WO2025141105A1 - Specific probes for the detection of nucleic acids, methods and uses - Google Patents
Specific probes for the detection of nucleic acids, methods and uses Download PDFInfo
- Publication number
- WO2025141105A1 WO2025141105A1 PCT/EP2024/088487 EP2024088487W WO2025141105A1 WO 2025141105 A1 WO2025141105 A1 WO 2025141105A1 EP 2024088487 W EP2024088487 W EP 2024088487W WO 2025141105 A1 WO2025141105 A1 WO 2025141105A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- probe
- oligonucleotide
- probes
- nucleic acid
- compound
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09B—ORGANIC DYES OR CLOSELY-RELATED COMPOUNDS FOR PRODUCING DYES, e.g. PIGMENTS; MORDANTS; LAKES
- C09B23/00—Methine or polymethine dyes, e.g. cyanine dyes
- C09B23/02—Methine or polymethine dyes, e.g. cyanine dyes the polymethine chain containing an odd number of >CH- or >C[alkyl]- groups
- C09B23/04—Methine or polymethine dyes, e.g. cyanine dyes the polymethine chain containing an odd number of >CH- or >C[alkyl]- groups one >CH- group, e.g. cyanines, isocyanines, pseudocyanines
Definitions
- the invention is related to new compounds that can be used as labeling emitting fluorescence in detection probes.
- the detection probes of the invention allow the detection of nucleic acids, and in particular of singlestrand DNA (ssDNA) and typically ssDNA of a target micro-organism or ssDNA of a subject (in particular a human subject).
- ssDNA singlestrand DNA
- ssDNA typically ssDNA of a target micro-organism or ssDNA of a subject (in particular a human subject).
- the invention finds application in various sectors, particularly in the field of diagnosis.
- target nucleic acid sequences serves various purposes such as detecting or identifying pathogenic organisms, finding bacterial contamination in a food processing chain or diagnosing mutations that are responsible for genetic diseases or cancers.
- the primary challenges faced in these approaches concern the specificity, sensitivity, rapidity and reproducibility of the testing method used.
- Detection probes have been developed to offer the following possibilities:
- fluorescent nucleic acid probes which use energy transfer, exist. They are known as molecular beacons, Taqman® probes, adjacent hybridization probes, Scorpions® probes, HyBeacons® .... They are, for instance, described in Annu. Rev. Biomed. Eng. (2007) 9:289-320, Anal. Bioanal. Chem. (2011) 399:3157-3176 and IF. Medrano et al. BioTechniques (July 2005) 39:75-85.
- the adjacent hybridization probes using the FRET technique are characterized by two single-stranded hybridization probes which are used simultaneously and are complementary to adjacent sites of the same strand of the amplified target nucleic acid. Both probes are labelled with different fluorescent components. When excited with light of a suitable wavelength, a first component transfers the absorbed energy to the second component according to the FRET principle such that a fluorescence emission of the second component can be measured when both hybridization probes bind to adjacent positions of the target molecule to be detected.
- FRET usually occurs over distances comparable to the dimensions of most biological macromolecules, that is, about 10 to 100A or about twice the helix repeat distance in base-paired nucleic acids. This approach is described for instance by R.A. Cardullo et al. in Proc. Natl. Acad. Sci. USA (1988) 85: 8790-8794.
- the patent application US 2009/111100 describes the use of minor groove binder in FRET strategy to reduce background fluorescence of a FRET probe or a pair of probes and increase the Signal/Background ratios.
- the patent US 6,902,900 describes novel methods and strategies to detect analytes.
- a fluorescent intercalator or groove binder and a donor or acceptor dye are both used in a FRET system.
- the following documents can also be cited: US 8,663,923 and US 7,348,141 focusing on the choice of the nucleotide strand included in the probes.
- the nucleotide strand of the probe is attached to a fluorophore (a molecule that emits a fluorescence signal when it is excited by light of a suitable wavelength).
- fluorophores are a rhodamine or a derivative such as Texas Red, a fluorescein or a derivative, a fluorophore of the Alexa family such as Alexa 532 and Alexa 647, Alexa 405, Alexa 700 or Alexa 680.
- the present invention proposes bright fluorescent probes for the detection of a target nucleic acid and corresponding compound at the origin of the obtained fluorescence.
- the detection probes of the invention are particularly suitable for PCR monitoring, melting experiments for the determination of the melting temperature of amplicon, genotyping studies, detection of Single Nucleotide Polymorphisms (SNPs) ...
- SNPs Single Nucleotide Polymorphisms
- the chemical stability of the nucleic acid binding dye attached to the oligonucleotide is surprisingly increased in comparison with the dye alone.
- the probes of the invention offer also the advantage to stabilize the formed hybridization duplex obtained after hybridization with a target nucleic acid due to the presence of the dye that is covalently attached to the oligonucleotide. As a result, they offer possibilities to reduce the length of the attached oligonucleotide, for a better discrimination.
- the invention proposes alternative new detection probes which are suitable for hybridization to nucleic acids and are specific to a target nucleic acid sequence, including both deoxyribonucleic acids (DNA) and ribonucleic acids (RNA). So, they can be used to demonstrate the presence of specific target sequences in a sample, even in the form of complex mixtures.
- the detection probes of the invention can be used to detect a target nucleic sequence, and typically after formation of an amplicon, by real-time PCR (rtPCR) and/or by melting temperature (Tm) measurement. When used, the detection probes of the invention lead to emission of fluorescence and are also called fluorogenic probes.
- the detection probes of the invention are suitable to be used in any type of amplification technologies, such as PCR, including RT-PCR (PCR with reverse transcription) and asymmetric PCR.
- PCR PCR with reverse transcription
- asymmetric PCR PCR with reverse transcription
- the detection probes of the invention are very powerful tools to identify single-nucleotide polymorphisms (SNPs).
- the first object of the invention is to propose new compounds which are useful as fluorescent labeling of detection probes.
- the invention concerns compounds FC having the formula (I): wherein:
- - n is equal to 0, 1 or 2;
- - X is oxygen, sulfur, selenium, tellurium or C(CH 3 ) 2 ,
- ⁇ ql is 1, 2, 3, 4, 5 or 6 and
- ⁇ R"e is a Ci- 6 alkoxy group
- ⁇ q2 is 1, 2, 3, 4, 5 or 6 and
- ⁇ A' is a Ci-ealkoxy group
- R 3 is selected among hydrogen and the groups alkyl, cycloalkyl, aryl and -(CH 2 )q3-Y 3 , in which:
- ⁇ Y 3 is an aryl or Ci-ealkoxy group; or R 3 and R4 are bonded together and form a -(CH 2 ) r - chain with r being equal to 3, 4, 5 or 6, and
- Rk selected from hydrogen and the groups alkyl, cycloalkyl, aryl and in which:
- ⁇ q4 is 1, 2, 3
- ⁇ Y 4 is an aryl group
- - Ri is chosen among the groups alkyl, cycloalkyl, alkenyl, alkynyl, aryl, said groups being unsubstituted or substituted by one or several substituents selected from -CF 3 , -CN, alkyl, -Oalkyl, C(O)alkyl, - C(O)Oalkyl, -Salkyl, -Oalkyl -NHC(O)H,
- R 2 is Y 2 -L 2 -R' 2 , in which:
- ⁇ Y 2 is CH 2 , CHalkyl, C(alkyl) 2 , S or 0,
- ⁇ L 2 is a linker, in particular an alkylidenyl
- Ri, Rj and Rk are hydrogen.
- Nitrogen containing aromatic ring used in the definition of Z refers to pyrrolo, pyrazolo, isoxazolyl, isothiazolyl, oxazolyl, thiazolyl, pyridinyl, pyridazinyl, pyrimidinyl, pyrazinyl, quinolinyl, isoquinolinyl, quinoxalinyl, quinazalinyl, and the like.
- the compounds FC corresponding to compounds of formula (I), (la), (lb), (Ic), (Id) or (le) and their salts herein may contain geometric centers.
- all geometric isomers are understood to be included in the description of the compounds FC corresponding to formula (I), (la), (lb), (Ic), (Id) or (le) and their salts, unless otherwise indicated.
- Such geometric isomers include cis, trans, E and Z isomers, either in pure form or in various mixtures of geometric configurations.
- the compounds FC according to the invention are in the form of a salt: the compounds of formula (I), (la), (lb), (Ic), (Id), (le) and 1.1 to 1.6 are positively charged due to the N + (Re). They may include one or several negative charges, but they do not include other positive charges.
- the salts of the compounds of formula (I), (la), (lb), (Ic), (Id) or (le) include an anion or a number of cations (typically which are identical) corresponding to the global charge of the compound of formula (I), (la), (lb), (Ic), (Id) or (le).
- linkers Li and L 2 are -(CH 2 ) p i-Ph- (CH 2 ) p2 -, with for instance pl and p2 being independently 0, 1, 2, 3, 4, 5 or 6, -(CH 2 )p 3 -, -(CH 3 CH)p 3 -, -(CH 2 CH 2 O)p 3 -, -(CH 3 CHCH 2 O) p3 - with for instance p3 being 1, 2, 3, 4, 5 or 6.
- when present Li and/or L 2 are/is short and are/is, in particular, a chain (in particular an unsubstituted chain) defined by from 2 to 6 successive atoms forming the chain.
- nucleic acid or “nucleotide sequence” or “polynucleotide” means a chain of at least two deoxyribonucleotides or ribonucleotides (nucleotide units) optionally comprising at least one modified nucleotide, for example at least one nucleotide having a modified nucleic acid base, such as inosine, methyl-5-deoxycytidine, dimethylamino-5-deoxyuridine, deoxyuridine, diamino-2,6-purine, bromo-5 -deoxyuridine or any other modified base permitting hybridization.
- modified nucleic acid base such as inosine, methyl-5-deoxycytidine, dimethylamino-5-deoxyuridine, deoxyuridine, diamino-2,6-purine, bromo-5 -deoxyuridine or any other modified base permitting hybridization.
- Such polynucleotides can also be modified at the level of the internucleotide bond for example phosphorothioates, H- phosphonates, alkyl phosphonates, at the level of the backbone for example alpha-oligonucleotides (as described in FR 2 607 507) or PNAs (for peptide nucleic acid, as described by M. Egholm et al., in J. Am. Chem. Soc., 114, 1895-1897, 1992) or 2'-O-alkyl ribose and LNAs (for locked nucleic acid, as described by B.W. Sun et a!., in Biochemistry, 4160-4169, 43, 2004).
- alpha-oligonucleotides as described in FR 2 607 507
- PNAs for peptide nucleic acid, as described by M. Egholm et al., in J. Am. Chem. Soc., 114, 1895
- the nucleic acid can be natural or synthetic, an oligonucleotide or a longer polynucleotide, a nucleic acid fragment, a ribosomal RNA, a messenger RNA, a transfer RNA, a nucleic acid obtained by an amplification technique, and in particular an enzymatic amplification technique.
- target sequence or “target nucleic acid” intends to mean any nucleic acid whose presence is to be detected or measured or whose function, interactions or properties are to be studied.
- a target nucleic acid has a nucleotidic sequence in which at least one part of the chain of nucleotide units is specific and complementary to the nucleotide sequence of the oligonucleotide of the detection probe PR used.
- the target nucleic acid can be natural or obtained from a reaction of amplification in vitro.
- the target sequence results from a reaction of enzymatic amplification in vitro, the sequences produced by this amplification are called "amplicons”.
- the target nucleic acid generally has a length that exceeds the length of the oligonucleotide of the detection probe PR. This is classical in the technical field of detection probes.
- complementarity corresponds to the degree to which the sequences of two single-stranded nucleic acids can form a double-stranded complex according to Watson and Crick's hybridizing or pairing rule, where the base A (Adenine) hybridizes (or pairs) with the base T (Thymine) or U (Uracil) whereas the base C (Cytosine) hybridizes (or pairs) with the base G (Guanine).
- Two complementary sequences correspond to two sequences with 100% complementarity (100% complementary).
- oligonucleotide refers to a short nucleic acid sequence, typically of 10 to 100 nucleotide units, notably from 10 to 60 nucleotide units and in particular from 10 to 50 nucleotide units and even more particularly from 12 to 30 nucleotide units.
- the oligonucleotide of the detection probe PR is a single strand nucleic acid, and in particular a single-strand DNA.
- the oligonucleotide may be a Nucleic Acid Analog (NAAs) that differs from natural nucleic acid but can still recognize them through specific base pairing.
- NAAs Nucleic Acid Analog
- NNAs are usually derived from the naturally occurring nucleosides A, C, G and T modified e.g.
- the oligonucleotide of the detection probe PR does not include PNAs and LNAs.
- Nucleotide units of the oligonucleotide of the detection probe PR are usually naturally occurring nucleosides A, C, G and T or are derived from the naturally occurring nucleosides A, C, G and T to allow the covalent linkage of the compound FC.
- the oligonucleotide of the detection probe PR has a full sequence, or has at least a portion of sequence, chosen to specifically hybridize with the target nucleic acid sequence, called hybridization sequence or target-specific sequence.
- the hybridization sequence has preferentially a length of 10 to 30, and in particular of 12 to 25, nucleotide units.
- the oligonucleotide of the detection probe PR may also include additional portion(s) located at one end or both ends of the hybridization sequence.
- the hybridization sequence of the oligonucleotide of the detection probe PR is in general 100% complementary to the portion of the target nucleic acid sequence to which it is configured to hybridize, but not necessarily. There could be one mismatch (absence of complementarity between two nucleotide units due to base mispair) or few mismatches between the target nucleic acid and the hybridization sequence of the oligonucleotide of the detection probe PR that do not prevent the obtaining of a suitable specific hybridization. The position and the number of acceptable mismatches depend on the type of probes and will be adjusted by the skilled artisan in the art.
- the mismatch(es) will preferentially be out of the 3' and the 5' end zones of the oligonucleotide of the detection probe PR.
- the mismatch(es) will preferably be out of the internal zone of the oligonucleotide of the detection probe PR where the annealing/pairing with the target sequence occurs.
- the acceptable number of mismatches it will be adapted by the skilled person, considering, for instance, the following parameters: the sequence itself, the Tm of the detection probe, the hybridization temperature, the salts concentration during the hybridization and amplification.
- the hybridization sequence of the oligonucleotide of the detection probe PR may be not 100% complementary to the portion of the target nucleic acid sequence to which it hybridizes.
- the hybridization sequence of the oligonucleotide of the detection probe PR is at least 90% complementary to the portion of the target nucleic acid sequence to which it hybridizes. This will correspond to at most 5 mismatches between the hybridization sequence of the oligonucleotide of the detection probe PR and the portion of the target nucleic acid sequence to which it hybridizes, and in particular, there is(are) 1, 2, 3, 4 mismatches between the two sequences, depending on the length of the hybridization sequence of the oligonucleotide of the detection probe PR.
- the mismatch(es) correspond to a mutation in a target nucleic or to a specific genotype, in particular SNPs.
- End of an oligonucleotide means the starting point and the end point of synthesis of an oligonucleotide generally defined by the number carried by the free hydroxyls of the first or the last nucleoside, i.e. 3' or 5'.
- an oligonucleotide can be synthesized in the 3' to 5' direction or in the opposite direction, or the direction of elongation can even alternate during synthesis. This leads to oligonucleotides bearing 3'-5', 5'-3', 3'-3' or 5'-5' ends. According to the invention, the oligonucleotide has mainly 3'-5' ends.
- the internal region (also called middle region or zone) of an oligonucleotide means a region which is both at least one nucleotide away from the 5' end and at least one nucleotide away from the 3' end.
- the oligonucleotide of the detection probe PR may have a single-stranded conformation, meaning that it does not include a region that can hybridize with another region of the oligonucleotide.
- the oligonucleotide of the detection probe PR may have two regions which are complementary and can hybridize together and form a secondary structure (called self- complementary). But, advantageously, the oligonucleotide of the detection probe PR has a single-stranded conformation.
- the oligonucleotide of a detection probe PR may include a target-specific sequence (i.e. hybridization sequence) that specifically hybridize with at least a portion of the target nucleic acid and non-targetspecific sequence(s).
- target-specific sequence i.e. hybridization sequence
- non-target-specific sequences can include sequences which will confer a desired secondary or tertiary structure, such as a hairpin structure, as described in US 5,118,801, US 5,312,728, US 6,835,542, and US 6,849,412. More specifically, in that case, the oligonucleotide includes a central region which is complementary to the target nucleic acid and two extreme regions which are complementary to each other.
- detection probes PR including an oligonucleotide that forms a secondary or tertiary structure may be useful, for instance, when a quencher is present at the 5' or 3' end of the detection probe PR and a compound FC of the invention at the other end, 3' or 5' respectively.
- this detection probe PR hybridizes to a target sequence, it loses its configuration, the 5' and 3' ends move further apart and the quencher and the compound FC' are separated from one another.
- the fluorescence emitted then reflects the hybridization of the detection probe on the target, which is detected during its amplification and optionally quantified.
- detection probes PR including an oligonucleotide that forms a secondary or tertiary structure may also be useful, for instance, when two molecules of a compound FC of the invention are linked at both the 3' and 5' ends of the oligonucleotide.
- FC and FC' are "fluorescent dye” that means that they emit electromagnetic radiations of longer wavelength by a fluorescence mechanism upon irradiation by a source of electromagnetic radiation, including but not limited to a lamp, a photodiode or a laser.
- a quencher is a molecule that interferes with, and in particular is able to quench the detectable signal from a reporter moiety, typically the fluorescence of compound FC.
- a quencher can be selected from non- fluorescent aromatic molecules, to avoid parasitic emissions.
- said quencher is a Dabcyl or a "Black Hole QuencherTM" (BHQ), examples of non-fluorescent aromatic molecules that prevent the emission of fluorescence when they are physically near a fluorophore.
- BHQ Black Hole QuencherTM
- Any quencher molecule known in the art or easily designed by the skilled person can be used. Typical examples are methyl red, Eclipse® Quenchers (EDQ, MGB Eclipse%), Iowa Black® Dark Quenchers (IBRQ, IBFQ%), Black BerryTM Quencher (BBQ).
- a "donor” as defined herein is a dye that is part of a FRET couple in which the dye transfers energy to another dye by a nonradiative process. Therefore, in general, the fluorescence of the dye decreases when it is part of a FRET couple. FRET is described in detail in Yang et al., 1997, Methods Enzymol. 278:417-44.
- An "acceptor” as defined herein is a dye that is part of a FRET system in which the dye accepts energy from another dye by a nonradiative process. Therefore, in general, the fluorescence of the dye increases when excited at the wavelength of the corresponding donor of the FRET couple donor/acceptor.
- n By selecting the value of n, it is possible to adjust the properties of fluorescence obtained after the hybridization of a probe PR of the invention, with a target nucleic acid. Therefore, the features of the quencher or of the acceptor will be adapted by the skilled person, in function of the selected compound FC attached in the detection probe PR (called FC')- Typically, when a couple donor/acceptor including a compound FC is used, the following guidelines can be followed or adapted by the skilled person:
- n l
- the maximum excitation wavelength is in the range 510-580 nm
- the maximum emission wavelength is in the range 560-620 nm.
- Stringency can also be a function of the reaction variables, such as the concentration and type of ionic species present in the hybridization solution, the nature and concentration of denaturing agents and/or the hybridization temperature.
- the stringency of the conditions in which a hybridization reaction must be carried out will mainly depend on the hybridization probes used. All these data are well known and the appropriate conditions can be determined by a person skilled in the art. In particular, the conditions of hybridization used are classically adjusted by the skilled person to obtain an optimal specificity.
- a sample may include a specimen of natural origin, a specimen of non-natural origin or of synthetic origin.
- a sample can have various origins, such as swabs of food, environmental, human, veterinary, cosmetic origins ...
- Biological samples include blood (whole blood, serum, plasma), umbilical cord blood, chorionic villi, amniotic fluid, cerebrospinal fluid, spinal fluid, lavage fluid (e.g., bronchioalveolar, gastric, peritoneal, ductal, ear, arthroscopic), biopsy sample, urine, feces, sputum, saliva, nasal mucous, prostate fluid, semen, lymphatic fluid, bile, tears, sweat, breast milk, breast fluid, embryonic cells and fetal cells, in particular of human origin.
- the biological sample is blood, and more preferably plasma.
- the compounds FC according to the invention can be prepared according to conventional reactions known by the skilled person in organic chemistry.
- the compound FC In its free state, the compound FC is weakly fluorescent, but when it is covalently coupled to the oligonucleotide of the detection probe, it gives higher fluorescence intensity corresponding to the FC' moiety and this FC' moiety gives even higher fluorescence intensity when the oligonucleotide of the detection probe PR is hybridized to its target nucleic acid.
- Compounds (VI) are commercially available or are prepared illustratively by condensation in acidic conditions of appropriately substituted 1,3-diones (VII) and conveniently substituted urea or thioureas (VIII). Further, compounds (VI) having a thiol or alkoxy, at C(2) may be modified illustratively by reacting with alkylhalides, alkoxyhalides, or any reactant Xd-L 2 -R p (R p being a precursor of the reactive group R' 2 ) with a good leaving group Xd under neutral conditions to obtain compound (V). Compounds (III) may be prepared by reacting compounds (V) and compounds (IV) under basic conditions.
- the unsymmetrical cyanine (III) which may be purified by reverse phase chromatography is transformed to obtain a reactive group R'2 which is for example an activated ester.
- R'2 which is for example an activated ester.
- a phosphoramidite R'2 is easily obtainable from an unsymmetrical cyanine (III) in which Rp is a hydroxyl.
- R' 2 is a succinimidyl (NHS) ester, it can be obtained by activation of an ester Rp using bis NHS carbonate in pyridine.
- Exemplary compounds having this formula can be prepared as herein described, purified by HPLC using TFA water/acetonitrile/TFA as the mobile phase, and isolated as their corresponding TFA salts.
- Scheme 2 illustrates the case where compound (IV) is a benzothiazolium substituted in para by a group -NHC(O)R' (called (IVa)), with R' as defined for formula (I) and is obtained by acylation of an amino group of the corresponding position of the benzothiazolium.
- R' as defined for formula (I)
- Methyl, propyl, butyl, isopropyl, terbutyl and phenyl are typical examples of R'.
- Xf is a leaving group, for instance a Cl, or a OC(O)R' group.
- the pyrimidine molecule (XI) is selectively alkylated at the N1 position by reaction with an excess of an alkylating agent Xd-Ri, with Xd being a leaving group, like Cl, Br or I or a tosyl group and Ri as defined for formula (I), typically in acetonitrile at 50-90°C, in a closed tube for 1-6 days to give the pyrimidinium compound (V).
- alkylating agent Xd-Ri are Mel or EtI.
- the compound (V) is reacted with a compound (IV) (typically a benzothiazolium derivative), typically in a mixture of acetonitrile, ethanol and triethylamine at room temperature (typically 22°C), for a few minutes to yield the expected unsymmetrical cyanine (III), which may be purified by reverse phase chromatography.
- the unsymmetrical cyanine (III) is transformed to obtain a reactive group R'2 which is for example an activated ester, in the same way as explained for Scheme 1.
- the obtained compounds (IVa) can then react with a masked aldehyde as bis phenyl imine (XV), to yield the corresponding acetylated hemicyanines (XIV) in the presence of acetic anhydride and acetic acid, or only by fusing.
- the acetylated hemicyanines (XIV) can be purified by reverse phase chromatography using acetonitrile/water/TFA eluents.
- the acetylated hemicyanine (XIV) can then be reacted in slightly alkaline conditions with pyrimidinium (Vila), to obtain the compound (XIII).
- the compounds FC with other definition of Y2 or with a reactive group at the Ri position (and not at the R 2 position) will be prepared by the skilled person, by well-known organic reactions, using appropriately substituted thiourea in Ri for example.
- the compounds FC of the invention comprise a reactive group RG, at the Ri or R 2 position.
- a reactive group RG is a chemical moiety capable of reacting with a reaction partner (also called corresponding functional group) on a substrate molecule to form a covalent bond.
- a compound of the invention can be used to label a wide variety of molecules or substrates that contain a suitable reaction partner or are derivatized to contain a suitable reaction partner. So the reaction partner also comprises a reactive group, which is complementary to the reactive group of the compound FC of the invention.
- the reactive group RG and its reaction partner may be an electrophile and a nucleophile, respectively, that can form a covalent bond with or without a coupling agent or catalyst, with or without photoactivation.
- the reactive group RG may be one that will react with an amine, a thiol, a hydroxyl, an aldehyde or an alkyne.
- a precursor reactive group Rp is introduced into an intermediate during the synthesis, followed by conversion of the precursor reactive group Rp into the final reactive group RG at the last step of the synthesis.
- Various methods of introducing a reactive group RG that can be used to prepare the compounds FC according to the invention have been further described in the prior art, for instance in US 5,863,753 or US 2020/0407780. They can be adapted easily by the person skilled in the art.
- the introduction of one of these reactive groups on a compound FC can be carried out by techniques known in organic chemistry.
- the detection probes PR according to the invention can have various structures and conformation.
- the number of attached compounds FC, the position of the attachment, the presence or absence of labeling different from a compound FC (typically a quencher), the structure of the oligonucleotide may vary.
- the attachment is on the phosphate group of the corresponding nucleotide and is obtained by the intermediary of a linker. Only one compound FC may be attached at the 5' or 3' end of the oligonucleotide. It is also possible to have several (typically 2 or 3) compounds FC attached on the same end 5' or 3'. This is obtained by the use of a linker having several (typically 2 or 3) corresponding functional groups able to react with the reactive group of the compound FC, said linker being present at the 5' or 3' end (in particular covalently bonded to the phosphate group).
- Figure 14 shows the PCR amplification experiments of a gene sequence of Neisseria Gonorrhoeae micro-organism and the melting curve of its hybridization product with the probe 11.17 and its first derivative. It allows to discriminate between 3 mutants that differ only by 2 nucleotides versus the wild-type sequence on panels A and B.
- the panels C and D show for comparison the same experiment carried out in the presence of the commercial free dye LCG Plus only (BioFire Gen scanning reagent, Salt Lake City, USA).
- Figure 15 shows the melting curves in RFU and the first derivative of the melting curves that determines the Tm temperature for the probes 11.47, 11.48 and 11.49 in the presence or absence of the complementary DNA (COMP).
- the probes were labelled with the dye 1.1 of the invention and analyzed on the CFX (Agilent).
- Figure 23 shows on Panel A: Real time PCR amplification of a gene sequence of a Listeria micro-organism using the probes 11.22 and 11.23 in FRET technology.
- Panel B corresponding amplicon melting peaks.
- Figure 24 shows the evolution of the melting peak height intensity at 640 nm (Panel A) and 705 nm (Panel B) after real time PCR amplification of a Cronobacter micro-organism using the probes 11.50, 11.51, 11.53 to 11.57 of the invention and Comparative Probes 6, 7, 8, 9, 11 and 12, in FRET technology.
- N-phenylthiourea (9.6 g, 63.16 mmol) was put in a round bottomed flask of 250 mL and dissolved in 130 mL of ethanol (EtOH). Acetylacetone (2,4- pentandione) was added followed by HCI 37% (15 mL). The mixture was stirred and heated to 90 °C (reflux) for 5 h. The mixture was turned orange- red. The heating was turned off and was cooled down to room temperature. 180 mL of diethyl ether was added to precipitate the product. The precipitate was filtered and washed with diethyl ether two times.
- the precipitate was neutralized in an Erlenmeyer flask of 500 mL with an aqueous solution of NaOH (150 mL, 8.18 g, 180 mmol) and 80 mL of EtOH was added.
- the solution was poured in a 1 L separatory funnel and 200 mL of DCM was added.
- the separatory funnel was shaken vigorously for a few seconds. After few minutes the organic phase was recovered.
- the aqueous phase was washed two times with 50 mL of DCM.
- the organic phase was evaporated by using a rotary evaporator.
- SEQ ID N°18 also called SEQ ID N°1 (AUTO 2): CCAGGCCGCCAGAAGAGGAGCCCCAATGCCTGG (underlined means a self- complementary sequence)
- the underlined nucleotide corresponds to the mismatch with the target nucleic acid.
- sequences SEQ ID N°17 (SEQ ID N°1 (AUTO 1)) and SEQ ID N°18 (SEQ ID N°1 (AUTO 2)) were designated to evaluate the influence of two selfcomplementarity portions in the two 3' and 5' end zones on the fluorescent background of the probe alone that bears two coupled compound FC (so in the form of FC') at its 3' and 5' ends.
- the synthetic target nucleic acids of the sequence below are complementary or partially complementary (presence of one mismatch) to the sequences of the probes and were used to exemplify the invention. They correspond to a portion of a sequence of a gene belonging to a real micro-organism.
- SEQ ID N°21 comprising a sequence complementary to the oligonucleotide of SEQ ID N°3: ATACGGACGATGGTGTCGTAAACTGCGGAATCGCCGTGGGGGTGGTATTTACCGA TGACGT
- SEQ ID N°22 comprising a sequence complementary to the oligonucleotide of SEQ ID N°4: GTAGCAGTAGACGGCTGCGACAGAAGGCTAGCGGTAGGCGCGG
- SEQ ID N°23 comprising a sequence complementary to Probe SEQ ID N° 5: ACCGTTATGGATTTGGAGATGCAGCAGTCTAAAGTGAAGGATCGGTATGTCAATT TTCCT
- SEQ ID N°24 comprising a sequence complementary to the oligonucleotide of SEQ ID N° 6, and partially complementary (presence of one mismatch) to the oligonucleotides of SEQ ID N°7, 8 and 9: TGTAAAGGAAAGTAACAATTAAAACCTTCAACACCATTACAAGGTGTGCTACCGGC CTGA
- SEQ ID N°25 comprising a sequence complementary to the oligonucleotide of SEQ ID N°10: CAGTGCTTGCGGATGCGATAGTTGGAGCAGCAAATGCTGTAACCGCAATCCCAGC T
- SEQ ID N°26 comprising a sequence complementary to the oligonucleotide of SEQ ID N°ll and 15: AAAAATTAGACACTACTTATGCTGGTACCGCTGAGATTAAACAACCAGTTGTTAAA TCTC
- SEQ ID N°29 comprising a sequence complementary to the oligonucleotide of SEQ ID N°14: CTTTGATTTGTTCGACATAACTTTCCATGAAGGAAGCAATGTTTTCTTTACCGTTA GCGT
- SEQ ID N°30 comprising a sequence complementary to the oligonucleotide of SEQ ID N°16: CGCATTCCAGAAATTGTTCCCAGTGCATAGATATGAAGCGAACAGGCTACCAGACA CA
- SEQ ID N°32 comprising a sequence complementary to the oligonucleotide of SEQ ID N°31: I I I I I I CGATCGCCCTCCCACGTGC I I I I I I I
- SEQ ID N°35 comprising a sequence complementary to the oligonucleotides of SEQ ID N°33 and 34: CGCATTCCTTCCGAGACGGTTCTGAATGGCTTACATGGATCACTTCGACACA
- the quencher used in probes 11.47 to 11.49 is BHQ1 (Black Hole Quencher 1, 2-[N-(2-hydroxyethyl)-4-[[2-methoxy-5- methyl-4-[(4-methyl-2-nitrophenyl)diazenyl]phenyl]diazenyl]anilino]ethanol).
- Table 4 (continued 2) Table 4 (continued 3) Table 4 (continued 4) Table 4 (continued 5)
- the oligonucleotide was produced by incorporating in 5' twice an Amino-Modifier Serinol Phosphoramidite (Ref. 10-1997 from Glen
- the probe obtained bears two molecules of compound 1.1 at the 5' end of its oligonucleotide.
- the oligonucleotide was produced by using a Symmetric Doubler Phosphoramidite (Ref. 10-1920 from Glen Research, Sterling, USA).
- the two functions 4,4'-dimethoxytrityloxy were after converted to -NH 2 by reaction with an 5'-Amino-Modifier C6 Phosphoramidite (Ref. 10-1906 from Glen Research, Sterling, USA) as it is done for most of the probes described with an amino modification in 5' end for further reaction with the activated dye.
- Two molecules of compound 1.1 carrying an NHS group were after conjugated on these reactive groups -NH 2 . So, the probe obtained bears two molecules of compound 1.1 at the 5' end of its oligonucleotide.
- amino modified dT, dA or dG phosphoramidite was introduced instead of dT, dA or dG : respectively Ref. 10-1039 for the Amino-Modifier C6 dT phosphoramidite, Ref. 10-1089 for the Amino-Modifier C6 dA phosphoramidite and Ref. 10-1529 for the N2-Amino- Modifier C6 dG phosphoramidite from Glen Research, Sterling, USA.
- the generated amino groups were further conjugated with the activated NHS dye.
- the Table 4bis below shows the exact position of the internal modified nucleoside for dye attachment.
- the oligonucleotide was produced by using a symmetrical branching phosphoramidite (Ref. CLP-5215 from ChemGenes, USA). After, the same protocol as described under (3) was used: a probe bearing two molecules of compound 1.1 at the 5' end of its sequence was obtained.
- the probe corresponding to the conjugated oligonucleotide was precipitated by adding to the crude reaction 18 pL of lithium perchlorate 3 M, QSP 300 pL water and 900 pL acetone. The mixture was vortexed and was centrifugated at 10 000 rpm (rotation per minute) and the supernatant was discarded. The pellet was resuspended in 282 pL of water and 18 pL of lithium perchlorate 3M and vortexed. Acetone (900 pL) was added to get a cloudy mixture which was again centrifugated at 10 000 rpm and the supernatant was discarded. The same operation was repeated 2 more times until the supernatant became translucent.
- the probes of the invention were prepared at 1 pM, in the presence of their complementary DNA strand (COMP.) at 4 pM in a model hybridization buffer 2 (Tris pH 8.4 20 mM, NaCI 10 mM, dNTP (4x0.3 mM), MgCI2 4 mM, proprietary stabilization buffer IX, TAQ polymerase 0.2 U/pL and BSA 550 ng/pL) for a total volume of 20 pL. Then, the solutions were poured into a microplate and the maximum absorption (equivalent to the maximum excitation) on the spectra was measured. Thereafter, the maximum fluorescence emission was also measured upon excitation at: (X absorption max -30 nm) with a gain of 60 using a spectrophotometer/spectrofluorometer reader (TECAN, Austria).
- a model hybridization buffer 2 Tris pH 8.4 20 mM, NaCI 10 mM, dNTP (4x0.3 mM), MgCI2 4
- Tm peak height The difference in RFU at the Tm temperature between the COMP, and the NON COMP, or probe alone experiment can be linked to the sensitivity of the detection (Tm peak height). The higher the peak is, the easier the detection of this event is (represented by the double arrow in Figures 6A-F on the right panels). Since the Tm peak height is linked to the probe concentration, the normalized Tm peak height is corrected by the concentration of the probe and allows to compare all these experiments, the obtained results demonstrate the general behavior of these probes.
- Table 6 Analytical melting experiments using probes of the invention with the appropriate controls (Probe: synthesized detection probe; Dye: dye bearing an activated ester used for conjugation; SEQ.
- Number of dyes The increase of the number of dyes from 2 (probe 11.26), 3 (probe 11.27), 4 (probe 11.28), 5 (probe 11.29), 6 (probe 11.30) has in general a benefit on the sensitivity of the detection and on the fluorescence exaltation at 60°C upon hybridization of the probe.
- the optimum is around five dyes (probe 11.29) with a ratio of 2 and a Tm height of 380 versus a ratio of 1.1 and a Tm height of 160 for (probe 11.26) with 2 dyes only.
- Dve Localization The localization of the dye on the probe can slightly affect the fluorescence ratio at 60°C and the height of the Tm peak as it can be seen on the series of probes II.6-8, 11.24-25 and II.9 where different localization and number of dyes have been used.
- the specific beneficial effect of the internal modifications is in particular visible with the 11.24 probe which is almost as good as the triple labelled probe II.9 and far better than the mono-functionalized probe II.6.
- the double modification at 5' and 3' ends and the triple modification at 5' and 3' ends and internally are the favorite modes.
- linker Most of the different linkers used between the dye and the oligonucleotide sequence have a same behavior or lead to slightly lower functional performance in terms of exaltation ratio and Tm height after hybridization. The simplest one (Ce alkylidenyl linker) seems to be the most convenient. Unsuspectedly linkers that allow the incorporation of 2 dyes at the 5' are not more efficient and even less as only one (probes 11.34-35 versus probe II.6). Very long oligonucleotide linkers 11.36 and 11.37 are poorly efficient certainly due to the length of the linker.
- C12 PEG linker (probe II.2) > Ce alkylidenyl linker (most of the examples) > > auto complementary oligonucleotide in 11.36 and 11.37 » doubler (probe 11.35) > serinol linker (probe 11.34).
- Type of nucleotide modification whatever the type of nucleotide modification, the probes behave the same. This is the case for probes 11.38 and 11.39 with LNA modification inside the DNA sequence in comparison with the probe II.4 including an unmodified DNA sequence.
- the probes of the invention are specific to a given sequence and emit a stronger fluorescence once hybridized, whatever the type of dye used. These probes cannot recognize a non-target nucleic acid.
- the dye attachment position, the type of linker and the number of dyes are important factors and allow to modulate the sensitivity of the detection. It was also shown that the detection of the hybridization event is possible at another wavelength that allows multiplexing possibilities. In a general manner, the universality of the approach proposed by the invention was demonstrated. Some slight differences in performances come from the nature of the dye, the oligonucleotide sequence, the number and the localization of the dyes on the probe.
- the probes II.1-4 and 11.10-12 evaluated analytically in Part III were used to specifically detect an amplicon generated by a PCR amplification reaction.
- a sequence of the OC43 gene from a Coronavirus micro-organism was used as an amplified model target sequence.
- PCR reaction with real time fluorescence measurements was performed during 50 cycles (initial denaturation 95°C for 30 sec., alternating cycles of denaturation/annealing/extension of 95°C for 10 sec I 60°C for 20 sec I 72°C for 20 sec) followed by a melting experiment from 60 to 90°C (by 0.5°C steps) on a CFX Biorad thermocycler while monitoring fluorescence on the FAM, Texas Red and Quasar 705 channels during PCR and melting.
- Probe II.11 reached roughly the level of detection of probe II.4, but with another detection channel Texas Red (A RFU of 80 versus 110 respectively).
- the probe 11.12 synthesized with the dye 1.5 demonstrated excellent properties with high detection sensitivity on the other detection channel Quasar 705. Table 7
- the detection probes PR of the invention allow the detection of an amplicon generated during a PCR reaction with high specificity and sensitivity.
- the double dye attachment at 5' and 3' ends greatly increases dramatically the sensitivity of the detection, even to as low as 1 input copy /mL sometimes. Those results were non-expected. It was also shown that the detection of a specific PCR amplification reaction is possible at another wavelength that opens multiplexing possibilities.
- the Figure 9 shows a real time PCR reaction using probe II.5 targeted to different input of gene DNA from 1 Geq/PCR (genome equivalent/PCR reaction) to 10e5 Geq/PCR.
- the oligonucleotide of probe II.5 corresponds to a gene sequence of a Streptococcus micro-organism used as an amplified model target sequence. The same protocol as described in Part IV was followed.
- This example shows the generalization of the real time monitoring of PCR amplification and the subsequent Tm determination of the hybridization product of a given denatured amplicon with the probes of the invention.
- the protocol as described in Part IV was used, except that a first PCR was carried out on a fast mode using a MBS platform (MOLECULAR BIOLOGY SYSTEMS B.V, Scottweg, The Netherlands).
- the protocol was as following: initial denaturation for 30 sec at 102°C followed by 30-40 alternating cycles of denaturation at 102°C for 2 s and extension at 60°C for 4 s starting from an initial input of 10e5 copies/PCR of gBIocks corresponding to a gene sequence of Neisseria Gonorrhoeae micro-organism.
- the PCR formulation was optimized to enable the handling of the harsh fast PCR conditions.
- the second PCR followed by melting experiments from 60 to 90°C was realized on a CFX Biorad thermocycler while monitoring fluorescence on the FAM channel during melting.
- the probes II.6-9 were used at 1 pM to specifically detect the amplicon generated by fast PCR amplification reaction.
- Table 8 compares the effect of the localization of the dye on the sensitivity of the fluorescent detection (height of the Tm peak).
- the unexpected 5 times higher fluorescence of the double labeled 5'-3' labeled probe II.7 versus the mono labeled one (probe II.6) was again demonstrated. It can also be seen that a dye localized in the internal region of the probe sequence (probe II.8) was 3 times more efficient than a dye localized at the 5' end (probe II.6).
- the triple conjugated probe (II.9) shows the best performances.
- Table 8 Functional melting experiments using Probes II.6-9 with an initial input of 10e5 copies/PCR corresponding to an amplifiable gene sequence of the Neisseria Gonorrhoeae micro-organism.
- 2.5 pM probes solutions were used in a typical PCR mix formulation (Tris pH 8.4 20 mM, 10 mM NaCI, IX proprietary stabilization buffer, dNTP (4x0.3 mM), MgCL 4 mM, TAQ polymerase and BSA 550 ng/pL) in a total volume of 20 pL.
- a CFX Maestro from Biorad Laboratories was used with PCR polypropylene cuvettes of 100 pL to record the fluorescence in the SybRgreen Channel as a function of temperature from 20°C to 90°C (0.5°C/min) using a preliminary denaturing step during 1 min at 95°C.
- a typical PCR mix formulation (Tris pH 8.4 20 mM, 10 mM NaCI, IX proprietary stabilization buffer, dNTP (4x0.3 mM), MgCL 4 mM, TAQ polymerase 0.2 U/pL and BSA 550 ng/pL) were used in a total volume of 20 pL. The same conditions were used as described in Part III.
- Figure 15 shows particularly the strong fluorescence exaltation at 60°C and, as expected, the low fluorescence background of the quenched probes without complementary strand.
- the Comp. Probes 1-4 were synthesized by conjugation between triple amino linked oligonucleotides and comparative NHS dyes Comp. 1, Comp. 2 and Comp. 3. The obtained results were compared to probes 11.40 and 11.41 linked to dye LI. The probes were evaluated at 2 pM for their specificity as described in Part III.
- Part XIII PCR directed to a sequence of Streptococcus microorganism using probes of the invention conjugated with a mono positively charged dye and comparative probes conjugated to double positively charged dyes
- the Comp. Probes 4-5 were synthesized by conjugation between double and triple amino linked oligonucleotides and comparative NHS dyes Comp. 1.
- the probes were evaluated at 1 pM in a PCR reaction with initial input of 1, 10 and 100 Geq of gene DNA from a Streptococcus micro-organism per PCR reaction as described in the protocol of Part VI. After the PCR, the neoformed amplicons were submitted to a melting experiment as shown on Figure 18 for Comp. Probe 4 and on Figure 19 for Comp. Probe 5.
- the Comp. Probes with a double cationic charge on the dye detect their target as expected, but in an unexpected manner, detect also the full amplicon at a higher Tm.
- the following temperature ramp was followed: 80°C to 40°C (depending on Tm temperature) at 0.5°C/Minute with one UV measurement at 260 nm each 0,5°C (renaturation), stop for 3 min and 40°C to 80°C at 0.5°C/Minute with one UV measurement at 260 nm each 0.5°C (denaturation/melting).
- the melting temperature was calculated from the first derivative on the melting curve (more precise than the denaturation curve even if there is no hysteresis).
- the ATm between the native non modified duplex (20/60 mers) and the modified probes/60 mers was calculated and plotted in Table 12.
- the obtained results showed that the detection probes of the invention possess a strong ability to stabilize a duplex ( ⁇ 2.5°C I dye) leading to a stabilization around 4-5°C for the double conjugated probes.
- Dyes 1.1 and 1.4 have similar stabilization properties. That makes possible to shorten the length of the oligonucleotide and to increase even more the specificity of the hybridization.
- FAM usual donor group (Comp, probe 6).
- the commercially available acceptor groups LcRed640® and ATTO590TM were introduced as the binary probes (designated Comp. Probes 7 and 8). All the synthesis were realized internally as previously described.
- the final conjugated probes (probes 11.50 according to the invention and Comp. Probes 6, 7 and 8) were further purified to eliminate any unlabeled products.
- the quality control was realized by LC-MS (Waters).
- the concentration of the donor probes and their complementary strand were fixed at 0.1 pM whereas the concentration of acceptor probes varied from 0.05 pM to 0.4 pM.
- excitation wavelength was fixed at 455 nm, and the emission wavelength start at 500 nm and finished at 750 nm, whereas for the 1.1 donor group, excitation wavelength was fixed at 410 nm, and the emission wavelength start at 450 nm and finished at 700 nm.
- excitation wavelength triggers fluorescent emission of the donor probe (compound FC) which is transferred to the acceptor fluorophore on the acceptor probe. This probe emits fluorescence in return.
- Figure 20 shows the evolution of the fluorescence exaltation of the acceptor group based on a fixed amount of the donor and the complementary strand, and variable concentration of the acceptor group. It shows particularly the strong exaltation with 1.1 dye of the invention (Probe 11.50) whatever the commercial acceptor groups used (Comparative Probes 7 and 8).
- the fluorescent emission resulting from the conjugated dye 1.1 in the probe of the invention is transferred more efficiently to the acceptor probes (Comparative Probe 11 and Probe 11.53) than with the usual FAM donor (Comparative Probe 6 in panels A and B).
- the max FRET signal at 705 nm was more than five times better for the FRET pair I.l/Cy5.5TM (Panel C: Probe II.50/Comparative Probe 11) using a probe of the invention compared to the commercial combination FAM/Cy5.5TM (Panel A: Comparative Probe 6/ Comparative Probe 11).
- a modified commercial PCR kit (Gene Up® Cronobacter, bioMerieux, France) was used with a specific donor probe, Comp. Probe 6 labelled with FAM in 3' end, and with specific acceptor probes, acceptor probes 11.50, 11.51, 11.53, 11.54, 11.55, 11.56 and 11.57 of the invention and comparative acceptor probes, Comp. Probe 7 labelled with LcRed640® in 5' end, Comp. Probe 8 labelled with ATTO590TM in 5' end, Comp. Probe 9 labelled with LcRed640® in 3' end, Comp. Probe 11 labelled with Cy5.5TM in 5' end and Comp. Probe 12 labelled with ATTO590TM in 3' end.
- the melting peak height intensity at 640 nm was even six times better with the 1.1 dye of the invention (Probe II.51/Comparative Probe 12 and Probe II.51/Comparative Probe 9 at respectively 443.8 and 458.3 RFU).
- the melting peak height intensity at 705 nm was four to fifty times better for all the FRET pairs including a probe of the invention labelled with the dye LI, 1.5 or 1.6 (respectively FRET pairs Comparative Probe 6/Probe 11.53, Probe II.54/Probe 11.56, Probe II.57/Probe 11.53 and Probe II.55/Probe 11.56 at 11.6 to 147.5 RFU) compared to the comparative probes (circled Comparative Probe 6/Comparative Probe 11 at 3.1 RFU).
- the melting peak height intensity at 705 nm was two times better with the 1.1 dye of the invention (Probe II.51/Probe 11.56 at 123.8 RFU) compared to the single labelled probe (Probe II.54/Probe 11.56 at 59.6 RFU).
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
Abstract
The invention relates to compounds FC having the formula (I), wherein Z, X, Re, Ri, Rj, Rk, n, R1, R2, R3 and R4, are as defined in claim 1, including their salts. The invention concerns also a detection probe PR labelled with at least one molecule of a compound FC according to the invention, comprising an oligonucleotide covalently coupled on the reactive group RG of said compound FC, the methods and kits using them.
Description
Specific probes for the detection of nucleic acids, methods and uses
FIELD
The invention is related to new compounds that can be used as labeling emitting fluorescence in detection probes. The detection probes of the invention allow the detection of nucleic acids, and in particular of singlestrand DNA (ssDNA) and typically ssDNA of a target micro-organism or ssDNA of a subject (in particular a human subject). The invention finds application in various sectors, particularly in the field of diagnosis.
BACKGROUND OF THE INVENTION
Currently, the detection and/or quantification of target nucleic acids represent a major goal in numerous laboratories or industries and especially in the medical or food-processing field. Within these fields, finding target nucleic acid sequences serves various purposes such as detecting or identifying pathogenic organisms, finding bacterial contamination in a food processing chain or diagnosing mutations that are responsible for genetic diseases or cancers. The primary challenges faced in these approaches concern the specificity, sensitivity, rapidity and reproducibility of the testing method used.
Various methods based on hybridization are disclosed in the literature. Most of the time, the detection and/or quantification of target nucleic acid sequences imply to extract nucleic acids from a given sample, and if desired, to amplify the extracted nucleic acids, before identifying the specific sequence of interest. Over time, a plethora of amplification methods have been proposed, including PCR (polymerase chain reaction), LCR (Ligase Chain Reaction), NASBA (Nucleic Acid Sequence-Based Amplification), TMA (Transcription Mediated Amplification), and SDA (Strand Displacement Amplification).
As described by E. Navarro et al., in Clinica Acta, 439 (2015) 231-250, realtime PCR detection chemistry is the method of choice in many laboratories for diagnostic and food applications. This technology merges the polymerase chain reaction with the use of fluorescent reporter molecules, in order to monitor the production of amplification products during each cycle of the PCR reaction. The technologies of detection can be divided in two main groups: a first group comprising double-stranded (ds) DNA intercalating molecules named dyes, such as SYBR® Green I, thiazole orange and EvaGreen®, another second group named detection probes comprising oligonucleotides labeled with a molecule that can emit fluorescence at least after hybridization with the target nucleic acid. The first group only enables a
non-specific detection. As a result, the techniques of the first group are very flexible because one dye can be used for the detection of different target nucleic acids. But dyes do not bind in a sequence-specific manner and cannot differentiate sequences and multiplexing reactions are not possible. Fluorescence will be emitted whatever the type of amplicon generated by the amplification, as soon as a duplex is encountered by the dye. The specificity can only come from the use of a melting curve and the melting temperature (Tm) measurement which is specific of a given nucleic acid sequence and therefore of a dedicated amplicon.
On the contrary, the interest of the developed detection probes is to allow specific detection of a target nucleic acid. Detection probes have been developed to offer the following possibilities:
- detecting single-nucleotide polymorphisms (SNPs), which involve a variation of a single nucleotide at a specific location in the genome (SNPs are the most abundant form of sequence variation in the human genome) and corresponds to a site within the DNA sequence that varies by a single base (substitution, insertion or deletion) from person to person. These SNPs are often linked to phenotypic characteristics, such as certain diseases and are commonly used as genetic markers,
- detecting large amplicons with an higher specificity for the detection of variants,
- distinguishing different amplification products in the same tube/experiment by multiplex detection.
Different types of fluorescent nucleic acid probes, which use energy transfer, exist. They are known as molecular beacons, Taqman® probes, adjacent hybridization probes, Scorpions® probes, HyBeacons® .... They are, for instance, described in Annu. Rev. Biomed. Eng. (2007) 9:289-320, Anal. Bioanal. Chem. (2011) 399:3157-3176 and IF. Medrano et al. BioTechniques (July 2005) 39:75-85.
The adjacent hybridization probes using the FRET technique are characterized by two single-stranded hybridization probes which are used simultaneously and are complementary to adjacent sites of the same strand of the amplified target nucleic acid. Both probes are labelled with different fluorescent components. When excited with light of a suitable wavelength, a first component transfers the absorbed energy to the second component according to the FRET principle such that a fluorescence emission of the second component can be measured when both hybridization probes bind to adjacent positions of the target molecule to be detected. In practice, FRET usually occurs over distances comparable to the dimensions of most biological macromolecules, that is, about 10 to 100A or about twice the helix repeat distance in base-paired nucleic acids. This approach is described for
instance by R.A. Cardullo et al. in Proc. Natl. Acad. Sci. USA (1988) 85: 8790-8794.
Several embodiments of this technique have been described in the prior art. For instance, the patent application US 2009/111100 describes the use of minor groove binder in FRET strategy to reduce background fluorescence of a FRET probe or a pair of probes and increase the Signal/Background ratios. The patent US 6,902,900 describes novel methods and strategies to detect analytes. A fluorescent intercalator or groove binder and a donor or acceptor dye are both used in a FRET system.
As specific examples of detection probe disclosed in the prior art, the following documents can also be cited: US 8,663,923 and US 7,348,141 focusing on the choice of the nucleotide strand included in the probes. In these documents, the nucleotide strand of the probe is attached to a fluorophore (a molecule that emits a fluorescence signal when it is excited by light of a suitable wavelength). The cited examples of fluorophores are a rhodamine or a derivative such as Texas Red, a fluorescein or a derivative, a fluorophore of the Alexa family such as Alexa 532 and Alexa 647, Alexa 405, Alexa 700 or Alexa 680.
The patent US 5,863,753 of Molecular Probes describes cyanine dyes of
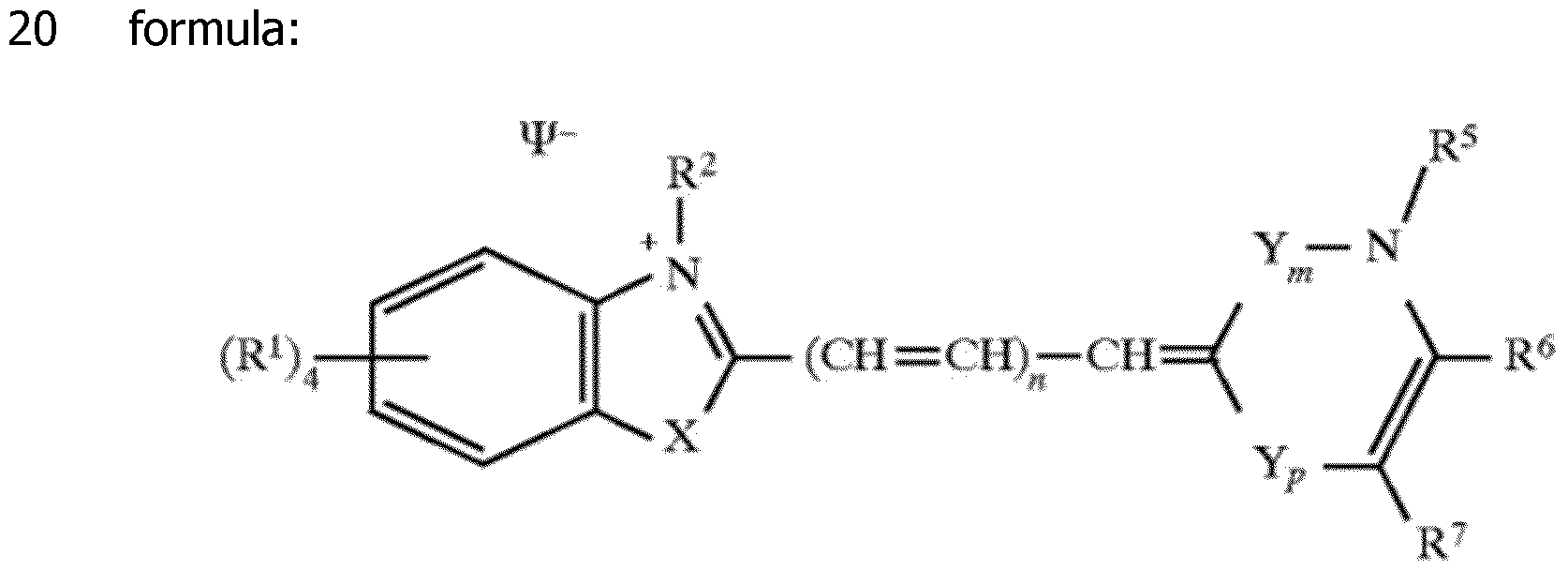
These dyes can bear a reactive moiety used to form a conjugate by covalent link with various entities including amino acid, peptide, protein, polysaccharide, a nucleic acid base, nucleoside, nucleotide or a nucleic acid polymer, lipid, lipophilic polymer, non-biological organic polymer, polymeric microparticle, animal cell, plant cell, bacterium, yeast, or virus.
The patent US 6,329,144 describes other types of probes which can include unsymmetrical cyanines. The attachment to the nucleic acid is made via the equivalent R2 group of the molecule shown above.
The patent US 9,682,970 in the name of Biotium describes a large class of compounds that can be used as DNA binding dyes or coupled to substrate (including a nucleic acid) by a reactive group. The inventors of the present invention have worked on detection probes comprising an oligonucleotide
covered by US 9,682,970 and found that some of those detection probes are not specific. They behave like a free dye and lead to fluorescence emission by interaction with any double stranded DNA that may be present in a PCR reaction.
In this context and despite the research progress and the commercial availability of a wide range of detection probes, there remains a need for improvement in various aspects of specific detection probes including but not limited to cost and ease of synthesis, design, chemical stability of the probe, stability of the hybridization products, detection limit, dynamic range of detection, compatibility with different detection formats and instruments, development of easily industrializable and even cheaper specific detection probes that are useful, in particular, for sensitive multiplex PCR assays. The aim of the present invention is to address some of these needs.
Particularly, most of probes of the prior art use dyes which belong to the thiazole orange family, with a quinolone ring. Certain of these dyes appear poorly fluorescent when excited by cheap and current blue laser diode that became a standard in most of the fluorescence readers used for molecular diagnostic assays. Certain solutions of the prior art describe PNA probes which are tremendously expensive and difficult to synthesize.
The present invention proposes bright fluorescent probes for the detection of a target nucleic acid and corresponding compound at the origin of the obtained fluorescence. The detection probes of the invention are particularly suitable for PCR monitoring, melting experiments for the determination of the melting temperature of amplicon, genotyping studies, detection of Single Nucleotide Polymorphisms (SNPs) ... The chemical stability of the nucleic acid binding dye attached to the oligonucleotide is surprisingly increased in comparison with the dye alone. The probes of the invention offer also the advantage to stabilize the formed hybridization duplex obtained after hybridization with a target nucleic acid due to the presence of the dye that is covalently attached to the oligonucleotide. As a result, they offer possibilities to reduce the length of the attached oligonucleotide, for a better discrimination.
SUMMARY OF THE INVENTION
The invention proposes alternative new detection probes which are suitable for hybridization to nucleic acids and are specific to a target nucleic acid sequence, including both deoxyribonucleic acids (DNA) and ribonucleic acids (RNA). So, they can be used to demonstrate the presence of specific target sequences in a sample, even in the form of complex mixtures. In particular, the detection probes of the invention can be used to detect a target nucleic sequence, and typically after formation of an amplicon, by real-time PCR
(rtPCR) and/or by melting temperature (Tm) measurement. When used, the detection probes of the invention lead to emission of fluorescence and are also called fluorogenic probes. More generally, the detection probes of the invention are suitable to be used in any type of amplification technologies, such as PCR, including RT-PCR (PCR with reverse transcription) and asymmetric PCR. The detection probes of the invention are very powerful tools to identify single-nucleotide polymorphisms (SNPs).
In this context, the first object of the invention is to propose new compounds which are useful as fluorescent labeling of detection probes. The invention concerns compounds FC having the formula (I):
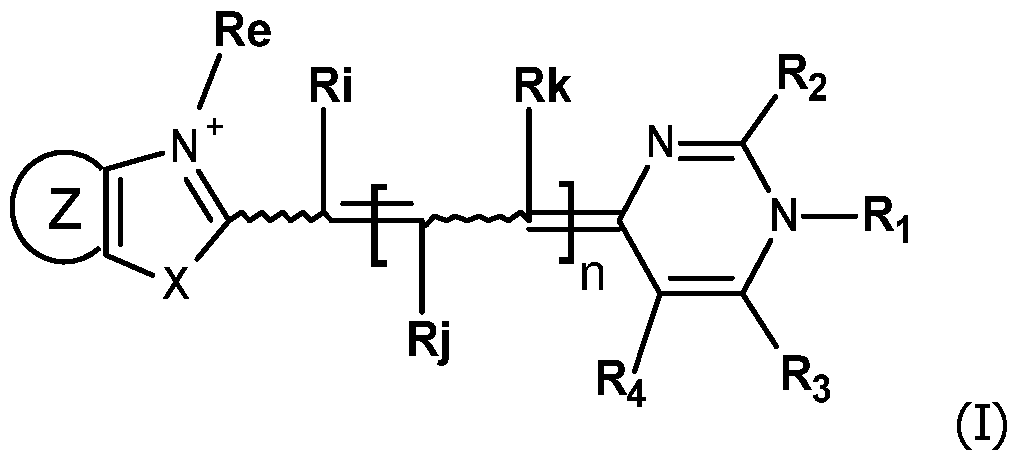 wherein:
wherein:
- n is equal to 0, 1 or 2;
- X is oxygen, sulfur, selenium, tellurium or C(CH3)2,
- Re is an alkyl, cycloalkyl, alkenyl, alkynyl, aryl, -(CH2)pR'e, where p is 1, 2, 3, 4, 5 or 6 and R'e is an aryl, -CF3, -CN, -C(O)alkyl, -C(O)Oalkyl, -Salkyl, - Oalkyl, -NHC(O)H, -S(O2)O’, -S(O2)Oalkyl, -P(O2)O’, -P(O2)Oalkyl, -CH=N-O-R, -C(CH3)=N-O-R, -CH=N-NH-C(O)-R, -C(CH3)=N-NH-C(O)-R, -CH=N-O-C(O)-R, -C(CH3)=N-O-C(O)-R, -NHCOR and -CONHR, -CONHR being preferred, R being either an alkyl or -(CH2)qi-R"e, in which:
■ ql is 1, 2, 3, 4, 5 or 6 and
■ R"e is a Ci-6alkoxy group,
- Z is a fused mono or polycyclic aromatic or nitrogen-containing heteroaromatic ring, optionally substituted by one or several substituent(s) A identical or different selected among the groups alkyl, cycloalkyl, alkenyl, alkynyl, aryl, -CF3, -CN, -C(O)alkyl, OAlkyl, -C(O)Oalkyl, -Salkyl, -Oalkyl, -NHC(O)H, -S(O2)O’, -S(O2)Oalkyl, -P(O2)O’, P(O2)Oalkyl, -CH=N-O-R', -C(CH3)=N-O-R', -CH=N-NH-C(O)-R', -C(CH3)=N-NH-C(O)-R', -CH=N-O-C(O)-R', -C(CH3)=N-O-C(O)-R', -NHCOR' and -CONHR', -NHCOR' and -CONHR' being preferred, R' being a phenyl, an alkyl or -(CH2)q2-A', in which:
■ q2 is 1, 2, 3, 4, 5 or 6 and
■ A' is a Ci-ealkoxy group;
- Ri, Rj, Rk, R3 and R4 are as defined hereinafter:
- Ri, Rj and Rk are identical or different and are independently selected from the group consisting of hydrogen and Ci-ealkyl or, when n=0, Ri and R4 are bonded together and form a -(CH2)r- chain with r being equal to 3, 4, 5 or 6, or, when n=l or 2, Rk and R4 are bonded together and form a -(CH2)r- chain with r being equal to 3, 4, 5 or 6,
- R3 is selected among hydrogen and the groups alkyl, cycloalkyl, aryl and -(CH2)q3-Y3, in which:
■ q3 is 1, 2, 3, 4, 5, 6 and
■ Y3 is an aryl or Ci-ealkoxy group; or R3 and R4 are bonded together and form a -(CH2)r- chain with r being equal to 3, 4, 5 or 6, and
- when R4 is not bonded to Ri, Rk selected from hydrogen and the groups alkyl, cycloalkyl, aryl and in which:
■ q4 is 1, 2, 3
■ Y4 is an aryl
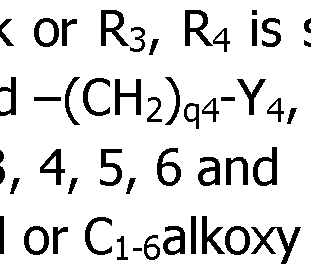 group;
group;
- Ri and R2 are as defined hereinafter: either:
- Ri is chosen among the groups alkyl, cycloalkyl, alkenyl, alkynyl, aryl, said groups being unsubstituted or substituted by one or several substituents selected from -CF3, -CN, alkyl, -Oalkyl, C(O)alkyl, - C(O)Oalkyl, -Salkyl, -Oalkyl -NHC(O)H,
-S(O2)O', -S(O2)Oalkyl, -P(O2)O' and -P(O2)Oalkyl, and
- R2 is Y2-L2-R'2, in which:
■ Y2 is CH2, CHalkyl, C(alkyl)2 , S or 0,
■ L2 is a linker, in particular an alkylidenyl,
-(CH2)mi-Ph-(CH2)m2-, with ml and m2 being independently 0, 1, 2, 3, 4, 5 or 6, or
-(CH2-CH2-O)m3-, with m3 being 1, 2, 3, 4, 5 or 6, and
■ R'2 is a reactive group RG, or:
- Ri is Li-R'i, in which:
■ Li is a linker, in particular an alkylidenyl,
-(CH2)pi-Ph-(CH2)p2-, with pl and p2 being independently 0, 1, 2, 3, 4, 5 or 6, or
-(CH2-CH2-O)p3-, with p3 being 1, 2, 3, 4, 5 or 6, and
■ R'i is a reactive group RG,
- R2 is -CH3, -CH2R"2, -CHalkylR"2, -C(alkylR"2)2, -SR"2 or -OR"2, in which R"2 is chosen among the groups alkyl, cycloalkyl, alkenyl, alkynyl, aryl, said groups being unsubstituted or substituted by one or several substituents selected from the groups -CF3, , -CN, alkyl, -
C(O)alkyl, -C(O)Oalkyl, -Salkyl, -Oalkyl -NHC(O)H, -S(O2)O’ , -S(O2)Oalkyl, -P(O2)O’ and -P(O2)Oalkyl, including their salts with at least one anion, in particular, chosen among halogenated anions, typically CP, Br' and r ; trifluoroacetate, acetate, formate ; sulfonates, such as methylsulfonate, trifluoromethylsulfonate and tosylate ; sulfates, such as methylsulfate ; phosphate, pyrophosphate and triphosphate or with at least one cation, in particular, chosen among Li+, Na+, K+, Cs+and triethyl ammonium.
The compounds FC behave as fluorophore (also called dye), as they emit fluorescence under solicitation. This fluorescence is very low when the compound FC is in its free state. The resulting moiety in the detection probe, coming from the covalent conjugation of compound FC on the oligonucleotide of the detection probe, is called FC'. FC' leads to the emission of a fluorescence signal when it is excited by light of a suitable wavelength, under certain conditions. As a result, in the detection probe PR of the invention, the compound FC that is covalently attached to the oligonucleotide (ie FC' moiety) confers to the detection probe a detectable fluorescence. This fluorescence is highly increased when the detection probe is hybridized to its target nucleic acid and acts as a reporter molecule. When the detection probe is hybridized to its target nucleic acid, this fluorescence is intensified.
The nature of the salt is, of course, dependent on the global charge of the compound of formula (I). This global charge can be neutral or equal to +1, -1 or -2, and will be determined by the substituents that are present on the molecule.
According to specific embodiments of the compounds FC of the invention, Ri, Rj and Rk are hydrogen.
According to specific embodiments of the compounds FC of the invention, there is no substituent A on Z ring.
In particular, the invention relates to the compounds FC having the formula
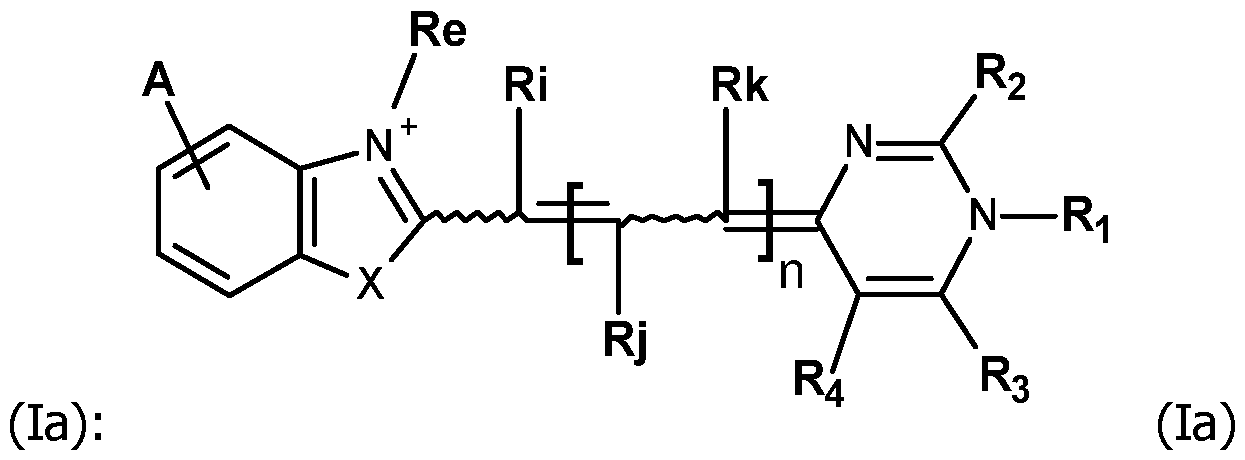 and in particular the formula (lb):
and in particular the formula (lb):

