CN100386310C - N,n'-二烷氧基-n,n'-二烷基草酰胺的制备方法 - Google Patents
N,n'-二烷氧基-n,n'-二烷基草酰胺的制备方法 Download PDFInfo
- Publication number
- CN100386310C CN100386310C CNB2004800258893A CN200480025889A CN100386310C CN 100386310 C CN100386310 C CN 100386310C CN B2004800258893 A CNB2004800258893 A CN B2004800258893A CN 200480025889 A CN200480025889 A CN 200480025889A CN 100386310 C CN100386310 C CN 100386310C
- Authority
- CN
- China
- Prior art keywords
- dialkoxy
- preparation
- oxamide
- group
- formula
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Landscapes
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
本发明提供一种通式(3)所示的N,N’-二烷氧基-N,N′-二烷基草酰胺的制备方法,其特征在于,在碱的存在下,使通式(1):式中:R1和R1’相同或不同,分别表示烃、所示的草酸二酯与通式(2):R2O-NHR3 (2)式中:R2和R3相同或不同,分别表示碳原子数为1~4的烷基、所示的N-烷基-O-烷基羟基胺或其酸盐反应,式中:R2和R3与上述意义相同。
Description
技术领域
本发明涉及作为医药、农药等的合成中间体有用的N,N’-二烷氧基-N,N′-二烷基草酰胺的新型制备方法。
背景技術
以往,作为N,N’-二烷氧基-N,N′-二烷基草酰胺的制备方法,公开了在吡啶的存在下,使草酰氯与N,O-二甲基羟基胺盐酸盐反应,制备出N,N′-二甲氧基-N,N′-二甲基草酰胺的方法(例如参照J.Org.Chem.,60,5016(1995))。但是,在该方法中,作为初始原料使用的是容易分解为毒性高的光气和一氧化碳的草酰氯,从而存在于目的物的分离操作中必须使用致癌性高的二氯甲烷的问题,而作为N,N’-二烷氧基-N,N′-二烷基草酰胺的工业制备方法是不合适的。
发明内容
本発明的课题是为了解决上述问题点,提供一种通过使用安全的初始原料,不需要复杂的操作,就能够以高收率制备出N,N’-二烷氧基-N,N′-二烷基草酰胺的适合工业化的N,N’-二烷氧基-N,N′-二烷基草酰胺的制备方法。
本发明涉及一种在碱的存在下,使通式(1):
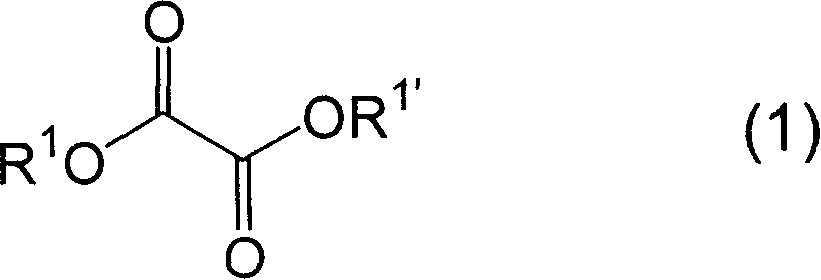
式中:R1和R1’可相同也可以不同,分别表示烃基、所示的草酸二酯与通式(2):
R2O-NHR3 (2)
式中:R2和R3可相同也可以不同,分别表示碳原子数为1~4的烷基、
所示的N-烷基-O-烷基羟基胺或其酸盐反应,生成通式(3):
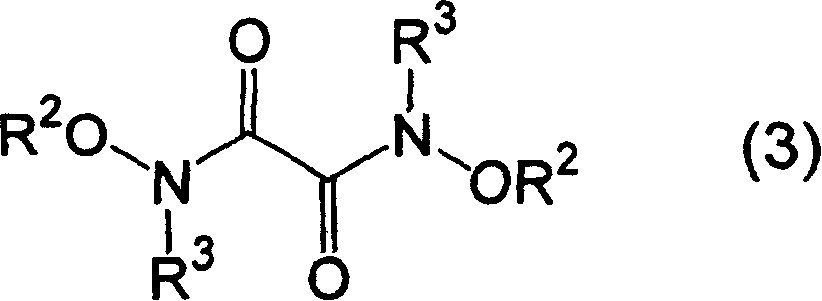
式中:R2和R3与上述意义相同、
所示的N,N’-二烷氧基-N,N′-二烷基草酰胺的制备方法。
具体实施方式
本发明的反应中使用的草酸二酯,如上述通式(1)所示。在该通式(1)中R1和R1’可相同也可以不同,分别为烃,例如可以举出甲基、乙基、丙基、丁基、戊基、己基、庚基、辛基等碳原子数为1~8的烷基;环丙基、环丁基、环戊基、环己基等环烷基;苄基、苯乙基等芳香烷基;苯基、甲苯基、萘基等芳基。其中优选甲基、乙基、异丙基、丁基、苯基。另外,这些基团也包括各种异构体。
