WO2025106910A1 - Degrader molecules targeting androgen receptor - Google Patents
Degrader molecules targeting androgen receptor Download PDFInfo
- Publication number
- WO2025106910A1 WO2025106910A1 PCT/US2024/056259 US2024056259W WO2025106910A1 WO 2025106910 A1 WO2025106910 A1 WO 2025106910A1 US 2024056259 W US2024056259 W US 2024056259W WO 2025106910 A1 WO2025106910 A1 WO 2025106910A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- methyl
- triazol
- trifluoro
- carboxamide
- phenyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/08—Bridged systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/10—Spiro-condensed systems
Definitions
- Inactive forms of androgen receptor are located in the cytoplasm, bound to heat shock proteins, which are responsible for proper protein folding, prevention of misfolding, and maintaining 3D protein structure during events of cellular stress. Androgen receptor is activated by the binding of androgen molecules, resulting in activation and transcription of a variety of downstream genes. Michmerhuizen, A.R.; npj Breast Cancer 6:47 (2020). Binding of androgen receptor may result in the activation of signaling pathways that have been implicated in cancer, including the PI3K/AKT pathway. Michmerhuizen, A.R.; npj Breast Cancer 6:47 (2020). Androgen receptor has been well characterized as a key driver for the development of prostate cancer in men.
- ubiquitin E3 ligase Heterobifunctional small molecules, which simultaneously bind to target proteins and recruit an ubiquitin ligase (e.g., ubiquitin E3 ligase) have been shown to result in the target protein’s ubiquitination and degradation (Bondeson, D. P., et al. Nat Chem Biol. 201511(8):611- 617).
- ubiquitin E3 ligase ubiquitination and degradation
- the present invention is further directed to a pharmaceutical composition comprising a compound of Formula I, or a pharmaceutically acceptable salt thereof, and at least one pharmaceutically acceptable carrier.
- the present invention is further directed to a method of treating a disease or disorder in a patient in need of treatment, where the disease or disorder is an androgen receptor-related disease, comprising administering to the patient a therapeutically effective amount of a compound Formula I, or a pharmaceutically acceptable salt thereof.
- the present invention is further directed to a method of treating cancer in a patient in need thereof comprising administering to said patient a therapeutically effective amount of a compound of Formula I, or a pharmaceutically acceptable salt thereof.
- the present invention also provides uses of the compounds described herein in the manufacture of a medicament for use in therapy.
- the present disclosure also provides the compounds described herein for use in therapy.
- DETAILED DESCRIPTION The present disclosure provides, inter alia, a compound of Formula I: or a pharmaceutically acceptable salt thereof, wherein: R 1 , R 2 , and R 3 are each independently selected from H, halo, SR A , OR A , C 1-4 alkyl, C 2-4 alkenyl, and C1-4 haloalkyl, wherein the C2-4 alkenyl is optionally substituted with 1, 2, or 3 substituents independently selected from halo, and wherein R A is H, C1-4 alkyl, or C1-4 haloalkyl; R 4 and R 5 are each independently selected from H, halo, SR A , OR A , C 1-4 alkyl, C 2-4 alkenyl, and C 1-4 haloalkyl, wherein the C 2-4 alkenyl is optionally substituted with 1, 2, or 3 substituents independently selected from halo, and wherein R A is H, C1-4 al
- R 1 , R 2 , and R 3 are each independently selected from H, halo, C1-4 alkyl, and C1-4 haloalkyl
- R 4 and R 5 are each independently selected from H, halo, OR A , C1-4 alkyl, and C1-4 haloalkyl, wherein R A is H, C 1-4 alkyl, or C 1-4 haloalkyl; or R 4 and R 5 together with the carbon atom to which they are attached form a C3-6 cycloalkyl group or a 3-6 membered heterocycloalkyl group, each optionally substituted with 1, 2, or 3 substituents independently selected from halo, OR A , C 1-4 alkyl, and C 1-4 haloalkyl
- R 6 and R 7 are each independently selected from H, halo, C1-4 alkyl, and C1-4 haloalkyl
- R 8 is H or C1-4 alkyl
- Cy 1 is selected from C 6-10 aryl, C 3
- R 1 , R 2 , and R 3 are each independently selected from H, F, methyl, and trifluoromethyl. In some embodiments, R 1 , R 2 , and R 3 are each F. In some embodiments, R 1 , R 2 , and R 3 are each selected from methyl and trifluoromethyl. In some embodiments, R 1 , R 2 , and R 3 are each H.
- R 4 and R 5 are each independently selected from H, halo, SR A , OR A , C 1-4 alkyl, C 2-4 alkenyl, and C 1-4 haloalkyl, wherein the C 2-4 alkenyl is optionally substituted with 1, 2, or 3 substituents independently selected from halo, and wherein R A is H, C 1-4 alkyl, or C1-4 haloalkyl.
- R 4 and R 5 are each independently selected from halo, SR A , OR A , C1-4 alkyl, C2-4 alkenyl, and C1-4 haloalkyl, wherein the C2-4 alkenyl is optionally substituted with 1, 2, or 3 substituents independently selected from halo, and wherein R A is H, C 1-4 alkyl, or C1-4 haloalkyl.
- R 4 and R 5 are each independently selected from H, SR A , C1- 4 alkyl, and C2-4 alkenyl, wherein the C2-4 alkenyl is optionally substituted with 1, 2, or 3 substituents independently selected from halo, and wherein R A is H, C 1-4 alkyl, or C 1-4 haloalkyl.
- R 4 and R 5 are each independently selected from SR A , C1-4 alkyl, and C2-4 alkenyl, wherein the C2-4 alkenyl is optionally substituted with 1, 2, or 3 substituents independently selected from halo, and wherein R A is H, C 1-4 alkyl, or C 1-4 haloalkyl.
- R 4 and R 5 are each independently selected from H, SR A , C 1-4 alkyl, and C 2-4 alkenyl, wherein the C 2-4 alkenyl is optionally substituted with 1, 2, or 3 substituents independently selected from halo, and wherein R A is C1-4 alkyl.
- R 4 and R 5 are each independently selected from SR A , C1-4 alkyl, and C2-4 alkenyl, wherein the C2-4 alkenyl is optionally substituted with 1, 2, or 3 substituents independently selected from halo, and wherein R A is C1-4 alkyl.
- R 4 and R 5 are each independently selected from OH, C1-4 alkyl, and C 1-4 haloalkyl; or R 4 and R 5 together with the carbon atom to which they are attached form a C 3-6 cycloalkyl group optionally substituted with 1, 2, or 3 substituents independently selected from halo, OR A , C1-4 alkyl, and C1-4 haloalkyl.
- R 4 and R 5 are each independently selected from OH, C 1-4 alkyl, and C 1-4 haloalkyl.
- R 4 and R 5 are each C 1-4 alkyl.
- R 4 and R 5 are each methyl.
- R 4 and R 5 together with the carbon atom to which they are attached form a C 3-6 cycloalkyl group optionally substituted with 1, 2, or 3 substituents independently selected from halo, OR A , C1-2 alkyl, and C1-2 haloalkyl.
- R 4 and R 5 together with the carbon atom to which they are attached form a C3-6 cycloalkyl group optionally substituted with 1 or 2 substituents independently selected from halo and C 1-2 alkyl.
- R 4 and R 5 together with the carbon atom to which they are attached form a C3-6 cycloalkyl group optionally substituted with 1 or 2 fluoro.
- R 6 and R 7 are each H.
- R 8 is H.
- Cy 1 is selected from C3-6 cycloalkyl, 5-6 membered heteroaryl, C5-6 cycloalkyl fused with phenyl to form a bicyclic ring, and 5-6 membered heterocycloalkyl fused with phenyl to form a bicyclic ring, each optionally substituted by 1 or 2 substituents independently selected from halo, C 1-6 alkyl, C 1-6 haloalkyl, CN, C 1-6 alkyl-NR c R d , C(O)NR c R d , and NR c R d .
- Cy 1 is C3-6 cycloalkyl optionally substituted by 1 or 2 substituents independently selected from halo, C 1-6 alkyl, and C 1-6 haloalkyl. In some embodiments, Cy 1 is C 3- 6 cycloalkyl. In some embodiments, Cy 1 is cyclopropyl or cyclobutyl. In some embodiments, Cy 1 is cyclopropyl. In some embodiments, Cy 1 is 5-6 membered heteroaryl, optionally substituted by 1 or 2 substituents independently selected from halo, C1-6 alkyl, and C(O)NR c R d .
- Cy 1 is 5 membered heteroaryl, optionally substituted by 1 or 2 substituents independently selected from halo, C 1-6 alkyl, and C(O)NR c R d .
- Cy 1 is pyrazolyl or pyrrolyl, optionally substituted with C(O)NR c R d .
- Cy 1 is pyrazolyl.
- Cy 1 is pyrazolyl, pyrrolyl, or isoxazolyl, each optionally substituted with C(O)NR c R d .
- Cy 1 is isoxazolyl.
- Cy 1 is pyrazolyl, pyrrolyl, or isoxazolyl.
- Cy 1 is C5-6 cycloalkyl fused with phenyl to form a bicyclic ring, optionally substituted by 1 or 2 substituents independently selected from halo, C1-6 alkyl, CN, C1-6 alkyl-NR c R d , and NR c R d .
- Cy 1 is cyclopentyl fused with phenyl to form a bicyclic ring, optionally substituted by methyl, CN, Cl, F, -CH2NHR d , and NH2.
- Cy 1 is 5-6 membered heterocycloalkyl fused with phenyl to form a bicyclic ring, optionally substituted by 1 or 2 substituents independently selected from halo and C 1-6 alkyl.
- Cy 1 is dihydrobenzofuran, optionally substituted by 1 or 2 substituents independently selected from chloro and methyl.
- Cy 1 is dihydrobenzofuran substituted by 1 chloro.
- Cy 2 is phenyl or 5-6 membered heteroaryl, optionally substituted by 1 or 2 substituents selected from halo and C 1-6 alkyl. In some embodiments, Cy 2 is C 6-10 aryl optionally substituted by 1 or 2 substituents independently selected from halo, C1-6 alkyl, and C1-6 haloalkyl. In some embodiments, Cy 2 is phenyl optionally substituted with 1 or 2 substituents each independently selected from C1-6 alkyl. In some embodiments, Cy 2 is phenyl, methylphenyl, methylchlorophenyl, or dimethylphenyl.
- Cy 2 is phenyl optionally substituted with 1 or 2 substituents selected from methyl and chloro. In some embodiments, Cy 2 is 5-10 membered heteroaryl optionally substituted by 1 or 2 substituents independently selected from halo, C1-6 alkyl, and C1-6 haloalkyl. In some embodiments, Cy 2 is 6 membered heteroaryl optionally substituted by 1 or 2 substituents each independently selected from C 1-6 alkyl. In some embodiments, Cy 2 is pyridinyl or methylpyridinyl. In some embodiments, Cy 2 is pyridinyl or pyrimidinyl, each optionally substituted by 1 or 2 substituents each independently selected from C1-6 alkyl.
- Cy 2 is pyridinyl or pyrimidinyl, each optionally substituted with methyl. In some embodiments, Cy 2 is pyridinyl, methylpyridinyl, or pyrimidinyl. In some embodiments, Cy 2 is pyridinyl or pyrimidinyl, each optionally substituted with 1 or 2 methyl groups.
- Cy 2 is bicyclo[1.1.1]pentyl.
- Cy A , Cy B , Cy C , and Cy D are each independently absent or independently selected from C6-10 aryl, 5-10 membered heteroaryl, and 4-10 membered heterocycloalkyl, each optionally substituted by 1, 2, or 3 substituents independently selected from halo and C1-6 alkyl.
- Cy A , Cy B , Cy C , and Cy D are each independently absent or independently selected from phenyl, 5-9 membered heteroaryl, and 4-7 membered heterocycloalkyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl.
- Cy A is absent or selected from C6-10 aryl, 5-10 membered heteroaryl, and 4-10 membered heterocycloalkyl, each optionally substituted by 1, 2, or 3 substituents independently selected from halo and C 1-6 alkyl. In some embodiments, Cy A is absent or selected from phenyl, 5-9 membered heteroaryl, and 4-7 membered heterocycloalkyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl.
- Cy A is absent or selected from phenyl, 5-6 membered heteroaryl, and 6-9 membered heterocycloalkyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, Cy A is absent or selected from phenyl, pyrazolyl, pyridinyl, indazolyl, and piperidinyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C 1-6 alkyl. In some embodiments, Cy A is phenyl or pyridinyl, each optionally substituted by halo or methyl. In some embodiments, Cy A is absent.
- Cy A is selected from C6-10 aryl, 5-10 membered heteroaryl, and 4- 10 membered heterocycloalkyl, each optionally substituted by 1, 2, or 3 substituents independently selected from halo and C 1-6 alkyl. In some embodiments, Cy A is selected from phenyl, 5-9 membered heteroaryl, and 4-7 membered heterocycloalkyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, Cy A is selected from phenyl, 5-6 membered heteroaryl, and 6-9 membered heterocycloalkyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl.
- Cy A is selected from phenyl, pyrazolyl, pyridinyl, indazolyl, and piperidinyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, Cy A is phenyl or pyridinyl, each optionally substituted by1 or 2 substituents selected from halo and methyl.
- Cy A is absent or selected from phenyl, pyrazolyl, pyridinyl, pyrazinyl, pyrimidinyl, indazolyl, 2,3-dihydrobenzofuranyl, and piperidinyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl.
- Cy A is selected from phenyl, pyrazolyl, pyridinyl, pyrazinyl, pyrimidinyl, indazolyl, 2,3-dihydrobenzofuranyl, and piperidinyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl.
- Cy B is absent or selected from C6-10 aryl, 5-10 membered heteroaryl, and 4-10 membered heterocycloalkyl, each optionally substituted by 1, 2, or 3 substituents independently selected from halo and C1-6 alkyl. In some embodiments, Cy B is absent or selected from phenyl, 5-9 membered heteroaryl, and 4-7 membered heterocycloalkyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C 1-6 alkyl.
- Cy B is absent or selected from phenyl, 5 membered heteroaryl, and 6-7 membered heterocycloalkyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, Cy B is absent or selected from phenyl, pyrazolyl, piperazinyl, piperidinyl, and azasprio[3.3]heptanyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C 1-6 alkyl. In some embodiments, Cy B is piperidinyl or piperazinyl. In some embodiments, Cy B is absent.
- Cy B is selected from C6-10 aryl, 5-10 membered heteroaryl, and 4- 10 membered heterocycloalkyl, each optionally substituted by 1, 2, or 3 substituents independently selected from halo and C1-6 alkyl. In some embodiments, Cy B is selected from phenyl, 5-9 membered heteroaryl, and 4-7 membered heterocycloalkyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C 1-6 alkyl. In some embodiments, Cy B is selected from phenyl, 5 membered heteroaryl, and 6-7 membered heterocycloalkyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl.
- Cy B is selected from phenyl, pyrazolyl, piperazinyl, piperidinyl, and azasprio[3.3]heptanyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl.
- Cy B is absent or selected from phenyl, cyclohexyl, pyrazolyl, azetidinyl, pyrrolidinyl, piperazinyl, piperidinyl, 2-azasprio[3.3]heptanyl, diazaspiro[3.5]nonanyl, 2,6-diazaspiro[3.3]heptanyl, 3,6-diazabicyclo[3.1.1]heptanyl, 2,5-diazabicyclo[2.2.1]heptanyl, 3,8-diazabicyclo[3.2.1]octanyl, 2-azaspiro[3.5]nonanyl, 7-azaspiro[3.5]nonanyl, 6- azaspiro[3.4]octanyl, and octahydropyrrolo[3,2-b]pyrrolyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C 1-6 alkyl.
- Cy B is selected from phenyl, cyclohexyl, pyrazolyl, azetidinyl, pyrrolidinyl, piperazinyl, piperidinyl, 2- azasprio[3.3]heptanyl, diazaspiro[3.5]nonanyl, 2,6-diazaspiro[3.3]heptanyl, 3,6- diazabicyclo[3.1.1]heptanyl, 2,5-diazabicyclo[2.2.1]heptanyl, 3,8-diazabicyclo[3.2.1]octanyl, 2- azaspiro[3.5]nonanyl, 7-azaspiro[3.5]nonanyl, 6-azaspiro[3.4]octanyl, and octahydropyrrolo[3,2- b]pyrrolyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C 1-6 alkyl.
- Cy B is absent or selected from phenyl, cyclohexyl, pyrazolyl, azetidinyl, pyrrolidinyl, piperazinyl, piperidinyl, azasprio[3.3]heptanyl, diazaspiro[3.5]nonanyl, 2,6-diazaspiro[3.3]heptanyl, 3,6-diazabicyclo[3.1.1]heptanyl, 2,5-diazabicyclo[2.2.1]heptanyl, 3,8-diazabicyclo[3.2.1]octanyl, 2-azaspiro[3.5]nonanyl, 7-azaspiro[3.5]nonanyl, 6- azaspiro[3.4]octanyl, and octahydropyrrolo[3,2-b]pyrrolyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl.
- Cy B is selected from phenyl, cyclohexyl, pyrazolyl, azetidinyl, pyrrolidinyl, piperazinyl, piperidinyl, azasprio[3.3]heptanyl, diazaspiro[3.5]nonanyl, 2,6-diazaspiro[3.3]heptanyl, 3,6- diazabicyclo[3.1.1]heptanyl, 2,5-diazabicyclo[2.2.1]heptanyl, 3,8-diazabicyclo[3.2.1]octanyl, 2- azaspiro[3.5]nonanyl, 7-azaspiro[3.5]nonanyl, 6-azaspiro[3.4]octanyl, and octahydropyrrolo[3,2- b]pyrrolyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl.
- Cy B is absent or selected from phenyl, cyclohexyl, pyrazolyl, azetidinyl, pyrrolidinyl, piperazinyl, piperidinyl, azasprio[3.3]heptanyl, diazaspiro[3.5]nonanyl, 2,6-diazaspiro[3.3]heptanyl, 3,6-diazabicyclo[3.1.1]heptanyl, 2,5-diazabicyclo[2.2.1]heptanyl, 3,8-diazabicyclo[3.2.1]octanyl, and azaspiro[3.5]nonanyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl.
- Cy B is selected from phenyl, cyclohexyl, pyrazolyl, azetidinyl, pyrrolidinyl, piperazinyl, piperidinyl, azasprio[3.3]heptanyl, diazaspiro[3.5]nonanyl, 2,6-diazaspiro[3.3]heptanyl, 3,6- diazabicyclo[3.1.1]heptanyl, 2,5-diazabicyclo[2.2.1]heptanyl, 3,8-diazabicyclo[3.2.1]octanyl, and azaspiro[3.5]nonanyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl.
- Cy B is 2-azasprio[3.3]heptanyl.
- Cy C is absent or selected from C 6-10 aryl, 5-10 membered heteroaryl, and 4-10 membered heterocycloalkyl, each optionally substituted by 1, 2, or 3 substituents independently selected from halo and C1-6 alkyl.
- Cy C is absent or selected from phenyl, 5-9 membered heteroaryl, and 4-7 membered heterocycloalkyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C 1-6 alkyl.
- Cy C is absent or selected from 9 membered heteroaryl and 4-6 membered heterocycloalkyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C 1-6 alkyl. In some embodiments, Cy C is absent or selected from indazolyl, piperidinyl, piperazinyl, azetidinyl, and pyrrolidinyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, Cy C is piperidinyl or piperazinyl. In some embodiments, Cy C is absent. In some embodiments, Cy C is piperidinyl, piperazinyl, or cyclobutyl.
- Cy C is cyclobutyl. In some embodiments, Cy C is selected from C 6-10 aryl, 5-10 membered heteroaryl, and 4- 10 membered heterocycloalkyl, each optionally substituted by 1, 2, or 3 substituents independently selected from halo and C1-6 alkyl. In some embodiments, Cy C is selected from phenyl, 5-9 membered heteroaryl, and 4-7 membered heterocycloalkyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl.
- Cy C is selected from 9 membered heteroaryl and 4-6 membered heterocycloalkyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C 1-6 alkyl. In some embodiments, Cy C is selected from indazolyl, piperidinyl, piperazinyl, azetidinyl, and pyrrolidinyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C 1-6 alkyl.
- Cy D is absent or selected from C 6-10 aryl, 5-10 membered heteroaryl, and 4-10 membered heterocycloalkyl, each optionally substituted by 1, 2, or 3 substituents independently selected from halo and C1-6 alkyl. In some embodiments, Cy D is absent or selected from phenyl, 5-9 membered heteroaryl, and 4-7 membered heterocycloalkyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, Cy D is absent or 5-6 membered heterocycloalkyl optionally substituted by 1 or 2 substituents independently selected from halo and C 1-6 alkyl.
- Cy D is absent or selected from piperazinyl and piperidinyl. In some embodiments, Cy D is absent or 6 membered heterocycloalkyl. In some embodiments, Cy D is absent or selected from piperazinyl and piperidinyl. In some embodiments, Cy D is absent or piperazinyl. In some embodiments, Cy D is absent. In some embodiments, Cy D is selected from C6-10 aryl, 5-10 membered heteroaryl, and 4- 10 membered heterocycloalkyl, each optionally substituted by 1, 2, or 3 substituents independently selected from halo and C 1-6 alkyl.
- Cy D is selected from phenyl, 5-9 membered heteroaryl, and 4-7 membered heterocycloalkyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, Cy D is 5-6 membered heterocycloalkyl optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, Cy D is selected from piperazinyl and piperidinyl. In some embodiments, Cy D is 6-membered heterocycloalkyl.
- L A , L B , and L C are each independently absent or independently selected from C1-6 alkylene, -C(O)-, -O-, -O-(C1-6 alkylene)-, -NR c23 -, -C(O)NR c23 -, -C(O)-(C1-6 alkylene)-, -C(O)NR c23 -(C1-6 alkylene)-, and -NR c23 C(O)-.
- L A , L B , and L C are each independently absent or independently selected from C1-6 alkylene, -C(O)-, -O-, -O-(C1-6 alkylene)-, -C(O)-(C1-6 alkylene)-, -C(O)NR c23 - (C1-6 alkylene)-, and -NR c23 C(O)-.
- R c23 is selected from H and C1-6 alkyl.
- L A , L B , and L C are each independently absent or independently selected from methylene, -C(O)-, -O-, -O-(C1-3 alkylene)-, -C(O)CH2-, -C(O)NH-(C1-3 alkylene).
- L A is absent or selected from methylene, -C(O)-, -C(O)CH2-, - C(O)NH-(C 1-3 alkylene)-, and -NHC(O)-.
- L A is absent or selected from methylene and -NHC(O)-.
- L A is absent.
- L A is selected from methylene and -NHC(O)-. In some embodiments, L A is methylene. In some embodiments, L A is -NHC(O)-. In some embodiments, L A is absent or selected from methylene, -C(O)-, -C(O)CH 2 -, - C(O)NH-(C1-3 alkylene)-, -NHC(O)-, -CH(CH3)-, -CH(CH(CH3)2)-, -CH(CH2CH2CH3)-, - CH(CH2OCH3)-, and -O-.
- L A is selected from methylene, -C(O)-, - C(O)CH 2 -, -C(O)NH-(C 1-3 alkylene)-, -NHC(O)-, -CH(CH 3 )-, -CH(CH(CH 3 ) 2 )-, - CH(CH2CH2CH3)-, -CH(CH2OCH3)-, and -O-.
- L B is absent or selected from methylene, -C(O)-, -OR a3 -, - C(O)CH 2 -, -C(O)NH-(C 1-3 alkylene)-, -NHC(O)-, and -O-(C 1-6 alkylene)-.
- L B is absent or methylene.
- L B is absent.
- L B is selected from methylene, -C(O)-, -OR a3 -, -C(O)CH2-, -C(O)NH-(C1-3 alkylene)-, and -NHC(O)-.
- L B is methylene.
- L B is selected from -C(O)-, -OR a3 - , -C(O)CH 2 -, -C(O)NH-(C 1-3 alkylene)-, and -NHC(O)-.
- L B is absent or selected from methylene, -C(O)-, -OR a3 -, - C(O)CH2-, -C(O)NH-(C1-3 alkylene)-, -NHC(O)-, -O-, -O-(C1-6 alkylene)-, and -NR c23 -(C1-6 alkylene)-.
- L B is selected from methylene, -C(O)-, -OR a3 -, -C(O)CH 2 -, - C(O)NH-(C1-3 alkylene)-, -NHC(O)-, -O-, -O-(C1-6 alkylene)-, and -NR c23 -(C1-6 alkylene)-.
- L C is absent or selected from methylene, -O-, -C(O)CH2-, and - NHC(O)-.
- L C is absent or methylene.
- L C is absent.
- L C is methylene.
- Ubiquitin ligase binding moieties and linkers are known and well-described in the art, for example: Bondeson, D. P., et al. Nat Chem Biol. 201511(8):611-617; An S, et al. EBioMedicine 201836:553-562; Paiva S-L. et al, Curr. Op. in Chem. Bio. 2010, 50:111-119; and International Patent Application Publication No. WO 2017/197056, each of which is incorporated by reference in its entirety.
- thalidomide derivatives such as lenalidomide or pomalidomide, have been reported to recruit potential protein substrates to cereblon, for example: WO 2019/099926 and WO 2020/023851.
- Z is a cereblon E3 ubiquitin ligase binding moiety. In some embodiments, Z is an E3 ubiquitin ligase binding moiety that binds to cereblon. In some embodiments, Z comprises a chemical group derived from an imide, a thioimide, an amide, or a thioamide. In some embodiments, Z is thalidomide, lenalidomide, pomalidomide, analogs thereof, isosteres thereof, or derivatives thereof.
- Z is a group having Formula III: wherein ring A is C6-10 aryl, 5-14 membered heteroaryl, C3-7 cycloalkyl fused with phenyl to form a bicyclic ring, 4-7 membered heterocycloalkyl fused with phenyl to form a bicyclic ring, each optionally substituted with 1, 2, or 3 substituents independently selected from halo, C1-6 alkyl, and C1-6 haloalkyl; L 1 is absent, CH 2 , NH, or O; and W is CH or N, wherein the wavy line represents the point of attachment to group L.
- ring A is 4-7 membered heterocycloalkyl fused with phenyl to form a bicyclic ring, optionally substituted with halo.
- ring A is phenyl or pyridinyl, each optionally substituted with 1, 2, or 3 substituents independently selected from halo, C1-6 alkyl, and C1-6 haloalkyl.
- ring A is phenyl optionally substituted with 1, 2, or 3 substituents independently selected from halo, C 1-6 alkyl, and C 1-6 haloalkyl.
- ring A is pyrimidinyl optionally substituted with 1, 2, or 3 substituents independently selected from halo, C 1-6 alkyl, and C 1-6 haloalkyl.
- ring A is phenyl or pyridinyl, each optionally substituted with halo.
- ring A is phenyl optionally substituted with halo.
- ring A is pyrimidinyl optionally substituted with halo.
- ring A is 4-7 membered heterocycloalkyl fused with phenyl to form a bicyclic ring, optionally substituted with halo.
- ring A is phenyl or pyridinyl. In some embodiments, ring A is phenyl. In some embodiments, ring A is pyrimidinyl. In some embodiments, L 1 is absent or NH. In some embodiments, L 1 is absent. In some embodiments, L 1 is NH. In some embodiments, W is CH. In some embodiments, W is N. In some embodiments, Z is selected from: ;
- V is independently selected from CH, C(CH 3 ), and N.
- Z is selected from:
- the compound has Formula IVA: , In some embodiments, the compound has Formula IVB:
- Cy 1 is selected from C3-6 cycloalkyl, 5-6 membered heteroaryl, C5-6 cycloalkyl fused with phenyl to form a bicyclic ring, and 5-6 membered heterocycloalkyl fused with phenyl to form a bicyclic ring, each optionally substituted by 1 or 2 substituents independently selected from halo, C1-6 alkyl, C1-6 haloalkyl, CN, C1-6 alkyl-NR c R d , C(O)NR c R d , and NR c R d ; Cy 2 is selected from C 6-10 aryl and 5-10 membered heteroaryl, optionally substituted by 1, 2, or 3 substituents independently selected from halo, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C1-6 haloalkyl, CN, NO2, OR a1 , SR a1 , C(O)R b1
- the compound has Formula IB: IB or a pharmaceutically acceptable salt thereof; wherein R 41 is selected from halo, C1-4 alkyl, and C1-4 haloalkyl; and p is 0, 1, or 2.
- R 41 is halo.
- R 41 is chloro.
- p is 0 or 1.
- R 41 is chloro and p is 0 or 1.
- the compound has Formula IB-1: In some embodiments, the compound has Formula IC: or a pharmaceutically acceptable salt thereof; wherein X 1 and X 2 are each independently selected from CH and N; each R 42 and R 43 is independently selected from halo, C1-4 alkyl, and C1-4 haloalkyl; and q and r are each independently 0, 1, or 2.
- X is CH. In some embodiments, X is N. In some embodiments, R 42 and R 43 are each independently C1-4 alkyl. In some embodiments, R 42 and R 43 are methyl.
- the compound has Formula ID: wherein X is CH or N; and R 44 is selected from H, halo, C 1-4 alkyl, and C 1-4 haloalkyl. In some embodiments, X is CH. In some embodiments, X is N. In some embodiments, R 44 is halo. In some embodiments, R 44 is chloro. In some embodiments, the compound has Formula ID-1: In some embodiments, the compound has Formula IE: or a pharmaceutically acceptable salt thereof; wherein each R 42 and R 43 is independently selected from halo, C 1-4 alkyl, and C 1-4 haloalkyl; X 1 and X 2 are each independently selected from CH and N; and q and r are each independently 0, 1, or 2.
- the compound has Formula IF: or a wherein each is independently selected from halo, C 1-4 alkyl, and C 1-4 haloalkyl; and q is 0, 1, or 2.
- the compound has Formula IG: IG, or a pharmaceutically acceptable salt thereof; wherein each R 42 and R 43 is independently selected from halo, C 1-4 alkyl, and C 1-4 haloalkyl; X 1 and X 2 are each independently selected from CH and N; and q and r are each independently 0, 1, or 2.
- the compound is selected from: (2R)-5-chloro-6-(4-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperazin-1-yl)methyl)phenyl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5- yl)methyl)-2,3-dihydrobenzofuran-2-carboxamide; (2R)-5-chloro-6-(4-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperazine-1-carbonyl)phenyl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol
- the compound is selected from: 3-(5-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2,6- diazaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluor
- a compound of Formula (A1) or a pharmaceutically acceptable salt thereof, wherein constituent members are defined herein.
- Compounds of Formula (A1) can be useful intermediates in the preparation of compounds of Formula (I).
- compounds of Formula (A1) can be useful as binders of androgen receptor.
- R 1 , R 2 , and R 3 are each independently selected from H, halo, C1-4 alkyl, and C1-4 haloalkyl
- R 4 and R 5 are each independently selected from H, halo, OR A , C 1-4 alkyl, and C 1-4 haloalkyl, wherein R A is H, C1-4 alkyl, or C1-4 haloalkyl; or R 4 and R 5 together with the carbon atom to which they are attached form a C3-6 cycloalkyl group or a 3-6 membered heterocycloalkyl group, each optionally substituted with 1, 2, or 3 substituents independently selected from H, halo, OR A , C1-4 alkyl, and C1-4 haloalkyl
- R 6 and R 7 are each independently selected from H, halo, C1-4 alkyl, and C1-4 haloalkyl
- R 8 is H or C or C
- R 1 , R 2 , and R 3 are each independently selected from H, F, methyl, and trifluoromethyl. In some embodiments, R 1 , R 2 , and R 3 are each F. In some embodiments, R 4 and R 5 are each C1-4 alkyl. In some embodiments, R 4 and R 5 are each methyl. In some embodiments, R 6 and R 7 are each H. In some embodiments, R 8 is H.
- Cy 1 is selected from C3-6 cycloalkyl, 5-6 membered heteroaryl, C5-6 cycloalkyl fused with phenyl to form a bicyclic ring, and 5-6 membered heterocycloalkyl fused with phenyl to form a bicyclic ring, each optionally substituted by 1 or 2 substituents independently selected from halo, C1-6 alkyl, C1-6 haloalkyl, CN, C1-6 alkyl-NR c R d , C(O)NR c R d , and NR c R d .
- Cy 1 is cyclopropyl, pyrazolyl, or isoxazolyl.
- Cy 1 is cyclopropyl or pyrazolyl. In some embodiments, Cy 1 is C 5-6 cycloalkyl fused with phenyl to form a bicyclic ring, optionally substituted by 1 or 2 substituents independently selected from halo, C 1-6 alkyl, CN, C 1-6 alkyl-NR c R d , and NR c R d . In some embodiments, Cy 1 is cyclopentyl fused with phenyl to form a bicyclic ring, optionally substituted by CN, Cl, and F.
- Cy 1 is 5-6 membered heterocycloalkyl fused with phenyl to form a bicyclic ring, optionally substituted by 1, 2, or 3 substituents independently selected from halo and C 1-6 alkyl.
- Cy 1 is dihydrobenzofuran, optionally substituted by 1, 2, or 3 substituents independently selected from Cl and I.
- A is phenyl or 5-6 membered heteroaryl, optionally substituted by 1, 2, or 3 substituents selected from halo and C1-6 alkyl. In some embodiments, A is phenyl optionally substituted by 1, 2, or 3 substituents selected from methyl, Br, and Cl. In some embodiments, A is H.
- the compound of Formula (A1) is selected from: 5-chloro-6-iodo-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]- 2,3-dihydrobenzofuran-2-carboxamide; (1S,2S)-2-(4-bromophenyl)-N-[[5-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-3- yl]methyl]cyclopropanecarboxamide; 1-(4-bromo-2-methyl-phenyl)-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol- 5-yl]methyl]pyrazole-4-carboxamide; 1-(3-bromo-2,4-dimethyl-phenyl)-N-[[[[3-
- the compound of Formula (A1) is 3-(4-methylpyrimidin-2-yl)-N- ((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide, or a pharmaceutically acceptable salt thereof. It is further appreciated that certain features of the invention, which are, for clarity, described in the context of separate embodiments, can also be provided in combination in a single embodiment. Conversely, various features of the invention which are, for brevity, described in the context of a single embodiment, can also be provided separately or in any suitable subcombination.
- substituents of compounds of the invention are disclosed in groups or in ranges. It is specifically intended that the invention include each and every individual subcombination of the members of such groups and ranges.
- C 1-6 alkyl is specifically intended to individually disclose methyl, ethyl, C 3 alkyl, C 4 alkyl, C 5 alkyl, and C 6 alkyl.
- aryl, heteroaryl, cycloalkyl, and heterocycloalkyl rings are described. Unless otherwise specified, these rings can be attached to the rest of the molecule at any ring member as permitted by valency.
- pyridinyl may refer to a pyridin-2-yl, pyridin-3-yl, or pyridin-4- yl ring.
- n-membered typically describes the number of ring- forming atoms in a moiety where the number of ring-forming atoms is “n”.
- piperidinyl is an example of a 6-membered heterocycloalkyl ring
- pyrazolyl is an example of a 5- membered heteroaryl ring
- pyridyl is an example of a 6-membered heteroaryl ring.
- each linking substituent include both the forward and backward forms of the linking substituent.
- -C(O)NR c23 -(C1-6 alkylene)- includes both -C(O)NR c23 -(C 1-6 alkylene)- and -(C 1-6 alkylene)-NR c23 (O)C- and is intended to disclose each of the forms individually.
- each variable can be a different moiety independently selected from the group defining the variable.
- the two R groups can represent different moieties independently selected from the group defined for R.
- the phrase “optionally substituted” means unsubstituted or substituted.
- substituted means that a hydrogen atom is replaced by a non- hydrogen group. It is to be understood that substitution at a given atom is limited by valency.
- C1-6 alkyl refers to an alkyl group having 1, 2, 3, 4, 5, or 6 carbon atoms.
- alkyl employed alone or in combination with other terms, refers to a saturated hydrocarbon group that may be straight-chain or branched. In some embodiments, the alkyl group contains 1 to 7, 1 to 6, 1 to 4, or 1 to 3 carbon atoms.
- alkyl moieties include, but are not limited to, chemical groups such as methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl, tert-butyl, n-pentyl, 2-methyl-1-butyl, 3-pentyl, n-hexyl, 1,2,2-trimethylpropyl, n-heptyl, and the like.
- the alkyl group is methyl, ethyl, or propyl.
- alkylene refers to a linking alkyl group.
- alkenyl refers to an alkyl group having one or more carbon-carbon double bonds. In some embodiments, the alkenyl moiety contains 2 to 6 or 2 to 4 carbon atoms.
- Example alkenyl groups include, but are not limited to, ethenyl, n-propenyl, isopropenyl, n-butenyl, sec-butenyl, and the like.
- alkenylene refers to a linking alkenyl group.
- alkynyl employed alone or in combination with other terms, refers to an alkyl group having one or more carbon-carbon triple bonds.
- Example alkynyl groups include, but are not limited to, ethynyl, propyn-1-yl, propyn-2-yl, and the like. In some embodiments, the alkynyl moiety contains 2 to 6 or 2 to 4 carbon atoms.
- alkynylene refers to a linking alkynyl group.
- halo or “halogen”, employed alone or in combination with other terms, includes fluoro, chloro, bromo, and iodo. In some embodiments, halo is F or Cl.
- haloalkyl employed alone or in combination with other terms, refers to an alkyl group having up to the full valency of halogen atom substituents, which may either be the same or different.
- the halogen atoms are fluoro atoms.
- the alkyl group has 1 to 6 or 1 to 4 carbon atoms.
- Example haloalkyl groups include CF 3 , C 2 F 5 , CHF 2 , CCl 3 , CHCl 2 , C 2 Cl 5 , and the like.
- alkoxy employed alone or in combination with other terms, refers to a group of formula -O-alkyl.
- Example alkoxy groups include methoxy, ethoxy, propoxy (e.g., n-propoxy and isopropoxy), t-butoxy, and the like.
- the alkyl group has 1 to 6 or 1 to 4 carbon atoms.
- haloalkoxy employed alone or in combination with other terms, refers to a group of formula -O-(haloalkyl).
- the alkyl group has 1 to 6 or 1 to 4 carbon atoms.
- An example haloalkoxy group is -OCF 3 .
- amino employed alone or in combination with other terms, refers to NH2.
- alkylamino employed alone or in combination with other terms, refers to a group of formula -NH(alkyl). In some embodiments, the alkylamino group has 1 to 6 or 1 to 4 carbon atoms.
- Example alkylamino groups include methylamino, ethylamino, propylamino (e.g., n-propylamino and isopropylamino), and the like.
- dialkylamino employed alone or in combination with other terms, refers to a group of formula -N(alkyl)2.
- Example dialkylamino groups include dimethylamino, diethylamino, dipropylamino (e.g., di(n-propyl)amino and di(isopropyl)amino), and the like.
- each alkyl group independently has 1 to 6 or 1 to 4 carbon atoms.
- the term “cycloalkyl,” employed alone or in combination with other terms, refers to a non-aromatic cyclic hydrocarbon including cyclized alkyl and alkenyl groups. Cycloalkyl groups can include mono- or polycyclic (e.g., having 2, 3, or 4 fused, bridged, or spiro rings) ring systems.
- cycloalkyl moieties that have one or more aromatic rings (e.g., aryl or heteroaryl rings) fused (i.e., having a bond in common with) to the cycloalkyl ring, for example, benzo derivatives of cyclopentane, cyclohexene, cyclohexane, and the like, or pyrido derivatives of cyclopentane or cyclohexane. Ring-forming carbon atoms of a cycloalkyl group can be optionally substituted by oxo. Cycloalkyl groups also include cycloalkylidenes.
- cycloalkyl also includes bridgehead cycloalkyl groups (e.g., non- aromatic cyclic hydrocarbon moieties containing at least one bridgehead carbon, such as admantan-1-yl) and spirocycloalkyl groups (e.g., non-aromatic hydrocarbon moieties containing at least two rings fused at a single carbon atom, such as spiro[2.5]octane and the like).
- the cycloalkyl group has 3 to 14 ring members, 3 to 10 ring members, 3 to 7 ring members, 3 to 6 ring members, or 5 to 6 ring members.
- the cycloalkyl group is monocyclic or bicyclic.
- the cycloalkyl group is monocyclic. In some embodiments, the cycloalkyl group is a C3-7 monocyclic cycloalkyl group.
- Example cycloalkyl groups include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclopentenyl, cyclohexenyl, cyclohexadienyl, cycloheptatrienyl, norbornyl, norpinyl, norcarnyl, tetrahydronaphthalenyl, octahydronaphthalenyl, indanyl, and the like.
- the cycloalkyl group is cyclopropyl, cyclobutyl, cyclopentyl, or cyclohexyl.
- the alkyl portion has 1 to 4, 1 to 3, 1 to 2, or 1 carbon atom(s).
- the alkyl portion is methylene.
- the cycloalkyl portion has 3 to 10 ring members or 3 to 7 ring members.
- the cycloalkyl group is monocyclic or bicyclic.
- the cycloalkyl portion is monocyclic. In some embodiments, the cycloalkyl portion is a C3-7 monocyclic cycloalkyl group.
- the term “heterocycloalkyl,” employed alone or in combination with other terms, refers to a non-aromatic ring or ring system, which may optionally contain one or more alkenylene or alkynylene groups as part of the ring structure, which has at least one heteroatom ring member independently selected from nitrogen, sulfur, oxygen, and phosphorus. Heterocycloalkyl groups can include mono- or polycyclic (e.g., having 2, 3 or 4 fused, bridged, or spiro rings) ring systems.
- the heterocycloalkyl group is a monocyclic or bicyclic group having 1, 2, 3, or 4 heteroatoms independently selected from nitrogen, sulfur and oxygen.
- moieties that have one or more aromatic rings (e.g., aryl or heteroaryl rings) fused (i.e., having a bond in common with) to the non-aromatic heterocycloalkyl ring, for example, 1,2,3,4-tetrahydro-quinoline and the like.
- Heterocycloalkyl groups can also include bridgehead heterocycloalkyl groups (e.g., a heterocycloalkyl moiety containing at least one bridgehead atom, such as azaadmantan-1-yl and the like) and spiroheterocycloalkyl groups (e.g., a heterocycloalkyl moiety containing at least two rings fused at a single atom, such as [1,4-dioxa-8-aza-spiro[4.5]decan-N-yl] and the like).
- bridgehead heterocycloalkyl groups e.g., a heterocycloalkyl moiety containing at least one bridgehead atom, such as azaadmantan-1-yl and the like
- spiroheterocycloalkyl groups e.g., a heterocycloalkyl moiety containing at least two rings fused at a single atom, such as [1,4-dioxa-8-
- the heterocycloalkyl group has 4 to 14 ring-forming atoms, 3 to 10 ring- forming atoms, 4 to 10 ring-forming atoms, 3 to 8 ring forming atoms, 4 to 7 ring forming atoms, 3 to 6 ring forming atoms, or 5 to 6 ring forming atoms.
- the heterocycloalkyl group has 2 to 20 carbon atoms, 2 to 15 carbon atoms, 2 to 10 carbon atoms, or about 2 to 8 carbon atoms.
- the heterocycloalkyl group has 1 to 5 heteroatoms, 1 to 4 heteroatoms, 1 to 3 heteroatoms, or 1 to 2 heteroatoms.
- the carbon atoms or heteroatoms in the ring(s) of the heterocycloalkyl group can be oxidized to form a carbonyl, an N- oxide, or a sulfonyl group (or other oxidized linkage) or a nitrogen atom can be quaternized.
- the heterocycloalkyl portion is a C2-7 monocyclic heterocycloalkyl group.
- the heterocycloalkyl group is a morpholine ring, pyrrolidine ring, piperazine ring, piperidine ring, tetrahydropyran ring, tetrahyropyridine, azetidine ring, or tetrahydrofuran ring.
- heterocycloalkylalkyl refers to a group of formula heterocycloalkyl-alkyl-.
- the alkyl portion has 1 to 4, 1 to 3, 1 to 2, or 1 carbon atom(s).
- the alkyl portion is methylene.
- the heterocycloalkyl portion has 3 to 10 ring members, 4 to 10 ring members, or 3 to 7 ring members.
- the heterocycloalkyl group is monocyclic or bicyclic. In some embodiments, the heterocycloalkyl portion is monocyclic.
- the heterocycloalkyl portion is a C2-7 monocyclic heterocycloalkyl group.
- aryl employed alone or in combination with other terms, refers to a monocyclic or polycyclic (e.g., a fused ring system) aromatic hydrocarbon moiety, such as, but not limited to, phenyl, 1-naphthyl, 2-naphthyl, and the like. In some embodiments, aryl groups have from 6 to 10 carbon atoms or 6 carbon atoms. In some embodiments, the aryl group is a monocyclic or bicyclic group. In some embodiments, the aryl group is phenyl or naphthyl.
- arylalkyl refers to a group of formula aryl-alkyl-.
- the alkyl portion has 1 to 4, 1 to 3, 1 to 2, or 1 carbon atom(s).
- the alkyl portion is methylene.
- the aryl portion is phenyl.
- the aryl group is a monocyclic or bicyclic group.
- the arylalkyl group is benzyl.
- heteroaryl refers to a monocyclic or polycyclic (e.g., a fused ring system) aromatic hydrocarbon moiety, having one or more heteroatom ring members independently selected from nitrogen, sulfur and oxygen.
- the heteroaryl group is a monocyclic or a bicyclic group having 1, 2, 3, or 4 heteroatoms independently selected from nitrogen, sulfur and oxygen.
- Example heteroaryl groups include, but are not limited to, pyridyl, pyrimidinyl, pyrazinyl, pyridazinyl, triazinyl, furyl, thienyl, imidazolyl, thiazolyl, indolyl, pyrryl, oxazolyl, benzofuryl, benzothienyl, benzthiazolyl, isoxazolyl, pyrazolyl, triazolyl, tetrazolyl, indazolyl, 1,2,4-thiadiazolyl, isothiazolyl, purinyl, carbazolyl, benzimidazolyl, indolinyl, pyrrolyl, azolyl, quinolinyl, isoquinolinyl, benzisoxazolyl, imidazo[1,2-b]thiazolyl or the like.
- the carbon atoms or heteroatoms in the ring(s) of the heteroaryl group can be oxidized to form a carbonyl, an N-oxide, or a sulfonyl group (or other oxidized linkage) or a nitrogen atom can be quaternized, provided the aromatic nature of the ring is preserved.
- the heteroaryl group has from 3 to 10 carbon atoms, from 3 to 8 carbon atoms, from 3 to 5 carbon atoms, from 1 to 5 carbon atoms, or from 5 to 10 carbon atoms.
- the heteroaryl group contains 3 to 14, 4 to 12, 4 to 8, 9 to 10, or 5 to 6 ring-forming atoms.
- the heteroaryl group has 5 to 14 ring-forming atoms, 5 to 10 ring-forming atoms, 5 to 9 ring-forming atoms, or 5 to 6 ring forming atoms. In some embodiments, the heteroaryl group has 1 to 4, 1 to 3, or 1 to 2 heteroatoms.
- the alkyl portion has 1 to 4, 1 to 3, 1 to 2, or 1 carbon atom(s). In some embodiments, the alkyl portion is methylene.
- the heteroaryl portion is a monocyclic or bicyclic group having 1, 2, 3, or 4 heteroatoms independently selected from nitrogen, sulfur and oxygen. In some embodiments, the heteroaryl portion has 5 to 10 carbon atoms.
- the compounds described herein can be asymmetric (e.g., having one or more stereocenters). All stereoisomers, such as enantiomers and diastereomers, are intended unless otherwise indicated.
- Compounds of the present invention that contain asymmetrically substituted carbon atoms can be isolated in optically active or racemic forms. Methods on how to prepare optically active forms from optically inactive starting materials are known in the art, such as by resolution of racemic mixtures or by stereoselective synthesis.
- Cis and trans geometric isomers of the compounds of the present invention may be isolated as a mixture of isomers or as separated isomeric forms.
- Compounds of the invention also include tautomeric forms. Tautomeric forms result from the swapping of a single bond with an adjacent double bond together with the concomitant migration of a proton. Tautomeric forms include prototropic tautomers which are isomeric protonation states having the same empirical formula and total charge.
- Example prototropic tautomers include ketone – enol pairs, amide - imidic acid pairs, lactam – lactim pairs, enamine – imine pairs, and annular forms where a proton can occupy two or more positions of a heterocyclic system, for example, 1H- and 3H-imidazole, 1H-, 2H- and 4H- 1,2,4-triazole, 1H- and 2H- isoindole, and 1H- and 2H-pyrazole.
- Tautomeric forms can be in equilibrium or sterically locked into one form by appropriate substitution.
- Compounds of the invention also include all isotopes of atoms occurring in the intermediates or final compounds.
- Isotopes include those atoms having the same atomic number but different mass numbers.
- isotopes of hydrogen include tritium and deuterium.
- the compounds of the invention include at least one deuterium atom.
- the term “compound,” as used herein, is meant to include all stereoisomers, geometric isomers, tautomers, and isotopes of the structures depicted, unless otherwise specified. All compounds, and pharmaceutically acceptable salts thereof, can be found together with other substances such as water and solvents (e.g., in the form of hydrates and solvates) or can be isolated. In some embodiments, the compounds of the invention, or salts thereof, are substantially isolated.
- substantially isolated is meant that the compound is at least partially or substantially separated from the environment in which it was formed or detected.
- Partial separation can include, for example, a composition enriched in the compounds of the invention.
- Substantial separation can include compositions containing at least about 50%, at least about 60%, at least about 70%, at least about 80%, at least about 90%, at least about 95%, at least about 97%, or at least about 99% by weight of the compounds of the invention, or salt thereof. Methods for isolating compounds and their salts are routine in the art.
- phrases “pharmaceutically acceptable” is employed herein to refer to those compounds, materials, compositions, and/or dosage forms which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of human beings and animals without excessive toxicity, irritation, allergic response, or other problem or complication, commensurate with a reasonable benefit/risk ratio.
- the present invention also includes pharmaceutically acceptable salts of the compounds described herein.
- pharmaceutically acceptable salts refers to derivatives of the disclosed compounds wherein the parent compound is modified by converting an existing acid or base moiety to its salt form.
- Examples of pharmaceutically acceptable salts include, but are not limited to, mineral or organic acid salts of basic residues such as amines; alkali or organic salts of acidic residues such as carboxylic acids; and the like.
- the pharmaceutically acceptable salts of the present invention include the non-toxic salts of the parent compound formed, for example, from non-toxic inorganic or organic acids.
- the pharmaceutically acceptable salts of the present invention can be synthesized from the parent compound which contains a basic or acidic moiety by conventional chemical methods. Generally, such salts can be prepared by reacting the free acid or base forms of these compounds with a stoichiometric amount of the appropriate base or acid in water or in an organic solvent, or in a mixture of the two.
- Suitable solvents can be substantially nonreactive with the starting materials (reactants), the intermediates, or products at the temperatures at which the reactions are carried out, e.g., temperatures which can range from the solvent's freezing temperature to the solvent's boiling temperature.
- a given reaction can be carried out in one solvent or a mixture of more than one solvent.
- suitable solvents for a particular reaction step can be selected by the skilled artisan.
- Preparation of compounds of the invention can involve the protection and deprotection of various chemical groups. The need for protection and deprotection, and the selection of appropriate protecting groups, can be readily determined by one skilled in the art. The chemistry of protecting groups can be found, for example, in T.W. Greene and P.G.M.
- reactions can be monitored according to any suitable method known in the art.
- product formation can be monitored by spectroscopic means, such as nuclear magnetic resonance spectroscopy (e.g., 1 H or 13 C), infrared spectroscopy, spectrophotometry (e.g., UV- visible), or mass spectrometry, or by chromatography such as high performance liquid chromatography.
- spectroscopic means such as nuclear magnetic resonance spectroscopy (e.g., 1 H or 13 C), infrared spectroscopy, spectrophotometry (e.g., UV- visible), or mass spectrometry, or by chromatography such as high performance liquid chromatography.
- Compounds of the invention can be prepared according to numerous preparatory routes known in the literature. Example synthetic methods for preparing compounds of the invention are provided in the Schemes below.
- Scheme 1 of formula 1-A can be coupled with compounds of Formula 1-B to provide compounds of formula 1-C, wherein X 1 is halogen.
- the coupling can be performed under peptide coupling conditions (e.g., EDCI, HOBt, and DIPEA; or HATU, DIPEA).
- Compounds of formula 1-C can be coupled with compounds of formula 1-D to provide compounds of formula 1-E (e.g., the compounds of the disclosure) under the appropriate palladium cross-coupling conditions, for example, in the presence of Pd(dppf)Cl2.
- the compounds provided herein can degrade androgen receptor in a cell, which comprises contacting the cell with the compound or a pharmaceutically acceptable salt or a stereoisomer thereof.
- a method for degrading androgen receptor in a patient where the method comprises administering to the patient an effective amount of a compound described herein or a pharmaceutically acceptable salt or a stereoisomer thereof.
- degradation androgen receptor rendering the androgen receptor inactive by, for example, altering its structure or breaking down androgen receptor into multiple peptide or amino acid fragments.
- the compounds of the invention are useful in the treatment of various diseases associated with abnormal expression or activity of androgen receptor.
- the compounds of the invention are useful in the treatment of cancer.
- the cancers treatable according to the present invention include prostate cancer, breast cancer, glioblastoma, bladder cancer, renal cell carcinoma, salivary gland cancer, colorectal cancer, esophageal cancer, pancreatic cancer, and stomach cancer.
- the cancers treatable according to the present invention include quamous-cell carcinoma, basal cell carcinoma, adenocarcinoma, hepatocellular carcinomas, and renal cell carcinomas, cancer of the bladder, bowel, breast, cervix, colon, esophagus, head, kidney, liver, lung, neck, ovary, pancreas, prostate, and stomach; leukemias; benign and malignant lymphomas, particularly Burkitt's lymphoma and Non-Hodgkin's lymphoma; benign and malignant melanomas; myeloproliferative diseases; sarcomas, including Ewing's sarcoma, hemangiosarcoma, Kaposi's sarcoma, liposarcoma, myosarcomas, peripheral neuroepithelioma, synovial sarcoma, gliomas, astrocytomas, oligodendrogliomas, ependymomas, gli
- the disease to be treated is cancer, e.g., prostate cancer, or Kennedy's Disease.
- the cancers treatable according to the present invention include hematopoietic malignancies such as leukemia and lymphoma.
- Example lymphomas include Hodgkin’s or non-Hodgkin’s lymphoma, multiple myeloma, B-cell lymphoma (e.g., diffuse large B-cell lymphoma (DLBCL)), chronic lymphocytic lymphoma (CLL), T-cell lymphoma, hairy cell lymphoma, and Burkett's lymphoma.
- B-cell lymphoma e.g., diffuse large B-cell lymphoma (DLBCL)
- CLL chronic lymphocytic lymphoma
- T-cell lymphoma hairy cell lymphoma
- Burkett's lymphoma Burkett's lymphoma.
- Example leukemias include acute lymphocytic leukemia (ALL), acute myelogenous leukemia (AML), chronic lymphocytic leukemia (CLL), and chronic myelogenous leukemia (CML).
- Other cancers treatable by the administration of the compounds of the invention include liver cancer (e.g., hepatocellular carcinoma), bladder cancer, bone cancer, glioma, breast cancer, cervical cancer, colon cancer, endometrial cancer, epithelial cancer, esophageal cancer, Ewing's sarcoma, pancreatic cancer, gallbladder cancer, gastric cancer, gastrointestinal tumors, head and neck cancer, intestinal cancers, Kaposi's sarcoma, kidney cancer, laryngeal cancer, liver cancer (e.g., hepatocellular carcinoma), lung cancer, prostate cancer, rectal cancer, skin cancer, stomach cancer, testicular cancer, thyroid cancer, and uterine cancer.
- liver cancer e.g., hepatocellular carcinoma
- the cancer treatable by administration of the compounds of the invention is multiple myeloma, DLBCL, hepatocellular carcinoma, bladder cancer, esophageal cancer, head and neck cancer, kidney cancer, prostate cancer, rectal cancer, stomach cancer, thyroid cancer, uterine cancer, breast cancer, glioma, follicular lymphoma, pancreatic cancer, lung cancer, colon cancer, or melanoma.
- the term “cell” is meant to refer to a cell that is in vitro, ex vivo or in vivo.
- an ex vivo cell can be part of a tissue sample excised from an organism such as a mammal.
- an in vitro cell can be a cell in a cell culture.
- an in vivo cell is a cell living in an organism such as a mammal.
- the term “contacting” refers to the bringing together of indicated moieties in an in vitro system or an in vivo system.
- “contacting” androgen receptor or “contacting” a cell with a compound of the invention includes the administration of a compound of the present invention to an individual or patient, such as a human, having androgen receptor, as well as, for example, introducing a compound of the invention into a sample containing a cellular or purified preparation containing androgen receptor.
- the phrase “therapeutically effective amount” refers to the amount of active compound or pharmaceutical agent that elicits the biological or medicinal response in a tissue, system, animal, individual or human that is being sought by a researcher, veterinarian, medical doctor or other clinician.
- treating refers to 1) inhibiting the disease in an individual who is experiencing or displaying the pathology or symptomatology of the disease (i.e., arresting further development of the pathology and/or symptomatology), or 2) ameliorating the disease in an individual who is experiencing or displaying the pathology or symptomatology of the disease (i.e., reversing the pathology and/or symptomatology).
- preventing or “prevention” refers to preventing the disease in an individual who may be predisposed to the disease but does not yet experience or display the pathology or symptomatology of the disease.
- reducing is with respect to the level in the patient prior to administration.
- a biomarker or symptom when a biomarker or symptom is reduced in a patient, the reduction is with respect to the level of or severity of the biomarker or symptom in the patient prior to administration of the compound of Formula (I), or a pharmaceutically acceptable salt thereof.
- Combination Therapy One or more additional pharmaceutical agents or treatment methods such as, for example, chemotherapeutics or other anti-cancer agents, immune enhancers, immunosuppressants, immunotherapies, radiation, anti-tumor and anti-viral vaccines, cytokine therapy (e.g., IL2, GM- CSF, etc.), and/or kinase (tyrosine or serine/threonine), epigenetic or signal transduction inhibitors can be used in combination with the compounds of the present invention.
- chemotherapeutics or other anti-cancer agents immune enhancers, immunosuppressants, immunotherapies, radiation, anti-tumor and anti-viral vaccines, cytokine therapy (e.g.,
- agents can be combined with the present compounds in a single dosage form, or the agents can be administered simultaneously or sequentially as separate dosage forms.
- Suitable agents for use in combination with the compounds of the present invention for the treatment of cancer include chemotherapeutic agents, targeted cancer therapies, immunotherapies or radiation therapy.
- Compounds of this invention may be effective in combination with anti- hormonal agents for treatment of breast cancer and other tumors.
- anti- estrogen agents including but not limited to tamoxifen and toremifene, aromatase inhibitors including but not limited to letrozole, anastrozole, and exemestane, adrenocorticosteroids (e.g. prednisone), progestins (e.g.
- Suitable anti-hormone agents used for treatment of prostate and other cancers may also be combined with compounds of the present invention. These include anti-androgens including but not limited to flutamide, bicalutamide, and nilutamide, luteinizing hormone- releasing hormone (LHRH) analogs including leuprolide, goserelin, triptorelin, and histrelin, LHRH antagonists (e.g. degarelix), androgen receptor blockers (e.g. enzalutamide) and agents that inhibit androgen production (e.g. abiraterone).
- anti-androgens including but not limited to flutamide, bicalutamide, and nilutamide, luteinizing hormone- releasing hormone (LHRH) analogs including leuprolide, goserelin, triptorelin, and histrelin, LHRH antagonists (e.g. degarelix), androgen receptor blockers (e.g. enzalutamide
- Angiogenesis inhibitors may be efficacious in some tumors in combination with FGFR inhibitors. These include antibodies against VEGF or VEGFR or kinase inhibitors of VEGFR. Antibodies or other therapeutic proteins against VEGF include bevacizumab and aflibercept.
- Inhibitors of VEGFR kinases and other anti-angiogenesis inhibitors include but are not limited to sunitinib, sorafenib, axitinib, cediranib, pazopanib, regorafenib, brivanib, and vandetanib
- Suitable chemotherapeutic or other anti-cancer agents include, for example, alkylating agents (including, without limitation, nitrogen mustards, ethylenimine derivatives, alkyl sulfonates, nitrosoureas and triazenes) such as uracil mustard, chlormethine, cyclophosphamide (Cytoxan TM ), ifosfamide, melphalan, chlorambucil, pipobroman, triethylene-melamine, triethylenethio- phosphoramine, busulfan, carmustine, lomustine, streptozocin, dacarbazine, and temozolomide.
- anti-cancer agent(s) include antibody therapeutics to costimulatory molecules such as CTLA-4, 4-1BB, PD-1, and PD-L1, or antibodies to cytokines (IL-10, TGF- ⁇ , etc.).
- exemplary cancer immunotherapy antibodies include pembrolizumab, alemtuzumab, ipilimumab, nivolumab, ofatumumab and rituximab. Methods for the safe and effective administration of most of these chemotherapeutic agents are known to those skilled in the art. In addition, their administration is described in the standard literature.
- ACN acetonitrile
- Boc tert-butyloxycarbonyl
- BOP benzotriazole-1-yl-oxy-tris- (dimethylamino)-phosphoniumhexafluorophosphate
- Cbz benzyloxycarbonyl
- DBA dibenzylideneacetone
- DCE 1,2-dichloroethane
- DIEA N,N-diisopropylethylamine
- DiBAl-H diisobutylaluminum hydride
- DMF N,N-dimethylformamide
- DMSO dimethylsulfoxide
- DPPF 1,1’--bis(diphenylphosphino)ferrocene
- DTBPF 1,1’-Bis(di-tert-butylphosphino)ferrocene
- EDCI 1-ethyl-3-(3-dimethylaminopropyl)carbodi
- Step B Synthesis of 3,3,3-trifluoro-2,2-dimethyl-propanehydrazide hydrochloride (1b)
- Intermediate 1a 210.0 g, 777.0 mmol, 1.0 equiv
- 4M HCl 4M HCl in dioxane (1500 mL).
- the mixture was stirred at 20°C for 2 h.
- the reaction mixture was concentrated under reduced pressure to afford 1b, which was used without further purification.
- 1 H NMR (400 MHz, DMSO-d6) ⁇ 11.37 (br s, 1H), 1.42 (s, 6H).
- 1b A mixture of 1b (75.0 g, 363.0 mmol, 1.0 equiv), benzyl-N-(2-amino-2-imino- ethyl)carbamate hydrochloride (88.4 g, 363.0 mmol, 1.0 equiv), NaOH (30.4 g, 762.3 mmol, 2.1 equiv) in 2-MeTHF (1500.0 mL) was three-fold degassed and purged with nitrogen and then the mixture was stirred at 110°C for 16 h under an atmosphere of nitrogen.
- Step D Synthesis of [3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methanamine (Intermediate 1)
- HBr acetic acid
- the mixture was stirred at 20°C for 1 h and then concentrated under reduced pressure.
- the residue was two-fold triturated with MTBE (500 mL) at 20°C for 20 min and the solids were collected by vacuum filtration to afford Intermediate 1 (Int 1).
- Step A Synthesis of tert-butyl ((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5- yl)methyl)carbamate (1d)
- tert-butyl (2-amino-2-iminoethyl)carbamate (1.90 g, 18.9 mmol, 1.0 equiv.) in 2-methyltetrahydrofuran (30 mL) was treated with NaOH (2.20 g, 56.9 mmol, 3.0 equiv.).
- Step C Synthesis of ethyl 2-[4-chloro-2-(hydroxymethyl)-5-iodo-phenoxy]acetate (2c)
- 2b 2.8 g, 9.8 mmol, 1.0 equiv
- K 2 CO 3 1.5 g, 10.8 mmol, 1.1 equiv
- DMF 24.0 mL
- ethyl 2-bromoacetate 1.6 g, 9.8 mmol, 1.0 mL, 1.0 equiv.
- the mixture was stirred at 20 °C for 4 h, diluted with water 15 mL and extracted with ethyl acetate (2 x 20 mL).
- Step D Synthesis of ethyl 2-[4-chloro-2-(chloromethyl)-5-iodo-phenoxy]acetate (2d)
- SOCl 2 5.5 g, 46.4 mmol, 3.3 mL, 4.0 equiv
- 2c 4.3 g, 11.6 mmol, 1.0 equiv
- toluene 40 mL
- the mixture was diluted with water 15 mL at 0°C and extracted with ethyl acetate (2 x 20 mL).
- Step E Synthesis of ethyl 5-chloro-6-iodo-2,3-dihydrobenzofuran-2-carboxylate (2e) NaH (380.9 mg, 9.5 mmol, 60.0% purity, 1.3 equiv) was added to the solution of 2d (2.8 g, 7.3 mmol, 1.0 equiv) in NMP (25.0 mL) at 0°C. The solution was stirred at 20°C for 3 h. The mixture was quenched with saturated aqueous NH4Cl (10 mL) and extracted with ethyl acetate (3 x 30 mL).
- Step F Synthesis of 5-chloro-6-iodo-2,3-dihydrobenzofuran-2-carboxylic acid (2f)
- 2e 3.0 g, 8.5 mmol, 1.0 equiv
- tetrahydrofuran 15.0 mL
- LiOH•H2O 642.7 mg, 15.3 mmol, 1.8 equiv
- water 15.0 mL
- the mixture was extracted with ethyl acetate (3 x 100 mL).
- Step B Synthesis of methyl 1-(4-amino-2-methylphenyl)pyrazole-4-carboxylate (4b)
- 4a 110.0 g, 421.0 mmol, 1 equiv
- methanol 100 ml
- Pd/C 11.00 g, 10 wt% Pd, 50 wt% in water
- methanol 100 ml
- the mixture was stirred at 25°C for 12 h under hydrogen (1 atm).
- the reaction mixture was filtered and the filtrate was concentrated under reduced pressure to afford methyl 4b.
- Step E Synthesis of 1-(4-bromo-2-methyl-phenyl)-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H- 1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Intermediate 4)
- BOP 17.9 g, 40.5 mmol, 1.2 equiv
- triethylamine 10.2 g, 101.3 mmol, 3.0 equiv.
- Example 5 Synthesis of Intermediate 5 Step – A mixture of 1,3-dibromo-2,4-dimethyl-benzene (1.00 g, 3.79 mmol, 1.0 equiv), methyl 1H-pyrazole-4-carboxylate (573 mg, 4.5 mmol, 1.2 equiv), (1R,2R)-N1,N2-dimethylcyclohexane- 1,2-diamine (216 mg, 1.5 mmol, 0.4 equiv), iodocopper tetrabutylammonium diiodide (848.3 mg, 0.76 mmol, 0.2 equiv) and cesium carbonate (2.5 g, 7.5 mmol, 2.0 equiv) in dioxane (10.0 mL) was three-fold degassed and purged with argon.
- 1,3-dibromo-2,4-dimethyl-benzene (1.00 g, 3.79 mmol, 1.0 equiv)
- Step B Synthesis of 1-(3-bromo-2,4-dimethyl-phenyl)pyrazole-4-carboxylic acid (5b)
- a solution of 5a 1.0 g, 3.2 mmol, 1.0 equiv
- methanol 4.0 mL
- tetrahydrofuran 4.0 mL
- water 2.0 mL
- LiOH•H2O 407.2 mg, 9.7 mmol, 3.0 equiv
- Step C Synthesis of 1-(3-bromo-2,4-dimethyl-phenyl)-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Intermediate 5)
- DIEA 946 mg, 7.3 mmol, 1.3 mL, 3.0 equiv
- HATU 928 mg, 2.4 mmol, 1.0 equiv
- Int 1 (916.8 mg, 3.1 mmol, 1.3 equiv).
- Step B Synthesis of (R)-5-chloro-6-(4-(piperazin-1-ylmethyl)phenyl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-2,3-dihydrobenzofuran-2-carboxamide (6b)
- 6a To a solution of 6a (30.0 mg, 45.24 ⁇ mol, 1.0 equiv) in dichloromethane (0.5 mL) was added TFA (45 ⁇ mol, 3 ⁇ L, 1.0 equiv). The mixture was stirred at 25°C for 2 h and then concentrated to afford 6b that was used without further purification.
- Example 7 Synthesis of Compound 7 2 and [4-(4-tert-butoxycarbonylpiperazine-1-carbonyl)phenyl]boronic acid.
- the intermediate corresponding to 6a was resolved by SFC (DAICEL CHIRALPAK IG (250mm x 30mm,10 ⁇ m); [CO 2 -EtOH(0.1% NH 3 H 2 O)]; 45% isocratic elution) and the early eluting enantiomer was carried forward to afford (2R)-5-chloro-6-(4-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3- dioxoisoindolin-5-yl)piperazine-1-carbonyl)phenyl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)- 1H-1,2,4-triazol-5-yl)methyl)-2,3-dihydrobenzofuran-2
- Step B Synthesis of [6-[4-[(4-tert-butoxycarbonylpiperazin-1-yl)methyl]-1-piperidyl]-3- pyridyl]boronic acid (8b)
- 8a 640 mg, 1.4 mmol, 1.0 equiv
- 4,4,5,5-tetramethyl-2-(4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3,2-dioxaborolane (1.11 g, 4.3 mmol, 3.0 equiv) in dioxane (10.0 mL) was added KOAc (714.7 mg, 7.2 mmol, 5.0 eq) Pd(PPh3)4 (168.3 mg, 145.6 ⁇ mol, 0.1 eq) and in one portion at 20°C under nitrogen.
- Step C Synthesis of tert-butyl (R)-4-((1-(5-(5-chloro-2-(((3-(1,1,1-trifluoro-2-methylpropan-2- yl)-1H-1,2,4-triazol-5-yl)methyl)carbamoyl)-2,3-dihydrobenzofuran-6-yl)pyridin-2-yl)piperidin- 4-yl)methyl)piperazine-1-carboxylate (8c) To a solution of 8b (300 mg, 742 ⁇ mol, 1.5 equiv), Intermediate 2 (254.6 mg, 495 ⁇ mol, 1.0 equiv), Pd(dppf)Cl 2 (54.3 mg, 74.2 ⁇ mol, 0.15 equiv), K 3 PO 4 (315.01 mg, 1.48 mmol, 3.0 equiv) in tetrahydrofuran (4.0 mL) and water (1.0 mL) was three-fold degassed and
- the reaction mixture was partitioned between ethyl acetate (40 mL) and water (40 mL). The organic phase was separated, washed with brine (2 x 15 ml), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure to give a residue.
- Step D and Step E were performed in a similar manner to Example 6 to afford (2R)-5-chloro-6- (6-(4-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1- yl)methyl)piperidin-1-yl)pyridin-3-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)-2,3-dihydrobenzofuran-2-carboxamide (Compound 8).
- Example 9 Synthesis of Compound 9 Step – carboxylate (9a) To a mixture of tert-butyl 4-(piperidin-4-ylmethyl)piperazine-1-carboxylate (500 mg, 1.7 mmol, 1.0 equiv) and DIEA (250.8 mg, 1.9 mmol, 338 ⁇ L, 1.1 equiv) in dichloromethane (5 mL) was added 2-bromoacetyl bromide (356 mg, 1.7 mmol, 154 ⁇ L, 1.0 equiv) dropwise at 0°C. The reaction mixture was agitated at 25oC for 1 h. The residue was poured into water (10 mL).
- Step B Synthesis of tert-butyl 4-((1-(2-(4-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)phenyl)-1H-pyrazol-1-yl)acetyl)piperidin-4-yl)methyl)piperazine-1-carboxylate (9b)
- 9a 160.0 mg, 395.7 ⁇ mol, 1.0 equiv
- 4-(4-(4,4,5,5-tetramethyl-1,3,2- dioxaborolan-2-yl)phenyl)-1H-pyrazole 106.9 mg, 395.7 ⁇ mol, 1.0 equiv
- K 2 CO 3 (164.1 mg, 1.2 mmol, 3.0 equiv) in one portion at 20°C under nitrogen.
- Step C Synthesis of tert-butyl 4-((1-(2-(4-(4'-((1S,2S)-2-(((3-(1,1,1-trifluoro-2-methylpropan-2- yl)-1H-1,2,4-triazol-5-yl)methyl)carbamoyl)cyclopropyl)-[1,1'-biphenyl]-4-yl)-1H-pyrazol-1- yl)acetyl)piperidin-4-yl)methyl)piperazine-1-carboxylate (9c) To a mixture of 9b (140.0 mg, 235.8 ⁇ mol, 1.2 equiv) and Intermediate 3 (84.7 mg, 196.5 ⁇ mol, 1.0 equiv) in dioxane (1.5 mL) and water (0.2 mL) was added K2CO3 (81.5 mg, 589.6 ⁇ mol, 3.0 equiv) and Pd(dppf)Cl2•CH2
- Step D Synthesis of (1S,2S)-2-(4'-(1-(2-oxo-2-(4-(piperazin-1-ylmethyl)piperidin-1-yl)ethyl)-1H- pyrazol-4-yl)-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol- 5-yl)methyl)cyclopropane-1-carboxamide (9d) To a mixture of 9c (150.0 mg, 183.4 ⁇ mol, 1.0 equiv) in HCl/ dioxane (5.0 mL, 4 M) was stirred at 20°C under nitrogen for 1 h.
- Step E Synthesis of (1S,2S)-2-(4'-(1-(2-(4-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3- dioxoisoindolin-5-yl)piperazin-1-yl)methyl)piperidin-1-yl)-2-oxoethyl)-1H-pyrazol-4-yl)-[1,1'- biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5- yl)methyl)cyclopropane-1-carboxamide (Compound 9) To a mixture of 9d (130.0 mg, 181.1 ⁇ mol, 1.0 equiv) in DMSO (3.0 mL) was added DIEA (70.2 mg, 543.3 ⁇ mol, 94.6 ⁇ L, 3.0 equiv) and 2-(2,6-
- Example 10 Synthesis of Compound 10 Compound 10 was prepared in a similar manner to Example 9. Final purification by preparative HPLC (Phenomenex Luna C18, 80mm x 30mm, 3 ⁇ m; H 2 O (0.04% HCl)-acetonitrile) afforded (1S,2S)-2-[4-[4-[1-[2-[4-[[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]piperazin-1-yl]methyl]-1-piperidyl]-2-oxo-ethyl]pyrazol-3-yl]phenyl]phenyl]phenyl]-N-[[3-(2,2,2- trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]cyclopropanecarboxamide hydrochloride (Compound 10).
- Step B Synthesis of tert-butyl 4-((1-(2-(4-(4-bromo-2-chlorophenyl)-1H-pyrazol-1- yl)acetyl)piperidin-4-yl)methyl)piperazine-1-carboxylate (11b)
- 11a 500.0 mg, 918.1 ⁇ mol, 1.0 equiv
- (4-bromo-2-chlorophenyl)boronic acid 216.0 mg, 918.1 ⁇ mol, 1.0 equiv
- Pd(dppf)Cl2.CH2Cl2 (75.0 mg, 91.8 ⁇ mol, 0.1 equiv)
- K 2 CO 3 380.6 mg, 2.8 mmol, 3.0 equiv) in water (1.0 mL) and dioxane (5.0 mL) was three-fold degassed and purged with nitrogen.
- Step C Synthesis of (4-(1-(2-(4-((4-(tert-butoxycarbonyl)piperazin-1-yl)methyl)piperidin-1-yl)- 2-oxoethyl)-1H-pyrazol-4-yl)-3-chlorophenyl)boronic acid (11c)
- 11b 400.0 mg, 688.5 ⁇ mol, 1.0 equiv
- 4,4,4',4',5,5,5',5'-octamethyl-2,2'- bi(1,3,2-dioxaborolane) 262.3 mg, 1.0 mmol, 1.5 equiv
- potassium acetate 202.7 mg, 2.1 mmol, 3.0 equiv
- Pd(dppf)Cl 2 50.4 mg, 68.9 ⁇ mol, 0.1 equiv) in dioxane (4.0 mL) was three-fold degassed and purged with nitrogen.
- Step D Synthesis of tert-butyl 4-((1-(2-(4-(3-chloro-4'-((1S,2S)-2-(((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)carbamoyl)cyclopropyl)-[1,1'-biphenyl]-4-yl)- 1H-pyrazol-1-yl)acetyl)piperidin-4-yl)methyl)piperazine-1-carboxylate (11d) A mixture of 11c (120.3 mg, 220.3 ⁇ mol, 1.0 equiv), Intermediate 3 (95.0 mg, 220.3 ⁇ mol, 1.0 equiv), K2CO3 (91.3 mg, 660.9 ⁇ mol, 3.0 equiv), Pd(dppf)Cl2•CH2Cl2 (18.0 mg, 22.0 ⁇ mol, 0.1 equiv) in diox
- Step E Synthesis of 4-((1-(2-(4-(3-chloro-4'-((1S,2S)-2-(((3-(1,1,1-trifluoro-2-methylpropan-2- yl)-1H-1,2,4-triazol-5-yl)methyl)carbamoyl)cyclopropyl)-[1,1'-biphenyl]-4-yl)-1H-pyrazol-1- yl)acetyl)piperidin-4-yl)methyl)piperazin-1-ium (11e) Intermediate 11d (90.0 mg, 105.6 ⁇ mol, 1.0 equiv) was dissolved in HCl/dioxane (2 mL, 4 M).
- Step F Synthesis of 5-(4-((1-(2-(4-(3-chloro-4'-((1S,2S)-2-(3-(3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)propanoyl)cyclopropyl)-[1,1'-biphenyl]-4-yl)-1H- pyrazol-1-yl)acetyl)piperidin-4-yl)methyl)piperazin-1-yl)-2-(2,6-dioxopiperidin-3-yl)-6- fluoroisoindoline-1,3-dione (Compound 11) To a solution of 11e (80.0 mg, 106.3 ⁇ mol, 1.0 equiv) in DMSO (1.0 mL) was added DIEA (20.6 mg, 159.5 ⁇ mol, 27.8 ⁇ L, 1.5 equiv) and 2-(2,6-dio
- Example 12 Synthesis of Compound 12 (2'- chloro-4'-(1-(2-(4-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1- yl)methyl)piperidin-1-yl)-2-oxoethyl)-1H-pyrazol-4-yl)-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)cyclopropane-1-carboxamide (Compound 12).
- Example 13 Synthesis of Compound 13 Step A – Synthesis of 4-[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]-1H-pyrazole (13a) To a solution of 4-(4-bromophenyl)-1H-pyrazole (9 g, 40.3 mmol, 1 equiv), 4,4,5,5- tetramethyl-2-(4,4,5,5-tetramethyl1,3,2-dioxaborolan-2-yl)-1,3,2-dioxaborolane (10.2 g, 40.3 mmol, 1 equiv) in dioxane (90 mL) was added potassium acetate (11.8 g, 121.0 mmol, 3 equiv) and Pd(dppf)Cl2•CH2Cl2 (3.2 g, 4.03 mmol, 0.1 equiv).
- Step B Synthesis of tert-butyl 2-[4-[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)phenyl]pyrazol-1-yl]acetate (13b)
- 4-[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]-1H-pyrazole (4 g, 14.8 mmol, 1 equiv) in DMF (40 mL) was added K 2 CO 3 (4.09 g, 29.6 mmol, 2 equiv) and tert- butyl 2-bromoacetate (3.47 g, 17.7 mmol, 2.62 mL, 1.2 equiv).
- Step C Synthesis of tert-butyl 2-[4-[4-[4-[4-[4-[(1S,2S)-2-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H- 1,2,4-triazol-5-yl]methylcarbamoyl]cyclopropyl]phenyl]phenyl]pyrazol-1-yl]acetate (13c)
- 13b 3.5 g, 9.0 mmol, 1.3 equiv
- Intermediate 3 (3 g, 6.9 mmol, 1 equiv)
- Pd(DPPF)Cl2 (906.8 mg, 1.3 mmol, 0.2 equiv)
- Cs2CO3 (6.8 g, 20.8 mmol, 3 equiv) in 2- methylbutan-2-ol (30 mL) and water (7 mL) was 3-fold degassed and purged with nitrogen.
- the mixture was stirred at 85 °C for 5 h under an atmosphere of nitrogen.
- the mixture was cooled to ambient temperature and poured into water (100 mL).
- the aqueous phase was extracted with ethyl acetate (40 mL x 3).
- the combined organic phase was washed with brine (30 mL x 3), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure.
- Step D Synthesis of 2-[4-[4-[4-[4-[(1S,2S)-2-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4- triazol-5-yl]methylcarbamoyl]cyclopropyl]phenyl]phenyl] pyrazol-1-yl]acetic acid (13d)
- 13c 2.2 g, 3.6 mmol, 1 equiv
- dichloromethane 22 mL
- trifluoroacetic acid 33.7 g, 296.1 mmol, 22.0 mL, 81.9 equiv
- Step E Synthesis of benzyl 4-[(1-tert-butoxycarbonylazetidin-3-yl)methyl]piperazine-1- carboxylate (13e)
- tert-butyl 3-(bromomethyl)azetidine-1-carboxylate 1 g, 4.0 mmol, 1.0 equiv
- benzyl piperazine-1-carboxylate 880.6 mg, 4.0 mmol, 771.1 ⁇ L, 1.0 equiv
- K 2 CO 3 1.1 g, 8.0 mmol, 2 equiv
- KI 132.7 mg, 799.5 ⁇ mol, 0.2 equiv
- Step F Synthesis of tert-butyl 3-(piperazin-1-ylmethyl)azetidine-1-carboxylate (13f)
- 13e 1.0 g, 2.5 mmol, 1.0 equiv
- Pd/C 1.2 g, 1.1 mmol, 10 wt%
- Step G Synthesis of tert-butyl 3-[[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]piperazin-1-yl]methyl]azetidine-1-carboxylate (13g)
- 13f 0.1 g, 391.6 ⁇ mol, 1 equiv
- 2-(2,6-dioxo-3-piperidyl)-5,6- difluoro-isoindoline-1,3-dione 115.2 mg, 391.6 ⁇ mol, 1 equiv
- DIEA 253.0 mg, 1.9 mmol, 341.0 ⁇ L, 5.0 equiv
- Example 14 Synthesis of Compound 14 carboxylate (14a) A solution of benzyl (3-bromopropyl)carbamate (3.9 g, 14.5 mmol, 1.1 equiv) and tetrabutylammonium hydrogen sulfate (449.6 mg, 1.3 mmol, 0.1 equiv) in dichloromethane (30.0 mL) was stirred at 25°C for 5 min, then tert-butyl 4-(2-hydroxyethyl)piperazine-1-carboxylate (3.0 g, 13.2 mmol, 1.0 equiv) and NaOH (1.6 g, 39.7 mmol, 3.0 equiv) were added.
- the reaction mixture was stirred at 40°C for 16 h.
- the mixture was poured into water (100 mL) and the aqueous phase was extracted with ethyl acetate (3 x 100 mL).
- the combined organic phase was washed with brine (3 x 100 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure.
- Step B Synthesis of tert-butyl 4-(2-(3-aminopropoxy)ethyl)piperazine-1-carboxylate (14b)
- 14a 550.0 mg, 1.3 mmol, 1.0 equiv
- 2,2,2-trifluoroethanol 5.0 mL
- Pd/C 694.2 mg, 10.0% Pd on carbon, 50% in water(w/w)
- the mixture was stirred at 50°C for 2 h under H 2 (1 atm).
- the reaction mixture was filtered and the filtrate was concentrated under reduced pressure to afford 14b.
- Step D Synthesis of tert-butyl 4-(2-(3-(3-chloro-4'-((1S,2S)-2-(((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)carbamoyl)cyclopropyl)-[1,1'-biphenyl]-4- carboxamido)propoxy)ethyl)piperazine-1-carboxylate (14d) To a solution of 14c (180.0 mg, 355.1 ⁇ mol, 1.0 equiv) in DMF (1.8 mL) was added DIEA (137.6 mg, 1.0 mmol, 185.5 ⁇ L, 3.0 equiv), BOP (188.4 mg, 426.1 ⁇ mol, 1.2 equiv) and 14b (122.4 mg, 426.1 ⁇ mol, 1.2 equiv).
- Step E Synthesis of 3-chloro-N-(3-(2-(piperazin-1-yl)ethoxy)propyl)-4'-((1S,2S)-2-(((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)carbamoyl)cyclopropyl)-[1,1'- biphenyl]-4-carboxamide (14e) To a solution of 14d (150.0 mg.0, 193 ⁇ mol, 1.0 equiv) in dioxane (0.5 mL) was added HCl (4.0 M, 145 ⁇ L, 3.0 equiv).
- Step F Synthesis of 3-chloro-N-(3-(2-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3- dioxoisoindolin-5-yl)piperazin-1-yl)ethoxy)propyl)-4'-((1S,2S)-2-(((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)carbamoyl)cyclopropyl)-[1,1'-biphenyl]-4- carboxamide (Compound 14) To a solution of 14e (70.0 mg, 103.5 ⁇ mol, 1.0 equiv) in DMSO (1 mL) was added DIEA (40.1 mg, 310.5 ⁇ mol, 54.1 ⁇ L, 3.0 equiv) and 2-(2,6-dioxopiperidin-3-yl)-5,6- difluo
- Example 15 Synthesis of Compound 15 To a solution of 2-chloro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoic acid (1.0 g, 3.5 mmol, 1.0 equiv), DIEA (1.4 g, 10.6 mmol, 1.9 mL, 3.0 equiv) and tert-butyl glycinate (557.1 mg, 4.2 mmol, 1.2 equiv) in DMF (10.0 mL) was added T 4 P (3.1 g, 4.3 mmol, 50.0% purity, 1.2 equiv).
- Step B Synthesis of (2-chloro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoyl)glycine
- Intermediate 15a 1.0 g, 2.5 mmol, 1.0 equiv
- HCl/dioxane 4.0 M, 10.0 mL, 15.8 equiv
- Step C Synthesis of tert-butyl 4-((1-((2-chloro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)benzoyl)glycyl)piperidin-4-yl)methyl)piperazine-1-carboxylate
- 15b 600.0 mg, 1.8 mmol, 1.0 equiv
- DIEA 685.1 mg, 5.3 mmol, 923.3 ⁇ L, 3.0 equiv
- tert-butyl 4-(piperidin-4-ylmethyl)piperazine-1-carboxylate 600.9 mg, 2.1 mmol, 1.2 equiv
- T 4 P 1.9 g, 2.7 mmol, 50% in DMF, 1.5 equiv).
- the reaction mixture was stirred at 25 °C for 1 h.
- the mixture was cooled to 25°C quenched by water (10 mL) and extracted with ethyl acetate (3 x 10 mL).
- the combined organic layers washed with brine (10 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure.
- Step D Synthesis of tert-butyl 4-((1-((3-chloro-4'-((1S,2S)-2-(((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)carbamoyl)cyclopropyl)-[1,1'-biphenyl]-4- carbonyl)glycyl)piperidin-4-yl)methyl)piperazine-1-carboxylate (15d) To solution of 15c (490.9 mg, 811.6 ⁇ mol, 1.4 equiv), Int 3 (250.0 mg, 579.7 ⁇ mol, 1.0 equiv) and Pd(dppf)Cl2•CH2Cl2 (47.3 mg, 57.9 ⁇ mol, 0.1 equiv) in dioxane (2.5 mL) and water (0.5 mL) was added K2CO3 (240.4 mg,
- Step E Synthesis of 3-chloro-N-(2-oxo-2-(4-(piperazin-1-ylmethyl)piperidin-1-yl)ethyl)-4'- ((1S,2S)-2-(((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5- yl)methyl)carbamoyl)cyclopropyl)-[1,1'-biphenyl]-4-carboxamide (15e) A solution of Intermediate 15d (230.0 mg, 277.3 ⁇ mol, 1.0 equiv) in HCl/dioxane (4.0 M, 3.0 mL, 43.2 equiv) was stirred at ambient temperature for 1 h.
- Step F Synthesis of 3-chloro-N-(2-(4-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3- dioxoisoindolin-5-yl)piperazin-1-yl)methyl)piperidin-1-yl)-2-oxoethyl)-4'-((1S,2S)-2-(((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)carbamoyl)cyclopropyl)-[1,1'- biphenyl]-4-carboxamide (Compound 15) To a solution of 15e (100.0 mg, 137.1 ⁇ mol, 1.0 equiv.) in DMSO (1.0 mL) was added DIEA (17.7 mg, 137.1 ⁇ mol, 23.9 ⁇ L, 1.0 equiv) and 2-(2,6-dioxopi
- Step A Synthesis of tert-butyl 4-[4-[[2-chloro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl) benzoyl]amino]-1- piperidyl]piperidine-1-carboxylate (16a) To a solution of 2-chloro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoic acid (0.5 g, 1.77 ⁇ mol, 1.0 equiv) in DMF (5 mL) was added DIEA (686.18 mg, 5.31 mmol, 924.77 ⁇ L, 3 equiv), HOBt (358.7 mg, 2.65 mmol, 1.5 equiv) and EDCI (508.9 mg, 2.65 mmol, 1.5 equiv) and the mixture was stirred at 25°C for 30 minutes.
- Step B Synthesis of tert-butyl 4-[4-[[2-chloro-4-[4-[(1S,2S)-2-[[3-(2,2,2-trifluoro-1,1-dimethyl- ethyl)-1H-1,2,4-triazol-5-yl]methylcarbamoyl]cyclopropyl]phenyl]benzoyl]amino]-1- piperidyl]piperidine-1-carboxylate (16b) To a solution of Intermediate 3 (104.9 mg, 243.3 ⁇ mol, 1 equiv) in dioxane (3 mL) and water (0.5 mL) was added 16a (200 mg, 365.0 ⁇ mol, 1.5 equiv), Pd(dppf)Cl2•CH2Cl2 (19.9 mg, 24.3 ⁇ mol, 0.1 equiv) and K2CO3 (100.9 mg, 730.0 ⁇ mol, 3 equiv) at 25°C.
- the mixture was three-fold degassed and purged with nitrogen, stirred at 100°C for 16 h under nitrogen.
- Step C Synthesis of 2-chloro-N-[1-(4-piperidyl)-4-piperidyl]-4-[4-[(1S,2S)-2-[[3-(2,2,2- trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methylcarbamoyl]cyclopropyl]phenyl]benzamide
- 16c To a solution of 16b (100 mg, 129.5 ⁇ mol, 1 equiv) in dichloromethane (1 mL) was added trifluoroacetic acid (0.2 mL) at 25°C. The mixture was stirred at 25°C for 1 h.
- Step D Synthesis of 2-chloro-N-[1-[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin- 5-yl]-4-piperidyl]-4-piperidyl]-4-[4-[(1S,2S)-2-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4- triazol-5-yl]methylcarbamoyl]cyclopropyl]phenyl]benzamide (Compound 16) To a solution of 16c (90 mg, 114.5 ⁇ mol, 1 equiv) in DMSO (1 mL) was added DIEA (74.0 mg, 572.4 ⁇ mol, 99.7 ⁇ L, 5 equiv) and 2-(2,6-dioxo-3-piperidyl)-5,6-difluoro-isoindoline- 1,3
- Step C Synthesis of tert-butyl 2-[3-methyl-5-[4-[(1S,2S)-2-[[3-(2,2,2-trifluoro-1,1-dimethyl- ethyl)-1H-1,2,4-triazol-5-yl]methylcarbamoyl]cyclopropyl]phenyl]indazol-1-yl]acetate
- 17b (258.9 mg, 695.6 ⁇ mol, 1.5 equiv) and Intermediate 3 (200.0 mg, 463.7 ⁇ mol, 1.0 equiv) in tetrahydrofuran (4 mL) and water (1.0 mL) was added K3PO4 (295.3 mg, 1.3 mmol, 3.0 equiv) and Pd(dppf)Cl2 (60.4 mg, 92.7 ⁇ mol, 0.2 equiv) under nitrogen.
- Step A Synthesis of tert-butyl 3-[4-(4-bromophenyl)pyrazol-1-yl]azetidine-1-carboxylate (18a)
- 4-(4-bromophenyl)-1H-pyrazole 1.2 g, 5.38 mmol, 1.0 equiv
- cesium carbonate 3.51 g, 10.76 mmol, 2.0 equiv
- tert-butyl 3- methylsulfonyloxyazetidine-1-carboxylate (2.03 g, 8.07 mmol, 1.5 equiv).
- Step B Synthesis of 1-(azetidin-3-yl)-4-(4-bromophenyl)pyrazole (18b)
- dichloromethane 25 mL
- trifluoroacetic acid 3.91 mL
- Step D Synthesis of tert-butyl 4-[2-[3-[4-[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)phenyl]pyrazol-1-yl]azetidin-1-yl]acetyl]piperazine-1-carboxylate (18d)
- 18c 200 mg, 396.50 ⁇ mol, 1.0 equiv
- 4,4,5,5-tetramethyl-2-(4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3,2-dioxaborolane 141.0 mg, 555.1 ⁇ mol, 1.4 equiv
- potassium acetate 116.7 mg, 1.19 mmol, 3.0 equiv
- Pd(dppf)Cl 2 32.4 mg, 39.7 ⁇ mol, 0.1 equiv) in dioxane (0.5 mL) was three-fold degassed and
- Step F Synthesis of (1S,2S)-2-[4-[4-[1-[1-(2-oxo-2-piperazin-1-yl-ethyl)azetidin-3-yl]pyrazol-4- yl]phenyl]phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methyl]cyclopropanecarboxamide (18f) To a solution of 18e (58.0 mg, 74.8 ⁇ mol, 1.0 equiv) in dichloromethane (0.5 mL) was added TFA (171 ⁇ L).
- Example 19 Synthesis of Compound 19 tert-butyl 4-(bromomethyl)piperidine-1-carboxylate in Step A to afford (1S,2S)-2-[4-[4-[1-[[1-[2-[4- [2- (2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl] piperazin-1-yl]-2-oxo-ethyl] -4- piperidyl]methyl]pyrazol-4-yl]phenyl]phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H- 1,2,4-triazol-5-yl]methyl]cyclopropanecarboxamide (Compound 19).
- the suspension was three-fold degassed and purged with hydrogen.
- the mixture was stirred under hydrogen (1 atm) at 25°C for 16 h.
- the reaction mixture was filtered to remove solids and the filtrate was poured into NaOH (1M). Volatile components were removed under reduced pressure and the resulting mixture was extracted with dichloromethane (3 x 10 mL).
- the combined organic phase was washed with brine (10 mL), dried with anhydrous sodium sulfate, filtered and filtrate was concentrated under reduced pressure to afford 20b.
- Step C Synthesis of tert-butyl (3S)-3-[[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo- isoindolin-5-yl]-4-piperidyl]oxy]pyrrolidine-1-carboxylate (20c)
- DMSO dimethyl sulfoxide
- 2- (2,6-dioxo-3-piperidyl)-5,6-difluoro-isoindoline-1,3-dione (200.0 mg, 679.7 ⁇ mol, 1.0 equiv) and DIEA (439.2 mg, 3.4 mmol, 592.0 ⁇ L, 5.0 equiv).
- Step D Synthesis of 2-(2,6-dioxo-3-piperidyl)-5-fluoro-6-[4-[(3S)-pyrrolidin-3-yl]oxy-1- piperidyl]isoindoline-1,3-dione (20d)
- the mixture of 20c (190.0 mg, 348.9 ⁇ mol, 1 equiv) in trifluoroacetic acid (5 mL) and dichloromethane (1 mL) was stirred at 25°C for 1 h. The mixture was concentrated under reduced pressure. The residue was triturated with MTBE (5 mL) at 25°C for 20 min to afford 20d.
- Example 21 Synthesis of Intermediate 21 (Int 21) a mg, acid (62.3 mg, 695.6 ⁇ mol, 3 equiv) in EtOH (1 mL) was added DIEA (299.6 mg, 2.32 mmol, 403.9 ⁇ L, 10 equiv) and Pd(dppf)Cl2 (3.02 mg, 4.64 ⁇ mol, 0.02 equiv). The mixture was three-fold degassed and purged with nitrogen and stirred at 50°C for 2 h under an atmosphere of nitrogen.
- Step C Synthesis of tert-butyl 4-[[1-[4-(4-bromopyrazol-1-yl)benzoyl]-4- piperidyl]methyl]piperazine-1-carboxylate (22c)
- HATU 7.49 mmol, 1.30 mL, 4 equiv
- DIEA 967.8 mg, 7.49 mmol, 1.30 mL, 4 equiv
- Step D Synthesis of tert-butyl 4-[[1-[4-[4-[4-[(1S,2S)-2-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H-1,2,4-triazol-5-yl]methylcarbamoyl]cyclopropyl]phenyl]pyrazol-1-yl]benzoyl]-4- piperidyl]methyl]piperazine-1-carboxylate (22d) To a solution of 22c (134.4 mg, 252.4 ⁇ mol, 1 equiv) and Int 21 (100 mg, 252.4 ⁇ mol, 1 equiv) in dioxane (2.5 mL) and water (0.5 mL) was added potassium carbonate (104.6 mg, 757.2 ⁇ mol, 3 equiv) and Pd(dppf)Cl 2 (20.6 mg, 25.2 ⁇ mol, 0.1 equiv).
- the mixture was three-fold degassed and purged with nitrogen and stirred at 100°C for 12 h under an atmosphere of nitrogen.
- the reaction mixture was quenched by addition water (20 mL), and then extracted with ethyl acetate (3 x 10 mL). The combined organic layers were washed with brine (3 x 10 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure.
- Step E Synthesis of (1S,2S)-2-[4-[1-[4-[4-(piperazin-1-ylmethyl)piperidine-1- carbonyl]phenyl]pyrazol-4-yl]phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4- triazol-5-yl]methyl] cyclopropanecarboxamide (22e) To a solution of 22d (80 mg, 99.5 ⁇ mol, 1 equiv) in dichloromethane (0.8 mL) was added trifluoroacetic acid (0.8 mL), the mixture was stirred at 25°C for 1 h and then concentrated under reduced pressure to afford 22e, which was used without further purification.
- Step F Synthesis of (1S,2S)-2-[4-[1-[4-[4-[[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo- isoindolin-5-yl]piperazin-1-yl]methyl]piperidine-1-carbonyl]phenyl]pyrazol-4-yl]phenyl]-N-[[3- (2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]cyclopropanecarboxamide (Compound 22) To a solution of 22e (0.14 g, 198.9 ⁇ mol, 1 equiv) and 2-(2,6-dioxo-3-piperidyl)-5,6- difluoro-isoindoline-1,3-dione (87.7 mg, 298.3 ⁇ mol, 1.5 equiv) in
- Example 23 Synthesis of Compound 23 1,2,4-triazol-5-yl]methylcarbamoyl]cyclopropyl]phenyl]phenyl]methyl]piperazine-1-carboxylate (23a) To a solution of Intermediate 3 (200 mg, 463.7 ⁇ mol, 1.0 equiv), tert-butyl 4-[[4- (4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]methyl]piperazine-1-carboxylate (223.9 mg, 556.5 ⁇ mol, 1.2 equiv) in dioxane (4.0 mL) and water (1.0 mL) was added potassium carbonate (192.2 mg, 1.3 mmol, 3.0 equiv) and Pd(dppf)Cl 2 •CH 2 Cl 2 (189.3 mg, 231.8 ⁇ mol, 0.5 equiv).
- Step B Synthesis of (1S,2S)-2-[4-[4-(piperazin-1-ylmethyl)phenyl]phenyl]-N-[[3-(2,2,2- trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]cyclopropanecarboxamide
- 23a A solution of 23a (115 mg, 183.50 ⁇ mol, 1.0 equiv) in HCl/dioxane (1.0 mL) was stirred at 25°C for 1 h and then concentrated to dryness to afford 23b, which was used without further purification.
- LCMS [M+1] 527.4.
- Step C Synthesis of (1S,2S)-2-(4'-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin- 5-yl)piperazin-1-yl)methyl)-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H- 1,2,4-triazol-5-yl)methyl)cyclopropane-1-carboxamide (Compound 23) To a solution of 23b (60 mg, 113.9 ⁇ mol, 1.0 equiv), 2-(2,6-dioxo-3-piperidyl)-5,6- difluoro-isoindoline-1,3-dione (50.2 mg, 170.9 ⁇ mol, 1.5 equiv) in DMSO (1.0 mL) was added DIEA (44.1 mg, 341.8 ⁇ mol, DM
- Example 24 Synthesis of Compound 24 was a manner tert-butyl 4-[[3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]methyl]piperazine-1-carboxylate in Step A to afford (1S,2S)-2-(4'-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperazin-1-yl)methyl)-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H- 1,2,4-triazol-5-yl)methyl)cyclopropane-1-carboxamide (Compound 24).
- Example 25 Synthesis of Compound 25 Step A – Synthesis of tert-butyl 4-[[4-[2,6-dimethyl-3-[4-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H-1,2,4-triazol-5-yl]methylcarbamoyl]pyrazol-1-yl]phenyl]phenyl]methyl]piperazine-1- carboxylate (25a) To a solution of Intermediate 5 (50 mg, 103.0 ⁇ mol, 1.0 equiv), tert-butyl 4-[[4-(4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]methyl]piperazine-1-carboxylate (62.1 mg, 154.5 ⁇ mol, 1.5 equiv) in tetrahydrofuran (0.4 mL) and water (0.1 mL) was added K 3 PO 4 (65.6 mg, 309.0 ⁇ mol,
- Step B Synthesis of 1-[2,4-dimethyl-3-[4-(piperazin-1-ylmethyl)phenyl]phenyl]-N-[[3-(2,2,2- trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (25b)
- 25a 75 mg, 110.1 ⁇ mol, 1.0 equiv
- dichloromethane 0.4 mL
- trifluoroacetic acid 0.4 mL
- Step C Synthesis of 1-[3-[4-[[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]piperazin-1-yl]methyl]phenyl]-2,4-dimethyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Compound 25) To a solution of 25b (85.0 mg, 52.9 ⁇ mol, 1.0 equiv) in DMSO (1.0 mL) was added 2- (2,6-dioxo-3-piperidyl)-5,6-difluoro-isoindoline-1,3-dione (23.3 mg, 79.3 ⁇ mol, 1.5 equiv) and DIEA (68.3 mg, 528.9
- Example 26 Synthesis of Compound 26 -1H- 1,2,4-triazol-5-yl]methylcarbamoyl]pyrazol-1-yl]phenyl]phenyl]methyl]piperazine-1-carboxylate (26a)
- Step B Synthesis of 1-[2-methyl-4-[4-(piperazin-1-ylmethyl)phenyl]phenyl]-N-[[3-(2,2,2- trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (26b)
- 26a To a solution of 26a (150.0 mg, 224.9 ⁇ mol, 1.0 equiv) in dichloromethane (2.0 mL) and trifluoroacetic acid (0.4 mL). The mixture was stirred at ambient temperature for 2 h. The mixture was concentrated under reduced pressure to afford 26b, which was used without further purification.
- LCMS: [M+1] 567.4.
- Step C Synthesis of 1-[4-[4-[[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]piperazin-1-yl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H- 1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Compound 26) A mixture of 26b (150.0 mg, 220.3 ⁇ mol, 1.0 equiv), 2-(2,6-dioxo-3-piperidyl)-5,6- difluoro-isoindoline-1,3-dione (129.6 mg, 440.7 ⁇ mol, 2.0 equiv) and DIEA (85.4 mg, 661.1 ⁇ mol, 115.1 ⁇ L, 3.0 equiv) in DMSO (2.0
- Example 28 Synthesis of Compound 28 yl)phenyl]methyl]-4-piperidyl]piperazine-1-carboxylate (28a) To a solution of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzaldehyde (215.3 mg, 928.0 ⁇ mol, 1.0 equiv) in dichloromethane (4.0 mL) was added tert-butyl-4-(4- piperidyl)piperazine-1-carboxylate (500.0 mg, 1.86 mmol, 2.0 equiv) and acetic acid (55.7 mg, 928.0 ⁇ mol, 53.1 ⁇ L, 1.0 equiv) the mixture was stirred at 20°C for 1 h and then treated with NaBH(OAc) 3 (393.3 mg, 1.86 mmol, 2.0 equiv).
- Step B Synthesis of tert-butyl 4-[1-[[4-[3-methyl-4-[4-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H-1,2,4-triazol-5-yl]methylcarbamoyl]pyrazol-1-yl]phenyl]phenyl]methyl]-4- piperidyl]piperazine-1-carboxylate (28b)
- a mixture of 28a 231.7 mg, 477.4 ⁇ mol, 1.5 equiv
- Intermediate 4 (150.0 mg, 318.2 ⁇ mol, 1.0 equiv)
- potassium carbonate 131.9 mg, 954.8 ⁇ mol, 3.0 equiv
- Pd(dppf)Cl 2 •CH 2 Cl 2 51.9 mg, 63.6 ⁇ mol, 0.2 equiv) in water (0.2 mL) and dioxane (1.0 mL) was three-fold degassed and purged with nitrogen, and then
- Step C Synthesis of 1-[2-methyl-4-[4-[(4-piperazin-1-yl-1-piperidyl)methyl]phenyl]phenyl]-N- [[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (28c)
- 28b A solution of 28b (210.0 mg, 280.0 ⁇ mol, 1.0 equiv) in trifluoracetic acid (0.2 mL) and dichloromethane (2.0 mL) was stirred at 20°C for 1 h. The mixture was concentrated under reduced pressure to afford 28c.
- LCMS: [M+1] 650.6.
- Step D Synthesis of 1-[4-[4-[[4-[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]piperazin-1-yl]-1-piperidyl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Compound 28).
- Example 29 Synthesis of Compound 29 tert-butyl 4-piperazin-1-ylpiperidine-1-carboxylate in Step A to afford 1-[4-[4-[[4-[1-[2-(2,6-dioxo-3- piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]-4-piperidyl]piperazin-1-yl]methyl]phenyl]-2- methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole- 4-carboxamide (Compound 29).
- Example 31 Synthesis of Compound 31 butyl 4-(piperidin-4-ylmethyl)piperazine-1-carboxylate in Step A to afford 1-(4'-((4-((4-(2-(2,6- dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1-yl)methyl)piperidin-1- yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide.
- Example 32 Synthesis of Compound 32 4-(4-piperidyloxy)piperidine-1-carboxylate in Step A to afford 1-(4'-((4-((1-(2-(2,6- dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperidin-4-yl)oxy)piperidin-1-yl)methyl)- 3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5- yl)methyl)-1H-pyrazole-4-carboxamide.
- Compound 33 was prepared in a similar manner to Example 28 by substituting tert-butyl 1,4-diazepane-1-carboxylate in Step A to afford 1-(4'-((4-(1-(2-(2,6-dioxopiperidin-3-yl)-6- fluoro-1,3-dioxoisoindolin-5-yl)piperidin-4-yl)-1,4-diazepan-1-yl)methyl)-3-methyl-[1,1'- biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H- pyrazole-4-carboxamide.
- Compound 34 was prepared in a similar manner to Example 28 by substituting tert-butyl 4-(4-piperidyl)piperidine-1-carboxylate in Step A to afford 1-(4'-((1'-(2-(2,6-dioxopiperidin-3-yl)- 6-fluoro-1,3-dioxoisoindolin-5-yl)-[4,4'-bipiperidin]-1-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)- N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4- carboxamide.
- Example 36 Synthesis of Compound 36 5-(4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine-2-carbaldehyde in Step A to afford 1-(4-(6-((4-(4- (2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1-yl)piperidin-1- yl)methyl)pyridin-3-yl)-2-methylphenyl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide.
- Example 37 Synthesis of Compound 37 3-chloro-4- (4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzaldehyde in Step A to afford 1-(2'-chloro-4'-((4- (4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1-yl)piperidin-1- yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide.
- Example 39 Synthesis of Compound 39-1 and 39-2 substituting 1-[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]ethenone in Step A. Separation of enantiomers after Step B by SFC (DAICEL CHIRALPAK AD (250 mm x 30 mm,10 ⁇ m); [CO2-IPA(0.1%NH3water)]; isocratic elution) afforded the early-eluting enantiomer, which was processed further to afford 39-1 (1-(4'-(1-(4-(4-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro- 1,3-dioxoisoindolin-5-yl)piperazin-1-yl)piperidin-1-yl)ethyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N- ((3-(1,1,1-
- Example 40 Synthesis of Compound 40 carboxylate (40a) A mixture of tert-butyl 4-(4-piperidyl)piperazine-1-carboxylate (792.3 mg, 2.9 mmol, 1.3 equiv.), 4-bromo-2,6-difluoro-benzaldehyde (500 mg, 2.2 mmol, 1.0 equiv.) and acetic acid (135.8 mg, 2.2 mmol, 129.5 ⁇ L, 1.0 equiv.) in dichloromethane (5 mL) was stirred at 30°C for 2 h.
- Step C Synthesis of tert-butyl 4-[1-[[2,6-difluoro-4-[3-methyl-4-[4-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methylcarbamoyl]pyrazol-1-yl]phenyl]phenyl]methyl]-4- piperidyl]piperazine-1-carboxylate (40c) To a solution of Int 4 (100 mg, 212.1 ⁇ mol, 1.0 equiv.), 40b (221.2 mg, 424.3 ⁇ mol, 2.0 equiv.) in dioxane (2 mL) and water (0.2 mL) was added potassium carbonate (87.9 mg, 636.5 ⁇ mol, 3.0 equiv.) and Pd(dppf)Cl 2 .CH 2 Cl 2 (34.6 mg, 42.4 ⁇ mol, 0.2 equiv.).
- Step D Synthesis of 1-[4-[3,5-difluoro-4-[(4-piperazin-1-yl-1-piperidyl)methyl]phenyl]-2- methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole- 4-carboxamide (40d) To a solution of 40c (80.0 mg, 101.8 ⁇ mol, 1.0 equiv.) in HCl in dioxane (4 M, 1 mL) under nitrogen. The mixture was stirred at 25°C for 1 h.
- the mixture was stirred at 100°C for 3 h.
- the mixture was cooled to ambient temperature and poured into water (100 mL).
- the aqueous phase was extracted with ethyl acetate (3 x 100 mL).
- the combined organic phase was washed with brine (3 x 50 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum.
- Step B Synthesis of tert-butyl 4-(3-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H- indazol-1-yl)-[1,4'-bipiperidine]-1'-carboxylate (41b)
- 41a 1.5 g, 3.1 mmol, 1.0 equiv.
- 4,4,5,5-tetramethyl-2-(4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3,2-dioxaborolane 1.2 g, 4.7 mmol, 1.5 equiv.
- potassium acetate 925.0 mg, 9.4 mmol, 3.0 equiv.
- Pd(dppf)Cl2 229.8 mg, 314.1 ⁇ mol, 0.1 equiv.
- Step C Synthesis of tert-butyl 4-(3-methyl-5-(3-methyl-4-(4-(((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)carbamoyl)-1H-pyrazol-1-yl)phenyl)-1H- indazol-1-yl)-[1,4'-bipiperidine]-1'-carboxylate (41c) To a solution of Int 4 (250.0 mg, 530.4 ⁇ mol, 1.0 equiv.) and 41b (333.88 mg, 636.57 ⁇ mol, 1.2 equiv.) in dioxane (2.5 mL) and water (0.4 mL) was added K2CO3 (219.9 mg, 1.5 mmol, 3.0 equiv.) and Pd(dppf)Cl2.CH2Cl2 (43.3 mg, 53.0 ⁇ mol, 0.1
- Step D Synthesis of 1-(4-(1-([1,4'-bipiperidin]-4-yl)-3-methyl-1H-indazol-5-yl)-2- methylphenyl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H- pyrazole-4-carboxamide (41d)
- 41c A solution of 41c (230.0 mg, 291.5 ⁇ mol, 1.0 equiv.) in HCl/dioxane (4 M, 2.5 mL) was stirred at ambient temperature for 0.5 h.
- Step E Synthesis of 1-(4-(1-(1'-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)- [1,4'-bipiperidin]-4-yl)-3-methyl-1H-indazol-5-yl)-2-methylphenyl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide (Compound 41) To a solution of 41d (150.0 mg, 197.0 ⁇ mol, 1.0 equiv.) in DMSO (1.5 mL) was added DIEA (84.4 mg, 653.3 ⁇ mol, 113.8 ⁇ L, 3.3 equiv.) and 2-(2,6-dioxopiperidin-3-yl)-5,6- difluoroisoindoline-1
- Example 42 Synthesis of Compound 42 (42a) To solution of tert-butyl 4-(4-hydroxycyclohexyl)piperazine-1-carboxylate (969.6 mg, 3.41 mmol, 1.2 equiv.) in DMF (10.0 mL) was added NaH (227.3 mg, 5.68 mmol, 60.0% purity, 2.0 equiv.) at 0°C and the mixture was stirred at 0°C for 1 h. 5-bromo-2-fluoro-pyridine (500.0 mg, 2.84 mmol, 292.40 ⁇ L, 1.0 equiv.) was added to the mixture. The mixture was stirred at 25°C for 12 h.
- Step C Synthesis of tert-butyl 4-[4-[[5-[3-methyl-4-[4-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H-1,2,4-triazol-5-yl]methylcarbamoyl]pyrazol-1-yl]phenyl]-2-pyridyl]oxy]-1- piperidyl]piperidine-1-carboxylate (42c) To a solution of 42b (150.0 mg, 184.64 ⁇ mol, 1.5 equiv.) and Int 4 (58.0 mg, 123.09 ⁇ mol, 1.0 equiv.) in THF (1.0 mL) and water (0.25 mL) was added K3PO4 (78.39 mg, 369.28 ⁇ mol, 3.0 equiv.) and PdCl 2 (DTBPF) (16.05 mg, 24.62 ⁇ mol, 0.2 equiv.) at 25°C under
- Step D Synthesis of 1-[2-methyl-4-[6-[[1-(4-piperidyl)-4-piperidyl]oxy]-3-pyridyl]phenyl]-N- [[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (42d)
- 42c A solution of 42c (100.0 mg, 79.80 ⁇ mol, 1 equiv.) in HCl/MeOH (3 M, 1.0 mL) was stirred at 25°C for 1 h. The reaction was monitored by LCMS. The reaction mixture was concentrated under reduced pressure to give 42d.
- Step E Synthesis of 1-[4-[6-[[1-[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]-4-piperidyl]-4-piperidyl]oxy]-3-pyridyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Compound 42) To a solution of 42d (50.0 mg, 69.1 ⁇ mol, 1 equiv.) and 2-(2,6-dioxo-3-piperidyl)-5,6- difluoro-isoindoline-1,3-dione (22.6 mg, 76.7 ⁇ mol, 1.1 equiv.) in DMSO (1.0 mL) was added DIEA (99
- the mixture was stirred at 90°C for 16 h.
- the reaction mixture was partitioned between ethyl acetate (100 mL) and water (100 mL) extracted with ethyl acetate (2 x 100 mL).
- the combined organic layers were washed with brine (5 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure.
- Step A Synthesis of 1-[1-(4-bromophenyl)-1-methyl-ethyl]piperidin-4-one (46a)
- 2-(4-bromophenyl)propan-2-amine 2.0 g, 9.3 mmol, 1.0 equiv.
- EtOH 20 mL
- water 8 mL
- potassium carbonate 129.1 mg, 934.1 ⁇ mol, 0.1 equiv.
- 1-ethyl-1-methyl-piperidin-1-ium-4-one iodide 2.0 g, 7.4 mmol, 0.8 equiv.
- Step B Synthesis of tert-butyl 4-[1-[1-(4-bromophenyl)-1-methyl-ethyl]-4-piperidyl]piperazine- 1-carboxylate (46b)
- acetic acid (223.0 mg, 3.7 mmol, 212.6 ⁇ L, 1.0 equiv.)
- NaBH(OAc) 3 3.1 g, 14.8 mmol, 4.0 equiv.
- tert-butyl piperazine-1-carboxylate 830.0 mg, 4.4 mmol, 1.2 equiv.
- Step C Synthesis of tert-butyl 4-[1-[1-methyl-1-[4-[3-methyl-4-[4-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methylcarbamoyl]pyrazol-1-yl]phenyl]phenyl]ethyl]-4- piperidyl]piperazine-1-carboxylate (46c) To a solution of 46b (100.0 mg, 214.3 ⁇ mol, 1.0 equiv.) in dioxane (1 mL), water (0.2 mL) was added potassium carbonate (59.2 mg, 428.7 ⁇ mol, 2.0 equiv.) and Pd(dppf)Cl2 (15.6 mg, 21.4 ⁇ mol, 0.1 equiv.), Int 45 (111.1 mg, 214.3 ⁇ mol, 1.0 equiv.) under a nitrogen atmosphere.
- Step D Synthesis of 1-[2-methyl-4-[4-[1-methyl-1-(4-piperazin-1-yl-1-piperidyl)ethyl]phenyl]- phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4- carboxamide (46d) To a solution of 46c (100.0 mg, 128.5 ⁇ mol, 1.0 equiv.) in CF3CwaterH (10 mL) was added chlorotrimethylsilane (856.0 mg, 7.8 mmol, 1 mL, 61.2 equiv.).
- Step B Synthesis of tert-butyl 4-[1-[[5-[3-methyl-4-[4-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H-1,2,4-triazol-5-yl]methylcarbamoyl]pyrazol-1-yl]phenyl]pyrazin-2-yl] methyl]-4-piperidyl] piperazine-1-carboxylate (47b) To a solution of 47a (130.7 mg, 330.1 ⁇ mol, 1.2 equiv.) and Int 45 (142.6 mg, 275.1 ⁇ mol, 1.0 equiv.) in dioxane (2.0 mL) and water (0.4 mL) was added potassium carbonate (114.0 mg, 825.3 ⁇ mol, 3.0 equiv.) and Pd(dppf)Cl2 (44.9 mg, 55.0 ⁇ mol, 0.2 equiv.).
- Step D Synthesis of 1-[4-[5-[[4-[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]piperazin-1-yl]-1-piperidyl]methyl]pyrazin-2-yl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Compound 47).
- Example 48 Synthesis of Compound 48 6- bromopyridine-3-carbaldehyde in Step A to afford 1-[4-[5-[[4-[4-[2-(2,6-dioxo-3-piperidyl)-6- fluoro-1,3-dioxo-isoindolin-5-yl]piperazin-1-yl]-1-piperidyl]methyl]-2-pyridyl]-2-methyl- phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4- carboxamide.
- LCMS [M+1] 925.2.
- a solution of tert-butyl 4-(6-bromo-3-pyridyl)piperazine-1-carboxylate (500.0 mg, 1.5 mmol, 1.0 equiv.) and 4-bromophenol (758.3 mg, 4.4 mmol, 3.0 equiv.) in DMSO (5.0 mL) was added K3PO4 (620.2 mg, 2.9 mmol, 2.0 equiv.), pyridine-2-carboxylic acid (18.0 mg, 146.1 ⁇ mol, 0.1 equiv.) and CuI (13.9 mg, 73.1 ⁇ mol, 0.05 equiv.).
- Step B Synthesis of tert-butyl 4-[6-[4-[3-methyl-4-[4-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H-1,2,4-triazol-5-yl]methylcarbamoyl]pyrazol-1-yl]phenyl]phenoxy]-3-pyridyl] piperazine-1- carboxylate (50b) To a solution of 50a (80.0 mg, 184.2 ⁇ mol, 1.0 equiv.) and Int 45 (100.3 mg, 193.4 ⁇ mol, 1.05 equiv.) in THF (0.8 mL) and water (0.2 mL) was added K3PO4 (78.2 mg, 368.4 ⁇ mol, 2.0 equiv.) and PdCl2(DTBPF) (24.0 mg, 36.8 ⁇ mol, 0.2 equiv.).
- Step C Synthesis of 1-[2-methyl-4-[4-[(5-piperazin-1-yl-2-pyridyl) oxy]phenyl]phenyl]-N-[[3- (2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl] methyl]pyrazole-4-carboxamide (50c)
- 50b To a solution of 50b (60.0 mg, 80.5 ⁇ mol, 1.0 equiv.) in dichloromethane (0.6 mL) was added TFA (0.2 mL). The mixture was stirred at 25°C for 1 h. The mixture was concentrated under reduced pressure to afford 50c, which was used without further purification.
- Example 51 Synthesis of Compound 51 Step A – Synthesis of tert-butyl 4-(6-oxo-2,3-dihydro-1H-pyridin-4-yl)piperazine-1-carboxylate (51a) To a mixture of piperidine-2,4-dione (303.6 mg, 2.6 mmol, 1.0 equiv.) and tert-butyl piperazine-1-carboxylate (500 mg, 2.6 mmol, 1.0 equiv.) in CHCl3 (1.5 mL) was added acetic acid (165.2 mg, 2.7 mmol, 157.5 ⁇ L, 1.0 equiv.), the mixture was stirred at 60°C for 16 h.
- reaction mixture was quenched with water 5 mL at 25°C, and then extracted with dichloromethane (3 mL x 3). The combined organic portions were dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure to afford 51a.
- Step D Synthesis of tert-butyl 4-[1-[[4-[3-methyl-4-[4-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H-1,2,4-triazol-5-yl]methylcarbamoyl]pyrazol-1-yl]phenyl]phenyl]methyl]-2-oxo-4- piperidyl]piperazine-1-carboxylate (51d) To a mixture of 51c (69.8 mg, 154.3 ⁇ mol, 1.0 equiv.) and Int 45 (80.0 mg, 154.3 ⁇ mol, 1.0 equiv.) and K2CO3 (63.9 mg, 463.0 ⁇ mol, 3.0 equiv.) in dioxane (1 mL) and water (0.2 mL) was three-fold degassed and purged with nitrogen, treated with Pd(DPPF)Cl2 (22.5 mg, 30.8 ⁇ mol, 0.2 e
- Step E Synthesis of 1-[2-methyl-4-[4-[(2-oxo-4-piperazin-1-yl-1-piperidyl)methyl]phenyl]- phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4- carboxamide (51e)
- 51d A mixture of 51d (40.0 mg, 52.3 ⁇ mol, 1.0 equiv.) in dichloromethane (0.5 mL) and TFA (0.1 mL) was stirred at 25°C for 0.5 h. The reaction mixture was concentrated under reduced pressure to afford (51e), which was used without further purification.
- Step A Synthesis of tert-butyl 4-(4-hydroxycyclohexyl) piperazine-1-carboxylate (52a)
- 4-aminocyclohexanol 5 g, 43.41 mmol, 1.05 equiv.
- tert-butyl N,N- bis(2-chloroethyl)carbamate 10.01 g, 41.35 mmol, 1 equiv.
- K2CO3 17.14 g, 124.04 mmol, 3 equiv.
- KI 20.59 g, 124.04 mmol, 3 equiv.
- Step B Synthesis of tert-butyl 4-[4-[(5-bromo-2-pyridyl)oxy]cyclohexyl]piperazine-1- carboxylate (52b)
- THF 10 mL
- NaH 211 mg, 5.3 mmol, 60% purity, 1.5 equiv.
- 5-bromo-2-fluoro-pyridine 618.81 mg, 3.52 mmol, 361.88 ⁇ L, 1 equiv.
- Step C Synthesis of tert-butyl4-[4-[[5-[3-methyl-4-[4-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H-1,2,4-triazol-5-yl]methylcarbamoyl]pyrazol-1-yl]phenyl]-2- pyridyl]oxy]cyclohexyl]piperazine-1-carboxylate (52c) To a solution of 52b (220 mg, 499.6 ⁇ mol, 1 equiv.) and Int 45 (258.9 mg, 499.6 ⁇ mol, 1.0 equiv.) in water (0.2 mL) and THF (0.8 mL) was added K 3 PO 4 (318.13 mg, 1.50 mmol, 3 equiv.) at 20°C, sequentially, was added PdCl 2 (DTBPF) (65.12 mg, 99.92 ⁇ mol, 0.2 e
- Step E Synthesis of 1-[4-[6-[4-[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]piperazin-1-yl]cyclohexoxy]-3-pyridyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl- ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Compound 52) To a solution of 52d (52 mg, 80 ⁇ mol, 1 equiv.) and 2-(2,6-dioxo-3-piperidyl)-5,6- difluoro-isoindoline-1,3-dione (35.2 mg, 120 ⁇ mol, 1.5 equiv.) in DMSO (5 mL) was added DIEA (51.6 mg, 399 ⁇ mol,
- Step B Synthesis of tert-butyl 4-[4-[[4-[3-methyl-4-[4-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H- 1,2,4-triazol-5-yl]methylcarbamoyl]pyrazol-1-yl]phenyl]pyrazol-1-yl]methyl]-1- piperidyl]piperidine-1-carboxylate (53b) A mixture of 53a (79.1 mg, 185.2 ⁇ mol, 1.2 equiv.), Int 45 (80.0 mg, 154.3 ⁇ mol, 1.0 equiv.), K3PO4 (98.3 mg, 463.0 ⁇ mol, 3.0 equiv.), PdCl2(DTBPF) (20.1 mg, 30.9 ⁇ mol, 0.2 equiv.) in water (0.16 mL) and THF (0.64 mL) was degassed and purged threefold with nitrogen.
- Step C Synthesis of 1-[2-methyl-4-[1-[[1-(4-piperidyl)-4-piperidyl] methyl]pyrazol-4- yl]phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4- carboxamide (53c) To a solution of 53b (43.0 mg, 58.2 ⁇ mol, 1.0 equiv.) in dichloromethane (0.4 mL) was added TFA (0.2 mL).
- Example 56 Synthesis of Compound 56 Step A -1H- 1,2,4-triazol-5-yl]methylcarbamoyl]pyrazol-1-yl]phenyl]phenyl]methyl]-2,7- diazaspiro[3.5]nonane-2-carboxylate (56a) To a solution of tert-butyl 2,7-diazaspiro[3.5]nonane-2-carboxylate (68.3 mg, 302.1 ⁇ mol, 1.5 equiv) and 1-[4-(4-formylphenyl)-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl- ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (100.0 mg, 201.4 ⁇ mol, 1.0 equiv) in DCE (2.0 mL) was added triethylamine (40.76 mg, 402.8 ⁇ mol, 5
- Step B Synthesis of 1-[4-[4-[[2-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]- 2,7-diazaspiro[3.5]nonan-7-yl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Compound 56) A solution of 56a (50.0 mg, 70.7 ⁇ mol, 1.0 equiv) in HCl/ dioxane (4.0 M, 1.0 mL) was stirred at 25°C for 30 mins.
- the reaction mixture was concentrated under reduced pressure. The residue was dissolved in DMSO (1.0 ml) and treated with 2-(2,6-dioxo-3-piperidyl)-5,6-difluoro- isoindoline-1,3-dione (29.1 mg, 98.9 ⁇ mol, 1.5 equiv) and DIEA (42.6 mg, 329.6 ⁇ mol, 57.4 ⁇ L, 5.0 equiv). The mixture was stirred at 70°C for 12 h. The reaction was cooled ambient temperature and diluted by water (2.0 mL). The mixture extracted with ethyl acetate (3 x 2 mL).
- Step A Synthesis of benzyl 4-(4-tert-butoxycarbonylpiperazin-1-yl) azepane-1-carboxylate (57a)
- benzyl 4-oxoazepane-1-carboxylate (2.00 g, 8.09 mmol, 1.0 equiv.)
- tert-butyl piperazine-1-carboxylate (1.66 g, 8.90 mmol, 1.1 equiv.) in THF (20 mL) was added AcOH (971 mg, 16.2 mmol, 926 ⁇ L, 2 equiv.).
- the mixture was stirred at 25 °C for 30 min, treated with NaBH(OAc)3 (3.43 g, 16.18 mmol, 2 equiv.) was added to the reaction mixture and stirred for 1.5 h at 25°C.
- the reaction mixture was quenched with water (10 mL) at 20 °C, diluted with water (100 mL) and extracted with ethyl acetate (100 mL x 2). The combined organic layers were dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure to afford 57a, which was used without further purification.
- Step B Synthesis of tert-butyl 4-(azepan-4-yl)piperazine-1-carboxylate (57b)
- 57a 3.2 g, 7.66 mmol, 1 equiv.
- THF 10 mL
- Pd/C 1.63 g, 1.53 mmol, 10% purity, 0.2 equiv.
- the mixture was degassed and purged three-fold with hydrogen and stirred at 25 °C for 16 h under a hydrogen atmosphere (15 Psi).
- the reaction mixture was filtered and the filtrate was concentrated under reduced pressure to afford 57b, which was used without further purification.
- Step C Synthesis of tert-butyl 4-[1-[[4-[3-methyl-4-[4-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H-1,2,4-triazol-5-yl]methylcarbamoyl]pyrazol-1-yl]phenyl]phenyl]methyl]azepan-4- yl]piperazine-1-carboxylate (57c) To a solution of 57b (300 mg, 1.06 mmol, 1 equiv.) and Int 55 (472.99 mg, 952.69 ⁇ mol, 0.9 equiv.) in THF (10 mL) was added AcOH (127.14 mg, 2.12 mmol, 121.20 ⁇ L, 2 equiv.).
- Step D Synthesis of 1-(4'-((4-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperazin-1-yl)azepan-1-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide (Compound 57-1) Intermediate 57c-1 (100 mg, 130.91 ⁇ mol, 1 equiv.) was dissolved into HCl/dioxane (4 M, 10 mL), and the mixture was stirred at 25 °C for 2 h.
- Step A olin-5-yl]- 1,4-diazepane-1-carboxylate (58a)
- 2-(2,6-dioxo-3-piperidyl)-5,6-difluoro-isoindoline-1,3-dione 500 mg, 1.70 mmol, 1 equiv.
- DMSO DMSO
- DIPEA tert-butyl-1,4-diazepane-1-carboxylate
- the mixture was stirred at 80 °C for 4 h.
- reaction mixture was quenched with water (100 mL) at 20 °C and extracted with ethyl acetate. The combined organic portions were washed with brine (10 mL x 3), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum to afford 58a which was used without further purification.
- Step B Synthesis of 1-(4'-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)- 1,4-diazepan-1-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan- 2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide (Compound 58) To a solution of 58a (450 mg, 948 ⁇ mol, 1 equiv.) in dichloromethane (10 mL) was added TFA (1.08 g, 9.48 mmol, 704 ⁇ L, 10 equiv.).
- the mixture was treated with NaBH(OAc) 3 (4.4 g, 21.2 mmol, 3.0 equiv.) and stirred at for 2 h at 25°C.
- the reaction mixture was quenched with aqueous.NaHCO3 (5 mL) at 25°C and extracted with dichloromethane (3 mL x 3), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure.
- Step C Synthesis of tert-butyl 8-[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]-4-piperidyl]-3,8-diazabicyclo[3.2.1]octane-3-carboxylate (59c)
- 59c To a mixture of 59b (150 mg, 507.7 ⁇ mol, 1.0 equiv.) and 2-(2,6-dioxo-3-piperidyl)-5,6- difluoro-isoindoline-1,3-dione (179.2 mg, 609.3 ⁇ mol, 1.2 equiv.) in DMSO (2 mL), was added DIEA (328.1 mg, 2.5 mmol, 442.2 ⁇ L, 5.0 equiv.).
- Step D Synthesis of 5-[4-(3,8-diazabicyclo[3.2.1]octan-8-yl)-1-piperidyl]-2-(2,6-dioxo-3- piperidyl)-6-fluoro-isoindoline-1,3-dione (59d)
- 59d A solution of 59c (70.0 mg, 122.8 ⁇ mol, 1 equiv.) in chlorotrimethylsilane (0.1 mL) and trifluoroethanol (0.9 mL) was stirred at 25°C for 30 min. The reaction mixture was concentrated under reduced pressure to afford 59c, which was used without further purification.
- LCMS: [M+1] 470.4.
- Step E Synthesis of 1-[4-[4-[[8-[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]-4-piperidyl]-3,8-diazabicyclo[3.2.1]octan-3-yl]methyl]phenyl]-2-methyl-phenyl]-N-[[3- (2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Compound 59) A mixture of 59d (41.6 mg, 71.3 ⁇ mol, 1.1 equiv.) and Int 55 (40.0 mg, 80.57 ⁇ mol, 1.1 equiv.) in DCE (0.5 mL) was treated with AcOH (9.6 mg, 161.1 ⁇ mol, 9.2 ⁇ L, 2.25 equiv.) and
- Example 60 Synthesis of Compound 60 Compound 60 was prepared in a similar manner to Example 59 by substituting tert-butyl 3,6-diazabicyclo[3.1.1]heptane-6-carboxylate in Step A to afford 1-(4'-((3-(1-(2-(2,6- dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperidin-4-yl)-3,6- diazabicyclo[3.1.1]heptan-6-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide.
- Example 61 Synthesis of Compound 61 tert-butyl (1S,4S)-2,5-diazabicyclo[2.2.1]heptane-2-carboxylate in Step A to afford 1-(4'-(((1S,4S)-5-(1-(2- (2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperidin-4-yl)-2,5- diazabicyclo[2.2.1]heptan-2-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide.
- Compound 63 was prepared in a similar manner to Example 59 by substituting tert-butyl 3,8-diazabicyclo[3.2.1]octane-8-carboxylate in Step A to afford 1-(4'-((3-(1-(2-(2,6- dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperidin-4-yl)-3,8- diazabicyclo[3.2.1]octan-8-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide .
- Example 64 Synthesis of Compound 64 butyl 3-(piperazin-1-yl)azetidine-1-carboxylate in Step C to afford 1-(4'-((3-(4-(2-(2,6-dioxopiperidin- 3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1-yl)azetidin-1-yl)methyl)-3-methyl-[1,1'- biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H- pyrazole-4-carboxamide.
- Step B Synthesis of 5-[4-(2,6-diazaspiro[3.3]heptan-2-yl)-1-piperidyl]-2-(2,6-dioxo-3- piperidyl)-6-fluoro-isoindoline-1,3-dione (65b)
- 65a 130 mg, 233.9 ⁇ mol, 1 equiv.
- dichloromethane 1 mL
- Step C Synthesis of 1-[4-[4-[[2-[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]-4-piperidyl]-2,6-diazaspiro[3.3]heptan-6-yl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2- trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Compound 65) A solution of 65b (60 mg, 105 ⁇ mol, 1 equiv.) and Int 55 (65.4 mg, 132 ⁇ mol, 1.25 equiv.) in THF (1 mL) was treated with Ti(Oi-Pr) 4 (300 mg, 1.1 mmol, 311 ⁇ L, 10.5 equiv.) and heated at
- Step B Synthesis of 2-(2,6-dioxo-3-piperidyl)-5-fluoro-6-piperazin-1-yl-isoindoline-1,3-dione (66b)
- 66a 2-(2,6-dioxo-3-piperidyl)-5-fluoro-6-piperazin-1-yl-isoindoline-1,3-dione
- the reaction mixture was cooled to room temperature, filtered through Celite and the filtrate was concentrated under vacuum. The residue was dissolved in methanol (2.5 mL) and DCE (2.5 mL) and treated with NaBH3CN (395.5 mg) and acetic acid (0.25 mL). The mixture was stirred at 40 °C for 16 h., cooled to ambient temperature, diluted with water (20 mL) and extracted with dichloromethane (15 mL x 3).
- Step D Synthesis of 1-[4-[4-[[4-[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]piperazin-1-yl]-3,3-difluoro-1-piperidyl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2- trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Compound 66) To a solution of 66c (300.0 mg, 518 ⁇ mol, 1.0 equiv.) in dichloromethane (3 mL) was added TFA (921.0 mg, 8.08 mmol, 600.0 ⁇ L, 15.6 equiv.).
- a solution of benzyl 4-oxopiperidine-1-carboxylate (500.0 mg, 2.1 mmol, 426.6 ⁇ L, 1.0 equiv.) in toluene (5.0 mL) was treated with tert-butyl piperazine-1-carboxylate (439.1 mg, 2.3 mmol, 1.1 equiv.) and 1,2,3-triazole (177.6 mg, 2.5 mmol, 1.2 equiv.) at 100 o C for 12 h, cooled to ambient temperature and treated with MeMgBr (641.2 mg, 5.3 mmol, 3 M, 2.5 equiv.), the solution was stirred at 25 o C for 3 h.
- Step B Synthesis of 5-(4-chloro-2-methyl-phenyl)-1-(2-methoxyethyl)-6-oxo-pyridine-3- carboxylic acid (67b)
- Pd/C 101.9 mg, 10 wt%) under nitrogen.
- the suspension was degassed under vacuum and purged with hydrogen.
- the mixture was stirred under hydrogen (15 Psi) at ambient temperature for 3 h.
- the mixture was filtered through a pad of Celite and the filter bed was washed with methanol (5 mL x 3).
- Step C Synthesis of tert-butyl 4-[4-methyl-1-[[4-[3-methyl-4-[4-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methylcarbamoyl]pyrazol-1-yl]phenyl]phenyl]methyl]-4- piperidyl]piperazine-1-carboxylate (67c)
- a solution of 67b (59.9 mg, 211 ⁇ mol, 3.0 equiv.) in DCE (1.0 mL) was treated with triethylamine (14.2 mg, 141 ⁇ mol, 19.6 ⁇ L, 2.0 equiv.), Int 55 (35.0 mg, 70.5 ⁇ mol, 1.0 equiv.) and NaBH(OAc) 3 (44.8 mg, 211 ⁇ mol, 3.0 equiv.).
- Step D Synthesis of 1-[4-[4-[[4-[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]piperazin-1-yl]-4-methyl-1-piperidyl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro- 1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Compound 67) A solution of 67c (70.0 mg, 92 ⁇ mol, 1.0 equiv.) in HCl/ MeOH (3M, 1.0 mL) was stirred at ambient temperature for 12 h.
- Step D Synthesis of 1-[4-[4-[[4-[6-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]-2,6-diazaspiro[3.3]heptan-2-yl]-1-piperidyl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2- trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Compound 68) A mixture of 68c (110 mg, 144.5 ⁇ mol, 1 equiv.) in TFA (0.2 mL) and dichloromethane (1 mL) was stirred at 20°C for 1 h.
- Step B Synthesis of tert-butyl 4-((2S)-2-methylpiperidin-4-yl) piperazine-1-carboxylate (69b)
- 69a 3.20 g, 7.6 mmol, 1.0 equiv.
- MeOH 40.0 mL
- Pd/C 3.00 g, 10 wt%.
- the mixture was stirred at 20°C for 4 h under hydrogen (1 atm).
- the solution was filtered and the filter cake was washed with MeOH (200 mL x 3).
- the combined filtrate was concentrated under vacuum to afford 69b.
- Step C Synthesis of tert-butyl 4-((2S,4S)-2-methyl-1-((3'-methyl-4'-(4-(((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)carbamoyl)-1H-pyrazol-1-yl)-[1,1'-biphenyl]-4- yl)methyl)piperidin-4-yl)piperazine-1-carboxylate (69c-1) and tert-butyl 4-[(2S,4R)-2-methyl-1- [[4-[3-methyl-4-[4-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methylcarbamoyl]pyrazol-1-yl]phenyl]phenyl]methyl]-4-piperidyl]piperazine-1
- Step A To a solution of 5-bromo-1H-indazole (2.1 g, 10.4 mmol, 1.0 equiv.) in DMF (20.0 mL) was added NaH (416.6 mg, 10.4 mmol, 60 wt%, 1.0 equiv.) at 0°C and the resulting mixture was stirred for 5 min, treated with 3-bromopiperidine-2, 6-dione (2.00 g, 10.4 mmol, 1.0 equiv.) and stirred at 20 °C for 1 h. The mixture was quenched by water (10 mL) and extracted with ethyl acetate (10 mL x 3).
- Step B Synthesis of tert-butyl 4-(1-(2,6-dioxopiperidin-3-yl)-1H-indazol-5-yl)piperidine-1- carboxylate (71b)
- 71a 400.0 mg, 1.3 mmol, 1.3 equiv.
- tert-butyl 4-bromopiperidine-1-carboxylate (445.8 mg, 1.7 mmol, 1.3 equiv.)
- Ir[dF(CF3)ppy]2(dtbpy)(PF6) (14.6 mg, 13 ⁇ mol, 0.1 equiv.
- NiCl2.dtbbpy 7.8 mg, 20 ⁇ mol, 0.1 equiv.
- tris(trimethylsilyl)silane 322.8 mg, 1.3 mmol, 400.5 ⁇ L, 1.
- Step D Synthesis of tert-butyl-4-(1-(2,6-dioxopiperidin-3-yl)-1H-indazol-5-yl)-[1,4'- bipiperidine]-1'-carboxylate (71d)
- 71d To a solution of tert-butyl-4-oxopiperidine-1-carboxylate (287.0 mg, 1.4 mmol, 1.6 equiv.) and 71c (300.0 mg, 0.86 mmol, 1.0 equiv.) in THF (1.0 mL) was added Ti(OEt) 4 (1.1 g, 4.8 mmol, 5.6 equiv.).
- Step E Synthesis of 1-(4'-((4-(1-(2,6-dioxopiperidin-3-yl)-1H-indazol-5-yl)-[1,4'-bipiperidin]- 1'-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H- 1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide (Compound 71) A solution of 71d (300 mg, 605 ⁇ mol, 1.0 equiv.) in HCl/dioxane (4.0 M, 5.1 mL, 33.4 equiv.) was stirred at 25°C for 0.5 h.
- Example 72 Synthesis of Compound 72 (3- piperazin-1-ylphenyl)piperidine-2,6-dione in Step D to afford 1-(4'-((4-(4-(3-(2,6-dioxopiperidin- 3-yl)phenyl)piperazin-1-yl)piperidin-1-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide.
- Example 73 Synthesis of Compound 73 carboxylate (73a) To a solution of tert-butyl 4-[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)phenyl]piperazine-1-carboxylate (2.0 g, 5.1 mmol, 1.0 equiv.) and 2,6-dibenzyloxy-3-bromo- pyridine (2.6 g, 7.2 mmol, 1.4 equiv.) in dioxane (100.0 mL) and water (20.0 mL) was added Pd(dppf)Cl 2 (376.8 mg, 515 ⁇ mol, 0.1 equiv.) and K 2 CO 3 (1.4 g, 10.3 mmol, 2.0 equiv.).
- Step B Synthesis of tert-butyl 4-[4-(2,6-dioxo-3-piperidyl)phenyl]piperazine-1-carboxylate (73b)
- 73a a solution of 73a (10.0 g, 18.1 mmol, 1.0 equiv.) in EtOH (60 mL) and ethyl acetate (60 mL) was added Pd/C (3.8 g, 3.6 mmol, 10 wt%, 0.2 equiv.).
- Pd/C 3.8 g, 3.6 mmol, 10 wt%, 0.2 equiv.
- Step C Synthesis of 3-(4-piperazin-1-ylphenyl)piperidine-2,6-dione (73c)
- Intermediate 73b (2.0 g, 5.3 mmol, 1.0 equiv.) was dissolved in HCl/ dioxane (4.0 M, 22.2 mL, 16.6 equiv.). The mixture was stirred at 20 °C for 1 h. The reaction mixture was concentrated under reduced pressure to afford 73c, which was used without further purification.
- Step D Synthesis of tert-butyl 4-[4-[4-(2,6-dioxo-3-piperidyl)phenyl]piperazin-1-yl]piperidine- 1-carboxylate (73d)
- 73c 250 mg, 915 ⁇ mol, 1 equiv.
- tert-butyl 4-oxopiperidine-1- carboxylate 182 mg, 915 ⁇ mol, 1 equiv.
- Ti(OEt)4 (1.04 g, 4.57 mmol, 948 ⁇ L, 5 equiv.
- Step E Synthesis of 3-[4-[4-(4-piperidyl) piperazin-1-yl] phenyl] piperidine-2, 6-dione (73e)
- Intermediate 73d (150 mg, 328.53 ⁇ mol, 1 equiv.) was stirred in a solution of TFA (614.00 mg, 5.38 mmol, 0.4 mL, 16.39 equiv.) in dichloromethane (2 mL) at 20°C for 1 h.
- the reaction mixture was concentrated under reduced pressure to afford 73e, which was used without further purification.
- Step F Synthesis of 1-[4-[4-[[4-[4-[4-[4-(2,6-dioxo-3-piperidyl)phenyl]piperazin-1-yl]-1- piperidyl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4- triazol-5-yl]methyl]pyrazole-4-carboxamide (Compound 73) To a solution of 73e in MeOH (1 mL) was added Na 2 CO 3 (45.5 mg, 429 ⁇ mol, 10 equiv.). The resulting mixture was stirred at 20°C for 5 min and filtered.
- Example 74 Synthesis of Compound 74 Synthesis of phenyl]-2- methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole- 4-carboxamide (Compound 74) To a solution of 3-(4-piperazin-1-ylphenyl)piperidine-2,6-dione (73c) (41.2 mg, 151 ⁇ mol, 1.5 equiv.) and Int 55 (50.0 mg, 101 ⁇ mol, 1.0 equiv.) in DCE (1.0 mL) was added acetic acid (12.1 mg, 201.4 ⁇ mol, 11.5 ⁇ L, 2.0 equiv.).
- Example 75 Synthesis of Compound 75 piperidine- 1-carboxylate (75a) To a solution of tert-butyl 4-(4-amino-2-fluoro-phenyl)piperidine-1-carboxylate (500.0 mg, 1.7 mmol, 1.0 equiv) and 3-bromopiperidine-2,6-dione (652.3 mg, 3.4 mmol, 2.0 equiv) in DMF (6.0 mL) was added NaHCO 3 (713.5 mg, 8.5 mmol, 330.5 ⁇ L, 5.0 equiv). The mixture was stirred at 60°C for 16 h, cooled to 25°C and poured into water (20 mL).
- the aqueous phase was extracted with ethyl acetate (20 mL x 3).
- the combined organic phase was washed with brine (20 mL x 3), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum.
- Step B Synthesis of 3-[3-fluoro-4-(4-piperidyl)anilino]piperidine-2,6-dione (75b)
- 75a A mixture of 75a (300.0 mg, 740 ⁇ mol, 1.0 equiv) in HCl/ dioxane (4 M, 3.0 mL) was stirred at 25°C for 1 h under a nitrogen atmosphere. The reaction mixture was concentrated under reduced pressure to afford 75b, which was used without further purification.
- Step C Synthesis of 1-[4-[4-[[4-[4-[(2,6-dioxo-3-piperidyl)amino]-2-fluoro-phenyl]-1- piperidyl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4- triazol-5-yl]methyl]pyrazole-4-carboxamide (Compound 75) To a solution of 75b (50.0 mg, 163.7 ⁇ mol, 1.0 equiv) and Int 55 (81.3 mg, 163.7 ⁇ mol, 1.0 equiv) in DMF (0.5 mL) was added MgSO 4 (197.1 mg, 1.6 mmol, 10.0 equiv) and triethylamine (99.4 mg, 983 ⁇ mol, 137 ⁇ L, 6.0 equiv).
- Example 76 Synthesis of Intermediate 76 (76a) To a solution of 1-bromo-2-chloro-4-fluoro-5-methylbenzene (3.80 g, 17.0 mmol, 1.0 equiv.) and methyl 1H-pyrazole-4-carboxylate (2.10 g, 17.0 mmol, 1.0 equiv.) in DMA (40 mL) was added K 2 CO 3 (4.7 g, 34.0 mmol, 2.0 equiv.). The mixture was stirred at 120°C for 20 h, cooled to ambient temperature, filtered and the filtrate was diluted with NH 4 Cl (100 mL) and extracted with ethyl acetate (100 mL x 2).
- Step B Synthesis of 1-(4-bromo-5-chloro-2-methylphenyl)-1H-pyrazole-4-carboxylic acid (76b)
- 76a (1.00 g, 3.0 mmol, 1.0 equiv.) in THF (10 mL)
- MeOH MeOH
- water 10 mL
- lithium hydroxide hydrate 255 mg, 6.0 mmol, 2.0 equiv.
- the solids were collected by vacuum filtration to afford 76b.
- Step C Synthesis of 1-(4-bromo-5-chloro-2-methyl-phenyl)-N-[[3-(2,2,2-trifluoro-1,1-dimethyl- ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide
- Intermediate 76 To a solution of 76b (100 mg, 316.9 ⁇ mol, 1.0 equiv.) and Int 1 (109.9 mg, 380 ⁇ mol, 1.2 equiv., HBr) in DMF (2.0 mL) was added triethylamine (320.6 mg, 3.1 mmol, 441 ⁇ L, 10 equiv.), then BOP (168.1 mg, 380.2 ⁇ mol, 1.2 equiv.) was added.
- Step A Synthesis of tert-butyl 4-[1-[[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)phenyl]methyl]-4-piperidyl]piperazine-1-carboxylate (77a)
- a solution of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzaldehyde (5.00 g, 21.5 mmol, 1.0 equiv.) in dichloromethane (70.0 mL) was treated with tert-butyl 4-(4- piperidyl)piperazine-1-carboxylate (6.96 g, 25.8 mmol, 1.2 equiv.) and acetic acid (1.29 g, 21.5 mmol, 1.23 mL, 1.0 equiv.).
- Step B Synthesis of tert-butyl 4-(1-((2'-chloro-5'-methyl-4'-(4-(((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)carbamoyl)-1H-pyrazol-1-yl)-[1,1'-biphenyl]-4- yl)methyl)piperidin-4-yl)piperazine-1-carboxylate (77b) To a solution of 77a (115.1 mg, 237.2 ⁇ mol, 1.2 equiv.) and Int 76 (100 mg, 198 ⁇ mol, 1.0 equiv.) in dioxane (2.0 mL) and water (0.4 mL), was added K2CO3 (81.9 mg, 593 ⁇ mol, 3.0 eq) and Pd(dppf)Cl 2 .CH 2 Cl 2 (32.3 mg, 40 ⁇ mol,
- Example 78 Synthesis of Compound 78 Step A – Synthesis of tert-butyl N-[(3S)-1-[[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)phenyl]methyl]pyrrolidin-3-yl]carbamate (78a)
- a solution of 2-[4-(bromomethyl)phenyl]-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (1.00 g, 3.37 mmol, 1.0 equiv.), K 2 CO 3 (1.16 g, 8.42 mmol, 2.5 equiv.) in DMF (16.0 mL) was added tert- butyl N-[(3S)-pyrrolidin-3-yl]carbamate (752.5 mg, 4.04 mmol, 1.2 equiv.).
- Step B Synthesis of tert-butyl N-[(3S)-1-[[4-[2,6-dimethyl-3-[4-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methylcarbamoyl]pyrazol-1- yl]phenyl]phenyl]methyl]pyrrolidin-3-yl]carbamate (78b) To a solution of 78a (99.4 mg, 247 ⁇ mol, 1.5 equiv.) and Int 5 (80.0 mg, 165 ⁇ mol, 1.0 equiv.) in THF (2.0 mL) and water (0.5 mL) was added PdCl2(DTBPF) (21.4 mg, 33 ⁇ mol, 0.2 equiv.) and K 3 PO 4 (104.9 mg, 494.5 ⁇ mol, 3.0 equiv.).
- Step C Synthesis of 1-[3-[4-[[(3S)-3-[[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin- 5-yl]amino]pyrrolidin-1-yl]methyl]phenyl]-2,4-dimethyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Compound 78).
- Step A a) A solution of tert-butyl 4-hydroxypiperidine-1-carboxylate (2.70 g, 13.6 mmol, 1.2 equiv.) in DMF (20.0 mL) was treated with NaH (909.1 mg, 22.7 mmol, 60% purity, 2.0 equiv.). The mixture was stirred at 0°C for 1 h. 5-bromo-2-fluoro-pyridine (2.0 g, 11.4 mmol, 1.17 mL, 1.0 equiv.) was added to the reaction mixture, and the mixture was stirred at 25 °C for 12 h. The reaction mixture was diluted with water (20 mL) and extracted with ethyl acetate (20 mL x 3).
- Step B Synthesis of tert-butyl 4-[[5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-2- pyridyl]oxy] piperidine-1-carboxylate (79b)
- a solution of 79a (3.00 g, 8.40 mmol, 1.0 equiv.) and bis(pinacolato)diboron (3.20 g, 12.6 mmol, 1.5 equiv.) in dioxane (50 mL) was treated with KOAc (2.5 g, 25.2 mmol, 3.0 equiv.) and Pd(dppf)Cl 2 (614.5 mg, 839.8 ⁇ mol, 0.1 equiv.).
- Step C Synthesis of tert-butyl 4- [[5-[2,6-dimethyl-3-[4-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H-1,2,4-triazol-5-yl]methylcarbamoyl]pyrazol-1-yl]phenyl]-2-pyridyl]oxy]piperidine-1- carboxylate (79c)
- a solution of 79b (74.9 mg, 185 ⁇ mol, 1.5 equiv.), Int 5 (60 mg, 124 ⁇ mol, 1.0 equiv.) in THF (1.2 mL) and water (0.3 mL) was treated with K3PO4 (78.7 mg, 370.9 ⁇ mol, 3.0 equiv.) and PdCl2(DTBPF) (16.1 mg, 24 ⁇ mol, 0.2 equiv.) at 20°C under nitrogen.
- Example 80 Synthesis of Compound 80 trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (80a)
- a solution of Int 5 200 mg, 412 ⁇ mol, 1.0 equiv.
- 2-fluoro-5-(4,4,5,5-tetramethyl- 1,3,2-dioxaborolan-2-yl)pyridine 137.8 mg, 618 ⁇ mol, 1.5 equiv.
- THF 4.0 mL
- water 1.0 mL
- K 3 PO 4 262.4 mg, 1.2 mmol, 3.0 equiv.
- PdC l2 DTBPF
- Step B Synthesis of tert-butyl 4-[[4-[5-[2,6-dimethyl-3-[4-[[3-(2,2,2-trifluoro-1,1-dimethyl- ethyl)-1H-1,2,4-triazol-5-yl]methylcarbamoyl]pyrazol-1-yl]phenyl]-2-pyridyl]piperazin-1- yl]methyl]piperidine-1-carboxylate (80b) A solution of 80a (100 mg, 199 ⁇ mol, 1.0 equiv.),tert-butyl 4-(piperazin-1- ylmethyl)piperidine-1-carboxylate (113.0 mg, 399 ⁇ mol, 2.0 equiv.) in DMSO (1.0 mL) was added DIEA (77.3 mg, 598 ⁇ mol, 104 ⁇ L, 3.0 equiv.).
- Step C Synthesis of 1-[3-[6-[4-[[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]-4-piperidyl]methyl]piperazin-1-yl]-3-pyridyl]-2,4-dimethyl-phenyl]-N-[[3-(2,2,2-trifluoro- 1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Compound 80) To a solution of 80b (60 mg, 78.4 ⁇ mol, 1.0 equiv.) in HCl/ dioxane (1 mL).
- Example 81 Synthesis of Compound 81 tert-butyl 4-(4-piperidylmethyl)piperazine-1-carboxylate in Step B to afford 1-(3-(6-(4-((4-(2-(2,6- dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1-yl)methyl)piperidin-1- yl)pyridin-3-yl)-2,4-dimethylphenyl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide.
- Step B Synthesis of tert-butyl 4-[1-[[4-[2,6-dimethyl-3-[4-[[3-(2,2,2-trifluoro-1,1-dimethyl- ethyl)-1H-1,2,4-triazol-5-yl]methylcarbamoyl]pyrazol-1-yl]phenyl]phenyl]methyl]-4- piperidyl]piperazine-1-carboxylate (82b)
- a solution of 82a (120.0 mg, 247 ⁇ mol, 1.5 equiv.), Int 5 (80 mg, 165 ⁇ mol, 1.0 equiv.) in THF (1.6 mL) and water (0.4 mL) was treated with K3PO4 (104.9 mg, 495 ⁇ mol, 3.0 equiv.) and PdCl2(DTBPF) (21.4 mg, 33 ⁇ mol, 0.2 equiv.) under a nitrogen atmosphere.
- Example 83 Synthesis of Compound 83 tert-butyl 4-piperazin-1-ylpiperidine-1-carboxylate in Step A to afford 1-[3-[4-[[4-[1-[2-(2,6-dioxo-3- piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]-4-piperidyl]piperazin-1-yl]methyl]phenyl]-2,4- dimethyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methyl]pyrazole-4-carboxamide.
- Step B Synthesis of [2-[4-[(1-tert-butoxycarbonyl-4-piperidyl)methyl]piperazin-1-yl]-4- pyridyl]boronic acid (84b)
- Pd(DPPF)Cl2 92.9 mg, 0.114 mmol, 0.1 equiv.
- KOAc 335.0 mg, 3.40 mmol, 3.0 equiv.
- 4,4,5,5-tetramethyl-2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3,2-dioxaborolane 577.9 mg, 2.20 mmol, 2.0 equiv.
- Step C Synthesis of tert-butyl 4-[[4-[4-[2,6-dimethyl-3-[4-[[3-(2,2,2-trifluoro-1,1-dimethyl- ethyl)-1H-1,2,4-triazol-5-yl]methylcarbamoyl]pyrazol-1-yl]phenyl]-2-pyridyl]piperazin-1- yl]methyl]piperidine-1-carboxylate (84c) A solution of 84b (73.31 mg, 181 ⁇ mol, 1.1 equiv.) in dioxane (2.0 mL), water (0.4 mL) was added K2CO3 (68.3 mg, 495 ⁇ mol, 3.0 equiv.) and Pd(dppf)Cl2 (24.1 mg, 32.9 ⁇ mol, 0.2 equiv.), Int 5 (80.0 mg, 165 ⁇ mol, 1.0 equiv.) under an atmosphere of nitrogen.
- Step D Synthesis of 1-[2,4-dimethyl-3-[2-[4-(4-piperidylmethyl)piperazin-1-yl]-4- pyridyl]phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole- 4-carboxamide (84d) To a solution of 84c (80.0 mg, 105 ⁇ mol, 1.0 equiv.) in trifluoroethanol (1 mL) was treated with chlorotrimethylsilane (85.6 mg, 788 ⁇ mol, 0.1 mL, 7.5 equiv.).
- Step A Synthesis of tert-butyl 6-[(1-benzyloxycarbonyl-4-piperidyl)methyl]-2,6- diazaspiro[3.3]heptane-2-carboxylate (86a)
- a solution of tert-butyl 2,6-diazaspiro[3.3]heptane-2-carboxylate, oxalic acid salt (1.0 g, 3.4 mmol, 1.0 equiv.) and benzyl 4-formylpiperidine-1-carboxylate (857.7 mg, 3.4 mmol, 1.0 equiv.) in DCE (10.0 mL) was treated with acetic acid (416.6 mg, 6.9 mmol, 397.1 ⁇ L, 2.0 equiv.), stirred at 20°C for 30 min and then treated with NaBH(OAc)3 (2.2 g, 10.4 mmol, 3 equiv.).
- Step B Synthesis of tert-butyl 6-(4-piperidylmethyl)-2,6-diazaspiro[3.3]heptane-2-carboxylate (86b)
- a slurry of Pd/C (745.0 mg, 700.0 ⁇ mol, 10.0% purity) in methanol (20.0 mL) was treated with 86a (1.4 g, 3.4 mmol, 1.0 equiv.).
- the mixture was degassed and purged threefold with hydrogen.
- the mixture was stirred at 40°C for 2 h under hydrogen (15 psi).
- the suspension was filtered through a pad of Celite and the pad was washed with ethanol (20mL ⁇ 2).
- the combined filtrates were concentrated to dryness to afford 86b.
- Step C Synthesis of tert-butyl 6-[[1-[5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-2-pyridyl]- 4-piperidyl]methyl]-2,6-diazaspiro[3.3]heptane-2-carboxylate (86c)
- 86b (1.20 g, 4.0 mmol, 1.0 equiv.)
- 2-fluoro-5-(4,4,5,5-tetramethyl-1,3,2- dioxaborolan-2-yl)pyridine 906.0 mg, 4.0 mmol, 1.0 equiv.
- DIEA 2.60 g, 20.3 mmol, 3.5 mL, 5.0 equiv.
- Step D Synthesis of tert-butyl 6-[[1-[5-[2,6-dimethyl-3-[4-[[3-(2,2,2-trifluoro-1,1-dimethyl- ethyl)-1H-1,2,4-triazol-5-yl]methylcarbamoyl]pyrazol-1-yl]phenyl]-2-pyridyl]-4- piperidyl]methyl]-2,6-diazaspiro[3.3]heptane-2-carboxylate (86d) A mixture of 86c (138.6 mg, 278 ⁇ mol, 1.5 equiv.), Int 5 (90.0 mg, 185.4 ⁇ mol, 1.0 equiv.), Pd(dppf)Cl2 (27.1 mg, 37.0 ⁇ mol, 0.2 equiv.) and K2CO3 (76.8 mg, 556.3 ⁇ mol, 3.0 equiv.) in dioxane (1.0 mL) and water (0.2 0.2
- Step E Synthesis of 1-[3-[6-[4-[[2-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]-2,6-diazaspiro[3.3]heptan-6-yl]methyl]-1-piperidyl]-3-pyridyl]-2,4-dimethyl-phenyl]-N-[[3- (2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Compound 86) A solution of 86d (120.0 mg, 154.4 ⁇ mol, 1.0 equiv.) in HCl/ dioxane (3.0 M, 1.0 mL) was stirred at 20°C for 2 h.
- Compound 87 was prepared in a similar manner to Example 86 by substituting benzyl 2,6- diazaspiro[3.3]heptane-2-carboxylate and tert-butyl 4-formylpiperidine-1-carboxylate in Step A to afford 1-[3-[6-[6-[[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]-4- piperidyl]methyl]-2,6-diazaspiro[3.3]heptan-2-yl]-3-pyridyl]-2,4-dimethyl-phenyl]-N-[[3-(2,2,2- trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide.
- Example 88 Synthesis of Compound 88 Compound 88 was prepared in a similar manner to Example 86 by substituting tert-butyl 4-(4-piperidylmethyl)piperazine-1-carboxylate and 2-chloro-5-(4,4,5,5-tetramethyl-1,3,2- dioxaborolan-2-yl)pyrimidine in Step C to afford 1-[3-[2-[4-[[4-[2-(2,6-dioxo-3-piperidyl)-6- fluoro-1,3-dioxo-isoindolin-5-yl]piperazin-1-yl]methyl]-1-piperidyl]pyrimidin-5-yl]-2,4- dimethyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methyl]pyrazole-4-carboxamide.
- Example 89 Synthesis of Compound 89 Step A – 1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]-2,3-dihydrobenzofuran-2-carboxamide (89a)
- Int 2 420.0 mg, 816 ⁇ mol, 1.0 equiv.
- [4-(4,4,5,5-tetramethyl-1,3,2- dioxaborolan-2-yl)phenyl]methanol (229.2 mg, 979 ⁇ mol, 1.2 equiv.) in water (0.8 mL) and THF (3.2 mL) was added K3PO4 (519.7 mg, 2.5 mmol, 3.0 equiv.) and PdCl2(DTBPF) (106.7 mg, 163 ⁇ mol, 0.2 equiv.).
- the mixture was degassed and purged threefold with nitrogen, and then stirred at 85°C for 4 h under a nitrogen atmosphere.
- Step B Synthesis of (2R)-5-chloro-6-(4-formylphenyl)-N-[[3-(2, 2, 2-trifluoro-1, 1-dimethyl- ethyl)-1H-1, 2, 4-triazol-5-yl] methyl]-2, 3-dihydrobenzofuran-2-carboxamide
- 89b A solution of 89a (50.0 mg, 101.0 ⁇ mol, 1.0 equiv.) in dichloromethane (2.0 mL) was added MnO2 (87.8 mg, 1.0 mmol, 10.0 equiv.). The mixture was stirred at 30°C for 12 h. The mixture was filtered and concentrated under reduced pressure to give 89b.
- Step yl) -1H- 1,2,4-triazol-5-yl]methylcarbamoyl]-2,3-dihydrobenzofuran-6-yl]phenyl]methyl]-4- piperidyl]piperazine-1-carboxylate (90a) A mixture of Int 2 (100 mg, 194 ⁇ mol, 1 equiv.) and tert-butyl 4-[1-[[4-(4,4,5,5-tetramethyl- 1,3,2-dioxaborolan-2-yl)phenyl]methyl]-4-piperidyl]piperazine-1-carboxylate (141.5 mg, 292 ⁇ mol, 1.5 equiv.), K3PO4 (123.7 mg, 583 ⁇ mol, 3 equiv.), PdCl2(DTBPF) (25.3 mg, 38.9 ⁇ mol, 0.2 equiv.) in THF (2 mL) and water (0.4 mL) was degassed and purged with nitrogen for
- Step B Synthesis of 5-chloro-6-[4-[(4-piperazin-1-yl-1-piperidyl)methyl]phenyl]-N-[[3-(2,2,2- trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]-2,3-dihydrobenzofuran-2- carboxamide (90b)
- a solution of 90a (140 mg, 188 ⁇ mol, 1 equiv.) in dichloromethane (3.0 mL) was treated with TFA (1.0 mL). The mixture was stirred at 20°C for 2 h. The reaction mixture was concentrated under reduced pressure to afford 90b, which was used without further purification.
- Step C Synthesis of (2R)-5-chloro-6-(4-((4-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3- dioxoisoindolin-5-yl)piperazin-1-yl)piperidin-1-yl)methyl)phenyl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-2,3-dihydrobenzofuran-2-carboxamide (90c) To a solution of 90b (200 mg) and 2-(2,6-dioxo-3-piperidyl)-5,6-difluoro-isoindoline-1,3- dione (91.1 mg) in DMSO (2 mL) was added DIEA (270 ⁇ L).
- Step A A mixture of [4,5-dichloro-2-(hydroxymethyl)phenyl]methanol (500.0 mg, 2.4 mmol, 1.0 equiv) in SOCl2 (5.0 mL) was degassed and purged three-fold with nitrogen. The mixture was stirred at 80°C for 2 h under a nitrogen atmosphere. The mixture was cooled to ambient temperature. The reaction mixture was treated with aqueous NaHCO3 (12.5 g per 100 mL H2O), dropwise and the mixture was extracted by MTBE (30 mL x 2). The combined organic layers were washed brine (20 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure.
- Step B Synthesis of ethyl 5,6-dichloro-2-cyano-indane-2-carboxylate (91b)
- 91a 140.0 mg, 574 ⁇ mol, 1.0 equiv
- ethyl 2-cyanoacetate 64.9 mg, 574 ⁇ mol, 61.2 ⁇ L, 1.0 equiv
- DMF 2.0 mL
- potassium carbonate 198.3 mg, 1.40 mmol, 2.5 equiv
- 91b 40.0 mg, 140.8 ⁇ mol, 1.0 equiv
- water 0.3 mL
- LiOH•H2O 11.8 mg, 281.5 ⁇ mol, 2.0 equiv
- the mixture was stirred at 50°C for 2 h.
- the reaction mixture was concentrated under reduced pressure to remove THF and the remainder was poured into water.
- the mixture was adjusted to pH 4 with 1 M HCl.
- the mixture was extracted with ethyl acetate.
- Step D Synthesis of 5,6-dichloro-2-cyano-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4- triazol-5-yl]methyl]indane-2-carboxamide (Intermediate 91)
- 91c 35.0 mg, 136.7 ⁇ mol, 1.0 equiv
- Int 1 42.7 mg, 205.0 ⁇ mol, 1.5 equiv
- BOP 72.5 mg, 164.0 ⁇ mol, 1.2 equiv
- triethylamine 138.3 mg, 1.4 mmol, 190.2 uL, 10.0 equiv).
- Step B Synthesis of tert-butyl 4-[[1-[5-[6-chloro-2-cyano-2-[[3-(2,2,2-trifluoro-1,1-dimethyl- ethyl)-1H-1,2,4-triazol-5-yl]methylcarbamoyl]indan-5-yl]-2-pyridyl]-4- piperidyl]methyl]piperazine-1-carboxylate (92b)
- 92a 523.2 mg, 1.00 mmol, 1.2 equiv.
- Int 91 400 mg, 896 ⁇ mol, 1.0 equiv.
- K3PO4 570.8 mg, 2.60 mmol, 3.0 equiv.
- ethanol 4 mL
- water 0.8 mL
- Step C Synthesis of (2R)-5-chloro-2-cyano-6-(6-(4-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro- 1,3-dioxoisoindolin-5-yl)piperazin-1-yl)methyl)piperidin-1-yl)pyridin-3-yl)-N-((3-(1,1,1-trifluoro- 2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-2,3-dihydro-1H-indene-2-carboxamide (Compound 92).
- Example 93 Synthesis of Compound 93 2-yl)-1H-1,2,4-triazol-5-yl)methyl) carbamoyl)-2,3-dihydro-1H-inden-5-yl)benzyl)piperidin-4- yl)piperazine-1-carboxylate (93a) A mixture of tert-butyl 4-[1-[[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)phenyl]methyl]-4-piperidyl]piperazine-1-carboxylate (90.0 mg, 185 ⁇ mol, 1.0 equiv.), Int 91 (118.2 mg, 185 ⁇ mol, 1.0 equiv.), K3PO4 (78.7 mg, 371 ⁇ mol, 2.0 equiv.) and PdCl2(DTBPF) (48.3 mg, 74 ⁇ mol, 0.4 equiv.) in THF (0.8 mL) and
- Step B Synthesis of 5-chloro-2-cyano-6-(4-((4-(piperazin-1-yl)piperidin-1-yl)methyl)phenyl)-N- ((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-2,3-dihydro-1H-indene-2- carboxamide (93b)
- a solution of trifluoroethanol (0.9 mL) and chlorotrimethylsilane (0.1 mL) was added to a solution of 93a (90.0 mg, 117 ⁇ mol, 1.0 equiv.) in trifluoroethanol (0.9 mL) and the mixture was stirred at ambient temperature for 0.5 h.
- Step C Synthesis of 5-chloro-2-cyano-6-(4-((4-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3- dioxoisoindolin-5-yl)piperazin-1-yl)piperidin-1-yl)methyl)phenyl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-2,3-dihydro-1H-indene-2-carboxamide (Compound 93) A mixture of 93b (70.0 mg, 99 ⁇ mol, 1.0 equiv.), 2-(2,6-dioxo-3-piperidyl)-5,6-difluoro- isoindoline-1,3-dione (43.8 mg, 149 ⁇ mol, 1.5 equiv.), DIEA (38.5 mg, 298 ⁇ mol, 51.8
- (94a) A mixture of Int 91 (500.0 mg, 1.12 mmol, 1.0 equiv.) and tert-butyl 4-[[4-(4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]methyl]piperazine-1-carboxylate (360.6 mg, 0.896 mmol, 0.8 equiv.) in ethanol (4.0 mL) and water (1.0 mL) was added K 3 PO 4 (475.6 mg, 2.2 ⁇ mol, 2.0 equiv.) and XPHOS-PD-G2
- Step B Synthesis of (2R)-5-chloro-2-cyano-6-[4-[[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3- dioxo-isoindolin-5-yl]piperazin-1-yl]methyl]phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H-1,2,4-triazol-5-yl]methyl]indane-2-carboxamide (Compound 94) A mixture of 94a (90.0 mg, 131 ⁇ mol, 1.0 equiv.) in TFA (0.1 mL) and dichloromethane (0.5 mL) was stirred at ambient temperature for 2 h.
- Step A Synthesis of tert-butyl 4-(4-aminopyrazol-1-yl)piperidine-1-carboxylate (95a)
- a solution of tert-butyl 4-(4-nitropyrazol-1-yl)piperidine-1-carboxylate (2.00 g, 6.70 mmol, 1 equiv.) in EtOH (20 mL) was treated with iron (1.10 g, 20.2 mmol, 3.0 equiv.) and NH4Cl (1.80 g, 33.7 mmol, 5.0 equiv.). The mixture was stirred at 80°C for 1 h, cooled to ambient temperature and filtered through Celite.
- Step B Synthesis of tert-butyl 4-[4-[(5-chloro-2-pyridyl)amino]pyrazol-1-yl]piperidine-1- carboxylate (95b)
- a solution of 95a (400 mg, 1.5 mmol, 1.0 equiv.) in DMF (2 mL) was treated with cesium carbonate (978.6 mg, 3.0 mmol, 2.0 equiv.) and BINAP (187.0 mg, 300 ⁇ mol, 0.2 equiv.), 2- bromo-5-chloro-pyridine (170.0 mg, 883.3 ⁇ mol, 0.6 equiv.), Pd2(dba)3 (137.5 mg, 150 ⁇ mol, 0.1 equiv.) under a nitrogen atmosphere.
- Step C Synthesis of tert-butyl 4-[4-[(5-chloro-2-pyridyl)amino]pyrazol-1-yl]piperidine-1- carboxylate (95c)
- a solution of 95b 402.6 mg, 1.1 mmol, 1.0 equiv.
- HCl/dioxane 4 M, 6 mL
- the mixture was filtered and the filtrate was concentrated under reduced pressure to afford 95c, which was used without further purification.
- LCMS [M+1] 278.0, 280.0.
- Step D Synthesis of tert-butyl 4-[[4-[4-[(5-chloro-2-pyridyl)amino]pyrazol-1-yl]-1- piperidyl]methyl]piperidine-1-carboxylate (95d)
- a solution of 95c 230.0 mg, 732 ⁇ mol, 1.0 equiv.
- dichloromethane 5 mL
- NaBH(OAc) 3 620.5 mg, 2.9 mmol, 4.0 equiv.
- DIEA 189.2 mg, 1.4 mmol, 255 ⁇ L, 2.0 equiv.
- tert-butyl 4-formylpiperidine-1-carboxylate 156.1 mg, 732 ⁇ mol, 1.0 equiv.).
- Step F Synthesis of tert-butyl 4-[[4-[4-[[5-[4-[(1S,2S)-2-[[3-(2,2,2-trifluoro-1,1-dimethyl- ethyl)-1H-1,2,4-triazol-5-yl]methylcarbamoyl]cyclopropyl]phenyl]-2-pyridyl]amino]pyrazol-1- yl]-1-piperidyl]methyl]piperidine-1-carboxylate (95f) A solution of 95e (216.7 mg, 383 ⁇ mol, 1.1 equiv.) in THF (2 mL), water (0.6 mL) was treated with PdCl2(DTBPF) (113.3 mg, 174 ⁇ mol, 0.5 equiv.), tripotassium;phosphate (221.5 mg, 1.0 mmol, 3.0 equiv.) and Int 3 (150.0 mg, 348 ⁇ mol, 1.0
- Step G Synthesis of (1S,2S)-2-[4-[6-[[1-[1-(4-piperidylmethyl)-4-piperidyl]pyrazol-4- yl]amino]-3-pyridyl]phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methyl]cyclopropanecarboxamide (95g) A solution of 95f (200.0 mg, 253 ⁇ mol, 1.0 equiv.) in HCl/dioxane (4 M, 3.0 mL) was stirred at 20°C for 1 h.
- Example 97 Synthesis of Intermediate 97 1-carboxylate (97a) A solution of tert-butyl 4-(4-amino-2-fluoro-phenyl)piperidine-1-carboxylate (5.0 g, 16.9 mmol, 1.0 equiv.) in DMF (50 mL) was treated with NaHCO3 (8.5 g, 101.9 mmol, 3.9 mL, 6.0 equiv.) and 3-bromopiperidine-2,6-dione (9.70 g, 50.9 mmol, 3.0 equiv.). The mixture was stirred at 70°C for 16 h.
- NaHCO3 8.5 g, 101.9 mmol, 3.9 mL, 6.0 equiv.
- 3-bromopiperidine-2,6-dione 9.70 g, 50.9 mmol, 3.0 equiv.
- Example 98 Synthesis of Intermediate 98 Step A – Synthesis of tert-butyl 4-(5-amino-3-fluoro-2-pyridyl)-3,6-dihydro-2H-pyridine-1- carboxylate (98a)
- 98a A mixture of 6-bromo-5-fluoro-pyridin-3-amine (50.0 g, 261.8 mmol, 1.0 equiv.) and tert- butyl 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-3,6-dihydro-2H-pyridine-1-carboxylate (97.13 g, 314.1 mmol, 1.2 equiv.) in water (90.0 mL) and dioxane (450.0 mL) was degassed and purged threefold with nitrogen.
- the mixture was treated with K 3 PO 4 (111.1 g, 523.6 mmol, 2.0 equiv.) and Pd(dppf)Cl2.CH2Cl2 (21.38 g, 26.18 mmol, 0.1 equiv.) and stirred at 100°C for 12 h under a nitrogen atmosphere.
- the reaction mixture was quenched with water (1 L) and extracted with ethyl acetate (500 mL x 3). The combined organic layers were washed with brine (1 L), dried over anhydrous sodium sulphate, filtered and the filtrate was concentrated under reduced pressure.
- Step C Synthesis of tert-butyl 4-[5-[(2, 6-dioxo-3-piperidyl) amino]-3-fluoro-2-pyridyl] piperidine-1-carboxylate (98c)
- Pd(OH)2/C 9.76 g, 13.90 mmol, 20 wt%, 0.3 equiv.
- ethanol 540 mL
- 98b 27 g, 46.34 mmol, 1.0 equiv.
- Step B Synthesis of tert-butyl 4-(5-amino-3-fluoropyridin-2-yl)piperazine-1-carboxylate (99b)
- 99a 17.7 g, 54.2 mmol, 1.0 equiv.
- ethanol 90.0 mL
- water 90.0 mL
- Fe 15.2 g, 271.2 mmol, 5.0 equiv.
- NH4Cl 17.4 g, 325.4 mmol, 6.0 equiv.
- the mixture was stirred at 80°C for 1 h, cooled to 25°C, filtered and the filter cake was washed with ethyl acetate (200 mL).
- Step D Synthesis of 3-[(5-fluoro-6-piperazin-1-yl-3-pyridyl)amino]piperidine-2,6-dione (Int 99)
- 99c 8.9 g, 21.9 mmol, 1.0 equiv.
- HCl/dioxane 2 M, 90.0 mL, 8.2 equiv.
- LCMS [M+1] 308.1.
- Step A Synthesis of 2,6-dibenzyloxy-3-(5, 6-difluoro-3-pyridyl) pyridine (100a)
- Step B Synthesis of 3-(5, 6-difluoro-3-pyridyl) piperidine-2, 6-dione (100b)
- 100a (10.0 g, 24.7 mmol, 1.0 equiv.) in methanol (200 mL)
- methanol 200 mL
- Pd/C 2.6 g, 10% purity
- Pd(OH)2/C 2.6 g, 20% purity
- the mixture was stirred at 50°C under hydrogen (1 atm) for 12 h.
- the mixture was filtered with diatomaceous earth and the filtrate was concentrated under vacuum.
- the mixture was sparged with nitrogen for 15 minutes, treated with CuI (632.9 mg, 3.3 mmol, 0.2 equiv.) and K 2 CO 3 (4.6 g, 33.2 mmol, 2.0 equiv.) and stirred at 100°C for 12 h under a nitrogen atmosphere.
- the reaction mixture was quenched with water (200 mL) and extracted with ethyl acetate (200 mL x 3). The combined organic layers were washed with brine (200 mL x 3), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure.
- Step C Synthesis of tert-butyl 4-(4-(2,6-dioxopiperidin-3-yl)-2-fluorophenyl)piperazine-1- carboxylate (101c)
- 101b 310.0 mg, 544 ⁇ mol, 1.0 equiv.
- ethanol 3.0 mL
- ethyl acetate 3.0 mL
- Pd/C 289.5 mg, 272.0 ⁇ mol, 10.0% purity, 0.5 equiv.
- the mixture was stirred at 20°C for 1 h under hydrogen (1 atm).
- the reaction mixture was filtered and the filtrate was concentrated under vacuum to afford 101c, which was used without further purification.
- the mixture was stirred at 100°C for 12 h under a nitrogen atmosphere.
- the reaction mixture was cooled to ambient temperature, diluted with water (500 mL) and extracted with ethyl acetate (500 mL x 2).
- the combined organic layers were washed with brine (400 mL x 2), dried over anhydrous sodium sulfate , filtered and the filtrate was concentrated under reduced pressure to afford 103a, which was used without further purification.
- Step A Synthesis of tert-butyl 4-(4-fluoro-2-pyridyl)piperazine-1-carboxylate (104a)
- a solution of 2-chloro-4-fluoro-pyridine (0.5 g, 3.80 mmol, 1.0 equiv.) and tert-butyl piperazine-1-carboxylate (849.6 mg, 4.56 mmol, 1.2 equiv.) in toluene (5 mL) was treated with cesium carbonate (1.86 g, 5.70 mmol, 1.5 equiv.) and BINAP (213.0 mg, 342 ⁇ mol, 0.09 equiv.). The mixture was degassed and purged threefold with nitrogen.
- Step B Synthesis of tert-butyl4-(4-((2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)methyl)-2- fluorophenyl)piperazine-1-carboxylate (105b)
- 105a 450.0 mg, 1.5 mmol, 1.0 equiv.
- DMSO 3.0 mL
- cesium carbonate 980.4 mg, 3.0 mmol, 2.0 equiv.
- [1,3-bis[2,6-bis(1-ethylpropyl)phenyl]-4,5- dichloroimidazol-2-ylidene]-dichloro-(2-methylpyridin-1-ium-1-yl)palladium 63.2 mg, 75 ⁇ mol, 0.05 equiv.
- Example 108 Synthesis of Compound 108 Compound Example 107 by substituting Int 100 to afford 1-[4-[4-[[4-[5-(2,6-dioxo-3-piperidyl)-3-fluoro-2-pyridyl]piperazin- 1-yl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4- triazol-5-yl]methyl]pyrazole-4-carboxamide.
- LCMS [M+1] 773.2.
- Example 109 Synthesis of Compound 109 Example 107 by substituting Int 101 to afford 1-(4'-((4-(4-(2,6-dioxopiperidin-3-yl)-2-fluorophenyl)piperazin-1- yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide.
- LCMS [M+1] 772.3.
- Example 110 Synthesis of Compound 110 Example 106 by substituting Int 102 to afford 1-(4'-((4-(5-((2,6-dioxopiperidin-3-yl)amino)pyrazin-2- yl)piperidin-1-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2- yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide.
- Example 113 Synthesis of Compound 113 Compound by substituting Int 104 to afford 1-[4-[4-[[4-[5-(2,6-dioxo-3-piperidyl)-4-fluoro-2-pyridyl]piperazin-1-yl]methyl]phenyl]- 2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methyl]pyrazole-4-carboxamide.
- LCMS [M+H] 773.3.
- Example 114 Synthesis of Compound 114 Step A – Synthesis of methyl 1-(5-bromopyrimidin-2-yl)pyrazole-4-carboxylate (114a) A solution of methyl 1H-pyrazole-4-carboxylate (2.80 g, 22.2 mmol, 1 equiv.) in DMF (50 mL) was treated with cesium carbonate (21.7 g, 66.6 mmol, 3 equiv.) and 5-bromo-2-chloro- pyrimidine (4.29 g, 22.2 mmol, 1 equiv.).
- the mixture was cooled to 20°C and treated with NaBH3CN (29.1 mg, 464 ⁇ mol, 4.5 equiv.) and stirred at 25°C for 3 h.
- the mixture was diluted in ethyl acetate (3 mL) and water (10 mL), filtered and the filtrate was extracted with ethyl acetate (5 mL x 3).
- the combined the organic layer was washed with brine (5 mL x 3), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum.
- Example 115 Synthesis of Intermediate 115 Step A – Synthesis of tert-butyl 1-(5-bromopyrimidin-2-yl)-1H-pyrazole-4-carboxylate (115a)
- a solution of tert-butyl 1H-pyrazole-4-carboxylate (10.0 g, 59.5 mmol, 1.0 equiv.) in DMF (180.0 mL) was treated with cesium carbonate (58.1 g, 178.4 mmol, 3.0 equiv.) and 5-bromo-2- chloro-pyrimidine (11.5 g, 59.5 mmol, 1.0 equiv.). The mixture was stirred at 100°C for 1 h under a nitrogen atmosphere.
- Step C Synthesis of 1-[5-(4-formylphenyl)pyrimidin-2-yl]pyrazole-4-carboxylic acid (115c)
- 115b 9.00 g, 25.7 mmol
- dichloromethane 90.0 mL
- trifluoroacetic acid 20.0 mL
- the mixture was stirred at 25°C for 4 h.
- the mixture was concentrated under vacuum.
- the residue was triturated with MTBE (40 mL) at 25 °C for 1 h and then the solids were collected by vacuum filtration and dried under vacuum to afford 115c, which was used without further purification.
- LCMS [M+1] 295.2.
- Step D 1-[5-(4-formylphenyl)pyrimidin-2-yl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H- 1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Int 115)
- a solution of 115c (2.5 g, 6.1 mmol) in DMF (25.0 mL) was treated with triethylamine (4.3 mL, 30.6 mmol, 5.0 equiv.), BOP (3.3 g, 7.4 mmol, 1.2 equiv).
- Example 116 Synthesis of Compound 116 A solution of Int 115 (50.0 mg, 103 ⁇ mol, 1.0 equiv.) and 3-(5-fluoro-6-piperazin-1-yl-3- pyridyl)piperidine-2,6-dione (40.7 mg, 139 ⁇ mol, 1.35 equiv.) in dichloromethane (1.0 mL) was treated with triethylamine (43 ⁇ L, 310 ⁇ mol, 3.0 equiv.) at 25°C.
- the mixture was stirred for 1 h, and then treated with NaBH(OAc) 3 (109.3 mg, 516 ⁇ mol, 5.0 equiv.). The resulting mixture was stirred at 25°C for 16 h.
- the reaction mixture was diluted with dichloromethane (20 mL), washed by water (20 mL x 2), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure.
- Step B Synthesis of 1-(5-bromopyridin-2-yl)-1H-pyrazole-4-carboxylic acid (119b)
- ethyl 1-(5-bromopyridin-2-yl)-1H-pyrazole-4-carboxylate 30.0 g, 101 mmol, 1.0 equiv.
- methanol 200.0 mL
- water 50.0 mL
- LiOH 4.8 g, 202.6 mmol, 2.0 equiv.
- the mixture was stirred at 50°C for 2 h, cooled to ambient temperature and extracted with ethyl acetate (30 mL x 3).
- Step D Synthesis of 1-[5-(4-formylphenyl)-2-pyridyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (119)
- a mixture of 119c (10.0 g, 21.8 mmol, 1.0 equiv.), 4-(4,4,5,5-tetramethyl-1,3,2- dioxaborolan-2-yl)benzaldehyde (7.60 g, 32.7 mmol, 1.5 equiv.), Pd(dppf)Cl 2 (1.6 g, 2.1 mmol, 0.1 equiv.) and K2CO3 (6.0 g, 43.6 mmol, 2.0 equiv.) in dioxane (100.0 mL) and water (20.0 mL) was degassed and purged threefold with nitrogen, and then the mixture was stirred
- Step A Synthesis of tert-butyl 1-(5-bromopyrimidin-2-yl)-1H-pyrazole-4-carboxylate (121a)
- a solution of tert-butyl 1H-pyrazole-4-carboxylate (40.0 g, 237.8 mmol, 1.0 equiv.) in DMF (400 mL) was treated with cesium carbonate (232.5 g, 713.5 mmol, 3.0 equiv.) and 5- bromo-2-chloropyrimidine (46.0 g, 237.8 mmol, 1.0 equiv.).
- the mixture was stirred at 80°C for 2 h under an atmosphere of nitrogen, cooled to ambient temperature and poured into water (3 L).
- Example 122 Synthesis of Intermediate 122 - 1H-pyrazole-4-carboxylate (122a) A mixture of tert-butyl 1-(5-bromopyrimidin-2-yl)-1H-pyrazole-4-carboxylate (30.0 g, 92.3 mmol, 1.0 equiv.), 4,4,5,5-tetramethyl-2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3,2- dioxaborolane (32.8 g, 129.2 mmol, 1.4 equiv.), potassium acetate (27.2 g, 276.8 mmol, 3.0 equiv.) and Pd(dppf)Cl 2 (3.40 g, 4.60 mmol, 0.05 equiv.) in dioxane (400 mL) was degassed and purged threefold with nitrogen.
- 122a A mixture of tert-butyl 1-(5-bromo
- the mixture was stirred at 100°C for 16 h under a nitrogen atmosphere.
- the mixture was poured into water (400 mL) and stirred for 2 min.
- the aqueous phase was extracted with ethyl acetate (400 mL x 3).
- the combined organic phase was washed with brine (400 mL x 3), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum.
- the mixture was stirred at 50°C for 1.5 h, treated with NaBH(OAc)3 (168.4 mg, 795 ⁇ mol, 2.0 equiv.) and stirred at 50°C for 0.5 h.
- the reaction mixture was quenched by water (30 ml), filtered and the filtrate was concentrated under vacuum.
- Step A Synthesis of benzyl 4-(4-tert-butoxycarbonyl-3-fluoro-phenyl)piperazine-1-carboxylate (Int 124)
- a solution of benzyl piperazine-1-carboxylate (2.00 g, 9.08 mmol, 1 equiv.) in toluene (150 mL) was treated with tert-butyl 4-bromo-2-fluoro-benzoate (2.5 g, 9.0 mmol, 1.0 equiv.), cesium carbonate (3.2 g, 9.9 mmol, 1.1 equiv.), BINAP (565.3 mg, 908 ⁇ mol, 0.1 equiv.) and Pd(OAc)2 (203.8 mg, 908 ⁇ mol, 0.1 equiv.) under an atmosphere of nitrogen.
- tert-butyl 4-bromo-2-fluoro-benzoate 2.5 g, 9.0 mmol, 1.0 equiv.
- the mixture was degassed and purged threefold with nitrogen and stirred at 60°C for 16 h under an atmosphere of nitrogen.
- the mixture was cooled to ambient temperature and concentrated under reduced pressure.
- the residue was diluted with water (20 mL) and extracted with ethyl acetate (30 mL x 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure.
- Example 128 Synthesis of Intermediate 128 Step A – Synthesis of tert-butyl 4-(3-fluoro-4-methoxycarbonyl-phenyl)-3,6-dihydro-2H-pyridine- 1-carboxylate (Int 128) A mixture of tert-butyl 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-3,6-dihydro-2H- pyridine-1-carboxylate (5.0 g, 16.1 mmol, 0.9 eq), methyl 4-bromo-2-fluoro-benzoate (4.1 g, 17.9 mmol, 1.0 eq) in dioxane (50.0 mL) and water (10.0 mL) was treated with potassium carbonate (7.4 g, 53.9 mmol, 3.0 eq) and Pd(dppf)Cl 2 (1.3 g, 1.8 mmol, 0.1 eq).
- Step A Synthesis of methyl 2-fluoro-4-[1-[[4-[3-methyl-4-[4-[[3-(2,2,2-trifluoro-1,1-dimethyl- ethyl)-1H-1,2,4-triazol-5-l]methylcarbamoyl]pyrazol-1-yl]phenyl]phenyl]methyl]-4- piperidyl]benzoate (129a)
- a solution of methyl 2-fluoro-4-(4-piperidyl)benzoate hydrochloride (200.0 mg, 843 ⁇ mol, 1.0 eq) and Int 55 (418.5 mg, 843 ⁇ mol, 1.0 eq) in THF (2.0 mL) was treated with Ti(Oi- Pr)4 (498 ⁇ L,1.6 mmol, 2.0 eq) and stirred at 60°C for 1 h.
- Example 131 Synthesis of Compound 131
- Step A Synthesis of 2-fluoro-4-(1-(4-(6-(4-(((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)carbamoyl)-1H-pyrazol-1-yl)pyridin-3-yl)benzyl)piperidin-4-yl)benzoic acid (131a)
- a solution of Int 130 (36.9 mg) and Int 119 (40.0 mg) in THF (1.0 mL) was treated with Ti(OEt)4 (172 ⁇ L). The mixture was stirred at 70°C for 1 h, and then treated with NaBH3CN (7.8 mg) at 25°C.
- Step B Synthesis of (S)-N-((3-(1,1-difluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)- 1-(5-(4-((4-(4-((2,6-dioxopiperidin-3-yl)carbamoyl)-3-fluorophenyl)piperidin-1- yl)methyl)phenyl)pyridin-2-yl)-1H-pyrazole-4-carboxamide (Compound 131)
- a solution of 131a (20.0 mg) and (3S)-3-aminopiperidine-2,6-dione hydrochloride (9.5 mg) in DMF (2.0 mL) was treated with NMI (23.8 mg, 289.6 ⁇ mol, 23.1 ⁇ L, 10.0 equiv.) and TCFH (16.2 mg, 57.9 ⁇ mol, 2.0 equiv.).
- Example 132 Synthesis of Compound 132 Int 115 in Step A to afford 1-[5-[4-[[4-[4-[[[(3S)-2,6-dioxo-3-piperidyl]carbamoyl]-3-fluoro-phenyl]-1- piperidyl]methyl]phenyl]pyrimidin-2-yl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4- triazol-5-yl]methyl]pyrazole-4-carboxamide.
- the mixture was stirred at 100°C for 1 h under an atmosphere of nitrogen.
- the mixture was cooled to 25°C and poured into water (40 mL).
- the aqueous phase was extracted with ethyl acetate (40 mL x 3) and the combined organic phase was washed with brine (40 mL x 3), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum.
- Step B Synthesis of tert-butyl 4-[[3-methyl-4-[4-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H- 1,2,4-triazol-5-yl]methylcarbamoyl]pyrazol-1-yl]phenyl]methyl]piperidine-1-carboxylate
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
The present invention relates to heterobifunctional compounds which cause androgen receptor degradation and are useful, for example, in the treatment of cancer.
Description
DEGRADER MOLECULES TARGETING ANDROGEN RECEPTOR CROSS-REFERENCE TO RELATED APPLICATIONS This application claims the benefit of U.S. Provisional Application Serial No. 63/600,190, filed November 17, 2023. The disclosure of the prior application is considered part of the disclosure of this application, and is incorporated in its entirety into this application. FIELD OF THE INVENTION The present invention relates to heterobifunctional compounds which cause degradation of androgen receptor and are useful in the treatment of cancer. BACKGROUND OF THE INVENTION Androgen receptor is a Type I class nuclear hormone transcription factor that may be important in the development of several different types of cancer, including prostate cancer and breast cancer. Inactive forms of androgen receptor are located in the cytoplasm, bound to heat shock proteins, which are responsible for proper protein folding, prevention of misfolding, and maintaining 3D protein structure during events of cellular stress. Androgen receptor is activated by the binding of androgen molecules, resulting in activation and transcription of a variety of downstream genes. Michmerhuizen, A.R.; npj Breast Cancer 6:47 (2020). Binding of androgen receptor may result in the activation of signaling pathways that have been implicated in cancer, including the PI3K/AKT pathway. Michmerhuizen, A.R.; npj Breast Cancer 6:47 (2020). Androgen receptor has been well characterized as a key driver for the development of prostate cancer in men. To this end, androgen deprivation therapy is a first line of therapy for men with metastatic prostate cancer. However, despite its effectiveness, resistance to androgen deprivation therapy is nearly universal. Because targeting androgen receptor as a monotherapy or in combination with other therapies has proven to be an effective clinical strategy in the treatment of prostate cancer, this strategy is increasingly being investigated for the treatment of additional types of cancer, such as, for example, bladder cancer, renal cell carcinoma, salivary gland cancer, colorectal cancer, esophageal cancer, pancreatic cancer, and stomach cancer. Hu, C.; Genomics 112(2):1926-1940 (2020). Data from multiple cancer models suggest that androgen receptor signaling may also be important in the development of breast cancer,
glioblastoma, and additional tumor types exhibiting androgen receptor expression. Schweizer, M. T.; Cancers 9, 1–19 (2017). Most clinically used pharmaceutical agents are based upon small-molecule inhibition of protein function. However, alternative approaches that provide for protein degradation, rather than inhibition, also have the potential to provide clinical efficacy. Accordingly, targeted protein degradation through ubiquitination of protein targets has emerged as an effective strategy in drug discovery. Heterobifunctional small molecules, which simultaneously bind to target proteins and recruit an ubiquitin ligase (e.g., ubiquitin E3 ligase) have been shown to result in the target protein’s ubiquitination and degradation (Bondeson, D. P., et al. Nat Chem Biol. 201511(8):611- 617). There is a need for the development of new drugs, such as small molecules that can bind to both androgen receptor and ubiquitin E3 ligase to cause androgen receptor degradation, which are useful in the treatment of various diseases, including cancer. SUMMARY OF THE INVENTION The present invention is directed to a compound of Formula I: or a pharmaceutically
 are defined herein. The present invention is further directed to a pharmaceutical composition comprising a compound of Formula I, or a pharmaceutically acceptable salt thereof, and at least one pharmaceutically acceptable carrier. The present invention is further directed to a method of treating a disease or disorder in a patient in need of treatment, where the disease or disorder is an androgen receptor-related disease, comprising administering to the patient a therapeutically effective amount of a compound Formula I, or a pharmaceutically acceptable salt thereof.
The present invention is further directed to a method of treating cancer in a patient in need thereof comprising administering to said patient a therapeutically effective amount of a compound of Formula I, or a pharmaceutically acceptable salt thereof. The present invention also provides uses of the compounds described herein in the manufacture of a medicament for use in therapy. The present disclosure also provides the compounds described herein for use in therapy. DETAILED DESCRIPTION The present disclosure provides, inter alia, a compound of Formula I:
are defined herein. The present invention is further directed to a pharmaceutical composition comprising a compound of Formula I, or a pharmaceutically acceptable salt thereof, and at least one pharmaceutically acceptable carrier. The present invention is further directed to a method of treating a disease or disorder in a patient in need of treatment, where the disease or disorder is an androgen receptor-related disease, comprising administering to the patient a therapeutically effective amount of a compound Formula I, or a pharmaceutically acceptable salt thereof.
