WO2025111297A1 - Polynucleotides encoding cystic fibrosis transmembrane conductance regulator for the treatment of cystic fibrosis - Google Patents
Polynucleotides encoding cystic fibrosis transmembrane conductance regulator for the treatment of cystic fibrosis Download PDFInfo
- Publication number
- WO2025111297A1 WO2025111297A1 PCT/US2024/056591 US2024056591W WO2025111297A1 WO 2025111297 A1 WO2025111297 A1 WO 2025111297A1 US 2024056591 W US2024056591 W US 2024056591W WO 2025111297 A1 WO2025111297 A1 WO 2025111297A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- mrna
- dose
- administered
- day
- lipid
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/17—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- A61K38/1703—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K48/00—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy
- A61K48/005—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy characterised by an aspect of the 'active' part of the composition delivered, i.e. the nucleic acid delivered
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K48/00—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy
- A61K48/0075—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy characterised by an aspect of the delivery route, e.g. oral, subcutaneous
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/007—Pulmonary tract; Aromatherapy
- A61K9/0073—Sprays or powders for inhalation; Aerolised or nebulised preparations generated by other means than thermal energy
- A61K9/0078—Sprays or powders for inhalation; Aerolised or nebulised preparations generated by other means than thermal energy for inhalation via a nebulizer such as a jet nebulizer, ultrasonic nebulizer, e.g. in the form of aqueous drug solutions or dispersions
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/50—Microcapsules having a gas, liquid or semi-solid filling; Solid microparticles or pellets surrounded by a distinct coating layer, e.g. coated microspheres, coated drug crystals
- A61K9/51—Nanocapsules; Nanoparticles
- A61K9/5107—Excipients; Inactive ingredients
- A61K9/5123—Organic compounds, e.g. fats, sugars
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/12—Mucolytics
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/79—Vectors or expression systems specially adapted for eukaryotic hosts
- C12N15/85—Vectors or expression systems specially adapted for eukaryotic hosts for animal cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/87—Introduction of foreign genetic material using processes not otherwise provided for, e.g. co-transformation
- C12N15/88—Introduction of foreign genetic material using processes not otherwise provided for, e.g. co-transformation using microencapsulation, e.g. using amphiphile liposome vesicle
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2300/00—Mixtures or combinations of active ingredients, wherein at least one active ingredient is fully defined in groups A61K31/00 - A61K41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/46—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates
- C07K14/47—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates from mammals
- C07K14/4701—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates from mammals not used
- C07K14/4712—Cystic fibrosis
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2830/00—Vector systems having a special element relevant for transcription
- C12N2830/50—Vector systems having a special element relevant for transcription regulating RNA stability, not being an intron, e.g. poly A signal
Definitions
- Cystic Fibrosis (“CF”) is a rare autosomal recessive disease with serious, chronically debilitating morbidities and high premature mortality for which there is currently no cure. Cystic Fibrosis affects more than 80,000 individuals worldwide, including over 31,000 individuals in the US and 49,000 individuals in the EU.
- Cystic Fibrosis is caused by decreased quantity and/or function of the cystic fibrosis transmembrane conductance regulator (CFTR) protein due to mutations in the CFTR gene.
- CFTR cystic fibrosis transmembrane conductance regulator
- CFTR is an ion channel that regulates the flow of chloride and other ions across epithelia in various tissues, including the lungs, pancreas and other gastrointestinal organs, and sweat glands. Decreased CFTR quantity or function results in the failure to regulate chloride transport in these tissues leading to the multisystem pathology associated with CF. Progressive loss of lung function is the leading cause of mortality.
- the present disclosure provides messenger RNA (mRNA) therapeutics for the treatment of cystic fibrosis.
- mRNA messenger RNA
- the mRNA therapeutics of the invention are particularly well-suited for the treatment of cystic fibrosis as the technology provides for the intracellular delivery of mRNA encoding a cystic fibrosis transmembrane conductance regulator (CFTR) polypeptide followed by de novo synthesis of functional CFTR polypeptide within target cells.
- CFTR cystic fibrosis transmembrane conductance regulator
- the disclosure features a method of treating cystic fibrosis in a human subject in need thereof, the method comprising administering to the human subject by inhalation a lipid nanoparticle comprising a mRNA comprising an open reading frame (ORF) encoding the CFTR polypeptide of SEQ ID NO: 1, wherein the mRNA is administered at a dose of 1 mg to 7 mg.
- a lipid nanoparticle comprising a mRNA comprising an open reading frame (ORF) encoding the CFTR polypeptide of SEQ ID NO: 1, wherein the mRNA is administered at a dose of 1 mg to 7 mg.
- ORF open reading frame
- the ORF is at least 80% identical to the nucleotide sequence of SEQ ID NO: 10.
- the ORF is at least 95% identical to the nucleotide sequence of SEQ ID NO: 10.
- the ORF is at least 99% identical to the nucleotide sequence of SEQ ID NO: 10. In some embodiments, the ORF is 100% identical to the nucleotide sequence of SEQ ID NO: 10.
- the mRNA comprises a 5' UTR comprising the nucleotide sequence of SEQ ID NO:50.
- the mRNA comprises a 3' UTR comprising the nucleotide sequence of SEQ ID NO: 141.
- the mRNA comprises the nucleic acid sequence of SEQ ID NO: 11.
- the mRNA comprises a 5' terminal cap.
- the 5' terminal cap comprises m 7 G-ppp-Gm.
- the mRNA comprises a poly-A region.
- the poly-A region comprises SEQ ID NO: 195.
- the poly-A region comprises A100-UCUAG-A20-inverted deoxy-thymidine (SEQ ID NO:211).
- all of the uracils of the mRNA are Nl- methylpseudouracils .
- the mRNA comprises the nucleotide sequence of SEQ ID NO: 13.
- the mRNA is administered at a dose of 1 mg.
- the mRNA is administered at a dose of 2 mg.
- the mRNA is administered at a dose of 3 mg.
- the mRNA is administered at a dose of 4 mg.
- the mRNA is administered at a dose of 5 mg.
- the mRNA is administered at a dose of 6 mg.
- the mRNA is administered at a dose of 7 mg.
- the method comprises multiple administrations of the dose.
- the dose is administered repeatedly once every day.
- the method comprises at least 28 consecutive daily administrations of the dose.
- the lipid nanoparticle is administered using a nebulizer.
- the human subject carries mutations in both alleles of the CFTR gene that result in no CFTR protein produced or a mutant CFTR protein that is not responsive to therapy with CFTR modulators.
- the CFTR modulators comprise ivacaftor, lumacaftor/ivacaftor, tezacaftor/ivacaftor, and elexacaftor/tezacaftor/ivacaftor.
- the mutations in both alleles of the CFTR gene are selected from the mutations depicted in Table 4.
- the method comprises co-administering a CFTR potentiator to the human subject.
- the CFTR potentiator is ivacaftor, lumacaftor/ivacaftor, tezacaftor/ivacaftor, or elexacaftor/tezacaftor/ivacaftor.
- the CFTR potentiator is ivacaftor.
- ivacaftor is administered orally.
- ivacaftor is administered orally every 12 hours at a dose of 150 mg.
- the mRNA is administered once every day at a dose of
- the mRNA is administered once every day at a dose of 1 mg and ivacaftor is administered orally. In some embodiments, the mRNA is administered once every day at a dose of 1 mg and ivacaftor is administered orally every 12 hours at a dose of 150 mg.
- the mRNA is administered once every day at a dose of
- the mRNA is administered once every day at a dose of 2 mg and ivacaftor is administered orally. In some embodiments, the mRNA is administered once every day at a dose of 2 mg and ivacaftor is administered orally every 12 hours at a dose of 150 mg.
- the mRNA is administered once every day at a dose of
- the mRNA is administered once every day at a dose of 3 mg and ivacaftor is administered orally. In some embodiments, the mRNA is administered once every day at a dose of 3 mg and ivacaftor is administered orally every 12 hours at a dose of 150 mg.
- the mRNA is administered once every day at a dose of
- the mRNA is administered once every day at a dose of 4 mg and ivacaftor is administered orally. In some embodiments, the mRNA is administered once every day at a dose of 4 mg and ivacaftor is administered orally every 12 hours at a dose of 150 mg.
- the mRNA is administered once every day at a dose of
- the mRNA is administered once every day at a dose of 5 mg and ivacaftor is administered orally. In some embodiments, the mRNA is administered once every day at a dose of 5 mg and ivacaftor is administered orally every 12 hours at a dose of 150 mg.
- the mRNA is administered once every day at a dose of
- the mRNA is administered once every day at a dose of 6 mg and ivacaftor is administered orally. In some embodiments, the mRNA is administered once every day at a dose of 6 mg and ivacaftor is administered orally every 12 hours at a dose of 150 mg.
- the mRNA is administered once every day at a dose of
- the mRNA is administered once every day at a dose of 7 mg and ivacaftor is administered orally. In some embodiments, the mRNA is administered once every day at a dose of 7 mg and ivacaftor is administered orally every 12 hours at a dose of 150 mg.
- the mRNA comprises the nucleotide sequence of SEQ ID NO: 13 and the mRNA is administered once every day at a dose of 1 mg. In some embodiments, the mRNA comprises the nucleotide sequence of SEQ ID NO: 13, the mRNA is administered once every day at a dose of 1 mg, and ivacaftor is administered orally. In some embodiments, the mRNA comprises the nucleotide sequence of SEQ ID NO: 13, the mRNA is administered once every day at a dose of 1 mg, and ivacaftor is administered orally every 12 hours at a dose of 150 mg.
- the mRNA comprises the nucleotide sequence of SEQ ID NO: 13 and the mRNA is administered once every day at a dose of 2 mg. In some embodiments, the mRNA comprises the nucleotide sequence of SEQ ID NO: 13, the mRNA is administered once every day at a dose of 2 mg, and ivacaftor is administered orally. In some embodiments, the mRNA comprises the nucleotide sequence of SEQ ID NO: 13, the mRNA is administered once every day at a dose of 2 mg, and ivacaftor is administered orally every 12 hours at a dose of 150 mg.
- the mRNA comprises the nucleotide sequence of SEQ ID NO: 13 and the mRNA is administered once every day at a dose of 3 mg. In some embodiments, the mRNA comprises the nucleotide sequence of SEQ ID NO: 13, the mRNA is administered once every day at a dose of 3 mg, and ivacaftor is administered orally. In some embodiments, the mRNA comprises the nucleotide sequence of SEQ ID NO: 13, the mRNA is administered once every day at a dose of 3 mg, and ivacaftor is administered orally every 12 hours at a dose of 150 mg.
- the mRNA comprises the nucleotide sequence of SEQ ID NO: 13 and the mRNA is administered once every day at a dose of 4 mg. In some embodiments, the mRNA comprises the nucleotide sequence of SEQ ID NO: 13, the mRNA is administered once every day at a dose of 4 mg, and ivacaftor is administered orally. In some embodiments, the mRNA comprises the nucleotide sequence of SEQ ID NO: 13, the mRNA is administered once every day at a dose of 4 mg, and ivacaftor is administered orally every 12 hours at a dose of 150 mg.
- the mRNA comprises the nucleotide sequence of SEQ ID NO: 13 and the mRNA is administered once every day at a dose of 5 mg. In some embodiments, the mRNA comprises the nucleotide sequence of SEQ ID NO: 13, the mRNA is administered once every day at a dose of 5 mg, and ivacaftor is administered orally. In some embodiments, the mRNA comprises the nucleotide sequence of SEQ ID NO: 13, the mRNA is administered once every day at a dose of 5 mg, and ivacaftor is administered orally every 12 hours at a dose of 150 mg.
- the mRNA comprises the nucleotide sequence of SEQ ID NO: 13 and the mRNA is administered once every day at a dose of 6 mg. In some embodiments, the mRNA comprises the nucleotide sequence of SEQ ID NO: 13, the mRNA is administered once every day at a dose of 6 mg, and ivacaftor is administered orally. In some embodiments, the mRNA comprises the nucleotide sequence of SEQ ID NO: 13, the mRNA is administered once every day at a dose of 6 mg, and ivacaftor is administered orally every 12 hours at a dose of 150 mg.
- the mRNA comprises the nucleotide sequence of SEQ ID NO: 13 and the mRNA is administered once every day at a dose of 7 mg. In some embodiments, the mRNA comprises the nucleotide sequence of SEQ ID NO: 13, the mRNA is administered once every day at a dose of 7 mg, and ivacaftor is administered orally. In some embodiments, the mRNA comprises the nucleotide sequence of SEQ ID NO: 13, the mRNA is administered once every day at a dose of 7 mg, and ivacaftor is administered orally every 12 hours at a dose of 150 mg.
- the treatment increases percent predicted forced expiratory volume in 1 second (ppFEVi) from baseline.
- the method comprises administering to the human subject long-acting p agonist (LABA)Zinhaled corticosteroid (ICS) therapy.
- the human subject is administered LABA/ICS therapy prior to administration of the lipid nanoparticle.
- the human subject is administered LABA/ICS therapy for at least 28 days before the first administration of the lipid nanoparticle.
- the method comprises administering to the human subject LABA/ICS therapy and short-acting p agonist (SABA) therapy.
- the human subject is administered LABA/ICS therapy and SABA therapy prior to administration of the lipid nanoparticle.
- the human subject is administered LABA/ICS therapy and SABA therapy on the same day as administration of the lipid nanoparticle.
- the human subject is administered LABA/ICS therapy prior to administration of the lipid nanoparticle and the human subject is administered LABA/ICS therapy and SABA therapy on the same day as administration of the lipid nanoparticle.
- the human subject is administered LABA/ICS therapy for at least 28 days before the first administration of the lipid nanoparticle and the human subject is administered LABA/ICS therapy and SABA therapy on the same day as administration of the lipid nanoparticle.
- the lipid nanoparticle comprises:
- the ionizable lipid is (Compound II) or a salt thereof.
- the cationic agent is salt thereof.
- the ionizable lipid is thereof, and the cationic agent is salt thereof.
- the ionizable lipid is (Compound II) or a salt thereof;
- the phospholipid is 1,2 distearoyl-sn-glycero-3-phosphocholine (DSPC);
- the structural lipid is cholesterol;
- the PEG lipid is l,2-dimyristoyl-rac-glycero-3 -methoxypolyethylene glycol-2000 (PEG2000-DMG);
- Fig. 1 shows a general process for preparing empty lipid nanoparticles (eLNPs) where nanoprecipitation is carried out at pH 4 followed by titration to pH 5.
- Fig. 2 shows a general process for preparing fdled lipid nanoparticles (fLNPs) where encapsulation is carried out at pH 5.
- Fig. 3A contains representative photomicrographs depicting CFTR mRNA (ISH) and CFTR protein (IHC) 18 hours after a single nebulized delivery of buffer or 3.8 pg/cm 2 VX-522 to the apical surface of G542X/K684SfsX38 CF-HBE cells.
- the cells were formalin-fixed paraffin-embedded, and nuclei were counter stained with hematoxylin.
- Fig. 3B contains representative photomicrographs demonstrating apical CFTR expression in different bronchial epithelial cell types including ionocytes, goblet cells and ciliated cells in VX-522 treated (3.8 pg/cm 2 ) G542X/K684SfsX38 CF-HBE cells but not in buffer-treated controls.
- Fig. 3C is a graph depicting morphometric analysis of CFTR protein in MF/MF-HBE cells from Figs. 3 A and 3B. Data indicate the total percentage of MF/MF HBE cells expressing CFTR protein 18 hours after a single treatment with VX-522 or buffer control. Numbers on the x-axis denote delivered VX-522-mRNA expressed as deposited pg/cm 2 . Data represents the mean of 3 to 4 replicate experiments, ⁇ SEM in CF-HBE cells from 2 MF/MF donors.
- Fig. 3D is a graph depicting dose-dependent increase in CFTR-mediated chloride transport in CF-HBE cells derived from 2 MF/MF donors 18 hours after a single treatment of either buffer or 2.1 to 6.1 pg/cm 2 VX-522 nebulized to the apical surface, or clinically relevant concentrations ELX/TEZ/IVA (TRI). Data represents the mean, ⁇ SEM of 4 replicate experiments in CF-HBE cells derived from 2 MF/MF donors.
- Fig. 3E is a graph depicting dose-dependent increase in CFTR-mediated chloride transport in CF-HBE cells derived from a single F508dellMF donor 18 hours after a single treatment of either buffer, 0.9 to 5.6 pg/cm 2 VX-522 nebulized to the apical surface, or clinically relevant concentrations of ELX/TEZ/IVA (TRI).
- Data represents the mean, ⁇ SEM of 6 replicate experiments in CF-HBE cells derived from one F508del/MF donor.
- Fig. 4A (upper panel) contains a representative photomicrograph of bronchial epithelium from VX-522 treated (2.4 pg/kg/day for 28 days) monkey lungs depicting VX-522-mRNA (ISH).
- Fig. 4B (upper panel) contains a representative photomicrograph of bronchial epithelium from VX-522 treated (2.4 pg/kg/day for 28 days) monkey lungs depicting CFTR protein (IHC).
- Fig. 5 is a graph depicting chloride transport in CF-HBE cells following treatment with VX-522 or VX-522 in combination with ivacaftor (IVA). Data represents the mean, ⁇ SEM of four replicate experiments in CF-HBE cells derived from two MF/MF donors, * denotes P ⁇ 0.05 when compared to VX-522 alone at the given dose. At each dose, VX-522 is the bar depicted on the left and VX-522+IVA is on the right. DETAILED DESCRIPTION
- Cystic fibrosis is a progressive, genetic disease that causes persistent lung infections and limits the ability to breathe overtime. This disease is characterized by the presence of mutations in both copies of the gene for the cystic fibrosis transmembrane conductance regulator (CFTR) protein. Without CFTR, which is involved in the production of sweat, digestive fluids and mucus, secretions that are usually thin instead become thick.
- the disclosed mRNA therapeutics provide for the intracellular delivery of mRNA encoding CFTR followed by de novo synthesis of functional CFTR protein within target cells. After delivery of mRNA to the target cells, the desired CFTR protein is expressed by the cells’ own translational machinery, and hence, fully functional CFTR protein replaces the defective or missing protein.
- Cystic Fibrosis Transmembrane Conductance Regulator (CFTR; EC 3.6.3.49) is an ABC transporter-class ion channel. It conducts chloride and thiocyanate ions across epithelial cell membranes.
- the structure of the approximately 168 kDa CFTR which is highly conserved amongst organisms, consists of seven domains.
- CFTR contains two transmembrane domains with six transmembrane helices each. Additionally, CFTR contains two nucleotide binding domains, two ABC transporter domains, and one PDZ-binding domain.
- the nucleotide binding domains are used for binding and hydrolyzing ATP, ABC transporters move ions across the plasma membrane, and the PDZ-binding domain which CFTR to anchor itself to the plasma membrane.
- CFTR usually exists in dimer units in the plasma membrane of the cell.
- a CFTR polypeptide The amino acid sequence of a CFTR polypeptide is provided in SEQ ID NO: 1.
- This CFTR polypeptide (and nucleic acids encoding it) is described in WO 2022/104131, the content of which is incorporated by reference.
- the disclosure provides a polynucleotide (e.g., a RNA, e.g., a mRNA) comprising a nucleotide sequence (e.g., an open reading frame) encoding a CFTR polypeptide.
- a polynucleotide disclosed herein comprises a sequence encoding the CFTR polypeptide of SEQ ID NO: 1.
- the instant invention features mRNAs for use in treating or preventing cystic fibrosis.
- the mRNAs featured for use in the invention are administered to subjects and encode human CFTR protein in vivo.
- the invention relates to polynucleotides, e.g., mRNA, comprising an open reading frame of linked nucleosides encoding a CFTR protein (SEQ ID NO: 1).
- the open reading frame is sequence-optimized.
- the polynucleotide (e.g., a RNA, e.g., an mRNA) of the invention comprises a nucleotide sequence (e.g., an ORF) encoding a CFTR polypeptide (e.g., SEQ ID NO: 1), wherein the nucleotide sequence has at least 70%, at least 71%, at least 72%, at least 73%, at least 74%, at least 75%, at least 76%, at least 77%, at least 78%, at least 79%, at least 80%, at least 81%, at least 82%, at least 83%, at least 84%, at least 85%, at least 86%, at least 87%, at least 88%, at least 89%, at least 90%, at least 91%, at least 92%, at least 93%, at least 94%, at least 95%, at least 96%, at least 97%, at least 98%, at least 99%, or 100% sequence identity to SEQ ID NOTO.
- the polynucleotide of the invention (e.g., a RNA, e.g., an mRNA) comprises a nucleotide sequence (e.g., an ORF, e.g., SEQ ID NOTO) encoding a CFTR polypeptide further comprises a 5'-UTR (e.g., SEQ ID NO:50) and a 3'-UTR (e.g., SEQ ID NO: 141).
- the polynucleotide (e.g., a RNA, e.g., an mRNA) of the invention comprises the sequence of SEQ ID NO: 10.
- the polynucleotide (e.g., a RNA, e.g., an mRNA) comprises a 5' terminal cap (e.g., m 7 G-ppp-Gm-AG, CapO, Capl, ARCA, inosine, N1 -methylguanosine, 2'-fluoro-guanosine, 7-deaza-guanosine, 8-oxo-guanosine, 2-amino- guanosine, LNA-guanosine, 2-azidoguanosine, Cap2, Cap4, 5' methylG cap, or an analog thereof) and a poly-A-tail region (e.g., about 100 nucleotides in length).
- a 5' terminal cap e.g., m 7 G-ppp-Gm-AG, CapO, Capl, ARCA, inosine, N1 -methylguanosine, 2'-fluoro-guanosine, 7-deaza-guanosine
- the mRNA comprises a polyA tail.
- the poly A tail is 50-150 (SEQ ID NO: 197), 75-150 (SEQ ID NO: 198), 85-150 (SEQ ID NO: 199), 90-120 (SEQ ID NO: 193), 90-130 (SEQ ID NO: 194), or 90-150 (SEQ ID NO: 192) nucleotides in length.
- the poly A tail is 100 nucleotides in length (SEQ ID NO: 195).
- the poly A tail is protected (e.g., with an inverted deoxy-thymidine).
- the poly A tail comprises A 100- UCUAG-A20-inverted deoxy-thymidine (SEQ ID NO:211). In some instances, the poly A tail is A100-UCUAG-A20-inverted deoxy-thymidine (SEQ ID NO: 211).
- the polynucleotide of the invention e.g., a RNA, e.g., an mRNA
- a nucleotide sequence e.g., an ORF
- a CFTR polypeptide is single stranded or double stranded.
- the polynucleotide of the invention comprising a nucleotide sequence (e.g., an ORF) encoding a CFTR polypeptide is DNA or RNA.
- the polynucleotide of the invention is RNA.
- the polynucleotide of the invention is, or functions as, an mRNA.
- the mRNA comprises a nucleotide sequence (e.g., an ORF) that encodes at least one CFTR polypeptide, and is capable of being translated to produce the encoded CFTR polypeptide in vitro, in vivo, in situ or ex vivo.
- the polynucleotide of the invention (e.g., a RNA, e.g., an mRNA) comprises a sequence-optimized nucleotide sequence (e.g., an ORF) encoding a CFTR polypeptide, wherein the polynucleotide comprises at least one chemically modified nucleobase, e.g., N1 -methylpseudouracil or 5 -methoxyuracil. In certain embodiments, all uracils in the polynucleotide are N1 -methylpseudouracils. In other embodiments, all uracils in the polynucleotide are 5 -methoxyuracils. In some embodiments, the polynucleotide further comprises a miRNA binding site, e.g., a miRNA binding site that binds to miR-142 and/or a miRNA binding site that binds to miR-126.
- the polynucleotide (e.g., a RNA, e.g., a mRNA) disclosed herein is formulated with a delivery agent comprising, e.g., a compound having the Formula (I), e.g., Compound II; a compound having the Formula (III), (IV), (V), or (VI), e.g., Compound VI; a compound having the Formula (VIII), e.g., any of Compounds 419-428, e.g., Compound I, or a compound having the Formula Al, A2, A3, A4, or A5, e.g., any one of SA1-SA41, or any combination thereof.
- a delivery agent comprising, e.g., a compound having the Formula (I), e.g., Compound II; a compound having the Formula (III), (IV), (V), or (VI), e.g., Compound VI; a compound having the Formula (VIII), e.g., any of
- the delivery agent comprises Compound II, DSPC, Cholesterol, Compound I or PEG-DMG, and GL-67 or a salt thereof, e.g., with a mole ratio of about 47.6 ⁇ 25:9.5 ⁇ 8:36.6 ⁇ 20: 1.4 ⁇ 1.25:4.9 ⁇ 2.5.
- the delivery agent comprises Compound II, DSPC, Cholesterol, Compound I or PEG-DMG, and GL-67 or a salt thereof, e.g., with a mole ratio of about
- the delivery agent comprises Compound II, DSPC, Cholesterol, and Compound I or PEG-DMG, e.g., with a mole ratio of about 47.6 ⁇ 6.25:9.5 ⁇ 2:36.6 ⁇ 5: 1.4 ⁇ 0.375:4.9 ⁇ 0.625.
- the delivery agent comprises Compound II, DSPC, Cholesterol, Compound I or PEG-DMG, and GL-67 or a salt thereof, e.g., with a mole ratio of about 47.6:9.5:36.6: 1.4:4.9.
- the delivery agent comprises Compound VI, DSPC, Cholesterol, Compound I or PEG-DMG, and GL-67 or a salt thereof, e.g., with a mole ratio of about 47.6 ⁇ 25:9.5 ⁇ 8:36.6 ⁇ 20: 1.4 ⁇ 1.25:4.9 ⁇ 2.5.
- the delivery agent comprises Compound VI, DSPC, Cholesterol, Compound I or PEG-DMG, and GL-67 or a salt thereof, e.g., with a mole ratio of about 47.6 ⁇ 12.5:9.5 ⁇ 4:36.6 ⁇ 10: 1.4 ⁇ 0.75:4.9 ⁇ 1.25.
- the delivery agent comprises Compound VI, DSPC, Cholesterol, and Compound I or PEG-DMG, e.g., with a mole ratio of about
- the delivery agent comprises Compound VI, DSPC, Cholesterol, Compound I or PEG-DMG, and GL-67 or a salt thereof, e.g., with a mole ratio of about 47.6:9.5:36.6: 1.4:4.9.
- the delivery agent comprises Compound II, DSPC, Cholesterol, Compound I or PEG-DMG, and GL-67 or a salt thereof, e.g., with a mole ratio of about 47.3 ⁇ 25:9.5 ⁇ 8:36.4 ⁇ 20: 1.4 ⁇ 1.25:5.5 ⁇ 2.5.
- the delivery agent comprises Compound II, DSPC, Cholesterol, Compound I or PEG-DMG, and GL-67 or a salt thereof, e.g., with a mole ratio of about
- the delivery agent comprises Compound II, DSPC, Cholesterol, and Compound I or PEG-DMG, e.g., with a mole ratio of about 47.3 ⁇ 6.25:9.5 ⁇ 2:36.4 ⁇ 5: 1.4 ⁇ 0.375:5.5 ⁇ 0.625.
- the delivery agent comprises Compound II, DSPC, Cholesterol, Compound I or PEG-DMG, and GL-67 or a salt thereof, e.g., with a mole ratio of about 47.3:9.5:36.4: 1.4:5.5.
- the delivery agent comprises Compound VI, DSPC, Cholesterol, Compound I or PEG-DMG, and GL-67 or a salt thereof, e.g., with a mole ratio of about 47.3 ⁇ 25:9.5 ⁇ 8:36.4 ⁇ 20: 1.4 ⁇ 1.25:5.5 ⁇ 2.5.
- the delivery agent comprises Compound VI, DSPC, Cholesterol, Compound I or PEG-DMG, and GL-67 or a salt thereof, e.g., with a mole ratio of about 47.3 ⁇ 12.5:9.5 ⁇ 4:36.4 ⁇ 10: 1.4 ⁇ 0.75:5.5 ⁇ 1.25.
- the delivery agent comprises Compound VI, DSPC, Cholesterol, and Compound I or PEG-DMG, e.g., with a mole ratio of about
- the delivery agent comprises Compound VI, DSPC, Cholesterol, Compound I or PEG-DMG, and GL-67 or a salt thereof, e.g., with a mole ratio of about 47.3:9.5:36.4: 1.4:5.5.
- the delivery agent comprises Compound II, DSPC, Cholesterol, Compound I or PEG-DMG, and GL-67 or a salt thereof, e.g., with a mole ratio of about 45.8 ⁇ 25: 10.5 ⁇ 8:36.8 ⁇ 20: 1.4 ⁇ 1.25:5.5 ⁇ 2.5.
- the delivery agent comprises Compound II, DSPC, Cholesterol, Compound I or PEG-DMG, and GL-67 or a salt thereof, e.g., with a mole ratio of about
- the delivery agent comprises Compound II, DSPC, Cholesterol, and Compound I or PEG-DMG, e.g., with a mole ratio of about 45.8 ⁇ 6.25: 10.5 ⁇ 2:36.8 ⁇ 5: 1.4 ⁇ 0.375:5.5 ⁇ 0.625.
- the delivery agent comprises Compound II, DSPC, Cholesterol, Compound I or PEG-DMG, and GL-67 or a salt thereof, e.g., with a mole ratio of about 45.8: 10.5:36.8: 1.4:5.5.
- the delivery agent comprises Compound VI, DSPC, Cholesterol, Compound I or PEG-DMG, and GL-67 or a salt thereof, e.g., with a mole ratio of about 45.8 ⁇ 25: 10.5 ⁇ 8:36.8 ⁇ 20: 1.4 ⁇ 1.25:5.5 ⁇ 2.5.
- the delivery agent comprises Compound VI, DSPC, Cholesterol, Compound I or PEG-DMG, and GL-67 or a salt thereof, e.g., with a mole ratio of about 45.8 ⁇ 12.5: 10.5 ⁇ 4:36.8 ⁇ 10: 1.4 ⁇ 0.75:5.5 ⁇ 1.25.
- the delivery agent comprises Compound VI, DSPC, Cholesterol, and Compound I or PEG-DMG, e.g., with a mole ratio of about 45.8 ⁇ 6.25: 10.5 ⁇ 2:36.8 ⁇ 5: 1.4 ⁇ 0.375:5.5 ⁇ 0.625.
- the delivery agent comprises Compound VI, DSPC, Cholesterol, Compound I or PEG-DMG, and GL-67 or a salt thereof, e.g., with a mole ratio of about 45.8: 10.5:36.8: 1.4:5.5.
- the delivery agent comprises Compound II or VI, DSPC, Cholesterol, Compound I or PEG-DMG, and GL-67 or a salt thereof, e.g., with a mole ratio in the range of about 30 to about 60 mol% Compound II or VI (or related suitable amino lipid) (e.g., 30-40, 40-45, 45-50, 50-55 or 55-60 mol% Compound II or VI (or related suitable amino lipid)), about 5 to about 20 mol% phospholipid (or related suitable phospholipid or “helper lipid”) (e.g., 5-10, 10-15, or 15-20 mol% phospholipid (or related suitable phospholipid or “helper lipid”)), about 20 to about 50 mol% cholesterol (or related sterol or “non-cationic” lipid) (e.g., about 20-30, 30- 35, 35-40, 40-45, or 45-50 mol% cholesterol (or related sterol or “non-cationic” lipid)), about 0.05 to
- an exemplary delivery agent can comprise mole ratios of, for example, 48:9.5:35.5: 1.5:5.5; 47: 10:36: 1.5:5.5;
- the delivery agent comprises Compound II or VI, DSPC, Cholesterol, Compound I or PEG-DMG, and GL-67 or a salt thereof, e.g., with a mole ratio of about 47.6:9.5:36.6: 1.4:4.9.
- the delivery agent comprises Compound II or VI, DSPC, Cholesterol, Compound I or PEG-DMG, and GL-67 or a salt thereof, e.g., with a mole ratio of about 47.3:9.5:36.4: 1.4:5.5.
- the delivery agent comprises Compound II or VI, DSPC, Cholesterol, Compound I or PEG-DMG, and GL-67 or a salt thereof, e.g., with a mole ratio ofabout 45.8: 10.5:36.8: 1.4:5.5.
- the polynucleotide (e.g., a RNA, e.g., a mRNA) disclosed herein is formulated with a delivery agent comprising, e.g., a compound having the Formula (I), e.g., any of Compounds 1-232, e.g., Compound II; a compound having the Formula (III), (IV), (V), or (VI), e.g., any of Compounds 233- 342, e.g., Compound VI; or a compound having the Formula (VIII), e.g., any of Compounds 419-428, e.g., Compound I, or any combination thereof.
- the delivery agent comprises Compound II, DSPC, Cholesterol, and Compound I or PEG-DMG, e.g., with a mole ratio of about
- the delivery agent comprises Compound VI, DSPC, Cholesterol, and Compound I or PEG-DMG, e.g., with a mole ratio of about 49.5 ⁇ 3: 10.5 ⁇ 2:39 ⁇ 3: l ⁇ 0.75.
- the delivery agent comprises about 48-52 mol % Compound II or VI (or related suitable amino lipid) (e.g., 48-51, 48-50, 49-52, or 49-51 mol % Compound II or VI (or related suitable amino lipid)), about 9-12 mol % phospholipid (or related suitable phospholipid or “helper lipid”) (e.g., 9-11, 9-10, 10-12, 10-11.5, 10-11 mol %phospholipid (or related suitable phospholipid or “helper lipid”)), about 36-42 mol% cholesterol (or related sterol or “non-cationic” lipid) (e.g., about 36-41, 36-40, 37-40, or 38-40 mol% cholesterol (or related sterol or “non-cationic” lipid)) and about 0.25-2.5 mol% PEG lipid (or other suitable PEG lipid) (e.g., 0.25-2, 0.25-1.5, 0.25-2, or 0.5-1.5 mol% PEG lipid
- the polynucleotide (e.g., a RNA, e.g., a mRNA) disclosed herein is formulated with a delivery agent comprising, e.g., a compound having the Formula (I), e.g., any of Compounds 1-232, e.g., Compound II; a compound having the Formula (III), (IV), (V), or (VI), e.g., any of Compounds 233- 342, e.g., Compound VI; a compound having the Formula (VIII), e.g., any of Compounds 419-428, e.g., Compound I, or a compound having the Formula Al, A2, A3, A4, or A5, e.g., any one of SA1-SA41, or any combination thereof.
- a delivery agent comprising, e.g., a compound having the Formula (I), e.g., any of Compounds 1-232, e.g., Compound II;
- the delivery agent comprises Compound II, DSPC, Cholesterol, and Compound I or PEG-DMG, e.g., with a mole ratio of about 46.5 ⁇ 3: 10 ⁇ 2:36 ⁇ 3: 1.25 ⁇ 0.75:4.5 ⁇ 1.5.
- the delivery agent comprises Compound VI, DSPC, Cholesterol, and Compound I or PEG-DMG, e.g., with a mole ratio of about 46.5 ⁇ 3: 10 ⁇ 2:36 ⁇ 3: 1.25 ⁇ 0.75:4.5 ⁇ 1.5.
- the delivery agent comprises about 43-49 mol % Compound II or VI (or related suitable amino lipid) (e.g., 43-48, 44-48, 45-48, or 45.5-48 mol % Compound II or VI (or related suitable amino lipid)), about 8-12 mol % phospholipid (or related suitable phospholipid or “helper lipid”) (e.g., 8-11, 8-10, 9-12, 9-11, 9.5- 10.5 mol %phospholipid (or related suitable phospholipid or “helper lipid”)), about
- mol% cholesterol or related sterol or “non-cationic” lipid (e.g., about 33-38,
- the delivery agent comprises Compound II, DSPC, Cholesterol, DMG-PEG-2k, and GL-67.
- the delivery agent comprises about 45-48 mol% Compound II, about 9-11 mol% DSPC, about 35-38 mol% cholesterol, about 1-3 mol% DMG-PEG-2k, and about 4-6 mol% GL-67. In further embodiments, the delivery agent comprises about 45-48 mol% Compound II, about 9-11 mol% DSPC, about 35-38 mol% cholesterol, about 1-3 mol% DMG-PEG- 2k, and about 4-6 mol% GL-67.
- the delivery agent comprises about 45.8-47.6 mol% Compound II, about 9.5-10.5 mol% DSPC, about 36.4-36.8 mol% cholesterol, about 1.4 mol% DMG-PEG-2k, and about 4.9-5.5 mol% GL-67.
- a polynucleotide e.g., a RNA, e.g., a mRNA
- a delivery agent comprising, e.g., a compound having the Formula (I), e.g., any of Compounds 1-232, e.g., Compound II; a compound having the Formula (III), (IV), (V), or (VI), e.g., any of Compounds 233- 342, e.g., Compound VI; a compound having the Formula (VIII), e.g., any of Compounds 419-428, e.g., Compound I, or a compound having the Formula Al, A2, A3, A4, or A5, e.g., any one of SA1-SA41, or any combination thereof.
- a delivery agent comprising, e.g., a compound having the Formula (I), e.g., any of Compounds 1-232, e.g., Compound II; a compound having the Formula
- the delivery agent comprises Compound II, DSPC, Cholesterol, and Compound I or PEG-DMG, e.g., with a mole ratio of about 47 ⁇ 3: 10 ⁇ 2:36 ⁇ 3: 1.25 ⁇ 0.75:4.5 ⁇ 1.5.
- the delivery agent comprises Compound VI, DSPC, Cholesterol, and Compound I or PEG-DMG, e.g., with a mole ratio of about 46.5 ⁇ 3: 10 ⁇ 2:36 ⁇ 3: 1.25 ⁇ 0.75:4.5 ⁇ 1.5.
- the delivery agent comprises about 43-49 mol % Compound II or VI (or related suitable amino lipid) (e.g., 43-48, 44-48, 45-48, or 45.5-48 mol % Compound II or VI (or related suitable amino lipid)), about 8-12 mol % phospholipid (or related suitable phospholipid or “helper lipid”) (e.g., 8-11, 8-10, 9-12, 9-11, 9.5- 10.5 mol %phospholipid (or related suitable phospholipid or “helper lipid”)), about
- mol% cholesterol or related sterol or “non-cationic” lipid (e.g., about 33-38,
- mol% cholesterol or related sterol or “non-cationic” lipid
- about 0.5-2 mol% PEG lipid or other suitable PEG lipid
- about 3-6 mol% cationic agent e.g., sterol amine
- e.g., 3-5, 3-4.5, 4-6, or 5-6 mol% cationic agent e.g., sterol amine
- the polynucleotide of the disclosure is an mRNA that comprises a 5'-terminal cap (e.g., Capl, e.g., m 7 Gp-ppGm-A), a 5'UTR (e.g., SEQ ID NO:50), an ORF sequence of SEQ ID NO: 10, a 3'UTR (e.g., SEQ ID NO: 141), and a poly A tail (e.g., about 100 nt in length, e.g., SEQ ID NO: 195), wherein all uracils in the polynucleotide are N1 -methylpseudouracils.
- a 5'-terminal cap e.g., Capl, e.g., m 7 Gp-ppGm-A
- a 5'UTR e.g., SEQ ID NO:50
- an ORF sequence of SEQ ID NO: 10 e.g., SEQ ID NO: 10
- the polynucleotide of the disclosure is an mRNA that comprises a 5'-terminal cap (e.g., Capl, e.g., m 7 Gp-ppGm-A), a 5'UTR (e.g., SEQ ID NO:50), an ORF sequence of SEQ ID NO: 10, a 3'UTR (e.g., SEQ ID NO: 141), and a poly A tail (e.g., A100-UCUAG-A20-inverted deoxy-thymidine (SEQ ID NO:211)), wherein all uracils in the polynucleotide are N1 -methylpseudouracils.
- a 5'-terminal cap e.g., Capl, e.g., m 7 Gp-ppGm-A
- a 5'UTR e.g., SEQ ID NO:50
- an ORF sequence of SEQ ID NO: 10 e.g., SEQ ID NO:
- the polynucleotide of the disclosure is an mRNA that comprises a 5'-terminal cap (e.g., Capl, e.g., m 7 Gp-ppGm-A), a 5'UTR (e.g., any one of SEQ ID N0s:50-80), an ORF sequence of SEQ ID NO: 10, a 3'UTR (e.g., any one of SEQ ID NOs: 100-141), and a poly A tail (e.g., about 100 nt in length, e.g., SEQ ID NO: 195), wherein all uracils in the polynucleotide are N1 -methylpseudouracils.
- the delivery agent comprises Compound II as the ionizable amino lipid and DMG-PEG 2k as the PEG lipid.
- the polynucleotide of the disclosure is an mRNA that comprises a 5'-terminal cap (e.g., Cap 1, e.g., m 7 Gp-ppGm-A), a 5'UTR (e.g., SEQ ID NO:50), an ORF sequence of SEQ ID NO: 10, a 3'UTR (e.g., SEQ ID NO: 141), and a poly A tail (e.g., about 100 nt in length), wherein all uracils in the polynucleotide are N1 -methylpseudouracils.
- a 5'-terminal cap e.g., Cap 1, e.g., m 7 Gp-ppGm-A
- a 5'UTR e.g., SEQ ID NO:50
- an ORF sequence of SEQ ID NO: 10 e.g., SEQ ID NO: 10
- a 3'UTR e.g., SEQ ID NO: 141
- the delivery agent comprises Compound II or Compound VI as the ionizable amino lipid and DMG-PEG 2k or Compound I as the PEG lipid. In some embodiments, the delivery agent comprises Compound II as the ionizable amino lipid and DMG-PEG 2k as the PEG lipid.
- the polynucleotide of the disclosure is an mRNA that comprises an ORF sequence of SEQ ID NO: 10. In some embodiments, the polynucleotide of the disclosure is an mRNA that comprises an ORF sequence of SEQ ID NO: 10, wherein all uracils in the polynucleotide are N1 -methylpseudouracils. In some embodiments, the polynucleotide of the disclosure is an mRNA that comprises an ORF sequence of SEQ ID NO: 10, wherein the mRNA is formulated with a delivery agent comprising Compound II as the ionizable amino lipid and DMG-PEG 2k as the PEG lipid.
- the polynucleotide of the disclosure is an mRNA that comprises an ORF sequence of SEQ ID NO: 10, wherein all uracils in the polynucleotide are N1 -methylpseudouracils, wherein the mRNA is formulated with a delivery agent comprising Compound II as the ionizable amino lipid and DMG-PEG 2k as the PEG lipid.
- the polynucleotide of the disclosure is an mRNA that comprises an ORF sequence of SEQ ID NO: 10, wherein the mRNA is formulated with a delivery agent comprising Compound II as the ionizable amino lipid, DMG-PEG 2k as the PEG lipid, and GL-67 as the cationic agent.
- the polynucleotide of the disclosure is an mRNA that comprises an ORF sequence of SEQ ID NO: 10, wherein all uracils in the polynucleotide are N1 -methylpseudouracils, wherein the mRNA is formulated with a delivery agent comprising Compound II as the ionizable amino lipid, DMG-PEG 2k as the PEG lipid, and GL-67 as the cationic agent.
- the polynucleotide of the disclosure is an mRNA that comprises an ORF sequence of SEQ ID NO: 11. In some embodiments, the polynucleotide of the disclosure is an mRNA that comprises an ORF sequence of SEQ ID NO: 11, wherein all uracils in the polynucleotide are N1 -methylpseudouracils. In some embodiments, the polynucleotide of the disclosure is an mRNA that comprises an ORF sequence of SEQ ID NO: 11, wherein the mRNA is formulated with a delivery agent comprising Compound II as the ionizable amino lipid and DMG-PEG 2k as the PEG lipid.
- the polynucleotide of the disclosure is an mRNA that comprises an ORF sequence of SEQ ID NO: 11, wherein all uracils in the polynucleotide are N1 -methylpseudouracils, wherein the mRNA is formulated with a delivery agent comprising Compound II as the ionizable amino lipid and DMG-PEG 2k as the PEG lipid.
- the polynucleotide of the disclosure is an mRNA that comprises an ORF sequence of SEQ ID NO: 11, wherein the mRNA is formulated with a delivery agent comprising Compound II as the ionizable amino lipid, DMG-PEG 2k as the PEG lipid, and GL-67 as the cationic agent.
- the polynucleotide of the disclosure is an mRNA that comprises an ORF sequence of SEQ ID NO: 11, wherein all uracils in the polynucleotide are N1 -methylpseudouracils, wherein the mRNA is formulated with a delivery agent comprising Compound II as the ionizable amino lipid, DMG-PEG 2k as the PEG lipid, and GL-67 as the cationic agent.
- the polynucleotide of the disclosure is an mRNA that comprises the sequence of SEQ ID NO: 13. In some embodiments, the polynucleotide of the disclosure is an mRNA that comprises the sequence of SEQ ID NO: 13, wherein the mRNA is formulated with a delivery agent comprising Compound II as the ionizable amino lipid and DMG-PEG 2k as the PEG lipid.
- the polynucleotide of the disclosure is an mRNA that comprises the sequence of SEQ ID NO: 13, wherein the mRNA is formulated with a delivery agent comprising Compound II as the ionizable amino lipid, DMG-PEG 2k as the PEG lipid, and GL-67 as the cationic agent.
- the polynucleotides e.g., a RNA, e.g., an mRNA
- One such feature that aids in protein trafficking is the signal sequence, or targeting sequence.
- the peptides encoded by these signal sequences are known by a variety of names, including targeting peptides, transit peptides, and signal peptides.
- the polynucleotide (e.g., a RNA, e.g., an mRNA) comprises a nucleotide sequence (e.g., an ORF) that encodes a signal peptide operably linked to a nucleotide sequence that encodes a CFTR polypeptide described herein.
- a nucleotide sequence e.g., an ORF
- the "signal sequence” or “signal peptide” is a polynucleotide or polypeptide, respectively, which is from about 30-210, e.g., about 45-80 or 15-60 nucleotides (e.g., about 20, 30, 40, 50, 60, or 70 amino acids) in length that, optionally, is incorporated at the 5' (or N-terminus) of the coding region or the polypeptide, respectively. Addition of these sequences results in trafficking the encoded polypeptide to a desired site, such as the endoplasmic reticulum or the mitochondria through one or more targeting pathways. Some signal peptides are cleaved from the protein, for example by a signal peptidase after the proteins are transported to the desired site.
- the polynucleotide of the invention comprises a nucleotide sequence encoding a CFTR polypeptide, wherein the nucleotide sequence further comprises a 5' nucleic acid sequence encoding a heterologous signal peptide.
- the polynucleotide of the invention comprises a sequence-optimized nucleotide sequence encoding a CFTR polypeptide disclosed herein. In some embodiments, the polynucleotide of the invention comprises an open reading frame (ORF) encoding a CFTR polypeptide, wherein the ORF has been sequence optimized.
- ORF open reading frame
- An exemplary sequence-optimized nucleotide sequence encoding CFTR is set forth as SEQ ID NO: 10.
- a further exemplary sequence-optimized nucleotide sequence encoding CFTR is set forth as SEQ ID NO: 11.
- a further exemplary sequence-optimized nucleotide sequence encoding CFTR is set forth as SEQ ID NO: 13.
- the sequence optimized CFTR sequences, fragments, and variants thereof are used to practice the methods disclosed herein.
- a polynucleotide of the present disclosure for example a polynucleotide comprising an mRNA nucleotide sequence encoding a CFTR polypeptide, comprises from 5' to 3' end:
- a 5' cap provided herein, for example, Capl
- a 5' UTR such as the sequences provided herein, for example, SEQ ID NO:50
- CFTR polypeptide e.g., a sequence optimized nucleic acid sequence encoding CFTR set forth as SEQ ID NO: 10;
- a 3' UTR such as the sequences provided herein, for example, SEQ ID NO: 141;
- all uracils in the polynucleotide are N1 -methylpseudouracil. In certain embodiments, all uracils in the polynucleotide are 5 -methoxyuracil .
- sequence-optimized nucleotide sequences disclosed herein are distinct from the corresponding wild type nucleotide acid sequences and from other known sequence-optimized nucleotide sequences, e.g., these sequence-optimized nucleic acids have unique compositional characteristics.
- the percentage of uracil or thymine nucleobases in a sequence-optimized nucleotide sequence is modified (e.g., reduced) with respect to the percentage of uracil or thymine nucleobases in the reference wild-type nucleotide sequence.
- a sequence is referred to as a uracil-modified or thymine -modified sequence.
- the percentage of uracil or thymine content in a nucleotide sequence can be determined by dividing the number of uracils or thymines in a sequence by the total number of nucleotides and multiplying by 100.
- the sequence-optimized nucleotide sequence has a lower uracil or thymine content than the uracil or thymine content in the reference wild-type sequence.
- the uracil or thymine content in a sequence -optimized nucleotide sequence of the invention is greater than the uracil or thymine content in the reference wild-type sequence and still maintain beneficial effects, e.g., increased expression and/or reduced Toll-Like Receptor (TLR) response when compared to the reference wild-type sequence.
- TLR Toll-Like Receptor
- an ORF of any one or more of the sequences provided herein may be codon optimized.
- Codon optimization in some embodiments, may be used to match codon frequencies in target and host organisms to ensure proper folding; bias GC content to increase mRNA stability or reduce secondary structures; minimize tandem repeat codons or base runs that may impair gene construction or expression; customize transcriptional and translational control regions; insert or remove protein trafficking sequences; remove/add post translation modification sites in encoded protein (e.g., glycosylation sites); add, remove or shuffle protein domains; insert or delete restriction sites; modify ribosome binding sites and mRNA degradation sites; adjust translational rates to allow the various domains of the protein to fold properly; or reduce or eliminate problem secondary structures within the polynucleotide.
- Codon optimization tools, algorithms and services are known in the art - non-limiting examples include services from GeneArt (Life Technologies), DNA2.0 (Menlo Park CA) and/or proprietary methods.
- the open reading frame (ORF) sequence is optimized using optimization algorithms.
- An Identification and Ratio Determination (IDR) sequence is a sequence of a biological molecule (e.g., nucleic acid or protein) that, when combined with the sequence of a target biological molecule, serves to identify the target biological molecule.
- an IDR sequence is a heterologous sequence that is incorporated within or appended to a sequence of a target biological molecule and can be used as a reference to identify the target molecule.
- a nucleic acid e.g., mRNA
- a target sequence of interest e.g., a coding sequence encoding a therapeutic and/or antigenic peptide or protein
- a unique IDR sequence e.g., a unique IDR sequence.
- RNA species may comprise an IDR sequence that differs from the IDR sequence of other RNA species (e.g., RNA(s) having different coding sequence(s)).
- Each IDR sequence thus identifies a particular RNA species, and so the abundance of IDR sequences may be measured to determine the abundance of each RNA species in a composition.
- Use of distinct IDR sequences to identify RNA species allows for analysis of multivalent RNA compositions (e.g., containing multiple RNA species) containing RNA species with similar coding sequences and/or lengths, which could otherwise be difficult to distinguish using PCR- or chromatography-based analysis of full-length RNAs.
- Each RNA species in a multivalent RNA composition may comprise an IDR sequence that is not a sequence isomer of an IDR sequence of another RNA species in a multivalent RNA composition (e.g., the IDR sequence does not have the same number of adenosine nucleotides, the same number of cytosine nucleotides, the same number of guanine nucleotides, and the same number of uracil nucleotides, as another IDR sequence in the composition, even if those sequences have different sequences).
- Having identical nucleotide compositions causes sequence isomers to have the same mass, presenting a challenge to distinguishing sequence isomers using mass-based identification methods (e.g., mass spectrometry).
- Each RNA species in a multivalent RNA composition may comprise an IDR sequence having a mass that differs from the mass of IDR sequences of each other RNA species in a multivalent RNA composition.
- the mass of each IDR sequence may differ from the mass of other IDR sequences by at least 9 Da, at least 25 Da, at least 25 Da, or at least 50 Da.
- Use of IDR sequences with distinct masses allows RNA fragments comprising different IDR sequences to be distinguished using mass-based analysis methods (e.g., mass spectrometry), which do not require reverse transcription, amplification, or sequencing of RNAs.
- Each RNA species in an RNA composition may comprises an IDR sequence with a different length.
- each IDR sequence may have a length independently selected from 0 to 25 nucleotides.
- the length of a nucleic acid influences the rate at which the nucleic acid traverses a chromatography column, and so the use of IDR sequences of different lengths on different RNA species allows RNA fragments having different IDR sequences to be distinguished using chromatography-based methods (e.g., LC-UV).
- IDR sequences may be chosen such that no IDR sequence comprises a start codon, ‘AUG’ . Lack of a start codon in an IDR sequence prevents undesired translation of nucleotide sequences within and/or downstream from the IDR sequence.
- IDR sequences may be chosen such that no IDR sequence comprises a recognition site for a restriction enzyme.
- no IDR sequence comprises a recognition site for Xbal, ‘UCUAG’.
- Lack of a recognition site for a restriction enzyme e.g., Xbal recognition site ‘UCUAG’) allows the restriction enzyme to be used in generating and modifying a DNA template for in vitro transcription, without affecting the IDR sequence or sequence of the transcribed RNA.
- the polynucleotide (e.g., a RNA, e.g., an mRNA) of the invention comprises a chemically modified nucleobase, for example, a chemically modified uracil, e.g., pseudouracil, N1 -methylpseudouracil, 5 -methoxyuracil, or the like.
- a chemically modified uracil e.g., pseudouracil, N1 -methylpseudouracil, 5 -methoxyuracil, or the like.
- the mRNA is a uracil -modified sequence comprising an ORF encoding a CFTR polypeptide, wherein the mRNA comprises a chemically modified nucleobase, for example, a chemically modified uracil, e.g., pseudouracil, N1 -methylpseudouracil, or 5 -methoxyuracil.
- a chemically modified uracil e.g., pseudouracil, N1 -methylpseudouracil, or 5 -methoxyuracil.
- modified uracil in the polynucleotide is at least about 25%, at least about 30%, at least about 40%, at least about 50%, at least about 60%, at least about 70%, at least about 80%, at least 90%, at least 95%, at least 99%, or about 100% modified uracil.
- uracil in the polynucleotide is at least 95% modified uracil.
- uracil in the polynucleotide is 100% modified uracil.
- modified uracil content of the ORF is between about 100% and about 150%, between about 100% and about 110%, between about 105% and about 115%, between about 110% and about 120%, between about 115% and about 125%, between about 120% and about 130%, between about 125% and about 135%, between about 130% and about 140%, between about 135% and about 145%, between about 140% and about 150% of the theoretical minimum uracil content in the corresponding wild-type ORF (%UTM).
- the uracil content of the ORF is between about 121% and about 136% or between 123% and 134% of the %UTM. In some embodiments, the uracil content of the ORF encoding a CFTR polypeptide is about 115%, about 120%, about 125%, about 130%, about 135%, about 140%, about 145%, or about 150% of the %UTM.
- uracil can refer to modified uracil and/or naturally occurring uracil.
- the uracil content in the ORF of the mRNA encoding a CFTR polypeptide of the invention is less than about 30%, about 25%, about 20%, about 15%, or about 10% of the total nucleobase content in the ORF. In some embodiments, the uracil content in the ORF is between about 10% and about 20% of the total nucleobase content in the ORF. In other embodiments, the uracil content in the ORF is between about 10% and about 25% of the total nucleobase content in the ORF. In one embodiment, the uracil content in the ORF of the mRNA encoding a CFTR polypeptide is less than about 20% of the total nucleobase content in the open reading frame. In this context, the term "uracil" can refer to modified uracil and/or naturally occurring uracil.
- the ORF of the mRNA encoding a CFTR polypeptide having modified uracil and adjusted uracil content has increased Cytosine (C), Guanine (G), or Guanine/Cytosine (G/C) content (absolute or relative).
- the overall increase in C, G, or G/C content (absolute or relative) of the ORF is at least about 2%, at least about 3%, at least about 4%, at least about 5%, at least about 6%, at least about 7%, at least about 10%, at least about 15%, at least about 20%, at least about 40%, at least about 50%, at least about 60%, at least about 70%, at least about 80%, at least about 90%, at least about 95%, or at least about 100% relative to the G/C content (absolute or relative) of the wild-type ORF.
- the G, the C, or the G/C content in the ORF is less than about 100%, less than about 90%, less than about 85%, or less than about 80% of the theoretical maximum G, C, or G/C content of the corresponding wild type nucleotide sequence encoding the CFTR polypeptide (%GTMX; %CTMX, or %G/CTMX).
- the increases in G and/or C content (absolute or relative) described herein can be conducted by replacing synonymous codons with low G, C, or G/C content with synonymous codons having higher G, C, or G/C content.
- the increase in G and/or C content (absolute or relative) is conducted by replacing a codon ending with U with a synonymous codon ending with G or C.
- the ORF of the mRNA encoding a CFTR polypeptide of the invention comprises modified uracil and has an adjusted uracil content containing less uracil pairs (UU) and/or uracil triplets (UUU) and/or uracil quadruplets (UUUU) than the corresponding wild-type nucleotide sequence encoding the CFTR polypeptide.
- the ORF of the mRNA encoding a CFTR polypeptide of the invention contains no uracil pairs and/or uracil triplets and/or uracil quadruplets.
- uracil pairs and/or uracil triplets and/or uracil quadruplets are reduced below a certain threshold, e.g., no more than 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, or 20 occurrences in the ORF of the mRNA encoding the CFTR polypeptide.
- the ORF of the mRNA encoding the CFTR polypeptide of the invention contains less than 20, 19, 18, 17, 16, 15, 14, 13, 12, 11, 10, 9, 8, 7, 6, 5, 4, 3, 2, or 1 nonphenylalanine uracil pairs and/or triplets.
- the ORF of the mRNA encoding the CFTR polypeptide contains no non-phenylalanine uracil pairs and/or triplets.
- the ORF of the mRNA encoding a CFTR polypeptide of the invention comprises modified uracil and has an adjusted uracil content containing less uracil-rich clusters than the corresponding wild-type nucleotide sequence encoding the CFTR polypeptide.
- the ORF of the mRNA encoding the CFTR polypeptide of the invention contains uracil-rich clusters that are shorter in length than corresponding uracil-rich clusters in the corresponding wild-type nucleotide sequence encoding the CFTR polypeptide.
- alternative lower frequency codons are employed. At least about 5%, at least about 10%, at least about 15%, at least about 20%, at least about 25%, at least about 30%, at least about 35%, at least about 40%, at least about 45%, at least about 50%, at least about 55%, at least about 60%, at least about 65%, at least about 70%, at least about 75%, at least about 80%, at least about 85%, at least about 90%, at least about 95%, at least about 99%, or 100% of the codons in the CFTR polypeptide-encoding ORF of the modified uracil-comprising mRNA are substituted with alternative codons, each alternative codon having a codon frequency lower than the codon frequency of the substituted codon in the synonymous codon set.
- the ORF also has adjusted uracil content, as described above.
- at least one codon in the ORF of the mRNA encoding the CFTR polypeptide is substituted with an alternative codon having a codon frequency lower than the codon frequency of the substituted codon in the synonymous codon set.
- the adjusted uracil content, CFTR polypeptide- encoding ORF of the modified uracil-comprising mRNA exhibits expression levels of CFTR when administered to a mammalian cell that are higher than expression levels of CFTR from the corresponding wild-type mRNA.
- the mammalian cell is a mouse cell, a rat cell, or a rabbit cell.
- the mammalian cell is a monkey cell or a human cell.
- the human cell is a HeLa cell, a BJ fibroblast cell, or a peripheral blood mononuclear cell (PBMC).
- PBMC peripheral blood mononuclear cell
- CFTR is expressed at a level higher than expression levels of CFTR from the corresponding wild-type mRNA when the mRNA is administered to a mammalian cell in vivo.
- the mRNA is administered to mice, rabbits, rats, monkeys, or humans.
- mice are null mice.
- the mRNA is administered intravenously or intramuscularly.
- the CFTR polypeptide is expressed when the mRNA is administered to a mammalian cell in vitro.
- the expression is increased by at least about 2-fold, at least about 5 -fold, at least about 10- fold, at least about 50-fold, at least about 500-fold, at least about 1500-fold, or at least about 3000-fold. In other embodiments, the expression is increased by at least about 10%, about 20%, about 30%, about 40%, about 50%, 60%, about 70%, about 80%, about 90%, or about 100%.
- adjusted uracil content, CFTR polypeptide-encoding ORF of the modified uracil-comprising mRNA exhibits increased stability.
- the mRNA exhibits increased stability in a cell relative to the stability of a corresponding wild-type mRNA under the same conditions.
- the mRNA exhibits increased stability including resistance to nucleases, thermal stability, and/or increased stabilization of secondary structure.
- increased stability exhibited by the mRNA is measured by determining the half-life of the mRNA (e.g., in a plasma, serum, cell, or tissue sample) and/or determining the area under the curve (AUC) of the protein expression by the mRNA over time (e.g., in vitro or in vivo).
- An mRNA is identified as having increased stability if the half-life and/or the AUC is greater than the half-life and/or the AUC of a corresponding wild-type mRNA under the same conditions.
- the mRNA of the present invention induces a detectably lower immune response (e.g., innate or acquired) relative to the immune response induced by a corresponding wild-type mRNA under the same conditions.
- the mRNA of the present disclosure induces a detectably lower immune response (e.g., innate or acquired) relative to the immune response induced by an mRNA that encodes for a CFTR polypeptide but does not comprise modified uracil under the same conditions, or relative to the immune response induced by an mRNA that encodes for a CFTR polypeptide and that comprises modified uracil but that does not have adjusted uracil content under the same conditions.
- the innate immune response can be manifested by increased expression of pro-inflammatory cytokines, activation of intracellular PRRs (RIG-I, MDA5, etc.), cell death, and/or termination or reduction in protein translation.
- a reduction in the innate immune response can be measured by expression or activity level of Type 1 interferons (e.g., IFN-a, IFN-P, IFN-K, IFN-5, IFN-s. IFN-r, IFN-co, and IFN-Q or the expression of interferon-regulated genes such as the toll-like receptors (e.g., TUR7 and TUR8), and/or by decreased cell death following one or more administrations of the mRNA of the invention into a cell.
- Type 1 interferons e.g., IFN-a, IFN-P, IFN-K, IFN-5, IFN-s. IFN-r, IFN-co, and IFN-Q
- interferon-regulated genes such as the toll-like receptor
- the expression of Type- 1 interferons by a mammalian cell in response to the mRNA of the present disclosure is reduced by at least 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 95%, 99%, 99.9%, or greater than 99.9% relative to a corresponding wild-type mRNA, to an mRNA that encodes a CFTR polypeptide but does not comprise modified uracil, or to an mRNA that encodes a CFTR polypeptide and that comprises modified uracil but that does not have adjusted uracil content.
- the interferon is IFN-p.
- cell death frequency caused by administration of mRNA of the present disclosure to a mammalian cell is 10%, 25%, 50%, 75%, 85%, 90%, 95%, or over 95% less than the cell death frequency observed with a corresponding wild-type mRNA, an mRNA that encodes for a CFTR polypeptide but does not comprise modified uracil, or an mRNA that encodes for a CFTR polypeptide and that comprises modified uracil but that does not have adjusted uracil content.
- the mammalian cell is a BJ fibroblast cell. In other embodiments, the mammalian cell is a splenocyte.
- the mammalian cell is that of a mouse or a rat. In other embodiments, the mammalian cell is that of a human. In one embodiment, the mRNA of the present disclosure does not substantially induce an innate immune response of a mammalian cell into which the mRNA is introduced.
- modified polynucleotides comprising a polynucleotide described herein (e.g., a polynucleotide, e.g. mRNA, comprising a nucleotide sequence encoding a CFTR polypeptide).
- the modified polynucleotides can be chemically modified and/or structurally modified.
- modified polynucleotides can be referred to as "modified polynucleotides.”
- nucleosides and nucleotides of a polynucleotide e.g., RNA polynucleotides, such as mRNA polynucleotides
- a “nucleoside” refers to a compound containing a sugar molecule (e.g., a pentose or ribose) or a derivative thereof in combination with an organic base (e.g. , a purine or pyrimidine) or a derivative thereof (also referred to herein as "nucleobase”).
- a “nucleotide” refers to a nucleoside including a phosphate group.
- Modified nucleotides can be synthesized by any useful method, such as, for example, chemically, enzymatically, or recombinantly, to include one or more modified or non-natural nucleosides.
- Polynucleotides can comprise a region or regions of linked nucleosides. Such regions can have variable backbone linkages. The linkages can be standard phosphodiester linkages, in which case the polynucleotides would comprise regions of nucleotides.
- the modified polynucleotides disclosed herein can comprise various distinct modifications. In some embodiments, the modified polynucleotides contain one, two, or more (optionally different) nucleoside or nucleotide modifications.
- a modified polynucleotide, introduced to a cell can exhibit one or more desirable properties, e.g., improved protein expression, reduced immunogenicity, or reduced degradation in the cell, as compared to an unmodified polynucleotide.
- a polynucleotide of the present invention e.g., a polynucleotide comprising a nucleotide sequence encoding a CFTR polypeptide
- a "structural" modification is one in which two or more linked nucleosides are inserted, deleted, duplicated, inverted or randomized in a polynucleotide without significant chemical modification to the nucleotides themselves. Because chemical bonds will necessarily be broken and reformed to effect a structural modification, structural modifications are of a chemical nature and hence are chemical modifications. However, structural modifications will result in a different sequence of nucleotides.
- the polynucleotide "ATCG” can be chemically modified to "AT-5meC-G".
- the same polynucleotide can be structurally modified from “ATCG” to "ATCCCG”.
- the dinucleotide "CC” has been inserted, resulting in a structural modification to the polynucleotide.
- compositions of the present disclosure comprise, in some embodiments, at least one nucleic acid (e.g., RNA) having an open reading frame encoding CFTR (e.g., SEQ ID NO: 10), wherein the nucleic acid comprises nucleotides and/or nucleosides that can be standard (unmodified) or modified as is known in the art.
- nucleotides and nucleosides of the present disclosure comprise modified nucleotides or nucleosides.
- modified nucleotides and nucleosides can be naturally-occurring modified nucleotides and nucleosides or non-naturally occurring modified nucleotides and nucleosides.
- modifications can include those at the sugar, backbone, or nucleobase portion of the nucleotide and/or nucleoside as are recognized in the art.
- a naturally-occurring modified nucleotide or nucleotide of the disclosure is one as is generally known or recognized in the art.
- Non-limiting examples of such naturally occurring modified nucleotides and nucleotides can be found, inter alia, in the widely recognized MODOMICS database.
- a non-naturally occurring modified nucleotide or nucleoside of the disclosure is one as is generally known or recognized in the art.
- Non-limiting examples of such non-naturally occurring modified nucleotides and nucleosides can be found, inter alia, in published US application Nos. PCI7US2012/058519; PCI7US2013/075177; PCT/US2014/058897;
- RNA e.g., mRNA
- nucleotides and nucleosides of the present disclosure comprise standard nucleoside residues such as those present in transcribed RNA (e.g. A, G, C, or U).
- nucleotides and nucleosides of the present disclosure comprise standard deoxyribonucleosides such as those present in DNA (e.g. dA, dG, dC, or dT).
- nucleic acids of the disclosure can comprise standard nucleotides and nucleosides, naturally-occurring nucleotides and nucleosides, non-naturally-occurring nucleotides and nucleosides, or any combination thereof.
- Nucleic acids of the disclosure e.g., DNA nucleic acids and RNA nucleic acids, such as mRNA nucleic acids
- Nucleic acids of the disclosure comprise various (more than one) different types of standard and/or modified nucleotides and nucleosides.
- a particular region of a nucleic acid contains one, two or more (optionally different) types of standard and/or modified nucleotides and nucleosides.
- a modified RNA nucleic acid e.g. , a modified mRNA nucleic acid
- a modified RNA nucleic acid introduced to a cell or organism, exhibits reduced degradation in the cell or organism, respectively, relative to an unmodified nucleic acid comprising standard nucleotides and nucleosides.
- a modified RNA nucleic acid e.g. , a modified mRNA nucleic acid
- introduced into a cell or organism may exhibit reduced immunogenicity in the cell or organism, respectively (e.g., a reduced innate response) relative to an unmodified nucleic acid comprising standard nucleotides and nucleosides.
- Nucleic acids e.g., RNA nucleic acids, such as mRNA nucleic acids
- Nucleic acids in some embodiments, comprise non-natural modified nucleotides that are introduced during synthesis or post-synthesis of the nucleic acids to achieve desired functions or properties.
- the modifications may be present on intemucleotide linkages, purine or pyrimidine bases, or sugars.
- the modification may be introduced with chemical synthesis or with a polymerase enzyme at the terminal of a chain or anywhere else in the chain. Any of the regions of a nucleic acid may be chemically modified.
- nucleic acid e.g., RNA nucleic acids, such as mRNA nucleic acids.
- a “nucleoside” refers to a compound containing a sugar molecule (e.g. , a pentose or ribose) or a derivative thereof in combination with an organic base (e.g. , a purine or pyrimidine) or a derivative thereof (also referred to herein as “nucleobase”).
- nucleotide refers to a nucleoside, including a phosphate group.
- Modified nucleotides may by synthesized by any useful method, such as, for example, chemically, enzymatically, or recombinantly, to include one or more modified or non-natural nucleosides.
- Nucleic acids can comprise a region or regions of linked nucleosides. Such regions may have variable backbone linkages. The linkages can be standard phosphodiester linkages, in which case the nucleic acids would comprise regions of nucleotides.
- Modified nucleotide base pairing encompasses not only the standard adenosine-thymine, adenosine-uracil, or guanosine-cytosine base pairs, but also base pairs formed between nucleotides and/or modified nucleotides comprising nonstandard or modified bases, wherein the arrangement of hydrogen bond donors and hydrogen bond acceptors permits hydrogen bonding between a non-standard base and a standard base or between two complementary non-standard base structures, such as, for example, in those nucleic acids having at least one chemical modification.
- One example of such non-standard base pairing is the base pairing between the modified nucleotide inosine and adenine, cytosine or uracil. Any combination of base/sugar or linker may be incorporated into nucleic acids of the present disclosure.
- modified nucleobases in nucleic acids comprise N1 -methyl -pseudouridine (ml ⁇ ). 1 -ethyl -pseudouridine (e ly), 5 -methoxy-uridine (mo5U), 5-methyl-cytidine (m5C), and/or pseudouridine (v)-
- modified nucleobases in nucleic acids comprise 5- methoxymethyl uridine, 5 -methylthio uridine, 1 -methoxymethyl pseudouridine, 5- methyl cytidine, and/or 5 -methoxy cytidine.
- the polyribonucleotide includes a combination of at least two (e.g., 2, 3, 4 or more) of any of the aforementioned modified nucle
- a RNA nucleic acid of the disclosure comprises Nl- methyl -pseudouridine (ml ⁇ ) substitutions at one or more or all uridine positions of the nucleic acid.
- a RNA nucleic acid of the disclosure comprises Nl- methyl-pseudouridine (ml ⁇ ) substitutions at one or more or all uridine positions of the nucleic acid and 5 -methyl cytidine substitutions at one or more or all cytidine positions of the nucleic acid.
- a RNA nucleic acid of the disclosure comprises pseudouridine (v) substitutions at one or more or all uridine positions of the nucleic acid.
- a RNA nucleic acid of the disclosure comprises pseudouridine (v) substitutions at one or more or all uridine positions of the nucleic acid and 5-methyl cytidine substitutions at one or more or all cytidine positions of the nucleic acid.
- a RNA nucleic acid of the disclosure comprises uridine at one or more or all uridine positions of the nucleic acid.
- nucleic acids e.g., RNA nucleic acids, such as mRNA nucleic acids
- RNA nucleic acids are uniformly modified (e.g., fully modified, modified throughout the entire sequence) for a particular modification.
- a nucleic acid can be uniformly modified with N1 -methyl -pseudouridine, meaning that all uridine residues in the mRNA sequence are replaced with N1 -methyl -pseudouridine.
- a nucleic acid can be uniformly modified for any type of nucleoside residue present in the sequence by replacement with a modified residue such as those set forth above.
- the nucleic acids of the present disclosure may be partially or fully modified along the entire length of the molecule.
- one or more or all or a given type of nucleotide e.g. , purine or pyrimidine, or any one or more or all of A, G, U, C
- nucleotides X in a nucleic acid of the present disclosure are modified nucleotides, wherein X may be any one of nucleotides A, G, U, C, or any one of the combinations A+G, A+U, A+C, G+U, G+C, U+C, A+G+U, A+G+C, G+U+C or A+G+C.
- the nucleic acid may contain from about 1% to about 100% modified nucleotides (either in relation to overall nucleotide content, or in relation to one or more types of nucleotide, i.e., any one or more of A, G, U or C) or any intervening percentage (e.g., from l% to 20%, from l% to 25%, from l% to 50%, from l% to 60%, from l% to 70%, from l% to 80%, from l% to 90%, from l% to 95%, from 10% to 20%, from 10% to 25%, from 10% to 50%, from 10% to 60%, from 10% to 70%, from 10% to 80%, from 10% to 90%, from 10% to 95%, from 10% to 100%, from 20% to 25%, from 20% to 50%, from 20% to 60%, from 20% to 70%, from 20% to 80%, from 20% to 90%, from 20% to 95%, from 20% to 100%, from 50% to 60%, from 50% to 70%, from 50% to 80%, from 50% to 90%, from 20% to 95%, from
- the nucleic acids may contain at a minimum 1% and at maximum 100% modified nucleotides, or any intervening percentage, such as at least 5% modified nucleotides, at least 10% modified nucleotides, at least 25% modified nucleotides, at least 50% modified nucleotides, at least 80% modified nucleotides, or at least 90% modified nucleotides.
- the nucleic acids may contain a modified pyrimidine such as a modified uracil or cytosine.
- At least 5%, at least 10%, at least 25%, at least 50%, at least 80%, at least 90% or 100% of the uracil in the nucleic acid is replaced with a modified uracil (e.g., a 5-substituted uracil).
- the modified uracil can be replaced by a compound having a single unique structure, or can be replaced by a plurality of compounds having different structures (e.g., 2, 3, 4 or more unique structures).
- cytosine in the nucleic acid is replaced with a modified cytosine (e.g., a 5-substituted cytosine).
- the modified cytosine can be replaced by a compound having a single unique structure, or can be replaced by a plurality of compounds having different structures (e.g., 2, 3, 4 or more unique structures).
- Untranslated regions are nucleic acid sections of a polynucleotide before a start codon (5' UTR) and after a stop codon (3' UTR) that are not translated.
- a polynucleotide e.g., a ribonucleic acid (RNA), e.g., a messenger RNA (mRNA)
- RNA e.g., a messenger RNA (mRNA)
- mRNA messenger RNA
- ORF open reading frame
- UTR e.g., a 5' UTR or functional fragment thereof, a 3' UTR or functional fragment thereof, or a combination thereof.
- a UTR (e.g., 5' UTR or 3' UTR) can be homologous or heterologous to the coding region in a polynucleotide.
- the UTR is homologous to the ORF encoding the CFTR polypeptide.
- the UTR is heterologous to the ORF encoding the CFTR polypeptide.
- the polynucleotide comprises two or more 5' UTRs or functional fragments thereof, each of which has the same or different nucleotide sequences. In some embodiments, the polynucleotide comprises two or more 3' UTRs or functional fragments thereof, each of which has the same or different nucleotide sequences.
- the 5' UTR or functional fragment thereof, 3' UTR or functional fragment thereof, or any combination thereof is sequence optimized.
- the 5'UTR or functional fragment thereof, 3' UTR or functional fragment thereof, or any combination thereof comprises at least one chemically modified nucleobase, e.g., N1 -methylpseudouracil or 5 -methoxyuracil.
- UTRs can have features that provide a regulatory role, e.g., increased or decreased stability, localization and/or translation efficiency.
- a polynucleotide comprising a UTR can be administered to a cell, tissue, or organism, and one or more regulatory features can be measured using routine methods.
- a functional fragment of a 5' UTR or 3' UTR comprises one or more regulatory features of a full length 5' or 3' UTR, respectively.
- Natural 5' UTRs bear features that play roles in translation initiation. They harbor signatures like Kozak sequences that are commonly known to be involved in the process by which the ribosome initiates translation of many genes. Kozak sequences have the consensus CCR(A/G)CCAUGG, where R is a purine (adenine or guanine) three bases upstream of the start codon (AUG), which is followed by another ‘G’. 5' UTRs also have been known to form secondary structures that are involved in elongation factor binding.
- liver-expressed mRNA such as albumin, serum amyloid A, Apolipoprotein A/B/E, transferrin, alpha fetoprotein, erythropoietin, or Factor VIII, can enhance expression of polynucleotides in hepatic cell lines or liver.
- 5'UTR from other tissue-specific mRNA to improve expression in that tissue is possible for muscle (e.g., MyoD, Myosin, Myoglobin, Myogenin, Herculin), for endothelial cells (e.g., Tie-1, CD36), for myeloid cells (e.g, C/EBP, AML1, G-CSF, GM-CSF, CDl lb, MSR, Fr-1, i-NOS), for leukocytes (e.g., CD45, CD 18), for adipose tissue (e.g., CD36, GLUT4, ACRP30, adiponectin) and for lung epithelial cells (e.g., SP-A/B/C/D).
- muscle e.g., MyoD, Myosin, Myoglobin, Myogenin, Herculin
- endothelial cells e.g., Tie-1, CD36
- myeloid cells e.g, C/E
- UTRs are selected from a family of transcripts whose proteins share a common function, structure, feature or property.
- an encoded polypeptide can belong to a family of proteins (i. e. , that share at least one function, structure, feature, localization, origin, or expression pattern), which are expressed in a particular cell, tissue or at some time during development.
- the UTRs from any of the genes or mRNA can be swapped for any other UTR of the same or different family of proteins to create a new polynucleotide.
- the 5' UTR and the 3' UTR can be heterologous. In some embodiments, the 5' UTR can be derived from a different species than the 3' UTR. In some embodiments, the 3' UTR can be derived from a different species than the 5' UTR.
- Additional exemplary UTRs of the application include, but are not limited to, one or more 5 'UTR and/or 3 'UTR derived from the nucleic acid sequence of: a globin, such as an a- or P-globin (e.g., aXenopus, mouse, rabbit, or human globin); a strong Kozak translational initiation signal; a CYBA (e.g., human cytochrome b-245 a polypeptide); an albumin (e.g., human albumin?); a HSD17B4 (hydroxysteroid (17-P) dehydrogenase); a virus (e.g., a tobacco etch virus (TEV), a Venezuelan equine encephalitis virus (VEEV), a Dengue virus, a cytomegalovirus (CMV) (e.g., CMV immediate early 1 (IE 1)), a hepatitis virus (e.g., hepatitis B virus), a Sindbis virus
- hsp70 a translation initiation factor (e.g., elF4G); a glucose transporter (e.g., hGLUTl (human glucose transporter 1)); an actin (e.g., human a or P actin); a GAPDH; a tubulin; a histone; a citric acid cycle enzyme; a topoisomerase (e.g., a 5'UTR of a TOP gene lacking the 5' TOP motif (the oligopyrimidine tract)); a ribosomal protein Large 32 (L32); a ribosomal protein (e.g., human or mouse ribosomal protein, such as, for example, rps9); an ATP synthase (e.g., ATP5A1 or the P subunit of mitochondrial H + -ATP synthase); a growth hormone e (e.g, bovine (bGH) or human (hGH)); an elongation factor (e.g,
- the 5' UTR is selected from the group consisting of a P-globin 5' UTR; a 5 'UTR containing a strong Kozak translational initiation signal; a cytochrome b-245 a polypeptide (CYBA) 5' UTR; a hydroxysteroid (17-P) dehydrogenase (HSD17B4) 5' UTR; a Tobacco etch virus (TEV) 5' UTR; a Vietnamese etch virus (TEV) 5' UTR; a decielen equine encephalitis virus (TEEV) 5' UTR; a 5' proximal open reading frame of rubella virus (RV) RNA encoding nonstructural proteins; a Dengue virus (DEN) 5' UTR; a heat shock protein 70 (Hsp70) 5' UTR; a eIF4G 5' UTR; a GLUT1 5' UTR; functional fragments thereof and any combination thereof.
- CYBA cytochrome b-2
- the 3' UTR is selected from the group consisting of a P-globin 3' UTR; a CYBA 3' UTR; an albumin 3' UTR; a growth hormone (GH) 3' UTR; a VEEV 3' UTR; a hepatitis B virus (HBV) 3' UTR; a-globin 3 'UTR; a DEN 3' UTR; a PAV barley yellow dwarf virus (BYDV-PAV) 3' UTR; an elongation factor 1 al (EEF1A1) 3' UTR; a manganese superoxide dismutase (MnSOD) 3' UTR; a P subunit of mitochondrial H(+)-ATP synthase (P-mRNA) 3' UTR; a GLUT1 3' UTR; a MEF2A 3' UTR; a P-Fl-ATPase 3' UTR; functional fragments thereof and combinations thereof.
- a P-globin 3' UTR
- Wild-type UTRs derived from any gene or mRNA can be incorporated into the polynucleotides of the invention.
- a UTR can be altered relative to a wild type or native UTR to produce a variant UTR, e.g., by changing the orientation or location of the UTR relative to the ORF; or by inclusion of additional nucleotides, deletion of nucleotides, swapping or transposition of nucleotides.
- variants of 5' or 3' UTRs can be utilized, for example, mutants of wild type UTRs, or variants wherein one or more nucleotides are added to or removed from a terminus of the UTR.
- one or more synthetic UTRs can be used in combination with one or more non-synthetic UTRs. See, e.g., Mandal and Rossi, Nat. Protoc. 2013 8(3):568-82, the contents of which are incorporated herein by reference in their entirety.
- UTRs or portions thereof can be placed in the same orientation as in the transcript from which they were selected or can be altered in orientation or location. Hence, a 5' and/or 3' UTR can be inverted, shortened, lengthened, or combined with one or more other 5' UTRs or 3' UTRs.
- the polynucleotide comprises multiple UTRs, e.g., a double, a triple or a quadruple 5' UTR or 3' UTR.
- a double UTR comprises two copies of the same UTR either in series or substantially in series.
- a double beta-globin 3'UTR can be used (see US2010/0129877, the contents of which are incorporated herein by reference in its entirety).
- the polynucleotides of the invention can comprise combinations of features.
- the ORF can be flanked by a 5 'UTR that comprises a strong Kozak translational initiation signal and/or a 3'UTR comprising an oligo(dT) sequence for templated addition of a poly-A tail.
- a 5 'UTR can comprise a first polynucleotide fragment and a second polynucleotide fragment from the same and/or different UTRs (see, e.g., US2010/0293625, herein incorporated by reference in its entirety).
- non-UTR sequences can be used as regions or subregions within the polynucleotides of the invention.
- introns or portions of intron sequences can be incorporated into the polynucleotides of the invention. Incorporation of intronic sequences can increase protein production as well as polynucleotide expression levels.
- the polynucleotide of the invention comprises an internal ribosome entry site (IRES) instead of or in addition to a UTR (see, e.g., Yakubov et al., Biochem. Biophys. Res. Commun. 2010 394(1): 189-193, the contents of which are incorporated herein by reference in their entirety).
- ITR internal ribosome entry site
- the polynucleotide comprises an IRES instead of a 5' UTR sequence. In some embodiments, the polynucleotide comprises an ORF and a viral capsid sequence. In some embodiments, the polynucleotide comprises a synthetic 5' UTR in combination with a non-synthetic 3' UTR.
- the UTR can also include at least one translation enhancer polynucleotide, translation enhancer element, or translational enhancer elements (collectively, "TEE," which refers to nucleic acid sequences that increase the amount of polypeptide or protein produced from a polynucleotide.
- TEE translation enhancer polynucleotide, translation enhancer element, or translational enhancer elements
- the TEE can be located between the transcription promoter and the start codon.
- the 5' UTR comprises a TEE.
- a TEE is a conserved element in a UTR that can promote translational activity of a nucleic acid such as, but not limited to, cap-dependent or cap-independent translation. a. 5' UTR sequences
- a polynucleotide e.g., mRNA
- a CFTR polypeptide e.g., SEQ ID NO: 1 or SEQ ID NO:2
- SEQ ID NO: 1 or SEQ ID NO:2 CFTR polypeptide
- 5' UTR confers an increased half-life, increased expression and/or increased activity of the polypeptide encoded by said polynucleotide, or of the polynucleotide itself.
- a polynucleotide disclosed herein comprises: (a) a 5'-UTR (e.g., as provided in Table 1 or a variant or fragment thereof); (b) a coding region comprising a stop element (e.g., as described herein); and (c) a 3'-UTR (e.g., as described herein), and LNP compositions comprising the same.
- the polynucleotide comprises a 5'-UTR comprising a sequence provided in Table 1 or a variant or fragment thereof (e.g., a functional variant or fragment thereof).
- the polynucleotide having a 5' UTR sequence provided in Table 1 or a variant or fragment thereof has an increase in the half-life of the polynucleotide, e.g., about 1.5-20-fold increase in half-life of the polynucleotide.
- the increase in half-life is about 1.5, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 or 20-fold, or more.
- the increase in half life is about 1.5-fold or more.
- the increase in half life is about 2- fold or more.
- the increase in half life is about 3-fold or more.
- the increase in half life is about 4-fold or more.
- the increase in half life is about 5-fold or more.
- the polynucleotide having a 5' UTR sequence provided in Table 1 or a variant or fragment thereof results in an increased level and/or activity, e.g., output, of the polypeptide encoded by the polynucleotide.
- the 5 'UTR results in about 1.5-20-fold increase in level and/or activity, e.g., output, of the polypeptide encoded by the polynucleotide.
- the increase in level and/or activity is about 1.5, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 or 20-fold, or more. In an embodiment, the increase in level and/or activity is about 1.5-fold or more.
- the increase in level and/or activity is about 2- fold or more. In an embodiment, the increase in level and/or activity is about 3-fold or more. In an embodiment, the increase in level and/or activity is about 4-fold or more. In an embodiment, the increase in level and/or activity is about 5 -fold or more.
- the increase is compared to an otherwise similar polynucleotide which does not have a 5' UTR, has a different 5' UTR, or does not have a 5' UTR described in Table 1 or a variant or fragment thereof.
- the increase in half-life of the polynucleotide is measured according to an assay that measures the half-life of a polynucleotide.
- the increase in level and/or activity, e.g., output, of the polypeptide encoded by the polynucleotide is measured according to an assay that measures the level and/or activity of a polypeptide.
- the 5' UTR comprises a sequence provided in Table 1 or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to a 5' UTR sequence provided in Table 1, or a variant or a fragment thereof.
- the 5' UTR comprises a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 50, SEQ ID NO: 51, SEQ ID NO: 52, SEQ ID NO: 53, SEQ ID NO: 54, SEQ ID NO: 55, SEQ ID NO: 56, SEQ ID NO: 57, SEQ ID NO: 58, or SEQ ID NO: 78.
- the 5' UTR comprises the sequence of SEQ ID NO:50. In an embodiment, the 5' UTR consists of the sequence of SEQ ID NO:50. In an embodiment, a 5' UTR sequence provided in Table 1 has a first nucleotide which is an A. In an embodiment, a 5' UTR sequence provided in Table 1 has a first nucleotide which is a G. Table 1: 5' UTR sequences In an embodiment, the 5' UTR comprises a variant of SEQ ID NO:50. In an embodiment, the variant of SEQ ID NO:50 comprises a nucleic acid sequence of Formula A:
- (N 3 ) x is a guanine and x is an integer from 0 to 1 ;
- X is a cytosine and x is an integer from 0 to 1 ;
- Ne is a uracil or cytosine
- N? is a uracil or guanine
- Ns is adenine or guanine and x is an integer from 0 to 1.
- N 2 x is a uracil and x is 0. In an embodiment (N 2 ) x is a uracil and x is 1. In an embodiment (N 2 ) x is a uracil and x is 2. In an embodiment (N 2 ) X is a uracil and x is 3. In an embodiment, (N 2 ) x is a uracil and x is 4. In an embodiment (N 2 ) x is a uracil and x is 5.
- (N 3 ) x is a guanine and x is 0. In an embodiment, (N 3 ) x is a guanine and x is 1.
- (N 4 ) x is a cytosine and x is 0. In an embodiment, (N 4 ) x is a cytosine and x is 1.
- Ns) x is a uracil and x is 0. In an embodiment (Ns) x is a uracil and x is 1. In an embodiment (Ns)x is a uracil and x is 2. In an embodiment (Ns)x is a uracil and x is 3. In an embodiment, (Ns)x is a uracil and x is 4. In an embodiment (Ns) x is a uracil and x is 5.
- N6 is a uracil. In an embodiment, N6 is a cytosine.
- N7 is a uracil. In an embodiment, N7 is a guanine.
- N8 is an adenine and x is 0. In an embodiment, N8 is an adenine and x is 1. In an embodiment, N8 is a guanine and x is 0. In an embodiment, N8 is a guanine and x is 1.
- the 5' UTR comprises a variant of SEQ ID NO: 50.
- the variant of SEQ ID NO: 50 comprises a sequence with at least 50%, 60%, 70%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, or 99% identity to SEQ ID NO: 50.
- the variant of SEQ ID NO: 50 comprises a sequence with at least 50% identity to SEQ ID NO: 50.
- the variant of SEQ ID NO: 50 comprises a sequence with at least 60% identity to SEQ ID NO: 50.
- the variant of SEQ ID NO: 50 comprises a sequence with at least 70% identity to SEQ ID NO: 50.
- the variant of SEQ ID NO: 50 comprises a sequence with at least 80% identity to SEQ ID NO: 50. In an embodiment, the variant of SEQ ID NO: 50 comprises a sequence with at least 90% identity to SEQ ID NO: 50. In an embodiment, the variant of SEQ ID NO: 50 comprises a sequence with at least 95% identity to SEQ ID NO: 50. In an embodiment, the variant of SEQ ID NO: 50 comprises a sequence with at least 96% identity to SEQ ID NO: 50. In an embodiment, the variant of SEQ ID NO: 50 comprises a sequence with at least 97% identity to SEQ ID NO: 50. In an embodiment, the variant of SEQ ID NO: 50 comprises a sequence with at least 98% identity to SEQ ID NO: 50. In an embodiment, the variant of SEQ ID NO: 50 comprises a sequence with at least 99% identity to SEQ ID NO: 50.
- the variant of SEQ ID NO: 50 comprises a uridine content of at least 5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, or 80%. In an embodiment, the variant of SEQ ID NO: 50 comprises a uridine content of at least 5%. In an embodiment, the variant of SEQ ID NO: 50 comprises a uridine content of at least 10%. In an embodiment, the variant of SEQ ID NO: 50 comprises a uridine content of at least 20%. In an embodiment, the variant of SEQ ID NO: 50 comprises a uridine content of at least 30%. In an embodiment, the variant of SEQ ID NO: 50 comprises a uridine content of at least 40%.
- the variant of SEQ ID NO: 50 comprises a uridine content of at least 50%. In an embodiment, the variant of SEQ ID NO: 50 comprises a uridine content of at least 60%. In an embodiment, the variant of SEQ ID NO: 50 comprises a uridine content of at least 70%. In an embodiment, the variant of SEQ ID NO: 50 comprises a uridine content of at least 80%.
- the variant of SEQ ID NO: 50 comprises at least 2, 3, 4, 5, 6 or 7 consecutive uridines (e.g, a polyuridine tract).
- the polyuridine tract in the variant of SEQ ID NO: 50 comprises at least 1-7, 2-7, 3-7, 4-7, 5-7, 6-7, 1-6, 1-5, 1-4, 1-3, 1-2, 2-6, or 3-5 consecutive uridines.
- the polyuridine tract in the variant of SEQ ID NO: 50 comprises 4 consecutive uridines.
- the polyuridine tract in the variant of SEQ ID NO: 50 comprises 5 consecutive uridines.
- the variant of SEQ ID NO: 50 comprises 1, 2, 3, 4, 5, 6, 7,
- the variant of SEQ ID NO: 50 comprises 3 polyuridine tracts. In an embodiment, the variant of SEQ ID NO: 50 comprises 4 polyuridine tracts. In an embodiment, the variant of SEQ ID NO: 50 comprises 5 polyuridine tracts.
- one or more of the polyuridine tracts are adjacent to a different polyuridine tract.
- each of, e.g., all, the polyuridine tracts are adjacent to each other, e.g., all of the polyuridine tracts are contiguous.
- one or more of the polyuridine tracts are separated by 1, 2,
- each of, e.g., all of, the polyuridine tracts are separated by 1, 2, 3,
- a first polyuridine tract and a second polyuridine tract are adjacent to each other.
- a subsequent, e.g., third, fourth, fifth, sixth or seventh, eighth, ninth, or tenth, polyuridine tract is separated by 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 2, 13, 14, 15, 16, 17, 18. 19, 20, 30, 40, 50 or 60 nucleotides from the first polyuridine tract, the second polyuridine tract, or any one of the subsequent polyuridine tracts.
- a first polyuridine tract is separated by 1, 2, 3, 4, 5, 6, 7, 8,
- a subsequent polyuridine tract e.g., a second, third, fourth, fifth, sixth or seventh, eighth, ninth, or tenth polyuridine tract.
- one or more of the subsequent polyuridine tracts are adjacent to a different polyuridine tract.
- the 5' UTR comprises a Kozak sequence, e.g., a GCCRCC nucleotide sequence wherein R is an adenine or guanine.
- the Kozak sequence is disposed at the 3' end of the 5'UTR sequence.
- the polynucleotide e.g., mRNA
- the polynucleotide comprising an open reading frame encoding a CFTR polypeptide (e.g., SEQ ID NO: 1) and comprising a 5' UTR sequence disclosed herein is formulated as an LNP.
- the LNP composition comprises: (i) an ionizable lipid, e.g., an amino lipid; (ii) a sterol or other structural lipid; (iii) a non-cationic helper lipid or phospholipid; (iv) a PEG-lipid; and (v) a cationic agent.
- the LNP compositions of the disclosure are used in a method of treating cystic fibrosis in a subject.
- an LNP composition comprising a polynucleotide disclosed herein encoding a CFTR polypeptide, e.g., as described herein, can be administered with an additional agent, e.g., as described herein. b. 3' UTR sequences
- 3 'UTR sequences have been shown to influence translation, half-life, and subcellular localization of mRNAs (Mayr C., Cold Spring Harb Persp Biol 2019 Oct l;l l(10):a034728).
- a polynucleotide e.g., mRNA
- a CFTR polypeptide e.g., SEQ ID NO: 1
- SEQ ID NO: 1 CFTR polypeptide
- a polynucleotide disclosed herein comprises: (a) a 5'-UTR (e.g., as described herein); (b) a coding region comprising a stop element (e.g., as described herein); and (c) a 3'-UTR (e.g., as provided in Table 2 or a variant or fragment thereof), and LNP compositions comprising the same.
- the polynucleotide comprises a 3'-UTR comprising a sequence provided in Table 2 or a variant or fragment thereof.
- the polynucleotide having a 3' UTR sequence provided in Table 2 or a variant or fragment thereof results in an increased half-life of the polynucleotide, e.g., about 1.5-10-fold increase in half-life of the polynucleotide.
- the increase in half-life is about 1.5, 2, 3, 4, 5, 6, 7, 8, 9, or 10-fold, or more.
- the increase in half-life is about 1.5-fold or more.
- the increase in half-life is about 2-fold or more.
- the increase in half-life is about 3 -fold or more.
- the increase in halflife is about 4-fold or more.
- the increase in half-life is about 5 -fold or more. In an embodiment, the increase in half-life is about 6-fold or more. In an embodiment, the increase in half-life is about 7-fold or more. In an embodiment, the increase in half-life is about 8-fold. In an embodiment, the increase in half-life is about 9-fold or more. In an embodiment, the increase in half-life is about 10-fold or more.
- the polynucleotide having a 3' UTR sequence provided in Table 2 or a variant or fragment thereof results in a polynucleotide with a mean halflife score of greater than 10.
- the polynucleotide having a 3' UTR sequence provided in Table 2 or a variant or fragment thereof results in an increased level and/or activity, e.g., output, of the polypeptide encoded by the polynucleotide.
- the increase is compared to an otherwise similar polynucleotide which does not have a 3' UTR, has a different 3' UTR, or does not have a 3' UTR of Table 2 or a variant or fragment thereof.
- the polynucleotide comprises a 3' UTR sequence provided in Table 2 or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to a 3' UTR sequence provided in Table 2, or a fragment thereof.
- the 3' UTR comprises a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 100, SEQ ID NO: 101, SEQ ID NO: 102, SEQ ID NO: 103, SEQ ID NO: 104, SEQ ID NO: 105, SEQ ID NO: 106, SEQ ID NO: 107, SEQ ID NO: 108, SEQ ID NO: 109, SEQ ID NO: 110, SEQ ID NO: 111, SEQ ID NO: 112, SEQ ID NO: 113, SEQ ID NO: 114, SEQ ID NO: 115, or SEQ ID NO: 141.
- the 3' UTR comprises the sequence of SEQ ID NO: 141, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 141.
- Table 2 3' UTR sequences
- the 3' UTR comprises a micro RNA (miRNA) binding site, e.g., as described herein, which binds to a miR present in a human cell.
- the 3' UTR comprises a miRNA binding site of SEQ ID NO:212, SEQ ID NO: 174, SEQ ID NO: 152 or a combination thereof.
- the 3' UTR comprises a plurality of miRNA binding sites, e.g., 2, 3, 4, 5, 6, 7 or 8 miRNA binding sites.
- the plurality of miRNA binding sites comprises the same or different miRNA binding sites.
- miR122 bs CAAACACCAUUGUCACACUCCA (SEQ ID NO: 212)
- miR-142-3p bs UCCAUAAAGUAGGAAACACUACA (SEQ ID NO: 174)
- miR- 126 bs CGCAUUAUUACUCACGGUACGA (SEQ ID NO: 152)
- the disclosure also includes a polynucleotide that comprises both a 5' Cap and a polynucleotide of the present invention (e.g., a polynucleotide comprising a nucleotide sequence encoding a CFTR polypeptide to be expressed).
- the 5' cap structure of a natural mRNA is involved in nuclear export, increasing mRNA stability and binds the mRNA Cap Binding Protein (CBP), which is responsible for mRNA stability in the cell and translation competency through the association of CBP with poly(A) binding protein to form the mature cyclic mRNA species.
- CBP mRNA Cap Binding Protein
- the cap further assists the removal of 5' proximal introns during mRNA splicing.
- Endogenous mRNA molecules can be 5'-end capped generating a 5'-ppp-5'- triphosphate linkage between a terminal guanosine cap residue and the 5 '-terminal transcribed sense nucleotide of the mRNA molecule.
- This 5 '-guanylate cap can then be methylated to generate an N7-methyl -guanylate residue.
- the ribose sugars of the terminal and/or anteterminal transcribed nucleotides of the 5' end of the mRNA can optionally also be 2'-O-methylated.
- 5 '-decapping through hydrolysis and cleavage of the guanylate cap structure can target a nucleic acid molecule, such as an mRNA molecule, for degradation.
- the polynucleotides of the present invention incorporate a cap moiety.
- polynucleotides of the present invention comprise a non-hydrolyzable cap structure preventing decapping and thus increasing mRNA halflife.
- modified nucleotides can be used during the capping reaction.
- a Vaccinia Capping Enzyme from New England Biolabs (Ipswich, MA) can be used with a-thio-guanosine nucleotides according to the manufacturer's instructions to create a phosphorothioate linkage in the 5 '-ppp-5' cap.
- Additional modified guanosine nucleotides can be used such as a-methyl-phosphonate and seleno-phosphate nucleotides.
- Additional modifications include, but are not limited to, 2'-O-methylation of the ribose sugars of 5 '-terminal and/or 5 '-anteterminal nucleotides of the polynucleotide (as mentioned above) on the 2'-hydroxyl group of the sugar ring.
- Multiple distinct 5 '-cap structures can be used to generate the 5 '-cap of a nucleic acid molecule, such as a polynucleotide that functions as an mRNA molecule.
- Cap analogs which herein are also referred to as synthetic cap analogs, chemical caps, chemical cap analogs, or structural or functional cap analogs, differ from natural (i.e., endogenous, wild-type or physiological) 5 '-caps in their chemical structure, while retaining cap function.
- Cap analogs can be chemically (i. e. , non-enzymatically) or enzymatically synthesized and/or linked to the polynucleotides of the invention.
- the Anti-Reverse Cap Analog (ARCA) cap contains two guanines linked by a 5 '-5 '-triphosphate group, wherein one guanine contains an N7 methyl group as well as a 3'-O-methyl group (i.e., N7,3'-O-dimethyl-guanosine-5'- triphosphate-5 '-guanosine (m 7 G-3'mppp-G; which can equivalently be designated 3' O-Me-m 7 G(5')ppp(5')G).
- the 3'-0 atom of the other, unmodified, guanine becomes linked to the 5'-terminal nucleotide of the capped polynucleotide.
- the N7- and 3'-O- methlyated guanine provides the terminal moiety of the capped polynucleotide.
- mCAP is similar to ARCA but has a 2'-O- methyl group on guanosine (i.e., N7,2'-O-dimethyl-guanosine-5 '-triphosphate-5 '- guanosine, m 7 Gm-ppp-G).
- Another exemplary cap is m 7 G-ppp-Gm-A (i.e., N7, guanosine-5 '-triphosphate- 2'-O-dimethyl-guanosine-adenosine).
- the cap is a dinucleotide cap analog.
- the dinucleotide cap analog can be modified at different phosphate positions with a boranophosphate group or a phosphoroselenoate group such as the dinucleotide cap analogs described in U.S. Patent No. US 8,519, 110, the contents of which are herein incorporated by reference in its entirety.
- the cap is a cap analog is a N7-(4- chlorophenoxy ethyl) substituted dinucleotide form of a cap analog known in the art and/or described herein.
- Non-limiting examples of a N7-(4-chlorophenoxyethyl) substituted dinucleotide form of a cap analog include a N7-(4-chlorophenoxyethyl)- G(5')ppp(5')G and aN7-(4-chlorophenoxyethyl)-m 3 '°G(5')ppp(5')G cap analog (See, e.g., the various cap analogs and the methods of synthesizing cap analogs described in Kore et al.
- a cap analog of the present invention is a 4-chloro/bromophenoxyethyl analog.
- Polynucleotides of the invention can also be capped post-manufacture (whether IVT or chemical synthesis), using enzymes, in order to generate more authentic 5'-cap structures.
- the phrase "more authentic" refers to a feature that closely mirrors or mimics, either structurally or functionally, an endogenous or wild type feature.
- a "more authentic" feature is better representative of an endogenous, wild-type, natural or physiological cellular function and/or structure as compared to synthetic features or analogs, etc., of the prior art, or which outperforms the corresponding endogenous, wild-type, natural or physiological feature in one or more respects.
- Non-limiting examples of more authentic 5'cap structures of the present invention are those that, among other things, have enhanced binding of cap binding proteins, increased half-life, reduced susceptibility to 5' endonucleases and/or reduced 5 'decapping, as compared to synthetic 5'cap structures known in the art (or to a wild-type, natural or physiological 5'cap structure).
- recombinant Vaccinia Virus Capping Enzyme and recombinant 2'-O- methyltransferase enzyme can create a canonical 5 '-5 '-triphosphate linkage between the 5 '-terminal nucleotide of a polynucleotide and a guanine cap nucleotide wherein the cap guanine contains an N7 methylation and the 5 '-terminal nucleotide of the mRNA contains a 2'-O-methyl.
- Capl structure Such a structure is termed the Capl structure.
- Cap structures include, but are not limited to, 7mG(5')ppp(5')NlpN2p (cap 0), 7mG(5')ppp(5')NlmpNp (cap 1), and 7mG(5')- ppp(5')NlmpN2mp (cap 2).
- capping chimeric polynucleotides postmanufacture can be more efficient as nearly 100% of the chimeric polynucleotides can be capped. This is in contrast to -80% when a cap analog is linked to a chimeric polynucleotide in the course of an in vitro transcription reaction.
- 5' terminal caps can include endogenous caps or cap analogs.
- a 5' terminal cap can comprise a guanine analog.
- Useful guanine analogs include, but are not limited to, inosine, N1 -methyl -guanosine, 2'fluoro-guanosine, 7-deaza-guanosine, 8-oxo- guanosine, 2-amino-guanosine, LNA-guanosine, and 2-azido-guanosine.
- caps including those that can be used in co-transcriptional capping methods for ribonucleic acid (RNA) synthesis, using RNA polymerase, e.g., wild type RNA polymerase or variants thereof, e.g., such as those variants described herein.
- RNA polymerase e.g., wild type RNA polymerase or variants thereof, e.g., such as those variants described herein.
- caps can be added when RNA is produced in a “one-pot” reaction, without the need for a separate capping reaction.
- the methods in some embodiments, comprise reacting a polynucleotide template with an RNA polymerase variant, nucleoside triphosphates, and a cap analog under in vitro transcription reaction conditions to produce RNA transcript.
- cap includes the inverted G nucleotide and can comprise one or more additional nucleotides 3’ of the inverted G nucleotide, e.g., 1, 2, 3, or more nucleotides 3’ of the inverted G nucleotide and 5’ to the 5’ UTR, e.g., a 5’ UTR described herein.
- Exemplary caps comprise a sequence of GG, GA, or GGA, wherein the underlined, italicized G is an in inverted G nucleotide followed by a 5 ’-5’- triphosphate group.
- a cap comprises a compound of formula (I) stereoisomer, tautomer or salt thereof, wherein
- ring Bi is a modified or unmodified Guanine
- ring B2 and ring B3 each independently is a nucleobase or a modified nucleobase
- X 2 is O, S(O) P , NR 24 or CR 25 R 26 in which p is 0, 1, or 2;
- Y 0 is O or CR 6 R 7 ;
- Y1 is O, S(O) n , CR 6 R 7 , or NR, in which n is 0, 1 , or 2; each — is a single bond or absent, wherein when each — is a single bond, Yi is o, S(O) n, CR 6 R 7 , or NRs; and when each — is absent, Y 1 is void;
- Y 2 is (0P(0)R 4 )m in which m is 0, 1, or 2, or -0-(CR4oR4i)u-Qo-(CR42R43)v-, in which Qo is a bond, O, S(O) r , NR44, or CR45R46, r is 0, 1 , or 2, and each of u and v independently is 1, 2, 3 or 4; each R2 and R2' independently is halo, LNA, or OR3; each R3 independently is H, C 1 -C 6 alkyl, C 2 -C 6 alkenyl, or C 2 -C 6 alkynyl and R 3 , when being C 1 -C 6 alkyl, C 2 -C 6 alkenyl, or C 2 -C 6 alkynyl, is optionally substituted with one or more of halo, OH and C 1 -C 6 alkoxyl that is optionally substituted with one or more OH or OC(O)-C 1 -C
- R30 is C 1 -C 6 alkylene optionally substituted with one or more of halo, OH and C 1 -C 6 alkoxyl; each of R 31 R 32 , and R 33 , independently is H, C 1 -C 6 alkyl, C 3 -C 8 cycloalkyl, C 6 -C 10 aryl, 4 to 12-membered heterocycloalkyl, or 5- or 6-membered heteroaryl; each of R40, R41, R42, and R43 independently is H, halo, OH, cyano, N3, OP(O)R 47 R 48 , or C 1 -C 6 alkyl optionally substituted with one or more OP(O)R 47 R 48 , or one R41 and one R43, together with the carbon atoms to which they are attached and Qo, form C4-C 10 cycloalkyl, 4- to 14-membered heterocycloalkyl, C 6 -C 10 aryl, or 5- to 14-membered
- R44 is H, C 1 -C 6 alkyl, or an amine protecting group; each of R 45 and R 46 independently is H, OP(O)R 47 R 48 , or C 1 -C 6 alkyl optionally substituted with one or more OP(O)R 47 R 48 , and each ofR 47 and R 48 , independently is H, halo, C 1 -C 6 alkyl, OH, SH, SeH, or
- a cap analog may include any of the cap analogs described in international publication WO 2017/066797, published on 20 April 2017, incorporated by reference herein in its entirety.
- the B2 middle position can be a non-ribose molecule, such as arabinose.
- R2 is ethyl-based.
- a cap comprises the following structure: In other embodiments, a cap comprises the following structure:
- a cap comprises the following structure:
- a cap comprises the following structure:
- R is an alkyl (e.g., C 1 -C 6 alkyl). In some embodiments, R is a methyl group (e.g., Ci alkyl). In some embodiments, R is an ethyl group (e.g, C 2 alkyl).
- a cap comprises a sequence selected from the following sequences: GAA, GAC, GAG, GAU, GCA, GCC, GCG, GCU, GGA , GGC, GGG, GGU, GUA, GUC, GUG, and GUU.
- a cap comprises GAA.
- a cap comprises GAC.
- a cap comprises GAG.
- a cap comprises GAU.
- a cap comprises GCA.
- a cap comprises GCC.
- a cap comprises GCG.
- a cap comprises GCU.
- a cap comprises GGA.
- a cap comprises GGC.
- a cap comprises a sequence selected from the following sequences: m 7 GpppApA, m 7 GpppApC, m 7 GpppApG, m 7 GpppApU, m 7 GpppCpA, m 7 GpppCpC, m 7 GpppCpG, m 7 GpppCpU, m 7 GpppGpA, m 7 GpppGpC, m 7 GpppGpG, m 7 GpppGpU, m 7 GpppUpA, m 7 GpppUpC, m 7 GpppUpG, and m 7 GpppUpU.
- a cap comprises m 7 GpppApA. In some embodiments, a cap comprises m 7 GpppApC. In some embodiments, a cap comprises m 7 GpppApG. In some embodiments, a cap comprises m 7 GpppApU. In some embodiments, a cap comprises m 7 GpppCpA. In some embodiments, a cap comprises m 7 GpppCpC. In some embodiments, a cap comprises m 7 GpppCpG. In some embodiments, a cap comprises m 7 GpppCpU. In some embodiments, a cap comprises m 7 GpppGpA. In some embodiments, a cap comprises m 7 GpppGpC.
- a cap comprises m 7 GpppGpG. In some embodiments, a cap comprises m 7 GpppGpU. In some embodiments, a cap comprises m 7 GpppUpA. In some embodiments, a cap comprises m 7 GpppUpC. In some embodiments, a cap comprises m 7 GpppUpG. In some embodiments, a cap comprises m 7 GpppUpU.
- a cap in some embodiments, comprises a sequence selected from the following sequences: m 7 G3'oM e pppApA, m 7 G3'oM e pppApC, m 7 G3'oM e pppApG, m 7 G3'oMePPpApU, m 7 G3'oMePPpCpA, m 7 G3'oMePPpCpC, m 7 G3'oMePPpCpG, m 7 G3'OMePPpCpU, m 7 G3'OMePPpGpA, m 7 G3'OMePPpGpC, m 7 G3'OMePPpGpG, m 7 G3'OM e pppGpU, m 7 G3'OMepppUpA, m 7 G3'OMepppUpC, m 7 G3'OMepppUpG, and m 7 G3'OM e pppUp
- a cap comprises m 7 G3'oMePPpApA. In some embodiments, a cap comprises m 7 G3'oM e pppApC. In some embodiments, a cap comprises m 7 G3'oM e pppApG. In some embodiments, a cap comprises m 7 G3'OM e pppApU. In some embodiments, a cap comprises m 7 G3'OMepppCpA. In some embodiments, a cap comprises m 7 G3'oM e pppCpC. In some embodiments, a cap comprises m 7 G3'oM e pppCpG.
- a cap comprises m 7 G3'oMePPpCpU. In some embodiments, a cap comprises m 7 G3'oMePPpGpA. In some embodiments, a cap comprises m 7 G3'oM e pppGpC. In some embodiments, a cap comprises m 7 G3'oM e pppGpG. In some embodiments, a cap comprises m 7 G3'oMePPpGpU. In some embodiments, a cap comprises m 7 G3'oMePPpUpA. In some embodiments, a cap comprises m 7 G3'oM e pppUpC.
- a cap comprises m 7 G3'oM e pppUpG. In some embodiments, a cap comprises m 7 G3'OM e pppUpU.
- a cap in other embodiments, comprises a sequence selected from the following sequences: m’Gs'oMePPpA/oMepA, m’Gs'oMePPpA/oMepC, m 7 G3'OM e pppA2'OMepG, m’Gs'OMePPpA/OMepU, m’Gs'OMePPpC/OMepA, m 7 G3'OMePPpC2'OMepC, nfGs'OMePPpC/OMepG, nfGs'OMePPpC/OMepU, m 7 G3'OMePPpG2'OMepA, nfGs'OMePPpG/OMepC, m’Gs'OMePPpG
- a cap comprises m 7 G3'OMepppA2'oMepA. In some embodiments, a cap comprises m 7 G3'oMePPpA2'oMepC. In some embodiments, a cap comprises m 7 G3'oMePPpA2'oMepG. In some embodiments, a cap comprises m 7 G3'oMePPpA2'oMepU. In some embodiments, a cap comprises m 7 G3'oMePPpC2'oMepA. In some embodiments, a cap comprises m 7 G3'oMePPpC2'oMepC.
- a cap comprises m 7 G3'OMepppC2'OMepG. In some embodiments, a cap comprises m 7 G3'oMePPpC2'oMepU. In some embodiments, a cap comprises m 7 G3'oMePPpG2'oMepA. In some embodiments, a cap comprises m 7 G3'oMePPpG2'oMepC. In some embodiments, a cap comprises m 7 G3'oMePPpG2'oMepG. In some embodiments, a cap comprises m 7 G3'oMePPpG2'oMepU.
- a cap comprises m 7 G3'oMePPpU2'oMepA. In some embodiments, a cap comprises m 7 G3'oMePPpU2'oMepC. In some embodiments, a cap comprises m 7 G3'oMePPpU2'oMepG. In some embodiments, a cap comprises m 7 G3'OM e pppU2'OMepU.
- a cap in still other embodiments, comprises a sequence selected from the following sequences: m 7 GpppA2'oM e pA, m 7 GpppA2'oM e pC, m 7 GpppA2'oM e pG, m 7 GpppA2'oMepU, m 7 GpppC2'oMepA, m 7 GpppC2'oMepC, m 7 GpppC2'oMepG, m 7 GpppC2'OMepU, m 7 GpppG2'OMepA, m 7 GpppG2'OMepC, m 7 GpppG2'OMepG, m 7 GpppG2'0MepU, m 7 GpppU2'0MepA, m 7 GpppU2'0MepC, m 7 GpppU2'0MepC, m 7 GpppU2
- a cap comprises m 7 GpppA2'oMepA. In some embodiments, a cap comprises m 7 GpppA2'oM e pC. In some embodiments, a cap comprises m 7 GpppA2'oM e pG. In some embodiments, a cap comprises m 7 GpppA2'oMepU. In some embodiments, a cap comprises m 7 GpppC2'OMepA. In some embodiments, a cap comprises m 7 GpppC2'OM e pC. In some embodiments, a cap comprises m 7 GpppC2'oM e pG.
- a trinucleotide cap comprises m 7 GpppC2'oMepU. In some embodiments, a cap comprises m 7 GpppG2'oMepA. In some embodiments, a cap comprises m 7 GpppG2'oM e pC. In some embodiments, a cap comprises m 7 GpppG2'OM e pG. In some embodiments, a cap comprises m 7 GpppG2'oMepU. In some embodiments, a cap comprises m 7 GpppU2'oMepA. In some embodiments, a cap comprises m 7 GpppU2'oM e pC. In some embodiments, a cap comprises m 7 GpppU2'oM e pG. In some embodiments, a cap comprises m 7 GpppU2'oMepU.
- a cap comprises m 7 Gpppm 6 A2’Om e pG. In some embodiments, a cap comprises m 7 Gpppe 6 A2’Om e pG.
- a cap comprises GAG. In some embodiments, a cap comprises GCG. In some embodiments, a cap comprises GUG. In some embodiments, a cap comprises GGG.
- a cap comprises any one of the following structures:
- the cap comprises m7 GpppNiN2N3, where Ni, N2, and N3 are optional (i.e., can be absent or one or more can be present) and are independently a natural, a modified, or an unnatural nucleoside base.
- m7 G is further methylated, e.g., at the 3’ position.
- the m7 G comprises an O-methyl at the 3’ position.
- Ni, N2, and N3 if present, optionally, are independently an adenine, a uracil, a guanidine, a thymine, or a cytosine.
- one or more (or all) of Ni, N2, and N3, if present are methylated, e.g., at the 2’ position. In some embodiments, one or more (or all) of Ni, N2, and N3, if present have an O-methyl at the 2’ position.
- the cap comprises the following structure:
- Bi, B2, and B3 are independently a natural, a modified, or an unnatural nucleoside based; and Ri, R2, R3, and R4 are independently OH or O- methyl.
- R3 is O-methyl and R4 is OH.
- R3 and R4 are O-methyl.
- R4 is O-methyl.
- Ri is OH, R2 is OH, R3 is O-methyl, and R4 is OH.
- Ri is OH, R2 is OH, R3 is O-methyl, and R4 is O-methyl.
- at least one of Ri and R2 is O-methyl, R3 is O-methyl, and R4 is OH.
- at least one of Ri and R2 is O-methyl, R3 is O-methyl, and R4 is O-methyl.
- Bi, B3, and B3 are natural nucleoside bases. In some embodiments, at least one of Bi, B2, and B3 is a modified or unnatural base. In some embodiments, at least one of Bi, B2, and B3 is N6-methyladenine. In some embodiments, Bi is adenine, cytosine, thymine, or uracil. In some embodiments, Bi is adenine, B2 is uracil, and B3 is adenine. In some embodiments, Ri and R2 are OH, R3 and R4 are O-methyl, Bi is adenine, B2 is uracil, and B3 is adenine.
- the cap comprises a sequence selected from the following sequences: GAAA, GACA, GAGA, GAUA, GCAA, GCCA, GCGA, GCUA, GGAA, GGCA, GGGA, GGUA, GUCA, and GUUA.
- the cap comprises a sequence selected from the following sequences: GAAG, GACG, GAGG, GAUG, GCAG, GCCG, GCGG, GCUG, GGAG, GGCG, GGGG, GGUG, GUCG, GUGG, and GUUG.
- the cap comprises a sequence selected from the following sequences: GAAU, GACU, GAGU, GAUU, GCAU, GCCU, GCGU, GCUU, GGAU, GGCU, GGGU, GGUU, GUAU, GUCU, GUGU, and GUUU.
- the cap comprises a sequence selected from the following sequences: GAAC, GACC, GAGC, GAUC, GCAC, GCCC, GCGC, GCUC, GGAC, GGCC, GGGC, GGUC, GUAC, GUCC, GUGC, and GUUC.
- a cap in some embodiments, comprises a sequence selected from the following sequences: m 7 G3'oM e pppApApN, m 7 G3'oM e pppApCpN, m 7 G3'oMePPpApGpN, m 7 G3'oM e pppApUpN, m 7 G3'oM e pppCpApN, m 7 G3'oMePPpCpCpN, m 7 G3'oMePPpCpGpN, m 7 G3'oM e pppCpUpN, m 7 G3'OM e pppGpApN, m 7 G3'OM e pppGpCpN, m 7 G3'OM e pppGpGpN, m 7 G3'OM e pppGpGpN, m 7 G3'OM e pppGpGpN, m
- a cap in other embodiments, comprises a sequence selected from the following sequences: m’Gs'oMePPpA/oMepApN, m’Gs'oMePPpA/oMepCpN, m 7 G3'OMePPpA2'OMepGpN, m’Gs'oMePPpA/oMepUpN, m’Gs'oMePPpCzoMepApN, m 7 G3'OMePPpC2'OMepCpN, m 7 G3'OM e pppC2'OMepGpN, m 7 G3'OM e pppC2'OMepUpN, m 7 G3'OMePPpG2'OMepApN, nfGs'oMePPpGzoMepCpN, nfGs'oMePPpGzoMepGpN, m 7 G3'OMe
- a cap in still other embodiments, comprises a sequence selected from the following sequences: m 7 GpppA2'OM e pApN, m 7 GpppA2'OM e pCpN, m 7 GpppA2'oM e pGpN, m 7 GpppA2'oM e pUpN, m 7 GpppC2'OM e pApN, m 7 GpppC2'OM e pCpN, m 7 GpppC2'OM e pGpN, m 7 GpppC2'OM e pUpN, m 7 GpppG2'OM e pApN, m 7 GpppG2'OM e pCpN, m 7 GpppG2'OM e pGpN, m 7 GpppG2'OM e pCpN, m 7 GpppG2'OM e pGpN,
- a cap in other embodiments, comprises a sequence selected from the following sequences: m’Gs'oMePPpA/oMepAzoMepN, m’Gs'oMePPpA/oMepCzoMepN, m 7 G3'OM e pppA2'OMepG2'OMepN, m 7 G3'OM e pppA2'OMepU2'OMepN, m 7 G3'OMePPpC2'OMepA2'OMepN, m’Gs'OMePPpC/OMepCzOMepN, m 7 G3'OMePPpC2'OMepG2'OMepN, m 7 G3'OM e pppC2'OMepU2'OM e pN, m 7 G3'OMePPpG2'OMepN, m 7 G3'OM e pppC2'OMep
- a cap in still other embodiments, comprises a sequence selected from the following sequences: m’GpppA/oMepAzoMepN, m’GpppA/oMepCzoMepN, m 7 GpppA2'OMepG2'OMepN, m 7 GpppA2'OM e pU2'OM e pN, m 7 GpppC2'OM e pA2'OM e pN, m 7 GpppC2'OMepC2'OMepN, m 7 GpppC2'OM e pG2'OM e pN, m’GpppC/OMepUzOMepN, m 7 GpppG2'OMepA2'OMepN, m 7 GpppG2'OM e pC2'OM e pN, m 7 GpppG2'OM e pG2'OM e
- a cap comprises GGAG. In some embodiments, a cap comprises the following structure:
- the polynucleotides of the present disclosure e.g., a polynucleotide comprising a nucleotide sequence encoding a CFTR polypeptide
- the polynucleotides of the present disclosure further comprise a poly-A tail.
- terminal groups on the poly-A tail can be incorporated for stabilization.
- a poly-A tail comprises des-3' hydroxyl tails.
- a long chain of adenine nucleotides can be added to a polynucleotide such as an mRNA molecule in order to increase stability.
- a polynucleotide such as an mRNA molecule
- the 3' end of the transcript can be cleaved to free a 3' hydroxyl.
- poly-A polymerase adds a chain of adenine nucleotides to the RNA.
- polyadenylation adds a poly-A tail that can be between, for example, approximately 80 to approximately 250 residues long, including approximately 80, 90, 100, 110, 120, 130, 140, 150, 160, 170, 180, 190, 200, 210, 220, 230, 240 or 250 residues long.
- the poly-A tail is 100 nucleotides in length (SEQ ID NO: 195).
- PolyA tails can also be added after the construct is exported from the nucleus.
- terminal groups on the poly A tail can be incorporated for stabilization.
- Polynucleotides of the present invention can include des-3' hydroxyl tails. They can also include structural moieties or 2'-Omethyl modifications as taught by Junjie Li, et al. (Current Biology, Vol. 15, 1501-1507, August 23, 2005, the contents of which are incorporated herein by reference in its entirety).
- the polynucleotides of the present invention can be designed to encode transcripts with alternative polyA tail structures including histone mRNA. According to Norbury, "Terminal uridylation has also been detected on human replicationdependent histone mRNAs. The turnover of these mRNAs is thought to be important for the prevention of potentially toxic histone accumulation following the completion or inhibition of chromosomal DNA replication.
- mRNAs are distinguished by their lack of a 3 ' poly(A) tail, the function of which is instead assumed by a stable stem-loop structure and its cognate stem-loop binding protein (SLBP); the latter carries out the same functions as those of PABP on polyadenylated mRNAs" (Norbury, "Cytoplasmic RNA: a case of the tail wagging the dog," Nature Reviews Molecular Cell Biology; AOP, published online 29 August 2013; doi: 10.1038/nrm3645) the contents of which are incorporated herein by reference in its entirety.
- SLBP stem-loop binding protein
- the length of a poly-A tail when present, is greater than 30 nucleotides in length (SEQ ID NO:215). In another embodiment, the poly-A tail is greater than 35 nucleotides in length (SEQ ID NO:216) (e.g., at least or greater than about 35, 40, 45, 50, 55, 60, 70, 80, 90, 100, 120, 140, 160, 180, 200, 250, 300, 350, 400, 450, 500, 600, 700, 800, 900, 1,000, 1,100, 1,200, 1,300, 1,400, 1,500, 1,600, 1,700, 1,800, 1,900, 2,000, 2,500, and 3,000 nucleotides).
- the polynucleotide or region thereof includes from about 30 to about 3,000 nucleotides (e.g., from 30 to 50, from 30 to 100, from 30 to 250, from 30 to 500, from 30 to 750, from 30 to 1,000, from 30 to 1,500, from 30 to 2,000, from 30 to 2,500, from 50 to 100, from 50 to 250, from 50 to 500, from 50 to 750, from 50 to 1,000, from 50 to 1,500, from 50 to 2,000, from 50 to 2,500, from 50 to 3,000, from 100 to 500, from 100 to 750, from 100 to 1,000, from 100 to 1,500, from 100 to 2,000, from 100 to 2,500, from 100 to 3,000, from 500 to 750, from 500 to 1,000, from 500 to 1,500, from 500 to 2,000, from 500 to 2,500, from 500 to 3,000, from 1,000 to 1,500, from 1,000 to 2,000, from 1,000 to 2,500, from 1,000 to 3,000, from 1,500 to 2,000, from 1,500 to 2,500, from 1,500 to 3,000, from from about 30 to
- the poly-A tail is designed relative to the length of the overall polynucleotide or the length of a particular region of the polynucleotide. This design can be based on the length of a coding region, the length of a particular feature or region or based on the length of the ultimate product expressed from the polynucleotides.
- the poly-A tail can be 10, 20, 30, 40, 50, 60, 70, 80, 90, or 100% greater in length than the polynucleotide or feature thereof.
- the poly-A tail can also be designed as a fraction of the polynucleotides to which it belongs.
- the poly-A tail can be 10, 20, 30, 40, 50, 60, 70, 80, or 90% or more of the total length of the construct, a construct region or the total length of the construct minus the poly-A tail.
- engineered binding sites and conjugation of polynucleotides for Poly-A binding protein can enhance expression.
- multiple distinct polynucleotides can be linked together via the PABP (Poly-A binding protein) through the 3 '-end using modified nucleotides at the 3 '-terminus of the poly-A tail.
- Transfection experiments can be conducted in relevant cell lines at and protein production can be assayed by ELISA at 12hr, 24hr, 48hr, 72hr and day 7 post-transfection.
- the polynucleotides of the present invention are designed to include a polyA-G quartet region.
- the G-quartet is a cyclic hydrogen bonded array of four guanine nucleotides that can be formed by G-rich sequences in both DNA and RNA.
- the G-quartet is incorporated at the end of the poly-A tail.
- the resultant polynucleotide is assayed for stability, protein production and other parameters including half-life at various time points. It has been discovered that the polyA-G quartet results in protein production from an mRNA equivalent to at least 75% of that seen using a poly-A tail of 120 nucleotides alone (SEQ ID NO: 196).
- the polyA tail comprises an alternative nucleoside, e.g., inverted thymidine.
- PolyA tails comprising an alternative nucleoside, e.g., inverted thymidine may be generated as described herein. For instance, mRNA constructs may be modified by ligation to stabilize the poly (A) tail.
- Ligation may be performed using 0.5-1.5 mg/mL mRNA (5' Capl, 3' A100), 50 mM Tris-HCl pH 7.5, 10 mM MgCh, 1 mM TCEP, 1000 units/mL T4 RNA Ligase 1, 1 mM ATP, 20% w/v polyethylene glycol 8000, and 5: 1 molar ratio of modifying oligo to mRNA.
- Modifying oligo has a sequence of 5’-phosphate-AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA- (see below). Ligation reactions are mixed and incubated at room temperature ( ⁇ 22°C) for, e.g., 4 hours.
- Stable tail mRNA are purified by, e.g., dT purification, reverse phase purification, hydroxyapatite purification, ultrafiltration into water, and sterile filtration.
- the resulting stable tail-containing mRNAs contain the following structure at the 3 ’end, starting with the polyA region: Aioo-UCUAGAAAAAAAAAAAAAAAAAA- inverted deoxythymidine (SEQ ID NO:211).
- the polyA tail comprises A100-UCUAG-A20-inverted deoxy-thymidine (SEQ ID NO:211). In some instances, the polyA tail consists of A100-UCUAG-A20-inverted deoxy-thymidine (SEQ ID NO:211). Start codon region
- the invention also includes a polynucleotide that comprises both a start codon region and the polynucleotide described herein (e.g., a polynucleotide comprising a nucleotide sequence encoding a CFTR polypeptide).
- a polynucleotide that comprises both a start codon region and the polynucleotide described herein (e.g., a polynucleotide comprising a nucleotide sequence encoding a CFTR polypeptide).
- the polynucleotides of the present invention can have regions that are analogous to or function like a start codon region.
- the translation of a polynucleotide can initiate on a codon that is not the start codon AUG.
- Translation of the polynucleotide can initiate on an alternative start codon such as, but not limited to, ACG, AGG, AAG, CTG/CUG, GTG/GUG, ATA/AUA, ATT/AUU, TTG/UUG (see Touriol et al. Biology of the Cell 95 (2003) 169-178 and Matsuda and Mauro PLoS ONE, 2010 5 : 11 ; the contents of each of which are herein incorporated by reference in its entirety).
- the translation of a polynucleotide begins on the alternative start codon ACG.
- polynucleotide translation begins on the alternative start codon CTG or CUG.
- the translation of a polynucleotide begins on the alternative start codon GTG or GUG.
- Nucleotides flanking a codon that initiates translation such as, but not limited to, a start codon or an alternative start codon, are known to affect the translation efficiency, the length and/or the structure of the polynucleotide. (See, e.g., Matsuda and Mauro PLoS ONE, 2010 5: 11; the contents of which are herein incorporated by reference in its entirety). Masking any of the nucleotides flanking a codon that initiates translation can be used to alter the position of translation initiation, translation efficiency, length and/or structure of a polynucleotide.
- a masking agent can be used near the start codon or alternative start codon in order to mask or hide the codon to reduce the probability of translation initiation at the masked start codon or alternative start codon.
- masking agents include antisense locked nucleic acids (LNA) polynucleotides and exon-junction complexes (EJCs) (See, e.g., Matsuda and Mauro describing masking agents LNA polynucleotides and EJCs (PLoS ONE, 2010 5: 11); the contents of which are herein incorporated by reference in its entirety).
- a masking agent can be used to mask a start codon of a polynucleotide in order to increase the likelihood that translation will initiate on an alternative start codon.
- a masking agent can be used to mask a first start codon or alternative start codon in order to increase the chance that translation will initiate on a start codon or alternative start codon downstream to the masked start codon or alternative start codon.
- a start codon or alternative start codon can be located within a perfect complement for a miRNA binding site.
- the perfect complement of a miRNA binding site can help control the translation, length and/or structure of the polynucleotide similar to a masking agent.
- the start codon or alternative start codon can be located in the middle of a perfect complement for a miRNA binding site.
- the start codon or alternative start codon can be located after the first nucleotide, second nucleotide, third nucleotide, fourth nucleotide, fifth nucleotide, sixth nucleotide, seventh nucleotide, eighth nucleotide, ninth nucleotide, tenth nucleotide, eleventh nucleotide, twelfth nucleotide, thirteenth nucleotide, fourteenth nucleotide, fifteenth nucleotide, sixteenth nucleotide, seventeenth nucleotide, eighteenth nucleotide, nineteenth nucleotide, twentieth nucleotide or twenty-first nucleotide.
- the start codon of a polynucleotide can be removed from the polynucleotide sequence in order to have the translation of the polynucleotide begin on a codon that is not the start codon.
- Translation of the polynucleotide can begin on the codon following the removed start codon or on a downstream start codon or an alternative start codon.
- the start codon ATG or AUG is removed as the first 3 nucleotides of the polynucleotide sequence in order to have translation initiate on a downstream start codon or alternative start codon.
- the polynucleotide sequence where the start codon was removed can further comprise at least one masking agent for the downstream start codon and/or alternative start codons in order to control or attempt to control the initiation of translation, the length of the polynucleotide and/or the structure of the polynucleotide.
- the invention also includes a polynucleotide that comprises both a stop codon region and the polynucleotide described herein (e.g., a polynucleotide comprising a nucleotide sequence encoding a CFTR polypeptide).
- the polynucleotides of the present invention can include at least two stop codons before the 3' untranslated region (UTR).
- the stop codon can be selected from TGA, TAA and TAG in the case of DNA, or from UGA, UAA and UAG in the case of RNA.
- the polynucleotides of the present invention include the stop codon TGA in the case or DNA, or the stop codon UGA in the case of RNA, and one additional stop codon.
- the addition stop codon can be TAA or UAA.
- the polynucleotides of the present invention include three consecutive stop codons, four stop codons, or more.
- any of the polynucleotides disclosed herein can comprise one, two, three, or all of the following elements: (a) a 5 ’-UTR, e.g., as described herein; (b) a coding region comprising a stop element (e.g., as described herein); (c) a 3 ’-UTR (e.g., as described herein) and; optionally (d) a 3’ stabilizing region, e.g., as described herein. Also disclosed herein are UNP compositions comprising the same.
- a polynucleotide of the disclosure comprises (a) a 5’ UTR described in Table 1 or a variant or fragment thereof and (b) a coding region comprising a stop element provided herein.
- the polynucleotide further comprises a cap structure, e.g., as described herein, or a poly A tail, e.g., as described herein.
- the polynucleotide further comprises a 3’ stabilizing region, e.g., as described herein.
- a polynucleotide of the disclosure comprises (a) a 5’ UTR described in Table 1 or a variant or fragment thereof and (c) a 3’ UTR described in Table 2 or a variant or fragment thereof.
- the polynucleotide further comprises a cap structure, e.g., as described herein, or a poly A tail, e.g., as described herein.
- the polynucleotide further comprises a 3’ stabilizing region, e.g., as described herein.
- a polynucleotide of the disclosure comprises (c) a 3’ UTR described in Table 2 or a variant or fragment thereof and (b) a coding region comprising a stop element provided herein.
- the polynucleotide comprises a sequence provided in Table 3.
- the polynucleotide further comprises a cap structure, e.g., as described herein, or a poly A tail, e.g., as described herein.
- the polynucleotide further comprises a 3’ stabilizing region, e.g. , as described herein.
- a polynucleotide of the disclosure comprises (a) a 5’ UTR described in Table 1 or a variant or fragment thereof; (b) a coding region comprising a stop element provided herein; and (c) a 3’ UTR described in Table 2 or a variant or fragment thereof.
- the polynucleotide further comprises a cap structure, e.g., as described herein, or a poly A tail, e.g., as described herein.
- the polynucleotide further comprises a 3’ stabilizing region, e.g., as described herein.
- a polynucleotide of the present disclosure for example a polynucleotide comprising an mRNA nucleotide sequence encoding a CFTR polypeptide, comprises from 5' to 3' end:
- an ORF encoding a CFTR polypeptide (e.g., SEQ ID NO: 1), wherein the ORF has at least 90%, at least 95%, at least 97%, at least 98%, at least 99%, or 100% sequence identity to the sequence of SEQ ID NO: 10;
- the polynucleotide further comprises a miRNA binding site, e.g., a miRNA binding site that binds to miRNA-142.
- the 5' UTR comprises the miRNA binding site.
- the 3' UTR comprises the miRNA binding site.
- a polynucleotide of the present disclosure comprises a nucleotide sequence encoding a polypeptide sequence at least 70%, at least 80%, at least 81%, at least 82%, at least 83%, at least 84%, at least 85%, at least 86%, at least 87%, at least 88%, at least 89%, at least 90%, at least 91%, at least 92%, at least 93%, at least 94%, at least 95%, at least 96% , at least 97%, at least 98%, at least 99%, or 100% identical to the protein sequence of a human CFTR having the amino acid sequence of SEQ ID NO: 1.
- a polynucleotide of the present disclosure for example a polynucleotide comprising an mRNA nucleotide sequence encoding a polypeptide, comprises (1) a 5' cap such as provided above, for example, m 7 Gp-ppGm-A, (2) a 5' UTR, (3) a nucleotide sequence ORF of SEQ ID NO: 10, (3) a stop codon, (4) a 3 'UTR, and (5) a poly-A tail provided above, for example, a poly-A tail of SEQ ID NO: 195 or A100-UCUAG-A20-inverted deoxy-thymidine (SEQ ID NO:211).
- SEQ ID NO: 11 consists from 5’ to 3’ end: 5' UTR of SEQ ID NO:50, ORF Sequence of SEQ ID NO: 10, and 3' UTR of SEQ ID NO: 141.
- An exemplary CFTR nucleotide construct is SEQ ID NO: 13.
- a polynucleotide of the present disclosure for example a polynucleotide comprising an mRNA nucleotide sequence encoding a CFTR polypeptide, comprises (1) a 5' cap such as provided above, for example, m 7 Gp- ppGm-A, (2) a nucleotide sequence of SEQ ID NO: 11, and (3) a poly-A tail provided above, for example, a poly A tail of -100 residues, e.g., SEQ ID NO: 195 or A100- UCUAG-A20-inverted deoxy-thymidine (SEQ ID NO:211).
- SEQ ID NO: 11 in constructs with SEQ ID NO: 11, all uracils therein are replaced by Nl- methylpseudouracil .
- a polynucleotide of the invention e.g., a polynucleotide comprising a nucleotide sequence encoding a CFTR polypeptide
- a polynucleotide e.g., a RNA, e.g., an mRNA
- IVT in vitro transcription
- a polynucleotide e.g., a RNA, e.g., an mRNA
- a CFTR polypeptide can be constructed by chemical synthesis using an oligonucleotide synthesizer.
- a polynucleotide e.g., a RNA, e.g., an mRNA
- encoding a CFTR polypeptide is made by using a host cell.
- a polynucleotide e.g., a RNA, e.g., an mRNA
- encoding a CFTR polypeptide is made by one or more combination of the IVT, chemical synthesis, host cell expression, or any other methods known in the art.
- Naturally occurring nucleosides, non-naturally occurring nucleosides, or combinations thereof, can totally or partially naturally replace occurring nucleosides present in the candidate nucleotide sequence and can be incorporated into a sequence- optimized nucleotide sequence (e.g., a RNA, e.g., an mRNA) encoding a CFTR polypeptide.
- a sequence- optimized nucleotide sequence e.g., a RNA, e.g., an mRNA
- the resultant polynucleotides, e.g., mRNAs can then be examined for their ability to produce protein and/or produce a therapeutic outcome.
- compositions and formulations that comprise any of the polynucleotides described above.
- the composition or formulation further comprises a delivery agent.
- the composition or formulation can contain a polynucleotide comprising a sequence optimized nucleic acid sequence disclosed herein which encodes a CFTR polypeptide.
- the composition or formulation can contain a polynucleotide (e.g., a RNA, e.g., an mRNA) comprising a polynucleotide (e.g., an ORF) having significant sequence identity to a sequence optimized nucleic acid sequence disclosed herein which encodes a CFTR polypeptide.
- the polynucleotide further comprises a miRNA binding site, e.g., a miRNA binding site that binds miR-126, miR-142, miR-144, miR-146, miR- 150, miR-155, miR-16, miR-21, miR-223, miR-24, miR-27 and miR-26a.
- a miRNA binding site e.g., a miRNA binding site that binds miR-126, miR-142, miR-144, miR-146, miR- 150, miR-155, miR-16, miR-21, miR-223, miR-24, miR-27 and miR-26a.
- Pharmaceutical compositions or formulation can optionally comprise one or more additional active substances, e.g., therapeutically and/or prophylactically active substances.
- Pharmaceutical compositions or formulation of the present invention can be sterile and/or pyrogen -free.
- compositions are administered to humans, human patients or subjects.
- active ingredient generally refers to polynucleotides to be delivered as described herein.
- Formulations and pharmaceutical compositions described herein can be prepared by any method known or hereafter developed in the art of pharmacology.
- such preparatory methods include the step of associating the active ingredient with an excipient and/or one or more other accessory ingredients, and then, if necessary and/or desirable, dividing, shaping and/or packaging the product into a desired single- or multi-dose unit.
- a pharmaceutical composition or formulation in accordance with the present disclosure can be prepared, packaged, and/or sold in bulk, as a single unit dose, and/or as a plurality of single unit doses.
- a "unit dose" refers to a discrete amount of the pharmaceutical composition comprising a predetermined amount of the active ingredient.
- the amount of the active ingredient is generally equal to the dosage of the active ingredient that would be administered to a subject and/or a convenient fraction of such a dosage such as, for example, one-half or one-third of such a dosage.
- Relative amounts of the active ingredient, the pharmaceutically acceptable excipient, and/or any additional ingredients in a pharmaceutical composition in accordance with the present disclosure can vary, depending upon the identity, size, and/or condition of the subject being treated and further depending upon the route by which the composition is to be administered.
- the compositions and formulations described herein can contain at least one polynucleotide of the invention.
- the composition or formulation can contain 1, 2, 3, 4 or 5 polynucleotides of the invention.
- the compositions or formulations described herein can comprise more than one type of polynucleotide.
- the composition or formulation can comprise a polynucleotide in linear and circular form.
- the composition or formulation can comprise a circular polynucleotide and an in vitro transcribed (IVT) polynucleotide.
- the composition or formulation can comprise an IVT polynucleotide, a chimeric polynucleotide and a circular polynucleotide.
- compositions and formulations are principally directed to pharmaceutical compositions and formulations that are suitable for administration to humans, it will be understood by the skilled artisan that such compositions are generally suitable for administration to any other animal, e.g., to non-human animals, e.g. non-human mammals.
- the present invention provides pharmaceutical formulations that comprise a polynucleotide described herein (e.g., a polynucleotide comprising a nucleotide sequence encoding a CFTR polypeptide).
- the polynucleotides described herein can be Formulated using one or more excipients to: (1) increase stability; (2) increase cell transfection; (3) permit the sustained or delayed release (e.g., from a depot formulation of the polynucleotide); (4) alter the biodistribution (e.g., target the polynucleotide to specific tissues or cell types); (5) increase the translation of encoded protein in vivo,' and/or (6) alter the release profile of encoded protein in vivo.
- the pharmaceutical formulation further comprises a delivery agent comprising, e.g., a compound having the Formula (I), e.g., Compound II; or a compound having the Formula (III), (IV), (V), or (VI), e.g., Compound I or VI, or any combination thereof.
- a delivery agent comprising, e.g., a compound having the Formula (I), e.g., Compound II; or a compound having the Formula (III), (IV), (V), or (VI), e.g., Compound I or VI, or any combination thereof.
- the delivery agent comprises an ionizable amino lipid (e.g., Compound II or VI), a helper lipid (e.g., DSPC), a sterol (e.g., Cholesterol), and a PEG lipid (e.g., Compound I or PEG-DMG), e.g., with a mole ratio in the range of about (i) 40-50 mol% ionizable amino lipid (e.g., Compound II, VI, or A), optionally 45-50 mol% ionizable amino lipid, for example, 45-46 mol%, 46-47 mol%, 47-48 mol%, 48-49 mol%, or 49-50 mol% for example about 45 mol%, 45.5 mol%, 46 mol%, 46.5 mol%, 47 mol%, 47.5 mol%, 48 mol%, 48.5 mol%, 49 mol%, or 49.5 mol%; (ii) 30-45 mol%%
- a pharmaceutically acceptable excipient includes, but are not limited to, any and all solvents, dispersion media, or other liquid vehicles, dispersion or suspension aids, diluents, granulating and/or dispersing agents, surface active agents, isotonic agents, thickening or emulsifying agents, preservatives, binders, lubricants or oil, coloring, sweetening or flavoring agents, stabilizers, antioxidants, antimicrobial or antifungal agents, osmolality adjusting agents, pH adjusting agents, buffers, chelants, cyoprotectants, and/or bulking agents, as suited to the particular dosage form desired.
- Exemplary diluents include, but are not limited to, calcium or sodium carbonate, calcium phosphate, calcium hydrogen phosphate, sodium phosphate, lactose, sucrose, cellulose, microcrystalline cellulose, kaolin, mannitol, sorbitol, etc., and/or combinations thereof.
- Exemplary surface active agents and/or emulsifiers include, but are not limited to, natural emulsifiers (e.g., acacia, agar, alginic acid, sodium alginate, tragacanth, chondrux, cholesterol, xanthan, pectin, gelatin, egg yolk, casein, wool fat, cholesterol, wax, and lecithin), sorbitan fatty acid esters (e.g., polyoxyethylene sorbitan monooleate [TWEEN®80], sorbitan monopalmitate [SPAN®40], glyceryl monooleate, polyoxyethylene esters, polyethylene glycol fatty acid esters (e.g., CREMOPHOR®), polyoxyethylene ethers (e.g., polyoxyethylene lauryl ether [BRU®30]), PLUORINC®F 68, POLOXAMER®188, etc. and/or combinations thereof.
- natural emulsifiers e.g., a
- Exemplary binding agents include, but are not limited to, starch, gelatin, sugars (e.g., sucrose, glucose, dextrose, dextrin, molasses, lactose, lactitol, mannitol), amino acids (e.g., glycine), natural and synthetic gums (e.g., acacia, sodium alginate), ethylcellulose, hydroxyethylcellulose, hydroxypropyl methylcellulose, etc., and combinations thereof.
- sugars e.g., sucrose, glucose, dextrose, dextrin, molasses, lactose, lactitol, mannitol
- amino acids e.g., glycine
- natural and synthetic gums e.g., acacia, sodium alginate
- ethylcellulose hydroxyethylcellulose, hydroxypropyl methylcellulose, etc., and combinations thereof.
- Oxidation is a potential degradation pathway for mRNA, especially for liquid mRNA formulations.
- antioxidants can be added to the formulations.
- Exemplary antioxidants include, but are not limited to, alpha tocopherol, ascorbic acid, ascorbyl palmitate, benzyl alcohol, butylated hydroxyanisole, m-cresol, methionine, butylated hydroxytoluene, monothioglycerol, sodium or potassium metabisulfite, propionic acid, propyl gallate, sodium ascorbate, etc., and combinations thereof.
- Exemplary chelating agents include, but are not limited to, ethylenediaminetetraacetic acid (EDTA), citric acid monohydrate, disodium edetate, fumaric acid, malic acid, phosphoric acid, sodium edetate, tartaric acid, trisodium edetate, etc., and combinations thereof.
- EDTA ethylenediaminetetraacetic acid
- citric acid monohydrate disodium edetate
- fumaric acid malic acid
- phosphoric acid sodium edetate
- tartaric acid trisodium edetate, etc.
- antimicrobial or antifungal agents include, but are not limited to, benzalkonium chloride, benzethonium chloride, methyl paraben, ethyl paraben, propyl paraben, butyl paraben, benzoic acid, hydroxybenzoic acid, potassium or sodium benzoate, potassium or sodium sorbate, sodium propionate, sorbic acid, etc., and combinations thereof.
- Exemplary preservatives include, but are not limited to, vitamin A, vitamin C, vitamin E, beta-carotene, citric acid, ascorbic acid, butylated hydroxyanisol, ethylenediamine, sodium lauryl sulfate (SLS), sodium lauryl ether sulfate (SLES), etc., and combinations thereof.
- the pH of polynucleotide solutions is maintained between pH 5 and pH 8 to improve stability.
- exemplary buffers to control pH can include, but are not limited to sodium phosphate, sodium citrate, sodium succinate, histidine (or histidine-HCl), sodium malate, sodium carbonate, etc., and/or combinations thereof.
- Exemplary lubricating agents include, but are not limited to, magnesium stearate, calcium stearate, stearic acid, silica, talc, malt, hydrogenated vegetable oils, polyethylene glycol, sodium benzoate, sodium or magnesium lauryl sulfate, etc., and combinations thereof.
- the pharmaceutical composition or formulation described here can contain a cryoprotectant to stabilize a polynucleotide described herein during freezing.
- cryoprotectants include, but are not limited to mannitol, sucrose, trehalose, lactose, glycerol, dextrose, etc., and combinations thereof.
- the pharmaceutical composition or formulation described here can contain a bulking agent in lyophilized polynucleotide formulations to yield a "pharmaceutically elegant" cake, stabilize the lyophilized polynucleotides during long term (e.g., 36 month) storage.
- exemplary bulking agents of the present invention can include, but are not limited to sucrose, trehalose, mannitol, glycine, lactose, raffinose, and combinations thereof.
- the pharmaceutical composition or formulation further comprises a delivery agent.
- the delivery agent of the present disclosure can include, without limitation, liposomes, lipid nanoparticles, lipidoids, polymers, lipoplexes, microvesicles, exosomes, peptides, proteins, cells transfected with polynucleotides, hyaluronidase, nanoparticle mimics, nanotubes, conjugates, and combinations thereof.
- the present disclosure provides pharmaceutical compositions with advantageous properties.
- the lipid compositions described herein may be advantageously used in lipid nanoparticle compositions for the delivery of therapeutic and/or prophylactic agents, e.g., mRNAs, to mammalian cells or organs.
- the lipids described herein have little or no immunogenicity.
- the lipid compounds disclosed herein have a lower immunogenicity as compared to a reference lipid (e.g., MC3, KC2, or DLinDMA).
- a formulation comprising a lipid disclosed herein and a therapeutic or prophylactic agent, e.g., mRNA, has an increased therapeutic index as compared to a corresponding formulation which comprises a reference lipid (e.g., MC3, KC2, or DLinDMA) and the same therapeutic or prophylactic agent.
- a reference lipid e.g., MC3, KC2, or DLinDMA
- compositions comprising:
- nucleic acids of the invention are Formulated in a lipid nanoparticle (LNP).
- LNP lipid nanoparticle
- Lipid nanoparticles typically comprise ionizable cationic lipid, non-cationic lipid, sterol and PEG lipid components along with the nucleic acid cargo of interest.
- the lipid nanoparticles of the invention can be generated using components, compositions, and methods as are generally known in the art, see for example PCT/US2016/052352; PCT/US2016/068300; PCT/US2017/037551; PCT/US2015/027400; PCT/US2016/047406;
- PCT/US2016/52117; PCT/US2012/069610; PCT/US2017/027492; PCT/US2016/059575 and PCT/US2016/069491 all of which are incorporated by reference herein in their entirety.
- Nucleic acids of the present disclosure are typically Formulated in lipid nanoparticle.
- the lipid nanoparticle comprises at least one ionizable cationic lipid, at least one non-cationic lipid, at least one sterol, and/or at least one polyethylene glycol (PEG)-modified lipid.
- PEG polyethylene glycol
- the lipid nanoparticle comprises a molar ratio of 20- 60% ionizable cationic lipid.
- the lipid nanoparticle may comprise a molar ratio of 20-50%, 20-40%, 20-30%, 30-60%, 30-50%, 30-40%, 40-60%, 40- 50%, or 50-60% ionizable cationic lipid.
- the lipid nanoparticle comprises a molar ratio of 20%, 30%, 40%, 50, or 60% ionizable cationic lipid.
- the lipid nanoparticle comprises a molar ratio of 5-25% non-cationic lipid.
- the lipid nanoparticle may comprise a molar ratio of 5-20%, 5-15%, 5-10%, 10-25%, 10-20%, 10-25%, 15-25%, 15-20%, or 20-25% noncationic lipid.
- the lipid nanoparticle comprises a molar ratio of 5%, 10%, 15%, 20%, or 25% non-cationic lipid.
- the lipid nanoparticle comprises a molar ratio of 25- 55% sterol.
- the lipid nanoparticle may comprise a molar ratio of 25- 50%, 25-45%, 25-40%, 25-35%, 25-30%, 30-55%, 30-50%, 30-45%, 30-40%, 30- 35%, 35-55%, 35-50%, 35-45%, 35-40%, 40-55%, 40-50%, 40-45%, 45-55%, 45- 50%, or 50-55% sterol.
- the lipid nanoparticle comprises a molar ratio of 25%, 30%, 35%, 40%, 45%, 50%, or 55% sterol.
- the lipid nanoparticle comprises a molar ratio of 0.5- 15% PEG-modified lipid.
- the lipid nanoparticle may comprise a molar ratio of 0.5-10%, 0.5-5%, 1-15%, 1-10%, 1-5%, 2-15%, 2-10%, 2-5%, 5-15%, 5-10%, or 10-15%.
- the lipid nanoparticle comprises a molar ratio of 0.5%, 1%, 2%, 3%, 4%, 5%, 6%, 7%, 8%, 9%, 10%, 11%, 12%, 13%, 14%, or 15% PEG-modified lipid.
- the lipid nanoparticle core comprises a molar ratio of 20-60% ionizable cationic lipid, 5-25% non-cationic lipid, 25-55% sterol, and 0.5- 15% PEG-modified lipid. In some embodiments, the lipid nanoparticle core comprises a molar ratio of 40-60% ionizable cationic lipid, 5-15% non-cationic lipid, 30-50% sterol, and 0.5-10% PEG-modified lipid.
- the lipid nanoparticle core comprises a molar ratio of 45-55% ionizable cationic lipid, 7.5- 12.5% non-cationic lipid, 35-45% sterol, and 0.5-5% PEG-modified lipid.
- the LNP provided herein comprises lipid nanoparticle core, a polynucleotide (e.g., CFTR mRNA) encapsulated within the core for delivery into a cell, and a cationic agent disposed primarily on the outer surface of the core.
- LNP with cationic agent disposed primarily on the outer surface of the core can have improved accumulation of the LNP in cells such as human bronchial epithelial (HBE) and also improved function of the polynucleotide, e.g., as measured mRNA expression in cells, e.g., airway epithelial cells.
- HBE human bronchial epithelial
- the cationic agent can comprise any aqueous soluble molecule or substance that has a net positive charge at physiologic pH and can adhere to the surface of a lipid nanoparticle core. Such agent may also be lipid soluble, but will also be soluble in aqueous solution. Generally speaking, the cationic agent features a net positive charge at physiologic pH because it contains one or more basic functional groups that is protonated at physiologic pH in aqueous media.
- the cationic agent can contain one or more amine groups, e.g. primary, secondary, or tertiary amines each having a pKa of 8.0 or greater. The pKa can be greater than about 9.
- the cationic agent can be a cationic lipid which is a water-soluble, amphiphilic molecule in which one portion of the molecule is hydrophobic comprising, for example, a lipid moiety, and where the other portion of the molecule is hydrophilic, containing one or more functional groups which is typically charged at physiologic pH.
- the hydrophobic portion comprising the lipid moiety, can serve to anchor the cationic agent to a lipid nanoparticle core.
- the hydrophilic portion can serve to increase the charge on the surface of a lipid nanoparticle core.
- the cationic agent can have a solubility of greater than about 1 mg/mL in alcohol.
- the solubility in alcohol can be greater than about 5 mg/mL.
- the solubility in alcohol can be greater than about 10 mg/mL.
- the solubility in alcohol can be greater than about 20 mg/mL in alcohol.
- the alcohol can be C 1-6 alcohol such as ethanol.
- the lipid portion of the molecule can be, for example, a structural lipid, fatty acid, or similar hydrocarbyl group.
- the structural lipid can be selected from, but is not limited to, a steroid, diterpeniod, triterpenoid, cholestane, ursolic acid, or derivatives thereof.
- the structural lipid is a steroid selected from, but not limited to, cholesterol or a phystosterol. In some embodiments, the structural lipid is an analog of cholesterol. In some embodiments, the structural lipid is a sitosterol, campesterol, or stigmasterol. In some embodiments, the structural lipid is an analog of sitosterol, campesterol, or stigmasterol.
- the fatty acid comprises 1 to 4 C6-20 hydrocarbon chains.
- the fatty acid can be fully saturated or can contain 1 to 7 double bonds.
- the fatty acid can contain 1 to 5 heteroatoms either along the main chain or pendent to the main chain.
- the fatty acid comprises two Cio-18 hydrocarbon chains. In some embodiments, the fatty acid comprises two Cio-18 saturated hydrocarbon chains. In some embodiments, the fatty acid comprises two Ci6 saturated hydrocarbon chain. In some embodiments, the fatty acid comprises two C14 saturated hydrocarbon chain. In some embodiments, the fatty acid comprises two unsaturated Cio-18 hydrocarbon chains. In some embodiments, the fatty acid comprises two C16-18 hydrocarbon chains, each with one double bond. In some embodiments, the fatty acid comprises three C 8 -18 saturated hydrocarbon chains.
- the hydrocarbyl group consists of 1 to 4 C6-20 alkyl, alkenyl, or alkynyl chains or 3 to 10 membered cycloalkyl, cycloalkenyl, or cycloalkynyl groups.
- the hydrocarbyl chain is a C 8 -io alkyl. In some embodiments, the hydrocarbyl chain is C 8 -io alkenyl.
- the hydrophilic portion can comprise 1 to 5 functional groups that would be charged at physiologic pH, 7.3 to 7.4.
- the hydrophilic group can comprise a basic functional group that would be protonated and positively charged at physiologic pH. At least one of the basic functional groups has a pKa of 8 or greater.
- the hydrophilic portion comprises an amine group.
- the amine group can comprise one to four primary, secondary, or tertiary amines and mixtures thereof.
- the amine can be contained in a three to eight membered heteroalkyl or heteroaryl ring.
- the amine group comprises one or two terminal primary amines. In some embodiments, the amine group comprises one or two terminal primary amines and one internal secondary amine. In some embodiments, the amine group comprises one or two tertiary amine. In some embodiments, the tertiary amine is (CH 3 ) 2 N-. In some embodiments, amine group comprises one to two terminal (CH 3 ) 2 N-.
- the hydrophilic portion can comprise a phosphonium group.
- the counter ion of the phosphonium ion consists of an anion with a charge of one.
- the counter ion is a halo, hydrogen sulfate, nitrite, chlorate, or hydrogen carbonate. In some embodiments, the counter ion is a bromide.
- the cationic agent is a cationic lipid which is a sterol amine.
- a sterol amine has, for its hydrophobic portion, a sterol, and for its hydrophilic portion, an amine group.
- the sterol group is selected from, but not limited to, cholesterol, sitosterol, campesterol, stigmasterol or derivatives thereof.
- the amine group can comprise one to five primary, secondary, tertiary amines, or mixtures thereof. At least one of the amines has a pKa of 8 or greater and is charged at physiological pH.
- the amine can be contained in a three to eight membered heteroalkyl or heteroaryl ring.
- the amine group of the sterol amine comprises one or two terminal primary amines. In some embodiments, the amine group comprises one or two terminal primary amines and one internal secondary amine. In some embodiments, the amine group comprises one or two tertiary amine. In some embodiments, the tertiary amine is (C’H 3 ) 2 N-. In some embodiments, amine group comprises one to two terminal (CH 3 ) 2 N-.
- Sterol amines useful in the nanoparticles of the invention include molecules having Formula (Al):
- A is an amine group
- L is an optional linker
- B is a sterol.
- the amine group is an alkyl (e.g., C1-14 alkyl, C1-12 alkyl, C 1 -10 alkyl, etc.), 3 to 8 membered heterocycloalkyl, 5 to 6 membered heteroaryl, C1-6 alkyl-(3 to 8 membered heterocycloalkyl), or C1-6 alkyl-(5 to 6 membered heteroaryl), wherein the alkyl, 3 to 8 membered heterocycloalkyl, 5 to 6 membered heteroaryl, C1-6 alkyl-(3 to 8 membered heterocycloalkyl), and C1-6 alkyl- (5 to 6 membered heteroaryl) comprises one to five primary, secondary, or tertiary amines or combination thereof, wherein the alkyl, 3 to 8 membered heterocycloalkyl, 5 to 6 membered heteroaryl, C1-6 alkyl-(3
- the sterol group is a cholesterol, sitosterol, campesterol, stigmasterol or derivatives thereof.
- the sterol amine has Formula A2: or a salt thereof, wherein:
- R 1 is C 1 -14 alkyl or C1-14 alkenyl
- the sterol amine has Formula A3: or a salt thereof, wherein:
- — is a single or double bond
- R 2 is H or C1-6 alkyl
- Y 2 is selected from:
- the sterol amine has Formula A4: or a salt thereof, wherein:
- Z 1 is OH or C 3-6 alkyl;
- Y 1 is C 1-10 alkyl, 3 to 8 membered heterocycloalkyl, 5 to 6 membered heteroaryl, C 1-6 alkyl-(3 to 8 membered heterocycloalkyl), or C 1-6 alkyl-(5 to 6 membered heteroaryl), wherein the alkyl, 3 to 8 membered heterocycloalkyl, 5 to 6 membered heteroaryl, C 1-6 alkyl-(3 to 8 membered heterocycloalkyl), and C 1-6 alkyl-(5 to 6 membered heteroaryl) comprises one to five primary, secondary, or tertiary amines or combination thereof, wherein the alkyl, 3 to 8 membered heterocycloalkyl, 5 to 6 membered heteroaryl, C 1-6 alkyl-(3 to 8 membered heterocycloalkyl), and C 1-6 alkyl-(5 to 6 membered heteroaryl) are each optionally substituted with 1, 2, 3, or 4 substituents selected from C
- the sterol amine has Formula A5: or a salt thereof, wherein:
- Z 2 is OH or isopropyl
- L 1 is -CH 2 -NH-C(O)-, -C(O)NH-, or -C(O)O-.
- the sterol amine is selected from:
- the sterol amine is SA3 : salt thereof, which is also referred to as GL-67.
- SA3 or GL-67 can be prepared according to known processes in the art or purchased from a commercial vendor such as Avanti® Polar Lipids, Inc. (SKU 890893).
- the cationic lipid is a modified amino acid, such as a modified arginine, in which an amino acid residue having an amine-containing side chain is appended to a hydrophobic group such as a sterol (e.g., cholesterol or derivative thereof), fatty acid, or similar hydrocarbyl group.
- a hydrophobic group such as a sterol (e.g., cholesterol or derivative thereof), fatty acid, or similar hydrocarbyl group.
- At least one amine of the modified amino acid portion has a pKa of 8.0 or greater.
- At least one amine of the modified amino acid portion is positively charger at physiological pH.
- the amino acid residue can include but is not limited to arginine, histidine, lysine, tryptophan, ornithine, and 5 -hydroxy lysine.
- the amino acid is bonded to the hydrophobic group through a linker.
- the modified amino acid is a modified arginine.
- the cationic agent is a non-lipid cationic agent.
- non-lipid cationic agent include e.g., benzalkonium chloride, cetylpyridium chloride, L-lysine monohydrate, or tromethamine.
- the lipid nanoparticle comprises a cationic agent (e.g., a sterol amine) at a molar ratio of 2-15%, 3-10%, 4-10%, 5-10%, 6-10%, 2-3%, 2-4%, 2-5%, 2-6%, 2-7%, 2-8%, 3-4%, 3-5%, 3-6%, 3-7%, 3-8%, 4-5%, 4-6%, 4-7%, 4-8%, 5-6%, 5-7%, 5-8%, 6-7%, 6-8%, 2%, 2.5%, 3%, 3.5%, 4%, 4.5%, 5%, 5.5%, 6%, 6.5%, 7%, 7.5%, 8%, 8.5%, 9%, 9.5%, 10%, at least 2%, at least 3%, at least 4%, at least 5%, at least 6%, at least 7%, at least 8%, less than 15%, less than 14%, less than 13%, less than 12%, less than 11%, or less than 10%.
- a cationic agent e.g., a sterol amine
- the lipid nanoparticle comprises a molar ratio of 20-60% ionizable cationic lipid, 5-25% noncationic lipid, 25-55% sterol, 0.5-15% PEG-modified lipid, and 2-10% cationic agent (e.g., a sterol amine). In some embodiments, the lipid nanoparticle comprises a molar ratio of 40-60% ionizable cationic lipid, 5-15% non-cationic lipid, 30-50% sterol, 0.5- 10% PEG-modified lipid, and 3-7% cationic agent.
- the lipid nanoparticle comprises a molar ratio of 45-55% ionizable cationic lipid, 7.5-12.5% non-cationic lipid, 35-45% sterol, 0.5-5% PEG-modified lipid, and 4.5-6% cationic agent.
- the cationic agent is GL-67 or a salt thereof.
- a weight ratio of the cationic agent to polynucleotide is about 0.1 : 1 to about 15: 1. In some embodiments, a weight ratio of the cationic agent to polynucleotide is about 0.2: 1 to about 10: 1. In some embodiments, a weight ratio of the cationic agent to polynucleotide is about 1 : 1 to about 10: 1. In some embodiments, a weight ratio of the cationic agent to polynucleotide is about 1 : 1 to about 8: 1. In some embodiments, a weight ratio of the cationic agent to polynucleotide is about 1 : 1 to about 7: 1.
- a weight ratio of the cationic agent to polynucleotide is about 1 : 1 to about 6: 1. In some embodiments, a weight ratio of the cationic agent to polynucleotide is about 1 : 1 to about 5: 1. In some embodiments, a weight ratio of the cationic agent to polynucleotide is about 1 : 1 to about 4: 1. In some embodiments, a weight ratio of the cationic agent to polynucleotide is about 1.25: 1 to about 3.75 : 1. In some embodiments, a weight ratio of the cationic agent to polynucleotide is about 1.25 : 1. In some embodiments, a weight ratio of the cationic agent to polynucleotide is about 2.5 : 1. In some embodiments, a weight ratio of the cationic agent to polynucleotide is about 3.75: 1.
- a molar ratio of the cationic agent to polynucleotide is about 0.1 : 1 to about 20: 1. In some embodiments, a molar ratio of the cationic agent to polynucleotide is about 1.5: 1 to about 10: 1. In some embodiments, a molar ratio of the cationic agent to polynucleotide is about 1.5 : 1 to about 9: 1. In some embodiments, a molar ratio of the cationic agent to polynucleotide is about 1.5: 1 to about 8: 1. In some embodiments, a molar ratio of the cationic agent to polynucleotide is about 1.5 : 1 to about 7: 1.
- a molar ratio of the cationic agent to polynucleotide is about 1.5 : 1 to about 6: 1. In some embodiments, a molar ratio of the cationic agent to polynucleotide is about 1.5: 1 to about 5: 1. In some embodiments, a molar ratio of the cationic agent to polynucleotide is about 1.5 : 1. In some embodiments, a molar ratio of the cationic agent to polynucleotide is about 2: 1. In some embodiments, a molar ratio of the cationic agent to polynucleotide is about 3: 1. In some embodiments, a molar ratio of the cationic agent to polynucleotide is about 4: 1. In some embodiments, a molar ratio of the cationic agent to polynucleotide is about 5: 1.
- the nanoparticle has a zeta potential of about 5 mV to about 20 mV. In some embodiments, the nanoparticle has a zeta potential of about 5 mV to about 20 mV. In some embodiments, the nanoparticle has a zeta potential of about 5 mV to about 15 mV. In some embodiments, the nanoparticle has a zeta potential of about 5 mV to about 10 mV.
- the lipid nanoparticle core has a neutral charge at a neutral pH.
- greater than about 80% of the cationic agent is on the surface on the nanoparticle. In some embodiments, greater than about 90% of the cationic agent is on the surface on the nanoparticle. In some embodiments, greater than about 95% of the cationic agent is on the surface on the nanoparticle.
- the ionizable lipids of the present disclosure may be one or more of compounds of Formula (I): or their N-oxides, or salts or isomers thereof, wherein:
- Ri is selected from the group consisting of C5-30 alkyl, C5-20 alkenyl, -R*YR”, -YR”, and -R ’M’R’;
- R.2 and R3 are independently selected from the group consisting of H, C 1 -14 alkyl, C2-14 alkenyl, -R*YR”, -YR”, and -R*OR”, or R2 and R3, together with the atom to which they are attached, form a heterocycle or carbocycle;
- R4 is selected from the group consisting of hydrogen, a C 3-6 carbocycle, -(CH2) n Q, -(CH2) n CHQR
- -CHQR -CQ(R)2, and unsubstituted C1-6 alkyl, where Q is selected from a carbocycle, heterocycle, -OR, -O(CH2) n N(R)2, -C(O)OR, - OC(O)R -CX 3 , -CX 2 H, -CXH 2 , -CN,
- M and M’ are independently selected from -C(O)O-, -OC(O)-, -OC(O)-M”-C(O)O-, -C(O)N(R’)-,
- R7 is selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H;
- R 8 is selected from the group consisting of C 3-6 carbocycle and heterocycle;
- R 9 is selected from the group consisting of H, CN, NO2, C1-6 alkyl, -
- each R is independently selected from the group consisting of C 1-3 alkyl, C 2-3 alkenyl, and H; each R’ is independently selected from the group consisting of C 1-18 alkyl, C 2-18 alkenyl, -R*YR”, -YR”, and H; each R” is independently selected from the group consisting of C 3-15 alkyl and C 3-15 alkenyl; each R* is independently selected from the group consisting of C 1-12 alkyl and
- m is 5, 7, or 9.
- Q is OH, -NHC(S)N(R)2, or -NHC(0)N(R)2.
- Q is -N(R)C(O)R, or -N(R)S(0)2R.
- a subset of compounds of Formula (I) includes those of Formula (IB), or its N-oxide, or a salt or isomer thereof in which all variables are as defined herein.
- m is selected from 5, 6, 7, 8, and 9;
- R4 is hydrogen, unsubstituted C1-3 alkyl, or -(CH2) n Q, in which Q is
- M and M’ are independently selected from -C(O)O-, -OC(O)-, -OC(O)-M”-C(O)O-, -C(O)N(R’)-, -P(O)(OR’)O-, -S -S-, an aryl group, and a heteroaryl group; and R2 and R3 are independently selected from the group consisting of H, C1-14 alkyl, and C2-14 alkenyl.
- m is 5, 7, or 9.
- Q is OH, -NHC(S)N(R)2, or -NHC(0)N(R)2.
- Q is -N(R)C(O)R, or -N(R)S(0)2R.
- a subset of compounds of Formula (I) includes those of Formula (II): salt or isomer thereof, wherein 1 is selected from 1, 2, 3, 4, and 5; Mi is a bond or M’; R4 is hydrogen, unsubstituted C1-3 alkyl, or -(CH2) n Q, in which n is 2, 3, or 4, and Q is
- M and M’ are independently selected from -C(O)O-, -OC(O)-, -OC(O)-M”-C(O)O-, -C(O)N(R’)-, -P(O)(OR’)O-, -S -S-, an aryl group, and a heteroaryl group; and R 2 and R3 are independently selected from the group consisting of H, C1-14 alkyl, and C2-14 alken
- the compounds of Formula (I) are of Formula (lib), or their N-oxides, or salts or isomers thereof, wherein R4 is as described herein.
- the compounds of Formula (I) are of Formula (lie) or (lie):
- the compounds of Formula (I) are of Formula (Ilf): (Ilf) or their N-oxides, or salts or isomers thereof, wherein M is -C(O)O- or -OC(O)-, M” is C 1-6 alkyl or C2-6 alkenyl, R2 and R3 are independently selected from the group consisting of C5-14 alkyl and
- n is selected from 2, 3, and 4.
- the compounds of Formula (I) are of Formula (lid), (lid), or their N-oxides, or salts or isomers thereof, wherein n is 2, 3, or 4; and m, R’, R”, and R2 through R$ are as described herein.
- each of R2 and R3 may be independently selected from the group consisting of C5-14 alkyl and C5-14 alkenyl.
- the compounds of Formula (I) are of Formula (Ilg), , , , , , -S-S-, an aryl group, and a heteroaryl group; and R2 and R3 are independently selected from the group consisting of H, C1-14 alkyl, and C2-14 alkenyl.
- M is C1-6 alkyl (e.g., C1-4 alkyl) or C2-6 alkenyl (e.g. C2-4 alkenyl).
- R2 and R3 are independently selected from the group consisting of C5-14 alkyl and C5-14 alkenyl.
- the ionizable lipids are one or more of the compounds described in U.S. Application Nos. 62/220,091, 62/252,316, 62/253,433, 62/266,460, 62/333,557, 62/382,740, 62/393,940, 62/471,937, 62/471,949, 62/475,140, and
- the ionizable lipids are selected from Compounds 1- 280 described in U.S. Application No. 62/475,166.
- the ionizable lipid is N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N
- the ionizable lipid is In some embodiments, the ionizable lipid is salt thereof.
- the ionizable lipid is salt thereof.
- a lipid may have a positive or partial positive charge at physiological pH.
- Such lipids may be referred to as cationic or ionizable (amino)lipids.
- Lipids may also be zwitterionic, i.e., neutral molecules having both a positive and a negative charge.
- the ionizable lipids of the present disclosure may be one or more of compounds of formula (III), or salts or isomers thereof, wherein ring t is 1 or 2;
- Ai and A2 are each independently selected from CH or N;
- Z is CH2 or absent wherein when Z is CH2, the dashed lines (1) and (2) each represent a single bond; and when Z is absent, the dashed lines (1) and (2) are both absent;
- Ri, R2, R3, R4, and Rs are independently selected from the group consisting of C5-20 alkyl, C5-20 alkenyl, -R”MR’, -R*YR”, -YR”, and -R*OR”;
- Rxi and Rx2 are each independently H or C1-3 alkyl; each M is independently selected from the group consisting of -C(O)O-, -OC(O)-, -OC(O)O-, -C(O)N(R’)-, -N(R’)C(O)-, -C(O)-, -C(S)-, - C(S)S-, -SC(S)-,
- M* is C 1 -C 6 alkyl
- W 1 and W 2 are each independently selected from the group consisting of -O- and -N(Re)-; each R5 is independently selected from the group consisting of H and C1-5 alkyl;
- X 1 , X 2 , and X 3 are independently selected from the group consisting of a bond, -CH2-,
- the compound is of any of formulae (IIIal)-(IIIa8):
- the ionizable lipids are one or more of the compounds described in U.S. Application Nos. 62/271,146, 62/338,474, 62/413,345, and 62/519,826, and PCT Application No. PCT/US2016/068300.
- the ionizable lipids are selected from Compounds 1- 156 described in U.S. Application No. 62/519,826.
- the ionizable lipids are selected from Compounds 1-16, 42-66, 68-76, and 78-156 described in U.S. Application No. 62/519,826.
- the ionizable lipid is (Compound
- the ionizable lipid is N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N
- the central amine moiety of a lipid according to Formula (III), (Illal ), (IIIa2), (IIIa3), (IIIa4), (IIIa5), (IIIa6), (IIIa7), or (IIIa8) may be protonated at a physiological pH.
- a lipid may have a positive or partial positive charge at physiological pH.
- Such lipids may be referred to as cationic or ionizable (amino)lipids.
- Lipids may also be zwitterionic, i.e., neutral molecules having both a positive and a negative charge.
- the lipid composition of the lipid nanoparticle composition disclosed herein can comprise one or more phospholipids, for example, one or more saturated or (poly)unsaturated phospholipids or a combination thereof.
- phospholipids comprise a phospholipid moiety and one or more fatty acid moieties.
- a phospholipid moiety can be selected, for example, from the non-limiting group consisting of phosphatidyl choline, phosphatidyl ethanolamine, phosphatidyl glycerol, phosphatidyl serine, phosphatidic acid, 2-lysophosphatidyl choline, and a sphingomyelin.
- a fatty acid moiety can be selected, for example, from the non-limiting group consisting of lauric acid, myristic acid, myristoleic acid, palmitic acid, palmitoleic acid, stearic acid, oleic acid, linoleic acid, alpha-linolenic acid, erucic acid, phytanoic acid, arachidic acid, arachidonic acid, eicosapentaenoic acid, behenic acid, docosapentaenoic acid, and docosahexaenoic acid.
- Particular phospholipids can facilitate fusion to a membrane.
- a cationic phospholipid can interact with one or more negatively charged phospholipids of a membrane (e.g., a cellular or intracellular membrane). Fusion of a phospholipid to a membrane can allow one or more elements (e.g., a therapeutic agent) of a lipid- containing composition (e.g., LNPs) to pass through the membrane permitting, e.g., delivery of the one or more elements to a target tissue.
- a membrane e.g., a cellular or intracellular membrane.
- Non-natural phospholipid species including natural species with modifications and substitutions including branching, oxidation, cyclization, and alkynes are also contemplated.
- a phospholipid can be functionalized with or cross-linked to one or more alkynes (e.g., an alkenyl group in which one or more double bonds is replaced with a triple bond).
- an alkyne group can undergo a copper-catalyzed cycloaddition upon exposure to an azide.
- Such reactions can be useful in functionalizing a lipid bilayer of a nanoparticle composition to facilitate membrane permeation or cellular recognition or in conjugating a nanoparticle composition to a useful component such as a targeting or imaging moiety (e.g., a dye).
- Phospholipids include, but are not limited to, glycerophospholipids such as phosphatidylcholines, phosphatidylethanolamines, phosphatidylserines, phosphatidylinositols, phosphatidy glycerols, and phosphatidic acids. Phospholipids also include phosphosphingolipid, such as sphingomyelin.
- a phospholipid of the invention comprises 1,2- distearoyl-sn-glycero-3-phosphocholine (DSPC), l,2-dioleoyl-sn-glycero-3- phosphoethanolamine (DOPE), l,2-dilinoleoyl-sn-glycero-3 -phosphocholine (DLPC), 1,2-dimyristoyl-sn-gly cero-phosphocholine (DMPC), l,2-dioleoyl-sn-glycero-3- phosphocholine (DOPC), l,2-dipalmitoyl-sn-glycero-3 -phosphocholine (DPPC), 1,2- diundecanoyl-sn-glycero-phosphocholine (DUPC), 1 -palmitoyl -2 -oleoyl-sn-glycero- 3 -phosphocholine (POPC), l,2-di-O-octade
- DOPE 1,
- DOPG 1.2-dioleoyl-sn-glycero-3-phospho-rac-(l -glycerol) sodium salt
- DOPG 1.2-dioleoyl-sn-glycero-3-phospho-rac-(l -glycerol) sodium salt
- a phospholipid useful or potentially useful in the present invention is an analog or variant of DSPC. In certain embodiments, a phospholipid useful or potentially useful in the present invention is a compound of Formula (IV):
- each R 1 is independently optionally substituted alkyl; or optionally two R 1 are joined together with the intervening atoms to form optionally substituted monocyclic carbocyclyl or optionally substituted monocyclic heterocyclyl; or optionally three R 1 are joined together with the intervening atoms to form optionally substituted bicyclic carbocyclyl or optionally substitute bicyclic heterocyclyl; n is 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10; m is 0, 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10;
- each instance of L 2 is independently a bond or optionally substituted C 1-6 alkylene, wherein one methylene unit of the optionally substituted C 1-6 alkylene is optionally replaced with O, N(R N ), S, C(O), C(O)N(R N ), - NR N C(O), C(O)O, OC(O), OC(O)O, OC(O)N(R N ), NR N C(O)O, or - NR N C(O)N(R N ); each instance of R 2 is independently optionally substituted C1-30 alkyl, optionally substituted C1-30 alkenyl, or optionally substituted C1-30 alkynyl; optionally wherein one or more methylene units of R 2 are independently replaced with optionally substituted carbocyclylene, optionally substituted heterocyclylene, optionally substituted arylene, optionally substituted heteroarylene, N(R N ), O, S, C(O), C(O)N
- Ring B is optionally substituted carbocyclyl, optionally substituted heterocyclyl, optionally substituted aryl, or optionally substituted heteroaryl; and p is 1 or 2; provided that the compound is not of the formula: wherein each instance of R 2 is independently unsubstituted alkyl, unsubstituted alkenyl, or unsubstituted alkynyl.
- the phospholipids may be one or more of the phospholipids described in U.S. Application No. 62/520,530. i) Phospholipid Head Modifications
- a phospholipid useful or potentially useful in the present invention comprises a modified phospholipid head (e.g., a modified choline group).
- a phospholipid with a modified head is DSPC, or analog thereof, with a modified quaternary amine.
- at least one of R 1 is not methyl.
- at least one of R 1 is not hydrogen or methyl.
- the compound of Formula (IV) is of one of the following formulae: or a salt thereof, wherein: each t is independently 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10; each u is independently 0, 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10; and each v is independently 1, 2, or 3.
- a compound of Formula (IV) is of Formula
- a phospholipid useful or potentially useful in the present invention comprises a cyclic moiety in place of the glyceride moiety.
- a phospholipid useful in the present invention is DSPC, or analog thereof, with a cyclic moiety in place of the glyceride moiety.
- the compound of Formula (IV) is of Formula (IV-b):
- a phospholipid useful or potentially useful in the present invention comprises a modified tail.
- a phospholipid useful or potentially useful in the present invention is DSPC, or analog thereof, with a modified tail.
- a “modified tail” may be a tail with shorter or longer aliphatic chains, aliphatic chains with branching introduced, aliphatic chains with substituents introduced, aliphatic chains wherein one or more methylenes are replaced by cyclic or heteroatom groups, or any combination thereof.
- a phospholipid useful or potentially useful in the present invention comprises a modified phosphocholine moiety, wherein the alkyl chain linking the quaternary amine to the phosphoryl group is not ethylene (e.g., n is not 2). Therefore, in certain embodiments, a phospholipid useful or potentially useful in the present invention is a compound of Formula (IV), wherein n is 1, 3, 4, 5, 6, 7, 8, 9, or 10. For example, in certain embodiments, a compound of Formula (IV) is of one of the following formulae: or a salt thereof.
- a phospholipid useful or potentially useful in the present invention comprises a modified phosphocholine moiety, wherein the alkyl chain linking the quaternary amine to the phosphoryl group is not ethylene (e.g., n is not 2). Therefore, in certain embodiments, a phospholipid useful.
- an alternative lipid is used in place of a phospholipid of the present disclosure.
- an alternative lipid of the invention is oleic acid.
- the alternative lipid is one of the following:
- the lipid composition of a pharmaceutical composition disclosed herein can comprise one or more structural lipids.
- structural lipid refers to sterols and also to lipids containing sterol moieties.
- Structural lipids can be selected from the group including but not limited to, cholesterol, fecosterol, sitosterol, ergosterol, campesterol, stigmasterol, brassicasterol, tomatidine, tomatine, ursolic acid, alphatocopherol, hopanoids, phytosterols, steroids, and mixtures thereof.
- the structural lipid is a sterol.
- sterols are a subgroup of steroids consisting of steroid alcohols.
- the structural lipid is a steroid.
- the structural lipid is cholesterol.
- the structural lipid is an analog of cholesterol.
- the structural lipid is alpha-tocopherol.
- the structural lipids may be one or more of the structural lipids described in U.S. Application No. 62 /520,530.
- the lipid composition of a pharmaceutical composition disclosed herein can comprise one or more a polyethylene glycol (PEG) lipid.
- PEG polyethylene glycol
- PEG-lipid refers to polyethylene glycol (PEG)- modified lipids.
- PEG-lipids include PEG-modified phosphatidylethanolamine and phosphatidic acid, PEG-ceramide conjugates (e.g., PEG-CerC14 or PEG-CerC20), PEG-modified dialkylamines and PEG-modified 1,2- diacyloxypropan-3 -amines.
- PEGylated lipids PEGylated lipids.
- a PEG lipid can be PEG-c-DOMG, PEG-DMG, PEG-DLPE, PEG-DMPE, PEG-DPPC, or a PEG-DSPE lipid.
- the PEG-lipid includes, but not limited to 1,2- dimyristoyl-sn-glycerol methoxypolyethylene glycol (PEG-DMG), 1,2-distearoyl-sn- glycero-3-phosphoethanolamine-N-[amino(polyethylene glycol)] (PEG-DSPE), PEG- disteryl glycerol (PEG-DSG), PEG-dipalmetoleyl, PEG-dioleyl, PEG-distearyl, PEG- diacylglycamide (PEG-DAG), PEG-dipalmitoyl phosphatidylethanolamine (PEG- DPPE), or PEG-1, 2-dimyristyloxlpropyl-3 -amine (PEG-c-DMA).
- PEG-DMG 1,2- dimyristoyl-sn-glycerol methoxypolyethylene glycol
- PEG-DSPE 1,2-distearoyl-sn- g
- the PEG-lipid is selected from the group consisting of a PEG-modified phosphatidylethanolamine, a PEG-modified phosphatidic acid, a PEG- modified ceramide, a PEG-modified dialkylamine, a PEG-modified diacylglycerol, a PEG-modified dialkylglycerol, and mixtures thereof.
- the lipid moiety of the PEG-lipids includes those having lengths of from about Ci4to about C22, preferably from about Cuto about Ci6.
- a PEG moiety for example an mPEG-NEE, has a size of about 1000, 2000, 5000, 10,000, 15,000 or 20,000 daltons.
- the PEG- lipid is PEG 2k -DMG.
- the lipid nanoparticles described herein can comprise a PEG lipid which is a non-diffusible PEG.
- PEG lipid which is a non-diffusible PEG.
- non-diffusible PEGs include PEG-DSG and PEG-DSPE.
- PEG-lipids are known in the art, such as those described in U.S. Patent No. 8158601 and International Publ. No. WO 2015/130584 A2, which are incorporated herein by reference in their entirety.
- lipid components e.g., PEG lipids
- PEG lipids lipid components of various formulae, described herein may be synthesized as described International Patent Application No. PCT/US2016/000129, filed December 10, 2016, entitled “Compositions and Methods for Delivery of Therapeutic Agents,” which is incorporated by reference in its entirety.
- the lipid component of a lipid nanoparticle composition may include one or more molecules comprising polyethylene glycol, such as PEG or PEG-modified lipids. Such species may be alternately referred to as PEGylated lipids.
- a PEG lipid is a lipid modified with polyethylene glycol.
- a PEG lipid may be selected from the non-limiting group including PEG-modified phosphatidylethanolamines, PEG- modified phosphatidic acids, PEG-modified ceramides, PEG-modified dialkylamines, PEG-modified diacylglycerols, PEG-modified dialkylglycerols, and mixtures thereof.
- a PEG lipid may be PEG-c-DOMG, PEG-DMG, PEG-DLPE, PEG-DMPE, PEG-DPPC, or a PEG-DSPE lipid.
- PEG-modified lipids are a modified form of PEG DMG.
- PEG-DMG has the following structure:
- PEG lipids useful in the present invention can be PEGylated lipids described in International Publication No. WO2012099755, the contents of which is herein incorporated by reference in its entirety. Any of these exemplary PEG lipids described herein may be modified to comprise a hydroxyl group on the PEG chain.
- the PEG lipid is a PEG-OH lipid.
- a “PEG-OH lipid” (also referred to herein as “hydroxy- PEGylated lipid”) is a PEGylated lipid having one or more hydroxyl (-OH) groups on the lipid.
- the PEG-OH lipid includes one or more hydroxyl groups on the PEG chain.
- a PEG-OH or hydroxy-PEGylated lipid comprises an -OH group at the terminus of the PEG chain.
- a PEG lipid useful in the present invention is a compound of Formula (V).
- R 3 is -OR°
- R° is hydrogen, optionally substituted alkyl, or an oxygen protecting group; r is an integer between 1 and 100, inclusive; L 1 is optionally substituted C 1 -io alkylene, wherein at least one methylene of the optionally substituted C 1 -io alkylene is independently replaced with optionally substituted carbocyclylene, optionally substituted heterocyclylene, optionally substituted arylene, optionally substituted heteroarylene, O, N(R N ), S, C(O), C(O)N(R N ), NR N C(O), C(O)O, OC(O), - OC(O)O, OC(O)N(R N ), NR N C(O)O, or NR N C(O)N(R N );
- D is a moiety obtained by click chemistry or a moiety cleavable under physiological conditions; m is 0, 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10;
- each instance of L 2 is independently a bond or optionally substituted C 1-6 alkylene, wherein one methylene unit of the optionally substituted C 1-6 alkylene is optionally replaced with O, N(R N ), S, C(O), C(O)N(R N ), - NR N C(O), C(O)O, OC(O), OC(O)O, OC(O)N(R N ), NR N C(O)O, or - NR N C(O)N(R N ); each instance of R 2 is independently optionally substituted C1-30 alkyl, optionally substituted C1-30 alkenyl, or optionally substituted C1-30 alkynyl; optionally wherein one or more methylene units of R 2 are independently replaced with optionally substituted carbocyclylene, optionally substituted heterocyclylene, optionally substituted arylene, optionally substituted heteroarylene, N(R N ), O, S, C(O), C(O)N
- the compound of Fomula (V) is a PEG-OH lipid (i. e. , R 3 is -OR°, and R° is hydrogen). In certain embodiments, the compound of Formula (V) is of Formula (V-OH): or a salt thereof.
- a PEG lipid useful in the present invention is a PEGylated fatty acid.
- a PEG lipid useful in the present invention is a compound of Formula (VI).
- R 3 is-OR°
- R° is hydrogen, optionally substituted alkyl or an oxygen protecting group; r is an integer between 1 and 100, inclusive;
- the compound of Formula (VI) is of Formula (VI-
- the compound of Formula (VI) is: or a salt thereof.
- the compound of Formula (VI) is N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl
- the lipid composition of the pharmaceutical compositions disclosed herein does not comprise a PEG-lipid.
- the PEG-lipids may be one or more of the PEG lipids described in U.S. Application No. 62/520,530.
- a PEG lipid of the invention comprises a PEG- modified phosphatidylethanolamine, a PEG-modified phosphatidic acid, a PEG- modified ceramide, a PEG-modified dialkylamine, a PEG-modified diacylglycerol, a PEG-modified dialkylglycerol, and mixtures thereof.
- the PEG-modified lipid is PEG-DMG, PEG-c-DOMG (also referred to as PEG-DOMG), PEG-DSG and/or PEG-DPG.
- a LNP of the invention comprises an ionizable cationic lipid of any of Formula I, II or III, a phospholipid comprising DSPC, a structural lipid, and a PEG lipid comprising PEG-DMG. In some embodiments, a LNP of the invention comprises an ionizable cationic lipid of any of Formula I, II or III, a phospholipid comprising DSPC, a structural lipid, and a PEG lipid comprising a compound having Formula VI.
- a LNP of the invention comprises an ionizable cationic lipid of Formula I, II or III, a phospholipid comprising a compound having Formula IV, a structural lipid, and the PEG lipid comprising a compound having Formula V or VI.
- a LNP of the invention comprises an ionizable cationic lipid of Formula I, II or III, a phospholipid comprising a compound having Formula IV, a structural lipid, and the PEG lipid comprising a compound having Formula V or VI.
- a LNP of the invention comprises an ionizable cationic lipid of Formula I, II or III, a phospholipid having Formula IV, a structural lipid, and a PEG lipid comprising a compound having Formula VI.
- a LNP of the invention comprises an ionizable cationic lipid of and a PEG lipid comprising Formula VI.
- a LNP of the invention comprises an ionizable cationic lipid of and an alternative lipid comprising oleic acid.
- a LNP of the invention comprises an ionizable cationic lipid of an alternative lipid comprising oleic acid, a structural lipid comprising cholesterol, and a PEG lipid comprising a compound having Formula VI.
- a LNP of the invention comprises an ionizable cationic a phospholipid comprising DOPE, a structural lipid comprising cholesterol, and a PEG lipid comprising a compound having Formula VI.
- a LNP of the invention comprises an ionizable cationic a phospholipid comprising DOPE, a structural lipid comprising cholesterol, and a PEG lipid comprising a compound having Formula VIE
- a LNP of the invention comprises an N:P ratio of from about 2: 1 to about 30:1.
- a LNP of the invention comprises an N:P ratio of about 6: 1.
- a LNP of the invention comprises an N:P ratio of about In some embodiments, a LNP of the invention comprises a wt/wt ratio of the ionizable cationic lipid component to the RNA of from about 10: 1 to about 100: 1.
- a LNP of the invention comprises a wt/wt ratio of the ionizable cationic lipid component to the RNA of about 20: 1.
- a LNP of the invention comprises a wt/wt ratio of the ionizable cationic lipid component to the RNA of about 10: 1.
- a LNP of the invention has a mean diameter from about 50nm to about 150nm.
- a LNP of the invention has a mean diameter from about 70nm to about 120nm.
- alkyl As used herein, the term “alkyl”, “alkyl group”, or “alkylene” means a linear or branched, saturated hydrocarbon including one or more carbon atoms (e.g., one, two, three, four, five, six, seven, eight, nine, ten, eleven, twelve, thirteen, fourteen, fifteen, sixteen, seventeen, eighteen, nineteen, twenty, or more carbon atoms), which is optionally substituted.
- C1-14 alkyl means an optionally substituted linear or branched, saturated hydrocarbon including 1-14 carbon atoms. Unless otherwise specified, an alkyl group described herein refers to both unsubstituted and substituted alkyl groups.
- alkenyl means a linear or branched hydrocarbon including two or more carbon atoms (e.g., two, three, four, five, six, seven, eight, nine, ten, eleven, twelve, thirteen, fourteen, fifteen, sixteen, seventeen, eighteen, nineteen, twenty, or more carbon atoms) and at least one double bond, which is optionally substituted.
- C2-14 alkenyl means an optionally substituted linear or branched hydrocarbon including 2-14 carbon atoms and at least one carbon-carbon double bond.
- An alkenyl group may include one, two, three, four, or more carbon-carbon double bonds.
- Cis alkenyl may include one or more double bonds.
- a Cis alkenyl group including two double bonds may be a linoleyl group.
- an alkenyl group described herein refers to both unsubstituted and substituted alkenyl groups.
- alkynyl means a linear or branched hydrocarbon including two or more carbon atoms (e.g., two, three, four, five, six, seven, eight, nine, ten, eleven, twelve, thirteen, fourteen, fifteen, sixteen, seventeen, eighteen, nineteen, twenty, or more carbon atoms) and at least one carbon-carbon triple bond, which is optionally substituted.
- the notation "C2-14 alkynyl” means an optionally substituted linear or branched hydrocarbon including 2- 14 carbon atoms and at least one carbon-carbon triple bond.
- An alkynyl group may include one, two, three, four, or more carbon-carbon triple bonds.
- Cis alkynyl may include one or more carbon-carbon triple bonds.
- an alkynyl group described herein refers to both unsubstituted and substituted alkynyl groups.
- Carbocycle or “carbocyclic group” means an optionally substituted mono- or multi-cyclic system including one or more rings of carbon atoms. Rings may be three, four, five, six, seven, eight, nine, ten, eleven, twelve, thirteen, fourteen, fifteen, sixteen, seventeen, eighteen, nineteen, or twenty membered rings.
- the notation "C 3-6 carbocycle” means a carbocycle including a single ring having 3-6 carbon atoms. Carbocycles may include one or more carboncarbon double or triple bonds and may be non-aromatic or aromatic (e.g., cycloalkyl or aryl groups).
- carbocycles include cyclopropyl, cyclopentyl, cyclohexyl, phenyl, naphthyl, and 1,2 dihydronaphthyl groups.
- cycloalkyl as used herein means a non-aromatic carbocycle and may or may not include any double or triple bond.
- carbocycles described herein refers to both unsubstituted and substituted carbocycle groups, i.e., optionally substituted carbocycles.
- heterocycle or “heterocyclic group” means an optionally substituted mono- or multi-cyclic system including one or more rings, where at least one ring includes at least one heteroatom.
- Heteroatoms may be, for example, nitrogen, oxygen, or sulfur atoms. Rings may be three, four, five, six, seven, eight, nine, ten, eleven, twelve, thirteen, or fourteen membered rings.
- Heterocycles may include one or more double or triple bonds and may be non- aromatic or aromatic (e.g., heterocycloalkyl or heteroaryl groups).
- heterocycles include imidazolyl, imidazolidinyl, oxazolyl, oxazolidinyl, thiazolyl, thiazolidinyl, pyrazolidinyl, pyrazolyl, isoxazolidinyl, isoxazolyl, isothiazolidinyl, isothiazolyl, morpholinyl, pyrrolyl, pyrrolidinyl, furyl, tetrahydrofuryl, thiophenyl, pyridinyl, piperidinyl, quinolyl, and isoquinolyl groups.
- heterocycloalkyl as used herein means a non-aromatic heterocycle and may or may not include any double or triple bond. Unless otherwise specified, heterocycles described herein refers to both unsubstituted and substituted heterocycle groups, i.e., optionally substituted heterocycles.
- heteroalkyl refers respectively to an alkyl, alkenyl, alkynyl group, as defined herein, which further comprises one or more (e.g., 1, 2, 3, or 4) heteroatoms (e.g., oxygen, sulfur, nitrogen, boron, silicon, phosphorus) wherein the one or more heteroatoms is inserted between adjacent carbon atoms within the parent carbon chain and/or one or more heteroatoms is inserted between a carbon atom and the parent molecule, i.e., between the point of attachment.
- heteroatoms e.g., oxygen, sulfur, nitrogen, boron, silicon, phosphorus
- heteroalkyls, heteroalkenyls, or heteroalkynyls described herein refers to both unsubstituted and substituted heteroalkyls, heteroalkenyls, or heteroalkynyls, i.e., optionally substituted heteroalkyls, heteroalkenyls, or heteroalkynyls.
- a “biodegradable group” is a group that may facilitate faster metabolism of a lipid in a mammalian entity.
- a biodegradable group may be selected from the group consisting of, but is not limited to, -C(O)O-, -OC(O)-, -C(O)N(R')-, -N(R')C(O)-, -C(O)-, -C(S)-, -C(S)S-, -SC(S)-, -CH(OH)-, -P(O)(OR)O-, -S(O)2-, an aryl group, and a heteroaryl group.
- an "aryl group” is an optionally substituted carbocyclic group including one or more aromatic rings.
- aryl groups include phenyl and naphthyl groups.
- a "heteroaryl group” is an optionally substituted heterocyclic group including one or more aromatic rings.
- heteroaryl groups include pyrrolyl, furyl, thiophenyl, imidazolyl, oxazolyl, and thiazolyl. Both aryl and heteroaryl groups may be optionally substituted.
- M and M' can be selected from the nonlimiting group consisting of optionally substituted phenyl, oxazole, and thiazole.
- M and M' can be independently selected from the list of biodegradable groups above.
- aryl or heteroaryl groups described herein refers to both unsubstituted and substituted groups, i.e., optionally substituted aryl or heteroaryl groups.
- Alkyl, alkenyl, and cyclyl (e.g., carbocyclyl and heterocyclyl) groups may be optionally substituted unless otherwise specified.
- R is an alkyl or alkenyl group, as defined herein.
- the substituent groups themselves may be further substituted with, for example, one, two, three, four, five, or six substituents as defined herein.
- a C1-6 alkyl group may be further substituted with one, two, three, four, five, or six substituents as described herein.
- N-oxidizing agent e.g., 3 -chloroperoxybenzoic acid (mCPBA) and/or hydrogen peroxides
- mCPBA 3 -chloroperoxybenzoic acid
- hydrogen peroxides hydrogen peroxides
- all shown and claimed nitrogen-containing compounds are considered, when allowed by valency and structure, to include both the compound as shown and its N- oxide derivative (which can be designated as N->0 or N+-O-).
- the nitrogens in the compounds of the disclosure can be converted to N- hydroxy or N-alkoxy compounds.
- N-hydroxy compounds can be prepared by oxidation of the parent amine by an oxidizing agent such as m CPBA.
- nitrogen-containing compounds are also considered, when allowed by valency and structure, to cover both the compound as shown and its N- hydroxy (i.e., N-OH) and N-alkoxy (i.e., N-OR, wherein R is substituted or unsubstituted C 1 -C 6 alkyl, C 1 -C 6 alkenyl, C 1 -C 6 alkynyl, 3-14-membered carbocycle or 3-14-membered heterocycle) derivatives.
- N-OH N-hydroxy
- N-alkoxy i.e., N-OR, wherein R is substituted or unsubstituted C 1 -C 6 alkyl, C 1 -C 6 alkenyl, C 1 -C 6 alkynyl, 3-14-membered carbocycle or 3-14-membered heterocycle
- the lipid composition of a pharmaceutical composition disclosed herein can include one or more components in addition to those described above.
- the lipid composition can include one or more permeability enhancer molecules, carbohydrates, polymers, surface altering agents (e.g., surfactants), or other components.
- a permeability enhancer molecule can be a molecule described by U.S. Patent Application Publication No. 2005/0222064.
- Carbohydrates can include simple sugars (e.g., glucose) and polysaccharides (e.g., glycogen and derivatives and analogs thereof).
- a polymer can be included in and/or used to encapsulate or partially encapsulate a pharmaceutical composition disclosed herein (e.g., a pharmaceutical composition in lipid nanoparticle form).
- a polymer can be biodegradable and/or biocompatible.
- a polymer can be selected from, but is not limited to, polyamines, polyethers, polyamides, polyesters, polycarbamates, polyureas, polycarbonates, polystyrenes, polyimides, polysulfones, polyurethanes, polyacetylenes, polyethylenes, polyethyleneimines, polyisocyanates, polyacrylates, polymethacrylates, polyacrylonitriles, and polyarylates.
- the ratio between the lipid composition and the polynucleotide range can be from about 10: 1 to about 60: 1 (wt/wt).
- the ratio between the lipid composition and the polynucleotide can be about 10: 1, 11: 1, 12: 1, 13: 1, 14: 1, 15: 1, 16: 1, 17: 1, 18: 1, 19: 1, 20: 1, 21: 1, 22: 1, 23: 1, 24: 1, 25: 1, 26: 1, 27: 1, 28: 1, 29: 1, 30: 1, 31: 1, 32: 1, 33: 1, 34: 1, 35:1,36:1,37:1,38:1,39:1,40:1,41:1,42:1,43:1,44:1,45:1,46:1,47:1,48:1,49:1, 50:1, 51:1, 52:1, 53:1, 54:1, 55:1, 56:1, 57:1, 58:1, 59:1 or 60:1 (wt/wt). In some embodiments, the wt/wt ratio of the lipid composition to the polynucleotide encoding a therapeutic agent is about 20 : 1 or about 15:1.
- the pharmaceutical composition disclosed herein can contain more than one polypeptides.
- a pharmaceutical composition disclosed herein can contain two or more polynucleotides (e.g., RNA, e.g., mRNA).
- the lipid nanoparticles described herein can comprise polynucleotides (e.g., mRNA) in a lipid: polynucleotide weight ratio of 5: 1, 10: 1, 15:1, 20: 1, 25:1, 30: 1, 35:1, 40: 1, 45:1, 50:1, 55:1, 60: 1 or 70:1, or a range or any of these ratios such as, but not limited to, 5: 1 to about 10:1, from about 5: 1 to about 15:1, from about 5: 1 to about 20: 1, from about 5: 1 to about 25: 1, from about 5: 1 to about 30: 1, from about 5: 1 to about 35: 1, from about 5: 1 to about 40: 1, from about 5: 1 to about 45:1, from about 5: 1 to about 50:1, from about 5: 1 to about 55:1, from about 5: 1 to about 60: 1, from about 5: 1 to about 70: 1, from about 10: 1 to about 15:1, from about 10: 1 to about 20: 1, from about 10: 1 to about 25:1,
- the lipid nanoparticles described herein can comprise the polynucleotide in a concentration from approximately 0.1 mg/ml to 2 mg/ml such as, but not limited to, 0.1 mg/ml, 0.2 mg/ml, 0.3 mg/ml, 0.4 mg/ml, 0.5 mg/ml, 0.6 mg/ml, 0.7 mg/ml, 0.8 mg/ml, 0.9 mg/ml, 1.0 mg/ml, 1.1 mg/ml, 1.2 mg/ml, 1.3 mg/ml, 1.4 mg/ml, 1.5 mg/ml, 1.6 mg/ml, 1.7 mg/ml, 1.8 mg/ml, 1.9 mg/ml, 2.0 mg/ml or greater than 2.0 mg/ml.
- the pharmaceutical compositions disclosed herein are formulated as lipid nanoparticles (LNP). Accordingly, the present disclosure also provides nanoparticle compositions comprising (i) a lipid composition comprising a delivery agent such as compound as described herein, and (ii) a polynucleotide encoding a CFTR polypeptide. In such nanoparticle composition, the lipid composition disclosed herein can encapsulate the polynucleotide encoding a CFTR polypeptide.
- Nanoparticle compositions are typically sized on the order of micrometers or smaller and can include a lipid bilayer.
- Nanoparticle compositions encompass lipid nanoparticles (LNPs), liposomes (e.g., lipid vesicles), and lipoplexes.
- LNPs lipid nanoparticles
- liposomes e.g., lipid vesicles
- lipoplexes e.g., lipoplexes.
- a nanoparticle composition can be a liposome having a lipid bilayer with a diameter of 500 nm or less.
- Nanoparticle compositions include, for example, lipid nanoparticles (LNPs), liposomes, and lipoplexes.
- LNPs lipid nanoparticles
- nanoparticle compositions are vesicles including one or more lipid bilayers.
- a nanoparticle composition includes two or more concentric bilayers separated by aqueous compartments.
- Lipid bilayers can be functionalized and/or crosslinked to one another.
- Lipid bilayers can include one or more ligands, proteins, or channels.
- a lipid nanoparticle comprises an ionizable lipid, a structural lipid, a phospholipid, and mRNA.
- the LNP comprises an ionizable lipid, a PEG-modified lipid, a sterol and a structural lipid.
- the LNP has a molar ratio of about 20-60% ionizable lipid: about 5-25% structural lipid: about 25-55% sterol; and about 0.5-15% PEG-modified lipid.
- the LNP has a polydispersity value of less than 0.4. In some embodiments, the LNP has a net neutral charge at a neutral pH. In some embodiments, the LNP has a mean diameter of 50-150 nm. In some embodiments, the LNP has a mean diameter of 80-100 nm.
- lipid refers to a small molecule that has hydrophobic or amphiphilic properties. Lipids may be naturally occurring or synthetic. Examples of classes of lipids include, but are not limited to, fats, waxes, sterol-containing metabolites, vitamins, fatty acids, glycerolipids, glycerophospholipids, sphingolipids, saccharolipids, and polyketides, and prenol lipids. In some instances, the amphiphilic properties of some lipids leads them to form liposomes, vesicles, or membranes in aqueous media.
- a lipid nanoparticle may comprise an ionizable lipid.
- ionizable lipid has its ordinary meaning in the art and may refer to a lipid comprising one or more charged moieties.
- an ionizable lipid may be positively charged or negatively charged.
- An ionizable lipid may be positively charged, in which case it can be referred to as “cationic lipid”.
- an ionizable lipid molecule may comprise an amine group, and can be referred to as an ionizable amino lipid.
- a “charged moiety” is a chemical moiety that carries a formal electronic charge, e.g., monovalent (+1, or -1), divalent (+2, or -2), trivalent (+3, or -3), etc.
- the charged moiety may be anionic (i.e., negatively charged) or cationic (i.e., positively charged).
- positively-charged moieties include amine groups (e.g., primary, secondary, and/or tertiary amines), ammonium groups, pyridinium group, guanidine groups, and imidizolium groups.
- the charged moieties comprise amine groups.
- negatively- charged groups or precursors thereof include carboxylate groups, sulfonate groups, sulfate groups, phosphonate groups, phosphate groups, hydroxyl groups, and the like.
- the charge of the charged moiety may vary, in some cases, with the environmental conditions, for example, changes in pH may alter the charge of the moiety, and/or cause the moiety to become charged or uncharged. In general, the charge density of the molecule may be selected as desired.
- charge does not refer to a “partial negative charge” or “partial positive charge” on a molecule.
- the terms “partial negative charge” and “partial positive charge” are given its ordinary meaning in the art.
- a “partial negative charge” may result when a functional group comprises a bond that becomes polarized such that electron density is pulled toward one atom of the bond, creating a partial negative charge on the atom.
- the ionizable lipid is an ionizable amino lipid, sometimes referred to in the art as an “ionizable cationic lipid”.
- the ionizable amino lipid may have a positively charged hydrophilic head and a hydrophobic tail that are connected via a linker structure.
- an ionizable lipid may also be a lipid including a cyclic amine group.
- the ionizable lipid may be selected from, but not limited to, an ionizable lipid described in International Publication Nos. WO2013086354 and WO2013116126; the contents of each of which are herein incorporated by reference in their entirety.
- the ionizable lipid may be selected from, but not limited to, formula CLI-CLXXXII of US Patent No. 7,404,969; each of which is herein incorporated by reference in their entirety.
- the lipid may be a cleavable lipid such as those described in International Publication No. WO2012170889, herein incorporated by reference in its entirety.
- the lipid may be synthesized by methods known in the art and/or as described in International Publication Nos. WO2013086354; the contents of each of which are herein incorporated by reference in their entirety.
- Nanoparticle compositions can be characterized by a variety of methods. For example, microscopy (e.g., transmission electron microscopy or scanning electron microscopy) can be used to examine the morphology and size distribution of a nanoparticle composition. Dynamic light scattering or potentiometry (e.g., potentiometric titrations) can be used to measure zeta potentials. Dynamic light scattering can also be utilized to determine particle sizes. Instruments such as the Zetasizer Nano ZS (Malvern Instruments Ltd, Malvern, Worcestershire, UK) can also be used to measure multiple characteristics of a nanoparticle composition, such as particle size, polydispersity index, and zeta potential. The size of the nanoparticles can help counter biological reactions such as, but not limited to, inflammation, or can increase the biological effect of the polynucleotide.
- microscopy e.g., transmission electron microscopy or scanning electron microscopy
- Dynamic light scattering or potentiometry
- size or “mean size” in the context of nanoparticle compositions refers to the mean diameter of a nanoparticle composition.
- the polynucleotide encoding a CFTR polypeptide are formulated in lipid nanoparticles having a diameter from about 10 to about 100 nm such as, but not limited to, about 10 to about 20 nm, about 10 to about 30 nm, about 10 to about 40 nm, about 10 to about 50 nm, about 10 to about 60 nm, about 10 to about 70 nm, about 10 to about 80 nm, about 10 to about 90 nm, about 20 to about 30 nm, about 20 to about 40 nm, about 20 to about 50 nm, about 20 to about 60 nm, about 20 to about 70 nm, about 20 to about 80 nm, about 20 to about 90 nm, about 20 to about 100 nm, about 30 to about 40 nm, about 30 to about 50 nm, about 30 to about 60 nm, about 30 to about 70 nm, about 30 to about 80 nm, about 30 to about 90 nm, about 30 to about 100
- the nanoparticles have a diameter from about 10 to 500 nm. In one embodiment, the nanoparticle has a diameter greater than 100 nm, greater than 150 nm, greater than 200 nm, greater than 250 nm, greater than 300 nm, greater than 350 nm, greater than 400 nm, greater than 450 nm, greater than 500 nm, greater than 550 nm, greater than 600 nm, greater than 650 nm, greater than 700 nm, greater than 750 nm, greater than 800 nm, greater than 850 nm, greater than 900 nm, greater than 950 nm or greater than 1000 nm.
- the largest dimension of a nanoparticle composition is 1 pm or shorter (e.g., 1 pm, 900 nm, 800 nm, 700 nm, 600 nm, 500 nm, 400 nm, 300 nm, 200 nm, 175 nm, 150 nm, 125 nm, 100 nm, 75 nm, 50 nm, or shorter).
- a nanoparticle composition can be relatively homogenous.
- a polydispersity index can be used to indicate the homogeneity of a nanoparticle composition, e.g., the particle size distribution of the nanoparticle composition.
- a small (e.g., less than 0.3) poly dispersity index generally indicates a narrow particle size distribution.
- a nanoparticle composition can have a polydispersity index from about 0 to about 0.25, such as 0.01, 0.02, 0.03, 0.04, 0.05, 0.06, 0.07, 0.08, 0.09, 0.10, 0.11, 0.12, 0.13, 0.14, 0.15, 0.16, 0.17, 0.18, 0.19, 0.20, 0.21, 0.22, 0.23, 0.24, or 0.25.
- the polydispersity index of a nanoparticle composition disclosed herein can be from about 0.10 to about 0.20.
- the zeta potential of a nanoparticle composition can be used to indicate the electrokinetic potential of the composition.
- the zeta potential can describe the surface charge of a nanoparticle composition.
- Nanoparticle compositions with relatively low charges, positive or negative, are generally desirable, as more highly charged species can interact undesirably with cells, tissues, and other elements in the body.
- the zeta potential of a nanoparticle composition disclosed herein can be from about -10 mV to about +20 mV, from about -10 mV to about +15 mV, from about 10 mV to about +10 mV, from about -10 mV to about +5 mV, from about -10 mV to about 0 mV, from about -10 mV to about -5 mV, from about -5 mV to about +20 mV, from about -5 mV to about +15 mV, from about -5 mV to about +10 mV, from about -5 mV to about +5 mV, from about -5 mV to about 0 mV, from about 0 mV to about +20 mV, from about 0 mV to about +15 mV, from about 0 mV to about +10 mV, from about 0 mV to about +5 mV, from about 0 mV to about +20
- the zeta potential of the lipid nanoparticles can be from about 0 mV to about 100 mV, from about 0 mV to about 90 mV, from about 0 mV to about 80 mV, from about 0 mV to about 70 mV, from about 0 mV to about 60 mV, from about 0 mV to about 50 mV, from about 0 mV to about 40 mV, from about 0 mV to about 30 mV, from about 0 mV to about 20 mV, from about 0 mV to about 10 mV, from about 10 mV to about 100 mV, from about 10 mV to about 90 mV, from about 10 mV to about 80 mV, from about 10 mV to about 70 mV, from about 10 mV to about 60 mV, from about 10 mV to about 50 mV, from about 10 mV to about 40 mV, from about 10
- the zeta potential of the lipid nanoparticles can be from about 10 mV to about 50 mV, from about 15 mV to about 45 mV, from about 20 mV to about 40 mV, and from about 25 mV to about 35 mV. In some embodiments, the zeta potential of the lipid nanoparticles can be about 10 mV, about 20 mV, about 30 mV, about 40 mV, about 50 mV, about 60 mV, about 70 mV, about 80 mV, about 90 mV, and about 100 mV.
- encapsulation efficiency of a polynucleotide describes the amount of the polynucleotide that is encapsulated by or otherwise associated with a nanoparticle composition after preparation, relative to the initial amount provided.
- encapsulation can refer to complete, substantial, or partial enclosure, confinement, surrounding, or encasement.
- Encapsulation efficiency is desirably high (e.g., close to 100%).
- the encapsulation efficiency can be measured, for example, by comparing the amount of the polynucleotide in a solution containing the nanoparticle composition before and after breaking up the nanoparticle composition with one or more organic solvents or detergents.
- the encapsulation efficiency of a polynucleotide can be at least 50%, for example 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, or 100%. In some embodiments, the encapsulation efficiency can be at least 80%. In certain embodiments, the encapsulation efficiency can be at least 90%.
- the amount of a polynucleotide present in a pharmaceutical composition disclosed herein can depend on multiple factors such as the size of the polynucleotide, desired target and/or application, or other properties of the nanoparticle composition as well as on the properties of the polynucleotide.
- the amount of an mRNA useful in a nanoparticle composition can depend on the size (expressed as length, or molecular mass), sequence, and other characteristics of the mRNA.
- the relative amounts of a polynucleotide in a nanoparticle composition can also vary.
- the relative amounts of the lipid composition and the polynucleotide present in a lipid nanoparticle composition of the present disclosure can be optimized according to considerations of efficacy and tolerability.
- the N:P ratio can serve as a useful metric.
- N:P ratio of a nanoparticle composition controls both expression and tolerability
- nanoparticle compositions with low N:P ratios and strong expression are desirable.
- N:P ratios vary according to the ratio of lipids to RNA in a nanoparticle composition.
- RNA, lipids, and amounts thereof can be selected to provide an N:P ratio from about 2: 1 to about 30: 1, such as 2: 1, 3: 1, 4: 1, 5: 1, 6: 1, 7: 1, 8: 1, 9: 1, 10: 1, 12: 1, 14: 1, 16: 1, 18: 1, 20: 1, 22: 1, 24: 1, 26: 1, 28: 1, or 30: 1.
- the N:P ratio can be from about 2: 1 to about 8: 1.
- the N:P ratio is from about 5: 1 to about 8: 1.
- the N:P ratio is between 5: 1 and 6: 1.
- the N:P ratio is about is about 5.67: 1.
- the present disclosure also provides methods of producing lipid nanoparticles comprising encapsulating a polynucleotide.
- Such method comprises using any of the pharmaceutical compositions disclosed herein and producing lipid nanoparticles in accordance with methods of production of lipid nanoparticles known in the art. See, e.g., Wang et al. (2015) “Delivery of oligonucleotides with lipid nanoparticles” Adv. Drug Deliv. Rev. 87:68- 80; Silva et al. (2015) “Delivery Systems for Biopharmaceuticals. Part I: Nanoparticles and Microparticles” Curr. Pharm. Technol. 16: 940-954; Naseri et al.
- an LNP described herein comprises a LNP core and a cationic agent disposed primarily on the outer surface of the core.
- Such LNPs have a greater than neutral zeta potential at physiologic pH
- Core lipid nanoparticles typically comprise one or more of the following components: lipids (which may include ionizable amino lipids, phospholipids, helper lipids which may be neutral lipids, zwitterionic lipid, anionic lipids, and the like), structural lipids such as cholesterol or cholesterol analogs, fatty acids, polymers, stabilizers, salts, buffers, solvent, and the like.
- lipids which may include ionizable amino lipids, phospholipids, helper lipids which may be neutral lipids, zwitterionic lipid, anionic lipids, and the like
- structural lipids such as cholesterol or cholesterol analogs, fatty acids, polymers, stabilizers, salts, buffers, solvent, and the like.
- LNP cores comprise an ionizable lipid, such as an ionizable lipid, e.g., an ionizable amino lipid, a phospholipid, a structural lipid, and optionally a stabilizer (e.g., a molecule comprising polyethylene glycol) which may or may not be provided conjugated to another lipid.
- an ionizable lipid such as an ionizable lipid, e.g., an ionizable amino lipid, a phospholipid, a structural lipid, and optionally a stabilizer (e.g., a molecule comprising polyethylene glycol) which may or may not be provided conjugated to another lipid.
- the structural lipid may be but is not limited to a sterol such as for example cholesterol.
- the helper lipid is a non-cationic lipid.
- the helper lipid may comprise at least one fatty acid chain of at least 8C and at least one polar headgroup moiety.
- a molecule comprising polyethylene glycol i.e., PEG
- the molecule comprising polyethylene glycol may be polyethylene glycol conjugated to a lipid and thus may be provided as PEG-c-DOMG or PEG-DMG, for example.
- Certain of the LNPs provided herein comprise no or low levels of PEGylated lipids, including no or low levels of alkyl - PEGylated lipids, and may be referred to herein as being free of PEG or PEGylated lipid. Thus, some LNPs comprise less than 0.5 mol % PEGylated lipid.
- PEG may be an alkyl-PEG such as methoxy-PEG.
- Still other LNPs comprise non-alkyl-PEG such as hydroxy-PEG, and/or non-alkyl-PEGylated lipids such as hydroxy-PEGylated lipids.
- a core nanoparticle composition can have the formulation of Compound II:Phospholipid:Chol:a PEG lipid with a mole ratio of 50: 10:38.5: 1.5.
- a nanoparticle core composition can have the formulation of Compound II:DSPC:Chol:Compound 428 with a mole ratio of 50: 10:38.5: 1.5.
- Core nanoparticle compositions of the present disclosure comprise at least one compound according to Formula (I).
- Nanoparticle compositions can also include a variety of other components.
- the nanoparticle composition can include one or more other lipids in addition to a lipid according to Formula (I) or (II), for example (i) at least one phospholipid, (ii) at least one structural lipid, (iii) at least one PEG-lipid, or (iv) any combination thereof.
- the nanoparticle composition comprises a compound of Formula (I), (e.g., Compounds II, III, or V). In some embodiments, the nanoparticle composition comprises a compound of Formula (I) (e.g., Compounds II, III, or V) and a phospholipid (e.g., DSPC, DOP, or MSPC).
- a compound of Formula (I) e.g., Compounds II, III, or V
- a phospholipid e.g., DSPC, DOP, or MSPC
- the present disclosure also provides process of preparing a nanoparticle comprising contacting a lipid nanoparticle core with a cationic agent, wherein the lipid nanoparticle comprises:
- lipid nanoparticle core comprising:
- a polynucleotide e.g., mRNA
- the contacting of the lipid nanoparticle core with a cationic agent comprises dissolving the cationic agent in a non-ionic excipient.
- the non-ionic excipient is selected from macrogol 15 hydroxystearate (HS 15), l,2-dimyristoyl-rac-glycero-3 -methoxypolyethylene glycol- 2000 (DMG-PEG2K), Compound 428 , polyoxyethylene sorbitan monooleate [TWEEN®80], and d-a-Tocopherol polyethylene glycol succinate (TPGS).
- the non-ionic excipient is macrogol 15 hydroxystearate (HS 15).
- the contacting of the lipid nanoparticle with a cationic agent comprises the cationic agent dissolved in a buffer solution.
- the buffer solution is a phosphate buffered saline (PBS).
- the buffer solution is a Tris-based buffer.
- nanoparticles prepared by the process as described herein, e.g., by contacting the core lipid nanoparticle with a cationic agent.
- the cationic agent can be a sterol amine such as GL-67.
- the lipid nanoparticle core of the lipid nanoparticle optionally comprises a PEG-lipid.
- the lipid nanoparticle core forming the lipid nanoparticle which is contacted with the cationic agent is substantially free of PEG-lipid.
- the PEG-lipid is added to the lipid nanoparticle together with the cationic agent, prior to the contacting with the cationic agent, or after the contacting with the cationic agent.
- an LNP of the invention can be made using traditional mixing technology in which the nucleic acid payload is mixed with core LNP components to create the core LNP plus payload. Once this loaded core LNP is prepared, the cationic agent is contacted with the loaded core LNP.
- an LNP of the invention can be made using empty LNPs as the starting point.
- empty LNPs can be made prior to loading in the nucleic acid payload.
- the cationic agent can be added to form an LNP of the invention.
- empty LNPs are formulated first in a nanoprecipitation step, and buffer exchanged into a low pH buffer (i.e. pH 5).
- a low pH buffer i.e. pH 5
- these empty LNPs are introduced to mRNA (also acidified at low pH) through a mixing event.
- a pH adjustment method is used to neutralize the pH.
- a PEG lipid e.g., DMG- PEG-2k is added to stabilize the particle.
- a cationic agent e.g., GL-67 is added.
- the lipids of the LNP are used to form an empty LNP, but the PEG lipid is not included in that step.
- the nucleic acid solution is contacted with the empty LNPs, forming loaded LNPs.
- the PEG lipids are added at one or two points during further processing of the loaded LNPs and the cationic agent can be added at any point during that further processing.
- the cationic agent can be added at any point during the further processing of the LNP.
- an LNP of the invention can be prepared using nanoprecipitation, which is the unit operation in which the LNPs are self-assembled from their individual lipid components by way of kinetic mixing and subsequent maturation and continuous dilution.
- This unit operation includes three individual steps, which are: mixing of the aqueous and organic inputs, maturation of the LNPs, and dilution after a controlled residence time. Due to the continuous nature of these steps, they are considered one unit operation.
- the unit operation includes the continuous inline combination of three liquid streams with one inline maturation step: mixing of the aqueous buffer with lipid stock solution, maturation via controlled residence time, and dilution of the nanoparticles.
- the nanoprecipitation itself occurs in the scale- appropriate mixer, which is designed to allow continuous, high-energy, combination of the aqueous solution with the lipid stock solution dissolved in ethanol.
- the particles are thus self-assembled in the mixing chamber.
- an LNP of the invention can be prepared using nanoprecipitation, which is the unit operation in which the LNPs are self-assembled from their individual lipid components by way of kinetic mixing and subsequent maturation and continuous dilution. This unit operation includes three individual steps, which are: mixing of the aqueous and organic inputs, maturation of the LNPs, and dilution after a controlled residence time.
- the unit operation includes the continuous inline combination of three liquid streams with one inline maturation step: mixing of the aqueous buffer with lipid stock solution, maturation via controlled residence time, and dilution of the nanoparticles.
- the nanoprecipitation itself occurs in the scale- appropriate mixer, which is designed to allow continuous, high-energy, combination of the aqueous solution with the lipid stock solution dissolved in ethanol.
- the aqueous solution and the lipid stock solution both flow simultaneously into the mixing hardware continuously throughout this operation.
- the ethanol content which keeps the lipids dissolved, is abruptly reduced and the lipids all precipitate with each other.
- the particles are thus self-assembled in the mixing chamber.
- One of the objectives of unit operation is to exchange the solution into a fully aqueous buffer, free of ethanol, and to reach a target concentration of LNP. This can be achieved by first reaching a target processing concentration, then diafiltering, and then (if necessary) a final concentration step, once the ethanol has been completely removed.
- polynucleotides, pharmaceutical compositions and formulations described above are used in the preparation, manufacture and therapeutic use of to treat and/or prevent CFTR-related diseases, disorders or conditions.
- the polynucleotides, compositions and formulations of the present disclosure are used to treat and/or prevent cystic fibrosis.
- the polynucleotides, pharmaceutical compositions and formulations of the present disclosure are used in methods for reducing cellular sodium levels in a subject in need thereof.
- one aspect of the present disclosure provides a method of alleviating the signs and symptoms of cystic fibrosis in a subject comprising the administration of a composition or formulation comprising a polynucleotide encoding CFTRto that subject (e.g., an mRNA encoding a CFTR polypeptide).
- the administration of an effective amount of a polynucleotide, pharmaceutical composition or formulation of the invention reduces the levels of a biomarker of cystic fibrosis, e.g., intracellular sodium levels.
- the administration of the polynucleotide, pharmaceutical composition or formulation of the present disclosure results in reduction in the level of one or more biomarkers of cystic fibrosis, e.g., intracellular sodium levels, within a short period of time after administration of the polynucleotide, pharmaceutical composition or formulation of the present disclosure.
- the polynucleotides, e.g., mRNA, disclosed herein comprise one or more sequences encoding an CFTR polypeptide that is suitable for use in gene replacement therapy for cystic fibrosis.
- the present disclosure treats a lack of CFTR or CFTR activity, or decreased or abnormal CFTR activity in a subject by providing a polynucleotide, e.g., mRNA, that encodes an CFTR polypeptide to the subject.
- the polynucleotide is sequence-optimized.
- the polynucleotide (e.g., an mRNA) comprises a nucleic acid sequence (e.g., an ORF) encoding an CFTR polypeptide, wherein the nucleic acid is sequence-optimized, e.g., by modifying its G/C, uridine, or thymidine content, and/or the polynucleotide comprises at least one chemically modified nucleoside.
- the polynucleotide comprises a miRNA binding site, e.g., a miRNA binding site that binds miRNA-142.
- the administration of a composition or formulation comprising polynucleotide, pharmaceutical composition or formulation of the present disclosure to a subject results in a decrease in intracellular sodium levels in cells to a level at least 10%, at least 15%, at least 20%, at least 25%, at least 30%, at least 35%, at least 40%, at least 45%, at least 50%, at least 55%, at least 60%, at least 65%, at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, at least 95%, or to 100% lower than the level observed prior to the administration of the composition or formulation.
- the administration of the polynucleotide, pharmaceutical composition or formulation of the present disclosure results in expression of CFTR in cells of the subject.
- administering the polynucleotide, pharmaceutical composition or formulation of the present disclosure results in an increase of CFTR activity in the subject.
- the polynucleotides of the present disclosure are used in methods of administering a composition or formulation comprising an mRNA encoding an CFTR polypeptide to a subject, wherein the method results in an increase of CFTR activity in at least some cells of a subject.
- the administration of a composition or formulation comprising an mRNA encoding an CFTR polypeptide to a subject results in an increase of CFTR activity in cells subject to a level at least 10%, at least 15%, at least 20%, at least 25%, at least 30%, at least 35%, at least 40%, at least 45%, at least 50%, at least 55%, at least 60%, at least 65%, at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, at least 95%, or to 100% or more of the activity level expected in a normal subject, e.g., a human not suffering from cystic fibrosis.
- polynucleotides, pharmaceutical compositions, or formulations of the present disclosure can be repeatedly administered such that CFTR protein is expressed at a therapeutic level for a period of time sufficient to have a beneficial biological effect as described herein.
- the expression of the encoded polypeptide is increased.
- the polynucleotide increases CFTR expression levels in cells when introduced into those cells, e.g., by at least 10%, at least 15%, at least 20%, at least 25%, at least 30%, at least 35%, at least 40%, at least 45%, at least 50%, at least 55%, at least 60%, at least 65%, at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, at least 95%, or to 100% with respect to the CFTR expression level in the cells before the polypeptide is introduced in the cells.
- the method or use comprises administering a polynucleotide, e.g., mRNA, comprising a nucleotide sequence having sequence similarity to a polynucleotide of SEQ ID NO: 10, wherein the polynucleotide encodes an CFTR polypeptide (e.g., SEQ ID NO: 1).
- a polynucleotide e.g., mRNA
- the method or use comprises administering a polynucleotide, e.g., mRNA, comprising a nucleotide sequence having sequence similarity to a polynucleotide of SEQ ID NO: 10, wherein the polynucleotide encodes an CFTR polypeptide (e.g., SEQ ID NO: 1).
- the present disclosure also provides methods to increase CFTR activity in a subject in need thereof, e.g., a subject with cystic fibrosis, comprising administering to the subject a therapeutically effective amount of a composition or formulation comprising mRNA encoding an CFTR polypeptide disclosed herein.
- the CFTR activity measured after administration to a subject in need thereof is at least the normal CFTR activity level observed in healthy human subjects.
- the CFTR activity measured after administration is at higher than the CFTR activity level observed in cystic fibrosis patients, e.g., untreated cystic fibrosis patients.
- the increase in CFTR activity in a subject in need thereof, e.g., a subject with cystic fibrosis, after administering to the subject a therapeutically effective amount of a composition or formulation comprising mRNA encoding an CFTR polypeptide disclosed herein is at least about 5, at least about 10, at least about 15, at least about 20, at least about 25, at least about 30, at least about 35, at least about 40, at least about 45, at least about 50, at least about 55, at least about 60, at least about 65, at least about 70, at least about 75, at least about 80, at least about 85, at least about 90, at least about 95, at least about 100, or greater than 100 percent of the normal CFTR activity level observed in healthy human subjects.
- the increase in CFTR activity above the CFTR activity level observed in cystic fibrosis patients after administering to the subject a composition or formulation comprising an mRNA encoding an CFTR polypeptide disclosed herein (e.g., after a single dose administration) is maintained for at least 1 day, at least 2 days, at least 3 days, at least 4 days, at least 5 days, at least 6 days, at least 7 days, at least 8 days, at least 9 days, at least 10 days, at least 12 days, at least 14 days, at least 21 days, or at least 28 days.
- the present disclosure also provides a method to treat, prevent, or ameliorate the symptoms of cystic fibrosis (e.g., persistent coughing, lung infection, wheezing, shortness of breath, poor growth, poor weight gain, frequent greasy, bulky stools) in an cystic fibrosis patient comprising administering to the subject a therapeutically effective amount of a composition or formulation comprising mRNA encoding an CFTR polypeptide disclosed herein.
- the administration of a therapeutically effective amount of a composition or formulation comprising mRNA encoding an CFTR polypeptide disclosed herein to subject in need of treatment for cystic fibrosis results in reducing the symptoms of cystic fibrosis.
- the therapeutic effectiveness of a drug or a treatment of the instant invention can be characterized or determined by measuring the level of expression of an encoded protein (e.g., enzyme) in a sample or in samples taken from a subject (e.g., from a preclinical test subject (rodent, primate, etc.) or from a clinical subject (human).
- an encoded protein e.g., enzyme
- the therapeutic effectiveness of a drug or a treatment of the instant invention can be characterized or determined by measuring the level of activity of an encoded protein (e.g., enzyme) in a sample or in samples taken from a subject (e.g., from a preclinical test subject (rodent, primate, etc.) or from a clinical subject (human).
- the therapeutic effectiveness of a drug or a treatment of the instant invention can be characterized or determined by measuring the level of an appropriate biomarker in sample(s) taken from a subject.
- Levels of protein and/or biomarkers can be determined post-administration with a single dose of an mRNA therapeutic of the invention or can be determined and/or monitored at several time points following administration with a single dose or can be determined and/or monitored throughout a course of treatment, e.g., a multi -dose treatment.
- Certain aspects of the invention feature measurement, determination and/or monitoring of the expression level or levels of CFTR protein in a subject.
- CFTR protein expression levels can be measured or determined by any art- recognized method for determining protein levels in biological samples, e.g., from blood samples or a needle biopsy.
- level or “level of a protein” as used herein, preferably means the weight, mass or concentration of the protein within a sample or a subject. It will be understood by the skilled artisan that in certain embodiments the sample may be subjected, e.g., to any of the following: purification, precipitation, separation, e.g. centrifugation and/or HPLC, and subsequently subjected to determining the level of the protein, e.g., using mass and/or spectrometric analysis.
- enzyme-linked immunosorbent assay can be used to determine protein expression levels.
- protein purification, separation and LC-MS can be used as a means for determining the level of a protein according to the invention.
- an mRNA therapy of the invention results in increased CFTR protein expression levels in the tissue (e.g., heart, liver, brain, or skeletal muscle) of the subject (e.g., 2-fold, 3-fold, 4-fold, 5-fold, 6-fold, 7-fold, 8-fold, 9-fold, 10-fold, 20- fold, 30-fold, 40-fold, 50-fold increase and/or increased to at least 50%, at least 60%, at least 70%, at least 75%, 80%, at least 85%, at least 90%, at least 95%, or at least 100% of normal levels) for at least 6 hours, at least 12 hours, at least 24 hours, at least 36 hours, at least 48 hours, at least 60 hours, at least 72 hours, at least 84 hours, at least 96 hours, at least 108 hours, at least 122 hours after administration of a single dose of the mRNA therapy.
- tissue e.g., heart, liver, brain, or skeletal muscle
- the subject e.g., 2-fold, 3-fold, 4-fold, 5-fold, 6-fold
- the administration of an effective amount of a polynucleotide, pharmaceutical composition or formulation of the invention reduces the levels of a biomarker of CFTR, e.g., intracellular sodium levels.
- the administration of the polynucleotide, pharmaceutical composition or formulation of the invention results in reduction in the level of one or more biomarkers of CFTR, e.g., intracellular sodium levels, within a short period of time after administration of the polynucleotide, pharmaceutical composition or formulation of the invention.
- Further aspects of the invention feature determining the level (or levels) of a biomarker determined in a sample as compared to a level (e.g., a reference level) of the same or another biomarker in another sample, e.g., from the same patient, from another patient, from a control and/or from the same or different time points, and/or a physiologic level, and/or an elevated level, and/or a supraphysiologic level, and/or a level of a control.
- a level e.g., a reference level
- physiologic levels for example, levels in normal or wild type animals, normal or healthy subjects, and the like, in particular, the level or levels characteristic of subjects who are healthy and/or normal functioning.
- the phrase “elevated level” means amounts greater than normally found in a normal or wild type preclinical animal or in a normal or healthy subject, e.g. a human subject.
- the term “supraphysiologic” means amounts greater than normally found in a normal or wild type preclinical animal or in a normal or healthy subject, e.g. a human subject, optionally producing a significantly enhanced physiologic response.
- the term “comparing” or “compared to” preferably means the mathematical comparison of the two or more values, e.g., of the levels of the biomarker(s).
- Comparing or comparison to can be in the context, for example, of comparing to a control value, e.g., as compared to a reference blood, serum, plasma, and/or tissue (e.g., liver) intracellular sodium level, in said subject prior to administration (e.g., in a person suffering from cystic fibrosis) or in a normal or healthy subject.
- a control value e.g., as compared to a reference blood, serum, plasma, and/or tissue (e.g., liver) intracellular sodium level
- Comparing or comparison to can also be in the context, for example, of comparing to a control value, e.g., as compared to a reference blood, serum, plasma and/or tissue (e.g., liver) intracellular sodium level in said subject prior to administration (e.g., in a person suffering from cystic fibrosis) or in a normal or healthy subject.
- a “control” is preferably a sample from a subject wherein the cystic fibrosis status of said subject is known.
- a control is a sample of a healthy patient.
- control is a sample from at least one subject having a known cystic fibrosis status, for example, a severe, mild, or healthy cystic fibrosis status, e.g. a control patient.
- control is a sample from a subject not being treated for cystic fibrosis.
- control is a sample from a single subject or a pool of samples from different subjects and/or samples taken from the subject(s) at different time points.
- level or “level of a biomarker” as used herein, preferably means the mass, weight or concentration of a biomarker of the invention within a sample or a subject. It will be understood by the skilled artisan that in certain embodiments the sample may be subjected to, e.g., one or more of the following: substance purification, precipitation, separation, e.g. centrifugation and/or HPLC and subsequently subjected to determining the level of the biomarker, e.g. using mass spectrometric analysis. In certain embodiments, LC-MS can be used as a means for determining the level of a biomarker according to the invention.
- determining the level of a biomarker can mean methods which include quantifying an amount of at least one substance in a sample from a subject, for example, in a bodily fluid from the subject (e.g., serum, plasma, urine, lymph, etc.) or in a tissue of the subject (e.g., liver, etc.).
- a bodily fluid e.g., serum, plasma, urine, lymph, etc.
- a tissue of the subject e.g., liver, etc.
- reference level can refer to levels (e.g., of a biomarker) in a subject prior to administration of an mRNA therapy of the invention (e.g., in a person suffering from cystic fibrosis) or in a normal or healthy subject.
- normal subject or “healthy subject” refers to a subject not suffering from symptoms associated with cystic fibrosis. Moreover, a subject will be considered to be normal (or healthy) if it has no mutation of the functional portions or domains of the CFTR gene and/or no mutation of the CFTR gene resulting in a reduction of or deficiency of CFTR or the activity thereof, resulting in symptoms associated with CF. Said mutations will be detected if a sample from the subject is subjected to a genetic testing for such CFTR mutations. In certain embodiments of the present invention, a sample from a healthy subject is used as a control sample, or the known or standardized value for the level of biomarker from samples of healthy or normal subjects is used as a control.
- comparing the level of the biomarker in a sample from a subject in need of treatment for cystic fibrosis or in a subject being treated for cystic fibrosis to a control level of the biomarker comprises comparing the level of the biomarker in the sample from the subject (in need of treatment or being treated for cystic fibrosis) to a baseline or reference level, wherein if a level of the biomarker in the sample from the subject (in need of treatment or being treated for cystic fibrosis) is elevated, increased or higher compared to the baseline or reference level, this is indicative that the subject is suffering from cystic fibrosis and/or is in need of treatment; and/or wherein if a level of the biomarker in the sample from the subject (in need of treatment or being treated for cystic fibrosis) is decreased or lower compared to the baseline level this is indicative that the subject is not suffering from, is successfully being treated for cystic fibrosis, or is not in need of treatment for cystic fibrosis.
- the stronger the reduction e.g., at least 2-fold, at least 3-fold, at least 4-fold, at least 5 -fold, at least 6-fold, at least 7-fold, at least 8-fold, at least 10-fold, at least 20-fold, at least-30 fold, at least 40-fold, at least 50-fold reduction and/or at least 10%, at least 20%, at least 30% at least 40%, at least 50%, at least 60%, at least 70%, at least 80%, at least 90%, or at least 100% reduction) of the level of a biomarker, within a certain time period, e.g., within 6 hours, within 12 hours, 24 hours, 36 hours, 48 hours, 60 hours, or 72 hours, and/or for a certain duration of time, e.g., 48 hours, 72 hours, 96 hours, 120 hours, 144 hours, 1 week, 2 weeks, 3 weeks, 4 weeks, 1 month, 2 months, 3 months, 4 months, 5 months, 6 months, 7 months, 8 months, 9 months, 10 months, 11 months, 12 months, 18 months,
- Exemplary time periods include 12, 24, 48, 72, 96, 120 or 144 hours post administration, in particular 24, 48, 72 or 96 hours post administration.
- a sustained reduction in substrate levels is particularly indicative of mRNA therapeutic dosing and/or administration regimens successful for treatment of cystic fibrosis.
- Such sustained reduction can be referred to herein as “duration” of effect.
- a bodily fluid e.g., plasma, serum, urine,
- sustained reduction in substrate (e.g., biomarker) levels in one or more samples is preferred.
- substrate e.g., biomarker
- samples e.g., fluids and/or tissues
- sustained reduction in substrate e.g., biomarker
- each dose of an mRNA therapy of the invention is about 1 mg to about 7 mg. In some embodiments, each dose of an mRNA therapy of the invention is about 1 mg. In some embodiments, each dose of an mRNA therapy of the invention is about 2 mg. In some embodiments, each dose of an mRNA therapy of the invention is about 3 mg. In some embodiments, each dose of an mRNA therapy of the invention is about 4 mg. In some embodiments, each dose of an mRNA therapy of the invention is about 5 mg. In some embodiments, each dose of an mRNA therapy of the invention is about 6 mg. In some embodiments, each dose of an mRNA therapy of the invention is about 7 mg.
- each dose of an mRNA therapy of the invention is administered about once every day. In some embodiments, each dose of an mRNA therapy of the invention is administered once every day.
- one or more bronchodilators are administered to a subject prior to and/or concurrent with administration of a polynucleotide, pharmaceutical composition or formulation of the invention to so as to minimize possible airway irritation.
- bronchodilators include long -acting P agonist (LABA)/inhaled corticosteroid (ICS) therapy and short-acting p agonist (SABA) therapy.
- LABA/ICS therapies include salmeterol/fluticasone, formoterol/budesonide, formoterol/mometasone, and vilanterol/fluticasone.
- SABA therapies include albuterol, levalbuterol, and salbutamol.
- a formulation for a route of administration can include at least one inactive ingredient.
- polynucleotides, pharmaceutical compositions and formulations described above can be administered via the buccal cavity.
- a formulation may comprise dry particles which comprise the active ingredient and which have a diameter in the range from about 0.5 nm to about 7 nm or from about 1 nm to about 6 nm.
- such a formulation may comprise dry particles which have a diameter in the range from about 1 pm to about 5 pm or from about 1 pm to about 6 pm.
- compositions are suitably in the form of dry powders for administration using a device comprising a dry powder reservoir to which a stream of propellant may be directed to disperse the powder and/or using a self propelling solvent/powder dispensing container such as a device comprising the active ingredient dissolved and/or suspended in a low-boiling propellant in a sealed container.
- a self propelling solvent/powder dispensing container such as a device comprising the active ingredient dissolved and/or suspended in a low-boiling propellant in a sealed container.
- Such powders comprise particles wherein at least 98% of the particles by weight have a diameter greater than 0.5 nm and at least 95% of the particles by number have a diameter less than 7 nm. Alternatively, at least 95% of the particles by weight have a diameter greater than 1 nm and at least 90% of the particles by number have a diameter less than 6 nm.
- Dry powder compositions may include a solid fine powder diluent such as sugar and are conveniently provided in a unit dose form.
- Low boiling propellants generally include liquid propellants having a boiling point of below 65° F at atmospheric pressure. Generally the propellant may constitute 50% to 99.9% (w/w) of the composition, and active ingredient may constitute 0.1% to 20% (w/w) of the composition.
- a propellant may further comprise additional ingredients such as a liquid non-ionic and/or solid anionic surfactant and/or a solid diluent (which may have a particle size of the same order as particles comprising the active ingredient).
- polynucleotides, pharmaceutical compositions and formulations of the invention described above may be formulated for pulmonary delivery by the methods described in U.S. Pat. No. 8,257,685; herein incorporated by reference in its entirety.
- Polynucleotides, pharmaceutical compositions and formulations of the invention described above formulated for pulmonary delivery may provide an active ingredient in the form of droplets of a solution and/or suspension.
- Such formulations may be prepared, packaged, and/or sold as aqueous and/or dilute alcoholic solutions and/or suspensions, optionally sterile, comprising active ingredient, and may conveniently be administered using any nebulization and/or atomization device.
- Suitable nebulizers are known in the art, including, e.g., ulstrasonic nebulizers, jet nebulizers, and vibrating -mesh nebulizers.
- the nebulizer is a vibrating-mesh nebulizer.
- Such formulations for pulmonary delivery may further comprise one or more additional ingredients including, but not limited to, a flavoring agent such as saccharin sodium, a volatile oil, a buffering agent, a surface active agent, and/or a preservative such as methylhydroxybenzoate. Droplets provided by this route of administration may have an average diameter in the range from about 0. 1 nm to about 200 nm.
- polynucleotides, pharmaceutical compositions, and formulations described above can be administered via intranasal, nasal, or buccal administration for pulmonary delivery.
- polynucleotides, pharmaceutical compositions, and formulations described herein as being useful for pulmonary delivery are useful for intranasal delivery of a pharmaceutical composition.
- Another formulation suitable for intranasal administration is a coarse powder comprising the active ingredient and having an average particle from about 0.2 pm to 500 pm.
- such a formulation may comprise dry particles which have a diameter in the range from about 1 pm to about 5 pm or from about 1 pm to about 6 pm.
- such a formulation is contained in a capsule or blister.
- Such a formulation is administered in the manner in which snuff is taken, i.e. by rapid inhalation through the nasal passage from a container of the powder held close to the nose.
- Polynucleotides, pharmaceutical compositions, and formulations suitable for nasal administration may, for example, comprise from about as little as 0.1% (w/w) and as much as 100% (w/w) of active ingredient, and may comprise one or more of the additional ingredients described herein.
- Polynucleotides, pharmaceutical compositions, and formulations may be prepared, packaged, and/or sold in a formulation suitable for buccal administration.
- formulations may, for example, be in the form of tablets and/or lozenges made using conventional methods, and may, for example, 0.1% to 20% (w/w) active ingredient, the balance comprising an orally dissolvable and/or degradable composition and, optionally, one or more of the additional ingredients described herein.
- formulations suitable for buccal administration may comprise a powder and/or an aerosolized and/or atomized solution and/or suspension comprising active ingredient.
- Such powdered, aerosolized, and/or aerosolized formulations, when dispersed may have an average particle and/or droplet size in the range from about 0.1 nm to about 200 nm, and may further comprise one or more of any additional ingredients described herein.
- the polynucleotides described herein can be formulated, using the LNPs and methods described herein.
- the formulations can contain polynucleotides that can be modified and/or unmodified.
- the formulations can further include, but are not limited to, cell penetration agents, a pharmaceutically acceptable carrier, a delivery agent, a bioerodible or biocompatible polymer, a solvent, and a sustained-release delivery depot.
- the formulated polynucleotides can be delivered to the cell using routes of administration known in the art and described herein.
- a pharmaceutical composition for parenteral administration can comprise at least one inactive ingredient. Any or none of the inactive ingredients used can have been approved by the US Food and Drug Administration (FDA).
- FDA US Food and Drug Administration
- a non-exhaustive list of inactive ingredients for use in pharmaceutical compositions for parenteral administration includes hydrochloric acid, mannitol, nitrogen, sodium acetate, sodium chloride and sodium hydroxide.
- Formulations can be sterilized, for example, by filtration through a bacterial- retaining filter, and/or by incorporating sterilizing agents in the form of sterile solid compositions that can be dissolved or dispersed in sterile water or other sterile injectable medium prior to use.
- Formulations can be aerosolized using methods known in the art for delivery to the lung.
- the polynucleotides, pharmaceutical compositions and formulations of the invention described above may be formulated for pulmonary delivery by the methods described in U.S. Pat. No. 8,257,685; herein incorporated by reference in its entirety.
- each dose of an mRNA therapy of the invention is administered about once every day using a nebulizer. In some embodiments, each dose of an mRNA therapy of the invention is administered once every day using a nebulizer.
- each dose of an mRNA therapy of the invention is about 1 mg to about 7 mg and administered using a nebulizer. In some embodiments, each dose of an mRNA therapy of the invention is about 1 mg and administered using a nebulizer. In some embodiments, each dose of an mRNA therapy of the invention is about 2 mg and administered using a nebulizer. In some embodiments, each dose of an mRNA therapy of the invention is about 3 mg and administered using a nebulizer. In some embodiments, each dose of an mRNA therapy of the invention is about 4 mg and administered using a nebulizer. In some embodiments, each dose of an mRNA therapy of the invention is about 5 mg and administered using a nebulizer. In some embodiments, each dose of an mRNA therapy of the invention is about 6 mg and administered using a nebulizer. In some embodiments, each dose of an mRNA therapy of the invention is about 7 mg and administered using a nebulizer.
- each dose of an mRNA therapy of the invention is about 1 mg to about 7 mg and administered daily using a nebulizer. In some embodiments, each dose of an mRNA therapy of the invention is about 1 mg and administered daily using a nebulizer. In some embodiments, each dose of an mRNA therapy of the invention is about 2 mg and administered daily using a nebulizer. In some embodiments, each dose of an mRNA therapy of the invention is about 3 mg and administered daily using a nebulizer. In some embodiments, each dose of an mRNA therapy of the invention is about 4 mg and administered daily using a nebulizer.
- each dose of an mRNA therapy of the invention is about 5 mg and administered daily using a nebulizer. In some embodiments, each dose of an mRNA therapy of the invention is about 6 mg and administered daily using a nebulizer. In some embodiments, each dose of an mRNA therapy of the invention is about 7 mg and administered daily using a nebulizer.
- the invention includes embodiments in which exactly one member of the group is present in, employed in, or otherwise relevant to a given product or process.
- the invention includes embodiments in which more than one, or all of the group members are present in, employed in, or otherwise relevant to a given product or process.
- the singular forms “a”, “an” and “the” include plural referents unless the context clearly dictates otherwise.
- the terms “a” (or “an”), as well as the terms “one or more,” and “at least one” can be used interchangeably herein.
- the term “a” or “an” means “single.”
- the term “a” or “an” includes “two or more” or “multiple.”
- Nucleotides are referred to by their commonly accepted single-letter codes. Unless otherwise indicated, nucleic acids are written left to right in 5' to 3' orientation. Nucleobases are referred to herein by their commonly known one-letter symbols recommended by the IUPAC-IUB Biochemical Nomenclature Commission. Accordingly, A represents adenine, C represents cytosine, G represents guanine, T represents thymine, U represents uracil.
- Amino acids are referred to herein by either their commonly known three letter symbols or by the one-letter symbols recommended by the IUPAC-IUB Biochemical Nomenclature Commission. Unless otherwise indicated, amino acid sequences are written left to right in amino to carboxy orientation.
- the term “approximately,” as applied to one or more values of interest, refers to a value that is similar to a stated reference value. In certain embodiments, the term “approximately” refers to a range of values that fall within 25%, 20%, 19%, 18%, 17%, 16%, 15%, 14%, 13%, 12%, 11%, 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1%, or less in either direction (greater than or less than) of the stated reference value unless otherwise stated or otherwise evident from the context (except where such number would exceed 100% of a possible value).
- Dosing regime is a schedule of administration or physician determined regimen of treatment, prophylaxis, or palliative care.
- an effective amount of an agent is that amount sufficient to effect beneficial or desired results, for example, clinical results, and, as such, an "effective amount” depends upon the context in which it is being applied.
- an effective amount of an agent is, for example, an amount of mRNA expressing sufficient CFTR to ameliorate, reduce, eliminate, or prevent the symptoms associated with the CFTR deficiency, as compared to the severity of the symptom observed without administration of the agent.
- the term "effective amount” can be used interchangeably with “effective dose,” “therapeutically effective amount,” or “therapeutically effective dose.”
- CFTR-associated disease or "CFTR-associated disorder” refer to diseases or disorders, respectively, which result from aberrant CFTR activity (e.g., decreased activity or increased activity).
- cystic fibrosis is a CFTR-associated disease. Numerous clinical variants of cystic fibrosis are known in the art. See, e.g., www.omim .org/entry/219700.
- CFTR activity refers to CFTR’s ability to transport chloride ions through the cellular membrane. Accordingly, a polypeptide having CFTR activity refers to a polypeptide that elicits measurable chloride transport across the cell membrane.
- Ionizable amino lipid' includes those lipids having one, two, three, or more fatty acid or fatty alkyl chains and a pH-titratable amino head group (e.g., an alkylamino or dialkylamino head group).
- An ionizable amino lipid is typically protonated (i.e., positively charged) at a pH below the pKa of the amino head group and is substantially not charged at a pH above the pKa.
- Such ionizable amino lipids include, but are not limited to DLin-MC 3 -DMA (MC3) and ( 13Z, 165Z)-N,N-dimethyl-3 -nonydocosa- 13-16-dien- 1 -amine (L608) .
- Methods of Administration can include intravenous, intramuscular, intradermal, subcutaneous, or other methods of delivering a composition to a subject.
- a method of administration can be selected to target delivery (e.g. , to specifically deliver) to a specific region or system of a body.
- Nanoparticle Composition is a composition comprising one or more lipids. Nanoparticle compositions are typically sized on the order of micrometers or smaller and can include a lipid bilayer. Nanoparticle compositions encompass lipid nanoparticles (LNPs), liposomes (e.g., lipid vesicles), and lipoplexes. For example, a nanoparticle composition can be a liposome having a lipid bilayer with a diameter of 500 nm or less.
- LNPs lipid nanoparticles
- liposomes e.g., lipid vesicles
- lipoplexes e.g., lipoplexes.
- a nanoparticle composition can be a liposome having a lipid bilayer with a diameter of 500 nm or less.
- nucleotide sequence encoding refers to the nucleic acid (e.g., an mRNA or DNA molecule) coding sequence which encodes a polypeptide.
- the coding sequence can further include initiation and termination signals operably linked to regulatory elements including a promoter and polyadenylation signal capable of directing expression in the cells of an individual or mammal to which the nucleic acid is administered.
- the coding sequence can further include sequences that encode signal peptides.
- patient refers to a subject who can seek or be in need of treatment, requires treatment, is receiving treatment, will receive treatment, or a subject who is under care by a trained professional for a particular disease or condition.
- the treatment is needed, required, or received to prevent or decrease the risk of developing acute disease, i.e., it is a prophylactic treatment.
- pseudouridine refers to the C-glycoside isomer of the nucleoside uridine.
- a "pseudouridine analog” is any modification, variant, isoform or derivative of pseudouridine.
- pseudouridine analogs include but are not limited to 1 -carboxymethyl -pseudouridine, 1-propynyl- pseudouridine, 1 -taurinomethyl -pseudouridine, 1 -taurinomethyl-4-thio-pseudouridine,
- 1 -methylpseudouridine (m 1 ⁇
- therapeutically effective amount means an amount of an agent to be delivered (e.g., nucleic acid, drug, therapeutic agent, diagnostic agent, prophylactic agent, etc.) that is sufficient, when administered to a subject suffering from or susceptible to an infection, disease, disorder, and/or condition, to treat, improve symptoms of, diagnose, prevent, and/or delay the onset of the infection, disease, disorder, and/or condition.
- an agent to be delivered e.g., nucleic acid, drug, therapeutic agent, diagnostic agent, prophylactic agent, etc.
- Uracil is one of the four nucleobases in the nucleic acid of RNA, and it is represented by the letter U.
- Uracil can be attached to a ribose ring, or more specifically, a ribofuranose via a p-Ni-glycosidic bond to yield the nucleoside uridine.
- the nucleoside uridine is also commonly abbreviated according to the one letter code of its nucleobase, i.e., U.
- U when a monomer in a polynucleotide sequence is U, such U is designated interchangeably as a "uracil” or a "uridine.”
- Uridine Content refers to the amount of uracil or uridine present in a certain nucleic acid sequence. Uridine content or uracil content can be expressed as an absolute value (total number of uridine or uracil in the sequence) or relative (uridine or uracil percentage respect to the total number of nucleobases in the nucleic acid sequence).
- Uridine-Modified Sequence refers to a sequence optimized nucleic acid (e.g., a synthetic mRNA sequence) with a different overall or local uridine content (higher or lower uridine content) or with different uridine patterns (e.g., gradient distribution or clustering) with respect to the uridine content and/or uridine patterns of a candidate nucleic acid sequence.
- nucleobase refers to a purine or pyrimidine heterocyclic compound found in nucleic acids, including any derivatives or analogs of the naturally occurring purines and pyrimidines that confer improved properties (e.g., binding affinity, nuclease resistance, chemical stability) to a nucleic acid or a portion or segment thereof.
- Adenine, cytosine, guanine, thymine, and uracil are the nucleobases predominately found in natural nucleic acids.
- nucleobase sequence of a SEQ ID NO described herein encompasses both natural nucleobases and chemically modified nucleobases (e.g., a “U” designation in a SEQ ID NO encompasses both uracil and chemically modified uracil).
- nucleoside refers to a compound containing a sugar molecule (e.g., a ribose in RNA or a deoxyribose in DNA), or derivative or analog thereof, covalently linked to a nucleobase (e.g., a purine or pyrimidine), or a derivative or analog thereof (also referred to herein as “nucleobase”), but lacking an intemucleoside linking group (e.g., a phosphate group).
- a sugar molecule e.g., a ribose in RNA or a deoxyribose in DNA
- nucleobase e.g., a purine or pyrimidine
- intemucleoside linking group e.g., a phosphate group
- nucleotide refers to a nucleoside covalently bonded to an intemucleoside linking group (e.g., a phosphate group), or any derivative, analog, or modification thereof that confers improved chemical and/or functional properties (e.g., binding affinity, nuclease resistance, chemical stability) to a nucleic acid or a portion or segment thereof.
- an intemucleoside linking group e.g., a phosphate group
- any derivative, analog, or modification thereof e.g., binding affinity, nuclease resistance, chemical stability
- nucleic acid As used herein, the term “nucleic acid” is used in its broadest sense and encompasses any compound and/or substance that includes a polymer of nucleotides, or derivatives or analogs thereof. These polymers are often referred to as “polynucleotides”. Accordingly, as used herein the terms “nucleic acid” and “polynucleotide” are equivalent and are used interchangeably.
- nucleic acids or polynucleotides of the disclosure include, but are not limited to, ribonucleic acids (RNAs), deoxyribonucleic acids (DNAs), DNA-RNA hybrids, RNAi-inducing agents, RNAi agents, siRNAs, shRNAs, mRNAs, modified mRNAs, miRNAs, antisense RNAs, ribozymes, catalytic DNA, RNAs that induce triple helix formation, threose nucleic acids (TNAs), glycol nucleic acids (GNAs), peptide nucleic acids (PNAs), locked nucleic acids (LNAs, including LNA having a P-D-ribo configuration, a-LNA having an a-L-ribo configuration (a diastereomer of LNA), 2'-amino-LNA having a 2'-amino functionalization, and 2'-amino-a-LNA having a 2'-amino functionalization) or hybrid
- Open Reading Frame As used herein, the term “open reading frame”, abbreviated as “ORF”, refers to a segment or region of an mRNA molecule that encodes a polypeptide.
- the ORF comprises a continuous stretch of non-overlapping, in-frame codons, beginning with the initiation codon and ending with a stop codon, and is translated by the ribosome.
- any particular embodiment of the present invention that falls within the prior art can be explicitly excluded from any one or more of the claims. Since such embodiments are deemed to be known to one of ordinary skill in the art, they can be excluded even if the exclusion is not set forth explicitly herein. Any particular embodiment of the compositions of the invention (e.g., any nucleic acid or protein encoded thereby; any method of production; any method of use; etc.) can be excluded from any one or more claims, for any reason, whether or not related to the existence of prior art.
- An mRNA encoding human CFTR (SEQ ID NO: 1) is constructed by using the ORF sequence (nucleotide) provided in SEQ ID NO: 10.
- the mRNA sequence includes both 5' and 3' UTR regions flanking the ORF sequence.
- the 5' UTR and 3' UTR sequences contain SEQ ID NO:50 and SEQ ID NO: 141, respectively.
- the CFTRmRNA sequence is prepared as modified mRNA. Specifically, during in vitro transcription, modified mRNA can be generated using Nl- methylpseudouridine-5'-Triphosphate to ensure that the mRNAs contain 100% Nl- methylpseudouridine instead of uridine.
- modified mRNA can be generated using Nl-methoxyuridine-5 '-Triphosphate to ensure that the mRNAs contain 100% 5 -methoxyuridine instead of uridine.
- CFTR-mRNA can be synthesized with a primer that introduces a polyA-tail, and a cap structure is generated on both mRNAs using co-transcriptional capping to incorporate a m 7 G-ppp-Gm-AG 5' capl.
- CFTR-mRNA can be synthesized and the polyA-tail introduced during Gibson assembly of the DNA template.
- CFTR mRNA can be synthesized with a primer that introduces a polyA-tail. mRNA constructs are modified by ligation to stabilize the polyA tail. Ligation is performed using 0.5-1.5 mg/mL mRNA (5’ Capl, 3’ A100), 50 mM Tris-HCl pH 7.5, 10 mM MgCh, 1 mM TCEP, 1000 units/mL T4 RNA Ligase 1, 1 mM ATP, 20% w/v polyethylene glycol 8000, and 5: 1 molar ratio of modifying oligo to mRNA.
- 5’ Capl, 3’ A100 50 mM Tris-HCl pH 7.5, 10 mM MgCh, 1 mM TCEP, 1000 units/mL T4 RNA Ligase 1, 1 mM ATP, 20% w/v polyethylene glycol 8000, and 5: 1 molar ratio of modifying oligo to mRNA.
- Modifying oligo has a sequence of 5’-phosphate-AAAAAAAAAAAAAAAAAAAA- (inverted deoxythymidine (idT)) (SEQ ID NO:209) (see below). Ligation reactions are mixed and incubated at room temperature ( ⁇ 22°C) for 4 hours. Stable tail mRNA are purified by dT purification, reverse phase purification, hydroxyapatite purification, ultrafiltration into water, and sterile filtration. Ligation efficiency for each mRNA is >80% as assessed by UPLC separation of ligated and unligated mRNA.
- idT inverted deoxythymidine
- the resulting stable tail-containing mRNAs contain the following structure at the 3 ’end, starting with the polyA region: Aioo- UCUAGAAAAAAAAAAAAAAAAAAAAAA-inverted deoxythymidine (SEQ ID NO:211).
- lipid nanoparticles are prepared according to the process outlined in Fig. 1.
- Lipids ionizable lipid: DSPC: cholesterol: DMG-PEG 2000 lipid
- the acidification buffer 45 mM acetate buffer at pH 4
- the lipid solution and acidification buffer are mixed using a multi -inlet vortex mixer at a 3:7 volumetric ratio of lipid: buffer for mixer 1 and mixer 2 and a 1:3 volumetric ratio of lipid:buffer (25% ethanol) for mixer 3.
- the resulting eLNPs are mixed with 55 mM sodium acetate at pH 5.6 at a volumetric ratio of 5:7 of eLNP:buffer.
- the resulting dilute eLNPs are then buffer exchanged and concentrated using tangential flow filtration (TFF) into a final buffer containing 5 mM sodium acetate pH 5.0. Then a 70% sucrose solution in 5 mM acetate buffer at pH 5 is subsequently added.
- lipid nanoparticles prepared according to the procedures above are filled with nucleic acid (mRNA) according to the process depicted in Fig. 2.
- Loading of the mRNA takes place using a post-hoc loading (PHL) process.
- eLNP at a lipid concentration of 11.72 mg/mL in 5 mM acetate (pH 5) and 75 g/L sucrose is mixed with mRNA at a concentration of 1.0 mg/mL in 42.5 mM sodium acetate pH 5.0.
- the eLNP solution and mRNA are mixed using a multi-inlet vortex mixer at a 3 :2 volumetric ratio of eLNP:mRNA.
- the eLNP’s are loaded with mRNA, they undergo a 60 s residence time prior to mixing in-line with a neutralization buffer containing 120 mM TRIS pH 8.12 at a volumetric ratio of 5: 1 of nanoparticle:buffer.
- the nanoparticle formulation is mixed in-line with a buffer containing 20 mM TRIS (pH 7.5), 1.42 mg/mL DMG-PEG 2000, and 2.5 mg/mL GL- 67 (a sterol amine) at a volumetric ratio of 6: 1 of nanoparticle :buffer.
- the resulting nanoparticle suspension undergoes concentration using tangential flow fdtration (TFF) and is diluted in running buffer (20 mM TRIS, 14.3 mM sodium acetate, and 32 g/L sucrose, pH 7.5) with a 300 nM NaCl solution to a final buffer matrix containing 70 mM NaCl.
- the resulting nanoparticle suspension is filtered through a 0.8/0.2 pm capsule filter and filled into glass vials at a mRNA strength of about 1 mg/mL (e.g., 0.5 - 2 mg/mL).
- the clinical plan is to dose into the predicted clinically efficacious range, starting with 1 mg and with a maximum planned nominal dose of 7 mg.
- VX-522 is a modified hCFTR mRNA formulated into a lipid nanoparticle comprised of five lipid components: (1) Compound II; (2) GL-67; (3) 1,2 distearoyl - sn-glycero-3 -phosphocholine (DSPC); (4) cholesterol; and (5) 1,2-dimyristoyl-rac- glycero-3 -methoxypolyethylene gly col-2000 (PEG2000-DMG).
- the eFlow PARI Nebulizer System a medical device manufactured by PARI Respiratory Equipment, will be used to deliver VX-522.
- the SAD study includes two sequential parts.
- Part A a single dose of VX-522 is administered to each subject on Day 1, with daily follow-up through Day 7, and a clinic visit at Day 14.
- the starting dose is 1 mg. All subjects transition from Part A to Part B upon completion of the Day 14 Visit.
- Part B includes follow-up visits on Day 28 and Week 12, and a Safety Followup Visit 24 ( ⁇ 1) weeks after Day 1.
- the SAD comprises approximately 9 subjects (3 cohorts with 3 subjects in each cohort). Based on a 3 + 3 study design, 3 additional subjects may be added per cohort based on emerging data, resulting in up to 18 subjects total.
- Each cohort will include sequential dosing of subjects.
- the previous subject’s safety and tolerability data through the Day 7 Visit will be reviewed before the next subject in the cohort is dosed.
- Previous cohorts may be expanded to 6 subjects, or an intermediate dose may be enrolled with 3 new subjects to identify the VX-522 maximum tolerated dose.
- the IDMC will assess the death’s relationship to VX- 522. Dose escalation may only proceed if there are no deaths attributable to the study drug following review by the IDMC.
- the initiation of successive cohorts and dose selection will be based on evaluation of available safety and tolerability data from the previous cohort. If 2 or more subjects in a cohort have dose-limiting toxicity or there is a death attributed to study drug, the dose may be de-escalated (i.e., all subsequent cohorts will evaluate lower doses of VX-522).
- the starting dose in the MAD study will be at or below dose levels for which safety and tolerability have been established in the SAD study.
- Subjects who participate in the SAD study may enroll in the MAD study after completion of the Safety Follow-up Visit.
- the MAD study includes two treatment arms, T1 and T2.
- T1 evaluates multiple ascending doses of VX-522.
- T1 evaluates MAD of VX- 522 in 2 cohorts with approximately 9 subjects per cohort dosed daily with VX-522 for 28 days and a Safety Follow-up Visit 28 ( ⁇ 3) days after the last dose of VX-522. Up to 3 additional cohorts may be added, the size of each cohort may be increased or decreased (with a minimum of 6 subjects), and some cohorts may dose VX 522 less frequently than daily dosing, based on emerging data.
- 1 sentinel subject will be dosed. Before the remaining subjects in the cohort are dosed, the sentinel subject’s safety and tolerability data through the Day 15 Visit (hereafter referred to as the sentinel data) will be reviewed.
- T2 evaluates multiple doses of VX-522 while also receiving oral CFTR potentiator (ivacaftor; IVA) treatment.
- T2 includes 1 cohort of 9 subjects.
- One additional cohort may be enrolled T2, the size of each cohort may be increased or decreased (with a minimum of 6 subjects), or some cohorts may dose VX-522 less frequently than daily dosing, based on emerging data.
- T2 may be conducted concurrently with T1 Cohort 2 after the completion of sentinel data review for T1 Cohort 2. Sentinel data from T1 Cohort 2 must be reviewed before the first subject in T2 receives the first dose of VX-522. Run-in dosing with IVA in T2 may occur before review of the T1 Cohort 2 sentinel data.
- Subjects in T2 will receive IVA (150 mg) every 12 hours for 28 days during the Run-in Period. During the Treatment Period, subjects will receive daily VX-522 while also receiving IVA for 28 days. The last dose of IVA will be on the morning of the Day 29 Visit, with a Safety Follow-up Visit 28 ( ⁇ 3) days after the last dose of VX-522.
- CFTR mutations that are considered not responsive to CFTR modulator therapy on both alleles to be eligible for the study.
- a CFTR mutation is considered not responsive to CFTR modulator therapy if it meets at least one of the following two criteria:
- Table 4 includes acceptable mutations, which are detectable by an FDA- cleared genotyping assay or other method (e.g., sequencing). This list does not include every eligible mutation.
- the primary objectives are of the study are to evaluate the safety and tolerability of: (1) single ascending doses of VX-522 (SAD); (2) multiple ascending doses of VX-522 (MAD, Tl); and (3) multiple doses of VX-522 coadministered with ivacaftor treatment (MAD, T2).
- the primary objectives are evaluated by measuring safety and tolerability, based on the assessment of adverse events (AEs), clinical laboratory values (serum chemistry, hematology, coagulation, and urinalysis), standard 12-lead ECGs, vital signs, pulse oximetry, spirometry, and immune response to VX-522 components and CFTR protein.
- AEs adverse events
- clinical laboratory values serum chemistry, hematology, coagulation, and urinalysis
- standard 12-lead ECGs vital signs
- pulse oximetry pulse oximetry
- spirometry immune response to VX-522 components and CFTR protein.
- the secondary objectives of the study are: (1) to evaluate the efficacy of administration of multiple ascending doses of VX-522 (MAD, Tl); and (2) to evaluate the efficacy of multiple doses of VX-522 co-administered with ivacaftor treatment (MAD, T2).
- the secondary objectives are evaluated by measuring: (1) change from baseline in percent predicted forced expiratory volume in 1 second (ppFEVi) at Day 29 (MAD, Tl); and (2) change from baseline and pre-Run-in baseline in ppFEVi at Day 29 (MAD, T2).
- VX-522 components mRNA and lipid nanoparticle constituents
- SAD single ascending doses
- MAD multiple ascending doses
- T2 multiple doses of VX-522 co-administered with ivacaftor treatment
- the other objectives are evaluated by measuring: (1) concentrations of VX- 522 components (mRNA and lipid nanoparticle constituents) in blood (SAD); (2) change from baseline in Cystic Fibrosis Questionnaire - Revised respiratory domain (CFQ-R RD) score at Day 29, concentrations of VX-522 components (mRNA and lipid nanoparticle constituents) in blood, exogenous CFTR mRNA in airway tissue (bronchoscopy substudy only), CFTR protein expression in airway tissue (bronchoscopy substudy only), and VX-522 components (mRNA and lipid nanoparticle constituents) in bronchoalveolar lavage fluid (bronchoscopy substudy only) (MAD, Tl); and (3) change from baseline and pre-Run-in baseline in CFQ-R RD score at Day 29, concentrations of VX-522 components (mRNA and LNP constituents) in blood, exogenous CFTR mRNA in airway tissue (bronchoscopy substudy only), CFTR protein expression in airway
- bronchodilators including a metered dose inhaler (MDI) long-acting P agonist (LABA)Zinhaled corticosteroid (ICS) therapy and short-acting p agonist (SABA) therapy.
- MDI metered dose inhaler
- LABA long-acting P agonist
- SABA short-acting p agonist
- Table 5 Examples of Acceptable LABA/ICS and SABA Therapies Run-in Period: All subjects will be dosed with LABA/ICS therapy for at least 28 days in the Run-in Period before the first dose of VX-522. Subjects who are not already taking a LABA/ICS therapy and a SABA before participation in the MAD will be prescribed these therapies before the Run-in Period begins. Subjects who are already taking medications listed in Table 5 before study participation (as part of their routine medical care) will continue those medications and will still be required to complete all Run-in Period visits.
- Treatment Period Subjects will be treated with LABA/ICS and SABA therapies before each VX-522 dosing occasion during the Treatment Period.
- CF-HBE cell cultures derived from donor cystic fibrosis lungs are a well- established, clinically validated translational model that predicts the level of clinical benefit associated with improved CFTR function by orally available small molecule CFTR modulators.
- CFHBE cell cultures display a striking phenotypic and functional resemblance to the surface airway epithelium in people with CF. They form a pseudostratified epithelium composed of secretory cells (goblet and club cells), ciliated cells, ionocytes, and basal cells.
- VX-522-mediated CFTR protein expression in lung surface epithelial cells was evaluated in vitro using CF-HBE cell cultures derived from donor cystic fibrosis lungs, and in vivo using monkeys. CF-HBE cells were also used to assess CFTR- mediated chloride transport resulting from VX-522-mediated CFTR protein expression and to benchmark efficacy of VX-522 to CFTR modulators known to provide clinical benefit in people with CF.
- VX-522 or a buffer control were nebulized to the apical surface of CF-HBE cell cultures derived from one F508del/MF (F508del/3905insT) and two MF/MF (G542X/R553X and G542X/ K684SfsX38) donor bronchi.
- G542X and R553X, nonsense mutations, and K684SfsX38, a frameshift mutation lead to no CFTR protein production or CFTR function.
- Ussing chamber electrophysiology was used to directly measure CFTR-mediated chloride transport.
- VX-522- mRNA and CFTR protein were assessed in VX-522-nebulized CF-HBE cells by standardized in situ hybridization (ISH) using a VX-522-mRNA-specific RNAScope probe, and immunohistochemistry (IHC) respectively.
- ISH in situ hybridization
- IHC immunohistochemistry
- Nebulized delivery of VX-522 to F508del/MF-HBE and MF/MF-HBE cells resulted in VX-522-mRNA deposition and CFTR protein expression in multiple epithelial cell types, including ionocytes and goblet cells which are known to endogenously express CFTR.
- Ionocytes were identified by labeling epithelia with Barttin (BSND), an ionocyte marker, while goblet cells and ciliated cells were identified by morphological analysis.
- BSND Barttin
- goblet cells and ciliated cells were identified by morphological analysis.
- the increase in % CFTR-positive cells was associated with a dose-dependent increase in CFTR function to levels known to result in clinical benefit in people with CF.
- the VX-522-mediated increase in CFTR-mediated chloride transport declines over time with an estimated functional half-life of 36 hours, supporting a daily dosing regimen to maintain CFTR function at levels required for clinical benefit in people with CF.
- VX-522-mRNA deposition and CFTR protein expression was demonstrated in vivo following daily nebulized delivery of VX-522 to the lung of monkeys via oronasal inhalation for up to 28 days.
- the increase in the percentage of CFTR proteinexpressing monkey lung epithelial cells was comparable to that observed in CF-HBE cells associated with levels of CFTR function known to result in clinical benefit.
- VX- 522-mRNA deposition and CFTR protein expression was widely distributed throughout the lung and observed primarily in bronchial surface epithelial cells.
- the luminal surface of bronchial epithelial cells exhibited strong apical CFTR expression that correlated with VX-522 -mRNA deposition. Histopathological examination showed apical CFTR expression in multiple luminally exposed cell types.
- No VX- 522-related effects on safety pharmacology parameters were observed following inhalation exposure in vivo up to 19 pg/kg/day.
- CF-HBE cell cultures derived from donor cystic fibrosis lungs is a well- established, clinically validated translational model that predicts the level of clinical benefit associated with improved CFTR function by therapeutic intervention.
- HBE cell cultures display a striking phenotypic and functional resemblance to the conducting airway epithelium. They form pseudostratified epithelium composed of secretory cells (goblet and club cells), ciliated cells, ionocytes, and progenitor cells, lonocytes express the highest levels of CFTR but are rare (approximately 1% of airway epithelial cells). The more abundant secretory cells express low to medium levels of CFTR.
- Buffer or VX-522 was nebulized to the apical surface of CF-HBE cell cultures derived from 1 F50SJe//minimal function (MF) (F508del/3905insT) and 2 MF/MF (G542X/R553X and G542X/ K684SfsX38) donor bronchi.
- MF F508del/3905insT
- MF MF/MF
- G542X/R553X and G542X/ K684SfsX38 donor bronchi.
- G542X and R553X nonsense mutations
- K684SfsX38 a frameshift mutation
- VX-522- mRNA, CFTR protein, and CFTR function were typically analyzed approximately 18 hours after treatment by standardized in situ hybridization (ISH), immunohistochemistry (IHC), and Ussing chamber electrophysiology methods, respectively.
- Nebulization of VX-522 to the apical surface of F50&/e//MF-HBE and MF/MF-HBE cells resulted in delivery of VX-522-mRNA and exogenous expression of CFTR protein in multiple epithelial cell types, including those known to endogenously express CFTR (Figs. 3A-3C).
- CFTR expression was associated with a dose-dependent increase in CFTR function to levels comparable to ELX/TEZ/IVA treatment in F508 de//MF-HBE cells (Fig. 3E).
- This level of CFTR function in the HBE cell assay is known to result in clinical benefit in people with cystic fibrosis.
- CFTR function returned to baseline levels with a half-life of approximately 36 hours, supporting a daily dosing regimen to sustain CFTR-mediated chloride transport.
- VX-522-mRNA delivery to airway epithelial cells that results in increased CFTR protein expression
- a nebulized formulation of VX-522 or buffer control was administered daily to monkeys (cynomolgus macaques) via oronasal mask inhalation for 1 to 28 days using vibrating mesh nebulizers.
- VX- 522-mRNA delivery was assessed by ISH using a VX-522-mRNA-specific RNAScope probe.
- CFTR protein expression was assessed by IHC using an anti-CFTR antibody that detects human and monkey CFTR.
- Buffer, VX-522, or VX-522 in combination with clinically relevant concentrations of ivacaftor was nebulized to the apical surface of CF-HBE cell cultures derived from two MF/MF (G542X/R553X and G542X/ K684SfsX38) donor bronchi.
- MF mutations such as G542X, R553X, and K684SfsX38, lead to no CFTR protein production or CFTR function.
- CFTR-mediated chloride transport was quantified by Ussing chamber electrophysiology .
- VX-522 can increase Cl" current in MF/MF CF-HBE cells and that the addition of ivacaftor can further increase the VX-522-mediated increase in Cl" current.
- VX-522 in combination with ivacaftor increased Cl" currents in MF/MF CF-HBE cells to levels similar to or above those induced by elexacaftor/tezacaftor/ivacaftor in F508del/MF CF-HBE cells, levels that have previously been shown to translate into clinically meaningful benefits for cystic fibrosis patients.
- VX-522 may provide clinical benefit in people with cystic fibrosis caused by minimal function mutations on both alleles that are otherwise unresponsive to CFTR modulators and combining VX-522 with ivacaftor may provide additional clinical benefit versus VX-522 monotherapy.
- EXAMPLE 8 Human Dose Selection
- nebulized VX-522 (1) produces functional CFTR protein when administered to HBEs from cystic fibrosis donors; (2) can deliver VX-522-mRNA to monkey lung epithelial cells that results in increased CFTR protein expression at doses well below the no-observed-adverse-effect-level (NOAEL); (3) have a concordant safety profile in rats and monkeys consisting of non-adverse changes that are consistent with a local adaptive inflammatory response to an inhaled foreign agent; and (4) are effective for local delivery of VX-522 components, with negligible systemic exposure and systemic toxicity risk.
- NOAEL no-observed-adverse-effect-level
- the estimated achieved pulmonary (deposited) doses from the 28-day good laboratory practice (GLP) rat and monkey studies were converted from a pg/kg body weight dose to a pg/g lung tissue dose. This conversion was conducted because (1) the lung is the pharmacological target organ; (2) there was no evidence of systemic toxicity associated with VX-522 in either species; and (3) VX-522-related non-adverse changes were limited to the lung and associated lymphoid tissue in both species. This approach is consistent with recommendations in the literature.
- the achieved pulmonary (deposited) doses were 2.9 pg/g lung (rat) and 4.12 pg/g lung (monkeys).
- the monkey was considered as the most relevant species for subsequent calculations of the safe starting dose in the FIH study because human respiratory physiology, airway geometry, and cellular composition of the lower respiratory tract is more similar to monkeys than rats.
- the deposition fraction of inhaled aerosols is assumed to be the same for monkeys and human.
- proof of delivery of hCFTR mRNA and CFTR protein was demonstrated in monkeys following oronasal inhalation of VX- 522.
- hCFTR is more similar to monkey CFTR than rat CFTR (based on peptide sequence), which may have influenced the immunogenicity profile of VX-522 in vivo.
- the achieved pulmonary (deposited) dose at the NOAEL in monkeys of 4.12 pg/g lung was multiplied by the weight of a human lung (assumed to be 1000 g in a 60 kg human) to derive a 4.12 mg achieved pulmonary (deposited) human dose.
- the PARI nebulizer utilizing eFlow technology was demonstrated to deliver approximately 40% of the nominal VX-522 dose into the lung.
- the human equivalent pulmonary dose of 4. 12 mg at the NOAEL in monkeys was divided by 0.40 to account for the fraction of delivery and deposition (device efficiency) using the PARI nebulizer, leading to the human equivalent nominal dose of 10 mg.
- An approximate 10-fold safety margin was applied to the 10 mg nominal dose to arrive at the 1 mg nominal safe starting FIH dose.
- lung deposition modeling was used to calculate the nominal dose needed to achieve the equivalent amount of VX-522 at the lung surface that resulted in Trikafta (TRI)-like levels of CFTR function in F508del/minimal function-HBE cells.
- TRI Trikafta
- the CF-HBE cell model, and the comparison to Trikafta, were selected as benchmarks based on the well-established translation of this assay and treatment to clinical benefit. This approach estimated a daily nominal dose of 6 to 12 mg of VX- 522 would be efficacious.
- lung-weight-based scaling was used to calculate the human equivalent dose that would correspond to the deposited dose in monkeys that led to a 2% to 4% increase in the proportion of CFTR protein-positive epithelial cells.
- the 2% to 4% increase in CFTR-protein-positive cells is based on the proportion of CFTR proteinpositive cells observed in VX-522 -treated CF-HBE cells that resulted in CFTR- mediated chloride transport increases known to translate into clinical benefit. This approach estimated a daily nominal dose of approximately 1 mg of VX-522 would be efficacious.
- the clinical plan for Phase 1/2 is to dose into the predicted clinically efficacious range, with a maximum planned nominal dose of 7 mg.
- the monkey NOAEL provides an approximate 1.5-fold safety margin over 7 mg clinical nominal dose.
- the selection of the starting nominal dose was based on the totality of nonclinical toxicology and pharmacology data, in accordance with EMA and FDA guidance on FIH starting dose selection.
- the starting dose was selected based on NOAEL established for monkeys in the GLP-compliant non-clinical safety studies.
- the clinically efficacious dose range was estimated to provide assurance that the starting dose and maximum dose will be appropriate for evaluation in a Phase 1/Phase 2 study of patients with cystic fibrosis.
- the starting nominal dose of 1 mg was selected as follows:
- NOAELs were established in GLP-compliant nonclinical safety studies of VX-522 dosing in monkeys for 28 days (19 pg/kg/day).
- the NOAELs were first converted into human equivalent lung-deposited doses using lung weight scaling.
- the monkey NOAEL of 19 pg/kg/day corresponds to a human equivalent lung -deposited dose of 4. 1 mg.
- the amount of VX-522 required in the nebulizer (called the “nominal dose”) to achieve this human-equivalent lung dose was then calculated based on nebulizer efficiency measured with specific configuration for VX-522.
- a dose of 10 mg in the nebulizer (nominal dose) is predicted to result in a lung-deposited dose of 4.1 mg.
- An exposure margin of approximately 10-fold relative to the human equivalent nominal dose of the monkey NOAEL was applied to select the starting (nominal) dose of 1 mg.
- the planned maximum dose will be 7 mg nominal dose, which is predicted to provide an approximately 1.5-fold safety margin under the human equivalent of the monkey NOAEL.
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Chemical & Material Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Genetics & Genomics (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Organic Chemistry (AREA)
- Biotechnology (AREA)
- Epidemiology (AREA)
- Biomedical Technology (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Molecular Biology (AREA)
- General Engineering & Computer Science (AREA)
- Pulmonology (AREA)
- Physics & Mathematics (AREA)
- General Chemical & Material Sciences (AREA)
- Plant Pathology (AREA)
- Biochemistry (AREA)
- Biophysics (AREA)
- Microbiology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nanotechnology (AREA)
- Optics & Photonics (AREA)
- Dispersion Chemistry (AREA)
- Otolaryngology (AREA)
- Marine Sciences & Fisheries (AREA)
- Gastroenterology & Hepatology (AREA)
- Immunology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
This disclosure relates to mRNA therapy for the treatment of cystic fibrosis. mRNAs for use in the invention, when administered in vivo, encode cystic fibrosis transmembrane conductance regulator (CFTR). mRNA therapies of the disclosure increase and/or restore deficient levels of CFTR expression and/or activity in subjects.
Description
POLYNUCLEOTIDES ENCODING CYSTIC FIBROSIS TRANSMEMBRANE CONDUCTANCE REGULATOR FOR THE TREATMENT OF CYSTIC FIBROSIS
CROSS-REFERENCE TO RELATED APPLICATIONS
This application claims the benefit of priority of U.S. Provisional Application No. 63/601,353, filed on November 21, 2023, the contents of which is incorporated by reference in its entirety herein.
SEQUENCE LISTING
The instant application contains a Sequence Listing which has been submitted electronically in XML format and is hereby incorporated by reference in its entirety. Said XML copy, created on November 18, 2024, is named 45817-0131WOl_SL.xml and is 607,429 bytes in size.
BACKGROUND
Cystic Fibrosis (“CF”) is a rare autosomal recessive disease with serious, chronically debilitating morbidities and high premature mortality for which there is currently no cure. Cystic Fibrosis affects more than 80,000 individuals worldwide, including over 31,000 individuals in the US and 49,000 individuals in the EU.
Cystic Fibrosis is caused by decreased quantity and/or function of the cystic fibrosis transmembrane conductance regulator (CFTR) protein due to mutations in the CFTR gene. CFTR is an ion channel that regulates the flow of chloride and other ions across epithelia in various tissues, including the lungs, pancreas and other gastrointestinal organs, and sweat glands. Decreased CFTR quantity or function results in the failure to regulate chloride transport in these tissues leading to the multisystem pathology associated with CF. Progressive loss of lung function is the leading cause of mortality. People with cystic fibrosis who have mutations in both alleles of CFTR that result in complete or near complete loss of CFTR-mediated chloride transport (e.g., F508del or Class I mutations which make no CFTR protein) demonstrate severe cystic fibrosis characterized by early onset and relatively rapid disease progression.
The most common disease-causing mutation is F5O8del: approximately 85% of individuals in the US and 80% of individuals in Europe have at least one F508del mutation. People with at least one F508del mutation, or at least one of over 170 other CFTR mutations, can be treated with the CFTR modulators. With extended use, restoration of CFTR function by CFTR modulators has been shown to modify the course of cystic fibrosis disease by improving survival and reducing the rate of decline in lung function.
However, approximately 7% of people with cystic fibrosis have mutations in the CFTR gene that result in no CFTR protein or a mutant CFTR protein that is not responsive to CFTR modulators. These people are unable to benefit from the substantial advances provided by CFTR modulator therapies.
SUMMARY
The present disclosure provides messenger RNA (mRNA) therapeutics for the treatment of cystic fibrosis. The mRNA therapeutics of the invention are particularly well-suited for the treatment of cystic fibrosis as the technology provides for the intracellular delivery of mRNA encoding a cystic fibrosis transmembrane conductance regulator (CFTR) polypeptide followed by de novo synthesis of functional CFTR polypeptide within target cells.
In one aspect, the disclosure features a method of treating cystic fibrosis in a human subject in need thereof, the method comprising administering to the human subject by inhalation a lipid nanoparticle comprising a mRNA comprising an open reading frame (ORF) encoding the CFTR polypeptide of SEQ ID NO: 1, wherein the mRNA is administered at a dose of 1 mg to 7 mg.
In some embodiments, the ORF is at least 80% identical to the nucleotide sequence of SEQ ID NO: 10.
In some embodiments, the ORF is at least 95% identical to the nucleotide sequence of SEQ ID NO: 10.
In some embodiments, the ORF is at least 99% identical to the nucleotide sequence of SEQ ID NO: 10.
In some embodiments, the ORF is 100% identical to the nucleotide sequence of SEQ ID NO: 10.
In some embodiments, the mRNA comprises a 5' UTR comprising the nucleotide sequence of SEQ ID NO:50.
In some embodiments, the mRNA comprises a 3' UTR comprising the nucleotide sequence of SEQ ID NO: 141.
In some embodiments, the mRNA comprises the nucleic acid sequence of SEQ ID NO: 11.
In some embodiments, the mRNA comprises a 5' terminal cap. In some embodiments, the 5' terminal cap comprises m7G-ppp-Gm.
In some embodiments, the mRNA comprises a poly-A region. In some embodiments, the poly-A region comprises SEQ ID NO: 195. In some embodiments, the poly-A region comprises A100-UCUAG-A20-inverted deoxy-thymidine (SEQ ID NO:211).
In some embodiments, all of the uracils of the mRNA are Nl- methylpseudouracils .
In some embodiments, the mRNA comprises the nucleotide sequence of SEQ ID NO: 13.
In some embodiments, the mRNA is administered at a dose of 1 mg.
In some embodiments, the mRNA is administered at a dose of 2 mg.
In some embodiments, the mRNA is administered at a dose of 3 mg.
In some embodiments, the mRNA is administered at a dose of 4 mg.
In some embodiments, the mRNA is administered at a dose of 5 mg.
In some embodiments, the mRNA is administered at a dose of 6 mg.
In some embodiments, the mRNA is administered at a dose of 7 mg.
In some embodiments, the method comprises multiple administrations of the dose.
In some embodiments, the dose is administered repeatedly once every day.
In some embodiments, the method comprises at least 28 consecutive daily administrations of the dose.
In some embodiments, the lipid nanoparticle is administered using a nebulizer.
In some embodiments, the human subject carries mutations in both alleles of the CFTR gene that result in no CFTR protein produced or a mutant CFTR protein that is not responsive to therapy with CFTR modulators. In some embodiments, the CFTR modulators comprise ivacaftor, lumacaftor/ivacaftor, tezacaftor/ivacaftor, and elexacaftor/tezacaftor/ivacaftor. In some embodiments, the mutations in both alleles of the CFTR gene are selected from the mutations depicted in Table 4.
In some embodiments, the method comprises co-administering a CFTR potentiator to the human subject. In some embodiments, the CFTR potentiator is ivacaftor, lumacaftor/ivacaftor, tezacaftor/ivacaftor, or elexacaftor/tezacaftor/ivacaftor. In some embodiments, the CFTR potentiator is ivacaftor. In some embodiments, ivacaftor is administered orally. In some embodiments, ivacaftor is administered orally every 12 hours at a dose of 150 mg.
In some embodiments, the mRNA is administered once every day at a dose of
1 mg. In some embodiments, the mRNA is administered once every day at a dose of 1 mg and ivacaftor is administered orally. In some embodiments, the mRNA is administered once every day at a dose of 1 mg and ivacaftor is administered orally every 12 hours at a dose of 150 mg.
In some embodiments, the mRNA is administered once every day at a dose of
2 mg. In some embodiments, the mRNA is administered once every day at a dose of 2 mg and ivacaftor is administered orally. In some embodiments, the mRNA is administered once every day at a dose of 2 mg and ivacaftor is administered orally every 12 hours at a dose of 150 mg.
In some embodiments, the mRNA is administered once every day at a dose of
3 mg. In some embodiments, the mRNA is administered once every day at a dose of 3 mg and ivacaftor is administered orally. In some embodiments, the mRNA is administered once every day at a dose of 3 mg and ivacaftor is administered orally every 12 hours at a dose of 150 mg.
In some embodiments, the mRNA is administered once every day at a dose of
4 mg. In some embodiments, the mRNA is administered once every day at a dose of 4 mg and ivacaftor is administered orally. In some embodiments, the mRNA is
administered once every day at a dose of 4 mg and ivacaftor is administered orally every 12 hours at a dose of 150 mg.
In some embodiments, the mRNA is administered once every day at a dose of
5 mg. In some embodiments, the mRNA is administered once every day at a dose of 5 mg and ivacaftor is administered orally. In some embodiments, the mRNA is administered once every day at a dose of 5 mg and ivacaftor is administered orally every 12 hours at a dose of 150 mg.
In some embodiments, the mRNA is administered once every day at a dose of
6 mg. In some embodiments, the mRNA is administered once every day at a dose of 6 mg and ivacaftor is administered orally. In some embodiments, the mRNA is administered once every day at a dose of 6 mg and ivacaftor is administered orally every 12 hours at a dose of 150 mg.
In some embodiments, the mRNA is administered once every day at a dose of
7 mg. In some embodiments, the mRNA is administered once every day at a dose of 7 mg and ivacaftor is administered orally. In some embodiments, the mRNA is administered once every day at a dose of 7 mg and ivacaftor is administered orally every 12 hours at a dose of 150 mg.
In some embodiments, the mRNA comprises the nucleotide sequence of SEQ ID NO: 13 and the mRNA is administered once every day at a dose of 1 mg. In some embodiments, the mRNA comprises the nucleotide sequence of SEQ ID NO: 13, the mRNA is administered once every day at a dose of 1 mg, and ivacaftor is administered orally. In some embodiments, the mRNA comprises the nucleotide sequence of SEQ ID NO: 13, the mRNA is administered once every day at a dose of 1 mg, and ivacaftor is administered orally every 12 hours at a dose of 150 mg.
In some embodiments, the mRNA comprises the nucleotide sequence of SEQ ID NO: 13 and the mRNA is administered once every day at a dose of 2 mg. In some embodiments, the mRNA comprises the nucleotide sequence of SEQ ID NO: 13, the mRNA is administered once every day at a dose of 2 mg, and ivacaftor is administered orally. In some embodiments, the mRNA comprises the nucleotide sequence of SEQ ID NO: 13, the mRNA is administered once every day at a dose of 2 mg, and ivacaftor is administered orally every 12 hours at a dose of 150 mg.
In some embodiments, the mRNA comprises the nucleotide sequence of SEQ ID NO: 13 and the mRNA is administered once every day at a dose of 3 mg. In some embodiments, the mRNA comprises the nucleotide sequence of SEQ ID NO: 13, the mRNA is administered once every day at a dose of 3 mg, and ivacaftor is administered orally. In some embodiments, the mRNA comprises the nucleotide sequence of SEQ ID NO: 13, the mRNA is administered once every day at a dose of 3 mg, and ivacaftor is administered orally every 12 hours at a dose of 150 mg.
In some embodiments, the mRNA comprises the nucleotide sequence of SEQ ID NO: 13 and the mRNA is administered once every day at a dose of 4 mg. In some embodiments, the mRNA comprises the nucleotide sequence of SEQ ID NO: 13, the mRNA is administered once every day at a dose of 4 mg, and ivacaftor is administered orally. In some embodiments, the mRNA comprises the nucleotide sequence of SEQ ID NO: 13, the mRNA is administered once every day at a dose of 4 mg, and ivacaftor is administered orally every 12 hours at a dose of 150 mg.
In some embodiments, the mRNA comprises the nucleotide sequence of SEQ ID NO: 13 and the mRNA is administered once every day at a dose of 5 mg. In some embodiments, the mRNA comprises the nucleotide sequence of SEQ ID NO: 13, the mRNA is administered once every day at a dose of 5 mg, and ivacaftor is administered orally. In some embodiments, the mRNA comprises the nucleotide sequence of SEQ ID NO: 13, the mRNA is administered once every day at a dose of 5 mg, and ivacaftor is administered orally every 12 hours at a dose of 150 mg.
In some embodiments, the mRNA comprises the nucleotide sequence of SEQ ID NO: 13 and the mRNA is administered once every day at a dose of 6 mg. In some embodiments, the mRNA comprises the nucleotide sequence of SEQ ID NO: 13, the mRNA is administered once every day at a dose of 6 mg, and ivacaftor is administered orally. In some embodiments, the mRNA comprises the nucleotide sequence of SEQ ID NO: 13, the mRNA is administered once every day at a dose of 6 mg, and ivacaftor is administered orally every 12 hours at a dose of 150 mg.
In some embodiments, the mRNA comprises the nucleotide sequence of SEQ ID NO: 13 and the mRNA is administered once every day at a dose of 7 mg. In some embodiments, the mRNA comprises the nucleotide sequence of SEQ ID NO: 13, the
mRNA is administered once every day at a dose of 7 mg, and ivacaftor is administered orally. In some embodiments, the mRNA comprises the nucleotide sequence of SEQ ID NO: 13, the mRNA is administered once every day at a dose of 7 mg, and ivacaftor is administered orally every 12 hours at a dose of 150 mg.
In some embodiments, the treatment increases percent predicted forced expiratory volume in 1 second (ppFEVi) from baseline.
In some embodiments, the method comprises administering to the human subject long-acting p agonist (LABA)Zinhaled corticosteroid (ICS) therapy. In some embodiments, the human subject is administered LABA/ICS therapy prior to administration of the lipid nanoparticle. In some embodiments, the human subject is administered LABA/ICS therapy for at least 28 days before the first administration of the lipid nanoparticle.
In some embodiments, the method comprises administering to the human subject LABA/ICS therapy and short-acting p agonist (SABA) therapy. In some embodiments, the human subject is administered LABA/ICS therapy and SABA therapy prior to administration of the lipid nanoparticle. In some embodiments, the human subject is administered LABA/ICS therapy and SABA therapy on the same day as administration of the lipid nanoparticle. In some embodiments, the human subject is administered LABA/ICS therapy prior to administration of the lipid nanoparticle and the human subject is administered LABA/ICS therapy and SABA therapy on the same day as administration of the lipid nanoparticle. In some embodiments, the human subject is administered LABA/ICS therapy for at least 28 days before the first administration of the lipid nanoparticle and the human subject is administered LABA/ICS therapy and SABA therapy on the same day as administration of the lipid nanoparticle.
In some embodiments, the lipid nanoparticle comprises:
(i) an ionizable lipid,
(ii) a phospholipid;
(iii) a structural lipid;
(iv) a PEG-lipid; and
(v) a cationic agent.
In some embodiments, the ionizable lipid is
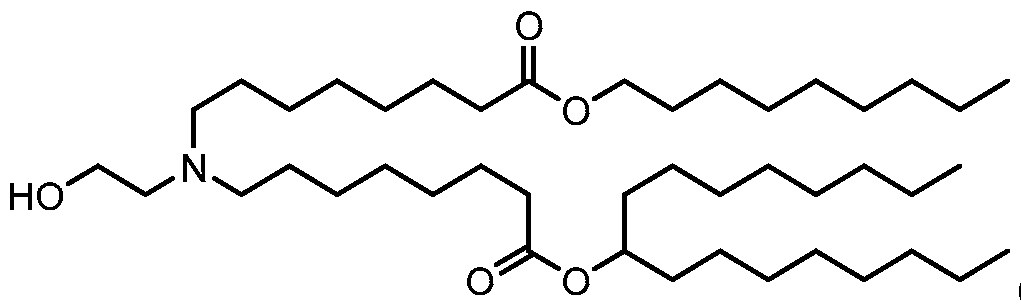 (Compound II) or a salt thereof.
(Compound II) or a salt thereof.
In some embodiments, the cationic agent is
 salt thereof.
salt thereof.
In some embodiments, the ionizable lipid is
 thereof, and the cationic agent is
thereof, and the cationic agent is
 salt thereof.
salt thereof.
In some embodiments,
(i) the ionizable lipid is
 (Compound II) or a salt thereof;
(Compound II) or a salt thereof;
(ii) the phospholipid is 1,2 distearoyl-sn-glycero-3-phosphocholine (DSPC);
(iii) the structural lipid is cholesterol;
(iv) the PEG lipid is l,2-dimyristoyl-rac-glycero-3 -methoxypolyethylene glycol-2000 (PEG2000-DMG); and
(v) the cationic agent is
 salt thereof.
salt thereof.
BRIEF DESCRIPTION OF THE DRAWINGS
Fig. 1 shows a general process for preparing empty lipid nanoparticles (eLNPs) where nanoprecipitation is carried out at pH 4 followed by titration to pH 5.
Fig. 2 shows a general process for preparing fdled lipid nanoparticles (fLNPs) where encapsulation is carried out at pH 5.
Fig. 3A contains representative photomicrographs depicting CFTR mRNA (ISH) and CFTR protein (IHC) 18 hours after a single nebulized delivery of buffer or 3.8 pg/cm2 VX-522 to the apical surface of G542X/K684SfsX38 CF-HBE cells. The cells were formalin-fixed paraffin-embedded, and nuclei were counter stained with hematoxylin.
Fig. 3B contains representative photomicrographs demonstrating apical CFTR expression in different bronchial epithelial cell types including ionocytes, goblet cells and ciliated cells in VX-522 treated (3.8 pg/cm2) G542X/K684SfsX38 CF-HBE cells but not in buffer-treated controls.
Fig. 3C is a graph depicting morphometric analysis of CFTR protein in MF/MF-HBE cells from Figs. 3 A and 3B. Data indicate the total percentage of MF/MF HBE cells expressing CFTR protein 18 hours after a single treatment with VX-522 or buffer control. Numbers on the x-axis denote delivered VX-522-mRNA expressed as deposited pg/cm2. Data represents the mean of 3 to 4 replicate experiments, ± SEM in CF-HBE cells from 2 MF/MF donors.
Fig. 3D is a graph depicting dose-dependent increase in CFTR-mediated chloride transport in CF-HBE cells derived from 2 MF/MF donors 18 hours after a single treatment of either buffer or 2.1 to 6.1 pg/cm2 VX-522 nebulized to the apical
surface, or clinically relevant concentrations ELX/TEZ/IVA (TRI). Data represents the mean, ± SEM of 4 replicate experiments in CF-HBE cells derived from 2 MF/MF donors.
Fig. 3E is a graph depicting dose-dependent increase in CFTR-mediated chloride transport in CF-HBE cells derived from a single F508dellMF donor 18 hours after a single treatment of either buffer, 0.9 to 5.6 pg/cm2 VX-522 nebulized to the apical surface, or clinically relevant concentrations of ELX/TEZ/IVA (TRI). Data represents the mean, ± SEM of 6 replicate experiments in CF-HBE cells derived from one F508del/MF donor.
Fig. 4A (upper panel) contains a representative photomicrograph of bronchial epithelium from VX-522 treated (2.4 pg/kg/day for 28 days) monkey lungs depicting VX-522-mRNA (ISH). Fig. 4A (lower panel) is a graph depicting morphometric analysis of exogenously delivered VX-522-mRNA in monkey bronchial epithelium following daily dosing of VX-522 or buffer for 28 days. Data represent the mean ± SEM (n=6 monkeys/treatment group).
Fig. 4B (upper panel) contains a representative photomicrograph of bronchial epithelium from VX-522 treated (2.4 pg/kg/day for 28 days) monkey lungs depicting CFTR protein (IHC). Fig. 4B (lower panel) is a graph depicting morphometric analysis of CFTR protein in monkey bronchial epithelium following daily dosing of VX-522 or buffer for 28 days. Data in the lower panel indicate the total percentage of cells expressing CFTR protein (lower left panel) or the percentage of cells with strong CFTR expression (defined by ionocyte-level thresholds) (lower right panel). Data represent the mean ± SEM (n=6 monkeys/treatment group).
Fig. 5 is a graph depicting chloride transport in CF-HBE cells following treatment with VX-522 or VX-522 in combination with ivacaftor (IVA). Data represents the mean, ± SEM of four replicate experiments in CF-HBE cells derived from two MF/MF donors, * denotes P<0.05 when compared to VX-522 alone at the given dose. At each dose, VX-522 is the bar depicted on the left and VX-522+IVA is on the right.
DETAILED DESCRIPTION
The present disclosure provides therapeutics for the treatment of cystic fibrosis. Cystic fibrosis is a progressive, genetic disease that causes persistent lung infections and limits the ability to breathe overtime. This disease is characterized by the presence of mutations in both copies of the gene for the cystic fibrosis transmembrane conductance regulator (CFTR) protein. Without CFTR, which is involved in the production of sweat, digestive fluids and mucus, secretions that are usually thin instead become thick. The disclosed mRNA therapeutics provide for the intracellular delivery of mRNA encoding CFTR followed by de novo synthesis of functional CFTR protein within target cells. After delivery of mRNA to the target cells, the desired CFTR protein is expressed by the cells’ own translational machinery, and hence, fully functional CFTR protein replaces the defective or missing protein.
Cystic Fibrosis Transmembrane Conductance Regulator (CFTR)
Cystic Fibrosis Transmembrane Conductance Regulator (CFTR; EC 3.6.3.49) is an ABC transporter-class ion channel. It conducts chloride and thiocyanate ions across epithelial cell membranes. The structure of the approximately 168 kDa CFTR, which is highly conserved amongst organisms, consists of seven domains. CFTR contains two transmembrane domains with six transmembrane helices each. Additionally, CFTR contains two nucleotide binding domains, two ABC transporter domains, and one PDZ-binding domain. The nucleotide binding domains are used for binding and hydrolyzing ATP, ABC transporters move ions across the plasma membrane, and the PDZ-binding domain which CFTR to anchor itself to the plasma membrane. CFTR usually exists in dimer units in the plasma membrane of the cell.
The amino acid sequence of a CFTR polypeptide is provided in SEQ ID NO: 1. This CFTR polypeptide (and nucleic acids encoding it) is described in WO 2022/104131, the content of which is incorporated by reference.
In certain aspects, the disclosure provides a polynucleotide (e.g., a RNA, e.g., a mRNA) comprising a nucleotide sequence (e.g., an open reading frame) encoding a CFTR polypeptide. In some embodiments, a polynucleotide disclosed herein comprises a sequence encoding the CFTR polypeptide of SEQ ID NO: 1.
Polynucleotides and Open Reading Frames (ORFs)
The instant invention features mRNAs for use in treating or preventing cystic fibrosis. The mRNAs featured for use in the invention are administered to subjects and encode human CFTR protein in vivo. Accordingly, the invention relates to polynucleotides, e.g., mRNA, comprising an open reading frame of linked nucleosides encoding a CFTR protein (SEQ ID NO: 1). In some embodiments, the open reading frame is sequence-optimized.
In some embodiments, the polynucleotide (e.g., a RNA, e.g., an mRNA) of the invention comprises a nucleotide sequence (e.g., an ORF) encoding a CFTR polypeptide (e.g., SEQ ID NO: 1), wherein the nucleotide sequence has at least 70%, at least 71%, at least 72%, at least 73%, at least 74%, at least 75%, at least 76%, at least 77%, at least 78%, at least 79%, at least 80%, at least 81%, at least 82%, at least 83%, at least 84%, at least 85%, at least 86%, at least 87%, at least 88%, at least 89%, at least 90%, at least 91%, at least 92%, at least 93%, at least 94%, at least 95%, at least 96%, at least 97%, at least 98%, at least 99%, or 100% sequence identity to SEQ ID NOTO.
In some embodiments, the polynucleotide of the invention (e.g., a RNA, e.g., an mRNA) comprises a nucleotide sequence (e.g., an ORF, e.g., SEQ ID NOTO) encoding a CFTR polypeptide further comprises a 5'-UTR (e.g., SEQ ID NO:50) and a 3'-UTR (e.g., SEQ ID NO: 141). In some embodiments, the polynucleotide (e.g., a RNA, e.g., an mRNA) of the invention comprises the sequence of SEQ ID NO: 10. In a further embodiment, the polynucleotide (e.g., a RNA, e.g., an mRNA) comprises a 5' terminal cap (e.g., m7G-ppp-Gm-AG, CapO, Capl, ARCA, inosine, N1 -methylguanosine, 2'-fluoro-guanosine, 7-deaza-guanosine, 8-oxo-guanosine, 2-amino- guanosine, LNA-guanosine, 2-azidoguanosine, Cap2, Cap4, 5' methylG cap, or an analog thereof) and a poly-A-tail region (e.g., about 100 nucleotides in length). In
some embodiments, the mRNA comprises a polyA tail. In some instances, the poly A tail is 50-150 (SEQ ID NO: 197), 75-150 (SEQ ID NO: 198), 85-150 (SEQ ID NO: 199), 90-120 (SEQ ID NO: 193), 90-130 (SEQ ID NO: 194), or 90-150 (SEQ ID NO: 192) nucleotides in length. In some instances, the poly A tail is 100 nucleotides in length (SEQ ID NO: 195). In some instances, the poly A tail is protected (e.g., with an inverted deoxy-thymidine). In some instances, the poly A tail comprises A 100- UCUAG-A20-inverted deoxy-thymidine (SEQ ID NO:211). In some instances, the poly A tail is A100-UCUAG-A20-inverted deoxy-thymidine (SEQ ID NO: 211).
In some embodiments, the polynucleotide of the invention (e.g., a RNA, e.g., an mRNA) comprises a nucleotide sequence (e.g., an ORF) encoding a CFTR polypeptide is single stranded or double stranded.
In some embodiments, the polynucleotide of the invention comprising a nucleotide sequence (e.g., an ORF) encoding a CFTR polypeptide is DNA or RNA. In some embodiments, the polynucleotide of the invention is RNA. In some embodiments, the polynucleotide of the invention is, or functions as, an mRNA. In some embodiments, the mRNA comprises a nucleotide sequence (e.g., an ORF) that encodes at least one CFTR polypeptide, and is capable of being translated to produce the encoded CFTR polypeptide in vitro, in vivo, in situ or ex vivo.
In some embodiments, the polynucleotide of the invention (e.g., a RNA, e.g., an mRNA) comprises a sequence-optimized nucleotide sequence (e.g., an ORF) encoding a CFTR polypeptide, wherein the polynucleotide comprises at least one chemically modified nucleobase, e.g., N1 -methylpseudouracil or 5 -methoxyuracil. In certain embodiments, all uracils in the polynucleotide are N1 -methylpseudouracils. In other embodiments, all uracils in the polynucleotide are 5 -methoxyuracils. In some embodiments, the polynucleotide further comprises a miRNA binding site, e.g., a miRNA binding site that binds to miR-142 and/or a miRNA binding site that binds to miR-126.
In some embodiments, the polynucleotide (e.g., a RNA, e.g., a mRNA) disclosed herein is formulated with a delivery agent comprising, e.g., a compound having the Formula (I), e.g., Compound II; a compound having the Formula (III), (IV), (V), or (VI), e.g., Compound VI; a compound having the Formula (VIII), e.g.,
any of Compounds 419-428, e.g., Compound I, or a compound having the Formula Al, A2, A3, A4, or A5, e.g., any one of SA1-SA41, or any combination thereof. In some embodiments, the delivery agent comprises Compound II, DSPC, Cholesterol, Compound I or PEG-DMG, and GL-67 or a salt thereof, e.g., with a mole ratio of about 47.6±25:9.5±8:36.6±20: 1.4±1.25:4.9±2.5. In some embodiments, the delivery agent comprises Compound II, DSPC, Cholesterol, Compound I or PEG-DMG, and GL-67 or a salt thereof, e.g., with a mole ratio of about
47.6±12.5:9.5±4:36.6±10: 1.4±0.75:4.9±1.25. In some embodiments, the delivery agent comprises Compound II, DSPC, Cholesterol, and Compound I or PEG-DMG, e.g., with a mole ratio of about 47.6±6.25:9.5±2:36.6±5: 1.4±0.375:4.9±0.625. In some embodiments, the delivery agent comprises Compound II, DSPC, Cholesterol, Compound I or PEG-DMG, and GL-67 or a salt thereof, e.g., with a mole ratio of about 47.6:9.5:36.6: 1.4:4.9. In some embodiments, the delivery agent comprises Compound VI, DSPC, Cholesterol, Compound I or PEG-DMG, and GL-67 or a salt thereof, e.g., with a mole ratio of about 47.6±25:9.5±8:36.6±20: 1.4±1.25:4.9±2.5. In some embodiments, the delivery agent comprises Compound VI, DSPC, Cholesterol, Compound I or PEG-DMG, and GL-67 or a salt thereof, e.g., with a mole ratio of about 47.6±12.5:9.5±4:36.6±10: 1.4±0.75:4.9±1.25. In some embodiments, the delivery agent comprises Compound VI, DSPC, Cholesterol, and Compound I or PEG-DMG, e.g., with a mole ratio of about
47.6±6.25:9.5±2:36.6±5: 1.4±0.375:4.9±0.625. In some embodiments, the delivery agent comprises Compound VI, DSPC, Cholesterol, Compound I or PEG-DMG, and GL-67 or a salt thereof, e.g., with a mole ratio of about 47.6:9.5:36.6: 1.4:4.9. In some embodiments, the delivery agent comprises Compound II, DSPC, Cholesterol, Compound I or PEG-DMG, and GL-67 or a salt thereof, e.g., with a mole ratio of about 47.3±25:9.5±8:36.4±20: 1.4±1.25:5.5±2.5. In some embodiments, the delivery agent comprises Compound II, DSPC, Cholesterol, Compound I or PEG-DMG, and GL-67 or a salt thereof, e.g., with a mole ratio of about
47.3±12.5:9.5±4:36.4±10: 1.4±0.75:5.5±1.25. In some embodiments, the delivery agent comprises Compound II, DSPC, Cholesterol, and Compound I or PEG-DMG, e.g., with a mole ratio of about 47.3±6.25:9.5±2:36.4±5: 1.4±0.375:5.5±0.625. In
some embodiments, the delivery agent comprises Compound II, DSPC, Cholesterol, Compound I or PEG-DMG, and GL-67 or a salt thereof, e.g., with a mole ratio of about 47.3:9.5:36.4: 1.4:5.5. In some embodiments, the delivery agent comprises Compound VI, DSPC, Cholesterol, Compound I or PEG-DMG, and GL-67 or a salt thereof, e.g., with a mole ratio of about 47.3±25:9.5±8:36.4±20: 1.4±1.25:5.5±2.5. In some embodiments, the delivery agent comprises Compound VI, DSPC, Cholesterol, Compound I or PEG-DMG, and GL-67 or a salt thereof, e.g., with a mole ratio of about 47.3±12.5:9.5±4:36.4±10: 1.4±0.75:5.5±1.25. In some embodiments, the delivery agent comprises Compound VI, DSPC, Cholesterol, and Compound I or PEG-DMG, e.g., with a mole ratio of about
47.3±6.25:9.5±2:36.4±5: 1.4±0.375:5.5±0.625. In some embodiments, the delivery agent comprises Compound VI, DSPC, Cholesterol, Compound I or PEG-DMG, and GL-67 or a salt thereof, e.g., with a mole ratio of about 47.3:9.5:36.4: 1.4:5.5. In some embodiments, the delivery agent comprises Compound II, DSPC, Cholesterol, Compound I or PEG-DMG, and GL-67 or a salt thereof, e.g., with a mole ratio of about 45.8±25: 10.5±8:36.8±20: 1.4±1.25:5.5±2.5. In some embodiments, the delivery agent comprises Compound II, DSPC, Cholesterol, Compound I or PEG-DMG, and GL-67 or a salt thereof, e.g., with a mole ratio of about
45.8±12.5: 10.5±4:36.8±10: 1.4±0.75:5.5±1.25. In some embodiments, the delivery agent comprises Compound II, DSPC, Cholesterol, and Compound I or PEG-DMG, e.g., with a mole ratio of about 45.8±6.25: 10.5±2:36.8±5: 1.4±0.375:5.5±0.625. In some embodiments, the delivery agent comprises Compound II, DSPC, Cholesterol, Compound I or PEG-DMG, and GL-67 or a salt thereof, e.g., with a mole ratio of about 45.8: 10.5:36.8: 1.4:5.5. In some embodiments, the delivery agent comprises Compound VI, DSPC, Cholesterol, Compound I or PEG-DMG, and GL-67 or a salt thereof, e.g., with a mole ratio of about 45.8±25: 10.5±8:36.8±20: 1.4±1.25:5.5±2.5. In some embodiments, the delivery agent comprises Compound VI, DSPC, Cholesterol, Compound I or PEG-DMG, and GL-67 or a salt thereof, e.g., with a mole ratio of about 45.8±12.5: 10.5±4:36.8±10: 1.4±0.75:5.5±1.25. In some embodiments, the delivery agent comprises Compound VI, DSPC, Cholesterol, and Compound I or PEG-DMG, e.g., with a mole ratio of about
45.8±6.25: 10.5±2:36.8±5: 1.4±0.375:5.5±0.625. In some embodiments, the delivery agent comprises Compound VI, DSPC, Cholesterol, Compound I or PEG-DMG, and GL-67 or a salt thereof, e.g., with a mole ratio of about 45.8: 10.5:36.8: 1.4:5.5. In some embodiments, the delivery agent comprises Compound II or VI, DSPC, Cholesterol, Compound I or PEG-DMG, and GL-67 or a salt thereof, e.g., with a mole ratio in the range of about 30 to about 60 mol% Compound II or VI (or related suitable amino lipid) (e.g., 30-40, 40-45, 45-50, 50-55 or 55-60 mol% Compound II or VI (or related suitable amino lipid)), about 5 to about 20 mol% phospholipid (or related suitable phospholipid or “helper lipid”) (e.g., 5-10, 10-15, or 15-20 mol% phospholipid (or related suitable phospholipid or “helper lipid”)), about 20 to about 50 mol% cholesterol (or related sterol or “non-cationic” lipid) (e.g., about 20-30, 30- 35, 35-40, 40-45, or 45-50 mol% cholesterol (or related sterol or “non-cationic” lipid)), about 0.05 to about 10 mol% PEG lipid (or other suitable PEG lipid) (e.g., 0.05-1, 1-2, 2-3, 3-4, 4-5, 5-7, or 7-10 mol% PEG lipid (or other suitable PEG lipid)), and about 1 to about 10 mol% GL-67 or a salt thereof (e.g., 1-3, 3-5, 5-7, 7-10, 3-8, 3.5-6.5 mol% GL-67 or a salt thereof). An exemplary delivery agent can comprise mole ratios of, for example, 47.6:9.5:36.6: 1.4:4.9, 47.3:9.5:36.4: 1.4:5.5, or
45.8: 10.5:36.8: 1.4:5.5. In certain instances, an exemplary delivery agent can comprise mole ratios of, for example, 48:9.5:35.5: 1.5:5.5; 47: 10:36: 1.5:5.5;
46: 10.5:36.5: 1.5:5.5; 45: 10.5:37.5: 1.5:5.5; 48:9.5:36: 1.5:5; 47: 10:36.5: 1.5:5;
46: 10.5:37: 1.5:5; or 45: 10.5:38: 1.5:5. In some embodiments, the delivery agent comprises Compound II or VI, DSPC, Cholesterol, Compound I or PEG-DMG, and GL-67 or a salt thereof, e.g., with a mole ratio of about 47.6:9.5:36.6: 1.4:4.9. In some embodiments, the delivery agent comprises Compound II or VI, DSPC, Cholesterol, Compound I or PEG-DMG, and GL-67 or a salt thereof, e.g., with a mole ratio of about 47.3:9.5:36.4: 1.4:5.5. In some embodiments, the delivery agent comprises Compound II or VI, DSPC, Cholesterol, Compound I or PEG-DMG, and GL-67 or a salt thereof, e.g., with a mole ratio ofabout 45.8: 10.5:36.8: 1.4:5.5.
In some embodiments, the polynucleotide (e.g., a RNA, e.g., a mRNA) disclosed herein is formulated with a delivery agent comprising, e.g., a compound having the Formula (I), e.g., any of Compounds 1-232, e.g., Compound II; a
compound having the Formula (III), (IV), (V), or (VI), e.g., any of Compounds 233- 342, e.g., Compound VI; or a compound having the Formula (VIII), e.g., any of Compounds 419-428, e.g., Compound I, or any combination thereof. In some embodiments, the delivery agent comprises Compound II, DSPC, Cholesterol, and Compound I or PEG-DMG, e.g., with a mole ratio of about
49.5±3: 10.5±2:39±3: l±0.75. In some embodiments, the delivery agent comprises Compound VI, DSPC, Cholesterol, and Compound I or PEG-DMG, e.g., with a mole ratio of about 49.5±3: 10.5±2:39±3: l±0.75. In some embodiments, the delivery agent comprises about 48-52 mol % Compound II or VI (or related suitable amino lipid) (e.g., 48-51, 48-50, 49-52, or 49-51 mol % Compound II or VI (or related suitable amino lipid)), about 9-12 mol % phospholipid (or related suitable phospholipid or “helper lipid”) (e.g., 9-11, 9-10, 10-12, 10-11.5, 10-11 mol %phospholipid (or related suitable phospholipid or “helper lipid”)), about 36-42 mol% cholesterol (or related sterol or “non-cationic” lipid) (e.g., about 36-41, 36-40, 37-40, or 38-40 mol% cholesterol (or related sterol or “non-cationic” lipid)) and about 0.25-2.5 mol% PEG lipid (or other suitable PEG lipid) (e.g., 0.25-2, 0.25-1.5, 0.25-2, or 0.5-1.5 mol% PEG lipid (or other suitable PEG lipid)).
In some embodiments, the polynucleotide (e.g., a RNA, e.g., a mRNA) disclosed herein is formulated with a delivery agent comprising, e.g., a compound having the Formula (I), e.g., any of Compounds 1-232, e.g., Compound II; a compound having the Formula (III), (IV), (V), or (VI), e.g., any of Compounds 233- 342, e.g., Compound VI; a compound having the Formula (VIII), e.g., any of Compounds 419-428, e.g., Compound I, or a compound having the Formula Al, A2, A3, A4, or A5, e.g., any one of SA1-SA41, or any combination thereof. In some embodiments, the delivery agent comprises Compound II, DSPC, Cholesterol, and Compound I or PEG-DMG, e.g., with a mole ratio of about 46.5±3: 10±2:36±3: 1.25±0.75:4.5±1.5. In some embodiments, the delivery agent comprises Compound VI, DSPC, Cholesterol, and Compound I or PEG-DMG, e.g., with a mole ratio of about 46.5±3: 10±2:36±3: 1.25±0.75:4.5±1.5. In some embodiments, the delivery agent comprises about 43-49 mol % Compound II or VI (or related suitable amino lipid) (e.g., 43-48, 44-48, 45-48, or 45.5-48 mol %
Compound II or VI (or related suitable amino lipid)), about 8-12 mol % phospholipid (or related suitable phospholipid or “helper lipid”) (e.g., 8-11, 8-10, 9-12, 9-11, 9.5- 10.5 mol %phospholipid (or related suitable phospholipid or “helper lipid”)), about
33-39 mol% cholesterol (or related sterol or “non-cationic” lipid) (e.g., about 33-38,
34-38, 35-38, or 36-37 mol% cholesterol (or related sterol or “non-cationic” lipid)), about 0.5-2 mol% PEG lipid (or other suitable PEG lipid) (e.g., 0.5-1.5, 0.75-1.5, or 1-1.5 mol% PEG lipid (or other suitable PEG lipid)), and about 3-6 mol% cationic agent (e.g., sterol amine) (e.g., 3-5, 3-4.5, 4-6, or 5-6 mol% cationic agent (e.g., sterol amine)). In some embodiments, the delivery agent comprises Compound II, DSPC, Cholesterol, DMG-PEG-2k, and GL-67. In further embodiments, the delivery agent comprises about 45-48 mol% Compound II, about 9-11 mol% DSPC, about 35-38 mol% cholesterol, about 1-3 mol% DMG-PEG-2k, and about 4-6 mol% GL-67. In further embodiments, the delivery agent comprises about 45-48 mol% Compound II, about 9-11 mol% DSPC, about 35-38 mol% cholesterol, about 1-3 mol% DMG-PEG- 2k, and about 4-6 mol% GL-67. In further embodiments, the delivery agent comprises about 45.8-47.6 mol% Compound II, about 9.5-10.5 mol% DSPC, about 36.4-36.8 mol% cholesterol, about 1.4 mol% DMG-PEG-2k, and about 4.9-5.5 mol% GL-67.
In some embodiments, a polynucleotide (e.g., a RNA, e.g., a mRNA) disclosed herein is formulated with a delivery agent comprising, e.g., a compound having the Formula (I), e.g., any of Compounds 1-232, e.g., Compound II; a compound having the Formula (III), (IV), (V), or (VI), e.g., any of Compounds 233- 342, e.g., Compound VI; a compound having the Formula (VIII), e.g., any of Compounds 419-428, e.g., Compound I, or a compound having the Formula Al, A2, A3, A4, or A5, e.g., any one of SA1-SA41, or any combination thereof. In some embodiments, the delivery agent comprises Compound II, DSPC, Cholesterol, and Compound I or PEG-DMG, e.g., with a mole ratio of about 47±3: 10±2:36±3: 1.25±0.75:4.5±1.5. In some embodiments, the delivery agent comprises Compound VI, DSPC, Cholesterol, and Compound I or PEG-DMG, e.g., with a mole ratio of about 46.5±3: 10±2:36±3: 1.25±0.75:4.5±1.5. In some embodiments, the delivery agent comprises about 43-49 mol % Compound II or VI
(or related suitable amino lipid) (e.g., 43-48, 44-48, 45-48, or 45.5-48 mol % Compound II or VI (or related suitable amino lipid)), about 8-12 mol % phospholipid (or related suitable phospholipid or “helper lipid”) (e.g., 8-11, 8-10, 9-12, 9-11, 9.5- 10.5 mol %phospholipid (or related suitable phospholipid or “helper lipid”)), about
33-39 mol% cholesterol (or related sterol or “non-cationic” lipid) (e.g., about 33-38,
34-38, 35-38, or 36-37 mol% cholesterol (or related sterol or “non-cationic” lipid)), about 0.5-2 mol% PEG lipid (or other suitable PEG lipid) (e.g., 0.5-1.5, 0.75-1.5, or 1-1.5 mol% PEG lipid (or other suitable PEG lipid)), and about 3-6 mol% cationic agent (e.g., sterol amine) (e.g., 3-5, 3-4.5, 4-6, or 5-6 mol% cationic agent (e.g., sterol amine)).
In some embodiments, the polynucleotide of the disclosure is an mRNA that comprises a 5'-terminal cap (e.g., Capl, e.g., m7Gp-ppGm-A), a 5'UTR (e.g., SEQ ID NO:50), an ORF sequence of SEQ ID NO: 10, a 3'UTR (e.g., SEQ ID NO: 141), and a poly A tail (e.g., about 100 nt in length, e.g., SEQ ID NO: 195), wherein all uracils in the polynucleotide are N1 -methylpseudouracils.
In some embodiments, the polynucleotide of the disclosure is an mRNA that comprises a 5'-terminal cap (e.g., Capl, e.g., m7Gp-ppGm-A), a 5'UTR (e.g., SEQ ID NO:50), an ORF sequence of SEQ ID NO: 10, a 3'UTR (e.g., SEQ ID NO: 141), and a poly A tail (e.g., A100-UCUAG-A20-inverted deoxy-thymidine (SEQ ID NO:211)), wherein all uracils in the polynucleotide are N1 -methylpseudouracils.
In some embodiments, the polynucleotide of the disclosure is an mRNA that comprises a 5'-terminal cap (e.g., Capl, e.g., m7Gp-ppGm-A), a 5'UTR (e.g., any one of SEQ ID N0s:50-80), an ORF sequence of SEQ ID NO: 10, a 3'UTR (e.g., any one of SEQ ID NOs: 100-141), and a poly A tail (e.g., about 100 nt in length, e.g., SEQ ID NO: 195), wherein all uracils in the polynucleotide are N1 -methylpseudouracils. In some embodiments, the delivery agent comprises Compound II as the ionizable amino lipid and DMG-PEG 2k as the PEG lipid.
In some embodiments, the polynucleotide of the disclosure is an mRNA that comprises a 5'-terminal cap (e.g., Cap 1, e.g., m7Gp-ppGm-A), a 5'UTR (e.g., SEQ ID NO:50), an ORF sequence of SEQ ID NO: 10, a 3'UTR (e.g., SEQ ID NO: 141), and a poly A tail (e.g., about 100 nt in length), wherein all uracils in the polynucleotide are
N1 -methylpseudouracils. In some embodiments, the delivery agent comprises Compound II or Compound VI as the ionizable amino lipid and DMG-PEG 2k or Compound I as the PEG lipid. In some embodiments, the delivery agent comprises Compound II as the ionizable amino lipid and DMG-PEG 2k as the PEG lipid.
In some embodiments, the polynucleotide of the disclosure is an mRNA that comprises an ORF sequence of SEQ ID NO: 10. In some embodiments, the polynucleotide of the disclosure is an mRNA that comprises an ORF sequence of SEQ ID NO: 10, wherein all uracils in the polynucleotide are N1 -methylpseudouracils. In some embodiments, the polynucleotide of the disclosure is an mRNA that comprises an ORF sequence of SEQ ID NO: 10, wherein the mRNA is formulated with a delivery agent comprising Compound II as the ionizable amino lipid and DMG-PEG 2k as the PEG lipid. In some embodiments, the polynucleotide of the disclosure is an mRNA that comprises an ORF sequence of SEQ ID NO: 10, wherein all uracils in the polynucleotide are N1 -methylpseudouracils, wherein the mRNA is formulated with a delivery agent comprising Compound II as the ionizable amino lipid and DMG-PEG 2k as the PEG lipid. In some embodiments, the polynucleotide of the disclosure is an mRNA that comprises an ORF sequence of SEQ ID NO: 10, wherein the mRNA is formulated with a delivery agent comprising Compound II as the ionizable amino lipid, DMG-PEG 2k as the PEG lipid, and GL-67 as the cationic agent. In some embodiments, the polynucleotide of the disclosure is an mRNA that comprises an ORF sequence of SEQ ID NO: 10, wherein all uracils in the polynucleotide are N1 -methylpseudouracils, wherein the mRNA is formulated with a delivery agent comprising Compound II as the ionizable amino lipid, DMG-PEG 2k as the PEG lipid, and GL-67 as the cationic agent.
In some embodiments, the polynucleotide of the disclosure is an mRNA that comprises an ORF sequence of SEQ ID NO: 11. In some embodiments, the polynucleotide of the disclosure is an mRNA that comprises an ORF sequence of SEQ ID NO: 11, wherein all uracils in the polynucleotide are N1 -methylpseudouracils. In some embodiments, the polynucleotide of the disclosure is an mRNA that comprises an ORF sequence of SEQ ID NO: 11, wherein the mRNA is formulated with a delivery agent comprising Compound II as the ionizable amino lipid and
DMG-PEG 2k as the PEG lipid. In some embodiments, the polynucleotide of the disclosure is an mRNA that comprises an ORF sequence of SEQ ID NO: 11, wherein all uracils in the polynucleotide are N1 -methylpseudouracils, wherein the mRNA is formulated with a delivery agent comprising Compound II as the ionizable amino lipid and DMG-PEG 2k as the PEG lipid. In some embodiments, the polynucleotide of the disclosure is an mRNA that comprises an ORF sequence of SEQ ID NO: 11, wherein the mRNA is formulated with a delivery agent comprising Compound II as the ionizable amino lipid, DMG-PEG 2k as the PEG lipid, and GL-67 as the cationic agent. In some embodiments, the polynucleotide of the disclosure is an mRNA that comprises an ORF sequence of SEQ ID NO: 11, wherein all uracils in the polynucleotide are N1 -methylpseudouracils, wherein the mRNA is formulated with a delivery agent comprising Compound II as the ionizable amino lipid, DMG-PEG 2k as the PEG lipid, and GL-67 as the cationic agent.
In some embodiments, the polynucleotide of the disclosure is an mRNA that comprises the sequence of SEQ ID NO: 13. In some embodiments, the polynucleotide of the disclosure is an mRNA that comprises the sequence of SEQ ID NO: 13, wherein the mRNA is formulated with a delivery agent comprising Compound II as the ionizable amino lipid and DMG-PEG 2k as the PEG lipid. In some embodiments, the polynucleotide of the disclosure is an mRNA that comprises the sequence of SEQ ID NO: 13, wherein the mRNA is formulated with a delivery agent comprising Compound II as the ionizable amino lipid, DMG-PEG 2k as the PEG lipid, and GL-67 as the cationic agent.
Signal Sequences
The polynucleotides (e.g., a RNA, e.g., an mRNA) of the invention can also comprise nucleotide sequences that encode additional features that facilitate trafficking of the encoded polypeptides to therapeutically relevant sites. One such feature that aids in protein trafficking is the signal sequence, or targeting sequence. The peptides encoded by these signal sequences are known by a variety of names, including targeting peptides, transit peptides, and signal peptides. In some embodiments, the polynucleotide (e.g., a RNA, e.g., an mRNA) comprises a
nucleotide sequence (e.g., an ORF) that encodes a signal peptide operably linked to a nucleotide sequence that encodes a CFTR polypeptide described herein.
In some embodiments, the "signal sequence" or "signal peptide" is a polynucleotide or polypeptide, respectively, which is from about 30-210, e.g., about 45-80 or 15-60 nucleotides (e.g., about 20, 30, 40, 50, 60, or 70 amino acids) in length that, optionally, is incorporated at the 5' (or N-terminus) of the coding region or the polypeptide, respectively. Addition of these sequences results in trafficking the encoded polypeptide to a desired site, such as the endoplasmic reticulum or the mitochondria through one or more targeting pathways. Some signal peptides are cleaved from the protein, for example by a signal peptidase after the proteins are transported to the desired site.
In some embodiments, the polynucleotide of the invention comprises a nucleotide sequence encoding a CFTR polypeptide, wherein the nucleotide sequence further comprises a 5' nucleic acid sequence encoding a heterologous signal peptide.
Sequence-Optimized Nucleotide Sequences Encoding CFTR polypeptides
In some embodiments, the polynucleotide of the invention comprises a sequence-optimized nucleotide sequence encoding a CFTR polypeptide disclosed herein. In some embodiments, the polynucleotide of the invention comprises an open reading frame (ORF) encoding a CFTR polypeptide, wherein the ORF has been sequence optimized.
An exemplary sequence-optimized nucleotide sequence encoding CFTR is set forth as SEQ ID NO: 10. A further exemplary sequence-optimized nucleotide sequence encoding CFTR is set forth as SEQ ID NO: 11. A further exemplary sequence-optimized nucleotide sequence encoding CFTR is set forth as SEQ ID NO: 13. In some embodiments, the sequence optimized CFTR sequences, fragments, and variants thereof are used to practice the methods disclosed herein.
In some embodiments, a polynucleotide of the present disclosure, for example a polynucleotide comprising an mRNA nucleotide sequence encoding a CFTR polypeptide, comprises from 5' to 3' end:
(i) a 5' cap provided herein, for example, Capl;
(ii) a 5' UTR, such as the sequences provided herein, for example, SEQ ID NO:50;
(iii) an open reading frame encoding a CFTR polypeptide, e.g., a sequence optimized nucleic acid sequence encoding CFTR set forth as SEQ ID NO: 10;
(iv) at least one stop codon (if not present at 5' terminus of 3 'UTR);
(v) a 3' UTR, such as the sequences provided herein, for example, SEQ ID NO: 141; and
(vi) a poly-A tail provided above.
In certain embodiments, all uracils in the polynucleotide are N1 -methylpseudouracil. In certain embodiments, all uracils in the polynucleotide are 5 -methoxyuracil .
The sequence-optimized nucleotide sequences disclosed herein are distinct from the corresponding wild type nucleotide acid sequences and from other known sequence-optimized nucleotide sequences, e.g., these sequence-optimized nucleic acids have unique compositional characteristics.
In some embodiments, the percentage of uracil or thymine nucleobases in a sequence-optimized nucleotide sequence (e.g., encoding a CFTR polypeptide) is modified (e.g., reduced) with respect to the percentage of uracil or thymine nucleobases in the reference wild-type nucleotide sequence. Such a sequence is referred to as a uracil-modified or thymine -modified sequence. The percentage of uracil or thymine content in a nucleotide sequence can be determined by dividing the number of uracils or thymines in a sequence by the total number of nucleotides and multiplying by 100. In some embodiments, the sequence-optimized nucleotide sequence has a lower uracil or thymine content than the uracil or thymine content in the reference wild-type sequence. In some embodiments, the uracil or thymine content in a sequence -optimized nucleotide sequence of the invention is greater than the uracil or thymine content in the reference wild-type sequence and still maintain beneficial effects, e.g., increased expression and/or reduced Toll-Like Receptor (TLR) response when compared to the reference wild-type sequence.
Methods for optimizing codon usage are known in the art. For example, an ORF of any one or more of the sequences provided herein may be codon optimized. Codon optimization, in some embodiments, may be used to match codon frequencies
in target and host organisms to ensure proper folding; bias GC content to increase mRNA stability or reduce secondary structures; minimize tandem repeat codons or base runs that may impair gene construction or expression; customize transcriptional and translational control regions; insert or remove protein trafficking sequences; remove/add post translation modification sites in encoded protein (e.g., glycosylation sites); add, remove or shuffle protein domains; insert or delete restriction sites; modify ribosome binding sites and mRNA degradation sites; adjust translational rates to allow the various domains of the protein to fold properly; or reduce or eliminate problem secondary structures within the polynucleotide. Codon optimization tools, algorithms and services are known in the art - non-limiting examples include services from GeneArt (Life Technologies), DNA2.0 (Menlo Park CA) and/or proprietary methods. In some embodiments, the open reading frame (ORF) sequence is optimized using optimization algorithms.
Identification and Ratio Determination (IDR) Sequences
An Identification and Ratio Determination (IDR) sequence is a sequence of a biological molecule (e.g., nucleic acid or protein) that, when combined with the sequence of a target biological molecule, serves to identify the target biological molecule. Typically, an IDR sequence is a heterologous sequence that is incorporated within or appended to a sequence of a target biological molecule and can be used as a reference to identify the target molecule. Thus, in some embodiments, a nucleic acid (e.g., mRNA) comprises (i) a target sequence of interest (e.g., a coding sequence encoding a therapeutic and/or antigenic peptide or protein); and (ii) a unique IDR sequence.
An RNA species (e.g., RNA having a given coding sequence) may comprise an IDR sequence that differs from the IDR sequence of other RNA species (e.g., RNA(s) having different coding sequence(s)). Each IDR sequence thus identifies a particular RNA species, and so the abundance of IDR sequences may be measured to determine the abundance of each RNA species in a composition. Use of distinct IDR sequences to identify RNA species allows for analysis of multivalent RNA compositions (e.g., containing multiple RNA species) containing RNA species with
similar coding sequences and/or lengths, which could otherwise be difficult to distinguish using PCR- or chromatography-based analysis of full-length RNAs.
Each RNA species in a multivalent RNA composition may comprise an IDR sequence that is not a sequence isomer of an IDR sequence of another RNA species in a multivalent RNA composition (e.g., the IDR sequence does not have the same number of adenosine nucleotides, the same number of cytosine nucleotides, the same number of guanine nucleotides, and the same number of uracil nucleotides, as another IDR sequence in the composition, even if those sequences have different sequences). Having identical nucleotide compositions causes sequence isomers to have the same mass, presenting a challenge to distinguishing sequence isomers using mass-based identification methods (e.g., mass spectrometry).
Each RNA species in a multivalent RNA composition may comprise an IDR sequence having a mass that differs from the mass of IDR sequences of each other RNA species in a multivalent RNA composition. For example, the mass of each IDR sequence may differ from the mass of other IDR sequences by at least 9 Da, at least 25 Da, at least 25 Da, or at least 50 Da. Use of IDR sequences with distinct masses allows RNA fragments comprising different IDR sequences to be distinguished using mass-based analysis methods (e.g., mass spectrometry), which do not require reverse transcription, amplification, or sequencing of RNAs.
Each RNA species in an RNA composition may comprises an IDR sequence with a different length. For example, each IDR sequence may have a length independently selected from 0 to 25 nucleotides. The length of a nucleic acid influences the rate at which the nucleic acid traverses a chromatography column, and so the use of IDR sequences of different lengths on different RNA species allows RNA fragments having different IDR sequences to be distinguished using chromatography-based methods (e.g., LC-UV).
IDR sequences may be chosen such that no IDR sequence comprises a start codon, ‘AUG’ . Lack of a start codon in an IDR sequence prevents undesired translation of nucleotide sequences within and/or downstream from the IDR sequence.
IDR sequences may be chosen such that no IDR sequence comprises a recognition site for a restriction enzyme. In one example, no IDR sequence comprises
a recognition site for Xbal, ‘UCUAG’. Lack of a recognition site for a restriction enzyme (e.g., Xbal recognition site ‘UCUAG’) allows the restriction enzyme to be used in generating and modifying a DNA template for in vitro transcription, without affecting the IDR sequence or sequence of the transcribed RNA.
Modified Nucleotide Sequences Encoding CFTR polypeptides
In some embodiments, the polynucleotide (e.g., a RNA, e.g., an mRNA) of the invention comprises a chemically modified nucleobase, for example, a chemically modified uracil, e.g., pseudouracil, N1 -methylpseudouracil, 5 -methoxyuracil, or the like. In some embodiments, the mRNA is a uracil -modified sequence comprising an ORF encoding a CFTR polypeptide, wherein the mRNA comprises a chemically modified nucleobase, for example, a chemically modified uracil, e.g., pseudouracil, N1 -methylpseudouracil, or 5 -methoxyuracil.
In certain aspects of the invention, when the modified uracil base is connected to a ribose sugar, as it is in polynucleotides, the resulting modified nucleoside or nucleotide is referred to as modified uridine. In some embodiments, uracil in the polynucleotide is at least about 25%, at least about 30%, at least about 40%, at least about 50%, at least about 60%, at least about 70%, at least about 80%, at least 90%, at least 95%, at least 99%, or about 100% modified uracil. In one embodiment, uracil in the polynucleotide is at least 95% modified uracil. In another embodiment, uracil in the polynucleotide is 100% modified uracil.
In embodiments where uracil in the polynucleotide is at least 95% modified uracil overall uracil content can be adjusted such that an mRNA provides suitable protein expression levels while inducing little to no immune response. In some embodiments, the uracil content of the ORF is between about 100% and about 150%, between about 100% and about 110%, between about 105% and about 115%, between about 110% and about 120%, between about 115% and about 125%, between about 120% and about 130%, between about 125% and about 135%, between about 130% and about 140%, between about 135% and about 145%, between about 140% and about 150% of the theoretical minimum uracil content in the corresponding wild-type ORF (%UTM). In other embodiments, the uracil content of the ORF is between about
121% and about 136% or between 123% and 134% of the %UTM. In some embodiments, the uracil content of the ORF encoding a CFTR polypeptide is about 115%, about 120%, about 125%, about 130%, about 135%, about 140%, about 145%, or about 150% of the %UTM. In this context, the term "uracil" can refer to modified uracil and/or naturally occurring uracil.
In some embodiments, the uracil content in the ORF of the mRNA encoding a CFTR polypeptide of the invention is less than about 30%, about 25%, about 20%, about 15%, or about 10% of the total nucleobase content in the ORF. In some embodiments, the uracil content in the ORF is between about 10% and about 20% of the total nucleobase content in the ORF. In other embodiments, the uracil content in the ORF is between about 10% and about 25% of the total nucleobase content in the ORF. In one embodiment, the uracil content in the ORF of the mRNA encoding a CFTR polypeptide is less than about 20% of the total nucleobase content in the open reading frame. In this context, the term "uracil" can refer to modified uracil and/or naturally occurring uracil.
In further embodiments, the ORF of the mRNA encoding a CFTR polypeptide having modified uracil and adjusted uracil content has increased Cytosine (C), Guanine (G), or Guanine/Cytosine (G/C) content (absolute or relative). In some embodiments, the overall increase in C, G, or G/C content (absolute or relative) of the ORF is at least about 2%, at least about 3%, at least about 4%, at least about 5%, at least about 6%, at least about 7%, at least about 10%, at least about 15%, at least about 20%, at least about 40%, at least about 50%, at least about 60%, at least about 70%, at least about 80%, at least about 90%, at least about 95%, or at least about 100% relative to the G/C content (absolute or relative) of the wild-type ORF. In some embodiments, the G, the C, or the G/C content in the ORF is less than about 100%, less than about 90%, less than about 85%, or less than about 80% of the theoretical maximum G, C, or G/C content of the corresponding wild type nucleotide sequence encoding the CFTR polypeptide (%GTMX; %CTMX, or %G/CTMX). In some embodiments, the increases in G and/or C content (absolute or relative) described herein can be conducted by replacing synonymous codons with low G, C, or G/C content with synonymous codons having higher G, C, or G/C content. In other
embodiments, the increase in G and/or C content (absolute or relative) is conducted by replacing a codon ending with U with a synonymous codon ending with G or C.
In further embodiments, the ORF of the mRNA encoding a CFTR polypeptide of the invention comprises modified uracil and has an adjusted uracil content containing less uracil pairs (UU) and/or uracil triplets (UUU) and/or uracil quadruplets (UUUU) than the corresponding wild-type nucleotide sequence encoding the CFTR polypeptide. In some embodiments, the ORF of the mRNA encoding a CFTR polypeptide of the invention contains no uracil pairs and/or uracil triplets and/or uracil quadruplets. In some embodiments, uracil pairs and/or uracil triplets and/or uracil quadruplets are reduced below a certain threshold, e.g., no more than 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, or 20 occurrences in the ORF of the mRNA encoding the CFTR polypeptide. In a particular embodiment, the ORF of the mRNA encoding the CFTR polypeptide of the invention contains less than 20, 19, 18, 17, 16, 15, 14, 13, 12, 11, 10, 9, 8, 7, 6, 5, 4, 3, 2, or 1 nonphenylalanine uracil pairs and/or triplets. In another embodiment, the ORF of the mRNA encoding the CFTR polypeptide contains no non-phenylalanine uracil pairs and/or triplets.
In further embodiments, the ORF of the mRNA encoding a CFTR polypeptide of the invention comprises modified uracil and has an adjusted uracil content containing less uracil-rich clusters than the corresponding wild-type nucleotide sequence encoding the CFTR polypeptide. In some embodiments, the ORF of the mRNA encoding the CFTR polypeptide of the invention contains uracil-rich clusters that are shorter in length than corresponding uracil-rich clusters in the corresponding wild-type nucleotide sequence encoding the CFTR polypeptide.
In further embodiments, alternative lower frequency codons are employed. At least about 5%, at least about 10%, at least about 15%, at least about 20%, at least about 25%, at least about 30%, at least about 35%, at least about 40%, at least about 45%, at least about 50%, at least about 55%, at least about 60%, at least about 65%, at least about 70%, at least about 75%, at least about 80%, at least about 85%, at least about 90%, at least about 95%, at least about 99%, or 100% of the codons in the CFTR polypeptide-encoding ORF of the modified uracil-comprising mRNA are
substituted with alternative codons, each alternative codon having a codon frequency lower than the codon frequency of the substituted codon in the synonymous codon set. The ORF also has adjusted uracil content, as described above. In some embodiments, at least one codon in the ORF of the mRNA encoding the CFTR polypeptide is substituted with an alternative codon having a codon frequency lower than the codon frequency of the substituted codon in the synonymous codon set.
In some embodiments, the adjusted uracil content, CFTR polypeptide- encoding ORF of the modified uracil-comprising mRNA exhibits expression levels of CFTR when administered to a mammalian cell that are higher than expression levels of CFTR from the corresponding wild-type mRNA. In some embodiments, the mammalian cell is a mouse cell, a rat cell, or a rabbit cell. In other embodiments, the mammalian cell is a monkey cell or a human cell. In some embodiments, the human cell is a HeLa cell, a BJ fibroblast cell, or a peripheral blood mononuclear cell (PBMC). In some embodiments, CFTR is expressed at a level higher than expression levels of CFTR from the corresponding wild-type mRNA when the mRNA is administered to a mammalian cell in vivo. In some embodiments, the mRNA is administered to mice, rabbits, rats, monkeys, or humans. In one embodiment, mice are null mice. In some embodiments, the mRNA is administered intravenously or intramuscularly. In other embodiments, the CFTR polypeptide is expressed when the mRNA is administered to a mammalian cell in vitro. In some embodiments, the expression is increased by at least about 2-fold, at least about 5 -fold, at least about 10- fold, at least about 50-fold, at least about 500-fold, at least about 1500-fold, or at least about 3000-fold. In other embodiments, the expression is increased by at least about 10%, about 20%, about 30%, about 40%, about 50%, 60%, about 70%, about 80%, about 90%, or about 100%.
In some embodiments, adjusted uracil content, CFTR polypeptide-encoding ORF of the modified uracil-comprising mRNA exhibits increased stability. In some embodiments, the mRNA exhibits increased stability in a cell relative to the stability of a corresponding wild-type mRNA under the same conditions. In some embodiments, the mRNA exhibits increased stability including resistance to nucleases, thermal stability, and/or increased stabilization of secondary structure. In
some embodiments, increased stability exhibited by the mRNA is measured by determining the half-life of the mRNA (e.g., in a plasma, serum, cell, or tissue sample) and/or determining the area under the curve (AUC) of the protein expression by the mRNA over time (e.g., in vitro or in vivo). An mRNA is identified as having increased stability if the half-life and/or the AUC is greater than the half-life and/or the AUC of a corresponding wild-type mRNA under the same conditions.
In some embodiments, the mRNA of the present invention induces a detectably lower immune response (e.g., innate or acquired) relative to the immune response induced by a corresponding wild-type mRNA under the same conditions. In other embodiments, the mRNA of the present disclosure induces a detectably lower immune response (e.g., innate or acquired) relative to the immune response induced by an mRNA that encodes for a CFTR polypeptide but does not comprise modified uracil under the same conditions, or relative to the immune response induced by an mRNA that encodes for a CFTR polypeptide and that comprises modified uracil but that does not have adjusted uracil content under the same conditions. The innate immune response can be manifested by increased expression of pro-inflammatory cytokines, activation of intracellular PRRs (RIG-I, MDA5, etc.), cell death, and/or termination or reduction in protein translation. In some embodiments, a reduction in the innate immune response can be measured by expression or activity level of Type 1 interferons (e.g., IFN-a, IFN-P, IFN-K, IFN-5, IFN-s. IFN-r, IFN-co, and IFN-Q or the expression of interferon-regulated genes such as the toll-like receptors (e.g., TUR7 and TUR8), and/or by decreased cell death following one or more administrations of the mRNA of the invention into a cell.
In some embodiments, the expression of Type- 1 interferons by a mammalian cell in response to the mRNA of the present disclosure is reduced by at least 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 95%, 99%, 99.9%, or greater than 99.9% relative to a corresponding wild-type mRNA, to an mRNA that encodes a CFTR polypeptide but does not comprise modified uracil, or to an mRNA that encodes a CFTR polypeptide and that comprises modified uracil but that does not have adjusted uracil content. In some embodiments, the interferon is IFN-p. In some embodiments, cell death frequency caused by administration of mRNA of the present
disclosure to a mammalian cell is 10%, 25%, 50%, 75%, 85%, 90%, 95%, or over 95% less than the cell death frequency observed with a corresponding wild-type mRNA, an mRNA that encodes for a CFTR polypeptide but does not comprise modified uracil, or an mRNA that encodes for a CFTR polypeptide and that comprises modified uracil but that does not have adjusted uracil content. In some embodiments, the mammalian cell is a BJ fibroblast cell. In other embodiments, the mammalian cell is a splenocyte. In some embodiments, the mammalian cell is that of a mouse or a rat. In other embodiments, the mammalian cell is that of a human. In one embodiment, the mRNA of the present disclosure does not substantially induce an innate immune response of a mammalian cell into which the mRNA is introduced.
Methods for Modifying Polynucleotides
The disclosure includes modified polynucleotides comprising a polynucleotide described herein (e.g., a polynucleotide, e.g. mRNA, comprising a nucleotide sequence encoding a CFTR polypeptide). The modified polynucleotides can be chemically modified and/or structurally modified. When the polynucleotides of the present invention are chemically and/or structurally modified the polynucleotides can be referred to as "modified polynucleotides."
The present disclosure provides for modified nucleosides and nucleotides of a polynucleotide (e.g., RNA polynucleotides, such as mRNA polynucleotides) encoding a CFTR polypeptide. A "nucleoside" refers to a compound containing a sugar molecule (e.g., a pentose or ribose) or a derivative thereof in combination with an organic base (e.g. , a purine or pyrimidine) or a derivative thereof (also referred to herein as "nucleobase"). A “nucleotide" refers to a nucleoside including a phosphate group. Modified nucleotides can be synthesized by any useful method, such as, for example, chemically, enzymatically, or recombinantly, to include one or more modified or non-natural nucleosides. Polynucleotides can comprise a region or regions of linked nucleosides. Such regions can have variable backbone linkages. The linkages can be standard phosphodiester linkages, in which case the polynucleotides would comprise regions of nucleotides.
The modified polynucleotides disclosed herein can comprise various distinct modifications. In some embodiments, the modified polynucleotides contain one, two, or more (optionally different) nucleoside or nucleotide modifications. In some embodiments, a modified polynucleotide, introduced to a cell can exhibit one or more desirable properties, e.g., improved protein expression, reduced immunogenicity, or reduced degradation in the cell, as compared to an unmodified polynucleotide.
In some embodiments, a polynucleotide of the present invention (e.g., a polynucleotide comprising a nucleotide sequence encoding a CFTR polypeptide) is structurally modified. As used herein, a "structural" modification is one in which two or more linked nucleosides are inserted, deleted, duplicated, inverted or randomized in a polynucleotide without significant chemical modification to the nucleotides themselves. Because chemical bonds will necessarily be broken and reformed to effect a structural modification, structural modifications are of a chemical nature and hence are chemical modifications. However, structural modifications will result in a different sequence of nucleotides. For example, the polynucleotide "ATCG" can be chemically modified to "AT-5meC-G". The same polynucleotide can be structurally modified from "ATCG" to "ATCCCG". Here, the dinucleotide "CC" has been inserted, resulting in a structural modification to the polynucleotide.
Therapeutic compositions of the present disclosure comprise, in some embodiments, at least one nucleic acid (e.g., RNA) having an open reading frame encoding CFTR (e.g., SEQ ID NO: 10), wherein the nucleic acid comprises nucleotides and/or nucleosides that can be standard (unmodified) or modified as is known in the art. In some embodiments, nucleotides and nucleosides of the present disclosure comprise modified nucleotides or nucleosides. Such modified nucleotides and nucleosides can be naturally-occurring modified nucleotides and nucleosides or non-naturally occurring modified nucleotides and nucleosides. Such modifications can include those at the sugar, backbone, or nucleobase portion of the nucleotide and/or nucleoside as are recognized in the art.
In some embodiments, a naturally-occurring modified nucleotide or nucleotide of the disclosure is one as is generally known or recognized in the art. Non-limiting
examples of such naturally occurring modified nucleotides and nucleotides can be found, inter alia, in the widely recognized MODOMICS database.
In some embodiments, a non-naturally occurring modified nucleotide or nucleoside of the disclosure is one as is generally known or recognized in the art. Non-limiting examples of such non-naturally occurring modified nucleotides and nucleosides can be found, inter alia, in published US application Nos. PCI7US2012/058519; PCI7US2013/075177; PCT/US2014/058897;
PCT7US2014/058891; PCT7US2014/070413; PCT/US2015/36773;
PCI7US2015/36759; PCI7US2015/36771; or PCT/IB2017/051367 all of which are incorporated by reference herein.
In some embodiments, at least one RNA (e.g., mRNA) of the present disclosure is not chemically modified and comprises the standard ribonucleotides consisting of adenosine, guanosine, cytosine and uridine. In some embodiments, nucleotides and nucleosides of the present disclosure comprise standard nucleoside residues such as those present in transcribed RNA (e.g. A, G, C, or U). In some embodiments, nucleotides and nucleosides of the present disclosure comprise standard deoxyribonucleosides such as those present in DNA (e.g. dA, dG, dC, or dT).
Hence, nucleic acids of the disclosure (e.g., DNA nucleic acids and RNA nucleic acids, such as mRNA nucleic acids) can comprise standard nucleotides and nucleosides, naturally-occurring nucleotides and nucleosides, non-naturally-occurring nucleotides and nucleosides, or any combination thereof.
Nucleic acids of the disclosure (e.g., DNA nucleic acids and RNA nucleic acids, such as mRNA nucleic acids), in some embodiments, comprise various (more than one) different types of standard and/or modified nucleotides and nucleosides. In some embodiments, a particular region of a nucleic acid contains one, two or more (optionally different) types of standard and/or modified nucleotides and nucleosides.
In some embodiments, a modified RNA nucleic acid (e.g. , a modified mRNA nucleic acid), introduced to a cell or organism, exhibits reduced degradation in the cell or organism, respectively, relative to an unmodified nucleic acid comprising standard nucleotides and nucleosides.
In some embodiments, a modified RNA nucleic acid (e.g. , a modified mRNA nucleic acid), introduced into a cell or organism, may exhibit reduced immunogenicity in the cell or organism, respectively (e.g., a reduced innate response) relative to an unmodified nucleic acid comprising standard nucleotides and nucleosides.
Nucleic acids (e.g., RNA nucleic acids, such as mRNA nucleic acids), in some embodiments, comprise non-natural modified nucleotides that are introduced during synthesis or post-synthesis of the nucleic acids to achieve desired functions or properties. The modifications may be present on intemucleotide linkages, purine or pyrimidine bases, or sugars. The modification may be introduced with chemical synthesis or with a polymerase enzyme at the terminal of a chain or anywhere else in the chain. Any of the regions of a nucleic acid may be chemically modified.
The present disclosure provides for modified nucleosides and nucleotides of a nucleic acid (e.g., RNA nucleic acids, such as mRNA nucleic acids). A “nucleoside” refers to a compound containing a sugar molecule (e.g. , a pentose or ribose) or a derivative thereof in combination with an organic base (e.g. , a purine or pyrimidine) or a derivative thereof (also referred to herein as “nucleobase”). A “nucleotide” refers to a nucleoside, including a phosphate group. Modified nucleotides may by synthesized by any useful method, such as, for example, chemically, enzymatically, or recombinantly, to include one or more modified or non-natural nucleosides. Nucleic acids can comprise a region or regions of linked nucleosides. Such regions may have variable backbone linkages. The linkages can be standard phosphodiester linkages, in which case the nucleic acids would comprise regions of nucleotides.
Modified nucleotide base pairing encompasses not only the standard adenosine-thymine, adenosine-uracil, or guanosine-cytosine base pairs, but also base pairs formed between nucleotides and/or modified nucleotides comprising nonstandard or modified bases, wherein the arrangement of hydrogen bond donors and hydrogen bond acceptors permits hydrogen bonding between a non-standard base and a standard base or between two complementary non-standard base structures, such as, for example, in those nucleic acids having at least one chemical modification. One example of such non-standard base pairing is the base pairing between the modified
nucleotide inosine and adenine, cytosine or uracil. Any combination of base/sugar or linker may be incorporated into nucleic acids of the present disclosure.
In some embodiments, modified nucleobases in nucleic acids (e.g., RNA nucleic acids, such as mRNA nucleic acids) comprise N1 -methyl -pseudouridine (mlψ). 1 -ethyl -pseudouridine (e ly), 5 -methoxy-uridine (mo5U), 5-methyl-cytidine (m5C), and/or pseudouridine (v)- In some embodiments, modified nucleobases in nucleic acids (e.g., RNA nucleic acids, such as mRNA nucleic acids) comprise 5- methoxymethyl uridine, 5 -methylthio uridine, 1 -methoxymethyl pseudouridine, 5- methyl cytidine, and/or 5 -methoxy cytidine. In some embodiments, the polyribonucleotide includes a combination of at least two (e.g., 2, 3, 4 or more) of any of the aforementioned modified nucleobases, including but not limited to chemical modifications.
In some embodiments, a RNA nucleic acid of the disclosure comprises Nl- methyl -pseudouridine (mlψ) substitutions at one or more or all uridine positions of the nucleic acid.
In some embodiments, a RNA nucleic acid of the disclosure comprises Nl- methyl-pseudouridine (mlψ) substitutions at one or more or all uridine positions of the nucleic acid and 5 -methyl cytidine substitutions at one or more or all cytidine positions of the nucleic acid.
In some embodiments, a RNA nucleic acid of the disclosure comprises pseudouridine (v) substitutions at one or more or all uridine positions of the nucleic acid.
In some embodiments, a RNA nucleic acid of the disclosure comprises pseudouridine (v) substitutions at one or more or all uridine positions of the nucleic acid and 5-methyl cytidine substitutions at one or more or all cytidine positions of the nucleic acid.
In some embodiments, a RNA nucleic acid of the disclosure comprises uridine at one or more or all uridine positions of the nucleic acid.
In some embodiments, nucleic acids (e.g., RNA nucleic acids, such as mRNA nucleic acids) are uniformly modified (e.g., fully modified, modified throughout the entire sequence) for a particular modification. For example, a nucleic acid can be
uniformly modified with N1 -methyl -pseudouridine, meaning that all uridine residues in the mRNA sequence are replaced with N1 -methyl -pseudouridine. Similarly, a nucleic acid can be uniformly modified for any type of nucleoside residue present in the sequence by replacement with a modified residue such as those set forth above.
The nucleic acids of the present disclosure may be partially or fully modified along the entire length of the molecule. For example, one or more or all or a given type of nucleotide (e.g. , purine or pyrimidine, or any one or more or all of A, G, U, C) may be uniformly modified in a nucleic acid of the disclosure, or in a predetermined sequence region thereof (e.g., in the mRNA including or excluding the polyA tail). In some embodiments, all nucleotides X in a nucleic acid of the present disclosure (or in a sequence region thereof) are modified nucleotides, wherein X may be any one of nucleotides A, G, U, C, or any one of the combinations A+G, A+U, A+C, G+U, G+C, U+C, A+G+U, A+G+C, G+U+C or A+G+C.
The nucleic acid may contain from about 1% to about 100% modified nucleotides (either in relation to overall nucleotide content, or in relation to one or more types of nucleotide, i.e., any one or more of A, G, U or C) or any intervening percentage (e.g., from l% to 20%, from l% to 25%, from l% to 50%, from l% to 60%, from l% to 70%, from l% to 80%, from l% to 90%, from l% to 95%, from 10% to 20%, from 10% to 25%, from 10% to 50%, from 10% to 60%, from 10% to 70%, from 10% to 80%, from 10% to 90%, from 10% to 95%, from 10% to 100%, from 20% to 25%, from 20% to 50%, from 20% to 60%, from 20% to 70%, from 20% to 80%, from 20% to 90%, from 20% to 95%, from 20% to 100%, from 50% to 60%, from 50% to 70%, from 50% to 80%, from 50% to 90%, from 50% to 95%, from 50% to 100%, from 70% to 80%, from 70% to 90%, from 70% to 95%, from 70% to 100%, from 80% to 90%, from 80% to 95%, from 80% to 100%, from 90% to 95%, from 90% to 100%, and from 95% to 100%). It will be understood that any remaining percentage is accounted for by the presence of unmodified A, G, U, or C.
The nucleic acids may contain at a minimum 1% and at maximum 100% modified nucleotides, or any intervening percentage, such as at least 5% modified nucleotides, at least 10% modified nucleotides, at least 25% modified nucleotides, at least 50% modified nucleotides, at least 80% modified nucleotides, or at least 90%
modified nucleotides. For example, the nucleic acids may contain a modified pyrimidine such as a modified uracil or cytosine. In some embodiments, at least 5%, at least 10%, at least 25%, at least 50%, at least 80%, at least 90% or 100% of the uracil in the nucleic acid is replaced with a modified uracil (e.g., a 5-substituted uracil). The modified uracil can be replaced by a compound having a single unique structure, or can be replaced by a plurality of compounds having different structures (e.g., 2, 3, 4 or more unique structures). In some embodiments, at least 5%, at least 10%, at least 25%, at least 50%, at least 80%, at least 90% or 100% of the cytosine in the nucleic acid is replaced with a modified cytosine (e.g., a 5-substituted cytosine). The modified cytosine can be replaced by a compound having a single unique structure, or can be replaced by a plurality of compounds having different structures (e.g., 2, 3, 4 or more unique structures).
Untranslated Regions (UTRs)
Untranslated regions (UTRs) are nucleic acid sections of a polynucleotide before a start codon (5' UTR) and after a stop codon (3' UTR) that are not translated. In some embodiments, a polynucleotide (e.g., a ribonucleic acid (RNA), e.g., a messenger RNA (mRNA)) of the invention comprising an open reading frame (ORF) encoding a CFTR polypeptide further comprises UTR (e.g., a 5' UTR or functional fragment thereof, a 3' UTR or functional fragment thereof, or a combination thereof).
A UTR (e.g., 5' UTR or 3' UTR) can be homologous or heterologous to the coding region in a polynucleotide. In some embodiments, the UTR is homologous to the ORF encoding the CFTR polypeptide. In some embodiments, the UTR is heterologous to the ORF encoding the CFTR polypeptide.
In some embodiments, the polynucleotide comprises two or more 5' UTRs or functional fragments thereof, each of which has the same or different nucleotide sequences. In some embodiments, the polynucleotide comprises two or more 3' UTRs or functional fragments thereof, each of which has the same or different nucleotide sequences.
In some embodiments, the 5' UTR or functional fragment thereof, 3' UTR or functional fragment thereof, or any combination thereof is sequence optimized.
In some embodiments, the 5'UTR or functional fragment thereof, 3' UTR or functional fragment thereof, or any combination thereof comprises at least one chemically modified nucleobase, e.g., N1 -methylpseudouracil or 5 -methoxyuracil.
UTRs can have features that provide a regulatory role, e.g., increased or decreased stability, localization and/or translation efficiency. A polynucleotide comprising a UTR can be administered to a cell, tissue, or organism, and one or more regulatory features can be measured using routine methods. In some embodiments, a functional fragment of a 5' UTR or 3' UTR comprises one or more regulatory features of a full length 5' or 3' UTR, respectively.
Natural 5' UTRs bear features that play roles in translation initiation. They harbor signatures like Kozak sequences that are commonly known to be involved in the process by which the ribosome initiates translation of many genes. Kozak sequences have the consensus CCR(A/G)CCAUGG, where R is a purine (adenine or guanine) three bases upstream of the start codon (AUG), which is followed by another ‘G’. 5' UTRs also have been known to form secondary structures that are involved in elongation factor binding.
By engineering the features typically found in abundantly expressed genes of specific target organs, one can enhance the stability and protein production of a polynucleotide. For example, introduction of 5' UTR of liver-expressed mRNA, such as albumin, serum amyloid A, Apolipoprotein A/B/E, transferrin, alpha fetoprotein, erythropoietin, or Factor VIII, can enhance expression of polynucleotides in hepatic cell lines or liver. Uikewise, use of 5'UTR from other tissue-specific mRNA to improve expression in that tissue is possible for muscle (e.g., MyoD, Myosin, Myoglobin, Myogenin, Herculin), for endothelial cells (e.g., Tie-1, CD36), for myeloid cells (e.g, C/EBP, AML1, G-CSF, GM-CSF, CDl lb, MSR, Fr-1, i-NOS), for leukocytes (e.g., CD45, CD 18), for adipose tissue (e.g., CD36, GLUT4, ACRP30, adiponectin) and for lung epithelial cells (e.g., SP-A/B/C/D).
In some embodiments, UTRs are selected from a family of transcripts whose proteins share a common function, structure, feature or property. For example, an encoded polypeptide can belong to a family of proteins (i. e. , that share at least one function, structure, feature, localization, origin, or expression pattern), which are
expressed in a particular cell, tissue or at some time during development. The UTRs from any of the genes or mRNA can be swapped for any other UTR of the same or different family of proteins to create a new polynucleotide.
In some embodiments, the 5' UTR and the 3' UTR can be heterologous. In some embodiments, the 5' UTR can be derived from a different species than the 3' UTR. In some embodiments, the 3' UTR can be derived from a different species than the 5' UTR.
Co-owned International Patent Application No. PCT/US2014/021522 (Publ. No. WO/2014/164253, incorporated herein by reference in its entirety) provides a listing of exemplary UTRs that can be utilized in the polynucleotide of the present invention as flanking regions to an ORF.
Additional exemplary UTRs of the application include, but are not limited to, one or more 5 'UTR and/or 3 'UTR derived from the nucleic acid sequence of: a globin, such as an a- or P-globin (e.g., aXenopus, mouse, rabbit, or human globin); a strong Kozak translational initiation signal; a CYBA (e.g., human cytochrome b-245 a polypeptide); an albumin (e.g., human albumin?); a HSD17B4 (hydroxysteroid (17-P) dehydrogenase); a virus (e.g., a tobacco etch virus (TEV), a Venezuelan equine encephalitis virus (VEEV), a Dengue virus, a cytomegalovirus (CMV) (e.g., CMV immediate early 1 (IE 1)), a hepatitis virus (e.g., hepatitis B virus), a sindbis virus, or a PAV barley yellow dwarf virus); a heat shock protein (e.g. , hsp70); a translation initiation factor (e.g., elF4G); a glucose transporter (e.g., hGLUTl (human glucose transporter 1)); an actin (e.g., human a or P actin); a GAPDH; a tubulin; a histone; a citric acid cycle enzyme; a topoisomerase (e.g., a 5'UTR of a TOP gene lacking the 5' TOP motif (the oligopyrimidine tract)); a ribosomal protein Large 32 (L32); a ribosomal protein (e.g., human or mouse ribosomal protein, such as, for example, rps9); an ATP synthase (e.g., ATP5A1 or the P subunit of mitochondrial H+-ATP synthase); a growth hormone e (e.g, bovine (bGH) or human (hGH)); an elongation factor (e.g, elongation factor 1 al (EEF1A1)); a manganese superoxide dismutase (MnSOD); a myocyte enhancer factor 2A (MEF2A); a P-Fl-ATPase, a creatine kinase, a myoglobin, a granulocyte-colony stimulating factor (G-CSF); a collagen (e.g., collagen type I, alpha 2 (CollA2), collagen type I, alpha 1 (CollAl), collagen
type VI, alpha 2 (Col6A2), collagen type VI, alpha 1 (C0I6AI)); a ribophorin (e.g., ribophorin I (RPNI)); a low density lipoprotein receptor-related protein (e.g., LRP1); a cardiotrophin-like cytokine factor (e.g., Nntl); calreticulin (Calr); a procollagenlysine, 2-oxoglutarate 5-dioxygenase 1 (Plodl); and a nucleobindin (e.g., Nucbl).
In some embodiments, the 5' UTR is selected from the group consisting of a P-globin 5' UTR; a 5 'UTR containing a strong Kozak translational initiation signal; a cytochrome b-245 a polypeptide (CYBA) 5' UTR; a hydroxysteroid (17-P) dehydrogenase (HSD17B4) 5' UTR; a Tobacco etch virus (TEV) 5' UTR; a Venezuelen equine encephalitis virus (TEEV) 5' UTR; a 5' proximal open reading frame of rubella virus (RV) RNA encoding nonstructural proteins; a Dengue virus (DEN) 5' UTR; a heat shock protein 70 (Hsp70) 5' UTR; a eIF4G 5' UTR; a GLUT1 5' UTR; functional fragments thereof and any combination thereof.
In some embodiments, the 3' UTR is selected from the group consisting of a P-globin 3' UTR; a CYBA 3' UTR; an albumin 3' UTR; a growth hormone (GH) 3' UTR; a VEEV 3' UTR; a hepatitis B virus (HBV) 3' UTR; a-globin 3 'UTR; a DEN 3' UTR; a PAV barley yellow dwarf virus (BYDV-PAV) 3' UTR; an elongation factor 1 al (EEF1A1) 3' UTR; a manganese superoxide dismutase (MnSOD) 3' UTR; a P subunit of mitochondrial H(+)-ATP synthase (P-mRNA) 3' UTR; a GLUT1 3' UTR; a MEF2A 3' UTR; a P-Fl-ATPase 3' UTR; functional fragments thereof and combinations thereof.
Wild-type UTRs derived from any gene or mRNA can be incorporated into the polynucleotides of the invention. In some embodiments, a UTR can be altered relative to a wild type or native UTR to produce a variant UTR, e.g., by changing the orientation or location of the UTR relative to the ORF; or by inclusion of additional nucleotides, deletion of nucleotides, swapping or transposition of nucleotides. In some embodiments, variants of 5' or 3' UTRs can be utilized, for example, mutants of wild type UTRs, or variants wherein one or more nucleotides are added to or removed from a terminus of the UTR.
Additionally, one or more synthetic UTRs can be used in combination with one or more non-synthetic UTRs. See, e.g., Mandal and Rossi, Nat. Protoc. 2013
8(3):568-82, the contents of which are incorporated herein by reference in their entirety.
UTRs or portions thereof can be placed in the same orientation as in the transcript from which they were selected or can be altered in orientation or location. Hence, a 5' and/or 3' UTR can be inverted, shortened, lengthened, or combined with one or more other 5' UTRs or 3' UTRs.
In some embodiments, the polynucleotide comprises multiple UTRs, e.g., a double, a triple or a quadruple 5' UTR or 3' UTR. For example, a double UTR comprises two copies of the same UTR either in series or substantially in series. For example, a double beta-globin 3'UTR can be used (see US2010/0129877, the contents of which are incorporated herein by reference in its entirety).
The polynucleotides of the invention can comprise combinations of features. For example, the ORF can be flanked by a 5 'UTR that comprises a strong Kozak translational initiation signal and/or a 3'UTR comprising an oligo(dT) sequence for templated addition of a poly-A tail. A 5 'UTR can comprise a first polynucleotide fragment and a second polynucleotide fragment from the same and/or different UTRs (see, e.g., US2010/0293625, herein incorporated by reference in its entirety).
Other non-UTR sequences can be used as regions or subregions within the polynucleotides of the invention. For example, introns or portions of intron sequences can be incorporated into the polynucleotides of the invention. Incorporation of intronic sequences can increase protein production as well as polynucleotide expression levels. In some embodiments, the polynucleotide of the invention comprises an internal ribosome entry site (IRES) instead of or in addition to a UTR (see, e.g., Yakubov et al., Biochem. Biophys. Res. Commun. 2010 394(1): 189-193, the contents of which are incorporated herein by reference in their entirety). In some embodiments, the polynucleotide comprises an IRES instead of a 5' UTR sequence. In some embodiments, the polynucleotide comprises an ORF and a viral capsid sequence. In some embodiments, the polynucleotide comprises a synthetic 5' UTR in combination with a non-synthetic 3' UTR.
In some embodiments, the UTR can also include at least one translation enhancer polynucleotide, translation enhancer element, or translational enhancer
elements (collectively, "TEE," which refers to nucleic acid sequences that increase the amount of polypeptide or protein produced from a polynucleotide. As a non -limiting example, the TEE can be located between the transcription promoter and the start codon. In some embodiments, the 5' UTR comprises a TEE.
In one aspect, a TEE is a conserved element in a UTR that can promote translational activity of a nucleic acid such as, but not limited to, cap-dependent or cap-independent translation. a. 5' UTR sequences
5' UTR sequences are important for ribosome recruitment to the mRNA and have been reported to play a role in translation (Hinnebusch A, et al., (2016) Science, 352:6292: 1413-6).
Disclosed herein, inter alia, is a polynucleotide, e.g., mRNA, comprising an open reading frame encoding a CFTR polypeptide (e.g., SEQ ID NO: 1 or SEQ ID NO:2), which polynucleotide has a 5' UTR that confers an increased half-life, increased expression and/or increased activity of the polypeptide encoded by said polynucleotide, or of the polynucleotide itself. In an embodiment, a polynucleotide disclosed herein comprises: (a) a 5'-UTR (e.g., as provided in Table 1 or a variant or fragment thereof); (b) a coding region comprising a stop element (e.g., as described herein); and (c) a 3'-UTR (e.g., as described herein), and LNP compositions comprising the same. In an embodiment, the polynucleotide comprises a 5'-UTR comprising a sequence provided in Table 1 or a variant or fragment thereof (e.g., a functional variant or fragment thereof).
In an embodiment, the polynucleotide having a 5' UTR sequence provided in Table 1 or a variant or fragment thereof, has an increase in the half-life of the polynucleotide, e.g., about 1.5-20-fold increase in half-life of the polynucleotide. In an embodiment, the increase in half-life is about 1.5, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 or 20-fold, or more. In an embodiment, the increase in half life is about 1.5-fold or more. In an embodiment, the increase in half life is about 2- fold or more. In an embodiment, the increase in half life is about 3-fold or more. In an
embodiment, the increase in half life is about 4-fold or more. In an embodiment, the increase in half life is about 5-fold or more.
In an embodiment, the polynucleotide having a 5' UTR sequence provided in Table 1 or a variant or fragment thereof, results in an increased level and/or activity, e.g., output, of the polypeptide encoded by the polynucleotide. In an embodiment, the 5 'UTR results in about 1.5-20-fold increase in level and/or activity, e.g., output, of the polypeptide encoded by the polynucleotide. In an embodiment, the increase in level and/or activity is about 1.5, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 or 20-fold, or more. In an embodiment, the increase in level and/or activity is about 1.5-fold or more. In an embodiment, the increase in level and/or activity is about 2- fold or more. In an embodiment, the increase in level and/or activity is about 3-fold or more. In an embodiment, the increase in level and/or activity is about 4-fold or more. In an embodiment, the increase in level and/or activity is about 5 -fold or more.
In an embodiment, the increase is compared to an otherwise similar polynucleotide which does not have a 5' UTR, has a different 5' UTR, or does not have a 5' UTR described in Table 1 or a variant or fragment thereof.
In an embodiment, the increase in half-life of the polynucleotide is measured according to an assay that measures the half-life of a polynucleotide.
In an embodiment, the increase in level and/or activity, e.g., output, of the polypeptide encoded by the polynucleotide is measured according to an assay that measures the level and/or activity of a polypeptide.
In an embodiment, the 5' UTR comprises a sequence provided in Table 1 or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to a 5' UTR sequence provided in Table 1, or a variant or a fragment thereof. In an embodiment, the 5' UTR comprises a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 50, SEQ ID NO: 51, SEQ ID NO: 52, SEQ ID NO: 53, SEQ ID NO: 54, SEQ ID NO: 55, SEQ ID NO: 56, SEQ ID NO: 57, SEQ ID NO: 58, or SEQ ID NO: 78.
In an embodiment, the 5' UTR comprises the sequence of SEQ ID NO:50. In an embodiment, the 5' UTR consists of the sequence of SEQ ID NO:50.
In an embodiment, a 5' UTR sequence provided in Table 1 has a first nucleotide which is an A. In an embodiment, a 5' UTR sequence provided in Table 1 has a first nucleotide which is a G. Table 1: 5' UTR sequences