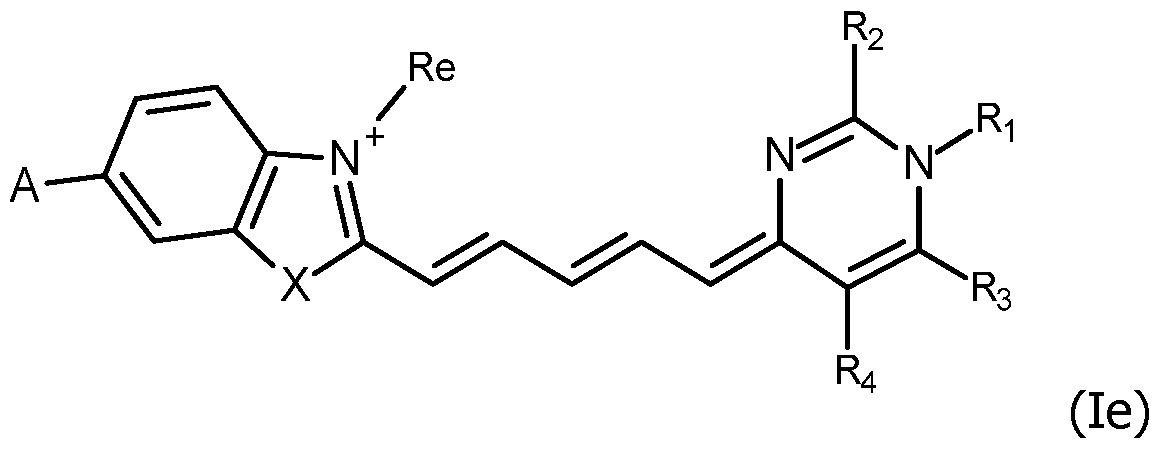
According to specific embodiments of the compounds FC of the invention, whatever they correspond to formula (I), (la), (lb), (Ic), (Id), or (le), they include one of the following features or any combination of the following features, and advantageously, when they do not exclude each other, all the following features:
- Re is Ci-6alkyl, in particular methyl;
- R3 is Ci-ealkyl, in particular methyl;
- R4 is a hydrogen atom;
- A is a hydrogen atom or -NHC(O)CH3;
- Ri is Ci-6alkyl, in particular methyl or Ri is aryl, in particular a phenyl and -R2 is -Y2-l_2-R'2, in which Y2, L2 and R'2 are as defined for formula (I); More specifically, Y2 is CH2 or S and/or L2 is an alkylidenyl, in particular - (CH2)3-, -(CH2)4- or -(CH2)5-. In particular, Y2-L2 is -S-(CH2)5- or -CH2- (CH2)3- ; As particularly suitable RG group, R'2 is an activated ester, and in particular a succinimidyl ester;
- R2 is -SCi-salkyl, in particular -SCH3, or Ci-ealkyl, in particular methyl, and -Ri is -Li-R'i, in which Li and R'i are as defined for formula (I); More specifically, Li is a phenyl substituted in meta or para by R'i as defined for formula (I); As particularly suitable RG group, R'i is an activated ester, and in particular a succinimidyl ester;
- X is oxygen or sulfur.
As way of examples, the compounds FC according to the invention are selected among:
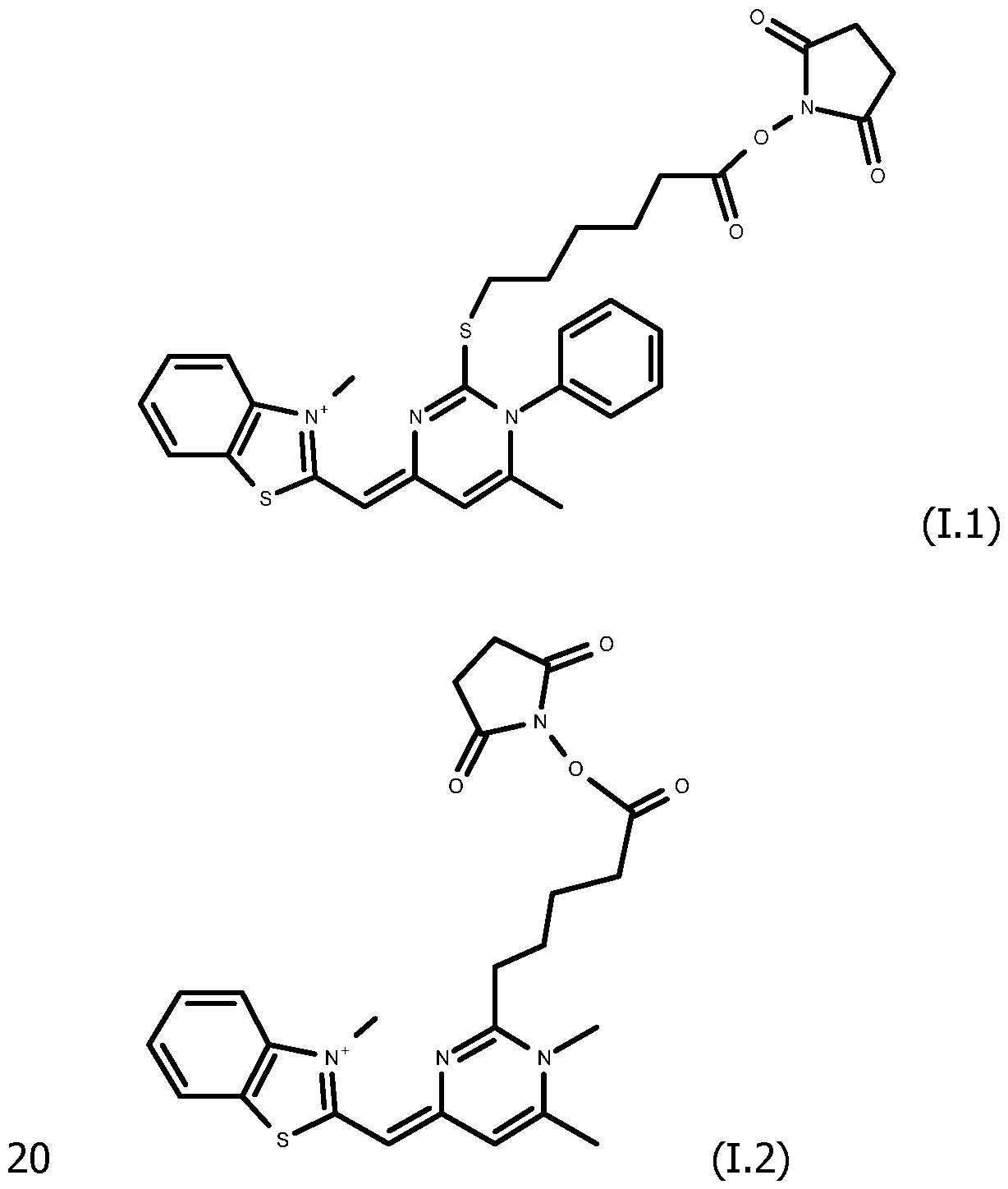
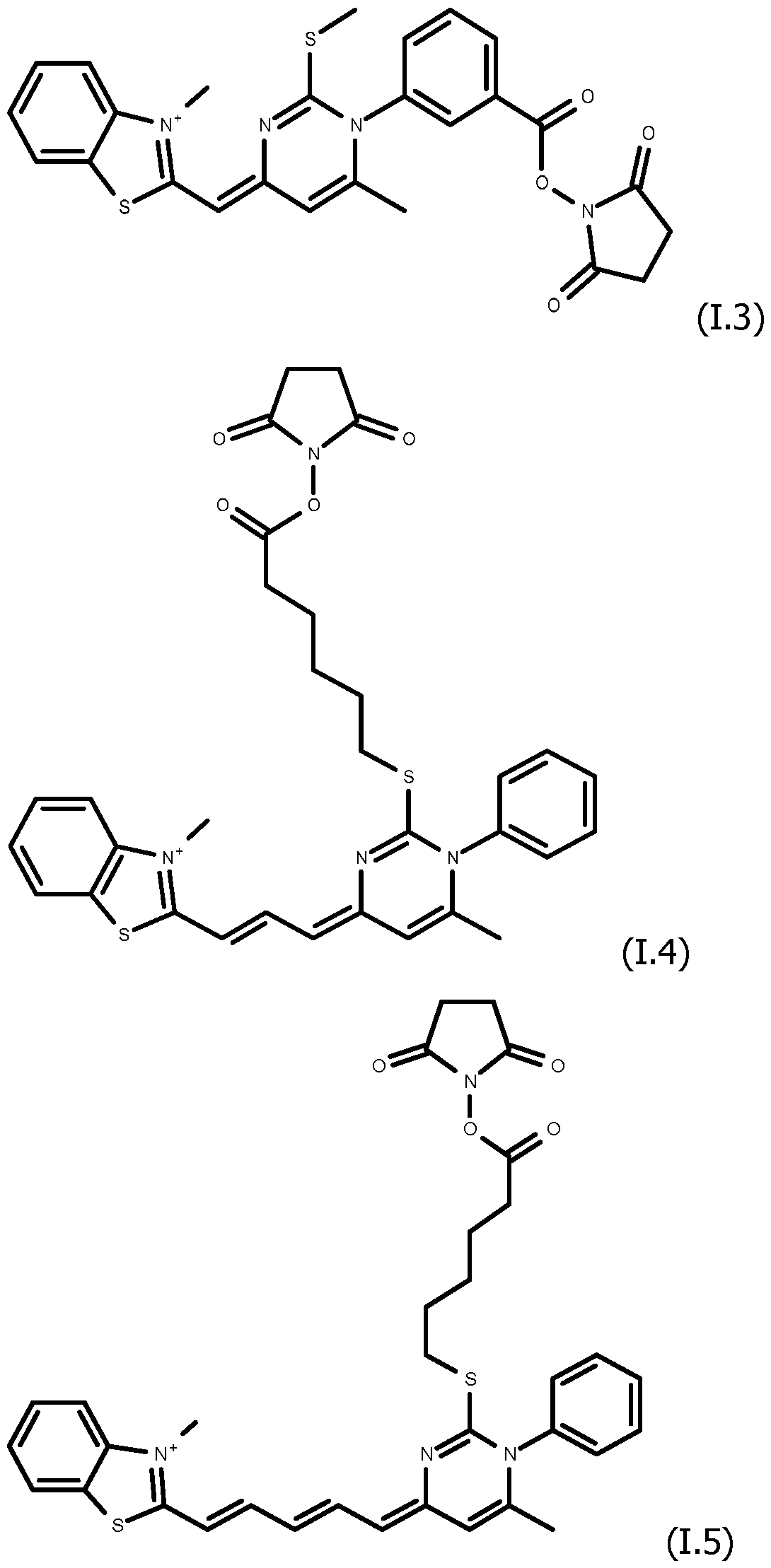
 including their salts with one anion, in particular, chosen among halogenated anions, typically CP, Br' and r ; trifluoroacetate, acetate, formate ; sulfonates, such as methylsulfonate, trifluoromethylsulfonate and tosylate ; sulfates, such as methylsulfate ; phosphate, pyrophosphate and triphosphate, and typically their trifluoroacetate salts.
including their salts with one anion, in particular, chosen among halogenated anions, typically CP, Br' and r ; trifluoroacetate, acetate, formate ; sulfonates, such as methylsulfonate, trifluoromethylsulfonate and tosylate ; sulfates, such as methylsulfate ; phosphate, pyrophosphate and triphosphate, and typically their trifluoroacetate salts.
Another aspect of the invention concerns detection probes PR labelled with at least one molecule of a compound FC according to the invention, comprising an oligonucleotide covalently coupled on the reactive group RG of said compound FC.
More specifically, in the detection probes PR of the invention, the compound FC is coupled to said oligonucleotide by attachment to at least one nucleotide of said oligonucleotide. That involves that the attachment is not obtained by replacing a base of the oligonucleotide.
In particular, at least one region of the oligonucleotide selected from the 5' end, the 3' end, and the internal region of the oligonucleotide is bonded to a molecule of the compound FC.
According to specific embodiments, in the detection probes PR of the invention, the compound FC is coupled to said oligonucleotide at its 5' or 3' end, by attachment to the phosphate group of the corresponding nucleotide. The attachment is in general obtained via a linker. These kinds of coupling are well known for the skilled person.
According to specific embodiments, in the detection probes PR of the invention, the compound FC is coupled to said oligonucleotide in its internal region. The attachment is in general obtained via a linker. These kinds of coupling are also well known for the skilled person.
According to specific embodiments, in the detection probes PR of the invention, the oligonucleotide is bonded to at least two molecules of the same compound FC, with one being attached at its 5' end and another one being attached at its 3' end.
According to specific embodiments, in the detection probes PR of the
invention, the oligonucleotide is bonded to at least three molecules of the same compound FC (in particular 3, 4 or 5 molecules), with at least one molecule (typically 1, 2 or 3 and in particular one), of the said compound FC attached in the internal region of the oligonucleotide.
According to specific embodiments, in the detection probes PR of the invention, the oligonucleotide is bonded to the compound FC in the internal region of the oligonucleotide by attachment to a base A, T or G of the oligonucleotide.
In general, in the detection probes PR of the invention, the oligonucleotide is composed of 10 to 60 nucleotides, preferentially of 10 to 30 nucleotides, and more preferably of 12 to 25 nucleotides.
In the detection probes PR of the invention, typically, the oligonucleotide may be a single-strand nucleic acid comprising or composed of a complementary sequence to a DNA gene sequence of a target microorganism or a complementary sequence to a DNA gene sequence of a subject.
Another aspect of the invention relates to a method for the preparation of a detection probe PR of the invention, comprising i) the synthesis of the oligonucleotide bearing at least one functional group Q suitable to react with the reactive group RG that is present in Ri or R2 of the compound FC, eventually in a protected form, preferentially on a solid phase and ii) the covalent coupling of the compound FC, by reaction of the reactive group RG and the functional group Q.
Several embodiments of the probes of the invention and different ways of implementing the probes of the invention are presented on Figure 1. In specific embodiments, the detection probes of the invention are useful in detection methods using the FRET technique, and more specifically the detection probes of the invention can be a member of a pair of adjacent hybridization probes using the FRET technique. According to Theodor Forster's theory, FRET is defined as a non-radiative energy transfer (without light emission) resulting from a dipole - dipole interaction between two molecules (energy donor and acceptor). This physical phenomenon requires energetic compatibility between these two molecules. This means that the emission spectrum of the donor must overlap, at least partially, the excitation spectrum of the acceptor. Therefore, in such cases, a pair of probes is used: a donor dye is covalently conjugated to the oligonucleotide of one of the probe (designated donor probe) and an acceptor dye is covalently conjugated to the oligonucleotide of other of the probe (designated acceptor probe). The acceptor probe and the donor probe hybridize to two close portions of the target nucleic acid so that, after hybridization, the donor dye and the acceptor dye are close to each other to enable the fluorescent resonance energy transfer (FRET). The detection probes PR of the invention
can be used either as donor probe and/or as acceptor probe where both the donor probe and the acceptor probe are probes PR of the invention. Several embodiments using the probes of the invention in FRET techniques are illustrated by Figure 1.
By selecting the value of n, it is possible to adjust the properties of fluorescence obtained after the hybridization of a probe PR of the invention, with a target nucleic acid. For instance, by comparison with compounds wherein n=0, compounds wherein n=l or 2 have higher excitation and emission wavelengths. Typically:
- for compounds FC' wherein n=0, the maximum excitation wavelength is in the range 410-490 nm and the maximum emission wavelength is in the range 460-540 nm. Those compounds will be used as donor only in probes bearing one dye or several dyes.
- for compounds FC' wherein n=l, the maximum excitation wavelength is in the range 510-580 nm and the maximum emission wavelength is in the range 560-620 nm. Those compounds will be used as donor or acceptor in probes bearing one dye or several dyes.
- for compounds FC' wherein n=2, the maximum excitation wavelength is in the range 620-710 nm and the maximum emission wavelength is in the range 660-740 nm. Those compounds will be used as acceptor only in probes bearing one dye or several dyes.
When a donor is used in combination with an acceptor or a quencher, the couple donor/acceptor or donor/quencher will be chosen to ensure that the emission spectrum of the donor overlaps, at least partially, with the excitation spectrum of the acceptor or of the quencher.
As way of examples, the excitation and emission wavelengths (in nm) of compounds FC' in the probes of the invention are presented in Table 1 hereinafter, for examples of compounds FC (dyes 1.1 to 1.6).
Table 1
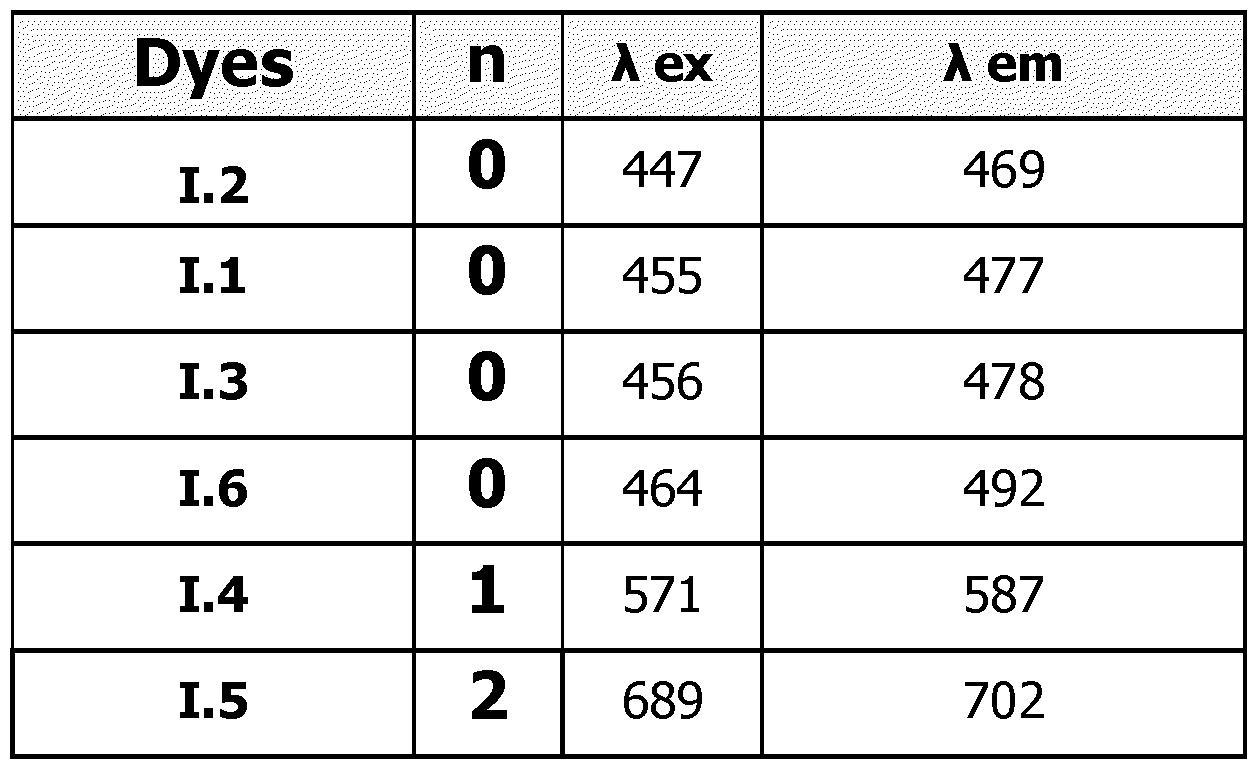 As shown hereinafter in the experimental part, with the use of a probe PR of the invention, either as donor probe or acceptor probe, or as both donor probe and acceptor probe, the transfer of fluorescence is higher, in comparison with the use of conventional donor probes and/or acceptor probes of the same emission/ excitation wavelength ranges. It is believed that this beneficial effect is due to the behavior of moiety FC' that is present in the probe PR of the invention as minor groove binder in the double strand formed after hybridization of the probe PR on its target nucleic acid.
As shown hereinafter in the experimental part, with the use of a probe PR of the invention, either as donor probe or acceptor probe, or as both donor probe and acceptor probe, the transfer of fluorescence is higher, in comparison with the use of conventional donor probes and/or acceptor probes of the same emission/ excitation wavelength ranges. It is believed that this beneficial effect is due to the behavior of moiety FC' that is present in the probe PR of the invention as minor groove binder in the double strand formed after hybridization of the probe PR on its target nucleic acid.
Therefore, the invention also relates to the use of a detection probe PR according to the invention as a member of a pair of adjacent hybridization probes including a donor probe and an acceptor probe in a detection method of a target nucleic acid by FRET technique. The detection probes PR of the invention that are particularly useful in those detection methods (pair of adjacent hybridization probes) using the FRET technique are those in which the 5' end and/or the 3' end is bonded to a molecule of a compound FC.
The invention also concerns a method for detecting a target nucleic acid in a sample, comprising incubating the sample with a detection probe PR as described in the invention, in conditions suitable to obtain the hybridization of the target nucleic acid with the oligonucleotide of the detection probe PR, when the target nucleic acid is present in the sample, and detecting the hybridization obtained.
Most of the time, the method for detecting a target nucleic acid includes an amplification step of the target nucleic acid.
The hybridization of the target nucleic acid with the oligonucleotide of the detection probe PR may be detected during and/or after amplification of the target nucleic acid.
In particular, the amplification step is carried out by PCR, and in particular by real time PCR. In that case, the method for detecting a target nucleic acid comprises the steps of:
- mixing a detection probe PR according to any the invention with a sample comprising a target nucleic acid, a polymerase, and a pair of primers suitable to amplify a portion of the target nucleic acid and generate at least an amplicon, leading to a PCR mixture,
- amplifying the target nucleic acid from the PCR mixture and generating at least an amplicon, and
- monitoring the fluorescence from the compound FC, in its bonded form in the detection probe PR of the invention (corresponding to FC' moiety), during or subsequent to the amplifying step.
In that case, the method may further comprise detecting the presence of the amplicon from the monitored fluorescence.
The monitoring step may occur subsequent to amplification and may include
generating a melting curve. In a specific embodiment, the melting curve is used to identify the genotype of the target nucleic acid, to detect or identify at least one mutation, polymorphism, preferentially single nucleotide polymorphism, and/or epigenetic variation.
The invention also concerns a method of performing a multiplex assay on a sample, comprising incubating the sample with a plurality of detection probes PR according to the invention, said detection probes PR being labelled with different compounds FC. In such a case, it is preferable to use several compounds FC, each corresponding to a different value of n. Therefore, the excitation and emission wavelengths of those compounds FC will differ and their fluorescence will be detected at different emission wavelengths.
Another aspect of the invention is related to test kits comprising at least one detection probe PR of the invention and at least one additional reagent. In particular, the test kit may comprise an amplification enzyme and/or primer sequence for the amplification of a target nucleic acid sequence.
DEFINITION
The following definitions provide a better understanding of the invention.
In the compounds FC according to the invention, the meaning of the substituents is usual, if it is not specified otherwise.
The term "alkyl" as used in the invention refers to a monovalent saturated hydrocarbon moiety, typically comprising from 1 to about 12 carbon atoms, in particular 1 to 6 carbon atoms. An alkyl group may be linear or branched and illustratively includes methyl (Me), ethyl, propyl, butyl, dodecyl, 4- ethylpentyl, and the like. Ci-ealkyl refers to alkyl comprising 1, 2, 3, 4, 5 or 6 carbon atoms and typically to methyl.
The term "alkenyl", as used herein, refers to monovalent unsaturated hydrocarbon moieties, typically comprising from 1 to about 12 carbon atoms, in particular 1 to 6 carbon atoms, which contain at least one carbon-carbon double bond, wherein each double bond can have E- or Z-configuration. The term "alkynyl", as used herein, refers to monovalent unsaturated hydrocarbon moieties, typically comprising from 1 to about 12 carbon atoms, in particular 1 to 6 carbon atoms, which contain at least one carbon-carbon triple bond. The alkenyl and alkynyl groups can be linear or branched. Double bonds and triple bonds in alkenyl groups and alkynyl groups respectively can be present in any positions. Examples of alkenyl and alkynyl are ethenyl, prop-l-enyl, prop-2-enyl, but-2-enyl, 2-methylprop- 2-enyl, 3-methylbut-2-enyl, hex-3-enyl, hex-4-enyl, prop-2-ynyl, but-2-ynyl, but-3-ynyl, hex-4-ynyl or hex-5-ynyl.
The term "alkylidenyl" as used in the invention refers to a bivalent saturated hydrocarbon moiety comprising from 1 to about 20 carbon atoms, typically 1
to 12 carbon atoms or 1 to 6 carbon atoms. Typical examples of alkylidenyl moiety are -(CH2)3-, -(CH2)4-, -(CH2)5- and -(CH2)6-.
The term "alkoxy" as used in the invention refers to -O-alkyl.
The term cycloalkyl refers to cyclic alkyl, alkenyl and alkynyl, excluding the aromatic groups. In particular, a cycloalkyl is a cyclic alkyl, typically a cyclopropyl or cyclohexyl.
The term heteroalkyl refers to a saturated cycloalkyl where the cyclic hydrocarbon chain is interrupted by one or several heteroatoms, such as tetrahydrofuranyl, dioxanyl, piperidinyl and pyrerazinyl.
The term "aryl" or aromatic cyclic ring (that can be mono or polycyclic and which is fused in the definition of Z) as used in the invention refers to a cyclic aromatic hydrocarbonated moiety, illustratively including but not limited to phenyl (Ph) and naphthyl. Phenyl is the illustrative aryl group and in Z is named as fused-benzo. Nitrogen containing aromatic ring used in the definition of Z refers to pyrrolo, pyrazolo, isoxazolyl, isothiazolyl, oxazolyl, thiazolyl, pyridinyl, pyridazinyl, pyrimidinyl, pyrazinyl, quinolinyl, isoquinolinyl, quinoxalinyl, quinazalinyl, and the like. In particular, according to the definition of Z, typically when fused with the other cycle ring presented in formula (I), Z can form an optionally substituted benzoxazolium or benzothiazolium ring, or an optionally substituted naphthoxazolium or naphthothiazolium ring.
The compounds FC of the invention are cyanine derivatives, having a pyrimidinium core structure, wherein, in particular, X is oxygen or sulfur, and the moiety Z represents an optionally substituted fused benzo, forming an optionally substituted benzoxazolium or benzothiazolium ring, or the moiety Z represents an optionally substituted fused naphtho, forming an optionally substituted naphthoxazolium or naphthothiazolium ring.
It is appreciated that the compounds FC of formula (I), (la), (lb), (Ic), (Id) or (le) and their salts described herein may contain one or several chiral centers. In those cases, all stereoisomers are understood to be included in the description of these compounds FC, unless otherwise indicated. Such stereoisomers include pure enantiomers, racemic mixtures, mixtures of enantiomers in any relative amount, pure diastereoisomers and mixtures of diastereoisomers containing any relative amount of one or more stereoisomeric configurations.
It is also appreciated that the compounds FC corresponding to compounds of formula (I), (la), (lb), (Ic), (Id) or (le) and their salts herein may contain geometric centers. In those cases, all geometric isomers are understood to be included in the description of the compounds FC corresponding to formula (I), (la), (lb), (Ic), (Id) or (le) and their salts, unless otherwise indicated. Such geometric isomers include cis, trans, E and Z isomers, either in pure
form or in various mixtures of geometric configurations. It is also understood that depending upon the nature of the double bond contained in the compounds FC corresponding to formula (I), (la), (lb), (Ic) , (Id) or (le) and their salts, such double bond isomers may interconvert between cis and trans, or between E and Z configurations depending upon the conditions, such as solvent composition, solvent polarity, ionic strength, and the like. So, the two forms cis/trans or E/Z are most of the time, in equilibrium. In the presented formula (Ic), (Id), (le), 1.1 to 1.6, even if the double bond is presented in a specific geometric form, all possible isomeric forms are in equilibrium.
Most of the time, the compounds FC according to the invention are in the form of a salt: the compounds of formula (I), (la), (lb), (Ic), (Id), (le) and 1.1 to 1.6 are positively charged due to the N+(Re). They may include one or several negative charges, but they do not include other positive charges. The salts of the compounds of formula (I), (la), (lb), (Ic), (Id) or (le) include an anion or a number of cations (typically which are identical) corresponding to the global charge of the compound of formula (I), (la), (lb), (Ic), (Id) or (le). When the compounds of formula (I), (la), (lb), (Ic), (Id) or (le) (this is the case of 1.1 to 1.6) are positively charged, several resonance structures of these compounds may exist. Typically, a positive charge may be formally localized on the nitrogen atom N+(Re) as depicted in Formula (I), (la), (lb), (Ic), (Id), (le) and 1.1 to 1.6, or alternatively, the charge may be localized on the pyrimidinyl group.
In the compound FC and in the detection probe PR of the invention, many linkers can be constructed to join the concerned groups. Advantageously, the linkers Li and L2 in the compounds FC do not bear any positive charge, and in particular are not charged at all. Li and L2 are identical or different. Li and L2 may be a single bond or an alkylidenyl, eventually interrupted by one or several 0, S, phenyl (Ph), -NRf-, -N(O)NRf, with Rf being H, Ci-salkyl, heteroalkyl, aryl or heteroaryl. Examples of linkers Li and L2 are -(CH2)pi-Ph- (CH2)p2-, with for instance pl and p2 being independently 0, 1, 2, 3, 4, 5 or 6, -(CH2)p3-, -(CH3CH)p3-, -(CH2CH2O)p3-, -(CH3CHCH2O)p3- with for instance p3 being 1, 2, 3, 4, 5 or 6. According to specific embodiments, when present Li and/or L2 are/is short and are/is, in particular, a chain (in particular an unsubstituted chain) defined by from 2 to 6 successive atoms forming the chain.
The term "nucleic acid" or "nucleotide sequence" or "polynucleotide" means a chain of at least two deoxyribonucleotides or ribonucleotides (nucleotide units) optionally comprising at least one modified nucleotide, for example at least one nucleotide having a modified nucleic acid base, such as inosine, methyl-5-deoxycytidine, dimethylamino-5-deoxyuridine, deoxyuridine,
diamino-2,6-purine, bromo-5 -deoxyuridine or any other modified base permitting hybridization. Such polynucleotides can also be modified at the level of the internucleotide bond for example phosphorothioates, H- phosphonates, alkyl phosphonates, at the level of the backbone for example alpha-oligonucleotides (as described in FR 2 607 507) or PNAs (for peptide nucleic acid, as described by M. Egholm et al., in J. Am. Chem. Soc., 114, 1895-1897, 1992) or 2'-O-alkyl ribose and LNAs (for locked nucleic acid, as described by B.W. Sun et a!., in Biochemistry, 4160-4169, 43, 2004). Other types of modifications are also described in US 2018/0073056. The nucleic acid can be natural or synthetic, an oligonucleotide or a longer polynucleotide, a nucleic acid fragment, a ribosomal RNA, a messenger RNA, a transfer RNA, a nucleic acid obtained by an amplification technique, and in particular an enzymatic amplification technique.
In the sense of the present invention, "target sequence" or "target nucleic acid" intends to mean any nucleic acid whose presence is to be detected or measured or whose function, interactions or properties are to be studied.
A target nucleic acid has a nucleotidic sequence in which at least one part of the chain of nucleotide units is specific and complementary to the nucleotide sequence of the oligonucleotide of the detection probe PR used.
The target nucleic acid can be natural or obtained from a reaction of amplification in vitro. When the target sequence results from a reaction of enzymatic amplification in vitro, the sequences produced by this amplification are called "amplicons". The target nucleic acid generally has a length that exceeds the length of the oligonucleotide of the detection probe PR. This is classical in the technical field of detection probes.
The term "complementarity" corresponds to the degree to which the sequences of two single-stranded nucleic acids can form a double-stranded complex according to Watson and Crick's hybridizing or pairing rule, where the base A (Adenine) hybridizes (or pairs) with the base T (Thymine) or U (Uracil) whereas the base C (Cytosine) hybridizes (or pairs) with the base G (Guanine). Two complementary sequences correspond to two sequences with 100% complementarity (100% complementary).
The term "oligonucleotide" refers to a short nucleic acid sequence, typically of 10 to 100 nucleotide units, notably from 10 to 60 nucleotide units and in particular from 10 to 50 nucleotide units and even more particularly from 12 to 30 nucleotide units.
The oligonucleotide of the detection probe PR is a single strand nucleic acid, and in particular a single-strand DNA. The oligonucleotide may be a Nucleic Acid Analog (NAAs) that differs from natural nucleic acid but can still recognize them through specific base pairing. NNAs are usually derived from the naturally occurring nucleosides A, C, G and T modified e.g. in the base
portion and/or the sugar portion and/or the triphosphate link, for instance, propynyl dU (dT-analogue), 2-amino dA (dA analogue), peptide nucleic acid (PNA), locked nucleic acid (LNA), 2'-O-methyl RNA, phosphoramidate DNA, phosphorothioate DNA, methyl phosphonate DNA, phosphotriester DNA or DNA base analogues may be employed to form more stable interactions with target sequences. Nevertheless, advantageously, the oligonucleotide of the detection probe PR does not include PNAs and LNAs. Nucleotide units of the oligonucleotide of the detection probe PR are usually naturally occurring nucleosides A, C, G and T or are derived from the naturally occurring nucleosides A, C, G and T to allow the covalent linkage of the compound FC. The oligonucleotide of the detection probe PR has a full sequence, or has at least a portion of sequence, chosen to specifically hybridize with the target nucleic acid sequence, called hybridization sequence or target-specific sequence. The hybridization sequence has preferentially a length of 10 to 30, and in particular of 12 to 25, nucleotide units. The oligonucleotide of the detection probe PR may also include additional portion(s) located at one end or both ends of the hybridization sequence. Each of these additional portions is, for instance, 1 to 10 and more particularly 4 to 8 nucleotide units long. The hybridization sequence of the oligonucleotide of the detection probe PR is in general 100% complementary to the portion of the target nucleic acid sequence to which it is configured to hybridize, but not necessarily. There could be one mismatch (absence of complementarity between two nucleotide units due to base mispair) or few mismatches between the target nucleic acid and the hybridization sequence of the oligonucleotide of the detection probe PR that do not prevent the obtaining of a suitable specific hybridization. The position and the number of acceptable mismatches depend on the type of probes and will be adjusted by the skilled artisan in the art. In some cases, the mismatch(es) will preferentially be out of the 3' and the 5' end zones of the oligonucleotide of the detection probe PR. For other detection probes, in particular for molecular beacon type detection probes, the mismatch(es) will preferably be out of the internal zone of the oligonucleotide of the detection probe PR where the annealing/pairing with the target sequence occurs. As regard to the acceptable number of mismatches, it will be adapted by the skilled person, considering, for instance, the following parameters: the sequence itself, the Tm of the detection probe, the hybridization temperature, the salts concentration during the hybridization and amplification. The hybridization sequence of the oligonucleotide of the detection probe PR may be not 100% complementary to the portion of the target nucleic acid sequence to which it hybridizes. In general, the hybridization sequence of the oligonucleotide of the detection probe PR is at least 90% complementary to the portion of the target nucleic
acid sequence to which it hybridizes. This will correspond to at most 5 mismatches between the hybridization sequence of the oligonucleotide of the detection probe PR and the portion of the target nucleic acid sequence to which it hybridizes, and in particular, there is(are) 1, 2, 3, 4 mismatches between the two sequences, depending on the length of the hybridization sequence of the oligonucleotide of the detection probe PR. In particular, the mismatch(es) correspond to a mutation in a target nucleic or to a specific genotype, in particular SNPs. According to specific embodiments, there is only one mismatch between the two sequences, depending on the length of the hybridization sequence of the oligonucleotide of the detection probe PR. "End" of an oligonucleotide means the starting point and the end point of synthesis of an oligonucleotide generally defined by the number carried by the free hydroxyls of the first or the last nucleoside, i.e. 3' or 5'. It is understood that with an appropriate choice of elongation units (phosphoramidites of alpha or beta nucleosides, inverted or not), an oligonucleotide can be synthesized in the 3' to 5' direction or in the opposite direction, or the direction of elongation can even alternate during synthesis. This leads to oligonucleotides bearing 3'-5', 5'-3', 3'-3' or 5'-5' ends. According to the invention, the oligonucleotide has mainly 3'-5' ends.
The internal region (also called middle region or zone) of an oligonucleotide means a region which is both at least one nucleotide away from the 5' end and at least one nucleotide away from the 3' end.
According to specific embodiments, the oligonucleotide of the detection probe PR may have a single-stranded conformation, meaning that it does not include a region that can hybridize with another region of the oligonucleotide. According to other embodiments, the oligonucleotide of the detection probe PR may have two regions which are complementary and can hybridize together and form a secondary structure (called self- complementary). But, advantageously, the oligonucleotide of the detection probe PR has a single-stranded conformation.
More generally, the oligonucleotide of a detection probe PR may include a target-specific sequence (i.e. hybridization sequence) that specifically hybridize with at least a portion of the target nucleic acid and non-targetspecific sequence(s). Such non-target-specific sequences can include sequences which will confer a desired secondary or tertiary structure, such as a hairpin structure, as described in US 5,118,801, US 5,312,728, US 6,835,542, and US 6,849,412. More specifically, in that case, the oligonucleotide includes a central region which is complementary to the target nucleic acid and two extreme regions which are complementary to each other. When hybridized, these two extreme regions confer to the oligonucleotide a hairpin conformation, also called secondary structure, like
in beacon probes known in the prior art. Such detection probes PR including an oligonucleotide that forms a secondary or tertiary structure may be useful, for instance, when a quencher is present at the 5' or 3' end of the detection probe PR and a compound FC of the invention at the other end, 3' or 5' respectively. When this detection probe PR hybridizes to a target sequence, it loses its configuration, the 5' and 3' ends move further apart and the quencher and the compound FC' are separated from one another. The fluorescence emitted then reflects the hybridization of the detection probe on the target, which is detected during its amplification and optionally quantified. Such detection probes PR including an oligonucleotide that forms a secondary or tertiary structure may also be useful, for instance, when two molecules of a compound FC of the invention are linked at both the 3' and 5' ends of the oligonucleotide.
FC and FC' are "fluorescent dye" that means that they emit electromagnetic radiations of longer wavelength by a fluorescence mechanism upon irradiation by a source of electromagnetic radiation, including but not limited to a lamp, a photodiode or a laser.
A quencher is a molecule that interferes with, and in particular is able to quench the detectable signal from a reporter moiety, typically the fluorescence of compound FC. A quencher can be selected from non- fluorescent aromatic molecules, to avoid parasitic emissions. For instance, said quencher is a Dabcyl or a "Black Hole Quencher™" (BHQ), examples of non-fluorescent aromatic molecules that prevent the emission of fluorescence when they are physically near a fluorophore. Any quencher molecule known in the art or easily designed by the skilled person can be used. Typical examples are methyl red, Eclipse® Quenchers (EDQ, MGB Eclipse...), Iowa Black® Dark Quenchers (IBRQ, IBFQ...), Black Berry™ Quencher (BBQ).
A "donor" as defined herein is a dye that is part of a FRET couple in which the dye transfers energy to another dye by a nonradiative process. Therefore, in general, the fluorescence of the dye decreases when it is part of a FRET couple. FRET is described in detail in Yang et al., 1997, Methods Enzymol. 278:417-44. An "acceptor" as defined herein is a dye that is part of a FRET system in which the dye accepts energy from another dye by a nonradiative process. Therefore, in general, the fluorescence of the dye increases when excited at the wavelength of the corresponding donor of the FRET couple donor/acceptor.
By selecting the value of n, it is possible to adjust the properties of fluorescence obtained after the hybridization of a probe PR of the invention, with a target nucleic acid. Therefore, the features of the quencher or of the acceptor will be adapted by the skilled person, in function of the selected
compound FC attached in the detection probe PR (called FC')- Typically, when a couple donor/acceptor including a compound FC is used, the following guidelines can be followed or adapted by the skilled person:
- for compounds FC' wherein n=0, the maximum excitation wavelength is in the range 410-490 nm and the maximum emission wavelength is in the range 460-560 nm. Those compounds will be used as donor only in probes bearing one or multiple dyes. Suitable acceptors have, in particular, a maximum excitation wavelength in the range 550-710 nm, with an overlap between the range of the emission spectrum of the donor and the range of the excitation spectrum of the acceptor. Examples of acceptors that can be used in that case are ATTO590™, LcRed640®, Cy5.5™ or a compound FC' wherein n=l or 2.
- for compounds FC' wherein n=l, the maximum excitation wavelength is in the range 510-580 nm and the maximum emission wavelength is in the range 560-620 nm. Those compounds will be used as donor or acceptor in probes bearing one or multiple dyes. Suitable acceptors have, in particular, a maximum excitation wavelength in the range 600-710 nm, with an overlap between the range of the emission spectrum of the donor and the range of the excitation spectrum of the acceptor. Examples of acceptors that can be used in that case are LcRed640®, Cy5.5™ or a compound FC' wherein n=2. Suitable donors have, in particular, a maximum emission wavelength in the range 460-540 nm, with an overlap between the range of the emission spectrum of the donor and the range of the excitation spectrum of the acceptor. Examples of donors that can be use in that case are 6FAM or a compound FC' wherein n=0.
- for compounds FC' wherein n=2, the maximum excitation wavelength is in the range 620-710 nm and the maximum emission wavelength is in the range 660-740 nm. Those compounds will be used as acceptor only in probes bearing one or multiple dyes. Suitable donors have, in particular, a maximum emission wavelength in the range 460-620 nm, with an overlap between the range of the emission spectrum of the donor and the range of the excitation spectrum of the acceptor. Examples of donors that can be use in that case are 6FAM or a compound FC' wherein n=0 or 1.
When a donor is used in combination with an acceptor or a quencher, the couple donor/acceptor or donor/quencher will be chosen to ensure that the emission spectrum of the donor overlaps, at least partially, with the excitation spectrum of the acceptor or of the quencher. Those having an ordinary level of skill in the art will understand that when donor and acceptor dyes are compatible, energy transfer can be detected by the appearance of increased fluorescence of the acceptor or by decreased donor fluorescence.
The term "detection probe" refers to probe including an oligonucleotide sequence capable of hybridizing to a target nucleic acid of interest and allows for the detection of the target nucleic acid and, in particular the specific detection of the target nucleic acid. The detection concludes of the presence or absence of a target nucleic acid and can be measured quantitatively or qualitatively.
According to the invention, the expressions "bonded", "covalently attached", "conjugated", "coupled", used to describe the detection probes PR, have the same meaning and mean that there is a covalent link between the oligonucleotide and the compound FC (giving FC' moiety in the probe PR) obtained by the reaction of the reactive group RG. The compound FC includes a reactive group RG for its conjugation to the oligonucleotide of the probe PR, which is functionalized with a suitable functional group (corresponding functional group Xa, X , Xc in Table 2 hereinafter) that is able to react with the reactive group RG. This reactive group is no longer present in the probe PR of the invention and it is transformed in a covalent linkage (resulting covalent linkage Qa, Qb, Qc in Table 2 hereinafter) in the probe PR where the compound FC is in its conjugated form (designated by FC', FC'i or FC'2 later in the specification).
"Hybridization" or "hybridizing" means the process during which, in suitable conditions, two single-stranded nucleotide fragments having, wholly or partly, sequences that are sufficiently complementary, can form a double strand or "duplex" stabilized by hydrogen bonds between nucleic acid bases. The hybridization conditions are determined by the stringency and low salinity of the operating conditions. Hybridization is the more specific when it is carried out under greater stringency. Stringency is notably defined in relation to the base composition of a probe/target duplex, as well as by the degree of mispairing between two nucleic acids. Stringency can also be a function of the reaction variables, such as the concentration and type of ionic species present in the hybridization solution, the nature and concentration of denaturing agents and/or the hybridization temperature. The stringency of the conditions in which a hybridization reaction must be carried out will mainly depend on the hybridization probes used. All these data are well known and the appropriate conditions can be determined by a person skilled in the art. In particular, the conditions of hybridization used are classically adjusted by the skilled person to obtain an optimal specificity.
The term "specific" or "specificity" in reference to the hybridization of a detection probe PR to a target nucleic acid refers to the recognition, contact, and formation of a stable complex (called duplex) between the two entities (at least a portion of the oligonucleotide of the detection probe PR with at least a portion of the target nucleic acid) together with substantially less
recognition, contact, or complex formation of that detection probe with other molecules than the target nucleic acid. The specificity of a detection probe depends on its pairing degree to its target nucleic acid to be detected. This pairing degree follows the complementarity rules according to Watson and Crick where A anneals (or pairs) with T or U, and C anneals (or pairs) with G. As mentioned above, a specific hybridization can be obtained either with 100% complementary of the hybridization sequence of the oligonucleotide of the detection probe PR and the portion of the target nucleic acid sequence to which it is configured to hybridize, or with one mismatch or few mismatches leading to a complementary of the hybridization sequence of the oligonucleotide of the detection probe PR and the portion of the target nucleic acid sequence to which it is configured to hybridize lower than 100%. The term "amplification reaction" refers to any in vitro means for multiplying the copies of a target sequence of nucleic acid. "Amplification" or "amplifying" refers to a step of submitting a mixture comprising a nucleic acid to conditions sufficient to allow for amplification.
The term "sample" as used herein includes a target nucleic acid that can be present in a variety of different forms including, for example, simple or complex mixtures, or in substantially purified forms, most of the time the target nucleic acid being in a liquid. For example, a target nucleic acid can be part of a sample that contains other components or can be the sole or major component of the sample. Therefore, a target nucleic acid can be a component of a whole cell or tissue, a cell or tissue extract, a fractionated lysate thereof or a substantially purified molecule. Additionally, a target nucleic acid can have either a known or unknown sequence or structure. A sample includes a specimen or culture (e.g., microbiological cultures) that includes nucleic acids. The detection can be carried out on biological and environmental samples. A sample may include a specimen of natural origin, a specimen of non-natural origin or of synthetic origin. A sample can have various origins, such as swabs of food, environmental, human, veterinary, cosmetic origins ... Biological samples include blood (whole blood, serum, plasma), umbilical cord blood, chorionic villi, amniotic fluid, cerebrospinal fluid, spinal fluid, lavage fluid (e.g., bronchioalveolar, gastric, peritoneal, ductal, ear, arthroscopic), biopsy sample, urine, feces, sputum, saliva, nasal mucous, prostate fluid, semen, lymphatic fluid, bile, tears, sweat, breast milk, breast fluid, embryonic cells and fetal cells, in particular of human origin. In a preferred embodiment, the biological sample is blood, and more preferably plasma. These biological samples, or more generally the sample taken from one or more entities for analytical purpose, can have undergone a pretreatment, for example a pretreatment with an agent (an anticoagulant agent for instance in the case of blood sample), a mixing, a separation, an
enrichment, a culture, a dilution ... These examples are not limiting.
PREPARATION OF THE COMPOUNDS ACCORDING TO THE INVENTION The compounds FC according to the invention (also called dyes or reactive dyes) can be prepared according to conventional reactions known by the skilled person in organic chemistry. In its free state, the compound FC is weakly fluorescent, but when it is covalently coupled to the oligonucleotide of the detection probe, it gives higher fluorescence intensity corresponding to the FC' moiety and this FC' moiety gives even higher fluorescence intensity when the oligonucleotide of the detection probe PR is hybridized to its target nucleic acid.
By way of example, when n=0, the preparation used for the compounds LI, 1.3 and 1.6 is described hereafter. While there are many ways of preparing compounds FC, one method is shown on Scheme 1 where X, Z, Re, Ri, Ri, R'2, R3, R^ and L2 are as defined for formula (I), n=0 and Y2 is S or 0.
Scheme 1 where n=0, Y2 is 0 or S
 kylation kylating agent Xd-I_2-Rp group Rp, precusor of R'2 aving group, such as an halogen atom
kylation kylating agent Xd-I_2-Rp group Rp, precusor of R'2 aving group, such as an halogen atom
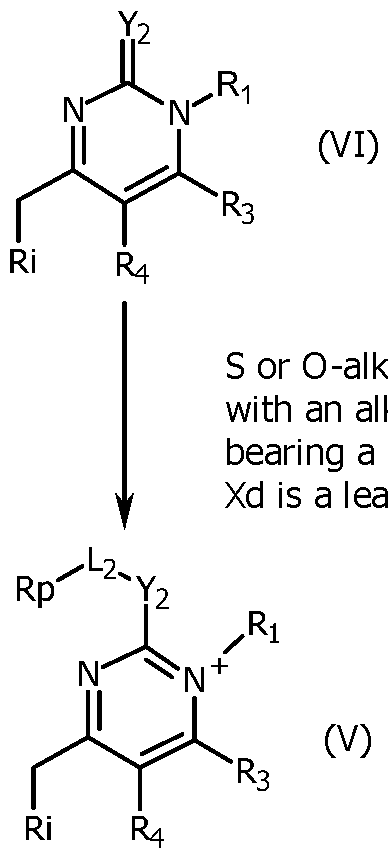
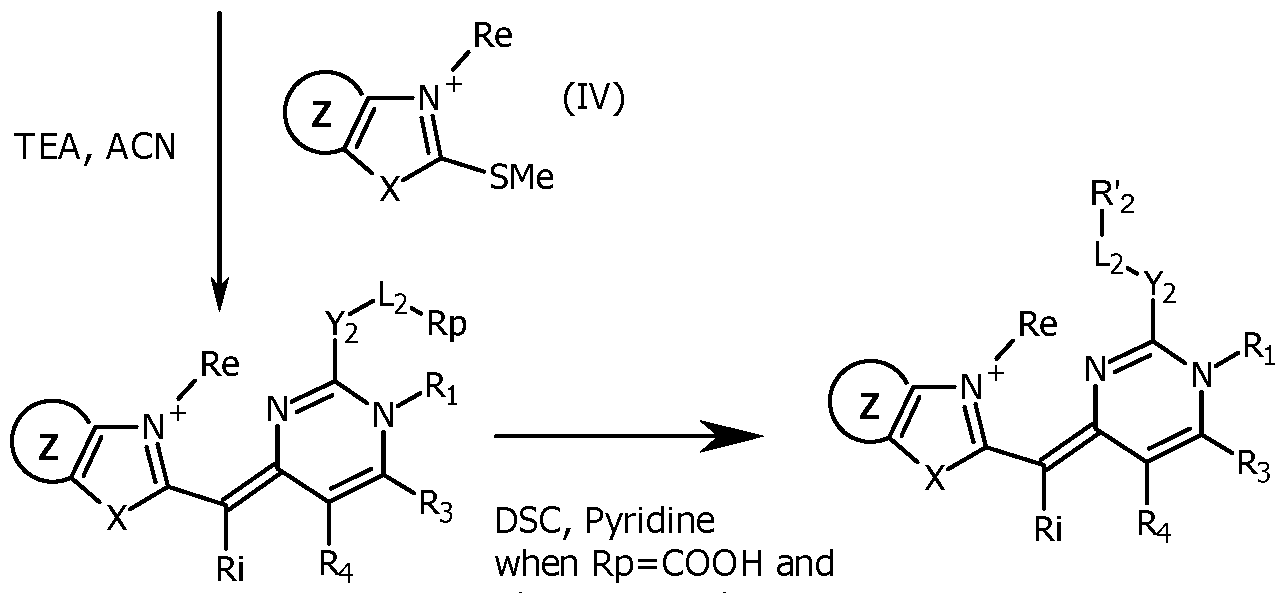
R'2 = activated ester (i) with with n=0
(HI) and R2 being -Y2-L2-R2
Compounds (IV) are commercially available, or may be prepared by conventional methods by alkylation of the precursor at N(3) using alkylating agents such as alkylhalides, alkylsulfates, and the like.
Compounds (VI) are commercially available or are prepared illustratively by condensation in acidic conditions of appropriately substituted 1,3-diones (VII) and conveniently substituted urea or thioureas (VIII). Further, compounds (VI) having a thiol or alkoxy, at C(2) may be modified illustratively by reacting with alkylhalides, alkoxyhalides, or any reactant Xd-L2-Rp (Rp being a precursor of the reactive group R'2) with a good leaving group Xd under
neutral conditions to obtain compound (V). Compounds (III) may be prepared by reacting compounds (V) and compounds (IV) under basic conditions. The unsymmetrical cyanine (III) which may be purified by reverse phase chromatography is transformed to obtain a reactive group R'2 which is for example an activated ester. Various other methods of functionalization are available. For instance, a phosphoramidite R'2 is easily obtainable from an unsymmetrical cyanine (III) in which Rp is a hydroxyl. When R'2 is a succinimidyl (NHS) ester, it can be obtained by activation of an ester Rp using bis NHS carbonate in pyridine. As another example, an azide or alkyne group can, respectively, be introduced as Rp on an unsymmetrical cyanine (III), by reaction of compound (VI) with a compound halogen— (CH2)3— N3/C=CH or halogen— (CH2—CH2—O)3—(CH2)2—N3/C=CH that are commercially available.
Exemplary compounds having this formula can be prepared as herein described, purified by HPLC using TFA water/acetonitrile/TFA as the mobile phase, and isolated as their corresponding TFA salts.
When the compounds (IV) are not commercially available, in particular, when they are substituted on the Z cycle, they can be prepared by conventional technique. Scheme 2 illustrates the case where compound (IV) is a benzothiazolium substituted in para by a group -NHC(O)R' (called (IVa)), with R' as defined for formula (I) and is obtained by acylation of an amino group of the corresponding position of the benzothiazolium. Methyl, propyl, butyl, isopropyl, terbutyl and phenyl are typical examples of R'. In Scheme 2, Xf is a leaving group, for instance a Cl, or a OC(O)R' group.