The present invention is further directed to a method of treating cancer in a patient in need thereof comprising administering to said patient a therapeutically effective amount of a compound of Formula I, or a pharmaceutically acceptable salt thereof. The present invention also provides uses of the compounds described herein in the manufacture of a medicament for use in therapy. The present disclosure also provides the compounds described herein for use in therapy. DETAILED DESCRIPTION The present disclosure provides, inter alia, a compound of Formula I:
 or a pharmaceutically acceptable salt thereof, wherein: R1, R2, and R3 are each independently selected from H, halo, SRA, ORA, C1-4 alkyl, C2-4 alkenyl, and C1-4 haloalkyl, wherein the C2-4 alkenyl is optionally substituted with 1, 2, or 3 substituents independently selected from halo, and wherein RA is H, C1-4 alkyl, or C1-4 haloalkyl; R4 and R5 are each independently selected from H, halo, SRA, ORA, C1-4 alkyl, C2-4 alkenyl, and C1-4 haloalkyl, wherein the C2-4 alkenyl is optionally substituted with 1, 2, or 3 substituents independently selected from halo, and wherein RA is H, C1-4 alkyl, or C1-4 haloalkyl; or R4 and R5 together with the carbon atom to which they are attached form a C3-6 cycloalkyl group or a 3-6 membered heterocycloalkyl group, each optionally substituted with 1, 2, or 3 substituents independently selected from H, halo, ORA, C1-4 alkyl, and C1-4 haloalkyl; R6 and R7 are each independently selected from H, halo, C1-4 alkyl, and C1-4 haloalkyl; R8 is H or C1-4 alkyl; Cy1 is selected from C6-10 aryl, C3-14 cycloalkyl, 5-14 membered heteroaryl, 4-14 membered heterocycloalkyl, C3-7 cycloalkyl fused with phenyl to form a bicyclic ring, C4-7 cycloalkyl fused with 5-6 membered heteroaryl to form a bicyclic ring, 4-7 membered
heterocycloalkyl fused with phenyl to form a bicyclic ring, and 4-7 membered heterocycloalkyl fused with 5-6 membered heteroaryl to form a bicyclic ring, each optionally substituted by 1, 2, 3, or 4 substituents independently selected from halo, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C1-6 haloalkyl, CN, NO2, ORa, SRa, C1-6 alkyl-NRcRd, C(O)Rb, C(O)NRcRd, C(O)ORa, OC(O)Rb, OC(O)NRcRd, C(=NRe)NRcRd, NRcC(=NRe)NRcRd, NRcRd, NRcC(O)Rb, NRcC(O)ORa, NRcC(O)NRcRd, NRcS(O)Rb, NRcS(O)2Rb, NRcS(O)2NRcRd, S(O)Rb, S(O)NRcRd, S(O)2Rb, and S(O)2NRcRd; Cy2 is absent or selected from C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, and 4- 7 membered heterocycloalkyl, each optionally substituted by 1, 2, 3, or 4 substituents independently selected from 5-6 membered heteroaryl, halo, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C1-6 haloalkyl, CN, NO2, ORa1, SRa1, C(O)Rb1, C(O)NRc1Rd1, C(O)ORa1, OC(O)Rb1, OC(O)NRc1Rd1, C(=NRe1)NRc1Rd1, NRc1C(=NRe1)NRc1Rd1, NRc1Rd1, NRc1C(O)Rb1, NRc1C(O)ORa1, NRc1C(O)NRc1Rd1, NRc1S(O)Rb1, NRc1S(O)2Rb1, NRc1S(O)2NRc1Rd1, S(O)Rb1, S(O)NRc1Rd1, S(O)2Rb1, and S(O)2NRc1Rd1, wherein said 5-6 membered heteroaryl substituent of Cy2 is optionally substituted with 1 or 2 substituents independently selected from halo and C1-6 alkyl; L is a linker; and Z is a binder of cereblon E3 ligase; each Ra, Rb, Rc, Rd, Ra1, Rb1, Rc1, and Rd1 is independently selected from H, C1-6 alkyl, C1-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl-C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl, wherein said C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl-C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl of Ra, Rb, Rc, Rd, Ra1, Rb1, Rc1, and Rd1 is optionally substituted with 1, 2, 3, 4, or 5 substituents independently selected from halo, C1-4 alkyl, C1-4 haloalkyl, C1-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, CN, ORa3, SRa3, C(O)Rb3, C(O)NRc3Rd3, C(O)ORa3, OC(O)Rb3, OC(O)NRc3Rd3, NRc3Rd3, NRc3C(O)Rb3, NRc3C(O)NRc3Rd3,
or Rc and Rd together with the N atom to which they are attached form a 4-7 membered heterocycloalkyl group optionally substituted with 1, 2, or 3 substituents independently selected from halo, C1-4 alkyl, C1-4 haloalkyl, CN, ORa3, SRa3, C(O)Rb3, C(O)NRc3Rd3, C(O)ORa3, OC(O)Rb3, OC(O)NRc3Rd3, NRc3Rd3, NRc3C(O)Rb3, NRc3C(O)NRc3Rd3, NRc3C(O)ORa3, C(=NRe3)NRc3Rd3, NRc3C(=NRe3)NRc3Rd3, S(O)Rb3, S(O)NRc3Rd3, S(O)2Rb3, NRc3S(O)2Rb3, NRc3S(O)2NRc3Rd3, and S(O)2NRc3Rd3; or Rc1 and Rd1 together with the N atom to which they are attached form a 4-7 membered heterocycloalkyl group optionally substituted with 1, 2, or 3 substituents independently selected from halo, C1-4 alkyl, C1-4 haloalkyl, CN, ORa3, SRa3, C(O)Rb3, C(O)NRc3Rd3, C(O)ORa3, OC(O)Rb3, OC(O)NRc3Rd3, NRc3Rd3, NRc3C(O)Rb3, NRc3C(O)NRc3Rd3, NRc3C(O)ORa3, C(=NRe3)NRc3Rd3, NRc3C(=NRe3)NRc3Rd3, S(O)Rb3, S(O)NRc3Rd3, S(O)2Rb3, NRc3S(O)2Rb3, NRc3S(O)2NRc3Rd3, and S(O)2NRc3Rd3; each Ra3, Rb3, Rc3, and Rd3 is independently selected from H, C1-6 alkyl, C1-6 haloalkyl, C2- 6 alkenyl, C2-6 alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl-C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl, wherein said C1-6 alkyl, C1-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl-C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl are each optionally substituted with 1, 2, or 3 substituents independently selected from OH, CN, amino, halo, C1-6 alkyl, C1-6 alkylamino, di(C1-6 alkyl)amino, C1-6 alkoxy, C1-6 haloalkyl, and C1-6 haloalkoxy; and each Re, Re1, Re2, and Re3 is independently selected from H, C1-4 alkyl, and CN. The present disclosure further provides a compound of Formula I:
or a pharmaceutically acceptable salt thereof, wherein: R1, R2, and R3 are each independently selected from H, halo, SRA, ORA, C1-4 alkyl, C2-4 alkenyl, and C1-4 haloalkyl, wherein the C2-4 alkenyl is optionally substituted with 1, 2, or 3 substituents independently selected from halo, and wherein RA is H, C1-4 alkyl, or C1-4 haloalkyl; R4 and R5 are each independently selected from H, halo, SRA, ORA, C1-4 alkyl, C2-4 alkenyl, and C1-4 haloalkyl, wherein the C2-4 alkenyl is optionally substituted with 1, 2, or 3 substituents independently selected from halo, and wherein RA is H, C1-4 alkyl, or C1-4 haloalkyl; or R4 and R5 together with the carbon atom to which they are attached form a C3-6 cycloalkyl group or a 3-6 membered heterocycloalkyl group, each optionally substituted with 1, 2, or 3 substituents independently selected from H, halo, ORA, C1-4 alkyl, and C1-4 haloalkyl; R6 and R7 are each independently selected from H, halo, C1-4 alkyl, and C1-4 haloalkyl; R8 is H or C1-4 alkyl; Cy1 is selected from C6-10 aryl, C3-14 cycloalkyl, 5-14 membered heteroaryl, 4-14 membered heterocycloalkyl, C3-7 cycloalkyl fused with phenyl to form a bicyclic ring, C4-7 cycloalkyl fused with 5-6 membered heteroaryl to form a bicyclic ring, 4-7 membered
heterocycloalkyl fused with phenyl to form a bicyclic ring, and 4-7 membered heterocycloalkyl fused with 5-6 membered heteroaryl to form a bicyclic ring, each optionally substituted by 1, 2, 3, or 4 substituents independently selected from halo, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C1-6 haloalkyl, CN, NO2, ORa, SRa, C1-6 alkyl-NRcRd, C(O)Rb, C(O)NRcRd, C(O)ORa, OC(O)Rb, OC(O)NRcRd, C(=NRe)NRcRd, NRcC(=NRe)NRcRd, NRcRd, NRcC(O)Rb, NRcC(O)ORa, NRcC(O)NRcRd, NRcS(O)Rb, NRcS(O)2Rb, NRcS(O)2NRcRd, S(O)Rb, S(O)NRcRd, S(O)2Rb, and S(O)2NRcRd; Cy2 is absent or selected from C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, and 4- 7 membered heterocycloalkyl, each optionally substituted by 1, 2, 3, or 4 substituents independently selected from 5-6 membered heteroaryl, halo, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C1-6 haloalkyl, CN, NO2, ORa1, SRa1, C(O)Rb1, C(O)NRc1Rd1, C(O)ORa1, OC(O)Rb1, OC(O)NRc1Rd1, C(=NRe1)NRc1Rd1, NRc1C(=NRe1)NRc1Rd1, NRc1Rd1, NRc1C(O)Rb1, NRc1C(O)ORa1, NRc1C(O)NRc1Rd1, NRc1S(O)Rb1, NRc1S(O)2Rb1, NRc1S(O)2NRc1Rd1, S(O)Rb1, S(O)NRc1Rd1, S(O)2Rb1, and S(O)2NRc1Rd1, wherein said 5-6 membered heteroaryl substituent of Cy2 is optionally substituted with 1 or 2 substituents independently selected from halo and C1-6 alkyl; L is a linker; and Z is a binder of cereblon E3 ligase; each Ra, Rb, Rc, Rd, Ra1, Rb1, Rc1, and Rd1 is independently selected from H, C1-6 alkyl, C1-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl-C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl, wherein said C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl-C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl of Ra, Rb, Rc, Rd, Ra1, Rb1, Rc1, and Rd1 is optionally substituted with 1, 2, 3, 4, or 5 substituents independently selected from halo, C1-4 alkyl, C1-4 haloalkyl, C1-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, CN, ORa3, SRa3, C(O)Rb3, C(O)NRc3Rd3, C(O)ORa3, OC(O)Rb3, OC(O)NRc3Rd3, NRc3Rd3, NRc3C(O)Rb3, NRc3C(O)NRc3Rd3,
or Rc and Rd together with the N atom to which they are attached form a 4-7 membered heterocycloalkyl group optionally substituted with 1, 2, or 3 substituents independently selected from halo, C1-4 alkyl, C1-4 haloalkyl, CN, ORa3, SRa3, C(O)Rb3, C(O)NRc3Rd3, C(O)ORa3, OC(O)Rb3, OC(O)NRc3Rd3, NRc3Rd3, NRc3C(O)Rb3, NRc3C(O)NRc3Rd3, NRc3C(O)ORa3, C(=NRe3)NRc3Rd3, NRc3C(=NRe3)NRc3Rd3, S(O)Rb3, S(O)NRc3Rd3, S(O)2Rb3, NRc3S(O)2Rb3, NRc3S(O)2NRc3Rd3, and S(O)2NRc3Rd3; or Rc1 and Rd1 together with the N atom to which they are attached form a 4-7 membered heterocycloalkyl group optionally substituted with 1, 2, or 3 substituents independently selected from halo, C1-4 alkyl, C1-4 haloalkyl, CN, ORa3, SRa3, C(O)Rb3, C(O)NRc3Rd3, C(O)ORa3, OC(O)Rb3, OC(O)NRc3Rd3, NRc3Rd3, NRc3C(O)Rb3, NRc3C(O)NRc3Rd3, NRc3C(O)ORa3, C(=NRe3)NRc3Rd3, NRc3C(=NRe3)NRc3Rd3, S(O)Rb3, S(O)NRc3Rd3, S(O)2Rb3, NRc3S(O)2Rb3, NRc3S(O)2NRc3Rd3, and S(O)2NRc3Rd3; each Ra3, Rb3, Rc3, and Rd3 is independently selected from H, C1-6 alkyl, C1-6 haloalkyl, C2- 6 alkenyl, C2-6 alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl-C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl, wherein said C1-6 alkyl, C1-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl-C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl are each optionally substituted with 1, 2, or 3 substituents independently selected from OH, CN, amino, halo, C1-6 alkyl, C1-6 alkylamino, di(C1-6 alkyl)amino, C1-6 alkoxy, C1-6 haloalkyl, and C1-6 haloalkoxy; and each Re, Re1, Re2, and Re3 is independently selected from H, C1-4 alkyl, and CN. The present disclosure further provides a compound of Formula I:
or a pharmaceutically acceptable salt thereof, wherein: R1, R2, and R3 are each independently selected from H, halo, C1-4 alkyl, and C1-4 haloalkyl; R4 and R5 are each independently selected from H, halo, ORA, C1-4 alkyl, and C1-4 haloalkyl, wherein RA is H, C1-4 alkyl, or C1-4 haloalkyl; or R4 and R5 together with the carbon atom to which they are attached form a C3-6 cycloalkyl group or a 3-6 membered heterocycloalkyl group, each optionally substituted with 1, 2, or 3 substituents independently selected from halo, ORA, C1-4 alkyl, and C1-4 haloalkyl; R6 and R7 are each independently selected from H, halo, C1-4 alkyl, and C1-4 haloalkyl; R8 is H or C1-4 alkyl; Cy1 is selected from C6-10 aryl, C3-14 cycloalkyl, 5-14 membered heteroaryl, 4-14 membered heterocycloalkyl, C3-7 cycloalkyl fused with phenyl to form a bicyclic ring, C3-7 cycloalkyl fused with 5-6 membered heteroaryl to form a bicyclic ring, 4-7 membered heterocycloalkyl fused with phenyl to form a bicyclic ring, and 4-7 membered heterocycloalkyl fused with 5-6 membered heteroaryl to form a bicyclic ring, each optionally substituted by 1, 2, 3, or 4 substituents independently selected from halo, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C1-6 haloalkyl, CN, NO2, ORa, SRa, C1-6 alkyl-NRcRd, C(O)Rb, C(O)NRcRd, C(O)ORa, OC(O)Rb, OC(O)NRcRd, C(=NRe)NRcRd, NRcC(=NRe)NRcRd, NRcRd, NRcC(O)Rb, NRcC(O)ORa,
 4- 7 membered heterocycloalkyl, each optionally substituted by 1, 2, 3, or 4 substituents independently selected from halo, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C1-6 haloalkyl, CN, NO2, ORa1, SRa1, C(O)Rb1, C(O)NRc1Rd1, C(O)ORa1, OC(O)Rb1, OC(O)NRc1Rd1, C(=NRe1)NRc1Rd1, NRc1C(=NRe1)NRc1Rd1, NRc1Rd1, NRc1C(O)Rb1, NRc1C(O)ORa1, NRc1C(O)NRc1Rd1, NRc1S(O)Rb1, NRc1S(O)2Rb1, NRc1S(O)2NRc1Rd1, S(O)Rb1, S(O)NRc1Rd1, S(O)2Rb1, and S(O)2NRc1Rd1; L is a linker; and Z is a binder of cereblon E3 ligase; each Ra, Rb, Rc, Rd, Ra1, Rb1, Rc1, and Rd1 is independently selected from H, C1-6 alkyl, C1-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl-C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered
heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl, wherein said C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl-C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl of Ra, Rb, Rc, Rd, Ra1, Rb1, Rc1, and Rd1 is optionally substituted with 1, 2, 3, 4, or 5 substituents independently selected from halo, C1-4 alkyl, C1-4 haloalkyl, C1-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, CN, ORa3, SRa3, C(O)Rb3, C(O)NRc3Rd3, C(O)ORa3, OC(O)Rb3, OC(O)NRc3Rd3, NRc3Rd3, NRc3C(O)Rb3, NRc3C(O)NRc3Rd3, NRc3C(O)ORa3, C(=NRe3)NRc3Rd3, NRc3C(=NRe3)NRc3Rd3, S(O)Rb3, S(O)NRc3Rd3, S(O)2Rb3, NRc3S(O)2Rb3, NRc3S(O)2NRc3Rd3, and S(O)2NRc3Rd3; or Rc and Rd together with the N atom to which they are attached form a 4-7 membered heterocycloalkyl group optionally substituted with 1, 2, or 3 substituents independently selected from halo, C1-4 alkyl, C1-4 haloalkyl, CN, ORa3, SRa3, C(O)Rb3, C(O)NRc3Rd3, C(O)ORa3, OC(O)Rb3, OC(O)NRc3Rd3, NRc3Rd3, NRc3C(O)Rb3, NRc3C(O)NRc3Rd3, NRc3C(O)ORa3,
4- 7 membered heterocycloalkyl, each optionally substituted by 1, 2, 3, or 4 substituents independently selected from halo, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C1-6 haloalkyl, CN, NO2, ORa1, SRa1, C(O)Rb1, C(O)NRc1Rd1, C(O)ORa1, OC(O)Rb1, OC(O)NRc1Rd1, C(=NRe1)NRc1Rd1, NRc1C(=NRe1)NRc1Rd1, NRc1Rd1, NRc1C(O)Rb1, NRc1C(O)ORa1, NRc1C(O)NRc1Rd1, NRc1S(O)Rb1, NRc1S(O)2Rb1, NRc1S(O)2NRc1Rd1, S(O)Rb1, S(O)NRc1Rd1, S(O)2Rb1, and S(O)2NRc1Rd1; L is a linker; and Z is a binder of cereblon E3 ligase; each Ra, Rb, Rc, Rd, Ra1, Rb1, Rc1, and Rd1 is independently selected from H, C1-6 alkyl, C1-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl-C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered
heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl, wherein said C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl-C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl of Ra, Rb, Rc, Rd, Ra1, Rb1, Rc1, and Rd1 is optionally substituted with 1, 2, 3, 4, or 5 substituents independently selected from halo, C1-4 alkyl, C1-4 haloalkyl, C1-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, CN, ORa3, SRa3, C(O)Rb3, C(O)NRc3Rd3, C(O)ORa3, OC(O)Rb3, OC(O)NRc3Rd3, NRc3Rd3, NRc3C(O)Rb3, NRc3C(O)NRc3Rd3, NRc3C(O)ORa3, C(=NRe3)NRc3Rd3, NRc3C(=NRe3)NRc3Rd3, S(O)Rb3, S(O)NRc3Rd3, S(O)2Rb3, NRc3S(O)2Rb3, NRc3S(O)2NRc3Rd3, and S(O)2NRc3Rd3; or Rc and Rd together with the N atom to which they are attached form a 4-7 membered heterocycloalkyl group optionally substituted with 1, 2, or 3 substituents independently selected from halo, C1-4 alkyl, C1-4 haloalkyl, CN, ORa3, SRa3, C(O)Rb3, C(O)NRc3Rd3, C(O)ORa3, OC(O)Rb3, OC(O)NRc3Rd3, NRc3Rd3, NRc3C(O)Rb3, NRc3C(O)NRc3Rd3, NRc3C(O)ORa3,
 heterocycloalkyl group optionally substituted with 1, 2, or 3 substituents independently selected from halo, C1-4 alkyl, C1-4 haloalkyl, CN, ORa3, SRa3, C(O)Rb3, C(O)NRc3Rd3, C(O)ORa3, OC(O)Rb3, OC(O)NRc3Rd3, NRc3Rd3, NRc3C(O)Rb3, NRc3C(O)NRc3Rd3, NRc3C(O)ORa3,
heterocycloalkyl group optionally substituted with 1, 2, or 3 substituents independently selected from halo, C1-4 alkyl, C1-4 haloalkyl, CN, ORa3, SRa3, C(O)Rb3, C(O)NRc3Rd3, C(O)ORa3, OC(O)Rb3, OC(O)NRc3Rd3, NRc3Rd3, NRc3C(O)Rb3, NRc3C(O)NRc3Rd3, NRc3C(O)ORa3,
 C2- 6 alkenyl, C2-6 alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl-C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl, wherein said C1-6 alkyl, C1-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl-C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl are each optionally substituted with 1, 2, or 3 substituents independently selected from OH, CN, amino, halo, C1-6 alkyl, C1-6 alkylamino, di(C1-6 alkyl)amino, C1-6 alkoxy, C1-6 haloalkyl, and C1-6 haloalkoxy; and each Re, Re1, Re2, and Re3 is independently selected from H, C1-4 alkyl, and CN.
In some embodiments, L is a linker having formula II: II wherein CyA, CyB, CyC, and CyD are each independently absent or independently selected from C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, and 4-10 membered heterocycloalkyl, each optionally substituted by 1, 2, 3, or 4 substituents independently selected from halo, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C1-6 haloalkyl, CN, NO2, ORa11, SRa11, C(O)Rb11, C(O)NRc11Rd11, C(O)ORa11, OC(O)Rb11, OC(O)NRc11Rd11, C(=NRe11)NRc11Rd11, NRc11C(=NRe11)NRc11Rd11, NRc11Rd11, NRc11C(O)Rb11, NRc11C(O)ORa11, NRc11C(O)NRc11Rd11, NRc11S(O)Rb11, NRc11S(O)2Rb11, NRc11S(O)2NRc11Rd11, S(O)Rb11, S(O)NRc11Rd11, S(O)2Rb11, and S(O)2NRc11Rd11; and LA, LB, and LC are each independently absent or independently selected from C1-6 alkylene, C2-6 alkenylene, C2-6 alkynylene, -C(O)-, -O-, -O-(C1-6 alkylene)-, -NRc23-, -S-(C1-6 alkylene)-, -C(O)-(C1-6 alkylene)-, -C(O)NRc23-(C1-6 alkylene)-, -C(O)O-, -C(O)O-(C1-6 alkylene)- , -OC(O)-(C1-6 alkylene)-, -OC(O)NRc23-(C1-6 alkylene)-, -NRc23-(C1-6 alkylene)-, -N-(C1-6 alkylene)-C(O)-, -NRc23C(O)-(C1-6 alkylene)-, -NRc23C(O)NRc23-(C1-6 alkylene)-, -NRc23C(O)O- (C1-6 alkylene)-, -C(=NRe23)NRc23-(C1-6 alkylene)-, -NRc23C(=NRe23)NRc23-(C1-6 alkylene)-, - S(O)-(C1-6 alkylene)-, -S(O)NRc23-(C1-6 alkylene)-, -S(O)2-(C1-6 alkylene)-, -NRc23S(O)2-(C1-6 alkylene)-, -NRc23S(O)2NRc23-(C1-6 alkylene)-, and -S(O)2NRc23-(C1-6 alkylene)-, wherein the C1-6 alkylene, C2-6 alkenylene, or C2-6 alkynylene is optionally substituted by 1, 2, or 3 substituents independently selected from C1-4 alkyl, C1-4 haloalkyl, C1-4 alkoxy, halo, and OH; each Ra11, Rb11, Rc11, and Rd11 is independently selected from H, C1-6 alkyl, C1-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl-C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl, wherein said C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl-C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl of Ra11, Rb11, Rc11, and Rd11 is optionally substituted with 1, 2, 3, 4, or 5 substituents independently selected from halo, C1-4 alkyl, C1-4 haloalkyl, C1-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, CN, ORa3, SRa3, C(O)Rb3, C(O)NRc3Rd3, C(O)ORa3,
OC(O)Rb3, OC(O)NRc3Rd3, NRc3Rd3, NRc3C(O)Rb3, NRc3C(O)NRc3Rd3, NRc3C(O)ORa3, C(=NRe3)NRc3Rd3, NRc3C(=NRe3)NRc3Rd3, S(O)Rb3, S(O)NRc3Rd3, S(O)2Rb3, NRc3S(O)2Rb3, NRc3S(O)2NRc3Rd3, and S(O)2NRc3Rd3; Rc23 is independently selected from H, C1-6 alkyl, C1-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl- C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl, wherein said C1-6 alkyl, C1-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl-C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl are each optionally substituted with 1, 2, or 3 substituents independently selected from OH, CN, amino, halo, C1-6 alkyl, C1-6 alkylamino, di(C1-6 alkyl)amino, C1-6 alkoxy, C1-6 haloalkyl, and C1-6 haloalkoxy; and Re23 is independently selected from H, C1-4 alkyl, and CN. In some embodiments, at least one of LA, LB, LC, CyA, CyB, CyC, and CyD is not absent In some embodiments, R1, R2, and R3 are each independently selected from H, F, methyl, and trifluoromethyl. In some embodiments, R1, R2, and R3 are each F. In some embodiments, R1, R2, and R3 are each selected from methyl and trifluoromethyl. In some embodiments, R1, R2, and R3 are each H. In some embodiments, R4 and R5 are each independently selected from H, halo, SRA, ORA, C1-4 alkyl, C2-4 alkenyl, and C1-4 haloalkyl, wherein the C2-4 alkenyl is optionally substituted with 1, 2, or 3 substituents independently selected from halo, and wherein RA is H, C1-4 alkyl, or C1-4 haloalkyl. In some embodiments, R4 and R5 are each independently selected from halo, SRA, ORA, C1-4 alkyl, C2-4 alkenyl, and C1-4 haloalkyl, wherein the C2-4 alkenyl is optionally substituted with 1, 2, or 3 substituents independently selected from halo, and wherein RA is H, C1-4 alkyl, or C1-4 haloalkyl. In some embodiments, R4 and R5 are each independently selected from H, SRA, C1- 4 alkyl, and C2-4 alkenyl, wherein the C2-4 alkenyl is optionally substituted with 1, 2, or 3 substituents independently selected from halo, and wherein RA is H, C1-4 alkyl, or C1-4 haloalkyl. In some embodiments, R4 and R5 are each independently selected from SRA, C1-4 alkyl, and C2-4 alkenyl, wherein the C2-4 alkenyl is optionally substituted with 1, 2, or 3 substituents independently selected from halo, and wherein RA is H, C1-4 alkyl, or C1-4 haloalkyl. In some embodiments, R4 and R5 are each independently selected from H, SRA, C1-4 alkyl, and C2-4
alkenyl, wherein the C2-4 alkenyl is optionally substituted with 1, 2, or 3 substituents independently selected from halo, and wherein RA is C1-4 alkyl. In some embodiments, R4 and R5 are each independently selected from SRA, C1-4 alkyl, and C2-4 alkenyl, wherein the C2-4 alkenyl is optionally substituted with 1, 2, or 3 substituents independently selected from halo, and wherein RA is C1-4 alkyl. In some embodiments, R4 and R5 are each independently selected from OH, C1-4 alkyl, and C1-4 haloalkyl; or R4 and R5 together with the carbon atom to which they are attached form a C3-6 cycloalkyl group optionally substituted with 1, 2, or 3 substituents independently selected from halo, ORA, C1-4 alkyl, and C1-4 haloalkyl. In some embodiments, R4 and R5 are each independently selected from OH, C1-4 alkyl, and C1-4 haloalkyl. In some embodiments, R4 and R5 are each C1-4 alkyl. In some embodiments, R4 and R5 are each methyl. In some embodiments, R4 and R5 together with the carbon atom to which they are attached form a C3-6 cycloalkyl group optionally substituted with 1, 2, or 3 substituents independently selected from halo, ORA, C1-2 alkyl, and C1-2 haloalkyl. In some embodiments, R4 and R5 together with the carbon atom to which they are attached form a C3-6 cycloalkyl group optionally substituted with 1 or 2 substituents independently selected from halo and C1-2 alkyl. In some embodiments, R4 and R5 together with the carbon atom to which they are attached form a C3-6 cycloalkyl group optionally substituted with 1 or 2 fluoro. In some embodiments, R6 and R7 are each H. In some embodiments, R8 is H. In some embodiments, Cy1 is selected from C3-14 cycloalkyl, 5-14 membered heteroaryl, C4-7 cycloalkyl fused with phenyl to form a bicyclic ring, and 4-7 membered heterocycloalkyl fused with phenyl to form a bicyclic ring, each optionally substituted by 1 or 2 substituents independently selected from halo, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C1-6 haloalkyl, CN, NO2, ORa, SRa, C1-6 alkyl-NRcRd, C(O)Rb, C(O)NRcRd, C(O)ORa, OC(O)Rb, OC(O)NRcRd, C(=NRe)NRcRd, NRcC(=NRe)NRcRd, NRcRd, NRcC(O)Rb, NRcC(O)ORa, NRcC(O)NRcRd, NRcS(O)Rb, NRcS(O)2Rb, NRcS(O)2NRcRd, S(O)Rb, S(O)NRcRd, S(O)2Rb, and S(O)2NRcRd. In some embodiments, Cy1 is selected from C3-6 cycloalkyl, 5-6 membered heteroaryl, C5-6 cycloalkyl fused with phenyl to form a bicyclic ring, and 5-6 membered heterocycloalkyl fused with phenyl to form a bicyclic ring, each optionally substituted by 1 or 2 substituents
independently selected from halo, C1-6 alkyl, C1-6 haloalkyl, CN, C1-6 alkyl-NRcRd, C(O)NRcRd, and NRcRd. In some embodiments, Cy1 is C3-6 cycloalkyl optionally substituted by 1 or 2 substituents independently selected from halo, C1-6 alkyl, and C1-6 haloalkyl. In some embodiments, Cy1 is C3- 6 cycloalkyl. In some embodiments, Cy1 is cyclopropyl or cyclobutyl. In some embodiments, Cy1 is cyclopropyl. In some embodiments, Cy1 is 5-6 membered heteroaryl, optionally substituted by 1 or 2 substituents independently selected from halo, C1-6 alkyl, and C(O)NRcRd. In some embodiments, Cy1 is 5 membered heteroaryl, optionally substituted by 1 or 2 substituents independently selected from halo, C1-6 alkyl, and C(O)NRcRd. In some embodiments, Cy1 is pyrazolyl or pyrrolyl, optionally substituted with C(O)NRcRd. In some embodiments, Cy1 is pyrazolyl. In some embodiments, Cy1 is pyrazolyl, pyrrolyl, or isoxazolyl, each optionally substituted with C(O)NRcRd. In some embodiments, Cy1 is isoxazolyl. In some embodiments, Cy1 is pyrazolyl, pyrrolyl, or isoxazolyl. In some embodiments, Cy1 is C5-6 cycloalkyl fused with phenyl to form a bicyclic ring, optionally substituted by 1 or 2 substituents independently selected from halo, C1-6 alkyl, CN, C1-6 alkyl-NRcRd, and NRcRd. In some embodiments, Cy1 is cyclopentyl fused with phenyl to form a bicyclic ring, optionally substituted by methyl, CN, Cl, F, -CH2NHRd, and NH2. In some embodiments, Cy1 is 5-6 membered heterocycloalkyl fused with phenyl to form a bicyclic ring, optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, Cy1 is dihydrobenzofuran, optionally substituted by 1 or 2 substituents independently selected from chloro and methyl. In some embodiments, Cy1 is dihydrobenzofuran substituted by 1 chloro. In some embodiments, Cy2 is selected from C6-10 aryl and 5-10 membered heteroaryl, optionally substituted by 1, 2, or 3 substituents independently selected from halo, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C1-6 haloalkyl, CN, NO2, ORa1, SRa1, C(O)Rb1, C(O)NRc1Rd1, C(O)ORa1, OC(O)Rb1, OC(O)NRc1Rd1, C(=NRe1)NRc1Rd1, NRc1C(=NRe1)NRc1Rd1, NRc1Rd1, NRc1C(O)Rb1, NRc1C(O)ORa1, NRc1C(O)NRc1Rd1, NRc1S(O)Rb1, NRc1S(O)2Rb1, NRc1S(O)2NRc1Rd1, S(O)Rb1, S(O)NRc1Rd1, S(O)2Rb1, and S(O)2NRc1Rd1. In some embodiments, Cy2 is phenyl or 5-6 membered heteroaryl, optionally substituted by 1 or 2 substituents selected from halo and C1-6 alkyl.
In some embodiments, Cy2 is C6-10 aryl optionally substituted by 1 or 2 substituents independently selected from halo, C1-6 alkyl, and C1-6 haloalkyl. In some embodiments, Cy2 is phenyl optionally substituted with 1 or 2 substituents each independently selected from C1-6 alkyl. In some embodiments, Cy2 is phenyl, methylphenyl, methylchlorophenyl, or dimethylphenyl. In some embodiments, Cy2 is phenyl optionally substituted with 1 or 2 substituents selected from methyl and chloro. In some embodiments, Cy2 is 5-10 membered heteroaryl optionally substituted by 1 or 2 substituents independently selected from halo, C1-6 alkyl, and C1-6 haloalkyl. In some embodiments, Cy2 is 6 membered heteroaryl optionally substituted by 1 or 2 substituents each independently selected from C1-6 alkyl. In some embodiments, Cy2 is pyridinyl or methylpyridinyl. In some embodiments, Cy2 is pyridinyl or pyrimidinyl, each optionally substituted by 1 or 2 substituents each independently selected from C1-6 alkyl. In some embodiments, Cy2 is pyridinyl or pyrimidinyl, each optionally substituted with methyl. In some embodiments, Cy2 is pyridinyl, methylpyridinyl, or pyrimidinyl. In some embodiments, Cy2 is pyridinyl or pyrimidinyl, each optionally substituted with 1 or 2 methyl groups. In some embodiments, Cy2 is C3-7 cycloalkyl optionally substituted by 1 or 2 substituents independently selected from halo, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C1-6 haloalkyl, CN, NO2, ORa1, SRa1, C(O)Rb1, C(O)NRc1Rd1, C(O)ORa1, OC(O)Rb1, OC(O)NRc1Rd1, C(=NRe1)NRc1Rd1, NRc1C(=NRe1)NRc1Rd1, NRc1Rd1, NRc1C(O)Rb1, NRc1C(O)ORa1, NRc1C(O)NRc1Rd1, NRc1S(O)Rb1, NRc1S(O)2Rb1, NRc1S(O)2NRc1Rd1, S(O)Rb1, S(O)NRc1Rd1, S(O)2Rb1, and S(O)2NRc1Rd1. In some embodiments, Cy2 is bicyclo[1.1.1]pentyl. In some embodiments, CyA, CyB, CyC, and CyD are each independently absent or independently selected from C6-10 aryl, 5-10 membered heteroaryl, and 4-10 membered heterocycloalkyl, each optionally substituted by 1, 2, or 3 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyA, CyB, CyC, and CyD are each independently absent or independently selected from phenyl, 5-9 membered heteroaryl, and 4-7 membered heterocycloalkyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyA is absent or selected from C6-10 aryl, 5-10 membered heteroaryl, and 4-10 membered heterocycloalkyl, each optionally substituted by 1, 2, or 3 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyA is absent
or selected from phenyl, 5-9 membered heteroaryl, and 4-7 membered heterocycloalkyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyA is absent or selected from phenyl, 5-6 membered heteroaryl, and 6-9 membered heterocycloalkyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyA is absent or selected from phenyl, pyrazolyl, pyridinyl, indazolyl, and piperidinyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyA is phenyl or pyridinyl, each optionally substituted by halo or methyl. In some embodiments, CyA is absent. In some embodiments, CyA is selected from C6-10 aryl, 5-10 membered heteroaryl, and 4- 10 membered heterocycloalkyl, each optionally substituted by 1, 2, or 3 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyA is selected from phenyl, 5-9 membered heteroaryl, and 4-7 membered heterocycloalkyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyA is selected from phenyl, 5-6 membered heteroaryl, and 6-9 membered heterocycloalkyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyA is selected from phenyl, pyrazolyl, pyridinyl, indazolyl, and piperidinyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyA is phenyl or pyridinyl, each optionally substituted by1 or 2 substituents selected from halo and methyl. In some embodiments, CyA is absent or selected from phenyl, pyrazolyl, pyridinyl, pyrazinyl, pyrimidinyl, indazolyl, 2,3-dihydrobenzofuranyl, and piperidinyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyA is selected from phenyl, pyrazolyl, pyridinyl, pyrazinyl, pyrimidinyl, indazolyl, 2,3-dihydrobenzofuranyl, and piperidinyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyB is absent or selected from C6-10 aryl, 5-10 membered heteroaryl, and 4-10 membered heterocycloalkyl, each optionally substituted by 1, 2, or 3 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyB is absent or selected from phenyl, 5-9 membered heteroaryl, and 4-7 membered heterocycloalkyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyB is absent or selected from phenyl, 5 membered heteroaryl, and 6-7
membered heterocycloalkyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyB is absent or selected from phenyl, pyrazolyl, piperazinyl, piperidinyl, and azasprio[3.3]heptanyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyB is piperidinyl or piperazinyl. In some embodiments, CyB is absent. In some embodiments, CyB is selected from C6-10 aryl, 5-10 membered heteroaryl, and 4- 10 membered heterocycloalkyl, each optionally substituted by 1, 2, or 3 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyB is selected from phenyl, 5-9 membered heteroaryl, and 4-7 membered heterocycloalkyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyB is selected from phenyl, 5 membered heteroaryl, and 6-7 membered heterocycloalkyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyB is selected from phenyl, pyrazolyl, piperazinyl, piperidinyl, and azasprio[3.3]heptanyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyB is absent or selected from phenyl, cyclohexyl, pyrazolyl, azetidinyl, pyrrolidinyl, piperazinyl, piperidinyl, 2-azasprio[3.3]heptanyl, diazaspiro[3.5]nonanyl, 2,6-diazaspiro[3.3]heptanyl, 3,6-diazabicyclo[3.1.1]heptanyl, 2,5-diazabicyclo[2.2.1]heptanyl, 3,8-diazabicyclo[3.2.1]octanyl, 2-azaspiro[3.5]nonanyl, 7-azaspiro[3.5]nonanyl, 6- azaspiro[3.4]octanyl, and octahydropyrrolo[3,2-b]pyrrolyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyB is selected from phenyl, cyclohexyl, pyrazolyl, azetidinyl, pyrrolidinyl, piperazinyl, piperidinyl, 2- azasprio[3.3]heptanyl, diazaspiro[3.5]nonanyl, 2,6-diazaspiro[3.3]heptanyl, 3,6- diazabicyclo[3.1.1]heptanyl, 2,5-diazabicyclo[2.2.1]heptanyl, 3,8-diazabicyclo[3.2.1]octanyl, 2- azaspiro[3.5]nonanyl, 7-azaspiro[3.5]nonanyl, 6-azaspiro[3.4]octanyl, and octahydropyrrolo[3,2- b]pyrrolyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyB is absent or selected from phenyl, cyclohexyl, pyrazolyl, azetidinyl, pyrrolidinyl, piperazinyl, piperidinyl, azasprio[3.3]heptanyl, diazaspiro[3.5]nonanyl, 2,6-diazaspiro[3.3]heptanyl, 3,6-diazabicyclo[3.1.1]heptanyl, 2,5-diazabicyclo[2.2.1]heptanyl, 3,8-diazabicyclo[3.2.1]octanyl, 2-azaspiro[3.5]nonanyl, 7-azaspiro[3.5]nonanyl, 6-
azaspiro[3.4]octanyl, and octahydropyrrolo[3,2-b]pyrrolyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyB is selected from phenyl, cyclohexyl, pyrazolyl, azetidinyl, pyrrolidinyl, piperazinyl, piperidinyl, azasprio[3.3]heptanyl, diazaspiro[3.5]nonanyl, 2,6-diazaspiro[3.3]heptanyl, 3,6- diazabicyclo[3.1.1]heptanyl, 2,5-diazabicyclo[2.2.1]heptanyl, 3,8-diazabicyclo[3.2.1]octanyl, 2- azaspiro[3.5]nonanyl, 7-azaspiro[3.5]nonanyl, 6-azaspiro[3.4]octanyl, and octahydropyrrolo[3,2- b]pyrrolyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyB is absent or selected from phenyl, cyclohexyl, pyrazolyl, azetidinyl, pyrrolidinyl, piperazinyl, piperidinyl, azasprio[3.3]heptanyl, diazaspiro[3.5]nonanyl, 2,6-diazaspiro[3.3]heptanyl, 3,6-diazabicyclo[3.1.1]heptanyl, 2,5-diazabicyclo[2.2.1]heptanyl, 3,8-diazabicyclo[3.2.1]octanyl, and azaspiro[3.5]nonanyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyB is selected from phenyl, cyclohexyl, pyrazolyl, azetidinyl, pyrrolidinyl, piperazinyl, piperidinyl, azasprio[3.3]heptanyl, diazaspiro[3.5]nonanyl, 2,6-diazaspiro[3.3]heptanyl, 3,6- diazabicyclo[3.1.1]heptanyl, 2,5-diazabicyclo[2.2.1]heptanyl, 3,8-diazabicyclo[3.2.1]octanyl, and azaspiro[3.5]nonanyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyB is 2-azasprio[3.3]heptanyl. In some embodiments, CyC is absent or selected from C6-10 aryl, 5-10 membered heteroaryl, and 4-10 membered heterocycloalkyl, each optionally substituted by 1, 2, or 3 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyC is absent or selected from phenyl, 5-9 membered heteroaryl, and 4-7 membered heterocycloalkyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyC is absent or selected from 9 membered heteroaryl and 4-6 membered heterocycloalkyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyC is absent or selected from indazolyl, piperidinyl, piperazinyl, azetidinyl, and pyrrolidinyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyC is piperidinyl or piperazinyl. In some embodiments, CyC is absent. In some embodiments, CyC is piperidinyl, piperazinyl, or cyclobutyl. In some embodiments, CyC is cyclobutyl.
In some embodiments, CyC is selected from C6-10 aryl, 5-10 membered heteroaryl, and 4- 10 membered heterocycloalkyl, each optionally substituted by 1, 2, or 3 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyC is selected from phenyl, 5-9 membered heteroaryl, and 4-7 membered heterocycloalkyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyC is selected from 9 membered heteroaryl and 4-6 membered heterocycloalkyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyC is selected from indazolyl, piperidinyl, piperazinyl, azetidinyl, and pyrrolidinyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyD is absent or selected from C6-10 aryl, 5-10 membered heteroaryl, and 4-10 membered heterocycloalkyl, each optionally substituted by 1, 2, or 3 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyD is absent or selected from phenyl, 5-9 membered heteroaryl, and 4-7 membered heterocycloalkyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyD is absent or 5-6 membered heterocycloalkyl optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyD is absent or selected from piperazinyl and piperidinyl. In some embodiments, CyD is absent or 6 membered heterocycloalkyl. In some embodiments, CyD is absent or selected from piperazinyl and piperidinyl. In some embodiments, CyD is absent or piperazinyl. In some embodiments, CyD is absent. In some embodiments, CyD is selected from C6-10 aryl, 5-10 membered heteroaryl, and 4- 10 membered heterocycloalkyl, each optionally substituted by 1, 2, or 3 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyD is selected from phenyl, 5-9 membered heteroaryl, and 4-7 membered heterocycloalkyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyD is 5-6 membered heterocycloalkyl optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyD is selected from piperazinyl and piperidinyl. In some embodiments, CyD is 6-membered heterocycloalkyl.
In some embodiments, LA, LB, and LC are each independently absent or independently selected from C1-6 alkylene, -C(O)-, -O-, -O-(C1-6 alkylene)-, -NRc23-, -C(O)NRc23-, -C(O)-(C1-6 alkylene)-, -C(O)NRc23-(C1-6 alkylene)-, and -NRc23C(O)-. In some embodiments, LA, LB, and LC are each independently absent or independently selected from C1-6 alkylene, -C(O)-, -O-, -O-(C1-6 alkylene)-, -C(O)-(C1-6 alkylene)-, -C(O)NRc23- (C1-6 alkylene)-, and -NRc23C(O)-. In some embodiments, Rc23 is selected from H and C1-6 alkyl. In some embodiments, LA, LB, and LC are each independently absent or independently selected from methylene, -C(O)-, -O-, -O-(C1-3 alkylene)-, -C(O)CH2-, -C(O)NH-(C1-3 alkylene). In some embodiments, LA is absent or selected from methylene, -C(O)-, -C(O)CH2-, - C(O)NH-(C1-3 alkylene)-, and -NHC(O)-. In some embodiments, LA is absent or selected from methylene and -NHC(O)-. In some embodiments, LA is absent. In some embodiments, LA is selected from methylene and -NHC(O)-. In some embodiments, LA is methylene. In some embodiments, LA is -NHC(O)-. In some embodiments, LA is absent or selected from methylene, -C(O)-, -C(O)CH2-, - C(O)NH-(C1-3 alkylene)-, -NHC(O)-, -CH(CH3)-, -CH(CH(CH3)2)-, -CH(CH2CH2CH3)-, - CH(CH2OCH3)-, and -O-. In some embodiments, LA is selected from methylene, -C(O)-, - C(O)CH2-, -C(O)NH-(C1-3 alkylene)-, -NHC(O)-, -CH(CH3)-, -CH(CH(CH3)2)-, - CH(CH2CH2CH3)-, -CH(CH2OCH3)-, and -O-. In some embodiments, LB is absent or selected from methylene, -C(O)-, -ORa3-, - C(O)CH2-, -C(O)NH-(C1-3 alkylene)-, -NHC(O)-, and -O-(C1-6 alkylene)-. In some embodiments, LB is absent or methylene. In some embodiments, LB is absent. In some embodiments, LB is selected from methylene, -C(O)-, -ORa3-, -C(O)CH2-, -C(O)NH-(C1-3 alkylene)-, and -NHC(O)-. In some embodiments, LB is methylene. In some embodiments, LB is selected from -C(O)-, -ORa3- , -C(O)CH2-, -C(O)NH-(C1-3 alkylene)-, and -NHC(O)-. In some embodiments, LB is absent or selected from methylene, -C(O)-, -ORa3-, - C(O)CH2-, -C(O)NH-(C1-3 alkylene)-, -NHC(O)-, -O-, -O-(C1-6 alkylene)-, and -NRc23-(C1-6 alkylene)-. In some embodiments, LB is selected from methylene, -C(O)-, -ORa3-, -C(O)CH2-, - C(O)NH-(C1-3 alkylene)-, -NHC(O)-, -O-, -O-(C1-6 alkylene)-, and -NRc23-(C1-6 alkylene)-. In some embodiments, LC is absent or selected from methylene, -O-, -C(O)CH2-, and - NHC(O)-. In some embodiments, LC is absent or methylene. In some embodiments, LC is absent. In some embodiments, LC is methylene.
Ubiquitin ligase binding moieties and linkers are known and well-described in the art, for example: Bondeson, D. P., et al. Nat Chem Biol. 201511(8):611-617; An S, et al. EBioMedicine 201836:553-562; Paiva S-L. et al, Curr. Op. in Chem. Bio. 2010, 50:111-119; and International Patent Application Publication No. WO 2017/197056, each of which is incorporated by reference in its entirety. For example, thalidomide derivatives, such as lenalidomide or pomalidomide, have been reported to recruit potential protein substrates to cereblon, for example: WO 2019/099926 and WO 2020/023851. In some embodiments, Z is a cereblon E3 ubiquitin ligase binding moiety. In some embodiments, Z is an E3 ubiquitin ligase binding moiety that binds to cereblon. In some embodiments, Z comprises a chemical group derived from an imide, a thioimide, an amide, or a thioamide. In some embodiments, Z is thalidomide, lenalidomide, pomalidomide, analogs thereof, isosteres thereof, or derivatives thereof. In some embodiments, Z is a group having Formula III:
C2- 6 alkenyl, C2-6 alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl-C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl, wherein said C1-6 alkyl, C1-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl-C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl are each optionally substituted with 1, 2, or 3 substituents independently selected from OH, CN, amino, halo, C1-6 alkyl, C1-6 alkylamino, di(C1-6 alkyl)amino, C1-6 alkoxy, C1-6 haloalkyl, and C1-6 haloalkoxy; and each Re, Re1, Re2, and Re3 is independently selected from H, C1-4 alkyl, and CN.
In some embodiments, L is a linker having formula II: II wherein CyA, CyB, CyC, and CyD are each independently absent or independently selected from C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, and 4-10 membered heterocycloalkyl, each optionally substituted by 1, 2, 3, or 4 substituents independently selected from halo, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C1-6 haloalkyl, CN, NO2, ORa11, SRa11, C(O)Rb11, C(O)NRc11Rd11, C(O)ORa11, OC(O)Rb11, OC(O)NRc11Rd11, C(=NRe11)NRc11Rd11, NRc11C(=NRe11)NRc11Rd11, NRc11Rd11, NRc11C(O)Rb11, NRc11C(O)ORa11, NRc11C(O)NRc11Rd11, NRc11S(O)Rb11, NRc11S(O)2Rb11, NRc11S(O)2NRc11Rd11, S(O)Rb11, S(O)NRc11Rd11, S(O)2Rb11, and S(O)2NRc11Rd11; and LA, LB, and LC are each independently absent or independently selected from C1-6 alkylene, C2-6 alkenylene, C2-6 alkynylene, -C(O)-, -O-, -O-(C1-6 alkylene)-, -NRc23-, -S-(C1-6 alkylene)-, -C(O)-(C1-6 alkylene)-, -C(O)NRc23-(C1-6 alkylene)-, -C(O)O-, -C(O)O-(C1-6 alkylene)- , -OC(O)-(C1-6 alkylene)-, -OC(O)NRc23-(C1-6 alkylene)-, -NRc23-(C1-6 alkylene)-, -N-(C1-6 alkylene)-C(O)-, -NRc23C(O)-(C1-6 alkylene)-, -NRc23C(O)NRc23-(C1-6 alkylene)-, -NRc23C(O)O- (C1-6 alkylene)-, -C(=NRe23)NRc23-(C1-6 alkylene)-, -NRc23C(=NRe23)NRc23-(C1-6 alkylene)-, - S(O)-(C1-6 alkylene)-, -S(O)NRc23-(C1-6 alkylene)-, -S(O)2-(C1-6 alkylene)-, -NRc23S(O)2-(C1-6 alkylene)-, -NRc23S(O)2NRc23-(C1-6 alkylene)-, and -S(O)2NRc23-(C1-6 alkylene)-, wherein the C1-6 alkylene, C2-6 alkenylene, or C2-6 alkynylene is optionally substituted by 1, 2, or 3 substituents independently selected from C1-4 alkyl, C1-4 haloalkyl, C1-4 alkoxy, halo, and OH; each Ra11, Rb11, Rc11, and Rd11 is independently selected from H, C1-6 alkyl, C1-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl-C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl, wherein said C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl-C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl of Ra11, Rb11, Rc11, and Rd11 is optionally substituted with 1, 2, 3, 4, or 5 substituents independently selected from halo, C1-4 alkyl, C1-4 haloalkyl, C1-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, CN, ORa3, SRa3, C(O)Rb3, C(O)NRc3Rd3, C(O)ORa3,
OC(O)Rb3, OC(O)NRc3Rd3, NRc3Rd3, NRc3C(O)Rb3, NRc3C(O)NRc3Rd3, NRc3C(O)ORa3, C(=NRe3)NRc3Rd3, NRc3C(=NRe3)NRc3Rd3, S(O)Rb3, S(O)NRc3Rd3, S(O)2Rb3, NRc3S(O)2Rb3, NRc3S(O)2NRc3Rd3, and S(O)2NRc3Rd3; Rc23 is independently selected from H, C1-6 alkyl, C1-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl- C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl, wherein said C1-6 alkyl, C1-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl-C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl are each optionally substituted with 1, 2, or 3 substituents independently selected from OH, CN, amino, halo, C1-6 alkyl, C1-6 alkylamino, di(C1-6 alkyl)amino, C1-6 alkoxy, C1-6 haloalkyl, and C1-6 haloalkoxy; and Re23 is independently selected from H, C1-4 alkyl, and CN. In some embodiments, at least one of LA, LB, LC, CyA, CyB, CyC, and CyD is not absent In some embodiments, R1, R2, and R3 are each independently selected from H, F, methyl, and trifluoromethyl. In some embodiments, R1, R2, and R3 are each F. In some embodiments, R1, R2, and R3 are each selected from methyl and trifluoromethyl. In some embodiments, R1, R2, and R3 are each H. In some embodiments, R4 and R5 are each independently selected from H, halo, SRA, ORA, C1-4 alkyl, C2-4 alkenyl, and C1-4 haloalkyl, wherein the C2-4 alkenyl is optionally substituted with 1, 2, or 3 substituents independently selected from halo, and wherein RA is H, C1-4 alkyl, or C1-4 haloalkyl. In some embodiments, R4 and R5 are each independently selected from halo, SRA, ORA, C1-4 alkyl, C2-4 alkenyl, and C1-4 haloalkyl, wherein the C2-4 alkenyl is optionally substituted with 1, 2, or 3 substituents independently selected from halo, and wherein RA is H, C1-4 alkyl, or C1-4 haloalkyl. In some embodiments, R4 and R5 are each independently selected from H, SRA, C1- 4 alkyl, and C2-4 alkenyl, wherein the C2-4 alkenyl is optionally substituted with 1, 2, or 3 substituents independently selected from halo, and wherein RA is H, C1-4 alkyl, or C1-4 haloalkyl. In some embodiments, R4 and R5 are each independently selected from SRA, C1-4 alkyl, and C2-4 alkenyl, wherein the C2-4 alkenyl is optionally substituted with 1, 2, or 3 substituents independently selected from halo, and wherein RA is H, C1-4 alkyl, or C1-4 haloalkyl. In some embodiments, R4 and R5 are each independently selected from H, SRA, C1-4 alkyl, and C2-4
alkenyl, wherein the C2-4 alkenyl is optionally substituted with 1, 2, or 3 substituents independently selected from halo, and wherein RA is C1-4 alkyl. In some embodiments, R4 and R5 are each independently selected from SRA, C1-4 alkyl, and C2-4 alkenyl, wherein the C2-4 alkenyl is optionally substituted with 1, 2, or 3 substituents independently selected from halo, and wherein RA is C1-4 alkyl. In some embodiments, R4 and R5 are each independently selected from OH, C1-4 alkyl, and C1-4 haloalkyl; or R4 and R5 together with the carbon atom to which they are attached form a C3-6 cycloalkyl group optionally substituted with 1, 2, or 3 substituents independently selected from halo, ORA, C1-4 alkyl, and C1-4 haloalkyl. In some embodiments, R4 and R5 are each independently selected from OH, C1-4 alkyl, and C1-4 haloalkyl. In some embodiments, R4 and R5 are each C1-4 alkyl. In some embodiments, R4 and R5 are each methyl. In some embodiments, R4 and R5 together with the carbon atom to which they are attached form a C3-6 cycloalkyl group optionally substituted with 1, 2, or 3 substituents independently selected from halo, ORA, C1-2 alkyl, and C1-2 haloalkyl. In some embodiments, R4 and R5 together with the carbon atom to which they are attached form a C3-6 cycloalkyl group optionally substituted with 1 or 2 substituents independently selected from halo and C1-2 alkyl. In some embodiments, R4 and R5 together with the carbon atom to which they are attached form a C3-6 cycloalkyl group optionally substituted with 1 or 2 fluoro. In some embodiments, R6 and R7 are each H. In some embodiments, R8 is H. In some embodiments, Cy1 is selected from C3-14 cycloalkyl, 5-14 membered heteroaryl, C4-7 cycloalkyl fused with phenyl to form a bicyclic ring, and 4-7 membered heterocycloalkyl fused with phenyl to form a bicyclic ring, each optionally substituted by 1 or 2 substituents independently selected from halo, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C1-6 haloalkyl, CN, NO2, ORa, SRa, C1-6 alkyl-NRcRd, C(O)Rb, C(O)NRcRd, C(O)ORa, OC(O)Rb, OC(O)NRcRd, C(=NRe)NRcRd, NRcC(=NRe)NRcRd, NRcRd, NRcC(O)Rb, NRcC(O)ORa, NRcC(O)NRcRd, NRcS(O)Rb, NRcS(O)2Rb, NRcS(O)2NRcRd, S(O)Rb, S(O)NRcRd, S(O)2Rb, and S(O)2NRcRd. In some embodiments, Cy1 is selected from C3-6 cycloalkyl, 5-6 membered heteroaryl, C5-6 cycloalkyl fused with phenyl to form a bicyclic ring, and 5-6 membered heterocycloalkyl fused with phenyl to form a bicyclic ring, each optionally substituted by 1 or 2 substituents
independently selected from halo, C1-6 alkyl, C1-6 haloalkyl, CN, C1-6 alkyl-NRcRd, C(O)NRcRd, and NRcRd. In some embodiments, Cy1 is C3-6 cycloalkyl optionally substituted by 1 or 2 substituents independently selected from halo, C1-6 alkyl, and C1-6 haloalkyl. In some embodiments, Cy1 is C3- 6 cycloalkyl. In some embodiments, Cy1 is cyclopropyl or cyclobutyl. In some embodiments, Cy1 is cyclopropyl. In some embodiments, Cy1 is 5-6 membered heteroaryl, optionally substituted by 1 or 2 substituents independently selected from halo, C1-6 alkyl, and C(O)NRcRd. In some embodiments, Cy1 is 5 membered heteroaryl, optionally substituted by 1 or 2 substituents independently selected from halo, C1-6 alkyl, and C(O)NRcRd. In some embodiments, Cy1 is pyrazolyl or pyrrolyl, optionally substituted with C(O)NRcRd. In some embodiments, Cy1 is pyrazolyl. In some embodiments, Cy1 is pyrazolyl, pyrrolyl, or isoxazolyl, each optionally substituted with C(O)NRcRd. In some embodiments, Cy1 is isoxazolyl. In some embodiments, Cy1 is pyrazolyl, pyrrolyl, or isoxazolyl. In some embodiments, Cy1 is C5-6 cycloalkyl fused with phenyl to form a bicyclic ring, optionally substituted by 1 or 2 substituents independently selected from halo, C1-6 alkyl, CN, C1-6 alkyl-NRcRd, and NRcRd. In some embodiments, Cy1 is cyclopentyl fused with phenyl to form a bicyclic ring, optionally substituted by methyl, CN, Cl, F, -CH2NHRd, and NH2. In some embodiments, Cy1 is 5-6 membered heterocycloalkyl fused with phenyl to form a bicyclic ring, optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, Cy1 is dihydrobenzofuran, optionally substituted by 1 or 2 substituents independently selected from chloro and methyl. In some embodiments, Cy1 is dihydrobenzofuran substituted by 1 chloro. In some embodiments, Cy2 is selected from C6-10 aryl and 5-10 membered heteroaryl, optionally substituted by 1, 2, or 3 substituents independently selected from halo, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C1-6 haloalkyl, CN, NO2, ORa1, SRa1, C(O)Rb1, C(O)NRc1Rd1, C(O)ORa1, OC(O)Rb1, OC(O)NRc1Rd1, C(=NRe1)NRc1Rd1, NRc1C(=NRe1)NRc1Rd1, NRc1Rd1, NRc1C(O)Rb1, NRc1C(O)ORa1, NRc1C(O)NRc1Rd1, NRc1S(O)Rb1, NRc1S(O)2Rb1, NRc1S(O)2NRc1Rd1, S(O)Rb1, S(O)NRc1Rd1, S(O)2Rb1, and S(O)2NRc1Rd1. In some embodiments, Cy2 is phenyl or 5-6 membered heteroaryl, optionally substituted by 1 or 2 substituents selected from halo and C1-6 alkyl.
In some embodiments, Cy2 is C6-10 aryl optionally substituted by 1 or 2 substituents independently selected from halo, C1-6 alkyl, and C1-6 haloalkyl. In some embodiments, Cy2 is phenyl optionally substituted with 1 or 2 substituents each independently selected from C1-6 alkyl. In some embodiments, Cy2 is phenyl, methylphenyl, methylchlorophenyl, or dimethylphenyl. In some embodiments, Cy2 is phenyl optionally substituted with 1 or 2 substituents selected from methyl and chloro. In some embodiments, Cy2 is 5-10 membered heteroaryl optionally substituted by 1 or 2 substituents independently selected from halo, C1-6 alkyl, and C1-6 haloalkyl. In some embodiments, Cy2 is 6 membered heteroaryl optionally substituted by 1 or 2 substituents each independently selected from C1-6 alkyl. In some embodiments, Cy2 is pyridinyl or methylpyridinyl. In some embodiments, Cy2 is pyridinyl or pyrimidinyl, each optionally substituted by 1 or 2 substituents each independently selected from C1-6 alkyl. In some embodiments, Cy2 is pyridinyl or pyrimidinyl, each optionally substituted with methyl. In some embodiments, Cy2 is pyridinyl, methylpyridinyl, or pyrimidinyl. In some embodiments, Cy2 is pyridinyl or pyrimidinyl, each optionally substituted with 1 or 2 methyl groups. In some embodiments, Cy2 is C3-7 cycloalkyl optionally substituted by 1 or 2 substituents independently selected from halo, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C1-6 haloalkyl, CN, NO2, ORa1, SRa1, C(O)Rb1, C(O)NRc1Rd1, C(O)ORa1, OC(O)Rb1, OC(O)NRc1Rd1, C(=NRe1)NRc1Rd1, NRc1C(=NRe1)NRc1Rd1, NRc1Rd1, NRc1C(O)Rb1, NRc1C(O)ORa1, NRc1C(O)NRc1Rd1, NRc1S(O)Rb1, NRc1S(O)2Rb1, NRc1S(O)2NRc1Rd1, S(O)Rb1, S(O)NRc1Rd1, S(O)2Rb1, and S(O)2NRc1Rd1. In some embodiments, Cy2 is bicyclo[1.1.1]pentyl. In some embodiments, CyA, CyB, CyC, and CyD are each independently absent or independently selected from C6-10 aryl, 5-10 membered heteroaryl, and 4-10 membered heterocycloalkyl, each optionally substituted by 1, 2, or 3 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyA, CyB, CyC, and CyD are each independently absent or independently selected from phenyl, 5-9 membered heteroaryl, and 4-7 membered heterocycloalkyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyA is absent or selected from C6-10 aryl, 5-10 membered heteroaryl, and 4-10 membered heterocycloalkyl, each optionally substituted by 1, 2, or 3 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyA is absent
or selected from phenyl, 5-9 membered heteroaryl, and 4-7 membered heterocycloalkyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyA is absent or selected from phenyl, 5-6 membered heteroaryl, and 6-9 membered heterocycloalkyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyA is absent or selected from phenyl, pyrazolyl, pyridinyl, indazolyl, and piperidinyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyA is phenyl or pyridinyl, each optionally substituted by halo or methyl. In some embodiments, CyA is absent. In some embodiments, CyA is selected from C6-10 aryl, 5-10 membered heteroaryl, and 4- 10 membered heterocycloalkyl, each optionally substituted by 1, 2, or 3 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyA is selected from phenyl, 5-9 membered heteroaryl, and 4-7 membered heterocycloalkyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyA is selected from phenyl, 5-6 membered heteroaryl, and 6-9 membered heterocycloalkyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyA is selected from phenyl, pyrazolyl, pyridinyl, indazolyl, and piperidinyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyA is phenyl or pyridinyl, each optionally substituted by1 or 2 substituents selected from halo and methyl. In some embodiments, CyA is absent or selected from phenyl, pyrazolyl, pyridinyl, pyrazinyl, pyrimidinyl, indazolyl, 2,3-dihydrobenzofuranyl, and piperidinyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyA is selected from phenyl, pyrazolyl, pyridinyl, pyrazinyl, pyrimidinyl, indazolyl, 2,3-dihydrobenzofuranyl, and piperidinyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyB is absent or selected from C6-10 aryl, 5-10 membered heteroaryl, and 4-10 membered heterocycloalkyl, each optionally substituted by 1, 2, or 3 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyB is absent or selected from phenyl, 5-9 membered heteroaryl, and 4-7 membered heterocycloalkyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyB is absent or selected from phenyl, 5 membered heteroaryl, and 6-7
membered heterocycloalkyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyB is absent or selected from phenyl, pyrazolyl, piperazinyl, piperidinyl, and azasprio[3.3]heptanyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyB is piperidinyl or piperazinyl. In some embodiments, CyB is absent. In some embodiments, CyB is selected from C6-10 aryl, 5-10 membered heteroaryl, and 4- 10 membered heterocycloalkyl, each optionally substituted by 1, 2, or 3 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyB is selected from phenyl, 5-9 membered heteroaryl, and 4-7 membered heterocycloalkyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyB is selected from phenyl, 5 membered heteroaryl, and 6-7 membered heterocycloalkyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyB is selected from phenyl, pyrazolyl, piperazinyl, piperidinyl, and azasprio[3.3]heptanyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyB is absent or selected from phenyl, cyclohexyl, pyrazolyl, azetidinyl, pyrrolidinyl, piperazinyl, piperidinyl, 2-azasprio[3.3]heptanyl, diazaspiro[3.5]nonanyl, 2,6-diazaspiro[3.3]heptanyl, 3,6-diazabicyclo[3.1.1]heptanyl, 2,5-diazabicyclo[2.2.1]heptanyl, 3,8-diazabicyclo[3.2.1]octanyl, 2-azaspiro[3.5]nonanyl, 7-azaspiro[3.5]nonanyl, 6- azaspiro[3.4]octanyl, and octahydropyrrolo[3,2-b]pyrrolyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyB is selected from phenyl, cyclohexyl, pyrazolyl, azetidinyl, pyrrolidinyl, piperazinyl, piperidinyl, 2- azasprio[3.3]heptanyl, diazaspiro[3.5]nonanyl, 2,6-diazaspiro[3.3]heptanyl, 3,6- diazabicyclo[3.1.1]heptanyl, 2,5-diazabicyclo[2.2.1]heptanyl, 3,8-diazabicyclo[3.2.1]octanyl, 2- azaspiro[3.5]nonanyl, 7-azaspiro[3.5]nonanyl, 6-azaspiro[3.4]octanyl, and octahydropyrrolo[3,2- b]pyrrolyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyB is absent or selected from phenyl, cyclohexyl, pyrazolyl, azetidinyl, pyrrolidinyl, piperazinyl, piperidinyl, azasprio[3.3]heptanyl, diazaspiro[3.5]nonanyl, 2,6-diazaspiro[3.3]heptanyl, 3,6-diazabicyclo[3.1.1]heptanyl, 2,5-diazabicyclo[2.2.1]heptanyl, 3,8-diazabicyclo[3.2.1]octanyl, 2-azaspiro[3.5]nonanyl, 7-azaspiro[3.5]nonanyl, 6-
azaspiro[3.4]octanyl, and octahydropyrrolo[3,2-b]pyrrolyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyB is selected from phenyl, cyclohexyl, pyrazolyl, azetidinyl, pyrrolidinyl, piperazinyl, piperidinyl, azasprio[3.3]heptanyl, diazaspiro[3.5]nonanyl, 2,6-diazaspiro[3.3]heptanyl, 3,6- diazabicyclo[3.1.1]heptanyl, 2,5-diazabicyclo[2.2.1]heptanyl, 3,8-diazabicyclo[3.2.1]octanyl, 2- azaspiro[3.5]nonanyl, 7-azaspiro[3.5]nonanyl, 6-azaspiro[3.4]octanyl, and octahydropyrrolo[3,2- b]pyrrolyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyB is absent or selected from phenyl, cyclohexyl, pyrazolyl, azetidinyl, pyrrolidinyl, piperazinyl, piperidinyl, azasprio[3.3]heptanyl, diazaspiro[3.5]nonanyl, 2,6-diazaspiro[3.3]heptanyl, 3,6-diazabicyclo[3.1.1]heptanyl, 2,5-diazabicyclo[2.2.1]heptanyl, 3,8-diazabicyclo[3.2.1]octanyl, and azaspiro[3.5]nonanyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyB is selected from phenyl, cyclohexyl, pyrazolyl, azetidinyl, pyrrolidinyl, piperazinyl, piperidinyl, azasprio[3.3]heptanyl, diazaspiro[3.5]nonanyl, 2,6-diazaspiro[3.3]heptanyl, 3,6- diazabicyclo[3.1.1]heptanyl, 2,5-diazabicyclo[2.2.1]heptanyl, 3,8-diazabicyclo[3.2.1]octanyl, and azaspiro[3.5]nonanyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyB is 2-azasprio[3.3]heptanyl. In some embodiments, CyC is absent or selected from C6-10 aryl, 5-10 membered heteroaryl, and 4-10 membered heterocycloalkyl, each optionally substituted by 1, 2, or 3 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyC is absent or selected from phenyl, 5-9 membered heteroaryl, and 4-7 membered heterocycloalkyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyC is absent or selected from 9 membered heteroaryl and 4-6 membered heterocycloalkyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyC is absent or selected from indazolyl, piperidinyl, piperazinyl, azetidinyl, and pyrrolidinyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyC is piperidinyl or piperazinyl. In some embodiments, CyC is absent. In some embodiments, CyC is piperidinyl, piperazinyl, or cyclobutyl. In some embodiments, CyC is cyclobutyl.
In some embodiments, CyC is selected from C6-10 aryl, 5-10 membered heteroaryl, and 4- 10 membered heterocycloalkyl, each optionally substituted by 1, 2, or 3 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyC is selected from phenyl, 5-9 membered heteroaryl, and 4-7 membered heterocycloalkyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyC is selected from 9 membered heteroaryl and 4-6 membered heterocycloalkyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyC is selected from indazolyl, piperidinyl, piperazinyl, azetidinyl, and pyrrolidinyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyD is absent or selected from C6-10 aryl, 5-10 membered heteroaryl, and 4-10 membered heterocycloalkyl, each optionally substituted by 1, 2, or 3 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyD is absent or selected from phenyl, 5-9 membered heteroaryl, and 4-7 membered heterocycloalkyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyD is absent or 5-6 membered heterocycloalkyl optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyD is absent or selected from piperazinyl and piperidinyl. In some embodiments, CyD is absent or 6 membered heterocycloalkyl. In some embodiments, CyD is absent or selected from piperazinyl and piperidinyl. In some embodiments, CyD is absent or piperazinyl. In some embodiments, CyD is absent. In some embodiments, CyD is selected from C6-10 aryl, 5-10 membered heteroaryl, and 4- 10 membered heterocycloalkyl, each optionally substituted by 1, 2, or 3 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyD is selected from phenyl, 5-9 membered heteroaryl, and 4-7 membered heterocycloalkyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyD is 5-6 membered heterocycloalkyl optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl. In some embodiments, CyD is selected from piperazinyl and piperidinyl. In some embodiments, CyD is 6-membered heterocycloalkyl.
In some embodiments, LA, LB, and LC are each independently absent or independently selected from C1-6 alkylene, -C(O)-, -O-, -O-(C1-6 alkylene)-, -NRc23-, -C(O)NRc23-, -C(O)-(C1-6 alkylene)-, -C(O)NRc23-(C1-6 alkylene)-, and -NRc23C(O)-. In some embodiments, LA, LB, and LC are each independently absent or independently selected from C1-6 alkylene, -C(O)-, -O-, -O-(C1-6 alkylene)-, -C(O)-(C1-6 alkylene)-, -C(O)NRc23- (C1-6 alkylene)-, and -NRc23C(O)-. In some embodiments, Rc23 is selected from H and C1-6 alkyl. In some embodiments, LA, LB, and LC are each independently absent or independently selected from methylene, -C(O)-, -O-, -O-(C1-3 alkylene)-, -C(O)CH2-, -C(O)NH-(C1-3 alkylene). In some embodiments, LA is absent or selected from methylene, -C(O)-, -C(O)CH2-, - C(O)NH-(C1-3 alkylene)-, and -NHC(O)-. In some embodiments, LA is absent or selected from methylene and -NHC(O)-. In some embodiments, LA is absent. In some embodiments, LA is selected from methylene and -NHC(O)-. In some embodiments, LA is methylene. In some embodiments, LA is -NHC(O)-. In some embodiments, LA is absent or selected from methylene, -C(O)-, -C(O)CH2-, - C(O)NH-(C1-3 alkylene)-, -NHC(O)-, -CH(CH3)-, -CH(CH(CH3)2)-, -CH(CH2CH2CH3)-, - CH(CH2OCH3)-, and -O-. In some embodiments, LA is selected from methylene, -C(O)-, - C(O)CH2-, -C(O)NH-(C1-3 alkylene)-, -NHC(O)-, -CH(CH3)-, -CH(CH(CH3)2)-, - CH(CH2CH2CH3)-, -CH(CH2OCH3)-, and -O-. In some embodiments, LB is absent or selected from methylene, -C(O)-, -ORa3-, - C(O)CH2-, -C(O)NH-(C1-3 alkylene)-, -NHC(O)-, and -O-(C1-6 alkylene)-. In some embodiments, LB is absent or methylene. In some embodiments, LB is absent. In some embodiments, LB is selected from methylene, -C(O)-, -ORa3-, -C(O)CH2-, -C(O)NH-(C1-3 alkylene)-, and -NHC(O)-. In some embodiments, LB is methylene. In some embodiments, LB is selected from -C(O)-, -ORa3- , -C(O)CH2-, -C(O)NH-(C1-3 alkylene)-, and -NHC(O)-. In some embodiments, LB is absent or selected from methylene, -C(O)-, -ORa3-, - C(O)CH2-, -C(O)NH-(C1-3 alkylene)-, -NHC(O)-, -O-, -O-(C1-6 alkylene)-, and -NRc23-(C1-6 alkylene)-. In some embodiments, LB is selected from methylene, -C(O)-, -ORa3-, -C(O)CH2-, - C(O)NH-(C1-3 alkylene)-, -NHC(O)-, -O-, -O-(C1-6 alkylene)-, and -NRc23-(C1-6 alkylene)-. In some embodiments, LC is absent or selected from methylene, -O-, -C(O)CH2-, and - NHC(O)-. In some embodiments, LC is absent or methylene. In some embodiments, LC is absent. In some embodiments, LC is methylene.
Ubiquitin ligase binding moieties and linkers are known and well-described in the art, for example: Bondeson, D. P., et al. Nat Chem Biol. 201511(8):611-617; An S, et al. EBioMedicine 201836:553-562; Paiva S-L. et al, Curr. Op. in Chem. Bio. 2010, 50:111-119; and International Patent Application Publication No. WO 2017/197056, each of which is incorporated by reference in its entirety. For example, thalidomide derivatives, such as lenalidomide or pomalidomide, have been reported to recruit potential protein substrates to cereblon, for example: WO 2019/099926 and WO 2020/023851. In some embodiments, Z is a cereblon E3 ubiquitin ligase binding moiety. In some embodiments, Z is an E3 ubiquitin ligase binding moiety that binds to cereblon. In some embodiments, Z comprises a chemical group derived from an imide, a thioimide, an amide, or a thioamide. In some embodiments, Z is thalidomide, lenalidomide, pomalidomide, analogs thereof, isosteres thereof, or derivatives thereof. In some embodiments, Z is a group having Formula III:
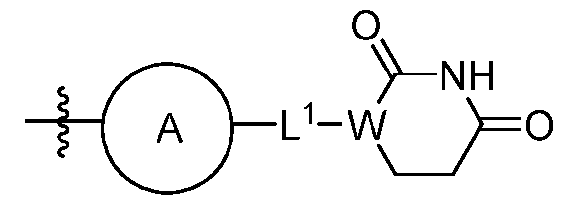 wherein ring A is C6-10 aryl, 5-14 membered heteroaryl, C3-7 cycloalkyl fused with phenyl to form a bicyclic ring, 4-7 membered heterocycloalkyl fused with phenyl to form a bicyclic ring, each optionally substituted with 1, 2, or 3 substituents independently selected from halo, C1-6 alkyl, and C1-6 haloalkyl; L1 is absent, CH2, NH, or O; and W is CH or N, wherein the wavy line represents the point of attachment to group L. In some embodiments, ring A is 4-7 membered heterocycloalkyl fused with phenyl to form a bicyclic ring, optionally substituted with halo. In some embodiments, ring A is phenyl or pyridinyl, each optionally substituted with 1, 2, or 3 substituents independently selected from halo, C1-6 alkyl, and C1-6 haloalkyl. In some embodiments, ring A is phenyl optionally substituted with 1, 2, or 3 substituents independently selected from halo, C1-6 alkyl, and C1-6 haloalkyl. In some embodiments, ring A is pyrimidinyl optionally substituted with 1, 2, or 3 substituents
independently selected from halo, C1-6 alkyl, and C1-6 haloalkyl. In some embodiments, ring A is phenyl or pyridinyl, each optionally substituted with halo. In some embodiments, ring A is phenyl optionally substituted with halo. In some embodiments, ring A is pyrimidinyl optionally substituted with halo. In some embodiments, ring A is 4-7 membered heterocycloalkyl fused with phenyl to form a bicyclic ring, optionally substituted with halo. In some embodiments, ring A is phenyl or pyridinyl. In some embodiments, ring A is phenyl. In some embodiments, ring A is pyrimidinyl. In some embodiments, L1 is absent or NH. In some embodiments, L1 is absent. In some embodiments, L1 is NH. In some embodiments, W is CH. In some embodiments, W is N. In some embodiments, Z is selected from: ;
wherein ring A is C6-10 aryl, 5-14 membered heteroaryl, C3-7 cycloalkyl fused with phenyl to form a bicyclic ring, 4-7 membered heterocycloalkyl fused with phenyl to form a bicyclic ring, each optionally substituted with 1, 2, or 3 substituents independently selected from halo, C1-6 alkyl, and C1-6 haloalkyl; L1 is absent, CH2, NH, or O; and W is CH or N, wherein the wavy line represents the point of attachment to group L. In some embodiments, ring A is 4-7 membered heterocycloalkyl fused with phenyl to form a bicyclic ring, optionally substituted with halo. In some embodiments, ring A is phenyl or pyridinyl, each optionally substituted with 1, 2, or 3 substituents independently selected from halo, C1-6 alkyl, and C1-6 haloalkyl. In some embodiments, ring A is phenyl optionally substituted with 1, 2, or 3 substituents independently selected from halo, C1-6 alkyl, and C1-6 haloalkyl. In some embodiments, ring A is pyrimidinyl optionally substituted with 1, 2, or 3 substituents
independently selected from halo, C1-6 alkyl, and C1-6 haloalkyl. In some embodiments, ring A is phenyl or pyridinyl, each optionally substituted with halo. In some embodiments, ring A is phenyl optionally substituted with halo. In some embodiments, ring A is pyrimidinyl optionally substituted with halo. In some embodiments, ring A is 4-7 membered heterocycloalkyl fused with phenyl to form a bicyclic ring, optionally substituted with halo. In some embodiments, ring A is phenyl or pyridinyl. In some embodiments, ring A is phenyl. In some embodiments, ring A is pyrimidinyl. In some embodiments, L1 is absent or NH. In some embodiments, L1 is absent. In some embodiments, L1 is NH. In some embodiments, W is CH. In some embodiments, W is N. In some embodiments, Z is selected from: ;
; ; ; 5 Cl, C(CH3), and N; and each V is independently selected from CH, C(CH3), and N. In some embodiments, Z is selected from:
O U V NH ; ;
 N; and each V is independently selected from CH and N. In some embodiments, Z is selected from: O
. A:
N; and each V is independently selected from CH and N. In some embodiments, Z is selected from: O
. A:
 or a pharmaceutically acceptable salt thereof. In some embodiments, the compound has Formula IVA: ,
or a pharmaceutically acceptable salt thereof. In some embodiments, the compound has Formula IVA: ,
 In some embodiments, the compound has Formula IVB:
In some embodiments, the compound has Formula IVB:
In some embodiments, Cy1 is selected from C3-6 cycloalkyl, 5-6 membered heteroaryl, C5-6 cycloalkyl fused with phenyl to form a bicyclic ring, and 5-6 membered heterocycloalkyl fused with phenyl to form a bicyclic ring, each optionally substituted by 1 or 2 substituents independently selected from halo, C1-6 alkyl, C1-6 haloalkyl, CN, C1-6 alkyl-NRcRd, C(O)NRcRd, and NRcRd; Cy2 is selected from C6-10 aryl and 5-10 membered heteroaryl, optionally substituted by 1, 2, or 3 substituents independently selected from halo, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C1-6 haloalkyl, CN, NO2, ORa1, SRa1, C(O)Rb1, C(O)NRc1Rd1, C(O)ORa1, OC(O)Rb1, OC(O)NRc1Rd1, C(=NRe1)NRc1Rd1, NRc1C(=NRe1)NRc1Rd1, NRc1Rd1, NRc1C(O)Rb1, NRc1C(O)ORa1, NRc1C(O)NRc1Rd1, NRc1S(O)Rb1, NRc1S(O)2Rb1, NRc1S(O)2NRc1Rd1, S(O)Rb1, S(O)NRc1Rd1, S(O)2Rb1, and S(O)2NRc1Rd1; CyA, CyB, CyC, and CyD are each independently absent or independently selected from C6- 10 aryl, 5-10 membered heteroaryl, and 4-10 membered heterocycloalkyl, each optionally substituted by 1, 2, or 3 substituents independently selected from halo and C1-6 alkyl; and LA, LB, and LC are each independently absent or independently selected from C1-6 alkylene, -C(O)-, -O-, -O-(C1-6 alkylene)-, -C(O)-(C1-6 alkylene)-, -C(O)NRc23-(C1-6 alkylene)-, and -NRc23C(O)-. In some embodiments, the compound has Formula IB:
IB or a pharmaceutically acceptable salt thereof; wherein R41 is selected from halo, C1-4 alkyl, and C1-4 haloalkyl; and p is 0, 1, or 2. In some embodiments, R41 is halo. In some embodiments, R41 is chloro. In some embodiments, p is 0 or 1. In some embodiments, R41 is chloro and p is 0 or 1. In some embodiments, the compound has Formula IB-1:
 In some embodiments, the compound has Formula IC:
In some embodiments, the compound has Formula IC:
 or a pharmaceutically acceptable salt thereof; wherein X1 and X2 are each independently selected from CH and N; each R42 and R43 is independently selected from halo, C1-4 alkyl, and C1-4 haloalkyl; and q and r are each independently 0, 1, or 2. In some embodiments, X is CH. In some embodiments, X is N. In some embodiments, R42 and R43 are each independently C1-4 alkyl. In some embodiments, R42 and R43 are methyl. In some embodiments, the compound has Formula ID:
wherein X is CH or N; and R44 is selected from H, halo, C1-4 alkyl, and C1-4 haloalkyl. In some embodiments, X is CH. In some embodiments, X is N. In some embodiments, R44 is halo. In some embodiments, R44 is chloro. In some embodiments, the compound has Formula ID-1:
or a pharmaceutically acceptable salt thereof; wherein X1 and X2 are each independently selected from CH and N; each R42 and R43 is independently selected from halo, C1-4 alkyl, and C1-4 haloalkyl; and q and r are each independently 0, 1, or 2. In some embodiments, X is CH. In some embodiments, X is N. In some embodiments, R42 and R43 are each independently C1-4 alkyl. In some embodiments, R42 and R43 are methyl. In some embodiments, the compound has Formula ID:
wherein X is CH or N; and R44 is selected from H, halo, C1-4 alkyl, and C1-4 haloalkyl. In some embodiments, X is CH. In some embodiments, X is N. In some embodiments, R44 is halo. In some embodiments, R44 is chloro. In some embodiments, the compound has Formula ID-1:
 In some embodiments, the compound has Formula IE:
In some embodiments, the compound has Formula IE:
 or a pharmaceutically acceptable salt thereof; wherein each R42 and R43 is independently selected from halo, C1-4 alkyl, and C1-4 haloalkyl; X1 and X2 are each independently selected from CH and N;
and q and r are each independently 0, 1, or 2. In some embodiments, the compound has Formula IF: or a
or a pharmaceutically acceptable salt thereof; wherein each R42 and R43 is independently selected from halo, C1-4 alkyl, and C1-4 haloalkyl; X1 and X2 are each independently selected from CH and N;
and q and r are each independently 0, 1, or 2. In some embodiments, the compound has Formula IF: or a
 wherein each is independently selected from halo, C1-4 alkyl, and C1-4 haloalkyl; and q is 0, 1, or 2. In some embodiments, the compound has Formula IG:
wherein each is independently selected from halo, C1-4 alkyl, and C1-4 haloalkyl; and q is 0, 1, or 2. In some embodiments, the compound has Formula IG:
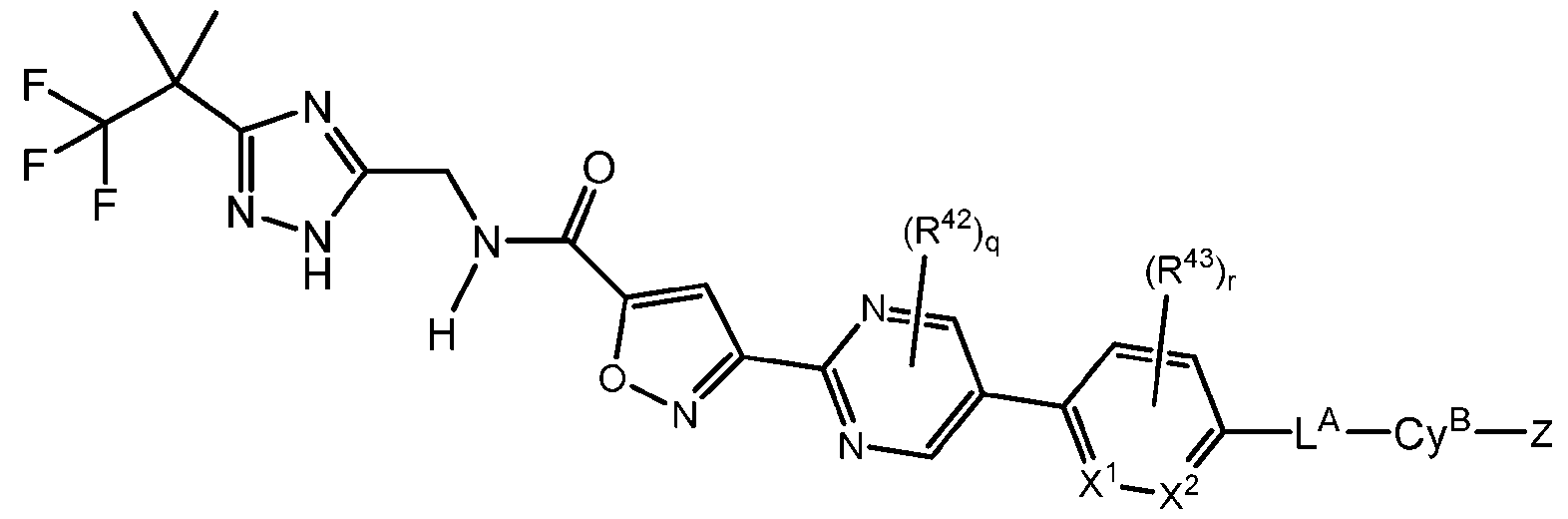 IG, or a pharmaceutically acceptable salt thereof; wherein each R42 and R43 is independently selected from halo, C1-4 alkyl, and C1-4 haloalkyl; X1 and X2 are each independently selected from CH and N; and q and r are each independently 0, 1, or 2. In some embodiments, the compound is selected from: (2R)-5-chloro-6-(4-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperazin-1-yl)methyl)phenyl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5- yl)methyl)-2,3-dihydrobenzofuran-2-carboxamide;
(2R)-5-chloro-6-(4-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperazine-1-carbonyl)phenyl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5- yl)methyl)-2,3-dihydrobenzofuran-2-carboxamide; (2R)-5-chloro-6-(6-(4-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperazin-1-yl)methyl)piperidin-1-yl)pyridin-3-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2- yl)-1H-1,2,4-triazol-5-yl)methyl)-2,3-dihydrobenzofuran-2-carboxamide; (1S,2S)-2-(4'-(1-(2-(4-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperazin-1-yl)methyl)piperidin-1-yl)-2-oxoethyl)-1H-pyrazol-4-yl)-[1,1'-biphenyl]-4-yl)-N- ((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)cyclopropane-1- carboxamide; (1S,2S)-2-[4-[4-[1-[2-[4-[[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]piperazin-1-yl]methyl]-1-piperidyl]-2-oxo-ethyl]pyrazol-3-yl]phenyl]phenyl]-N-[[3-(2,2,2- trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]cyclopropanecarboxamide hydrochloride; 5-(4-((1-(2-(4-(3-chloro-4'-((1S,2S)-2-(3-(3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H- 1,2,4-triazol-5-yl)propanoyl)cyclopropyl)-[1,1'-biphenyl]-4-yl)-1H-pyrazol-1-yl)acetyl)piperidin- 4-yl)methyl)piperazin-1-yl)-2-(2,6-dioxopiperidin-3-yl)-6-fluoroisoindoline-1,3-dione; (1S,2S)-2-(2'-chloro-4'-(1-(2-(4-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3- dioxoisoindolin-5-yl)piperazin-1-yl)methyl)piperidin-1-yl)-2-oxoethyl)-1H-pyrazol-4-yl)-[1,1'- biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5- yl)methyl)cyclopropane-1-carboxamide; (1S,2S)-2-[4-[4-[1-[2-[3-[[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]piperazin-1-yl]methyl]azetidin-1-yl]-2-oxo-ethyl]pyrazol-4-yl]phenyl]phenyl]-N-[[3-(2,2,2- trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]cyclopropanecarboxamide; 3-chloro-N-(3-(2-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperazin-1-yl)ethoxy)propyl)-4'-((1S,2S)-2-(((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H- 1,2,4-triazol-5-yl)methyl)carbamoyl)cyclopropyl)-[1,1'-biphenyl]-4-carboxamide; 3-chloro-N-(2-(4-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperazin-1-yl)methyl)piperidin-1-yl)-2-oxoethyl)-4'-((1S,2S)-2-(((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)carbamoyl)cyclopropyl)-[1,1'-biphenyl]-4- carboxamide;
2-chloro-N-[1-[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]-4- piperidyl]-4-piperidyl]-4-[4-[(1S,2S)-2-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol- 5-yl]methylcarbamoyl]cyclopropyl]phenyl]benzamide; (1S,2S)-2-[4-[1-[2-[4-[[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]piperazin-1-yl]methyl]-1-piperidyl]-2-oxo-ethyl]-3-methyl-indazol-5-yl]phenyl]-N-[[3-(2,2,2- trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]cyclopropanecarboxamide; (1S,2S)-2-[4-[4-[1-[1-[2-[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]piperazin-1-yl]-2-oxo-ethyl]azetidin-3-yl]pyrazol-4-yl]phenyl]phenyl]-N-[[3-(2,2,2-trifluoro- 1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]cyclopropanecarboxamide; (1S,2S)-2-[4-[4-[1-[[1-[2-[4- [2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl] piperazin-1-yl]-2-oxo-ethyl] -4-piperidyl]methyl]pyrazol-4-yl]phenyl]phenyl]-N-[[3-(2,2,2- trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]cyclopropanecarboxamide; (1S,2S)-2-[4-[4-[1-[2-[(3S)-3-[[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo- isoindolin-5-yl]-4-piperidyl] oxy]pyrrolidin-1-yl]-2-oxo-ethyl]pyrazol-4-yl]phenyl]phenyl]-N-[[3- (2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]cyclopropanecarboxamide; (1S,2S)-2-[4-[1-[4-[4-[[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]piperazin-1-yl]methyl]piperidine-1-carbonyl]phenyl]pyrazol-4-yl]phenyl]-N-[[3-(2,2,2- trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]cyclopropanecarboxamide; (1S,2S)-2-(4'-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperazin-1-yl)methyl)-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H- 1,2,4-triazol-5-yl)methyl)cyclopropane-1-carboxamide; (1S,2S)-2-(4'-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperazin-1-yl)methyl)-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H- 1,2,4-triazol-5-yl)methyl)cyclopropane-1-carboxamide; 1-[3-[4-[[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]piperazin-1- yl]methyl]phenyl]-2,4-dimethyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4- triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-[4-[4-[[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]piperazin-1- yl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol- 5-yl]methyl]pyrazole-4-carboxamide;
1-[4-[3-[[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]piperazin-1- yl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol- 5-yl]methyl]pyrazole-4-carboxamide; 1-[4-[4-[[4-[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]piperazin-1- yl]-1-piperidyl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H- 1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-[4-[4-[[4-[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]-4- piperidyl]piperazin-1-yl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl- ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-(4'-((4-((1-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperidin-4- yl)methyl)piperazin-1-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-(4'-((4-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1- yl)methyl)piperidin-1-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-(4'-((4-((1-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperidin-4- yl)oxy)piperidin-1-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-(4'-((4-(1-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperidin-4- yl)-1,4-diazepan-1-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-(4'-((1'-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)-[4,4'- bipiperidin]-1-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2- yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-(4'-((3-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1- yl)pyrrolidin-1-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan- 2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-(4-(6-((4-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1- yl)piperidin-1-yl)methyl)pyridin-3-yl)-2-methylphenyl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2- yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide;
1-(2'-chloro-4'-((4-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperazin-1-yl)piperidin-1-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-(3'-((4-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1- yl)piperidin-1-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2- yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-(4'-(1-(4-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1- yl)piperidin-1-yl)ethyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2- yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-[4-[4-[[4-[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]piperazin-1- yl]-1-piperidyl]methyl]-3,5-difluoro-phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-(4-(1-(1'-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)-[1,4'- bipiperidin]-4-yl)-3-methyl-1H-indazol-5-yl)-2-methylphenyl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-[4-[6-[[1-[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]-4-piperidyl]- 4-piperidyl]oxy]-3-pyridyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H- 1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-[4-[4-[2-[4-[[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]piperazin- 1-yl]methyl]-1-piperidyl]ethoxy]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl- ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-(3'-(2-(4-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1- yl)methyl)piperidin-1-yl)ethoxy)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-[4-[4-[1-[4-[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]piperazin- 1-yl]-1-piperidyl]-1-methyl-ethyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl- ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-[4-[5-[[4-[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]piperazin-1- yl]-1-piperidyl]methyl]pyrazin-2-yl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide;
1-[4-[5-[[4-[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]piperazin-1- yl]-1-piperidyl]methyl]-2-pyridyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-(3'-chloro-4'-((4-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperazin-1-yl)piperidin-1-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-[4-[4-[[5-[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl] piperazin-1- yl]-2-pyridyl]oxy]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4- triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-[4-[4-[[4-[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]piperazin-1- yl]-2-oxo-1-piperidyl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl- ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-[4-[6-[4-[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]piperazin-1- yl]cyclohexoxy]-3-pyridyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H- 1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-[4-[1-[[1-[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]-4- piperidyl]-4-piperidyl]methyl]pyrazol-4-yl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-[4-[6-[[1-[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]-4-piperidyl]- 4-piperidyl]amino]-3-pyridyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H- 1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-[4-[4-[[2-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]-2,7- diazaspiro[3.5]nonan-7-yl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl- ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-(4'-((4-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1- yl)azepan-1-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2- yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-(4'-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)-1,4-diazepan-1- yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide;
1-[4-[4-[[8-[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]-4-piperidyl]- 3,8-diazabicyclo[3.2.1]octan-3-yl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-(4'-((3-(1-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperidin-4- yl)-3,6-diazabicyclo[3.1.1]heptan-6-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-(4'-(((1S,4S)-5-(1-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperidin-4-yl)-2,5-diazabicyclo[2.2.1]heptan-2-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N- ((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4- carboxamide; 1-(4'-(((1R,4R)-5-(1-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperidin-4-yl)-2,5-diazabicyclo[2.2.1]heptan-2-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N- ((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4- carboxamide; 1-(4'-((3-(1-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperidin-4- yl)-3,8-diazabicyclo[3.2.1]octan-8-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-(4'-((3-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1- yl)azetidin-1-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2- yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-[4-[4-[[2-[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]-4-piperidyl]- 2,6-diazaspiro[3.3]heptan-6-yl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-[4-[4-[[4-[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]piperazin-1- yl]-3,3-difluoro-1-piperidyl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-[4-[4-[[4-[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]piperazin-1- yl]-4-methyl-1-piperidyl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl- ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide;
1-[4-[4-[[4-[6-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]-2,6- diazaspiro[3.3]heptan-2-yl]-1-piperidyl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro- 1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-(4'-(((2S,4S)-4-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperazin-1-yl)-2-methylpiperidin-1-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-(4'-(((2S,4R)-4-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperazin-1-yl)-2-methylpiperidin-1-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-(4'-(((2R,4R)-4-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperazin-1-yl)-2-methylpiperidin-1-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-(4'-((4-(1-(2,6-dioxopiperidin-3-yl)-1H-indazol-5-yl)-[1,4'-bipiperidin]-1'-yl)methyl)-3- methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5- yl)methyl)-1H-pyrazole-4-carboxamide; 1-(4'-((4-(4-(3-(2,6-dioxopiperidin-3-yl)phenyl)piperazin-1-yl)piperidin-1-yl)methyl)-3- methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5- yl)methyl)-1H-pyrazole-4-carboxamide; 1-[4-[4-[[4-[4-[4-(2,6-dioxo-3-piperidyl)phenyl]piperazin-1-yl]-1- piperidyl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4- triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-[4-[4-[[4-[4-(2,6-dioxo-3-piperidyl)phenyl]piperazin-1-yl]methyl]phenyl]-2-methyl- phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4- carboxamide; 1-[4-[4-[[4-[4-[(2,6-dioxo-3-piperidyl)amino]-2-fluoro-phenyl]-1- piperidyl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4- triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-(2-chloro-4'-((4-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperazin-1-yl)piperidin-1-yl)methyl)-5-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide;
1-[3-[4-[[(3S)-3-[[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]amino]pyrrolidin-1-yl]methyl]phenyl]-2,4-dimethyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-[3-[6-[[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]-4- piperidyl]oxy]-3-pyridyl]-2,4-dimethyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H- 1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-[3-[6-[4-[[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]-4- piperidyl]methyl]piperazin-1-yl]-3-pyridyl]-2,4-dimethyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-(3-(6-(4-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1- yl)methyl)piperidin-1-yl)pyridin-3-yl)-2,4-dimethylphenyl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-[3-[4-[[4-[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]piperazin-1- yl]-1-piperidyl]methyl]phenyl]-2,4-dimethyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-[3-[4-[[4-[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]-4- piperidyl]piperazin-1-yl]methyl]phenyl]-2,4-dimethyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-[3-[2-[4-[[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]-4- piperidyl]methyl]piperazin-1-yl]-4-pyridyl]-2,4-dimethyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-(3-(2-(4-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1- yl)methyl)piperidin-1-yl)pyridin-4-yl)-2,4-dimethylphenyl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-[3-[6-[4-[[2-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]-2,6- diazaspiro[3.3]heptan-6-yl]methyl]-1-piperidyl]-3-pyridyl]-2,4-dimethyl-phenyl]-N-[[3-(2,2,2- trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-[3-[6-[6-[[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]-4- piperidyl]methyl]-2,6-diazaspiro[3.3]heptan-2-yl]-3-pyridyl]-2,4-dimethyl-phenyl]-N-[[3-(2,2,2- trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide;
1-[3-[2-[4-[[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]piperazin-1- yl]methyl]-1-piperidyl]pyrimidin-5-yl]-2,4-dimethyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl- ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; (2S)-5-chloro-6-[4-[[4-[4-(2,6-dioxo-3-piperidyl)phenyl]piperazin-1-yl]methyl]phenyl]-N- [[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl] -2,3-dihydrobenzofuran-2- carboxamide; (2R)-5-chloro-6-(4-((4-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperazin-1-yl)piperidin-1-yl)methyl)phenyl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H- 1,2,4-triazol-5-yl)methyl)-2,3-dihydrobenzofuran-2-carboxamide; (2R)-5-chloro-2-cyano-6-(6-(4-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3- dioxoisoindolin-5-yl)piperazin-1-yl)methyl)piperidin-1-yl)pyridin-3-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-2,3-dihydro-1H-indene-2-carboxamide; 5-chloro-2-cyano-6-(4-((4-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperazin-1-yl)piperidin-1-yl)methyl)phenyl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H- 1,2,4-triazol-5-yl)methyl)-2,3-dihydro-1H-indene-2-carboxamide; (2R)-5-chloro-2-cyano-6-[4-[[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin- 5-yl]piperazin-1-yl]methyl]phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methyl]indane-2-carboxamide; (1S,2S)-2-[4-[6-[[1-[1-[[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]- 4-piperidyl]methyl]-4-piperidyl]pyrazol-4-yl]amino]-3-pyridyl]phenyl]-N-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]cyclopropanecarboxamide; (1S,2S)-2-(4-(6-((1-(1-((1-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperidin-4-yl)methyl)piperidin-4-yl)-1H-pyrazol-4-yl)oxy)pyridin-3-yl)phenyl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)cyclopropane-1-carboxamide; 1-[4-[4-[[4-[5-[(2,6-dioxo-3-piperidyl)amino]-3-fluoro-2-pyridyl]-1-piperidyl]methyl]- phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methyl]pyrazole-4-carboxamide; 1-[4-[4-[[4-[4-[(3S)-2,6-dioxo-3-piperidyl]phenyl]piperazin-1-yl]methyl]phenyl]-2- methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole- 4-carboxamide;
1-[4-[4-[[4-[5-(2,6-dioxo-3-piperidyl)-3-fluoro-2-pyridyl]piperazin-1-yl]methyl]phenyl]- 2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methyl]pyrazole-4-carboxamide; 1-(4'-((4-(4-(2,6-dioxopiperidin-3-yl)-2-fluorophenyl)piperazin-1-yl)methyl)-3-methyl- [1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)- 1H-pyrazole-4-carboxamide; 1-(4'-((4-(5-((2,6-dioxopiperidin-3-yl)amino)pyrazin-2-yl)piperidin-1-yl)methyl)-3- methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5- yl)methyl)-1H-pyrazole-4-carboxamide; 1-[4-[4-[[4-[5-[(2,6-dioxo-3-piperidyl)amino]-3-fluoro-2-pyridyl]piperazin-1- yl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol- 5-yl]methyl]pyrazole-4-carboxamide; 1-[4-[4-[[4-[5-(2,6-dioxo-3-piperidyl)-3-fluoro-2-pyridyl]-1-piperidyl]methyl]phenyl]-2- methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole- 4-carboxamide; 1-[4-[4-[[4-[5-(2,6-dioxo-3-piperidyl)-4-fluoro-2-pyridyl]piperazin-1-yl]methyl]phenyl]- 2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methyl]pyrazole-4-carboxamide; 1-[5-[4-[[4-[4-[(2,6-dioxo-3-piperidyl)amino]-2-fluoro-phenyl]-1- piperidyl]methyl]phenyl]pyrimidin-2-yl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4- triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-[5-[4-[[4-[5-(2,6-dioxo-3-piperidyl)-3-fluoro-2-pyridyl]piperazin-1- yl]methyl]phenyl]pyrimidin-2-yl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methyl]pyrazole-4-carboxamide; 1-[5-[4-[[4-[5-[(2,6-dioxo-3-piperidyl)amino]-3-fluoro-2-pyridyl]piperazin-1- yl]methyl]phenyl]pyrimidin-2-yl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methyl]pyrazole-4-carboxamide; 1-[5-[4-[[4-[4-[(2,4-dioxohexahydropyrimidin-1-yl)methyl]-2-fluoro-phenyl]piperazin-1- yl]methyl]phenyl]pyrimidin-2-yl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methyl]pyrazole-4-carboxamide;
1-[5-[4-[[4-[5-[(2,6-dioxo-3-piperidyl)amino]-3-fluoro-2-pyridyl]piperazin-1- yl]methyl]phenyl]-2-pyridyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methyl]pyrazole-4-carboxamide; 1-(5-(5-((4-(5-((2,6-dioxopiperidin-3-yl)amino)-3-fluoropyridin-2-yl)piperazin-1- yl)methyl)pyridin-2-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-(5-(5-((4-(5-((2,6-dioxopiperidin-3-yl)amino)-3-fluoropyridin-2-yl)piperazin-1- yl)methyl)-3-fluoropyridin-2-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H- 1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-[5-[4-[[4-[4-[[(3S)-2,6-dioxo-3-piperidyl]carbamoyl]-3-fluoro-phenyl]piperazin-1- yl]methyl]phenyl]pyrimidin-2-yl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methyl]pyrazole-4-carboxamide; 1-[4-[4-[[4-[4-[[(3S)-2,6-dioxo-3-piperidyl]carbamoyl]-3-fluoro-phenyl]piperazin-1- yl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol- 5-yl]methyl]pyrazole-4-carboxamide hydrochloride; 1-[4-[4-[[4-[4-[[(3S)-2,6-dioxo-3-piperidyl]carbamoyl]-3-fluoro-phenyl]-1- piperidyl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4- triazol-5-yl]methyl]pyrazole-4-carboxamide; (S)-N-((3-(1,1-difluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1-(5-(4-((4- (4-((2,6-dioxopiperidin-3-yl)carbamoyl)-3-fluorophenyl)piperidin-1-yl)methyl)phenyl)pyridin-2- yl)-1H-pyrazole-4-carboxamide; 1-[5-[4-[[4-[4-[[(3S)-2,6-dioxo-3-piperidyl]carbamoyl]-3-fluoro-phenyl]-1-piperidyl]- methyl]phenyl]pyrimidin-2-yl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methyl]pyrazole-4-carboxamide; 1-[5-[4-[[4-[5-(2,6-dioxo-3-piperidyl)-3-fluoro-2-pyridyl]-1-piperidyl]methyl]phenyl]- pyrimidin-2-yl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole- 4-carboxamide; 1-(4-((4-(5-(2,6-dioxopiperidin-3-yl)-3-fluoropyridin-2-yl)piperazin-1-yl)methyl)-2- methylphenyl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H- pyrazole-4-carboxamide;
1-[4-[[1-[5-(2,6-dioxo-3-piperidyl)-3-fluoro-2-pyridyl]-4-piperidyl]methyl]-2-methyl- phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4- carboxamide; 1-[4-[[2-[5-(2,6-dioxo-3-piperidyl)-3-fluoro-2-pyridyl]-2-azaspiro[3.5]nonan-7-yl]oxy]-2- methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole- 4-carboxamide; and 3-[5-[4-[[4-[5-[(2,6-dioxo-3-piperidyl)amino]-3-fluoro-2-pyridyl]piperazin-1- yl]methyl]phenyl]pyrimidin-2-yl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methyl]isoxazole-5-carboxamide, or a pharmaceutically acceptable salt of any of the aforementioned. In some embodiments, the compound is selected from: 3-(5-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2,6- diazaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-1-methyl-1H-indazol-6- yl)piperidin-1-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan- 2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5-a]pyridin-6- yl)piperazin-1-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan- 2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5-a]pyrimidin-6- yl)piperazin-1-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan- 2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5-a]pyrazin-6- yl)piperazin-1-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan- 2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide;
3-(5-(4-((4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5-a]pyrazin-6- yl)piperidin-1-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan- 2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(2-fluoro-4-((6-(3-fluoro-4-(3-methyl-2,6-dioxopiperidin-3-yl)phenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)phenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2- yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5-a]pyridin-6- yl)piperidin-1-yl)methyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-(1-(4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5-a]pyridin-6- yl)piperidin-1-yl)ethyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-5-fluorobenzo[d]isoxazol-6- yl)piperidin-1-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan- 2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-(1-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)ethyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro- 2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; N-((3-(3,3-difluoro-2-methylbutan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-3-(5-(4-(1-(6-(4- (2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2-azaspiro[3.3]heptan-2-yl)ethyl)-2- fluorophenyl)-4-methylpyrimidin-2-yl)isoxazole-5-carboxamide; 3-(5-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro- 2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2-azaspiro- [3.3]heptan-2-yl)methyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(3-fluoro-2-methylbut- 3-en-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; N-((3-(1-(1,1-difluoroethyl)cyclopropyl)-1H-1,2,4-triazol-5-yl)methyl)-3-(5-(4-((6-(4- (2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2-azaspiro[3.3]heptan-2-yl)methyl)-2- fluorophenyl)-4-methylpyrimidin-2-yl)isoxazole-5-carboxamide;
3-(5-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1- (trifluoromethyl)cyclobutyl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-[5-[4-[1-[6-[4-(2,4-dioxohexahydropyrimidin-1-yl)-3-fluoro-phenyl]-2- azaspiro[3.3]heptan-2-yl]ethyl]-2-fluoro-phenyl]-4,6-dimethyl-pyrimidin-2-yl]-N-[[3-(2,2,2- trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]isoxazole-5-carboxamide; 3-[5-[5-[1-[6-[4- (2,4-dioxohexahydropyrimidin-1-yl)-3-fluoro-phenyl]-2- azaspiro[3.3]heptan-2-yl]ethyl]-3-fluoro-2-pyridyl]-4-methyl-pyrimidin-2-yl]-N-[[3-(2,2,2- trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]isoxazole-5-carboxamide; 3-(5-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)piperidin-1-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((4-(4-((2,4-dioxotetrahydropyrimidin-1(2H)-yl)methyl)-2,5- difluorophenyl)piperidin-1-yl)methyl)phenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((6-(2-chloro-3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)phenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)piperidin-1-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(2-(4-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)piperazin-1-yl)-7- azaspiro[3.5]nonan-7-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)isoxazole-5-carboxamide; 1-(4-((2-(5-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluoropyridin-2-yl)-2- azaspiro[3.5]nonan-7-yl)oxy)-2-(1-methyl-1H-pyrazol-4-yl)phenyl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-(4-((2-(5-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyridin-2-yl)-2-azaspiro[3.5]nonan-7- yl)oxy)-2-methylphenyl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5- yl)methyl)-1H-pyrazole-4-carboxamide; 1-[4-[[2-[5-(2,4-dioxohexahydropyrimidin-1-yl)-3-methyl-2-pyridyl]-2- azaspiro[3.5]nonan-7-yl]oxy]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H- 1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide;
(R)-1-(4-((4-(4-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-3- methylpiperazin-1-yl)cyclohexyl)oxy)-2-methylphenyl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2- yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; (S)-1-(4-((4-(4-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-3- methylpiperazin-1-yl)cyclohexyl)oxy)-2-methylphenyl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2- yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-(3-(2-(5-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-methylpyridin-2-yl)-2- azaspiro[3.5]nonan-7-yl)-5-fluoro-2-methylphenyl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)- 1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 3-(3-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2,6- diazaspiro[3.3]heptan-2-yl)methyl)phenyl)bicyclo[1.1.1]pentan-1-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-(1-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)ethyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(2- (methylthio)propan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(2-fluoro-4-((6-(3-fluoro-4-(3-methyl-2,6-dioxopiperidin-3-yl)phenyl)-2,6- diazaspiro[3.3]heptan-2-yl)methyl)phenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan- 2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((4-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3,5-difluorophenyl)piperazin-1- yl)methyl)piperidin-1-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(5-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)-3-fluoropyridin-2-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-2,3-difluorophenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-2,5-difluorophenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide;
3-(5-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-2-fluoro-5-methylphenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((6-(5-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluoro-6-methylpyridin-2-yl)-2- azaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(5-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3,5-difluorophenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)pyridin-2-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(5-((4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5-a]pyrazin-6- yl)piperazin-1-yl)methyl)-3-fluoropyridin-2-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(5-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)pyrazin-2-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; (R)-3-(5-(4-((4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5-a]pyrazin-6-yl)- 3-methylpiperazin-1-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5-a]pyridin-6- yl)piperazin-1-yl)methyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5-a]pyrazin-6- yl)piperidin-1-yl)methyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-(1-(4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5-a]pyrazin-6- yl)piperazin-1-yl)ethyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2- yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5-a]pyrazin-6- yl)piperazin-1-yl)methyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide;
3-(5-(4-(1-(4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5-a]pyrimidin-6- yl)piperazin-1-yl)ethyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(5-(1-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3,5-difluorophenyl)-2- azaspiro[3.3]heptan-2-yl)ethyl)-3-fluoropyridin-2-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(3-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)-2,3-dihydrobenzofuran-6-yl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(5-(1-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)ethyl)pyridin-2-yl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; N-((3-(3,3-difluoro-2-methylbutan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-3-(5-(4-(1-(6-(4- (2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2-azaspiro[3.3]heptan-2-yl)propyl)-2- fluorophenyl)-4-methylpyrimidin-2-yl)isoxazole-5-carboxamide; 3-(5-(1-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)-2,3-dihydro-1H-inden-5-yl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-(1-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)propyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro- 2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(5-((2-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-6- azaspiro[3.4]octan-6-yl)methyl)-3-fluoropyridin-2-yl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(1-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)ethyl)-4'-methyl-[2,5'-bipyrimidin]-2'-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; N-((3-(3,3-difluoro-2-methylbutan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-3-(5-(1-(6-(4-(2,4- dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2-azaspiro[3.3]heptan-2-yl)ethyl)-4'-methyl- [2,5'-bipyrimidin]-2'-yl)isoxazole-5-carboxamide;
3-(5-(4-(1-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)butyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro- 2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-(1-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)-2-methylpropyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3- (1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-(1-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)-2-methoxyethyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3- (1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(7-(4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5-a]pyrazin-6- yl)piperazin-1-yl)-5-oxa-2-azaspiro[3.4]octan-2-yl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; N-((3-(tert-butyl)-1H-1,2,4-triazol-5-yl)methyl)-3-(5-(4-((6-(4-(2,4- dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2-azaspiro[3.3]heptan-2-yl)methyl)-2- fluorophenyl)-4-methylpyrimidin-2-yl)isoxazole-5-carboxamide; N-((3-(3,3-difluoro-2-methylbutan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-3-(5-(5-(1-(6-(4- (2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2-azaspiro[3.3]heptan-2-yl)ethyl)-3- fluoropyridin-2-yl)-4-methylpyrimidin-2-yl)isoxazole-5-carboxamide; 3-(5-(5-(1-(6-(2-chloro-3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)phenyl)-2- azaspiro[3.3]heptan-2-yl)ethyl)-3-fluoropyridin-2-yl)-4-methylpyrimidin-2-yl)-N-((3-(3,3- difluoro-2-methylbutan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(1-(6-(2-chloro-3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)phenyl)-2- azaspiro[3.3]heptan-2-yl)ethyl)-4'-methyl-[2,5'-bipyrimidin]-2'-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 1-(4-((4-(4-(5-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-4-methylpyridin-2-yl)piperazin-1- yl)cyclohexyl)oxy)-2-methylphenyl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 3-(5-(5-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)pyridin-2-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide;
3-(5-(4-(1-(6-(2-chloro-3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)phenyl)-2- azaspiro[3.3]heptan-2-yl)ethyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro- 2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((6-(2-chloro-3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)phenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-(1-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)ethyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(5-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3,5-difluorophenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)-3-fluoropyridin-2-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((((3-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3,5- difluorophenyl)cyclobutyl)methyl)amino)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-(((3-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3,5- difluorophenyl)cyclobutyl)amino)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro- 2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3,5-difluorophenyl)-2,6- diazaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((4-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3,5-difluorophenyl)piperazin-1- yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((4-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3,5-difluorophenoxy)piperidin-1- yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((3-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3,5-difluorophenoxy)azetidin-1- yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)isoxazole-5-carboxamide;
3-(5-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3,5-difluorophenoxy)-2- azaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro- 2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(3-(4-((4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5-a]pyrimidin-6- yl)piperazin-1-yl)methyl)phenyl)bicyclo[1.1.1]pentan-1-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(3-(4-((4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5-a]pyrazin-6- yl)piperazin-1-yl)methyl)phenyl)bicyclo[1.1.1]pentan-1-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)phenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2- yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-2-methylphenyl)piperidin-1- yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-2-methylphenyl)piperidin-1- yl)methyl)piperidin-1-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((4-(2-chloro-3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)phenyl)piperidin-1- yl)methyl)piperidin-1-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-(((3aS*,6aS*)-4-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3- fluorophenyl)hexahydropyrrolo[3,2-b]pyrrol-1(2H)-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)- N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5- carboxamide; 3-(5-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3,5-difluorophenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro- 2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(5-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)-3-fluoropyridin-2-yl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide;
3-(5-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)-[2,5'-bipyrimidin]-2'-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan- 2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2,6- diazaspiro[3.3]heptan-2-yl)methyl)phenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan- 2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; or a pharmaceutically acceptable salt of any of the aforementioned. Intermediates Also provided herein is a compound of Formula (A1):
IG, or a pharmaceutically acceptable salt thereof; wherein each R42 and R43 is independently selected from halo, C1-4 alkyl, and C1-4 haloalkyl; X1 and X2 are each independently selected from CH and N; and q and r are each independently 0, 1, or 2. In some embodiments, the compound is selected from: (2R)-5-chloro-6-(4-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperazin-1-yl)methyl)phenyl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5- yl)methyl)-2,3-dihydrobenzofuran-2-carboxamide;
(2R)-5-chloro-6-(4-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperazine-1-carbonyl)phenyl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5- yl)methyl)-2,3-dihydrobenzofuran-2-carboxamide; (2R)-5-chloro-6-(6-(4-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperazin-1-yl)methyl)piperidin-1-yl)pyridin-3-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2- yl)-1H-1,2,4-triazol-5-yl)methyl)-2,3-dihydrobenzofuran-2-carboxamide; (1S,2S)-2-(4'-(1-(2-(4-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperazin-1-yl)methyl)piperidin-1-yl)-2-oxoethyl)-1H-pyrazol-4-yl)-[1,1'-biphenyl]-4-yl)-N- ((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)cyclopropane-1- carboxamide; (1S,2S)-2-[4-[4-[1-[2-[4-[[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]piperazin-1-yl]methyl]-1-piperidyl]-2-oxo-ethyl]pyrazol-3-yl]phenyl]phenyl]-N-[[3-(2,2,2- trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]cyclopropanecarboxamide hydrochloride; 5-(4-((1-(2-(4-(3-chloro-4'-((1S,2S)-2-(3-(3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H- 1,2,4-triazol-5-yl)propanoyl)cyclopropyl)-[1,1'-biphenyl]-4-yl)-1H-pyrazol-1-yl)acetyl)piperidin- 4-yl)methyl)piperazin-1-yl)-2-(2,6-dioxopiperidin-3-yl)-6-fluoroisoindoline-1,3-dione; (1S,2S)-2-(2'-chloro-4'-(1-(2-(4-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3- dioxoisoindolin-5-yl)piperazin-1-yl)methyl)piperidin-1-yl)-2-oxoethyl)-1H-pyrazol-4-yl)-[1,1'- biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5- yl)methyl)cyclopropane-1-carboxamide; (1S,2S)-2-[4-[4-[1-[2-[3-[[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]piperazin-1-yl]methyl]azetidin-1-yl]-2-oxo-ethyl]pyrazol-4-yl]phenyl]phenyl]-N-[[3-(2,2,2- trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]cyclopropanecarboxamide; 3-chloro-N-(3-(2-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperazin-1-yl)ethoxy)propyl)-4'-((1S,2S)-2-(((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H- 1,2,4-triazol-5-yl)methyl)carbamoyl)cyclopropyl)-[1,1'-biphenyl]-4-carboxamide; 3-chloro-N-(2-(4-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperazin-1-yl)methyl)piperidin-1-yl)-2-oxoethyl)-4'-((1S,2S)-2-(((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)carbamoyl)cyclopropyl)-[1,1'-biphenyl]-4- carboxamide;
2-chloro-N-[1-[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]-4- piperidyl]-4-piperidyl]-4-[4-[(1S,2S)-2-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol- 5-yl]methylcarbamoyl]cyclopropyl]phenyl]benzamide; (1S,2S)-2-[4-[1-[2-[4-[[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]piperazin-1-yl]methyl]-1-piperidyl]-2-oxo-ethyl]-3-methyl-indazol-5-yl]phenyl]-N-[[3-(2,2,2- trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]cyclopropanecarboxamide; (1S,2S)-2-[4-[4-[1-[1-[2-[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]piperazin-1-yl]-2-oxo-ethyl]azetidin-3-yl]pyrazol-4-yl]phenyl]phenyl]-N-[[3-(2,2,2-trifluoro- 1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]cyclopropanecarboxamide; (1S,2S)-2-[4-[4-[1-[[1-[2-[4- [2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl] piperazin-1-yl]-2-oxo-ethyl] -4-piperidyl]methyl]pyrazol-4-yl]phenyl]phenyl]-N-[[3-(2,2,2- trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]cyclopropanecarboxamide; (1S,2S)-2-[4-[4-[1-[2-[(3S)-3-[[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo- isoindolin-5-yl]-4-piperidyl] oxy]pyrrolidin-1-yl]-2-oxo-ethyl]pyrazol-4-yl]phenyl]phenyl]-N-[[3- (2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]cyclopropanecarboxamide; (1S,2S)-2-[4-[1-[4-[4-[[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]piperazin-1-yl]methyl]piperidine-1-carbonyl]phenyl]pyrazol-4-yl]phenyl]-N-[[3-(2,2,2- trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]cyclopropanecarboxamide; (1S,2S)-2-(4'-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperazin-1-yl)methyl)-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H- 1,2,4-triazol-5-yl)methyl)cyclopropane-1-carboxamide; (1S,2S)-2-(4'-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperazin-1-yl)methyl)-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H- 1,2,4-triazol-5-yl)methyl)cyclopropane-1-carboxamide; 1-[3-[4-[[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]piperazin-1- yl]methyl]phenyl]-2,4-dimethyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4- triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-[4-[4-[[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]piperazin-1- yl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol- 5-yl]methyl]pyrazole-4-carboxamide;
1-[4-[3-[[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]piperazin-1- yl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol- 5-yl]methyl]pyrazole-4-carboxamide; 1-[4-[4-[[4-[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]piperazin-1- yl]-1-piperidyl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H- 1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-[4-[4-[[4-[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]-4- piperidyl]piperazin-1-yl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl- ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-(4'-((4-((1-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperidin-4- yl)methyl)piperazin-1-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-(4'-((4-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1- yl)methyl)piperidin-1-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-(4'-((4-((1-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperidin-4- yl)oxy)piperidin-1-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-(4'-((4-(1-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperidin-4- yl)-1,4-diazepan-1-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-(4'-((1'-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)-[4,4'- bipiperidin]-1-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2- yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-(4'-((3-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1- yl)pyrrolidin-1-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan- 2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-(4-(6-((4-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1- yl)piperidin-1-yl)methyl)pyridin-3-yl)-2-methylphenyl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2- yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide;
1-(2'-chloro-4'-((4-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperazin-1-yl)piperidin-1-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-(3'-((4-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1- yl)piperidin-1-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2- yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-(4'-(1-(4-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1- yl)piperidin-1-yl)ethyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2- yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-[4-[4-[[4-[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]piperazin-1- yl]-1-piperidyl]methyl]-3,5-difluoro-phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-(4-(1-(1'-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)-[1,4'- bipiperidin]-4-yl)-3-methyl-1H-indazol-5-yl)-2-methylphenyl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-[4-[6-[[1-[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]-4-piperidyl]- 4-piperidyl]oxy]-3-pyridyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H- 1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-[4-[4-[2-[4-[[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]piperazin- 1-yl]methyl]-1-piperidyl]ethoxy]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl- ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-(3'-(2-(4-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1- yl)methyl)piperidin-1-yl)ethoxy)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-[4-[4-[1-[4-[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]piperazin- 1-yl]-1-piperidyl]-1-methyl-ethyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl- ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-[4-[5-[[4-[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]piperazin-1- yl]-1-piperidyl]methyl]pyrazin-2-yl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide;
1-[4-[5-[[4-[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]piperazin-1- yl]-1-piperidyl]methyl]-2-pyridyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-(3'-chloro-4'-((4-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperazin-1-yl)piperidin-1-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-[4-[4-[[5-[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl] piperazin-1- yl]-2-pyridyl]oxy]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4- triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-[4-[4-[[4-[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]piperazin-1- yl]-2-oxo-1-piperidyl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl- ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-[4-[6-[4-[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]piperazin-1- yl]cyclohexoxy]-3-pyridyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H- 1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-[4-[1-[[1-[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]-4- piperidyl]-4-piperidyl]methyl]pyrazol-4-yl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-[4-[6-[[1-[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]-4-piperidyl]- 4-piperidyl]amino]-3-pyridyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H- 1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-[4-[4-[[2-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]-2,7- diazaspiro[3.5]nonan-7-yl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl- ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-(4'-((4-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1- yl)azepan-1-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2- yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-(4'-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)-1,4-diazepan-1- yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide;
1-[4-[4-[[8-[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]-4-piperidyl]- 3,8-diazabicyclo[3.2.1]octan-3-yl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-(4'-((3-(1-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperidin-4- yl)-3,6-diazabicyclo[3.1.1]heptan-6-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-(4'-(((1S,4S)-5-(1-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperidin-4-yl)-2,5-diazabicyclo[2.2.1]heptan-2-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N- ((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4- carboxamide; 1-(4'-(((1R,4R)-5-(1-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperidin-4-yl)-2,5-diazabicyclo[2.2.1]heptan-2-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N- ((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4- carboxamide; 1-(4'-((3-(1-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperidin-4- yl)-3,8-diazabicyclo[3.2.1]octan-8-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-(4'-((3-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1- yl)azetidin-1-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2- yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-[4-[4-[[2-[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]-4-piperidyl]- 2,6-diazaspiro[3.3]heptan-6-yl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-[4-[4-[[4-[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]piperazin-1- yl]-3,3-difluoro-1-piperidyl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-[4-[4-[[4-[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]piperazin-1- yl]-4-methyl-1-piperidyl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl- ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide;
1-[4-[4-[[4-[6-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]-2,6- diazaspiro[3.3]heptan-2-yl]-1-piperidyl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro- 1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-(4'-(((2S,4S)-4-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperazin-1-yl)-2-methylpiperidin-1-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-(4'-(((2S,4R)-4-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperazin-1-yl)-2-methylpiperidin-1-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-(4'-(((2R,4R)-4-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperazin-1-yl)-2-methylpiperidin-1-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-(4'-((4-(1-(2,6-dioxopiperidin-3-yl)-1H-indazol-5-yl)-[1,4'-bipiperidin]-1'-yl)methyl)-3- methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5- yl)methyl)-1H-pyrazole-4-carboxamide; 1-(4'-((4-(4-(3-(2,6-dioxopiperidin-3-yl)phenyl)piperazin-1-yl)piperidin-1-yl)methyl)-3- methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5- yl)methyl)-1H-pyrazole-4-carboxamide; 1-[4-[4-[[4-[4-[4-(2,6-dioxo-3-piperidyl)phenyl]piperazin-1-yl]-1- piperidyl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4- triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-[4-[4-[[4-[4-(2,6-dioxo-3-piperidyl)phenyl]piperazin-1-yl]methyl]phenyl]-2-methyl- phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4- carboxamide; 1-[4-[4-[[4-[4-[(2,6-dioxo-3-piperidyl)amino]-2-fluoro-phenyl]-1- piperidyl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4- triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-(2-chloro-4'-((4-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperazin-1-yl)piperidin-1-yl)methyl)-5-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide;
1-[3-[4-[[(3S)-3-[[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]amino]pyrrolidin-1-yl]methyl]phenyl]-2,4-dimethyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-[3-[6-[[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]-4- piperidyl]oxy]-3-pyridyl]-2,4-dimethyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H- 1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-[3-[6-[4-[[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]-4- piperidyl]methyl]piperazin-1-yl]-3-pyridyl]-2,4-dimethyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-(3-(6-(4-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1- yl)methyl)piperidin-1-yl)pyridin-3-yl)-2,4-dimethylphenyl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-[3-[4-[[4-[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]piperazin-1- yl]-1-piperidyl]methyl]phenyl]-2,4-dimethyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-[3-[4-[[4-[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]-4- piperidyl]piperazin-1-yl]methyl]phenyl]-2,4-dimethyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-[3-[2-[4-[[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]-4- piperidyl]methyl]piperazin-1-yl]-4-pyridyl]-2,4-dimethyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-(3-(2-(4-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1- yl)methyl)piperidin-1-yl)pyridin-4-yl)-2,4-dimethylphenyl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-[3-[6-[4-[[2-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]-2,6- diazaspiro[3.3]heptan-6-yl]methyl]-1-piperidyl]-3-pyridyl]-2,4-dimethyl-phenyl]-N-[[3-(2,2,2- trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-[3-[6-[6-[[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]-4- piperidyl]methyl]-2,6-diazaspiro[3.3]heptan-2-yl]-3-pyridyl]-2,4-dimethyl-phenyl]-N-[[3-(2,2,2- trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide;
1-[3-[2-[4-[[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]piperazin-1- yl]methyl]-1-piperidyl]pyrimidin-5-yl]-2,4-dimethyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl- ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; (2S)-5-chloro-6-[4-[[4-[4-(2,6-dioxo-3-piperidyl)phenyl]piperazin-1-yl]methyl]phenyl]-N- [[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl] -2,3-dihydrobenzofuran-2- carboxamide; (2R)-5-chloro-6-(4-((4-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperazin-1-yl)piperidin-1-yl)methyl)phenyl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H- 1,2,4-triazol-5-yl)methyl)-2,3-dihydrobenzofuran-2-carboxamide; (2R)-5-chloro-2-cyano-6-(6-(4-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3- dioxoisoindolin-5-yl)piperazin-1-yl)methyl)piperidin-1-yl)pyridin-3-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-2,3-dihydro-1H-indene-2-carboxamide; 5-chloro-2-cyano-6-(4-((4-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperazin-1-yl)piperidin-1-yl)methyl)phenyl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H- 1,2,4-triazol-5-yl)methyl)-2,3-dihydro-1H-indene-2-carboxamide; (2R)-5-chloro-2-cyano-6-[4-[[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin- 5-yl]piperazin-1-yl]methyl]phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methyl]indane-2-carboxamide; (1S,2S)-2-[4-[6-[[1-[1-[[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]- 4-piperidyl]methyl]-4-piperidyl]pyrazol-4-yl]amino]-3-pyridyl]phenyl]-N-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]cyclopropanecarboxamide; (1S,2S)-2-(4-(6-((1-(1-((1-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperidin-4-yl)methyl)piperidin-4-yl)-1H-pyrazol-4-yl)oxy)pyridin-3-yl)phenyl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)cyclopropane-1-carboxamide; 1-[4-[4-[[4-[5-[(2,6-dioxo-3-piperidyl)amino]-3-fluoro-2-pyridyl]-1-piperidyl]methyl]- phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methyl]pyrazole-4-carboxamide; 1-[4-[4-[[4-[4-[(3S)-2,6-dioxo-3-piperidyl]phenyl]piperazin-1-yl]methyl]phenyl]-2- methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole- 4-carboxamide;
1-[4-[4-[[4-[5-(2,6-dioxo-3-piperidyl)-3-fluoro-2-pyridyl]piperazin-1-yl]methyl]phenyl]- 2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methyl]pyrazole-4-carboxamide; 1-(4'-((4-(4-(2,6-dioxopiperidin-3-yl)-2-fluorophenyl)piperazin-1-yl)methyl)-3-methyl- [1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)- 1H-pyrazole-4-carboxamide; 1-(4'-((4-(5-((2,6-dioxopiperidin-3-yl)amino)pyrazin-2-yl)piperidin-1-yl)methyl)-3- methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5- yl)methyl)-1H-pyrazole-4-carboxamide; 1-[4-[4-[[4-[5-[(2,6-dioxo-3-piperidyl)amino]-3-fluoro-2-pyridyl]piperazin-1- yl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol- 5-yl]methyl]pyrazole-4-carboxamide; 1-[4-[4-[[4-[5-(2,6-dioxo-3-piperidyl)-3-fluoro-2-pyridyl]-1-piperidyl]methyl]phenyl]-2- methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole- 4-carboxamide; 1-[4-[4-[[4-[5-(2,6-dioxo-3-piperidyl)-4-fluoro-2-pyridyl]piperazin-1-yl]methyl]phenyl]- 2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methyl]pyrazole-4-carboxamide; 1-[5-[4-[[4-[4-[(2,6-dioxo-3-piperidyl)amino]-2-fluoro-phenyl]-1- piperidyl]methyl]phenyl]pyrimidin-2-yl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4- triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-[5-[4-[[4-[5-(2,6-dioxo-3-piperidyl)-3-fluoro-2-pyridyl]piperazin-1- yl]methyl]phenyl]pyrimidin-2-yl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methyl]pyrazole-4-carboxamide; 1-[5-[4-[[4-[5-[(2,6-dioxo-3-piperidyl)amino]-3-fluoro-2-pyridyl]piperazin-1- yl]methyl]phenyl]pyrimidin-2-yl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methyl]pyrazole-4-carboxamide; 1-[5-[4-[[4-[4-[(2,4-dioxohexahydropyrimidin-1-yl)methyl]-2-fluoro-phenyl]piperazin-1- yl]methyl]phenyl]pyrimidin-2-yl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methyl]pyrazole-4-carboxamide;
1-[5-[4-[[4-[5-[(2,6-dioxo-3-piperidyl)amino]-3-fluoro-2-pyridyl]piperazin-1- yl]methyl]phenyl]-2-pyridyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methyl]pyrazole-4-carboxamide; 1-(5-(5-((4-(5-((2,6-dioxopiperidin-3-yl)amino)-3-fluoropyridin-2-yl)piperazin-1- yl)methyl)pyridin-2-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-(5-(5-((4-(5-((2,6-dioxopiperidin-3-yl)amino)-3-fluoropyridin-2-yl)piperazin-1- yl)methyl)-3-fluoropyridin-2-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H- 1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-[5-[4-[[4-[4-[[(3S)-2,6-dioxo-3-piperidyl]carbamoyl]-3-fluoro-phenyl]piperazin-1- yl]methyl]phenyl]pyrimidin-2-yl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methyl]pyrazole-4-carboxamide; 1-[4-[4-[[4-[4-[[(3S)-2,6-dioxo-3-piperidyl]carbamoyl]-3-fluoro-phenyl]piperazin-1- yl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol- 5-yl]methyl]pyrazole-4-carboxamide hydrochloride; 1-[4-[4-[[4-[4-[[(3S)-2,6-dioxo-3-piperidyl]carbamoyl]-3-fluoro-phenyl]-1- piperidyl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4- triazol-5-yl]methyl]pyrazole-4-carboxamide; (S)-N-((3-(1,1-difluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1-(5-(4-((4- (4-((2,6-dioxopiperidin-3-yl)carbamoyl)-3-fluorophenyl)piperidin-1-yl)methyl)phenyl)pyridin-2- yl)-1H-pyrazole-4-carboxamide; 1-[5-[4-[[4-[4-[[(3S)-2,6-dioxo-3-piperidyl]carbamoyl]-3-fluoro-phenyl]-1-piperidyl]- methyl]phenyl]pyrimidin-2-yl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methyl]pyrazole-4-carboxamide; 1-[5-[4-[[4-[5-(2,6-dioxo-3-piperidyl)-3-fluoro-2-pyridyl]-1-piperidyl]methyl]phenyl]- pyrimidin-2-yl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole- 4-carboxamide; 1-(4-((4-(5-(2,6-dioxopiperidin-3-yl)-3-fluoropyridin-2-yl)piperazin-1-yl)methyl)-2- methylphenyl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H- pyrazole-4-carboxamide;
1-[4-[[1-[5-(2,6-dioxo-3-piperidyl)-3-fluoro-2-pyridyl]-4-piperidyl]methyl]-2-methyl- phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4- carboxamide; 1-[4-[[2-[5-(2,6-dioxo-3-piperidyl)-3-fluoro-2-pyridyl]-2-azaspiro[3.5]nonan-7-yl]oxy]-2- methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole- 4-carboxamide; and 3-[5-[4-[[4-[5-[(2,6-dioxo-3-piperidyl)amino]-3-fluoro-2-pyridyl]piperazin-1- yl]methyl]phenyl]pyrimidin-2-yl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methyl]isoxazole-5-carboxamide, or a pharmaceutically acceptable salt of any of the aforementioned. In some embodiments, the compound is selected from: 3-(5-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2,6- diazaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-1-methyl-1H-indazol-6- yl)piperidin-1-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan- 2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5-a]pyridin-6- yl)piperazin-1-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan- 2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5-a]pyrimidin-6- yl)piperazin-1-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan- 2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5-a]pyrazin-6- yl)piperazin-1-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan- 2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide;
3-(5-(4-((4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5-a]pyrazin-6- yl)piperidin-1-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan- 2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(2-fluoro-4-((6-(3-fluoro-4-(3-methyl-2,6-dioxopiperidin-3-yl)phenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)phenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2- yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5-a]pyridin-6- yl)piperidin-1-yl)methyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-(1-(4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5-a]pyridin-6- yl)piperidin-1-yl)ethyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-5-fluorobenzo[d]isoxazol-6- yl)piperidin-1-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan- 2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-(1-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)ethyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro- 2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; N-((3-(3,3-difluoro-2-methylbutan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-3-(5-(4-(1-(6-(4- (2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2-azaspiro[3.3]heptan-2-yl)ethyl)-2- fluorophenyl)-4-methylpyrimidin-2-yl)isoxazole-5-carboxamide; 3-(5-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro- 2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2-azaspiro- [3.3]heptan-2-yl)methyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(3-fluoro-2-methylbut- 3-en-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; N-((3-(1-(1,1-difluoroethyl)cyclopropyl)-1H-1,2,4-triazol-5-yl)methyl)-3-(5-(4-((6-(4- (2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2-azaspiro[3.3]heptan-2-yl)methyl)-2- fluorophenyl)-4-methylpyrimidin-2-yl)isoxazole-5-carboxamide;
3-(5-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1- (trifluoromethyl)cyclobutyl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-[5-[4-[1-[6-[4-(2,4-dioxohexahydropyrimidin-1-yl)-3-fluoro-phenyl]-2- azaspiro[3.3]heptan-2-yl]ethyl]-2-fluoro-phenyl]-4,6-dimethyl-pyrimidin-2-yl]-N-[[3-(2,2,2- trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]isoxazole-5-carboxamide; 3-[5-[5-[1-[6-[4- (2,4-dioxohexahydropyrimidin-1-yl)-3-fluoro-phenyl]-2- azaspiro[3.3]heptan-2-yl]ethyl]-3-fluoro-2-pyridyl]-4-methyl-pyrimidin-2-yl]-N-[[3-(2,2,2- trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]isoxazole-5-carboxamide; 3-(5-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)piperidin-1-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((4-(4-((2,4-dioxotetrahydropyrimidin-1(2H)-yl)methyl)-2,5- difluorophenyl)piperidin-1-yl)methyl)phenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((6-(2-chloro-3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)phenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)piperidin-1-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(2-(4-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)piperazin-1-yl)-7- azaspiro[3.5]nonan-7-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)isoxazole-5-carboxamide; 1-(4-((2-(5-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluoropyridin-2-yl)-2- azaspiro[3.5]nonan-7-yl)oxy)-2-(1-methyl-1H-pyrazol-4-yl)phenyl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-(4-((2-(5-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyridin-2-yl)-2-azaspiro[3.5]nonan-7- yl)oxy)-2-methylphenyl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5- yl)methyl)-1H-pyrazole-4-carboxamide; 1-[4-[[2-[5-(2,4-dioxohexahydropyrimidin-1-yl)-3-methyl-2-pyridyl]-2- azaspiro[3.5]nonan-7-yl]oxy]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H- 1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide;
(R)-1-(4-((4-(4-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-3- methylpiperazin-1-yl)cyclohexyl)oxy)-2-methylphenyl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2- yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; (S)-1-(4-((4-(4-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-3- methylpiperazin-1-yl)cyclohexyl)oxy)-2-methylphenyl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2- yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-(3-(2-(5-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-methylpyridin-2-yl)-2- azaspiro[3.5]nonan-7-yl)-5-fluoro-2-methylphenyl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)- 1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 3-(3-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2,6- diazaspiro[3.3]heptan-2-yl)methyl)phenyl)bicyclo[1.1.1]pentan-1-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-(1-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)ethyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(2- (methylthio)propan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(2-fluoro-4-((6-(3-fluoro-4-(3-methyl-2,6-dioxopiperidin-3-yl)phenyl)-2,6- diazaspiro[3.3]heptan-2-yl)methyl)phenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan- 2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((4-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3,5-difluorophenyl)piperazin-1- yl)methyl)piperidin-1-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(5-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)-3-fluoropyridin-2-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-2,3-difluorophenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-2,5-difluorophenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide;
3-(5-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-2-fluoro-5-methylphenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((6-(5-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluoro-6-methylpyridin-2-yl)-2- azaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(5-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3,5-difluorophenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)pyridin-2-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(5-((4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5-a]pyrazin-6- yl)piperazin-1-yl)methyl)-3-fluoropyridin-2-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(5-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)pyrazin-2-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; (R)-3-(5-(4-((4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5-a]pyrazin-6-yl)- 3-methylpiperazin-1-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5-a]pyridin-6- yl)piperazin-1-yl)methyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5-a]pyrazin-6- yl)piperidin-1-yl)methyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-(1-(4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5-a]pyrazin-6- yl)piperazin-1-yl)ethyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2- yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5-a]pyrazin-6- yl)piperazin-1-yl)methyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide;
3-(5-(4-(1-(4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5-a]pyrimidin-6- yl)piperazin-1-yl)ethyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(5-(1-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3,5-difluorophenyl)-2- azaspiro[3.3]heptan-2-yl)ethyl)-3-fluoropyridin-2-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(3-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)-2,3-dihydrobenzofuran-6-yl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(5-(1-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)ethyl)pyridin-2-yl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; N-((3-(3,3-difluoro-2-methylbutan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-3-(5-(4-(1-(6-(4- (2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2-azaspiro[3.3]heptan-2-yl)propyl)-2- fluorophenyl)-4-methylpyrimidin-2-yl)isoxazole-5-carboxamide; 3-(5-(1-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)-2,3-dihydro-1H-inden-5-yl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-(1-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)propyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro- 2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(5-((2-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-6- azaspiro[3.4]octan-6-yl)methyl)-3-fluoropyridin-2-yl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(1-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)ethyl)-4'-methyl-[2,5'-bipyrimidin]-2'-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; N-((3-(3,3-difluoro-2-methylbutan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-3-(5-(1-(6-(4-(2,4- dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2-azaspiro[3.3]heptan-2-yl)ethyl)-4'-methyl- [2,5'-bipyrimidin]-2'-yl)isoxazole-5-carboxamide;
3-(5-(4-(1-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)butyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro- 2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-(1-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)-2-methylpropyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3- (1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-(1-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)-2-methoxyethyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3- (1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(7-(4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5-a]pyrazin-6- yl)piperazin-1-yl)-5-oxa-2-azaspiro[3.4]octan-2-yl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; N-((3-(tert-butyl)-1H-1,2,4-triazol-5-yl)methyl)-3-(5-(4-((6-(4-(2,4- dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2-azaspiro[3.3]heptan-2-yl)methyl)-2- fluorophenyl)-4-methylpyrimidin-2-yl)isoxazole-5-carboxamide; N-((3-(3,3-difluoro-2-methylbutan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-3-(5-(5-(1-(6-(4- (2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2-azaspiro[3.3]heptan-2-yl)ethyl)-3- fluoropyridin-2-yl)-4-methylpyrimidin-2-yl)isoxazole-5-carboxamide; 3-(5-(5-(1-(6-(2-chloro-3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)phenyl)-2- azaspiro[3.3]heptan-2-yl)ethyl)-3-fluoropyridin-2-yl)-4-methylpyrimidin-2-yl)-N-((3-(3,3- difluoro-2-methylbutan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(1-(6-(2-chloro-3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)phenyl)-2- azaspiro[3.3]heptan-2-yl)ethyl)-4'-methyl-[2,5'-bipyrimidin]-2'-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 1-(4-((4-(4-(5-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-4-methylpyridin-2-yl)piperazin-1- yl)cyclohexyl)oxy)-2-methylphenyl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 3-(5-(5-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)pyridin-2-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide;
3-(5-(4-(1-(6-(2-chloro-3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)phenyl)-2- azaspiro[3.3]heptan-2-yl)ethyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro- 2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((6-(2-chloro-3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)phenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-(1-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)ethyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(5-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3,5-difluorophenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)-3-fluoropyridin-2-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((((3-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3,5- difluorophenyl)cyclobutyl)methyl)amino)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-(((3-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3,5- difluorophenyl)cyclobutyl)amino)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro- 2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3,5-difluorophenyl)-2,6- diazaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((4-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3,5-difluorophenyl)piperazin-1- yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((4-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3,5-difluorophenoxy)piperidin-1- yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((3-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3,5-difluorophenoxy)azetidin-1- yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)isoxazole-5-carboxamide;
3-(5-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3,5-difluorophenoxy)-2- azaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro- 2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(3-(4-((4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5-a]pyrimidin-6- yl)piperazin-1-yl)methyl)phenyl)bicyclo[1.1.1]pentan-1-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(3-(4-((4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5-a]pyrazin-6- yl)piperazin-1-yl)methyl)phenyl)bicyclo[1.1.1]pentan-1-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)phenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2- yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-2-methylphenyl)piperidin-1- yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-2-methylphenyl)piperidin-1- yl)methyl)piperidin-1-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((4-(2-chloro-3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)phenyl)piperidin-1- yl)methyl)piperidin-1-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-(((3aS*,6aS*)-4-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3- fluorophenyl)hexahydropyrrolo[3,2-b]pyrrol-1(2H)-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)- N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5- carboxamide; 3-(5-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3,5-difluorophenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro- 2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(5-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)-3-fluoropyridin-2-yl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide;
3-(5-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)-[2,5'-bipyrimidin]-2'-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan- 2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2,6- diazaspiro[3.3]heptan-2-yl)methyl)phenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan- 2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; or a pharmaceutically acceptable salt of any of the aforementioned. Intermediates Also provided herein is a compound of Formula (A1):
 or a pharmaceutically acceptable salt thereof, wherein constituent members are defined herein. Compounds of Formula (A1) can be useful intermediates in the preparation of compounds of Formula (I). Moreover, compounds of Formula (A1) can be useful as binders of androgen receptor. In some embodiments, provided herein is a compound of Formula (A1), or a pharmaceutically acceptable salt thereof, wherein: R1, R2, and R3 are each independently selected from H, halo, C1-4 alkyl, and C1-4 haloalkyl; R4 and R5 are each independently selected from H, halo, ORA, C1-4 alkyl, and C1-4 haloalkyl, wherein RA is H, C1-4 alkyl, or C1-4 haloalkyl; or R4 and R5 together with the carbon atom to which they are attached form a C3-6 cycloalkyl group or a 3-6 membered heterocycloalkyl group, each optionally substituted with 1, 2, or 3 substituents independently selected from H, halo, ORA, C1-4 alkyl, and C1-4 haloalkyl; R6 and R7 are each independently selected from H, halo, C1-4 alkyl, and C1-4 haloalkyl; R8 is H or C1-4 alkyl;
Cy1 is selected from C6-10 aryl, C3-14 cycloalkyl, 5-14 membered heteroaryl, 4-14 membered heterocycloalkyl, C3-7 cycloalkyl fused with phenyl to form a bicyclic ring, C4-7 cycloalkyl fused with 5-6 membered heteroaryl to form a bicyclic ring, 4-7 membered heterocycloalkyl fused with phenyl to form a bicyclic ring, and 4-7 membered heterocycloalkyl fused with 5-6 membered heteroaryl to form a bicyclic ring, each optionally substituted by 1, 2, 3, or 4 substituents independently selected from halo, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C1-6 haloalkyl, CN, NO2, ORa, SRa, C1-6 alkyl-NRcRd, C(O)Rb, C(O)NRcRd, C(O)ORa, OC(O)Rb, OC(O)NRcRd, C(=NRe)NRcRd, NRcC(=NRe)NRcRd, NRcRd, NRcC(O)Rb, NRcC(O)ORa, NRcC(O)NRcRd, NRcS(O)Rb, NRcS(O)2Rb, NRcS(O)2NRcRd, S(O)Rb, S(O)NRcRd, S(O)2Rb, and S(O)2NRcRd; A is selected from H, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, and 4-7 membered heterocycloalkyl, each optionally substituted by 1, 2, 3, or 4 substituents independently selected from halo, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C1-6 haloalkyl, CN, NO2, ORa1, SRa1, C(O)Rb1, C(O)NRc1Rd1, C(O)ORa1, OC(O)Rb1, OC(O)NRc1Rd1, C(=NRe1)NRc1Rd1, NRc1C(=NRe1)NRc1Rd1, NRc1Rd1, NRc1C(O)Rb1, NRc1C(O)ORa1, NRc1C(O)NRc1Rd1, NRc1S(O)Rb1, NRc1S(O)2Rb1, NRc1S(O)2NRc1Rd1, S(O)Rb1, S(O)NRc1Rd1, S(O)2Rb1, and S(O)2NRc1Rd1; each Ra, Rb, Rc, Rd, Ra1, Rb1, Rc1, and Rd1 is independently selected from H, C1-6 alkyl, C1-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl-C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl, wherein said C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl-C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl of Ra, Rb, Rc, Rd, Ra1, Rb1, Rc1, and Rd1 is optionally substituted with 1, 2, 3, 4, or 5 substituents independently selected from halo, C1-4 alkyl, C1-4 haloalkyl, C1-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, CN, ORa3, SRa3, C(O)Rb3, C(O)NRc3Rd3, C(O)ORa3, OC(O)Rb3, OC(O)NRc3Rd3, NRc3Rd3, NRc3C(O)Rb3, NRc3C(O)NRc3Rd3,
or a pharmaceutically acceptable salt thereof, wherein constituent members are defined herein. Compounds of Formula (A1) can be useful intermediates in the preparation of compounds of Formula (I). Moreover, compounds of Formula (A1) can be useful as binders of androgen receptor. In some embodiments, provided herein is a compound of Formula (A1), or a pharmaceutically acceptable salt thereof, wherein: R1, R2, and R3 are each independently selected from H, halo, C1-4 alkyl, and C1-4 haloalkyl; R4 and R5 are each independently selected from H, halo, ORA, C1-4 alkyl, and C1-4 haloalkyl, wherein RA is H, C1-4 alkyl, or C1-4 haloalkyl; or R4 and R5 together with the carbon atom to which they are attached form a C3-6 cycloalkyl group or a 3-6 membered heterocycloalkyl group, each optionally substituted with 1, 2, or 3 substituents independently selected from H, halo, ORA, C1-4 alkyl, and C1-4 haloalkyl; R6 and R7 are each independently selected from H, halo, C1-4 alkyl, and C1-4 haloalkyl; R8 is H or C1-4 alkyl;
Cy1 is selected from C6-10 aryl, C3-14 cycloalkyl, 5-14 membered heteroaryl, 4-14 membered heterocycloalkyl, C3-7 cycloalkyl fused with phenyl to form a bicyclic ring, C4-7 cycloalkyl fused with 5-6 membered heteroaryl to form a bicyclic ring, 4-7 membered heterocycloalkyl fused with phenyl to form a bicyclic ring, and 4-7 membered heterocycloalkyl fused with 5-6 membered heteroaryl to form a bicyclic ring, each optionally substituted by 1, 2, 3, or 4 substituents independently selected from halo, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C1-6 haloalkyl, CN, NO2, ORa, SRa, C1-6 alkyl-NRcRd, C(O)Rb, C(O)NRcRd, C(O)ORa, OC(O)Rb, OC(O)NRcRd, C(=NRe)NRcRd, NRcC(=NRe)NRcRd, NRcRd, NRcC(O)Rb, NRcC(O)ORa, NRcC(O)NRcRd, NRcS(O)Rb, NRcS(O)2Rb, NRcS(O)2NRcRd, S(O)Rb, S(O)NRcRd, S(O)2Rb, and S(O)2NRcRd; A is selected from H, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, and 4-7 membered heterocycloalkyl, each optionally substituted by 1, 2, 3, or 4 substituents independently selected from halo, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C1-6 haloalkyl, CN, NO2, ORa1, SRa1, C(O)Rb1, C(O)NRc1Rd1, C(O)ORa1, OC(O)Rb1, OC(O)NRc1Rd1, C(=NRe1)NRc1Rd1, NRc1C(=NRe1)NRc1Rd1, NRc1Rd1, NRc1C(O)Rb1, NRc1C(O)ORa1, NRc1C(O)NRc1Rd1, NRc1S(O)Rb1, NRc1S(O)2Rb1, NRc1S(O)2NRc1Rd1, S(O)Rb1, S(O)NRc1Rd1, S(O)2Rb1, and S(O)2NRc1Rd1; each Ra, Rb, Rc, Rd, Ra1, Rb1, Rc1, and Rd1 is independently selected from H, C1-6 alkyl, C1-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl-C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl, wherein said C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl-C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl of Ra, Rb, Rc, Rd, Ra1, Rb1, Rc1, and Rd1 is optionally substituted with 1, 2, 3, 4, or 5 substituents independently selected from halo, C1-4 alkyl, C1-4 haloalkyl, C1-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, CN, ORa3, SRa3, C(O)Rb3, C(O)NRc3Rd3, C(O)ORa3, OC(O)Rb3, OC(O)NRc3Rd3, NRc3Rd3, NRc3C(O)Rb3, NRc3C(O)NRc3Rd3,
 heterocycloalkyl group optionally substituted with 1, 2, or 3 substituents independently selected
from halo, C1-4 alkyl, C1-4 haloalkyl, CN, ORa3, SRa3, C(O)Rb3, C(O)NRc3Rd3, C(O)ORa3, OC(O)Rb3, OC(O)NRc3Rd3, NRc3Rd3, NRc3C(O)Rb3, NRc3C(O)NRc3Rd3, NRc3C(O)ORa3,
heterocycloalkyl group optionally substituted with 1, 2, or 3 substituents independently selected
from halo, C1-4 alkyl, C1-4 haloalkyl, CN, ORa3, SRa3, C(O)Rb3, C(O)NRc3Rd3, C(O)ORa3, OC(O)Rb3, OC(O)NRc3Rd3, NRc3Rd3, NRc3C(O)Rb3, NRc3C(O)NRc3Rd3, NRc3C(O)ORa3,
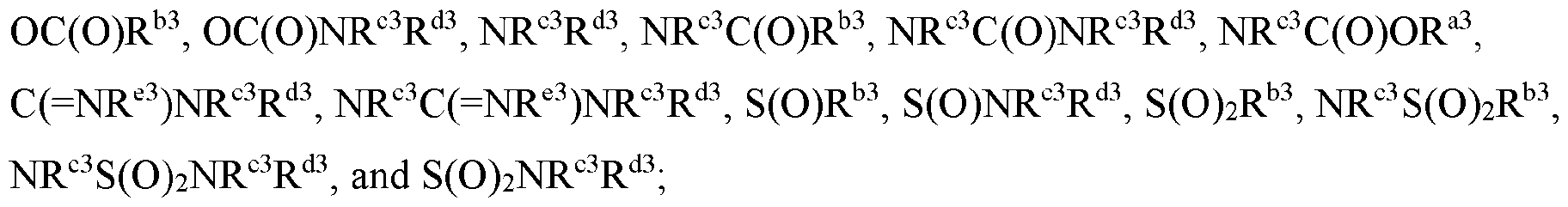 from halo, C1-4 alkyl, C1-4 haloalkyl, CN, ORa3, SRa3, C(O)Rb3, C(O)NRc3Rd3, C(O)ORa3, OC(O)Rb3, OC(O)NRc3Rd3, NRc3Rd3, NRc3C(O)Rb3, NRc3C(O)NRc3Rd3, NRc3C(O)ORa3, C2-
from halo, C1-4 alkyl, C1-4 haloalkyl, CN, ORa3, SRa3, C(O)Rb3, C(O)NRc3Rd3, C(O)ORa3, OC(O)Rb3, OC(O)NRc3Rd3, NRc3Rd3, NRc3C(O)Rb3, NRc3C(O)NRc3Rd3, NRc3C(O)ORa3, C2-
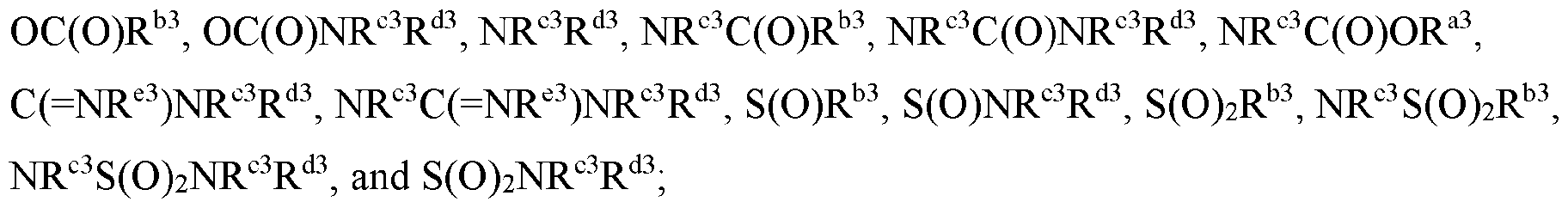 6 alkenyl, C2-6 alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl-C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl, wherein said C1-6 alkyl, C1-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl-C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl are each optionally substituted with 1, 2, or 3 substituents independently selected from OH, CN, amino, halo, C1-6 alkyl, C1-6 alkylamino, di(C1-6 alkyl)amino, C1-6 alkoxy, C1-6 haloalkyl, and C1-6 haloalkoxy; and each Re, Re1, Re2, and Re3 is independently selected from H, C1-4 alkyl, and CN. In some embodiments, R1, R2, and R3 are each independently selected from H, F, methyl, and trifluoromethyl. In some embodiments, R1, R2, and R3 are each F. In some embodiments, R4 and R5 are each C1-4 alkyl. In some embodiments, R4 and R5 are each methyl. In some embodiments, R6 and R7 are each H. In some embodiments, R8 is H. In some embodiments, Cy1 is selected from C3-6 cycloalkyl, 5-6 membered heteroaryl, C5-6 cycloalkyl fused with phenyl to form a bicyclic ring, and 5-6 membered heterocycloalkyl fused with phenyl to form a bicyclic ring, each optionally substituted by 1 or 2 substituents independently selected from halo, C1-6 alkyl, C1-6 haloalkyl, CN, C1-6 alkyl-NRcRd, C(O)NRcRd, and NRcRd. In some embodiments, Cy1 is cyclopropyl, pyrazolyl, or isoxazolyl. In some embodiments, Cy1 is cyclopropyl or pyrazolyl. In some embodiments, Cy1 is C5-6 cycloalkyl fused with phenyl to form a bicyclic ring, optionally substituted by 1 or 2 substituents independently
selected from halo, C1-6 alkyl, CN, C1-6 alkyl-NRcRd, and NRcRd. In some embodiments, Cy1 is cyclopentyl fused with phenyl to form a bicyclic ring, optionally substituted by CN, Cl, and F. In some embodiments, Cy1 is 5-6 membered heterocycloalkyl fused with phenyl to form a bicyclic ring, optionally substituted by 1, 2, or 3 substituents independently selected from halo and C1-6 alkyl. In some embodiments, Cy1 is dihydrobenzofuran, optionally substituted by 1, 2, or 3 substituents independently selected from Cl and I. In some embodiments, A is selected from H, C6-10 aryl and 5-10 membered heteroaryl, optionally substituted by 1, 2, or 3 substituents independently selected from halo, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C1-6 haloalkyl, CN, NO2, ORa1, SRa1, C(O)Rb1, C(O)NRc1Rd1, C(O)ORa1, OC(O)Rb1, OC(O)NRc1Rd1, C(=NRe1)NRc1Rd1, NRc1C(=NRe1)NRc1Rd1, NRc1Rd1, NRc1C(O)Rb1, NRc1C(O)ORa1, NRc1C(O)NRc1Rd1, NRc1S(O)Rb1, NRc1S(O)2Rb1, NRc1S(O)2NRc1Rd1, S(O)Rb1, S(O)NRc1Rd1, S(O)2Rb1, and S(O)2NRc1Rd1. In some embodiments, A is phenyl or 5-6 membered heteroaryl, optionally substituted by 1, 2, or 3 substituents selected from halo and C1-6 alkyl. In some embodiments, A is phenyl optionally substituted by 1, 2, or 3 substituents selected from methyl, Br, and Cl. In some embodiments, A is H. In some embodiments, the compound of Formula (A1) is selected from: 5-chloro-6-iodo-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]- 2,3-dihydrobenzofuran-2-carboxamide; (1S,2S)-2-(4-bromophenyl)-N-[[5-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-3- yl]methyl]cyclopropanecarboxamide; 1-(4-bromo-2-methyl-phenyl)-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol- 5-yl]methyl]pyrazole-4-carboxamide; 1-(3-bromo-2,4-dimethyl-phenyl)-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4- triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-(4-bromo-5-chloro-2-methyl-phenyl)-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H- 1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 5,6-dichloro-2-cyano-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methyl]indane-2-carboxamide; 1-[5-(4-formylphenyl)pyrimidin-2-yl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H- 1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide;
1-(5-bromo-2-pyridyl)-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methyl]pyrazole-4-carboxamide; 1-[5-(4-formylphenyl)-2-pyridyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4- triazol-5-yl]methyl]pyrazole-4-carboxamide; and 1-(5-(3-fluoro-5-formylpyridin-2-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide, or a pharmaceutically acceptable salt of any of the aforementioned. In some embodiments, the compound of Formula (A1) is 3-(4-methylpyrimidin-2-yl)-N- ((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide, or a pharmaceutically acceptable salt thereof. It is further appreciated that certain features of the invention, which are, for clarity, described in the context of separate embodiments, can also be provided in combination in a single embodiment. Conversely, various features of the invention which are, for brevity, described in the context of a single embodiment, can also be provided separately or in any suitable subcombination. At various places in the present specification, substituents of compounds of the invention are disclosed in groups or in ranges. It is specifically intended that the invention include each and every individual subcombination of the members of such groups and ranges. For example, the term “C1-6 alkyl” is specifically intended to individually disclose methyl, ethyl, C3 alkyl, C4 alkyl, C5 alkyl, and C6 alkyl. At various places in the present specification various aryl, heteroaryl, cycloalkyl, and heterocycloalkyl rings are described. Unless otherwise specified, these rings can be attached to the rest of the molecule at any ring member as permitted by valency. For example, the term “pyridinyl,” “pyridyl,” or “a pyridine ring” may refer to a pyridin-2-yl, pyridin-3-yl, or pyridin-4- yl ring. The term “n-membered,” where “n” is an integer, typically describes the number of ring- forming atoms in a moiety where the number of ring-forming atoms is “n”. For example, piperidinyl is an example of a 6-membered heterocycloalkyl ring, pyrazolyl is an example of a 5- membered heteroaryl ring, and pyridyl is an example of a 6-membered heteroaryl ring.
At various places in the present specification, variables defining divalent linking groups may be described. It is specifically intended that each linking substituent include both the forward and backward forms of the linking substituent. For example, -C(O)NRc23-(C1-6 alkylene)- includes both -C(O)NRc23-(C1-6 alkylene)- and -(C1-6 alkylene)-NRc23(O)C- and is intended to disclose each of the forms individually. For compounds of the invention in which a variable appears more than once, each variable can be a different moiety independently selected from the group defining the variable. For example, where a structure is described having two R groups that are simultaneously present on the same compound, the two R groups can represent different moieties independently selected from the group defined for R. As used herein, the phrase “optionally substituted” means unsubstituted or substituted. As used herein, the term “substituted” means that a hydrogen atom is replaced by a non- hydrogen group. It is to be understood that substitution at a given atom is limited by valency. As used herein, the term “C1-j,” where i and j are integers, employed in combination with a chemical group, designates a range of the number of carbon atoms in the chemical group with i-j defining the range. For example, C1-6 alkyl refers to an alkyl group having 1, 2, 3, 4, 5, or 6 carbon atoms. As used herein, the term “alkyl,” employed alone or in combination with other terms, refers to a saturated hydrocarbon group that may be straight-chain or branched. In some embodiments, the alkyl group contains 1 to 7, 1 to 6, 1 to 4, or 1 to 3 carbon atoms. Examples of alkyl moieties include, but are not limited to, chemical groups such as methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl, tert-butyl, n-pentyl, 2-methyl-1-butyl, 3-pentyl, n-hexyl, 1,2,2-trimethylpropyl, n-heptyl, and the like. In some embodiments, the alkyl group is methyl, ethyl, or propyl. The term “alkylene” refers to a linking alkyl group. As used herein, “alkenyl,” employed alone or in combination with other terms, refers to an alkyl group having one or more carbon-carbon double bonds. In some embodiments, the alkenyl moiety contains 2 to 6 or 2 to 4 carbon atoms. Example alkenyl groups include, but are not limited to, ethenyl, n-propenyl, isopropenyl, n-butenyl, sec-butenyl, and the like. The term “alkenylene” refers to a linking alkenyl group. As used herein, “alkynyl,” employed alone or in combination with other terms, refers to an alkyl group having one or more carbon-carbon triple bonds. Example alkynyl groups include, but
are not limited to, ethynyl, propyn-1-yl, propyn-2-yl, and the like. In some embodiments, the alkynyl moiety contains 2 to 6 or 2 to 4 carbon atoms. The term “alkynylene” refers to a linking alkynyl group. As used herein, “halo” or “halogen”, employed alone or in combination with other terms, includes fluoro, chloro, bromo, and iodo. In some embodiments, halo is F or Cl. As used herein, the term “haloalkyl,” employed alone or in combination with other terms, refers to an alkyl group having up to the full valency of halogen atom substituents, which may either be the same or different. In some embodiments, the halogen atoms are fluoro atoms. In some embodiments, the alkyl group has 1 to 6 or 1 to 4 carbon atoms. Example haloalkyl groups include CF3, C2F5, CHF2, CCl3, CHCl2, C2Cl5, and the like. As used herein, the term “alkoxy,” employed alone or in combination with other terms, refers to a group of formula -O-alkyl. Example alkoxy groups include methoxy, ethoxy, propoxy (e.g., n-propoxy and isopropoxy), t-butoxy, and the like. In some embodiments, the alkyl group has 1 to 6 or 1 to 4 carbon atoms. As used herein, “haloalkoxy,” employed alone or in combination with other terms, refers to a group of formula -O-(haloalkyl). In some embodiments, the alkyl group has 1 to 6 or 1 to 4 carbon atoms. An example haloalkoxy group is -OCF3. As used herein, “amino,” employed alone or in combination with other terms, refers to NH2. As used herein, the term “alkylamino,” employed alone or in combination with other terms, refers to a group of formula -NH(alkyl). In some embodiments, the alkylamino group has 1 to 6 or 1 to 4 carbon atoms. Example alkylamino groups include methylamino, ethylamino, propylamino (e.g., n-propylamino and isopropylamino), and the like. As used herein, the term “dialkylamino,” employed alone or in combination with other terms, refers to a group of formula -N(alkyl)2. Example dialkylamino groups include dimethylamino, diethylamino, dipropylamino (e.g., di(n-propyl)amino and di(isopropyl)amino), and the like. In some embodiments, each alkyl group independently has 1 to 6 or 1 to 4 carbon atoms. As used herein, the term “cycloalkyl,” employed alone or in combination with other terms, refers to a non-aromatic cyclic hydrocarbon including cyclized alkyl and alkenyl groups. Cycloalkyl groups can include mono- or polycyclic (e.g., having 2, 3, or 4 fused, bridged, or spiro
rings) ring systems. Also included in the definition of cycloalkyl are moieties that have one or more aromatic rings (e.g., aryl or heteroaryl rings) fused (i.e., having a bond in common with) to the cycloalkyl ring, for example, benzo derivatives of cyclopentane, cyclohexene, cyclohexane, and the like, or pyrido derivatives of cyclopentane or cyclohexane. Ring-forming carbon atoms of a cycloalkyl group can be optionally substituted by oxo. Cycloalkyl groups also include cycloalkylidenes. The term “cycloalkyl” also includes bridgehead cycloalkyl groups (e.g., non- aromatic cyclic hydrocarbon moieties containing at least one bridgehead carbon, such as admantan-1-yl) and spirocycloalkyl groups (e.g., non-aromatic hydrocarbon moieties containing at least two rings fused at a single carbon atom, such as spiro[2.5]octane and the like). In some embodiments, the cycloalkyl group has 3 to 14 ring members, 3 to 10 ring members, 3 to 7 ring members, 3 to 6 ring members, or 5 to 6 ring members. In some embodiments, the cycloalkyl group is monocyclic or bicyclic. In some embodiments, the cycloalkyl group is monocyclic. In some embodiments, the cycloalkyl group is a C3-7 monocyclic cycloalkyl group. Example cycloalkyl groups include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclopentenyl, cyclohexenyl, cyclohexadienyl, cycloheptatrienyl, norbornyl, norpinyl, norcarnyl, tetrahydronaphthalenyl, octahydronaphthalenyl, indanyl, and the like. In some embodiments, the cycloalkyl group is cyclopropyl, cyclobutyl, cyclopentyl, or cyclohexyl. As used herein, the term “cycloalkylalkyl,” employed alone or in combination with other terms, refers to a group of formula cycloalkyl-alkyl-. In some embodiments, the alkyl portion has 1 to 4, 1 to 3, 1 to 2, or 1 carbon atom(s). In some embodiments, the alkyl portion is methylene. In some embodiments, the cycloalkyl portion has 3 to 10 ring members or 3 to 7 ring members. In some embodiments, the cycloalkyl group is monocyclic or bicyclic. In some embodiments, the cycloalkyl portion is monocyclic. In some embodiments, the cycloalkyl portion is a C3-7 monocyclic cycloalkyl group. As used herein, the term “heterocycloalkyl,” employed alone or in combination with other terms, refers to a non-aromatic ring or ring system, which may optionally contain one or more alkenylene or alkynylene groups as part of the ring structure, which has at least one heteroatom ring member independently selected from nitrogen, sulfur, oxygen, and phosphorus. Heterocycloalkyl groups can include mono- or polycyclic (e.g., having 2, 3 or 4 fused, bridged, or spiro rings) ring systems. In some embodiments, the heterocycloalkyl group is a monocyclic or bicyclic group having 1, 2, 3, or 4 heteroatoms independently selected from nitrogen, sulfur and
oxygen. Also included in the definition of heterocycloalkyl are moieties that have one or more aromatic rings (e.g., aryl or heteroaryl rings) fused (i.e., having a bond in common with) to the non-aromatic heterocycloalkyl ring, for example, 1,2,3,4-tetrahydro-quinoline and the like. Heterocycloalkyl groups can also include bridgehead heterocycloalkyl groups (e.g., a heterocycloalkyl moiety containing at least one bridgehead atom, such as azaadmantan-1-yl and the like) and spiroheterocycloalkyl groups (e.g., a heterocycloalkyl moiety containing at least two rings fused at a single atom, such as [1,4-dioxa-8-aza-spiro[4.5]decan-N-yl] and the like). In some embodiments, the heterocycloalkyl group has 4 to 14 ring-forming atoms, 3 to 10 ring- forming atoms, 4 to 10 ring-forming atoms, 3 to 8 ring forming atoms, 4 to 7 ring forming atoms, 3 to 6 ring forming atoms, or 5 to 6 ring forming atoms. In some embodiments, the heterocycloalkyl group has 2 to 20 carbon atoms, 2 to 15 carbon atoms, 2 to 10 carbon atoms, or about 2 to 8 carbon atoms. In some embodiments, the heterocycloalkyl group has 1 to 5 heteroatoms, 1 to 4 heteroatoms, 1 to 3 heteroatoms, or 1 to 2 heteroatoms. The carbon atoms or heteroatoms in the ring(s) of the heterocycloalkyl group can be oxidized to form a carbonyl, an N- oxide, or a sulfonyl group (or other oxidized linkage) or a nitrogen atom can be quaternized. In some embodiments, the heterocycloalkyl portion is a C2-7 monocyclic heterocycloalkyl group. In some embodiments, the heterocycloalkyl group is a morpholine ring, pyrrolidine ring, piperazine ring, piperidine ring, tetrahydropyran ring, tetrahyropyridine, azetidine ring, or tetrahydrofuran ring. As used herein, the term “heterocycloalkylalkyl,” employed alone or in combination with other terms, refers to a group of formula heterocycloalkyl-alkyl-. In some embodiments, the alkyl portion has 1 to 4, 1 to 3, 1 to 2, or 1 carbon atom(s). In some embodiments, the alkyl portion is methylene. In some embodiments, the heterocycloalkyl portion has 3 to 10 ring members, 4 to 10 ring members, or 3 to 7 ring members. In some embodiments, the heterocycloalkyl group is monocyclic or bicyclic. In some embodiments, the heterocycloalkyl portion is monocyclic. In some embodiments, the heterocycloalkyl portion is a C2-7 monocyclic heterocycloalkyl group. As used herein, the term “aryl,” employed alone or in combination with other terms, refers to a monocyclic or polycyclic (e.g., a fused ring system) aromatic hydrocarbon moiety, such as, but not limited to, phenyl, 1-naphthyl, 2-naphthyl, and the like. In some embodiments, aryl groups have from 6 to 10 carbon atoms or 6 carbon atoms. In some embodiments, the aryl group is a monocyclic or bicyclic group. In some embodiments, the aryl group is phenyl or naphthyl.
As used herein, the term “arylalkyl,” employed alone or in combination with other terms, refers to a group of formula aryl-alkyl-. In some embodiments, the alkyl portion has 1 to 4, 1 to 3, 1 to 2, or 1 carbon atom(s). In some embodiments, the alkyl portion is methylene. In some embodiments, the aryl portion is phenyl. In some embodiments, the aryl group is a monocyclic or bicyclic group. In some embodiments, the arylalkyl group is benzyl. As used herein, the term “heteroaryl,” employed alone or in combination with other terms, refers to a monocyclic or polycyclic (e.g., a fused ring system) aromatic hydrocarbon moiety, having one or more heteroatom ring members independently selected from nitrogen, sulfur and oxygen. In some embodiments, the heteroaryl group is a monocyclic or a bicyclic group having 1, 2, 3, or 4 heteroatoms independently selected from nitrogen, sulfur and oxygen. Example heteroaryl groups include, but are not limited to, pyridyl, pyrimidinyl, pyrazinyl, pyridazinyl, triazinyl, furyl, thienyl, imidazolyl, thiazolyl, indolyl, pyrryl, oxazolyl, benzofuryl, benzothienyl, benzthiazolyl, isoxazolyl, pyrazolyl, triazolyl, tetrazolyl, indazolyl, 1,2,4-thiadiazolyl, isothiazolyl, purinyl, carbazolyl, benzimidazolyl, indolinyl, pyrrolyl, azolyl, quinolinyl, isoquinolinyl, benzisoxazolyl, imidazo[1,2-b]thiazolyl or the like. The carbon atoms or heteroatoms in the ring(s) of the heteroaryl group can be oxidized to form a carbonyl, an N-oxide, or a sulfonyl group (or other oxidized linkage) or a nitrogen atom can be quaternized, provided the aromatic nature of the ring is preserved. In some embodiments, the heteroaryl group has from 3 to 10 carbon atoms, from 3 to 8 carbon atoms, from 3 to 5 carbon atoms, from 1 to 5 carbon atoms, or from 5 to 10 carbon atoms. In some embodiments, the heteroaryl group contains 3 to 14, 4 to 12, 4 to 8, 9 to 10, or 5 to 6 ring-forming atoms. In some embodiments, the heteroaryl group has 5 to 14 ring-forming atoms, 5 to 10 ring-forming atoms, 5 to 9 ring-forming atoms, or 5 to 6 ring forming atoms. In some embodiments, the heteroaryl group has 1 to 4, 1 to 3, or 1 to 2 heteroatoms. As used herein, the term “heteroarylalkyl,” employed alone or in combination with other terms, refers to a group of formula heteroaryl-alkyl-. In some embodiments, the alkyl portion has 1 to 4, 1 to 3, 1 to 2, or 1 carbon atom(s). In some embodiments, the alkyl portion is methylene. In some embodiments, the heteroaryl portion is a monocyclic or bicyclic group having 1, 2, 3, or 4 heteroatoms independently selected from nitrogen, sulfur and oxygen. In some embodiments, the heteroaryl portion has 5 to 10 carbon atoms.
The compounds described herein can be asymmetric (e.g., having one or more stereocenters). All stereoisomers, such as enantiomers and diastereomers, are intended unless otherwise indicated. Compounds of the present invention that contain asymmetrically substituted carbon atoms can be isolated in optically active or racemic forms. Methods on how to prepare optically active forms from optically inactive starting materials are known in the art, such as by resolution of racemic mixtures or by stereoselective synthesis. Geometric isomers of olefins, C=N double bonds, and the like can also be present in the compounds described herein, and all such stable isomers are contemplated in the present invention. Cis and trans geometric isomers of the compounds of the present invention may be isolated as a mixture of isomers or as separated isomeric forms. Compounds of the invention also include tautomeric forms. Tautomeric forms result from the swapping of a single bond with an adjacent double bond together with the concomitant migration of a proton. Tautomeric forms include prototropic tautomers which are isomeric protonation states having the same empirical formula and total charge. Example prototropic tautomers include ketone – enol pairs, amide - imidic acid pairs, lactam – lactim pairs, enamine – imine pairs, and annular forms where a proton can occupy two or more positions of a heterocyclic system, for example, 1H- and 3H-imidazole, 1H-, 2H- and 4H- 1,2,4-triazole, 1H- and 2H- isoindole, and 1H- and 2H-pyrazole. Tautomeric forms can be in equilibrium or sterically locked into one form by appropriate substitution. Compounds of the invention also include all isotopes of atoms occurring in the intermediates or final compounds. Isotopes include those atoms having the same atomic number but different mass numbers. For example, isotopes of hydrogen include tritium and deuterium. In some embodiments, the compounds of the invention include at least one deuterium atom. The term “compound,” as used herein, is meant to include all stereoisomers, geometric isomers, tautomers, and isotopes of the structures depicted, unless otherwise specified. All compounds, and pharmaceutically acceptable salts thereof, can be found together with other substances such as water and solvents (e.g., in the form of hydrates and solvates) or can be isolated. In some embodiments, the compounds of the invention, or salts thereof, are substantially isolated. By “substantially isolated” is meant that the compound is at least partially or substantially separated from the environment in which it was formed or detected. Partial
separation can include, for example, a composition enriched in the compounds of the invention. Substantial separation can include compositions containing at least about 50%, at least about 60%, at least about 70%, at least about 80%, at least about 90%, at least about 95%, at least about 97%, or at least about 99% by weight of the compounds of the invention, or salt thereof. Methods for isolating compounds and their salts are routine in the art. The phrase “pharmaceutically acceptable” is employed herein to refer to those compounds, materials, compositions, and/or dosage forms which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of human beings and animals without excessive toxicity, irritation, allergic response, or other problem or complication, commensurate with a reasonable benefit/risk ratio. The present invention also includes pharmaceutically acceptable salts of the compounds described herein. As used herein, "pharmaceutically acceptable salts" refers to derivatives of the disclosed compounds wherein the parent compound is modified by converting an existing acid or base moiety to its salt form. Examples of pharmaceutically acceptable salts include, but are not limited to, mineral or organic acid salts of basic residues such as amines; alkali or organic salts of acidic residues such as carboxylic acids; and the like. The pharmaceutically acceptable salts of the present invention include the non-toxic salts of the parent compound formed, for example, from non-toxic inorganic or organic acids. The pharmaceutically acceptable salts of the present invention can be synthesized from the parent compound which contains a basic or acidic moiety by conventional chemical methods. Generally, such salts can be prepared by reacting the free acid or base forms of these compounds with a stoichiometric amount of the appropriate base or acid in water or in an organic solvent, or in a mixture of the two. Lists of suitable salts are found in Remington's Pharmaceutical Sciences, 17th ed., Mack Publishing Company, Easton, Pa., 1985, p. 1418 and Journal of Pharmaceutical Science, 66, 2 (1977), each of which is incorporated herein by reference in its entirety. Synthesis Compounds of the invention, including salts thereof, can be prepared using known organic synthesis techniques and can be synthesized according to any of numerous possible synthetic routes.
The reactions for preparing compounds of the invention can be carried out in suitable solvents which can be readily selected by one of skill in the art of organic synthesis. Suitable solvents can be substantially nonreactive with the starting materials (reactants), the intermediates, or products at the temperatures at which the reactions are carried out, e.g., temperatures which can range from the solvent's freezing temperature to the solvent's boiling temperature. A given reaction can be carried out in one solvent or a mixture of more than one solvent. Depending on the particular reaction step, suitable solvents for a particular reaction step can be selected by the skilled artisan. Preparation of compounds of the invention can involve the protection and deprotection of various chemical groups. The need for protection and deprotection, and the selection of appropriate protecting groups, can be readily determined by one skilled in the art. The chemistry of protecting groups can be found, for example, in T.W. Greene and P.G.M. Wuts, Protective Groups in Organic Synthesis, 3rd. Ed., Wiley & Sons, Inc., New York (1999), which is incorporated herein by reference in its entirety. Reactions can be monitored according to any suitable method known in the art. For example, product formation can be monitored by spectroscopic means, such as nuclear magnetic resonance spectroscopy (e.g., 1H or 13C), infrared spectroscopy, spectrophotometry (e.g., UV- visible), or mass spectrometry, or by chromatography such as high performance liquid chromatography. Compounds of the invention can be prepared according to numerous preparatory routes known in the literature. Example synthetic methods for preparing compounds of the invention are provided in the Schemes below.
6 alkenyl, C2-6 alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl-C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl, wherein said C1-6 alkyl, C1-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl-C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl are each optionally substituted with 1, 2, or 3 substituents independently selected from OH, CN, amino, halo, C1-6 alkyl, C1-6 alkylamino, di(C1-6 alkyl)amino, C1-6 alkoxy, C1-6 haloalkyl, and C1-6 haloalkoxy; and each Re, Re1, Re2, and Re3 is independently selected from H, C1-4 alkyl, and CN. In some embodiments, R1, R2, and R3 are each independently selected from H, F, methyl, and trifluoromethyl. In some embodiments, R1, R2, and R3 are each F. In some embodiments, R4 and R5 are each C1-4 alkyl. In some embodiments, R4 and R5 are each methyl. In some embodiments, R6 and R7 are each H. In some embodiments, R8 is H. In some embodiments, Cy1 is selected from C3-6 cycloalkyl, 5-6 membered heteroaryl, C5-6 cycloalkyl fused with phenyl to form a bicyclic ring, and 5-6 membered heterocycloalkyl fused with phenyl to form a bicyclic ring, each optionally substituted by 1 or 2 substituents independently selected from halo, C1-6 alkyl, C1-6 haloalkyl, CN, C1-6 alkyl-NRcRd, C(O)NRcRd, and NRcRd. In some embodiments, Cy1 is cyclopropyl, pyrazolyl, or isoxazolyl. In some embodiments, Cy1 is cyclopropyl or pyrazolyl. In some embodiments, Cy1 is C5-6 cycloalkyl fused with phenyl to form a bicyclic ring, optionally substituted by 1 or 2 substituents independently
selected from halo, C1-6 alkyl, CN, C1-6 alkyl-NRcRd, and NRcRd. In some embodiments, Cy1 is cyclopentyl fused with phenyl to form a bicyclic ring, optionally substituted by CN, Cl, and F. In some embodiments, Cy1 is 5-6 membered heterocycloalkyl fused with phenyl to form a bicyclic ring, optionally substituted by 1, 2, or 3 substituents independently selected from halo and C1-6 alkyl. In some embodiments, Cy1 is dihydrobenzofuran, optionally substituted by 1, 2, or 3 substituents independently selected from Cl and I. In some embodiments, A is selected from H, C6-10 aryl and 5-10 membered heteroaryl, optionally substituted by 1, 2, or 3 substituents independently selected from halo, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C1-6 haloalkyl, CN, NO2, ORa1, SRa1, C(O)Rb1, C(O)NRc1Rd1, C(O)ORa1, OC(O)Rb1, OC(O)NRc1Rd1, C(=NRe1)NRc1Rd1, NRc1C(=NRe1)NRc1Rd1, NRc1Rd1, NRc1C(O)Rb1, NRc1C(O)ORa1, NRc1C(O)NRc1Rd1, NRc1S(O)Rb1, NRc1S(O)2Rb1, NRc1S(O)2NRc1Rd1, S(O)Rb1, S(O)NRc1Rd1, S(O)2Rb1, and S(O)2NRc1Rd1. In some embodiments, A is phenyl or 5-6 membered heteroaryl, optionally substituted by 1, 2, or 3 substituents selected from halo and C1-6 alkyl. In some embodiments, A is phenyl optionally substituted by 1, 2, or 3 substituents selected from methyl, Br, and Cl. In some embodiments, A is H. In some embodiments, the compound of Formula (A1) is selected from: 5-chloro-6-iodo-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]- 2,3-dihydrobenzofuran-2-carboxamide; (1S,2S)-2-(4-bromophenyl)-N-[[5-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-3- yl]methyl]cyclopropanecarboxamide; 1-(4-bromo-2-methyl-phenyl)-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol- 5-yl]methyl]pyrazole-4-carboxamide; 1-(3-bromo-2,4-dimethyl-phenyl)-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4- triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-(4-bromo-5-chloro-2-methyl-phenyl)-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H- 1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 5,6-dichloro-2-cyano-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methyl]indane-2-carboxamide; 1-[5-(4-formylphenyl)pyrimidin-2-yl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H- 1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide;
1-(5-bromo-2-pyridyl)-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methyl]pyrazole-4-carboxamide; 1-[5-(4-formylphenyl)-2-pyridyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4- triazol-5-yl]methyl]pyrazole-4-carboxamide; and 1-(5-(3-fluoro-5-formylpyridin-2-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide, or a pharmaceutically acceptable salt of any of the aforementioned. In some embodiments, the compound of Formula (A1) is 3-(4-methylpyrimidin-2-yl)-N- ((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide, or a pharmaceutically acceptable salt thereof. It is further appreciated that certain features of the invention, which are, for clarity, described in the context of separate embodiments, can also be provided in combination in a single embodiment. Conversely, various features of the invention which are, for brevity, described in the context of a single embodiment, can also be provided separately or in any suitable subcombination. At various places in the present specification, substituents of compounds of the invention are disclosed in groups or in ranges. It is specifically intended that the invention include each and every individual subcombination of the members of such groups and ranges. For example, the term “C1-6 alkyl” is specifically intended to individually disclose methyl, ethyl, C3 alkyl, C4 alkyl, C5 alkyl, and C6 alkyl. At various places in the present specification various aryl, heteroaryl, cycloalkyl, and heterocycloalkyl rings are described. Unless otherwise specified, these rings can be attached to the rest of the molecule at any ring member as permitted by valency. For example, the term “pyridinyl,” “pyridyl,” or “a pyridine ring” may refer to a pyridin-2-yl, pyridin-3-yl, or pyridin-4- yl ring. The term “n-membered,” where “n” is an integer, typically describes the number of ring- forming atoms in a moiety where the number of ring-forming atoms is “n”. For example, piperidinyl is an example of a 6-membered heterocycloalkyl ring, pyrazolyl is an example of a 5- membered heteroaryl ring, and pyridyl is an example of a 6-membered heteroaryl ring.
At various places in the present specification, variables defining divalent linking groups may be described. It is specifically intended that each linking substituent include both the forward and backward forms of the linking substituent. For example, -C(O)NRc23-(C1-6 alkylene)- includes both -C(O)NRc23-(C1-6 alkylene)- and -(C1-6 alkylene)-NRc23(O)C- and is intended to disclose each of the forms individually. For compounds of the invention in which a variable appears more than once, each variable can be a different moiety independently selected from the group defining the variable. For example, where a structure is described having two R groups that are simultaneously present on the same compound, the two R groups can represent different moieties independently selected from the group defined for R. As used herein, the phrase “optionally substituted” means unsubstituted or substituted. As used herein, the term “substituted” means that a hydrogen atom is replaced by a non- hydrogen group. It is to be understood that substitution at a given atom is limited by valency. As used herein, the term “C1-j,” where i and j are integers, employed in combination with a chemical group, designates a range of the number of carbon atoms in the chemical group with i-j defining the range. For example, C1-6 alkyl refers to an alkyl group having 1, 2, 3, 4, 5, or 6 carbon atoms. As used herein, the term “alkyl,” employed alone or in combination with other terms, refers to a saturated hydrocarbon group that may be straight-chain or branched. In some embodiments, the alkyl group contains 1 to 7, 1 to 6, 1 to 4, or 1 to 3 carbon atoms. Examples of alkyl moieties include, but are not limited to, chemical groups such as methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl, tert-butyl, n-pentyl, 2-methyl-1-butyl, 3-pentyl, n-hexyl, 1,2,2-trimethylpropyl, n-heptyl, and the like. In some embodiments, the alkyl group is methyl, ethyl, or propyl. The term “alkylene” refers to a linking alkyl group. As used herein, “alkenyl,” employed alone or in combination with other terms, refers to an alkyl group having one or more carbon-carbon double bonds. In some embodiments, the alkenyl moiety contains 2 to 6 or 2 to 4 carbon atoms. Example alkenyl groups include, but are not limited to, ethenyl, n-propenyl, isopropenyl, n-butenyl, sec-butenyl, and the like. The term “alkenylene” refers to a linking alkenyl group. As used herein, “alkynyl,” employed alone or in combination with other terms, refers to an alkyl group having one or more carbon-carbon triple bonds. Example alkynyl groups include, but
are not limited to, ethynyl, propyn-1-yl, propyn-2-yl, and the like. In some embodiments, the alkynyl moiety contains 2 to 6 or 2 to 4 carbon atoms. The term “alkynylene” refers to a linking alkynyl group. As used herein, “halo” or “halogen”, employed alone or in combination with other terms, includes fluoro, chloro, bromo, and iodo. In some embodiments, halo is F or Cl. As used herein, the term “haloalkyl,” employed alone or in combination with other terms, refers to an alkyl group having up to the full valency of halogen atom substituents, which may either be the same or different. In some embodiments, the halogen atoms are fluoro atoms. In some embodiments, the alkyl group has 1 to 6 or 1 to 4 carbon atoms. Example haloalkyl groups include CF3, C2F5, CHF2, CCl3, CHCl2, C2Cl5, and the like. As used herein, the term “alkoxy,” employed alone or in combination with other terms, refers to a group of formula -O-alkyl. Example alkoxy groups include methoxy, ethoxy, propoxy (e.g., n-propoxy and isopropoxy), t-butoxy, and the like. In some embodiments, the alkyl group has 1 to 6 or 1 to 4 carbon atoms. As used herein, “haloalkoxy,” employed alone or in combination with other terms, refers to a group of formula -O-(haloalkyl). In some embodiments, the alkyl group has 1 to 6 or 1 to 4 carbon atoms. An example haloalkoxy group is -OCF3. As used herein, “amino,” employed alone or in combination with other terms, refers to NH2. As used herein, the term “alkylamino,” employed alone or in combination with other terms, refers to a group of formula -NH(alkyl). In some embodiments, the alkylamino group has 1 to 6 or 1 to 4 carbon atoms. Example alkylamino groups include methylamino, ethylamino, propylamino (e.g., n-propylamino and isopropylamino), and the like. As used herein, the term “dialkylamino,” employed alone or in combination with other terms, refers to a group of formula -N(alkyl)2. Example dialkylamino groups include dimethylamino, diethylamino, dipropylamino (e.g., di(n-propyl)amino and di(isopropyl)amino), and the like. In some embodiments, each alkyl group independently has 1 to 6 or 1 to 4 carbon atoms. As used herein, the term “cycloalkyl,” employed alone or in combination with other terms, refers to a non-aromatic cyclic hydrocarbon including cyclized alkyl and alkenyl groups. Cycloalkyl groups can include mono- or polycyclic (e.g., having 2, 3, or 4 fused, bridged, or spiro
rings) ring systems. Also included in the definition of cycloalkyl are moieties that have one or more aromatic rings (e.g., aryl or heteroaryl rings) fused (i.e., having a bond in common with) to the cycloalkyl ring, for example, benzo derivatives of cyclopentane, cyclohexene, cyclohexane, and the like, or pyrido derivatives of cyclopentane or cyclohexane. Ring-forming carbon atoms of a cycloalkyl group can be optionally substituted by oxo. Cycloalkyl groups also include cycloalkylidenes. The term “cycloalkyl” also includes bridgehead cycloalkyl groups (e.g., non- aromatic cyclic hydrocarbon moieties containing at least one bridgehead carbon, such as admantan-1-yl) and spirocycloalkyl groups (e.g., non-aromatic hydrocarbon moieties containing at least two rings fused at a single carbon atom, such as spiro[2.5]octane and the like). In some embodiments, the cycloalkyl group has 3 to 14 ring members, 3 to 10 ring members, 3 to 7 ring members, 3 to 6 ring members, or 5 to 6 ring members. In some embodiments, the cycloalkyl group is monocyclic or bicyclic. In some embodiments, the cycloalkyl group is monocyclic. In some embodiments, the cycloalkyl group is a C3-7 monocyclic cycloalkyl group. Example cycloalkyl groups include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclopentenyl, cyclohexenyl, cyclohexadienyl, cycloheptatrienyl, norbornyl, norpinyl, norcarnyl, tetrahydronaphthalenyl, octahydronaphthalenyl, indanyl, and the like. In some embodiments, the cycloalkyl group is cyclopropyl, cyclobutyl, cyclopentyl, or cyclohexyl. As used herein, the term “cycloalkylalkyl,” employed alone or in combination with other terms, refers to a group of formula cycloalkyl-alkyl-. In some embodiments, the alkyl portion has 1 to 4, 1 to 3, 1 to 2, or 1 carbon atom(s). In some embodiments, the alkyl portion is methylene. In some embodiments, the cycloalkyl portion has 3 to 10 ring members or 3 to 7 ring members. In some embodiments, the cycloalkyl group is monocyclic or bicyclic. In some embodiments, the cycloalkyl portion is monocyclic. In some embodiments, the cycloalkyl portion is a C3-7 monocyclic cycloalkyl group. As used herein, the term “heterocycloalkyl,” employed alone or in combination with other terms, refers to a non-aromatic ring or ring system, which may optionally contain one or more alkenylene or alkynylene groups as part of the ring structure, which has at least one heteroatom ring member independently selected from nitrogen, sulfur, oxygen, and phosphorus. Heterocycloalkyl groups can include mono- or polycyclic (e.g., having 2, 3 or 4 fused, bridged, or spiro rings) ring systems. In some embodiments, the heterocycloalkyl group is a monocyclic or bicyclic group having 1, 2, 3, or 4 heteroatoms independently selected from nitrogen, sulfur and
oxygen. Also included in the definition of heterocycloalkyl are moieties that have one or more aromatic rings (e.g., aryl or heteroaryl rings) fused (i.e., having a bond in common with) to the non-aromatic heterocycloalkyl ring, for example, 1,2,3,4-tetrahydro-quinoline and the like. Heterocycloalkyl groups can also include bridgehead heterocycloalkyl groups (e.g., a heterocycloalkyl moiety containing at least one bridgehead atom, such as azaadmantan-1-yl and the like) and spiroheterocycloalkyl groups (e.g., a heterocycloalkyl moiety containing at least two rings fused at a single atom, such as [1,4-dioxa-8-aza-spiro[4.5]decan-N-yl] and the like). In some embodiments, the heterocycloalkyl group has 4 to 14 ring-forming atoms, 3 to 10 ring- forming atoms, 4 to 10 ring-forming atoms, 3 to 8 ring forming atoms, 4 to 7 ring forming atoms, 3 to 6 ring forming atoms, or 5 to 6 ring forming atoms. In some embodiments, the heterocycloalkyl group has 2 to 20 carbon atoms, 2 to 15 carbon atoms, 2 to 10 carbon atoms, or about 2 to 8 carbon atoms. In some embodiments, the heterocycloalkyl group has 1 to 5 heteroatoms, 1 to 4 heteroatoms, 1 to 3 heteroatoms, or 1 to 2 heteroatoms. The carbon atoms or heteroatoms in the ring(s) of the heterocycloalkyl group can be oxidized to form a carbonyl, an N- oxide, or a sulfonyl group (or other oxidized linkage) or a nitrogen atom can be quaternized. In some embodiments, the heterocycloalkyl portion is a C2-7 monocyclic heterocycloalkyl group. In some embodiments, the heterocycloalkyl group is a morpholine ring, pyrrolidine ring, piperazine ring, piperidine ring, tetrahydropyran ring, tetrahyropyridine, azetidine ring, or tetrahydrofuran ring. As used herein, the term “heterocycloalkylalkyl,” employed alone or in combination with other terms, refers to a group of formula heterocycloalkyl-alkyl-. In some embodiments, the alkyl portion has 1 to 4, 1 to 3, 1 to 2, or 1 carbon atom(s). In some embodiments, the alkyl portion is methylene. In some embodiments, the heterocycloalkyl portion has 3 to 10 ring members, 4 to 10 ring members, or 3 to 7 ring members. In some embodiments, the heterocycloalkyl group is monocyclic or bicyclic. In some embodiments, the heterocycloalkyl portion is monocyclic. In some embodiments, the heterocycloalkyl portion is a C2-7 monocyclic heterocycloalkyl group. As used herein, the term “aryl,” employed alone or in combination with other terms, refers to a monocyclic or polycyclic (e.g., a fused ring system) aromatic hydrocarbon moiety, such as, but not limited to, phenyl, 1-naphthyl, 2-naphthyl, and the like. In some embodiments, aryl groups have from 6 to 10 carbon atoms or 6 carbon atoms. In some embodiments, the aryl group is a monocyclic or bicyclic group. In some embodiments, the aryl group is phenyl or naphthyl.
As used herein, the term “arylalkyl,” employed alone or in combination with other terms, refers to a group of formula aryl-alkyl-. In some embodiments, the alkyl portion has 1 to 4, 1 to 3, 1 to 2, or 1 carbon atom(s). In some embodiments, the alkyl portion is methylene. In some embodiments, the aryl portion is phenyl. In some embodiments, the aryl group is a monocyclic or bicyclic group. In some embodiments, the arylalkyl group is benzyl. As used herein, the term “heteroaryl,” employed alone or in combination with other terms, refers to a monocyclic or polycyclic (e.g., a fused ring system) aromatic hydrocarbon moiety, having one or more heteroatom ring members independently selected from nitrogen, sulfur and oxygen. In some embodiments, the heteroaryl group is a monocyclic or a bicyclic group having 1, 2, 3, or 4 heteroatoms independently selected from nitrogen, sulfur and oxygen. Example heteroaryl groups include, but are not limited to, pyridyl, pyrimidinyl, pyrazinyl, pyridazinyl, triazinyl, furyl, thienyl, imidazolyl, thiazolyl, indolyl, pyrryl, oxazolyl, benzofuryl, benzothienyl, benzthiazolyl, isoxazolyl, pyrazolyl, triazolyl, tetrazolyl, indazolyl, 1,2,4-thiadiazolyl, isothiazolyl, purinyl, carbazolyl, benzimidazolyl, indolinyl, pyrrolyl, azolyl, quinolinyl, isoquinolinyl, benzisoxazolyl, imidazo[1,2-b]thiazolyl or the like. The carbon atoms or heteroatoms in the ring(s) of the heteroaryl group can be oxidized to form a carbonyl, an N-oxide, or a sulfonyl group (or other oxidized linkage) or a nitrogen atom can be quaternized, provided the aromatic nature of the ring is preserved. In some embodiments, the heteroaryl group has from 3 to 10 carbon atoms, from 3 to 8 carbon atoms, from 3 to 5 carbon atoms, from 1 to 5 carbon atoms, or from 5 to 10 carbon atoms. In some embodiments, the heteroaryl group contains 3 to 14, 4 to 12, 4 to 8, 9 to 10, or 5 to 6 ring-forming atoms. In some embodiments, the heteroaryl group has 5 to 14 ring-forming atoms, 5 to 10 ring-forming atoms, 5 to 9 ring-forming atoms, or 5 to 6 ring forming atoms. In some embodiments, the heteroaryl group has 1 to 4, 1 to 3, or 1 to 2 heteroatoms. As used herein, the term “heteroarylalkyl,” employed alone or in combination with other terms, refers to a group of formula heteroaryl-alkyl-. In some embodiments, the alkyl portion has 1 to 4, 1 to 3, 1 to 2, or 1 carbon atom(s). In some embodiments, the alkyl portion is methylene. In some embodiments, the heteroaryl portion is a monocyclic or bicyclic group having 1, 2, 3, or 4 heteroatoms independently selected from nitrogen, sulfur and oxygen. In some embodiments, the heteroaryl portion has 5 to 10 carbon atoms.
The compounds described herein can be asymmetric (e.g., having one or more stereocenters). All stereoisomers, such as enantiomers and diastereomers, are intended unless otherwise indicated. Compounds of the present invention that contain asymmetrically substituted carbon atoms can be isolated in optically active or racemic forms. Methods on how to prepare optically active forms from optically inactive starting materials are known in the art, such as by resolution of racemic mixtures or by stereoselective synthesis. Geometric isomers of olefins, C=N double bonds, and the like can also be present in the compounds described herein, and all such stable isomers are contemplated in the present invention. Cis and trans geometric isomers of the compounds of the present invention may be isolated as a mixture of isomers or as separated isomeric forms. Compounds of the invention also include tautomeric forms. Tautomeric forms result from the swapping of a single bond with an adjacent double bond together with the concomitant migration of a proton. Tautomeric forms include prototropic tautomers which are isomeric protonation states having the same empirical formula and total charge. Example prototropic tautomers include ketone – enol pairs, amide - imidic acid pairs, lactam – lactim pairs, enamine – imine pairs, and annular forms where a proton can occupy two or more positions of a heterocyclic system, for example, 1H- and 3H-imidazole, 1H-, 2H- and 4H- 1,2,4-triazole, 1H- and 2H- isoindole, and 1H- and 2H-pyrazole. Tautomeric forms can be in equilibrium or sterically locked into one form by appropriate substitution. Compounds of the invention also include all isotopes of atoms occurring in the intermediates or final compounds. Isotopes include those atoms having the same atomic number but different mass numbers. For example, isotopes of hydrogen include tritium and deuterium. In some embodiments, the compounds of the invention include at least one deuterium atom. The term “compound,” as used herein, is meant to include all stereoisomers, geometric isomers, tautomers, and isotopes of the structures depicted, unless otherwise specified. All compounds, and pharmaceutically acceptable salts thereof, can be found together with other substances such as water and solvents (e.g., in the form of hydrates and solvates) or can be isolated. In some embodiments, the compounds of the invention, or salts thereof, are substantially isolated. By “substantially isolated” is meant that the compound is at least partially or substantially separated from the environment in which it was formed or detected. Partial
separation can include, for example, a composition enriched in the compounds of the invention. Substantial separation can include compositions containing at least about 50%, at least about 60%, at least about 70%, at least about 80%, at least about 90%, at least about 95%, at least about 97%, or at least about 99% by weight of the compounds of the invention, or salt thereof. Methods for isolating compounds and their salts are routine in the art. The phrase “pharmaceutically acceptable” is employed herein to refer to those compounds, materials, compositions, and/or dosage forms which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of human beings and animals without excessive toxicity, irritation, allergic response, or other problem or complication, commensurate with a reasonable benefit/risk ratio. The present invention also includes pharmaceutically acceptable salts of the compounds described herein. As used herein, "pharmaceutically acceptable salts" refers to derivatives of the disclosed compounds wherein the parent compound is modified by converting an existing acid or base moiety to its salt form. Examples of pharmaceutically acceptable salts include, but are not limited to, mineral or organic acid salts of basic residues such as amines; alkali or organic salts of acidic residues such as carboxylic acids; and the like. The pharmaceutically acceptable salts of the present invention include the non-toxic salts of the parent compound formed, for example, from non-toxic inorganic or organic acids. The pharmaceutically acceptable salts of the present invention can be synthesized from the parent compound which contains a basic or acidic moiety by conventional chemical methods. Generally, such salts can be prepared by reacting the free acid or base forms of these compounds with a stoichiometric amount of the appropriate base or acid in water or in an organic solvent, or in a mixture of the two. Lists of suitable salts are found in Remington's Pharmaceutical Sciences, 17th ed., Mack Publishing Company, Easton, Pa., 1985, p. 1418 and Journal of Pharmaceutical Science, 66, 2 (1977), each of which is incorporated herein by reference in its entirety. Synthesis Compounds of the invention, including salts thereof, can be prepared using known organic synthesis techniques and can be synthesized according to any of numerous possible synthetic routes.
The reactions for preparing compounds of the invention can be carried out in suitable solvents which can be readily selected by one of skill in the art of organic synthesis. Suitable solvents can be substantially nonreactive with the starting materials (reactants), the intermediates, or products at the temperatures at which the reactions are carried out, e.g., temperatures which can range from the solvent's freezing temperature to the solvent's boiling temperature. A given reaction can be carried out in one solvent or a mixture of more than one solvent. Depending on the particular reaction step, suitable solvents for a particular reaction step can be selected by the skilled artisan. Preparation of compounds of the invention can involve the protection and deprotection of various chemical groups. The need for protection and deprotection, and the selection of appropriate protecting groups, can be readily determined by one skilled in the art. The chemistry of protecting groups can be found, for example, in T.W. Greene and P.G.M. Wuts, Protective Groups in Organic Synthesis, 3rd. Ed., Wiley & Sons, Inc., New York (1999), which is incorporated herein by reference in its entirety. Reactions can be monitored according to any suitable method known in the art. For example, product formation can be monitored by spectroscopic means, such as nuclear magnetic resonance spectroscopy (e.g., 1H or 13C), infrared spectroscopy, spectrophotometry (e.g., UV- visible), or mass spectrometry, or by chromatography such as high performance liquid chromatography. Compounds of the invention can be prepared according to numerous preparatory routes known in the literature. Example synthetic methods for preparing compounds of the invention are provided in the Schemes below.
