WO2025122754A1 - Gaa knockout non-human animals - Google Patents
Gaa knockout non-human animals Download PDFInfo
- Publication number
- WO2025122754A1 WO2025122754A1 PCT/US2024/058681 US2024058681W WO2025122754A1 WO 2025122754 A1 WO2025122754 A1 WO 2025122754A1 US 2024058681 W US2024058681 W US 2024058681W WO 2025122754 A1 WO2025122754 A1 WO 2025122754A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- human animal
- gaa
- cell
- gene
- endogenous
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01K—ANIMAL HUSBANDRY; AVICULTURE; APICULTURE; PISCICULTURE; FISHING; REARING OR BREEDING ANIMALS, NOT OTHERWISE PROVIDED FOR; NEW BREEDS OF ANIMALS
- A01K67/00—Rearing or breeding animals, not otherwise provided for; New or modified breeds of animals
- A01K67/027—New or modified breeds of vertebrates
- A01K67/0275—Genetically modified vertebrates, e.g. transgenic
- A01K67/0276—Knock-out vertebrates
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01K—ANIMAL HUSBANDRY; AVICULTURE; APICULTURE; PISCICULTURE; FISHING; REARING OR BREEDING ANIMALS, NOT OTHERWISE PROVIDED FOR; NEW BREEDS OF ANIMALS
- A01K2217/00—Genetically modified animals
- A01K2217/07—Animals genetically altered by homologous recombination
- A01K2217/072—Animals genetically altered by homologous recombination maintaining or altering function, i.e. knock in
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01K—ANIMAL HUSBANDRY; AVICULTURE; APICULTURE; PISCICULTURE; FISHING; REARING OR BREEDING ANIMALS, NOT OTHERWISE PROVIDED FOR; NEW BREEDS OF ANIMALS
- A01K2217/00—Genetically modified animals
- A01K2217/07—Animals genetically altered by homologous recombination
- A01K2217/075—Animals genetically altered by homologous recombination inducing loss of function, i.e. knock out
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01K—ANIMAL HUSBANDRY; AVICULTURE; APICULTURE; PISCICULTURE; FISHING; REARING OR BREEDING ANIMALS, NOT OTHERWISE PROVIDED FOR; NEW BREEDS OF ANIMALS
- A01K2227/00—Animals characterised by species
- A01K2227/10—Mammal
- A01K2227/105—Murine
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01K—ANIMAL HUSBANDRY; AVICULTURE; APICULTURE; PISCICULTURE; FISHING; REARING OR BREEDING ANIMALS, NOT OTHERWISE PROVIDED FOR; NEW BREEDS OF ANIMALS
- A01K2267/00—Animals characterised by purpose
- A01K2267/03—Animal model, e.g. for test or diseases
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01K—ANIMAL HUSBANDRY; AVICULTURE; APICULTURE; PISCICULTURE; FISHING; REARING OR BREEDING ANIMALS, NOT OTHERWISE PROVIDED FOR; NEW BREEDS OF ANIMALS
- A01K2267/00—Animals characterised by purpose
- A01K2267/03—Animal model, e.g. for test or diseases
- A01K2267/0306—Animal model for genetic diseases
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/79—Vectors or expression systems specially adapted for eukaryotic hosts
- C12N15/85—Vectors or expression systems specially adapted for eukaryotic hosts for animal cells
- C12N15/8509—Vectors or expression systems specially adapted for eukaryotic hosts for animal cells for producing genetically modified animals, e.g. transgenic
Definitions
- a genetically modified non-human animal e.g., a rodent, e.g., a mouse, or a rat
- GAA a-glucosidase locus
- Pompe disease is an autosomal recessive disorder in which glycogen accumulates in the body’s cells due to a deficiency of acid alpha-glucosidase (GAA). GAA breaks down complex sugars in the body. A deficiency of GAA can result from mutations in the GAA gene. Without GAA, glycogen accumulation occurs in organs and tissues, especially in muscles, causing the muscles to break down.
- Mouse models of Pompe disease are useful for testing candidate therapies aimed at restoring GAA activity in patients in need thereof.
- a-glucosidase locus having a knockout mutation in the a-glucosidase (GAA) locus, and exhibiting ocular phenotypes associated with an accumulation of glycogen in ocular cells or tissues.
- the ocular phenotypes exhibited by the non-human animals described herein provide a useful biomarker to evaluate when testing the ability of a candidate therapeutic modality and/or compound to increase or restore GAA activity and/or reduce symptoms of Pompe disease.
- GAA a-glucosidase
- non-human animal, non-human animal cell, or non-human animal genome comprising a knockout mutation of an endogenous a-glucosidase (Gaa) gene.
- the knockout mutation comprises a deletion of the Gaa gene or a portion thereof.
- the knockout mutation comprises a deletion of the entire coding sequence of the Gaa gene.
- a non-human animal, a non-human animal cell, or a non-human animal genome in the Pompe disease model does not express GAA protein.
- a non-human animal, a non-human animal cell, or a non-human animal genome comprises an endogenous Gaa locus that comprises a nucleotide sequence selected from the group consisting of: the nucleotide sequence as set forth in SEQ ID NO: 26, the nucleotide sequence as set forth in SEQ ID NO: 27, the nucleotide sequence as set forth in SEQ ID NO: 29, or the nucleotide sequence as set forth in SEQ ID NO: 30.
- a non-human animal and/or Pompe disease model comprising a knockout mutation of an endogenous Gaa gene exhibits an ocular phenotype compared to a wildtype control non-human animal comprising a wildtype Gaa gene.
- the ocular phenotype is: (i) an accumulation of, e.g., substantially more, glycogen in ocular tissue (e.g., ciliary body, lens epithelium, neural retina) compared to a wildtype control non-human animal comprising a wildtype Gaa gene; (ii) a decrease in, e.g., significantly smaller, pupil size compared to a wildtype control non-human animal; (iii) vacuolar myopathy in the extraocular muscle; (iv) elevated glycogen levels in the iris; or (v) any combination thereof.
- ocular tissue e.g., ciliary body, lens epithelium, neural retina
- a genetically modified non-human animal disclosed herein has a significantly smaller pupil size when the non-human animal and/or Pompe disease model comprising a knockout mutation of an endogenous Gaa gene is at least 6 months in age. In some embodiments, a significantly smaller pupil size is seen when the non-human animal and/or Pompe disease model comprising a knockout mutation of an endogenous Gaa gene is at least 8 months in age. In some embodiments, a significantly smaller pupil size is seen when the non-human animal and/or Pompe disease model comprising a knockout mutation of an endogenous Gaa gene is at least 11 months in age.
- a non-human cell comprising a knockout mutation of an endogenous Gaa gene comprises an accumulation of glycogen in a cellular compartment (e.g., a lysosome) compared to a wildtype control non-human animal cell comprising a wildtype Gaa gene.
- Methods of knocking out an endogenous Gaa gene are also provided.
- the methods comprise modifying an endogenous Gaa locus of the non-human animal to comprise a knockout mutation of the Gaa gene.
- a non-human animal as described herein comprising a knockout mutation of an endogenous Gaa gene can be used for testing a candidate therapeutic modality and/or compound, e.g., a candidate therapeutic modality and/or compound comprising a- glucosidase.
- the candidate therapeutic modality and/or modality can increase GAA expression and/or activity in a subject.
- the therapeutic agent comprises encodes or comprises a protein having a-glucosidase activity, e.g., a-glucosidase or a biologically active portion thereof.
- a method utilizing a non-human animal comprising a knockout mutation of a Gaa gene and exhibiting an ocular phenotype as described herein comprises administering a candidate therapeutic agent to the non-human animal, and measuring the effect of the candidate therapeutic agent on the ocular phenotype of the non- human animal.
- measuring the effect comprises determining: (i) glycogen levels in ocular tissue (e.g., ciliary body, lens epithelium, neural retina, iris, etc.); (ii) pupil size; (iii) the absence or presence of vacuolar myopathy in the extraocular muscle; or (iv) any combination thereof.
- a therapeutic agent may be useful for increasing GAA activity in a patient in need thereof, e.g., in a patient with Pompe disease, if the therapeutic agent is able to reduce one or more of the ocular phenotypes exhibited by a non-human animal comprising a Gaa knockout mutation as described herein.
- a reduction of one or more of the ocular phenotypes exhibited by a non-human animal comprising a Gaa knockout mutation comprises (i) a decrease in glycogen levels in ocular tissue (e.g., ciliary body, lens epithelium, neural retina, iris) compared to the glycogen levels in the ocular tissue (e.g., ciliary body, lens epithelium, neural retina, iris) of the non-human animal comprising a knockout mutation of an endogenous Gaa gene prior to administration of the candidate therapeutic agent; (ii) an increase in pupil size of the non-human animal comprising a knockout mutation of an endogenous Gaa gene compared to pupil size of the non-human animal comprising a knockout mutation of an endogenous Gaa gene prior to administration of the candidate therapeutic agent; (iii) decreased vacuolar myopathy in the extraocular muscle of the non-human animal comprising a knockout mutation of an endogenous Gaa
- Figure 1 provides a schematic (not to scale) for the insertion of a Gaa knockout mutation comprising a replacement of an endogenous sequence spanning coding exon 1 (cExonl) and coding exon 19 (cExon 19) of an endogenous Gaa gene (SEQ ID NO: 28) with a Zen-Ubl targeting vector comprising a LacZ reporter gene (SEQ ID NO: 29 or SEQ ID NO: 30).
- the Zen-Ubl cassette also comprises a selection cassette comprising a promotor from the human ubiquitin C gene operably linked to a neomycin phosphotransferase (Neo) resistance gene.
- Figure 2A provides identifying information for a mouse a-glucosidase Gaa) gene (SEQ ID NO: 1) and the GAA proteins (SEQ ID NO: 2) so encoded, and four SpCas9 guide RNAs (gRNAs) that can be used to collapse the Gaa allele in a mouse embryonic stem cell.
- SEQ ID NO: 1 mouse a-glucosidase Gaa gene
- SEQ ID NO: 2 GAA proteins
- gRNAs SpCas9 guide RNAs
- Figure 2B provides a schematic (not to scale) of a Gaa knockout allele comprising a deletion of the Gaa gene sequence using guide RNAs that directed SpCas9 cleavage close to the Gaa ATG start codon (guide 9251mGU; SEQ ID NO: 5), cut site 38bp upstream from the ATG; guide 9251mGU3; SEQ ID NO: 6), cut site 18bp downstream of the ATG start codon) and after the stop codon (guide 9251mGD3; SEQ ID NO: 7], cut site 677bp downstream of the stop; guide 9251mGD4; SEQ ID NO: 8], cut site 705bp downstream of the stop codon).
- Approximate locations of the BHQ probes used to assay for loss-of-allele via TaqMan RT- PCR are shown with asterisks (*).
- Figure 3 provides the body weight (B.W. (gram), y-axis) of mice expressing wildtype GAA (GAA WT) or mice homozygous for the GAA knockout mutation described in Figure 1 (GAA KO) at 6, 8, and 11 months of age (x-axis).
- Figure 4 provides the cornea thicknesses (pm, y-axis) of the eyes of mice expressing wildtype GAA (GAA WT) or mice homozygous for the GAA knockout mutation described in Figure 1 (GAA KO).
- Figure 5 provides the intraocular pressure (1OP, y-axis) of the eyes of 7-month- old mice expressing wildtype GAA (GAA WT) or 7-month-old mice homozygous for the GAA knockout mutation described in Figure 1 (GAA KO).
- WT 9 mice (18 eyes);
- KO 15 mice (30 eyes).
- Figure 6 provides optical coherence tomography (OCT) images of a retina from a mouse expressing wildtype GAA (GAA WT) or a mouse homozygous for the GAA knockout mutation described in Figure 1 (GAA KO).
- OCT optical coherence tomography
- Figure 7 provides optical coherence tomography (OCT) images of a retina from a mouse expressing wildtype GAA (GAA WT) to depict the measurements used to determine corneal pupil diameters and areas.
- OCT optical coherence tomography
- Figure 8 provides graphs showing central pupil diameters (pm, y-axis) of the eyes of (A) 6-, (B) 8-, or (C) 11 month old mice expressing wildtype GAA (GAA WT) or mice homozygous for the GAA knockout mutation described in Figure 1 (GAA KO) with dilation (first two bars of each graph; x-axis) or with dilation with tropicamide (last two bars of each graph; x-axis).
- Figure 9 provides graphs showing central pupil areas (pm 2 , y-axis) of the eyes of (A) 6-, (B) 8-, or (C) 11 month old mice expressing wildtype GAA (GAA WT) or mice homozygous for the GAA knockout mutation described in Figure 1 (GAA KO) with dilation (first two bars of each graph; x-axis) or with dilation with tropicamide (last two bars of each graph; x-axis).
- Figure 10 provides histological images of eye sections isolated from a mouse expressing wildtype GAA (GAA WT) or a mouse homozygous for the GAA knockout mutation described in Figure 1 (GAA KO) and stained with Hematoxylin to show cell nuclei, and Eosin to show the extracellular matrix, cytoplasm, and other structures (which appears light grey in a black and white image). Arrows point to muscle fibers.
- Figure 11 provides histological images of extraocular muscle isolated from a mouse expressing wildtype GAA (GAA WT) or a mouse homozygous for the GAA knockout mutation described in Figure 1 (GAA KO) and stained with Hematoxylin to show cell nuclei, and Eosin to show the extracellular matrix, cytoplasm, and other structures (which appears light grey in a black and white image). Arrows point to muscle fibers.
- Figures 12A-12F provide images of (A) corneas, (B) irises, (C) ciliary bodies, (D) lens epithelial cells, (E) retinas, and (F) extraocular muscles isolated from mouse homozygous for the GAA knockout mutation described in Figure 1 (GAA KO) and incubated with Periodic Acid Schiff (PAS) stain. Stronger or darker PAS staining indicates more glycogen accumulation.
- A corneas
- B irises
- C ciliary bodies
- D lens epithelial cells
- E retinas
- F extraocular muscles isolated from mouse homozygous for the GAA knockout mutation described in Figure 1 (GAA KO) and incubated with Periodic Acid Schiff (PAS) stain. Stronger or darker PAS staining indicates more glycogen accumulation.
- PAS Periodic Acid Schiff
- Figure 13 provides graphs showing the levels of glycogen (pg/mg protein) in the hearts or irises of mice expressing wildtype GAA (GAA WT) or mice homozygous for the GAA knockout mutation described in Figure 1 (GAA KO), and a correlation graph and chart providing the correlation of glycogen levels in iris and heart.
- Figure 14 provides a western blot showing anti-TfR scFv:GAA expression in the serum of GAA KO mice one month after AAV8 anti-hTfR scFv:GAA (3E13vg/kg)/LNP (3mg/kg) treatment (lanes 4-25). Serum from untreated GAA KO mice is also provided as negative controls (lanes 2 and 3). Each lane represents an individual mouse.
- Pompe disease is an autosomal recessive disorder resulting from a deficiency of acid alpha-glucosidase (GAA), leading to progressive intralysosomal glycogen accumulation in tissues due to mutations in the GAA gene.
- GAA acid alpha-glucosidase
- Some patients have ocular phenotypes such as bilateral ptosis, strabismus, myopia, and astigmatism.
- the iris is the tissue with the second highest expression of GAA in a mouse.
- determining whether a Pompe disease model in mice also demonstrates ocular phenotypes may be useful in validating the model as a platform to screen candidate therapeutics for treating Pompe disease.
- OCT optical coherence tomography
- Gaa' 7 ' knockout mice exhibited significantly smaller pupil sizes (p ⁇ 0.01 ) at 6 and 8 months of age, a trend that was also observed at 11 months age ( Figure 8 and Figure 9).
- the electroretinogram (ERG) test (an assay to measure changes in retinal function) did not show remarkable change in Gaa' 7 ' mice. Vacuolar myopathy was observed in the extraocular muscle of Gaa 7 ' mice ( Figure 11).
- the complete gene sequence for mouse Gaa can be found from the NCBI database (Gene ID: 14387).
- An example coding sequence for mGaa is set forth in SEQ ID NO: 1.
- non-human animal cells, non-human animals, and non-human genomes comprising a Gaa knockout mutation, and exhibiting an ocular phenotype.
- the knockout mutation comprises a deletion of the entire coding sequence of an endogenous Gaa gene.
- a non-human animal cell, non-human animal, or non-human genome described herein comprises, at an endogenous Gaa allele, a nucleotide sequence set forth as SEQ ID NO: 26, a nucleotide sequence set forth as SEQ ID NO: 27, a nucleotide sequence set forth as SEQ ID NO:29, or a nucleotide sequence set forth as SEQ ID NO:30, e.g., as a replacement for the wildtype endogenous Gaa allele.
- a non-human animal cell, non-human animal, or non- human genome described herein comprises, at an endogenous Gaa allele, a nucleotide sequence set forth as SEQ ID NO: 26, e.g., as a replacement for the wildtype endogenous Gaa allele.
- a non-human animal cell, non-human animal, or non- human genome described herein comprises, at an endogenous Gaa allele, a nucleotide sequence set forth as SEQ ID NO: 27, e.g., as a replacement for the wildtype endogenous Gaa allele.
- a non-human animal cell, non-human animal, or non- human genome described herein comprises, at an endogenous Gaa allele, a nucleotide sequence set forth as SEQ ID NO: 29, e.g., as a replacement for the wildtype endogenous Gaa allele.
- a non-human animal cell, non-human animal, or non- human genome described herein comprises, at an endogenous Gaa allele, a nucleotide sequence set forth as SEQ ID NO: 30, e.g., as a replacement for the wildtype endogenous Gaa allele.
- the Gaa knockout mutation sequence can be inserted into an endogenous Gaa locus, thus providing non-human animal cells and non-human animals having a genetically modified endogenous Gaa locus.
- a non-human animal and/or Pompe disease model comprising a knockout mutation of an endogenous Gaa gene comprises an ocular phenotype compared to a wildtype control non-human animal comprising a wildtype Gaa gene.
- the ocular phenotype is: (i) an accumulation of, e.g., substantially more, glycogen in ocular tissue (e.g., ciliary body, lens epithelium, neural retina) compared to a wildtype control non-human animal comprising a wildtype Gaa gene; (ii) a decrease in, e.g., significantly smaller, pupil size compared to a wildtype control non-human animal; (iii) vacuolar myopathy in the extraocular muscle; (iv) elevated glycogen levels in the iris; or (v) any combination thereof.
- ocular tissue e.g., ciliary body, lens epithelium, neural retina
- a genetically modified non-human animal disclosed herein has a significantly smaller pupil size when the non-human animal and/or Pompe disease model comprising a knockout mutation of an endogenous Gaa gene is at least 6 months in age. In some embodiments, a significantly smaller pupil size is seen when the non-human animal and/or Pompe disease model comprising a knockout mutation of an endogenous Gaa gene is at least 8 months in age. In some embodiments, a significantly smaller pupil size is seen when the non-human animal and/or Pompe disease model comprising a knockout mutation of an endogenous Gaa gene is at least 11 months in age.
- a non-human cell comprising a knockout mutation of an endogenous Gaa gene comprises an accumulation of glycogen in a cellular compartment (e.g., a lysosome) compared to a wildtype control non-human animal cell comprising a wildtype Gaa gene.
- a cellular compartment e.g., a lysosome
- Such animals may be used in methods of screening drugs that are candidates for increasing GAA activity in a patient in need thereof, e.g., in a patient with Pompe disease.
- the non-human animal is a mammal, or the non-human animal genome is a mammalian genome.
- the non-human animal can be a rodent, or the non-human animal genome can be a rodent genome.
- the non-human animal can be a rat or mouse, or the non-human animal genome can be a rat genome or a mouse genome.
- protein polypeptide
- polypeptide polymeric forms of amino acids of any length, including coded and noncoded amino acids and chemically or biochemically modified or derivatized amino acids.