When Y2 is CH2 or CHalkyl, the compounds FC can be obtained as illustrated in Scheme 3.
Scheme 3 where Y2 is CH2 or CHalkyl
(XII) with Y2 being CH2 or CHalkyl
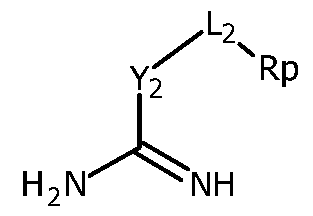
Acid or Base

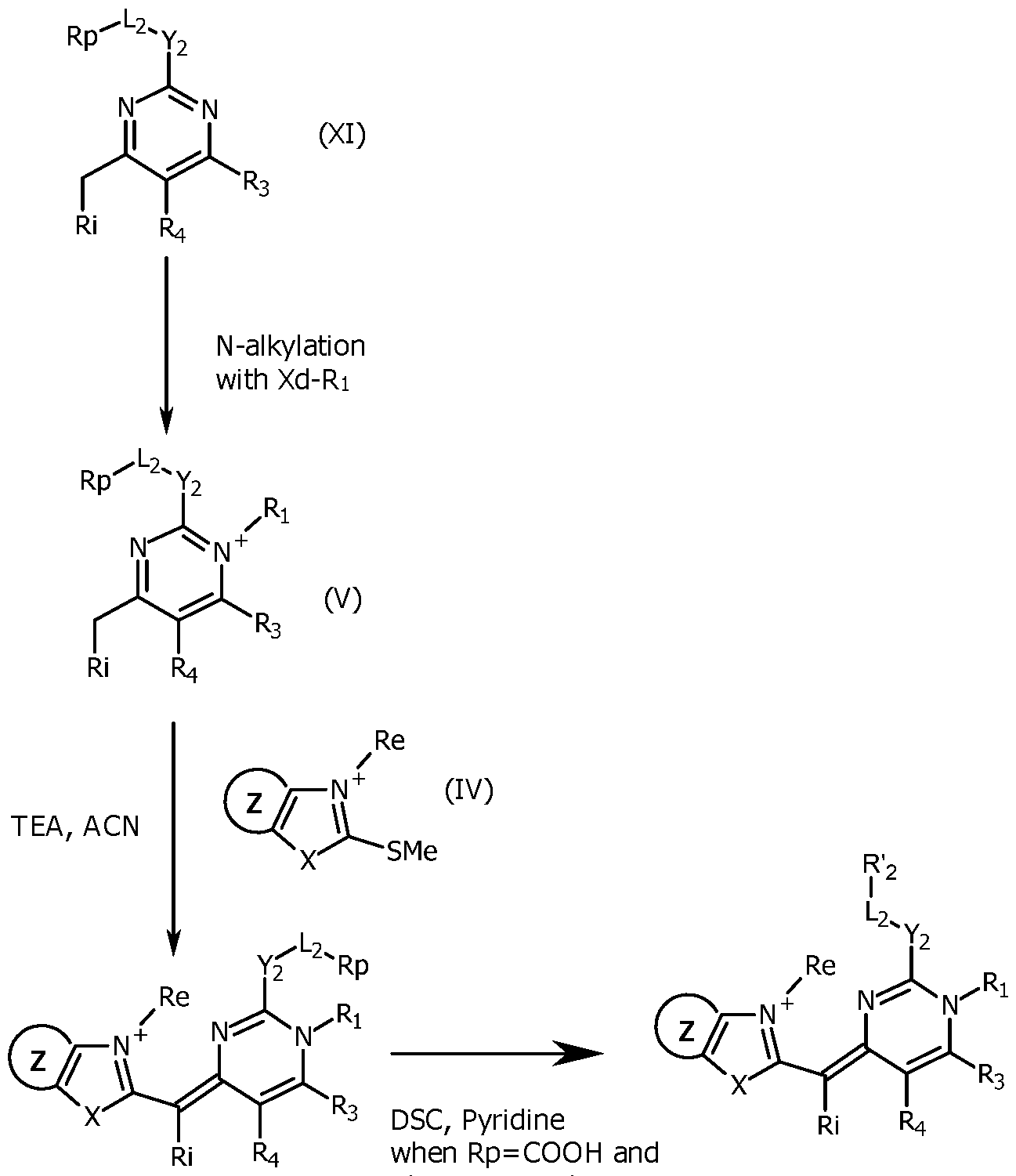
R'2 = activated ester (I) with with n=0
(HI) and R2 being -Y2-L2- '2 In Scheme 3, where X, Z, Re, Ri, Ri, R'2, R3, R4 and L2 are as defined for formula (I), n=0 and Y2 is CH2 or CHalkyl, a conveniently substituted amidine (XII) is reacted with a conveniently substituted dicetone (VII) in strongly alkaline conditions (for instance, in the presence of potassium carbonate), typically in a mixture of water/ethanol for 1 week at room temperature (typically 22°C) to obtain the pyrimidine molecule (XI). The pyrimidine molecule (XI) can be purified by column chromatography or by liquid/liquid
extraction, before further reaction. The pyrimidine molecule (XI) is selectively alkylated at the N1 position by reaction with an excess of an alkylating agent Xd-Ri, with Xd being a leaving group, like Cl, Br or I or a tosyl group and Ri as defined for formula (I), typically in acetonitrile at 50-90°C, in a closed tube for 1-6 days to give the pyrimidinium compound (V). Examples of the alkylating agent Xd-Ri are Mel or EtI.
After evaporation of the remaining alkylating agent, the compound (V) is reacted with a compound (IV) (typically a benzothiazolium derivative), typically in a mixture of acetonitrile, ethanol and triethylamine at room temperature (typically 22°C), for a few minutes to yield the expected unsymmetrical cyanine (III), which may be purified by reverse phase chromatography. The unsymmetrical cyanine (III) is transformed to obtain a reactive group R'2 which is for example an activated ester, in the same way as explained for Scheme 1.
Scheme 4 illustrates a general route to synthesize compounds FC, with different value of n from 1 to 2 and where Ri=Rj=Rk=H, X, Z, Re, Ri, R'2, R3, R4, Y2 and L2 are as defined for formula (I).
Scheme 4

As shown in Scheme 4, conveniently substituted compounds (XVI) (typically
benzothiazoles) are N-functionalized (typically N-alkylated) to obtain the compounds (IVa). In particular, an N-alkylation is carried out at high temperature (for instance from 130 to 160°C) without solvent in the presence of an alkylating agent Re-Xd with Xd being a leaving group, for instance a Cl, Br or I or a tosyl group and Re as defined for formula (I). The leaving group the most suitable for the Re group will be chosen. When Re=Me, MeOTs is particularly suitable. The obtained compounds (IVa) can then react with a masked aldehyde as bis phenyl imine (XV), to yield the corresponding acetylated hemicyanines (XIV) in the presence of acetic anhydride and acetic acid, or only by fusing. The acetylated hemicyanines (XIV) can be purified by reverse phase chromatography using acetonitrile/water/TFA eluents. The acetylated hemicyanine (XIV) can then be reacted in slightly alkaline conditions with pyrimidinium (Vila), to obtain the compound (XIII). Compound (XIII) is transformed to obtain a reactive group R'2 which is for example an activated ester, in the same way as explained for Scheme 1. Various other methods of functionalization, for instance leading to a phosphoramidite, alkyne or azide group, are available.
In Schemes 1 to 4, when X is S, the starting compound is a benzothiazole, and when X=O or when X= CHs , the same route can be followed, except that the starting benzothiazole compound is replaced by respectively a benzoxazole or a dimethyl indole as starting compound.
The compounds FC with other definition of Y2 or with a reactive group at the Ri position (and not at the R2 position) will be prepared by the skilled person, by well-known organic reactions, using appropriately substituted thiourea in Ri for example.
The compounds FC of the invention comprise a reactive group RG, at the Ri or R2 position. A reactive group RG is a chemical moiety capable of reacting with a reaction partner (also called corresponding functional group) on a substrate molecule to form a covalent bond. A compound of the invention can be used to label a wide variety of molecules or substrates that contain a suitable reaction partner or are derivatized to contain a suitable reaction partner. So the reaction partner also comprises a reactive group, which is complementary to the reactive group of the compound FC of the invention. The reactive group RG and its reaction partner may be an electrophile and a nucleophile, respectively, that can form a covalent bond with or without a coupling agent or catalyst, with or without photoactivation.
In particular, the reactive group RG may be one that will react with an amine, a thiol, a hydroxyl, an aldehyde or an alkyne. The reactive group may be an amine-reactive group, typically an activated ester, such as a succinimidyl ester; a thiol-reactive group, such as a maleimide, a haloacetamide, or a methanethiosulfonate; an aldehyde-reactive group, such
as an amine, an aminooxy, or a hydrazide ; an alkyne-reactive group, such an azide (N3); or an azide-reactive group, such an alkyne (-C=CH). More examples are given in Table 2 hereafter.
In general, to prepare a compound comprising a reactive group RG according to the invention, a precursor reactive group Rp is introduced into an intermediate during the synthesis, followed by conversion of the precursor reactive group Rp into the final reactive group RG at the last step of the synthesis. Various methods of introducing a reactive group RG that can be used to prepare the compounds FC according to the invention have been further described in the prior art, for instance in US 5,863,753 or US 2020/0407780. They can be adapted easily by the person skilled in the art. The introduction of one of these reactive groups on a compound FC can be carried out by techniques known in organic chemistry.
PREPARATION OF THE PROBES PR ACCORDING TO THE INVENTION Compounds FC of the invention comprising a reactive group can easily be covalently conjugated to another substrate molecule (typically an oligonucleotide) comprising a suitable corresponding functional group able to react with the reactive group and to form a covalent attachment. The preparation of the detection probes PR of the invention comprises i) the synthesis of the oligonucleotide bearing at least one functional group Q corresponding to the reactive group RG that is present in Ri or R2 of the compound FC, eventually in a protective form, preferentially on a solid phase and ii) the covalent coupling of the compound FC, by reaction of the reactive group RG and the functional group Q.
For example, a compound FC of the invention comprising an activated ester group, such as a succinimidyl ester, can be conjugated to an oligonucleotide comprising an amine group via an amide bond. The Table 2 hereafter (taken from US 9,682,970) presents several examples of reactive groups RG that can be present on compounds FC, corresponding functional groups that can be present on the substrate molecule (typically the oligonucleotide) and the resulting covalent linkages obtained after the reaction of the reactive group with its corresponding functional group.
Table 2
 Table 2 (continued 1)
Table 2 (continued 1)

*Activated esters, as understood in the art, generally have the formula -CORh, where Rh is a good leaving group, such as succinimidyloxy (-OC6H4O2), sulfosuccinimidyloxy (-OC4HO2-SO3H), or 1-oxybenzotriazolyl (-OC6H4N3); or an aryloxy group or aryloxy substituted one or more times by electron-withdrawing substituent(s), such as nitro, fluoro, chloro, cyano, trifluoromethyl, or combinations thereof, used to form activated aryl esters; or a carboxylic acid activated by a carbodiimide to form an anhydride or mixed anhydride -OCORa or -OCNRaNHRb, where Ra and Rb, which may be the same or different, are independently Ci-C6alkyl, Cx-C6 perfluoroalkyl, or CrC6alkoxy; or cyclohexyl, 3-dimethylaminopropyl, or N- morpholinoethyl.
**Acyl azides can also rearrange to isocyanates.
Most of the time, the suitable functional group will be attached to a nucleotide unit of the oligonucleotide by a linker. The main function of the linker is to allow the attachment of the compound FC, via the functional group, on the oligonucleotide in a way that does not obstruct the hybridization of the oligonucleotide on its target nucleic acid. The linker may be uncomplicated, such as a chemical single bond, but may also be complex containing, for example, a long chain, cycle and/or various groups. The examples show that suitable detection probes PR may be obtained by using
very different linkers, in particular short and/or stiff linkers, and linkers containing bulky groups.
The detection probes PR according to the invention can have various structures and conformation. In particular, the number of attached compounds FC, the position of the attachment, the presence or absence of labeling different from a compound FC (typically a quencher), the structure of the oligonucleotide may vary.
In some embodiments, a detection probe PR according to the invention further includes a labeling that interacts with a compound FC. In particular, this labeling is a quencher. Detection probes PR comprising both a compound FC and a quencher are particularly useful in fluorescence resonance energy transfer (FRET) assays. Specific variations of such detection probes include TaqMan® probes (Roche Molecular Diagnostics) and "molecular beacons" probes (for instance described in S.Tyagi et al., Nature Biotechnol. 16:49-53, 1998; US 5,118,801 and US 5,312,728).
In other embodiments, a detection probe PR according to the invention does not include a labeling that interacts with the compound(s) FC that is(are) attached to the oligonucleotide. Therefore, in some embodiments, a detection probe PR according to the invention does not include a quencher of the fluorescence of the compound(s) FC that is(are) attached to the oligonucleotide, in particular of the fluorescence of the compound(s) FC when the oligonucleotide of the probe is hybridized to the target nucleic acid. In particular, a detection probe PR according to the invention only includes one or several molecules of compound(s) FC as label or dye. When in a detection probe PR according to the invention, several compounds FC are covalently coupled to an oligonucleotide, said compounds FC may be identical or different. When several molecules of the same compound FC are covalently attached to the oligonucleotide of the detection probe PR, their introduction will use the same pair RG/complementary functional group on the oligonucleotide. When different compounds FC are covalently attached to the oligonucleotide of the detection probe PR, their introduction will use different pairs RG/complementary functional group on the oligonucleotide. In the detection probe PR according to the invention, a compound FC may be attached at the extremity of the oligonucleotide (typically at its 5' and/or 3' end). It is also possible that a compound FC is attached in an internal region (also called middle region) of the oligonucleotide.
If only one compound FC is present on the oligonucleotide, it may be at one of the extremity of the oligonucleotide or in its internal region. Conjugation of the compound FC at the 3' end may be preferred, for convenient reason, as it is not necessary in this case to block the 3' end. Conjugation of the compound FC on a nucleotide of the internal region may be preferred, for
fluorescent properties of the probe. If there is no compound FC or other labeling at the 3' end, this end is blocked to prevent extension during PCR, as well known in the art (el91 Nucleic Acids Research, 2013, Vol. 41, No. 20). The blocking of the 3' end can be performed by techniques known in the art, in particular by attachment of a -(Chh -OH, an alkyl, an amino linker, a PEG or by the presence at the 3' end of an inverted nucleoside.
When the compound FC is coupled to the oligonucleotide of the detection probe at its 5' or 3' end, the attachment is on the phosphate group of the corresponding nucleotide and is obtained by the intermediary of a linker. Only one compound FC may be attached at the 5' or 3' end of the oligonucleotide. It is also possible to have several (typically 2 or 3) compounds FC attached on the same end 5' or 3'. This is obtained by the use of a linker having several (typically 2 or 3) corresponding functional groups able to react with the reactive group of the compound FC, said linker being present at the 5' or 3' end (in particular covalently bonded to the phosphate group).
Some examples of detection probes PR according to the invention correspond to:
- detection probes PR bearing two molecules of a compound FC, one at the 3' end and one at the 5' end ; according to specific embodiments, these types of detection probes do not bear a quencher, or more generally do not bear a labeling that interacts with the compound(s) FC that is(are) attached to the oligonucleotide,
- detection probes PR bearing three molecules of a compound FC, one at the 3' end, one at the 5' end and one at an internal nucleotide; according to specific embodiments, these types of detection probes do not bear a quencher, or more generally do not bear a labeling that interacts with the compound(s) FC that is(are) attached to the oligonucleotide,
- detection probes PR bearing four molecules of compound FC, one at the 3' end, one at the 5' end and two in the middle region, each attached to an internal and different nucleotide; according to specific embodiments, these types of detection probes do not bear a quencher, or more generally do not bear a labeling that interacts with the compound(s) FC that is(are) attached to the oligonucleotide,
- detection probes PR bearing five molecules of compound FC, one at the 3' end, one at the 5' end and three in the middle region, each attached to an internal and different nucleotide; according to specific embodiments, these types of detection probes do not bear a quencher, or more generally do not bear a labeling that interacts with the compound(s) FC that is(are) attached to the oligonucleotide,
- detection probes PR bearing one molecule of compound FC at the 5' end,
and a quencher at the 3' end,
- detection probes PR bearing one molecule of compound FC at the 3' end, and a quencher at the 5' end,
- detection probes PR bearing two molecules of compound FC, one at the 5' end, one in the middle region and a quencher at the 3' end,
- detection probes PR bearing two molecules of compound FC, one at the 3' end, one in the middle region and a quencher at the 5' end,
- detection probes PR bearing three molecules of compound FC, one at the 5' end, two in the middle region and a quencher at the 3' end,
- detection probes PR bearing two molecules of compound FC, one at the 5' end, one in the middle region and a quencher at the 3' end
- detection probes PR bearing one molecule of compound FC at the 5' end, acting as a donor, and an acceptor dye at the 3' end,
- detection probes PR bearing one molecule of compound FC at the 3' end, acting as a donor, and an acceptor dye at the 5' end,
- detection probes PR bearing two molecules of compound FC, acting as donors, one at the 5' end, one in the middle region and an acceptor dye at the 3' end,
- detection probes PR bearing two molecules of compound FC, acting as donors, one at the 3' end, one in the middle region and an acceptor dye at the 5' end,
- detection probes PR bearing three molecules of compound FC, acting as donors, one at the 5' end, two in the middle region and an acceptor dye at the 3' end,
- detection probes PR bearing two molecules of compound FC, acting as donors, one at the 5' end, one in the middle region and an acceptor dye at the 3' end.
When the compound FC is coupled to said oligonucleotide in its middle region, the attachment is on the base of an nucleotide located in the middle region. In particular, the compound FC is coupled by attachment to a base A, T or G of the oligonucleotide. A coupling on a C base is less satisfactory.
The oligonucleotide can have different sequences. Most of the time, it is composed of 10 to 60 nucleotides. The invention offers the possibility to use oligonucleotides of short sequence, in particular the detection probes PR of the invention include an oligonucleotide of 10 to 30 nucleotides, and more preferably of 12 to 25 nucleotides. Advantageously, the detection probes PR of the invention include an oligonucleotide of 10 to 30 nucleotides, and more preferably of 12 to 25 nucleotides, and at least two molecules of compound FC coupled to the oligonucleotide, typically 2, 3, 4 or 5.
The oligonucleotide may be a single-stranded nucleic acid, without self- complementary sequences (that does not substantially form conformations
held by intramolecular bonds) or with self-complementary sequences (that form a secondary structure).
The oligonucleotide may be composed of a complementary sequence to a DNA gene sequence of a target microorganism or a complementary sequence to a DNA gene sequence of a subject (called target-specific sequence).
In the detection probe PR of the invention, the oligonucleotide may also comprise a target-specific sequence, also called hybridization sequence (i.e. a complementary sequence to a DNA gene sequence of a target microorganism or a complementary sequence to a DNA gene sequence of a subject), with in addition one or several non-target-specific sequence. This is particularly the case when the oligonucleotide of the detection probe PR exhibits some degree of self-complementarity. Specific embodiments of such detection probes PR include, for example, probes that form conformations held by intramolecular hybridization, such as conformations generally referred to as hairpins. Suitable hairpin probes include "molecular torches" (see, e.g., U.S. Pat. Nos. 6,849,412; 6,835,542; 6,534,274; and 6,361,945) and a "molecular beacons" (see, e.g., U.S. Pat. No. 5,118,801 and U.S. Pat. No. 5,312,728).
Therefore, the detection probes PR of the invention can have one or more molecules of a compound FC attached by a covalent bond to an oligonucleotide (OLIGO). In the detection probes FC and in the representation given hereafter, as illustrative but non limiting examples, the coupled compound FC is symbolized as -FC' as the reactive group present at the R2 or Ri position forms a covalent linkage (Qa at the 5' end, Qb at the 3' end, Qc in the middle region of the oligonucleotide) with the corresponding functional group (Xa at the 5' end, Xb at the 3' end, Xc in the middle region of the oligonucleotide) that is present on the oligonucleotide (OLIGO). Qa, Qb, Qc may be identical or different, but most of the time, they are identical when several molecules of a compound FC are attached to the oligonucleotide.
In the detection probes PR of the invention, -FC' corresponds to one of the following formula:

-FC'i when in compound FC (and therefore the corresponding group in -FC'i):
- Ri is Li-R'i, in which:
■ Li is a linker, in particular a linker defined previously in the DEFINITION section, and
■ R'i is a reactive group RG as defined for formula (I) and, in particular corresponding to one of the groups RG of Table 2, and
- R2 is -CH3, -CH2R"2, -CHalkylR"2, -C(alkylR"2)2, -SR"2 or -OR"2, in which R"2 is chosen among the groups alkyl, cycloalkyl, alkenyl, alkynyl, aryl, said groups being unsubstituted or substituted by one or several substituents chosen among, the groups, , -CF3, , -CN, alkyl, -C(O)alkyl, -C(O)Oalkyl, -Salkyl, -Oalkyl -NHC(O)H, , -S(O2)O’, -S(O2)Oalkyl, -P(O2)O’ and

-FC'2 when in compound FC (and therefore the corresponding group in -FC'2):
- Ri is chosen among the groups alkyl, cycloalkyl, alkenyl, alkynyl, aryl, said groups being unsubstituted or substituted by one or several substituents chosen among, the groups, -, -CF3, -CN, alkyl, -C(O)alkyl, -C(O)Oalkyl, -Salkyl, -Oalkyl -NHC(O)H, -S(O2)O’, -S(O2)Oalkyl, -P(O2)O’ and -P(O2)Oalkyl, and
- R2 is Y2-L2-R'2, in which:
■ Y2 is CH2, CHalkyl, C(alkyl)2, S or 0,
■ L2 is a linker, in particular a linker defined previously
in the DEFINITION section, and
■ R'2 is a reactive group RG as defined for formula (I) and, in particular corresponding to the groups RG of Table 2,
X, Z, n, Ri, Rj, Rk, Re, R3 and R4 being as defined for formula (I).
Of course, all the specific definitions and embodiments given for compounds FC apply to -FC', -FC'i and -FC'2.
A family of detection probes PR of the invention corresponds to formula (II) hereafter:
[Tc - (Qc - FC')mc]nc
Za - 5 OLIGO 3 PO 4 - Zb (Ii: with Za that represents -OH or -P0'4-Ta-(Qa-FC)ma ; nc that represents the number of -Tc-(Qc-FC')mc attached to nucleotide(s) of the middle region of the oligonucleotide and is equal to 0, 1, 2, 3, 4 or 5; Zb being -Tb-(Qb-FC')mb or a blocking group preventing extension during an amplification by PCR; ma, mb and me that represent the number of -Qa, b or c-FC' attached to Ta, b or c respectively and are the same or different and are equal to 1, 2 or 3 (preferentially is equal to 1); FC', Qa, Qb and Qc being as defined previously; and, Ta, Tb and Tc being a linker; with the proviso that if Za is OH and nc=0, then Zb is -Tb-(Qb-FC')mb. In the detection probes PR of formula (II), when there are several moieties FC', they can be identical or different.
When nc is 2, 3, 4 or 5, the groups -^-(Qc-F mc are attached on different nucleotides of the middle region of the oligonucleotide.
According to specific embodiments, in the detection probes PR of the invention corresponding to formula (II), when present, ma is 1, when present, mb is 1 and when present, me is 1.
Another family of detection probes PR of the invention including a quencher corresponds to formula (III) hereafter:
 , -FC)ma or -PO'4-Ta-(Qa-D)ma ; nc that represents the number of -Tc-(Qc-FC')mc attached to nucleotide(s) of the middle region of the oligonucleotide and is equal to 0, 1, 2, 3, 4 or 5; Ze being -Tb-(Qb-FC')mb, -Tb-(Qb-D)mb or a blocking group preventing extension
during an amplification by PCR; nf that represents the number of -Tr(Qf-D)mf attached to nucleotide(s) of the middle region of the oligonucleotide and is equal to 0, 1, 2, 3, 4 or 5; D being a quencher ; ma, mb and me that represent respectively the number of -Qa, b or c-FC' or the number of -Qa, b or c-D attached to Ta, b or c and are the same or different and are equal to 1, 2 or 3 (preferentially is equal to 1); FC', Qa, Qb and Qc being as defined previously; Qf being a resulting covalent linkage as defined previously in Table 2 and, Ta, Tb, Tc and Tf being a linker; with the proviso that :
, -FC)ma or -PO'4-Ta-(Qa-D)ma ; nc that represents the number of -Tc-(Qc-FC')mc attached to nucleotide(s) of the middle region of the oligonucleotide and is equal to 0, 1, 2, 3, 4 or 5; Ze being -Tb-(Qb-FC')mb, -Tb-(Qb-D)mb or a blocking group preventing extension
during an amplification by PCR; nf that represents the number of -Tr(Qf-D)mf attached to nucleotide(s) of the middle region of the oligonucleotide and is equal to 0, 1, 2, 3, 4 or 5; D being a quencher ; ma, mb and me that represent respectively the number of -Qa, b or c-FC' or the number of -Qa, b or c-D attached to Ta, b or c and are the same or different and are equal to 1, 2 or 3 (preferentially is equal to 1); FC', Qa, Qb and Qc being as defined previously; Qf being a resulting covalent linkage as defined previously in Table 2 and, Ta, Tb, Tc and Tf being a linker; with the proviso that :
- if Zd is OH and nc=0, then Ze is -Tb-(Qb-FC')mb and
- nf is not 0 or Zd represents -PO'4-Ta-(Qa-D)ma or Ze represents -Tb-(Qb- D) robin the detection probes PR of formula (III), when there are several moieties FC', they can be identical or different. When there are several quenchers D, they can be identical or different.
When nc is 2, 3, 4 or 5, the groups -TcXQc-F mc are attached on different nucleotides of the middle region of the oligonucleotide.
In the definition of formula (II) and (III), FC' corresponds to FC'l or FC'2, as previously defined.
In particular, in the definition of formula (II) and (III), the linkers Ta, Tb, Tc and Tf, when present, can include or can be composed of an alkylidenyl chain comprising 3 to 20 carbon atoms, said alkylidenyl chain being potentially interrupted by a double bond, a triple bond, S, 0, PO4 and/or - C(O)-NH-, -NH-C(O)- -S-S-, an heterocycle, such as a triazole group, or a group :
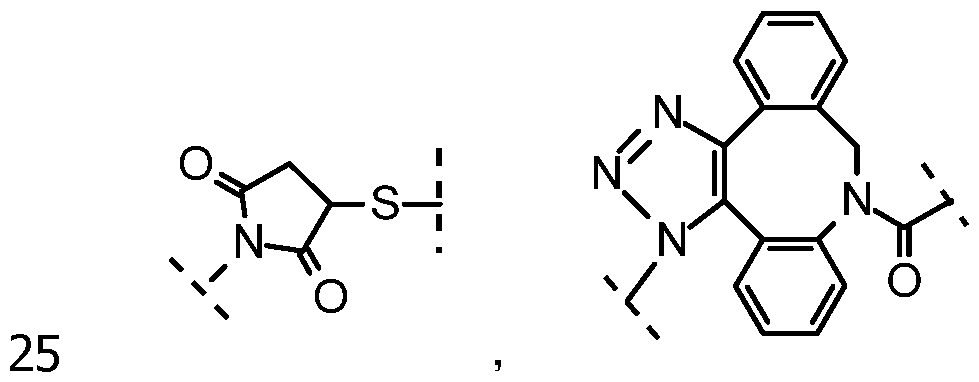 and/or being potentially substituted. The definition of the linkers Ta, Tb, Tc and Tf is given in the sense going towards the oligonucleotide. "Potentially substituted" refers in particular to one or several substituents on the alkylidenyl chain chosen among, aryl, -CF3, --CN, -C(O)alkyl, -C(O)Oalkyl, - Salkyl, -Oalkyl, -NHC(O)H, -S(O2)O’, -S(O2)Oalkyl, -P(O2)O’, -P(O2)Oalkyl, - CH=N-O-alkyl, -C(CH3)=N-O-alkyl, -CH=N-NH-C(O)-alkyl, -C(CH3)=N-NH- C(O)-alkyl, -CH=N-O-C(O)-alkyl, -C(CH3)=N-O-C(O)-alkyl, -NHCOalkyl and - CONHalkyl.
and/or being potentially substituted. The definition of the linkers Ta, Tb, Tc and Tf is given in the sense going towards the oligonucleotide. "Potentially substituted" refers in particular to one or several substituents on the alkylidenyl chain chosen among, aryl, -CF3, --CN, -C(O)alkyl, -C(O)Oalkyl, - Salkyl, -Oalkyl, -NHC(O)H, -S(O2)O’, -S(O2)Oalkyl, -P(O2)O’, -P(O2)Oalkyl, - CH=N-O-alkyl, -C(CH3)=N-O-alkyl, -CH=N-NH-C(O)-alkyl, -C(CH3)=N-NH- C(O)-alkyl, -CH=N-O-C(O)-alkyl, -C(CH3)=N-O-C(O)-alkyl, -NHCOalkyl and - CONHalkyl.
Typical examples of linker Ta (in the sense FC' -> oligo) are -(CH2)4-, - (CH2)5-, -(CH2)6-, -(CH2)6-PO4-(CH2CH2O)6- ; typical examples of linker Tb (in the sense FC' -> oligo) are -(CH2)4-, -(CH2)5-, -(CH2)6-, -(CH2)4-CH(CH2OH)-
CH2- ; and typical examples of linker Tc or Tf (in the sense FC -> oligo) are - (CH2)6-NH-C(O)-CH=CH- and -(CH2)6-NH-.
A typical example of Qa, Qb, Qc and Qf (in the sense FC' -> oligo) is -C(O)- NH-.
The detection probes PR may be obtained from a precursor oligonucleotide having the suitable corresponding functional group (Xa, Xb, Xc...) at the desired position. Scheme 5 illustrates the coupling reaction in the case of the detection probes of formula (Ila) that comprises a compound FC covalently coupled at the 5' and 3' ends and in the middle region of the oligonucleotide (OLIGO) resulting in the FC' part in the formula (Ila).
Scheme 5

