washed with 0.1M Na2CO3 (2X 20mL), water (1X 20mL) and then saturated NaCl (1X 20 mL). With 2g MgSO4And 1g of activated carbon the ethyl acetate was treated for 10 minutes. The solids were removed by filtration through a pad of celite and the solvent was removed on a rotary evaporator. The residue starts to crystallize. Fresh ethyl acetate (10mL) was added and the solution was then warmed with a heat gun to redissolve the solids. The product crystallized at room temperature overnight. The crystalline material was collected and washed with ethyl acetate (5mL) followed by ether (10mL)Washed and then dried in vacuo to a constant weight of 3.59 g.
1-tosyl-4 (5) -acetoxymethylimidazole
1-tosyl-4 (5) -hydroxymethylimidazole (2.52g, 10mmole) was dissolved in chloroform (10 ml). To this solution was added triethylamine (2.02g, 20mmole) dropwise at room temperature, followed by acetic anhydride (1.33g, 13mmole) dropwise over a period of 15 minutes. The mixture was stirred at room temperature and monitored by LC/MS for four days. The chloroform was removed by reduced pressure and the residue was dissolved in ethyl acetate (60 ml). The organic layer was washed with 1X 40ml each of 0.1M sodium bicarbonate, water then saturated sodium chloride. The organic layer was treated with activated carbon and magnesium sulfate at the same time and then filtered through a celite pad. The solvent was removed by reduced pressure and the resulting residue was dissolved in warm ethyl acetate (10 ml). To this solution was slowly added 20ml of diethyl ether. The solution was allowed to crystallize overnight at room temperature. The crystals were collected, washed with diethyl ether (2X 10ml) and dried overnight in vacuo to give 1.55 g.
Alpha-carbomethoxy-alpha-methyl-beta-4- (1-tosylimidazole) -propionic acid methyl ester
The following steps were taken from Synthetic Communications, 19 (7)&8),1157-1165, 1989. A solution of 1-tosyl-4 (5) -acetoxymethylimidazole (0.3516g, 1.2mmole) and dimethyl methylmalonate (0.1485g, 1.0mmole) in acetonitrile (2ml) was added to a stirred suspension of powdered KOH (0.1694g, 3.0mmole) and tetrabutylammonium bromide (0.0496 g, 0.15mmole) in acetonitrile (1 ml). The reaction was complete after 40mins as determined by HPLC analysis. The reaction mixture was poured into diethyl ether (100ml), filtered through a pad of celite and the solvent was removed by evaporation under reduced pressure. The remaining oil was dissolved in 30ml ethyl acetate and washed with 0.1M NaHCO3(1X 15ml), saturated NaCl (1X 15ml) and then MgSO4And (5) drying. The solvent was removed under reduced pressure and the resulting oil was placed in a desiccator in vacuo for 3 days to give 0.207 g.
Alpha-methyl-beta-4-imidazole propionic acid
Alpha-carbomethoxy-alpha-methyl-beta-4- (1-tosylimidazole) -propionic acid methyl ester (0.186g, 0.5mmole) was dissolved in 2ml of methanol. 1.5ml of 1.0N NaOH was added thereto and the reaction was allowed to stir overnight. After purification by preparative HPLC, the product obtained by freeze-drying (0.1366g) was dissolved with 5ml of 1.0N NaOH and heated at 100 ℃ for 2 hours in a 16X 100mm screw-cap tube sealed with a PTFE-lined cap, followed by addition of 2ml of concentrated HCl and subsequent heating at 145 ℃ for 6 hours. The desired decarboxylated product is thus formed. The entire solution was filtered and loaded onto a YMC G-340-10P ODS 50X 20mm preparative HPLC column. The product was eluted with a gradient of 0.1% TFA/MeCN in 0.1% TFA/water over 60 minutes. Fractions corresponding to 11 to 13 minutes in the gradient were pooled, frozen and lyophilized to give 32mg of product.
Alpha-methyl-beta- [1- (2, 4-dinitrophenyl) -imidazol-4-yl]Propionic acid
To a solution of α -methyl- β -4-imidazolopropionic acid (0.0305g, 0.114mmoles) and sodium bicarbonate (0.0617g, 0.734mmole) in water (1mL) (pH 8.04) was added a solution of 2, 4-dinitrofluorobenzene (0.0323g, 0.174mmole) in MeCN (1.0 mL). The reaction mixture was vortexed overnight. The MeCN was removed under reduced pressure and the residue was redissolved in 2mL water, filtered and loaded in two aliquots of 1.5 and 0.5mL each onto a Phenomenex Luna C18(2)5 μm 100 × 21.2mm preparative HPLC column. The product was eluted with a gradient of 0.1% TFA/MeCN in 0.1% TFA/water from 0% to 80% over a 40 minute period. Fractions corresponding to 12.5 to 14.5 min in the gradient were pooled and pooled in a Savant SpeedVacTMDried overnight. Additional product was recovered by dissolving the water insoluble crude product in DMSO, followed by preparative HPLC as described above. The combined fractions yielded 31mg of pure product after freeze-drying.
Example 19
SEQ ID NO: 77. synthesis of 78, 79 and 80 peptides
(R, S) -3- (1- (2, 4-dinitrophenyl) -imidazol-4-yl) -2-methylpropionic acid was coupled to the relevant X as followsaa2-Xaa11-peptidyl-Sieber resin:
to a solution of (R, S) -3- (1- (2, 4-dinitrophenyl) -imidazol-4-yl) -2-methylpropanoic acid (0.0267g, 0.083mmoles), 6-Cl-HOBt (0.0151g, 0.089mmoles) and HCTU (0.0360g, 0.087mmoles) in 1mL of NMP/DCM (3: 1) was added DIEA (0.0315g, 0.244 mmoles); the solution was vortexed slightly and then added to the relevant Fmoc deprotected X prepared as described in example 19aa2-Xaa11peptidyl-Sieber resin. The coupling was allowed to proceed for 16 hours. The peptidyl-resin was washed with NMP followed by DCM (3X 1.5mL X1 min), then treated with 10% acetic anhydride in DCM for 1X 2mL X90 min, followed by washing with DCM followed by DMF (3X 1.5mL X1 min). The peptidyl resin was treated with 10% thiophenol in DMF (1.5mL) for 1 hour and then washed with DMF and DCM (4X 1.5mL X1 min). The peptidyl resin was then treated with TFA/DCM/TIS (3: 1.9: 0.1) (1mL) for 10min and filtered. The filtrate was collected and vortexed gently for an additional 1 hour. The TFA mixture was concentrated to about 0.5mL in a flash vacuum and added to 4mL MTBE. After 1 hour, the precipitated product was collected by centrifugation, washed and then dried to obtain 0.0841g of a crude product. It was purified by preparative HPLC as follows: the crude peptide was dissolved and injected into a Phenomenex Luna C18(2) (5 μm, 250 x 30mm) column and eluted with a linear gradient of 0.1% TFA/MeCN in 0.1% TFA/water at a flow rate of 15mL/min from 20% to 50% over a 40min period and the effluent UV detection was performed at 217 nm. The fractions containing the desired product were pooled and lyophilized to give 26.7mg of 97.5% pure peptide; HPLC retention time 21.2min under the following conditions: gradient from 10% to 60% of a solution of solvent A in solvent B at 1mL/min during 25 minutes. Solvent A: 0.1% TFAThe aqueous solution of (1), solvent B: 0.1% TFA in MeCN. Column: YMC ODS-A150X 4.6m m, particle size 3 μm, pore size 12 nm. Mass spectrum: ESI (M + H)+1527.9 and (M +2H)/2 764.9.