- the terms also include polymers that have been modified, such as polypeptides having modified peptide backbones.
- domain can refer to any part of a protein or polypeptide having a particular function or structure.
- Proteins are said to have an “N-terminus” and a “C-terminus.”
- N- terminus relates to the start of a protein or polypeptide, terminated by an amino acid with a free amine group (-NH2).
- C-terminus relates to the end of an amino acid chain (protein or polypeptide), terminated by a free carboxyl group (-COOH).
- nucleic acid and polynucleotide include polymeric forms of nucleotides of any length, including ribonucleotides, deoxyribonucleotides, or analogs or modified versions thereof.
- Nucleic acids and polynucleotides can include single-, double-, and multi-stranded DNA or RNA, genomic DNA, cDNA, DNA-RNA hybrids, and polymers comprising purine bases, pyrimidine bases, or other natural, chemically modified, biochemically modified, non-natural, or derivatized nucleotide bases.
- Nucleic acids are said to have “5’ ends” and “3’ ends” because mononucleotides are reacted to make oligonucleotides in a manner such that the 5 ’ phosphate of one mononucleotide pentose ring is attached to the 3’ oxygen of its neighbor in one direction via a phosphodiester linkage.
- An end of an oligonucleotide is referred to as the “5’ end” if its 5’ phosphate is not linked to the 3’ oxygen of a mononucleotide pentose ring.
- An end of an oligonucleotide is referred to as the “3’ end” if its 3’ oxygen is not linked to a 5’ phosphate of another mononucleotide pentose ring.
- a nucleic acid sequence even if internal to a larger oligonucleotide, also may be said to have 5’ and 3’ ends.
- discrete elements are referred to as being “upstream” or 5’ of the “downstream” or 3’ elements.
- the term “genomically integrated” refers to a nucleic acid that has been introduced into a cell such that the nucleotide sequence integrates into the genome of the cell and is capable of being inherited by progeny thereof. Any protocol may be used for the stable incorporation of a nucleic acid into the genome of a cell.
- targeting vector refers to a recombinant nucleic acid that can be introduced by homologous recombination, non-homologous-end-joining-mediated ligation, or any other means of recombination to a target position in the genome of a cell.
- viral vector refers to a recombinant nucleic acid that includes at least one element of viral origin and includes elements sufficient for or permissive of packaging into a viral vector particle.
- the vector and/or particle can be utilized for the purpose of transferring DNA, RNA, or other nucleic acids into cells either ex vivo or in vivo. Numerous forms of viral vectors are known.
- wild type includes entities having a structure and/or activity as found in a normal (as contrasted with mutant, diseased, altered, or so forth) state or context. Wild type genes and polypeptides often exist in multiple different forms (e.g., alleles).
- gross mutant phenotype refers to a significant difference or variation in phenotype between an engineered non-human mouse of the disclosure and a “wild type.”
- endogenous refers to a nucleic acid sequence that occurs naturally within a cell or non-human animal.
- Exogenous molecules or sequences include molecules or sequences that are not normally present in a cell in that form. Normal presence includes presence with respect to the particular developmental stage and environmental conditions of the cell.
- An exogenous molecule or sequence for example, can include a mutated version of a corresponding endogenous sequence within the cell, such as a humanized version of the endogenous sequence, or can include a sequence corresponding to an endogenous sequence within the cell but in a different form (i.e., not within a chromosome).
- endogenous molecules or sequences include molecules or sequences that are normally present in that form in a particular cell at a particular developmental stage under particular environmental conditions.
- heterologous when used in the context of a nucleic acid or a protein indicates that the nucleic acid or protein comprises at least two portions that do not naturally occur together in the same molecule.
- a “heterologous” region of a nucleic acid vector is a segment of nucleic acid within or attached to another nucleic acid molecule that is not found in association with the other molecule in nature.
- a heterologous region of a nucleic acid vector could include a coding sequence flanked by sequences not found in association with the coding sequence in nature.
- a “heterologous” region of a protein is a segment of amino acids within or attached to another peptide molecule that is not found in association with the other peptide molecule in nature (e.g., a fusion protein, or a protein with a tag).
- a nucleic acid or protein can comprise a heterologous label or a heterologous secretion or localization sequence.
- Codon optimization takes advantage of the degeneracy of codons, as exhibited by the multiplicity of three-base pair codon combinations that specify an amino acid, and generally includes a process of modifying a nucleic acid sequence for enhanced expression in particular host cells by replacing at least one codon of the native sequence with a codon that is more frequently or most frequently used in the genes of the host cell while maintaining the native amino acid sequence.
- a nucleic acid encoding a Cas9 protein can be modified to substitute codons having a higher frequency of usage in a given prokaryotic or eukaryotic cell, including a bacterial cell, a yeast cell, a human cell, a non-human cell, a mammalian cell, a rodent cell, a mouse cell, a rat cell, a hamster cell, or any other host cell, as compared to the naturally occurring nucleic acid sequence.
- Codon usage tables are readily available, for example, at the “Codon Usage Database.” These tables can be adapted in a number of ways. See Nakamura et al. (2000) Nucleic Acids Research 28:292, herein incorporated by reference in its entirety for all purposes.
- nucleic acid sequence as disclosed herein encompasses variants thereof, including those variants that differ due to degeneracy of the genetic code and/or codon optimization, and that encode the same or substantially similar amino acid sequence of a biologically active polypeptide.
- locus refers to a specific location of a gene (or significant sequence), DNA sequence, polypeptide-encoding sequence, or position on a chromosome of the genome of an organism.
- an “Gaa locus” may refer to the specific location of a Gaa gene, Gaa DNA sequence, Gaa-encoding sequence, or Gaa position on a chromosome of the genome of an organism that has been identified as to where such a sequence resides.
- a “Gaa locus” may comprise a regulatory element of an Gaa gene, including, for example, an enhancer, a promoter, 5’ and/or 3’ untranslated region (UTR), or a combination thereof.
- the term “gene” refers to a DNA sequence in a chromosome that codes for a product (e.g., an RNA product and/or a polypeptide product) and includes the coding region interrupted with non-coding introns and sequence located adjacent to the coding region on both the 5’ and 3’ ends such that the gene corresponds to the full-length mRNA (including the 5’ and 3’ untranslated sequences).
- the term “gene” also includes other non-coding sequences including regulatory sequences (e.g., promoters, enhancers, and transcription factor binding sites), polyadenylation signals, internal ribosome entry sites, silencers, insulating sequence, and matrix attachment regions. These sequences may be close to the coding region of the gene (e.g., within 10 kb) or at distant sites, and they influence the level or rate of transcription and translation of the gene.
- allele refers to a variant form of a gene. Some genes have a variety of different forms, which are located at the same position, or genetic locus, on a chromosome. A diploid organism has two alleles at each genetic locus. Each pair of alleles represents the genotype of a specific genetic locus. Genotypes are described as homozygous if there are two identical alleles at a particular locus and as heterozygous if the two alleles differ.
- a “promoter” is a regulatory region of DNA usually comprising a TATA box capable of directing RNA polymerase II to initiate RNA synthesis at the appropriate transcription initiation site for a particular polynucleotide sequence.
- a promoter may additionally comprise other regions which influence the transcription initiation rate.
- the promoter sequences disclosed herein modulate transcription of an operably linked polynucleotide.
- a promoter can be active in one or more of the cell types disclosed herein (e.g., a eukaryotic cell, a non-human mammalian cell, a human cell, a rodent cell, a pluripotent cell, a one-cell stage embryo, a differentiated cell, or a combination thereof).
- a promoter can be, for example, a constitutively active promoter, a conditional promoter, an inducible promoter, a temporally restricted promoter (e.g., a developmentally regulated promoter), or a spatially restricted promoter (e.g., a cell-specific or tissue-specific promoter). Examples of promoters can be found, for example, in WO 2013/176772, herein incorporated by reference in its entirety for all purposes.
- “Operable linkage” or being “operably linked” includes juxtaposition of two or more components (e.g., a promoter and another sequence element) such that both components function normally and allow the possibility that at least one of the components can mediate a function that is exerted upon at least one of the other components.
- a promoter can be operably linked to a coding sequence if the promoter controls the level of transcription of the coding sequence in response to the presence or absence of one or more transcriptional regulatory factors.
- Operable linkage can include such sequences being contiguous with each other or acting in trans (e.g., a regulatory sequence can act at a distance to control transcription of the coding sequence).
- variant refers to a nucleotide sequence differing from the sequence most prevalent in a population (e.g., by one nucleotide) or a protein sequence different from the sequence most prevalent in a population (e.g., by one amino acid).
- fragment when referring to a protein means a protein that is shorter or has fewer amino acids than the full-length protein.
- fragment when referring to a nucleic acid means a nucleic acid that is shorter or has fewer nucleotides than the full-length nucleic acid.
- a fragment can be, for example, an N-terminal fragment (i.e., removal of a portion of the C-terminal end of the protein), a C-terminal fragment (i.e., removal of a portion of the N-terminal end of the protein), or an internal fragment.
- sequence identity in the context of two polynucleotides or polypeptide sequences makes reference to the residues in the two sequences that are the same when aligned for maximum correspondence over a specified comparison window.
- residue positions which are not identical often differ by conservative amino acid substitutions, where amino acid residues are substituted for other amino acid residues with similar chemical properties (e.g., charge or hydrophobicity) and therefore do not change the functional properties of the molecule.
- sequences differ in conservative substitutions the percent sequence identity may be adjusted upwards to correct for the conservative nature of the substitution.
- Sequences that differ by such conservative substitutions are said to have “sequence similarity” or “similarity.” Means for making this adjustment are well known. Typically, this involves scoring a conservative substitution as a partial rather than a full mismatch, thereby increasing the percentage sequence identity. Thus, for example, where an identical amino acid is given a score of 1 and a non-conservative substitution is given a score of zero, a conservative substitution is given a score between zero and 1. The scoring of conservative substitutions is calculated, e.g., as implemented in the program PC/GENE (Intelligenetics, Mountain View, California).
- Percentage of sequence identity includes the value determined by comparing two optimally aligned sequences (greatest number of perfectly matched residues) over a comparison window, wherein the portion of the polynucleotide sequence in the comparison window may comprise additions or deletions (i.e., gaps) as compared to the reference sequence (which does not comprise additions or deletions) for optimal alignment of the two sequences. The percentage is calculated by determining the number of positions at which the identical nucleic acid base or amino acid residue occurs in both sequences to yield the number of matched positions, dividing the number of matched positions by the total number of positions in the window of comparison, and multiplying the result by 100 to yield the percentage of sequence identity. Unless otherwise specified (e.g., the shorter sequence includes a linked heterologous sequence), the comparison window is the full length of the shorter of the two sequences being compared.
- sequence identity/similarity values include the value obtained using GAP Version 10 using the following parameters: % identity and % similarity for a nucleotide sequence using GAP Weight of 50 and Length Weight of 3, and the nwsgapdna.cmp scoring matrix; % identity and % similarity for an amino acid sequence using GAP Weight of 8 and Length Weight of 2, and the BLOSUM62 scoring matrix; or any equivalent program thereof.
- “Equivalent program” includes any sequence comparison program that, for any two sequences in question, generates an alignment having identical nucleotide or amino acid residue matches and an identical percent sequence identity when compared to the corresponding alignment generated by GAP Version 10.
- conservative amino acid substitution refers to the substitution of an amino acid that is normally present in the sequence with a different amino acid of similar size, charge, or polarity.
- conservative substitutions include the substitution of a non-polar (hydrophobic) residue such as isoleucine, valine, or leucine for another non-polar residue.
- conservative substitutions include the substitution of one polar (hydrophilic) residue for another such as between arginine and lysine, between glutamine and asparagine, or between glycine and serine.
- substitution of a basic residue such as lysine, arginine, or histidine for another, or the substitution of one acidic residue such as aspartic acid or glutamic acid for another acidic residue are additional examples of conservative substitutions.
- non-conservative substitutions include the substitution of a non-polar (hydrophobic) amino acid residue such as isoleucine, valine, leucine, alanine, or methionine for a polar (hydrophilic) residue such as cysteine, glutamine, glutamic acid or lysine and/or a polar residue for a non-polar residue.
- Typical amino acid categorizations are summarized below.
- a “homologous” sequence includes a sequence that is either identical or substantially similar to a known reference sequence, such that it is, for example, at least 50%, at least 55%, at least 60%, at least 65%, at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, at least 95%, at least 96%, at least 97%, at least 98%, at least 99%, or 100% identical to the known reference sequence.
- Homologous sequences can include, for example, orthologous sequence and paralogous sequences.
- Homologous genes typically descend from a common ancestral DNA sequence, either through a speciation event (orthologous genes) or a genetic duplication event (paralogous genes).
- Orthologous genes include genes in different species that evolved from a common ancestral gene by speciation. Orthologs typically retain the same function in the course of evolution.
- Parentous genes include genes related by duplication within a genome. Paralogs can evolve new functions in the course of evolution.
- in vitro includes artificial environments and to processes or reactions that occur within an artificial environment (e.g., a test tube).
- in vivo includes natural environments (e.g., a cell or organism or body) and to processes or reactions that occur within a natural environment.
- ex vivo includes cells that have been removed from the body of an individual and to processes or reactions that occur within such cells.
- reporter gene refers to a nucleic acid having a sequence encoding a gene product (typically an enzyme) that is easily and quantifiably assayed when a construct comprising the reporter gene sequence operably linked to a heterologous promoter and/or enhancer element is introduced into cells containing (or which can be made to contain) the factors necessary for the activation of the promoter and/or enhancer elements.
- a gene product typically an enzyme
- reporter genes include, but are not limited, to genes encoding beta-galactosidase (lacZ), the bacterial chloramphenicol acetyltransferase (cat) genes, firefly luciferase genes, genes encoding beta-glucuronidase (GUS), and genes encoding fluorescent proteins.
- lacZ beta-galactosidase
- cat bacterial chloramphenicol acetyltransferase
- GUS beta-glucuronidase
- fluorescent proteins include, but are not limited, to genes encoding beta-galactosidase (lacZ), the bacterial chloramphenicol acetyltransferase (cat) genes, firefly luciferase genes, genes encoding beta-glucuronidase (GUS), and genes encoding fluorescent proteins.
- a “reporter protein” refers to a protein encoded by a reporter gene.
- fluorescent reporter protein means a reporter protein that is detectable based on fluorescence wherein the fluorescence may be either from the reporter protein directly, activity of the reporter protein on a fluorogenic substrate, or a protein with affinity for binding to a fluorescent tagged compound.
- fluorescent proteins examples include green fluorescent proteins (e.g., GFP, GFP-2, tagGFP, turboGFP, eGFP, Emerald, Azami Green, Monomeric Azami Green, CopGFP, AceGFP, and ZsGreenl), yellow fluorescent proteins (e.g., YFP, eYFP, Citrine, Venus, YPet, PhiYFP, and ZsYellowl), blue fluorescent proteins (e.g., BFP, eBFP, eBFP2, Azurite, mKalamal, GFPuv, Sapphire, and T- sapphire), cyan fluorescent proteins (e.g., CFP, eCFP, Cerulean, CyPet, AmCyanl, and Midoriishi-Cyan), red fluorescent proteins (e.g., RFP, mKate, mKate2, mPlum, DsRed monomer, mCherry, mRFPl, DsRed-Express, DsRed2, DsRed
- the term “recombination” includes any process of exchange of genetic information between two polynucleotides and can occur by any mechanism. Recombination in response to double-strand breaks (DSBs) occurs principally through two conserved DNA repair pathways: non-homologous end joining (NHEJ) and homologous recombination (HR). See Kasparek & Humphrey (2011) Seminars in Cell & Dev. Biol. 22:886-897, herein incorporated by reference in its entirety for all purposes. Likewise, repair of a target nucleic acid mediated by an exogenous donor nucleic acid can include any process of exchange of genetic information between the two polynucleotides.
- NHEJ includes the repair of double-strand breaks in a nucleic acid by direct ligation of the break ends to one another or to an exogenous sequence without the need for a homologous template. Ligation of non-contiguous sequences by NHEJ can often result in deletions, insertions, or translocations near the site of the double-strand break. For example, NHEJ can also result in the targeted integration of an exogenous donor nucleic acid through direct ligation of the break ends with the ends of the exogenous donor nucleic acid (i.e., NHEJ-based capture).
- NHEJ-mediated targeted integration can be preferred for insertion of an exogenous donor nucleic acid when homology directed repair (HDR) pathways are not readily usable (e.g., in non-dividing cells, primary cells, and cells which perform homology-based DNA repair poorly).
- HDR homology directed repair
- knowledge concerning large regions of sequence identity flanking the cleavage site is not needed, which can be beneficial when attempting targeted insertion into organisms that have genomes for which there is limited knowledge of the genomic sequence.
- the integration can proceed via ligation of blunt ends between the exogenous donor nucleic acid and the cleaved genomic sequence, or via ligation of sticky ends (i.e., having 5’ or 3’ overhangs) using an exogenous donor nucleic acid that is flanked by overhangs that are compatible with those generated by a nuclease agent in the cleaved genomic sequence.
- blunt ends are ligated, target and/or donor resection may be needed to generation regions of microhomology needed for fragment joining, which may create unwanted alterations in the target sequence.
- HDR or HR includes a form of nucleic acid repair that can require nucleotide sequence homology, uses a “donor” molecule as a template for repair of a “target” molecule (i.e., the one that experienced the double-strand break), and leads to transfer of genetic information from the donor to target.
- donor a template for repair of a “target” molecule
- target i.e., the one that experienced the double-strand break
- transfer can involve mismatch correction of heteroduplex DNA that forms between the broken target and the donor, and/or synthesis -dependent strand annealing, in which the donor is used to resynthesize genetic information that will become part of the target, and/or related processes.
- the donor polynucleotide, a portion of the donor polynucleotide, a copy of the donor polynucleotide, or a portion of a copy of the donor polynucleotide integrates into the target DNA. See Wang et al. (2013) Cell 153:910-918; Mandalos et al. (2012) PLOS ONE 7:e45768: 1-9; and Wang et al. (2013) Nat Biotechnol. 31:530-532, each of which is herein incorporated by reference in its entirety for all purposes. [0064] “Optional” or “optionally” means that the subsequently described event or circumstance may or may not occur and that the description includes instances in which the event or circumstance occurs and instances in which it does not.
- Designation of a range of values includes all integers within or defining the range, and all subranges defined by integers within the range.
- a protein or “at least one protein” can include a plurality of proteins, including mixtures thereof.
- Non-human animal cells, non-human animal genomes (e.g., non-human animal cell nuclei), and non-human animals comprising a knockout Gaa mutation as described herein are provided, wherein the non-human animals exhibit an ocular phenotype, such as but not limited to: (i) an accumulation of, e.g., substantially more, glycogen in ocular tissue (e.g., ciliary body, lens epithelium, neural retina) compared to a wildtype control non-human animal comprising a wildtype Gaa gene; (ii) a decrease in, e.g., significantly smaller, pupil size compared to a wildtype control non-human animal; (iii) vacuolar myopathy in the extraocular muscle; (iv) elevated glycogen levels in the iris; or (v) any combination thereof.
- an ocular phenotype such as but not limited to: (i) an accumulation of, e.g., substantially more, glycogen in ocular tissue (e.g.,
- a non-human animal cell or tissue e.g., an ocular cell or tissue
- a knockout Gaa mutation as described herein, wherein the non- human cell or tissue exhibits a phenotype not seen in a corresponding cell or tissue expressing a wildtype GAA or having wildtype GAA activity.