在本发明的反应中使用的N-烷基-O-烷基羟基胺,如上述通式(2)所示。在该通式(2)中,R2和R3可以相同也可以不同,分别表示碳原子数1~4的烷基,例如可以举出甲基、乙基、丙基、丁基。另外,这些基团也包括各种异构体。另外,该N-烷基-O-烷基羟基胺,既可以使用游离的N-烷基-O-烷基羟基胺(也包括水合物),也可以作为盐酸盐、硫酸盐、硝酸盐、磷酸盐等酸性盐使用,或者也可以以其水溶液的方式使用。此外,这些N-烷基-O-烷基羟基胺可单独使用或两种以上混合使用。
上述N-烷基-O-烷基羟基胺或其酸盐的使用量,相对于1摩尔草酸二酯优选为0.5~5.0摩尔,更优选为1.5~3.0摩尔。
作为本发明的反应中使用的碱,例如可以举出甲醇锂、甲醇钠、乙醇钠、甲醇钾、乙醇钾、叔丁醇钾等碱金属烷氧化物(也包括相应的醇溶液);氢氧化钠、氢氧化钾等碱金属氢氧化物;碳酸锂、碳酸钠、碳酸钾等碱金属碳酸盐;碳酸氢钠、碳酸氢钾等碱金属碳酸氢盐;三乙胺、三丁胺等胺类,但是优选使用碱金属烷氧化物、更优选使用甲醇钠、乙醇钠。另外,这些碱可单独使用或两种以上混合使用。
上述碱的使用量,相对于1摩尔草酸二酯,优选为0.5~10摩尔,更优选为1.5~6摩尔。
本发明的反应,优选在溶剂中进行。作为使用的溶剂,只要不阻碍反应则没有特别限制,例如可以举出甲醇、乙醇、正丙醇、异丙醇、正丁醇、仲丁醇、叔丁醇等醇类;乙腈、丙腈等腈类;己烷、庚烷等脂肪族烃类:二氯甲烷、氯仿、四氯化碳等卤代脂肪族烃;苯、甲苯、二甲苯、均三甲苯等芳香族烃类;氯代苯等卤代芳香族烃类;二乙醚、四氢呋喃、二异丙醚、二氧杂环己烷等醚类;N,N-二甲基甲酰胺、N,N-二甲基乙酰胺等酰胺类;醋酸乙酯、醋酸丁酯、丙酸乙酯等羧酸酯类:二甲基亚砜等亚砜类;碳酸二甲酯、碳酸二乙酯等碳酸二酯类,优选醇类、腈类、芳香族烃类,更优选为甲醇、乙醇、异丙醇。另外,这些溶剂可单独使用或两种以上混合使用。
所述溶剂的使用量可以根据反应液的均匀性或搅拌性等进行适宜调节,但是优选相对于草酸二酯1g为0.1~100g、更优选为0.5~50g。
本发明的反应例如可以通过混合草酸二酯、N-烷基-O-烷基羟基胺或其酸盐、碱及溶剂,边搅拌边使反应等方法进行。此时的反应温度优选-40~100℃、更优选为-20~50℃,反応圧力以及反应时间没有特别限制。
作为本发明中所得的N,N’-二烷氧基-N’N’-二烷基草酰胺,例如可以举出R2和R3相同或不同、分别表示碳原子数1~4烷基的化合物。
另外,作为最终生成物的N,N’-二烷氧基-N,N′-二烷基草酰胺,例如可以通过反应结束后,过滤、浓缩、蒸馏、重结晶、柱层析色谱等一般方法进行分离精制。
本发明中所得的N,N’-二烷氧基-N,N′-二烷基草酰胺,例如可以通过日本公报特表2002-541104、特表2004-509059或Bioorg.Med.Chem.Lett.12(2002)pp.3001-3004等中记载的方法,制备出有用的医药或农药。
下面,通过实施例对本发明进行具体说明,但是本发明的范围不受这些实施例的限制。
实施例
实施例1(N,N′-二甲氧基-N,N′-二甲基草酰胺的合成)
在备有搅拌装置、温度计以及滴液漏斗的内容积1000ml的烧瓶中,加入98质量%草酸二乙酯126.8g(0.85mol)、98.1质量%N,O-二甲基羟基胺盐酸盐169.0g(1.70mol)以及甲醇100ml之后,在保持液温5~10℃的同时,缓慢滴下28质量%甲醇钠甲醇溶液656.0g(3.40mol),边搅拌边使其在该温度下反应3小时。反应结束后,在备有搅拌装置、温度计以及滴液漏斗的内容积2000ml的烧瓶中,加入醋酸107.1g(1.79mol)以及水893g,在冷却到5℃之后,边保持液温5~15℃,边缓慢滴下上述反应液并进行搅拌。然后,在减压下浓缩反应液,然后用醋酸乙酯1500ml进行萃取。所得有机层(醋酸乙酯层)再次减压浓缩,然后加入正庚烷240g,在保持液温5~10℃的同时,搅拌30分钟。析出的结晶过滤,用冷却的正庚烷清洗后减压干燥,作为白色结晶得到N,N′-二甲氧基-N,N′-二甲基草酰胺92.3g分离收率:61.6%)。