Scheme 1
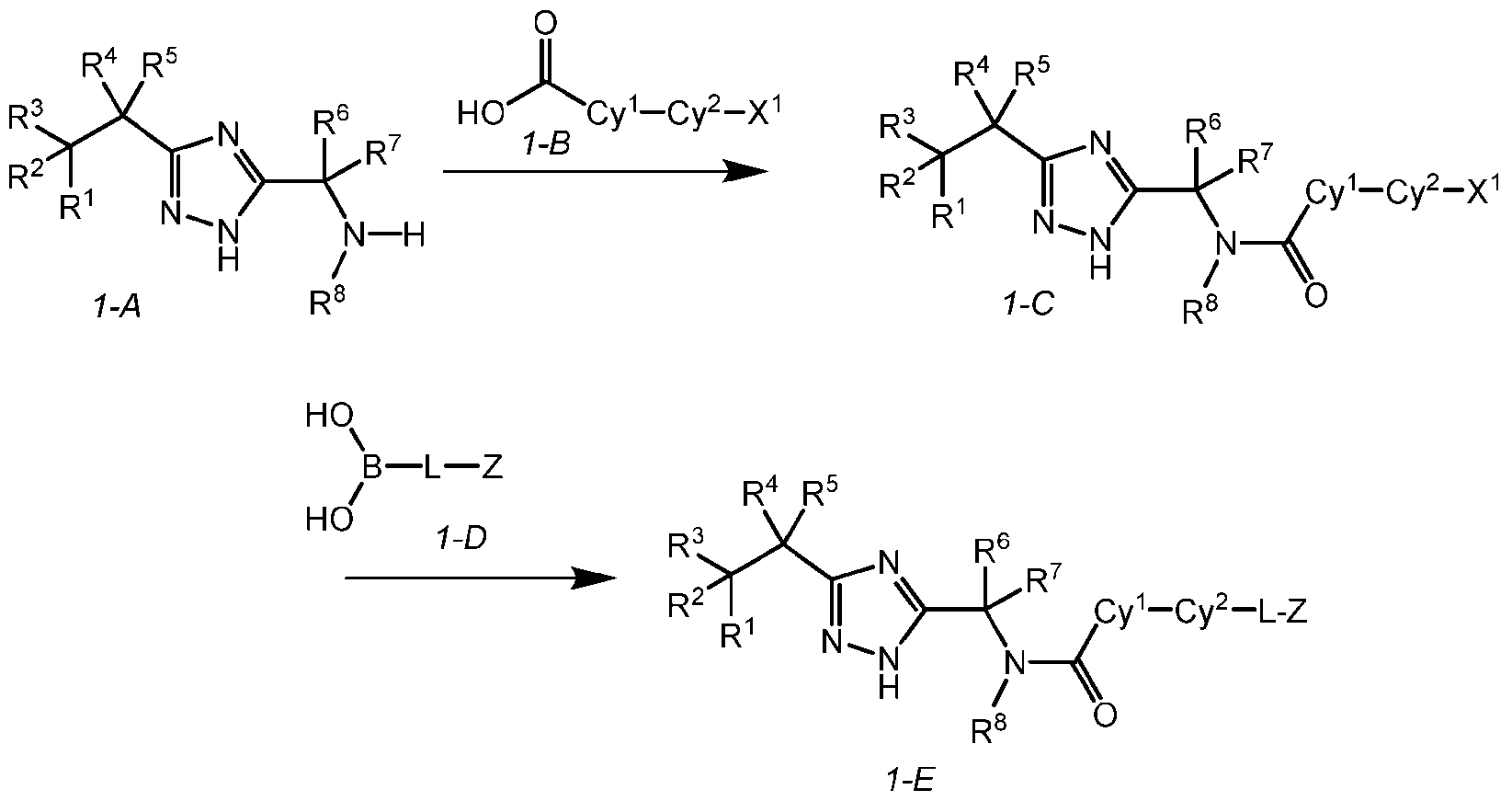 of formula 1-A, many of which are commercially available or can be made via routes known to one skill in the art, can be coupled with compounds of Formula 1-B to provide compounds of formula 1-C, wherein X1 is halogen. The coupling can be performed under peptide coupling conditions (e.g., EDCI, HOBt, and DIPEA; or HATU, DIPEA). Compounds of formula 1-C can be coupled with compounds of formula 1-D to provide compounds of formula 1-E (e.g., the compounds of the disclosure) under the appropriate palladium cross-coupling conditions, for example, in the presence of Pd(dppf)Cl2. Methods of Use Compounds of the present disclosure can bind to both androgen receptor and ubiquitin E3 ligase to cause androgen receptor degradation, which is useful in the treatment of various diseases including cancer. In some embodiments, the compounds provided herein can degrade androgen receptor in a cell, which comprises contacting the cell with the compound or a pharmaceutically acceptable salt or a stereoisomer thereof. In some embodiments, provided herein is a method for degrading androgen receptor in a patient, where the method comprises administering to the patient an effective amount of a compound described herein or a pharmaceutically acceptable salt or a stereoisomer thereof. By “degrading androgen receptor,” it is meant rendering the androgen
receptor inactive by, for example, altering its structure or breaking down androgen receptor into multiple peptide or amino acid fragments. The compounds of the invention are useful in the treatment of various diseases associated with abnormal expression or activity of androgen receptor. For example, the compounds of the invention are useful in the treatment of cancer. In some embodiments, the cancers treatable according to the present invention include prostate cancer, breast cancer, glioblastoma, bladder cancer, renal cell carcinoma, salivary gland cancer, colorectal cancer, esophageal cancer, pancreatic cancer, and stomach cancer. In some embodiments, the cancers treatable according to the present invention include quamous-cell carcinoma, basal cell carcinoma, adenocarcinoma, hepatocellular carcinomas, and renal cell carcinomas, cancer of the bladder, bowel, breast, cervix, colon, esophagus, head, kidney, liver, lung, neck, ovary, pancreas, prostate, and stomach; leukemias; benign and malignant lymphomas, particularly Burkitt's lymphoma and Non-Hodgkin's lymphoma; benign and malignant melanomas; myeloproliferative diseases; sarcomas, including Ewing's sarcoma, hemangiosarcoma, Kaposi's sarcoma, liposarcoma, myosarcomas, peripheral neuroepithelioma, synovial sarcoma, gliomas, astrocytomas, oligodendrogliomas, ependymomas, gliobastomas, neuroblastomas, ganglioneuromas, gangliogliomas, medulloblastomas, pineal cell tumors, meningiomas, meningeal sarcomas, neurofibromas, and Schwannomas; bowel cancer, breast cancer, prostate cancer, cervical cancer, uterine cancer, lung cancer, ovarian cancer, testicular cancer, thyroid cancer, astrocytoma, esophageal cancer, pancreatic cancer, stomach cancer, liver cancer, colon cancer, melanoma; carcinosarcoma, Hodgkin's disease, Wilms' tumor or teratocarcinomas. In certain embodiments, the disease to be treated is cancer, e.g., prostate cancer, or Kennedy's Disease. In some embodiments, the cancers treatable according to the present invention include hematopoietic malignancies such as leukemia and lymphoma. Example lymphomas include Hodgkin’s or non-Hodgkin’s lymphoma, multiple myeloma, B-cell lymphoma (e.g., diffuse large B-cell lymphoma (DLBCL)), chronic lymphocytic lymphoma (CLL), T-cell lymphoma, hairy cell lymphoma, and Burkett's lymphoma. Example leukemias include acute lymphocytic leukemia (ALL), acute myelogenous leukemia (AML), chronic lymphocytic leukemia (CLL), and chronic myelogenous leukemia (CML).
Other cancers treatable by the administration of the compounds of the invention include liver cancer (e.g., hepatocellular carcinoma), bladder cancer, bone cancer, glioma, breast cancer, cervical cancer, colon cancer, endometrial cancer, epithelial cancer, esophageal cancer, Ewing's sarcoma, pancreatic cancer, gallbladder cancer, gastric cancer, gastrointestinal tumors, head and neck cancer, intestinal cancers, Kaposi's sarcoma, kidney cancer, laryngeal cancer, liver cancer (e.g., hepatocellular carcinoma), lung cancer, prostate cancer, rectal cancer, skin cancer, stomach cancer, testicular cancer, thyroid cancer, and uterine cancer. In some embodiments, the cancer treatable by administration of the compounds of the invention is multiple myeloma, DLBCL, hepatocellular carcinoma, bladder cancer, esophageal cancer, head and neck cancer, kidney cancer, prostate cancer, rectal cancer, stomach cancer, thyroid cancer, uterine cancer, breast cancer, glioma, follicular lymphoma, pancreatic cancer, lung cancer, colon cancer, or melanoma. As used herein, the term “cell” is meant to refer to a cell that is in vitro, ex vivo or in vivo. In some embodiments, an ex vivo cell can be part of a tissue sample excised from an organism such as a mammal. In some embodiments, an in vitro cell can be a cell in a cell culture. In some embodiments, an in vivo cell is a cell living in an organism such as a mammal. As used herein, the term “contacting” refers to the bringing together of indicated moieties in an in vitro system or an in vivo system. For example, “contacting” androgen receptor or “contacting” a cell with a compound of the invention includes the administration of a compound of the present invention to an individual or patient, such as a human, having androgen receptor, as well as, for example, introducing a compound of the invention into a sample containing a cellular or purified preparation containing androgen receptor. As used herein, the term “individual” or “patient,” used interchangeably, refers to mammals, and particularly humans. As used herein, the phrase “therapeutically effective amount” refers to the amount of active compound or pharmaceutical agent that elicits the biological or medicinal response in a tissue, system, animal, individual or human that is being sought by a researcher, veterinarian, medical doctor or other clinician. As used herein the term “treating” or “treatment” refers to 1) inhibiting the disease in an individual who is experiencing or displaying the pathology or symptomatology of the disease (i.e., arresting further development of the pathology and/or symptomatology), or 2) ameliorating
the disease in an individual who is experiencing or displaying the pathology or symptomatology of the disease (i.e., reversing the pathology and/or symptomatology). As used herein the term “preventing” or “prevention” refers to preventing the disease in an individual who may be predisposed to the disease but does not yet experience or display the pathology or symptomatology of the disease. As used herein, the term “reducing” is with respect to the level in the patient prior to administration. More specifically, when a biomarker or symptom is reduced in a patient, the reduction is with respect to the level of or severity of the biomarker or symptom in the patient prior to administration of the compound of Formula (I), or a pharmaceutically acceptable salt thereof. Combination Therapy One or more additional pharmaceutical agents or treatment methods such as, for example, chemotherapeutics or other anti-cancer agents, immune enhancers, immunosuppressants, immunotherapies, radiation, anti-tumor and anti-viral vaccines, cytokine therapy (e.g., IL2, GM- CSF, etc.), and/or kinase (tyrosine or serine/threonine), epigenetic or signal transduction inhibitors can be used in combination with the compounds of the present invention. The agents can be combined with the present compounds in a single dosage form, or the agents can be administered simultaneously or sequentially as separate dosage forms. Suitable agents for use in combination with the compounds of the present invention for the treatment of cancer include chemotherapeutic agents, targeted cancer therapies, immunotherapies or radiation therapy. Compounds of this invention may be effective in combination with anti- hormonal agents for treatment of breast cancer and other tumors. Suitable examples are anti- estrogen agents including but not limited to tamoxifen and toremifene, aromatase inhibitors including but not limited to letrozole, anastrozole, and exemestane, adrenocorticosteroids (e.g. prednisone), progestins (e.g. megastrol acetate), and estrogen receptor antagonists (e.g. fulvestrant). Suitable anti-hormone agents used for treatment of prostate and other cancers may also be combined with compounds of the present invention. These include anti-androgens including but not limited to flutamide, bicalutamide, and nilutamide, luteinizing hormone- releasing hormone (LHRH) analogs including leuprolide, goserelin, triptorelin, and histrelin,
LHRH antagonists (e.g. degarelix), androgen receptor blockers (e.g. enzalutamide) and agents that inhibit androgen production (e.g. abiraterone). Angiogenesis inhibitors may be efficacious in some tumors in combination with FGFR inhibitors. These include antibodies against VEGF or VEGFR or kinase inhibitors of VEGFR. Antibodies or other therapeutic proteins against VEGF include bevacizumab and aflibercept. Inhibitors of VEGFR kinases and other anti-angiogenesis inhibitors include but are not limited to sunitinib, sorafenib, axitinib, cediranib, pazopanib, regorafenib, brivanib, and vandetanib Suitable chemotherapeutic or other anti-cancer agents include, for example, alkylating agents (including, without limitation, nitrogen mustards, ethylenimine derivatives, alkyl sulfonates, nitrosoureas and triazenes) such as uracil mustard, chlormethine, cyclophosphamide (CytoxanTM), ifosfamide, melphalan, chlorambucil, pipobroman, triethylene-melamine, triethylenethio- phosphoramine, busulfan, carmustine, lomustine, streptozocin, dacarbazine, and temozolomide. Other anti-cancer agent(s) include antibody therapeutics to costimulatory molecules such as CTLA-4, 4-1BB, PD-1, and PD-L1, or antibodies to cytokines (IL-10, TGF-β, etc.). Exemplary cancer immunotherapy antibodies include pembrolizumab, alemtuzumab, ipilimumab, nivolumab, ofatumumab and rituximab. Methods for the safe and effective administration of most of these chemotherapeutic agents are known to those skilled in the art. In addition, their administration is described in the standard literature. For example, the administration of many of the chemotherapeutic agents is described in the "Physicians' Desk Reference" (PDR, e.g., 1996 edition, Medical Economics Company, Montvale, NJ), the disclosure of which is incorporated herein by reference as if set forth in its entirety. EXAMPLES Abbreviations: ACN: acetonitrile; Boc: tert-butyloxycarbonyl; BOP: benzotriazole-1-yl-oxy-tris- (dimethylamino)-phosphoniumhexafluorophosphate; Cbz: benzyloxycarbonyl; DBA: dibenzylideneacetone; DCE: 1,2-dichloroethane; DIEA: N,N-diisopropylethylamine; DiBAl-H: diisobutylaluminum hydride; DMF: N,N-dimethylformamide; DMSO: dimethylsulfoxide; DPPF: 1,1’--bis(diphenylphosphino)ferrocene; DTBPF: 1,1’-Bis(di-tert-butylphosphino)ferrocene; EDCI: 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide; FA: formic acid; HATU: 1-
[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide; hexafluorophosphate; HPLC: high performance liquid chromatography or high-pressure liquid chromatography; HOBt: 1-Hydroxybenzotriazole hydrate; h: hour(s); LCMS: liquid chromatography mass spectrometer; 2-MeTHF: 2-methyltetrahydrofuran; min: minute(s); MTBE: tert-butyl methyl ether; NCS: N-chlorosuccinimide; NMI: N-methyl imidazole; NMP: N-methyl pyrrolidinone; SFC: supercritical fluid chromatography; TLC: thin layer chromatography; T3P: n- propylphosphonic anhydride; T4P: n-butylphosphonic anhydride; TCFH: chloro-N,N,N′,N′- tetramethylformamidinium hexafluorophosphate; TFA: trifluoroacetic acid Example 1: Synthesis of Intermediate 1 Step
of formula 1-A, many of which are commercially available or can be made via routes known to one skill in the art, can be coupled with compounds of Formula 1-B to provide compounds of formula 1-C, wherein X1 is halogen. The coupling can be performed under peptide coupling conditions (e.g., EDCI, HOBt, and DIPEA; or HATU, DIPEA). Compounds of formula 1-C can be coupled with compounds of formula 1-D to provide compounds of formula 1-E (e.g., the compounds of the disclosure) under the appropriate palladium cross-coupling conditions, for example, in the presence of Pd(dppf)Cl2. Methods of Use Compounds of the present disclosure can bind to both androgen receptor and ubiquitin E3 ligase to cause androgen receptor degradation, which is useful in the treatment of various diseases including cancer. In some embodiments, the compounds provided herein can degrade androgen receptor in a cell, which comprises contacting the cell with the compound or a pharmaceutically acceptable salt or a stereoisomer thereof. In some embodiments, provided herein is a method for degrading androgen receptor in a patient, where the method comprises administering to the patient an effective amount of a compound described herein or a pharmaceutically acceptable salt or a stereoisomer thereof. By “degrading androgen receptor,” it is meant rendering the androgen
receptor inactive by, for example, altering its structure or breaking down androgen receptor into multiple peptide or amino acid fragments. The compounds of the invention are useful in the treatment of various diseases associated with abnormal expression or activity of androgen receptor. For example, the compounds of the invention are useful in the treatment of cancer. In some embodiments, the cancers treatable according to the present invention include prostate cancer, breast cancer, glioblastoma, bladder cancer, renal cell carcinoma, salivary gland cancer, colorectal cancer, esophageal cancer, pancreatic cancer, and stomach cancer. In some embodiments, the cancers treatable according to the present invention include quamous-cell carcinoma, basal cell carcinoma, adenocarcinoma, hepatocellular carcinomas, and renal cell carcinomas, cancer of the bladder, bowel, breast, cervix, colon, esophagus, head, kidney, liver, lung, neck, ovary, pancreas, prostate, and stomach; leukemias; benign and malignant lymphomas, particularly Burkitt's lymphoma and Non-Hodgkin's lymphoma; benign and malignant melanomas; myeloproliferative diseases; sarcomas, including Ewing's sarcoma, hemangiosarcoma, Kaposi's sarcoma, liposarcoma, myosarcomas, peripheral neuroepithelioma, synovial sarcoma, gliomas, astrocytomas, oligodendrogliomas, ependymomas, gliobastomas, neuroblastomas, ganglioneuromas, gangliogliomas, medulloblastomas, pineal cell tumors, meningiomas, meningeal sarcomas, neurofibromas, and Schwannomas; bowel cancer, breast cancer, prostate cancer, cervical cancer, uterine cancer, lung cancer, ovarian cancer, testicular cancer, thyroid cancer, astrocytoma, esophageal cancer, pancreatic cancer, stomach cancer, liver cancer, colon cancer, melanoma; carcinosarcoma, Hodgkin's disease, Wilms' tumor or teratocarcinomas. In certain embodiments, the disease to be treated is cancer, e.g., prostate cancer, or Kennedy's Disease. In some embodiments, the cancers treatable according to the present invention include hematopoietic malignancies such as leukemia and lymphoma. Example lymphomas include Hodgkin’s or non-Hodgkin’s lymphoma, multiple myeloma, B-cell lymphoma (e.g., diffuse large B-cell lymphoma (DLBCL)), chronic lymphocytic lymphoma (CLL), T-cell lymphoma, hairy cell lymphoma, and Burkett's lymphoma. Example leukemias include acute lymphocytic leukemia (ALL), acute myelogenous leukemia (AML), chronic lymphocytic leukemia (CLL), and chronic myelogenous leukemia (CML).
Other cancers treatable by the administration of the compounds of the invention include liver cancer (e.g., hepatocellular carcinoma), bladder cancer, bone cancer, glioma, breast cancer, cervical cancer, colon cancer, endometrial cancer, epithelial cancer, esophageal cancer, Ewing's sarcoma, pancreatic cancer, gallbladder cancer, gastric cancer, gastrointestinal tumors, head and neck cancer, intestinal cancers, Kaposi's sarcoma, kidney cancer, laryngeal cancer, liver cancer (e.g., hepatocellular carcinoma), lung cancer, prostate cancer, rectal cancer, skin cancer, stomach cancer, testicular cancer, thyroid cancer, and uterine cancer. In some embodiments, the cancer treatable by administration of the compounds of the invention is multiple myeloma, DLBCL, hepatocellular carcinoma, bladder cancer, esophageal cancer, head and neck cancer, kidney cancer, prostate cancer, rectal cancer, stomach cancer, thyroid cancer, uterine cancer, breast cancer, glioma, follicular lymphoma, pancreatic cancer, lung cancer, colon cancer, or melanoma. As used herein, the term “cell” is meant to refer to a cell that is in vitro, ex vivo or in vivo. In some embodiments, an ex vivo cell can be part of a tissue sample excised from an organism such as a mammal. In some embodiments, an in vitro cell can be a cell in a cell culture. In some embodiments, an in vivo cell is a cell living in an organism such as a mammal. As used herein, the term “contacting” refers to the bringing together of indicated moieties in an in vitro system or an in vivo system. For example, “contacting” androgen receptor or “contacting” a cell with a compound of the invention includes the administration of a compound of the present invention to an individual or patient, such as a human, having androgen receptor, as well as, for example, introducing a compound of the invention into a sample containing a cellular or purified preparation containing androgen receptor. As used herein, the term “individual” or “patient,” used interchangeably, refers to mammals, and particularly humans. As used herein, the phrase “therapeutically effective amount” refers to the amount of active compound or pharmaceutical agent that elicits the biological or medicinal response in a tissue, system, animal, individual or human that is being sought by a researcher, veterinarian, medical doctor or other clinician. As used herein the term “treating” or “treatment” refers to 1) inhibiting the disease in an individual who is experiencing or displaying the pathology or symptomatology of the disease (i.e., arresting further development of the pathology and/or symptomatology), or 2) ameliorating
the disease in an individual who is experiencing or displaying the pathology or symptomatology of the disease (i.e., reversing the pathology and/or symptomatology). As used herein the term “preventing” or “prevention” refers to preventing the disease in an individual who may be predisposed to the disease but does not yet experience or display the pathology or symptomatology of the disease. As used herein, the term “reducing” is with respect to the level in the patient prior to administration. More specifically, when a biomarker or symptom is reduced in a patient, the reduction is with respect to the level of or severity of the biomarker or symptom in the patient prior to administration of the compound of Formula (I), or a pharmaceutically acceptable salt thereof. Combination Therapy One or more additional pharmaceutical agents or treatment methods such as, for example, chemotherapeutics or other anti-cancer agents, immune enhancers, immunosuppressants, immunotherapies, radiation, anti-tumor and anti-viral vaccines, cytokine therapy (e.g., IL2, GM- CSF, etc.), and/or kinase (tyrosine or serine/threonine), epigenetic or signal transduction inhibitors can be used in combination with the compounds of the present invention. The agents can be combined with the present compounds in a single dosage form, or the agents can be administered simultaneously or sequentially as separate dosage forms. Suitable agents for use in combination with the compounds of the present invention for the treatment of cancer include chemotherapeutic agents, targeted cancer therapies, immunotherapies or radiation therapy. Compounds of this invention may be effective in combination with anti- hormonal agents for treatment of breast cancer and other tumors. Suitable examples are anti- estrogen agents including but not limited to tamoxifen and toremifene, aromatase inhibitors including but not limited to letrozole, anastrozole, and exemestane, adrenocorticosteroids (e.g. prednisone), progestins (e.g. megastrol acetate), and estrogen receptor antagonists (e.g. fulvestrant). Suitable anti-hormone agents used for treatment of prostate and other cancers may also be combined with compounds of the present invention. These include anti-androgens including but not limited to flutamide, bicalutamide, and nilutamide, luteinizing hormone- releasing hormone (LHRH) analogs including leuprolide, goserelin, triptorelin, and histrelin,
LHRH antagonists (e.g. degarelix), androgen receptor blockers (e.g. enzalutamide) and agents that inhibit androgen production (e.g. abiraterone). Angiogenesis inhibitors may be efficacious in some tumors in combination with FGFR inhibitors. These include antibodies against VEGF or VEGFR or kinase inhibitors of VEGFR. Antibodies or other therapeutic proteins against VEGF include bevacizumab and aflibercept. Inhibitors of VEGFR kinases and other anti-angiogenesis inhibitors include but are not limited to sunitinib, sorafenib, axitinib, cediranib, pazopanib, regorafenib, brivanib, and vandetanib Suitable chemotherapeutic or other anti-cancer agents include, for example, alkylating agents (including, without limitation, nitrogen mustards, ethylenimine derivatives, alkyl sulfonates, nitrosoureas and triazenes) such as uracil mustard, chlormethine, cyclophosphamide (CytoxanTM), ifosfamide, melphalan, chlorambucil, pipobroman, triethylene-melamine, triethylenethio- phosphoramine, busulfan, carmustine, lomustine, streptozocin, dacarbazine, and temozolomide. Other anti-cancer agent(s) include antibody therapeutics to costimulatory molecules such as CTLA-4, 4-1BB, PD-1, and PD-L1, or antibodies to cytokines (IL-10, TGF-β, etc.). Exemplary cancer immunotherapy antibodies include pembrolizumab, alemtuzumab, ipilimumab, nivolumab, ofatumumab and rituximab. Methods for the safe and effective administration of most of these chemotherapeutic agents are known to those skilled in the art. In addition, their administration is described in the standard literature. For example, the administration of many of the chemotherapeutic agents is described in the "Physicians' Desk Reference" (PDR, e.g., 1996 edition, Medical Economics Company, Montvale, NJ), the disclosure of which is incorporated herein by reference as if set forth in its entirety. EXAMPLES Abbreviations: ACN: acetonitrile; Boc: tert-butyloxycarbonyl; BOP: benzotriazole-1-yl-oxy-tris- (dimethylamino)-phosphoniumhexafluorophosphate; Cbz: benzyloxycarbonyl; DBA: dibenzylideneacetone; DCE: 1,2-dichloroethane; DIEA: N,N-diisopropylethylamine; DiBAl-H: diisobutylaluminum hydride; DMF: N,N-dimethylformamide; DMSO: dimethylsulfoxide; DPPF: 1,1’--bis(diphenylphosphino)ferrocene; DTBPF: 1,1’-Bis(di-tert-butylphosphino)ferrocene; EDCI: 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide; FA: formic acid; HATU: 1-
[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide; hexafluorophosphate; HPLC: high performance liquid chromatography or high-pressure liquid chromatography; HOBt: 1-Hydroxybenzotriazole hydrate; h: hour(s); LCMS: liquid chromatography mass spectrometer; 2-MeTHF: 2-methyltetrahydrofuran; min: minute(s); MTBE: tert-butyl methyl ether; NCS: N-chlorosuccinimide; NMI: N-methyl imidazole; NMP: N-methyl pyrrolidinone; SFC: supercritical fluid chromatography; TLC: thin layer chromatography; T3P: n- propylphosphonic anhydride; T4P: n-butylphosphonic anhydride; TCFH: chloro-N,N,N′,N′- tetramethylformamidinium hexafluorophosphate; TFA: trifluoroacetic acid Example 1: Synthesis of Intermediate 1 Step
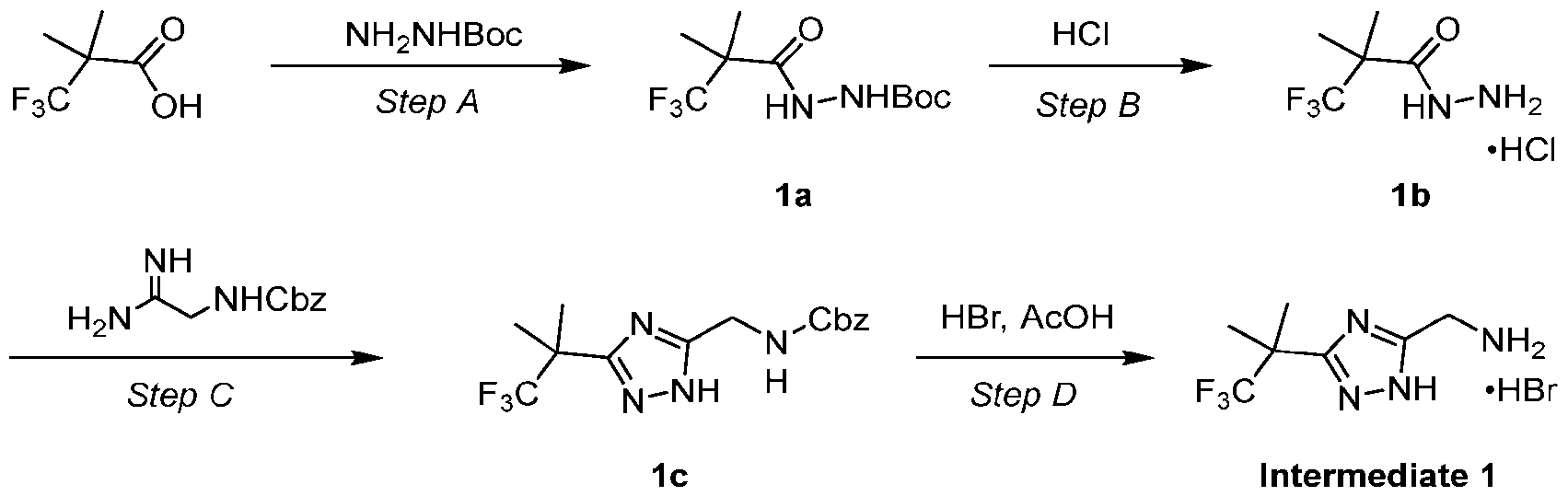 – (1a) To a solution of 3,3,3-trifluoro-2,2-dimethyl-propanoic acid (220.0 g, 1.4 mol, 1.0 equiv) and tert-butyl-N-aminocarbamate (204.8 g, 1.5 mol, 1.1 equiv) in DMF (2000 mL) was added HATU (589.4 g, 1.5 mol, 1.1 equiv) and DIEA (728.5 g, 5.6 mol, 981.9 mL, 4.0 equiv). The mixture was stirred at 20°C for 16 h. The reaction mixture was diluted with water (3 L). The solids were collected by vacuum filtration and the filter cake was washed with water and dried under vacuum to afford 1a. LCMS: [M-1] = 269.1; 1H NMR (400 MHz, DMSO-d6) δ = 9.75 (br s, 1H), 8.81 (s, 1H), 1.46 - 1.29 (m, 15H). Step B – Synthesis of 3,3,3-trifluoro-2,2-dimethyl-propanehydrazide hydrochloride (1b) Intermediate 1a (210.0 g, 777.0 mmol, 1.0 equiv) was added to a solution of 4M HCl in dioxane (1500 mL). The mixture was stirred at 20°C for 2 h. The reaction mixture was
concentrated under reduced pressure to afford 1b, which was used without further purification. 1H NMR (400 MHz, DMSO-d6) δ = 11.37 (br s, 1H), 1.42 (s, 6H). Step C – benzyl ((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)carbamate (1c) A mixture of 1b (75.0 g, 363.0 mmol, 1.0 equiv), benzyl-N-(2-amino-2-imino- ethyl)carbamate hydrochloride (88.4 g, 363.0 mmol, 1.0 equiv), NaOH (30.4 g, 762.3 mmol, 2.1 equiv) in 2-MeTHF (1500.0 mL) was three-fold degassed and purged with nitrogen and then the mixture was stirred at 110°C for 16 h under an atmosphere of nitrogen. The reaction mixture was cooled to ambient temperature, diluted with water (500 mL) and extracted with ethyl acetate (2 x 500 m). The combined organic layers were washed with brine (100 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, dichloromethane : methanol=100/1 to 30/1). The crude product was triturated with MTBE (200 mL) at 20°C for 20 min and the solids were collected by vacuum filtration to afford 1c. LCMS [M+1] =343.2; 1H NMR (400 MHz, DMSO- d6) δ 13.85 (br s, 1H), 7.92 - 7.84 (m, 1H), 7.41 - 7.27 (m, 5H), 5.05 (s, 2H), 4.31 (br d, J = 3.4 Hz, 2H), 1.52 (br s, 6H). Step D – Synthesis of [3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methanamine (Intermediate 1) To a mixture of 1c (70.0 g, 204.4 mmol, 1.0 equiv) in acetic acid (500.0 mL) was added HBr (300.0 mL, 33% purity). The mixture was stirred at 20°C for 1 h and then concentrated under reduced pressure. The residue was two-fold triturated with MTBE (500 mL) at 20°C for 20 min and the solids were collected by vacuum filtration to afford Intermediate 1 (Int 1). LCMS [M- 1] = 207.2; 1H NMR (400 MHz, DMSO-d6) δ = 8.39 (s, 3H), 4.22 - 4.06 (m, 2H), 1.59 (s, 6H). The hydrochloride salt of Intermediate 1 was prepared in the following manner. Either the hydrobromide and hydrochloride salts of Intermediate 1 can be used for the preparation of the following examples.
– (1a) To a solution of 3,3,3-trifluoro-2,2-dimethyl-propanoic acid (220.0 g, 1.4 mol, 1.0 equiv) and tert-butyl-N-aminocarbamate (204.8 g, 1.5 mol, 1.1 equiv) in DMF (2000 mL) was added HATU (589.4 g, 1.5 mol, 1.1 equiv) and DIEA (728.5 g, 5.6 mol, 981.9 mL, 4.0 equiv). The mixture was stirred at 20°C for 16 h. The reaction mixture was diluted with water (3 L). The solids were collected by vacuum filtration and the filter cake was washed with water and dried under vacuum to afford 1a. LCMS: [M-1] = 269.1; 1H NMR (400 MHz, DMSO-d6) δ = 9.75 (br s, 1H), 8.81 (s, 1H), 1.46 - 1.29 (m, 15H). Step B – Synthesis of 3,3,3-trifluoro-2,2-dimethyl-propanehydrazide hydrochloride (1b) Intermediate 1a (210.0 g, 777.0 mmol, 1.0 equiv) was added to a solution of 4M HCl in dioxane (1500 mL). The mixture was stirred at 20°C for 2 h. The reaction mixture was
concentrated under reduced pressure to afford 1b, which was used without further purification. 1H NMR (400 MHz, DMSO-d6) δ = 11.37 (br s, 1H), 1.42 (s, 6H). Step C – benzyl ((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)carbamate (1c) A mixture of 1b (75.0 g, 363.0 mmol, 1.0 equiv), benzyl-N-(2-amino-2-imino- ethyl)carbamate hydrochloride (88.4 g, 363.0 mmol, 1.0 equiv), NaOH (30.4 g, 762.3 mmol, 2.1 equiv) in 2-MeTHF (1500.0 mL) was three-fold degassed and purged with nitrogen and then the mixture was stirred at 110°C for 16 h under an atmosphere of nitrogen. The reaction mixture was cooled to ambient temperature, diluted with water (500 mL) and extracted with ethyl acetate (2 x 500 m). The combined organic layers were washed with brine (100 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, dichloromethane : methanol=100/1 to 30/1). The crude product was triturated with MTBE (200 mL) at 20°C for 20 min and the solids were collected by vacuum filtration to afford 1c. LCMS [M+1] =343.2; 1H NMR (400 MHz, DMSO- d6) δ 13.85 (br s, 1H), 7.92 - 7.84 (m, 1H), 7.41 - 7.27 (m, 5H), 5.05 (s, 2H), 4.31 (br d, J = 3.4 Hz, 2H), 1.52 (br s, 6H). Step D – Synthesis of [3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methanamine (Intermediate 1) To a mixture of 1c (70.0 g, 204.4 mmol, 1.0 equiv) in acetic acid (500.0 mL) was added HBr (300.0 mL, 33% purity). The mixture was stirred at 20°C for 1 h and then concentrated under reduced pressure. The residue was two-fold triturated with MTBE (500 mL) at 20°C for 20 min and the solids were collected by vacuum filtration to afford Intermediate 1 (Int 1). LCMS [M- 1] = 207.2; 1H NMR (400 MHz, DMSO-d6) δ = 8.39 (s, 3H), 4.22 - 4.06 (m, 2H), 1.59 (s, 6H). The hydrochloride salt of Intermediate 1 was prepared in the following manner. Either the hydrobromide and hydrochloride salts of Intermediate 1 can be used for the preparation of the following examples.
Step A – Synthesis of tert-butyl ((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5- yl)methyl)carbamate (1d) A solution of 3,3,3-trifluoro-2,2-dimethyl-propanehydrazide hydrochloride (3.80 g, 18.9 mmol, 1.0 equiv.), tert-butyl (2-amino-2-iminoethyl)carbamate (1.90 g, 18.9 mmol, 1.0 equiv.) in 2-methyltetrahydrofuran (30 mL) was treated with NaOH (2.20 g, 56.9 mmol, 3.0 equiv.). The mixture was stirred at 100 °C for 48 h, cooled to room temperature, diluted with water (30 mL) and extracted with ethyl acetate (3 x 50 mL). The combined organic layers were washed with brine (10 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, 90% ethyl acetate/ petroleum ether) to afford 1d. LCMS [M+1] = 309.1 Step B – Synthesis of [3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methanamine hydrochloride (Int 1•HCl) A mixture of tert-butyl ((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5- yl)methyl)carbamate (1.20 g, 3.9 mmol, 1.0 equiv.) in methanol (3 mL) was cooled to 0 °C and treated with 4 M HCl in dioxane (15 mL). The mixture was stirred at ambient temperature for 16 h. The reaction mixture was concentrated under reduced pressure to afford Int 1 as the hydrochloride salt, which was used without further purification. LCMS [M+1] = 209.1 1H NMR (400 MHz, DMSO-d6) δ = 8.62 (br s, 3H), 4.15-4.11 (m, 2H), 1.59 (s, 6H). Example 2: Synthesis of Intermediate 2
To a solution of methyl 2-hydroxy-4-iodo-benzoate (15.0 g, 53.9 mmol, 1.0 equiv) in acetic acid (150.0 mL), was added NCS (7.20 g, 53.9 mmol, 1.0 equiv) and the mixture was stirred at 110°C for 1 h, cooled to 20°C and poured into 1000 mL water. The solids were collected by vacuum filtration. The filter cake was triturated with MTBE and the solids were collected by vacuum filtration to afford 2a. LCMS [M-1] - =310.9; 1H NMR (400 MHz, CHLOROFORM-d) δ 10.59 (br s, 1H), 7.86 (s, 1H), 7.57 (s, 1H), 3.97 (s, 3H). Step B – Synthesis of 4-chloro-2-(hydroxymethyl)-5-iodo-phenol (2b) DIBAL-H (1.0 M, 57.4 mL, 3.0 equiv) was added to the solution of 2a (5.9 g, 19.1 mmol, 1.0 equiv) in toluene (60.0 mL) at -5°C. The mixture was stirred at 20°C for 4 h. The mixture was quenched with saturated aqueous HCl (1M, 100.0 mL) and extracted with ethyl acetate (3 x 60.0 mL). The combined ethyl acetate was washed with brine (40 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by recrystallization from MTBE (30 mL) to give 2b. LCMS [M+1] = 284.9; 1H NMR (400 MHz, DMSO-d6) δ 9.97 (br s, 1H), 7.38 (s, 1H), 7.27 (s, 1H), 5.17 (br s, 1H), 4.40 (s, 2H). Step C – Synthesis of ethyl 2-[4-chloro-2-(hydroxymethyl)-5-iodo-phenoxy]acetate (2c)
A mixture of 2b (2.8 g, 9.8 mmol, 1.0 equiv) and K2CO3 (1.5 g, 10.8 mmol, 1.1 equiv) in DMF (24.0 mL) at 20°C was treated with ethyl 2-bromoacetate (1.6 g, 9.8 mmol, 1.0 mL, 1.0 equiv). The mixture was stirred at 20 °C for 4 h, diluted with water 15 mL and extracted with ethyl acetate (2 x 20 mL). The combined organic layers were washed with brine (2 x 30 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether/ethyl acetate=80:1 to 20:1) to afford 2c. 1H NMR (400 MHz, DMSO-d6) δ 7.47 (s, 1H), 7.40 (s, 1H), 5.27 (t, J = 5.6 Hz, 1H), 4.87 (s, 2H), 4.47 (d, J = 5.6 Hz, 2H), 4.16 (q, J = 7.2 Hz, 2H), 1.21 (t, J = 7.2 Hz, 3H). Step D – Synthesis of ethyl 2-[4-chloro-2-(chloromethyl)-5-iodo-phenoxy]acetate (2d) SOCl2 (5.5 g, 46.4 mmol, 3.3 mL, 4.0 equiv) was added to the solution of 2c (4.3 g, 11.6 mmol, 1.0 equiv) in toluene (40 mL) at 20°C and stirred at 20°C for 1 h. The mixture was diluted with water 15 mL at 0°C and extracted with ethyl acetate (2 x 20 mL). The combined organic layers were washed with brine (2 x 15 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether/ethyl acetate=80:1 to 20:1) to give 2d. 1H NMR (400 MHz, DMSO-d6) δ 7.65 (s, 1H), 7.55 (s, 1H), 4.96 (s, 2H), 4.70 (s, 2H), 4.18 (q, J = 7.2 Hz, 2H), 1.21 (t, J = 7.2 Hz, 3H). Step E – Synthesis of ethyl 5-chloro-6-iodo-2,3-dihydrobenzofuran-2-carboxylate (2e) NaH (380.9 mg, 9.5 mmol, 60.0% purity, 1.3 equiv) was added to the solution of 2d (2.8 g, 7.3 mmol, 1.0 equiv) in NMP (25.0 mL) at 0°C. The solution was stirred at 20°C for 3 h. The mixture was quenched with saturated aqueous NH4Cl (10 mL) and extracted with ethyl acetate (3 x 30 mL). The combined organic portions were washed with brine (50 mL), dried over sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether/ ethyl acetate = 80:1 to 20:1) to afford 2e. 1H NMR (400 MHz, Methanol-d4) δ 7.34 (s, 2H), 5.29 (dd, J = 6.2, 10.6 Hz, 1H), 4.24 (q, J = 7.2 Hz, 2H), 3.56 (dd, J = 10.6, 16.4 Hz, 1H), 3.27 (br d, J = 6.2 Hz, 1H), 1.29 (t, J = 7.2 Hz, 3H). Step F – Synthesis of 5-chloro-6-iodo-2,3-dihydrobenzofuran-2-carboxylic acid (2f)
A solution of 2e (3.0 g, 8.5 mmol, 1.0 equiv) in tetrahydrofuran (15.0 mL) was treated with a solution of LiOH•H2O (642.7 mg, 15.3 mmol, 1.8 equiv) in water (15.0 mL) at 20°C. The mixture was stirred at 20°C for 2 h. The pH if the mixture was adjusted to pH=6 with citric acid. The mixture was extracted with ethyl acetate (3 x 100 mL). The combined ethyl acetate was washed with brine (15 mL), dried over sodium sulfate, filtered and the filtrate was concentrated. The residue was purified by column chromatography (silica gel, petroleum ether/ ethyl acetate=10/1 to 5/1) to afford 2f. LCMS [M-1] = 322.9; 1H NMR (400 MHz, DMSO-d6) δ 7.43 (s, 1H), 7.41 (s, 1H), 5.28 (dd, J = 6.0 , 10.8 Hz, 1H), 3.52 (dd, J = 10.8, 16.8 Hz, 1H), 3.21 (dd, J = 6.0, 16.8 Hz, 1H) Step G – Synthesis of 5-chloro-6-iodo-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol- 5-yl]methyl]-2,3-dihydrobenzofuran-2-carboxamide (Intermediate 2) T3P (10.7 g, 16.9 mmol, 10.0 mL, 50% in DMF, 2.5 equiv) was added in one portion to a solution of DIEA (7.0 g, 54.2 mmol, 9.4 mL, 8.0 equiv), 2f (2.2 g, 6.7 mmol, 1.0 equiv), Intermediate 1 (2.5 g, 8.8 mmol, 1.3 equiv) in DMF (27.0 mL) at 20°C. Then the solution was stirred at 20 °C for 2 h. The mixture was quenched by water (40 mL) and extracted with ethyl acetate (3 x 50 mL). The combined organic layers were washed with brine (3 x 30 mL) dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, dichloromethane:methanol =100/1 to 5/1) to afford Intermediate 2 (Int 2). LCMS [M+1] =515.0, 517.0; 1H NMR (400 MHz, DMSO- d6) δ 13.82 (s, 1H), 8.82 (br t, J = 5.6 Hz, 1H), 7.45 (s, 1H), 7.37 (s, 1H), 5.28 (dd, J = 6.0, 10.4 Hz, 1H), 4.50 - 4.42 (m, 1H), 4.33 (dd, J = 5.6, 16.0 Hz, 1H), 3.49 (dd, J = 10.4, 16.8 Hz, 1H), 3.24 (dd, J = 5.6, 16.4 Hz, 1H), 1.50 (s, 6H) Example 3: Synthesis of Intermediate 3
 Step A – Synthesis of (1S,2S)-2-(4-bromophenyl)-N-[[5-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H- 1,2,4-triazol-3-yl]methyl]cyclopropanecarboxamide (Intermediate 3)
To a solution of (1S,2S)-2-(4-bromophenyl)cyclopropanecarboxylic acid (160.0 mg, 663.7 µmol, 1 equiv) and Intermediate 1 (268.6 mg, 929.2 umol, 1.4 equiv) in DMF (1.6 mL) was added DIEA (686.2 mg, 5.3 mmol, 924.8 μL, 8.0 equiv), followed by T3P (844.7 mg, 1.3 mmol, 789.4 μL, 50% in DMF, 2.0 equiv). The solution was stirred at 25°C for 16 h. The mixture was quenched with water (20 mL) and extracted with ethyl acetate (10 mL x 2). The combined organic layers were washed with brine (10 mL x 2), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative TLC (silica gel, petroleum ether: ethyl acetate = 1:1) to afford Intermediate 3 (Int 3). LCMS: [M+1] = 433.2, 435.2; 1H NMR (400 MHz, CHLOROFORM-d) δ 7.38 (d, J = 8.4 Hz, 2H), 7.03 (br s, 1H), 6.92 (d, J = 8.4 Hz, 2H), 4.69 - 4.43 (m, 2H), 2.56 - 2.39 (m, 1H), 1.71 - 1.62 (m, 2H), 1.62 - 1.57 (m, 6H), 1.31 - 1.20 (m, 2H) Example 4: Synthesis of Intermediate 4
Step A – Synthesis of (1S,2S)-2-(4-bromophenyl)-N-[[5-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H- 1,2,4-triazol-3-yl]methyl]cyclopropanecarboxamide (Intermediate 3)
To a solution of (1S,2S)-2-(4-bromophenyl)cyclopropanecarboxylic acid (160.0 mg, 663.7 µmol, 1 equiv) and Intermediate 1 (268.6 mg, 929.2 umol, 1.4 equiv) in DMF (1.6 mL) was added DIEA (686.2 mg, 5.3 mmol, 924.8 μL, 8.0 equiv), followed by T3P (844.7 mg, 1.3 mmol, 789.4 μL, 50% in DMF, 2.0 equiv). The solution was stirred at 25°C for 16 h. The mixture was quenched with water (20 mL) and extracted with ethyl acetate (10 mL x 2). The combined organic layers were washed with brine (10 mL x 2), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative TLC (silica gel, petroleum ether: ethyl acetate = 1:1) to afford Intermediate 3 (Int 3). LCMS: [M+1] = 433.2, 435.2; 1H NMR (400 MHz, CHLOROFORM-d) δ 7.38 (d, J = 8.4 Hz, 2H), 7.03 (br s, 1H), 6.92 (d, J = 8.4 Hz, 2H), 4.69 - 4.43 (m, 2H), 2.56 - 2.39 (m, 1H), 1.71 - 1.62 (m, 2H), 1.62 - 1.57 (m, 6H), 1.31 - 1.20 (m, 2H) Example 4: Synthesis of Intermediate 4
 – To a solution of methyl 1H-pyrazole-4-carboxylate (125.0 g, 991.1 mmol, 1.0 equiv) and 1-fluoro-2-methyl-4-nitrobenzene (153.7 g, 991.2 mmol, 1 equiv) in acetonitrile (1.5 L) was added potassium carbonate (232.8 g, 1.6 mol, 1.7 equiv). The reaction mixture was stirred at 80°C for 16 h. Water (2 L) was added to the reaction mixture and the solids were collected by vacuum filtration and dried under reduced pressure to afford 4a.1H NMR (400 MHz, DMSO-d6) δ 8.85 (s, 1H), 8.35 (s, 1H), 8.25 - 8.13 (m, 2H), 7.75 (d, J = 8.8 Hz, 1H), 3.80 (s, 3H), 2.39 (s, 3H). Step B – Synthesis of methyl 1-(4-amino-2-methylphenyl)pyrazole-4-carboxylate (4b)
To a mixture of 4a (110.0 g, 421.0 mmol, 1 equiv) in methanol (2 L) was added a slurry of Pd/C (11.00 g, 10 wt% Pd, 50 wt% in water) in methanol (100 ml). The mixture was stirred at 25°C for 12 h under hydrogen (1 atm). The reaction mixture was filtered and the filtrate was concentrated under reduced pressure to afford methyl 4b. 1H NMR (400 MHz, DMSO-d6) δ 8.40 (s, 1H), 8.01 (s, 1H), 6.98 (d, J = 8.4 Hz, 1H), 6.63 - 6.35 (m, 2H), 5.37 (s, 2H), 3.76 (s, 3H), 1.97 (s, 3H). Step C – Synthesis of methyl 1-(4-bromo-2-methylphenyl)pyrazole-4-carboxylate (4c) To a solution of 4b (110.0 g, 475.6 mmol, 1 equiv) in CH3CN (1.5 L) at 0°C was added t- BuONO (68.6 g, 665.9 mmol, 1.4 equiv). The reaction mixture was stirred at 20 °C for 1 h. CuBr2 (148.74 g, 665.95 mmol, 31.18 mL, 1.4 equiv) was added portion wise. The reaction mixture was stirred at 20°C for 3 h. Water (4 L) was added and the mixture was extracted with ethyl acetate (2 x 3 L). The combined organic layers were washed with brine (2 L), dried over anhydrous MgSO4, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether/ethyl acetate=1/0 to 5/1) to afford 4c. 1H NMR (400 MHz, DMSO-d6) δ 8.68 (s, 1H), 8.13 (s, 1H), 7.69 (s, 1H), 7.56 (m, 1H), 7.37 (m, 1H), 3.78 (s, 3H), 2.19 (s, 3H). Step D – Synthesis of 1-(4-bromo-2-methyl-phenyl) pyrazole-4-carboxylic acid (4d) To a solution of 4c (110.0 g, 372.7 mmol, 1 equiv) in methanol (0.2 L), tetrahydrofuran (0.4 L) and water (0.4 L) at 0°C was added LiOH•H2O (93.8 g, 2.24 mol, 6.0 equiv). The reaction mixture was stirred at 20°C for 18 h. The reaction mixture was concentrated under reduced pressure to remove most of the methanol and tetrahydrofuran. The concentrated mixture was cooled to 0°C and adjusted to pH 5 with 1N aqueous HCl. The precipitate was collected by vacuum filtration and dried under vacuum to afford 4d.1H NMR (400 MHz, DMSO-d6) δ 8.40 (s, 1H), 7.97 (s, 1H), 7.66 (s, 1H), 7.54 (d, J = 8.4 Hz, 1H), 7.35 (d, J = 8.4 Hz, 1H), 2.20 (s, 3H). Step E – Synthesis of 1-(4-bromo-2-methyl-phenyl)-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H- 1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Intermediate 4) To a mixture of 4d (9.5 g, 33.8 mmol, 1 equiv) and Intermediate 1 (10.7 g, 37.17 mmol, 1.1 equiv) in DMF (90 mL) was added BOP (17.9 g, 40.5 mmol, 1.2 equiv) and triethylamine (10.2 g, 101.3 mmol, 3.0 equiv). The mixture was stirred at 20°C for 1 h. The residue was
poured into water (200 mL). The aqueous phase was extracted with ethyl acetate (3 x 200 mL). The combined organic phase was washed with brine (3 x 200 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether/ethyl acetate=3/1 to 0/1) to afford Intermediate 4 (Int 4). 1H NMR (400 MHz, Methanol-d4) δ 8.32 (s, 1H), 8.15 (s, 1H), 7.61 (s, 1H), 7.52 (dd, J = 2.0, 8.4 Hz, 1H),7.29 (d, J = 8.4 Hz, 1H), 4.69 (s, 2H), 2.22 (s, 3H), 1.60 (s, 6H). Example 5: Synthesis of Intermediate 5 Step
– To a solution of methyl 1H-pyrazole-4-carboxylate (125.0 g, 991.1 mmol, 1.0 equiv) and 1-fluoro-2-methyl-4-nitrobenzene (153.7 g, 991.2 mmol, 1 equiv) in acetonitrile (1.5 L) was added potassium carbonate (232.8 g, 1.6 mol, 1.7 equiv). The reaction mixture was stirred at 80°C for 16 h. Water (2 L) was added to the reaction mixture and the solids were collected by vacuum filtration and dried under reduced pressure to afford 4a.1H NMR (400 MHz, DMSO-d6) δ 8.85 (s, 1H), 8.35 (s, 1H), 8.25 - 8.13 (m, 2H), 7.75 (d, J = 8.8 Hz, 1H), 3.80 (s, 3H), 2.39 (s, 3H). Step B – Synthesis of methyl 1-(4-amino-2-methylphenyl)pyrazole-4-carboxylate (4b)
To a mixture of 4a (110.0 g, 421.0 mmol, 1 equiv) in methanol (2 L) was added a slurry of Pd/C (11.00 g, 10 wt% Pd, 50 wt% in water) in methanol (100 ml). The mixture was stirred at 25°C for 12 h under hydrogen (1 atm). The reaction mixture was filtered and the filtrate was concentrated under reduced pressure to afford methyl 4b. 1H NMR (400 MHz, DMSO-d6) δ 8.40 (s, 1H), 8.01 (s, 1H), 6.98 (d, J = 8.4 Hz, 1H), 6.63 - 6.35 (m, 2H), 5.37 (s, 2H), 3.76 (s, 3H), 1.97 (s, 3H). Step C – Synthesis of methyl 1-(4-bromo-2-methylphenyl)pyrazole-4-carboxylate (4c) To a solution of 4b (110.0 g, 475.6 mmol, 1 equiv) in CH3CN (1.5 L) at 0°C was added t- BuONO (68.6 g, 665.9 mmol, 1.4 equiv). The reaction mixture was stirred at 20 °C for 1 h. CuBr2 (148.74 g, 665.95 mmol, 31.18 mL, 1.4 equiv) was added portion wise. The reaction mixture was stirred at 20°C for 3 h. Water (4 L) was added and the mixture was extracted with ethyl acetate (2 x 3 L). The combined organic layers were washed with brine (2 L), dried over anhydrous MgSO4, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether/ethyl acetate=1/0 to 5/1) to afford 4c. 1H NMR (400 MHz, DMSO-d6) δ 8.68 (s, 1H), 8.13 (s, 1H), 7.69 (s, 1H), 7.56 (m, 1H), 7.37 (m, 1H), 3.78 (s, 3H), 2.19 (s, 3H). Step D – Synthesis of 1-(4-bromo-2-methyl-phenyl) pyrazole-4-carboxylic acid (4d) To a solution of 4c (110.0 g, 372.7 mmol, 1 equiv) in methanol (0.2 L), tetrahydrofuran (0.4 L) and water (0.4 L) at 0°C was added LiOH•H2O (93.8 g, 2.24 mol, 6.0 equiv). The reaction mixture was stirred at 20°C for 18 h. The reaction mixture was concentrated under reduced pressure to remove most of the methanol and tetrahydrofuran. The concentrated mixture was cooled to 0°C and adjusted to pH 5 with 1N aqueous HCl. The precipitate was collected by vacuum filtration and dried under vacuum to afford 4d.1H NMR (400 MHz, DMSO-d6) δ 8.40 (s, 1H), 7.97 (s, 1H), 7.66 (s, 1H), 7.54 (d, J = 8.4 Hz, 1H), 7.35 (d, J = 8.4 Hz, 1H), 2.20 (s, 3H). Step E – Synthesis of 1-(4-bromo-2-methyl-phenyl)-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H- 1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Intermediate 4) To a mixture of 4d (9.5 g, 33.8 mmol, 1 equiv) and Intermediate 1 (10.7 g, 37.17 mmol, 1.1 equiv) in DMF (90 mL) was added BOP (17.9 g, 40.5 mmol, 1.2 equiv) and triethylamine (10.2 g, 101.3 mmol, 3.0 equiv). The mixture was stirred at 20°C for 1 h. The residue was
poured into water (200 mL). The aqueous phase was extracted with ethyl acetate (3 x 200 mL). The combined organic phase was washed with brine (3 x 200 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether/ethyl acetate=3/1 to 0/1) to afford Intermediate 4 (Int 4). 1H NMR (400 MHz, Methanol-d4) δ 8.32 (s, 1H), 8.15 (s, 1H), 7.61 (s, 1H), 7.52 (dd, J = 2.0, 8.4 Hz, 1H),7.29 (d, J = 8.4 Hz, 1H), 4.69 (s, 2H), 2.22 (s, 3H), 1.60 (s, 6H). Example 5: Synthesis of Intermediate 5 Step
 – A mixture of 1,3-dibromo-2,4-dimethyl-benzene (1.00 g, 3.79 mmol, 1.0 equiv), methyl 1H-pyrazole-4-carboxylate (573 mg, 4.5 mmol, 1.2 equiv), (1R,2R)-N1,N2-dimethylcyclohexane- 1,2-diamine (216 mg, 1.5 mmol, 0.4 equiv), iodocopper tetrabutylammonium diiodide (848.3 mg, 0.76 mmol, 0.2 equiv) and cesium carbonate (2.5 g, 7.5 mmol, 2.0 equiv) in dioxane (10.0 mL) was three-fold degassed and purged with argon. The mixture was stirred at 120°C for 16 h under argon. The reaction was cooled to 20°C and partitioned between ethyl acetate (120 mL) and water (120 mL). The organic phase was separated, washed with brine (2 x 100 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether/ ethyl acetate=100:0 to 96:4) to afford 5a. LCMS [M+1] = 309.1, 311.1; 1H NMR (400 MHz, CHLOROFORM-d) δ 8.11 (s, 1H), 8.04 (s, 1H), 7.20 (app s, 2H), 3.88 (s, 3H), 2.50 (s, 3H), 2.25 (s, 3H). Step B – Synthesis of 1-(3-bromo-2,4-dimethyl-phenyl)pyrazole-4-carboxylic acid (5b)
To a solution of 5a (1.0 g, 3.2 mmol, 1.0 equiv) in methanol (4.0 mL), tetrahydrofuran (4.0 mL) and water (2.0 mL) was added LiOH•H2O (407.2 mg, 9.7 mmol, 3.0 equiv). The mixture was stirred at 25°C for 16 h. The reaction mixture was concentrated under reduced pressure. The residue was diluted with water (10 mL) and pH adjusted to 3 with 1 M aqueous HCl. The solids were collected by vacuum filtration, the filter cake was dried under reduced pressure to afford 5b. LCMS [M+1] = 295.1, 297.1; 1H NMR (400 MHz, CHLOROFORM-d) δ 8.18 (s, 1H), 8.11 (s, 1H), 7.22 (app s, 2H), 2.51 (s, 3H), 2.27 (s, 3H). Step C – Synthesis of 1-(3-bromo-2,4-dimethyl-phenyl)-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Intermediate 5) To a solution of 5b (720 mg, 2.4 mmol, 1.0 equiv) in DMF (6.5 mL) was added DIEA (946 mg, 7.3 mmol, 1.3 mL, 3.0 equiv), HATU (928 mg, 2.4 mmol, 1.0 equiv) and Int 1 (916.8 mg, 3.1 mmol, 1.3 equiv). The mixture was stirred at 25°C for 2 h. The reaction mixture was partitioned between ethyl acetate (300 mL) and water (300 mL). The organic phase was separated, washed with brine (2 x 50 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, dichloromethane:methanol=100/0 to 96/4) to afford Intermediate 5 (Int 5). LCMS [M+1] = 485.3, 487.2; 1H NMR (400 MHz, DMSO-d6) δ 13.88 (br s, 1H), 8.91 (br t, J = 5.4 Hz, 1H), 8.45 (s, 1H), 8.16 (s, 1H), 7.43 - 7.27 (m, 2H), 4.56 (br d, J = 5.6 Hz, 2H), 2.45 (s, 3H), 2.19 (s, 3H), 1.53 (s, 6H). Example 6: Synthesis of Compound 6
– A mixture of 1,3-dibromo-2,4-dimethyl-benzene (1.00 g, 3.79 mmol, 1.0 equiv), methyl 1H-pyrazole-4-carboxylate (573 mg, 4.5 mmol, 1.2 equiv), (1R,2R)-N1,N2-dimethylcyclohexane- 1,2-diamine (216 mg, 1.5 mmol, 0.4 equiv), iodocopper tetrabutylammonium diiodide (848.3 mg, 0.76 mmol, 0.2 equiv) and cesium carbonate (2.5 g, 7.5 mmol, 2.0 equiv) in dioxane (10.0 mL) was three-fold degassed and purged with argon. The mixture was stirred at 120°C for 16 h under argon. The reaction was cooled to 20°C and partitioned between ethyl acetate (120 mL) and water (120 mL). The organic phase was separated, washed with brine (2 x 100 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether/ ethyl acetate=100:0 to 96:4) to afford 5a. LCMS [M+1] = 309.1, 311.1; 1H NMR (400 MHz, CHLOROFORM-d) δ 8.11 (s, 1H), 8.04 (s, 1H), 7.20 (app s, 2H), 3.88 (s, 3H), 2.50 (s, 3H), 2.25 (s, 3H). Step B – Synthesis of 1-(3-bromo-2,4-dimethyl-phenyl)pyrazole-4-carboxylic acid (5b)
To a solution of 5a (1.0 g, 3.2 mmol, 1.0 equiv) in methanol (4.0 mL), tetrahydrofuran (4.0 mL) and water (2.0 mL) was added LiOH•H2O (407.2 mg, 9.7 mmol, 3.0 equiv). The mixture was stirred at 25°C for 16 h. The reaction mixture was concentrated under reduced pressure. The residue was diluted with water (10 mL) and pH adjusted to 3 with 1 M aqueous HCl. The solids were collected by vacuum filtration, the filter cake was dried under reduced pressure to afford 5b. LCMS [M+1] = 295.1, 297.1; 1H NMR (400 MHz, CHLOROFORM-d) δ 8.18 (s, 1H), 8.11 (s, 1H), 7.22 (app s, 2H), 2.51 (s, 3H), 2.27 (s, 3H). Step C – Synthesis of 1-(3-bromo-2,4-dimethyl-phenyl)-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Intermediate 5) To a solution of 5b (720 mg, 2.4 mmol, 1.0 equiv) in DMF (6.5 mL) was added DIEA (946 mg, 7.3 mmol, 1.3 mL, 3.0 equiv), HATU (928 mg, 2.4 mmol, 1.0 equiv) and Int 1 (916.8 mg, 3.1 mmol, 1.3 equiv). The mixture was stirred at 25°C for 2 h. The reaction mixture was partitioned between ethyl acetate (300 mL) and water (300 mL). The organic phase was separated, washed with brine (2 x 50 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, dichloromethane:methanol=100/0 to 96/4) to afford Intermediate 5 (Int 5). LCMS [M+1] = 485.3, 487.2; 1H NMR (400 MHz, DMSO-d6) δ 13.88 (br s, 1H), 8.91 (br t, J = 5.4 Hz, 1H), 8.45 (s, 1H), 8.16 (s, 1H), 7.43 - 7.27 (m, 2H), 4.56 (br d, J = 5.6 Hz, 2H), 2.45 (s, 3H), 2.19 (s, 3H), 1.53 (s, 6H). Example 6: Synthesis of Compound 6
Step A – Synthesis of tert-butyl (R)-4-(4-(5-chloro-2-(((3-(1,1,1-trifluoro-2-methylpropan-2-yl)- 1H-1,2,4-triazol-5-yl)methyl)carbamoyl)-2,3-dihydrobenzofuran-6-yl)benzyl)piperazine-1- carboxylate (6a) To a solution of Intermediate 2 (200.0 mg, 388.6 μmol, 1 equiv), [4-[(4-tert- butoxycarbonylpiperazin-1-yl)methyl]phenyl]boronic acid (186.6 mg, 582.9 μmol, 1.5 equiv), K3PO4 (247.4 mg, 1.17 mmol, 3.0 equiv) in tetrahydrofuran (4.0 mL) and water (1.0 mL) was three-fold degassed and purged with nitrogen gas and treated with Pd(dppf)Cl2 (43 mg, 0.06 mmol, 0.15 equiv). The mixture was stirred at 80°C for 16 h under a nitrogen atmosphere. The reaction mixture was cooled to ambient temperature and partitioned between ethyl acetate (10 mL) and water (10 mL). The organic phase was separated, washed with brine (5 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated. The residue was purified by preparative TLC (silica gel, dichloromethane: methanol = 10:1). The enantiomers were separated with SFC (CHIRALCEL-OJ(250 mm x 30mm,10µm, CO2-methanol(0.1%NH3H2O)], gradient:10%-30% over 11 min) to afford 6a as the early eluting enantiomer. LCMS [M+1] = 663.2. Step B – Synthesis of (R)-5-chloro-6-(4-(piperazin-1-ylmethyl)phenyl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-2,3-dihydrobenzofuran-2-carboxamide (6b) To a solution of 6a (30.0 mg, 45.24 μmol, 1.0 equiv) in dichloromethane (0.5 mL) was added TFA (45 μmol, 3 μL, 1.0 equiv). The mixture was stirred at 25°C for 2 h and then concentrated to afford 6b that was used without further purification. LCMS [M+1] + = 563.3. Step C – Synthesis of (2R)-5-chloro-6-(4-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3- dioxoisoindolin-5-yl)piperazin-1-yl)methyl)phenyl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)- 1H-1,2,4-triazol-5-yl)methyl)-2,3-dihydrobenzofuran-2-carboxamide (Compound 6) To a solution of 6b (30.0 mg, 44.3 μmol, 1.0 equiv) and 2-(2,6-dioxo-3-piperidyl)-5,6- difluoro-isoindoline-1,3-dione (13 mg, 44 μmol, 1.0 equiv) in DMSO (0.5 mL) was added DIEA (17 mg, 133 μmol, 23 μL, 3.0 equiv). The mixture was stirred at 70°C for 16 h. The reaction mixture was partitioned between ethyl acetate (30.0 mL) and water (30.0 mL). The organic phase was separated, washed with brine (30.0 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative
TLC (silica gel, dichloromethane: methanol = 10:1) to afford Compound 6. LCMS [M+1] = 837.3; 1H NMR (400 MHz, CHLOROFORM-d) δ 8.00 (br s, 1H), 7.40 (d, J = 11.0 Hz, 1H), 7.36 - 7.24 (m, 6H), 7.22 (s, 1H), 6.78 (s, 1H), 5.22 - 5.12 (m, 1H), 4.86 (dd, J = 5.4, 12.2 Hz, 1H), 4.59 - 4.43 (m, 2H), 3.63 - 3.54 (m, 3H), 3.39 (dd, J = 6.6, 16.8 Hz, 1H), 3.28 - 3.22 (m, 4H), 2.88 - 2.65 (m, 3H), 2.63 - 2.59 (m, 4H), 2.07 (br dd, J = 4.8, 7.6 Hz, 1H), 1.94 (s, 1H), 1.53 (s, 6H). Example 7: Synthesis of Compound 7
 2 and [4-(4-tert-butoxycarbonylpiperazine-1-carbonyl)phenyl]boronic acid. The intermediate corresponding to 6a was resolved by SFC (DAICEL CHIRALPAK IG (250mm x 30mm,10 µm); [CO2-EtOH(0.1% NH3H2O)]; 45% isocratic elution) and the early eluting enantiomer was carried forward to afford (2R)-5-chloro-6-(4-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3- dioxoisoindolin-5-yl)piperazine-1-carbonyl)phenyl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)- 1H-1,2,4-triazol-5-yl)methyl)-2,3-dihydrobenzofuran-2-carboxamide (Compound 7). LCMS: [M+1]+ = 851.3; 1H NMR (400 MHz, DMSO-d6) δ = 14.00 - 13.61 (m, 1H), 11.10 (d, J = 2 Hz, 1H), 8.93 - 8.69 (m, 1H), 7.78 (d, J = 11.2 Hz, 1H), 7.62 - 7.36 (m, 6H), 6.89 (s, 1H), 5.33 - 5.32 (m, 1H), 5.31 (dd, J = 6.4, 10.4 Hz, 1H), 5.19 - 5.01 (m, 1H), 4.56 - 4.41 (m, 1H), 4.39 - 4.28 (m, 1H), 3.90 - 3.71 (m, 1H), 3.68 - 3.49 (m, 3H), 3.37 (s, 1H), 2.95 - 2.81 (m, 1H), 2.71 - 2.56 (m, 2H), 2.54 (s, 3H), 2.09 - 1.96 (m, 1H), 2.11 - 1.95 (m, 1H), 1.52 (s, 6H). Example 8: Synthesis of Compound 8
2 and [4-(4-tert-butoxycarbonylpiperazine-1-carbonyl)phenyl]boronic acid. The intermediate corresponding to 6a was resolved by SFC (DAICEL CHIRALPAK IG (250mm x 30mm,10 µm); [CO2-EtOH(0.1% NH3H2O)]; 45% isocratic elution) and the early eluting enantiomer was carried forward to afford (2R)-5-chloro-6-(4-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3- dioxoisoindolin-5-yl)piperazine-1-carbonyl)phenyl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)- 1H-1,2,4-triazol-5-yl)methyl)-2,3-dihydrobenzofuran-2-carboxamide (Compound 7). LCMS: [M+1]+ = 851.3; 1H NMR (400 MHz, DMSO-d6) δ = 14.00 - 13.61 (m, 1H), 11.10 (d, J = 2 Hz, 1H), 8.93 - 8.69 (m, 1H), 7.78 (d, J = 11.2 Hz, 1H), 7.62 - 7.36 (m, 6H), 6.89 (s, 1H), 5.33 - 5.32 (m, 1H), 5.31 (dd, J = 6.4, 10.4 Hz, 1H), 5.19 - 5.01 (m, 1H), 4.56 - 4.41 (m, 1H), 4.39 - 4.28 (m, 1H), 3.90 - 3.71 (m, 1H), 3.68 - 3.49 (m, 3H), 3.37 (s, 1H), 2.95 - 2.81 (m, 1H), 2.71 - 2.56 (m, 2H), 2.54 (s, 3H), 2.09 - 1.96 (m, 1H), 2.11 - 1.95 (m, 1H), 1.52 (s, 6H). Example 8: Synthesis of Compound 8
carboxylate (8a) To a mixture of 5-bromo-2-fluoro-pyridine (619.4 mg, 3.5 mmol, 362.2 μL, 1.05 equiv) and tert-butyl 4-(4-piperidylmethyl)piperazine-1-carboxylate (0.95 g, 3.3 mmol, 1.0 equiv) in DMF (10.0 mL) was added K2CO3 (926.5 mg, 6.7 mmol, 2.0 equiv) in one portion at 20°C under nitrogen The mixture was stirred at 90°C for 12 h. The reaction mixture was cooled to 20°C and the residue was poured into water (10 mL). The aqueous phase was extracted with ethyl acetate (20 mL). The combined organic phase was washed with brine (20 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether/ethyl acetate 75/25 to 1/1) to afford 8a. LCMS [M+1] = 439.1; 1H NMR (400 MHz, CHLOROFORM-d) δ 8.17 (d, J = 2.4 Hz, 1H), 7.49 (dd, J = 2.4, 8.8 Hz, 1H), 6.55 (d, J = 9.2 Hz, 1H), 4.22 (br d, J = 13.2 Hz, 2H), 3.48 - 3.39 (m, 4H), 2.81 (dt, J = 2.4, 12.6 Hz, 2H), 2.42 - 2.29 (m, 4H), 2.20 (d, J = 7.2 Hz, 2H), 1.84 (br d, J = 13.2 Hz, 2H), 1.79 - 1.68 (m, 1H), 1.47 (s, 9H), 1.32 - 1.13 (m, 2H). Step B – Synthesis of [6-[4-[(4-tert-butoxycarbonylpiperazin-1-yl)methyl]-1-piperidyl]-3- pyridyl]boronic acid (8b)
To a mixture of 8a (640 mg, 1.4 mmol, 1.0 equiv) and 4,4,5,5-tetramethyl-2-(4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3,2-dioxaborolane (1.11 g, 4.3 mmol, 3.0 equiv) in dioxane (10.0 mL) was added KOAc (714.7 mg, 7.2 mmol, 5.0 eq) Pd(PPh3)4 (168.3 mg, 145.6 μmol, 0.1 eq) and in one portion at 20°C under nitrogen. The mixture was stirred at 80°C for 12 h, cooled to rt, filtered and the filtrate was concentrated under reduced pressure. The crude product was triturated with ethyl acetate to afford 8b. LCMS: [M+1] = 405.4. Step C – Synthesis of tert-butyl (R)-4-((1-(5-(5-chloro-2-(((3-(1,1,1-trifluoro-2-methylpropan-2- yl)-1H-1,2,4-triazol-5-yl)methyl)carbamoyl)-2,3-dihydrobenzofuran-6-yl)pyridin-2-yl)piperidin- 4-yl)methyl)piperazine-1-carboxylate (8c) To a solution of 8b (300 mg, 742 μmol, 1.5 equiv), Intermediate 2 (254.6 mg, 495 μmol, 1.0 equiv), Pd(dppf)Cl2 (54.3 mg, 74.2 μmol, 0.15 equiv), K3PO4 (315.01 mg, 1.48 mmol, 3.0 equiv) in tetrahydrofuran (4.0 mL) and water (1.0 mL) was three-fold degassed and purged with nitrogen, and then the mixture was stirred at 80°C for 12 h under a nitrogen atmosphere. The reaction mixture was partitioned between ethyl acetate (40 mL) and water (40 mL). The organic phase was separated, washed with brine (2 x 15 ml), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure to give a residue. The residue was purified by column chromatography (silica gel, dichloromethane/methanol=1/0 to 96/4) and then the residue was purified by preparative TLC (silica gel, dichloromethane: methanol = 10:1). The enantiomers were resolved by SFC (DAICEL CHIRALPAK IG (250 mm x 30mm, 10µm); [CO2- IPA(0.1%NH3H2O)]; 40% isocratic elution) to afford the early eluting isomer 8c. LCMS [M+1] = 747.3. Step D and Step E were performed in a similar manner to Example 6 to afford (2R)-5-chloro-6- (6-(4-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1- yl)methyl)piperidin-1-yl)pyridin-3-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)-2,3-dihydrobenzofuran-2-carboxamide (Compound 8). LCMS [M+1] = 920.9; 1H NMR (400 MHz, CHLOROFORM-d) δ 11.87 - 11.33 (m, 1H), 8.27 - 8.18 (m, 2H), 7.57 (dd, J = 2.4, 8.9 Hz, 1H), 7.48 (d, J = 11.1 Hz, 1H), 7.44 - 7.44 (m, 1H), 7.44 - 7.38 (m, 1H), 7.29 - 7.27 (m, 1H), 6.81 (s, 1H), 6.72 (d, J = 8.9 Hz, 1H), 5.25 (dd, J = 6.4, 10.7 Hz, 1H), 4.95 (dd, J = 5.3, 12.3 Hz, 1H), 4.70 - 4.49 (m, 2H), 4.38 (br d, J = 12.8 Hz, 2H), 3.64 (dd, J = 11.1, 16.4 Hz, 1H), 3.45 (br dd, J = 6.2, 16.7 Hz, 1H), 3.33 - 3.28 (m, 4H), 2.94 - 2.72 (m, 5H), 2.63 (br
d, J = 1.1 Hz, 4H), 2.31 (br d, J = 6.8 Hz, 2H), 2.23 - 2.06 (m, 1H), 1.91 (br d, J = 12.1 Hz, 2H), 1.86 - 1.67 (m, 3H), 1.60 (s, 6H). Example 9: Synthesis of Compound 9
 Step – carboxylate (9a) To a mixture of tert-butyl 4-(piperidin-4-ylmethyl)piperazine-1-carboxylate (500 mg, 1.7 mmol, 1.0 equiv) and DIEA (250.8 mg, 1.9 mmol, 338 μL, 1.1 equiv) in dichloromethane (5 mL) was added 2-bromoacetyl bromide (356 mg, 1.7 mmol, 154 μL, 1.0 equiv) dropwise at 0°C. The reaction mixture was agitated at 25ºC for 1 h. The residue was poured into water (10 mL). The
aqueous phase was extracted with dichloromethane (3 x 10 mL). The combined organic phase was washed with brine (10 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated in vacuum to afford 9a, which was used without further purification. LCMS: [M+1] = 404.1, 406.1; 1H NMR (400 MHz, Methanol-d4) δ 4.46 (d, J = 12.0 Hz, 1H), 4.15 - 3.88 (m, 3H), 3.42 (s, 4H), 3.21 - 3.09 (m, 1H), 2.71 (dt, J = 2.6, 12.8 Hz, 1H), 2.38 (t, J = 4.8 Hz, 4H), 2.23 (d, J = 6.6 Hz, 2H), 1.93 - 1.79(m, 3H), 1.45 (s, 9H), 1.29 (s, 1H), 1.14 - 1.01 (m, 1H). Step B – Synthesis of tert-butyl 4-((1-(2-(4-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)phenyl)-1H-pyrazol-1-yl)acetyl)piperidin-4-yl)methyl)piperazine-1-carboxylate (9b) To a mixture of 9a (160.0 mg, 395.7 μmol, 1.0 equiv) and 4-(4-(4,4,5,5-tetramethyl-1,3,2- dioxaborolan-2-yl)phenyl)-1H-pyrazole (106.9 mg, 395.7 μmol, 1.0 equiv) in DMF (2.0 mL) was added K2CO3 (164.1 mg, 1.2 mmol, 3.0 equiv) in one portion at 20°C under nitrogen. The mixture was stirred at 20°C for 16 h. The reaction mixture was poured into water (10 mL). The aqueous phase was extracted with ethyl acetate (3 x 10 mL). The combined organic phase was washed with brine (3 x 10 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, ethyl acetate/ methanol=0/1 to 0/1 to give 9b. LCMS: [M+1] + = 594.3; 1H NMR (400 MHz, Methanol-d4) δ 8.02 (s, 1H), 7.90 (s, 1H), 7.73 (d, J = 7.6 Hz, 1H), 7.57 (d, J = 7.4 Hz, 2H), 5.26 - 5.07 (m, 2H), 4.47 (d, J = 14.0 Hz, 1H), 4.03 - 3.95 (m, 1H), 3.42 (s, 4H), 3.22 - 3.11 (m, 1H), 2.79 - 2.67 (m, 1H), 2.38 (s, 3H), 2.23 (d, J = 5.8 Hz, 2H), 1.94 - 1.78 (m, 4H), 1.46 (s, 9H), 1.38 - 1.27 (m, 12H), 1.27 - 1.17 (m, 2H), 1.15 - 1.04 (m, 1H). Step C – Synthesis of tert-butyl 4-((1-(2-(4-(4'-((1S,2S)-2-(((3-(1,1,1-trifluoro-2-methylpropan-2- yl)-1H-1,2,4-triazol-5-yl)methyl)carbamoyl)cyclopropyl)-[1,1'-biphenyl]-4-yl)-1H-pyrazol-1- yl)acetyl)piperidin-4-yl)methyl)piperazine-1-carboxylate (9c) To a mixture of 9b (140.0 mg, 235.8 μmol, 1.2 equiv) and Intermediate 3 (84.7 mg, 196.5 μmol, 1.0 equiv) in dioxane (1.5 mL) and water (0.2 mL) was added K2CO3 (81.5 mg, 589.6 μmol, 3.0 equiv) and Pd(dppf)Cl2•CH2Cl2 (80.3 mg, 98.3 μmol, 0.5 equiv) in one portion at 20°C under nitrogen. The mixture was stirred at 100°C for 2 h. The mixture was poured into water (10 mL). The aqueous phase was extracted with ethyl acetate (3 x 10 mL). The combined organic phase was washed with brine (10 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated in vacuum. The residue was purified by column chromatography
(silica gel, ethyl acetate /methanol =0/1 to 0/1) to give 9c. LCMS: [M+1] = 818.4; 1H NMR (400 MHz, Methanol-d4) δ 8.00 (s, 1H), 7.89 (s, 1H), 7.67 - 7.51 (m, 6H), 7.21 (d, J = 8.0 Hz, 2H), 5.25 - 5.08 (m, 2H), 4.62 - 4.42 (m, 3H), 4.00 (d, J = 13.2 Hz, 1H), 3.43 (s, 4H), 3.17 (t, J = 12.8 Hz, 1H), 2.73 (t, J = 12.2 Hz, 1H), 2.55 - 2.33 (m, 5H), 2.25 (d, J = 6.4 Hz, 2H), 1.98 (s, 1H), 1.94 - 1.78 (m, 3H), 1.60 (s, 5H), 1.46 (s, 9H), 1.38 - 1.28 (m, 2H), 1.12 (s, 2H), 0.99 - 0.84 (m, 1H). Step D – Synthesis of (1S,2S)-2-(4'-(1-(2-oxo-2-(4-(piperazin-1-ylmethyl)piperidin-1-yl)ethyl)-1H- pyrazol-4-yl)-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol- 5-yl)methyl)cyclopropane-1-carboxamide (9d) To a mixture of 9c (150.0 mg, 183.4 μmol, 1.0 equiv) in HCl/ dioxane (5.0 mL, 4 M) was stirred at 20°C under nitrogen for 1 h. The reaction mixture was concentrated under reduced pressure to afford 9d which was used into next step without further purification. LCMS: [M+1] + = 718.3; 1H NMR (400 MHz, Methanol-d4) δ 8.11 - 8.01 (m, 1H), 7.95 (dt, J = 2.8, 5.5 Hz, 1H), 7.67 - 7.54 (m, 6H), 7.32 - 7.13 (m, 2H), 5.22 - 5.19 (m, 2H), 4.66 - 4.61 (m, 3H), 4.10 - 4.05 (m, 1H), 3.66 (s, 4H), 3.20 (d, J = 4.0 Hz, 1H), 2.82 - 2.79 (m, 1H), 2.65 (d, J = 9.8 Hz, 5H), 2.53 - 2.44 (m, 2H), 2.29 - 2.21 (m, 1H), 2.06 - 1.95 (m, 3H), 1.69 - 1.59 (m, 6H), 1.40 - 1.26 (m, 5H). Step E – Synthesis of (1S,2S)-2-(4'-(1-(2-(4-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3- dioxoisoindolin-5-yl)piperazin-1-yl)methyl)piperidin-1-yl)-2-oxoethyl)-1H-pyrazol-4-yl)-[1,1'- biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5- yl)methyl)cyclopropane-1-carboxamide (Compound 9) To a mixture of 9d (130.0 mg, 181.1 μmol, 1.0 equiv) in DMSO (3.0 mL) was added DIEA (70.2 mg, 543.3 μmol, 94.6 μL, 3.0 equiv) and 2-(2,6-dioxopiperidin-3-yl)-5,6- difluoroisoindoline-1,3-dione (79.9 mg, 271.6 μmol, 1.5 equiv) in one portion at 20°C under nitrogen. The mixture was stirred at 80°C for 16 h. The reaction mixture was concentrated under reduced pressure. The residue was purified by preparative HPLC (Phenomenex C18150mm x 25mm, 10µm; mobile phase: [water( NH4HCO3)-acetonitrile]) to give Compound 9. LCMS: [M+1] = 992.4; 1H NMR (400 MHz, Methanol-d4) δ 8.03 (s, 1H), 7.93 (s, 1H), 7.67 - 7.58 (m, 6H), 7.56 (d, J = 8.4 Hz, 2H), 7.22 (d, J = 8.2 Hz, 2H), 5.29 - 5.09 (m, 3H), 4.57 (d, J = 2.6 Hz, 3H), 4.10 (d, J = 14.8 Hz, 1H), 3.88 - 3.72 (m, 4H), 3.38 (d, J = 9.0 Hz, 4H), 3.26 (d, J = 12.4 Hz,
1H), 3.21 (d, J = 6.8 Hz, 2H), 2.93 - 2.65 (m, 4H), 2.51 - 2.44 (m, 1H), 2.34 - 2.22 (m, 1H), 2.17 - 2.08 (m, 1H), 2.03 - 1.88 (m, 3H), 1.61 (s, 6H), 1.60 - 1.54 (m, 1H), 1.50 - 1.23 (m, 4H). Example 10: Synthesis of Compound 10
Step – carboxylate (9a) To a mixture of tert-butyl 4-(piperidin-4-ylmethyl)piperazine-1-carboxylate (500 mg, 1.7 mmol, 1.0 equiv) and DIEA (250.8 mg, 1.9 mmol, 338 μL, 1.1 equiv) in dichloromethane (5 mL) was added 2-bromoacetyl bromide (356 mg, 1.7 mmol, 154 μL, 1.0 equiv) dropwise at 0°C. The reaction mixture was agitated at 25ºC for 1 h. The residue was poured into water (10 mL). The
aqueous phase was extracted with dichloromethane (3 x 10 mL). The combined organic phase was washed with brine (10 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated in vacuum to afford 9a, which was used without further purification. LCMS: [M+1] = 404.1, 406.1; 1H NMR (400 MHz, Methanol-d4) δ 4.46 (d, J = 12.0 Hz, 1H), 4.15 - 3.88 (m, 3H), 3.42 (s, 4H), 3.21 - 3.09 (m, 1H), 2.71 (dt, J = 2.6, 12.8 Hz, 1H), 2.38 (t, J = 4.8 Hz, 4H), 2.23 (d, J = 6.6 Hz, 2H), 1.93 - 1.79(m, 3H), 1.45 (s, 9H), 1.29 (s, 1H), 1.14 - 1.01 (m, 1H). Step B – Synthesis of tert-butyl 4-((1-(2-(4-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)phenyl)-1H-pyrazol-1-yl)acetyl)piperidin-4-yl)methyl)piperazine-1-carboxylate (9b) To a mixture of 9a (160.0 mg, 395.7 μmol, 1.0 equiv) and 4-(4-(4,4,5,5-tetramethyl-1,3,2- dioxaborolan-2-yl)phenyl)-1H-pyrazole (106.9 mg, 395.7 μmol, 1.0 equiv) in DMF (2.0 mL) was added K2CO3 (164.1 mg, 1.2 mmol, 3.0 equiv) in one portion at 20°C under nitrogen. The mixture was stirred at 20°C for 16 h. The reaction mixture was poured into water (10 mL). The aqueous phase was extracted with ethyl acetate (3 x 10 mL). The combined organic phase was washed with brine (3 x 10 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, ethyl acetate/ methanol=0/1 to 0/1 to give 9b. LCMS: [M+1] + = 594.3; 1H NMR (400 MHz, Methanol-d4) δ 8.02 (s, 1H), 7.90 (s, 1H), 7.73 (d, J = 7.6 Hz, 1H), 7.57 (d, J = 7.4 Hz, 2H), 5.26 - 5.07 (m, 2H), 4.47 (d, J = 14.0 Hz, 1H), 4.03 - 3.95 (m, 1H), 3.42 (s, 4H), 3.22 - 3.11 (m, 1H), 2.79 - 2.67 (m, 1H), 2.38 (s, 3H), 2.23 (d, J = 5.8 Hz, 2H), 1.94 - 1.78 (m, 4H), 1.46 (s, 9H), 1.38 - 1.27 (m, 12H), 1.27 - 1.17 (m, 2H), 1.15 - 1.04 (m, 1H). Step C – Synthesis of tert-butyl 4-((1-(2-(4-(4'-((1S,2S)-2-(((3-(1,1,1-trifluoro-2-methylpropan-2- yl)-1H-1,2,4-triazol-5-yl)methyl)carbamoyl)cyclopropyl)-[1,1'-biphenyl]-4-yl)-1H-pyrazol-1- yl)acetyl)piperidin-4-yl)methyl)piperazine-1-carboxylate (9c) To a mixture of 9b (140.0 mg, 235.8 μmol, 1.2 equiv) and Intermediate 3 (84.7 mg, 196.5 μmol, 1.0 equiv) in dioxane (1.5 mL) and water (0.2 mL) was added K2CO3 (81.5 mg, 589.6 μmol, 3.0 equiv) and Pd(dppf)Cl2•CH2Cl2 (80.3 mg, 98.3 μmol, 0.5 equiv) in one portion at 20°C under nitrogen. The mixture was stirred at 100°C for 2 h. The mixture was poured into water (10 mL). The aqueous phase was extracted with ethyl acetate (3 x 10 mL). The combined organic phase was washed with brine (10 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated in vacuum. The residue was purified by column chromatography
(silica gel, ethyl acetate /methanol =0/1 to 0/1) to give 9c. LCMS: [M+1] = 818.4; 1H NMR (400 MHz, Methanol-d4) δ 8.00 (s, 1H), 7.89 (s, 1H), 7.67 - 7.51 (m, 6H), 7.21 (d, J = 8.0 Hz, 2H), 5.25 - 5.08 (m, 2H), 4.62 - 4.42 (m, 3H), 4.00 (d, J = 13.2 Hz, 1H), 3.43 (s, 4H), 3.17 (t, J = 12.8 Hz, 1H), 2.73 (t, J = 12.2 Hz, 1H), 2.55 - 2.33 (m, 5H), 2.25 (d, J = 6.4 Hz, 2H), 1.98 (s, 1H), 1.94 - 1.78 (m, 3H), 1.60 (s, 5H), 1.46 (s, 9H), 1.38 - 1.28 (m, 2H), 1.12 (s, 2H), 0.99 - 0.84 (m, 1H). Step D – Synthesis of (1S,2S)-2-(4'-(1-(2-oxo-2-(4-(piperazin-1-ylmethyl)piperidin-1-yl)ethyl)-1H- pyrazol-4-yl)-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol- 5-yl)methyl)cyclopropane-1-carboxamide (9d) To a mixture of 9c (150.0 mg, 183.4 μmol, 1.0 equiv) in HCl/ dioxane (5.0 mL, 4 M) was stirred at 20°C under nitrogen for 1 h. The reaction mixture was concentrated under reduced pressure to afford 9d which was used into next step without further purification. LCMS: [M+1] + = 718.3; 1H NMR (400 MHz, Methanol-d4) δ 8.11 - 8.01 (m, 1H), 7.95 (dt, J = 2.8, 5.5 Hz, 1H), 7.67 - 7.54 (m, 6H), 7.32 - 7.13 (m, 2H), 5.22 - 5.19 (m, 2H), 4.66 - 4.61 (m, 3H), 4.10 - 4.05 (m, 1H), 3.66 (s, 4H), 3.20 (d, J = 4.0 Hz, 1H), 2.82 - 2.79 (m, 1H), 2.65 (d, J = 9.8 Hz, 5H), 2.53 - 2.44 (m, 2H), 2.29 - 2.21 (m, 1H), 2.06 - 1.95 (m, 3H), 1.69 - 1.59 (m, 6H), 1.40 - 1.26 (m, 5H). Step E – Synthesis of (1S,2S)-2-(4'-(1-(2-(4-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3- dioxoisoindolin-5-yl)piperazin-1-yl)methyl)piperidin-1-yl)-2-oxoethyl)-1H-pyrazol-4-yl)-[1,1'- biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5- yl)methyl)cyclopropane-1-carboxamide (Compound 9) To a mixture of 9d (130.0 mg, 181.1 μmol, 1.0 equiv) in DMSO (3.0 mL) was added DIEA (70.2 mg, 543.3 μmol, 94.6 μL, 3.0 equiv) and 2-(2,6-dioxopiperidin-3-yl)-5,6- difluoroisoindoline-1,3-dione (79.9 mg, 271.6 μmol, 1.5 equiv) in one portion at 20°C under nitrogen. The mixture was stirred at 80°C for 16 h. The reaction mixture was concentrated under reduced pressure. The residue was purified by preparative HPLC (Phenomenex C18150mm x 25mm, 10µm; mobile phase: [water( NH4HCO3)-acetonitrile]) to give Compound 9. LCMS: [M+1] = 992.4; 1H NMR (400 MHz, Methanol-d4) δ 8.03 (s, 1H), 7.93 (s, 1H), 7.67 - 7.58 (m, 6H), 7.56 (d, J = 8.4 Hz, 2H), 7.22 (d, J = 8.2 Hz, 2H), 5.29 - 5.09 (m, 3H), 4.57 (d, J = 2.6 Hz, 3H), 4.10 (d, J = 14.8 Hz, 1H), 3.88 - 3.72 (m, 4H), 3.38 (d, J = 9.0 Hz, 4H), 3.26 (d, J = 12.4 Hz,
1H), 3.21 (d, J = 6.8 Hz, 2H), 2.93 - 2.65 (m, 4H), 2.51 - 2.44 (m, 1H), 2.34 - 2.22 (m, 1H), 2.17 - 2.08 (m, 1H), 2.03 - 1.88 (m, 3H), 1.61 (s, 6H), 1.60 - 1.54 (m, 1H), 1.50 - 1.23 (m, 4H). Example 10: Synthesis of Compound 10
 Compound 10 was prepared in a similar manner to Example 9. Final purification by preparative HPLC (Phenomenex Luna C18, 80mm x 30mm, 3 µm; H2O (0.04% HCl)-acetonitrile) afforded (1S,2S)-2-[4-[4-[1-[2-[4-[[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]piperazin-1-yl]methyl]-1-piperidyl]-2-oxo-ethyl]pyrazol-3-yl]phenyl]phenyl]-N-[[3-(2,2,2- trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]cyclopropanecarboxamide hydrochloride (Compound 10). LCMS: [M+1]+ = 992.5; 1H NMR (400MHz, DMSO-d6) δ 11.12 (s, 1H), 8.76 (s, 1H), 7.88 - 7.81 (m, 3H), 7.72 - 7.67 (m, 3H), 7.65 - 7.59 (m, 3H), 7.23 (d, J = 8.4 Hz, 2H), 6.77 (d, J = 2.4 Hz, 1H), 5.27 - 5.09 (m, 3H), 4.41 (d, J = 5.6 Hz, 2H), 4.37 - 4.30 (m, 1H), 4.02 - 3.95 (m, 1H), 3.79 (d, J = 12.8 Hz, 2H), 3.64 (d, J = 12.4 Hz, 2H), 3.26 - 3.19 (m, 2H), 3.14 - 3.08 (m, 2H), 2.93 - 2.84 (m, 1H), 2.71 - 2.55 (m, 4H), 2.35 - 2.30 (m, 1H), 2.15 (s, 1H), 2.07 - 1.98 (m, 3H), 1.85 (t, J =12.4 Hz, 2H), 1.53 (s, 6H), 1.45 - 1.38 (m, 1H), 1.33 - 1.21 (m, 3H), 1.16 - 1.06 (m, 1H). Example 11: Synthesis of Compound 11
Compound 10 was prepared in a similar manner to Example 9. Final purification by preparative HPLC (Phenomenex Luna C18, 80mm x 30mm, 3 µm; H2O (0.04% HCl)-acetonitrile) afforded (1S,2S)-2-[4-[4-[1-[2-[4-[[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]piperazin-1-yl]methyl]-1-piperidyl]-2-oxo-ethyl]pyrazol-3-yl]phenyl]phenyl]-N-[[3-(2,2,2- trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]cyclopropanecarboxamide hydrochloride (Compound 10). LCMS: [M+1]+ = 992.5; 1H NMR (400MHz, DMSO-d6) δ 11.12 (s, 1H), 8.76 (s, 1H), 7.88 - 7.81 (m, 3H), 7.72 - 7.67 (m, 3H), 7.65 - 7.59 (m, 3H), 7.23 (d, J = 8.4 Hz, 2H), 6.77 (d, J = 2.4 Hz, 1H), 5.27 - 5.09 (m, 3H), 4.41 (d, J = 5.6 Hz, 2H), 4.37 - 4.30 (m, 1H), 4.02 - 3.95 (m, 1H), 3.79 (d, J = 12.8 Hz, 2H), 3.64 (d, J = 12.4 Hz, 2H), 3.26 - 3.19 (m, 2H), 3.14 - 3.08 (m, 2H), 2.93 - 2.84 (m, 1H), 2.71 - 2.55 (m, 4H), 2.35 - 2.30 (m, 1H), 2.15 (s, 1H), 2.07 - 1.98 (m, 3H), 1.85 (t, J =12.4 Hz, 2H), 1.53 (s, 6H), 1.45 - 1.38 (m, 1H), 1.33 - 1.21 (m, 3H), 1.16 - 1.06 (m, 1H). Example 11: Synthesis of Compound 11
Step A – Synthesis of tert-butyl 4-((1-(2-(4-iodo-1H-pyrazol-1-yl)acetyl)piperidin-4- yl)methyl)piperazine-1-carboxylate (11a) To a solution of 2-(4-iodo-1H-pyrazol-1-yl)acetic acid (1.0 g, 4.0 mmol, 1.1 equiv) and tert-butyl 4-((1-(2-bromoacetyl)piperidin-4-yl)methyl)piperazine-1-carboxylate (1.0 g, 3.6 mmol, 1.0 equiv) in DMF (10.0 mL) was added HATU (1.4 g,3.6 mmol, 1.0 equiv) and DIPEA (2.3 g, 18.0 mmol, 3.1 mL, 5.0 equiv). The mixture was stirred at 25°C for 1 h. The residue was purified by preparative HPLC (Phenomenex Luna C18; 250mm x 70mm, 10µm; water( NH4HCO3)-acetonitrile) to afford 11a. LCMS [M+1] = 518.1; 1H NMR (400 MHz, DMSO-d6) δ 7.80 (s, 1H), 7.50 (s, 1H), 5.12 (d, J = 4.2 Hz, 2H), 4.27 (d, J = 13.2 Hz, 1H), 3.85 (d, J = 13.6
Hz, 1H), 3.29 (s, 4H), 2.70 - 2.52 (m, 2H), 2.28 (t, J = 5.0 Hz, 4H), 2.13 (d, J = 7.2 Hz, 2H), 1.81 - 1.67 (m, 3H), 1.39 (s, 9H), 1.15 - 0.87 (m, 2H). Step B – Synthesis of tert-butyl 4-((1-(2-(4-(4-bromo-2-chlorophenyl)-1H-pyrazol-1- yl)acetyl)piperidin-4-yl)methyl)piperazine-1-carboxylate (11b) A mixture of 11a (500.0 mg, 918.1 μmol, 1.0 equiv), (4-bromo-2-chlorophenyl)boronic acid (216.0 mg, 918.1 μmol, 1.0 equiv), Pd(dppf)Cl2.CH2Cl2 (75.0 mg, 91.8 μmol, 0.1 equiv) and K2CO3 (380.6 mg, 2.8 mmol, 3.0 equiv) in water (1.0 mL) and dioxane (5.0 mL) was three-fold degassed and purged with nitrogen. The mixture was stirred at 100°C for 1 h under nitrogen atmosphere. The aqueous phase was extracted with ethyl acetate (3 x 50 mL). The combined organic phase was washed with brine (3x50 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated in vacuum to give residue. The residue was purified by column chromatography (silica gel, petroleum ether/ethyl acetate=10:1 to 0:1) to afford 11b. LCMS [M+1] = 580.0, 582.0; 1H NMR (400 MHz, DMSO-d6) δ 8.18 (s, 1H), 7.89 (s, 1H), 7.80 - 7.74 (m, 1H), 7.58 - 7.55 (m, 2H), 5.17 (d, J = 5.9 Hz, 2H), 3.30 (s, 4H), 2.70 - 2.56 (m, 4H), 2.28 (br t, J = 4.4 Hz, 4H), 2.13 (br d, J = 7.0 Hz, 3H), 1.81 - 1.65 (m, 4H), 1.39 (s, 9H). Step C – Synthesis of (4-(1-(2-(4-((4-(tert-butoxycarbonyl)piperazin-1-yl)methyl)piperidin-1-yl)- 2-oxoethyl)-1H-pyrazol-4-yl)-3-chlorophenyl)boronic acid (11c) A mixture of 11b (400.0 mg, 688.5 μmol, 1.0 equiv), 4,4,4',4',5,5,5',5'-octamethyl-2,2'- bi(1,3,2-dioxaborolane) (262.3 mg, 1.0 mmol, 1.5 equiv), potassium acetate (202.7 mg, 2.1 mmol, 3.0 equiv) and Pd(dppf)Cl2 (50.4 mg, 68.9 μmol, 0.1 equiv) in dioxane (4.0 mL) was three-fold degassed and purged with nitrogen. The mixture was stirred at 100°C for 6 h under nitrogen atmosphere, cooled to ambient temperature and poured into water (50 mL). The aqueous phase was extracted with ethyl acetate (3 x 50 mL). The combined organic phase was washed with brine (3 x 50 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative HPLC (Phenomenex Luna C18 150mm x 25mm, 10µm; water(formic acid)-acetonitrile];gradient:23%-53% B over 10 min) to give 11c. LCMS [M+1] = 546.3, 548.3; 1H NMR (400 MHz, DMSO-d6) δ 8.23 (s, 1H), 8.20 - 8.13 (m, 1H), 7.92 (d, J = 9.6 Hz, 1H), 7.69 - 7.65 (m, 1H), 7.63 - 7.56 (m, 1H), 5.17 (dd, J = 2.5, 5.6 Hz, 2H), 4.34 - 4.25 (m, 1H), 3.96 - 3.84 (m, 1H), 3.05 (br t, J = 11.9 Hz, 2H), 2.52 (br s, 6H), 2.28 (br t, J = 4.8 Hz, 4H), 2.14 (br d, J = 7.0 Hz, 2H), 1.89 - 1.64 (m, 4H), 1.39 (s, 10H).
Step D – Synthesis of tert-butyl 4-((1-(2-(4-(3-chloro-4'-((1S,2S)-2-(((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)carbamoyl)cyclopropyl)-[1,1'-biphenyl]-4-yl)- 1H-pyrazol-1-yl)acetyl)piperidin-4-yl)methyl)piperazine-1-carboxylate (11d) A mixture of 11c (120.3 mg, 220.3 μmol, 1.0 equiv), Intermediate 3 (95.0 mg, 220.3 μmol, 1.0 equiv), K2CO3 (91.3 mg, 660.9 μmol, 3.0 equiv), Pd(dppf)Cl2•CH2Cl2 (18.0 mg, 22.0 μmol, 0.1 equiv) in dioxane (1.0 mL) and water (0.2 mL) was three-fold degassed and purged with nitrogen, and then the mixture was stirred at 100°C for 2 h. The residue was poured into water (10 mL) and extracted with ethyl acetate (3 x 130 mL). The combined organic phase was washed with brine (3 x 10 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative TLC (silica gel, ethyl acetate) to afford 11d. LCMS [M+1] = 852.4, 853.8; 1H NMR (400 MHz, Methanol-d4) δ 8.12 (s, 1H), 7.92 (s, 1H), 7.70 (d, J = 1.8 Hz, 1H), 7.66 - 7.61 (m, 1H), 7.59 - 7.53 (m, 3H), 7.24 (d, J = 8.4 Hz, 2H), 5.30 - 5.12 (m, 2H), 4.55 (d, J = 2.4 Hz, 2H), 4.51 - 4.43 (m, 1H), 4.00 (d, J = 13.4 Hz, 1H), 3.42 (s, 4H), 3.17 (s, 1H), 2.73 (t, J = 11.8 Hz, 1H), 2.53 - 2.44 (m, 1H), 2.39 ( t, J = 5.0 Hz, 4H), 2.23 (d, J = 6.6 Hz, 2H), 2.01 - 1.96 (m, 1H), 1.94 - 1.71 (m, 3H), 1.60 (s, 6H), 1.58 - 1.52 (m, 1H), 1.46 (s, 9H), 1.34 (ddd, J = 4.6, 6.3, 8.2 Hz, 1H), 1.22 - 0.97 (m, 2H). Step E – Synthesis of 4-((1-(2-(4-(3-chloro-4'-((1S,2S)-2-(((3-(1,1,1-trifluoro-2-methylpropan-2- yl)-1H-1,2,4-triazol-5-yl)methyl)carbamoyl)cyclopropyl)-[1,1'-biphenyl]-4-yl)-1H-pyrazol-1- yl)acetyl)piperidin-4-yl)methyl)piperazin-1-ium (11e) Intermediate 11d (90.0 mg, 105.6 μmol, 1.0 equiv) was dissolved in HCl/dioxane (2 mL, 4 M). The mixture was stirred at 20°C for 1 h and then concentrated under reduced pressure to afford 11e which was used without further purification. LCMS [M+1] = 752.7, 754.7; 1H NMR (400 MHz, Methanol-d4) δ 8.01 (s, 1H), 7.73 - 7.55 (m, 5H), 7.25 (d, J = 7.8 Hz, 2H), 5.39 - 5.12 (m, 2H), 4.70 (s, 3H), 4.62 - 4.49 (m, 1H), 4.14 - 4.00 (m, 1H), 3.75 - 3.56 (m, 10H), 3.27 - 3.16 (m, 3H), 2.94 - 2.74 (m, 1H), 2.53 - 2.42 (m, 1H), 2.35 - 2.23 (m, 1H), 2.12 - 1.94 (m, 3H), 1.68 (s, 6H), 1.61 - 1.54 (m, 1H), 1.43 - 1.23 (m, 3H). Step F – Synthesis of 5-(4-((1-(2-(4-(3-chloro-4'-((1S,2S)-2-(3-(3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)propanoyl)cyclopropyl)-[1,1'-biphenyl]-4-yl)-1H- pyrazol-1-yl)acetyl)piperidin-4-yl)methyl)piperazin-1-yl)-2-(2,6-dioxopiperidin-3-yl)-6- fluoroisoindoline-1,3-dione (Compound 11)
To a solution of 11e (80.0 mg, 106.3 μmol, 1.0 equiv) in DMSO (1.0 mL) was added DIEA (20.6 mg, 159.5 μmol, 27.8 μL, 1.5 equiv) and 2-(2,6-dioxopiperidin-3-yl)-5,6- difluoroisoindoline-1,3-dione (46.9 mg, 159.5 μmol, 1.5 equiv). The mixture was stirred at 80°C for 3 h. The mixture was purified by preparative HPLC (Phenomenex Luna C18150mm x 25mm, 10µm; water(NH4HCO3)-acetonitrile]; gradient:44%-74% B over 10 min) to afford Compound 11. LCMS: [M+1] = 1025.5; 1H NMR (400 MHz, DMSO-d6) δ 14.08 - 13.65 (m, 1H), 11.23 - 10.99 (m, 1H), 8.75 (br s, 1H), 8.20 (s, 1H), 7.92 (s, 1H), 7.78 (s, 1H), 7.76 - 7.60 (m, 5H), 7.45 (d, J = 7.4 Hz, 1H), 7.23 (d, J = 8.4 Hz, 2H), 5.19 (d, J = 6.8 Hz, 2H), 5.10 (dd, J = 5.4, 12.8 Hz, 1H), 4.41 (d, J = 5.4 Hz, 2H), 4.33 (d, J = 12.8 Hz, 1H), 4.01 - 3.85 (m, 1H), 3.26 (s, 4H), 3.08 (t, J = 11.6 Hz, 2H), 2.93 - 2.84 (m, 1H), 2.72 - 2.55 (m, 4H), 2.46 - 2.26 (m, 2H), 2.26 - 2.18 (m, 2H), 2.08 - 1.97 (m, 2H), 1.89 - 1.73 (m, 3H), 1.53 (s, 6H), 1.42 (td, J = 4.6, 9.0 Hz, 1H), 1.37 - 1.19 (m, 2H), 1.16 - 0.95 (m, 2H). Example 12: Synthesis of Compound 12
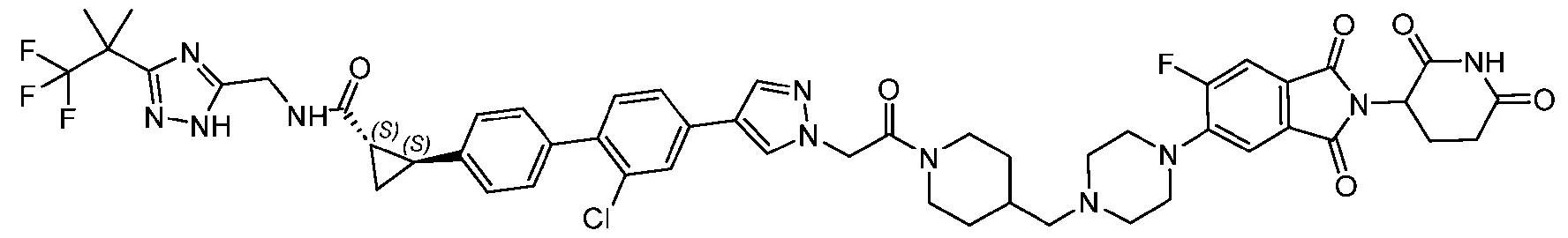 (2'- chloro-4'-(1-(2-(4-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1- yl)methyl)piperidin-1-yl)-2-oxoethyl)-1H-pyrazol-4-yl)-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)cyclopropane-1-carboxamide (Compound 12). LCMS [M+1, M+3] + = 1026.4, 1028.4; 1H NMR (400 MHz, DMSO-d6) δ 14.08 - 13.76 (m, 1H), 11.11 (s, 1H), 8.77 (br s, 1H), 8.22 (s, 1H), 7.99 (s, 1H), 7.78 (d, J = 1.4 Hz, 1H), 7.73 (d, J = 11.4 Hz, 1H), 7.61 (dd, J = 1.4, 7.8 Hz, 1H), 7.45 (d, J = 7.4 Hz, 1H), 7.40 - 7.33 (m, 3H), 7.22 (d, J = 8.4 Hz, 2H), 5.18 - 5.06 (m, 3H), 4.41 (br d, J = 5.8 Hz, 2H), 4.32 (br d, J = 12.4 Hz, 1H), 3.93 (br d, J = 12.6 Hz, 1H), 3.25 (br s, 4H), 3.08 (br t, J = 12.2 Hz, 1H), 2.95 - 2.82 (m, 1H), 2.71 - 2.60 (m, 2H), 2.59 - 2.51 (m, 5H), 2.38 - 2.31 (m, 1H), 2.22 (br d, J = 6.6 Hz, 2H), 2.08 - 1.98 (m, 2H), 1.90 - 1.71 (m, 3H), 1.53 (s, 6H), 1.42 (td, J = 4.4, 9.4 Hz, 1H), 1.35 - 1.27 (m, 1H), 1.15 - 0.94 (m, 2H). Example 13: Synthesis of Compound 13
Step A – Synthesis of 4-[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]-1H-pyrazole (13a) To a solution of 4-(4-bromophenyl)-1H-pyrazole (9 g, 40.3 mmol, 1 equiv), 4,4,5,5- tetramethyl-2-(4,4,5,5-tetramethyl1,3,2-dioxaborolan-2-yl)-1,3,2-dioxaborolane (10.2 g, 40.3 mmol, 1 equiv) in dioxane (90 mL) was added potassium acetate (11.8 g, 121.0 mmol, 3 equiv) and Pd(dppf)Cl2•CH2Cl2 (3.2 g, 4.03 mmol, 0.1 equiv). The mixture was stirred at 80°C for 16 h. The mixture was cooled to ambient temperature and quenched with water (600 mL). The mixture was extracted with ethyl acetate (2 x 500 mL). The combined organic layers were washed with brine (300 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography silica gel, petroleum ether/ethyl acetate=10/1 to 4/1) to afford 13a. LCMS [M+1] + = 271.2; 1H NMR (400 MHz, DMSO-d6) δ 13.00 (s, 1H), 8.26 (s, 1H), 7.96 (d, J = 11.4 Hz, 1H), 7.70 - 7.50 (m, 4H), 1.29 (s, 12H). Step B – Synthesis of tert-butyl 2-[4-[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)phenyl]pyrazol-1-yl]acetate (13b) To a solution of 4-[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]-1H-pyrazole (4 g, 14.8 mmol, 1 equiv) in DMF (40 mL) was added K2CO3 (4.09 g, 29.6 mmol, 2 equiv) and tert- butyl 2-bromoacetate (3.47 g, 17.7 mmol, 2.62 mL, 1.2 equiv). The mixture was stirred at 25°C for 3 h under a nitrogen atmosphere. The mixture was poured into saturated ammonium chloride (200 mL). The aqueous phase was extracted with ethyl acetate (3 x 60 mL). The combined organic phase was washed with brine (3 x 60 mL), dried with anhydrous Na2SO4, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether/ethyl acetate = 1/0 to 3/1) to afford 13b. 1H NMR (400 MHz, DMSO-d6) δ 8.22 (s, 1H), 7.96 (s, 1H), 7.68 - 7.63 (m, 2H), 7.62 - 7.56 (m, 2H), 4.96 (s, 2H), 1.44 (s, 9H), 1.30 (s, 12H). Step C – Synthesis of tert-butyl 2-[4-[4-[4-[(1S,2S)-2-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H- 1,2,4-triazol-5-yl]methylcarbamoyl]cyclopropyl]phenyl]phenyl]pyrazol-1-yl]acetate (13c) A mixture of 13b (3.5 g, 9.0 mmol, 1.3 equiv), Intermediate 3 (3 g, 6.9 mmol, 1 equiv), Pd(DPPF)Cl2 (906.8 mg, 1.3 mmol, 0.2 equiv), Cs2CO3 (6.8 g, 20.8 mmol, 3 equiv) in 2-
methylbutan-2-ol (30 mL) and water (7 mL) was 3-fold degassed and purged with nitrogen. The mixture was stirred at 85 °C for 5 h under an atmosphere of nitrogen. The mixture was cooled to ambient temperature and poured into water (100 mL). The aqueous phase was extracted with ethyl acetate (40 mL x 3). The combined organic phase was washed with brine (30 mL x 3), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether/ethyl acetate = 1/0 to 0/1) 13c. LCMS [M+1] = 609.4; 1H NMR (400 MHz, DMSO-d6) δ 13.87 (s, 1H), 8.75 (s, 1H), 8.20 (s, 1H), 7.96 (s, 1H), 7.65 (s, 3H), 7.61 (d, J = 8.4 Hz, 2H), 7.45 (d, J = 8.6 Hz, 1H), 7.22 (d, J = 8.4 Hz, 2H), 4.97 (s, 2H), 4.41 (d, J = 5.8 Hz, 2H), 2.38 - 2.29 (m, 1H), 1.53 (s, 6H), 1.44 (s, 9H), 1.42 - 1.35 (m, 1H), 1.32 - 1.21 (m, 2H). Step D – Synthesis of 2-[4-[4-[4-[(1S,2S)-2-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4- triazol-5-yl]methylcarbamoyl]cyclopropyl]phenyl]phenyl] pyrazol-1-yl]acetic acid (13d) To a solution of 13c (2.2 g, 3.6 mmol, 1 equiv) in dichloromethane (22 mL) was added trifluoroacetic acid (33.7 g, 296.1 mmol, 22.0 mL, 81.9 equiv) at 0°C, the mixture was stirred at 25°C for 1.5 h. The mixture was concentrated under reduced pressure. The mixture was treated with water (20 mL) and the solids were collected by vacuum filtration. The filter cake was wash with water (10 mL x 3) and dried under vacuum to afford 13d, which was used to next step directly. LCMS [M+1] + = 553.2; 1H NMR (400 MHz, DMSO-d6) δ 14.37 - 13.44 (m, 1H), 13.41 - 12.79 (m, 1H), 8.75 (s, 1H), 8.20 (s, 1H), 7.95 (s, 1H), 7.65 (s, 3H), 7.61 (d, J = 8.4 Hz, 2H), 7.22 (d, J = 8.4 Hz, 2H), 4.98 (s, 2H), 4.41 (d, J = 5.6 Hz, 2H), 4.31 - 4.14 (m, 1H), 2.37 - 2.29 (m, 1H), 2.04 - 1.95 (m, 1H), 1.53 (s, 6H), 1.45 - 1.38 (m, 1H), 1.29 (ddd, J = 4.2, 5.8, 8.2 Hz, 1H). Step E – Synthesis of benzyl 4-[(1-tert-butoxycarbonylazetidin-3-yl)methyl]piperazine-1- carboxylate (13e) To a mixture of tert-butyl 3-(bromomethyl)azetidine-1-carboxylate (1 g, 4.0 mmol, 1.0 equiv) and benzyl piperazine-1-carboxylate (880.6 mg, 4.0 mmol, 771.1 μL, 1.0 equiv) in DMF (10.0 mL) was added K2CO3 (1.1 g, 8.0 mmol, 2 equiv) and KI (132.7 mg, 799.5 μmol, 0.2 equiv) in one portion at 20°C under nitrogen. The mixture was stirred at 50°C for 4 h, cooled to ambient temperature and poured into water (10 mL). The aqueous phase was extracted with ethyl acetate (20 mL). The combined organic phase was washed with brine (20 mL), dried with anhydrous
sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether/ethyl acetate=9/1 to 67/33) to afford 13e. LCMS [M+1] = 390.3; 1H NMR (400 MHz, CHLOROFORM-d) δ 7.43 - 7.29 (m, 5H), 5.14 (s, 2H), 4.01 (t, J = 8.4 Hz, 2H), 3.59 (dd, J = 5.4, 8.4 Hz, 2H), 3.53 - 3.46 (m, 4H), 2.77 - 2.67 (m, 1H), 2.58 (d, J = 7.6 Hz, 2H), 2.37 (br s, 4H), 1.44 (s, 9H). Step F – Synthesis of tert-butyl 3-(piperazin-1-ylmethyl)azetidine-1-carboxylate (13f) To a solution of 13e (1.0 g, 2.5 mmol, 1.0 equiv) in methanol (10.0 mL) was added Pd/C (1.2 g, 1.1 mmol, 10 wt%) under a nitrogen atmosphere. The suspension was three-fold degassed and purged with H2. The mixture was stirred under hydrogen (1 atm) at 30°C for 2 h, filtered and the filtrate was concentrated to afford 13f. LCMS: [M+1] = 256.3; 1H NMR (400 MHz, Methanol- d4) δ 4.03 - 3.92 (m, 2H), 3.60 - 3.51 (m, 2H), 3.27 (td, J = 1.6, 3.2 Hz, 3H), 2.84 - 2.75 (m, 4H), 2.55 (d, J = 7.2 Hz, 2H), 2.39 (br s, 3H), 1.39 (s, 9H). Step G – Synthesis of tert-butyl 3-[[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]piperazin-1-yl]methyl]azetidine-1-carboxylate (13g) To a mixture of 13f (0.1 g, 391.6 μmol, 1 equiv) and 2-(2,6-dioxo-3-piperidyl)-5,6- difluoro-isoindoline-1,3-dione (115.2 mg, 391.6 μmol, 1 equiv) in DMSO (1.0 mL) was added DIEA (253.0 mg, 1.9 mmol, 341.0 μL, 5.0 equiv) in one portion at 20°C under nitrogen. The mixture was stirred at 70°C for 12 h, cooled to ambient temperature and poured into water (10.0 mL). The aqueous phase was extracted with ethyl acetate (20.0 mL). The combined organic phase was washed with brine (10.0 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated in vacuum. The residue was purified by preparative TLC (silica gel, dichloromethane: methanol = 10:1) to afford 13g. LCMS [M+1] + = 530.4. Step H – Synthesis of 5-[4-(azetidin-3-ylmethyl)piperazin-1-yl]-2-(2,6-dioxo-3-piperidyl)-6- fluoro-isoindoline-1,3-dione (13h) To a mixture of 13g (170 mg, 321.0 μmol, 1.0 eq) in dichloromethane (0.5 mL) was added trifluoroacetic acid (261 mg, 2.2 mmol, 170.0 μL, 7.1 equiv) in one portion at ambient temperature, stirred for 1 h and concentrated under reduced pressure. The residue was triturated with MTBE (5.0 mL) to afford 13h. LCMS [M+1] + = 430.3.
Step I – Synthesis of (1S,2S)-2-[4-[4-[1-[2-[3-[[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo- isoindolin-5-yl]piperazin-1-yl]methyl]azetidin-1-yl]-2-oxo-ethyl]pyrazol-4-yl]phenyl]phenyl]-N- [[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]cyclopropanecarboxamide (Compound 13) To a mixture of 13d (80 mg, 144.7 μmol, 1 equiv) and 13h (118.1 mg, 275.0 μmol, 1.9 equiv) and DIEA (93.5 mg, 723.9 μmol, 126.0 μL, 5.0 equiv) in DMF (0.5 mL) was treated with T4P (156.4 mg, 217.1 μmol, 50% purity, 1.5 equiv) in one portion at 20°C under N2. The mixture was stirred at 20 °C for 1 h, and then concentrated under reduced pressure. The residue was purified by preparative HPLC (Phenomenex Luna C18100mm x 30mm, 5µm; water (0.2% formic acid)-acetonitrile]) to give Compound 13. LCMS [M+1] =964.3; 1H NMR (400 MHz, DMSO-d6) δ 14.05 - 13.53 (m, 1H), 11.11 (s, 1H), 8.75 (br s, 1H), 8.17 (s, 1H), 7.95 (s, 1H), 7.73 (d, J = 11.6 Hz, 1H), 7.69 - 7.57 (m, 6H), 7.46 (br d, J = 6.8 Hz, 1H), 7.21 (d, J = 8.4 Hz, 2H), 5.11 (dd, J = 5.6, 12.8 Hz, 1H), 4.87 (s, 2H), 4.41 (br d, J = 5.2 Hz, 2H), 4.24 (br t, J = 8.4 Hz, 1H), 4.01 (br t, J = 8.8 Hz, 1H), 3.85 (br d, J = 6.4 Hz, 1H), 3.59 (br s, 1H), 3.48 - 3.36 (m, 2H), 2.96 - 2.82 (m, 2H), 2.67 - 2.53 (m, 6H), 2.36 - 2.28 (m, 1H), 2.09 - 1.94 (m, 2H), 1.90 - 1.58 (m, 1H), 1.53 (s, 6H), 1.49 - 1.36 (m, 2H), 1.36 - 1.22 (m, 2H), 0.90 - 0.81 (m, 1H)
(2'- chloro-4'-(1-(2-(4-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1- yl)methyl)piperidin-1-yl)-2-oxoethyl)-1H-pyrazol-4-yl)-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)cyclopropane-1-carboxamide (Compound 12). LCMS [M+1, M+3] + = 1026.4, 1028.4; 1H NMR (400 MHz, DMSO-d6) δ 14.08 - 13.76 (m, 1H), 11.11 (s, 1H), 8.77 (br s, 1H), 8.22 (s, 1H), 7.99 (s, 1H), 7.78 (d, J = 1.4 Hz, 1H), 7.73 (d, J = 11.4 Hz, 1H), 7.61 (dd, J = 1.4, 7.8 Hz, 1H), 7.45 (d, J = 7.4 Hz, 1H), 7.40 - 7.33 (m, 3H), 7.22 (d, J = 8.4 Hz, 2H), 5.18 - 5.06 (m, 3H), 4.41 (br d, J = 5.8 Hz, 2H), 4.32 (br d, J = 12.4 Hz, 1H), 3.93 (br d, J = 12.6 Hz, 1H), 3.25 (br s, 4H), 3.08 (br t, J = 12.2 Hz, 1H), 2.95 - 2.82 (m, 1H), 2.71 - 2.60 (m, 2H), 2.59 - 2.51 (m, 5H), 2.38 - 2.31 (m, 1H), 2.22 (br d, J = 6.6 Hz, 2H), 2.08 - 1.98 (m, 2H), 1.90 - 1.71 (m, 3H), 1.53 (s, 6H), 1.42 (td, J = 4.4, 9.4 Hz, 1H), 1.35 - 1.27 (m, 1H), 1.15 - 0.94 (m, 2H). Example 13: Synthesis of Compound 13
Step A – Synthesis of 4-[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]-1H-pyrazole (13a) To a solution of 4-(4-bromophenyl)-1H-pyrazole (9 g, 40.3 mmol, 1 equiv), 4,4,5,5- tetramethyl-2-(4,4,5,5-tetramethyl1,3,2-dioxaborolan-2-yl)-1,3,2-dioxaborolane (10.2 g, 40.3 mmol, 1 equiv) in dioxane (90 mL) was added potassium acetate (11.8 g, 121.0 mmol, 3 equiv) and Pd(dppf)Cl2•CH2Cl2 (3.2 g, 4.03 mmol, 0.1 equiv). The mixture was stirred at 80°C for 16 h. The mixture was cooled to ambient temperature and quenched with water (600 mL). The mixture was extracted with ethyl acetate (2 x 500 mL). The combined organic layers were washed with brine (300 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography silica gel, petroleum ether/ethyl acetate=10/1 to 4/1) to afford 13a. LCMS [M+1] + = 271.2; 1H NMR (400 MHz, DMSO-d6) δ 13.00 (s, 1H), 8.26 (s, 1H), 7.96 (d, J = 11.4 Hz, 1H), 7.70 - 7.50 (m, 4H), 1.29 (s, 12H). Step B – Synthesis of tert-butyl 2-[4-[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)phenyl]pyrazol-1-yl]acetate (13b) To a solution of 4-[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]-1H-pyrazole (4 g, 14.8 mmol, 1 equiv) in DMF (40 mL) was added K2CO3 (4.09 g, 29.6 mmol, 2 equiv) and tert- butyl 2-bromoacetate (3.47 g, 17.7 mmol, 2.62 mL, 1.2 equiv). The mixture was stirred at 25°C for 3 h under a nitrogen atmosphere. The mixture was poured into saturated ammonium chloride (200 mL). The aqueous phase was extracted with ethyl acetate (3 x 60 mL). The combined organic phase was washed with brine (3 x 60 mL), dried with anhydrous Na2SO4, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether/ethyl acetate = 1/0 to 3/1) to afford 13b. 1H NMR (400 MHz, DMSO-d6) δ 8.22 (s, 1H), 7.96 (s, 1H), 7.68 - 7.63 (m, 2H), 7.62 - 7.56 (m, 2H), 4.96 (s, 2H), 1.44 (s, 9H), 1.30 (s, 12H). Step C – Synthesis of tert-butyl 2-[4-[4-[4-[(1S,2S)-2-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H- 1,2,4-triazol-5-yl]methylcarbamoyl]cyclopropyl]phenyl]phenyl]pyrazol-1-yl]acetate (13c) A mixture of 13b (3.5 g, 9.0 mmol, 1.3 equiv), Intermediate 3 (3 g, 6.9 mmol, 1 equiv), Pd(DPPF)Cl2 (906.8 mg, 1.3 mmol, 0.2 equiv), Cs2CO3 (6.8 g, 20.8 mmol, 3 equiv) in 2-
methylbutan-2-ol (30 mL) and water (7 mL) was 3-fold degassed and purged with nitrogen. The mixture was stirred at 85 °C for 5 h under an atmosphere of nitrogen. The mixture was cooled to ambient temperature and poured into water (100 mL). The aqueous phase was extracted with ethyl acetate (40 mL x 3). The combined organic phase was washed with brine (30 mL x 3), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether/ethyl acetate = 1/0 to 0/1) 13c. LCMS [M+1] = 609.4; 1H NMR (400 MHz, DMSO-d6) δ 13.87 (s, 1H), 8.75 (s, 1H), 8.20 (s, 1H), 7.96 (s, 1H), 7.65 (s, 3H), 7.61 (d, J = 8.4 Hz, 2H), 7.45 (d, J = 8.6 Hz, 1H), 7.22 (d, J = 8.4 Hz, 2H), 4.97 (s, 2H), 4.41 (d, J = 5.8 Hz, 2H), 2.38 - 2.29 (m, 1H), 1.53 (s, 6H), 1.44 (s, 9H), 1.42 - 1.35 (m, 1H), 1.32 - 1.21 (m, 2H). Step D – Synthesis of 2-[4-[4-[4-[(1S,2S)-2-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4- triazol-5-yl]methylcarbamoyl]cyclopropyl]phenyl]phenyl] pyrazol-1-yl]acetic acid (13d) To a solution of 13c (2.2 g, 3.6 mmol, 1 equiv) in dichloromethane (22 mL) was added trifluoroacetic acid (33.7 g, 296.1 mmol, 22.0 mL, 81.9 equiv) at 0°C, the mixture was stirred at 25°C for 1.5 h. The mixture was concentrated under reduced pressure. The mixture was treated with water (20 mL) and the solids were collected by vacuum filtration. The filter cake was wash with water (10 mL x 3) and dried under vacuum to afford 13d, which was used to next step directly. LCMS [M+1] + = 553.2; 1H NMR (400 MHz, DMSO-d6) δ 14.37 - 13.44 (m, 1H), 13.41 - 12.79 (m, 1H), 8.75 (s, 1H), 8.20 (s, 1H), 7.95 (s, 1H), 7.65 (s, 3H), 7.61 (d, J = 8.4 Hz, 2H), 7.22 (d, J = 8.4 Hz, 2H), 4.98 (s, 2H), 4.41 (d, J = 5.6 Hz, 2H), 4.31 - 4.14 (m, 1H), 2.37 - 2.29 (m, 1H), 2.04 - 1.95 (m, 1H), 1.53 (s, 6H), 1.45 - 1.38 (m, 1H), 1.29 (ddd, J = 4.2, 5.8, 8.2 Hz, 1H). Step E – Synthesis of benzyl 4-[(1-tert-butoxycarbonylazetidin-3-yl)methyl]piperazine-1- carboxylate (13e) To a mixture of tert-butyl 3-(bromomethyl)azetidine-1-carboxylate (1 g, 4.0 mmol, 1.0 equiv) and benzyl piperazine-1-carboxylate (880.6 mg, 4.0 mmol, 771.1 μL, 1.0 equiv) in DMF (10.0 mL) was added K2CO3 (1.1 g, 8.0 mmol, 2 equiv) and KI (132.7 mg, 799.5 μmol, 0.2 equiv) in one portion at 20°C under nitrogen. The mixture was stirred at 50°C for 4 h, cooled to ambient temperature and poured into water (10 mL). The aqueous phase was extracted with ethyl acetate (20 mL). The combined organic phase was washed with brine (20 mL), dried with anhydrous
sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether/ethyl acetate=9/1 to 67/33) to afford 13e. LCMS [M+1] = 390.3; 1H NMR (400 MHz, CHLOROFORM-d) δ 7.43 - 7.29 (m, 5H), 5.14 (s, 2H), 4.01 (t, J = 8.4 Hz, 2H), 3.59 (dd, J = 5.4, 8.4 Hz, 2H), 3.53 - 3.46 (m, 4H), 2.77 - 2.67 (m, 1H), 2.58 (d, J = 7.6 Hz, 2H), 2.37 (br s, 4H), 1.44 (s, 9H). Step F – Synthesis of tert-butyl 3-(piperazin-1-ylmethyl)azetidine-1-carboxylate (13f) To a solution of 13e (1.0 g, 2.5 mmol, 1.0 equiv) in methanol (10.0 mL) was added Pd/C (1.2 g, 1.1 mmol, 10 wt%) under a nitrogen atmosphere. The suspension was three-fold degassed and purged with H2. The mixture was stirred under hydrogen (1 atm) at 30°C for 2 h, filtered and the filtrate was concentrated to afford 13f. LCMS: [M+1] = 256.3; 1H NMR (400 MHz, Methanol- d4) δ 4.03 - 3.92 (m, 2H), 3.60 - 3.51 (m, 2H), 3.27 (td, J = 1.6, 3.2 Hz, 3H), 2.84 - 2.75 (m, 4H), 2.55 (d, J = 7.2 Hz, 2H), 2.39 (br s, 3H), 1.39 (s, 9H). Step G – Synthesis of tert-butyl 3-[[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]piperazin-1-yl]methyl]azetidine-1-carboxylate (13g) To a mixture of 13f (0.1 g, 391.6 μmol, 1 equiv) and 2-(2,6-dioxo-3-piperidyl)-5,6- difluoro-isoindoline-1,3-dione (115.2 mg, 391.6 μmol, 1 equiv) in DMSO (1.0 mL) was added DIEA (253.0 mg, 1.9 mmol, 341.0 μL, 5.0 equiv) in one portion at 20°C under nitrogen. The mixture was stirred at 70°C for 12 h, cooled to ambient temperature and poured into water (10.0 mL). The aqueous phase was extracted with ethyl acetate (20.0 mL). The combined organic phase was washed with brine (10.0 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated in vacuum. The residue was purified by preparative TLC (silica gel, dichloromethane: methanol = 10:1) to afford 13g. LCMS [M+1] + = 530.4. Step H – Synthesis of 5-[4-(azetidin-3-ylmethyl)piperazin-1-yl]-2-(2,6-dioxo-3-piperidyl)-6- fluoro-isoindoline-1,3-dione (13h) To a mixture of 13g (170 mg, 321.0 μmol, 1.0 eq) in dichloromethane (0.5 mL) was added trifluoroacetic acid (261 mg, 2.2 mmol, 170.0 μL, 7.1 equiv) in one portion at ambient temperature, stirred for 1 h and concentrated under reduced pressure. The residue was triturated with MTBE (5.0 mL) to afford 13h. LCMS [M+1] + = 430.3.
Step I – Synthesis of (1S,2S)-2-[4-[4-[1-[2-[3-[[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo- isoindolin-5-yl]piperazin-1-yl]methyl]azetidin-1-yl]-2-oxo-ethyl]pyrazol-4-yl]phenyl]phenyl]-N- [[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]cyclopropanecarboxamide (Compound 13) To a mixture of 13d (80 mg, 144.7 μmol, 1 equiv) and 13h (118.1 mg, 275.0 μmol, 1.9 equiv) and DIEA (93.5 mg, 723.9 μmol, 126.0 μL, 5.0 equiv) in DMF (0.5 mL) was treated with T4P (156.4 mg, 217.1 μmol, 50% purity, 1.5 equiv) in one portion at 20°C under N2. The mixture was stirred at 20 °C for 1 h, and then concentrated under reduced pressure. The residue was purified by preparative HPLC (Phenomenex Luna C18100mm x 30mm, 5µm; water (0.2% formic acid)-acetonitrile]) to give Compound 13. LCMS [M+1] =964.3; 1H NMR (400 MHz, DMSO-d6) δ 14.05 - 13.53 (m, 1H), 11.11 (s, 1H), 8.75 (br s, 1H), 8.17 (s, 1H), 7.95 (s, 1H), 7.73 (d, J = 11.6 Hz, 1H), 7.69 - 7.57 (m, 6H), 7.46 (br d, J = 6.8 Hz, 1H), 7.21 (d, J = 8.4 Hz, 2H), 5.11 (dd, J = 5.6, 12.8 Hz, 1H), 4.87 (s, 2H), 4.41 (br d, J = 5.2 Hz, 2H), 4.24 (br t, J = 8.4 Hz, 1H), 4.01 (br t, J = 8.8 Hz, 1H), 3.85 (br d, J = 6.4 Hz, 1H), 3.59 (br s, 1H), 3.48 - 3.36 (m, 2H), 2.96 - 2.82 (m, 2H), 2.67 - 2.53 (m, 6H), 2.36 - 2.28 (m, 1H), 2.09 - 1.94 (m, 2H), 1.90 - 1.58 (m, 1H), 1.53 (s, 6H), 1.49 - 1.36 (m, 2H), 1.36 - 1.22 (m, 2H), 0.90 - 0.81 (m, 1H)
Example 14: Synthesis of Compound 14
 carboxylate (14a) A solution of benzyl (3-bromopropyl)carbamate (3.9 g, 14.5 mmol, 1.1 equiv) and tetrabutylammonium hydrogen sulfate (449.6 mg, 1.3 mmol, 0.1 equiv) in dichloromethane (30.0 mL) was stirred at 25°C for 5 min, then tert-butyl 4-(2-hydroxyethyl)piperazine-1-carboxylate (3.0 g, 13.2 mmol, 1.0 equiv) and NaOH (1.6 g, 39.7 mmol, 3.0 equiv) were added. The reaction mixture was stirred at 40°C for 16 h. The mixture was poured into water (100 mL) and the aqueous phase was extracted with ethyl acetate (3 x 100 mL). The combined organic phase was washed with brine (3 x 100 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel chromatography (silica gel, petroleum ether/ethyl acetate=1/0, 3/2) to 14a. LCMS [M+1] = 422.3; 1H NMR (400
MHz, DMSO-d6) δ 7.39 - 7.29 (m, 4H), 7.21 (br s, 1H), 5.00 (s, 2H), 3.45 (br s, 2H), 3.37 (br t, J = 5.8 Hz, 2H), 3.27 (br s, 4H), 3.04 (br d, J = 6.0 Hz, 2H), 2.46 (br d, J = 5.6 Hz, 2H), 2.34 (br s, 4H), 1.66 - 1.57 (m, 2H), 1.39 (s, 9H). Step B – Synthesis of tert-butyl 4-(2-(3-aminopropoxy)ethyl)piperazine-1-carboxylate (14b) To a solution of 14a (550.0 mg, 1.3 mmol, 1.0 equiv) in 2,2,2-trifluoroethanol (5.0 mL) was added Pd/C (694.2 mg, 10.0% Pd on carbon, 50% in water(w/w)). The mixture was stirred at 50°C for 2 h under H2 (1 atm). The reaction mixture was filtered and the filtrate was concentrated under reduced pressure to afford 14b. LCMS [M+1] = 288.2; 1H NMR (400 MHz, DMSO-d6) δ 3.63 - 3.53 (m, 4H), 2.75 - 2.58 (m, 14H), 1.70 (quin, J = 6.6 Hz, 2H), 1.54 (s, 9H). Step C – Synthesis of 3-chloro-4'-((1S,2S)-2-(((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)carbamoyl)cyclopropyl)-[1,1'-biphenyl]-4-carboxylic acid (14c). To a solution of Intermediate 3 (300.0 mg, 695.6 μmol, 1.0 equiv) and 2-chloro-4- (4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoic acid (294.8 mg, 1.0 mmol, 1.5 equiv) in dioxane (3.0 mL) and water (0.3 mL) added K2CO3 (288.4 mg, 2.1 mmol, 3.0 equiv) and Pd(dppf)Cl2•CH2Cl2 (56.8 mg, 69.5 μmol, 0.1 equiv). The mixture was stirred at 120°C for 2 h. The mixture was cooled to 20°C and poured into water (30 mL), the aqueous phase was extracted with ethyl acetate (3 x 30 mL). The combined organic phase was washed with brine (3 x 30 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative TLC (ethyl acetate: methanol=1:1) to afford 14c. LCMS [M+1] = 507.1, 509.1; 1H NMR (400 MHz, DMSO-d6) δ 7.52 - 7.45 (m, 3H), 7.30 (br s, 2H), 7.18 (br d, J = 7.6 Hz, 2H), 4.62 - 4.52 (m, 1H), 4.42 - 4.33 (m, 1H), 3.16 (br d, J = 4.8 Hz, 2H), 1.53 (s, 6H), 1.39 (br d, J = 4.8 Hz, 1H), 1.25 (br d, J = 4.4 Hz, 2H). Step D – Synthesis of tert-butyl 4-(2-(3-(3-chloro-4'-((1S,2S)-2-(((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)carbamoyl)cyclopropyl)-[1,1'-biphenyl]-4- carboxamido)propoxy)ethyl)piperazine-1-carboxylate (14d) To a solution of 14c (180.0 mg, 355.1 μmol, 1.0 equiv) in DMF (1.8 mL) was added DIEA (137.6 mg, 1.0 mmol, 185.5 μL, 3.0 equiv), BOP (188.4 mg, 426.1 μmol, 1.2 equiv) and 14b (122.4 mg, 426.1 μmol, 1.2 equiv). The mixture was stirred at 20°C for 2 h. The mixture was poured into water (50 mL) and the aqueous phase was extracted with ethyl acetate (3 x 50 mL).
The combined organic phase was washed with brine (3 x 50 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative TLC(ethyl acetate : methanol=1:1) to afford 14d. LCMS [M+1] = 776.3, 778.3; 1H NMR (400 MHz, DMSO-d6) δ 8.82 - 8.75 (m, 1H), 8.41 (br d, J = 5.8 Hz, 1H), 7.75 (s, 1H), 7.69 - 7.60 (m, 3H), 7.47 (br d, J = 8.0 Hz, 1H), 7.28 - 7.20 (m, 2H), 4.40 (br d, J = 4.8 Hz, 2H), 3.53 - 3.40 (m, 6H), 2.73 (s, 8H), 2.35 (br s, 4H), 1.78 - 1.70 (m, 2H), 1.52 (s, 6H), 1.38 (s, 9H), 1.23 (br s, 2H). Step E – Synthesis of 3-chloro-N-(3-(2-(piperazin-1-yl)ethoxy)propyl)-4'-((1S,2S)-2-(((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)carbamoyl)cyclopropyl)-[1,1'- biphenyl]-4-carboxamide (14e) To a solution of 14d (150.0 mg.0, 193 μmol, 1.0 equiv) in dioxane (0.5 mL) was added HCl (4.0 M, 145 μL, 3.0 equiv). The mixture was stirred at 20°C for 4 h. The reaction mixture was concentrated under reduced pressure to afford 14e, which was used without further purification. LCMS [M+1, M+3] + = 676.3, 678.3; 1H NMR (400 MHz, DMSO-d6) δ 8.80 (br t, J = 5.0 Hz, 1H), 8.55 - 8.49 (m, 1H), 7.75 (s, 1H), 7.68 - 7.61 (m, 3H), 7.49 (br d, J = 8.0 Hz, 1H), 7.24 (br d, J = 8.2 Hz, 2H), 4.41 (br d, J = 5.6 Hz, 6H), 3.80 (br s, 2H), 3.39 (br d, J = 3.4 Hz, 5H), 2.73 (s, 5H), 2.35 (br dd, J = 4.8, 9.6 Hz, 2H), 1.82 - 1.73 (m, 2H), 1.53 (s, 6H), 1.41 (br dd, J = 4.4, 9.0 Hz, 2H). Step F – Synthesis of 3-chloro-N-(3-(2-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3- dioxoisoindolin-5-yl)piperazin-1-yl)ethoxy)propyl)-4'-((1S,2S)-2-(((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)carbamoyl)cyclopropyl)-[1,1'-biphenyl]-4- carboxamide (Compound 14) To a solution of 14e (70.0 mg, 103.5 μmol, 1.0 equiv) in DMSO (1 mL) was added DIEA (40.1 mg, 310.5 μmol, 54.1 μL, 3.0 equiv) and 2-(2,6-dioxopiperidin-3-yl)-5,6- difluoroisoindoline-1,3-dione (45.7 mg, 155.3 μmol, 1.5 equiv). The mixture was stirred at 120°C for 6 h, cooled to rt and filtered. The filtrate was purified by preparative HPLC (Waters Xbridge 150mm x 25mm 10µm; water( NH4HCO3)-acetonitrile) to afford Compound 14. LCMS [M+1] = 950.3, 952.3; 1H NMR (400 MHz, DMSO-d6) δ 11.11 (br d, J = 1.0 Hz, 1H), 8.80 (br d, J = 5.8 Hz, 1H), 8.41 (br t, J = 5.6 Hz, 1H), 7.77 - 7.69 (m, 2H), 7.67 - 7.60 (m, 3H), 7.50 - 7.45 (m, 1H), 7.45 - 7.41 (m, 1H), 7.23 (br d, J = 8.4 Hz, 2H), 5.10 (dd, J = 5.2, 12.8 Hz, 1H), 4.41 (br d, J =
5.6 Hz, 2H), 3.58 - 3.48 (m, 4H), 3.21 (br s, 5H), 2.98 - 2.78 (m, 2H), 2.60 - 2.53 (m, 7H), 2.39 - 2.31 (m, 1H), 2.08 - 1.92 (m, 3H), 1.76 (quin, J = 6.2 Hz, 2H), 1.52 (s, 6H), 1.41 (td, J = 4.6, 9.2 Hz, 1H), 1.32 - 1.21 (m, 2H). Example 15: Synthesis of Compound 15
carboxylate (14a) A solution of benzyl (3-bromopropyl)carbamate (3.9 g, 14.5 mmol, 1.1 equiv) and tetrabutylammonium hydrogen sulfate (449.6 mg, 1.3 mmol, 0.1 equiv) in dichloromethane (30.0 mL) was stirred at 25°C for 5 min, then tert-butyl 4-(2-hydroxyethyl)piperazine-1-carboxylate (3.0 g, 13.2 mmol, 1.0 equiv) and NaOH (1.6 g, 39.7 mmol, 3.0 equiv) were added. The reaction mixture was stirred at 40°C for 16 h. The mixture was poured into water (100 mL) and the aqueous phase was extracted with ethyl acetate (3 x 100 mL). The combined organic phase was washed with brine (3 x 100 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel chromatography (silica gel, petroleum ether/ethyl acetate=1/0, 3/2) to 14a. LCMS [M+1] = 422.3; 1H NMR (400
MHz, DMSO-d6) δ 7.39 - 7.29 (m, 4H), 7.21 (br s, 1H), 5.00 (s, 2H), 3.45 (br s, 2H), 3.37 (br t, J = 5.8 Hz, 2H), 3.27 (br s, 4H), 3.04 (br d, J = 6.0 Hz, 2H), 2.46 (br d, J = 5.6 Hz, 2H), 2.34 (br s, 4H), 1.66 - 1.57 (m, 2H), 1.39 (s, 9H). Step B – Synthesis of tert-butyl 4-(2-(3-aminopropoxy)ethyl)piperazine-1-carboxylate (14b) To a solution of 14a (550.0 mg, 1.3 mmol, 1.0 equiv) in 2,2,2-trifluoroethanol (5.0 mL) was added Pd/C (694.2 mg, 10.0% Pd on carbon, 50% in water(w/w)). The mixture was stirred at 50°C for 2 h under H2 (1 atm). The reaction mixture was filtered and the filtrate was concentrated under reduced pressure to afford 14b. LCMS [M+1] = 288.2; 1H NMR (400 MHz, DMSO-d6) δ 3.63 - 3.53 (m, 4H), 2.75 - 2.58 (m, 14H), 1.70 (quin, J = 6.6 Hz, 2H), 1.54 (s, 9H). Step C – Synthesis of 3-chloro-4'-((1S,2S)-2-(((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)carbamoyl)cyclopropyl)-[1,1'-biphenyl]-4-carboxylic acid (14c). To a solution of Intermediate 3 (300.0 mg, 695.6 μmol, 1.0 equiv) and 2-chloro-4- (4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoic acid (294.8 mg, 1.0 mmol, 1.5 equiv) in dioxane (3.0 mL) and water (0.3 mL) added K2CO3 (288.4 mg, 2.1 mmol, 3.0 equiv) and Pd(dppf)Cl2•CH2Cl2 (56.8 mg, 69.5 μmol, 0.1 equiv). The mixture was stirred at 120°C for 2 h. The mixture was cooled to 20°C and poured into water (30 mL), the aqueous phase was extracted with ethyl acetate (3 x 30 mL). The combined organic phase was washed with brine (3 x 30 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative TLC (ethyl acetate: methanol=1:1) to afford 14c. LCMS [M+1] = 507.1, 509.1; 1H NMR (400 MHz, DMSO-d6) δ 7.52 - 7.45 (m, 3H), 7.30 (br s, 2H), 7.18 (br d, J = 7.6 Hz, 2H), 4.62 - 4.52 (m, 1H), 4.42 - 4.33 (m, 1H), 3.16 (br d, J = 4.8 Hz, 2H), 1.53 (s, 6H), 1.39 (br d, J = 4.8 Hz, 1H), 1.25 (br d, J = 4.4 Hz, 2H). Step D – Synthesis of tert-butyl 4-(2-(3-(3-chloro-4'-((1S,2S)-2-(((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)carbamoyl)cyclopropyl)-[1,1'-biphenyl]-4- carboxamido)propoxy)ethyl)piperazine-1-carboxylate (14d) To a solution of 14c (180.0 mg, 355.1 μmol, 1.0 equiv) in DMF (1.8 mL) was added DIEA (137.6 mg, 1.0 mmol, 185.5 μL, 3.0 equiv), BOP (188.4 mg, 426.1 μmol, 1.2 equiv) and 14b (122.4 mg, 426.1 μmol, 1.2 equiv). The mixture was stirred at 20°C for 2 h. The mixture was poured into water (50 mL) and the aqueous phase was extracted with ethyl acetate (3 x 50 mL).
The combined organic phase was washed with brine (3 x 50 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative TLC(ethyl acetate : methanol=1:1) to afford 14d. LCMS [M+1] = 776.3, 778.3; 1H NMR (400 MHz, DMSO-d6) δ 8.82 - 8.75 (m, 1H), 8.41 (br d, J = 5.8 Hz, 1H), 7.75 (s, 1H), 7.69 - 7.60 (m, 3H), 7.47 (br d, J = 8.0 Hz, 1H), 7.28 - 7.20 (m, 2H), 4.40 (br d, J = 4.8 Hz, 2H), 3.53 - 3.40 (m, 6H), 2.73 (s, 8H), 2.35 (br s, 4H), 1.78 - 1.70 (m, 2H), 1.52 (s, 6H), 1.38 (s, 9H), 1.23 (br s, 2H). Step E – Synthesis of 3-chloro-N-(3-(2-(piperazin-1-yl)ethoxy)propyl)-4'-((1S,2S)-2-(((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)carbamoyl)cyclopropyl)-[1,1'- biphenyl]-4-carboxamide (14e) To a solution of 14d (150.0 mg.0, 193 μmol, 1.0 equiv) in dioxane (0.5 mL) was added HCl (4.0 M, 145 μL, 3.0 equiv). The mixture was stirred at 20°C for 4 h. The reaction mixture was concentrated under reduced pressure to afford 14e, which was used without further purification. LCMS [M+1, M+3] + = 676.3, 678.3; 1H NMR (400 MHz, DMSO-d6) δ 8.80 (br t, J = 5.0 Hz, 1H), 8.55 - 8.49 (m, 1H), 7.75 (s, 1H), 7.68 - 7.61 (m, 3H), 7.49 (br d, J = 8.0 Hz, 1H), 7.24 (br d, J = 8.2 Hz, 2H), 4.41 (br d, J = 5.6 Hz, 6H), 3.80 (br s, 2H), 3.39 (br d, J = 3.4 Hz, 5H), 2.73 (s, 5H), 2.35 (br dd, J = 4.8, 9.6 Hz, 2H), 1.82 - 1.73 (m, 2H), 1.53 (s, 6H), 1.41 (br dd, J = 4.4, 9.0 Hz, 2H). Step F – Synthesis of 3-chloro-N-(3-(2-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3- dioxoisoindolin-5-yl)piperazin-1-yl)ethoxy)propyl)-4'-((1S,2S)-2-(((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)carbamoyl)cyclopropyl)-[1,1'-biphenyl]-4- carboxamide (Compound 14) To a solution of 14e (70.0 mg, 103.5 μmol, 1.0 equiv) in DMSO (1 mL) was added DIEA (40.1 mg, 310.5 μmol, 54.1 μL, 3.0 equiv) and 2-(2,6-dioxopiperidin-3-yl)-5,6- difluoroisoindoline-1,3-dione (45.7 mg, 155.3 μmol, 1.5 equiv). The mixture was stirred at 120°C for 6 h, cooled to rt and filtered. The filtrate was purified by preparative HPLC (Waters Xbridge 150mm x 25mm 10µm; water( NH4HCO3)-acetonitrile) to afford Compound 14. LCMS [M+1] = 950.3, 952.3; 1H NMR (400 MHz, DMSO-d6) δ 11.11 (br d, J = 1.0 Hz, 1H), 8.80 (br d, J = 5.8 Hz, 1H), 8.41 (br t, J = 5.6 Hz, 1H), 7.77 - 7.69 (m, 2H), 7.67 - 7.60 (m, 3H), 7.50 - 7.45 (m, 1H), 7.45 - 7.41 (m, 1H), 7.23 (br d, J = 8.4 Hz, 2H), 5.10 (dd, J = 5.2, 12.8 Hz, 1H), 4.41 (br d, J =
5.6 Hz, 2H), 3.58 - 3.48 (m, 4H), 3.21 (br s, 5H), 2.98 - 2.78 (m, 2H), 2.60 - 2.53 (m, 7H), 2.39 - 2.31 (m, 1H), 2.08 - 1.92 (m, 3H), 1.76 (quin, J = 6.2 Hz, 2H), 1.52 (s, 6H), 1.41 (td, J = 4.6, 9.2 Hz, 1H), 1.32 - 1.21 (m, 2H). Example 15: Synthesis of Compound 15
 To a solution of 2-chloro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoic acid (1.0 g, 3.5 mmol, 1.0 equiv), DIEA (1.4 g, 10.6 mmol, 1.9 mL, 3.0 equiv) and tert-butyl glycinate (557.1 mg, 4.2 mmol, 1.2 equiv) in DMF (10.0 mL) was added T4P (3.1 g, 4.3 mmol, 50.0% purity, 1.2 equiv). The reaction mixture was stirred at 25 °C for 1 h, quenched by water (10 mL)
and extracted with ethyl acetate (3 x 10 mL). The combined organic layers were washed with brine (10 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether: ethyl acetate=1:0 to 3:1) to afford 15a. LCMS [M-55] = 340.0; 1H NMR (400 MHz, DMSO-d6) δ 8.81 (bs, 1H), 7.70 - 7.60 (m, 2H), 7.47 (d, J = 7.6 Hz, 1H), 3.87 (d, J = 6.0 Hz, 2H), 1.44 (s, 9H), 1.31 (s, 12H). Step B – Synthesis of (2-chloro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoyl)glycine (15b) Intermediate 15a (1.0 g, 2.5 mmol, 1.0 equiv) was dissolved in HCl/dioxane (4.0 M, 10.0 mL, 15.8 equiv) and the reaction was stirred at 25°C for 30 min. The reaction mixture was filtered and the filtrate was concentrated under reduced pressure to afford 15b which was used without additional purification. LCMS [M+1] = 340.0; 1H NMR (400 MHz, DMSO-d6) δ 8.79 (t, J = 6.0 Hz, 1H), 7.70 - 7.61 (m, 2H), 7.47 (d, J = 7.6 Hz, 1H), 3.91 (d, J = 6.0 Hz, 2H), 1.31 (s, 12H). Step C – Synthesis of tert-butyl 4-((1-((2-chloro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)benzoyl)glycyl)piperidin-4-yl)methyl)piperazine-1-carboxylate (15c) To a solution of 15b (600.0 mg, 1.8 mmol, 1.0 equiv), DIEA (685.1 mg, 5.3 mmol, 923.3 μL, 3.0 equiv) and tert-butyl 4-(piperidin-4-ylmethyl)piperazine-1-carboxylate (600.9 mg, 2.1 mmol, 1.2 equiv) in DMF (6.0 mL) was added T4P (1.9 g, 2.7 mmol, 50% in DMF, 1.5 equiv). The reaction mixture was stirred at 25 °C for 1 h. The mixture was cooled to 25°C quenched by water (10 mL) and extracted with ethyl acetate (3 x 10 mL). The combined organic layers washed with brine (10 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether: ethyl acetate=1:0 to 1:1) to give 15c. LCMS [M+1] = 605.2; 1H NMR (400 MHz, DMSO-d6) δ 8.48 (t, J = 5.6 Hz, 1H), 7.68 - 7.60 (m, 2H), 7.51 (d, J = 7.6 Hz, 1H), 4.33 (d, J = 13.2 Hz, 1H), 4.10 (d, J = 6.0 Hz, 2H), 3.84 (d, J = 12.4 Hz, 1H), 3.29 (s, 4H), 3.06 - 2.96 (m, 1H), 2.64 - 2.58 (m, 1H), 2.32 - 2.24 (m, 5H), 2.13 (d, J = 7.2 Hz, 2H), 1.83 - 1.67 (m, 4H), 1.39 (s, 9H), 1.31 (s, 12H).
Step D – Synthesis of tert-butyl 4-((1-((3-chloro-4'-((1S,2S)-2-(((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)carbamoyl)cyclopropyl)-[1,1'-biphenyl]-4- carbonyl)glycyl)piperidin-4-yl)methyl)piperazine-1-carboxylate (15d) To solution of 15c (490.9 mg, 811.6 μmol, 1.4 equiv), Int 3 (250.0 mg, 579.7 μmol, 1.0 equiv) and Pd(dppf)Cl2•CH2Cl2 (47.3 mg, 57.9 μmol, 0.1 equiv) in dioxane (2.5 mL) and water (0.5 mL) was added K2CO3 (240.4 mg, 1.7 mmol, 3.0 equiv) under nitrogen at 25°C. The reaction was stirred at 80°C for 2 h, cooled to ambient temperature quenched by water (5 mL) and extracted with ethyl acetate (3 x 5 mL). The combined organic layers were washed with brine (5 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, ethyl acetate: methanol=1:0 to 10:1) to afford 15d. LCMS [M+1] = 829.4; 1H NMR (400 MHz, DMSO-d6) δ 8.75 (br t, J = 4.6 Hz, 1H), 8.49 - 8.41 (m, 1H), 7.76 (s, 1H), 7.72 - 7.62 (m, 3H), 7.57 (br d, J = 8.0 Hz, 2H), 7.28 - 7.20 (m, 2H), 4.44 - 4.30 (m, 3H), 3.95 - 3.80 (m, 2H), 3.07 - 2.95 (m, 2H), 2.38 - 2.24 (m, 9H), 2.14 (br d, J = 6.6 Hz, 3H), 1.84 - 1.66 (m, 5H), 1.52 (s, 6H), 1.39 (s, 9H), 1.07 (s, 3H). Step E – Synthesis of 3-chloro-N-(2-oxo-2-(4-(piperazin-1-ylmethyl)piperidin-1-yl)ethyl)-4'- ((1S,2S)-2-(((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5- yl)methyl)carbamoyl)cyclopropyl)-[1,1'-biphenyl]-4-carboxamide (15e) A solution of Intermediate 15d (230.0 mg, 277.3 μmol, 1.0 equiv) in HCl/dioxane (4.0 M, 3.0 mL, 43.2 equiv) was stirred at ambient temperature for 1 h. Concentrated under reduced pressure afforded 15e, which was used without further purification. LCMS [M+1] = 729.3; 1H NMR (400 MHz, DMSO-d6) δ 11.53 - 11.31 (m, 1H), 9.88 - 9.60 (m, 1H), 8.78 (br t, J = 5.4 Hz, 1H), 8.53 - 8.48 (m, 1H), 7.76 (d, J = 1.4 Hz, 1H), 7.72 - 7.63 (m, 3H), 7.57 (d, J = 8.0 Hz, 1H), 7.25 (d, J = 8.4 Hz, 2H), 4.44 - 4.30 (m, 6H), 4.15 - 4.10 (m, 2H), 3.94 - 3.85 (m, 1H), 3.79 - 3.67 (m, 2H), 3.40 - 3.35 (m, 1H), 3.33 - 3.21 (m, 2H), 3.12 - 2.98 (m, 3H), 2.68 - 2.59 (m, 1H), 2.38 - 2.30 (m, 2H), 2.05 - 1.97 (m, 2H), 1.53 (s, 6H), 1.45 - 1.38 (m, 1H), 1.33 - 1.26 (m, 1H), 1.25 - 1.17 (m, 1H), 1.13 - 1.01 (m, 2H). Step F – Synthesis of 3-chloro-N-(2-(4-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3- dioxoisoindolin-5-yl)piperazin-1-yl)methyl)piperidin-1-yl)-2-oxoethyl)-4'-((1S,2S)-2-(((3-(1,1,1-
trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)carbamoyl)cyclopropyl)-[1,1'- biphenyl]-4-carboxamide (Compound 15) To a solution of 15e (100.0 mg, 137.1 μmol, 1.0 equiv.) in DMSO (1.0 mL) was added DIEA (17.7 mg, 137.1 μmol, 23.9 μL, 1.0 equiv) and 2-(2,6-dioxopiperidin-3-yl)-5,6- difluoroisoindoline-1,3-dione (60.5 mg, 205.7 μmol, 1.5 equiv). The mixture was stirred at 80°C for 1hr, cooled to ambient temperature and filtered. The filtrate was purified by prep HPLC(Phenomenex Luna C18150mm x 25mm, 10µm; water(formic acid)-acetonitrile) to give Compound 15. LCMS [M+1] = 1003.4; 1H NMR (400 MHz, DMSO-d6) δ 14.05 - 13.77 (m, 1H), 11.11 (s, 1H), 8.77 (br s, 1H), 8.47 (t, J = 5.6 Hz, 1H), 8.17 (s, 1H), 7.78 - 7.74 (m, 1H), 7.73 - 7.69 (m, 1H), 7.67 (d, J = 8.4 Hz, 2H), 7.58 (d, J = 7.8 Hz, 1H), 7.46 (d, J = 7.4 Hz, 1H), 7.26 (d, J = 8.4 Hz, 2H), 5.11 (dd, J = 5.4, 12.8 Hz, 1H), 4.45 - 4.34 (m, 3H), 4.13 (br d, J = 5.8 Hz, 2H), 3.88 (br d, J = 13.4 Hz, 1H), 3.05 (br t, J = 11.8 Hz, 2H), 2.89 (ddd, J = 5.2, 13.8, 17.0 Hz, 1H), 2.69 - 2.60 (m, 2H), 2.59 - 2.53 (m, 5H), 2.40 - 2.31 (m, 2H), 2.22 (br d, J = 6.8 Hz, 2H), 2.11 - 1.96 (m, 3H), 1.89 - 1.73 (m, 3H), 1.54 (s, 6H), 1.47 - 1.40 (m, 1H), 1.34 - 1.28 (m, 1H), 1.20 - 1.04 (m, 2H), 1.04 - 0.92 (m, 1H). Example 16: Synthesis of Compound 16
To a solution of 2-chloro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoic acid (1.0 g, 3.5 mmol, 1.0 equiv), DIEA (1.4 g, 10.6 mmol, 1.9 mL, 3.0 equiv) and tert-butyl glycinate (557.1 mg, 4.2 mmol, 1.2 equiv) in DMF (10.0 mL) was added T4P (3.1 g, 4.3 mmol, 50.0% purity, 1.2 equiv). The reaction mixture was stirred at 25 °C for 1 h, quenched by water (10 mL)
and extracted with ethyl acetate (3 x 10 mL). The combined organic layers were washed with brine (10 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether: ethyl acetate=1:0 to 3:1) to afford 15a. LCMS [M-55] = 340.0; 1H NMR (400 MHz, DMSO-d6) δ 8.81 (bs, 1H), 7.70 - 7.60 (m, 2H), 7.47 (d, J = 7.6 Hz, 1H), 3.87 (d, J = 6.0 Hz, 2H), 1.44 (s, 9H), 1.31 (s, 12H). Step B – Synthesis of (2-chloro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoyl)glycine (15b) Intermediate 15a (1.0 g, 2.5 mmol, 1.0 equiv) was dissolved in HCl/dioxane (4.0 M, 10.0 mL, 15.8 equiv) and the reaction was stirred at 25°C for 30 min. The reaction mixture was filtered and the filtrate was concentrated under reduced pressure to afford 15b which was used without additional purification. LCMS [M+1] = 340.0; 1H NMR (400 MHz, DMSO-d6) δ 8.79 (t, J = 6.0 Hz, 1H), 7.70 - 7.61 (m, 2H), 7.47 (d, J = 7.6 Hz, 1H), 3.91 (d, J = 6.0 Hz, 2H), 1.31 (s, 12H). Step C – Synthesis of tert-butyl 4-((1-((2-chloro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)benzoyl)glycyl)piperidin-4-yl)methyl)piperazine-1-carboxylate (15c) To a solution of 15b (600.0 mg, 1.8 mmol, 1.0 equiv), DIEA (685.1 mg, 5.3 mmol, 923.3 μL, 3.0 equiv) and tert-butyl 4-(piperidin-4-ylmethyl)piperazine-1-carboxylate (600.9 mg, 2.1 mmol, 1.2 equiv) in DMF (6.0 mL) was added T4P (1.9 g, 2.7 mmol, 50% in DMF, 1.5 equiv). The reaction mixture was stirred at 25 °C for 1 h. The mixture was cooled to 25°C quenched by water (10 mL) and extracted with ethyl acetate (3 x 10 mL). The combined organic layers washed with brine (10 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether: ethyl acetate=1:0 to 1:1) to give 15c. LCMS [M+1] = 605.2; 1H NMR (400 MHz, DMSO-d6) δ 8.48 (t, J = 5.6 Hz, 1H), 7.68 - 7.60 (m, 2H), 7.51 (d, J = 7.6 Hz, 1H), 4.33 (d, J = 13.2 Hz, 1H), 4.10 (d, J = 6.0 Hz, 2H), 3.84 (d, J = 12.4 Hz, 1H), 3.29 (s, 4H), 3.06 - 2.96 (m, 1H), 2.64 - 2.58 (m, 1H), 2.32 - 2.24 (m, 5H), 2.13 (d, J = 7.2 Hz, 2H), 1.83 - 1.67 (m, 4H), 1.39 (s, 9H), 1.31 (s, 12H).
Step D – Synthesis of tert-butyl 4-((1-((3-chloro-4'-((1S,2S)-2-(((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)carbamoyl)cyclopropyl)-[1,1'-biphenyl]-4- carbonyl)glycyl)piperidin-4-yl)methyl)piperazine-1-carboxylate (15d) To solution of 15c (490.9 mg, 811.6 μmol, 1.4 equiv), Int 3 (250.0 mg, 579.7 μmol, 1.0 equiv) and Pd(dppf)Cl2•CH2Cl2 (47.3 mg, 57.9 μmol, 0.1 equiv) in dioxane (2.5 mL) and water (0.5 mL) was added K2CO3 (240.4 mg, 1.7 mmol, 3.0 equiv) under nitrogen at 25°C. The reaction was stirred at 80°C for 2 h, cooled to ambient temperature quenched by water (5 mL) and extracted with ethyl acetate (3 x 5 mL). The combined organic layers were washed with brine (5 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, ethyl acetate: methanol=1:0 to 10:1) to afford 15d. LCMS [M+1] = 829.4; 1H NMR (400 MHz, DMSO-d6) δ 8.75 (br t, J = 4.6 Hz, 1H), 8.49 - 8.41 (m, 1H), 7.76 (s, 1H), 7.72 - 7.62 (m, 3H), 7.57 (br d, J = 8.0 Hz, 2H), 7.28 - 7.20 (m, 2H), 4.44 - 4.30 (m, 3H), 3.95 - 3.80 (m, 2H), 3.07 - 2.95 (m, 2H), 2.38 - 2.24 (m, 9H), 2.14 (br d, J = 6.6 Hz, 3H), 1.84 - 1.66 (m, 5H), 1.52 (s, 6H), 1.39 (s, 9H), 1.07 (s, 3H). Step E – Synthesis of 3-chloro-N-(2-oxo-2-(4-(piperazin-1-ylmethyl)piperidin-1-yl)ethyl)-4'- ((1S,2S)-2-(((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5- yl)methyl)carbamoyl)cyclopropyl)-[1,1'-biphenyl]-4-carboxamide (15e) A solution of Intermediate 15d (230.0 mg, 277.3 μmol, 1.0 equiv) in HCl/dioxane (4.0 M, 3.0 mL, 43.2 equiv) was stirred at ambient temperature for 1 h. Concentrated under reduced pressure afforded 15e, which was used without further purification. LCMS [M+1] = 729.3; 1H NMR (400 MHz, DMSO-d6) δ 11.53 - 11.31 (m, 1H), 9.88 - 9.60 (m, 1H), 8.78 (br t, J = 5.4 Hz, 1H), 8.53 - 8.48 (m, 1H), 7.76 (d, J = 1.4 Hz, 1H), 7.72 - 7.63 (m, 3H), 7.57 (d, J = 8.0 Hz, 1H), 7.25 (d, J = 8.4 Hz, 2H), 4.44 - 4.30 (m, 6H), 4.15 - 4.10 (m, 2H), 3.94 - 3.85 (m, 1H), 3.79 - 3.67 (m, 2H), 3.40 - 3.35 (m, 1H), 3.33 - 3.21 (m, 2H), 3.12 - 2.98 (m, 3H), 2.68 - 2.59 (m, 1H), 2.38 - 2.30 (m, 2H), 2.05 - 1.97 (m, 2H), 1.53 (s, 6H), 1.45 - 1.38 (m, 1H), 1.33 - 1.26 (m, 1H), 1.25 - 1.17 (m, 1H), 1.13 - 1.01 (m, 2H). Step F – Synthesis of 3-chloro-N-(2-(4-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3- dioxoisoindolin-5-yl)piperazin-1-yl)methyl)piperidin-1-yl)-2-oxoethyl)-4'-((1S,2S)-2-(((3-(1,1,1-
trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)carbamoyl)cyclopropyl)-[1,1'- biphenyl]-4-carboxamide (Compound 15) To a solution of 15e (100.0 mg, 137.1 μmol, 1.0 equiv.) in DMSO (1.0 mL) was added DIEA (17.7 mg, 137.1 μmol, 23.9 μL, 1.0 equiv) and 2-(2,6-dioxopiperidin-3-yl)-5,6- difluoroisoindoline-1,3-dione (60.5 mg, 205.7 μmol, 1.5 equiv). The mixture was stirred at 80°C for 1hr, cooled to ambient temperature and filtered. The filtrate was purified by prep HPLC(Phenomenex Luna C18150mm x 25mm, 10µm; water(formic acid)-acetonitrile) to give Compound 15. LCMS [M+1] = 1003.4; 1H NMR (400 MHz, DMSO-d6) δ 14.05 - 13.77 (m, 1H), 11.11 (s, 1H), 8.77 (br s, 1H), 8.47 (t, J = 5.6 Hz, 1H), 8.17 (s, 1H), 7.78 - 7.74 (m, 1H), 7.73 - 7.69 (m, 1H), 7.67 (d, J = 8.4 Hz, 2H), 7.58 (d, J = 7.8 Hz, 1H), 7.46 (d, J = 7.4 Hz, 1H), 7.26 (d, J = 8.4 Hz, 2H), 5.11 (dd, J = 5.4, 12.8 Hz, 1H), 4.45 - 4.34 (m, 3H), 4.13 (br d, J = 5.8 Hz, 2H), 3.88 (br d, J = 13.4 Hz, 1H), 3.05 (br t, J = 11.8 Hz, 2H), 2.89 (ddd, J = 5.2, 13.8, 17.0 Hz, 1H), 2.69 - 2.60 (m, 2H), 2.59 - 2.53 (m, 5H), 2.40 - 2.31 (m, 2H), 2.22 (br d, J = 6.8 Hz, 2H), 2.11 - 1.96 (m, 3H), 1.89 - 1.73 (m, 3H), 1.54 (s, 6H), 1.47 - 1.40 (m, 1H), 1.34 - 1.28 (m, 1H), 1.20 - 1.04 (m, 2H), 1.04 - 0.92 (m, 1H). Example 16: Synthesis of Compound 16
Step A – Synthesis of tert-butyl 4-[4-[[2-chloro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl) benzoyl]amino]-1- piperidyl]piperidine-1-carboxylate (16a) To a solution of 2-chloro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoic acid (0.5 g, 1.77 μmol, 1.0 equiv) in DMF (5 mL) was added DIEA (686.18 mg, 5.31 mmol, 924.77 μL, 3 equiv), HOBt (358.7 mg, 2.65 mmol, 1.5 equiv) and EDCI (508.9 mg, 2.65 mmol, 1.5 equiv) and the mixture was stirred at 25°C for 30 minutes. The mixture was treated with tert-butyl 4-(4- amino-1-piperidyl)piperidine-1-carboxylate (501.6 mg, 1.77 mmol, 1 equiv) and stirred at 25°C for 12 h. The reaction mixture was diluted with water (8 mL) and extracted with ethyl acetate (3 x 5 mL). The combined organic portions were washed with brine (5 mL), dried with anhydrous
sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, dichloromethane/ methanol = 100: 1 to 100: 10) to afford 16a. LCMS: [M+1] = 548.3, 550.3; 1H NMR (400MHz, methanol-d4): δ 7.73 (s, 1H), 7.68 (d, J = 7.2 Hz, 1H), 7.39 (d, J = 7.2 Hz, 1H), 4.16 (d, J = 13.2 Hz, 2H), 3.85-3.96 (m, 1H), 3.02- 3.12 (m, 2H), 2.69-2.84 (m, 2H), 2.67- 2.57 (m, 1H), 2.50 (t, J = 11.6 Hz, 2H), 1.99 - 2.11 (m, 2H), 1.91 (br d, J = 11.6 Hz, 2H), 1.59-1.74 (m, 2H), 1.46 (s, 9H), 1.39-1.44 (m, 2H), 1.32-1.39 (m, 12H). Step B – Synthesis of tert-butyl 4-[4-[[2-chloro-4-[4-[(1S,2S)-2-[[3-(2,2,2-trifluoro-1,1-dimethyl- ethyl)-1H-1,2,4-triazol-5-yl]methylcarbamoyl]cyclopropyl]phenyl]benzoyl]amino]-1- piperidyl]piperidine-1-carboxylate (16b) To a solution of Intermediate 3 (104.9 mg, 243.3 μmol, 1 equiv) in dioxane (3 mL) and water (0.5 mL) was added 16a (200 mg, 365.0 μmol, 1.5 equiv), Pd(dppf)Cl2•CH2Cl2 (19.9 mg, 24.3 μmol, 0.1 equiv) and K2CO3 (100.9 mg, 730.0 μmol, 3 equiv) at 25°C. The mixture was three-fold degassed and purged with nitrogen, stirred at 100°C for 16 h under nitrogen. The reaction mixture was diluted with water (5 mL) and extracted with ethyl acetate (3 x 5 mL). The combined organic phases were washed with brine (5 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, dichloromethane: methanol = 100: 1 to 100: 10) to afford 16b. LCMS: [M+1] = 772.5, 773.7l; 1H NMR (400MHz, methanol-d4): δ 7.69 (d, J = 1.6 Hz, 1H), 7.54-7.64 (m, 3H), 7.48 (d, J = 8.0 Hz, 1H), 7.25 (d, J = 8.4 Hz, 2H), 5.49 (s, 2H), 4.55 - 4.50 (m, 2H), 4.16 (d, J = 13.2 Hz, 2H), 3.98 - 3.86 (m, 1H), 3.07 (br d, J = 12.4 Hz, 2H), 2.76 (br s, 2H), 2.65 - 2.56 (m, 1H), 2.42-2.55 (m, 3H), 2.06 (d, J = 11.2 Hz, 2H), 1.96-2.02 (m, 1H), 1.91 (d, J = 11.6 Hz, 2H), 1.64-1.73 (m, 2H), 1.60 (s, 6H), 1.46 (s, 9H), 1.27-1.42 (m, 4H). Step C – Synthesis of 2-chloro-N-[1-(4-piperidyl)-4-piperidyl]-4-[4-[(1S,2S)-2-[[3-(2,2,2- trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methylcarbamoyl]cyclopropyl]phenyl]benzamide (16c) To a solution of 16b (100 mg, 129.5 μmol, 1 equiv) in dichloromethane (1 mL) was added trifluoroacetic acid (0.2 mL) at 25°C. The mixture was stirred at 25°C for 1 h. The mixture was concentrated under reduced pressure to give 16c, which was used without further purification. LCMS: [M+1] = 672.5, 674.5; 1H NMR (Methanol-d4, 400MHz): δ 7.72 (d, J = 1.6 Hz, 1H), 7.63
(d, J = 8.0 Hz, 1H), 7.58 (d, J = 8.0 Hz, 2H), 7.51 (d, J = 7.2 Hz, 1H), 7.26 (d, J = 8.4 Hz, 2H), 4.55 (d, J = 2.0 Hz, 2H), 4.25 - 4.10 (m, 1H), 3.53-3.77 (m, 6H), 3.07-3.16 (m, 2H), 2.45-2.53 (m, 1H), 2.32-2.44 (m, 4H), 1.77-2.17 (m, 6H), 1.60 (s, 6H), 1.39 - 1.27 (m, 2H). Step D – Synthesis of 2-chloro-N-[1-[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin- 5-yl]-4-piperidyl]-4-piperidyl]-4-[4-[(1S,2S)-2-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4- triazol-5-yl]methylcarbamoyl]cyclopropyl]phenyl]benzamide (Compound 16) To a solution of 16c (90 mg, 114.5 μmol, 1 equiv) in DMSO (1 mL) was added DIEA (74.0 mg, 572.4 μmol, 99.7 μL, 5 equiv) and 2-(2,6-dioxo-3-piperidyl)-5,6-difluoro-isoindoline- 1,3-dione (50.5 mg, 171.7 μmol, 1.5 equiv). The mixture was three-fold degassed and purged with nitrogen and then stirred at 70°C for 12 h under a nitrogen atmosphere. The reaction mixture was purified directly by preparative HPLC (Waters Xbridge C18150mm x 50mm, 10µm; H2O (10 mM NH4HCO3) - acetonitrile) to give Compound 16. LCMS: [M+1] = 946.5, 948.5; 1H NMR (Methanol-d4, 400MHz): δ 7.70 (d, J = 1.6 Hz, 1H), 7.45-7.65 (m, 6H), 7.25 (d, J = 8.4 Hz, 2H), 5.09 (dd, J = 2.0, 8.4 Hz, 1H), 4.55 (d, J = 2.0 Hz, 2H), 3.98 - 3.87 (m, 1H), 3.76 (d, J = 12.0 Hz, 2H), 3.09 (d, J = 12.0 Hz, 2H), 2.92 (t, J = 11.6 Hz, 2H), 2.81-2.88 (m, 1H), 2.65-2.78 (m, 2H), 2.52-2.62 (m, 1H), 2.40-2.52 (m, 3H), 1.97-2.16 (m, 6H), 1.65-1.80 (m, 4H), 1.60 (s, 6H), 1.58 - 1.54 (m, 1H), 1.32-1.37 (m, 1H). Example 17: Synthesis of Compound 17
To a solution of 5-bromo-3-methyl-1H-indazole (1.0 g, 4.7 mmol, 1.0 equiv) and tert- butyl-2-bromoacetate (1.3 g, 7.1 mmol, 1.0 mL, 1.5 equiv) in DMF (10.0 mL) was added Cs2CO3 (2.3 g, 7.1 mmol, 1.5 equiv). The mixture was stirred at 20°C for 1 h, diluted with water (15 mL) and extracted with ethyl acetate (2 x 50 mL). The combined organic layers were washed with brine (2 x 15 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether/ethyl acetate=10:1 to 0:1) to afford 17a. LCMS [M-55] = 269.0, 271.0; 1H NMR (400 MHz, CHLOROFORM-d) δ 7.81 (d, J = 1.2 Hz, 1H), 7.46 (dd, J = 1.8, 8.8 Hz, 1H), 7.16 (d, J = 8.8 Hz, 1H), 4.96 (s, 2H), 2.55 (s, 3H), 1.44 (s, 9H). Step B – Synthesis of tert-butyl 2-[3-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)indazol-1-yl]acetate (17b) To a solution of 17a (600.0 mg, 1.8 mmol, 1.0 equiv) and 4,4,5,5-tetramethyl-2-(4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3,2-dioxaborolane (702.7 mg, 2.7 mmol, 1.5 equiv) in dioxane (6.0 mL) was added potassium acetate (543.2 mg, 5.5 mmol, 3.0 equiv) and Pd(dppf)Cl2
(135.0 mg, 184.5 μmol, 0.1 equiv). The mixture was stirred at 100°C for 16 h cooled to ambient temperature, diluted with water (3 mL) and extracted with ethyl acetate (2 x 30 mL). The combined organic layers were washed with brine (2 x 2 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, dichloromethane : methanol=100 :1 to 0 :1) to afford 17b. LCMS [M+1] = 373.3; 1H NMR (400 MHz, CHLOROFORM-d) δ 8.21 (s, 1H), 7.81 (dd, J = 0.6, 8.5 Hz, 1H), 7.25 (d, J = 8.0 Hz, 1H), 4.98 (s, 2H), 2.60 (s, 3H), 1.40 (d, J = 17.2 Hz, 21H). Step C – Synthesis of tert-butyl 2-[3-methyl-5-[4-[(1S,2S)-2-[[3-(2,2,2-trifluoro-1,1-dimethyl- ethyl)-1H-1,2,4-triazol-5-yl]methylcarbamoyl]cyclopropyl]phenyl]indazol-1-yl]acetate (17c) To a solution of 17b (258.9 mg, 695.6 μmol, 1.5 equiv) and Intermediate 3 (200.0 mg, 463.7 μmol, 1.0 equiv) in tetrahydrofuran (4 mL) and water (1.0 mL) was added K3PO4 (295.3 mg, 1.3 mmol, 3.0 equiv) and Pd(dppf)Cl2 (60.4 mg, 92.7 μmol, 0.2 equiv) under nitrogen. The mixture was stirred at 80°C for 16 h, cooled to ambient temperature, treated with water (3 mL) and extracted with ethyl acetate (2 x 30 mL). The combined organic layers were washed with brine (2 x 2 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, dichloromethane : methanol=100 :1 to 0 : 1) afford 17c. LCMS [M+1] = 597.4 Step D – Synthesis of 2-[3-methyl-5-[4-[(1S,2S)-2-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H- 1,2,4-triazol-5-yl]methylcarbamoyl]cyclopropyl]phenyl]indazol-1-yl]acetic acid (17d) To a solution of 17c (200.0 mg, 335.2 μmol, 1.0 equiv) in dichloromethane (2.0 mL) was added trifluoroacetic acid (614.0 mg, 5.3 mmol, 0.4 mL, 16.0 equiv) . The mixture was stirred at 20°C for 16 h. The mixture was concentrated under reduced pressure to afford 17d, which was used without further purification. LCMS [M+1] = 541.4. Step E – Synthesis of (1S,2S)-2-[4-[1-[2-[4-[[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo- isoindolin-5-yl]piperazin-1-yl]methyl]-1-piperidyl]-2-oxo-ethyl]-3-methyl-indazol-5-yl]phenyl]- N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methyl]cyclopropanecarboxamide (Compound 17)
To a solution of 17d (180.0 mg, 333.0 μmol, 1.0 equiv) in DMF (2.0 mL) was added T4P (527.8 mg, 732.6 μmol, 50% purity, 2.2 equiv) and DIEA (215.1 mg, 1.6 mmol, 290.0 μL, 5.0 eq). The reaction was stirred at 20°C for 15 min. Then 2-(2,6-dioxo-3-piperidyl)-5-fluoro-6-[4-(4- piperidylmethyl)piperazin-1-yl]isoindoline-1,3-dione (285.6 mg, 499.5 μmol, 1.5 equiv) was added and the resulting mixture was stirred at 20°C for 1 h. The reaction mixture was diluted with water (3 mL) and extracted with ethyl acetate (2 x 30 mL). The combined organic layers were washed with brine (2 x 2 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative HPLC (Phenomenex Luna C18100mm x 30mm, 5µm; H2O (0.2% formic acid)-acetonitrile) to afford Compound 17. LCMS [M+1] = 980.4; 1H NMR (400 MHz, CHLOROFORM-d) δ 8.52 - 8.44 (m, 1H), 8.40 (br s, 1H), 7.75 (s, 1H), 7.57 (d, J = 8.8 Hz, 1H), 7.52 - 7.47 (m, 2H), 7.46 - 7.40 (m, 1H), 7.39 - 7.35 (m, 1H), 7.11 (d, J = 8.0 Hz, 2H), 6.84 (br s, 1H), 5.30 - 5.12 (dd, J = 16.4, 16.0 Hz, 2H), 4.94 (dd, J = 5.2, 12.2 Hz, 1H), 4.61 - 4.50 (m, 3H), 4.05 (br d, J = 13.4 Hz, 1H), 3.29 (br s, 4H), 3.14 - 3.03 (m, 1H), 2.98 - 2.86 (m, 1H), 2.86 - 2.69 (m, 2H), 2.65 (br s, 4H), 2.61 - 2.53 (m, 4H), 2.30 (br s, 2H), 2.20 - 2.11 (m, 1H), 1.92 - 1.74 (m, 3H), 1.74 - 1.65 (m, 2H), 1.62 (s, 6H), 1.44 - 1.25 (m, 2H), 1.17 - 0.95 (m, 2H), 0.93 - 0.83 (m, 1H). Example 18: Synthesis of Compound 18
Step A – Synthesis of tert-butyl 3-[4-(4-bromophenyl)pyrazol-1-yl]azetidine-1-carboxylate (18a) To a solution of 4-(4-bromophenyl)-1H-pyrazole (1.2 g, 5.38 mmol, 1.0 equiv) in DMF (15 mL) was added cesium carbonate (3.51 g, 10.76 mmol, 2.0 equiv) and tert-butyl 3- methylsulfonyloxyazetidine-1-carboxylate (2.03 g, 8.07 mmol, 1.5 equiv). The mixture was heated at 70°C for 16 h, cooled to ambient temperature and partitioned between ethyl acetate (60.0 mL) and water (60.0 mL). The organic phase was separated, washed with brine (3 x 20 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel,
dichloromethane : methanol=1/0 to 95/5) to afford 18a. LCMS [M-55] = 322.0, 324.0; 1H NMR (400 MHz, CHLOROFORM-d) δ 7.83 (s, 1H), 7.77 (s, 1H), 7.53 - 7.45 (m, 2H), 7.39 - 7.33 (m, 2H), 5.08 (tt, J = 5.5, 7.8 Hz, 1H), 4.45 - 4.29 (m, 4H), 1.47 (s, 9H). Step B – Synthesis of 1-(azetidin-3-yl)-4-(4-bromophenyl)pyrazole (18b) To a solution of 18a (1.8 g, 4.76 mmol, 1.0 equiv) in dichloromethane (25 mL) was added trifluoroacetic acid (3.91 mL). The mixture was stirred at 25°C for 2 h and then concentrated under reduced pressure to afford 18b, which was used without further purification. LCMS [M+1] = 278.0, 280.0; 1H NMR (400 MHz, DMSO-d6) δ 9.10 (bs, 2H), 8.36 (s, 1H), 8.16 (s, 1H), 7.57 (s, 4H), 5.44 - 5.34 (m, 1H), 4.46 - 4.34 (m, 4H). Step C – Synthesis of tert-butyl 4-[2-[3-[4-(4-bromophenyl)pyrazol-1-yl]azetidin-1- yl]acetyl]piperazine-1-carboxylate (18c) To a solution of 18b (500 mg, 1.27 mmol, 1.0 eq, TFA) and tert-butyl 4-(2- bromoacetyl)piperazine-1-carboxylate (352.48 mg, 1.15 mmol, 0.9 eq) in acetonitrile (5.0 mL) was added DIEA (329.56 mg, 2.55 mmol, 444.15 μL, 2.0 eq). The mixture was stirred at 20°C for 4 h and then partitioned between ethyl acetate (40 mL) and water (40 mL). The organic phase was separated, washed with brine (3 x 10 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, dichloromethane : methanol=1/0 to 97/3) to afford 18c. LCMS [M+1] = 504.2, 506.2. Step D – Synthesis of tert-butyl 4-[2-[3-[4-[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)phenyl]pyrazol-1-yl]azetidin-1-yl]acetyl]piperazine-1-carboxylate (18d) To a solution of 18c (200 mg, 396.50 μmol, 1.0 equiv), 4,4,5,5-tetramethyl-2-(4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3,2-dioxaborolane (141.0 mg, 555.1 μmol, 1.4 equiv), potassium acetate (116.7 mg, 1.19 mmol, 3.0 equiv), Pd(dppf)Cl2 (32.4 mg, 39.7 μmol, 0.1 equiv) in dioxane (0.5 mL) was three-fold degassed and purged with nitrogen and then the mixture was stirred at 100°C for 16 h under an atmosphere of nitrogen. The reaction mixture was partitioned between ethyl acetate (30 mL) and water (30mL). The organic phase was separated, washed with brine (2 x 10 mL), dried over sodium sulfate, filtered and the filtrate was concentrated under
reduced pressure. The residue was purified by column chromatography by preparative TLC (silica gel, dichloromethane: methanol = 10:1) to afford 18d. LCMS [M+1] = 552.3. Step E – Synthesis of tert-butyl 4-[2-[3-[4-[4-[4-[(1S,2S)-2-[[3-(2,2,2-trifluoro-1,1-dimethyl- ethyl)-1H-1,2,4-triazol-5-yl]methylcarbamoyl]cyclopropyl]phenyl]phenyl]pyrazol-1-yl]azetidin- 1-yl]acetyl]piperazine-1-carboxylate (18e) To a solution of 18d (80 mg, 145 μmol, 1.2 equiv), Intermediate 3 (52.1 mg, 120.9 μmol, 1.0 equiv), Pd(dppf)Cl2 (49.4 mg, 60.4 μmol, 0.5 equiv), potassium carbonate (50.1 mg, 362.7 μmol, 3.0 equiv) in dioxane (0.4 mL) and water (0.1 mL) was three-fold degassed and purged with nitrogen. The mixture was stirred at 100°C for 2 h under N2 atmosphere. The reaction mixture was partitioned between ethyl acetate (30 mL) and water (30 mL). The organic phase was separated, washed with brine 30 mL (3 x 10 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by prep-TLC (silica gel, dichloromethane: methanol = 10:1) to afford 18e. LCMS [M+1] + = 776.6. Step F – Synthesis of (1S,2S)-2-[4-[4-[1-[1-(2-oxo-2-piperazin-1-yl-ethyl)azetidin-3-yl]pyrazol-4- yl]phenyl]phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methyl]cyclopropanecarboxamide (18f) To a solution of 18e (58.0 mg, 74.8 μmol, 1.0 equiv) in dichloromethane (0.5 mL) was added TFA (171 μL). The mixture was stirred at 20°C for 1 h and then concentrated under reduced pressure to afford 18f, which was used without further purification. LCMS [M+1] = 676.6. Step G – Synthesis of (1S,2S)-2-[4-[4-[1-[1-[2-[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo- isoindolin-5-yl]piperazin-1-yl]-2-oxo-ethyl]azetidin-3-yl]pyrazol-4-yl]phenyl]phenyl]-N-[[3- (2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]cyclopropanecarboxamide (Compound 18) To a solution of 18f (56 mg, 70.91 μmol, 1.0 equiv) and 2-(2,6-dioxo-3-piperidyl)-5,6- difluoro-isoindoline-1,3-dione (31.3 mg, 106 μmol, 1.5 equiv) in DMSO (0.5 mL) was added DIEA (27.5 mg, 212 μmol, 37 μL, 3.0 equiv). The mixture was stirred at 70°C for 16 h, cooled to ambient temperature and partitioned between ethyl acetate (20 mL) and water (20 mL). The organic phase was separated, washed with brine (2 x 10 mL), dried over anhydrous sodium
sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative TLC (silica gel, dichloromethane: methanol = 10:1) to afford Compound 18. LCMS [M+1] = 950.4; 1H NMR (400 MHz, DMSO-d6) δ 14.04 - 13.79 (br s, 1H), 11.21 - 10.99 (br s, 1H), 8.77 (br s, 1H), 8.42 (s, 1H), 8.00 (s, 1H), 7.77 (d, J = 11.2 Hz, 1H), 7.71 - 7.56 (m, 6H), 7.50 (d, J = 7.4 Hz, 1H), 7.22 (d, J = 8.2 Hz, 2H), 5.16 - 4.97 (m, 2H), 4.41 (br d, J = 5.4 Hz, 2H), 3.85 (t, J = 7.4 Hz, 2H), 3.67 - 3.60 (m, 4H), 3.60 - 3.46 (m, 4H), 3.24 (br s, 3H), 2.96 - 2.81 (m, 1H), 2.70 - 2.54 (m, 2H), 2.33 (br s, 2H), 2.08 - 1.94 (m, 2H), 1.53 (s, 6H), 1.46 - 1.37 (m, 1H), 1.33 - 1.20 (m, 1H). Example 19: Synthesis of Compound 19
 tert-butyl 4-(bromomethyl)piperidine-1-carboxylate in Step A to afford (1S,2S)-2-[4-[4-[1-[[1-[2-[4- [2- (2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl] piperazin-1-yl]-2-oxo-ethyl] -4- piperidyl]methyl]pyrazol-4-yl]phenyl]phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H- 1,2,4-triazol-5-yl]methyl]cyclopropanecarboxamide (Compound 19). LCMS: [M+1] = 992.5; 1H NMR (400 MHz, DMSO-d6) δ 13.91 (s, 1H), 11.11 (s, 1H), 8.78 (s, 1H), 8.19 (s, 1H), 8.15 (s, 1H), 7.91 (s, 1H), 7.76 (d, J = 11.2 Hz, 1H), 7.65 - 7.57 (m, 6H), 7.49 (d, J = 7.2 Hz, 1H), 7.21 (d, J = 8.4 Hz, 2H), 5.12 (dd, J = 5.2, 12.8 Hz, 1H), 4.41 (d, J = 4.4 Hz, 2H), 4.01 (d, J = 6.8 Hz, 2H), 3.76 - 3.71 (m, 2H), 3.63 - 3.58 (s, 2H), 3.27 - 3.14 (m, 6H), 2.93 - 2.76 (m, 3H), 2.64 - 2.53 (m, 2H), 2.36 - 2.29 (m, 1H), 2.09 - 1.91 (m, 4H), 1.88 - 1.77 (m, 1H), 1.53 (s, 6H), 1.49 (s, 1H), 1.44 - 1.38 (m, 1H), 1.32 - 1.22 (m, 3H). Example 20: Synthesis of Compound 20
tert-butyl 4-(bromomethyl)piperidine-1-carboxylate in Step A to afford (1S,2S)-2-[4-[4-[1-[[1-[2-[4- [2- (2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl] piperazin-1-yl]-2-oxo-ethyl] -4- piperidyl]methyl]pyrazol-4-yl]phenyl]phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H- 1,2,4-triazol-5-yl]methyl]cyclopropanecarboxamide (Compound 19). LCMS: [M+1] = 992.5; 1H NMR (400 MHz, DMSO-d6) δ 13.91 (s, 1H), 11.11 (s, 1H), 8.78 (s, 1H), 8.19 (s, 1H), 8.15 (s, 1H), 7.91 (s, 1H), 7.76 (d, J = 11.2 Hz, 1H), 7.65 - 7.57 (m, 6H), 7.49 (d, J = 7.2 Hz, 1H), 7.21 (d, J = 8.4 Hz, 2H), 5.12 (dd, J = 5.2, 12.8 Hz, 1H), 4.41 (d, J = 4.4 Hz, 2H), 4.01 (d, J = 6.8 Hz, 2H), 3.76 - 3.71 (m, 2H), 3.63 - 3.58 (s, 2H), 3.27 - 3.14 (m, 6H), 2.93 - 2.76 (m, 3H), 2.64 - 2.53 (m, 2H), 2.36 - 2.29 (m, 1H), 2.09 - 1.91 (m, 4H), 1.88 - 1.77 (m, 1H), 1.53 (s, 6H), 1.49 (s, 1H), 1.44 - 1.38 (m, 1H), 1.32 - 1.22 (m, 3H). Example 20: Synthesis of Compound 20
To a solution of tert-butyl (3R)-3-hydroxypyrrolidine-1-carboxylate (5.0 g, 26.7 mmol, 1.0 equiv) in tetrahydrofuran (50.0 mL) was added PPh3 (8.4 g, 32.0 mmol, 1.2 equiv) and pyridin-4- ol (3.0 g, 32.0 mmol, 1.2 equiv). DIAD (6.4 g, 32.0 mmol, 6.2 mL, 1.2 equiv) was added at 0°C. The mixture was stirred at 55°C for 16 h, cooled to ambient temperature and treated with water (5 mL). The mixture was extracted with ethyl acetate (3 x 10 mL) and the combined organic phase was washed with brine (10 mL). Then the organic phase was dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure to give the residue. The residue was purified by column chromatography (silica gel, dichloromethane/methanol=15/1 to 10/1) to afford 20a. LCMS [M+1] =265.0; 1H NMR (400 MHz, DMSO-d6) δ = 8.42 - 8.36 (m, 2H), 7.01 - 6.95 (m, 2H), 5.12 (br s, 1H), 3.64 - 3.52 (m, 1H), 3.49 - 3.35 (m, 2H), 3.31 - 3.25 (m, 1H), 2.26 - 1.98 (m, 2H), 1.39 (br d, J = 5.6 Hz, 9H) Step B – Synthesis of tert-butyl (3S)-3-(4-piperidyloxy)pyrrolidine-1-carboxylate (20b) To a solution of 20a (1.0 g, 3.7 mmol, 1.0 equiv) in ethanol (10.0 mL) was added H2SO4 (371.0 mg, 3.7 mmol, 201.6 μL, 1.0 equiv) and PtO2 (257.7 mg, 1.1 mmol, 0.3 equiv) under a nitrogen atmosphere. The suspension was three-fold degassed and purged with hydrogen. The mixture was stirred under hydrogen (1 atm) at 25°C for 16 h. The reaction mixture was filtered to remove solids and the filtrate was poured into NaOH (1M). Volatile components were removed
under reduced pressure and the resulting mixture was extracted with dichloromethane (3 x 10 mL). The combined organic phase was washed with brine (10 mL), dried with anhydrous sodium sulfate, filtered and filtrate was concentrated under reduced pressure to afford 20b. LCMS [M+1] =271.1; 1H NMR (400 MHz, DMSO-d6) δ = 4.25 - 4.12 (m, 1H), 3.39 - 3.11 (m, 6H), 2.87 (br d, J = 12.4 Hz, 2H), 2.41 (br t, J = 11.2 Hz, 2H), 1.89 - 1.72 (m, 4H), 1.39 (s, 9H), 1.30 - 1.12 (m, 2H). Step C – Synthesis of tert-butyl (3S)-3-[[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo- isoindolin-5-yl]-4-piperidyl]oxy]pyrrolidine-1-carboxylate (20c) To a solution of 20b (183.7 mg, 679.7 μmol, 1.0 equiv) in DMSO (3.0 mL) was added 2- (2,6-dioxo-3-piperidyl)-5,6-difluoro-isoindoline-1,3-dione (200.0 mg, 679.7 μmol, 1.0 equiv) and DIEA (439.2 mg, 3.4 mmol, 592.0 μL, 5.0 equiv). The suspension was three-fold degassed and purged with nitrogen, stirred at 70°C for 12 h, cooled to ambient temperature and treated with water (5 mL). The mixture was extracted with ethyl acetate (3 x 10 mL). The combined organic portions were washed with brine (10 mL). The organic phase was dried by anhydrous sodium sulfate, filtered and filtrate was concentrated under reduced pressure. The residue was purified by preparative TLC (silica gel, dichloromethane: methanol = 10:1) to afford 20c. LCMS [M+1] = 545.3; 1H NMR (400 MHz, DMSO-d6) δ = 11.20 - 11.03 (m, 1H), 7.71 (d, J = 11.6 Hz, 1H), 7.45 (d, J = 7.2 Hz, 1H), 5.10 (dd, J = 5.6, 12.8 Hz, 1H), 4.23 (br s, 1H), 3.66 - 3.56 (m, 1H), 3.48 (br d, J = 12.8 Hz, 2H), 3.42 - 3.34 (m, 1H), 3.29 - 3.20 (m, 2H), 3.05 (br t, J = 10.0 Hz, 2H), 2.94 - 2.83 (m, 1H), 2.65 - 2.53 (m, 2H), 2.12 - 1.83 (m, 6H), 1.66 - 1.50 (m, 2H), 1.40 (s, 9H). Step D – Synthesis of 2-(2,6-dioxo-3-piperidyl)-5-fluoro-6-[4-[(3S)-pyrrolidin-3-yl]oxy-1- piperidyl]isoindoline-1,3-dione (20d) The mixture of 20c (190.0 mg, 348.9 μmol, 1 equiv) in trifluoroacetic acid (5 mL) and dichloromethane (1 mL) was stirred at 25°C for 1 h. The mixture was concentrated under reduced pressure. The residue was triturated with MTBE (5 mL) at 25°C for 20 min to afford 20d. LCMS [M+1] = 445.3; 1H NMR (400 MHz, DMSO-d6) δ 11.11 (s, 1H), 9.09 - 8.67 (m, 2H), 7.72 (d, J = 11.6 Hz, 1H), 7.46 (d, J = 7.2 Hz, 1H), 5.10 (dd, J = 5.6, 12.8 Hz, 1H), 4.40 (br s, 1H), 3.66 - 3.62 (m, 2H), 3.52 - 3.45 (m, 2H), 3.31 - 3.16 (m, 4H), 3.06 - 3.02 (m, 1H), 2.92 - 2.84 (m, 1H), 2.65 - 2.52 (m, 2H), 2.12 - 1.91 (m, 6H), 1.66 - 1.50 (m, 2H).