- a non- human animal cell or tissue as described herein comprises: (i) an ocular ciliary body, lens epithelium, and/or neural retina comprising a knockout mutation of an endogenous Gaa gene and exhibiting an accumulation of, e.g., substantially more, glycogen compared to a wildtype control non-human animal ocular ciliary body, lens epithelium, neural retina, and/or iris comprising a wildtype Gaa gene or activity; (ii) a pupil comprising a knockout mutation in a Gaa gene and exhibiting a decrease in, e.g., significantly smaller, 1 size (e.g., diameter and/or area) compared to a wildtype pupil control non-human animal pupil comprising a wildtype Gaa gene or activity; (iii) extraocular muscle comprising a knockout mutation of an endogenous Gaa gene and exhibiting increased vacuolar myopathy compared to a wildtype extraocular muscle comprising a wildtype Gaa gene or activity;
- the cells, genomes (nuclei), or non-human animals can be heterozygous or homozygous for the knockout Gaa mutation.
- a diploid organism has two alleles at each genetic locus. Each pair of alleles represents the genotype of a specific genetic locus. Genotypes are described as homozygous if there are two identical alleles at a particular locus and as heterozygous if the two alleles differ.
- the disclosure further provides methods for making any non-human animal, or reagents required for making the non-human animal as described herein.
- the non-human animal cells and genomes (e.g., nuclei) provided herein can be, for example, any non-human animal cell comprising a knockout mutation in an endogenous Gaa gene.
- the cells and genomes (e.g., nuclei) can be eukaryotic, which include, for example, fungal (e.g., yeast) cells and genomes, plant cells and genomes, animal cells and genomes, mammalian cells and genomes, and non-human mammalian cells and genomes.
- a non-human animal can be, for example, a mammal, fish, or bird.
- a mammalian cell can be, for example, a non-human mammalian cell, a rodent cell, a rat cell, a mouse cell, or a hamster cell.
- Other non-human mammals include, for example, non-human primates, monkeys, apes, orangutans, cats, dogs, rabbits, horses, bulls, deer, bison, livestock (e.g., bovine species such as cows, steer, and so forth; ovine species such as sheep, goats, and so forth; and porcine species such as pigs and boars).
- Birds include, for example, chickens, turkeys, ostrich, geese, ducks, and so forth. Domesticated animals and agricultural animals are also included.
- the term “non-human” excludes humans.
- the cells can also be any type of undifferentiated or differentiated state.
- a cell can be a totipotent cell, a pluripotent cell (e.g., a human pluripotent cell or a non-human pluripotent cell such as a mouse embryonic stem (ES) cell or a rat ES cell), or a non-pluripotent cell.
- Totipotent cells include undifferentiated cells that can give rise to any cell type, and pluripotent cells include undifferentiated cells that possess the ability to develop into more than one differentiated cell types.
- pluripotent and/or totipotent cells can be, for example, ES cells or ES-like cells, such as an induced pluripotent stem (iPS) cells.
- iPS induced pluripotent stem
- ES cells include embryo-derived totipotent or pluripotent cells that can contribute to any tissue of the developing embryo upon introduction into an embryo.
- ES cells can be derived from the inner cell mass of a blastocyst and can differentiate into cells of any of the three vertebrate germ layers (endoderm, ectoderm, and mesoderm).
- the cells provided herein can also be germ cells (e.g., sperm or oocytes).
- the cells can be mitotically competent cells or mitotically-inactive cells, meiotically competent cells or meiotically-inactive cells.
- the cells disclosed herein can also be primary somatic cells or cells that are not a primary somatic cell.
- Somatic cells include any cell that is not a gamete, germ cell, gametocyte, or undifferentiated stem cell.
- Suitable cells provided herein also include primary cells.
- Primary cells include cells or cultures of cells that have been isolated directly from an organism, organ, or tissue.
- Primary cells include cells that are neither transformed nor immortal.
- Primary cells include any cell obtained from an organism, organ, or tissue which was not previously passed in tissue culture or has been previously passed in tissue culture but is incapable of being indefinitely passed in tissue culture. Such cells can be isolated by conventional techniques.
- immortalized cells include cells from a multicellular organism that would normally not proliferate indefinitely but, due to mutation or alteration, have evaded normal cellular senescence and instead can keep undergoing division. Such mutations or alterations can occur naturally or be intentionally induced. Examples of immortalized cell lines are myofiber cell lines.
- Immortalized or primary cells include cells that can be used for culturing or for expressing recombinant genes or proteins.
- the cells provided herein also include one-cell stage embryos (i.e., fertilized oocytes or zygotes).
- Such one-cell stage embryos can be from any genetic background (e.g., BALB/c, C57BL/6, 129, or a combination thereof for mice), can be fresh or frozen, and can be derived from natural breeding or in vitro fertilization.
- the cells provided herein can be normal, healthy cells, or can be diseased or mutant- bearing cells.
- Non-human animals comprising a knockout mutation of an endogenous Gaa gene as described herein can be made by the methods described elsewhere herein.
- An animal can be, for example, a mammal, fish, or bird.
- Non-human mammals include, for example, non- human primates, monkeys, apes, orangutans, cats, dogs, horses, bulls, deer, bison, sheep, rabbits, rodents (e.g., mice, rats, hamsters, and guinea pigs), and livestock (e.g., bovine species such as cows and steer; ovine species such as sheep and goats; and porcine species such as pigs and boars).
- livestock e.g., bovine species such as cows and steer; ovine species such as sheep and goats; and porcine species such as pigs and boars.
- Birds include, for example, chickens, turkeys, ostrich, geese, and ducks. Domesticated animals and agricultural animals are also included.
- Preferred non-human animals include, for example, rodents, such as mice and rats.
- the non-human animals can be from any genetic background.
- suitable mice can be from a 129 strain, a C57BL/6 strain, a mix of 129 and C57BL/6, a BALB/c strain, or a Swiss Webster strain.
- 129 strains include 129P1, 129P2, 129P3, 129X1, 129S 1 (e.g., 129S1/SV, 129Sl/Svlm), 129S2, 129S4, 129S5, 129S9/SvEvH, 129S6 (129/SvEvTac), 129S7, 129S8, 129T1, and 129T2. See, e.g., esting et al.
- C57BL strains include C57BL/A, C57BL/An, C57BL/GrFa, C57BL/Kal_wN, C57BL/6, C57BL/6J, C57BL/6ByJ, C57BL/6NJ, C57BL/10, C57BL/10ScSn, C57BL/10Cr, and C57BL/01a.
- Suitable mice can also be from a mix of an aforementioned 129 strain and an aforementioned C57BL/6 strain (e.g., 50% 129 and 50% C57BL/6).
- suitable mice can be from a mix of aforementioned 129 strains or a mix of aforementioned BL/6 strains (e.g., the 129S6 (129/SvEvTac) strain).
- rats can be from any rat strain, including, for example, an ACI rat strain, a Dark Agouti (DA) rat strain, a Wistar rat strain, a LEA rat strain, a Sprague Dawley (SD) rat strain, or a Fischer rat strain such as Fisher F344 or Fisher F6.
- Rats can also be obtained from a strain derived from a mix of two or more strains recited above.
- a suitable rat can be from a DA strain or an ACI strain.
- the ACI rat strain is characterized as having black agouti, with white belly and feet and an RTl avI haplotype.
- Such strains are available from a variety of sources including Harlan Laboratories.
- the Dark Agouti (DA) rat strain is characterized as having an agouti coat and an RTl avI haplotype.
- Such rats are available from a variety of sources including Charles River and Harlan Laboratories.
- Some suitable rats can be from an inbred rat strain. See, e.g., US 2014/0235933, herein incorporated by reference in its entirety for all purposes.
- Non-Human Animals Comprising a Gaa Knockout Mutation
- Any convenient method or protocol for producing a genetically modified organism is suitable for producing such a genetically modified non-human animal. See, e.g. , Cho et al. (2009) Current Protocols in Cell Biology 42:19.11:19.11.1-19.11.22 and Gama Sosa et al. (2010) Brain Struct. Fund. 214(2-3):91-109, each of which is herein incorporated by reference in its entirety for all purposes.
- Such genetically modified non-human animals can be generated, for example, through gene knock-out at a targeted Gaa locus.
- the method of producing a non-human animal comprising a knockout mutation of a Gaa gene can comprise: (1) modifying the genome of a pluripotent cell to comprise the knockout mutation; (2) identifying or selecting the genetically modified pluripotent cell comprising the knockout mutation; (3) introducing the genetically modified pluripotent cell into a non-human animal host embryo cells in vitro', and (4) implanting and gestating the host embryo cells in a surrogate mother.
- the host embryo comprising the modified pluripotent cell e.g., a non-human ES cell
- the surrogate mother can then produce an F0 generation non-human animal comprising the knockout mutation.
- the methods described herein can further comprise identifying a cell or animal having a modified target genomic locus.
- Various methods can be used to identify cells and animals having a targeted genetic modification.
- the screening step can comprise, for example, a quantitative assay for assessing modification of allele (MOA) of a parental chromosome.
- the quantitative assay can be carried out via a quantitative PCR, such as a real-time PCR (qPCR).
- the real-time PCR can utilize a first primer set that recognizes the target locus and a second primer set that recognizes a non-targeted reference locus.
- the primer set can comprise a fluorescent probe that recognizes the amplified sequence.
- FISH fluorescence-mediated in situ hybridization
- comparative genomic hybridization isothermal DNA amplification
- quantitative hybridization to an immobilized probe(s)
- INVADER® Probes to an immobilized probe(s)
- TAQMAN® Molecular Beacon probes to an immobilized probe(s)
- ECLIPSETM probe technology see, e.g., US 2005/0144655, incorporated herein by reference in its entirety for all purposes.
- An example of a suitable pluripotent cell is an embryonic stem (ES) cell (e.g., a mouse ES cell or a rat ES cell).
- the modified pluripotent cell can be generated, for example, through recombination by (a) introducing into the cell one or more targeting vectors comprising an insert nucleic acid flanked by 5’ and 3’ homology arms corresponding to 5’ and 3’ target sites, wherein the insert nucleic acid comprises a Gaa knockout mutation; and (b) identifying at least one cell comprising in its genome the insert nucleic acid integrated at the target genomic locus.
- the modified pluripotent cell can be generated by (a) introducing into the cell: (i) a nuclease agent, wherein the nuclease agent induces a nick or double-strand break at a recognition site within the target genomic locus; and (ii) one or more targeting vectors comprising an insert nucleic acid flanked by 5’ and 3’ homology arms corresponding to 5 ’ and 3 ’ target sites located in sufficient proximity to the recognition site, wherein the insert nucleic acid comprises the Gaa knockout mutation; and (c) identifying at least one cell comprising a modification (e.g., integration of the insert nucleic acid) at the target genomic locus.
- a nuclease agent wherein the nuclease agent induces a nick or double-strand break at a recognition site within the target genomic locus
- one or more targeting vectors comprising an insert nucleic acid flanked by 5’ and 3’ homology arms corresponding to 5 ’ and 3 ’ target sites located in sufficient proximity
- nuclease agent that induces a nick or double-strand break into a desired recognition site
- suitable nucleases include a Transcription Activator-Like Effector Nuclease (TALEN), a zinc-finger nuclease (ZFN), a meganuclease, and Clustered Regularly Interspersed Short Palindromic Repeats (CRISPR)/CRISPR- associated (Cas) systems or components of such systems (e.g., CRISPR/Cas9).
- TALEN Transcription Activator-Like Effector Nuclease
- ZFN zinc-finger nuclease
- meganuclease a meganuclease
- CRISPR Clustered Regularly Interspersed Short Palindromic Repeats
- Cas Clustered Regularly Interspersed Short Palindromic Repeats
- the donor cell can be introduced into a host embryo at any stage, such as the blastocyst stage or the pre-morula stage (i.e., the 4 cell stage or the 8 cell stage).
- Progeny that are capable of transmitting the genetic modification though the germline is generated. See, e.g., US Patent No. 7,294,754, herein incorporated by reference in its entirety for all purposes.
- Nuclear transfer techniques can also be used to generate the non-human mammalian animals.
- methods for nuclear transfer can include the steps of: (1) enucleating an oocyte or providing an enucleated oocyte; (2) isolating or providing a donor cell or nucleus to be combined with the enucleated oocyte; (3) inserting the cell or nucleus into the enucleated oocyte to form a reconstituted cell; (4) implanting the reconstituted cell into the womb of an animal to form an embryo; and (5) allowing the embryo to develop.
- oocytes are generally retrieved from deceased animals, although they may be isolated also from either oviducts and/or ovaries of live animals.
- Insertion of the donor cell or nucleus into the enucleated oocyte to form a reconstituted cell can be by microinjection of a donor cell under the zona pellucida prior to fusion. Fusion may be induced by application of a DC electrical pulse across the contact/fusion plane (electrofusion), by exposure of the cells to fusion-promoting chemicals, such as polyethylene glycol, or by way of an inactivated virus, such as the Sendai virus.
- a reconstituted cell can be activated by electrical and/or nonelectrical means before, during, and/or after fusion of the nuclear donor and recipient oocyte.
- Activation methods include electric pulses, chemically induced shock, penetration by sperm, increasing levels of divalent cations in the oocyte, and reducing phosphorylation of cellular proteins (as by way of kinase inhibitors) in the oocyte.
- the activated reconstituted cells, or embryos can be cultured in media and then transferred to the womb of an animal. See, e.g., US 2008/0092249, WO 1999/005266, US 2004/0177390, WO 2008/017234, and US Patent No. 7,612,250, each of which is herein incorporated by reference in its entirety for all purposes.
- the various methods provided herein allow for the generation of a genetically modified non-human F0 animal wherein the cells of the genetically modified F0 animal comprise the Gaa knockout mutation. It is recognized that depending on the method used to generate the F0 animal, the number of cells within the F0 animal that have the Gaa knockout mutation will vary.
- the introduction of the donor ES cells into a pre-morula stage embryo from a corresponding organism (e.g., an 8-cell stage mouse embryo) via for example, the VELOCIMOUSE® method allows for a greater percentage of the cell population of the F0 animal to comprise cells having the nucleotide sequence of interest comprising the targeted genetic modification.
- the disclosure provides a method of making a non-human animal, a non-human animal cell, or a non-human animal genome described herein, comprising inserting a nucleic acid sequence encoding a Gaa knockout mutation into the genome of the non-human animal, the genome of the non-human animal cell, or the non- human animal genome.
- the non-human animals and cells described herein may be useful as models for preclinical testing of therapeutic modalities or compounds e.g., gene therapy with a Gaa transgene, that aim to increase GAA expression levels or activity in a patient in need thereof, e.g., a subject with Pompe disease.
- therapeutic modalities or compounds e.g., gene therapy with a Gaa transgene
- described herein are non-human animal models for testing therapeutic modalities and/or compounds useful for increasing GAA expression or activity.
- a non-human animal model for testing therapeutic modalities or compounds useful for increasing GAA expression or activity comprising a non-human animal as described herein, e.g., a mouse comprising a knockout mutation of an endogenous Gaa gene and a therapeutic agent.
- the testing of the therapeutic modalities and/or compounds for usefulness in increasing GAA expression or activity involves performing an assay or a study that allows determination of the effect of the therapeutic modalities and/or compounds on one or more ocular phenotypes described herein.
- the determination of the effect comprises measuring the pupil diameter or area of a non-human animal as described herein before and after administration of the candidate therapeutic modality and/or compound, wherein an increase in pupil diameter or area indicates the therapeutic modality and/or compound may be useful in increasing GAA expression or activity in a subject in need thereof.
- the determination of the effect of therapeutic modalities and/or compounds comprises measuring the level of glycogen in an ocular cell or tissue, e.g., ciliary body, lens epithelium, neural retina, iris) before and after administration of the candidate therapeutic modality and/or compound to a non-human animal model as described herein, wherein a decrease in the level of glycogen indicates the candidate therapeutic modality and/or compound may be useful in increasing GAA expression or activity in a subject in need thereof.
- an ocular cell or tissue e.g., ciliary body, lens epithelium, neural retina, iris
- the candidate therapeutic modality and/or compound may be introduced into a non-human animal as described herein.
- the candidate therapeutic modality and/or compound may be introduced into a non-human animal as described herein by several methods known to those skilled in the art. Some nonlimiting methods include transgenesis, hydrodynamic delivery (HDD), lipid nanoparticle (LNP) delivery, intravenous injection, parenteral administration, tissue or cell transplantation, etc.
- Nucleotides encoding, e.g., a protein comprising GAA activity may be targeted to be expressed by particular cell types, e.g., the liver, or to be expressed by a particular locus, e.g., a safe harbor locus, according to well-known methods.
- the LNP when administering nucleotides encoding a protein comprising GAA activity by LNP delivery, the LNP may contain one or more or all of the following: (i) a lipid for encapsulation and for endosomal escape; (ii) a neutral lipid for stabilization; (iii) a helper lipid for stabilization; and (iv) a stealth lipid.
- the cargo can include a guide RNA or a nucleic acid encoding a guide RNA.
- the cargo can include an mRNA encoding a Cas nuclease, such as Cas9, and a guide RNA or a nucleic acid encoding a guide RNA.
- the cargo can include an exogenous donor sequence (e.g., encoding a protein comprising GAA activity).
- the cargo can include a nuclease agent (or a nucleic acid encoding the nuclease agent or one or more nucleic acids encoding the nuclease agent) and an exogenous donor sequence (e.g., encoding a protein comprising GAA activity).
- the cargo can include an mRNA encoding a Cas nuclease, such as Cas9, a guide RNA or a nucleic acid encoding a guide RNA, and an exogenous donor sequence (e.g., encoding a protein comprising GAA activity) for CRISPR-mediated insertion of the exogenous donor sequence (e.g., encoding a protein comprising GAA activity) into a safe harbor locus of the animal, such as but not limited to a safe harbor locus, e.g., albumin, e.g., the first intron of the albumin locus. See, e.g., W02020206162, incorporated herein in its entirety by reference.
- a safe harbor locus e.g., albumin, e.g., the first intron of the albumin locus.
- nucleotide and amino acid sequences listed in the accompanying sequence listing are shown using standard letter abbreviations for nucleotide bases, and three-letter code for amino acids.
- the nucleotide sequences follow the standard convention of beginning at the 5’ end of the sequence and proceeding forward (i.e., from left to right in each line) to the 3’ end. Only one strand of each nucleotide sequence is shown, but the complementary strand is understood to be included by any reference to the displayed strand.
- the amino acid sequences follow the standard convention of beginning at the amino terminus of the sequence and proceeding forward (i.e., from left to right in each line) to the carboxy terminus. Table 1. Description of Sequences.
- embryonic stem cells may be genetically modified to delete and/or replace an endogenous coding sequence encoding Gaa (see Figure 1), or to collapse the GAA gene in the embryonic stem cells (see Figure 2).
- mice used in Example 2 described herein were generated using VelociGene® methods, as described previously, which allows for the rapid and high-throughput generation of custom gene mutations in mice (Valenzuela, D.M., et al. (2003b), Nat Biotechnol 21:652- 659). Briefly, a large targeting vectors (LTVEC) were generated using BAC clones. The lacZ/neo' reporter/selection cassette ( Figure 1) was introduced into ES cells (Poueymirou et al. (2007); Valenzuela et al. (2003a)). Selection medium containing G418 was added to the cultures.
- Guides direct SpCas9 cleavage close to the Gaa start ATG (guide 9251mGU (SEQ ID NO:5), cut site 38bp upstream from the ATG; guide 9251mGU3 (SEQ ID NO:6), cut site 18bp downstream of the ATG) and after the stop codon (guide 9251mGD3 (SEQ ID NO:7), cut site 677bp downstream of the stop; guide 9251mGD4 (SEQ ID NO:8), cut site 705bp downstream of the stop). See, e.g., Figure 2A.
- a mixture of 125 pmol of each guide is complexed with 31.25pmol SpCas9 and electroporated into 2 x 10 6 hybrid 129S6/SvEvTac:C57Bl/6NTac Fl embryonic stems cells (ESC). Resulting clonal colonies are screened first via TaqMan for deletion of both copies of Gaa, using loss-of-allele assays. See, Table 2.