另外,N,N′-二甲氧基-N,N′-二甲基草酰胺的物性值如下所示。
熔点:89.5~92.0℃
1H-NMR(CDCl3,δ(ppm));3.24(6H,s)、3.74(6H,s)
实施例2(N,N′-二甲氧基-N,N′-二甲基草酰胺的合成)
在备有搅拌装置、温度计以及滴液漏斗的内容积25ml的烧瓶中,加入99质量%草酸二甲酯1.01g(8.47mmol)、98.1质量%N,O-二甲基羟基胺盐酸盐1.68g(16.94mmol)以及甲醇1ml之后,边保持液温5~10℃边缓慢滴下28质量%甲醇钠甲醇溶液6.54g(33.88mmol),边搅拌边使其在该温度下反应3小时。反应结束后,在备有搅拌装置、温度计以及滴液漏斗的内容积25ml的烧瓶中,加入2mol/l酢酸10ml(2.00mmol)冷却到5℃后,边保持液温5~15℃边缓慢滴下上述反应液并进行搅拌。通过高效液相色谱法分析该溶液(绝对定量法),结果生成N,N′-二甲氧基-N,N′-二甲基草酰胺1.21g(反应收率:80.9%)。
实施例3(N,N′-二甲氧基-N,N′-二甲基草酰胺的合成)
除了将实施例2中的99质量%草酸二甲酯1.01g(8.47mmol)替换为98质量%草酸二乙酯1.26g(8.47mmol),将甲醇替换为乙醇之外,其余的通过与实施例2相同的方式进行反应。其结果生成N,N′-二甲氧基-N,N′-二甲基草酰胺1.29g(反应收率:86.4%)。
实施例4(N,N′-二甲氧基-N,N′-二甲基草酰胺的合成)
除了将实施例2中的99质量%革酸二甲酯1.01g(8.47mmol)替换为99质量%草酸二异丙酯1.49g(8.47mmol)、将甲醇替换为乙醇之外,其余的通过与实施例2同样的方式进行反应。其结果生成N,N′-二甲氧基-N,N′-二甲基草酰胺1.23g(反应收率:82.4%)。
实施例5(N,N′-二甲氧基-N,N′-二甲基草酰胺的合成)
除了将实施例2中的99质量%草酸二甲酯1.01g(8.47mmol)替换为99质量%草酸二丁酯1.73g(8.47mmol)、将甲醇替换为乙醇之外,其余的通过与实施例2同样的方式进行反应。其结果生成N,N′-二甲氧基-N,N′-二甲基草酰胺1.24g(反应收率:83.1%)。
实施例6(N,N′-二甲氧基-N,N′-二甲基草酰胺的合成)
除了将实施例2中的99质量%草酸二甲酯1.01g(8.47mmol)替换为99质量%草酸二苯酯2.07g(8.47mmol)、将甲醇替换为乙醇之外,其余的通过与实施例2同样的方式进行反应。其结果生成N,N′-二甲氧基-N,N′-二甲基草酰胺0.85g(反应收率:57.1%)。
实施例7(N,N′-二甲氧基-N,N′-二甲基草酰胺的合成)
除了将实施例3中的乙醇替换为异丙醇之外,其余的通过与实施例3同样的方式进行反应。其结果生成N,N′-二甲氧基-N,N′-二甲基草酰胺1.21g(反应收率:80.8%)。
实施例8(N,N′-二甲氧基-N,N′-二甲基草酰胺的合成)
除了将实施例3中的乙醇替换为乙腈之外,其余的通过与实施例3同样的方式进行反应。其结果生成N,N′-二甲氧基-N,N′-二甲基草酰胺1.18g生成(反应收率:78.8%)。
实施例9(N,N′-二甲氧基-N,N′-二甲基草酰胺的合成)
除了将实施例3中的乙醇替换为碳酸二甲酯之外,其余的通过与实施例3同样的方式进行反应。其结果生成N,N′-二甲氧基-N,N′-二甲基草酰胺1.13g生成(反应收率:75.7%)。
实施例10(N,N′-二甲氧基-N,N′-二甲基草酰胺的合成)
除了将实施例3中的乙醇替换为二甲基甲酰胺之外,其余的通过与实施例3同样的方式进行反应。其结果生成N,N′-二甲氧基-N,N′-二甲基草酰胺1.16g(反应收率:77.5%)。