Step E – Synthesis of (1S,2S)-2-[4-[4-[1-[2-[(3S)-3-[[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3- dioxo-isoindolin-5-yl]-4-piperidyl] oxy]pyrrolidin-1-yl]-2-oxo-ethyl]pyrazol-4-yl]phenyl]phenyl]- N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methyl]cyclopropanecarboxamide (Compound 20) To a solution of 20d (67.5 mg, 152.0 μmol, 1.2 equiv) in DMF (1.5 mL) was added 13d (70.0 mg, 126.6 μmol, 1.0 equiv) and DIEA (81.8 mg, 633.4 μmol, 110.3 μL, 5.0 equiv). The mixture was treated with T4P (136.9 mg, 190.0 μmol, 50% in DMF, 1.5 equiv). The mixture was stirred at 25°C for 1 h. The mixture was treated with water (5 mL), extracted with ethyl acetate (3 x 10 mL). The combined organic portions were washed with brine (10 mL), dried with anhydrous sodium sulfate, filtered and filtrate was concentrated under reduced pressure. The residue was purified by preparative HPLC Waters Xbridge BEH C18100mm x 30mm, 10µm; water(10mM NH4HCO3)-acetonitrile) to afford Compound 20. LCMS [M+1] = 979.3; 1H NMR (400 MHz, Methanol-d4) δ 8.05 (d, J = 5.2 Hz, 1H), 7.91 (d, J = 4.4 Hz, 1H), 7.69 - 7.41 (m, 8H), 7.25 - 7.18 (m, 2H), 5.18 - 5.01 (m, 3H), 4.66 - 4.48 (m, 2H), 4.48 - 4.30 (m, 1H), 3.81 - 3.41 (m, 8H), 3.17 - 2.99 (m, 3H), 2.93 - 2.80 (m, 1H), 2.80 - 2.64 (m, 2H), 2.52 - 2.43 (m, 1H), 2.29 - 1.94 (m, 6H), 1.82 - 1.68 (m, 2H), 1.66 - 1.53 (m, 6H), 1.42 - 1.25 (m, 2H). Example 21: Synthesis of Intermediate 21 (Int 21)
 a mg, acid (62.3 mg, 695.6 μmol, 3 equiv) in EtOH (1 mL) was added DIEA (299.6 mg, 2.32 mmol, 403.9 μL, 10 equiv) and Pd(dppf)Cl2 (3.02 mg, 4.64 μmol, 0.02 equiv). The mixture was three-fold degassed and purged with nitrogen and stirred at 50°C for 2 h under an atmosphere of nitrogen. The mixture was concentrated under reduced pressure to give [4-[(1S,2S)-2-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methylcarbamoyl]cyclopropyl]phenyl]boronic acid (Intermediate 21), which was used without further purification. Example 22: Synthesis of Compound 22
a mg, acid (62.3 mg, 695.6 μmol, 3 equiv) in EtOH (1 mL) was added DIEA (299.6 mg, 2.32 mmol, 403.9 μL, 10 equiv) and Pd(dppf)Cl2 (3.02 mg, 4.64 μmol, 0.02 equiv). The mixture was three-fold degassed and purged with nitrogen and stirred at 50°C for 2 h under an atmosphere of nitrogen. The mixture was concentrated under reduced pressure to give [4-[(1S,2S)-2-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methylcarbamoyl]cyclopropyl]phenyl]boronic acid (Intermediate 21), which was used without further purification. Example 22: Synthesis of Compound 22
S ep – Syn es s o me y -( -bromopyrazo - -y ) benzoa e ( a) To a solution of 4-bromo-1H-pyrazole (980.0 mg, 6.67 mmol, 1.2 equiv), (4- methoxycarbonylphenyl)boronic acid (1 g, 5.56 mmol, 1 equiv) in dichloromethane (20 mL) was added pyridine (879.0 mg, 11.1 mmol, 897.0 μL, 2 equiv) and Cu(OAc)2 (2.02 g, 11.1 mmol, 2 equiv) and purged with oxygen. The mixture was stirred at 20°C for 16 h under oxygen (1 atm). The mixture was quench with water (100 mL) and extracted with dichloromethane (2 x 50 mL). The combined organic layers were washed with brine (50 mL), dried with anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether/ethyl acetate = 100/0 to 10/1) to afford 22a. LCMS [M+1] = 281.2, 283.2; 1H NMR (400 MHz, DMSO-d6) δ 8.93 (s, 1H), 8.10 - 8.04 (m, 2H), 8.01 - 7.94 (m, 3H), 3.86 (s, 3H). Step B – Synthesis of 4-(4-bromopyrazol-1-yl)benzoic acid (22b)
To a solution of 22a (800 mg, 2.85 mmol, 1 equiv) in tetrahydrofuran (4 mL), methanol (4 mL) and water (2 mL) was added LiOH•H2O (358.2 mg, 8.54 mmol, 3 equiv). The mixture was stirred at 20°C for 16 h. The reaction mixture was concentrated under reduced pressure to remove volatile components and then the pH was adjusted to approximately 3 by progressively adding HCl (1 mol/L). The solids were collected by vacuum filtration and the filter cake was dried under vacuum to afford 22b. LCMS [M+1] = 266.9, 268.9; 1H NMR (400 MHz, DMSO-d6) δ 13.09 (br s, 1H), 8.92 (s, 1H), 8.08 - 8.03 (m, 2H), 7.98 - 7.94 (m, 3H). Step C – Synthesis of tert-butyl 4-[[1-[4-(4-bromopyrazol-1-yl)benzoyl]-4- piperidyl]methyl]piperazine-1-carboxylate (22c) To a solution of 22b (500 mg, 1.87 mmol, 1 equiv) in DMF (5 mL) was added HATU (783.0 mg, 2.06 mmol, 1.1 equiv) and DIEA (967.8 mg, 7.49 mmol, 1.30 mL, 4 equiv). The reaction was stirred for 5 min at 20°C and then treated with tert-butyl 4-(4- piperidylmethyl)piperazine-1-carboxylate (530.5 mg, 1.87 mmol, 1 equiv). The mixture was stirred at 20°C for 1 h and then partitioned between ethyl acetate (50 mL) and water (50 mL). The organic phase was separated, washed with brine (20 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, dichloromethane: methanol = 100/0 to 96/4) to 22c. LCMS [M+1] + = 532.2, 534.2; 1H NMR (400 MHz, DMSO-d6) δ 8.86 (s, 1H), 8.01 - 7.79 (m, 3H), 7.51 (d, J = 8.5 Hz, 2H), 4.45 (br dd, J = 3.8, 8.3 Hz, 1H), 3.61 (br s, 1H), 3.30 (br s, 4H), 3.20 - 2.95 (m, 2H), 2.31 (br s, 4H), 2.19 (br d, J = 5.5 Hz, 2H), 1.88 - 1.61 (m, 3H), 1.39 (s, 9H), 1.14 - 1.00 (m, 2H). Step D – Synthesis of tert-butyl 4-[[1-[4-[4-[4-[(1S,2S)-2-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H-1,2,4-triazol-5-yl]methylcarbamoyl]cyclopropyl]phenyl]pyrazol-1-yl]benzoyl]-4- piperidyl]methyl]piperazine-1-carboxylate (22d) To a solution of 22c (134.4 mg, 252.4 μmol, 1 equiv) and Int 21 (100 mg, 252.4 μmol, 1 equiv) in dioxane (2.5 mL) and water (0.5 mL) was added potassium carbonate (104.6 mg, 757.2 μmol, 3 equiv) and Pd(dppf)Cl2 (20.6 mg, 25.2 μmol, 0.1 equiv). The mixture was three-fold degassed and purged with nitrogen and stirred at 100°C for 12 h under an atmosphere of nitrogen. The reaction mixture was quenched by addition water (20 mL), and then extracted with ethyl acetate (3 x 10 mL). The combined organic layers were washed with brine (3 x 10 mL), dried
over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative TLC (silica gel, dichloromethane: methanol = 10: 1) to afford 22d. LCMS [M+1] = 804.5; 1H NMR (400 MHz, DMSO-d6) δ 13.87 (br s, 1H), 9.03 (s, 1H), 8.78 (br s, 1H), 8.24 (s, 1H), 7.94 (d, J = 8.6 Hz, 2H), 7.64 (d, J = 8.1 Hz, 2H), 7.52 (d, J = 8.6 Hz, 2H), 7.18 (d, J = 8.1 Hz, 2H), 4.41 (br s, 3H), 3.73 - 3.57 (m, 1H), 3.29 (br s, 4H), 3.11 - 2.93 (m, 1H), 2.89 - 2.72 (m, 1H), 2.29 (br d, J = 3.9 Hz, 4H), 2.16 (br d, J = 6.8 Hz, 2H), 1.99 - 1.92 (m, 1H), 2.04 - 1.92 (m, 1H), 1.89 - 1.62 (m, 3H), 1.52 (br s, 6H), 1.39 (s, 9H), 1.33 - 1.20 (m, 2H), 1.14 - 1.00 (m, 2H). Step E – Synthesis of (1S,2S)-2-[4-[1-[4-[4-(piperazin-1-ylmethyl)piperidine-1- carbonyl]phenyl]pyrazol-4-yl]phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4- triazol-5-yl]methyl] cyclopropanecarboxamide (22e) To a solution of 22d (80 mg, 99.5 μmol, 1 equiv) in dichloromethane (0.8 mL) was added trifluoroacetic acid (0.8 mL), the mixture was stirred at 25°C for 1 h and then concentrated under reduced pressure to afford 22e, which was used without further purification. Step F – Synthesis of (1S,2S)-2-[4-[1-[4-[4-[[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo- isoindolin-5-yl]piperazin-1-yl]methyl]piperidine-1-carbonyl]phenyl]pyrazol-4-yl]phenyl]-N-[[3- (2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]cyclopropanecarboxamide (Compound 22) To a solution of 22e (0.14 g, 198.9 μmol, 1 equiv) and 2-(2,6-dioxo-3-piperidyl)-5,6- difluoro-isoindoline-1,3-dione (87.7 mg, 298.3 μmol, 1.5 equiv) in DMSO (1 mL) was added DIEA (77.1 mg, 596.7 μmol, 103.9 μL, 3 equiv). The mixture was stirred at 70°C for 12 h. The reaction mixture was quenched by addition water (20 mL) and then extracted with ethyl acetate (3 x 10 mL). The combined organic layers were washed with brine (5 x 10 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative-HPLC (column: Phenomenex Luna C18100mm x 30 mm, 5 µm; (H2O (0.2% formic acid)-acetonitrile) to afford Compound 22. LCMS [M+1] = 978.5; 1H NMR (400 MHz, DMSO-d6) δ 13.97 - 13.84 (m, 1H), 11.10 (s, 1H), 9.03 (s, 1H), 8.83 - 8.69 (m, 1H), 8.24 (s, 1H), 7.94 (d, J = 8.5 Hz, 2H), 7.73 (d, J = 11.6 Hz, 1H), 7.64 (d, J = 8.4 Hz, 2H), 7.53 (d, J = 8.6 Hz, 2H), 7.45 (d, J = 7.4 Hz, 1H), 7.18 (d, J = 8.4 Hz, 2H), 5.10 (dd, J = 5.4, 12.8 Hz, 1H), 4.40 (d, J = 4.6 Hz, 2H), 3.25 (s, 4H), 3.15 - 2.98 (m, 2H), 2.86 (dd, J = 4.8, 16.6 Hz,
2H), 2.69 - 2.60 (m, 1H), 2.58 - 2.53 (m, 4H), 2.35 - 2.21 (m, 4H), 2.09 - 1.93 (m, 3H), 1.92 - 1.68 (m, 3H), 1.53 (s, 6H), 1.39 (td, J = 4.6, 9.2 Hz, 1H), 1.32 - 1.22 (m, 1H), 1.20 - 1.06 (m, 2H). Example 23: Synthesis of Compound 23
 1,2,4-triazol-5-yl]methylcarbamoyl]cyclopropyl]phenyl]phenyl]methyl]piperazine-1-carboxylate (23a) To a solution of Intermediate 3 (200 mg, 463.7 μmol, 1.0 equiv), tert-butyl 4-[[4- (4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]methyl]piperazine-1-carboxylate (223.9 mg, 556.5 μmol, 1.2 equiv) in dioxane (4.0 mL) and water (1.0 mL) was added potassium carbonate (192.2 mg, 1.3 mmol, 3.0 equiv) and Pd(dppf)Cl2•CH2Cl2 (189.3 mg, 231.8 μmol, 0.5 equiv). The mixture was stirred at 100°C for 2 h, cooled to ambient temperature, treated with water 10 mL and extracted with ethyl acetate (2 x 5 mL). The combined organic layers were washed with brine (5 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative TLC (silica gel, dichloromethane: methanol = 10:1) to afford 23a. LCMS [M+1] =627.5; 1H NMR (400 MHz, DMSO-d6) δ 13.87 (br s, 1H), 8.79 (br s, 1H), 7.58 (t, J = 8.8 Hz, 4H), 7.36 (d, J = 8.2 Hz, 2H), 7.21 (br d, J = 8.2 Hz, 2H), 4.49 - 4.32 (m, 2H), 3.50 (s, 2H), 3.32 - 3.23 (m, 4H), 2.32 (br t, J = 4.6 Hz, 4H), 2.28 - 2.23 (m, 1H), 1.99 (br s, 1H), 1.52 (br s, 6H), 1.39 (s, 9H), 1.33 - 1.21 (m, 2H). Step B – Synthesis of (1S,2S)-2-[4-[4-(piperazin-1-ylmethyl)phenyl]phenyl]-N-[[3-(2,2,2- trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]cyclopropanecarboxamide (23b)
A solution of 23a (115 mg, 183.50 μmol, 1.0 equiv) in HCl/dioxane (1.0 mL) was stirred at 25°C for 1 h and then concentrated to dryness to afford 23b, which was used without further purification. LCMS [M+1] = 527.4. Step C – Synthesis of (1S,2S)-2-(4'-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin- 5-yl)piperazin-1-yl)methyl)-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H- 1,2,4-triazol-5-yl)methyl)cyclopropane-1-carboxamide (Compound 23) To a solution of 23b (60 mg, 113.9 μmol, 1.0 equiv), 2-(2,6-dioxo-3-piperidyl)-5,6- difluoro-isoindoline-1,3-dione (50.2 mg, 170.9 μmol, 1.5 equiv) in DMSO (1.0 mL) was added DIEA (44.1 mg, 341.8 μmol, 59.54 μL, 3.0 equiv). The mixture was stirred at 70°C for 16 h, cooled to ambient temperature and partitioned between ethyl acetate (50 mL) and water (50 mL). The organic phase was separated, washed with brine (30 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative TLC (silica gel, dichloromethane: methanol = 10:1) to give Compound 23. LCMS [M+1] = 801.3; 1H NMR (400 MHz, DMSO-d6) δ 3.89 (br s, 1H), 11.10 (br s, 1H), 8.76 (br d, J = 2.0 Hz, 1H), 7.72 (d, J = 11.2 Hz, 1H), 7.60 (dd, J = 8.0, 12.4 Hz, 4H), 7.48 - 7.37 (m, 3H), 7.21 (d, J = 8.4 Hz, 2H), 5.10 (dd, J = 5.6, 12.8 Hz, 1H), 4.41 (br d, J = 4.4 Hz, 2H), 3.58 (s, 2H), 3.27 (br s, 4H), 2.88 (br d, J = 2.0 Hz, 1H), 2.63 - 2.52 (m, 6H), 2.37 - 2.29 (m, 1H), 2.08 - 1.95 (m, 2H), 1.53 (s, 6H), 1.44 - 1.36 (m, 1H), 1.33 - 1.21 (m, 1H). Example 24: Synthesis of Compound 24
1,2,4-triazol-5-yl]methylcarbamoyl]cyclopropyl]phenyl]phenyl]methyl]piperazine-1-carboxylate (23a) To a solution of Intermediate 3 (200 mg, 463.7 μmol, 1.0 equiv), tert-butyl 4-[[4- (4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]methyl]piperazine-1-carboxylate (223.9 mg, 556.5 μmol, 1.2 equiv) in dioxane (4.0 mL) and water (1.0 mL) was added potassium carbonate (192.2 mg, 1.3 mmol, 3.0 equiv) and Pd(dppf)Cl2•CH2Cl2 (189.3 mg, 231.8 μmol, 0.5 equiv). The mixture was stirred at 100°C for 2 h, cooled to ambient temperature, treated with water 10 mL and extracted with ethyl acetate (2 x 5 mL). The combined organic layers were washed with brine (5 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative TLC (silica gel, dichloromethane: methanol = 10:1) to afford 23a. LCMS [M+1] =627.5; 1H NMR (400 MHz, DMSO-d6) δ 13.87 (br s, 1H), 8.79 (br s, 1H), 7.58 (t, J = 8.8 Hz, 4H), 7.36 (d, J = 8.2 Hz, 2H), 7.21 (br d, J = 8.2 Hz, 2H), 4.49 - 4.32 (m, 2H), 3.50 (s, 2H), 3.32 - 3.23 (m, 4H), 2.32 (br t, J = 4.6 Hz, 4H), 2.28 - 2.23 (m, 1H), 1.99 (br s, 1H), 1.52 (br s, 6H), 1.39 (s, 9H), 1.33 - 1.21 (m, 2H). Step B – Synthesis of (1S,2S)-2-[4-[4-(piperazin-1-ylmethyl)phenyl]phenyl]-N-[[3-(2,2,2- trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]cyclopropanecarboxamide (23b)
A solution of 23a (115 mg, 183.50 μmol, 1.0 equiv) in HCl/dioxane (1.0 mL) was stirred at 25°C for 1 h and then concentrated to dryness to afford 23b, which was used without further purification. LCMS [M+1] = 527.4. Step C – Synthesis of (1S,2S)-2-(4'-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin- 5-yl)piperazin-1-yl)methyl)-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H- 1,2,4-triazol-5-yl)methyl)cyclopropane-1-carboxamide (Compound 23) To a solution of 23b (60 mg, 113.9 μmol, 1.0 equiv), 2-(2,6-dioxo-3-piperidyl)-5,6- difluoro-isoindoline-1,3-dione (50.2 mg, 170.9 μmol, 1.5 equiv) in DMSO (1.0 mL) was added DIEA (44.1 mg, 341.8 μmol, 59.54 μL, 3.0 equiv). The mixture was stirred at 70°C for 16 h, cooled to ambient temperature and partitioned between ethyl acetate (50 mL) and water (50 mL). The organic phase was separated, washed with brine (30 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative TLC (silica gel, dichloromethane: methanol = 10:1) to give Compound 23. LCMS [M+1] = 801.3; 1H NMR (400 MHz, DMSO-d6) δ 3.89 (br s, 1H), 11.10 (br s, 1H), 8.76 (br d, J = 2.0 Hz, 1H), 7.72 (d, J = 11.2 Hz, 1H), 7.60 (dd, J = 8.0, 12.4 Hz, 4H), 7.48 - 7.37 (m, 3H), 7.21 (d, J = 8.4 Hz, 2H), 5.10 (dd, J = 5.6, 12.8 Hz, 1H), 4.41 (br d, J = 4.4 Hz, 2H), 3.58 (s, 2H), 3.27 (br s, 4H), 2.88 (br d, J = 2.0 Hz, 1H), 2.63 - 2.52 (m, 6H), 2.37 - 2.29 (m, 1H), 2.08 - 1.95 (m, 2H), 1.53 (s, 6H), 1.44 - 1.36 (m, 1H), 1.33 - 1.21 (m, 1H). Example 24: Synthesis of Compound 24
 was a manner tert-butyl 4-[[3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]methyl]piperazine-1-carboxylate in Step A to afford (1S,2S)-2-(4'-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperazin-1-yl)methyl)-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H- 1,2,4-triazol-5-yl)methyl)cyclopropane-1-carboxamide (Compound 24). LCMS [M+1] =801.3; 1H
NMR (400 MHz, DMSO-d6) δ 13.90 (br s, 1H), 11.10 (br s, 1H), 8.73 (br s, 1H), 7.72 (d, J = 11.2 Hz, 1H), 7.62 - 7.56 (m, 3H), 7.54 (br d, J = 8.0 Hz, 1H), 7.48 - 7.38 (m, 2H), 7.32 (d, J = 7.6 Hz, 1H), 7.22 (d, J = 8.0 Hz, 2H), 5.10 (dd, J = 5.2, 12.8 Hz, 1H), 4.40 (br d, J = 5.2 Hz, 2H), 3.61 (s, 2H), 3.27 (br s, 4H), 2.94 - 2.82 (m, 1H), 2.58 (br s, 5H), 2.37 - 2.30 (m, 2H), 2.06 - 1.96 (m, 2H), 1.53 (s, 6H), 1.41 (td, J = 4.8, 9.2 Hz, 1H), 1.32 - 1.24 (m, 1H). Example 25: Synthesis of Compound 25
was a manner tert-butyl 4-[[3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]methyl]piperazine-1-carboxylate in Step A to afford (1S,2S)-2-(4'-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperazin-1-yl)methyl)-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H- 1,2,4-triazol-5-yl)methyl)cyclopropane-1-carboxamide (Compound 24). LCMS [M+1] =801.3; 1H
NMR (400 MHz, DMSO-d6) δ 13.90 (br s, 1H), 11.10 (br s, 1H), 8.73 (br s, 1H), 7.72 (d, J = 11.2 Hz, 1H), 7.62 - 7.56 (m, 3H), 7.54 (br d, J = 8.0 Hz, 1H), 7.48 - 7.38 (m, 2H), 7.32 (d, J = 7.6 Hz, 1H), 7.22 (d, J = 8.0 Hz, 2H), 5.10 (dd, J = 5.2, 12.8 Hz, 1H), 4.40 (br d, J = 5.2 Hz, 2H), 3.61 (s, 2H), 3.27 (br s, 4H), 2.94 - 2.82 (m, 1H), 2.58 (br s, 5H), 2.37 - 2.30 (m, 2H), 2.06 - 1.96 (m, 2H), 1.53 (s, 6H), 1.41 (td, J = 4.8, 9.2 Hz, 1H), 1.32 - 1.24 (m, 1H). Example 25: Synthesis of Compound 25
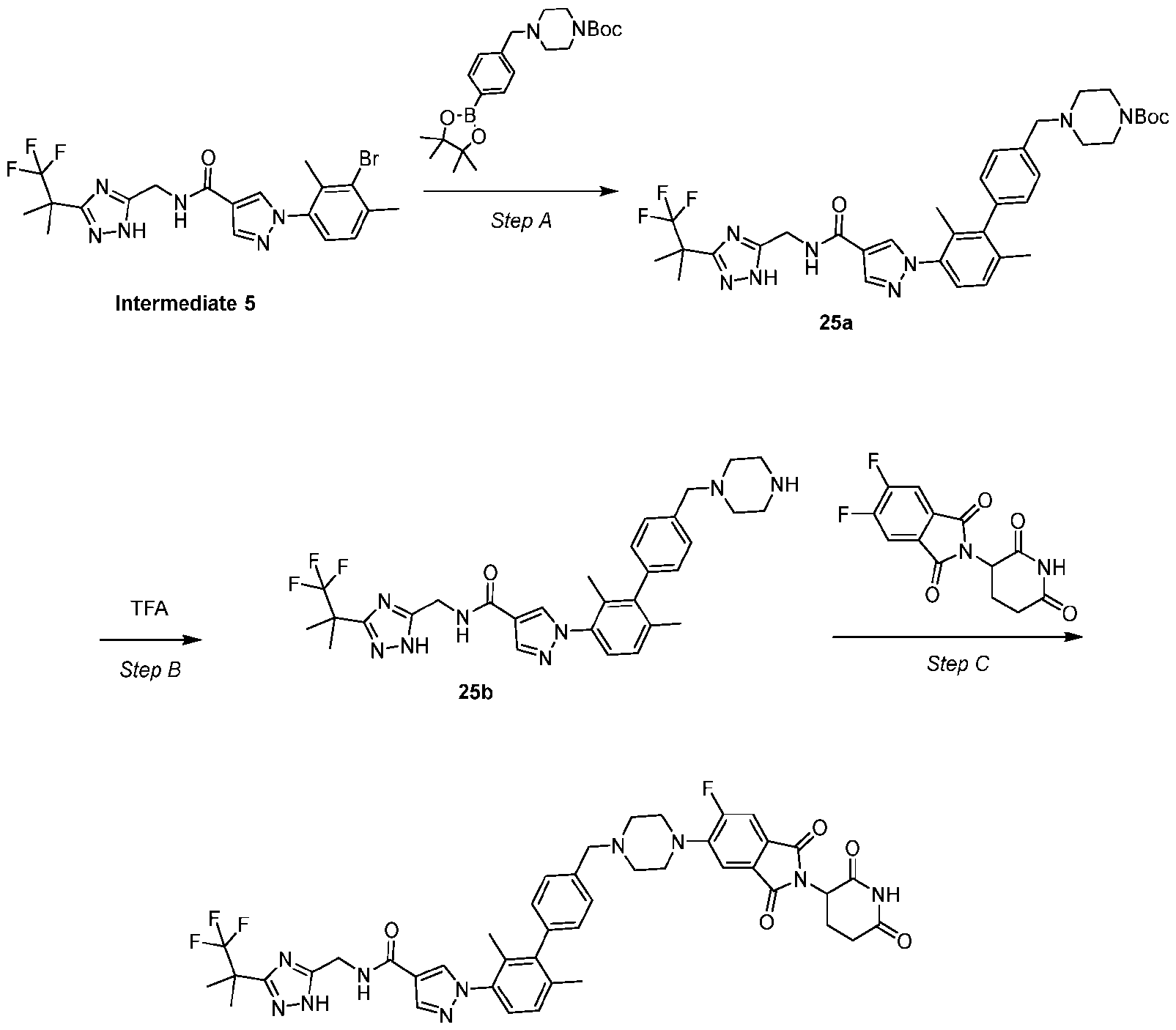 Step A – Synthesis of tert-butyl 4-[[4-[2,6-dimethyl-3-[4-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H-1,2,4-triazol-5-yl]methylcarbamoyl]pyrazol-1-yl]phenyl]phenyl]methyl]piperazine-1- carboxylate (25a) To a solution of Intermediate 5 (50 mg, 103.0 μmol, 1.0 equiv), tert-butyl 4-[[4-(4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]methyl]piperazine-1-carboxylate (62.1 mg, 154.5 μmol, 1.5 equiv) in tetrahydrofuran (0.4 mL) and water (0.1 mL) was added K3PO4 (65.6 mg,
309.0 μmol, 3 equiv) and Pd(dppf)Cl2 (13.4 mg, 20.6 μmol, 0.2 equiv). The mixture was stirred at 80°C for 16 h, cooled to ambient temperature and treated with water (1.0 mL). The mixture was extracted with ethyl acetate (3 x 1 mL). The combined organic portions were washed with brine (1 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative TLC (silica gel, dichloromethane: methanol = 10: 1) to afford 25a. LCMS [M+1] = 681.4; 1H NMR (400 MHz, DMSO-d6) δ 8.47 - 8.42 (m, 1H), 8.13 (s, 1H), 7.42 (d, J = 8.0 Hz, 2H), 7.29 (d, J = 2.0 Hz, 2H), 7.15 (d, J = 8.0 Hz, 2H), 4.58 - 4.51 (m, 2H), 3.55 (s, 2H), 3.33 (d, J = 3.6 Hz, 4H), 2.35 (d, J = 4.8 Hz, 4H), 2.02 (s, 3H), 1.76 (s, 3H), 1.51 - 1.50 (m, 1H), 1.53 (s, 6H), 1.39 (s, 9H). Step B – Synthesis of 1-[2,4-dimethyl-3-[4-(piperazin-1-ylmethyl)phenyl]phenyl]-N-[[3-(2,2,2- trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (25b) To a solution of 25a (75 mg, 110.1 μmol, 1.0 equiv) in dichloromethane (0.4 mL) was added trifluoroacetic acid (0.4 mL) the mixture was stirred at ambient temperature for 1 h and then concentrated under reduced pressure to afford 25b, which was used without further purification. LCMS [M+1] = 581.3; 1H NMR (400 MHz, DMSO-d6) δ 9.05 (br s, 1H), 8.84 (br s, 1H), 8.46 - 8.39 (s, 1H), 8.14 (s, 1H), 7.60 (s, 2H), 7.36 - 7.29 (m, 4H), 4.54 (d, J = 5.6 Hz, 2H), 4.33 (d, J = 6.4 Hz, 2H), 3.35 - 3.22 (m, 8H), 2.03 (s, 3H), 1.77 (s, 3H), 1.53 (s, 6H). Step C – Synthesis of 1-[3-[4-[[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]piperazin-1-yl]methyl]phenyl]-2,4-dimethyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Compound 25) To a solution of 25b (85.0 mg, 52.9 μmol, 1.0 equiv) in DMSO (1.0 mL) was added 2- (2,6-dioxo-3-piperidyl)-5,6-difluoro-isoindoline-1,3-dione (23.3 mg, 79.3 μmol, 1.5 equiv) and DIEA (68.3 mg, 528.9 μmol, 10.0 equiv) at 100 °C for 16 h. The mixture was cooled to ambient temperature and was purified by preparative HPLC (Waters Xbridge Prep OBD C18150mm x 40 mm, 10 µm; water (10mM NH4HCO3)-acetonitrile;) to afford Compound 25. LCMS [M+1] = 855.4; 1H NMR (400 MHz, Methanol-d4) δ 8.30 (s, 1H), 8.14 (s, 1H), 7.57 - 7.48 (m, 4H), 7.28 (d, J = 4.8 Hz, 2H), 7.17 (d, J = 8.0 Hz, 2H), 5.09 (dd, J = 5.6, 12.4 Hz, 1H), 4.68 (s, 2H), 3.69 (s, 2H), 3.37 - 3.33 (m, 4H), 2.89 - 2.79 (m, 1H), 2.77 - 2.68 (m, 6H), 2.13 (td, J = 2.8, 5.6 Hz, 1H), 2.09 (s, 3H), 1.80 (s, 3H), 1.61 (s, 6H).
Example 26: Synthesis of Compound 26
Step A – Synthesis of tert-butyl 4-[[4-[2,6-dimethyl-3-[4-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H-1,2,4-triazol-5-yl]methylcarbamoyl]pyrazol-1-yl]phenyl]phenyl]methyl]piperazine-1- carboxylate (25a) To a solution of Intermediate 5 (50 mg, 103.0 μmol, 1.0 equiv), tert-butyl 4-[[4-(4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]methyl]piperazine-1-carboxylate (62.1 mg, 154.5 μmol, 1.5 equiv) in tetrahydrofuran (0.4 mL) and water (0.1 mL) was added K3PO4 (65.6 mg,
309.0 μmol, 3 equiv) and Pd(dppf)Cl2 (13.4 mg, 20.6 μmol, 0.2 equiv). The mixture was stirred at 80°C for 16 h, cooled to ambient temperature and treated with water (1.0 mL). The mixture was extracted with ethyl acetate (3 x 1 mL). The combined organic portions were washed with brine (1 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative TLC (silica gel, dichloromethane: methanol = 10: 1) to afford 25a. LCMS [M+1] = 681.4; 1H NMR (400 MHz, DMSO-d6) δ 8.47 - 8.42 (m, 1H), 8.13 (s, 1H), 7.42 (d, J = 8.0 Hz, 2H), 7.29 (d, J = 2.0 Hz, 2H), 7.15 (d, J = 8.0 Hz, 2H), 4.58 - 4.51 (m, 2H), 3.55 (s, 2H), 3.33 (d, J = 3.6 Hz, 4H), 2.35 (d, J = 4.8 Hz, 4H), 2.02 (s, 3H), 1.76 (s, 3H), 1.51 - 1.50 (m, 1H), 1.53 (s, 6H), 1.39 (s, 9H). Step B – Synthesis of 1-[2,4-dimethyl-3-[4-(piperazin-1-ylmethyl)phenyl]phenyl]-N-[[3-(2,2,2- trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (25b) To a solution of 25a (75 mg, 110.1 μmol, 1.0 equiv) in dichloromethane (0.4 mL) was added trifluoroacetic acid (0.4 mL) the mixture was stirred at ambient temperature for 1 h and then concentrated under reduced pressure to afford 25b, which was used without further purification. LCMS [M+1] = 581.3; 1H NMR (400 MHz, DMSO-d6) δ 9.05 (br s, 1H), 8.84 (br s, 1H), 8.46 - 8.39 (s, 1H), 8.14 (s, 1H), 7.60 (s, 2H), 7.36 - 7.29 (m, 4H), 4.54 (d, J = 5.6 Hz, 2H), 4.33 (d, J = 6.4 Hz, 2H), 3.35 - 3.22 (m, 8H), 2.03 (s, 3H), 1.77 (s, 3H), 1.53 (s, 6H). Step C – Synthesis of 1-[3-[4-[[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]piperazin-1-yl]methyl]phenyl]-2,4-dimethyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Compound 25) To a solution of 25b (85.0 mg, 52.9 μmol, 1.0 equiv) in DMSO (1.0 mL) was added 2- (2,6-dioxo-3-piperidyl)-5,6-difluoro-isoindoline-1,3-dione (23.3 mg, 79.3 μmol, 1.5 equiv) and DIEA (68.3 mg, 528.9 μmol, 10.0 equiv) at 100 °C for 16 h. The mixture was cooled to ambient temperature and was purified by preparative HPLC (Waters Xbridge Prep OBD C18150mm x 40 mm, 10 µm; water (10mM NH4HCO3)-acetonitrile;) to afford Compound 25. LCMS [M+1] = 855.4; 1H NMR (400 MHz, Methanol-d4) δ 8.30 (s, 1H), 8.14 (s, 1H), 7.57 - 7.48 (m, 4H), 7.28 (d, J = 4.8 Hz, 2H), 7.17 (d, J = 8.0 Hz, 2H), 5.09 (dd, J = 5.6, 12.4 Hz, 1H), 4.68 (s, 2H), 3.69 (s, 2H), 3.37 - 3.33 (m, 4H), 2.89 - 2.79 (m, 1H), 2.77 - 2.68 (m, 6H), 2.13 (td, J = 2.8, 5.6 Hz, 1H), 2.09 (s, 3H), 1.80 (s, 3H), 1.61 (s, 6H).
Example 26: Synthesis of Compound 26
 -1H- 1,2,4-triazol-5-yl]methylcarbamoyl]pyrazol-1-yl]phenyl]phenyl]methyl]piperazine-1-carboxylate (26a) A mixture of Intermediate 4 (200.0 mg, 424.3 μmol, 1.0 equiv), tert-butyl 4-[[4-(4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]methyl]piperazine-1-carboxylate (341.4 mg, 848.7 μmol, 2.0 equiv), Pd(dppf)Cl2.CH2Cl2 (69.3 mg, 84.8 μmol, 0.2 equiv), potassium carbonate (175.9 mg, 1.27 mmol, 3.0 equiv) in dioxane (2.0 mL) and water (0.4 mL) was three-fold degassed and purged with nitrogen. The mixture was stirred at 100°C for 2 h under a nitrogen atmosphere. The reaction mixture was cooled to ambient temperature diluted with water (10 mL) and extracted with ethyl acetate (3 x15 mL). The combined organic layers were washed with brine (15 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, dichloromethane : methanol=50/1 to 13/1) to afford 26a. LCMS [M+1] = 667.5; 1H NMR (400 MHz, CHLOROFORM-d) δ 12.00 (br s, 1H), 8.16 (s, 1H), 8.02 (s, 1H), 7.59 - 7.52 (m, 4H), 7.42 (d, J = 8.0 Hz, 3H), 7.01 - 6.87 (m, 1H), 4.72 (d, J = 5.6 Hz, 2H), 3.57 (s, 2H), 3.46 (s, 4H), 2.44 (s, 4H), 2.30 (s, 3H), 1.63 - 1.60 (m, 6H), 1.47 (s, 9H).
Step B – Synthesis of 1-[2-methyl-4-[4-(piperazin-1-ylmethyl)phenyl]phenyl]-N-[[3-(2,2,2- trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (26b) To a solution of 26a (150.0 mg, 224.9 μmol, 1.0 equiv) in dichloromethane (2.0 mL) and trifluoroacetic acid (0.4 mL). The mixture was stirred at ambient temperature for 2 h. The mixture was concentrated under reduced pressure to afford 26b, which was used without further purification. LCMS: [M+1] = 567.4. Step C – Synthesis of 1-[4-[4-[[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]piperazin-1-yl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H- 1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Compound 26) A mixture of 26b (150.0 mg, 220.3 μmol, 1.0 equiv), 2-(2,6-dioxo-3-piperidyl)-5,6- difluoro-isoindoline-1,3-dione (129.6 mg, 440.7 μmol, 2.0 equiv) and DIEA (85.4 mg, 661.1 μmol, 115.1 μL, 3.0 equiv) in DMSO (2.0 mL) was three-fold degassed and purged with nitrogen. The mixture was stirred at 100°C for 16 h under a nitrogen atmosphere, cooled to ambient temperature. Purification by reverse-phase HPLC. (Waters Xbridge BEH C18100mm x 30mm, 10µm; H2O(10mM NH4HCO3)-acetonitrile) afforded Compound 26. LCMS: [M+1] = 841.3; 1H NMR (400 MHz, Methanol-d4) δ 8.36 (s, 1H), 8.18 (s, 1H), 7.68 (d, J = 7.2 Hz, 3H), 7.64 - 7.60 (m, 1H), 7.57 - 7.42 (m, 5H), 5.09 (dd, J = 5.4, 12.6 Hz, 1H), 4.70 (s, 2H), 3.68 (s, 2H), 3.36 - 3.32 (m, 4H), 2.91 - 2.80 (m, 1H), 2.79 - 2.64 (m, 6H), 2.31 (s, 3H), 2.17 - 2.09 (m, 1H), 1.66 - 1.54 (m, 6H). Example 27: Synthesis of Compound 27
-1H- 1,2,4-triazol-5-yl]methylcarbamoyl]pyrazol-1-yl]phenyl]phenyl]methyl]piperazine-1-carboxylate (26a) A mixture of Intermediate 4 (200.0 mg, 424.3 μmol, 1.0 equiv), tert-butyl 4-[[4-(4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]methyl]piperazine-1-carboxylate (341.4 mg, 848.7 μmol, 2.0 equiv), Pd(dppf)Cl2.CH2Cl2 (69.3 mg, 84.8 μmol, 0.2 equiv), potassium carbonate (175.9 mg, 1.27 mmol, 3.0 equiv) in dioxane (2.0 mL) and water (0.4 mL) was three-fold degassed and purged with nitrogen. The mixture was stirred at 100°C for 2 h under a nitrogen atmosphere. The reaction mixture was cooled to ambient temperature diluted with water (10 mL) and extracted with ethyl acetate (3 x15 mL). The combined organic layers were washed with brine (15 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, dichloromethane : methanol=50/1 to 13/1) to afford 26a. LCMS [M+1] = 667.5; 1H NMR (400 MHz, CHLOROFORM-d) δ 12.00 (br s, 1H), 8.16 (s, 1H), 8.02 (s, 1H), 7.59 - 7.52 (m, 4H), 7.42 (d, J = 8.0 Hz, 3H), 7.01 - 6.87 (m, 1H), 4.72 (d, J = 5.6 Hz, 2H), 3.57 (s, 2H), 3.46 (s, 4H), 2.44 (s, 4H), 2.30 (s, 3H), 1.63 - 1.60 (m, 6H), 1.47 (s, 9H).
Step B – Synthesis of 1-[2-methyl-4-[4-(piperazin-1-ylmethyl)phenyl]phenyl]-N-[[3-(2,2,2- trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (26b) To a solution of 26a (150.0 mg, 224.9 μmol, 1.0 equiv) in dichloromethane (2.0 mL) and trifluoroacetic acid (0.4 mL). The mixture was stirred at ambient temperature for 2 h. The mixture was concentrated under reduced pressure to afford 26b, which was used without further purification. LCMS: [M+1] = 567.4. Step C – Synthesis of 1-[4-[4-[[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]piperazin-1-yl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H- 1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Compound 26) A mixture of 26b (150.0 mg, 220.3 μmol, 1.0 equiv), 2-(2,6-dioxo-3-piperidyl)-5,6- difluoro-isoindoline-1,3-dione (129.6 mg, 440.7 μmol, 2.0 equiv) and DIEA (85.4 mg, 661.1 μmol, 115.1 μL, 3.0 equiv) in DMSO (2.0 mL) was three-fold degassed and purged with nitrogen. The mixture was stirred at 100°C for 16 h under a nitrogen atmosphere, cooled to ambient temperature. Purification by reverse-phase HPLC. (Waters Xbridge BEH C18100mm x 30mm, 10µm; H2O(10mM NH4HCO3)-acetonitrile) afforded Compound 26. LCMS: [M+1] = 841.3; 1H NMR (400 MHz, Methanol-d4) δ 8.36 (s, 1H), 8.18 (s, 1H), 7.68 (d, J = 7.2 Hz, 3H), 7.64 - 7.60 (m, 1H), 7.57 - 7.42 (m, 5H), 5.09 (dd, J = 5.4, 12.6 Hz, 1H), 4.70 (s, 2H), 3.68 (s, 2H), 3.36 - 3.32 (m, 4H), 2.91 - 2.80 (m, 1H), 2.79 - 2.64 (m, 6H), 2.31 (s, 3H), 2.17 - 2.09 (m, 1H), 1.66 - 1.54 (m, 6H). Example 27: Synthesis of Compound 27
 was a manner tert-butyl 4-[[3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]methyl]piperazine-1-carboxylate in Step A to afford 1-[4-[3-[[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]piperazin-1-yl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Compound 27). LCMS [M+1] = 841.3; 1H
NMR (400 MHz, DMSO-d6) δ 13.90 (s, 1H), 11.10 (s, 1H), 9.06 - 8.82 (m, 1H), 8.53 (s, 1H), 8.18 (s, 1H), 7.80 - 7.70 (m, 2H), 7.69 - 7.62 (m, 3H), 7.53 - 7.44 (m, 3H), 7.39 (d, J = 7.6 Hz, 1H), 5.10 (dd, J = 5.6, 12.8 Hz, 1H), 4.57 (d, J = 4.0 Hz, 2H), 3.64 (s, 2H), 3.27 (s, 4H), 2.94 - 2.82 (m, 1H), 2.64 - 2.55 (m, 6H), 2.31 (s, 3H), 2.10 - 1.94 (m, 1H), 1.64 - 1.47 (m, 6H). Example 28: Synthesis of Compound 28
was a manner tert-butyl 4-[[3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]methyl]piperazine-1-carboxylate in Step A to afford 1-[4-[3-[[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]piperazin-1-yl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Compound 27). LCMS [M+1] = 841.3; 1H
NMR (400 MHz, DMSO-d6) δ 13.90 (s, 1H), 11.10 (s, 1H), 9.06 - 8.82 (m, 1H), 8.53 (s, 1H), 8.18 (s, 1H), 7.80 - 7.70 (m, 2H), 7.69 - 7.62 (m, 3H), 7.53 - 7.44 (m, 3H), 7.39 (d, J = 7.6 Hz, 1H), 5.10 (dd, J = 5.6, 12.8 Hz, 1H), 4.57 (d, J = 4.0 Hz, 2H), 3.64 (s, 2H), 3.27 (s, 4H), 2.94 - 2.82 (m, 1H), 2.64 - 2.55 (m, 6H), 2.31 (s, 3H), 2.10 - 1.94 (m, 1H), 1.64 - 1.47 (m, 6H). Example 28: Synthesis of Compound 28
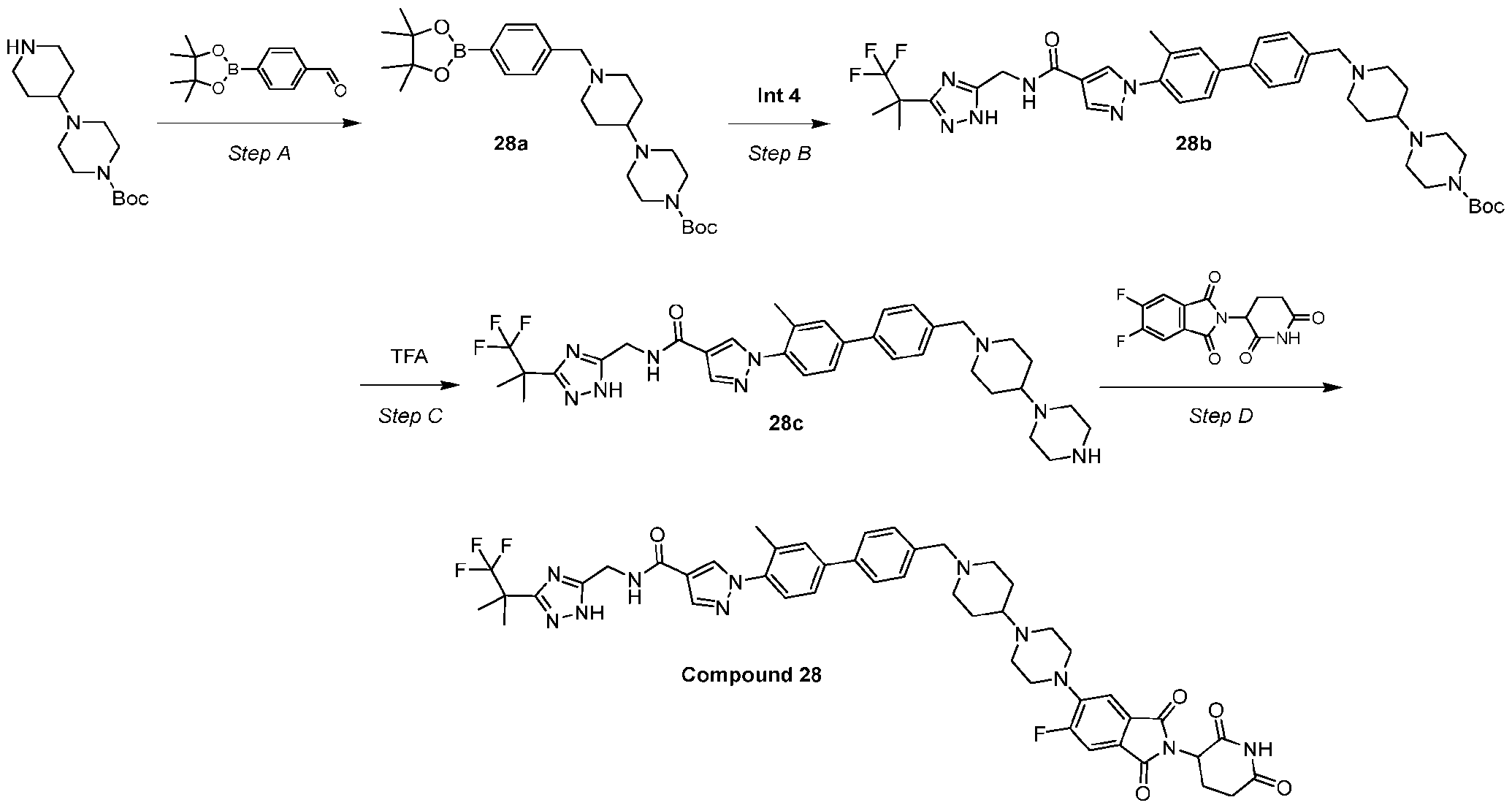 yl)phenyl]methyl]-4-piperidyl]piperazine-1-carboxylate (28a) To a solution of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzaldehyde (215.3 mg, 928.0 μmol, 1.0 equiv) in dichloromethane (4.0 mL) was added tert-butyl-4-(4- piperidyl)piperazine-1-carboxylate (500.0 mg, 1.86 mmol, 2.0 equiv) and acetic acid (55.7 mg, 928.0 μmol, 53.1 μL, 1.0 equiv) the mixture was stirred at 20°C for 1 h and then treated with NaBH(OAc)3 (393.3 mg, 1.86 mmol, 2.0 equiv). The resulting mixture was stirred at 20°C for 2 h. The reaction mixture was quenched with saturated aqueous NaHCO3 (15 mL), then extracted with ethyl acetate (3 x 10 mL). The combined organic layers were washed with brine (15 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced
pressure. The residue was purified by column chromatography (silica gel, dichloromethane ethanol=15/1 to 12/1) to afford 28a. LCMS: [M+1] = 486.2. Step B – Synthesis of tert-butyl 4-[1-[[4-[3-methyl-4-[4-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H-1,2,4-triazol-5-yl]methylcarbamoyl]pyrazol-1-yl]phenyl]phenyl]methyl]-4- piperidyl]piperazine-1-carboxylate (28b) A mixture of 28a (231.7 mg, 477.4 μmol, 1.5 equiv), Intermediate 4 (150.0 mg, 318.2 μmol, 1.0 equiv), potassium carbonate (131.9 mg, 954.8 μmol, 3.0 equiv), Pd(dppf)Cl2•CH2Cl2 (51.9 mg, 63.6 μmol, 0.2 equiv) in water (0.2 mL) and dioxane (1.0 mL) was three-fold degassed and purged with nitrogen, and then the mixture was stirred at 100°C for 2 h under a nitrogen atmosphere. The reaction mixture was cooled to ambient temperature, treated with water (15 mL) and extracted with ethyl acetate (3 x 15 mL). The combined organic portions were washed with brine (15 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, dichloromethane:methanol=10/1) to afford 28b. LCMS: [M+1] = 750.5; 1H NMR (400 MHz, CHLOROFORM-d) δ 8.16 (s, 1H), 8.03 (s, 1H), 7.62 - 7.46 (m, 4H), 7.44 - 7.33 (m, 3H), 7.00 (br s, 1H), 4.72 (br d, J = 5.5 Hz, 2H), 3.55 (s, 2H), 3.47 - 3.39 (m, 4H), 3.07 - 2.95 (m, 3H), 2.51 (br s, 4H), 2.36 - 2.25 (m, 3H), 2.02 (br t, J = 10.9 Hz, 2H), 1.78 (br d, J = 11.0 Hz, 2H), 1.68 - 1.55 (m, 6H), 1.51 - 1.43 (m, 9H), 1.38 - 1.29 (m, 2H). Step C – Synthesis of 1-[2-methyl-4-[4-[(4-piperazin-1-yl-1-piperidyl)methyl]phenyl]phenyl]-N- [[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (28c) A solution of 28b (210.0 mg, 280.0 μmol, 1.0 equiv) in trifluoracetic acid (0.2 mL) and dichloromethane (2.0 mL) was stirred at 20°C for 1 h. The mixture was concentrated under reduced pressure to afford 28c. LCMS: [M+1] = 650.6. Step D – Synthesis of 1-[4-[4-[[4-[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]piperazin-1-yl]-1-piperidyl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Compound 28). To a solution of 28c (200.0 mg, 261.8 μmol, 1.0 equiv, TFA) in DMSO (2.0 mL) was added DIEA (101.5 mg, 785.5 μmol, 136.8 μL, 3.0 eq) and 2-(2,6-dioxo-3-piperidyl)-5,6-
difluoro-isoindoline-1,3-dione (154.0 mg, 523.7 μmol, 2.0 equiv). The mixture was stirred at 100°C for 16 h. The mixture was concentrated under reduced pressure. The residue was purified by preparative HPLC (Waters Xbridge BEH C18, 100mm x 30mm, 10 µm; water (10mM NH4HCO3)-acetonitrile) to afford Compound 28. LCMS: [M+1] = 924.5; 1H NMR (400 MHz, CHLOROFORM-d) δ 12.25 (br s, 1H), 8.46 - 8.36 (m, 1H), 8.24 - 8.13 (m, 1H), 8.02 (s, 1H), 7.59 - 7.53 (m, 3H), 7.52 - 7.45 (m, 2H), 7.44 - 7.34 (m, 4H), 6.94 (br s, 1H), 4.94 (dd, J = 5.2, 12.4 Hz, 1H), 4.72 (d, J = 5.6 Hz, 2H), 3.68 - 3.48 (m, 2H), 3.29 (s, 4H), 3.01 (d, J = 10.4 Hz, 2H), 2.95 - 2.70 (m, 7H), 2.42 - 2.32 (m, 1H), 2.32 - 2.27 (m, 3H), 2.20 - 2.10 (m, 1H), 2.05 (t, J = 10.8 Hz, 2H), 1.84 (d, J = 10.8 Hz, 2H), 1.66 (s, 2H), 1.61 (s, 6H). Example 29: Synthesis of Compound 29
yl)phenyl]methyl]-4-piperidyl]piperazine-1-carboxylate (28a) To a solution of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzaldehyde (215.3 mg, 928.0 μmol, 1.0 equiv) in dichloromethane (4.0 mL) was added tert-butyl-4-(4- piperidyl)piperazine-1-carboxylate (500.0 mg, 1.86 mmol, 2.0 equiv) and acetic acid (55.7 mg, 928.0 μmol, 53.1 μL, 1.0 equiv) the mixture was stirred at 20°C for 1 h and then treated with NaBH(OAc)3 (393.3 mg, 1.86 mmol, 2.0 equiv). The resulting mixture was stirred at 20°C for 2 h. The reaction mixture was quenched with saturated aqueous NaHCO3 (15 mL), then extracted with ethyl acetate (3 x 10 mL). The combined organic layers were washed with brine (15 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced
pressure. The residue was purified by column chromatography (silica gel, dichloromethane ethanol=15/1 to 12/1) to afford 28a. LCMS: [M+1] = 486.2. Step B – Synthesis of tert-butyl 4-[1-[[4-[3-methyl-4-[4-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H-1,2,4-triazol-5-yl]methylcarbamoyl]pyrazol-1-yl]phenyl]phenyl]methyl]-4- piperidyl]piperazine-1-carboxylate (28b) A mixture of 28a (231.7 mg, 477.4 μmol, 1.5 equiv), Intermediate 4 (150.0 mg, 318.2 μmol, 1.0 equiv), potassium carbonate (131.9 mg, 954.8 μmol, 3.0 equiv), Pd(dppf)Cl2•CH2Cl2 (51.9 mg, 63.6 μmol, 0.2 equiv) in water (0.2 mL) and dioxane (1.0 mL) was three-fold degassed and purged with nitrogen, and then the mixture was stirred at 100°C for 2 h under a nitrogen atmosphere. The reaction mixture was cooled to ambient temperature, treated with water (15 mL) and extracted with ethyl acetate (3 x 15 mL). The combined organic portions were washed with brine (15 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, dichloromethane:methanol=10/1) to afford 28b. LCMS: [M+1] = 750.5; 1H NMR (400 MHz, CHLOROFORM-d) δ 8.16 (s, 1H), 8.03 (s, 1H), 7.62 - 7.46 (m, 4H), 7.44 - 7.33 (m, 3H), 7.00 (br s, 1H), 4.72 (br d, J = 5.5 Hz, 2H), 3.55 (s, 2H), 3.47 - 3.39 (m, 4H), 3.07 - 2.95 (m, 3H), 2.51 (br s, 4H), 2.36 - 2.25 (m, 3H), 2.02 (br t, J = 10.9 Hz, 2H), 1.78 (br d, J = 11.0 Hz, 2H), 1.68 - 1.55 (m, 6H), 1.51 - 1.43 (m, 9H), 1.38 - 1.29 (m, 2H). Step C – Synthesis of 1-[2-methyl-4-[4-[(4-piperazin-1-yl-1-piperidyl)methyl]phenyl]phenyl]-N- [[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (28c) A solution of 28b (210.0 mg, 280.0 μmol, 1.0 equiv) in trifluoracetic acid (0.2 mL) and dichloromethane (2.0 mL) was stirred at 20°C for 1 h. The mixture was concentrated under reduced pressure to afford 28c. LCMS: [M+1] = 650.6. Step D – Synthesis of 1-[4-[4-[[4-[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]piperazin-1-yl]-1-piperidyl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Compound 28). To a solution of 28c (200.0 mg, 261.8 μmol, 1.0 equiv, TFA) in DMSO (2.0 mL) was added DIEA (101.5 mg, 785.5 μmol, 136.8 μL, 3.0 eq) and 2-(2,6-dioxo-3-piperidyl)-5,6-
difluoro-isoindoline-1,3-dione (154.0 mg, 523.7 μmol, 2.0 equiv). The mixture was stirred at 100°C for 16 h. The mixture was concentrated under reduced pressure. The residue was purified by preparative HPLC (Waters Xbridge BEH C18, 100mm x 30mm, 10 µm; water (10mM NH4HCO3)-acetonitrile) to afford Compound 28. LCMS: [M+1] = 924.5; 1H NMR (400 MHz, CHLOROFORM-d) δ 12.25 (br s, 1H), 8.46 - 8.36 (m, 1H), 8.24 - 8.13 (m, 1H), 8.02 (s, 1H), 7.59 - 7.53 (m, 3H), 7.52 - 7.45 (m, 2H), 7.44 - 7.34 (m, 4H), 6.94 (br s, 1H), 4.94 (dd, J = 5.2, 12.4 Hz, 1H), 4.72 (d, J = 5.6 Hz, 2H), 3.68 - 3.48 (m, 2H), 3.29 (s, 4H), 3.01 (d, J = 10.4 Hz, 2H), 2.95 - 2.70 (m, 7H), 2.42 - 2.32 (m, 1H), 2.32 - 2.27 (m, 3H), 2.20 - 2.10 (m, 1H), 2.05 (t, J = 10.8 Hz, 2H), 1.84 (d, J = 10.8 Hz, 2H), 1.66 (s, 2H), 1.61 (s, 6H). Example 29: Synthesis of Compound 29
 tert-butyl 4-piperazin-1-ylpiperidine-1-carboxylate in Step A to afford 1-[4-[4-[[4-[1-[2-(2,6-dioxo-3- piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]-4-piperidyl]piperazin-1-yl]methyl]phenyl]-2- methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole- 4-carboxamide (Compound 29). LCMS: [M+1] = 924.5; 1H NMR (400 MHz, DMSO-d6) δ 13.90 (br s, 1 H), 11.10 (br s, 1 H), 8.87 (br s, 1 H), 8.52 (s, 1 H), 8.17 (s, 1 H), 7.59 - 7.78 (m, 5 H), 7.35 - 7.51 (m, 4 H), 5.10 (m, 1 H), 4.55 (br d, J = 4.8 Hz, 2 H), 3.64 (br d, J = 11.0 Hz, 2 H), 3.50 (br s, 2 H), 2.84 - 2.95 (m, 3 H), 2.61 (br s, 4 H), 2.41 (br d, J = 1.8 Hz, 5 H), 2.30 (s, 4 H), 1.97 - 2.09 (m, 1 H), 1.88 (br d, J = 11.2 Hz, 2 H), 1.54 (s, 9 H). Example 30: Synthesis of Compound 30
tert-butyl 4-piperazin-1-ylpiperidine-1-carboxylate in Step A to afford 1-[4-[4-[[4-[1-[2-(2,6-dioxo-3- piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]-4-piperidyl]piperazin-1-yl]methyl]phenyl]-2- methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole- 4-carboxamide (Compound 29). LCMS: [M+1] = 924.5; 1H NMR (400 MHz, DMSO-d6) δ 13.90 (br s, 1 H), 11.10 (br s, 1 H), 8.87 (br s, 1 H), 8.52 (s, 1 H), 8.17 (s, 1 H), 7.59 - 7.78 (m, 5 H), 7.35 - 7.51 (m, 4 H), 5.10 (m, 1 H), 4.55 (br d, J = 4.8 Hz, 2 H), 3.64 (br d, J = 11.0 Hz, 2 H), 3.50 (br s, 2 H), 2.84 - 2.95 (m, 3 H), 2.61 (br s, 4 H), 2.41 (br d, J = 1.8 Hz, 5 H), 2.30 (s, 4 H), 1.97 - 2.09 (m, 1 H), 1.88 (br d, J = 11.2 Hz, 2 H), 1.54 (s, 9 H). Example 30: Synthesis of Compound 30
utyl 4-(piperazin-1-ylmethyl)piperidine-1-carboxylate in Step A to afford 1-(4'-((4-((1-(2-(2,6- dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperidin-4-yl)methyl)piperazin-1- yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide. LCMS [M+1] =938.7; 1H NMR (400 MHz, DMSO-d6) δ 13.93 ((br s, 1H), 11.10 (s, 1H), 8.88 (br s, 1H), 8.52 (s, 1H), 8.17 (s, 1H), 7.74 - 7.61 (m, 5H), 7.49 - 7.39 (m, 4H), 5.09 (dd, J = 5.6, 5.2 Hz, 1H), 4.55 (br d, J = 5.6 Hz, 2H), 3.65 - 3.46 (m, 5H), 2.95 - 2.83 (m, 3H), 2.69 - 2.54 (m, 2H), 2.45 - 2.29 (m, 10H), 2.18 (br d, J = 7.2 Hz, 2H), 2.10 - 1.98 (m, 1H), 1.87 - 1.67 (m, 3H), 1.54 (s, 6H), 1.32 - 1.18 (m, 2H). Example 31: Synthesis of Compound 31
 butyl 4-(piperidin-4-ylmethyl)piperazine-1-carboxylate in Step A to afford 1-(4'-((4-((4-(2-(2,6- dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1-yl)methyl)piperidin-1- yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide. LCMS [M+1] = 938.7; 1H NMR (400 MHz, METHANOL-d4) δ(ppm) = 8.40 (s, 1H), 8.20 (s, 1H), 7.84 (d, J = 8.1 Hz, 2H), 7.76 - 7.58 (m, 6H), 7.49 (d, J = 8.3 Hz, 1H), 5.11 (dd, J = 5.5, 12.5 Hz, 1H), 4.75 (s, 2H), 4.42 (s, 2H), 3.89 -
3.72 (m, 4H), 3.62 (br d, J = 12.1 Hz, 2H), 3.51 - 3.43 (m, 2H), 3.42 - 3.35 (m, 2H), 3.27 - 3.21 (m, 2H), 3.16 (br t, J = 11.8 Hz, 2H), 2.93 - 2.81 (m, 1H), 2.79 - 2.65 (m, 2H), 2.33 (s, 4H), 2.26 - 2.17 (m, 2H), 2.13 (br dd, J = 4.8, 10.1 Hz, 1H), 1.79 - 1.58 (m, 8H). Example 32: Synthesis of Compound 32
butyl 4-(piperidin-4-ylmethyl)piperazine-1-carboxylate in Step A to afford 1-(4'-((4-((4-(2-(2,6- dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1-yl)methyl)piperidin-1- yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide. LCMS [M+1] = 938.7; 1H NMR (400 MHz, METHANOL-d4) δ(ppm) = 8.40 (s, 1H), 8.20 (s, 1H), 7.84 (d, J = 8.1 Hz, 2H), 7.76 - 7.58 (m, 6H), 7.49 (d, J = 8.3 Hz, 1H), 5.11 (dd, J = 5.5, 12.5 Hz, 1H), 4.75 (s, 2H), 4.42 (s, 2H), 3.89 -
3.72 (m, 4H), 3.62 (br d, J = 12.1 Hz, 2H), 3.51 - 3.43 (m, 2H), 3.42 - 3.35 (m, 2H), 3.27 - 3.21 (m, 2H), 3.16 (br t, J = 11.8 Hz, 2H), 2.93 - 2.81 (m, 1H), 2.79 - 2.65 (m, 2H), 2.33 (s, 4H), 2.26 - 2.17 (m, 2H), 2.13 (br dd, J = 4.8, 10.1 Hz, 1H), 1.79 - 1.58 (m, 8H). Example 32: Synthesis of Compound 32
 4-(4-piperidyloxy)piperidine-1-carboxylate in Step A to afford 1-(4'-((4-((1-(2-(2,6- dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperidin-4-yl)oxy)piperidin-1-yl)methyl)- 3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5- yl)methyl)-1H-pyrazole-4-carboxamide. LCMS [M+1] = 939.3; 1H NMR (400 MHz, DMSO-d6) δ 13.94 (br s, 1H), 11.10 (s, 1H), 8.88 (s, 1H), 8.52 (s, 1H), 8.18 - 8.15 (m, 1H), 7.74 - 7.62 (m, 5H), 7.48 - 7.39 (m, 4H), 5.10 (dd, J = 5.4, 12.6 Hz, 1H), 4.55 (d, J = 5.0 Hz, 2H), 3.65 (s, 2H), 3.51 (s, 3H), 3.05 (t, J = 10.0 Hz, 3H), 2.95 - 2.81 (m, 2H), 2.75 - 2.66 (m, 2H), 2.61 (s, 1H), 2.56 (s, 1H), 2.30 (s, 3H), 2.13 (t, J = 9.8 Hz, 2H), 2.08 - 2.00 (m, 1H), 1.93 (d, J = 10.8 Hz, 2H), 1.83 (d, J = 9.4 Hz, 2H), 1.61 - 1.57 (m, 1H), 1.54 (s, 6H), 1.47 (d, J = 9.4 Hz, 2H). Example 33: Synthesis of Compound 33
4-(4-piperidyloxy)piperidine-1-carboxylate in Step A to afford 1-(4'-((4-((1-(2-(2,6- dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperidin-4-yl)oxy)piperidin-1-yl)methyl)- 3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5- yl)methyl)-1H-pyrazole-4-carboxamide. LCMS [M+1] = 939.3; 1H NMR (400 MHz, DMSO-d6) δ 13.94 (br s, 1H), 11.10 (s, 1H), 8.88 (s, 1H), 8.52 (s, 1H), 8.18 - 8.15 (m, 1H), 7.74 - 7.62 (m, 5H), 7.48 - 7.39 (m, 4H), 5.10 (dd, J = 5.4, 12.6 Hz, 1H), 4.55 (d, J = 5.0 Hz, 2H), 3.65 (s, 2H), 3.51 (s, 3H), 3.05 (t, J = 10.0 Hz, 3H), 2.95 - 2.81 (m, 2H), 2.75 - 2.66 (m, 2H), 2.61 (s, 1H), 2.56 (s, 1H), 2.30 (s, 3H), 2.13 (t, J = 9.8 Hz, 2H), 2.08 - 2.00 (m, 1H), 1.93 (d, J = 10.8 Hz, 2H), 1.83 (d, J = 9.4 Hz, 2H), 1.61 - 1.57 (m, 1H), 1.54 (s, 6H), 1.47 (d, J = 9.4 Hz, 2H). Example 33: Synthesis of Compound 33
Compound 33 was prepared in a similar manner to Example 28 by substituting tert-butyl 1,4-diazepane-1-carboxylate in Step A to afford 1-(4'-((4-(1-(2-(2,6-dioxopiperidin-3-yl)-6- fluoro-1,3-dioxoisoindolin-5-yl)piperidin-4-yl)-1,4-diazepan-1-yl)methyl)-3-methyl-[1,1'- biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H- pyrazole-4-carboxamide. LCMS [M+H] = 938.5; 1H NMR (400 MHz, DMSO-d6) δ 13.92 (s, 1H), 11.10 (s, 1H), 8.87 (t, J = 4.4 Hz, 1H), 8.52 (s, 1H), 8.17 (s, 1H), 7.72 - 7.62 (m, 5H), 7.47 - 7.42 (m, 4H), 5.10 (dd, J = 5.6, 12.8 Hz, 1H), 4.55 (d, J = 6.0 Hz, 2H), 3.66 - 3.64 (m, 4H), 2.91 - 2.84 (m, 3H), 2.80 - 2.77 (m, 4H), 2.66 - 2.58 (m, 6H), 2.30 (s, 3H), 2.07 - 2.01 (m, 2H), 1.83 - 1.80 (m, 2H), 1.73 - 1.70 (m, 2H), 1.63 - 1.57 (m, 2H), 1.54 (s, 6H). Example 34. Synthesis of Compound 34
Compound 34 was prepared in a similar manner to Example 28 by substituting tert-butyl 4-(4-piperidyl)piperidine-1-carboxylate in Step A to afford 1-(4'-((1'-(2-(2,6-dioxopiperidin-3-yl)- 6-fluoro-1,3-dioxoisoindolin-5-yl)-[4,4'-bipiperidin]-1-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)- N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4- carboxamide. LCMS [M+1] = 923.3; 1H NMR (400 MHz, DMSO-d6) δ 13.92 (br s, 1H), 11.10 (s, 1H), 8.88 (br s, 1H), 8.53 (s, 1H), 8.17 (s, 1H), 7.69 - 7.66 (m, 5H), 7.44 - 7.36 (m, 4H), 5.09 (dd, J = 5.4, 12.7 Hz, 1H), 4.55 (br d, J = 5.4 Hz, 2H), 3.63 (br d, J = 11.2 Hz, 2H), 3.49 (br s, 3H), 2.89 - 2.79 (m, 4H), 2.68 - 2.56 (m, 2H), 2.30 (s, 3H), 2.08 - 1.98 (m, 1H), 1.95 - 1.87 (m, 3H), 1.79 (br d, J = 10.2 Hz, 2H), 1.68 (br d, J = 11.8 Hz, 2H), 1.54 (s, 6H), 1.36 - 1.21 (m, 4H), 1.16 - 1.05 (m, 1H) Example 35: Synthesis of Compound 35-1 and 35-2
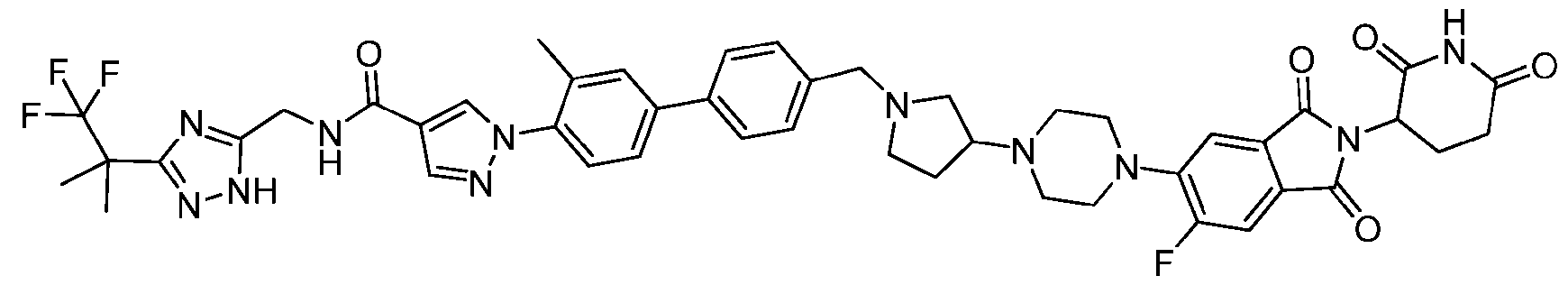 Compounds 35-1 and 35-2 were prepared in a similar manner to Example 28 by substituting tert-butyl 4-pyrrolidin-3-ylpiperazine-1-carboxylate in Step A. Chiral resolution of the enantiomers from Step C by SFC (DAICEL CHIRALPAK IG (250mm x 30mm, 10µm), [CO2-MeOH(0.1%NH3.water)]; isocratic elution) afforded the early-eluting isomer that was processed further to afford Compound 35-1, 1-(4'-((3-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro- 1,3-dioxoisoindolin-5-yl)piperazin-1-yl)pyrrolidin-1-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)- N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4- carboxamide (absolute stereochemistry was not confirmed). LCMS: [M+1] = 910.3; 1H NMR (400 MHz, METHANOL-d4) δ 8.36 (s, 1H), 8.18 (s, 1H), 7.70 -7.64 (m, 3H), 7.62 (dd, J = 2.0, 8.0 Hz, 1H), 7.54 (d, J = 11.2 Hz, 1H), 7.51 - 7.41 (m, 4H), 5.08 (dd, J = 5.4, 12.4 Hz, 1H), 4.70 (s, 2H), 3.81 - 3.65 (m, 2H), 3.04 - 2.92 (m, 2H), 2.89 - 2.58 (m, 9H), 2.52 - 2.43 (m, 1H), 2.31 (s, 3H), 2.17 - 2.03 (m, 2H), 1.93 - 1.76 (m, 1H), 1.61 (s, 6H).
The late-eluting enantiomer was processed further to afford Compound 35-2, 1-(4'-((3-(4- (2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1-yl)pyrrolidin-1- yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide (absolute stereochemistry was not confirmed) LCMS: [M+1] = 910.3; 1H NMR (400 MHz, METHANOL-d4) δ 8.36 (s, 1H), 8.18 (s, 1H), 7.70 - 7.64 (m, 3H), 7.62 (dd, J = 2.0, 8.0 Hz, 1H), 7.54 (d, J = 11.2 Hz, 1H), 7.51 - 7.41 (m, 4H), 5.08 (dd, J = 5.4, 12.4 Hz, 1H), 4.70 (s, 2H), 3.81 - 3.65 (m, 2H), 3.04 - 2.92 (m, 2H), 2.89 - 2.58 (m, 9H), 2.52 - 2.43 (m, 1H), 2.31 (s, 3H), 2.17 - 2.03 (m, 2H), 1.93 - 1.76 (m, 1H), 1.61 (s, 6H). Example 36: Synthesis of Compound 36
Compounds 35-1 and 35-2 were prepared in a similar manner to Example 28 by substituting tert-butyl 4-pyrrolidin-3-ylpiperazine-1-carboxylate in Step A. Chiral resolution of the enantiomers from Step C by SFC (DAICEL CHIRALPAK IG (250mm x 30mm, 10µm), [CO2-MeOH(0.1%NH3.water)]; isocratic elution) afforded the early-eluting isomer that was processed further to afford Compound 35-1, 1-(4'-((3-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro- 1,3-dioxoisoindolin-5-yl)piperazin-1-yl)pyrrolidin-1-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)- N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4- carboxamide (absolute stereochemistry was not confirmed). LCMS: [M+1] = 910.3; 1H NMR (400 MHz, METHANOL-d4) δ 8.36 (s, 1H), 8.18 (s, 1H), 7.70 -7.64 (m, 3H), 7.62 (dd, J = 2.0, 8.0 Hz, 1H), 7.54 (d, J = 11.2 Hz, 1H), 7.51 - 7.41 (m, 4H), 5.08 (dd, J = 5.4, 12.4 Hz, 1H), 4.70 (s, 2H), 3.81 - 3.65 (m, 2H), 3.04 - 2.92 (m, 2H), 2.89 - 2.58 (m, 9H), 2.52 - 2.43 (m, 1H), 2.31 (s, 3H), 2.17 - 2.03 (m, 2H), 1.93 - 1.76 (m, 1H), 1.61 (s, 6H).
The late-eluting enantiomer was processed further to afford Compound 35-2, 1-(4'-((3-(4- (2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1-yl)pyrrolidin-1- yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide (absolute stereochemistry was not confirmed) LCMS: [M+1] = 910.3; 1H NMR (400 MHz, METHANOL-d4) δ 8.36 (s, 1H), 8.18 (s, 1H), 7.70 - 7.64 (m, 3H), 7.62 (dd, J = 2.0, 8.0 Hz, 1H), 7.54 (d, J = 11.2 Hz, 1H), 7.51 - 7.41 (m, 4H), 5.08 (dd, J = 5.4, 12.4 Hz, 1H), 4.70 (s, 2H), 3.81 - 3.65 (m, 2H), 3.04 - 2.92 (m, 2H), 2.89 - 2.58 (m, 9H), 2.52 - 2.43 (m, 1H), 2.31 (s, 3H), 2.17 - 2.03 (m, 2H), 1.93 - 1.76 (m, 1H), 1.61 (s, 6H). Example 36: Synthesis of Compound 36
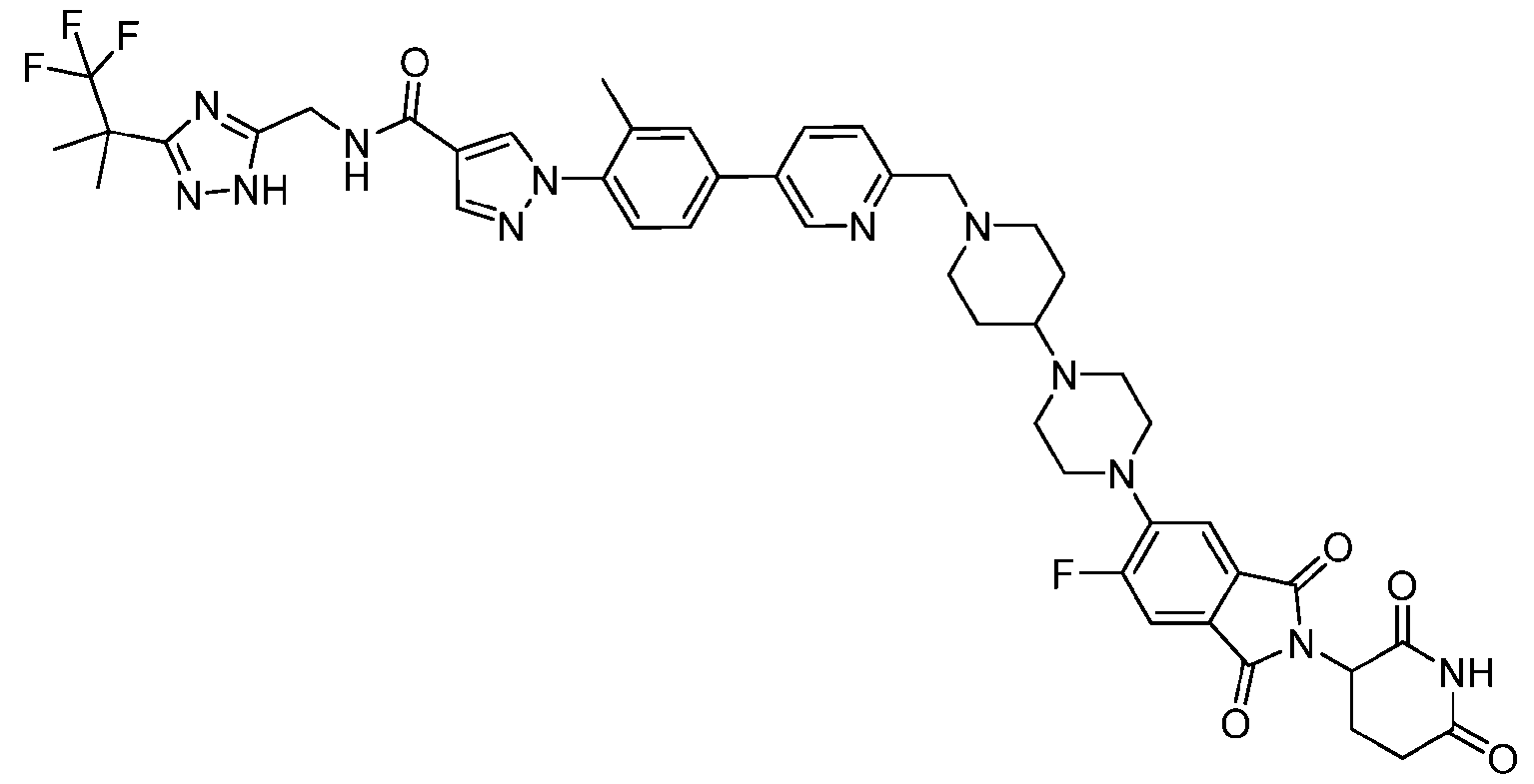 5-(4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine-2-carbaldehyde in Step A to afford 1-(4-(6-((4-(4- (2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1-yl)piperidin-1- yl)methyl)pyridin-3-yl)-2-methylphenyl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide. LCMS: [M+1] = 925.5; 1H NMR (400 MHz, DMSO-d6) δ 13.90 (br s, 1H), 11.10 (s, 1H), 8.95 - 8.88 (m, 1H), 8.86 (d, J = 2.3 Hz, 1H), 8.54 (s, 1H), 8.18 (s, 1H), 8.12 (dd, J = 2.4, 8.2 Hz, 1H), 7.80 (s, 1H), 7.74 - 7.68 (m, 2H), 7.53 (dd, J = 8.2, 14.4 Hz, 2H), 7.44 (d, J = 7.4 Hz, 1H), 5.10 (dd, J = 5.6, 12.8 Hz, 1H), 4.55 (br d, J = 2.4 Hz, 2H), 3.64 (s, 2H), 3.24 (br s, 4H), 2.95 - 2.86 (m, 3H), 2.69 - 2.64 (m, 4H), 2.34 - 2.30 (m, 4H),
2.28 - 2.22 (m, 1H), 2.11 - 1.99 (m, 4H), 1.79 (br d, J = 11.0 Hz, 2H), 1.54 (s, 6H), 1.51 - 1.42 (m, 2H). Example 37: Synthesis of Compound 37
5-(4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine-2-carbaldehyde in Step A to afford 1-(4-(6-((4-(4- (2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1-yl)piperidin-1- yl)methyl)pyridin-3-yl)-2-methylphenyl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide. LCMS: [M+1] = 925.5; 1H NMR (400 MHz, DMSO-d6) δ 13.90 (br s, 1H), 11.10 (s, 1H), 8.95 - 8.88 (m, 1H), 8.86 (d, J = 2.3 Hz, 1H), 8.54 (s, 1H), 8.18 (s, 1H), 8.12 (dd, J = 2.4, 8.2 Hz, 1H), 7.80 (s, 1H), 7.74 - 7.68 (m, 2H), 7.53 (dd, J = 8.2, 14.4 Hz, 2H), 7.44 (d, J = 7.4 Hz, 1H), 5.10 (dd, J = 5.6, 12.8 Hz, 1H), 4.55 (br d, J = 2.4 Hz, 2H), 3.64 (s, 2H), 3.24 (br s, 4H), 2.95 - 2.86 (m, 3H), 2.69 - 2.64 (m, 4H), 2.34 - 2.30 (m, 4H),
2.28 - 2.22 (m, 1H), 2.11 - 1.99 (m, 4H), 1.79 (br d, J = 11.0 Hz, 2H), 1.54 (s, 6H), 1.51 - 1.42 (m, 2H). Example 37: Synthesis of Compound 37
 3-chloro-4- (4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzaldehyde in Step A to afford 1-(2'-chloro-4'-((4- (4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1-yl)piperidin-1- yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide. LCMS [M+1] = 958.4;1H NMR (400 MHz, DMSO-d6) δ 14.12 - 13.80 (br s, 1H), 11.10 (br s, 1H), 8.88 (br s, 1H), 8.55 (s, 1H), 8.18 (s, 1H), 7.81 - 7.63 (m, 1H), 7.55 - 7.34 (m, 7H), 5.10 (dd, J = 5.4, 12.8 Hz, 1H), 4.55 (d, J = 5.8 Hz, 2H), 3.51 (s, 2H), 3.23 (br s, 4H), 2.96 - 2.81 (m, 4H), 2.66 (br d, J = 1.5 Hz, 5H), 2.37 - 2.20 (m, 4H), 2.04 -1.97 (m, 3H), 1.87 - 1.69 (m, 2H), 1.54 (s, 6H), 1.51 - 1.41 (m, 2H) Example 38: Synthesis of Compound 38
3-chloro-4- (4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzaldehyde in Step A to afford 1-(2'-chloro-4'-((4- (4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1-yl)piperidin-1- yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide. LCMS [M+1] = 958.4;1H NMR (400 MHz, DMSO-d6) δ 14.12 - 13.80 (br s, 1H), 11.10 (br s, 1H), 8.88 (br s, 1H), 8.55 (s, 1H), 8.18 (s, 1H), 7.81 - 7.63 (m, 1H), 7.55 - 7.34 (m, 7H), 5.10 (dd, J = 5.4, 12.8 Hz, 1H), 4.55 (d, J = 5.8 Hz, 2H), 3.51 (s, 2H), 3.23 (br s, 4H), 2.96 - 2.81 (m, 4H), 2.66 (br d, J = 1.5 Hz, 5H), 2.37 - 2.20 (m, 4H), 2.04 -1.97 (m, 3H), 1.87 - 1.69 (m, 2H), 1.54 (s, 6H), 1.51 - 1.41 (m, 2H) Example 38: Synthesis of Compound 38
4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)benzaldehyde in Step A to afford 1-(3'-((4-(4-(2-(2,6- dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1-yl)piperidin-1-yl)methyl)-3- methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5- yl)methyl)-1H-pyrazole-4-carboxamide. LCMS: [M+1] = 924.5; 1H NMR (400 MHz, DMSO-d6) δ 13.93 (br s, 1H), 11.09 (s, 1H), 8.88 (s, 1H), 8.53 (s, 1H), 8.18 (br s, 1H), 7.76 - 7.69 (m, 2H), 7.67 - 7.56 (m, 3H), 7.46 (dd, J = 8.4, 16.0 Hz, 3H), 7.34 (d, J = 8.0 Hz, 1H), 5.10 (dd, J = 5.2, 12.8 Hz, 1H), 4.55 (d, J = 5.6 Hz, 2H), 3.54 (s, 2H), 3.23 (s, 5H), 2.96 - 2.82 (m, 3H), 2.68 - 2.55 (m, 5H), 2.31 (s, 3H), 2.26 - 2.12 (m, 1H), 2.08 - 1.92 (m, 3H), 1.78 - 1.75 (m, 2H), 1.54 (s, 6H), 1.50 - 1.36 (m, 2H). Example 39: Synthesis of Compound 39-1 and 39-2
 substituting 1-[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]ethenone in Step A. Separation of enantiomers after Step B by SFC (DAICEL CHIRALPAK AD (250 mm x 30 mm,10µm); [CO2-IPA(0.1%NH3water)]; isocratic elution) afforded the early-eluting enantiomer, which was processed further to afford 39-1 (1-(4'-(1-(4-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro- 1,3-dioxoisoindolin-5-yl)piperazin-1-yl)piperidin-1-yl)ethyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-
((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4- carboxamide). LCMS [M+1] =938.4; 1H NMR (400 MHz, DMSO-d6) δ 13.93 (br s, 1H), 11.10 (s, 1H), 8.88 (br s, 1H), 8.53 (s, 1H), 8.17 (s, 1H), 7.79 - 7.58 (m, 5H), 7.53 - 7.31 (m, 4H), 5.10 (dd, J = 5.4, 12.8 Hz, 1H), 4.55 (br s, 2H), 3.56 - 3.48 (m, 1H), 3.22 (br s, 4H), 3.04 (br d, J = 9.0 Hz, 1H), 2.93 - 2.79 (m, 2H), 2.64 (br s, 5H), 2.30 (s, 3H), 2.23 - 2.10 (m, 1H), 2.08 - 1.68 (m, 6H), 1.54 (s, 6H), 1.43 - 1.27 (m, 5H). The late-eluting isomer from the SFC separation was further processed to afford 39-2 LCMS [M+1] = 938.5; 1H NMR (400 MHz, DMSO-d6) δ 13.93 (br s, 1H), 11.10 (s, 1H), 8.89 (br s, 1H), 8.53 (s, 1H), 8.17 (s, 1H), 7.76 - 7.58 (m, 5H), 7.52 - 7.35 (m, 4H), 5.10 (dd, J = 5.4, 12.8 Hz, 1H), 4.55 (br d, J = 5.4 Hz, 2H), 3.55 - 3.46 (m, 2H), 3.22 (br s, 4H), 3.04 (br d, J = 9.0 Hz, 1H), 2.96 - 2.80 (m, 2H), 2.71 - 2.58 (m, 6H), 2.30 (s, 3H), 2.23 - 2.12 (m, 1H), 2.08 - 1.68 (m, 6H), 1.54 (s, 6H), 1.33 (br d, J = 6.6 Hz, 3H). Example 40: Synthesis of Compound 40
substituting 1-[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]ethenone in Step A. Separation of enantiomers after Step B by SFC (DAICEL CHIRALPAK AD (250 mm x 30 mm,10µm); [CO2-IPA(0.1%NH3water)]; isocratic elution) afforded the early-eluting enantiomer, which was processed further to afford 39-1 (1-(4'-(1-(4-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro- 1,3-dioxoisoindolin-5-yl)piperazin-1-yl)piperidin-1-yl)ethyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-
((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4- carboxamide). LCMS [M+1] =938.4; 1H NMR (400 MHz, DMSO-d6) δ 13.93 (br s, 1H), 11.10 (s, 1H), 8.88 (br s, 1H), 8.53 (s, 1H), 8.17 (s, 1H), 7.79 - 7.58 (m, 5H), 7.53 - 7.31 (m, 4H), 5.10 (dd, J = 5.4, 12.8 Hz, 1H), 4.55 (br s, 2H), 3.56 - 3.48 (m, 1H), 3.22 (br s, 4H), 3.04 (br d, J = 9.0 Hz, 1H), 2.93 - 2.79 (m, 2H), 2.64 (br s, 5H), 2.30 (s, 3H), 2.23 - 2.10 (m, 1H), 2.08 - 1.68 (m, 6H), 1.54 (s, 6H), 1.43 - 1.27 (m, 5H). The late-eluting isomer from the SFC separation was further processed to afford 39-2 LCMS [M+1] = 938.5; 1H NMR (400 MHz, DMSO-d6) δ 13.93 (br s, 1H), 11.10 (s, 1H), 8.89 (br s, 1H), 8.53 (s, 1H), 8.17 (s, 1H), 7.76 - 7.58 (m, 5H), 7.52 - 7.35 (m, 4H), 5.10 (dd, J = 5.4, 12.8 Hz, 1H), 4.55 (br d, J = 5.4 Hz, 2H), 3.55 - 3.46 (m, 2H), 3.22 (br s, 4H), 3.04 (br d, J = 9.0 Hz, 1H), 2.96 - 2.80 (m, 2H), 2.71 - 2.58 (m, 6H), 2.30 (s, 3H), 2.23 - 2.12 (m, 1H), 2.08 - 1.68 (m, 6H), 1.54 (s, 6H), 1.33 (br d, J = 6.6 Hz, 3H). Example 40: Synthesis of Compound 40
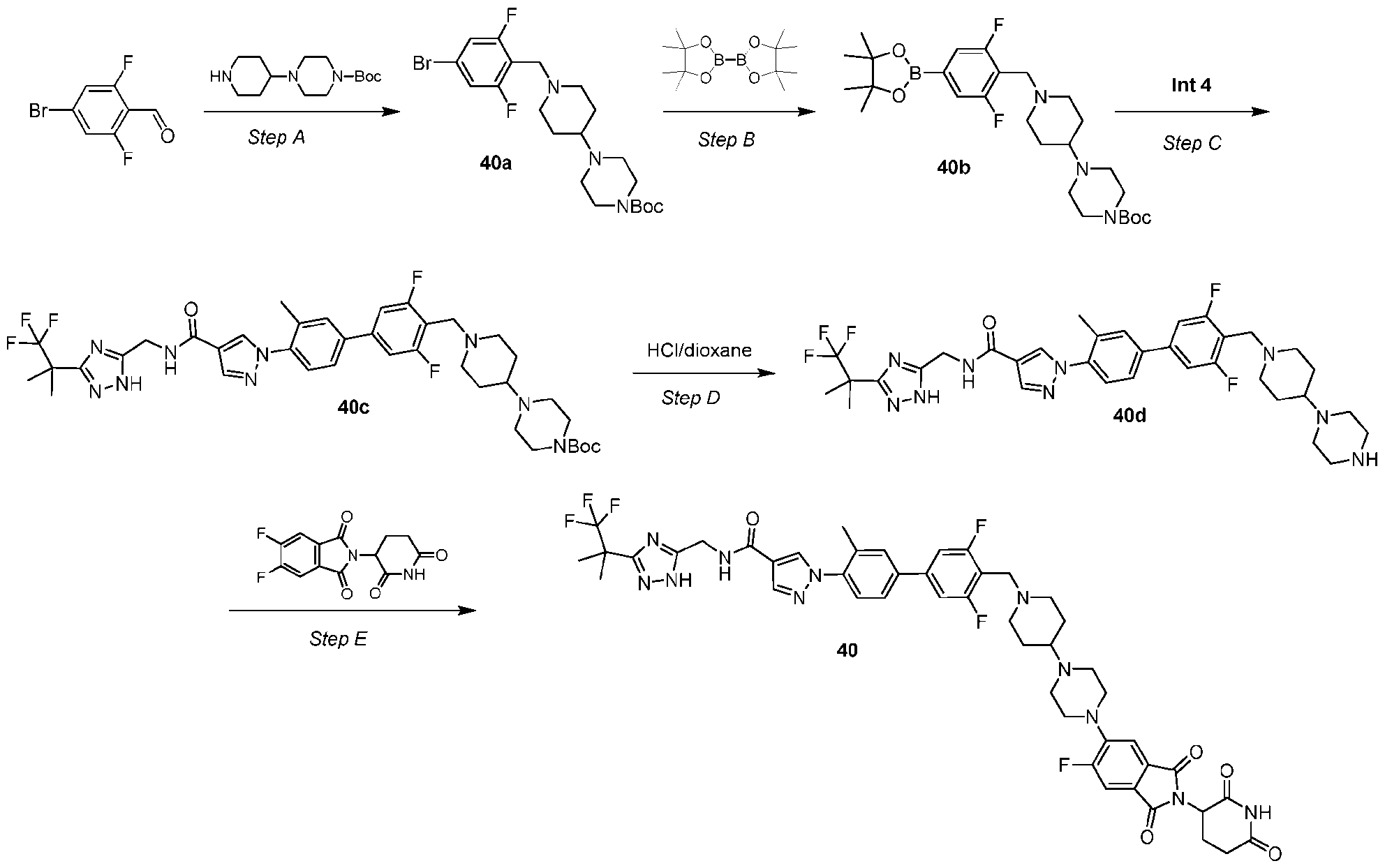 carboxylate (40a)
A mixture of tert-butyl 4-(4-piperidyl)piperazine-1-carboxylate (792.3 mg, 2.9 mmol, 1.3 equiv.), 4-bromo-2,6-difluoro-benzaldehyde (500 mg, 2.2 mmol, 1.0 equiv.) and acetic acid (135.8 mg, 2.2 mmol, 129.5 μL, 1.0 equiv.) in dichloromethane (5 mL) was stirred at 30°C for 2 h. Then the mixture was added NaBH(OAc)3 (959.0 mg, 4.5 mmol, 2.0 equiv.) and the mixture was stirred at 30°C for 16 h under a nitrogen atmosphere. The reaction mixture was partitioned between ethyl acetate (100 mL) and aqueous sodium bicarbonate (100 mL). The organic phase was separated, washed with brine 30 mL, dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, dichloromethane: methanol = 100:0 to 96:4) to afford 40a. LCMS [M+1] =474.2, 476.0; 1H NMR (400 MHz, DMSO-d6) δ 7.46 (d, J = 7.0 Hz, 2H), 3.50 (s, 2H), 3.25 (br s, 4H), 2.80 (br d, J = 11.2 Hz, 2H), 2.37 (br t, J = 4.6 Hz, 4H), 2.18 - 2.08 (m, 1H), 1.95 (br t, J = 11.2 Hz, 2H), 1.65 (br d, J = 11.8 Hz, 2H), 1.33-1.40 (m, 11H) Step B – Synthesis of tert-butyl 4-[1-[[2,6-difluoro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)phenyl]methyl]-4-piperidyl]piperazine-1-carboxylate (40b) To a solution of 40a (500 mg, 1.05 mmol, 1.0 equiv.), 4,4,5,5-tetramethyl-2-(4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3,2-dioxaborolane (401.4 mg, 1.5 mmol, 1.5 equiv.) in dioxane (5 mL) was added potassium acetate (310.3 mg, 3.1 mmol, 3.0 equiv.) and Pd(dppf)Cl2.CH2Cl2 (172.1 mg, 210.8 μmol, 0.2 equiv.) under nitrogen. The mixture was stirred at 80°C for 4 h. The reaction mixture was cooled to ambient temperature and quenched with water (100 mL) and extracted with ethyl acetate (3 x 30 mL). The combined organic layers were washed with brine (50 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, dichloromethane: methanol = 100:0 to 0:100) to afford 40b, which was used without further purification. Step C – Synthesis of tert-butyl 4-[1-[[2,6-difluoro-4-[3-methyl-4-[4-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methylcarbamoyl]pyrazol-1-yl]phenyl]phenyl]methyl]-4- piperidyl]piperazine-1-carboxylate (40c) To a solution of Int 4 (100 mg, 212.1 μmol, 1.0 equiv.), 40b (221.2 mg, 424.3 μmol, 2.0 equiv.) in dioxane (2 mL) and water (0.2 mL) was added potassium carbonate (87.9 mg, 636.5
μmol, 3.0 equiv.) and Pd(dppf)Cl2.CH2Cl2 (34.6 mg, 42.4 μmol, 0.2 equiv.). The mixture was stirred at 100°C for 2 h., cooled to ambient temperature and quenched with water (80 mL) and extracted with ethyl acetate (3 x 30 mL). The combined organic layers were washed with brine (50 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative-TLC (silica gel, dichloromethane: MeOH = 10:1) to afford 40c. LCMS [M+1] =786.5; 1H NMR (400 MHz, DMSO-d6) δ 13.90 (br s, 1H), 8.89 (br s, 1H), 8.55 (s, 1H), 8.18 (s, 1H), 7.85 (d, J = 1.6 Hz, 1H), 7.74 (dd, J = 1.8, 8.3 Hz, 1H), 7.59 - 7.45 (m, 3H), 4.56 (br d, J = 3.6 Hz, 2H), 3.58 (s, 2H), 3.26 (br s, 4H), 2.86 (br s, 2H), 2.39 (br s, 4H), 2.31 (s, 3H), 2.17 (br t, J = 11.4 Hz, 1H), 2.01 (br t, J = 11.0 Hz, 2H), 1.68 (br d, J = 11.4 Hz, 2H), 1.54 (s, 6H), 1.34-1.40 (m, 11H). Step D – Synthesis of 1-[4-[3,5-difluoro-4-[(4-piperazin-1-yl-1-piperidyl)methyl]phenyl]-2- methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole- 4-carboxamide (40d) To a solution of 40c (80.0 mg, 101.8 μmol, 1.0 equiv.) in HCl in dioxane (4 M, 1 mL) under nitrogen. The mixture was stirred at 25°C for 1 h. The reaction mixture was concentrated to dryness to afford 40d, which was used without further purification. LCMS [M+1] =686.5. Step E – Synthesis of 1-[4-[4-[[4-[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]piperazin-1-yl]-1-piperidyl]methyl]-3,5-difluoro-phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2- trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Compound 40) To a solution of 40d (75 mg, 109.3 μmol, 1.0 equiv.) ,2-(2,6-dioxo-3-piperidyl)-5,6- difluoro-isoindoline-1,3-dione (48.2 mg, 164.0 μmol, 1.5 equiv.) in DMSO (1 mL) was added DIEA (42.4 mg, 328.1 μmol, 57.1 μL, 3.0 equiv.). The mixture was stirred at 70°C for 16 h, cooled t ambient temperature and partitioned between ethyl acetate (40 mL) and water (40 mL). The organic phase was separated, washed with brine (30 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative TLC (silica gel, dichloromethane: MeOH = 10:1) to afford Compound 40. LCMS [M+1] =960.3; 1H NMR (400 MHz, DMSO-d6) δ 13.92 (br s, 1H), 11.10 (s, 1H), 8.90 (br s, 1H), 8.55 (s, 1H), 8.18 (s, 1H), 7.85 (s, 1H), 7.79 - 7.67 (m, 2H), 7.63 - 7.38 (m, 4H), 5.10 (dd,
J = 5.2, 12.8 Hz, 1H), 4.55 (br d, J = 4.6 Hz, 2H), 3.60 (br s, 2H), 3.27 - 3.16 (m, 4H), 2.88 (br s, 3H), 2.70 - 2.53 (m, 6H), 2.32 (s, 3H), 2.26 - 2.14 (m, 1H), 2.09 - 1.96 (m, 3H), 1.76 (br d, J = 10.8 Hz, 2H), 1.54 (s, 6H), 1.48 - 1.34 (m, 2H) Example 41: Synthesis of Compound 41
carboxylate (40a)
A mixture of tert-butyl 4-(4-piperidyl)piperazine-1-carboxylate (792.3 mg, 2.9 mmol, 1.3 equiv.), 4-bromo-2,6-difluoro-benzaldehyde (500 mg, 2.2 mmol, 1.0 equiv.) and acetic acid (135.8 mg, 2.2 mmol, 129.5 μL, 1.0 equiv.) in dichloromethane (5 mL) was stirred at 30°C for 2 h. Then the mixture was added NaBH(OAc)3 (959.0 mg, 4.5 mmol, 2.0 equiv.) and the mixture was stirred at 30°C for 16 h under a nitrogen atmosphere. The reaction mixture was partitioned between ethyl acetate (100 mL) and aqueous sodium bicarbonate (100 mL). The organic phase was separated, washed with brine 30 mL, dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, dichloromethane: methanol = 100:0 to 96:4) to afford 40a. LCMS [M+1] =474.2, 476.0; 1H NMR (400 MHz, DMSO-d6) δ 7.46 (d, J = 7.0 Hz, 2H), 3.50 (s, 2H), 3.25 (br s, 4H), 2.80 (br d, J = 11.2 Hz, 2H), 2.37 (br t, J = 4.6 Hz, 4H), 2.18 - 2.08 (m, 1H), 1.95 (br t, J = 11.2 Hz, 2H), 1.65 (br d, J = 11.8 Hz, 2H), 1.33-1.40 (m, 11H) Step B – Synthesis of tert-butyl 4-[1-[[2,6-difluoro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)phenyl]methyl]-4-piperidyl]piperazine-1-carboxylate (40b) To a solution of 40a (500 mg, 1.05 mmol, 1.0 equiv.), 4,4,5,5-tetramethyl-2-(4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3,2-dioxaborolane (401.4 mg, 1.5 mmol, 1.5 equiv.) in dioxane (5 mL) was added potassium acetate (310.3 mg, 3.1 mmol, 3.0 equiv.) and Pd(dppf)Cl2.CH2Cl2 (172.1 mg, 210.8 μmol, 0.2 equiv.) under nitrogen. The mixture was stirred at 80°C for 4 h. The reaction mixture was cooled to ambient temperature and quenched with water (100 mL) and extracted with ethyl acetate (3 x 30 mL). The combined organic layers were washed with brine (50 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, dichloromethane: methanol = 100:0 to 0:100) to afford 40b, which was used without further purification. Step C – Synthesis of tert-butyl 4-[1-[[2,6-difluoro-4-[3-methyl-4-[4-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methylcarbamoyl]pyrazol-1-yl]phenyl]phenyl]methyl]-4- piperidyl]piperazine-1-carboxylate (40c) To a solution of Int 4 (100 mg, 212.1 μmol, 1.0 equiv.), 40b (221.2 mg, 424.3 μmol, 2.0 equiv.) in dioxane (2 mL) and water (0.2 mL) was added potassium carbonate (87.9 mg, 636.5
μmol, 3.0 equiv.) and Pd(dppf)Cl2.CH2Cl2 (34.6 mg, 42.4 μmol, 0.2 equiv.). The mixture was stirred at 100°C for 2 h., cooled to ambient temperature and quenched with water (80 mL) and extracted with ethyl acetate (3 x 30 mL). The combined organic layers were washed with brine (50 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative-TLC (silica gel, dichloromethane: MeOH = 10:1) to afford 40c. LCMS [M+1] =786.5; 1H NMR (400 MHz, DMSO-d6) δ 13.90 (br s, 1H), 8.89 (br s, 1H), 8.55 (s, 1H), 8.18 (s, 1H), 7.85 (d, J = 1.6 Hz, 1H), 7.74 (dd, J = 1.8, 8.3 Hz, 1H), 7.59 - 7.45 (m, 3H), 4.56 (br d, J = 3.6 Hz, 2H), 3.58 (s, 2H), 3.26 (br s, 4H), 2.86 (br s, 2H), 2.39 (br s, 4H), 2.31 (s, 3H), 2.17 (br t, J = 11.4 Hz, 1H), 2.01 (br t, J = 11.0 Hz, 2H), 1.68 (br d, J = 11.4 Hz, 2H), 1.54 (s, 6H), 1.34-1.40 (m, 11H). Step D – Synthesis of 1-[4-[3,5-difluoro-4-[(4-piperazin-1-yl-1-piperidyl)methyl]phenyl]-2- methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole- 4-carboxamide (40d) To a solution of 40c (80.0 mg, 101.8 μmol, 1.0 equiv.) in HCl in dioxane (4 M, 1 mL) under nitrogen. The mixture was stirred at 25°C for 1 h. The reaction mixture was concentrated to dryness to afford 40d, which was used without further purification. LCMS [M+1] =686.5. Step E – Synthesis of 1-[4-[4-[[4-[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]piperazin-1-yl]-1-piperidyl]methyl]-3,5-difluoro-phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2- trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Compound 40) To a solution of 40d (75 mg, 109.3 μmol, 1.0 equiv.) ,2-(2,6-dioxo-3-piperidyl)-5,6- difluoro-isoindoline-1,3-dione (48.2 mg, 164.0 μmol, 1.5 equiv.) in DMSO (1 mL) was added DIEA (42.4 mg, 328.1 μmol, 57.1 μL, 3.0 equiv.). The mixture was stirred at 70°C for 16 h, cooled t ambient temperature and partitioned between ethyl acetate (40 mL) and water (40 mL). The organic phase was separated, washed with brine (30 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative TLC (silica gel, dichloromethane: MeOH = 10:1) to afford Compound 40. LCMS [M+1] =960.3; 1H NMR (400 MHz, DMSO-d6) δ 13.92 (br s, 1H), 11.10 (s, 1H), 8.90 (br s, 1H), 8.55 (s, 1H), 8.18 (s, 1H), 7.85 (s, 1H), 7.79 - 7.67 (m, 2H), 7.63 - 7.38 (m, 4H), 5.10 (dd,
J = 5.2, 12.8 Hz, 1H), 4.55 (br d, J = 4.6 Hz, 2H), 3.60 (br s, 2H), 3.27 - 3.16 (m, 4H), 2.88 (br s, 3H), 2.70 - 2.53 (m, 6H), 2.32 (s, 3H), 2.26 - 2.14 (m, 1H), 2.09 - 1.96 (m, 3H), 1.76 (br d, J = 10.8 Hz, 2H), 1.54 (s, 6H), 1.48 - 1.34 (m, 2H) Example 41: Synthesis of Compound 41
 Step A – Synthesis of tert-butyl 4-(5-bromo-3-methyl-1H-indazol-1-yl)-[1, 4'-bipiperidine]-1'- carboxylate (41a)
To a solution of tert-butyl 4-hydroxy-[1, 4'-bipiperidine]-1'-carboxylate (3.0 g, 10.5 mmol, 1.0 equiv.) and 5-bromo-3-methyl-1H-indazole (2.2 g, 10.5 mmol, 1.0 equiv.) in toluene (30.0 mL) was added 2-(tributyl-phosphanylidene)acetonitrile (5.0 g, 21.1 mmol, 2.0 equiv.). The mixture was stirred at 100°C for 3 h. The mixture was cooled to ambient temperature and poured into water (100 mL). The aqueous phase was extracted with ethyl acetate (3 x 100 mL). The combined organic phase was washed with brine (3 x 50 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum. The residue was purified by column chromatography (silica gel, petroleum ether/ ethyl acetate=1/0 to 4/1) to give 41a. LCMS [M+1]= 477.2, 479.1; 1H NMR (400 MHz, CHLOROFORM-d) δ 7.78 (s, 1H), 7.40 (dd, J = 1.6, 8.9 Hz, 1H), 7.30 (d, J = 8.9 Hz, 1H), 4.34 - 4.25 (m, 1H), 4.23 - 4.17 (m, 1H), 4.15 (m, 1H), 3.17 - 3.07 (m, 2H), 2.67 (m, 3H), 2.52 (s, 3H), 2.46 - 2.27 (m, 4H), 1.99 (m, 2H), 1.90 - 1.75 (m, 4H), 1.72 - 1.67 (m, 2H), 1.47 (s, 9H). Step B – Synthesis of tert-butyl 4-(3-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H- indazol-1-yl)-[1,4'-bipiperidine]-1'-carboxylate (41b) To a solution of 41a (1.5 g, 3.1 mmol, 1.0 equiv.) and 4,4,5,5-tetramethyl-2-(4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3,2-dioxaborolane (1.2 g, 4.7 mmol, 1.5 equiv.) in dioxane (15.0 mL) was added potassium acetate (925.0 mg, 9.4 mmol, 3.0 equiv.) and Pd(dppf)Cl2 (229.8 mg, 314.1 μmol, 0.1 equiv.). The mixture was stirred at 100 °C for 1 h. The mixture was cooled to ambient temperature and poured into water (100 mL). The aqueous phase was extracted with ethyl acetate (3 x 100 mL). The combined organic phase was washed with brine (3 x 50 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum. The residue was purified by column chromatography (silica gel, petroleum ether/ ethyl acetate=1/0 to 3/1) afford (41b). LCMS [M+1] = 525.2; 1H NMR (400 MHz, CHLOROFORM-d) δ 8.19 (s, 1H), 7.75 (d, J = 8.0 Hz, 1H), 7.40 (d, J = 8.0 Hz, 1H), 4.42 - 4.29 (m, 1H), 4.26 - 4.12 (m, 2H), 3.10 (m, 2H), 2.83 - 2.63 (m, 3H), 2.57 (s, 3H), 2.55 (m, 1H), 2.50 - 2.28 (m, 5H), 2.02 (m, 2H), 1.83 (m, 2H), 1.49 (m, 2H), 1.47 (s, 9H), 1.38 (s, 12H). Step C – Synthesis of tert-butyl 4-(3-methyl-5-(3-methyl-4-(4-(((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)carbamoyl)-1H-pyrazol-1-yl)phenyl)-1H- indazol-1-yl)-[1,4'-bipiperidine]-1'-carboxylate (41c)
To a solution of Int 4 (250.0 mg, 530.4 μmol, 1.0 equiv.) and 41b (333.88 mg, 636.57 μmol, 1.2 equiv.) in dioxane (2.5 mL) and water (0.4 mL) was added K2CO3 (219.9 mg, 1.5 mmol, 3.0 equiv.) and Pd(dppf)Cl2.CH2Cl2 (43.3 mg, 53.0 μmol, 0.1 equiv.) under a nitrogen atmosphere at ambient temperature. The mixture was stirred at 100°C for 16 h. The mixture was cooled to ambient temperature and poured into water (30 mL). The aqueous phase was extracted with ethyl acetate (3 x 30 mL). The combined organic phase was washed with brine (3 x 10 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum. The residue was purified by column chromatography (silica gel, dichloromethane: methanol =1/0 to 20/1) to afford 41c. LCMS [M+1] = 789.5; 1H NMR (400 MHz, METHANOL-d4) δ 8.36 (s, 1H), 8.18 (s, 1H), 7.96 (s, 1H), 7.75 - 7.70 (m, 2H), 7.68 - 7.63 (m, 2H), 7.44 (d, J = 8.0 Hz, 1H), 4.70 (s, 2H), 4.55 - 4.54 (m, 1H), 4.22 - 4.16 (m, 2H), 3.24- 3.21 (m, 2H), 2.82 – 2.80 (m, 2H), 2.71 - 2.62 (m, 2H), 2.59 (s, 3H), 2.58 - 2.52 (m, 1H), 2.43 - 2.33 (m, 2H), 2.31 (s, 3H), 2.10 - 2.00 (m, 2H), 1.99 - 1.90 (m, 2H), 1.61 (s, 6H), 1.50 - 1.49 (m, 2H), 1.47 (s, 9H). Step D – Synthesis of 1-(4-(1-([1,4'-bipiperidin]-4-yl)-3-methyl-1H-indazol-5-yl)-2- methylphenyl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H- pyrazole-4-carboxamide (41d) A solution of 41c (230.0 mg, 291.5 μmol, 1.0 equiv.) in HCl/dioxane (4 M, 2.5 mL) was stirred at ambient temperature for 0.5 h. The mixture was concentrated in vacuum to afford 41d, which was used without further purification.LCMS [M+1] = 689.5; 1H NMR (400 MHz, DMSO- d6) δ 11.38 (s, 1H), 9.33 - 9.22 (m, 1H), 9.16 - 9.14 (m, 1H), 8.96 (t, J = 5.3 Hz, 1H), 8.58 (s, 1H), 8.19 (s, 1H), 8.10 (s, 1H), 7.81 (br s, 2H), 7.72 (br d, J = 8.2 Hz, 1H), 7.47 (br d, J = 8.2 Hz, 1H), 5.13 - 5.10 (m, 1H), 5.03 - 4.92 (m, 2H), 4.56 (d, J = 5.5 Hz, 2H), 3.47 - 3.43 (m, 2H), 3.32 - 3.27 (m, 2H), 2.97 - 2.92 (m, 2H), 2.70 - 2.65 (m, 2H), 2.60 - 2.59 (m, 1H), 2.56 (s, 3H), 2.39 - 2.28 (m, 5H), 2.20 - 1.94 (m, 4H), 1.54 (s, 6H). Step E – Synthesis of 1-(4-(1-(1'-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)- [1,4'-bipiperidin]-4-yl)-3-methyl-1H-indazol-5-yl)-2-methylphenyl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide (Compound 41) To a solution of 41d (150.0 mg, 197.0 μmol, 1.0 equiv.) in DMSO (1.5 mL) was added DIEA (84.4 mg, 653.3 μmol, 113.8 μL, 3.3 equiv.) and 2-(2,6-dioxopiperidin-3-yl)-5,6-
difluoroisoindoline-1,3-dione (96.1 mg, 326.6 μmol, 1.7 equiv.). The mixture was stirred at 100°C for 1 h. The mixture was diluted with acetonitrile (1 mL) and filtered to remove the insoluble portion. The filtrate was purified by preparative HPLC (Phenomenex Luna C18, 150 mm x 25mm; 10 µm; [water(FA)-ACN]) to give afford Compound 41. LCMS [M+1] = 963.5; 1H NMR (400 MHz, DMSO-d6) δ 13.93 (s, 1H), 11.12 (s, 1H), 8.89 (s, 1H), 8.53 (s, 1H), 8.18 (s, 1H), 8.14 (s, 1H), 8.06 (s, 1H), 7.80 (s, 1H), 7.77 - 7.67 (m, 4H), 7.50 - 7.43 (m, 2H), 5.11 (dd, J = 5.6, 12.6 Hz, 1H), 4.63 - 4.51 (m, 3H), 3.72 - 3.67 (m, 2H), 3.12 - 3.07 (m, 2H), 3.00 - 2.83 (m, 3H), 2.71 - 2.57 (m, 3H), 2.56 - 2.55 (m, 4H), 2.32 (s, 3H), 2.23 - 1.98 (m, 4H), 1.94 - 1.02 (m, 4H), 1.70 - 1.64 (m, 2H), 1.54 (s, 6H). Example 42: Synthesis of Compound 42
Step A – Synthesis of tert-butyl 4-(5-bromo-3-methyl-1H-indazol-1-yl)-[1, 4'-bipiperidine]-1'- carboxylate (41a)
To a solution of tert-butyl 4-hydroxy-[1, 4'-bipiperidine]-1'-carboxylate (3.0 g, 10.5 mmol, 1.0 equiv.) and 5-bromo-3-methyl-1H-indazole (2.2 g, 10.5 mmol, 1.0 equiv.) in toluene (30.0 mL) was added 2-(tributyl-phosphanylidene)acetonitrile (5.0 g, 21.1 mmol, 2.0 equiv.). The mixture was stirred at 100°C for 3 h. The mixture was cooled to ambient temperature and poured into water (100 mL). The aqueous phase was extracted with ethyl acetate (3 x 100 mL). The combined organic phase was washed with brine (3 x 50 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum. The residue was purified by column chromatography (silica gel, petroleum ether/ ethyl acetate=1/0 to 4/1) to give 41a. LCMS [M+1]= 477.2, 479.1; 1H NMR (400 MHz, CHLOROFORM-d) δ 7.78 (s, 1H), 7.40 (dd, J = 1.6, 8.9 Hz, 1H), 7.30 (d, J = 8.9 Hz, 1H), 4.34 - 4.25 (m, 1H), 4.23 - 4.17 (m, 1H), 4.15 (m, 1H), 3.17 - 3.07 (m, 2H), 2.67 (m, 3H), 2.52 (s, 3H), 2.46 - 2.27 (m, 4H), 1.99 (m, 2H), 1.90 - 1.75 (m, 4H), 1.72 - 1.67 (m, 2H), 1.47 (s, 9H). Step B – Synthesis of tert-butyl 4-(3-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H- indazol-1-yl)-[1,4'-bipiperidine]-1'-carboxylate (41b) To a solution of 41a (1.5 g, 3.1 mmol, 1.0 equiv.) and 4,4,5,5-tetramethyl-2-(4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3,2-dioxaborolane (1.2 g, 4.7 mmol, 1.5 equiv.) in dioxane (15.0 mL) was added potassium acetate (925.0 mg, 9.4 mmol, 3.0 equiv.) and Pd(dppf)Cl2 (229.8 mg, 314.1 μmol, 0.1 equiv.). The mixture was stirred at 100 °C for 1 h. The mixture was cooled to ambient temperature and poured into water (100 mL). The aqueous phase was extracted with ethyl acetate (3 x 100 mL). The combined organic phase was washed with brine (3 x 50 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum. The residue was purified by column chromatography (silica gel, petroleum ether/ ethyl acetate=1/0 to 3/1) afford (41b). LCMS [M+1] = 525.2; 1H NMR (400 MHz, CHLOROFORM-d) δ 8.19 (s, 1H), 7.75 (d, J = 8.0 Hz, 1H), 7.40 (d, J = 8.0 Hz, 1H), 4.42 - 4.29 (m, 1H), 4.26 - 4.12 (m, 2H), 3.10 (m, 2H), 2.83 - 2.63 (m, 3H), 2.57 (s, 3H), 2.55 (m, 1H), 2.50 - 2.28 (m, 5H), 2.02 (m, 2H), 1.83 (m, 2H), 1.49 (m, 2H), 1.47 (s, 9H), 1.38 (s, 12H). Step C – Synthesis of tert-butyl 4-(3-methyl-5-(3-methyl-4-(4-(((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)carbamoyl)-1H-pyrazol-1-yl)phenyl)-1H- indazol-1-yl)-[1,4'-bipiperidine]-1'-carboxylate (41c)
To a solution of Int 4 (250.0 mg, 530.4 μmol, 1.0 equiv.) and 41b (333.88 mg, 636.57 μmol, 1.2 equiv.) in dioxane (2.5 mL) and water (0.4 mL) was added K2CO3 (219.9 mg, 1.5 mmol, 3.0 equiv.) and Pd(dppf)Cl2.CH2Cl2 (43.3 mg, 53.0 μmol, 0.1 equiv.) under a nitrogen atmosphere at ambient temperature. The mixture was stirred at 100°C for 16 h. The mixture was cooled to ambient temperature and poured into water (30 mL). The aqueous phase was extracted with ethyl acetate (3 x 30 mL). The combined organic phase was washed with brine (3 x 10 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum. The residue was purified by column chromatography (silica gel, dichloromethane: methanol =1/0 to 20/1) to afford 41c. LCMS [M+1] = 789.5; 1H NMR (400 MHz, METHANOL-d4) δ 8.36 (s, 1H), 8.18 (s, 1H), 7.96 (s, 1H), 7.75 - 7.70 (m, 2H), 7.68 - 7.63 (m, 2H), 7.44 (d, J = 8.0 Hz, 1H), 4.70 (s, 2H), 4.55 - 4.54 (m, 1H), 4.22 - 4.16 (m, 2H), 3.24- 3.21 (m, 2H), 2.82 – 2.80 (m, 2H), 2.71 - 2.62 (m, 2H), 2.59 (s, 3H), 2.58 - 2.52 (m, 1H), 2.43 - 2.33 (m, 2H), 2.31 (s, 3H), 2.10 - 2.00 (m, 2H), 1.99 - 1.90 (m, 2H), 1.61 (s, 6H), 1.50 - 1.49 (m, 2H), 1.47 (s, 9H). Step D – Synthesis of 1-(4-(1-([1,4'-bipiperidin]-4-yl)-3-methyl-1H-indazol-5-yl)-2- methylphenyl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H- pyrazole-4-carboxamide (41d) A solution of 41c (230.0 mg, 291.5 μmol, 1.0 equiv.) in HCl/dioxane (4 M, 2.5 mL) was stirred at ambient temperature for 0.5 h. The mixture was concentrated in vacuum to afford 41d, which was used without further purification.LCMS [M+1] = 689.5; 1H NMR (400 MHz, DMSO- d6) δ 11.38 (s, 1H), 9.33 - 9.22 (m, 1H), 9.16 - 9.14 (m, 1H), 8.96 (t, J = 5.3 Hz, 1H), 8.58 (s, 1H), 8.19 (s, 1H), 8.10 (s, 1H), 7.81 (br s, 2H), 7.72 (br d, J = 8.2 Hz, 1H), 7.47 (br d, J = 8.2 Hz, 1H), 5.13 - 5.10 (m, 1H), 5.03 - 4.92 (m, 2H), 4.56 (d, J = 5.5 Hz, 2H), 3.47 - 3.43 (m, 2H), 3.32 - 3.27 (m, 2H), 2.97 - 2.92 (m, 2H), 2.70 - 2.65 (m, 2H), 2.60 - 2.59 (m, 1H), 2.56 (s, 3H), 2.39 - 2.28 (m, 5H), 2.20 - 1.94 (m, 4H), 1.54 (s, 6H). Step E – Synthesis of 1-(4-(1-(1'-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)- [1,4'-bipiperidin]-4-yl)-3-methyl-1H-indazol-5-yl)-2-methylphenyl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide (Compound 41) To a solution of 41d (150.0 mg, 197.0 μmol, 1.0 equiv.) in DMSO (1.5 mL) was added DIEA (84.4 mg, 653.3 μmol, 113.8 μL, 3.3 equiv.) and 2-(2,6-dioxopiperidin-3-yl)-5,6-
difluoroisoindoline-1,3-dione (96.1 mg, 326.6 μmol, 1.7 equiv.). The mixture was stirred at 100°C for 1 h. The mixture was diluted with acetonitrile (1 mL) and filtered to remove the insoluble portion. The filtrate was purified by preparative HPLC (Phenomenex Luna C18, 150 mm x 25mm; 10 µm; [water(FA)-ACN]) to give afford Compound 41. LCMS [M+1] = 963.5; 1H NMR (400 MHz, DMSO-d6) δ 13.93 (s, 1H), 11.12 (s, 1H), 8.89 (s, 1H), 8.53 (s, 1H), 8.18 (s, 1H), 8.14 (s, 1H), 8.06 (s, 1H), 7.80 (s, 1H), 7.77 - 7.67 (m, 4H), 7.50 - 7.43 (m, 2H), 5.11 (dd, J = 5.6, 12.6 Hz, 1H), 4.63 - 4.51 (m, 3H), 3.72 - 3.67 (m, 2H), 3.12 - 3.07 (m, 2H), 3.00 - 2.83 (m, 3H), 2.71 - 2.57 (m, 3H), 2.56 - 2.55 (m, 4H), 2.32 (s, 3H), 2.23 - 1.98 (m, 4H), 1.94 - 1.02 (m, 4H), 1.70 - 1.64 (m, 2H), 1.54 (s, 6H). Example 42: Synthesis of Compound 42
 (42a) To solution of tert-butyl 4-(4-hydroxycyclohexyl)piperazine-1-carboxylate (969.6 mg, 3.41 mmol, 1.2 equiv.) in DMF (10.0 mL) was added NaH (227.3 mg, 5.68 mmol, 60.0% purity, 2.0
equiv.) at 0°C and the mixture was stirred at 0°C for 1 h. 5-bromo-2-fluoro-pyridine (500.0 mg, 2.84 mmol, 292.40 μL, 1.0 equiv.) was added to the mixture. The mixture was stirred at 25°C for 12 h. The reaction mixture was diluted with water (10.0 mL) and extracted with ethyl acetate (10 mL x 3). The combined organic layers were washed with brine (10.0 mL), dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether: ethyl acetate = 1: 0 to 0: 1) to afford 42a. LCMS [M+1] = 440.2; 1H NMR (400 MHz, DMSO-d6) δ ppm 8.25 (d, J = 2.4 Hz, 1H), 7.86 (dd, J = 2.8, 8.8 Hz, 1H), 6.78 (d, J = 8.8 Hz, 1H), 4.96 - 4.82 (m, 1H), 3.97-3.93 (m, 2H), 2.80 - 2.73 (m, 3H), 2.70 - 2.66 (m, 1H), 2.45 - 2.38 (m, 1H), 2.37 - 2.29 (m, 2H), 1.99 - 1.91 (m, 2H), 1.70 (d, J = 12.4 Hz, 2H), 1.64 - 1.54 (m, 2H), 1.38 (s, 9H), 1.31 - 1.21 (m, 2H) Step B – Synthesis of tert-butyl 4-[4-[[5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-2- pyridyl]oxy]-1-piperidyl]piperidine-1-carboxylate (42b) To a solution of 42a (1.0 g, 1.36 mmol, 1.0 equiv.) and bis(pinacolato)diboron (346.0 mg, 1.36 mmol, 1 equiv.) in dioxane (10.0 mL) was added potassium acetate (267.4 mg, 2.72 mmol, 2.0 equiv.) and Pd(dppf)Cl2 (99.7 mg, 136.25 μmol, 0.1 equiv.) at 25°C under a nitrogen atmosphere. The mixture was stirred at 100°C for 12 h. The reaction mixture was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether: ethyl acetate = 1: 0 to 0: 1) to afford 42b. LCMS [M+1] = 488.3. Step C – Synthesis of tert-butyl 4-[4-[[5-[3-methyl-4-[4-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H-1,2,4-triazol-5-yl]methylcarbamoyl]pyrazol-1-yl]phenyl]-2-pyridyl]oxy]-1- piperidyl]piperidine-1-carboxylate (42c) To a solution of 42b (150.0 mg, 184.64 μmol, 1.5 equiv.) and Int 4 (58.0 mg, 123.09 μmol, 1.0 equiv.) in THF (1.0 mL) and water (0.25 mL) was added K3PO4 (78.39 mg, 369.28 μmol, 3.0 equiv.) and PdCl2(DTBPF) (16.05 mg, 24.62 μmol, 0.2 equiv.) at 25°C under a nitrogen atmosphere. .The mixture was stirred at 80°C for 12 h. The reaction mixture was diluted with water (1.0 mL) and extracted with ethyl acetate (1.0 mL x 3). The combined organic layers were washed with brine (1.0 mL), dried over sodium sulfate, filtered and concentrated under reduced
pressure. The residue was purified by preparative TLC (silica gel, petroleum ether: ethyl acetate= 0: 1) to afford 42c. LCMS [M+1] = 752.2. Step D – Synthesis of 1-[2-methyl-4-[6-[[1-(4-piperidyl)-4-piperidyl]oxy]-3-pyridyl]phenyl]-N- [[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (42d) A solution of 42c (100.0 mg, 79.80 μmol, 1 equiv.) in HCl/MeOH (3 M, 1.0 mL) was stirred at 25°C for 1 h. The reaction was monitored by LCMS. The reaction mixture was concentrated under reduced pressure to give 42d. Step E – Synthesis of 1-[4-[6-[[1-[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]-4-piperidyl]-4-piperidyl]oxy]-3-pyridyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Compound 42) To a solution of 42d (50.0 mg, 69.1 μmol, 1 equiv.) and 2-(2,6-dioxo-3-piperidyl)-5,6- difluoro-isoindoline-1,3-dione (22.6 mg, 76.7 μmol, 1.1 equiv.) in DMSO (1.0 mL) was added DIEA (99.1 mg, 767 μmol, 133.6 μL, 11.0 equiv.).The mixture was stirred at 100 °C for 12 h. The solution was filtered to give a residue and the residue purified by preparative HPLC (Phenomenex Luna C18, 100 mm x 40 mm, 5 µm; [water(0.04% HCl)-ACN]) to afford Compound 42. LCMS [M+1] = 926.4; 1H NMR (400 MHz, MeOD) δ ppm 8.48 (d, J = 11.2 Hz, 1H), 8.37 (s, 1H), 8.19 (s, 1H), 8.06 (t, J = 8.8 Hz, 1H), 7.66 (s, 1H), 7.62 - 7.57 (m, 2H), 7.54 (d, J = 7.2 Hz, 1H), 7.47 (d, J = 8.4 Hz, 1H), 7.05 - 6.91 (m, 1H), 5.52 - 5.29 (m, 1H), 5.10 (dd, J = 5.2, 12.4 Hz, 1H), 4.72 (d, J = 3.2 Hz, 2H), 3.87 (d, J = 12.0 Hz, 2H), 3.76 (d, J = 12.4 Hz, 1H), 3.61 (d, J = 11.2 Hz, 1H), 3.50 - 3.35 (m, 4H), 3.02 (t, J = 12.0 Hz, 2H), 2.92 - 2.82 (m, 1H), 2.80 - 2.65 (m, 2H), 2.59 (dd, J = 16.0 Hz, 1H), 2.43 (d, J = 15.2 Hz, 1H), 2.35 (s, 1H) 2.31 (s, 3H), 2.26 - 2.18 (m, 1H), 2.15 - 2.09 (m, 1H), 2.08 - 1.95 (m, 3H), 1.63 (s, 6H). Example 43: Synthesis of Compound 43
(42a) To solution of tert-butyl 4-(4-hydroxycyclohexyl)piperazine-1-carboxylate (969.6 mg, 3.41 mmol, 1.2 equiv.) in DMF (10.0 mL) was added NaH (227.3 mg, 5.68 mmol, 60.0% purity, 2.0
equiv.) at 0°C and the mixture was stirred at 0°C for 1 h. 5-bromo-2-fluoro-pyridine (500.0 mg, 2.84 mmol, 292.40 μL, 1.0 equiv.) was added to the mixture. The mixture was stirred at 25°C for 12 h. The reaction mixture was diluted with water (10.0 mL) and extracted with ethyl acetate (10 mL x 3). The combined organic layers were washed with brine (10.0 mL), dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether: ethyl acetate = 1: 0 to 0: 1) to afford 42a. LCMS [M+1] = 440.2; 1H NMR (400 MHz, DMSO-d6) δ ppm 8.25 (d, J = 2.4 Hz, 1H), 7.86 (dd, J = 2.8, 8.8 Hz, 1H), 6.78 (d, J = 8.8 Hz, 1H), 4.96 - 4.82 (m, 1H), 3.97-3.93 (m, 2H), 2.80 - 2.73 (m, 3H), 2.70 - 2.66 (m, 1H), 2.45 - 2.38 (m, 1H), 2.37 - 2.29 (m, 2H), 1.99 - 1.91 (m, 2H), 1.70 (d, J = 12.4 Hz, 2H), 1.64 - 1.54 (m, 2H), 1.38 (s, 9H), 1.31 - 1.21 (m, 2H) Step B – Synthesis of tert-butyl 4-[4-[[5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-2- pyridyl]oxy]-1-piperidyl]piperidine-1-carboxylate (42b) To a solution of 42a (1.0 g, 1.36 mmol, 1.0 equiv.) and bis(pinacolato)diboron (346.0 mg, 1.36 mmol, 1 equiv.) in dioxane (10.0 mL) was added potassium acetate (267.4 mg, 2.72 mmol, 2.0 equiv.) and Pd(dppf)Cl2 (99.7 mg, 136.25 μmol, 0.1 equiv.) at 25°C under a nitrogen atmosphere. The mixture was stirred at 100°C for 12 h. The reaction mixture was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether: ethyl acetate = 1: 0 to 0: 1) to afford 42b. LCMS [M+1] = 488.3. Step C – Synthesis of tert-butyl 4-[4-[[5-[3-methyl-4-[4-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H-1,2,4-triazol-5-yl]methylcarbamoyl]pyrazol-1-yl]phenyl]-2-pyridyl]oxy]-1- piperidyl]piperidine-1-carboxylate (42c) To a solution of 42b (150.0 mg, 184.64 μmol, 1.5 equiv.) and Int 4 (58.0 mg, 123.09 μmol, 1.0 equiv.) in THF (1.0 mL) and water (0.25 mL) was added K3PO4 (78.39 mg, 369.28 μmol, 3.0 equiv.) and PdCl2(DTBPF) (16.05 mg, 24.62 μmol, 0.2 equiv.) at 25°C under a nitrogen atmosphere. .The mixture was stirred at 80°C for 12 h. The reaction mixture was diluted with water (1.0 mL) and extracted with ethyl acetate (1.0 mL x 3). The combined organic layers were washed with brine (1.0 mL), dried over sodium sulfate, filtered and concentrated under reduced
pressure. The residue was purified by preparative TLC (silica gel, petroleum ether: ethyl acetate= 0: 1) to afford 42c. LCMS [M+1] = 752.2. Step D – Synthesis of 1-[2-methyl-4-[6-[[1-(4-piperidyl)-4-piperidyl]oxy]-3-pyridyl]phenyl]-N- [[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (42d) A solution of 42c (100.0 mg, 79.80 μmol, 1 equiv.) in HCl/MeOH (3 M, 1.0 mL) was stirred at 25°C for 1 h. The reaction was monitored by LCMS. The reaction mixture was concentrated under reduced pressure to give 42d. Step E – Synthesis of 1-[4-[6-[[1-[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]-4-piperidyl]-4-piperidyl]oxy]-3-pyridyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Compound 42) To a solution of 42d (50.0 mg, 69.1 μmol, 1 equiv.) and 2-(2,6-dioxo-3-piperidyl)-5,6- difluoro-isoindoline-1,3-dione (22.6 mg, 76.7 μmol, 1.1 equiv.) in DMSO (1.0 mL) was added DIEA (99.1 mg, 767 μmol, 133.6 μL, 11.0 equiv.).The mixture was stirred at 100 °C for 12 h. The solution was filtered to give a residue and the residue purified by preparative HPLC (Phenomenex Luna C18, 100 mm x 40 mm, 5 µm; [water(0.04% HCl)-ACN]) to afford Compound 42. LCMS [M+1] = 926.4; 1H NMR (400 MHz, MeOD) δ ppm 8.48 (d, J = 11.2 Hz, 1H), 8.37 (s, 1H), 8.19 (s, 1H), 8.06 (t, J = 8.8 Hz, 1H), 7.66 (s, 1H), 7.62 - 7.57 (m, 2H), 7.54 (d, J = 7.2 Hz, 1H), 7.47 (d, J = 8.4 Hz, 1H), 7.05 - 6.91 (m, 1H), 5.52 - 5.29 (m, 1H), 5.10 (dd, J = 5.2, 12.4 Hz, 1H), 4.72 (d, J = 3.2 Hz, 2H), 3.87 (d, J = 12.0 Hz, 2H), 3.76 (d, J = 12.4 Hz, 1H), 3.61 (d, J = 11.2 Hz, 1H), 3.50 - 3.35 (m, 4H), 3.02 (t, J = 12.0 Hz, 2H), 2.92 - 2.82 (m, 1H), 2.80 - 2.65 (m, 2H), 2.59 (dd, J = 16.0 Hz, 1H), 2.43 (d, J = 15.2 Hz, 1H), 2.35 (s, 1H) 2.31 (s, 3H), 2.26 - 2.18 (m, 1H), 2.15 - 2.09 (m, 1H), 2.08 - 1.95 (m, 3H), 1.63 (s, 6H). Example 43: Synthesis of Compound 43
Step A – Synthesis of tert-butyl 4-[[1-[2-[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)phenoxy]ethyl]-4-piperidyl]methyl]piperazine-1-carboxylate (43a) To a solution of 2-[4-(2-bromoethoxy)phenyl]-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (2 g, 6.1 mmol, 1 equiv.), tert-butyl 4-(4-piperidylmethyl)piperazine-1-carboxylate (1.7 g, 6.1 mmol, 1 equiv.) in DMF (2 mL) was added cesium carbonate (4.0 g, 12.2 mmol, 2 equiv.). The mixture was stirred at 60°C for 2 h. The reaction mixture was cooled to ambient temperature, partitioned between ethyl acetate (30 mL) and water (30 mL). The aqueous phase was extracted with ethyl acetate (2 x 30 mL). The organic phase was combined and washed with brine (2 x 20 mL), dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether : ethyl acetate = 5/1 to 0/1) to afford 43a. 1H NMR (400 MHz, CHLOROFORM-d) δ = 7.74 (d, J = 8.5 Hz, 2H), 6.90 (d, J = 8.5 Hz, 2H), 4.24 - 4.11 (m, 2H), 3.44 - 3.39 (m, 4H), 3.07 - 3.00 (m, 2H), 2.84 (br s, 2H), 2.33 (br t,
J = 4.5 Hz, 4H), 2.23 - 2.10 (m, 4H), 1.77 (br d, J = 12.4 Hz, 2H), 1.51 - 1.48 (m, 1H), 1.46 (s, 9H), 1.34 (s, 12H) Step B – Synthesis of tert-butyl 4-((1-(2-((3'-methyl-4'-(4-(((3-(1,1,1-trifluoro-2-methylpropan-2- yl)-1H-1,2,4-triazol-5-yl)methyl)carbamoyl)-1H-pyrazol-1-yl)-[1,1'-biphenyl]-4- yl)oxy)ethyl)piperidin-4-yl)methyl)piperazine-1-carboxylate (43b) To a mixture of 43a (247.2 mg, 466.8 μmol, 1.1 equiv.), Int 4 (0.2 g, 424.4 μmol, 1 equiv.) and potassium carbonate (176.0 mg, 1.3 mmol, 3 equiv.) in dioxane (2 mL) and water (0.4 mL) was added Pd(dppf)Cl2.CH2Cl2 (69.3 mg, 84.9 μmol, 0.2 eq) in one portion at ambient temperature under nitrogen. The mixture was stirred at 100°C for 12 h. The mixture was cooled to ambient temperature, treated with water (5 mL) and extracted with ethyl acetate (3 x 5 mL). The combined organic phase was washed with brine (5 mL), dried with anhydrous sodium sulfate, filtered and concentrated in vacuum. The residue was purified by preparative TLC (silica gel, ethyl acetate: methanol = 10:1) to afford 43b. LCMS [M+1] = 794.5; 1H NMR (400 MHz, CHLOROFORM-d) δ = 8.12 (s, 1H), 7.99 (s, 1H), 7.55 - 7.45 (m, 4H), 7.34 (d, J = 8.3 Hz, 1H), 7.00 (d, J = 8.6 Hz, 2H), 4.72 (d, J = 5.8 Hz, 2H), 4.19 (br t, J = 5.3 Hz, 2H), 3.41 (br d, J = 4.3 Hz, 4H), 3.11 - 3.00 (m, 2H), 2.93 - 2.82 (m, 2H), 2.33 (br d, J = 4.5 Hz, 4H), 2.30 (s, 3H), 2.19 (br d, J = 7.3 Hz, 4H), 1.84 - 1.74 (m, 3H), 1.62 (s, 6H), 1.47 (s, 9H), 1.37 - 1.30 (m, 2H) Step C – Synthesis of 1-(3-methyl-4'-(2-(4-(piperazin-1-ylmethyl)piperidin-1-yl)ethoxy)-[1,1'- biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H- pyrazole-4-carboxamide (43c) To a solution of 43b (130 mg, 163.7 μmol, 1 equiv.) in TFA (0.4 mL)and dichloromethane (2 mL).The mixture was stirred at ambient temperature for 1 h. The reaction mixture was concentrated under reduced pressure to afford 43c. LCMS [M+2H] = 347.9. Step D – Synthesis of 1-[4-[4-[2-[4-[[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin- 5-yl]piperazin-1-yl]methyl]-1-piperidyl]ethoxy]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro- 1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Compound 43)
To a solution of 2-(2,6-dioxo-3-piperidyl)-5,6-difluoro-isoindoline-1,3-dione (71.9 mg, 244 μmol, 2.0 equiv.) in DMSO (2 mL) was added 43c (113 mg, 123 μmol, 1 equiv.) and DIEA (105 mg, 814 μmol, 142 μL, 6.6 equiv.) .The mixture was stirred at 70°C for 16 h, cooled to ambient temperature and partitioned between ethyl acetate (10 mL) and water (10 mL) extracted with ethyl acetate (20 mL x 2). The combined organic layers were washed with brine (5 mL), dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to give a residue. The residue was purified by preparative HPLC (Phenomenex Luna C1875mm x 30mm, 3 µm; [water(0.04% HCl)-ACN]). to afford Compound 43. LCMS [M+1] = 968.3; 1H NMR (400 MHz, METHANOL-d4) δ = 8.37 (s, 1H), 8.18 (s, 1H), 7.54 - 7.44 (m, 5H), 7.44 - 7.35 (m, 3H), 6.95 (d, J = 8.8 Hz, 2H), 5.08 (dd, J = 5.4, 12.8 Hz, 1H), 4.73 (s, 2H), 4.21 - 4.15 (m, 2H), 3.92 - 3.87 (m, 4H), 3.80 - 3.68 (m, 13H), 3.47 - 3.42 (m, 2H), 3.28 - 3.19 (m, 2H), 2.91 - 2.80 (m, 1H), 2.78 - 2.62 (m, 2H), 2.27 (s, 3H), 2.05 (tdd, J = 2.8, 5.1, 10.3 Hz, 1H), 1.63 (s, 6H) Example 44: Synthesis of Compound 44
 Compound 44 was prepared in a similar manner to Example 43 by substituting 2-[3-(2- bromoethoxy)phenyl]-4,4,5,5-tetramethyl-1,3,2-dioxaborolane in Step A to afford 1-(3'-(2-(4-((4- (2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1-yl)methyl)piperidin-1- yl)ethoxy)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide. LCMS: [M+1] =968.6; 1H NMR (400 MHz, DMSO-d6) δ 13.93 (br s, 1H), 11.11 (s, 1H), 8.89 (br s, 1H), 8.52 (s, 1H), 8.16 (d, J = 6.2 Hz, 1H), 7.78 - 7.69 (m, 2H), 7.66 (dd, J = 1.6, 8.4 Hz, 1H), 7.48 - 7.42 (m, 2H), 7.41 - 7.35 (m, 1H), 7.32 - 7.21 (m, 2H), 6.98 (d, J = 8.6 Hz, 1H), 5.10 (dd, J = 5.4, 12.8 Hz, 1H), 4.55 (d, J = 5.6 Hz, 2H), 4.17 (br t, J = 5.6 Hz, 2H), 3.23 (br s, 5H), 3.01 - 2.81 (m, 3H), 2.79 - 2.66 (m, 2H), 2.63 - 2.52 (m, 2H), 2.52 - 2.50 (m, 4H), 2.30 (s, 3H), 2.18 (br d, J = 7.0 Hz, 2H), 2.13 - 1.96 (m, 3H), 1.70 (br d, J = 11.8 Hz, 2H), 1.53 (s, 6H), 1.20 - 1.07 (m, 2H)
Example 45: Synthesis of Intermediate 45
Compound 44 was prepared in a similar manner to Example 43 by substituting 2-[3-(2- bromoethoxy)phenyl]-4,4,5,5-tetramethyl-1,3,2-dioxaborolane in Step A to afford 1-(3'-(2-(4-((4- (2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1-yl)methyl)piperidin-1- yl)ethoxy)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide. LCMS: [M+1] =968.6; 1H NMR (400 MHz, DMSO-d6) δ 13.93 (br s, 1H), 11.11 (s, 1H), 8.89 (br s, 1H), 8.52 (s, 1H), 8.16 (d, J = 6.2 Hz, 1H), 7.78 - 7.69 (m, 2H), 7.66 (dd, J = 1.6, 8.4 Hz, 1H), 7.48 - 7.42 (m, 2H), 7.41 - 7.35 (m, 1H), 7.32 - 7.21 (m, 2H), 6.98 (d, J = 8.6 Hz, 1H), 5.10 (dd, J = 5.4, 12.8 Hz, 1H), 4.55 (d, J = 5.6 Hz, 2H), 4.17 (br t, J = 5.6 Hz, 2H), 3.23 (br s, 5H), 3.01 - 2.81 (m, 3H), 2.79 - 2.66 (m, 2H), 2.63 - 2.52 (m, 2H), 2.52 - 2.50 (m, 4H), 2.30 (s, 3H), 2.18 (br d, J = 7.0 Hz, 2H), 2.13 - 1.96 (m, 3H), 1.70 (br d, J = 11.8 Hz, 2H), 1.53 (s, 6H), 1.20 - 1.07 (m, 2H)
Example 45: Synthesis of Intermediate 45
 To a solution of Int 4 (500 mg, 1.0 mmol, 1.0 equiv.) in 1,2-dimethoxyethane (9 mL) was added potassium acetate (416.4 mg, 4.2 mmol, 4.0 equiv.) and PdDPPF (86.6 mg, 106.1 μmol, 0.1 equiv.), bis(pinacolato)diboron (538.8 mg, 2.1 mmol, 2 equiv.) under a nitrogen atmosphere. The mixture was stirred at 90°C for 16 h. The reaction mixture was partitioned between ethyl acetate (100 mL) and water (100 mL) extracted with ethyl acetate (2 x 100 mL). The combined organic layers were washed with brine (5 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, dichloromethane : methanol=100/1 to 90/10) to afford 1-[2-methyl-4- (4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Intermediate 45). LCMS [M+1] = 519.3. Example 46: Synthesis of Compound 46
To a solution of Int 4 (500 mg, 1.0 mmol, 1.0 equiv.) in 1,2-dimethoxyethane (9 mL) was added potassium acetate (416.4 mg, 4.2 mmol, 4.0 equiv.) and PdDPPF (86.6 mg, 106.1 μmol, 0.1 equiv.), bis(pinacolato)diboron (538.8 mg, 2.1 mmol, 2 equiv.) under a nitrogen atmosphere. The mixture was stirred at 90°C for 16 h. The reaction mixture was partitioned between ethyl acetate (100 mL) and water (100 mL) extracted with ethyl acetate (2 x 100 mL). The combined organic layers were washed with brine (5 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, dichloromethane : methanol=100/1 to 90/10) to afford 1-[2-methyl-4- (4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Intermediate 45). LCMS [M+1] = 519.3. Example 46: Synthesis of Compound 46
Step A – Synthesis of 1-[1-(4-bromophenyl)-1-methyl-ethyl]piperidin-4-one (46a) To a solution of 2-(4-bromophenyl)propan-2-amine (2.0 g, 9.3 mmol, 1.0 equiv.) in EtOH (20 mL) and water (8 mL) was added potassium carbonate (129.1 mg, 934.1 μmol, 0.1 equiv.) and 1-ethyl-1-methyl-piperidin-1-ium-4-one iodide (2.0 g, 7.4 mmol, 0.8 equiv.). The mixture was stirred at 80 °C for 2 h . The reaction mixture was partitioned between ethyl acetate (200 mL) and water (200 mL) extracted with ethyl acetate (2 x 200 mL). The combined organic layers were washed with brine (50 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether/ ethyl acetate = 80/20 to 50/50) to afford 46a. LCMS [M+1] = 297.0, 299.0; 1H NMR (400 MHz, DMSO-d6) δ = 7.64 - 7.40 (m, 4H), 2.65 (t, J = 6.0 Hz, 4H), 2.30 (t, J = 6.0 Hz, 4H), 1.32 (s, 6H).
Step B – Synthesis of tert-butyl 4-[1-[1-(4-bromophenyl)-1-methyl-ethyl]-4-piperidyl]piperazine- 1-carboxylate (46b) To a solution of 46a (1.1 g, 3.7 mmol, 1 equiv.) in dichloromethane (15 mL) was added acetic acid (223.0 mg, 3.7 mmol, 212.6 μL, 1.0 equiv.) and NaBH(OAc)3 (3.1 g, 14.8 mmol, 4.0 equiv.), tert-butyl piperazine-1-carboxylate (830.0 mg, 4.4 mmol, 1.2 equiv.). The mixture was stirred at 20°C for 2 h. The reaction mixture was partitioned between ethyl acetate (200 mL) and water (200 mL) extracted with ethyl acetate (2 x 200 mL). The combined organic layers were washed with brine (50 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure to afford 46b. LCMS [M+1] = 468.2, 470.2. Step C – Synthesis of tert-butyl 4-[1-[1-methyl-1-[4-[3-methyl-4-[4-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methylcarbamoyl]pyrazol-1-yl]phenyl]phenyl]ethyl]-4- piperidyl]piperazine-1-carboxylate (46c) To a solution of 46b (100.0 mg, 214.3 μmol, 1.0 equiv.) in dioxane (1 mL), water (0.2 mL) was added potassium carbonate (59.2 mg, 428.7 μmol, 2.0 equiv.) and Pd(dppf)Cl2 (15.6 mg, 21.4 μmol, 0.1 equiv.), Int 45 (111.1 mg, 214.3 μmol, 1.0 equiv.) under a nitrogen atmosphere. The mixture was stirred at 90°C for 1 h, cooled to ambient temperature and partitioned between ethyl acetate (20 mL) and water (20 mL) extracted with ethyl acetate (2 x 20 mL). The combined organic layers were washed with brine (5 mL), dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to afford 46c. LCMS [M+1] =778.6. Step D – Synthesis of 1-[2-methyl-4-[4-[1-methyl-1-(4-piperazin-1-yl-1-piperidyl)ethyl]phenyl]- phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4- carboxamide (46d) To a solution of 46c (100.0 mg, 128.5 μmol, 1.0 equiv.) in CF3CwaterH (10 mL) was added chlorotrimethylsilane (856.0 mg, 7.8 mmol, 1 mL, 61.2 equiv.). The mixture was stirred at 20 °C for 1 h concentrated under reduced pressure to afford 46d. LCMS [M+1] = 704.5.
Step E – Synthesis of 1-[4-[4-[1-[4-[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]piperazin-1-yl]-1-piperidyl]-1-methyl-ethyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro- 1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Compound 46) To a solution of 46d (80.0 mg, 88 μmol, 1.0 equiv.) in DMSO (1 mL) was added DIEA (61.0 mg, 472 μmol, 82.2 μL, 5.3 equiv.) and 2-(2,6-dioxo-3-piperidyl)-5,6-difluoro-isoindoline-1,3-dione (52.1 mg, 177 μmol, 2.0 equiv.). The mixture was stirred at 80°C for 4 h. The reaction mixture was cooled to ambient temperature, partitioned between ethyl acetate (10 mL) and water (10 mL), extracted with ethyl acetate (2 x 10 mL). The combined organic layers were washed with brine (5 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative HPLC (Waters Xbridge C18150 mm x 50mm, 10µm; [water(10mM NH4HCO3)-ACN]) to afford Compound 46. LCMS [M+1] =952.6; 1H NMR (400 MHz, DMSO-d6) δ = 13.91 (br s, 1H), 11.10 (s, 1H), 8.72 (s, 1H), 8.53 (s, 1H), 8.17 (s, 1H), 7.80 - 7.56 (m, 8H), 7.50 - 7.37 (m, 2H), 5.10 (dd, J = 5.2, 12.4 Hz, 1H), 4.56 (s, 2H), 3.23 (s, 4H), 2.94 - 2.78 (m, 3H), 2.72 - 2.56 (m, 2H), 2.37 - 2.25 (m, 4H), 2.25 - 2.14 (m, 1H), 2.12 - 1.98 (m, 4H), 1.77 (d, J = 8.8 Hz, 2H), 1.54 (s, 6H), 1.43 - 1.40 (m, 2H), 1.34 (s, 2H). Example 47: Synthesis of Compound 47
carboxylate (47a) To a solution of tert-butyl 4-(4-piperidyl)piperazine-1-carboxylate (1.2 g, 4.5 mmol, 1.3 equiv.) and 5-chloropyrazine-2-carbaldehyde (500.0 mg, 3.5 mmol, 1.0 equiv.) in dichloromethane (5.0 mL) was added acetic acid (210.6 mg, 3.5 mmol, 200.8 μL, 1.0 equiv.). The mixture was stirred at ambient temperature for 1.5 h, treated with NaBH(OAc)3 (1.4 g, 7.0 mmol, 2.0 equiv.) and stirred at ambient temperature for 16 h. The reaction mixture was diluted with water (15.0 mL) and extracted with ethyl acetate (2 x 30.0 mL). The combined organic layers were washed with brine (2 x 10.0 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, dichloromethane : methanol=100:1 to 0:1) to afford 47a. LCMS [M+1] = 396.3, 398.3; 1H NMR (400 MHz, CHLOROFORM-d) δ 8.53 (d, J = 1.1 Hz, 1H), 8.44 (d, J = 1.1 Hz, 1H), 3.66 (s, 2H), 3.49 - 3.36 (m, 4H), 2.93 (br d, J = 11.8 Hz, 2H), 2.61 - 2.42 (m, 4H), 2.38 - 2.24 (m, 1H), 2.11 (dt, J = 1.8, 11.6 Hz, 2H), 1.78 (br d, J = 12.2 Hz, 2H), 1.60 (br dd, J = 3.4, 12.0 Hz, 2H), 1.45 (s, 9H).
Step B – Synthesis of tert-butyl 4-[1-[[5-[3-methyl-4-[4-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H-1,2,4-triazol-5-yl]methylcarbamoyl]pyrazol-1-yl]phenyl]pyrazin-2-yl] methyl]-4-piperidyl] piperazine-1-carboxylate (47b) To a solution of 47a (130.7 mg, 330.1 μmol, 1.2 equiv.) and Int 45 (142.6 mg, 275.1 μmol, 1.0 equiv.) in dioxane (2.0 mL) and water (0.4 mL) was added potassium carbonate (114.0 mg, 825.3 μmol, 3.0 equiv.) and Pd(dppf)Cl2 (44.9 mg, 55.0 μmol, 0.2 equiv.). The mixture was threefold degassed and purged with nitrogen for 3 times, and then the mixture was stirred at 100 °C for 16 h under a nitrogen atmosphere. LCMS showed starting material was consumed completely and one main peak with desired mass was detected. The reaction mixture was diluted with water (10.0 mL) and extracted with ethyl acetate (15 mL x 2). The combined organic layers were washed with brine (5.0 mL), dried with anhydrous sodium sulfate and filtered. The filtrate was concentrated under reduced pressure to give a residue. The residue was purified by column chromatography (silica gel, Dichloromethane : Methanol=100 :1 to 0 :1) to afford 47b. LCMS [M+1] = 752.7. Step C – Synthesis of 1-[2-methyl-4-[5-[(4-piperazin-1-yl-1-piperidyl)methyl]pyrazin-2- yl]phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4- carboxamide (47c) A solution of 47b (50.0 mg, 66.5 μmol, 1.0 equiv.) in HCl/dioxane (1.0 mL) was stirred at 20°C for 1 h. The reaction mixture was concentrated under reduced pressure to afford 47c, which was used without further purification. LCMS [M+1] = 652.6. Step D – Synthesis of 1-[4-[5-[[4-[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]piperazin-1-yl]-1-piperidyl]methyl]pyrazin-2-yl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Compound 47). To a solution of 47c (50.0 mg, 72.6 μmol, 1.0 equiv) and DIEA (56.3 mg, 435.9 μmol, 75.9 μL, 6.0 equiv.) in DMSO (1.0 mL) was added 2-(2,6-dioxo-3-piperidyl)-5,6-difluoro- isoindoline-1,3-dione (25.6 mg, 87.1 μmol, 1.2 equiv.). The mixture was stirred at 70 °C for 16 h, cooled to ambient temperature, diluted with water (3.0 mL) and extracted with ethyl acetate (2 x 10 mL). The combined organic layers were washed with brine (2 x 2mL), dried with anhydrous
sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by reversed-phase HPLC (Waters Xbridge BEH C18, 100 mm x 30 mm, 10 µm; [water(10mM NH4HCO3)-ACN]) to afford Compound 47. LCMS [M+1] = 926.4; 1H NMR (400 MHz, DMSO-d6) δ 13.90 (br s, 1H), 11.10 (br s, 1H), 9.24 (d, J = 1.1 Hz, 1H), 9.00 (br s, 1H), 8.75 (d, J = 1.1 Hz, 1H), 8.58 (s, 1H), 8.25 - 8.16 (m, 2H), 8.12 (dd, J = 1.6, 8.2 Hz, 1H), 7.72 (d, J = 11.4 Hz, 1H), 7.56 (d, J = 8.2 Hz, 1H), 7.44 (d, J = 7.4 Hz, 1H), 5.10 (dd, J = 5.6, 12.8 Hz, 1H), 4.56 (br d, J = 4.8 Hz, 2H), 3.71 (s, 2H), 3.24 (br s, 5H), 3.01 - 2.78 (m, 3H), 2.67 (br d, J = 1.8 Hz, 5H), 2.36 (s, 3H), 2.34 - 2.31 (m, 1H), 2.26 (br s, 1H), 2.16 - 1.98 (m, 4H), 1.83 - 1.75 (m, 2H), 1.54 (s, 6H). Example 48: Synthesis of Compound 48
 6- bromopyridine-3-carbaldehyde in Step A to afford 1-[4-[5-[[4-[4-[2-(2,6-dioxo-3-piperidyl)-6- fluoro-1,3-dioxo-isoindolin-5-yl]piperazin-1-yl]-1-piperidyl]methyl]-2-pyridyl]-2-methyl- phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4- carboxamide. LCMS [M+1] = 925.2. 1H NMR (400 MHz, CHLOROFORM-d) δ 8.63 (s, 1H), 8.38 (br s, 1H), 8.27 (s, 1H), 8.19 (s, 1H), 8.01 (d, J = 12.0 Hz, 2H), 7.94 - 7.79 (m, 2H), 7.79 - 7.69 (m, 1H), 7.48 (d, J = 11.0 Hz, 1H), 7.40 (dd, J = 7.8, 11.8 Hz, 2H), 6.98 (s, 1H), 4.94 (dd, J = 5.2, 12.0 Hz, 1H), 4.73 (d, J = 6.0 Hz, 2H), 3.68 (s, 2H), 3.31 (s, 4H), 3.07 (d, J = 10.8 Hz, 2H),
2.98 - 2.86 (m, 2H), 2.81 (s, 5H), 2.51 - 2.41 (m, 1H), 2.34 (s, 3H), 2.25 - 2.11 (m, 3H), 1.92 (d, J = 11.0 Hz, 2H), 1.81 - 1.69 (m, 2H), 1.62 (s, 6H) Example 49: Synthesis of Compound 49
6- bromopyridine-3-carbaldehyde in Step A to afford 1-[4-[5-[[4-[4-[2-(2,6-dioxo-3-piperidyl)-6- fluoro-1,3-dioxo-isoindolin-5-yl]piperazin-1-yl]-1-piperidyl]methyl]-2-pyridyl]-2-methyl- phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4- carboxamide. LCMS [M+1] = 925.2. 1H NMR (400 MHz, CHLOROFORM-d) δ 8.63 (s, 1H), 8.38 (br s, 1H), 8.27 (s, 1H), 8.19 (s, 1H), 8.01 (d, J = 12.0 Hz, 2H), 7.94 - 7.79 (m, 2H), 7.79 - 7.69 (m, 1H), 7.48 (d, J = 11.0 Hz, 1H), 7.40 (dd, J = 7.8, 11.8 Hz, 2H), 6.98 (s, 1H), 4.94 (dd, J = 5.2, 12.0 Hz, 1H), 4.73 (d, J = 6.0 Hz, 2H), 3.68 (s, 2H), 3.31 (s, 4H), 3.07 (d, J = 10.8 Hz, 2H),
2.98 - 2.86 (m, 2H), 2.81 (s, 5H), 2.51 - 2.41 (m, 1H), 2.34 (s, 3H), 2.25 - 2.11 (m, 3H), 1.92 (d, J = 11.0 Hz, 2H), 1.81 - 1.69 (m, 2H), 1.62 (s, 6H) Example 49: Synthesis of Compound 49
 bromo- 2-chloro-benzaldehyde in Step A to afford 1-(3'-chloro-4'-((4-(4-(2-(2,6-dioxopiperidin-3-yl)-6- fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1-yl)piperidin-1-yl)methyl)-3-methyl-[1,1'-biphenyl]-4- yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4- carboxamide. LCMS [M+1] = 958.2;1H NMR (400 MHz, DMSO-d6) δ = 13.92 (br s, 1H), 11.11 (s, 1H), 8.90 (br s, 1H), 8.54 (s, 1H), 8.18 (s, 1H), 7.81 - 7.79 (m, 2H), 7.74 - 7.69 (m, 3H), 7.60 (d, J = 8.0 Hz, 1H), 7.49 (d, J = 8.4 Hz, 1H), 7.45 (d, J = 7.6 Hz, 1H), 5.11 (dd, J = 5.6, 12.8 Hz, 1H), 4.56 (d, J = 4.8 Hz, 2H), 3.61 (s, 2H), 3.25 (s, 4H), 2.94 - 2.84 (m, 3H), 2.68 (s, 4H), 2.62 - 2.57 (m, 2H), 2.32 (s, 3H), 2.29 – 2.26 (m, 1H), 2.12 - 2.02 (m, 3H), 1.80 (d, J = 10.8 Hz, 2H), 1.55 (s, 6H), 1.51 - 1.44 (m, 2H). Example 50: Synthesis of Compound 50
bromo- 2-chloro-benzaldehyde in Step A to afford 1-(3'-chloro-4'-((4-(4-(2-(2,6-dioxopiperidin-3-yl)-6- fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1-yl)piperidin-1-yl)methyl)-3-methyl-[1,1'-biphenyl]-4- yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4- carboxamide. LCMS [M+1] = 958.2;1H NMR (400 MHz, DMSO-d6) δ = 13.92 (br s, 1H), 11.11 (s, 1H), 8.90 (br s, 1H), 8.54 (s, 1H), 8.18 (s, 1H), 7.81 - 7.79 (m, 2H), 7.74 - 7.69 (m, 3H), 7.60 (d, J = 8.0 Hz, 1H), 7.49 (d, J = 8.4 Hz, 1H), 7.45 (d, J = 7.6 Hz, 1H), 5.11 (dd, J = 5.6, 12.8 Hz, 1H), 4.56 (d, J = 4.8 Hz, 2H), 3.61 (s, 2H), 3.25 (s, 4H), 2.94 - 2.84 (m, 3H), 2.68 (s, 4H), 2.62 - 2.57 (m, 2H), 2.32 (s, 3H), 2.29 – 2.26 (m, 1H), 2.12 - 2.02 (m, 3H), 1.80 (d, J = 10.8 Hz, 2H), 1.55 (s, 6H), 1.51 - 1.44 (m, 2H). Example 50: Synthesis of Compound 50
Step A – Synthesis of tert-butyl 4-[6-(4-bromophenoxy)-3-pyridyl] piperazine-1-carboxylate (50a) To a solution of tert-butyl 4-(6-bromo-3-pyridyl)piperazine-1-carboxylate (500.0 mg, 1.5 mmol, 1.0 equiv.) and 4-bromophenol (758.3 mg, 4.4 mmol, 3.0 equiv.) in DMSO (5.0 mL) was added K3PO4 (620.2 mg, 2.9 mmol, 2.0 equiv.), pyridine-2-carboxylic acid (18.0 mg, 146.1 μmol, 0.1 equiv.) and CuI (13.9 mg, 73.1 μmol, 0.05 equiv.). The mixture was three-fold degassed and purged with nitrogen, and then the mixture was stirred at 100°C for 12 h under a nitrogen atmosphere. The reaction mixture was quenched by addition water (10 mL) and extracted with ethyl acetate (5 mL x 3). The combined organic layers were washed with brine (10 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative HPLC (Waters Xbridge BEH C18100 mm x 30 mm, 10µm; [water (10mM NH4HCO3)-ACN]) to afford 50a. LCMS [M+1] = 434.2, 436.2.
Step B – Synthesis of tert-butyl 4-[6-[4-[3-methyl-4-[4-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H-1,2,4-triazol-5-yl]methylcarbamoyl]pyrazol-1-yl]phenyl]phenoxy]-3-pyridyl] piperazine-1- carboxylate (50b) To a solution of 50a (80.0 mg, 184.2 μmol, 1.0 equiv.) and Int 45 (100.3 mg, 193.4 μmol, 1.05 equiv.) in THF (0.8 mL) and water (0.2 mL) was added K3PO4 (78.2 mg, 368.4 μmol, 2.0 equiv.) and PdCl2(DTBPF) (24.0 mg, 36.8 μmol, 0.2 equiv.). The mixture was three-fold degassed and purged with nitrogen. The mixture was stirred at 80°C for 3 h under a nitrogen atmosphere. The reaction mixture was quenched with water (2 mL) and extracted with ethyl acetate (1 mL x 3). The combined organic layers were washed with brine (2 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative TLC (silica gel, ethyl acetate: MeOH = 10: 1) to afford 50b. LCMS [M+1] = 746.5. Step C – Synthesis of 1-[2-methyl-4-[4-[(5-piperazin-1-yl-2-pyridyl) oxy]phenyl]phenyl]-N-[[3- (2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl] methyl]pyrazole-4-carboxamide (50c) To a solution of 50b (60.0 mg, 80.5 μmol, 1.0 equiv.) in dichloromethane (0.6 mL) was added TFA (0.2 mL). The mixture was stirred at 25°C for 1 h. The mixture was concentrated under reduced pressure to afford 50c, which was used without further purification. LCMS [M+1] = 646.5. Step D – Synthesis of 1-[4-[4-[[5-[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl] piperazin-1-yl]-2-pyridyl]oxy]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl- ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Compound 50) To a solution of 50c (60.0 mg, 78.9 μmol, 1.0 equiv) and 2-(2,6-dioxo-3-piperidyl)-5,6- difluoro-isoindoline-1,3-dione (34.9 mg, 118.5 μmol, 1.5 equiv.) in DMSO (0.5 mL) was added DIEA (153.1 mg, 1.2 mmol, 206.4 μL, 15.0 equiv.). The mixture was stirred at 70°C for 12 h, cooled to ambient temperature and the filtered. Direct purification by preparative HPLC (column: Phenomenex Luna C18100mm x 30mm, 5µm; [water(0.2% FA)-ACN]) to afford Compound 50. LCMS [M+1] = 920.2; 1H NMR (400 MHz, METHANOL-d4) δ = 8.36 (s, 1H), 8.18 (s, 1H), 7.92 (d, J = 2.8 Hz, 1H), 7.71 (d, J = 8.8 Hz, 2H), 7.65 - 7.55 (m, 4H), 7.44 (d, J = 8.0 Hz, 1H), 7.15 (d, J = 8.8 Hz, 2H), 6.98 (d, J = 8.8 Hz, 1H), 5.13 - 5.08 (m, 1H), 4.71 - 4.67 (m, 2H), 3.48 - 3.46
(m, 4H), 3.38 - 3.36 (m, 4H), 2.91 - 2.82 (m, 1H), 2.78 - 2.65 (m, 2H), 2.31 (s, 3H), 2.15 - 2.09 (m, 1H), 1.60 (s, 6H). Example 51: Synthesis of Compound 51
 Step A – Synthesis of tert-butyl 4-(6-oxo-2,3-dihydro-1H-pyridin-4-yl)piperazine-1-carboxylate (51a)
To a mixture of piperidine-2,4-dione (303.6 mg, 2.6 mmol, 1.0 equiv.) and tert-butyl piperazine-1-carboxylate (500 mg, 2.6 mmol, 1.0 equiv.) in CHCl3 (1.5 mL) was added acetic acid (165.2 mg, 2.7 mmol, 157.5 μL, 1.0 equiv.), the mixture was stirred at 60°C for 16 h. The reaction mixture was quenched with water 5 mL at 25°C, and then extracted with dichloromethane (3 mL x 3). The combined organic portions were dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure to afford 51a. LCMS: [M+1] = 282.5; 1H NMR (400 MHz, DMSO-d6) δ 6.58 (s, 1H), 4.61 (s, 1H), 3.35 (s, 3H), 3.23 - 3.09 (m, 5H), 2.61 (t, J = 4.8 Hz, 1H), 2.37 (t, J = 6.8 Hz, 2H), 1.47 - 1.34 (m, 9H) Step B – Synthesis of tert-butyl 4-(2-oxo-4-piperidyl)piperazine-1-carboxylate (51b) To a mixture of 51a (700 mg, 2.4 mmol, 1.0 equiv.) in MeOH (7 mL) was added NaBH3CN (781.7 mg, 12.4 mmol, 5.0 equiv.) and the mixture was stirred at 60°C for 16 h. The reaction mixture was filtered to remove the insolubles and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, ethyl acetate: methanol=1/0 to 1/5) to afford 51b. LCMS: [M+1] = 284.5; 1H NMR (400 MHz, DMSO-d6) δ 7.46 (s, 1H), 3.28 (t, J = 4.4 Hz, 4H), 3.18 - 3.11 (m, 1H), 3.03 (td, J = 4.0, 11.2 Hz, 1H), 2.71 - 2.59 (m, 1H), 2.48 - 2.34 (m, 4H), 2.29 - 2.22 (m, 1H), 2.18 - 2.07 (m, 1H), 1.92 - 1.84 (m, 1H), 1.56 - 1.44 (m, 1H), 1.39 (s, 9H) Step C – Synthesis of tert-butyl 4-[1-[(4-bromophenyl)methyl]-2-oxo-4-piperidyl]piperazine-1- carboxylate (51c) To a solution of 51b (700 mg, 2.4 mmol, 1.0 equiv.) in THF (7 mL) was added NaH (296.4 mg, 7.4 mmol, 60% purity, 3.0 equiv.) at 0°C under nitrogen. The mixture was stirred at 25°C for 0.5 h, then the mixture was treated with 1-bromo-4-(bromomethyl)benzene (679.1 mg, 2.7 mmol, 1.1 equiv.) and the mixture was stirred at 25°C for 2 h, quenched with water (20 mL) and extracted with ethyl acetate (10 mL x 3). The combined organic layers were washed with brine (10 mL x 3), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure to give a residue. The residue was purified by column chromatography (silica gel, ethyl acetate: methanol=1/0 to 1/1) to afford 51c. LCMS: [M+1] = 452.2, 454.2; 1H NMR (400 MHz, DMSO-d6) δ 7.52 (d, J = 8.4 Hz, 2H), 7.19 (d, J = 8.0 Hz, 2H), 4.55 (d, J = 15.2
Hz, 1H), 4.36 (d, J = 15.2 Hz, 1H), 3.30 - 3.06 (m, 6H), 2.72 - 2.64 (m, 1H), 2.47 - 2.31 (m, 6H), 1.93 (s, 1H), 1.71 - 1.61 (m, 1H), 1.39 (s, 9H). Step D – Synthesis of tert-butyl 4-[1-[[4-[3-methyl-4-[4-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H-1,2,4-triazol-5-yl]methylcarbamoyl]pyrazol-1-yl]phenyl]phenyl]methyl]-2-oxo-4- piperidyl]piperazine-1-carboxylate (51d) To a mixture of 51c (69.8 mg, 154.3 μmol, 1.0 equiv.) and Int 45 (80.0 mg, 154.3 μmol, 1.0 equiv.) and K2CO3 (63.9 mg, 463.0 μmol, 3.0 equiv.) in dioxane (1 mL) and water (0.2 mL) was three-fold degassed and purged with nitrogen, treated with Pd(DPPF)Cl2 (22.5 mg, 30.8 μmol, 0.2 equiv.) and stirred at 80°C for 16 h. The reaction mixture was cooled to ambient temperature, quenched with water (10 mL) and then extracted with ethyl acetate (5 mL x 3). The combined organic portions were dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative TLC (silica gel, dichloromethane: MeOH = 10:1) to afford 51d. LCMS: [M+1] = 764.6. Step E – Synthesis of 1-[2-methyl-4-[4-[(2-oxo-4-piperazin-1-yl-1-piperidyl)methyl]phenyl]- phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4- carboxamide (51e) A mixture of 51d (40.0 mg, 52.3 μmol, 1.0 equiv.) in dichloromethane (0.5 mL) and TFA (0.1 mL) was stirred at 25°C for 0.5 h. The reaction mixture was concentrated under reduced pressure to afford (51e), which was used without further purification. LCMS: [M+1] =664.5. Step F – Synthesis of 1-[4-[4-[[4-[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]piperazin-1-yl]-2-oxo-1-piperidyl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Compound 51) To a mixture of 51e (40.0 mg, 51.4 μmol, 1.0 equiv) and 2-(2,6-dioxo-3-piperidyl)-5,6-difluoro- isoindoline-1,3-dione (18.1 mg, 61.7 μmol, 1.2equiv.) in DMSO (0.5 mL) was added DIEA (33.2 mg, 257.1 μmol, 44.7 μL, 5.0 equiv.) and the mixture was stirred at 70°C for 16 h. The reaction mixture was filtered to remove the insoluble. The residue was purified by preparative HPLC (Phenomenex Luna C18100mm x 30mm, 5 µm, [water(0.2% FA)-ACN]) to afford Compound 51. LCMS: [M+1] = 938.2; 1H NMR (400 MHz, DMSO-d6 METHANOL-d4) δ 8.49 (s, 1H), 8.16
(s, 1H), 7.72 - 7.66 (m, 4H), 7.61 (d, J = 8.4 Hz, 1H), 7.45 (t, J = 7.2 Hz, 2H), 7.37 (d, J = 8.0 Hz, 2H), 5.09 (dd, J = 5.2, 12.8 Hz, 1H), 4.69 (dd, J = 4.0, 14.8 Hz, 1H), 4.60 - 4.41 (m, 3H), 3.35 - 3.16 (m, 7H), 3.12 (d, J = 1.6 Hz, 3H), 2.93 - 2.81 (m, 1H), 2.80 - 2.59 (m, 7H), 2.28 (s, 3H), 2.08 - 1.98 (m, 2H), 1.75 (dd, J = 4.8, 9.6 Hz, 1H), 1.53 (s, 6H). Example 52: Synthesis of Compound 52
Step A – Synthesis of tert-butyl 4-(6-oxo-2,3-dihydro-1H-pyridin-4-yl)piperazine-1-carboxylate (51a)
To a mixture of piperidine-2,4-dione (303.6 mg, 2.6 mmol, 1.0 equiv.) and tert-butyl piperazine-1-carboxylate (500 mg, 2.6 mmol, 1.0 equiv.) in CHCl3 (1.5 mL) was added acetic acid (165.2 mg, 2.7 mmol, 157.5 μL, 1.0 equiv.), the mixture was stirred at 60°C for 16 h. The reaction mixture was quenched with water 5 mL at 25°C, and then extracted with dichloromethane (3 mL x 3). The combined organic portions were dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure to afford 51a. LCMS: [M+1] = 282.5; 1H NMR (400 MHz, DMSO-d6) δ 6.58 (s, 1H), 4.61 (s, 1H), 3.35 (s, 3H), 3.23 - 3.09 (m, 5H), 2.61 (t, J = 4.8 Hz, 1H), 2.37 (t, J = 6.8 Hz, 2H), 1.47 - 1.34 (m, 9H) Step B – Synthesis of tert-butyl 4-(2-oxo-4-piperidyl)piperazine-1-carboxylate (51b) To a mixture of 51a (700 mg, 2.4 mmol, 1.0 equiv.) in MeOH (7 mL) was added NaBH3CN (781.7 mg, 12.4 mmol, 5.0 equiv.) and the mixture was stirred at 60°C for 16 h. The reaction mixture was filtered to remove the insolubles and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, ethyl acetate: methanol=1/0 to 1/5) to afford 51b. LCMS: [M+1] = 284.5; 1H NMR (400 MHz, DMSO-d6) δ 7.46 (s, 1H), 3.28 (t, J = 4.4 Hz, 4H), 3.18 - 3.11 (m, 1H), 3.03 (td, J = 4.0, 11.2 Hz, 1H), 2.71 - 2.59 (m, 1H), 2.48 - 2.34 (m, 4H), 2.29 - 2.22 (m, 1H), 2.18 - 2.07 (m, 1H), 1.92 - 1.84 (m, 1H), 1.56 - 1.44 (m, 1H), 1.39 (s, 9H) Step C – Synthesis of tert-butyl 4-[1-[(4-bromophenyl)methyl]-2-oxo-4-piperidyl]piperazine-1- carboxylate (51c) To a solution of 51b (700 mg, 2.4 mmol, 1.0 equiv.) in THF (7 mL) was added NaH (296.4 mg, 7.4 mmol, 60% purity, 3.0 equiv.) at 0°C under nitrogen. The mixture was stirred at 25°C for 0.5 h, then the mixture was treated with 1-bromo-4-(bromomethyl)benzene (679.1 mg, 2.7 mmol, 1.1 equiv.) and the mixture was stirred at 25°C for 2 h, quenched with water (20 mL) and extracted with ethyl acetate (10 mL x 3). The combined organic layers were washed with brine (10 mL x 3), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure to give a residue. The residue was purified by column chromatography (silica gel, ethyl acetate: methanol=1/0 to 1/1) to afford 51c. LCMS: [M+1] = 452.2, 454.2; 1H NMR (400 MHz, DMSO-d6) δ 7.52 (d, J = 8.4 Hz, 2H), 7.19 (d, J = 8.0 Hz, 2H), 4.55 (d, J = 15.2
Hz, 1H), 4.36 (d, J = 15.2 Hz, 1H), 3.30 - 3.06 (m, 6H), 2.72 - 2.64 (m, 1H), 2.47 - 2.31 (m, 6H), 1.93 (s, 1H), 1.71 - 1.61 (m, 1H), 1.39 (s, 9H). Step D – Synthesis of tert-butyl 4-[1-[[4-[3-methyl-4-[4-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H-1,2,4-triazol-5-yl]methylcarbamoyl]pyrazol-1-yl]phenyl]phenyl]methyl]-2-oxo-4- piperidyl]piperazine-1-carboxylate (51d) To a mixture of 51c (69.8 mg, 154.3 μmol, 1.0 equiv.) and Int 45 (80.0 mg, 154.3 μmol, 1.0 equiv.) and K2CO3 (63.9 mg, 463.0 μmol, 3.0 equiv.) in dioxane (1 mL) and water (0.2 mL) was three-fold degassed and purged with nitrogen, treated with Pd(DPPF)Cl2 (22.5 mg, 30.8 μmol, 0.2 equiv.) and stirred at 80°C for 16 h. The reaction mixture was cooled to ambient temperature, quenched with water (10 mL) and then extracted with ethyl acetate (5 mL x 3). The combined organic portions were dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative TLC (silica gel, dichloromethane: MeOH = 10:1) to afford 51d. LCMS: [M+1] = 764.6. Step E – Synthesis of 1-[2-methyl-4-[4-[(2-oxo-4-piperazin-1-yl-1-piperidyl)methyl]phenyl]- phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4- carboxamide (51e) A mixture of 51d (40.0 mg, 52.3 μmol, 1.0 equiv.) in dichloromethane (0.5 mL) and TFA (0.1 mL) was stirred at 25°C for 0.5 h. The reaction mixture was concentrated under reduced pressure to afford (51e), which was used without further purification. LCMS: [M+1] =664.5. Step F – Synthesis of 1-[4-[4-[[4-[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]piperazin-1-yl]-2-oxo-1-piperidyl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Compound 51) To a mixture of 51e (40.0 mg, 51.4 μmol, 1.0 equiv) and 2-(2,6-dioxo-3-piperidyl)-5,6-difluoro- isoindoline-1,3-dione (18.1 mg, 61.7 μmol, 1.2equiv.) in DMSO (0.5 mL) was added DIEA (33.2 mg, 257.1 μmol, 44.7 μL, 5.0 equiv.) and the mixture was stirred at 70°C for 16 h. The reaction mixture was filtered to remove the insoluble. The residue was purified by preparative HPLC (Phenomenex Luna C18100mm x 30mm, 5 µm, [water(0.2% FA)-ACN]) to afford Compound 51. LCMS: [M+1] = 938.2; 1H NMR (400 MHz, DMSO-d6 METHANOL-d4) δ 8.49 (s, 1H), 8.16
(s, 1H), 7.72 - 7.66 (m, 4H), 7.61 (d, J = 8.4 Hz, 1H), 7.45 (t, J = 7.2 Hz, 2H), 7.37 (d, J = 8.0 Hz, 2H), 5.09 (dd, J = 5.2, 12.8 Hz, 1H), 4.69 (dd, J = 4.0, 14.8 Hz, 1H), 4.60 - 4.41 (m, 3H), 3.35 - 3.16 (m, 7H), 3.12 (d, J = 1.6 Hz, 3H), 2.93 - 2.81 (m, 1H), 2.80 - 2.59 (m, 7H), 2.28 (s, 3H), 2.08 - 1.98 (m, 2H), 1.75 (dd, J = 4.8, 9.6 Hz, 1H), 1.53 (s, 6H). Example 52: Synthesis of Compound 52
Step A – Synthesis of tert-butyl 4-(4-hydroxycyclohexyl) piperazine-1-carboxylate (52a) To a solution of 4-aminocyclohexanol (5 g, 43.41 mmol, 1.05 equiv.) and tert-butyl N,N- bis(2-chloroethyl)carbamate (10.01 g, 41.35 mmol, 1 equiv.) in dioxane (200 mL) was added K2CO3 (17.14 g, 124.04 mmol, 3 equiv.), KI (20.59 g, 124.04 mmol, 3 equiv.) at 20 °C. The mixture was stirred at 115°C for 17 h. The reaction was quenched with NH4Cl (250 mL) and extracted with ethyl acetate (500 mL x 3). The organic layer was concentrated under reduced pressure to give a residue. The residue was purified by silica gel chromatography (silica gel, Dichloromethane: Methanol= 1: 0 to 0: 1) to give (1 g, 3.52 mmol, 8.50% yield) was obtained as a white solid. 1H NMR (400 MHz, CHLOROFORM-d) δ(ppm) = 3.77 - 3.67 (m, 1H), 3.65 - 3.54 (m, 2H), 3.42 (br d, J = 4.4 Hz, 3H), 2.50 (br s, 4H), 2.29 (br s, 1H), 2.03 (br d, J = 3.8 Hz, 2H), 1.88 (br s, 2H), 1.46 (s, 9H), 1.36 - 1.23 (m, 4H). Step B – Synthesis of tert-butyl 4-[4-[(5-bromo-2-pyridyl)oxy]cyclohexyl]piperazine-1- carboxylate (52b) To a solution of 52a (1 g, 3.52 mmol, 1 equiv.) in THF (10 mL) was added NaH (211 mg, 5.3 mmol, 60% purity, 1.5 equiv.) at 0°C, sequentially, was added 5-bromo-2-fluoro-pyridine (618.81 mg, 3.52 mmol, 361.88 μL, 1 equiv.) in THF (5 mL) at 0°C. The mixture was stirred at 50°C for 16 h, quenched with NH4Cl (50 mL) and extracted with ethyl acetate (250 mL x 3). The combined organic portions were concentrated under reduced pressure. The residue was purified by silica gel chromatography (silica gel, petroleum ether: ethyl acetate = 1: 0 to 0: 1) to afford (52b).1H NMR (400 MHz, CHLOROFORM-d) δ = 8.16 (d, J = 2.6 Hz, 1H), 7.62 (dd, J = 2.4, 8.6 Hz, 1H), 6.59 (d, J = 8.8 Hz, 1H), 4.87 (br s, 1H), 3.44 (br s, 4H), 2.52 (br s, 4H), 2.38 (br d, J = 1.6 Hz, 1H), 2.21 (br s, 2H), 1.94 (br s, 2H), 1.52 - 1.41 (m, 13H). Step C – Synthesis of tert-butyl4-[4-[[5-[3-methyl-4-[4-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H-1,2,4-triazol-5-yl]methylcarbamoyl]pyrazol-1-yl]phenyl]-2- pyridyl]oxy]cyclohexyl]piperazine-1-carboxylate (52c) To a solution of 52b (220 mg, 499.6 μmol, 1 equiv.) and Int 45 (258.9 mg, 499.6 μmol, 1.0 equiv.) in water (0.2 mL) and THF (0.8 mL) was added K3PO4 (318.13 mg, 1.50 mmol, 3 equiv.) at 20°C, sequentially, was added PdCl2(DTBPF) (65.12 mg, 99.92 μmol, 0.2 equiv.) at
20°C. The mixture was stirred at 80°C for 12 h. The mixture was poured into ethyl acetate (25 mL) and washed with water (25 mL) and brine (25 mL x 3). The organic phase was dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum. The residue was purified by silica gel chromatography (silica gel, petroleum ether: ethyl acetate= 1: 0 to 0: 1) to afford 52c. LCMS [M+1] = 752.5. Step D – Synthesis of 1-[2-methyl-4-[6-(4-piperazin-1-ylcyclohexoxy)-3-pyridyl]phenyl]-N-[[3- (2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (52d) To a solution of 52c (60 mg, 79.8 μmol, 1 equiv.) in TFA (0.4 mL) and dichloromethane (2 mL) at 20°C. The mixture was stirred at 20°C for 1 h. The reaction mixture was concentrated under reduced pressure to afford 52d. LCMS [M+1] = 652.5. Step E – Synthesis of 1-[4-[6-[4-[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]piperazin-1-yl]cyclohexoxy]-3-pyridyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl- ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Compound 52) To a solution of 52d (52 mg, 80 μmol, 1 equiv.) and 2-(2,6-dioxo-3-piperidyl)-5,6- difluoro-isoindoline-1,3-dione (35.2 mg, 120 μmol, 1.5 equiv.) in DMSO (5 mL) was added DIEA (51.6 mg, 399 μmol, 70 μL, 5 equiv.) at 20°C. The mixture was stirred at 70°C for 12 h, cooled to ambient temperature and concentrated under reduced pressure. The residue was purified by preparative HPLC(Phenomenex Luna C18, 100 mm x 40mm, 3 µm; [water(0.2% FA)-ACN]) to afford Compound 52. LCMS [M+1] = 926.3; 1H NMR (400 MHz, METHANOL- d4) δ(ppm) = 13.93 (br s, 1H), 11.11 (s, 1H), 8.88 (br s, 1H), 8.56 - 8.49 (m, 2H), 8.17 (s, 1H), 8.06 (dd, J = 2.4, 8.8 Hz, 1H), 7.76 - 7.69 (m, 2H), 7.67 - 7.58 (m, 1H), 7.50 - 7.42 (m, 2H), 6.87 (d, J = 8.6 Hz, 1H), 5.11 (dd, J = 5.4, 12.8 Hz, 1H), 4.99 (br d, J = 3.4 Hz, 1H), 4.55 (br d, J = 5.6 Hz, 2H), 3.25 (br s, 4H), 2.95 - 2.81 (m, 1H), 2.74 - 2.66 (m, 4H), 2.57 (br s, 2H), 2.43 (br d, J = 10.0 Hz, 2H), 2.32 (br d, J = 1.6 Hz, 1H), 2.29 (s, 3H), 2.23 - 2.13 (m, 2H), 2.09 - 1.99 (m, 1H), 1.96 - 1.86 (m, 2H), 1.54 (s, 6H), 1.48 - 1.43 (m, 3H). Example 53: Synthesis of Compound 53
carboxylate (53a) To a solution of tert-butyl 4-[(4-bromopyrazol-1-yl)methyl]piperidine-1-carboxylate (500 mg, 1.45 mmol, 1.0 equiv.) in dichloromethane (4.0 mL) was added TFA (1.0 mL) at 20°C. The resulting mixture was stirred at 20°C for 2 h. The reaction mixture was concentrated under reduced pressure. The residue was suspended in THF (4.0 mL) and treated with tert-butyl 4- oxopiperidine-1-carboxylate (129.8 mg, 651.5 μmol, 1.0 equiv.) and tetraisopropoxytitanium (1.11 g, 3.91 mmol, 1.15 mL, 6.0 equiv.) and heated at 60 °C for 1 h and then cooled to ambient temperature. Sodium triacetoxyborohydride (690.4 mg, 3.26 mmol, 5.0 equiv.) was added portion wise and the mixture was stirred at ambient temperature for 1 h. The reaction mixture was quenched with water (10 mL) and filtered to remove the precipitate. The filtrate was extracted with ethyl acetate (5 mL x3). The combined organic layers were washed with brine (7 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether: ethyl acetate = 1:0 to dichloromethane: methanol = 10:1) to afford 53a. LCMS: [M+1] = 427.3, 429.3; 1H NMR (400 MHz, DMSO-d6) δ 7.99 (s, 1H), 7.56 (s, 1H), 4.14 - 3.96 (m, 4H), 3.53 - 3.34 (m, 2H), 3.17 (d, J = 5.2 Hz, 2H), 3.05 - 2.60 (m, 4H), 2.14 - 1.95 (m, 2H), 1.91 (s, 1H), 1.67 (s, 2H), 1.54 (s, 2H), 1.39 (s, 9H), 1.30 - 1.19 (m, 1H).
Step B – Synthesis of tert-butyl 4-[4-[[4-[3-methyl-4-[4-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H- 1,2,4-triazol-5-yl]methylcarbamoyl]pyrazol-1-yl]phenyl]pyrazol-1-yl]methyl]-1- piperidyl]piperidine-1-carboxylate (53b) A mixture of 53a (79.1 mg, 185.2 μmol, 1.2 equiv.), Int 45 (80.0 mg, 154.3 μmol, 1.0 equiv.), K3PO4 (98.3 mg, 463.0 μmol, 3.0 equiv.), PdCl2(DTBPF) (20.1 mg, 30.9 μmol, 0.2 equiv.) in water (0.16 mL) and THF (0.64 mL) was degassed and purged threefold with nitrogen. The mixture was stirred at 80°C for 4 h under a nitrogen atmosphere. The reaction mixture was quenched with water (1 mL) at 20°C and then extracted with ethyl acetate (1 mL x 3). The combined organic layers were washed with brine (2 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative TLC (silica gel, dichloromethane : methanol = 10:1) to give (53b). LCMS: [M+1] = 739.6; 1H NMR (400 MHz, DMSO-d6) δ 14.16 - 13.62 (br s, 1H), 8.84 (br s, 1H), 8.47 (s, 1H), 8.22 (s, 1H), 8.14 (s, 1H), 7.94 (s, 1H), 7.63 (s, 1H), 7.53 (dd, J = 6.4 Hz, 1H), 7.35 (d, J = 8.4 Hz, 1H), 4.54 (d, J = 5.6 Hz, 2H), 4.03 - 3.91 (m, 4H), 2.82 (d, J = 9.6 Hz, 2H), 2.23 (s, 3H), 2.15 - 2.04 (m, 2H), 1.86 - 1.74 (m, 2H), 1.71 - 1.62 (m, 3H), 1.54 (s, 6H), 1.38 (s, 9H), 1.28 - 1.13 (m, 8H). Step C – Synthesis of 1-[2-methyl-4-[1-[[1-(4-piperidyl)-4-piperidyl] methyl]pyrazol-4- yl]phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4- carboxamide (53c) To a solution of 53b (43.0 mg, 58.2 μmol, 1.0 equiv.) in dichloromethane (0.4 mL) was added TFA (0.2 mL). The mixture was stirred at 20°C for 1 h and then concentrated under reduced pressure to afford LCMS: [M+1] = 639.5. Step D – Synthesis of 1-[4-[1-[[1-[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]-4- piperidyl]-4-piperidyl]methyl]pyrazol-4-yl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Compound 53) To a solution of 53c (94.3 mg, 62.6 μmol, 1.0 equiv., TFA) and 2-(2,6-dioxo-3-piperidyl)- 5,6 -difluoro-isoindoline-1,3-dione (27.6 mg, 93.9 μmol, 1.5 equiv.) in DMSO (0.9 mL) was
added DIEA (24.3 mg, 188 μmol, 33 μL, 3.0 equiv.). The mixture was stirred at 70°C for 16 h cooled to ambient temperature, diluted with DMSO (0.9 mL) and filtered. The filtrate was purified directly by preparative HPLC (Phenomenex Luna C18, 100mm x 40 mm, 5 µm; [water(0.04% HCl)-ACN]) to afford Compound 53. LCMS: [M+1] = 913.5; 1H NMR (400 MHz, DMSO-d6) δ 13.88 (br s, 1H), 11.11 (s, 1H), 9.55 (br s, 1H), 8.86 (br s, 1H), 8.48 (s, 1H), 8.29 (s, 1H), 8.15 (s, 1H), 8.00 (s, 1H), 7.76 (d, J = 11.2 Hz, 1H), 7.64 (s, 1H), 7.55 (d, J = 8.0 Hz, 1H), 7.50 (d, J = 7.6 Hz, 1H), 7.37 (d, J = 8.4 Hz, 1H), 5.14 - 5.06 (m, 1H), 4.55 (d, J = 4.8 Hz, 2H), 4.10 (d, J = 6.4 Hz, 2H), 3.75 (d, J = 11.6 Hz, 2H), 3.52 – 3.49 (m, 2H), 3.06 - 2.81 (m, 8H), 2.61 (s, 1H), 2.57 (s, 1H), 2.24 (s, 3H), 2.17 - 2.12 (m, 2H), 2.08 - 1.96 (m, 2H), 1.84 - 1.74 (m, 4H), 1.54 (s, 6H) Example 54: Synthesis of Compound 54
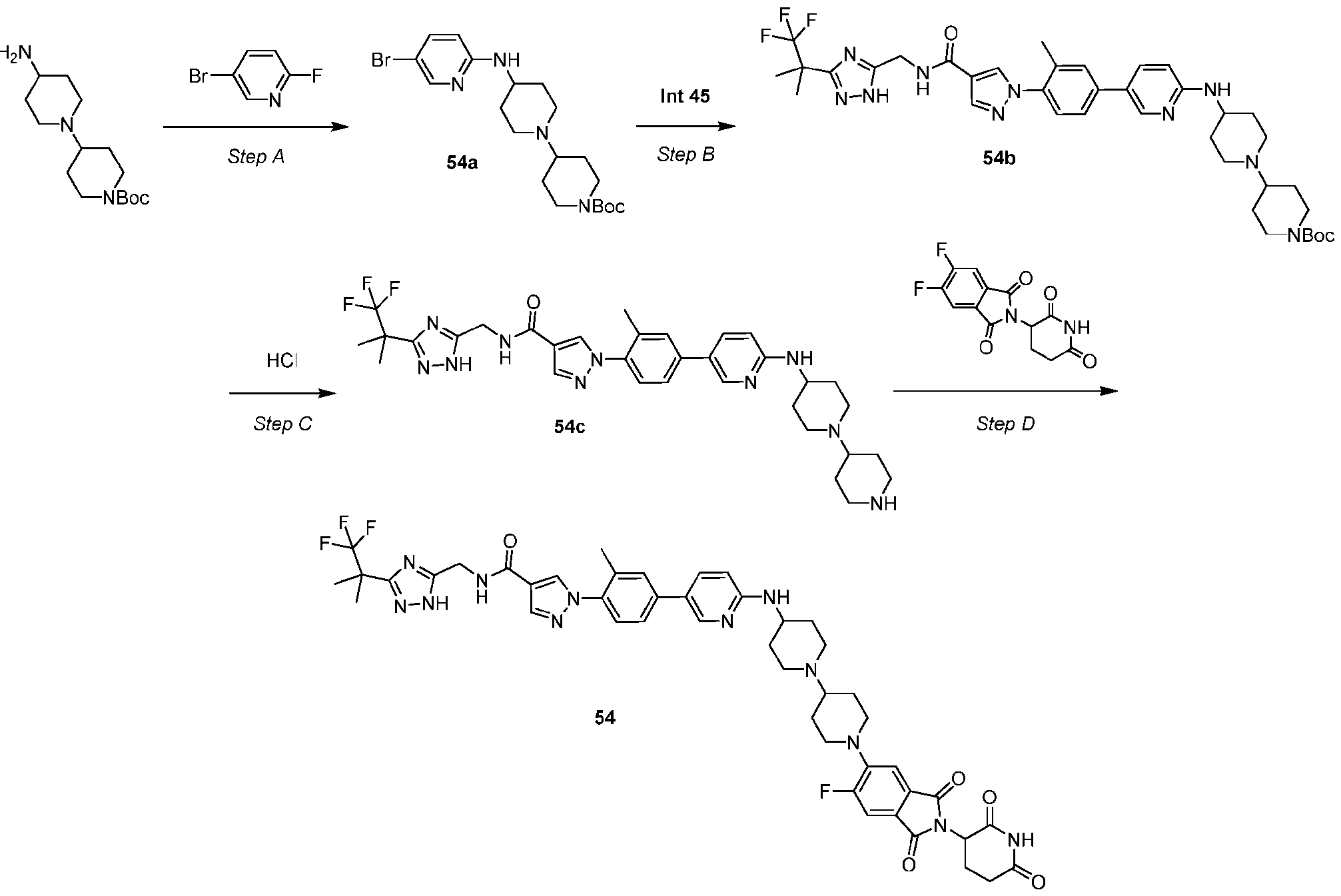 carboxylate (54a)
To a solution of tert-butyl 4-(4-amino-1-piperidyl)piperidine-1-carboxylate (1.0 g, 3.5 mmol, 1.0 equiv.) in DMSO (10 mL) was added 5-bromo-2-fluoro-pyridine (620.9 mg, 3.5 mmol, 363.1 μL, 1.0 equiv.) and DIEA (912.0 mg, 7.0 mmol, 1.2 mL, 2.0 equiv.). The mixture was stirred at 70°C for 16 h. The reaction mixture was quenched with water (20 mL) at 20 °C, and then diluted with water (20 mL) and extracted with ethyl acetate 120 mL (60 mL x 2). The combined organic layers were washed with brine (30 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, dichloromethane : methanol=99/1 to 94/6) to afford 54a. LCMS [M+1] = 439.1, 441.1; 1H NMR (400 MHz, CHLOROFORM-d) δ 8.09 (d, J = 2.4 Hz, 1H), 7.45 (dd, J = 2.4, 8.8 Hz, 1H), 6.28 (d, J = 8.8 Hz, 1H), 4.41 (br d, J = 8.0 Hz, 1H), 4.16 (br s, 2H), 3.60 (br s, 1H), 2.91 (br d, J = 11.2 Hz, 2H), 2.70 (br t, J = 11.6 Hz, 2H), 2.55 - 2.30 (m, 3H), 2.07 (br d, J = 11.6 Hz, 2H), 1.81 (br d, J = 12.0 Hz, 2H), 1.60 - 1.36 (m, 13H) Step B – Synthesis of tert-butyl 4-[4-[[5-[3-methyl-4-[4-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H-1,2,4-triazol-5-yl]methylcarbamoyl]pyrazol-1-yl]phenyl]-2-pyridyl]amino]-1- piperidyl]piperidine-1-carboxylate (54b) To a solution of 54a (101.7 mg, 231.5 μmol, 1.0 equiv.) and Int 45 (150.0 mg, 231.5 μmol, 1.0 equiv.) in dioxane (2.0 mL) and water (0.4 mL) was added K2CO3 (95.9 mg, 694.5 μmol, 3.0 equiv.), Pd(DPPF)Cl2.CH2Cl2 (37.8 mg, 46.3 μmol, 0.2 equiv.). The mixture was stirred at 100°C under a nitrogen atmosphere for 16 h. The reaction mixture was filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative HPLC (Phenomenex Luna C18100 mm x 30mm, 5 µm; [water(0.2% FA)-ACN]) to afford 54b. LCMS [M+1] = 751.6; 1H NMR (400 MHz, CHLOROFORM-d) δ 8.41 - 8.28 (s, 1H), 8.16 (s, 1H), 8.05 (s, 1H), 7.61 (br d, J = 7.2 Hz, 2H), 7.50 - 7.34 (m, 3H), 7.30 (m, 1H), 7.13 - 6.98 (m, 1H), 6.43 (br d, J = 8.8 Hz, 1H), 4.73 (br d, J = 5.6 Hz, 3H), 4.22 (br s, 2H), 3.92 - 3.69 (br s, 1H), 3.50 (s, 2H), 3.05 (br s, 2H), 2.71 (br t, J = 8.8 Hz, 3H), 2.61 - 2.44 (br s, 2H), 2.27 (s, 3H), 2.22 - 2.07 (m, 2H), 1.91 (br s, 3H), 1.61 (s, 6H), 1.48 (s, 9H) Step C – Synthesis of 1-[2-methyl-4-[6-[[1-(4-piperidyl)-4-piperidyl]amino]-3-pyridyl]phenyl]-N- [[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (54c)
A solution of 54b (35.0 mg, 46.6 μmol, 1.0 equiv.) in HCl/ dioxane (4 M, 0.5 mL) was stirred at 20°C for 0.5 h. The reaction mixture was concentrated under reduced pressure to afford 54c. LCMS [M+1] = 651.4; Step D – Synthesis of 1-[4-[6-[[1-[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]-4-piperidyl]-4-piperidyl]amino]-3-pyridyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Compound 54) To a solution of 54c (29.0 mg, 44.5 μmol, 1.0 equiv.) in DMSO (1.0 mL) was added 2- (2,6-dioxo-3-piperidyl)-5,6-difluoro-isoindoline-1,3-dione (19.6 mg, 66.8 μmol, 1.5 equiv.) and DIEA (28.8 mg, 222.8 μmol, 38.8 μL, 5.0 equiv.). The mixture was stirred at 70°C for 16 h. The reaction mixture was cooled to ambient temperature, quenched with water (1 mL) and concentrated under reduced pressure. The mixture was purified by preparative HPLC (Phenomenex Luna C18100mm x 30mm, 5µm; [water(0.2% FA)-ACN]) to afford Compound 54. LCMS [M+1] = 925.6; 1H NMR (400 MHz, DMSO-d6) δ 13.91 (br s, 1H), 11.11 (s, 1H), 8.87 (br s, 1H), 8.49 (s, 1H), 8.38 (d, J = 2.2 Hz, 1H), 8.22 - 8.10 (m, 1H), 7.87 - 7.68 (m, 2H), 7.62 (s, 1H), 7.54 (br d, J = 8.2 Hz, 1H), 7.46 (d, J = 7.6 Hz, 1H), 7.40 (d, J = 8.4 Hz, 1H), 6.69 (br d, J = 7.6 Hz, 1H), 6.57 (d, J = 8.8 Hz, 1H), 5.11 (dd, J = 5.4, 12.8 Hz, 1H), 4.55 (br d, J = 5.2 Hz, 2H), 3.84 - 3.73 (m, 1H), 3.68 (br d, J = 11.4 Hz, 2H), 3.06 - 2.79 (m, 5H), 2.66 - 2.52 (m, 3H), 2.45 - 2.35 (m, 2H), 2.26 (s, 3H), 2.10 - 1.84 (m, 5H), 1.73 - 1.58 (m, 2H), 1.54 (s, 6H), 1.50 - 1.34 (m, 2H) Example 55: Synthesis of Intermediate 55
carboxylate (54a)
To a solution of tert-butyl 4-(4-amino-1-piperidyl)piperidine-1-carboxylate (1.0 g, 3.5 mmol, 1.0 equiv.) in DMSO (10 mL) was added 5-bromo-2-fluoro-pyridine (620.9 mg, 3.5 mmol, 363.1 μL, 1.0 equiv.) and DIEA (912.0 mg, 7.0 mmol, 1.2 mL, 2.0 equiv.). The mixture was stirred at 70°C for 16 h. The reaction mixture was quenched with water (20 mL) at 20 °C, and then diluted with water (20 mL) and extracted with ethyl acetate 120 mL (60 mL x 2). The combined organic layers were washed with brine (30 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, dichloromethane : methanol=99/1 to 94/6) to afford 54a. LCMS [M+1] = 439.1, 441.1; 1H NMR (400 MHz, CHLOROFORM-d) δ 8.09 (d, J = 2.4 Hz, 1H), 7.45 (dd, J = 2.4, 8.8 Hz, 1H), 6.28 (d, J = 8.8 Hz, 1H), 4.41 (br d, J = 8.0 Hz, 1H), 4.16 (br s, 2H), 3.60 (br s, 1H), 2.91 (br d, J = 11.2 Hz, 2H), 2.70 (br t, J = 11.6 Hz, 2H), 2.55 - 2.30 (m, 3H), 2.07 (br d, J = 11.6 Hz, 2H), 1.81 (br d, J = 12.0 Hz, 2H), 1.60 - 1.36 (m, 13H) Step B – Synthesis of tert-butyl 4-[4-[[5-[3-methyl-4-[4-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H-1,2,4-triazol-5-yl]methylcarbamoyl]pyrazol-1-yl]phenyl]-2-pyridyl]amino]-1- piperidyl]piperidine-1-carboxylate (54b) To a solution of 54a (101.7 mg, 231.5 μmol, 1.0 equiv.) and Int 45 (150.0 mg, 231.5 μmol, 1.0 equiv.) in dioxane (2.0 mL) and water (0.4 mL) was added K2CO3 (95.9 mg, 694.5 μmol, 3.0 equiv.), Pd(DPPF)Cl2.CH2Cl2 (37.8 mg, 46.3 μmol, 0.2 equiv.). The mixture was stirred at 100°C under a nitrogen atmosphere for 16 h. The reaction mixture was filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative HPLC (Phenomenex Luna C18100 mm x 30mm, 5 µm; [water(0.2% FA)-ACN]) to afford 54b. LCMS [M+1] = 751.6; 1H NMR (400 MHz, CHLOROFORM-d) δ 8.41 - 8.28 (s, 1H), 8.16 (s, 1H), 8.05 (s, 1H), 7.61 (br d, J = 7.2 Hz, 2H), 7.50 - 7.34 (m, 3H), 7.30 (m, 1H), 7.13 - 6.98 (m, 1H), 6.43 (br d, J = 8.8 Hz, 1H), 4.73 (br d, J = 5.6 Hz, 3H), 4.22 (br s, 2H), 3.92 - 3.69 (br s, 1H), 3.50 (s, 2H), 3.05 (br s, 2H), 2.71 (br t, J = 8.8 Hz, 3H), 2.61 - 2.44 (br s, 2H), 2.27 (s, 3H), 2.22 - 2.07 (m, 2H), 1.91 (br s, 3H), 1.61 (s, 6H), 1.48 (s, 9H) Step C – Synthesis of 1-[2-methyl-4-[6-[[1-(4-piperidyl)-4-piperidyl]amino]-3-pyridyl]phenyl]-N- [[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (54c)
A solution of 54b (35.0 mg, 46.6 μmol, 1.0 equiv.) in HCl/ dioxane (4 M, 0.5 mL) was stirred at 20°C for 0.5 h. The reaction mixture was concentrated under reduced pressure to afford 54c. LCMS [M+1] = 651.4; Step D – Synthesis of 1-[4-[6-[[1-[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]-4-piperidyl]-4-piperidyl]amino]-3-pyridyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Compound 54) To a solution of 54c (29.0 mg, 44.5 μmol, 1.0 equiv.) in DMSO (1.0 mL) was added 2- (2,6-dioxo-3-piperidyl)-5,6-difluoro-isoindoline-1,3-dione (19.6 mg, 66.8 μmol, 1.5 equiv.) and DIEA (28.8 mg, 222.8 μmol, 38.8 μL, 5.0 equiv.). The mixture was stirred at 70°C for 16 h. The reaction mixture was cooled to ambient temperature, quenched with water (1 mL) and concentrated under reduced pressure. The mixture was purified by preparative HPLC (Phenomenex Luna C18100mm x 30mm, 5µm; [water(0.2% FA)-ACN]) to afford Compound 54. LCMS [M+1] = 925.6; 1H NMR (400 MHz, DMSO-d6) δ 13.91 (br s, 1H), 11.11 (s, 1H), 8.87 (br s, 1H), 8.49 (s, 1H), 8.38 (d, J = 2.2 Hz, 1H), 8.22 - 8.10 (m, 1H), 7.87 - 7.68 (m, 2H), 7.62 (s, 1H), 7.54 (br d, J = 8.2 Hz, 1H), 7.46 (d, J = 7.6 Hz, 1H), 7.40 (d, J = 8.4 Hz, 1H), 6.69 (br d, J = 7.6 Hz, 1H), 6.57 (d, J = 8.8 Hz, 1H), 5.11 (dd, J = 5.4, 12.8 Hz, 1H), 4.55 (br d, J = 5.2 Hz, 2H), 3.84 - 3.73 (m, 1H), 3.68 (br d, J = 11.4 Hz, 2H), 3.06 - 2.79 (m, 5H), 2.66 - 2.52 (m, 3H), 2.45 - 2.35 (m, 2H), 2.26 (s, 3H), 2.10 - 1.84 (m, 5H), 1.73 - 1.58 (m, 2H), 1.54 (s, 6H), 1.50 - 1.34 (m, 2H) Example 55: Synthesis of Intermediate 55
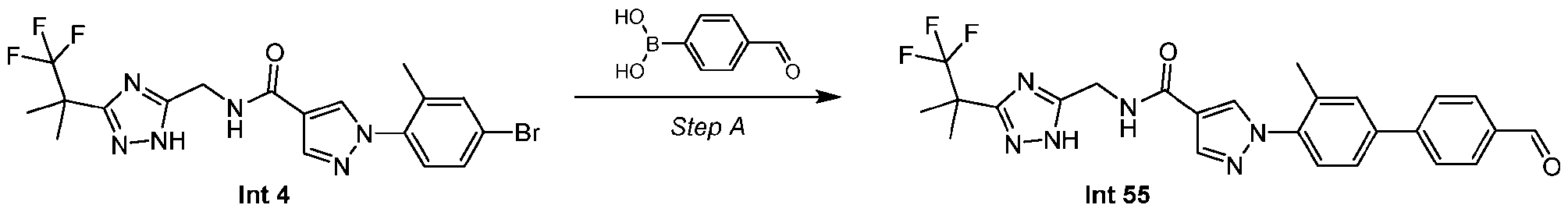 Step A – Synthesis of 1-[4-(4-formylphenyl)-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Intermediate 55) To a mixture of Int 4 (8.0 g, 16.9 mmol, 1 equiv.), (4-formylphenyl)boronic acid (3.0 g, 20.3 mmol, 1.2 equiv.) and K2CO3 (4.6 g, 33.9 mmol, 2.0 equiv.) in dioxane (68 mL) and water
(17 mL) was degassed and purged three-fold with nitrogen and then treated with Pd(DPPF)Cl2.CH2Cl2 (1.3 g, 1.7 mmol, 0.1 equiv.). The mixture was stirred at 100 °C for 5 h and then cooled to ambient temperature. The reaction mixture was quenched with water (200 mL) and then extracted with ethyl acetate (3 x 150 mL ). The combined organic layer was washed with brine (3 x 150 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether/ethyl acetate=3/1 to 0/1) to give Intermediate 55. LCMS: [M+1] = 497.4; 1H NMR (400 MHz, DMSO-d6) δ 13.90 (br s, 1H), 10.07 (s, 1H), 8.92 (t, J = 6.0 Hz, 1H), 8.56 (s, 1H), 8.19 (s, 1H), 8.00 (q, J = 8.0 Hz, 4H), 7.85 (s, 1H), 7.76 (dd, J = 2.0, 8.4 Hz, 1H), 7.53 (d, J = 8.0 Hz, 1H), 4.57 (d, J = 5.6 Hz, 2H), 2.34 (s, 3H), 1.56 (s, 6H). Example 56: Synthesis of Compound 56 Step A
Step A – Synthesis of 1-[4-(4-formylphenyl)-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Intermediate 55) To a mixture of Int 4 (8.0 g, 16.9 mmol, 1 equiv.), (4-formylphenyl)boronic acid (3.0 g, 20.3 mmol, 1.2 equiv.) and K2CO3 (4.6 g, 33.9 mmol, 2.0 equiv.) in dioxane (68 mL) and water
(17 mL) was degassed and purged three-fold with nitrogen and then treated with Pd(DPPF)Cl2.CH2Cl2 (1.3 g, 1.7 mmol, 0.1 equiv.). The mixture was stirred at 100 °C for 5 h and then cooled to ambient temperature. The reaction mixture was quenched with water (200 mL) and then extracted with ethyl acetate (3 x 150 mL ). The combined organic layer was washed with brine (3 x 150 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether/ethyl acetate=3/1 to 0/1) to give Intermediate 55. LCMS: [M+1] = 497.4; 1H NMR (400 MHz, DMSO-d6) δ 13.90 (br s, 1H), 10.07 (s, 1H), 8.92 (t, J = 6.0 Hz, 1H), 8.56 (s, 1H), 8.19 (s, 1H), 8.00 (q, J = 8.0 Hz, 4H), 7.85 (s, 1H), 7.76 (dd, J = 2.0, 8.4 Hz, 1H), 7.53 (d, J = 8.0 Hz, 1H), 4.57 (d, J = 5.6 Hz, 2H), 2.34 (s, 3H), 1.56 (s, 6H). Example 56: Synthesis of Compound 56 Step A
 -1H- 1,2,4-triazol-5-yl]methylcarbamoyl]pyrazol-1-yl]phenyl]phenyl]methyl]-2,7- diazaspiro[3.5]nonane-2-carboxylate (56a) To a solution of tert-butyl 2,7-diazaspiro[3.5]nonane-2-carboxylate (68.3 mg, 302.1 μmol, 1.5 equiv) and 1-[4-(4-formylphenyl)-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl- ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (100.0 mg, 201.4 μmol, 1.0 equiv) in DCE (2.0 mL) was added triethylamine (40.76 mg, 402.8 μmol, 56.0 μL, 2.0 equiv). The mixture was stirred at 25°C for 0.5 h and then treated with NaBH(OAc)3 (128.0 mg, 604.2 μmol, 3.0
equiv). The mixture was stirred at 25°C for 1 hr, diluted by water (3.0 mL). The mixture extracted with dichloromethane (3 × 3 mL). The combined organic phase was washed with brine (3 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative TLC (silica gel, petroleum ether: ethyl acetate = 3:1) to afford 56a. LCMS [M+1] = 707.6; 1H NMR (400 MHz, CHLOROFORM-d) δ 8.16 (s, 1H), 8.03 (s, 1H), 7.57 - 7.51 (m, 3H), 7.49 (br d, J = 8.1 Hz, 1H), 7.39 (d, J = 8.1 Hz, 2H), 7.35 (d, J = 8.1 Hz, 1H), 7.02 (br d, J = 3.1 Hz, 1H), 4.71 (br d, J = 5.6 Hz, 2H), 3.61 (s, 4H), 3.50 (s, 2H), 2.52 - 2.31 (m, 4H), 2.29 (s, 3H), 1.76 (br t, J = 5.1 Hz, 4H), 1.59 (s, 6H), 1.44 (s, 9H). Step B – Synthesis of 1-[4-[4-[[2-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]- 2,7-diazaspiro[3.5]nonan-7-yl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Compound 56) A solution of 56a (50.0 mg, 70.7 μmol, 1.0 equiv) in HCl/ dioxane (4.0 M, 1.0 mL) was stirred at 25°C for 30 mins. The reaction mixture was concentrated under reduced pressure. The residue was dissolved in DMSO (1.0 ml) and treated with 2-(2,6-dioxo-3-piperidyl)-5,6-difluoro- isoindoline-1,3-dione (29.1 mg, 98.9 μmol, 1.5 equiv) and DIEA (42.6 mg, 329.6 μmol, 57.4 μL, 5.0 equiv). The mixture was stirred at 70°C for 12 h. The reaction was cooled ambient temperature and diluted by water (2.0 mL). The mixture extracted with ethyl acetate (3 x 2 mL). The combined organic phase was washed with brine (2 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative HPLC (Phenomenex Luna C18, 100 mm x 30 mm; 5 µm; [water(0.2% FA)-ACN]) to afford Compound 56. LCMS [M+1] = 881.6; 1H NMR (400 MHz, DMSO-d6) δ 13.91 (br s, 1H), 11.08 (s, 1H), 8.88 (br s, 1H), 8.53 (s, 1H), 8.24 - 8.08 (m, 1H), 7.73 (s, 1H), 7.69 (d, J = 8.3 Hz, 2H), 7.64 (br d, J = 8.1 Hz, 1H), 7.59 (d, J = 11.3 Hz, 1H), 7.46 (d, J = 8.3 Hz, 1H), 7.42 (d, J = 8.0 Hz, 2H), 6.90 (d, J = 7.9 Hz, 1H), 5.06 (dd, J = 5.3, 13.0 Hz, 1H), 4.55 (br d, J = 4.3 Hz, 2H), 3.91 (s, 4H), 3.51 (s, 2H), 2.94 - 2.80 (m, 1H), 2.62 - 2.52 (m, 3H), 2.36 (br d, J = 1.6 Hz, 3H), 2.30 (s, 3H), 2.04 - 1.95 (m, 1H), 1.80 (br s, 4H), 1.54 (s, 6H) Example 57: Synthesis of Compound 57-1 and 57-2
-1H- 1,2,4-triazol-5-yl]methylcarbamoyl]pyrazol-1-yl]phenyl]phenyl]methyl]-2,7- diazaspiro[3.5]nonane-2-carboxylate (56a) To a solution of tert-butyl 2,7-diazaspiro[3.5]nonane-2-carboxylate (68.3 mg, 302.1 μmol, 1.5 equiv) and 1-[4-(4-formylphenyl)-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl- ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (100.0 mg, 201.4 μmol, 1.0 equiv) in DCE (2.0 mL) was added triethylamine (40.76 mg, 402.8 μmol, 56.0 μL, 2.0 equiv). The mixture was stirred at 25°C for 0.5 h and then treated with NaBH(OAc)3 (128.0 mg, 604.2 μmol, 3.0
equiv). The mixture was stirred at 25°C for 1 hr, diluted by water (3.0 mL). The mixture extracted with dichloromethane (3 × 3 mL). The combined organic phase was washed with brine (3 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative TLC (silica gel, petroleum ether: ethyl acetate = 3:1) to afford 56a. LCMS [M+1] = 707.6; 1H NMR (400 MHz, CHLOROFORM-d) δ 8.16 (s, 1H), 8.03 (s, 1H), 7.57 - 7.51 (m, 3H), 7.49 (br d, J = 8.1 Hz, 1H), 7.39 (d, J = 8.1 Hz, 2H), 7.35 (d, J = 8.1 Hz, 1H), 7.02 (br d, J = 3.1 Hz, 1H), 4.71 (br d, J = 5.6 Hz, 2H), 3.61 (s, 4H), 3.50 (s, 2H), 2.52 - 2.31 (m, 4H), 2.29 (s, 3H), 1.76 (br t, J = 5.1 Hz, 4H), 1.59 (s, 6H), 1.44 (s, 9H). Step B – Synthesis of 1-[4-[4-[[2-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]- 2,7-diazaspiro[3.5]nonan-7-yl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Compound 56) A solution of 56a (50.0 mg, 70.7 μmol, 1.0 equiv) in HCl/ dioxane (4.0 M, 1.0 mL) was stirred at 25°C for 30 mins. The reaction mixture was concentrated under reduced pressure. The residue was dissolved in DMSO (1.0 ml) and treated with 2-(2,6-dioxo-3-piperidyl)-5,6-difluoro- isoindoline-1,3-dione (29.1 mg, 98.9 μmol, 1.5 equiv) and DIEA (42.6 mg, 329.6 μmol, 57.4 μL, 5.0 equiv). The mixture was stirred at 70°C for 12 h. The reaction was cooled ambient temperature and diluted by water (2.0 mL). The mixture extracted with ethyl acetate (3 x 2 mL). The combined organic phase was washed with brine (2 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative HPLC (Phenomenex Luna C18, 100 mm x 30 mm; 5 µm; [water(0.2% FA)-ACN]) to afford Compound 56. LCMS [M+1] = 881.6; 1H NMR (400 MHz, DMSO-d6) δ 13.91 (br s, 1H), 11.08 (s, 1H), 8.88 (br s, 1H), 8.53 (s, 1H), 8.24 - 8.08 (m, 1H), 7.73 (s, 1H), 7.69 (d, J = 8.3 Hz, 2H), 7.64 (br d, J = 8.1 Hz, 1H), 7.59 (d, J = 11.3 Hz, 1H), 7.46 (d, J = 8.3 Hz, 1H), 7.42 (d, J = 8.0 Hz, 2H), 6.90 (d, J = 7.9 Hz, 1H), 5.06 (dd, J = 5.3, 13.0 Hz, 1H), 4.55 (br d, J = 4.3 Hz, 2H), 3.91 (s, 4H), 3.51 (s, 2H), 2.94 - 2.80 (m, 1H), 2.62 - 2.52 (m, 3H), 2.36 (br d, J = 1.6 Hz, 3H), 2.30 (s, 3H), 2.04 - 1.95 (m, 1H), 1.80 (br s, 4H), 1.54 (s, 6H) Example 57: Synthesis of Compound 57-1 and 57-2
Step A – Synthesis of benzyl 4-(4-tert-butoxycarbonylpiperazin-1-yl) azepane-1-carboxylate (57a) To a solution of benzyl 4-oxoazepane-1-carboxylate (2.00 g, 8.09 mmol, 1.0 equiv.) and tert-butyl piperazine-1-carboxylate (1.66 g, 8.90 mmol, 1.1 equiv.) in THF (20 mL) was added AcOH (971 mg, 16.2 mmol, 926 μL, 2 equiv.). The mixture was stirred at 25 °C for 30 min, treated with NaBH(OAc)3 (3.43 g, 16.18 mmol, 2 equiv.) was added to the reaction mixture and stirred for 1.5 h at 25°C. The reaction mixture was quenched with water (10 mL) at 20 °C, diluted with water (100 mL) and extracted with ethyl acetate (100 mL x 2). The combined organic layers were dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure to afford 57a, which was used without further purification. LCMS [M+1] = 418.2; 1H NMR (400 MHz, CHLOROFORM-d) δ 7.29 - 7.26 (m, 5H), 5.06 (s, 2H), 3.62 - 3.51 (m, 4H), 3.51 - 3.45 (m, 1H), 3.41 - 3.31 (m, 4H), 2.61 - 2.55 (m, 2H), 2.45 - 2.40 (m, 2H), 1.88 - 1.79 (m, 2H), 1.78 - 1.71 (m, 2H), 1.60 - 1.44 (m, 2H), 1.38 (s, 9H). Step B – Synthesis of tert-butyl 4-(azepan-4-yl)piperazine-1-carboxylate (57b)
A solution of 57a (3.2 g, 7.66 mmol, 1 equiv.) in THF (10 mL) was degassed and purged three-fold with nitrogen, treated with Pd/C (1.63 g, 1.53 mmol, 10% purity, 0.2 equiv.). The mixture was degassed and purged three-fold with hydrogen and stirred at 25 °C for 16 h under a hydrogen atmosphere (15 Psi). The reaction mixture was filtered and the filtrate was concentrated under reduced pressure to afford 57b, which was used without further purification. 1H NMR (400 MHz, CHLOROFORM-d) δ 4.79 (s, 1H), 3.46 - 3.36 (m, 4H), 3.13 - 3.08 (M, 1H), 3.05 - 2.93 (m, 2H), 2.72 - 2.61 (m, 2H), 2.48 - 2.45 (m, 4H), 1.89 - 1.77 (m, 4H), 1.71 - 1.63 (m, 2H), 1.46 (s, 9H). Step C – Synthesis of tert-butyl 4-[1-[[4-[3-methyl-4-[4-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H-1,2,4-triazol-5-yl]methylcarbamoyl]pyrazol-1-yl]phenyl]phenyl]methyl]azepan-4- yl]piperazine-1-carboxylate (57c) To a solution of 57b (300 mg, 1.06 mmol, 1 equiv.) and Int 55 (472.99 mg, 952.69 μmol, 0.9 equiv.) in THF (10 mL) was added AcOH (127.14 mg, 2.12 mmol, 121.20 μL, 2 equiv.). The mixture was stirred at 25 °C for 30 min, treated with NaBH(OAc)3 (673.05 mg, 3.18 mmol, 3 equiv.) and stirred at 25°C for 1.5 h. The reaction mixture was quenched by addition of water (10 mL), diluted with water (100 mL) and extracted with ethyl acetate (50 mL x 2). The combined organic layers were washed with saturated aqueous NaHCO3 (50 mL x 2), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative HPLC (water/ACN= 95/5 to 25/75, 35 min) to afford 57c. LCMS [M+1, M+2] = 764.3, 765.3; 1H NMR (400 MHz, CHLOROFORM-d) δ 8.17 (s, 1H), 8.03 (s, 1H), 7.58 - 7.43 (m, 6H), 7.36 (d, J = 8.0 Hz, 1H), 6.92 (s, 1H), 4.73 (d, J = 6.0 Hz, 2H), 3.71 (s, 2H), 3.44 - 3.42 (m, 4H), 2.89 - 2.56 (m, 5H), 2.52 - 2.49 (m, 4H), 2.30 (s, 3H), 1.95 - 1.81 (m, 4H), 1.76 - 1.71 (m, 2H), 1.62 (s, 6H), 1.46 (s, 9H). Intermediate 57c was resolved by preparative SFC (DAICEL CHIRALPAK AD 250 mm x 30 mm,10µm); [CO2-ACN/i-PrOH(0.1%: NH3water)]) to afford 57c-1 as the early-eluting enantiomer and 57c-2 as the late-eluting enantiomer.