Preparative chiral HPLC purification of the peptide: the diastereomeric peptide mixture (10mg) was dissolved in MeCN/MeOH. The solution was loaded onto a Chirobiotic V2.2X 50cm, 5 μm column and then eluted with MeCN/MeOH/N (CH)2CH3)3/CH3COOH: 65/35/0.5/0.5 was eluted at 20 mL/min. Isomer a was collected between 29 and 35 minutes. Isomer B was collected between 36 and 44 min. The second round (operation) is performed as described above. The fractions containing isomer a were combined, concentrated to approximately 5mL, diluted with water/MeCN (4: 1) and the solution was then freeze dried. Isomer B was treated in the same manner. The resulting residue was converted to a TFA salt using preparative HPLC. Each peptide was injected into a Phenomenex Luna C18(2)5 μm 100 x 21.2mm column and eluted with a linear gradient of 0.1% TFA/MeCN in 0.1% TFA/water at a flow rate of 10mL/min from 20% to 50% during 40min and the effluent UV detected at 217 nm. The fractions containing the desired product were pooled, frozen and lyophilized to give 6.0mg of 100% pure isomer a with an HPLC retention time of 21.28min under the following conditions: gradient, from 10% to 60% solvent B in solvent a solution at 1mL/min during 25 minutes. Solvent A: aqueous 0.1% TFA, solvent B: 0.1% TFA in MeCN. Column: YMC ODS-A150X 4.6mm, particle size 3 μm, pore size 12 nm. Mass spectrum: ESI (M + H)+1527.6 and (M +2H)/2 764.7. In a similar manner, 4.9mg of 100% pure peptide isomer B were obtained; HPLC retention time under the following conditions was 21.3 min.: gradient, from 10% to 60% solvent B in solvent a solution at 1mL/min during 25 minutes. Solvent A: aqueous 0.1% TFA, solvent B: 0.1% TFA in MeCN. Column: YMC ODS-A150X 4.6mm, particle size 3 μm, pore size 12 nm. Mass spectrum: ESI (M + H)+1527.5 and (M +2H)/2 764.6
Example 20
The 11-mer peptides listed in table I were prepared using the synthetic methods described herein. X for each compound listed in Table 3aa1-Xaa11Reference is made to the following formula I:
Xaa1-Xaa2-Xaa3-Xaa4-Xaa5-Xaa6-Xaa7-Xaa8-Xaa9-Xaa10-Xaa11
I
TABLE 3


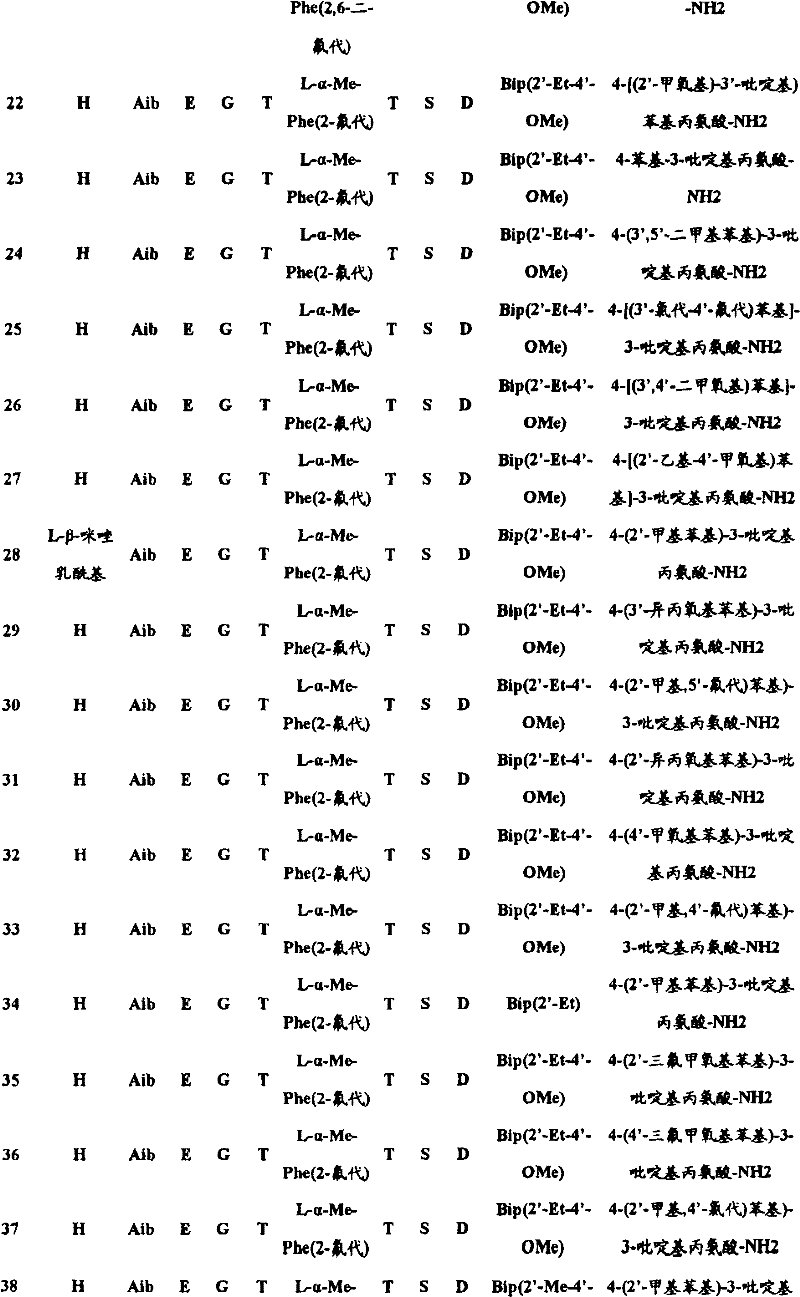


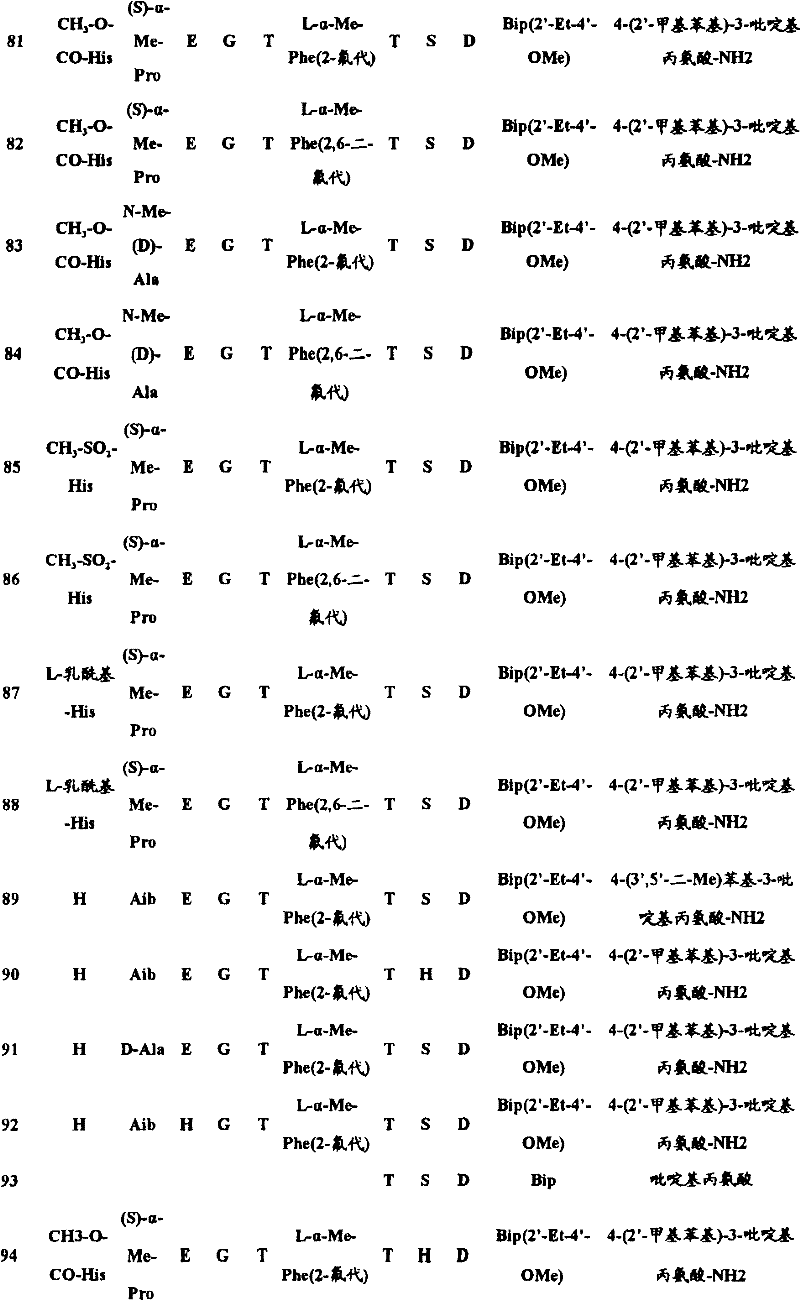
Amino acid abbreviations and structures
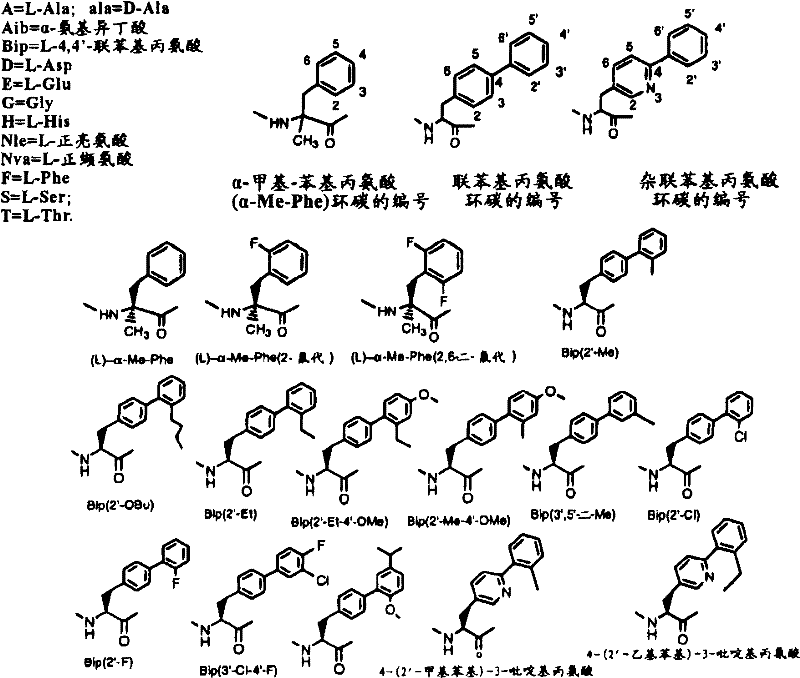
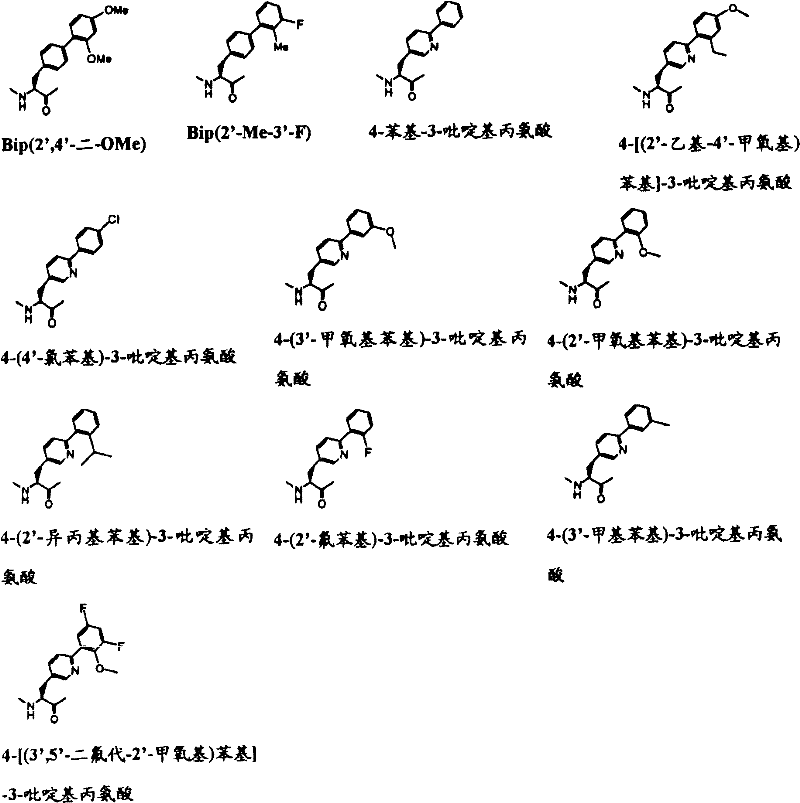

As is known to those skilled in the art of amino acid and peptide chemistry, a phenylalanine amino acid with a phenyl substituent at the 4 or para position can be alternatively defined as 4- (phenyl) phenylalanine or 4, 4' -biphenylalanine, and thus may be referred to simply as "Bip". For the purpose of the "amino acid abbreviations and structures" section and abbreviations shown in the tables herein, a biphenylalanine amino acid may be abbreviated as, for example, "Bip (2 ' -Me)" which is meant to denote a phenylalanine substituted at the 4-position with a2 ' -methylphenyl group and wherein the 2 ' -methyl group is located at an ortho position relative to the point of attachment to the phenyl ring.
Example 21
(2(R & S) - ((((9H-fluoren-9-yl) methoxy) carbonyl) -3- (2-o-tolylpyrimidin-5-yl) propionic acid]
And (4) synthesizing.
The following scheme 21 depicts the synthesis of (2S) -2- (((9H-fluoren-9-yl) methoxy) carbonylamino) -3- (6-o-tolylpyrimidin-5-yl) propionic acid hydrochloride:
flow chart 21

1.N' -hydroxy-2-methylbenzamidine.
To a stirred solution of 9.92g (84.7mmol) o-tolunitrile and 7.82g (93.2mmol) sodium bicarbonate in water (20mL) was added isopropanol (100 mL). The reaction mixture was heated to reflux under argon. After 20h, the reaction mixture was cooled and evaporated to remove most of the isopropanol. The semi-solid residue was partitioned between water and hexanes and the resulting solid was collected and dried to give a white solid, 8.03g, 83% yield.
2.2-methylbenzamidine acetate.
To a stirred solution of 7.82g of the above compound (52.1mmol) in acetic acid (100mL) was added 5.65mL of acetic anhydride (5.65mmol, 1.15 equivalents) and 1.0g of 10% palladium on carbon at room temperature under argon. The mixture was stirred and purged twice with argon. The mixture was then stirred under a hydrogen atmosphere (-1 atm) for 3 h. The reaction mixture was purified and filtered through celite. The filtrate was evaporated to give the product as a brown solid, 10.32g, 99% yield.
3.4-hydroxy-2-o-tolylpyrimidine-5-carboxylic acid ethyl ester.
To stirred ethanol (57mL) at 0-5 ℃ under argon was added portionwise 2.35g of an oil dispersion of sodium hydride (60%, 58.7mmol) over a period of 10 min. After a further 10min, a solution of 5.70g of 2-methylbenzamidine acetate (29.3mmol) was added. When 5.93mL (2-ethoxy-methylene) diethyl malonate (29.3mmol) was added during 5min14mL of a solution in ethanol, the resulting orange slurry was stirred. The mixture was heated to reflux to form a solution. After 15h, the solution was cooled and poured into 300mL of ice water. The resulting solution was stirred and 2.3mL of concentrated HCl (27.6mmol) was added to bring the solution to pH 7. The resulting off-white flocculated slurry was filtered and washed with N2Drying under air flow gave the title compound as a white solid, 6.87g, 91% yield.