 In an embodiment, the 5' UTR comprises a variant of SEQ ID NO:50. In an embodiment, the variant of SEQ ID NO:50 comprises a nucleic acid sequence of Formula A:
In an embodiment, the 5' UTR comprises a variant of SEQ ID NO:50. In an embodiment, the variant of SEQ ID NO:50 comprises a nucleic acid sequence of Formula A:
GGAAAUCGCAAAA (N2)x(N3)xC U (N4)x(N5)xC G CGUUAGAUU UCUUUUAGUUUUCUN6N7C AACUAGCAAGCUUUUUGU UCUCGCC (Ns C C)x (SEQ ID NO: 59), wherein:
(N2)x is a uracil and x is an integer from 0 to 5, e.g., wherein x =3 or 4;
(N3)x is a guanine and x is an integer from 0 to 1 ;
(N4)X is a cytosine and x is an integer from 0 to 1 ;
(Ns)x is a uracil and x is an integer from 0 to 5, e.g., wherein x =2 or 3;
Ne is a uracil or cytosine;
N? is a uracil or guanine;
Ns is adenine or guanine and x is an integer from 0 to 1.
In an embodiment (N2)x is a uracil and x is 0. In an embodiment (N2)x is a uracil and x is 1. In an embodiment (N2)x is a uracil and x is 2. In an embodiment (N2)X is a uracil and x is 3. In an embodiment, (N2)x is a uracil and x is 4. In an embodiment (N2)x is a uracil and x is 5.
In an embodiment, (N3)x is a guanine and x is 0. In an embodiment, (N3)x is a guanine and x is 1.
In an embodiment, (N4)x is a cytosine and x is 0. In an embodiment, (N4)x is a cytosine and x is 1.
In an embodiment (Ns)x is a uracil and x is 0. In an embodiment (Ns)x is a uracil and x is 1. In an embodiment (Ns)x is a uracil and x is 2. In an embodiment (Ns)x is a uracil and x is 3. In an embodiment, (Ns)x is a uracil and x is 4. In an embodiment (Ns)x is a uracil and x is 5.
In an embodiment, N6 is a uracil. In an embodiment, N6 is a cytosine.
In an embodiment, N7 is a uracil. In an embodiment, N7 is a guanine.
In an embodiment, N8 is an adenine and x is 0. In an embodiment, N8 is an adenine and x is 1.
In an embodiment, N8 is a guanine and x is 0. In an embodiment, N8 is a guanine and x is 1.
In an embodiment, the 5' UTR comprises a variant of SEQ ID NO: 50. In an embodiment, the variant of SEQ ID NO: 50 comprises a sequence with at least 50%, 60%, 70%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, or 99% identity to SEQ ID NO: 50. In an embodiment, the variant of SEQ ID NO: 50 comprises a sequence with at least 50% identity to SEQ ID NO: 50. In an embodiment, the variant of SEQ ID NO: 50 comprises a sequence with at least 60% identity to SEQ ID NO: 50. In an embodiment, the variant of SEQ ID NO: 50 comprises a sequence with at least 70% identity to SEQ ID NO: 50. In an embodiment, the variant of SEQ ID NO: 50 comprises a sequence with at least 80% identity to SEQ ID NO: 50. In an embodiment, the variant of SEQ ID NO: 50 comprises a sequence with at least 90% identity to SEQ ID NO: 50. In an embodiment, the variant of SEQ ID NO: 50 comprises a sequence with at least 95% identity to SEQ ID NO: 50. In an embodiment, the variant of SEQ ID NO: 50 comprises a sequence with at least 96% identity to SEQ ID NO: 50. In an embodiment, the variant of SEQ ID NO: 50 comprises a sequence with at least 97% identity to SEQ ID NO: 50. In an embodiment, the variant of SEQ ID NO: 50 comprises a sequence with at least 98% identity to SEQ ID NO: 50. In an embodiment, the variant of SEQ ID NO: 50 comprises a sequence with at least 99% identity to SEQ ID NO: 50.
In an embodiment, the variant of SEQ ID NO: 50 comprises a uridine content of at least 5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, or 80%. In an embodiment, the variant of SEQ ID NO: 50 comprises a uridine content of at least 5%. In an embodiment, the variant of SEQ ID NO: 50 comprises a uridine content of at least 10%. In an embodiment, the variant of SEQ ID NO: 50 comprises a uridine content of at least 20%. In an embodiment, the variant of SEQ ID NO: 50 comprises a uridine content of at least 30%. In an embodiment, the variant of SEQ ID NO: 50 comprises a uridine content of at least 40%. In an embodiment, the variant of SEQ ID NO: 50 comprises a uridine content of at least 50%. In an embodiment, the variant of SEQ ID NO: 50 comprises a uridine content of at least 60%. In an embodiment, the variant of
SEQ ID NO: 50 comprises a uridine content of at least 70%. In an embodiment, the variant of SEQ ID NO: 50 comprises a uridine content of at least 80%.
In an embodiment, the variant of SEQ ID NO: 50 comprises at least 2, 3, 4, 5, 6 or 7 consecutive uridines (e.g, a polyuridine tract). In an embodiment, the polyuridine tract in the variant of SEQ ID NO: 50 comprises at least 1-7, 2-7, 3-7, 4-7, 5-7, 6-7, 1-6, 1-5, 1-4, 1-3, 1-2, 2-6, or 3-5 consecutive uridines. In an embodiment, the polyuridine tract in the variant of SEQ ID NO: 50 comprises 4 consecutive uridines. In an embodiment, the polyuridine tract in the variant of SEQ ID NO: 50 comprises 5 consecutive uridines.
In an embodiment, the variant of SEQ ID NO: 50 comprises 1, 2, 3, 4, 5, 6, 7,
8, 9, 10, 11, 12, 13, 14, or 15 polyuridine tracts. In an embodiment, the variant of SEQ ID NO: 50 comprises 3 polyuridine tracts. In an embodiment, the variant of SEQ ID NO: 50 comprises 4 polyuridine tracts. In an embodiment, the variant of SEQ ID NO: 50 comprises 5 polyuridine tracts.
In an embodiment, one or more of the polyuridine tracts are adjacent to a different polyuridine tract. In an embodiment, each of, e.g., all, the polyuridine tracts are adjacent to each other, e.g., all of the polyuridine tracts are contiguous.
In an embodiment, one or more of the polyuridine tracts are separated by 1, 2,
3, 4, 5, 6, 7, 8, 9, 10, 11, 2, 13, 14, 15, 16, 17, 18. 19, 20, 30, 40, 50 or 60 nucleotides. In an embodiment, each of, e.g., all of, the polyuridine tracts are separated by 1, 2, 3,
4, 5, 6, 7, 8, 9, 10, 11, 2, 13, 14, 15, 16, 17, 18. 19, 20, 30, 40, 50 or 60 nucleotides.
In an embodiment, a first polyuridine tract and a second polyuridine tract are adjacent to each other.
In an embodiment, a subsequent, e.g., third, fourth, fifth, sixth or seventh, eighth, ninth, or tenth, polyuridine tract is separated by 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 2, 13, 14, 15, 16, 17, 18. 19, 20, 30, 40, 50 or 60 nucleotides from the first polyuridine tract, the second polyuridine tract, or any one of the subsequent polyuridine tracts.
In an embodiment, a first polyuridine tract is separated by 1, 2, 3, 4, 5, 6, 7, 8,
9, 10, 11, 2, 13, 14, 15, 16, 17, 18. 19, 20, 30, 40, 50 or 60 nucleotides from a subsequent polyuridine tract, e.g., a second, third, fourth, fifth, sixth or seventh,
eighth, ninth, or tenth polyuridine tract. In an embodiment, one or more of the subsequent polyuridine tracts are adjacent to a different polyuridine tract.
In an embodiment, the 5' UTR comprises a Kozak sequence, e.g., a GCCRCC nucleotide sequence wherein R is an adenine or guanine. In an embodiment, the Kozak sequence is disposed at the 3' end of the 5'UTR sequence.
In an aspect, the polynucleotide (e.g., mRNA) comprising an open reading frame encoding a CFTR polypeptide (e.g., SEQ ID NO: 1) and comprising a 5' UTR sequence disclosed herein is formulated as an LNP. In an embodiment, the LNP composition comprises: (i) an ionizable lipid, e.g., an amino lipid; (ii) a sterol or other structural lipid; (iii) a non-cationic helper lipid or phospholipid; (iv) a PEG-lipid; and (v) a cationic agent.
In another aspect, the LNP compositions of the disclosure are used in a method of treating cystic fibrosis in a subject.
In an aspect, an LNP composition comprising a polynucleotide disclosed herein encoding a CFTR polypeptide, e.g., as described herein, can be administered with an additional agent, e.g., as described herein. b. 3' UTR sequences
3 'UTR sequences have been shown to influence translation, half-life, and subcellular localization of mRNAs (Mayr C., Cold Spring Harb Persp Biol 2019 Oct l;l l(10):a034728).
Disclosed herein, inter alia, is a polynucleotide, e.g., mRNA, comprising an open reading frame encoding a CFTR polypeptide (e.g., SEQ ID NO: 1), which polynucleotide has a 3' UTR that confers an increased half-life, increased expression and/or increased activity of the polypeptide encoded by said polynucleotide, or of the polynucleotide itself. In an embodiment, a polynucleotide disclosed herein comprises: (a) a 5'-UTR (e.g., as described herein); (b) a coding region comprising a stop element (e.g., as described herein); and (c) a 3'-UTR (e.g., as provided in Table 2 or a variant or fragment thereof), and LNP compositions comprising the same. In an embodiment, the polynucleotide comprises a 3'-UTR comprising a sequence provided in Table 2 or a variant or fragment thereof.
In an embodiment, the polynucleotide having a 3' UTR sequence provided in Table 2 or a variant or fragment thereof, results in an increased half-life of the polynucleotide, e.g., about 1.5-10-fold increase in half-life of the polynucleotide. In an embodiment, the increase in half-life is about 1.5, 2, 3, 4, 5, 6, 7, 8, 9, or 10-fold, or more. In an embodiment, the increase in half-life is about 1.5-fold or more. In an embodiment, the increase in half-life is about 2-fold or more. In an embodiment, the increase in half-life is about 3 -fold or more. In an embodiment, the increase in halflife is about 4-fold or more. In an embodiment, the increase in half-life is about 5 -fold or more. In an embodiment, the increase in half-life is about 6-fold or more. In an embodiment, the increase in half-life is about 7-fold or more. In an embodiment, the increase in half-life is about 8-fold. In an embodiment, the increase in half-life is about 9-fold or more. In an embodiment, the increase in half-life is about 10-fold or more.
In an embodiment, the polynucleotide having a 3' UTR sequence provided in Table 2 or a variant or fragment thereof, results in a polynucleotide with a mean halflife score of greater than 10.
In an embodiment, the polynucleotide having a 3' UTR sequence provided in Table 2 or a variant or fragment thereof, results in an increased level and/or activity, e.g., output, of the polypeptide encoded by the polynucleotide.
In an embodiment, the increase is compared to an otherwise similar polynucleotide which does not have a 3' UTR, has a different 3' UTR, or does not have a 3' UTR of Table 2 or a variant or fragment thereof.
In an embodiment, the polynucleotide comprises a 3' UTR sequence provided in Table 2 or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to a 3' UTR sequence provided in Table 2, or a fragment thereof. In an embodiment, the 3' UTR comprises a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 100, SEQ ID NO: 101, SEQ ID NO: 102, SEQ ID NO: 103, SEQ ID NO: 104, SEQ ID NO: 105, SEQ ID NO: 106, SEQ ID NO: 107, SEQ ID NO: 108, SEQ ID NO: 109, SEQ ID NO: 110, SEQ ID NO: 111, SEQ ID NO: 112, SEQ ID NO: 113, SEQ ID NO: 114, SEQ ID NO: 115, or SEQ ID NO: 141.
In an embodiment, the 3' UTR comprises the sequence of SEQ ID NO: 141, or a sequence with at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% or 100% identity to SEQ ID NO: 141. Table 2: 3' UTR sequences