Compound covalent coupling of the reactive group RG of FC on Xa, Xb and Xc of Ila’

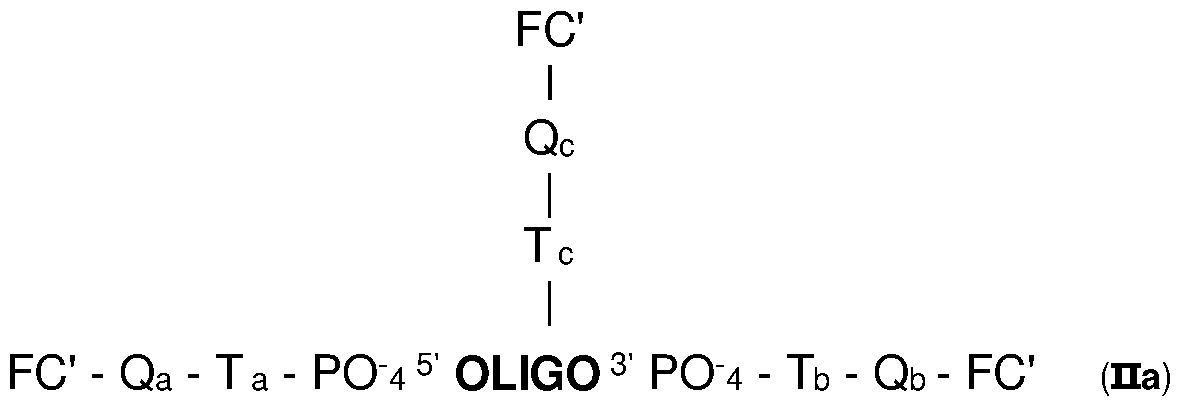
The coupling reaction can be carried out in DMSO at pH8 when the reactive group on the compound FC is an activated ester (in particular NHS) and the functional group Xa, Xb, Xc are an amine. The conditions of coupling are adapted by the skilled person, in function of the selected reactive group RG and the corresponding functional group (i.e. Xa, Xb, Xc).
The oligonucleotide (Ila7) functionalized with the suitable corresponding functional groups Xa, Xb and Xc and other analogues functionalized with the suitable corresponding functional group(s) (Xa, Xb, and/or Xc) are obtained by conventional methods. The functionalization introduces a linker (Ta at the 5' end, Tb at the 3' end, Tc in the internal region of the oligonucleotide) either between the corresponding functional group (Xa, Xb) and the PO4‘ of the
nucleotide at the 5' end or a modified nucleotide bearing a PO4" at the 3' end, when the compound FC is coupled to the 5' or 3' end respectively, or between the corresponding functional group (Xc) and the base of an internal nucleotide, when the compound FC is coupled to the internal region of the oligonucleotide. Most of the time, when the oligonucleotide bears several functional groups (Xa at the 5' end, Xb at the 3' end and/or Xc in the internal region), these functional groups are, preferably, identical and result in the same covalent linkage (meaning that Qa=Qb=Qc). But, it is not excluded to have different functional groups Xa, Xb and/or Xc, in particular for the introduction of different compounds FC or of a compound FC and a quencher.
Scheme 6 hereinafter illustrates an example of detection probe PR according to the invention, with specific linkers Ta, Tb and Tc, obtained with the use of commercial products, and where the covalent bond Qa, Qb and Qc are a carboxamide -CONH- in the sense compound FC' -> oligo and the OLIGO is
SEQ ID N°31: AGCACGTGGGAGGGCGATCG
Scheme 6
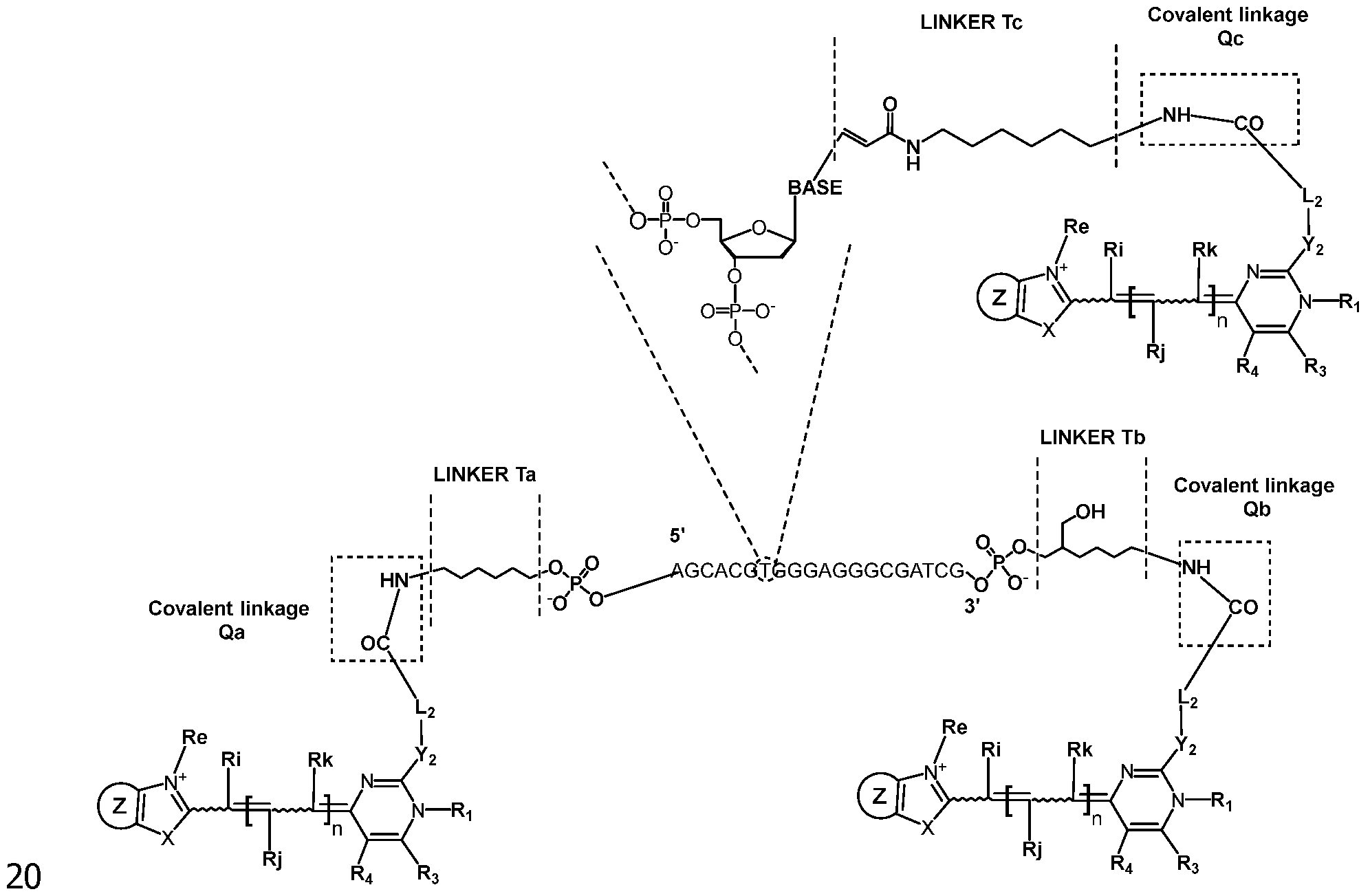
Other types of functionalized oligonucleotides can be used. The synthesis of the oligonucleotide is carried out, according to conventional methods known
in the art, and in particular on solid phase. The functionalized oligonucleotides can be obtained by classical protocols using available modifier of nucleoside. These kinds of modifier designed to be used in DNA synthetiser to functionalize the 5'-end, 3'-end or an internal nucleoside are, for instance distributed, by Glen research (https://www.glenresearch.com/products/labels-and-modifiers/oligo- modification/amino-modifier-phosphoramidites-and-supports.html). They can result in a protected functional group (for instance an amino group protected by a Fmoc group - fluorenyl methoxycarbonyle group or MMT group - monomethoxytrityl group) that can easily be deprotected before conjugation. In some embodiments, modifiers can be used to obtain 2 or 3 functional groups on the same nucleotide and as a result to covalently couple two compounds FC on the same nucleotide.
USES OF THE PROBES PR OF THE INVENTION
The detection probes PR of the present invention may be used for a variety of different applications. In particular, they may be used for the detection of a target nucleic acid which is a single-stranded nucleic acid. In some embodiments of the invention, the probes PR allow improved detection of a target nucleic acid, increasing sensitivity, improving detection limit, range of linear detection, or range of dynamic response of the detection probes PR of the invention to the concentration of a target nucleic acid.
A detection probe PR of the invention may recognize single-stranded (ss) nucleic acid as well as it may be used to detect double-stranded (ds) nucleic acid. It may form specific complexes with ss nucleic acids and can be used to probe ds nucleic acids at a temperature where their strands are separated. So, the invention encompasses uses and methods for detecting a target nucleic acid which is a ss or ds nucleic acid comprising a step of mixing a detection probe PR according to the invention, with a sample comprising the target nucleic acid.
Thus, the present invention proposes a method for detecting specifically a target nucleic acid that may be present in a sample, in which a sample is contacted with a detection probe according to the present invention, the formation of a specific hybridization product (based on complementarity) between the detection probe PR and the target nucleic acid is detected, indicating the presence of said target nucleic acid in said sample.
In one embodiment, the present invention provides a method of using the detection probes PR of the invention for detecting and/or quantifying the presence or absence of a target nucleic acid in a sample. The method typically comprises the steps of:
(a) providing at least one detection probe PR of the invention;
(b) allowing said at least one detection probe PR to interact specifically with nucleic acids contained within said sample, the whole forming a reaction mixture, under conditions such that at least one detection probe PR-target nucleic acid complex (also called duplex or hybridization product) is formed; and
(c) detecting an optical signal in said reaction mixture, said optical signal being indicative of the presence of said nucleic acid.
The optical signal results from the fluorescence caused by the presence of the covalently coupled compound FC (resulting in the moiety FC') on the detection probe PR and resulting from the specific formation of the detection probe PR-target nucleic acid complex.
Most of the time, the method of the invention includes a reaction of amplification of the target nucleic acid. In specific embodiments, detection is carried out together with a reaction of amplification of the target nucleic acid; this corresponds to the so-called real-time detection. In other specific embodiments, detection is carried out after a reaction of amplification of the target sequence; this corresponds to the so called "end-point" detection.
The amplification reaction can be based on any methods of amplification, linear or exponential, such as PCR, LCR, NASBA, TMA, SDA, RCA, LAMP. As way of example, the method of amplification is a method of amplification producing single-stranded DNA or RNA nucleic acids, such as NASBA, TMA or asymmetric PCR.
Whatever the method used, the detection probe PR will interact with a single strand target nucleic acid that may imply a previous step of denaturation, in particular after amplification. The conditions for the amplification, the denaturation, and the formation of the hybridization product are classical for the skilled person. In particular conditions used in amplification techniques by PCR can be used. A suitable buffer, enzyme, primers, optional salts (such as NaCI or MgCL) and/or dNTPs will be adapted by the skilled person, according to his general knowledge. Advantageously, an enzyme having a nuclease activity is not used. In particular, suitable enzyme are polymerase enzyme, in particular thermostable polymerases such as Taq polymerases, mutant Taq polymerases engineered to bring resistance to common inhibitors found in blood. Examples of mutant polymerase commercially available are HemoKlenTaq™ (New England Biolabs, Inc., Ipswich, MA), OmniTaq™ and OmniKlenTaq™ (DNA Polymerase Technology, Inc., St. Louis, MO).
According to preferred mode, an asymmetric PCR is used for amplifying the initial nucleic acid ie DNA. In that case, one strand of the original DNA is more amplified than the other and will be qualified as the target nucleic acid. Asymmetric amplification is well known in the art and required the excessive
amount of primers for a chosen strand (Multiplex-Ready PCR: A new method for multiplexed SSR and SNP genotyping », BMC Genomics, vol. 9, 2008, p. 80 (PMID 18282271, PMCID 2275739, DOI 10.118 6/1471-2164-9-80), « DNA sequencing with Thermus aquaticus DNA polymerase and direct sequencing of polymerase chain reaction-amplified DNA », Proc. Natl. Acad. Sci. U.S.A., vol. 85, n° 24, decembre 1988, p. 9436- 40 (PMID 3200828, PMCID 282767, DOI 10.1073/pnas.85.24.9436, Bibcode 1988PNAS...85.9436I), Linear-after-the-exponential polymerase chain reaction and allied technologies Real-time detection strategies for rapid, reliable diagnosis from single cells, vol. 132, coll. « Methods in Molecular Medicine™ », 2007, 65-85 p. (ISBN 978-1-58829-578-
1, PMID 17876077, DOI 10.1007/978-l-59745-298-4_7).
Therefore, the present invention provides methods for performing a nucleic acid amplification reaction and method for detecting a target nucleic acid after amplification.
The method of detecting a target nucleic acid of the invention typically comprises:
(a) conducting a nucleic acid amplification reaction in the presence of at least one detection probe PR of the invention, which reaction results in an apparition or an increase in optical signal that is indicative of the presence of amplified target nucleic acids;
(b) detecting said optical signal.
In one aspect, the increase in optical signal is proportional to the increase in the amount of amplified nucleic acids resulted from said amplification.
The detection probes PR of the invention are particularly useful in a realtime polymerase chain reaction (rtPCR) and the detection probe PR of the invention detects the amplified PCR products in real-time as the polymerization reaction proceeds. Because the detection probes PR of the invention are highly specific and sensitive in detecting nucleic acid, the number of time cycles required for detecting a target nucleic acid (ie. Ct value) may be reduced and the end-point fluorescence signal is high.
The rtPCR instrument consists of a thermal cycler with an integrated excitation light source (lamp, laser, or LED), a fluorescence detection system or fluorimeter, and software that displays the recorded fluorescence data as a DNA amplification curve. To perform rtPCR, a detection probe PR according to the invention needs to be added to the reaction mixture.
In the uses and method of the invention, the detection of an optical signal (i.e. fluorescence) may be performed on a variety of fluorescence based detection systems, including but not limited to microplate readers, hand-held portable meters, bench-top spectrofluorometers, or instruments that employ microfluidic chips.
In other embodiments of the invention, the detection probes PR of the invention can be used in a DNA melt curve analysis, a technique commonly used to analyze the products of a DNA amplification reaction. The DNA melt curve analysis can be performed on the same reaction mixture used in the nucleic acid amplification reaction. Thus, according to other embodiments, a detection probe PR according to the invention can be used in melting curve analysis. A melting curve analysis involves applying heat to the reaction mixture and monitoring fluorescence emission as the temperature is increased, typically from 40°C to 95 °C. The temperature at which DNA denaturation occurs is indicated by a sharp drop in the fluorescence signal, resulting from the dissociation of the double strand. Non-specific products and primer-dimers denature at lower temperatures compared to specific products. PCR products with different lengths or nucleotide content exhibit distinct peaks when plotting the derivative of fluorescence with respect to temperature (-dT/dF) since they denature at different temperatures.
A detection probe PR according to the invention has the ability to hybridize to its target nucleic acid. The moiety FC' corresponding to compound FC in detection probes PR according to the invention exhibits, in general, a significant change in fluorescence resulting from the hybridization to the target nucleic acid, typically ssDNA. As a result, they are useful tools for studying nucleic acids. After hybridization of the detection probes PR to their target nucleic acids, the compounds FC according to the invention behave as strong fluorescent dyes. Of course, the conditions of the method and of the fluorescence measurements will be adapted by the skilled person in function of the design of the detection probe PR (use of a quencher, of FRET, or hairpin or molecular beacon structures ...).
The invention includes method for detecting a target nucleic acid which is a single-stranded or double-stranded nucleic acid comprising a step of mixing a a detection probe PR of the invention, with a sample comprising the target nucleic acid or an amplicon of the target nucleic acid.
In particular embodiments, the detection probe PR according to the present invention are used for the detection of double stranded nucleic acids during a nucleic acid amplification reaction in real time and/or subsequent to amplification via a melting curve analysis or end-point analysis. In this context, the detection probe PR of the invention will be a part of an amplification, preferably a PCR, reaction mixture and it can already be present at the beginning of the amplification reaction. As it has been shown by the inventors, the detection probes PR according to the invention do not significantly interfere with the efficiency of such an amplification (preferably PCR) reaction. In particular, the detection probes PR according to the invention do not significantly inhibit amplification (preferably PCR) when
present at concentrations that provide high fluorescence signal.
In the use or the method according to the invention, the following steps may be carried out:
- amplifying the target nucleic acid to generate an amplicon,
- adding a detection probe PR according to the invention to the sample or a reaction mixture comprising the target nucleic acid and/or the amplicon, before, during or after the amplifying step,
- monitoring fluorescence from the detection probe PR according to the invention during or subsequent to the amplifying step.
According to particular embodiments, in the use and the method according to the invention, the following steps are carried out:
-amplifying the target nucleic acid, in the presence of the detection probe PR according to the invention, in particular by PCR, to generate an amplicon, and
- during the amplification, monitoring the fluorescence of the detection probe PR according to the invention, resulting from the hybridization of the detection probe PR to the amplicon.
According to specific embodiments, subsequent to the amplification step, a step of melting the generated amplicon is carried out, while monitoring the fluorescence resulting from the detection probe PR according to the invention, to obtain a melting curve.
Of course, in the methods and uses according to the invention, the detection probe PR of the invention and the amplicon are placed in a reaction mixture, which is suitable for their hybridization and the fluorescence obtaining. Typically, this corresponds to an aqueous medium of pH 7.5-9.5, more specifically of pH 8-9. So, the methods and uses according to the invention include a step of contacting the generated amplicon and the detection probe PR according to the invention, in conditions which enable the hybridization after denaturation of the amplicon in a single strand, which are known in the art. When the detection probe PR is present during the amplification, the usual conditions of amplification, in particular of PCR, enable this hybridization.
According to specific embodiments, in the use or the method according to the invention, the following steps may be carried out:
- amplifying of the target nucleic acid, in the presence of the detection probe PR according to the invention, in particular by PCR, and typically by real time PCR, to produce an amplicon,
- optionally during the amplifying step, monitoring the fluorescence of the detection probe PR according to the invention, resulting from the hybridization of the detection probe PR to the amplicon after its denaturation in a single strand, and
- optionally subsequent to the amplifying step, monitoring the fluorescence resulting from the hybridization of the detection probe PR to the amplicon after its denaturation in a single strand, via an end-point analysis or while melting the amplicon to obtain a melting curve.
It could also be possible to carry out the amplifying step first and to add the detection probe PR according to the invention later, in particular if the purpose is to obtain a melting curve or if an end-point analysis of the fluorescence is aimed.
Whether the detection probe PR according to the invention is present during amplification or is added subsequently, the melting step allows the analysis of the target nucleic acid, for instance the identification of a specific genotype or polymorphism. According to specific embodiments, the said melting curve is used to identify the genotype of the target nucleic acid, to detect or identify at least one mutation, polymorphism, preferentially single nucleotide polymorphism, and/or epigenetic variation.
A melting curve (also called melt curve) is generated by slowly denaturing (melting) the generated amplicon. The generation of melting curves and the use for analysis of nucleic acid are known in the art. More precisely, when a melting curve analysis is used for the detection or the quantification of a target double stranded nucleic acid (preferably dsDNA), the mixture containing the generated amplicon hybridized to the detection probe PR according to the invention is subjected to a thermal gradient. Preferably, the gradient is a continuous gradient, but step gradients are also possible. Most preferably, the gradient is a linear gradient. In one particular embodiment, the reaction mixture is subjected to a temperature increase which results in the generation of a dissociation curve. According to other embodiments, the double stranded nucleic acid (preferably dsDNA) is first thermally denatured at a temperature around 90°C into single strands, the cooling of the reaction mixture around 60°C allows the hybridization of the primers and initiate the polymerization process, the probes hybridize also at this temperature and allow the fluorescence emission to eventually monitor real time amplification. Temperature dependence of fluorescence of the hybridized probe is monitored during a final denaturation step that allow eventually the determination of a specific melting temperature. It is understood that the probes of the invention will have a better efficiency when an asymmetric PCR reaction is conducted to avoid the hybridization of the longer amplicon before the probe. An asymmetric PCR is routinely conducted by using an excess of one of the primers.
In the method and uses according to the invention, the amplification of the target nucleic acid, and in particular of the target DNA, can be carried out by different techniques, and in particular by enzymatic amplification reaction. By
«enzymatic amplification reaction*, it should be understood a process generating multiple copies of a target nucleotide fragment, by the action of at least one enzyme. According to the invention, the detection probes PR of the invention are not used in techniques that lead to the hydrolysis of the probes. Therefore, during the amplification and/or detection step, the detection probe PR is not in contact with an enzyme having a nuclease activity. Such amplification reactions (isothermal or not) are well known to one skilled in the art and the following techniques may be mentioned in particular: PCR (Polymerase Chain Reaction) of all kind (for instance, standard PCR, RealTime-PCR, quantitative PCR, digital PCR, multiplex PCR, asymmetric PCR, nested PCR, semi-nested PCR, LATE-PCR, Touchdown PCR, Hot-Start PCR, COLD-PCR, assembly PCR, Reverse Transcriptase-PCR (RT- PCR)), LCR (Ligase Chain Reaction), RCR (Repair Chain Reaction), 3SR (Self Sustained Sequence Replication) with the patent application WO-A-90/06995, NASBA (Nucleic Acid Sequence-Based Amplification), SDA (Strand Displacement Amplification), MDA (Multiple Displacement Amplification), RPA (Recombinase Polymerase Amplification), HDA (Helicase Dependent Amplification, RCA (Rolling Circle Amplification), TMA (Transcription Mediated Amplification) with U.S. Pat. No. 5,399,491, and LAMP (Loop mediated isothermal amplification) with the U.S. Pat. No. 6,410,278, RT-LAMP (Reverse Transcription-Loop-mediated isothermal Amplification). When the enzymatic amplification reaction is a PCR, RT-PCR (RT standing for «reverse transcription*) is used when the amplification step is preceded by a messenger RNA reverse-transcription step (mRNA) into complementary DNA (cDNA), and qPCR or RT-qPCR is used when PCR is quantitative.
These techniques use a pair of primers (consisting of two primers), typically one forward primer and one reverse primer. Herein, when in the methods, uses, kits and mixtures, it is mentioned that a pair of primer is used, that means that one or several pairs of primers can be used. A primer is a nucleotide fragment which may consist of 5 to 100 nucleotides, preferably of 10 to 40 nucleotides, more preferably of 15 to 30 nucleotides, and possesses a specificity of hybridization with a target nucleic acid sequence, under conditions determined for the initiation of an enzymatic polymerization, for example in an enzymatic amplification reaction of the target nucleic acid sequence. For instance, when one reverse primer and several forward primers or alternatively one forward primer and several reverse primers are used in an amplification, they form several pairs of primers. When it is desired to detect variants or different target nucleic acids simultaneously from the same sample and the same amplification, several pairs of primers will be used and so several amplicons are produced. This is the multiplex amplification and generally this is used with the PCR method. For this
purpose, the methods, uses, kits and mixtures according to the invention will include more than one pair of primers: one for each target nucleic acid.
Methods of PCR analysis using a detection probe PR according to the invention are particularly interesting.
The invention also concerns a method of PCR analysis of a target nucleic acid comprising the steps of:
- mixing a detection probe PR according to the invention with a sample comprising a target nucleic acid, a polymerase, and a pair of primers suitable to amplify a portion of the target nucleic acid and generate at least an amplicon, leading to a PCR mixture,
- amplifying the target nucleic acid from the PCR mixture and generating at least an amplicon,
- hybridizing the generated amplicon with the detection probe PR and generate a specific duplex,
- monitoring the fluorescence from the specific duplex during and/or subsequent to the amplifying step.
Of course, to enable the hybridization, the obtained amplicon is denatured to obtain a single stranded nucleic acid that hybridize with the detection probe PR. Advantageously, the amplifying step is carried out by asymmetric PCR and leads to a single stranded amplicon after denaturation, which is hybridized with the detection probe PR. According to some embodiments, such a method of PCR analysis further comprises detecting the presence of the amplicon from the monitored fluorescence.
In these methods of PCR analysis, the monitoring step may occur during and/or subsequent to amplification and may include generating a melting curve or end-point analysis of the fluorescence. According to specific embodiments, the said melting curve, and in particular its shape, is used to identify the genotype of the target nucleic acid, to detect or identify at least one mutation, polymorphism, preferentially single nucleotide polymorphism (SNP), and/or epigenetic variation. When end-point analysis of the fluorescence is done, the total amount of fluorescence is analyzed at the end of the amplification phase, when all the amplification cycles are completed and not during the amplification as it is generally done in Real-Time PCR where the amount of fluorescence is monitored and analyzed after each cycle of amplification.
It is also possible that a method of the invention includes the quantification of the target nucleic acid, which is initially present in the sample or reaction mixture. The quantity of target nucleic acid, which is initially present corresponds to the amount of nucleic acid which is present at the beginning, that means the sample used before any step of amplification.
Quantification of the initial amount of nucleic acid in the sample could be
carried out by any method classically known by those in the art and it could be applied during or after any amplification method, preferably PCR, qPCR, rtPCR, digital PCR, droplet digital PCR or LAMP.
One method for quantifying a target nucleic acid is by determining Cp (Crossing point - also named Ct for Cycle Threshold) and comparing the Cp to a standard or to a control.
Absolute quantification, including amplification by qPCR, frequently uses a standard curve approach. In this approach, a standard curve generated from plotting the Cp values obtained from amplification, preferentially real-time PCR, against known quantities of a single reference template (also called standard or control) provides a regression line that can be used to extrapolate the quantities of the target nucleic acid in a sample of interest. Serial dilutions (generally 10-fold dilutions) of the reference template are set up alongside samples containing the specific target nucleic acid that needs to be quantified. Various separate reactions are run, usually one for each level of the reference target and one each for the samples of interest. Also, since assay-specific differences in PCR efficiencies often affect quantification, separate standard curves, with separate reference templates, may be set up to quantify different gene targets.
It is also possible to use a single point of Cp of a single quantification standard as it is described in WO 2017/165269 using an imported calibration or quantification curve. When a single point of Cp is used, these methods could comprise only one quantification standard nucleic acid provided at a known concentration, the Cp for this standard is obtained and then a regression line corresponding to this standard (standard curve) is imported and placed on the single point Cp for the calibration and then the quantification of the target nucleic acid can be operated using the calibrated standard curve.
So, the method for detecting a target nucleic acid or the method of PCR analysis of a target nucleic acid may correspond to a method of performing quantitative amplification, preferably PCR, on a sample. In that cases, the methods may comprise amplifying a sample in an amplification mixture, the amplification mixture comprising a pair of target primers configured to amplify a target that may be present in the sample, the amplification mixture further comprising a plurality of quantification standard nucleic acids each provided at a different known concentration and at least one pair of quantification standard primers, the quantification standard primers configured to amplify quantification standard nucleic acids, generating a standard curve from the quantification standard amplicons, and quantifying the target nucleic acid using the standard curve.
In the present invention, when quantitative amplification is performed, either
external or internal quantification standard nucleic acids maybe be used for the quantification of the target nucleic acid. This means that if the standard nucleic acid is external, it is separated and not in the same reaction mixture (also called sample) as the one containing the target nucleic acid to quantify. If the standard nucleic acid is internal, it is in the same reaction mixture (also called sample) as the target nucleic acid to be quantified. The internal standard nucleic acid(s) is(are) generally amplified at the same time as the target nucleic acid to quantify but this can also be done previously, and the standard curve obtained can be stored and imported at the moment of the quantification of the target nucleic acid.
Quantification standard can be synthetic or natural. The calibration or quantification can be performed against a known natural microorganism with known concentrations or against other naturally occurring nucleic acid templates. It could be for example a yeast or bacteriophages for viruses and/or synthetic particles able to mimic membrane and/or capsid and/or envelope structures but also housekeeping genes.
As an alternative to determining Cp by absolute or normalized amplification data, the quantification of the target nucleic acid could also be done using the melting curves.
In particular, the quantification may imply the generation of a melting curve, and more precisely of several melting curves. In particular, the melting curve is generated by the hybridization product of the detection probe PR with the target nucleic acid.
Methods of quantification using a melting curve are known from those skilled in the art. Livak method for example is usable. It is also possible to use the maximum of the negative first derivative of the intensity of the fluorescence and of the temperature (max of - (dintensity of fluorescence/dTemperature) which gives the melting temperature and then the quantity of the target nucleic acid. The term "Tm" can be used in reference to the melting temperature. The melting temperature can be the temperature at which one half of hybridization products (i.e. detection probe PR/target nucleic acid), become dissociated into single strands. The prediction of a Tm of a duplex polynucleotide can take into account the base sequence as well as other factors including structural and sequence characteristics and nature of the oligomeric linkages. A Tm can be determined from a melting curve. Exemplary methods for the experimental determination of Tm are described in a variety of sources, e.g., Liew et al., "Genotyping of Single-Nucleotide Polymorphism by High-Resolution Melting of Small Amplicons," Clinical Chemistry 50(7): 1156-1164 (2004); Reed and Wittwer, "Sensitivity and Specificity of Single-Nucleotide Polymorphism Scanning by High-Resolution Melting Analysis," Clinical Chemistry 50(10): 1748-1754 (2004); Zhou et a!.,
"Closed-Tube Genotyping with Unlabeled Oligonucleotide Probes and a Saturating DNA Dye," Clinical Chemistry 50(8):1328-1335 (2004); and Zhou et a!., "High-resolution DNA melting curve analysis to establish HLA genotypic identity," Tissue Antigens 64:156-164 (2004). Melting/annealing curve analysis instrumentation is commercially available from a variety of manufacturers. The Tm value can be used to differentiate or identify a mutant, a SNPs, a specific genotype ...
This method may further include determining a value for the melting curve, and determining a Cp by identifying the amplification cycle in which the value for the melting curve exceeds a predetermined value. The value may be determined by peak height or peak area of a negative derivative of the melting curve. A set of negative derivative melting curves can be used, wherein the flattest curves represent the earliest cycles and the area under the curve increases through a number of cycles. It is expected that such derivative melting curves acquired at a plurality of cycles during amplification can be used to determine Cp. The height of the transition for each melting curve or the area under the negative first derivative of the melt curve can be determined for each cycle. The Cp may then be assigned to the cycle at which this value exceeds a pre-determined threshold.
Other methods for determining Cp may be applied. For example, a melt detector may be used (see U.S. 6,387,621; US 6,730,501; and US 7,373,253, herein incorporated by reference). The detector would interrogate curve shape and background noise to determine if the produced amplicon, preferably the amplicon obtained by PCR, is present in the sample. The use of a melt detector could be used to increase the sensitivity of the system (See Poritz, et al., PLos One 6(10):e2604 7). Optionally, additional filters could be applied to the melting curve analysis to window the melt transition to increase the specificity of the system, by analyzing only those melting curves having a melting transition, displayed as a melt peak, within a set temperature range. It is expected that such methods would result in a more accurate Cp (see WO 2014/039963).
Methods of continuous monitoring of temperature and fluorescence are used for relative quantification, illustratively using the detection probe PR according to the invention, in a single reaction with a control or standard nucleic acid. A multiplexed amplification (preferably PCR) reaction is provided, containing a control or standard nucleic acid at a known initial concentration and a target nucleic acid at an unknown concentration. Primers for amplification of the control or standard nucleic acid are present at the same initial concentration as primers for amplification of the target nucleic acid. In addition, it is desirable if the control or standard nucleic acid is selected such that its melting temperature is sufficiently well separated
from the melting temperature of the target nucleic acid, so that melting of each of these nucleic acids is discernable from melting of the other. It is understood that multiple target nucleic acids of unknown concentration may be multiplexed in the reaction, noting that it is desirable that the melting curve for each nucleic acid is distinguishable from the others and from the control or standard nucleic acid.
In PCR, the signals of fluorescence obtained from the hybridization of the detection probe PR according to the invention to the ssDNA allow to produce amplification curves for the standard or control nucleic acid and the target nucleic acid to quantify. However, sometimes signals from the control and the target combine to generate a single amplification curve and information about the amplification of the individual nucleic acids is not discernable. To avoid this, with continuous data acquisition, a series of melting curves can be generated during PCR cycling. Provided that the melting temperatures of the control or standard nucleic acid and the target nucleic acid are sufficiently separated, the melting profile of each of the two reactions can be distinguished. To generate a corrected amplification curve for the control or standard nucleic acid, at each cycle the integral of the negative first derivative of the melt curve over a pre-defined melt window can be computed and plotted as a function of the cycle number, with the Cp determined as the cycle at which each value exceeds a predetermined value. Similarly, a corrected amplification curve for the target nucleic acid may be generated by integrating the negative first derivative of the melting curve over the pre-defined melt window for the target as it is described in WO 2014/039963 which is incorporated by reference.
Other methods for converting the melt curve to a value are known in the art, such as using peak height of the negative first derivative. It is understood that the predetermined value should be selected according to method used.
For instance, a method of PCR analysis, according to the invention, may comprise the steps of mixing the detection probe PR according to the invention, with a sample comprising an unknown initial quantity of a target nucleic acid and primers configured for amplifying the target nucleic acid, to form a mixture, amplifying the target nucleic acid in the presence of the detection probe PR according to the invention to generate an amplicon and obtain its hybridization with the amplicon after its denaturation, monitoring fluorescence of the detection probe PR according to the invention, resulting from the hybridization of the detection probe PR according to the invention with the denatured amplicon throughout a temperature range during a plurality of amplification cycles to generate a plurality of melting curves, and using the melting curves to quantify the initial quantity of the target nucleic acid.
Illustratively, a method of PCR analysis of a target nucleic acid according to the invention may comprise the steps of:
- mixing a detection probe PR according to the invention with a sample comprising a target nucleic acid and at least a pair of primers suitable to amplify a portion of the target nucleic acid and generate an amplicon, leading to a PCR mixture,
- amplifying the target nucleic acid from the PCR mixture, and generating at least an amplicon, and hybridizing the generated amplicon after denaturation with the detection probe PR and generating a specific duplex,
- during the amplifying step, monitoring the fluorescence of the detection probe PR resulting from the duplex generated,
- at the end of the amplifying step, melting the generated duplex, to obtain a melting curve, and
- identifying the genotype or polymorphism of the target nucleic acid using a shape of the melting curve.
According to particular embodiments, whatever the methods according to the invention, the generated fluorescence may be generated by FRET with the use of a pair of adjacent hybridization probes. In that case, with the detection probe PR, used as the donor probe and/or as the acceptor probe of the pair of adjacent hybridization probes, the other member of the pair of adjacent hybridization probe is also introduced. The pair of adjacent hybridization probes may be detection probe PR as donor probe/acceptor probe (corresponding or not to a detection probe PR of the invention), donor probe (corresponding or not to a detection probe PR of the invention)/ detection probe PR as acceptor probe or detection probe PR as donor probe/quenching probe.
According to particular embodiments, whatever the methods according to the invention, the amplifying step may include a plurality of temperature cycles including at least a denaturation temperature and an extension temperature, wherein each cycle has a cycle time of less than 90 seconds per cycle, and wherein the polymerase is provided at a concentration of at least 0.005 pM and primers are each provided at a concentration of at least 0.1 pM. According to specific embodiment, the amplifying step includes a plurality of temperature cycles including at least a denaturation temperature and an extension temperature, wherein each cycle has a cycle time of less than 20 seconds per cycle, and wherein the polymerase is provided at a concentration of at least 0.5 pM and primers are each provided at a concentration of at least 2 pM. These concentrations are related to the amplification mixture, in particular to the PCR mixture.
The PCR techniques are often classified according to the time required for the PCR and according to the quantity of primers which is used. More details
are given in US 7,387,887 and US 9,932,634. Classical or standard PCR are quite slow and occur in approximately 90 seconds or less per cycles, rapid PCR occur in less than 60 seconds per cycle, for example between 20 and 60 seconds per cycle, fast, ultra-fast and extreme PCR occur in less than 20 seconds, preferentially less than 12 seconds for fast PCR, less than 6 seconds for ultrafast PCR and in less than 2 seconds for extreme PCR. As the PCR speeds become faster, the concentrations of primers and polymerase are increased. This allows to maintain PCR efficiency and yield. The concentrations of primers range from at least 0.1 pM for the classical or standard PCR to at least 2 pM (typically in the range 2-6 pM) for extreme PCR, that is at least 0.1 pM, at least 0.2 pM, at least 0.4 pM, at least 0.6 pM, at least 0.8 pM, at least 1 pM, at least 1.2 pM, at least 1.4 pM, at least 1.6 pM, at least 1.8 pM or at least 2 pM. The concentrations of polymerase range from at least 0.005 pM for classical or standard PCR to at least 0.5 pM for extreme PCR, that is at least 0.005 pM, at least 0.01 pM, at least 0.02 pM, at least 0.04 pM, at least 0.06 pM, at least 0.08 pM, at least 0.1 pM, at least 0.2 pM, at least 0.3 pM, at least 0.4 pM or at least 0.5 pM. Any kind of these PCR can be used, according to the invention. Herein, 1 pM of polymerase corresponds to 3.8 U/pL. All these concentrations are related to the PCR mixture.
More details on the method for the analysis of nucleic acids which can be used with the detection probes PR according to the invention can be found in US 9,682,970, WO 2008/052742, WO 2006/121423, US 7,456,281, US 7,387,887 and US 7,582,429.
The invention also concerns a PCR reaction mixture, also named PCR mixture, comprising:
- a target nucleic acid,
- a pair of primers suitable to amplify a portion of the target nucleic acid, to generate an amplicon,
- a polymerase, in particular a thermostable polymerase,
- a detection probe PR according to the invention.
Said pair of primers is designed to amplify a specific sequence of interest in the target nucleic acid according to standard methods known in the art of molecular biology. More than one pair of primers can be used, in particular for multiplex PCR, where more than one target sequence must be amplified. The pair of primers may be a pair of primers for asymmetric PCR.
The target nucleic acid is typically genomic DNA or alternatively cellular RNA or cellular mRNA. In case of RNA, the thermostable DNA polymerase may be a DNA polymerase or a mixture of polymerases comprising reverse transcriptase activity.
Typically, such a PCR reaction mixture also includes a mix of deoxynucleoside triphosphates which is usually dA, dG, dC and dT, or dA, dG, dC and dU.
Such a PCR reaction mixture classically includes a buffer. In particular, the PCR reaction mixture is buffered at pH from 7.5 to 9.5, preferentially from 8 to 9.
The classical components of the mixture will be used in a concentration, easily determined by the person skilled in the art, according to common practice.
In the method, use and mixture of the invention, the concentration of the detection probe PR according to the present invention is, typically, from 0.5 to 20 pM, and preferably from 1 to 5 pM. That concentration corresponds to the concentration of the detection probe PR according to the invention in the sample which is used for monitoring the fluorescence of the detection probe PR of the invention resulting from the hybridization with the denatured amplicon.
The methods of nucleic acid detection can be associated with a variety of practical applications. According to one embodiment, the method is associated with routine quantification of nucleic acid in solution. Examples of such application include quantifying yields of purified DNA fragments for subcloning or for use as transcription templates, quantifying yields from cDNA library production, quantifying DNA amplification products or DNA input for PCR, detecting DNA contamination in protein drug preparation produced from recombinant organisms, and quantifying forensic DNA samples extracted from various biological samples.
Another object of the invention relates to a kit for detecting a target sequence comprising at least one detection probe PR according to the present invention and at least one additional reagent. Examples of additional reagents are the primers suitable to amplify a portion of the target nucleic acid, to generate an amplicon, a polymerase, in particular a thermostable polymerase, as described previously.
EXAMPLES
The following examples illustrate the invention.
The examples refer to the attached figures.
BRIEF DESCRIPTION OF THE FIGURES
Figure 1 illustrates different examples of probes PR according to the invention and the way they hybridize to their target nucleic acid sequence. A: the probe PR bears three FC' moieties (resulting from the conjugation of a compound FC of the invention): one at the 5' end, one in the internal region and one at the 3' end. B: the probe PR bears two FC' moieties (resulting
from the conjugation of a compound FC of the invention): one at the 5' end, one in the internal region and a quencher at the 3' end. The FC' and the quencher interact by FRET. C to F illustrate various pairs of adjacent hybridization probes including at least one FC' moieties (resulting from the conjugation of a compound FC of the invention). FC' may be a donor (D in Figure 1) and/or an acceptor (A in Figure 1). E: the probe PR bears two FC' moieties (resulting from the conjugation of a compound FC of the invention): one at the 5' end and one at the 3' end and the oligonucleotide forms a secondary structure (hairpin like) before hybridization.
Figure 2 illustrates the monitoring of a typical example of a conjugation reaction by reverse phase chromatography (example of detection probe II.10). A represents the oligonucleotide before conjugation, B represents the crude conjugation reaction and C represents the purified conjugate (i.e. the detection probe PR).
Figure 3 shows the fluorescence excitation and emission maxima of a selection of synthesized detection probes PR of the invention determined in the presence of their complementary target in PCR conditions (4 mM MgCL containing PCR buffer).
Figure 4 shows the fluorescence exaltation when the detection probe 11.33 of the invention hybridizes selectively to its complementary DNA target (at room temperature).
Figure 5 illustrates the principle of the experimental set up used to assess the specificity and the selectivity of detection probes PR of the invention. A typical example corresponding to a doubly conjugated probe is represented. Figure 6A to 6F show: in Panels A, C and E, the fluorescence recorded in RFU in function of the temperature (°C) during the melt of the hybridization product of probes II.6, II.4 or 11.33, in the presence of a model complementary DNA, a non-complementary DNA or a non-complementary random DNA duplex, respectively (+ COMP., + NON COMP., + RANDOM). The double arrow indicates the ratio of fluorescence exaltation at 60°C between the hybridized and the non-hybridized probe (= Exaltation factor at 60°C; in these cases, it is 1.1, 1.6 and 2.6 respectively). In Panels B, D and F, the first derivative of the melting curve (-d(RFU)/dT) in function of the temperature of the probes II.6, II.4 and 11.33. The double arrow indicates the height of the peak of the first derivative (-d(RFU)/dT) at the Tm in RFU/°C (= Tm peak height; in these cases, it is 250, 1080 and 1800 respectively).
Figure 7 illustrates the real time PCR detection of a specific sequence of a gene of a Coronavirus micro-organism using II.4 probe for different target inputs. The fluorescence was recorded as a function of the number of PCR cycles (Cp). The dotted line indicates the maximum fluorescence obtained at
the end of the amplification. Only one replicate out of 3 is shown.
Figure 8 shows: in Panel A, the fluorescence recorded in RFU as a function of the temperature (°C) during the melt of the hybridization product resulting from probe II.4 in the presence of a specific amplicon generated after PCR amplification (a sequence of a gene from a Coronavirus micro-organism). Only one replicate out of 3 is shown; In Panel B, the first derivative of the melting curve (-d(RFU)/dT) as a function of the temperature. Only one replicate out of 3 is shown.
Figure 9 shows the real time PCR amplification experiments using the probe II.5 that targets a sequence of a gene of a Streptococcus micro-organism.
Figure 10 shows the melting experiments and Tm determination of the hybridization product resulting from an amplicon, with the use of probe II.5 targeting a gene sequence of a Streptococcus micro-organism.
Figure 11 presents the melting experiments of the hybridization product resulting from a PCR amplicon and the probe II.7 targeted to a gene sequence of a Neisseria Gonorrhoeae micro-organism.
Figures 12A and 12B show: in Panel A, the detection of probe II.11 that is specific to a gene sequence of a Coronavirus micro-organism, on Texas red channel, with from the left to the right, the real time PCR amplification curve, the melting curve of the amplicon and the first derivative of the melting curve; In panel B, the detection of probe 11.16 that is specific to a gene sequence of a S. Pombe micro-organism on Quasar 705 channel, with from the left to the right, the real time PCR amplification curve, the melting curve of the amplicon and the first derivative of the melting curve. Both amplifications were done in the same tube.
Figure 13 shows the melting curve of the hybridization product and the first derivative of the melting curve that determines the Tm temperature for probes 11.18, 11.19, 11.20 and 11.21. Probes 11.18 to 11.21 include only one single nucleotide mismatch versus their complementary sequence and allow to detect mutants due to the strong destabilization of the duplex versus the wild-type sequence that is present in probe 11.18. The targeted sequence corresponds to a portion of a gene of a Corona Virus.
Figure 14 shows the PCR amplification experiments of a gene sequence of Neisseria Gonorrhoeae micro-organism and the melting curve of its hybridization product with the probe 11.17 and its first derivative. It allows to discriminate between 3 mutants that differ only by 2 nucleotides versus the wild-type sequence on panels A and B. The panels C and D show for comparison the same experiment carried out in the presence of the commercial free dye LCG Plus only (BioFire Gen scanning reagent, Salt Lake City, USA).
Figure 15 shows the melting curves in RFU and the first derivative of the melting curves that determines the Tm temperature for the probes 11.47, 11.48 and 11.49 in the presence or absence of the complementary DNA (COMP). The probes were labelled with the dye 1.1 of the invention and analyzed on the CFX (Agilent).
Figure 16 shows the melting curves obtained with three different probes in the presence of their target nucleic acid (COMP). The probes were labelled internally with the dye 1.1 on T, G or A respectively in probes 11.13, 11.14 and 11.15. The probes were analyzed on the CFX (Agilent).
Figure 17 shows the melting curves of the Comp. Probe 1 in the presence of its target sequence and in the presence of a random duplex that is not complementary to the probe. The non-specificity of this probe is evident. The plain double arrow corresponds to the specific Tm peak height and the double dotted arrow corresponds to the non-specific Tm peak height.
Figure 18 shows the melting curves of the hybridization product obtained from an amplicon generated from a gene sequence of a Streptococcus microorganism after PCR amplification in the presence of Comp. Probe 4 comprising two dyes that have two positive charges that shows the nonspecificity of this type of probe. The plain double arrow corresponds to the specific Tm peak height and the double dotted arrow corresponds to the non-specific Tm peak height.
Figure 19 shows the melting curves of the hybridization product obtained from an amplicon generated from a gene sequence of a Streptococcus microorganism after PCR amplification in the presence of Comp. Probe 5 that comprises 3 dyes having two positive charges. That shows the non-specificity of this type of probes. The plain double arrow corresponds to the specific Tm peak height and the double dotted arrow corresponds to the non-specific Tm peak height.
Figure 20 shows the evolution of the fluorescence exaltation of the acceptor group for the Probe 11.50 and Comparative Probe 6 (donor probes) and Comparative Probes 7 and 8 (acceptor probes) in the presence or absence of the complementary DNA. The concentration of the donor probes and their complementary strand were fixed at 0.1 pM whereas the concentration of the acceptor probes varied from 0.05 pM to 0.4 pM. The analyses were performed on a spectrofluorometer Spark (Tecan, USA). The FRET signal at 640 nm was followed. Panel A describes the FRET signal obtained with Comparative Probe 6/Comparative Probe 7, Panel B with Comparative Probe 6/Comparative Probe 8, Panel C with the Probe II.50/Comparative Probe 7 and Panel D with the Probe II.50/Comparative Probe 8.
Figure 21 shows the variation of the fluorescence exaltation of the acceptor group at 640 nm (Comparative Probe 9) with the donor probes 11.51 and
11.52 of the invention and the Comparative Probe 10 in the presence or absence of the complementary DNA. The oligonucleotide of the donor probes was doubly labelled with either the dye 1.1 or 1.6 (respectively Probe 11.51 and Probe 11.52), or the classical FAM group (Comparative Probe 10). The FRET signal at 640 nm was followed. Panel A describes the FRET signal obtained with the probes Comparative Probe 10/Comparative Probe 9, Panel B with the Probe II.51/Comparative Probe 9 and Panel C with the Probe II.52/Comparative Probe 9.
Figure 22 represents the modulation of the fluorescence intensity obtained with probes of the invention with conjugated dye 1.1 (Probe 11.50) or 1.5 (Probe 11.53) compared to the combination of probes using the pair FAM/Cy5.5™ (Comparative Probe 6/Comparative Probe 11). The concentration of the donor probes and their complementary strand were fixed at 0.1 pM whereas the concentration of the acceptor probes varied from 0.05 pM to 0.4 pM. The analyses were performed on a spectrofluorometer Spark (Tecan, USA). The FRET signal at 705 nm was followed. Panel A describes the FRET signal obtained with Comparative Probe 6/Comparative Probe 11, Panel B with Comparative Probe 6/Probe 11.53, Panel C with the Probe II.50/Comparative Probe 11 and Panel D with the Probe 11.50 /Probe 11.53.
Figure 23 shows on Panel A: Real time PCR amplification of a gene sequence of a Listeria micro-organism using the probes 11.22 and 11.23 in FRET technology. Panel B: corresponding amplicon melting peaks.
Figure 24 shows the evolution of the melting peak height intensity at 640 nm (Panel A) and 705 nm (Panel B) after real time PCR amplification of a Cronobacter micro-organism using the probes 11.50, 11.51, 11.53 to 11.57 of the invention and Comparative Probes 6, 7, 8, 9, 11 and 12, in FRET technology.
ABREVIATIONS ACN: acetonitrile; TFA': trifluoroacetate salt; TsO': tosylate salt ; DMSO: dimethylsulfoxyde; Me: Methyl; UPLC: ultra-performance liquid chromatography; HPLC: high performance liquid chromatography; Ph: phenyl; NAP column: sephadex gel filtration (GE, USA) ; QSP: quantitate sufficient per; DCM: dichloromethane; TFA: trifluoroacetic acid; TEAAc: triethylammonium acetate; TEA: triethylamine; Ac: acetyl; RT: room temperature; D/A: Donor/ Acceptor.
A. SYNTHESIS OF COMPOUNDS OF FORMULA (If) The compounds of the following formula (If), with the definition of A, Rb R2 and n given in Table 3, have been synthetized.