- deletion sizes are limited to within the region indicated by retention TaqMan assays. Clones with both Gaa copies deleted are subject to Illumina technology to fully characterize the knockout sequence using primers mm_Gaa_AmpF3 and mm_Gaa_AmpRl. See Table 3.
- iris is the second highest tissue expressing GAA in a mouse body atlas, and since GAA mRNA expression in ocular tissues are similar in mice and human, phenotypic analysis of Gaa' 7 ' animals comprising the Gaa knockout mutation depicted in Figure 1 were performed to assess whether the knockout mutation in these animals would produce any ocular phenotypes.
- IOP intraocular pressure
- OCT anterior and posterior optical coherence tomography
- PAS Periodic Acid Shiff
- OCT Heidelberg Spectralis optical coherence tomography
- Comeal thickness and pupil size were measured at various ages.
- Electroretinogram (ERG) was conducted to assess the retina function change. After sacrificing of the mice, the eyeballs were collected for Periodic Acid-Schiff (PAS) staining to evaluate glycogen accumulation, while hearts and irises were harvested for glycogen measurement with a bioassay kit.
- PAS Periodic Acid-Schiff
- OCT of the corneas, irises, and lens of each mouse were analyzed for pupil diameter and area using standard methodology (see Figure 7).
- Gaa ⁇ 'mice exhibited statistically smaller pupil diameters and areas without dilation at 6- and 8- months of age compared to similarly aged wildtype mice ( Figure 8 and Figure 9). There was no significant difference at 11 months of age without dilation or at any age with dilation between the two groups. ( Figure 8 and Figure 9).
- Vacuolar myopathy is seen in Gaa ' mice compared to wildtype control mice ( Figure 11).
- Periodic Acid-Schiff (PAS) staining of corneas, irises, ciliary bodies, lens epithelial cells, retinas and extraocular muscles was performed to measure glycogen accumulation in these tissues/cells by wildtype control mice or Gaa 1 ' mice.
- the PAS Stain is used for the demonstration of carbohydrates and carbohydrate rich compounds in tissues.
- Glycogen in the iris and heart were also measured.
- the tissues were isolated, homogenized, heated to boiling for 10 minutes, centrifuged for 10 minutes at 4 °C and 18000xg, and measured using a glycogen assay kit (Abeam).
- Example 2 the pupil phenotype as observed in Example 2 for Gaa ' ⁇ mice (GAA KO) was confirmed and changes following CRISPR/Cas9-mediated liver insertion by AAV8 hTfR:GAA/LNP in humanized TfRl mice were measured.
- mice were matched with wild type mice of the similar age. All mice were negative for Rd8 mutation of the Crbl gene. Only male mice were used in current Example. Tail vein injection of AAV8 hTfR:GAA (3E13vg/kg)/LNP (3mg/kg) were performed with 2-3 month old mice.
- mice were tested in a manner counterbalanced by genotype and age to avoid confounding effects of time of day, or testing sequence. In detail, mice were anesthetized with an intra-peritoneal (i.p.) dose of 70-80 mg/kg ketamine, and 15 mg/kg xylazine in sterile saline.
- i.p. intra-peritoneal
- Akorn anti-muscarinic pupil dilator tropicamide
- Non-treated GAA KO mice exhibited significantly smaller pupil size compared to non-treated wild type (WT) mice at 2 and 3 months old without tropicamide (Figure 15A).
- WT wild type
- Figure 15A One month post CRISPR/Cas9-mediated liver insertion, TfR:GAA expression were confirmed by Western blot in the serum from treated GAA KO mice ( Figure 14; lanes 4-25). Pupil size without dilation for both GAA KO groups is significantly smaller than non-treated WT control mice.
- Two months post CRISPR/Cas9-mediated liver insertion of TfR:GAA, without pupil dilation there is a significant difference between AAV8 treated GAA KO and non-treated GAA KO mice ( Figure 15A; p ⁇ 0.001, Two-way ANOVA). With pupil dilation, there is a significant difference between treated GAA KO mice and non-treated GAA KO mice only at the 3-month timepoint ( Figure 15B; p ⁇ 0.05; Two-way ANOVA).
- This Example indicates that glycogen accumulation in ocular tissues, resulting from GAA gene mutation, can instigate pathological changes in the eye, such as reduced pupil size. Enzyme replacement therapy to reverse these ocular alterations might offer a potential biomarker for therapeutic interventions in Pompe disease.
Landscapes
- Life Sciences & Earth Sciences (AREA)
- Environmental Sciences (AREA)
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Biotechnology (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- Zoology (AREA)
- Animal Husbandry (AREA)
- Biodiversity & Conservation Biology (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
Abstract
Non-human animal cells and non-human animals comprising a knockout mutation in a Gaa gene, e.g., at an endogenous Gaa locus, and exhibiting ocular phenotypes that correlate with an accumulation of glycogen are provided. Methods of using such non-human animal cells and non-human animals are also provided. Such animals are useful to screen therapies based on increasing Gaa expression or activity for the treatment of, e.g., Pompe disease.
Description
GAA KNOCKOUT NON-HUMAN ANIMALS
CROSS-REFERENCE TO RELATED APPLICATION
[0001] This application claims benefit of priority to U.S. Provisional Application No. 63/607,184, filed December 7, 2023, which is incorporated herein by reference in its entirety.
FIELD OF THE INVENTION
[0002] The measurable phenotype of a genetically modified non-human animal (e.g., a rodent, e.g., a mouse, or a rat) comprising in its genome a knockout mutation at an a-glucosidase (GAA) locus is described. Also described are methods of using the genetically modified non-human animals that lack GAA as models for preclinical testing of a candidate therapeutic modality and/or compound and determining whether such candidate therapeutic modality and/or compound may be useful for increasing GAA activity in subjects in need thereof.
SEQUENCE LISTING
[0003] A Sequence Listing in xml format entitled “11691WO01.xml,” which was created on December 5, 2024, and is 69 Kb, is incorporated herein by reference in its entirety.
BACKGROUND
[0004] Pompe disease is an autosomal recessive disorder in which glycogen accumulates in the body’s cells due to a deficiency of acid alpha-glucosidase (GAA). GAA breaks down complex sugars in the body. A deficiency of GAA can result from mutations in the GAA gene. Without GAA, glycogen accumulation occurs in organs and tissues, especially in muscles, causing the muscles to break down. Mouse models of Pompe disease are useful for testing candidate therapies aimed at restoring GAA activity in patients in need thereof.
SUMMARY
[0005] Provided herein are genetically modified non-human animals having a knockout mutation in the a-glucosidase (GAA) locus, and exhibiting ocular phenotypes associated with an accumulation of glycogen in ocular cells or tissues. The ocular phenotypes exhibited by the non-human animals described herein provide a useful biomarker to evaluate when testing the ability of a candidate therapeutic modality and/or compound to increase or restore GAA activity and/or reduce symptoms of Pompe disease.
[0006] Thus, described herein is an animal model of Pompe disease, wherein the animal model exhibits an ocular phenotype. Accordingly, described herein is a non-human animal, non-human animal cell, or non-human animal genome comprising a knockout mutation of an endogenous a-glucosidase (Gaa) gene. In some embodiments, the knockout mutation comprises a deletion of the Gaa gene or a portion thereof. In some embodiments, the knockout mutation comprises a deletion of the entire coding sequence of the Gaa gene. In some embodiments, a non-human animal, a non-human animal cell, or a non-human animal genome in the Pompe disease model does not express GAA protein. In some Pompe disease model embodiments disclosed herein, a non-human animal, a non-human animal cell, or a non-human animal genome comprises an endogenous Gaa locus that comprises a nucleotide sequence selected from the group consisting of: the nucleotide sequence as set forth in SEQ ID NO: 26, the nucleotide sequence as set forth in SEQ ID NO: 27, the nucleotide sequence as set forth in SEQ ID NO: 29, or the nucleotide sequence as set forth in SEQ ID NO: 30. [0007] In some embodiments, a non-human animal and/or Pompe disease model comprising a knockout mutation of an endogenous Gaa gene exhibits an ocular phenotype compared to a wildtype control non-human animal comprising a wildtype Gaa gene. In some embodiments, the ocular phenotype is: (i) an accumulation of, e.g., substantially more, glycogen in ocular tissue (e.g., ciliary body, lens epithelium, neural retina) compared to a wildtype control non-human animal comprising a wildtype Gaa gene; (ii) a decrease in, e.g., significantly smaller, pupil size compared to a wildtype control non-human animal; (iii) vacuolar myopathy in the extraocular muscle; (iv) elevated glycogen levels in the iris; or (v) any combination thereof. In some embodiments, a genetically modified non-human animal disclosed herein has a significantly smaller pupil size when the non-human animal and/or Pompe disease model comprising a knockout mutation of an endogenous Gaa gene is at least 6 months in age. In some embodiments, a significantly smaller pupil size is seen when the non-human animal and/or Pompe disease model comprising a knockout mutation of an endogenous Gaa gene is at least 8 months in age. In some embodiments, a significantly smaller pupil size is seen when the non-human animal and/or Pompe disease model comprising a knockout mutation of an endogenous Gaa gene is at least 11 months in age. In some embodiments, a non-human cell comprising a knockout mutation of an endogenous Gaa gene comprises an accumulation of glycogen in a cellular compartment (e.g., a lysosome) compared to a wildtype control non-human animal cell comprising a wildtype Gaa gene.
[0008] Methods of knocking out an endogenous Gaa gene are also provided. In some embodiments, the methods comprise modifying an endogenous Gaa locus of the non-human animal to comprise a knockout mutation of the Gaa gene.
[0009] A non-human animal as described herein comprising a knockout mutation of an endogenous Gaa gene can be used for testing a candidate therapeutic modality and/or compound, e.g., a candidate therapeutic modality and/or compound comprising a- glucosidase. The candidate therapeutic modality and/or modality can increase GAA expression and/or activity in a subject. In some embodiments, the therapeutic agent comprises encodes or comprises a protein having a-glucosidase activity, e.g., a-glucosidase or a biologically active portion thereof.
[0010] In some embodiments, a method utilizing a non-human animal comprising a knockout mutation of a Gaa gene and exhibiting an ocular phenotype as described herein comprises administering a candidate therapeutic agent to the non-human animal, and measuring the effect of the candidate therapeutic agent on the ocular phenotype of the non- human animal. In some embodiments, measuring the effect comprises determining: (i) glycogen levels in ocular tissue (e.g., ciliary body, lens epithelium, neural retina, iris, etc.); (ii) pupil size; (iii) the absence or presence of vacuolar myopathy in the extraocular muscle; or (iv) any combination thereof. In some embodiments, a therapeutic agent may be useful for increasing GAA activity in a patient in need thereof, e.g., in a patient with Pompe disease, if the therapeutic agent is able to reduce one or more of the ocular phenotypes exhibited by a non-human animal comprising a Gaa knockout mutation as described herein. In some embodiments, after administration of the candidate therapeutic agent, a reduction of one or more of the ocular phenotypes exhibited by a non-human animal comprising a Gaa knockout mutation comprises (i) a decrease in glycogen levels in ocular tissue (e.g., ciliary body, lens epithelium, neural retina, iris) compared to the glycogen levels in the ocular tissue (e.g., ciliary body, lens epithelium, neural retina, iris) of the non-human animal comprising a knockout mutation of an endogenous Gaa gene prior to administration of the candidate therapeutic agent; (ii) an increase in pupil size of the non-human animal comprising a knockout mutation of an endogenous Gaa gene compared to pupil size of the non-human animal comprising a knockout mutation of an endogenous Gaa gene prior to administration of the candidate therapeutic agent; (iii) decreased vacuolar myopathy in the extraocular muscle of the non-human animal comprising a knockout mutation of an endogenous Gaa gene compared to that of the non-human animal comprising a knockout mutation of an
endogenous Gaa gene prior to administration of the candidate therapeutic agent; or (iv) any combination thereof.
BRIEF DESCRIPTION OF THE FIGURES
[0011 ] Figure 1 provides a schematic (not to scale) for the insertion of a Gaa knockout mutation comprising a replacement of an endogenous sequence spanning coding exon 1 (cExonl) and coding exon 19 (cExon 19) of an endogenous Gaa gene (SEQ ID NO: 28) with a Zen-Ubl targeting vector comprising a LacZ reporter gene (SEQ ID NO: 29 or SEQ ID NO: 30). The Zen-Ubl cassette also comprises a selection cassette comprising a promotor from the human ubiquitin C gene operably linked to a neomycin phosphotransferase (Neo) resistance gene.
[0012] Figure 2A provides identifying information for a mouse a-glucosidase Gaa) gene (SEQ ID NO: 1) and the GAA proteins (SEQ ID NO: 2) so encoded, and four SpCas9 guide RNAs (gRNAs) that can be used to collapse the Gaa allele in a mouse embryonic stem cell. Figure 2B provides a schematic (not to scale) of a Gaa knockout allele comprising a deletion of the Gaa gene sequence using guide RNAs that directed SpCas9 cleavage close to the Gaa ATG start codon (guide 9251mGU; SEQ ID NO: 5), cut site 38bp upstream from the ATG; guide 9251mGU3; SEQ ID NO: 6), cut site 18bp downstream of the ATG start codon) and after the stop codon (guide 9251mGD3; SEQ ID NO: 7], cut site 677bp downstream of the stop; guide 9251mGD4; SEQ ID NO: 8], cut site 705bp downstream of the stop codon). Approximate locations of the BHQ probes used to assay for loss-of-allele via TaqMan RT- PCR are shown with asterisks (*).
[0013] Figure 3 provides the body weight (B.W. (gram), y-axis) of mice expressing wildtype GAA (GAA WT) or mice homozygous for the GAA knockout mutation described in Figure 1 (GAA KO) at 6, 8, and 11 months of age (x-axis). WT = 9 mice; KO = 15 mice. [0014] Figure 4 provides the cornea thicknesses (pm, y-axis) of the eyes of mice expressing wildtype GAA (GAA WT) or mice homozygous for the GAA knockout mutation described in Figure 1 (GAA KO). WT = 9 mice (18 eyes); KO = 15 mice (30 eyes).
[0015] Figure 5 provides the intraocular pressure (1OP, y-axis) of the eyes of 7-month- old mice expressing wildtype GAA (GAA WT) or 7-month-old mice homozygous for the GAA knockout mutation described in Figure 1 (GAA KO). WT = 9 mice (18 eyes); KO = 15 mice (30 eyes).
[0016] Figure 6 provides optical coherence tomography (OCT) images of a retina from a mouse expressing wildtype GAA (GAA WT) or a mouse homozygous for the GAA knockout
mutation described in Figure 1 (GAA KO).
[0017] Figure 7 provides optical coherence tomography (OCT) images of a retina from a mouse expressing wildtype GAA (GAA WT) to depict the measurements used to determine corneal pupil diameters and areas.
[0018] Figure 8 provides graphs showing central pupil diameters (pm, y-axis) of the eyes of (A) 6-, (B) 8-, or (C) 11 month old mice expressing wildtype GAA (GAA WT) or mice homozygous for the GAA knockout mutation described in Figure 1 (GAA KO) with dilation (first two bars of each graph; x-axis) or with dilation with tropicamide (last two bars of each graph; x-axis). WT = 9 mice (18 eyes); KO = 15 mice (30 eyes).
[0019] Figure 9 provides graphs showing central pupil areas (pm2, y-axis) of the eyes of (A) 6-, (B) 8-, or (C) 11 month old mice expressing wildtype GAA (GAA WT) or mice homozygous for the GAA knockout mutation described in Figure 1 (GAA KO) with dilation (first two bars of each graph; x-axis) or with dilation with tropicamide (last two bars of each graph; x-axis). WT = 9 mice (18 eyes); KO = 15 mice (30 eyes).
[0020] Figure 10 provides histological images of eye sections isolated from a mouse expressing wildtype GAA (GAA WT) or a mouse homozygous for the GAA knockout mutation described in Figure 1 (GAA KO) and stained with Hematoxylin to show cell nuclei, and Eosin to show the extracellular matrix, cytoplasm, and other structures (which appears light grey in a black and white image). Arrows point to muscle fibers.
[0021 ] Figure 11 provides histological images of extraocular muscle isolated from a mouse expressing wildtype GAA (GAA WT) or a mouse homozygous for the GAA knockout mutation described in Figure 1 (GAA KO) and stained with Hematoxylin to show cell nuclei, and Eosin to show the extracellular matrix, cytoplasm, and other structures (which appears light grey in a black and white image). Arrows point to muscle fibers.
[0022] Figures 12A-12F provide images of (A) corneas, (B) irises, (C) ciliary bodies, (D) lens epithelial cells, (E) retinas, and (F) extraocular muscles isolated from mouse homozygous for the GAA knockout mutation described in Figure 1 (GAA KO) and incubated with Periodic Acid Schiff (PAS) stain. Stronger or darker PAS staining indicates more glycogen accumulation.
[0023] Figure 13 provides graphs showing the levels of glycogen (pg/mg protein) in the hearts or irises of mice expressing wildtype GAA (GAA WT) or mice homozygous for the GAA knockout mutation described in Figure 1 (GAA KO), and a correlation graph and chart providing the correlation of glycogen levels in iris and heart.
[0024] Figure 14 provides a western blot showing anti-TfR scFv:GAA expression in the
serum of GAA KO mice one month after AAV8 anti-hTfR scFv:GAA (3E13vg/kg)/LNP (3mg/kg) treatment (lanes 4-25). Serum from untreated GAA KO mice is also provided as negative controls (lanes 2 and 3). Each lane represents an individual mouse.
[0025] Figures ISA and 15B provide graphs showing pupil diameter (i.e., pupil size) in non- treated WT mice, non-treated GAA KO mice, and GAA KO mice treated with AAV8 anti-hTfR scFv:GAA (3E13vg/kg)/LNP (3mg/kg) (Figure 15A) before tropicamide (without dilation) and (Figure 15B) after tropicamide (with dilation). Values are mean ± SEM; n=20- 22 eyes for each group. **p < 0.05, ***p<0.001 , Two Way ANOVA, GAA KO treated vs GAA KO non-treated.
DETAILED DESCRIPTION
I. Overview
[0026] Pompe disease is an autosomal recessive disorder resulting from a deficiency of acid alpha-glucosidase (GAA), leading to progressive intralysosomal glycogen accumulation in tissues due to mutations in the GAA gene. Some patients have ocular phenotypes such as bilateral ptosis, strabismus, myopia, and astigmatism. There are multiple mouse models of Pompe disease; however, no ocular phenotypes have ever been reported for said mouse models. The iris is the tissue with the second highest expression of GAA in a mouse. Thus, determining whether a Pompe disease model in mice also demonstrates ocular phenotypes may be useful in validating the model as a platform to screen candidate therapeutics for treating Pompe disease.
[0027] Ocular pathological changes that may occur due to glycogen accumulation in tissues are shown for a Gaa-I- knockout mouse as described herein. Although no difference was observed in anterior and posterior segment via optical coherence tomography (OCT), Gaa'7' knockout mice exhibited significantly smaller pupil sizes (p<0.01 ) at 6 and 8 months of age, a trend that was also observed at 11 months age (Figure 8 and Figure 9). The electroretinogram (ERG) test (an assay to measure changes in retinal function) did not show remarkable change in Gaa'7' mice. Vacuolar myopathy was observed in the extraocular muscle of Gaa 7' mice (Figure 11). PAS staining revealed substantial glycogen accumulation in the ciliary body, lens epithelium, and neural retina of Gaa'7' out mice compared to wildtype control mice (Figure 12C, Figure 12D, Figure 12E). Additionally, glycogen levels in the iris of GAA knockout mice were significantly elevated (Figure 13). Such changes are consistent with establishing an ocular biomarker for Pompe disease therapy.