实施例11(N,N′-二甲氧基-N,N′-二甲基草酰胺的合成)
除了将实施例3中的乙醇替换为四氢呋喃之外,其余的通过与实施例3同样的方式进行反应。其结果生成N,N′-二甲氧基-N,N′-二甲基草酰胺1.25g(反应收率:83.6%)。
实施例12(N,N′-二甲氧基-N,N′-二甲基草酰胺的合成)
除了将实施例3中的乙醇替换为甲苯之外,其余的通过与实施例3同样的方式进行反应。其结果生成N,N′-二甲氧基-N,N′-二甲基草酰胺1.05g(反应收率:71.0%)。
实施例13(N,N′-二甲氧基-N,N′-二甲基草酰胺的合成)
在备有搅拌装置以及温度计的内容积25ml的烧瓶中,加入99质量%草酸二甲酯1.01g(8.47mmol)、98.1质量%N,O-二甲基羟基胺盐酸盐1.68g(16.94mmol)以及二甲基亚砜5ml之后,在保持液温5~25℃的同时,分数次加入95质量%甲醇钠粉末1.93g(33.88mmol),边搅拌边使其在该温度下反应1.5小时。反应结束后,在备有搅拌装置、温度计以及滴液漏斗的内容积25ml的烧瓶中,加入2mol/l醋酸10ml(2.00mmol)冷却至5℃后,边保持液温5~15℃边缓慢滴下上述反应液并进行搅拌。通过高效液相色谱法分析该溶液(绝对定量法),结果生成N,N′-二甲氧基-N,N′-二甲基草酰胺0.91g(反应收率:61.0%)。
实施例14(N,N′-二甲氧基-N,N′-二甲基草酰胺的合成)
在备有搅拌装置、温度计以及滴液漏斗的内容积200ml的烧瓶中,加入98质量%草酸二乙酯12.63g(84.7mmol)、98.8质量%N,O-二甲基羟基胺盐酸盐16.72g(169.4mmol)以及甲醇10ml后,边保持液温3~7℃,边缓慢滴下28质量%甲醇钠甲醇溶液65.36g(338.8mmol),边搅拌边使其在3~8℃反应3小时。反应结束后,在备有搅拌装置、温度计以及滴液漏斗的内容积300ml的烧瓶中,加入2mol/l盐酸90ml(180mmol)并冷却5℃后,边保持液温5~15℃边缓慢滴下上述反应液并进行搅拌。通过高效液相色谱法分析该溶液(绝对定量法),结果生成N,N′-二甲氧基-N,N′-二甲基草酰胺13.25g(反应收率:88.8%)。
实施例15(N,N′-二甲氧基-N,N′-二甲基草酰胺的合成)
在备有搅拌装置、温度计以及滴液漏斗的内容积200ml的烧瓶中,加入98质量%草酸二乙酯12.63g(84.7mmol)、98.8质量%N,O-二甲基羟基胺盐酸盐16.72g(169.4mmol)以及甲醇10ml后,在保持液温-10~-7℃的同时缓慢滴下28质量%甲醇钠甲醇溶液65.36g(338.8mmol),边搅拌边使其在-9~-7℃反应3小时。反应结束后,在备有搅拌装置、温度计以及滴液漏斗的内容积300ml的烧瓶中,加入2mol/l盐酸90ml(180mmol),冷却到5℃后,边保持液温5~15℃边缓慢滴下上述反应液并进行搅拌。通过高效液相色谱法分析该溶液(绝对定量法),结果生成N,N′-二甲氧基-N,N′-二甲基草酰胺13.12g(反应收率:87.9%)。
实施例16(N,N′-二甲氧基-N,N′-二甲基草酰胺的合成)
在备有搅拌装置、温度计以及滴液漏斗的内容积200ml的烧瓶中,加入98质量%革酸二乙酯12.63g(84.7mmol)、98.8质量%N,O-二甲基羟基胺盐酸盐20.07g(203.3mmol)以及甲醇10ml后,边保持液温3~7℃边缓慢滴下28质量%甲醇钠甲醇溶液71.90g(372.7mmol),边搅拌边使其在3~7℃反应3小时。反应结束后,在备有搅拌装置、温度计以及滴液漏斗的内容积300ml的烧瓶中,加入1.7mol/l盐酸110ml(187mmol),冷却到5℃后,边保持液温5~15℃边缓慢滴下上述反应液并进行搅拌。通过高效液相色谱法分析该溶液(绝对定量法),结果生成N,N′-二甲氧基-N,N′-二甲基草酰胺14.32g(反应收率:96.0%)。