4.4-chloro-2-o-tolylpyrimidine-5-carboxylic acid ethyl ester.
A solution of 4.00g of ethyl 4-hydroxy-2-o-tolylpyrimidine-5-carboxylate (15.5mmol) in 11.6mL of phosphorus oxychloride (124mmol) was heated to reflux (with a tube filled with calcium chloride as a protection against the outside air). After 3h, the excess POCl was distilled (. about.20 torr, 90-100 ℃ oil bath)3And (5) removing. The remaining red oil solidified upon cooling. The solids were ground to a powder and covered with 100mL EtOAc. The slurry was cooled to-5 ℃ and stirred vigorously with 25mL potassium carbonate solution (2M, 50mmol) for 10 min. The resulting organic phase was separated and dried (MgSO)4) Filtration and evaporation to triturate in hexanes gave the title compound as a pale orange solid, 3.58g, 84% yield.
5.2-o-tolylpyrimidine-5-carboxylic acid ethyl ester.
A solution of 2.33g of the above compound (8.42mmol) and 1.17mL triethylamine (8.42mmol) in 35mL ethyl acetate was treated with 210mg of 10% palladium on carbon at room temperature. The rapidly stirred mixture was purged twice with argon and then exposed to a hydrogen atmosphere (-3 atm) for 3 h. The reaction mixture was purified, filtered through celite and evaporated. Re-evaporation from hexanes gave the title compound as a yellow solid, 1.88g, 92% yield.
6.(2-o-tolylpyrimidin-5-yl) methanol.
To a solution of 2.10g of the above compound (8.67mmol) in 45mL of dichloromethane stirred at-78 deg.C under argon was added 12.7mL of diisobutylaluminumSolution in toluene (1.5M, 19.1 mmol). After 90min, 19.1mL of a solution of sodium potassium tartrate (1M, 19.1mmol) was added dropwise and the precision-frozen dispersion was stirred and allowed to warm to room temperature. The mixture was partitioned between water and dichloromethane. The organic phase was separated and dried (MgSO)4) Filtered and then evaporated to give a yellow oil. Purification by silica gel chromatography (5X 15cm column, 1: 1 EtOAc/hexanes) provided the title compound as a white solid, 975mg, 56% yield.
7.(2-o-tolylpyrimidin-5-yl) methyl bromide with (R, S)2- (((9H-fluoren-9-yl) methoxy) Carbonyl) -3- (2-o-tolylpyrimidin-5-yl) propionic acid tert-butyl ester.
A mixture of 918mg of the above-mentioned compound (4.58mmol) and 2.20g of phosphorus oxybromide (7.67mmol) was heated to 120 ℃ with a calcium chloride-filled drying tube as protection from the outside air. After 1h, the reaction mixture was distilled (. about.20 torr, 90-100 ℃ oil bath). The resulting black resinous residue was cooled to room temperature, covered with EtOAc and cooled to 0 ℃ and then finely treated with 10mL of sodium carbonate solution (2N, 10 mmol). Drying the organic extract, unstabilized 2-o-tolylpyrimidin-5-yl) methyl bromide (MgSO4) Filtered and evaporated at < 30 ℃ to give 1.35g of a yellow glass which was used directly in the following reaction.
To a mixture of 1.31g (3.64mmol) of the above compound, 1.075g (3.64mmol, 1.0 eq) of tert-butyl 2- (diphenylmethyleneamino) acetate and 117mg (0.36mmol, 0.1 eq) of tetrabutylammonium bromide in 16mL of dichloromethane stirred at-78 ℃ under argon was added 1.58mL (5.46mmol, 1.5 eq) of 2-tert-butylimino-2-diethylamino-1, 3-dimethyl-perhydro-1, 3, 2-diazaphosphorine during 5min. The reaction mixture was stirred at-78 ℃ for 1h and then allowed to warm in situ to room temperature. After 17h, the mixture was directly purified by silica gel chromatography (5X 20cm column) using ether/dichloromethane (3: 47) as eluent to give tert-butyl 2- (diphenylmethyleneamino) -3- (6-o-tolylpyrimidin-5-yl) propionate as a yellow oil, 1.45g, 78% yield.
To a stirred solution of 1.45g (2.33mmol) tert-butyl 2- (diphenylmethyleneamino) -3- (6-o-tolylpyrimidin-5-yl) propionate in 12mL THF at room temperature under argon was added 12mL of an aqueous solution of 3.40g (17.7mmol, 5.8 equivalents) citric acid. After 3h, the reaction mixture was diluted with water and washed twice with diethyl ether. The aqueous phase was then adjusted to pH 9 with solid sodium carbonate and then extracted twice with dichloromethane.
The dichloromethane extracts were combined, dried over sodium sulfate and concentrated. The resulting oil was dissolved in 6mL of THF and then treated successively with 4.2mL of 10% sodium carbonate solution and 864mg (3.34mmol, 1.1 equiv.) of 9-fluorenylmethoxycarbonyl chloride at room temperature. After 2h, the reaction mixture was extracted twice with EtOAc, dried over sodium sulfate, filtered, concentrated and purified by chromatography on silica gel (5 × 15cm column) using ethyl acetate/dichloromethane (1: 19) as eluent to give a colorless oil, 1.61g, 99% yield.
The product was dissolved in 1: 1EtOH/MeOH (130 mL). After 10min, a precipitate formed. After filtration, the filtrate was subjected to chiral chromatography (Chiralpak AD column, 5X 50cm, 20. mu. packing; 2: 96 MeOH/EtOH/hexane as eluent at a flow rate of 50mL/min) to give, after collection, concentration and evaporation, two fractions which were analyzed by manual HPLC (4.6X 250mm AD column, 2: 96 heptane: methanol: ethanol as eluent at a flow rate of 1mL/min) both:
(S) tert-butyl 2- (((9H-fluoren-9-yl) methoxy) carbonyl) -3- (2-o-tolylpyrimidin-5-yl) propionate (identified by comparison to the pyridine analogue), 221mg, > 98% ee; and (R) tert-butyl 2- (((9H-fluoren-9-yl) methoxy) carbonyl) -3- (2-o-tolylpyrimidin-5-yl) propionate, 295mg, 96% ee.
8a.(2S) -2- (((9H-fluoren-9-yl) methoxy) carbonylamino) -3- (6-o-tolylpyrimidine- 5-yl) propionic acid hydrochloride.
A solution of 220mg (0.41mmol) of tert-butyl (2S) -2- (((9H-fluoren-9-yl) methoxy) carbonylamino) -3- (6-o-tolylpyrimidin-5-yl) propionate in TFA (2.1mL) in a drying tube filled with calcium chloride was stirred at room temperature for 4 hours with exclusion of air. The reaction mixture was concentrated in vacuo at below 40 ℃ and the resulting orange oil was redissolved in toluene twice and evaporated to give the title compound as a white powder, 195mg, 99% yield.
8b.(2R) -2- (((9H-fluoren-9-yl) methoxy) carbonylamino) -3- (6-o-tolylpyrimidine- 5-yl) propionic acid hydrochloride.
A solution of 290mg (0.54mmol) of tert-butyl (2R) -2- (((9H-fluoren-9-yl) methoxy) carbonylamino) -3- (6-o-tolylpyrimidin-5-yl) propionate in TFA (2.7mL) in a drying tube filled with calcium chloride to exclude air was stirred at room temperature for 4 hours. The reaction mixture was concentrated in vacuo at below 40 ℃ and the resulting orange oil was redissolved in toluene twice and evaporated to give the title compound as a white powder, 255mg, 98% yield.
Example 22
(2S) -2- (((9H-fluoren-9-yl) methoxy) carbonyl) -2-methyl-3- (6-o-tolylpyridine-3-
Yl) propionic acid synthesis.
The following scheme 22 describes the synthesis of ((2S) -2- (((9H-fluoren-9-yl) methoxy) carbonyl) -2-methyl-3- (6-o-tolylpyridin-3-yl) propionic acid:
flow chart 22
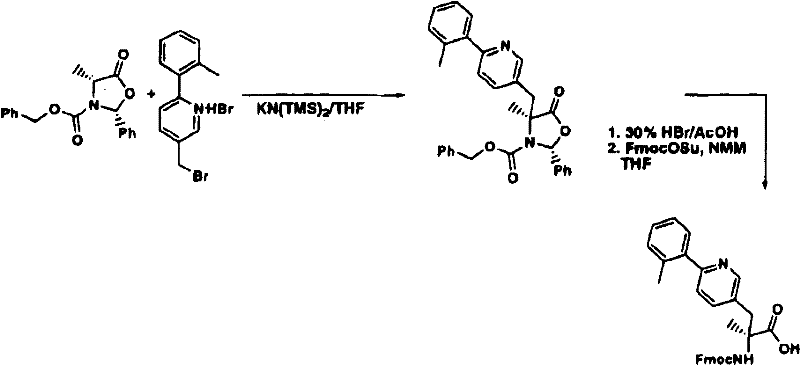
1.(2R, 4S) -4-methyl-5-oxo-2-phenyl-4- ((6-o-tolylpyridin-3-yl) methyl Base)  Oxazolidine-3-carboxylic acid benzyl ester.
Oxazolidine-3-carboxylic acid benzyl ester.
To 515mg (1.50mmol) of the slurry of example 11.4 stirred at-78 ℃ under argon was added 3.08mL of a solution of potassium hexamethyldisilazide (0.5M in toluene, 1.54 mmol). After 20min, similar amounts of potassium hexamethyldisilazide and 467mg (1.50mmol) (2R, 4R) -4-methyl-5-oxo-2-phenyl-amine in a separate syringe were added A solution of oxazolidine-3-carboxylic acid benzyl ester (s.r. kappa, j.org.chem.661903(2001)) in 2mL THF was added dropwise alternately during 30 min. The reaction mixture was allowed to warm to room temperature. After 14h, the reaction mixture was poured into saturated sodium bicarbonate solution and extracted twice with diethyl ether. The organic extracts were combined and dried (Na)2SO4) Filtration and evaporation. Purification by silica gel chromatography (5X 15cm column, 1: 3 EtOAc/hexane as eluent) afforded the title compound as a white foam, 240mg, 33% yield.
A solution of oxazolidine-3-carboxylic acid benzyl ester (s.r. kappa, j.org.chem.661903(2001)) in 2mL THF was added dropwise alternately during 30 min. The reaction mixture was allowed to warm to room temperature. After 14h, the reaction mixture was poured into saturated sodium bicarbonate solution and extracted twice with diethyl ether. The organic extracts were combined and dried (Na)2SO4) Filtration and evaporation. Purification by silica gel chromatography (5X 15cm column, 1: 3 EtOAc/hexane as eluent) afforded the title compound as a white foam, 240mg, 33% yield.
2.(2R, 4S) -4-methyl-5-oxo-2-phenyl-4- ((6-o-tolylpyridin-3-yl) methyl Base)  Oxazolidine-3-carboxylic acid benzyl ester.
Oxazolidine-3-carboxylic acid benzyl ester.
A stirred solution of 240mg (0.49mmol) of the above compound and 4mL of 30% HBr in acetic acid was heated to reflux for 24 h. The resulting solution was evaporated to dryness and then redissolved in 9mL of water. The solution was extracted once with ether. The pH of the aqueous phase was adjusted to 8 with solid sodium bicarbonate (750mg) and then stirred at room temperature when 216mg (0.84mmol) of FmocOSu in THF (3mL) was added. After 60h, the reaction mixture was quenched with 5% citric acid solution and extracted twice with EtOAc. The combined organic extracts were dried (MgSO)4) Filtration and evaporation. Crystallization from 9: 1 acetonitrile/water gave a white solidTitle compound in the form of a solid, 52mg, 22%.