Step D – Synthesis of 1-(4'-((4-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperazin-1-yl)azepan-1-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide (Compound 57-1) Intermediate 57c-1 (100 mg, 130.91 μmol, 1 equiv.) was dissolved into HCl/dioxane (4 M, 10 mL), and the mixture was stirred at 25 °C for 2 h. The reaction mixture was concentrated to afford a residue which was used without further purification (LCMS [M+1] = 664.4). The residue (90 mg, 129 μmol, 1 equiv) was dissolved in DMSO (2 mL) and treated with 2-(2,6- dioxopiperidin-3-yl)-5,6-difluoroisoindoline-1,3-dione (37.8 mg, 128.7 μmol, 1 equiv.) and DIPEA (83.01 mg, 643.45 μmol, 5 equiv.). The mixture was stirred at 80°C for 12 h. The residue was purified by preparative HPLC (Phenomenex Luna C18, 150mm x 25mm, 10 µm; [water(FA)- ACN]) to give Compound 57-1. LCMS [M+1] = 938.5; 1H NMR (400 MHz, DMSO-d6) δ11.10 (s, 1H), 8.94 (s, 1H), 8.53 (s, 1H), 8.18 (d, J = 4.8 Hz, 2H), 7.75 - 7.59 (m, 5H), 7.50 - 7.40 (m, 4H), 5.10 (dd, J = 5.6, 12.8 Hz, 1H), 4.55 (d, J = 6.0 Hz, 2H), 3.70 - 3.61 (m, 3H), 3.28 - 3.17 (m, 6H), 2.91 - 2.83 (m, 1H), 2.75 - 2.72 (m, 2H), 2.67 - 2.63 (m, 7H), 2.30 (s, 3H), 2.09 - 1.97 (m, 1H), 1.87 - 1.76 (m, 2H), 1.75 - 1.61 (m, 3H), 1.54 (s, 6H). Intermediate 57c-2 was processed in a similar manner to afford Compound 57-2. LCMS [M+1, M+2] = 938.5, 939.5; 1H NMR (400 MHz, DMSO-d6) δ 13.74 (s, 1H), 11.10 (s, 1H), 8.87 (s, 1H), 8.52 (s, 1H), 8.16 (d, J = 7.2 Hz, 2H), 7.73 - 7.67 (m, 4H), 7.66 - 7.62 (m, 1H), 7.48 - 7.42 (m, 4H), 5.10 (dd, J = 5.6, 12.8 Hz, 1H), 4.55 (d, J = 6.0 Hz, 2H), 3.67 (s, 2H), 3.25 - 3.18 (m, 8H), 2.92 - 2.85 (m, 1H), 2.78 - 2.71 (m, 2H), 2.68 - 2.60 (m, 8H), 2.30 (s, 3H), 2.06 - 1.99 (m, 1H), 1.82 - 1.80 (m, 2H), 1.68 - 1.64 (m, 1H), 1.54 (s, 6H). Example 58: Synthesis of Compound 58
Step A olin-5-yl]- 1,4-diazepane-1-carboxylate (58a) To a solution of 2-(2,6-dioxo-3-piperidyl)-5,6-difluoro-isoindoline-1,3-dione (500 mg, 1.70 mmol, 1 equiv.) in DMSO (10 mL) was added DIPEA (659 mg, 5.10 mmol, 888 μL, 3 equiv.) and tert-butyl-1,4-diazepane-1-carboxylate (340 mg, 1.70 mmol, 335 μL, 1 equiv.). The mixture was stirred at 80 °C for 4 h. The reaction mixture was quenched with water (100 mL) at 20 °C and extracted with ethyl acetate. The combined organic portions were washed with brine (10 mL x 3), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum to afford 58a which was used without further purification. LCMS [M+Na] = 497.1; 1H NMR (400 MHz, CHLOROFORM-d) δ 7.94 (br s, 1 H), 7.37 (d, J = 12.8 Hz, 1 H), 7.18 (s, 1 H), 4.85 (dd, J = 12.4, 5.6 Hz, 1 H), 3.60 – 3.54 (m, 4 H), 3.40 – 3.39 (m, 1 H), 3.33 - 3.31 (m, 1 H), 2.89 - 2.66 (m, 3 H), 2.12 - 2.01 (m, 1 H), 1.97 - 1.85 (m, 2 H), 1.67 - 1.45 (m, 2 H), 1.40 - 1.32 (m, 9 H). Step B – Synthesis of 1-(4'-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)- 1,4-diazepan-1-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan- 2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide (Compound 58) To a solution of 58a (450 mg, 948 μmol, 1 equiv.) in dichloromethane (10 mL) was added TFA (1.08 g, 9.48 mmol, 704 μL, 10 equiv.). The mixture was stirred at 25 °C for 16 h. The reaction mixture was quenched with saturated aqueous NaHCO3 (20 mL) at 20 °C and extracted with dichloromethane (50 mL x 2). The combined organic layers were washed with brine (50
mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure to afford a residue (LCMS [M+1] = 375.1). The residue (113.1 mg, 275.3 μmol, 1.4 equiv) in THF (1 mL) was treated with triethylamine (82 mg, 806 μmol, 112 μL, 4.0 equiv.), AcOH (72.6 mg, 1.2 mmol, 69.2 μL, 6.0 equiv.) and Int 55 (100.0 mg, 201.4 μmol, 1.0 equiv.). The mixture was stirred at 50°C for 12 h, and then treated with NaBH(OAc)3 (85.4 mg, 402.8 μmol, 2.0 equiv.) and stirred at 50°C for 1 h. The mixture was cooled to 20°C and poured into water (10 mL), the aqueous phase was extracted with ethyl acetate (10 mL x 3). The combined organic phase was washed with brine (10 mL x 3), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum. The residue was purified by preparative HPLC (Phenomenex Luna C18, 150 mm x 25 mm, 10 µm; [water(FA)-ACN];) to afford Compound 58. LCMS [M+1] = 855.3; 1H NMR (400 MHz, DMSO-d6) δ 13.92 (br s, 1H), 11.09 (s, 1H), 8.92 (br s, 1H), 8.52 (s, 1H), 8.16 (d, J = 6.8 Hz, 1H), 7.73 - 7.60 (m, 5H), 7.46 (d, J = 8.4 Hz, 1H), 7.39 (d, J = 8.0 Hz, 2H), 7.29 (d, J = 7.6 Hz, 1H), 5.08 (dd, J = 5.2, 12.8 Hz, 1H), 4.55 (d, J= 5.6 Hz, 2H), 3.69 (s, 2H), 3.66 - 3.63 (m, 4H), 2.93 - 2.83 (m, 1H), 2.81 - 2.78 (m, 2H), 2.69 - 2.66 (m, 2H), 2.62 - 2.52 (m, 2H), 2.30 (s, 3H), 2.05 - 1.98 (m, 1H), 1.93 - 1.91 (m, 2H), 1.54 (s, 6H). Example 59: Synthesis of Compound 59
diazabicyclo[3.2.1]octane-3-carboxylate (59a) A mixture of tert-butyl 3,8-diazabicyclo[3.2.1]octane-3-carboxylate (1.5 g, 7.0 mmol, 1.0 equiv.) and benzyl 4-oxopiperidine-1-carboxylate (1.6 g, 7.0 mmol, 1.4 mL, 1.0 equiv.) in DCE (15 mL) was treated with acetic acid (848.6 mg, 14.1 mmol, 808.9 μL, 2.0 equiv.)and stirred for 30 min at. The mixture was treated with NaBH(OAc)3 (4.4 g, 21.2 mmol, 3.0 equiv.) and stirred at for 2 h at 25°C. The reaction mixture was quenched with aqueous.NaHCO3 (5 mL) at 25°C and extracted with dichloromethane (3 mL x 3), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether/ethyl acetate=10/1 to 3/1) to afford 59a. LCMS: [M+1] = 430.2 Step B – Synthesis of tert-butyl 8-(4-piperidyl)-3,8-diazabicyclo[3.2.1]octane-3-carboxylate (59b) To a suspension of Pd/C (1.0 g, 939.6 μmol, 10 wt% purity, 0.2 equiv.) in methanol (5 mL) under a nitrogen atmosphere was added 59a (2.0 g, 4.6 mmol, 1.0 equiv.). The mixture was degassed and purged three-fold with hydrogen and stirred at 40°C under hydrogen (15 psi) for 2
h. The reaction mixture was filtered through diatomaceous earth and the filtrate was concentrated under reduced pressure to afford 59b. LCMS: [M+1] = 296.2; 1H NMR (400 MHz, DMSO-d6) δ 3.56 - 3.35 (m, 4H), 3.00 - 2.79 (m, 5H), 2.44 - 2.34 (m, 3H), 1.75 - 1.63 (m, 5H), 1.38 (s, 9H), 1.23 - 1.02 (m, 3H). Step C – Synthesis of tert-butyl 8-[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]-4-piperidyl]-3,8-diazabicyclo[3.2.1]octane-3-carboxylate (59c) To a mixture of 59b (150 mg, 507.7 μmol, 1.0 equiv.) and 2-(2,6-dioxo-3-piperidyl)-5,6- difluoro-isoindoline-1,3-dione (179.2 mg, 609.3 μmol, 1.2 equiv.) in DMSO (2 mL), was added DIEA (328.1 mg, 2.5 mmol, 442.2 μL, 5.0 equiv.). The mixture was stirred at 70°C for 16 h. The reaction mixture was cooled to ambient temperature, quenched with water (10 mL) and extracted with ethyl acetate (15 mL x 3). The combined organic layers were washed with brine (15 mL x 3), dried over sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative TLC (silica gel, ethyl acetate: MeOH = 10:1) to afford 59c. LCMS: [M+1] = 570.4. 1H NMR (400 MHz, DMSO-d6) δ 11.10 (s, 1H), 7.70 (d, J = 11.6 Hz, 1H), 7.43 (d, J = 7.6 Hz, 1H), 5.10 (dd, J = 5.2, 12.8 Hz, 1H), 3.65 - 3.40 (m, 6H), 3.06 - 2.82 (m, 5H), 2.64 - 2.52 (m, 2H), 2.48 - 2.40 (m, 1H), 2.07 - 2.00 (m, 1H), 1.95 (s, 1H), 1.81 - 1.72 (m, 2H), 1.55 - 1.42 (m, 4H), 1.39 (s, 9H). Step D – Synthesis of 5-[4-(3,8-diazabicyclo[3.2.1]octan-8-yl)-1-piperidyl]-2-(2,6-dioxo-3- piperidyl)-6-fluoro-isoindoline-1,3-dione (59d) A solution of 59c (70.0 mg, 122.8 μmol, 1 equiv.) in chlorotrimethylsilane (0.1 mL) and trifluoroethanol (0.9 mL) was stirred at 25°C for 30 min. The reaction mixture was concentrated under reduced pressure to afford 59c, which was used without further purification. LCMS: [M+1] =470.4. Step E – Synthesis of 1-[4-[4-[[8-[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]-4-piperidyl]-3,8-diazabicyclo[3.2.1]octan-3-yl]methyl]phenyl]-2-methyl-phenyl]-N-[[3- (2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Compound 59)
A mixture of 59d (41.6 mg, 71.3 μmol, 1.1 equiv.) and Int 55 (40.0 mg, 80.57 μmol, 1.1 equiv.) in DCE (0.5 mL) was treated with AcOH (9.6 mg, 161.1 μmol, 9.2 μL, 2.25 equiv.) and stirred at 25°C for 30 mins. The mixture was treated with NaBH(OAc)3 (51.2 mg, 241.7 μmol, 3.4 equiv.) and stirred at 25°C for 2 h, quenched with water (5 mL) at 25°C and extracted with ethyl acetate (3 mL x 3). The combined organic layers were washed with brine ( 3 mL x 3), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated. The residue was purified by preparative HPLC (Waters Xbridge C18150 mm x 50mm, 10 μm, [water(10mM NH4HCO3)-ACN]) to afford Compound 59. LCMS: [M+1] = 950.5; 1H NMR (400 MHz, DMSO- d6) δ 13.90 (br s, 1H), 11.10 (s, 1H), 8.89 (br s, 1H), 8.52 (s, 1H), 8.17 (s, 1H), 7.75 - 7.60 (m, 5H), 7.49 - 7.36 (m, 4H), 5.10 (dd, J = 6.0, 12.8 Hz, 1H), 4.56 (s, 2H), 3.66 - 3.57 (m, 2H), 3.50 (s, 2H), 3.42 (s, 2H), 2.99 - 2.82 (m, 3H), 2.31 - 2.24 (m, 5H), 2.08 - 1.90 (m, 4H), 1.82 - 1.69 (m, 4H), 1.54 (s, 6H), 1.48 - 1.34 (m, 1H), 1.23 (s, 3H). Example 60: Synthesis of Compound 60
 Compound 60 was prepared in a similar manner to Example 59 by substituting tert-butyl 3,6-diazabicyclo[3.1.1]heptane-6-carboxylate in Step A to afford 1-(4'-((3-(1-(2-(2,6- dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperidin-4-yl)-3,6- diazabicyclo[3.1.1]heptan-6-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide. LCMS: [M+1] = 936.3; 1H NMR (400 MHz, METHANOL-d4) δ = 8.38 (s, 1H), 8.19 (s, 1H), 7.86 - 7.81 (m, 2H), 7.79 - 7.68 (m, 3H), 7.65 (dd, J = 2.0, 8.4 Hz, 1H), 7.60 - 7.45 (m, 3H), 5.09 (dd, J = 5.6, 12.4 Hz, 1H), 4.77 - 4.69 (m, 3H), 4.57 - 4.48 (m, 2H), 4.11 - 3.91 (m, 3H), 3.81 (br d, J =
12.4 Hz, 3H), 3.67 - 3.53 (m, 2H), 3.02 (br t, J = 12.0 Hz, 2H), 2.93 - 2.63 (m, 4H), 2.60 - 2.45 (m, 1H), 2.37 - 2.25 (m, 5H), 2.17 - 2.07 (m, 1H), 2.05 - 1.92 (m, 2H), 1.63 (s, 6H). Example 61: Synthesis of Compound 61
Compound 60 was prepared in a similar manner to Example 59 by substituting tert-butyl 3,6-diazabicyclo[3.1.1]heptane-6-carboxylate in Step A to afford 1-(4'-((3-(1-(2-(2,6- dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperidin-4-yl)-3,6- diazabicyclo[3.1.1]heptan-6-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide. LCMS: [M+1] = 936.3; 1H NMR (400 MHz, METHANOL-d4) δ = 8.38 (s, 1H), 8.19 (s, 1H), 7.86 - 7.81 (m, 2H), 7.79 - 7.68 (m, 3H), 7.65 (dd, J = 2.0, 8.4 Hz, 1H), 7.60 - 7.45 (m, 3H), 5.09 (dd, J = 5.6, 12.4 Hz, 1H), 4.77 - 4.69 (m, 3H), 4.57 - 4.48 (m, 2H), 4.11 - 3.91 (m, 3H), 3.81 (br d, J =
12.4 Hz, 3H), 3.67 - 3.53 (m, 2H), 3.02 (br t, J = 12.0 Hz, 2H), 2.93 - 2.63 (m, 4H), 2.60 - 2.45 (m, 1H), 2.37 - 2.25 (m, 5H), 2.17 - 2.07 (m, 1H), 2.05 - 1.92 (m, 2H), 1.63 (s, 6H). Example 61: Synthesis of Compound 61
 tert-butyl (1S,4S)-2,5-diazabicyclo[2.2.1]heptane-2-carboxylate in Step A to afford 1-(4'-(((1S,4S)-5-(1-(2- (2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperidin-4-yl)-2,5- diazabicyclo[2.2.1]heptan-2-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide. LCMS:[M+1] = 936.4; 1H NMR (400 MHz, DMSO-d6) δ = 13.89 (br s, 1H), 11.09 (s, 1H), 8.89 (br s, 1H), 8.52 (s, 1H), 8.17 (s, 1H), 7.76 - 7.61 (m, 5H), 7.55 - 7.41 (m, 4H), 5.17 - 5.04 (m, 1H), 4.55 ( d, J = 4.0 Hz, 2H), 3.81 - 3.50 (m, 6H), 3.08 - 2.82 (m, 5H), 2.64 - 2.57 (m, 4H), 2.30 (s, 3H), 2.12 - 1.92 (m, 3H), 1.54 (s, 6H), 1.30 - 1.20 (m, 5H). Example 62: Synthesis of Compound 62
tert-butyl (1S,4S)-2,5-diazabicyclo[2.2.1]heptane-2-carboxylate in Step A to afford 1-(4'-(((1S,4S)-5-(1-(2- (2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperidin-4-yl)-2,5- diazabicyclo[2.2.1]heptan-2-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide. LCMS:[M+1] = 936.4; 1H NMR (400 MHz, DMSO-d6) δ = 13.89 (br s, 1H), 11.09 (s, 1H), 8.89 (br s, 1H), 8.52 (s, 1H), 8.17 (s, 1H), 7.76 - 7.61 (m, 5H), 7.55 - 7.41 (m, 4H), 5.17 - 5.04 (m, 1H), 4.55 ( d, J = 4.0 Hz, 2H), 3.81 - 3.50 (m, 6H), 3.08 - 2.82 (m, 5H), 2.64 - 2.57 (m, 4H), 2.30 (s, 3H), 2.12 - 1.92 (m, 3H), 1.54 (s, 6H), 1.30 - 1.20 (m, 5H). Example 62: Synthesis of Compound 62
mple 59 by substituting tert-butyl (1R,4R)-2,5-diazabicyclo[2.2.1]heptane-2-carboxylate in Step A to afford 1-(4'-(((1R,4R)-5-(1- (2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperidin-4-yl)-2,5- diazabicyclo[2.2.1]heptan-2-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide. LCMS: [M+1] = 936.2; 1H NMR (400 MHz, DMSO-d6) δ 13.89 (br s, 1H), 12.21(br s, 1H), 11.45 (br s, 1H), 11.11 (s, 1H), 8.90 (br s, 1H), 8.56 (s, 1H), 8.19 (s, 1H), 7.85 (s, 3H), 7.80 - 7.73 (m, 2H), 7.70 (d, J = 8.4 Hz, 1H), 7.50 (d, J = 8.0 Hz, 2H), 5.11 (dd, J = 5.2, 12.4 Hz, 1H), 4.85 - 4.29 (m, 6H), 4.26 - 3.91 (m, 2H), 3.85 - 3.61 (m, 4H), 2.99 - 2.76 (m, 4H), 2.64 - 2.52 (m, 3H), 2.32 (s, 4H), 2.23 - 1.80 (m, 5H), 1.54 (s, 6H) Example 63: Synthesis of Compound 63
Compound 63 was prepared in a similar manner to Example 59 by substituting tert-butyl 3,8-diazabicyclo[3.2.1]octane-8-carboxylate in Step A to afford 1-(4'-((3-(1-(2-(2,6- dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperidin-4-yl)-3,8- diazabicyclo[3.2.1]octan-8-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide . LCMS [M+1] = 950.6; 1H NMR (400 MHz, DMSO-d6) δ = 13.90 (br s, 1H), 11.10 (s, 1H), 8.92 (br s, 1H), 8.52 (s, 1H), 8.17 (s, 1H), 7.72 - 7.63 (m, 5H), 7.49 - 7.43 (m, 4H), 5.10 (dd, J = 5.4, 12.8 Hz, 1H), 4.56 - 4.55 (br s, 2H), 3.60 (d, J = 12.0 Hz, 2H), 3.53 (s, 2H), 3.11 (s, 2H), 2.95 - 2.84 (m, 3H), 2.63 - 2.56 (m, 4H), 2.40 - 2.36 (m, 3H), 2.30 (s, 3H), 2.04 - 2.01 (m, 1H), 1.91 - 1.83 (m, 4H), 1.71 - 1.70 (m, 2H), 1.60 - 1.58 (m, 2H), 1.54 (s, 6H). Example 64: Synthesis of Compound 64
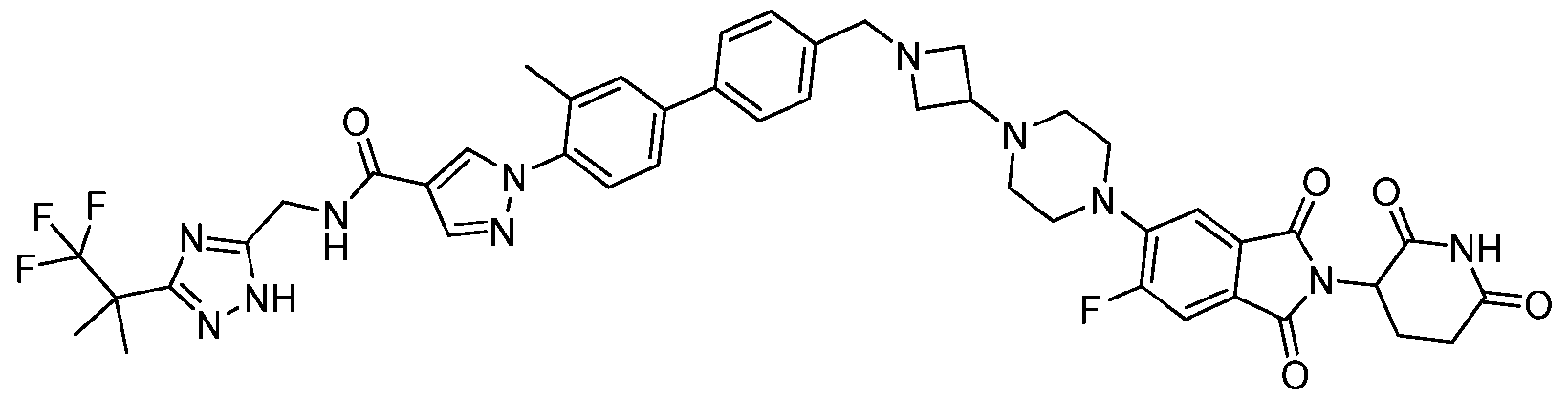 butyl 3-(piperazin-1-yl)azetidine-1-carboxylate in Step C to afford 1-(4'-((3-(4-(2-(2,6-dioxopiperidin- 3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1-yl)azetidin-1-yl)methyl)-3-methyl-[1,1'- biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H- pyrazole-4-carboxamide. LCMS [M+1] = 896.5; 1H NMR (400 MHz, MeOD) δ ppm 8.40 (s, 1H), 8.20 (s, 1H), 7.84 (d, J = 8.4 Hz, 2H), 7.73 - 7.57 (m, 6H), 7.48 (d, J = 8.4 Hz, 1H), 5.12 (dd, J = 5.6, 12.4 Hz, 1H), 4.83 - 4.77 (m, 2H), 4.75 (s, 3H), 4.73 (s, 1H), 4.54 - 4.45 (t, 2H), 4.23 (s, 1H), 3.63 (s, 4H), 3.30 - 3.24 (m, 4H), 2.88 - 2.82 (m, 1H), 2.77 - 2.69 (m, 2H), 2.33 (s, 3H), 2.15 - 2.11 (m, 1H), 1.65 (s, 6H). Example 65: Synthesis of Compound 65
butyl 3-(piperazin-1-yl)azetidine-1-carboxylate in Step C to afford 1-(4'-((3-(4-(2-(2,6-dioxopiperidin- 3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1-yl)azetidin-1-yl)methyl)-3-methyl-[1,1'- biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H- pyrazole-4-carboxamide. LCMS [M+1] = 896.5; 1H NMR (400 MHz, MeOD) δ ppm 8.40 (s, 1H), 8.20 (s, 1H), 7.84 (d, J = 8.4 Hz, 2H), 7.73 - 7.57 (m, 6H), 7.48 (d, J = 8.4 Hz, 1H), 5.12 (dd, J = 5.6, 12.4 Hz, 1H), 4.83 - 4.77 (m, 2H), 4.75 (s, 3H), 4.73 (s, 1H), 4.54 - 4.45 (t, 2H), 4.23 (s, 1H), 3.63 (s, 4H), 3.30 - 3.24 (m, 4H), 2.88 - 2.82 (m, 1H), 2.77 - 2.69 (m, 2H), 2.33 (s, 3H), 2.15 - 2.11 (m, 1H), 1.65 (s, 6H). Example 65: Synthesis of Compound 65
olin-5- yl]-4-piperidyl]-2,6-diazaspiro[3.3]heptane-2-carboxylate (65a) To a solution of tert-butyl 6-(4-piperidyl)-2,6-diazaspiro[3.3]heptane-2-carboxylate (130 mg, 462 μmol, 1 equiv.) in DMSO (1.5 mL) was added DIEA (179.1 mg, 1.4 mmol, 241.4 μL, 3 equiv.) and 2-(2,6-dioxo-3-piperidyl)-5,6-difluoro-isoindoline-1,3-dione (203.8 mg, 693 μmol, 1.5 equiv.). The mixture was stirred at 70°C for 16 h, cooled to ambient temperature, poured into water (5 mL) and extracted with ethyl acetate (5 mL x 2). The combined organic phase was washed with brine (5 mL x 2), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum. The residue was purified by preparative TLC (silica gel, dichloromethane: MeOH = 10:1) to afford 65a. LCMS [M+1]=556.3;1H NMR (400 MHz, CHLOROFORM-d) δ 8.41 (br s, 1H), 7.44 (d, J = 11.0 Hz, 1H), 7.37 (d, J = 7.4 Hz, 1H), 4.92 (dd, J = 5.2, 12.3 Hz, 1H), 4.01 (s, 4H), 3.57 (br d, J = 12.4 Hz, 2H), 3.34 (br s, 3H), 2.97 - 2.85 (m, 3H), 2.84 - 2.71 (m, 2H), 2.18 - 2.07 (m, 1H), 1.87 - 1.67 (m, 6H), 1.56 - 1.47 (m, 1H), 1.43 (s, 9H). Step B – Synthesis of 5-[4-(2,6-diazaspiro[3.3]heptan-2-yl)-1-piperidyl]-2-(2,6-dioxo-3- piperidyl)-6-fluoro-isoindoline-1,3-dione (65b)
A solution of 65a (130 mg, 233.9 μmol, 1 equiv.) in TFA (0.2 mL) and dichloromethane (1 mL) was stirred at 20 °C for 1 h and then concentrated under reduced pressure to afford 65b, which was used without further purification. LCMS [M+1]=456.2. Step C – Synthesis of 1-[4-[4-[[2-[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]-4-piperidyl]-2,6-diazaspiro[3.3]heptan-6-yl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2- trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Compound 65) A solution of 65b (60 mg, 105 μmol, 1 equiv.) and Int 55 (65.4 mg, 132 μmol, 1.25 equiv.) in THF (1 mL) was treated with Ti(Oi-Pr)4 (300 mg, 1.1 mmol, 311 μL, 10.5 equiv.) and heated at 60°C for 1 h. The mixture was cooled to ambient temperature, treated with NaBH(OAc)3 (83.7 mg, 395 μmol, 3.8 equiv.), stirred at 60°C for 5 h, cooled to ambient temperature, poured into water (5 mL) and filtered. The filtrate was extracted with ethyl acetate (5 mL x 2). The combined organic phase was washed with brine (5 mL x 2), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum. The residue was purified by preparative HPLC (Phenomenex Luna C18, 100 mm x 30mm, 5 µm; [water (0.2% FA)-ACN]) to afford Compound 65. LCMS [M+1]=936.6; 1H NMR (400 MHz, METHANOL-d4) δ 8.36 (s, 1H), 8.18 (s, 1H), 7.72 - 7.65 (m, 3H), 7.61 (dd, J = 1.8, 8.0 Hz, 1H), 7.57 - 7.39 (m, 5H), 5.08 (dd, J = 5.4, 12.4 Hz, 1H), 4.70 (s, 2H), 3.78 (s, 2H), 3.66 (br d, J = 12.4 Hz, 2H), 3.59 - 3.45 (m, 8H), 2.97 - 2.64 (m, 5H), 2.44 - 2.35 (m, 1H), 2.31 (s, 3H), 2.17 - 2.06 (m, 1H), 1.98 - 1.83 (m, 2H), 1.61 (s, 6H), 1.51 - 1.34 (m, 2H). Example 66: Synthesis of Compound 66
O O Step A –
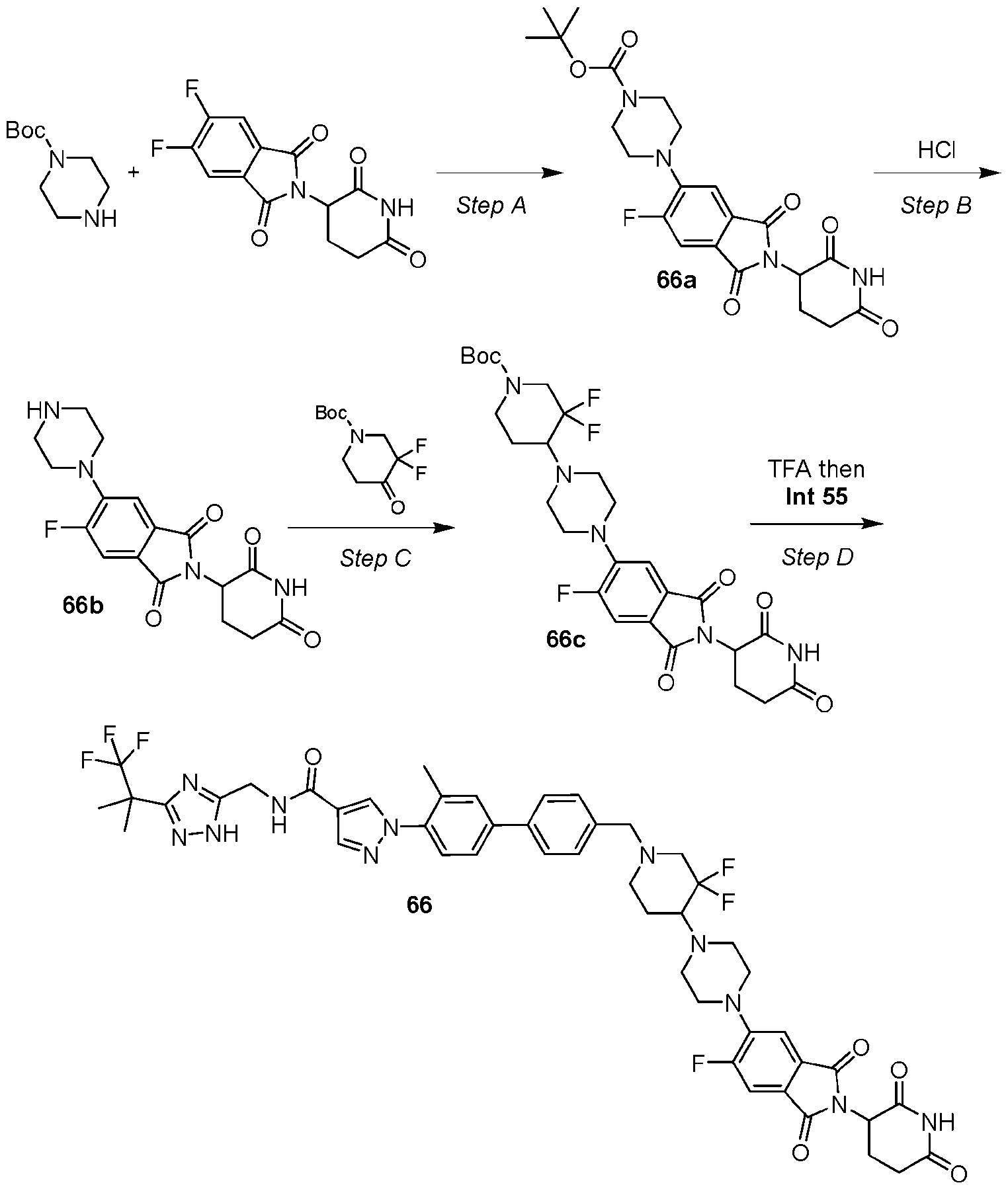 5- yl]piperazine-1-carboxylate (66a) A solution of tert-butyl piperazine-1-carboxylate (696.3 mg, 3.74 mmol, 1.1 equiv.) and 2- (2,6-dioxo-3-piperidyl)-5,6-difluoro-isoindoline-1,3-dione (1.00 g, 3.40 mmol, 1.0 equiv.) in NMP (5.0 mL) was treated with NaHCO3 (342.6 mg, 4.08 mmol, 1.2 equiv.). The mixture was stirred at 90°C for 16 h cooled to ambient temperature and treated with water (9.6 mL) and acetonitrile (0.4 mL). The mixture was stirred for 1 h. The solids were collected by vacuum filtration and the filter cake was rinsed with water (10 mL x 2) and dried to afford 66a.
LCMS [M-55] = 405; 1H NMR (400 MHz, CHLOROFORM-d) δ 7.96 (s, 1H), 7.51 (d, J = 10.8 Hz, 1H), 7.39 (d, J = 7.6 Hz, 1H), 4.94 (dd, J = 5.2, 12.0 Hz, 1H), 3.68 - 3.56 (m, 4H), 3.39 (t, J = 7.2 Hz, 1H), 3.28 - 3.17 (m, 4H), 2.95 - 2.87 (m, 1H), 2.82 - 2.72 (m, 1H), 2.20 - 2.11 (m, 1H), 2.08 - 1.97 (m, 1H), 1.50 (s, 9H). Step B – Synthesis of 2-(2,6-dioxo-3-piperidyl)-5-fluoro-6-piperazin-1-yl-isoindoline-1,3-dione (66b) A solution of 66a (2.0 g, 4.3 mmol, 1.0 equiv.) in HCl/ dioxane (4 M, 20 mL, 18.4 equiv.) was stirred at 20°C for 2 h. The reaction mixture was concentrated under reduced pressure afford 66b, which was used without further purification. LCMS [M+1] = 361.0; 1H NMR (400 MHz, DMSO-d6) δ 11.12 (s, 1H), 9.23 (br s, 2H), 7.80 (d, J = 11.2 Hz, 1H), 7.59 (d, J = 7.6 Hz, 1H), 5.12 (dd, J = 5.6, 13.2 Hz, 1H), 3.52 - 3.45 (m, 4H), 3.26 (s, 4H), 2.96 - 2.82 (m, 1H), 2.66 - 2.52 (m, 2H), 2.10 - 1.99 (m, 1H). Step C – Synthesis of tert-butyl 4-[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]piperazin-1-yl]-3,3-difluoro-piperidine-1-carboxylate (66c) A solution of 66b (500.0 mg, 1.26 mmol, 1.0 equiv) in toluene (8 mL) and acetonitrile (4 mL) was treated with sodium acetate (258.4 mg, 3.15 mmol, 2.5 equiv.), acetic acid (0.4 mL) and 4Å molecular sieves (500 mg), stirred for 15 min at ambient temperature, treated with tert-butyl 3,3-difluoro-4-oxo-piperidine-1-carboxylate (444.6 mg, 1.89 mmol, 1.5 equiv.) and stirred at 135°C for 12 h. The reaction mixture was cooled to room temperature, filtered through Celite and the filtrate was concentrated under vacuum. The residue was dissolved in methanol (2.5 mL) and DCE (2.5 mL) and treated with NaBH3CN (395.5 mg) and acetic acid (0.25 mL). The mixture was stirred at 40 °C for 16 h., cooled to ambient temperature, diluted with water (20 mL) and extracted with dichloromethane (15 mL x 3). The combined organic layers were washed with brine (30 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure The residue was purified by column chromatography (silica gel, petroleum ether/ ethyl acetate=1/0 to 0/1) to afford 66c. LCMS [M+1] = 580.3; 1H NMR (400 MHz, DMSO-d6) δ = 11.09 (s, 1H), 7.71 (d, J = 11.2 Hz, 1H), 7.43 (d, J = 7.2 Hz, 1H), 5.09 (dd, J = 5.2, 12.8 Hz, 1H), 4.16 - 3.93 (m, 2H), 3.23 - 3.17 (m, 4H), 3.16 - 3.00 (m, 2H), 2.93 - 2.78 (m, 6H), 2.62 - 2.58 (m, 1H), 2.57 - 2.54 (m, 1H), 2.07 - 1.98 (m, 1H), 1.85 - 1.77 (m, 1H), 1.76 - 1.63 (m, 1H), 1.39 (s, 9H).
Step D – Synthesis of 1-[4-[4-[[4-[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]piperazin-1-yl]-3,3-difluoro-1-piperidyl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2- trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Compound 66) To a solution of 66c (300.0 mg, 518 μmol, 1.0 equiv.) in dichloromethane (3 mL) was added TFA (921.0 mg, 8.08 mmol, 600.0 μL, 15.6 equiv.). The mixture was stirred at 20°C for 2 h and then concentrated under reduced pressure (LCMS [M+1] = 480.3). A solution of the resulting residue (71.7 mg, 120.8 μmol, 1.2 equiv) in THF (1.0 ml) was treated with Int 55 (50.0 mg, 100.7 μmol, 1.0 equiv.), Ti(i-PrO)4 (171.7 mg, 604.2 μmol, 178.3 μL, 6.0 equiv.) and NaBH(OAc)3 (85.3 mg, 402.8 μmol, 4.0 equiv.). The mixture was stirred at 60°C for 1 h. The reaction mixture was diluted with water (5 mL) and filtered. The filtrate was extracted with ethyl acetate (5 mL x 3). The combined organic layers were washed with brine (20 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative HPLC (Phenomenex Luna C18, 100mm x 40mm, 3 µm; [water(0.2% FA)-ACN]) to afford Compound 66. LCMS [M+1] = 960.6; 1H NMR (400 MHz, DMSO-d6) δ 13.92 (br s, 1H), 11.10 (s, 1H), 8.88 (br s, 1H), 8.53 (s, 1H), 8.17 (s, 1H), 7.79 - 7.61 (m, 5H), 7.44 (dd, J = 8.4, 18.8 Hz, 4H), 5.10 (dd, J = 5.6, 12.8 Hz, 1H), 4.55 (d, J = 5.2 Hz, 2H), 3.63 (s, 2H), 3.21 (d, J = 4.0 Hz, 4H), 3.08 - 2.98 (m, 1H), 2.90 (d, J = 4.8 Hz, 7H), 2.65 - 2.55 (m, 3H), 2.34 - 2.27 (m, 4H), 2.22 - 2.12 (m, 1H), 2.03 (d, J = 5.6 Hz, 1H), 1.82 (d, J = 4.4 Hz, 2H), 1.54 (s, 6H). Example 67: Synthesis of Compound 67
5- yl]piperazine-1-carboxylate (66a) A solution of tert-butyl piperazine-1-carboxylate (696.3 mg, 3.74 mmol, 1.1 equiv.) and 2- (2,6-dioxo-3-piperidyl)-5,6-difluoro-isoindoline-1,3-dione (1.00 g, 3.40 mmol, 1.0 equiv.) in NMP (5.0 mL) was treated with NaHCO3 (342.6 mg, 4.08 mmol, 1.2 equiv.). The mixture was stirred at 90°C for 16 h cooled to ambient temperature and treated with water (9.6 mL) and acetonitrile (0.4 mL). The mixture was stirred for 1 h. The solids were collected by vacuum filtration and the filter cake was rinsed with water (10 mL x 2) and dried to afford 66a.
LCMS [M-55] = 405; 1H NMR (400 MHz, CHLOROFORM-d) δ 7.96 (s, 1H), 7.51 (d, J = 10.8 Hz, 1H), 7.39 (d, J = 7.6 Hz, 1H), 4.94 (dd, J = 5.2, 12.0 Hz, 1H), 3.68 - 3.56 (m, 4H), 3.39 (t, J = 7.2 Hz, 1H), 3.28 - 3.17 (m, 4H), 2.95 - 2.87 (m, 1H), 2.82 - 2.72 (m, 1H), 2.20 - 2.11 (m, 1H), 2.08 - 1.97 (m, 1H), 1.50 (s, 9H). Step B – Synthesis of 2-(2,6-dioxo-3-piperidyl)-5-fluoro-6-piperazin-1-yl-isoindoline-1,3-dione (66b) A solution of 66a (2.0 g, 4.3 mmol, 1.0 equiv.) in HCl/ dioxane (4 M, 20 mL, 18.4 equiv.) was stirred at 20°C for 2 h. The reaction mixture was concentrated under reduced pressure afford 66b, which was used without further purification. LCMS [M+1] = 361.0; 1H NMR (400 MHz, DMSO-d6) δ 11.12 (s, 1H), 9.23 (br s, 2H), 7.80 (d, J = 11.2 Hz, 1H), 7.59 (d, J = 7.6 Hz, 1H), 5.12 (dd, J = 5.6, 13.2 Hz, 1H), 3.52 - 3.45 (m, 4H), 3.26 (s, 4H), 2.96 - 2.82 (m, 1H), 2.66 - 2.52 (m, 2H), 2.10 - 1.99 (m, 1H). Step C – Synthesis of tert-butyl 4-[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]piperazin-1-yl]-3,3-difluoro-piperidine-1-carboxylate (66c) A solution of 66b (500.0 mg, 1.26 mmol, 1.0 equiv) in toluene (8 mL) and acetonitrile (4 mL) was treated with sodium acetate (258.4 mg, 3.15 mmol, 2.5 equiv.), acetic acid (0.4 mL) and 4Å molecular sieves (500 mg), stirred for 15 min at ambient temperature, treated with tert-butyl 3,3-difluoro-4-oxo-piperidine-1-carboxylate (444.6 mg, 1.89 mmol, 1.5 equiv.) and stirred at 135°C for 12 h. The reaction mixture was cooled to room temperature, filtered through Celite and the filtrate was concentrated under vacuum. The residue was dissolved in methanol (2.5 mL) and DCE (2.5 mL) and treated with NaBH3CN (395.5 mg) and acetic acid (0.25 mL). The mixture was stirred at 40 °C for 16 h., cooled to ambient temperature, diluted with water (20 mL) and extracted with dichloromethane (15 mL x 3). The combined organic layers were washed with brine (30 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure The residue was purified by column chromatography (silica gel, petroleum ether/ ethyl acetate=1/0 to 0/1) to afford 66c. LCMS [M+1] = 580.3; 1H NMR (400 MHz, DMSO-d6) δ = 11.09 (s, 1H), 7.71 (d, J = 11.2 Hz, 1H), 7.43 (d, J = 7.2 Hz, 1H), 5.09 (dd, J = 5.2, 12.8 Hz, 1H), 4.16 - 3.93 (m, 2H), 3.23 - 3.17 (m, 4H), 3.16 - 3.00 (m, 2H), 2.93 - 2.78 (m, 6H), 2.62 - 2.58 (m, 1H), 2.57 - 2.54 (m, 1H), 2.07 - 1.98 (m, 1H), 1.85 - 1.77 (m, 1H), 1.76 - 1.63 (m, 1H), 1.39 (s, 9H).
Step D – Synthesis of 1-[4-[4-[[4-[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]piperazin-1-yl]-3,3-difluoro-1-piperidyl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2- trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Compound 66) To a solution of 66c (300.0 mg, 518 μmol, 1.0 equiv.) in dichloromethane (3 mL) was added TFA (921.0 mg, 8.08 mmol, 600.0 μL, 15.6 equiv.). The mixture was stirred at 20°C for 2 h and then concentrated under reduced pressure (LCMS [M+1] = 480.3). A solution of the resulting residue (71.7 mg, 120.8 μmol, 1.2 equiv) in THF (1.0 ml) was treated with Int 55 (50.0 mg, 100.7 μmol, 1.0 equiv.), Ti(i-PrO)4 (171.7 mg, 604.2 μmol, 178.3 μL, 6.0 equiv.) and NaBH(OAc)3 (85.3 mg, 402.8 μmol, 4.0 equiv.). The mixture was stirred at 60°C for 1 h. The reaction mixture was diluted with water (5 mL) and filtered. The filtrate was extracted with ethyl acetate (5 mL x 3). The combined organic layers were washed with brine (20 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative HPLC (Phenomenex Luna C18, 100mm x 40mm, 3 µm; [water(0.2% FA)-ACN]) to afford Compound 66. LCMS [M+1] = 960.6; 1H NMR (400 MHz, DMSO-d6) δ 13.92 (br s, 1H), 11.10 (s, 1H), 8.88 (br s, 1H), 8.53 (s, 1H), 8.17 (s, 1H), 7.79 - 7.61 (m, 5H), 7.44 (dd, J = 8.4, 18.8 Hz, 4H), 5.10 (dd, J = 5.6, 12.8 Hz, 1H), 4.55 (d, J = 5.2 Hz, 2H), 3.63 (s, 2H), 3.21 (d, J = 4.0 Hz, 4H), 3.08 - 2.98 (m, 1H), 2.90 (d, J = 4.8 Hz, 7H), 2.65 - 2.55 (m, 3H), 2.34 - 2.27 (m, 4H), 2.22 - 2.12 (m, 1H), 2.03 (d, J = 5.6 Hz, 1H), 1.82 (d, J = 4.4 Hz, 2H), 1.54 (s, 6H). Example 67: Synthesis of Compound 67
Step A – Synthesis of tert-butyl 4-(1-benzyloxycarbonyl-4-methyl-4-piperidyl)piperazine-1- carboxylate (67a) A solution of benzyl 4-oxopiperidine-1-carboxylate (500.0 mg, 2.1 mmol, 426.6 μL, 1.0 equiv.) in toluene (5.0 mL) was treated with tert-butyl piperazine-1-carboxylate (439.1 mg, 2.3 mmol, 1.1 equiv.) and 1,2,3-triazole (177.6 mg, 2.5 mmol, 1.2 equiv.) at 100oC for 12 h, cooled to ambient temperature and treated with MeMgBr (641.2 mg, 5.3 mmol, 3 M, 2.5 equiv.), the solution was stirred at 25oC for 3 h. The reaction mixture was diluted with water (10 mL) and extracted with dichloromethane (10 mL x 3). The combined organic layers were washed with brine (10.0 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether: Ethyl acetate = 1: 0 to 5: 1) afford 67a. LCMS [M+1] = 418.2; 1H NMR (400 MHz, DMSO-d6) δ ppm 7.35 - 7.28 (m, 5H), 5.05 (s, 2H), 3.49 (d, J = 12.8 Hz, 2H), 3.32 (s, 2H),
3.27 (s, 4H), 2.37 (s, 4H), 1.74 (d, J = 13.2 Hz, 2H), 1.40 - 1.36 (m, 9H), 1.34 – 1.25 (m, 2H), 0.83 (s, 3H). Step B – Synthesis of 5-(4-chloro-2-methyl-phenyl)-1-(2-methoxyethyl)-6-oxo-pyridine-3- carboxylic acid (67b) To a solution of 67a (200.0 mg, 479 μmol, 1.0 equiv.) in methanol (3.0 mL) was added Pd/C (101.9 mg, 10 wt%) under nitrogen. The suspension was degassed under vacuum and purged with hydrogen. The mixture was stirred under hydrogen (15 Psi) at ambient temperature for 3 h. The mixture was filtered through a pad of Celite and the filter bed was washed with methanol (5 mL x 3). The filtrate was concentrated to afford 67b, which was used in the next step without further purification. LCMS [M+1] = 284.2; 1H NMR (400 MHz, DMSO-d6) δ ppm 3.27 (s, 4H), 2.85 - 2.73 (m, 4H), 2.39 (m, 4H), 1.99 - 1.97 (br s, 1H), 1.59 - 1.51 (m, 2H), 1.41 - 1.38 (m, 9H), 1.35 - 1.28 (m, 2H), 0.83 (s, 3H). Step C – Synthesis of tert-butyl 4-[4-methyl-1-[[4-[3-methyl-4-[4-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methylcarbamoyl]pyrazol-1-yl]phenyl]phenyl]methyl]-4- piperidyl]piperazine-1-carboxylate (67c) A solution of 67b (59.9 mg, 211 μmol, 3.0 equiv.) in DCE (1.0 mL) was treated with triethylamine (14.2 mg, 141 μmol, 19.6 μL, 2.0 equiv.), Int 55 (35.0 mg, 70.5 μmol, 1.0 equiv.) and NaBH(OAc)3 (44.8 mg, 211 μmol, 3.0 equiv.). The reaction mixture was stirred at ambient temperature for 30 min and then diluted with saturated aqueous sodium thiosulfate (10 mL). The mixture was extracted with ethyl acetate (10 mL x 3). The combined organic layers were washed with water (10 mL x 2), brine (1 mL) dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, ethyl acetate: methanol = 1: 0 to 5: 1) to afford 67c. LCMS [M+1] = 764.2; 1H NMR (400 MHz, DMSO-d6) δ ppm 9.04 (br s, 1H), 8.56 - 8.53 (m, 1H), 8.17 (s, 1H), 7.74 - 7.71 (m, 1H), 7.69 - 7.61 (m, 3H), 7.48 - 7.38 (m, 3H), 4.56 (d, J = 4.4 Hz, 2H), 3.50 (s, 2H), 2.39 (s, 2H), 2.30 (s, 4H), 1.81 (d, J = 1.2 Hz, 3H), 1.77 (s, 6H), 1.72 - 1.63 (m, 2H), 1.54 (s, 6H), 1.49 - 1.42 (m, 2H), 1.39 (s, 9H), 0.84 (s, 3H).
Step D – Synthesis of 1-[4-[4-[[4-[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]piperazin-1-yl]-4-methyl-1-piperidyl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro- 1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Compound 67) A solution of 67c (70.0 mg, 92 μmol, 1.0 equiv.) in HCl/ MeOH (3M, 1.0 mL) was stirred at ambient temperature for 12 h. The reaction was filtered and concentrated under reduced pressure to give a residue. LCMS [M +1] = 664.4. The residue was dissolved in DMSO (1.0 mL) and treated with 2-(2,6-dioxo-3-piperidyl)-5,6-difluoro-isoindoline-1,3-dione (40.6 mg, 138 μmol, 1.5 equiv.) and DIEA (119 mg, 0.92 mmol, 160 μL, 10.0 equiv.), the solution was stirred at 100oC for 12 h. The reaction mixture was cooled to ambient temperature and diluted with water (10.0 mL) and extracted with dichloromethane (10 mL x 3). The combined organic layers were washed with brine (10 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative HPLC (Waters X bridge BEH, C18, 100 mm x 30 mm , 10 µm; [water (10 mM NH4HCO3) - ACN]) to afford Compound 67. LCMS [M+1] = 938.5; 1H NMR (400 MHz, METHANOL-d4) δ ppm 8.36 (s, 1H), 8.17 (s, 1H), 7.69 - 7.62 (m, 3H), 7.56 (s, 1H), 7.54 - 7.44 (m, 3H), 7.40 (m, 2H), 5.08 (dd, J = 5.2, 12.8 Hz, 1H), 4.69 (s, 2H), 3.71 (s, 2H), 3.27 - 3.13 (m, 4H), 2.93 - 2.62 (m, 10H), 2.49 (s, 2H), 2.27 (s, 3H), 2.08 (m, 1H), 1.96 - 1.86 (m, 2H), 1.63 - 1.60 (s, 6H), 1.57 (s, 1H), 0.99 (s, 3H) Example 68: Synthesis of Compound 68
[3.3]heptane-2-carboxylate (68a) A solution of tert-butyl 2,6-diazaspiro[3.3]heptane-2-carboxylate (3.00 g, 10.4 mmol, 1 equiv.) in methanol (100 mL) was treated with benzyl 4-oxopiperidine-1-carboxylate (4.80 g, 20.8 mmol, 4.1 mL, 2 equiv.) and acetic acid (1.20 g, 20.8 mmol, 1.2 mL, 2 equiv.). The solution was stirred for 1 h and then treated with NaBH3CN (784.7 mg, 12.4 mmol, 1.2 equiv.). The mixture was stirred at 20°C for 16 h. The mixture was poured into 5% aqueous K2CO3 (100mL) and methanol was removed under reduced pressure. The resulting aqueous was extracted with ethyl acetate (100 mL x 2). and the combined organic phase was washed with brine (50 mL x 2), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum. The residue was purified by preparative HPLC (Waters Xbridge C18, 150mm x 50mm, 10 µm; [water (0.05% NH3water+10mM NH4HCO3)-ACN]) to afford 68a. LCMS [M+1] =416.2; 1H NMR (400 MHz, DMSO-d6) δ 7.43 - 7.26 (m, 5H), 5.05 (s, 2H), 3.86 (s, 4H), 3.80 - 3.72 (m, 2H), 3.17 (s, 4H), 2.94 (br d, J = 0.8 Hz, 2H), 2.17 - 2.02 (m, 1H), 1.56 (br dd, J = 4.0, 9.2 Hz, 2H), 1.35 (s, 9H), 1.08 - 0.95 (m, 2H) Step B – Synthesis of tert-butyl 6-(4-piperidyl)-2,6-diazaspiro[3.3]heptane-2-carboxylate (68b)
A mixture of 68a (600 mg, 1.44 mmol, 1 equiv.), Pd/C (10 wt%, 600 mg, 564 μmol, 0.39 equiv.) in trifluoroethanol (12 mL) was degassed and purged threefold with hydrogen, and then the mixture was stirred at 30°C for 2 h under a hydrogen atmosphere. The mixture was filtered and the filtrate was concentrated under reduced pressure to give 68b. LCMS [M+1]=282.2; 1H NMR (400 MHz, DMSO-d6) δ 6.08 (br s, 1H), 3.86 (s, 4H), 3.13 (s, 4H), 2.87 (td, J = 3.4, 12.4 Hz, 2H), 2.44 - 2.32 (m, 2H), 1.96 - 1.85 (m, 1H), 1.56 - 1.46 (m, 2H), 1.35 (s, 9H), 1.03 - 0.89 (m, 2H) Step C – Synthesis of tert-butyl 6-[1-[[4-[3-methyl-4-[4-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H-1,2,4-triazol-5-yl]methylcarbamoy l]pyrazol-1-yl]phenyl]phenyl]methyl]-4-piperidyl]-2,6- diazaspiro[3.3]heptane-2-carboxylate (68c) To a solution of 68b (85.0 mg, 302.1 μmol, 1 equiv.) and Int 55 (150 mg, 302.1 μmol, 1 equiv.) in THF (4 mL) was added Ti(Oi-Pr)4 (686.9 mg, 2.4 mmol, 713.3 μL, 8 equiv.) at 60°C for 1 h. The mixture was cooled to 20°C and treated with NaBH(OAc)3 (192.1 mg, 906.3 μmol, 3 equiv.), The mixture was stirred at 60°C for 5 h. The reaction mixture was poured into water (5 mL) and extracted with ethyl acetate (10 mL x 2). The combined organic portions were washed with brine (5 mL x 2), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated in vacuum. The residue was purified by preparative TLC (silica gel, petroleum ether: ethyl acetate = 0:1) to afford 68c. LCMS [M+1] =762.5. Step D – Synthesis of 1-[4-[4-[[4-[6-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]-2,6-diazaspiro[3.3]heptan-2-yl]-1-piperidyl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2- trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Compound 68) A mixture of 68c (110 mg, 144.5 μmol, 1 equiv.) in TFA (0.2 mL) and dichloromethane (1 mL) was stirred at 20°C for 1 h. The reaction mixture was concentrated under reduced pressure to afford a residue (LCMS [M+1] =662.6). A portion of this residue (95 mg) in DMSO (1 mL) was treated with DIEA (56 mg) and 2-(2,6-dioxo-3-piperidyl)-5,6-difluoro-isoindoline-1,3-dione (63 mg). The mixture was stirred at 70°C for 16 h. The reaction mixture was concentrated under reduced pressure to give an intermediate. The residue was purified by preparative-HPLC (Phenomenex Luna C18, 100 mm x 30 mm, 5 µm; [water(0.2% FA)-ACN]) to afford Compound
68. LCMS [M+1] =936.2. 1H NMR (400 MHz, DMSO-d6) δ 13.92 (br s, 1H), 11.08 (s, 1H), 8.87 (br s, 1H), 8.16 (d, J = 7.8 Hz, 2H), 7.74 - 7.56 (m, 5H), 7.46 (d, J = 8.1 Hz, 1H), 7.40 (d, J = 8.4 Hz, 2H), 6.92 (d, J = 7.6 Hz, 1H), 5.06 (dd, J = 5.4, 12.8 Hz, 1H), 4.55 (br d, J = 5.6 Hz, 2H), 4.23 (s, 4H), 3.50 (s, 2H), 3.29 (br s, 4H), 2.93 - 2.81 (m, 1H), 2.74 (br d, J = 10.0 Hz, 2H), 2.63 - 2.54 (m, 2H), 2.30 (s, 3H), 2.00 (br t, J = 8.9 Hz, 4H), 1.61 (br d, J = 10.0 Hz, 2H), 1.54 (s, 6H), 1.25 - 1.10 (m, 2H) Example 69: Synthesis of Compound 69-1 and 69-2
 Step A – Synthesis of tert-butyl 4-((2S)-1-((benzyloxy) carbonyl)-2-methylpiperidin-4-yl) piperazine-1-carboxylate (69a) To a solution of tert-butyl piperazine-1-carboxylate (2.20 g, 12.1 mmol, 1.5 equiv.) in DCE (20.0 mL) was added benzyl (S)-2-methyl-4-oxopiperidine-1-carboxylate (2.00 g, 8.0 mmol, 1.0 equiv.) and AcOH (2.0 mL) at 20°C for 16 h. The mixture was treated with NaBH(OAc)3 (5.1 g, 24.2 mmol, 3.0 equiv.) and stirred at 20°C for 2 h. tert - Butyl piperazine-1-carboxylate (2.20 g, 12.1 mmol, 1.5 equiv.) was added and the mixture was stirred at 20°C for 1 h, poured into a mixture of ice and water (20 mL). The aqueous was extracted with dichloromethane (15 mL x 3).
The combined organic phase was washed with brine (10 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum. The residue was purified by column chromatography (silica gel, petroleum ether: ethyl acetate = 1: 0 to 0: 1) to afford 69a. 1H NMR (400 MHz, MeOD) δ ppm 7.36 - 7.29 (m, 5H), 5.13 – 5.09 (m, 2H), 3.98 (td, J = 6.4, 8.8 Hz, 1H), 3.85 - 3.77 (m, 1H), 3.44 (t, J = 5.6 Hz, 4H), 3.27 - 3.20 (m, 1H), 2.68 - 2.41 (m, 5H), 1.95 - 1.85 (m, 2H), 1.71 - 1.59 (m, 2H), 1.45 (s, 9H), 1.27 (s, 1H), 1.26 (d, J = 1.6 Hz, 2H). Step B – Synthesis of tert-butyl 4-((2S)-2-methylpiperidin-4-yl) piperazine-1-carboxylate (69b) A solution of 69a (3.20 g, 7.6 mmol, 1.0 equiv.) in MeOH (40.0 mL) was added to a suspension of MeOH (20.0 mL) in Pd/C (3.00 g, 10 wt%.) under Ar. The mixture was stirred at 20°C for 4 h under hydrogen (1 atm). The solution was filtered and the filter cake was washed with MeOH (200 mL x 3). The combined filtrate was concentrated under vacuum to afford 69b. 1H NMR (400 MHz, MeOD) δ 3.42 (t, J = 4.4 Hz, 4H), 3.27 - 3.21 (m, 1H), 2.98 - 2.88 (m, 1H), 2.85 - 2.76 (m, 1H), 2.58 - 2.52 (m, 4H), 2.47 (t, J = 5.2 Hz, 1H), 2.04 - 1.93 (m, 2H), 1.45 (s, 9H), 1.30 - 1.21 (m, 5H). Step C – Synthesis of tert-butyl 4-((2S,4S)-2-methyl-1-((3'-methyl-4'-(4-(((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)carbamoyl)-1H-pyrazol-1-yl)-[1,1'-biphenyl]-4- yl)methyl)piperidin-4-yl)piperazine-1-carboxylate (69c-1) and tert-butyl 4-[(2S,4R)-2-methyl-1- [[4-[3-methyl-4-[4-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methylcarbamoyl]pyrazol-1-yl]phenyl]phenyl]methyl]-4-piperidyl]piperazine-1-carboxylate (69c-2) To a solution of Int 55 (200.0 mg, 403 μmol, 1.0 equiv.) and 69b (285.4 mg, 1.0 mmol, 2.5 equiv.) in DCE (4.0 mL) was added tetraisopropoxytitanium (343.4 mg, 1.20 mmol, 3.0 equiv.) at 20°C. The mixture was stirred at 20°C for 16 h, treated with NaBH3CN (139.2 mg, 2.20 mmol, 5.5 equiv.) and MeOH (2.0 mL) was added to the reaction solution stirred at 20°C for 2 h. The residue was poured into water (0.1 mL) and filtered to remove the solids. The filtrate was concentrated under vacuum. The residue was purified by preparative HPLC (Phenomenex Gemini NX C18 (75 mm x 30 mm, 3 µm); [water (0.05% NH3•water 10 mM NH4HCO3) - ACN];) to afford 69c-1 as the early-eluting isomer and 69c-2 as the late-eluting isomer. LCMS [M+1] = 764.6.
Step D – Synthesis of 1-(4'-(((2S,4S)-4-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3- dioxoisoindolin-5-yl)piperazin-1-yl)-2-methylpiperidin-1-yl)methyl)-3-methyl-[1,1'-biphenyl]-4- yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4- carboxamide (Compound 69-1) A solution of 69c-1 (95.0 mg, 124.3 μmol, 1.0 equiv.) in HCl/MeOH (3 M, 1.0 mL) was stirred at 20°C for 1 h and then concentrated under vacuum (LCMS [M+1] = 664.4). The residue was dissolved in DMSO (1.0 mL) and treated with 2-(2,6-dioxopiperidin-3-yl)-5,6- difluoroisoindoline-1,3-dione (53.6 mg, 182 μmol, 1.5 equiv.) and DIEA (157.1 mg, 1.2 mmol, 211 μL, 10.0 equiv.). The mixture was stirred at 100°C for 16 h, cooled to ambient temperature and purified directly by preparative HPLC (Phenomenex Luna C18, 100 mm x 40 mm, 5 µm; [water (0.04% HCl) - ACN]) to afford Compound 69-1. LCMS [M+1] = 938.4; 1H NMR (400 MHz, MeOD) δ ppm 8.39 (s, 1H), 8.19 (s, 1H), 7.85 (d, J = 8.0 Hz, 2H), 7.75 - 7.59 (m, 6H), 7.49 (d, J = 8.0 Hz, 1H), 5.11 (dd, J = 5.6, 12.4 Hz, 1H), 4.91 (d, J = 13.2 Hz, 1H), 4.72 (s, 2H), 4.20 (d, J = 13.4 Hz, 1H), 3.95 - 3.65 (m, 5H), 3.61 - 3.35 (m, 6H), 3.19 - 3.13 (t, J = 12.4 Hz, 1H), 2.92 - 2.80 (m, 1H), 2.79 - 2.62 (m, 3H), 2.49 (d, J = 13.2 Hz, 1H), 2.33 (s, 3H), 2.28 - 2.06 (m, 3H), 1.77 (d, J = 6.0 Hz, 3H), 1.63 (s, 6H). Intermediate 69c-2 was processed in a similar manner to afford 1-(4'-(((2S,4R)-4-(4-(2- (2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1-yl)-2-methylpiperidin-1- yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide (Compound 69-2). LCMS [M +1] = 938.4; 1H NMR (400 MHz, MeOD) δ ppm 8.38 (s, 1H), 8.19 (s, 1H), 7.86 (d, J = 6.4 Hz, 2H), 7.78 (d, J = 8.0 Hz, 2H), 7.72 (s, 1H), 7.69 - 7.57 (m, 3H), 7.49 (d, J = 8.4 Hz, 1H), 5.12 (dd, J = 5.2, 12.4 Hz, 1H), 4.78 (d, J = 11.2 Hz, 1H), 4.70 (s, 2H), 4.43 (s, 1H), 4.03 (s, 1H), 3.97 - 3.63 (m, 5H), 3.63 - 3.36 (m, 6H), 2.93 - 2.56 (m, 4H), 2.54 - 2.43 (m, 2H), 2.33 (s, 3H), 2.31 - 2.18 (m, 1H), 2.16 - 2.09 (m, 1H), 1.62 (s, 6H), 1.59 (d, J = 6.8 Hz, 3H). Example 70: Synthesis of Compound 70
Step A – Synthesis of tert-butyl 4-((2S)-1-((benzyloxy) carbonyl)-2-methylpiperidin-4-yl) piperazine-1-carboxylate (69a) To a solution of tert-butyl piperazine-1-carboxylate (2.20 g, 12.1 mmol, 1.5 equiv.) in DCE (20.0 mL) was added benzyl (S)-2-methyl-4-oxopiperidine-1-carboxylate (2.00 g, 8.0 mmol, 1.0 equiv.) and AcOH (2.0 mL) at 20°C for 16 h. The mixture was treated with NaBH(OAc)3 (5.1 g, 24.2 mmol, 3.0 equiv.) and stirred at 20°C for 2 h. tert - Butyl piperazine-1-carboxylate (2.20 g, 12.1 mmol, 1.5 equiv.) was added and the mixture was stirred at 20°C for 1 h, poured into a mixture of ice and water (20 mL). The aqueous was extracted with dichloromethane (15 mL x 3).
The combined organic phase was washed with brine (10 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum. The residue was purified by column chromatography (silica gel, petroleum ether: ethyl acetate = 1: 0 to 0: 1) to afford 69a. 1H NMR (400 MHz, MeOD) δ ppm 7.36 - 7.29 (m, 5H), 5.13 – 5.09 (m, 2H), 3.98 (td, J = 6.4, 8.8 Hz, 1H), 3.85 - 3.77 (m, 1H), 3.44 (t, J = 5.6 Hz, 4H), 3.27 - 3.20 (m, 1H), 2.68 - 2.41 (m, 5H), 1.95 - 1.85 (m, 2H), 1.71 - 1.59 (m, 2H), 1.45 (s, 9H), 1.27 (s, 1H), 1.26 (d, J = 1.6 Hz, 2H). Step B – Synthesis of tert-butyl 4-((2S)-2-methylpiperidin-4-yl) piperazine-1-carboxylate (69b) A solution of 69a (3.20 g, 7.6 mmol, 1.0 equiv.) in MeOH (40.0 mL) was added to a suspension of MeOH (20.0 mL) in Pd/C (3.00 g, 10 wt%.) under Ar. The mixture was stirred at 20°C for 4 h under hydrogen (1 atm). The solution was filtered and the filter cake was washed with MeOH (200 mL x 3). The combined filtrate was concentrated under vacuum to afford 69b. 1H NMR (400 MHz, MeOD) δ 3.42 (t, J = 4.4 Hz, 4H), 3.27 - 3.21 (m, 1H), 2.98 - 2.88 (m, 1H), 2.85 - 2.76 (m, 1H), 2.58 - 2.52 (m, 4H), 2.47 (t, J = 5.2 Hz, 1H), 2.04 - 1.93 (m, 2H), 1.45 (s, 9H), 1.30 - 1.21 (m, 5H). Step C – Synthesis of tert-butyl 4-((2S,4S)-2-methyl-1-((3'-methyl-4'-(4-(((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)carbamoyl)-1H-pyrazol-1-yl)-[1,1'-biphenyl]-4- yl)methyl)piperidin-4-yl)piperazine-1-carboxylate (69c-1) and tert-butyl 4-[(2S,4R)-2-methyl-1- [[4-[3-methyl-4-[4-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methylcarbamoyl]pyrazol-1-yl]phenyl]phenyl]methyl]-4-piperidyl]piperazine-1-carboxylate (69c-2) To a solution of Int 55 (200.0 mg, 403 μmol, 1.0 equiv.) and 69b (285.4 mg, 1.0 mmol, 2.5 equiv.) in DCE (4.0 mL) was added tetraisopropoxytitanium (343.4 mg, 1.20 mmol, 3.0 equiv.) at 20°C. The mixture was stirred at 20°C for 16 h, treated with NaBH3CN (139.2 mg, 2.20 mmol, 5.5 equiv.) and MeOH (2.0 mL) was added to the reaction solution stirred at 20°C for 2 h. The residue was poured into water (0.1 mL) and filtered to remove the solids. The filtrate was concentrated under vacuum. The residue was purified by preparative HPLC (Phenomenex Gemini NX C18 (75 mm x 30 mm, 3 µm); [water (0.05% NH3•water 10 mM NH4HCO3) - ACN];) to afford 69c-1 as the early-eluting isomer and 69c-2 as the late-eluting isomer. LCMS [M+1] = 764.6.
Step D – Synthesis of 1-(4'-(((2S,4S)-4-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3- dioxoisoindolin-5-yl)piperazin-1-yl)-2-methylpiperidin-1-yl)methyl)-3-methyl-[1,1'-biphenyl]-4- yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4- carboxamide (Compound 69-1) A solution of 69c-1 (95.0 mg, 124.3 μmol, 1.0 equiv.) in HCl/MeOH (3 M, 1.0 mL) was stirred at 20°C for 1 h and then concentrated under vacuum (LCMS [M+1] = 664.4). The residue was dissolved in DMSO (1.0 mL) and treated with 2-(2,6-dioxopiperidin-3-yl)-5,6- difluoroisoindoline-1,3-dione (53.6 mg, 182 μmol, 1.5 equiv.) and DIEA (157.1 mg, 1.2 mmol, 211 μL, 10.0 equiv.). The mixture was stirred at 100°C for 16 h, cooled to ambient temperature and purified directly by preparative HPLC (Phenomenex Luna C18, 100 mm x 40 mm, 5 µm; [water (0.04% HCl) - ACN]) to afford Compound 69-1. LCMS [M+1] = 938.4; 1H NMR (400 MHz, MeOD) δ ppm 8.39 (s, 1H), 8.19 (s, 1H), 7.85 (d, J = 8.0 Hz, 2H), 7.75 - 7.59 (m, 6H), 7.49 (d, J = 8.0 Hz, 1H), 5.11 (dd, J = 5.6, 12.4 Hz, 1H), 4.91 (d, J = 13.2 Hz, 1H), 4.72 (s, 2H), 4.20 (d, J = 13.4 Hz, 1H), 3.95 - 3.65 (m, 5H), 3.61 - 3.35 (m, 6H), 3.19 - 3.13 (t, J = 12.4 Hz, 1H), 2.92 - 2.80 (m, 1H), 2.79 - 2.62 (m, 3H), 2.49 (d, J = 13.2 Hz, 1H), 2.33 (s, 3H), 2.28 - 2.06 (m, 3H), 1.77 (d, J = 6.0 Hz, 3H), 1.63 (s, 6H). Intermediate 69c-2 was processed in a similar manner to afford 1-(4'-(((2S,4R)-4-(4-(2- (2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1-yl)-2-methylpiperidin-1- yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide (Compound 69-2). LCMS [M +1] = 938.4; 1H NMR (400 MHz, MeOD) δ ppm 8.38 (s, 1H), 8.19 (s, 1H), 7.86 (d, J = 6.4 Hz, 2H), 7.78 (d, J = 8.0 Hz, 2H), 7.72 (s, 1H), 7.69 - 7.57 (m, 3H), 7.49 (d, J = 8.4 Hz, 1H), 5.12 (dd, J = 5.2, 12.4 Hz, 1H), 4.78 (d, J = 11.2 Hz, 1H), 4.70 (s, 2H), 4.43 (s, 1H), 4.03 (s, 1H), 3.97 - 3.63 (m, 5H), 3.63 - 3.36 (m, 6H), 2.93 - 2.56 (m, 4H), 2.54 - 2.43 (m, 2H), 2.33 (s, 3H), 2.31 - 2.18 (m, 1H), 2.16 - 2.09 (m, 1H), 1.62 (s, 6H), 1.59 (d, J = 6.8 Hz, 3H). Example 70: Synthesis of Compound 70
Compou rd 1-(4'-(((2R,4R)- 4-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1-yl)-2- methylpiperidin-1-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide. LCMS [M+1] = 938.5; 1H NMR (400 MHz, MeOD) δ ppm 8.38 (s, 1H), 8.19 (s, 1H), 7.86 (d, J = 8.0 Hz, 2H), 7.77 - 7.75 (m, 2H), 7.73 (d, J = 1.2 Hz, 1H), 7.67 - 7.64 (m, 2H), 7.60 (d, J = 1.2 Hz, 1H), 7.49 (d, J = 8.4 Hz, 1H), 5.12 (dd, J = 12.4 Hz, 1H), 4.91 - 4.89 (m, 1H), 4.70 (s, 2H), 4.43 (s, 1H), 4.08 - 4.03 (m, 1H), 3.96 - 3.72 (m, 5H), 3.62 - 3.37 (m, 6H), 2.93 - 2.83 (m, 1H), 2.77 - 2.68 (m, 2H), 2.67 - 2.35 (m, 3H), 2.33 (s, 3H), 2.29 - 2.19 (m, 1H), 2.15 - 2.09 (m, 1H), 1.62 - 1.58 (m, 9H) Example 71: Synthesis of Compound 71
Step A – To a solution of 5-bromo-1H-indazole (2.1 g, 10.4 mmol, 1.0 equiv.) in DMF (20.0 mL) was added NaH (416.6 mg, 10.4 mmol, 60 wt%, 1.0 equiv.) at 0°C and the resulting mixture was stirred for 5 min, treated with 3-bromopiperidine-2, 6-dione (2.00 g, 10.4 mmol, 1.0 equiv.) and stirred at 20 °C for 1 h. The mixture was quenched by water (10 mL) and extracted with ethyl acetate (10 mL x 3). The combined organic portions were washed with brine (10 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether: ethyl acetate=1:0 to 3:1) to afford 71a. LCMS [M+1] = 307.8, 309.8; 1H NMR (400 MHz, DMSO-d6) δ 11.12 (s, 1H), 8.13 (s, 1H), 8.04 (d, J = 0.8 Hz, 1H), 7.66 - 7.61 (m, 1H), 7.55 (dd, J = 1.6, 8.8 Hz, 1H), 5.87 (dd, J = 4.0, 12.0 Hz, 1H), 2.92 - 2.66 (m, 3H), 2.34 - 2.24 (m, 1H). Step B– Synthesis of tert-butyl 4-(1-(2,6-dioxopiperidin-3-yl)-1H-indazol-5-yl)piperidine-1- carboxylate (71b) A 15 mL vial equipped with a stir bar was charged with 71a (400.0 mg, 1.3 mmol, 1.3 equiv.), tert-butyl 4-bromopiperidine-1-carboxylate (445.8 mg, 1.7 mmol, 1.3 equiv.), Ir[dF(CF3)ppy]2(dtbpy)(PF6) (14.6 mg, 13 μmol, 0.1 equiv.), NiCl2.dtbbpy (7.8 mg, 20 μmol, 0.1 equiv.), tris(trimethylsilyl)silane (322.8 mg, 1.3 mmol, 400.5 μL, 1. equiv.), 2,6-lutidine (1.3 g,
11.7 mmol, 1.4 mL, 9.0 equiv.) and DME (2.0 mL). The vial was sealed and placed under nitrogen. The reaction was stirred and irradiated with a blue 10 W LED lamp (3 cm distance) with water cooling to maintain the reaction temperature at 25°C for 14 h. The mixture was quenched by water (5 mL) and extracted with ethyl acetate (5 mL x 3). The combined organic layers were washed with brine (5 mL), dried over sodium sulfate, concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, ethyl acetate: methanol = 1:0 to 10:1) to afford 71b. LCMS [M+1] = 413.1; 1H NMR (400 MHz, DMSO-d6) δ 11.09 (s, 1H), 8.04 (s, 1H), 7.59 (s, 1H), 7.54 (d, J = 8.6 Hz, 1H), 7.33 (d, J = 8.6 Hz, 1H), 5.81 (m, 1H), 2.88 - 2.69 (m, 6H), 2.28 - 2.21 (m, 1H), 1.81 - 1.78 (m, 2H), 1.64 - 1.45 (m, 4H), 1.42 (s, 9H) Step C – Synthesis of 3-(5-(piperidin-4-yl)-1H-indazol-1-yl)piperidine-2,6-dione (71c) A solution of 71b (500.0 mg, 1.2 mmol, 1.0 equiv.) in HCl/dioxane (4.0 M, 10.1 mL, 33.4 equiv.) was stirred at 25°C for 0.5 h. The reaction mixture was concentrated under reduced pressure to afford 71c. LCMS [M+1] = 313.2; 1H NMR (400 MHz, DMSO-d6) δ 11.09 (s, 1H), 8.09 (s, 1H), 7.65 - 7.56 (m, 2H), 7.31 (d, J = 12.0 Hz, 1H), 5.83 (dd, J = 4.0, 12.0 Hz, 1H), 3.42 - 3.32 (m, 2H), 3.06 - 2.97 (m, 2H), 2.82 - 2.66 (m, 4H), 2.28 - 2.22 (m, 1H), 2.01 - 1.87 (m, 4H). Step D – Synthesis of tert-butyl-4-(1-(2,6-dioxopiperidin-3-yl)-1H-indazol-5-yl)-[1,4'- bipiperidine]-1'-carboxylate (71d) To a solution of tert-butyl-4-oxopiperidine-1-carboxylate (287.0 mg, 1.4 mmol, 1.6 equiv.) and 71c (300.0 mg, 0.86 mmol, 1.0 equiv.) in THF (1.0 mL) was added Ti(OEt)4 (1.1 g, 4.8 mmol, 5.6 equiv.). The mixture was stirred at 20°C for 0.5 h, treated with NaBH3CN (90.5 mg, 1.4 mmol, 1.5 equiv.), stirred at 20°C for 1 h, quenched by water (3 mL) and extracted with dichloromethane (3 mL x 3). The combined organic portions were washed with brine (3 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure to afford 71d. LCMS [M+1] = 496.3; 1H NMR (400 MHz, DMSO-d6) δ 11.10 (s, 1H), 8.12 - 8.05 (m, 1H), 7.64 - 7.54 (m, 2H), 7.32 (d, J = 10.4 Hz, 1H), 5.82 (dd, J = 4.8, 11.6 Hz, 1H), 3.68 - 3.55 (m, 7H), 2.99 - 2.92 (m, 3H), 2.77 - 2.71 (m, 4H), 2.18 - 2.03 (m, 4H), 1.70 - 1.62 (m, 4H), 1.38 (s, 9H).
Step E – Synthesis of 1-(4'-((4-(1-(2,6-dioxopiperidin-3-yl)-1H-indazol-5-yl)-[1,4'-bipiperidin]- 1'-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H- 1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide (Compound 71) A solution of 71d (300 mg, 605 μmol, 1.0 equiv.) in HCl/dioxane (4.0 M, 5.1 mL, 33.4 equiv.) was stirred at 25°C for 0.5 h. The reaction mixture was concentrated under reduced pressure The residue (50.0 mg, 126.4 μmol, 1.0 equiv.) was dissolved in DMF (1.0 mL) and treated with Int 55 (62.8 mg, 126.4 μmol, 1.0 equiv.), triethylamine (127.9 mg, 1.3 mmol, 175.9 μL, 10.0 equiv.), MgSO4 (152.2 mg, 1.3 mmol, 10 equiv.), and stirred at 20°C for 0.5 h. The mixture was treated with NaBH(OAc)3 (66.9 mg, 316.1 μmol, 2.5 equiv.), stirred at 20°C for 1 h, quenched with water (5 mL) and extracted with ethyl acetate (5 mL x 3). The combined organic layers were washed with brine (5 mL), dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated under reduced pressure. The residue was purified by preparative HPLC (Phenomenex Luna C18150mm x 25mm, 10µm; [water(FA)-ACN]) to afford Compound 71. LCMS [M+1] = 876.6; 1H NMR (400 MHz, METHANOL-d4) δ 8.48 (s, 1H), 8.36 (s, 1H), 8.18 (s, 1H), 8.05 (s, 1H), 7.71 - 7.64 (m, 4H), 7.62 (dd, J = 1.6, 8.0 Hz, 1H), 7.57 (d, J = 8.4 Hz, 1H), 7.50 - 7.43 (m, 3H), 7.42 - 7.35 (m, 1H), 5.72 (dd, J = 5.2, 11.6 Hz, 1H), 4.70 (s, 2H), 4.58 (s, 2H), 3.70 - 3.68 (m, 2H), 3.64 - 3.55 (m, 2H), 3.20 - 3.11 (m, 4H), 2.97 - 2.84 (m, 3H), 2.42 - 2.35 (m, 1H), 2.31 (s, 3H), 2.28 - 2.11 (m, 6H), 2.06 - 1.97 (m, 2H), 1.86 - 1.82 (m, 2H), 1.62 (s, 6H). Example 72: Synthesis of Compound 72
 (3- piperazin-1-ylphenyl)piperidine-2,6-dione in Step D to afford 1-(4'-((4-(4-(3-(2,6-dioxopiperidin-
3-yl)phenyl)piperazin-1-yl)piperidin-1-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide. LCMS [M+1] = 837.6; 1H NMR (400 MHz, METHANOL-d4) δ 8.50 (br s, 1H), 8.36 (s, 1H), 8.18 (s, 1H), 7.74 - 7.67 (m, 3H), 7.65 - 7.60 (m, 1H), 7.50 (d, J = 8.0 Hz, 2H), 7.46 (d, J = 8.0 Hz, 1H), 7.28 - 7.19 (m, 1H), 6.91 (dd, J = 1.8, 8.0 Hz, 1H), 6.86 (s, 1H), 6.75 (d, J = 8.0 Hz, 1H), 4.70 (s, 2H), 3.87 - 3.76 (m, 3H), 3.27 - 3.17 (m, 6H), 2.86 (br s, 4H), 2.75 - 2.61 (m, 2H), 2.60 - 2.48 (m, 1H), 2.31 (s, 3H), 2.26 - 2.14 (m, 2H), 2.05 (m, 2H), 1.79 - 1.66 (m, 2H), 1.62 (s, 6H). Example 73: Synthesis of Compound 73
(3- piperazin-1-ylphenyl)piperidine-2,6-dione in Step D to afford 1-(4'-((4-(4-(3-(2,6-dioxopiperidin-
3-yl)phenyl)piperazin-1-yl)piperidin-1-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide. LCMS [M+1] = 837.6; 1H NMR (400 MHz, METHANOL-d4) δ 8.50 (br s, 1H), 8.36 (s, 1H), 8.18 (s, 1H), 7.74 - 7.67 (m, 3H), 7.65 - 7.60 (m, 1H), 7.50 (d, J = 8.0 Hz, 2H), 7.46 (d, J = 8.0 Hz, 1H), 7.28 - 7.19 (m, 1H), 6.91 (dd, J = 1.8, 8.0 Hz, 1H), 6.86 (s, 1H), 6.75 (d, J = 8.0 Hz, 1H), 4.70 (s, 2H), 3.87 - 3.76 (m, 3H), 3.27 - 3.17 (m, 6H), 2.86 (br s, 4H), 2.75 - 2.61 (m, 2H), 2.60 - 2.48 (m, 1H), 2.31 (s, 3H), 2.26 - 2.14 (m, 2H), 2.05 (m, 2H), 1.79 - 1.66 (m, 2H), 1.62 (s, 6H). Example 73: Synthesis of Compound 73
 carboxylate (73a)
To a solution of tert-butyl 4-[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)phenyl]piperazine-1-carboxylate (2.0 g, 5.1 mmol, 1.0 equiv.) and 2,6-dibenzyloxy-3-bromo- pyridine (2.6 g, 7.2 mmol, 1.4 equiv.) in dioxane (100.0 mL) and water (20.0 mL) was added Pd(dppf)Cl2 (376.8 mg, 515 μmol, 0.1 equiv.) and K2CO3 (1.4 g, 10.3 mmol, 2.0 equiv.). The mixture was stirred at 100°C for 2 h. The reaction mixture was quenched with addition water (150 mL), and then extracted with ethyl acetate (200 mL x 2). The combined organic layers were washed with brine (100 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, 0 to 100% ethyl acetate/ petroleum ether) to afford 73a. LCMS [M+1] = 552.3; 1H NMR (400 MHz, DMSO-d6) δ 7.68 (d, J = 8.0 Hz, 1H), 7.44 - 7.26 (m, 12H), 6.96 (br d, J = 8.6 Hz, 2H), 6.52 (d, J = 8.0 Hz, 1H), 5.39 - 5.35 (m, 4H), 3.45 (br d, J = 4.4 Hz, 4H), 3.18 - 3.07 (m, 4H), 1.42 - 1.41 (m, 1H), 1.42 (s, 9H). Step B – Synthesis of tert-butyl 4-[4-(2,6-dioxo-3-piperidyl)phenyl]piperazine-1-carboxylate (73b) To a solution of 73a (10.0 g, 18.1 mmol, 1.0 equiv.) in EtOH (60 mL) and ethyl acetate (60 mL) was added Pd/C (3.8 g, 3.6 mmol, 10 wt%, 0.2 equiv.). The suspension was degassed under vacuum and purged with hydrogen threefold. The mixture was stirred under hydrogen (50 psi) at 20°C for 8 h. The reaction mixture was filtered through a Celite pad and the filter cake was rinsed with ethyl acetate (500 ml x 3). The filtrate was concentrated under vacuum to afford 73b, which was used without further purification. LCMS [M+1] = 374.2; 1H NMR (400 MHz, DMSO-d6) δ 10.77 (s, 1H), 7.07 (d, J = 8.0 Hz, 2H), 6.91 (d, J = 7.8 Hz, 2H), 3.74 (dd, J = 4.8, 10.8 Hz, 1H), 3.45 (br s, 4H), 3.07 (br t, J = 4.6 Hz, 4H), 2.69 - 2.58 (m, 1H), 2.48 - 2.41 (m, 1H), 2.13 (dq, J = 3.8, 11.8 Hz, 1H), 2.05 - 1.95 (m, 1H), 1.42 (s, 9H). Step C – Synthesis of 3-(4-piperazin-1-ylphenyl)piperidine-2,6-dione (73c) Intermediate 73b (2.0 g, 5.3 mmol, 1.0 equiv.) was dissolved in HCl/ dioxane (4.0 M, 22.2 mL, 16.6 equiv.). The mixture was stirred at 20 °C for 1 h. The reaction mixture was concentrated under reduced pressure to afford 73c, which was used without further purification.
LCMS [M+1] = 274.1; 1H NMR (400 MHz, METHANOL-d4) δ 7.31 - 7.25 (m, 2H), 7.25 - 7.19 (m, 2H), 3.86 (br dd, J = 5.4, 10.4 Hz, 1H), 3.57 (br d, J = 4.2 Hz, 4H), 3.50 (br d, J = 4.4 Hz, 4H), 2.81 - 2.52 (m, 2H), 2.34 - 2.10 (m, 2H). Step D – Synthesis of tert-butyl 4-[4-[4-(2,6-dioxo-3-piperidyl)phenyl]piperazin-1-yl]piperidine- 1-carboxylate (73d) To a solution of 73c (250 mg, 915 μmol, 1 equiv.) and tert-butyl 4-oxopiperidine-1- carboxylate (182 mg, 915 μmol, 1 equiv.) in THF (3 mL) was added Ti(OEt)4 (1.04 g, 4.57 mmol, 948 μL, 5 equiv.) at 20°C. The mixture was stirred at 20°C for 30 min, treated with NaBH3CN (86.2 mg, 1.37 mmol, 1.5 equiv.), stirred at 20°C for 1 h and treated with MeOH (5 mL) and water (5 mL). The resulting mixture was filtered and the filtrate was extracted with ethyl acetate. The combined organic portions were concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether: ethyl acetate = 10:3 - 0:1) afford (73d). 1H NMR (400 MHz, METHANOL-d4) δ 7.15 (d, J = 8.8 Hz, 2H), 6.98 (d, J = 8.8 Hz, 2H), 4.15 (d, J = 13.2 Hz, 2H), 3.80 (dd, J = 7.2, 8.4 Hz, 1H), 3.32 - 3.09 (m, 5H), 2.87 - 2.74 (m, 5H), 2.73 - 2.58 (m, 2H), 2.57 - 2.46 (m, 1H), 2.30 - 2.14 (m, 2H), 1.97 (d, J = 12.8 Hz, 2H), 1.48 (s, 9H), 1.45 - 1.30 (m, 2H). Step E – Synthesis of 3-[4-[4-(4-piperidyl) piperazin-1-yl] phenyl] piperidine-2, 6-dione (73e) Intermediate 73d (150 mg, 328.53 μmol, 1 equiv.) was stirred in a solution of TFA (614.00 mg, 5.38 mmol, 0.4 mL, 16.39 equiv.) in dichloromethane (2 mL) at 20°C for 1 h. The reaction mixture was concentrated under reduced pressure to afford 73e, which was used without further purification. LCMS [M+1]=357.2; 1H NMR (400 MHz, METHANOL-d4) δ 7.20 (d, J = 8.4 Hz, 2H), 7.03 (d, J = 8.8 Hz, 2H), 3.81 (dd, J = 5.6, 9.6 Hz, 1H), 3.70 - 3.58 (m, 4H), 3.56 - 3.38 (m, 4H), 3.33 - 3.20 (m, 2H), 3.12 (dt, J = 2.0, 13.2 Hz, 2H), 3.05 - 2.80 (m, 1H), 2.77 - 2.55 (m, 2H), 2.47 (s, J = 13.6 Hz, 2H), 2.31 - 2.11 (m, 2H), 2.09 - 1.90 (m, 2H). Step F – Synthesis of 1-[4-[4-[[4-[4-[4-(2,6-dioxo-3-piperidyl)phenyl]piperazin-1-yl]-1- piperidyl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4- triazol-5-yl]methyl]pyrazole-4-carboxamide (Compound 73)
To a solution of 73e in MeOH (1 mL) was added Na2CO3 (45.5 mg, 429 μmol, 10 equiv.). The resulting mixture was stirred at 20°C for 5 min and filtered. To the filtrate was added Int 55 (10.7 mg, 21 μmol, 0.5 equiv.) and Ti(OEt)4 (19.6 mg, 85 μmol, 17.8 μL, 2 equiv.) at 20°C. The resulting mixture was stirred at 20°C for 0.5 h, treated with NaBH3CN (4 mg, 64 μmol, 1.5 equiv.) and stirred at 20°C for 0.5 h. MeOH (5 mL) and water (5 mL) was added into the reaction mixture. The resulting mixture was filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative HPLC (Welch Xtimate C18, 150mm x 25mm, 5 µm; [water(FA)-ACN]) to afford Compound 73. LCMS [M+1] = 837.5; 1H NMR (400 MHz, DMSO-d6) δ 13,92 (s, 1H), 10.76 (s, 1H), 8.87 (s, 1H), 8.52 (s, 1H), 8.17 (s, 2H), 7.72 (d, J = 1.6 Hz, 1H), 7.68 (d, J = 8.4 Hz, 2H), 7.64 (dd, J = 2.0, 8.4 Hz, 1H), 7.46 (d, J = 8.4 Hz, 1H), 7.41 (d, J = 8.4 Hz, 2H), 7.04 (d, J = 8.8 Hz, 2H), 6.88 (d, J = 8.8 Hz, 2H), 4.55 (d, J = 5.6 Hz, 2H), 3.72 (dd, J = 4.8, 10.8 Hz, 1H), 3.51 (s, 2H), 3.11 - 3.07 (m, 4H), 2.91 - 2,97 (m, 2H), 2.66 - 2.57 (m, 5H), 2.30 (s, 3H), 2.25 - 2.07 (m, 2H), 2.04 - 1.91 (m, 3H), 1.81 - 1.74 (m, 2H), 1.54 (s, 6H), 1.50 - 1.37 (m, 2H). Example 74: Synthesis of Compound 74 Synthesis of
carboxylate (73a)
To a solution of tert-butyl 4-[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)phenyl]piperazine-1-carboxylate (2.0 g, 5.1 mmol, 1.0 equiv.) and 2,6-dibenzyloxy-3-bromo- pyridine (2.6 g, 7.2 mmol, 1.4 equiv.) in dioxane (100.0 mL) and water (20.0 mL) was added Pd(dppf)Cl2 (376.8 mg, 515 μmol, 0.1 equiv.) and K2CO3 (1.4 g, 10.3 mmol, 2.0 equiv.). The mixture was stirred at 100°C for 2 h. The reaction mixture was quenched with addition water (150 mL), and then extracted with ethyl acetate (200 mL x 2). The combined organic layers were washed with brine (100 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, 0 to 100% ethyl acetate/ petroleum ether) to afford 73a. LCMS [M+1] = 552.3; 1H NMR (400 MHz, DMSO-d6) δ 7.68 (d, J = 8.0 Hz, 1H), 7.44 - 7.26 (m, 12H), 6.96 (br d, J = 8.6 Hz, 2H), 6.52 (d, J = 8.0 Hz, 1H), 5.39 - 5.35 (m, 4H), 3.45 (br d, J = 4.4 Hz, 4H), 3.18 - 3.07 (m, 4H), 1.42 - 1.41 (m, 1H), 1.42 (s, 9H). Step B – Synthesis of tert-butyl 4-[4-(2,6-dioxo-3-piperidyl)phenyl]piperazine-1-carboxylate (73b) To a solution of 73a (10.0 g, 18.1 mmol, 1.0 equiv.) in EtOH (60 mL) and ethyl acetate (60 mL) was added Pd/C (3.8 g, 3.6 mmol, 10 wt%, 0.2 equiv.). The suspension was degassed under vacuum and purged with hydrogen threefold. The mixture was stirred under hydrogen (50 psi) at 20°C for 8 h. The reaction mixture was filtered through a Celite pad and the filter cake was rinsed with ethyl acetate (500 ml x 3). The filtrate was concentrated under vacuum to afford 73b, which was used without further purification. LCMS [M+1] = 374.2; 1H NMR (400 MHz, DMSO-d6) δ 10.77 (s, 1H), 7.07 (d, J = 8.0 Hz, 2H), 6.91 (d, J = 7.8 Hz, 2H), 3.74 (dd, J = 4.8, 10.8 Hz, 1H), 3.45 (br s, 4H), 3.07 (br t, J = 4.6 Hz, 4H), 2.69 - 2.58 (m, 1H), 2.48 - 2.41 (m, 1H), 2.13 (dq, J = 3.8, 11.8 Hz, 1H), 2.05 - 1.95 (m, 1H), 1.42 (s, 9H). Step C – Synthesis of 3-(4-piperazin-1-ylphenyl)piperidine-2,6-dione (73c) Intermediate 73b (2.0 g, 5.3 mmol, 1.0 equiv.) was dissolved in HCl/ dioxane (4.0 M, 22.2 mL, 16.6 equiv.). The mixture was stirred at 20 °C for 1 h. The reaction mixture was concentrated under reduced pressure to afford 73c, which was used without further purification.
LCMS [M+1] = 274.1; 1H NMR (400 MHz, METHANOL-d4) δ 7.31 - 7.25 (m, 2H), 7.25 - 7.19 (m, 2H), 3.86 (br dd, J = 5.4, 10.4 Hz, 1H), 3.57 (br d, J = 4.2 Hz, 4H), 3.50 (br d, J = 4.4 Hz, 4H), 2.81 - 2.52 (m, 2H), 2.34 - 2.10 (m, 2H). Step D – Synthesis of tert-butyl 4-[4-[4-(2,6-dioxo-3-piperidyl)phenyl]piperazin-1-yl]piperidine- 1-carboxylate (73d) To a solution of 73c (250 mg, 915 μmol, 1 equiv.) and tert-butyl 4-oxopiperidine-1- carboxylate (182 mg, 915 μmol, 1 equiv.) in THF (3 mL) was added Ti(OEt)4 (1.04 g, 4.57 mmol, 948 μL, 5 equiv.) at 20°C. The mixture was stirred at 20°C for 30 min, treated with NaBH3CN (86.2 mg, 1.37 mmol, 1.5 equiv.), stirred at 20°C for 1 h and treated with MeOH (5 mL) and water (5 mL). The resulting mixture was filtered and the filtrate was extracted with ethyl acetate. The combined organic portions were concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether: ethyl acetate = 10:3 - 0:1) afford (73d). 1H NMR (400 MHz, METHANOL-d4) δ 7.15 (d, J = 8.8 Hz, 2H), 6.98 (d, J = 8.8 Hz, 2H), 4.15 (d, J = 13.2 Hz, 2H), 3.80 (dd, J = 7.2, 8.4 Hz, 1H), 3.32 - 3.09 (m, 5H), 2.87 - 2.74 (m, 5H), 2.73 - 2.58 (m, 2H), 2.57 - 2.46 (m, 1H), 2.30 - 2.14 (m, 2H), 1.97 (d, J = 12.8 Hz, 2H), 1.48 (s, 9H), 1.45 - 1.30 (m, 2H). Step E – Synthesis of 3-[4-[4-(4-piperidyl) piperazin-1-yl] phenyl] piperidine-2, 6-dione (73e) Intermediate 73d (150 mg, 328.53 μmol, 1 equiv.) was stirred in a solution of TFA (614.00 mg, 5.38 mmol, 0.4 mL, 16.39 equiv.) in dichloromethane (2 mL) at 20°C for 1 h. The reaction mixture was concentrated under reduced pressure to afford 73e, which was used without further purification. LCMS [M+1]=357.2; 1H NMR (400 MHz, METHANOL-d4) δ 7.20 (d, J = 8.4 Hz, 2H), 7.03 (d, J = 8.8 Hz, 2H), 3.81 (dd, J = 5.6, 9.6 Hz, 1H), 3.70 - 3.58 (m, 4H), 3.56 - 3.38 (m, 4H), 3.33 - 3.20 (m, 2H), 3.12 (dt, J = 2.0, 13.2 Hz, 2H), 3.05 - 2.80 (m, 1H), 2.77 - 2.55 (m, 2H), 2.47 (s, J = 13.6 Hz, 2H), 2.31 - 2.11 (m, 2H), 2.09 - 1.90 (m, 2H). Step F – Synthesis of 1-[4-[4-[[4-[4-[4-(2,6-dioxo-3-piperidyl)phenyl]piperazin-1-yl]-1- piperidyl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4- triazol-5-yl]methyl]pyrazole-4-carboxamide (Compound 73)
To a solution of 73e in MeOH (1 mL) was added Na2CO3 (45.5 mg, 429 μmol, 10 equiv.). The resulting mixture was stirred at 20°C for 5 min and filtered. To the filtrate was added Int 55 (10.7 mg, 21 μmol, 0.5 equiv.) and Ti(OEt)4 (19.6 mg, 85 μmol, 17.8 μL, 2 equiv.) at 20°C. The resulting mixture was stirred at 20°C for 0.5 h, treated with NaBH3CN (4 mg, 64 μmol, 1.5 equiv.) and stirred at 20°C for 0.5 h. MeOH (5 mL) and water (5 mL) was added into the reaction mixture. The resulting mixture was filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative HPLC (Welch Xtimate C18, 150mm x 25mm, 5 µm; [water(FA)-ACN]) to afford Compound 73. LCMS [M+1] = 837.5; 1H NMR (400 MHz, DMSO-d6) δ 13,92 (s, 1H), 10.76 (s, 1H), 8.87 (s, 1H), 8.52 (s, 1H), 8.17 (s, 2H), 7.72 (d, J = 1.6 Hz, 1H), 7.68 (d, J = 8.4 Hz, 2H), 7.64 (dd, J = 2.0, 8.4 Hz, 1H), 7.46 (d, J = 8.4 Hz, 1H), 7.41 (d, J = 8.4 Hz, 2H), 7.04 (d, J = 8.8 Hz, 2H), 6.88 (d, J = 8.8 Hz, 2H), 4.55 (d, J = 5.6 Hz, 2H), 3.72 (dd, J = 4.8, 10.8 Hz, 1H), 3.51 (s, 2H), 3.11 - 3.07 (m, 4H), 2.91 - 2,97 (m, 2H), 2.66 - 2.57 (m, 5H), 2.30 (s, 3H), 2.25 - 2.07 (m, 2H), 2.04 - 1.91 (m, 3H), 1.81 - 1.74 (m, 2H), 1.54 (s, 6H), 1.50 - 1.37 (m, 2H). Example 74: Synthesis of Compound 74 Synthesis of
 phenyl]-2- methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole- 4-carboxamide (Compound 74) To a solution of 3-(4-piperazin-1-ylphenyl)piperidine-2,6-dione (73c) (41.2 mg, 151 μmol, 1.5 equiv.) and Int 55 (50.0 mg, 101 μmol, 1.0 equiv.) in DCE (1.0 mL) was added acetic acid
(12.1 mg, 201.4 μmol, 11.5 μL, 2.0 equiv.). The mixture was stirred at 50°C for 0.5 h, treated with NaBH(OAc)3 (42.6 mg, 201 μmol, 2.0 equiv.) and the mixture was stirred at 50°C for 1 h. The reaction mixture was quenched with water (5 mL) and extracted with ethyl acetate (20 mL). The combined organic layers were washed with brine (5 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative HPLC (Welch Ultimate C18, 150mm x 25mm, 5 µm, [water(FA)-ACN]) to afford Compound 74. LCMS [M+1] =754.4; 1H NMR (400 MHz, DMSO-d6) δ 10.77 (s, 1H), 8.88 (br s, 1H), 8.53 (s, 1H), 8.19 (s, 1H), 7.76 - 7.68 (m, 3H), 7.65 (dd, J = 1.6, 8.0 Hz, 1H), 7.48 - 7.44 (m, 3H), 7.05 (d, J = 8.8 Hz, 2H), 6.89 (d, J = 8.8 Hz, 2H), 4.55 (d, J = 5.6 Hz, 2H), 3.72 (dd, J = 4.8, 11.2 Hz, 1H), 3.58 (s, 2H), 3.14 (s, 4H), 2.68 - 2.58 (m, 1H), 2.54 (d, J = 4.0 Hz, 4H), 2.45 - 2.42 (m, 1H), 2.30 (s, 3H), 2.18 - 2.07 (m, 1H), 2.04 - 1.97 (m, 1H), 1.54 (s, 6H). Example 75: Synthesis of Compound 75
phenyl]-2- methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole- 4-carboxamide (Compound 74) To a solution of 3-(4-piperazin-1-ylphenyl)piperidine-2,6-dione (73c) (41.2 mg, 151 μmol, 1.5 equiv.) and Int 55 (50.0 mg, 101 μmol, 1.0 equiv.) in DCE (1.0 mL) was added acetic acid
(12.1 mg, 201.4 μmol, 11.5 μL, 2.0 equiv.). The mixture was stirred at 50°C for 0.5 h, treated with NaBH(OAc)3 (42.6 mg, 201 μmol, 2.0 equiv.) and the mixture was stirred at 50°C for 1 h. The reaction mixture was quenched with water (5 mL) and extracted with ethyl acetate (20 mL). The combined organic layers were washed with brine (5 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative HPLC (Welch Ultimate C18, 150mm x 25mm, 5 µm, [water(FA)-ACN]) to afford Compound 74. LCMS [M+1] =754.4; 1H NMR (400 MHz, DMSO-d6) δ 10.77 (s, 1H), 8.88 (br s, 1H), 8.53 (s, 1H), 8.19 (s, 1H), 7.76 - 7.68 (m, 3H), 7.65 (dd, J = 1.6, 8.0 Hz, 1H), 7.48 - 7.44 (m, 3H), 7.05 (d, J = 8.8 Hz, 2H), 6.89 (d, J = 8.8 Hz, 2H), 4.55 (d, J = 5.6 Hz, 2H), 3.72 (dd, J = 4.8, 11.2 Hz, 1H), 3.58 (s, 2H), 3.14 (s, 4H), 2.68 - 2.58 (m, 1H), 2.54 (d, J = 4.0 Hz, 4H), 2.45 - 2.42 (m, 1H), 2.30 (s, 3H), 2.18 - 2.07 (m, 1H), 2.04 - 1.97 (m, 1H), 1.54 (s, 6H). Example 75: Synthesis of Compound 75
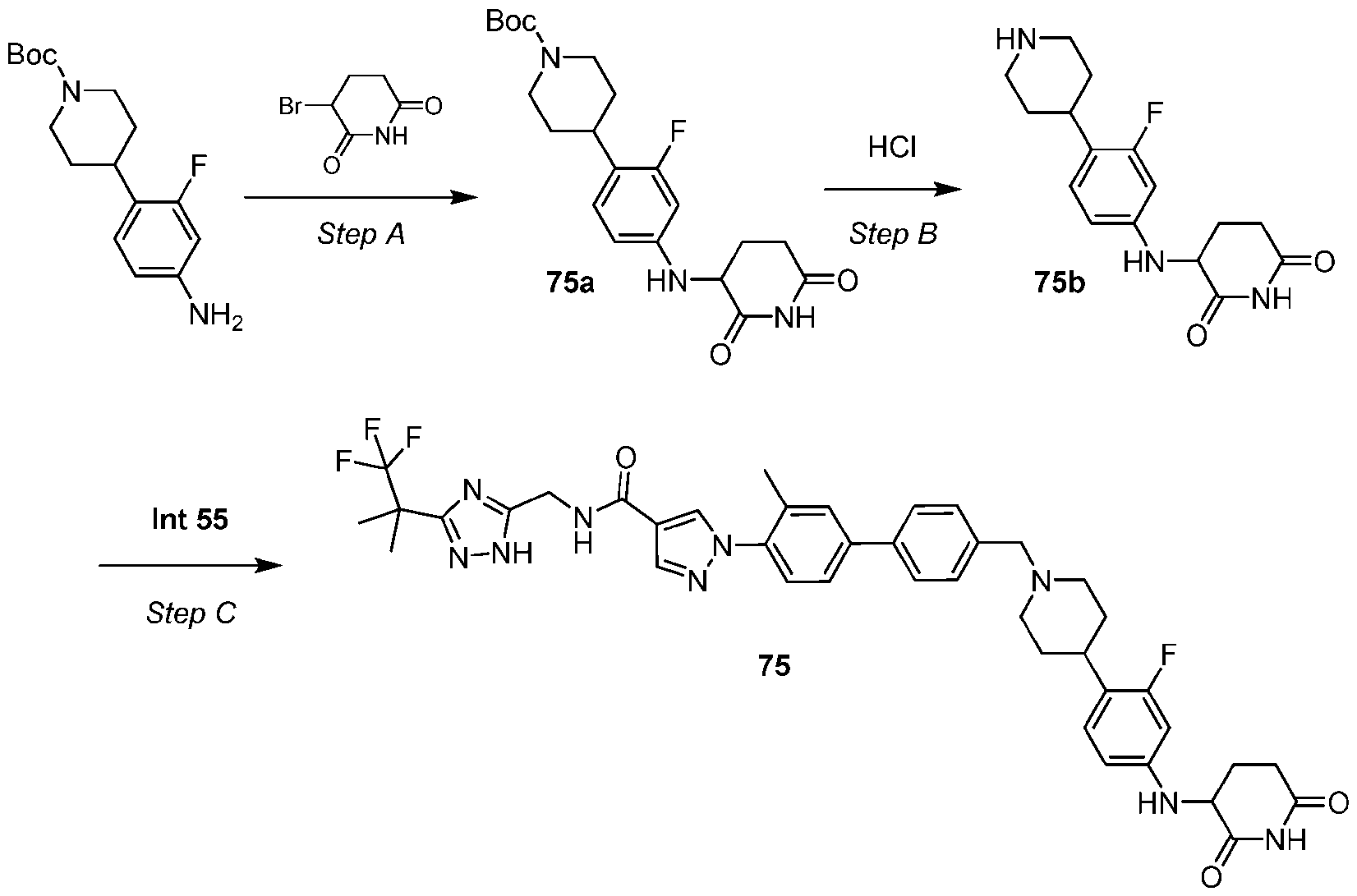 piperidine- 1-carboxylate (75a) To a solution of tert-butyl 4-(4-amino-2-fluoro-phenyl)piperidine-1-carboxylate (500.0 mg, 1.7 mmol, 1.0 equiv) and 3-bromopiperidine-2,6-dione (652.3 mg, 3.4 mmol, 2.0 equiv) in
DMF (6.0 mL) was added NaHCO3 (713.5 mg, 8.5 mmol, 330.5 μL, 5.0 equiv). The mixture was stirred at 60°C for 16 h, cooled to 25°C and poured into water (20 mL). The aqueous phase was extracted with ethyl acetate (20 mL x 3). The combined organic phase was washed with brine (20 mL x 3), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum. The residue was purified by column chromatography (silica gel, petroleum ether / ethyl acetate = 100/0 to 3/1) to afford 75a. LCMS [M - 55] = 350.2; 1H NMR (400 MHz, DMSO-d6) δ 10.78 (s, 1H), 6.97 (t, J = 8.8, 1H), 6.48 - 6.40 (m, 2H), 6.02 (d, J = 8.0 Hz, 1H), 4.32 - 4.28 (m, 1H), 4.11 - 3.97 (m, 2H), 2.81 - 2.65 (m, 4H), 2.62 - 2.56 (m, 1H), 2.13 - 2.03 (m, 1H), 1.88 - 1.85 (m, 1H), 1.67 - 1.63 (m, 2H), 1.53 - 1.44 (m, 2H), 1.41 (s, 9H). Step B – Synthesis of 3-[3-fluoro-4-(4-piperidyl)anilino]piperidine-2,6-dione (75b) A mixture of 75a (300.0 mg, 740 μmol, 1.0 equiv) in HCl/ dioxane (4 M, 3.0 mL) was stirred at 25°C for 1 h under a nitrogen atmosphere. The reaction mixture was concentrated under reduced pressure to afford 75b, which was used without further purification. LCMS [M+1] = 306.2; 1H NMR (400 MHz, DMSO-d6) δ 10.79 (s, 1H), 9.04 - 8.90 (s, 1H), 8.87 - 8.68 (m, 1H), 7.00 - 6.88 (s, 1H), 6.53 - 6.40 (m, 2H), 4.32 (dd, J = 4.8, 11.6 Hz, 1H), 3.34 - 2.98 (m, 2H), 3.04 - 2.87 (m, 3H), 2.80 - 2.65 (m, 1H), 2.60-2.56 (m, 2H), 2.12 - 2.03 (m, 1H), 1.95 - 1.76 (m, 5H). Step C – Synthesis of 1-[4-[4-[[4-[4-[(2,6-dioxo-3-piperidyl)amino]-2-fluoro-phenyl]-1- piperidyl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4- triazol-5-yl]methyl]pyrazole-4-carboxamide (Compound 75) To a solution of 75b (50.0 mg, 163.7 μmol, 1.0 equiv) and Int 55 (81.3 mg, 163.7 μmol, 1.0 equiv) in DMF (0.5 mL) was added MgSO4 (197.1 mg, 1.6 mmol, 10.0 equiv) and triethylamine (99.4 mg, 983 μmol, 137 μL, 6.0 equiv). The mixture was stirred at 25°C for 0.5 h, treated with NaBH(OAc)3 (86.8 mg, 409 μmol, 2.5 equiv), the reaction mixture was stirred at 25°C for 2 h. The mixture was filtered and filtrate was concentrated under reduce pressure to give a residue. The residue was purified by preparative HPLC (Waters Xbridge 150mm x 25mm, 10 µm; [water(NH3water+NH4HCO3)-ACN) to afford Compound 75. LCMS [M+1] = 786.4; 1H NMR (400 MHz, DMSO-d6) δ 13.87 (br s, 1H), 10.78 (br s, 1H), 8.86 (br s, 1H), 8.52 (s, 1H), 8.17 (s, 1H), 7.75 - 7.61 (m, 4H), 7.47 - 7.42 (m, 3H), 7.01 (t, J = 9.2 Hz, 1H), 6.49 - 6.40 (m,
2H), 5.99 (d, J = 7.6 Hz, 1H), 4.55 (d, J = 5.2 Hz, 2H), 4.37 - 4.23 (m, 1H), 3.54 (s, 2H), 2.94 - 2.92 (m, 2H), 2.73 - 2.70 (m, 1H), 2.62- 2.56 (m, 2H), 2.30 (s, 3H), 2.13 - 1.99 (m, 3H), 1.93 - 1.78 (m, 1H), 1.68 - 2.65 (m, 4H), 1.54 (s, 6H). Example 76: Synthesis of Intermediate 76
piperidine- 1-carboxylate (75a) To a solution of tert-butyl 4-(4-amino-2-fluoro-phenyl)piperidine-1-carboxylate (500.0 mg, 1.7 mmol, 1.0 equiv) and 3-bromopiperidine-2,6-dione (652.3 mg, 3.4 mmol, 2.0 equiv) in
DMF (6.0 mL) was added NaHCO3 (713.5 mg, 8.5 mmol, 330.5 μL, 5.0 equiv). The mixture was stirred at 60°C for 16 h, cooled to 25°C and poured into water (20 mL). The aqueous phase was extracted with ethyl acetate (20 mL x 3). The combined organic phase was washed with brine (20 mL x 3), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum. The residue was purified by column chromatography (silica gel, petroleum ether / ethyl acetate = 100/0 to 3/1) to afford 75a. LCMS [M - 55] = 350.2; 1H NMR (400 MHz, DMSO-d6) δ 10.78 (s, 1H), 6.97 (t, J = 8.8, 1H), 6.48 - 6.40 (m, 2H), 6.02 (d, J = 8.0 Hz, 1H), 4.32 - 4.28 (m, 1H), 4.11 - 3.97 (m, 2H), 2.81 - 2.65 (m, 4H), 2.62 - 2.56 (m, 1H), 2.13 - 2.03 (m, 1H), 1.88 - 1.85 (m, 1H), 1.67 - 1.63 (m, 2H), 1.53 - 1.44 (m, 2H), 1.41 (s, 9H). Step B – Synthesis of 3-[3-fluoro-4-(4-piperidyl)anilino]piperidine-2,6-dione (75b) A mixture of 75a (300.0 mg, 740 μmol, 1.0 equiv) in HCl/ dioxane (4 M, 3.0 mL) was stirred at 25°C for 1 h under a nitrogen atmosphere. The reaction mixture was concentrated under reduced pressure to afford 75b, which was used without further purification. LCMS [M+1] = 306.2; 1H NMR (400 MHz, DMSO-d6) δ 10.79 (s, 1H), 9.04 - 8.90 (s, 1H), 8.87 - 8.68 (m, 1H), 7.00 - 6.88 (s, 1H), 6.53 - 6.40 (m, 2H), 4.32 (dd, J = 4.8, 11.6 Hz, 1H), 3.34 - 2.98 (m, 2H), 3.04 - 2.87 (m, 3H), 2.80 - 2.65 (m, 1H), 2.60-2.56 (m, 2H), 2.12 - 2.03 (m, 1H), 1.95 - 1.76 (m, 5H). Step C – Synthesis of 1-[4-[4-[[4-[4-[(2,6-dioxo-3-piperidyl)amino]-2-fluoro-phenyl]-1- piperidyl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4- triazol-5-yl]methyl]pyrazole-4-carboxamide (Compound 75) To a solution of 75b (50.0 mg, 163.7 μmol, 1.0 equiv) and Int 55 (81.3 mg, 163.7 μmol, 1.0 equiv) in DMF (0.5 mL) was added MgSO4 (197.1 mg, 1.6 mmol, 10.0 equiv) and triethylamine (99.4 mg, 983 μmol, 137 μL, 6.0 equiv). The mixture was stirred at 25°C for 0.5 h, treated with NaBH(OAc)3 (86.8 mg, 409 μmol, 2.5 equiv), the reaction mixture was stirred at 25°C for 2 h. The mixture was filtered and filtrate was concentrated under reduce pressure to give a residue. The residue was purified by preparative HPLC (Waters Xbridge 150mm x 25mm, 10 µm; [water(NH3water+NH4HCO3)-ACN) to afford Compound 75. LCMS [M+1] = 786.4; 1H NMR (400 MHz, DMSO-d6) δ 13.87 (br s, 1H), 10.78 (br s, 1H), 8.86 (br s, 1H), 8.52 (s, 1H), 8.17 (s, 1H), 7.75 - 7.61 (m, 4H), 7.47 - 7.42 (m, 3H), 7.01 (t, J = 9.2 Hz, 1H), 6.49 - 6.40 (m,
2H), 5.99 (d, J = 7.6 Hz, 1H), 4.55 (d, J = 5.2 Hz, 2H), 4.37 - 4.23 (m, 1H), 3.54 (s, 2H), 2.94 - 2.92 (m, 2H), 2.73 - 2.70 (m, 1H), 2.62- 2.56 (m, 2H), 2.30 (s, 3H), 2.13 - 1.99 (m, 3H), 1.93 - 1.78 (m, 1H), 1.68 - 2.65 (m, 4H), 1.54 (s, 6H). Example 76: Synthesis of Intermediate 76
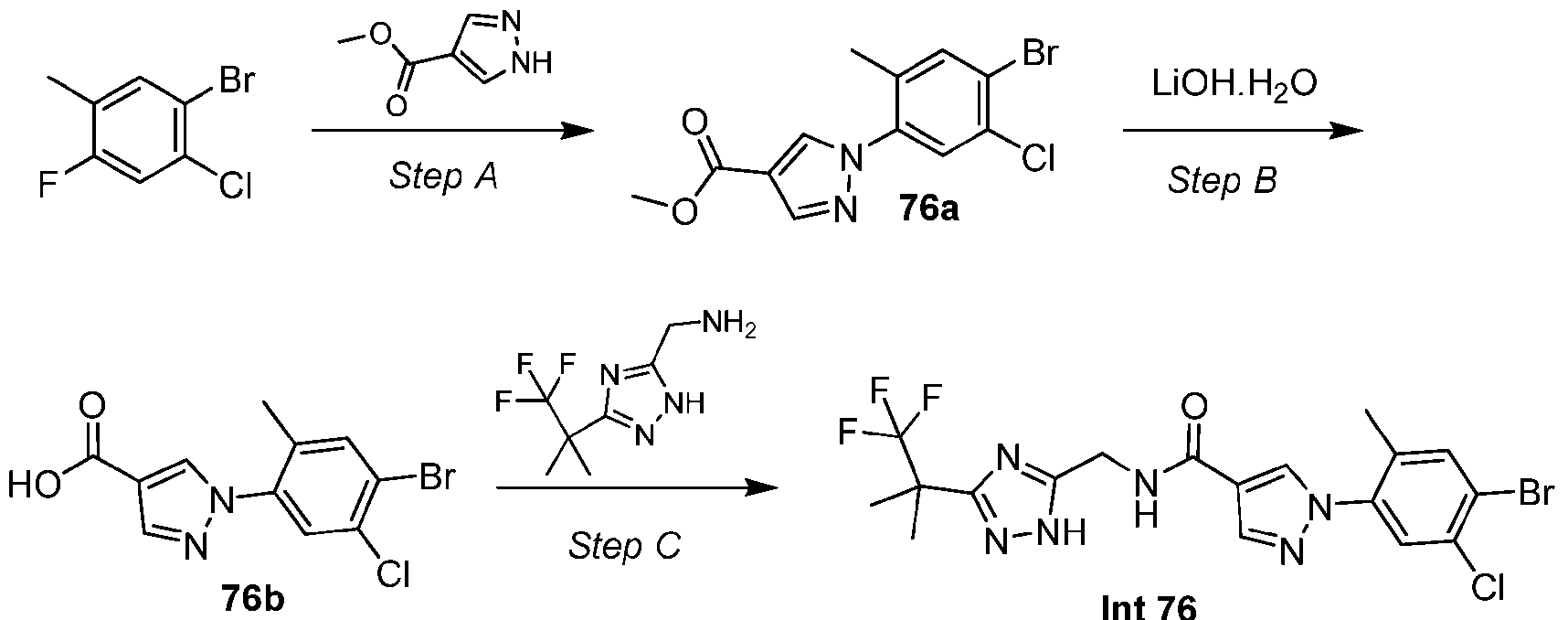 (76a) To a solution of 1-bromo-2-chloro-4-fluoro-5-methylbenzene (3.80 g, 17.0 mmol, 1.0 equiv.) and methyl 1H-pyrazole-4-carboxylate (2.10 g, 17.0 mmol, 1.0 equiv.) in DMA (40 mL) was added K2CO3 (4.7 g, 34.0 mmol, 2.0 equiv.). The mixture was stirred at 120°C for 20 h, cooled to ambient temperature, filtered and the filtrate was diluted with NH4Cl (100 mL) and extracted with ethyl acetate (100 mL x 2). The combined organic portions were dried over sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether/ ethyl acetate=4/1) to afford 76a. LCMS: [M+1] = 329.0, 331.0; 1H NMR (400 MHz, DMSO-d6) δ 8.74 (s, 1H), 8.15 (s, 1H), 7.90 (s, 1H), 7.77 (s, 1H), 3.79 (s, 3H), 2.20 (s, 3H). Step B – Synthesis of 1-(4-bromo-5-chloro-2-methylphenyl)-1H-pyrazole-4-carboxylic acid (76b) To a solution of 76a (1.00 g, 3.0 mmol, 1.0 equiv.) in THF (10 mL), MeOH (10 mL) and water (10 mL) was added lithium hydroxide hydrate (255 mg, 6.0 mmol, 2.0 equiv.). The mixture solution was stirred at 50°C for 1 h, cooled to ambient temperature and adjusted to pH = 2 with 1 M aqueous HCl. The solids were collected by vacuum filtration to afford 76b.
LCMS: [M+1] = 315.0, 317.0; 1H NMR (400 MHz, DMSO-d6) δ 12.62 (br s, 1H), 8.63 (s, 1H), 8.08 (s, 1H), 7.88 (s, 1H), 7.76 (s, 1H), 2.20 (s, 3H). Step C – Synthesis of 1-(4-bromo-5-chloro-2-methyl-phenyl)-N-[[3-(2,2,2-trifluoro-1,1-dimethyl- ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Intermediate 76) To a solution of 76b (100 mg, 316.9 µmol, 1.0 equiv.) and Int 1 (109.9 mg, 380 µmol, 1.2 equiv., HBr) in DMF (2.0 mL) was added triethylamine (320.6 mg, 3.1 mmol, 441 µL, 10 equiv.), then BOP (168.1 mg, 380.2 µmol, 1.2 equiv.) was added. The mixture was stirred at 20°C for 16 h. The reaction mixture was partitioned between ethyl acetate (10 mL) and water (8 mL). The organic phase was separated, washed with NaCl 10 mL, dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure to give a residue. The residue was purified by column chromatography (silica gel, dichloromethane: MeOH =94/6) to afford Intermediate 76. LCMS: [M+1] = 504.9, 506.9; 1H NMR (400 MHz, DMSO-d6) δ 13.94 (br s, 1H), 8.90 (br s, 1H), 8.58 (s, 1H), 8.19 (s, 1H), 7.90 (s, 1H), 7.73 (s, 1H), 4.54 (br d, J = 5.6 Hz, 2H), 3.33 (s, 3H), 2.23 (s, 3H), 1.53 (s, 6H). Example 77: Synthesis of Compound 77
(76a) To a solution of 1-bromo-2-chloro-4-fluoro-5-methylbenzene (3.80 g, 17.0 mmol, 1.0 equiv.) and methyl 1H-pyrazole-4-carboxylate (2.10 g, 17.0 mmol, 1.0 equiv.) in DMA (40 mL) was added K2CO3 (4.7 g, 34.0 mmol, 2.0 equiv.). The mixture was stirred at 120°C for 20 h, cooled to ambient temperature, filtered and the filtrate was diluted with NH4Cl (100 mL) and extracted with ethyl acetate (100 mL x 2). The combined organic portions were dried over sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether/ ethyl acetate=4/1) to afford 76a. LCMS: [M+1] = 329.0, 331.0; 1H NMR (400 MHz, DMSO-d6) δ 8.74 (s, 1H), 8.15 (s, 1H), 7.90 (s, 1H), 7.77 (s, 1H), 3.79 (s, 3H), 2.20 (s, 3H). Step B – Synthesis of 1-(4-bromo-5-chloro-2-methylphenyl)-1H-pyrazole-4-carboxylic acid (76b) To a solution of 76a (1.00 g, 3.0 mmol, 1.0 equiv.) in THF (10 mL), MeOH (10 mL) and water (10 mL) was added lithium hydroxide hydrate (255 mg, 6.0 mmol, 2.0 equiv.). The mixture solution was stirred at 50°C for 1 h, cooled to ambient temperature and adjusted to pH = 2 with 1 M aqueous HCl. The solids were collected by vacuum filtration to afford 76b.
LCMS: [M+1] = 315.0, 317.0; 1H NMR (400 MHz, DMSO-d6) δ 12.62 (br s, 1H), 8.63 (s, 1H), 8.08 (s, 1H), 7.88 (s, 1H), 7.76 (s, 1H), 2.20 (s, 3H). Step C – Synthesis of 1-(4-bromo-5-chloro-2-methyl-phenyl)-N-[[3-(2,2,2-trifluoro-1,1-dimethyl- ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Intermediate 76) To a solution of 76b (100 mg, 316.9 µmol, 1.0 equiv.) and Int 1 (109.9 mg, 380 µmol, 1.2 equiv., HBr) in DMF (2.0 mL) was added triethylamine (320.6 mg, 3.1 mmol, 441 µL, 10 equiv.), then BOP (168.1 mg, 380.2 µmol, 1.2 equiv.) was added. The mixture was stirred at 20°C for 16 h. The reaction mixture was partitioned between ethyl acetate (10 mL) and water (8 mL). The organic phase was separated, washed with NaCl 10 mL, dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure to give a residue. The residue was purified by column chromatography (silica gel, dichloromethane: MeOH =94/6) to afford Intermediate 76. LCMS: [M+1] = 504.9, 506.9; 1H NMR (400 MHz, DMSO-d6) δ 13.94 (br s, 1H), 8.90 (br s, 1H), 8.58 (s, 1H), 8.19 (s, 1H), 7.90 (s, 1H), 7.73 (s, 1H), 4.54 (br d, J = 5.6 Hz, 2H), 3.33 (s, 3H), 2.23 (s, 3H), 1.53 (s, 6H). Example 77: Synthesis of Compound 77
Step A – Synthesis of tert-butyl 4-[1-[[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)phenyl]methyl]-4-piperidyl]piperazine-1-carboxylate (77a) A solution of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzaldehyde (5.00 g, 21.5 mmol, 1.0 equiv.) in dichloromethane (70.0 mL) was treated with tert-butyl 4-(4- piperidyl)piperazine-1-carboxylate (6.96 g, 25.8 mmol, 1.2 equiv.) and acetic acid (1.29 g, 21.5 mmol, 1.23 mL, 1.0 equiv.). The mixture was stirred at 20°C for 2 h, treated with NaBH(OAc)3 (11.4 g, 53.8 mmol, 2.5 equiv.), stirred at 20°C for 2 h, quenched with saturated aqueous NaHCO3 (25 mL), then extracted with ethyl acetate (20 mL x 3). The combined organic portions were washed with brine (25 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, dichloromethane : methanol=15/1 to 12/1) to afford 77a. LCMS: [M+1] = 486.3; 1H NMR (400 MHz, CHLOROFORM-d) δ 7.78 (d, J = 7.8 Hz, 2H), 7.35 (d, J = 7.6 Hz, 2H), 3.63 (s, 2H), 3.47 (d, J = 14.4 Hz, 4H), 3.02 (d, J = 10.0 Hz, 2H), 2.54 (s, 4H), 2.36 (s, 1H), 2.10 (d, J = 2.4 Hz, 1H), 1.89 - 1.60 (m, 4H), 1.46 (s, 9H), 1.35 (s, 12H).
Step B – Synthesis of tert-butyl 4-(1-((2'-chloro-5'-methyl-4'-(4-(((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)carbamoyl)-1H-pyrazol-1-yl)-[1,1'-biphenyl]-4- yl)methyl)piperidin-4-yl)piperazine-1-carboxylate (77b) To a solution of 77a (115.1 mg, 237.2 µmol, 1.2 equiv.) and Int 76 (100 mg, 198 µmol, 1.0 equiv.) in dioxane (2.0 mL) and water (0.4 mL), was added K2CO3 (81.9 mg, 593 µmol, 3.0 eq) and Pd(dppf)Cl2.CH2Cl2 (32.3 mg, 40 µmol, 0.2 equiv.). The mixture solution was stirred at 100°C for 2 h cooled to ambient temperature and partitioned between water (8 mL) and ethyl acetate (10 mL). The organic phase was separated, dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative TLC (silica gel, dichloromethane: MeOH = 10:1) to afford 77b. LCMS: [M+1] = 784.5; 1H NMR (400 MHz, DMSO-d6) δ 13.94 (br s, 1H), 8.90 (br s, 1H), 8.61 (s, 1H), 8.21 (s, 1H), 7.65 (s, 1H), 7.51 (s, 1H), 7.43 (q, J = 8.2 Hz, 4H), 4.56 (br d, J = 5.6 Hz, 2H), 3.51 (s, 2H), 3.28 (br s, 4H), 2.88 (br d, J = 11.4 Hz, 2H), 2.45 - 2.39 (m, 3H), 2.28 (s, 4H), 1.95 (br s, 2H), 1.70 (br s, 2H), 1.55 (s, 6H), 1.49 - 1.33 (m, 9H). Step C – Synthesis of 1-(2-chloro-4'-((4-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3- dioxoisoindolin-5-yl)piperazin-1-yl)piperidin-1-yl)methyl)-5-methyl-[1,1'-biphenyl]-4-yl)-N-((3- (1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide (Compound 77) A solution of 77b (50 mg, 64 µmol, 1.0 equiv.) in HCl/ dioxane (2.0 mL) was stirred at 20 °C for 1 h and then concentrated (LCMS: [M+1] = 684.4). The residue was dissolved in DMSO (1 mL) was treated with DIEA (51 µL) and 2-(2,6-dioxo-3-piperidyl)-5,6-difluoro-isoindoline- 1,3-dione (25.8 mg). The mixture solution was stirred at 70°C for 16 h, cooled to ambient temperature, diluted with water (5 mL) and extracted with ethyl acetate (4 mL x 2). The combined organic layers were washed with brine (5 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative TLC (silica gel, dichloromethane: MeOH = 10:1) to afford Compound 77. LCMS: [M+1] = 958.2; 1H NMR (400 MHz, DMSO-d6) δ 13.96 (br s, 1H), 11.11 (br s, 1H), 8.93 (br d, J = 5.2 Hz, 1H), 8.62 (s, 1H), 8.21 (s, 1H), 7.73 (d, J = 11.4 Hz, 1H), 7.65 (s, 1H), 7.52 (s, 1H), 7.49 - 7.39 (m, 4H), 5.11 (dd, J = 5.4, 12.8 Hz, 1H), 4.56 (br d, J = 5.8 Hz, 2H), 3.53 (s, 2H),
3.25 (br s, 4H), 2.91 (br d, J = 11.0 Hz, 2H), 2.68 (br d, J = 1.6 Hz, 5H), 2.63 - 2.55 (m, 3H), 2.28 (s, 4H), 2.10 - 1.94 (m, 3H), 1.79 (br d, J = 10.8 Hz, 2H), 1.55 (s, 6H), 1.48 (br d, J = 9.0 Hz, 2H). Example 78: Synthesis of Compound 78
 Step A – Synthesis of tert-butyl N-[(3S)-1-[[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)phenyl]methyl]pyrrolidin-3-yl]carbamate (78a) A solution of 2-[4-(bromomethyl)phenyl]-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (1.00 g, 3.37 mmol, 1.0 equiv.), K2CO3 (1.16 g, 8.42 mmol, 2.5 equiv.) in DMF (16.0 mL) was added tert- butyl N-[(3S)-pyrrolidin-3-yl]carbamate (752.5 mg, 4.04 mmol, 1.2 equiv.). The mixture was stirred at 20°C for 2 h, poured into saturated aqueous ammonium chloride (30 mL) and extracted with ethyl acetate (20 mL x 3). The combined organic layers were washed with brine (40 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether/ ethyl acetate=1/0 to 1/2) to afford 78a. LCMS: [M+1] = 403.3. Step B – Synthesis of tert-butyl N-[(3S)-1-[[4-[2,6-dimethyl-3-[4-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methylcarbamoyl]pyrazol-1- yl]phenyl]phenyl]methyl]pyrrolidin-3-yl]carbamate (78b) To a solution of 78a (99.4 mg, 247 μmol, 1.5 equiv.) and Int 5 (80.0 mg, 165 μmol, 1.0 equiv.) in THF (2.0 mL) and water (0.5 mL) was added PdCl2(DTBPF) (21.4 mg, 33 μmol, 0.2
equiv.) and K3PO4 (104.9 mg, 494.5 μmol, 3.0 equiv.). The mixture was stirred at 80°C for 16 h under a nitrogen atmosphere. The mixture was cooled to ambient temperature poured into water (10 mL) and extracted with ethyl acetate (10 mL×3). The combined organic layers were dried over sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative TLC (silica gel, dichloromethane : methanol = 10:1, Rf = 0.38) to afford 78b. LCMS: [M+1] = 681.5. Step C – Synthesis of 1-[3-[4-[[(3S)-3-[[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin- 5-yl]amino]pyrrolidin-1-yl]methyl]phenyl]-2,4-dimethyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Compound 78). A solution of 78b (50.0 mg, 73 μmol, 1.0 equiv.) in dichloromethane (0.5 mL) and TFA (0.1 mL) was stirred at 20°C for 1 h and then concentrated under vacuum (LCMS: [M+1] = 581.5). The residue was dissolved in DMSO (0.5 mL) was treated with DIEA (46.5 mg) and 2- (2,6-dioxo-3-piperidyl)-5,6-difluoro-isoindoline-1,3-dione (31.7 mg). The mixture was stirred at 100°C for 16 h, cooled to ambient temperature and then extracted with ethyl acetate (10 mL x 3) and water (10 mL). The combined organic layers were washed with brine (5 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative HPLC. (Phenomenex Luna C18, 100 mm x 40mm, 3 µm; [water(0.2% FA)-ACN]) to afford Compound 78. LCMS: [M+1] = 855.2; 1H NMR (400 MHz, CHLOROFORM-d) δ 9.18 - 8.87 (m, 1H), 8.24 - 8.13 (m, 1H), 7.95 (s, 1H), 7.49 - 7.33 (m, 3H), 7.26 - 7.14 (m, 2H), 7.12 - 6.98 (m, 3H), 6.95 - 6.81 (m, 1H), 5.27 - 5.11 (m, 1H), 4.91 (ddd, J = 5.4, 7.6, 12.6 Hz, 1H), 4.79 - 4.63 (m, 2H), 4.22 - 4.09 (m, 1H), 3.94 - 3.82 (m, 1H), 3.77 - 3.64 (m, 1H), 3.21 - 3.09 (m, 1H), 2.95 - 2.80 (m, 3H), 2.73 (br dd, J = 3.8, 12.2 Hz, 2H), 2.54 - 2.46 (m, 1H), 2.12 (ddd, J = 2.4, 5.2, 7.6 Hz, 1H), 2.05 (d, J = 4.2 Hz, 3H), 1.95 - 1.85 (m, 1H), 1.81 - 1.72 (m, 3H), 1.62 (s, 6H). Example 79: Synthesis of Compound 79
Step A – Synthesis of tert-butyl N-[(3S)-1-[[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)phenyl]methyl]pyrrolidin-3-yl]carbamate (78a) A solution of 2-[4-(bromomethyl)phenyl]-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (1.00 g, 3.37 mmol, 1.0 equiv.), K2CO3 (1.16 g, 8.42 mmol, 2.5 equiv.) in DMF (16.0 mL) was added tert- butyl N-[(3S)-pyrrolidin-3-yl]carbamate (752.5 mg, 4.04 mmol, 1.2 equiv.). The mixture was stirred at 20°C for 2 h, poured into saturated aqueous ammonium chloride (30 mL) and extracted with ethyl acetate (20 mL x 3). The combined organic layers were washed with brine (40 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether/ ethyl acetate=1/0 to 1/2) to afford 78a. LCMS: [M+1] = 403.3. Step B – Synthesis of tert-butyl N-[(3S)-1-[[4-[2,6-dimethyl-3-[4-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methylcarbamoyl]pyrazol-1- yl]phenyl]phenyl]methyl]pyrrolidin-3-yl]carbamate (78b) To a solution of 78a (99.4 mg, 247 μmol, 1.5 equiv.) and Int 5 (80.0 mg, 165 μmol, 1.0 equiv.) in THF (2.0 mL) and water (0.5 mL) was added PdCl2(DTBPF) (21.4 mg, 33 μmol, 0.2
equiv.) and K3PO4 (104.9 mg, 494.5 μmol, 3.0 equiv.). The mixture was stirred at 80°C for 16 h under a nitrogen atmosphere. The mixture was cooled to ambient temperature poured into water (10 mL) and extracted with ethyl acetate (10 mL×3). The combined organic layers were dried over sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative TLC (silica gel, dichloromethane : methanol = 10:1, Rf = 0.38) to afford 78b. LCMS: [M+1] = 681.5. Step C – Synthesis of 1-[3-[4-[[(3S)-3-[[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin- 5-yl]amino]pyrrolidin-1-yl]methyl]phenyl]-2,4-dimethyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Compound 78). A solution of 78b (50.0 mg, 73 μmol, 1.0 equiv.) in dichloromethane (0.5 mL) and TFA (0.1 mL) was stirred at 20°C for 1 h and then concentrated under vacuum (LCMS: [M+1] = 581.5). The residue was dissolved in DMSO (0.5 mL) was treated with DIEA (46.5 mg) and 2- (2,6-dioxo-3-piperidyl)-5,6-difluoro-isoindoline-1,3-dione (31.7 mg). The mixture was stirred at 100°C for 16 h, cooled to ambient temperature and then extracted with ethyl acetate (10 mL x 3) and water (10 mL). The combined organic layers were washed with brine (5 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative HPLC. (Phenomenex Luna C18, 100 mm x 40mm, 3 µm; [water(0.2% FA)-ACN]) to afford Compound 78. LCMS: [M+1] = 855.2; 1H NMR (400 MHz, CHLOROFORM-d) δ 9.18 - 8.87 (m, 1H), 8.24 - 8.13 (m, 1H), 7.95 (s, 1H), 7.49 - 7.33 (m, 3H), 7.26 - 7.14 (m, 2H), 7.12 - 6.98 (m, 3H), 6.95 - 6.81 (m, 1H), 5.27 - 5.11 (m, 1H), 4.91 (ddd, J = 5.4, 7.6, 12.6 Hz, 1H), 4.79 - 4.63 (m, 2H), 4.22 - 4.09 (m, 1H), 3.94 - 3.82 (m, 1H), 3.77 - 3.64 (m, 1H), 3.21 - 3.09 (m, 1H), 2.95 - 2.80 (m, 3H), 2.73 (br dd, J = 3.8, 12.2 Hz, 2H), 2.54 - 2.46 (m, 1H), 2.12 (ddd, J = 2.4, 5.2, 7.6 Hz, 1H), 2.05 (d, J = 4.2 Hz, 3H), 1.95 - 1.85 (m, 1H), 1.81 - 1.72 (m, 3H), 1.62 (s, 6H). Example 79: Synthesis of Compound 79
Step A a) A solution of tert-butyl 4-hydroxypiperidine-1-carboxylate (2.70 g, 13.6 mmol, 1.2 equiv.) in DMF (20.0 mL) was treated with NaH (909.1 mg, 22.7 mmol, 60% purity, 2.0 equiv.). The mixture was stirred at 0°C for 1 h. 5-bromo-2-fluoro-pyridine (2.0 g, 11.4 mmol, 1.17 mL, 1.0 equiv.) was added to the reaction mixture, and the mixture was stirred at 25 °C for 12 h. The reaction mixture was diluted with water (20 mL) and extracted with ethyl acetate (20 mL x 3). The combined organic layers were washed with brine (20 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether: ethyl acetate = 1: 0 to 0: 1) to afford 79a. LCMS [M+1] = 357.1, 359.1; 1H NMR (400 MHz, DMSO-d6) δ 8.26 (s, 1H), 7.97 - 7.78 (m, 1H), 6.80 (dd, J = 8.6 Hz, 1H), 5.11 (s, 1H), 7.61 (s, 1H), 3.76 – 3.62 (m, 2H), 3.15 (s, 2H), 1.92 (s, 2H), 1.58 - 1.50 (m, 2H), 1.40 (s, 9H). Step B – Synthesis of tert-butyl 4-[[5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-2- pyridyl]oxy] piperidine-1-carboxylate (79b) A solution of 79a (3.00 g, 8.40 mmol, 1.0 equiv.) and bis(pinacolato)diboron (3.20 g, 12.6 mmol, 1.5 equiv.) in dioxane (50 mL) was treated with KOAc (2.5 g, 25.2 mmol, 3.0 equiv.) and Pd(dppf)Cl2 (614.5 mg, 839.8 μmol, 0.1 equiv.). The mixture was stirred at 85 °C for 12 h, cooled to ambient temperature, diluted with water (40.0 mL) and extracted with ethyl acetate
(40.0 mL x 3). The combined organic layers were washed with brine (40.0 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure to afford 79b, which was used without further purification. LCMS [M+1] = 405.3; 1H NMR (400 MHz, DMSO-d6) δ 8.37 (s, 1H), 7.86 (dd, J = 8.0 Hz, 1H), 6.77 (dd, J = 8.0 Hz, 1H), 5.23 (s, 1H), 3.68 (dd, J = 12.8 Hz, 2H), 3.17 (s, 2H), 1.93 (dd, J = 8.0 Hz, 2H), 1.59 – 1.50 (m, 2H), 1.31 - 1.13 (m, 21H). Step C – Synthesis of tert-butyl 4- [[5-[2,6-dimethyl-3-[4-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H-1,2,4-triazol-5-yl]methylcarbamoyl]pyrazol-1-yl]phenyl]-2-pyridyl]oxy]piperidine-1- carboxylate (79c) A solution of 79b (74.9 mg, 185 μmol, 1.5 equiv.), Int 5 (60 mg, 124 μmol, 1.0 equiv.) in THF (1.2 mL) and water (0.3 mL) was treated with K3PO4 (78.7 mg, 370.9 μmol, 3.0 equiv.) and PdCl2(DTBPF) (16.1 mg, 24 μmol, 0.2 equiv.) at 20°C under nitrogen. The mixture was stirred at 80°C under nitrogen for 16 h, cooled to ambient temperature, quenched with water (30 mL) and extracted with ethyl acetate (10 mL x 3). The combined organic portions were washed with brine (10 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure to give a residue. The residue was purified by column chromatography (silica gel, dichloromethane: methanol = 10:1) to afford 79c. LCMS [M+1] =683.6; 1H NMR (400 MHz, DMSO-d6) δ 13.87 (br s, 1H), 8.88 (br s, 1H), 8.42 (s, 1H), 8.14 (s, 1H), 8.00 (d, J = 2.4 Hz, 1H), 7.59 (dd, J = 2.4, 8.4 Hz, 1H), 7.32 (s, 2H), 6.91 (d, J = 8.6 Hz, 1H), 5.27 - 5.15 (m, 1H), 4.60 - 4.46 (m, 2H), 3.75 (br d, J = 13.4 Hz, 2H), 3.22 - 3.10 (m, 2H), 2.06 (s, 3H), 2.03 - 1.94 (m, 2H), 1.79 (s, 3H), 1.64 - 1.48 (m, 8H), 1.41 (s, 9H). Step D – Synthesis of 1-[3-[6-[[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]- 4-piperidyl]oxy]-3-pyridyl]-2,4-dimethyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H- 1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Compound 79) A solution of 79c (55 mg, 81 μmol, 1.0 equiv.) in HCl/dioxane (1 mL) was stirred at 25°C for 1 h and then concentrated under reduced pressure (LCMS [M+1] =583.4). The residue was dissolved in DMSO (1 mL) and treated with 2-(2,6-dioxo-3-piperidyl)-5,6-difluoro-isoindoline- 1,3-dione (37.8 mg) and DIEA (45 μL). The mixture was stirred at 70°C for 16 h, cooled to ambient temperature and partitioned between ethyl acetate (40 mL) and water (40 mL). The
organic phase was separated, washed with brine (30 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative TLC (silica gel, dichloromethane: methanol = 10:1) to afford Compound 79. LCMS [M+1] =857.3; 1H NMR (400 MHz, DMSO-d6) δ 13.90 (br s, 1H), 11.11 (br s, 1H), 8.86 (br s, 1H), 8.43 (s, 1H), 8.14 (s, 1H), 8.03 (d, J = 2.4 Hz, 1H), 7.74 (d, J = 11.4 Hz, 1H), 7.61 (dd, J = 2.4, 8.4 Hz, 1H), 7.52 (d, J = 7.4 Hz, 1H), 7.32 (s, 2H), 6.95 (d, J = 8.6 Hz, 1H), 5.29 (tt, J = 4.0, 8.2 Hz, 1H), 5.11 (dd, J = 5.4, 12.8 Hz, 1H), 4.60 - 4.47 (m, 2H), 3.64 - 3.52 (m, 2H), 3.23 (br t, J = 10.4 Hz, 2H), 2.95 - 2.82 (m, 1H), 2.64 - 2.53 (m, 2H), 2.19 (br d, J = 2.4 Hz, 2H), 2.08 (s, 4H), 1.92 - 1.77 (m, 5H), 1.53 (s, 6H). Example 80: Synthesis of Compound 80
 trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (80a) A solution of Int 5 (200 mg, 412 μmol, 1.0 equiv.) and 2-fluoro-5-(4,4,5,5-tetramethyl- 1,3,2-dioxaborolan-2-yl)pyridine (137.8 mg, 618 μmol, 1.5 equiv.) in THF (4.0 mL) and water (1.0 mL) was treated with K3PO4 (262.4 mg, 1.2 mmol, 3.0 equiv.) and PdCl2(DTBPF) (53.7 mg, 82 μmol, 0.2 equiv.). The mixture was stirred at 80°C under nitrogen for 16 h, cooled to ambient
temperature, quenched with water (10mL) and extracted with ethyl acetate (5 mL x 2). The combined organic portions were washed with brine (5 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, dichloromethane: methanol=100/0 to 96/4) to afford 80a. LCMS [M+1] =502.3; 1H NMR (400 MHz, DMSO-d6) δ 13.87 (s, 1H), 8.89 (br t, J = 5.8 Hz, 1H), 8.43 (s, 1H), 8.21 - 8.04 (m, 2H), 7.92 (dt, J = 2.4, 8.2 Hz, 1H), 7.42 - 7.21 (m, 3H), 4.55 (d, J = 5.8 Hz, 2H), 2.05 (s, 3H), 1.78 (s, 3H), 1.52 (s, 6H). Step B – Synthesis of tert-butyl 4-[[4-[5-[2,6-dimethyl-3-[4-[[3-(2,2,2-trifluoro-1,1-dimethyl- ethyl)-1H-1,2,4-triazol-5-yl]methylcarbamoyl]pyrazol-1-yl]phenyl]-2-pyridyl]piperazin-1- yl]methyl]piperidine-1-carboxylate (80b) A solution of 80a (100 mg, 199 μmol, 1.0 equiv.),tert-butyl 4-(piperazin-1- ylmethyl)piperidine-1-carboxylate (113.0 mg, 399 μmol, 2.0 equiv.) in DMSO (1.0 mL) was added DIEA (77.3 mg, 598 μmol, 104 μL, 3.0 equiv.). The mixture was stirred at 80°C for 16 h, cooled to ambient temperature, quenched with water (50 mL) and extracted with ethyl acetate (25 mL x 2). The combined organic portions were washed with brine (5 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography by preparative TLC (silica gel, dichloromethane: MeOH = 10:1) to afford 80b. LCMS [M+1] =765.7; 1H NMR (400 MHz, DMSO-d6) δ 13.88 (br s, 1H), 8.87 (br s, 1H), 8.46 - 8.38 (m, 1H), 8.17 - 8.08 (m, 1H), 7.94 (d, J = 2.4 Hz, 1H), 7.40 (dd, J = 2.4, 8.7 Hz, 1H), 7.36 - 7.24 (m, 2H), 6.92 (d, J = 8.8 Hz, 1H), 4.54 (br s, 2H), 3.93 (br d, J = 10.4 Hz, 2H), 3.52 (br s, 4H), 2.82 - 2.60 (m, 2H), 2.48 - 2.39 (m, 4H), 2.18 (br d, J = 6.8 Hz, 2H), 2.11 - 2.02 (m, 3H), 1.82 - 1.75 (m, 3H), 1.71 (br d, J = 12.0 Hz, 3H), 1.53 (br s, 6H), 1.39 (s, 9H), 1.06 - 0.86 (m, 2H). Step C – Synthesis of 1-[3-[6-[4-[[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]-4-piperidyl]methyl]piperazin-1-yl]-3-pyridyl]-2,4-dimethyl-phenyl]-N-[[3-(2,2,2-trifluoro- 1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Compound 80) To a solution of 80b (60 mg, 78.4 μmol, 1.0 equiv.) in HCl/ dioxane (1 mL). The mixture was stirred at 25°C for 1 h and concentrated under reduced pressure (LCMS [M+1] =665.6). The residue was dissolved in DMSO (0.6 mL) was treated with DIEA (43 μL) and 2-(2,6-dioxo-3-
piperidyl)-5,6-difluoro-isoindoline-1,3-dione (36.5 mg). The mixture was stirred at 70°C for 16 h, cooled to ambient temperature. The mixture was partitioned between ethyl acetate (40 mL) and water (40 mL). The organic phase was separated, washed with brine (30 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative TLC (silica gel, dichloromethane: methanol = 10:1) to afford Compound 80. LCMS [M+1] =939.4; 1H NMR (400 MHz, DMSO-d6) δ 13.88 (br d, J = 1.2 Hz, 1H), 11.10 (br s, 1H), 8.94 - 8.78 (m, 1H), 8.42 (s, 1H), 8.13 (s, 1H), 7.95 (d, J = 2.4 Hz, 1H), 7.71 (d, J = 11.4 Hz, 1H), 7.47 - 7.38 (m, 2H), 7.33 - 7.24 (m, 2H), 6.93 (d, J = 8.8 Hz, 1H), 5.10 (dd, J = 5.4, 12.8 Hz, 1H), 4.54 (br d, J = 4.5 Hz, 2H), 3.63 (br d, J = 11.6 Hz, 2H), 3.55 (br s, 4H), 2.99 - 2.79 (m, 3H), 2.70 - 2.51 (m, 5H), 2.35 - 2.19 (m, 2H), 2.12 - 1.96 (m, 4H), 1.92 - 1.76 (m, 6H), 1.53 (s, 6H), 1.38 - 1.18 (m, 3H). Example 81: Synthesis of Compound 81
trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (80a) A solution of Int 5 (200 mg, 412 μmol, 1.0 equiv.) and 2-fluoro-5-(4,4,5,5-tetramethyl- 1,3,2-dioxaborolan-2-yl)pyridine (137.8 mg, 618 μmol, 1.5 equiv.) in THF (4.0 mL) and water (1.0 mL) was treated with K3PO4 (262.4 mg, 1.2 mmol, 3.0 equiv.) and PdCl2(DTBPF) (53.7 mg, 82 μmol, 0.2 equiv.). The mixture was stirred at 80°C under nitrogen for 16 h, cooled to ambient
temperature, quenched with water (10mL) and extracted with ethyl acetate (5 mL x 2). The combined organic portions were washed with brine (5 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, dichloromethane: methanol=100/0 to 96/4) to afford 80a. LCMS [M+1] =502.3; 1H NMR (400 MHz, DMSO-d6) δ 13.87 (s, 1H), 8.89 (br t, J = 5.8 Hz, 1H), 8.43 (s, 1H), 8.21 - 8.04 (m, 2H), 7.92 (dt, J = 2.4, 8.2 Hz, 1H), 7.42 - 7.21 (m, 3H), 4.55 (d, J = 5.8 Hz, 2H), 2.05 (s, 3H), 1.78 (s, 3H), 1.52 (s, 6H). Step B – Synthesis of tert-butyl 4-[[4-[5-[2,6-dimethyl-3-[4-[[3-(2,2,2-trifluoro-1,1-dimethyl- ethyl)-1H-1,2,4-triazol-5-yl]methylcarbamoyl]pyrazol-1-yl]phenyl]-2-pyridyl]piperazin-1- yl]methyl]piperidine-1-carboxylate (80b) A solution of 80a (100 mg, 199 μmol, 1.0 equiv.),tert-butyl 4-(piperazin-1- ylmethyl)piperidine-1-carboxylate (113.0 mg, 399 μmol, 2.0 equiv.) in DMSO (1.0 mL) was added DIEA (77.3 mg, 598 μmol, 104 μL, 3.0 equiv.). The mixture was stirred at 80°C for 16 h, cooled to ambient temperature, quenched with water (50 mL) and extracted with ethyl acetate (25 mL x 2). The combined organic portions were washed with brine (5 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography by preparative TLC (silica gel, dichloromethane: MeOH = 10:1) to afford 80b. LCMS [M+1] =765.7; 1H NMR (400 MHz, DMSO-d6) δ 13.88 (br s, 1H), 8.87 (br s, 1H), 8.46 - 8.38 (m, 1H), 8.17 - 8.08 (m, 1H), 7.94 (d, J = 2.4 Hz, 1H), 7.40 (dd, J = 2.4, 8.7 Hz, 1H), 7.36 - 7.24 (m, 2H), 6.92 (d, J = 8.8 Hz, 1H), 4.54 (br s, 2H), 3.93 (br d, J = 10.4 Hz, 2H), 3.52 (br s, 4H), 2.82 - 2.60 (m, 2H), 2.48 - 2.39 (m, 4H), 2.18 (br d, J = 6.8 Hz, 2H), 2.11 - 2.02 (m, 3H), 1.82 - 1.75 (m, 3H), 1.71 (br d, J = 12.0 Hz, 3H), 1.53 (br s, 6H), 1.39 (s, 9H), 1.06 - 0.86 (m, 2H). Step C – Synthesis of 1-[3-[6-[4-[[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]-4-piperidyl]methyl]piperazin-1-yl]-3-pyridyl]-2,4-dimethyl-phenyl]-N-[[3-(2,2,2-trifluoro- 1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Compound 80) To a solution of 80b (60 mg, 78.4 μmol, 1.0 equiv.) in HCl/ dioxane (1 mL). The mixture was stirred at 25°C for 1 h and concentrated under reduced pressure (LCMS [M+1] =665.6). The residue was dissolved in DMSO (0.6 mL) was treated with DIEA (43 μL) and 2-(2,6-dioxo-3-
piperidyl)-5,6-difluoro-isoindoline-1,3-dione (36.5 mg). The mixture was stirred at 70°C for 16 h, cooled to ambient temperature. The mixture was partitioned between ethyl acetate (40 mL) and water (40 mL). The organic phase was separated, washed with brine (30 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative TLC (silica gel, dichloromethane: methanol = 10:1) to afford Compound 80. LCMS [M+1] =939.4; 1H NMR (400 MHz, DMSO-d6) δ 13.88 (br d, J = 1.2 Hz, 1H), 11.10 (br s, 1H), 8.94 - 8.78 (m, 1H), 8.42 (s, 1H), 8.13 (s, 1H), 7.95 (d, J = 2.4 Hz, 1H), 7.71 (d, J = 11.4 Hz, 1H), 7.47 - 7.38 (m, 2H), 7.33 - 7.24 (m, 2H), 6.93 (d, J = 8.8 Hz, 1H), 5.10 (dd, J = 5.4, 12.8 Hz, 1H), 4.54 (br d, J = 4.5 Hz, 2H), 3.63 (br d, J = 11.6 Hz, 2H), 3.55 (br s, 4H), 2.99 - 2.79 (m, 3H), 2.70 - 2.51 (m, 5H), 2.35 - 2.19 (m, 2H), 2.12 - 1.96 (m, 4H), 1.92 - 1.76 (m, 6H), 1.53 (s, 6H), 1.38 - 1.18 (m, 3H). Example 81: Synthesis of Compound 81
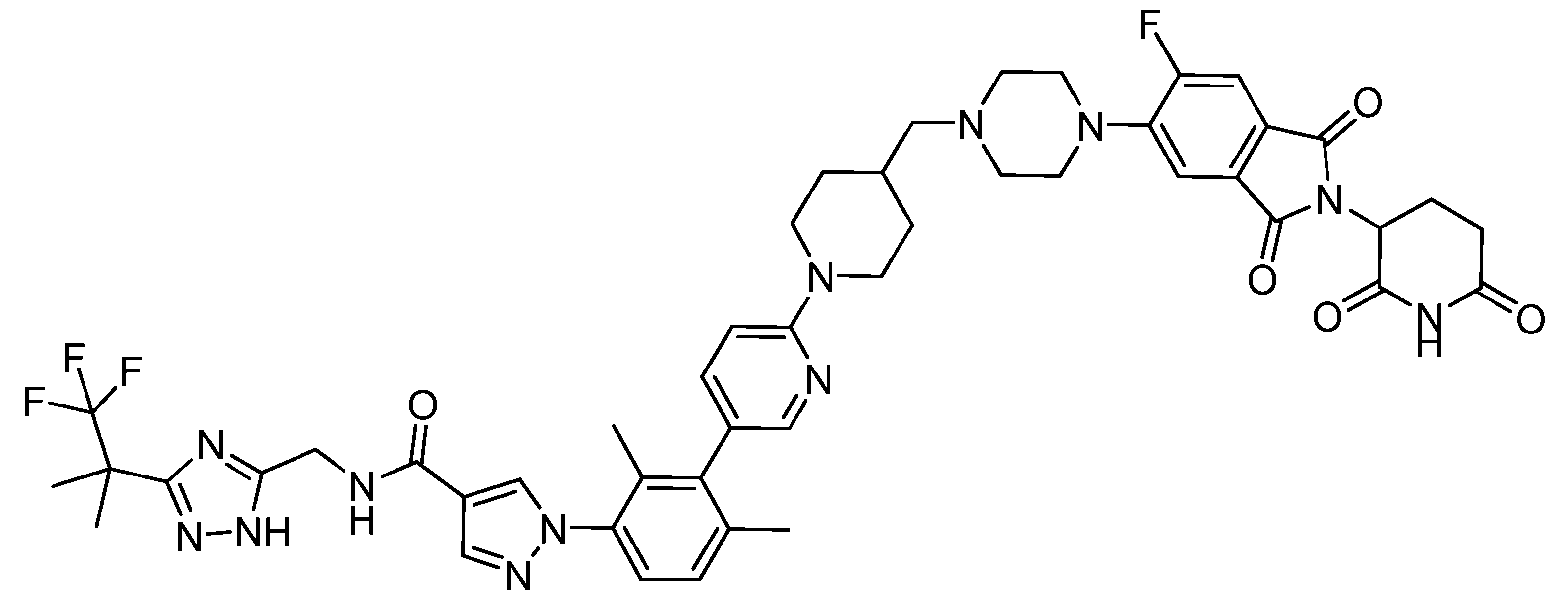 tert-butyl 4-(4-piperidylmethyl)piperazine-1-carboxylate in Step B to afford 1-(3-(6-(4-((4-(2-(2,6- dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1-yl)methyl)piperidin-1- yl)pyridin-3-yl)-2,4-dimethylphenyl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide. LCMS [M+1] =939.4; 1H NMR (400 MHz, DMSO-d6) δ 13.89 (br s, 1H), 11.11 (br s, 1H), 8.86 (br dd, J = 4.1, 8.3 Hz, 1H), 8.42 (s, 1H), 8.13 (s, 1H), 7.92 (d, J = 2.4 Hz, 1H), 7.73 (d, J = 11.5 Hz, 1H), 7.46 (d, J = 7.4 Hz, 1H), 7.37 (dd, J = 2.3, 8.7 Hz, 1H), 7.31 - 7.25 (m, 2H), 6.92 (d, J = 8.8 Hz, 1H), 5.11 (dd, J = 5.4, 12.9 Hz, 1H), 4.54 (br d, J = 4.5 Hz, 2H), 4.35 (br d, J = 12.8 Hz, 2H), 3.27 (br s, 4H), 2.95 - 2.77 (m, 3H), 2.63 - 2.54 (m, 4H), 2.54 (s, 1H), 2.25 (br d, J = 6.0 Hz, 2H), 2.09 (s, 4H), 1.81 (s, 6H), 1.53 (s, 6H), 1.23 (s, 3H)
Example 82: Synthesis of Compound 82
tert-butyl 4-(4-piperidylmethyl)piperazine-1-carboxylate in Step B to afford 1-(3-(6-(4-((4-(2-(2,6- dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1-yl)methyl)piperidin-1- yl)pyridin-3-yl)-2,4-dimethylphenyl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide. LCMS [M+1] =939.4; 1H NMR (400 MHz, DMSO-d6) δ 13.89 (br s, 1H), 11.11 (br s, 1H), 8.86 (br dd, J = 4.1, 8.3 Hz, 1H), 8.42 (s, 1H), 8.13 (s, 1H), 7.92 (d, J = 2.4 Hz, 1H), 7.73 (d, J = 11.5 Hz, 1H), 7.46 (d, J = 7.4 Hz, 1H), 7.37 (dd, J = 2.3, 8.7 Hz, 1H), 7.31 - 7.25 (m, 2H), 6.92 (d, J = 8.8 Hz, 1H), 5.11 (dd, J = 5.4, 12.9 Hz, 1H), 4.54 (br d, J = 4.5 Hz, 2H), 4.35 (br d, J = 12.8 Hz, 2H), 3.27 (br s, 4H), 2.95 - 2.77 (m, 3H), 2.63 - 2.54 (m, 4H), 2.54 (s, 1H), 2.25 (br d, J = 6.0 Hz, 2H), 2.09 (s, 4H), 1.81 (s, 6H), 1.53 (s, 6H), 1.23 (s, 3H)
Example 82: Synthesis of Compound 82
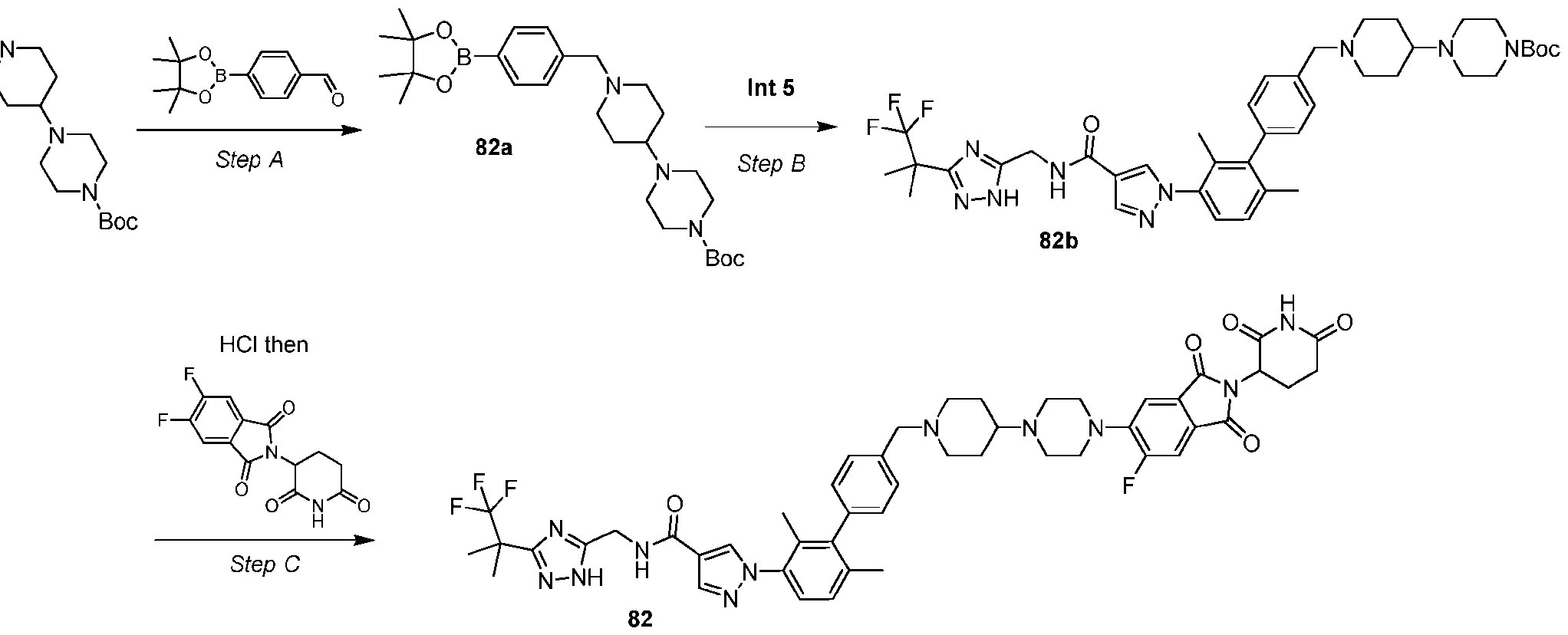 Step A – Synthesis of tert-butyl 4-[1-[[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)phenyl]methyl]-4-piperidyl]piperazine-1-carboxylate (82a) To a solution of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzaldehyde (5.00 g, 21.5 mmol, 1.0 equiv.) in dichloromethane (70.0 mL) was added tert-butyl 4-(4- piperidyl)piperazine-1-carboxylate (6.96 g, 25.8 mmol, 1.2 equiv.) and acetic acid (1.29 g, 21.5 mmol, 1.23 mL, 1.0 equiv.) the mixture was stirred at 20°C for 2 h and then treated with NaBH(OAc)3 (11.4 g, 53.8 mmol, 2.5 equiv.). The resulting mixture was stirred at 20°C for 2 h, quenched with saturated aqueous NaHCO3 (25 mL) and extracted with ethyl acetate (20 mL x 3). The combined organic layers were washed with brine (25 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, dichloromethane : methanol=15/1 to 12/1) to afford 82a. LCMS: [M+1] = 486.3; 1H NMR (400 MHz, CHLOROFORM-d) δ 7.78 (d, J = 7.8 Hz, 2H), 7.35 (d, J = 7.6 Hz, 2H), 3.63 (s, 2H), 3.50 - 3.45 (m, 4H), 3.02 (d, J = 10.0 Hz, 2H), 2.54 (s, 4H), 2.36 (s, 1H), 2.10 (d, J = 2.4 Hz, 1H), 1.89 - 1.60 (m, 4H), 1.46 (s, 9H), 1.35 (s, 12H).
Step B – Synthesis of tert-butyl 4-[1-[[4-[2,6-dimethyl-3-[4-[[3-(2,2,2-trifluoro-1,1-dimethyl- ethyl)-1H-1,2,4-triazol-5-yl]methylcarbamoyl]pyrazol-1-yl]phenyl]phenyl]methyl]-4- piperidyl]piperazine-1-carboxylate (82b) A solution of 82a (120.0 mg, 247 μmol, 1.5 equiv.), Int 5 (80 mg, 165 μmol, 1.0 equiv.) in THF (1.6 mL) and water (0.4 mL) was treated with K3PO4 (104.9 mg, 495 μmol, 3.0 equiv.) and PdCl2(DTBPF) (21.4 mg, 33 μmol, 0.2 equiv.) under a nitrogen atmosphere. The mixture was stirred at 80°C for 16 h, cooled to ambient temperature, quenched with water (80 mL) and extracted with ethyl acetate (30 mL x 3). The combined organic layers were washed with brine (50 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure to give a residue. The residue was purified by preparative TLC (silica gel, dichloromethane: methanol = 10:1) to afford 82b. LCMS [M+1] =764.6; 1H NMR (400 MHz, DMSO-d6) δ 13.90 (br s, 1H), 8.84 (br s, 1H), 8.43 (s, 1H), 8.13 (s, 1H), 7.39 (br d, J = 8.0 Hz, 2H), 7.32 - 7.24 (m, 2H), 7.13 (d, J = 7.8 Hz, 2H), 4.53 (br d, J = 5.6 Hz, 2H), 3.50 (s, 2H), 3.29 - 3.21 (m, 4H), 2.95 - 2.82 (m, 2H), 2.41 (br s, 4H), 2.26 - 2.15 (m, 1H), 2.09 - 1.88 (m, 5H), 1.82 - 1.64 (m, 5H), 1.53 (s, 6H), 1.47 - 1.34 (m, 11H). Step C – Synthesis of 1-[3-[4-[[4-[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]piperazin-1-yl]-1-piperidyl]methyl]phenyl]-2,4-dimethyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Compound 82) A solution of 82b (90 mg, 118 μmol, 1.0 equiv.) in HCl/dioxane (1 mL) was stirred at 20°C for 1 h and then concentrated (LCMS [M+1] =664.7). The residue was dissolved in DMSO (1 mL) and treated with 2-(2,6-dioxo-3-piperidyl)-5,6-difluoro-isoindoline-1,3-dione (53.1 mg) and DIEA (63 μL). The mixture was stirred at 70°C for 16 h, cooled to ambient temperature, partitioned between ethyl acetate (40 mL) and water (40 mL). The organic phase was separated, washed with brine (30 mL), dried over sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative TLC (silica gel, dichloromethane: methanol = 10:1) to afford Compound 82.. LCMS [M+1] =938.4; 1H NMR (400 MHz, DMSO-d6) δ 13.90 (br s, 1H), 11.10 (s, 1H), 8.83 (br s, 1H), 8.43 (s, 1H), 8.13 (s, 1H), 7.72 (d, J = 11.4 Hz, 1H), 7.43 (br dd, J = 7.6, 11.1 Hz, 3H), 7.33 - 7.24 (m, 2H), 7.14 (br d, J = 7.8 Hz, 2H), 5.10 (dd, J = 5.4, 12.8 Hz, 1H), 4.53 (br d, J = 5.6 Hz, 2H), 3.52 (br s, 2H), 3.23 (br
s, 4H), 2.95 - 2.80 (m, 3H), 2.72 - 2.55 (m, 6H), 2.32 - 2.17 (m, 1H), 2.03 (s, 6H), 1.76 (s, 5H), 1.53 (s, 8H). Example 83: Synthesis of Compound 83 tert-butyl
Step A – Synthesis of tert-butyl 4-[1-[[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)phenyl]methyl]-4-piperidyl]piperazine-1-carboxylate (82a) To a solution of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzaldehyde (5.00 g, 21.5 mmol, 1.0 equiv.) in dichloromethane (70.0 mL) was added tert-butyl 4-(4- piperidyl)piperazine-1-carboxylate (6.96 g, 25.8 mmol, 1.2 equiv.) and acetic acid (1.29 g, 21.5 mmol, 1.23 mL, 1.0 equiv.) the mixture was stirred at 20°C for 2 h and then treated with NaBH(OAc)3 (11.4 g, 53.8 mmol, 2.5 equiv.). The resulting mixture was stirred at 20°C for 2 h, quenched with saturated aqueous NaHCO3 (25 mL) and extracted with ethyl acetate (20 mL x 3). The combined organic layers were washed with brine (25 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, dichloromethane : methanol=15/1 to 12/1) to afford 82a. LCMS: [M+1] = 486.3; 1H NMR (400 MHz, CHLOROFORM-d) δ 7.78 (d, J = 7.8 Hz, 2H), 7.35 (d, J = 7.6 Hz, 2H), 3.63 (s, 2H), 3.50 - 3.45 (m, 4H), 3.02 (d, J = 10.0 Hz, 2H), 2.54 (s, 4H), 2.36 (s, 1H), 2.10 (d, J = 2.4 Hz, 1H), 1.89 - 1.60 (m, 4H), 1.46 (s, 9H), 1.35 (s, 12H).
Step B – Synthesis of tert-butyl 4-[1-[[4-[2,6-dimethyl-3-[4-[[3-(2,2,2-trifluoro-1,1-dimethyl- ethyl)-1H-1,2,4-triazol-5-yl]methylcarbamoyl]pyrazol-1-yl]phenyl]phenyl]methyl]-4- piperidyl]piperazine-1-carboxylate (82b) A solution of 82a (120.0 mg, 247 μmol, 1.5 equiv.), Int 5 (80 mg, 165 μmol, 1.0 equiv.) in THF (1.6 mL) and water (0.4 mL) was treated with K3PO4 (104.9 mg, 495 μmol, 3.0 equiv.) and PdCl2(DTBPF) (21.4 mg, 33 μmol, 0.2 equiv.) under a nitrogen atmosphere. The mixture was stirred at 80°C for 16 h, cooled to ambient temperature, quenched with water (80 mL) and extracted with ethyl acetate (30 mL x 3). The combined organic layers were washed with brine (50 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure to give a residue. The residue was purified by preparative TLC (silica gel, dichloromethane: methanol = 10:1) to afford 82b. LCMS [M+1] =764.6; 1H NMR (400 MHz, DMSO-d6) δ 13.90 (br s, 1H), 8.84 (br s, 1H), 8.43 (s, 1H), 8.13 (s, 1H), 7.39 (br d, J = 8.0 Hz, 2H), 7.32 - 7.24 (m, 2H), 7.13 (d, J = 7.8 Hz, 2H), 4.53 (br d, J = 5.6 Hz, 2H), 3.50 (s, 2H), 3.29 - 3.21 (m, 4H), 2.95 - 2.82 (m, 2H), 2.41 (br s, 4H), 2.26 - 2.15 (m, 1H), 2.09 - 1.88 (m, 5H), 1.82 - 1.64 (m, 5H), 1.53 (s, 6H), 1.47 - 1.34 (m, 11H). Step C – Synthesis of 1-[3-[4-[[4-[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]piperazin-1-yl]-1-piperidyl]methyl]phenyl]-2,4-dimethyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Compound 82) A solution of 82b (90 mg, 118 μmol, 1.0 equiv.) in HCl/dioxane (1 mL) was stirred at 20°C for 1 h and then concentrated (LCMS [M+1] =664.7). The residue was dissolved in DMSO (1 mL) and treated with 2-(2,6-dioxo-3-piperidyl)-5,6-difluoro-isoindoline-1,3-dione (53.1 mg) and DIEA (63 μL). The mixture was stirred at 70°C for 16 h, cooled to ambient temperature, partitioned between ethyl acetate (40 mL) and water (40 mL). The organic phase was separated, washed with brine (30 mL), dried over sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative TLC (silica gel, dichloromethane: methanol = 10:1) to afford Compound 82.. LCMS [M+1] =938.4; 1H NMR (400 MHz, DMSO-d6) δ 13.90 (br s, 1H), 11.10 (s, 1H), 8.83 (br s, 1H), 8.43 (s, 1H), 8.13 (s, 1H), 7.72 (d, J = 11.4 Hz, 1H), 7.43 (br dd, J = 7.6, 11.1 Hz, 3H), 7.33 - 7.24 (m, 2H), 7.14 (br d, J = 7.8 Hz, 2H), 5.10 (dd, J = 5.4, 12.8 Hz, 1H), 4.53 (br d, J = 5.6 Hz, 2H), 3.52 (br s, 2H), 3.23 (br
s, 4H), 2.95 - 2.80 (m, 3H), 2.72 - 2.55 (m, 6H), 2.32 - 2.17 (m, 1H), 2.03 (s, 6H), 1.76 (s, 5H), 1.53 (s, 8H). Example 83: Synthesis of Compound 83 tert-butyl
 4-piperazin-1-ylpiperidine-1-carboxylate in Step A to afford 1-[3-[4-[[4-[1-[2-(2,6-dioxo-3- piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]-4-piperidyl]piperazin-1-yl]methyl]phenyl]-2,4- dimethyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methyl]pyrazole-4-carboxamide. LCMS: [M+1] = 938.3; 1H NMR (400 MHz, CHLOROFORM-d) δ 8.30 (br s, 1H), 8.12 (s, 1H), 8.00 (s, 1H), 7.50 - 7.36 (m, 4H), 7.20 (s, 2H), 7.10 (d, J = 8.0 Hz, 2H), 6.92 (s, 1H), 4.94 (dd, J = 5.2, 12.0 Hz, 1H), 4.71 (d, J = 6.0 Hz, 2H), 3.73 (d, J = 12.0 Hz, 2H), 3.64 (s, 2H), 2.96 - 2.56 (m, 14H), 2.19 - 2.09 (m, 2H), 2.07 (s, 3H), 2.03 (d, J = 9.6 Hz, 2H), 1.78 (s, 4H), 1.61 (s, 6H). Example 84: Synthesis of Compound 84
4-piperazin-1-ylpiperidine-1-carboxylate in Step A to afford 1-[3-[4-[[4-[1-[2-(2,6-dioxo-3- piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]-4-piperidyl]piperazin-1-yl]methyl]phenyl]-2,4- dimethyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methyl]pyrazole-4-carboxamide. LCMS: [M+1] = 938.3; 1H NMR (400 MHz, CHLOROFORM-d) δ 8.30 (br s, 1H), 8.12 (s, 1H), 8.00 (s, 1H), 7.50 - 7.36 (m, 4H), 7.20 (s, 2H), 7.10 (d, J = 8.0 Hz, 2H), 6.92 (s, 1H), 4.94 (dd, J = 5.2, 12.0 Hz, 1H), 4.71 (d, J = 6.0 Hz, 2H), 3.73 (d, J = 12.0 Hz, 2H), 3.64 (s, 2H), 2.96 - 2.56 (m, 14H), 2.19 - 2.09 (m, 2H), 2.07 (s, 3H), 2.03 (d, J = 9.6 Hz, 2H), 1.78 (s, 4H), 1.61 (s, 6H). Example 84: Synthesis of Compound 84
carboxylate (84a) To a solution of 4-bromo-2-fluoro-pyridine (500.0 mg, 2.8 mmol, 1.0 equiv.) in DMSO (10 mL) was added DIEA (1.40 g, 11.3 mmol, 1.9 mL, 4.0 equiv.) and tert-butyl 4-(piperazin-1- ylmethyl)piperidine-1-carboxylate (966.2 mg, 3.4 mmol, 1.2 equiv.). The mixture was stirred at 30°C for 4 h. The reaction mixture was partitioned between ethyl acetate (100 mL) and water (100 mL), extracted with ethyl acetate (100 mL x 2). The combined organic layers were washed with brine (50 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure to afford 84a, which was used without further purification. LCMS [M+1] = 439.2, 441.2. Step B – Synthesis of [2-[4-[(1-tert-butoxycarbonyl-4-piperidyl)methyl]piperazin-1-yl]-4- pyridyl]boronic acid (84b) To a solution of 84a (500 mg, 1.1 mmol, 1.0 equiv.) in dioxane (9.0 mL) was added Pd(DPPF)Cl2 (92.9 mg, 0.114 mmol, 0.1 equiv.), KOAc (335.0 mg, 3.40 mmol, 3.0 equiv.), and 4,4,5,5-tetramethyl-2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3,2-dioxaborolane (577.9 mg, 2.20 mmol, 2.0 equiv.) under nitrogen. The mixture was stirred at 90°C for 3 h. The reaction
mixture was cooled to ambient temperature and partitioned between ethyl acetate (100 mL) and water (100 mL) and extracted with ethyl acetate (100 mL x 2). The combined organic portions were washed with brine (5 mL), dried over anhydrous sodium sulfate, filtered and filtrate was concentrated under reduced pressure. The residue was purified by preparative HPLC (Phenomenex Luna C18, 75mm x 30mm, 3 µm; [water(0.04% HCl)-ACN]) to afford 84b. LCMS [M+1] = 405.2. Step C – Synthesis of tert-butyl 4-[[4-[4-[2,6-dimethyl-3-[4-[[3-(2,2,2-trifluoro-1,1-dimethyl- ethyl)-1H-1,2,4-triazol-5-yl]methylcarbamoyl]pyrazol-1-yl]phenyl]-2-pyridyl]piperazin-1- yl]methyl]piperidine-1-carboxylate (84c) A solution of 84b (73.31 mg, 181 μmol, 1.1 equiv.) in dioxane (2.0 mL), water (0.4 mL) was added K2CO3 (68.3 mg, 495 μmol, 3.0 equiv.) and Pd(dppf)Cl2 (24.1 mg, 32.9 μmol, 0.2 equiv.), Int 5 (80.0 mg, 165 μmol, 1.0 equiv.) under an atmosphere of nitrogen. The mixture was stirred at 90°C for 2 h. The reaction mixture was partitioned between ethyl acetate (20 mL) and water (20 mL) and extracted with ethyl acetate (20 mL x 2). The combined organic portions were washed with brine (5 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure to afford 84c. LCMS [M+1] =765.5. Step D – Synthesis of 1-[2,4-dimethyl-3-[2-[4-(4-piperidylmethyl)piperazin-1-yl]-4- pyridyl]phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole- 4-carboxamide (84d) To a solution of 84c (80.0 mg, 105 μmol, 1.0 equiv.) in trifluoroethanol (1 mL) was treated with chlorotrimethylsilane (85.6 mg, 788 μmol, 0.1 mL, 7.5 equiv.). The mixture was stirred at 20°C for 1 h. The mixture was filtered and concentrated under reduced pressure to afford 84d, which was used without further purification. LCMS [M+1] = 665.5. Step E – Synthesis of 1-[3-[2-[4-[[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]-4-piperidyl]methyl]piperazin-1-yl]-4-pyridyl]-2,4-dimethyl-phenyl]-N-[[3-(2,2,2-trifluoro- 1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (84e)
A solution of 84d (60.0 mg, 90.2 μmol, 1.0 equiv.) in DMSO (1 mL) was treated with DIEA (94.3 μL) and 2-(2,6-dioxo-3-piperidyl)-5,6-difluoro-isoindoline-1,3-dione (31.8 mg). The mixture was stirred at 60°C for 16 h. The mixture was cooled to ambient temperature, treated with methanol (1 ml) and filtered. Direct purification with preparative HPLC (Phenomenex Luna C18, 100mm x 30mm, 5 µm; [water (0.2% FA)-ACN]) to afford Compound 84. LCMS [M+1] =939.2; 1H NMR (400 MHz, DMSO-d6) δ = 13.89 (s, 1H), 11.10 (s, 1H), 8.86 (br s, 1H), 8.42 (s, 1H), 8.21 (d, J = 5.2 Hz, 1H), 8.15 (s, 1H), 8.13 (s, 1H), 7.70 (d, J = 11.2 Hz, 1H), 7.44 (d, J = 7.2 Hz, 1H), 7.30 (s, 2H), 6.65 (s, 1H), 6.48 (d, J = 5.2 Hz, 1H), 5.10 (dd, J = 5.2, 12.8 Hz, 1H), 4.54 (d, J = 4.4 Hz, 2H), 3.62 (d, J = 12.0 Hz, 2H), 3.53 (s, 4H), 2.99 - 2.81 (m, 3H), 2.64 - 2.53 (m, 1H), 2.45 ( s, 4H), 2.23 ( d, J = 6.8 Hz, 2H), 2.08 (s, 3H), 2.05 - 1.98 (m, 1H), 1.85 ( d, J = 12.4 Hz, 2H), 1.81 - 1.73 (m, 2H), 1.53 (s, 6H), 1.36 - 1.21 (m, 2H) Example 85: Synthesis of Compound 85
 butyl 4-(4-piperidylmethyl)piperazine-1-carboxylate in Step A to afford 1-(3-(2-(4-((4-(2-(2,6- dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1-yl)methyl)piperidin-1- yl)pyridin-4-yl)-2,4-dimethylphenyl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide. LCMS [(M+1] = 939.2;1H NMR (400 MHz, DMSO-d6) δ 13.87 (br s, 1H), 11.10 (s, 1H), 8.95 - 8.76 (m, 1H), 8.42 (s, 1H), 8.19 (d, J = 4.9 Hz, 1H), 8.13 (s, 1H), 7.73 (d, J = 11.4 Hz, 1H), 7.45 (br d, J = 7.3 Hz, 1H), 7.30 (s, 2H), 6.63 (s, 1H), 6.43 (d, J = 5.0 Hz, 1H), 5.10 (dd, J = 5.2, 12.8 Hz, 1H), 4.55 (br d, J = 5.1 Hz, 2H), 4.39 - 4.28 (m, 2H), 3.26 (br s, 4H), 2.93 - 2.75 (m, 4H), 2.64 - 2.53 (m, 4H), 2.22 (br d, J = 4.1 Hz, 2H), 2.09 (s, 3H), 2.07 - 1.99 (m, 1H), 1.85 - 1.72 (m, 7H), 1.52 (s, 6H), 1.16 - 1.07 (m, 2H) Example 86: Synthesis of Compound 86
butyl 4-(4-piperidylmethyl)piperazine-1-carboxylate in Step A to afford 1-(3-(2-(4-((4-(2-(2,6- dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1-yl)methyl)piperidin-1- yl)pyridin-4-yl)-2,4-dimethylphenyl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide. LCMS [(M+1] = 939.2;1H NMR (400 MHz, DMSO-d6) δ 13.87 (br s, 1H), 11.10 (s, 1H), 8.95 - 8.76 (m, 1H), 8.42 (s, 1H), 8.19 (d, J = 4.9 Hz, 1H), 8.13 (s, 1H), 7.73 (d, J = 11.4 Hz, 1H), 7.45 (br d, J = 7.3 Hz, 1H), 7.30 (s, 2H), 6.63 (s, 1H), 6.43 (d, J = 5.0 Hz, 1H), 5.10 (dd, J = 5.2, 12.8 Hz, 1H), 4.55 (br d, J = 5.1 Hz, 2H), 4.39 - 4.28 (m, 2H), 3.26 (br s, 4H), 2.93 - 2.75 (m, 4H), 2.64 - 2.53 (m, 4H), 2.22 (br d, J = 4.1 Hz, 2H), 2.09 (s, 3H), 2.07 - 1.99 (m, 1H), 1.85 - 1.72 (m, 7H), 1.52 (s, 6H), 1.16 - 1.07 (m, 2H) Example 86: Synthesis of Compound 86
Step A – Synthesis of tert-butyl 6-[(1-benzyloxycarbonyl-4-piperidyl)methyl]-2,6- diazaspiro[3.3]heptane-2-carboxylate (86a) A solution of tert-butyl 2,6-diazaspiro[3.3]heptane-2-carboxylate, oxalic acid salt (1.0 g, 3.4 mmol, 1.0 equiv.) and benzyl 4-formylpiperidine-1-carboxylate (857.7 mg, 3.4 mmol, 1.0 equiv.) in DCE (10.0 mL) was treated with acetic acid (416.6 mg, 6.9 mmol, 397.1 μL, 2.0 equiv.), stirred at 20°C for 30 min and then treated with NaBH(OAc)3 (2.2 g, 10.4 mmol, 3 equiv.). The mixture was stirred at 20°C for 2 h and quenched with water (15 mL). The mixture was adjusted to pH 9 with solid NaHCO3 and extracted with ethyl acetate (25 mL x 2). The combined organic layers were washed with brine (15 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure to afford 86a. LCMS: [M+1] = 430.2; 1H NMR (400 MHz, DMSO-d6) δ 7.27 - 7.42 (m, 5 H), 5.05 (s, 2 H), 3.99 - 3.94 (m, 2 H), 3.89 - 3.93 (m, 4 H), 3.55 (s, 4 H), 2.86 - 2.74 (m, 2 H), 2.45 - 2.49 (m, 2 H), 1.61 (d, J = 12.0 Hz, 2 H), 1.42 - 1.55 (m, 1 H), 1.36 (s, 9 H), 0.91 - 1.06 (m, 2 H).