[0028] An example amino acid sequence of mouse (m) Gaa is set forth as SEQ ID NO:2, which is identical to the amino acid sequence of the mGaa protein represented as Uniprot P70699. The mouse Gaa gene is found on chromosome 11 in mice. The complete gene sequence for mouse Gaa, with annotated exons and introns, can be found from the NCBI database (Gene ID: 14387). An example coding sequence for mGaa is set forth in SEQ ID NO: 1. Disclosed herein are non-human animal cells, non-human animals, and non-human genomes (e.g., found in non-human animal cell nuclei) comprising a Gaa knockout mutation, and exhibiting an ocular phenotype. In some embodiments, the knockout mutation comprises a deletion of the entire coding sequence of an endogenous Gaa gene. In some embodiments, a non-human animal cell, non-human animal, or non-human genome described herein comprises, at an endogenous Gaa allele, a nucleotide sequence set forth as SEQ ID NO: 26, a nucleotide sequence set forth as SEQ ID NO: 27, a nucleotide sequence set forth as SEQ ID NO:29, or a nucleotide sequence set forth as SEQ ID NO:30, e.g., as a replacement for the wildtype endogenous Gaa allele.
[0029] In some embodiments, a non-human animal cell, non-human animal, or non- human genome described herein comprises, at an endogenous Gaa allele, a nucleotide sequence set forth as SEQ ID NO: 26, e.g., as a replacement for the wildtype endogenous Gaa allele. In some embodiments, a non-human animal cell, non-human animal, or non- human genome described herein comprises, at an endogenous Gaa allele, a nucleotide sequence set forth as SEQ ID NO: 27, e.g., as a replacement for the wildtype endogenous Gaa allele. In some embodiments, a non-human animal cell, non-human animal, or non- human genome described herein comprises, at an endogenous Gaa allele, a nucleotide sequence set forth as SEQ ID NO: 29, e.g., as a replacement for the wildtype endogenous Gaa allele. In some embodiments, a non-human animal cell, non-human animal, or non- human genome described herein comprises, at an endogenous Gaa allele, a nucleotide sequence set forth as SEQ ID NO: 30, e.g., as a replacement for the wildtype endogenous Gaa allele.
[0030] In some embodiments, provided herein are non-human animal cells and non- human animals having a heterologous Gaa knockout mutation sequence in the genomes (cellular nuclei) of the non-human animal cells or non-human animals provided herein. The Gaa knockout mutation sequence can be inserted into an endogenous Gaa locus, thus providing non-human animal cells and non-human animals having a genetically modified endogenous Gaa locus.
[0031] In some embodiments, a non-human animal and/or Pompe disease model comprising a knockout mutation of an endogenous Gaa gene comprises an ocular phenotype compared to a wildtype control non-human animal comprising a wildtype Gaa gene. In some embodiments, the ocular phenotype is: (i) an accumulation of, e.g., substantially more, glycogen in ocular tissue (e.g., ciliary body, lens epithelium, neural retina) compared to a wildtype control non-human animal comprising a wildtype Gaa gene; (ii) a decrease in, e.g., significantly smaller, pupil size compared to a wildtype control non-human animal; (iii) vacuolar myopathy in the extraocular muscle; (iv) elevated glycogen levels in the iris; or (v) any combination thereof. In some embodiments, a genetically modified non-human animal disclosed herein has a significantly smaller pupil size when the non-human animal and/or Pompe disease model comprising a knockout mutation of an endogenous Gaa gene is at least 6 months in age. In some embodiments, a significantly smaller pupil size is seen when the non-human animal and/or Pompe disease model comprising a knockout mutation of an endogenous Gaa gene is at least 8 months in age. In some embodiments, a significantly smaller pupil size is seen when the non-human animal and/or Pompe disease model comprising a knockout mutation of an endogenous Gaa gene is at least 11 months in age. In some embodiments, a non-human cell comprising a knockout mutation of an endogenous Gaa gene comprises an accumulation of glycogen in a cellular compartment (e.g., a lysosome) compared to a wildtype control non-human animal cell comprising a wildtype Gaa gene. Such animals may be used in methods of screening drugs that are candidates for increasing GAA activity in a patient in need thereof, e.g., in a patient with Pompe disease. [0032] In some embodiments, the non-human animal is a mammal, or the non-human animal genome is a mammalian genome. In some embodiments, the non-human animal can be a rodent, or the non-human animal genome can be a rodent genome. In some embodiments, the non-human animal can be a rat or mouse, or the non-human animal genome can be a rat genome or a mouse genome.
[0033] The terms “protein,” “polypeptide,” and “peptide,” are used interchangeably herein, and include polymeric forms of amino acids of any length, including coded and noncoded amino acids and chemically or biochemically modified or derivatized amino acids. The terms also include polymers that have been modified, such as polypeptides having modified peptide backbones. The term domain can refer to any part of a protein or polypeptide having a particular function or structure.
[0034] Proteins are said to have an “N-terminus” and a “C-terminus.” The term “N- terminus” relates to the start of a protein or polypeptide, terminated by an amino acid with a
free amine group (-NH2). The term “C-terminus” relates to the end of an amino acid chain (protein or polypeptide), terminated by a free carboxyl group (-COOH).
[0035] The terms “nucleic acid” and “polynucleotide,” used interchangeably herein, include polymeric forms of nucleotides of any length, including ribonucleotides, deoxyribonucleotides, or analogs or modified versions thereof. Nucleic acids and polynucleotides can include single-, double-, and multi-stranded DNA or RNA, genomic DNA, cDNA, DNA-RNA hybrids, and polymers comprising purine bases, pyrimidine bases, or other natural, chemically modified, biochemically modified, non-natural, or derivatized nucleotide bases.
[0036] Nucleic acids are said to have “5’ ends” and “3’ ends” because mononucleotides are reacted to make oligonucleotides in a manner such that the 5 ’ phosphate of one mononucleotide pentose ring is attached to the 3’ oxygen of its neighbor in one direction via a phosphodiester linkage. An end of an oligonucleotide is referred to as the “5’ end” if its 5’ phosphate is not linked to the 3’ oxygen of a mononucleotide pentose ring. An end of an oligonucleotide is referred to as the “3’ end” if its 3’ oxygen is not linked to a 5’ phosphate of another mononucleotide pentose ring. A nucleic acid sequence, even if internal to a larger oligonucleotide, also may be said to have 5’ and 3’ ends. In either a linear or circular DNA molecule, discrete elements are referred to as being “upstream” or 5’ of the “downstream” or 3’ elements.
[0037] The term “genomically integrated” refers to a nucleic acid that has been introduced into a cell such that the nucleotide sequence integrates into the genome of the cell and is capable of being inherited by progeny thereof. Any protocol may be used for the stable incorporation of a nucleic acid into the genome of a cell.
[0038] The term “targeting vector” refers to a recombinant nucleic acid that can be introduced by homologous recombination, non-homologous-end-joining-mediated ligation, or any other means of recombination to a target position in the genome of a cell.
[0039] The term “viral vector” refers to a recombinant nucleic acid that includes at least one element of viral origin and includes elements sufficient for or permissive of packaging into a viral vector particle. The vector and/or particle can be utilized for the purpose of transferring DNA, RNA, or other nucleic acids into cells either ex vivo or in vivo. Numerous forms of viral vectors are known.
[0040] The term “wild type” includes entities having a structure and/or activity as found in a normal (as contrasted with mutant, diseased, altered, or so forth) state or context. Wild type genes and polypeptides often exist in multiple different forms (e.g., alleles).
[0041] The expression “gross mutant phenotype” refers to a significant difference or variation in phenotype between an engineered non-human mouse of the disclosure and a “wild type.”
[0042] The term “endogenous” refers to a nucleic acid sequence that occurs naturally within a cell or non-human animal.
[0043] “Exogenous” molecules or sequences include molecules or sequences that are not normally present in a cell in that form. Normal presence includes presence with respect to the particular developmental stage and environmental conditions of the cell. An exogenous molecule or sequence, for example, can include a mutated version of a corresponding endogenous sequence within the cell, such as a humanized version of the endogenous sequence, or can include a sequence corresponding to an endogenous sequence within the cell but in a different form (i.e., not within a chromosome). In contrast, endogenous molecules or sequences include molecules or sequences that are normally present in that form in a particular cell at a particular developmental stage under particular environmental conditions. [0044] The term “heterologous” when used in the context of a nucleic acid or a protein indicates that the nucleic acid or protein comprises at least two portions that do not naturally occur together in the same molecule. For example, the term “heterologous,” when used with reference to portions of a nucleic acid or portions of a protein, indicates that the nucleic acid or protein comprises two or more sub-sequences that are not found in the same relationship to each other (e.g., joined together) in nature. As one example, a “heterologous” region of a nucleic acid vector is a segment of nucleic acid within or attached to another nucleic acid molecule that is not found in association with the other molecule in nature. For example, a heterologous region of a nucleic acid vector could include a coding sequence flanked by sequences not found in association with the coding sequence in nature. Likewise, a “heterologous” region of a protein is a segment of amino acids within or attached to another peptide molecule that is not found in association with the other peptide molecule in nature (e.g., a fusion protein, or a protein with a tag). Similarly, a nucleic acid or protein can comprise a heterologous label or a heterologous secretion or localization sequence.
[0045] “Codon optimization” takes advantage of the degeneracy of codons, as exhibited by the multiplicity of three-base pair codon combinations that specify an amino acid, and generally includes a process of modifying a nucleic acid sequence for enhanced expression in particular host cells by replacing at least one codon of the native sequence with a codon that is more frequently or most frequently used in the genes of the host cell while maintaining the native amino acid sequence. For example, a nucleic acid encoding a Cas9 protein can be
modified to substitute codons having a higher frequency of usage in a given prokaryotic or eukaryotic cell, including a bacterial cell, a yeast cell, a human cell, a non-human cell, a mammalian cell, a rodent cell, a mouse cell, a rat cell, a hamster cell, or any other host cell, as compared to the naturally occurring nucleic acid sequence. Codon usage tables are readily available, for example, at the “Codon Usage Database.” These tables can be adapted in a number of ways. See Nakamura et al. (2000) Nucleic Acids Research 28:292, herein incorporated by reference in its entirety for all purposes. Computer algorithms for codon optimization of a particular sequence for expression in a particular host are also available (see, e.g. , Gene Forge). A skilled artisan will understand that a nucleic acid sequence as disclosed herein encompasses variants thereof, including those variants that differ due to degeneracy of the genetic code and/or codon optimization, and that encode the same or substantially similar amino acid sequence of a biologically active polypeptide.
[0046] The term “locus” refers to a specific location of a gene (or significant sequence), DNA sequence, polypeptide-encoding sequence, or position on a chromosome of the genome of an organism. For example, an “Gaa locus” may refer to the specific location of a Gaa gene, Gaa DNA sequence, Gaa-encoding sequence, or Gaa position on a chromosome of the genome of an organism that has been identified as to where such a sequence resides. A “Gaa locus” may comprise a regulatory element of an Gaa gene, including, for example, an enhancer, a promoter, 5’ and/or 3’ untranslated region (UTR), or a combination thereof. [0047] The term “gene” refers to a DNA sequence in a chromosome that codes for a product (e.g., an RNA product and/or a polypeptide product) and includes the coding region interrupted with non-coding introns and sequence located adjacent to the coding region on both the 5’ and 3’ ends such that the gene corresponds to the full-length mRNA (including the 5’ and 3’ untranslated sequences). The term “gene” also includes other non-coding sequences including regulatory sequences (e.g., promoters, enhancers, and transcription factor binding sites), polyadenylation signals, internal ribosome entry sites, silencers, insulating sequence, and matrix attachment regions. These sequences may be close to the coding region of the gene (e.g., within 10 kb) or at distant sites, and they influence the level or rate of transcription and translation of the gene.
[0048] The term “allele” refers to a variant form of a gene. Some genes have a variety of different forms, which are located at the same position, or genetic locus, on a chromosome. A diploid organism has two alleles at each genetic locus. Each pair of alleles represents the genotype of a specific genetic locus. Genotypes are described as homozygous if there are two identical alleles at a particular locus and as heterozygous if the two alleles differ.
[0049] A “promoter” is a regulatory region of DNA usually comprising a TATA box capable of directing RNA polymerase II to initiate RNA synthesis at the appropriate transcription initiation site for a particular polynucleotide sequence. A promoter may additionally comprise other regions which influence the transcription initiation rate. The promoter sequences disclosed herein modulate transcription of an operably linked polynucleotide. A promoter can be active in one or more of the cell types disclosed herein (e.g., a eukaryotic cell, a non-human mammalian cell, a human cell, a rodent cell, a pluripotent cell, a one-cell stage embryo, a differentiated cell, or a combination thereof). A promoter can be, for example, a constitutively active promoter, a conditional promoter, an inducible promoter, a temporally restricted promoter (e.g., a developmentally regulated promoter), or a spatially restricted promoter (e.g., a cell-specific or tissue-specific promoter). Examples of promoters can be found, for example, in WO 2013/176772, herein incorporated by reference in its entirety for all purposes.
[0050] “Operable linkage” or being “operably linked” includes juxtaposition of two or more components (e.g., a promoter and another sequence element) such that both components function normally and allow the possibility that at least one of the components can mediate a function that is exerted upon at least one of the other components. For example, a promoter can be operably linked to a coding sequence if the promoter controls the level of transcription of the coding sequence in response to the presence or absence of one or more transcriptional regulatory factors. Operable linkage can include such sequences being contiguous with each other or acting in trans (e.g., a regulatory sequence can act at a distance to control transcription of the coding sequence).
[0051] The term “variant” refers to a nucleotide sequence differing from the sequence most prevalent in a population (e.g., by one nucleotide) or a protein sequence different from the sequence most prevalent in a population (e.g., by one amino acid).
[0052] The term “fragment” when referring to a protein means a protein that is shorter or has fewer amino acids than the full-length protein. The term “fragment” when referring to a nucleic acid means a nucleic acid that is shorter or has fewer nucleotides than the full-length nucleic acid. A fragment can be, for example, an N-terminal fragment (i.e., removal of a portion of the C-terminal end of the protein), a C-terminal fragment (i.e., removal of a portion of the N-terminal end of the protein), or an internal fragment.
[0053] “Sequence identity” or “identity” in the context of two polynucleotides or polypeptide sequences makes reference to the residues in the two sequences that are the same when aligned for maximum correspondence over a specified comparison window. When
percentage of sequence identity is used in reference to proteins, residue positions which are not identical often differ by conservative amino acid substitutions, where amino acid residues are substituted for other amino acid residues with similar chemical properties (e.g., charge or hydrophobicity) and therefore do not change the functional properties of the molecule. When sequences differ in conservative substitutions, the percent sequence identity may be adjusted upwards to correct for the conservative nature of the substitution. Sequences that differ by such conservative substitutions are said to have “sequence similarity” or “similarity.” Means for making this adjustment are well known. Typically, this involves scoring a conservative substitution as a partial rather than a full mismatch, thereby increasing the percentage sequence identity. Thus, for example, where an identical amino acid is given a score of 1 and a non-conservative substitution is given a score of zero, a conservative substitution is given a score between zero and 1. The scoring of conservative substitutions is calculated, e.g., as implemented in the program PC/GENE (Intelligenetics, Mountain View, California).
[0054] “Percentage of sequence identity” includes the value determined by comparing two optimally aligned sequences (greatest number of perfectly matched residues) over a comparison window, wherein the portion of the polynucleotide sequence in the comparison window may comprise additions or deletions (i.e., gaps) as compared to the reference sequence (which does not comprise additions or deletions) for optimal alignment of the two sequences. The percentage is calculated by determining the number of positions at which the identical nucleic acid base or amino acid residue occurs in both sequences to yield the number of matched positions, dividing the number of matched positions by the total number of positions in the window of comparison, and multiplying the result by 100 to yield the percentage of sequence identity. Unless otherwise specified (e.g., the shorter sequence includes a linked heterologous sequence), the comparison window is the full length of the shorter of the two sequences being compared.
[0055] Unless otherwise stated, sequence identity/similarity values include the value obtained using GAP Version 10 using the following parameters: % identity and % similarity for a nucleotide sequence using GAP Weight of 50 and Length Weight of 3, and the nwsgapdna.cmp scoring matrix; % identity and % similarity for an amino acid sequence using GAP Weight of 8 and Length Weight of 2, and the BLOSUM62 scoring matrix; or any equivalent program thereof. “Equivalent program” includes any sequence comparison program that, for any two sequences in question, generates an alignment having identical nucleotide or amino acid residue matches and an identical percent sequence identity when compared to the corresponding alignment generated by GAP Version 10.
[0056] The term “conservative amino acid substitution” refers to the substitution of an amino acid that is normally present in the sequence with a different amino acid of similar size, charge, or polarity. Examples of conservative substitutions include the substitution of a non-polar (hydrophobic) residue such as isoleucine, valine, or leucine for another non-polar residue. Likewise, examples of conservative substitutions include the substitution of one polar (hydrophilic) residue for another such as between arginine and lysine, between glutamine and asparagine, or between glycine and serine. Additionally, the substitution of a basic residue such as lysine, arginine, or histidine for another, or the substitution of one acidic residue such as aspartic acid or glutamic acid for another acidic residue are additional examples of conservative substitutions. Examples of non-conservative substitutions include the substitution of a non-polar (hydrophobic) amino acid residue such as isoleucine, valine, leucine, alanine, or methionine for a polar (hydrophilic) residue such as cysteine, glutamine, glutamic acid or lysine and/or a polar residue for a non-polar residue. Typical amino acid categorizations are summarized below.
Alanine Ala A Nonpolar Neutral 1.8
Arginine Arg R Polar Positive -4.5
Asparagine Asn N Polar Neutral -3.5
Aspartic acid Asp D Polar Negative -3.5
Cysteine Cys C Nonpolar Neutral 2.5
Glutamic acid Glu E Polar Negative -3.5
Glutamine Gin Q Polar Neutral -3.5
Glycine Gly G Nonpolar Neutral -0.4
Histidine His H Polar Positive -3.2
Isoleucine He I Nonpolar Neutral 4.5
Leucine Leu L Nonpolar Neutral 3.8
Lysine Lys K Polar Positive -3.9
Methionine Met M Nonpolar Neutral 1.9
Phenylalanine Phe F Nonpolar Neutral 2.8
Proline Pro P Nonpolar Neutral -1.6
Serine Ser S Polar Neutral -0.8
Threonine Thr T Polar Neutral -0.7
Tryptophan Trp W Nonpolar Neutral -0.9
Tyrosine Tyr Y Polar Neutral -1.3
Valine Vai V Nonpolar Neutral 4.2
[0057] A “homologous” sequence (e.g., nucleic acid sequence) includes a sequence that is either identical or substantially similar to a known reference sequence, such that it is, for example, at least 50%, at least 55%, at least 60%, at least 65%, at least 70%, at least 75%, at
least 80%, at least 85%, at least 90%, at least 95%, at least 96%, at least 97%, at least 98%, at least 99%, or 100% identical to the known reference sequence. Homologous sequences can include, for example, orthologous sequence and paralogous sequences. Homologous genes, for example, typically descend from a common ancestral DNA sequence, either through a speciation event (orthologous genes) or a genetic duplication event (paralogous genes). “Orthologous” genes include genes in different species that evolved from a common ancestral gene by speciation. Orthologs typically retain the same function in the course of evolution. “Paralogous” genes include genes related by duplication within a genome. Paralogs can evolve new functions in the course of evolution.
[0058] The term “in vitro” includes artificial environments and to processes or reactions that occur within an artificial environment (e.g., a test tube). The term “in vivo” includes natural environments (e.g., a cell or organism or body) and to processes or reactions that occur within a natural environment. The term “ex vivo” includes cells that have been removed from the body of an individual and to processes or reactions that occur within such cells.