产业上的利用可能性
本发明涉及作为医药、农药等合成中间体有用的N,N’-二烷氧基-N,N′-二烷基草酰胺的新型制备方法。
通过本発明可以提供一种适合工业化的N,N’-二烷氧基-N,N′-二烷基草酰胺的制备方法,该方法通过使用安全的初始原料而不需要复杂的操作,就能够以高收率制备出N,N-二烷氧基-N,N′-二烷基草酰胺。
Claims (16)
1.一种通式(3)所示的N,N’-二烷氧基-N,N′-二烷基草酰胺的制备方法,其特征在于,在碱的存在下,使通式(1):

式中:R1和R1’相同或不同,分别表示烃基、所示的草酸二酯与通式(2):
R2O-NHR3(2)
式中:R2和R3相同或不同,分别表示碳原子数为1~4的烷基、
所示的N-烷基-O-烷基羟基胺或其酸盐反应,

式中:R2和R3与上述意义相同。
2.根据权利要求1所述的N,N’-二烷氧基-N,N′-二烷基草酰胺的制备方法,其特征在于,碱是选自由碱金属烷氧化物、碱金属氢氧化物、碱金属碳酸盐、碱金属碳酸氢盐以及胺类构成的物质组中的至少一种。
3.根据权利要求1所述的N,N’-二烷氧基-N,N′-二烷基草酰胺的制备方法,其特征在于,碱是选自由甲醇锂、甲醇钠、乙醇钠、甲醇钾、乙醇钾、叔丁醇钾、氢氧化钠、氢氧化钾、碳酸锂、碳酸钠、碳酸钾、碳酸氢钠、碳酸氢钾、三乙胺以及三丁胺构成的物质组中的至少一种。
4.根据权利要求1所述的N,N’-二烷氧基-N,N′-二烷基草酰胺的制备方法,其特征在于,碱是选自由甲醇钠以及乙醇钠构成的物质组中的至少一种。
5.根据权利要求1所述的N,N’-二烷氧基-N,N′-二烷基草酰胺的制备方法,其特征在于,碱的使用量是相对于1摩尔草酸二酯为0.5~10摩尔。
6.根据权利要求1所述的N,N’-二烷氧基-N,N′-二烷基草酰胺的制备方法,其特征在于,碱的使用量是相对于1摩尔草酸二酯为1.5~6摩尔。
7.根据权利要求1所述的N,N’-二烷氧基-N,N′-二烷基草酰胺的制备方法,其特征在于,反应是在溶剂中进行。
8.根据权利要求7所述的N,N’-二烷氧基-N,N′-二烷基草酰胺的制备方法,其特征在于,溶剂是选自由醇类、腈类、脂肪族烃类、卤代脂肪族烃、芳香族烃类、卤代芳香族烃类、醚类、酰胺类、羧酸酯类、亚砜类以及碳酸二酯类构成的物质组中的至少一种。
9.根据权利要求7所述的N,N’-二烷氧基-N,N′-二烷基草酰胺的制备方法,其特征在于,溶剂是选自由甲醇、乙醇、正丙醇、异丙醇、正丁醇、仲丁醇、叔丁醇、乙腈、丙腈、己烷、庚烷、二氯甲烷、氯仿、四氯化碳、苯、甲苯、二甲苯、均三甲苯、氯代苯、二乙醚、四氢呋喃、二异丙醚、二氧杂环己烷、N,N-二甲基甲酰胺、N,N-二甲基乙酰胺、醋酸乙酯、醋酸丁酯、丙酸乙酯、二甲基亚砜、碳酸二甲酯以及碳酸二乙酯构成的物质组中的至少一种。
10.根据权利要求7所述的N,N’-二烷氧基-N,N′-二烷基草酰胺的制备方法,其特征在于,溶剂的使用量是相对于1g草酸二酯为0.1~100g。
11.根据权利要求7所述的N,N’-二烷氧基-N,N′-二烷基草酰胺的制备方法,其特征在于,溶剂的使用量是相对于1g草酸二酯为0.5~50g。
12.根据权利要求1所述的N,N’-二烷氧基-N,N′-二烷基草酰胺的制备方法,其特征在于,式(1)所示的草酸二酯中的R1以及R1’是选自由烷基、环烷基、芳香烷基、以及芳基构成的物质组中的至少一种。
13.根据权利要求1所述的N,N’-二烷氧基-N,N′-二烷基草酰胺的制备方法,其特征在于,式(1)所示的草酸二酯中的R1以及R1’是选自由甲基、乙基、丙基、丁基、戊基、己基、庚基、辛基、环丙基、环丁基、环戊基、环己基、苄基、苯乙基、苯基、甲苯基以及萘基构成的物质组中至少一种。