Step B – Synthesis of tert-butyl 6-(4-piperidylmethyl)-2,6-diazaspiro[3.3]heptane-2-carboxylate (86b) A slurry of Pd/C (745.0 mg, 700.0 μmol, 10.0% purity) in methanol (20.0 mL) was treated with 86a (1.4 g, 3.4 mmol, 1.0 equiv.). The mixture was degassed and purged threefold with hydrogen. The mixture was stirred at 40°C for 2 h under hydrogen (15 psi). The suspension was filtered through a pad of Celite and the pad was washed with ethanol (20mL × 2). The combined filtrates were concentrated to dryness to afford 86b. LCMS: [M+1] = 296.2; 1H NMR (400 MHz, DMSO-d6) δ 3.86 (s, 4 H), 3.18 (s, 4 H), 3.09 (d, J = 12.4 Hz, 2 H), 2.65 (t, J = 12.4 Hz, 2 H), 2.18 (d, J = 6.8 Hz, 2 H), 1.68 (d, J = 12.4 Hz, 2 H), 1.48 - 1.39 (m , 1 H), 1.35 (s, 9 H), 1.08 - 1.26 (m, 2 H). Step C – Synthesis of tert-butyl 6-[[1-[5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-2-pyridyl]- 4-piperidyl]methyl]-2,6-diazaspiro[3.3]heptane-2-carboxylate (86c) A solution of 86b (1.20 g, 4.0 mmol, 1.0 equiv.) and 2-fluoro-5-(4,4,5,5-tetramethyl-1,3,2- dioxaborolan-2-yl)pyridine (906.0 mg, 4.0 mmol, 1.0 equiv.) in DMSO (20.0 mL) was treated with DIEA (2.60 g, 20.3 mmol, 3.5 mL, 5.0 equiv.). The mixture was stirred at 80°C for 5 h. The reaction mixture was diluted with water (50 mL) and extracted with ethyl acetate (25 mL x 2). The combined organic portions were washed with brine (15 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure to give a residue. The residue was triturated with acetonitrile (5 mL) and the solids were collected by vacuum filtration. The filter cake was dried under vacuum to afford 86c. LCMS: [M+1] = 499.3. Step D – Synthesis of tert-butyl 6-[[1-[5-[2,6-dimethyl-3-[4-[[3-(2,2,2-trifluoro-1,1-dimethyl- ethyl)-1H-1,2,4-triazol-5-yl]methylcarbamoyl]pyrazol-1-yl]phenyl]-2-pyridyl]-4- piperidyl]methyl]-2,6-diazaspiro[3.3]heptane-2-carboxylate (86d) A mixture of 86c (138.6 mg, 278 μmol, 1.5 equiv.), Int 5 (90.0 mg, 185.4 μmol, 1.0 equiv.), Pd(dppf)Cl2 (27.1 mg, 37.0 μmol, 0.2 equiv.) and K2CO3 (76.8 mg, 556.3 μmol, 3.0 equiv.) in dioxane (1.0 mL) and water (0.2 mL) was degassed and purged threefold with nitrogen, and then the mixture was stirred at 80°C for 16 h under a nitrogen atmosphere. The reaction mixture was cooled to ambient temperature, diluted with water (20 mL) and extracted with ethyl acetate (15 mL
x 2). The combined organic portions were washed with brine (15 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative TLC (silica gel, dichloromethane: MeOH = 10:1) to afford 86d. LCMS: [M+1] = 777.4. Step E – Synthesis of 1-[3-[6-[4-[[2-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]-2,6-diazaspiro[3.3]heptan-6-yl]methyl]-1-piperidyl]-3-pyridyl]-2,4-dimethyl-phenyl]-N-[[3- (2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Compound 86) A solution of 86d (120.0 mg, 154.4 μmol, 1.0 equiv.) in HCl/ dioxane (3.0 M, 1.0 mL) was stirred at 20°C for 2 h. The reaction mixture was concentrated under reduced pressure (LCMS: [M+1] = 677.4). The residue (90.0 mg) in DMSO (1.0 mL) and 2-(2,6-dioxo-3- piperidyl)-5,6-difluoro-isoindoline-1,3-dione (55.6 mg) was treated with DIEA (110 μL). The mixture was stirred at 70°C for 16 h, cooled to ambient temperature and purified directly by preparative HPLC (Phenomenex Luna C18, 100mm x 40mm, 3 µm; [water(0.2% FA)-ACN]) to afford Compound 86. LCMS: [M+1] = 951.6; 1H NMR (400 MHz, DMSO-d6) δ = 13.90 (br s, 1H), 11.07 (s, 1H), 8.83 (br s, 1H), 8.41 (s, 1H), 8.12 (s, 1H), 7.91 (d, J = 2.4 Hz, 1H), 7.60 (d, J = 11.2 Hz, 1H), 7.36 (dd, J = 2.0, 8.4 Hz, 1H), 7.32 - 7.24 (m, 2H), 6.91 (t, J = 7.2 Hz, 2H), 5.06 (dd, J = 5.6, 12.8 Hz, 1H), 4.53 (d, J = 5.6 Hz, 2H), 4.34 - 4.28 (m, 2H), 4.24 (s, 4H), 2.92 - 2.73 (m, 3H), 2.62 - 2.51 (m, 2H), 2.46-2.45 (m, 1H), 2.28 (d, J = 6.8 Hz, 2H), 2.08 (s, 3H), 2.04 - 1.96 (m, 1H), 1.80 (s, 3H), 1.76 (d, J = 13.2 Hz, 2H), 1.55 - 1.50 (m, 7H), 1.27 - 1.19 (m, 1H), 1.18 - 1.05 (m, 2H). Example 87: Synthesis of Compound 87
Compound 87 was prepared in a similar manner to Example 86 by substituting benzyl 2,6- diazaspiro[3.3]heptane-2-carboxylate and tert-butyl 4-formylpiperidine-1-carboxylate in Step A to afford 1-[3-[6-[6-[[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]-4- piperidyl]methyl]-2,6-diazaspiro[3.3]heptan-2-yl]-3-pyridyl]-2,4-dimethyl-phenyl]-N-[[3-(2,2,2- trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide. LCMS: [M+1] = 951.3; 1H NMR (400 MHz, DMSO-d6) δ 13.91 (br s, 1H), 11.10 (s, 1H), 8.84 (br s, 1H), 8.41 (s, 1H), 8.20 (s, 1H), 8.13 (s, 1H), 7.88 (d, J = 2.4 Hz, 1H), 7.70 (d, J = 11.6 Hz, 1H), 7.43 (d, J = 7.6 Hz, 1H), 7.38 (dd, J = 2.4, 8.4 Hz, 1H), 7.34 - 7.22 (m, 2H), (d, J = 8.4 Hz,
 1H), 5.10 (dd, J = 5.6, 12.8 Hz, 1H), 4.53 (d, J = 6.8 Hz, 2H), 4.05 (s, 4H), 3.58 (br d, J = 12.4 Hz, 2H), 2.95 - 2.78 (m, 4H), 2.63 - 2.55 (m, 1H), 2.09 - 1.98 (m, 4H), 1.82 - 1.74 (m, 4H), 1.53 (s, 6H), 1.50 - 1.42 (m, 1H), 1.33 - 1.20 (m, 2H). Example 88: Synthesis of Compound 88
1H), 5.10 (dd, J = 5.6, 12.8 Hz, 1H), 4.53 (d, J = 6.8 Hz, 2H), 4.05 (s, 4H), 3.58 (br d, J = 12.4 Hz, 2H), 2.95 - 2.78 (m, 4H), 2.63 - 2.55 (m, 1H), 2.09 - 1.98 (m, 4H), 1.82 - 1.74 (m, 4H), 1.53 (s, 6H), 1.50 - 1.42 (m, 1H), 1.33 - 1.20 (m, 2H). Example 88: Synthesis of Compound 88
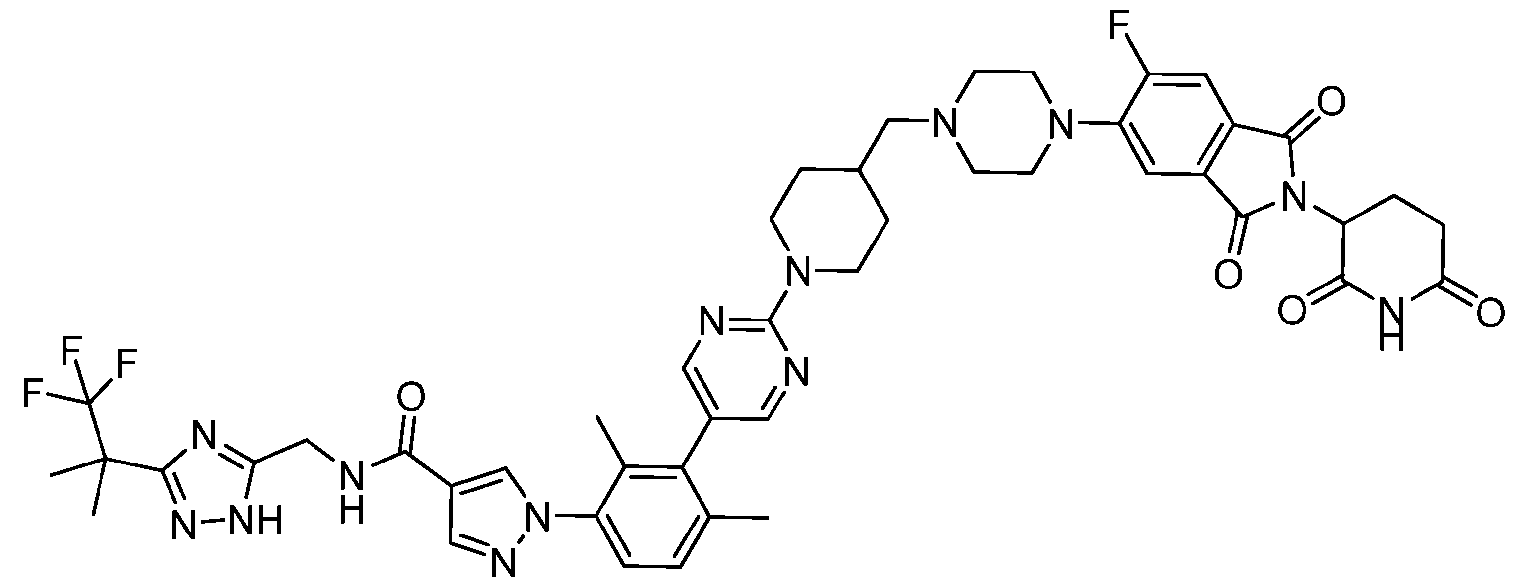 Compound 88 was prepared in a similar manner to Example 86 by substituting tert-butyl 4-(4-piperidylmethyl)piperazine-1-carboxylate and 2-chloro-5-(4,4,5,5-tetramethyl-1,3,2- dioxaborolan-2-yl)pyrimidine in Step C to afford 1-[3-[2-[4-[[4-[2-(2,6-dioxo-3-piperidyl)-6- fluoro-1,3-dioxo-isoindolin-5-yl]piperazin-1-yl]methyl]-1-piperidyl]pyrimidin-5-yl]-2,4- dimethyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methyl]pyrazole-4-carboxamide. LCMS: [M+1] = 940.2; 1H NMR (400 MHz, DMSO-d6) δ 13.90 (br s, 1H), 11.10 (br s, 1H), 8.86 (br s, 1H), 8.41 (s, 1H), 8.25 (s, 2H), 8.14 (s, 1H), 7.73 (d, J = 11.6 Hz, 1H), 7.46 (d, J = 7.4 Hz, 1H), 7.31 (s, 2H), 5.11 (dd, J = 5.4, 12.8 Hz, 1H), 4.72 (br d, J = 12.6 Hz, 2H), 4.54 (br d, J = 5.0 Hz, 2H), 3.29 - 3.22 (m, 4H), 2.94 (br t, J = 12.0 Hz, 2H),
2.69 - 2.57 (m, 2H), 2.54 (s, 6H), 2.34 - 2.31 (m, 1H), 2.25 (br d, J = 6.4 Hz, 2H), 2.12 (s, 3H), 2.08 - 2.01 (m, 1H), 1.84 (s, 5H), 1.53 (s, 6H), 1.18 - 1.07 (m, 2H). Example 89: Synthesis of Compound 89 Step A –
Compound 88 was prepared in a similar manner to Example 86 by substituting tert-butyl 4-(4-piperidylmethyl)piperazine-1-carboxylate and 2-chloro-5-(4,4,5,5-tetramethyl-1,3,2- dioxaborolan-2-yl)pyrimidine in Step C to afford 1-[3-[2-[4-[[4-[2-(2,6-dioxo-3-piperidyl)-6- fluoro-1,3-dioxo-isoindolin-5-yl]piperazin-1-yl]methyl]-1-piperidyl]pyrimidin-5-yl]-2,4- dimethyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methyl]pyrazole-4-carboxamide. LCMS: [M+1] = 940.2; 1H NMR (400 MHz, DMSO-d6) δ 13.90 (br s, 1H), 11.10 (br s, 1H), 8.86 (br s, 1H), 8.41 (s, 1H), 8.25 (s, 2H), 8.14 (s, 1H), 7.73 (d, J = 11.6 Hz, 1H), 7.46 (d, J = 7.4 Hz, 1H), 7.31 (s, 2H), 5.11 (dd, J = 5.4, 12.8 Hz, 1H), 4.72 (br d, J = 12.6 Hz, 2H), 4.54 (br d, J = 5.0 Hz, 2H), 3.29 - 3.22 (m, 4H), 2.94 (br t, J = 12.0 Hz, 2H),
2.69 - 2.57 (m, 2H), 2.54 (s, 6H), 2.34 - 2.31 (m, 1H), 2.25 (br d, J = 6.4 Hz, 2H), 2.12 (s, 3H), 2.08 - 2.01 (m, 1H), 1.84 (s, 5H), 1.53 (s, 6H), 1.18 - 1.07 (m, 2H). Example 89: Synthesis of Compound 89 Step A –
 1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]-2,3-dihydrobenzofuran-2-carboxamide (89a) A solution of Int 2 (420.0 mg, 816 μmol, 1.0 equiv.) and [4-(4,4,5,5-tetramethyl-1,3,2- dioxaborolan-2-yl)phenyl]methanol (229.2 mg, 979 μmol, 1.2 equiv.) in water (0.8 mL) and THF (3.2 mL) was added K3PO4 (519.7 mg, 2.5 mmol, 3.0 equiv.) and PdCl2(DTBPF) (106.7 mg, 163 μmol, 0.2 equiv.). The mixture was degassed and purged threefold with nitrogen, and then stirred at 85°C for 4 h under a nitrogen atmosphere. The reaction mixture was quenched by addition water (10 mL) and extracted with ethyl acetate (5 mL x 3). The combined organic layers were washed with brine (10.0 mL), dried over sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether: ethyl acetate = 1: 0 to 0: 1) to afford the racemate. Enantiomers were resolved by chiral SFC (DAICEL CHIRALCEL OJ, 250 mm x 30 mm,10 µm); [CO2- EtOH(0.1%NH3water)]), LCMS [M+1] = 495.1, 497.1. The early-eluting enantiomer was carried forward as 89a.
Step B – Synthesis of (2R)-5-chloro-6-(4-formylphenyl)-N-[[3-(2, 2, 2-trifluoro-1, 1-dimethyl- ethyl)-1H-1, 2, 4-triazol-5-yl] methyl]-2, 3-dihydrobenzofuran-2-carboxamide (89b) A solution of 89a (50.0 mg, 101.0 μmol, 1.0 equiv.) in dichloromethane (2.0 mL) was added MnO2 (87.8 mg, 1.0 mmol, 10.0 equiv.). The mixture was stirred at 30°C for 12 h. The mixture was filtered and concentrated under reduced pressure to give 89b. LCMS [M+1] = 493.1; 1H NMR (400 MHz, DMSO-d6) δ = 13.83 (s, 1H), 10.07 (s, 1H), 8.85 (t, J = 6.0 Hz, 1H), 7.99 (d, J = 8.4 Hz, 2H), 7.63 (d, J = 8.4 Hz, 2H), 7.46 (s, 1H), 6.91 (s, 1H), 5.35 - 5.30 (m, 1H), 4.52 - 4.46 (m, 1H), 4.38 - 4.32 (m, 1H), 3.62 - 3.55 (m, 1H), 3.38 (d, J = 6.8 Hz, 1H), 1.51 (s, 4H), 1.35 (s, 2H). Step C – Synthesis of (2S)-5-chloro-6-[4-[[4-[4-(2,6-dioxo-3-piperidyl)phenyl]piperazin-1- yl]methyl]phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl] -2,3- dihydrobenzofuran-2-carboxamide (Compound 89) To a solution of 89b (20.0 mg, 40.6 μmol, 1.0 equiv.) and 3-(4-piperazin-1- ylphenyl)piperidine-2,6-dione (30.8 mg, 81.2 μmol, 2.0 equiv.) in DCE (0.5 mL) was added AcOH (4.9 mg, 81.2 μmol, 4.7 μL, 2.0 equiv.) and the mixture was stirred at 50°C for 6 h. The mixture was treated with NaBH(OAc)3 (25.8 mg, 122 μmol, 3.0 equiv.) and stirred at 50°C for 2 h. The mixture was quenched by water (1.0 mL) and extracted with ethyl acetate (0.5 mL x 3). The combined organic layers were washed with brine (1.0 mL), dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to give a residue. The residue was purified by preparative HPLC (Phenomenex Luna C18, 80 mm x 30 mm, 3 µm; [water(0.04% HCl)-ACN];) to afford Compound 89. LCMS [M+1] = 750.3, 752.3; 1H NMR (400 MHz, METHANOL-d4) δ = 7.63 (d, J = 8.4 Hz, 2H), 7.57 (d, J = 8.4 Hz, 2H), 7.37 (s, 1H), 7.19 (d, J = 8.4 Hz, 2H), 7.02 (d, J = 8.4 Hz, 2H), 6.89 (s, 1H), 5.33 (dd, J = 6.4, 10.4 Hz, 1H), 4.62 (d, J = 16.0 Hz, 1H), 4.52 (s, 1H), 4.48 - 4.47 (m, 2H), 3.88 (d, J = 12.0 Hz, 2H), 3.81 (dd, J = 6.0, 10.0 Hz, 1H), 3.69 - 3.60 (m, 3H), 3.49 - 3.46 (m, 1H), 3.43 (d, J = 6.4 Hz, 1H), 3.39 - 3.36 (m, 2H), 3.06 (t, J = 12.6 Hz, 2H), 2.74 - 2.66 (m, 1H), 2.64 - 2.57 (m, 1H), 2.23 - 2.16 (m, 2H), 1.59 (s, 6H). Example 90: Synthesis of Intermediate 90
1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]-2,3-dihydrobenzofuran-2-carboxamide (89a) A solution of Int 2 (420.0 mg, 816 μmol, 1.0 equiv.) and [4-(4,4,5,5-tetramethyl-1,3,2- dioxaborolan-2-yl)phenyl]methanol (229.2 mg, 979 μmol, 1.2 equiv.) in water (0.8 mL) and THF (3.2 mL) was added K3PO4 (519.7 mg, 2.5 mmol, 3.0 equiv.) and PdCl2(DTBPF) (106.7 mg, 163 μmol, 0.2 equiv.). The mixture was degassed and purged threefold with nitrogen, and then stirred at 85°C for 4 h under a nitrogen atmosphere. The reaction mixture was quenched by addition water (10 mL) and extracted with ethyl acetate (5 mL x 3). The combined organic layers were washed with brine (10.0 mL), dried over sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether: ethyl acetate = 1: 0 to 0: 1) to afford the racemate. Enantiomers were resolved by chiral SFC (DAICEL CHIRALCEL OJ, 250 mm x 30 mm,10 µm); [CO2- EtOH(0.1%NH3water)]), LCMS [M+1] = 495.1, 497.1. The early-eluting enantiomer was carried forward as 89a.
Step B – Synthesis of (2R)-5-chloro-6-(4-formylphenyl)-N-[[3-(2, 2, 2-trifluoro-1, 1-dimethyl- ethyl)-1H-1, 2, 4-triazol-5-yl] methyl]-2, 3-dihydrobenzofuran-2-carboxamide (89b) A solution of 89a (50.0 mg, 101.0 μmol, 1.0 equiv.) in dichloromethane (2.0 mL) was added MnO2 (87.8 mg, 1.0 mmol, 10.0 equiv.). The mixture was stirred at 30°C for 12 h. The mixture was filtered and concentrated under reduced pressure to give 89b. LCMS [M+1] = 493.1; 1H NMR (400 MHz, DMSO-d6) δ = 13.83 (s, 1H), 10.07 (s, 1H), 8.85 (t, J = 6.0 Hz, 1H), 7.99 (d, J = 8.4 Hz, 2H), 7.63 (d, J = 8.4 Hz, 2H), 7.46 (s, 1H), 6.91 (s, 1H), 5.35 - 5.30 (m, 1H), 4.52 - 4.46 (m, 1H), 4.38 - 4.32 (m, 1H), 3.62 - 3.55 (m, 1H), 3.38 (d, J = 6.8 Hz, 1H), 1.51 (s, 4H), 1.35 (s, 2H). Step C – Synthesis of (2S)-5-chloro-6-[4-[[4-[4-(2,6-dioxo-3-piperidyl)phenyl]piperazin-1- yl]methyl]phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl] -2,3- dihydrobenzofuran-2-carboxamide (Compound 89) To a solution of 89b (20.0 mg, 40.6 μmol, 1.0 equiv.) and 3-(4-piperazin-1- ylphenyl)piperidine-2,6-dione (30.8 mg, 81.2 μmol, 2.0 equiv.) in DCE (0.5 mL) was added AcOH (4.9 mg, 81.2 μmol, 4.7 μL, 2.0 equiv.) and the mixture was stirred at 50°C for 6 h. The mixture was treated with NaBH(OAc)3 (25.8 mg, 122 μmol, 3.0 equiv.) and stirred at 50°C for 2 h. The mixture was quenched by water (1.0 mL) and extracted with ethyl acetate (0.5 mL x 3). The combined organic layers were washed with brine (1.0 mL), dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to give a residue. The residue was purified by preparative HPLC (Phenomenex Luna C18, 80 mm x 30 mm, 3 µm; [water(0.04% HCl)-ACN];) to afford Compound 89. LCMS [M+1] = 750.3, 752.3; 1H NMR (400 MHz, METHANOL-d4) δ = 7.63 (d, J = 8.4 Hz, 2H), 7.57 (d, J = 8.4 Hz, 2H), 7.37 (s, 1H), 7.19 (d, J = 8.4 Hz, 2H), 7.02 (d, J = 8.4 Hz, 2H), 6.89 (s, 1H), 5.33 (dd, J = 6.4, 10.4 Hz, 1H), 4.62 (d, J = 16.0 Hz, 1H), 4.52 (s, 1H), 4.48 - 4.47 (m, 2H), 3.88 (d, J = 12.0 Hz, 2H), 3.81 (dd, J = 6.0, 10.0 Hz, 1H), 3.69 - 3.60 (m, 3H), 3.49 - 3.46 (m, 1H), 3.43 (d, J = 6.4 Hz, 1H), 3.39 - 3.36 (m, 2H), 3.06 (t, J = 12.6 Hz, 2H), 2.74 - 2.66 (m, 1H), 2.64 - 2.57 (m, 1H), 2.23 - 2.16 (m, 2H), 1.59 (s, 6H). Example 90: Synthesis of Intermediate 90
Step yl) -1H- 1,2,4-triazol-5-yl]methylcarbamoyl]-2,3-dihydrobenzofuran-6-yl]phenyl]methyl]-4- piperidyl]piperazine-1-carboxylate (90a) A mixture of Int 2 (100 mg, 194 μmol, 1 equiv.) and tert-butyl 4-[1-[[4-(4,4,5,5-tetramethyl- 1,3,2-dioxaborolan-2-yl)phenyl]methyl]-4-piperidyl]piperazine-1-carboxylate (141.5 mg, 292 μmol, 1.5 equiv.), K3PO4 (123.7 mg, 583 μmol, 3 equiv.), PdCl2(DTBPF) (25.3 mg, 38.9 μmol, 0.2 equiv.) in THF (2 mL) and water (0.4 mL) was degassed and purged with nitrogen for 3 times, and then the mixture was stirred at 80°C for 12 h under a nitrogen atmosphere. The reaction mixture was poured into water (50 mL) and extracted with ethyl acetate (50 mL x 3). The combined organic portions were washed with brine (50 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative TLC(silica gel, dichloromethane: methanol = 10:1 ) to afford 90a. LCMS [M+1] = 746.2, 748.2; 1H NMR (400 MHz, DMSO-d6) δ = 13.87 (br s, 1H), 8.80 (br s, 1H), 7.45 - 7.25 (m, 4H), 6.84 (s, 1H), 5.39 - 5.24 (m, 1H), 4.10 (q, J = 5.2 Hz, 2H), 3.50 - 3.43 (m, 2H), 3.27 (s, 4H), 3.17 (d, J = 5.2 Hz, 6H), 2.86 (d, J = 10.4 Hz, 2H), 2.41 (s, 4H), 2.01 - 1.88 (m, 2H), 1.69 (d, J = 10.4 Hz, 2H), 1.52 (s, 6H), 1.38 (s, 9H). Step B – Synthesis of 5-chloro-6-[4-[(4-piperazin-1-yl-1-piperidyl)methyl]phenyl]-N-[[3-(2,2,2- trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]-2,3-dihydrobenzofuran-2- carboxamide (90b)
A solution of 90a (140 mg, 188 μmol, 1 equiv.) in dichloromethane (3.0 mL) was treated with TFA (1.0 mL). The mixture was stirred at 20°C for 2 h. The reaction mixture was concentrated under reduced pressure to afford 90b, which was used without further purification. LCMS [M+1] = 646.3, 648.3; 1H NMR (400 MHz, DMSO-d6) δ = 9.90 (br s, 1H), 8.92 (br s, 2H), 8.80 (t, J = 5.2 Hz, 1H), 7.59 (d, J = 8.4 Hz, 2H), 7.54 - 7.47 (m, 2H), 7.44 (s, 1H), 6.85 (s, 1H), 5.31 (dd, J = 6.4, 10.4 Hz, 1H), 4.54 - 4.42 (m, 1H), 4.40 - 4.28 (m, 3H), 3.63 - 3.49 (m, 3H), 3.34 (dd, J = 6.4, 16.8 Hz, 2H), 3.13 - 2.87 (m, 8H), 2.22 - 2.07 (m, 3H), 1.86 - 1.69 (m, 2H), 1.54 - 1.50 (m, 6H). Step C – Synthesis of (2R)-5-chloro-6-(4-((4-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3- dioxoisoindolin-5-yl)piperazin-1-yl)piperidin-1-yl)methyl)phenyl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-2,3-dihydrobenzofuran-2-carboxamide (90c) To a solution of 90b (200 mg) and 2-(2,6-dioxo-3-piperidyl)-5,6-difluoro-isoindoline-1,3- dione (91.1 mg) in DMSO (2 mL) was added DIEA (270 μL). The mixture was stirred at 70°C for 12 h. The reaction mixture was poured into water (5 mL) and extracted with ethyl acetate (5 mL x 3). The residue was purified by preparative HPLC (Phenomenex Luna C18, 100mm x 40mm, 3 µm; [water (0.2% FA) - ACN] to afford the racemate. Chiral resolution by SFC (DAICEL CHIRALPAK IE(50mm x 250mm, 10µm) [heptane-EtOH:ACN=4:1(0.1% IPA)] afforded Compound 90 as the early-eluting enantiomer. LCMS [M+1] = 920.3, 922.3; 1H NMR (400 MHz, DMSO-d6) δ = 13.87 (br s, 1H), 11.10 (s, 1H), 8.78 (s, 1H), 7.72 (d, J = 11.6 Hz, 1H), 7.47 - 7.28 (m, 6H), 6.84 (s, 1H), 5.30 (dd, J = 6.4, 10.4 Hz, 1H), 5.10 (dd, J = 5.6, 12.8 Hz, 1H), 4.52 - 4.40 (m, 1H), 4.39 - 4.29 (m, 1H), 3.62 - 3.47 (m, 3H), 3.27 - 3.15 (m, 5H), 2.96 - 2.81 (m, 3H), 2.71 - 2.53 (m, 6H), 2.26 (t, J = 10.8 Hz, 1H), 2.09 - 1.90 (m, 3H), 1.84 - 1.72 (m, 2H), 1.58 - 1.39 (m, 8H). Example 91: Synthesis of Intermediate 91
Step A – A mixture of [4,5-dichloro-2-(hydroxymethyl)phenyl]methanol (500.0 mg, 2.4 mmol, 1.0 equiv) in SOCl2 (5.0 mL) was degassed and purged three-fold with nitrogen. The mixture was stirred at 80°C for 2 h under a nitrogen atmosphere. The mixture was cooled to ambient temperature. The reaction mixture was treated with aqueous NaHCO3 (12.5 g per 100 mL H2O), dropwise and the mixture was extracted by MTBE (30 mL x 2). The combined organic layers were washed brine (20 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether / ethyl acetate = 1 / 0 to 50 / 1) afford 91a. 1H NMR (400 MHz, DMSO-d6) δ 7.82 (s, 2H), 4.89 (s, 4H). Step B – Synthesis of ethyl 5,6-dichloro-2-cyano-indane-2-carboxylate (91b) To a solution of 91a (140.0 mg, 574 μmol, 1.0 equiv) and ethyl 2-cyanoacetate (64.9 mg, 574 μmol, 61.2 μL, 1.0 equiv) in DMF (2.0 mL) was added potassium carbonate(198.3 mg, 1.40 mmol, 2.5 equiv). The mixture was stirred at 20°C for 16 h. The mixture was filtered and the filtrate was purified by preparative HPLC (Phenomenex Luna 80 mm x 30 mm, 3 µm; [water (TFA)-ACN]) to afford 91b. 1H NMR (400 MHz, DMSO-d6) δ 7.59 (s, 2H), 4.24 (q, J = 7.2 Hz, 2H), 3.59 (s, 4H), 1.24 (t, J = 6.4 Hz, 3H). Step C – Synthesis of 5,6-dichloro-2-cyano-indane-2-carboxylic acid (91c) To a solution of 91b (40.0 mg, 140.8 μmol, 1.0 equiv) in THF (1.0 mL) and water (0.3 mL) was added LiOH•H2O (11.8 mg, 281.5 μmol, 2.0 equiv). The mixture was stirred at 50°C for 2 h. The reaction mixture was concentrated under reduced pressure to remove THF and the
remainder was poured into water. The mixture was adjusted to pH 4 with 1 M HCl. The mixture was extracted with ethyl acetate. The combined organic portions were dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure to afford 91c. LCMS [M-1] -=254.1, 256.1; 1H NMR (400 MHz, DMSO-d6) δ = 7.58 (s, 2H), 3.61 - 3.51 (m, 4H). Step D – Synthesis of 5,6-dichloro-2-cyano-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4- triazol-5-yl]methyl]indane-2-carboxamide (Intermediate 91) To a solution of 91c (35.0 mg, 136.7 μmol, 1.0 equiv) and Int 1 (42.7 mg, 205.0 μmol, 1.5 equiv) in DMF (0.5 mL) was added BOP (72.5 mg, 164.0 μmol, 1.2 equiv) and triethylamine (138.3 mg, 1.4 mmol, 190.2 uL, 10.0 equiv). The mixture was stirred at 20°C for 1 h. The reaction mixture was diluted with water (15 mL) and extracted with ethyl acetate (2 x 10 mL). The combined organic layers were washed with brine (10 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative HPLC (Phenomenex Luna C18, 80 mm x 40 mm, 3 µm; [water(formic acid)-acetonitrile]) to afford Intermediate 91. LCMS [M+1]=446.0, 448.0; 1H NMR (400 MHz, DMSO-d6) δ 13.86 (s, 1H), 9.20 (t, J = 5.6 Hz, 1H), 7.60 (s, 2H), 4.44 (d, J = 5.6 Hz, 2H), 3.65 (d, J = 16.6 Hz, 2H), 3.52 (d, J = 16.6 Hz, 2H) 1.51 (s, 6H). Example 92: Synthesis of Compound 92
Step A – Synthesis of tert-butyl 4-[[1-[5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-2-pyridyl]- 4-piperidyl] methyl]piperazine-1-carboxylate (92a) To a solution of tert-butyl 4-(4-piperidylmethyl)piperazine-1-carboxylate (2.00 g, 7.0 mmol, 1.0 equiv.) in DMF (20.0 mL) was added DIEA (2.70 g, 21.1 mmol, 3.6 mL, 3.0 equiv.) and 2-fluoro-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine (1.50 g, 7.0 mmol, 1.0 equiv.). The mixture was stirred at 120°C for 16 h. The reaction mixture was quenched by addition of water (10 mL) at 20°C and filtered. The filtered cake was dried under vacuum to afford 92a. LCMS [M+1]=487.5;1H NMR (400 MHz, METHANOL-d4) δ 8.35 (s, 1H), 7.77 (dd, J = 1.2, 8.8 Hz, 1H), 6.77 (d, J = 8.8 Hz, 1H), 4.34 (d, J = 13.2 Hz, 2H), 3.49 - 3.36 (m, 4H), 2.98 - 2.77 (m, 2H), 2.39 (t, J = 4.8 Hz, 4H), 2.23 (d, J = 6.8 Hz, 2H), 1.97 - 1.78 (m, 3H), 1.46 (s, 9H), 1.32 (s, 12H), 1.25 - 1.07 (m, 2H). Step B – Synthesis of tert-butyl 4-[[1-[5-[6-chloro-2-cyano-2-[[3-(2,2,2-trifluoro-1,1-dimethyl- ethyl)-1H-1,2,4-triazol-5-yl]methylcarbamoyl]indan-5-yl]-2-pyridyl]-4- piperidyl]methyl]piperazine-1-carboxylate (92b) A mixture of 92a (523.2 mg, 1.00 mmol, 1.2 equiv.), Int 91 (400 mg, 896 μmol, 1.0 equiv.), K3PO4 (570.8 mg, 2.60 mmol, 3.0 equiv.) in ethanol (4 mL) and water (0.8 mL) was
degassed and purged threefold with nitrogen, treated with XPHOS-PD-G2 (211.5 mg, 269 μmol, 0.3 equiv.), the mixture was stirred at 80°C for 16 h under a nitrogen atmosphere. The mixture is quenched with water and extracted with ethyl acetate (20 mL). The combined organic portions were washed with brine (10 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative HPLC (Waters Xbridge BEH C18, 100mm x 30mm, 10 μm; [water(10mM NH4HCO3)-ACN]) to afford the racemate. LCMS [M+1] =770.5. Chiral resolution by SFC (DAICEL CHIRALPAK IG (250mm x 30mm,10 μm); [CO2-IPA(0.1%NH3water)]) afforded 92b as the early-eluting isomer. LCMS [M+1]=770.6; 1H NMR (400 MHz, DMSO-d6) δ 13.91 (br s, 1H), 9.20 (s, 1H), 8.13 (d, J = 2.4 Hz, 1H), 7.58 (dd, J = 2.4, 8.8 Hz, 1H), 7.49 (s, 1H), 7.33 (s, 1H), 6.88 (d, J = 8.8 Hz, 1H), 4.47 - 4.29 (m, 4H), 3.77 - 3.61 (m, 2H), 3.60 - 3.49 (m, 2H), 3.47 - 3.38 (m, 1H), 3.31 (s, 3H), 2.82 (t, J = 12.0 Hz, 2H), 2.29 (t, J = 4.4 Hz, 4H), 2.15 (d, J = 6.8 Hz, 2H), 1.78 (d, J = 10.4 Hz, 3H), 1.53 (s, 6H), 1.39 (s, 9H), 1.15 - 1.07 (m, 2H). Step C – Synthesis of (2R)-5-chloro-2-cyano-6-(6-(4-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro- 1,3-dioxoisoindolin-5-yl)piperazin-1-yl)methyl)piperidin-1-yl)pyridin-3-yl)-N-((3-(1,1,1-trifluoro- 2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-2,3-dihydro-1H-indene-2-carboxamide (Compound 92). A solution of 92b (80.0 mg, 103.8 μmol, 1.0 equiv.) in dichloromethane (0.5 mL) and trifluoroacetic acid (0.1 mL) was stirred at 20°C for 1 h. The reaction mixture was concentrated under reduced pressure LCMS [M+1] =670.4. The residue was dissolved in DMSO (1.0 mL) was treated with DIEA (155 μL) and 2-(2,6-dioxo-3-piperidyl)-5,6-difluoro-isoindoline-1,3-dione (105.0 mg). The mixture was stirred at 70°C for 5 h, cooled to ambient temperature quenched with water (20 mL) and extracted with ethyl acetate (20 mL). The combined organic portions were washed with brine (10 mL), dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to give a residue. The residue was purified by preparative HPLC (Phenomenex Luna C18, 100mm x 40mm, 3 µm; [water(0.2% FA)-ACN] to afford Compound 92. LCMS [M+1] =944.1, 946.2; 1H NMR (400 MHz, DMSO-d6) δ 13.88 (s, 0.6H), 12.74 (br s, 0.4H), 11.12 (s, 1H), 9.21 (br s, 1H), 9.14 (br s, 1H)8.14 (d, J = 2.4 Hz, 1H), 7.74 (d, J = 11.6 Hz, 1H), 7.59 (dd, J = 2.4, 9.2 Hz, 1H), 7.52 - 7.43 (m, 2H), 7.33 (s, 1H), 6.89 (d, J = 8.8 Hz, 1H), 5.11 (dd, J = 5.2, 12.8 Hz, 1H), 4.66 - 4.09 (m, 4H), 3.75 - 3.62 (m, 2H), 3.60 - 3.46 (m, 2H), 3.27
(s, 4H), 2.87 (d, J = 12.0 Hz, 3H), 2.61 - 2.50 (m, 4H), 2.23 (s, 2H), 2.07 - 1.98 (m, 1H), 1.82 (d, J = 11.6 Hz, 3H), 1.63 - 1.41 (m, 6H), 1.19 - 1.04 (m, 2H). Example 93: Synthesis of Compound 93
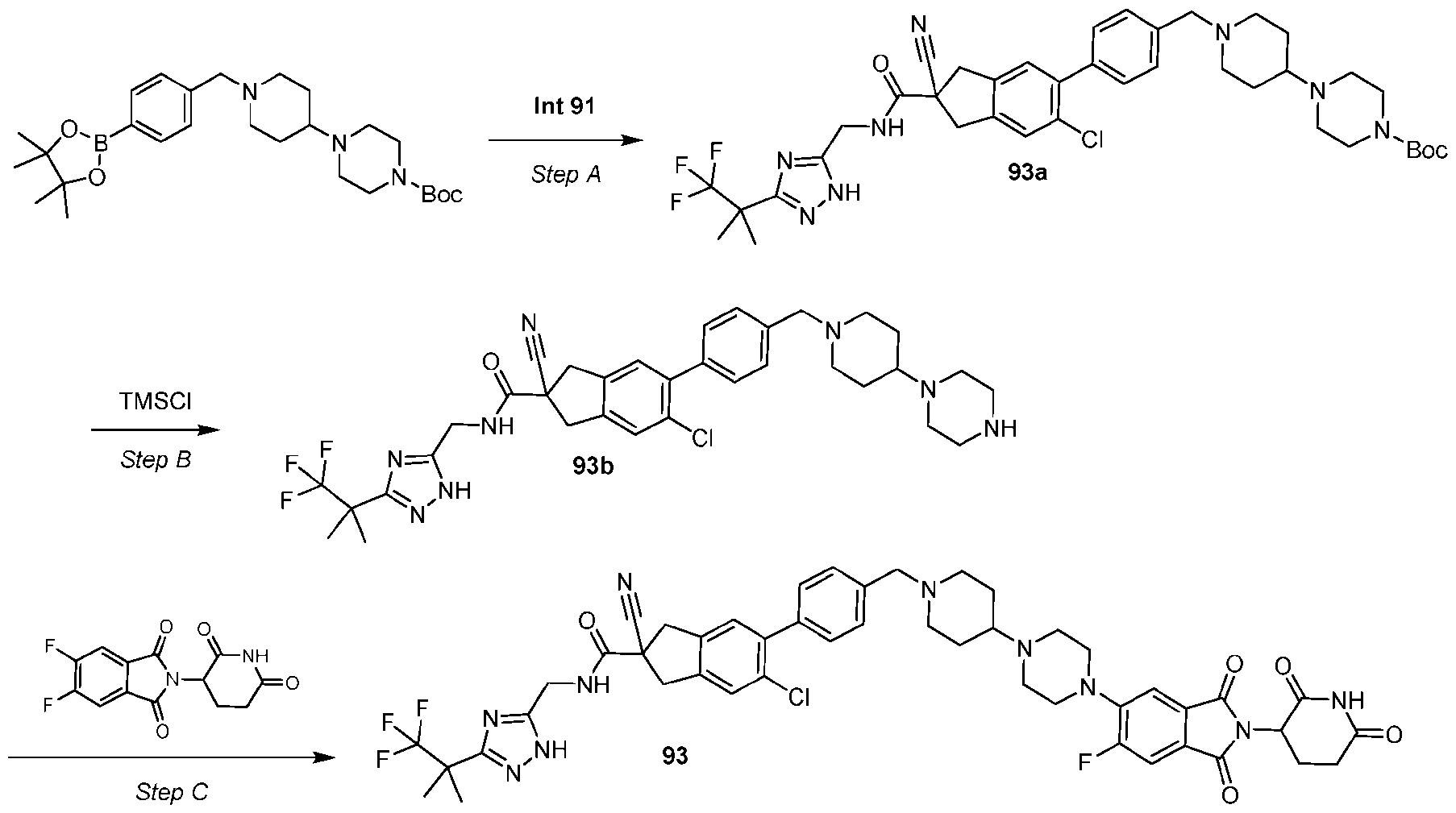 2-yl)-1H-1,2,4-triazol-5-yl)methyl) carbamoyl)-2,3-dihydro-1H-inden-5-yl)benzyl)piperidin-4- yl)piperazine-1-carboxylate (93a) A mixture of tert-butyl 4-[1-[[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)phenyl]methyl]-4-piperidyl]piperazine-1-carboxylate (90.0 mg, 185 μmol, 1.0 equiv.), Int 91 (118.2 mg, 185 μmol, 1.0 equiv.), K3PO4 (78.7 mg, 371 μmol, 2.0 equiv.) and PdCl2(DTBPF) (48.3 mg, 74 μmol, 0.4 equiv.) in THF (0.8 mL) and water (0.2 mL) was degassed and purged threefold with nitrogen. The mixture was stirred at 80°C for 16 h under a nitrogen atmosphere. The mixture was cooled to ambient temperature and poured into water (10 mL) and extracted with ethyl acetate (10 mL x 2). The combined organic portions were washed with brine (5 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum. The residue was purified by preparative HPLC (Waters Xbridge BEH C18, 100mm x 30mm, 10 μm; [water(10mM NH4HCO3)-ACN]) to afford 93a. LCMS [M-99] = 669.4, 671.4; 1H NMR (400 MHz, CHLOROFORM-d) δ 7.41 - 7.31 (m, 5H), 7.21 (s, 1H), 4.64 (s, 2H), 3.76 (t, J = 15.8 Hz, 2H), 3.61 - 3.49 (m, 4H), 3.47 - 3.39 (m, 3H), 3.07 - 2.91 (m, 3H), 2.66 - 2.46 (m, 4H), 2.37 -
2.24 (m, 1H), 2.03 - 1.95 (m, 2H), 1.85 - 1.72 (m, 2H), 1.62 - 1.55 (m, 2H) 1.47 (s, 6H), 1.28 - 1.20 (m, 9H). Step B – Synthesis of 5-chloro-2-cyano-6-(4-((4-(piperazin-1-yl)piperidin-1-yl)methyl)phenyl)-N- ((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-2,3-dihydro-1H-indene-2- carboxamide (93b) A solution of trifluoroethanol (0.9 mL) and chlorotrimethylsilane (0.1 mL) was added to a solution of 93a (90.0 mg, 117 μmol, 1.0 equiv.) in trifluoroethanol (0.9 mL) and the mixture was stirred at ambient temperature for 0.5 h. The mixture was concentrated under vacuum to afford 93b, which was used without further purification. LCMS [M+1] = 669.4, 671.4; 1H NMR (400 MHz, METHANOL-d4) δ 7.69 - 7.61 (m, 2H), 7.60 - 7.54 (m, 2H), 7.46 (s, 1H), 7.29 (s, 1H), 4.63 (s, 2H), 4.43 (s, 2H), 3.82 - 3.52 (m, 15H), 3.22 (t, J = 12.0 Hz, 2H), 2.58 - 2.44 (m, 2H), 2.28 - 2.11 (m, 2H), 1.64 (s, 6H). Step C – Synthesis of 5-chloro-2-cyano-6-(4-((4-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3- dioxoisoindolin-5-yl)piperazin-1-yl)piperidin-1-yl)methyl)phenyl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-2,3-dihydro-1H-indene-2-carboxamide (Compound 93) A mixture of 93b (70.0 mg, 99 μmol, 1.0 equiv.), 2-(2,6-dioxo-3-piperidyl)-5,6-difluoro- isoindoline-1,3-dione (43.8 mg, 149 μmol, 1.5 equiv.), DIEA (38.5 mg, 298 μmol, 51.8 μL, 3.0 equiv.) in DMSO (1.0 mL) was degassed and purged threefold with nitrogen. The mixture was stirred at 70°C for 2 h under a nitrogen atmosphere, cooled to ambient temperature and filtered. The filtrate was purified directly by preparative HPLC (Waters Xbridge BEH C18, 100mm x 30mm, 10 μm; [water(10mM NH4HCO3)-ACN]) to afford Compound 93. LCMS [M+1] = 943.3, 945.3; 1H NMR (400 MHz, CHLOROFORM-d) δ 8.45 (br s, 1H), 7.47 (d, J = 11.2 Hz, 1H), 7.43 - 7.30 (m, 6H), 7.20 (s, 1H), 4.94 (dd, J = 5.2, 12.4 Hz, 1H), 4.64 (d, J = 5.6 Hz, 2H), 3.84 - 3.69 (m, 2H), 3.64 - 3.46 (m, 4H), 3.29 (s, 4H), 3.04 (d, J = 10.8 Hz, 2H), 2.94 - 2.70 (m, 6H), 2.43 - 2.31 (m, 1H), 2.23 - 1.99 (m, 4H), 1.85 (d, J = 10.0 Hz, 2H), 1.73 - 1.54 (m, 8H). Example 94: Synthesis of Compound 94
2-yl)-1H-1,2,4-triazol-5-yl)methyl) carbamoyl)-2,3-dihydro-1H-inden-5-yl)benzyl)piperidin-4- yl)piperazine-1-carboxylate (93a) A mixture of tert-butyl 4-[1-[[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)phenyl]methyl]-4-piperidyl]piperazine-1-carboxylate (90.0 mg, 185 μmol, 1.0 equiv.), Int 91 (118.2 mg, 185 μmol, 1.0 equiv.), K3PO4 (78.7 mg, 371 μmol, 2.0 equiv.) and PdCl2(DTBPF) (48.3 mg, 74 μmol, 0.4 equiv.) in THF (0.8 mL) and water (0.2 mL) was degassed and purged threefold with nitrogen. The mixture was stirred at 80°C for 16 h under a nitrogen atmosphere. The mixture was cooled to ambient temperature and poured into water (10 mL) and extracted with ethyl acetate (10 mL x 2). The combined organic portions were washed with brine (5 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum. The residue was purified by preparative HPLC (Waters Xbridge BEH C18, 100mm x 30mm, 10 μm; [water(10mM NH4HCO3)-ACN]) to afford 93a. LCMS [M-99] = 669.4, 671.4; 1H NMR (400 MHz, CHLOROFORM-d) δ 7.41 - 7.31 (m, 5H), 7.21 (s, 1H), 4.64 (s, 2H), 3.76 (t, J = 15.8 Hz, 2H), 3.61 - 3.49 (m, 4H), 3.47 - 3.39 (m, 3H), 3.07 - 2.91 (m, 3H), 2.66 - 2.46 (m, 4H), 2.37 -
2.24 (m, 1H), 2.03 - 1.95 (m, 2H), 1.85 - 1.72 (m, 2H), 1.62 - 1.55 (m, 2H) 1.47 (s, 6H), 1.28 - 1.20 (m, 9H). Step B – Synthesis of 5-chloro-2-cyano-6-(4-((4-(piperazin-1-yl)piperidin-1-yl)methyl)phenyl)-N- ((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-2,3-dihydro-1H-indene-2- carboxamide (93b) A solution of trifluoroethanol (0.9 mL) and chlorotrimethylsilane (0.1 mL) was added to a solution of 93a (90.0 mg, 117 μmol, 1.0 equiv.) in trifluoroethanol (0.9 mL) and the mixture was stirred at ambient temperature for 0.5 h. The mixture was concentrated under vacuum to afford 93b, which was used without further purification. LCMS [M+1] = 669.4, 671.4; 1H NMR (400 MHz, METHANOL-d4) δ 7.69 - 7.61 (m, 2H), 7.60 - 7.54 (m, 2H), 7.46 (s, 1H), 7.29 (s, 1H), 4.63 (s, 2H), 4.43 (s, 2H), 3.82 - 3.52 (m, 15H), 3.22 (t, J = 12.0 Hz, 2H), 2.58 - 2.44 (m, 2H), 2.28 - 2.11 (m, 2H), 1.64 (s, 6H). Step C – Synthesis of 5-chloro-2-cyano-6-(4-((4-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3- dioxoisoindolin-5-yl)piperazin-1-yl)piperidin-1-yl)methyl)phenyl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-2,3-dihydro-1H-indene-2-carboxamide (Compound 93) A mixture of 93b (70.0 mg, 99 μmol, 1.0 equiv.), 2-(2,6-dioxo-3-piperidyl)-5,6-difluoro- isoindoline-1,3-dione (43.8 mg, 149 μmol, 1.5 equiv.), DIEA (38.5 mg, 298 μmol, 51.8 μL, 3.0 equiv.) in DMSO (1.0 mL) was degassed and purged threefold with nitrogen. The mixture was stirred at 70°C for 2 h under a nitrogen atmosphere, cooled to ambient temperature and filtered. The filtrate was purified directly by preparative HPLC (Waters Xbridge BEH C18, 100mm x 30mm, 10 μm; [water(10mM NH4HCO3)-ACN]) to afford Compound 93. LCMS [M+1] = 943.3, 945.3; 1H NMR (400 MHz, CHLOROFORM-d) δ 8.45 (br s, 1H), 7.47 (d, J = 11.2 Hz, 1H), 7.43 - 7.30 (m, 6H), 7.20 (s, 1H), 4.94 (dd, J = 5.2, 12.4 Hz, 1H), 4.64 (d, J = 5.6 Hz, 2H), 3.84 - 3.69 (m, 2H), 3.64 - 3.46 (m, 4H), 3.29 (s, 4H), 3.04 (d, J = 10.8 Hz, 2H), 2.94 - 2.70 (m, 6H), 2.43 - 2.31 (m, 1H), 2.23 - 1.99 (m, 4H), 1.85 (d, J = 10.0 Hz, 2H), 1.73 - 1.54 (m, 8H). Example 94: Synthesis of Compound 94
Step -1,3- dioxoisoindolin-5-yl)piperazin-1-yl)methyl)phenyl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)- 1H-1,2,4-triazol-5-yl)methyl)-2,3-dihydro-1H-indene-2-carboxamide (94a) A mixture of Int 91 (500.0 mg, 1.12 mmol, 1.0 equiv.) and tert-butyl 4-[[4-(4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]methyl]piperazine-1-carboxylate (360.6 mg, 0.896 mmol, 0.8 equiv.) in ethanol (4.0 mL) and water (1.0 mL) was added K3PO4 (475.6 mg, 2.2 μmol, 2.0 equiv.) and XPHOS-PD-G2 (88.1 mg, 112.0 μmol, 0.1 equiv.) was degassed and purged threefold with nitrogen and the mixture was stirred at 80°C for 16 h. The mixture was filtered through a Celite pad, and the filtrate was concentrated. The residue was purified by preparative HPLC (Waters Xbridge C18, 150 mm x 50mm, 10 µm; [water(10mM NH4HCO3)-ACN]) to afford the racemate. Chiral separation by SFC (DAICEL CHIRALPAK IG 250 mm x 30 mm,10 µm); [CO2-EtOH(0.1% NH3water)]) to afford 94a as the early-eluting enantiomer. LCMS: [M+1] =686.2, 688.2. Step B – Synthesis of (2R)-5-chloro-2-cyano-6-[4-[[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3- dioxo-isoindolin-5-yl]piperazin-1-yl]methyl]phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H-1,2,4-triazol-5-yl]methyl]indane-2-carboxamide (Compound 94) A mixture of 94a (90.0 mg, 131 μmol, 1.0 equiv.) in TFA (0.1 mL) and dichloromethane (0.5 mL) was stirred at ambient temperature for 2 h. The mixture was concentrated under vacuum. LCMS: [M+1] =586.3, 588.3. The residue (50.0 mg) in DMSO (0.5 mL) was treated with 2-(2,6- dioxo-3-piperidyl)-5,6-difluoro-isoindoline-1,3-dione (37.6 mg) and DIEA (75 μL) and the
mixture was stirred at 80°C for 2 h under a nitrogen atmosphere. The mixture cooled to ambient temperature, diluted with water (10 ml) was extracted with ethyl acetate (10 mL x 3). The combined organic portion was washed with brine (10 mL x 3), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum. The residue was purified by preparative HPLC (Phenomenex Luna C18, 100mm x 40mm, 3 µm; [water(0.2% FA)-ACN]) to afford Compound 94. LCMS: [M+1] =860.1; 1H NMR (400 MHz, DMSO-d6) δ = 13.93 (br s, 1H), 11.10 (s, 1H), 9.19 (t, J= 4.8 Hz, 1H), 7.73 (d, J = 11.6 Hz, 1H), 7.51 - 7.35 (m, 7H), 5.10 (dd, J = 5.6, 8.8 Hz, 1H), 4.44 (d, J = 5.2 Hz, 2H), 3.74 - 3.52 (m, 6H), 3.29 - 3.28 (m, 4H), 2.92 - 2.83 (m, 1H), 2.60 - 2.56 (m, 4H), 2.05 – 2.00 (m, 1H), 1.53 (s, 6H). Example 95: Synthesis of Compound 95
Step A – Synthesis of tert-butyl 4-(4-aminopyrazol-1-yl)piperidine-1-carboxylate (95a) A solution of tert-butyl 4-(4-nitropyrazol-1-yl)piperidine-1-carboxylate (2.00 g, 6.70 mmol, 1 equiv.) in EtOH (20 mL), water (6 mL) was treated with iron (1.10 g, 20.2 mmol, 3.0 equiv.) and NH4Cl (1.80 g, 33.7 mmol, 5.0 equiv.). The mixture was stirred at 80°C for 1 h, cooled to ambient temperature and filtered through Celite. The filtrate was concentrated to obtain 95a, which was used without further purification. =LCMS [M+1] = 267.1. Step B – Synthesis of tert-butyl 4-[4-[(5-chloro-2-pyridyl)amino]pyrazol-1-yl]piperidine-1- carboxylate (95b) A solution of 95a (400 mg, 1.5 mmol, 1.0 equiv.) in DMF (2 mL) was treated with cesium carbonate (978.6 mg, 3.0 mmol, 2.0 equiv.) and BINAP (187.0 mg, 300 μmol, 0.2 equiv.), 2- bromo-5-chloro-pyridine (170.0 mg, 883.3 μmol, 0.6 equiv.), Pd2(dba)3 (137.5 mg, 150 μmol, 0.1 equiv.) under a nitrogen atmosphere. The mixture was stirred at 90oC for 7 h, cooled to ambient temperature, partitioned between ethyl acetate (100 mL) and water (100 mL) and extracted with ethyl acetate (100 mL x 2). The combined organic layers were washed with brine (5 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative HPLC (Phenomenex Luna C18, 100mm x 30mm, 5 µm; [water (0.2% FA) - ACN]) to afford 95b. LCMS [M+1] = 378.2, 380.2; 1H NMR (400 MHz, DMSO-d6) δ = 8.97 (s, 1H), 8.07 (d, J = 2.4 Hz, 1H), 7.95 (s, 1H), 7.52 (dd, J = 2.8, 9.2 Hz, 1H), 7.43 (s, 1H), 6.67 (d, J = 8.8 Hz, 1H), 4.26 - 4.30 (m, 1H), 4.09 - 3.90 (m, 2H), 3.00 - 2.78 (m, 2H), 2.03 - 1.90 (m, 2H), 1.72 -1.79 (m, 2H), 1.41 (s, 9H). Step C – Synthesis of tert-butyl 4-[4-[(5-chloro-2-pyridyl)amino]pyrazol-1-yl]piperidine-1- carboxylate (95c) A solution of 95b (402.6 mg, 1.1 mmol, 1.0 equiv.) in HCl/dioxane (4 M, 6 mL) was stirred at 20 °C for 1 h. The mixture was filtered and the filtrate was concentrated under reduced pressure to afford 95c, which was used without further purification. LCMS [M+1] = 278.0, 280.0.
Step D – Synthesis of tert-butyl 4-[[4-[4-[(5-chloro-2-pyridyl)amino]pyrazol-1-yl]-1- piperidyl]methyl]piperidine-1-carboxylate (95d) To a solution of 95c (230.0 mg, 732 μmol, 1.0 equiv.) in dichloromethane (5 mL) was added NaBH(OAc)3 (620.5 mg, 2.9 mmol, 4.0 equiv.), DIEA (189.2 mg, 1.4 mmol, 255 μL, 2.0 equiv.), tert-butyl 4-formylpiperidine-1-carboxylate (156.1 mg, 732 μmol, 1.0 equiv.). The mixture was stirred at 20°C for 2 h. The reaction mixture was partitioned between ethyl acetate (100 mL) and water (100 mL) and extracted with ethyl acetate (20 mL x 2). The combined organic portions were washed with brine (5 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure afford 95d, which was used without further purification. LCMS [M+1] = 475.3, 477.3. Step E – Synthesis of tert-butyl 4-[[4-[4-[[5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-2- pyridyl]amino]pyrazol-1-yl]-1-piperidyl]methyl]piperidine-1-carboxylate (95e) A solution of 95d (300.0 mg, 632 μmol, 1.0 equiv.) in dioxane (4 mL) was treated with KOAc (185.9 mg, 1.8 mmol, 3.0 equiv.), XPhos (60.2 mg, 126 μmol, 0.2 equiv.), 4,4,5,5- tetramethyl-2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3,2-dioxaborolane (320.7 mg, 1.20 mmol, 2.0 equiv.), Pd2(dba)3 (115.6 mg, 126 μmol, 0.2 equiv.) under a nitrogen atmosphere. The mixture was stirred at 85°C for 16 h, cooled to ambient temperature, partitioned between ethyl acetate (50 mL) and water (50 mL) and extracted with ethyl acetate (20 mL x 2). The combined organic portion was washed with brine (5 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure to afford 95e, which was used without further purification. LCMS [M+1] = 567.5. Step F – Synthesis of tert-butyl 4-[[4-[4-[[5-[4-[(1S,2S)-2-[[3-(2,2,2-trifluoro-1,1-dimethyl- ethyl)-1H-1,2,4-triazol-5-yl]methylcarbamoyl]cyclopropyl]phenyl]-2-pyridyl]amino]pyrazol-1- yl]-1-piperidyl]methyl]piperidine-1-carboxylate (95f) A solution of 95e (216.7 mg, 383 μmol, 1.1 equiv.) in THF (2 mL), water (0.6 mL) was treated with PdCl2(DTBPF) (113.3 mg, 174 μmol, 0.5 equiv.), tripotassium;phosphate (221.5 mg, 1.0 mmol, 3.0 equiv.) and Int 3 (150.0 mg, 348 μmol, 1.0 equiv.) under an atmosphere of nitrogen. The mixture was stirred at 85°C for 16 h. The reaction mixture was partitioned between ethyl acetate (50 mL) and water (50 mL) extracted with ethyl acetate (20 mL x 2). The combined
organic layers were washed with brine (5 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure to afford 95f. LCMS [M+1] = 791.6. Step G – Synthesis of (1S,2S)-2-[4-[6-[[1-[1-(4-piperidylmethyl)-4-piperidyl]pyrazol-4- yl]amino]-3-pyridyl]phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methyl]cyclopropanecarboxamide (95g) A solution of 95f (200.0 mg, 253 μmol, 1.0 equiv.) in HCl/dioxane (4 M, 3.0 mL) was stirred at 20°C for 1 h. The mixture was filtered and concentrated under reduced pressure to afford 95g, which was used without further purification. Step H – Synthesis of (1S,2S)-2-[4-[6-[[1-[1-[[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo- isoindolin-5-yl]-4-piperidyl]methyl]-4-piperidyl]pyrazol-4-yl]amino]-3-pyridyl]phenyl]-N-[[3- (2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]cyclopropanecarboxamide (Compound 95) A solution of 95g (30.0 mg, 41 μmol, 1.0 equiv.) in DMSO (1 mL) was treated with DIEA (21.3 mg, 165 μmol, 29 μL, 4.0 equiv.) and 2-(2,6-dioxo-3-piperidyl)-5,6-difluoro-isoindoline- 1,3-dione (13.3 mg, 45.3 μmol, 1.1 equiv.). The mixture was stirred at 70°C for 16 h, cooled to ambient temperature, partitioned between ethyl acetate (10 mL ) and water (10 mL) and extracted with ethyl acetate (10 mL x 2). The combined organic layers were washed with brine (5 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative HPLC (Phenomenex Luna C18, 100mm x 30mm, 5 µm; [water(0.2% FA)-ACN]) to afford Compound 95. LCMS [M+1] = 965.6; 1H NMR (400 MHz, DMSO-d6) δ = 11.11 ( s, 1H), 8.89 (s, 1H), 8.75 (br s, 1H), 8.42 (d, J = 2.4 Hz, 1H), 8.03 (s, 1H), 7.85 - 7.76 (m, 1H), 7.71 (d, J = 11.2 Hz, 1H), 7.53 (d, J = 8.2 Hz, 2H), 7.47 - 7.40 (m, 2H), 7.18 (d, J = 8.4 Hz, 2H), 6.74 (d, J = 8.8 Hz, 1H), 5.10 (dd, J = 5.4, 12.8 Hz, 1H), 4.41 (d, J = 5.6 Hz, 2H), 4.20 - 4.03 (m, 1H), 3.62 (d, J = 11.6 Hz, 2H), 3.03 - 2.81 (m, 5H), 2.71 - 2.56 (m, 2H), 2.37 - 2.28 (m, 2H), 2.23 (d, J = 6.4 Hz, 2H), 2.13 - 2.02 (m, 3H), 2.00 - 1.91 (m, 4H), 1.89 - 1.81 (m, 2H), 1.79 - 1.67 (m, 1H), 1.53 (s, 6H), 1.45 - 1.36 (m, 1H), 1.34 - 1.19 (m, 4H), 1.05 - 0.94 (m, 1H)
Example 96: Synthesis of Compound 96
 4-[4-[(5-bromo-2-pyridyl)oxy]pyrazol-1-yl]piperidine-1-carboxylate (prepared by SNAr condensation of tert-butyl 4-(4-hydroxypyrazol-1-yl)piperidine-1-carboxylate and 5-bromo-2- fluoro-pyridine in DMF with potassium carbonate (2 equiv.), 80°C, 16 h) in Step C. Example 97: Synthesis of Intermediate 97
4-[4-[(5-bromo-2-pyridyl)oxy]pyrazol-1-yl]piperidine-1-carboxylate (prepared by SNAr condensation of tert-butyl 4-(4-hydroxypyrazol-1-yl)piperidine-1-carboxylate and 5-bromo-2- fluoro-pyridine in DMF with potassium carbonate (2 equiv.), 80°C, 16 h) in Step C. Example 97: Synthesis of Intermediate 97
 1-carboxylate (97a) A solution of tert-butyl 4-(4-amino-2-fluoro-phenyl)piperidine-1-carboxylate (5.0 g, 16.9 mmol, 1.0 equiv.) in DMF (50 mL) was treated with NaHCO3 (8.5 g, 101.9 mmol, 3.9 mL, 6.0 equiv.) and 3-bromopiperidine-2,6-dione (9.70 g, 50.9 mmol, 3.0 equiv.). The mixture was stirred at 70°C for 16 h. The reaction mixture was partitioned between water (100 mL) and ethyl acetate (200 mL.). The organic phase was separated, dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether/ethyl acetate=2/1) to afford 97a. LCMS [M- 55] =350.1; Step B – Synthesis of 3-[3-fluoro-4-(4-piperidyl)anilino]piperidine-2,6-dione (Int 97)
A solution of 97a (2.0 g, 4.9 mmol, 1.0 equiv.) in HCl/dioxane (4 M, 20 mL) was stirred at 20°C for 1 h. The mixture solution was concentrated under vacuum to give afford Int 97 as the hydrochloride salt, which was used without further purification. LCMS: [M+1] = 306.0. Example 98: Synthesis of Intermediate 98
1-carboxylate (97a) A solution of tert-butyl 4-(4-amino-2-fluoro-phenyl)piperidine-1-carboxylate (5.0 g, 16.9 mmol, 1.0 equiv.) in DMF (50 mL) was treated with NaHCO3 (8.5 g, 101.9 mmol, 3.9 mL, 6.0 equiv.) and 3-bromopiperidine-2,6-dione (9.70 g, 50.9 mmol, 3.0 equiv.). The mixture was stirred at 70°C for 16 h. The reaction mixture was partitioned between water (100 mL) and ethyl acetate (200 mL.). The organic phase was separated, dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether/ethyl acetate=2/1) to afford 97a. LCMS [M- 55] =350.1; Step B – Synthesis of 3-[3-fluoro-4-(4-piperidyl)anilino]piperidine-2,6-dione (Int 97)
A solution of 97a (2.0 g, 4.9 mmol, 1.0 equiv.) in HCl/dioxane (4 M, 20 mL) was stirred at 20°C for 1 h. The mixture solution was concentrated under vacuum to give afford Int 97 as the hydrochloride salt, which was used without further purification. LCMS: [M+1] = 306.0. Example 98: Synthesis of Intermediate 98
 Step A – Synthesis of tert-butyl 4-(5-amino-3-fluoro-2-pyridyl)-3,6-dihydro-2H-pyridine-1- carboxylate (98a) A mixture of 6-bromo-5-fluoro-pyridin-3-amine (50.0 g, 261.8 mmol, 1.0 equiv.) and tert- butyl 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-3,6-dihydro-2H-pyridine-1-carboxylate (97.13 g, 314.1 mmol, 1.2 equiv.) in water (90.0 mL) and dioxane (450.0 mL) was degassed and purged threefold with nitrogen. The mixture was treated with K3PO4 (111.1 g, 523.6 mmol, 2.0 equiv.) and Pd(dppf)Cl2.CH2Cl2 (21.38 g, 26.18 mmol, 0.1 equiv.) and stirred at 100°C for 12 h under a nitrogen atmosphere. The reaction mixture was quenched with water (1 L) and extracted with ethyl acetate (500 mL x 3). The combined organic layers were washed with brine (1 L), dried over anhydrous sodium sulphate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether: ethyl acetate=1: 0 to 0: 1) to afford 98a. LCMS [M+1] = 294.3.
Step B – Synthesis of tert-butyl 4-[5-[(2,6-dibenzyloxy-3-pyridyl)amino]-3-fluoro-2-pyridyl]-3,6- dihydro-2H-pyridine-1-carboxylate (98b) A mixture of 98a (67.0 g, 228.4 mmol, 1.0 equiv.), 2,6-dibenzyloxy-3-bromo-pyridine (101.5 g, 274.1 mmol, 1.2 equiv.) and cesium carbonate(148.8 g, 456.8 mmol, 2.0 equiv.) in toluene (700.0 mL) was degassed and purged threefold with nitrogen and treated with Pd(dba)2 (13.13 g, 22.84 mmol, 0.1 equiv.) and BRETTPHOS (24.52 g, 45.68 mmol, 0.2 equiv.). The mixture was stirred at 120°C for 12 h under a nitrogen atmosphere. The reaction mixture was quenched with water (1 L) and extracted with ethyl acetate (500 mL x 3). The combined organic layers were washed with brine (1 L), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether: ethyl acetate = 1: 0 to 0: 1) to afford 98b. LCMS [M+1] = 583.3. Step C – Synthesis of tert-butyl 4-[5-[(2, 6-dioxo-3-piperidyl) amino]-3-fluoro-2-pyridyl] piperidine-1-carboxylate (98c) A hydrogenation vessel was charged with Pd(OH)2/C (9.76 g, 13.90 mmol, 20 wt%, 0.3 equiv.), purged with argon and charged with ethanol (540 mL) and a solution of 98b (27 g, 46.34 mmol, 1.0 equiv.) in THF (30 mL). The mixture was purged and degassed threefold with hydrogen and stirred at 30 °C under hydrogen (1 atm) for 12 h. The mixture was filtered and concentrated under reduce pressure and the residue was purified by column chromatography (silica gel, petroleum ether: ethyl acetate = 1: 0 to 0: 1) to afford 98c. LCMS [M+1] = 407.2. Step D – Synthesis of 3-[[5-fluoro-6-(4-piperidyl)-3-pyridyl] amino] piperidine-2, 6-dione (Int 98) A solution of 98c (15.0 g, 36.90 mmol, 1.0 equiv.) in dichloromethane (6.0 mL) was treated with trifluoroacetic acid (3.0 mL). The mixture was stirred at 25°C for 1 h. The reaction mixture was quenched with water (10.0 mL) and extracted with dichloromethane (5.0 mL x 3). The aqueous phase was lyophilized to afford Int 98 as the trifluoroacetate salt. LCMS [M+1] = 307.1
Example 99: Synthesis of Intermediate 99
Step A – Synthesis of tert-butyl 4-(5-amino-3-fluoro-2-pyridyl)-3,6-dihydro-2H-pyridine-1- carboxylate (98a) A mixture of 6-bromo-5-fluoro-pyridin-3-amine (50.0 g, 261.8 mmol, 1.0 equiv.) and tert- butyl 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-3,6-dihydro-2H-pyridine-1-carboxylate (97.13 g, 314.1 mmol, 1.2 equiv.) in water (90.0 mL) and dioxane (450.0 mL) was degassed and purged threefold with nitrogen. The mixture was treated with K3PO4 (111.1 g, 523.6 mmol, 2.0 equiv.) and Pd(dppf)Cl2.CH2Cl2 (21.38 g, 26.18 mmol, 0.1 equiv.) and stirred at 100°C for 12 h under a nitrogen atmosphere. The reaction mixture was quenched with water (1 L) and extracted with ethyl acetate (500 mL x 3). The combined organic layers were washed with brine (1 L), dried over anhydrous sodium sulphate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether: ethyl acetate=1: 0 to 0: 1) to afford 98a. LCMS [M+1] = 294.3.
Step B – Synthesis of tert-butyl 4-[5-[(2,6-dibenzyloxy-3-pyridyl)amino]-3-fluoro-2-pyridyl]-3,6- dihydro-2H-pyridine-1-carboxylate (98b) A mixture of 98a (67.0 g, 228.4 mmol, 1.0 equiv.), 2,6-dibenzyloxy-3-bromo-pyridine (101.5 g, 274.1 mmol, 1.2 equiv.) and cesium carbonate(148.8 g, 456.8 mmol, 2.0 equiv.) in toluene (700.0 mL) was degassed and purged threefold with nitrogen and treated with Pd(dba)2 (13.13 g, 22.84 mmol, 0.1 equiv.) and BRETTPHOS (24.52 g, 45.68 mmol, 0.2 equiv.). The mixture was stirred at 120°C for 12 h under a nitrogen atmosphere. The reaction mixture was quenched with water (1 L) and extracted with ethyl acetate (500 mL x 3). The combined organic layers were washed with brine (1 L), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether: ethyl acetate = 1: 0 to 0: 1) to afford 98b. LCMS [M+1] = 583.3. Step C – Synthesis of tert-butyl 4-[5-[(2, 6-dioxo-3-piperidyl) amino]-3-fluoro-2-pyridyl] piperidine-1-carboxylate (98c) A hydrogenation vessel was charged with Pd(OH)2/C (9.76 g, 13.90 mmol, 20 wt%, 0.3 equiv.), purged with argon and charged with ethanol (540 mL) and a solution of 98b (27 g, 46.34 mmol, 1.0 equiv.) in THF (30 mL). The mixture was purged and degassed threefold with hydrogen and stirred at 30 °C under hydrogen (1 atm) for 12 h. The mixture was filtered and concentrated under reduce pressure and the residue was purified by column chromatography (silica gel, petroleum ether: ethyl acetate = 1: 0 to 0: 1) to afford 98c. LCMS [M+1] = 407.2. Step D – Synthesis of 3-[[5-fluoro-6-(4-piperidyl)-3-pyridyl] amino] piperidine-2, 6-dione (Int 98) A solution of 98c (15.0 g, 36.90 mmol, 1.0 equiv.) in dichloromethane (6.0 mL) was treated with trifluoroacetic acid (3.0 mL). The mixture was stirred at 25°C for 1 h. The reaction mixture was quenched with water (10.0 mL) and extracted with dichloromethane (5.0 mL x 3). The aqueous phase was lyophilized to afford Int 98 as the trifluoroacetate salt. LCMS [M+1] = 307.1
Example 99: Synthesis of Intermediate 99
 Step A – Synthesis of tert-butyl 4-(3-fluoro-5-nitro-2-pyridyl)piperazine-1-carboxylate (99a) A solution of tert-butyl piperazine-1-carboxylate (11.9 g, 63.7 mmol, 1.5 equiv.) and 2- chloro-3-fluoro-5-nitro-pyridine (7.5 g, 42.5 mmol, 1.0 equiv.) in DMF (75.0 mL) was treated with DIEA (22.2 mL, 127.4 mmol, 3.0 equiv.). The mixture was stirred at 80°C for 2 h, cooled to ambient temperature and quenched with water (50 mL). The mixture was diluted with water (50 mL) and extracted with ethyl acetate (150 mL x 2). The combined organic layers were washed with brine (170 mL x 2), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure to afford 99a, which was used without further purification. LCMS [M-99] = 227.0. Step B – Synthesis of tert-butyl 4-(5-amino-3-fluoropyridin-2-yl)piperazine-1-carboxylate (99b) A mixture of 99a (17.7 g, 54.2 mmol, 1.0 equiv.) in ethanol (90.0 mL) and water (90.0 mL) was added Fe (15.2 g, 271.2 mmol, 5.0 equiv.) was treated with NH4Cl (17.4 g, 325.4 mmol, 6.0 equiv.). The mixture was stirred at 80°C for 1 h, cooled to 25°C, filtered and the filter cake was washed with ethyl acetate (200 mL). The combined filtrate was concentrated under vacuum The residue was purified by silica gel chromatography (silica gel, ethyl acetate: petroleum =0:1 to 1:4) to afford 99b. LCMS [M-99] = 196.8.
Step C – Synthesis of tert-butyl 4-[5-[(2,6-dioxo-3-piperidyl)amino]-3-fluoro-2- pyridyl]piperazine-1-carboxylate (99c) To a solution of 99b (7.5 g, 25.3 mmol, 1.0 equiv.) and 3-bromopiperidine-2,6-dione (9.7 g, 50.6 mmol, 2.0 equiv.) in DMF (90.0 mL) was added NaHCO3 (6.4 g, 75.9 mmol, 2.95 mL, 3.0 equiv.). The mixture was stirred at 70°C for 16 h. The mixture was cooled to 25°C, quenched with water (200 mL), diluted with water (100 mL) and extracted with ethyl acetate (350mL x 2). The combined organic layers were washed with brine (350 mL x 2), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel chromatography (silica gel, ethyl acetate: petroleum =0: 1 to 1: 2) to afford 99c. LCMS [M-99] = 308.0. Step D – Synthesis of 3-[(5-fluoro-6-piperazin-1-yl-3-pyridyl)amino]piperidine-2,6-dione (Int 99) A solution of 99c (8.9 g, 21.9 mmol, 1.0 equiv.) in HCl/dioxane (2 M, 90.0 mL, 8.2 equiv.) was stirred at 25°C for 30 min. The mixture was concentrated under vacuum to afford Int 99 as the hydrochloride salt, which was used without further purification. LCMS [M+1] = 308.1. Example 100: Synthesis of Intermediate 100
Step A – Synthesis of tert-butyl 4-(3-fluoro-5-nitro-2-pyridyl)piperazine-1-carboxylate (99a) A solution of tert-butyl piperazine-1-carboxylate (11.9 g, 63.7 mmol, 1.5 equiv.) and 2- chloro-3-fluoro-5-nitro-pyridine (7.5 g, 42.5 mmol, 1.0 equiv.) in DMF (75.0 mL) was treated with DIEA (22.2 mL, 127.4 mmol, 3.0 equiv.). The mixture was stirred at 80°C for 2 h, cooled to ambient temperature and quenched with water (50 mL). The mixture was diluted with water (50 mL) and extracted with ethyl acetate (150 mL x 2). The combined organic layers were washed with brine (170 mL x 2), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure to afford 99a, which was used without further purification. LCMS [M-99] = 227.0. Step B – Synthesis of tert-butyl 4-(5-amino-3-fluoropyridin-2-yl)piperazine-1-carboxylate (99b) A mixture of 99a (17.7 g, 54.2 mmol, 1.0 equiv.) in ethanol (90.0 mL) and water (90.0 mL) was added Fe (15.2 g, 271.2 mmol, 5.0 equiv.) was treated with NH4Cl (17.4 g, 325.4 mmol, 6.0 equiv.). The mixture was stirred at 80°C for 1 h, cooled to 25°C, filtered and the filter cake was washed with ethyl acetate (200 mL). The combined filtrate was concentrated under vacuum The residue was purified by silica gel chromatography (silica gel, ethyl acetate: petroleum =0:1 to 1:4) to afford 99b. LCMS [M-99] = 196.8.
Step C – Synthesis of tert-butyl 4-[5-[(2,6-dioxo-3-piperidyl)amino]-3-fluoro-2- pyridyl]piperazine-1-carboxylate (99c) To a solution of 99b (7.5 g, 25.3 mmol, 1.0 equiv.) and 3-bromopiperidine-2,6-dione (9.7 g, 50.6 mmol, 2.0 equiv.) in DMF (90.0 mL) was added NaHCO3 (6.4 g, 75.9 mmol, 2.95 mL, 3.0 equiv.). The mixture was stirred at 70°C for 16 h. The mixture was cooled to 25°C, quenched with water (200 mL), diluted with water (100 mL) and extracted with ethyl acetate (350mL x 2). The combined organic layers were washed with brine (350 mL x 2), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel chromatography (silica gel, ethyl acetate: petroleum =0: 1 to 1: 2) to afford 99c. LCMS [M-99] = 308.0. Step D – Synthesis of 3-[(5-fluoro-6-piperazin-1-yl-3-pyridyl)amino]piperidine-2,6-dione (Int 99) A solution of 99c (8.9 g, 21.9 mmol, 1.0 equiv.) in HCl/dioxane (2 M, 90.0 mL, 8.2 equiv.) was stirred at 25°C for 30 min. The mixture was concentrated under vacuum to afford Int 99 as the hydrochloride salt, which was used without further purification. LCMS [M+1] = 308.1. Example 100: Synthesis of Intermediate 100
Step A – Synthesis of 2,6-dibenzyloxy-3-(5, 6-difluoro-3-pyridyl) pyridine (100a) A mixture of 5-bromo-2,3-difluoro-pyridine (10.0 g, 51.6 mmol, 1.0 equiv.), 2,6- dibenzyloxy-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine (21.5 g, 51.6 mmol, 1.0 equiv.), Pd(dppf)Cl2 (3.8 g, 5.2 mmol, 0.1 equiv.) and sodium carbonate (16.4 g, 154.7 mmol, 3.0 equiv.) in dioxane (100 mL) and water (25 mL) was degassed and purged threefold with nitrogen. The mixture was stirred at 100°C under a nitrogen atmosphere for 2 h. The reaction mixture was diluted with water (50 mL), then extracted with ethyl acetate (100 mL x 2). The combined organic layers were washed with brine (100 mL x 2). The combined organic layers were dried over anhydrous sodium sulfate and concentrated under vacuum. The residue was purified by silica gel chromatography (ethyl acetate: petroleum ether = 1/3) to afford 100a. LCMS [M+1] + = 405.0. Step B – Synthesis of 3-(5, 6-difluoro-3-pyridyl) piperidine-2, 6-dione (100b) A solution of 100a (10.0 g, 24.7 mmol, 1.0 equiv.) in methanol (200 mL) was treated with a methanol slurry of Pd/C (2.6 g, 10% purity) and Pd(OH)2/C (2.6 g, 20% purity). The mixture was stirred at 50°C under hydrogen (1 atm) for 12 h. The mixture was filtered with diatomaceous earth and the filtrate was concentrated under vacuum. The residue was purified by silica gel chromatography dichloromethane: methanol = 1/2) to afford 100b. LCMS [M+1] = 227.2 Step C – Synthesis of tert-butyl 4-(5-(2,6-dioxopiperidin-3-yl)-3-fluoropyridin-2-yl)piperazine-1- carboxylate (100c) A solution of tert-butyl piperazine-1-carboxylate (90.6 mg, 486.3 μmol, 1.1 equiv.) in DMSO (1.0 mL) was treated with DIEA (171.4 mg, 1.3 mmol, 231.0 μL, 3.0 equiv.) and 100b (100.0 mg, 442.1 μmol, 1.0 equiv.). The mixture was stirred at 80°C for 12 h. The mixture was cooled to 25°C and poured into water (20 mL). The aqueous phase was extracted with ethyl acetate (20 mL x 3). The combined organic phase was washed with brine (20 mL x 3), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum. The residue was purified by preparative HPLC (Phenomenex Luna C18, 150x25mm, 10µm; [water(FA)- ACN]) to afford 100c. LCMS [M+1] = 393.0
Step D – Synthesis of 3-(5-fluoro-6-(piperazin-1-yl) pyridin-3-yl) piperidine-2, 6-dione (Int 100) A solution of 100c (90.0 mg, 229.3 μmol, 1.0 equiv.) in HCl/dioxane (2M, 2.0 mL) was stirred at 25°C for 1 h. The mixture was concentrated under vacuum to afford Int 100 as the hydrochloride salt, which was used without further purification. LCMS [M+1] = 293.0 Example 101: Synthesis of Intermediate 101
 A solution of 4-bromo-2-fluoro-1-iodobenzene (5.0 g, 16.6 mmol, 1.0 equiv.) and tert- butyl piperazine-1-carboxylate (3.8 g, 17.3 mmol, 1.0 equiv.) in DMSO (50.0 mL) was treated with (2S)-pyrrolidine-2-carboxylic acid (765.2 mg, 6.7 mmol, 0.4 equiv.). The mixture was sparged with nitrogen for 15 minutes, treated with CuI (632.9 mg, 3.3 mmol, 0.2 equiv.) and K2CO3 (4.6 g, 33.2 mmol, 2.0 equiv.) and stirred at 100°C for 12 h under a nitrogen atmosphere. The reaction mixture was quenched with water (200 mL) and extracted with ethyl acetate (200 mL x 3). The combined organic layers were washed with brine (200 mL x 3), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether : ethyl acetate=1 : 0 to 96 : 4) to afford 101a. LCMS [M+1] = 359.0.
Step B – Synthesis of tert-butyl 4-(4-(2,6-bis(benzyloxy)pyridin-3-yl)-2-fluorophenyl)piperazine- 1-carboxylate (101b) A mixture of 101a (200.0 mg, 557 μmol, 1.0 equiv.) and 2,6-bis(benzyloxy)-3-(4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine (348.5 mg, 835 μmol, 1.5 equiv.) in dioxane (2.0 mL) and water (1.0 mL) was treated with sodium carbonate (177.0 mg, 1.6 mmol, 3.0 equiv.) and Pd(dppf)Cl2 (40.7 mg, 55.6 μmol, 0.1 equiv.). The mixture was stirred at 100°C for 3 h under a nitrogen atmosphere. The mixture was poured into water (20 mL) and extracted with ethyl acetate (20 mL x 3). The combined organic phase was washed with brine (20 mL x 2), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum. The residue was purified by preparative TLC (petroleum ether/ ethyl acetate=5/1) to afford 101b. LCMS [M+1] = 570.2. Step C – Synthesis of tert-butyl 4-(4-(2,6-dioxopiperidin-3-yl)-2-fluorophenyl)piperazine-1- carboxylate (101c) A solution of 101b (310.0 mg, 544 μmol, 1.0 equiv.) in ethanol (3.0 mL) and ethyl acetate (3.0 mL) under a nitrogen atmosphere was treated with Pd/C (289.5 mg, 272.0 μmol, 10.0% purity, 0.5 equiv.). The mixture was stirred at 20°C for 1 h under hydrogen (1 atm). The reaction mixture was filtered and the filtrate was concentrated under vacuum to afford 101c, which was used without further purification. LCMS [M+1] = 392.1. Step D – Synthesis of 3-(3-fluoro-4-(piperazin-1-yl)phenyl)piperidine-2,6-dione (Int 100) A solution of 101c (210.0 mg, 537 μmol, 1.0 equiv.) in dioxane (0.5 mL) was treated with HCl/dioxane (4.0 M, 134 μL, 10.0 equiv.). The mixture was stirred at 20°C for 2 h and then concentrated under vacuum to afford Int 100 as the HCl salt, which was used without further purification. LCMS [M+1] = 292.1. Example 102: Synthesis of Intermediate 102
A solution of 4-bromo-2-fluoro-1-iodobenzene (5.0 g, 16.6 mmol, 1.0 equiv.) and tert- butyl piperazine-1-carboxylate (3.8 g, 17.3 mmol, 1.0 equiv.) in DMSO (50.0 mL) was treated with (2S)-pyrrolidine-2-carboxylic acid (765.2 mg, 6.7 mmol, 0.4 equiv.). The mixture was sparged with nitrogen for 15 minutes, treated with CuI (632.9 mg, 3.3 mmol, 0.2 equiv.) and K2CO3 (4.6 g, 33.2 mmol, 2.0 equiv.) and stirred at 100°C for 12 h under a nitrogen atmosphere. The reaction mixture was quenched with water (200 mL) and extracted with ethyl acetate (200 mL x 3). The combined organic layers were washed with brine (200 mL x 3), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether : ethyl acetate=1 : 0 to 96 : 4) to afford 101a. LCMS [M+1] = 359.0.
Step B – Synthesis of tert-butyl 4-(4-(2,6-bis(benzyloxy)pyridin-3-yl)-2-fluorophenyl)piperazine- 1-carboxylate (101b) A mixture of 101a (200.0 mg, 557 μmol, 1.0 equiv.) and 2,6-bis(benzyloxy)-3-(4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine (348.5 mg, 835 μmol, 1.5 equiv.) in dioxane (2.0 mL) and water (1.0 mL) was treated with sodium carbonate (177.0 mg, 1.6 mmol, 3.0 equiv.) and Pd(dppf)Cl2 (40.7 mg, 55.6 μmol, 0.1 equiv.). The mixture was stirred at 100°C for 3 h under a nitrogen atmosphere. The mixture was poured into water (20 mL) and extracted with ethyl acetate (20 mL x 3). The combined organic phase was washed with brine (20 mL x 2), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum. The residue was purified by preparative TLC (petroleum ether/ ethyl acetate=5/1) to afford 101b. LCMS [M+1] = 570.2. Step C – Synthesis of tert-butyl 4-(4-(2,6-dioxopiperidin-3-yl)-2-fluorophenyl)piperazine-1- carboxylate (101c) A solution of 101b (310.0 mg, 544 μmol, 1.0 equiv.) in ethanol (3.0 mL) and ethyl acetate (3.0 mL) under a nitrogen atmosphere was treated with Pd/C (289.5 mg, 272.0 μmol, 10.0% purity, 0.5 equiv.). The mixture was stirred at 20°C for 1 h under hydrogen (1 atm). The reaction mixture was filtered and the filtrate was concentrated under vacuum to afford 101c, which was used without further purification. LCMS [M+1] = 392.1. Step D – Synthesis of 3-(3-fluoro-4-(piperazin-1-yl)phenyl)piperidine-2,6-dione (Int 100) A solution of 101c (210.0 mg, 537 μmol, 1.0 equiv.) in dioxane (0.5 mL) was treated with HCl/dioxane (4.0 M, 134 μL, 10.0 equiv.). The mixture was stirred at 20°C for 2 h and then concentrated under vacuum to afford Int 100 as the HCl salt, which was used without further purification. LCMS [M+1] = 292.1. Example 102: Synthesis of Intermediate 102
Step A – Synthesis of tert-butyl 4-(5-aminopyrazin-2-yl)-3,6-dihydro-2H-pyridine-1-carboxylate (102a) A solution of tert-butyl 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-3,6- dihydropyridine-1(2H)-carboxylate (5.33 g, 17.2 mmol, 1.5 equiv.) and 5-bromopyrazin-2-amine (2 g, 11.4 mmol, 1 equiv.) in dioxane (20 mL) and water (4 mL) was treated with K3PO4 (4.88 g, 22.9 mmol, 2 equiv.) and Pd(dppf)Cl2.CH2Cl2 (938.7 mg, 1.1 mmol, 0.1 equiv.). The mixture was degassed and purged threefold with nitrogen, stirred at 100°C for 4 h under nitrogen atmosphere, cooled to ambient temperature and diluted with water (50 mL) and ethyl acetate (20 mL). The mixture was extracted with ethyl acetate (20 mL x 3). The combined organic layers were washed with brine (50 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether : ethyl acetate=1:0 to 0:1) to afford 102a. LCMS [M+1] =277.5 Step B – Synthesis of tert-butyl 4-[5-[(2,6-dibenzyloxy-3-pyridyl)amino]pyrazin-2-yl]-3,6- dihydro-2H-pyridine-1-carboxylate (102b) A solution of 102a (1.50 g, 5.40 mmol, 1 equiv.) and 2,6-dibenzyloxy-3-bromo-pyridine (1.61 g, 4.3 mmol, 0.8 equiv.) in toluene (15 mL) was treated with cesium carbonate (3.54 g, 10.8 mmol, 2 equiv.), degassed and purged threefold with nitrogen and treated with Pd(dba)2 (312.13
mg, 543 μmol, 0.1 equiv.) and dicyclohexyl-[3,6-dimethoxy-2-(2,4,6- triisopropylphenyl)phenyl]phosphane (2.91 g, 5.4 mmol, 1 equiv.). The mixture was degassed and purged with nitrogen and stirred at 115°C for 12 h under nitrogen. The reaction was cooled to ambient temperature, quenched with water (20mL) and extracted with ethyl acetate (20 mL x 3). The combined organic phase was washed with brine (20 mL x 3), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum. The product was purified by preparative TLC(silica gel, petroleum ether : ethyl acetate = 0:1) afford 102b. LCMS [M+1] = 566.3 Step C – Synthesis of tert-butyl4-[5-[(2,6-dioxo-3-piperidyl)amino]pyrazin-2-yl]piperidine-1- carboxylate (102c) A hydrogenation vessel, purged with argon, was charged with Pd/C (1.5 g, 1.4 mmol, 10% purity) and a mixture of 102b (2.30 g, 4.00 mmol, 1 equiv.) in ethanol (20 mL) and THF (10 mL). The vessel was purged and degassed threefold with hydrogen and the mixture was stirred at 30 °C under hydrogen (15 psi) for 3 h. The mixture was filtered and the filtrate was concentrated under vacuum. The residue was purified by column chromatography (silica gel, petroleum ether: ethyl acetate = 1:0 to 1:1) to afford 102c. LCMS [M+1] = 390.4 Step D – Synthesis of 3-[[5-(4-piperidyl)pyrazin-2-yl]amino]piperidine-2,6-dione (Int 102) A solution of 102c (300 mg, 770 μmol, 1 equiv.) in dichloromethane (2 mL) was treated with trifluoroacetic acid (600 μL, 8.0 mmol, 10.4 equiv.), the reaction was stirred at 25°C for 1 h. and then concentrated under reduced pressure to afford Int 102 as the trifluoroacetate salt, which was used without further purification. LCMS [M+1] = 290.5 Example 103: Synthesis of Intermediate 103
Boc N Boc
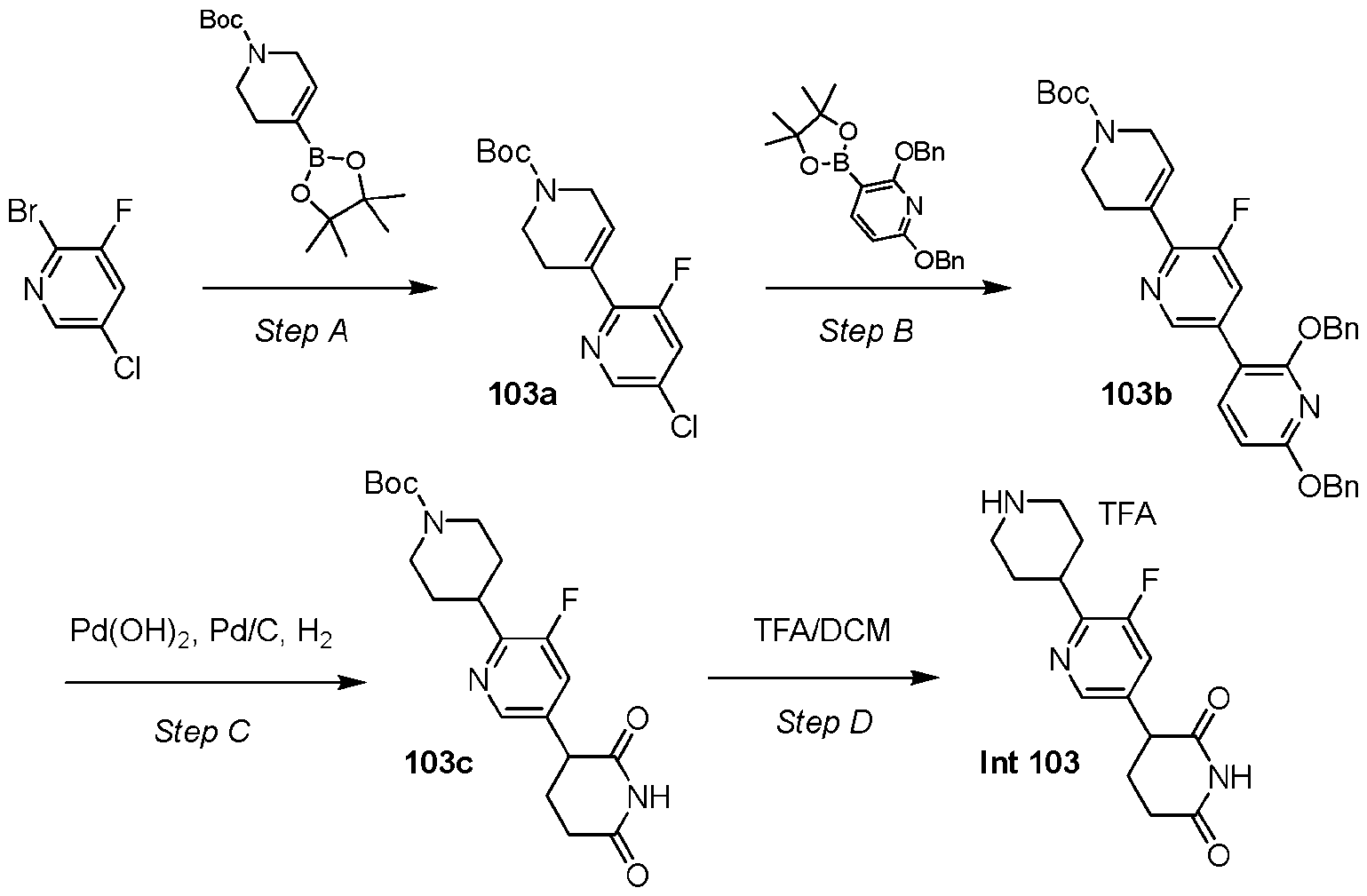 Step A – Synthesis of tert-butyl 4-(5-chloro-3-fluoro-2-pyridyl)-3,6-dihydro-2H-pyridine-1- carboxylate (103a) A mixture of 2-bromo-5-chloro-3-fluoro-pyridine (15.0 g, 71.3 mmol, 1.0 equiv.), tert- butyl 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-3,6-dihydro-2H-pyridine-1-carboxylate (22.0 g, 71.3 mmol, 1.0 equiv.), Pd(dppf)Cl2 (5.2 g, 7.1 mmol, 0.1 equiv.) and sodium carbonate (22.7 g, 213.9 mmol, 3.0 equiv.) in dioxane (500 mL) and water (125 mL) was degassed and purged threefold with nitrogen. The mixture was stirred at 100°C for 12 h under a nitrogen atmosphere. The reaction mixture was cooled to ambient temperature, diluted with water (500 mL) and extracted with ethyl acetate (500 mL x 2). The combined organic layers were washed with brine (400 mL x 2), dried over anhydrous sodium sulfate , filtered and the filtrate was concentrated under reduced pressure to afford 103a, which was used without further purification. LCMS [M+1] = 257.1, 259.0 Step B – Synthesis of tert-butyl 4-[5-(2,6-dibenzyloxy-3-pyridyl)-3-fluoro-2-pyridyl]-3,6-dihydro- 2H-pyridine-1-carboxylate (103b) A mixture of 103a (16.0 g, 51.2 mmol, 1.0 equiv.), 2,6-dibenzyloxy-3-(4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine (21.4 g, 51.2 mmol, 1.0 equiv.), Pd(dppf)Cl2 (3.7 g, 5.1 mmol), sodium carbonate (16.3 g, 153.5 mmol, 3.0 equiv.) in dioxane (300 mL) and water (75
mL) was degassed and purged threefold with nitrogen. The mixture was stirred at 100°C for 5 h under a nitrogen atmosphere, cooled to ambient temperature, diluted with water (500 mL) and extracted with ethyl acetate (500 mL x 2). The combined organic layers were washed with brine (300 mL x 2), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum. The residue was purified by column chromatography (silica gel, ethyl acetate : petroleum ether =0/1 to 1/1) to afford 103b. LCMS [M+1] = 568.3 Step C – Synthesis of tert-butyl 4-[5-(2,6-dioxo-3-piperidyl)-3-fluoro-2-pyridyl]piperidine-1- carboxylate (103c) A mixture of 103b (34.0 g, 59.9 mmol, 1.0 equiv.), Pd/C (12.8 g, 10%), and Pd(OH)2/C (12.7 g, 20%) in MeOH (20 mL) was degassed and purged threefold with nitrogen. The mixture was stirred at 50°C for 12 h under hydrogen (20 psi). cooled to ambient temperature and filtered through diatomaceous earth. The filtrate was concentrated under vacuum to afford 103c, which was used without further purification. LCMS [M+1] = 336.1 Step D – Synthesis of 3-[5-fluoro-6-(4-piperidyl)-3-pyridyl]piperidine-2,6-dione (Int 103) To a solution of 103c (15 g, 19.2 mmol, 1.0 equiv.) in dichloromethane (150 mL) was added TFA (50 mL), and the mixture was stirred at 25°C for 2 h. The mixture was concentrated under vacuum to afford Int 103 as the trifluoroacetate salt, which was used without further purification. LCMS [M+1] = 499.2. Example 104: Synthesis of Intermediate 104
Step A – Synthesis of tert-butyl 4-(5-chloro-3-fluoro-2-pyridyl)-3,6-dihydro-2H-pyridine-1- carboxylate (103a) A mixture of 2-bromo-5-chloro-3-fluoro-pyridine (15.0 g, 71.3 mmol, 1.0 equiv.), tert- butyl 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-3,6-dihydro-2H-pyridine-1-carboxylate (22.0 g, 71.3 mmol, 1.0 equiv.), Pd(dppf)Cl2 (5.2 g, 7.1 mmol, 0.1 equiv.) and sodium carbonate (22.7 g, 213.9 mmol, 3.0 equiv.) in dioxane (500 mL) and water (125 mL) was degassed and purged threefold with nitrogen. The mixture was stirred at 100°C for 12 h under a nitrogen atmosphere. The reaction mixture was cooled to ambient temperature, diluted with water (500 mL) and extracted with ethyl acetate (500 mL x 2). The combined organic layers were washed with brine (400 mL x 2), dried over anhydrous sodium sulfate , filtered and the filtrate was concentrated under reduced pressure to afford 103a, which was used without further purification. LCMS [M+1] = 257.1, 259.0 Step B – Synthesis of tert-butyl 4-[5-(2,6-dibenzyloxy-3-pyridyl)-3-fluoro-2-pyridyl]-3,6-dihydro- 2H-pyridine-1-carboxylate (103b) A mixture of 103a (16.0 g, 51.2 mmol, 1.0 equiv.), 2,6-dibenzyloxy-3-(4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine (21.4 g, 51.2 mmol, 1.0 equiv.), Pd(dppf)Cl2 (3.7 g, 5.1 mmol), sodium carbonate (16.3 g, 153.5 mmol, 3.0 equiv.) in dioxane (300 mL) and water (75
mL) was degassed and purged threefold with nitrogen. The mixture was stirred at 100°C for 5 h under a nitrogen atmosphere, cooled to ambient temperature, diluted with water (500 mL) and extracted with ethyl acetate (500 mL x 2). The combined organic layers were washed with brine (300 mL x 2), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum. The residue was purified by column chromatography (silica gel, ethyl acetate : petroleum ether =0/1 to 1/1) to afford 103b. LCMS [M+1] = 568.3 Step C – Synthesis of tert-butyl 4-[5-(2,6-dioxo-3-piperidyl)-3-fluoro-2-pyridyl]piperidine-1- carboxylate (103c) A mixture of 103b (34.0 g, 59.9 mmol, 1.0 equiv.), Pd/C (12.8 g, 10%), and Pd(OH)2/C (12.7 g, 20%) in MeOH (20 mL) was degassed and purged threefold with nitrogen. The mixture was stirred at 50°C for 12 h under hydrogen (20 psi). cooled to ambient temperature and filtered through diatomaceous earth. The filtrate was concentrated under vacuum to afford 103c, which was used without further purification. LCMS [M+1] = 336.1 Step D – Synthesis of 3-[5-fluoro-6-(4-piperidyl)-3-pyridyl]piperidine-2,6-dione (Int 103) To a solution of 103c (15 g, 19.2 mmol, 1.0 equiv.) in dichloromethane (150 mL) was added TFA (50 mL), and the mixture was stirred at 25°C for 2 h. The mixture was concentrated under vacuum to afford Int 103 as the trifluoroacetate salt, which was used without further purification. LCMS [M+1] = 499.2. Example 104: Synthesis of Intermediate 104
Step A – Synthesis of tert-butyl 4-(4-fluoro-2-pyridyl)piperazine-1-carboxylate (104a) A solution of 2-chloro-4-fluoro-pyridine (0.5 g, 3.80 mmol, 1.0 equiv.) and tert-butyl piperazine-1-carboxylate (849.6 mg, 4.56 mmol, 1.2 equiv.) in toluene (5 mL) was treated with cesium carbonate (1.86 g, 5.70 mmol, 1.5 equiv.) and BINAP (213.0 mg, 342 μmol, 0.09 equiv.). The mixture was degassed and purged threefold with nitrogen. Pd2(dba)3 (104.4 mg, 114.0 μmol, 0.03 equiv.) was added, the mixture was degassed and purged threefold with nitrogen and then heated at 110°C for 12 h. The mixture was cooled to ambient temperature and concentrated under vacuum. The residue was dissolved in ethyl acetate (15 mL) and water (5 mL). The organic phase was washed with brine (5 mL) and dried with anhydrous sodium sulfate and concentrated under vacuum. The residue was purified by column chromatography (silica gel, petroleum ether:ethyl acetate = 100:1 to 90:10) to afford 104a. LCMS [M+H] = 282.1; Step B – Synthesis of tert-butyl 4-(4-fluoro-5-iodo-2-pyridyl)piperazine-1-carboxylate (104b) To a solution of 104a (850 mg, 3.02 mmol, 1 equiv.) in DMF (15 mL) was added N- iodosuccinimide (1.36 g, 6.04 mmol, 2 equiv.) at 25°C. The mixture was stirred at 25°C for 12 h, quenched with water (5 mL) and diluted with ethyl acetate (25 mL). The organic phase was separated, dried with anhydrous sodium sulfate and concentrated under vacuum. The residue was
purified by column chromatography (silica gel, petroleum ether : ethyl acetate = 100:1 to 85:15) to afford 104b. LCMS [M-55] = 351.9; Step C – Synthesis of tert-butyl 4-[5-(2,6-dibenzyloxy-3-pyridyl)-4-fluoro-2-pyridyl]piperazine-1- carboxylate (104c) A mixture of 104b (0.4 g, 982.3 μmol, 1 equiv.) and 2,6-dibenzyloxy-3-(4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine (491.9 mg, 1.18 mmol, 1.2 equiv.) in dioxane (5 mL) and water (1 mL) was treated with potassium carbonate (407.3 mg, 2.95 mmol, 3 equiv.). The mixture was sparged with nitrogen for 5 min and treated with Pd(dppf)Cl2.CH2Cl2 (80.2 mg, 98.2 μmol, 0.1 equiv.). The reaction mixture was heated at 100 °C for 16 h under nitrogen, cooled to ambient temperature, filtered and the filtrate was concentrated under vacuum. The residue was partitioned between water (5 mL) and ethyl acetate (20 mL). The organic layer was washed with brine (5 mL), dried with anhydrous sodium sulfate and concentrated under vacuum. The residue was purified by column chromatography (silica gel, petroleum ether : ethyl acetate = 100 : 0 to 90 : 10) to afford 104c. LCMS [M+H] = 571.2; Step D – Synthesis of tert-butyl 4-[5-(2,6-dioxo-3-piperidyl)-4-fluoro-2-pyridyl]piperazine-1- carboxylate (104d) A solution of 104c (500 mg, 876.2 μmol, 1 equiv.) in THF (15 mL) and ethyl acetate (15 mL) under a nitrogen atmosphere was treated with Pd/C (100 mg, 10 wt %) and Pd(OH)2/C (100 mg, 20 wt%). The suspension was degassed and purged threefold with hydrogen. The mixture was stirred under hydrogen (15 psi.) at 25°C for 12 h. The mixture was filtered and the filtrate was concentrated under vacuum to afford 104d, which was used without further purification. LCMS [M+H] = 393.1; Step E – Synthesis of 3-(4-fluoro-6-piperazin-1-yl-3-pyridyl)piperidine-2,6-dione (Int 104) A solution of 104d (200 mg, 509.7 μmol, 1 equiv.) in HCl/ ethyl acetate (1M, 4 mL) was stirred at 30°C for 3 h. The mixture was concentrated under vacuum to afford Int 104, as the hydrochloride salt that was used without further purification. LCMS [M+H] = 293.2;
Example 105 Synthesis of Intermediate 105
 Step A – Synthesis of 1-(4-bromo-3-fluorobenzyl)pyrimidine-2,4(1H,3H)-dione (105a) A solution of 1-bromo-4-(bromomethyl)-2-fluorobenzene (836.7 mg, 7.4 mmol, 1.0 equiv.) in DMSO (20.0 mL) was treated with pyrimidine-2,4(1H,3H)-dione (2.0 g, 7.5 mmol, 1.0 equiv.) and potassium carbonate (3.1 g, 22.4 mmol, 3.0 equiv.) under nitrogen. The mixture was stirred at 80°C for 3 h, cooled to ambient temperature and filtered to remove the insoluble matter. The filtrate was treated with water (30 ml) and the solids were collected by vacuum filtration to afford 105a. LCMS [M+1] = 298.8, 300.8. 1HNMR (400 MHz, DMSO-d6) δ 11.33 (s, 1H), 7.76 (d, J = 7.6 Hz, 1H), 7.70 (t, J = 7.6 Hz, 1H), 7.33 (dd, J = 2.0, 9.6 Hz, 1H), 7.09 (dd, J = 1.6, 8.0 Hz, 1H), 5.60 (d, J = 7.6 Hz, 1H), 4.85 (s, 2H). Step B – Synthesis of tert-butyl4-(4-((2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)methyl)-2- fluorophenyl)piperazine-1-carboxylate (105b) A solution of 105a (450.0 mg, 1.5 mmol, 1.0 equiv.) in DMSO (3.0 mL) was treated with tert-butyl piperazine-1-carboxylate (1.30 g, 6.00 mmol, 4.0 equiv.), cesium carbonate (980.4 mg, 3.0 mmol, 2.0 equiv.) and [1,3-bis[2,6-bis(1-ethylpropyl)phenyl]-4,5- dichloroimidazol-2-ylidene]-dichloro-(2-methylpyridin-1-ium-1-yl)palladium (63.2 mg, 75 μmol, 0.05 equiv.) under a nitrogen atmosphere. The mixture was stirred at 100°C for 12 h
under nitrogen, cooled to ambient temperature and poured into water (60 mL). The aqueous phase was extracted with ethyl acetate (40 mL x 3). The combined organic phase was washed with brine (50 mL x 2), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum. The residue was purified by column chromatography (silica gel, petroleum ether: ethyl acetate=3:1 to 0:1) to afford 105b. LCMS [M-55] = 349.0, 405.1 Step C – Synthesis of 1-(3-fluoro-4-(piperazin-1-yl)benzyl)pyrimidine-2,4(1H,3H)-dione (105c) A solution of 105b (460.0 mg, 1.1 mmol, 1.0 equiv.) in HCl / dioxane (4M, 5.0 mL) was stirred at 20°C for 1 h. The reaction mixture was concentrated under reduced pressure to afford 105c as the hydrochloride salt, which was used without further purification. LCMS [M+1] = 304.9 Step D – Synthesis of 1-(3-fluoro-4-(piperazin-1-yl)benzyl)dihydropyrimidine-2,4(1H,3H)-dione (Int 105) A solution of 105c (50.0 mg, 146.7 μmol, 1.0 equiv., HCl) in water (0.3 mL) and THF (0.3 mL) under nitrogen was treated with Pd/C (34.9 mg, 10 wt%). The mixture was degassed under vacuum and purged threefold with hydrogen. The mixture was stirred under hydrogen (50 psi) at 20°C for 12 h, cooled to ambient temperature and filtered through a Celite filter pad. The filter pad was rinsed with THF (10 ml x 3) and the combined filtrate was concentrated under vacuum to afford Int 105 as the hydrochloride salt, which was used without further purification. LCMS [M+1] = 307.0 Example 106: Synthesis of Compound 106
Step A – Synthesis of 1-(4-bromo-3-fluorobenzyl)pyrimidine-2,4(1H,3H)-dione (105a) A solution of 1-bromo-4-(bromomethyl)-2-fluorobenzene (836.7 mg, 7.4 mmol, 1.0 equiv.) in DMSO (20.0 mL) was treated with pyrimidine-2,4(1H,3H)-dione (2.0 g, 7.5 mmol, 1.0 equiv.) and potassium carbonate (3.1 g, 22.4 mmol, 3.0 equiv.) under nitrogen. The mixture was stirred at 80°C for 3 h, cooled to ambient temperature and filtered to remove the insoluble matter. The filtrate was treated with water (30 ml) and the solids were collected by vacuum filtration to afford 105a. LCMS [M+1] = 298.8, 300.8. 1HNMR (400 MHz, DMSO-d6) δ 11.33 (s, 1H), 7.76 (d, J = 7.6 Hz, 1H), 7.70 (t, J = 7.6 Hz, 1H), 7.33 (dd, J = 2.0, 9.6 Hz, 1H), 7.09 (dd, J = 1.6, 8.0 Hz, 1H), 5.60 (d, J = 7.6 Hz, 1H), 4.85 (s, 2H). Step B – Synthesis of tert-butyl4-(4-((2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)methyl)-2- fluorophenyl)piperazine-1-carboxylate (105b) A solution of 105a (450.0 mg, 1.5 mmol, 1.0 equiv.) in DMSO (3.0 mL) was treated with tert-butyl piperazine-1-carboxylate (1.30 g, 6.00 mmol, 4.0 equiv.), cesium carbonate (980.4 mg, 3.0 mmol, 2.0 equiv.) and [1,3-bis[2,6-bis(1-ethylpropyl)phenyl]-4,5- dichloroimidazol-2-ylidene]-dichloro-(2-methylpyridin-1-ium-1-yl)palladium (63.2 mg, 75 μmol, 0.05 equiv.) under a nitrogen atmosphere. The mixture was stirred at 100°C for 12 h
under nitrogen, cooled to ambient temperature and poured into water (60 mL). The aqueous phase was extracted with ethyl acetate (40 mL x 3). The combined organic phase was washed with brine (50 mL x 2), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum. The residue was purified by column chromatography (silica gel, petroleum ether: ethyl acetate=3:1 to 0:1) to afford 105b. LCMS [M-55] = 349.0, 405.1 Step C – Synthesis of 1-(3-fluoro-4-(piperazin-1-yl)benzyl)pyrimidine-2,4(1H,3H)-dione (105c) A solution of 105b (460.0 mg, 1.1 mmol, 1.0 equiv.) in HCl / dioxane (4M, 5.0 mL) was stirred at 20°C for 1 h. The reaction mixture was concentrated under reduced pressure to afford 105c as the hydrochloride salt, which was used without further purification. LCMS [M+1] = 304.9 Step D – Synthesis of 1-(3-fluoro-4-(piperazin-1-yl)benzyl)dihydropyrimidine-2,4(1H,3H)-dione (Int 105) A solution of 105c (50.0 mg, 146.7 μmol, 1.0 equiv., HCl) in water (0.3 mL) and THF (0.3 mL) under nitrogen was treated with Pd/C (34.9 mg, 10 wt%). The mixture was degassed under vacuum and purged threefold with hydrogen. The mixture was stirred under hydrogen (50 psi) at 20°C for 12 h, cooled to ambient temperature and filtered through a Celite filter pad. The filter pad was rinsed with THF (10 ml x 3) and the combined filtrate was concentrated under vacuum to afford Int 105 as the hydrochloride salt, which was used without further purification. LCMS [M+1] = 307.0 Example 106: Synthesis of Compound 106
Synthesis of 1-[4-[4-[[4-[5-[(2,6-dioxo-3-piperidyl)amino]-3-fluoro-2-pyridyl]-1- piperidyl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4- triazol-5-yl]methyl]pyrazole-4-carboxamide (Compound 106) A solution of Int 98 (14.31 g, 34.04 mmol, 1.3 equiv) and Int 55 (13.0 g, 26.18 mmol, 1.0 equiv.) in dichloromethane (150 mL) was treated with triethylamine (13.25 g, 130.9 mmol, 18.22 mL, 5.0 equiv.) and NaBH(OAc)3 (27.75 g, 130.9 mmol, 5.0 equiv.). The mixture was stirred at 25°C for 12 h. The reaction mixture was quenched with water (300 mL) and extracted with dichloromethane (100 mL x 3). The combined organic layers were concentrated under reduced pressure to give a residue and then the blue solid was concentrated under reduced pressure to give a residue. The residue was purified by preparative HPLC (Phenomenex Luna C18, 250mm x 100mm, 15µm; [water (0.2%FA)-ACN]) to afford Compound 106. LCMS [M+1] = 787.3. 1H NMR (400 MHz, DMSO-d6) δ = 10.83 (s, 1H), 8.90 (br t, J = 5.6 Hz, 1H), 8.53 (s, 1H), 8.18 (s, 1H), 8.17 (s, 1H), 7.87 (br s, 1H), 7.73 - 7.63 (m, 3H), 7.65 (dd, J = 1.6, 8.4 Hz, 1H), 7.46 (dd, J = 3.6, 8.4 Hz, 3H), 6.88 (dd, J = 2.4, 13.2 Hz, 1H), 6.33 (d, J = 7.6 Hz, 1H), 4.56 (d, J = 5.6 Hz, 2H), 4.40 - 4.34 (m, 1H), 3.63 (s, 2H), 2.99 (br d, J = 10.8 Hz, 2H), 2.84 - 2.68 (m, 2H), 2.60 - 2.56 (m, 1H), 2.30 (s, 3H), 2.20 - 2.07 (m, 3H), 1.96 - 1.81 (m, 3H), 1.67 (br d, J = 11.6 Hz, 2H), 1.54 (s, 6H) Example 107: Synthesis of Compound 107-1 and Compound 107-2
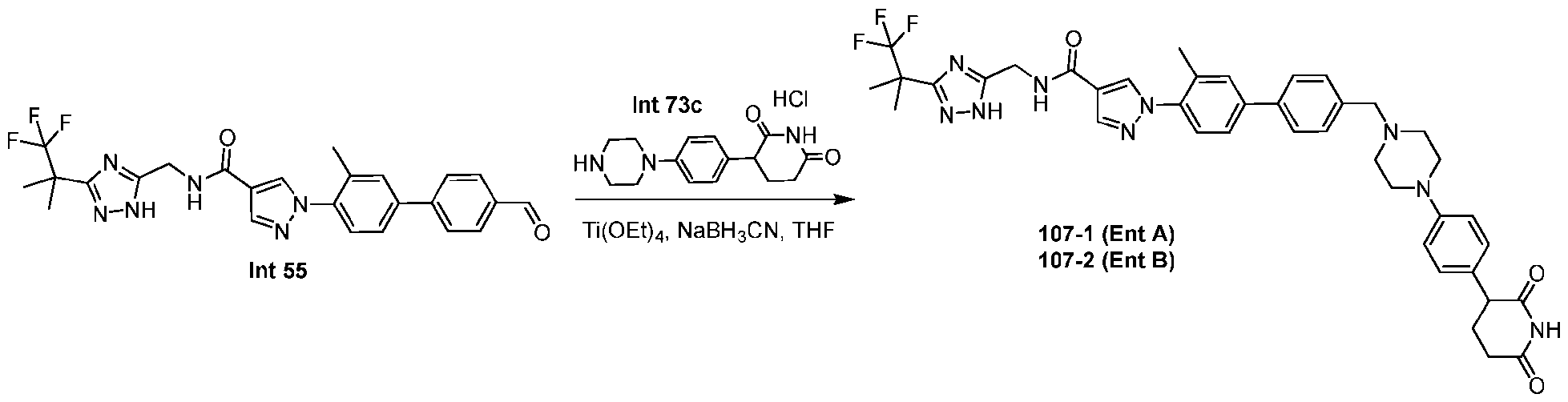 methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole- 4-carboxamide (Compound 107-1 and Compound 107-2) A solution of Int 73c (1.50 g, 4.80 mmol, 1.0 equiv, HCl) and Int 55 (2.40 g, 4.80 mmol, 1.0 equiv.) in THF (15.0 mL) was treated with Ti(OEt)4 (5.40 g, 24.3 mmol, 5.1 mL, 5.0 equiv.) and the mixture was stirred at 50°C for 0.5 h. The mixture was treated with NaBH3CN (608.4
mg, 9.6 mmol, 2.0 equiv.) and the mixture was stirred at 50°C for 2 h. The mixture was cooled to 25°C, poured into water (30 mL), filtered and the filter pad was washed with ethyl acetate (40 mL x 3). The filtrate was concentrated under reduced pressure. The residue was purified by preparative HPLC (Phenomenex Luna C18, 250x70mm,10 µm, [water(FA)-ACN]) to afford Compound 107 as the racemate. The enantiomers were resolved by SFC (REGIS (R,R)- WHELK-O1, 250 mm x 25 mm, 10 µm; [CO2-iPrOH/ACN]) to afford Compound 107-1 as the early eluting isomer and Compound 107-2 as the late eluting isomer. LCMS [M+1] = 754.4. 1H NMR (400 MHz, DMSO-d6) δ 10.78 (s, 2H), 8.95 - 8.85 (m, 1H), 8.57 (s, 1H), 8.19 (s, 1H), 7.86 (d, J = 8.4 Hz, 2H), 7.79 (s, 1H), 7.74 (d, J = 8.4 Hz, 2H), 7.72 - 7.68 (m, 1H), 7.51 (d, J = 8.4 Hz, 1H), 7.11 (d, J = 8.8 Hz, 2H), 6.95 (d, J = 8.8 Hz, 2H), 4.56 (d, J = 5.6 Hz, 2H), 4.45 (br d, J = 4.4 Hz, 2H), 3.87 - 3.72 (m, 4H), 3.42 (br d, J = 11.2 Hz, 2H), 3.26 - 3.07 (m, 4H), 2.72 - 2.59 (m, 1H), 2.32 (s, 3H), 2.21 - 2.07 (m, 1H), 2.05 - 1.94 (m, 1H), 1.54 (s, 6H). Example 108: Synthesis of Compound 108 Compound
methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole- 4-carboxamide (Compound 107-1 and Compound 107-2) A solution of Int 73c (1.50 g, 4.80 mmol, 1.0 equiv, HCl) and Int 55 (2.40 g, 4.80 mmol, 1.0 equiv.) in THF (15.0 mL) was treated with Ti(OEt)4 (5.40 g, 24.3 mmol, 5.1 mL, 5.0 equiv.) and the mixture was stirred at 50°C for 0.5 h. The mixture was treated with NaBH3CN (608.4
mg, 9.6 mmol, 2.0 equiv.) and the mixture was stirred at 50°C for 2 h. The mixture was cooled to 25°C, poured into water (30 mL), filtered and the filter pad was washed with ethyl acetate (40 mL x 3). The filtrate was concentrated under reduced pressure. The residue was purified by preparative HPLC (Phenomenex Luna C18, 250x70mm,10 µm, [water(FA)-ACN]) to afford Compound 107 as the racemate. The enantiomers were resolved by SFC (REGIS (R,R)- WHELK-O1, 250 mm x 25 mm, 10 µm; [CO2-iPrOH/ACN]) to afford Compound 107-1 as the early eluting isomer and Compound 107-2 as the late eluting isomer. LCMS [M+1] = 754.4. 1H NMR (400 MHz, DMSO-d6) δ 10.78 (s, 2H), 8.95 - 8.85 (m, 1H), 8.57 (s, 1H), 8.19 (s, 1H), 7.86 (d, J = 8.4 Hz, 2H), 7.79 (s, 1H), 7.74 (d, J = 8.4 Hz, 2H), 7.72 - 7.68 (m, 1H), 7.51 (d, J = 8.4 Hz, 1H), 7.11 (d, J = 8.8 Hz, 2H), 6.95 (d, J = 8.8 Hz, 2H), 4.56 (d, J = 5.6 Hz, 2H), 4.45 (br d, J = 4.4 Hz, 2H), 3.87 - 3.72 (m, 4H), 3.42 (br d, J = 11.2 Hz, 2H), 3.26 - 3.07 (m, 4H), 2.72 - 2.59 (m, 1H), 2.32 (s, 3H), 2.21 - 2.07 (m, 1H), 2.05 - 1.94 (m, 1H), 1.54 (s, 6H). Example 108: Synthesis of Compound 108 Compound
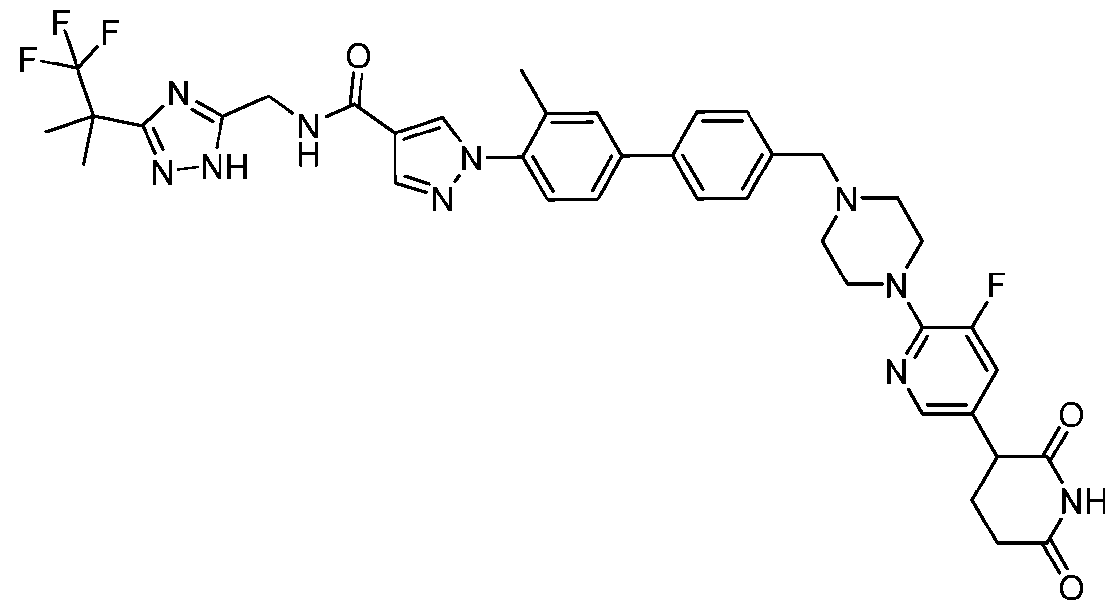 Example 107 by substituting Int 100 to afford 1-[4-[4-[[4-[5-(2,6-dioxo-3-piperidyl)-3-fluoro-2-pyridyl]piperazin- 1-yl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4- triazol-5-yl]methyl]pyrazole-4-carboxamide. LCMS [M+1] = 773.2. 1HNMR (400 MHz, DMSO- d6) δ 13.91 (br s, 1H), 10.86 (s, 1H), 8.88 (br s, 1H), 8.53 (s, 1H), 8.17 (s, 1H), 8.14 (s, 1H), 7.88 (s, 1H), 7.74 - 7.69 (m, 3H), 7.65 (dd, J = 1.8, 8.2 Hz, 1H), 7.50 - 7.42 (m, 4H), 4.56 (br d, J = 5.0 Hz, 2H), 3.85 (dd, J = 4.8, 12.4 Hz, 1H), 3.58 (s, 2H), 3.40 (br s, 4H), 2.75 - 2.63 (m, 1H), 2.55 (br s, 4H), 2.32 (br dd, J = 1.8, 3.6 Hz, 1H), 2.30 (s, 3H), 2.24 (br dd, J = 4.2, 12.8 Hz, 1H), 2.04 - 1.92 (m, 1H), 1.54 (s, 6H).
Example 109: Synthesis of Compound 109 Example 107 by
Example 107 by substituting Int 100 to afford 1-[4-[4-[[4-[5-(2,6-dioxo-3-piperidyl)-3-fluoro-2-pyridyl]piperazin- 1-yl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4- triazol-5-yl]methyl]pyrazole-4-carboxamide. LCMS [M+1] = 773.2. 1HNMR (400 MHz, DMSO- d6) δ 13.91 (br s, 1H), 10.86 (s, 1H), 8.88 (br s, 1H), 8.53 (s, 1H), 8.17 (s, 1H), 8.14 (s, 1H), 7.88 (s, 1H), 7.74 - 7.69 (m, 3H), 7.65 (dd, J = 1.8, 8.2 Hz, 1H), 7.50 - 7.42 (m, 4H), 4.56 (br d, J = 5.0 Hz, 2H), 3.85 (dd, J = 4.8, 12.4 Hz, 1H), 3.58 (s, 2H), 3.40 (br s, 4H), 2.75 - 2.63 (m, 1H), 2.55 (br s, 4H), 2.32 (br dd, J = 1.8, 3.6 Hz, 1H), 2.30 (s, 3H), 2.24 (br dd, J = 4.2, 12.8 Hz, 1H), 2.04 - 1.92 (m, 1H), 1.54 (s, 6H).
Example 109: Synthesis of Compound 109 Example 107 by
 substituting Int 101 to afford 1-(4'-((4-(4-(2,6-dioxopiperidin-3-yl)-2-fluorophenyl)piperazin-1- yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide. LCMS [M+1] = 772.3. 1H NMR (400 MHz, DMSO-d6) δ 13.90 (br s, 1H), 10.81 (s, 1H), 8.95 - 8.82 (m, 1H), 8.53 (s, 1H), 8.20 - 8.16 (m, 1H), 7.75 - 7.62 (m, 4H), 7.46 (m, 3H), 7.06 - 6.93 (m, 3H), 4.55 (d, J = 5.6 Hz, 2H), 3.80 (m, 1H), 3.59 (s, 2H), 3.03 (s, 4H), 2.71 - 2.53 (m, 5H), 2.47 (s, 1H), 2.30 (s, 3H), 2.19 (m, 1H), 1.99 (m, 1H), 1.54 (s, 6H). Example 110: Synthesis of Compound 110
substituting Int 101 to afford 1-(4'-((4-(4-(2,6-dioxopiperidin-3-yl)-2-fluorophenyl)piperazin-1- yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide. LCMS [M+1] = 772.3. 1H NMR (400 MHz, DMSO-d6) δ 13.90 (br s, 1H), 10.81 (s, 1H), 8.95 - 8.82 (m, 1H), 8.53 (s, 1H), 8.20 - 8.16 (m, 1H), 7.75 - 7.62 (m, 4H), 7.46 (m, 3H), 7.06 - 6.93 (m, 3H), 4.55 (d, J = 5.6 Hz, 2H), 3.80 (m, 1H), 3.59 (s, 2H), 3.03 (s, 4H), 2.71 - 2.53 (m, 5H), 2.47 (s, 1H), 2.30 (s, 3H), 2.19 (m, 1H), 1.99 (m, 1H), 1.54 (s, 6H). Example 110: Synthesis of Compound 110
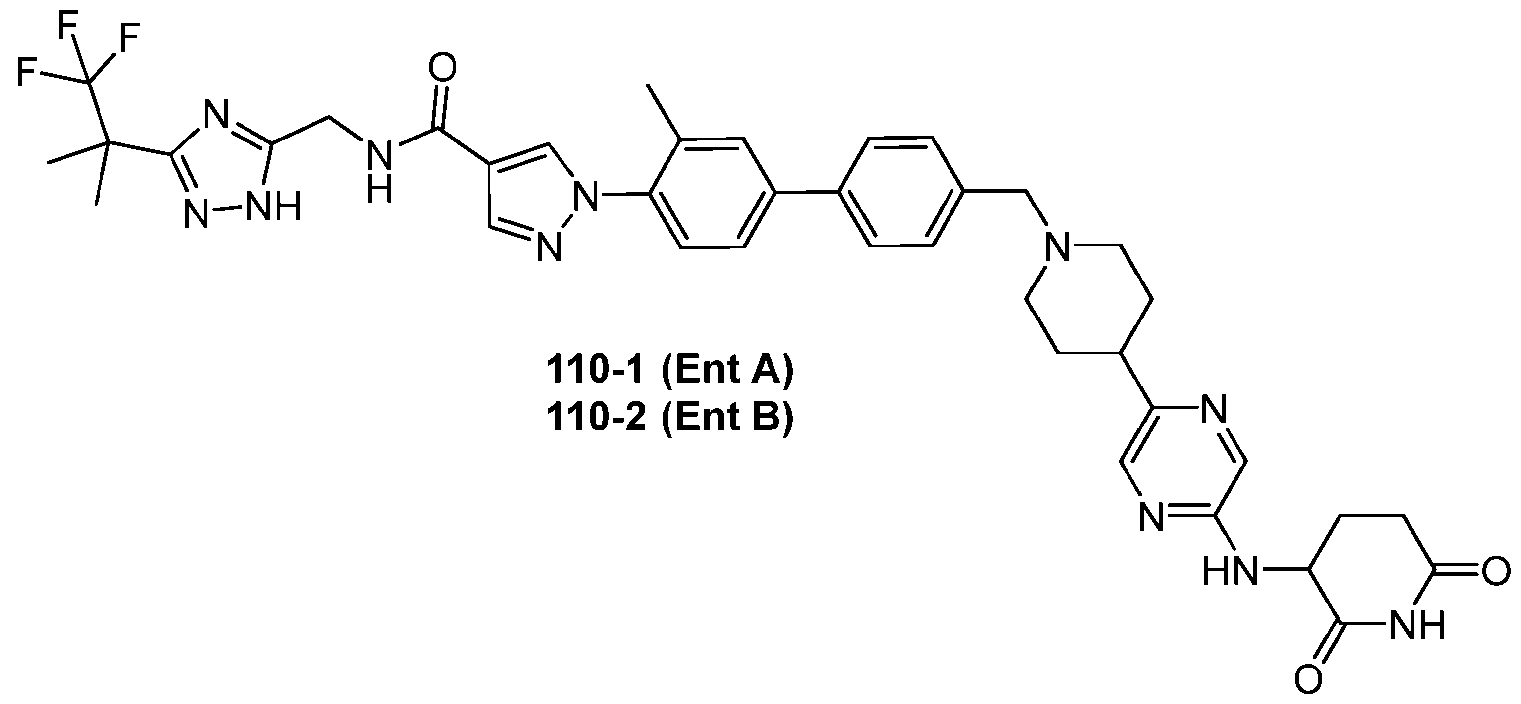 Example 106 by substituting Int 102 to afford 1-(4'-((4-(5-((2,6-dioxopiperidin-3-yl)amino)pyrazin-2-
yl)piperidin-1-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2- yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide. The enantiomers were resolved by SFC (ChiralPak IH, 250mm x 30mm, 10µm; [CO2-ethanol]) to afford Compound 110-1 as the early eluting isomer and Compound 110-2 as the late eluting isomer. LCMS [M+1] = 770.3. 1H NMR (400 MHz, DMSO-d6) δ = 10.80 (s, 1H), 10.31 (br s, 1H), 8.90 (t, J = 5.2 Hz, 1H), 8.56 (s, 1H), 8.19 (s, 1H), 8.00 (d, J = 1.2 Hz, 1H), 7.90 - 7.82 (m, 3H), 7.78 (s, 1H), 7.74 - 7.64 (m, 3H), 7.51 (d, J = 8.4 Hz, 1H), 7.40 - 7.24 (m, 1H), 4.76 - 4.64 (m, 1H), 4.56 (d, J = 5.6 Hz, 2H), 4.38 (d, J = 4.4 Hz, 1H), 3.51 - 3.40 (m, 3H), 3.17 - 3.00 (m, 2H), 2.90 - 2.70 (m, 2H), 2.59 - 2.55 (m, 1H), 2.32 (s, 3H), 2.12 - 1.94 (m, 6H), 1.54 (s, 6H) Example 111: Synthesis of Compound 111 Compound
Example 106 by substituting Int 102 to afford 1-(4'-((4-(5-((2,6-dioxopiperidin-3-yl)amino)pyrazin-2-
yl)piperidin-1-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2- yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide. The enantiomers were resolved by SFC (ChiralPak IH, 250mm x 30mm, 10µm; [CO2-ethanol]) to afford Compound 110-1 as the early eluting isomer and Compound 110-2 as the late eluting isomer. LCMS [M+1] = 770.3. 1H NMR (400 MHz, DMSO-d6) δ = 10.80 (s, 1H), 10.31 (br s, 1H), 8.90 (t, J = 5.2 Hz, 1H), 8.56 (s, 1H), 8.19 (s, 1H), 8.00 (d, J = 1.2 Hz, 1H), 7.90 - 7.82 (m, 3H), 7.78 (s, 1H), 7.74 - 7.64 (m, 3H), 7.51 (d, J = 8.4 Hz, 1H), 7.40 - 7.24 (m, 1H), 4.76 - 4.64 (m, 1H), 4.56 (d, J = 5.6 Hz, 2H), 4.38 (d, J = 4.4 Hz, 1H), 3.51 - 3.40 (m, 3H), 3.17 - 3.00 (m, 2H), 2.90 - 2.70 (m, 2H), 2.59 - 2.55 (m, 1H), 2.32 (s, 3H), 2.12 - 1.94 (m, 6H), 1.54 (s, 6H) Example 111: Synthesis of Compound 111 Compound
 by substituting Int 99 to afford 1-[4-[4-[[4-[5-[(2,6-dioxo-3-piperidyl)amino]-3-fluoro-2-pyridyl]piperazin-1- yl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol- 5-yl]methyl]pyrazole-4-carboxamide. LCMS [M+1] = 788.5. 1H NMR (400 MHz, DMSO-d6) δ 11.40 (br s, 1H), 10.82 (s, 1H), 8.98 (br t, J = 5.8 Hz, 1H), 8.60 (s, 1H), 8.20 (s, 1H), 7.87 - 7.82 (m, 2H), 7.81 - 7.75 (m, 3H), 7.70 (dd, J = 1.6, 8.2 Hz, 1H), 7.61 (d, J = 1.8 Hz, 1H), 7.50 (d, J = 8.2 Hz, 1H), 7.09 (dd, J = 2.2, 14.6 Hz, 1H), 4.56 (d, J = 5.8 Hz, 2H), 4.41 (br d, J = 3.8 Hz, 2H), 4.34 (dd, J = 4.8, 11.7 Hz, 1H), 3.75 - 3.57 (m, 2H), 3.43 - 3.25 (m, 4H), 3.25 - 3.13 (m, 2H), 2.80 - 2.63 (m, 1H), 2.63 - 2.54 (m, 1H), 2.32 (s, 3H), 2.18 - 2.03 (m, 1H), 1.98 - 1.81 (m, 1H), 1.59 - 1.49 (m, 6H) Example 112: Synthesis of Compound 112
by substituting Int 99 to afford 1-[4-[4-[[4-[5-[(2,6-dioxo-3-piperidyl)amino]-3-fluoro-2-pyridyl]piperazin-1- yl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol- 5-yl]methyl]pyrazole-4-carboxamide. LCMS [M+1] = 788.5. 1H NMR (400 MHz, DMSO-d6) δ 11.40 (br s, 1H), 10.82 (s, 1H), 8.98 (br t, J = 5.8 Hz, 1H), 8.60 (s, 1H), 8.20 (s, 1H), 7.87 - 7.82 (m, 2H), 7.81 - 7.75 (m, 3H), 7.70 (dd, J = 1.6, 8.2 Hz, 1H), 7.61 (d, J = 1.8 Hz, 1H), 7.50 (d, J = 8.2 Hz, 1H), 7.09 (dd, J = 2.2, 14.6 Hz, 1H), 4.56 (d, J = 5.8 Hz, 2H), 4.41 (br d, J = 3.8 Hz, 2H), 4.34 (dd, J = 4.8, 11.7 Hz, 1H), 3.75 - 3.57 (m, 2H), 3.43 - 3.25 (m, 4H), 3.25 - 3.13 (m, 2H), 2.80 - 2.63 (m, 1H), 2.63 - 2.54 (m, 1H), 2.32 (s, 3H), 2.18 - 2.03 (m, 1H), 1.98 - 1.81 (m, 1H), 1.59 - 1.49 (m, 6H) Example 112: Synthesis of Compound 112
Compound 112 6 by substituting Int 103 to afford 1-[4-[4-[[4-[5-(2,6-dioxo-3-piperidyl)-3-fluoro-2-pyridyl]-1-piperidyl]methyl]phenyl]-2- methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole- 4-carboxamide. LCMS [M+1] = 772.1. 1H NMR (400 MHz, DMSO-d6) δ 13.93 (br s, 1 H), 10.91 (s, 1 H), 9.06 - 9.07 (m, 1 H), 8.86 (d, J = 2.0 Hz, 1 H), 8.35 (dd, J = 8.8, 2.4 Hz, 1 H), 8.25 (d, J = 18.4 Hz, 3 H), 8.03 (d, J = 8.4 Hz, 1 H), 7.77 (d, J = 8.0 Hz, 2 H), 7.58 (dd, J = 11.44, 1.56 Hz, 1 H), 7.48 (d, J = 8.4 Hz, 2 H), 4.55 (d, J = 5.6 Hz, 2 H), 3.98 (dd, J = 12.4, 4.4 Hz, 1 H), 3.58 (s, 2 H), 2.97 (d, J = 10.8 Hz, 3 H), 2.64 - 2.71 (m, 1 H), 2.56 - 2.58 (m, 1 H), 2.27 - 2.34 (m, 1 H), 2.09 - 2.16 (m, 2 H), 2.01 - 2.07 (m, 1 H), 1.84 - 1.94 (m, 2 H), 1.73 (d, J = 12 Hz, 2 H), 1.54 (s, 6 H). Example 113: Synthesis of Compound 113
 Compound by substituting Int 104 to afford 1-[4-[4-[[4-[5-(2,6-dioxo-3-piperidyl)-4-fluoro-2-pyridyl]piperazin-1-yl]methyl]phenyl]- 2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methyl]pyrazole-4-carboxamide. LCMS [M+H] = 773.3. 1H NMR (400 MHz, METHANOL- d4) δ 8.36 (s, 1 H), 8.18 (s, 1 H), 7.99 (d, J = 10.4 Hz, 1 H), 7.75 - 7.65 (m, 3 H), 7.62 (br d, J =
8.0 Hz, 1 H), 7.54 - 7.41 (m, 3 H), 6.59 (br d, J = 14.0 Hz, 1 H), 4.70 (s, 2 H), 3.89 (br dd, J = 12.4, 4.8 Hz, 1 H), 3.69 (s, 2 H), 3.58 (br s, 4 H), 2.86 - 2.68 (m, 2 H), 2.65 (br s, 4 H), 2.38 - 2.19 (m, 4 H), 2.17 - 2.05 (m, 1 H), 1.61 (s, 6 H). Note: The “H” of amide in HNMR was deuterated by CD3OD. Example 114: Synthesis of Compound 114
Compound by substituting Int 104 to afford 1-[4-[4-[[4-[5-(2,6-dioxo-3-piperidyl)-4-fluoro-2-pyridyl]piperazin-1-yl]methyl]phenyl]- 2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methyl]pyrazole-4-carboxamide. LCMS [M+H] = 773.3. 1H NMR (400 MHz, METHANOL- d4) δ 8.36 (s, 1 H), 8.18 (s, 1 H), 7.99 (d, J = 10.4 Hz, 1 H), 7.75 - 7.65 (m, 3 H), 7.62 (br d, J =
8.0 Hz, 1 H), 7.54 - 7.41 (m, 3 H), 6.59 (br d, J = 14.0 Hz, 1 H), 4.70 (s, 2 H), 3.89 (br dd, J = 12.4, 4.8 Hz, 1 H), 3.69 (s, 2 H), 3.58 (br s, 4 H), 2.86 - 2.68 (m, 2 H), 2.65 (br s, 4 H), 2.38 - 2.19 (m, 4 H), 2.17 - 2.05 (m, 1 H), 1.61 (s, 6 H). Note: The “H” of amide in HNMR was deuterated by CD3OD. Example 114: Synthesis of Compound 114
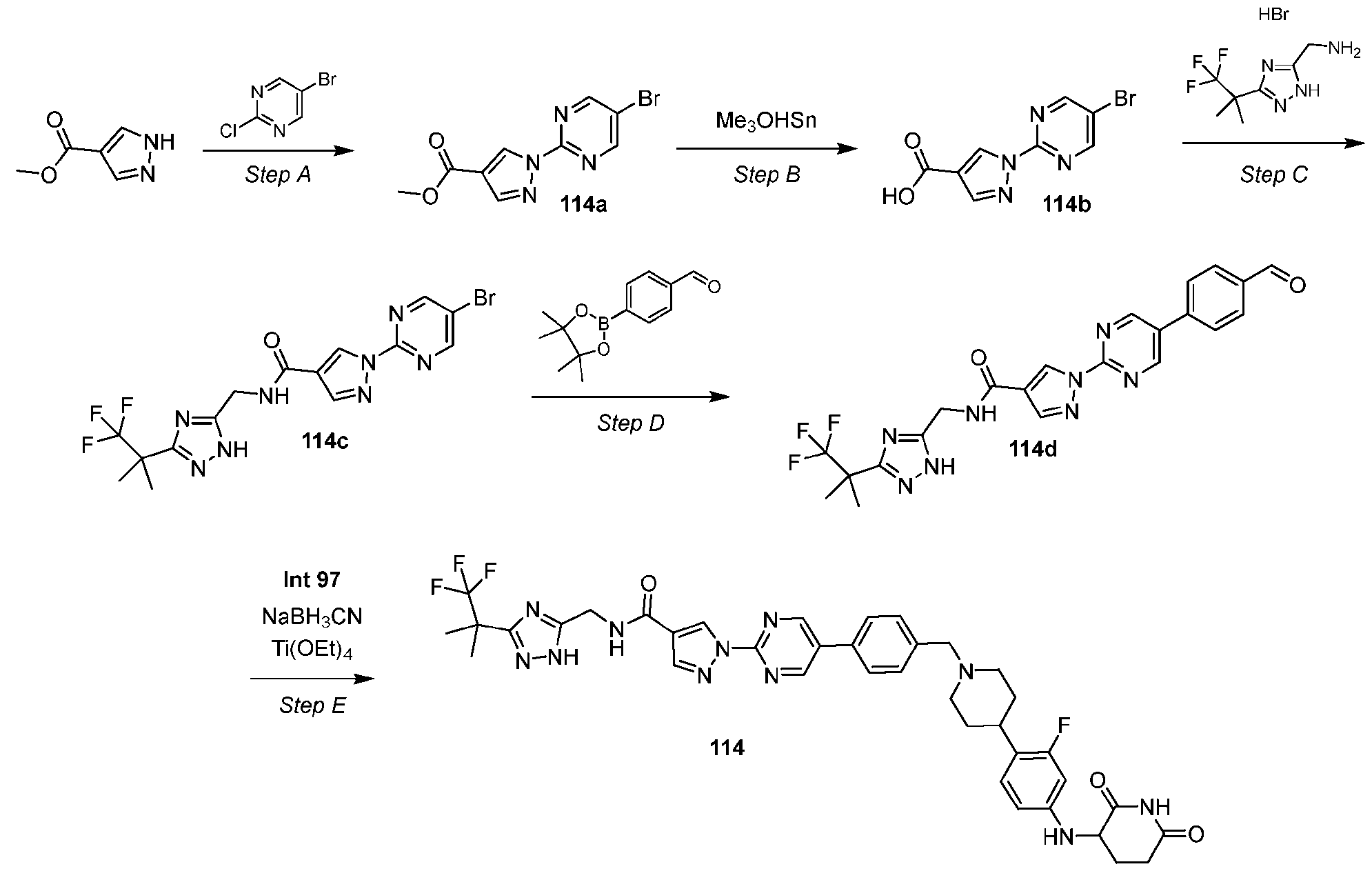 Step A – Synthesis of methyl 1-(5-bromopyrimidin-2-yl)pyrazole-4-carboxylate (114a) A solution of methyl 1H-pyrazole-4-carboxylate (2.80 g, 22.2 mmol, 1 equiv.) in DMF (50 mL) was treated with cesium carbonate (21.7 g, 66.6 mmol, 3 equiv.) and 5-bromo-2-chloro- pyrimidine (4.29 g, 22.2 mmol, 1 equiv.). The mixture was stirred at 100°C for 2 h under a nitrogen atmosphere, cooled to 20°C and diluted with saturated aqueous NH4Cl (100 mL). The mixture was extracted with ethyl acetate (40 mL x 3), washed with brine (40 mLx 3), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum. The residue
was dissolved in MeOH (10 mL) and stirred at 20°C for 30 min, filtered and the filtrate was concentrated under vacuum to afford 114a. LCMS [M+1] = 283.1; Step B – Synthesis of 1-(5-bromopyrimidin-2-yl)pyrazole-4-carboxylic acid (114b) A solution of 114a (800 mg, 2.83 mmol, 1 equiv.) in dichloromethane (16 mL) was added hydroxy(trimethyl)stannane (1.53 g, 8.48 mmol, 3 equiv.), the mixture was stirred at 80°C for 32 h. The mixture was adjusted to pH of 5 to 6 using 0.5 N HCl at 0 °C and extracted with dichloromethane (30 mL x 5). The combined organic layers were washed with brine (30 mL x 2), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure to afford 114b, which was used without further purification. LCMS [M+1] = 269.0 Step C – Synthesis of 1-(5-bromopyrimidin-2-yl)-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H- 1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (114c) A solution of Int 114b (350 mg, 1.30 mmol, 1 equiv.) and Int 1 (413.68 mg, 1.43 mmol, 1.1 equiv., HBr salt) in DMF (4 mL) was treated with triethylamine (1.32 g, 13.0 mmol, 1.81 mL, 10 equiv.) and BOP (690.4 mg, 1.56 mmol, 1.2 equiv.). The mixture was stirred at 25°C for 12 h, quenched with water (40 mL) and extracted with ethyl acetate (30 mL x 3). The combined the organic layer was washed with brine (30 mL x 3), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum. The residue was purified by column chromatography (silica gel, dichloromethane: methanol = 1/0 to 10/1) to afford 114c. LCMS [M+1] = 459.0 Step D – Synthesis of 1-[5-(4-formylphenyl)pyrimidin-2-yl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl- ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (114d). A solution of 114c (500 mg, 1.09 mmol, 1 equiv.) and 4-(4,4,5,5-tetramethyl-1,3,2- dioxaborolan-2-yl)benzaldehyde (303.2 mg, 1.31 mmol, 1.2 equiv.) in dioxane (5 mL) and water (1 mL) was treated with potassium carbonate (451.4 mg, 3.27 mmol, 3 equiv.) and Pd(dppf)Cl2.CH2Cl2 (177.8 mg, 218 μmol, 0.2 equiv.). The mixture was stirred at 100°C for 16 h under a nitrogen atmosphere. The reaction was cooled to 20°C, poured into water (15 mL) and extracted with ethyl acetate (15 mL x 2). The combined organic phase was washed with brine (5 mL x 2), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum. The residue was purified by column chromatography (silica gel, dichloromethane: MeOH =1/0 to 10/1) followed by preparative TLC (silica gel, dichloromethane: MeOH = 10: 1) to afford 114d. LCMS [M+1] = 485.1;
Step E – Synthesis of 1-[5-[4-[[4-[4-[(2,6-dioxo-3-piperidyl)amino]-2-fluoro-phenyl]-1- piperidyl]methyl]phenyl]pyrimidin-2-yl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4- triazol-5-yl]methyl]pyrazole-4-carboxamide (114) A solution of 114d (50 mg, 103.2 μmol,) and Int 97 (31.5 mg, 92 μmol) in THF (1 mL) was treated with Ti(OEt)4 (47.0 mg, 206 μmol) and the mixture was stirred at 60°C for 12 h. The mixture was cooled to 20°C and treated with NaBH3CN (29.1 mg, 464 μmol, 4.5 equiv.) and stirred at 25°C for 3 h. The mixture was diluted in ethyl acetate (3 mL) and water (10 mL), filtered and the filtrate was extracted with ethyl acetate (5 mL x 3). The combined the organic layer was washed with brine (5 mL x 3), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum. The residue was purified by preparative HPLC (Phenomenex Luna C18, 100 mm x 40 mm; 3 µm; [H2O (0.2% FA)-ACN]) to afford Compound 114. LCMS [M+1] = 774.6. 1H NMR (400 MHz, DMSO-d6) δ 13.94 (br s, 1H), 10.78 (s, 1H), 9.31 - 9.23 (m, 3H), 9.12 (s, 1H), 8.24 (s, 1H), 7.84 (d, J = 8.0 Hz, 2H), 7.50 (d, J = 8.0 Hz, 2H), 7.01 (t, J = 8.8 Hz, 1H), 6.49 - 6.40 (m, 2H), 6.00 (d, J = 7.6 Hz, 1H), 4.56 (d, J = 5.6 Hz, 2H), 4.35 - 4.25 (m, 1H), 3.56 (s, 2H), 2.93 (d, J = 10.8 Hz, 2H), 2.71 (dd, J = 5.4, 12.2 Hz, 1H), 2.64 - 2.58 (m, 2H), 2.12 - 2.02 (m, 3H), 1.93 - 1.82 (m, 1H), 1.66 (s, 4H), 1.54 (s, 6H). Example 115: Synthesis of Intermediate 115
Step A – Synthesis of methyl 1-(5-bromopyrimidin-2-yl)pyrazole-4-carboxylate (114a) A solution of methyl 1H-pyrazole-4-carboxylate (2.80 g, 22.2 mmol, 1 equiv.) in DMF (50 mL) was treated with cesium carbonate (21.7 g, 66.6 mmol, 3 equiv.) and 5-bromo-2-chloro- pyrimidine (4.29 g, 22.2 mmol, 1 equiv.). The mixture was stirred at 100°C for 2 h under a nitrogen atmosphere, cooled to 20°C and diluted with saturated aqueous NH4Cl (100 mL). The mixture was extracted with ethyl acetate (40 mL x 3), washed with brine (40 mLx 3), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum. The residue
was dissolved in MeOH (10 mL) and stirred at 20°C for 30 min, filtered and the filtrate was concentrated under vacuum to afford 114a. LCMS [M+1] = 283.1; Step B – Synthesis of 1-(5-bromopyrimidin-2-yl)pyrazole-4-carboxylic acid (114b) A solution of 114a (800 mg, 2.83 mmol, 1 equiv.) in dichloromethane (16 mL) was added hydroxy(trimethyl)stannane (1.53 g, 8.48 mmol, 3 equiv.), the mixture was stirred at 80°C for 32 h. The mixture was adjusted to pH of 5 to 6 using 0.5 N HCl at 0 °C and extracted with dichloromethane (30 mL x 5). The combined organic layers were washed with brine (30 mL x 2), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure to afford 114b, which was used without further purification. LCMS [M+1] = 269.0 Step C – Synthesis of 1-(5-bromopyrimidin-2-yl)-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H- 1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (114c) A solution of Int 114b (350 mg, 1.30 mmol, 1 equiv.) and Int 1 (413.68 mg, 1.43 mmol, 1.1 equiv., HBr salt) in DMF (4 mL) was treated with triethylamine (1.32 g, 13.0 mmol, 1.81 mL, 10 equiv.) and BOP (690.4 mg, 1.56 mmol, 1.2 equiv.). The mixture was stirred at 25°C for 12 h, quenched with water (40 mL) and extracted with ethyl acetate (30 mL x 3). The combined the organic layer was washed with brine (30 mL x 3), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum. The residue was purified by column chromatography (silica gel, dichloromethane: methanol = 1/0 to 10/1) to afford 114c. LCMS [M+1] = 459.0 Step D – Synthesis of 1-[5-(4-formylphenyl)pyrimidin-2-yl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl- ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (114d). A solution of 114c (500 mg, 1.09 mmol, 1 equiv.) and 4-(4,4,5,5-tetramethyl-1,3,2- dioxaborolan-2-yl)benzaldehyde (303.2 mg, 1.31 mmol, 1.2 equiv.) in dioxane (5 mL) and water (1 mL) was treated with potassium carbonate (451.4 mg, 3.27 mmol, 3 equiv.) and Pd(dppf)Cl2.CH2Cl2 (177.8 mg, 218 μmol, 0.2 equiv.). The mixture was stirred at 100°C for 16 h under a nitrogen atmosphere. The reaction was cooled to 20°C, poured into water (15 mL) and extracted with ethyl acetate (15 mL x 2). The combined organic phase was washed with brine (5 mL x 2), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum. The residue was purified by column chromatography (silica gel, dichloromethane: MeOH =1/0 to 10/1) followed by preparative TLC (silica gel, dichloromethane: MeOH = 10: 1) to afford 114d. LCMS [M+1] = 485.1;
Step E – Synthesis of 1-[5-[4-[[4-[4-[(2,6-dioxo-3-piperidyl)amino]-2-fluoro-phenyl]-1- piperidyl]methyl]phenyl]pyrimidin-2-yl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4- triazol-5-yl]methyl]pyrazole-4-carboxamide (114) A solution of 114d (50 mg, 103.2 μmol,) and Int 97 (31.5 mg, 92 μmol) in THF (1 mL) was treated with Ti(OEt)4 (47.0 mg, 206 μmol) and the mixture was stirred at 60°C for 12 h. The mixture was cooled to 20°C and treated with NaBH3CN (29.1 mg, 464 μmol, 4.5 equiv.) and stirred at 25°C for 3 h. The mixture was diluted in ethyl acetate (3 mL) and water (10 mL), filtered and the filtrate was extracted with ethyl acetate (5 mL x 3). The combined the organic layer was washed with brine (5 mL x 3), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum. The residue was purified by preparative HPLC (Phenomenex Luna C18, 100 mm x 40 mm; 3 µm; [H2O (0.2% FA)-ACN]) to afford Compound 114. LCMS [M+1] = 774.6. 1H NMR (400 MHz, DMSO-d6) δ 13.94 (br s, 1H), 10.78 (s, 1H), 9.31 - 9.23 (m, 3H), 9.12 (s, 1H), 8.24 (s, 1H), 7.84 (d, J = 8.0 Hz, 2H), 7.50 (d, J = 8.0 Hz, 2H), 7.01 (t, J = 8.8 Hz, 1H), 6.49 - 6.40 (m, 2H), 6.00 (d, J = 7.6 Hz, 1H), 4.56 (d, J = 5.6 Hz, 2H), 4.35 - 4.25 (m, 1H), 3.56 (s, 2H), 2.93 (d, J = 10.8 Hz, 2H), 2.71 (dd, J = 5.4, 12.2 Hz, 1H), 2.64 - 2.58 (m, 2H), 2.12 - 2.02 (m, 3H), 1.93 - 1.82 (m, 1H), 1.66 (s, 4H), 1.54 (s, 6H). Example 115: Synthesis of Intermediate 115
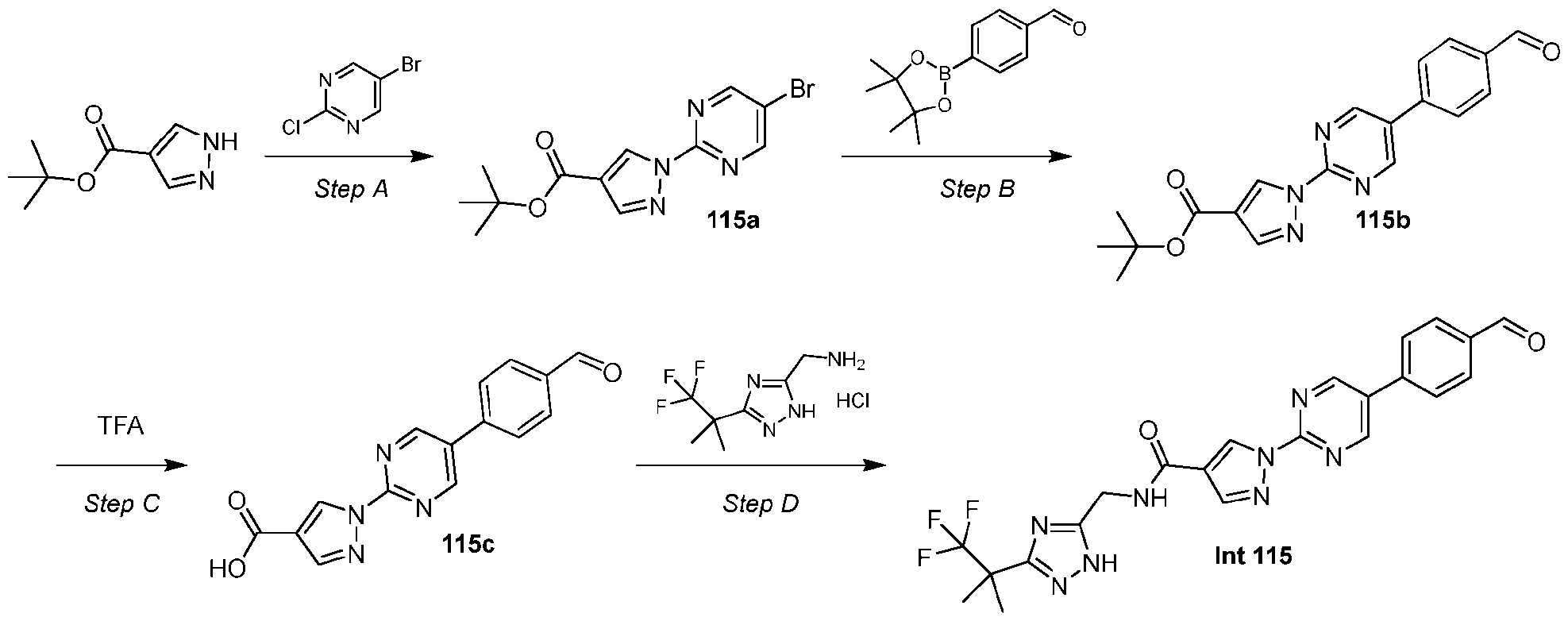 Step A – Synthesis of tert-butyl 1-(5-bromopyrimidin-2-yl)-1H-pyrazole-4-carboxylate (115a) A solution of tert-butyl 1H-pyrazole-4-carboxylate (10.0 g, 59.5 mmol, 1.0 equiv.) in DMF (180.0 mL) was treated with cesium carbonate (58.1 g, 178.4 mmol, 3.0 equiv.) and 5-bromo-2-
chloro-pyrimidine (11.5 g, 59.5 mmol, 1.0 equiv.). The mixture was stirred at 100°C for 1 h under a nitrogen atmosphere. The mixture was cooled to ambient temperature and added to water (1 L) with agitation. The solid was collected by vacuum filtration and the filter cake was dried under vacuum to afford 115a, which was used without further purification. LCMS [M+1] = 325.2, 327.2 Step B – Synthesis of tert-butyl 1-[5-(4-formylphenyl)pyrimidin-2-yl]pyrazole-4-carboxylate (115b) A mixture of 115a (54.0 g, 166.1 mmol, 1.0 equiv.), 4-(4,4,5,5-tetramethyl-1,3,2- dioxaborolan-2-yl)benzaldehyde (34.7 g, 149.5 mmol, 0.9 equiv.), potassium carbonate (68.9 g, 498 mmol, 3.0 equiv.) and Pd(dppf)Cl2 (6.08 g, 8.3 mmol, 0.05 equiv.) in dioxane (540 mL) and water (135 mL) was degassed and purged threefold with nitrogen. The mixture was stirred at 80°C for 2 h under a nitrogen atmosphere. The mixture was cooled to ambient temperature, diluted with water (550 mL) and extracted with ethyl acetate (550 mL x 3). The combined organic layers were washed with brine (550 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether: ethyl acetate = 1: 0 to 0: 1) to afford 115b. LCMS [M+1] = 351.2. Step C – Synthesis of 1-[5-(4-formylphenyl)pyrimidin-2-yl]pyrazole-4-carboxylic acid (115c) A solution of 115b (9.00 g, 25.7 mmol) in dichloromethane (90.0 mL) was treated with trifluoroacetic acid (20.0 mL), the mixture was stirred at 25°C for 4 h. The mixture was concentrated under vacuum. The residue was triturated with MTBE (40 mL) at 25 °C for 1 h and then the solids were collected by vacuum filtration and dried under vacuum to afford 115c, which was used without further purification. LCMS [M+1] = 295.2. Step D – 1-[5-(4-formylphenyl)pyrimidin-2-yl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H- 1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Int 115) A solution of 115c (2.5 g, 6.1 mmol) in DMF (25.0 mL) was treated with triethylamine (4.3 mL, 30.6 mmol, 5.0 equiv.), BOP (3.3 g, 7.4 mmol, 1.2 equiv). and [3-(2,2,2-trifluoro-1,1-
dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methanamine hydrochloride (1.5 g, 6.1 mmol). The resulting mixture was stirred at 25 °C for 0.5 h, diluted with water (25 mL) and ethyl acetate (25
Step A – Synthesis of tert-butyl 1-(5-bromopyrimidin-2-yl)-1H-pyrazole-4-carboxylate (115a) A solution of tert-butyl 1H-pyrazole-4-carboxylate (10.0 g, 59.5 mmol, 1.0 equiv.) in DMF (180.0 mL) was treated with cesium carbonate (58.1 g, 178.4 mmol, 3.0 equiv.) and 5-bromo-2-
chloro-pyrimidine (11.5 g, 59.5 mmol, 1.0 equiv.). The mixture was stirred at 100°C for 1 h under a nitrogen atmosphere. The mixture was cooled to ambient temperature and added to water (1 L) with agitation. The solid was collected by vacuum filtration and the filter cake was dried under vacuum to afford 115a, which was used without further purification. LCMS [M+1] = 325.2, 327.2 Step B – Synthesis of tert-butyl 1-[5-(4-formylphenyl)pyrimidin-2-yl]pyrazole-4-carboxylate (115b) A mixture of 115a (54.0 g, 166.1 mmol, 1.0 equiv.), 4-(4,4,5,5-tetramethyl-1,3,2- dioxaborolan-2-yl)benzaldehyde (34.7 g, 149.5 mmol, 0.9 equiv.), potassium carbonate (68.9 g, 498 mmol, 3.0 equiv.) and Pd(dppf)Cl2 (6.08 g, 8.3 mmol, 0.05 equiv.) in dioxane (540 mL) and water (135 mL) was degassed and purged threefold with nitrogen. The mixture was stirred at 80°C for 2 h under a nitrogen atmosphere. The mixture was cooled to ambient temperature, diluted with water (550 mL) and extracted with ethyl acetate (550 mL x 3). The combined organic layers were washed with brine (550 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether: ethyl acetate = 1: 0 to 0: 1) to afford 115b. LCMS [M+1] = 351.2. Step C – Synthesis of 1-[5-(4-formylphenyl)pyrimidin-2-yl]pyrazole-4-carboxylic acid (115c) A solution of 115b (9.00 g, 25.7 mmol) in dichloromethane (90.0 mL) was treated with trifluoroacetic acid (20.0 mL), the mixture was stirred at 25°C for 4 h. The mixture was concentrated under vacuum. The residue was triturated with MTBE (40 mL) at 25 °C for 1 h and then the solids were collected by vacuum filtration and dried under vacuum to afford 115c, which was used without further purification. LCMS [M+1] = 295.2. Step D – 1-[5-(4-formylphenyl)pyrimidin-2-yl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H- 1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (Int 115) A solution of 115c (2.5 g, 6.1 mmol) in DMF (25.0 mL) was treated with triethylamine (4.3 mL, 30.6 mmol, 5.0 equiv.), BOP (3.3 g, 7.4 mmol, 1.2 equiv). and [3-(2,2,2-trifluoro-1,1-
dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methanamine hydrochloride (1.5 g, 6.1 mmol). The resulting mixture was stirred at 25 °C for 0.5 h, diluted with water (25 mL) and ethyl acetate (25
 mL) and filtrate was separated and the aqueous layer was extracted with ethyl acetate (15 mL x 2). The combined organic layers were washed with brine (15.0 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was diluted in 2N HCl (25.0 mL) and stirred at 25°C for 1 h, the mixture was filtered. The solid was treated with saturated NaHCO3 (50.0mL) and stirred at 25°C for 1 h, filtered and the filter cake was washed with water (200 mL). The solid was triturated with MTBE to afford Int 115. LCMS [M+1] = 485.3. 1H NMR (400 MHz, DMSO-d6) δ = 10.09 (s, 1H), 9.34 (s, 2H), 9.28 (s, 1H), 9.19 – 9.16 (m, 1H), 8.25 (s, 1H), 8.14 - 8.06 (m, 4H), 4.54 (br s, 2H), 1.53 (s, 6H) Example 116: Synthesis of Compound 116
mL) and filtrate was separated and the aqueous layer was extracted with ethyl acetate (15 mL x 2). The combined organic layers were washed with brine (15.0 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was diluted in 2N HCl (25.0 mL) and stirred at 25°C for 1 h, the mixture was filtered. The solid was treated with saturated NaHCO3 (50.0mL) and stirred at 25°C for 1 h, filtered and the filter cake was washed with water (200 mL). The solid was triturated with MTBE to afford Int 115. LCMS [M+1] = 485.3. 1H NMR (400 MHz, DMSO-d6) δ = 10.09 (s, 1H), 9.34 (s, 2H), 9.28 (s, 1H), 9.19 – 9.16 (m, 1H), 8.25 (s, 1H), 8.14 - 8.06 (m, 4H), 4.54 (br s, 2H), 1.53 (s, 6H) Example 116: Synthesis of Compound 116
 A solution of Int 115 (50.0 mg, 103 μmol, 1.0 equiv.) and 3-(5-fluoro-6-piperazin-1-yl-3- pyridyl)piperidine-2,6-dione (40.7 mg, 139 μmol, 1.35 equiv.) in dichloromethane (1.0 mL) was treated with triethylamine (43 μL, 310 μmol, 3.0 equiv.) at 25°C. The mixture was stirred for 1 h, and then treated with NaBH(OAc)3 (109.3 mg, 516 μmol, 5.0 equiv.). The resulting mixture was stirred at 25°C for 16 h. The reaction mixture was diluted with dichloromethane (20 mL), washed by water (20 mL x 2), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative TLC (silica gel, dichloromethane: MeOH = 10:1) to afford 1-[5-[4-[[4-[5-(2,6-dioxo-3-piperidyl)-3-fluoro-2- pyridyl]piperazin-1-yl]methyl]phenyl]pyrimidin-2-yl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (116). LCMS [M+1] = 761.6.
1H NMR (400 MHz, DMSO-d6) δ 13.95 (br s, 1H), 10.86 (s, 1H), 9.30 - 9.21 (m, 3H), 9.11 (br s, 1H), 8.24 (s, 1H), 7.92 - 7.76 (m, 3H), 7.52 (d, J = 8.0 Hz, 2H), 7.46 (dd, J = 1.6, 14.4 Hz, 1H), 4.56 (br d, J = 5.6 Hz, 2H), 3.85 (dd, J = 4.8, 12.5 Hz, 1H), 3.61 (s, 2H), 3.40 (br s, 4H), 2.77 - 2.60 (m, 1H), 2.55 (br s, 4H), 2.24 (br dd, J = 4.4, 12.8 Hz, 2H), 2.12 - 1.86 (m, 1H), 1.54 (s, 6H) Example 117: Synthesis of Compound 117
A solution of Int 115 (50.0 mg, 103 μmol, 1.0 equiv.) and 3-(5-fluoro-6-piperazin-1-yl-3- pyridyl)piperidine-2,6-dione (40.7 mg, 139 μmol, 1.35 equiv.) in dichloromethane (1.0 mL) was treated with triethylamine (43 μL, 310 μmol, 3.0 equiv.) at 25°C. The mixture was stirred for 1 h, and then treated with NaBH(OAc)3 (109.3 mg, 516 μmol, 5.0 equiv.). The resulting mixture was stirred at 25°C for 16 h. The reaction mixture was diluted with dichloromethane (20 mL), washed by water (20 mL x 2), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative TLC (silica gel, dichloromethane: MeOH = 10:1) to afford 1-[5-[4-[[4-[5-(2,6-dioxo-3-piperidyl)-3-fluoro-2- pyridyl]piperazin-1-yl]methyl]phenyl]pyrimidin-2-yl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (116). LCMS [M+1] = 761.6.
1H NMR (400 MHz, DMSO-d6) δ 13.95 (br s, 1H), 10.86 (s, 1H), 9.30 - 9.21 (m, 3H), 9.11 (br s, 1H), 8.24 (s, 1H), 7.92 - 7.76 (m, 3H), 7.52 (d, J = 8.0 Hz, 2H), 7.46 (dd, J = 1.6, 14.4 Hz, 1H), 4.56 (br d, J = 5.6 Hz, 2H), 3.85 (dd, J = 4.8, 12.5 Hz, 1H), 3.61 (s, 2H), 3.40 (br s, 4H), 2.77 - 2.60 (m, 1H), 2.55 (br s, 4H), 2.24 (br dd, J = 4.4, 12.8 Hz, 2H), 2.12 - 1.86 (m, 1H), 1.54 (s, 6H) Example 117: Synthesis of Compound 117
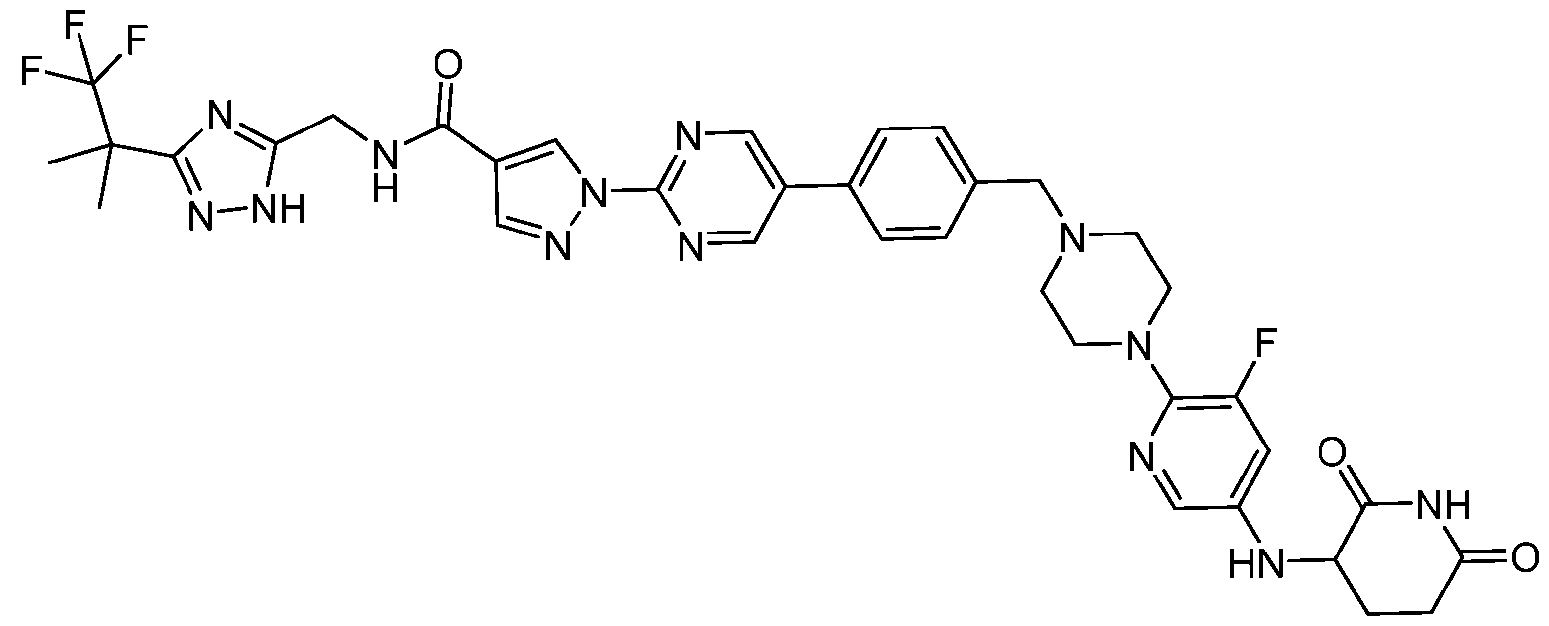 1-yl- 3-pyridyl)amino]piperidine-2,6-dione hydrochloride (4.32 g, 12.56 mmol, 1.2 equiv.) in dichloromethane (50 mL) was treated with triethylamine (5.22 g, 51.61 mmol, 7.18 mL, 5.0 equiv.) at 25°C. The mixture was stirred at ambient temperature for 1 h and then NaBH(OAc)3 (6.56 g, 30.96 mmol, 3.0 equiv.) was added at 25°C. The resulting mixture was stirred at 25°C for 16 h and then partitioned between dichloromethane (40 mL) and water (40 mL). The organic phase was separated, washed with water (10 mL x 3), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, dichloromethane : methanol=1/0 to 95/5) followed by preparative HPLC (Phenomenex Luna C18, 250mm x 70mm, 15 µm; [H2O(0.04%HCl)-ACN]). After concentration of the pooled eluant, the residue was dissolved in water and CH3CN, acidified with aqueous HCl (1 M, 26.30 mL, 4.0 equiv.) and lyophilized to afford 1-[5-[4-[[4-[5-[(2,6- dioxo-3-piperidyl)amino]-3-fluoro-2-pyridyl]piperazin-1-yl]methyl]phenyl]pyrimidin-2-yl]-N- [[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (117) as the hydrochloride salt. LCMS [M+1] = 776.5.1H NMR (400 MHz, DMSO-d6) δ 11.58 (br s, 1H), 10.82 (s, 1H), 9.35 - 9.26 (m, 3H), 9.17 (t, J = 5.6 Hz, 1H), 8.27 (s, 1H), 8.00 (d, J =
8.2 Hz, 2H), 7.86 (d, J = 8.3 Hz, 2H), 7.61 (d, J = 1.8 Hz, 1H), 7.09 (dd, J = 2.2, 14.6 Hz, 1H), 4.57 (d, J = 5.8 Hz, 2H), 4.44 (br d, J = 3.8 Hz, 2H), 4.34 (dd, J = 4.8, 11.6 Hz, 1H), 3.65 (br d, J = 12.4 Hz, 2H), 3.45 - 3.27 (m, 4H), 3.25 - 3.12 (m, 2H), 2.77 - 2.65 (m, 1H), 2.63 - 2.53 (m, 1H), 2.16 - 2.01 (m, 1H), 1.89 (dq, J = 4.8, 12.2 Hz, 1H), 1.54 (s, 6H) Example 118: Synthesis of Compound 118 F F F O N O A
1-yl- 3-pyridyl)amino]piperidine-2,6-dione hydrochloride (4.32 g, 12.56 mmol, 1.2 equiv.) in dichloromethane (50 mL) was treated with triethylamine (5.22 g, 51.61 mmol, 7.18 mL, 5.0 equiv.) at 25°C. The mixture was stirred at ambient temperature for 1 h and then NaBH(OAc)3 (6.56 g, 30.96 mmol, 3.0 equiv.) was added at 25°C. The resulting mixture was stirred at 25°C for 16 h and then partitioned between dichloromethane (40 mL) and water (40 mL). The organic phase was separated, washed with water (10 mL x 3), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, dichloromethane : methanol=1/0 to 95/5) followed by preparative HPLC (Phenomenex Luna C18, 250mm x 70mm, 15 µm; [H2O(0.04%HCl)-ACN]). After concentration of the pooled eluant, the residue was dissolved in water and CH3CN, acidified with aqueous HCl (1 M, 26.30 mL, 4.0 equiv.) and lyophilized to afford 1-[5-[4-[[4-[5-[(2,6- dioxo-3-piperidyl)amino]-3-fluoro-2-pyridyl]piperazin-1-yl]methyl]phenyl]pyrimidin-2-yl]-N- [[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (117) as the hydrochloride salt. LCMS [M+1] = 776.5.1H NMR (400 MHz, DMSO-d6) δ 11.58 (br s, 1H), 10.82 (s, 1H), 9.35 - 9.26 (m, 3H), 9.17 (t, J = 5.6 Hz, 1H), 8.27 (s, 1H), 8.00 (d, J =
8.2 Hz, 2H), 7.86 (d, J = 8.3 Hz, 2H), 7.61 (d, J = 1.8 Hz, 1H), 7.09 (dd, J = 2.2, 14.6 Hz, 1H), 4.57 (d, J = 5.8 Hz, 2H), 4.44 (br d, J = 3.8 Hz, 2H), 4.34 (dd, J = 4.8, 11.6 Hz, 1H), 3.65 (br d, J = 12.4 Hz, 2H), 3.45 - 3.27 (m, 4H), 3.25 - 3.12 (m, 2H), 2.77 - 2.65 (m, 1H), 2.63 - 2.53 (m, 1H), 2.16 - 2.01 (m, 1H), 1.89 (dq, J = 4.8, 12.2 Hz, 1H), 1.54 (s, 6H) Example 118: Synthesis of Compound 118 F F F O N O A
 1-yl- phenyl)methyl]hexahydropyrimidine-2,4-dione hydrochloride (126.5 mg, 412 μmol, 2 equiv.) in THF (2 mL) was treated with Ti(OEt)4 (428 μL, 2.06 mmol, 10 equiv.) and the mixture was stirred for 10 min at 70°C, cooled to ambient temperature and treated with NaBH3CN (19.46 mg, 309.64 μmol, 1.5 equiv.). The mixture was stirred at 70°C for 10 min, cooled to ambient temperature and then filtered and the filtrate was concentrated under vacuum. The residue was purified by preparative HPLC (Phenomenex Luna C18, 150 mm x 25mm, 10µm; [water(FA)- ACN]) to afford 1-[5-[4-[[4-[4-[(2,4-dioxohexahydropyrimidin-1-yl)methyl]-2-fluoro- phenyl]piperazin-1-yl]methyl]phenyl]pyrimidin-2-yl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (118). LCMS [M+1] = 775.4. 1H NMR (400 MHz, CHLOROFORM-d) δ 9.11 (s, 1 H), 8.98 (s, 2 H), 8.15 (s, 1 H), 7.50 - 7.63 (m, 4 H), 6.95 - 7.03 (m, 2 H), 6.87 - 6.95 (m, 2 H), 4.74 (d, J = 6.0 Hz, 2 H), 4.54 (s, 2 H), 3.69 (s, 2 H), 3.33 (t, J = 6.8 Hz, 2 H), 3.15 (br s, 4 H), 2.70 (br s, 4 H), 2.64 (t, J = 6.8 Hz, 2 H), 1.63 (s, 6 H). Example 119: Synthesis of Intermediate 119
1-yl- phenyl)methyl]hexahydropyrimidine-2,4-dione hydrochloride (126.5 mg, 412 μmol, 2 equiv.) in THF (2 mL) was treated with Ti(OEt)4 (428 μL, 2.06 mmol, 10 equiv.) and the mixture was stirred for 10 min at 70°C, cooled to ambient temperature and treated with NaBH3CN (19.46 mg, 309.64 μmol, 1.5 equiv.). The mixture was stirred at 70°C for 10 min, cooled to ambient temperature and then filtered and the filtrate was concentrated under vacuum. The residue was purified by preparative HPLC (Phenomenex Luna C18, 150 mm x 25mm, 10µm; [water(FA)- ACN]) to afford 1-[5-[4-[[4-[4-[(2,4-dioxohexahydropyrimidin-1-yl)methyl]-2-fluoro- phenyl]piperazin-1-yl]methyl]phenyl]pyrimidin-2-yl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (118). LCMS [M+1] = 775.4. 1H NMR (400 MHz, CHLOROFORM-d) δ 9.11 (s, 1 H), 8.98 (s, 2 H), 8.15 (s, 1 H), 7.50 - 7.63 (m, 4 H), 6.95 - 7.03 (m, 2 H), 6.87 - 6.95 (m, 2 H), 4.74 (d, J = 6.0 Hz, 2 H), 4.54 (s, 2 H), 3.69 (s, 2 H), 3.33 (t, J = 6.8 Hz, 2 H), 3.15 (br s, 4 H), 2.70 (br s, 4 H), 2.64 (t, J = 6.8 Hz, 2 H), 1.63 (s, 6 H). Example 119: Synthesis of Intermediate 119
A solution of ethyl 1H-pyrazole-4-carboxylate (25.0 g, 178.3 mmol, 1.0 equiv.) and 5- bromo-2-fluoropyridine (31.3 g, 178.3 mmol, 18.3 mL, 1.0 equiv.) in DMF (500 mL) was treated with cesium carbonate (87.1 g, 267.5 mmol, 1.5 equiv.). The mixture was stirred at 100°C for 8 h. The reaction mixture was extracted with ethyl acetate (100.0 x 3) and water (500 mL). The combined organic layers were washed with brine (100 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether/ ethyl acetate=100/1 to 5/1) to afford 119a. LCMS [M+1]= 295.8, 297.8. Step B– Synthesis of 1-(5-bromopyridin-2-yl)-1H-pyrazole-4-carboxylic acid (119b) A solution of ethyl 1-(5-bromopyridin-2-yl)-1H-pyrazole-4-carboxylate (30.0 g, 101 mmol, 1.0 equiv.) in methanol (200.0 mL) and water (50.0 mL) was added LiOH (4.8 g, 202.6 mmol, 2.0 equiv.). The mixture was stirred at 50°C for 2 h, cooled to ambient temperature and extracted with ethyl acetate (30 mL x 3). The combined organic layers were washed with brine (30 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure to afford 119b. LCMS [M+1] = 268.0, 270.0. Step C – Synthesis of 1-(5-bromo-2-pyridyl)-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4- triazol-5-yl]methyl]pyrazole-4-carboxamide (119c) To a solution of 1-(5-bromo-2-pyridyl)pyrazole-4-carboxylic acid (12.0 g, 44.7 mmol, 1.0 equiv.) and [3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methanamine (10.2 g, 49.2 mmol, 1.1 equiv.) in DMF (120.0 mL) was added TEA (13.5 g, 134.2 mmol, 18.6 mL, 3.0 equiv.) to adjust pH to 9-10. The mixture was stirred at 25°C for 0.5 h. Then BOP (23.7 g, 53.7 mmol, 1.2 equiv.) was added, the resulting mixture was stirred at 25°C for 1 h.
LCMS showed starting material was consumed completely and desired mass was detected. The mixture was poured into water (300 mL). The aqueous phase was extracted with ethyl acetate (100 mL x 3). The combined organic phase was washed with brine (100 mL x 3), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum to give residue. The residue was purified by column chromatography (silica gel, petroleum ether/ethyl acetate=1/0 to 3/1) to give (9.7 g, 21.1 mmol, 47.3% yield) as white solid. LCMS [M+1] = 458.0, 460.0. Step D – Synthesis of 1-[5-(4-formylphenyl)-2-pyridyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (119) A mixture of 119c (10.0 g, 21.8 mmol, 1.0 equiv.), 4-(4,4,5,5-tetramethyl-1,3,2- dioxaborolan-2-yl)benzaldehyde (7.60 g, 32.7 mmol, 1.5 equiv.), Pd(dppf)Cl2 (1.6 g, 2.1 mmol, 0.1 equiv.) and K2CO3 (6.0 g, 43.6 mmol, 2.0 equiv.) in dioxane (100.0 mL) and water (20.0 mL) was degassed and purged threefold with nitrogen, and then the mixture was stirred at 100°C for 3 h under a nitrogen atmosphere. The mixture was cooled to ambient temperature and concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether/ ethyl acetate=1/0 to 0/1) to afford Int 119. LCMS [M+1] = 484.1. 1H NMR (400 MHz, DMSO-d6) δ 13.91 (br s, 1H), 10.09 (s, 1H), 9.25 (s, 1H), 9.11 (br t, J = 5.2 Hz, 1H), 8.97 (d, J = 2.0 Hz, 1H), 8.46 (dd, J = 2.4, 8.6 Hz, 1H), 8.24 (s, 1H), 8.10 - 8.03 (m, 5H), 4.57 (br d, J = 5.6 Hz, 2H), 1.53 (s, 6H) Example 120: Synthesis of Compound 120 A
 1-yl-3- pyridyl)amino]piperidine-2,6-dione hydrochloride (63.5 mg, 207 μmol, 2.0 equiv.) in THF (2 mL) was treated with Ti(OEt)4 (214 μL,, 1.0 mmol, 10 equiv.), stirred for 10 min at 70°C, cooled to ambient temperature and treated with NaBH3CN (9.7 mg, 155 μmol, 1.5 equiv.) and stirred for 15
min. The mixture was dissolved in water (2 mL) and MeOH (5 mL), filtered to remove the insoluble portion and the filtrate was concentrated. The residue was purified by preparative HPLC (Welch Xtimate C18, 150 mm x 25mm, 5µm [water(FA)-ACN]) to give 1-[5-[4-[[4-[5- [(2,6-dioxo-3-piperidyl)amino]-3-fluoro-2-pyridyl]piperazin-1-yl]methyl]phenyl]-2-pyridyl]-N- [[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (120). LCMS [M+1] = 775.1. 1H NMR (400 MHz, DMSO-d6) δ 13.90 (br s, 1H), 10.80 (s, 1H), 9.24 (s, 1H), 9.02 - 9.11 (m, 1H), 8.87 (d, J = 2.0 Hz, 1H), 8.35 (dd, J = 8.8, 2.4 Hz, 1H), 8.23 (s, 1H), 8.21 (s, 1H), 8.04 (d, J = 8.4 Hz, 1H), 7.78 (d, J = 8.0 Hz, 2H), 7.58 (d, J = 1.6 Hz, 1H), 7.49 (d, J = 8.0 Hz, 2H), 7.00 (dd, J = 14.8, 2.4 Hz, 1H), 5.93 (d, J = 8.0 Hz, 1H), 4.56 (d, J = 5.6 Hz, 2H), 4.24 - 4.35 (m, 1H), 3.60 (s, 4H), 3.14 (s, 6H), 2.66 - 2.78 (m, 2H), 2.10 (td, J = 8.4, 4.0 Hz, 1H), 1.88 (qd, J = 12.0, 4.4 Hz, 1H), 1.55 (s, 6H). Example 121: Synthesis of Compound 121
1-yl-3- pyridyl)amino]piperidine-2,6-dione hydrochloride (63.5 mg, 207 μmol, 2.0 equiv.) in THF (2 mL) was treated with Ti(OEt)4 (214 μL,, 1.0 mmol, 10 equiv.), stirred for 10 min at 70°C, cooled to ambient temperature and treated with NaBH3CN (9.7 mg, 155 μmol, 1.5 equiv.) and stirred for 15
min. The mixture was dissolved in water (2 mL) and MeOH (5 mL), filtered to remove the insoluble portion and the filtrate was concentrated. The residue was purified by preparative HPLC (Welch Xtimate C18, 150 mm x 25mm, 5µm [water(FA)-ACN]) to give 1-[5-[4-[[4-[5- [(2,6-dioxo-3-piperidyl)amino]-3-fluoro-2-pyridyl]piperazin-1-yl]methyl]phenyl]-2-pyridyl]-N- [[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (120). LCMS [M+1] = 775.1. 1H NMR (400 MHz, DMSO-d6) δ 13.90 (br s, 1H), 10.80 (s, 1H), 9.24 (s, 1H), 9.02 - 9.11 (m, 1H), 8.87 (d, J = 2.0 Hz, 1H), 8.35 (dd, J = 8.8, 2.4 Hz, 1H), 8.23 (s, 1H), 8.21 (s, 1H), 8.04 (d, J = 8.4 Hz, 1H), 7.78 (d, J = 8.0 Hz, 2H), 7.58 (d, J = 1.6 Hz, 1H), 7.49 (d, J = 8.0 Hz, 2H), 7.00 (dd, J = 14.8, 2.4 Hz, 1H), 5.93 (d, J = 8.0 Hz, 1H), 4.56 (d, J = 5.6 Hz, 2H), 4.24 - 4.35 (m, 1H), 3.60 (s, 4H), 3.14 (s, 6H), 2.66 - 2.78 (m, 2H), 2.10 (td, J = 8.4, 4.0 Hz, 1H), 1.88 (qd, J = 12.0, 4.4 Hz, 1H), 1.55 (s, 6H). Example 121: Synthesis of Compound 121

Step A – Synthesis of tert-butyl 1-(5-bromopyrimidin-2-yl)-1H-pyrazole-4-carboxylate (121a) A solution of tert-butyl 1H-pyrazole-4-carboxylate (40.0 g, 237.8 mmol, 1.0 equiv.) in DMF (400 mL) was treated with cesium carbonate (232.5 g, 713.5 mmol, 3.0 equiv.) and 5- bromo-2-chloropyrimidine (46.0 g, 237.8 mmol, 1.0 equiv.). The mixture was stirred at 80°C for 2 h under an atmosphere of nitrogen, cooled to ambient temperature and poured into water (3 L). The solid was collected by vacuum filtration and the filter cake was dried under vacuum to afford 121a. LCMS [M+1] = 324.9, 326.9 Step B – Synthesis of tert-butyl 1-(5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrimidin-2-yl)- 1H-pyrazole-4-carboxylate (121b) A mixture of 121a (30.0 g, 92.3 mmol, 1.0 equiv.), 4,4,5,5-tetramethyl-2-(4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3,2-dioxaborolane (32.8 g, 129.2 mmol, 1.4 equiv.), potassium acetate (27.2 g, 276.8 mmol, 3.0 equiv.) and Pd(dppf)Cl2 (3.4 g, 4.6 mmol, 0.05 equiv.) in dioxane (400.0 mL) was degassed and purged threefold with nitrogen, stirred at 100°C for 16 h
under a nitrogen atmosphere. The residue was poured into water (400 mL) and extracted with ethyl acetate (400 mL x 3). The combined organic phase was washed with brine (400 mL x 3), dried with anhydrous sodium sulfate, filtered and concentrated under vacuum. The crude product was triturated with ethyl acetate : petroleum ether =1:1(100 mL)at 25oC for 10 minutes and the solid was collected by vacuum filtration to afford 121b. LCMS [M-82] = 290.9 Step C – Synthesis of tert-butyl 1-(5-(5-formylpyridin-2-yl)pyrimidin-2-yl)-1H-pyrazole-4- carboxylate (121c) A mixture of 121b (1.0 g, 2.7 mmol, 1.0 equiv.), 6-bromonicotinaldehyde (550.0 mg, 2.9 mmol, 1.1 equiv.), potassium carbonate (1.11 g, 8.1 mmol, 3.0 equiv.), Pd(dppf)Cl2 (393.4 mg, 537.62 μmol, 0.2 equiv.) in dioxane (10.0 mL) and water (2.0 mL) was degassed and purged threefold with nitrogen and the mixture was stirred at 100°C for 2 h under a nitrogen atmosphere. The mixture was cooled to ambient temperature and poured into water (100 mL). The aqueous phase was extracted with ethyl acetate (100 mL x 3). The combined organic phase was washed with brine (100mL x 3), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum. The residue was triturated with ethyl acetate (80ml) at 25oC and the solid was collected by vacuum filtration to afford 121c. LCMS [M+19] = 370.1 Step D – Synthesis of 1-(5-(5-formylpyridin-2-yl)pyrimidin-2-yl)-1H-pyrazole-4-carboxylic acid (121d) To a solution of 121c (300.0 mg, 853.8 μmol, 1.0 equiv.) in dichloromethane (4.0 mL) and trifluoroacetic acid (2.0 mL). The mixture was stirred at 25°C for 3h and then concentrated under vacuum to afford 121d, which was used without further purification. LCMS [M+1] = 295.9 Step E – Synthesis of 1-(5-(5-formylpyridin-2-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide (121e) To a solution of 121d (280.0 mg, 948 μmol, 1.0 equiv.) and (3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methanamine hydrobromide (274.2 mg, 948 μmol, 1.0 equiv.) in DMF (5.0 mL) was added BOP (503.3 mg, 1.1 mmol, 1.2 equiv.) and triethylamine (287.9 mg, 2.8 mmol, 396 μL, 3.0 equiv.). The mixture was stirred at 25°C for 1 h. The residue was poured into water (20 mL) and stirred for 0.5 h, the mixture was filtered to afford 121e.