[0059] The term “reporter gene” refers to a nucleic acid having a sequence encoding a gene product (typically an enzyme) that is easily and quantifiably assayed when a construct comprising the reporter gene sequence operably linked to a heterologous promoter and/or enhancer element is introduced into cells containing (or which can be made to contain) the factors necessary for the activation of the promoter and/or enhancer elements. Examples of reporter genes include, but are not limited, to genes encoding beta-galactosidase (lacZ), the bacterial chloramphenicol acetyltransferase (cat) genes, firefly luciferase genes, genes encoding beta-glucuronidase (GUS), and genes encoding fluorescent proteins. A “reporter protein” refers to a protein encoded by a reporter gene.
[0060] The term “fluorescent reporter protein” as used herein means a reporter protein that is detectable based on fluorescence wherein the fluorescence may be either from the reporter protein directly, activity of the reporter protein on a fluorogenic substrate, or a protein with affinity for binding to a fluorescent tagged compound. Examples of fluorescent proteins include green fluorescent proteins (e.g., GFP, GFP-2, tagGFP, turboGFP, eGFP, Emerald, Azami Green, Monomeric Azami Green, CopGFP, AceGFP, and ZsGreenl), yellow fluorescent proteins (e.g., YFP, eYFP, Citrine, Venus, YPet, PhiYFP, and ZsYellowl), blue fluorescent proteins (e.g., BFP, eBFP, eBFP2, Azurite, mKalamal, GFPuv, Sapphire, and T- sapphire), cyan fluorescent proteins (e.g., CFP, eCFP, Cerulean, CyPet, AmCyanl, and Midoriishi-Cyan), red fluorescent proteins (e.g., RFP, mKate, mKate2, mPlum, DsRed
monomer, mCherry, mRFPl, DsRed-Express, DsRed2, DsRed-Monomer, HcRed-Tandem, HcRedl, AsRed2, eqFP611, mRaspberry, mStrawberry, and Jred), orange fluorescent proteins (e.g., mOrange, mKO, Kus abira- Orange, Monomeric Kusabira-Orange, mTangerine, and tdTomato), and any other suitable fluorescent protein whose presence in cells can be detected by flow cytometry methods.
[0061] The term “recombination” includes any process of exchange of genetic information between two polynucleotides and can occur by any mechanism. Recombination in response to double-strand breaks (DSBs) occurs principally through two conserved DNA repair pathways: non-homologous end joining (NHEJ) and homologous recombination (HR). See Kasparek & Humphrey (2011) Seminars in Cell & Dev. Biol. 22:886-897, herein incorporated by reference in its entirety for all purposes. Likewise, repair of a target nucleic acid mediated by an exogenous donor nucleic acid can include any process of exchange of genetic information between the two polynucleotides.
[0062] NHEJ includes the repair of double-strand breaks in a nucleic acid by direct ligation of the break ends to one another or to an exogenous sequence without the need for a homologous template. Ligation of non-contiguous sequences by NHEJ can often result in deletions, insertions, or translocations near the site of the double-strand break. For example, NHEJ can also result in the targeted integration of an exogenous donor nucleic acid through direct ligation of the break ends with the ends of the exogenous donor nucleic acid (i.e., NHEJ-based capture). Such NHEJ-mediated targeted integration can be preferred for insertion of an exogenous donor nucleic acid when homology directed repair (HDR) pathways are not readily usable (e.g., in non-dividing cells, primary cells, and cells which perform homology-based DNA repair poorly). In addition, in contrast to homology-directed repair, knowledge concerning large regions of sequence identity flanking the cleavage site is not needed, which can be beneficial when attempting targeted insertion into organisms that have genomes for which there is limited knowledge of the genomic sequence. The integration can proceed via ligation of blunt ends between the exogenous donor nucleic acid and the cleaved genomic sequence, or via ligation of sticky ends (i.e., having 5’ or 3’ overhangs) using an exogenous donor nucleic acid that is flanked by overhangs that are compatible with those generated by a nuclease agent in the cleaved genomic sequence. See, e.g., US 2011/020722, WO 2014/033644, WO 2014/089290, and Maresca et al. (2013) Genome Res. 23(3) :539-546, each of which is herein incorporated by reference in its entirety for all purposes. If blunt ends are ligated, target and/or donor resection may be needed to
generation regions of microhomology needed for fragment joining, which may create unwanted alterations in the target sequence.
[0063] Recombination can also occur via homology directed repair (HDR) or homologous recombination (HR). HDR or HR includes a form of nucleic acid repair that can require nucleotide sequence homology, uses a “donor” molecule as a template for repair of a “target” molecule (i.e., the one that experienced the double-strand break), and leads to transfer of genetic information from the donor to target. Without wishing to be bound by any particular theory, such transfer can involve mismatch correction of heteroduplex DNA that forms between the broken target and the donor, and/or synthesis -dependent strand annealing, in which the donor is used to resynthesize genetic information that will become part of the target, and/or related processes. In some cases, the donor polynucleotide, a portion of the donor polynucleotide, a copy of the donor polynucleotide, or a portion of a copy of the donor polynucleotide integrates into the target DNA. See Wang et al. (2013) Cell 153:910-918; Mandalos et al. (2012) PLOS ONE 7:e45768: 1-9; and Wang et al. (2013) Nat Biotechnol. 31:530-532, each of which is herein incorporated by reference in its entirety for all purposes. [0064] “Optional” or “optionally” means that the subsequently described event or circumstance may or may not occur and that the description includes instances in which the event or circumstance occurs and instances in which it does not.
[0065] Designation of a range of values includes all integers within or defining the range, and all subranges defined by integers within the range.
[0066] Unless otherwise apparent from the context, the term “about” encompasses values within a standard margin of error of measurement (e.g., SEM) of a stated value.
[0067] The term “and/or” refers to and encompasses any and all possible combinations of one or more of the associated listed items, as well as the lack of combinations when interpreted in the alternative (“or”).
[0068] The term “or” refers to any one member of a particular list and also includes any combination of members of that list.
[0069] The singular forms of the articles “a,” “an,” and “the” include plural references unless the context clearly dictates otherwise. For example, the term “a protein” or “at least one protein” can include a plurality of proteins, including mixtures thereof.
[0070] Statistically significant means p <0.05.
II. Non-Human Cells and Non-Human Animals Comprising a Gaa Knockout Gene [0071] Non-human animal cells, non-human animal genomes (e.g., non-human animal cell nuclei), and non-human animals comprising a knockout Gaa mutation as described herein are provided, wherein the non-human animals exhibit an ocular phenotype, such as but not limited to: (i) an accumulation of, e.g., substantially more, glycogen in ocular tissue (e.g., ciliary body, lens epithelium, neural retina) compared to a wildtype control non-human animal comprising a wildtype Gaa gene; (ii) a decrease in, e.g., significantly smaller, pupil size compared to a wildtype control non-human animal; (iii) vacuolar myopathy in the extraocular muscle; (iv) elevated glycogen levels in the iris; or (v) any combination thereof. In some embodiments, provided herein is a non-human animal cell or tissue (e.g., an ocular cell or tissue) comprising a knockout Gaa mutation as described herein, wherein the non- human cell or tissue exhibits a phenotype not seen in a corresponding cell or tissue expressing a wildtype GAA or having wildtype GAA activity. In some embodiments, a non- human animal cell or tissue as described herein comprises: (i) an ocular ciliary body, lens epithelium, and/or neural retina comprising a knockout mutation of an endogenous Gaa gene and exhibiting an accumulation of, e.g., substantially more, glycogen compared to a wildtype control non-human animal ocular ciliary body, lens epithelium, neural retina, and/or iris comprising a wildtype Gaa gene or activity; (ii) a pupil comprising a knockout mutation in a Gaa gene and exhibiting a decrease in, e.g., significantly smaller, 1 size (e.g., diameter and/or area) compared to a wildtype pupil control non-human animal pupil comprising a wildtype Gaa gene or activity; (iii) extraocular muscle comprising a knockout mutation of an endogenous Gaa gene and exhibiting increased vacuolar myopathy compared to a wildtype extraocular muscle comprising a wildtype Gaa gene or activity; or (iv) any combination thereof. The cells, genomes (nuclei), or non-human animals can be heterozygous or homozygous for the knockout Gaa mutation. A diploid organism has two alleles at each genetic locus. Each pair of alleles represents the genotype of a specific genetic locus. Genotypes are described as homozygous if there are two identical alleles at a particular locus and as heterozygous if the two alleles differ.
[0072] In some embodiments, the disclosure further provides methods for making any non-human animal, or reagents required for making the non-human animal as described herein.
[0073] The non-human animal cells and genomes (e.g., nuclei) provided herein can be, for example, any non-human animal cell comprising a knockout mutation in an endogenous Gaa gene. The cells and genomes (e.g., nuclei) can be eukaryotic, which include, for
example, fungal (e.g., yeast) cells and genomes, plant cells and genomes, animal cells and genomes, mammalian cells and genomes, and non-human mammalian cells and genomes. A non-human animal can be, for example, a mammal, fish, or bird. A mammalian cell can be, for example, a non-human mammalian cell, a rodent cell, a rat cell, a mouse cell, or a hamster cell. Other non-human mammals include, for example, non-human primates, monkeys, apes, orangutans, cats, dogs, rabbits, horses, bulls, deer, bison, livestock (e.g., bovine species such as cows, steer, and so forth; ovine species such as sheep, goats, and so forth; and porcine species such as pigs and boars). Birds include, for example, chickens, turkeys, ostrich, geese, ducks, and so forth. Domesticated animals and agricultural animals are also included. The term “non-human” excludes humans.
[0074] The cells can also be any type of undifferentiated or differentiated state. For example, a cell can be a totipotent cell, a pluripotent cell (e.g., a human pluripotent cell or a non-human pluripotent cell such as a mouse embryonic stem (ES) cell or a rat ES cell), or a non-pluripotent cell. Totipotent cells include undifferentiated cells that can give rise to any cell type, and pluripotent cells include undifferentiated cells that possess the ability to develop into more than one differentiated cell types. Such pluripotent and/or totipotent cells can be, for example, ES cells or ES-like cells, such as an induced pluripotent stem (iPS) cells. ES cells include embryo-derived totipotent or pluripotent cells that can contribute to any tissue of the developing embryo upon introduction into an embryo. ES cells can be derived from the inner cell mass of a blastocyst and can differentiate into cells of any of the three vertebrate germ layers (endoderm, ectoderm, and mesoderm).
[0075] The cells provided herein can also be germ cells (e.g., sperm or oocytes). The cells can be mitotically competent cells or mitotically-inactive cells, meiotically competent cells or meiotically-inactive cells. Similarly, the cells disclosed herein can also be primary somatic cells or cells that are not a primary somatic cell. Somatic cells include any cell that is not a gamete, germ cell, gametocyte, or undifferentiated stem cell. Suitable cells provided herein also include primary cells. Primary cells include cells or cultures of cells that have been isolated directly from an organism, organ, or tissue. Primary cells include cells that are neither transformed nor immortal. Primary cells include any cell obtained from an organism, organ, or tissue which was not previously passed in tissue culture or has been previously passed in tissue culture but is incapable of being indefinitely passed in tissue culture. Such cells can be isolated by conventional techniques.
[0076] Other suitable cells provided herein include immortalized cells. Immortalized cells include cells from a multicellular organism that would normally not proliferate
indefinitely but, due to mutation or alteration, have evaded normal cellular senescence and instead can keep undergoing division. Such mutations or alterations can occur naturally or be intentionally induced. Examples of immortalized cell lines are myofiber cell lines.
Immortalized or primary cells include cells that can be used for culturing or for expressing recombinant genes or proteins.
[0077] The cells provided herein also include one-cell stage embryos (i.e., fertilized oocytes or zygotes). Such one-cell stage embryos can be from any genetic background (e.g., BALB/c, C57BL/6, 129, or a combination thereof for mice), can be fresh or frozen, and can be derived from natural breeding or in vitro fertilization.
[0078] The cells provided herein can be normal, healthy cells, or can be diseased or mutant- bearing cells.
[0079] Non-human animals comprising a knockout mutation of an endogenous Gaa gene as described herein can be made by the methods described elsewhere herein. An animal can be, for example, a mammal, fish, or bird. Non-human mammals include, for example, non- human primates, monkeys, apes, orangutans, cats, dogs, horses, bulls, deer, bison, sheep, rabbits, rodents (e.g., mice, rats, hamsters, and guinea pigs), and livestock (e.g., bovine species such as cows and steer; ovine species such as sheep and goats; and porcine species such as pigs and boars). Birds include, for example, chickens, turkeys, ostrich, geese, and ducks. Domesticated animals and agricultural animals are also included. The term “non- human animal” excludes humans. Preferred non-human animals include, for example, rodents, such as mice and rats.
[0080] The non-human animals can be from any genetic background. For example, suitable mice can be from a 129 strain, a C57BL/6 strain, a mix of 129 and C57BL/6, a BALB/c strain, or a Swiss Webster strain. Examples of 129 strains include 129P1, 129P2, 129P3, 129X1, 129S 1 (e.g., 129S1/SV, 129Sl/Svlm), 129S2, 129S4, 129S5, 129S9/SvEvH, 129S6 (129/SvEvTac), 129S7, 129S8, 129T1, and 129T2. See, e.g., esting et al. (1999) Mammalian Genome 10:836, herein incorporated by reference in its entirety for all purposes. Examples of C57BL strains include C57BL/A, C57BL/An, C57BL/GrFa, C57BL/Kal_wN, C57BL/6, C57BL/6J, C57BL/6ByJ, C57BL/6NJ, C57BL/10, C57BL/10ScSn, C57BL/10Cr, and C57BL/01a. Suitable mice can also be from a mix of an aforementioned 129 strain and an aforementioned C57BL/6 strain (e.g., 50% 129 and 50% C57BL/6). Likewise, suitable mice can be from a mix of aforementioned 129 strains or a mix of aforementioned BL/6 strains (e.g., the 129S6 (129/SvEvTac) strain).
[0081 ] Similarly, rats can be from any rat strain, including, for example, an ACI rat
strain, a Dark Agouti (DA) rat strain, a Wistar rat strain, a LEA rat strain, a Sprague Dawley (SD) rat strain, or a Fischer rat strain such as Fisher F344 or Fisher F6. Rats can also be obtained from a strain derived from a mix of two or more strains recited above. For example, a suitable rat can be from a DA strain or an ACI strain. The ACI rat strain is characterized as having black agouti, with white belly and feet and an RTlavI haplotype. Such strains are available from a variety of sources including Harlan Laboratories. The Dark Agouti (DA) rat strain is characterized as having an agouti coat and an RTlavI haplotype. Such rats are available from a variety of sources including Charles River and Harlan Laboratories. Some suitable rats can be from an inbred rat strain. See, e.g., US 2014/0235933, herein incorporated by reference in its entirety for all purposes.
III. Methods of Making Non-Human Animals Comprising a Gaa Knockout Mutation [0082] Various methods are provided for making a non-human animal comprising a knockout mutation of an endogenous Gaa gene as disclosed elsewhere herein. Any convenient method or protocol for producing a genetically modified organism is suitable for producing such a genetically modified non-human animal. See, e.g. , Cho et al. (2009) Current Protocols in Cell Biology 42:19.11:19.11.1-19.11.22 and Gama Sosa et al. (2010) Brain Struct. Fund. 214(2-3):91-109, each of which is herein incorporated by reference in its entirety for all purposes. Such genetically modified non-human animals can be generated, for example, through gene knock-out at a targeted Gaa locus.
[0083] For example, the method of producing a non-human animal comprising a knockout mutation of a Gaa gene can comprise: (1) modifying the genome of a pluripotent cell to comprise the knockout mutation; (2) identifying or selecting the genetically modified pluripotent cell comprising the knockout mutation; (3) introducing the genetically modified pluripotent cell into a non-human animal host embryo cells in vitro', and (4) implanting and gestating the host embryo cells in a surrogate mother. Optionally, the host embryo comprising the modified pluripotent cell (e.g., a non-human ES cell) can be incubated until the blastocyst stage before being implanted into and gestated in the surrogate mother to produce an F0 non-human animal. The surrogate mother can then produce an F0 generation non-human animal comprising the knockout mutation.
[0084] The methods described herein can further comprise identifying a cell or animal having a modified target genomic locus. Various methods can be used to identify cells and animals having a targeted genetic modification.
[0085] The screening step can comprise, for example, a quantitative assay for assessing
modification of allele (MOA) of a parental chromosome. For example, the quantitative assay can be carried out via a quantitative PCR, such as a real-time PCR (qPCR). The real-time PCR can utilize a first primer set that recognizes the target locus and a second primer set that recognizes a non-targeted reference locus. The primer set can comprise a fluorescent probe that recognizes the amplified sequence.
[0086] Other examples of suitable quantitative assays include fluorescence-mediated in situ hybridization (FISH), comparative genomic hybridization, isothermal DNA amplification, quantitative hybridization to an immobilized probe(s), INVADER® Probes, TAQMAN® Molecular Beacon probes, or ECLIPSE™ probe technology (see, e.g., US 2005/0144655, incorporated herein by reference in its entirety for all purposes).
[0087] An example of a suitable pluripotent cell is an embryonic stem (ES) cell (e.g., a mouse ES cell or a rat ES cell). The modified pluripotent cell can be generated, for example, through recombination by (a) introducing into the cell one or more targeting vectors comprising an insert nucleic acid flanked by 5’ and 3’ homology arms corresponding to 5’ and 3’ target sites, wherein the insert nucleic acid comprises a Gaa knockout mutation; and (b) identifying at least one cell comprising in its genome the insert nucleic acid integrated at the target genomic locus. Alternatively, the modified pluripotent cell can be generated by (a) introducing into the cell: (i) a nuclease agent, wherein the nuclease agent induces a nick or double-strand break at a recognition site within the target genomic locus; and (ii) one or more targeting vectors comprising an insert nucleic acid flanked by 5’ and 3’ homology arms corresponding to 5 ’ and 3 ’ target sites located in sufficient proximity to the recognition site, wherein the insert nucleic acid comprises the Gaa knockout mutation; and (c) identifying at least one cell comprising a modification (e.g., integration of the insert nucleic acid) at the target genomic locus. Any nuclease agent that induces a nick or double-strand break into a desired recognition site can be used. Examples of suitable nucleases include a Transcription Activator-Like Effector Nuclease (TALEN), a zinc-finger nuclease (ZFN), a meganuclease, and Clustered Regularly Interspersed Short Palindromic Repeats (CRISPR)/CRISPR- associated (Cas) systems or components of such systems (e.g., CRISPR/Cas9). See, e.g., US 2013/0309670 and US 2015/0159175, each of which is herein incorporated by reference in its entirety for all purposes.
[0088] The donor cell can be introduced into a host embryo at any stage, such as the blastocyst stage or the pre-morula stage (i.e., the 4 cell stage or the 8 cell stage). Progeny that are capable of transmitting the genetic modification though the germline is generated. See, e.g., US Patent No. 7,294,754, herein incorporated by reference in its entirety for all
purposes.
[0089] Nuclear transfer techniques can also be used to generate the non-human mammalian animals. Briefly, methods for nuclear transfer can include the steps of: (1) enucleating an oocyte or providing an enucleated oocyte; (2) isolating or providing a donor cell or nucleus to be combined with the enucleated oocyte; (3) inserting the cell or nucleus into the enucleated oocyte to form a reconstituted cell; (4) implanting the reconstituted cell into the womb of an animal to form an embryo; and (5) allowing the embryo to develop. In such methods, oocytes are generally retrieved from deceased animals, although they may be isolated also from either oviducts and/or ovaries of live animals. Insertion of the donor cell or nucleus into the enucleated oocyte to form a reconstituted cell can be by microinjection of a donor cell under the zona pellucida prior to fusion. Fusion may be induced by application of a DC electrical pulse across the contact/fusion plane (electrofusion), by exposure of the cells to fusion-promoting chemicals, such as polyethylene glycol, or by way of an inactivated virus, such as the Sendai virus. A reconstituted cell can be activated by electrical and/or nonelectrical means before, during, and/or after fusion of the nuclear donor and recipient oocyte. Activation methods include electric pulses, chemically induced shock, penetration by sperm, increasing levels of divalent cations in the oocyte, and reducing phosphorylation of cellular proteins (as by way of kinase inhibitors) in the oocyte. The activated reconstituted cells, or embryos, can be cultured in media and then transferred to the womb of an animal. See, e.g., US 2008/0092249, WO 1999/005266, US 2004/0177390, WO 2008/017234, and US Patent No. 7,612,250, each of which is herein incorporated by reference in its entirety for all purposes.