In an embodiment, the 3' UTR comprises a micro RNA (miRNA) binding site, e.g., as described herein, which binds to a miR present in a human cell. In an embodiment, the 3' UTR comprises a miRNA binding site of SEQ ID NO:212, SEQ ID NO: 174, SEQ ID NO: 152 or a combination thereof. In an embodiment, the 3' UTR comprises a plurality of miRNA binding sites, e.g., 2, 3, 4, 5, 6, 7 or 8 miRNA binding sites. In an embodiment, the plurality of miRNA binding sites comprises the same or different miRNA binding sites. miR122 bs = CAAACACCAUUGUCACACUCCA (SEQ ID NO: 212) miR-142-3p bs = UCCAUAAAGUAGGAAACACUACA (SEQ ID NO: 174) miR- 126 bs = CGCAUUAUUACUCACGGUACGA (SEQ ID NO: 152)
Regions having a 5' Cap
The disclosure also includes a polynucleotide that comprises both a 5' Cap and a polynucleotide of the present invention (e.g., a polynucleotide comprising a nucleotide sequence encoding a CFTR polypeptide to be expressed).
The 5' cap structure of a natural mRNA is involved in nuclear export, increasing mRNA stability and binds the mRNA Cap Binding Protein (CBP), which is responsible for mRNA stability in the cell and translation competency through the association of CBP with poly(A) binding protein to form the mature cyclic mRNA species. The cap further assists the removal of 5' proximal introns during mRNA splicing.
Endogenous mRNA molecules can be 5'-end capped generating a 5'-ppp-5'- triphosphate linkage between a terminal guanosine cap residue and the 5 '-terminal transcribed sense nucleotide of the mRNA molecule. This 5 '-guanylate cap can then be methylated to generate an N7-methyl -guanylate residue. The ribose sugars of the terminal and/or anteterminal transcribed nucleotides of the 5' end of the mRNA can optionally also be 2'-O-methylated. 5 '-decapping through hydrolysis and cleavage of the guanylate cap structure can target a nucleic acid molecule, such as an mRNA molecule, for degradation.
In some embodiments, the polynucleotides of the present invention (e.g., a polynucleotide comprising a nucleotide sequence encoding a CFTR polypeptide) incorporate a cap moiety.
In some embodiments, polynucleotides of the present invention comprise a non-hydrolyzable cap structure preventing decapping and thus increasing mRNA halflife. Because cap structure hydrolysis requires cleavage of 5'-ppp-5' phosphorodiester linkages, modified nucleotides can be used during the capping reaction. For example, a Vaccinia Capping Enzyme from New England Biolabs (Ipswich, MA) can be used with a-thio-guanosine nucleotides according to the manufacturer's instructions to create a phosphorothioate linkage in the 5 '-ppp-5' cap. Additional modified guanosine nucleotides can be used such as a-methyl-phosphonate and seleno-phosphate nucleotides.
Additional modifications include, but are not limited to, 2'-O-methylation of the ribose sugars of 5 '-terminal and/or 5 '-anteterminal nucleotides of the polynucleotide (as mentioned above) on the 2'-hydroxyl group of the sugar ring. Multiple distinct 5 '-cap structures can be used to generate the 5 '-cap of a nucleic acid molecule, such as a polynucleotide that functions as an mRNA molecule. Cap
analogs, which herein are also referred to as synthetic cap analogs, chemical caps, chemical cap analogs, or structural or functional cap analogs, differ from natural (i.e., endogenous, wild-type or physiological) 5 '-caps in their chemical structure, while retaining cap function. Cap analogs can be chemically (i. e. , non-enzymatically) or enzymatically synthesized and/or linked to the polynucleotides of the invention.
For example, the Anti-Reverse Cap Analog (ARCA) cap contains two guanines linked by a 5 '-5 '-triphosphate group, wherein one guanine contains an N7 methyl group as well as a 3'-O-methyl group (i.e., N7,3'-O-dimethyl-guanosine-5'- triphosphate-5 '-guanosine (m7G-3'mppp-G; which can equivalently be designated 3' O-Me-m7G(5')ppp(5')G). The 3'-0 atom of the other, unmodified, guanine becomes linked to the 5'-terminal nucleotide of the capped polynucleotide. The N7- and 3'-O- methlyated guanine provides the terminal moiety of the capped polynucleotide.
Another exemplary cap is mCAP, which is similar to ARCA but has a 2'-O- methyl group on guanosine (i.e., N7,2'-O-dimethyl-guanosine-5 '-triphosphate-5 '- guanosine, m7Gm-ppp-G).
Another exemplary cap is m7G-ppp-Gm-A (i.e., N7, guanosine-5 '-triphosphate- 2'-O-dimethyl-guanosine-adenosine).
In some embodiments, the cap is a dinucleotide cap analog. As a non-limiting example, the dinucleotide cap analog can be modified at different phosphate positions with a boranophosphate group or a phosphoroselenoate group such as the dinucleotide cap analogs described in U.S. Patent No. US 8,519, 110, the contents of which are herein incorporated by reference in its entirety.
In another embodiment, the cap is a cap analog is a N7-(4- chlorophenoxy ethyl) substituted dinucleotide form of a cap analog known in the art and/or described herein. Non-limiting examples of a N7-(4-chlorophenoxyethyl) substituted dinucleotide form of a cap analog include a N7-(4-chlorophenoxyethyl)- G(5')ppp(5')G and aN7-(4-chlorophenoxyethyl)-m3 '°G(5')ppp(5')G cap analog (See, e.g., the various cap analogs and the methods of synthesizing cap analogs described in Kore et al. Bioorganic & Medicinal Chemistry 2013 21:4570-4574; the contents of which are herein incorporated by reference in its entirety). In another embodiment, a cap analog of the present invention is a 4-chloro/bromophenoxyethyl analog.
Polynucleotides of the invention can also be capped post-manufacture (whether IVT or chemical synthesis), using enzymes, in order to generate more authentic 5'-cap structures. As used herein, the phrase "more authentic" refers to a feature that closely mirrors or mimics, either structurally or functionally, an endogenous or wild type feature. That is, a "more authentic" feature is better representative of an endogenous, wild-type, natural or physiological cellular function and/or structure as compared to synthetic features or analogs, etc., of the prior art, or which outperforms the corresponding endogenous, wild-type, natural or physiological feature in one or more respects. Non-limiting examples of more authentic 5'cap structures of the present invention are those that, among other things, have enhanced binding of cap binding proteins, increased half-life, reduced susceptibility to 5' endonucleases and/or reduced 5 'decapping, as compared to synthetic 5'cap structures known in the art (or to a wild-type, natural or physiological 5'cap structure). For example, recombinant Vaccinia Virus Capping Enzyme and recombinant 2'-O- methyltransferase enzyme can create a canonical 5 '-5 '-triphosphate linkage between the 5 '-terminal nucleotide of a polynucleotide and a guanine cap nucleotide wherein the cap guanine contains an N7 methylation and the 5 '-terminal nucleotide of the mRNA contains a 2'-O-methyl. Such a structure is termed the Capl structure. This cap results in a higher translational-competency and cellular stability and a reduced activation of cellular pro-inflammatory cytokines, as compared, e.g., to other 5'cap analog structures known in the art. Cap structures include, but are not limited to, 7mG(5')ppp(5')NlpN2p (cap 0), 7mG(5')ppp(5')NlmpNp (cap 1), and 7mG(5')- ppp(5')NlmpN2mp (cap 2).
As a non-limiting example, capping chimeric polynucleotides postmanufacture can be more efficient as nearly 100% of the chimeric polynucleotides can be capped. This is in contrast to -80% when a cap analog is linked to a chimeric polynucleotide in the course of an in vitro transcription reaction.
According to the present invention, 5' terminal caps can include endogenous caps or cap analogs. According to the present invention, a 5' terminal cap can comprise a guanine analog. Useful guanine analogs include, but are not limited to,
inosine, N1 -methyl -guanosine, 2'fluoro-guanosine, 7-deaza-guanosine, 8-oxo- guanosine, 2-amino-guanosine, LNA-guanosine, and 2-azido-guanosine.
Also provided herein are exemplary caps including those that can be used in co-transcriptional capping methods for ribonucleic acid (RNA) synthesis, using RNA polymerase, e.g., wild type RNA polymerase or variants thereof, e.g., such as those variants described herein. In one embodiment, caps can be added when RNA is produced in a “one-pot” reaction, without the need for a separate capping reaction. Thus, the methods, in some embodiments, comprise reacting a polynucleotide template with an RNA polymerase variant, nucleoside triphosphates, and a cap analog under in vitro transcription reaction conditions to produce RNA transcript.
As used here the term “cap” includes the inverted G nucleotide and can comprise one or more additional nucleotides 3’ of the inverted G nucleotide, e.g., 1, 2, 3, or more nucleotides 3’ of the inverted G nucleotide and 5’ to the 5’ UTR, e.g., a 5’ UTR described herein.
Exemplary caps comprise a sequence of GG, GA, or GGA, wherein the underlined, italicized G is an in inverted G nucleotide followed by a 5 ’-5’- triphosphate group.
In one embodiment, a cap comprises a compound of formula (I)
 stereoisomer, tautomer or salt thereof, wherein
stereoisomer, tautomer or salt thereof, wherein