Table 3
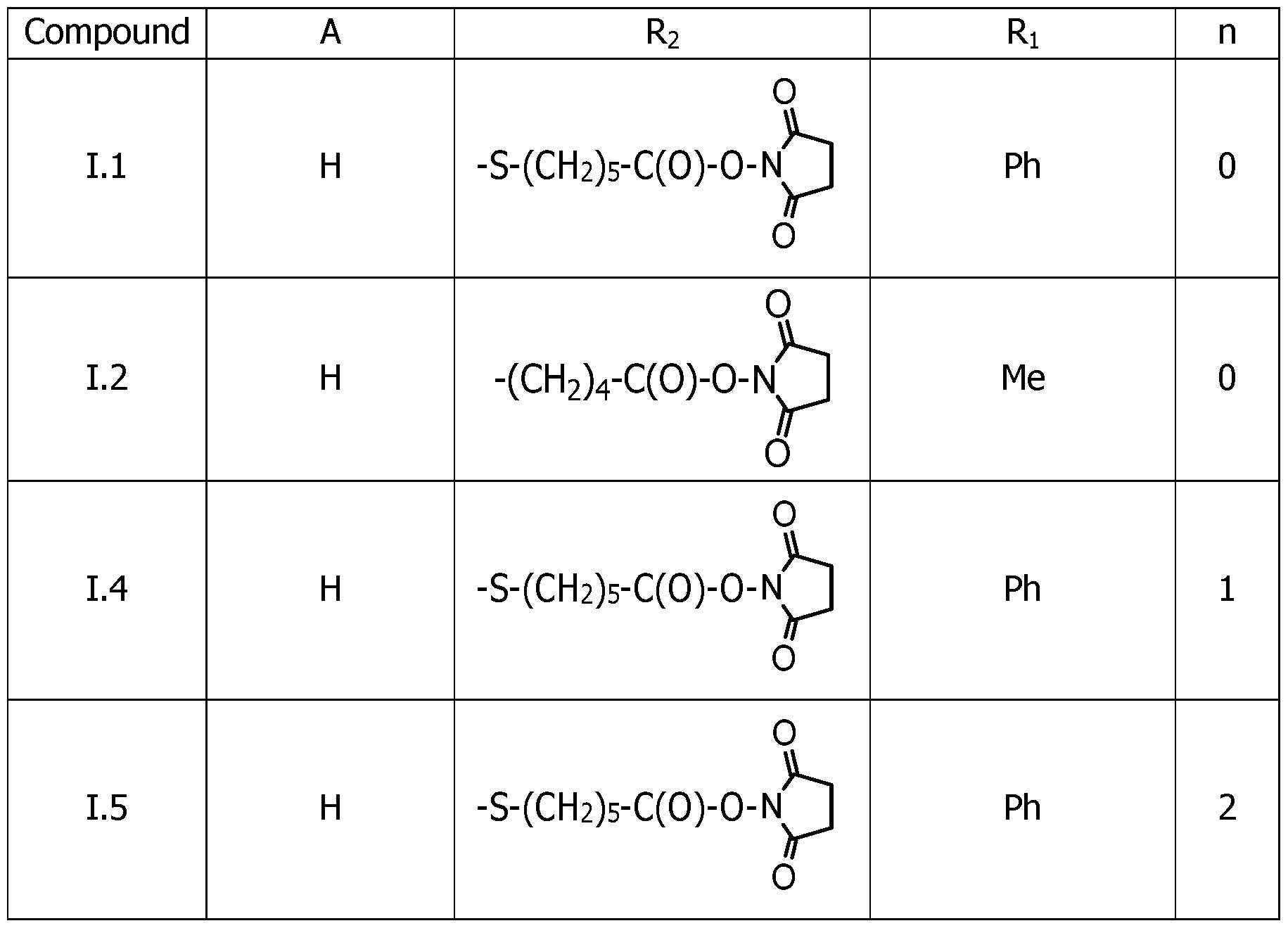 Table 3 (continued 1)
Table 3 (continued 1)

Example A 2-((2-((6-((2,5-dioxopyrrolidin-l-yl)oxy)-6-oxohexyl)thio)-6- methyl-l-phenylpyrimidin-4(lH)-ylidene)methyl)-3- methylbenzo[d]thiazol-3-ium trifluoroacetate 1.1, TFA
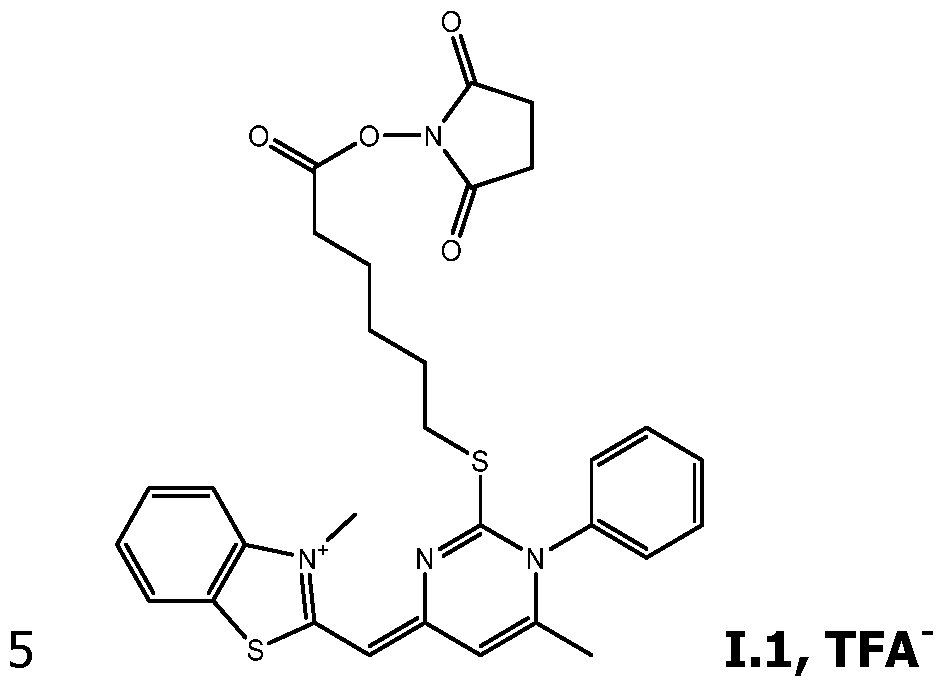 a) Preparation of 3-methyl-2-(methylthio)benzo[d]thiazol-3- ium tosylate, (1)
a) Preparation of 3-methyl-2-(methylthio)benzo[d]thiazol-3- ium tosylate, (1)
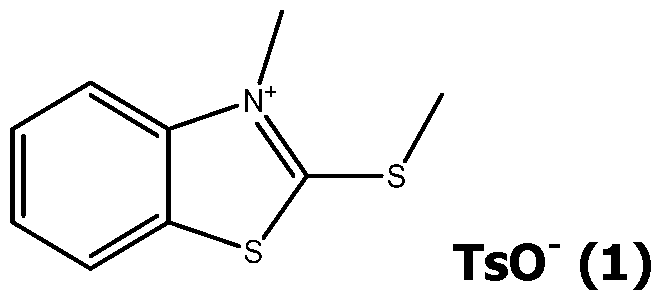
2-(Methylthio)benzo[d]thiazole (10 g, 55.16 mmol) was put in a round bottomed flask of 250 mL and methyl p-toluenesulfonate (75 mL, 496.47 mmol) was added. The mixture was stirred and heated at 145°C (fusion reaction) for 2 hours. The mixture turned orange. The mixture was then left to cool down to room temperature. 180 mL of diethyl ether was added to crude product and a white precipitate was obtained. The precipitate was filtered and washed with diethyl ether two times. The compound (1) was obtained, as a solid, by evaporating the rest of diethyl ether using a rotary evaporator. Yield = 90 %.
Mass spectrometry (ESI-Q (+)): M/Z = 196.02
XH NMR (400 MHz, D2O) 5 (ppm): 8.5(d,lH) ; 8.1(t,lH) ; 7.87(t,lH) ;
7.25(d,lH) ; 4.3(s,3H) ; 2.44(s,3H)
b) Preparation of 4,6-dimethyl-l-phenylpynmidine-2(lH)-thione

N-phenylthiourea (9.6 g, 63.16 mmol) was put in a round bottomed flask of 250 mL and dissolved in 130 mL of ethanol (EtOH). Acetylacetone (2,4- pentandione) was added followed by HCI 37% (15 mL). The mixture was stirred and heated to 90 °C (reflux) for 5 h. The mixture was turned orange- red. The heating was turned off and was cooled down to room temperature. 180 mL of diethyl ether was added to precipitate the product. The precipitate was filtered and washed with diethyl ether two times. Then, the precipitate was neutralized in an Erlenmeyer flask of 500 mL with an aqueous solution of NaOH (150 mL, 8.18 g, 180 mmol) and 80 mL of EtOH was added. The solution was poured in a 1 L separatory funnel and 200 mL of DCM was added. The separatory funnel was shaken vigorously for a few seconds. After few minutes the organic phase was recovered. The aqueous phase was washed two times with 50 mL of DCM. The organic phase was evaporated by using a rotary evaporator.
Mass spectrometry (ESI-Q (+)): M/Z = 216.07
NMR (400 MHz, D2O) 5 (ppm): 7.49(t,lH) ; 7.43(t,2H) ; 7.26(d,2H) ; 5.99(s,lH) ; 2.15(s,3H) ; 2.13(s,3H). c) Preparation of 2-((5-carboxypentyl)thio)-4,6-dimethyl-l- phenylpyrimidin-l-ium, Br (3)
 , mmol) was put in a round bottomed flask of 250 mL with 6-bromohexanoic acid (2.029 g, 10.4 mmol) and 100 mL of acetonitrile. The mixture was stirred and heated at 92°C during 2 h. The
heating was turned off and was cooled down to room temperature then the crude product was evaporated by using a rotary evaporator. The residue was extracted in a mixture of DCM/water and the product was recovered in aqueous phase. The solvent was evaporated and co-evaporation was carried out 2 times with ACN by using a rotary evaporator, to obtain compound (3). Yield = 100 %.
, mmol) was put in a round bottomed flask of 250 mL with 6-bromohexanoic acid (2.029 g, 10.4 mmol) and 100 mL of acetonitrile. The mixture was stirred and heated at 92°C during 2 h. The
heating was turned off and was cooled down to room temperature then the crude product was evaporated by using a rotary evaporator. The residue was extracted in a mixture of DCM/water and the product was recovered in aqueous phase. The solvent was evaporated and co-evaporation was carried out 2 times with ACN by using a rotary evaporator, to obtain compound (3). Yield = 100 %.
Mass spectrometry (ESI-Q (+)): M/Z = 331.15 d) Preparation of 2-((2-((5-carboxypentyl)thio)-6-methyl-l- phenylpyrimidin-4(lH)-ylidene)methyl)-3- methylbenzo[d]thiazol-3-ium, TFA" (4)
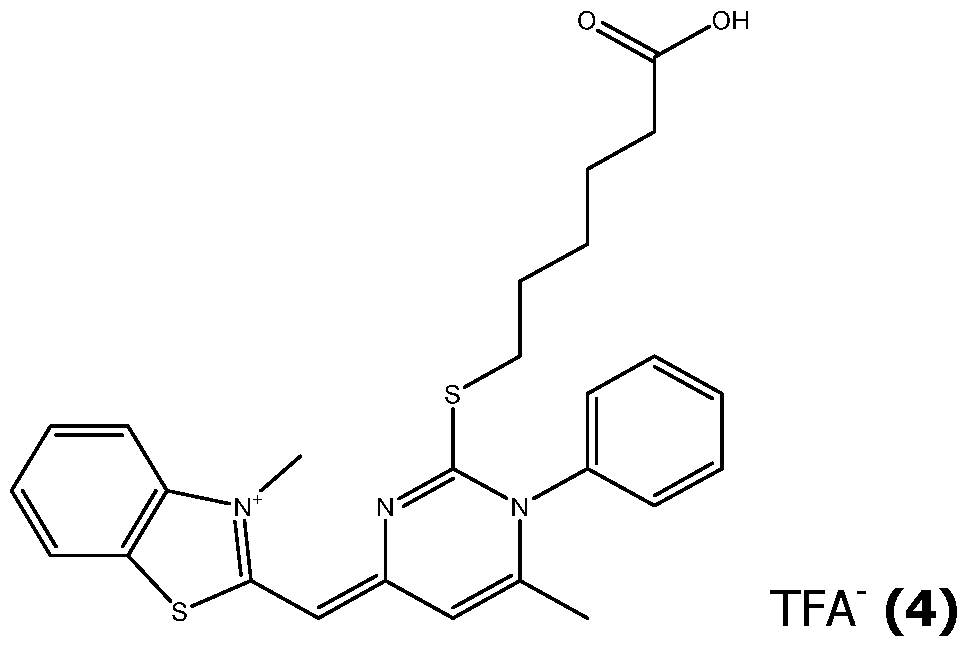
3-methyl-2-(methylthio)benzo[d]thiazol-3-ium tosylate (compound (1), 196 mg, 530 pmol) was poured in a round bottomed flask of 100 mL with 10 mL of a solution of 2-((5-carboxypentyl)thio)-4,6-dimethyl-l-phenylpyrimidin-l- ium Bromide at 20 mg/mL in acetonitrile (compound (3), 200 mg, 486 pmol). 52 mL of acetonitrile and 4.78 mL of ethyl alcohol were successively added in the round bottomed flask. Finally, 210 pL of triethylamine (152.4 mg, 1.5 mmol) were dropped. The mixture was then stirred at room temperature for 25 minutes. The solvents were evaporated and the crude product obtained was purified by flash chromatography with C18 column of 40 g (eluent A 10 mM TFA in water, eluent B 10 mM TFA in 90% ACN and 10 % water, gradient 0 to 70 % of eluent B in 21 min), leading to compound (4), as a trifluoroacetate salt. Yield = 12 %.
Mass spectrometry (ESI-Q (+)): M/Z = 478.1
XH NMR (400 MHz, D2O) 5 (ppm): 7.98 (d,lH) ; 7.82 (d,lH) ; 7.68 (m,7H) ; 7.46 (t,lH) ; 7 (s,lH) ; 6.45 (s,lH) ; 3.93 (s,3H) ; 3.49 (t,2H) ; 2.21 (t,2H) ; 1.97 (s,3H) ; 1.77 (Qu,2H) ; 1.51 (m,4H).
e) Preparation of compound LI, TFA

Compound (4) (51.0 mg, 78.5 pmol) was poured in a round bottomed flask of 10 mL with 2 mL of anhydrous acetonitrile, N,N'-disuccinimidyl carbonate (301.6 mg, 1.18 mmol) and pyridine ( 95.8 pL, 94.1 mg, 1.18 mmol). The mixture was then stirred at 90°C for 2 hours. The mixture was evaporated by using a rotary evaporator. The crude product was extracted in mix of DCM/water and the product was recovered in organic phase. The solvent was evaporated and co-evaporation was carried out 2 times with ACN by using a rotary evaporator. Then, the residue of organic phase was purified by flash chromatography with C18 column of 12 g (eluent A : 10 mM TFA in water, eluent B : 10 mM TFA in 90% ACN and 10 % water, gradient 20 to 100 % of eluent B in 23 min) and leaded to compound 1.1, TFA'. Yield = 27 %.
Mass spectrometry (ESI-Q (+)): M/Z= 575.2
XH NMR (400 MHz, D2O) 5 (ppm): 7.98 (d,lH) ; 7.81 (d,lH) ; 7.68 (m,6H) ; 7.46 (t,lH) ; 7.00 (s,lH) ; 6.46 (s,lH) ; 3.94 (s,3H) ; 3.51 (t,2H) ; 2.79 (m,4H) ; 2.68 (t,2H) ; 1.97 (s,3H) ; 1.81 (Qu,2H) ; 1.66 (Qu,2H) ) ; 1.54 (Qu,2H).
Example B (Z)-2-((2-(5-((2,5-dioxopyrrolidin-l-yl)oxy)-5-oxopentyl)-l,6- dimethylpyrimidin-4(lH)-ylidene)methyl)-3- methylbenzo[d]thiazol-3-ium trifluoroacetate, 1.2, TFA
 a) Preparation of 5-(4,6-dimethylpyrimidin-2-yl)pentanoic acid
a) Preparation of 5-(4,6-dimethylpyrimidin-2-yl)pentanoic acid

Pentane-2, 4-dione (6.0 g, 20.6 mmol) was poured in a round bottomed flask of 250 mL with 150 mL of water, potassium carbonate (12.0 g, 86.8 mmol) and the methyl 6-amino-6-iminohexanoate (6.0 g, 60.0 mmol). The mixture was then stirred at room temperature for 6 days. The solvent was evaporated and the crude product obtained was purified by flash chromatography with C18 column of 80 g (eluent A 10 mM TFA in water, eluent B 10 mM TFA in 90% ACN and 10 % water, gradient 10 to 22 % of eluent B in 17 min), leading to compound (5). Yield = 10 %.
Mass spectrometry (ESI-Q (+)): M/Z= 209.13.
b) Preparation of 2-(4-carboxybutyl)-l,4,6-trimethylpyrimidin- 1-ium, I’ (6)
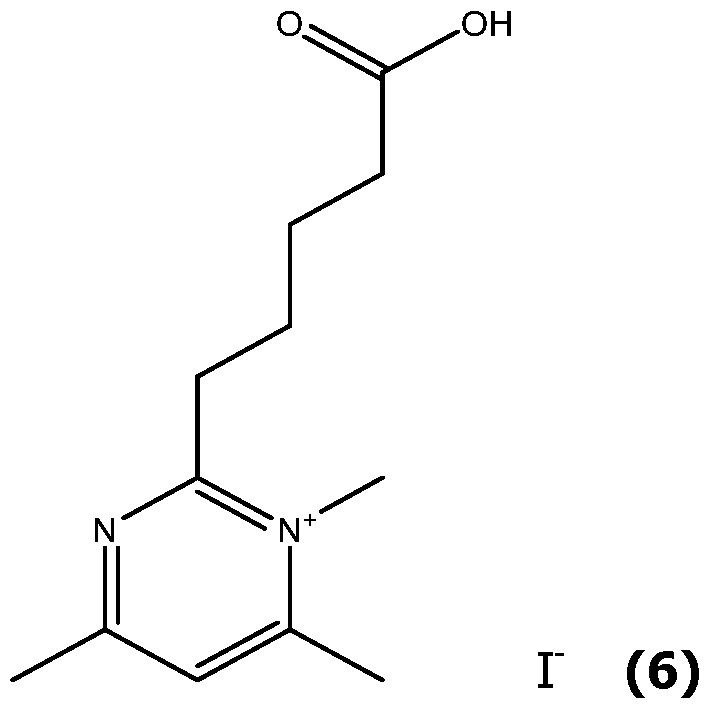
Compound (5) (100 mg, 480 pmol) was poured in a screwed glass tube of 15 mL with 2 mL of acetonitrile and methyl iodide (800 pL, 1.824 g, 12.8 mmol). The tube was screwed tightly and the mixture was then stirred at 85 °C during 67 h. The solvent was evaporated and the compound (6) was obtained. The selectivity of the methylation comes from the acidic pH used during the reaction brought by compound (5) which was purified with TFA eluents. Yield = 98 %.
Mass spectrometry (ESI-Q (+)): M/Z= 223.14 c) Preparation of 2-((2-(4-carboxybutyl)-l,6-dimethylpyrimidin- 4(lH)-ylidene)methyl)-3-methylbenzo[d]thiazol-3-ium, TFA'
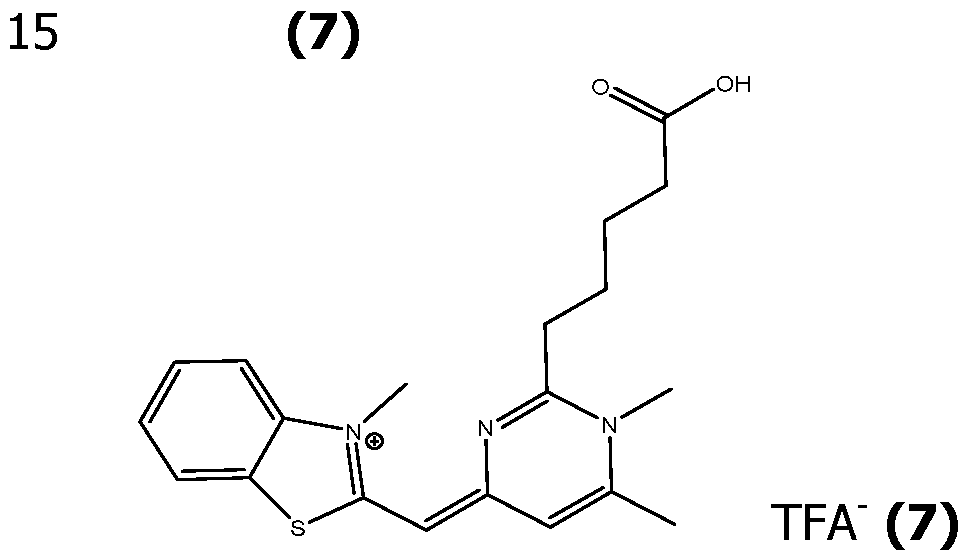
3-Methyl-2-(methylthio)benzo[d]thiazol-3-ium tosylate (compound (1), 150 mg, 765 pmol) was poured in a round bottomed flask of 50 mL with 10 mL of a solution of 2-(4-carboxybutyl)-l,4,6-trimethylpyrimidin-l-ium at 20 mg/mL in acetonitrile (compound (6), 200 mg, 486 pmol). 52 mL of acetonitrile and 4.78 mL of ethyl alcohol were successively added in the round bottomed flask. Finally, 210 pL of triethylamine (152.4 mg, 1.5 mmol) were dropped. The mixture was then stirred at room temperature for 25 minutes. The solvents were evaporated and the crude product obtained was purified by flash chromatography with C18 column of 40 g (eluent A 10 mM
TFA in water, eluent B 10 mM TFA in 90% ACN and 10 % water, gradient 0 to 70 % of eluent B in 21 min), resulting compound (7), as trifluoracetate salt. Yield = 12 %.
Mass spectrometry (ESI-Q (+)): M/Z= 478.1.

Compound (7) (52.0 mg, 151 pmol) was poured in a round bottomed flask of 25 mL with 5.6 mL of anhydrous acetonitrile, N,N'-disuccinimidyl carbonate (602 mg, 2.35 mmol) and pyridine ( 189 pL, 186 mg, 2.35 mmol). The mixture was then stirred at 90°C for 2 hours. The solvent was evaporated by using a rotary evaporator. Then, the crude product was purified by flash chromatography with C18 column of 4 g (eluent A : 10 mM TFA in water, eluent B : 10 mM TFA in 90% ACN and 10 % water, gradient 10 to 80 % of eluent B in 30 min) and led to compound 1.2, TFA'. Yield = 70 %.
Mass spectrometry (ESI-Q (+)): M/Z= 467.2.
Example C 6-acetamido-2-((2-((6-((2,5-dioxopyrrolidin-l-yl)oxy)-6- oxohexyl)thio)-6-methyl-l-phenylpyrimidin-4(lH)- ylidene)methyl)-3-methylbenzo[d]thiazol-3-ium, 1.6, TFA
 1.6, TFA a) Preparation of N-(2-(methylthio)benzo[d]thiazol-6- yl)acetamide (8)
1.6, TFA a) Preparation of N-(2-(methylthio)benzo[d]thiazol-6- yl)acetamide (8)
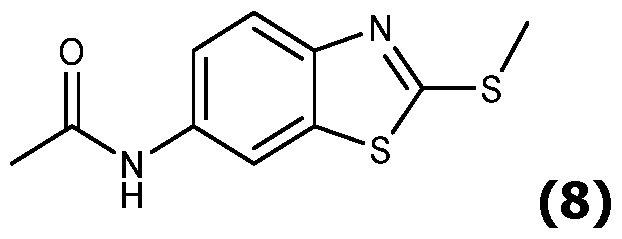
2-(Methylthio)benzo[d]thiazol-6-amine-methane (1.842 g, 9.4 mmol) was poured in a round bottomed flask of 50 mL with 30 mL of DCM and acetic anhydride (1.151 g, 11.3 mmol). The mixture was then stirred at room temperature for 24 h. The mixture was evaporated by using a rotary evaporator. Yield = 92 %.
Mass spectrometry (ESI-Q (+)): M/Z= 239.1 7.75 (d,lH) ; 7.17
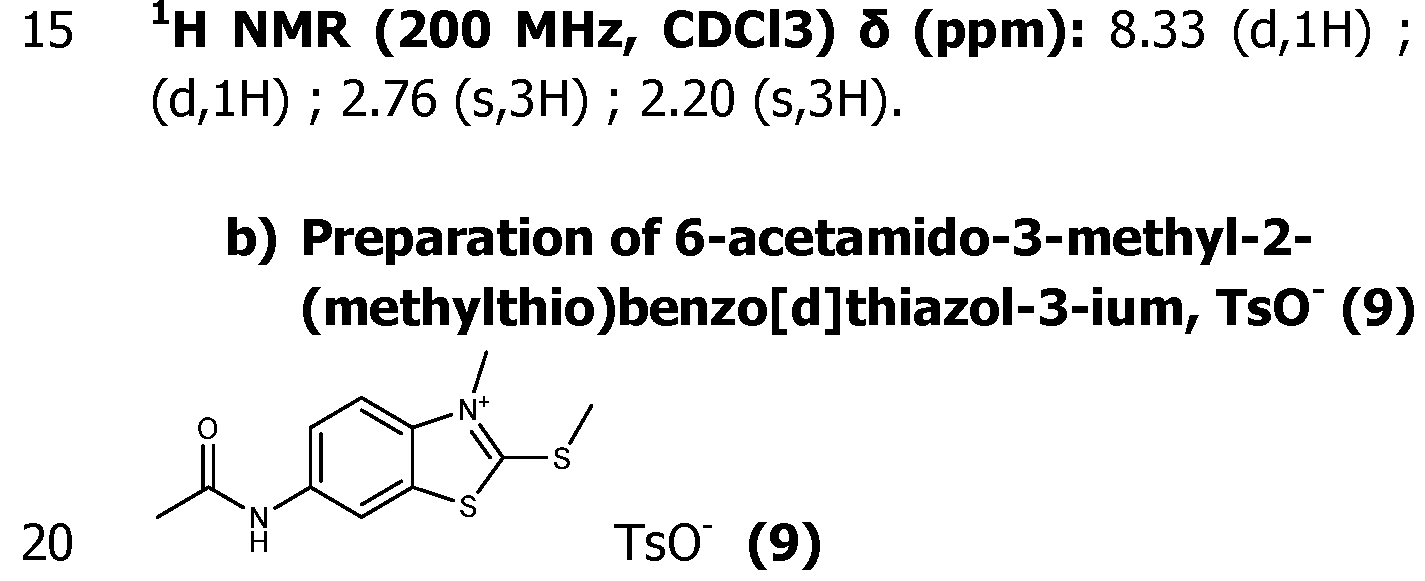
Compound (8) (2.065 g, 8.66 mmol) was poured in a round bottomed flask with p-toluenesulfonic acid monohydrate (1.780 g, 9.53 mmol). The mixture was then stirred at 130 °C for 3 h. The heating was turned off and was cooled down to room temperature then the crude product was triturated with 20 mL of acetone and was triturated 4 more times until obtaining a red powder. The residue of solvent was evaporated by using a rotary evaporator. Yield = 74 %.
Mass spectrometry (ESI-Q (+)): M/Z= 253.1
XH NMR (200 MHz, D2O) 5 (ppm): 8.1 (d,lH) ; 7.57 (d,lH) ; 7.44 (d, 1H) ; 7. 38 (d, 2H) ; 7.04 (d, 2H) ; 3.79 (s, 3H) ; 2.86 (s, 3H) ; 2.12 (s, 3H) ; 2.06 (s, 3H).
RMN 13C (50 MHz ; D2O) 5 (ppm): 180.07 ; 172.69 ; 142.02 ; 139.37 ; 138.56 ; 136.63 ; 129.12 ; 128.68 ; 125.06 ; 121.80 ; 115.15 ; 113.50 ; 35.80 ; 23.10 ; 20.34 ; 17.49. c) Preparation of 6-acetamido-2-((2-((5-carboxypentyl)thio)-6- methyl-l-phenylpyrimidin-4(lH)-ylidene)methyl)-3- methylbenzo[d]thiazol-3-ium, TFA' (10)
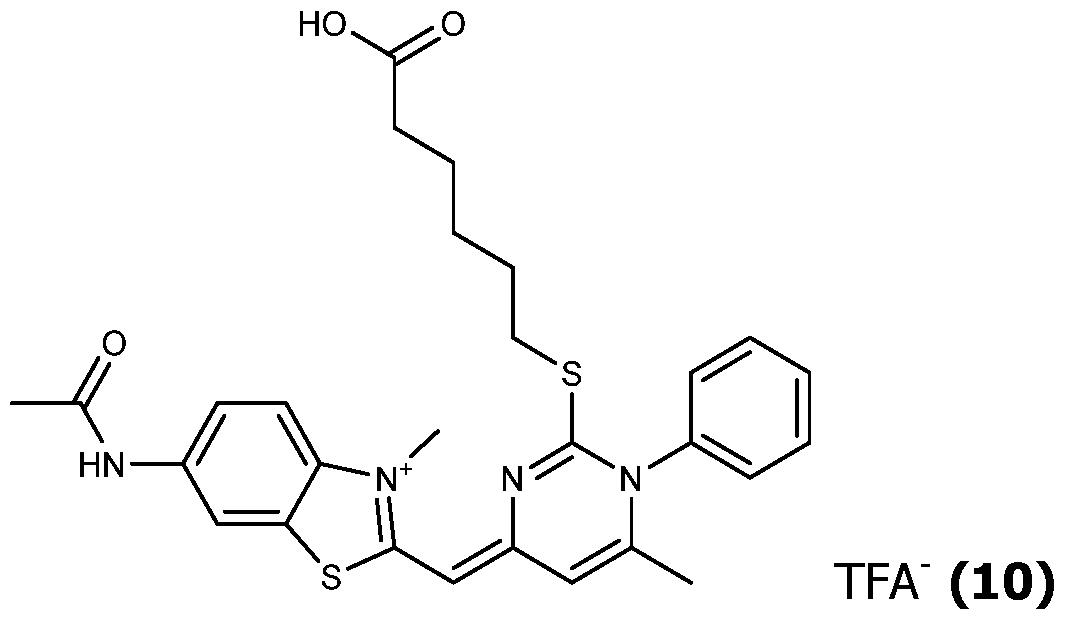
2-((5-Carboxypentyl)thio)-4,6-dimethyl-l-phenylpyrimidin-l-ium (compound (3), 200 mg, 603 pmol) was poured in a round bottomed flask of 25 mL with compound (9) (200 mg, 789 pmol). 8 mL of acetonitrile and 0.5 mL of ethyl alcohol were successively added in the round bottomed flask. Finally, 135.5 pL of triethylamine (98.4 mg, 2 mmol) were dropped. The mixture was then stirred at room temperature for 20 minutes. The solvents were evaporated and the crude product obtained was purified by flash chromatography with C18 column of 20 g (eluent A 10 mM TFA in water, eluent B 10 mM TFA in 90% ACN and 10 % water, gradient 10 to 20 % of eluent B in 10 min, then 20 to 100 % of eluent B in 20 min), was leaded to compound (10), as a trifluoroacetate salt. Yield = 40 %.
Mass spectrometry (ESI-Q (+)): M/Z= 535.2
d) Preparation of 1.6, TFA

Compound (10) (98 mg, 151 pmol) was poured in a screwed glass tube of 50 mL with 9.8 mL of anhydrous acetonitrile, N,N'-disuccinimidyl carbonate (580 mg, 2.27 mmol) and pyridine (182.5 pL, 179 mg, 2.27 mmol). The mixture was then stirred at 90°C for 40 min. The solvents were evaporated by using a rotary evaporator. Then, the crude product was purified by flash chromatography with C18 column of 12 g (eluent A 10 mM TFA in water, eluent B 10 mM TFA in 90% ACN and 10 % water, gradient 10 to 30 % of eluent B in 10 min, then 30 to 80 % of eluent B in 9 min), leading to compound 1.6, TFA'. Yield = 76 %.
Mass spectrometry (ESI-Q (+)) : M/Z= 632.2.
Example D 2-((lE,3Z)-3-(2-((6-((2,5-dioxopyrrolidin-l-yl)oxy)-6- oxohexyl)thio)-6-methyl-l-phenylpyrimidin-4(lH)-ylidene)prop-l- en-l-yl)-3-methylbenzo[d]thiazol-3-ium trifluoroacetate, 1.4, TFA'

2-Methylbenzothiazole (1 g, 6.63 mmol) was put in a round bottomed flask
of 100 mL and methyl p-toluenesulfonate (9.9 g, 52.8 mmol) was added. The mixture was stirred and heated at 145°C for 2 hours. The mixture was then left to cool down to room temperature. 50 mL of diethyl ether were added, the crude product was precipitated, supernatant was discarded and the obtained solid was washed 3 more times in 50 mL of diethyl ether. Then, the obtained precipitate was washed 3 times with 50 mL of acetone. The obtained solid was dried by evaporating the rest of solvent with a rotary evaporator to obtain a grey powder (2.15 g, yield = 96 %), corresponding to compound (11) as a tosylate salt.
XH NMR (D2O, 400 MHz) : 5 (ppm) 8.05 (d, 1H, Hl), 7.88 (d, 1H, H2), 7.76 (td, 1H, H3), 7.64 (td, 1H, H4), 7.58 (d, 2H, HorthoTsO ), 7.26 (d, 2H, Hmeta TsO ), 4.02 (s, 3H, H6), 3.00 (s, 3H, H5), 2.30 (s, 3H, CH3-TsO ).
Mass spectrometry (ESI-Q (+)) : M/Z= 164.1 b) Preparation of 3-methyl-2-(2-
(phenylamino)vinyl)benzo[d]thiazol-3-ium, TsO' (12)
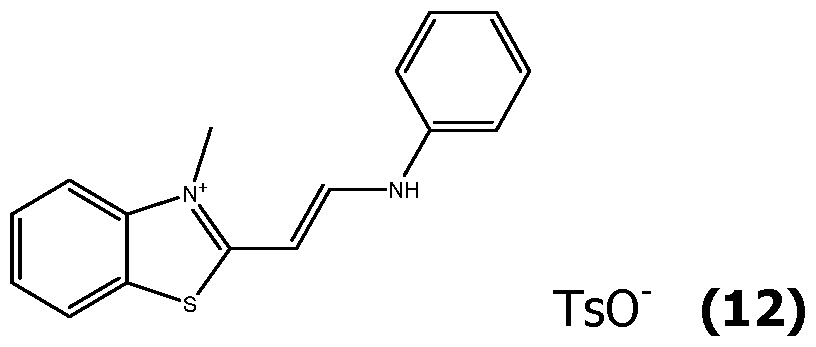
2,3-Dimethylbenzo[d]thiazol-3-ium, tosylate (11) (167.9 mg, 0.50 mmol) was put in a round bottomed flask of 50 mL and N,N'-diphenylformamidine (196.2 mg, 1.00 mmol) was added. The mixture was stirred and heated at 150°C (fusion reaction) for 2 hours and 30 minutes. The mixture was then left to cool down to room temperature. Compound (12) as a tosylate salt was obtained after purification, by liquid extraction in water I dichloromethane and evaporation of the organic phase before final purification by reverse flash chromatography (eluent A 10 mM TFA in water, eluent B 10 mM TFA in 90% ACN and 10 % water. Gradient 20 to 100 % of eluent B in 30 minutes). Yield = 67 %.
Mass spectrometry (ESI-Q (+)): M/Z= 267.2
c) Preparation of 2-((lE,3Z)-3-(2-((5-carboxypentyl)thio)-6- methyl-l-phenylpyrimidin-4(lH)-ylidene)prop-l-en-l-yl)-3- methylbenzo[d]thiazol-3-ium, TFA (13)
 , poured in a round bottomed flask of 50 mL with compound (3) (20.6 mg, 50 pmol), 600 pL of acetic anhydride and 600 pL of pyridine. The mixture was then stirred at 50 °C for 10 min. The mixture was evaporated by using a rotary evaporator. Then, the crude product was purified by flash chromatography with C18 column of 4 g (eluent A : 10 mM TFA in water, eluent B : 10 mM TFA in 90% ACN and 10 % water, gradient 20 to 100 % of eluent B in 20 min) and leaded to compound (13) as a trifluoacetate salt. Yield = 37 %.
, poured in a round bottomed flask of 50 mL with compound (3) (20.6 mg, 50 pmol), 600 pL of acetic anhydride and 600 pL of pyridine. The mixture was then stirred at 50 °C for 10 min. The mixture was evaporated by using a rotary evaporator. Then, the crude product was purified by flash chromatography with C18 column of 4 g (eluent A : 10 mM TFA in water, eluent B : 10 mM TFA in 90% ACN and 10 % water, gradient 20 to 100 % of eluent B in 20 min) and leaded to compound (13) as a trifluoacetate salt. Yield = 37 %.
Mass spectrometry (ESI-Q (+)) : M/Z= 504.2

100 mL with of anhydrous acetonitrile, N,N'-disuccinimidyl carbonate (71.5 mg, 279 pmol) and pyridine ( 22.5 pL, 22.1 mg, 279 pmol). The mixture was then stirred at 90°C for 2 hours. The solvent was evaporated by using a rotary evaporator for tube. Then, the crude product was purified by flash chromatography with C18 column of 4 g (eluent A : 10 mM TFA in water, eluent B : 10 mM TFA in 90% ACN and 10 % water, gradient 10 to 80 % of eluent B in 10 min and then 100 % of eluent B during 10 min) and leaded to compound 1.4, TFA'. Yield = 37 %.
Mass spectrometry (ESI-Q (+)): M/Z= 601.1
Example E
Preparation of 2-(-5-(2-((6-((2,5-dioxopyrrolidin-l-yl)oxy)-6- oxohexyl)thio)-6-methyl-l-phenylpyrimidin-4(lH)-ylidene)penta- l,3-dien-l-yl)-3-methylbenzo[d]thiazol-3-ium, 1.5, TFA
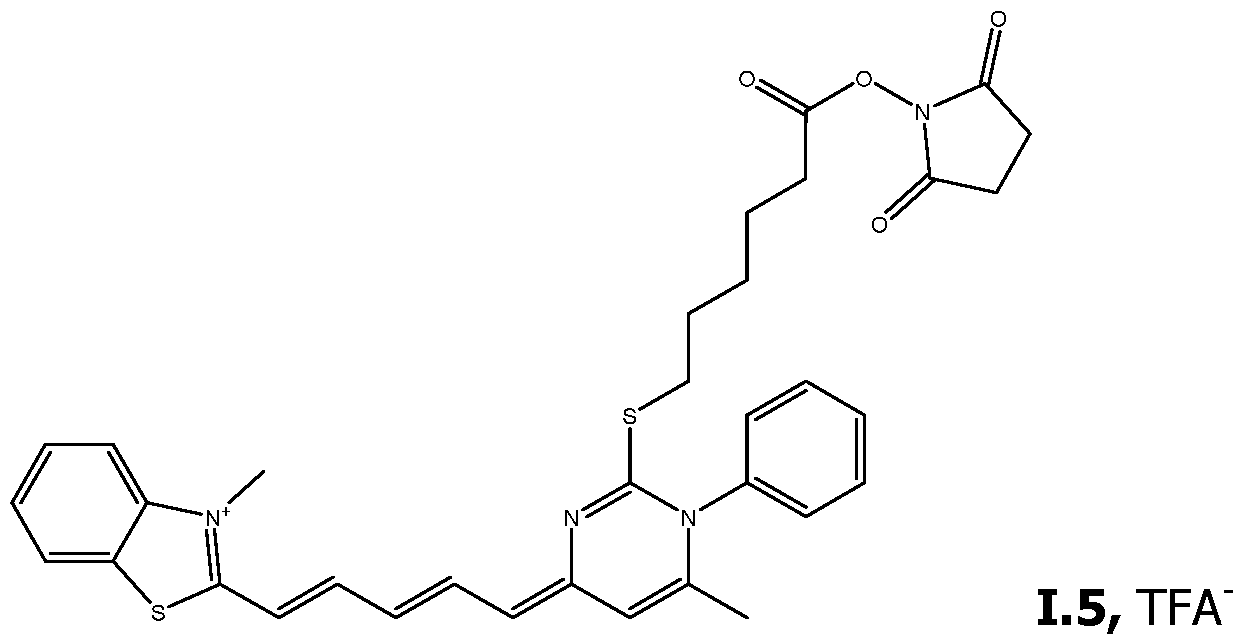 a) Preparation of 3-methyl-2-(4-(N-phenylacetamido)buta-l,3~ zol-3-ium, TsO (14)
a) Preparation of 3-methyl-2-(4-(N-phenylacetamido)buta-l,3~ zol-3-ium, TsO (14)
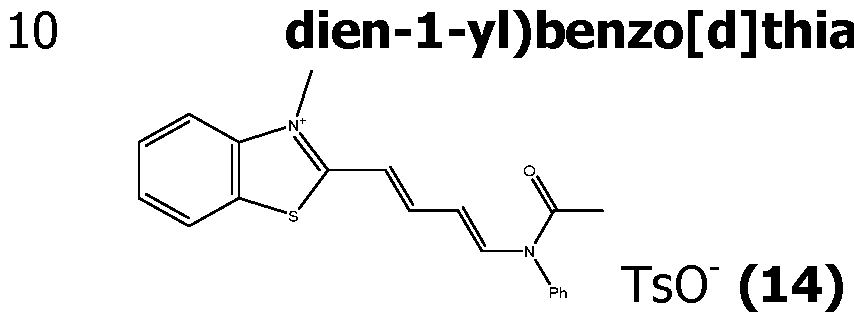
Compound (11) (1 g, 3.0 mmol) prepared previously was put in a round bottomed flask of 25 mL with malonaldehyde bis(phenylimine) monohydrochloride (1.1 g, 4.3 mmol). The mixture was stirred and heated at 115 °C during 2 h. The mixture was then left to cool down to room temperature and the crude product was purified by liquid extraction in water I ACN 90/10 I ethyl acetate. The organic phase was washed several times with water and then was evaporated with a rotary evaporator.
Mass spectrometry (ESI-Q (+)): M/Z= 335.1
b) Preparation of 2-(5-(2-((5-carboxypentyl)thio)-6-methyl-l- phenylpyrimidin-4(lH)-ylidene)penta-l,3-dien-l-yl)-3- methylbenzo[d]thiazol-3-ium, TFA' (15)
 , oured in a round bottomed flask of 25 mL with compound (3) (314 mg, 743 pmol), 500 pL of ethanol and 5 mL of acetonitrile. Finally, triethylamine (194 pL, 141 mg, 139 pmol) was dropped. The mixture was then stirred at room temperature for 20 minutes. Then trifluoroacetic acid solution at 500 mM (3.35 mL, 1.67 mmol) was added for neutralizing triethylamine. The solvents were evaporated and the crude product obtained was purified by flash chromatography with C18 column of 20 g (eluent A 10 mM TFA in water, eluent B 10 mM TFA in 90% ACN and 10 % water, gradient 20 to 90 % of eluent B in 18 min) was leaded to compound (15), trifluoroacetate salt. Yield = 48 %.
, oured in a round bottomed flask of 25 mL with compound (3) (314 mg, 743 pmol), 500 pL of ethanol and 5 mL of acetonitrile. Finally, triethylamine (194 pL, 141 mg, 139 pmol) was dropped. The mixture was then stirred at room temperature for 20 minutes. Then trifluoroacetic acid solution at 500 mM (3.35 mL, 1.67 mmol) was added for neutralizing triethylamine. The solvents were evaporated and the crude product obtained was purified by flash chromatography with C18 column of 20 g (eluent A 10 mM TFA in water, eluent B 10 mM TFA in 90% ACN and 10 % water, gradient 20 to 90 % of eluent B in 18 min) was leaded to compound (15), trifluoroacetate salt. Yield = 48 %.
Mass spectrometry (ESI-Q (+)): M/Z= 530.19 c) Preparation of 1.5, TFA"
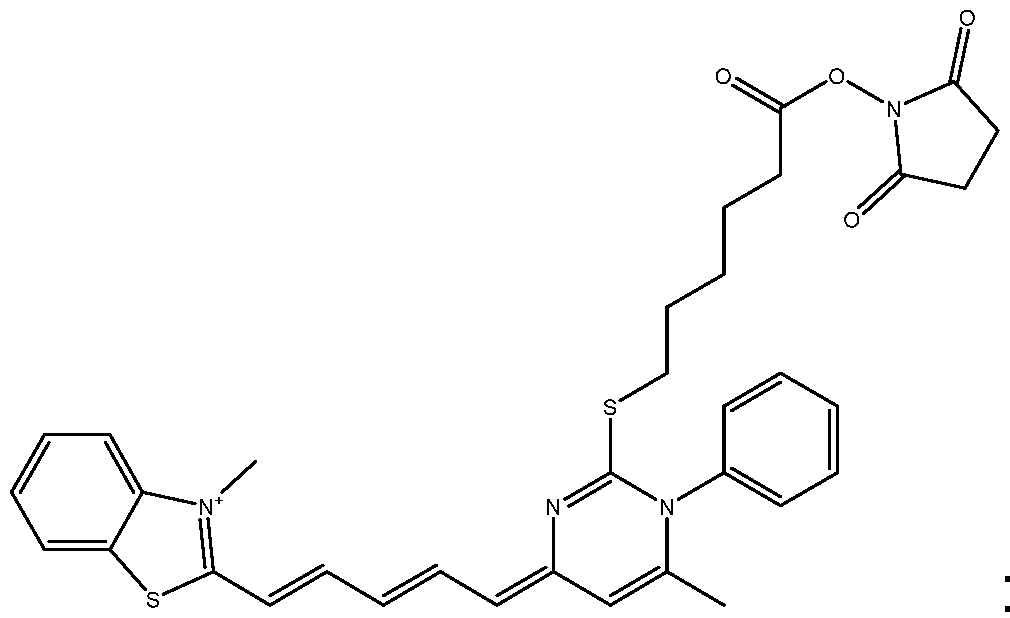 .5, TFA
.5, TFA
Compound (15) (206.5 mg ; 321 pmol) was poured in a round bottomed flask of 25 mL with 10 mL of anhydrous acetonitrile, N,N'-disuccinimidyl carbonate (1.23 g ; 4.81 pmol) and pyridine ( 387 pL ; 380 mg ; 4.81 mmol). The mixture was then stirred at 90°C for 2 hours. The solvent was evaporated by using a rotary evaporator for tube. Then, the crude product was purified by flash chromatography with C18 column of 4 g (eluent A : 10 mM TFA in water, eluent B : 10 mM TFA in 90% ACN and 10 % water, gradient 10 to 100 % of eluent B in 15 min and then 100 % of eluent B
during 10 min) leading to compound 1.5, TFA'. Yield = 15 %.
Mass spectrometry (ESI-Q (+)): M/Z= 627.2.
Example F Preparation of 2-((l-(3-(((2,5-dioxopyrrolidin-l- yl)oxy)carbonyl)phenyl)-6-methyl-2-(methylthio)pyrimidin-4(lH)- ylidene)methyl)-3-methylbenzo[d]thiazol-3-ium, 1.3, TFA
 a) Preparation of ethyl 3-(4,6-dimethyl-2-thioxopyrimidin-
a) Preparation of ethyl 3-(4,6-dimethyl-2-thioxopyrimidin-