Example 23
Determination of cyclic AMP
The GLP-1 receptor is a G-protein coupled receptor. A biologically active form of GLP-1(7-36) -amide binds to the GLP-1 receptor and via signal transduction causes activation of adenylate cyclase and increases intracellular cAMP concentration. To monitor the agonistic effect of peptide compounds in stimulating the GLP-1 receptor, adenylate cyclase activity was monitored by measuring intracellular cAMP levels. The full-length human glucagon-like peptide 1 receptor was stably expressed in CHO-K1 cells and clonal lines were established. Clonal lines with the greatest cAMP content increase in response to saturating doses of GLP-1 are selected from said clonal lines to select the clonal line CHO-GLP 1R-19.
Cells were cultured in Ham's F12 nutrient medium (Gibco # 11765-054), 10% FBS, 1x L-glutamine, 1x Pen/Strep and 0.4mg/ml G418. CHO-GLP-1R-19 cells (20,000 in 100. mu.l medium) were plated into each well of a 96-well tissue culture microtiter plate and then plated at 37 ℃ in 5% CO2The culture was carried out overnight under an atmosphere. On the day of the assay, cells were washed once with 100 μ Ι phosphate-buffered saline (PBS). All peptides were serially diluted with Biomek 2000 before the assay was started. Serial dilutions were performed in 100% DMSO. Peptide plates were generated prior to starting the assay using platimate Plus; 1.5uL of the compound was transferred into a V-bottom plate and then 150uL of assay buffer supplemented with 100. mu.M 3-isobutyl-1-methylxanthine (a non-selective phosphodiesterase inhibitor) was added to the plate to give a 1: 100 dilution and a final concentration of DMSO of 1%.
To generate a cAMP standard curve, cAMP was serially diluted to a range of 0.2-25.6 pmol/well in lysis reagent 1(Amersham cAMP SPA kit). 50 μ l of each cAMP standard was added manually and 70 μ l of the pooled reagent (Amersham cAMP SPA kit) was added with a multidrop. The plates were then sealed and counted on a Trilux counter after 15 hours. CPM was converted to pmol cAMP using this standard curve.
1.Procedure for cAMP determination on Platemate Plus.
Cell plates and peptide plates were loaded onto the platimate. The medium was aspirated from the wells and discarded. 100uL of peptide/buffer mixture from the peptide plate per well was then added to start the assay. After incubation for 30 minutes the peptide/buffer was removed and 50uL of the lysis reagent 1 solution was added to each well. The plates were kept at room temperature for one hour, overnight if frozen and sealed. 70uL of the cAMP detection reagent was added to the Multidrop (premixed with125I-CAMP analogue, anti-CAMP antibody and anti-rabbit antibody conjugated to SPA beads-all from the Amersham CAMP SPA kit) and the plates were sealed. After 15 hours the plates were counted on a Trilux scintillation counter.
The dose dependence of the compounds was determined in duplicate at half-log concentration. Ten nMGLP-1 was used as a reference standard for determining maximal activity. The standard curve was determined with known amounts of cyclic AMP. The amount of cAMP synthesized by treated cells was measured from the cyclic AMP standard curve, calculated as a percentage of maximal GLP-1 stimulating activity and plotted against compound log concentration. The EC of the compound was determined by analyzing the data using a non-linear regression curve fitting method (4-parameter sigmoidal dose-response curve)50The value is obtained. For example, the peptides of the invention have an EC in the range of 0.0005nM to 10nM, more preferably in the range of 0.0005nM to 0.200nM50The value is obtained.
Example 24
In vivo studies
The peptide is dissolved in a suitable vehicle at a concentration of nmol/ml which corresponds to the dose administered at nmol/kg such that each mouse will receive the same volume/weight of the dose solution. Male C57BL/6J-ob/ob mice (10 weeks old) were randomly assigned to groups of 6 mice each based on plasma glucose and body weight fed. After an overnight fast, mice were weighed and placed in the laboratory. After 30min in the environment, the mice were bled caudally at-30 min and immediately injected subcutaneously (sc) with vehicle or peptide dissolved in vehicle (0.1ml solution/100 g body weight). At time 0, the mice were bled and then injected intraperitoneally with 50% glucose (2g/kg) to begin the intraperitoneal glucose tolerance test (ipGTT). The mice were bled at 30, 60, 120 and 180min after the glucose injection. Blood samples were drawn into potassium EDTA, placed on ice during the study and then centrifuged at 3000rpm for 10min at 4 ℃. Blood samples were diluted 11-fold for glucose analysis in the Cobas system. Another 5 μ l blood sample was diluted 5-fold with 20 μ l sample diluent (insulin ELISA assay kit, Crystal Chem Inc.) and stored at-20 ℃ for subsequent analysis using the hypersensitive mouse insulin ELISA kit (Crystal Chem Inc.).
The in vivo glucose lowering properties of compound I, compound II and III in ob/ob mice (insulin resistant mouse model) are as described above. Subcutaneous injection administration of peptide I attenuated the postprandial glucose excursion curve in the intraperitoneal glucose tolerance test (ipGTT) and the plasma glucose area under the curve (AUC) decreased in a dose-dependent manner between 0 and 180 minutes (figure 1). Determination of ED of Compound I50The value was 50 nmoles/kg. The postprandial plasma insulin levels in these animals were also accompanied by a statistically significant dose-dependent increase (fig. 2) the correlation between plasma glucose and insulin changes in animals treated with compound I (fig. 1 and 2) shows that the glucose lowering effect is mediated by compound I promoting the release of insulin.
More significantly and unexpectedly, when SEQ ID NO's are: 1 and 58 into ob/ob mice subcutaneously, postprandial plasma glucose production is time-dependent (between 0 and 180 points)Between clocks) was statistically significant (fig. 3 and 4). The effect of compound II on postprandial glucose was dose-dependent between 1-100nmol/kg and the plasma glucose AUC decreased by 85.8% at the 100nmol/kg dose (figure 3). More significantly and unexpectedly, seq id NO: 1 ED of peptides50At 5nmoles/kg, compound II was shown to be about 10 times more potent than compound I on a dose basis. Determined by the determination of SEQ ID NO: ED of glucose lowering Activity of 58 peptides50It was 2.5nmol/kg (FIG. 4).
Example 25
Pharmacokinetic study in dogs
SEQ ID NO: pharmacokinetic parameters for the 1 peptide were determined in male beagle dogs (n ═ 4, 14 ± 1 kg). After an overnight fast, each animal received the sequence of SEQ id no: 1 peptide. Following the crossover design, each animal received intravenous and subcutaneous doses with a one week washout period between doses. The dosage excipient for both routes of administration is propylene glycol phosphate buffer (50: 50). A series of blood samples were collected in microcentrifuge tubes containing EDTA before dosing, 0.083, 0.25, 0.5, 0.75, 1,2, 4,6, 8, 24 and 30 hours after intravenous administration, and 0.25, 0.5, 0.75, 1,2, 4,6, 8, 24 and 30 hours before dosing, after subcutaneous administration. Approximately 0.3mL of blood was collected at each time point. Blood samples were immediately centrifuged at 4 ℃. The resulting plasma was then frozen using dry ice and stored at-20 ℃. Plasma drug levels were determined using the LC-MS/MS assay as described above.
By LC-MS/MSSEQ ID NO: 1 peptideQuantitative determination of
Plasma samples from in vivo dog studies were prepared for analysis by precipitating plasma proteins with two volumes of acetonitrile containing an internal standard. Vortexing the sample and removing the precipitated proteins by centrifugation. The resulting supernatant was then transferred to a 96-well plate and 10 μ Ι _ was injected for analysis. Samples were prepared using a Packard Multiprobe II and Quadra 96 liquid handling system.
The HPLC system consisted of two Shimadzu LC10AD pumps (Columbia, MD), a CTC PAL autosampler (Leap Technologies, Switzerland). The column used was YMC Hydrosphere C18 (2.0X 50mm, 3 μm) (YMC, Inc., Milford, MA). The column temperature was maintained at 50 ℃ and the flow rate was 0.3 mL/min. The mobile phase a consisted of 10mM ammonium formate and 0.1% formic acid in water and the mobile phase B consisted of 0.1% formic acid in acetonitrile. The starting mobile phase composition was 5% B and held at 5% B for one minute to allow the column to equilibrate. The composition was ramped to 95% B over a two minute period and held for an additional minute. The mobile phase was then returned to the initial state within one minute. The total analysis time was five minutes. And a switching valve is used. The eluate is transferred between 0-1 minute to waste.
The HPLC was connected to a Sciex API 4000 mass spectrometer, (applied biosystems, Foster City, Calif.) and equipped with a Turbolonspray ion source. Ultra-high purity nitrogen was used as the atomizing and pressurizing (turbo) gas. The pressurized gas temperature was set to 300 ℃ and the interface heater was set to 60 ℃. Data acquisition used selective reaction detection (SRM). Selecting in Q1 a nucleic acid sequence representing SEQ ID NO: 1 of peptide (M +2H)2+Ions and (M +2H) representing BMS-501143(IS)2+The ions were then treated with high purity nitrogen at 3.5X 10-3Which is collisionally dissociated at pressure to form specific product ions which are then monitored by Q3. The migration and voltage are summarized in table 4.
Table 4.SEQ ID NO: 1 peptideAnd MS/MS analysis parameters of internal standard substance
| SEQ ID NO: 1 peptide | Internal standard substance | |
| SMR migration (mz) | 765.1→195.2 | 740.7→210.0 |
| Declustering voltage | 60 | 60 |
| Collision energy (V) | 45 | 30 |
Standard curves at concentrations ranging from 1 to 1000nM and 4 to 5000nM were used for in vivo samples from low and high doses, respectively. Fitting the curve to the inverse concentration (1/x)2) Weighted quadratic regression. Standards were analyzed in duplicate. Quality Control (QC) samples prepared in a blank matrix at the same concentration as the standards were also analyzed in each analysis group. For SEQ id no: peptide 1, with QC calculated at concentrations above 80% within 20% of the nominal concentration, showed acceptable assay performance.
Data analysis
Using KINETICA according to non-compartmental methodTMThe software program analyzed SEQ ID NO: 1 plasma concentration of peptide versus time. The Cmax and Tmax values were recorded directly from experimental observations. The AUC0-n and AUCtot values were calculated using a combined linear and logarithmic trapezoidal summation.Total plasma Clearance (CL) was calculated after intra-arterial or intravenous administrationP) Ultimate half-life (t)1/2) Average residence time (MRT), and steady state volume of distribution (Vss). Total blood Clearance (CL) was calculated using the total plasma clearance and the ratio of blood to plasma concentrationB). CLB and Vss values were compared to standard liver blood flow and total body water values, respectively, reported in the literature. The absolute subcutaneous bioavailability (expressed as%) was determined by finding the sequence of SEQ ID NO: 1 peptide is calculated as the ratio of the dose normalized AUC value obtained after subcutaneous administration to the dose normalized AUC value obtained after intravenous administration.
Pharmacokinetic results in dogs
Following Intravenous (IV) and Subcutaneous (SC) administration, the amino acid sequence of SEQ ID NO: pharmacokinetic parameters for peptide 1 are summarized in table 5.
SEQ ID NO: peptide 1 exhibited low systemic clearance (0.9. + -. 0.2 mL/min/kg; 3.2% liver blood flow, 31 mL/min/kg). The steady state distribution volume (Vss) was 0.10. + -. 0.03L/kg (2 times of vascular fluid (0.05L/kg); 71% of extracellular fluid (0.14L/kg)), showing limited extravascular distribution. The estimated clearance half-life was 5.1 + -0.5 h and the mean residence time was 3.0 + -1.0 h. The time to peak concentration (Tmax) after subcutaneous administration of 67. mu.g/kg occurred at 5.0. + -. 1.0 h. The maximum plasma concentration (Cmax) after subcutaneous administration was 90 ± 29 nM. SEQ ID NO: the subcutaneous bioavailability of peptide 1 in dogs was 93 ± 22%.