X2 is O, S(O)P, NR24 or CR25R26 in which p is 0, 1, or 2;
Y0 is O or CR6R7;
Y1 is O, S(O)n, CR6R7, or NR, in which n is 0, 1 , or 2; each — is a single bond or absent, wherein when each — is a single bond, Yi is o, S(O) n, CR6R7, or NRs; and when each — is absent, Y 1 is void;
Y2 is (0P(0)R4)m in which m is 0, 1, or 2, or -0-(CR4oR4i)u-Qo-(CR42R43)v-, in which Qo is a bond, O, S(O)r, NR44, or CR45R46, r is 0, 1 , or 2, and each of u and v independently is 1, 2, 3 or 4; each R2 and R2' independently is halo, LNA, or OR3; each R3 independently is H, C1-C6 alkyl, C2-C6 alkenyl, or C2-C6 alkynyl and R3, when being C1-C6 alkyl, C2-C6 alkenyl, or C2-C6 alkynyl, is optionally substituted with one or more of halo, OH and C1-C6 alkoxyl that is optionally substituted with one or more OH or OC(O)-C1-C6 alkyl; each R4 and R' independently is H, halo, C1-C6 alkyl, OH, SH, SeH, or BHs"; each of R, R7, and Rs, independently, is -Q1-T1, in which Qi is a bond or C1- C3 alkyl linker optionally substituted with one or more of halo, cyano, OH and C1-C6 alkoxy, and T1 is H, halo, OH, COOH, cyano, or Rs1, in which Rs1 is C1-C3 alkyl, C2- C6 alkenyl, C2-C6 alkynyl, C1-C6 alkoxyl, C(O)O- C1-C6 alkyl, C3-C8 cycloalkyl, C6- C10 aryl, NR31R32, ( NR31R32R33) , 4 to 12- membered heterocycloalkyl, or 5- or 6- membered heteroaryl, and Rs1 is optionally substituted with one or more substituents selected from the group consisting of halo, OH, oxo, C1-C6 alkyl, COOH, C(O)O-C1- C6 alkyl, cyano, C1-C6 alkoxyl, NR31R32, ( NR31R32R33) , C3-C8 cycloalkyl, C6- C10 aryl, 4 to 12-membered heterocycloalkyl, and 5- or 6-membered heteroaryl;
each of R10, R11, R12, R13 R14, and R15, independently, is -Q2-T2, in which Q2 is a bond or C1-C3 alkyl linker optionally substituted with one or more of halo, cyano, OH and C1-C6 alkoxy, and T2 is H, halo, OH, NH2, cyano, NO2, N3, Rs2, or O Rs2, in which Rs2 is C1-C6 alkyl, C2-C6 alkenyl, C2-C6 alkynyl, C3-C8 cycloalkyl, C6-C10 aryl, NHC(O)-C1-C6 alkyl, NR31R32, (NR31R32R33) , 4 to 12-membered heterocycloalkyl, or 5- or 6-membered heteroaryl, and R$2 is optionally substituted with one or more substituents selected from the group consisting of halo, OH, oxo, C1-C6 alkyl, COOH, C(O)O- C1-C6 alkyl, cyano, Ci - G> alkoxyl, NR31R32, (NR31R32R33) , C3-C8 cycloalkyl, C6-C10 aryl, 4 to 12-membered heterocycloalkyl, and 5- or 6- membered heteroaryl; or alternatively R12 together with R14 is oxo, or R13 together with R15 is oxo, each of R20, R21, R22, and R23 independently is -Q3-T3, in which Q3 is a bond or C1-C3 alkyl linker optionally substituted with one or more of halo, cyano, OH and C1-C6 alkoxy, and T3 is H, halo, OH, NH2, cyano, NO2, N3, Rs3, or O Rs3, in which Rs3 is C1-C6 alkyl, C2-C6 alkenyl, C2-C6 alkynyl, C3-C8 cycloalkyl, C6-C10 aryl, NHC(O)-C1-C6 alkyl, mono- C1-C6 alkylamino, di- C1-C6, alkylamino, 4 to 12- membered heterocycloalkyl, or 5- or 6-membered heteroaryl, and Rs3 is optionally substituted with one or more substituents selected from the group consisting of halo, OH, oxo, C1-C6 alkyl, COOH, C(O)O-C1-C6 alkyl, cyano, C1-C6 alkoxyl, amino, mono-C1-C6 alkylamino, di- C1-C6 alkylamino, C3-C8 cycloalkyl, C6-C10 aryl, 4 to 12- membered heterocycloalkyl, and 5- or 6-membered heteroaryl; each of R24, R25, and R26 independently is H or C1-C6 alkyl; each of R27 and R28 independently is H or OR29; or R27 and R28 together form O-R30-O; each R29 independently is H, C1-C6 alkyl, C2-C6 alkenyl, or C2-C6 alkynyl and R29, when being C1-C6 alkyl, C2-C6 alkenyl, or C2-C6 alkynyl, is optionally substituted with one or more of halo, OH and C1-C6 alkoxyl that is optionally substituted with one or more OH or OC(O)-C1-C6 alkyl;
R30 is C1-C6 alkylene optionally substituted with one or more of halo, OH and C1-C6 alkoxyl; each of R31R32 , and R33, independently is H, C1-C6 alkyl, C3-C8 cycloalkyl, C6-C10 aryl, 4 to 12-membered heterocycloalkyl, or 5- or 6-membered heteroaryl;
each of R40, R41, R42, and R43 independently is H, halo, OH, cyano, N3, OP(O)R47R48, or C1-C6 alkyl optionally substituted with one or more OP(O)R47R48, or one R41 and one R43, together with the carbon atoms to which they are attached and Qo, form C4-C10 cycloalkyl, 4- to 14-membered heterocycloalkyl, C6-C10 aryl, or 5- to 14-membered heteroaryl, and each of the cycloalkyl, heterocycloalkyl, phenyl, or 5- to 6-membered heteroaryl is optionally substituted with one or more of OH, halo, cyano, N3, oxo, OP(O)R47R48, C1-C6 alkyl, C1-C6 haloalkyl, COOH, C(O)O- C1-C6 alkyl, C1-C6 alkoxyl, C1-C6 haloalkoxyl, amino, mono-C1-C6 alkylamino, and di-C1-C6 alkylamino;
R44 is H, C1-C6 alkyl, or an amine protecting group; each of R45 and R46 independently is H, OP(O)R47R48, or C1-C6 alkyl optionally substituted with one or more OP(O)R47R48, and each ofR47 and R48, independently is H, halo, C1-C6 alkyl, OH, SH, SeH, or
BH3-.
It should be understood that a cap analog, as provided herein, may include any of the cap analogs described in international publication WO 2017/066797, published on 20 April 2017, incorporated by reference herein in its entirety.
In some embodiments, the B2 middle position can be a non-ribose molecule, such as arabinose.
In some embodiments R2 is ethyl-based.
Thus, in some embodiments, a cap comprises the following structure:
 In other embodiments, a cap comprises the following structure:
In other embodiments, a cap comprises the following structure:

In yet other embodiments, a cap comprises the following structure:

In still other embodiments, a cap comprises the following structure:

In some embodiments, R is an alkyl (e.g., C1-C6 alkyl). In some embodiments, R is a methyl group (e.g., Ci alkyl). In some embodiments, R is an ethyl group (e.g, C2 alkyl).
In some embodiments, a cap comprises a sequence selected from the following sequences: GAA, GAC, GAG, GAU, GCA, GCC, GCG, GCU, GGA , GGC, GGG, GGU, GUA, GUC, GUG, and GUU. In some embodiments, a cap comprises GAA. In some embodiments, a cap comprises GAC. In some embodiments, a cap comprises GAG. In some embodiments, a cap comprises GAU. In some embodiments, a cap comprises GCA. In some embodiments, a cap comprises GCC. In some embodiments, a cap comprises GCG. In some embodiments, a cap comprises GCU. In some embodiments, a cap comprises GGA. In some embodiments, a cap comprises GGC. In some embodiments, a cap comprises GGG. In some embodiments, a cap comprises GGU. In some embodiments, a cap comprises GUA. In some embodiments, a cap comprises GUC. In some embodiments, a cap comprises GUG. In some embodiments, a cap comprises GUU.
In some embodiments, a cap comprises a sequence selected from the following sequences: m7GpppApA, m7GpppApC, m7GpppApG, m7GpppApU, m7GpppCpA, m7GpppCpC, m7GpppCpG, m7GpppCpU, m7GpppGpA, m7GpppGpC, m7GpppGpG, m7GpppGpU, m7GpppUpA, m7GpppUpC, m7GpppUpG, and m7GpppUpU.
In some embodiments, a cap comprises m7GpppApA. In some embodiments, a cap comprises m7GpppApC. In some embodiments, a cap comprises m7GpppApG. In some embodiments, a cap comprises m7GpppApU. In some embodiments, a cap comprises m7GpppCpA. In some embodiments, a cap comprises m7GpppCpC. In some embodiments, a cap comprises m7GpppCpG. In some embodiments, a cap comprises m7GpppCpU. In some embodiments, a cap comprises m7GpppGpA. In some embodiments, a cap comprises m7GpppGpC. In some embodiments, a cap comprises m7GpppGpG. In some embodiments, a cap comprises m7GpppGpU. In some embodiments, a cap comprises m7GpppUpA. In some embodiments, a cap comprises m7GpppUpC. In some embodiments, a cap comprises m7GpppUpG. In some embodiments, a cap comprises m7GpppUpU.
A cap, in some embodiments, comprises a sequence selected from the following sequences: m7G3'oMepppApA, m7G3'oMepppApC, m7G3'oMepppApG, m7G3'oMePPpApU, m7G3'oMePPpCpA, m7G3'oMePPpCpC, m7G3'oMePPpCpG, m7G3'OMePPpCpU, m7G3'OMePPpGpA, m7G3'OMePPpGpC, m7G3'OMePPpGpG, m7G3'OMepppGpU, m7G3'OMepppUpA, m7G3'OMepppUpC, m7G3'OMepppUpG, and m7G3'OMepppUpU.
In some embodiments, a cap comprises m7G3'oMePPpApA. In some embodiments, a cap comprises m7G3'oMepppApC. In some embodiments, a cap comprises m7G3'oMepppApG. In some embodiments, a cap comprises m7G3'OMepppApU. In some embodiments, a cap comprises m7G3'OMepppCpA. In some embodiments, a cap comprises m7G3'oMepppCpC. In some embodiments, a cap comprises m7G3'oMepppCpG. In some embodiments, a cap comprises m7G3'oMePPpCpU. In some embodiments, a cap comprises m7G3'oMePPpGpA. In some embodiments, a cap comprises m7G3'oMepppGpC. In some embodiments, a cap comprises m7G3'oMepppGpG. In some embodiments, a cap comprises m7G3'oMePPpGpU. In some embodiments, a cap comprises m7G3'oMePPpUpA. In some embodiments, a cap comprises m7G3'oMepppUpC. In some embodiments, a cap comprises m7G3'oMepppUpG. In some embodiments, a cap comprises m7G3'OMepppUpU.
A cap, in other embodiments, comprises a sequence selected from the following sequences: m’Gs'oMePPpA/oMepA, m’Gs'oMePPpA/oMepC, m7G3'OMepppA2'OMepG, m’Gs'OMePPpA/OMepU, m’Gs'OMePPpC/OMepA, m7G3'OMePPpC2'OMepC, nfGs'OMePPpC/OMepG, nfGs'OMePPpC/OMepU, m7G3'OMePPpG2'OMepA, nfGs'OMePPpG/OMepC, m’Gs'OMePPpG/OMepG, m7G3'OMePPpG2'OMepU, nfGs'OMePPpU/OMepA, m’Gs'OMePPpU/OMepC, m7G3'OMePPpU2'OMepG, and m7G3'OMePPpU2'OMepU.
In some embodiments, a cap comprises m7G3'OMepppA2'oMepA. In some embodiments, a cap comprises m7G3'oMePPpA2'oMepC. In some embodiments, a cap comprises m7G3'oMePPpA2'oMepG. In some embodiments, a cap comprises m7G3'oMePPpA2'oMepU. In some embodiments, a cap comprises m7G3'oMePPpC2'oMepA. In some embodiments, a cap comprises m7G3'oMePPpC2'oMepC. In some embodiments, a cap comprises m7G3'OMepppC2'OMepG. In some embodiments, a cap comprises m7G3'oMePPpC2'oMepU. In some embodiments, a cap comprises m7G3'oMePPpG2'oMepA. In some embodiments, a cap comprises m7G3'oMePPpG2'oMepC. In some embodiments, a cap comprises m7G3'oMePPpG2'oMepG. In some embodiments, a cap comprises m7G3'oMePPpG2'oMepU. In some embodiments, a cap comprises m7G3'oMePPpU2'oMepA. In some embodiments, a cap comprises m7G3'oMePPpU2'oMepC. In some embodiments, a cap comprises m7G3'oMePPpU2'oMepG. In some embodiments, a cap comprises m7G3'OMepppU2'OMepU.
A cap, in still other embodiments, comprises a sequence selected from the following sequences: m7GpppA2'oMepA, m7GpppA2'oMepC, m7GpppA2'oMepG, m7GpppA2'oMepU, m7GpppC2'oMepA, m7GpppC2'oMepC, m7GpppC2'oMepG, m7GpppC2'OMepU, m7GpppG2'OMepA, m7GpppG2'OMepC, m7GpppG2'OMepG, m7GpppG2'0MepU, m7GpppU2'0MepA, m7GpppU2'0MepC, m7GpppU2'0MepG, and m7GpppU2'OMepU.
In some embodiments, a cap comprises m7GpppA2'oMepA. In some embodiments, a cap comprises m7GpppA2'oMepC. In some embodiments, a cap comprises m7GpppA2'oMepG. In some embodiments, a cap comprises m7GpppA2'oMepU. In some embodiments, a cap comprises m7GpppC2'OMepA. In some embodiments, a cap comprises m7GpppC2'OMepC. In some embodiments, a cap comprises m7GpppC2'oMepG. In some embodiments, a trinucleotide cap comprises m7GpppC2'oMepU. In some embodiments, a cap comprises m7GpppG2'oMepA. In some embodiments, a cap comprises m7GpppG2'oMepC. In some embodiments, a cap comprises m7GpppG2'OMepG. In some embodiments, a cap comprises m7GpppG2'oMepU. In some embodiments, a cap comprises m7GpppU2'oMepA. In some embodiments, a cap comprises m7GpppU2'oMepC. In some embodiments, a cap comprises m7GpppU2'oMepG. In some embodiments, a cap comprises m7GpppU2'oMepU.
In some embodiments, a cap comprises m7Gpppm6A2’OmepG. In some embodiments, a cap comprises m7Gpppe6A2’OmepG.
In some embodiments, a cap comprises GAG. In some embodiments, a cap comprises GCG. In some embodiments, a cap comprises GUG. In some embodiments, a cap comprises GGG.
In some embodiments, a cap comprises any one of the following structures:


In some embodiments, the cap comprises m7GpppNiN2N3, where Ni, N2, and N3 are optional (i.e., can be absent or one or more can be present) and are independently a natural, a modified, or an unnatural nucleoside base. In some embodiments, m7G is further methylated, e.g., at the 3’ position. In some embodiments, the m7G comprises an O-methyl at the 3’ position. In some embodiments Ni, N2, and N3 if present, optionally, are independently an adenine, a uracil, a guanidine, a thymine, or a cytosine. In some embodiments, one or more (or all) of Ni, N2, and N3, if present, are methylated, e.g., at the 2’ position. In some embodiments, one or more (or all) of Ni, N2, and N3, if present have an O-methyl at the 2’ position.
In some embodiments, the cap comprises the following structure:

In some embodiments, Bi, B3, and B3 are natural nucleoside bases. In some embodiments, at least one of Bi, B2, and B3 is a modified or unnatural base. In some embodiments, at least one of Bi, B2, and B3 is N6-methyladenine. In some embodiments, Bi is adenine, cytosine, thymine, or uracil. In some embodiments, Bi is adenine, B2 is uracil, and B3 is adenine. In some embodiments, Ri and R2 are OH, R3 and R4 are O-methyl, Bi is adenine, B2 is uracil, and B3 is adenine.
In some embodiments the cap comprises a sequence selected from the following sequences: GAAA, GACA, GAGA, GAUA, GCAA, GCCA, GCGA, GCUA, GGAA, GGCA, GGGA, GGUA, GUCA, and GUUA. In some embodiments the cap comprises a sequence selected from the following sequences: GAAG, GACG, GAGG, GAUG, GCAG, GCCG, GCGG, GCUG, GGAG, GGCG, GGGG, GGUG, GUCG, GUGG, and GUUG. In some embodiments the cap comprises a sequence
selected from the following sequences: GAAU, GACU, GAGU, GAUU, GCAU, GCCU, GCGU, GCUU, GGAU, GGCU, GGGU, GGUU, GUAU, GUCU, GUGU, and GUUU. In some embodiments the cap comprises a sequence selected from the following sequences: GAAC, GACC, GAGC, GAUC, GCAC, GCCC, GCGC, GCUC, GGAC, GGCC, GGGC, GGUC, GUAC, GUCC, GUGC, and GUUC.
A cap, in some embodiments, comprises a sequence selected from the following sequences: m7G3'oMepppApApN, m7G3'oMepppApCpN, m7G3'oMePPpApGpN, m7G3'oMepppApUpN, m7G3'oMepppCpApN, m7G3'oMePPpCpCpN, m7G3'oMePPpCpGpN, m7G3'oMepppCpUpN, m7G3'OMepppGpApN, m7G3'OMepppGpCpN, m7G3'OMepppGpGpN, m7G3'OMepppGpUpN, m7G3'OMepppUpApN, m7G3'OMepppUpCpN, m7G3'oMepppUpGpN, and m7G3'oMepppUpUpN, where N is a natural, a modified, or an unnatural nucleoside base.
A cap, in other embodiments, comprises a sequence selected from the following sequences: m’Gs'oMePPpA/oMepApN, m’Gs'oMePPpA/oMepCpN, m7G3'OMePPpA2'OMepGpN, m’Gs'oMePPpA/oMepUpN, m’Gs'oMePPpCzoMepApN, m7G3'OMePPpC2'OMepCpN, m7G3'OMepppC2'OMepGpN, m7G3'OMepppC2'OMepUpN, m7G3'OMePPpG2'OMepApN, nfGs'oMePPpGzoMepCpN, nfGs'oMePPpGzoMepGpN, m7G3'OMePPpG2'OMepUpN, m’Gs'OMePPpU/OMepApN, m7G3'OMepppU2'OMepCpN, m7G3'oMepppU2'oMepGpN, and m7G3'oMepppU2'oMepUpN, where N is a natural, a modified, or an unnatural nucleoside base.
A cap, in still other embodiments, comprises a sequence selected from the following sequences: m7GpppA2'OMepApN, m7GpppA2'OMepCpN, m7GpppA2'oMepGpN, m7GpppA2'oMepUpN, m7GpppC2'OMepApN, m7GpppC2'OMepCpN, m7GpppC2'OMepGpN, m7GpppC2'OMepUpN, m7GpppG2'OMepApN, m7GpppG2'OMepCpN, m7GpppG2'OMepGpN, m7GpppG2'OMepUpN, m7GpppU2'OMepApN, m7GpppU2'OMepCpN, m7GpppU2'oMepGpN, and m7GpppU2'oMepUpN, where N is a natural, a modified, or an unnatural nucleoside base.
A cap, in other embodiments, comprises a sequence selected from the following sequences: m’Gs'oMePPpA/oMepAzoMepN, m’Gs'oMePPpA/oMepCzoMepN, m7G3'OMepppA2'OMepG2'OMepN, m7G3'OMepppA2'OMepU2'OMepN, m7G3'OMePPpC2'OMepA2'OMepN, m’Gs'OMePPpC/OMepCzOMepN, m7G3'OMePPpC2'OMepG2'OMepN, m7G3'OMepppC2'OMepU2'OMepN, m7G3'OMePPpG2'OMepA2'OMepN, m’Gs'OMePPpG/OMepCzOMepN, m7G3'OMePPpG2'OMepG2'OMepN, m’Gs'OMePPpG/OMeplh'OMepN, m7G3'OMepppU2'OMepA2'OMepN, m’Gs'OMePPpU/OMepCzOMepN, m7G3'0MePPpU2'0MepG2'0MepN, and m7G3'0MePPpU2'0MepU2'0MepN, where N is a natural, a modified, or an unnatural nucleoside base.
A cap, in still other embodiments, comprises a sequence selected from the following sequences: m’GpppA/oMepAzoMepN, m’GpppA/oMepCzoMepN, m7GpppA2'OMepG2'OMepN, m7GpppA2'OMepU2'OMepN, m7GpppC2'OMepA2'OMepN, m7GpppC2'OMepC2'OMepN, m7GpppC2'OMepG2'OMepN, m’GpppC/OMepUzOMepN, m7GpppG2'OMepA2'OMepN, m7GpppG2'OMepC2'OMepN, m7GpppG2'OMepG2'OMepN, m7GpppG2'OMepU2'OMepN, m7GpppU2'OMepA2'OMepN, m’GpppU/OMepCzOMepN, m7GpppU2'oMepG2'oMepN, and m7GpppU2'oMepU2'oMepN, where N is a natural, a modified, or an unnatural nucleoside base.
In some embodiments, a cap comprises GGAG. In some embodiments, a cap comprises the following structure:
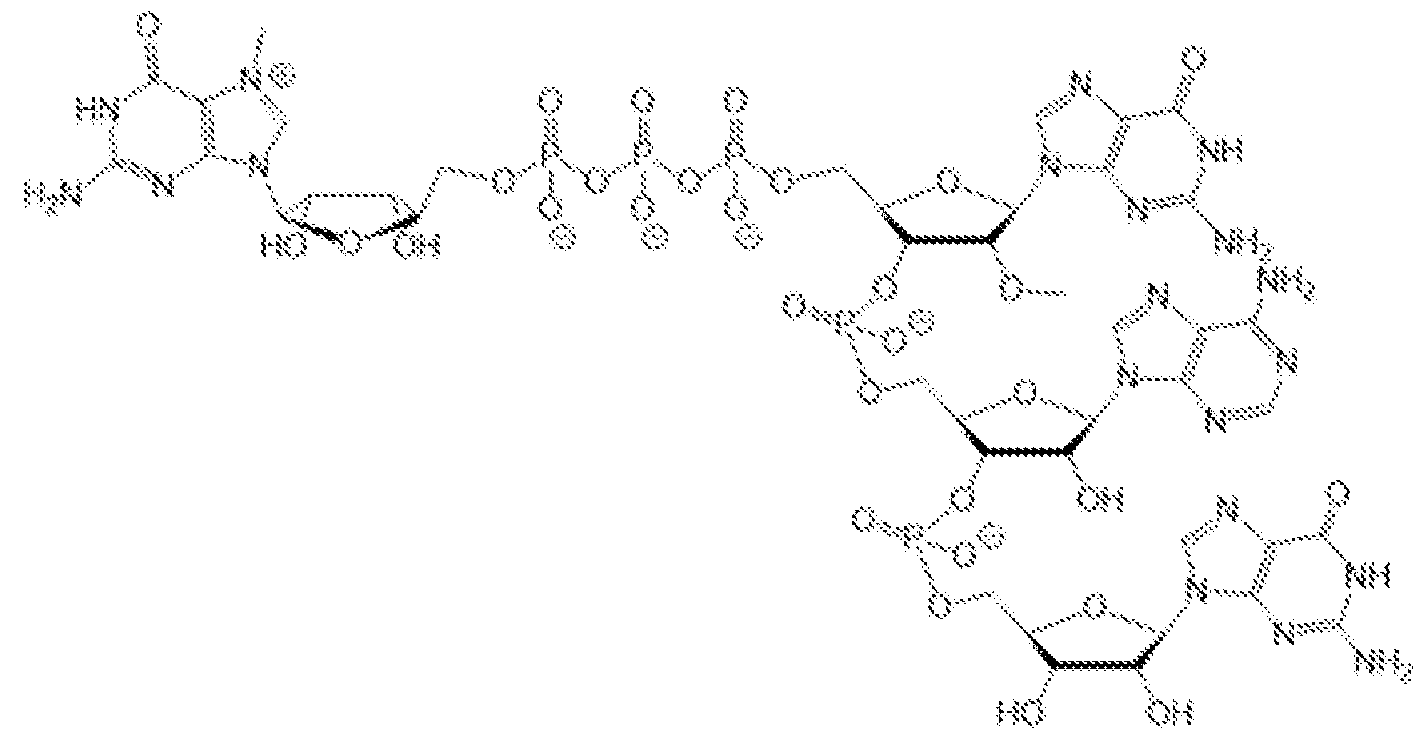
(X).
Poly-A Tails
In some embodiments, the polynucleotides of the present disclosure (e.g., a polynucleotide comprising a nucleotide sequence encoding a CFTR polypeptide) further comprise a poly-A tail. In further embodiments, terminal groups on the poly-A tail can be incorporated for stabilization. In other embodiments, a poly-A tail comprises des-3' hydroxyl tails.
During RNA processing, a long chain of adenine nucleotides (poly-A tail) can be added to a polynucleotide such as an mRNA molecule in order to increase stability. Immediately after transcription, the 3' end of the transcript can be cleaved to free a 3' hydroxyl. Then poly-A polymerase adds a chain of adenine nucleotides to the RNA. The process, called polyadenylation, adds a poly-A tail that can be between, for example, approximately 80 to approximately 250 residues long, including approximately 80, 90, 100, 110, 120, 130, 140, 150, 160, 170, 180, 190, 200, 210, 220, 230, 240 or 250 residues long. In one embodiment, the poly-A tail is 100 nucleotides in length (SEQ ID NO: 195).
PolyA tails can also be added after the construct is exported from the nucleus.
According to the present invention, terminal groups on the poly A tail can be incorporated for stabilization. Polynucleotides of the present invention can include
des-3' hydroxyl tails. They can also include structural moieties or 2'-Omethyl modifications as taught by Junjie Li, et al. (Current Biology, Vol. 15, 1501-1507, August 23, 2005, the contents of which are incorporated herein by reference in its entirety).
The polynucleotides of the present invention can be designed to encode transcripts with alternative polyA tail structures including histone mRNA. According to Norbury, "Terminal uridylation has also been detected on human replicationdependent histone mRNAs. The turnover of these mRNAs is thought to be important for the prevention of potentially toxic histone accumulation following the completion or inhibition of chromosomal DNA replication. These mRNAs are distinguished by their lack of a 3 ' poly(A) tail, the function of which is instead assumed by a stable stem-loop structure and its cognate stem-loop binding protein (SLBP); the latter carries out the same functions as those of PABP on polyadenylated mRNAs" (Norbury, "Cytoplasmic RNA: a case of the tail wagging the dog," Nature Reviews Molecular Cell Biology; AOP, published online 29 August 2013; doi: 10.1038/nrm3645) the contents of which are incorporated herein by reference in its entirety.
Unique poly-A tail lengths provide certain advantages to the polynucleotides of the present invention. Generally, the length of a poly-A tail, when present, is greater than 30 nucleotides in length (SEQ ID NO:215). In another embodiment, the poly-A tail is greater than 35 nucleotides in length (SEQ ID NO:216) (e.g., at least or greater than about 35, 40, 45, 50, 55, 60, 70, 80, 90, 100, 120, 140, 160, 180, 200, 250, 300, 350, 400, 450, 500, 600, 700, 800, 900, 1,000, 1,100, 1,200, 1,300, 1,400, 1,500, 1,600, 1,700, 1,800, 1,900, 2,000, 2,500, and 3,000 nucleotides).
In some embodiments, the polynucleotide or region thereof includes from about 30 to about 3,000 nucleotides (e.g., from 30 to 50, from 30 to 100, from 30 to 250, from 30 to 500, from 30 to 750, from 30 to 1,000, from 30 to 1,500, from 30 to 2,000, from 30 to 2,500, from 50 to 100, from 50 to 250, from 50 to 500, from 50 to 750, from 50 to 1,000, from 50 to 1,500, from 50 to 2,000, from 50 to 2,500, from 50 to 3,000, from 100 to 500, from 100 to 750, from 100 to 1,000, from 100 to 1,500, from 100 to 2,000, from 100 to 2,500, from 100 to 3,000, from 500 to 750, from 500
to 1,000, from 500 to 1,500, from 500 to 2,000, from 500 to 2,500, from 500 to 3,000, from 1,000 to 1,500, from 1,000 to 2,000, from 1,000 to 2,500, from 1,000 to 3,000, from 1,500 to 2,000, from 1,500 to 2,500, from 1,500 to 3,000, from 2,000 to 3,000, from 2,000 to 2,500, and from 2,500 to 3,000).
In some embodiments, the poly-A tail is designed relative to the length of the overall polynucleotide or the length of a particular region of the polynucleotide. This design can be based on the length of a coding region, the length of a particular feature or region or based on the length of the ultimate product expressed from the polynucleotides.
In this context, the poly-A tail can be 10, 20, 30, 40, 50, 60, 70, 80, 90, or 100% greater in length than the polynucleotide or feature thereof. The poly-A tail can also be designed as a fraction of the polynucleotides to which it belongs. In this context, the poly-A tail can be 10, 20, 30, 40, 50, 60, 70, 80, or 90% or more of the total length of the construct, a construct region or the total length of the construct minus the poly-A tail. Further, engineered binding sites and conjugation of polynucleotides for Poly-A binding protein can enhance expression.
Additionally, multiple distinct polynucleotides can be linked together via the PABP (Poly-A binding protein) through the 3 '-end using modified nucleotides at the 3 '-terminus of the poly-A tail. Transfection experiments can be conducted in relevant cell lines at and protein production can be assayed by ELISA at 12hr, 24hr, 48hr, 72hr and day 7 post-transfection.
In some embodiments, the polynucleotides of the present invention are designed to include a polyA-G Quartet region. The G-quartet is a cyclic hydrogen bonded array of four guanine nucleotides that can be formed by G-rich sequences in both DNA and RNA. In this embodiment, the G-quartet is incorporated at the end of the poly-A tail. The resultant polynucleotide is assayed for stability, protein production and other parameters including half-life at various time points. It has been discovered that the polyA-G quartet results in protein production from an mRNA equivalent to at least 75% of that seen using a poly-A tail of 120 nucleotides alone (SEQ ID NO: 196).
In some embodiments, the polyA tail comprises an alternative nucleoside, e.g., inverted thymidine. PolyA tails comprising an alternative nucleoside, e.g., inverted thymidine, may be generated as described herein. For instance, mRNA constructs may be modified by ligation to stabilize the poly (A) tail. Ligation may be performed using 0.5-1.5 mg/mL mRNA (5' Capl, 3' A100), 50 mM Tris-HCl pH 7.5, 10 mM MgCh, 1 mM TCEP, 1000 units/mL T4 RNA Ligase 1, 1 mM ATP, 20% w/v polyethylene glycol 8000, and 5: 1 molar ratio of modifying oligo to mRNA. Modifying oligo has a sequence of 5’-phosphate-AAAAAAAAAAAAAAAAAAAA- (inverted deoxythymidine (idT) (SEQ ID NO:209)) (see below). Ligation reactions are mixed and incubated at room temperature (~22°C) for, e.g., 4 hours. Stable tail mRNA are purified by, e.g., dT purification, reverse phase purification, hydroxyapatite purification, ultrafiltration into water, and sterile filtration. The resulting stable tail-containing mRNAs contain the following structure at the 3 ’end, starting with the polyA region: Aioo-UCUAGAAAAAAAAAAAAAAAAAAAA- inverted deoxythymidine (SEQ ID NO:211).
Modifying oligo to stabilize tail (5’-phosphate- AAAAAAAAAAAAAAAAAAAA-(inverted deoxythymidine)(SEQ ID NO: 209) (shown below)):

In some instances, the polyA tail comprises A100-UCUAG-A20-inverted deoxy-thymidine (SEQ ID NO:211). In some instances, the polyA tail consists of A100-UCUAG-A20-inverted deoxy-thymidine (SEQ ID NO:211).
Start codon region
The invention also includes a polynucleotide that comprises both a start codon region and the polynucleotide described herein (e.g., a polynucleotide comprising a nucleotide sequence encoding a CFTR polypeptide). In some embodiments, the polynucleotides of the present invention can have regions that are analogous to or function like a start codon region.
In some embodiments, the translation of a polynucleotide can initiate on a codon that is not the start codon AUG. Translation of the polynucleotide can initiate on an alternative start codon such as, but not limited to, ACG, AGG, AAG, CTG/CUG, GTG/GUG, ATA/AUA, ATT/AUU, TTG/UUG (see Touriol et al. Biology of the Cell 95 (2003) 169-178 and Matsuda and Mauro PLoS ONE, 2010 5 : 11 ; the contents of each of which are herein incorporated by reference in its entirety).
As a non-limiting example, the translation of a polynucleotide begins on the alternative start codon ACG. As another non-limiting example, polynucleotide translation begins on the alternative start codon CTG or CUG. As yet another nonlimiting example, the translation of a polynucleotide begins on the alternative start codon GTG or GUG.
Nucleotides flanking a codon that initiates translation such as, but not limited to, a start codon or an alternative start codon, are known to affect the translation efficiency, the length and/or the structure of the polynucleotide. (See, e.g., Matsuda and Mauro PLoS ONE, 2010 5: 11; the contents of which are herein incorporated by reference in its entirety). Masking any of the nucleotides flanking a codon that initiates translation can be used to alter the position of translation initiation, translation efficiency, length and/or structure of a polynucleotide.
In some embodiments, a masking agent can be used near the start codon or alternative start codon in order to mask or hide the codon to reduce the probability of translation initiation at the masked start codon or alternative start codon. Non-limiting examples of masking agents include antisense locked nucleic acids (LNA) polynucleotides and exon-junction complexes (EJCs) (See, e.g., Matsuda and Mauro
describing masking agents LNA polynucleotides and EJCs (PLoS ONE, 2010 5: 11); the contents of which are herein incorporated by reference in its entirety).
In another embodiment, a masking agent can be used to mask a start codon of a polynucleotide in order to increase the likelihood that translation will initiate on an alternative start codon. In some embodiments, a masking agent can be used to mask a first start codon or alternative start codon in order to increase the chance that translation will initiate on a start codon or alternative start codon downstream to the masked start codon or alternative start codon.
In some embodiments, a start codon or alternative start codon can be located within a perfect complement for a miRNA binding site. The perfect complement of a miRNA binding site can help control the translation, length and/or structure of the polynucleotide similar to a masking agent. As a non-limiting example, the start codon or alternative start codon can be located in the middle of a perfect complement for a miRNA binding site. The start codon or alternative start codon can be located after the first nucleotide, second nucleotide, third nucleotide, fourth nucleotide, fifth nucleotide, sixth nucleotide, seventh nucleotide, eighth nucleotide, ninth nucleotide, tenth nucleotide, eleventh nucleotide, twelfth nucleotide, thirteenth nucleotide, fourteenth nucleotide, fifteenth nucleotide, sixteenth nucleotide, seventeenth nucleotide, eighteenth nucleotide, nineteenth nucleotide, twentieth nucleotide or twenty-first nucleotide.
In another embodiment, the start codon of a polynucleotide can be removed from the polynucleotide sequence in order to have the translation of the polynucleotide begin on a codon that is not the start codon. Translation of the polynucleotide can begin on the codon following the removed start codon or on a downstream start codon or an alternative start codon. In a non-limiting example, the start codon ATG or AUG is removed as the first 3 nucleotides of the polynucleotide sequence in order to have translation initiate on a downstream start codon or alternative start codon. The polynucleotide sequence where the start codon was removed can further comprise at least one masking agent for the downstream start codon and/or alternative start codons in order to control or attempt to control the
initiation of translation, the length of the polynucleotide and/or the structure of the polynucleotide.
Stop Codon Region
The invention also includes a polynucleotide that comprises both a stop codon region and the polynucleotide described herein (e.g., a polynucleotide comprising a nucleotide sequence encoding a CFTR polypeptide). In some embodiments, the polynucleotides of the present invention can include at least two stop codons before the 3' untranslated region (UTR). The stop codon can be selected from TGA, TAA and TAG in the case of DNA, or from UGA, UAA and UAG in the case of RNA. In some embodiments, the polynucleotides of the present invention include the stop codon TGA in the case or DNA, or the stop codon UGA in the case of RNA, and one additional stop codon. In a further embodiment the addition stop codon can be TAA or UAA. In another embodiment, the polynucleotides of the present invention include three consecutive stop codons, four stop codons, or more.
Combination of mRNA elements
Any of the polynucleotides disclosed herein can comprise one, two, three, or all of the following elements: (a) a 5 ’-UTR, e.g., as described herein; (b) a coding region comprising a stop element (e.g., as described herein); (c) a 3 ’-UTR (e.g., as described herein) and; optionally (d) a 3’ stabilizing region, e.g., as described herein. Also disclosed herein are UNP compositions comprising the same.
In an embodiment, a polynucleotide of the disclosure comprises (a) a 5’ UTR described in Table 1 or a variant or fragment thereof and (b) a coding region comprising a stop element provided herein. In an embodiment, the polynucleotide further comprises a cap structure, e.g., as described herein, or a poly A tail, e.g., as described herein. In an embodiment, the polynucleotide further comprises a 3’ stabilizing region, e.g., as described herein.
In an embodiment, a polynucleotide of the disclosure comprises (a) a 5’ UTR described in Table 1 or a variant or fragment thereof and (c) a 3’ UTR described in Table 2 or a variant or fragment thereof. In an embodiment, the polynucleotide
further comprises a cap structure, e.g., as described herein, or a poly A tail, e.g., as described herein. In an embodiment, the polynucleotide further comprises a 3’ stabilizing region, e.g., as described herein.
In an embodiment, a polynucleotide of the disclosure comprises (c) a 3’ UTR described in Table 2 or a variant or fragment thereof and (b) a coding region comprising a stop element provided herein. In an embodiment, the polynucleotide comprises a sequence provided in Table 3. In an embodiment, the polynucleotide further comprises a cap structure, e.g., as described herein, or a poly A tail, e.g., as described herein. In an embodiment, the polynucleotide further comprises a 3’ stabilizing region, e.g. , as described herein.
In an embodiment, a polynucleotide of the disclosure comprises (a) a 5’ UTR described in Table 1 or a variant or fragment thereof; (b) a coding region comprising a stop element provided herein; and (c) a 3’ UTR described in Table 2 or a variant or fragment thereof. In an embodiment, the polynucleotide further comprises a cap structure, e.g., as described herein, or a poly A tail, e.g., as described herein. In an embodiment, the polynucleotide further comprises a 3’ stabilizing region, e.g., as described herein.
Table 3: Exemplary 3’ UTR and stop element sequences