3-(Carbamothioylamino)benzoic acid (0.50 g, 2.55 mmol) was poured in ethanol (5.04 mL, 0.5053 M) and pentane-2, 4-dione (0.31 mL, 3.02 mmol) in a round bottomed flask of 25 mL. HCI 37% (0.64 mL, 7.68 mmol) was added drop by drop in the mixture. Then the reaction mixture was heated to reflux for 4.5 h then allowed to cool down to room temperature overnight. Reaction mixture was poured into Et20 and stirred for 30 min. Yellow solid was filtered, washed with Et20. Solid was taken up in DCM and treated with aq. 2N NaOH and DCM. Aqueous layer was separated and washed with DCM (2x). Combined organics were dried (Na2SO4), filtered off and concentrated under reduced pressure. The compound (16) was obtained after drying (382 mg, 52 %, Brown Solid).
Mass spectrometry (ESI-Q (+)) : [M+H+]/Z= 289.1 b) Preparation of l-(3-(ethoxycarbonyl)phenyl)-4,6-dimethyl-2- ((3-trimethylammonio)propyl)thio) pyrimidin-l-ium, 2 Br-
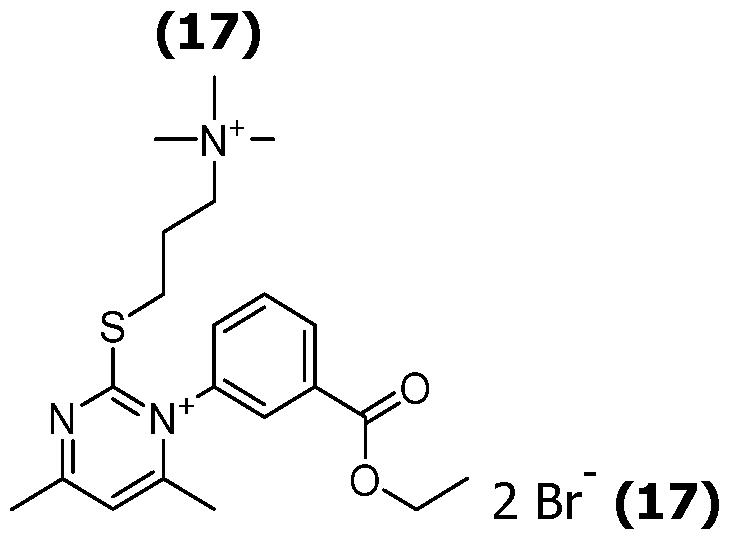 A mixture of and (3-bromopropyl)trimethylammonium bromide (300 mg ; 1.15 mmol) and compound (16) (328.7 mg ; 1.14 mmol) in acetonitrile ( 7.66 mL, 0.1500 M) was stirred at reflux (heat block at 90-95°C) for 4 h. Reaction mixture was concentrated under vacuum and partitioned between water and DCM. Aqueous layer was washed 3 times with DCM. DCM was back extracted with water (3x). Aqueous layer was concentrated under reduced pressure and co-evaporated twice with acetonitrile to give a sticky brown oil/solid. The compound (17) (642 mg, 1.07 mmol) was obtained with the purity of 91.5 %.
A mixture of and (3-bromopropyl)trimethylammonium bromide (300 mg ; 1.15 mmol) and compound (16) (328.7 mg ; 1.14 mmol) in acetonitrile ( 7.66 mL, 0.1500 M) was stirred at reflux (heat block at 90-95°C) for 4 h. Reaction mixture was concentrated under vacuum and partitioned between water and DCM. Aqueous layer was washed 3 times with DCM. DCM was back extracted with water (3x). Aqueous layer was concentrated under reduced pressure and co-evaporated twice with acetonitrile to give a sticky brown oil/solid. The compound (17) (642 mg, 1.07 mmol) was obtained with the purity of 91.5 %.
Mass spectrometry (ESI-Q (+)): M/Z= 194.6 c) Preparation of l-(3-carboxyphenyl)-4,6-dimethyl-2-((3- (trirnethylammonio)propyl)thio) pyrimidin-l-ium, 2 Br (18)
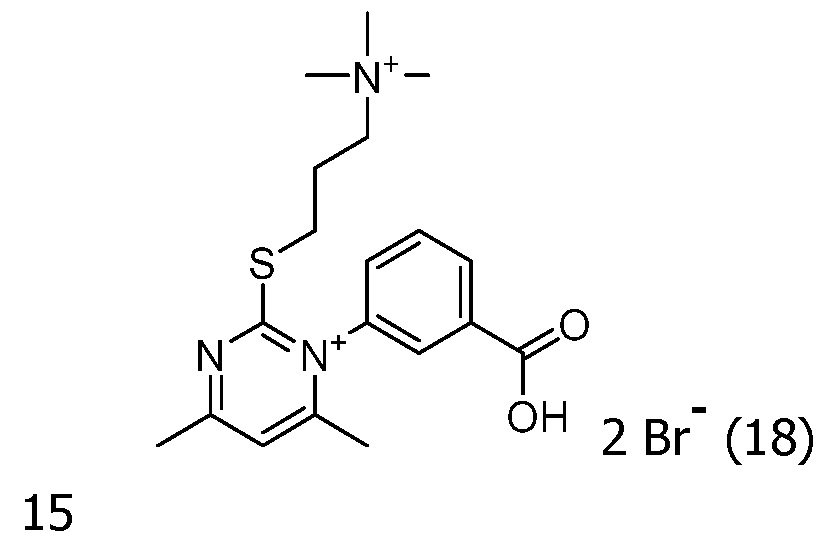
A mixture of compound (17) (120 mg, 0.197 mmol) in 1/1 of 1,4-dioxane (1 mL, 0.0983 M) and water (1 mL, 0.0983 M) was treated with HCI 37% (0.25 mL, 2.95 mmol) and heated at 100°C for 12 h. The residue was diluted with water and washed with DCM. The aqueous layer was evaporated under reduce pressure to give compound (18).
Mass spectrometry (ESI-Q (+)): M/Z= 181.6 d) Preparation of 2-((l-(3-carboxyphenyl)-6-methyl-2- (methylthio)pyrimidin-4(lH)-ylidene)methyl)-3- methylbenzo[d]thiazol-3-ium, TFA (19)

Compound (19) (13.2 mg; yield 15 %) was obtained and purified in the same way as for compound (4) using the benzothiazole (1) and the pyrimidinium salt (18). During the reaction, the trimethylammonium arm was substituted in situ by the methane thiol released during the cyanine
synthesis.
Mass spectrometry (ESI-Q (+)): M/Z= 422.1 d) Preparation of 1.3, TFA'

Compound 1.3, TFA" (13.9 mg; yield 89 %) was prepared from compound (19) in the same way as for compound 1.1.
Mass spectrometry (ESI-Q (+)): M/Z= 519.2
The comparative compounds (Comp.) 1 to 3 were prepared according to similar protocols and obtained as trifluoroacetate salts.
B. PREPARATION OF PROBES PR
The probes PR presented in Table 4 were synthesized. They include an oligonucleotide covalently linked to one or several compounds FC (called dyes hereafter) according to the invention. Comparative probes obtained by coupling to the multi-cationic dyes (comparatives 1 to 3 in Table 4) were also prepared.
The following oligonucleotides sequences represented from 5'-> 3' were used (Oligo SEQ ID° in Table 4):
SEQ ID N°l : CGCCAGAAGAGGAGCCCCAAT
SEQ ID N°lbis also called SEQ ID N°1 (LNA 1): CGCCAAAAAAGAAGCCCCAAT (underlined means a LNA nucleoside)
SEQ ID N°lter also called SEQ ID N°1 (LNA 2): CGCCAGAAGAGGAGCCCCAAT (underlined means a LNA nucleoside)
SEQ ID N°2: CTGGTACCGCTGAGATTAAACAACC
SEQ ID N°3: ACGGCGATTCCGCAGTTTACGA
SEQ ID N°4: TAGCCTTCTGTCGCAGCCGTCT
SEQ ID N°5: ATTGACATACCGATCCTTCAC
SEQ ID N°6: ATGGTGTTGAAGGTTT
SEQ ID N°7: ATGGTGTTAAAGGTTT
SEQ ID N°8: ATGGTGTTAAAGGTTT
SEQ ID N°9 : ATGGTGTTGAAGGTTT
SEQ ID N°10: GGATTGCGGTTACAGCATT
SEQ ID N°ll: GGTTGTTTAATCTCAGCGGTACCAG
SEQ ID N°12: CAACGGGACGGAAAGACCCCG
SEQ ID N°13: CAACGGGACGGGAAGACCCCG
>
SEQ ID N°14: TGCTTCCTTCATGGAAAGTTATGTCGA
SEQ ID N°15: TTGTTTAATCTCAGCGGTACCAGC
SEQ ID N°16: CTGGTAGCCTGTTCGCTTCAT
SEQ ID N°17 also called SEQ ID N°1 (AUTO 1): GCATGCCCGCCAGAAGAGGAGCCCCAATGGCATGC (underlined means a self- complementary sequence)
SEQ ID N°18 also called SEQ ID N°1 (AUTO 2): CCAGGCCGCCAGAAGAGGAGCCCCAATGCCTGG (underlined means a self- complementary sequence)
SEQ ID N°31: AGCACGTGGGAGGGCGATCG
SEQ ID N°33: CGAAGTGATCCATGTAAGCC
SEQ ID N°34: TCAGAACCGTCTCGGAAGG
In the sequences corresponding to SEQ ID N°7, 8 and 9, the underlined nucleotide corresponds to the mismatch with the target nucleic acid.
The sequences SEQ ID N°17 (SEQ ID N°1 (AUTO 1)) and SEQ ID N°18 (SEQ ID N°1 (AUTO 2)) were designated to evaluate the influence of two selfcomplementarity portions in the two 3' and 5' end zones on the fluorescent background of the probe alone that bears two coupled compound FC (so in the form of FC') at its 3' and 5' ends.
The synthetic target nucleic acids of the sequence below are complementary or partially complementary (presence of one mismatch) to the sequences of the probes and were used to exemplify the invention. They correspond to a portion of a sequence of a gene belonging to a real micro-organism.
SEQ ID N°19 comprising a sequence complementary to the oligonucleotides of SEQ ID N°l, Ibis, Iter, and complementary to a portion of the oligonucleotides of SEQ ID N°17 and 18:
AGATTGGTCCTCCATTCGCTTATTGGGGCTCCTCTTCTGGCGTGTCACACTCTGCA TGTT
SEQ ID N°20 comprising a sequence complementary to the oligonucleotide of SEQ ID N°2:
> AAGGAGAGATTTAACAACTGGTTGTTTAATCTCAGCGGTACCAGCATAAGTAGTG
TCTAA
SEQ ID N°21 comprising a sequence complementary to the oligonucleotide of SEQ ID N°3:
ATACGGACGATGGTGTCGTAAACTGCGGAATCGCCGTGGGGGTGGTATTTACCGA TGACGT
SEQ ID N°22 comprising a sequence complementary to the oligonucleotide of SEQ ID N°4: GTAGCAGTAGACGGCTGCGACAGAAGGCTAGCGGTAGGCGCGG SEQ ID N°23 comprising a sequence complementary to Probe SEQ ID N° 5: ACCGTTATGGATTTGGAGATGCAGCAGTCTAAAGTGAAGGATCGGTATGTCAATT TTCCT
SEQ ID N°24 comprising a sequence complementary to the oligonucleotide of SEQ ID N° 6, and partially complementary (presence of one mismatch) to the oligonucleotides of SEQ ID N°7, 8 and 9: TGTAAAGGAAAGTAACAATTAAAACCTTCAACACCATTACAAGGTGTGCTACCGGC CTGA
SEQ ID N°25 comprising a sequence complementary to the oligonucleotide of SEQ ID N°10: CAGTGCTTGCGGATGCGATAGTTGGAGCAGCAAATGCTGTAACCGCAATCCCAGC T
SEQ ID N°26 comprising a sequence complementary to the oligonucleotide of SEQ ID N°ll and 15: AAAAATTAGACACTACTTATGCTGGTACCGCTGAGATTAAACAACCAGTTGTTAAA TCTC
SEQ ID N°27 comprising a sequence complementary to the oligonucleotide of SEQ ID N°12: TACAGTAAAGCTTCACGGGGTCTTTCCGTCCCGTTGCGCCTAACGGGTGTCT SEQ ID N°28 complementary to Probe SEQ ID N°13: TACAGTAAAGCTTCACGGGGTCTTCCCGTCCCGTTGCGCCTAACGGGTGTCT
SEQ ID N°29 comprising a sequence complementary to the oligonucleotide of SEQ ID N°14: CTTTGATTTGTTCGACATAACTTTCCATGAAGGAAGCAATGTTTTCTTTACCGTTA GCGT
SEQ ID N°30 comprising a sequence complementary to the oligonucleotide of SEQ ID N°16: CGCATTCCAGAAATTGTTCCCAGTGCATAGATATGAAGCGAACAGGCTACCAGACA CA
SEQ ID N°32 comprising a sequence complementary to the oligonucleotide of SEQ ID N°31: I I I I I CGATCGCCCTCCCACGTGC I I I I I I
SEQ ID N°35 comprising a sequence complementary to the oligonucleotides of SEQ ID N°33 and 34: CGCATTCCTTCCGAGACGGTTCTGAATGGCTTACATGGATCACTTCGACACA
Therefore, in the experiments described hereinafter, the complementary target nucleic acids are used, for hybridization to the corresponding probes
(according to the invention or outside the invention and designated as Comparative Probes).
In Table 4, the covalent linkage resulting from the coupling of the reactive function (activated ester) present on Ri or R2 position of the compound FC (also named dye hereafter) and the corresponding reactive function present on the oligo are given in the sense dye->oligo. For an attachment in position 5', the linker corresponds to the chain between the covalent linkage and the phosphate in 5' (in the sense dye->oligo). For an attachment in position 3', the linker corresponds to the chain between the covalent linkage and the phosphate in 3' (in the sense dye->oligo). For an attachment in the internal region of the oligo, the linker corresponds to the chain between the covalent linkage and the base bearing the dye (in the sense dye->oligo). The covalent linkage between the dye and the linker (in the sense dye -> oligo) is always -CO-NH- in the prepared probes.
In Table 4, linker 1: -(CH2)6- ; linker 2: -(CH2)6-PO4— (CH2CH2O)e- ; linker 3: - (CH2)4- ; linker 4: -(CH2)4-CH(CH2OH)-CH2- ; linker 5: -(CH2)6-NH-C(O)- CH=CH- ; linker 6: -(CH2)6-NH-. The quencher used in probes 11.47 to 11.49 is BHQ1 (Black Hole Quencher 1, 2-[N-(2-hydroxyethyl)-4-[[2-methoxy-5- methyl-4-[(4-methyl-2-nitrophenyl)diazenyl]phenyl]diazenyl]anilino]ethanol).
Table 4
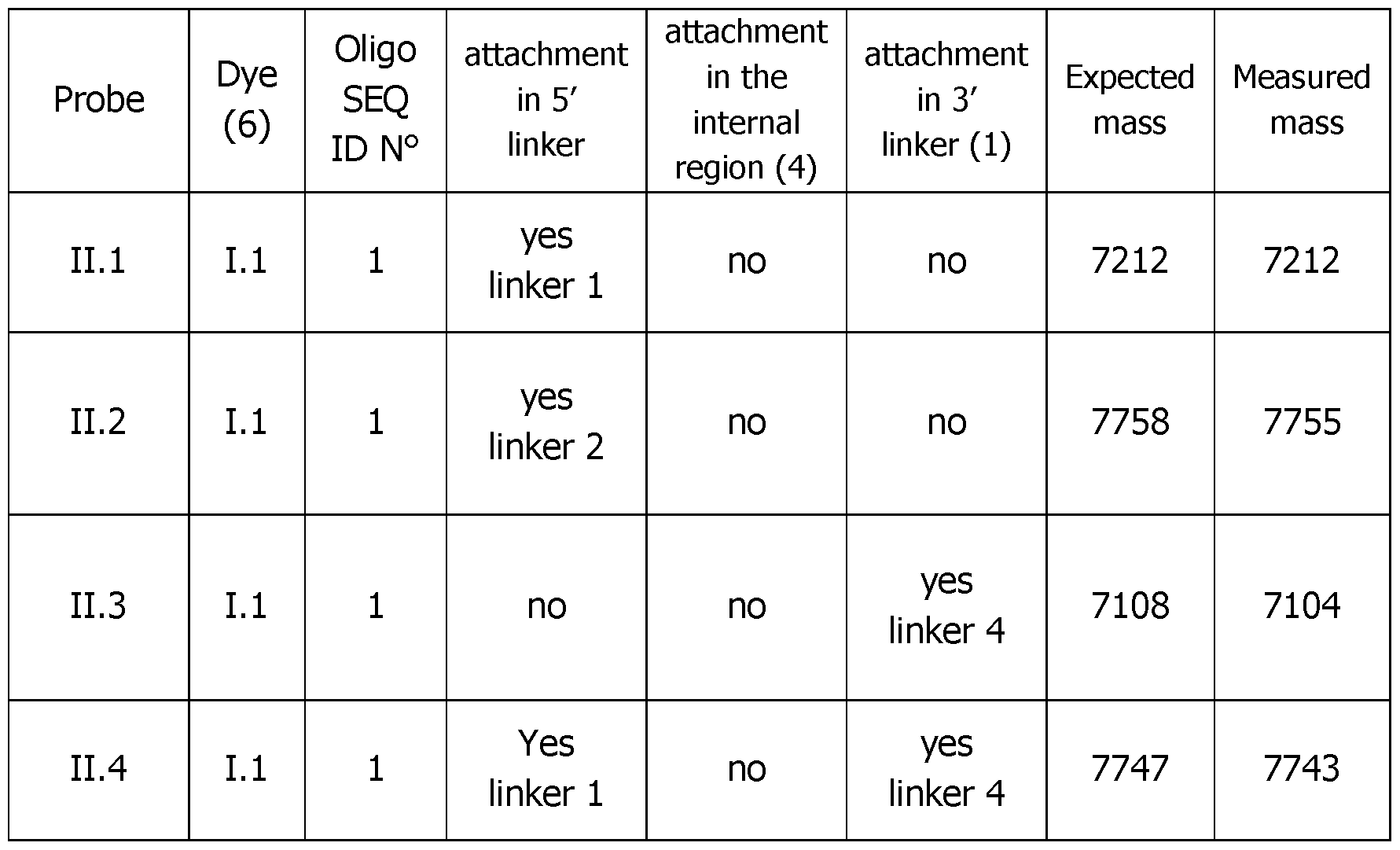
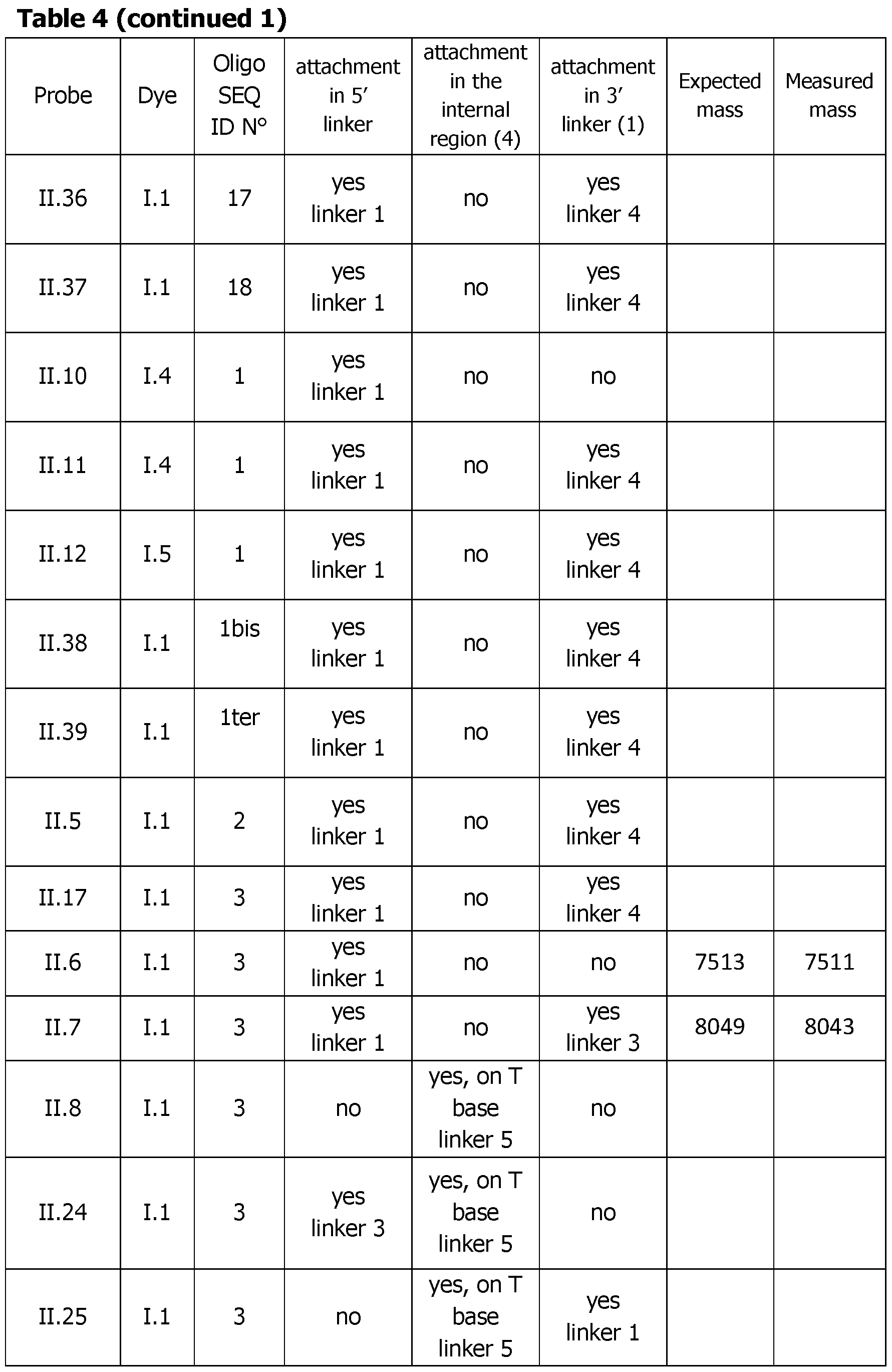 Table 4 (continued 2)
Table 4 (continued 2)
 Table 4 (continued 3)
Table 4 (continued 3)
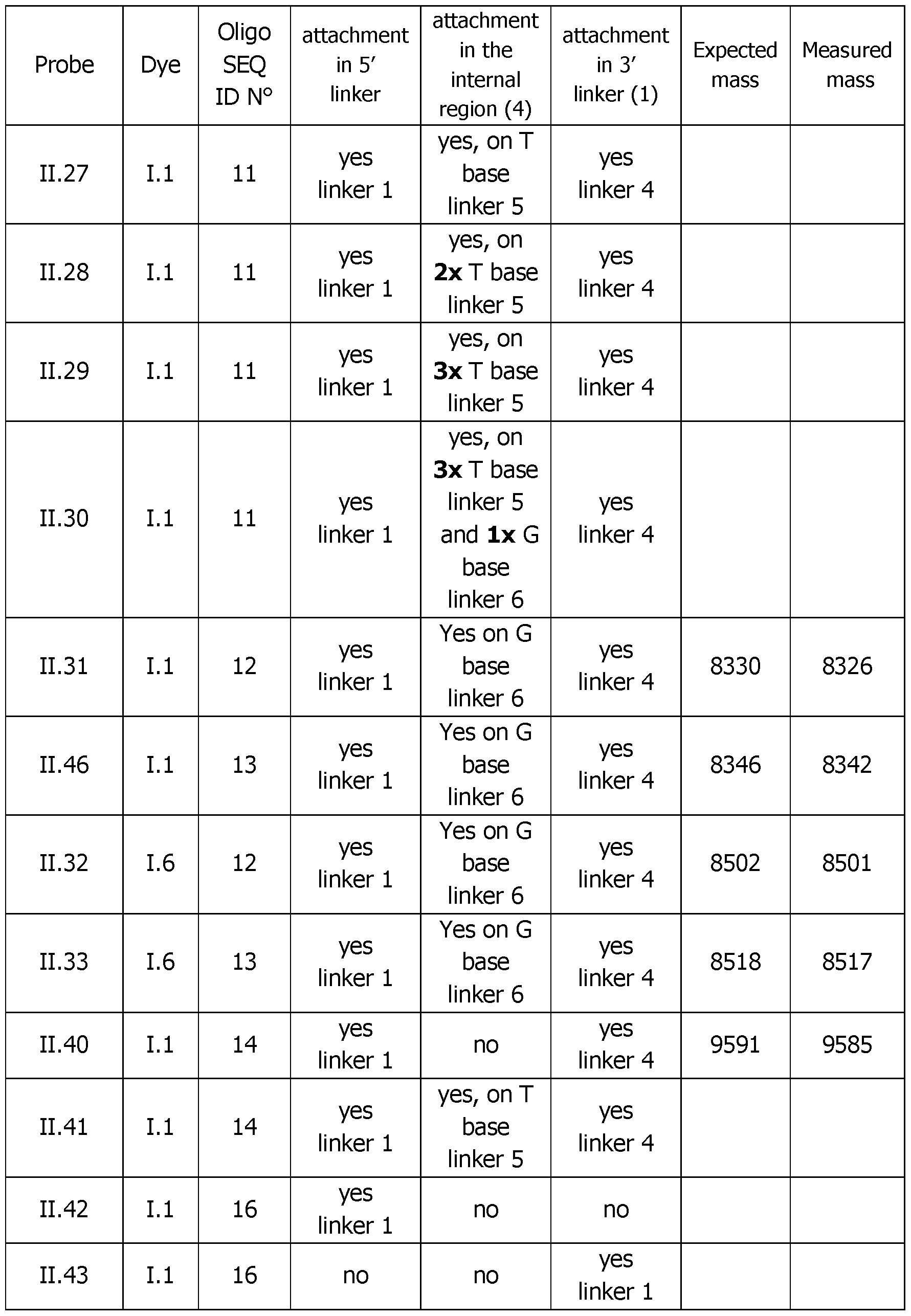 Table 4 (continued 4)
Table 4 (continued 4)
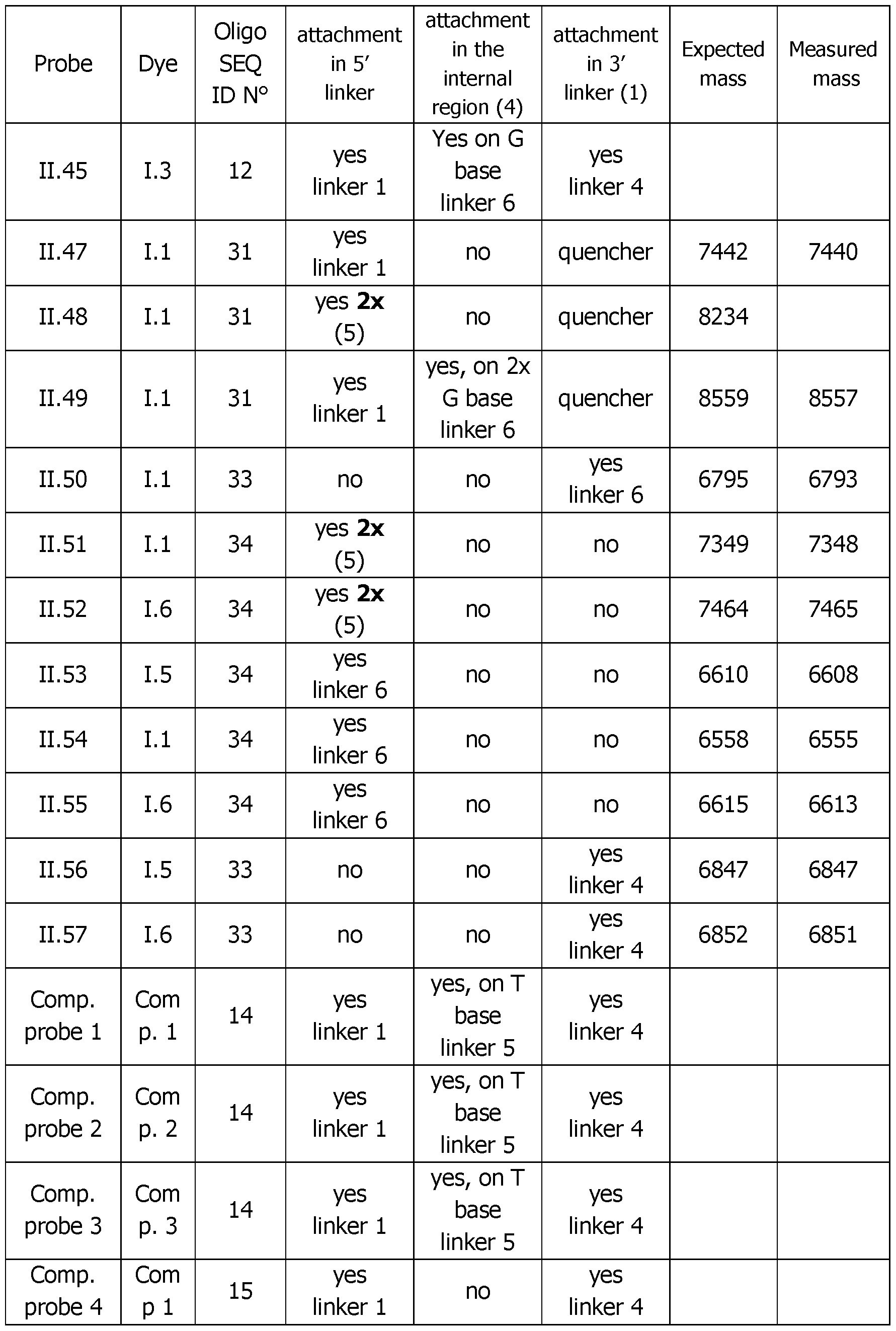 Table 4 (continued 5)
Table 4 (continued 5)

(1) when there is no dye attached in the 3' end, the 3' end is blocked to prevent extension during PCR. In that case, a blocking group was present on the 3' end, as previously described in the art (el91 Nucleic Acids Research, 2013, Vol. 41, No. 20 PAGE 4) by using a modified solid support (either a 3 - Spacer C3 CPG, Ref. 20-2913 from Glen Research, Sterling , USA ; or a 3 - Phosphate CPG, Ref. 20-2900 from Glen Research, Sterling , USA). When there is a dye attached at the 3' end, a modified solid support (3'-Amine C7 CPG, Ref. 20-2958 from Glen Research, Sterling, USA) was used to generate the amino group that will be further reacted with the NHS activated dye. When there is a quencher attached at the 3' end, the 3' BHQ1-CPG (Ref. 20- 5931 from Glen Research, Sterling, USA) was used.
(2) in that case, the oligonucleotide was produced by incorporating in 5' twice an Amino-Modifier Serinol Phosphoramidite (Ref. 10-1997 from Glen
Research, Sterling, USA) in order to generate two amino groups in 5'. Two
molecules of compound LI carrying an NHS group were then conjugated on these reactive groups. So, the probe obtained bears two molecules of compound 1.1 at the 5' end of its oligonucleotide.
(3) in that case, the oligonucleotide was produced by using a Symmetric Doubler Phosphoramidite (Ref. 10-1920 from Glen Research, Sterling, USA). The two functions 4,4'-dimethoxytrityloxy were after converted to -NH2 by reaction with an 5'-Amino-Modifier C6 Phosphoramidite (Ref. 10-1906 from Glen Research, Sterling, USA) as it is done for most of the probes described with an amino modification in 5' end for further reaction with the activated dye. Two molecules of compound 1.1 carrying an NHS group were after conjugated on these reactive groups -NH2. So, the probe obtained bears two molecules of compound 1.1 at the 5' end of its oligonucleotide.
(4) when there is an internal labeling, amino modified dT, dA or dG phosphoramidite was introduced instead of dT, dA or dG : respectively Ref. 10-1039 for the Amino-Modifier C6 dT phosphoramidite, Ref. 10-1089 for the Amino-Modifier C6 dA phosphoramidite and Ref. 10-1529 for the N2-Amino- Modifier C6 dG phosphoramidite from Glen Research, Sterling, USA. The generated amino groups were further conjugated with the activated NHS dye. The Table 4bis below shows the exact position of the internal modified nucleoside for dye attachment.
(5) in that case, the oligonucleotide was produced by using a symmetrical branching phosphoramidite (Ref. CLP-5215 from ChemGenes, USA). After, the same protocol as described under (3) was used: a probe bearing two molecules of compound 1.1 at the 5' end of its sequence was obtained.
(6) used as TFA salt.
Table 4bis

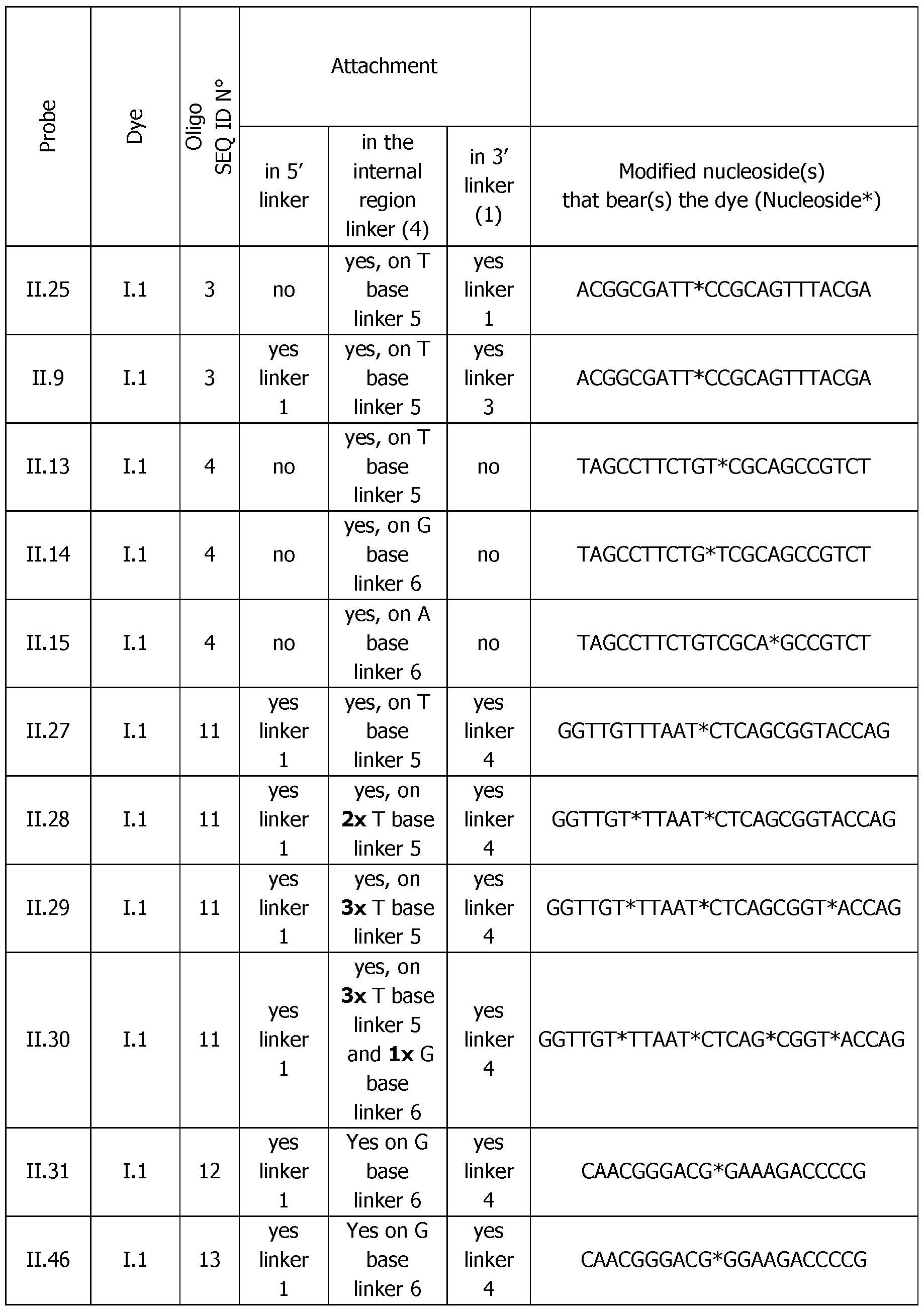
Table 4bis (continued 2)
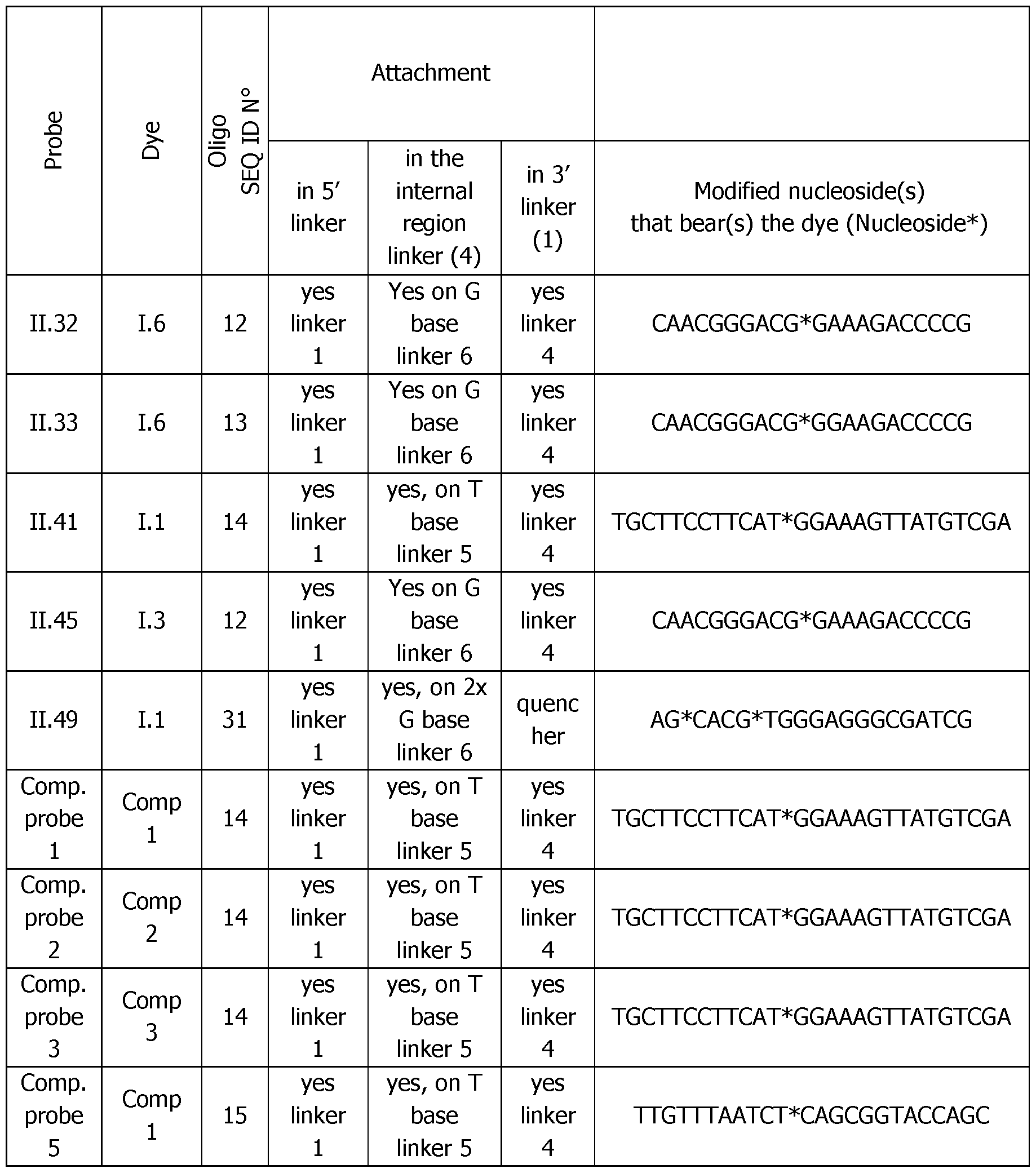
Protocol for the conjugation of compounds of formula (I) to oligonucleotides bearing one or several amino linkers The compounds of formula (LI) to (1.6) bearing an activated ester and in the form of a trifluoroacetate salt were linked in one step to the oligonucleotides bearing one or several aminolinkers arms at their 5' or/and 3' ends or/and internally in the oligonucleotidic sequence obtained from regular suppliers like Integrated DNA Technologies (Leuven, Belgium), Eurogentec (Seraing, Belgium). This is illustrated by Scheme 7 showing the protocol for the synthesis of probe II.5 synthesized from compound FC of
formula LI (TFA salt) and from an appropriately functionalized oligonucleotide with amino linkers in 3' and 5' ends. When the probe carries only one compound FC attached internally or in 5' end, a blocking group was introduced at the 3' end to prevent extension during PCR. The same protocol is applied for the other probes.
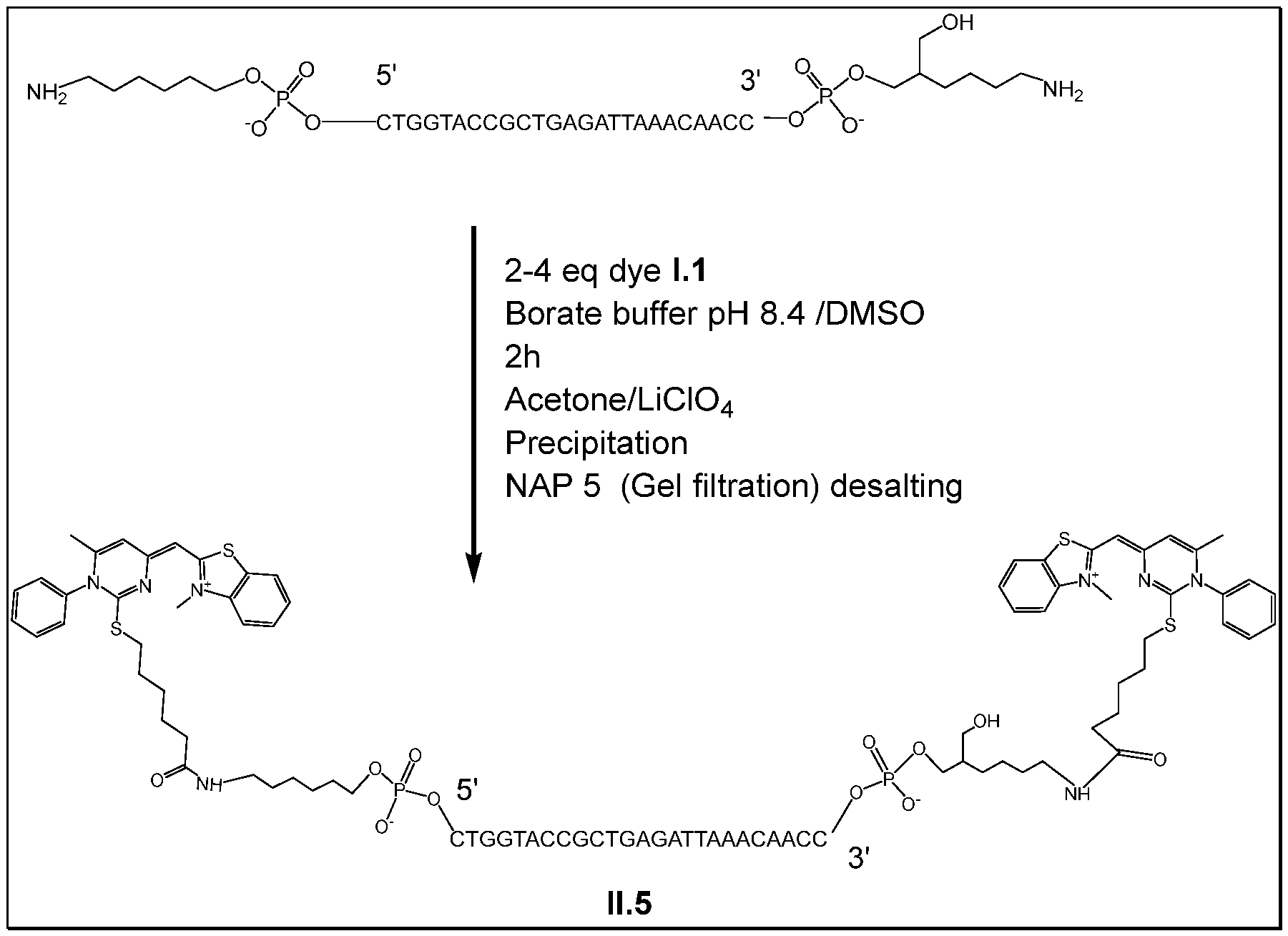
The following protocol was followed to synthesize the probes II.1 to 11.46 (their obtained TFA salt) described in Tables 4 and 4bis. Dried amino-linked oligonucleotides (40 nmol) were solubilized into 80 pL borate buffer 100 mM pH 8.5. The activated ester solution (1.1 to 1.6) was added under vortex (16 pL (160 nmol, 4 eq)) from a 10 mM dye solution in DMSO to the oligonucleotide. The conjugation reaction is monitored by UPLC (method B) until completion in around 2 hours as shown on Figure 2 for probe II.10. The probe corresponding to the conjugated oligonucleotide was precipitated by adding to the crude reaction 18 pL of lithium perchlorate 3 M, QSP 300 pL water and 900 pL acetone. The mixture was vortexed and was centrifugated at 10 000 rpm (rotation per minute) and the supernatant was discarded. The pellet was resuspended in 282 pL of water and 18 pL of lithium perchlorate 3M and vortexed. Acetone (900 pL) was added to get a cloudy mixture which was again centrifugated at 10 000 rpm and the supernatant was discarded. The same operation was repeated 2 more times until the supernatant
became translucent. Then, a final wash of the pellet with 900 pL of acetone was done before evaporating residual traces of acetone with a GeneVac centrifuge evaporator (Ipswich, UK). The dried pellet was resuspended in 200 pL of water and purified on a NAP 5 column (Cytiva) following manufacturer procedure in order to remove the residual alkaline borate salts. Final NAP 5 water eluate was neutral and was adjusted to 200 pL in water. The obtained probes were used without further purification after quantification by UV spectrophotometry at 260 nm (Denovix), UPLC mass analysis (Waters) and suspension at 100 pM in water. Alternatively, the probes can be further purified by preparative UPLC
Analytical UPLC Method B used: 0 to 95% acetonitrile in TEAAc 10 mM pH 7 for 5 min at 0.5 mL/min on a BEH C18 column (Waters). Max plot detection was carried out.
C. Results
Part I. Demonstration that the probes of the invention have an excitation and an emission spectra brought by the attached dye
The probes of the invention were prepared at 1 pM, in the presence of their complementary DNA strand (COMP.) at 4 pM in a model hybridization buffer 2 (Tris pH 8.4 20 mM, NaCI 10 mM, dNTP (4x0.3 mM), MgCI2 4 mM, proprietary stabilization buffer IX, TAQ polymerase 0.2 U/pL and BSA 550 ng/pL) for a total volume of 20 pL. Then, the solutions were poured into a microplate and the maximum absorption (equivalent to the maximum excitation) on the spectra was measured. Thereafter, the maximum fluorescence emission was also measured upon excitation at: (X absorption max -30 nm) with a gain of 60 using a spectrophotometer/spectrofluorometer reader (TECAN, Austria).
The Figure 3 represents these data for some probes of the invention and shows that the oligonucleotides became fluorescent upon conjugation to the dyes of the invention. The same fluorescent behavior was observed with all the probes according to the invention.
Part II. The probes of the invention demonstrate specific fluorescence exaltation upon hybridization with their complementary DNA target
The probes of the invention were prepared at 1 pM, alone or in the presence of their complementary DNA strand (COMP.) or with a non-complementary 60 mer duplex as specificity control (RANDOM) all at 4 pM in the model hybridization buffer 2 for a total volume of 20 pL. The fluorescence emission spectra was recorded at room temperature as in the previous Part I. As a
representative example, the results obtained with probe 11.33 are shown on the Figure 4: the specific fluorescence exaltation (X3) of the probe 11.33 occurred only in the presence of its complementary DNA target demonstrating the strong and specific interaction of the dye with DNA once after hybridization.
Table 5 represents the fluorescence exaltation ratio between the hybridized probe and the free probe obtained at room temperature for some probes of the invention. These data's demonstrate the general behavior of these probes in terms of fluorescence exaltation once hybridized to their target.
Table 5 (5': dye attached at 5' end; Internal: dye attached in the internal region of the oligonucleotide; 3': dye attached at 3' end)
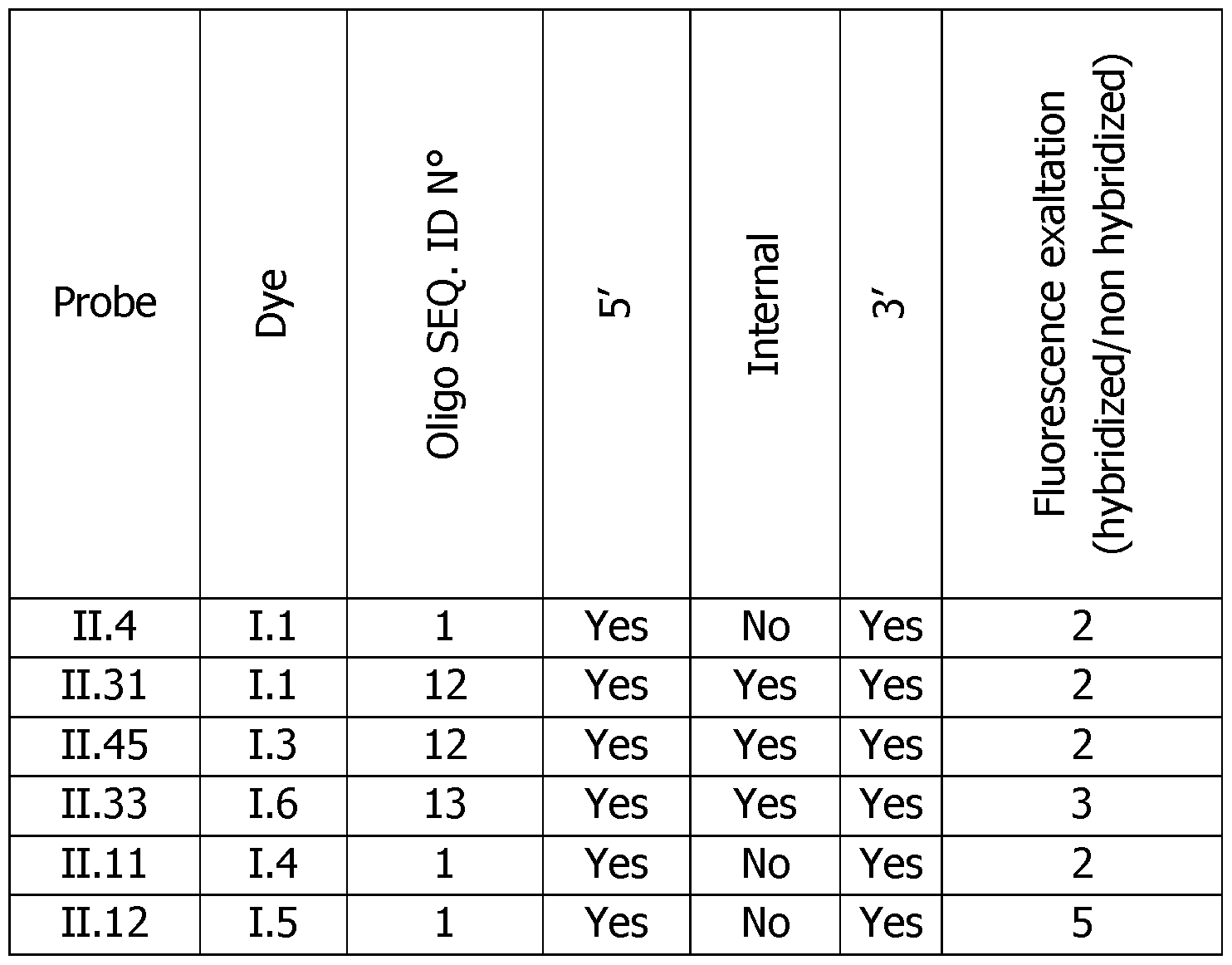
Part III. Demonstration that the probes allow to detect a specific sequence by melting measurement
The various conjugated probes synthesized were assayed in melting experiments versus their complementary targets. 1-10 pM probes solutions were used in a model hybridization buffer 1 (20 mM Tris-HCI, pH 9.0 containing 10 mM NaCI and 2 mM MgCL) or buffer 2 in a total volume of 20 pL. The probes were analyzed alone, with a non-complementary strand (NON. COMP.) as a control or as duplexes with their complementary DNA strand (COMP.) or with a non-complementary 60 mers duplex (RANDOM) also as a specificity control. The DNA target strands were used at 4 pM when
the probes were at 1 pM and at 10 pM when the probes were at 10 pM. These different experiments are summarized in Figure 5, in the case of a probe bearing a dye at both its 5' and 3' ends.
A CFX Maestro from Biorad Laboratories was used with PCR polypropylene cuvettes of 100 pL to record fluorescence in the SybRgreen Channel as a function of temperature from 20°C to 90°C (0.5°C/min) using a preliminary denaturing step during 1 min at 95°C. Duplex stabilities were determined by monitoring the fluorescence of the probe as a function of temperature. The thermal denaturation and determination of the Tm were done by calculating the first derivatives of the relation RFU = f Temperature) as illustrated on Figures 6A to 6F for probes II.4, II.6 and 11.33 taken as typical examples. With compound II.6, a weak transition, but that is easily detected, is observed with the first derivative of the fluorescence.
The hybridization of the probe allows to monitor a melting curve and to determine a melting temperature which is characteristic of the sequence of the duplex. No melting temperature was detected for the control experiments (NON COMP., RANDOM) demonstrating the specificity of all the probes of Table 6.
It also appears that the fluorescence increased when the probe was hybridized with its complementary target versus in the presence of the non- complementary target. This fluorescence exaltation already occurred at room temperature but particularly around 60°C (similarly as it is shown on the Figures 6A-F and represented by the double arrow on the left panels) where it varies from 1.1 to 2.6 and even more. The monitoring of real time PCR is generally done around 60°C and this exaltation could be maximized in order to have a better sensitivity when monitoring PCR in real time.
The difference in RFU at the Tm temperature between the COMP, and the NON COMP, or probe alone experiment can be linked to the sensitivity of the detection (Tm peak height). The higher the peak is, the easier the detection of this event is (represented by the double arrow in Figures 6A-F on the right panels). Since the Tm peak height is linked to the probe concentration, the normalized Tm peak height is corrected by the concentration of the probe and allows to compare all these experiments, the obtained results demonstrate the general behavior of these probes.
Table 6: Analytical melting experiments using probes of the invention with the appropriate controls (Probe: synthesized detection probe; Dye: dye bearing an activated ester used for conjugation; SEQ. : Oligo SEQ ID N°; Cone.: probe cone, in pM; Buffer: buffer 1 or 2 used for the measurement; 5': dye attached at 5' end by covalent linkage; Internal: dye attached in the internal region by covalent linkage; 3': dye attached at 3' end by covalent linkage; Exaltation factor at 60°C: Fluorescence exaltation at 60°C upon hybridization of the probe in comparison with the probe alone or with the probe with a non-complementary strand; Tm peak height: Height of the peak of the first derivative (-d(RFU)/dT) at the Tm in RFU/°C; Normalized Tm peak height: Normalized Height peak of the first derivative (- d(RFU)/dT) at the Tm as the function of the probe concentration in RFU/°C/pM; Channel: Detection channel on the CFX); Y=Yes and N=No.