[0090] The various methods provided herein allow for the generation of a genetically modified non-human F0 animal wherein the cells of the genetically modified F0 animal comprise the Gaa knockout mutation. It is recognized that depending on the method used to generate the F0 animal, the number of cells within the F0 animal that have the Gaa knockout mutation will vary. The introduction of the donor ES cells into a pre-morula stage embryo from a corresponding organism (e.g., an 8-cell stage mouse embryo) via for example, the VELOCIMOUSE® method allows for a greater percentage of the cell population of the F0 animal to comprise cells having the nucleotide sequence of interest comprising the targeted genetic modification. For example, at least 50%, 60%, 65%, 70%, 75%, 85%, 86%, 87%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% or 100% of the cellular contribution of the non-human F0 animal can comprise a cell population having the targeted modification.
[0091] In some embodiments, the disclosure provides a method of making a non-human animal, a non-human animal cell, or a non-human animal genome described herein, comprising inserting a nucleic acid sequence encoding a Gaa knockout mutation into the genome of the non-human animal, the genome of the non-human animal cell, or the non- human animal genome.
IV. Mouse Model for Testing Human Therapies/ Methods of Using Non-Human Animals Comprising a Gaa Knockout Mutation
[0092] By exhibiting non- invasive biomarkers of GAA accumulation, e.g., decreased pupil diameters and sizes, the non-human animals and cells described herein may be useful as models for preclinical testing of therapeutic modalities or compounds e.g., gene therapy with a Gaa transgene, that aim to increase GAA expression levels or activity in a patient in need thereof, e.g., a subject with Pompe disease. Thus, in some embodiments, described herein are non-human animal models for testing therapeutic modalities and/or compounds useful for increasing GAA expression or activity. In some such embodiments, described is a non- human animal model for testing therapeutic modalities or compounds useful for increasing GAA expression or activity comprising a non-human animal as described herein, e.g., a mouse comprising a knockout mutation of an endogenous Gaa gene and a therapeutic agent. In some embodiments, the testing of the therapeutic modalities and/or compounds for usefulness in increasing GAA expression or activity involves performing an assay or a study that allows determination of the effect of the therapeutic modalities and/or compounds on one or more ocular phenotypes described herein. In some embodiments, the determination of the effect comprises measuring the pupil diameter or area of a non-human animal as described herein before and after administration of the candidate therapeutic modality and/or compound, wherein an increase in pupil diameter or area indicates the therapeutic modality and/or compound may be useful in increasing GAA expression or activity in a subject in need thereof. In some embodiments, the determination of the effect of therapeutic modalities and/or compounds comprises measuring the level of glycogen in an ocular cell or tissue, e.g., ciliary body, lens epithelium, neural retina, iris) before and after administration of the candidate therapeutic modality and/or compound to a non-human animal model as described herein, wherein a decrease in the level of glycogen indicates the candidate therapeutic modality and/or compound may be useful in increasing GAA expression or activity in a subject in need thereof.
[0093] The candidate therapeutic modality and/or compound may be introduced into a
non-human animal as described herein. The candidate therapeutic modality and/or compound may be introduced into a non-human animal as described herein by several methods known to those skilled in the art. Some nonlimiting methods include transgenesis, hydrodynamic delivery (HDD), lipid nanoparticle (LNP) delivery, intravenous injection, parenteral administration, tissue or cell transplantation, etc. Nucleotides encoding, e.g., a protein comprising GAA activity, may be targeted to be expressed by particular cell types, e.g., the liver, or to be expressed by a particular locus, e.g., a safe harbor locus, according to well-known methods. As a non-limiting example, when administering nucleotides encoding a protein comprising GAA activity by LNP delivery, the LNP may contain one or more or all of the following: (i) a lipid for encapsulation and for endosomal escape; (ii) a neutral lipid for stabilization; (iii) a helper lipid for stabilization; and (iv) a stealth lipid. In certain LNPs, the cargo can include a guide RNA or a nucleic acid encoding a guide RNA. In certain LNPs, the cargo can include an mRNA encoding a Cas nuclease, such as Cas9, and a guide RNA or a nucleic acid encoding a guide RNA. In certain LNPs, the cargo can include an exogenous donor sequence (e.g., encoding a protein comprising GAA activity). In certain LNPs, the cargo can include a nuclease agent (or a nucleic acid encoding the nuclease agent or one or more nucleic acids encoding the nuclease agent) and an exogenous donor sequence (e.g., encoding a protein comprising GAA activity). In certain LNPs, the cargo can include an mRNA encoding a Cas nuclease, such as Cas9, a guide RNA or a nucleic acid encoding a guide RNA, and an exogenous donor sequence (e.g., encoding a protein comprising GAA activity) for CRISPR-mediated insertion of the exogenous donor sequence (e.g., encoding a protein comprising GAA activity) into a safe harbor locus of the animal, such as but not limited to a safe harbor locus, e.g., albumin, e.g., the first intron of the albumin locus. See, e.g., W02020206162, incorporated herein in its entirety by reference.
BRIEF DESCRIPTION OF THE SEQUENCES
[0094] The nucleotide and amino acid sequences listed in the accompanying sequence listing are shown using standard letter abbreviations for nucleotide bases, and three-letter code for amino acids. The nucleotide sequences follow the standard convention of beginning at the 5’ end of the sequence and proceeding forward (i.e., from left to right in each line) to the 3’ end. Only one strand of each nucleotide sequence is shown, but the complementary strand is understood to be included by any reference to the displayed strand. The amino acid sequences follow the standard convention of beginning at the amino terminus of the sequence and proceeding forward (i.e., from left to right in each line) to the carboxy terminus.
Table 1. Description of Sequences.

EXAMPLES
Example 1. Generation of GAA'/_ Animals
[0095] To generate GAA knockout animals, embryonic stem cells may be genetically modified to delete and/or replace an endogenous coding sequence encoding Gaa (see Figure 1), or to collapse the GAA gene in the embryonic stem cells (see Figure 2).
[0096] The mice used in Example 2 described herein were generated using VelociGene® methods, as described previously, which allows for the rapid and high-throughput generation of custom gene mutations in mice (Valenzuela, D.M., et al. (2003b), Nat Biotechnol 21:652- 659). Briefly, a large targeting vectors (LTVEC) were generated using BAC clones. The lacZ/neo' reporter/selection cassette (Figure 1) was introduced into ES cells (Poueymirou et
al. (2007); Valenzuela et al. (2003a)). Selection medium containing G418 was added to the cultures. Drug-resistant colonies were chosen, and correctly targeted ES cell clones were identified by the modification-of-allele (MOA) assay (Frendewey et al. (2010), Methods in enzymology 476:295-307; Valenzuela et al. (2003a), Nat. Biotechnol. 21:652-659).
[0097] The VelociMouse® method (Dechiara, T.M., (2009), Methods Mol Biol 530:311-
324; Poueymirou et al. (2007), Nat. Biotechnol. 25:91-99) was used, in which targeted ES cells were injected into uncompacted 8-cell stage Swiss Webster embryos to produce fully ES cell-derived F0 generation mice carrying the Gaa knockout mutations. Male VelociMice® were used directly for lacZ expression profiling or mated with C57BL/129 females to produce embryos or adults for lacZ analysis or to produce Fl breeders and phenotypic studies as described in Example 2 were performed.
[0098] Alternatively, collapse of both Gaa alleles is achieved using a combination of four SpCas9 guide RNAs (gRNA), each consisting of invariant tracr RNA, the scaffold for binding to SpCas9 enzyme, and a 20bp guide sequence specific to Gaa that allows a precise double-stranded cut. Guides direct SpCas9 cleavage close to the Gaa start ATG (guide 9251mGU (SEQ ID NO:5), cut site 38bp upstream from the ATG; guide 9251mGU3 (SEQ ID NO:6), cut site 18bp downstream of the ATG) and after the stop codon (guide 9251mGD3 (SEQ ID NO:7), cut site 677bp downstream of the stop; guide 9251mGD4 (SEQ ID NO:8), cut site 705bp downstream of the stop). See, e.g., Figure 2A.
[0099] A mixture of 125 pmol of each guide is complexed with 31.25pmol SpCas9 and electroporated into 2 x 106 hybrid 129S6/SvEvTac:C57Bl/6NTac Fl embryonic stems cells (ESC). Resulting clonal colonies are screened first via TaqMan for deletion of both copies of Gaa, using loss-of-allele assays. See, Table 2.
Table 2

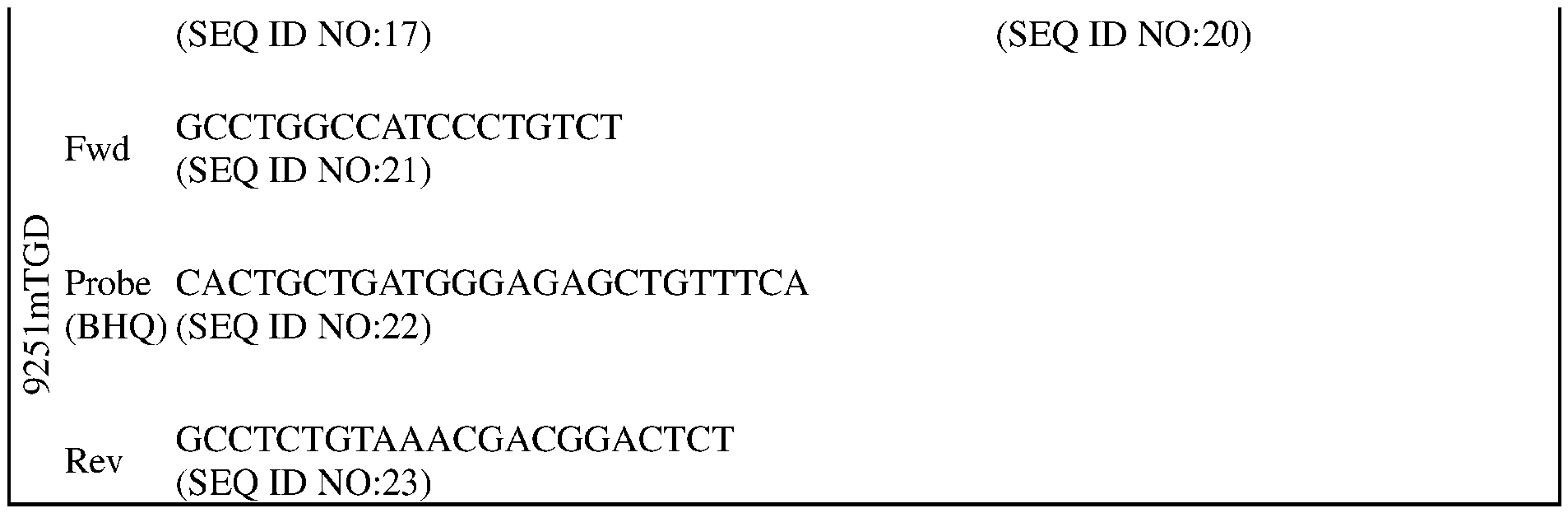
[00100] The deletion sizes are limited to within the region indicated by retention TaqMan assays. Clones with both Gaa copies deleted are subject to Illumina technology to fully characterize the knockout sequence using primers mm_Gaa_AmpF3 and mm_Gaa_AmpRl. See Table 3.
Table 3
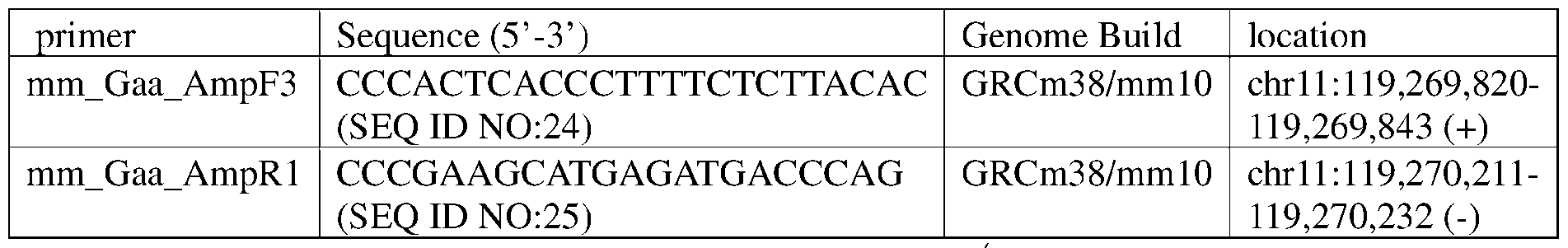
00101 ] F0 mice were bred to homozygosity to generate Gaa'7' mice.
Example 2. Analysis of GAA-/- Animals
[00102] Since the iris is the second highest tissue expressing GAA in a mouse body atlas, and since GAA mRNA expression in ocular tissues are similar in mice and human, phenotypic analysis of Gaa'7' animals comprising the Gaa knockout mutation depicted in Figure 1 were performed to assess whether the knockout mutation in these animals would produce any ocular phenotypes. Specifically, in addition to body weight, the following were assessed for Gaa '7' male mice (62.5% C57BL/6Tac; 37.5% 129SvTac; n = 15) that were at least 6 months old and compared to control mice expressing wildtype GAA (75% C57BL/6Tac 25% 129SvTac; n = 9): intraocular pressure (IOP); anterior and posterior optical coherence tomography (OCT) images for cornea, iris, and lens; histological staining of the eyes; and Periodic Acid Shiff (PAS) staining to measure glycogen accumulation.
[00103] Ocular morphology in GAA wild type and Gaa'7' knockout mice as described in Example 1 was evaluated with Heidelberg Spectralis optical coherence tomography (OCT). Comeal thickness and pupil size were measured at various ages. Electroretinogram (ERG)
was conducted to assess the retina function change. After sacrificing of the mice, the eyeballs were collected for Periodic Acid-Schiff (PAS) staining to evaluate glycogen accumulation, while hearts and irises were harvested for glycogen measurement with a bioassay kit.
[00104] There were no differences in body weights, cornea thicknesses, intraocular pressures, or in the pathologies of the retinas between wildtype and Gaa ^ mice (Figure 3, Figure 4, Figure 5, Figure 6).
[00105] OCT of the corneas, irises, and lens of each mouse were analyzed for pupil diameter and area using standard methodology (see Figure 7). Gaa^'mice exhibited statistically smaller pupil diameters and areas without dilation at 6- and 8- months of age compared to similarly aged wildtype mice (Figure 8 and Figure 9). There was no significant difference at 11 months of age without dilation or at any age with dilation between the two groups. (Figure 8 and Figure 9).
[00106] Vacuolar myopathy is seen in Gaa ' mice compared to wildtype control mice (Figure 11).
[00107] Periodic Acid-Schiff (PAS) staining of corneas, irises, ciliary bodies, lens epithelial cells, retinas and extraocular muscles was performed to measure glycogen accumulation in these tissues/cells by wildtype control mice or Gaa 1' mice. The PAS Stain is used for the demonstration of carbohydrates and carbohydrate rich compounds in tissues.
[00108] Based on images shown in Figures 12A-12F, there was no significant difference of glycogen accumulation in the cornea (Figure 12A) of a Gaa ' mouse (GAA KO) compared to the wildtype control mouse (GAA WT). Glycogen accumulation was difficult to observe in the irises due the pigmentation of these tissues (Figure 12B). Stronger PAS staining, indicating more glycogen accumulation, was seen in the ciliary body (Figure 12C), lens epithelial (Figure 12D), retina (Figure 12E), and extraocular muscle (Figure 12F) of a Gaaf' mouse (GAA KO) compared to the wildtype control mouse (GAA WT).
[00109] Glycogen in the iris and heart were also measured. The tissues were isolated, homogenized, heated to boiling for 10 minutes, centrifuged for 10 minutes at 4 °C and 18000xg, and measured using a glycogen assay kit (Abeam).
[00110] While there were no significant differences in body weight (Figure 3) cornea thickness (Figure 4), or intraocular pressure (Figure 5) in Gaa ' mice compared to wildtype control mice at exam date (6-7 months old), smaller pupil diameters and cornea areas were measured in Gua /-mice (Figure 8 and Figure 9). Histology also shows more glycogen accumulation in the ciliary body (Figure 12C), lens epithelial cell (Figure 12D), retina (Figure 12E), and extraocular muscle (Figure 12F) in Gaa'/' mice compared to wildtype
control mice. Moreover, glycogen measurement showed significant glycogen accumulation in the heart and irises of Gaa mice compared to wildtype control mice (Figure 13).
Example 3. CRISPR-mediated liver insertion of anti-Tfr:GAA in mouse model of Pompe disease
[00111] In the present Example, the pupil phenotype as observed in Example 2 for Gaa '~ mice (GAA KO) was confirmed and changes following CRISPR/Cas9-mediated liver insertion by AAV8 hTfR:GAA/LNP in humanized TfRl mice were measured.
[00112] GAA KO and humanized TfRl mice were matched with wild type mice of the similar age. All mice were negative for Rd8 mutation of the Crbl gene. Only male mice were used in current Example. Tail vein injection of AAV8 hTfR:GAA (3E13vg/kg)/LNP (3mg/kg) were performed with 2-3 month old mice.
[00113] Pupil size was measured before and various times after CRISPR/Cas9-mediated liver insertion. Anterior segment optical coherence tomography images were acquired by Optical Coherence Tomography (Heidelberg Engineering, Germany), both with and without the use of tropicamide. Mice were tested in a manner counterbalanced by genotype and age to avoid confounding effects of time of day, or testing sequence. In detail, mice were anesthetized with an intra-peritoneal (i.p.) dose of 70-80 mg/kg ketamine, and 15 mg/kg xylazine in sterile saline.
[00114] To measure pupil diameter without dilation, eyes were treated without the anti- muscarinic pupil dilator tropicamide. PBS was applied to the eyes immediately post ketamine/ xylazine injection, after 5 minutes ketamine/ xylazine injection, mice were secured on an adjustable holding platform and pictures of the anterior segment of the eye were taken by Optical Coherence Tomography.
[00115] On the second day, to measure dilated pupils, eyes were treated with a topical drop of 1 % solution of the anti-muscarinic pupil dilator tropicamide (Akorn) immediately post ketamine/ xylazine injection. After 5 minutes of ketamine/ xylazine injection, mice were secured on an adjustable holding platform, and pictures of the anterior segment of the eye, were taken by Optical Coherence Tomography.
[00116] Non-treated GAA KO mice exhibited significantly smaller pupil size compared to non- treated wild type (WT) mice at 2 and 3 months old without tropicamide (Figure 15A). One month post CRISPR/Cas9-mediated liver insertion, TfR:GAA expression were confirmed by Western blot in the serum from treated GAA KO mice (Figure 14; lanes 4-25). Pupil size without dilation for both GAA KO groups is significantly smaller than non-treated
WT control mice. Two months post CRISPR/Cas9-mediated liver insertion of TfR:GAA, without pupil dilation, there is a significant difference between AAV8 treated GAA KO and non-treated GAA KO mice (Figure 15A; p<0.001, Two-way ANOVA). With pupil dilation, there is a significant difference between treated GAA KO mice and non-treated GAA KO mice only at the 3-month timepoint (Figure 15B; p<0.05; Two-way ANOVA).