Table 5.SEQ ID NO: 1 peptidePharmacokinetic parameters in dogs.
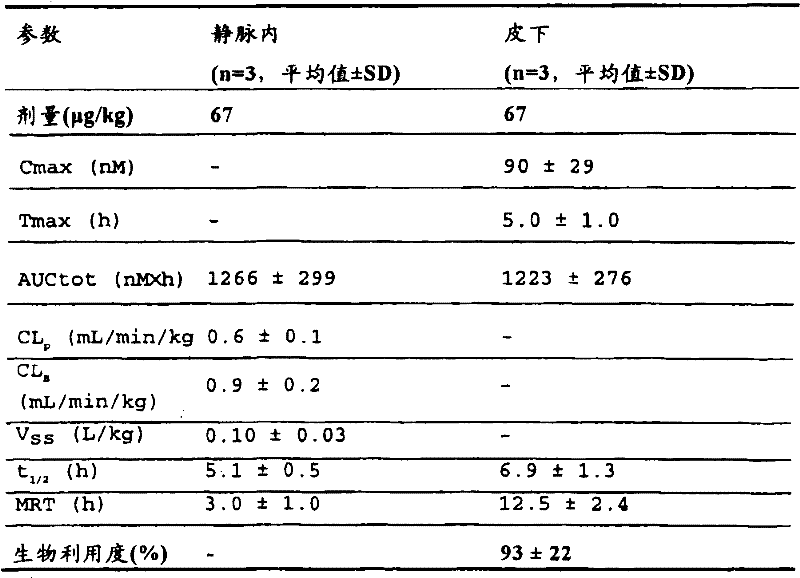
Example 26
Intestines and stomachRoute of external administration
A. Liquid formulations for pulmonary/inhalation or nasal delivery having the following composition were prepared following the methods described below.
| Components | Measurement of |
| 11-mer peptide drugs | 10mg |
| HCl or NaOH | Adjusting pH to 5-8 |
| SBE-Cyclodextrin (Captisol) | 50mg |
| Pure water | Adding into 1ml |
The weighed 11-mer peptide was dissolved in a portion of the optimal pH water. Captisol was added to the drug solution and stirred for about 5min. NaOH and HCL were added to adjust the pH to the desired value (between 5 and 8). Pure water was added to make the final volume 1 ml. Other inactive ingredients such as preservatives, antioxidants, buffer salts and co-solvents may be added as required prior to adjusting the pH. Water is added to achieve the desired target volume.
The above solution preparation can be administered to the lung in the form of a fine mist using a syringe micro-nebulizer or an air jet or an ultrasonic nebulizer. The solution can be delivered to the nasal cavity using a metered nasal spray pump or a syringe micro-nebulizer.
B. A dry powder formulation for pulmonary/inhalation or nasal delivery having the following composition was prepared as described below.
| Components | Measurement of |
| 11-mer peptide drugs | 10mg |
| Lactose | 90mg |
A weighed mass of 11-mer peptide, preferably having a Mass Median Aerodynamic Diameter (MMAD) of less than 5 microns, is mixed with 30-100 μm inhalation grade lactose (DMV International)Mix in the mixer for 5min. The dry powder mixture may be delivered to the lungs using a powder insufflator or a dry powder inhaler.
C. Suspension formulations for pulmonary/inhalation or nasal delivery having the following compositions were prepared as described below.
| Components | Measurement of |
| 11-mer peptide drugs | 10mg |
| Lecithin | 0.1% |
| Gas propellant | 1ml |
The micronized 11-mer peptide is homogeneously suspended in a mixture of lecithin and gaseous propellants such as hydrofluorocarbons (HFA's). The suspension is transferred to a pressurized metered dose inhaler.
D. Uptake of 11-mer peptide from solution formulations in rats

The 11-mer peptide was administered as a solution (as described above) to male Sprague-Dawley rats anesthetized by intraperitoneal injection of pentobarbital. Drugs were either infused into the trachea using a syringe micro nebulizer to assess pulmonary delivery or instilled into each nostril using a pipette for intranasal delivery. Blood samples were collected from the cannulated carotid artery into heparinized vacutainers over a 4 hour period. The blood samples were centrifuged and the separated plasma was stored at-80 ℃ until analysis by LC/MS. The pharmacokinetic parameters were calculated from the plasma-time concentration curves and reported in the table. Three rats were used for each route of administration. Data are presented as mean ± standard deviation. Tmax is reported as median.
Use and combination
A.Use of
The present invention provides novel 11-mer peptides which have excellent properties and are useful as GLP-1 receptor modulators, e.g., such 11-mer peptides have agonist activity for the GLP-1 receptor. Further, the 11-mer peptides of the invention show enhanced stability to proteolytic cleavage compared to the GLP-1 native sequence.
Thus, the compounds of the present invention may be administered to mammals, preferably humans, for the treatment of a variety of conditions and disorders, including, but not limited to, treating or delaying the progression or onset of diabetes (preferably type II, glucose intolerance, insulin resistance, and diabetic complications such as nephropathy, retinopathy, neuropathy and cataracts), hyperglycemia, hyperinsulinemia, hypercholesterolemia, elevated blood levels of free fatty acids or glycerol, hyperlipidemia, hypertriglyceridemia, obesity, wound healing, tissue ischemia, atherosclerosis, hypertension, AIDS, intestinal disease (such as necrotic enteritis, microvilli inclusion body disease or celiac disease), inflammatory bowel syndrome, chemotherapy-induced atrophy or damage of the intestinal mucosa, anorexia nervosa, osteoporosis, metabolic disturbance syndrome, and inflammatory bowel disease (such as Crohn's disease and ulcerative colitis). The compounds of the present invention may also be used to increase the blood level of High Density Lipoproteins (HDL).
Additionally, the compounds of the present invention may be used to treat conditions, diseases and disorders collectively referred to as "syndrome X" or metabolic syndrome as detailed by Johannsson j.clin.endocrinol.metab., 82, 727-34 (1997).
B.Combination of
The scope of the present invention includes pharmaceutical compositions comprising as an active ingredient a therapeutically effective amount of at least one compound of formula I, alone or in combination with a pharmaceutical carrier or diluent. Optionally, the compounds of the present invention may be used alone, in combination with other compounds of the present invention, or in combination with one or more other therapeutic agents, such as antidiabetic agents or other pharmaceutically active substances.
The compounds of the invention may be used in combination with other GLP-1 receptor modulators (e.g., agonists or partial agonists, such as peptide agonists) or other suitable therapeutic agents useful in the treatment of the above-mentioned conditions, including: antidiabetic agents, antihyperglycemic agents, hypolipidemic/lipid-lowering agents, anti-obesity agents (including appetite suppressants/modulators), and antihypertensive agents. In addition, the compounds of the present invention may be used in combination with one or more of the following therapeutic agents; an infertility agent, an agent for the treatment of polycystic ovary syndrome, an agent for the treatment of growth disorders, an agent for the treatment of frailty, an agent for the treatment of arthritis, an agent for the prevention of allograft rejection in transplants, an agent for the treatment of autoimmune diseases, an anti-AIDS agent, an anti-osteoporosis agent, an agent for the treatment of immunomodulatory diseases, an anti-thrombotic agent, an agent for the treatment of cardiovascular diseases, an antibiotic agent, an antipsychotic agent, an agent for the treatment of chronic inflammatory bowel disease or syndrome, and/or an agent for the treatment of anorexia nervosa.
Examples of suitable antidiabetic agents for use in combination with the compounds of the present invention include biguanides (e.g., metformin or phenformin), glucosidase inhibitors (e.g., acarbose or miglitol), insulin (including insulin secretagogues or insulin sensitizers), meglitinides (meglitinides) (e.g., repaglinide), sulfonylureas (e.g., glimepiride, glyburide, gliclazide, chlorpropamide, and glipizide), biguanide/glyburide combinations (e.g., ) Thiazolidinediones (e.g., troglitazone, rosiglitazone and pioglitazone), PPAR-alpha agonists, PPAR-gamma agonistsAgonists, PPAR α/γ dual agonists, glycogen phosphorylase inhibitors, fatty acid binding protein (aP2) inhibitors, DPP-IV inhibitors, and SGLT2 inhibitors.
) Thiazolidinediones (e.g., troglitazone, rosiglitazone and pioglitazone), PPAR-alpha agonists, PPAR-gamma agonistsAgonists, PPAR α/γ dual agonists, glycogen phosphorylase inhibitors, fatty acid binding protein (aP2) inhibitors, DPP-IV inhibitors, and SGLT2 inhibitors.
Other suitable thiazolidinediones include MCC-555 (disclosed in U.S. Pat. No.5,594,016) from Mitsubishi, GL-262570 from Glaxo-Welcom, englitazone (CP-68722, Pfizer) or darlitazone (CP-86325, Pfizer), eaglitazone (MIT/J & J), JTT-501(JPNT/P & U), L-895645(Merck), R-119702(San kyo/WL), NN-2344(Dr. Reddy/NN), or YM-440 (Yamanouchi).
Suitable dual PPAR α/γ agonists include mogrositazar (Bristol-Myers Squibb), AR-HO39242(Astra/Zeneca), GW-409544(Glaxo-Wellcome), KRP297(Kyorin Merck), and the compounds disclosed by Murakami et al in "A Novel Insulin transducer assays As a collagen and for peroxisome promotion-Activated Receptor Alpha (PPAR Alpha) and PPAR gamma. effect on PPAR Alpha Activation on Absolute lipid Metabolism of ZucFatty Rats", Diabetes 47, 1841-1847(1998), and the US series of US applications 09/644,598 of the year 2000 application No. 18, the disclosure of which is hereby incorporated by reference As if set forth As preferred compounds.
Suitable aP2 inhibitors include those disclosed in U.S. application serial No.09/391,053, filed on 7/9/1999 and U.S. application serial No.09/519,079, filed on 6/3/2000, using the dosages as given herein.
Suitable DPP4 inhibitors which may be used in combination with the compounds of the invention include those disclosed in WO99/38501, WO99/46272, WO99/67279(PROBIODRUG), WO99/67278(PROBIODRUG), WO99/61431(PROBIODRUG), NVP-728A (1- [ [ [2- [ (5-cyanopyridin-2-yl) amino ] ethyl ] amino ] acetyl ] -2-cyano- (S) -pyrrolidine) (Novartis), LAF237, saxagliptin, MK0431, TSL-225 (tryptophanyl-1, 2,3, 4-tetrahydroisoquinoline-3-carboxylic acid (disclosed in Yaorg et al Bioorg. & gt. chem. Lett.8(1998)1537, Med. tetrahydroisoquinoline-3-carboxylic acid (Biorr. & gt., Chevritn. & gt, 2748, Biodlork. & gt, Biorr. & gt, Biodlork. & gt, 38 (38), (36), 11597,11603,11603,1999), the dosages used are as given in the above references.
Suitable meglitinide drugs include nateglinide (Novartis) or KAD1229 (PF/Kissei).
Examples of other suitable glucagon-like peptide-1 (GLP-1) compounds that can be used in combination with a GLP-1 receptor modulator (e.g., agonist or partial agonist) of the present invention include GLP-1(1-36) amide, GLP-1(7-37) (as disclosed in U.S. Pat. No.5,614,492 to Habener), and AC2993(Amylin), LY-315902(Lilly), and NN2211(Novo Nordisk).