LCMS [M+1] = 485.9 Step F – Synthesis of 1-(5-(5-((4-(5-((2,6-dioxopiperidin-3-yl)amino)-3-fluoropyridin-2- yl)piperazin-1-yl)methyl)pyridin-2-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2- yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide (121) A solution of 3-((5-fluoro-6-(piperazin-1-yl)pyridin-3-yl)amino)piperidine-2,6-dione hydrochloride (106.2 mg, 309 μmol, 1.5 equiv) in THF (3.0 mL) was added Ti(OEt)4 (70.4 mg, 309 μmol, 64.0 μL, 10.0 equiv.) and 121e (100.0 mg, 206 μmol, 1.0 equiv.), the mixture was stirred at 70°C for 1 h and then treated with NaBH(OAc)3 (436.6 mg, 2.0 mmol, 1.5 equiv.). The mixture was stirred at 70°C for 2 h, cooled to 25°C, quenched with water (1 mL), filtered and the filtrate concentrated under reduced pressure. The residue was purified by preparative HPLC (Phenomenex Luna C18, 150mm x 25mm, 10µm; [water(FA)-ACN]) to afford 121. LCMS [M+1] = 777.4. 1H NMR (400 MHz, DMSO-d6) δ 14.00 (br s, 1H), 10.79 (s, 1H), 9.54 (s, 2H), 9.30 (s, 1H), 9.12 (s, 1H), 8.70 (s, 1H), 8.25 (s, 1H), 8.23 - 8.12 (m, 2H), 7.957.92 (m, 1H), 7.56 (s, 1H), 7.02 - 6.97 (m, 1H), 5.92 (d, J = 7.6 Hz, 1H), 4.56 (d, J = 5.6 Hz, 2H), 4.33 - 4.23 (m, 1H), 3.64 (s, 2H), 3.13 (s, 4H), 2.74 - 2.68 (m, 1H), 2.59 - 2.54 (m, 5H), 2.12 - 2.08 (m, 1H), 1.88 (s, 1H), 1.54 (s, 6H). Example 122: Synthesis of Intermediate 122
 - 1H-pyrazole-4-carboxylate (122a) A mixture of tert-butyl 1-(5-bromopyrimidin-2-yl)-1H-pyrazole-4-carboxylate (30.0 g, 92.3 mmol, 1.0 equiv.), 4,4,5,5-tetramethyl-2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3,2- dioxaborolane (32.8 g, 129.2 mmol, 1.4 equiv.), potassium acetate (27.2 g, 276.8 mmol, 3.0
equiv.) and Pd(dppf)Cl2 (3.40 g, 4.60 mmol, 0.05 equiv.) in dioxane (400 mL) was degassed and purged threefold with nitrogen. The mixture was stirred at 100°C for 16 h under a nitrogen atmosphere. The mixture was poured into water (400 mL) and stirred for 2 min. The aqueous phase was extracted with ethyl acetate (400 mL x 3). The combined organic phase was washed with brine (400 mL x 3), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum. The residue was triturated with ethyl acetate:petroleum ether =1:1 (100 mL) at 25oC for 10 minutes and the solids were collected by vacuum filtration to afford 122a. LCMS [M-82] = 290.9 Step B – Synthesis of tert-butyl 1-(5-(3-fluoro-5-formylpyridin-2-yl)pyrimidin-2-yl)-1H-pyrazole- 4-carboxylate (122b) A mixture of 122a (2.30 g, 6.20 mmol, 1.4 equiv.), 6-bromo-5-fluoronicotinaldehyde (900 mg, 4.40 mmol, 1.0 equiv.), Pd(dppf)Cl2 (322.8 mg, 441.2 μmol, 0.1 equiv.) and K2CO3 (1.8 g, 13.2 mmol, 3.0 equiv.) in dioxane (10.0 mL) and water (1.0 mL) was degassed and purged threefold with nitrogen. The mixture was stirred at 100°C for 2 h under nitrogen atmosphere. The residue was poured into water (50 mL) and the aqueous phase was extracted with ethyl acetate (50 mL x 3). The combined organic phase was washed with brine (50 mL x 3), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum. The residue was triturated with ethyl acetate (30ml) at 25oC for 10 min and the solids were collected by vacuum filtration to afford 122b. LCMS [M+1] = 370.1 Step C – Synthesis of 1-(5-(3-fluoro-5-formylpyridin-2-yl)pyrimidin-2-yl)-1H-pyrazole-4- carboxylic acid (122c) To a solution of 122b (600 mg, 1.60 mmol, 1.0 equiv.) in dichloromethane (9.0 mL) was added TFA (3.0 mL). The mixture was stirred at 25°C for 3 h and was concentrated under vacuum to afford 122c, which was used without further purification. LCMS [M+1] = 313.9 Step D – Synthesis of 1-(5-(3-fluoro-5-formylpyridin-2-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro- 2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide (Int 121) A solution of 122c (380.0 mg, 1.20 mmol, 1.0 equiv.) and (3-(3,3-difluoro-2-methylbutan- 2-yl)-1H-1,2,4-triazol-5-yl)methanamine hydrobromide (455.9 mg, 1.6 mmol, 1.3 equiv) in DMF
(5.0 mL) was treated with DIEA (845 μL, 4.80 mmol, 4.0 equiv.) and BOP (643.8 mg, 1.50 mmol, 1.2 equiv.). The mixture was stirred at 25°C for 0.5 h, diluted with water (20 mL). The mixture was acidified with1 M aqueous HCl to an adjusted of pH to 5-6. The mixture was filtered to afford Int 122, which was used without further purification. LCMS [M+1] = 504.0 Example 123: Synthesis of Compound 123 A
- 1H-pyrazole-4-carboxylate (122a) A mixture of tert-butyl 1-(5-bromopyrimidin-2-yl)-1H-pyrazole-4-carboxylate (30.0 g, 92.3 mmol, 1.0 equiv.), 4,4,5,5-tetramethyl-2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3,2- dioxaborolane (32.8 g, 129.2 mmol, 1.4 equiv.), potassium acetate (27.2 g, 276.8 mmol, 3.0
equiv.) and Pd(dppf)Cl2 (3.40 g, 4.60 mmol, 0.05 equiv.) in dioxane (400 mL) was degassed and purged threefold with nitrogen. The mixture was stirred at 100°C for 16 h under a nitrogen atmosphere. The mixture was poured into water (400 mL) and stirred for 2 min. The aqueous phase was extracted with ethyl acetate (400 mL x 3). The combined organic phase was washed with brine (400 mL x 3), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum. The residue was triturated with ethyl acetate:petroleum ether =1:1 (100 mL) at 25oC for 10 minutes and the solids were collected by vacuum filtration to afford 122a. LCMS [M-82] = 290.9 Step B – Synthesis of tert-butyl 1-(5-(3-fluoro-5-formylpyridin-2-yl)pyrimidin-2-yl)-1H-pyrazole- 4-carboxylate (122b) A mixture of 122a (2.30 g, 6.20 mmol, 1.4 equiv.), 6-bromo-5-fluoronicotinaldehyde (900 mg, 4.40 mmol, 1.0 equiv.), Pd(dppf)Cl2 (322.8 mg, 441.2 μmol, 0.1 equiv.) and K2CO3 (1.8 g, 13.2 mmol, 3.0 equiv.) in dioxane (10.0 mL) and water (1.0 mL) was degassed and purged threefold with nitrogen. The mixture was stirred at 100°C for 2 h under nitrogen atmosphere. The residue was poured into water (50 mL) and the aqueous phase was extracted with ethyl acetate (50 mL x 3). The combined organic phase was washed with brine (50 mL x 3), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum. The residue was triturated with ethyl acetate (30ml) at 25oC for 10 min and the solids were collected by vacuum filtration to afford 122b. LCMS [M+1] = 370.1 Step C – Synthesis of 1-(5-(3-fluoro-5-formylpyridin-2-yl)pyrimidin-2-yl)-1H-pyrazole-4- carboxylic acid (122c) To a solution of 122b (600 mg, 1.60 mmol, 1.0 equiv.) in dichloromethane (9.0 mL) was added TFA (3.0 mL). The mixture was stirred at 25°C for 3 h and was concentrated under vacuum to afford 122c, which was used without further purification. LCMS [M+1] = 313.9 Step D – Synthesis of 1-(5-(3-fluoro-5-formylpyridin-2-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro- 2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide (Int 121) A solution of 122c (380.0 mg, 1.20 mmol, 1.0 equiv.) and (3-(3,3-difluoro-2-methylbutan- 2-yl)-1H-1,2,4-triazol-5-yl)methanamine hydrobromide (455.9 mg, 1.6 mmol, 1.3 equiv) in DMF
(5.0 mL) was treated with DIEA (845 μL, 4.80 mmol, 4.0 equiv.) and BOP (643.8 mg, 1.50 mmol, 1.2 equiv.). The mixture was stirred at 25°C for 0.5 h, diluted with water (20 mL). The mixture was acidified with1 M aqueous HCl to an adjusted of pH to 5-6. The mixture was filtered to afford Int 122, which was used without further purification. LCMS [M+1] = 504.0 Example 123: Synthesis of Compound 123 A
 added Ti(OEt)4 (823.9 μL, 4.0 mmol, 10.0 equiv.) and 3-((5-fluoro-6-(piperazin-1-yl)pyridin-3- yl)amino)piperidine-2,6-dione (366.3 mg, 1.2 mmol, 3.0 equiv.). The mixture was stirred at 50°C for 1.5 h, treated with NaBH(OAc)3 (168.4 mg, 795 μmol, 2.0 equiv.) and stirred at 50°C for 0.5 h. The reaction mixture was quenched by water (30 ml), filtered and the filtrate was concentrated under vacuum. The residue was purified by preparative HPLC (Phenomenex Luna C18, 150 mm x 25 mm, 10 µm; [water(FA)-ACN]) to afford 1-(5-(5-((4-(5-((2,6-dioxopiperidin-3-yl)amino)-3- fluoropyridin-2-yl)piperazin-1-yl)methyl)-3-fluoropyridin-2-yl)pyrimidin-2-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide (123). LCMS [M+1] = 795.4. 1H NMR (400 MHz, DMSO-d6) δ 13.97 (br s, 1H), 10.81 (s, 1H), 9.39 (s, 2H), 9.31 (s, 1H), 9.14 (t, J = 4.8 Hz, 1H), 8.61 (s, 1H), 8.29 (s, 1H), 8.27 (s, 1H), 7.91 (d, J = 11.6 Hz, 1H), 7.57 (d, J = 1.6 Hz, 1H), 7.00 (dd, J = 2.4, 14.8 Hz, 1H), 5.94 (d, J = 8.0 Hz, 1H), 4.56 (d, J = 5.6 Hz, 2H), 4.29 (s, 1H), 3.70 (s, 2H), 3.09 - 3.09 (m, 1H), 3.14 (s, 3H), 2.76 - 2.66 (m, 1H), 2.64 - 2.52 (m, 5H), 2.09 (dt, J = 4.4, 8.8 Hz, 1H), 1.94 - 1.83 (m, 1H), 1.54 (s, 6H). Example 124: Synthesis of Intermediate 124
added Ti(OEt)4 (823.9 μL, 4.0 mmol, 10.0 equiv.) and 3-((5-fluoro-6-(piperazin-1-yl)pyridin-3- yl)amino)piperidine-2,6-dione (366.3 mg, 1.2 mmol, 3.0 equiv.). The mixture was stirred at 50°C for 1.5 h, treated with NaBH(OAc)3 (168.4 mg, 795 μmol, 2.0 equiv.) and stirred at 50°C for 0.5 h. The reaction mixture was quenched by water (30 ml), filtered and the filtrate was concentrated under vacuum. The residue was purified by preparative HPLC (Phenomenex Luna C18, 150 mm x 25 mm, 10 µm; [water(FA)-ACN]) to afford 1-(5-(5-((4-(5-((2,6-dioxopiperidin-3-yl)amino)-3- fluoropyridin-2-yl)piperazin-1-yl)methyl)-3-fluoropyridin-2-yl)pyrimidin-2-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide (123). LCMS [M+1] = 795.4. 1H NMR (400 MHz, DMSO-d6) δ 13.97 (br s, 1H), 10.81 (s, 1H), 9.39 (s, 2H), 9.31 (s, 1H), 9.14 (t, J = 4.8 Hz, 1H), 8.61 (s, 1H), 8.29 (s, 1H), 8.27 (s, 1H), 7.91 (d, J = 11.6 Hz, 1H), 7.57 (d, J = 1.6 Hz, 1H), 7.00 (dd, J = 2.4, 14.8 Hz, 1H), 5.94 (d, J = 8.0 Hz, 1H), 4.56 (d, J = 5.6 Hz, 2H), 4.29 (s, 1H), 3.70 (s, 2H), 3.09 - 3.09 (m, 1H), 3.14 (s, 3H), 2.76 - 2.66 (m, 1H), 2.64 - 2.52 (m, 5H), 2.09 (dt, J = 4.4, 8.8 Hz, 1H), 1.94 - 1.83 (m, 1H), 1.54 (s, 6H). Example 124: Synthesis of Intermediate 124
Step A – Synthesis of benzyl 4-(4-tert-butoxycarbonyl-3-fluoro-phenyl)piperazine-1-carboxylate (Int 124) A solution of benzyl piperazine-1-carboxylate (2.00 g, 9.08 mmol, 1 equiv.) in toluene (150 mL) was treated with tert-butyl 4-bromo-2-fluoro-benzoate (2.5 g, 9.0 mmol, 1.0 equiv.), cesium carbonate (3.2 g, 9.9 mmol, 1.1 equiv.), BINAP (565.3 mg, 908 μmol, 0.1 equiv.) and Pd(OAc)2 (203.8 mg, 908 μmol, 0.1 equiv.) under an atmosphere of nitrogen. The mixture was stirred at 80°C for 4 h. The reaction solution was cooled to ambient temperature, filtered and the filtrate was concentrated under vacuum. The residue was purified by column chromatography (silica gel, petroleum ether/ ethyl acetate=100: 1 to 50 : 50) to afford 124a. LCMS [M+1] = 415.2 Step B – Synthesis of tert-butyl 2-fluoro-4-piperazin-1-yl-benzoate A suspension of Pd/C (1.0 g, 10 wt% ) in ethanol (30 mL) under a nitrogen atmosphere was treated with Intermediate 124a (3.3 g, 7.9 mmol, 1.0 equiv.) and the mixture was degassed and purged threefold with hydrogen. The mixture was stirred at 40°C for 2 h under hydrogen (15 psi). The mixture was filtered through a pad of Celite and filter cake was washed with MeOH (100 mL×2). The combined filtrate was concentrated under vacuum to afford Intermediate 124, which was used without further purification. LCMS [M+1] =281.1 Example 125: Synthesis of Compound 125
1H-1,2,4-triazol-5-yl]methylcarbamoyl]pyrazol-1-yl]pyrimidin-5-yl]phenyl]methyl]piperazin-1- yl]benzoate (125a) A solution of Int 115 (600 mg, 1.2 mmol, 1.0 equiv.) in dichloromethane (10 mL) was treated with tert-butyl 2-fluoro-4-piperazin-1-yl-benzoate (381.9 mg, 1.36 mmol, 1.1 equiv.) and triethylamine (1.0 mL, 7.4 mmol, 6.0 equiv). The mixture was stirred at 20°C for 10 min and then treated with NaBH(OAc)3 (918.7 mg, 4.3 mmol, 3.5 equiv.). The mixture was stirred at 20°C for 2 h. The reaction mixture was partitioned between ethyl acetate (100 mL) and water (100 mL) and extracted with ethyl acetate (100 mL x 2). The combined organic layers were washed with brine (50 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative TLC (silica gel, dichloromethane: methanol = 10:1) to afford 125a. LCMS [M+1] =749.4
Step B – Synthesis of 2-fluoro-4-[4-[[4-[2-[4-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4- triazol-5-yl]methylcarbamoyl]pyrazol-1-yl]pyrimidin-5-yl]phenyl]methyl]piperazin-1-yl]benzoic acid (125b) A solution of 125a (900.0 mg, 1.2 mmol, 1.0 equiv.) in dichloromethane (10 mL) was treated with trifluoroacetic acid (4.50 mL, 60.5 mmol, 50 equiv.). The mixture was stirred at 20°C for 1 h and then concentrated under reduced pressure to afford 125b as the trifluoroacetate salt, which was used without further purification. LCMS [M+1] =693.3 Step C – Synthesis of 1-[5-[4-[[4-[4-[[(3S)-2,6-dioxo-3-piperidyl]carbamoyl]-3-fluoro-phenyl]- piperazin-1-yl]methyl]phenyl]pyrimidin-2-yl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H- 1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (125) A solution of 125b (550.0 mg, 682 μmol, 1.0 equiv) in DMF (7.0 mL) was treated with EDCI (261.4 mg, 1.3 mmol, 2.0 equiv.), HOBt (184.2 mg, 1.3 mmol, 2.0 equiv.) followed by (3S)-3-aminopiperidine-2,6-dione hydrochloride (134.6 mg, 818.1 μmol, 1.2 equiv.) and DIEA (528.7 mg, 4.1 mmol, 712.5 μL, 6.0 equiv.). The mixture was stirred at 20°C for 1 h. The mixture was quenched with MeOH (0.5 mL) and concentrated under vacuum. The residue was purified by preparative HPLC (Phenomenex Luna C18, 100 mm x 40mm, 3 µm; [H2O(0.2% FA)- ACN]) to afford Compound 125. LCMS [M+1] = 803.5.1H NMR (400 MHz, DMSO-d6) δ = 13.91 (s, 1H), 10.81 (s, 1H), 9.32 - 9.17 (m, 3H), 9.08 (br s, 1H), 8.22 (s, 1H), 8.02 (t, J = 7.2 Hz, 1H), 7.83 (d, J = 8.0 Hz, 2H), 7.60 (t, J = 9.2 Hz, 1H), 7.50 (d, J = 8.4 Hz, 2H), 6.88 - 6.68 (m, 2H), 4.68-4.72 (m, 1H), 4.53 (d, J = 5.2 Hz, 2H), 3.59 (s, 2H), 3.45 - 3.33 (m, 4H), 2.84 - 2.67 (m, 1H), 2.50 (br s, 4H), 2.19 - 2.04 (m, 1H), 2.01 - 1.87 (m, 1H), 1.51 (s, 6H) Example 126: Synthesis of Intermediate 126
 Step A – Synthesis of tert-butyl 4-(3-fluoro-4-(methoxycarbonyl)phenyl)piperazine-1-carboxylate (126a)
A mixture of tert-butyl piperazine-1-carboxylate (2.00 g, 10.7 mmol, 1.0 equiv.), methyl 4- bromo-2-fluorobenzoate (2.50 g, 10.7 mmol, 1.0 equiv.) in toluene (160 mL) was treated with cesium carbonate (3.80 g, 11.8 mmol, 1.1 equiv.), Pd(OAc)2 (241.1 mg, 1.1 mmol, 0.1 equiv.) and BINAP (668.6 mg, 1.1 mmol, 0.1 equiv.). The mixture was degassed and purged threefold with nitrogen and stirred at 60°C for 16 h under an atmosphere of nitrogen. The mixture was cooled to ambient temperature and concentrated under reduced pressure. The residue was diluted with water (20 mL) and extracted with ethyl acetate (30 mL x 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether : ethyl acetate = 1 : 0 to 4 : 1) to afford 126a. LCMS: [M+1] = 339.1 Step B – Synthesis of methyl 2-fluoro-4-piperazin-1-yl-benzoate (126) A mixture of 126a (6.7 g, 19.8 mmol, 1.0 equiv.) in a solution of 4 M HCl in ethyl acetate was stirred at 25°C for 16 h. The reaction mixture was concentrated under reduced pressure to afford 126 as the hydrochloride salt, which was used without further purification. LCMS: [M+1] =239.1 Example 127: Synthesis of Compound 127
Step A – Synthesis of tert-butyl 4-(3-fluoro-4-(methoxycarbonyl)phenyl)piperazine-1-carboxylate (126a)
A mixture of tert-butyl piperazine-1-carboxylate (2.00 g, 10.7 mmol, 1.0 equiv.), methyl 4- bromo-2-fluorobenzoate (2.50 g, 10.7 mmol, 1.0 equiv.) in toluene (160 mL) was treated with cesium carbonate (3.80 g, 11.8 mmol, 1.1 equiv.), Pd(OAc)2 (241.1 mg, 1.1 mmol, 0.1 equiv.) and BINAP (668.6 mg, 1.1 mmol, 0.1 equiv.). The mixture was degassed and purged threefold with nitrogen and stirred at 60°C for 16 h under an atmosphere of nitrogen. The mixture was cooled to ambient temperature and concentrated under reduced pressure. The residue was diluted with water (20 mL) and extracted with ethyl acetate (30 mL x 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether : ethyl acetate = 1 : 0 to 4 : 1) to afford 126a. LCMS: [M+1] = 339.1 Step B – Synthesis of methyl 2-fluoro-4-piperazin-1-yl-benzoate (126) A mixture of 126a (6.7 g, 19.8 mmol, 1.0 equiv.) in a solution of 4 M HCl in ethyl acetate was stirred at 25°C for 16 h. The reaction mixture was concentrated under reduced pressure to afford 126 as the hydrochloride salt, which was used without further purification. LCMS: [M+1] =239.1 Example 127: Synthesis of Compound 127
Step A – Synthesis of methyl 2-fluoro-4-[4-[[4-[3-methyl-4-[4-[[3-(2,2,2-trifluoro-1,1-dimethyl- ethyl)-1H-1,2,4-triazol-5-yl]methylcarbamoyl]pyrazol-1-yl]phenyl]phenyl]methyl]piperazin-1- yl]benzoate (127a) A solution of Int 55 (1.5 g, 3.0 mmol, 1.0 equiv.) in dichloromethane (25.5 mL) was treated with methyl 2-fluoro-4-piperazin-1-yl-benzoate hydrochloride (1.08 g, 3.9 mmol, 1.3 equiv.) and triethylamine (917.1 mg, 9.0 mmol, 1.2 mL, 3.0 equiv.). The mixture was stirred at 25°C for 2 h, treated with sodium triacetoxyborohydride (1.2 g, 6.0 mmol, 2.0 equiv.) and then stirred at 25°C for another 1 h. The mixture was treated with saturated NH4Cl (20 mL), diluted with water (30 mL) and extracted with ethyl acetate (20 mL x 3). The combined organic layers were washed with saturated NaCl (20 mL x3), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether: ethyl acetate = 1 : 0 to 35 : 65) to afford 127a. LCMS: [M+1] = 719.3 Step B – Synthesis of 2-fluoro-4-[4-[[4-[3-methyl-4-[4-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H-1,2,4-triazol-5-yl]methylcarbamoyl]pyrazol-1-yl]phenyl]phenyl]methyl]piperazin-1- yl]benzoic acid (127b) A solution of 127a (1.5 g, 2.0 mmol, 1.0 equiv.) in MeOH (1.7 mL) and THF (5.3 mL) was treated with 1 M aqueous LiOH·H2O (8.4 mL, 4.0 equiv.). The mixture was stirred at 25°C for 4 h. The mixture was concentrated under reduced pressure. The residue was diluted with water (30 mL), pH adjusted to 4 with HCl (1M) and filtered. The solid was collected by vacuum filtration and dried under vacuum to afford 127b. LCMS: [M+1] = 705.3 Step C – Synthesis of 1-[4-[4-[[4-[4-[[(3S)-2,6-dioxo-3-piperidyl]carbamoyl]-3-fluoro-phenyl]- piperazin-1-yl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H- 1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide hydrochloride (127) A solution of 127b (1.0g, 1.4 mmol, 1.0 equiv.) and (3S)-3-aminopiperidine-2,6-dione hydrochloride (256.9 mg, 1.5 mmol, 1.1 equiv.) in DMF (10 mL) was treated with DIEA (1.4 g, 11.3 mmol, 1.9 mL, 8.0 equiv.) at 0°C. The mixture was stirred at 0°C for 30 mins, and then T4P (2.0 g, 2.8 mmol, 50%, 2.0 equiv.) was added at 0°C. The resulting mixture was stirred at 25°C
for 1.5 h, quenched by addition water (30 mL) and extracted with ethyl acetate (30 mL x3). The combined organic layers were washed with saturated NH4Cl (20 mL x 3), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative HPLC (Phenomenex Luna C18, 100mm x 40mm, 5 µm [H2O(0.04% HCl)- ACN]), to afford Compound 127. LCMS [M+1] = 815.5; 1H NMR (400 MHz, DMSO-d6) δ ppm 13.92 (br s, 1 H), 10.85 (s, 2 H), 8.90 (br s, 1 H), 8.56 (s, 1 H), 8.09 - 8.22 (m, 2 H), 7.87 (d, J = 8.2 Hz, 2 H), 7.79 (s, 1 H), 7.62 - 7.75 (m, 4 H), 7.51 (d, J = 8.2 Hz, 1 H), 6.83 - 6.94 (m, 2 H), 4.66 - 4.80 (m, 1 H), 4.56 (br d, J = 5.6 Hz, 2 H), 4.44 (br s, 2 H), 4.05 (br d, J = 12.8 Hz, 2 H), 3.09 - 3.31 (m, 6 H), 2.71 - 2.85 (m, 1 H), 2.32 (s, 3 H), 1.93 - 2.20 (m, 2 H), 1.54 (s, 6 H). Example 128: Synthesis of Intermediate 128
 Step A – Synthesis of tert-butyl 4-(3-fluoro-4-methoxycarbonyl-phenyl)-3,6-dihydro-2H-pyridine- 1-carboxylate (Int 128) A mixture of tert-butyl 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-3,6-dihydro-2H- pyridine-1-carboxylate (5.0 g, 16.1 mmol, 0.9 eq), methyl 4-bromo-2-fluoro-benzoate (4.1 g, 17.9 mmol, 1.0 eq) in dioxane (50.0 mL) and water (10.0 mL) was treated with potassium carbonate (7.4 g, 53.9 mmol, 3.0 eq) and Pd(dppf)Cl2 (1.3 g, 1.8 mmol, 0.1 eq). The mixture was degassed and purged threefold with nitrogen and stirred at 80°C for 12 h under an atmosphere of nitrogen. The reaction was cooled to ambient temperature, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether: ethyl acetate=1: 0 to 10: 1) to afford 128a. LCMS [M-99] = 236.1 Step B – Synthesis of tert-butyl 4-(3-fluoro-4-methoxycarbonyl-phenyl)piperidine-1-carboxylate (128b)
A solution of 128a (5.2 g, 15.5 mmol, 1.0 eq) in MeOH (50.0 mL) was added Pd(OH)2 (2.1 g, 3.1 mmol, 20.0% purity, 0.2 eq) under N2 atmosphere. The suspension was degassed and purged with H2. The mixture was stirred under H2 (15 psi) at 25°C for 1 h. LCMS showed starting material was consumed completely and desired mass was detected. The reaction mixture was filtered and the filtrate was concentrated under reduced pressure to give (4.8 g, 13.6 mmol, 88.0% yield, 96.0% purity) as a white solid. LCMS [(M-56)+1] + = 282.1 Step C – Synthesis of methyl 2-fluoro-4-(4-piperidyl)benzoate (128) A solution of 128b (2.0 g, 5.9 mmol, 1.0 eq) in HCl/dioxane (4.0 M, 20.0 mL) was stirred at 25°C for 1 h. The mixture was concentrated under reduced pressure to afford Intermediate 128 as the hydrochloride salt, which was used without further purification. LCMS [M+1] = 238.2 Example 129: Synthesis of Compound 129
Step A – Synthesis of tert-butyl 4-(3-fluoro-4-methoxycarbonyl-phenyl)-3,6-dihydro-2H-pyridine- 1-carboxylate (Int 128) A mixture of tert-butyl 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-3,6-dihydro-2H- pyridine-1-carboxylate (5.0 g, 16.1 mmol, 0.9 eq), methyl 4-bromo-2-fluoro-benzoate (4.1 g, 17.9 mmol, 1.0 eq) in dioxane (50.0 mL) and water (10.0 mL) was treated with potassium carbonate (7.4 g, 53.9 mmol, 3.0 eq) and Pd(dppf)Cl2 (1.3 g, 1.8 mmol, 0.1 eq). The mixture was degassed and purged threefold with nitrogen and stirred at 80°C for 12 h under an atmosphere of nitrogen. The reaction was cooled to ambient temperature, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether: ethyl acetate=1: 0 to 10: 1) to afford 128a. LCMS [M-99] = 236.1 Step B – Synthesis of tert-butyl 4-(3-fluoro-4-methoxycarbonyl-phenyl)piperidine-1-carboxylate (128b)
A solution of 128a (5.2 g, 15.5 mmol, 1.0 eq) in MeOH (50.0 mL) was added Pd(OH)2 (2.1 g, 3.1 mmol, 20.0% purity, 0.2 eq) under N2 atmosphere. The suspension was degassed and purged with H2. The mixture was stirred under H2 (15 psi) at 25°C for 1 h. LCMS showed starting material was consumed completely and desired mass was detected. The reaction mixture was filtered and the filtrate was concentrated under reduced pressure to give (4.8 g, 13.6 mmol, 88.0% yield, 96.0% purity) as a white solid. LCMS [(M-56)+1] + = 282.1 Step C – Synthesis of methyl 2-fluoro-4-(4-piperidyl)benzoate (128) A solution of 128b (2.0 g, 5.9 mmol, 1.0 eq) in HCl/dioxane (4.0 M, 20.0 mL) was stirred at 25°C for 1 h. The mixture was concentrated under reduced pressure to afford Intermediate 128 as the hydrochloride salt, which was used without further purification. LCMS [M+1] = 238.2 Example 129: Synthesis of Compound 129
Step A – Synthesis of methyl 2-fluoro-4-[1-[[4-[3-methyl-4-[4-[[3-(2,2,2-trifluoro-1,1-dimethyl- ethyl)-1H-1,2,4-triazol-5-l]methylcarbamoyl]pyrazol-1-yl]phenyl]phenyl]methyl]-4- piperidyl]benzoate (129a) A solution of methyl 2-fluoro-4-(4-piperidyl)benzoate hydrochloride (200.0 mg, 843 μmol, 1.0 eq) and Int 55 (418.5 mg, 843 μmol, 1.0 eq) in THF (2.0 mL) was treated with Ti(Oi- Pr)4 (498 μL,1.6 mmol, 2.0 eq) and stirred at 60°C for 1 h. The mixture was cooled to ambient temperature and NaBH(OAc)3 (535.9 mg, 2.5 mmol, 3.0 eq) was added. The mixture was stirred at 25°C for 12 h, diluted with water (3.0 mL) and extracted with ethyl acetate (3.0 mL x 3). The combined organic phase was washed with brine (3.0 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure to afford 129a, which was used without further purification. LCMS [M+1] =718.4 Step B – Synthesis of 2-fluoro-4-[1-[[4-[3-methyl-4-[4-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H-1,2,4-triazol-5-yl]methylcarbamoyl]pyrazol-1-yl]phenyl]phenyl]methyl]-4-piperidyl]benzoic acid (129b) A solution of 129a (600.0 mg, 835.9 μmol, 1.0 eq) in methanol (2.0 mL), THF (2.0 mL) and water (2.0 mL) was treated with LiOH (40.0 mg, 1.6 mmol, 2.0 eq). The mixture was stirred at 25°C for 3 h and then concentrated under reduced pressure to afford 129b, which was used without further purification. LCMS [M+1] = 704.5 Step C – Synthesis of 1-[4-[4-[[4-[4-[[(3S)-2,6-dioxo-3-piperidyl]carbamoyl]-3-fluoro-phenyl]- 1-piperidyl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4- triazol-5-yl]methyl]pyrazole-4-carboxamide (129) A mixture of 129b (200.0 mg, 284.2 μmol, 1.0 eq) and (3S)-3-aminopiperidine-2,6-dione hydrochloride (40.0 mg, 312.6 μmol, 1.1 eq, HCl) in DMF (2.0 mL) was treated with EDCI (136.2 mg, 710.5 μmol, 2.5 eq), HOBt (57.6 mg, 426.3 μmol, 1.5 eq) and DIEA (148.5 μL, 852.6 μmol, , 3.0 eq). The mixture was degassed and purged threefold with nitrogen and then stirred at 25°C for 12 h under an atmosphere of nitrogen. The residue was purified by preparative HPLC (Phenomenex Luna C18, 100 mm x 40mm, 5 µm; [H2O(0.04% HCl)-ACN]) to afford Compound 129. LCMS [M +1] = 814.6. 1H NMR (400 MHz, DMSO-d6) δ 10.86 (br s, 1H), 9.99 (br s, 1H), 8.89 (br d, J = 1.0 Hz, 1H), 8.55 (s, 1H), 8.50 (dd, J = 3.2, 8.1 Hz, 1H), 8.19 (s, 1H), 7.88 (d, J =
8.4 Hz, 2H), 7.79 (s, 1H), 7.70 (br d, J = 8.4 Hz, 3H), 7.68 - 7.64 (m, 1H), 7.51 (d, J = 8.2 Hz, 1H), 7.20 (s, 1H), 7.17 (d, J = 3.6 Hz, 1H), 4.80 - 4.71 (m, 1H), 4.56 (br d, J = 5.4 Hz, 2H), 4.41 (br d, J = 4.4 Hz, 2H), 3.51 (br d, J = 10.8 Hz, 3H), 3.16 - 3.04 (m, 2H), 2.97 - 2.89 (m, 1H), 2.83 - 2.73 (m, 1H), 2.55 - 2.53 (m, 2H), 2.32 (s, 3H), 2.08 (br d, J = 3.8 Hz, 1H), 2.04 - 1.97 (m, 3H), 1.54 (s, 6H) Example 130: Synthesis of Intermediate 130 Step A
 A solution of tert-butyl 4-(3-fluoro-4-(methoxycarbonyl)phenyl)piperidine-1-carboxylate (9.6 g, 28.4 mmol, 1.0 equiv.) in dichloromethane (100.0 mL) was treated with trifluoroacetic acid (10.6 mL, 142.3 mmol, 5.0 equiv.). The mixture was stirred at 25°C for 16 h. The reaction mixture was concentrated under reduced pressure to afford 130a as the trifluoroacetate salt, which was used without further purification. LCMS [M+1] = 238.0. Step B – Synthesis of 2-fluoro-4-(piperidin-4-yl)benzoic acid (Int 130) To a solution of 130a (500.0 mg, 1.4 mmol, 1.0 equiv.) in THF (6.0 mL), MeOH (2.0 mL) and water (2.0 mL) treated with LiOH.H2O (298.6 mg, 7.1 mmol, 5.0 equiv.). The mixture was stirred at 25°C for 1 h. The reaction mixture was concentrated under reduced pressure to afford Int 130, which was used without further purification.LCMS [M+1] =224.0. Example 131: Synthesis of Compound 131
A solution of tert-butyl 4-(3-fluoro-4-(methoxycarbonyl)phenyl)piperidine-1-carboxylate (9.6 g, 28.4 mmol, 1.0 equiv.) in dichloromethane (100.0 mL) was treated with trifluoroacetic acid (10.6 mL, 142.3 mmol, 5.0 equiv.). The mixture was stirred at 25°C for 16 h. The reaction mixture was concentrated under reduced pressure to afford 130a as the trifluoroacetate salt, which was used without further purification. LCMS [M+1] = 238.0. Step B – Synthesis of 2-fluoro-4-(piperidin-4-yl)benzoic acid (Int 130) To a solution of 130a (500.0 mg, 1.4 mmol, 1.0 equiv.) in THF (6.0 mL), MeOH (2.0 mL) and water (2.0 mL) treated with LiOH.H2O (298.6 mg, 7.1 mmol, 5.0 equiv.). The mixture was stirred at 25°C for 1 h. The reaction mixture was concentrated under reduced pressure to afford Int 130, which was used without further purification.LCMS [M+1] =224.0. Example 131: Synthesis of Compound 131
Step A – Synthesis of 2-fluoro-4-(1-(4-(6-(4-(((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)carbamoyl)-1H-pyrazol-1-yl)pyridin-3-yl)benzyl)piperidin-4-yl)benzoic acid (131a) A solution of Int 130 (36.9 mg) and Int 119 (40.0 mg) in THF (1.0 mL) was treated with Ti(OEt)4 (172 μL). The mixture was stirred at 70°C for 1 h, and then treated with NaBH3CN (7.8 mg) at 25°C. The resulting mixture was stirred at 70°C for 1 h. The mixture was cooled to 25°C and quenched with water (1 mL) and acetonitrile (3 mL) at 25°C. The mixture was filtered and the filtrate was concentrated under reduced pressure to afford 131a, which was used without further purification. LCMS [M+1] = 691.3. Step B – Synthesis of (S)-N-((3-(1,1-difluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)- 1-(5-(4-((4-(4-((2,6-dioxopiperidin-3-yl)carbamoyl)-3-fluorophenyl)piperidin-1- yl)methyl)phenyl)pyridin-2-yl)-1H-pyrazole-4-carboxamide (Compound 131) A solution of 131a (20.0 mg) and (3S)-3-aminopiperidine-2,6-dione hydrochloride (9.5 mg) in DMF (2.0 mL) was treated with NMI (23.8 mg, 289.6 μmol, 23.1 μL, 10.0 equiv.) and TCFH (16.2 mg, 57.9 μmol, 2.0 equiv.). The mixture was stirred at 25°C for 1 h. The reaction mixture was diluted with water (5 mL) and extracted with ethyl acetate (10 mL x 3). The combined organic layers were washed with brine (10 mL x 2), dried over anhydrous sodium sulfate, filtered
and concentrated under reduced pressure. The residue was purified by preparative HPLC (Phenomenex Luna C18, 150mm x 25mm, 10 µm [water(FA)-ACN]) to afford Compound 131. LCMS [M+1] = 801.4. 1H NMR (400 MHz, DMSO-d6) δ 13.92 (br s, 1H), 10.84 (s, 1H), 9.22 (s, 1H), 9.05 (br s, 1H), 8.86 (d, J = 2.0 Hz, 1H), 8.46 (dd, J = 3.2, 8.0 Hz, 1H), 8.34 (dd, J = 2.4, 8.4 Hz, 1H), 8.22 (s, 1H), 8.14 (s, 1H), 8.03 (d, J = 8.4 Hz, 1H), 7.77 (d, J = 8.4 Hz, 2H), 7.62 (t, J = 8.0 Hz, 1H), 7.49 (d, J = 8.0 Hz, 2H), 7.21 (d, J = 10.0 Hz, 2H), 4.80 - 4.69 (m, 1H), 4.55 (br d, J = 5.2 Hz, 2H), 3.60 (br s, 2H), 2.97 (br d, J = 11.2 Hz, 2H), 2.82 - 2.73 (m, 1H), 2.65 - 2.58 (m, 1H), 2.54 (br d, J = 4.0 Hz, 1H), 2.16 - 2.06 (m, 3H), 2.05 - 1.97 (m, 1H), 1.81 - 1.75 (m, 2H), 1.74 - 1.64 (m, 2H), 1.54 (s, 6H). Example 132: Synthesis of Compound 132
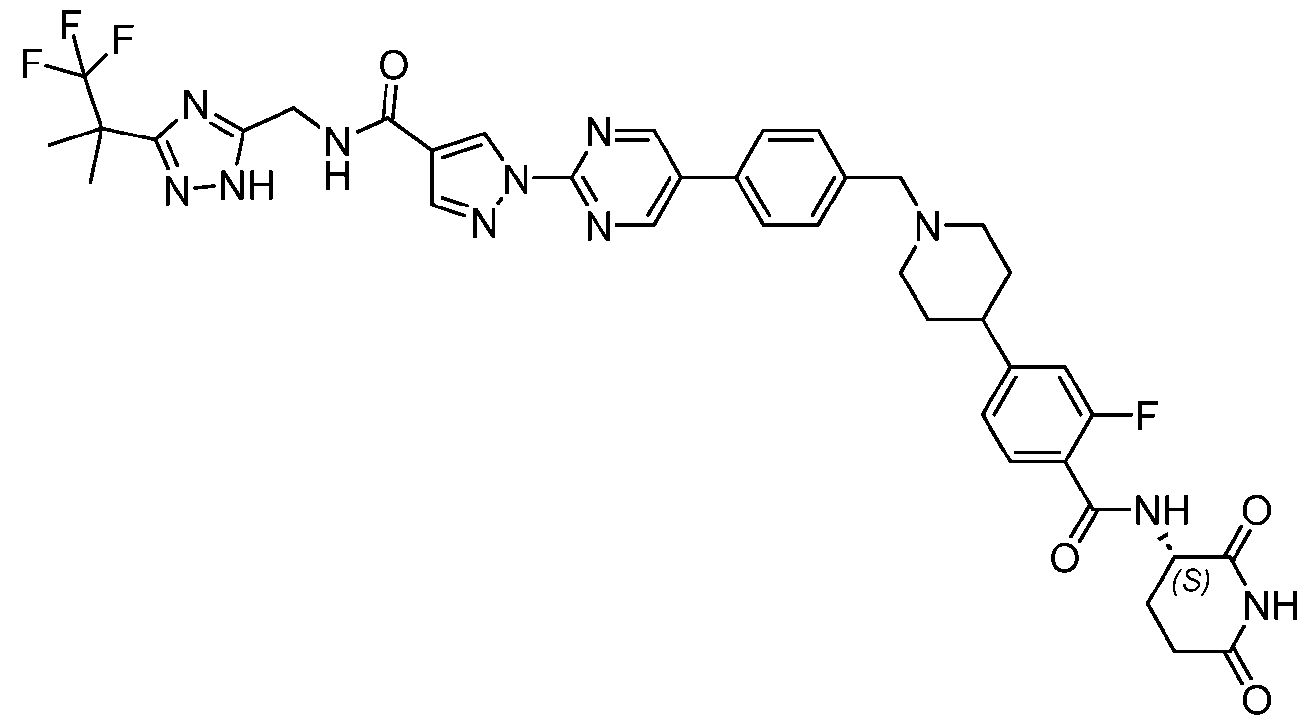 Int 115 in Step A to afford 1-[5-[4-[[4-[4-[[(3S)-2,6-dioxo-3-piperidyl]carbamoyl]-3-fluoro-phenyl]-1- piperidyl]methyl]phenyl]pyrimidin-2-yl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4- triazol-5-yl]methyl]pyrazole-4-carboxamide. LCMS [M+1] = 802.4; 1H NMR (400 MHz, METHANOL-d4) δ 9.26 (s, 1H), 9.16 (s, 2H), 8.29 (s, 1H), 7.86 (d, J = 8.4 Hz, 2H), 7.79 (d, J = 8.0 Hz, 1H), 7.66 (d, J = 8.4 Hz, 2H), 7.22 (d, J = 8.4 Hz, 1H), 7.16 (d, J = 12.0 Hz, 1H), 4.71 (s, 3H), 4.10 - 4.06 (m, 2H), 2.87 - 2.81 (m, 2H), 2.79 (d, J = 5.6 Hz, 1H), 2.75 - 2.71 (m, 2H), 2.40 - 2.24 (m, 2H), 2.23 - 2.11 (m, 2H), 2.05 (s, 1H), 2.02 (s, 1H), 1.93 - 1.86 (m, 2H), 1.62 (s, 6H). Example 133: Synthesis of Compound 133
Int 115 in Step A to afford 1-[5-[4-[[4-[4-[[(3S)-2,6-dioxo-3-piperidyl]carbamoyl]-3-fluoro-phenyl]-1- piperidyl]methyl]phenyl]pyrimidin-2-yl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4- triazol-5-yl]methyl]pyrazole-4-carboxamide. LCMS [M+1] = 802.4; 1H NMR (400 MHz, METHANOL-d4) δ 9.26 (s, 1H), 9.16 (s, 2H), 8.29 (s, 1H), 7.86 (d, J = 8.4 Hz, 2H), 7.79 (d, J = 8.0 Hz, 1H), 7.66 (d, J = 8.4 Hz, 2H), 7.22 (d, J = 8.4 Hz, 1H), 7.16 (d, J = 12.0 Hz, 1H), 4.71 (s, 3H), 4.10 - 4.06 (m, 2H), 2.87 - 2.81 (m, 2H), 2.79 (d, J = 5.6 Hz, 1H), 2.75 - 2.71 (m, 2H), 2.40 - 2.24 (m, 2H), 2.23 - 2.11 (m, 2H), 2.05 (s, 1H), 2.02 (s, 1H), 1.93 - 1.86 (m, 2H), 1.62 (s, 6H). Example 133: Synthesis of Compound 133
Compound 133 was prepared in a similar manner to Example 131 by substituting Int 121e in Step A to afford 1-[5-[4-[[4-[5-(2,6-dioxo-3-piperidyl)-3-fluoro-2-pyridyl]-1- piperidyl]methyl]phenyl]pyrimidin-2-yl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4- triazol-5-yl]methyl]pyrazole-4-carboxamide. LCMS [M+1] = 803.4. 1HNMR (400 MHz, METHANOL-d4) δ 9.47 (s, 2H), 9.28 (s, 1H), 8.72 (s, 1H), 8.29 (s, 1H), 8.08 - 7.98 (m, 2H), 7.79 - 7.75 (m, 1H), 7.21 (br d, J = 8.0 Hz, 1H), 7.14 (br d, J = 12.8 Hz, 1H), 4.71 (s, 2H), 3.71 (s, 2H), 3.07 (br d, J = 10.8 Hz, 2H), 2.80 (br dd, J = 5.2,
 Hz, 1H), 2.74 - 2.67 (m, 2H), 2.30 - 2.26 (m, 2H), 2.23 (br d, J = 1.6 Hz, 1H), 2.16 (br dd, J = 4.8, 13.6 Hz, 1H), 1.92 - 1.85 (m, 3H), 1.84 - 1.77 (m, 2H), 1.62 (s, 6H). Example 134: Synthesis of Compound 134
Hz, 1H), 2.74 - 2.67 (m, 2H), 2.30 - 2.26 (m, 2H), 2.23 (br d, J = 1.6 Hz, 1H), 2.16 (br dd, J = 4.8, 13.6 Hz, 1H), 1.92 - 1.85 (m, 3H), 1.84 - 1.77 (m, 2H), 1.62 (s, 6H). Example 134: Synthesis of Compound 134
F F F O O B F F O NaIO4 N O F K N 2OsO4 O NH O 1H-
 1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide (134a) A mixture of 1-(4-bromo-2-methylphenyl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)- 1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide (500.0 mg, 1.1 mmol, 1.0 equiv.), 4,4,5,5-tetramethyl-2-vinyl-1,3,2-dioxaborolane (270 μL, 1.6 mmol, 1.5 equiv.), Pd(dppf)Cl2.CH2Cl2 (86.6 mg, 106.1 μmol, 0.1 equiv.), K2CO3 (439.9 mg, 3.2 mmol, 3.0 equiv.) in dioxane (5.0 mL) and water (1.0 mL) was degassed and purged threefold with nitrogen. The mixture was stirred at 100°C for 1 h under an atmosphere of nitrogen. The mixture was cooled to 25°C and poured into water (40 mL). The aqueous phase was extracted with ethyl acetate (40 mL x 3) and the combined organic phase was washed with brine (40 mL x 3), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum. The residue was purified by column chromatography (silica gel, petroleum ether/ethyl acetate = 10/1 to 2/1) to afford 134a. LCMS [M+1] = 418.9 Step B – Synthesis of 1-(4-formyl-2-methylphenyl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)- 1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide (134b) A solution of 134a (250.0 mg, 598 μmol, 1.0 equiv.), NaIO4 (511.2 mg, 2.4 mmol, 4.0 equiv.), K2OsO4 (22.0 mg, 60 μmol, 0.1 equiv.) and 2,6-dimethylpyridine (128.0 mg, 1.19 mmol, 139.18 μL, 2.0 equiv.) in dioxane (2.5 mL) and water (1.25 mL). The reaction was stirred at 25°C for 0.5 h. The residue was poured into water (30 mL) and stirred for 2 min. The aqueous phase
was extracted with ethyl acetate (30 mL x 3). The combined organic phase was washed with brine (30 mL x 3), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum to afford 134b, which was used without further purification. LCMS [M+1] = 420.9 Step C – Synthesis of 1-(4-((4-(5-(2,6-dioxopiperidin-3-yl)-3-fluoropyridin-2-yl)piperazin-1- yl)methyl)-2-methylphenyl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5- yl)methyl)-1H-pyrazole-4-carboxamide (134) A solution of 134b (60.0 mg, 143 μmol, 1.0 equiv.) was treated with Ti(OEt)4 (148 μL, 714 μmol, 5.0 equiv.) and 3-(5-fluoro-6-(piperazin-1-yl)pyridin-3-yl)piperidine-2,6-dione hydrochloride (62.6 mg, 214.1 μmol, 1.5 equiv) in THF (1.0 mL) and DMSO (0.34 mL). The mixture was stirred at 50°C for 7.5 h, treated with NaBH3CN (60.5 mg, 286 μmol, 2.0 equiv.) at 25°C and stirred at 25°C for 0.5 h. The mixture was quenched with water (0.5 ml), filtered and the filtrate was concentrated. The residue was purified by preparative HPLC (Phenomenex Luna C18, 150 mm x 25mm, 10µm; [water(FA)-ACN]) to afford 134. LCMS [M+1] = 697.3 1H NMR (400 MHz, METHANOL-d4) δ 8.32 (s, 1H), 8.16 (s, 1H), 7.89 (s, 1H), 7.45 (s, 1H), 7.42 - 7.30 (m, 3H), 4.69 (s, 2H), 3.87 (dd, J = 5.2, 12.1 Hz, 1H), 3.72 (s, 2H), 3.52 (s, 4H), 2.83 - 2.74 (m, 1H), 2.74 - 2.70 (m, 4H), 2.70 - 2.62 (m, 1H), 2.27 (d, J = 5.2 Hz, 1H), 2.24 (s, 3H), 2.17 (ddd, J = 4.4, 9.2, 13.6 Hz, 1H), 1.61 (s, 6H). Example 135: Synthesis of Compound 135
1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide (134a) A mixture of 1-(4-bromo-2-methylphenyl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)- 1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide (500.0 mg, 1.1 mmol, 1.0 equiv.), 4,4,5,5-tetramethyl-2-vinyl-1,3,2-dioxaborolane (270 μL, 1.6 mmol, 1.5 equiv.), Pd(dppf)Cl2.CH2Cl2 (86.6 mg, 106.1 μmol, 0.1 equiv.), K2CO3 (439.9 mg, 3.2 mmol, 3.0 equiv.) in dioxane (5.0 mL) and water (1.0 mL) was degassed and purged threefold with nitrogen. The mixture was stirred at 100°C for 1 h under an atmosphere of nitrogen. The mixture was cooled to 25°C and poured into water (40 mL). The aqueous phase was extracted with ethyl acetate (40 mL x 3) and the combined organic phase was washed with brine (40 mL x 3), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum. The residue was purified by column chromatography (silica gel, petroleum ether/ethyl acetate = 10/1 to 2/1) to afford 134a. LCMS [M+1] = 418.9 Step B – Synthesis of 1-(4-formyl-2-methylphenyl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)- 1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide (134b) A solution of 134a (250.0 mg, 598 μmol, 1.0 equiv.), NaIO4 (511.2 mg, 2.4 mmol, 4.0 equiv.), K2OsO4 (22.0 mg, 60 μmol, 0.1 equiv.) and 2,6-dimethylpyridine (128.0 mg, 1.19 mmol, 139.18 μL, 2.0 equiv.) in dioxane (2.5 mL) and water (1.25 mL). The reaction was stirred at 25°C for 0.5 h. The residue was poured into water (30 mL) and stirred for 2 min. The aqueous phase
was extracted with ethyl acetate (30 mL x 3). The combined organic phase was washed with brine (30 mL x 3), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum to afford 134b, which was used without further purification. LCMS [M+1] = 420.9 Step C – Synthesis of 1-(4-((4-(5-(2,6-dioxopiperidin-3-yl)-3-fluoropyridin-2-yl)piperazin-1- yl)methyl)-2-methylphenyl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5- yl)methyl)-1H-pyrazole-4-carboxamide (134) A solution of 134b (60.0 mg, 143 μmol, 1.0 equiv.) was treated with Ti(OEt)4 (148 μL, 714 μmol, 5.0 equiv.) and 3-(5-fluoro-6-(piperazin-1-yl)pyridin-3-yl)piperidine-2,6-dione hydrochloride (62.6 mg, 214.1 μmol, 1.5 equiv) in THF (1.0 mL) and DMSO (0.34 mL). The mixture was stirred at 50°C for 7.5 h, treated with NaBH3CN (60.5 mg, 286 μmol, 2.0 equiv.) at 25°C and stirred at 25°C for 0.5 h. The mixture was quenched with water (0.5 ml), filtered and the filtrate was concentrated. The residue was purified by preparative HPLC (Phenomenex Luna C18, 150 mm x 25mm, 10µm; [water(FA)-ACN]) to afford 134. LCMS [M+1] = 697.3 1H NMR (400 MHz, METHANOL-d4) δ 8.32 (s, 1H), 8.16 (s, 1H), 7.89 (s, 1H), 7.45 (s, 1H), 7.42 - 7.30 (m, 3H), 4.69 (s, 2H), 3.87 (dd, J = 5.2, 12.1 Hz, 1H), 3.72 (s, 2H), 3.52 (s, 4H), 2.83 - 2.74 (m, 1H), 2.74 - 2.70 (m, 4H), 2.70 - 2.62 (m, 1H), 2.27 (d, J = 5.2 Hz, 1H), 2.24 (s, 3H), 2.17 (ddd, J = 4.4, 9.2, 13.6 Hz, 1H), 1.61 (s, 6H). Example 135: Synthesis of Compound 135
Step A – Synthesis of tert-butyl 4-[[3-methyl-4-[4-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H- 1,2,4-triazol-5-yl]methylcarbamoyl]pyrazol-1-yl]phenyl]methylene]piperidine-1-carboxylate (135a) A stirred suspension of tert-butyl 4-[(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)methylene]piperidine-1-carboxylate (2.0 g, 6.2 mmol, 1.0 equiv.) and 1-(4-bromo-2-methyl- phenyl)-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4- carboxamide (3.5 g, 7.4 mmol, 1.2 equiv.) in dioxane (20.0 mL) and water (7.0 mL) was treated with K3PO4 (3.9 g, 18.6 mmol, 3.0 equiv.) and Pd(dppf)Cl2 (452.7 mg, 619 μmol, 0.1 equiv.), the mixture was stirred at 90°C for 1 h under an atmosphere of nitrogen. The reaction mixture was diluted with water (25.0 mL) and extracted with ethyl acetate (25.0 mL x 3). The combined organic layers were washed with brine (25.0 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative HPLC (Phenomenex Luna C18, 250 x 70mm, 15 µm); [H2O(0.2%FA)-ACN] to afford 135a. LCMS [M+1] = 588.5.
Step B – Synthesis of tert-butyl 4-[[3-methyl-4-[4-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H- 1,2,4-triazol-5-yl]methylcarbamoyl]pyrazol-1-yl]phenyl]methyl]piperidine-1-carboxylate (135b) A mixture of Pd/C (1.0 g, 10% purity) and 135a (1.0 g, 1.7 mmol, 1.0 equiv.) in THF (15.0 mL) stirred under hydrogen (15 psi) at 25°C for 5 h. The mixture was filtered and the filtrate was concentrated under reduced pressure to afford 135b, which was used without further purification. LCMS [M+1] = 590.4 Step C – Synthesis of 1-[2-methyl-4-(4-piperidylmethyl)phenyl]-N-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (135c) A solution of 135b (980.0 mg, 1.7 mmol, 1.0 equiv.) in dichloromethane (9.0 mL) was treated with trifluoroacetic acid (3.0 mL). The mixture was stirred at 25°C for 2 h. The resulting mixture was concentrated under reduced pressure to afford 135c as the trifluoroacetate salt, which was used without further purification. LCMS [M+1] = 490.4. Step D– Synthesis of 1-[4-[[1-[5-(2,6-dioxo-3-piperidyl)-3-fluoro-2-pyridyl]-4-piperidyl]methyl]- 2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methyl]pyrazole-4-carboxamide (135) A solution of 135c (100.0 mg, 204 μmol, 1.0 equiv.) in DMSO (1 mL) was treated with DIEA (105.6 mg, 817.1 μmol, 142.32 μL, 4.0 equiv.) and 3-(5,6-difluoro-3-pyridyl)piperidine- 2,6-dione (55.4 mg, 245 μmol, 1.2 equiv.). The mixture was stirred at 120°C for 8 h, cooled to ambient temperature, treated with acetonitrile (0.5 mL) and filtered. The filtrate was purified by preparative HPLC (Phenomenex Luna C18, 100mm x 40mm, 5 µm; [H2O (0.2% FA)-ACN]) to afford 135. LCMS [M+1] = 696.5. 1H NMR (400 MHz, DMSO-d6) δ = 13.87 (br s, 1H), 10.84 (s, 1H), 8.85 (br s, 1H), 8.43 (s, 1H), 8.12 (s, 1H), 7.85 (s, 1H), 7.42 (dd, J = 1.6, 14.0 Hz, 1H), 7.28 (d, J = 8.0 Hz, 1H), 7.24 (br s, 1H), 7.20 - 7.13 (m, 1H), 4.54 (br s, 2H), 3.93 (br d, J = 12.8 Hz, 2H), 3.83 (dd, J = 4.8, 12.4 Hz, 1H), 3.37 - 3.33 (m, 2H), 2.78 (t, J = 11.6 Hz, 2H), 2.70 - 2.63 (m, 1H), 2.58 (d, J = 7.2 Hz, 2H), 2.54 (d, J = 4.0 Hz, 1H), 2.27 - 2.21 (m, 1H), 2.18 (s, 3H), 2.03 - 1.94 (m, 1H), 1.77-1.79 (m, 1H), 1.68 (d, J = 11.8 Hz, 2H), 1.53 (s, 6H), 1.38 - 1.25 (m, 2H)
Example 136: Synthesis of Intermediate 136 Step
 A solution of 5-bromo-2,3-difluoro-pyridine (2 g, 10.31 mmol, 1 equiv.) in DMF (20 mL) was treated with piperidin-4-ol (1.15 g, 11.34 mmol, 1.1 equiv.) and DIEA (2.67 g, 20.62 mmol, 3.59 mL, 2 equiv.) at 25°C. The mixture was stirred at 80°C for 2 h, cooled to ambient temperature, diluted with water (20 mL) and extracted with ethyl acetate (20 mL x 3). The combined organic phases were washed with brine (20 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum to afford 136a, which was used without further purification. LCMS: [M+1] = 275.2, 277.2 Step B – Synthesis of 1-[5-(2, 6-dibenzyloxy-3-pyridyl)-3-fluoro- 2-pyridyl] piperidin-4-ol (136) A solution of 1-(5-bromo-3-fluoro-2-pyridyl)piperidin-4-ol (2.2 g, 8.00 mmol, 1 equiv.) in dioxane (20 mL) and water (4 mL) was treated with 2,6-dibenzyloxy-3-(4,4,5,5-tetramethyl- 1,3,2-dioxaborolan-2-yl)pyridine (4.00 g, 9.60 mmol, 1.2 equiv.), K2CO3 (3.32 g, 23.99 mmol, 3 equiv.) and Pd(dppf)Cl2 (585.12 mg, 799.66 μmol, 0.1 equiv.) at 25°C. The mixture was degassed and purged threefold with nitrogen and stirred at 80°C for 12 h under a nitrogen atmosphere. The reaction mixture was diluted with water (25 mL) and the resulting mixture was extracted with ethyl acetate (25 mL x 3). The combined organic phases were washed with brine (25 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum to afford a yellow residue. The residue was purified by column chromatography (silica gel, petroleum ether: ethyl acetate = 100: 1 to 100: 50) to afford Intermediate 136. LCMS: [M+1] = 486.3
Example 137: Synthesis of Intermediate 137
A solution of 5-bromo-2,3-difluoro-pyridine (2 g, 10.31 mmol, 1 equiv.) in DMF (20 mL) was treated with piperidin-4-ol (1.15 g, 11.34 mmol, 1.1 equiv.) and DIEA (2.67 g, 20.62 mmol, 3.59 mL, 2 equiv.) at 25°C. The mixture was stirred at 80°C for 2 h, cooled to ambient temperature, diluted with water (20 mL) and extracted with ethyl acetate (20 mL x 3). The combined organic phases were washed with brine (20 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum to afford 136a, which was used without further purification. LCMS: [M+1] = 275.2, 277.2 Step B – Synthesis of 1-[5-(2, 6-dibenzyloxy-3-pyridyl)-3-fluoro- 2-pyridyl] piperidin-4-ol (136) A solution of 1-(5-bromo-3-fluoro-2-pyridyl)piperidin-4-ol (2.2 g, 8.00 mmol, 1 equiv.) in dioxane (20 mL) and water (4 mL) was treated with 2,6-dibenzyloxy-3-(4,4,5,5-tetramethyl- 1,3,2-dioxaborolan-2-yl)pyridine (4.00 g, 9.60 mmol, 1.2 equiv.), K2CO3 (3.32 g, 23.99 mmol, 3 equiv.) and Pd(dppf)Cl2 (585.12 mg, 799.66 μmol, 0.1 equiv.) at 25°C. The mixture was degassed and purged threefold with nitrogen and stirred at 80°C for 12 h under a nitrogen atmosphere. The reaction mixture was diluted with water (25 mL) and the resulting mixture was extracted with ethyl acetate (25 mL x 3). The combined organic phases were washed with brine (25 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum to afford a yellow residue. The residue was purified by column chromatography (silica gel, petroleum ether: ethyl acetate = 100: 1 to 100: 50) to afford Intermediate 136. LCMS: [M+1] = 486.3
Example 137: Synthesis of Intermediate 137
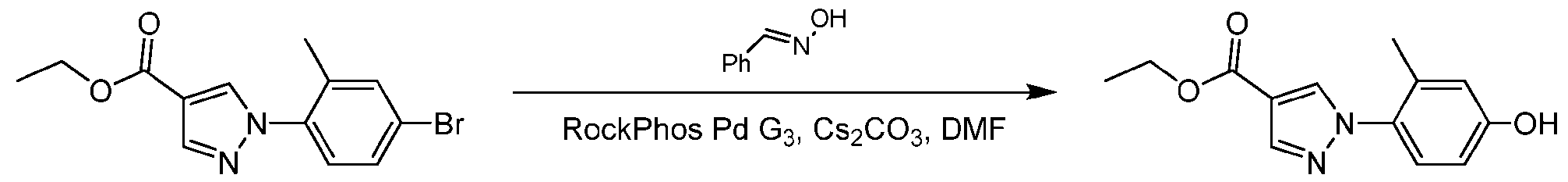 was - g, equiv.), Cs2CO3 (5.80 g, 17.79 mmol, 2.2 equiv.) and RockPhos Pd G3 (677.99 mg, 808.65 μmol, 0.1 equiv.) at 25°C. The mixture was degassed and purged threefold with nitrogen and stirred at 80°C for 12 h under an atmosphere of nitrogen. The reaction mixture was diluted with water (25 mL) and the resulting mixture was extracted with ethyl acetate (20 mL x 3). The combined organic phases were washed with brine (20 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum. The residue was purified by column chromatography (silica gel, petroleum ether: ethyl acetate = 100: 1 to 100:10) to afford ethyl 1-(4-hydroxy- 2-methyl-phenyl) pyrazole-4-carboxylate (Intermediate 137). LCMS: [M+1] = 247.2 Example 138: Synthesis of Compound 138
was - g, equiv.), Cs2CO3 (5.80 g, 17.79 mmol, 2.2 equiv.) and RockPhos Pd G3 (677.99 mg, 808.65 μmol, 0.1 equiv.) at 25°C. The mixture was degassed and purged threefold with nitrogen and stirred at 80°C for 12 h under an atmosphere of nitrogen. The reaction mixture was diluted with water (25 mL) and the resulting mixture was extracted with ethyl acetate (20 mL x 3). The combined organic phases were washed with brine (20 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum. The residue was purified by column chromatography (silica gel, petroleum ether: ethyl acetate = 100: 1 to 100:10) to afford ethyl 1-(4-hydroxy- 2-methyl-phenyl) pyrazole-4-carboxylate (Intermediate 137). LCMS: [M+1] = 247.2 Example 138: Synthesis of Compound 138
O OH O N O O NH O
 -4- piperidyl]oxy]-2-methyl-phenyl]pyrazole-4-carboxylate (138a) A solution of ethyl 1-(4-hydroxy-2-methyl-phenyl) pyrazole-4-carboxylate (700 mg, 2.84 mmol, 1 equiv.) in toluene. (10 mL) was treated with 1-[5-(2,6-dibenzyloxy-3-pyridyl) -3-fluoro- 2-pyridyl]piperidin-4-ol (1.38 g, 2.84 mmol, 1 equiv.) and triphenylphosphine (1.49 g, 5.69 mmol, 2 equiv.) at 25°C. The mixture was degassed and purged threefold with nitrogen and then cooled to 0°C. A solution of DIAD (1.15 g, 5.69 mmol, 1.10 mL, 2 equiv.) in toluene (10 mL) was added and the mixture was stirred at 80°C for 12 h. The mixture was cooled to ambient temperature and diluted with water (20 mL) and the resulting mixture was extracted with ethyl acetate (20 mL x 3). The combined organic phases were washed with brine (20 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum. The residue was purified by column chromatography (silica gel, petroleum ether : ethyl acetate = 100 : 1 to 100 : 30) to afford LCMS: [M+1] = 714.4
Step B – Synthesis of 1-[4-[[1-[5-(2,6-dibenzyloxy -3-pyridyl)-3-fluoro-2-pyridyl]-4- piperidyl]oxy]-2-methyl-phenyl]pyrazole-4-carboxylic acid (138b) A solution of 138a (750 mg, 1.05 mmol, 1 equiv.) in THF (7 mL) and water (1 mL) was treated with LiOH.H2O (176.4 mg, 4.20 mmol, 4 equiv.) at 25°C and the mixture was stirred at 25°C for 2 h. The reaction mixture was acidified to pH = 3 with aqueous HCl (1 M) and extracted with ethyl acetate (10 mL x 3). The combined organic phase was washed with brine (10 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum to afford 138b, which was used without further purification. LCMS: [M+1] = 686.4 Step C – Synthesis of 1-[4-[[1-[5-(2,6- dibenzyloxy-3-pyridyl)-3-fluoro-2-pyridyl]-4- piperidyl]oxy]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methyl]pyrazole-4-carboxamide (138c) A solution of 138b (700 mg, 1.02 mmol, 1 equiv.) in DMF (7 mL) was treated with [3- (2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methanamine hydrochloride (249.7 mg, 1.02 mmol, 1 equiv.), triethylamine (1.14 mL, 8.17 mmol, 8 equiv.) and BOP (677.2 mg, 1.53 mmol, 1.5 equiv.) at 25°C and the mixture was stirred at 25°C for 2 h. The mixture was diluted with water (0.5 mL) and extracted with ethyl acetate (0.5 mL x 3). The combined organic phases were washed with brine (5 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum. The residue was purified by column chromatography (silica gel, petroleum ether : ethyl acetate = 100 : 1 to 100 : 50) to afford 138c. LCMS: [M+1] = 876.5 Step D – Synthesis of 1-[4-[[1-[5-(2,6-dioxo- 3-piperidyl)-3-fluoro-2-pyridyl]-4-piperidyl]oxy]-2- methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole- 4-carboxamide (138) A suspension of Pd/C (200 mg, 10% purity) and Pd(OH)2/C (200 mg, 20 wt%) in ethyl acetate (3 mL) under a nitrogen atmosphere was treated with a solution of 138c (500 mg, 571 μmol, 1 equiv.) in THF (2 mL) at ambient temperature. The suspension was degassed and purged threefold with hydrogen. The mixture was stirred at 25 °C for 2 h under hydrogen (15 Psi). The reaction mixture was filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative HPLC (Phenomenex Luna C18, 100 mm x 40 mm, 5 µm; [H2O (0.04% HCl)-ACN]) to afford Compound 138 as the hydrochloride salt. LCMS: [M+1] = 698.4. 1H NMR
(400 MHz, DMSO-d6) δ 8.37 (s, 1H), 8.10 (s, 1H), 7.88 (s, 1H), 7.54 (dd, J = 1.8, 14.2 Hz, 1H), 7.26 (d, J = 8.8 Hz, 1H), 7.02 (d, J = 2.8 Hz, 1H), 6.95 (dd, J = 2.8, 8.8 Hz, 1H), 4.71 - 4.67 (m, 1H), 4.52 (s, 2H), 3.88 (dd, J = 4.8, 12.4 Hz, 1H), 3.78 -3.75 (m, 2H), 3.35 - 3.29 (m, 2H), 2.73 - 2.64 (m, 1H), 2.56 - 2.53 (m, 1H), 2.28 - 2.18 (m, 1H), 2.12 (s, 3H), 2.10 - 2.04 (m, 2H), 2.02 - 1.97 (m, 1H), 1.78 - 1.70 (m, 2H), 1.52 (s, 6H) Example 139: Synthesis of Compound 139 F F HO N O OBn O NH O
Step A – Synthesis of 2-azaspiro[3.5]nonan-7-ol hydrochloride (139a) A mixture of tert-butyl 7-hydroxy-2-azaspiro[3.5]nonane-2-carboxylate (900.0 mg, 3.20 mmol, 1.0 equiv.) in HCl/ dioxane (2 M, 7.8 mL, 4.8 equiv.) was stirred at 25°C for 0.5 h. The mixture was concentrated under reduced pressure to afford 139a as the hydrochloride salt. LCMS [M+1] = 142.2 Step B – Synthesis of 2-[5-(2,6-dibenzyloxy-3-pyridyl)-3-fluoro-2-pyridyl]-2-azaspiro[3.5]nonan- 7-ol (139b) A solution of 139a (553.5 mg, 3.1 mmol, 1.4 equiv.) and 2,6-dibenzyloxy-3-(5,6-difluoro- 3-pyridyl)pyridine (900.0 mg, 2.2 mmol, 1.0 equiv.) in DMSO (18.0 mL) was treated with DIEA (862.8 mg, 6.6 mmol, 1.1 mL, 3.0 equiv.). The mixture was stirred at 80°C for 16 h, cooled to ambient temperature and poured into water (60 mL). The aqueous phase was extracted with ethyl acetate (30 mL x 3). The combined organic phase was washed with brine (40 mL x 3), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether/ ethyl acetate=1/0 to 0/1) to afford 139b. LCMS [M+1] = 526.2 Step C – Synthesis of ethyl 1-[4-[[2-[5-(2,6-dibenzyloxy-3-pyridyl)-3-fluoro-2-pyridyl]-2- azaspiro[3.5]nonan-7-yl]oxy]-2-methyl-phenyl]pyrazole-4-carboxylate (139c) A solution of 139b (533.5 mg, 1.0 mmol, 1.0 equiv.) and ethyl 1-(4-hydroxy-2-methyl- phenyl)pyrazole-4-carboxylate (250.0 mg, 1.0 mmol, 1.0 equiv.) in toluene (6.0 mL) was treated with triphenylphosphine (532.5 mg, 2.0 mmol, 2.0 equiv.) and DIAD (410.5 mg, 2.0 mmol, 393.6 μL, 2.0 equiv.) at 0°C under an atmosphere of nitrogen. The mixture was stirred at 80°C for 16 h. The mixture was cooled to 25°C and poured into water (20 mL). The aqueous phase was extracted with ethyl acetate (10 mL x 3). The combined organic phase was washed with brine (10 mL x 3), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative HPLC (Phenomenex Luna C18, 250 mm x 50mm, 15 µm; [water(FA)-ACN]) to afford 139c. LCMS [M+1] = 754.3
Step D – Synthesis of 1-[4-[[2-[5-(2,6-dibenzyloxy-3-pyridyl)-3-fluoro-2-pyridyl]-2- azaspiro[3.5]nonan-7-yl]oxy]-2-methyl-phenyl]pyrazole-4-carboxylic acid (139d) A solution of 139c (600.0 mg, 795.9 μmol, 1.0 equiv.) in THF (6.0 mL), H2O (3.0 mL) and MeOH (1.5 mL) was treated with LiOH.H2O (66.8 mg, 1.5 mmol, 2.0 equiv.). The mixture was stirred at 25°C for 16 h. The mixture pH was adjusted with 1 M sulfuric acid, diluted with water (20 mL), and extracted with ethyl acetate (10 mL x 3). The combined organic layer was washed with brine (20 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure to afford 139c, which was used without further purification. LCMS [M+1] = 726.0 Step E – Synthesis of 1-[4-[[2-[5-(2,6-dibenzyloxy-3-pyridyl)-3-fluoro-2-pyridyl]-2- azaspiro[3.5]nonan-7-yl]oxy]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H- 1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (139e) A solution of 139d (400.0 mg, 551.1 μmol, 1.0 equiv.) and triethylamine (278.8 mg, 2.7 mmol, 383.5 μL, 5.0 equiv.) in DMF (4.0 mL) was treated with BOP (365.6 mg, 826.6 μmol, 1.5 equiv.) and [3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methanamine hydrochloride (134.8 mg, 551.1 μmol, 1.0 equiv., HCl salt). The mixture was stirred at 25°C for 1 h, poured into water (20 mL) and extracted with ethyl acetate (10 mL x 3). The combined organic phase was washed with brine (10 mL x 3), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether/ ethyl acetate=1/0 to 1/1) to afford 139e. LCMS [M+1] = 916.4 Step F – Synthesis of 1-[4-[[2-[5-(2,6-dioxo-3-piperidyl)-3-fluoro-2-pyridyl]-2- azaspiro[3.5]nonan-7-yl]oxy]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H- 1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (139) A mixture of 139e (390.0 mg, 425.7 μmol, 1.0 equiv.) and Pd/C (226.5 mg, 10 wt%) in THF (15.0 mL) was degassed and purged threefold with hydrogen. The mixture was stirred under hydrogen (50 psi) at 30°C for 4 h. The mixture was filtered at ambient temperature and the filtrate was concentrated under reduced pressure. The residue was purified by preparative HPLC (Phenomenex Luna C18, 150 mm x 25 mm, 10µm; [water(FA)-ACN]) to afford Compound 139.
LCMS [M+1] = 738.4. 1H NMR (400 MHz, DMSO-d6) δ 13.91 (br s, 1H), 10.83 (s, 1H), 8.87 - 8.78 (m, 1H), 8.38 (s, 1H), 8.10 (s, 1H), 7.77 (s, 1H), 7.36 (d, J = 13.6 Hz, 1H), 7.26 (d, J = 8.4 Hz, 1H), 6.98 (s, 1H), 6.93 - 6.90 (m, 1H), 4.54 (d, J = 5.2 Hz, 2H), 4.46 (s, 1H), 3.81 - 3.78 (m, 5H), 2.73 - 2.64 (m, 1H), 2.54 (d, J = 3.6 Hz, 1H), 2.22 (dq, J = 4.4, 12.8 Hz, 1H), 2.13 (s, 3H), 1.97 - 1.89 (m, 5H), 1.72 - 1.67 (m, 2H), 1.61 - 1.56 (m, 2H), 1.53 (s, 6H). Example 140: Synthesis of Compound 140
-4- piperidyl]oxy]-2-methyl-phenyl]pyrazole-4-carboxylate (138a) A solution of ethyl 1-(4-hydroxy-2-methyl-phenyl) pyrazole-4-carboxylate (700 mg, 2.84 mmol, 1 equiv.) in toluene. (10 mL) was treated with 1-[5-(2,6-dibenzyloxy-3-pyridyl) -3-fluoro- 2-pyridyl]piperidin-4-ol (1.38 g, 2.84 mmol, 1 equiv.) and triphenylphosphine (1.49 g, 5.69 mmol, 2 equiv.) at 25°C. The mixture was degassed and purged threefold with nitrogen and then cooled to 0°C. A solution of DIAD (1.15 g, 5.69 mmol, 1.10 mL, 2 equiv.) in toluene (10 mL) was added and the mixture was stirred at 80°C for 12 h. The mixture was cooled to ambient temperature and diluted with water (20 mL) and the resulting mixture was extracted with ethyl acetate (20 mL x 3). The combined organic phases were washed with brine (20 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum. The residue was purified by column chromatography (silica gel, petroleum ether : ethyl acetate = 100 : 1 to 100 : 30) to afford LCMS: [M+1] = 714.4
Step B – Synthesis of 1-[4-[[1-[5-(2,6-dibenzyloxy -3-pyridyl)-3-fluoro-2-pyridyl]-4- piperidyl]oxy]-2-methyl-phenyl]pyrazole-4-carboxylic acid (138b) A solution of 138a (750 mg, 1.05 mmol, 1 equiv.) in THF (7 mL) and water (1 mL) was treated with LiOH.H2O (176.4 mg, 4.20 mmol, 4 equiv.) at 25°C and the mixture was stirred at 25°C for 2 h. The reaction mixture was acidified to pH = 3 with aqueous HCl (1 M) and extracted with ethyl acetate (10 mL x 3). The combined organic phase was washed with brine (10 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum to afford 138b, which was used without further purification. LCMS: [M+1] = 686.4 Step C – Synthesis of 1-[4-[[1-[5-(2,6- dibenzyloxy-3-pyridyl)-3-fluoro-2-pyridyl]-4- piperidyl]oxy]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methyl]pyrazole-4-carboxamide (138c) A solution of 138b (700 mg, 1.02 mmol, 1 equiv.) in DMF (7 mL) was treated with [3- (2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methanamine hydrochloride (249.7 mg, 1.02 mmol, 1 equiv.), triethylamine (1.14 mL, 8.17 mmol, 8 equiv.) and BOP (677.2 mg, 1.53 mmol, 1.5 equiv.) at 25°C and the mixture was stirred at 25°C for 2 h. The mixture was diluted with water (0.5 mL) and extracted with ethyl acetate (0.5 mL x 3). The combined organic phases were washed with brine (5 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under vacuum. The residue was purified by column chromatography (silica gel, petroleum ether : ethyl acetate = 100 : 1 to 100 : 50) to afford 138c. LCMS: [M+1] = 876.5 Step D – Synthesis of 1-[4-[[1-[5-(2,6-dioxo- 3-piperidyl)-3-fluoro-2-pyridyl]-4-piperidyl]oxy]-2- methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole- 4-carboxamide (138) A suspension of Pd/C (200 mg, 10% purity) and Pd(OH)2/C (200 mg, 20 wt%) in ethyl acetate (3 mL) under a nitrogen atmosphere was treated with a solution of 138c (500 mg, 571 μmol, 1 equiv.) in THF (2 mL) at ambient temperature. The suspension was degassed and purged threefold with hydrogen. The mixture was stirred at 25 °C for 2 h under hydrogen (15 Psi). The reaction mixture was filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative HPLC (Phenomenex Luna C18, 100 mm x 40 mm, 5 µm; [H2O (0.04% HCl)-ACN]) to afford Compound 138 as the hydrochloride salt. LCMS: [M+1] = 698.4. 1H NMR
(400 MHz, DMSO-d6) δ 8.37 (s, 1H), 8.10 (s, 1H), 7.88 (s, 1H), 7.54 (dd, J = 1.8, 14.2 Hz, 1H), 7.26 (d, J = 8.8 Hz, 1H), 7.02 (d, J = 2.8 Hz, 1H), 6.95 (dd, J = 2.8, 8.8 Hz, 1H), 4.71 - 4.67 (m, 1H), 4.52 (s, 2H), 3.88 (dd, J = 4.8, 12.4 Hz, 1H), 3.78 -3.75 (m, 2H), 3.35 - 3.29 (m, 2H), 2.73 - 2.64 (m, 1H), 2.56 - 2.53 (m, 1H), 2.28 - 2.18 (m, 1H), 2.12 (s, 3H), 2.10 - 2.04 (m, 2H), 2.02 - 1.97 (m, 1H), 1.78 - 1.70 (m, 2H), 1.52 (s, 6H) Example 139: Synthesis of Compound 139 F F HO N O OBn O NH O
Step A – Synthesis of 2-azaspiro[3.5]nonan-7-ol hydrochloride (139a) A mixture of tert-butyl 7-hydroxy-2-azaspiro[3.5]nonane-2-carboxylate (900.0 mg, 3.20 mmol, 1.0 equiv.) in HCl/ dioxane (2 M, 7.8 mL, 4.8 equiv.) was stirred at 25°C for 0.5 h. The mixture was concentrated under reduced pressure to afford 139a as the hydrochloride salt. LCMS [M+1] = 142.2 Step B – Synthesis of 2-[5-(2,6-dibenzyloxy-3-pyridyl)-3-fluoro-2-pyridyl]-2-azaspiro[3.5]nonan- 7-ol (139b) A solution of 139a (553.5 mg, 3.1 mmol, 1.4 equiv.) and 2,6-dibenzyloxy-3-(5,6-difluoro- 3-pyridyl)pyridine (900.0 mg, 2.2 mmol, 1.0 equiv.) in DMSO (18.0 mL) was treated with DIEA (862.8 mg, 6.6 mmol, 1.1 mL, 3.0 equiv.). The mixture was stirred at 80°C for 16 h, cooled to ambient temperature and poured into water (60 mL). The aqueous phase was extracted with ethyl acetate (30 mL x 3). The combined organic phase was washed with brine (40 mL x 3), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether/ ethyl acetate=1/0 to 0/1) to afford 139b. LCMS [M+1] = 526.2 Step C – Synthesis of ethyl 1-[4-[[2-[5-(2,6-dibenzyloxy-3-pyridyl)-3-fluoro-2-pyridyl]-2- azaspiro[3.5]nonan-7-yl]oxy]-2-methyl-phenyl]pyrazole-4-carboxylate (139c) A solution of 139b (533.5 mg, 1.0 mmol, 1.0 equiv.) and ethyl 1-(4-hydroxy-2-methyl- phenyl)pyrazole-4-carboxylate (250.0 mg, 1.0 mmol, 1.0 equiv.) in toluene (6.0 mL) was treated with triphenylphosphine (532.5 mg, 2.0 mmol, 2.0 equiv.) and DIAD (410.5 mg, 2.0 mmol, 393.6 μL, 2.0 equiv.) at 0°C under an atmosphere of nitrogen. The mixture was stirred at 80°C for 16 h. The mixture was cooled to 25°C and poured into water (20 mL). The aqueous phase was extracted with ethyl acetate (10 mL x 3). The combined organic phase was washed with brine (10 mL x 3), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by preparative HPLC (Phenomenex Luna C18, 250 mm x 50mm, 15 µm; [water(FA)-ACN]) to afford 139c. LCMS [M+1] = 754.3
Step D – Synthesis of 1-[4-[[2-[5-(2,6-dibenzyloxy-3-pyridyl)-3-fluoro-2-pyridyl]-2- azaspiro[3.5]nonan-7-yl]oxy]-2-methyl-phenyl]pyrazole-4-carboxylic acid (139d) A solution of 139c (600.0 mg, 795.9 μmol, 1.0 equiv.) in THF (6.0 mL), H2O (3.0 mL) and MeOH (1.5 mL) was treated with LiOH.H2O (66.8 mg, 1.5 mmol, 2.0 equiv.). The mixture was stirred at 25°C for 16 h. The mixture pH was adjusted with 1 M sulfuric acid, diluted with water (20 mL), and extracted with ethyl acetate (10 mL x 3). The combined organic layer was washed with brine (20 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure to afford 139c, which was used without further purification. LCMS [M+1] = 726.0 Step E – Synthesis of 1-[4-[[2-[5-(2,6-dibenzyloxy-3-pyridyl)-3-fluoro-2-pyridyl]-2- azaspiro[3.5]nonan-7-yl]oxy]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H- 1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (139e) A solution of 139d (400.0 mg, 551.1 μmol, 1.0 equiv.) and triethylamine (278.8 mg, 2.7 mmol, 383.5 μL, 5.0 equiv.) in DMF (4.0 mL) was treated with BOP (365.6 mg, 826.6 μmol, 1.5 equiv.) and [3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methanamine hydrochloride (134.8 mg, 551.1 μmol, 1.0 equiv., HCl salt). The mixture was stirred at 25°C for 1 h, poured into water (20 mL) and extracted with ethyl acetate (10 mL x 3). The combined organic phase was washed with brine (10 mL x 3), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether/ ethyl acetate=1/0 to 1/1) to afford 139e. LCMS [M+1] = 916.4 Step F – Synthesis of 1-[4-[[2-[5-(2,6-dioxo-3-piperidyl)-3-fluoro-2-pyridyl]-2- azaspiro[3.5]nonan-7-yl]oxy]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H- 1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (139) A mixture of 139e (390.0 mg, 425.7 μmol, 1.0 equiv.) and Pd/C (226.5 mg, 10 wt%) in THF (15.0 mL) was degassed and purged threefold with hydrogen. The mixture was stirred under hydrogen (50 psi) at 30°C for 4 h. The mixture was filtered at ambient temperature and the filtrate was concentrated under reduced pressure. The residue was purified by preparative HPLC (Phenomenex Luna C18, 150 mm x 25 mm, 10µm; [water(FA)-ACN]) to afford Compound 139.
LCMS [M+1] = 738.4. 1H NMR (400 MHz, DMSO-d6) δ 13.91 (br s, 1H), 10.83 (s, 1H), 8.87 - 8.78 (m, 1H), 8.38 (s, 1H), 8.10 (s, 1H), 7.77 (s, 1H), 7.36 (d, J = 13.6 Hz, 1H), 7.26 (d, J = 8.4 Hz, 1H), 6.98 (s, 1H), 6.93 - 6.90 (m, 1H), 4.54 (d, J = 5.2 Hz, 2H), 4.46 (s, 1H), 3.81 - 3.78 (m, 5H), 2.73 - 2.64 (m, 1H), 2.54 (d, J = 3.6 Hz, 1H), 2.22 (dq, J = 4.4, 12.8 Hz, 1H), 2.13 (s, 3H), 1.97 - 1.89 (m, 5H), 1.72 - 1.67 (m, 2H), 1.61 - 1.56 (m, 2H), 1.53 (s, 6H). Example 140: Synthesis of Compound 140
 – - Chloro(methyl)magnesium (3 M, 35.1 mL, 1.5 equiv.) was added slowly to a solution of 5-bromo-2-iodo-pyrimidine (20.0 g, 70.2 mmol, 1.0 equiv.) in THF (180.0 mL) at -78°C. The resulting mixture was stirred for 30 min, then treated with DMF (51.3 g, 702.0 mmol, 54.0 mL, 10.0 equiv.). The solution was slowly warmed to ambient temperature and stirred for 2 h. MeOH (120.0 mL) and H2O (120.0 mL) were added follow by NH2OH•HCl (9.7 g, 140.4 mmol, 2.0 equiv.) and NaHCO3 (3.5 g, 42.1 mmol, 1.6 mL, 0.6 equiv.). The mixture was stirred at 25°C for 1 h. Water (20 ml) was added to the mixture slowly at 0°C and then stirred for an additional 30
min. The reaction mixture was filtered and the filtrate was concentrated under reduced pressure. The residue was diluted with water (40 mL) and extracted with EtOAc (40 mL x 3). The combined organic layers were washed with brine (40 mL x 2), dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure to afford 140a. LCMS [M+1] = 201.9. Step B – Synthesis of methyl 3-(5-bromopyrimidin-2-yl)isoxazole-5-carboxylate (140b) A solution of methyl prop-2-ynoate (12.4 g, 148.5 mmol, 12.4 mL, 2.5 equiv.), 140a (12.0 g, 59.4 mmol, 1.0 equiv.) in water (100.0 mL) was added KCl (4.4 g, 59.4 mmol, 1.0 equiv.) and Oxone (54.7 g, 89.1 mmol, 1.5 equiv.). The mixture was stirred at 25°C for 1 h. The mixture was extracted with dichloromethane (2 × 4 mL). The combined organic phase was dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether/ ethyl acetate=5/1) to afford 140b. LCMS [M+1] = 284.0. Step C – Synthesis of 3-[5-(4-formylphenyl)pyrimidin-2-yl]isoxazole-5-carboxylic acid (140c) A mixture of 140b (1.0 g, 3.5 mmol, 1 equiv.), 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan- 2-yl)benzaldehyde (817.0 mg, 3.5 mmol, 1.0 equiv.), Pd(dppf)Cl2 (257.5 mg, 352 μmol, 0.1 equiv.) and sodium carbonate (1.1 g, 10.5 mmol, 3.0 equiv.) in dioxane (10.0 mL) and water (3.0 mL) was degassed and purged threefold with nitrogen. The mixture was stirred at 80°C for 1 h under an atmosphere of nitrogen. The residue was diluted with water (40 mL) and extracted with ethyl acetate (40 mL x 3). The combined organic layers were washed with brine (40 mL x 2), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure to afford 140c. LCMS [M+1] = 296.0. Step D – Synthesis of 3-[5-(4-formylphenyl)pyrimidin-2-yl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl- ethyl)-1H-1,2,4-triazol-5-yl]methyl]isoxazole-5-carboxamide (140d) A solution of 140c (1.0 g, 3.3 mmol, 1.0 equiv.) and [3-(2,2,2-trifluoro-1,1-dimethyl- ethyl)-1H-1,2,4-triazol-5-yl]methanamine hydrochloride (828.6 mg, 3.3 mmol, 1.0 equiv) in DMF (20.0 mL) was treated with BOP (3.0 g, 6.7 mmol, 2.0 equiv.) and triethylamine (1.0 g, 10.1 mmol, 1.4 mL, 3.0 equiv.). The mixture was stirred at 25°C for 1 h. The reaction mixture was
concentrated under reduced pressure. The residue was purified by preparative HPLC (Phenomenex Luna C18, 250 mm x 50mm, 15µm; [n-Heptane-IPA(0.1% FA)]) to afford 140d. LCMS [M+1] = 486.1. Step E – Synthesis of 3-[5-[4-[[4-[5-[(2,6-dioxo-3-piperidyl)amino]-3-fluoro-2- pyridyl]piperazin-1-yl]methyl]phenyl]pyrimidin-2-yl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H-1,2,4-triazol-5-yl]methyl]isoxazole-5-carboxamide (140) A solution of 140d (70.0 mg, 144 μmol, 1.0 equiv.) and 3-[(5-fluoro-6-piperazin-1-yl-3- pyridyl)amino]piperidine-2,6-dione hydrochloride (182.2 mg, 530.2 μmol, 3.68 eq) in dichloromethane (2.0 mL) was treated with triethylamine (60.2 μL, 433 μmol, 3.0 equiv.). The mixture was stirred at 25°C for 2 h, treated with NaBH(OAc)3 (91.6 mg, 433 μmol, 3.0 equiv.), stirred at 25°C for 12 h. The reaction mixture was concentrated under reduced pressure. The residue was purified by preparative HPLC (Phenomenex Luna C18, 150 mm x 25mm, 10µm) to afford Compound 140. LCMS [M+1] = 777.2. 1H NMR (400 MHz, DMSO-d6) δ = 10.80 (s, 1H), 9.73 (br t, J = 5.6 Hz, 1H), 9.36 (s, 2H), 8.19 (s, 1H), 7.89 (d, J = 8.2 Hz, 2H), 7.72 (s, 1H), 7.60 - 7.47 (m, 3H), 6.99 (dd, J = 2.4, 14.8 Hz, 1H), 5.92 (d, J = 7.6 Hz, 1H), 4.61 (br d, J = 5.6 Hz, 2H), 4.29 (s, 1H), 3.61 (s, 2H), 3.13 (br s, 5H), 2.76 - 2.66 (m, 1H), 2.63 - 2.52 (m, 5H), 2.17 - 2.04 (m, 1H), 1.94 - 1.80 (m, 1H), 1.54 (s, 6H). Example 141: Synthesis of Compound 141
– - Chloro(methyl)magnesium (3 M, 35.1 mL, 1.5 equiv.) was added slowly to a solution of 5-bromo-2-iodo-pyrimidine (20.0 g, 70.2 mmol, 1.0 equiv.) in THF (180.0 mL) at -78°C. The resulting mixture was stirred for 30 min, then treated with DMF (51.3 g, 702.0 mmol, 54.0 mL, 10.0 equiv.). The solution was slowly warmed to ambient temperature and stirred for 2 h. MeOH (120.0 mL) and H2O (120.0 mL) were added follow by NH2OH•HCl (9.7 g, 140.4 mmol, 2.0 equiv.) and NaHCO3 (3.5 g, 42.1 mmol, 1.6 mL, 0.6 equiv.). The mixture was stirred at 25°C for 1 h. Water (20 ml) was added to the mixture slowly at 0°C and then stirred for an additional 30
min. The reaction mixture was filtered and the filtrate was concentrated under reduced pressure. The residue was diluted with water (40 mL) and extracted with EtOAc (40 mL x 3). The combined organic layers were washed with brine (40 mL x 2), dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure to afford 140a. LCMS [M+1] = 201.9. Step B – Synthesis of methyl 3-(5-bromopyrimidin-2-yl)isoxazole-5-carboxylate (140b) A solution of methyl prop-2-ynoate (12.4 g, 148.5 mmol, 12.4 mL, 2.5 equiv.), 140a (12.0 g, 59.4 mmol, 1.0 equiv.) in water (100.0 mL) was added KCl (4.4 g, 59.4 mmol, 1.0 equiv.) and Oxone (54.7 g, 89.1 mmol, 1.5 equiv.). The mixture was stirred at 25°C for 1 h. The mixture was extracted with dichloromethane (2 × 4 mL). The combined organic phase was dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether/ ethyl acetate=5/1) to afford 140b. LCMS [M+1] = 284.0. Step C – Synthesis of 3-[5-(4-formylphenyl)pyrimidin-2-yl]isoxazole-5-carboxylic acid (140c) A mixture of 140b (1.0 g, 3.5 mmol, 1 equiv.), 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan- 2-yl)benzaldehyde (817.0 mg, 3.5 mmol, 1.0 equiv.), Pd(dppf)Cl2 (257.5 mg, 352 μmol, 0.1 equiv.) and sodium carbonate (1.1 g, 10.5 mmol, 3.0 equiv.) in dioxane (10.0 mL) and water (3.0 mL) was degassed and purged threefold with nitrogen. The mixture was stirred at 80°C for 1 h under an atmosphere of nitrogen. The residue was diluted with water (40 mL) and extracted with ethyl acetate (40 mL x 3). The combined organic layers were washed with brine (40 mL x 2), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure to afford 140c. LCMS [M+1] = 296.0. Step D – Synthesis of 3-[5-(4-formylphenyl)pyrimidin-2-yl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl- ethyl)-1H-1,2,4-triazol-5-yl]methyl]isoxazole-5-carboxamide (140d) A solution of 140c (1.0 g, 3.3 mmol, 1.0 equiv.) and [3-(2,2,2-trifluoro-1,1-dimethyl- ethyl)-1H-1,2,4-triazol-5-yl]methanamine hydrochloride (828.6 mg, 3.3 mmol, 1.0 equiv) in DMF (20.0 mL) was treated with BOP (3.0 g, 6.7 mmol, 2.0 equiv.) and triethylamine (1.0 g, 10.1 mmol, 1.4 mL, 3.0 equiv.). The mixture was stirred at 25°C for 1 h. The reaction mixture was
concentrated under reduced pressure. The residue was purified by preparative HPLC (Phenomenex Luna C18, 250 mm x 50mm, 15µm; [n-Heptane-IPA(0.1% FA)]) to afford 140d. LCMS [M+1] = 486.1. Step E – Synthesis of 3-[5-[4-[[4-[5-[(2,6-dioxo-3-piperidyl)amino]-3-fluoro-2- pyridyl]piperazin-1-yl]methyl]phenyl]pyrimidin-2-yl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H-1,2,4-triazol-5-yl]methyl]isoxazole-5-carboxamide (140) A solution of 140d (70.0 mg, 144 μmol, 1.0 equiv.) and 3-[(5-fluoro-6-piperazin-1-yl-3- pyridyl)amino]piperidine-2,6-dione hydrochloride (182.2 mg, 530.2 μmol, 3.68 eq) in dichloromethane (2.0 mL) was treated with triethylamine (60.2 μL, 433 μmol, 3.0 equiv.). The mixture was stirred at 25°C for 2 h, treated with NaBH(OAc)3 (91.6 mg, 433 μmol, 3.0 equiv.), stirred at 25°C for 12 h. The reaction mixture was concentrated under reduced pressure. The residue was purified by preparative HPLC (Phenomenex Luna C18, 150 mm x 25mm, 10µm) to afford Compound 140. LCMS [M+1] = 777.2. 1H NMR (400 MHz, DMSO-d6) δ = 10.80 (s, 1H), 9.73 (br t, J = 5.6 Hz, 1H), 9.36 (s, 2H), 8.19 (s, 1H), 7.89 (d, J = 8.2 Hz, 2H), 7.72 (s, 1H), 7.60 - 7.47 (m, 3H), 6.99 (dd, J = 2.4, 14.8 Hz, 1H), 5.92 (d, J = 7.6 Hz, 1H), 4.61 (br d, J = 5.6 Hz, 2H), 4.29 (s, 1H), 3.61 (s, 2H), 3.13 (br s, 5H), 2.76 - 2.66 (m, 1H), 2.63 - 2.52 (m, 5H), 2.17 - 2.04 (m, 1H), 1.94 - 1.80 (m, 1H), 1.54 (s, 6H). Example 141: Synthesis of Compound 141
, , 141a) A mixture of 4-bromo-2-fluoroaniline (40.0 g, 210.5 mmol, 1.0 equiv.) in acrylic acid (45.5 g, 631.5 mmol, 43.3 mL, 3.0 equiv.) was stirred at 100°C for 4 h to afford a suspension, which was treated with urea (80.9 g, 1.3 mol, 72.4 mL, 6.4 equiv.) and acetic acid (300.0 mL). The mixture was stirred at 120°C for 12 h, cooled to 20°C and poured into water (500 mL). The solid was collected by vacuum filtration and the filter cake was triturated with ethyl acetate (200 mL) at 20oC for 30 min to give 141a. 1H NMR (400 MHz, DMSO-d6) δ 10.55 (s, 1H), 7.67 (dd, J = 2.4, 9.8 Hz, 1H), 7.52 - 7.38 (m, 2H), 3.72 (t, J = 6.8 Hz, 2H), 2.71 (t, J = 6.8 Hz, 2H). Step B – Synthesis of tert-butyl 6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptane-2-carboxylate (141b) A mixture of 1-(4-bromo-2-fluorophenyl)dihydropyrimidine-2,4(1H,3H)-dione (9.0 g, 31.3 mmol, 1.0 equiv.), tert-butyl 6-iodo-2-azaspiro[3.3]heptane-2-carboxylate (12.1 g, 37.6 mmol, 1.2 equiv.), TFA (233 μL, 0.1 equiv.), 4-methoxypyridine-2-carboxamidine hydrochloride (588 mg, 3.1 mmol, 0.1 equiv.), NiCl2∙glyme (689 mg, 3.1 mmol, 0.1 equiv.) and Zn (4.1 g, 62.7 mmol, 2.0 equiv.) in DMA (100.0 mL) was degassed and purged threefold with nitrogen, and then
the mixture was stirred at 80°C for 12 h under a nitrogen atmosphere. The mixture was cooled to 25°C and filtered. The filtrate was diluted with water (200 mL), extracted with ethyl acetate (200 mL x3). The combined organic layers were washed with brine (200 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The crude product was triturated with (petroleum ether: ethyl acetate=4:1) (100 mL) at 25 °C for 30 min to give 141b. 1H NMR (400 MHz, DMSO-d6) δ 10.45 (s, 1H), 7.33 (t, J = 8.0 Hz, 1H), 7.20 - 7.04 (m, 2H), 3.97 (s, 2H), 3.76 (s, 2H), 3.67 (t, J = 6.8 Hz, 2H), 3.44 - 3.35 (m, 1H), 2.70 (t, J = 6.8 Hz, 2H), 2.57 - 2.51 (m, 2H), 2.30 - 2.20 (m, 2H), 1.42 - 1.33 (m, 9H). Step C – Synthesis of 1-(2-fluoro-4-(2-azaspiro[3.3]heptan-6-yl)phenyl)dihydropyrimidine- 2,4(1H,3H)-dione trifluoroacetate salt (141c) To a solution of tert-butyl 6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)- 2-azaspiro[3.3]heptane-2-carboxylate (1.5 g, 3.7 mmol, 1.0 equiv.) in dichloromethane (12 mL) was added TFA (4.0 mL), the mixture was stirred at 20°C for 1 h. The mixture was concentrated and triturated with MTBE (20 mL) at 20oC for 30 min to give 141c. 1H NMR (400 MHz, DMSO-d6) δ 10.46 (s, 1H), 8.71 (s, 1H), 7.34 (t, J = 8.4 Hz, 1H), 7.19 (dd, J = 1.6, 11.6 Hz, 1H), 7.08 (d, J = 8.4 Hz, 1H), 4.11 (t, J = 6.4 Hz, 2H), 3.89 (t, J = 6.4 Hz, 2H), 3.67 (t, J = 6.8 Hz, 2H), 3.39 (quin, J = 8.8 Hz, 1H), 2.70 (t, J = 6.8 Hz, 2H), 2.60 (dt, J = 2.8, 9.2 Hz, 2H), 2.35 - 2.27 (m, 2H). Step D – Synthesis of 3-(5-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (141) To a solution of 1-(2-fluoro-4-(2-azaspiro[3.3]heptan-6-yl)phenyl)dihydropyrimidine- 2,4(1H,3H)-dione trifluoracetic acid (1.0 g, 2.4 mmol, 1.0 equiv.) and 3-(5-(2-fluoro-4- formylphenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5- yl)methyl)isoxazole-5-carboxamide (1.2 g, 2.4 mmol, 1.0 equiv.) in DMF (10 mL) was added triethylamine (2.4 g, 23.9 mmol, 3.3 mL, 10.0 equiv.) at 20°C. The mixture was aged for 2 h, treated with NaBH(OAc)3 (1.0 g, 4.7 mmol, 2.0 equiv.) and stirred at 20°C for 14 h. The mixture was diluted with water (30 mL) and extracted with ethyl acetate (20 mL x 3). The combined organic layers were washed with brine (40 mL x 2), dried over anhydrous sodium sulfate, filtered
and the filtrate was concentrated under reduced pressure. The residue was purified by prep-HPLC (WePure Biotech XP C18250x70mm; 10 µm; [water(10mM NH4HCO3)-ACN];gradient:35%- 60% B over 20 min ) to give 141. LCMS [M+1] = 791.3; 1H NMR (400 MHz, DMSO-d6) δ 13.51 (br s, 1H), 10.44 (br s, 1H), 9.72 (br t, J = 5.6 Hz, 1H), 9.23 (s, 2H), 7.77 - 7.69 (m, 2H), 7.36 - 7.28 (m, 3H), 7.14 (d, J = 11.8 Hz, 1H), 7.07 (d, J = 8.1 Hz, 1H), 4.60 (br d, J = 5.2 Hz, 2H), 3.67 (t, J = 6.8 Hz, 2H), 3.63 (s, 2H), 3.43 - 3.37 (m, 1H), 3.34 (s, 2H), 3.13 (s, 2H), 2.70 (t, J = 6.8 Hz, 2H), 2.54 - 2.51 (m, 2H), 2.25 - 2.16 (m, 2H), 1.54 (s, 6H). Example 142: Synthesis of Compound 142 Step
 - 2,6-diazaspiro[3.3]heptane-2-carboxylate (142a) To a solution of 1-(4-bromo-2-fluoro-phenyl)hexahydropyrimidine-2,4-dione (3 g, 10.4 mmol, 1.0 equiv.) in dioxane (40 mL) was added tert-butyl 2,6-diazaspiro[3.3]heptane-2- carboxylate (4.1 g, 20.9 mmol, 2.0 equiv.), cesium carbonate (10.2 g, 31.3 mmol, 3.0 equiv.) and [1,3-bis[2,6-bis(1-ethylpropyl)phenyl]-4,5-dichloroimidazol-2-ylidene]-dichloro-(2- methylpyridin1-ium-1-yl)palladium (877.9 mg, 1.04 mmol, 0.1 equiv.). The mixture was stirred at 100°C for 12 h. The mixture was poured into water (60 mL), extracted with ethyl acetate (20
mL x 3). The organic phase was washed with brine (60 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by prep-TLC (silica gel, Petroleum ether: Ethyl acetate = 2:1) to give 142a. LCMS [M+1] = 405.2. Step B – Synthesis of 1-(2-fluoro-4-(2,6-diazaspiro[3.3]heptan-2-yl)phenyl)dihydropyrimidine- 2,4(1H,3H)-dione trifluoroacetate salt (142b) To a solution of tert-butyl 6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)- 2,6-diazaspiro[3.3]heptane-2-carboxylate (2.6 g, 6.4 mmol, 1.0 equiv.) in dichloromethane (26 mL) was added TFA (4.78 mL, 10 equiv.). The mixture was stirred at 25°C for 1 h. The mixture was concentrated under reduced pressure to give 142b, which was used without purification. LCMS [M+1] = 305.2. Step C – Synthesis of 3-(5-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)- 2,6-diazaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (142) To a solution of 1-[4-(2,6-diazaspiro[3.3]heptan-2-yl)-2-fluoro-phenyl] hexahydropyrimidine-2,4-dione trifluoroacetate salt (2.6 g, 6.22 mmol, 1.0 equiv.) and triethylamine (2.52 g, 24.86 mmol, 3.46 mL, 4.0 equiv.) in dichloromethane (26 mL) was added 3-[5-(2-fluoro-4-formyl-phenyl) pyrimidin-2-yl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H- 1,2,4-triazol-5-yl]methyl]isoxazole-5-carboxamide (1.88 g, 3.7 mmol, 0.6 equiv.), then the reaction was stirred for 2 h, added NaBH(OAc)3 (3.29 g, 15.54 mmol, 2.5 equiv.), the mixture was stirred at 25°C for 3 h. The mixture was poured into NaHCO3 (200 mL) and extracted with ethyl acetate (100 mL x 3). The combined organic layers were washed with brine (200 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by prep-HPLC (Welch Xtimate C18180x70mm; 10 µm; [water(10mM NH4HCO3)-ACN];gradient:30%-60% B over 17.0 min) to give 142. LCMS [M+1] = 792.3; 1H NMR (400 MHz, DMSO-d6) δ = 13.93 (br s, 1H), 10.36 (s, 1H), 9.74 (br t, J = 4.8 Hz,
- 2,6-diazaspiro[3.3]heptane-2-carboxylate (142a) To a solution of 1-(4-bromo-2-fluoro-phenyl)hexahydropyrimidine-2,4-dione (3 g, 10.4 mmol, 1.0 equiv.) in dioxane (40 mL) was added tert-butyl 2,6-diazaspiro[3.3]heptane-2- carboxylate (4.1 g, 20.9 mmol, 2.0 equiv.), cesium carbonate (10.2 g, 31.3 mmol, 3.0 equiv.) and [1,3-bis[2,6-bis(1-ethylpropyl)phenyl]-4,5-dichloroimidazol-2-ylidene]-dichloro-(2- methylpyridin1-ium-1-yl)palladium (877.9 mg, 1.04 mmol, 0.1 equiv.). The mixture was stirred at 100°C for 12 h. The mixture was poured into water (60 mL), extracted with ethyl acetate (20
mL x 3). The organic phase was washed with brine (60 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by prep-TLC (silica gel, Petroleum ether: Ethyl acetate = 2:1) to give 142a. LCMS [M+1] = 405.2. Step B – Synthesis of 1-(2-fluoro-4-(2,6-diazaspiro[3.3]heptan-2-yl)phenyl)dihydropyrimidine- 2,4(1H,3H)-dione trifluoroacetate salt (142b) To a solution of tert-butyl 6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)- 2,6-diazaspiro[3.3]heptane-2-carboxylate (2.6 g, 6.4 mmol, 1.0 equiv.) in dichloromethane (26 mL) was added TFA (4.78 mL, 10 equiv.). The mixture was stirred at 25°C for 1 h. The mixture was concentrated under reduced pressure to give 142b, which was used without purification. LCMS [M+1] = 305.2. Step C – Synthesis of 3-(5-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)- 2,6-diazaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (142) To a solution of 1-[4-(2,6-diazaspiro[3.3]heptan-2-yl)-2-fluoro-phenyl] hexahydropyrimidine-2,4-dione trifluoroacetate salt (2.6 g, 6.22 mmol, 1.0 equiv.) and triethylamine (2.52 g, 24.86 mmol, 3.46 mL, 4.0 equiv.) in dichloromethane (26 mL) was added 3-[5-(2-fluoro-4-formyl-phenyl) pyrimidin-2-yl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H- 1,2,4-triazol-5-yl]methyl]isoxazole-5-carboxamide (1.88 g, 3.7 mmol, 0.6 equiv.), then the reaction was stirred for 2 h, added NaBH(OAc)3 (3.29 g, 15.54 mmol, 2.5 equiv.), the mixture was stirred at 25°C for 3 h. The mixture was poured into NaHCO3 (200 mL) and extracted with ethyl acetate (100 mL x 3). The combined organic layers were washed with brine (200 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by prep-HPLC (Welch Xtimate C18180x70mm; 10 µm; [water(10mM NH4HCO3)-ACN];gradient:30%-60% B over 17.0 min) to give 142. LCMS [M+1] = 792.3; 1H NMR (400 MHz, DMSO-d6) δ = 13.93 (br s, 1H), 10.36 (s, 1H), 9.74 (br t, J = 4.8 Hz,
 , (s, 2H), 7.80 - 7.68 (m, 2H), 7.39 - 7.28 (m, 2H), 7.17 (t, J = 8.8 Hz, 1H), 6.38 - 6.20 (m, 2H), 4.61 (br d, J = 5.2 Hz, 2H), 3.93 (s, 4H), 3.65 (s, 2H), 3.59 (t, J = 6.8 Hz, 2H), 3.37 (s, 4H) 3.41 - 3.35 (m, 4H), 2.67 (t, J = 6.8 Hz, 2H), 1.54 (s, 6H).
Example 143: Synthesis of Compound 143
, (s, 2H), 7.80 - 7.68 (m, 2H), 7.39 - 7.28 (m, 2H), 7.17 (t, J = 8.8 Hz, 1H), 6.38 - 6.20 (m, 2H), 4.61 (br d, J = 5.2 Hz, 2H), 3.93 (s, 4H), 3.65 (s, 2H), 3.59 (t, J = 6.8 Hz, 2H), 3.37 (s, 4H) 3.41 - 3.35 (m, 4H), 2.67 (t, J = 6.8 Hz, 2H), 1.54 (s, 6H).
Example 143: Synthesis of Compound 143
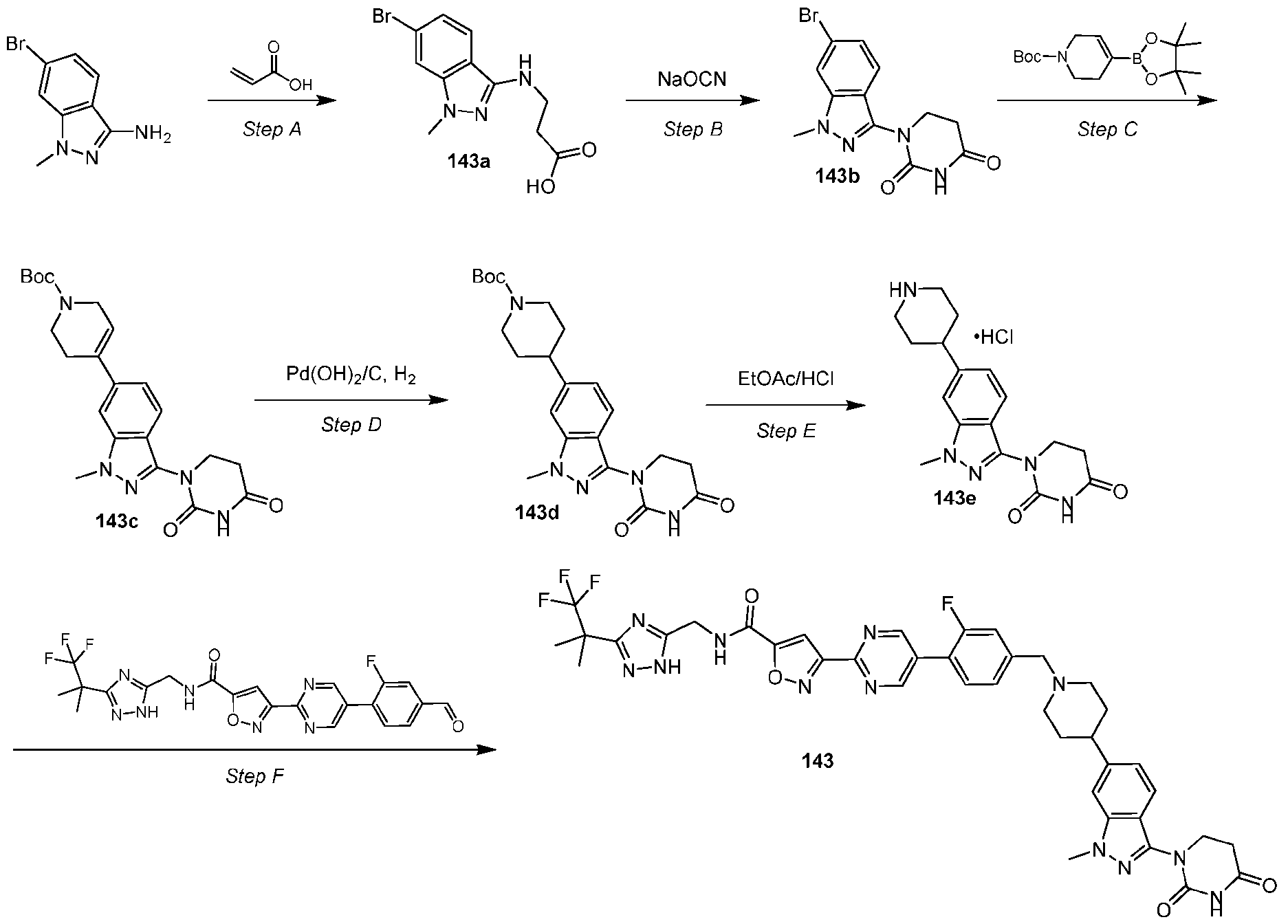 To a solution of 6-bromo-1-methyl-indazol-3-amine (25.0 g, 110.5 mmol, 1.0 equiv.) and acrylic acid (11.9 g, 165.8 mmol, 11.3 mL, 1.5 equiv.) in water (250 mL) was added TBAB (3.5 g, 11.0 mmol, 0.1 equiv.) and HCl (12 M, 50.0 mL, 5.4 equiv.) at 25°C. The mixture was stirred at 100°C for 12 h. After the reaction was complete, the mixture was cooled to 25°C and diluted with ice-cold water (200 mL). The solids were collected with vacuum filtration to afford 143a. LCMS [M+1] =298.0, 300.0. Step B – Synthesis of 1-(6-bromo-1-methyl-1H-indazol-3-yl)dihydropyrimidine-2,4(1H,3H)-dione (143b) To a solution of 3-((6-bromo-1-methyl-1H-indazol-3-yl)amino)propanoic acid (14.0 g, 46.9 mmol, 1.0 equiv.) in acetic acid (140.0 mL) was added sodium cyanate (9.1 g, 140.8 mmol, 3.0 equiv.). The mixture was stirred at 100°C for 12 h. After the reaction was complete, the
mixture was cooled to 25°C. The reaction was diluted with water (300 mL) and filtered. The filtrate was washed with water (2 x 30 mL) and concentrated under reduced pressure to afford 143b. LCMS [M+1] = 323.1, 325.1. Step C – Synthesis of tert-butyl 4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-1-methyl-1H- indazol-6-yl)-3,6-dihydropyridine-1(2H)-carboxylate (143c) To a solution of 1-(6-bromo-1-methyl-indazol-3-yl)hexahydropyrimidine-2,4-dione (2.3 g, 7.1 mmol, 1.0 equiv.) in dioxane (16.0 mL) and water (4.0 mL) was added tert-butyl 4-(4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)-3,6-dihydro-2H-pyridine-1-carboxylate (3.3 g, 10.6 mmol, 1.5 equiv.), Na2CO3 (2.3 g, 21.3 mmol, 3.0 equiv.) and Pd(dppf)Cl2.CH2Cl2 (581.2 mg, 711.7 μmol, 0.1 equiv.). The mixture was degassed and purged threefold with nitrogen. The mixture was stirred at 90°C for 2 h. The mixture was cooled to 25°C. The solution was diluted with water (30 mL) and extracted with ethyl acetate (20 mL x 3). The combined organic layer was washed with brine (50 mL) and dried with sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, Petroleum ether/Ethyl acetate = 1/0 to 1/1) to give 143c. LCMS [M+1] = 426.2. Step D – Synthesis of tert-butyl 4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-1-methyl-1H- indazol-6-yl)piperidine-1-carboxylate (143d) A reactor was purged with argon and charged with Pd(OH)2/C (1.2 g, 1.7 mmol, 20.0% purity, 0.3 equiv.), MeOH (10.0 mL) and solution of tert-butyl 4-(3-(2,4- dioxotetrahydropyrimidin-1(2H)-yl)-1-methyl-1H-indazol-6-yl)-3,6-dihydropyridine-1(2H)- carboxylate (2.5 g, 5.8 mmol, 1.0 equiv.) in THF (50.0 mL) and MeOH (50.0 mL). The mixture was degassed and purged threefold with hydrogen gas. The mixture was stirred at 25°C under hydrogen (15 Psi) for 36 h. The reaction was filtered and the filtrate was concentrated under reduced pressure to give 143d. LCMS [M+Na] = 450.3. Step E – Synthesis of 1-(1-methyl-6-(piperidin-4-yl)-1H-indazol-3-yl)dihydropyrimidine- 2,4(1H,3H)-dione hydrochloride (143e) A solution of tert-butyl 4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-1-methyl-1H- indazol-6-yl)piperidine-1-carboxylate (3.3 g, 7.7 mmol, 1.0 equiv.) in HCl (4 M in ethyl acetate,
35.0 mL) was stirred at 25°C for 12 h. The reaction was concentrated under reduced pressure to give 143e. LCMS [M+1] =328.2. Step F – Synthesis of 3-(5-(4-((4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-1-methyl-1H- indazol-6-yl)piperidin-1-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (143) To a solution of 1-(1-methyl-6-(piperidin-4-yl)-1H-indazol-3-yl)dihydropyrimidine- 2,4(1H,3H)-dione hydrochloride (2.8 g, 7.9 mmol, 2.0 equiv.) in dichloromethane (30.0 mL) and THF (30.0 mL) was added 3-[5-(2-fluoro-4-formyl-phenyl)pyrimidin-2-yl]-N-[[3-(2,2,2-trifluoro- 1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]isoxazole-5-carboxamide (2.0 g, 3.9 mmol, 1.0 equiv.) and triethylamine (4.8 mg, 47.6 mmol, 6.6 mL, 12.0 equiv.). The mixture was stirred at 25°C for 12 h, treated with NaBH(OAc)3 (2.5 g, 11.9 mmol, 3.0 equiv.) at 25°C and stirred at 25°C for 2 h. The mixture was concentrated under reduced pressure. The residue was purified by prep-HPLC (Phenomenex Luna C18100x40mm; 5 µm; [water (0.2% FA)-ACN]; gradient: 15%- 50% B over 8.0 min) to give 143. LCMS [M+1] = 815.7; 1H NMR (400 MHz, DMSO-d6) δ 13.98 (br s, 1H), 10.54 (s, 1H), 9.74 (br s, 1H), 9.26 (d, J = 1.2 Hz, 2H), 7.77 (t, J = 8.2 Hz, 1H), 7.73 (s, 1H), 7.55 (d, J = 8.4 Hz, 1H), 7.47 (s, 1H), 7.44 - 7.40 (m, 2H), 7.06 (d, J = 8.0 Hz, 1H), 4.61 (d, J = 5.6 Hz, 2H), 3.97 (s, 3H), 3.90 (t, J = 6.6 Hz, 2H), 3.64 (s, 2H), 3.00 (d, J = 10.8 Hz, 2H), 2.75 (t, J = 6.6 Hz, 2H), 2.70 - 2.64 (m, 1H), 2.20 - 2.14 (m, 2H), 1.88 - 1.76 (m, 4H), 1.54 (s, 6H). Example 144: Synthesis of Compound 144
To a solution of 6-bromo-1-methyl-indazol-3-amine (25.0 g, 110.5 mmol, 1.0 equiv.) and acrylic acid (11.9 g, 165.8 mmol, 11.3 mL, 1.5 equiv.) in water (250 mL) was added TBAB (3.5 g, 11.0 mmol, 0.1 equiv.) and HCl (12 M, 50.0 mL, 5.4 equiv.) at 25°C. The mixture was stirred at 100°C for 12 h. After the reaction was complete, the mixture was cooled to 25°C and diluted with ice-cold water (200 mL). The solids were collected with vacuum filtration to afford 143a. LCMS [M+1] =298.0, 300.0. Step B – Synthesis of 1-(6-bromo-1-methyl-1H-indazol-3-yl)dihydropyrimidine-2,4(1H,3H)-dione (143b) To a solution of 3-((6-bromo-1-methyl-1H-indazol-3-yl)amino)propanoic acid (14.0 g, 46.9 mmol, 1.0 equiv.) in acetic acid (140.0 mL) was added sodium cyanate (9.1 g, 140.8 mmol, 3.0 equiv.). The mixture was stirred at 100°C for 12 h. After the reaction was complete, the
mixture was cooled to 25°C. The reaction was diluted with water (300 mL) and filtered. The filtrate was washed with water (2 x 30 mL) and concentrated under reduced pressure to afford 143b. LCMS [M+1] = 323.1, 325.1. Step C – Synthesis of tert-butyl 4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-1-methyl-1H- indazol-6-yl)-3,6-dihydropyridine-1(2H)-carboxylate (143c) To a solution of 1-(6-bromo-1-methyl-indazol-3-yl)hexahydropyrimidine-2,4-dione (2.3 g, 7.1 mmol, 1.0 equiv.) in dioxane (16.0 mL) and water (4.0 mL) was added tert-butyl 4-(4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)-3,6-dihydro-2H-pyridine-1-carboxylate (3.3 g, 10.6 mmol, 1.5 equiv.), Na2CO3 (2.3 g, 21.3 mmol, 3.0 equiv.) and Pd(dppf)Cl2.CH2Cl2 (581.2 mg, 711.7 μmol, 0.1 equiv.). The mixture was degassed and purged threefold with nitrogen. The mixture was stirred at 90°C for 2 h. The mixture was cooled to 25°C. The solution was diluted with water (30 mL) and extracted with ethyl acetate (20 mL x 3). The combined organic layer was washed with brine (50 mL) and dried with sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, Petroleum ether/Ethyl acetate = 1/0 to 1/1) to give 143c. LCMS [M+1] = 426.2. Step D – Synthesis of tert-butyl 4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-1-methyl-1H- indazol-6-yl)piperidine-1-carboxylate (143d) A reactor was purged with argon and charged with Pd(OH)2/C (1.2 g, 1.7 mmol, 20.0% purity, 0.3 equiv.), MeOH (10.0 mL) and solution of tert-butyl 4-(3-(2,4- dioxotetrahydropyrimidin-1(2H)-yl)-1-methyl-1H-indazol-6-yl)-3,6-dihydropyridine-1(2H)- carboxylate (2.5 g, 5.8 mmol, 1.0 equiv.) in THF (50.0 mL) and MeOH (50.0 mL). The mixture was degassed and purged threefold with hydrogen gas. The mixture was stirred at 25°C under hydrogen (15 Psi) for 36 h. The reaction was filtered and the filtrate was concentrated under reduced pressure to give 143d. LCMS [M+Na] = 450.3. Step E – Synthesis of 1-(1-methyl-6-(piperidin-4-yl)-1H-indazol-3-yl)dihydropyrimidine- 2,4(1H,3H)-dione hydrochloride (143e) A solution of tert-butyl 4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-1-methyl-1H- indazol-6-yl)piperidine-1-carboxylate (3.3 g, 7.7 mmol, 1.0 equiv.) in HCl (4 M in ethyl acetate,
35.0 mL) was stirred at 25°C for 12 h. The reaction was concentrated under reduced pressure to give 143e. LCMS [M+1] =328.2. Step F – Synthesis of 3-(5-(4-((4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-1-methyl-1H- indazol-6-yl)piperidin-1-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (143) To a solution of 1-(1-methyl-6-(piperidin-4-yl)-1H-indazol-3-yl)dihydropyrimidine- 2,4(1H,3H)-dione hydrochloride (2.8 g, 7.9 mmol, 2.0 equiv.) in dichloromethane (30.0 mL) and THF (30.0 mL) was added 3-[5-(2-fluoro-4-formyl-phenyl)pyrimidin-2-yl]-N-[[3-(2,2,2-trifluoro- 1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]isoxazole-5-carboxamide (2.0 g, 3.9 mmol, 1.0 equiv.) and triethylamine (4.8 mg, 47.6 mmol, 6.6 mL, 12.0 equiv.). The mixture was stirred at 25°C for 12 h, treated with NaBH(OAc)3 (2.5 g, 11.9 mmol, 3.0 equiv.) at 25°C and stirred at 25°C for 2 h. The mixture was concentrated under reduced pressure. The residue was purified by prep-HPLC (Phenomenex Luna C18100x40mm; 5 µm; [water (0.2% FA)-ACN]; gradient: 15%- 50% B over 8.0 min) to give 143. LCMS [M+1] = 815.7; 1H NMR (400 MHz, DMSO-d6) δ 13.98 (br s, 1H), 10.54 (s, 1H), 9.74 (br s, 1H), 9.26 (d, J = 1.2 Hz, 2H), 7.77 (t, J = 8.2 Hz, 1H), 7.73 (s, 1H), 7.55 (d, J = 8.4 Hz, 1H), 7.47 (s, 1H), 7.44 - 7.40 (m, 2H), 7.06 (d, J = 8.0 Hz, 1H), 4.61 (d, J = 5.6 Hz, 2H), 3.97 (s, 3H), 3.90 (t, J = 6.6 Hz, 2H), 3.64 (s, 2H), 3.00 (d, J = 10.8 Hz, 2H), 2.75 (t, J = 6.6 Hz, 2H), 2.70 - 2.64 (m, 1H), 2.20 - 2.14 (m, 2H), 1.88 - 1.76 (m, 4H), 1.54 (s, 6H). Example 144: Synthesis of Compound 144
To a solution of 6-bromopyrazolo[1,5-a]pyridine (2.0 g, 10.1 mmol, 1.0 equiv.) and tert- butyl piperazine-1-carboxylate (1.8 g, 10.1 mmol, 1.0 equiv.) in DMSO (20.0 mL) was added [1,3-bis[2,6-bis(1-ethylpropyl)phenyl]-4,5-dichloro-imidazol-2-ylidene]-dichloro-(2- methylpyridin-1-ium-1-yl)palladium (426.4 mg, 507.5 μmol, 0.05 equiv.) and cesium carbonate (9.9 g, 30.4 mmol, 3.0 equiv.) under a nitrogen atmosphere. The mixture was stirred at 100°C for 12 h. The mixture was cooled to 25°C and poured into water (60 mL). The aqueous phase was extracted with ethyl acetate (30 mLx3). The combined organic phase was washed with brine (50 mLx3), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure to give 144a. LCMS [M+1] = 303.1. Step B – Synthesis of tert-butyl 4-(3-iodopyrazolo[1,5-a]pyridin-6-yl)piperazine-1-carboxylate (144b) To a solution of tert-butyl 4-(pyrazolo[1,5-a]pyridin-6-yl)piperazine-1-carboxylate (3.0 g, 9.9 mmol, 1.0 equiv.) in ACN (30.0 mL) was added NIS (2.3 g, 10.4 mmol, 1.0 equiv.). The mixture was stirred at 25°C for 1 h. The mixture was poured into Na2S2O3 aqueous (50 mL). The aqueous phase was extracted with ethyl acetate (30 mLx3). The combined organic phase was washed with brine (50 mLx3), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether/ethyl acetate=1/0 to 1/1) to give 144b. LCMS [M+1] = 429.0.
Step C – Synthesis of tert-butyl 4-(3-(3-(4-methoxybenzyl)-2,4-dioxotetrahydropyrimidin-1(2H)- yl)pyrazolo[1,5-a]pyridin-6-yl)piperazine-1-carboxylate (144c) To a solution of tert-butyl 4-(3-iodopyrazolo[1,5-a]pyridin-6-yl)piperazine-1-carboxylate (1.0 g, 2.3 mmol, 1.0 equiv.) and 3-(4-methoxybenzyl)dihydropyrimidine-2,4(1H,3H)-dione (546.9 mg, 2.3 mmol, 1.0 equiv.) in dioxane (10.0 mL) was added CuI (44.4 mg, 233.5 μmol, 0.1 equiv.), (1S,2S)-N1,N2-dimethylcyclohexane-1,2-diamine (66.4 mg, 467.0 μmol, 0.2 equiv.) and K3PO4 (991.2 mg, 4.6 mmol, 2.0 equiv.). The mixture was stirred at 80°C for 3 h. The mixture was cooled to 25°C and poured into water (30 mL). The aqueous phase was extracted with ethyl acetate (15 mLx3). The combined organic phase was washed with brine (50 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, Petroleum ether/Ethyl acetate=1/0 to 0/0) to give 144c. LCMS [M+1] = 535.2. Step D – Synthesis of 1-(6-(piperazin-1-yl)pyrazolo[1,5-a]pyridin-3-yl)dihydropyrimidine- 2,4(1H,3H)-dione (144d) To a solution of tert-butyl 4-(3-(3-(4-methoxybenzyl)-2,4-dioxotetrahydropyrimidin- 1(2H)-yl)pyrazolo[1,5-a]pyridin-6-yl)piperazine-1-carboxylate (600.0 mg, 1.1 mmol, 1.0 equiv.) in TFA (6.0 mL) was added trifluoromethanesulfonic acid (1.7 g, 11.3 mmol, 1.0 mL, 10.0 equiv.). The mixture was stirred at 70°C for 30 min. The mixture was concentrated under reduced pressure. The crude product was triturated with MBTE at 25 C for 5 min, then filtered to give 144d. LCMS [M+1] = 315.1. Step E – Synthesis of 3-(5-(4-((4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5- a]pyridin-6-yl)piperazin-1-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (144) To a solution of 1-(6-(piperazin-1-yl)pyrazolo[1,5-a]pyridin-3-yl)dihydropyrimidine- 2,4(1H,3H)-dione trifluoroacetate salt (255.2 mg, 595.9 μmol, 1.5 equiv.) and 3-(5-(2-fluoro-4- formylphenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5- yl)methyl)isoxazole-5-carboxamide (200.0 mg, 397.2 μmol, 1.0 equiv.) in dichloromethane (4.0 mL) was added triethylamine (201.0 mg, 1.9 mmol, 276.4 μL, 5.0 equiv.), the mixture was stirred
at 25°C for 30 min and treated with NaBH(OAc)3 (252.6 mg, 1.1 mmol, 3.0 equiv.), the mixture was stirred at 25°C for 1.5 h. The mixture was poured into water (10 mL). The aqueous phase was extracted with ethyl acetate (5 mLx3). The combined organic phase was washed with brine (5 mLx3), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by prep-HPLC (CD01-Phenomenex Luna C18 150x25mm; 10 µm; [water(FA)-ACN];gradient:10%-40% B over 10 min) to give 144. LCMS [M+1] = 802.4; 1H NMR (400 MHz, METHANOL-d4) δ 9.20 (s, 2H), 8.01 (s, 1H), 7.91 (s, 1H), 7.81 - 7.76 (m, 1H), 7.69 (s, 1H), 7.52 - 7.47 (m, 3H), 7.28 (dd, J = 2.0, 10.0 Hz, 1H), 4.75 (s, 2H), 4.07 (s, 2H), 3.88 (t, J = 6.8 Hz, 2H), 3.30 - 3.27 (m, 4H), 3.08 (s, 4H), 2.87 (t, J = 6.8 Hz, 2H), 1.62 (s, 6H). Example 145: Synthesis of Compound 145
 pyrimidine-2,4(1H,3H)-dione (145a)
To a solution of 3-(4-methoxybenzyl)dihydropyrimidine-2,4(1H,3H)-dione (10.0 g, 42.6 mmol, 1.0 equiv.) and 6-bromo-3-iodopyrazolo[1,5-a]pyrimidine (15.2 g, 46.9 mmol, 1.1 equiv.) in dioxane (250.0 mL) was added K3PO4 (18.1 g, 85.3 mmol, 2.0 equiv.), CuI (4.0 g, 21.3 mmol, 0.5 equiv.) and cesium carbonate (1.7 g, 5.2 mmol, 2.0 equiv.) in one portion at 20°C under a nitrogen atmosphere. The mixture was stirred at 100°C for 6 h. The mixture was quenched by addition water (400 mL) at 25°C. The aqueous phase was extracted with ethyl acetate (400 mL x 3). The combined organic phase was washed with brine (400 mL x 3), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether: ethyl acetate=1:0 to 1:3) to give 145a. LCMS [M+1] = 429.9, 431.8. Step B – Synthesis of tert-butyl 4-(3-(3-(4-methoxybenzyl)-2,4-dioxotetrahydropyrimidin-1(2H)- yl)pyrazolo[1,5-a]pyrimidin-6-yl)piperazine-1-carboxylate (145b) A mixture of 1-(6-bromopyrazolo[1,5-a]pyrimidin-3-yl)-3-(4- methoxybenzyl)dihydropyrimidine-2,4(1H,3H)-dione (4.0 g, 4.0 mmol, 1.0 equiv.), tert-butyl piperazine-1-carboxylate (748.0 mg, 4.0 mmol, 1.0 equiv.), cesium carbonate (3.9 g, 12.0 mmol, 3.0 equiv.) and Pd-PEPPSI-iPentCl (o-picoline) (337.4 mg, 401.6 μmol, 0.1 equiv.) in dioxane (40.0 mL) was degassed and purged threefold with nitrogen, and then the mixture was stirred at 90°C for 1 h under a nitrogen atmosphere. The mixture was quenched with water (100 mL) at 25°C. The aqueous phase was extracted with ethyl acetate (100 mL x 3). The combined organic phase was washed with brine (100 mL x 3), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by prep-HPLC (Phenomenex Luna C18 (250x70mm, 10 µm); [water (FA)-ACN]; gradient:40%-70% B over 20 min) to give 145b. LCMS [M+1] = 536.1. Step C – Synthesis of 1-(6-(piperazin-1-yl)pyrazolo[1,5-a]pyrimidin-3-yl)dihydropyrimidine- 2,4(1H,3H)-dione formic acid salt (145c) To a solution of tert-butyl 4-(3-(3-(4-methoxybenzyl)-2,4-dioxotetrahydropyrimidin- 1(2H)-yl)pyrazolo[1,5-a]pyrimidin-6-yl)piperazine-1-carboxylate (750.0 mg, 1.4 mmol, 1.0 equiv.) in TFA (6.0 mL) and TfOH (1.0 mL). The mixture was stirred at 80°C for 10 min. The mixture was concentrated under reduced pressure. The residue was purified by prep-HPLC
(Phenomenex Luna C18 (250x70mm,10 µm); [water (FA)-ACN]; gradient:1%-15% B over 20 min) to give 145c. LCMS [M+1] = 315.9; 1H NMR (400 MHz, DMSO-d6) δ 10.46 (s, 1H), 8.67 (s, 3H), 8.17 (s, 1H), 3.88 (t, J = 6.7 Hz, 2H), 3.40 - 3.20 (m, 8H), 2.75 (t, J = 6.8 Hz, 2H). Step D – Synthesis of 3-(5-(4-((4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5- a]pyrimidin-6-yl)piperazin-1-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (145) To a solution of 1-(6-(piperazin-1-yl)pyrazolo[1,5-a]pyrimidin-3-yl)dihydropyrimidine- 2,4(1H,3H)-dione formic acid salt (150.0 mg, 415.1 μmol, 1.0 equiv.) and 3-(5-(2-fluoro-4- formylphenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5- yl)methyl)isoxazole-5-carboxamide (208.9 mg, 415.1 μmol, 1.0 equiv.) in THF (2.0 mL) and DMSO (2.0 mL) was added Ti(OEt)4 (473.4 mg, 2.0 mmol, 430.4 μL, 5.0 equiv.) and triethylamine (289 μL, 5.0 equiv.), the mixture was stirred at 50°C for 30 min. Then was added NaBH3CN (39.1 mg, 622.6 μmol, 1.5 equiv.), the mixture was stirred at 20°C for 30 min. The mixture is filtered and the filtrate was purified by prep-HPLC (Phenomenex Kinetex EVO C18 150x30mm, 5µm; [water(FA)-ACN];gradient:0%-30% B over 20 min) to 145. LCMS [M+1] = 803.6; 1H NMR (400 MHz, DMSO-d6) δ 14.05 (s, 1H), 10.45 (s, 1H), 9.77 (s, 1H), 9.25 (d, J = 1.2 Hz, 2H), 8.68 (d, J = 2.4 Hz, 1H), 8.47 (d, J = 2.4 Hz, 1H), 8.35 (s, 1H), 8.12 (s, 1H), 7.78 (t, J = 8.0 Hz, 1H), 7.73 (s, 1H), 7.47 - 7.34 (m, 2H), 4.61 (d, J = 5.2 Hz, 2H), 3.88 (t, J = 6.8 Hz, 2H), 3.67 (s, 2H), 3.18 (s, 4H), 2.78 (t, J = 6.8 Hz, 2H), 2.62 (s, 4H), 1.54 (s, 6H). Example 146: Synthesis of Compound 146
pyrimidine-2,4(1H,3H)-dione (145a)
To a solution of 3-(4-methoxybenzyl)dihydropyrimidine-2,4(1H,3H)-dione (10.0 g, 42.6 mmol, 1.0 equiv.) and 6-bromo-3-iodopyrazolo[1,5-a]pyrimidine (15.2 g, 46.9 mmol, 1.1 equiv.) in dioxane (250.0 mL) was added K3PO4 (18.1 g, 85.3 mmol, 2.0 equiv.), CuI (4.0 g, 21.3 mmol, 0.5 equiv.) and cesium carbonate (1.7 g, 5.2 mmol, 2.0 equiv.) in one portion at 20°C under a nitrogen atmosphere. The mixture was stirred at 100°C for 6 h. The mixture was quenched by addition water (400 mL) at 25°C. The aqueous phase was extracted with ethyl acetate (400 mL x 3). The combined organic phase was washed with brine (400 mL x 3), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether: ethyl acetate=1:0 to 1:3) to give 145a. LCMS [M+1] = 429.9, 431.8. Step B – Synthesis of tert-butyl 4-(3-(3-(4-methoxybenzyl)-2,4-dioxotetrahydropyrimidin-1(2H)- yl)pyrazolo[1,5-a]pyrimidin-6-yl)piperazine-1-carboxylate (145b) A mixture of 1-(6-bromopyrazolo[1,5-a]pyrimidin-3-yl)-3-(4- methoxybenzyl)dihydropyrimidine-2,4(1H,3H)-dione (4.0 g, 4.0 mmol, 1.0 equiv.), tert-butyl piperazine-1-carboxylate (748.0 mg, 4.0 mmol, 1.0 equiv.), cesium carbonate (3.9 g, 12.0 mmol, 3.0 equiv.) and Pd-PEPPSI-iPentCl (o-picoline) (337.4 mg, 401.6 μmol, 0.1 equiv.) in dioxane (40.0 mL) was degassed and purged threefold with nitrogen, and then the mixture was stirred at 90°C for 1 h under a nitrogen atmosphere. The mixture was quenched with water (100 mL) at 25°C. The aqueous phase was extracted with ethyl acetate (100 mL x 3). The combined organic phase was washed with brine (100 mL x 3), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by prep-HPLC (Phenomenex Luna C18 (250x70mm, 10 µm); [water (FA)-ACN]; gradient:40%-70% B over 20 min) to give 145b. LCMS [M+1] = 536.1. Step C – Synthesis of 1-(6-(piperazin-1-yl)pyrazolo[1,5-a]pyrimidin-3-yl)dihydropyrimidine- 2,4(1H,3H)-dione formic acid salt (145c) To a solution of tert-butyl 4-(3-(3-(4-methoxybenzyl)-2,4-dioxotetrahydropyrimidin- 1(2H)-yl)pyrazolo[1,5-a]pyrimidin-6-yl)piperazine-1-carboxylate (750.0 mg, 1.4 mmol, 1.0 equiv.) in TFA (6.0 mL) and TfOH (1.0 mL). The mixture was stirred at 80°C for 10 min. The mixture was concentrated under reduced pressure. The residue was purified by prep-HPLC
(Phenomenex Luna C18 (250x70mm,10 µm); [water (FA)-ACN]; gradient:1%-15% B over 20 min) to give 145c. LCMS [M+1] = 315.9; 1H NMR (400 MHz, DMSO-d6) δ 10.46 (s, 1H), 8.67 (s, 3H), 8.17 (s, 1H), 3.88 (t, J = 6.7 Hz, 2H), 3.40 - 3.20 (m, 8H), 2.75 (t, J = 6.8 Hz, 2H). Step D – Synthesis of 3-(5-(4-((4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5- a]pyrimidin-6-yl)piperazin-1-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (145) To a solution of 1-(6-(piperazin-1-yl)pyrazolo[1,5-a]pyrimidin-3-yl)dihydropyrimidine- 2,4(1H,3H)-dione formic acid salt (150.0 mg, 415.1 μmol, 1.0 equiv.) and 3-(5-(2-fluoro-4- formylphenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5- yl)methyl)isoxazole-5-carboxamide (208.9 mg, 415.1 μmol, 1.0 equiv.) in THF (2.0 mL) and DMSO (2.0 mL) was added Ti(OEt)4 (473.4 mg, 2.0 mmol, 430.4 μL, 5.0 equiv.) and triethylamine (289 μL, 5.0 equiv.), the mixture was stirred at 50°C for 30 min. Then was added NaBH3CN (39.1 mg, 622.6 μmol, 1.5 equiv.), the mixture was stirred at 20°C for 30 min. The mixture is filtered and the filtrate was purified by prep-HPLC (Phenomenex Kinetex EVO C18 150x30mm, 5µm; [water(FA)-ACN];gradient:0%-30% B over 20 min) to 145. LCMS [M+1] = 803.6; 1H NMR (400 MHz, DMSO-d6) δ 14.05 (s, 1H), 10.45 (s, 1H), 9.77 (s, 1H), 9.25 (d, J = 1.2 Hz, 2H), 8.68 (d, J = 2.4 Hz, 1H), 8.47 (d, J = 2.4 Hz, 1H), 8.35 (s, 1H), 8.12 (s, 1H), 7.78 (t, J = 8.0 Hz, 1H), 7.73 (s, 1H), 7.47 - 7.34 (m, 2H), 4.61 (d, J = 5.2 Hz, 2H), 3.88 (t, J = 6.8 Hz, 2H), 3.67 (s, 2H), 3.18 (s, 4H), 2.78 (t, J = 6.8 Hz, 2H), 2.62 (s, 4H), 1.54 (s, 6H). Example 146: Synthesis of Compound 146
St The 4,6-dichloropyrazolo[1,5-a]pyrazine (45.0 g, 1.0 equiv.) in toluene (225.0 mL) was pumped by Pump 1 S1,P1,6.793 mL/min to flow reactor 1 FLR1,PFA,Coils reactor,3.175(1/8’’)mm, 68.06 mL, 25°C under a nitrogen atmosphere. The tributylstannane (153.2 g, 2.2 equiv.) in toluene. (225.0 mL) was pumped by Pump 2 S2, P2, 6.819 mL/min to flow reactor 1 FLR1, PFA,Coils reactor,3.175(1/8’’)mm, 68.06 mL, 25°C under a nitrogen atmosphere. The residence time of flow reactor 1 was FLR1, 5 min. The mixture was collected with a bottle. The Pump 1 and Pump 2 was started at the same time. The mixture was collected after running 3 mins. Collection of the mixture was stopped after 30 mins. The combined mixture was treated with KF aqueous (300 mL). The aqueous phase was extracted with ethyl acetate (500 mLx3). The combined organic phase was washed with brine (300 mLx2), dried with anhydrous sodium sulfate, filtered and filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether: ethyl acetate = 5:1) to give 146a. LCMS [M+1] = 153.9, 155.9.
Step B – Synthesis of 6-chloro-3-iodopyrazolo[1,5-a]pyrazine (146b) To a solution of 6-chloropyrazolo-[1,5-a]-pyrazine (30.0 g, 195.3 mmol, 1.0 equiv.) in DMF (2.0 mL) was added NIS (87.9 g, 390.7 mmol, 2.0 equiv.). The mixture was stirred at 100°C for 3 h. The mixture was quenched by the addition of Na2S2O3 (300 mL). The aqueous phase was extracted with ethyl acetate (200 mLx3). The combined organic phase was washed with NaHCO3 (200 mLx2) and brine (300 mLx2), dried with anhydrous sodium sulfate, filtered and filtrate was concentrated under reduced pressure to give 146b. LCMS [M+1] = 279.8, 281.8. Step C – Synthesis of 1-(6-chloropyrazolo[1,5-a]pyrazin-3-yl)-3-(4-methoxybenzyl)- dihydropyrimidine-2,4(1H,3H)-dione (146c) To a solution of 6-chloro-3-iodopyrazolo[1,5-a]pyrazine (49.0 g, 175.3 mmol, 1.0 equiv.) and 3-(4-methoxybenzyl)dihydropyrimidine-2,4(1H,3H)-dione (40.0 g, 170.7 mmol, 1.0 equiv.) in DMF (500.0 mL) was added K3PO4 (72.4 g, 341.5 mmol, 2.0 equiv.), (1S,2S)-N1,N2- dimethylcyclohexane-1,2-diamine (12.1 g, 85.3 mmol, 0.5 equiv.) and CuI (16.2 g, 85.3 mmol, 0.5 equiv.). The mixture was stirred at 60°C for 1 h. The mixture was cooled to 25°C treated with water (500 mL). The aqueous phase was extracted with ethyl acetate (300 mLx3). The combined organic phase was washed with brine (500 mLx2), dried with anhydrous sodium sulfate, filtered and filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether: ethyl acetate=1:1) to give 146c. LCMS [M+1] = 385.9, 387.9. Step D – Synthesis of 1-(6-chloropyrazolo[1,5-a]pyrazin-3-yl)dihydropyrimidine-2,4(1H,3H)- dione (146d) A solution of 1-(6-chloropyrazolo[1,5-a]pyrazin-3-yl)-3-(4- methoxybenzyl)dihydropyrimidine-2,4(1H,3H)-dione (20.0 g, 51.8 mmol, 1.0 equiv.) and TFA (33.0 mL) in TfOH (200.0 mL) was stirred at 70°C for 1 h. The mixture was concentrated to dryness. The residue was triturated with MTBE (100 mL) and the solids were collected by vacuum filtration to afford 146d. LCMS [M+1] = 265.8, 267.8. Step E – Synthesis of tert-butyl 4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5- a]pyrazin-6-yl)piperazine-1-carboxylate (146e)
To solution of tert-butyl piperazine-1-carboxylate (2.1 g, 11.3 mmol, 1.5 equiv.), 1-(6- chloropyrazolo[1,5-a]pyrazin-3-yl)dihydropyrimidine-2,4(1H,3H)-dione (2.0 g, 7.5 mmol, 1.0 equiv.) and cesium carbonate (7.3 g, 22.5 mmol, 3.0 equiv.) in dioxane (20.0 mL) and was added Pd-PEPPSI-iPentCl (o-picoline) (316.2 mg, 376.4 μmol, 0.1 equiv.) under a nitrogen atmosphere at 25°C. The reaction was stirred at 90°C for 1 h. The mixture was cooled to 25°C quenched by water (100 mL) and extracted with ethyl acetate (50 mL x 3). The combined organic layers were washed with brine (50 mL), dried over anhydrous sodium sulfate, concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, Petroleum ether: Ethyl acetate=1:0 to 0:1) to give 146e. LCMS [M+1] = 415.8. Step F – Synthesis of 1-(6-(piperazin-1-yl)pyrazolo[1,5-a]pyrazin-3-yl)dihydropyrimidine- 2,4(1H,3H)-dione trifluoroacetate salt (146f) A solution of tert-butyl 4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5- a]pyrazin-6-yl)piperazine-1-carboxylate (1.3 g, 3.1 mmol, 1.0 equiv.), in dichloromethane (10.0 mL) and TFA (3.0 mL) under a nitrogen atmosphere at 25°C. The mixture was stirred at this temperature for 30 min. The mixture was filtered and the filtrate was concentrated under reduced pressure. The residue was purified by prep-HPLC (05-Phenomenex Luna C18150x40mm, 10 µm; [water (FA)-ACN]; gradient:0%-10% B over 10 min) to give 146f. LCMS [M+1] = 316.0; 1H NMR (400 MHz, DMSO-d6) δ 10.54 (s, 1H), 8.93 (d, J = 1.2 Hz, 1H), 8.22 (d, J = 1.2 Hz, 1H), 8.00 (s, 1H), 3.87 (t, J = 6.8 Hz, 2H), 3.62 - 3.54 (m, 4H), 3.29 - 3.23 (m, 4H), 2.78 (t, J = 6.8 Hz, 2H). Step G – Synthesis of 3-(5-(4-((4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5- a]pyrazin-6-yl)piperazin-1-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (146) To a solution of 1-(6-piperazin-1-ylpyrazolo[1,5-a]pyrazin-3-yl)hexahydropyrimidine-2,4- dione trifluoroacetate salt (1.0 g, 2.3 mmol, 1.2 equiv., TFA) and 3-(5-(2-fluoro-4- formylphenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5- yl)methyl)isoxazole-5-carboxamide (1.0 g, 1.9 mmol, 1 equiv.) in DMSO (10.0 mL) was added Ti(OEt)4 (906.2 mg, 3.9 mmol, 823.8 μL, 2.0 equiv.), the mixture was stirred at 20°C for 30 min, treated with NaBH3CN (187.2 mg, 2.9 mmol, 1.5 equiv.) and stirred at 20°C for 30 min. The
mixture was quenched by water (50 mL) and extracted with dichloromethane (20 mL x 3). The combined organic layers were washed with brine (20 mL x 3), dried over anhydrous sodium sulfate, concentrated under reduced pressure. The residue was purified by prep-HPLC (Phenomenex Luna C18 (250x70mm,10 µm); [water (FA)-ACN]; gradient:12%-42% B over 64 min) to give 146. LCMS [M+1] = 803.4; 1H NMR (400 MHz, METHANOL-d4) δ 9.18 (d, J = 1.2 Hz, 2H), 8.86 (d, J = 1.2 Hz, 1H), 8.26 (s, 1H), 7.92 (s, 1H), 7.86 (d, J = 1.2 Hz, 1H), 7.74 - 7.66 (m, 2H), 7.46 - 7.37 (m, 2H), 4.75 (s, 2H), 3.97 (t, J = 6.8 Hz, 2H), 3.73 (s, 2H), 3.50 - 3.36 (m, 4H), 2.89 (t, J = 6.8 Hz, 2H), 2.77 - 2.64 (m, 4H), 1.62 (s, 6H). Example 147: Synthesis of Compound 147
 - yl)pyrazolo[1,5-a]pyrazin-6-yl)-3,6-dihydropyridine-1(2H)-carboxylate (147a) To a solution of 1-(6-chloropyrazolo[1,5-a]pyrazin-3-yl)-3-(4- methoxybenzyl)dihydropyrimidine-2,4(1H,3H)-dione (5.0 g, 12.9 mmol, 1.0 equiv.) and tert- butyl 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-5,6-dihydropyridine-1(2H)-carboxylate (4.8 g, 15.6 mmol, 1.2 equiv.) in dioxane (50.0 mL) and water (10.0 mL) was added Pd(dppf)Cl2 (948.3 mg, 1.3 mmol, 0.1 equiv.) and K2CO3 (5.4 g, 38.9 mmol, 3.0 equiv.) under a nitrogen atmosphere. The reaction was stirred at 100°C for 2 h. The mixture was diluted with water (100 mL) and extracted with ethyl acetate (100 mLx3). The combined organic layers were dried over
anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, Petroleum ether/Ethyl acetate = 1/0 to 1/1) to give 147a. 1HNMR (400 MHz, DMSO-d6) δ 9.06 (s, 1H), 8.64 (s, 1H), 8.19 (s, 1H), 7.26 (d, J = 8.4 Hz, 2H), 6.87 (d, J = 8.4 Hz, 2H), 6.83 (s, 1H), 4.85 (s, 2H), 4.09 - 4.05 (m, 2H), 3.93 (t, J = 6.4 Hz, 2H), 3.72 (s, 3H), 3.57 (t, J = 4.8 Hz, 2H), 3.21 (dd, J = 4.4, 6.6 Hz, 1H), 2.98 (t, J = 6.4 Hz, 2H), 2.62 (t, J = 6.8 Hz, 1H), 1.43 (s, 9H). Step B – Synthesis of tert-butyl 4-(3-(3-(4-methoxybenzyl)-2,4-dioxotetrahydropyrimidin-1(2H)- yl)pyrazolo[1,5-a]pyrazin-6-yl)piperidine-1-carboxylate (147b) To a solution of tert-butyl 4-(3-(3-(4-methoxybenzyl)-2,4-dioxotetrahydropyrimidin- 1(2H)-yl)pyrazolo[1,5-a]pyrazin-6-yl)-3,6-dihydropyridine-1(2H)-carboxylate (500.0 mg, 938.8 μmol, 1.0 equiv.) in ethyl acetate (100.0 mL) was added Pd/C (99.9 mg, 10 wt%) under a nitrogen atmosphere. The reaction was stirred at 25°C for 30 min under hydrogen gas (15 Psi). The mixture was filtered and the filtrate was concentrated under reduced pressure. The residue was purified by reversed phase flash chromatography (C18, 0.1% FA in acetonitrile/ water) to give 147b. LCMS [M+1] = 535.3. Step C – Synthesis of 1-(6-(piperidin-4-yl)pyrazolo[1,5-a]pyrazin-3-yl)dihydropyrimidine- 2,4(1H,3H)-dione trifluoroacetate salt (147c) To a solution of tert-butyl 4-(3-(3-(4-methoxybenzyl)-2,4-dioxotetrahydropyrimidin- 1(2H)-yl)pyrazolo[1,5-a]pyrazin-6-yl)piperidine-1-carboxylate (160.0 mg, 299.3 μmol, 1.0 equiv.) in TFA (7.7 g, 67.3 mmol, 5.0 mL, 224.9 equiv.) and TfOH (1.3 g, 8.5 mmol, 0.7 mL, 28.3 equiv.). The reaction was stirred at 80°C for 30 min. The reaction was concentrated under reduced pressure. The reaction was triturated with MTBE (25.0 mL) and the solids were collected by vacuum filtration to afford 147c. LCMS [M+1] = 315.1. Step D – Synthesis of 3-(5-(4-((4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5- a]pyrazin-6-yl)piperidin-1-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (147) To a solution of 3-(5-(2-fluoro-4-formylphenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (90.0 mg, 178.8 μmol,
1.0 equiv.) and 1-(6-(piperidin-4-yl)pyrazolo[1,5-a]pyrazin-3-yl)dihydropyrimidine-2,4(1H,3H)- dione trifluoroacetate salt (107.2 mg, 250.3 μmol, 1.4 equiv.) in dichloromethane (2.0 mL) and DMSO (0.5 mL) was added triethylamine (124 μL, 5.0 equiv.). The reaction was stirred at 25°C for 12 h, treated with NaBH(OAc)3 (151.6 mg, 715.1 μmol, 4.0 equiv.) and stirred at 25°C for 1 h. The mixture was concentrated under reduced pressure and the residue was purified by prep-HPLC (Phenomenex Luna C18150x25mm, 10 µm; [water (FA)-ACN]; gradient: 16%-36% B over 10 min) to give 147. LCMS [M+1] = 802.2; 1H NMR (400 MHz, METHANOL-d4) δ 9.20 (s, 2H), 9.08 (s, J = 0.8 Hz, 1H), 8.43 (s, 1H), 8.11 (s, 1H), 7.82 (t, J = 7.6 Hz, 1H), 7.69 (s, 1H), 7.53 (d, J = 9.6 Hz, 2H), 4.75 (s, 2H), 4.21 (s, 2H), 4.00 (t, J = 6.8 Hz, 2H), 3.47 (d, J = 12.0 Hz, 2H), 3.04 - 2.96 (m, 1H), 2.91 (t, J = 6.8 Hz, 4H), 2.21 - 2.07 (m, 4H), 1.62 (s, 6H). Example 148: Synthesis of Compound 148-1 and 148-2
- yl)pyrazolo[1,5-a]pyrazin-6-yl)-3,6-dihydropyridine-1(2H)-carboxylate (147a) To a solution of 1-(6-chloropyrazolo[1,5-a]pyrazin-3-yl)-3-(4- methoxybenzyl)dihydropyrimidine-2,4(1H,3H)-dione (5.0 g, 12.9 mmol, 1.0 equiv.) and tert- butyl 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-5,6-dihydropyridine-1(2H)-carboxylate (4.8 g, 15.6 mmol, 1.2 equiv.) in dioxane (50.0 mL) and water (10.0 mL) was added Pd(dppf)Cl2 (948.3 mg, 1.3 mmol, 0.1 equiv.) and K2CO3 (5.4 g, 38.9 mmol, 3.0 equiv.) under a nitrogen atmosphere. The reaction was stirred at 100°C for 2 h. The mixture was diluted with water (100 mL) and extracted with ethyl acetate (100 mLx3). The combined organic layers were dried over
anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, Petroleum ether/Ethyl acetate = 1/0 to 1/1) to give 147a. 1HNMR (400 MHz, DMSO-d6) δ 9.06 (s, 1H), 8.64 (s, 1H), 8.19 (s, 1H), 7.26 (d, J = 8.4 Hz, 2H), 6.87 (d, J = 8.4 Hz, 2H), 6.83 (s, 1H), 4.85 (s, 2H), 4.09 - 4.05 (m, 2H), 3.93 (t, J = 6.4 Hz, 2H), 3.72 (s, 3H), 3.57 (t, J = 4.8 Hz, 2H), 3.21 (dd, J = 4.4, 6.6 Hz, 1H), 2.98 (t, J = 6.4 Hz, 2H), 2.62 (t, J = 6.8 Hz, 1H), 1.43 (s, 9H). Step B – Synthesis of tert-butyl 4-(3-(3-(4-methoxybenzyl)-2,4-dioxotetrahydropyrimidin-1(2H)- yl)pyrazolo[1,5-a]pyrazin-6-yl)piperidine-1-carboxylate (147b) To a solution of tert-butyl 4-(3-(3-(4-methoxybenzyl)-2,4-dioxotetrahydropyrimidin- 1(2H)-yl)pyrazolo[1,5-a]pyrazin-6-yl)-3,6-dihydropyridine-1(2H)-carboxylate (500.0 mg, 938.8 μmol, 1.0 equiv.) in ethyl acetate (100.0 mL) was added Pd/C (99.9 mg, 10 wt%) under a nitrogen atmosphere. The reaction was stirred at 25°C for 30 min under hydrogen gas (15 Psi). The mixture was filtered and the filtrate was concentrated under reduced pressure. The residue was purified by reversed phase flash chromatography (C18, 0.1% FA in acetonitrile/ water) to give 147b. LCMS [M+1] = 535.3. Step C – Synthesis of 1-(6-(piperidin-4-yl)pyrazolo[1,5-a]pyrazin-3-yl)dihydropyrimidine- 2,4(1H,3H)-dione trifluoroacetate salt (147c) To a solution of tert-butyl 4-(3-(3-(4-methoxybenzyl)-2,4-dioxotetrahydropyrimidin- 1(2H)-yl)pyrazolo[1,5-a]pyrazin-6-yl)piperidine-1-carboxylate (160.0 mg, 299.3 μmol, 1.0 equiv.) in TFA (7.7 g, 67.3 mmol, 5.0 mL, 224.9 equiv.) and TfOH (1.3 g, 8.5 mmol, 0.7 mL, 28.3 equiv.). The reaction was stirred at 80°C for 30 min. The reaction was concentrated under reduced pressure. The reaction was triturated with MTBE (25.0 mL) and the solids were collected by vacuum filtration to afford 147c. LCMS [M+1] = 315.1. Step D – Synthesis of 3-(5-(4-((4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5- a]pyrazin-6-yl)piperidin-1-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (147) To a solution of 3-(5-(2-fluoro-4-formylphenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (90.0 mg, 178.8 μmol,
1.0 equiv.) and 1-(6-(piperidin-4-yl)pyrazolo[1,5-a]pyrazin-3-yl)dihydropyrimidine-2,4(1H,3H)- dione trifluoroacetate salt (107.2 mg, 250.3 μmol, 1.4 equiv.) in dichloromethane (2.0 mL) and DMSO (0.5 mL) was added triethylamine (124 μL, 5.0 equiv.). The reaction was stirred at 25°C for 12 h, treated with NaBH(OAc)3 (151.6 mg, 715.1 μmol, 4.0 equiv.) and stirred at 25°C for 1 h. The mixture was concentrated under reduced pressure and the residue was purified by prep-HPLC (Phenomenex Luna C18150x25mm, 10 µm; [water (FA)-ACN]; gradient: 16%-36% B over 10 min) to give 147. LCMS [M+1] = 802.2; 1H NMR (400 MHz, METHANOL-d4) δ 9.20 (s, 2H), 9.08 (s, J = 0.8 Hz, 1H), 8.43 (s, 1H), 8.11 (s, 1H), 7.82 (t, J = 7.6 Hz, 1H), 7.69 (s, 1H), 7.53 (d, J = 9.6 Hz, 2H), 4.75 (s, 2H), 4.21 (s, 2H), 4.00 (t, J = 6.8 Hz, 2H), 3.47 (d, J = 12.0 Hz, 2H), 3.04 - 2.96 (m, 1H), 2.91 (t, J = 6.8 Hz, 4H), 2.21 - 2.07 (m, 4H), 1.62 (s, 6H). Example 148: Synthesis of Compound 148-1 and 148-2
 To a solution of 2-(4-bromo-2-fluorophenyl)acetonitrile (5.0 g, 23.3 mmol, 1.0 equiv.) and CH3I (3.3 g, 23.3 mmol, 1.4 mL, 1.0 equiv.) in THF (100.0 mL) was cooled to -78°C and the NaHMDS (1.0 M, 23.36 mL, 1.0 equiv.) was added dropwise. The mixture was stirred at -78°C for 15 min under nitrogen and warmed gradually to 25°C, stirred at 25°C for 10 h. The mixture was quenched with ice-water (20 mL) at 0°C and extracted with ethyl acetate (20 mLx3). Combined organic layers were washed with brine (40.0 mLx2), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure to give 148a.
1H NMR (400 MHz, DMSO-d6) δ 7.71 - 7.54 (m, 1H), 7.52 - 7.39 (m, 2H), 4.45 (q, J = 7.2 Hz, 1H), 1.54 (d, J = 7.2 Hz, 3H). Step B – Synthesis of tert-butyl 4-(4-bromo-2-fluorophenyl)-4-cyanopentanoate (148b) To a solution of 2-(4-bromo-2-fluorophenyl)propanenitrile (3.0 g, 13.1 mmol, 1.0 equiv.), tert-butyl acrylate (3.3 g, 26.3 mmol, 3.8 mL, 2.0 equiv.), K2CO3 (3.6 g, 26.3 mmol, 2.0 equiv.) and BTEAC (599.2 mg, 2.6 mmol, 0.2 equiv.) in toluene (30 mL) was stirred at 90°C for 16 h. The mixture was cooled to 25 °C, diluted with water (20 mL) and extracted with ethyl acetate (20.0 mLx3). The combined organic layers were washed with brine (60 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel (I 0~15% Ethyl acetate/Petroleum ether) to give 148b. LCMS [M+Na+] =377.9, 379.9. Step C – Synthesis of 3-(4-bromo-2-fluorophenyl)-3-methylpiperidine-2,6-dione (148c) To a solution of tert-butyl 4-(4-bromo-2-fluorophenyl)-4-cyanopentanoate (3.7 g, 10.3 mmol, 1.0 equiv.) in acetic acid (37.0 mL) was added H2SO4 (1.1 mL, 2.0 equiv.). The mixture was stirred at 120°C for 2 h. The mixture was cooled to 25 °C and purified by prep-HPLC (Phenomenex Luna C18, 150mm x 25mm, 10 µm; [water(FA)-ACN];gradient:25%-55% B over 11 min ) to give 148c. LCMS [M+1] =299.8. Step D – Synthesis of tert-butyl 6-(3-fluoro-4-(3-methyl-2,6-dioxopiperidin-3-yl)phenyl)-2- azaspiro[3.3]heptane-2-carboxylate (148d) A mixture of Zn (261.4 mg, 4.0 mmol, 2.0 equiv.) in DMA (2.0 mL) was degassed and purged threefold with nitrogen and stirred at 25°C for 10 min. Then the 3-(4-bromo-2- fluorophenyl)-3-methylpiperidine-2,6-dione (600.0 mg, 2.0 mmol, 1.0 equiv.), tert-butyl 6-iodo- 2-azaspiro[3.3]heptane-2-carboxylate (775.2 mg, 2.4 mmol, 1.2 equiv.), 4-methoxypyridine-2- carboxamidine hydrochloride (37.5 mg, 199.9 μmol, 0.1 equiv.) and dichloronickel•1,2- dimethoxyethane (43.9 mg, 199.9 μmol, 0.1 equiv.) in DMA (4.0 mL) was added and threefold purged with nitrogen. TFA (15 μL, 0.1 equiv.) was added and the mixture was stirred at 80°C for 1 h under a nitrogen atmosphere. The mixture was cooled to 25°C, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel chromatography
(0~25% Ethyl acetate/Petroleum ether) to give 148d. LCMS [M+1] =361.3; 1H NMR (400 MHz, DMSO-d6) δ 10.87 (s, 1H), 7.27 (t, J = 8.0 Hz, 1H), 7.12 - 6.97 (m, 2H), 3.96 (s, 2H), 3.75 (s, 2H), 3.41 - 3.34 (m, 1H), 2.77 - 2.62 (m, 1H), 2.56 - 2.52 (m, 1H), 2.48 (s, 1H), 2.38 - 2.27 (m, 1H), 2.26 - 2.18 (m, 3H), 1.81 (td, J = 5.2, 13.2 Hz, 1H), 1.58 (s, 3H), 1.37 (s, 9H). Step E – Synthesis of 3-(2-fluoro-4-(2-azaspiro[3.3]heptan-6-yl)phenyl)-3-methylpiperidine-2,6- dione trifluoroacetate salt (148e) A mixture of tert-butyl 6-(3-fluoro-4-(3-methyl-2,6-dioxopiperidin-3-yl)phenyl)-2- azaspiro[3.3]heptane-2-carboxylate (440.0 mg, 1.0 mmol, 1.0 equiv.) in TFA (1.0 mL) and dichloromethane (3.0 mL) was stirred at 25°C for 1 h. The mixture was concentrated under reduced pressure to give 148e, which was used without purification. LCMS [M+1] =317.1. Step F – Synthesis of 3-(5-(2-fluoro-4-((6-(3-fluoro-4-(3-methyl-2,6-dioxopiperidin-3-yl)phenyl)- 2-azaspiro[3.3]heptan-2-yl)methyl)phenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan- 2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (148-1 and 148-2) To a solution of 3-(2-fluoro-4-(2-azaspiro[3.3]heptan-6-yl)phenyl)-3-methylpiperidine- 2,6-dione trifluoroacetate salt (300.0 mg, 948.2 μmol, 1.2 equiv.) and 3-(5-(2-fluoro-4- formylphenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5- yl)methyl)isoxazole-5-carboxamide (397.8 mg, 790.2 μmol, 1.0 equiv.) in DMSO (3.0 mL) was added triethylamine (220 μL, 2.0 equiv.). The mixture was stirred for 30 min and then treated with NaBH(OAc)3 (502.4 mg, 2.3 mmol, 3.0 equiv.). The mixture was stirred at 25°C for 1 h. The crude product was purified by prep-HPLC (Phenomenex Luna C18 (250x70 mm, 10 µm); [water(FA)-ACN];gradient:20%-50% B over 20 min ) to afford the product as the racemate. LCMS [M+1] =804.6; 1H NMR (400 MHz, DMSO-d6) δ 14.00 (s, 1H), 10.87 (s, 1H), 9.74 (t, J = 5.6 Hz, 1H), 9.24 (s, 2H), 7.79 - 7.70 (m, 2H), 7.38 - 7.31 (m, 2H), 7.29 - 7.24 (m, 1H), 7.08 - 7.00 (m, 2H), 4.61 (d, J = 6.0 Hz, 2H), 3.80 - 3.57 (m, 2H), 3.43 - 3.36 (m, 2H), 3.24 - 3.08 (m, 2H), 2.77 - 2.65 (m, 1H), 2.54 (s, 2H), 2.41 - 2.12 (m, 5H), 1.85 - 1.76 (m, 1H), 1.58 (s, 3H), 1.55 (s, 6H).
The enantiomers were separated with SFC (S,S-WHELK-O1 (250x30mm, 10 µm); [CO2- isopropanol/ACN];B%:40%, isocratic elution) to afford 148-1 as the early eluting isomer and 148-2 as the late eluting isomer. Example 149: Synthesis of Intermediate 149
To a solution of 2-(4-bromo-2-fluorophenyl)acetonitrile (5.0 g, 23.3 mmol, 1.0 equiv.) and CH3I (3.3 g, 23.3 mmol, 1.4 mL, 1.0 equiv.) in THF (100.0 mL) was cooled to -78°C and the NaHMDS (1.0 M, 23.36 mL, 1.0 equiv.) was added dropwise. The mixture was stirred at -78°C for 15 min under nitrogen and warmed gradually to 25°C, stirred at 25°C for 10 h. The mixture was quenched with ice-water (20 mL) at 0°C and extracted with ethyl acetate (20 mLx3). Combined organic layers were washed with brine (40.0 mLx2), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure to give 148a.
1H NMR (400 MHz, DMSO-d6) δ 7.71 - 7.54 (m, 1H), 7.52 - 7.39 (m, 2H), 4.45 (q, J = 7.2 Hz, 1H), 1.54 (d, J = 7.2 Hz, 3H). Step B – Synthesis of tert-butyl 4-(4-bromo-2-fluorophenyl)-4-cyanopentanoate (148b) To a solution of 2-(4-bromo-2-fluorophenyl)propanenitrile (3.0 g, 13.1 mmol, 1.0 equiv.), tert-butyl acrylate (3.3 g, 26.3 mmol, 3.8 mL, 2.0 equiv.), K2CO3 (3.6 g, 26.3 mmol, 2.0 equiv.) and BTEAC (599.2 mg, 2.6 mmol, 0.2 equiv.) in toluene (30 mL) was stirred at 90°C for 16 h. The mixture was cooled to 25 °C, diluted with water (20 mL) and extracted with ethyl acetate (20.0 mLx3). The combined organic layers were washed with brine (60 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel (I 0~15% Ethyl acetate/Petroleum ether) to give 148b. LCMS [M+Na+] =377.9, 379.9. Step C – Synthesis of 3-(4-bromo-2-fluorophenyl)-3-methylpiperidine-2,6-dione (148c) To a solution of tert-butyl 4-(4-bromo-2-fluorophenyl)-4-cyanopentanoate (3.7 g, 10.3 mmol, 1.0 equiv.) in acetic acid (37.0 mL) was added H2SO4 (1.1 mL, 2.0 equiv.). The mixture was stirred at 120°C for 2 h. The mixture was cooled to 25 °C and purified by prep-HPLC (Phenomenex Luna C18, 150mm x 25mm, 10 µm; [water(FA)-ACN];gradient:25%-55% B over 11 min ) to give 148c. LCMS [M+1] =299.8. Step D – Synthesis of tert-butyl 6-(3-fluoro-4-(3-methyl-2,6-dioxopiperidin-3-yl)phenyl)-2- azaspiro[3.3]heptane-2-carboxylate (148d) A mixture of Zn (261.4 mg, 4.0 mmol, 2.0 equiv.) in DMA (2.0 mL) was degassed and purged threefold with nitrogen and stirred at 25°C for 10 min. Then the 3-(4-bromo-2- fluorophenyl)-3-methylpiperidine-2,6-dione (600.0 mg, 2.0 mmol, 1.0 equiv.), tert-butyl 6-iodo- 2-azaspiro[3.3]heptane-2-carboxylate (775.2 mg, 2.4 mmol, 1.2 equiv.), 4-methoxypyridine-2- carboxamidine hydrochloride (37.5 mg, 199.9 μmol, 0.1 equiv.) and dichloronickel•1,2- dimethoxyethane (43.9 mg, 199.9 μmol, 0.1 equiv.) in DMA (4.0 mL) was added and threefold purged with nitrogen. TFA (15 μL, 0.1 equiv.) was added and the mixture was stirred at 80°C for 1 h under a nitrogen atmosphere. The mixture was cooled to 25°C, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel chromatography
(0~25% Ethyl acetate/Petroleum ether) to give 148d. LCMS [M+1] =361.3; 1H NMR (400 MHz, DMSO-d6) δ 10.87 (s, 1H), 7.27 (t, J = 8.0 Hz, 1H), 7.12 - 6.97 (m, 2H), 3.96 (s, 2H), 3.75 (s, 2H), 3.41 - 3.34 (m, 1H), 2.77 - 2.62 (m, 1H), 2.56 - 2.52 (m, 1H), 2.48 (s, 1H), 2.38 - 2.27 (m, 1H), 2.26 - 2.18 (m, 3H), 1.81 (td, J = 5.2, 13.2 Hz, 1H), 1.58 (s, 3H), 1.37 (s, 9H). Step E – Synthesis of 3-(2-fluoro-4-(2-azaspiro[3.3]heptan-6-yl)phenyl)-3-methylpiperidine-2,6- dione trifluoroacetate salt (148e) A mixture of tert-butyl 6-(3-fluoro-4-(3-methyl-2,6-dioxopiperidin-3-yl)phenyl)-2- azaspiro[3.3]heptane-2-carboxylate (440.0 mg, 1.0 mmol, 1.0 equiv.) in TFA (1.0 mL) and dichloromethane (3.0 mL) was stirred at 25°C for 1 h. The mixture was concentrated under reduced pressure to give 148e, which was used without purification. LCMS [M+1] =317.1. Step F – Synthesis of 3-(5-(2-fluoro-4-((6-(3-fluoro-4-(3-methyl-2,6-dioxopiperidin-3-yl)phenyl)- 2-azaspiro[3.3]heptan-2-yl)methyl)phenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan- 2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (148-1 and 148-2) To a solution of 3-(2-fluoro-4-(2-azaspiro[3.3]heptan-6-yl)phenyl)-3-methylpiperidine- 2,6-dione trifluoroacetate salt (300.0 mg, 948.2 μmol, 1.2 equiv.) and 3-(5-(2-fluoro-4- formylphenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5- yl)methyl)isoxazole-5-carboxamide (397.8 mg, 790.2 μmol, 1.0 equiv.) in DMSO (3.0 mL) was added triethylamine (220 μL, 2.0 equiv.). The mixture was stirred for 30 min and then treated with NaBH(OAc)3 (502.4 mg, 2.3 mmol, 3.0 equiv.). The mixture was stirred at 25°C for 1 h. The crude product was purified by prep-HPLC (Phenomenex Luna C18 (250x70 mm, 10 µm); [water(FA)-ACN];gradient:20%-50% B over 20 min ) to afford the product as the racemate. LCMS [M+1] =804.6; 1H NMR (400 MHz, DMSO-d6) δ 14.00 (s, 1H), 10.87 (s, 1H), 9.74 (t, J = 5.6 Hz, 1H), 9.24 (s, 2H), 7.79 - 7.70 (m, 2H), 7.38 - 7.31 (m, 2H), 7.29 - 7.24 (m, 1H), 7.08 - 7.00 (m, 2H), 4.61 (d, J = 6.0 Hz, 2H), 3.80 - 3.57 (m, 2H), 3.43 - 3.36 (m, 2H), 3.24 - 3.08 (m, 2H), 2.77 - 2.65 (m, 1H), 2.54 (s, 2H), 2.41 - 2.12 (m, 5H), 1.85 - 1.76 (m, 1H), 1.58 (s, 3H), 1.55 (s, 6H).
The enantiomers were separated with SFC (S,S-WHELK-O1 (250x30mm, 10 µm); [CO2- isopropanol/ACN];B%:40%, isocratic elution) to afford 148-1 as the early eluting isomer and 148-2 as the late eluting isomer. Example 149: Synthesis of Intermediate 149
 Step A – Synthesis of 5-bromo-N'-hydroxy-4-methylpyrimidine-2-carboximidamide (149a) To a mixture of 1-bromo-2-fluoro-4-iodobenzene (180 g, 0.909 mol, 1.0 equiv.) and NH2OH.HCl (66.32 g, 0.954 mol, 1.05 equiv.) in MeOH (3.6 L) was added sodium methoxide (171.88 g, 0.954 mol, 1.0 equiv.) and the mixture was stirred at 70°C for 2 h. The mixture was concentrated under reduce pressure. The residue was diluted with water (1500 mL) and filtered to afford 149a. LCMS [M+1] = 230.8, 232.8. Step B – Synthesis of 5-bromo-N-hydroxy-4-methylpyrimidine-2-carbimidoyl chloride (149b) To a mixture of 5-bromo-N'-hydroxy-4-methylpyrimidine-2-carboximidamide (195 g, 0.843 mol, 1.0 equiv.) in HCl aqueous (4 M, 1.9 L, 9.0 equiv.) was added NaNO2 (72.78 g, 1.056 mol, 1.2 equiv.) in water (1.95 L) at 0°C and stirred at 0°C for 2 h. The mixture was diluted with water (400 mL) and filtered. The filtrate was washed with water (500 mL) and concentrated under reduce pressure to give 149b. LCMS [M+1] = 249.9, 251.9. Step C – Synthesis of tert-butyl 3-(5-bromo-4-methylpyrimidin-2-yl)isoxazole-5-carboxylate (149) A mixture of tert-butyl propiolate (302.2 g, 2.4 mol, 329 mL, 3.0 equiv.) and triethylamine (13.8 mL, 1.0 equiv.) in acetonitrile (4 L) was treated with a solution of 5-bromo-N-hydroxy-4- methylpyrimidine-2-carbimidoyl chloride (200 g, 0.8 mol, 1.0 equiv.) in acetonitrile (4 L). The
mixture was stirred at 20°C for 2 h. The mixture was concentrated under reduce pressure. The residue was triturated with water (1 L) for 15 min and filtered. The filter cake was triturated with hexane (750 mL), then filtered to give 149. LCMS [M+1] =339.9, 341.9; 1H NMR (400 MHz, DMSO-d6) δ 9.07 (s, 1H), 7.52 (s, 1H), 2.66 (s, 3H), 1.57 (s, 9H). Example 150: Synthesis of Compound 150 Step
Step A – Synthesis of 5-bromo-N'-hydroxy-4-methylpyrimidine-2-carboximidamide (149a) To a mixture of 1-bromo-2-fluoro-4-iodobenzene (180 g, 0.909 mol, 1.0 equiv.) and NH2OH.HCl (66.32 g, 0.954 mol, 1.05 equiv.) in MeOH (3.6 L) was added sodium methoxide (171.88 g, 0.954 mol, 1.0 equiv.) and the mixture was stirred at 70°C for 2 h. The mixture was concentrated under reduce pressure. The residue was diluted with water (1500 mL) and filtered to afford 149a. LCMS [M+1] = 230.8, 232.8. Step B – Synthesis of 5-bromo-N-hydroxy-4-methylpyrimidine-2-carbimidoyl chloride (149b) To a mixture of 5-bromo-N'-hydroxy-4-methylpyrimidine-2-carboximidamide (195 g, 0.843 mol, 1.0 equiv.) in HCl aqueous (4 M, 1.9 L, 9.0 equiv.) was added NaNO2 (72.78 g, 1.056 mol, 1.2 equiv.) in water (1.95 L) at 0°C and stirred at 0°C for 2 h. The mixture was diluted with water (400 mL) and filtered. The filtrate was washed with water (500 mL) and concentrated under reduce pressure to give 149b. LCMS [M+1] = 249.9, 251.9. Step C – Synthesis of tert-butyl 3-(5-bromo-4-methylpyrimidin-2-yl)isoxazole-5-carboxylate (149) A mixture of tert-butyl propiolate (302.2 g, 2.4 mol, 329 mL, 3.0 equiv.) and triethylamine (13.8 mL, 1.0 equiv.) in acetonitrile (4 L) was treated with a solution of 5-bromo-N-hydroxy-4- methylpyrimidine-2-carbimidoyl chloride (200 g, 0.8 mol, 1.0 equiv.) in acetonitrile (4 L). The
mixture was stirred at 20°C for 2 h. The mixture was concentrated under reduce pressure. The residue was triturated with water (1 L) for 15 min and filtered. The filter cake was triturated with hexane (750 mL), then filtered to give 149. LCMS [M+1] =339.9, 341.9; 1H NMR (400 MHz, DMSO-d6) δ 9.07 (s, 1H), 7.52 (s, 1H), 2.66 (s, 3H), 1.57 (s, 9H). Example 150: Synthesis of Compound 150 Step
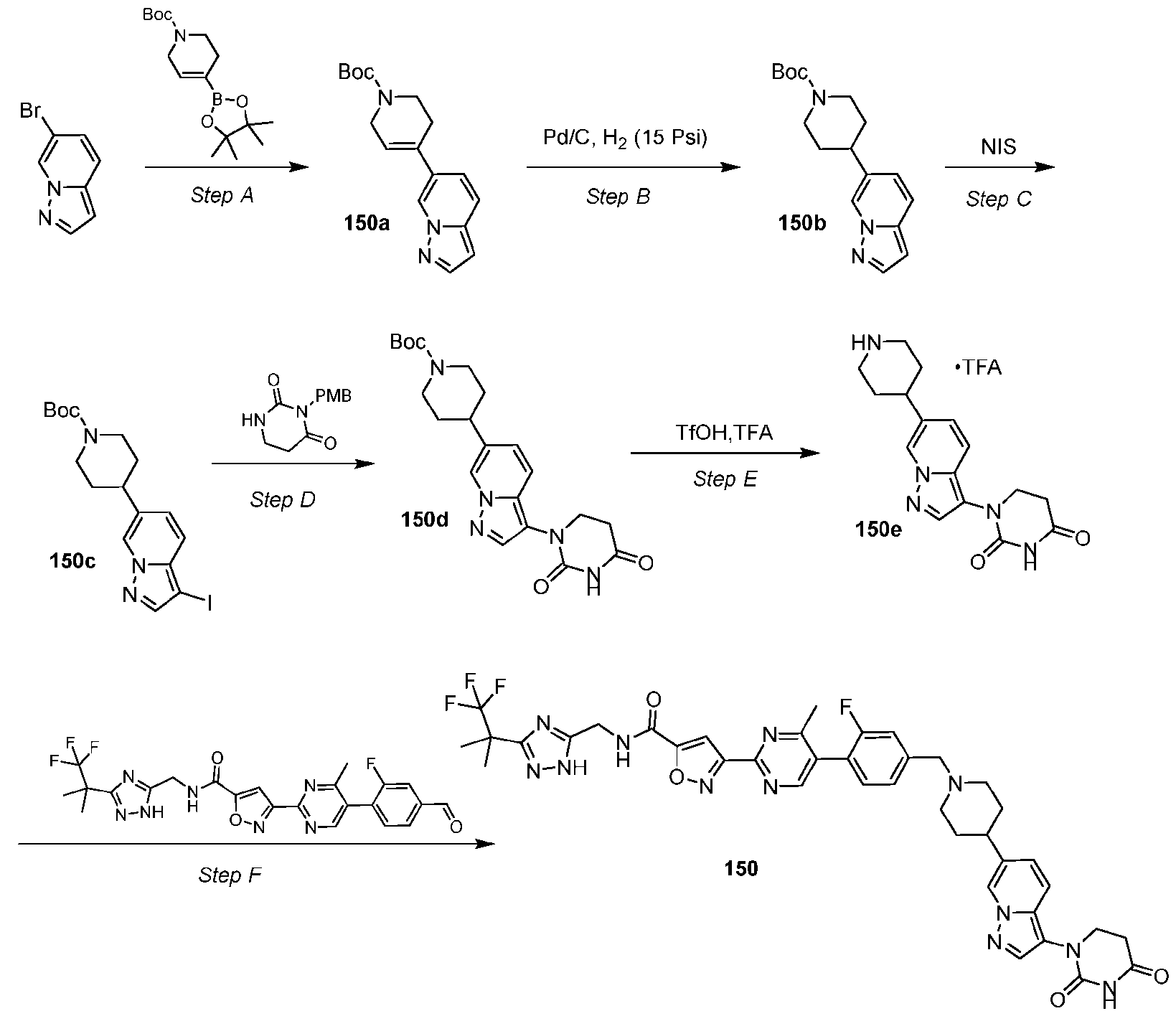 - carboxylate (150a) A mixture of 6-bromopyrazolo[1,5-a]pyridine (2.0 g, 10.1 mmol, 1.0 equiv.), tert-butyl 4- (4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-3,6-dihydropyridine-1(2H)-carboxylate (3.1 g, 10.1 mmol, 1.0 equiv.), K2CO3 (4.2 g, 30.4 mmol, 3.0 equiv.), Pd(dppf)Cl2 (742.7 mg, 1.0 mmol, 0.1 equiv.) and Pd(dppf)Cl2 (742.7 mg, 1.0 mmol, 0.1 equiv.) in dioxane (20.0 mL) and water (4.0 mL) was degassed and purged threefold with nitrogen. The mixture was stirred at 100°C for 1 h under a nitrogen atmosphere. The mixture was cooled to 25°C and poured into water (100 mL).
The aqueous phase was extracted with ethyl acetate (150 mLx3). The combined organic phase was washed with brine (100 mLx3), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, Petroleum ether/Ethyl acetate=1/0 to 9/1) to give 150a. LCMS [M+1] = 300.2. Step B – Synthesis of tert-butyl 4-(pyrazolo[1,5-a]pyridin-6-yl)piperidine-1-carboxylate (150b) To a solution of tert-butyl 4-(pyrazolo[1,5-a]pyridin-6-yl)-3,6-dihydropyridine-1(2H)- carboxylate (2.3 g, 7.6 mmol, 1.0 equiv.) in THF (230 mL) was added Pd/C (817.6 mg, 10.0 wt%) under a nitrogen atmosphere. The suspension was degassed and purged threefold with hydrogen gas. The mixture was stirred under hydrogen gas (30 psi) at 25°C for 2 h. The mixture was filtered and the filtrate was concentrated under reduced pressure to give 150b. LCMS [M+1] = 302.1. Step C – Synthesis of tert-butyl 4-(3-iodopyrazolo[1,5-a]pyridin-6-yl)piperidine-1-carboxylate (150c) To a solution of tert-butyl 4-(pyrazolo[1,5-a]pyridin-6-yl)piperidine-1-carboxylate (1.5 g, 4.9 mmol, 1.0 equiv.) in DMF (15.0 mL) was added NIS (1.1 g, 4.9 mmol, 1.0 equiv.). The mixture was stirred at 25°C for 1 h. The mixture was poured into water (80 mL). The aqueous phase was extracted with ethyl acetate (100 mLx3). The combined organic phase was washed with brine (80 mLx3), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, Petroleum ether/Ethyl acetate=1/0 to 5/1) to give 150c. LCMS [M-55] = 372.1. Step D – Synthesis of tert-butyl 4-(3-(3-(4-methoxybenzyl)-2,4-dioxotetrahydropyrimidin-1(2H)- yl)pyrazolo[1,5-a]pyridin-6-yl)piperidine-1-carboxylate (150d) A mixture of tert-butyl 4-(3-iodopyrazolo[1,5-a]pyridin-6-yl)piperidine-1-carboxylate (6.1 g, 14.2 mmol, 1.0 equiv.), 3-(4-methoxybenzyl)dihydropyrimidine-2,4(1H,3H)-dione (3.5 g, 14.9 mmol, 1.0 equiv.), K3PO4 (6.0 g, 28.5 mmol, 2.0 equiv.), CuI (1.3 g, 7.1 mmol, 0.5 equiv.) and (1S,2S)-N1,N2-dimethylcyclohexane-1,2-diamine (2.0 g, 14.2 mmol, 1.0 equiv.) in dioxane (60.0 mL) was degassed and purged threefold with nitrogen. The mixture was stirred at 80°C for 1 h under a nitrogen atmosphere. The mixture was cooled to 25°C and poured into water (150 mL).
The aqueous phase was extracted with ethyl acetate (200 mLx2). The combined organic phase was washed with brine (100 mLx3), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, Petroleum ether/Ethyl acetate=1/0 to 5/1) to give 150d. LCMS [M-55] = 478.3. Step E – Synthesis of 1-(6-(piperidin-4-yl)pyrazolo[1,5-a]pyridin-3-yl)dihydropyrimidine- 2,4(1H,3H)-dione (150e) To a solution of tert-butyl 4-(3-(3-(4-methoxybenzyl)-2,4-dioxotetrahydropyrimidin- 1(2H)-yl)pyrazolo[1,5-a]pyridin-6-yl)piperidine-1-carboxylate (100.0 mg, 187.4 μmol, 1.0 equiv.) in TFA (0.8 mL) and TfOH (0.1 mL). The mixture was stirred at 70°C for 10 min. The mixture was filtered and the filtrate was concentrated under reduced pressure to give 150e. LCMS [M+1] = 314.0; 1H NMR (400 MHz, METHANOL-d4) δ 8.39 (s, 1H), 8.03 (s, 1H), 7.60 (d, J = 9.2 Hz, 1H), 7.27 (d, J = 9.2 Hz, 1H), 3.89 (t, J = 6.8 Hz, 2H), 3.54 (d, J = 12.8 Hz, 2H), 3.22 - 3.14 (m, 2H), 3.05 - 2.98 (m, 1H), 2.88 (t, J = 6.8 Hz, 2H), 2.18 (d, J = 15.2 Hz, 2H), 1.99 - 1.88 (m, 2H). Step F – Synthesis of 3-(5-(4-((4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5- a]pyridin-6-yl)piperidin-1-yl)methyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (150) To a solution of 1-(6-(piperidin-4-yl)pyrazolo[1,5-a]pyridin-3-yl)dihydropyrimidine- 2,4(1H,3H)-dione (214.7 mg) and 3-(5-(2-fluoro-4-formylphenyl)-4-methylpyrimidin-2-yl)-N- ((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (200.0 mg) in THF (3.0 mL) was added Ti(OEt)4 (1.1 mL) at 50°C. After 60 min, NaBH(OAc)3 (245.7 mg) was added at 25°C and the mixture was stirred at this temperature for 30 min. The mixture was filtered and the filtrate was concentrated under reduced pressure. The residue was purified by prep-HPLC (Phenomenex Luna C18150x25mm, 10 µm; [water(FA)- ACN];gradient:5%-35% B over 8 min) to give 150. LCMS [M+1] = 815.0; 1H NMR (400 MHz, METHANOL-d4) δ 8.75 (s, 1H), 8.37 (s, 1H), 8.33 (s, 1H), 7.99 (s, 1H), 7.67 (s, 1H), 7.57 (d, J = 9.2 Hz, 1H), 7.54 - 7.49 (m, 1H), 7.47 (s, 1H), 7.44 (d, J = 2.8 Hz, 1H), 7.28 (d, J = 8.8 Hz, 1H), 4.74 (s, 2H), 3.97 (s, 2H), 3.88 (t, J = 6.4 Hz, 2H), 3.30 - 3.27 (m, 1H), 2.88 (t, J = 6.8 Hz, 2H),
2.84 - 2.76 (m, 1H), 2.68 - 2.57 (m, 3H), 2.55 (s, 3H), 2.08 - 2.02 (m, 2H), 1.97 - 1.87 (m, 2H), 1.62 (s, 6H). Example 151: Synthesis of Compound 151-1 and 151-2
- carboxylate (150a) A mixture of 6-bromopyrazolo[1,5-a]pyridine (2.0 g, 10.1 mmol, 1.0 equiv.), tert-butyl 4- (4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-3,6-dihydropyridine-1(2H)-carboxylate (3.1 g, 10.1 mmol, 1.0 equiv.), K2CO3 (4.2 g, 30.4 mmol, 3.0 equiv.), Pd(dppf)Cl2 (742.7 mg, 1.0 mmol, 0.1 equiv.) and Pd(dppf)Cl2 (742.7 mg, 1.0 mmol, 0.1 equiv.) in dioxane (20.0 mL) and water (4.0 mL) was degassed and purged threefold with nitrogen. The mixture was stirred at 100°C for 1 h under a nitrogen atmosphere. The mixture was cooled to 25°C and poured into water (100 mL).
The aqueous phase was extracted with ethyl acetate (150 mLx3). The combined organic phase was washed with brine (100 mLx3), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, Petroleum ether/Ethyl acetate=1/0 to 9/1) to give 150a. LCMS [M+1] = 300.2. Step B – Synthesis of tert-butyl 4-(pyrazolo[1,5-a]pyridin-6-yl)piperidine-1-carboxylate (150b) To a solution of tert-butyl 4-(pyrazolo[1,5-a]pyridin-6-yl)-3,6-dihydropyridine-1(2H)- carboxylate (2.3 g, 7.6 mmol, 1.0 equiv.) in THF (230 mL) was added Pd/C (817.6 mg, 10.0 wt%) under a nitrogen atmosphere. The suspension was degassed and purged threefold with hydrogen gas. The mixture was stirred under hydrogen gas (30 psi) at 25°C for 2 h. The mixture was filtered and the filtrate was concentrated under reduced pressure to give 150b. LCMS [M+1] = 302.1. Step C – Synthesis of tert-butyl 4-(3-iodopyrazolo[1,5-a]pyridin-6-yl)piperidine-1-carboxylate (150c) To a solution of tert-butyl 4-(pyrazolo[1,5-a]pyridin-6-yl)piperidine-1-carboxylate (1.5 g, 4.9 mmol, 1.0 equiv.) in DMF (15.0 mL) was added NIS (1.1 g, 4.9 mmol, 1.0 equiv.). The mixture was stirred at 25°C for 1 h. The mixture was poured into water (80 mL). The aqueous phase was extracted with ethyl acetate (100 mLx3). The combined organic phase was washed with brine (80 mLx3), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, Petroleum ether/Ethyl acetate=1/0 to 5/1) to give 150c. LCMS [M-55] = 372.1. Step D – Synthesis of tert-butyl 4-(3-(3-(4-methoxybenzyl)-2,4-dioxotetrahydropyrimidin-1(2H)- yl)pyrazolo[1,5-a]pyridin-6-yl)piperidine-1-carboxylate (150d) A mixture of tert-butyl 4-(3-iodopyrazolo[1,5-a]pyridin-6-yl)piperidine-1-carboxylate (6.1 g, 14.2 mmol, 1.0 equiv.), 3-(4-methoxybenzyl)dihydropyrimidine-2,4(1H,3H)-dione (3.5 g, 14.9 mmol, 1.0 equiv.), K3PO4 (6.0 g, 28.5 mmol, 2.0 equiv.), CuI (1.3 g, 7.1 mmol, 0.5 equiv.) and (1S,2S)-N1,N2-dimethylcyclohexane-1,2-diamine (2.0 g, 14.2 mmol, 1.0 equiv.) in dioxane (60.0 mL) was degassed and purged threefold with nitrogen. The mixture was stirred at 80°C for 1 h under a nitrogen atmosphere. The mixture was cooled to 25°C and poured into water (150 mL).
The aqueous phase was extracted with ethyl acetate (200 mLx2). The combined organic phase was washed with brine (100 mLx3), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, Petroleum ether/Ethyl acetate=1/0 to 5/1) to give 150d. LCMS [M-55] = 478.3. Step E – Synthesis of 1-(6-(piperidin-4-yl)pyrazolo[1,5-a]pyridin-3-yl)dihydropyrimidine- 2,4(1H,3H)-dione (150e) To a solution of tert-butyl 4-(3-(3-(4-methoxybenzyl)-2,4-dioxotetrahydropyrimidin- 1(2H)-yl)pyrazolo[1,5-a]pyridin-6-yl)piperidine-1-carboxylate (100.0 mg, 187.4 μmol, 1.0 equiv.) in TFA (0.8 mL) and TfOH (0.1 mL). The mixture was stirred at 70°C for 10 min. The mixture was filtered and the filtrate was concentrated under reduced pressure to give 150e. LCMS [M+1] = 314.0; 1H NMR (400 MHz, METHANOL-d4) δ 8.39 (s, 1H), 8.03 (s, 1H), 7.60 (d, J = 9.2 Hz, 1H), 7.27 (d, J = 9.2 Hz, 1H), 3.89 (t, J = 6.8 Hz, 2H), 3.54 (d, J = 12.8 Hz, 2H), 3.22 - 3.14 (m, 2H), 3.05 - 2.98 (m, 1H), 2.88 (t, J = 6.8 Hz, 2H), 2.18 (d, J = 15.2 Hz, 2H), 1.99 - 1.88 (m, 2H). Step F – Synthesis of 3-(5-(4-((4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5- a]pyridin-6-yl)piperidin-1-yl)methyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (150) To a solution of 1-(6-(piperidin-4-yl)pyrazolo[1,5-a]pyridin-3-yl)dihydropyrimidine- 2,4(1H,3H)-dione (214.7 mg) and 3-(5-(2-fluoro-4-formylphenyl)-4-methylpyrimidin-2-yl)-N- ((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (200.0 mg) in THF (3.0 mL) was added Ti(OEt)4 (1.1 mL) at 50°C. After 60 min, NaBH(OAc)3 (245.7 mg) was added at 25°C and the mixture was stirred at this temperature for 30 min. The mixture was filtered and the filtrate was concentrated under reduced pressure. The residue was purified by prep-HPLC (Phenomenex Luna C18150x25mm, 10 µm; [water(FA)- ACN];gradient:5%-35% B over 8 min) to give 150. LCMS [M+1] = 815.0; 1H NMR (400 MHz, METHANOL-d4) δ 8.75 (s, 1H), 8.37 (s, 1H), 8.33 (s, 1H), 7.99 (s, 1H), 7.67 (s, 1H), 7.57 (d, J = 9.2 Hz, 1H), 7.54 - 7.49 (m, 1H), 7.47 (s, 1H), 7.44 (d, J = 2.8 Hz, 1H), 7.28 (d, J = 8.8 Hz, 1H), 4.74 (s, 2H), 3.97 (s, 2H), 3.88 (t, J = 6.4 Hz, 2H), 3.30 - 3.27 (m, 1H), 2.88 (t, J = 6.8 Hz, 2H),
2.84 - 2.76 (m, 1H), 2.68 - 2.57 (m, 3H), 2.55 (s, 3H), 2.08 - 2.02 (m, 2H), 1.97 - 1.87 (m, 2H), 1.62 (s, 6H). Example 151: Synthesis of Compound 151-1 and 151-2
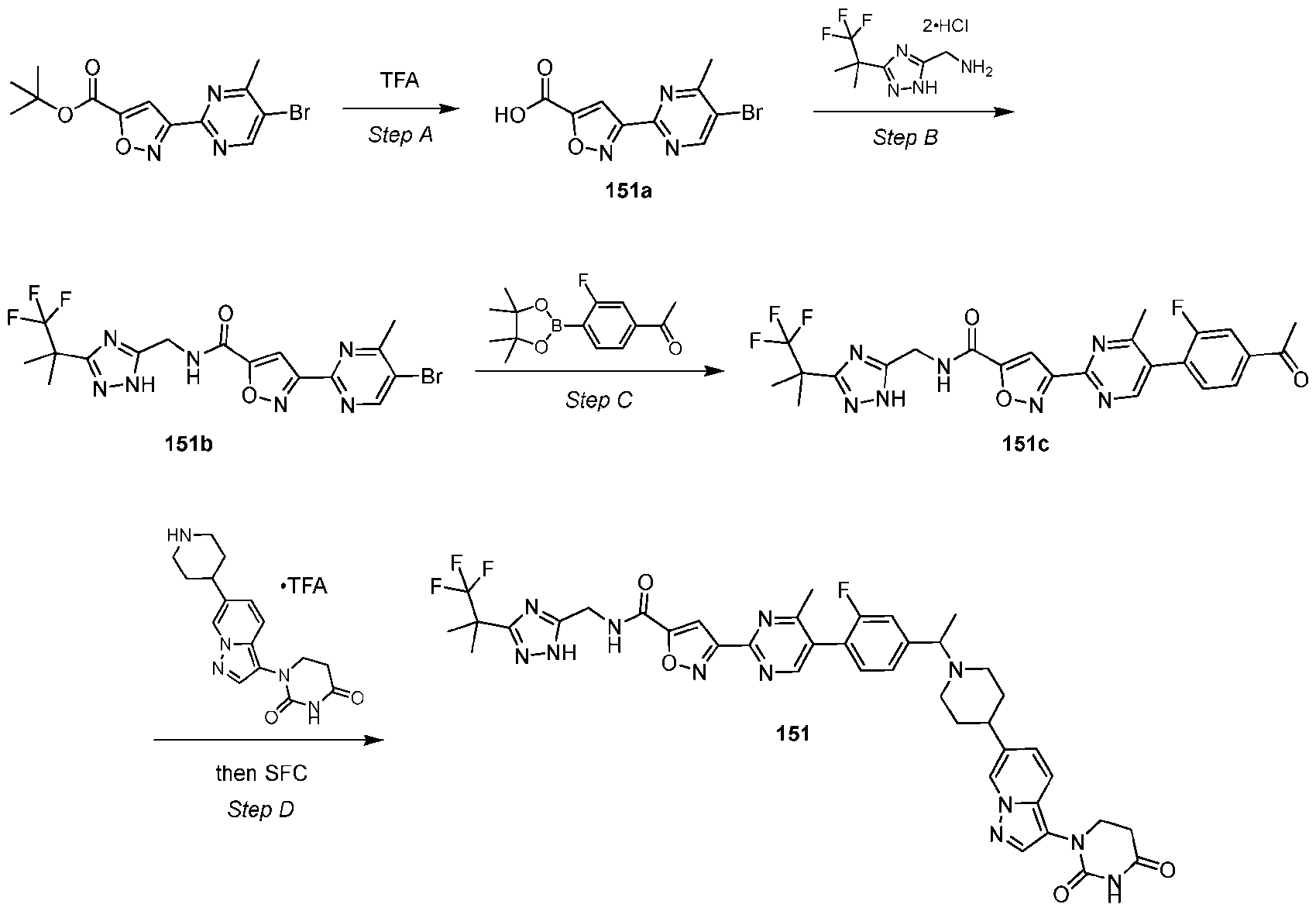 Step A – Synthesis of 3-(5-bromo-4-methylpyrimidin-2-yl)isoxazole-5-carboxylic acid (151a) To a solution of tert-butyl 3-(5-bromo-4-methylpyrimidin-2-yl)isoxazole-5-carboxylate (2.8 g, 8.2 mmol, 1.0 equiv.) in TFA (20.0 mL). The mixture was stirred at 25°C for 30 min. The mixture was concentrated under reduced pressure to give 151a. LCMS [M+1] = 286.0, 288.0. Step B – Synthesis of 3-(5-bromo-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan- 2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (151b) To a solution of 3-(5-bromo-4-methylpyrimidin-2-yl)isoxazole-5-carboxylic acid (1.8 g, 6.3 mmol, 1.0 equiv.) and (3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5- yl)methanamine hydrochloride (3.1 g, 12.6 mmol, 2.0 equiv.) in DMF (20.0 mL) was added BOP (4.2 g, 9.5 mmol, 1.5 equiv.) and triethylamine (2.6 mL, 3.0 equiv.), the mixture was stirred at 25°C for 30 min. The mixture was partitioned between ethyl acetate (200 mL) and water (150
mL). The organic phase was separated, dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, Petroleum ether/Ethyl acetate=1/1 to 0/1) to give 151b. LCMS [M+1] = 474.0, 476.0. Step C – Synthesis of 3-(5-(4-acetyl-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (151c) A mixture of 3-(5-bromo-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan- 2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (0.4 g, 948.9 μmol, 1.0 equiv.), 1-(3- fluoro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)ethan-1-one (375.9 mg, 1.4 mmol, 1.5 equiv.), Pd(dppf)Cl2 (69.4 mg, 94.8 μmol, 0.1 equiv.) and cesium carbonate (927.5 mg, 2.8 mmol, 3.0 equiv.) in dioxane (5.0 mL) and water (1.0 mL) was degassed and purged threefold with nitrogen, and then the mixture was stirred at 100°C for 1 h under a nitrogen atmosphere. The mixture was cooled to 25°C, the mixture was partitioned between ethyl acetate (100 mL) and water (50 mL). The organic phase was separated, dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, Petroleum ether/Ethyl acetate=1/1 to 10/7) to give 151c. LCMS [M+1] = 532.2. Step D – Synthesis of 3-(5-(4-(1-(4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5- a]pyridin-6-yl)piperidin-1-yl)ethyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (151) To a solution of 3-(5-(4-acetyl-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (0.1 g, 188.1 μmol, 1.0 equiv.) and 1-(6-(piperidin-4-yl)pyrazolo[1,5-a]pyridin-3-yl)dihydropyrimidine- 2,4(1H,3H)-dione trifluoroacetate (104.5 mg, 245 μmol, 1.3 equiv.) in THF (5.0 mL) was added Ti(OEt)4 (333 μL, 6.0 equiv.) and NaBH(OAc)3 (199.3 mg, 941 μmol, 5.0 equiv.). The mixture was stirred at 50°C for 1 h. The mixture was filtered and the filtrate was concentrated under reduced pressure. The residue was purified by prep-HPLC (Phenomenex Luna C18150x25mm, 10 µm; [water (FA)-ACN]; gradient: 7%-37% B over 8 min) to give 151 as the racemate. LCMS [M+1] = 829.1; 1H NMR (400 MHz, METHANOL-d4) δ 8.76 (s, 1H), 8.53 (s, 1H), 8.34 (s, 1H),
7.98 (s, 1H), 7.68 (s, 1H), 7.55 (d, J = 9.2 Hz, 1H), 7.45 (d, J = 7.2 Hz, 1H), 7.42 - 7.40 (m, 1H), 7.39 - 7.34 (m, 1H), 7.28 (dd, J = 1.6, 9.6 Hz, 1H), 4.75 (s, 2H), 3.89 (t, J = 6.8 Hz, 2H), 3.68 (d, J = 6.8 Hz, 1H), 3.53 - 3.46 (m, 1H), 3.09 - 3.03 (m, 1H), 2.89 (t, J = 6.8 Hz, 2H), 2.63 (t, J = 3.6 Hz, 1H), 2.56 (d, J = 1.2 Hz, 3H), 2.31 - 2.23 (m, 1H), 2.22 - 2.16 (m, 1H), 2.00 (d, J = 10.8 Hz, 1H), 1.95 - 1.87 (m, 2H), 1.80 (dt, J = 3.2, 12.4 Hz, 1H), 1.63 (s, 6H), 1.51 (d, J = 6.8 Hz, 3H). The racemate was separated by SFC (DAICEL CHIRALPAK IF(250x50mm,10 µm); [Hexane-EtOH/CAN (4:1)];B%:75%, isocratic elution mode) 151-1 as the early eluting isomer and 151-2 as the late eluting isomer. Example 152: Synthesis of Compound 152
Step A – Synthesis of 3-(5-bromo-4-methylpyrimidin-2-yl)isoxazole-5-carboxylic acid (151a) To a solution of tert-butyl 3-(5-bromo-4-methylpyrimidin-2-yl)isoxazole-5-carboxylate (2.8 g, 8.2 mmol, 1.0 equiv.) in TFA (20.0 mL). The mixture was stirred at 25°C for 30 min. The mixture was concentrated under reduced pressure to give 151a. LCMS [M+1] = 286.0, 288.0. Step B – Synthesis of 3-(5-bromo-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan- 2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (151b) To a solution of 3-(5-bromo-4-methylpyrimidin-2-yl)isoxazole-5-carboxylic acid (1.8 g, 6.3 mmol, 1.0 equiv.) and (3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5- yl)methanamine hydrochloride (3.1 g, 12.6 mmol, 2.0 equiv.) in DMF (20.0 mL) was added BOP (4.2 g, 9.5 mmol, 1.5 equiv.) and triethylamine (2.6 mL, 3.0 equiv.), the mixture was stirred at 25°C for 30 min. The mixture was partitioned between ethyl acetate (200 mL) and water (150
mL). The organic phase was separated, dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, Petroleum ether/Ethyl acetate=1/1 to 0/1) to give 151b. LCMS [M+1] = 474.0, 476.0. Step C – Synthesis of 3-(5-(4-acetyl-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (151c) A mixture of 3-(5-bromo-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan- 2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (0.4 g, 948.9 μmol, 1.0 equiv.), 1-(3- fluoro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)ethan-1-one (375.9 mg, 1.4 mmol, 1.5 equiv.), Pd(dppf)Cl2 (69.4 mg, 94.8 μmol, 0.1 equiv.) and cesium carbonate (927.5 mg, 2.8 mmol, 3.0 equiv.) in dioxane (5.0 mL) and water (1.0 mL) was degassed and purged threefold with nitrogen, and then the mixture was stirred at 100°C for 1 h under a nitrogen atmosphere. The mixture was cooled to 25°C, the mixture was partitioned between ethyl acetate (100 mL) and water (50 mL). The organic phase was separated, dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, Petroleum ether/Ethyl acetate=1/1 to 10/7) to give 151c. LCMS [M+1] = 532.2. Step D – Synthesis of 3-(5-(4-(1-(4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5- a]pyridin-6-yl)piperidin-1-yl)ethyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (151) To a solution of 3-(5-(4-acetyl-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (0.1 g, 188.1 μmol, 1.0 equiv.) and 1-(6-(piperidin-4-yl)pyrazolo[1,5-a]pyridin-3-yl)dihydropyrimidine- 2,4(1H,3H)-dione trifluoroacetate (104.5 mg, 245 μmol, 1.3 equiv.) in THF (5.0 mL) was added Ti(OEt)4 (333 μL, 6.0 equiv.) and NaBH(OAc)3 (199.3 mg, 941 μmol, 5.0 equiv.). The mixture was stirred at 50°C for 1 h. The mixture was filtered and the filtrate was concentrated under reduced pressure. The residue was purified by prep-HPLC (Phenomenex Luna C18150x25mm, 10 µm; [water (FA)-ACN]; gradient: 7%-37% B over 8 min) to give 151 as the racemate. LCMS [M+1] = 829.1; 1H NMR (400 MHz, METHANOL-d4) δ 8.76 (s, 1H), 8.53 (s, 1H), 8.34 (s, 1H),
7.98 (s, 1H), 7.68 (s, 1H), 7.55 (d, J = 9.2 Hz, 1H), 7.45 (d, J = 7.2 Hz, 1H), 7.42 - 7.40 (m, 1H), 7.39 - 7.34 (m, 1H), 7.28 (dd, J = 1.6, 9.6 Hz, 1H), 4.75 (s, 2H), 3.89 (t, J = 6.8 Hz, 2H), 3.68 (d, J = 6.8 Hz, 1H), 3.53 - 3.46 (m, 1H), 3.09 - 3.03 (m, 1H), 2.89 (t, J = 6.8 Hz, 2H), 2.63 (t, J = 3.6 Hz, 1H), 2.56 (d, J = 1.2 Hz, 3H), 2.31 - 2.23 (m, 1H), 2.22 - 2.16 (m, 1H), 2.00 (d, J = 10.8 Hz, 1H), 1.95 - 1.87 (m, 2H), 1.80 (dt, J = 3.2, 12.4 Hz, 1H), 1.63 (s, 6H), 1.51 (d, J = 6.8 Hz, 3H). The racemate was separated by SFC (DAICEL CHIRALPAK IF(250x50mm,10 µm); [Hexane-EtOH/CAN (4:1)];B%:75%, isocratic elution mode) 151-1 as the early eluting isomer and 151-2 as the late eluting isomer. Example 152: Synthesis of Compound 152
 carboxylate (152a) To a solution of 4-bromo-2,5-difluoro-benzonitrile (5.0 g, 22.9 mmol, 1.0 equiv.) in dioxane (50.0 mL) and water (50.0 mL) was added tert-butyl 4-(4,4,5,5-tetramethyl-1,3,2- dioxaborolan-2-yl)-3,6-dihydro-2H-pyridine-1-carboxylate (7.8 g, 25.2 mmol, 1.1 equiv.) and
Pd(dppf)Cl2.CH2Cl2 (1.87 g, 2.2 mmol, 0.1 equiv.) and cesium carbonate (22.4 g, 68.8 mmol, 3.0 equiv.) at 25°C. The mixture was purged threefold with nitrogen and heated at 100°C for 2 h. The mixture was diluted with water (100 mL) and extracted with ethyl acetate (100 mL x 3). The combined organic layers were washed with brine (100 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, Petroleum ether: Ethyl acetate = 1: 0 to 0: 1) to give 152a. LCMS [M-56+CH3CN+H] = 306.3; 1H NMR (400 MHz, DMSO-d6) δ 7.96 (dd, J = 5.6 Hz, 10.4 Hz, 1H), 7.59 (dd, J = 6.0 Hz, 10.0 Hz, 1H), 6.24 (s, 1H), 4.03 (s, 2H), 3.52 (d, J = 5.2 Hz, 2H), 2.43 (s, 2H), 1.42 (s, 9H). Step B – Synthesis of tert-butyl 4-(4-cyano-2,5-difluorophenyl)piperidine-1-carboxylate (152b) Pd/C (300.0 mg, 10% purity) was added to a reaction bottle under a nitrogen atmosphere, and ethyl acetate (10.0 mL) was added to the reaction bottle. Then added tert-butyl 4-(4-cyano- 2,5-difluorophenyl)-3,6-dihydropyridine-1(2H)-carboxylate (2.5 g, 7.80 mmol, 1.0 equiv.) in ethyl acetate (25.0 mL) was degassed and purged threefold with hydrogen gas. The mixture was stirred under hydrogen gas (15.0 Psi) at 25°C for 0.3 h. The mixture was filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, Petroleum ether: Ethyl acetate = 1: 0 to 0: 1) to give 152b. LCMS [M-56+CH3CN+H] = 308.3; 1H NMR (400 MHz, DMSO-d6) δ 7.93 (dd, J = 5.2 Hz, 9.2 Hz, 1H), 7.62 (dd, J = 6.0 Hz, 10.4 Hz, 1H), 4.08 - 4.04 (m, 2H), 3.07 - 3.01 (m, 1H), 2.88 - 2.77 (m, 2H), 1.72 - 1.69 (m, 2H), 1.61 - 1.51 (m, 2H), 1.41 (s, 9H). Step C – Synthesis of tert-butyl 4-(3-amino-5-fluorobenzo[d]isoxazol-6-yl)piperidine-1- carboxylate (152c) To a solution of ethanehydroxamic acid (768.4 mg, 10.2 mmol, 3.0 equiv.) in DMF (10.0 mL) was added t-BuOK (1.15 g, 10.2 mmol, 3.0 equiv.) at 25°C, and purged threefold with nitrogen. The mixture was stirred at 25°C for 2 h under a nitrogen atmosphere. A solution of tert-butyl 4-(4-cyano-2,5-difluorophenyl)piperidine-1-carboxylate (1.1 g, 3.4 mmol, 1.0 equiv.) in DMF (10.0 mL) was added to the mixture and the mixture was stirred at 100°C for 16 h. The mixture was diluted with water (30 mL) and extracted with ethyl acetate (30 mL x 3). The combined organic layers were washed with brine (30.0 mL), dried over anhydrous sodium sulfate,
filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, Petroleum ether: Ethyl acetate = 1: 0 to 0: 1) to give 152c. LCMS [M+1] = 336.3; 1H NMR (400 MHz, DMSO-d6) δ 7.57 (d, J = 9.6 Hz, 1H), 7.44 (d, J = 5.6 Hz, 1H), 6.38 (s, 2H), 4.09 (d, J = 9.6 Hz, 2H), 3.06 - 3.00 (m, 1H), 2.95 - 2.75 (m, 2H), 1.76 (d, J = 12.8 Hz, 2H), 1.64 - 1.52 (m, 2H), 1.42 (s, 9H). Step D – Synthesis of tert-butyl 4-(3-((3-amino-3-oxopropyl)amino)-5-fluorobenzo[d]isoxazol-6- yl)piperidine-1-carboxylate (152d) A solution of tert-butyl 4-(3-amino-5-fluorobenzo[d]isoxazol-6-yl)piperidine-1- carboxylate (830.0 mg, 2.47 mmol, 1.0 equiv.) and 2-propenamide (263.8 mg, 3.7 mmol, 256.1 μL, 1.5 equiv.) and cesium carbonate (1.21 g, 3.7 mmol, 1.5 equiv.) in DMA (9.0 mL) was stirred at 80°C for 24 h under a nitrogen atmosphere. The mixture was diluted with water (15.0 mL) and extracted with ethyl acetate (15.0 mL x 3). The combined organic layers were washed with brine (15.0 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, Petroleum ether: Ethyl acetate = 1: 0 to 0: 1) to give 152d. LCMS [M+1] = 407.4; 1H NMR (400 MHz, DMSO-d6) δ 7.62 (d, J = 9.6 Hz, 1H), 7.44 (d, J = 5.2 Hz, 1H), 7.35 (br s, 1H), 6.97 (t, J = 6.0 Hz, 1H), 6.84 (br s, 1H), 4.10 - 4.04 (m, 2H), 3.42 (q, J = 6.4 Hz, 2H), 3.06 - 3.00 (m, 1H), 2.89 - 2.81 (m, 2H), 2.44 (t, J = 6.8 Hz, 2H), 1.75 (d, J = 12.0 Hz, 2H), 1.62 - 1.52 (m, 2H), 1.42 (s, 9H). Step E – Synthesis of tert-butyl 4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-5- fluorobenzo[d]isoxazol-6-yl)piperidine-1-carboxylate (152e) A solution of tert-butyl 4-(3-((3-amino-3-oxopropyl)amino)-5-fluorobenzo[d]isoxazol-6- yl)piperidine-1-carboxylate (420.0 mg, 1.03 mmol, 1.0 equiv.), CDI (335.1 mg, 2.07 mmol, 2.0 equiv.) and cesium carbonate (1.35 g, 4.13 mmol, 4.0 equiv.) in DMA (5.0 mL) was stirred at 90°C for 6 h under a nitrogen atmosphere. The mixture was diluted with water (10.0 mL) and extracted with ethyl acetate (10.0 mL x 3). The combined organic layers were washed with brine (10.0 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced. The residue was purified by column chromatography (silica gel, Petroleum ether: Ethyl acetate = 1: 0 to 0: 1) to give 152e. LCMS [M+1] = 433.4; 1H NMR (400 MHz, DMSO-d6) δ
10.91 (br s, 1H), 7.75 (d, J = 5.6 Hz, 1H), 7.58 (d, J = 10.0 Hz, 1H), 4.12 - 4.03 (m, 4H), 3.12 - 3.06 (m, 1H), 2.87 - 2.76 (m, 4H), 1.79 (d, J = 12.0 Hz, 2H), 1.66 - 1.56 (m, 2H), 1.42 (s, 9H). Step F – Synthesis of 1-(5-fluoro-6-(piperidin-4-yl)benzo[d]isoxazol-3-yl)dihydropyrimidine- 2,4(1H,3H)-dione formic acid salt (152f) To a solution of tert-butyl 4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-5- fluorobenzo[d]isoxazol-6-yl)piperidine-1-carboxylate (150.0 mg, 346.8 μmol, 1.0 equiv.) in ethyl acetate (0.8 mL) was added HCOOH (0.8 mL). The mixture was stirred at 40°C for 12 h. The resulting mixture was concentrated under reduced pressure to give 152f. LCMS [M+1] = 333.3. Step G – Synthesis of 3-(5-(4-((4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-5- fluorobenzo[d]isoxazol-6-yl)piperidin-1-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (152) To a solution of 1-(5-fluoro-6-(piperidin-4-yl)benzo[d]isoxazol-3-yl)dihydropyrimidine- 2,4(1H,3H)-dione formic acid salt (150.0 mg, 396.4 μmol, 1.0 equiv.) and 3-[5-(2-fluoro-4- formyl-phenyl)pyrimidin-2-yl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methyl]isoxazole-5-carboxamide (199.5 mg, 396.4 μmol, 1.0 equiv.) in dichloromethane (2.0 mL) was added triethylamine (275.9 μL, 5.0 equiv.) under a nitrogen atmosphere. The mixture was stirred at 25°C for 1 h, treated with NaBH(OAc)3 (168.0 mg, 792.9 μmol, 2.0 equiv) for 25°C and the mixture was stirred at 25°C for 2 h. The mixture was diluted with water (4.0 mL) and extracted with dichloromethane (4.0 mL x 3). The combined organic layers were washed with brine (4.0 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by prep-HPLC (Waters Xbridge Prep OBD C18150 x 40 mm; 10 µm; [water (10mM NH4HCO3) - ACN]; gradient:35% - 65% B over 8.0 min) to give 152. LCMS [M+1] = 820.8; 1H NMR (400 MHz, DMSO-d6) δ 13.88 (br s, 1H), 10.96 (br s, 1H), 9.72 (br s, 1H), 9.25 (d, J = 0.8 Hz, 2H), 7.79 - 7.72 (m, 3H), 7.57 (d, J = 10.4 Hz, 1H), 7.43 - 7.39 (m, 2H), 4.60 (s, 2H), 4.05 (t, J = 6.6 Hz, 2H), 3.64 (s, 2H), 3.00 (d, J = 11.2 Hz, 2H), 2.94 - 2.89 (m, 1H), 2.77 (t, J = 6.6 Hz, 2H), 2.21 - 2.15 (m, 2H), 1.82 (s, 4H), 1.54 (s, 6H).