14.根据权利要求1所述的N,N’-二烷氧基-N,N′-二烷基草酰胺的制备方法,其特征在于,式(2)所示的N-烷基-O-烷基羟基胺中的R2以及R3分别是选自由甲基、乙基、丙基以及丁基构成的物质组中的至少一种。
15.根据权利要求1所述的N,N’-二烷氧基-N,N′-二烷基草酰胺的制备方法,其特征在于,式(2)所示的N-烷基-O-烷基羟基胺或其酸盐的使用量,相对于式(1)所示的草酸二酯1摩尔为0.5~5.0摩尔。
16.根据权利要求1所述的N,N’-二烷氧基-N,N′-二烷基草酰胺的制备方法,其特征在于,式(2)所示的N-烷基-O-烷基羟基胺或其酸盐的使用量,相对于式(1)所示的草酸二酯1摩尔为1.5~3.0摩尔。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2003316377 | 2003-09-09 | ||
| JP316377/2003 | 2003-09-09 | ||
| JP217301/2004 | 2004-07-26 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1849300A CN1849300A (zh) | 2006-10-18 |
| CN100386310C true CN100386310C (zh) | 2008-05-07 |
Family
ID=37078394
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNB2004800258893A Expired - Fee Related CN100386310C (zh) | 2003-09-09 | 2004-09-09 | N,n'-二烷氧基-n,n'-二烷基草酰胺的制备方法 |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN100386310C (zh) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102686643A (zh) * | 2009-12-22 | 2012-09-19 | 陶氏环球技术有限责任公司 | 聚双草酰胺 |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2002541104A (ja) * | 1999-04-06 | 2002-12-03 | メルク エンド カムパニー インコーポレーテッド | ピロリジン系ケモカイン受容体活性調節剤 |
-
2004
- 2004-09-09 CN CNB2004800258893A patent/CN100386310C/zh not_active Expired - Fee Related
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2002541104A (ja) * | 1999-04-06 | 2002-12-03 | メルク エンド カムパニー インコーポレーテッド | ピロリジン系ケモカイン受容体活性調節剤 |
Non-Patent Citations (1)
| Title |
|---|
| 新实验化学讲座14 有机化物的合成と反应II. 1227,日本化学会. 1997 * |
Also Published As
| Publication number | Publication date |
|---|---|
| CN1849300A (zh) | 2006-10-18 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN101535274B (zh) | 制备二氟甲基吡唑基羧酸酯的方法 | |
| KR101397352B1 (ko) | 4-치환된 페녹시페닐아세트산 유도체 | |