Suitable hypolipidemic/lipid-lowering agents for use in combination with the compounds of the invention include one or more MTP inhibitors, HMG CoA reductase inhibitors, squalene synthetase inhibitors, fibric acid derivatives, ACAT inhibitors, lipoxygenase inhibitors, cholesterol absorption inhibitors, ileal Na+Bile acid cotransporter inhibitors, upregulators of LDL receptor activity, bile acid sequestrants, cholesteryl ester transfer protein inhibitors (e.g., CP-529414(Pfizer)), and/or niacin and derivatives thereof.
MTP inhibitors that may be used as described above include those disclosed in U.S. patent No.5,595,872, U.S. patent No.5,739,135, U.S. patent No.5,712,279, U.S. patent No.5,760,246, U.S. patent No.5,827,875, U.S. patent No.5,885,983, and U.S. patent No.5,962,440, all of which are incorporated herein by reference.
HMG CoA reductase inhibitors that may be used in combination with one or more compounds of formula I include mevastatin and related compounds as disclosed in U.S. patent No.3,983,140, lovastatin (mevinolin) and related compounds as disclosed in U.S. patent No.4,231,938, pravastatin and related compounds as disclosed in U.S. patent No.4,346,227, and simvastatin and related compounds as disclosed in U.S. patent nos.4,448,784 and 4,450,171. Other HMG CoA reductase inhibitors that may be used herein include, but are not limited to, fluvastatin as disclosed in U.S. Pat. No.5,354,772, cerivastatin as disclosed in U.S. Pat. Nos.5,006,530 and 5,177,080, atorvastatin as disclosed in U.S. Pat. Nos.4,681,893, 5,273,995, 5,385,929 and 5,686,104, atavastatin as disclosed in U.S. Pat. No.5,011,930 (Nissan/Sankyo's Nivastatin (NK-104)), visastatin as disclosed in U.S. Pat. No.5,260,440 (Shionogi-Astra/Zeneca (ZD-4522)), and related statin compounds as disclosed in U.S. Pat. No.5,753,675, pyrazole analogs of mevalonolactone derivatives as disclosed in U.S. Pat. No.4,613,610, mevalonolactone derivatives as disclosed in PCT application WO 86/03488, indane derivatives as disclosed in U.S. Pat. No. 2-4,647,576-alkyl-substituted pyrrolidone derivatives thereof, searle's SC-45355 (a 3-substituted glutaric acid derivative) dichloroacetate, imidazole analogs of mevalonolactone as disclosed in PCT application WO 86/07054, 3-carboxy-2-hydroxy-propane-phosphonic acid derivatives as disclosed in French patent No.2,596,393, 2, 3-disubstituted pyrazine, furan and thiophene derivatives as disclosed in European patent application No.0221025, naphthyl analogs of mevalonolactone as disclosed in U.S. Pat. No.4,686,237, octahydronaphthalenes as disclosed in U.S. Pat. No.4,499,289, ketone analogs of mevinolin (lovastatin) as disclosed in European patent application No. 0142146A 2 and quinoline and pyridine derivatives as disclosed in U.S. Pat. Nos.5,506,219 and 5,691,322.
The ideal blood lipid lowering agent is pravastatin, lovastatin, simvastatin, atorvastatin, fluvastatin, cerivastatin, atavastatin and ZD-4522.
In addition, hypophosphorous acid compounds for inhibiting HMG CoA reductase, such as those disclosed in GB2205837, are also suitable for use in combination with the compounds of the present invention.
Squalene synthase inhibitors suitable for use herein include, but are not limited to, the α -phosphono-sulfonates disclosed in U.S. Pat. No.5,712,396, those disclosed by Biller et al in J.Med.chem., 1988, Vol.31, No.10, pp1869-1871, including isoprenoid (phosphinyl-methyl) phosphonates, as well as other known squalene synthase inhibitors, e.g., as in U.S. Pat. Nos.4,871,721 and 4,924,024 and Biller, S.A., Neuenschwander, KCurrent Pharmaceutical Design2, 1-40 (1996).
In addition, other squalene synthetase inhibitors suitable for use herein include the terpene pyrophosphates disclosed by P.Ortiz de Montelano et al in J.Med.chem., 1977, 20, 243- & 249, such as by Corey and Volante inJ.Am.Chem.SocFarnesyl diphosphate analogue A and the anterior squalene pyrophosphate (PSQ-PP) analogue disclosed in 1976, 98, 1291-1293, the phosphinyl phosphates reported by McClard, R.W. et al, J.A.C.S., 1987, 109, 5544 and the cyclopropanes reported by Capson, T.L. in PhD delivery, June, 1987, Dept.Med.chem.U of Utah, Abstract, Table of contacts, PP16, 17, 40-43, 48-51, Summary.
Fibric acid derivatives which may be used in combination with one or more compounds of formula I include fenofibrate, gemfibrozil, clofibrate, bezafibrate, ciprofibrate, clinofibrate and the like, probucol and related compounds, preferably probucol and gemfibrozil, as disclosed in U.S. Pat. No.3,674,836, bile acid sequestrants such as cholestyramine, colestipol and DEAE-SephadexAnd lipostabil (Rhone-Poulenc), Eisai E-5050 (an N-substituted ethanolamine derivative), imatinib (HOE-402), Tetrahydrolipstatin (THL), istigmastanylphos-phoylcholine (SPC, Roche), aminocyclodextrin (TanabeSeiyoku), Ajinomoto AJ-814 (azulene derivative)Biologics), melinamide (melinamide) (Sumitomo), Sandoz 58-035, American Cyanamid CL-277, 082 and CL-283, 546 (disubstituted urea derivatives), nicotinic acid, acipimox, acifran, neomycin, para-aminosalicylic acid, aspirin, poly (diallylmethylamine) derivatives such as disclosed in U.S. patent No.4,759,923, quaternary amine poly (diallyldimethylammonium chloride) and ionenes (ionenes) such as disclosed in U.S. patent No.4,027,009, among other known serum cholesterol lowering agents.
ACAT inhibitors that may be used in combination with one or more compounds of formula I include those described in Drugsof the Future 24, 9-15(1999), (Avasimibe); "The ACAT inhibitor, C1-1011 is effective in The preservation and regression of atomic failure area in The Hamsters", Nicolosi et al, Atherosclerosis (Shannon, Irel), (1998), 137(1), 77-85; "The pharmacological profile of FCE 27677: a novel ACAT inhibitor with a pore lipophilic activity mediated by selective administration of the hydrophobic interaction of ApoB 100-stabilizing lipoprotein ", Ghiselli, Giancaro, Cardiovasc. DrugRev. (1998), 16(1), 16-30; "RP 73163: a bioavailable alkylsulfinyl-diphenylimide ACAT inhibitor ", Smith, C. et al, bioorg. Med. chem. Lett. (1996), 6(1), 47-50; "ACAT inhibitors: physiologic mechanisms for hyprolipidemic and anti-osteolytic in experimental animals ", Krause et al, Editor(s): ruffolo, Robert r., jr.; hollinger, Mannfred a., inflimation: mediatorsPathways (1995), 173-98, Publisher: CRC, Boca Raton, fia; "ACATINHIBITORs: (1994), 1(3), 204-25, pharmaceutical agents, and pharmaceutical compositions containing said agents; "Inhibitors of acyl-CoA: cholesterol O-acyl transferase (ACAT) as a hypercholesterolemic agent.6. the first water-soluble ACAT inhibitor with lipid-regulating activity. Cholesterol Acyl Transferase (ACAT).7.Development of a series of substistuted N-phenyl-N' - [ (1-phenyl cyclic) methyl ] ureas with enhanced hypercholesterolemic activity ", Stout et al, ChemtFacts: chem. (1995), 8(6), 359-62, or TS-962(Taisho Pharmaceutical co.ltd).
The hypolipidemic agent may be an up-regulator of LD2 receptor activity, such as MD-700(Taisho pharmaceutical Co. Ltd.) and LY295427(Eli Lilly).
Examples of suitable cholesterol absorption inhibitors for use in combination with the compounds of the present invention include those disclosed in SCH48461(Schering-Plough) and in Atherosclerosis 115, 45-63(1995) and J.Med.chem.41, 973 (1998).
Suitable ileal Na for use in combination with the compounds of the invention+Examples of bile acid cotransporter inhibitors include compounds as disclosed in Drugs of the Future, 24, 425-430 (1999).
Lipoxygenase inhibitors which may be used in combination with one or more compounds of formula I include 15-Lipoxygenase (15-LO) inhibitors, such as benzimidazole derivatives disclosed in WO 97/12615, 15-LO inhibitors disclosed in WO 97/12613, isothiazolinones (isothiazolinones) disclosed in WO 96/38144, and Lipoxygenase inhibiting peptides disclosed by Sendory et al in "introduction of di-induced atherosclerosis in collagen with a high yield selective 15-Lipoxygenase inhibitor activating peptides, Brit.J. Pharmacology (1997)120, Cor1199-1206 and Cornicelli et al in" 15-Lipoxygenase inhibitors: 15-LO inhibitors disclosed in NovelTherapeutic Target for Vascular Disease ", Current pharmaceutical design, 1999, 5, 11-20.
Examples of suitable antihypertensive agents for use in combination with the compounds of the present invention include beta adrenergic blockers, calcium channel blockers (L-and T-forms; e.g., diltiazem Verapamil, nifedipine and ammoniaChlorodipine and mybefradil), diuretics such as chlorothiazide, hydrochlorothiazide, flumethiazide, hydroflumethiazide, bendroflumethiazide, methyclothiazide, trichlorthiazide, polythiazide, benthiazide, tricrynafen, chlorthalidone, furosemide, musolimine, bumetanide, triarene, amiloride and spironolactone, renin inhibitors, ACE inhibitors (e.g., captopril, zofenopril, fosinopril, enalapril, ceranopril, cilazopril, delapril, pentopril, quinapril, ramipril, lisinopril), AT-1 receptor antagonists (e.g., flusartan, irbesartan and valsartan), ET receptor antagonists (e.g., sitaxsentan), atan, and the compounds disclosed in U.S. Pat. Nos.5,612,359 and 6,043,265), dual antagonists (e.g. WO I), neutral peptide inhibitors (e.g., the compounds disclosed in WO 00/01389), NEP (NEP), vasopepsidase inhibitors (dual NEP-ACE inhibitors) (e.g., omatralat (omapatrilat) and gemopatrilat) and nitrate.
Verapamil, nifedipine and ammoniaChlorodipine and mybefradil), diuretics such as chlorothiazide, hydrochlorothiazide, flumethiazide, hydroflumethiazide, bendroflumethiazide, methyclothiazide, trichlorthiazide, polythiazide, benthiazide, tricrynafen, chlorthalidone, furosemide, musolimine, bumetanide, triarene, amiloride and spironolactone, renin inhibitors, ACE inhibitors (e.g., captopril, zofenopril, fosinopril, enalapril, ceranopril, cilazopril, delapril, pentopril, quinapril, ramipril, lisinopril), AT-1 receptor antagonists (e.g., flusartan, irbesartan and valsartan), ET receptor antagonists (e.g., sitaxsentan), atan, and the compounds disclosed in U.S. Pat. Nos.5,612,359 and 6,043,265), dual antagonists (e.g. WO I), neutral peptide inhibitors (e.g., the compounds disclosed in WO 00/01389), NEP (NEP), vasopepsidase inhibitors (dual NEP-ACE inhibitors) (e.g., omatralat (omapatrilat) and gemopatrilat) and nitrate.
Examples of suitable anti-obesity agents for use in combination with the compounds of the present invention include NPY receptor antagonists, NPY-Y2 or NPY-Y4 receptor agonists, MCH antagonists, GHSR antagonists, CRH antagonists, beta 3 adrenergic agonists, lipase inhibitors, serotonin (and dopamine) reuptake inhibitors, thyroid receptor beta drugs, CB-1 antagonists and/or anorectics.
Examples of lipase inhibitors which may optionally be used in combination with the compounds of the present invention include orlistat or ATL-962(Alizyme), and orlistat is preferred.