Polynucleotide Comprising an mRNA Encoding a CFTR Polypeptide
In certain embodiments, a polynucleotide of the present disclosure, for example a polynucleotide comprising an mRNA nucleotide sequence encoding a CFTR polypeptide, comprises from 5' to 3' end:
(i) a 5' cap such as provided above;
(ii) a 5' UTR, such as the sequences provided above;
(iii) an ORF encoding a CFTR polypeptide (e.g., SEQ ID NO: 1), wherein the ORF has at least 90%, at least 95%, at least 97%, at least 98%, at least 99%, or 100% sequence identity to the sequence of SEQ ID NO: 10;
(iv) at least one stop codon;
(v) a 3' UTR, such as the sequences provided above; and
(vi) a poly-A tail provided above.
In some embodiments, the polynucleotide further comprises a miRNA binding site, e.g., a miRNA binding site that binds to miRNA-142. In some embodiments, the 5' UTR comprises the miRNA binding site. In some embodiments, the 3' UTR comprises the miRNA binding site.
In some embodiments, a polynucleotide of the present disclosure comprises a nucleotide sequence encoding a polypeptide sequence at least 70%, at least 80%, at
least 81%, at least 82%, at least 83%, at least 84%, at least 85%, at least 86%, at least 87%, at least 88%, at least 89%, at least 90%, at least 91%, at least 92%, at least 93%, at least 94%, at least 95%, at least 96% , at least 97%, at least 98%, at least 99%, or 100% identical to the protein sequence of a human CFTR having the amino acid sequence of SEQ ID NO: 1.
In some embodiments, a polynucleotide of the present disclosure, for example a polynucleotide comprising an mRNA nucleotide sequence encoding a polypeptide, comprises (1) a 5' cap such as provided above, for example, m7Gp-ppGm-A, (2) a 5' UTR, (3) a nucleotide sequence ORF of SEQ ID NO: 10, (3) a stop codon, (4) a 3 'UTR, and (5) a poly-A tail provided above, for example, a poly-A tail of SEQ ID NO: 195 or A100-UCUAG-A20-inverted deoxy-thymidine (SEQ ID NO:211).
An exemplary CFTR nucleotide construct is described: SEQ ID NO: 11 consists from 5’ to 3’ end: 5' UTR of SEQ ID NO:50, ORF Sequence of SEQ ID NO: 10, and 3' UTR of SEQ ID NO: 141.
In certain embodiments, in a construct with SEQ ID NO: 11, all uracils therein are replaced by N1 -methylpseudouracil.
An exemplary CFTR nucleotide construct is SEQ ID NO: 13.
In some embodiments, a polynucleotide of the present disclosure, for example a polynucleotide comprising an mRNA nucleotide sequence encoding a CFTR polypeptide, comprises (1) a 5' cap such as provided above, for example, m7Gp- ppGm-A, (2) a nucleotide sequence of SEQ ID NO: 11, and (3) a poly-A tail provided above, for example, a poly A tail of -100 residues, e.g., SEQ ID NO: 195 or A100- UCUAG-A20-inverted deoxy-thymidine (SEQ ID NO:211). In certain embodiments, in constructs with SEQ ID NO: 11, all uracils therein are replaced by Nl- methylpseudouracil .
Methods of Making Polynucleotides
The present disclosure also provides methods for making a polynucleotide of the invention (e.g., a polynucleotide comprising a nucleotide sequence encoding a CFTR polypeptide) or a complement thereof.
In some aspects, a polynucleotide (e.g., a RNA, e.g., an mRNA) disclosed herein, and encoding a CFTR polypeptide, can be constructed using in vitro transcription (IVT). In other aspects, a polynucleotide (e.g., a RNA, e.g., an mRNA) disclosed herein, and encoding a CFTR polypeptide, can be constructed by chemical synthesis using an oligonucleotide synthesizer.
In other aspects, a polynucleotide (e.g., a RNA, e.g., an mRNA) disclosed herein, and encoding a CFTR polypeptide is made by using a host cell. In certain aspects, a polynucleotide (e.g., a RNA, e.g., an mRNA) disclosed herein, and encoding a CFTR polypeptide is made by one or more combination of the IVT, chemical synthesis, host cell expression, or any other methods known in the art.
Naturally occurring nucleosides, non-naturally occurring nucleosides, or combinations thereof, can totally or partially naturally replace occurring nucleosides present in the candidate nucleotide sequence and can be incorporated into a sequence- optimized nucleotide sequence (e.g., a RNA, e.g., an mRNA) encoding a CFTR polypeptide. The resultant polynucleotides, e.g., mRNAs, can then be examined for their ability to produce protein and/or produce a therapeutic outcome.
Pharmaceutical Compositions and Formulations
The present invention provides pharmaceutical compositions and formulations that comprise any of the polynucleotides described above. In some embodiments, the composition or formulation further comprises a delivery agent.
In some embodiments, the composition or formulation can contain a polynucleotide comprising a sequence optimized nucleic acid sequence disclosed herein which encodes a CFTR polypeptide. In some embodiments, the composition or formulation can contain a polynucleotide (e.g., a RNA, e.g., an mRNA) comprising a polynucleotide (e.g., an ORF) having significant sequence identity to a sequence optimized nucleic acid sequence disclosed herein which encodes a CFTR polypeptide. In some embodiments, the polynucleotide further comprises a miRNA binding site, e.g., a miRNA binding site that binds miR-126, miR-142, miR-144, miR-146, miR- 150, miR-155, miR-16, miR-21, miR-223, miR-24, miR-27 and miR-26a.
Pharmaceutical compositions or formulation can optionally comprise one or more additional active substances, e.g., therapeutically and/or prophylactically active substances. Pharmaceutical compositions or formulation of the present invention can be sterile and/or pyrogen -free. General considerations in the formulation and/or manufacture of pharmaceutical agents can be found, for example, in Remington : The Science and Practice of Pharmacy 21st ed., Lippincott Williams & Wilkins, 2005 (incorporated herein by reference in its entirety). In some embodiments, compositions are administered to humans, human patients or subjects. For the purposes of the present disclosure, the phrase "active ingredient" generally refers to polynucleotides to be delivered as described herein.
Formulations and pharmaceutical compositions described herein can be prepared by any method known or hereafter developed in the art of pharmacology. In general, such preparatory methods include the step of associating the active ingredient with an excipient and/or one or more other accessory ingredients, and then, if necessary and/or desirable, dividing, shaping and/or packaging the product into a desired single- or multi-dose unit.
A pharmaceutical composition or formulation in accordance with the present disclosure can be prepared, packaged, and/or sold in bulk, as a single unit dose, and/or as a plurality of single unit doses. As used herein, a "unit dose" refers to a discrete amount of the pharmaceutical composition comprising a predetermined amount of the active ingredient. The amount of the active ingredient is generally equal to the dosage of the active ingredient that would be administered to a subject and/or a convenient fraction of such a dosage such as, for example, one-half or one-third of such a dosage.
Relative amounts of the active ingredient, the pharmaceutically acceptable excipient, and/or any additional ingredients in a pharmaceutical composition in accordance with the present disclosure can vary, depending upon the identity, size, and/or condition of the subject being treated and further depending upon the route by which the composition is to be administered.
In some embodiments, the compositions and formulations described herein can contain at least one polynucleotide of the invention. As a non-limiting example, the composition or formulation can contain 1, 2, 3, 4 or 5 polynucleotides of the
invention. In some embodiments, the compositions or formulations described herein can comprise more than one type of polynucleotide. In some embodiments, the composition or formulation can comprise a polynucleotide in linear and circular form. In another embodiment, the composition or formulation can comprise a circular polynucleotide and an in vitro transcribed (IVT) polynucleotide. In yet another embodiment, the composition or formulation can comprise an IVT polynucleotide, a chimeric polynucleotide and a circular polynucleotide.
Although the descriptions of pharmaceutical compositions and formulations provided herein are principally directed to pharmaceutical compositions and formulations that are suitable for administration to humans, it will be understood by the skilled artisan that such compositions are generally suitable for administration to any other animal, e.g., to non-human animals, e.g. non-human mammals.
The present invention provides pharmaceutical formulations that comprise a polynucleotide described herein (e.g., a polynucleotide comprising a nucleotide sequence encoding a CFTR polypeptide). The polynucleotides described herein can be Formulated using one or more excipients to: (1) increase stability; (2) increase cell transfection; (3) permit the sustained or delayed release (e.g., from a depot formulation of the polynucleotide); (4) alter the biodistribution (e.g., target the polynucleotide to specific tissues or cell types); (5) increase the translation of encoded protein in vivo,' and/or (6) alter the release profile of encoded protein in vivo. In some embodiments, the pharmaceutical formulation further comprises a delivery agent comprising, e.g., a compound having the Formula (I), e.g., Compound II; or a compound having the Formula (III), (IV), (V), or (VI), e.g., Compound I or VI, or any combination thereof. In some embodiments, the delivery agent comprises an ionizable amino lipid (e.g., Compound II or VI), a helper lipid (e.g., DSPC), a sterol (e.g., Cholesterol), and a PEG lipid (e.g., Compound I or PEG-DMG), e.g., with a mole ratio in the range of about (i) 40-50 mol% ionizable amino lipid (e.g., Compound II, VI, or A), optionally 45-50 mol% ionizable amino lipid, for example, 45-46 mol%, 46-47 mol%, 47-48 mol%, 48-49 mol%, or 49-50 mol% for example about 45 mol%, 45.5 mol%, 46 mol%, 46.5 mol%, 47 mol%, 47.5 mol%, 48 mol%, 48.5 mol%, 49 mol%, or 49.5 mol%; (ii) 30-45 mol% sterol (e.g., cholesterol),
optionally 35-42 mol% sterol, for example, 30-31 mol%, 31-32 mol%, 32-33 mol%, 33-34 mol%, 35-35 mol%, 35-36 mol%, 36-37 mol%, 37-38 mol%, 38-39 mol%, or 39-40 mol%, or 40-42 mol% sterol; (iii) 5-15 mol% helper lipid (e.g., DSPC), optionally 10-15 mol% helper lipid, for example, 5-6 mol%, 6-7 mol%, 7-8 mol%, 8- 9 mol%, 9-10 mol%, 10-11 mol%, 11-12 mol%, 12-13 mol%, 13-14 mol%, or 14-15 mol% helper lipid; and (iv) 1-5% PEG lipid (e.g., Compound I or PEG-DMG), optionally 1-5 mol% PEG lipid, for example 1.5 to 2.5 mol%, 1-2 mol%, 2-3 mol%, 3-4 mol%, or 4-5 mol% PEG lipid.
A pharmaceutically acceptable excipient, as used herein, includes, but are not limited to, any and all solvents, dispersion media, or other liquid vehicles, dispersion or suspension aids, diluents, granulating and/or dispersing agents, surface active agents, isotonic agents, thickening or emulsifying agents, preservatives, binders, lubricants or oil, coloring, sweetening or flavoring agents, stabilizers, antioxidants, antimicrobial or antifungal agents, osmolality adjusting agents, pH adjusting agents, buffers, chelants, cyoprotectants, and/or bulking agents, as suited to the particular dosage form desired. Various excipients for Formulating pharmaceutical compositions and techniques for preparing the composition are known in the art (see Remington: The Science and Practice of Pharmacy, 21st Edition, A. R. Gennaro (Lippincott, Williams & Wilkins, Baltimore, MD, 2006; incorporated herein by reference in its entirety).
Exemplary diluents include, but are not limited to, calcium or sodium carbonate, calcium phosphate, calcium hydrogen phosphate, sodium phosphate, lactose, sucrose, cellulose, microcrystalline cellulose, kaolin, mannitol, sorbitol, etc., and/or combinations thereof.
Exemplary surface active agents and/or emulsifiers include, but are not limited to, natural emulsifiers (e.g., acacia, agar, alginic acid, sodium alginate, tragacanth, chondrux, cholesterol, xanthan, pectin, gelatin, egg yolk, casein, wool fat, cholesterol, wax, and lecithin), sorbitan fatty acid esters (e.g., polyoxyethylene sorbitan monooleate [TWEEN®80], sorbitan monopalmitate [SPAN®40], glyceryl monooleate, polyoxyethylene esters, polyethylene glycol fatty acid esters (e.g., CREMOPHOR®), polyoxyethylene ethers (e.g., polyoxyethylene lauryl ether
[BRU®30]), PLUORINC®F 68, POLOXAMER®188, etc. and/or combinations thereof.
Exemplary binding agents include, but are not limited to, starch, gelatin, sugars (e.g., sucrose, glucose, dextrose, dextrin, molasses, lactose, lactitol, mannitol), amino acids (e.g., glycine), natural and synthetic gums (e.g., acacia, sodium alginate), ethylcellulose, hydroxyethylcellulose, hydroxypropyl methylcellulose, etc., and combinations thereof.
Oxidation is a potential degradation pathway for mRNA, especially for liquid mRNA formulations. In order to prevent oxidation, antioxidants can be added to the formulations. Exemplary antioxidants include, but are not limited to, alpha tocopherol, ascorbic acid, ascorbyl palmitate, benzyl alcohol, butylated hydroxyanisole, m-cresol, methionine, butylated hydroxytoluene, monothioglycerol, sodium or potassium metabisulfite, propionic acid, propyl gallate, sodium ascorbate, etc., and combinations thereof.
Exemplary chelating agents include, but are not limited to, ethylenediaminetetraacetic acid (EDTA), citric acid monohydrate, disodium edetate, fumaric acid, malic acid, phosphoric acid, sodium edetate, tartaric acid, trisodium edetate, etc., and combinations thereof.
Exemplary antimicrobial or antifungal agents include, but are not limited to, benzalkonium chloride, benzethonium chloride, methyl paraben, ethyl paraben, propyl paraben, butyl paraben, benzoic acid, hydroxybenzoic acid, potassium or sodium benzoate, potassium or sodium sorbate, sodium propionate, sorbic acid, etc., and combinations thereof.
Exemplary preservatives include, but are not limited to, vitamin A, vitamin C, vitamin E, beta-carotene, citric acid, ascorbic acid, butylated hydroxyanisol, ethylenediamine, sodium lauryl sulfate (SLS), sodium lauryl ether sulfate (SLES), etc., and combinations thereof.
In some embodiments, the pH of polynucleotide solutions is maintained between pH 5 and pH 8 to improve stability. Exemplary buffers to control pH can include, but are not limited to sodium phosphate, sodium citrate, sodium succinate,
histidine (or histidine-HCl), sodium malate, sodium carbonate, etc., and/or combinations thereof.
Exemplary lubricating agents include, but are not limited to, magnesium stearate, calcium stearate, stearic acid, silica, talc, malt, hydrogenated vegetable oils, polyethylene glycol, sodium benzoate, sodium or magnesium lauryl sulfate, etc., and combinations thereof.
The pharmaceutical composition or formulation described here can contain a cryoprotectant to stabilize a polynucleotide described herein during freezing. Exemplary cryoprotectants include, but are not limited to mannitol, sucrose, trehalose, lactose, glycerol, dextrose, etc., and combinations thereof.
The pharmaceutical composition or formulation described here can contain a bulking agent in lyophilized polynucleotide formulations to yield a "pharmaceutically elegant" cake, stabilize the lyophilized polynucleotides during long term (e.g., 36 month) storage. Exemplary bulking agents of the present invention can include, but are not limited to sucrose, trehalose, mannitol, glycine, lactose, raffinose, and combinations thereof.
In some embodiments, the pharmaceutical composition or formulation further comprises a delivery agent. The delivery agent of the present disclosure can include, without limitation, liposomes, lipid nanoparticles, lipidoids, polymers, lipoplexes, microvesicles, exosomes, peptides, proteins, cells transfected with polynucleotides, hyaluronidase, nanoparticle mimics, nanotubes, conjugates, and combinations thereof.
Delivery Agents
Lipid Compound
The present disclosure provides pharmaceutical compositions with advantageous properties. The lipid compositions described herein may be advantageously used in lipid nanoparticle compositions for the delivery of therapeutic and/or prophylactic agents, e.g., mRNAs, to mammalian cells or organs. For example, the lipids described herein have little or no immunogenicity. For example, the lipid compounds disclosed herein have a lower immunogenicity as compared to a reference
lipid (e.g., MC3, KC2, or DLinDMA). For example, a formulation comprising a lipid disclosed herein and a therapeutic or prophylactic agent, e.g., mRNA, has an increased therapeutic index as compared to a corresponding formulation which comprises a reference lipid (e.g., MC3, KC2, or DLinDMA) and the same therapeutic or prophylactic agent.
In certain embodiments, the present application provides pharmaceutical compositions comprising:
(a) a polynucleotide comprising a nucleotide sequence encoding a CFTR polypeptide; and
(b) a delivery agent.
Lipid Nanoparticle Formulations
In some embodiments, nucleic acids of the invention (e.g., CFTR mRNA) are Formulated in a lipid nanoparticle (LNP). Lipid nanoparticles typically comprise ionizable cationic lipid, non-cationic lipid, sterol and PEG lipid components along with the nucleic acid cargo of interest. The lipid nanoparticles of the invention can be generated using components, compositions, and methods as are generally known in the art, see for example PCT/US2016/052352; PCT/US2016/068300; PCT/US2017/037551; PCT/US2015/027400; PCT/US2016/047406;
PCT/US2016000129; PCT/US2016/014280; PCT/US2016/014280;
PCT/US2017/038426; PCT/US2014/027077; PCT/US2014/055394;
PCT/US2016/52117; PCT/US2012/069610; PCT/US2017/027492; PCT/US2016/059575 and PCT/US2016/069491 all of which are incorporated by reference herein in their entirety.
Nucleic acids of the present disclosure (e.g., CFTR mRNA) are typically Formulated in lipid nanoparticle. In some embodiments, the lipid nanoparticle comprises at least one ionizable cationic lipid, at least one non-cationic lipid, at least one sterol, and/or at least one polyethylene glycol (PEG)-modified lipid.
In some embodiments, the lipid nanoparticle comprises a molar ratio of 20- 60% ionizable cationic lipid. For example, the lipid nanoparticle may comprise a molar ratio of 20-50%, 20-40%, 20-30%, 30-60%, 30-50%, 30-40%, 40-60%, 40-
50%, or 50-60% ionizable cationic lipid. In some embodiments, the lipid nanoparticle comprises a molar ratio of 20%, 30%, 40%, 50, or 60% ionizable cationic lipid.
In some embodiments, the lipid nanoparticle comprises a molar ratio of 5-25% non-cationic lipid. For example, the lipid nanoparticle may comprise a molar ratio of 5-20%, 5-15%, 5-10%, 10-25%, 10-20%, 10-25%, 15-25%, 15-20%, or 20-25% noncationic lipid. In some embodiments, the lipid nanoparticle comprises a molar ratio of 5%, 10%, 15%, 20%, or 25% non-cationic lipid.
In some embodiments, the lipid nanoparticle comprises a molar ratio of 25- 55% sterol. For example, the lipid nanoparticle may comprise a molar ratio of 25- 50%, 25-45%, 25-40%, 25-35%, 25-30%, 30-55%, 30-50%, 30-45%, 30-40%, 30- 35%, 35-55%, 35-50%, 35-45%, 35-40%, 40-55%, 40-50%, 40-45%, 45-55%, 45- 50%, or 50-55% sterol. In some embodiments, the lipid nanoparticle comprises a molar ratio of 25%, 30%, 35%, 40%, 45%, 50%, or 55% sterol.
In some embodiments, the lipid nanoparticle comprises a molar ratio of 0.5- 15% PEG-modified lipid. For example, the lipid nanoparticle may comprise a molar ratio of 0.5-10%, 0.5-5%, 1-15%, 1-10%, 1-5%, 2-15%, 2-10%, 2-5%, 5-15%, 5-10%, or 10-15%. In some embodiments, the lipid nanoparticle comprises a molar ratio of 0.5%, 1%, 2%, 3%, 4%, 5%, 6%, 7%, 8%, 9%, 10%, 11%, 12%, 13%, 14%, or 15% PEG-modified lipid.
In some embodiments, the lipid nanoparticle core comprises a molar ratio of 20-60% ionizable cationic lipid, 5-25% non-cationic lipid, 25-55% sterol, and 0.5- 15% PEG-modified lipid. In some embodiments, the lipid nanoparticle core comprises a molar ratio of 40-60% ionizable cationic lipid, 5-15% non-cationic lipid, 30-50% sterol, and 0.5-10% PEG-modified lipid. In some embodiments, the lipid nanoparticle core comprises a molar ratio of 45-55% ionizable cationic lipid, 7.5- 12.5% non-cationic lipid, 35-45% sterol, and 0.5-5% PEG-modified lipid.
Cationic Agent
In some embodiments, the LNP provided herein comprises lipid nanoparticle core, a polynucleotide (e.g., CFTR mRNA) encapsulated within the core for delivery into a cell, and a cationic agent disposed primarily on the outer surface of the core.
Without being bound by a particular theory, LNP with cationic agent disposed primarily on the outer surface of the core can have improved accumulation of the LNP in cells such as human bronchial epithelial (HBE) and also improved function of the polynucleotide, e.g., as measured mRNA expression in cells, e.g., airway epithelial cells.
The cationic agent can comprise any aqueous soluble molecule or substance that has a net positive charge at physiologic pH and can adhere to the surface of a lipid nanoparticle core. Such agent may also be lipid soluble, but will also be soluble in aqueous solution. Generally speaking, the cationic agent features a net positive charge at physiologic pH because it contains one or more basic functional groups that is protonated at physiologic pH in aqueous media. For example the cationic agent can contain one or more amine groups, e.g. primary, secondary, or tertiary amines each having a pKa of 8.0 or greater. The pKa can be greater than about 9.
In some embodiments, the cationic agent can be a cationic lipid which is a water-soluble, amphiphilic molecule in which one portion of the molecule is hydrophobic comprising, for example, a lipid moiety, and where the other portion of the molecule is hydrophilic, containing one or more functional groups which is typically charged at physiologic pH. The hydrophobic portion, comprising the lipid moiety, can serve to anchor the cationic agent to a lipid nanoparticle core. The hydrophilic portion can serve to increase the charge on the surface of a lipid nanoparticle core. For example, the cationic agent can have a solubility of greater than about 1 mg/mL in alcohol. The solubility in alcohol can be greater than about 5 mg/mL. The solubility in alcohol can be greater than about 10 mg/mL. The solubility in alcohol can be greater than about 20 mg/mL in alcohol. The alcohol can be C1-6 alcohol such as ethanol.
The lipid portion of the molecule can be, for example, a structural lipid, fatty acid, or similar hydrocarbyl group.
The structural lipid can be selected from, but is not limited to, a steroid, diterpeniod, triterpenoid, cholestane, ursolic acid, or derivatives thereof.
In some embodiments, the structural lipid is a steroid selected from, but not limited to, cholesterol or a phystosterol. In some embodiments, the structural lipid is
an analog of cholesterol. In some embodiments, the structural lipid is a sitosterol, campesterol, or stigmasterol. In some embodiments, the structural lipid is an analog of sitosterol, campesterol, or stigmasterol.
The fatty acid comprises 1 to 4 C6-20 hydrocarbon chains. The fatty acid can be fully saturated or can contain 1 to 7 double bonds. The fatty acid can contain 1 to 5 heteroatoms either along the main chain or pendent to the main chain.
In some embodiments, the fatty acid comprises two Cio-18 hydrocarbon chains. In some embodiments, the fatty acid comprises two Cio-18 saturated hydrocarbon chains. In some embodiments, the fatty acid comprises two Ci6 saturated hydrocarbon chain. In some embodiments, the fatty acid comprises two C14 saturated hydrocarbon chain. In some embodiments, the fatty acid comprises two unsaturated Cio-18 hydrocarbon chains. In some embodiments, the fatty acid comprises two C16-18 hydrocarbon chains, each with one double bond. In some embodiments, the fatty acid comprises three C8-18 saturated hydrocarbon chains.
The hydrocarbyl group consists of 1 to 4 C6-20 alkyl, alkenyl, or alkynyl chains or 3 to 10 membered cycloalkyl, cycloalkenyl, or cycloalkynyl groups.
In some embodiments, the hydrocarbyl chain is a C8-io alkyl. In some embodiments, the hydrocarbyl chain is C8-io alkenyl.
The hydrophilic portion can comprise 1 to 5 functional groups that would be charged at physiologic pH, 7.3 to 7.4. The hydrophilic group can comprise a basic functional group that would be protonated and positively charged at physiologic pH. At least one of the basic functional groups has a pKa of 8 or greater.
In some embodiments, the hydrophilic portion comprises an amine group. The amine group can comprise one to four primary, secondary, or tertiary amines and mixtures thereof. The primary, secondary, or tertiary amines can be part of larger amine containing functional group selected from, but not limited to, -C(=N-)-N-, - C=C-N-, -C=N-, or -N-C(=N-)-N-. The amine can be contained in a three to eight membered heteroalkyl or heteroaryl ring.
In some embodiments, the amine group comprises one or two terminal primary amines. In some embodiments, the amine group comprises one or two terminal primary amines and one internal secondary amine. In some embodiments, the
amine group comprises one or two tertiary amine. In some embodiments, the tertiary amine is (CH3)2N-. In some embodiments, amine group comprises one to two terminal (CH3)2N-.
The hydrophilic portion can comprise a phosphonium group. The counter ion of the phosphonium ion consists of an anion with a charge of one.
In some embodiments, three of the substituents on the phoshonium are isopropyl groups. In some embodiments, the counter ion is a halo, hydrogen sulfate, nitrite, chlorate, or hydrogen carbonate. In some embodiments, the counter ion is a bromide.
In some embodiments, the cationic agent is a cationic lipid which is a sterol amine. A sterol amine has, for its hydrophobic portion, a sterol, and for its hydrophilic portion, an amine group. The sterol group is selected from, but not limited to, cholesterol, sitosterol, campesterol, stigmasterol or derivatives thereof. The amine group can comprise one to five primary, secondary, tertiary amines, or mixtures thereof. At least one of the amines has a pKa of 8 or greater and is charged at physiological pH. The primary, secondary, or tertiary amines can be part of a larger amine containing functional group selected from, but not limited to -C(=N-)-N-, - C=C-N-, -C=N-, or -N-C(=N-)-N-. The amine can be contained in a three to eight membered heteroalkyl or heteroaryl ring.
In some embodiments, the amine group of the sterol amine comprises one or two terminal primary amines. In some embodiments, the amine group comprises one or two terminal primary amines and one internal secondary amine. In some embodiments, the amine group comprises one or two tertiary amine. In some embodiments, the tertiary amine is (C’H3)2N-. In some embodiments, amine group comprises one to two terminal (CH3)2N-.
Sterol amines useful in the nanoparticles of the invention include molecules having Formula (Al):
A-L-B (Al) or a salt thereof, wherein:
A is an amine group, L is an optional linker, and B is a sterol.
In some embodiments, the amine group is an alkyl (e.g., C1-14 alkyl, C1-12 alkyl, C1-10 alkyl, etc.), 3 to 8 membered heterocycloalkyl, 5 to 6 membered heteroaryl, C1-6 alkyl-(3 to 8 membered heterocycloalkyl), or C1-6 alkyl-(5 to 6 membered heteroaryl), wherein the alkyl, 3 to 8 membered heterocycloalkyl, 5 to 6 membered heteroaryl, C1-6 alkyl-(3 to 8 membered heterocycloalkyl), and C1-6 alkyl- (5 to 6 membered heteroaryl) comprises one to five primary, secondary, or tertiary amines or combination thereof, wherein the alkyl, 3 to 8 membered heterocycloalkyl, 5 to 6 membered heteroaryl, C1-6 alkyl-(3 to 8 membered heterocycloalkyl), and C1-6 alkyl-(5 to 6 membered heteroaryl) are each optionally substituted with 1, 2, 3, or 4 substituents selected from C1-6 alkyl, halo, OH, O(C1-6 alkyl), C1-6 alkyl-OH, NH2, NH(C1-6 alkyl), N(C1-6 alkyl)2, 3 to 8 membered heterocycloalkyl (optionally substituted with C1-14 alkyl comprising one to five primary, secondary, or tertiary amines or combination thereof), 5 to 6 membered heteroaryl, NH(3 to 8 membered heterocycloalkyl), and NH(5 to 6 membered heteroaryl). In some embodiments, the linker is absent, -O-, -S-S-, -OC(=O), -C(=O)N-, -OC(=O)N-, CH2-NH-C(O)-, - C(O)O-, -OC(O)-CH2-CH2-C(=O)N-, -S-S-CH2, or -SS-CH2-CH2-C(O)N-. In some embodiments, the sterol group is a cholesterol, sitosterol, campesterol, stigmasterol or derivatives thereof.
In some embodiments, the sterol amine has Formula A2:
 or a salt thereof, wherein:
or a salt thereof, wherein:
— is a single or double bond
R1 is C1-14 alkyl or C1-14 alkenyl;
L is absent, -O-, -S-S-, -OC(=O), -C(=O)N-, -OC(=O)N-, CH2-NH- C(O)-, -C(O)O-, -OC(O)-CH2-CH2-C(=O)N-, -S-S-CH2, or -SS-CH2-CH2- C(O)N-;
YHS CI -10 alkyl, 3 to 8 membered heterocycloalkyl, 5 to 6 membered heteroaryl, C1-6 alkyl-(3 to 8 membered heterocycloalkyl), or C1-6 alkyl-(5 to 6 membered heteroaryl), wherein the alkyl, 3 to 8 membered heterocycloalkyl, 5 to 6 membered heteroaryl, C1-6 alkyl-(3 to 8 membered heterocycloalkyl), and C1-6 alkyl-(5 to 6 membered heteroaryl) comprises one to five primary, secondary, or tertiary amines or combination thereof, wherein the alkyl, 3 to 8 membered heterocycloalkyl, 5 to 6 membered heteroaryl, C1-6 alkyl-(3 to 8 membered heterocycloalkyl), and C1-6 alkyl-(5 to 6 membered heteroaryl) are each optionally substituted with 1, 2, 3, or 4 substituents selected from C1-6 alkyl, halo, OH, O(C1-6 alkyl), C1-6 alkyl-OH, NH2, NH(C1-6 alkyl), N(C1-6 alkyl)2, 3 to 8 membered heterocycloalkyl (optionally substituted with C1-14 alkyl comprising one to five primary, secondary, or tertiary amines or combination thereof), 5 to 6 membered heteroaryl, NH(3 to 8 membered heterocycloalkyl), and NH(5 to 6 membered heteroaryl); and n = 1 or 2.
In some embodiments, the sterol amine has Formula A3:
 or a salt thereof, wherein:
or a salt thereof, wherein:
— is a single or double bond;
R2 is H or C1-6 alkyl;
L is absent, -O-, -S-S-, -OC(=O), -C(=O)N-, -OC(=O)N-, CH2-NH- C(O)-, -C(O)O-, -OC(O)-CH2-CH2-C(=O)N-, -S-S-CH2, or -SS-CH2-CH2- C(O)N-;
YHS CI -10 alkyl, 3 to 8 membered heterocycloalkyl, 5 to 6 membered heteroaryl, C1-6 alkyl-(3 to 8 membered heterocycloalkyl), or C1-6 alkyl-(5 to 6
membered heteroaryl), wherein the alkyl, 3 to 8 membered heterocycloalkyl, 5 to 6 membered heteroaryl, C1-6 alkyl-(3 to 8 membered heterocycloalkyl), and C1-6 alkyl-(5 to 6 membered heteroaryl) comprises one to five primary, secondary, or tertiary amines or combination thereof, wherein the alkyl, 3 to 8 membered heterocycloalkyl, 5 to 6 membered heteroaryl, C1-6 alkyl-(3 to 8 membered heterocycloalkyl), and C1-6 alkyl-(5 to 6 membered heteroaryl) are each optionally substituted with 1, 2, 3, or 4 substituents selected from C1-6 alkyl, halo, OH, O(C1-6 alkyl), C1-6 alkyl-OH, NH2, NH(C1-6 alkyl), N(C1-6 alkyl)2, 3 to 8 membered heterocycloalkyl (optionally substituted with C1-14 alkyl comprising one to five primary, secondary, or tertiary amines or combination thereof), 5 to 6 membered heteroaryl, NH(3 to 8 membered heterocycloalkyl), and NH(5 to 6 membered heteroaryl); and n = 1 or 2.
In some embodiments Y2 is selected from:


N(CH3)2.
In some embodiments, the sterol amine has Formula A4:
 or a salt thereof, wherein:
or a salt thereof, wherein:
Z1 is OH or C3-6 alkyl;
L is absent, -0-, -S-S-, -0C(=0), -C(=O)N-, -OC(=O)N-, CH2-NH- C(0)-, -C(0)0-, -OC(O)-CH2-CH2-C(=O)N-, -S-S-CH2, or -SS-CH2-CH2- C(O)N-;
Y1 is C1-10 alkyl, 3 to 8 membered heterocycloalkyl, 5 to 6 membered heteroaryl, C1-6 alkyl-(3 to 8 membered heterocycloalkyl), or C1-6 alkyl-(5 to 6 membered heteroaryl), wherein the alkyl, 3 to 8 membered heterocycloalkyl, 5 to 6 membered heteroaryl, C1-6 alkyl-(3 to 8 membered heterocycloalkyl), and C1-6 alkyl-(5 to 6 membered heteroaryl) comprises one to five primary, secondary, or tertiary amines or combination thereof, wherein the alkyl, 3 to 8 membered heterocycloalkyl, 5 to 6 membered heteroaryl, C1-6 alkyl-(3 to 8 membered heterocycloalkyl), and C1-6 alkyl-(5 to 6 membered heteroaryl) are each optionally substituted with 1, 2, 3, or 4 substituents selected from C1-6 alkyl, halo, OH, O(C1-6 alkyl), C1-6 alkyl-OH, NH2, NH(C1-6 alkyl), N(C1-6 alkyl)2, 3 to 8 membered heterocycloalkyl (optionally substituted with C1-i4 alkyl comprising one to five primary, secondary, or tertiary amines or combination thereof), 5 to 6 membered heteroaryl, NH(3 to 8 membered heterocycloalkyl), and NH(5 to 6 membered heteroaryl); and n = 1 or 2.
In some embodiments, the sterol amine has Formula A5:
 or a salt thereof, wherein:
or a salt thereof, wherein:
Z2 is OH or isopropyl;
L1 is -CH2-NH-C(O)-, -C(O)NH-, or -C(O)O-.
In some embodiments, the sterol amine is selected from:
Table SAI
Sterolamine Structure no.



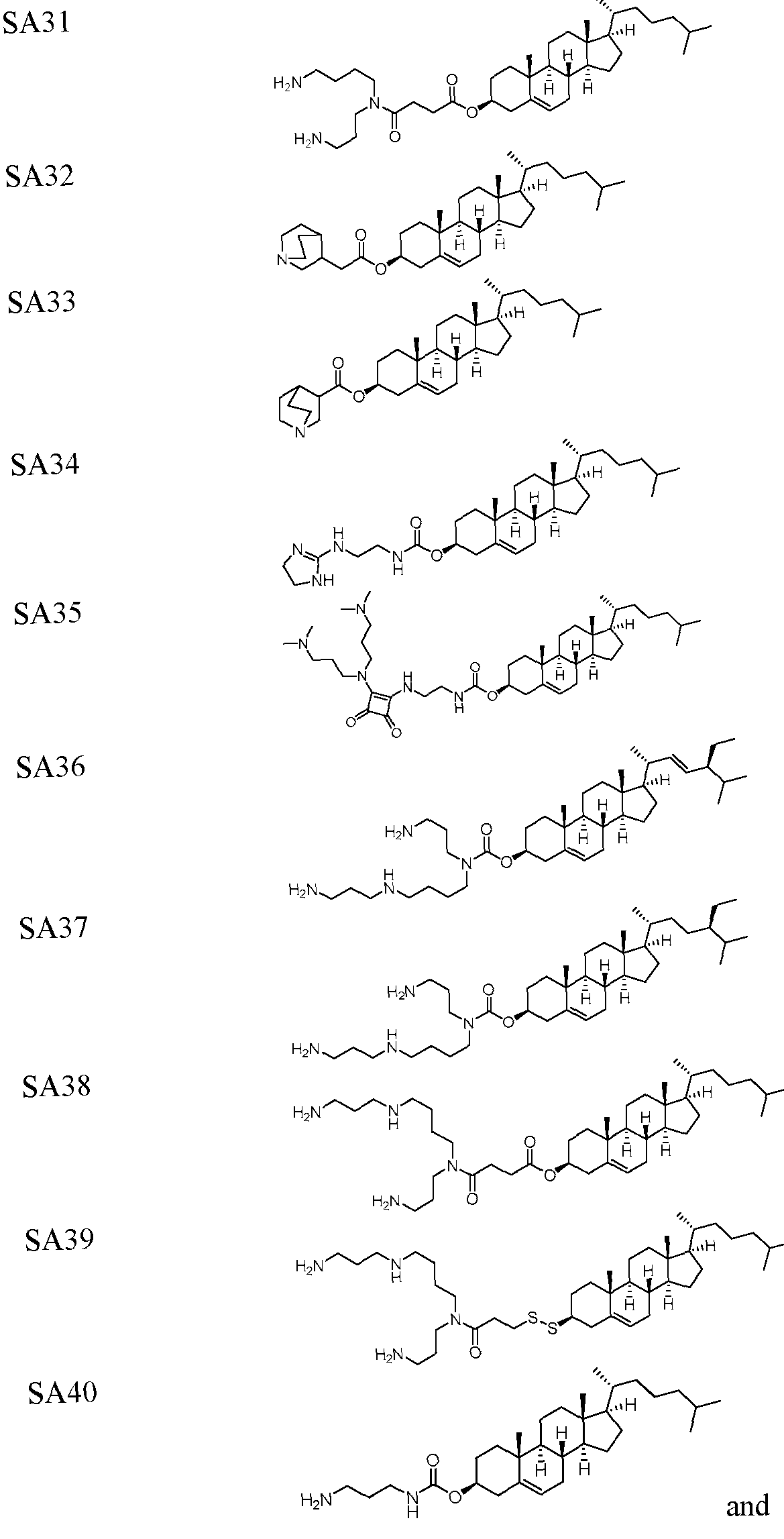 SA41
SA41
 or a salt thereof.
or a salt thereof.
In some embodiments, the sterol amine is SA3 :
 salt thereof, which is also referred to as GL-67. SA3 or GL-67 can be prepared according to known processes in the art or purchased from a commercial vendor such as Avanti® Polar Lipids, Inc. (SKU 890893).
salt thereof, which is also referred to as GL-67. SA3 or GL-67 can be prepared according to known processes in the art or purchased from a commercial vendor such as Avanti® Polar Lipids, Inc. (SKU 890893).
In some embodiments, the cationic lipid is a modified amino acid, such as a modified arginine, in which an amino acid residue having an amine-containing side chain is appended to a hydrophobic group such as a sterol (e.g., cholesterol or derivative thereof), fatty acid, or similar hydrocarbyl group. At least one amine of the modified amino acid portion has a pKa of 8.0 or greater. At least one amine of the modified amino acid portion is positively charger at physiological pH. The amino acid residue can include but is not limited to arginine, histidine, lysine, tryptophan, ornithine, and 5 -hydroxy lysine. The amino acid is bonded to the hydrophobic group through a linker.
In some embodiments, the modified amino acid is a modified arginine.
In some embodiments, the cationic agent is a non-lipid cationic agent. Examples of non-lipid cationic agent include e.g., benzalkonium chloride, cetylpyridium chloride, L-lysine monohydrate, or tromethamine.
In some embodiments, the lipid nanoparticle comprises a cationic agent (e.g., a sterol amine) at a molar ratio of 2-15%, 3-10%, 4-10%, 5-10%, 6-10%, 2-3%, 2-4%, 2-5%, 2-6%, 2-7%, 2-8%, 3-4%, 3-5%, 3-6%, 3-7%, 3-8%, 4-5%, 4-6%, 4-7%, 4-8%, 5-6%, 5-7%, 5-8%, 6-7%, 6-8%, 2%, 2.5%, 3%, 3.5%, 4%, 4.5%, 5%, 5.5%, 6%, 6.5%, 7%, 7.5%, 8%, 8.5%, 9%, 9.5%, 10%, at least 2%, at least 3%, at least 4%, at least 5%, at least 6%, at least 7%, at least 8%, less than 15%, less than 14%, less than 13%, less than 12%, less than 11%, or less than 10%. In some embodiments, the lipid
nanoparticle comprises a molar ratio of 20-60% ionizable cationic lipid, 5-25% noncationic lipid, 25-55% sterol, 0.5-15% PEG-modified lipid, and 2-10% cationic agent (e.g., a sterol amine). In some embodiments, the lipid nanoparticle comprises a molar ratio of 40-60% ionizable cationic lipid, 5-15% non-cationic lipid, 30-50% sterol, 0.5- 10% PEG-modified lipid, and 3-7% cationic agent. In some embodiments, the lipid nanoparticle comprises a molar ratio of 45-55% ionizable cationic lipid, 7.5-12.5% non-cationic lipid, 35-45% sterol, 0.5-5% PEG-modified lipid, and 4.5-6% cationic agent. In some instances, the cationic agent is GL-67 or a salt thereof.
In some embodiments, a weight ratio of the cationic agent to polynucleotide is about 0.1 : 1 to about 15: 1. In some embodiments, a weight ratio of the cationic agent to polynucleotide is about 0.2: 1 to about 10: 1. In some embodiments, a weight ratio of the cationic agent to polynucleotide is about 1 : 1 to about 10: 1. In some embodiments, a weight ratio of the cationic agent to polynucleotide is about 1 : 1 to about 8: 1. In some embodiments, a weight ratio of the cationic agent to polynucleotide is about 1 : 1 to about 7: 1. In some embodiments, a weight ratio of the cationic agent to polynucleotide is about 1 : 1 to about 6: 1. In some embodiments, a weight ratio of the cationic agent to polynucleotide is about 1 : 1 to about 5: 1. In some embodiments, a weight ratio of the cationic agent to polynucleotide is about 1 : 1 to about 4: 1. In some embodiments, a weight ratio of the cationic agent to polynucleotide is about 1.25: 1 to about 3.75 : 1. In some embodiments, a weight ratio of the cationic agent to polynucleotide is about 1.25 : 1. In some embodiments, a weight ratio of the cationic agent to polynucleotide is about 2.5 : 1. In some embodiments, a weight ratio of the cationic agent to polynucleotide is about 3.75: 1.
In some embodiments, a molar ratio of the cationic agent to polynucleotide is about 0.1 : 1 to about 20: 1. In some embodiments, a molar ratio of the cationic agent to polynucleotide is about 1.5: 1 to about 10: 1. In some embodiments, a molar ratio of the cationic agent to polynucleotide is about 1.5 : 1 to about 9: 1. In some embodiments, a molar ratio of the cationic agent to polynucleotide is about 1.5: 1 to about 8: 1. In some embodiments, a molar ratio of the cationic agent to polynucleotide is about 1.5 : 1 to about 7: 1. In some embodiments, a molar ratio of the cationic agent to polynucleotide is about 1.5 : 1 to about 6: 1. In some embodiments, a molar ratio of
the cationic agent to polynucleotide is about 1.5: 1 to about 5: 1. In some embodiments, a molar ratio of the cationic agent to polynucleotide is about 1.5 : 1. In some embodiments, a molar ratio of the cationic agent to polynucleotide is about 2: 1. In some embodiments, a molar ratio of the cationic agent to polynucleotide is about 3: 1. In some embodiments, a molar ratio of the cationic agent to polynucleotide is about 4: 1. In some embodiments, a molar ratio of the cationic agent to polynucleotide is about 5: 1.
In some embodiments, the nanoparticle has a zeta potential of about 5 mV to about 20 mV. In some embodiments, the nanoparticle has a zeta potential of about 5 mV to about 20 mV. In some embodiments, the nanoparticle has a zeta potential of about 5 mV to about 15 mV. In some embodiments, the nanoparticle has a zeta potential of about 5 mV to about 10 mV.
In some embodiments, the lipid nanoparticle core has a neutral charge at a neutral pH.
In some embodiments, greater than about 80% of the cationic agent is on the surface on the nanoparticle. In some embodiments, greater than about 90% of the cationic agent is on the surface on the nanoparticle. In some embodiments, greater than about 95% of the cationic agent is on the surface on the nanoparticle.
Ionizable Lipids
In some aspects, the ionizable lipids of the present disclosure may be one or more of compounds of Formula (I):
 or their N-oxides, or salts or isomers thereof, wherein:
or their N-oxides, or salts or isomers thereof, wherein:
Ri is selected from the group consisting of C5-30 alkyl, C5-20 alkenyl, -R*YR”, -YR”, and -R ’M’R’;
R.2 and R3 are independently selected from the group consisting of H, C1-14 alkyl, C2-14 alkenyl, -R*YR”, -YR”, and -R*OR”, or R2 and R3, together with the atom to which they are attached, form a heterocycle or carbocycle;
R4 is selected from the group consisting of hydrogen, a C3-6 carbocycle, -(CH2)nQ, -(CH2)nCHQR
-CHQR, -CQ(R)2, and unsubstituted C1-6 alkyl, where Q is selected from a carbocycle, heterocycle, -OR, -O(CH2)nN(R)2, -C(O)OR, - OC(O)R -CX3, -CX2H, -CXH2, -CN,
-N(R)2, -C(O)N(R)2, -N(R)C(O)R, -N(R)S(O)2R, -N(R)C(O)N(R)2, -N(R)C(S) N(R)2, -N(R)R8,
-N(R)S(O)2R8, -O(CH2)nOR, -N(R)C(=NR9)N(R)2, -N(R)C(=CHR9)N(R)2, -O C(0)N(R)2,
-N(R)C(O)OR, -N(OR)C(O)R, -N(OR)S(O)2R, -N(OR)C(O)OR, -N(OR)C(O) N(R)2,
-N(OR)C(S)N(R)2, -N(OR)C(=NR9)N(R)2, -N(OR)C(=CHR9)N(R)2, -C(=NR9 )N(R)2,
-C(=NR9)R -C(O)N(R)OR, and -C(R)N(R)2C(O)OR and each n is independently selected from 1, 2, 3, 4, and 5; each Rs is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; each R$ is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H;
M and M’ are independently selected from -C(O)O-, -OC(O)-, -OC(O)-M”-C(O)O-, -C(O)N(R’)-,
-N(R’)C(O)-, -C(O)-, -C(S)-, -C(S)S-, -SC(S)-, -CH(OH)-, -P(O)(OR’) O-, -S(O)2-, -S-S-, an aryl group, and a heteroaryl group, in which M” is a bond, C1-13 alkyl or C2-13 alkenyl;
R7 is selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H;
R8 is selected from the group consisting of C3-6 carbocycle and heterocycle;
R9 is selected from the group consisting of H, CN, NO2, C1-6 alkyl, -
OR, -S(O)2R, -S(O)2N(R)2, C2-6 alkenyl, C3-6 carbocycle and heterocycle; each R is independently selected from the group consisting of C1-3 alkyl, C2-3 alkenyl, and H; each R’ is independently selected from the group consisting of C1-18 alkyl, C2-18 alkenyl, -R*YR”, -YR”, and H; each R” is independently selected from the group consisting of C3-15 alkyl and C3-15 alkenyl; each R* is independently selected from the group consisting of C1-12 alkyl and
C2-12 alkenyl; each Y is independently a C3-6 carbocycle; each X is independently selected from the group consisting of F, Cl, Br, and I; and m is selected from 5, 6, 7, 8, 9, 10, 11, 12, and 13; and wherein when R4 is -(CH2)nQ, -(CH2)nCHQR, -CHQR, or -CQ(R)2, then (i) Q is not -N(R)2 when n is 1, 2, 3, 4 or 5, or (ii) Q is not 5, 6, or 7-membered heterocycloalkyl when n is 1 or 2.
In certain embodiments, a subset of compounds of Formula (I) includes those of Formula (IA):
 or its N-oxide, or a salt or isomer thereof, wherein 1 is selected from 1, 2, 3, 4, and 5; m is selected from 5, 6, 7, 8, and 9; Mi is a bond or M’; R4 is hydrogen, unsubstituted C1-3 alkyl, or -(CH2)nQ, in which Q is OH, -NHC(S)N(R)2, -NHC(O)N(R)2, -N(R)C(O)R, -N(R)S(O)2R, -N(R)RS, -NHC(=NR9)N(R)2, -NHC(=CHR9)N(R)2, -OC(O)N(R)2, -N(R)C(O)OR,
heteroaryl or heterocycloalkyl; M and M’ are independently selected from -C(O)O-, -OC(O)-, -OC(O)-M”-C(O)O-, -C(O)N(R’)-, -P(O)(OR’)O-, - S-S-, an aryl group, and a heteroaryl group,; and R2 and R3 are independently selected from the group consisting of H, C1-14 alkyl, and C2-14 alkenyl. For example, m is 5, 7, or 9. For example, Q is OH, -NHC(S)N(R)2, or -NHC(0)N(R)2. For example, Q is -N(R)C(O)R, or -N(R)S(0)2R.
or its N-oxide, or a salt or isomer thereof, wherein 1 is selected from 1, 2, 3, 4, and 5; m is selected from 5, 6, 7, 8, and 9; Mi is a bond or M’; R4 is hydrogen, unsubstituted C1-3 alkyl, or -(CH2)nQ, in which Q is OH, -NHC(S)N(R)2, -NHC(O)N(R)2, -N(R)C(O)R, -N(R)S(O)2R, -N(R)RS, -NHC(=NR9)N(R)2, -NHC(=CHR9)N(R)2, -OC(O)N(R)2, -N(R)C(O)OR,
heteroaryl or heterocycloalkyl; M and M’ are independently selected from -C(O)O-, -OC(O)-, -OC(O)-M”-C(O)O-, -C(O)N(R’)-, -P(O)(OR’)O-, - S-S-, an aryl group, and a heteroaryl group,; and R2 and R3 are independently selected from the group consisting of H, C1-14 alkyl, and C2-14 alkenyl. For example, m is 5, 7, or 9. For example, Q is OH, -NHC(S)N(R)2, or -NHC(0)N(R)2. For example, Q is -N(R)C(O)R, or -N(R)S(0)2R.
In certain embodiments, a subset of compounds of Formula (I) includes those of Formula (
 (IB), or its N-oxide, or a salt or isomer thereof in which all variables are as defined herein. For example, m is selected from 5, 6, 7, 8, and 9; R4 is hydrogen, unsubstituted C1-3 alkyl, or -(CH2)nQ, in which Q is
(IB), or its N-oxide, or a salt or isomer thereof in which all variables are as defined herein. For example, m is selected from 5, 6, 7, 8, and 9; R4 is hydrogen, unsubstituted C1-3 alkyl, or -(CH2)nQ, in which Q is
OH, -NHC(S)N(R)2, -NHC(O)N(R)2, -N(R)C(O)R, -N(R)S(O)2R, -N(R)RS, -NHC(=NR9)N(R)2, -NHC(=CHR9)N(R)2, -0C(0)N(R)2, -N(R)C(O)OR, heteroaryl or heterocycloalkyl; M and M’ are independently selected from -C(O)O-, -OC(O)-, -OC(O)-M”-C(O)O-, -C(O)N(R’)-, -P(O)(OR’)O-, -S -S-, an aryl group, and a heteroaryl group; and R2 and R3 are independently selected from the group consisting of H, C1-14 alkyl, and C2-14 alkenyl. For example, m is 5, 7, or 9. For example, Q is OH, -NHC(S)N(R)2, or -NHC(0)N(R)2. For example, Q is -N(R)C(O)R, or -N(R)S(0)2R.
In certain embodiments, a subset of compounds of Formula (I) includes those of Formula (II):
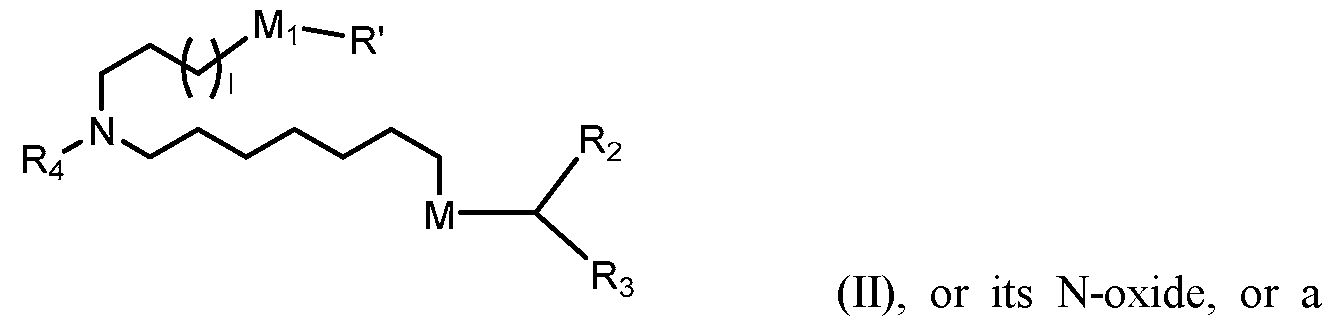 salt or isomer thereof, wherein 1 is selected from 1, 2, 3, 4, and 5; Mi is a bond
or M’; R4 is hydrogen, unsubstituted C1-3 alkyl, or -(CH2)nQ, in which n is 2, 3, or 4, and Q is
salt or isomer thereof, wherein 1 is selected from 1, 2, 3, 4, and 5; Mi is a bond
or M’; R4 is hydrogen, unsubstituted C1-3 alkyl, or -(CH2)nQ, in which n is 2, 3, or 4, and Q is
OH, -NHC(S)N(R)2, -NHC(O)N(R)2, -N(R)C(O)R, -N(R)S(O)2R, -N(R)RS, -NHC(=NR9)N(R)2, -NHC(=CHR9)N(R)2, -OC(O)N(R)2, -N(R)C(O)OR, heteroaryl or heterocycloalkyl; M and M’ are independently selected from -C(O)O-, -OC(O)-, -OC(O)-M”-C(O)O-, -C(O)N(R’)-, -P(O)(OR’)O-, -S -S-, an aryl group, and a heteroaryl group; and R2 and R3 are independently selected from the group consisting of H, C1-14 alkyl, and C2-14 alkenyl. In one embodiment, the compounds of Formula (I) are of Formula (Ila),
 or their N-oxides, or salts or isomers thereof, wherein R4 is as described herein.
or their N-oxides, or salts or isomers thereof, wherein R4 is as described herein.
In another embodiment, the compounds of Formula (I) are of Formula (lib),
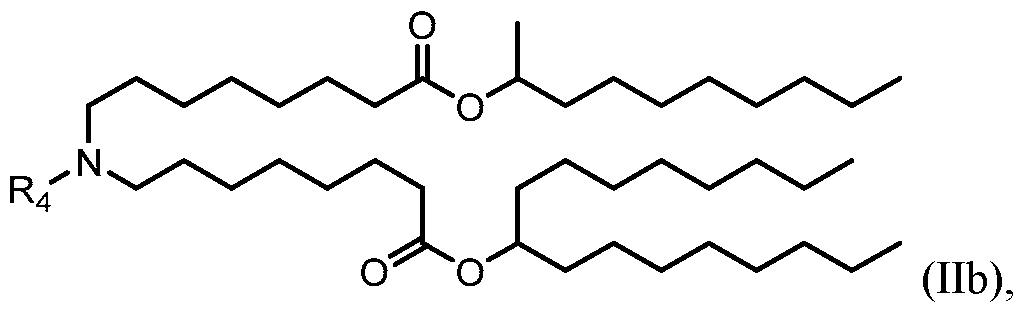 or their N-oxides, or salts or isomers thereof, wherein R4 is as described herein.
or their N-oxides, or salts or isomers thereof, wherein R4 is as described herein.
In another embodiment, the compounds of Formula (I) are of Formula (lie) or (lie):
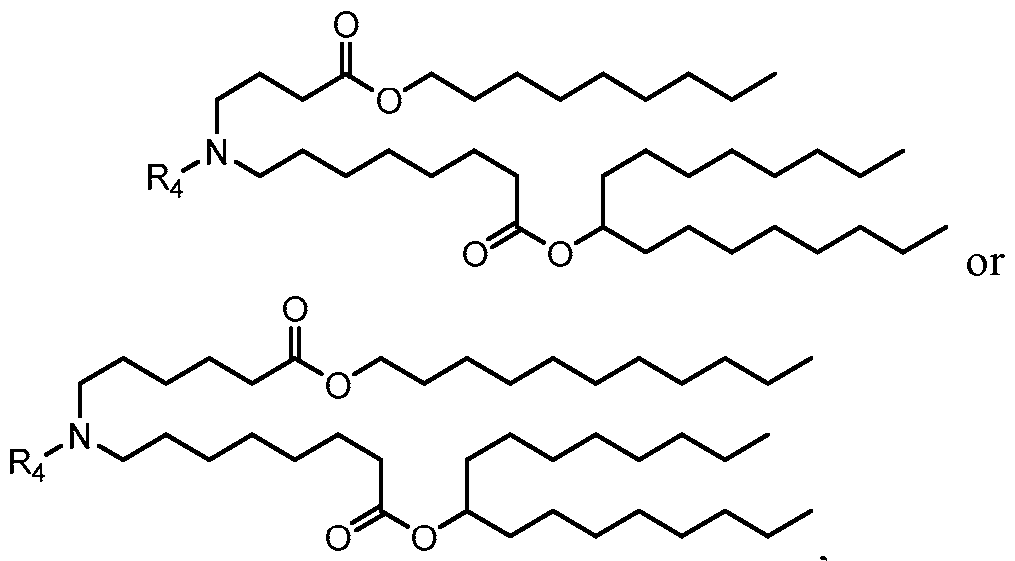
(lie) (lie) or their N-oxides, or salts or isomers thereof, wherein R4 is as described herein.
In another embodiment, the compounds of Formula (I) are of Formula (Ilf):
 (Ilf) or their N-oxides, or salts or isomers thereof, wherein M is -C(O)O- or -OC(O)-, M” is C1-6 alkyl or C2-6 alkenyl, R2 and R3 are independently selected from the group consisting of C5-14 alkyl and
(Ilf) or their N-oxides, or salts or isomers thereof, wherein M is -C(O)O- or -OC(O)-, M” is C1-6 alkyl or C2-6 alkenyl, R2 and R3 are independently selected from the group consisting of C5-14 alkyl and
C5-14 alkenyl, and n is selected from 2, 3, and 4.
In a further embodiment, the compounds of Formula (I) are of Formula (lid),
 (lid), or their N-oxides, or salts or isomers thereof, wherein n is 2, 3, or 4; and m, R’, R”, and R2 through R$ are as described herein. For example, each of R2 and R3 may be independently selected from the group consisting of C5-14 alkyl and C5-14 alkenyl.
In a further embodiment, the compounds of Formula (I) are of Formula (Ilg),
(lid), or their N-oxides, or salts or isomers thereof, wherein n is 2, 3, or 4; and m, R’, R”, and R2 through R$ are as described herein. For example, each of R2 and R3 may be independently selected from the group consisting of C5-14 alkyl and C5-14 alkenyl.
In a further embodiment, the compounds of Formula (I) are of Formula (Ilg),
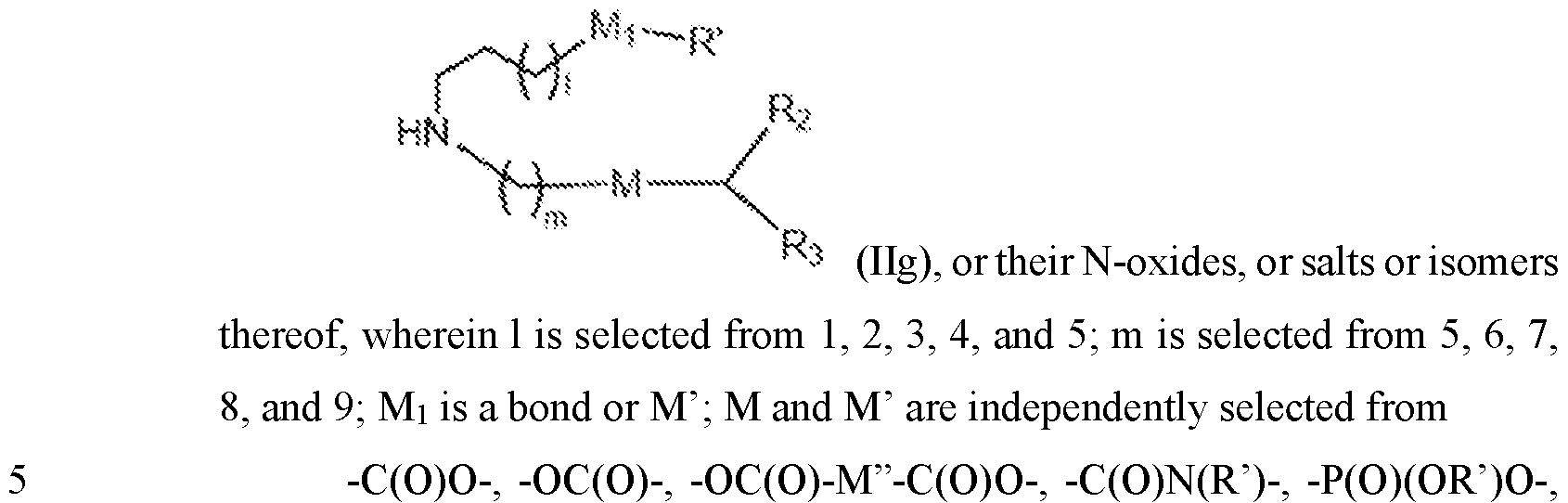 , , , , , -S-S-, an aryl group, and a heteroaryl group; and R2 and R3 are independently selected from the group consisting of H, C1-14 alkyl, and C2-14 alkenyl. For example, M” is C1-6 alkyl (e.g., C1-4 alkyl) or C2-6 alkenyl (e.g. C2-4 alkenyl). For example, R2 and R3 are independently selected from the group consisting of C5-14 alkyl and C5-14 alkenyl.
, , , , , -S-S-, an aryl group, and a heteroaryl group; and R2 and R3 are independently selected from the group consisting of H, C1-14 alkyl, and C2-14 alkenyl. For example, M” is C1-6 alkyl (e.g., C1-4 alkyl) or C2-6 alkenyl (e.g. C2-4 alkenyl). For example, R2 and R3 are independently selected from the group consisting of C5-14 alkyl and C5-14 alkenyl.
In some embodiments, the ionizable lipids are one or more of the compounds described in U.S. Application Nos. 62/220,091, 62/252,316, 62/253,433, 62/266,460, 62/333,557, 62/382,740, 62/393,940, 62/471,937, 62/471,949, 62/475,140, and
62/475,166, and PCT Application No. PCT/US2016/052352.
In some embodiments, the ionizable lipids are selected from Compounds 1- 280 described in U.S. Application No. 62/475,166.
In some embodiments, the ionizable lipid is

In some embodiments, the ionizable lipid is
 In some embodiments, the ionizable lipid is
In some embodiments, the ionizable lipid is
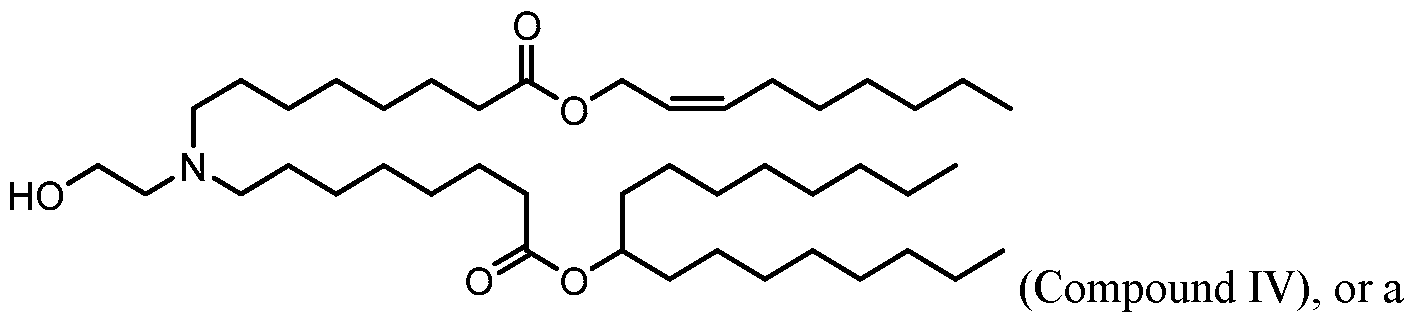 salt thereof.
salt thereof.
In some embodiments, the ionizable lipid is
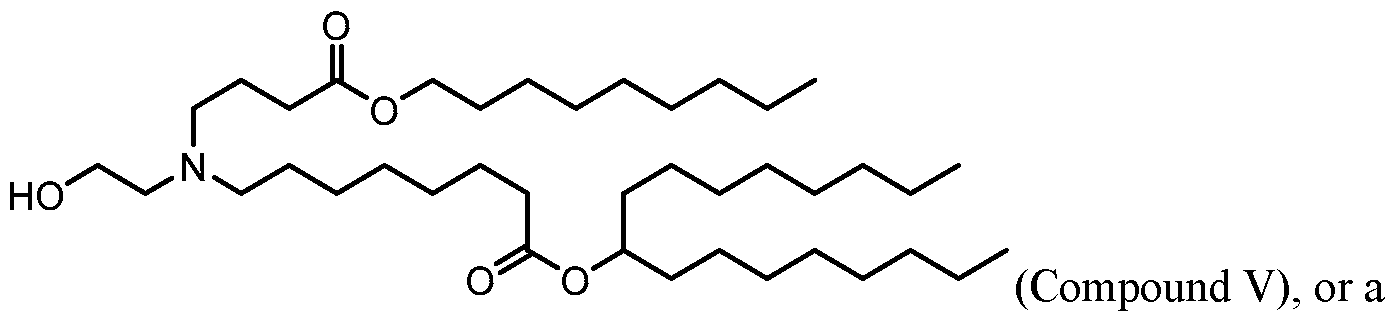 salt thereof.
salt thereof.
The central amine moiety of a lipid according to Formula (I), (IA), (IB), (II), (Ila), (lib), (lie), (lid), (lie), (Ilf), or (Ilg) may be protonated at a physiological pH. Thus, a lipid may have a positive or partial positive charge at physiological pH. Such lipids may be referred to as cationic or ionizable (amino)lipids. Lipids may also be zwitterionic, i.e., neutral molecules having both a positive and a negative charge.
In some aspects, the ionizable lipids of the present disclosure may be one or more of compounds of formula (III),
 or salts or isomers thereof, wherein
or salts or isomers thereof, wherein
 ring
ring
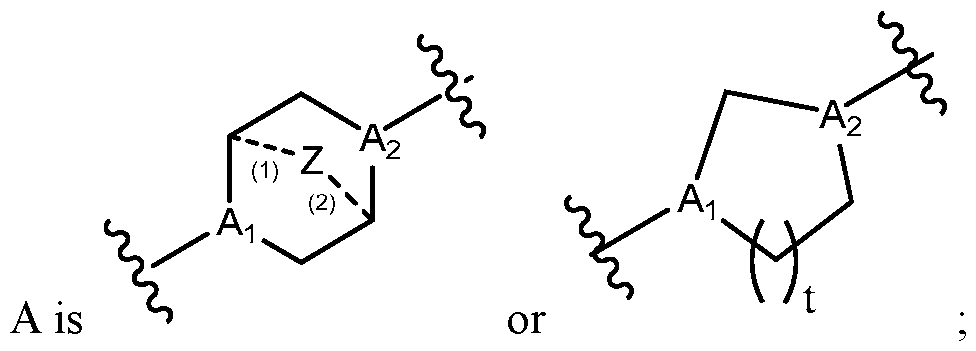 t is 1 or 2;
t is 1 or 2;
Ai and A2 are each independently selected from CH or N;
Z is CH2 or absent wherein when Z is CH2, the dashed lines (1) and (2) each represent a single bond; and when Z is absent, the dashed lines (1) and (2) are both absent;
Ri, R2, R3, R4, and Rs are independently selected from the group consisting of C5-20 alkyl, C5-20 alkenyl, -R”MR’, -R*YR”, -YR”, and -R*OR”;
Rxi and Rx2 are each independently H or C1-3 alkyl; each M is independently selected from the group consisting of -C(O)O-, -OC(O)-, -OC(O)O-, -C(O)N(R’)-, -N(R’)C(O)-, -C(O)-, -C(S)-, - C(S)S-, -SC(S)-,
-CH(OH)-, -P(O)(OR’)O-, -S(O)2-, -C(O)S-, -SC(O)-, an aryl group, and a heteroaryl group;
M* is C1-C6 alkyl,
W1 and W2 are each independently selected from the group consisting of -O- and -N(Re)-; each R5 is independently selected from the group consisting of H and C1-5 alkyl;
X1, X2, and X3 are independently selected from the group consisting of a bond, -CH2-,
-(CH2)2-, -CHR-, -CHY-, -C(O)-, -C(O)O-, -OC(O)-, -(CH2)n-C(O)-, -C(O)- (CH2)n-,
-(CH2)n-C(O)O-, -OC(O)-(CH2)n-, -(CH2)n-OC(O)-, -C(O)O-
(CH2)n-, -CH(OH)-, -C(S)-, and -CH(SH)-; each Y is independently a C3-6 carbocycle; each R* is independently selected from the group consisting of C1-12 alkyl and C2-12 alkenyl;
each R is independently selected from the group consisting of C1-3 alkyl and a C3-6 carbocycle; each R’ is independently selected from the group consisting of C1-12 alkyl, C2-12 alkenyl, and H; each R” is independently selected from the group consisting of C3-12 alkyl, C3-12 alkenyl and -R*MR’; and n is an integer from 1-6;
 i) at least one of X1, X2, and X3 is not -CH2-; and/or ii) at least one of R1, R2, R3, R4, and R5 is -R”MR’.
i) at least one of X1, X2, and X3 is not -CH2-; and/or ii) at least one of R1, R2, R3, R4, and R5 is -R”MR’.
In some embodiments, the compound is of any of formulae (IIIal)-(IIIa8):


In some embodiments, the ionizable lipids are one or more of the compounds described in U.S. Application Nos. 62/271,146, 62/338,474, 62/413,345, and 62/519,826, and PCT Application No. PCT/US2016/068300.
In some embodiments, the ionizable lipids are selected from Compounds 1- 156 described in U.S. Application No. 62/519,826.
In some embodiments, the ionizable lipids are selected from Compounds 1-16, 42-66, 68-76, and 78-156 described in U.S. Application No. 62/519,826.
In some embodiments, the ionizable lipid is
 (Compound
(Compound
VI), or a salt thereof.
In some embodiments, the ionizable lipid is
(Compound VII), or a salt thereof.
The central amine moiety of a lipid according to Formula (III), (Illal ), (IIIa2), (IIIa3), (IIIa4), (IIIa5), (IIIa6), (IIIa7), or (IIIa8) may be protonated at a physiological pH. Thus, a lipid may have a positive or partial positive charge at physiological pH. Such lipids may be referred to as cationic or ionizable (amino)lipids. Lipids may also be zwitterionic, i.e., neutral molecules having both a positive and a negative charge.
Phospholipids
The lipid composition of the lipid nanoparticle composition disclosed herein can comprise one or more phospholipids, for example, one or more saturated or (poly)unsaturated phospholipids or a combination thereof. In general, phospholipids comprise a phospholipid moiety and one or more fatty acid moieties.
A phospholipid moiety can be selected, for example, from the non-limiting group consisting of phosphatidyl choline, phosphatidyl ethanolamine, phosphatidyl glycerol, phosphatidyl serine, phosphatidic acid, 2-lysophosphatidyl choline, and a sphingomyelin.
A fatty acid moiety can be selected, for example, from the non-limiting group consisting of lauric acid, myristic acid, myristoleic acid, palmitic acid, palmitoleic acid, stearic acid, oleic acid, linoleic acid, alpha-linolenic acid, erucic acid, phytanoic acid, arachidic acid, arachidonic acid, eicosapentaenoic acid, behenic acid, docosapentaenoic acid, and docosahexaenoic acid.
Particular phospholipids can facilitate fusion to a membrane. For example, a cationic phospholipid can interact with one or more negatively charged phospholipids of a membrane (e.g., a cellular or intracellular membrane). Fusion of a phospholipid to a membrane can allow one or more elements (e.g., a therapeutic agent) of a lipid- containing composition (e.g., LNPs) to pass through the membrane permitting, e.g., delivery of the one or more elements to a target tissue.
Non-natural phospholipid species including natural species with modifications and substitutions including branching, oxidation, cyclization, and alkynes are also contemplated. For example, a phospholipid can be functionalized with or cross-linked to one or more alkynes (e.g., an alkenyl group in which one or more double bonds is replaced with a triple bond). Under appropriate reaction conditions, an alkyne group can undergo a copper-catalyzed cycloaddition upon exposure to an azide. Such reactions can be useful in functionalizing a lipid bilayer of a nanoparticle composition to facilitate membrane permeation or cellular recognition or in conjugating a nanoparticle composition to a useful component such as a targeting or imaging moiety (e.g., a dye).
Phospholipids include, but are not limited to, glycerophospholipids such as phosphatidylcholines, phosphatidylethanolamines, phosphatidylserines, phosphatidylinositols, phosphatidy glycerols, and phosphatidic acids. Phospholipids also include phosphosphingolipid, such as sphingomyelin.
In some embodiments, a phospholipid of the invention comprises 1,2- distearoyl-sn-glycero-3-phosphocholine (DSPC), l,2-dioleoyl-sn-glycero-3- phosphoethanolamine (DOPE), l,2-dilinoleoyl-sn-glycero-3 -phosphocholine (DLPC), 1,2-dimyristoyl-sn-gly cero-phosphocholine (DMPC), l,2-dioleoyl-sn-glycero-3- phosphocholine (DOPC), l,2-dipalmitoyl-sn-glycero-3 -phosphocholine (DPPC), 1,2- diundecanoyl-sn-glycero-phosphocholine (DUPC), 1 -palmitoyl -2 -oleoyl-sn-gly cero- 3 -phosphocholine (POPC), l,2-di-O-octadecenyl-sn-glycero-3-phosphocholine (18:0 Diether PC), l-oleoyl-2 cholesterylhemisuccinoyl-sn-glycero-3-phosphocholine (OChemsPC), l-hexadecyl-sn-glycero-3 -phosphocholine (C16 Lyso PC), 1,2- dilinolenoyl-sn-glycero-3-phosphocholine,l,2-diarachidonoyl-sn-glycero-3- phosphocholine, 1 ,2-didocosahexaenoyl-sn-glycero-3-phosphocholine, 1,2-
diphytanoyl-sn-glycero-3 -phosphoethanolamine (ME 16.0 PE), 1,2-distearoyl-sn- glycero-3 -phosphoethanolamine, l,2-dilinoleoyl-sn-glycero-3 -phosphoethanolamine,
1.2-dilinolenoyl-sn-glycero-3 -phosphoethanolamine, 1,2-diarachidonoyl-sn-glycero- 3 -phosphoethanolamine, 1 ,2-didocosahexaenoyl-sn-glycero-3 -phosphoethanolamine,
1.2-dioleoyl-sn-glycero-3-phospho-rac-(l -glycerol) sodium salt (DOPG), sphingomyelin, and mixtures thereof.
In certain embodiments, a phospholipid useful or potentially useful in the present invention is an analog or variant of DSPC. In certain embodiments, a phospholipid useful or potentially useful in the present invention is a compound of Formula (IV):

(IV), or a salt thereof, wherein: each R1 is independently optionally substituted alkyl; or optionally two R1 are joined together with the intervening atoms to form optionally substituted monocyclic carbocyclyl or optionally substituted monocyclic heterocyclyl; or optionally three R1 are joined together with the intervening atoms to form optionally substituted bicyclic carbocyclyl or optionally substitute bicyclic heterocyclyl; n is 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10; m is 0, 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10;
A is of the formula:
 each instance of L2 is independently a bond or optionally substituted C1-6 alkylene, wherein one methylene unit of the optionally substituted C1-6 alkylene is optionally replaced with O, N(RN), S, C(O), C(O)N(RN), - NRNC(O), C(O)O, OC(O), OC(O)O, OC(O)N(RN), NRNC(O)O, or - NRNC(O)N(RN);
each instance of R2 is independently optionally substituted C1-30 alkyl, optionally substituted C1-30 alkenyl, or optionally substituted C1-30 alkynyl; optionally wherein one or more methylene units of R2 are independently replaced with optionally substituted carbocyclylene, optionally substituted heterocyclylene, optionally substituted arylene, optionally substituted heteroarylene, N(RN), O, S, C(O), C(O)N(RN), NRNC(O), NRNC(O)N(RN), - C(O)O, OC(O), OC(O)O, OC(O)N(RN), NRNC(O)O, C(O)S, SC(O), - C(=NRN), C(=NRN)N(RN), NRNC(=NRN), NRNC(=NRN)N(RN), C(S), - C(S)N(RN), NRNC(S), NRNC(S)N(RN), S(O), OS(O), S(O)O, OS(O)O, - OS(O)2, S(O)2O, OS(O)2O, N(RN)S(O), S(O)N(RN), N(RN)S(O)N(RN), - OS(O)N(RN), N(RN)S(O)O, S(O)2, N(RN)S(O)2, S(O)2N(RN), - N(RN)S(O)2N(RN), OS(O)2N(RN), orN(RN)S(O)2O; each instance of RN is independently hydrogen, optionally substituted alkyl, or a nitrogen protecting group;
each instance of L2 is independently a bond or optionally substituted C1-6 alkylene, wherein one methylene unit of the optionally substituted C1-6 alkylene is optionally replaced with O, N(RN), S, C(O), C(O)N(RN), - NRNC(O), C(O)O, OC(O), OC(O)O, OC(O)N(RN), NRNC(O)O, or - NRNC(O)N(RN);
each instance of R2 is independently optionally substituted C1-30 alkyl, optionally substituted C1-30 alkenyl, or optionally substituted C1-30 alkynyl; optionally wherein one or more methylene units of R2 are independently replaced with optionally substituted carbocyclylene, optionally substituted heterocyclylene, optionally substituted arylene, optionally substituted heteroarylene, N(RN), O, S, C(O), C(O)N(RN), NRNC(O), NRNC(O)N(RN), - C(O)O, OC(O), OC(O)O, OC(O)N(RN), NRNC(O)O, C(O)S, SC(O), - C(=NRN), C(=NRN)N(RN), NRNC(=NRN), NRNC(=NRN)N(RN), C(S), - C(S)N(RN), NRNC(S), NRNC(S)N(RN), S(O), OS(O), S(O)O, OS(O)O, - OS(O)2, S(O)2O, OS(O)2O, N(RN)S(O), S(O)N(RN), N(RN)S(O)N(RN), - OS(O)N(RN), N(RN)S(O)O, S(O)2, N(RN)S(O)2, S(O)2N(RN), - N(RN)S(O)2N(RN), OS(O)2N(RN), orN(RN)S(O)2O; each instance of RN is independently hydrogen, optionally substituted alkyl, or a nitrogen protecting group;
Ring B is optionally substituted carbocyclyl, optionally substituted heterocyclyl, optionally substituted aryl, or optionally substituted heteroaryl; and p is 1 or 2; provided that the compound is not of the formula:
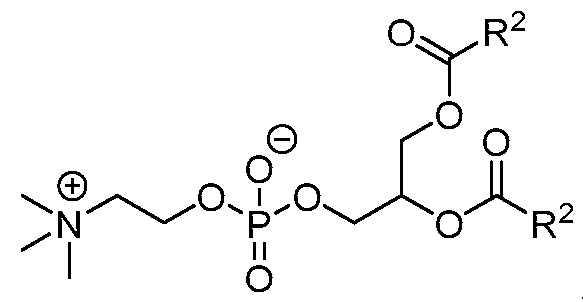 wherein each instance of R2 is independently unsubstituted alkyl, unsubstituted alkenyl, or unsubstituted alkynyl.
wherein each instance of R2 is independently unsubstituted alkyl, unsubstituted alkenyl, or unsubstituted alkynyl.
In some embodiments, the phospholipids may be one or more of the phospholipids described in U.S. Application No. 62/520,530. i) Phospholipid Head Modifications
In certain embodiments, a phospholipid useful or potentially useful in the present invention comprises a modified phospholipid head (e.g., a modified choline group). In certain embodiments, a phospholipid with a modified head is DSPC, or
analog thereof, with a modified quaternary amine. For example, in embodiments of Formula (IV), at least one of R1 is not methyl. In certain embodiments, at least one of R1 is not hydrogen or methyl. In certain embodiments, the compound of Formula (IV) is of one of the following formulae:
 or a salt thereof, wherein: each t is independently 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10; each u is independently 0, 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10; and each v is independently 1, 2, or 3.
or a salt thereof, wherein: each t is independently 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10; each u is independently 0, 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10; and each v is independently 1, 2, or 3.
In certain embodiments, a compound of Formula (IV) is of Formula
(IV-a):

In certain embodiments, a phospholipid useful or potentially useful in the present invention comprises a cyclic moiety in place of the glyceride moiety. In certain embodiments, a phospholipid useful in the present invention is DSPC, or analog thereof, with a cyclic moiety in place of the glyceride moiety. In certain embodiments, the compound of Formula (IV) is of Formula (IV-b):

(IV-b), or a salt thereof.
(it) Phospholipid Tail Modifications
In certain embodiments, a phospholipid useful or potentially useful in the present invention comprises a modified tail. In certain embodiments, a phospholipid useful or potentially useful in the present invention is DSPC, or analog thereof, with a modified tail. As described herein, a “modified tail” may be a tail with shorter or longer aliphatic chains, aliphatic chains with branching introduced, aliphatic chains with substituents introduced, aliphatic chains wherein one or more methylenes are replaced by cyclic or heteroatom groups, or any combination thereof. For example, in certain embodiments, the compound of (IV) is of Formula (IV-a), or a salt thereof, wherein at least one instance of R2 is each instance of R2 is optionally substituted C1- 30 alkyl, wherein one or more methylene units of R2 are independently replaced with optionally substituted carbocyclylene, optionally substituted heterocyclylene, optionally substituted arylene, optionally substituted heteroarylene, N(RN), O, S, - C(O), C(O)N(RN), NRNC(O), NRNC(O)N(RN), C(O)O, OC(O), OC(O)O, - OC(O)N(RN), NRNC(O)O, C(O)S, SC(O), C(=NRN), C(=NRN)N(RN), NRNC(=NRN), NRNC(=NRN)N(RN), C(S), C(S)N(RN), NRNC(S), NRNC(S)N(RN), S(O), OS(O), - S(O)O, OS(O)O, OS(O)2, S(O)2O, OS(O)2O, N(RN)S(O), S(O)N(RN), - N(RN)S(O)N(RN), OS(O)N(RN), N(RN)S(O)O, S(O)2, N(RN)S(O)2, S(O)2N(RN), - N(RN)S(O)2N(RN), OS(O)2N(RN), orN(RN)S(O)2O.
In certain embodiments, the compound of Formula (IV) is of Formula (IV-c):
 or a salt thereof, wherein: each x is independently an integer between 0-30, inclusive; and each instance is G is independently selected from the group consisting of optionally substituted carbocyclylene, optionally substituted heterocyclylene, optionally substituted arylene, optionally substituted
heteroarylene, N(RN), O, S, C(O), C(O)N(RN), NRNC(O), NRNC(O)N(RN), - C(O)O, OC(O), OC(O)O, OC(O)N(RN), NRNC(O)O, C(O)S, SC(O), - C(=NRN), C(=NRN)N(RN), NRNC(=NRN), NRNC(=NRN)N(RN), C(S), - C(S)N(RN), NRNC(S), NRNC(S)N(RN), S(O), OS(O), S(O)O, OS(O)O, - OS(O)2, S(O)2O, OS(O)2O, N(RN)S(O), S(O)N(RN), N(RN)S(O)N(RN), - OS(O)N(RN), N(RN)S(O)O, S(O)2, N(RN)S(O)2, S(O)2N(RN), - N(RN)S(O)2N(RN), OS(O)2N(RN), orN(RN)S(O)2O. Each possibility represents a separate embodiment of the present invention.
or a salt thereof, wherein: each x is independently an integer between 0-30, inclusive; and each instance is G is independently selected from the group consisting of optionally substituted carbocyclylene, optionally substituted heterocyclylene, optionally substituted arylene, optionally substituted
heteroarylene, N(RN), O, S, C(O), C(O)N(RN), NRNC(O), NRNC(O)N(RN), - C(O)O, OC(O), OC(O)O, OC(O)N(RN), NRNC(O)O, C(O)S, SC(O), - C(=NRN), C(=NRN)N(RN), NRNC(=NRN), NRNC(=NRN)N(RN), C(S), - C(S)N(RN), NRNC(S), NRNC(S)N(RN), S(O), OS(O), S(O)O, OS(O)O, - OS(O)2, S(O)2O, OS(O)2O, N(RN)S(O), S(O)N(RN), N(RN)S(O)N(RN), - OS(O)N(RN), N(RN)S(O)O, S(O)2, N(RN)S(O)2, S(O)2N(RN), - N(RN)S(O)2N(RN), OS(O)2N(RN), orN(RN)S(O)2O. Each possibility represents a separate embodiment of the present invention.
In certain embodiments, a phospholipid useful or potentially useful in the present invention comprises a modified phosphocholine moiety, wherein the alkyl chain linking the quaternary amine to the phosphoryl group is not ethylene (e.g., n is not 2). Therefore, in certain embodiments, a phospholipid useful or potentially useful in the present invention is a compound of Formula (IV), wherein n is 1, 3, 4, 5, 6, 7, 8, 9, or 10. For example, in certain embodiments, a compound of Formula (IV) is of one of the following formulae:
 or a salt thereof.
or a salt thereof.
Alternative Lipids
In certain embodiments, a phospholipid useful or potentially useful in the present invention comprises a modified phosphocholine moiety, wherein the alkyl chain linking the quaternary amine to the phosphoryl group is not ethylene (e.g., n is not 2). Therefore, in certain embodiments, a phospholipid useful.
In certain embodiments, an alternative lipid is used in place of a phospholipid of the present disclosure.
In certain embodiments, an alternative lipid of the invention is oleic acid.
In certain embodiments, the alternative lipid is one of the following:

Structural Lipids
The lipid composition of a pharmaceutical composition disclosed herein can comprise one or more structural lipids. As used herein, the term "structural lipid" refers to sterols and also to lipids containing sterol moieties.
Incorporation of structural lipids in the lipid nanoparticle may help mitigate aggregation of other lipids in the particle. Structural lipids can be selected from the group including but not limited to, cholesterol, fecosterol, sitosterol, ergosterol, campesterol, stigmasterol, brassicasterol, tomatidine, tomatine, ursolic acid, alphatocopherol, hopanoids, phytosterols, steroids, and mixtures thereof. In some embodiments, the structural lipid is a sterol. As defined herein, "sterols" are a subgroup of steroids consisting of steroid alcohols. In certain embodiments, the structural lipid is a steroid. In certain embodiments, the structural lipid is cholesterol. In certain embodiments, the structural lipid is an analog of cholesterol. In certain embodiments, the structural lipid is alpha-tocopherol.
In some embodiments, the structural lipids may be one or more of the structural lipids described in U.S. Application No. 62 /520,530.
Polyethylene Glycol (PEG)-Lipids
The lipid composition of a pharmaceutical composition disclosed herein can comprise one or more a polyethylene glycol (PEG) lipid.
As used herein, the term “PEG-lipid” refers to polyethylene glycol (PEG)- modified lipids. Non-limiting examples of PEG-lipids include PEG-modified phosphatidylethanolamine and phosphatidic acid, PEG-ceramide conjugates (e.g., PEG-CerC14 or PEG-CerC20), PEG-modified dialkylamines and PEG-modified 1,2- diacyloxypropan-3 -amines. Such lipids are also referred to as PEGylated lipids. For
example, a PEG lipid can be PEG-c-DOMG, PEG-DMG, PEG-DLPE, PEG-DMPE, PEG-DPPC, or a PEG-DSPE lipid.
In some embodiments, the PEG-lipid includes, but not limited to 1,2- dimyristoyl-sn-glycerol methoxypolyethylene glycol (PEG-DMG), 1,2-distearoyl-sn- glycero-3-phosphoethanolamine-N-[amino(polyethylene glycol)] (PEG-DSPE), PEG- disteryl glycerol (PEG-DSG), PEG-dipalmetoleyl, PEG-dioleyl, PEG-distearyl, PEG- diacylglycamide (PEG-DAG), PEG-dipalmitoyl phosphatidylethanolamine (PEG- DPPE), or PEG-1, 2-dimyristyloxlpropyl-3 -amine (PEG-c-DMA).
In one embodiment, the PEG-lipid is selected from the group consisting of a PEG-modified phosphatidylethanolamine, a PEG-modified phosphatidic acid, a PEG- modified ceramide, a PEG-modified dialkylamine, a PEG-modified diacylglycerol, a PEG-modified dialkylglycerol, and mixtures thereof.
In some embodiments, the lipid moiety of the PEG-lipids includes those having lengths of from about Ci4to about C22, preferably from about Cuto about Ci6. In some embodiments, a PEG moiety, for example an mPEG-NEE, has a size of about 1000, 2000, 5000, 10,000, 15,000 or 20,000 daltons. In one embodiment, the PEG- lipid is PEG2k-DMG.
In one embodiment, the lipid nanoparticles described herein can comprise a PEG lipid which is a non-diffusible PEG. Non-limiting examples of non-diffusible PEGs include PEG-DSG and PEG-DSPE.
PEG-lipids are known in the art, such as those described in U.S. Patent No. 8158601 and International Publ. No. WO 2015/130584 A2, which are incorporated herein by reference in their entirety.
In general, some of the other lipid components (e.g., PEG lipids) of various formulae, described herein may be synthesized as described International Patent Application No. PCT/US2016/000129, filed December 10, 2016, entitled “Compositions and Methods for Delivery of Therapeutic Agents,” which is incorporated by reference in its entirety.
The lipid component of a lipid nanoparticle composition may include one or more molecules comprising polyethylene glycol, such as PEG or PEG-modified lipids. Such species may be alternately referred to as PEGylated lipids. A PEG lipid
is a lipid modified with polyethylene glycol. A PEG lipid may be selected from the non-limiting group including PEG-modified phosphatidylethanolamines, PEG- modified phosphatidic acids, PEG-modified ceramides, PEG-modified dialkylamines, PEG-modified diacylglycerols, PEG-modified dialkylglycerols, and mixtures thereof. For example, a PEG lipid may be PEG-c-DOMG, PEG-DMG, PEG-DLPE, PEG-DMPE, PEG-DPPC, or a PEG-DSPE lipid.
In some embodiments the PEG-modified lipids are a modified form of PEG DMG. PEG-DMG has the following structure:

In one embodiment, PEG lipids useful in the present invention can be PEGylated lipids described in International Publication No. WO2012099755, the contents of which is herein incorporated by reference in its entirety. Any of these exemplary PEG lipids described herein may be modified to comprise a hydroxyl group on the PEG chain. In certain embodiments, the PEG lipid is a PEG-OH lipid. As generally defined herein, a “PEG-OH lipid” (also referred to herein as “hydroxy- PEGylated lipid”) is a PEGylated lipid having one or more hydroxyl (-OH) groups on the lipid. In certain embodiments, the PEG-OH lipid includes one or more hydroxyl groups on the PEG chain. In certain embodiments, a PEG-OH or hydroxy-PEGylated lipid comprises an -OH group at the terminus of the PEG chain. Each possibility represents a separate embodiment of the present invention.
In certain embodiments, a PEG lipid useful in the present invention is a compound of Formula (V). Provided herein are compounds of Formula (V):
 or salts thereof, wherein:
or salts thereof, wherein:
R3 is -OR°;
R° is hydrogen, optionally substituted alkyl, or an oxygen protecting group; r is an integer between 1 and 100, inclusive;
L1 is optionally substituted C1-io alkylene, wherein at least one methylene of the optionally substituted C1-io alkylene is independently replaced with optionally substituted carbocyclylene, optionally substituted heterocyclylene, optionally substituted arylene, optionally substituted heteroarylene, O, N(RN), S, C(O), C(O)N(RN), NRNC(O), C(O)O, OC(O), - OC(O)O, OC(O)N(RN), NRNC(O)O, or NRNC(O)N(RN);
D is a moiety obtained by click chemistry or a moiety cleavable under physiological conditions; m is 0, 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10;
A is of the formula:
 each instance of L2 is independently a bond or optionally substituted C1-6 alkylene, wherein one methylene unit of the optionally substituted C1-6 alkylene is optionally replaced with O, N(RN), S, C(O), C(O)N(RN), - NRNC(O), C(O)O, OC(O), OC(O)O, OC(O)N(RN), NRNC(O)O, or - NRNC(O)N(RN); each instance of R2 is independently optionally substituted C1-30 alkyl, optionally substituted C1-30 alkenyl, or optionally substituted C1-30 alkynyl; optionally wherein one or more methylene units of R2 are independently replaced with optionally substituted carbocyclylene, optionally substituted heterocyclylene, optionally substituted arylene, optionally substituted heteroarylene, N(RN), O, S, C(O), C(O)N(RN), NRNC(O), NRNC(O)N(RN), - C(O)O, OC(O), OC(O)O, OC(O)N(RN), NRNC(O)O, C(O)S, SC(O), - C(=NRN), C(=NRN)N(RN), NRNC(=NRN), NRNC(=NRN)N(RN), C(S), - C(S)N(RN), NRNC(S), NRNC(S)N(RN), S(O) , OS(O), S(O)O, OS(O)O, - OS(O)2, S(O)2O, OS(O)2O, N(RN)S(O), S(O)N(RN), N(RN)S(O)N(RN), - OS(O)N(RN), N(RN)S(O)O, S(O)2, N(RN)S(O)2, S(O)2N(RN), - N(RN)S(O)2N(RN), OS(O)2N(RN), orN(RN)S(O)2O; each instance of RN is independently hydrogen, optionally substituted alkyl, or a nitrogen protecting group;
Ring B is optionally substituted carbocyclyl, optionally substituted heterocyclyl, optionally substituted aryl, or optionally substituted heteroaryl; and p is 1 or 2.
each instance of L2 is independently a bond or optionally substituted C1-6 alkylene, wherein one methylene unit of the optionally substituted C1-6 alkylene is optionally replaced with O, N(RN), S, C(O), C(O)N(RN), - NRNC(O), C(O)O, OC(O), OC(O)O, OC(O)N(RN), NRNC(O)O, or - NRNC(O)N(RN); each instance of R2 is independently optionally substituted C1-30 alkyl, optionally substituted C1-30 alkenyl, or optionally substituted C1-30 alkynyl; optionally wherein one or more methylene units of R2 are independently replaced with optionally substituted carbocyclylene, optionally substituted heterocyclylene, optionally substituted arylene, optionally substituted heteroarylene, N(RN), O, S, C(O), C(O)N(RN), NRNC(O), NRNC(O)N(RN), - C(O)O, OC(O), OC(O)O, OC(O)N(RN), NRNC(O)O, C(O)S, SC(O), - C(=NRN), C(=NRN)N(RN), NRNC(=NRN), NRNC(=NRN)N(RN), C(S), - C(S)N(RN), NRNC(S), NRNC(S)N(RN), S(O) , OS(O), S(O)O, OS(O)O, - OS(O)2, S(O)2O, OS(O)2O, N(RN)S(O), S(O)N(RN), N(RN)S(O)N(RN), - OS(O)N(RN), N(RN)S(O)O, S(O)2, N(RN)S(O)2, S(O)2N(RN), - N(RN)S(O)2N(RN), OS(O)2N(RN), orN(RN)S(O)2O; each instance of RN is independently hydrogen, optionally substituted alkyl, or a nitrogen protecting group;
Ring B is optionally substituted carbocyclyl, optionally substituted heterocyclyl, optionally substituted aryl, or optionally substituted heteroaryl; and p is 1 or 2.
In certain embodiments, the compound of Fomula (V) is a PEG-OH lipid (i. e. , R3 is -OR°, and R° is hydrogen). In certain embodiments, the compound of Formula (V) is of Formula (V-OH):
 or a salt thereof.
or a salt thereof.
In certain embodiments, a PEG lipid useful in the present invention is a PEGylated fatty acid. In certain embodiments, a PEG lipid useful in the present invention is a compound of Formula (VI). Provided herein are compounds of Formula (VI):
 or a salts thereof, wherein:
or a salts thereof, wherein:
R3 is-OR°;
R° is hydrogen, optionally substituted alkyl or an oxygen protecting group; r is an integer between 1 and 100, inclusive;
R5 is optionally substituted Cio-40 alkyl, optionally substituted Cio-40 alkenyl, or optionally substituted Cio-40 alkynyl; and optionally one or more methylene groups of R5 are replaced with optionally substituted carbocyclylene, optionally substituted heterocyclylene, optionally substituted arylene, optionally substituted heteroarylene, N(RN), O, S, C(O), C(O)N(RN), NRNC(O), NRNC(O)N(RN), C(O)O, OC(O), OC(O)O, OC(O)N(RN), - NRNC(O)O, C(O)S, SC(O), C(=NRN), C(=NRN)N(RN), NRNC(=NRN), - NRNC(=NRN)N(RN), C(S), C(S)N(RN), NRNC(S), NRNC(S)N(RN), S(O), - OS(O), S(O)O, OS(O)O, OS(O)2, S(O)2O, OS(O)2O, N(RN)S(O), S(O)N(RN),
N(RN)S(O)N(RN), OS(O)N(RN), N(RN)S(O)O, S(0)2, N(RN)S(O)2, - S(O)2N(RN), N(RN)S(O)2N(RN), OS(O)2N(RN), orN(RN)S(O)2O; and each instance of RN is independently hydrogen, optionally substituted alkyl, or a nitrogen protecting group.
In certain embodiments, the compound of Formula (VI) is of Formula (VI-
OH):
 (VI-OH), or a salt thereof. In some embodiments, r is 45.
(VI-OH), or a salt thereof. In some embodiments, r is 45.
In yet other embodiments the compound of Formula (VI) is:
 or a salt thereof.
or a salt thereof.
In one embodiment, the compound of Formula (VI) is

(Compound I).
In some aspects, the lipid composition of the pharmaceutical compositions disclosed herein does not comprise a PEG-lipid.
In some embodiments, the PEG-lipids may be one or more of the PEG lipids described in U.S. Application No. 62/520,530.
In some embodiments, a PEG lipid of the invention comprises a PEG- modified phosphatidylethanolamine, a PEG-modified phosphatidic acid, a PEG- modified ceramide, a PEG-modified dialkylamine, a PEG-modified diacylglycerol, a PEG-modified dialkylglycerol, and mixtures thereof. In some embodiments, the PEG-modified lipid is PEG-DMG, PEG-c-DOMG (also referred to as PEG-DOMG), PEG-DSG and/or PEG-DPG.
In some embodiments, a LNP of the invention comprises an ionizable cationic lipid of any of Formula I, II or III, a phospholipid comprising DSPC, a structural lipid, and a PEG lipid comprising PEG-DMG.
In some embodiments, a LNP of the invention comprises an ionizable cationic lipid of any of Formula I, II or III, a phospholipid comprising DSPC, a structural lipid, and a PEG lipid comprising a compound having Formula VI.
In some embodiments, a LNP of the invention comprises an ionizable cationic lipid of Formula I, II or III, a phospholipid comprising a compound having Formula IV, a structural lipid, and the PEG lipid comprising a compound having Formula V or VI.
In some embodiments, a LNP of the invention comprises an ionizable cationic lipid of Formula I, II or III, a phospholipid comprising a compound having Formula IV, a structural lipid, and the PEG lipid comprising a compound having Formula V or VI.
In some embodiments, a LNP of the invention comprises an ionizable cationic lipid of Formula I, II or III, a phospholipid having Formula IV, a structural lipid, and a PEG lipid comprising a compound having Formula VI.
In some embodiments, a LNP of the invention comprises an ionizable cationic lipid of
 and a PEG lipid comprising Formula VI.
and a PEG lipid comprising Formula VI.
In some embodiments, a LNP of the invention comprises an ionizable cationic lipid of
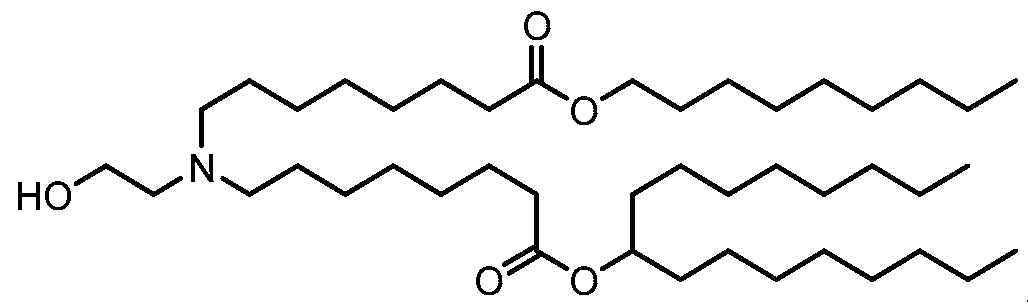 and an alternative lipid comprising oleic acid.
and an alternative lipid comprising oleic acid.
In some embodiments, a LNP of the invention comprises an ionizable cationic lipid of
 an alternative lipid comprising oleic acid, a structural lipid comprising cholesterol, and a PEG lipid comprising a compound having Formula VI.
an alternative lipid comprising oleic acid, a structural lipid comprising cholesterol, and a PEG lipid comprising a compound having Formula VI.
In some embodiments, a LNP of the invention comprises an ionizable cationic
 a phospholipid comprising DOPE, a structural lipid comprising cholesterol, and a PEG lipid comprising a compound having Formula VI.
a phospholipid comprising DOPE, a structural lipid comprising cholesterol, and a PEG lipid comprising a compound having Formula VI.
In some embodiments, a LNP of the invention comprises an ionizable cationic
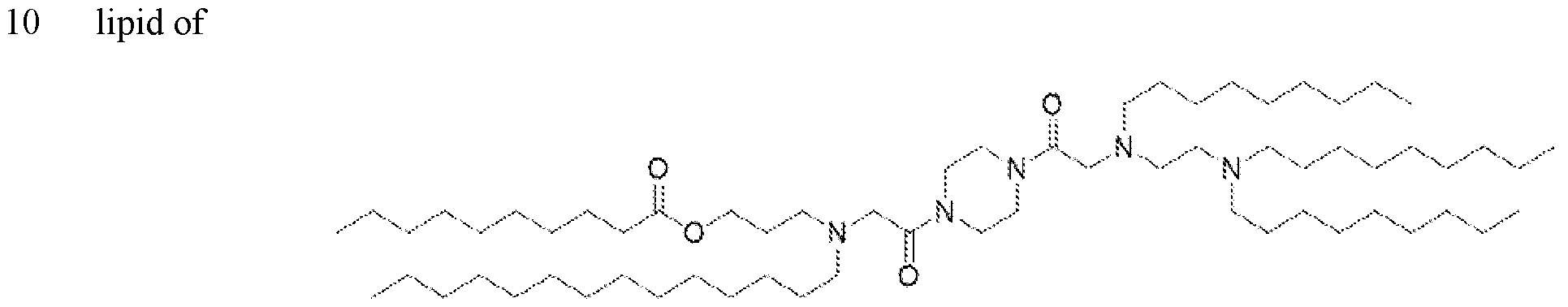 a phospholipid comprising DOPE, a structural lipid comprising cholesterol, and a PEG lipid comprising a compound having Formula VIE
a phospholipid comprising DOPE, a structural lipid comprising cholesterol, and a PEG lipid comprising a compound having Formula VIE
In some embodiments, a LNP of the invention comprises an N:P ratio of from about 2: 1 to about 30:1.
In some embodiments, a LNP of the invention comprises an N:P ratio of about 6: 1.
In some embodiments, a LNP of the invention comprises an N:P ratio of about
 In some embodiments, a LNP of the invention comprises a wt/wt ratio of the ionizable cationic lipid component to the RNA of from about 10: 1 to about 100: 1.
In some embodiments, a LNP of the invention comprises a wt/wt ratio of the ionizable cationic lipid component to the RNA of from about 10: 1 to about 100: 1.
In some embodiments, a LNP of the invention comprises a wt/wt ratio of the ionizable cationic lipid component to the RNA of about 20: 1.
In some embodiments, a LNP of the invention comprises a wt/wt ratio of the ionizable cationic lipid component to the RNA of about 10: 1.
In some embodiments, a LNP of the invention has a mean diameter from about 50nm to about 150nm.
In some embodiments, a LNP of the invention has a mean diameter from about 70nm to about 120nm.
As used herein, the term "alkyl", "alkyl group", or "alkylene" means a linear or branched, saturated hydrocarbon including one or more carbon atoms (e.g., one, two, three, four, five, six, seven, eight, nine, ten, eleven, twelve, thirteen, fourteen, fifteen, sixteen, seventeen, eighteen, nineteen, twenty, or more carbon atoms), which is optionally substituted. The notation "C1-14 alkyl" means an optionally substituted linear or branched, saturated hydrocarbon including 1-14 carbon atoms. Unless otherwise specified, an alkyl group described herein refers to both unsubstituted and substituted alkyl groups.
As used herein, the term "alkenyl", "alkenyl group", or "alkenylene" means a linear or branched hydrocarbon including two or more carbon atoms (e.g., two, three, four, five, six, seven, eight, nine, ten, eleven, twelve, thirteen, fourteen, fifteen, sixteen, seventeen, eighteen, nineteen, twenty, or more carbon atoms) and at least one double bond, which is optionally substituted. The notation "C2-14 alkenyl" means an optionally substituted linear or branched hydrocarbon including 2-14 carbon atoms and at least one carbon-carbon double bond. An alkenyl group may include one, two, three, four, or more carbon-carbon double bonds. For example, Cis alkenyl may include one or more double bonds. A Cis alkenyl group including two double bonds may be a linoleyl group. Unless otherwise specified, an alkenyl group described herein refers to both unsubstituted and substituted alkenyl groups.
As used herein, the term "alkynyl", "alkynyl group", or "alkynylene" means a linear or branched hydrocarbon including two or more carbon atoms (e.g., two, three,
four, five, six, seven, eight, nine, ten, eleven, twelve, thirteen, fourteen, fifteen, sixteen, seventeen, eighteen, nineteen, twenty, or more carbon atoms) and at least one carbon-carbon triple bond, which is optionally substituted. The notation "C2-14 alkynyl" means an optionally substituted linear or branched hydrocarbon including 2- 14 carbon atoms and at least one carbon-carbon triple bond. An alkynyl group may include one, two, three, four, or more carbon-carbon triple bonds. For example, Cis alkynyl may include one or more carbon-carbon triple bonds. Unless otherwise specified, an alkynyl group described herein refers to both unsubstituted and substituted alkynyl groups.
As used herein, the term "carbocycle" or "carbocyclic group" means an optionally substituted mono- or multi-cyclic system including one or more rings of carbon atoms. Rings may be three, four, five, six, seven, eight, nine, ten, eleven, twelve, thirteen, fourteen, fifteen, sixteen, seventeen, eighteen, nineteen, or twenty membered rings. The notation "C3-6 carbocycle" means a carbocycle including a single ring having 3-6 carbon atoms. Carbocycles may include one or more carboncarbon double or triple bonds and may be non-aromatic or aromatic (e.g., cycloalkyl or aryl groups). Examples of carbocycles include cyclopropyl, cyclopentyl, cyclohexyl, phenyl, naphthyl, and 1,2 dihydronaphthyl groups. The term "cycloalkyl" as used herein means a non-aromatic carbocycle and may or may not include any double or triple bond. Unless otherwise specified, carbocycles described herein refers to both unsubstituted and substituted carbocycle groups, i.e., optionally substituted carbocycles.
As used herein, the term "heterocycle" or "heterocyclic group" means an optionally substituted mono- or multi-cyclic system including one or more rings, where at least one ring includes at least one heteroatom. Heteroatoms may be, for example, nitrogen, oxygen, or sulfur atoms. Rings may be three, four, five, six, seven, eight, nine, ten, eleven, twelve, thirteen, or fourteen membered rings. Heterocycles may include one or more double or triple bonds and may be non- aromatic or aromatic (e.g., heterocycloalkyl or heteroaryl groups). Examples of heterocycles include imidazolyl, imidazolidinyl, oxazolyl, oxazolidinyl, thiazolyl, thiazolidinyl, pyrazolidinyl, pyrazolyl, isoxazolidinyl, isoxazolyl, isothiazolidinyl,
isothiazolyl, morpholinyl, pyrrolyl, pyrrolidinyl, furyl, tetrahydrofuryl, thiophenyl, pyridinyl, piperidinyl, quinolyl, and isoquinolyl groups. The term "heterocycloalkyl" as used herein means a non-aromatic heterocycle and may or may not include any double or triple bond. Unless otherwise specified, heterocycles described herein refers to both unsubstituted and substituted heterocycle groups, i.e., optionally substituted heterocycles.
As used herein, the term "heteroalkyl", "heteroalkenyl", or "heteroalkynyl", refers respectively to an alkyl, alkenyl, alkynyl group, as defined herein, which further comprises one or more (e.g., 1, 2, 3, or 4) heteroatoms (e.g., oxygen, sulfur, nitrogen, boron, silicon, phosphorus) wherein the one or more heteroatoms is inserted between adjacent carbon atoms within the parent carbon chain and/or one or more heteroatoms is inserted between a carbon atom and the parent molecule, i.e., between the point of attachment. Unless otherwise specified, heteroalkyls, heteroalkenyls, or heteroalkynyls described herein refers to both unsubstituted and substituted heteroalkyls, heteroalkenyls, or heteroalkynyls, i.e., optionally substituted heteroalkyls, heteroalkenyls, or heteroalkynyls.
As used herein, a "biodegradable group" is a group that may facilitate faster metabolism of a lipid in a mammalian entity. A biodegradable group may be selected from the group consisting of, but is not limited to, -C(O)O-, -OC(O)-, -C(O)N(R')-, -N(R')C(O)-, -C(O)-, -C(S)-, -C(S)S-, -SC(S)-, -CH(OH)-, -P(O)(OR)O-, -S(O)2-, an aryl group, and a heteroaryl group. As used herein, an "aryl group" is an optionally substituted carbocyclic group including one or more aromatic rings. Examples of aryl groups include phenyl and naphthyl groups. As used herein, a "heteroaryl group" is an optionally substituted heterocyclic group including one or more aromatic rings. Examples of heteroaryl groups include pyrrolyl, furyl, thiophenyl, imidazolyl, oxazolyl, and thiazolyl. Both aryl and heteroaryl groups may be optionally substituted. For example, M and M' can be selected from the nonlimiting group consisting of optionally substituted phenyl, oxazole, and thiazole. In the Formulas herein, M and M' can be independently selected from the list of biodegradable groups above. Unless otherwise specified, aryl or heteroaryl groups
described herein refers to both unsubstituted and substituted groups, i.e., optionally substituted aryl or heteroaryl groups.
Alkyl, alkenyl, and cyclyl (e.g., carbocyclyl and heterocyclyl) groups may be optionally substituted unless otherwise specified. Optional substituents may be selected from the group consisting of, but are not limited to, a halogen atom (e.g., a chloride, bromide, fluoride, or iodide group), a carboxylic acid (e.g., C(O)OH), an alcohol (e.g., a hydroxyl, OH), an ester (e.g., C(O)OR OC(O)R), an aldehyde (e.g., C(O)H), a carbonyl (e.g., C(O)R, alternatively represented by C=O), an acyl halide (e.g., C(O)X, in which X is a halide selected from bromide, fluoride, chloride, and iodide), a carbonate (e.g., OC(O)OR), an alkoxy (e.g., OR), an acetal (e.g., C(0R)2R"", in which each OR are alkoxy groups that can be the same or different and R"" is an alkyl or alkenyl group), a phosphate (e.g., P(O)43'), a thiol (e.g., SH), a sulfoxide (e.g., S(O)R), a sulfinic acid (e.g., S(O)OH), a sulfonic acid (e.g., S(O)2OH), a thial (e.g., C(S)H), a sulfate (e.g., S(O)42'), a sulfonyl (e.g., S(O)2 ), an amide (e.g., C(0)NR2, or N(R)C(O)R), an azido (e.g., N3), a nitro (e.g., NO2), a cyano (e.g., CN), an isocyano (e.g., NC), an acyloxy (e.g., OC(O)R), an amino (e.g., NR2, NRH, or NH2), a carbamoyl (e g., OC(O)NR2, OC(O)NRH, or OC(O)NH2), a sulfonamide (e.g., S(O)2NR2, S(O)2NRH, S(O)2NH2, N(R)S(O)2R, N(H)S(0)2R, N(R)S(0)2H, or N(H)S(0)2H), an alkyl group, an alkenyl group, and a cyclyl (e.g., carbocyclyl or heterocyclyl) group. In any of the preceding, R is an alkyl or alkenyl group, as defined herein. In some embodiments, the substituent groups themselves may be further substituted with, for example, one, two, three, four, five, or six substituents as defined herein. For example, a C1-6 alkyl group may be further substituted with one, two, three, four, five, or six substituents as described herein.
Compounds of the disclosure that contain nitrogens can be converted to N- oxides by treatment with an oxidizing agent (e.g., 3 -chloroperoxybenzoic acid (mCPBA) and/or hydrogen peroxides) to afford other compounds of the disclosure. Thus, all shown and claimed nitrogen-containing compounds are considered, when allowed by valency and structure, to include both the compound as shown and its N- oxide derivative (which can be designated as N->0 or N+-O-). Furthermore, in other instances, the nitrogens in the compounds of the disclosure can be converted to N-
hydroxy or N-alkoxy compounds. For example, N-hydroxy compounds can be prepared by oxidation of the parent amine by an oxidizing agent such as m CPBA. All shown and claimed nitrogen-containing compounds are also considered, when allowed by valency and structure, to cover both the compound as shown and its N- hydroxy (i.e., N-OH) and N-alkoxy (i.e., N-OR, wherein R is substituted or unsubstituted C1-C6 alkyl, C1-C6 alkenyl, C1-C6 alkynyl, 3-14-membered carbocycle or 3-14-membered heterocycle) derivatives.
Other Lipid Composition Components
The lipid composition of a pharmaceutical composition disclosed herein can include one or more components in addition to those described above. For example, the lipid composition can include one or more permeability enhancer molecules, carbohydrates, polymers, surface altering agents (e.g., surfactants), or other components. For example, a permeability enhancer molecule can be a molecule described by U.S. Patent Application Publication No. 2005/0222064. Carbohydrates can include simple sugars (e.g., glucose) and polysaccharides (e.g., glycogen and derivatives and analogs thereof).
A polymer can be included in and/or used to encapsulate or partially encapsulate a pharmaceutical composition disclosed herein (e.g., a pharmaceutical composition in lipid nanoparticle form). A polymer can be biodegradable and/or biocompatible. A polymer can be selected from, but is not limited to, polyamines, polyethers, polyamides, polyesters, polycarbamates, polyureas, polycarbonates, polystyrenes, polyimides, polysulfones, polyurethanes, polyacetylenes, polyethylenes, polyethyleneimines, polyisocyanates, polyacrylates, polymethacrylates, polyacrylonitriles, and polyarylates.
The ratio between the lipid composition and the polynucleotide range can be from about 10: 1 to about 60: 1 (wt/wt).
In some embodiments, the ratio between the lipid composition and the polynucleotide can be about 10: 1, 11: 1, 12: 1, 13: 1, 14: 1, 15: 1, 16: 1, 17: 1, 18: 1, 19: 1, 20: 1, 21: 1, 22: 1, 23: 1, 24: 1, 25: 1, 26: 1, 27: 1, 28: 1, 29: 1, 30: 1, 31: 1, 32: 1, 33: 1, 34: 1,
35:1,36:1,37:1,38:1,39:1,40:1,41:1,42:1,43:1,44:1,45:1,46:1,47:1,48:1,49:1, 50:1, 51:1, 52:1, 53:1, 54:1, 55:1, 56:1, 57:1, 58:1, 59:1 or 60:1 (wt/wt). In some embodiments, the wt/wt ratio of the lipid composition to the polynucleotide encoding a therapeutic agent is about 20 : 1 or about 15:1.
In some embodiments, the pharmaceutical composition disclosed herein can contain more than one polypeptides. For example, a pharmaceutical composition disclosed herein can contain two or more polynucleotides (e.g., RNA, e.g., mRNA).
In one embodiment, the lipid nanoparticles described herein can comprise polynucleotides (e.g., mRNA) in a lipid: polynucleotide weight ratio of 5: 1, 10: 1, 15:1, 20: 1, 25:1, 30: 1, 35:1, 40: 1, 45:1, 50:1, 55:1, 60: 1 or 70:1, or a range or any of these ratios such as, but not limited to, 5: 1 to about 10:1, from about 5: 1 to about 15:1, from about 5: 1 to about 20: 1, from about 5: 1 to about 25: 1, from about 5: 1 to about 30: 1, from about 5: 1 to about 35: 1, from about 5: 1 to about 40: 1, from about 5: 1 to about 45:1, from about 5: 1 to about 50:1, from about 5: 1 to about 55:1, from about 5: 1 to about 60: 1, from about 5: 1 to about 70: 1, from about 10: 1 to about 15:1, from about 10: 1 to about 20: 1, from about 10: 1 to about 25:1, from about 10: 1 to about 30: 1, from about 10: 1 to about 35:1, from about 10: 1 to about 40: 1, from about 10: 1 to about 45:1, from about 10: 1 to about 50: 1, from about 10: 1 to about 55:1, from about 10: 1 to about 60: 1, from about 10: 1 to about 70: 1, from about 15: 1 to about 20: 1, from about 15: 1 to about 25: 1, from about 15: 1 to about 30:1, from about 15: 1 to about 35:1, from about 15: 1 to about 40: 1, from about 15: 1 to about 45:1, from about 15: 1 to about 50:1, from about 15: 1 to about 55:1, from about 15: 1 to about 60: 1 or from about 15: 1 to about 70: 1.
In one embodiment, the lipid nanoparticles described herein can comprise the polynucleotide in a concentration from approximately 0.1 mg/ml to 2 mg/ml such as, but not limited to, 0.1 mg/ml, 0.2 mg/ml, 0.3 mg/ml, 0.4 mg/ml, 0.5 mg/ml, 0.6 mg/ml, 0.7 mg/ml, 0.8 mg/ml, 0.9 mg/ml, 1.0 mg/ml, 1.1 mg/ml, 1.2 mg/ml, 1.3 mg/ml, 1.4 mg/ml, 1.5 mg/ml, 1.6 mg/ml, 1.7 mg/ml, 1.8 mg/ml, 1.9 mg/ml, 2.0 mg/ml or greater than 2.0 mg/ml.
Nanoparticle Compositions
In some embodiments, the pharmaceutical compositions disclosed herein are formulated as lipid nanoparticles (LNP). Accordingly, the present disclosure also provides nanoparticle compositions comprising (i) a lipid composition comprising a delivery agent such as compound as described herein, and (ii) a polynucleotide encoding a CFTR polypeptide. In such nanoparticle composition, the lipid composition disclosed herein can encapsulate the polynucleotide encoding a CFTR polypeptide.
Nanoparticle compositions are typically sized on the order of micrometers or smaller and can include a lipid bilayer. Nanoparticle compositions encompass lipid nanoparticles (LNPs), liposomes (e.g., lipid vesicles), and lipoplexes. For example, a nanoparticle composition can be a liposome having a lipid bilayer with a diameter of 500 nm or less.
Nanoparticle compositions include, for example, lipid nanoparticles (LNPs), liposomes, and lipoplexes. In some embodiments, nanoparticle compositions are vesicles including one or more lipid bilayers. In certain embodiments, a nanoparticle composition includes two or more concentric bilayers separated by aqueous compartments. Lipid bilayers can be functionalized and/or crosslinked to one another. Lipid bilayers can include one or more ligands, proteins, or channels.
In one embodiment, a lipid nanoparticle comprises an ionizable lipid, a structural lipid, a phospholipid, and mRNA. In some embodiments, the LNP comprises an ionizable lipid, a PEG-modified lipid, a sterol and a structural lipid. In some embodiments, the LNP has a molar ratio of about 20-60% ionizable lipid: about 5-25% structural lipid: about 25-55% sterol; and about 0.5-15% PEG-modified lipid.
In some embodiments, the LNP has a polydispersity value of less than 0.4. In some embodiments, the LNP has a net neutral charge at a neutral pH. In some embodiments, the LNP has a mean diameter of 50-150 nm. In some embodiments, the LNP has a mean diameter of 80-100 nm.
As generally defined herein, the term “lipid” refers to a small molecule that has hydrophobic or amphiphilic properties. Lipids may be naturally occurring or
synthetic. Examples of classes of lipids include, but are not limited to, fats, waxes, sterol-containing metabolites, vitamins, fatty acids, glycerolipids, glycerophospholipids, sphingolipids, saccharolipids, and polyketides, and prenol lipids. In some instances, the amphiphilic properties of some lipids leads them to form liposomes, vesicles, or membranes in aqueous media.
In some embodiments, a lipid nanoparticle (LNP) may comprise an ionizable lipid. As used herein, the term “ionizable lipid” has its ordinary meaning in the art and may refer to a lipid comprising one or more charged moieties. In some embodiments, an ionizable lipid may be positively charged or negatively charged. An ionizable lipid may be positively charged, in which case it can be referred to as “cationic lipid”. In certain embodiments, an ionizable lipid molecule may comprise an amine group, and can be referred to as an ionizable amino lipid. As used herein, a “charged moiety” is a chemical moiety that carries a formal electronic charge, e.g., monovalent (+1, or -1), divalent (+2, or -2), trivalent (+3, or -3), etc. The charged moiety may be anionic (i.e., negatively charged) or cationic (i.e., positively charged). Examples of positively-charged moieties include amine groups (e.g., primary, secondary, and/or tertiary amines), ammonium groups, pyridinium group, guanidine groups, and imidizolium groups. In a particular embodiment, the charged moieties comprise amine groups. Examples of negatively- charged groups or precursors thereof, include carboxylate groups, sulfonate groups, sulfate groups, phosphonate groups, phosphate groups, hydroxyl groups, and the like. The charge of the charged moiety may vary, in some cases, with the environmental conditions, for example, changes in pH may alter the charge of the moiety, and/or cause the moiety to become charged or uncharged. In general, the charge density of the molecule may be selected as desired.
It should be understood that the terms “charged” or “charged moiety” does not refer to a “partial negative charge" or “partial positive charge" on a molecule. The terms “partial negative charge" and “partial positive charge" are given its ordinary meaning in the art. A “partial negative charge" may result when a functional group comprises a bond that becomes polarized such that electron density is pulled toward one atom of the bond, creating a partial negative charge on the atom. Those of
ordinary skill in the art will, in general, recognize bonds that can become polarized in this way.
In some embodiments, the ionizable lipid is an ionizable amino lipid, sometimes referred to in the art as an “ionizable cationic lipid”. In one embodiment, the ionizable amino lipid may have a positively charged hydrophilic head and a hydrophobic tail that are connected via a linker structure.
In addition to these, an ionizable lipid may also be a lipid including a cyclic amine group.
In one embodiment, the ionizable lipid may be selected from, but not limited to, an ionizable lipid described in International Publication Nos. WO2013086354 and WO2013116126; the contents of each of which are herein incorporated by reference in their entirety.
In yet another embodiment, the ionizable lipid may be selected from, but not limited to, formula CLI-CLXXXXII of US Patent No. 7,404,969; each of which is herein incorporated by reference in their entirety.
In one embodiment, the lipid may be a cleavable lipid such as those described in International Publication No. WO2012170889, herein incorporated by reference in its entirety. In one embodiment, the lipid may be synthesized by methods known in the art and/or as described in International Publication Nos. WO2013086354; the contents of each of which are herein incorporated by reference in their entirety.
Nanoparticle compositions can be characterized by a variety of methods. For example, microscopy (e.g., transmission electron microscopy or scanning electron microscopy) can be used to examine the morphology and size distribution of a nanoparticle composition. Dynamic light scattering or potentiometry (e.g., potentiometric titrations) can be used to measure zeta potentials. Dynamic light scattering can also be utilized to determine particle sizes. Instruments such as the Zetasizer Nano ZS (Malvern Instruments Ltd, Malvern, Worcestershire, UK) can also be used to measure multiple characteristics of a nanoparticle composition, such as particle size, polydispersity index, and zeta potential.
The size of the nanoparticles can help counter biological reactions such as, but not limited to, inflammation, or can increase the biological effect of the polynucleotide.
As used herein, “size” or “mean size” in the context of nanoparticle compositions refers to the mean diameter of a nanoparticle composition.
In one embodiment, the polynucleotide encoding a CFTR polypeptide are formulated in lipid nanoparticles having a diameter from about 10 to about 100 nm such as, but not limited to, about 10 to about 20 nm, about 10 to about 30 nm, about 10 to about 40 nm, about 10 to about 50 nm, about 10 to about 60 nm, about 10 to about 70 nm, about 10 to about 80 nm, about 10 to about 90 nm, about 20 to about 30 nm, about 20 to about 40 nm, about 20 to about 50 nm, about 20 to about 60 nm, about 20 to about 70 nm, about 20 to about 80 nm, about 20 to about 90 nm, about 20 to about 100 nm, about 30 to about 40 nm, about 30 to about 50 nm, about 30 to about 60 nm, about 30 to about 70 nm, about 30 to about 80 nm, about 30 to about 90 nm, about 30 to about 100 nm, about 40 to about 50 nm, about 40 to about 60 nm, about 40 to about 70 nm, about 40 to about 80 nm, about 40 to about 90 nm, about 40 to about 100 nm, about 50 to about 60 nm, about 50 to about 70 nm, about 50 to about 80 nm, about 50 to about 90 nm, about 50 to about 100 nm, about 60 to about 70 nm, about 60 to about 80 nm, about 60 to about 90 nm, about 60 to about 100 nm, about 70 to about 80 nm, about 70 to about 90 nm, about 70 to about 100 nm, about 80 to about 90 nm, about 80 to about 100 nm and/or about 90 to about 100 nm.
In one embodiment, the nanoparticles have a diameter from about 10 to 500 nm. In one embodiment, the nanoparticle has a diameter greater than 100 nm, greater than 150 nm, greater than 200 nm, greater than 250 nm, greater than 300 nm, greater than 350 nm, greater than 400 nm, greater than 450 nm, greater than 500 nm, greater than 550 nm, greater than 600 nm, greater than 650 nm, greater than 700 nm, greater than 750 nm, greater than 800 nm, greater than 850 nm, greater than 900 nm, greater than 950 nm or greater than 1000 nm.
In some embodiments, the largest dimension of a nanoparticle composition is 1 pm or shorter (e.g., 1 pm, 900 nm, 800 nm, 700 nm, 600 nm, 500 nm, 400 nm, 300 nm, 200 nm, 175 nm, 150 nm, 125 nm, 100 nm, 75 nm, 50 nm, or shorter).
A nanoparticle composition can be relatively homogenous. A polydispersity index can be used to indicate the homogeneity of a nanoparticle composition, e.g., the particle size distribution of the nanoparticle composition. A small (e.g., less than 0.3) poly dispersity index generally indicates a narrow particle size distribution. A nanoparticle composition can have a polydispersity index from about 0 to about 0.25, such as 0.01, 0.02, 0.03, 0.04, 0.05, 0.06, 0.07, 0.08, 0.09, 0.10, 0.11, 0.12, 0.13, 0.14, 0.15, 0.16, 0.17, 0.18, 0.19, 0.20, 0.21, 0.22, 0.23, 0.24, or 0.25. In some embodiments, the polydispersity index of a nanoparticle composition disclosed herein can be from about 0.10 to about 0.20.
The zeta potential of a nanoparticle composition can be used to indicate the electrokinetic potential of the composition. For example, the zeta potential can describe the surface charge of a nanoparticle composition. Nanoparticle compositions with relatively low charges, positive or negative, are generally desirable, as more highly charged species can interact undesirably with cells, tissues, and other elements in the body. In some embodiments, the zeta potential of a nanoparticle composition disclosed herein can be from about -10 mV to about +20 mV, from about -10 mV to about +15 mV, from about 10 mV to about +10 mV, from about -10 mV to about +5 mV, from about -10 mV to about 0 mV, from about -10 mV to about -5 mV, from about -5 mV to about +20 mV, from about -5 mV to about +15 mV, from about -5 mV to about +10 mV, from about -5 mV to about +5 mV, from about -5 mV to about 0 mV, from about 0 mV to about +20 mV, from about 0 mV to about +15 mV, from about 0 mV to about +10 mV, from about 0 mV to about +5 mV, from about +5 mV to about +20 mV, from about +5 mV to about +15 mV, or from about +5 mV to about +10 mV.
In some embodiments, the zeta potential of the lipid nanoparticles can be from about 0 mV to about 100 mV, from about 0 mV to about 90 mV, from about 0 mV to about 80 mV, from about 0 mV to about 70 mV, from about 0 mV to about 60 mV, from about 0 mV to about 50 mV, from about 0 mV to about 40 mV, from about 0 mV to about 30 mV, from about 0 mV to about 20 mV, from about 0 mV to about 10 mV, from about 10 mV to about 100 mV, from about 10 mV to about 90 mV, from about 10 mV to about 80 mV, from about 10 mV to about 70 mV, from about 10 mV
to about 60 mV, from about 10 mV to about 50 mV, from about 10 mV to about 40 mV, from about 10 mV to about 30 mV, from about 10 mV to about 20 mV, from about 20 mV to about 100 mV, from about 20 mV to about 90 mV, from about 20 mV to about 80 mV, from about 20 mV to about 70 mV, from about 20 mV to about 60 mV, from about 20 mV to about 50 mV, from about 20 mV to about 40 mV, from about 20 mV to about 30 mV, from about 30 mV to about 100 mV, from about 30 mV to about 90 mV, from about 30 mV to about 80 mV, from about 30 mV to about 70 mV, from about 30 mV to about 60 mV, from about 30 mV to about 50 mV, from about 30 mV to about 40 mV, from about 40 mV to about 100 mV, from about 40 mV to about 90 mV, from about 40 mV to about 80 mV, from about 40 mV to about 70 mV, from about 40 mV to about 60 mV, and from about 40 mV to about 50 mV. In some embodiments, the zeta potential of the lipid nanoparticles can be from about 10 mV to about 50 mV, from about 15 mV to about 45 mV, from about 20 mV to about 40 mV, and from about 25 mV to about 35 mV. In some embodiments, the zeta potential of the lipid nanoparticles can be about 10 mV, about 20 mV, about 30 mV, about 40 mV, about 50 mV, about 60 mV, about 70 mV, about 80 mV, about 90 mV, and about 100 mV.
The term “encapsulation efficiency” of a polynucleotide describes the amount of the polynucleotide that is encapsulated by or otherwise associated with a nanoparticle composition after preparation, relative to the initial amount provided. As used herein, “encapsulation” can refer to complete, substantial, or partial enclosure, confinement, surrounding, or encasement.
Encapsulation efficiency is desirably high (e.g., close to 100%). The encapsulation efficiency can be measured, for example, by comparing the amount of the polynucleotide in a solution containing the nanoparticle composition before and after breaking up the nanoparticle composition with one or more organic solvents or detergents.
Fluorescence can be used to measure the amount of free polynucleotide in a solution. For the nanoparticle compositions described herein, the encapsulation efficiency of a polynucleotide can be at least 50%, for example 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, or
100%. In some embodiments, the encapsulation efficiency can be at least 80%. In certain embodiments, the encapsulation efficiency can be at least 90%.
The amount of a polynucleotide present in a pharmaceutical composition disclosed herein can depend on multiple factors such as the size of the polynucleotide, desired target and/or application, or other properties of the nanoparticle composition as well as on the properties of the polynucleotide.
For example, the amount of an mRNA useful in a nanoparticle composition can depend on the size (expressed as length, or molecular mass), sequence, and other characteristics of the mRNA. The relative amounts of a polynucleotide in a nanoparticle composition can also vary.
The relative amounts of the lipid composition and the polynucleotide present in a lipid nanoparticle composition of the present disclosure can be optimized according to considerations of efficacy and tolerability. For compositions including an mRNA as a polynucleotide, the N:P ratio can serve as a useful metric.
As the N:P ratio of a nanoparticle composition controls both expression and tolerability, nanoparticle compositions with low N:P ratios and strong expression are desirable. N:P ratios vary according to the ratio of lipids to RNA in a nanoparticle composition.
In general, a lower N:P ratio is preferred. The one or more RNA, lipids, and amounts thereof can be selected to provide an N:P ratio from about 2: 1 to about 30: 1, such as 2: 1, 3: 1, 4: 1, 5: 1, 6: 1, 7: 1, 8: 1, 9: 1, 10: 1, 12: 1, 14: 1, 16: 1, 18: 1, 20: 1, 22: 1, 24: 1, 26: 1, 28: 1, or 30: 1. In certain embodiments, the N:P ratio can be from about 2: 1 to about 8: 1. In other embodiments, the N:P ratio is from about 5: 1 to about 8: 1. In certain embodiments, the N:P ratio is between 5: 1 and 6: 1. In one specific aspect, the N:P ratio is about is about 5.67: 1.
In addition to providing nanoparticle compositions, the present disclosure also provides methods of producing lipid nanoparticles comprising encapsulating a polynucleotide. Such method comprises using any of the pharmaceutical compositions disclosed herein and producing lipid nanoparticles in accordance with methods of production of lipid nanoparticles known in the art. See, e.g., Wang et al. (2015) “Delivery of oligonucleotides with lipid nanoparticles” Adv. Drug Deliv. Rev. 87:68-
80; Silva et al. (2015) “Delivery Systems for Biopharmaceuticals. Part I: Nanoparticles and Microparticles” Curr. Pharm. Technol. 16: 940-954; Naseri et al. (2015) “Solid Lipid Nanoparticles and Nanostructured Lipid Carriers: Structure, Preparation and Application” Adv. Pharm. Bull. 5:305-13; Silva et al. (2015) “Lipid nanoparticles for the delivery of biopharmaceuticals” Curr. Pharm. Biotechnol. 16:291-302, and references cited therein.
LNPs Comprising Cationic Agents
In some instances, an LNP described herein comprises a LNP core and a cationic agent disposed primarily on the outer surface of the core. Such LNPs have a greater than neutral zeta potential at physiologic pH
Core lipid nanoparticles typically comprise one or more of the following components: lipids (which may include ionizable amino lipids, phospholipids, helper lipids which may be neutral lipids, zwitterionic lipid, anionic lipids, and the like), structural lipids such as cholesterol or cholesterol analogs, fatty acids, polymers, stabilizers, salts, buffers, solvent, and the like.
Certain of the LNP cores provided herein comprise an ionizable lipid, such as an ionizable lipid, e.g., an ionizable amino lipid, a phospholipid, a structural lipid, and optionally a stabilizer (e.g., a molecule comprising polyethylene glycol) which may or may not be provided conjugated to another lipid.
The structural lipid may be but is not limited to a sterol such as for example cholesterol.
The helper lipid is a non-cationic lipid. The helper lipid may comprise at least one fatty acid chain of at least 8C and at least one polar headgroup moiety.
When a molecule comprising polyethylene glycol (i.e., PEG) is used, it may be used as a stabilizer. In some embodiments, the molecule comprising polyethylene glycol may be polyethylene glycol conjugated to a lipid and thus may be provided as PEG-c-DOMG or PEG-DMG, for example. Certain of the LNPs provided herein comprise no or low levels of PEGylated lipids, including no or low levels of alkyl - PEGylated lipids, and may be referred to herein as being free of PEG or PEGylated lipid. Thus, some LNPs comprise less than 0.5 mol % PEGylated lipid. In some
instances, PEG may be an alkyl-PEG such as methoxy-PEG. Still other LNPs comprise non-alkyl-PEG such as hydroxy-PEG, and/or non-alkyl-PEGylated lipids such as hydroxy-PEGylated lipids.
In some embodiments, a core nanoparticle composition can have the formulation of Compound II:Phospholipid:Chol:a PEG lipid with a mole ratio of 50: 10:38.5: 1.5. In some embodiments, a nanoparticle core composition can have the formulation of Compound II:DSPC:Chol:Compound 428 with a mole ratio of 50: 10:38.5: 1.5.
Compound 428:

Core nanoparticle compositions of the present disclosure comprise at least one compound according to Formula (I). Nanoparticle compositions can also include a variety of other components. For example, the nanoparticle composition can include one or more other lipids in addition to a lipid according to Formula (I) or (II), for example (i) at least one phospholipid, (ii) at least one structural lipid, (iii) at least one PEG-lipid, or (iv) any combination thereof.
In some embodiments, the nanoparticle composition comprises a compound of Formula (I), (e.g., Compounds II, III, or V). In some embodiments, the nanoparticle composition comprises a compound of Formula (I) (e.g., Compounds II, III, or V) and a phospholipid (e.g., DSPC, DOP, or MSPC).
The present disclosure also provides process of preparing a nanoparticle comprising contacting a lipid nanoparticle core with a cationic agent, wherein the lipid nanoparticle comprises:
(a) a lipid nanoparticle core comprising:
(i) an ionizable lipid,
(ii) a phospholipid,
(iii) a structural lipid, and
(iv) a PEG-lipid, and
(b) a polynucleotide (e.g., mRNA) encoding a CFTR polypeptide.
In some embodiments, the contacting of the lipid nanoparticle core with a cationic agent comprises dissolving the cationic agent in a non-ionic excipient. In some embodiments, the non-ionic excipient is selected from macrogol 15 hydroxystearate (HS 15), l,2-dimyristoyl-rac-glycero-3 -methoxypolyethylene glycol- 2000 (DMG-PEG2K), Compound 428 , polyoxyethylene sorbitan monooleate [TWEEN®80], and d-a-Tocopherol polyethylene glycol succinate (TPGS). In some embodiments, the non-ionic excipient is macrogol 15 hydroxystearate (HS 15). In some embodiments, the contacting of the lipid nanoparticle with a cationic agent comprises the cationic agent dissolved in a buffer solution. In some embodiments, the buffer solution is a phosphate buffered saline (PBS). In some embodiments, the buffer solution is a Tris-based buffer.
Provided are nanoparticles prepared by the process as described herein, e.g., by contacting the core lipid nanoparticle with a cationic agent. In some embodiments, the cationic agent can be a sterol amine such as GL-67. In some embodiments, the lipid nanoparticle core of the lipid nanoparticle optionally comprises a PEG-lipid. In some embodiments, the lipid nanoparticle core forming the lipid nanoparticle which is contacted with the cationic agent is substantially free of PEG-lipid. In some embodiments, the PEG-lipid is added to the lipid nanoparticle together with the cationic agent, prior to the contacting with the cationic agent, or after the contacting with the cationic agent.
In one embodiment, an LNP of the invention can be made using traditional mixing technology in which the nucleic acid payload is mixed with core LNP components to create the core LNP plus payload. Once this loaded core LNP is prepared, the cationic agent is contacted with the loaded core LNP.
In another embodiment, an LNP of the invention can be made using empty LNPs as the starting point. For example, empty LNPs can be made prior to loading in the nucleic acid payload. Once the nucleic acid payload is contacted with the LNP, the cationic agent can be added to form an LNP of the invention.
For example, in one embodiment, in the post-hoc loading (PHL) method, empty LNPs are formulated first in a nanoprecipitation step, and buffer exchanged into a low pH buffer (i.e. pH 5). Next, these empty LNPs are introduced to mRNA
(also acidified at low pH) through a mixing event. After the mixing step, a pH adjustment method is used to neutralize the pH. Finally, a PEG lipid, e.g., DMG- PEG-2k is added to stabilize the particle. These particles are then concentrated to the target concentration and filtered. A cationic agent, e.g., GL-67 is added.
In a variation of the empty LNP starting point, the lipids of the LNP are used to form an empty LNP, but the PEG lipid is not included in that step. In the next step, the nucleic acid solution is contacted with the empty LNPs, forming loaded LNPs. The PEG lipids are added at one or two points during further processing of the loaded LNPs and the cationic agent can be added at any point during that further processing. The cationic agent can be added at any point during the further processing of the LNP.
In some embodiments, an LNP of the invention can be prepared using nanoprecipitation, which is the unit operation in which the LNPs are self-assembled from their individual lipid components by way of kinetic mixing and subsequent maturation and continuous dilution. This unit operation includes three individual steps, which are: mixing of the aqueous and organic inputs, maturation of the LNPs, and dilution after a controlled residence time. Due to the continuous nature of these steps, they are considered one unit operation. The unit operation includes the continuous inline combination of three liquid streams with one inline maturation step: mixing of the aqueous buffer with lipid stock solution, maturation via controlled residence time, and dilution of the nanoparticles. The nanoprecipitation itself occurs in the scale- appropriate mixer, which is designed to allow continuous, high-energy, combination of the aqueous solution with the lipid stock solution dissolved in ethanol. The aqueous solution and the lipid stock solution both flow simultaneously into the mixing hardware continuously throughout this operation. The ethanol content, which keeps the lipids dissolved, is abruptly reduced and the lipids all precipitate with each other. The particles are thus self-assembled in the mixing chamber.
One of the objectives of unit operation is to exchange the solution into a fully aqueous buffer, free of ethanol, and to reach a target concentration of LNP. This can be achieved by first reaching a target processing concentration, then diafiltering, and then (if necessary) a final concentration step, once the ethanol has been completely removed.
In some embodiments, an LNP of the invention can be prepared using nanoprecipitation, which is the unit operation in which the LNPs are self-assembled from their individual lipid components by way of kinetic mixing and subsequent maturation and continuous dilution. This unit operation includes three individual steps, which are: mixing of the aqueous and organic inputs, maturation of the LNPs, and dilution after a controlled residence time. Due to the continuous nature of these steps, they are considered one unit operation. The unit operation includes the continuous inline combination of three liquid streams with one inline maturation step: mixing of the aqueous buffer with lipid stock solution, maturation via controlled residence time, and dilution of the nanoparticles. The nanoprecipitation itself occurs in the scale- appropriate mixer, which is designed to allow continuous, high-energy, combination of the aqueous solution with the lipid stock solution dissolved in ethanol. The aqueous solution and the lipid stock solution both flow simultaneously into the mixing hardware continuously throughout this operation. The ethanol content, which keeps the lipids dissolved, is abruptly reduced and the lipids all precipitate with each other. The particles are thus self-assembled in the mixing chamber.
One of the objectives of unit operation is to exchange the solution into a fully aqueous buffer, free of ethanol, and to reach a target concentration of LNP. This can be achieved by first reaching a target processing concentration, then diafiltering, and then (if necessary) a final concentration step, once the ethanol has been completely removed.
Certain aspects methods of preparing lipid nanoparticles used in the methods described herein are described in WO 2020/160397, WO 2022/104131, and WO 2023/009422, the contents of which are incorporated herein by reference in their entirety.
Methods of Use
The polynucleotides, pharmaceutical compositions and formulations described above are used in the preparation, manufacture and therapeutic use of to treat and/or prevent CFTR-related diseases, disorders or conditions. In some embodiments, the
polynucleotides, compositions and formulations of the present disclosure are used to treat and/or prevent cystic fibrosis.
In some embodiments, the polynucleotides, pharmaceutical compositions and formulations of the present disclosure are used in methods for reducing cellular sodium levels in a subject in need thereof. For instance, one aspect of the present disclosure provides a method of alleviating the signs and symptoms of cystic fibrosis in a subject comprising the administration of a composition or formulation comprising a polynucleotide encoding CFTRto that subject (e.g., an mRNA encoding a CFTR polypeptide).
In some embodiments, the administration of an effective amount of a polynucleotide, pharmaceutical composition or formulation of the invention reduces the levels of a biomarker of cystic fibrosis, e.g., intracellular sodium levels. In some embodiments, the administration of the polynucleotide, pharmaceutical composition or formulation of the present disclosure results in reduction in the level of one or more biomarkers of cystic fibrosis, e.g., intracellular sodium levels, within a short period of time after administration of the polynucleotide, pharmaceutical composition or formulation of the present disclosure.
Replacement therapy is a potential treatment for cystic fibrosis. Thus, in certain aspects of the present disclosure, the polynucleotides, e.g., mRNA, disclosed herein comprise one or more sequences encoding an CFTR polypeptide that is suitable for use in gene replacement therapy for cystic fibrosis. In some embodiments, the present disclosure treats a lack of CFTR or CFTR activity, or decreased or abnormal CFTR activity in a subject by providing a polynucleotide, e.g., mRNA, that encodes an CFTR polypeptide to the subject. In some embodiments, the polynucleotide is sequence-optimized. In some embodiments, the polynucleotide (e.g., an mRNA) comprises a nucleic acid sequence (e.g., an ORF) encoding an CFTR polypeptide, wherein the nucleic acid is sequence-optimized, e.g., by modifying its G/C, uridine, or thymidine content, and/or the polynucleotide comprises at least one chemically modified nucleoside. In some embodiments, the polynucleotide comprises a miRNA binding site, e.g., a miRNA binding site that binds miRNA-142.
In some embodiments, the administration of a composition or formulation comprising polynucleotide, pharmaceutical composition or formulation of the present disclosure to a subject results in a decrease in intracellular sodium levels in cells to a level at least 10%, at least 15%, at least 20%, at least 25%, at least 30%, at least 35%, at least 40%, at least 45%, at least 50%, at least 55%, at least 60%, at least 65%, at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, at least 95%, or to 100% lower than the level observed prior to the administration of the composition or formulation.
In some embodiments, the administration of the polynucleotide, pharmaceutical composition or formulation of the present disclosure results in expression of CFTR in cells of the subject. In some embodiments, administering the polynucleotide, pharmaceutical composition or formulation of the present disclosure results in an increase of CFTR activity in the subject. For example, in some embodiments, the polynucleotides of the present disclosure are used in methods of administering a composition or formulation comprising an mRNA encoding an CFTR polypeptide to a subject, wherein the method results in an increase of CFTR activity in at least some cells of a subject.
In some embodiments, the administration of a composition or formulation comprising an mRNA encoding an CFTR polypeptide to a subject results in an increase of CFTR activity in cells subject to a level at least 10%, at least 15%, at least 20%, at least 25%, at least 30%, at least 35%, at least 40%, at least 45%, at least 50%, at least 55%, at least 60%, at least 65%, at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, at least 95%, or to 100% or more of the activity level expected in a normal subject, e.g., a human not suffering from cystic fibrosis.
In another embodiment, the polynucleotides, pharmaceutical compositions, or formulations of the present disclosure can be repeatedly administered such that CFTR protein is expressed at a therapeutic level for a period of time sufficient to have a beneficial biological effect as described herein.
In some embodiments, the expression of the encoded polypeptide is increased. In some embodiments, the polynucleotide increases CFTR expression levels in cells when introduced into those cells, e.g., by at least 10%, at least 15%, at least 20%, at
least 25%, at least 30%, at least 35%, at least 40%, at least 45%, at least 50%, at least 55%, at least 60%, at least 65%, at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, at least 95%, or to 100% with respect to the CFTR expression level in the cells before the polypeptide is introduced in the cells.
In some embodiments, the method or use comprises administering a polynucleotide, e.g., mRNA, comprising a nucleotide sequence having sequence similarity to a polynucleotide of SEQ ID NO: 10, wherein the polynucleotide encodes an CFTR polypeptide (e.g., SEQ ID NO: 1).
The present disclosure also provides methods to increase CFTR activity in a subject in need thereof, e.g., a subject with cystic fibrosis, comprising administering to the subject a therapeutically effective amount of a composition or formulation comprising mRNA encoding an CFTR polypeptide disclosed herein.
In some aspects, the CFTR activity measured after administration to a subject in need thereof, e.g., a subject with cystic fibrosis, is at least the normal CFTR activity level observed in healthy human subjects. In some aspects, the CFTR activity measured after administration is at higher than the CFTR activity level observed in cystic fibrosis patients, e.g., untreated cystic fibrosis patients. In some aspects, the increase in CFTR activity in a subject in need thereof, e.g., a subject with cystic fibrosis, after administering to the subject a therapeutically effective amount of a composition or formulation comprising mRNA encoding an CFTR polypeptide disclosed herein is at least about 5, at least about 10, at least about 15, at least about 20, at least about 25, at least about 30, at least about 35, at least about 40, at least about 45, at least about 50, at least about 55, at least about 60, at least about 65, at least about 70, at least about 75, at least about 80, at least about 85, at least about 90, at least about 95, at least about 100, or greater than 100 percent of the normal CFTR activity level observed in healthy human subjects. In some aspects, the increase in CFTR activity above the CFTR activity level observed in cystic fibrosis patients after administering to the subject a composition or formulation comprising an mRNA encoding an CFTR polypeptide disclosed herein (e.g., after a single dose administration) is maintained for at least 1 day, at least 2 days, at least 3 days, at least
4 days, at least 5 days, at least 6 days, at least 7 days, at least 8 days, at least 9 days, at least 10 days, at least 12 days, at least 14 days, at least 21 days, or at least 28 days.
The present disclosure also provides a method to treat, prevent, or ameliorate the symptoms of cystic fibrosis (e.g., persistent coughing, lung infection, wheezing, shortness of breath, poor growth, poor weight gain, frequent greasy, bulky stools) in an cystic fibrosis patient comprising administering to the subject a therapeutically effective amount of a composition or formulation comprising mRNA encoding an CFTR polypeptide disclosed herein. In some aspects, the administration of a therapeutically effective amount of a composition or formulation comprising mRNA encoding an CFTR polypeptide disclosed herein to subject in need of treatment for cystic fibrosis results in reducing the symptoms of cystic fibrosis.
The skilled artisan will appreciate that the therapeutic effectiveness of a drug or a treatment of the instant invention can be characterized or determined by measuring the level of expression of an encoded protein (e.g., enzyme) in a sample or in samples taken from a subject (e.g., from a preclinical test subject (rodent, primate, etc.) or from a clinical subject (human). Likewise, the therapeutic effectiveness of a drug or a treatment of the instant invention can be characterized or determined by measuring the level of activity of an encoded protein (e.g., enzyme) in a sample or in samples taken from a subject (e.g., from a preclinical test subject (rodent, primate, etc.) or from a clinical subject (human). Furthermore, the therapeutic effectiveness of a drug or a treatment of the instant invention can be characterized or determined by measuring the level of an appropriate biomarker in sample(s) taken from a subject. Levels of protein and/or biomarkers can be determined post-administration with a single dose of an mRNA therapeutic of the invention or can be determined and/or monitored at several time points following administration with a single dose or can be determined and/or monitored throughout a course of treatment, e.g., a multi -dose treatment.
CFTR Protein Expression Levels
Certain aspects of the invention feature measurement, determination and/or monitoring of the expression level or levels of CFTR protein in a subject.
CFTR protein expression levels can be measured or determined by any art- recognized method for determining protein levels in biological samples, e.g., from blood samples or a needle biopsy. The term "level" or "level of a protein" as used herein, preferably means the weight, mass or concentration of the protein within a sample or a subject. It will be understood by the skilled artisan that in certain embodiments the sample may be subjected, e.g., to any of the following: purification, precipitation, separation, e.g. centrifugation and/or HPLC, and subsequently subjected to determining the level of the protein, e.g., using mass and/or spectrometric analysis. In exemplary embodiments, enzyme-linked immunosorbent assay (ELISA) can be used to determine protein expression levels. In other exemplary embodiments, protein purification, separation and LC-MS can be used as a means for determining the level of a protein according to the invention. In some embodiments, an mRNA therapy of the invention (e.g., a single intravenous dose) results in increased CFTR protein expression levels in the tissue (e.g., heart, liver, brain, or skeletal muscle) of the subject (e.g., 2-fold, 3-fold, 4-fold, 5-fold, 6-fold, 7-fold, 8-fold, 9-fold, 10-fold, 20- fold, 30-fold, 40-fold, 50-fold increase and/or increased to at least 50%, at least 60%, at least 70%, at least 75%, 80%, at least 85%, at least 90%, at least 95%, or at least 100% of normal levels) for at least 6 hours, at least 12 hours, at least 24 hours, at least 36 hours, at least 48 hours, at least 60 hours, at least 72 hours, at least 84 hours, at least 96 hours, at least 108 hours, at least 122 hours after administration of a single dose of the mRNA therapy.
CFTR Biomarkers
In some embodiments, the administration of an effective amount of a polynucleotide, pharmaceutical composition or formulation of the invention reduces the levels of a biomarker of CFTR, e.g., intracellular sodium levels. In some embodiments, the administration of the polynucleotide, pharmaceutical composition or formulation of the invention results in reduction in the level of one or more
biomarkers of CFTR, e.g., intracellular sodium levels, within a short period of time after administration of the polynucleotide, pharmaceutical composition or formulation of the invention.
Further aspects of the invention feature determining the level (or levels) of a biomarker determined in a sample as compared to a level (e.g., a reference level) of the same or another biomarker in another sample, e.g., from the same patient, from another patient, from a control and/or from the same or different time points, and/or a physiologic level, and/or an elevated level, and/or a supraphysiologic level, and/or a level of a control. The skilled artisan will be familiar with physiologic levels of biomarkers, for example, levels in normal or wild type animals, normal or healthy subjects, and the like, in particular, the level or levels characteristic of subjects who are healthy and/or normal functioning. As used herein, the phrase “elevated level” means amounts greater than normally found in a normal or wild type preclinical animal or in a normal or healthy subject, e.g. a human subject. As used herein, the term “supraphysiologic” means amounts greater than normally found in a normal or wild type preclinical animal or in a normal or healthy subject, e.g. a human subject, optionally producing a significantly enhanced physiologic response. As used herein, the term "comparing" or "compared to" preferably means the mathematical comparison of the two or more values, e.g., of the levels of the biomarker(s). It will thus be readily apparent to the skilled artisan whether one of the values is higher, lower or identical to another value or group of values if at least two of such values are compared with each other. Comparing or comparison to can be in the context, for example, of comparing to a control value, e.g., as compared to a reference blood, serum, plasma, and/or tissue (e.g., liver) intracellular sodium level, in said subject prior to administration (e.g., in a person suffering from cystic fibrosis) or in a normal or healthy subject. Comparing or comparison to can also be in the context, for example, of comparing to a control value, e.g., as compared to a reference blood, serum, plasma and/or tissue (e.g., liver) intracellular sodium level in said subject prior to administration (e.g., in a person suffering from cystic fibrosis) or in a normal or healthy subject.
As used herein, a “control” is preferably a sample from a subject wherein the cystic fibrosis status of said subject is known. In one embodiment, a control is a sample of a healthy patient. In another embodiment, the control is a sample from at least one subject having a known cystic fibrosis status, for example, a severe, mild, or healthy cystic fibrosis status, e.g. a control patient. In another embodiment, the control is a sample from a subject not being treated for cystic fibrosis. In a still further embodiment, the control is a sample from a single subject or a pool of samples from different subjects and/or samples taken from the subject(s) at different time points.
The term "level" or "level of a biomarker" as used herein, preferably means the mass, weight or concentration of a biomarker of the invention within a sample or a subject. It will be understood by the skilled artisan that in certain embodiments the sample may be subjected to, e.g., one or more of the following: substance purification, precipitation, separation, e.g. centrifugation and/or HPLC and subsequently subjected to determining the level of the biomarker, e.g. using mass spectrometric analysis. In certain embodiments, LC-MS can be used as a means for determining the level of a biomarker according to the invention.
The term "determining the level" of a biomarker as used herein can mean methods which include quantifying an amount of at least one substance in a sample from a subject, for example, in a bodily fluid from the subject (e.g., serum, plasma, urine, lymph, etc.) or in a tissue of the subject (e.g., liver, etc.).
The term "reference level" as used herein can refer to levels (e.g., of a biomarker) in a subject prior to administration of an mRNA therapy of the invention (e.g., in a person suffering from cystic fibrosis) or in a normal or healthy subject.
As used herein, the term “normal subject” or “healthy subject” refers to a subject not suffering from symptoms associated with cystic fibrosis. Moreover, a subject will be considered to be normal (or healthy) if it has no mutation of the functional portions or domains of the CFTR gene and/or no mutation of the CFTR gene resulting in a reduction of or deficiency of CFTR or the activity thereof, resulting in symptoms associated with CF. Said mutations will be detected if a sample from the subject is subjected to a genetic testing for such CFTR mutations. In certain embodiments of the present invention, a sample from a healthy subject is used as a
control sample, or the known or standardized value for the level of biomarker from samples of healthy or normal subjects is used as a control.
In some embodiments, comparing the level of the biomarker in a sample from a subject in need of treatment for cystic fibrosis or in a subject being treated for cystic fibrosis to a control level of the biomarker comprises comparing the level of the biomarker in the sample from the subject (in need of treatment or being treated for cystic fibrosis) to a baseline or reference level, wherein if a level of the biomarker in the sample from the subject (in need of treatment or being treated for cystic fibrosis) is elevated, increased or higher compared to the baseline or reference level, this is indicative that the subject is suffering from cystic fibrosis and/or is in need of treatment; and/or wherein if a level of the biomarker in the sample from the subject (in need of treatment or being treated for cystic fibrosis) is decreased or lower compared to the baseline level this is indicative that the subject is not suffering from, is successfully being treated for cystic fibrosis, or is not in need of treatment for cystic fibrosis. The stronger the reduction (e.g., at least 2-fold, at least 3-fold, at least 4-fold, at least 5 -fold, at least 6-fold, at least 7-fold, at least 8-fold, at least 10-fold, at least 20-fold, at least-30 fold, at least 40-fold, at least 50-fold reduction and/or at least 10%, at least 20%, at least 30% at least 40%, at least 50%, at least 60%, at least 70%, at least 80%, at least 90%, or at least 100% reduction) of the level of a biomarker, within a certain time period, e.g., within 6 hours, within 12 hours, 24 hours, 36 hours, 48 hours, 60 hours, or 72 hours, and/or for a certain duration of time, e.g., 48 hours, 72 hours, 96 hours, 120 hours, 144 hours, 1 week, 2 weeks, 3 weeks, 4 weeks, 1 month, 2 months, 3 months, 4 months, 5 months, 6 months, 7 months, 8 months, 9 months, 10 months, 11 months, 12 months, 18 months, 24 months, etc. the more successful is a therapy, such as for example an mRNA therapy of the invention (e.g., a single dose or a multiple regimen).
A reduction of at least about 20%, at least about 30%, at least about 40%, at least about 50%, at least about 60%, at least about 70%, at least about 80%, at least about 90%, at least 100% or more of the level of biomarker, in particular, in bodily fluid (e.g., plasma, serum, urine, e.g., urinary sediment) or in tissue(s) in a subject (e.g., liver), within 1, 2, 3, 4, 5, 6 or more days following administration is indicative
of a dose suitable for successful treatment cystic fibrosis, wherein reduction as used herein, preferably means that the level of biomarker determined at the end of a specified time period (e.g., post-administration, for example, of a single intravenous dose) is compared to the level of the same biomarker determined at the beginning of said time period (e.g., pre-administration of said dose). Exemplary time periods include 12, 24, 48, 72, 96, 120 or 144 hours post administration, in particular 24, 48, 72 or 96 hours post administration.
A sustained reduction in substrate levels (e.g., biomarkers) is particularly indicative of mRNA therapeutic dosing and/or administration regimens successful for treatment of cystic fibrosis. Such sustained reduction can be referred to herein as “duration” of effect. In exemplary embodiments, a reduction of at least about 20%, at least about 30%, at least about 40%, at least about 50%, at least about 60%, at least about 65%, at least about 70%, at least about 75%, at least about 80%, at least about 85%, at least about 90%, or at least about 95% at least about 96%, at least about 97%, at least about 98%, at least about 99%, at least about 100% or more of the level of biomarker, in particular, in a bodily fluid (e.g., plasma, serum, urine, e.g., urinary sediment) or in tissue(s) in a subject (e.g., liver), within 1, 2, 3, 4, 5, 6, 7, 8 or more days following administration is indicative of a successful therapeutic approach. In exemplary embodiments, sustained reduction in substrate (e.g., biomarker) levels in one or more samples (e.g., fluids and/or tissues) is preferred. For example, mRNA therapies resulting in sustained reduction in a biomarker, optionally in combination with sustained reduction of said biomarker in at least one tissue, preferably two, three, four, five or more tissues, is indicative of successful treatment.
In some embodiments, each dose of an mRNA therapy of the invention is about 1 mg to about 7 mg. In some embodiments, each dose of an mRNA therapy of the invention is about 1 mg. In some embodiments, each dose of an mRNA therapy of the invention is about 2 mg. In some embodiments, each dose of an mRNA therapy of the invention is about 3 mg. In some embodiments, each dose of an mRNA therapy of the invention is about 4 mg. In some embodiments, each dose of an mRNA therapy of the invention is about 5 mg. In some embodiments, each dose of an mRNA therapy of
the invention is about 6 mg. In some embodiments, each dose of an mRNA therapy of the invention is about 7 mg.
In some embodiments, each dose of an mRNA therapy of the invention is administered about once every day. In some embodiments, each dose of an mRNA therapy of the invention is administered once every day.
Bronchodilators
In some embodiments, one or more bronchodilators are administered to a subject prior to and/or concurrent with administration of a polynucleotide, pharmaceutical composition or formulation of the invention to so as to minimize possible airway irritation. Examples of bronchodilators include long -acting P agonist (LABA)/inhaled corticosteroid (ICS) therapy and short-acting p agonist (SABA) therapy. Examples of LABA/ICS therapies include salmeterol/fluticasone, formoterol/budesonide, formoterol/mometasone, and vilanterol/fluticasone. Examples of SABA therapies include albuterol, levalbuterol, and salbutamol.
Forms of Administration
The polynucleotides, pharmaceutical compositions and formulations described above can be administered by any route that results in a therapeutically effective outcome, e.g., pulmonary delivery. These also include, but are not limited to nasal administration (through the nose), insufflation (snorting), buccal (directed toward the cheek), intrapulmonary (within the lungs or its bronchi), intrasinal (within the nasal or periorbital sinuses), or respiratory (within the respiratory tract by inhaling orally or nasally for local or systemic effect). In some embodiments, a formulation for a route of administration can include at least one inactive ingredient.
In some instances, polynucleotides, pharmaceutical compositions and formulations described above can be administered via the buccal cavity. Such a formulation may comprise dry particles which comprise the active ingredient and which have a diameter in the range from about 0.5 nm to about 7 nm or from about 1 nm to about 6 nm. In some instances, such a formulation may comprise dry particles which have a diameter in the range from about 1 pm to about 5 pm or from about 1 pm to about 6 pm. Such compositions are suitably in the form of dry powders for
administration using a device comprising a dry powder reservoir to which a stream of propellant may be directed to disperse the powder and/or using a self propelling solvent/powder dispensing container such as a device comprising the active ingredient dissolved and/or suspended in a low-boiling propellant in a sealed container. Such powders comprise particles wherein at least 98% of the particles by weight have a diameter greater than 0.5 nm and at least 95% of the particles by number have a diameter less than 7 nm. Alternatively, at least 95% of the particles by weight have a diameter greater than 1 nm and at least 90% of the particles by number have a diameter less than 6 nm. Dry powder compositions may include a solid fine powder diluent such as sugar and are conveniently provided in a unit dose form. Low boiling propellants generally include liquid propellants having a boiling point of below 65° F at atmospheric pressure. Generally the propellant may constitute 50% to 99.9% (w/w) of the composition, and active ingredient may constitute 0.1% to 20% (w/w) of the composition. A propellant may further comprise additional ingredients such as a liquid non-ionic and/or solid anionic surfactant and/or a solid diluent (which may have a particle size of the same order as particles comprising the active ingredient). As a non-limiting example, the polynucleotides, pharmaceutical compositions and formulations of the invention described above may be formulated for pulmonary delivery by the methods described in U.S. Pat. No. 8,257,685; herein incorporated by reference in its entirety. Polynucleotides, pharmaceutical compositions and formulations of the invention described above formulated for pulmonary delivery may provide an active ingredient in the form of droplets of a solution and/or suspension. Such formulations may be prepared, packaged, and/or sold as aqueous and/or dilute alcoholic solutions and/or suspensions, optionally sterile, comprising active ingredient, and may conveniently be administered using any nebulization and/or atomization device. Suitable nebulizers are known in the art, including, e.g., ulstrasonic nebulizers, jet nebulizers, and vibrating -mesh nebulizers. In some instances, the nebulizer is a vibrating-mesh nebulizer. Such formulations for pulmonary delivery may further comprise one or more additional ingredients including, but not limited to, a flavoring agent such as saccharin sodium, a volatile oil, a buffering agent, a surface active agent, and/or a preservative such as
methylhydroxybenzoate. Droplets provided by this route of administration may have an average diameter in the range from about 0. 1 nm to about 200 nm.
In some instances, polynucleotides, pharmaceutical compositions, and formulations described above can be administered via intranasal, nasal, or buccal administration for pulmonary delivery. For instance, polynucleotides, pharmaceutical compositions, and formulations described herein as being useful for pulmonary delivery are useful for intranasal delivery of a pharmaceutical composition. Another formulation suitable for intranasal administration is a coarse powder comprising the active ingredient and having an average particle from about 0.2 pm to 500 pm. In some instances, such a formulation may comprise dry particles which have a diameter in the range from about 1 pm to about 5 pm or from about 1 pm to about 6 pm. In some instances, such a formulation is contained in a capsule or blister. Such a formulation is administered in the manner in which snuff is taken, i.e. by rapid inhalation through the nasal passage from a container of the powder held close to the nose. Polynucleotides, pharmaceutical compositions, and formulations suitable for nasal administration may, for example, comprise from about as little as 0.1% (w/w) and as much as 100% (w/w) of active ingredient, and may comprise one or more of the additional ingredients described herein. Polynucleotides, pharmaceutical compositions, and formulations may be prepared, packaged, and/or sold in a formulation suitable for buccal administration. Such formulations may, for example, be in the form of tablets and/or lozenges made using conventional methods, and may, for example, 0.1% to 20% (w/w) active ingredient, the balance comprising an orally dissolvable and/or degradable composition and, optionally, one or more of the additional ingredients described herein. Alternately, formulations suitable for buccal administration may comprise a powder and/or an aerosolized and/or atomized solution and/or suspension comprising active ingredient. Such powdered, aerosolized, and/or aerosolized formulations, when dispersed, may have an average particle and/or droplet size in the range from about 0.1 nm to about 200 nm, and may further comprise one or more of any additional ingredients described herein.
The polynucleotides described herein can be formulated, using the LNPs and methods described herein. The formulations can contain polynucleotides that can be
modified and/or unmodified. The formulations can further include, but are not limited to, cell penetration agents, a pharmaceutically acceptable carrier, a delivery agent, a bioerodible or biocompatible polymer, a solvent, and a sustained-release delivery depot. The formulated polynucleotides can be delivered to the cell using routes of administration known in the art and described herein.
A pharmaceutical composition for parenteral administration can comprise at least one inactive ingredient. Any or none of the inactive ingredients used can have been approved by the US Food and Drug Administration (FDA). A non-exhaustive list of inactive ingredients for use in pharmaceutical compositions for parenteral administration includes hydrochloric acid, mannitol, nitrogen, sodium acetate, sodium chloride and sodium hydroxide.
Formulations can be sterilized, for example, by filtration through a bacterial- retaining filter, and/or by incorporating sterilizing agents in the form of sterile solid compositions that can be dissolved or dispersed in sterile water or other sterile injectable medium prior to use.
Formulations can be aerosolized using methods known in the art for delivery to the lung. As a non-limiting example, the polynucleotides, pharmaceutical compositions and formulations of the invention described above may be formulated for pulmonary delivery by the methods described in U.S. Pat. No. 8,257,685; herein incorporated by reference in its entirety.
In some embodiments, each dose of an mRNA therapy of the invention is administered about once every day using a nebulizer. In some embodiments, each dose of an mRNA therapy of the invention is administered once every day using a nebulizer.
In some embodiments, each dose of an mRNA therapy of the invention is about 1 mg to about 7 mg and administered using a nebulizer. In some embodiments, each dose of an mRNA therapy of the invention is about 1 mg and administered using a nebulizer. In some embodiments, each dose of an mRNA therapy of the invention is about 2 mg and administered using a nebulizer. In some embodiments, each dose of an mRNA therapy of the invention is about 3 mg and administered using a nebulizer. In some embodiments, each dose of an mRNA therapy of the invention is about 4 mg
and administered using a nebulizer. In some embodiments, each dose of an mRNA therapy of the invention is about 5 mg and administered using a nebulizer. In some embodiments, each dose of an mRNA therapy of the invention is about 6 mg and administered using a nebulizer. In some embodiments, each dose of an mRNA therapy of the invention is about 7 mg and administered using a nebulizer.
In some embodiments, each dose of an mRNA therapy of the invention is about 1 mg to about 7 mg and administered daily using a nebulizer. In some embodiments, each dose of an mRNA therapy of the invention is about 1 mg and administered daily using a nebulizer. In some embodiments, each dose of an mRNA therapy of the invention is about 2 mg and administered daily using a nebulizer. In some embodiments, each dose of an mRNA therapy of the invention is about 3 mg and administered daily using a nebulizer. In some embodiments, each dose of an mRNA therapy of the invention is about 4 mg and administered daily using a nebulizer. In some embodiments, each dose of an mRNA therapy of the invention is about 5 mg and administered daily using a nebulizer. In some embodiments, each dose of an mRNA therapy of the invention is about 6 mg and administered daily using a nebulizer. In some embodiments, each dose of an mRNA therapy of the invention is about 7 mg and administered daily using a nebulizer.
Definitions
In order that the present disclosure can be more readily understood, certain terms are first defined. As used in this application, except as otherwise expressly provided herein, each of the following terms shall have the meaning set forth below. Additional definitions are set forth throughout the application.
The invention includes embodiments in which exactly one member of the group is present in, employed in, or otherwise relevant to a given product or process. The invention includes embodiments in which more than one, or all of the group members are present in, employed in, or otherwise relevant to a given product or process.
In this specification and the appended claims, the singular forms "a", "an" and "the" include plural referents unless the context clearly dictates otherwise. The terms "a" (or "an"), as well as the terms "one or more," and "at least one" can be used interchangeably herein. In certain aspects, the term "a" or "an" means "single." In other aspects, the term "a" or "an" includes "two or more" or "multiple."
Furthermore, "and/or" where used herein is to be taken as specific disclosure of each of the two specified features or components with or without the other. Thus, the term "and/or" as used in a phrase such as "A and/or B" herein is intended to include "A and B," "A or B," "A" (alone), and "B" (alone). Likewise, the term "and/or" as used in a phrase such as "A, B, and/or C" is intended to encompass each of the following aspects: A, B, and C; A, B, or C; A or C; A or B; B or C; A and C; A and B; B and C; A (alone); B (alone); and C (alone).
Unless defined otherwise, all technical and scientific terms used herein have the same meaning as commonly understood by one of ordinary skill in the art to which this disclosure is related. For example, the Concise Dictionary of Biomedicine and Molecular Biology, Juo, Pei-Show, 2nd ed., 2002, CRC Press; The Dictionary of Cell and Molecular Biology, 3rd ed., 1999, Academic Press; and the Oxford Dictionary Of Biochemistry And Molecular Biology, Revised, 2000, Oxford University Press, provide one of skill with a general dictionary of many of the terms used in this disclosure.
Wherever aspects are described herein with the language "comprising," otherwise analogous aspects described in terms of "consisting of' and/or "consisting essentially of' are also provided.
Units, prefixes, and symbols are denoted in their Systeme International de Unites (SI) accepted form. Numeric ranges are inclusive of the numbers defining the range. Where a range of values is recited, it is to be understood that each intervening integer value, and each fraction thereof, between the recited upper and lower limits of that range is also specifically disclosed, along with each subrange between such values. The upper and lower limits of any range can independently be included in or excluded from the range, and each range where either, neither or both limits are included is also encompassed within the invention. Where a value is explicitly recited,
it is to be understood that values which are about the same quantity or amount as the recited value are also within the scope of the invention. Where a combination is disclosed, each subcombination of the elements of that combination is also specifically disclosed and is within the scope of the invention. Conversely, where different elements or groups of elements are individually disclosed, combinations thereof are also disclosed. Where any element of an invention is disclosed as having a plurality of alternatives, examples of that invention in which each alternative is excluded singly or in any combination with the other alternatives are also hereby disclosed; more than one element of an invention can have such exclusions, and all combinations of elements having such exclusions are hereby disclosed.
Nucleotides are referred to by their commonly accepted single-letter codes. Unless otherwise indicated, nucleic acids are written left to right in 5' to 3' orientation. Nucleobases are referred to herein by their commonly known one-letter symbols recommended by the IUPAC-IUB Biochemical Nomenclature Commission. Accordingly, A represents adenine, C represents cytosine, G represents guanine, T represents thymine, U represents uracil.
Amino acids are referred to herein by either their commonly known three letter symbols or by the one-letter symbols recommended by the IUPAC-IUB Biochemical Nomenclature Commission. Unless otherwise indicated, amino acid sequences are written left to right in amino to carboxy orientation.
About: The term "about" as used in connection with a numerical value throughout the specification and the claims denotes an interval of accuracy, familiar and acceptable to a person skilled in the art, such interval of accuracy is ± 10 %.
Where ranges are given, endpoints are included. Furthermore, unless otherwise indicated or otherwise evident from the context and understanding of one of ordinary skill in the art, values that are expressed as ranges can assume any specific value or subrange within the stated ranges in different embodiments of the invention, to the tenth of the unit of the lower limit of the range, unless the context clearly dictates otherwise.
Approximately: As used herein, the term "approximately," as applied to one or more values of interest, refers to a value that is similar to a stated reference value. In
certain embodiments, the term "approximately" refers to a range of values that fall within 25%, 20%, 19%, 18%, 17%, 16%, 15%, 14%, 13%, 12%, 11%, 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1%, or less in either direction (greater than or less than) of the stated reference value unless otherwise stated or otherwise evident from the context (except where such number would exceed 100% of a possible value).
Dosing regimew. As used herein, a "dosing regimen" or a "dosing regimen" is a schedule of administration or physician determined regimen of treatment, prophylaxis, or palliative care.
Effective Amount: As used herein, the term "effective amount" of an agent is that amount sufficient to effect beneficial or desired results, for example, clinical results, and, as such, an "effective amount" depends upon the context in which it is being applied. For example, in the context of administering an agent that treats a protein deficiency (e.g., a CFTR deficiency), an effective amount of an agent is, for example, an amount of mRNA expressing sufficient CFTR to ameliorate, reduce, eliminate, or prevent the symptoms associated with the CFTR deficiency, as compared to the severity of the symptom observed without administration of the agent. The term "effective amount" can be used interchangeably with "effective dose," "therapeutically effective amount," or "therapeutically effective dose."
CFTR Associated Disease: As used herein the terms "CFTR-associated disease" or "CFTR-associated disorder" refer to diseases or disorders, respectively, which result from aberrant CFTR activity (e.g., decreased activity or increased activity). As a non-limiting example, cystic fibrosis is a CFTR-associated disease. Numerous clinical variants of cystic fibrosis are known in the art. See, e.g., www.omim .org/entry/219700.
The term "CFTR activity" as used in the present disclosure refers to CFTR’s ability to transport chloride ions through the cellular membrane. Accordingly, a polypeptide having CFTR activity refers to a polypeptide that elicits measurable chloride transport across the cell membrane.
Ionizable amino lipid'. The term “ionizable amino lipid” includes those lipids having one, two, three, or more fatty acid or fatty alkyl chains and a pH-titratable amino head group (e.g., an alkylamino or dialkylamino head group). An ionizable
amino lipid is typically protonated (i.e., positively charged) at a pH below the pKa of the amino head group and is substantially not charged at a pH above the pKa. Such ionizable amino lipids include, but are not limited to DLin-MC3-DMA (MC3) and ( 13Z, 165Z)-N,N-dimethyl-3 -nonydocosa- 13-16-dien- 1 -amine (L608) .
Methods of Administration'. As used herein, “methods of administration” can include intravenous, intramuscular, intradermal, subcutaneous, or other methods of delivering a composition to a subject. A method of administration can be selected to target delivery (e.g. , to specifically deliver) to a specific region or system of a body.
Nanoparticle Composition'. As used herein, a “nanoparticle composition” is a composition comprising one or more lipids. Nanoparticle compositions are typically sized on the order of micrometers or smaller and can include a lipid bilayer. Nanoparticle compositions encompass lipid nanoparticles (LNPs), liposomes (e.g., lipid vesicles), and lipoplexes. For example, a nanoparticle composition can be a liposome having a lipid bilayer with a diameter of 500 nm or less.
The phrase "nucleotide sequence encoding" refers to the nucleic acid (e.g., an mRNA or DNA molecule) coding sequence which encodes a polypeptide. The coding sequence can further include initiation and termination signals operably linked to regulatory elements including a promoter and polyadenylation signal capable of directing expression in the cells of an individual or mammal to which the nucleic acid is administered. The coding sequence can further include sequences that encode signal peptides.
Patient: As used herein, "patient" refers to a subject who can seek or be in need of treatment, requires treatment, is receiving treatment, will receive treatment, or a subject who is under care by a trained professional for a particular disease or condition. In some embodiments, the treatment is needed, required, or received to prevent or decrease the risk of developing acute disease, i.e., it is a prophylactic treatment.
Pseudouridine: As used herein, pseudouridine (y) refers to the C-glycoside isomer of the nucleoside uridine. A "pseudouridine analog" is any modification, variant, isoform or derivative of pseudouridine. For example, pseudouridine analogs include but are not limited to 1 -carboxymethyl -pseudouridine, 1-propynyl-
pseudouridine, 1 -taurinomethyl -pseudouridine, 1 -taurinomethyl-4-thio-pseudouridine,
1 -methylpseudouridine (m 1 \|/) (also known as Nl-methyl-pseudouridine), 1 -methyl -4- thio-pseudouridine (m 1 s4\|/)_ 4-thio-l -methyl -pseudouridine, 3-methyl-pseudouridine (m\|/). 2 -thio- 1 -methyl -pseudouridine, 1 -methyl- 1 -deaza-pseudouridine, 2-thio-l- methyl- 1 -deaza-pseudouridine, dihydropseudouridine, 2-thio-dihydropseudouridine,
2 -methoxyuridine, 2-methoxy-4-thio-uridine, 4-methoxy-pseudouridine, 4-methoxy- 2-thio-pseudouridine, l-methyl-3-(3-amino-3-carboxypropyl)pseudouridine (acp3 \|/), and 2'-O-methyl-pseudouridine (\|/m ) .
Therapeutically effective amount: As used herein, the term "therapeutically effective amount" means an amount of an agent to be delivered (e.g., nucleic acid, drug, therapeutic agent, diagnostic agent, prophylactic agent, etc.) that is sufficient, when administered to a subject suffering from or susceptible to an infection, disease, disorder, and/or condition, to treat, improve symptoms of, diagnose, prevent, and/or delay the onset of the infection, disease, disorder, and/or condition.
Uracil'. Uracil is one of the four nucleobases in the nucleic acid of RNA, and it is represented by the letter U. Uracil can be attached to a ribose ring, or more specifically, a ribofuranose via a p-Ni-glycosidic bond to yield the nucleoside uridine. The nucleoside uridine is also commonly abbreviated according to the one letter code of its nucleobase, i.e., U. Thus, in the context of the present disclosure, when a monomer in a polynucleotide sequence is U, such U is designated interchangeably as a "uracil" or a "uridine."
Uridine Content'. The terms "uridine content" or "uracil content" are interchangeable and refer to the amount of uracil or uridine present in a certain nucleic acid sequence. Uridine content or uracil content can be expressed as an absolute value (total number of uridine or uracil in the sequence) or relative (uridine or uracil percentage respect to the total number of nucleobases in the nucleic acid sequence).
Uridine-Modified Sequence'. The terms "uridine-modified sequence" refers to a sequence optimized nucleic acid (e.g., a synthetic mRNA sequence) with a different overall or local uridine content (higher or lower uridine content) or with different uridine patterns (e.g., gradient distribution or clustering) with respect to the uridine
content and/or uridine patterns of a candidate nucleic acid sequence. In the content of the present disclosure, the terms "uridine-modified sequence" and "uracil-modified sequence" are considered equivalent and interchangeable.
Nucleobase-. As used herein, the term “nucleobase” (alternatively “nucleotide base” or “nitrogenous base”) refers to a purine or pyrimidine heterocyclic compound found in nucleic acids, including any derivatives or analogs of the naturally occurring purines and pyrimidines that confer improved properties (e.g., binding affinity, nuclease resistance, chemical stability) to a nucleic acid or a portion or segment thereof. Adenine, cytosine, guanine, thymine, and uracil are the nucleobases predominately found in natural nucleic acids. Other natural, non-natural, and/or synthetic nucleobases, as known in the art and/or described herein, can be incorporated into nucleic acids. Unless otherwise specified, the nucleobase sequence of a SEQ ID NO described herein encompasses both natural nucleobases and chemically modified nucleobases (e.g., a “U” designation in a SEQ ID NO encompasses both uracil and chemically modified uracil).
Nucleoside/Nucleotide'. As used herein, the term “nucleoside” refers to a compound containing a sugar molecule (e.g., a ribose in RNA or a deoxyribose in DNA), or derivative or analog thereof, covalently linked to a nucleobase (e.g., a purine or pyrimidine), or a derivative or analog thereof (also referred to herein as “nucleobase”), but lacking an intemucleoside linking group (e.g., a phosphate group). As used herein, the term “nucleotide” refers to a nucleoside covalently bonded to an intemucleoside linking group (e.g., a phosphate group), or any derivative, analog, or modification thereof that confers improved chemical and/or functional properties (e.g., binding affinity, nuclease resistance, chemical stability) to a nucleic acid or a portion or segment thereof.
Nucleic acid: As used herein, the term “nucleic acid” is used in its broadest sense and encompasses any compound and/or substance that includes a polymer of nucleotides, or derivatives or analogs thereof. These polymers are often referred to as “polynucleotides”. Accordingly, as used herein the terms “nucleic acid” and “polynucleotide” are equivalent and are used interchangeably. Exemplary nucleic acids or polynucleotides of the disclosure include, but are not limited to, ribonucleic
acids (RNAs), deoxyribonucleic acids (DNAs), DNA-RNA hybrids, RNAi-inducing agents, RNAi agents, siRNAs, shRNAs, mRNAs, modified mRNAs, miRNAs, antisense RNAs, ribozymes, catalytic DNA, RNAs that induce triple helix formation, threose nucleic acids (TNAs), glycol nucleic acids (GNAs), peptide nucleic acids (PNAs), locked nucleic acids (LNAs, including LNA having a P-D-ribo configuration, a-LNA having an a-L-ribo configuration (a diastereomer of LNA), 2'-amino-LNA having a 2'-amino functionalization, and 2'-amino-a-LNA having a 2'-amino functionalization) or hybrids thereof.
Open Reading Frame: As used herein, the term “open reading frame”, abbreviated as “ORF”, refers to a segment or region of an mRNA molecule that encodes a polypeptide. The ORF comprises a continuous stretch of non-overlapping, in-frame codons, beginning with the initiation codon and ending with a stop codon, and is translated by the ribosome.
Equivalents and Scope
Those skilled in the art will recognize, or be able to ascertain using no more than routine experimentation, many equivalents to the specific embodiments in accordance with the invention described herein. The scope of the present invention is not intended to be limited to the above Description, but rather is as set forth in the appended claims.
In the claims, articles such as "a," "an," and "the" can mean one or more than one unless indicated to the contrary or otherwise evident from the context. Claims or descriptions that include "or" between one or more members of a group are considered satisfied if one, more than one, or all of the group members are present in, employed in, or otherwise relevant to a given product or process unless indicated to the contrary or otherwise evident from the context. The invention includes embodiments in which exactly one member of the group is present in, employed in, or otherwise relevant to a given product or process. The invention includes embodiments in which more than one, or all of the group members are present in, employed in, or otherwise relevant to a given product or process.
It is also noted that the term "comprising" is intended to be open and permits but does not require the inclusion of additional elements or steps. When the term "comprising" is used herein, the term "consisting of is thus also encompassed and disclosed.
Where ranges are given, endpoints are included. Furthermore, it is to be understood that unless otherwise indicated or otherwise evident from the context and understanding of one of ordinary skill in the art, values that are expressed as ranges can assume any specific value or subrange within the stated ranges in different embodiments of the invention, to the tenth of the unit of the lower limit of the range, unless the context clearly dictates otherwise.
In addition, it is to be understood that any particular embodiment of the present invention that falls within the prior art can be explicitly excluded from any one or more of the claims. Since such embodiments are deemed to be known to one of ordinary skill in the art, they can be excluded even if the exclusion is not set forth explicitly herein. Any particular embodiment of the compositions of the invention (e.g., any nucleic acid or protein encoded thereby; any method of production; any method of use; etc.) can be excluded from any one or more claims, for any reason, whether or not related to the existence of prior art.
All cited sources, for example, references, publications, databases, database entries, and art cited herein, are incorporated into this application by reference, even if not expressly stated in the citation. In case of conflicting statements of a cited source and the instant application, the statement in the instant application shall control.
Section and table headings are not intended to be limiting.
EXAMPLES
EXAMPLE 1: Synthesis of mRNA Encoding CFTR
An mRNA encoding human CFTR (SEQ ID NO: 1) is constructed by using the ORF sequence (nucleotide) provided in SEQ ID NO: 10. The mRNA sequence includes both 5' and 3' UTR regions flanking the ORF sequence. In an exemplary construct, the 5' UTR and 3' UTR sequences contain SEQ ID NO:50 and SEQ ID NO: 141, respectively.
The CFTRmRNA sequence is prepared as modified mRNA. Specifically, during in vitro transcription, modified mRNA can be generated using Nl- methylpseudouridine-5'-Triphosphate to ensure that the mRNAs contain 100% Nl- methylpseudouridine instead of uridine. Alternatively, during in vitro transcription, modified mRNA can be generated using Nl-methoxyuridine-5 '-Triphosphate to ensure that the mRNAs contain 100% 5 -methoxyuridine instead of uridine. Further, CFTR-mRNA can be synthesized with a primer that introduces a polyA-tail, and a cap structure is generated on both mRNAs using co-transcriptional capping to incorporate a m7G-ppp-Gm-AG 5' capl. Alternatively, CFTR-mRNA can be synthesized and the polyA-tail introduced during Gibson assembly of the DNA template.
CFTR mRNA can be synthesized with a primer that introduces a polyA-tail. mRNA constructs are modified by ligation to stabilize the polyA tail. Ligation is performed using 0.5-1.5 mg/mL mRNA (5’ Capl, 3’ A100), 50 mM Tris-HCl pH 7.5, 10 mM MgCh, 1 mM TCEP, 1000 units/mL T4 RNA Ligase 1, 1 mM ATP, 20% w/v polyethylene glycol 8000, and 5: 1 molar ratio of modifying oligo to mRNA. Modifying oligo has a sequence of 5’-phosphate-AAAAAAAAAAAAAAAAAAAA- (inverted deoxythymidine (idT)) (SEQ ID NO:209) (see below). Ligation reactions are mixed and incubated at room temperature (~22°C) for 4 hours. Stable tail mRNA are purified by dT purification, reverse phase purification, hydroxyapatite purification, ultrafiltration into water, and sterile filtration. Ligation efficiency for each mRNA is >80% as assessed by UPLC separation of ligated and unligated mRNA. The resulting stable tail-containing mRNAs contain the following structure at the 3 ’end, starting with the polyA region: Aioo- UCUAGAAAAAAAAAAAAAAAAAAAA-inverted deoxythymidine (SEQ ID NO:211).
Modifying oligo to stabilize tail (5’-phosphate- AAAAAAAAAAAAAAAAAAAA-(inverted deoxythymidine) (SEQ ID NO:209)):
S'-phosphate-AAAAAAAAAAAAAAA
3' -3’ linkage
Inverted deoxythymidine (idT)