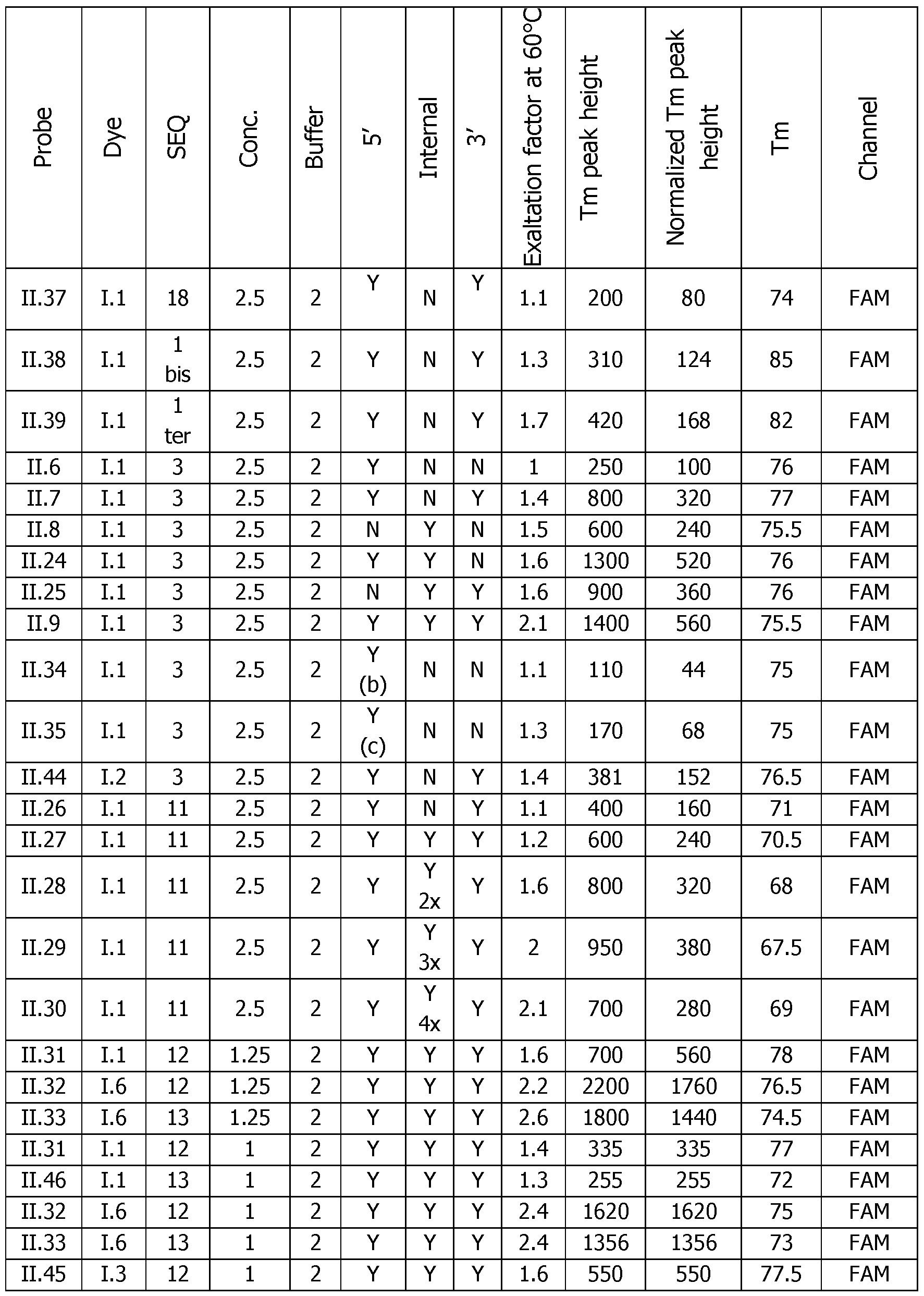
General observations:
Number of dyes: The increase of the number of dyes from 2 (probe 11.26), 3 (probe 11.27), 4 (probe 11.28), 5 (probe 11.29), 6 (probe 11.30) has in general a benefit on the sensitivity of the detection and on the fluorescence exaltation at 60°C upon hybridization of the probe. The optimum is around five dyes (probe 11.29) with a ratio of 2 and a Tm height of 380 versus a ratio of 1.1 and a Tm height of 160 for (probe 11.26) with 2 dyes only.
Another comparison on another sequence shows the same trend between 1 dye (probes II.1 and II.3) and 2 dyes (probe II.4) with a beneficial effect on the ratio (from 1.2-1.4 to 1.6) and on the Tm height (from 57-80 to 152).
Dve Localization: The localization of the dye on the probe can slightly affect the fluorescence ratio at 60°C and the height of the Tm peak as it can be seen on the series of probes II.6-8, 11.24-25 and II.9 where different localization and number of dyes have been used. The specific beneficial effect of the internal modifications is in particular visible with the 11.24 probe which is almost as good as the triple labelled probe II.9 and far better than the mono-functionalized probe II.6. The double modification at 5' and 3' ends and the triple modification at 5' and 3' ends and internally are the favorite modes.
Type of linker: Most of the different linkers used between the dye and the oligonucleotide sequence have a same behavior or lead to slightly lower functional performance in terms of exaltation ratio and Tm height after hybridization. The simplest one (Ce alkylidenyl linker) seems to be the most convenient. Unsuspectedly linkers that allow the incorporation of 2 dyes at the 5' are not more efficient and even less as only one (probes 11.34-35 versus probe II.6). Very long oligonucleotide linkers 11.36 and 11.37 are poorly efficient certainly due to the length of the linker.
A rough performance level can be established: C12 PEG linker (probe II.2) > Ce alkylidenyl linker (most of the examples) > > auto complementary oligonucleotide in 11.36 and 11.37 » doubler (probe 11.35) > serinol linker (probe 11.34).
Type of dve: The dye used selected among compounds FC of the invention does not affect the observations made for the dye 1.4. that the best attachment position to increase the sensitivity of the detection is the double labeling in 3' and 5' ends, versus mono labeling (II.11 versus II.10). The fluorescence exaltation at 60°C and the Tm peak height can vary as a function of the dye. For example, the dye 1.2 provides a twice less efficient probe 11.44 versus the corresponding probe II.7 made with dye LI. At the opposite, probes 11.12 or 11.16 made with dye 1.5 give excellent performances (exaltation of 3 with a Tm height of 2800). Similarly, dye 1.6
gives probes (11.32 and 11.33) with excellent performances (exaltation factor of 2.4 with Tm height > 1500). These results and differences come from the fluorescence properties of the dye, as well as the excitation/emission maxima of the dye. Despite the observed differences, the results show the generalization of the approach described in the invention as regards to the specificity of recognition of a target sequence.
Type of nucleotide modification: whatever the type of nucleotide modification, the probes behave the same. This is the case for probes 11.38 and 11.39 with LNA modification inside the DNA sequence in comparison with the probe II.4 including an unmodified DNA sequence.
Conclusions: The probes of the invention are specific to a given sequence and emit a stronger fluorescence once hybridized, whatever the type of dye used. These probes cannot recognize a non-target nucleic acid. The dye attachment position, the type of linker and the number of dyes are important factors and allow to modulate the sensitivity of the detection. It was also shown that the detection of the hybridization event is possible at another wavelength that allows multiplexing possibilities. In a general manner, the universality of the approach proposed by the invention was demonstrated. Some slight differences in performances come from the nature of the dye, the oligonucleotide sequence, the number and the localization of the dyes on the probe.
Part IV. Demonstration that the probes allow to detect a specific sequence of a Corona virus by real time PCR and melting measurement.
The probes II.1-4 and 11.10-12 evaluated analytically in Part III were used to specifically detect an amplicon generated by a PCR amplification reaction. A sequence of the OC43 gene from a Coronavirus micro-organism was used as an amplified model target sequence.
Protocol
A solution of 20 pL was prepared containing at final concentration in the PCR tube:
Tris pH 8.5 20 mM, 10 mM NaCI, 0,36X proprietary stabilization buffer, dNTP (0,45 mM x 4) , MgCL 5 mM, KT:Ab TAQ polymerase 0,2 U/pL and BSA 500 ng/pL, Different input concentration from 1, 10, 10e2, 10e3, 10e4 copies/PCR of corresponding gBIocks , 0,4 pM FWD primer and 2 pM REV primer (1/5), 1 pM of fluorogenic probe.
Then a PCR reaction with real time fluorescence measurements was performed during 50 cycles (initial denaturation 95°C for 30 sec., alternating cycles of denaturation/annealing/extension of 95°C for 10 sec I 60°C for 20 sec I 72°C for 20 sec) followed by a melting experiment from 60 to 90°C (by 0.5°C steps) on a CFX Biorad thermocycler while monitoring fluorescence on the FAM, Texas Red and Quasar 705 channels during PCR and melting.
Real Time experiments
As representative example, the results obtained with probe II.4 are presented in Figure 7. It appears that this probe, as the other ones, enables the real time monitoring of the PCR amplification reaction with a high sensitivity since all inputs are detected and even the lowest at 1 copy/PCR (NTC means no template control).
Melting experiments
The graphics in Figure 8 show the corresponding melting curve and Tm determination after amplification for all initial inputs. It appears that a precise Tm determination of the hybridization product denatured amplicon/probe was possible for any initial input as low as 1 copy/ reaction.
A more convenient view with only an initial input of 10e4 copies is shown in Table 7. The obtained results demonstrate the beneficial effect on the sensitivity of detection of the probe II.2 with a PEG linker at 5' end and above all the dramatic sensitivity enhancement of the probes II.4 when two dyes are present at 5' and 3' ends (A RFU = 170) versus the presence of only one dye at 5' end in probe II.1 (A RFU = 60) or in probe II.3 at 3' end (ARFU = 70). The enhancement was unexpectedly about 3 times. The best dye attachment position to increase the sensitivity of the detection is 5'-3'DYE >> 5'DYE PEG > 3'PEG DYE >5'PEG DYE. Unexpectedly, the double DYE conjugation 5'-3' increases by more than twice the signal emitted during real time PCR. The maximum of fluorescence (Max fluorescence in Table 7) data follows the same trend.
With dye 1.4 the sensitivity was increased 5 times between probes II.10 and 11.11. Surprisingly, adding two dyes at 5' and 3' ends provided more than twice the expected fluorescence.
Probe II.11 reached roughly the level of detection of probe II.4, but with another detection channel Texas Red (A RFU of 80 versus 110 respectively). The probe 11.12 synthesized with the dye 1.5 demonstrated excellent properties with high detection sensitivity on the other detection channel Quasar 705.
Table 7

Conclusions: Whatever the dye position, the detection probes PR of the invention allow the detection of an amplicon generated during a PCR reaction with high specificity and sensitivity. The double dye attachment at 5' and 3' ends greatly increases dramatically the sensitivity of the detection, even to as low as 1 input copy /mL sometimes. Those results were non-expected. It was also shown that the detection of a specific PCR amplification reaction is possible at another wavelength that opens multiplexing possibilities.
Part V. Detection of a sequence of a gene of Streptococcus microorganism by Real Time PCR amplification followed by a melting experiment
Real Time PCR
The Figure 9 shows a real time PCR reaction using probe II.5 targeted to different input of gene DNA from 1 Geq/PCR (genome equivalent/PCR reaction) to 10e5 Geq/PCR. The oligonucleotide of probe II.5 corresponds to a gene sequence of a Streptococcus micro-organism used as an amplified model target sequence. The same protocol as described in Part IV was followed. It can be seen that around 3 CT separate each concentration showing the efficiency of the PCR in the presence of the probe II.5 (synthesized as described before from 5'-3'NH2 oligonucleotide conjugated to the 1.1 NHS ester) to give the double conjugated probe and the high quality
of the fluorescence curves obtained with these probes (NTC= no template control). The experiments were carried out on a CFX (BioRad) with 0.2 pM Primer FWD, 1 pM Primer REV and 1 pM of Probe II.5.
Melting experiments
Melting experiments were realized on the obtained hybridization products. The melting curves and the curves of Tm determination are shown in Figure 10. The obtained results show once again how easy it was to determine the melting temperature of corresponding to specific amplicons even at very low initial input.
Conclusions: This example shows the generalization of the real time monitoring of PCR amplification and the subsequent Tm determination of the hybridization product of a given denatured amplicon with the probes of the invention.
Part VI. Ultrafast nested PCR directed to a gene sequence of a Neisseria Gonorrhoeae micro-organism using the probes of the invention
The protocol as described in Part IV was used, except that a first PCR was carried out on a fast mode using a MBS platform (MOLECULAR BIOLOGY SYSTEMS B.V, Scottweg, The Netherlands). The protocol was as following: initial denaturation for 30 sec at 102°C followed by 30-40 alternating cycles of denaturation at 102°C for 2 s and extension at 60°C for 4 s starting from an initial input of 10e5 copies/PCR of gBIocks corresponding to a gene sequence of Neisseria Gonorrhoeae micro-organism. The PCR formulation was optimized to enable the handling of the harsh fast PCR conditions.
The second PCR followed by melting experiments from 60 to 90°C (by 0,5°C steps) was realized on a CFX Biorad thermocycler while monitoring fluorescence on the FAM channel during melting. The probes II.6-9 were used at 1 pM to specifically detect the amplicon generated by fast PCR amplification reaction.
Melting experiments
Melting experiments of probe II.7, as a typical example, are presented in Figure 11. The obtained results show that the probe allowed the specific detection of the amplicon with a high sensitivity vs. the no template control (NTC) experiment on another biological target.
Table 8 compares the effect of the localization of the dye on the sensitivity of the fluorescent detection (height of the Tm peak). The unexpected 5 times higher fluorescence of the double labeled 5'-3' labeled probe II.7 versus the mono labeled one (probe II.6) was again demonstrated. It can also be seen that a dye localized in the internal region of the probe sequence (probe II.8)
was 3 times more efficient than a dye localized at the 5' end (probe II.6).
The triple conjugated probe (II.9) shows the best performances.
Table 8: Functional melting experiments using Probes II.6-9 with an initial input of 10e5 copies/PCR corresponding to an amplifiable gene sequence of the Neisseria Gonorrhoeae micro-organism.

Conclusions: The increase in the number of dyes up to 3 on the probe results in a dramatic increase of the detection sensitivity of the PCR product. The internal position is particularly efficient.
Part VII. Multiplex PCR detection with 2 probes labeled with two different dyes and each directed to the gene sequence of a different micro-organism S, Pombe and Coronavirus
In this experiment, a multiplex PCR was performed with the probe II.11 specific to a sequence of the OC43 gene of a Coronavirus micro-organism and the probe 11.16 specific to a sequence of S. Pombe micro-organism. The two probes were mixed together at 1 pM in the same tube with the corresponding specific primers and with an input of 10-100 and 1000 copy/reaction in the PCR conditions of as disclosed in Part VI.
The Figures 12A and 12B present the results after Real Time PCR and the melting experiments of the duplex PCR. The fluorescence reading on the CFX platform (BioRAD) was done at the same time on the Texas Red channel for probe II.11 on panel A and on the Quasar 705 channel for probe 11.16 on panel B. In the two cases, both amplicons were perfectly detected in the same time.
Conclusion: During multiplex PCR reaction, no cross detection occurs and it can be seen that even if the Tm of the two species have been rather closed, the detection at a specific wavelength makes possible to precisely determine a specific specie. The possibility to detect different specific hybridization events with the probes in the same time has been then demonstrated. Two fluorogenic probes tethered to two different dyes allow to detect two specific sequences by real time PCR and melting measurement.
Part VIII. Genotyping experiments and mutant detection (1 single nucleotide) targeting a sequence of a Corona Virus micro-organism
The probes 11.18 comprising a wild type oligonucleotide, 11.19 comprising mutant 1 oligonucleotide with a G base substituted by a A base, 11.20 comprising mutant 2 with a G base substituted by a C base and 11.21 comprising mutant 3 with a G base substituted by a A base were assayed in melting experiments versus one single complementary 60 mers target at 10 pM. 2.5 pM probes solutions were used in a typical PCR mix formulation (Tris pH 8.4 20 mM, 10 mM NaCI, IX proprietary stabilization buffer, dNTP (4x0.3 mM), MgCL 4 mM, TAQ polymerase and BSA 550 ng/pL) in a total volume of 20 pL. A CFX Maestro from Biorad Laboratories was used with PCR polypropylene cuvettes of 100 pL to record the fluorescence in the SybRgreen Channel as a function of temperature from 20°C to 90°C (0.5°C/min) using a preliminary denaturing step during 1 min at 95°C. Duplex stabilities were determined by thermal denaturation and determination of the Tm by calculating the first derivatives of the relation RFU= f(Temperature). Figure 13 shows the difference in Tm between the different duplexes and particularly the strong destabilization by more than 10 °C brought by one mismatch versus the wild type duplex.
Part IX. Genotyping experiments and mutant detection (2 nucleotides different from the wild type target) by a PCR targeting a sequence of Neisseria Gonorrhoeae micro-organism
In these experiments, the ability of probe 11.17 to detect 2 nucleotides mutations of various targets in comparison with the wild sequence was assessed. A set of GyrA gene mutants (Neisseria Gonorrhoeae microorganism) were chosen from literature: Mutant 1: C-> T, G -> A; Mutant 2: C-> T, A -> G; Mutant 3: C-> T, A -> C. The PCR protocol was the same as described in Part VI. It is apparent on the melting experiments done with the generated amplicon and presented in Figure 14 (panels A and B) that the probe 11.17 of the present invention was able to easily detect at least 3 different mutants based on melting experiments, with a high sensitivity.
Around 10°C of difference were observed between the Tm of the different mutants (Mutant 1, 2 or 3) versus the wild type (WT) depending on the destabilization brought by the mutation. It is understood that a more optimized design of the probes (length of the sequence and targeted region) could provide an even better discrimination. For comparison, the panels C and D show the same experiment done only in the presence of the commercial free dye LCG Plus (BioFire Gen scanning reagent, Salt Lake City, USA) and the inability of the free dye to detect the 3 mutants with sufficient discrimination versus the wild type sequence.
Part X. Analytical comparison between quenched probes with mono, double and triple labelled 1.1 dye of the invention, using FRET technique
To obtain the probe 11.49 of the invention with an oligonucleotide labelled internally with dye 1.1 and having the 3' end quenched, the commercially available amino-modified dG nucleotide was introduced on the oligonucleotide synthesizer. The modified oligonucleotides were deprotected and purified as classically used (Protocols for Oligonucleotide Conjugates Methods in Molecular Biology (volume 26) Sudhir Agrawal 1994) before conjugation with the dye 1.1 bearing an activated ester for the synthesis of the corresponding modified probe on G bases in the internal region of the sequence (probe 11.49). To doubly labelled the 5' end of probe 11.48, a commercially available modified symmetrical branching phosphoramidite was used before adding the usual C6-amino-linked form. The modified oligonucleotides were deprotected in the same way as previously described and purified before conjugation with the dye LI. All the final conjugated probes (11.47, 11.48 and 11.49) were further purified by HPLC to eliminate any unlabeled products. The probes at 1 pM were analyzed on CFX (Biorad, USA) alone or versus their complementary strand at 5 pM in order to evaluate the fluorescence exaltation, the Tm of the duplex and the fluorescence response at the Tm. A typical PCR mix formulation (Tris pH 8.4 20 mM, 10 mM NaCI, IX proprietary stabilization buffer, dNTP (4x0.3 mM), MgCL 4 mM, TAQ polymerase 0.2 U/pL and BSA 550 ng/pL) were used in a total volume of 20 pL. The same conditions were used as described in Part III. Figure 15 shows particularly the strong fluorescence exaltation at 60°C and, as expected, the low fluorescence background of the quenched probes without complementary strand.
All the results are presented in Table 9. Particularly strong fluorescence exaltation is observed at 60°C for the probe 11.49 (ratio of 3.7) due to the low fluorescence background brought by the quencher.
Table 9