| US9388168B2 (en) | Process for the preparation of 1-([1,3]dioxolan-4-ylmethyl)-1H-pyrazol-3-ylamine | |
| CN100528821C (zh) | 反式-4-氨基-1-环己烷羧酸衍生物的制备方法 | |
| CN107848987B (zh) | 含氮杂环化合物的制造方法及其中间体 | |
| CN102020634A (zh) | 一种n-(氰烷基)苯甲酰胺类化合物的制备方法 | |
| CN100386310C (zh) | N,n'-二烷氧基-n,n'-二烷基草酰胺的制备方法 | |
| CN101107228B (zh) | (z)-1-苯基-1-(n,n-二乙氨基羰基)-2-邻苯二甲酰亚氨甲基环丙烷的制造方法 | |
| CN102311447B (zh) | 杂环并嘧啶酮类dpp-iv抑制剂 | |
| CN103288699A (zh) | 脯氨酸类似物的制备方法 | |
| US8030512B2 (en) | Polycyclic pentafluorosulfanylbenzene compound and process for producing the compound | |
| CN116332906A (zh) | 一种氯虫苯甲酰胺关键中间体以及氯虫苯甲酰胺或其衍生物的合成方法 | |
| JP4929717B2 (ja) | N,n’−ジアルコキシ−n,n’−ジアルキルオキサミドの製法 | |
| US20040171863A1 (en) | Process for poducing beta-oxonitrile compound or alkali metal salt thereof | |
| US10774093B2 (en) | Synthetic processes and synthetic intermediates | |
| CN109988070B (zh) | 反式-1-羟基-1-(三氟甲基)-3-氨基环丁烷盐酸盐的中间体及制备方法和应用 | |
| US8163903B2 (en) | Process for the preparation of N-[5-(3-dimethylamino-acryloyl)-2-fluoro-phenyl]-N-methyl-acetamide | |
| CN101397291B (zh) | 2-氰乙酰-5-取代噻吩类化合物的制备方法 | |
| CN110003111A (zh) | 一种2-芳基-3-醚基-3-吡唑丙烯腈类化合物的制备方法 | |
| CN108358780B (zh) | 合成高非对映选择性的α-酰氧化环酮类化合物的方法 | |
| CN104151278B (zh) | 1-苯基-2,3-萘二羧酸酐及其衍生物的合成方法 | |
| JP4968602B2 (ja) | ベンズアミド誘導体の製造方法 | |
| CN105111084A (zh) | 一种4-取代-3-甲基-2,4-二氧代丁酸酯的合成方法 | |
| JP2637113B2 (ja) | (4―ヒドロキシフェニル)―シクロヘキセンカルボン酸及びその製造方法 | |
| CN1830977B (zh) | 制备2-氨基噻唑甲酰胺衍生物的方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| C17 | Cessation of patent right | ||
| CF01 | Termination of patent right due to non-payment of annual fee |
Granted publication date: 20080507 Termination date: 20100909 |