Serotonin (and dopamine) reuptake inhibitors which may optionally be used in combination with the compounds of formula I may be sibutramine, topiramate (Johnson & Johnson) or axokine (Regeneron), and are preferably sibutramine and topiramate.
Examples of thyroid receptor beta compounds that may optionally be used in combination with the compounds of the present invention include thyroid receptor ligands such as those disclosed in WO97/21993(U.Cal SF), WO99/00353(KaroBio) and GB98/284425(KaroBio), and preferably the compounds of the KaroBio application.
Examples of CB-1 antagonists that may optionally be used in combination with the compounds of the present invention include CB-1 antagonists and rimonabant (rimonabant) (SR 141716A).
Examples of NPY-Y2 and NPY-Y4 receptor agonists include PYY (3-36) and Pancreatic Polypeptide (PP), respectively.
Anorectics which may optionally be used in combination with the compounds of the present invention include dextroamphetamine, phentermine, phenylpropanolamine or mazindol, and preferably dextroamphetamine.
Examples of suitable antipsychotic agents include clozapine, haloperidol, olanzapine And aripiprazole
And aripiprazole
The above patents and patent applications are incorporated herein by reference.
Such other therapeutic agents, when used in combination with the compounds of the present invention, are indicated, for example, in terms of amounts by the Physician's Desk Reference, as set forth in the above patents or otherwise determined by one of ordinary skill in the art.
Dosage and formulation
Suitable 11-mer peptides of formula I can be administered to a patient as compounds alone and/or in pharmaceutical formulations in admixture with an acceptable carrier to treat diabetes and other related disorders. The dosage and route of administration of the compounds to mammals, including humans, in need of such treatment can be readily determined by one skilled in the art. The route of administration may include, but is not limited to, oral, intraoral, rectal, transdermal, buccal (buccal), intranasal, pulmonary, subcutaneous, intramuscular, intradermal, sublingual, intracolonic, intraocular, intravenous, or enteral administration. The compounds are formulated according to The route of administration on The basis of accepted pharmaceutical practice (described in Fingl et al, in The pharmaceutical basic of Therapeutics, Ch.1, p.1, 1975; "Remington's pharmaceutical sciences", 18th ed., Mack Publishing Co, Easton, Pa, 1990).
The pharmaceutically acceptable 11-mer peptide compositions of the invention can be administered in a variety of dosage forms such as tablets, capsules (each containing a sustained release or timed release dosage form), pills, powders, granules, elixirs, in situ gels, microspheres, crystalline complexes, liposomes, microemulsions, tinctures, suspensions, syrups, aerosol sprays, and emulsions. The compositions of the present invention may also be administered orally, intravenously (bolus or infusion), intraperitoneally, subcutaneously, transdermally, or intramuscularly, all using dosage forms well known to those of ordinary skill in the pharmaceutical arts. The compositions may be administered alone, but will generally be administered with a pharmaceutical carrier selected according to the chosen route of administration and standard pharmaceutical practice.
Of course, the dosage regimen of the compositions of the present invention will vary depending upon known factors, such as the pharmacokinetic properties of the particular agent and its mode and route of administration; species, age, sex, health status, medical status, and weight of the recipient; the nature and extent of the symptoms; the kind of concurrent therapy; the frequency of treatment; the route of administration, the renal and hepatic function of the patient and the desired effect. A physician or veterinarian can determine and prescribe the amount of the drug required to be effective in preventing, countering or inhibiting the progression of the disease state.
In general guidelines, the daily oral dosage of the active ingredient, when used to achieve the specified effect, will be in the range of about 0.001 to 1000mg/kg body weight, preferably between about 0.01 to 100 mg/kg body weight per day, and most preferably between about 0.6 to 20 mg/kg/day. When used to achieve the specified effect, the daily intravenous dose of the active ingredient will be between 0.001ng to 100.0ng per minute per Kg of body weight during a constant rate infusion such a continuous intravenous infusion may preferably be administered at a rate of 0.01ng to 50ng per minute per Kg of body weight and most preferably at a rate of 0.01ng to 10.0mg per minute per Kg of body weight. The compositions of the present invention may be administered in a single daily dose, or the total daily dose may be divided into two, three or four daily doses. The compositions of the present invention may also be administered in a long acting (depot) formulation which will allow the drug to be released slowly over a period of days/weeks/months as required.
The compositions of the present invention may be administered in intranasal form via topical use of suitable intranasal excipients or via the transdermal route using transdermal skin peptide patches. When administered in the form of a transdermal delivery system, of course, the dosage will be administered continuously rather than intermittently throughout the dosage regimen.
The compositions are generally administered in admixture with suitable pharmaceutical diluents, excipients or carriers (collectively referred to herein as pharmaceutical carriers) suitably selected with respect to the intended form of administration, i.e., oral tablets, capsules, elixirs, aerosol sprays with or without a propellant, and syrups, and in accordance with conventional pharmaceutical practice.
For example, for oral administration in the form of a tablet or capsule, the active pharmaceutical ingredient may be combined with an oral, non-toxic, pharmaceutically acceptable inert carrier such as, but not limited to, lactose, starch, sucrose, glucose, methylcellulose, magnesium stearate, dicalcium phosphate, calcium sulfate, mannitol, and sorbitol; for oral administration in liquid form, the oral pharmaceutical composition may be combined with any oral, non-toxic, pharmaceutically acceptable inert carrier such as, but not limited to, ethanol, glycerol, and water. Furthermore, when desired or necessary, suitable binders, lubricants, disintegrating agents and coloring agents may also be incorporated into the mixture. Suitable binders include, but are not limited to, starch, gelatin, natural sugars such as, but not limited to, glucose or beta-lactose, corn sweeteners, natural and synthetic gums such as acacia, tragacanth or sodium alginate, carboxymethylcellulose, polyethylene glycol and waxes. Lubricants used in these dosage forms include sodium oleate, sodium stearate, magnesium stearate, sodium benzoate, sodium acetate, and sodium chloride. Disintegrants include, but are not limited to, starch, methylcellulose, agar, bentonite, and xanthan gum.
The compositions of the invention may also be administered in the form of mixed micellar or liposomal delivery systems, such as small unilamellar vesicles, large unilamellar vesicles, and multilamellar vesicles. Liposomes can be formed from a variety of phospholipids, such as cholesterol, stearylamine or phosphatidylcholines. Penetration enhancers may be added to enhance drug absorption.
Since prodrugs are known to enhance many of the desired properties of drugs (i.e., solubility, bioavailability, manufacturing, etc.), the compounds of the present invention may be delivered in prodrug form. Accordingly, the present invention is intended to encompass prodrugs of the presently claimed compounds, methods of delivering the same, and compositions comprising the same.
The compositions of the invention may also be coupled to soluble polymers such as drug carriers that may be targeted. Such polymers may include polyvinyl-pyrrolidone, pyran copolymer, polyhydroxypropyl-methacrylamide-phenol, polyhydroxyethylaspartamide phenol, or polyethylene oxide-polylysine substituted with palmitoyl residues. Furthermore, the compositions of the present invention may be combined with biodegradable polymers useful in achieving controlled release of a drug, such as polylactic acid, polyglycolic acid, copolymers of polylactic and polyglycolic acid, polyepsilon caprolactone, polyhydroxy butyric acid, polyorthoesters, polyacetals, polydihydropyrans, polycyanoacylates, and crosslinked or amphiphilic block copolymers of hydrogels.
Dosage forms (pharmaceutical compositions) suitable for administration may contain from about 0.01 mg to about 500 mg of the active ingredient per dosage unit. In these pharmaceutical compositions, the active ingredient will generally be present in an amount of from about 0.5 to 95% by weight, based on the total weight of the composition.
Gelatin capsules may contain the active ingredient in association with powdered carriers such as lactose, starch, cellulose derivatives, magnesium stearate and stearic acid. Similar diluents can be used to make compressed tablets. Both tablets and capsules can be manufactured in the form of sustained release products to provide continuous release of the drug over a period of hours. Compressed tablets may be sugar coated or film coated to mask any unpleasant taste and keep the tablet from air, or enteric coated for selective disintegration in the gastrointestinal tract.
Liquid dosage forms for oral administration may contain coloring and flavoring agents to improve patient acceptance.
Generally, water, suitable oils, saline, aqueous dextrose (glucose) and related sugar solutions, and glycols such as propylene glycol or polyethylene glycol are suitable carriers for parenteral solutions. Solutions for parenteral administration preferably comprise water-soluble salts of the active ingredients, suitable stabilizers and, if necessary, buffer substances. Antioxidants such as sodium bisulfite, sodium sulfite or ascorbic acid, alone or in combination, are suitable stabilizers. Also useful are citric acid and its salts and sodium EDTA. In addition, parenteral solutions may contain preservatives such as benzalkonium chloride, methyl or propyl hydroxybenzoate and chlorobutanol.
Suitable pharmaceutical carriers are described in Remington: "The Science and Practice of pharmacy", Nineteenth Edition, Mack Publishing Company, 1995, a standard reference in The art.
Representative pharmaceutical dosage forms useful for the administration of the compounds of the present invention can be illustrated as follows:
capsule
A large batch of unit capsules can be prepared by filling standard two-piece hard gelatin capsules with 100mg of powdered active ingredient, 150 mg of lactose, 50mg of cellulose, and 6mg of magnesium stearate.
Soft gelatin capsule
A mixture of the active ingredient in a digestible oil such as soybean oil, cottonseed oil or olive oil may be prepared and injected with a positive displacement pump into gelatin to form soft gelatin capsules containing 100mg of the active ingredient. The capsules should be washed and dried.
Tablet formulation
Tablets may be prepared by conventional procedures so that the dosage unit is, for example, 100mg of active ingredient, 0.2 mg of colloidal silicon dioxide, 5mg of magnesium stearate, 275 mg of microcrystalline cellulose, 11 mg of starch and 98.8 mg of lactose. Appropriate coatings may be used to enhance mouthfeel or delay absorption.
Injection preparation
Injectable dosage forms of the 11-mer peptide compositions of the invention may or may not require the use of excipients such as those already approved by regulatory agencies. These excipients include, but are not limited to, solvents and co-solvents, solubilizers, emulsifiers or thickeners, chelating agents, antioxidants and reducing agents, antimicrobial preservatives, buffers and pH adjusting agents, bulking agents, protectants and tonicity adjusting agents and special additives. Injectable formulations must be sterile, pyrogen-free, and particulate matter free in the case of solutions.
Parenteral compositions suitable for administration by injection may be prepared by stirring, for example, 1.5% by weight of the active ingredient in a pharmaceutically acceptable buffer, which may or may not contain a cosolvent or other excipients. The solution should be made isotonic with sodium chloride and then sterilized.
Suspension
Aqueous suspensions for oral and/or parenteral administration may be prepared so as to contain, for example, 100mg of the finely divided active ingredient, 20mg of sodium carboxymethylcellulose, 5mg of sodium benzoate, 1.0g of sorbitol solution u.s.p., and 0.025 mL of vanillin or other palatable flavoring agent per 5 mL.
Biodegradable microparticles
Sustained release parenteral compositions suitable for administration by injection may be prepared, for example, by dissolving a suitable biodegradable polymer in a solvent, adding the active agent to be incorporated into the polymer solution, and then removing the solvent from the matrix thereby forming a matrix of the polymer with the active agent distributed throughout the matrix.
Obviously, many modifications and variations of the present invention are possible in light of the above teachings. It is, therefore, to be understood that within the scope of the appended claims, the invention may be practiced otherwise than as specifically described herein.
The present invention is not to be limited in scope by the single illustrated particular embodiments of the individual aspects of the invention. Functionally equivalent methods and components, in addition to those illustrated and described herein, will be apparent to those skilled in the art from the foregoing description and accompanying drawings. Such modifications are intended to fall within the scope of the appended claims. All references cited herein are incorporated herein by reference in their entirety.