Part XI: Dye attached internally in the sequence on a T, G or A nucleotide
To label internally the probes of the invention on the oligonucleotide synthesizer, the commercially available modified T, G or A nucleotide as their amino-linked form were introduced in the oligonucleotide used. The modified oligonucleotides were deprotected and purified according to an usual method before conjugation with the dye 1.1 bearing an activated ester for the synthesis of the corresponding probes modified on T base (probe 11.13), G base (probe 11.14) or A base (probe 11.15) in the internal region of the sequence. The probes were analyzed on the CFX (Biorad, USA) in the same conditions as in Part VIII, alone or versus their complementary strand in order to evaluate the fluorescence exaltation, the Tm of the duplex and the fluorescence response at the Tm. All the obtained results are shown in Figure 16.
The results presented in Table 10 show that whatever the type of base that bears the dye, the melting point determination can be performed with rather the same sensitivity. The possibility to label internally brings more flexibility to the design of the probes. It is understood that this sensitivity can be increased even further by adding dyes at the 5' and 3' ends and also by increasing the number of internal dyes.
Table 10
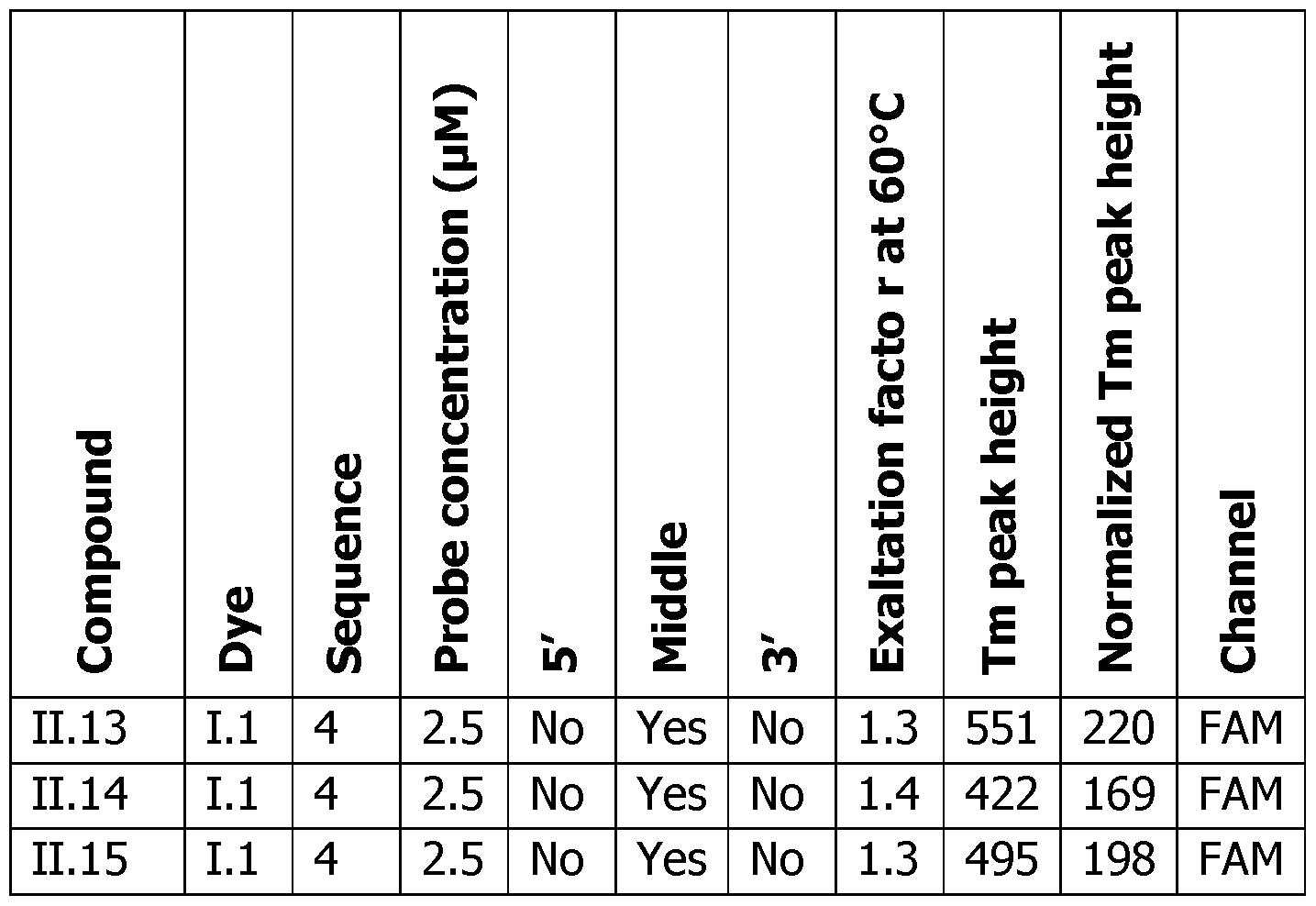
Part XII. Analytical comparison between probes of the invention conjugated with mono charged dye and comparative probes conjugated to double charged dyes
The Comp. Probes 1-4 were synthesized by conjugation between triple amino linked oligonucleotides and comparative NHS dyes Comp. 1, Comp. 2 and Comp. 3. The obtained results were compared to probes 11.40 and 11.41 linked to dye LI. The probes were evaluated at 2 pM for their specificity as described in Part III.
The probes of the invention 11.40 and 11.41 linked to dye 1.1 are specific as demonstrated before. Unsuspectedly the presence of an additional charge on the dye that is present in the comparative probes 1-3 makes them nonspecific since they detect also a non-complementary random duplex used as a control. This means that when this type of dye is used, the affinity of the dye itself for a double strand DNA prevails on the specificity of the oligonucleotide sequence. The Figure 17 shows the results obtained with the Comp. Probe 1 taken as a representative example. With these probes, the target sequence is detected but also the non-specific random duplex is detected with a height of the non-specific transition at 1500 RFU/°C. Table 11 summarizes the results obtained with these different comparative probes evaluated and shows the height in RFU/°C of the specific and the nonspecific transition. The obtained results show that these comparative probes are not useful. Therefore, the performances and the specificity of the probes depend on the dye attachment and on the structure of the dye, mainly on the number of positive charges on the dye.
Table 11 (Y=yes and N=no)
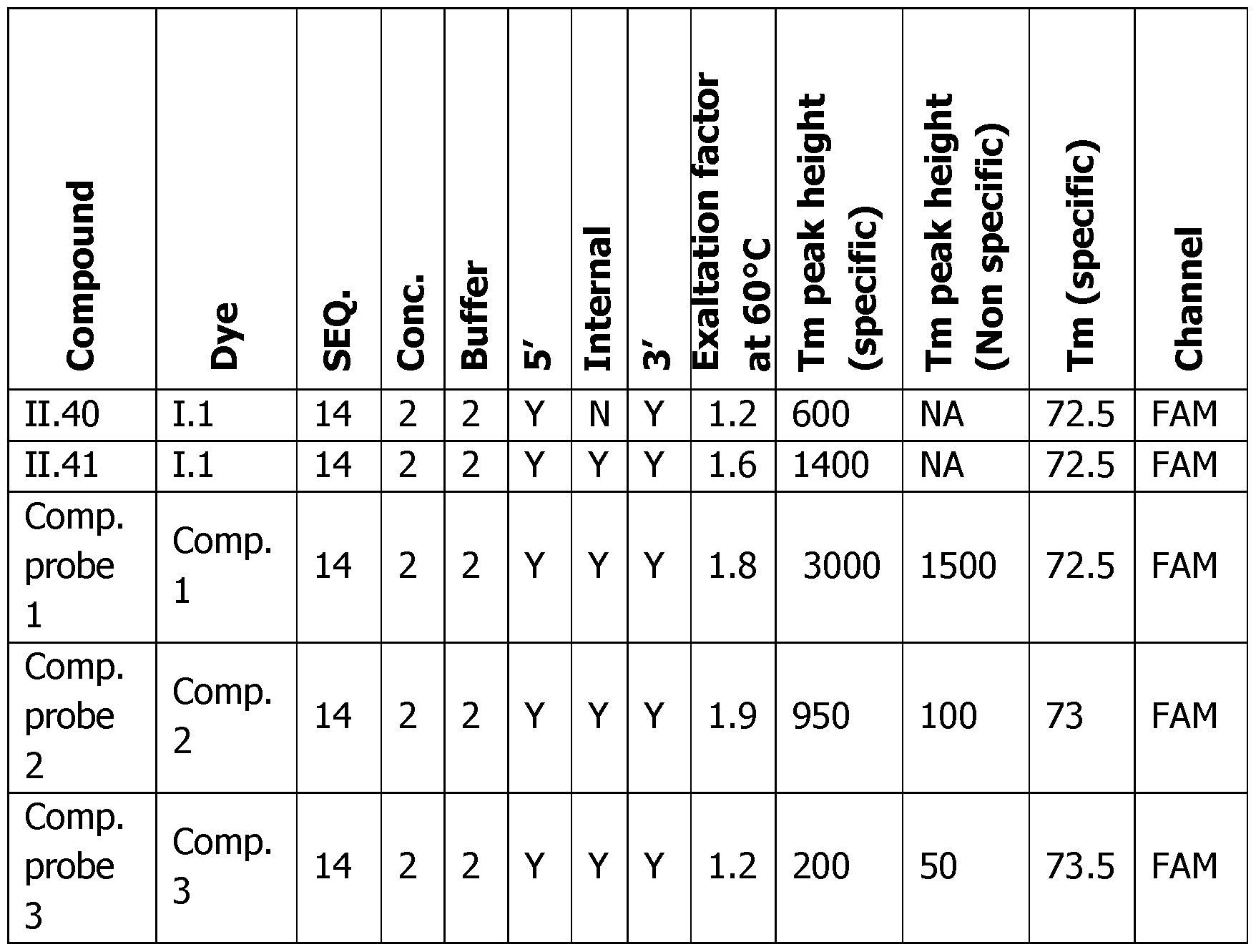
Part XIII: PCR directed to a sequence of Streptococcus microorganism using probes of the invention conjugated with a mono positively charged dye and comparative probes conjugated to double positively charged dyes
The Comp. Probes 4-5 were synthesized by conjugation between double and triple amino linked oligonucleotides and comparative NHS dyes Comp. 1. The probes were evaluated at 1 pM in a PCR reaction with initial input of 1, 10 and 100 Geq of gene DNA from a Streptococcus micro-organism per PCR reaction as described in the protocol of Part VI. After the PCR, the neoformed amplicons were submitted to a melting experiment as shown on Figure 18 for Comp. Probe 4 and on Figure 19 for Comp. Probe 5. As demonstrated before, the Comp. Probes with a double cationic charge on the dye detect their target as expected, but in an unexpected manner, detect also the full amplicon at a higher Tm. These results show the non-specificity of the probe that behaves as a free dye. The multi-cationic dyes are not suitable for probe conjugation.
Part XIV. Benefits of the probes of the invention that strongly stabilize duplex and allow to shorten the length of the oligonucleotide in the probes to increase their specificity
To assess the potential stabilization of duplex formed between the described probes and their target, melting experiments were realized using UV spectrophotometry that results in a direct Tm measurement. The probes 11.42-43, II.1-4, II.6-7, II.10-11 were evaluated versus their native duplexes. A Quartz cuvette of 1 mL was filled with 1 ml of a 1 M solution of probe (21 mers) and 1 pM complementary target (60 mers) in Tris 20 mM, NaCI 10 mM, MgCL 4 mM pH 8.4. The cuvette was equipped with a stopper. A Carry Multicell Peltier (Agilent) was used to record UV measurement during melting. The following temperature ramp was followed: 80°C to 40°C (depending on Tm temperature) at 0.5°C/Minute with one UV measurement at 260 nm each 0,5°C (renaturation), stop for 3 min and 40°C to 80°C at 0.5°C/Minute with one UV measurement at 260 nm each 0.5°C (denaturation/melting). The melting temperature was calculated from the first derivative on the melting curve (more precise than the denaturation curve even if there is no hysteresis). The ATm between the native non modified duplex (20/60 mers) and the modified probes/60 mers was calculated and plotted in Table 12.
Table 12
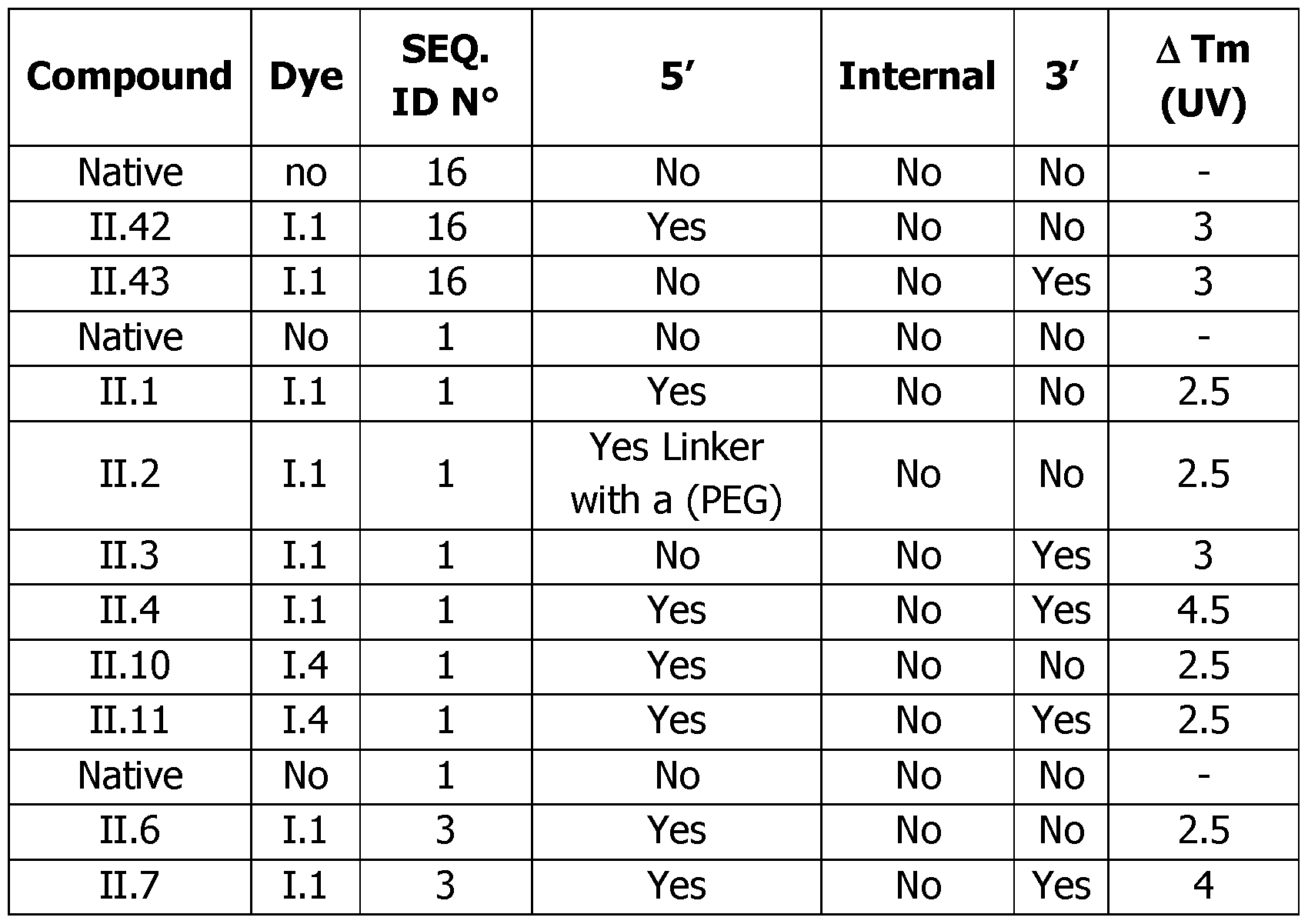
Conclusion: Surprisingly, the obtained results showed that the detection
probes of the invention possess a strong ability to stabilize a duplex (± 2.5°C I dye) leading to a stabilization around 4-5°C for the double conjugated probes. Dyes 1.1 and 1.4 have similar stabilization properties. That makes possible to shorten the length of the oligonucleotide and to increase even more the specificity of the hybridization.
Part XV. Detection using FRET adjacent hybridization probes technique
The following Table 13 describes the dyes of the invention and the commercial dyes used in FRET adjacent hybridization probes strategy:
Table 13
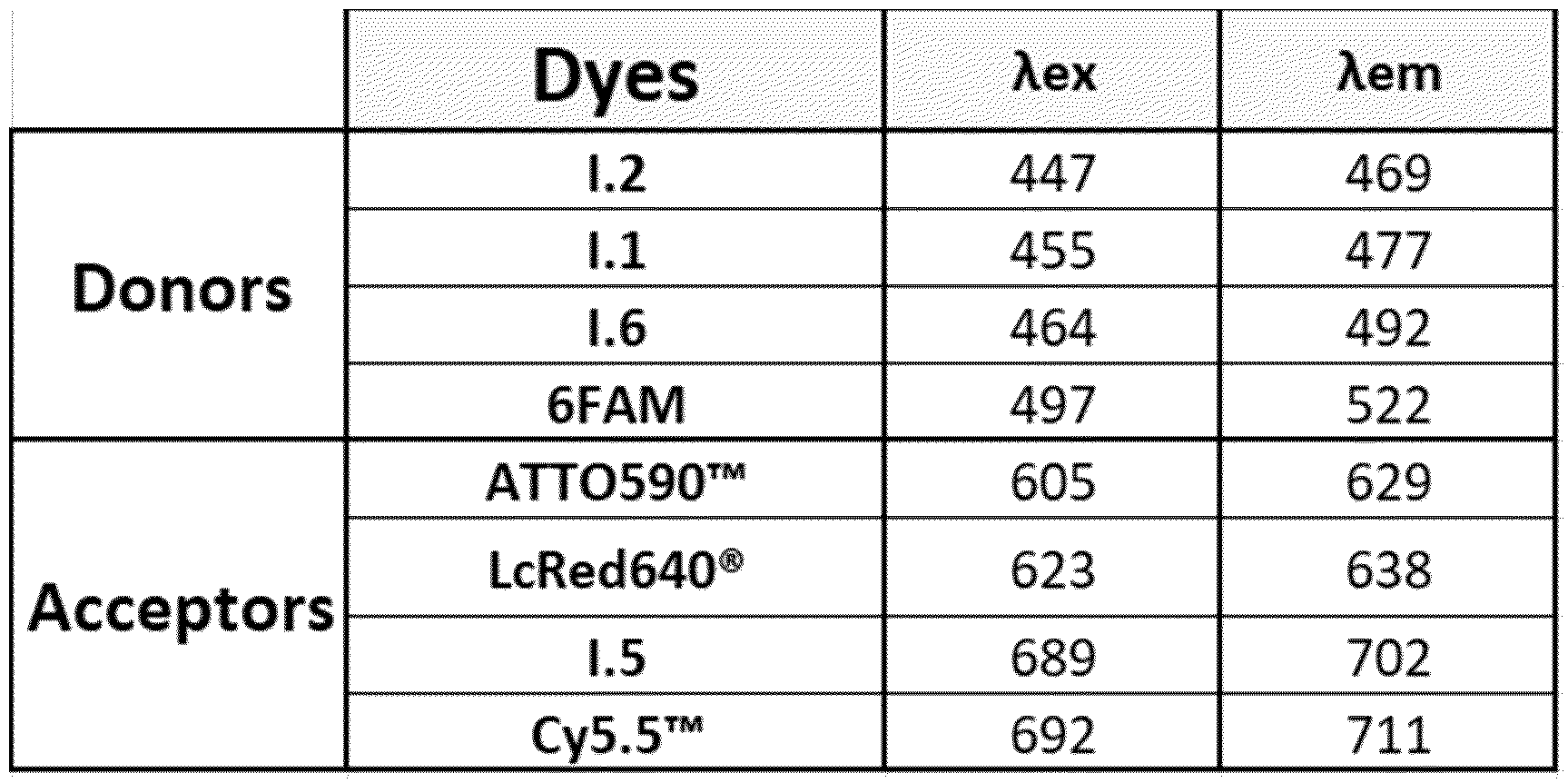
XV.l. Modulation of FRET intensity of 1.1 versus FAM Donor dyes with two different commercial Acceptor groups
To evaluate the FRET capacity of the 1.1 dye of the invention, a comparative study was initiated with FAM usual donor group (Comp, probe 6). The commercially available acceptor groups LcRed640® and ATTO590™ were introduced as the binary probes (designated Comp. Probes 7 and 8). All the synthesis were realized internally as previously described. The final conjugated probes (probes 11.50 according to the invention and Comp. Probes 6, 7 and 8) were further purified to eliminate any unlabeled products. The quality control was realized by LC-MS (Waters). The concentration of the donor probes and their complementary strand were fixed at 0.1 pM whereas the concentration of acceptor probes varied from 0.05 pM to 0.4 pM. The samples were analyzed on a spectrofluorometer (Tecan, USA) at room temperature in order to evaluate the fluorescence emission exaltation of the
Ill acceptor probe at 640 nm. For the FAM donor group, excitation wavelength was fixed at 455 nm, and the emission wavelength start at 500 nm and finished at 750 nm, whereas for the 1.1 donor group, excitation wavelength was fixed at 410 nm, and the emission wavelength start at 450 nm and finished at 700 nm. The same conditions were used as described in Part III. According to FRET principle, the excitation wavelength triggers fluorescent emission of the donor probe (compound FC) which is transferred to the acceptor fluorophore on the acceptor probe. This probe emits fluorescence in return.
Figure 20 shows the evolution of the fluorescence exaltation of the acceptor group based on a fixed amount of the donor and the complementary strand, and variable concentration of the acceptor group. It shows particularly the strong exaltation with 1.1 dye of the invention (Probe 11.50) whatever the commercial acceptor groups used (Comparative Probes 7 and 8).
As shown by Figure 20 (Panels C and D), the fluorescence emission resulting from the conjugation of the dye 1.1 on the oligonucleotide of the probes of the invention (Probe 11.50) is transferred more efficiently to the commercial acceptor dyes (Comparative Probes 7 and 8) than with the usual FAM dye (Comparative Probe 6). The max FRET signal at 640 nm with the probe of the invention obtained with dye 1.1 was twice better with Comparative Probe 7 as the acceptor probe (Panel C), and almost seven times better with Comparative Probe 8 as the acceptor probe (Panel D), compared respectively to the commercial Comparative Probe 6 (Panels A and B).
XV.2. Modulation of FRET intensity of probes bearing 2 conjugated dyes 1.1 or L6, as donor dye, versus 2x FAM Donor dyes with a commercial Acceptor group
To evaluate the FRET capacity of probes of the invention, doubly labelled with dye 1.1 or dye 1.6, a study was initiated with a doubly labelled FAM as usual comparative donor group (Comparative Probe 10). The commercially available acceptor dye LcRed640® was introduced as the corresponding binary probe (Comparative Probe 9). All the syntheses were realized as previously described. The final conjugated probes (Probes 11.51 and 52, Comparative Probes 9 and 10) were further purified by HPLC to eliminate any unlabeled by-products. The quality control was realized by LC-MS (Waters). The same conditions were used as described in Part XV.1. The samples were analyzed on a spectrofluorometer (Tecan, USA). Figure 21 shows the evolution of the fluorescence exaltation of the acceptor group at 640 nm. A particularly strong exaltation with the probes of the invention that are doubly labelled with dye 1.1 or dye 1.6 is shown.
As it is shown in Figure 21, the fluorescence emission resulting from the
presence of the dye 1.1 (Panel B: Probe 11.51) or 1.6 (Panel C: Probe 11.52) conjugated on the probes of the invention is transferred more efficiently to the commercial acceptor dye LcRed640® (Comparative Probe 9) at 640 nm than with the regular probe that is doubly labelled with FAM dye (Panel A: Comparative Probe 10). The max FRET signal at 640 nm was about three times better for the probe of the invention with two conjugated molecules of dye 1.1 (Panel B: Probe 11.51) compared to the commercial FAM donor. The max FRET signal at 640 nm was more than four times better for the probe of the invention with two conjugated molecules of dye 1.6 (Panel C: Probe 11.52) of the invention compared to the commercial FAM donor.
XV.3. Modulation of FRET intensity of probes bearing conjugated dyes 1.1 and 1.5 of the invention
A comparative study was initiated with the commercial FRET pair FAM/Cy5.5™ to evaluate the FRET capacity of the probes of the invention obtained by conjugation of dyes 1.1 and 1.5. All the syntheses were realized, as previously described. The final conjugated probes (Comparative Probes 6 and 11, Probes 11.50 and 53) were further purified by HPLC and quality controlled by LC-MS (Waters). The same conditions were used as described in Part XV.1. Figure 22 shows the evolution of the fluorescence exaltation of the acceptor group at 705 nm. A significant fluorescence exaltation is shown with the probes of the invention obtained by conjugation of dyes 1.1 or 1.5 on the oligonucleotide.
As it is shown in Figure 22, the fluorescent emission resulting from the conjugated dye 1.1 in the probe of the invention (Probe 11.50 in panels C and D) is transferred more efficiently to the acceptor probes (Comparative Probe 11 and Probe 11.53) than with the usual FAM donor (Comparative Probe 6 in panels A and B). For the Donor, the max FRET signal at 705 nm was more than five times better for the FRET pair I.l/Cy5.5™ (Panel C: Probe II.50/Comparative Probe 11) using a probe of the invention compared to the commercial combination FAM/Cy5.5™ (Panel A: Comparative Probe 6/ Comparative Probe 11). For the Acceptor, the max FRET signal at 705 nm was more than four times better for the FRET pair FAM/I.5 (Panel B: Comparative Probe 6/Probe 11.53) using a probe of the invention compared to the commercial combination FAM/Cy5.5™ (Panel A: Comparative Probe 6/Comparative Probe 11). For Donor and Acceptor, the max FRET signal at 705 nm was more than ten times better for the FRET pair 1.1/1.5 (Panel D: Probe II.50/Probe 11.53) of the invention compared to the commercial combination FAM/Cy5.5™ (Panel A: Comparative Probe 6/Comparative Probe !!)■
XV.4. Real Time PCR amplification directed to a sequence of Listeria micro-organism using FRET with a probe of the invention as a fluorescent donor and an acceptor probe
A modified commercial PCR kit (Gene Up® Listeria 2, bioMerieux, France) was used with specific donor probes: donor probes 11.22 or 11.23 of the invention labelled respectively with the dye 1.1 of the invention in 3' end or in 3' and 5' ends and comparative donor probe Comp. Probe 13 labelled with FAM in 3' end. According to FRET principle, the excitation wavelength triggers fluorescent emission of the donor probe (compound FC) which is transferred to the acceptor fluorophore on the acceptor probe. This probe emits fluorescence in return. This fluorescence was monitored on the Gene Up® thermocycler (bioMerieux, France).
Figure 23 shows that the fluorescent emission of the probes of the invention is transferred more efficiently to the acceptor probe than with the comparative donor probe. For both probes 11.22 and 11.23, the maximum of fluorescence signal at 640 nm was almost twice better than for the regular probe. It is worthy to notice also a lower background of fluorescence with the NTC of the probes 11.22 and 11.23 compared to the comparative donor probe. Therefore, it was demonstrated that the probes of the invention can be used advantageously in FRET technologies.
XV.5. Real Time PCR amplification directed to a sequence of Cronobacter micro-organism using FRET with dyes of the invention as fluorescent Donor and/or Acceptor
A modified commercial PCR kit (Gene Up® Cronobacter, bioMerieux, France) was used with a specific donor probe, Comp. Probe 6 labelled with FAM in 3' end, and with specific acceptor probes, acceptor probes 11.50, 11.51, 11.53, 11.54, 11.55, 11.56 and 11.57 of the invention and comparative acceptor probes, Comp. Probe 7 labelled with LcRed640® in 5' end, Comp. Probe 8 labelled with ATTO590™ in 5' end, Comp. Probe 9 labelled with LcRed640® in 3' end, Comp. Probe 11 labelled with Cy5.5™ in 5' end and Comp. Probe 12 labelled with ATTO590™ in 3' end. The fluorescence at 530 nm, 640 nm and 705 nm was monitored on the Gene Up® thermocycler (bioMerieux, France). Figure 24 shows the evolution of the melting peak height intensity at 640 nm (Panel A) and at 705 nm (Panel B). A significant fluorescence exaltation is shown on both channels with the probes obtained by conjugation of the dyes of the invention.
Figure 24 shows that the fluorescent emission of the probes of the invention is transferred more efficiently to the acceptor probe at 640 nm (Panel A) and 705 nm (Panel B) than with comparative probes (circled).
In Panel A, the melting peak height intensity at 640 nm was two to three
times better for all the FRET pairs including a probe labelled with either the dye 1.1 or the dye 1.6 of the invention (respectively Probe II.50/Comparative Probe 7, Probe II.54/Comparative Probe 9, Probe II.50/Comparative Probe 8, Probe II.55/Comparative Probe 9 and Probe II.55/Comparative Probe 12 at 157.5 to 247.5 RFU) compared to the comparative probes (circled Comparative Probe 6/Comparative Probe 7 at 76.2 RFU). For the doubly labelled probes strategy, the melting peak height intensity at 640 nm was even six times better with the 1.1 dye of the invention (Probe II.51/Comparative Probe 12 and Probe II.51/Comparative Probe 9 at respectively 443.8 and 458.3 RFU).
In Panel B, the melting peak height intensity at 705 nm was four to fifty times better for all the FRET pairs including a probe of the invention labelled with the dye LI, 1.5 or 1.6 (respectively FRET pairs Comparative Probe 6/Probe 11.53, Probe II.54/Probe 11.56, Probe II.57/Probe 11.53 and Probe II.55/Probe 11.56 at 11.6 to 147.5 RFU) compared to the comparative probes (circled Comparative Probe 6/Comparative Probe 11 at 3.1 RFU). For the doubly labelled probe, the melting peak height intensity at 705 nm was two times better with the 1.1 dye of the invention (Probe II.51/Probe 11.56 at 123.8 RFU) compared to the single labelled probe (Probe II.54/Probe 11.56 at 59.6 RFU).
Claims
1. Compounds FC having the formula (I):
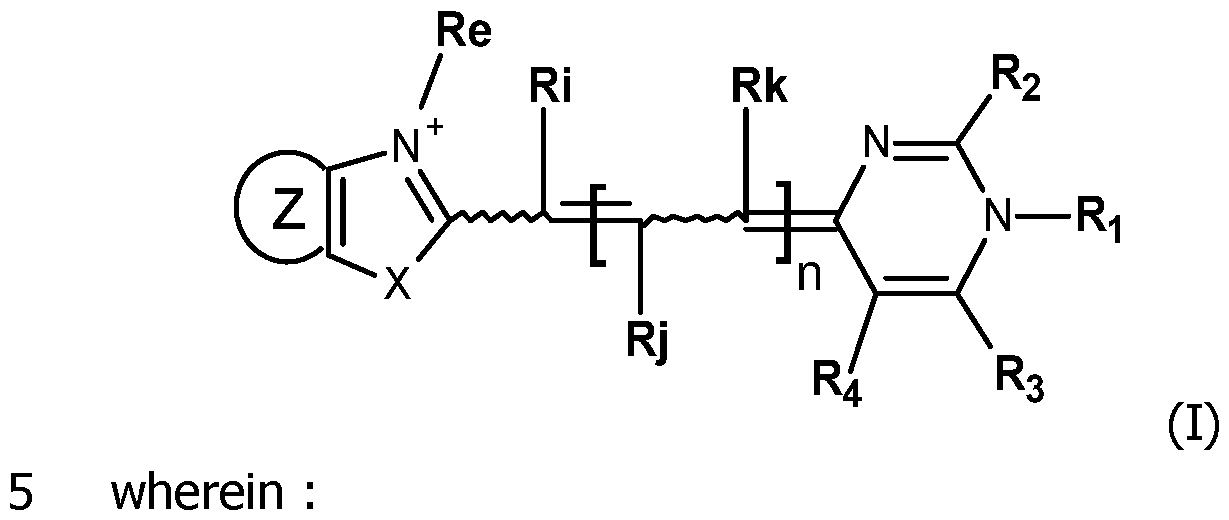
- n is equal to 0, 1 or 2;
- X is oxygen, sulfur, selenium, tellurium or C(CH3)2,
- Re is an alkyl, cycloalkyl, alkenyl, alkynyl, aryl, -(CH2)pR'e, where p is 1, 2, 3, 4, 5 or 6 and R'e is an aryl, -CF3, -CN, -C(O)alkyl, -C(O)Oalkyl, -Salkyl, - Oalkyl, -NHC(O)H, , -S(O2)O’, -S(O2)Oalkyl, -P(O2)O’, -P(O2)Oalkyl, -CH=N-O-R, -C(CH3)=N-O-R, -CH=N-NH-C(O)-R, -C(CH3)=N-NH-C(O)-R, -CH=N-O-C(O)-R, -C(CH3)=N-O-C(O)-R, -NHCOR and -CONHR, -CONHR being preferred, R being either an alkyl or -(CH2)qi-R"e, in which:
■ ql is 1, 2, 3, 4, 5 or 6 and
■ R"e is a Ci-ealkoxy group,
- Z is a fused mono or polycyclic aromatic or nitrogen-containing heteroaromatic ring, optionally substituted by one or several substituent(s) A identical or different selected among the groups alkyl, cycloalkyl, alkenyl, alkynyl, aryl, , -CF3, -, -CN, -C(O)alkyl, OAlkyl, -C(O)Oalkyl, -Salkyl, -Oalkyl, -NHC(O)H, -S(O2)O’, -S(O2)Oalkyl, -P(O2)O’, P(O2)Oalkyl, -CH=N-O-R', -C(CH3)=N-O-R', -CH=N-NH-C(O)-R', -C(CH3)=N-NH-C(O)-R', -CH=N-O- C(O)-R', -C(CH3)=N-O-C(O)-R', -NHCOR' and -CONHR', -NHCOR' and - CONHR' being preferred, R' being a phenyl, an alkyl or -(CH2)q2-A', in which:
■ q2 is 1, 2, 3, 4, 5 or 6 and
■ A' is a Ci-ealkoxy group;
- Ri, Rj, Rk, R3 and R4 are as defined hereinafter :
- Ri, Rj and Rk are identical or different and are independently selected from the group consisting of hydrogen and Ci-ealkyl or, when n=0, Ri and R4 are bonded together and form a -(CH2)r- chain with r being equal to 3, 4, 5 or 6, or, when n=l, 2, 3 or 4, Rk and R4 are bonded together and form a -(CH2)r- chain with r being equal to 3, 4, 5 or 6,
-R3 is selected among hydrogen, and the groups alkyl, cycloalkyl, aryl and -(CH2)q3-Y3, in which:
■ q3 is 1, 2, 3, 4, 5, 6 and
■ Y3 is an aryl or Ci-ealkoxy group; or R3 and R4 are bonded together and form a -(CH2)r- chain with r being equal to 3, 4, 5 or 6, and
- when R4 is not bonded to Ri, Rk or R3, R4 is selected from hydrogen, and the groups alkyl, cycloalkyl, aryl and -(CH2)q4-Y4, in which:
■ q4 is 1, 2, 3, 4, 5, 6 and
■ Y4 is an aryl or Ci^alkoxy group;
- Ri and R2 are as defined hereinafter: either:
- Ri is chosen among the groups alkyl, cycloalkyl, alkenyl, alkynyl, aryl, said groups being unsubstituted or substituted by one or several selected from the groups, , -CF3, , -CN, alkyl, -Oalkyl, C(O)alkyl, -C(O)Oalkyl, -Salkyl, -Oalkyl -NHC(O)H, -S(O2)O’ , -S(O2)Oalkyl, -P(O2)O' and -P(O2)Oalkyl, and
- R2 is Y2-L2-R'2, in which:
■ Y2 is CH2, CHalkyl, C(alkyl)2 , S or 0,
■ L2 is a linker, in particular an alkylidenyl, -(CH2)mi-Ph-(CH2)m2-, with ml and m2 being independently 0, 1, 2, 3, 4, 5 or 6, or -(CH2-CH2-O)m3-, with m3 being 1, 2, 3, 4, 5 or 6, and
■ R'2 is a reactive group RG, or:
- Ri is Li-R'i, in which:
■ Li is a linker, in particular an alkylidenyl,
-(CH2)pi-Ph-(CH2)p2-, with pl and p2 being independently 0, 1, 2, 3, 4, 5 or 6, or
-(CH2-CH2-O)p3-, with p3 being 1, 2, 3, 4, 5 or 6, and
■ R'i is a reactive group RG,
- R2 is -CH3, -CH2R"2, -CHalkylR"2, -C(alkylR"2)2, -SR"2 or -OR"2, in which R"2 is chosen among the groups alkyl, cycloalkyl, alkenyl, alkynyl, aryl, said groups being unsubstituted or substituted by one or several substituents selected from the groups -CF3, , -CN, alkyl, -C(O)alkyl, -C(O)Oalkyl, -Salkyl, -Oalkyl -NHC(O)H,
-S(O2)O’, -S(O2)Oalkyl, -P(O2)O’ and -P(O2)Oalkyl, including their salts with at least one anion, in particular, chosen among halogenated anions, typically CP, Br' and r ; trifluoroacetate, acetate, formate ; sulfonates, such as methylsulfonate, trifluoromethylsulfonate and tosylate ; sulfates, such as methylsulfate ; phosphate, pyrophosphate and triphosphate or with at least one cation, in particular, chosen among Li+, Na+, K+, Cs+ and triethyl ammonium.
2. The compounds FC according to claim 1, wherein Ri, Rj and Rk are hydrogen.
3. The compounds FC according to claim 1 or 2, wherein there is no substituent A on Z ring.
4. The compounds FC according to any one of claims 1 to 3, having the
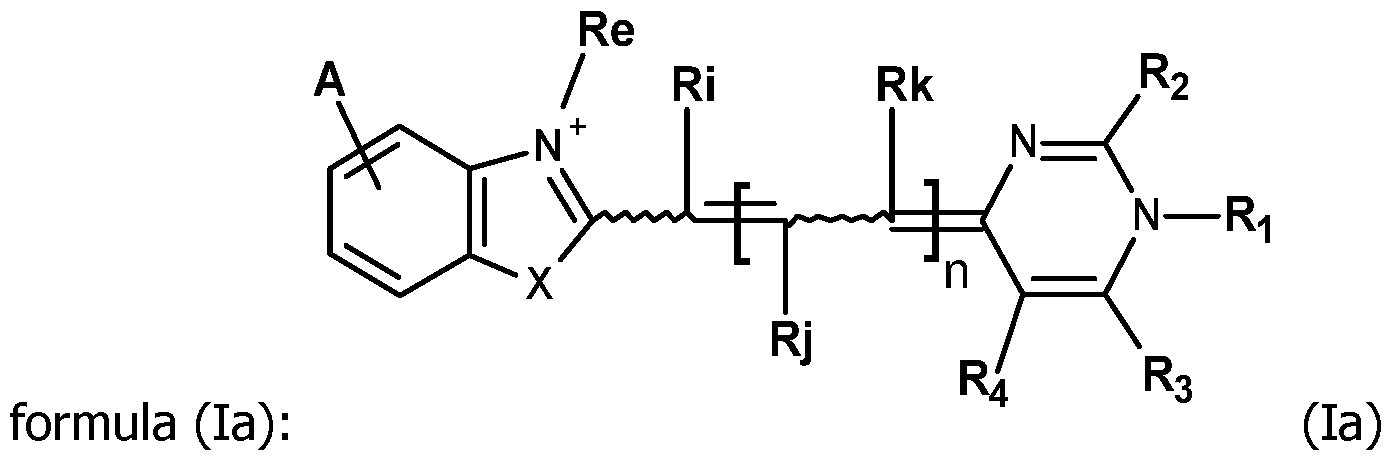 and in particular the formula (lb):
and in particular the formula (lb):
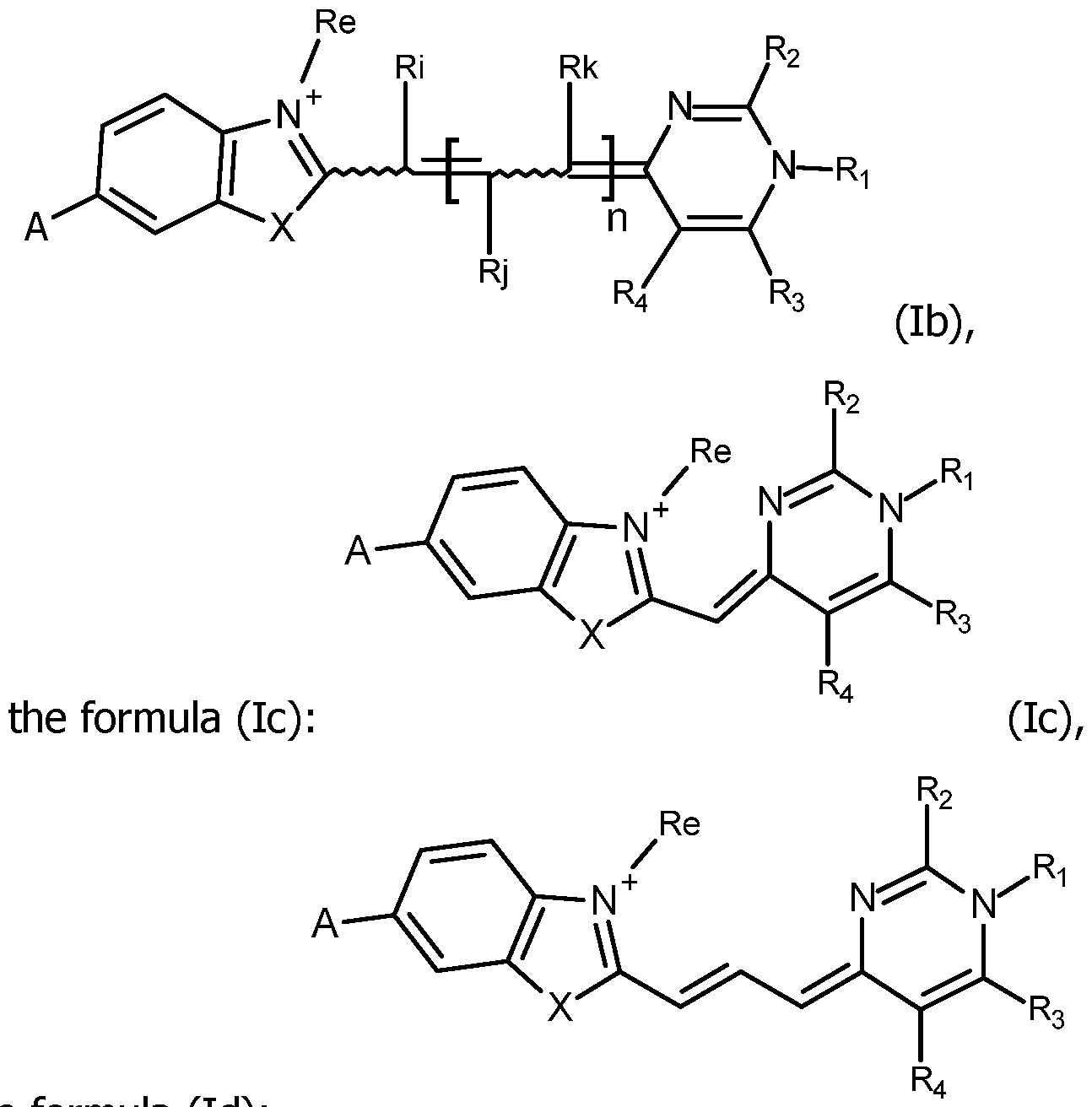 the formula (Id):
the formula (Id):
 or the formula (le):
or the formula (le):
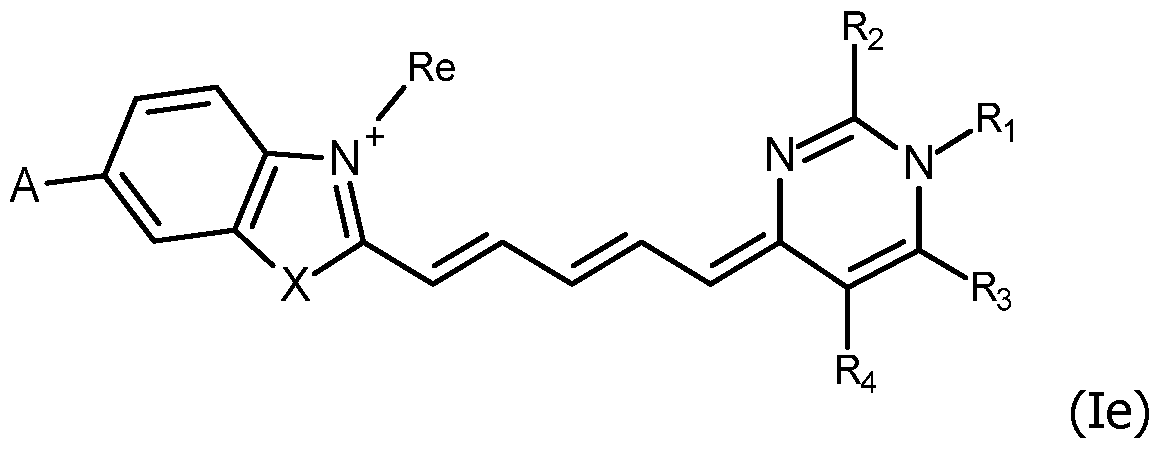 wherein Ri, Rj, Rk, Ri, R2, R3, R4, A, n and Re are as defined for claims 1 to
including their salts with at least one anion, in particular, chosen among halogenated anions, typically CP, Br' and r ; trifluoroacetate, acetate, formate ; sulfonates, such as methylsulfonate, trifluoromethylsulfonate and tosylate ; sulfates, such as methylsulfate ; phosphate, pyrophosphate and triphosphate or with at least one cation, in particular, chosen among Li+, Na+, K+, Cs+ and triethyl ammonium.
wherein Ri, Rj, Rk, Ri, R2, R3, R4, A, n and Re are as defined for claims 1 to
including their salts with at least one anion, in particular, chosen among halogenated anions, typically CP, Br' and r ; trifluoroacetate, acetate, formate ; sulfonates, such as methylsulfonate, trifluoromethylsulfonate and tosylate ; sulfates, such as methylsulfate ; phosphate, pyrophosphate and triphosphate or with at least one cation, in particular, chosen among Li+, Na+, K+, Cs+ and triethyl ammonium.
5. The compounds FC according to any one of claims 1 to 4, wherein Re is Ci-ealkyl, in particular methyl.
6. The compounds FC according to any one of claims 1 to 5, wherein R3 is Ci-6alkyl, in particular methyl.
7. The compounds FC according to any one of claims 1 to 6, wherein R4 is a hydrogen atom.
8. The compounds FC according to any one of claims 1 to 7, wherein A is a hydrogen atom or -NHC(O)CH3.
9. The compounds FC according to any one of claims 1 to 8, wherein Ri is Ci-ealkyl, in particular methyl or Ri is aryl, in particular a phenyl and -R2 is - Y2-L2-R'2, in which Y2, L2 and R'2 are as defined in claim 1.
10. The compounds FC according to claim 9, wherein Y2 is CH2 or S.
11. The compounds FC according to claim 9 or 10, wherein L2 is an alkylidenyl, in particular -(CH2)3-, -(CH2)4- or -(CH2)5.
12. The compounds FC according to any one of claims 9 to 11, wherein R'2 is an activated ester, and in particular a succinimidyl ester.
13. The compounds FC according to any one of claims 1 to 8, wherein R2 is -SCi-salkyl, in particular -SCH3, or Ci-ealkyl, in particular methyl, and -Ri is -Li-R'i, in which Li and R'i are as defined in claim 1.
14. The compounds FC according to claim 13, wherein Li is a phenyl substituted in meta or para by R'i as defined in claim 1.
15. The compounds FC according to claim 13 or 14, wherein R'i is an activated ester, and in particular a succinimidyl ester.
16.The compounds FC according to any one of claims 1 to 15, wherein X is oxygen or sulfur.
17. The compounds FC according to claim 1 selected among:
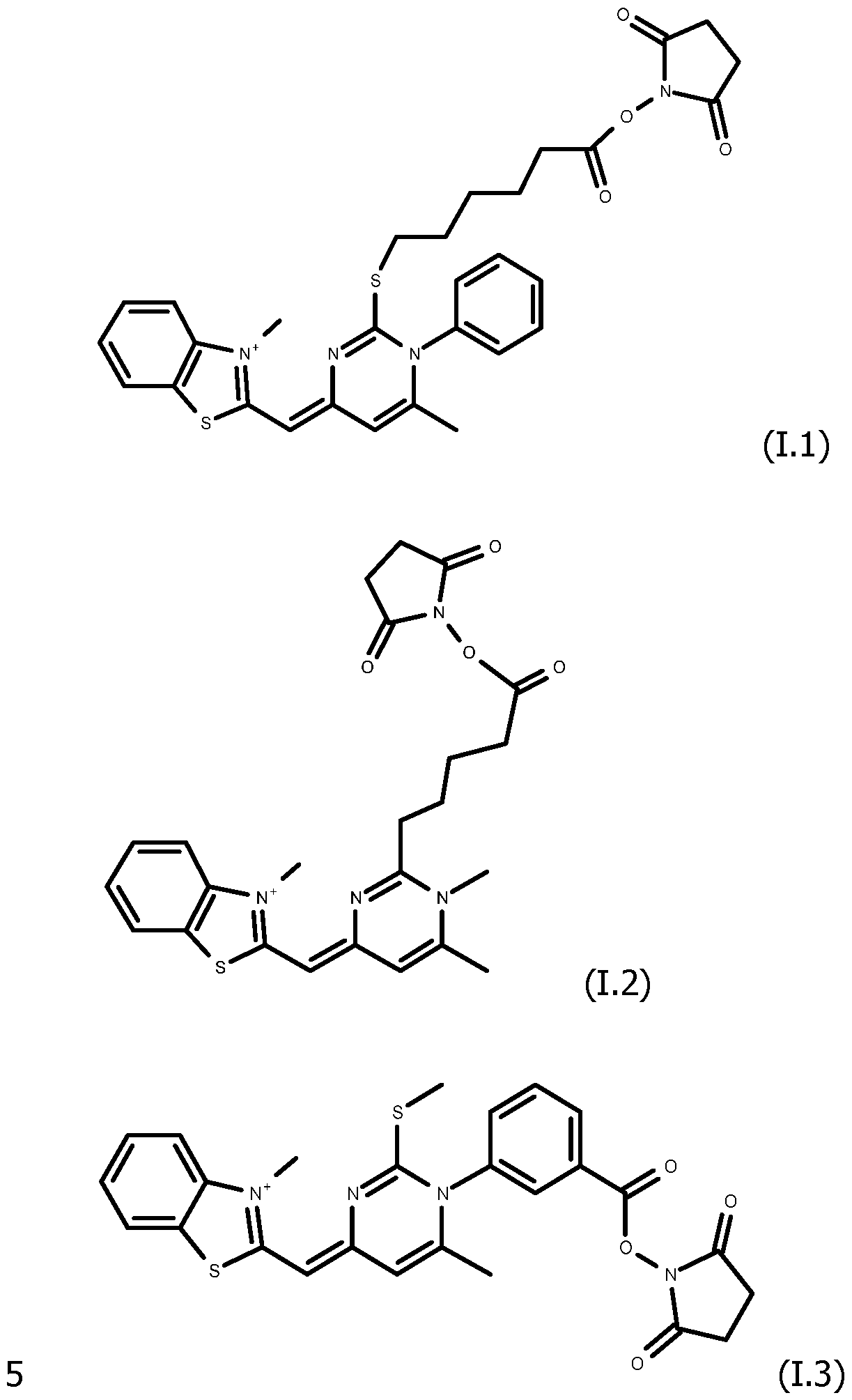

18. A detection probe PR labelled with at least one molecule of a compound FC according to any one of claims 1 to 17, comprising an oligonucleotide covalently coupled on the reactive group RG of said compound FC.
19.The detection probe PR according to claim 18, wherein the compound FC is coupled to said oligonucleotide by attachment to at least one nucleotide of said oligonucleotide.
20.The detection probe PR according to claim 18 or 19, wherein at least one region of the oligonucleotide selected from the 5' end, the 3' end, and the internal region of the oligonucleotide is bonded to a molecule of the compound FC.
Zl.The detection probe PR according to claim 20, wherein the compound FC is coupled to said oligonucleotide at its 5' or 3' end, by attachment to the phosphate group of the corresponding nucleotide.
22.The detection probe PR according to claim 20 or 21, wherein the compound FC is coupled to said oligonucleotide in its internal region.
23.The detection probe PR according to anyone of claims 18 to 22 wherein the oligonucleotide is bonded to at least two molecules of the same compound FC, with one being attached at its 5' end and another one being attached at its 3' end.
24.The detection probe PR according to claim 23 wherein the oligonucleotide is bonded to at least three molecules of the same compound FC, with at least one molecule of the said compound FC attached in the internal region of the oligonucleotide.
25.The detection probe PR according to claim 24 wherein the oligonucleotide is bonded to the compound FC in the internal region of the oligonucleotide by attachment to a base A, T or G of the oligonucleotide.
26.The detection probe PR according to anyone of claims 18 to 25, wherein the oligonucleotide is composed of 10 to 60 nucleotides, preferentially of 10 to 30 nucleotides, and more preferably of 12 to 25 nucleotides.
27. The detection probe PR according to anyone of claims 18 to 26, wherein the oligonucleotide is a single-strand nucleic acid comprising or composed of a complementary sequence to a DNA gene sequence of a target microorganism or a complementary sequence to a DNA gene sequence of a subject.
28.A method for the preparation of a detection probe PR as claimed in any of claims 18 to 27, comprising i) the synthesis of the oligonucleotide bearing at least one functional group Q suitable to react with the reactive
group RG that is present in Ri or R2 of the compound FC, eventually in a protected form, preferentially on a solid phase and ii) the covalent coupling of the compound FC, by reaction of the reactive group RG and the functional group Q.
29.A method for detecting a target nucleic acid in a sample, comprising incubating the sample with a detection probe PR as claimed in any of claims 18 to 28, in conditions suitable to obtain the hybridization of the target nucleic acid with the oligonucleotide of the detection probe PR, when the target nucleic acid is present in the sample, and detecting the hybridization obtained.
30.The method according to claim 29, characterized in that it includes an amplification step of the target nucleic acid.
31.The method according to claim 29 or 30 characterized in that the hybridization of the target nucleic acid with the oligonucleotide of the detection probe PR is detected during and/or after amplification of the target nucleic acid.
32.The method according to claim 30 or 31, characterized in that the amplification step is carried out by PCR, and in particular by real time PCR.
33.The method according to claim 32, characterized in that it comprises the steps of:
- mixing a detection probe PR according to any one of claims 18 to 27 with a sample comprising a target nucleic acid, a polymerase, and a pair of primers suitable to amplify a portion of the target nucleic acid and generate at least an amplicon, leading to a PCR mixture,
- amplifying the target nucleic acid from the PCR mixture and generating at least an amplicon, and
- monitoring the fluorescence from the compound FC, in its bonded form FC' in the detection probe PR according to any one of claims 18 to 27 during and/or subsequent to the amplifying step.
34.The method of claim 33, further comprising detecting the presence of the amplicon from the monitored fluorescence.
35.The method of claim 33 or 34, wherein the monitoring step occurs subsequent to amplification and includes generating a melting curve.
36.The method of claim 35 wherein the melting curve is used to identify the genotype of the target nucleic acid, to detect or identify at least one mutation, polymorphism, preferentially single nucleotide polymorphism, and/or epigenetic variation.
37.A method of performing a multiplex assay on a sample, comprising incubating the sample with a plurality of detection probes PR as claimed in any one of claims 18 to 27, said detection probes PR being labelled with different compounds FC.
38.A test kit comprising the detection probe PR as claimed in any one of claims 18 to 27 and at least one additional reagent.
39.A test kit according to claim 38 wherein it comprises an amplification enzyme and/or primer sequence for the amplification of a target nucleic acid sequence.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP23307400 | 2023-12-27 | ||
| EP23307400.4 | 2023-12-27 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2025141105A1 true WO2025141105A1 (en) | 2025-07-03 |
Family
ID=89620027
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2024/088487 Pending WO2025141105A1 (en) | 2023-12-27 | 2024-12-26 | Specific probes for the detection of nucleic acids, methods and uses |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2025141105A1 (en) |
Citations (25)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2607507A1 (en) | 1986-12-02 | 1988-06-03 | Centre Nat Rech Scient | NOVEL A-D-OLIGONUCLEOTIDE DERIVATIVES, THEIR PREPARATION AND THEIR USE |
| WO1990006995A1 (en) | 1988-12-16 | 1990-06-28 | Siska Diagnostics, Inc. | Self-sustained sequence replication system |
| US5118801A (en) | 1988-09-30 | 1992-06-02 | The Public Health Research Institute | Nucleic acid process containing improved molecular switch |
| US5399491A (en) | 1989-07-11 | 1995-03-21 | Gen-Probe Incorporated | Nucleic acid sequence amplification methods |
| US5863753A (en) | 1994-10-27 | 1999-01-26 | Molecular Probes, Inc. | Chemically reactive unsymmetrical cyanine dyes and their conjugates |
| US6329144B1 (en) | 1996-05-31 | 2001-12-11 | FORSKARPATENT I VäSTSVERIGE AB | Probe for analysis of target nucleic acids |
| US6387621B1 (en) | 1999-04-27 | 2002-05-14 | University Of Utah Research Foundation | Automated analysis of real-time nucleic acid amplification |
| US6410278B1 (en) | 1998-11-09 | 2002-06-25 | Eiken Kagaku Kabushiki Kaisha | Process for synthesizing nucleic acid |
| US6730501B2 (en) | 2002-02-12 | 2004-05-04 | University Of Utah Research Foundation | Multi-test analysis of real-time nucleic acid amplification |
| US6835542B2 (en) | 1998-07-02 | 2004-12-28 | Gen-Probe Incorporated | Molecular torches |
| US6902900B2 (en) | 2001-10-19 | 2005-06-07 | Prolico, Llc | Nucleic acid probes and methods to detect and/or quantify nucleic acid analytes |
| WO2006121423A2 (en) | 2004-04-20 | 2006-11-16 | University Of Utah Research Foundation | Nucleic acid melting analysis with saturation dyes |
| US7348141B2 (en) | 2000-03-29 | 2008-03-25 | Lgc Limited | Hybridization beacon and method of rapid sequence detection and discrimination |
| WO2008052742A1 (en) | 2006-11-02 | 2008-05-08 | Roche Diagnostics Gmbh | New ds dna binding fluorescent dyes |
| US7456281B2 (en) | 2005-04-20 | 2008-11-25 | Idaho Technology, Inc. | Nucleic acid melting analysis with saturation dyes |
| US20090111100A1 (en) | 2007-10-30 | 2009-04-30 | Eugene Lukhtanov | Minor groove binder - energy transfer oligonucleotides and methods for their use |
| US7582429B2 (en) | 2002-10-23 | 2009-09-01 | University Of Utah Research Foundation | Amplicon melting analysis with saturation dyes |
| WO2014005125A2 (en) * | 2012-06-29 | 2014-01-03 | Biotium, Inc. | Fluorescent compounds and uses thereof |
| US8663923B2 (en) | 2008-07-04 | 2014-03-04 | Biomerieux | Detection probe |
| WO2014039963A1 (en) | 2012-09-10 | 2014-03-13 | Biofire Diagnostics, Inc. | Multiple amplification cycle detection |
| WO2017165269A1 (en) | 2016-03-24 | 2017-09-28 | Biofire Diagnostics, Llc | Methods for quantitative amplification |
| US20180073056A1 (en) | 2016-09-15 | 2018-03-15 | Roche Molecular Systems, Inc. | Methods for performing multiplexed pcr |
| US9932634B2 (en) | 2012-05-24 | 2018-04-03 | University Of Utah Research Foundation | Methods for fast nucleic acid amplification |
| US20200407780A1 (en) | 2018-03-02 | 2020-12-31 | Life Technologies Corporation | Novel quencher and reporter dye combinations |
| WO2024110392A1 (en) * | 2022-11-22 | 2024-05-30 | bioMérieux | New nucleic acid binding compounds and uses |
-
2024
- 2024-12-26 WO PCT/EP2024/088487 patent/WO2025141105A1/en active Pending
Patent Citations (30)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2607507A1 (en) | 1986-12-02 | 1988-06-03 | Centre Nat Rech Scient | NOVEL A-D-OLIGONUCLEOTIDE DERIVATIVES, THEIR PREPARATION AND THEIR USE |
| US5118801A (en) | 1988-09-30 | 1992-06-02 | The Public Health Research Institute | Nucleic acid process containing improved molecular switch |
| US5312728A (en) | 1988-09-30 | 1994-05-17 | Public Health Research Institute Of The City Of New York, Inc. | Assays and kits incorporating nucleic acid probes containing improved molecular switch |
| WO1990006995A1 (en) | 1988-12-16 | 1990-06-28 | Siska Diagnostics, Inc. | Self-sustained sequence replication system |
| US5399491A (en) | 1989-07-11 | 1995-03-21 | Gen-Probe Incorporated | Nucleic acid sequence amplification methods |
| US5863753A (en) | 1994-10-27 | 1999-01-26 | Molecular Probes, Inc. | Chemically reactive unsymmetrical cyanine dyes and their conjugates |
| US6329144B1 (en) | 1996-05-31 | 2001-12-11 | FORSKARPATENT I VäSTSVERIGE AB | Probe for analysis of target nucleic acids |
| US6849412B2 (en) | 1998-07-02 | 2005-02-01 | Gen-Probe Incorporated | Molecular torches |
| US6835542B2 (en) | 1998-07-02 | 2004-12-28 | Gen-Probe Incorporated | Molecular torches |
| US6410278B1 (en) | 1998-11-09 | 2002-06-25 | Eiken Kagaku Kabushiki Kaisha | Process for synthesizing nucleic acid |
| US6387621B1 (en) | 1999-04-27 | 2002-05-14 | University Of Utah Research Foundation | Automated analysis of real-time nucleic acid amplification |
| US7348141B2 (en) | 2000-03-29 | 2008-03-25 | Lgc Limited | Hybridization beacon and method of rapid sequence detection and discrimination |
| US6902900B2 (en) | 2001-10-19 | 2005-06-07 | Prolico, Llc | Nucleic acid probes and methods to detect and/or quantify nucleic acid analytes |
| US7373253B2 (en) | 2002-02-12 | 2008-05-13 | Idaho Technology | Multi-test analysis of real-time nucleic acid amplification |
| US6730501B2 (en) | 2002-02-12 | 2004-05-04 | University Of Utah Research Foundation | Multi-test analysis of real-time nucleic acid amplification |
| US7582429B2 (en) | 2002-10-23 | 2009-09-01 | University Of Utah Research Foundation | Amplicon melting analysis with saturation dyes |
| US7387887B2 (en) | 2004-04-20 | 2008-06-17 | University Of Utah Research Foundation | Nucleic acid melting analysis with saturation dyes |
| WO2006121423A2 (en) | 2004-04-20 | 2006-11-16 | University Of Utah Research Foundation | Nucleic acid melting analysis with saturation dyes |
| US7456281B2 (en) | 2005-04-20 | 2008-11-25 | Idaho Technology, Inc. | Nucleic acid melting analysis with saturation dyes |
| WO2008052742A1 (en) | 2006-11-02 | 2008-05-08 | Roche Diagnostics Gmbh | New ds dna binding fluorescent dyes |
| US20090111100A1 (en) | 2007-10-30 | 2009-04-30 | Eugene Lukhtanov | Minor groove binder - energy transfer oligonucleotides and methods for their use |
| US8663923B2 (en) | 2008-07-04 | 2014-03-04 | Biomerieux | Detection probe |
| US9932634B2 (en) | 2012-05-24 | 2018-04-03 | University Of Utah Research Foundation | Methods for fast nucleic acid amplification |
| WO2014005125A2 (en) * | 2012-06-29 | 2014-01-03 | Biotium, Inc. | Fluorescent compounds and uses thereof |
| US9682970B2 (en) | 2012-06-29 | 2017-06-20 | Biotium, Inc. | Fluorescent compounds and uses thereof |
| WO2014039963A1 (en) | 2012-09-10 | 2014-03-13 | Biofire Diagnostics, Inc. | Multiple amplification cycle detection |
| WO2017165269A1 (en) | 2016-03-24 | 2017-09-28 | Biofire Diagnostics, Llc | Methods for quantitative amplification |
| US20180073056A1 (en) | 2016-09-15 | 2018-03-15 | Roche Molecular Systems, Inc. | Methods for performing multiplexed pcr |
| US20200407780A1 (en) | 2018-03-02 | 2020-12-31 | Life Technologies Corporation | Novel quencher and reporter dye combinations |
| WO2024110392A1 (en) * | 2022-11-22 | 2024-05-30 | bioMérieux | New nucleic acid binding compounds and uses |
Non-Patent Citations (17)
| Title |
|---|
| "DNA sequencing with Thermus aquaticus DNA polymerase and direct sequencing of polymerase chain reaction-amplified DNA", PROC. NATL. ACAD. SCI. U.S.A., vol. 85, pages 9436 - 40 |
| "Multiplex-Ready PCR: A new method for multiplexed SSR and SNP genotyping", BMC GENOMICS, vol. 9, 2008, pages 80 |
| ANAL. BIOANAL. CHEM., vol. 399, 2011, pages 3157 - 3176 |
| ANNU. REV. BIOMED. ENG., vol. 9, 2007, pages 289 - 320 |
| B.W. SUN ET AL., BIOCHEMISTRY, vol. 43, 2004, pages 4160 - 4169 |
| E. NAVARRO ET AL., CLINICA ACTA, vol. 439, 2015, pages 231 - 250 |
| J.F. MEDRANO ET AL., BIOTECHNIQUES, vol. 39, July 2005 (2005-07-01), pages 75 - 85 |
| LIEW ET AL.: "Genotyping of Single-Nucleotide Polymorphism by High-Resolution Melting of Small Amplicons", CLINICAL CHEMISTRY, vol. 50, no. 7, 2004, pages 1156 - 1164, XP008129908, DOI: 10.1373/clinchem.2004.032136 |
| M. EGHOLM, J. AM. CHEM. SOC., vol. 114, 1992, pages 1895 - 1897 |
| NUCLEIC ACIDS RESEARCH, vol. 41, no. 20, 2013, pages 4 |
| PORITZ ET AL., PLOS ONE, vol. 6, no. 10, pages 2604 7 |
| R.A. CARDULLO ET AL., PROC. NATL. ACAD. SCI. USA, vol. 85, 1988, pages 8790 - 8794 |
| REEDWITTWER: "Sensitivity and Specificity of Single-Nucleotide Polymorphism Scanning by High-Resolution Melting Analysis", CLINICAL CHEMISTRY, vol. 50, no. 10, 2004, pages 1748 - 1754, XP055113350, DOI: 10.1373/clinchem.2003.029751 |
| S.TYAGI ET AL., NATURE BIOTECHNOL., vol. 16, 1998, pages 49 - 53 |
| YANG ET AL., METHODS ENZYMOL., vol. 278, 1997, pages 417 - 44 |
| ZHOU ET AL.: "Closed-Tube Genotyping with Unlabeled Oligonucleotide Probes and a Saturating DNA Dye", CLINICAL CHEMISTRY, vol. 50, no. 8, 2004, pages 1328 - 1335, XP055041287, DOI: 10.1373/clinchem.2004.034322 |
| ZHOU ET AL.: "High-resolution DNA melting curve analysis to establish HLA genotypic identity", TISSUE ANTIGENS, vol. 64, 2004, pages 156 - 164, XP055050914, DOI: 10.1111/j.1399-0039.2004.00248.x |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US12480152B2 (en) | Probes for improved melt discrimination and multiplexing in nucleic acid assays | |
| CN101831496B (en) | Multiplex quantitative nucleic acid amplification and melting assay | |
| CN104379763B (en) | Polymerase chain reaction detection system using oligonucleotides containing phosphorothioate groups | |
| CN105200097A (en) | Improved allele-specific amplification | |
| JP7634551B2 (en) | Loop primer and loop-de-loop method for detecting target nucleic acid | |
| CN111247252A (en) | Methods and kits for the detection of nucleic acid molecules | |
| JP4481656B2 (en) | Methods of using FET labeled oligonucleotides comprising a 3 'to 5' exonuclease resistant quencher domain and compositions for performing the same | |
| WO2012038049A2 (en) | Amplification of distant nucleic acid targets using engineered primers | |
| CN105051212A (en) | Polymerase chain reaction detection system | |
| WO2025141105A1 (en) | Specific probes for the detection of nucleic acids, methods and uses | |
| AU2023386451A1 (en) | New nucleic acid binding compounds and uses | |
| US9260746B2 (en) | Photoinduced electron transfer (PET) primer for nucleic acid amplification | |
| Ranasinghe | Novel techniques for genetic analysis | |
| NZ630915B2 (en) | Polymerase chain reaction detection system using oligonucleotides comprising a phosphorothioate group | |
| HK1204015B (en) | Polymerase chain reaction detection system using oligonucleotides comprising a phosphorothioate group | |
| HK1236579A1 (en) | Probes for improved melt discrimination and multiplexing in nucleic acid assays |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 24840801 Country of ref document: EP Kind code of ref document: A1 |