EXAMPLE 2: Production of CFTR mRNA Cationic Nanoparticle Compositions
Preparation of CFTR mRNA cationic nanoparticle compositions can be found in WO 2023/009422, the contents of which is incorporated herein by reference.
Empty lipid nanoparticles are prepared according to the process outlined in Fig. 1. Lipids (ionizable lipid: DSPC: cholesterol: DMG-PEG 2000 lipid) are dissolved in ethanol at a total concentration of 24 mg/mL and mixed with the acidification buffer (45 mM acetate buffer at pH 4). The lipid solution and acidification buffer are mixed using a multi -inlet vortex mixer at a 3:7 volumetric ratio of lipid: buffer for mixer 1 and mixer 2 and a 1:3 volumetric ratio of lipid:buffer (25% ethanol) for mixer 3. After a 5 second residence time, the resulting eLNPs are mixed with 55 mM sodium acetate at pH 5.6 at a volumetric ratio of 5:7 of eLNP:buffer. The resulting dilute eLNPs are then buffer exchanged and concentrated using tangential flow filtration (TFF) into a final buffer containing 5 mM sodium acetate pH 5.0. Then a 70% sucrose solution in 5 mM acetate buffer at pH 5 is subsequently added.
Empty lipid nanoparticles prepared according to the procedures above are filled with nucleic acid (mRNA) according to the process depicted in Fig. 2. Loading of the mRNA takes place using a post-hoc loading (PHL) process. eLNP at a lipid
concentration of 11.72 mg/mL in 5 mM acetate (pH 5) and 75 g/L sucrose is mixed with mRNA at a concentration of 1.0 mg/mL in 42.5 mM sodium acetate pH 5.0. The eLNP solution and mRNA are mixed using a multi-inlet vortex mixer at a 3 :2 volumetric ratio of eLNP:mRNA. Once the eLNP’s are loaded with mRNA, they undergo a 60 s residence time prior to mixing in-line with a neutralization buffer containing 120 mM TRIS pH 8.12 at a volumetric ratio of 5: 1 of nanoparticle:buffer. After this addition step, the nanoparticle formulation is mixed in-line with a buffer containing 20 mM TRIS (pH 7.5), 1.42 mg/mL DMG-PEG 2000, and 2.5 mg/mL GL- 67 (a sterol amine) at a volumetric ratio of 6: 1 of nanoparticle :buffer. The resulting nanoparticle suspension undergoes concentration using tangential flow fdtration (TFF) and is diluted in running buffer (20 mM TRIS, 14.3 mM sodium acetate, and 32 g/L sucrose, pH 7.5) with a 300 nM NaCl solution to a final buffer matrix containing 70 mM NaCl. The resulting nanoparticle suspension is filtered through a 0.8/0.2 pm capsule filter and filled into glass vials at a mRNA strength of about 1 mg/mL (e.g., 0.5 - 2 mg/mL).
EXAMPLE S: Clinical Trial
This is a 2-part, Phase 1/2 dose-escalation study to evaluate the safety, tolerability, and efficacy of inhaled (nebulized) VX-522 in subjects 18 through 65 years of age (inclusive) with a confirmed diagnosis of cystic fibrosis and CFTR mutations on both alleles that are not responsive to CFTR modulator therapy. The clinical plan is to dose into the predicted clinically efficacious range, starting with 1 mg and with a maximum planned nominal dose of 7 mg.
VX-522 is a modified hCFTR mRNA formulated into a lipid nanoparticle comprised of five lipid components: (1) Compound II; (2) GL-67; (3) 1,2 distearoyl - sn-glycero-3 -phosphocholine (DSPC); (4) cholesterol; and (5) 1,2-dimyristoyl-rac- glycero-3 -methoxypolyethylene gly col-2000 (PEG2000-DMG).
The eFlow PARI Nebulizer System, a medical device manufactured by PARI Respiratory Equipment, will be used to deliver VX-522.
The sequence of the modified hCFTR mRNA of VX-522 is provided below (all uracils (U) in the following sequence are N1 -methylpseudouracils):
5 7MeGpppG2 OmeAGGAAAUCGCAAAAUUUGCUCUUCGCGUUAGAUU UCUUUUAGUUUUCUCGCAACUAGCAAGCUUUUUGUUCUCGCCAUGCAG CGGAGCCCUCUGGAGAAGGCCAGCGUGGUGAGCAAGCUGUUCUUCAGC UGGACCAGGCCCAUCCUGCGAAAGGGCUACAGACAGAGGCUGGAGCUG UCCGACAUCUACCAGAUUCCAUCCGUGGACAGCGCCGACAAUCUGAGCG AGAAGCUGGAGAGGGAGUGGGACCGCGAGCUGGCCAGCAAGAAGAACC CUAAGCUGAUCAACGCCCUGCGCCGCUGCUUCUUCUGGAGGUUCAUGUU CUACGGCAUCUUCCUGUACCUGGGCGAGGUGACCAAGGCCGUCCAGCCU CUGCUGCUGGGCCGCAUCAUCGCCAGCUACGAUCCCGACAACAAGGAGG AACGCUCCAUCGCCAUCUACCUGGGCAUCGGCCUGUGUCUGCUGUUCAU CGUGCGGACCCUGCUGCUGCACCCCGCCAUCUUCGGCCUGCAUCACAUC GGCAUGCAGAUGAGGAUCGCCAUGUUCUCCCUGAUCUACAAGAAGACC CUGAAGCUGUCCAGCCGCGUGCUGGAUAAGAUCAGCAUCGGGCAGCUG GUGAGCCUGCUGAGCAACAACCUGAACAAGUUCGACGAGGGGUUGGCG CUGGCCCACUUCGUGUGGAUUGCCCCGCUGCAGGUGGCUCUGCUGAUGG GGCUGAUCUGGGAGCUGCUGCAGGCCUCUGCCUUCUGCGGGCUGGGGU UUCUGAUCGUGCUGGCCCUGUUCCAAGCUGGCCUGGGCCGCAUGAUGA UGAAGUACCGCGAUCAGAGGGCCGGCAAGAUCAGCGAGCGCCUGGUGA UCACUAGCGAGAUGAUAGAGAACAUCCAGAGCGUGAAGGCUUACUGUU GGGAGGAGGCCAUGGAGAAGAUGAUCGAGAACCUGAGGCAGACCGAGC UGAAGCUGACUAGAAAGGCAGCCUACGUGAGGUAUUUCAACUCCAGCG CCUUCUUCUUCAGCGGCUUCUUCGUGGUGUUCCUGAGCGUGCUGCCCUA CGCCCUGAUCAAGGGCAUCAUCCUGAGGAAGAUCUUCACCACCAUUAGC UUCUGCAUCGUGCUGCGCAUGGCCGUGACCAGGCAGUUCCCUUGGGCCG UGCAGACUUGGUACGACAGCCUGGGAGCCAUCAACAAGAUCCAGGACU UUCUGCAGAAGCAGGAAUAUAAGACCCUGGAGUACAACCUGACCACCA CCGAGGUGGUGAUGGAGAACGUGACCGCCUUCUGGGAGGAGGGCUUCG GCGAGCUGUUCGAGAAGGCCAAGCAGAACAAUAACAACCGCAAGACCA GCAACGGCGACGACUCCCUCUUCUUCAGCAACUUUAGCCUGCUGGGCAC CCCUGUGCUGAAGGACAUCAACUUCAAGAUCGAAAGAGGCCAACUGCU GGCCGUGGCCGGAUCUACCGGCGCCGGCAAGACCAGCCUGCUGAUGAUG AUCAUGGGCGAGCUGGAGCCCAGCGAGGGCAAGAUCAAGCACAGCGGC CGGAUCUCCUUCUGCUCCCAGUUCUCCUGGAUCAUGCCCGGCACCAUCA AGGAGAACAUCAUCUUCGGCGUGAGCUACGACGAGUACAGGUACCGGA GCGUGAUCAAGGCCUGCCAGCUGGAGGAGGACAUCUCCAAAUUUGCCG AGAAGGACAACAUUGUGCUGGGCGAAGGCGGGAUCACCCUGUCCGGUG GCCAGCGUGCACGCAUCUCCCUGGCCCGGGCUGUGUACAAGGACGCCGA CCUGUACCUGCUGGACAGCCCUUUUGGCUACCUGGACGUGCUGACCGAG AAGGAGAUCUUCGAGUCCUGCGUGUGUAAGCUGAUGGCCAACAAGACC AGAAUCCUGGUGACCAGCAAGAUGGAGCAUCUGAAGAAGGCCGACAAG AUCCUGAUCCUGCACGAGGGGUCCAGCUACUUCUACGGCACCUUCAGCG AGCUGCAGAACCUGCAGCCCGACUUCAGCUCCAAGCUGAUGGGCUGCGA UAGCUUCGACCAGUUCUCCGCCGAGAGAAGGAACUCCAUUCUGACCGAG ACCCUGCACCGAUUCUCCCUGGAGGGAGACGCCCCAGUGAGCUGGACCG AGACCAAGAAGCAGAGCUUCAAGCAGACCGGCGAGUUCGGAGAGAAGC GCAAGAACUCCAUCCUCAACCCCAUCAACAGCAUCCGGAAGUUCAGCAU
 CGAGCCCCUGGAACGGCGACUGUCCCUCGUGCCCGACAGCGAGCAGGGC
CGAGCCCCUGGAACGGCGACUGUCCCUCGUGCCCGACAGCGAGCAGGGC
GAGGCCAUCCUGCCCCGGAUCUCCGUGAUCUCCACUGGGCCCACCCUGC
AAGCCCGACGGCGGCAAAGCGUGCUGAACCUGAUGACCCACAGCGUGAA
CCAGGGCCAGAAUAUCCACCGCAAGACUACAGCCAGCACCCGCAAGGUG
AGCCUGGCUCCCCAGGCCAACCUGACCGAGCUGGACAUCUACAGCAGGA
GGCUGAGCCAGGAGACAGGCCUGGAGAUCAGCGAGGAGAUCAACGAGG
AGGACCUGAAGGAGUGCUUCUUCGACGACAUGGAGUCCAUCCCCGCCGU
GACCACCUGGAACACCUACCUGAGAUACAUCACCGUGCACAAGAGCCUG
AUCUUCGUGCUGAUCUGGUGCCUGGUGAUCUUCUUGGCCGAGGUAGCC
GCCUCACUGGUGGUGCUGUGGCUGCUGGGCAAUACCCCACUGCAGGACA
AGGGGAACUCCACCCACAGCCGGAACAACAGCUACGCCGUGAUCAUCAC
CUCCACCAGCAGCUACUACGUGUUCUACAUCUACGUGGGCGUGGCCGAC
ACACUGCUGGCCAUGGGCUUCUUCAGAGGCCUCCCUCUGGUGCACACAC
UGAUCACCGUGAGCAAGAUCCUGCACCACAAGAUGCUGCACAGCGUGCU
GCAGGCUCCCAUGUCAACCCUGAACACCCUGAAGGCCGGCGGCAUCCUG
AACAGGUUCAGCAAGGACAUCGCCAUUCUGGACGAUCUGCUGCCCCUGA
CCAUCUUCGACUUCAUCCAGCUGCUGCUGAUCGUGAUCGGGGCCAUCGC
CGUGGUGGCCGUGCUGCAGCCCUACAUCUUCGUGGCCACAGUGCCCGUG
AUCGUGGCCUUCAUCAUGCUGCGCGCCUACUUCCUGCAGACCUCCCAGC
AGCUGAAGCAGCUGGAGAGCGAAGGCCGCAGCCCCAUCUUCACCCACCU
GGUGACUAGCCUGAAGGGGCUGUGGACCCUGCGCGCCUUCGGCAGGCA
GCCCUACUUCGAGACCCUGUUCCACAAGGCUCUGAACCUGCACACCGCC
AACUGGUUCCUGUACCUCAGCACCCUGCGCUGGUUCCAGAUGCGGAUCG
AGAUGAUCUUCGUGAUCUUCUUCAUCGCCGUGACCUUCAUCUCCAUCCU
GACCACCGGGGAAGGCGAGGGACGGGUGGGAAUCAUCCUGACCCUGGC
CAUGAAUAUCAUGAGCACCCUGCAGUGGGCCGUGAACAGCAGCAUCGA
CGUGGACAGCCUGAUGCGGUCCGUGUCCCGGGUGUUCAAGUUCAUCGA
CAUGCCCACCGAGGGCAAGCCCACCAAGAGCACCAAGCCCUACAAGAAC
GGCCAGCUGAGCAAGGUGAUGAUUAUCGAGAACAGCCACGUGAAGAAG
GACGACAUCUGGCCUUCCGGCGGCCAGAUGACCGUGAAGGACCUGACCG
CCAAGUACACCGAGGGCGGCAACGCCAUCCUGGAGAACAUCAGCUUCUC
CAUUAGCCCUGGACAGCGGGUGGGCCUGUUGGGACGGACCGGCAGUGG
GAAGAGCACCCUGCUGUCCGCCUUCCUGCGGCUGCUGAACACGGAGGGC
GAGAUCCAGAUCGACGGGGUGAGCUGGGACAGCAUCACCCUGCAGCAG
UGGCGAAAGGCCUUCGGCGUGAUCCCACAGAAGGUGUUUAUUUUCUCU
GGCACCUUUAGGAAGAACCUGGACCCCUACGAGCAGUGGUCCGACCAGG
AGAUCUGGAAGGUGGCCGACGAGGUGGGCCUGCGGUCAGUGAUCGAGC
AGUUCCCCGGCAAGCUGGACUUCGUGCUGGUGGACGGCGGCUGCGUGC
UGAGCCACGGCCACAAGCAGCUGAUGUGCCUGGCACGCAGCGUGCUGAG
CAAGGCCAAGAUCCUGCUGCUGGACGAGCCCUCCGCGCACCUGGAUCCC
GUGACCUACCAGAUCAUCAGGCGGACCCUGAAGCAGGCCUUCGCCGACU
GCACCGUGAUCCUGUGCGAGCACAGAAUCGAGGCUAUGCUGGAGUGCC
AGCAGUUCCUGGUGAUCGAGGAGAACAAGGUGCGGCAGUACGACUCCA
UCCAGAAGCUGCUGAACGAGCGCAGCCUGUUCAGACAGGCUAUCUCUCC
CAGCGAUCGCGUGAAGCUGUUCCCUCACAGGAACAGCAGCAAGUGCAA
GUCUAAGCCACAGAUCGCCGCCCUGAAGGAGGAGACCGAGGAGGAGGU
 UGUGAACUAUUGAGAAGAUCGGAACAGCUCCUUACUCUGAGGAAGUUG
UGUGAACUAUUGAGAAGAUCGGAACAGCUCCUUACUCUGAGGAAGUUG
GUACCCCCGUGGUCUUUGAAUAAAGUCUGAGUGGGCGGCAAAAAAAAA
AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA
AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAUCU AGAAAAAAAAAAAAAAAAAAAA-inverted deoxy-thymidine (SEQ ID NO: 13)
Components of the modified hCFTRmRNA of VX-522 are described below.





Single Ascending Dose (SAD) Study:
The SAD study includes two sequential parts.
In Part A, a single dose of VX-522 is administered to each subject on Day 1, with daily follow-up through Day 7, and a clinic visit at Day 14. The starting dose is 1 mg. All subjects transition from Part A to Part B upon completion of the Day 14 Visit.
Part B includes follow-up visits on Day 28 and Week 12, and a Safety Followup Visit 24 (± 1) weeks after Day 1.
The SAD comprises approximately 9 subjects (3 cohorts with 3 subjects in each cohort). Based on a 3 + 3 study design, 3 additional subjects may be added per cohort based on emerging data, resulting in up to 18 subjects total.
Up to 3 additional cohorts (each with a 3 + 3 design as described above) may be added (up to 18 additional subjects, for a total of 36 subjects).
Each cohort will include sequential dosing of subjects. The previous subject’s safety and tolerability data through the Day 7 Visit will be reviewed before the next subject in the cohort is dosed.
Initially, 3 subjects will be treated in each cohort. Safety and tolerability data will be reviewed by the sponsor and the independent data monitoring committee (IDMC) Chair after the third subject completes the Day 7 Visit. If no dose-limiting toxicities (DLTs) are observed in the first 3 subjects in Cohort 1, dose escalation will proceed in a new cohort of 3 subjects. If a DLT is observed in 1 of the 3 subjects, then
the cohort will be expanded to enroll 3 additional subjects (6 in total). If no additional DLTs are reported with the addition of 3 subjects, dose escalation may continue.
If a DLT is observed in 2 or more subjects in the cohort, no additional subjects will be enrolled at that dose level and all subsequent VX-522 dosing will occur at a lower dose level. Previous cohorts may be expanded to 6 subjects, or an intermediate dose may be enrolled with 3 new subjects to identify the VX-522 maximum tolerated dose.
In the event of a death, the IDMC will assess the death’s relationship to VX- 522. Dose escalation may only proceed if there are no deaths attributable to the study drug following review by the IDMC.
The initiation of successive cohorts and dose selection will be based on evaluation of available safety and tolerability data from the previous cohort. If 2 or more subjects in a cohort have dose-limiting toxicity or there is a death attributed to study drug, the dose may be de-escalated (i.e., all subsequent cohorts will evaluate lower doses of VX-522).
Multiple Ascending Dose (MAD) Study:
The starting dose in the MAD study will be at or below dose levels for which safety and tolerability have been established in the SAD study. Subjects who participate in the SAD study may enroll in the MAD study after completion of the Safety Follow-up Visit.
The MAD study includes two treatment arms, T1 and T2.
T1 evaluates multiple ascending doses of VX-522. T1 evaluates MAD of VX- 522 in 2 cohorts with approximately 9 subjects per cohort dosed daily with VX-522 for 28 days and a Safety Follow-up Visit 28 (± 3) days after the last dose of VX-522. Up to 3 additional cohorts may be added, the size of each cohort may be increased or decreased (with a minimum of 6 subjects), and some cohorts may dose VX 522 less frequently than daily dosing, based on emerging data. In each cohort, 1 sentinel subject will be dosed. Before the remaining subjects in the cohort are dosed, the sentinel subject’s safety and tolerability data through the Day 15 Visit (hereafter referred to as the sentinel data) will be reviewed.
T2 evaluates multiple doses of VX-522 while also receiving oral CFTR potentiator (ivacaftor; IVA) treatment. T2 includes 1 cohort of 9 subjects. One additional cohort may be enrolled T2, the size of each cohort may be increased or decreased (with a minimum of 6 subjects), or some cohorts may dose VX-522 less frequently than daily dosing, based on emerging data.
T2 may be conducted concurrently with T1 Cohort 2 after the completion of sentinel data review for T1 Cohort 2. Sentinel data from T1 Cohort 2 must be reviewed before the first subject in T2 receives the first dose of VX-522. Run-in dosing with IVA in T2 may occur before review of the T1 Cohort 2 sentinel data.
Subjects in T2 will receive IVA (150 mg) every 12 hours for 28 days during the Run-in Period. During the Treatment Period, subjects will receive daily VX-522 while also receiving IVA for 28 days. The last dose of IVA will be on the morning of the Day 29 Visit, with a Safety Follow-up Visit 28 (± 3) days after the last dose of VX-522.
CFTR Mutations that are Considered not Responsive to CFTR Modulators
Subjects must have CFTR mutations that are considered not responsive to CFTR modulator therapy on both alleles to be eligible for the study. A CFTR mutation is considered not responsive to CFTR modulator therapy if it meets at least one of the following two criteria:
(1) biological plausibility of no translated protein (genetic sequence predicts the complete absence of CFTR protein); or
(2) in vitro testing that supports lack of responsiveness to ivacaftor, tezacaftor/ivacaftor, and elexacaftor/tezacaftor/ivacaftor. Mutations were considered to be not responsive to these therapies if they met the following criteria in in vitro experiments:
• baseline chloride transport that was <10% of wildtype CFTR; and
• an increase in chloride transport of <10% over baseline following the addition of ivacaftor, tezacaftor/ivacaftor, or elexacaftor/tezacaftor/ivacaftor in the assay.
Table 4 includes acceptable mutations, which are detectable by an FDA- cleared genotyping assay or other method (e.g., sequencing). This list does not include every eligible mutation. Table 4: Eligible CFTR Mutations
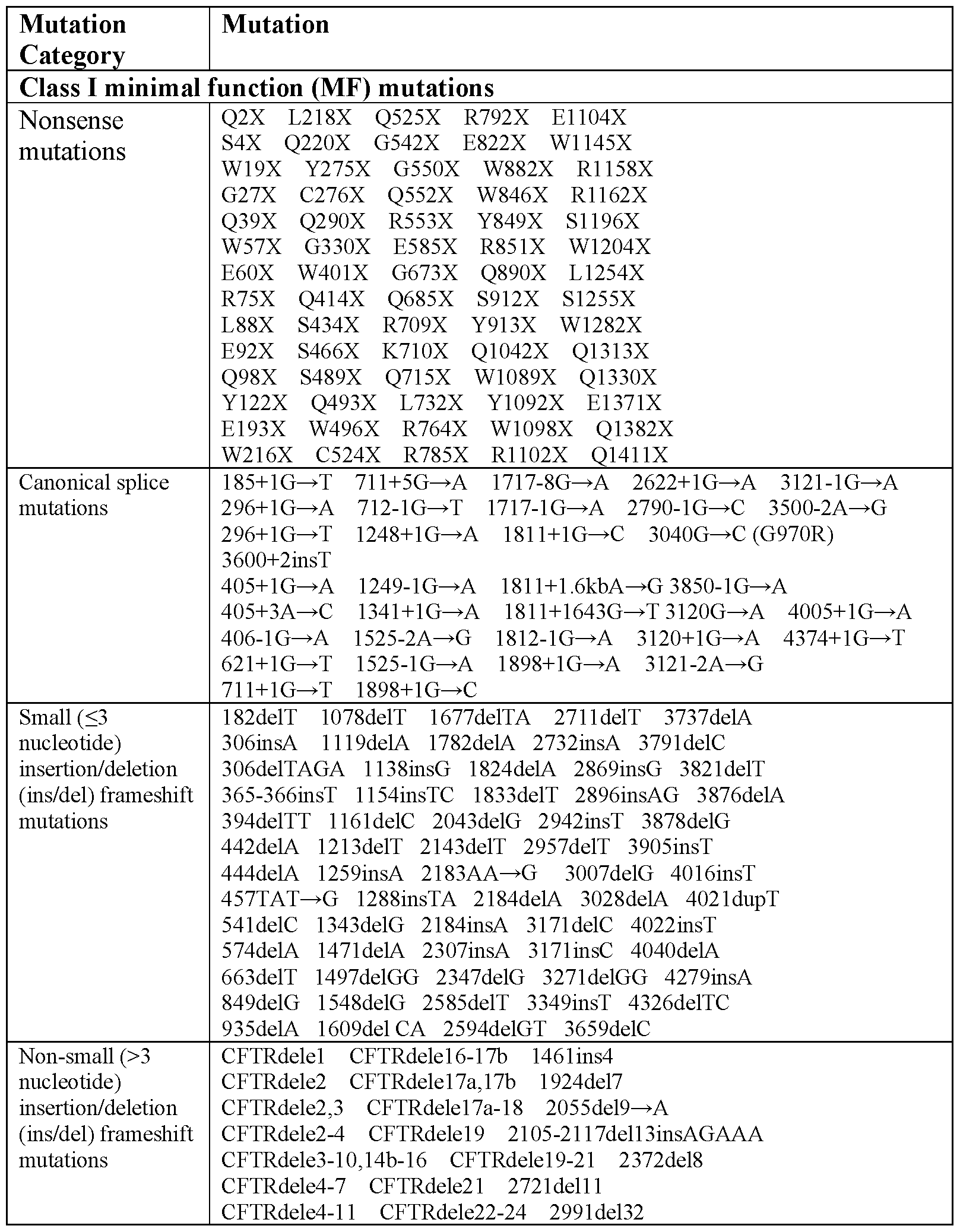
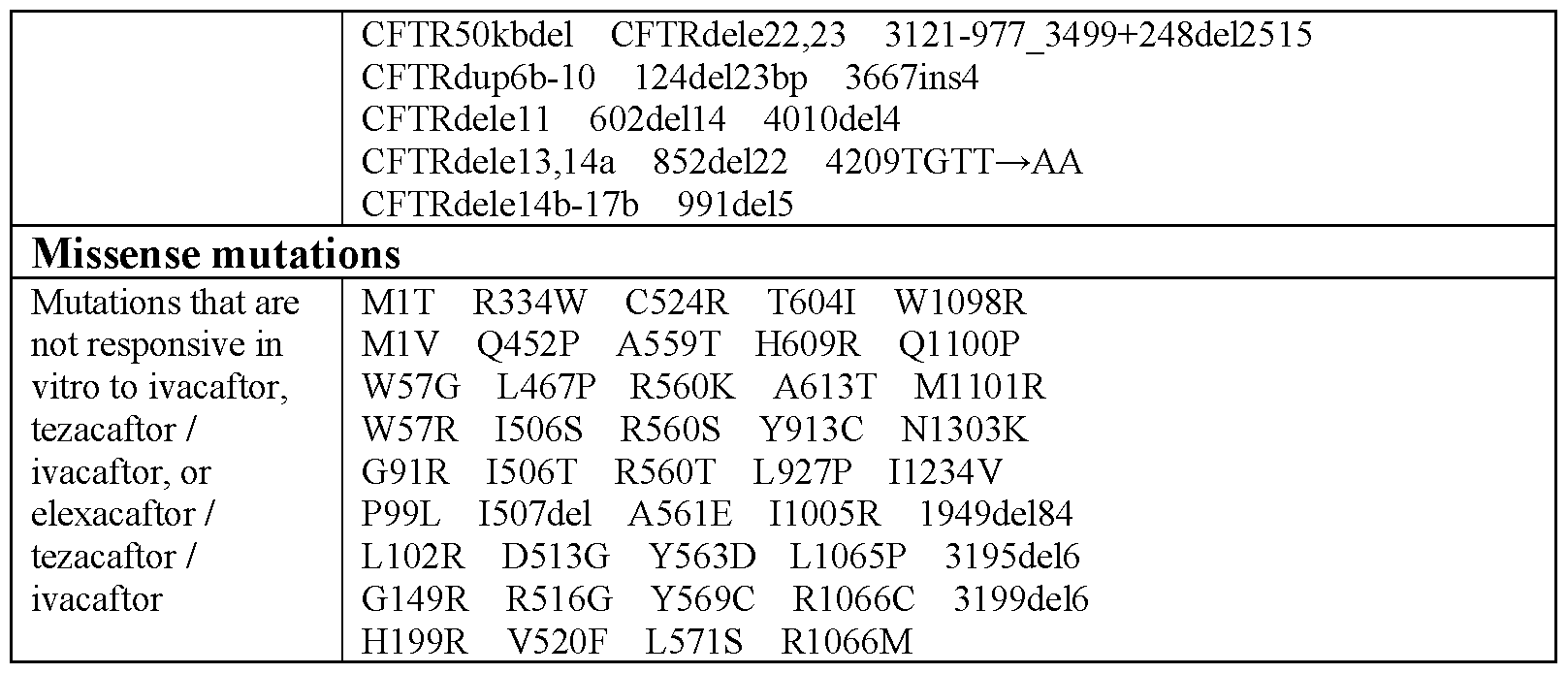
Objectives and Endpoints
The primary objectives are of the study are to evaluate the safety and tolerability of: (1) single ascending doses of VX-522 (SAD); (2) multiple ascending doses of VX-522 (MAD, Tl); and (3) multiple doses of VX-522 coadministered with ivacaftor treatment (MAD, T2). The primary objectives are evaluated by measuring safety and tolerability, based on the assessment of adverse events (AEs), clinical laboratory values (serum chemistry, hematology, coagulation, and urinalysis), standard 12-lead ECGs, vital signs, pulse oximetry, spirometry, and immune response to VX-522 components and CFTR protein.
The secondary objectives of the study are: (1) to evaluate the efficacy of administration of multiple ascending doses of VX-522 (MAD, Tl); and (2) to evaluate the efficacy of multiple doses of VX-522 co-administered with ivacaftor treatment (MAD, T2). The secondary objectives are evaluated by measuring: (1) change from baseline in percent predicted forced expiratory volume in 1 second (ppFEVi) at Day 29 (MAD, Tl); and (2) change from baseline and pre-Run-in baseline in ppFEVi at Day 29 (MAD, T2).
Other objectives of the study are to evaluate systemic exposure of VX-522 components (mRNA and lipid nanoparticle constituents) in blood after: (1) administration of single ascending doses (SAD); (2) administration of multiple ascending doses (MAD, Tl); and (3) multiple doses of VX-522 co-administered with ivacaftor treatment (MAD, T2).
The other objectives are evaluated by measuring: (1) concentrations of VX- 522 components (mRNA and lipid nanoparticle constituents) in blood (SAD); (2) change from baseline in Cystic Fibrosis Questionnaire - Revised respiratory domain (CFQ-R RD) score at Day 29, concentrations of VX-522 components (mRNA and lipid nanoparticle constituents) in blood, exogenous CFTR mRNA in airway tissue (bronchoscopy substudy only), CFTR protein expression in airway tissue (bronchoscopy substudy only), and VX-522 components (mRNA and lipid nanoparticle constituents) in bronchoalveolar lavage fluid (bronchoscopy substudy only) (MAD, Tl); and (3) change from baseline and pre-Run-in baseline in CFQ-R RD score at Day 29, concentrations of VX-522 components (mRNA and LNP constituents) in blood, exogenous CFTR mRNA in airway tissue (bronchoscopy substudy only), CFTR protein expression in airway tissue (bronchoscopy substudy only), VX-522 components (mRNA and LNP constituents) in bronchoalveolar lavage fluid (bronchoscopy substudy only), and change from baseline and pre-Run-in baseline in sweat chloride (SwCl) at Day 29 (MAD, T2).
EXAMPLE 4: Bronchodilators
To minimize possible airway irritation, all subjects will be required to take bronchodilators in the MAD, including a metered dose inhaler (MDI) long-acting P agonist (LABA)Zinhaled corticosteroid (ICS) therapy and short-acting p agonist (SABA) therapy. Table 5 includes examples of acceptable LABA/ICS therapies; however, this list does not include all acceptable medications.
Table 5: Examples of Acceptable LABA/ICS and SABA Therapies
 Run-in Period: All subjects will be dosed with LABA/ICS therapy for at least 28 days in the Run-in Period before the first dose of VX-522. Subjects who are not already taking a LABA/ICS therapy and a SABA before participation in the MAD will be prescribed these therapies before the Run-in Period begins. Subjects who are already taking medications listed in Table 5 before study participation (as part of their routine medical care) will continue those medications and will still be required to complete all Run-in Period visits.
Run-in Period: All subjects will be dosed with LABA/ICS therapy for at least 28 days in the Run-in Period before the first dose of VX-522. Subjects who are not already taking a LABA/ICS therapy and a SABA before participation in the MAD will be prescribed these therapies before the Run-in Period begins. Subjects who are already taking medications listed in Table 5 before study participation (as part of their routine medical care) will continue those medications and will still be required to complete all Run-in Period visits.
Treatment Period: Subjects will be treated with LABA/ICS and SABA therapies before each VX-522 dosing occasion during the Treatment Period.
Safety Follow-up Period: After the Day 43 Visit, subjects may discontinue LABA/ICS dosing, at the discretion of the investigator.
EXAMPLE 5: Pharmacology
CF-HBE cell cultures derived from donor cystic fibrosis lungs are a well- established, clinically validated translational model that predicts the level of clinical benefit associated with improved CFTR function by orally available small molecule CFTR modulators. CFHBE cell cultures display a striking phenotypic and functional resemblance to the surface airway epithelium in people with CF. They form a pseudostratified epithelium composed of secretory cells (goblet and club cells), ciliated cells, ionocytes, and basal cells.
VX-522-mediated CFTR protein expression in lung surface epithelial cells was evaluated in vitro using CF-HBE cell cultures derived from donor cystic fibrosis lungs, and in vivo using monkeys. CF-HBE cells were also used to assess CFTR- mediated chloride transport resulting from VX-522-mediated CFTR protein expression and to benchmark efficacy of VX-522 to CFTR modulators known to provide clinical benefit in people with CF.
VX-522 or a buffer control were nebulized to the apical surface of CF-HBE cell cultures derived from one F508del/MF (F508del/3905insT) and two MF/MF (G542X/R553X and G542X/ K684SfsX38) donor bronchi. G542X and R553X, nonsense mutations, and K684SfsX38, a frameshift mutation, lead to no CFTR protein production or CFTR function. Approximately 18 hours after VX-522
nebulized administration, Ussing chamber electrophysiology was used to directly measure CFTR-mediated chloride transport. In addition to CFTR function, VX-522- mRNA and CFTR protein were assessed in VX-522-nebulized CF-HBE cells by standardized in situ hybridization (ISH) using a VX-522-mRNA-specific RNAScope probe, and immunohistochemistry (IHC) respectively.
Nebulized delivery of VX-522 to F508del/MF-HBE and MF/MF-HBE cells resulted in VX-522-mRNA deposition and CFTR protein expression in multiple epithelial cell types, including ionocytes and goblet cells which are known to endogenously express CFTR. Ionocytes were identified by labeling epithelia with Barttin (BSND), an ionocyte marker, while goblet cells and ciliated cells were identified by morphological analysis.
The increase in % CFTR-positive cells was associated with a dose-dependent increase in CFTR function to levels known to result in clinical benefit in people with CF. The VX-522-mediated increase in CFTR-mediated chloride transport declines over time with an estimated functional half-life of 36 hours, supporting a daily dosing regimen to maintain CFTR function at levels required for clinical benefit in people with CF.
VX-522-mRNA deposition and CFTR protein expression was demonstrated in vivo following daily nebulized delivery of VX-522 to the lung of monkeys via oronasal inhalation for up to 28 days. The increase in the percentage of CFTR proteinexpressing monkey lung epithelial cells was comparable to that observed in CF-HBE cells associated with levels of CFTR function known to result in clinical benefit. VX- 522-mRNA deposition and CFTR protein expression was widely distributed throughout the lung and observed primarily in bronchial surface epithelial cells. The luminal surface of bronchial epithelial cells exhibited strong apical CFTR expression that correlated with VX-522 -mRNA deposition. Histopathological examination showed apical CFTR expression in multiple luminally exposed cell types. No VX- 522-related effects on safety pharmacology parameters were observed following inhalation exposure in vivo up to 19 pg/kg/day.
EXAMPLE 6: In Vitro and In Vivo Pharmacodynamics
CF-HBE In Vitro Cell Model
CF-HBE cell cultures derived from donor cystic fibrosis lungs is a well- established, clinically validated translational model that predicts the level of clinical benefit associated with improved CFTR function by therapeutic intervention. HBE cell cultures display a striking phenotypic and functional resemblance to the conducting airway epithelium. They form pseudostratified epithelium composed of secretory cells (goblet and club cells), ciliated cells, ionocytes, and progenitor cells, lonocytes express the highest levels of CFTR but are rare (approximately 1% of airway epithelial cells). The more abundant secretory cells express low to medium levels of CFTR.
Buffer or VX-522 was nebulized to the apical surface of CF-HBE cell cultures derived from 1 F50SJe//minimal function (MF) (F508del/3905insT) and 2 MF/MF (G542X/R553X and G542X/ K684SfsX38) donor bronchi. G542X and R553X (nonsense mutations) and K684SfsX38 (a frameshift mutation) lead to no protein production and minimal CFTR function. Following VX-522 nebulization, VX-522- mRNA, CFTR protein, and CFTR function were typically analyzed approximately 18 hours after treatment by standardized in situ hybridization (ISH), immunohistochemistry (IHC), and Ussing chamber electrophysiology methods, respectively.
In the absence of VX-522, little to no chloride transport was observed in F50&fe//MF-HBE or MF/MF-HBE cells, a finding consistent with minimal CFTR function and severe disease in people with cystic fibrosis with these CFTR genotypes. As previously shown, clinically relevant concentrations of elexacaftor/tezacaftor/ivacaftor (ELX/TEZ/IVA [TRI]) increased CFTR function in F50&fe//MF-HBE cells to levels known to provide high levels of clinical benefit in people with cystic fibrosis who carry at least one F508del CFTR variant. In contrast, no response was observed in MF/MF-HBE cells, as expected due to the lack of CFTR protein for ELX/TEZ/IVA to act on.
Nebulization of VX-522 to the apical surface of F50&/e//MF-HBE and MF/MF-HBE cells resulted in delivery of VX-522-mRNA and exogenous expression
of CFTR protein in multiple epithelial cell types, including those known to endogenously express CFTR (Figs. 3A-3C). In both MF/MF-HBE cells (Fig. 3D) and F50&fe//MF-HBE cells (Fig. 3E), this increase in CFTR expression was associated with a dose-dependent increase in CFTR function to levels comparable to ELX/TEZ/IVA treatment in F508 de//MF-HBE cells (Fig. 3E). This level of CFTR function in the HBE cell assay is known to result in clinical benefit in people with cystic fibrosis.
Following a single nebulized treatment of CF-HBE cells with VX-522, CFTR function returned to baseline levels with a half-life of approximately 36 hours, supporting a daily dosing regimen to sustain CFTR-mediated chloride transport.
In Vivo Pharmacodynamics
To demonstrate in vivo proof of VX-522-mRNA delivery to airway epithelial cells that results in increased CFTR protein expression, a nebulized formulation of VX-522 or buffer control was administered daily to monkeys (cynomolgus macaques) via oronasal mask inhalation for 1 to 28 days using vibrating mesh nebulizers. VX- 522-mRNA delivery was assessed by ISH using a VX-522-mRNA-specific RNAScope probe. CFTR protein expression was assessed by IHC using an anti-CFTR antibody that detects human and monkey CFTR.
After daily administration of nebulized VX-522 to monkeys for 14 or 28 days, exogenous VX-522-mRNA and CFTR protein expression was widely distributed throughout the lung and observed primarily in surface epithelial cells (Figs. 4A and 4B). After 28 days of daily VX-522 administration, the percent of total bronchial epithelial cells expressing CFTR increased by 8% to 11% increase over buffer-treated controls in bronchial epithelial cells expressing high levels of CFTR protein (similar to levels seen in ionocytes) (Fig. 4B). These levels of increased CFTR proteinpositive epithelial cells were similar to those observed in CF-HBE cells that resulted in levels of CFTR function known to result in clinical benefit (Figs. 3A-3E).
The increase in CFTR protein-positive epithelial cells following daily delivery of VX-522 for 14 days was similar to or better than a single treatment, suggesting that VX-522 results in durable CFTR protein expression in vivo. After a 28-day recovery
period there was no difference in CFTR protein-positive bronchial epithelial cells between monkeys dosed with VX-522 and control monkeys dosed with buffer, confirming the transient effects of VX-522.
EXAMPLE 7: VX-522 in Combination with Ivacaftor
Buffer, VX-522, or VX-522 in combination with clinically relevant concentrations of ivacaftor was nebulized to the apical surface of CF-HBE cell cultures derived from two MF/MF (G542X/R553X and G542X/ K684SfsX38) donor bronchi. MF mutations such as G542X, R553X, and K684SfsX38, lead to no CFTR protein production or CFTR function. Approximately 18 hours following nebulization, CFTR-mediated chloride transport was quantified by Ussing chamber electrophysiology .
In MF/MF-HBE cells, there was no response to treatment with clinically relevant concentrations of elexacaftor/tezacaftor/ivacaftor, a finding consistent with the lack of CFTR protein production in MF/MF-HBE cells. In contrast, nebulized delivery of VX-522 to MF/MF-HBE cells resulted in a dose-dependent increase in CFTR-mediated chloride transport 18 hours after a single treatment of 2.1 to 6.1 pg/cm2 VX-522 nebulized to the apical surface (Fig. 5). Treatment with VX-522 in combination with ivacaftor resulted in a greater increase in CFTR function than with VX-522 alone (Fig. 5).
These results demonstrate that aerosolized delivery of VX-522 can increase Cl" current in MF/MF CF-HBE cells and that the addition of ivacaftor can further increase the VX-522-mediated increase in Cl" current. VX-522 in combination with ivacaftor increased Cl" currents in MF/MF CF-HBE cells to levels similar to or above those induced by elexacaftor/tezacaftor/ivacaftor in F508del/MF CF-HBE cells, levels that have previously been shown to translate into clinically meaningful benefits for cystic fibrosis patients. These results suggest that VX-522 may provide clinical benefit in people with cystic fibrosis caused by minimal function mutations on both alleles that are otherwise unresponsive to CFTR modulators and combining VX-522 with ivacaftor may provide additional clinical benefit versus VX-522 monotherapy.
EXAMPLE 8: Human Dose Selection
The selection of the starting nominal dose of 1 mg was based on the totality of nonclinical toxicology and pharmacology data, in accordance with EMA and FDA guidance on first-in-human (FIH) starting dose selection. Overall, the nonclinical data demonstrate that nebulized VX-522: (1) produces functional CFTR protein when administered to HBEs from cystic fibrosis donors; (2) can deliver VX-522-mRNA to monkey lung epithelial cells that results in increased CFTR protein expression at doses well below the no-observed-adverse-effect-level (NOAEL); (3) have a concordant safety profile in rats and monkeys consisting of non-adverse changes that are consistent with a local adaptive inflammatory response to an inhaled foreign agent; and (4) are effective for local delivery of VX-522 components, with negligible systemic exposure and systemic toxicity risk.
To calculate a safe FIH starting dose, the estimated achieved pulmonary (deposited) doses from the 28-day good laboratory practice (GLP) rat and monkey studies were converted from a pg/kg body weight dose to a pg/g lung tissue dose. This conversion was conducted because (1) the lung is the pharmacological target organ; (2) there was no evidence of systemic toxicity associated with VX-522 in either species; and (3) VX-522-related non-adverse changes were limited to the lung and associated lymphoid tissue in both species. This approach is consistent with recommendations in the literature. At the NOAELs, the achieved pulmonary (deposited) doses were 2.9 pg/g lung (rat) and 4.12 pg/g lung (monkeys). The monkey was considered as the most relevant species for subsequent calculations of the safe starting dose in the FIH study because human respiratory physiology, airway geometry, and cellular composition of the lower respiratory tract is more similar to monkeys than rats. The deposition fraction of inhaled aerosols is assumed to be the same for monkeys and human. Furthermore, proof of delivery of hCFTR mRNA and CFTR protein was demonstrated in monkeys following oronasal inhalation of VX- 522. Additionally, hCFTR is more similar to monkey CFTR than rat CFTR (based on peptide sequence), which may have influenced the immunogenicity profile of VX-522 in vivo.
The achieved pulmonary (deposited) dose at the NOAEL in monkeys of 4.12 pg/g lung was multiplied by the weight of a human lung (assumed to be 1000 g in a 60 kg human) to derive a 4.12 mg achieved pulmonary (deposited) human dose.
The PARI nebulizer utilizing eFlow technology was demonstrated to deliver approximately 40% of the nominal VX-522 dose into the lung. Hence, the human equivalent pulmonary dose of 4. 12 mg at the NOAEL in monkeys was divided by 0.40 to account for the fraction of delivery and deposition (device efficiency) using the PARI nebulizer, leading to the human equivalent nominal dose of 10 mg. An approximate 10-fold safety margin was applied to the 10 mg nominal dose to arrive at the 1 mg nominal safe starting FIH dose.
To evaluate the potential efficacious dose range, two independent approaches were used to estimate the range of nominal doses of VX-522 that would be reasonably likely to result in clinical benefit. The two independent approaches — lung deposition modeling based on CFTR functional response in human bronchial epithelial (HBE) cells derived from the lungs of people with cystic fibrosis, and lung-weight based scaling from NHP — gave similar and consistent estimates that inform the proposed clinical doses.
First, lung deposition modeling was used to calculate the nominal dose needed to achieve the equivalent amount of VX-522 at the lung surface that resulted in Trikafta (TRI)-like levels of CFTR function in F508del/minimal function-HBE cells. The CF-HBE cell model, and the comparison to Trikafta, were selected as benchmarks based on the well-established translation of this assay and treatment to clinical benefit. This approach estimated a daily nominal dose of 6 to 12 mg of VX- 522 would be efficacious.
Second, lung-weight-based scaling was used to calculate the human equivalent dose that would correspond to the deposited dose in monkeys that led to a 2% to 4% increase in the proportion of CFTR protein-positive epithelial cells. The 2% to 4% increase in CFTR-protein-positive cells is based on the proportion of CFTR proteinpositive cells observed in VX-522 -treated CF-HBE cells that resulted in CFTR- mediated chloride transport increases known to translate into clinical benefit. This
approach estimated a daily nominal dose of approximately 1 mg of VX-522 would be efficacious.
Both independent approaches gave consistent estimates, suggesting that the proposed nominal daily dose range (starting dose of 1 mg to maximum dose of 7 mg) would be reasonably likely to result in clinical benefit.
The clinical plan for Phase 1/2 is to dose into the predicted clinically efficacious range, with a maximum planned nominal dose of 7 mg. The monkey NOAEL provides an approximate 1.5-fold safety margin over 7 mg clinical nominal dose.
EXAMPLE 9: Selection of VX-522 Starting Dose and Estimation of the Anticipated Therapeutic Dose Range
The selection of the starting nominal dose was based on the totality of nonclinical toxicology and pharmacology data, in accordance with EMA and FDA guidance on FIH starting dose selection. The starting dose was selected based on NOAEL established for monkeys in the GLP-compliant non-clinical safety studies. In addition, the clinically efficacious dose range was estimated to provide assurance that the starting dose and maximum dose will be appropriate for evaluation in a Phase 1/Phase 2 study of patients with cystic fibrosis.
The starting nominal dose of 1 mg was selected as follows:
(1) NOAELs were established in GLP-compliant nonclinical safety studies of VX-522 dosing in monkeys for 28 days (19 pg/kg/day).
(2) To adjust for differences in lung physiology between animal models and humans, the NOAELs were first converted into human equivalent lung-deposited doses using lung weight scaling. The monkey NOAEL of 19 pg/kg/day corresponds to a human equivalent lung -deposited dose of 4. 1 mg.
(3) The amount of VX-522 required in the nebulizer (called the “nominal dose”) to achieve this human-equivalent lung dose was then calculated based on nebulizer efficiency measured with specific configuration for VX-522. A dose of 10 mg in the nebulizer (nominal dose) is predicted to result in a lung-deposited dose of 4.1 mg.
(4) An exposure margin of approximately 10-fold relative to the human equivalent nominal dose of the monkey NOAEL was applied to select the starting (nominal) dose of 1 mg.
The planned maximum dose will be 7 mg nominal dose, which is predicted to provide an approximately 1.5-fold safety margin under the human equivalent of the monkey NOAEL.
To evaluate the potential efficacious dose range, two independent approaches were used to estimate the range of nominal doses of VX-522 that would be reasonably likely to result in clinical benefit. The two independent approaches — lung deposition modeling based on CFTR functional response in human bronchial epithelial (HBE) cells derived from the lungs of people with cystic fibrosis, and lung-weight based scaling from NHP — gave similar and consistent estimates that inform the proposed clinical doses. Specifically:
(1) Lung deposition modeling was used to calculate the nominal dose needed to achieve the equivalent amount of VX-522 at the lung surface that resulted in Trikafta (TRI)-like levels of CFTR function in F508de //minimal function-HBE cells. The CF-HBE cell model, and the comparison to Trikafta, were selected as benchmarks based on the well-established translation of this assay and treatment to clinical benefit. This approach estimated a daily nominal dose of 6 to 12 mg of VX- 522 would be efficacious.
(2) Lung-weight-based scaling was used to calculate the human equivalent dose that would correspond to the deposited dose in monkeys that led to a 2% to 4% increase in the proportion of CFTR protein-positive epithelial cells. The 2% to 4% increase in CFTR-protein-positive cells is based on the proportion of CFTR proteinpositive cells observed in VX-522 -treated CF-HBE cells that resulted in CFTR- mediated chloride transport increases known to translate into clinical benefit. This approach estimated a daily nominal dose of approximately 1 mg of VX-522 would be efficacious.
Both independent approaches gave consistent estimates, suggesting that the proposed nominal daily dose range (starting dose of 1 mg to maximum dose of 7 mg) would be reasonably likely to result in clinical benefit.
Claims
1. A method of treating cystic fibrosis in a human subject in need thereof, the method comprising administering to the human subject by inhalation a lipid nanoparticle comprising a messenger RNA (mRNA) comprising an open reading frame (ORF) encoding the cystic fibrosis transmembrane conductance regulator (CFTR) polypeptide of SEQ ID NO: 1, wherein the mRNA is administered at a dose of 1 mg to 7 mg.
2. The method of claim 1, wherein the ORF is at least 80% identical to the nucleotide sequence of SEQ ID NO: 10.
3. The method of claim 1, wherein the ORF is at least 95% identical to the nucleotide sequence of SEQ ID NO: 10.
4. The method of claim 1, wherein the ORF is at least 99% identical to the nucleotide sequence of SEQ ID NO: 10.
5. The method of claim 1, wherein the ORF is 100% identical to the nucleotide sequence of SEQ ID NO: 10.
6. The method of any one of claims 1 to 5, wherein the mRNA comprises a 5' UTR comprising the nucleotide sequence of SEQ ID NO:50.
7. The method of any one of claims 1 to 6, wherein the mRNA comprises a 3' UTR comprising the nucleotide sequence of SEQ ID NO: 141.
8. The method of claim 1, wherein the mRNA comprises the nucleic acid sequence of SEQ ID NO: 11.
9. The method of any one of claims 1 to 8, wherein the mRNA comprises a 5' terminal cap.
10. The method of claim 9, wherein the 5' terminal cap comprises m7G-ppp- Gm.
11. The method of any one of claims 1 to 10, wherein the mRNA comprises a poly-A region.
12. The method of claim 11, wherein the poly-A region comprises SEQ ID NO: 195.
13. The method of claim 11, wherein the poly-A region comprises A 100- UCUAG-A20-inverted deoxy-thymidine (SEQ ID NO:211).
14. The method of any one of claims 1 to 13, wherein all of the uracils of the mRNA are N1 -methylpseudouracils.
15. The method of claim 1, wherein the mRNA comprises the nucleotide sequence of SEQ ID NO : 13.
16. The method of any one of claims 1 to 15, wherein the mRNA is administered at a dose of 1 mg.
17. The method of any one of claims 1 to 15, wherein the mRNA is administered at a dose of 2 mg.
18. The method of any one of claims 1 to 15, wherein the mRNA is administered at a dose of 3 mg.
19. The method of any one of claims 1 to 15, wherein the mRNA is administered at a dose of 4 mg.
20. The method of any one of claims 1 to 15, wherein the mRNA is administered at a dose of 5 mg.
21. The method of any one of claims 1 to 15, wherein the mRNA is administered at a dose of 6 mg.
22. The method of any one of claims 1 to 15, wherein the mRNA is administered at a dose of 7 mg.
23. The method of any one of claims 1 to 22, comprising multiple administrations of the dose.
24. The method of any one of claims 1 to 23, wherein the dose is administered repeatedly once every day.
25. The method of claim 24, comprising at least 28 consecutive daily administrations of the dose.
26. The method of any one of claims 1 to 25, wherein the lipid nanoparticle is administered using a nebulizer.
27. The method of any one of claims 1 to 26, wherein the human subject carries mutations in both alleles of the CFTR gene that result in no CFTR protein produced or a mutant CFTR protein that is not responsive to therapy with CFTR modulators.
28. The method of claim 27, wherein the CFTR modulators comprise ivacaftor, lumacaftor/ivacaftor, tezacaftor/ivacaftor, and elexacaftor/tezacaftor/ivacaftor.
29. The method of claim 27, wherein the mutations in both alleles of the CFTR gene are selected from the mutations depicted in Table 4.
30. The method of any one of claims 1 to 29, comprising co-administering a CFTR potentiator to the human subject.
31. The method of claim 30, wherein the CFTR potentiator is ivacaftor, lumacaftor/ivacaftor, tezacaftor/ivacaftor, or elexacaftor/tezacaftor/ivacaftor.
32. The method of claim 30, wherein the CFTR potentiator is ivacaftor.
33. The method of claim 32, wherein ivacaftor is administered orally.
34. The method of claim 32, wherein ivacaftor is administered orally every 12 hours at a dose of 150 mg.
35. The method of any one of claims 1 to 14, wherein the mRNA is administered once every day at a dose of 1 mg.
36. The method of any one of claims 1 to 14, wherein the mRNA is administered once every day at a dose of 1 mg and wherein ivacaftor is administered orally.
37. The method of any one of claims 1 to 14, wherein the mRNA is administered once every day at a dose of 1 mg and wherein ivacaftor is administered orally every 12 hours at a dose of 150 mg.
38. The method of any one of claims 1 to 14, wherein the mRNA is administered once every day at a dose of 2 mg.
39. The method of any one of claims 1 to 14, wherein the mRNA is administered once every day at a dose of 2 mg and wherein ivacaftor is administered orally.
40. The method of any one of claims 1 to 14, wherein the mRNA is administered once every day at a dose of 2 mg and wherein ivacaftor is administered orally every 12 hours at a dose of 150 mg.
41. The method of any one of claims 1 to 14, wherein the mRNA is administered once every day at a dose of 3 mg.
42. The method of any one of claims 1 to 14, wherein the mRNA is administered once every day at a dose of 3 mg and wherein ivacaftor is administered orally.
43. The method of any one of claims 1 to 14, wherein the mRNA is administered once every day at a dose of 3 mg and wherein ivacaftor is administered orally every 12 hours at a dose of 150 mg.
44. The method of any one of claims 1 to 14, wherein the mRNA is administered once every day at a dose of 4 mg.
45. The method of any one of claims 1 to 14, wherein the mRNA is administered once every day at a dose of 4 mg and wherein ivacaftor is administered orally.
46. The method of any one of claims 1 to 14, wherein the mRNA is administered once every day at a dose of 4 mg and wherein ivacaftor is administered orally every 12 hours at a dose of 150 mg.
47. The method of any one of claims 1 to 14, wherein the mRNA is administered once every day at a dose of 5 mg.
48. The method of any one of claims 1 to 14, wherein the mRNA is administered once every day at a dose of 5 mg and wherein ivacaftor is administered orally.
49. The method of any one of claims 1 to 14, wherein the mRNA is administered once every day at a dose of 5 mg and wherein ivacaftor is administered orally every 12 hours at a dose of 150 mg.
50. The method of any one of claims 1 to 14, wherein the mRNA is administered once every day at a dose of 6 mg.
51. The method of any one of claims 1 to 14, wherein the mRNA is administered once every day at a dose of 6 mg and wherein ivacaftor is administered orally.
52. The method of any one of claims 1 to 14, wherein the mRNA is administered once every day at a dose of 6 mg and wherein ivacaftor is administered orally every 12 hours at a dose of 150 mg.
53. The method of any one of claims 1 to 14, wherein the mRNA is administered once every day at a dose of 7 mg.
54. The method of any one of claims 1 to 14, wherein the mRNA is administered once every day at a dose of 7 mg and wherein ivacaftor is administered orally.
55. The method of any one of claims 1 to 14, wherein the mRNA is administered once every day at a dose of 7 mg and wherein ivacaftor is administered orally every 12 hours at a dose of 150 mg.
56. The method of claim 15, wherein the mRNA is administered once every day at a dose of 1 mg.
57. The method of claim 15, wherein the mRNA is administered once every day at a dose of 1 mg and wherein ivacaftor is administered orally.
58. The method of claim 15, wherein the mRNA is administered once every day at a dose of 1 mg and wherein ivacaftor is administered orally every 12 hours at a dose of 150 mg.
59. The method of claim 15, wherein the mRNA is administered once every day at a dose of 2 mg.
60. The method of claim 15, wherein the mRNA is administered once every day at a dose of 2 mg and wherein ivacaftor is administered orally.
61. The method of claim 15, wherein the mRNA is administered once every day at a dose of 2 mg and wherein ivacaftor is administered orally every 12 hours at a dose of 150 mg.
62. The method of claim 15, wherein the mRNA is administered once every day at a dose of 3 mg.
63. The method of claim 15, wherein the mRNA is administered once every day at a dose of 3 mg and wherein ivacaftor is administered orally.
64. The method of claim 15, wherein the mRNA is administered once every day at a dose of 3 mg and wherein ivacaftor is administered orally every 12 hours at a dose of 150 mg.
65. The method of claim 15, wherein the mRNA is administered once every day at a dose of 4 mg.
66. The method of claim 15, wherein the mRNA is administered once every day at a dose of 4 mg and wherein ivacaftor is administered orally.
67. The method of claim 15, wherein the mRNA is administered once every day at a dose of 4 mg and wherein ivacaftor is administered orally every 12 hours at a dose of 150 mg.
68. The method of claim 15, wherein the mRNA is administered once every day at a dose of 5 mg.
69. The method of claim 15, wherein the mRNA is administered once every day at a dose of 5 mg and wherein ivacaftor is administered orally.
70. The method of claim 15, wherein the mRNA is administered once every day at a dose of 5 mg and wherein ivacaftor is administered orally every 12 hours at a dose of 150 mg.
71. The method of claim 15, wherein the mRNA is administered once every day at a dose of 6 mg.
72. The method of claim 15, wherein the mRNA is administered once every day at a dose of 6 mg and wherein ivacaftor is administered orally.
73. The method of claim 15, wherein the mRNA is administered once every day at a dose of 6 mg and wherein ivacaftor is administered orally every 12 hours at a dose of 150 mg.
74. The method of claim 15, wherein the mRNA is administered once every day at a dose of 7 mg.
75. The method of claim 15, wherein the mRNA is administered once every day at a dose of 7 mg and wherein ivacaftor is administered orally.
76. The method of claim 15, wherein the mRNA is administered once every day at a dose of 7 mg and wherein ivacaftor is administered orally every 12 hours at a dose of 150 mg.
77. The method of any one of claims 1 to 76, wherein the treatment increases percent predicted forced expiratory volume in 1 second (ppFEVi) from baseline.
78. The method of any one of claims 1 to 77, wherein the human subject is administered long-acting p agonist (LABA)Zinhaled corticosteroid (ICS) therapy.
79. The method of claim 78, wherein the human subject is administered LABA/ICS therapy prior to administration of the lipid nanoparticle.
80. The method of claim 78, wherein the human subject is administered LABA/ICS therapy for at least 28 days before the first administration of the lipid nanoparticle.
81. The method of any one of claims 1 to 80, wherein the human subject is administered LABA/ICS therapy and short-acting p agonist (SABA) therapy.
82. The method of claim 81, wherein the human subject is administered LABA/ICS therapy and SABA therapy prior to administration of the lipid nanoparticle.
83. The method of claim 81, wherein the human subject is administered LABA/ICS therapy and SABA therapy on the same day as administration of the lipid nanoparticle.
84. The method of any one of claims 1 to 83, wherein the lipid nanoparticle comprises:
(i) an ionizable lipid,
(ii) a phospholipid;
(iii) a structural lipid;
(iv) a PEG-lipid; and
(v) a cationic agent.
85. The method of claim 84, wherein the ionizable lipid is
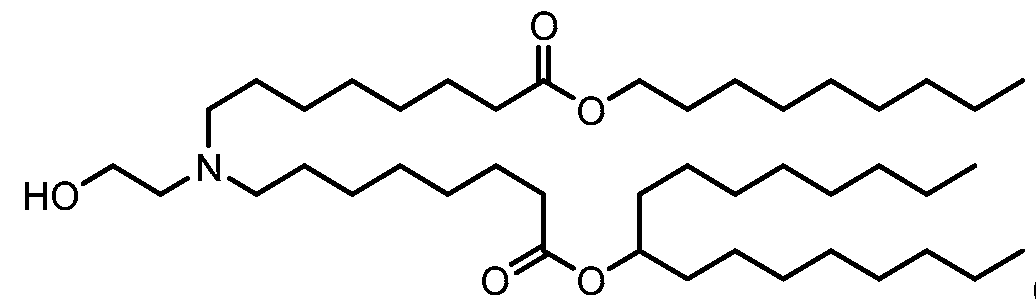 (Compound II) or a salt thereof.
(Compound II) or a salt thereof.
86. The method of claim 84, wherein the cationic agent is

87. The method of claim 84, wherein the ionizable lipid is
 (Compound II) or a salt thereof, and the cationic agent is
(Compound II) or a salt thereof, and the cationic agent is
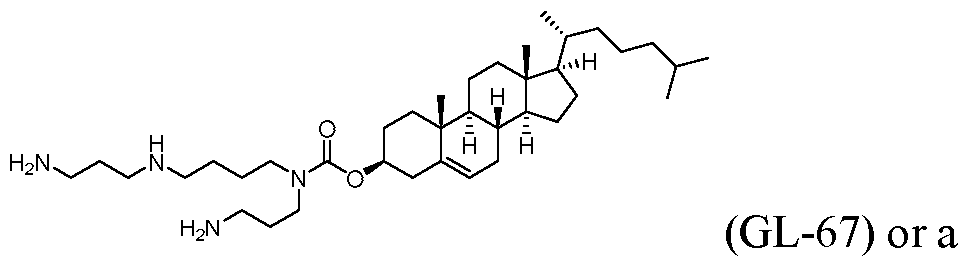 salt thereof.
salt thereof.
88. The method of claim 84, wherein
(i) the ionizable lipid is
 (Compound II) or a salt thereof;
(Compound II) or a salt thereof;
(ii) the phospholipid is 1,2 distearoyl-sn-glycero-3-phosphocholine (DSPC);
(iii) the structural lipid is cholesterol;
(iv) the PEG lipid is l,2-dimyristoyl-rac-glycero-3 -methoxypolyethylene glycol-2000 (PEG2000-DMG); and
(v) the cationic agent is

Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US202363601353P | 2023-11-21 | 2023-11-21 | |
| US63/601,353 | 2023-11-21 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2025111297A1 true WO2025111297A1 (en) | 2025-05-30 |
Family
ID=93893793
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2024/056591 Pending WO2025111297A1 (en) | 2023-11-21 | 2024-11-20 | Polynucleotides encoding cystic fibrosis transmembrane conductance regulator for the treatment of cystic fibrosis |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2025111297A1 (en) |
Citations (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20050222064A1 (en) | 2002-02-20 | 2005-10-06 | Sirna Therapeutics, Inc. | Polycationic compositions for cellular delivery of polynucleotides |
| US7404969B2 (en) | 2005-02-14 | 2008-07-29 | Sirna Therapeutics, Inc. | Lipid nanoparticle based compositions and methods for the delivery of biologically active molecules |
| US20100129877A1 (en) | 2005-09-28 | 2010-05-27 | Ugur Sahin | Modification of RNA, Producing an Increased Transcript Stability and Translation Efficiency |
| US20100293625A1 (en) | 2007-09-26 | 2010-11-18 | Interexon Corporation | Synthetic 5'UTRs, Expression Vectors, and Methods for Increasing Transgene Expression |
| US8158601B2 (en) | 2009-06-10 | 2012-04-17 | Alnylam Pharmaceuticals, Inc. | Lipid formulation |
| WO2012099755A1 (en) | 2011-01-11 | 2012-07-26 | Alnylam Pharmaceuticals, Inc. | Pegylated lipids and their use for drug delivery |
| US8257685B2 (en) | 2006-04-04 | 2012-09-04 | Stc.Unm | Swellable particles for drug delivery |
| WO2012170889A1 (en) | 2011-06-08 | 2012-12-13 | Shire Human Genetic Therapies, Inc. | Cleavable lipids |
| WO2013086354A1 (en) | 2011-12-07 | 2013-06-13 | Alnylam Pharmaceuticals, Inc. | Biodegradable lipids for the delivery of active agents |
| WO2013116126A1 (en) | 2012-02-01 | 2013-08-08 | Merck Sharp & Dohme Corp. | Novel low molecular weight, biodegradable cationic lipids for oligonucleotide delivery |
| US8519110B2 (en) | 2008-06-06 | 2013-08-27 | Board Of Supervisors Of Louisiana State University And Agricultural And Mechanical College | mRNA cap analogs |
| WO2014164253A1 (en) | 2013-03-09 | 2014-10-09 | Moderna Therapeutics, Inc. | Heterologous untranslated regions for mrna |
| US20150036759A1 (en) | 2013-08-03 | 2015-02-05 | Gregory Hubert Piesinger | COFDM Using Pseudo Orthogonal QPSK Coding |
| WO2015130584A2 (en) | 2014-02-25 | 2015-09-03 | Merck Sharp & Dohme Corp. | Lipid nanoparticle vaccine adjuvants and antigen delivery systems |
| WO2017051367A1 (en) | 2015-09-24 | 2017-03-30 | Medidata Sp. Z O.O. | Cryoapplicator for minimally invasive surgical cardiac ablation |
| WO2017066797A1 (en) | 2015-10-16 | 2017-04-20 | Modernatx, Inc. | Trinucleotide mrna cap analogs |
| WO2018213476A1 (en) * | 2017-05-16 | 2018-11-22 | Translate Bio, Inc. | Treatment of cystic fibrosis by delivery of codon-optimized mrna encoding cftr |
| WO2020106946A1 (en) * | 2018-11-21 | 2020-05-28 | Translate Bio, Inc. | TREATMENT OF CYSTIC FIBROSIS BY DELIVERY OF NEBULIZED mRNA ENCODING CFTR |
| WO2020160397A1 (en) | 2019-01-31 | 2020-08-06 | Modernatx, Inc. | Methods of preparing lipid nanoparticles |
| WO2022104131A1 (en) | 2020-11-13 | 2022-05-19 | Modernatx, Inc. | Polynucleotides encoding cystic fibrosis transmembrane conductance regulator for the treatment of cystic fibrosis |
| WO2023009422A1 (en) | 2021-07-26 | 2023-02-02 | Modernatx, Inc. | Processes for preparing lipid nanoparticle compositions for the delivery of payload molecules to airway epithelium |
| WO2023081776A1 (en) * | 2021-11-03 | 2023-05-11 | Arcturus Therapeutics, Inc. | Lipid formulations containing nucleic acids and methods of treatment for cystic fibrosis |
-
2024
- 2024-11-20 WO PCT/US2024/056591 patent/WO2025111297A1/en active Pending
Patent Citations (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20050222064A1 (en) | 2002-02-20 | 2005-10-06 | Sirna Therapeutics, Inc. | Polycationic compositions for cellular delivery of polynucleotides |
| US7404969B2 (en) | 2005-02-14 | 2008-07-29 | Sirna Therapeutics, Inc. | Lipid nanoparticle based compositions and methods for the delivery of biologically active molecules |
| US20100129877A1 (en) | 2005-09-28 | 2010-05-27 | Ugur Sahin | Modification of RNA, Producing an Increased Transcript Stability and Translation Efficiency |
| US8257685B2 (en) | 2006-04-04 | 2012-09-04 | Stc.Unm | Swellable particles for drug delivery |
| US20100293625A1 (en) | 2007-09-26 | 2010-11-18 | Interexon Corporation | Synthetic 5'UTRs, Expression Vectors, and Methods for Increasing Transgene Expression |
| US8519110B2 (en) | 2008-06-06 | 2013-08-27 | Board Of Supervisors Of Louisiana State University And Agricultural And Mechanical College | mRNA cap analogs |
| US8158601B2 (en) | 2009-06-10 | 2012-04-17 | Alnylam Pharmaceuticals, Inc. | Lipid formulation |
| WO2012099755A1 (en) | 2011-01-11 | 2012-07-26 | Alnylam Pharmaceuticals, Inc. | Pegylated lipids and their use for drug delivery |
| WO2012170889A1 (en) | 2011-06-08 | 2012-12-13 | Shire Human Genetic Therapies, Inc. | Cleavable lipids |
| WO2013086354A1 (en) | 2011-12-07 | 2013-06-13 | Alnylam Pharmaceuticals, Inc. | Biodegradable lipids for the delivery of active agents |
| WO2013116126A1 (en) | 2012-02-01 | 2013-08-08 | Merck Sharp & Dohme Corp. | Novel low molecular weight, biodegradable cationic lipids for oligonucleotide delivery |
| WO2014164253A1 (en) | 2013-03-09 | 2014-10-09 | Moderna Therapeutics, Inc. | Heterologous untranslated regions for mrna |
| US20150036759A1 (en) | 2013-08-03 | 2015-02-05 | Gregory Hubert Piesinger | COFDM Using Pseudo Orthogonal QPSK Coding |
| WO2015130584A2 (en) | 2014-02-25 | 2015-09-03 | Merck Sharp & Dohme Corp. | Lipid nanoparticle vaccine adjuvants and antigen delivery systems |
| WO2017051367A1 (en) | 2015-09-24 | 2017-03-30 | Medidata Sp. Z O.O. | Cryoapplicator for minimally invasive surgical cardiac ablation |
| WO2017066797A1 (en) | 2015-10-16 | 2017-04-20 | Modernatx, Inc. | Trinucleotide mrna cap analogs |
| WO2018213476A1 (en) * | 2017-05-16 | 2018-11-22 | Translate Bio, Inc. | Treatment of cystic fibrosis by delivery of codon-optimized mrna encoding cftr |
| WO2020106946A1 (en) * | 2018-11-21 | 2020-05-28 | Translate Bio, Inc. | TREATMENT OF CYSTIC FIBROSIS BY DELIVERY OF NEBULIZED mRNA ENCODING CFTR |
| WO2020160397A1 (en) | 2019-01-31 | 2020-08-06 | Modernatx, Inc. | Methods of preparing lipid nanoparticles |
| WO2022104131A1 (en) | 2020-11-13 | 2022-05-19 | Modernatx, Inc. | Polynucleotides encoding cystic fibrosis transmembrane conductance regulator for the treatment of cystic fibrosis |
| WO2023009422A1 (en) | 2021-07-26 | 2023-02-02 | Modernatx, Inc. | Processes for preparing lipid nanoparticle compositions for the delivery of payload molecules to airway epithelium |
| WO2023081776A1 (en) * | 2021-11-03 | 2023-05-11 | Arcturus Therapeutics, Inc. | Lipid formulations containing nucleic acids and methods of treatment for cystic fibrosis |
Non-Patent Citations (18)
| Title |
|---|
| "Dictionary of Cell and Molecular Biology", 1999, ACADEMIC PRESS |
| "Oxford Dictionary Of Biochemistry And Molecular Biology, Revised", 2000, OXFORD UNIVERSITY PRESS |
| "Solid Lipid Nanoparticles and Nanostructured Lipid Carriers: Structure, Preparation and Application", ADV. PHARM. BULL., vol. 5, 2015, pages 305 - 13 |
| A. R. GENNARO: "Remington: The Science and Practice of Pharmacy", 2006, LIPPINCOTT, WILLIAMS & WILKIN |
| HINNEBUSCH A ET AL., SCIENCE, vol. 352, no. 6292, 2016, pages 1413 - 6 |
| JUNJIE LI ET AL., CURRENT BIOLOGY, vol. 15, 23 August 2005 (2005-08-23), pages 1501 - 1507 |
| JUO, PEI-SHOW: "Concise Dictionary of Biomedicine and Molecular Biology", 2002, CRC PRESS |
| KORE ET AL., BIOORGANIC & MEDICINAL CHEMISTRY, vol. 21, 2013, pages 4570 - 4574 |
| MANDALROSSI, NAT. PROTOC., vol. 8, no. 3, 2013, pages 568 - 82 |
| MAYR C., COLD SPRING HARB PERSP BIOL, vol. 11, no. 10, 1 October 2019 (2019-10-01) |
| NORBURY: "Nature Reviews Molecular Cell Biology", 29 August 2013, AOP, article "Cytoplasmic RNA: a case of the tail wagging the dog" |
| ROBINSON EMA ET AL: "Lipid Nanoparticle-Delivered Chemically Modified mRNA Restores Chloride Secretion in Cystic Fibrosis", MOLECULAR THERAPY, vol. 26, no. 8, 1 August 2018 (2018-08-01), pages 2034 - 2046, XP093252260, ISSN: 1525-0016, Retrieved from the Internet <URL:https://pmc.ncbi.nlm.nih.gov/articles/PMC6094356/pdf/main.pdf> DOI: 10.1016/j.ymthe.2018.05.014 * |
| ROWE S.M. ET AL: "Inhaled mRNA therapy for treatment of cystic fibrosis: Interim results of a randomized, double-blind, placebo-controlled phase 1/2 clinical study", JOURNAL OF CYSTIC FIBROSIS, vol. 22, no. 4, 1 July 2023 (2023-07-01), NL, pages 656 - 664, XP093252222, ISSN: 1569-1993, Retrieved from the Internet <URL:https://pmc.ncbi.nlm.nih.gov/articles/PMC10524666/pdf/nihms-1896496.pdf> DOI: 10.1016/j.jcf.2023.04.008 * |
| SILVA ET AL.: "Delivery Systems for Biopharmaceuticals. Part I: Nanoparticles and Microparticles", CURR. PHARM. TECHNOL., vol. 16, 2015, pages 940 - 954, XP093229057, DOI: 10.2174/1389201016666150731112532 |
| SILVA ET AL.: "Lipid nanoparticles for the delivery of biopharmaceuticals", CURR. PHARM. BIOTECHNOL., vol. 16, 2015, pages 291 - 302, XP055602369 |
| TOURIOL ET AL., BIOLOGY OF THE CELL, vol. 95, 2003, pages 169 - 178 |
| WANG ET AL.: "Delivery of oligonucleotides with lipid nanoparticles", ADV. DRUG DELIV. REV., vol. 87, 2015, pages 68 - 80 |
| YAKUBOV ET AL., BIOCHEM. BIOPHYS. RES. COMMUN., vol. 394, no. 1, 2010, pages 189 - 193 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20230285310A1 (en) | Compositions for the delivery of payload molecules to airway epithelium | |
| EP3060257B1 (en) | Lipid formulations for delivery of messenger rna | |
| JP2023524071A (en) | Nucleic acids and methods for treating cystic fibrosis | |
| US20230242908A1 (en) | Lnp compositions comprising mrna therapeutics with extended half-life | |
| HUE029977T2 (en) | Oligonucleotides for making a change in the sequence of a target rna molecule present in a living cell | |
| US20230406895A1 (en) | Polynucleotides encoding cystic fibrosis transmembrane conductance regulator for the treatment of cystic fibrosis | |
| WO2022204390A1 (en) | Lipid nanoparticles containing polynucleotides encoding phenylalanine hydroxylase and uses thereof | |
| WO2023009422A1 (en) | Processes for preparing lipid nanoparticle compositions for the delivery of payload molecules to airway epithelium | |
| CN118319880B (en) | Lipid nanoparticles, inhalation preparations and applications for efficient delivery of nucleic acids to the lungs | |
| US20240123034A1 (en) | Mrnas encoding granulocyte-macrophage colony stimulating factor for treating parkinson's disease | |
| WO2022204371A1 (en) | Lipid nanoparticles containing polynucleotides encoding glucose-6-phosphatase and uses thereof | |
| CN118766874B (en) | Inhalation type preparation, iterative optimization flow and application thereof | |
| WO2025111297A1 (en) | Polynucleotides encoding cystic fibrosis transmembrane conductance regulator for the treatment of cystic fibrosis | |
| JP2025510777A (en) | Messenger ribonucleic acid with extended half-life | |
| WO2023287751A1 (en) | Polynucleotides encoding propionyl-coa carboxylase alpha and beta subunits for the treatment of propionic acidemia | |
| US20250090684A1 (en) | Polynucleotides encoding glucose-6-phosphatase for the treatment of glycogen storage disease type 1a (gsd1a) | |
| WO2025059290A1 (en) | Polynucleotides encoding phenylalanine hydroxylase for the treatment of phenylketonuria | |
| US20250177562A1 (en) | Lipid nanoparticle compositions and uses thereof | |
| US20240269248A1 (en) | Polynucleotides encoding methylmalonyl-coa mutase for the treatment of methylmalonic acidemia | |
| WO2025255199A1 (en) | Argininosuccinate synthase 1 and argininosuccinate lyase polypeptides and polynucleotides and uses thereof | |
| WO2024229321A1 (en) | Polynucleotides encoding cystic fibrosis transmembrane conductance regulator for the treatment of cystic fibrosis | |
| WO2025240355A1 (en) | Lipid nanoparticles for extrahepatic delivery | |
| KR20250157196A (en) | A Lipid Nanocomplex for Inhalation Delivery of Nucleic Acid Molecule | |
| WO2022271776A1 (en) | Polynucleotides encoding uridine diphosphate glycosyltransferase 1 family, polypeptide a1 for the treatment of crigler-najjar syndrome | |
| WO2024137953A2 (en) | Small nuclear ribonucleoprotein 13 polypeptides, polynucleotides, and uses thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 24822043 Country of ref document: EP Kind code of ref document: A1 |