Claims (20)
1. An isolated polypeptide selected from the group consisting of: as shown in SEQ ID NO: 1, as shown in SEQ ID NO: 58, as shown in SEQ ID NO: 70, as shown in SEQ ID NO: 91, as shown in SEQ ID NO: 82, as shown in SEQ ID NO: 60, as shown in SEQ ID NO: 61, as shown in SEQ ID NO: 81, as shown in SEQ ID NO: 59 and a compound as set forth in SEQ ID NO: 92, or a pharmaceutically acceptable salt thereof.
2. Use of the isolated polypeptide of claim 1 in the manufacture of a pharmaceutical composition for treating or delaying the progression or onset of diabetes, diabetic retinopathy, diabetic neuropathy, diabetic nephropathy, insulin resistance, diabetic complications or obesity.
3. The use of claim 2, further comprising the simultaneous or sequential administration of a therapeutically effective amount of one or more therapeutic agents selected from the group consisting of antidiabetic agents, antiobesity agents, antihypertensive agents, antiatherosclerotic agents and lipid-lowering agents.
4. The use according to any one of claims 2 to 3, wherein the pharmaceutical composition is a parenterally administrable formulation.
5. The use of any one of claims 2-3, wherein the pharmaceutical composition is a non-parenteral formulation.
6.The use of claim 4, wherein the parenteral administration is selected from the group consisting of Intravenous (IV) bolus, IV infusion, subcutaneous, intramuscular, intranasal, buccal, pulmonary, and ocular delivery.
7. The use of claim 6, wherein said subcutaneous administration comprises use of an immediate release formulation or a sustained release formulation.
8. The use of claim 6, wherein said intramuscular administration comprises the use of an immediate release formulation or a sustained release formulation.
9. The use of claim 4, wherein the formulation further comprises a pharmaceutically acceptable excipient selected from the group consisting of solvents, co-solvents, solubilizers, emulsifiers, thickeners, chelating agents, antioxidants, reducing agents, antimicrobial preservatives, buffers, pH adjusting agents, bulking agents, protectants, and tonicity adjusting agents.
10. The use of claim 4, wherein the formulation further comprises an encapsulated delivery system.
11. A pharmaceutical composition comprising the polypeptide of claim 1 and a pharmaceutically acceptable carrier.
12. A pharmaceutical combination comprising the polypeptide of claim 1 and at least one therapeutic agent selected from the group consisting of an antidiabetic agent, an antiobesity agent, an antihypertensive agent, an antiatherosclerotic agent and a lipid-lowering agent.
13. The combination of claim 12, wherein the antidiabetic agent is at least one agent selected from the group consisting of biguanides, sulfonylureas, glucosidase inhibitors, peroxisome proliferator-activated receptor (PPAR) gamma agonists, PPAR dual alpha/gamma agonists, adipocyte lipid binding protein (aP2) inhibitors, dipeptidyl peptidase 4(DP4) inhibitors, insulin sensitizers, glucagon-like peptide-1 (GLP-1), insulin, and meglitinides.
14. The combination of claim 13, wherein the antidiabetic agent is at least one agent selected from the group consisting of metformin, glyburide, glimepiride, glipizide, chlorpropamide, gliclazide, acarbose, miglitol, pioglitazone, troglitazone, rosiglitazone, insulin, iglione, repaglinide and nateglinide.
15. The combination of claim 12, wherein the anti-obesity agent is at least one agent selected from the group consisting of β 3 adrenergic agonists, lipase inhibitors, serotonin reuptake inhibitors, dopamine reuptake inhibitors, serotonin and dopamine reuptake inhibitors, thyroid receptor β compounds, and anorectics.
16. The combination of claim 15, wherein the anti-obesity agent is at least one agent selected from orlistat, sibutramine, topiramate, axolol, dextroamphetamine, phentermine, phenylpropanolamine, and mazindol.
17. The combination of claim 12, wherein the lipid-lowering agent is at least one agent selected from the group consisting of a microsomal triglyceride transfer protein (MTP) inhibitor, cholesterol ester transfer protein, hydroxy-3-methyl-glutaryl-coa (hmg coa) reductase inhibitor, squalene synthetase inhibitor, phenoxy acid derivative, up-regulator of Low Density Lipoprotein (LDL) receptor activity, lipoxygenase inhibitor or acyl coa-cholesterol acyltransferase (ACAT) inhibitor.
18. The combination of claim 17, wherein the lipid lowering agent is at least one agent selected from the group consisting of pravastatin, lovastatin, simvastatin, atorvastatin, cerivastatin, fluvastatin, nivastatin, visastatin, fenofibrate, gemfibrozil, clofibrate, and avasimibe.
19. A compound of formula VIa:
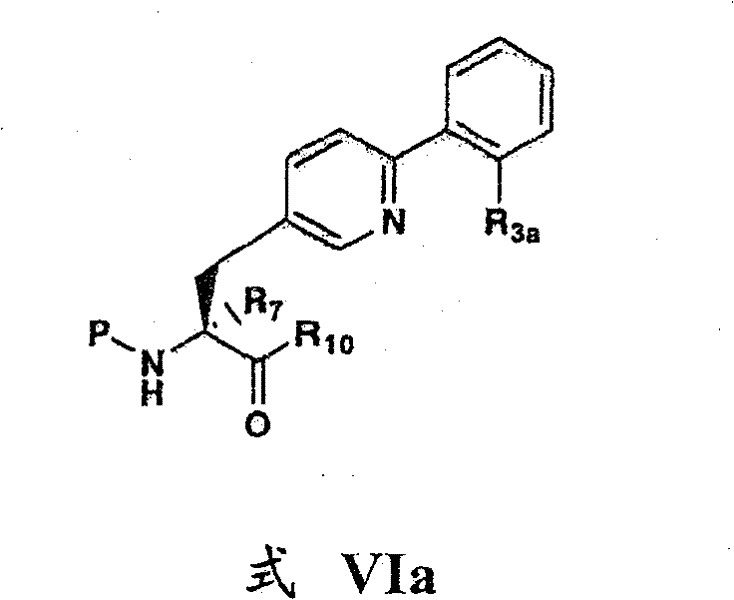
wherein P is hydrogen, fluorenylmethyloxycarbonyl or tert-butyloxycarbonyl; wherein R is3aSelected from methyl, ethyl and fluoro; wherein R is10Selected from OH and NH2(ii) a And wherein R7Selected from hydrogen and methyl.
20. A compound of formula VIIa:
wherein P is hydrogen, fluorenylmethyloxycarbonyl or tert-butyloxycarbonyl; wherein R is6aIs methoxy; wherein R is10Selected from OH and NH2(ii) a And wherein R7Selected from hydrogen and methyl.
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US58535804P | 2004-07-02 | 2004-07-02 | |
| US60/585,358 | 2004-07-02 | ||
| US68480505P | 2005-05-26 | 2005-05-26 | |
| US60/684,805 | 2005-05-26 | ||
| PCT/US2005/023076 WO2006014287A1 (en) | 2004-07-02 | 2005-06-30 | Human glucagon-like-peptide-1 modulators and their use in the treatment of diabetes and related conditions |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN101010339A CN101010339A (en) | 2007-08-01 |
| CN101010339B true CN101010339B (en) | 2011-11-09 |
Family
ID=38698080
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN2005800295435A Expired - Fee Related CN101010339B (en) | 2004-07-02 | 2005-06-30 | Human glucagon-like-peptide-1 modulators and their use in treatment of diabetes and related conditions |
Country Status (2)
| Country | Link |
|---|---|
| CN (1) | CN101010339B (en) |
| ZA (1) | ZA200610786B (en) |
Families Citing this family (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| RS56998B1 (en) | 2010-12-16 | 2018-05-31 | Novo Nordisk As | Solid compositions comprising a glp-1 agonist and a salt of n-(8-(2-hydroxybenzoyl)amino)caprylic acid |
| RU2602601C2 (en) | 2011-04-12 | 2016-11-20 | Ново Нордиск А/С | Double acylated glp-1 derivatives |
| TR201903918T4 (en) | 2012-03-22 | 2019-04-22 | Novo Nordisk As | Compositions containing a dispersing agent and their preparation. |
| WO2013139695A1 (en) | 2012-03-22 | 2013-09-26 | Novo Nordisk A/S | Compositions comprising a delivery agent and preparation thereof |
| CN104203266B (en) | 2012-03-22 | 2017-12-26 | 诺和诺德股份有限公司 | GLP-1 peptide composition and its preparation |
| EP2863895B1 (en) | 2012-06-20 | 2021-04-14 | Novo Nordisk A/S | Tablet formulation comprising a peptide and a delivery agent |
| RU2671406C2 (en) | 2013-05-02 | 2018-10-31 | Ново Нордиск А/С | Oral dosing of glucagon-like peptide-1 compounds |
| TWI726889B (en) * | 2015-06-10 | 2021-05-11 | 英商梅迪繆思有限公司 | Protease-resistant lipidated peptides |
| SG11201903938XA (en) * | 2016-12-09 | 2019-05-30 | Zealand Pharma As | Acylated glp-1/glp-2 dual agonists |
| BR112020014624A2 (en) | 2018-02-02 | 2020-12-08 | Novo Nordisk A/S | SOLID COMPOSITIONS UNDERSTANDING GLP-1 AGONIST, CAPRILIC AND LUBRICANT N- (8- (2-HYDROXYBENZOIL) AMINO ACID SALT) |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1329620A (en) * | 1998-12-07 | 2002-01-02 | 研究及应用科学协会股份有限公司 | Glucagon-like peptide-1 analogs |
-
2005
- 2005-06-30 CN CN2005800295435A patent/CN101010339B/en not_active Expired - Fee Related
-
2006
- 2006-12-20 ZA ZA200610786A patent/ZA200610786B/en unknown
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1329620A (en) * | 1998-12-07 | 2002-01-02 | 研究及应用科学协会股份有限公司 | Glucagon-like peptide-1 analogs |
Non-Patent Citations (2)
| Title |
|---|
| 宋宇宏等.胰高血糖素样肽.黑龙江医药科学26 6.2003,26(6),86-87. |
| 宋宇宏等.胰高血糖素样肽.黑龙江医药科学26 6.2003,26(6),86-87. * |
Also Published As
| Publication number | Publication date |
|---|---|
| ZA200610786B (en) | 2008-12-31 |
| CN101010339A (en) | 2007-08-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US7417028B2 (en) | Human glucagon-like-peptide-1 modulators and their use in treatment of diabetes and related conditions | |
| US7960349B2 (en) | N-terminally modified GLP-1 receptor modulators | |
| US20070021346A1 (en) | N-terminally modified GLP-1 receptor modulators | |
| AU2002348469B2 (en) | Human glucagon-like-peptide-1 mimics and their use in the treatment of diabetes and related conditions | |
| US20070238669A1 (en) | Human glucagon-like-peptide-1 modulators and their use in the treatment of diabetes related conditions | |
| AU2002348469A1 (en) | Human glucagon-like-peptide-1 mimics and their use in the treatment of diabetes and related conditions | |
| EP1615653A2 (en) | Human glucagon-like-peptide-1 mimics and their use in the treatment of diabetes and related conditions | |
| US7534763B2 (en) | Sustained release GLP-1 receptor modulators | |
| CN101010339B (en) | Human glucagon-like-peptide-1 modulators and their use in treatment of diabetes and related conditions | |
| CN101400699A (en) | Human glucagon-like-peptide-1 modulators and their use in the treatment of diabetes and related conditions |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| C17 | Cessation of patent right | ||
| CF01 | Termination of patent right due to non-payment of annual fee |
Granted publication date: 20111109 Termination date: 20130630 |