[00117] This Example indicates that glycogen accumulation in ocular tissues, resulting from GAA gene mutation, can instigate pathological changes in the eye, such as reduced pupil size. Enzyme replacement therapy to reverse these ocular alterations might offer a potential biomarker for therapeutic interventions in Pompe disease.
[00118] All references cited herein are incorporated by reference to the same extent as if each individual publication, database entry (e.g., Genbank sequences or GenelD entries), patent application, or patent, was specifically and individually indicated to be incorporated by reference. This statement of incorporation by reference is intended by Applicants to relate to each and every individual publication, database entry (e.g., Genbank sequences or GenelD entries), patent application, or patent, each of which is clearly identified in even if such citation is not immediately adjacent to a dedicated statement of incorporation by reference. The inclusion of dedicated statements of incorporation by reference, if any, within the specification does not in any way weaken this general statement of incorporation by reference. Citation of the references herein is not intended as an admission that the reference is pertinent prior art, nor does it constitute any admission as to the contents or date of these publications or documents.
Claims
1. A non-human animal comprising a knockout mutation of an endogenous a- glucosidase (Gad) gene, wherein the non-human animal exhibits an ocular phenotype.
2. The non-human animal of claim 1, wherein the knockout mutation comprises a deletion of the endogenous Gaa gene or a portion thereof.
3. The non-human animal of claim 1 or claim 2, wherein the knockout mutation comprises a deletion of the entire coding sequence of the endogenous Gaa gene.
4. The non-human animal of any one of claims 1-3, wherein the non-human animal does not express a-glucosidase (GAA) protein.
5. The non-human animal of any one of claims 1-4, wherein the non-human animal comprises an endogenous Gaa gene at an endogenous Gaa locus comprising a nucleotide sequence selected from the group consisting of: the nucleotide sequence as set forth in SEQ ID NO: 26, the nucleotide sequence as set forth in SEQ ID NO: 27, the nucleotide sequence as set forth in SEQ ID NO: 29, and the nucleotide sequence as set forth in SEQ ID NO: 30.
6. A non-human animal model for testing the efficacy of a candidate therapeutic agent for increasing a-glucosidase expression or activity, comprising: administering to the non-human animal according to any one of claims 1-5 the candidate therapeutic agent.
7. The non-human animal model of claim 6, wherein the candidate therapeutic agent is a-glucosidase.
8. A method of testing the efficacy of a candidate therapeutic agent for increasing a- glucosidase expression or activity, comprising: administering the candidate therapeutic agent to the non-human animal according to any one of claims 1 -5, evaluating the effect of the candidate therapeutic agent on the ocular phenotype of the non-human animal,
wherein after the evaluating, a reversal of the ocular phenotype in the non-human animal indicates that the candidate therapeutic agent increases a-glucosidase expression or activity in the non-human animal.
9. The method of claim 8, wherein the candidate therapeutic agent comprises a- glucosidase.
10. The non-human animal of any one of claims 1-5, the non-human animal model of claim 6 or claim 7, or the method of claim 8 or claim 9, wherein the ocular phenotype is (i) an accumulation of, e.g., substantially more, glycogen in ocular tissue (e.g., ciliary body, lens epithelium, neural retina) compared to a wildtype control non-human animal comprising a wildtype Gaa gene; (ii) a decrease in, e.g., significantly smaller, pupil size compared to a wildtype control non-human animal; (iii) vacuolar myopathy in the extraocular muscle; (iv) elevated glycogen levels in the iris; or (v) any combination thereof.
1 1. A non-human animal cell comprising a knockout mutation of an endogenous a- glucosidase Gaa) gene, wherein a non-human animal comprising the non-human animal cell exhibits an ocular phenotype.
12. The non-human animal cell of claim 11, wherein the knockout mutation comprises a deletion of the endogenous Gaa gene or a portion thereof.
13. The non-human animal cell of claim 11 or claim 12, wherein the knockout mutation comprises a deletion of the entire coding sequence of the endogenous Gaa gene.
14. The non-human animal cell of any one of claims 11-13, wherein the non-human animal cell does not express GAA protein.
15. The non-human animal cell of any one of claims 11-14, wherein the non-human animal cell comprises an endogenous Gaa gene at an endogenous Gaa locus comprising a nucleotide sequence selected from the group consisting of: the nucleotide sequence as set forth in SEQ ID NO: 26, the nucleotide sequence as set forth in SEQ ID NO: 27, the nucleotide sequence as set forth in SEQ ID NO: 29, and the nucleotide sequence as set forth in SEQ ID NO: 30.
16. The non-human animal cell of any one of claims 11-15, wherein the ocular phenotype is (i) an accumulation of, e.g., substantially more, glycogen in ocular tissue (e.g., ciliary body, lens epithelium, neural retina) compared to a wildtype control non-human animal comprising a wildtype Gaa gene; (ii) a decrease in, e.g., significantly smaller, pupil size compared to a wildtype control non-human animal; (iii) vacuolar myopathy in the extraocular muscle; (iv) elevated glycogen levels in the iris; or (v) any combination thereof.
17. A non-human animal genome comprising a knockout mutation of an endogenous a-glucosidase (Gaa) gene, wherein a non-human animal comprising the non-human animal genome exhibits an ocular phenotype.
18. The non-human animal genome of claim 17, wherein the knockout mutation comprises a deletion of the endogenous Gaa gene or a portion thereof.
19. The non-human animal genome of claim 17 or claim 18, wherein the knockout mutation comprises a deletion of the entire coding sequence of the endogenous Gaa gene.
20. The non-human animal genome of any one of claims 17-19, wherein a non-human animal or a non-human animal cell comprising the non-human animal genome does not express GAA protein.
21. The non-human animal genome of any one of claims 17-20, wherein the non- human animal genome comprises an endogenous Gaa gene at an endogenous Gaa locus comprising a nucleotide sequence selected from the group consisting of: the nucleotide sequence as set forth in SEQ ID NO: 26, the nucleotide sequence as set forth in SEQ ID NO: 27, the nucleotide sequence as set forth in SEQ ID NO: 29, and the nucleotide sequence as set forth in SEQ ID NO: 30.
22. The non-human animal genome of any one of claims 17-21, wherein the ocular phenotype is (i) an accumulation of, e.g., substantially more, glycogen in ocular tissue (e.g., ciliary body, lens epithelium, neural retina) compared to a wildtype control non-human animal comprising a wildtype Gaa gene; (ii) a decrease in, e.g., significantly smaller, pupil size compared to a wildtype control non-human animal; (iii) vacuolar myopathy in the extraocular muscle; (iv) elevated glycogen levels in the iris; or (v) any combination thereof.
23. A non-human animal, a non-human animal cell, or a non-human animal genome made according to any method described herein.
24. A method of making a non-human animal disease model of Pompe disease, the method comprising modifying the genome of a non-human animal to comprise a knockout mutation of an endogenous a-glucosidase (Gaa) gene, wherein the non-human animal exhibits an ocular phenotype,
25. A method of making a genetically modified non-human animal comprising a knockout mutation of an endogenous a-glucosidase (Gaa) gene, the method comprising:
(1) modifying the genome of a non-human animal pluripotent cell to comprise the knockout mutation;
(2) identifying or selecting the genetically modified non-human animal pluripotent cell comprising the knockout mutation;
(3) introducing the genetically modified non-human animal pluripotent cell into non-human animal host embryo cells in vitro; and
(4) implanting and gestating the non-human animal host embryo cells in a surrogate mother non-human animal to produce the genetically modified non- human animal, wherein the non-human animal exhibits an ocular phenotype.
26. The method of claim 25, wherein the non-human animal pluripotent cell is a non- human animal embryonic stem (ES) cell.
27. The method of any one of claims 24-26, wherein the knockout mutation comprises a deletion of the endogenous Gaa gene or a portion thereof.
28. The method of any one of claims 24-27, wherein the knockout mutation comprises a deletion of the entire coding sequence of the endogenous Gaa gene.
29. The method any one of claims 24-28, wherein the non-human animal does not express a-glucosidase (GAA) protein.
30. The method of any one of claims 24-29, wherein the non-human animal comprises an endogenous Gaa gene at an endogenous Gaa locus comprising a nucleotide sequence
selected from the group consisting of: the nucleotide sequence as set forth in SEQ ID NO: 26, the nucleotide sequence as set forth in SEQ ID NO: 27, the nucleotide sequence as set forth in SEQ ID NO: 29, and the nucleotide sequence as set forth in SEQ ID NO: 30.
31. The method of any one of claims 24-30, wherein the ocular phenotype is (i) an accumulation of, e.g., substantially more, glycogen in ocular tissue (e.g., ciliary body, lens epithelium, neural retina) compared to a wildtype control non-human animal comprising a wildtype Gaa gene; (ii) a decrease in, e.g., significantly smaller, pupil size compared to a wildtype control non-human animal; (iii) vacuolar myopathy in the extraocular muscle; (iv) elevated glycogen levels in the iris; or (v) any combination thereof.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US202363607184P | 2023-12-07 | 2023-12-07 | |
| US63/607,184 | 2023-12-07 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2025122754A1 true WO2025122754A1 (en) | 2025-06-12 |
Family
ID=94283969
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2024/058681 Pending WO2025122754A1 (en) | 2023-12-07 | 2024-12-05 | Gaa knockout non-human animals |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2025122754A1 (en) |
Citations (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1999005266A2 (en) | 1997-07-26 | 1999-02-04 | Wisconsin Alumni Research Foundation | Trans-species nuclear transfer |
| US20040177390A1 (en) | 2001-04-20 | 2004-09-09 | Ian Lewis | Method of nuclear transfer |
| US20050144655A1 (en) | 2000-10-31 | 2005-06-30 | Economides Aris N. | Methods of modifying eukaryotic cells |
| US7294754B2 (en) | 2004-10-19 | 2007-11-13 | Regeneron Pharmaceuticals, Inc. | Method for generating an animal homozygous for a genetic modification |
| WO2008017234A1 (en) | 2006-08-03 | 2008-02-14 | Shanghai Jiao Tong University Affiliated Children's Hospital | Cell nuclear transfer method |
| US7612250B2 (en) | 2002-07-29 | 2009-11-03 | Trustees Of Tufts College | Nuclear transfer embryo formation method |
| US20110020722A1 (en) | 2008-04-11 | 2011-01-27 | Lake Jeffrey G | Fuel cell and bipolar plate having manifold sump |
| US20130309670A1 (en) | 2012-04-25 | 2013-11-21 | Regeneron Pharmaceuticals, Inc. | Nuclease-Mediated Targeting With Large Targeting Vectors |
| WO2013176772A1 (en) | 2012-05-25 | 2013-11-28 | The Regents Of The University Of California | Methods and compositions for rna-directed target dna modification and for rna-directed modulation of transcription |
| WO2014033644A2 (en) | 2012-08-28 | 2014-03-06 | Novartis Ag | Methods of nuclease-based genetic engineering |
| WO2014089290A1 (en) | 2012-12-06 | 2014-06-12 | Sigma-Aldrich Co. Llc | Crispr-based genome modification and regulation |
| US20140235933A1 (en) | 2013-02-20 | 2014-08-21 | Regeneron Pharmaceuticals, Inc. | Genetic modification of rats |
| US20150159175A1 (en) | 2013-12-11 | 2015-06-11 | Regeneron Pharmaceutical, Inc. | Methods and compositions for the targeted modification of a genome |
| WO2020206162A1 (en) | 2019-04-03 | 2020-10-08 | Regeneron Pharmaceuticals, Inc. | Methods and compositions for insertion of antibody coding sequences into a safe harbor locus |
-
2024
- 2024-12-05 WO PCT/US2024/058681 patent/WO2025122754A1/en active Pending
Patent Citations (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1999005266A2 (en) | 1997-07-26 | 1999-02-04 | Wisconsin Alumni Research Foundation | Trans-species nuclear transfer |
| US20050144655A1 (en) | 2000-10-31 | 2005-06-30 | Economides Aris N. | Methods of modifying eukaryotic cells |
| US20080092249A1 (en) | 2001-04-20 | 2008-04-17 | Monash University | Method of nuclear transfer |
| US20040177390A1 (en) | 2001-04-20 | 2004-09-09 | Ian Lewis | Method of nuclear transfer |
| US7612250B2 (en) | 2002-07-29 | 2009-11-03 | Trustees Of Tufts College | Nuclear transfer embryo formation method |
| US7294754B2 (en) | 2004-10-19 | 2007-11-13 | Regeneron Pharmaceuticals, Inc. | Method for generating an animal homozygous for a genetic modification |
| WO2008017234A1 (en) | 2006-08-03 | 2008-02-14 | Shanghai Jiao Tong University Affiliated Children's Hospital | Cell nuclear transfer method |
| US20110020722A1 (en) | 2008-04-11 | 2011-01-27 | Lake Jeffrey G | Fuel cell and bipolar plate having manifold sump |
| US20130309670A1 (en) | 2012-04-25 | 2013-11-21 | Regeneron Pharmaceuticals, Inc. | Nuclease-Mediated Targeting With Large Targeting Vectors |
| WO2013176772A1 (en) | 2012-05-25 | 2013-11-28 | The Regents Of The University Of California | Methods and compositions for rna-directed target dna modification and for rna-directed modulation of transcription |
| WO2014033644A2 (en) | 2012-08-28 | 2014-03-06 | Novartis Ag | Methods of nuclease-based genetic engineering |
| WO2014089290A1 (en) | 2012-12-06 | 2014-06-12 | Sigma-Aldrich Co. Llc | Crispr-based genome modification and regulation |
| US20140235933A1 (en) | 2013-02-20 | 2014-08-21 | Regeneron Pharmaceuticals, Inc. | Genetic modification of rats |
| US20150159175A1 (en) | 2013-12-11 | 2015-06-11 | Regeneron Pharmaceutical, Inc. | Methods and compositions for the targeted modification of a genome |
| WO2020206162A1 (en) | 2019-04-03 | 2020-10-08 | Regeneron Pharmaceuticals, Inc. | Methods and compositions for insertion of antibody coding sequences into a safe harbor locus |
Non-Patent Citations (21)
| Title |
|---|
| ANAGNOSTOU E. ET AL: "Extraocular muscle function in adult-onset Pompe disease tested by saccadic eye movements", NEUROMUSCULAR DISORDERS, vol. 24, no. 12, 1 December 2014 (2014-12-01), GB, pages 1073 - 1078, XP093269053, ISSN: 0960-8966, Retrieved from the Internet <URL:https://pdf.sciencedirectassets.com/271174/1-s2.0-S0960896614X0012X/1-s2.0-S0960896614006312/main.pdf?hash=1f32d48ef5bcff05d9bc1ce909f4adce259ed64a4670fda8159dabbe91d33f45&host=68042c943591013ac2b2430a89b270f6af2c76d8dfd086a07176afe7c76c2c61&pii=S0960896614006312&tid=spdf-7d7780d7-b346-4194-9a0b-57b> [retrieved on 20250414], DOI: 10.1016/j.nmd.2014.08.004 * |
| ANDREW D BAIK ET AL: "Targeted delivery of acid alpha-glucosidase corrects skeletal muscle phenotypes in Pompe disease mice", 23 April 2020 (2020-04-23), XP055746512, Retrieved from the Internet <URL:https://www.biorxiv.org/content/10.1101/2020.04.22.051672v1.full.pdf> [retrieved on 20201103], DOI: 10.1101/2020.04.22.051672 * |
| BIJVOET A. G. A. ET AL: "Generalized glycogen storage and cardiomegaly in a knockout mouse model of Pompe disease", HUMAN MOLECULAR GENETICS, vol. 7, no. 1, 1 January 1998 (1998-01-01), GB, pages 53 - 62, XP093123441, ISSN: 0964-6906, DOI: 10.1093/hmg/7.1.53 * |
| CHAN JUSTIN ET AL: "The emerging phenotype of late-onset Pompe disease: A systematic literature review", MOLECULAR GENETICS AND METABOLISM, vol. 120, no. 3, 11 December 2016 (2016-12-11), pages 163 - 172, XP029942369, ISSN: 1096-7192, DOI: 10.1016/J.YMGME.2016.12.004 * |
| CHO ET AL., CURRENT PROTOCOLS IN CELL BIOLOGY, vol. 42, 2009 |
| COLELLA PASQUALINA ET AL: "Gene therapy with secreted acid alpha-glucosidase rescues Pompe disease in a novel mouse model with early-onset spinal cord and respiratory defects", EBIOMEDICINE, vol. 61, 1 November 2020 (2020-11-01), NL, pages 103052, XP093269471, ISSN: 2352-3964, Retrieved from the Internet <URL:https://www.thelancet.com/action/showPdf?pii=S2352-3964(20)30428-X> [retrieved on 20250414], DOI: 10.1016/j.ebiom.2020.103052 * |
| DECHIARA, T.M., METHODS MOL BIOL, vol. 530, 2009, pages 311 - 324 |
| FESTING ET AL., MAMMALIAN GENOME, vol. 10, 1999, pages 836 |
| FRENDEWEY ET AL., METHODS IN ENZYMOLOGY, vol. 476, 2010, pages 295 - 307 |
| GAMA SOSA ET AL., BRAIN STRUCT. FUNCT., vol. 214, no. 2-3, 2010, pages 91 - 109 |
| HUANG JEFFREY Y. ET AL: "CRISPR-Cas9 generated Pompe knock-in murine model exhibits early-onset hypertrophic cardiomyopathy and skeletal muscle weakness", SCIENTIFIC REPORTS, vol. 10, no. 1, 1 June 2020 (2020-06-01), US, pages 10321 - 10321, XP093123482, ISSN: 2045-2322, DOI: 10.1038/s41598-020-65259-8 * |
| KASPAREKHUMPHREY, SEMINARS IN CELL & DEV. BIOL., vol. 22, 2011, pages 886 - 897 |
| MANDALOS ET AL., PLOS ONE, vol. 7, no. 45768, 2012, pages 1 - 9 |
| MARESCA ET AL., GENOME RES., vol. 23, no. 3, 2013, pages 539 - 546 |
| NAKAMURA ET AL., NUCLEIC ACIDS RESEARCH, vol. 28, 2000, pages 292 |
| POUEYMIROU ET AL., NAT. BIOTECHNOL., vol. 25, 2007, pages 91 - 99 |
| VALENZUELA ET AL., NAT. BIOTECHNOL., vol. 21, 2003, pages 652 - 659 |
| VALENZUELA, D.M. ET AL., NAT BIOTECHNOL, vol. 21, 2003, pages 652 - 659 |
| WANG ET AL., CELL, vol. 153, 2013, pages 910 - 918 |
| WANG ET AL., NAT BIOTECHNOL., vol. 31, 2013, pages 530 - 532 |
| YUAN MING ET AL: "Ocular phenotypes in a mouse model of Pompe disease | IOVS | ARVO Journals", 1 June 2024 (2024-06-01), XP093269047, Retrieved from the Internet <URL:https://iovs.arvojournals.org/article.aspx?articleid=2799131> [retrieved on 20250410] * |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US12082565B2 (en) | Non-human animals models of retinoschisis | |
| JP6192865B2 (en) | lincRNA-deficient non-human animal | |
| US20210392865A1 (en) | Non-human animal exhibiting diminished upper and lower motor neuron function and sensory perception | |
| WO2025122754A1 (en) | Gaa knockout non-human animals | |
| US9974290B2 (en) | Animal model and method for studying gene-gene interactions | |
| US20250194571A1 (en) | Compositions and methods for defining optimal treatment timeframes in lysosomal disease | |
| CA3257739A1 (en) | Non-human animals comprising a modified transferrin receptor locus | |
| HK1238673A1 (en) | Pint knockout mouse exhibiting a premature aging-associated phenotype |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 24838078 Country of ref document: EP Kind code of ref document: A1 |