Example 153: Synthesis of Compound 153-1 and 153-2 -
carboxylate (152a) To a solution of 4-bromo-2,5-difluoro-benzonitrile (5.0 g, 22.9 mmol, 1.0 equiv.) in dioxane (50.0 mL) and water (50.0 mL) was added tert-butyl 4-(4,4,5,5-tetramethyl-1,3,2- dioxaborolan-2-yl)-3,6-dihydro-2H-pyridine-1-carboxylate (7.8 g, 25.2 mmol, 1.1 equiv.) and
Pd(dppf)Cl2.CH2Cl2 (1.87 g, 2.2 mmol, 0.1 equiv.) and cesium carbonate (22.4 g, 68.8 mmol, 3.0 equiv.) at 25°C. The mixture was purged threefold with nitrogen and heated at 100°C for 2 h. The mixture was diluted with water (100 mL) and extracted with ethyl acetate (100 mL x 3). The combined organic layers were washed with brine (100 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, Petroleum ether: Ethyl acetate = 1: 0 to 0: 1) to give 152a. LCMS [M-56+CH3CN+H] = 306.3; 1H NMR (400 MHz, DMSO-d6) δ 7.96 (dd, J = 5.6 Hz, 10.4 Hz, 1H), 7.59 (dd, J = 6.0 Hz, 10.0 Hz, 1H), 6.24 (s, 1H), 4.03 (s, 2H), 3.52 (d, J = 5.2 Hz, 2H), 2.43 (s, 2H), 1.42 (s, 9H). Step B – Synthesis of tert-butyl 4-(4-cyano-2,5-difluorophenyl)piperidine-1-carboxylate (152b) Pd/C (300.0 mg, 10% purity) was added to a reaction bottle under a nitrogen atmosphere, and ethyl acetate (10.0 mL) was added to the reaction bottle. Then added tert-butyl 4-(4-cyano- 2,5-difluorophenyl)-3,6-dihydropyridine-1(2H)-carboxylate (2.5 g, 7.80 mmol, 1.0 equiv.) in ethyl acetate (25.0 mL) was degassed and purged threefold with hydrogen gas. The mixture was stirred under hydrogen gas (15.0 Psi) at 25°C for 0.3 h. The mixture was filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, Petroleum ether: Ethyl acetate = 1: 0 to 0: 1) to give 152b. LCMS [M-56+CH3CN+H] = 308.3; 1H NMR (400 MHz, DMSO-d6) δ 7.93 (dd, J = 5.2 Hz, 9.2 Hz, 1H), 7.62 (dd, J = 6.0 Hz, 10.4 Hz, 1H), 4.08 - 4.04 (m, 2H), 3.07 - 3.01 (m, 1H), 2.88 - 2.77 (m, 2H), 1.72 - 1.69 (m, 2H), 1.61 - 1.51 (m, 2H), 1.41 (s, 9H). Step C – Synthesis of tert-butyl 4-(3-amino-5-fluorobenzo[d]isoxazol-6-yl)piperidine-1- carboxylate (152c) To a solution of ethanehydroxamic acid (768.4 mg, 10.2 mmol, 3.0 equiv.) in DMF (10.0 mL) was added t-BuOK (1.15 g, 10.2 mmol, 3.0 equiv.) at 25°C, and purged threefold with nitrogen. The mixture was stirred at 25°C for 2 h under a nitrogen atmosphere. A solution of tert-butyl 4-(4-cyano-2,5-difluorophenyl)piperidine-1-carboxylate (1.1 g, 3.4 mmol, 1.0 equiv.) in DMF (10.0 mL) was added to the mixture and the mixture was stirred at 100°C for 16 h. The mixture was diluted with water (30 mL) and extracted with ethyl acetate (30 mL x 3). The combined organic layers were washed with brine (30.0 mL), dried over anhydrous sodium sulfate,
filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, Petroleum ether: Ethyl acetate = 1: 0 to 0: 1) to give 152c. LCMS [M+1] = 336.3; 1H NMR (400 MHz, DMSO-d6) δ 7.57 (d, J = 9.6 Hz, 1H), 7.44 (d, J = 5.6 Hz, 1H), 6.38 (s, 2H), 4.09 (d, J = 9.6 Hz, 2H), 3.06 - 3.00 (m, 1H), 2.95 - 2.75 (m, 2H), 1.76 (d, J = 12.8 Hz, 2H), 1.64 - 1.52 (m, 2H), 1.42 (s, 9H). Step D – Synthesis of tert-butyl 4-(3-((3-amino-3-oxopropyl)amino)-5-fluorobenzo[d]isoxazol-6- yl)piperidine-1-carboxylate (152d) A solution of tert-butyl 4-(3-amino-5-fluorobenzo[d]isoxazol-6-yl)piperidine-1- carboxylate (830.0 mg, 2.47 mmol, 1.0 equiv.) and 2-propenamide (263.8 mg, 3.7 mmol, 256.1 μL, 1.5 equiv.) and cesium carbonate (1.21 g, 3.7 mmol, 1.5 equiv.) in DMA (9.0 mL) was stirred at 80°C for 24 h under a nitrogen atmosphere. The mixture was diluted with water (15.0 mL) and extracted with ethyl acetate (15.0 mL x 3). The combined organic layers were washed with brine (15.0 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, Petroleum ether: Ethyl acetate = 1: 0 to 0: 1) to give 152d. LCMS [M+1] = 407.4; 1H NMR (400 MHz, DMSO-d6) δ 7.62 (d, J = 9.6 Hz, 1H), 7.44 (d, J = 5.2 Hz, 1H), 7.35 (br s, 1H), 6.97 (t, J = 6.0 Hz, 1H), 6.84 (br s, 1H), 4.10 - 4.04 (m, 2H), 3.42 (q, J = 6.4 Hz, 2H), 3.06 - 3.00 (m, 1H), 2.89 - 2.81 (m, 2H), 2.44 (t, J = 6.8 Hz, 2H), 1.75 (d, J = 12.0 Hz, 2H), 1.62 - 1.52 (m, 2H), 1.42 (s, 9H). Step E – Synthesis of tert-butyl 4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-5- fluorobenzo[d]isoxazol-6-yl)piperidine-1-carboxylate (152e) A solution of tert-butyl 4-(3-((3-amino-3-oxopropyl)amino)-5-fluorobenzo[d]isoxazol-6- yl)piperidine-1-carboxylate (420.0 mg, 1.03 mmol, 1.0 equiv.), CDI (335.1 mg, 2.07 mmol, 2.0 equiv.) and cesium carbonate (1.35 g, 4.13 mmol, 4.0 equiv.) in DMA (5.0 mL) was stirred at 90°C for 6 h under a nitrogen atmosphere. The mixture was diluted with water (10.0 mL) and extracted with ethyl acetate (10.0 mL x 3). The combined organic layers were washed with brine (10.0 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced. The residue was purified by column chromatography (silica gel, Petroleum ether: Ethyl acetate = 1: 0 to 0: 1) to give 152e. LCMS [M+1] = 433.4; 1H NMR (400 MHz, DMSO-d6) δ
10.91 (br s, 1H), 7.75 (d, J = 5.6 Hz, 1H), 7.58 (d, J = 10.0 Hz, 1H), 4.12 - 4.03 (m, 4H), 3.12 - 3.06 (m, 1H), 2.87 - 2.76 (m, 4H), 1.79 (d, J = 12.0 Hz, 2H), 1.66 - 1.56 (m, 2H), 1.42 (s, 9H). Step F – Synthesis of 1-(5-fluoro-6-(piperidin-4-yl)benzo[d]isoxazol-3-yl)dihydropyrimidine- 2,4(1H,3H)-dione formic acid salt (152f) To a solution of tert-butyl 4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-5- fluorobenzo[d]isoxazol-6-yl)piperidine-1-carboxylate (150.0 mg, 346.8 μmol, 1.0 equiv.) in ethyl acetate (0.8 mL) was added HCOOH (0.8 mL). The mixture was stirred at 40°C for 12 h. The resulting mixture was concentrated under reduced pressure to give 152f. LCMS [M+1] = 333.3. Step G – Synthesis of 3-(5-(4-((4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-5- fluorobenzo[d]isoxazol-6-yl)piperidin-1-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (152) To a solution of 1-(5-fluoro-6-(piperidin-4-yl)benzo[d]isoxazol-3-yl)dihydropyrimidine- 2,4(1H,3H)-dione formic acid salt (150.0 mg, 396.4 μmol, 1.0 equiv.) and 3-[5-(2-fluoro-4- formyl-phenyl)pyrimidin-2-yl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methyl]isoxazole-5-carboxamide (199.5 mg, 396.4 μmol, 1.0 equiv.) in dichloromethane (2.0 mL) was added triethylamine (275.9 μL, 5.0 equiv.) under a nitrogen atmosphere. The mixture was stirred at 25°C for 1 h, treated with NaBH(OAc)3 (168.0 mg, 792.9 μmol, 2.0 equiv) for 25°C and the mixture was stirred at 25°C for 2 h. The mixture was diluted with water (4.0 mL) and extracted with dichloromethane (4.0 mL x 3). The combined organic layers were washed with brine (4.0 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by prep-HPLC (Waters Xbridge Prep OBD C18150 x 40 mm; 10 µm; [water (10mM NH4HCO3) - ACN]; gradient:35% - 65% B over 8.0 min) to give 152. LCMS [M+1] = 820.8; 1H NMR (400 MHz, DMSO-d6) δ 13.88 (br s, 1H), 10.96 (br s, 1H), 9.72 (br s, 1H), 9.25 (d, J = 0.8 Hz, 2H), 7.79 - 7.72 (m, 3H), 7.57 (d, J = 10.4 Hz, 1H), 7.43 - 7.39 (m, 2H), 4.60 (s, 2H), 4.05 (t, J = 6.6 Hz, 2H), 3.64 (s, 2H), 3.00 (d, J = 11.2 Hz, 2H), 2.94 - 2.89 (m, 1H), 2.77 (t, J = 6.6 Hz, 2H), 2.21 - 2.15 (m, 2H), 1.82 (s, 4H), 1.54 (s, 6H).
Example 153: Synthesis of Compound 153-1 and 153-2 -
 2-azaspiro[3.3]heptan-2-yl)ethyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro- 2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (153) To a solution of 3-(5-(4-acetyl-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (0.6 g, 1.1 mmol, 1.0 equiv.) and 1-(2-fluoro-4-(2-azaspiro[3.3]heptan-6-yl)phenyl)dihydropyrimidine- 2,4(1H,3H)-dione trifluoroacetate (706.7 mg, 1.6 mmol, 1.5 equiv.) in THF (15.0 mL) was added Ti(OEt)4 (1.4 mL), the mixture was stirred at 25°C for 30 min, and then NaBH(OAc)3 (1.2 g, 5.6 mmol, 5.0 equiv.) was added, the mixture was stirred at 25°C for 30 min. The mixture was filtered and the filtrate was concentrated under reduced pressure. The residue was purified by prep-HPLC (YMC Triart C18, 70x250 mm; 7µm; [water(FA)-ACN];gradient:20%-50% B over 25 min) to afford 153 as the racemate. LCMS [M+1] = 819.0; 1H NMR (400 MHz, METHANOL-d4) δ 8.73 (s, 1H), 8.45 (bs, 1H), 7.67 (s, 1H), 7.54 - 7.48 (m, 1H), 7.43 - 7.29 (m, 3H), 7.14 - 7.06 (m, 2H), 4.74 (s, 2H), 3.98 - 3.81 (m, 2H), 3.77 (t, J = 6.8 Hz, 2H), 3.69 - 3.75 (m, 1H), 3.67- 3.60 (m, 1H), 3.55 - 3.43 (m, 2H), 2.81 (t, J = 6.4 Hz, 2H), 2.71 - 2.59 (m, 2H), 2.54 (s, 3H), 2.34 (t, J = 10.0 Hz, 2H), 1.62 (s, 6H), 1.44 (d, J = 4.4 Hz, 3H). The enantiomers were separated by SFC (ChiralPak IH, 250x50mm, 10µm; [HEXANE- isopropanol/ACN(4:1)];B%:48%, isocratic elution mode) to afford 153-1 as the early eluting enantiomer and 153-2 as the late eluting enantiomer. Example 154: Synthesis of Compound 154-1 and 154-2
2-azaspiro[3.3]heptan-2-yl)ethyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro- 2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (153) To a solution of 3-(5-(4-acetyl-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (0.6 g, 1.1 mmol, 1.0 equiv.) and 1-(2-fluoro-4-(2-azaspiro[3.3]heptan-6-yl)phenyl)dihydropyrimidine- 2,4(1H,3H)-dione trifluoroacetate (706.7 mg, 1.6 mmol, 1.5 equiv.) in THF (15.0 mL) was added Ti(OEt)4 (1.4 mL), the mixture was stirred at 25°C for 30 min, and then NaBH(OAc)3 (1.2 g, 5.6 mmol, 5.0 equiv.) was added, the mixture was stirred at 25°C for 30 min. The mixture was filtered and the filtrate was concentrated under reduced pressure. The residue was purified by prep-HPLC (YMC Triart C18, 70x250 mm; 7µm; [water(FA)-ACN];gradient:20%-50% B over 25 min) to afford 153 as the racemate. LCMS [M+1] = 819.0; 1H NMR (400 MHz, METHANOL-d4) δ 8.73 (s, 1H), 8.45 (bs, 1H), 7.67 (s, 1H), 7.54 - 7.48 (m, 1H), 7.43 - 7.29 (m, 3H), 7.14 - 7.06 (m, 2H), 4.74 (s, 2H), 3.98 - 3.81 (m, 2H), 3.77 (t, J = 6.8 Hz, 2H), 3.69 - 3.75 (m, 1H), 3.67- 3.60 (m, 1H), 3.55 - 3.43 (m, 2H), 2.81 (t, J = 6.4 Hz, 2H), 2.71 - 2.59 (m, 2H), 2.54 (s, 3H), 2.34 (t, J = 10.0 Hz, 2H), 1.62 (s, 6H), 1.44 (d, J = 4.4 Hz, 3H). The enantiomers were separated by SFC (ChiralPak IH, 250x50mm, 10µm; [HEXANE- isopropanol/ACN(4:1)];B%:48%, isocratic elution mode) to afford 153-1 as the early eluting enantiomer and 153-2 as the late eluting enantiomer. Example 154: Synthesis of Compound 154-1 and 154-2
Step A A mixture of benzyl 3-oxobutanoate (120 g, 624.3 mmol, 107.7 mL, 1.0 equiv.) K2CO3 (258.8 g, 1.9 mol, 3.0 equiv.) in DMSO (1 L). The suspension was degassed and purged threefold with nitrogen. Then the mixture was cooled to 0°C. MeI (354.5 g, 2.5 mol, 155.5 mL, 4.0 equiv.) was added dropwise to the mixture. The mixture was stirred for 12 h at 20°C. The residue was poured into water (2 L). The aqueous phase was extracted with ethyl acetate (500 mLx2). The combined organic phase was washed with brine (50 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, Petroleum ether/Ethyl acetate=1/0 to 50/1) to give 154a. 1H NMR (400 MHz, CHLOROFORM-d) δ 7.41 - 7.30 (m, 5H), 5.18 (s, 2H), 2.09 (s, 3H), 1.39 (s, 6H). Step B – Synthesis of benzyl 3,3-difluoro-2,2-dimethyl-butanoate (154b-1) and benzyl 3-fluoro- 2,2-dimethyl-but-3-enoate (154b-2) A mixture of benzyl 2,2-dimethyl-3-oxo-butanoate (22.0 g, 99.9 mmol, 1.0 equiv.) in BAST (110 mL, 5.0 equiv.) was degassed and purged threefold with nitrogen and then the mixture was stirred at 60°C for 16 h under a nitrogen atmosphere. The mixture was diluted with dichloromethane (500 mL) and the residue was added slowly into water (500 mL). The aqueous phase was extracted with dichloromethane (300 mLx2). The combined organic phase was washed with brine (50 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica
gel, Petroleum ether/Ethyl acetate=1/0 to =10/1) to give a mixture of benzyl 3,3-difluoro-2,2- dimethyl-butanoate and benzyl 3-fluoro-2,2-dimethyl-but-3-enoate (37.0 g, crude, ratio = 4:1 base on HPLC). This mixture was resubjected to the reaction conditions as follows. The mixture (37.0 g, crude, 10:1 base on HPLC) in BAST (71 mL) was degassed and purged threefold with nitrogen, and then the mixture was stirred at 60°C for 16 h under a nitrogen atmosphere. The mixture was diluted with dichloromethane (500 mL) and the residue was added slowly into water (500 mL). The aqueous phase was extracted with dichloromethane (300 mLx2). The combined organic phase was washed with brine (100 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, Petroleum ether/Ethyl acetate=100/1 to 50/1) to give a mixture of 154b-1 and 154b-2 (ratio = 9:1 base on 1NMR). 1H NMR (400 MHz, CHLOROFORM-d) δ 7.43 - 7.30 (m, 5H), 5.17 (s, 2H), 1.65 (t, J = 19.2 Hz, 3H), 1.36 (s, 6H) Step C – Synthesis of 3,3-difluoro-2,2-dimethyl-butanoic acid (154c-1) and 3-fluoro-2,2-dimethyl- but-3-enoic acid (154c-2) To a solution of benzyl 3,3-difluoro-2,2-dimethyl-butanoate and benzyl 3-fluoro-2,2- dimethyl-but-3-enoate (34.0 g, crude, 9:1 base on HNMR) in MeOH (300 mL) water (30 mL) was added NaOH (10.0 M, 24.8 mL) at 20°C and stirred at 50°C for 30 min. The residue was poured into water (500 mL). The aqueous phase was extracted with ethyl acetate (500 mLx2). Then the pH of the aqueous phase was adjusted to 5-6 with aqueous HCl (2 M). The aqueous phase was extracted with ethyl acetate (500 mLx2). The combined organic phase was washed with brine (50 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure to give a mixture of 154c-1 and 154c-2. 1H NMR (400 MHz, CHLOROFORM- d) δ 1.74 (t, J = 19.2 Hz, 1H), 1.38 (s, 6H). Step D – Synthesis of tert-butyl N-[[3-(2,2-difluoro-1,1-dimethyl-propyl)-1H-1,2,4-triazol-5- yl]methyl]carbamate (154d-1) and tert-butyl N-[[3-(2-fluoro-1,1-dimethyl-allyl) -1H-1,2,4- triazol-5-yl] methyl] carbamate (154d-2) To a mixture of 3,3-difluoro-2,2-dimethyl-butanoic acid and 3-fluoro-2,2-dimethyl-but-3- enoic acid (18 g, crude, 9:1 base on HNMR) in DMF (90 mL) was added CDI (19.2 g.) and under nitrogen at 20°C. The mixture was stirred for 1.5 h at 20 °C under a nitrogen atmosphere. The
mixture was added dropwise to a solution of tert-butyl N-(2-amino-2-imino-ethyl)carbamate hydrochloride (24.8 g, 118.3 mmol, 1.0 equiv.) in DMF (90 mL) and triethylamine (32.9 mL, 2.0 equiv.) under nitrogen at 0°C. The mixture was stirred for 30 min at 0°C The mixture was cooled to 0-10°C and acetic acid (112.54 g, 1.87 mol, 107.29 mL, 16 equiv.) was added slowly at 0-10°C under a nitrogen atmosphere. Hydrazine hydrate (20.5 mL) was added dropwise to the mixture at 0°C and stirred at 0°C for 1 h. The mixture was added dropwise to Na2CO3(8.0 equiv) solution of water (500 mL). The mixture was extracted with ethyl acetate (500 mLx2). The combined organic phase was washed with brine (60 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, Petroleum ether/Ethyl acetate = 20/1 to 1/1) to give 154d-1: 1H NMR (400 MHz, CHLOROFORM-d) δ 5.45 (br s, 1H), 4.42 (br d, J = 5.6 Hz, 2H), 1.66 - 1.53 (m, 9H), 1.47 (s, 9H) and 154d-2, which was purified by prep-HPLC (Waters xBridge 150x25mm, 10 µm; [water(10mM NH4HCO3)-ACN];gradient:20%-50% B over 9.0 min) to give 154d-2: 1H NMR (400 MHz, CHLOROFORM-d) δ 5.33 (br s, 1H), 4.71 (dd, J = 3.6, 21.6 Hz, 1H), 4.51 (dd, J = 3.6, 21.6 Hz, 1H), 4.45 - 4.35 (m, 2H), 1.60 (s, 6H), 1.46 (s, 9H). Step E – Synthesis of [3-(2,2-difluoro-1,1-dimethyl-propyl)-1H-1,2,4-triazol-5-yl] methanamine hydrochloride (154-1 ) A mixture of tert-butyl N-[[3-(2,2-difluoro-1,1-dimethyl-propyl)-1H-1,2,4-triazol-5-yl] methyl] carbamate (1.5 g, 4.9 mmol, 1.0 equiv.) in HCl/ethyl acetate (10 mL) was degassed and purged threefold with nitrogen. The mixture was stirred at 20°C for 1 h under a nitrogen atmosphere. And then concentrated under reduced pressure. The residue was triturated with ethyl acetate (20 mLx3) at 20oC for 10 min to give 154-1.1H NMR (400 MHz, deuterium oxide) δ 4.26 (s, 2H), 1.56 (t, J = 20 Hz, 3H), 1.50 (s, 6H). Step F – Synthesis of [3-(2-fluoro-1,1-dimethyl-allyl)-1H-1,2,4-triazol-5-yl]methanamine hydrochloride (154-2) A mixture of tert-butyl N-[[3-(2-fluoro-1,1-dimethyl-allyl)-1H-1,2,4-triazol-5-yl]methyl] carbamate (40.0 mg, 140.7 μmol, 1.0 equiv.) in HCl/dioxane (1 mL) was degassed and purged threefold with nitrogen, and then the mixture was stirred at 20°C for 30 min under a nitrogen atmosphere. The mixture was concentrated under reduced pressure to give 154-2. 1H NMR (400
MHz, deuterium oxide) δ 4.82 - 4.80 (m, 1H), 4.59 (br d, J = 3.6 Hz, 1H), 4.25 (s, 2H), 3.72 (s, 4H), 1.56 (s, 6H). Example 155: Synthesis of Compound 155-1 and 155-2 Step
 5-carboxylate (155a) To a solution of 1-(4-bromo-3-fluorophenyl)ethan-1-one (2.5 g, 11.3 mmol, 1.1 equiv.) and tert-butyl 3-(4-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrimidin-2-yl) isoxazole-5-carboxylate (4.0 g, 10.3 mmol, 1.0 equiv.) in water (16 mL) and dioxane (80 mL) was added K2CO3 (4.3 g, 30.9 mmol, 3.0 equiv.) and Pd(dppf)Cl2.CH2Cl2 (1.3 g, 1.5 mmol, 0.15 equiv.) under Ar atmosphere. The mixture was stirred at 80°C for 3 h. The mixture was diluted with water (80 mL) and extracted with ethyl acetate (160 mL). The combined organic layers were
dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, Petroleum ether/Ethyl acetate=50/7 to 5/1) to afford 155a. LCMS [M+1] = 398.3. Step B – Synthesis of tert-butyl 3-[5-[4-[1-[6-[4-(2,4-dioxohexahydropyrimidin-1-yl)-3-fluoro- phenyl]-2-azaspiro[3.3]heptan-2-yl]ethyl]-2-fluoro-phenyl]-4-methyl-pyrimidin-2-yl]isoxazole-5- carboxylate (155b-1 and 155b-2) To a solution of 1-[4-(2-azaspiro[3.3]heptan-6-yl)-2-fluoro-phenyl]hexahydropyrimidine - 2,4-dione trifluoroacetate salt (1.58 g, 3.77 mmol, 1.5 equiv.) and tert-butyl 3-(5-(4-acetyl-2- fluorophenyl)-4-methylpyrimidin-2-yl)isoxazole-5-carboxylate (1 g, 2.52 mmol, 1 equiv.) in dichloromethane (20 mL) was added triethylamine (3.50 mL, 10 equiv.) and NaBH(OAc)3 (2.67 g, 12.58 mmol, 5 equiv.). The mixture was stirred at 25°C for 12 h. The mixture was poured into water (50 mL) and extracted with ethyl acetate (50 mL x 3). The combined organic portions were washed with brine (50 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, Petroleum ether/Ethyl acetate=100/1 to 10/1) to give tert-butyl 3-(5-(4-(1-(6-(4-(2,4- dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2-azaspiro[3.3]heptan-2-yl)ethyl)-2- fluorophenyl)-4-methylpyrimidin-2-yl)isoxazole-5-carboxylate (1.2 g, 1.75 mmol, 69.64% yield). LCMS [M+H] = 685.4 The enantiomers were separated by SFC (ChiralPak IH, 250 mm x 30mm; 10 µm; [CO2-EtOH:ACN=1:1 (0.1% isopropylamine)]; B%:50%, isocratic elution mode) to afford 155b-1 as the early eluting isomer and 155b-2 as the late eluting isomer. Each isomer was processed separately in the subsequent steps. LCMS [M+1] = 685.4. Step C – Synthesis of 3-(5-(4-(1-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)- 2-azaspiro[3.3]heptan-2-yl)ethyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)isoxazole-5-carboxylic acid trifluoroacetate salt (155c-1 and 155c-2) To a solution of tert-butyl 3-(5-(4-(1-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3- fluorophenyl)-2-azaspiro[3.3]heptan-2-yl)ethyl)-2-fluorophenyl)-4-methylpyrimidin-2- yl)isoxazole-5-carboxylate (500 mg) in dichloromethane (5 mL) was added TFA (1.54 g, 13.46 mmol, 1 mL, 18.44 equiv.). The mixture was stirred at 20 °C for 3 h. The mixture was concentrated under reduced pressure to give 155c-1 or 155c-2. LCMS [M+1] = 629.3.
Step D – Synthesis of N-((3-(3,3-difluoro-2-methylbutan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-3-(5- (4-(1-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2-azaspiro[3.3]heptan-2- yl)ethyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)isoxazole-5-carboxamide (155-1 and 155-2) To a solution of [3-(2,2-difluoro-1,1-dimethyl-propyl)-1H-1,2,4-triazol-5-yl]methanamine hydrochloride (31.11 mg, 112.26 μmol, 1.04 eq) in DMF (1 mL) was added BOP (95.29 mg, 215.45 μmol, 2 equiv.) and triethylamine (75 μL, 5 equiv.). 3-(5-(4-(1-(6-(4-(2,4- dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2-azaspiro[3.3]heptan-2-yl)ethyl)-2- fluorophenyl)-4-methylpyrimidin-2-yl)isoxazole-5-carboxylic acid (80 mg, 107.72 μmol, 1 equiv., TFA) was added to the mixture. The mixture was stirred at 20 °C for 2 h. The mixture was poured into water (0.5 mL) and extracted with ethyl acetate (0.5 mL x 3). The combined organic layers were washed with brine (0.5 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by prep-HPLC (Phenomenex Luna C18, 100x40mm, 5 µm; [water(0.2% FA)-ACN]; gradient:10%-40% B over 8.0 min) to 155-1 or 155-2. LCMS [M+1] = 815.5; 1H NMR (400 MHz, METHANOL-d4) δ = 8.73 (s, 1H), 7.66 (s, 1H), 7.53 - 7.44 (m, 1H), 7.42 - 7.28 (m, 3H), 7.14 - 7.01 (m, 2H), 4.71 (s, 2H), 3.77 (t, J = 6.8 Hz, 3H), 3.73 - 3.36 (m, 5H), 2.81 (t, J = 6.8 Hz, 2H), 2.71 - 2.59 (m, 2H), 2.53 (s, 3H), 2.31 (br t, J = 9.6 Hz, 2H), 1.62 - 1.50 (m, 9H), 1.38 (br d, J = 6.8 Hz, 3H). Example 156: Synthesis of Compound 156
5-carboxylate (155a) To a solution of 1-(4-bromo-3-fluorophenyl)ethan-1-one (2.5 g, 11.3 mmol, 1.1 equiv.) and tert-butyl 3-(4-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrimidin-2-yl) isoxazole-5-carboxylate (4.0 g, 10.3 mmol, 1.0 equiv.) in water (16 mL) and dioxane (80 mL) was added K2CO3 (4.3 g, 30.9 mmol, 3.0 equiv.) and Pd(dppf)Cl2.CH2Cl2 (1.3 g, 1.5 mmol, 0.15 equiv.) under Ar atmosphere. The mixture was stirred at 80°C for 3 h. The mixture was diluted with water (80 mL) and extracted with ethyl acetate (160 mL). The combined organic layers were
dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, Petroleum ether/Ethyl acetate=50/7 to 5/1) to afford 155a. LCMS [M+1] = 398.3. Step B – Synthesis of tert-butyl 3-[5-[4-[1-[6-[4-(2,4-dioxohexahydropyrimidin-1-yl)-3-fluoro- phenyl]-2-azaspiro[3.3]heptan-2-yl]ethyl]-2-fluoro-phenyl]-4-methyl-pyrimidin-2-yl]isoxazole-5- carboxylate (155b-1 and 155b-2) To a solution of 1-[4-(2-azaspiro[3.3]heptan-6-yl)-2-fluoro-phenyl]hexahydropyrimidine - 2,4-dione trifluoroacetate salt (1.58 g, 3.77 mmol, 1.5 equiv.) and tert-butyl 3-(5-(4-acetyl-2- fluorophenyl)-4-methylpyrimidin-2-yl)isoxazole-5-carboxylate (1 g, 2.52 mmol, 1 equiv.) in dichloromethane (20 mL) was added triethylamine (3.50 mL, 10 equiv.) and NaBH(OAc)3 (2.67 g, 12.58 mmol, 5 equiv.). The mixture was stirred at 25°C for 12 h. The mixture was poured into water (50 mL) and extracted with ethyl acetate (50 mL x 3). The combined organic portions were washed with brine (50 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, Petroleum ether/Ethyl acetate=100/1 to 10/1) to give tert-butyl 3-(5-(4-(1-(6-(4-(2,4- dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2-azaspiro[3.3]heptan-2-yl)ethyl)-2- fluorophenyl)-4-methylpyrimidin-2-yl)isoxazole-5-carboxylate (1.2 g, 1.75 mmol, 69.64% yield). LCMS [M+H] = 685.4 The enantiomers were separated by SFC (ChiralPak IH, 250 mm x 30mm; 10 µm; [CO2-EtOH:ACN=1:1 (0.1% isopropylamine)]; B%:50%, isocratic elution mode) to afford 155b-1 as the early eluting isomer and 155b-2 as the late eluting isomer. Each isomer was processed separately in the subsequent steps. LCMS [M+1] = 685.4. Step C – Synthesis of 3-(5-(4-(1-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)- 2-azaspiro[3.3]heptan-2-yl)ethyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)isoxazole-5-carboxylic acid trifluoroacetate salt (155c-1 and 155c-2) To a solution of tert-butyl 3-(5-(4-(1-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3- fluorophenyl)-2-azaspiro[3.3]heptan-2-yl)ethyl)-2-fluorophenyl)-4-methylpyrimidin-2- yl)isoxazole-5-carboxylate (500 mg) in dichloromethane (5 mL) was added TFA (1.54 g, 13.46 mmol, 1 mL, 18.44 equiv.). The mixture was stirred at 20 °C for 3 h. The mixture was concentrated under reduced pressure to give 155c-1 or 155c-2. LCMS [M+1] = 629.3.
Step D – Synthesis of N-((3-(3,3-difluoro-2-methylbutan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-3-(5- (4-(1-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2-azaspiro[3.3]heptan-2- yl)ethyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)isoxazole-5-carboxamide (155-1 and 155-2) To a solution of [3-(2,2-difluoro-1,1-dimethyl-propyl)-1H-1,2,4-triazol-5-yl]methanamine hydrochloride (31.11 mg, 112.26 μmol, 1.04 eq) in DMF (1 mL) was added BOP (95.29 mg, 215.45 μmol, 2 equiv.) and triethylamine (75 μL, 5 equiv.). 3-(5-(4-(1-(6-(4-(2,4- dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2-azaspiro[3.3]heptan-2-yl)ethyl)-2- fluorophenyl)-4-methylpyrimidin-2-yl)isoxazole-5-carboxylic acid (80 mg, 107.72 μmol, 1 equiv., TFA) was added to the mixture. The mixture was stirred at 20 °C for 2 h. The mixture was poured into water (0.5 mL) and extracted with ethyl acetate (0.5 mL x 3). The combined organic layers were washed with brine (0.5 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by prep-HPLC (Phenomenex Luna C18, 100x40mm, 5 µm; [water(0.2% FA)-ACN]; gradient:10%-40% B over 8.0 min) to 155-1 or 155-2. LCMS [M+1] = 815.5; 1H NMR (400 MHz, METHANOL-d4) δ = 8.73 (s, 1H), 7.66 (s, 1H), 7.53 - 7.44 (m, 1H), 7.42 - 7.28 (m, 3H), 7.14 - 7.01 (m, 2H), 4.71 (s, 2H), 3.77 (t, J = 6.8 Hz, 3H), 3.73 - 3.36 (m, 5H), 2.81 (t, J = 6.8 Hz, 2H), 2.71 - 2.59 (m, 2H), 2.53 (s, 3H), 2.31 (br t, J = 9.6 Hz, 2H), 1.62 - 1.50 (m, 9H), 1.38 (br d, J = 6.8 Hz, 3H). Example 156: Synthesis of Compound 156
Step A soxazole- 5-carboxylate (156a) To a solution of tert-butyl 3-(5-bromo-4-methylpyrimidin-2-yl)isoxazole-5-carboxylate (2.0 g, 5.8 mmol, 1.0 equiv.) and 3-fluoro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)benzaldehyde (1.9 g, 7.6 mmol, 1.3 equiv.) in dioxane (15.0 mL) and water (3.0 mL), was added Pd(dppf)Cl2 (430.1 mg, 587.9 μmol, 0.1 equiv.) and K2CO3 (2.4 g, 17.6 mmol, 3.0 equiv.) at 25°C. The mixture was stirred at 90°C for 2 h under a nitrogen atmosphere. The mixture was cooled to 25°C and quenched with water (100 mL) and extracted with ethyl acetate (50 mL ×3), the combined organic portions were dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated. The residue was purified by column chromatography (silica gel, Petroleum ether/Ethyl acetate=1/0 to 1/1) to give 156a. LCMS [M+1] = 384.0.
Step B – Synthesis of tert-butyl 3-(5-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3- fluorophenyl)-2-azaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)-4-methylpyrimidin-2- yl)isoxazole-5-carboxylate (156b) To a solution of tert-butyl 3-(5-(2-fluoro-4-formylphenyl)-4-methylpyrimidin-2- yl)isoxazole-5-carboxylate (1.5 g, 3.9 mmol, 1.0 equiv.) and 1-(2-fluoro-4-(2- azaspiro[3.3]heptan-6-yl)phenyl)dihydropyrimidine-2,4(1H,3H)-dione trifluoroacetate (1.6 g, 3.9 mmol, 1.0 equiv.) in dichloromethane (25.0 mL) was added triethylamine (2.7 mL, 5.0 equiv.). The mixture was stirred at 25°C for 1 h, treated with NaBH(OAc)3 (3.3 g, 15.6 mmol, 4.0 equiv.), the mixture was stirred at 25°C for 16 h. The mixture was quenched with water (100 mL) and extracted with dichloromethane (25 mL ×3), the combined organic portions were dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated. The residue was purified by column chromatography (silica gel, dichloromethane:MeOH= 1:0 to 8:1) to give 156b. LCMS [M+1] = 671.3. Step C – Synthesis of 3-(5-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)isoxazole-5-carboxylic acid trifluoroacetate salt (156c) To a solution of tert-butyl 3-(5-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3- fluorophenyl)-2-azaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)-4-methylpyrimidin-2- yl)isoxazole-5-carboxylate (2.0 g, 2.9 mmol, 1.0 equiv.) in dichloromethane (30.0 mL) was added TFA (10.0 mL). The mixture was stirred at 25°C for 4 h. The mixture was filtered and the filtrate was concentrated to give 156c. LCMS [M+1] = 615.2. Step D – Synthesis of 3-(5-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro- 2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (156) To a solution of 3-(5-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)- 2-azaspiro[3.3]¬heptan-2-yl)methyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)isoxazole-5- carboxylic acid trifluoroacetate salt (1.8 g, 2.4 mmol, 1.0 equiv.) and (3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methanamine methyl sulphonic acid salt (1.56 g, 3.9
mmol, 1.6 equiv.) in DMF (20.0 mL) was added NMI (1.0 g, 12.3 mmol, 984.5 μL, 5.0 equiv.) and TCFH (1.0 g, 3.7 mmol, 1.5 equiv.). The mixture was stirred at 25°C for 2 h. The mixture was filtered and the filtrate was purified by prep-HPLC (Phenomenex Luna C18 (250x70mm,10 µm); [water (FA)-ACN]; gradient: 15%-45% B over 20 min) to give 156. LCMS [M+1] = 805.2. 1H NMR (400 MHz, DMSO-d6) δ 14.0 (bs, 1H), 10.45 (s, 1H), 9.73 (t, J = 5.6 Hz, 1H), 8.85 (s, 1H), 7.73 (s, 1H), 7.50 (t, J = 8.0 Hz, 1H), 7.37 - 7.27 (m, 3H), 7.16 (d, J = 11.6 Hz, 1H), 7.12 - 7.05 (m, 1H), 4.61 (d, J = 6.0 Hz, 2H), 3.71 - 3.64 (m, 4H), 3.43 (d, J = 8.4 Hz, 2H), 3.18 (s, 3H), 2.71 (t, J = 6.8 Hz, 2H), 2.54 (d, J = 2.8 Hz, 2H), 2.48 (d, J = 0.8 Hz, 3H), 2.27 - 2.16 (m, 2H), 1.55 (s, 6H). Example 157: Synthesis of Compound 157
 [3.3]heptan-2-yl)methyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(3-fluoro-2-methylbut-3- en-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (157) To a solution of 3-(5-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)- 2-azaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)isoxazole-5- carboxylic acid trifluoroacetic acid salt (50.0g, 68.6 μmol, 1.0 equiv) in dichloromethane (1 mL) was added DIEA (133.0 mg, 1.0 mmol, 179.3 μL, 15.0 equiv.), BOP (33.4 mg, 75.5 μmol, 1.1 equiv.) at 20°C and stirred for 10 min. The mixture was treated with [3-(2-fluoro-1,1-dimethyl- allyl)-1H-1,2,4-triazol-5-yl] methanamine hydrochloride (35.3 mg, 137.3 μmol, 2.0 eq) at 20°C and stirred for 2 h. The residue was poured into water (20 mL). The aqueous phase was extracted with ethyl acetate (20 mL x 2). The combined organic phase was washed with brine (5 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by prep-HPLC (Phenomenex Luna C18100x30mm, 5µm;
[water(0.2% FA)-ACN];gradient:10%-40% B over 8.0 min) to give 157. LCMS [M+1] = 781.5; 1H NMR (400 MHz, CHLOROFORM-d) δ = 8.66 (s, 1H), 7.68 (s, 1H), 7.54 (br s, 1H), 7.44 (br t, J = 5.6 Hz, 1H), 7.26 - 7.17 (m, 3H), 7.07 - 6.95 (m, 2H), 4.85 - 4.72 (m, 3H), 4.67 - 4.48 (m, 1H), 3.85 - 3.69 (m, 4H), 3.54 (s, 2H), 3.49 - 3.38 (m, 1H), 3.34 (s, 2H), 2.86 (t, J = 6.4 Hz, 2H), 2.69 - 2.59 (m, 2H), 2.56 (s, 3H), 2.37 - 2.22 (m, 2H), 1.63 (s, 6H). Example 158: Synthesis of Compound 158
[3.3]heptan-2-yl)methyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(3-fluoro-2-methylbut-3- en-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (157) To a solution of 3-(5-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)- 2-azaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)isoxazole-5- carboxylic acid trifluoroacetic acid salt (50.0g, 68.6 μmol, 1.0 equiv) in dichloromethane (1 mL) was added DIEA (133.0 mg, 1.0 mmol, 179.3 μL, 15.0 equiv.), BOP (33.4 mg, 75.5 μmol, 1.1 equiv.) at 20°C and stirred for 10 min. The mixture was treated with [3-(2-fluoro-1,1-dimethyl- allyl)-1H-1,2,4-triazol-5-yl] methanamine hydrochloride (35.3 mg, 137.3 μmol, 2.0 eq) at 20°C and stirred for 2 h. The residue was poured into water (20 mL). The aqueous phase was extracted with ethyl acetate (20 mL x 2). The combined organic phase was washed with brine (5 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by prep-HPLC (Phenomenex Luna C18100x30mm, 5µm;
[water(0.2% FA)-ACN];gradient:10%-40% B over 8.0 min) to give 157. LCMS [M+1] = 781.5; 1H NMR (400 MHz, CHLOROFORM-d) δ = 8.66 (s, 1H), 7.68 (s, 1H), 7.54 (br s, 1H), 7.44 (br t, J = 5.6 Hz, 1H), 7.26 - 7.17 (m, 3H), 7.07 - 6.95 (m, 2H), 4.85 - 4.72 (m, 3H), 4.67 - 4.48 (m, 1H), 3.85 - 3.69 (m, 4H), 3.54 (s, 2H), 3.49 - 3.38 (m, 1H), 3.34 (s, 2H), 2.86 (t, J = 6.4 Hz, 2H), 2.69 - 2.59 (m, 2H), 2.56 (s, 3H), 2.37 - 2.22 (m, 2H), 1.63 (s, 6H). Example 158: Synthesis of Compound 158
 To a solution of benzyl 3-oxobutanoate (30.0 g, 156.0 mmol, 26.9 mL, 1.0 equiv.), 1,2- dibromoethane (58.6 g, 312.1 mmol, 23.5 mL, 2.0 equiv.) in DMF (60.0 mL) was added K2CO3 (53.9 g, 390.2 mmol, 2.5 equiv.). The mixture was stirred at 60°C for 32 h. The mixture was cooled to 25°C, diluted with water (300 mL) and extracted with ethyl acetate (200 mLx3). The organic phase was washed with brine (200 mLx2), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by prep-HPLC (Daisogel C18250x70mm, 10 µm; [water(NH3H2O)-ACN]; gradient:40%-70% B over 5 min ) to 158a. LCMS [M+1] = 219.0.
Step B – Synthesis of benzyl 1-(1,1-difluoroethyl)cyclopropane-1-carboxylate (158b) To a solution of benzyl 1-acetylcyclopropane-1-carboxylate (2.0 g, 9.1 mmol, 1.0 equiv.) in EtOH (21.1 mg, 458.1 μmol, 0.05 equiv.) was added BAST (20.2 g, 91.6 mmol, 20.0 mL, 10.0 equiv.) at 0°C. The mixture was stirred at 50 °C for 16 h. The mixture was cooled to 25°C, quenched with aqueous NaHCO3 (aq. 150.0 mL) and extracted with dichloromethane (100 mLx3). The combined organic portion was dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated. The residue was purified by column chromatography (silica gel, petroleum ether/ethyl acetate=1/0 to 10/1) to give 158b. 1H NMR (400 MHz, CHLOROFORM-d) δ 7.43 - 7.30 (m, 5H), 5.16 (s, 2H), 1.91 (J=18.8 Hz, 3H), 1.38 - 1.32 (m, 2H), 1.29 - 1.24 (m, 2H). Step C – Synthesis of 1-(1,1-difluoroethyl)cyclopropane-1-carboxylic acid (158c) To a solution of benzyl 1-(1,1-difluoroethyl)cyclopropane-1-carboxylate (1.0 gx2, 4.1 mmol, 1.0 equiv.) in ethyl acetate (20.0 mlx2) was added Pd/C (885.9 mgx2, 832.4 μmol, 10% purity, 0.2 equiv.) under a nitrogen atmosphere. The suspension was degassed and purged threefold with hydrogen. The mixture was stirred under hydrogen (15 Psi.) at 50°C for 2 h. The mixture was cooled to 25°C, filtered and the filtrate was concentrated under reduced pressure to give 158c, which was used without purification. 1H NMR (400 MHz, CHLOROFORM-d) δ 9.92 (s, 1H), 1.92 (t, J=18.8 Hz, 3H), 1.50 - 1.29 (m, 4H). Step D – Synthesis of tert-butyl (2-(1-(1,1-difluoroethyl)cyclopropane-1-carboxamido)-2- iminoethyl)carbamate (158d) To a solution of 1-(1,1-difluoroethyl)cyclopropane-1-carboxylic acid (1.4 g, 9.3 mmol, 1 equiv.) in DMF (7.0 mL) was added CDI (1.5 g, 9.3 mmol, 1.0 equiv.) and the mixture was stirred at 25°C for 50 min. The mixture was treated with triethylamine (1.3 mL, 1.0 equiv.) and tert- butyl (2-amino-2-iminoethyl)carbamate hydrochloride (1.9 g, 9.3 mmol, 1.0 equiv.) in DMF (7.0 mL). The mixture was stirred at 25°C for 30 min. The mixture was diluted with water (100 mL) and extracted with ethyl acetate (30 mLx3), the combined organic layer was washed with brine (30 mLx2), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether/ethyl acetate=1/0 to 10/1) to give 158d. LCMS [M+1] = 306.1.
Step E – Synthesis of tert-butyl ((3-(1-(1,1-difluoroethyl)cyclopropyl)-1H-1,2,4-triazol-5- yl)methyl)carbamate (158e) A solution of tert-butyl (2-(1-(1,1-difluoroethyl)cyclopropane-1-carboxamido)-2- iminoethyl)carbamate (1.4 g, 4.5 mmol, 1.0 equiv.) in DMF (7.0 mL) was treated with acetic acid (4.2 mL) at -10°C followed by hydrazine;hydrate (1.0 g, 18.3 mmol, 1.0 mL, 85% purity, 4 equiv.) was added at -10°C. The mixture was stirred at 25°C for 1 h under a nitrogen atmosphere. The mixture was added dropwise into a solution of Na2CO3 (80 mL) and extracted with ethyl acetate (30 mLx3), the combined organic layer was washed with brine (30 mLx2), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether/ethyl acetate=1/0 to 1/1) to give 158e. LCMS [M+1] = 303.2; 1H NMR (400 MHz, CHLOROFORM-d) δ 4.40 (d, J = 4.0 Hz, 2H) 1.78 (t, J = 18.8 Hz, 3H), 1.46 (s, 9H), 1.35 - 1.29 (m, 2 H), 1.28 - 1.23 (m, 2H). Step F – Synthesis of (3-(1-(1,1-difluoroethyl)cyclopropyl)-1H-1,2,4-triazol-5-yl)methanamine hydrochloride salt (158f) A mixture of tert-butyl ((3-(1-(1,1-difluoroethyl)cyclopropyl)-1H-1,2,4-triazol-5- yl)methyl)carbamate (200.0 mg, 662 μmol, 1.0 equiv.) in HCl/ethyl acetate (2 M, 331 μL, 1.0 equiv.) was stirred at 25°C for 1 h. The mixture was concentrated to give 158f, which was used without purification. LCMS [M+1] = 203.2; 1H NMR (400 MHz, DMSO-d6) δ 8.59 (s, 3H), 4.11 - 3.96 (m, 2 H) 1.79 (t, J = 18.8 Hz, 3H), 1.34 - 1.25 (m, 2H), 1.15 (s, 2H). Step G – Synthesis of N-((3-(1-(1,1-difluoroethyl)cyclopropyl)-1H-1,2,4-triazol-5-yl)methyl)-3-(5- (4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2-azaspiro[3.3]heptan-2- yl)methyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)isoxazole-5-carboxamide (158) To a solution of (3-(1-(1,1-difluoroethyl)cyclopropyl)-1H-1,2,4-triazol-5-yl)methanamine hydrochloride salt (33.4 mg, 122.0 μmol, 1.5 equiv), 3-(5-(4-((6-(4-(2,4- dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2-azaspiro[3.3]heptan-2-yl)methyl)-2- fluorophenyl)-4-methylpyrimidin-2-yl)isoxazole-5-carboxylic acid (50.0 mg, 81.3 μmol, 1.0 equiv.) in DMF (0.5 mL) was added NMI (20.0 mg, 244.0 μmol, 19.4 μL, 3.0 equiv.) and TCFH (68.4 mg, 244.0 μmol, 3 equiv.). The mixture was stirred at 25°C for 30 min. The mixture was
diluted with water (5 mL) and extracted ethyl acetate (10 mLx3). The organic phase was washed with brine (10 mLx2), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure to afford a residue. The residue was purified by prep-HPLC (Daisogel SP-100-8-ODS-PK 150x25mm, 10 µm; [water( NH4HCO3)-ACN]; gradient:26%-56% B over 10 min ) to give 158. LCMS [M+1] = 799.6; 1H NMR (400 MHz, DMSO-d6) δ 13.72 (bs, 1H), 10.44 (s, 1H), 9.65 (s, 1H), 8.84 (s, 1H), 7.71 (s, 1H), 7.25 - 7.45 (m, 1H), 7.37 - 7.30 (m, 2H), 7.29 (s, 1H), 7.18 - 7.13 (m, 1H), 7.11 - 7.06 (m, 1H), 4.57 - 4.51 (m, 2H), 3.70 - 3.66 (m, 2H), 3.65 - 3.60 (m, 2H), 3.46 - 3.38 (m, 1H) 3.36 - 3.34 (m, 2H), 3.15 (s, 2H), 2.70 (t, J = 6.8 Hz, 2H), 2.53 (s, 2H), 2.47 (s, 3H), 2.25 - 2.18 (m, 2H), 1.90 - 1.78 (m, 3H), 1.21 (s, 2H), 1.11 (s, 2H). Example 159: Synthesis of Compound 159
To a solution of benzyl 3-oxobutanoate (30.0 g, 156.0 mmol, 26.9 mL, 1.0 equiv.), 1,2- dibromoethane (58.6 g, 312.1 mmol, 23.5 mL, 2.0 equiv.) in DMF (60.0 mL) was added K2CO3 (53.9 g, 390.2 mmol, 2.5 equiv.). The mixture was stirred at 60°C for 32 h. The mixture was cooled to 25°C, diluted with water (300 mL) and extracted with ethyl acetate (200 mLx3). The organic phase was washed with brine (200 mLx2), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by prep-HPLC (Daisogel C18250x70mm, 10 µm; [water(NH3H2O)-ACN]; gradient:40%-70% B over 5 min ) to 158a. LCMS [M+1] = 219.0.
Step B – Synthesis of benzyl 1-(1,1-difluoroethyl)cyclopropane-1-carboxylate (158b) To a solution of benzyl 1-acetylcyclopropane-1-carboxylate (2.0 g, 9.1 mmol, 1.0 equiv.) in EtOH (21.1 mg, 458.1 μmol, 0.05 equiv.) was added BAST (20.2 g, 91.6 mmol, 20.0 mL, 10.0 equiv.) at 0°C. The mixture was stirred at 50 °C for 16 h. The mixture was cooled to 25°C, quenched with aqueous NaHCO3 (aq. 150.0 mL) and extracted with dichloromethane (100 mLx3). The combined organic portion was dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated. The residue was purified by column chromatography (silica gel, petroleum ether/ethyl acetate=1/0 to 10/1) to give 158b. 1H NMR (400 MHz, CHLOROFORM-d) δ 7.43 - 7.30 (m, 5H), 5.16 (s, 2H), 1.91 (J=18.8 Hz, 3H), 1.38 - 1.32 (m, 2H), 1.29 - 1.24 (m, 2H). Step C – Synthesis of 1-(1,1-difluoroethyl)cyclopropane-1-carboxylic acid (158c) To a solution of benzyl 1-(1,1-difluoroethyl)cyclopropane-1-carboxylate (1.0 gx2, 4.1 mmol, 1.0 equiv.) in ethyl acetate (20.0 mlx2) was added Pd/C (885.9 mgx2, 832.4 μmol, 10% purity, 0.2 equiv.) under a nitrogen atmosphere. The suspension was degassed and purged threefold with hydrogen. The mixture was stirred under hydrogen (15 Psi.) at 50°C for 2 h. The mixture was cooled to 25°C, filtered and the filtrate was concentrated under reduced pressure to give 158c, which was used without purification. 1H NMR (400 MHz, CHLOROFORM-d) δ 9.92 (s, 1H), 1.92 (t, J=18.8 Hz, 3H), 1.50 - 1.29 (m, 4H). Step D – Synthesis of tert-butyl (2-(1-(1,1-difluoroethyl)cyclopropane-1-carboxamido)-2- iminoethyl)carbamate (158d) To a solution of 1-(1,1-difluoroethyl)cyclopropane-1-carboxylic acid (1.4 g, 9.3 mmol, 1 equiv.) in DMF (7.0 mL) was added CDI (1.5 g, 9.3 mmol, 1.0 equiv.) and the mixture was stirred at 25°C for 50 min. The mixture was treated with triethylamine (1.3 mL, 1.0 equiv.) and tert- butyl (2-amino-2-iminoethyl)carbamate hydrochloride (1.9 g, 9.3 mmol, 1.0 equiv.) in DMF (7.0 mL). The mixture was stirred at 25°C for 30 min. The mixture was diluted with water (100 mL) and extracted with ethyl acetate (30 mLx3), the combined organic layer was washed with brine (30 mLx2), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether/ethyl acetate=1/0 to 10/1) to give 158d. LCMS [M+1] = 306.1.
Step E – Synthesis of tert-butyl ((3-(1-(1,1-difluoroethyl)cyclopropyl)-1H-1,2,4-triazol-5- yl)methyl)carbamate (158e) A solution of tert-butyl (2-(1-(1,1-difluoroethyl)cyclopropane-1-carboxamido)-2- iminoethyl)carbamate (1.4 g, 4.5 mmol, 1.0 equiv.) in DMF (7.0 mL) was treated with acetic acid (4.2 mL) at -10°C followed by hydrazine;hydrate (1.0 g, 18.3 mmol, 1.0 mL, 85% purity, 4 equiv.) was added at -10°C. The mixture was stirred at 25°C for 1 h under a nitrogen atmosphere. The mixture was added dropwise into a solution of Na2CO3 (80 mL) and extracted with ethyl acetate (30 mLx3), the combined organic layer was washed with brine (30 mLx2), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether/ethyl acetate=1/0 to 1/1) to give 158e. LCMS [M+1] = 303.2; 1H NMR (400 MHz, CHLOROFORM-d) δ 4.40 (d, J = 4.0 Hz, 2H) 1.78 (t, J = 18.8 Hz, 3H), 1.46 (s, 9H), 1.35 - 1.29 (m, 2 H), 1.28 - 1.23 (m, 2H). Step F – Synthesis of (3-(1-(1,1-difluoroethyl)cyclopropyl)-1H-1,2,4-triazol-5-yl)methanamine hydrochloride salt (158f) A mixture of tert-butyl ((3-(1-(1,1-difluoroethyl)cyclopropyl)-1H-1,2,4-triazol-5- yl)methyl)carbamate (200.0 mg, 662 μmol, 1.0 equiv.) in HCl/ethyl acetate (2 M, 331 μL, 1.0 equiv.) was stirred at 25°C for 1 h. The mixture was concentrated to give 158f, which was used without purification. LCMS [M+1] = 203.2; 1H NMR (400 MHz, DMSO-d6) δ 8.59 (s, 3H), 4.11 - 3.96 (m, 2 H) 1.79 (t, J = 18.8 Hz, 3H), 1.34 - 1.25 (m, 2H), 1.15 (s, 2H). Step G – Synthesis of N-((3-(1-(1,1-difluoroethyl)cyclopropyl)-1H-1,2,4-triazol-5-yl)methyl)-3-(5- (4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2-azaspiro[3.3]heptan-2- yl)methyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)isoxazole-5-carboxamide (158) To a solution of (3-(1-(1,1-difluoroethyl)cyclopropyl)-1H-1,2,4-triazol-5-yl)methanamine hydrochloride salt (33.4 mg, 122.0 μmol, 1.5 equiv), 3-(5-(4-((6-(4-(2,4- dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2-azaspiro[3.3]heptan-2-yl)methyl)-2- fluorophenyl)-4-methylpyrimidin-2-yl)isoxazole-5-carboxylic acid (50.0 mg, 81.3 μmol, 1.0 equiv.) in DMF (0.5 mL) was added NMI (20.0 mg, 244.0 μmol, 19.4 μL, 3.0 equiv.) and TCFH (68.4 mg, 244.0 μmol, 3 equiv.). The mixture was stirred at 25°C for 30 min. The mixture was
diluted with water (5 mL) and extracted ethyl acetate (10 mLx3). The organic phase was washed with brine (10 mLx2), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure to afford a residue. The residue was purified by prep-HPLC (Daisogel SP-100-8-ODS-PK 150x25mm, 10 µm; [water( NH4HCO3)-ACN]; gradient:26%-56% B over 10 min ) to give 158. LCMS [M+1] = 799.6; 1H NMR (400 MHz, DMSO-d6) δ 13.72 (bs, 1H), 10.44 (s, 1H), 9.65 (s, 1H), 8.84 (s, 1H), 7.71 (s, 1H), 7.25 - 7.45 (m, 1H), 7.37 - 7.30 (m, 2H), 7.29 (s, 1H), 7.18 - 7.13 (m, 1H), 7.11 - 7.06 (m, 1H), 4.57 - 4.51 (m, 2H), 3.70 - 3.66 (m, 2H), 3.65 - 3.60 (m, 2H), 3.46 - 3.38 (m, 1H) 3.36 - 3.34 (m, 2H), 3.15 (s, 2H), 2.70 (t, J = 6.8 Hz, 2H), 2.53 (s, 2H), 2.47 (s, 3H), 2.25 - 2.18 (m, 2H), 1.90 - 1.78 (m, 3H), 1.21 (s, 2H), 1.11 (s, 2H). Example 159: Synthesis of Compound 159
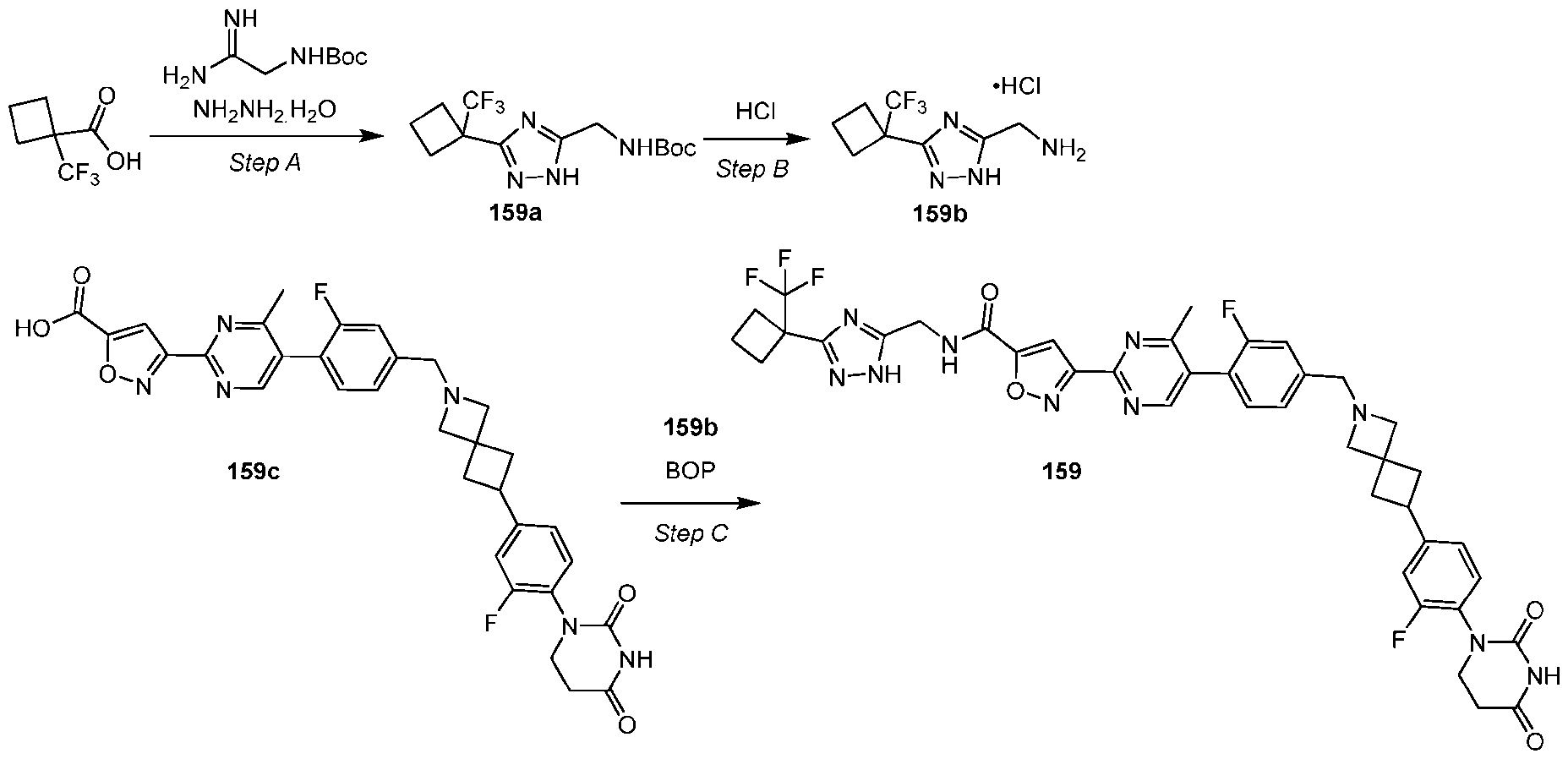 Step A – Synthesis of tert-butyl ((3-(1-(trifluoromethyl)cyclobutyl)-1H-1,2,4-triazol-5- yl)methyl)carbamate (159a) To a solution of 1-(trifluoromethyl)cyclobutanecarboxylic acid (2 g, 11.90 mmol, 1 equiv.) in DMF (10 mL) was added CDI (1.93 g, 11.90 mmol, 1 equiv.) at 25°C. The mixture was stirred at 25°C for 1 h. The mixture was added dropwise to a solution of tert-butyl (2-amino- 2-iminoethyl)carbamate hydrochloride (2.49 mg, 11.90 mmol, 1 equiv) and triethylamine (3.31 mL, 2 equiv.) in DMF (10 mL) at 0°C under a nitrogen atmosphere. The mixture was stirred at 0°C for 10 min. Acetic acid (11.29 g, 188.05 mmol, 10.77 mL, 16 equiv.) and hydrazine hydrate
(588.38 mg, 11.75 mmol, 85.52 μL, 1 equiv.) were added to the mixture at 0°C. The mixture was stirred at 0°C for 10 min. The mixture was quenched by addition of aq Na2CO3 (60 mL), filtered and the filtrate was dried under reduced pressure to give 159a. LCMS [M+1] = 321.3. Step B – Synthesis of (3-(1-(trifluoromethyl)cyclobutyl)-1H-1,2,4-triazol-5-yl)methanamine hydrochloride salt (159b) To a solution of tert-butyl ((3-(1-(trifluoromethyl)cyclobutyl)-1H-1,2,4-triazol-5- yl)methyl)carbamate (6.6 g, 20.61 mmol, 1 equiv.) in HCl/ethyl acetate (66 mL) at 25°C. The mixture was stirred at 25°C for 1 h. The mixture was concentrated under reduced pressure. The residue was triturated with ethyl acetate at 25oC for 5 min, then filtered and the filtrate was concentrated under reduced pressure to give 159b. LCMS [M+1] = 220.1; 1H NMR (400MHz, METHANOL-d4) δ 4.28 (s, 2H), 2.80 - 2.68 (m, 4H), 2.18 - 2.05 (m, 2H). Step C – Synthesis of 3-(5-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1- (trifluoromethyl)cyclobutyl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (159) A solution of 3-(5-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)isoxazole-5-carboxylic acid trifluoroacetate salt (100 mg, 137 μmol, 1 equiv.) in DMF (1 mL) was treated with BOP (182 mg, 412 μmol, 3 equiv.) and triethylamine (153 μL, 8 equiv.) and (3-(1- (trifluoromethyl)cyclobutyl)-1H-1,2,4-triazol-5-yl)methanamine hydrochloride (41.5 mg, 161 μmol, 1.5 equiv.) at 25°C. The mixture was stirred at 25°C for 1 h. The mixture was filtered to remove the insoluble portion. The filtrate was purified by prep-HPLC (Waters Xbridge Prep OBD C18150 x 40 mm, 10 µm; [water (10mM NH4HCO3) - ACN]; gradient: 23% - 63% B over 8.0 min) to give 159. LCMS [M+1] = 817.3; 1H NMR (400MHz, DMSO-d6) δ 14.01 (br s, 1H), 10.45 (br s, 1H), 9.73 (br s, 1H), 8.85 (s, 1H), 7.73 (s, 1H), 7.49 (t, J = 8.0 Hz, 1H), 7.35 - 7.33 (m, 1H), 7.31 - 7.29 (m, 2H), 7.15 (dd, J = 1.6 Hz, 11.6 Hz, 1H), 7.08 (dd, J = 1.2 Hz, 8.0 Hz, 1H), 4.62 (s, 2H), 3.69 - 3.64 (m, 4H), 3.43 - 3.41 (m, 1H), 3.35 (s, 2H), 3.15 (s, 2H), 2.70 (t, J = 6.8 Hz, 2H), 2.59 (t, J = 8.0 Hz, 4H), 2.53 (d, J = 2.4 Hz, 2H), 2.47 (d, J = 0.8 Hz, 3H), 2.24 - 2.19 (m, 2H), 2.06 - 1.89 (m, 2H).
Example 160: Synthesis of Compound 160-1 and 160-2
Step A – Synthesis of tert-butyl ((3-(1-(trifluoromethyl)cyclobutyl)-1H-1,2,4-triazol-5- yl)methyl)carbamate (159a) To a solution of 1-(trifluoromethyl)cyclobutanecarboxylic acid (2 g, 11.90 mmol, 1 equiv.) in DMF (10 mL) was added CDI (1.93 g, 11.90 mmol, 1 equiv.) at 25°C. The mixture was stirred at 25°C for 1 h. The mixture was added dropwise to a solution of tert-butyl (2-amino- 2-iminoethyl)carbamate hydrochloride (2.49 mg, 11.90 mmol, 1 equiv) and triethylamine (3.31 mL, 2 equiv.) in DMF (10 mL) at 0°C under a nitrogen atmosphere. The mixture was stirred at 0°C for 10 min. Acetic acid (11.29 g, 188.05 mmol, 10.77 mL, 16 equiv.) and hydrazine hydrate
(588.38 mg, 11.75 mmol, 85.52 μL, 1 equiv.) were added to the mixture at 0°C. The mixture was stirred at 0°C for 10 min. The mixture was quenched by addition of aq Na2CO3 (60 mL), filtered and the filtrate was dried under reduced pressure to give 159a. LCMS [M+1] = 321.3. Step B – Synthesis of (3-(1-(trifluoromethyl)cyclobutyl)-1H-1,2,4-triazol-5-yl)methanamine hydrochloride salt (159b) To a solution of tert-butyl ((3-(1-(trifluoromethyl)cyclobutyl)-1H-1,2,4-triazol-5- yl)methyl)carbamate (6.6 g, 20.61 mmol, 1 equiv.) in HCl/ethyl acetate (66 mL) at 25°C. The mixture was stirred at 25°C for 1 h. The mixture was concentrated under reduced pressure. The residue was triturated with ethyl acetate at 25oC for 5 min, then filtered and the filtrate was concentrated under reduced pressure to give 159b. LCMS [M+1] = 220.1; 1H NMR (400MHz, METHANOL-d4) δ 4.28 (s, 2H), 2.80 - 2.68 (m, 4H), 2.18 - 2.05 (m, 2H). Step C – Synthesis of 3-(5-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1- (trifluoromethyl)cyclobutyl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (159) A solution of 3-(5-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)isoxazole-5-carboxylic acid trifluoroacetate salt (100 mg, 137 μmol, 1 equiv.) in DMF (1 mL) was treated with BOP (182 mg, 412 μmol, 3 equiv.) and triethylamine (153 μL, 8 equiv.) and (3-(1- (trifluoromethyl)cyclobutyl)-1H-1,2,4-triazol-5-yl)methanamine hydrochloride (41.5 mg, 161 μmol, 1.5 equiv.) at 25°C. The mixture was stirred at 25°C for 1 h. The mixture was filtered to remove the insoluble portion. The filtrate was purified by prep-HPLC (Waters Xbridge Prep OBD C18150 x 40 mm, 10 µm; [water (10mM NH4HCO3) - ACN]; gradient: 23% - 63% B over 8.0 min) to give 159. LCMS [M+1] = 817.3; 1H NMR (400MHz, DMSO-d6) δ 14.01 (br s, 1H), 10.45 (br s, 1H), 9.73 (br s, 1H), 8.85 (s, 1H), 7.73 (s, 1H), 7.49 (t, J = 8.0 Hz, 1H), 7.35 - 7.33 (m, 1H), 7.31 - 7.29 (m, 2H), 7.15 (dd, J = 1.6 Hz, 11.6 Hz, 1H), 7.08 (dd, J = 1.2 Hz, 8.0 Hz, 1H), 4.62 (s, 2H), 3.69 - 3.64 (m, 4H), 3.43 - 3.41 (m, 1H), 3.35 (s, 2H), 3.15 (s, 2H), 2.70 (t, J = 6.8 Hz, 2H), 2.59 (t, J = 8.0 Hz, 4H), 2.53 (d, J = 2.4 Hz, 2H), 2.47 (d, J = 0.8 Hz, 3H), 2.24 - 2.19 (m, 2H), 2.06 - 1.89 (m, 2H).
Example 160: Synthesis of Compound 160-1 and 160-2
 To a solution of 5-bromo-2-chloro-4,6-dimethyl-pyrimidine (1.0 equiv.,4.0 g) and DABCO (0.3 equiv.,0.608 g) in DMSO (32 mL) was added a solution of NaCN (1.1 equiv., 0.974 g) in water (32 mL) and the mixture was stirred at 80°C for 30 min. The mixture was cooled to 25°C and poured into water (200 mL). The aqueous phase was extracted with ethyl acetate (200 mLx3). The combined organic phase was washed with brine (100 mLx3), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure to give residue. The residue was purified by column chromatography (silica gel, Petroleum ether/Ethyl acetate=1/0 to 98/2) to give 160a. LCMS [M+1] = 212.0, 214.0; 1H NMR (400 MHz, DMSO-d6) δ 2.64 (s, 6H). Step B – Synthesis of 5-bromo-N'-hydroxy-4,6-dimethyl-pyrimidine-2-carboxamidine (160b) To a solution of 5-bromo-4,6-dimethyl-pyrimidine-2-carbonitrile (2.0 g, 9.4 mmol, 1.0 equiv.) and NH2OH.HCl (688.2 mg, 9.9 mmol, 1.05 equiv.) in MeOH (20.0 mL) was added NaOMe (5.4 M, 1.8 mL, 30% purity, 1.05 equiv.). The mixture was stirred at 70°C for 2 h. The
mixture was concentrated under reduced pressure. The crude product was triturated with water (20 mL) at 25oC for 1 min. The mixture was filtered and the filter cake was washed with water (20 mL), dried under reduced pressure to give 160b. LCMS [M+1] = 245.0, 247.0. Step C – Synthesis of 5-bromo-N-hydroxy-4,6-dimethyl-pyrimidine-2-carboximidoyl chloride (160c) To a solution of 5-bromo-N'-hydroxy-4,6-dimethyl-pyrimidine-2-carboxamidine (2.1 g, 8.5 mmol, 1.0 equiv.) and aqueous HCl (4 M, 19.2 mL, 9.0 equiv.) in water (21.0 mL) was added NaNO2 (739.0 mg, 10.7 mmol, 1.2 equiv.). The mixture was stirred at 0°C for 2 h. The mixture was added water (50 ml), filtered and the filter cake was washed with water (50 mL), dried under reduced pressure to give 160c. LCMS [M+1] = 263.9, 265.9. Step D – Synthesis of tert-butyl 3-(5-bromo-4,6-dimethyl-pyrimidin-2-yl)isoxazole-5-carboxylate (160d) To a solution of 5-bromo-N-hydroxy-4,6-dimethyl-pyrimidine-2-carboximidoyl chloride (1.5 g, 5.6 mmol, 1.0 equiv.) and tert-butyl propiolate (2.15 g, 17.0 mmol, 2.3 mL, 3.0 equiv.) in acetonitrile (15.0 mL) was added triethylamine (789 μL, 1.0 equiv.). The mixture was stirred at 0°C for 1 h. The mixture was poured into water (50 mL). The aqueous phase was extracted with ethyl acetate (50 mLx3). The combined organic phase was washed with brine (50 mLx3), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure to give 160d. LCMS [M+1] = 354.0, 356.0; 1H NMR (400 MHz, DMSO-d6) δ 7.51 (s, 1H), 2.67 (s, 6H), 1.58 (s, 9H). Step E – Synthesis of tert-butyl 3-[5-(4-acetyl-2-fluoro-phenyl)-4,6-dimethyl-pyrimidin-2- yl]isoxazole-5-carboxylate (160e) A mixture of tert-butyl 3-(5-bromo-4,6-dimethyl-pyrimidin-2-yl)isoxazole-5-carboxylate (0.9 g, 2.6 mmol, 1.0 equiv.), 1-(3-fluoro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)phenyl)ethan-1-one (850.0 mg, 3.2 mmol, 1.2 equiv.), Pd(dppf)Cl2 (196.2 mg, 268.2 μmol, 0.1 equiv.) and K2CO3 (1.1 g, 8.0 mmol, 3.0 equiv.) in dioxane (10.0 mL) and water (2.0 mL) was degassed and purged threefold with nitrogen. The mixture was stirred at 100°C for 1 h under a nitrogen atmosphere, cooled to 25°C and poured into water (100 mL). The aqueous phase was
extracted with ethyl acetate (100 mLx3). The combined organic portion was washed with brine (50 mLx3), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, Petroleum ether/Ethyl acetate=1/0 to 93/7) to give 160e. LCMS [M+1] = 412.6; 1H NMR (400 MHz, DMSO-d6) δ 7.99 - 7.93 (m, 2H), 7.68 (t, J = 7.6 Hz, 1H), 7.58 (s, 1H), 2.67 (s, 3H), 2.32 (s, 6H), 1.59 (s, 9H). Step F – Synthesis of tert-butyl 3-[5-[4-[1-[6-[4-(2,4-dioxohexahydropyrimidin-1-yl)-3-fluoro- phenyl]-2-azaspiro[3.3]heptan-2-yl]ethyl]-2-fluoro-phenyl]-4,6-dimethyl-pyrimidin-2- yl]isoxazole-5-carboxylate (160f-1 and 160f-2) To a solution of tert-butyl 3-[5-(4-acetyl-2-fluoro-phenyl)-4,6-dimethyl-pyrimidin-2- yl]isoxazole-5-carboxylate (600.0 mg, 1.4 mmol, 1.0 equiv.) and 1-[4-(2-azaspiro[3.3]heptan-6- yl)-2-fluoro-phenyl]hexahydropyrimidine-2,4-dione trifluoroacetate salt (1.1 g, 2.6 mmol, 1.8 equiv.) in dichloromethane (6.0 mL) was added triethylamine (609 μL, 3.0 equiv.). The mixture was stirred at 40°C for 30 min and then treated with NaBH3CN (137.4 mg, 2.1 mmol, 1.5 equiv.). The mixture was stirred at 40°C for 30 min. The residue was purified by prep-HPLC (Phenomenex Luna C18150x40mm, 10 µm; [water(FA)-ACN];gradient:18%-48% B over 25 min) to give the product as the racemate. LCMS [M+1] = 699.3. The racemate was separated by SFC (ChiralPak IH, 250x50mm, 10µm, [hexane- isopropanol/ACN(0.1%isopropylamine)];B%:30%, isocratic elution) to give 160f-1 as the early eluting isomer and 160f-2 as the late eluting isomer. Step G – Synthesis of 3-[5-[4-[(1R)-1-[6-[4-(2,4-dioxohexahydropyrimidin-1-yl)-3-fluoro- phenyl]-2-azaspiro[3.3]heptan-2-yl]ethyl]-2-fluoro-phenyl]-4,6-dimethyl-pyrimidin-2- yl]isoxazole-5-carboxylic acid (160g-1) To a solution of tert-butyl 3-[5-[4-[(1R)-1-[6-[4-(2,4-dioxohexahydropyrimidin-1-yl)-3- fluoro-phenyl]-2-azaspiro[3.3]heptan-2-yl]ethyl]-2-fluoro-phenyl]-4,6-dimethyl-pyrimidin-2- yl]isoxazole-5-carboxylate (100.0 mg, 143.1 μmol, 1.0 equiv.) in dichloromethane (1.5 mL) was added TFA (0.5 mL). The mixture was stirred at 25°C for 2 h. The mixture was concentrated under reduced pressure to afford 160g-1, which was used without purification. LCMS [M+1] = 643.3.
Step H – Synthesis of 3-[5-[4-[1-[6-[4-(2,4-dioxohexahydropyrimidin-1-yl)-3-fluoro-phenyl]-2- azaspiro[3.3]heptan-2-yl]ethyl]-2-fluoro-phenyl]-4,6-dimethyl-pyrimidin-2-yl]-N-[[3-(2,2,2- trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]isoxazole-5-carboxamide (160-1) To a solution of 3-[5-[4-[1-[6-[4-(2,4-dioxohexahydropyrimidin-1-yl)-3-fluoro-phenyl]-2- azaspiro[3.3]heptan-2-yl]ethyl]-2-fluoro-phenyl]-4,6-dimethyl-pyrimidin-2-yl]isoxazole-5- carboxylic acid trifluoroacetic acid salt (110.0 mg, 145.3 μmol, 1.0 equiv.) and (3-(1,1,1-trifluoro- 2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methanamine methyl sulfonic acid salt (63.9 mg, 159.9 μmol, 1.1 equiv.) in DMF (1.0 mL) was added NMI (35.8 mg, 436.1 μmol, 34.7 μL, 3.0 equiv.) and TCFH (61.1 mg, 218.0 μmol, 1.5 equiv.). The mixture was stirred at 25°C for 1 h. The mixture was purified by prep-HPLC (Welch Xtimate C18150x25mm, 5µm; [water(FA)- ACN];gradient:11%-41% B over 14 min) to give 160-1. LCMS [M+1] = 833.3; 1H NMR (400 MHz, DMSO-d6) δ 14.03 (s, 1H), 10.45 (s, 1H), 9.71 (t, J = 5.6 Hz, 1H), 8.14 (s, 1H), 7.71 (s, 1H), 7.45 - 7.39 (m, 1H), 7.34 (q, J = 8.0 Hz, 3H), 7.15 (dd, J = 1.6, 11.6 Hz, 1H), 7.07 (dd, J = 1.6, 8.4 Hz, 1H), 4.60 (d, J = 5.6 Hz, 2H), 3.67 (t, J = 6.8 Hz, 2H), 3.49 - 3.42 (m, 1H), 3.41 - 3.38 (m, 2H), 3.23 - 3.15 (m, 2H), 3.14 - 3.06 (m, 1H), 2.70 (t, J = 6.8 Hz, 2H), 2.47 (s, 2H), 2.31 (s, 6H), 2.21 (t, J = 10.4 Hz, 2H), 1.54 (s, 6H), 1.22 (d, J = 6.4 Hz, 3H). Intermediate 160f-2 was processed in a similar manner to afford 160-2. Example 161: Synthesis of Intermediate 161
To a solution of 5-bromo-2-chloro-4,6-dimethyl-pyrimidine (1.0 equiv.,4.0 g) and DABCO (0.3 equiv.,0.608 g) in DMSO (32 mL) was added a solution of NaCN (1.1 equiv., 0.974 g) in water (32 mL) and the mixture was stirred at 80°C for 30 min. The mixture was cooled to 25°C and poured into water (200 mL). The aqueous phase was extracted with ethyl acetate (200 mLx3). The combined organic phase was washed with brine (100 mLx3), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure to give residue. The residue was purified by column chromatography (silica gel, Petroleum ether/Ethyl acetate=1/0 to 98/2) to give 160a. LCMS [M+1] = 212.0, 214.0; 1H NMR (400 MHz, DMSO-d6) δ 2.64 (s, 6H). Step B – Synthesis of 5-bromo-N'-hydroxy-4,6-dimethyl-pyrimidine-2-carboxamidine (160b) To a solution of 5-bromo-4,6-dimethyl-pyrimidine-2-carbonitrile (2.0 g, 9.4 mmol, 1.0 equiv.) and NH2OH.HCl (688.2 mg, 9.9 mmol, 1.05 equiv.) in MeOH (20.0 mL) was added NaOMe (5.4 M, 1.8 mL, 30% purity, 1.05 equiv.). The mixture was stirred at 70°C for 2 h. The
mixture was concentrated under reduced pressure. The crude product was triturated with water (20 mL) at 25oC for 1 min. The mixture was filtered and the filter cake was washed with water (20 mL), dried under reduced pressure to give 160b. LCMS [M+1] = 245.0, 247.0. Step C – Synthesis of 5-bromo-N-hydroxy-4,6-dimethyl-pyrimidine-2-carboximidoyl chloride (160c) To a solution of 5-bromo-N'-hydroxy-4,6-dimethyl-pyrimidine-2-carboxamidine (2.1 g, 8.5 mmol, 1.0 equiv.) and aqueous HCl (4 M, 19.2 mL, 9.0 equiv.) in water (21.0 mL) was added NaNO2 (739.0 mg, 10.7 mmol, 1.2 equiv.). The mixture was stirred at 0°C for 2 h. The mixture was added water (50 ml), filtered and the filter cake was washed with water (50 mL), dried under reduced pressure to give 160c. LCMS [M+1] = 263.9, 265.9. Step D – Synthesis of tert-butyl 3-(5-bromo-4,6-dimethyl-pyrimidin-2-yl)isoxazole-5-carboxylate (160d) To a solution of 5-bromo-N-hydroxy-4,6-dimethyl-pyrimidine-2-carboximidoyl chloride (1.5 g, 5.6 mmol, 1.0 equiv.) and tert-butyl propiolate (2.15 g, 17.0 mmol, 2.3 mL, 3.0 equiv.) in acetonitrile (15.0 mL) was added triethylamine (789 μL, 1.0 equiv.). The mixture was stirred at 0°C for 1 h. The mixture was poured into water (50 mL). The aqueous phase was extracted with ethyl acetate (50 mLx3). The combined organic phase was washed with brine (50 mLx3), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure to give 160d. LCMS [M+1] = 354.0, 356.0; 1H NMR (400 MHz, DMSO-d6) δ 7.51 (s, 1H), 2.67 (s, 6H), 1.58 (s, 9H). Step E – Synthesis of tert-butyl 3-[5-(4-acetyl-2-fluoro-phenyl)-4,6-dimethyl-pyrimidin-2- yl]isoxazole-5-carboxylate (160e) A mixture of tert-butyl 3-(5-bromo-4,6-dimethyl-pyrimidin-2-yl)isoxazole-5-carboxylate (0.9 g, 2.6 mmol, 1.0 equiv.), 1-(3-fluoro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)phenyl)ethan-1-one (850.0 mg, 3.2 mmol, 1.2 equiv.), Pd(dppf)Cl2 (196.2 mg, 268.2 μmol, 0.1 equiv.) and K2CO3 (1.1 g, 8.0 mmol, 3.0 equiv.) in dioxane (10.0 mL) and water (2.0 mL) was degassed and purged threefold with nitrogen. The mixture was stirred at 100°C for 1 h under a nitrogen atmosphere, cooled to 25°C and poured into water (100 mL). The aqueous phase was
extracted with ethyl acetate (100 mLx3). The combined organic portion was washed with brine (50 mLx3), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, Petroleum ether/Ethyl acetate=1/0 to 93/7) to give 160e. LCMS [M+1] = 412.6; 1H NMR (400 MHz, DMSO-d6) δ 7.99 - 7.93 (m, 2H), 7.68 (t, J = 7.6 Hz, 1H), 7.58 (s, 1H), 2.67 (s, 3H), 2.32 (s, 6H), 1.59 (s, 9H). Step F – Synthesis of tert-butyl 3-[5-[4-[1-[6-[4-(2,4-dioxohexahydropyrimidin-1-yl)-3-fluoro- phenyl]-2-azaspiro[3.3]heptan-2-yl]ethyl]-2-fluoro-phenyl]-4,6-dimethyl-pyrimidin-2- yl]isoxazole-5-carboxylate (160f-1 and 160f-2) To a solution of tert-butyl 3-[5-(4-acetyl-2-fluoro-phenyl)-4,6-dimethyl-pyrimidin-2- yl]isoxazole-5-carboxylate (600.0 mg, 1.4 mmol, 1.0 equiv.) and 1-[4-(2-azaspiro[3.3]heptan-6- yl)-2-fluoro-phenyl]hexahydropyrimidine-2,4-dione trifluoroacetate salt (1.1 g, 2.6 mmol, 1.8 equiv.) in dichloromethane (6.0 mL) was added triethylamine (609 μL, 3.0 equiv.). The mixture was stirred at 40°C for 30 min and then treated with NaBH3CN (137.4 mg, 2.1 mmol, 1.5 equiv.). The mixture was stirred at 40°C for 30 min. The residue was purified by prep-HPLC (Phenomenex Luna C18150x40mm, 10 µm; [water(FA)-ACN];gradient:18%-48% B over 25 min) to give the product as the racemate. LCMS [M+1] = 699.3. The racemate was separated by SFC (ChiralPak IH, 250x50mm, 10µm, [hexane- isopropanol/ACN(0.1%isopropylamine)];B%:30%, isocratic elution) to give 160f-1 as the early eluting isomer and 160f-2 as the late eluting isomer. Step G – Synthesis of 3-[5-[4-[(1R)-1-[6-[4-(2,4-dioxohexahydropyrimidin-1-yl)-3-fluoro- phenyl]-2-azaspiro[3.3]heptan-2-yl]ethyl]-2-fluoro-phenyl]-4,6-dimethyl-pyrimidin-2- yl]isoxazole-5-carboxylic acid (160g-1) To a solution of tert-butyl 3-[5-[4-[(1R)-1-[6-[4-(2,4-dioxohexahydropyrimidin-1-yl)-3- fluoro-phenyl]-2-azaspiro[3.3]heptan-2-yl]ethyl]-2-fluoro-phenyl]-4,6-dimethyl-pyrimidin-2- yl]isoxazole-5-carboxylate (100.0 mg, 143.1 μmol, 1.0 equiv.) in dichloromethane (1.5 mL) was added TFA (0.5 mL). The mixture was stirred at 25°C for 2 h. The mixture was concentrated under reduced pressure to afford 160g-1, which was used without purification. LCMS [M+1] = 643.3.
Step H – Synthesis of 3-[5-[4-[1-[6-[4-(2,4-dioxohexahydropyrimidin-1-yl)-3-fluoro-phenyl]-2- azaspiro[3.3]heptan-2-yl]ethyl]-2-fluoro-phenyl]-4,6-dimethyl-pyrimidin-2-yl]-N-[[3-(2,2,2- trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]isoxazole-5-carboxamide (160-1) To a solution of 3-[5-[4-[1-[6-[4-(2,4-dioxohexahydropyrimidin-1-yl)-3-fluoro-phenyl]-2- azaspiro[3.3]heptan-2-yl]ethyl]-2-fluoro-phenyl]-4,6-dimethyl-pyrimidin-2-yl]isoxazole-5- carboxylic acid trifluoroacetic acid salt (110.0 mg, 145.3 μmol, 1.0 equiv.) and (3-(1,1,1-trifluoro- 2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methanamine methyl sulfonic acid salt (63.9 mg, 159.9 μmol, 1.1 equiv.) in DMF (1.0 mL) was added NMI (35.8 mg, 436.1 μmol, 34.7 μL, 3.0 equiv.) and TCFH (61.1 mg, 218.0 μmol, 1.5 equiv.). The mixture was stirred at 25°C for 1 h. The mixture was purified by prep-HPLC (Welch Xtimate C18150x25mm, 5µm; [water(FA)- ACN];gradient:11%-41% B over 14 min) to give 160-1. LCMS [M+1] = 833.3; 1H NMR (400 MHz, DMSO-d6) δ 14.03 (s, 1H), 10.45 (s, 1H), 9.71 (t, J = 5.6 Hz, 1H), 8.14 (s, 1H), 7.71 (s, 1H), 7.45 - 7.39 (m, 1H), 7.34 (q, J = 8.0 Hz, 3H), 7.15 (dd, J = 1.6, 11.6 Hz, 1H), 7.07 (dd, J = 1.6, 8.4 Hz, 1H), 4.60 (d, J = 5.6 Hz, 2H), 3.67 (t, J = 6.8 Hz, 2H), 3.49 - 3.42 (m, 1H), 3.41 - 3.38 (m, 2H), 3.23 - 3.15 (m, 2H), 3.14 - 3.06 (m, 1H), 2.70 (t, J = 6.8 Hz, 2H), 2.47 (s, 2H), 2.31 (s, 6H), 2.21 (t, J = 10.4 Hz, 2H), 1.54 (s, 6H), 1.22 (d, J = 6.4 Hz, 3H). Intermediate 160f-2 was processed in a similar manner to afford 160-2. Example 161: Synthesis of Intermediate 161
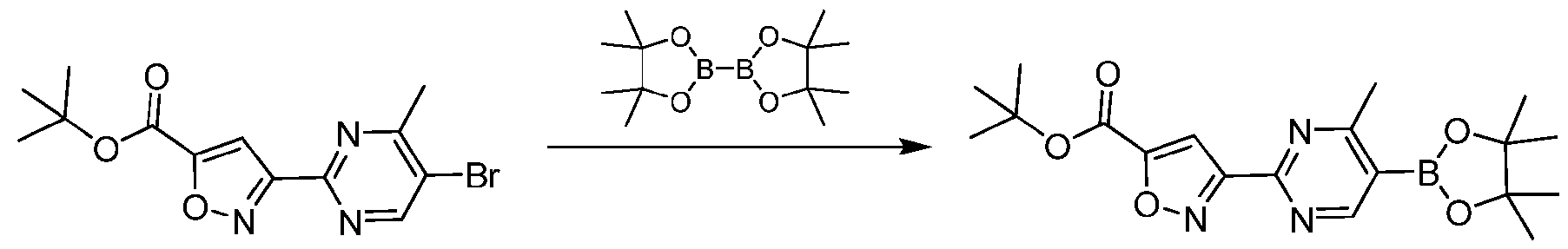 A mixture of tert-butyl 3-(5-bromo-4-methyl-pyrimidin-2-yl)isoxazole-5-carboxylate (10 g, 29.40 mmol, 1 equiv.) and 4,4,5,5-tetramethyl-2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)- 1,3,2-dioxaborolane (14.93 g, 58.79 mmol, 2 equiv.) in dioxane (100 mL) was treated with KOAc (8.66 g, 88.19 mmol, 3 eq) and Pd(dppf)Cl2 (2.15 g, 2.94 mmol, 0.1 equiv.) at 25 °C. The mixture was degassed and purged threefold with nitrogen. The mixture was stirred at 120 °C for 1 h under a nitrogen atmosphere. The mixture was diluted with water (200 mL) and extracted with ethyl acetate (100 mL x 3). The combined organic layers were washed with brine (100 mL x
2), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether: ethyl acetate= 10/0 to 9/1) to give 161. LCMS [M+1] = 388.0; 1H NMR (400 MHz, DMSO-d6) δ 8.93 (s, 1H), 7.55 (s, 1H), 2.71 (s, 3H), 1.58 (s, 9H), 1.35 (s, 12H). Example 162: Synthesis of Compound 162-1 and 162-1
A mixture of tert-butyl 3-(5-bromo-4-methyl-pyrimidin-2-yl)isoxazole-5-carboxylate (10 g, 29.40 mmol, 1 equiv.) and 4,4,5,5-tetramethyl-2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)- 1,3,2-dioxaborolane (14.93 g, 58.79 mmol, 2 equiv.) in dioxane (100 mL) was treated with KOAc (8.66 g, 88.19 mmol, 3 eq) and Pd(dppf)Cl2 (2.15 g, 2.94 mmol, 0.1 equiv.) at 25 °C. The mixture was degassed and purged threefold with nitrogen. The mixture was stirred at 120 °C for 1 h under a nitrogen atmosphere. The mixture was diluted with water (200 mL) and extracted with ethyl acetate (100 mL x 3). The combined organic layers were washed with brine (100 mL x
2), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether: ethyl acetate= 10/0 to 9/1) to give 161. LCMS [M+1] = 388.0; 1H NMR (400 MHz, DMSO-d6) δ 8.93 (s, 1H), 7.55 (s, 1H), 2.71 (s, 3H), 1.58 (s, 9H), 1.35 (s, 12H). Example 162: Synthesis of Compound 162-1 and 162-1
 To a solution of 6-chloro-5-fluoro-pyridine-3-carbaldehyde (5 g, 31.34 mmol, 1 equiv.) in THF (50 mL) was added dropwise MeMgBr (3 M, 10.45 mL, 1 equiv.) at 0°C. The mixture was stirred at 0°C for 2 h. The mixture was treated with NH4Cl (100 ml) and extracted with ethyl acetate (50mL x 3). The combined organic layers washed with brine (20 mL x 3), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, Petroleum ether/Ethyl acetate=100/1 to 1/1) to give 162a. LCMS [M+1] = 174.1, 176.0. Step B – Synthesis of 1-(6-chloro-5-fluoro-3-pyridyl)ethenone (162b)
To a solution of 1-(6-chloro-5-fluoro-3-pyridyl) ethanol (4.5 g, 25.63 mmol, 1.0 equiv.) in dichloromethane (45.0 mL) was added Dess-Martin (10.87 g, 25.63 mmol, 7.94 mL, 1.0 equiv.). The mixture was stirred at 25°C for 1 h. The mixture was quenched with aqueous Na2SO3 (100 mL), diluted with water (300 mL) and extracted with ethyl acetate (100 mL x 3). The combined organic layers were washed with brine (300 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was triturated with petroleum ether: ethyl acetate =3:1 at 25oC for 5 min to give 162b. LCMS [M+1] = 174.1, 176.0; Step C – Synthesis of tert-butyl 3-[5-(5-acetyl-3-fluoro-2-pyridyl)-4-methyl-pyrimidin-2- yl]isoxazole-5-carboxylate (162c) To a solution of 1-(6-chloro-5-fluoro-3-pyridyl)ethanone (1.5 g, 8.64 mmol, 1 equiv.) and tert-butyl 3-[4-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrimidin-2-yl] isoxazole- 5-carboxylate (3.35 g, 8.64 mmol, 1 equiv.) in dioxane (20.0 mL) and water (4.0 mL) was added K2CO3 (2.39 g, 17.28 mmol, 2.0 equiv.) and Pd(dppf)Cl2.CH2Cl2 (705.7 mg, 864.2 μmol, 0.1 equiv.). The mixture was stirred at 90°C for 4 h. The mixture was poured into water (100 mL) and extracted with ethyl acetate (100 mL x 3). The organic phase was washed with brine (100 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, Petroleum ether/Ethyl acetate=10/1 to 5/1) to give 162c. LCMS [M+1] = 399.2. Step D – Synthesis of tert-butyl 3-[5-[5-[1-[6-[4-(2,4-dioxohexahydropyrimidin-1-yl)-3-fluoro- phenyl]-2-azaspiro[3.3]heptan-2-yl]ethyl]-3-fluoro-2-pyridyl]-4-methyl-pyrimidin-2- yl]isoxazole-5-carboxylate (162d-1 and 162d-2) A solution of 1-[4-(2-azaspiro[3.3]heptan-6-yl)-2-fluoro-phenyl]hexahydropyrimidine - 2,4-dione trifluoroacetate salt (1.36 g, 3.26 mmol, 1.3 equiv.) and triethylamine (1.05 mL, 3.0 equiv.) in dichloromethane (15.0 mL) treated with tert-butyl 3-[5-(5-acetyl-3-fluoro-2-pyridyl)-4- methyl-pyrimidin-2-yl]isoxazole-5-carboxylate (1.0 g, 2.51 mmol, 1.0 equiv.). The mixture was stirred for 2 h, treated with NaBH(OAc)3 (1.33 g, 6.28 mmol, 2.5 equiv.) and stirred at 25°C for 12 h. The mixture was treated with water (50 mL) at 25°C and extracted with ethyl acetate (50 mL x 3). The combined organic portions were washed with brine (50 mL x 3), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The
residue was purified by prep-TLC (silica gel, Petroleum ether: Ethyl acetate = 0:1) to afford 162d as the racemate. The racemate was separated by SFC (ChiralPak IH, 250x50mm, 10µm; [CO2- isopropanol];B%:55%, isocratic elution) to give 162d-1 as the early eluting isomer and 162d-2 as the late eluting isomer. LCMS [M+1] = 686.4. Step E – Synthesis of 3-[5-[5-[-1-[6-[4-(2,4-dioxohexahydropyrimidin-1-yl)-3-fluoro-phenyl]-2- azaspiro[3.3]heptan-2-yl]ethyl]-3-fluoro-2-pyridyl]-4-methyl-pyrimidin-2-yl]isoxazole-5- carboxylic acid (162e-1) To a solution of tert-butyl 3-[5-[5-[1-[6-[4-(2,4-dioxohexahydropyrimidin-1-yl)-3- fluoro- phenyl]-2-azaspiro[3.3]heptan-2-yl]ethyl]-3-fluoro-2-pyridyl]-4-methyl-pyrimidin-2-yl]isoxazole- 5-carboxylate (400.0 mg, 583.3 μmol, 1.0 equiv.) in dichloromethane (4.0 mL) was added TFA (997.7 mg, 8.75 mmol, 649.9 μL, 15.0 equiv.). The mixture was stirred at 25°C for 12 h. The mixture was concentrated under reduced pressure to give 162e-1. LCMS [M+1] = 630.3. Step F – Synthesis of 3-[5-[5-[1-[6-[4- (2,4-dioxohexahydropyrimidin-1-yl)-3-fluoro-phenyl]-2- azaspiro[3.3]heptan-2-yl]ethyl]-3-fluoro-2-pyridyl]-4-methyl-pyrimidin-2-yl]-N-[[3-(2,2,2- trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]isoxazole-5-carboxamide (162-1) To a solution of 3-[5-[5-[1-[6-[4-(2,4-dioxohexahydropyrimidin-1-yl)-3-fluoro- phenyl]-2- azaspiro[3.3]heptan-2-yl]ethyl]-3-fluoro-2-pyridyl]-4-methyl-pyrimidin-2-yl]isoxazole-5- carboxylic acid (200.0 mg) in DMF (2.0 mL) was added [3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H-1,2,4-triazol-5-yl]methanamine (130 mg), DIEA (234) and BOP (237.9 mg). The mixture was stirred at 25°C for 2 h. The mixture was poured into water (10 mL) and extracted with ethyl acetate (10 mL x 3). The combined organic portion was washed with brine (20 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by prep-HPLC (Phenomenex Luna C18100x30mm, 5µm; [water(0.2% FA)- ACN];gradient:10%-40% B over 8.0 min) to give 162-1. LCMS [M+1] = 820.5; 1H NMR (400 MHz, METHANOL-d4) δ 8.92 (s, 1H), 8.65 (s, 1H), 7.91 - 7.85 (m, 1H), 7.69 (s, 1H), 7.33 (t, J = 8.0 Hz, 1H), 7.15 - 7.05 (m, 2H), 4.75 (s, 2H), 4.08 - 3.92 (m, 1H), 3.88 - 3.68 (m, 4H), 3.65 - 3.57 (m, 1H), 3.59 - 3.44 (m, 2H), 2.86 - 2.77 (m, 2H), 2.72 - 2.58 (m, 5H), 2.35 (br t, J = 10.0 Hz, 2H), 1.62 (s, 6H), 1.48 (d, J = 6.8 Hz, 3H). Intermediate 162d-2 was processed in a similar manner to afford 160-2.
Example 163: Synthesis of Compound 163 Step A
To a solution of 6-chloro-5-fluoro-pyridine-3-carbaldehyde (5 g, 31.34 mmol, 1 equiv.) in THF (50 mL) was added dropwise MeMgBr (3 M, 10.45 mL, 1 equiv.) at 0°C. The mixture was stirred at 0°C for 2 h. The mixture was treated with NH4Cl (100 ml) and extracted with ethyl acetate (50mL x 3). The combined organic layers washed with brine (20 mL x 3), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, Petroleum ether/Ethyl acetate=100/1 to 1/1) to give 162a. LCMS [M+1] = 174.1, 176.0. Step B – Synthesis of 1-(6-chloro-5-fluoro-3-pyridyl)ethenone (162b)
To a solution of 1-(6-chloro-5-fluoro-3-pyridyl) ethanol (4.5 g, 25.63 mmol, 1.0 equiv.) in dichloromethane (45.0 mL) was added Dess-Martin (10.87 g, 25.63 mmol, 7.94 mL, 1.0 equiv.). The mixture was stirred at 25°C for 1 h. The mixture was quenched with aqueous Na2SO3 (100 mL), diluted with water (300 mL) and extracted with ethyl acetate (100 mL x 3). The combined organic layers were washed with brine (300 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was triturated with petroleum ether: ethyl acetate =3:1 at 25oC for 5 min to give 162b. LCMS [M+1] = 174.1, 176.0; Step C – Synthesis of tert-butyl 3-[5-(5-acetyl-3-fluoro-2-pyridyl)-4-methyl-pyrimidin-2- yl]isoxazole-5-carboxylate (162c) To a solution of 1-(6-chloro-5-fluoro-3-pyridyl)ethanone (1.5 g, 8.64 mmol, 1 equiv.) and tert-butyl 3-[4-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrimidin-2-yl] isoxazole- 5-carboxylate (3.35 g, 8.64 mmol, 1 equiv.) in dioxane (20.0 mL) and water (4.0 mL) was added K2CO3 (2.39 g, 17.28 mmol, 2.0 equiv.) and Pd(dppf)Cl2.CH2Cl2 (705.7 mg, 864.2 μmol, 0.1 equiv.). The mixture was stirred at 90°C for 4 h. The mixture was poured into water (100 mL) and extracted with ethyl acetate (100 mL x 3). The organic phase was washed with brine (100 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, Petroleum ether/Ethyl acetate=10/1 to 5/1) to give 162c. LCMS [M+1] = 399.2. Step D – Synthesis of tert-butyl 3-[5-[5-[1-[6-[4-(2,4-dioxohexahydropyrimidin-1-yl)-3-fluoro- phenyl]-2-azaspiro[3.3]heptan-2-yl]ethyl]-3-fluoro-2-pyridyl]-4-methyl-pyrimidin-2- yl]isoxazole-5-carboxylate (162d-1 and 162d-2) A solution of 1-[4-(2-azaspiro[3.3]heptan-6-yl)-2-fluoro-phenyl]hexahydropyrimidine - 2,4-dione trifluoroacetate salt (1.36 g, 3.26 mmol, 1.3 equiv.) and triethylamine (1.05 mL, 3.0 equiv.) in dichloromethane (15.0 mL) treated with tert-butyl 3-[5-(5-acetyl-3-fluoro-2-pyridyl)-4- methyl-pyrimidin-2-yl]isoxazole-5-carboxylate (1.0 g, 2.51 mmol, 1.0 equiv.). The mixture was stirred for 2 h, treated with NaBH(OAc)3 (1.33 g, 6.28 mmol, 2.5 equiv.) and stirred at 25°C for 12 h. The mixture was treated with water (50 mL) at 25°C and extracted with ethyl acetate (50 mL x 3). The combined organic portions were washed with brine (50 mL x 3), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The
residue was purified by prep-TLC (silica gel, Petroleum ether: Ethyl acetate = 0:1) to afford 162d as the racemate. The racemate was separated by SFC (ChiralPak IH, 250x50mm, 10µm; [CO2- isopropanol];B%:55%, isocratic elution) to give 162d-1 as the early eluting isomer and 162d-2 as the late eluting isomer. LCMS [M+1] = 686.4. Step E – Synthesis of 3-[5-[5-[-1-[6-[4-(2,4-dioxohexahydropyrimidin-1-yl)-3-fluoro-phenyl]-2- azaspiro[3.3]heptan-2-yl]ethyl]-3-fluoro-2-pyridyl]-4-methyl-pyrimidin-2-yl]isoxazole-5- carboxylic acid (162e-1) To a solution of tert-butyl 3-[5-[5-[1-[6-[4-(2,4-dioxohexahydropyrimidin-1-yl)-3- fluoro- phenyl]-2-azaspiro[3.3]heptan-2-yl]ethyl]-3-fluoro-2-pyridyl]-4-methyl-pyrimidin-2-yl]isoxazole- 5-carboxylate (400.0 mg, 583.3 μmol, 1.0 equiv.) in dichloromethane (4.0 mL) was added TFA (997.7 mg, 8.75 mmol, 649.9 μL, 15.0 equiv.). The mixture was stirred at 25°C for 12 h. The mixture was concentrated under reduced pressure to give 162e-1. LCMS [M+1] = 630.3. Step F – Synthesis of 3-[5-[5-[1-[6-[4- (2,4-dioxohexahydropyrimidin-1-yl)-3-fluoro-phenyl]-2- azaspiro[3.3]heptan-2-yl]ethyl]-3-fluoro-2-pyridyl]-4-methyl-pyrimidin-2-yl]-N-[[3-(2,2,2- trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]isoxazole-5-carboxamide (162-1) To a solution of 3-[5-[5-[1-[6-[4-(2,4-dioxohexahydropyrimidin-1-yl)-3-fluoro- phenyl]-2- azaspiro[3.3]heptan-2-yl]ethyl]-3-fluoro-2-pyridyl]-4-methyl-pyrimidin-2-yl]isoxazole-5- carboxylic acid (200.0 mg) in DMF (2.0 mL) was added [3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H-1,2,4-triazol-5-yl]methanamine (130 mg), DIEA (234) and BOP (237.9 mg). The mixture was stirred at 25°C for 2 h. The mixture was poured into water (10 mL) and extracted with ethyl acetate (10 mL x 3). The combined organic portion was washed with brine (20 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by prep-HPLC (Phenomenex Luna C18100x30mm, 5µm; [water(0.2% FA)- ACN];gradient:10%-40% B over 8.0 min) to give 162-1. LCMS [M+1] = 820.5; 1H NMR (400 MHz, METHANOL-d4) δ 8.92 (s, 1H), 8.65 (s, 1H), 7.91 - 7.85 (m, 1H), 7.69 (s, 1H), 7.33 (t, J = 8.0 Hz, 1H), 7.15 - 7.05 (m, 2H), 4.75 (s, 2H), 4.08 - 3.92 (m, 1H), 3.88 - 3.68 (m, 4H), 3.65 - 3.57 (m, 1H), 3.59 - 3.44 (m, 2H), 2.86 - 2.77 (m, 2H), 2.72 - 2.58 (m, 5H), 2.35 (br t, J = 10.0 Hz, 2H), 1.62 (s, 6H), 1.48 (d, J = 6.8 Hz, 3H). Intermediate 162d-2 was processed in a similar manner to afford 160-2.
Example 163: Synthesis of Compound 163 Step A
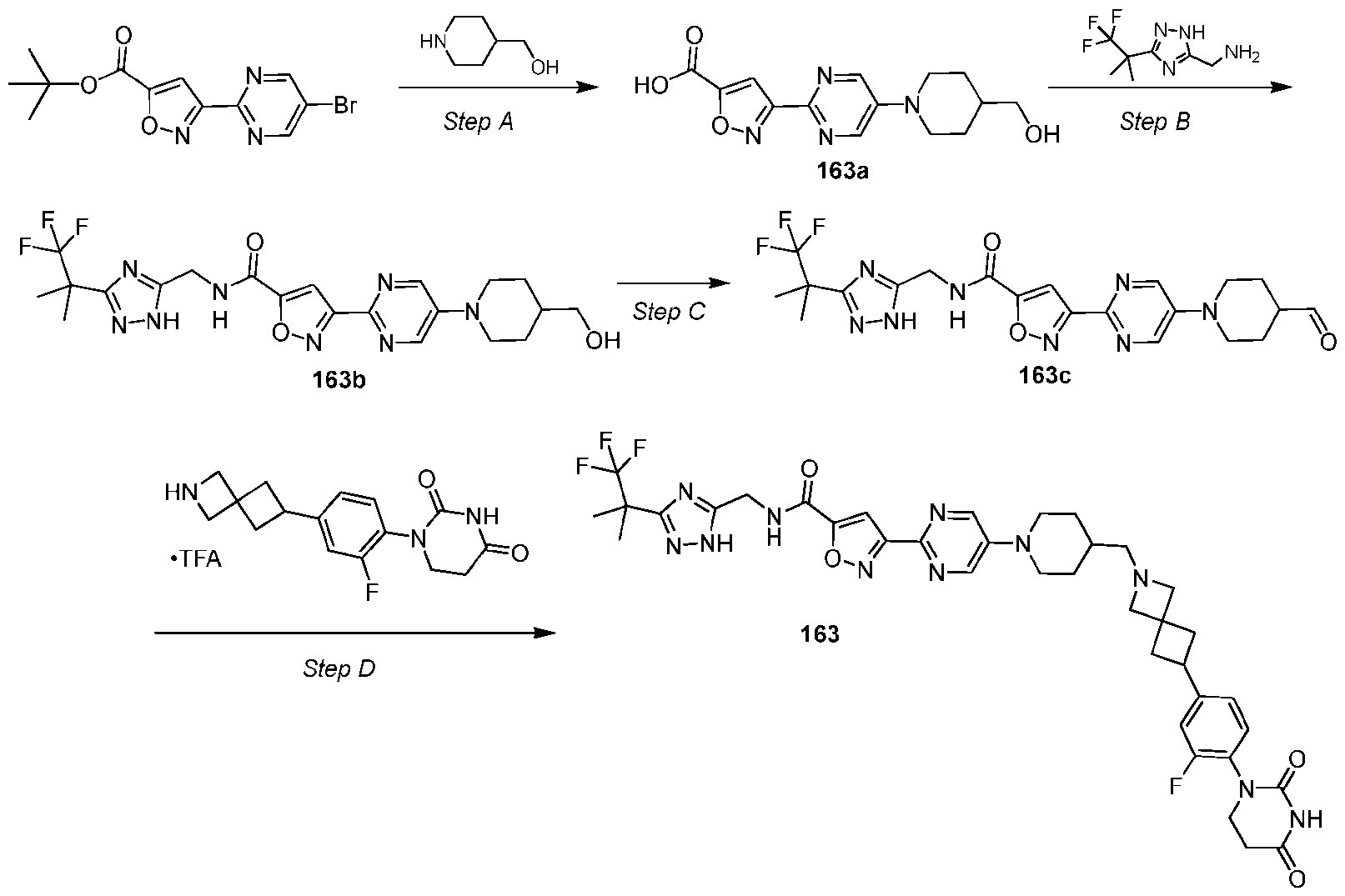 carboxylic acid (163a) A mixture of tert-butyl 3-(5-bromopyrimidin-2-yl)isoxazole-5-carboxylate (2.8 g, 24.5 mmol, 2.0 equiv.), piperidin-4-ylmethanol (1.0 g, 1.2 mmol, 0.1 equiv.), cesium carbonate (11.9 g, 36.7 mmol, 3.0 equiv.) in DMF (40.0 mL) was degassed and purged threefold with nitrogen. The mixture was stirred at 80°C for 1 h under a nitrogen atmosphere. The mixture was concentrated under reduced pressure. The residue was purified by prep-HPLC (Phenomenex Luna C18 (250x70mm, 10 µm); [water(FA)-ACN];gradient:6%-36% B over 20 min) to give 163a. LCMS [M+1] = 305.1. Step B – Synthesis of 3-(5-(4-(hydroxymethyl)piperidin-1-yl)pyrimidin-2-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (163b) To a solution of 3-(5-(4-(hydroxymethyl)piperidin-1-yl)pyrimidin-2-yl)isoxazole-5- carboxylic acid (14.0 g, 46.0 mmol, 1.0 equiv.) and (3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H- 1,2,4-triazol-5-yl)methanamine (14.2 g, 69.0 mmol, 1.5 equiv.) in DMF (60.0 mL) was added TCFH (28.6 g, 138.0 mmol, 3.0 equiv.) and NMI (11.2 g, 138.0 mmol, 11 mL, 3.0 equiv.). The
mixture was stirred at 25°C for 1 h. The mixture was filtered. The filtrate was purified by prep- HPLC (Phenomenex Luna C18 (250x70mm,10 µm); [water(FA)-ACN];gradient:16%-46% B over 20 min) to give 163b. LCMS [M+1] = 495.2. Step C – Synthesis of 3-(5-(4-formylpiperidin-1-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (163c) To a solution of 3-(5-(4-(hydroxymethyl)piperidin-1-yl)pyrimidin-2-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (8.0 g, 16.0 mmol, 1.0 equiv.) in DMF (20.0 mL) was added [acetoxy(phenyl)-iodanyl] acetate (10.44 g, 32.36 mmol, 2.0 equiv.) and 1-hydroxy-2,2,6,6-tetramethyl-piperidine (2.54 g, 16.0 mmol, 1.0 equiv.). The mixture was stirred at 20°C for 2 h. The mixture was concentrated under reduced pressure. The residue was purified by prep-HPLC (Phenomenex Luna C18, 250x70mm, 10 µm; [water(FA)-ACN];gradient:12%-42% B over 20 min) to give 163c. LCMS [M+1] = 493.1. 1H NMR (400 MHz, DMSO-d6) δ 13.90 (bs, 1H), 9.83 - 9.44 (m, 2H), 8.70 - 8.47 (m, 2H), 7.60 - 7.39 (m, 1H), 4.73 - 4.50 (m, 2H), 4.11 - 3.72 (m, 2H), 3.14 - 3.03 (m, 2H), 2.68 - 2.57 (m, 1H), 2.01 - 1.81 (m, 2H), 1.67 - 1.56 (m, 2H), 1.53 (s, 6H). Step D – Synthesis of 3-(5-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)piperidin-1-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (163) To a solution of 1-(2-fluoro-4-(2-azaspiro[3.3]heptan-6-yl)phenyl)dihydropyrimidine- 2,4(1H,3H)-dione trifluoroacetate salt (323.7 mg, 609.1 μmol, 2.0 equiv), 3-(5-(4- formylpiperidin-1-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)isoxazole-5-carboxamide (150.0 mg, 304.6 μmol, 1.0 equiv) in DMSO (1.5 mL) was added triethylamine (127 μL, 3.0 equiv) and NaBH(OAc)3 (193.6 mg, 913.7 μmol, 3.0 equiv). The mixture was stirred at 25°C for 1 h. The mixture was diluted with water (20 mL) and extracted with ethyl acetate (10 mLx3). The combined organic portion was washed with brine (10 mL x 2), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by prep-HPLC (Waters Xbridge BEH C18 150x25mm, 10 µm; [water( NH4HCO3)-ACN];gradient:19%-49% B over 10 min ) to give 163.
LCMS [M+1] = 780.5; 1H NMR (400 MHz, DMSO-d6) δ 13.96 (s, 1H), 10.45 (s, 1H), 9.63 (s, 1H), 8.61 (s, 2H), 7.52 (s, 1H), 7.33 (t, J = 8.0 Hz, 1H), 7.20 - 7.03 (m, 2H), 4.63 - 4.52 (m, 2H), 3.97 (d, J = 12.4 Hz, 2H), 3.68 (t, J = 6.8 Hz, 2H), 3.43 - 3.34 (m, 1H), 3.29 - 3.21 (m, 2H), 3.10 - 3.02 (m, 2H), 2.91 - 2.80 (m, 2H), 2.70 (t, J = 6.8 Hz, 2H), 2.47 - 2.43 (m, 2H), 2.30 - 2.14 (m, 4H), 1.81 - 1.71 (m, 2H), 1.56 - 1.49 (m, 7H), 1.26 - 1.13 (m, 2H). Example 164: Synthesis of Compound 164 Step
carboxylic acid (163a) A mixture of tert-butyl 3-(5-bromopyrimidin-2-yl)isoxazole-5-carboxylate (2.8 g, 24.5 mmol, 2.0 equiv.), piperidin-4-ylmethanol (1.0 g, 1.2 mmol, 0.1 equiv.), cesium carbonate (11.9 g, 36.7 mmol, 3.0 equiv.) in DMF (40.0 mL) was degassed and purged threefold with nitrogen. The mixture was stirred at 80°C for 1 h under a nitrogen atmosphere. The mixture was concentrated under reduced pressure. The residue was purified by prep-HPLC (Phenomenex Luna C18 (250x70mm, 10 µm); [water(FA)-ACN];gradient:6%-36% B over 20 min) to give 163a. LCMS [M+1] = 305.1. Step B – Synthesis of 3-(5-(4-(hydroxymethyl)piperidin-1-yl)pyrimidin-2-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (163b) To a solution of 3-(5-(4-(hydroxymethyl)piperidin-1-yl)pyrimidin-2-yl)isoxazole-5- carboxylic acid (14.0 g, 46.0 mmol, 1.0 equiv.) and (3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H- 1,2,4-triazol-5-yl)methanamine (14.2 g, 69.0 mmol, 1.5 equiv.) in DMF (60.0 mL) was added TCFH (28.6 g, 138.0 mmol, 3.0 equiv.) and NMI (11.2 g, 138.0 mmol, 11 mL, 3.0 equiv.). The
mixture was stirred at 25°C for 1 h. The mixture was filtered. The filtrate was purified by prep- HPLC (Phenomenex Luna C18 (250x70mm,10 µm); [water(FA)-ACN];gradient:16%-46% B over 20 min) to give 163b. LCMS [M+1] = 495.2. Step C – Synthesis of 3-(5-(4-formylpiperidin-1-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (163c) To a solution of 3-(5-(4-(hydroxymethyl)piperidin-1-yl)pyrimidin-2-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (8.0 g, 16.0 mmol, 1.0 equiv.) in DMF (20.0 mL) was added [acetoxy(phenyl)-iodanyl] acetate (10.44 g, 32.36 mmol, 2.0 equiv.) and 1-hydroxy-2,2,6,6-tetramethyl-piperidine (2.54 g, 16.0 mmol, 1.0 equiv.). The mixture was stirred at 20°C for 2 h. The mixture was concentrated under reduced pressure. The residue was purified by prep-HPLC (Phenomenex Luna C18, 250x70mm, 10 µm; [water(FA)-ACN];gradient:12%-42% B over 20 min) to give 163c. LCMS [M+1] = 493.1. 1H NMR (400 MHz, DMSO-d6) δ 13.90 (bs, 1H), 9.83 - 9.44 (m, 2H), 8.70 - 8.47 (m, 2H), 7.60 - 7.39 (m, 1H), 4.73 - 4.50 (m, 2H), 4.11 - 3.72 (m, 2H), 3.14 - 3.03 (m, 2H), 2.68 - 2.57 (m, 1H), 2.01 - 1.81 (m, 2H), 1.67 - 1.56 (m, 2H), 1.53 (s, 6H). Step D – Synthesis of 3-(5-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)piperidin-1-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (163) To a solution of 1-(2-fluoro-4-(2-azaspiro[3.3]heptan-6-yl)phenyl)dihydropyrimidine- 2,4(1H,3H)-dione trifluoroacetate salt (323.7 mg, 609.1 μmol, 2.0 equiv), 3-(5-(4- formylpiperidin-1-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)isoxazole-5-carboxamide (150.0 mg, 304.6 μmol, 1.0 equiv) in DMSO (1.5 mL) was added triethylamine (127 μL, 3.0 equiv) and NaBH(OAc)3 (193.6 mg, 913.7 μmol, 3.0 equiv). The mixture was stirred at 25°C for 1 h. The mixture was diluted with water (20 mL) and extracted with ethyl acetate (10 mLx3). The combined organic portion was washed with brine (10 mL x 2), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by prep-HPLC (Waters Xbridge BEH C18 150x25mm, 10 µm; [water( NH4HCO3)-ACN];gradient:19%-49% B over 10 min ) to give 163.
LCMS [M+1] = 780.5; 1H NMR (400 MHz, DMSO-d6) δ 13.96 (s, 1H), 10.45 (s, 1H), 9.63 (s, 1H), 8.61 (s, 2H), 7.52 (s, 1H), 7.33 (t, J = 8.0 Hz, 1H), 7.20 - 7.03 (m, 2H), 4.63 - 4.52 (m, 2H), 3.97 (d, J = 12.4 Hz, 2H), 3.68 (t, J = 6.8 Hz, 2H), 3.43 - 3.34 (m, 1H), 3.29 - 3.21 (m, 2H), 3.10 - 3.02 (m, 2H), 2.91 - 2.80 (m, 2H), 2.70 (t, J = 6.8 Hz, 2H), 2.47 - 2.43 (m, 2H), 2.30 - 2.14 (m, 4H), 1.81 - 1.71 (m, 2H), 1.56 - 1.49 (m, 7H), 1.26 - 1.13 (m, 2H). Example 164: Synthesis of Compound 164 Step
 A mixture of 1-bromo-2,5-difluoro-4-methylbenzene (40 g, 193.2 mmol, 1.0 equiv.), NBS (41.3 g, 231.9 mmol, 1.2 equiv.), AIBN (6.3 g, 38.6 mmol, 0.2 equiv.) in DCE (400 mL) was degassed and purged threefold with nitrogen. The mixture was stirred at 90°C for 16 h under a nitrogen atmosphere. The residue was poured into water (200 mL). The aqueous phase was extracted with dichloromethane (100 mLx2). The combined organic phase was washed with brine (5 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, Petroleum ether/Ethyl acetate=1/0 to 10/1) to give 164a. 1H NMR (400 MHz, CHLOROFORM- d) δ = 7.32 (dd, J = 5.6, 8.4 Hz, 1H), 7.19 (dd, J = 5.6, 8.0 Hz, 1H), 4.43 (s, 2H). Step B – Synthesis of 1-(4-bromo-2,5-difluorobenzyl) pyrimidine-2,4(1H,3H)-dione (164b) A mixture of 1-bromo-4-(bromomethyl)-2,5-difluoro-benzene (40 g, 139.9 mmol, 1.0 equiv.), pyrimidine-2,4(1H,3H)-dione (47.0 g, 419.7 mmol, 3.0 equiv.), K2CO3 (58.0 g, 419.7 mmol, 3.0 equiv.) in DMSO (800 mL) was degassed and purged threefold with nitrogen. The mixture was stirred at 80°C for 1 h under a nitrogen atmosphere. The mixture was treated with water (1.5 L) and stirred for 10 min, then aged for 8 h at ambient temperature. The solids were collected by vacuum filtration. The filtrate liquors were extracted with 2-methyltetrahydrofuran (500 mLx2). The combined organic phase was washed with brine (50 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was triturated with ethyl acetate (200 mL) at 20oC for 10 min to give 164b. LCMS [M+1] = 317.0, 319.0; 1H NMR (400 MHz, DMSO-d6) δ 11.3 (br s, 1H), 7.76 (dd, J = 5.6, 9.2 Hz, 1H), 7.67 (d, J = 8.0 Hz, 1H), 7.34 (dd, J = 6.8, 8.8 Hz, 1H), 5.59 (d, J = 7.6 Hz, 1H), 4.87 (s, 2H). Step C – Synthesis of tert-butyl 4-(4-((2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)methyl)-2,5- difluorophenyl)-3,6-dihydropyridine-1(2H)-carboxylate (164c) A mixture of 1-[(4-bromo-2,5-difluoro-phenyl)methyl]pyrimidine-2,4-dione (10.0 g, 31.5 mmol, 1.0 equiv.), tert-butyl 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-3,6- dihydropyridine-1(2H)-carboxylate (14.6 g, 47.3 mmol, 1.5 equiv.), Pd(dppf)Cl2 (2.3 g, 3.1 mmol, 0.1 equiv.), cesium carbonate (20.5 g, 63.1 mmol, 2.0 equiv.) in dioxane (80 mL) and water (20 mL) was degassed and purged threefold with nitrogen. The mixture was stirred at 100°C for 12 h under a nitrogen atmosphere. The residue was poured into water (200 mL). The
aqueous portion was extracted with ethyl acetate (100 mLx2). The combined organic portion was washed with brine (20 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, Petroleum ether/Ethyl acetate=10/1 to 0/1) to give 164c. LCMS [M-56+CH3CN+H] = 405.1. Step D – Synthesis of tert-butyl 4-(4-((2,4-dioxotetrahydropyrimidin-1(2H)-yl)methyl)-2,5- difluorophenyl)piperidine-1-carboxylate (164d) Pd(OH)2 (4.3 g, 31.0 mmol, 1.0 equiv.) was added to a flask purged with argon, then a solution of tert-butyl 4-(4-((2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)methyl)-2,5- difluorophenyl)-3,6-dihydropyridine-1(2H)-carboxylate (13.0 g, 31.0 mmol, 1.0 equiv.) in MeOH (150 mL) was added under argon. The mixture was degassed and purged threefold with hydrogen gas. The mixture was stirred at 50°C for 16 h under hydrogen gas (15 Psi). The mixture was filtered over a celite pad and the filtrate was concentrated. The residue was purified by column chromatography (silica gel, Petroleum ether/Ethyl acetate=5/1 to 1/1) to give 164d. LCMS [M- 56+1] = 368.1. Step E – Synthesis of 1-(2,5-difluoro-4-(piperidin-4-yl)benzyl)dihydropyrimidine-2,4(1H,3H)- dione hydrochloride (164e) A mixture of tert-butyl 4-(4-((2,4-dioxotetrahydropyrimidin-1(2H)-yl)methyl)-2,5- difluorophenyl)piperidine-1-carboxylate (11.0 g, 26.0 mmol, 1.0 equiv.) in HCl/ethyl acetate (100 mL) was degassed and purged threefold with nitrogen. The mixture was stirred at 20°C for 1 h under a nitrogen atmosphere and concentrated under reduced pressure. The residue was triturated with ethyl acetate (20 mL) at 20oC for 30 min to give 164e. LCMS [M+1] = 324.2. Step F – Synthesis of tert-butyl 3-(5-(4-((4-(4-((2,4-dioxotetrahydropyrimidin-1(2H)-yl)methyl)- 2,5-difluorophenyl)piperidin-1-yl)methyl)phenyl)pyrimidin-2-yl)isoxazole-5-carboxylate (164f) To a solution of tert-butyl 3-[5-(4-formylphenyl)pyrimidin-2-yl]isoxazole-5-carboxylate (0.5 g, 1.4 mmol, 1.0 equiv.) and 1-[[2,5-difluoro-4-(4- piperidyl)phenyl]methyl]hexahydropyrimidine-2,4-dione (614.4 mg, 1.7 mmol, 1.2 eq, HCl) in dichloromethane (5 mL) was added triethylamine (2.0 mL, 10.0 equiv.) at 20°C and stirred for 2 h, then NaBH(OAc)3 (603.2 mg, 2.9 mmol, 2.0 equiv.) was added to the mixture at 20°C and
stirred for 16 h. Water (20 mL) and ethyl acetate (20 mL) was added to the mixture and stirred for 10 min, then filtered under reduced pressure. The filter cake was triturated with acetonitrile (20 mL) at 60oC for 30 min to give 164f. LCMS [M+1] = 659.4. Step G – Synthesis of 3-(5-(4-((4-(4-((2,4-dioxotetrahydropyrimidin-1(2H)-yl)methyl)-2,5- difluorophenyl)piperidin-1-yl)methyl)phenyl)pyrimidin-2-yl)isoxazole-5-carboxylic acid hydrochloride salt (164g) A mixture of tert-butyl 3-(5-(4-((4-(4-((2,4-dioxotetrahydropyrimidin-1(2H)-yl)methyl)- 2,5-difluorophenyl)piperidin-1-yl)methyl)phenyl)pyrimidin-2-yl)isoxazole-5-carboxylate (700 mg, 1.1 mmol, 1.0 equiv.) in HCl/dioxane (10 mL) was degassed and purged threefold with nitrogen and then the mixture was heated at 60°C for 1 h under a nitrogen atmosphere. The mixture was concentrated under reduced pressure. The residue was triturated with acetonitrile (50 mL) at 60oC for 30 min to give 164g. LCMS [M+1] = 603.2. Step H – Synthesis of 3-(5-(4-((4-(4-((2,4-dioxotetrahydropyrimidin-1(2H)-yl)methyl)-2,5- difluorophenyl)piperidin-1-yl)methyl)phenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (164) To a suspension of 3-(5-(4-((4-(4-((2,4-dioxotetrahydropyrimidin-1(2H)-yl)methyl)-2,5- difluorophenyl)piperidin-1-yl)methyl)phenyl)pyrimidin-2-yl)isoxazole-5-carboxylic acid hydrochloride salt (660 mg, 1.10 mmol, 1 equiv.) in THF (7 mL) was added BOP (726.63 mg, 1.64 mmol, 1.5 equiv.) DIEA (707.78 mg, 5.48 mmol, 953.88 μL, 5 equiv.) in one portion at 20°C and stirred for 15 min, then [3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl] methanamine hydrochloride(401.93 mg, 1.43 mmol, 1.31 equiv) was added to the mixture at 60°C and stirred for 30 min. Water (50 mL) was added to the mixture and stirred for 10 min, then filtered under reduced pressure. The filter cake was triturated with acetonitrile (50 mL x 4) at 60oC for 1 h to give 164. LCMS [M+1] = 793.3; 1H NMR (400 MHz, DMSO-d6) δ = 14.00 (br s, 1H), 10.23 (s, 1H), 9.72 (br s, 1H), 9.36 (s, 2H), 7.89 (d, J = 8.0 Hz, 2H), 7.72 (s, 1H), 7.53 (d, J = 8.0 Hz, 2H), 7.23 (dd, J = 6.4, 10.8 Hz, 1H), 7.12 (dd, J = 6.4, 10.8 Hz, 1H), 4.60 (br s, 2H), 4.50 (s, 2H), 3.59 (s, 2H), 3.37 (t, J = 6.8 Hz, 2H), 2.95 (d, J = 10.8 Hz, 2H), 2.84 - 2.71 (m, 1H), 2.57 (t, J = 6.8 Hz, 3H), 2.17 - 2.05 (m, 2H), 1.08 - 1.65 (m, 4H), 1.54 (s, 6H).
Example 165: Synthesis of Compound 165 Step
A mixture of 1-bromo-2,5-difluoro-4-methylbenzene (40 g, 193.2 mmol, 1.0 equiv.), NBS (41.3 g, 231.9 mmol, 1.2 equiv.), AIBN (6.3 g, 38.6 mmol, 0.2 equiv.) in DCE (400 mL) was degassed and purged threefold with nitrogen. The mixture was stirred at 90°C for 16 h under a nitrogen atmosphere. The residue was poured into water (200 mL). The aqueous phase was extracted with dichloromethane (100 mLx2). The combined organic phase was washed with brine (5 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, Petroleum ether/Ethyl acetate=1/0 to 10/1) to give 164a. 1H NMR (400 MHz, CHLOROFORM- d) δ = 7.32 (dd, J = 5.6, 8.4 Hz, 1H), 7.19 (dd, J = 5.6, 8.0 Hz, 1H), 4.43 (s, 2H). Step B – Synthesis of 1-(4-bromo-2,5-difluorobenzyl) pyrimidine-2,4(1H,3H)-dione (164b) A mixture of 1-bromo-4-(bromomethyl)-2,5-difluoro-benzene (40 g, 139.9 mmol, 1.0 equiv.), pyrimidine-2,4(1H,3H)-dione (47.0 g, 419.7 mmol, 3.0 equiv.), K2CO3 (58.0 g, 419.7 mmol, 3.0 equiv.) in DMSO (800 mL) was degassed and purged threefold with nitrogen. The mixture was stirred at 80°C for 1 h under a nitrogen atmosphere. The mixture was treated with water (1.5 L) and stirred for 10 min, then aged for 8 h at ambient temperature. The solids were collected by vacuum filtration. The filtrate liquors were extracted with 2-methyltetrahydrofuran (500 mLx2). The combined organic phase was washed with brine (50 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was triturated with ethyl acetate (200 mL) at 20oC for 10 min to give 164b. LCMS [M+1] = 317.0, 319.0; 1H NMR (400 MHz, DMSO-d6) δ 11.3 (br s, 1H), 7.76 (dd, J = 5.6, 9.2 Hz, 1H), 7.67 (d, J = 8.0 Hz, 1H), 7.34 (dd, J = 6.8, 8.8 Hz, 1H), 5.59 (d, J = 7.6 Hz, 1H), 4.87 (s, 2H). Step C – Synthesis of tert-butyl 4-(4-((2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)methyl)-2,5- difluorophenyl)-3,6-dihydropyridine-1(2H)-carboxylate (164c) A mixture of 1-[(4-bromo-2,5-difluoro-phenyl)methyl]pyrimidine-2,4-dione (10.0 g, 31.5 mmol, 1.0 equiv.), tert-butyl 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-3,6- dihydropyridine-1(2H)-carboxylate (14.6 g, 47.3 mmol, 1.5 equiv.), Pd(dppf)Cl2 (2.3 g, 3.1 mmol, 0.1 equiv.), cesium carbonate (20.5 g, 63.1 mmol, 2.0 equiv.) in dioxane (80 mL) and water (20 mL) was degassed and purged threefold with nitrogen. The mixture was stirred at 100°C for 12 h under a nitrogen atmosphere. The residue was poured into water (200 mL). The
aqueous portion was extracted with ethyl acetate (100 mLx2). The combined organic portion was washed with brine (20 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, Petroleum ether/Ethyl acetate=10/1 to 0/1) to give 164c. LCMS [M-56+CH3CN+H] = 405.1. Step D – Synthesis of tert-butyl 4-(4-((2,4-dioxotetrahydropyrimidin-1(2H)-yl)methyl)-2,5- difluorophenyl)piperidine-1-carboxylate (164d) Pd(OH)2 (4.3 g, 31.0 mmol, 1.0 equiv.) was added to a flask purged with argon, then a solution of tert-butyl 4-(4-((2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)methyl)-2,5- difluorophenyl)-3,6-dihydropyridine-1(2H)-carboxylate (13.0 g, 31.0 mmol, 1.0 equiv.) in MeOH (150 mL) was added under argon. The mixture was degassed and purged threefold with hydrogen gas. The mixture was stirred at 50°C for 16 h under hydrogen gas (15 Psi). The mixture was filtered over a celite pad and the filtrate was concentrated. The residue was purified by column chromatography (silica gel, Petroleum ether/Ethyl acetate=5/1 to 1/1) to give 164d. LCMS [M- 56+1] = 368.1. Step E – Synthesis of 1-(2,5-difluoro-4-(piperidin-4-yl)benzyl)dihydropyrimidine-2,4(1H,3H)- dione hydrochloride (164e) A mixture of tert-butyl 4-(4-((2,4-dioxotetrahydropyrimidin-1(2H)-yl)methyl)-2,5- difluorophenyl)piperidine-1-carboxylate (11.0 g, 26.0 mmol, 1.0 equiv.) in HCl/ethyl acetate (100 mL) was degassed and purged threefold with nitrogen. The mixture was stirred at 20°C for 1 h under a nitrogen atmosphere and concentrated under reduced pressure. The residue was triturated with ethyl acetate (20 mL) at 20oC for 30 min to give 164e. LCMS [M+1] = 324.2. Step F – Synthesis of tert-butyl 3-(5-(4-((4-(4-((2,4-dioxotetrahydropyrimidin-1(2H)-yl)methyl)- 2,5-difluorophenyl)piperidin-1-yl)methyl)phenyl)pyrimidin-2-yl)isoxazole-5-carboxylate (164f) To a solution of tert-butyl 3-[5-(4-formylphenyl)pyrimidin-2-yl]isoxazole-5-carboxylate (0.5 g, 1.4 mmol, 1.0 equiv.) and 1-[[2,5-difluoro-4-(4- piperidyl)phenyl]methyl]hexahydropyrimidine-2,4-dione (614.4 mg, 1.7 mmol, 1.2 eq, HCl) in dichloromethane (5 mL) was added triethylamine (2.0 mL, 10.0 equiv.) at 20°C and stirred for 2 h, then NaBH(OAc)3 (603.2 mg, 2.9 mmol, 2.0 equiv.) was added to the mixture at 20°C and
stirred for 16 h. Water (20 mL) and ethyl acetate (20 mL) was added to the mixture and stirred for 10 min, then filtered under reduced pressure. The filter cake was triturated with acetonitrile (20 mL) at 60oC for 30 min to give 164f. LCMS [M+1] = 659.4. Step G – Synthesis of 3-(5-(4-((4-(4-((2,4-dioxotetrahydropyrimidin-1(2H)-yl)methyl)-2,5- difluorophenyl)piperidin-1-yl)methyl)phenyl)pyrimidin-2-yl)isoxazole-5-carboxylic acid hydrochloride salt (164g) A mixture of tert-butyl 3-(5-(4-((4-(4-((2,4-dioxotetrahydropyrimidin-1(2H)-yl)methyl)- 2,5-difluorophenyl)piperidin-1-yl)methyl)phenyl)pyrimidin-2-yl)isoxazole-5-carboxylate (700 mg, 1.1 mmol, 1.0 equiv.) in HCl/dioxane (10 mL) was degassed and purged threefold with nitrogen and then the mixture was heated at 60°C for 1 h under a nitrogen atmosphere. The mixture was concentrated under reduced pressure. The residue was triturated with acetonitrile (50 mL) at 60oC for 30 min to give 164g. LCMS [M+1] = 603.2. Step H – Synthesis of 3-(5-(4-((4-(4-((2,4-dioxotetrahydropyrimidin-1(2H)-yl)methyl)-2,5- difluorophenyl)piperidin-1-yl)methyl)phenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (164) To a suspension of 3-(5-(4-((4-(4-((2,4-dioxotetrahydropyrimidin-1(2H)-yl)methyl)-2,5- difluorophenyl)piperidin-1-yl)methyl)phenyl)pyrimidin-2-yl)isoxazole-5-carboxylic acid hydrochloride salt (660 mg, 1.10 mmol, 1 equiv.) in THF (7 mL) was added BOP (726.63 mg, 1.64 mmol, 1.5 equiv.) DIEA (707.78 mg, 5.48 mmol, 953.88 μL, 5 equiv.) in one portion at 20°C and stirred for 15 min, then [3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl] methanamine hydrochloride(401.93 mg, 1.43 mmol, 1.31 equiv) was added to the mixture at 60°C and stirred for 30 min. Water (50 mL) was added to the mixture and stirred for 10 min, then filtered under reduced pressure. The filter cake was triturated with acetonitrile (50 mL x 4) at 60oC for 1 h to give 164. LCMS [M+1] = 793.3; 1H NMR (400 MHz, DMSO-d6) δ = 14.00 (br s, 1H), 10.23 (s, 1H), 9.72 (br s, 1H), 9.36 (s, 2H), 7.89 (d, J = 8.0 Hz, 2H), 7.72 (s, 1H), 7.53 (d, J = 8.0 Hz, 2H), 7.23 (dd, J = 6.4, 10.8 Hz, 1H), 7.12 (dd, J = 6.4, 10.8 Hz, 1H), 4.60 (br s, 2H), 4.50 (s, 2H), 3.59 (s, 2H), 3.37 (t, J = 6.8 Hz, 2H), 2.95 (d, J = 10.8 Hz, 2H), 2.84 - 2.71 (m, 1H), 2.57 (t, J = 6.8 Hz, 3H), 2.17 - 2.05 (m, 2H), 1.08 - 1.65 (m, 4H), 1.54 (s, 6H).
Example 165: Synthesis of Compound 165 Step
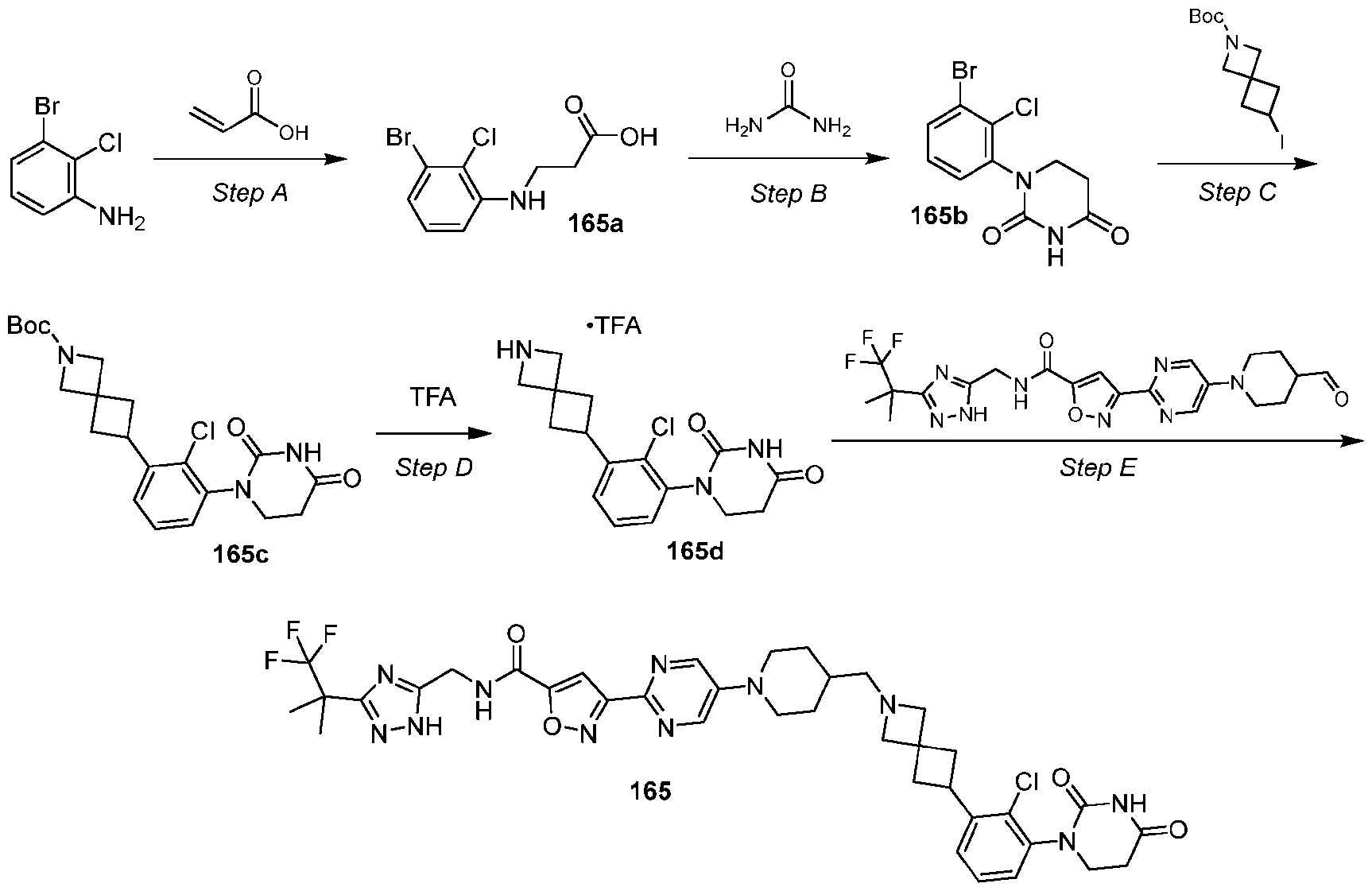 A solution of 3-bromo-2-chloro-aniline (10.0 g, 48.4 mmol, 1.0 equiv.) and acrylic acid (17.4 g, 242.1 mmol, 16.6 mL, 5.0 equiv.) in acetic acid (100 mL) was heated at 110°C for 4 h. The mixture was concentrated to dryness under reduced pressure to afford 165a, which was used without purification. LCMS [M+1] =277.9, 279.9. Step B – Synthesis of 1-(3-bromo-2-chlorophenyl)dihydropyrimidine-2,4(1H,3H)-dione (165b) A solution of 3-((3-bromo-2-chlorophenyl)amino)propanoic acid (13.5 g, 48.4 mmol, 1.0 equiv.) urea (18.6 g, 310.2 mmol, 16.6 mL, 6.4 equiv.) in acetic acid (100 mL) was stirred at 120°C for 16 h. The reaction was cooled to 20oC. The mixture was concentrated under reduced pressure. The mixture was diluted with water (20 mL) and extracted with ethyl acetate (60 mL). The combined organic layers were dried over Na2CO3, filtered, and concentrated under reduced pressure. The residue was triturated with ethyl acetate (40 ml) at 20 C for 30 min, the filter cake was collected to give 165b. LCMS [M+1] =302.9, 304.9; 1H NMR (400 MHz, DMSO-d6) δ 10.53 (s, 1H), 7.78 (d, J = 8.0 Hz, 1H), 7.54 (d, J = 8.0 Hz, 1H), 7.40 - 7.34 (m, 1H), 3.80 - 3.70 (m, 1H), 3.61 (m, 1H), 2.74 (t, J = 6.4 Hz, 2H).
Step C – Synthesis of tert-butyl 6-(2-chloro-3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)phenyl)-2- azaspiro[3.3]heptane-2-carboxylate (165c) To a solution of 1-(3-bromo-2-chlorophenyl)dihydropyrimidine-2,4(1H,3H)-dione (700.0 mg, 2.3 mmol, 1.0 equiv.) and tert-butyl 6-iodo-2-azaspiro[3.3]heptane-2-carboxylate (1.5 g, 4.6 mmol, 2.0 equiv.) in DMA (10 mL) was added methoxypyridine-2-carboxamidine; hydrochloride (43.2 mg, 230.6 μmol, 0.1 equiv.), dichloronickel•1,2-dimethoxyethane (50.6 mg, 230.6 μmol, 0.1 equiv.) ,TFA (26.3 mg, 230.6 μmol, 17.1 μL, 0.1 equiv.) and Zn (335.1 mg, 4.6 mmol, 90% purity, 2.0 equiv.) was added under an argon atmosphere. The mixture was stirred at 80°C for 12 h. The reaction was cooled to 20oC. The suspension was filtered through a pad of Celite and the pad was washed with ethyl acetate (50 ml) and THF (50 ml). The combined filtrate was extracted with ethyl acetate (50 mL x 2) and the combined organic extracts were washed with saturated aqueous sodium bicarbonate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by prep-HPLC (Phenomenex Luna C18, 100x40mm, 5 µm; [water(0.2% FA)-ACN];gradient:45%-65% B over 8.0 min) to give 165c. LCMS [M+1] =364.1, 366.1. Step D – Synthesis of 1-(2-chloro-3-(2-azaspiro[3.3]heptan-6-yl)phenyl)dihydropyrimidine- 2,4(1H,3H)-dione trifluoroacetic acid salt (165d) To a solution of tert-butyl-6-(2-chloro-3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)phenyl)- 2-azaspiro[3.3]heptane-2-carboxylate (50.0 mg, 119.1 μmol, 1.0 equiv.) in dichloromethane (1 mL) was added TFA (339.4 mg, 2.9 mmol, 221.1 μL, 25.0 equiv.). The mixture was stirred at 20°C for 12 h. The mixture was concentrated under reduced pressure to afford 165d. LCMS [M+1] =320.1, 322.1. Step E – Synthesis of 3-(5-(4-((6-(2-chloro-3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)phenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)piperidin-1-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (165) To a solution of 1-(2-chloro-3-(2-azaspiro[3.3]heptan-6-yl)phenyl)dihydropyrimidine- 2,4(1H,3H)-dione trifluoroacetic acid salt (45.0 mg, 140.7 μmol, 1.0 equiv.), 3-[5-(4-formyl-1- piperidyl)pyrimidin-2-yl] -N- [[3- (2,2,2 -trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methyl]isoxazole-5-carboxamide (69.3 mg, 140.7 μmol, 1.0 equiv.) in dichloromethane (1 mL)
was added triethylamine (98 μL, 5.0 equiv.) and NaBH(OAc)3 (89.5 mg, 422.1 μmol, 3.0 equiv.). The mixture was stirred at 25°C for 3 h. The mixture was concentrated under reduced pressure. The residue was purified by prep-HPLC (Phenomenex Luna C18100x40mm, 5 µm; [water(0.2% FA)-ACN];gradient:10%-40% B over 8.0 min) to give 165. LCMS [M+1] =796.4; 1H NMR (400 MHz, DMSO-d6) δ 13.98 (br s, 1H), 10.44 (s, 1H), 9.64 (t, J = 5.6 Hz, 1H), 8.61 (s, 2H), 7.52 (s, 1H), 7.41 - 7.37 (m, 2H), 7.36 - 7.31 (m, 1H), 4.58 (br d, J = 5.6 Hz, 2H), 3.97 (br d, J = 12.8 Hz, 2H), 3.72 - 3.51 (m, 4H), 3.36 (s, 2H), 3.13 (s, 2H), 2.86 (br t, J = 12.0 Hz, 2H), 2.72 (t, J = 6.0 Hz, 2H), 2.58 - 2.53 (m, 2H), 2.36 - 2.30 (m, 2H), 2.26 - 2.16 (m, 2H), 1.77 (br d, J = 11.6 Hz, 2H), 1.54 (s, 7H), 1.27 - 1.13 (m, 2H). Example 166: Synthesis of Compound 166
A solution of 3-bromo-2-chloro-aniline (10.0 g, 48.4 mmol, 1.0 equiv.) and acrylic acid (17.4 g, 242.1 mmol, 16.6 mL, 5.0 equiv.) in acetic acid (100 mL) was heated at 110°C for 4 h. The mixture was concentrated to dryness under reduced pressure to afford 165a, which was used without purification. LCMS [M+1] =277.9, 279.9. Step B – Synthesis of 1-(3-bromo-2-chlorophenyl)dihydropyrimidine-2,4(1H,3H)-dione (165b) A solution of 3-((3-bromo-2-chlorophenyl)amino)propanoic acid (13.5 g, 48.4 mmol, 1.0 equiv.) urea (18.6 g, 310.2 mmol, 16.6 mL, 6.4 equiv.) in acetic acid (100 mL) was stirred at 120°C for 16 h. The reaction was cooled to 20oC. The mixture was concentrated under reduced pressure. The mixture was diluted with water (20 mL) and extracted with ethyl acetate (60 mL). The combined organic layers were dried over Na2CO3, filtered, and concentrated under reduced pressure. The residue was triturated with ethyl acetate (40 ml) at 20 C for 30 min, the filter cake was collected to give 165b. LCMS [M+1] =302.9, 304.9; 1H NMR (400 MHz, DMSO-d6) δ 10.53 (s, 1H), 7.78 (d, J = 8.0 Hz, 1H), 7.54 (d, J = 8.0 Hz, 1H), 7.40 - 7.34 (m, 1H), 3.80 - 3.70 (m, 1H), 3.61 (m, 1H), 2.74 (t, J = 6.4 Hz, 2H).
Step C – Synthesis of tert-butyl 6-(2-chloro-3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)phenyl)-2- azaspiro[3.3]heptane-2-carboxylate (165c) To a solution of 1-(3-bromo-2-chlorophenyl)dihydropyrimidine-2,4(1H,3H)-dione (700.0 mg, 2.3 mmol, 1.0 equiv.) and tert-butyl 6-iodo-2-azaspiro[3.3]heptane-2-carboxylate (1.5 g, 4.6 mmol, 2.0 equiv.) in DMA (10 mL) was added methoxypyridine-2-carboxamidine; hydrochloride (43.2 mg, 230.6 μmol, 0.1 equiv.), dichloronickel•1,2-dimethoxyethane (50.6 mg, 230.6 μmol, 0.1 equiv.) ,TFA (26.3 mg, 230.6 μmol, 17.1 μL, 0.1 equiv.) and Zn (335.1 mg, 4.6 mmol, 90% purity, 2.0 equiv.) was added under an argon atmosphere. The mixture was stirred at 80°C for 12 h. The reaction was cooled to 20oC. The suspension was filtered through a pad of Celite and the pad was washed with ethyl acetate (50 ml) and THF (50 ml). The combined filtrate was extracted with ethyl acetate (50 mL x 2) and the combined organic extracts were washed with saturated aqueous sodium bicarbonate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by prep-HPLC (Phenomenex Luna C18, 100x40mm, 5 µm; [water(0.2% FA)-ACN];gradient:45%-65% B over 8.0 min) to give 165c. LCMS [M+1] =364.1, 366.1. Step D – Synthesis of 1-(2-chloro-3-(2-azaspiro[3.3]heptan-6-yl)phenyl)dihydropyrimidine- 2,4(1H,3H)-dione trifluoroacetic acid salt (165d) To a solution of tert-butyl-6-(2-chloro-3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)phenyl)- 2-azaspiro[3.3]heptane-2-carboxylate (50.0 mg, 119.1 μmol, 1.0 equiv.) in dichloromethane (1 mL) was added TFA (339.4 mg, 2.9 mmol, 221.1 μL, 25.0 equiv.). The mixture was stirred at 20°C for 12 h. The mixture was concentrated under reduced pressure to afford 165d. LCMS [M+1] =320.1, 322.1. Step E – Synthesis of 3-(5-(4-((6-(2-chloro-3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)phenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)piperidin-1-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (165) To a solution of 1-(2-chloro-3-(2-azaspiro[3.3]heptan-6-yl)phenyl)dihydropyrimidine- 2,4(1H,3H)-dione trifluoroacetic acid salt (45.0 mg, 140.7 μmol, 1.0 equiv.), 3-[5-(4-formyl-1- piperidyl)pyrimidin-2-yl] -N- [[3- (2,2,2 -trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methyl]isoxazole-5-carboxamide (69.3 mg, 140.7 μmol, 1.0 equiv.) in dichloromethane (1 mL)
was added triethylamine (98 μL, 5.0 equiv.) and NaBH(OAc)3 (89.5 mg, 422.1 μmol, 3.0 equiv.). The mixture was stirred at 25°C for 3 h. The mixture was concentrated under reduced pressure. The residue was purified by prep-HPLC (Phenomenex Luna C18100x40mm, 5 µm; [water(0.2% FA)-ACN];gradient:10%-40% B over 8.0 min) to give 165. LCMS [M+1] =796.4; 1H NMR (400 MHz, DMSO-d6) δ 13.98 (br s, 1H), 10.44 (s, 1H), 9.64 (t, J = 5.6 Hz, 1H), 8.61 (s, 2H), 7.52 (s, 1H), 7.41 - 7.37 (m, 2H), 7.36 - 7.31 (m, 1H), 4.58 (br d, J = 5.6 Hz, 2H), 3.97 (br d, J = 12.8 Hz, 2H), 3.72 - 3.51 (m, 4H), 3.36 (s, 2H), 3.13 (s, 2H), 2.86 (br t, J = 12.0 Hz, 2H), 2.72 (t, J = 6.0 Hz, 2H), 2.58 - 2.53 (m, 2H), 2.36 - 2.30 (m, 2H), 2.26 - 2.16 (m, 2H), 1.77 (br d, J = 11.6 Hz, 2H), 1.54 (s, 7H), 1.27 - 1.13 (m, 2H). Example 166: Synthesis of Compound 166
 Step A – Synthesis of tert-butyl 3-(5-(2-hydroxy-7-azaspiro[3.5]nonan-7-yl)pyrimidin-2- yl)isoxazole-5-carboxylate (166a) A mixture of tert-butyl 3-(5-bromopyrimidin-2-yl)isoxazole-5-carboxylate (6.0 g, 18.4 mmol, 1.0 equiv.), 7-azaspiro[3.5]nonan-2-ol hydrochloride (4.9 g, 27.6 mmol, 1.5 equiv.) ,
cesium carbonate (20.9 g, 64.3 mmol, 3.5 equiv.), [1,3-bis[2,6-bis(1-ethylpropyl)phenyl]-4,5- dichloro-imidazol-2-ylidene]-dichloro-(2-methylpyridin-1-ium-1-yl)palladium (1.5 g, 1.8 mmol, 0.1 equiv.) in dioxane (84.0 mL) was degassed and purged threefold with nitrogen. The mixture was stirred at 100°C for 16 h under a nitrogen atmosphere. The mixture was treated with water (10 mL) at 20°C and extracted with ethyl acetate (10 mL x 3). The combined organic portions were washed with brine (10 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, Petroleum ether/Ethyl acetate = 5/1 to 0/1) to give 166a. LCMS [M+1] = 387.3. Step B – Synthesis of tert-butyl 3-(5-(2-oxo-7-azaspiro[3.5]nonan-7-yl)pyrimidin-2-yl)isoxazole- 5-carboxylate (166b) To a solution of tert-butyl 3-(5-(2-hydroxy-7-azaspiro[3.5]nonan-7-yl)pyrimidin-2- yl)isoxazole-5-carboxylate (4.2 g, 11.0 mmol, 1.0 equiv.) in dichloromethane (43.0 mL) was added DMP (7.0 g, 16.6 mmol, 5.1 mL, 1.5 equiv.). The mixture was stirred at 15°C for 1 h. The mixture was treated with saturated aqueous NaHCO3 (50 mL) and saturated aqueous Na2SO3 solution (80 mL) and extracted with ethyl acetate (100 mL x 3). The combined organic portions were dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, Petroleum ether/THF = 3/1 to 0/1) to give 166b. LCMS [M+1] = 385.3; 1H NMR (400 MHz, DMSO-d6) δ = 8.66 (s, 2H), 7.43 (s, 1H), 3.49 - 3.41 (m, 4H), 2.88 (s, 4H), 1.85 - 1.78 (m, 4H), 1.57 (s, 9H). Step C – Synthesis of tert-butyl 3-(5-(2-(4-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3- fluorophenyl)piperazin-1-yl)-7-azaspiro[3.5]nonan-7-yl)pyrimidin-2-yl)isoxazole-5-carboxylate (166c) To a solution of tert-butyl 3-(5-(2-oxo-7-azaspiro[3.5]nonan-7-yl)pyrimidin-2- yl)isoxazole-5-carboxylate (300.0 mg, 780.3 μmol, 1.0 equiv.) and 1-(2-fluoro-4-piperazin-1-yl- phenyl)hexahydropyrimidine-2,4-dione (513.1 mg, 1.5 mmol, 2.0 equiv., HCl) in dichloromethane (4.0 mL) was added triethylamine (543 μL, 5.0 equiv.). The mixture was stirred at 15oC for 16 h. Then NaBH(OAc)3 (496.1 mg, 2.3 mmol, 3.0 equiv.) was added to the mixture and the mixture was stirred at 30oC for 1 h. The mixture was quenched by addition water (10 mL) at 20°C, and then extracted with ethyl acetate (10 mL x 3). The combined organic layers
were washed with brine (10 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by prep-TLC (silica gel, dichloromethane: MeOH = 10: 1) to give 166c. LCMS [M+1] = 661.4. Step D – Synthesis of 3-(5-(2-(4-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3- fluorophenyl)piperazin-1-yl)-7-azaspiro[3.5]nonan-7-yl)pyrimidin-2-yl)isoxazole-5-carboxylic acid (166d) A solution of tert-butyl 3-(5-(2-(4-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3- fluorophenyl)piperazin-1-yl)-7-azaspiro[3.5]nonan-7-yl)pyrimidin-2-yl)isoxazole-5-carboxylate (240.0 mg, 363.2 μmol, 1.0 equiv.) in HCl/ethyl acetate (20.0 mL) was stirred at 30°C for 16 h. The mixture was concentrated under reduced pressure to give 166d. LCMS [M+1] = 605.4. Step E – Synthesis of 3-(5-(2-(4-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3- fluorophenyl)piperazin-1-yl)-7-azaspiro[3.5]nonan-7-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (166) To a solution of 3-[5-[2-[4-[4-(2,4-dioxohexahydropyrimidin-1-yl)-3-fluoro-phenyl] piperazin-1-yl]-7-azaspiro[3.5]nonan-7-yl]pyrimidin-2-yl]isoxazole-5-carboxylic acid (160.0 mg, 249.5 μmol, 1.0 equiv., HCl) and [3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methanamine hydrochloride (105.24 mg, 374.36 μmol, 1.5 equiv., 2HCl) in DMF (2.0 mL) was added DIEA (161.2 mg, 1.2 mmol, 217.3 μL, 5.0 equiv.). Then T4P (359.6 mg, 499.1 μmol, 50% purity, 2.0 equiv.) was added to the mixture. The mixture was stirred at 15°C for 3 h. The mixture was quenched by addition water (10 mL) at 20°C, and then extracted with ethyl acetate (10 mL x 3). The combined organic layers were washed with brine (10 mL), dried over by sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by prep-HPLC (Phenomenex Luna C18100x40mm, 5 µm; [water (0.2% FA)-ACN]; gradient: 5%-35% B over 8.0 min) to give 166. LCMS [M+1] = 795.3; 1H NMR (400 MHz, DMSO-d6) δ 13.99 (br s, 1H), 10.37 (s, 1H), 9.63 (br t, J = 5.2 Hz, 1H), 8.63 (s, 2H), 7.52 (s, 1H), 7.21 (t, J = 8.8 Hz, 1H), 6.89 - 6.70 (m, 2H), 4.58 (d, J = 6.0 Hz, 2H), 3.62 (t, J = 6.4 Hz, 2H), 3.41 (s, 2H), 3.18 (s, 4H), 2.75 (t, J = 7.6 Hz,, 1H), 2.69 (t, J = 6.4 Hz, 3H), 2.38 (s, 4H), 2.03 (t, J = 9.6 Hz, 2H), 1.71 - 1.58 (m, 7H), 1.54 (s, 6H).
Example 167: Synthesis of Compound 167
Step A – Synthesis of tert-butyl 3-(5-(2-hydroxy-7-azaspiro[3.5]nonan-7-yl)pyrimidin-2- yl)isoxazole-5-carboxylate (166a) A mixture of tert-butyl 3-(5-bromopyrimidin-2-yl)isoxazole-5-carboxylate (6.0 g, 18.4 mmol, 1.0 equiv.), 7-azaspiro[3.5]nonan-2-ol hydrochloride (4.9 g, 27.6 mmol, 1.5 equiv.) ,
cesium carbonate (20.9 g, 64.3 mmol, 3.5 equiv.), [1,3-bis[2,6-bis(1-ethylpropyl)phenyl]-4,5- dichloro-imidazol-2-ylidene]-dichloro-(2-methylpyridin-1-ium-1-yl)palladium (1.5 g, 1.8 mmol, 0.1 equiv.) in dioxane (84.0 mL) was degassed and purged threefold with nitrogen. The mixture was stirred at 100°C for 16 h under a nitrogen atmosphere. The mixture was treated with water (10 mL) at 20°C and extracted with ethyl acetate (10 mL x 3). The combined organic portions were washed with brine (10 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, Petroleum ether/Ethyl acetate = 5/1 to 0/1) to give 166a. LCMS [M+1] = 387.3. Step B – Synthesis of tert-butyl 3-(5-(2-oxo-7-azaspiro[3.5]nonan-7-yl)pyrimidin-2-yl)isoxazole- 5-carboxylate (166b) To a solution of tert-butyl 3-(5-(2-hydroxy-7-azaspiro[3.5]nonan-7-yl)pyrimidin-2- yl)isoxazole-5-carboxylate (4.2 g, 11.0 mmol, 1.0 equiv.) in dichloromethane (43.0 mL) was added DMP (7.0 g, 16.6 mmol, 5.1 mL, 1.5 equiv.). The mixture was stirred at 15°C for 1 h. The mixture was treated with saturated aqueous NaHCO3 (50 mL) and saturated aqueous Na2SO3 solution (80 mL) and extracted with ethyl acetate (100 mL x 3). The combined organic portions were dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, Petroleum ether/THF = 3/1 to 0/1) to give 166b. LCMS [M+1] = 385.3; 1H NMR (400 MHz, DMSO-d6) δ = 8.66 (s, 2H), 7.43 (s, 1H), 3.49 - 3.41 (m, 4H), 2.88 (s, 4H), 1.85 - 1.78 (m, 4H), 1.57 (s, 9H). Step C – Synthesis of tert-butyl 3-(5-(2-(4-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3- fluorophenyl)piperazin-1-yl)-7-azaspiro[3.5]nonan-7-yl)pyrimidin-2-yl)isoxazole-5-carboxylate (166c) To a solution of tert-butyl 3-(5-(2-oxo-7-azaspiro[3.5]nonan-7-yl)pyrimidin-2- yl)isoxazole-5-carboxylate (300.0 mg, 780.3 μmol, 1.0 equiv.) and 1-(2-fluoro-4-piperazin-1-yl- phenyl)hexahydropyrimidine-2,4-dione (513.1 mg, 1.5 mmol, 2.0 equiv., HCl) in dichloromethane (4.0 mL) was added triethylamine (543 μL, 5.0 equiv.). The mixture was stirred at 15oC for 16 h. Then NaBH(OAc)3 (496.1 mg, 2.3 mmol, 3.0 equiv.) was added to the mixture and the mixture was stirred at 30oC for 1 h. The mixture was quenched by addition water (10 mL) at 20°C, and then extracted with ethyl acetate (10 mL x 3). The combined organic layers
were washed with brine (10 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by prep-TLC (silica gel, dichloromethane: MeOH = 10: 1) to give 166c. LCMS [M+1] = 661.4. Step D – Synthesis of 3-(5-(2-(4-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3- fluorophenyl)piperazin-1-yl)-7-azaspiro[3.5]nonan-7-yl)pyrimidin-2-yl)isoxazole-5-carboxylic acid (166d) A solution of tert-butyl 3-(5-(2-(4-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3- fluorophenyl)piperazin-1-yl)-7-azaspiro[3.5]nonan-7-yl)pyrimidin-2-yl)isoxazole-5-carboxylate (240.0 mg, 363.2 μmol, 1.0 equiv.) in HCl/ethyl acetate (20.0 mL) was stirred at 30°C for 16 h. The mixture was concentrated under reduced pressure to give 166d. LCMS [M+1] = 605.4. Step E – Synthesis of 3-(5-(2-(4-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3- fluorophenyl)piperazin-1-yl)-7-azaspiro[3.5]nonan-7-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (166) To a solution of 3-[5-[2-[4-[4-(2,4-dioxohexahydropyrimidin-1-yl)-3-fluoro-phenyl] piperazin-1-yl]-7-azaspiro[3.5]nonan-7-yl]pyrimidin-2-yl]isoxazole-5-carboxylic acid (160.0 mg, 249.5 μmol, 1.0 equiv., HCl) and [3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methanamine hydrochloride (105.24 mg, 374.36 μmol, 1.5 equiv., 2HCl) in DMF (2.0 mL) was added DIEA (161.2 mg, 1.2 mmol, 217.3 μL, 5.0 equiv.). Then T4P (359.6 mg, 499.1 μmol, 50% purity, 2.0 equiv.) was added to the mixture. The mixture was stirred at 15°C for 3 h. The mixture was quenched by addition water (10 mL) at 20°C, and then extracted with ethyl acetate (10 mL x 3). The combined organic layers were washed with brine (10 mL), dried over by sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by prep-HPLC (Phenomenex Luna C18100x40mm, 5 µm; [water (0.2% FA)-ACN]; gradient: 5%-35% B over 8.0 min) to give 166. LCMS [M+1] = 795.3; 1H NMR (400 MHz, DMSO-d6) δ 13.99 (br s, 1H), 10.37 (s, 1H), 9.63 (br t, J = 5.2 Hz, 1H), 8.63 (s, 2H), 7.52 (s, 1H), 7.21 (t, J = 8.8 Hz, 1H), 6.89 - 6.70 (m, 2H), 4.58 (d, J = 6.0 Hz, 2H), 3.62 (t, J = 6.4 Hz, 2H), 3.41 (s, 2H), 3.18 (s, 4H), 2.75 (t, J = 7.6 Hz,, 1H), 2.69 (t, J = 6.4 Hz, 3H), 2.38 (s, 4H), 2.03 (t, J = 9.6 Hz, 2H), 1.71 - 1.58 (m, 7H), 1.54 (s, 6H).
Example 167: Synthesis of Compound 167
 (167a) To a solution of 6-bromo-5-fluoro-pyridin-3-amine (10.0 g, 52.3 mmol, 1.0 equiv.) in acetic acid (100.0 mL) was added acrylic acid (11.3 g, 157.0 mmol, 10.7 mL, 3.0 equiv.) at 25°C.
The mixture was stirred at 110°C for 12 h to afford 3-[(6-bromo-5-fluoro-3- pyridyl)amino]propanoic acid (LCMS [M+1] = 263.0), which was used into the next step without purification. A solution of 3-[(6-bromo-5-fluoro-3-pyridyl)amino]propanoic acid (6.8 g, 26.1 mmol, 1.0 equiv.) and urea (10.0 g, 167.6 mmol, 9.0 mL, 6.4 equiv.) in acetic acid (50.0 mL) was heated at 120°C for 12 h. The mixture was diluted with water (50 mL). The mixture was extracted with ethyl acetate (10 mL × 3) and dried over by sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The crude product was triturated with MTBE (15 mL) at 25°C for 10 min, filtered and the filtrate was concentrated under reduced pressure to give 167a. LCMS [M+1] = 288.0, 290.0; 1H NMR (400 MHz, DMSO-d6) δ = 10.65 (s, 1H), 8.36 (d, J = 1.6 Hz, 1H), 7.97 (dd, J = 1.6, 9.2 Hz, 1H), 3.88 (t, J = 6.4 Hz, 2H), 2.73 (t, J = 6.8 Hz, 2H). Step B – Synthesis of tert-butyl 1-(4-bromo-2-iodophenyl)-1H-pyrazole-4-carboxylate (167b) To a solution of tert-butyl 1H-pyrazole-4-carboxylate (20.0 g, 118.9 mmol, 1.0 equiv.) and 4-bromo-1-fluoro-2-iodo-benzene (35.7 g, 118.9 mmol, 1.0 equiv.) in DMF (350.0 mL) was added cesium carbonate (58.1 g, 178.3 mmol, 1.5 equiv.) at 25°C, finally the mixture was stirred at 130°C for 16 h. The mixture was quenched by addition water 500 mL at 15°C, the mixture was filtered and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel chromatography (silica gel, Petroleum ether:Ethyl acetate = 92:8) followed with additional purification by prep-HPLC (Welch Xtimate C18250x100mm, 10µm; [water(10mM NH4HCO3)-ACN];gradient:58%-80% B over 20.0 min to give 167b. LCMS [M+1] = 449.0, 451.0. Step C – Synthesis of tert-butyl 1-(4-bromo-2-(1-methyl-1H-pyrazol-4-yl)phenyl)-1H-pyrazole-4- carboxylate (167c) To a solution of tert-butyl 1-(4-bromo-2-iodo-phenyl)pyrazole-4-carboxylate (24.0 g, 53.4 mmol, 1.0 equiv.) and 1-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrazole (16.6 g, 80.1 mmol, 1.5 equiv.) in DMF (200.0 mL) and water (40.0 mL) was added NaHCO3 (8.9 g, 106.8 mmol, 4.1 mL, 2.0 equiv.) and purged threefold with nitrogen, and then treated with dichloropalladium triphenylphosphane (1.8 g, 2.6 mmol, 0.05 equiv.) under a nitrogen atmosphere. The mixture was heated at 90°C for 12 h. The mixture was treated with water (500
mL) at 15°C and extracted ethyl acetate (500 mL), the mixture was filtered and extracted, the combined organic layers were washed with brine (50 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel chromatography (Petroleum ether : Ethyl acetate = 13:87 ) followed by prep-HPLC (WePure Biotech XP C18250x70mm, 10µm; [water(10mM NH4HCO3)-ACN];gradient:45%- 85% B over 20.0 min) to give 167c. LCMS [M+1] =403.2, 405.2. Step D – Synthesis of tert-butyl 1-(4-hydroxy-2-(1-methyl-1H-pyrazol-4-yl)phenyl)-1H-pyrazole- 4-carboxylate (167d) To a solution of tert-butyl 1-(4-bromo-2-(1-methyl-1H-pyrazol-4-yl)phenyl)-1H-pyrazole- 4-carboxylate (13.0 g, 32.2 mmol, 1.0 equiv.) in dioxane (130.0 mL) was added KOH (1.8 g, 32.2 mmol, 1.0 equiv.), water (11.6 g, 644.7 mmol, 11.6 mL, 20.0 equiv.), ditert-butyl-[3,6-dimethoxy- 2-(2,4,6-triisopropylphenyl)phenyl]phosphane (781.2 mg, 1.6 mmol, 0.05 equiv.) and 2-(2- aminophenyl)phenyl]-methylsulfonyloxy-palladium;ditert-butyl-[3,6-dimethoxy-2-(2,4,6 - triisopropylphenyl)phenyl]phosphane (1.3 g, 1.6 mmol, 0.05 equiv.). The mixture was stirred at 85°C for 12 h under a nitrogen atmosphere. The mixture was quenched by addition water 300 mL at 15°C and extracted with ethyl acetate 400 mL. the combined organic layers were washed with brine 500 mL, dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel chromatography (silica gel, Petroleum ether: Ethyl acetate = 1:0 to 6:4) to give 167d. LCMS [M+1] =341.2; 1H NMR (400 MHz, DMSO-d6) δ 9.97 (s, 1H), 8.18 (s, 1H), 7.96 (s, 1H), 7.39 (s, 1H), 7.19 (d, J = 8.4 Hz, 1H), 6.99 (d, J = 2.4 Hz, 1H), 6.75 (dd, J = 2.4, 8.4 Hz, 1H), 6.71 (s, 1H), 3.75 (s, 3H), 1.50 (s, 9H). Step E – Synthesis of tert-butyl 7-(4-(4-(tert-butoxycarbonyl)-1H-pyrazol-1-yl)-3-(1-methyl-1H- pyrazol-4-yl)phenoxy)-2-azaspiro[3.5]nonane-2-carboxylate (167e) To a solution of tert-butyl 1-(4-hydroxy-2-(1-methyl-1H-pyrazol-4-yl)phenyl)-1H- pyrazole-4-carboxylate (60 g, 17.6 mmol, 1.0 equiv.) and tert-butyl 7-hydroxy-2- azaspiro[3.5]nonane-2-carboxylate (8.5 g, 35.2 mmol, 2.0 equiv.) in toluene (60.0 mL) was added 2-(tributyl-phosphanylidene)acetonitrile (8.5 g, 35.2 mmol, 2.0 equiv.) and threefold purged with nitrogen gas, then the mixture was stirred at 100°C for 12 h. The mixture was concentrated under reduced pressure. The residue was purified by prep-HPLC (Welch Xtimate C18180x70mm,
10µm; [water(10mM NH4HCO3)-ACN];gradient:60%-90% B over 20.0 min) to give 167e. LCMS [M+1] =564.4. Step F – Synthesis of tert-butyl 1-(4-((2-azaspiro[3.5]nonan-7-yl)oxy)-2-(1-methyl-1H-pyrazol-4- yl)phenyl)-1H-pyrazole-4-carboxylate (167f) To a solution of tert-butyl 7-(4-(4-(tert-butoxycarbonyl)-1H-pyrazol-1-yl)-3-(1-methyl- 1H-pyrazol-4-yl)phenoxy)-2-azaspiro[3.5]nonane-2-carboxylate (3.0 g, 5.3 mmol, 1.0 equiv.) in dichloromethane (15.0 mL) was added TFA (4.8 g, 42.5 mmol, 3.1 mL, 8.0 equiv.) at 25°C for 12 h. The mixture was quenched by addition NaHCO3 and extracted with ethyl acetate (40 mL), the combined organic layers were dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure to give 167e. LCMS [M+1] =464.3. Step G – Synthesis of tert-butyl 1-(4-((2-(5-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3- fluoropyridin-2-yl)-2-azaspiro[3.5]nonan-7-yl)oxy)-2-(1-methyl-1H-pyrazol-4-yl)phenyl)-1H- pyrazole-4-carboxylate (167g) To a solution of tert-butyl 1-(4-((2-azaspiro[3.5]nonan-7-yl)oxy)-2-(1-methyl-1H-pyrazol- 4-yl)phenyl)-1H-pyrazole-4-carboxylate (2.0 g, 4.3 mmol, 1.0 equiv.) and 1-(6-bromo-5-fluoro-3- pyridyl)hexahydropyrimidine-2,4-dione (1.3 g, 4.7 mmol, 1.1 equiv.) in dioxane (20.0 mL) was added cesium carbonate (2.8 g, 8.6 mmol, 2.0 equiv.) and purged threefold with nitrogen and then treated with [1,3-bis[2,6-bis(1-ethylpropyl)phenyl]-4,5-dichloro-imidazol-2-ylidene]-dichloro-(2- methylpyridin-1-ium-1-yl)palladium (181.2 mg, 216 μmol, 0.05 equiv.). The mixture was heated at 100°C for 12 h under a nitrogen atmosphere. The mixture was quenched by addition water (30 mL) and extracted with ethyl acetate (40 mL). The combined organic layers were washed with brine (40 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was triturated with ethyl acetate to give 167g. LCMS [M+1] =671.5. Step H – Synthesis of 1-(4-((2-(5-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluoropyridin-2-yl)- 2-azaspiro[3.5]nonan-7-yl)oxy)-2-(1-methyl-1H-pyrazol-4-yl)phenyl)-1H-pyrazole-4-carboxylic acid (167h)
A solution of tert-butyl 1-(4-((2-(5-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3- fluoropyridin-2-yl)-2-azaspiro[3.5]nonan-7-yl)oxy)-2-(1-methyl-1H-pyrazol-4-yl)phenyl)-1H- pyrazole-4-carboxylate (1.2 g, 1.7 mmol, 1.0 equiv.) in HCl/ ethyl acetate (13.0 mL) was aged at 25°C for 12 h. The mixture was concentrated under reduced pressure to give 167h, which was used without purification. LCMS [M+1] =615.4. Step I – Synthesis of 1-(4-((2-(5-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluoropyridin-2-yl)-2- azaspiro[3.5]nonan-7-yl)oxy)-2-(1-methyl-1H-pyrazol-4-yl)phenyl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide (167) A mixture of 1-(4-((2-(5-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluoropyridin-2-yl)- 2-azaspiro[3.5]nonan-7-yl)oxy)-2-(1-methyl-1H-pyrazol-4-yl)phenyl)-1H-pyrazole-4-carboxylic acid (1.3 g, 2.0 mmol, 1.0 equiv), BOP (1.0 g, 2.4 mmol, 1.2 equiv.) and DIEA (774.1 mg, 5.9 mmol, 1.0 mL, 3.0 equiv.) in DMF (13.0 mL) was treated with [3-(2,2,2-trifluoro-1,1-dimethyl- ethyl)-1H-1,2,4-triazol-5-yl]methanamine hydrochloride (976.9 mg, 3.9 mmol, 2.0 equiv.). The mixture was stirred at ambient temperature for 2 h, treated with water (10 mL) and extracted with ethyl acetate (20 mL). The combined organic portions were washed with brine (20 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by prep-HPLC (Phenomenex Luna C18250x100mm, 15µm; [water(0.02%FA)-ACN]; gradient:20%-50% B over 25.0 min ) to give 167. LCMS [M+1] =805.4; 1H NMR (400 MHz, DMSO-d6) δ 13.89 (br s, 1H), 10.41 (s, 1H), 8.80 (br s, 1H), 8.18 (s, 1H), 8.12 (s, 1H), 7.91 (dd, J = 1.2, 2.0 Hz, 1H), 7.53 (dd, J = 2.0, 13.2 Hz, 1H), 7.46 (s, 1H), 7.29 (d, J = 8.8 Hz, 1H), 7.20 (d, J = 2.8 Hz, 1H), 6.96 (dd, J = 2.8, 8.8 Hz, 1H), 6.77 (s, 1H), 4.61 - 4.47 (m, 3H), 3.84 (br d, J = 7.2 Hz, 4H), 3.75 (s, 3H), 3.71 (t, J = 6.8 Hz, 2H), 2.70 (t, J = 6.4 Hz, 2H), 2.02 – 1.87 (m, 4H), 1.77 - 1.50 (m, 10H). Example 168: Synthesis of Compound 168
(167a) To a solution of 6-bromo-5-fluoro-pyridin-3-amine (10.0 g, 52.3 mmol, 1.0 equiv.) in acetic acid (100.0 mL) was added acrylic acid (11.3 g, 157.0 mmol, 10.7 mL, 3.0 equiv.) at 25°C.
The mixture was stirred at 110°C for 12 h to afford 3-[(6-bromo-5-fluoro-3- pyridyl)amino]propanoic acid (LCMS [M+1] = 263.0), which was used into the next step without purification. A solution of 3-[(6-bromo-5-fluoro-3-pyridyl)amino]propanoic acid (6.8 g, 26.1 mmol, 1.0 equiv.) and urea (10.0 g, 167.6 mmol, 9.0 mL, 6.4 equiv.) in acetic acid (50.0 mL) was heated at 120°C for 12 h. The mixture was diluted with water (50 mL). The mixture was extracted with ethyl acetate (10 mL × 3) and dried over by sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The crude product was triturated with MTBE (15 mL) at 25°C for 10 min, filtered and the filtrate was concentrated under reduced pressure to give 167a. LCMS [M+1] = 288.0, 290.0; 1H NMR (400 MHz, DMSO-d6) δ = 10.65 (s, 1H), 8.36 (d, J = 1.6 Hz, 1H), 7.97 (dd, J = 1.6, 9.2 Hz, 1H), 3.88 (t, J = 6.4 Hz, 2H), 2.73 (t, J = 6.8 Hz, 2H). Step B – Synthesis of tert-butyl 1-(4-bromo-2-iodophenyl)-1H-pyrazole-4-carboxylate (167b) To a solution of tert-butyl 1H-pyrazole-4-carboxylate (20.0 g, 118.9 mmol, 1.0 equiv.) and 4-bromo-1-fluoro-2-iodo-benzene (35.7 g, 118.9 mmol, 1.0 equiv.) in DMF (350.0 mL) was added cesium carbonate (58.1 g, 178.3 mmol, 1.5 equiv.) at 25°C, finally the mixture was stirred at 130°C for 16 h. The mixture was quenched by addition water 500 mL at 15°C, the mixture was filtered and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel chromatography (silica gel, Petroleum ether:Ethyl acetate = 92:8) followed with additional purification by prep-HPLC (Welch Xtimate C18250x100mm, 10µm; [water(10mM NH4HCO3)-ACN];gradient:58%-80% B over 20.0 min to give 167b. LCMS [M+1] = 449.0, 451.0. Step C – Synthesis of tert-butyl 1-(4-bromo-2-(1-methyl-1H-pyrazol-4-yl)phenyl)-1H-pyrazole-4- carboxylate (167c) To a solution of tert-butyl 1-(4-bromo-2-iodo-phenyl)pyrazole-4-carboxylate (24.0 g, 53.4 mmol, 1.0 equiv.) and 1-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrazole (16.6 g, 80.1 mmol, 1.5 equiv.) in DMF (200.0 mL) and water (40.0 mL) was added NaHCO3 (8.9 g, 106.8 mmol, 4.1 mL, 2.0 equiv.) and purged threefold with nitrogen, and then treated with dichloropalladium triphenylphosphane (1.8 g, 2.6 mmol, 0.05 equiv.) under a nitrogen atmosphere. The mixture was heated at 90°C for 12 h. The mixture was treated with water (500
mL) at 15°C and extracted ethyl acetate (500 mL), the mixture was filtered and extracted, the combined organic layers were washed with brine (50 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel chromatography (Petroleum ether : Ethyl acetate = 13:87 ) followed by prep-HPLC (WePure Biotech XP C18250x70mm, 10µm; [water(10mM NH4HCO3)-ACN];gradient:45%- 85% B over 20.0 min) to give 167c. LCMS [M+1] =403.2, 405.2. Step D – Synthesis of tert-butyl 1-(4-hydroxy-2-(1-methyl-1H-pyrazol-4-yl)phenyl)-1H-pyrazole- 4-carboxylate (167d) To a solution of tert-butyl 1-(4-bromo-2-(1-methyl-1H-pyrazol-4-yl)phenyl)-1H-pyrazole- 4-carboxylate (13.0 g, 32.2 mmol, 1.0 equiv.) in dioxane (130.0 mL) was added KOH (1.8 g, 32.2 mmol, 1.0 equiv.), water (11.6 g, 644.7 mmol, 11.6 mL, 20.0 equiv.), ditert-butyl-[3,6-dimethoxy- 2-(2,4,6-triisopropylphenyl)phenyl]phosphane (781.2 mg, 1.6 mmol, 0.05 equiv.) and 2-(2- aminophenyl)phenyl]-methylsulfonyloxy-palladium;ditert-butyl-[3,6-dimethoxy-2-(2,4,6 - triisopropylphenyl)phenyl]phosphane (1.3 g, 1.6 mmol, 0.05 equiv.). The mixture was stirred at 85°C for 12 h under a nitrogen atmosphere. The mixture was quenched by addition water 300 mL at 15°C and extracted with ethyl acetate 400 mL. the combined organic layers were washed with brine 500 mL, dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel chromatography (silica gel, Petroleum ether: Ethyl acetate = 1:0 to 6:4) to give 167d. LCMS [M+1] =341.2; 1H NMR (400 MHz, DMSO-d6) δ 9.97 (s, 1H), 8.18 (s, 1H), 7.96 (s, 1H), 7.39 (s, 1H), 7.19 (d, J = 8.4 Hz, 1H), 6.99 (d, J = 2.4 Hz, 1H), 6.75 (dd, J = 2.4, 8.4 Hz, 1H), 6.71 (s, 1H), 3.75 (s, 3H), 1.50 (s, 9H). Step E – Synthesis of tert-butyl 7-(4-(4-(tert-butoxycarbonyl)-1H-pyrazol-1-yl)-3-(1-methyl-1H- pyrazol-4-yl)phenoxy)-2-azaspiro[3.5]nonane-2-carboxylate (167e) To a solution of tert-butyl 1-(4-hydroxy-2-(1-methyl-1H-pyrazol-4-yl)phenyl)-1H- pyrazole-4-carboxylate (60 g, 17.6 mmol, 1.0 equiv.) and tert-butyl 7-hydroxy-2- azaspiro[3.5]nonane-2-carboxylate (8.5 g, 35.2 mmol, 2.0 equiv.) in toluene (60.0 mL) was added 2-(tributyl-phosphanylidene)acetonitrile (8.5 g, 35.2 mmol, 2.0 equiv.) and threefold purged with nitrogen gas, then the mixture was stirred at 100°C for 12 h. The mixture was concentrated under reduced pressure. The residue was purified by prep-HPLC (Welch Xtimate C18180x70mm,
10µm; [water(10mM NH4HCO3)-ACN];gradient:60%-90% B over 20.0 min) to give 167e. LCMS [M+1] =564.4. Step F – Synthesis of tert-butyl 1-(4-((2-azaspiro[3.5]nonan-7-yl)oxy)-2-(1-methyl-1H-pyrazol-4- yl)phenyl)-1H-pyrazole-4-carboxylate (167f) To a solution of tert-butyl 7-(4-(4-(tert-butoxycarbonyl)-1H-pyrazol-1-yl)-3-(1-methyl- 1H-pyrazol-4-yl)phenoxy)-2-azaspiro[3.5]nonane-2-carboxylate (3.0 g, 5.3 mmol, 1.0 equiv.) in dichloromethane (15.0 mL) was added TFA (4.8 g, 42.5 mmol, 3.1 mL, 8.0 equiv.) at 25°C for 12 h. The mixture was quenched by addition NaHCO3 and extracted with ethyl acetate (40 mL), the combined organic layers were dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure to give 167e. LCMS [M+1] =464.3. Step G – Synthesis of tert-butyl 1-(4-((2-(5-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3- fluoropyridin-2-yl)-2-azaspiro[3.5]nonan-7-yl)oxy)-2-(1-methyl-1H-pyrazol-4-yl)phenyl)-1H- pyrazole-4-carboxylate (167g) To a solution of tert-butyl 1-(4-((2-azaspiro[3.5]nonan-7-yl)oxy)-2-(1-methyl-1H-pyrazol- 4-yl)phenyl)-1H-pyrazole-4-carboxylate (2.0 g, 4.3 mmol, 1.0 equiv.) and 1-(6-bromo-5-fluoro-3- pyridyl)hexahydropyrimidine-2,4-dione (1.3 g, 4.7 mmol, 1.1 equiv.) in dioxane (20.0 mL) was added cesium carbonate (2.8 g, 8.6 mmol, 2.0 equiv.) and purged threefold with nitrogen and then treated with [1,3-bis[2,6-bis(1-ethylpropyl)phenyl]-4,5-dichloro-imidazol-2-ylidene]-dichloro-(2- methylpyridin-1-ium-1-yl)palladium (181.2 mg, 216 μmol, 0.05 equiv.). The mixture was heated at 100°C for 12 h under a nitrogen atmosphere. The mixture was quenched by addition water (30 mL) and extracted with ethyl acetate (40 mL). The combined organic layers were washed with brine (40 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was triturated with ethyl acetate to give 167g. LCMS [M+1] =671.5. Step H – Synthesis of 1-(4-((2-(5-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluoropyridin-2-yl)- 2-azaspiro[3.5]nonan-7-yl)oxy)-2-(1-methyl-1H-pyrazol-4-yl)phenyl)-1H-pyrazole-4-carboxylic acid (167h)
A solution of tert-butyl 1-(4-((2-(5-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3- fluoropyridin-2-yl)-2-azaspiro[3.5]nonan-7-yl)oxy)-2-(1-methyl-1H-pyrazol-4-yl)phenyl)-1H- pyrazole-4-carboxylate (1.2 g, 1.7 mmol, 1.0 equiv.) in HCl/ ethyl acetate (13.0 mL) was aged at 25°C for 12 h. The mixture was concentrated under reduced pressure to give 167h, which was used without purification. LCMS [M+1] =615.4. Step I – Synthesis of 1-(4-((2-(5-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluoropyridin-2-yl)-2- azaspiro[3.5]nonan-7-yl)oxy)-2-(1-methyl-1H-pyrazol-4-yl)phenyl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide (167) A mixture of 1-(4-((2-(5-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluoropyridin-2-yl)- 2-azaspiro[3.5]nonan-7-yl)oxy)-2-(1-methyl-1H-pyrazol-4-yl)phenyl)-1H-pyrazole-4-carboxylic acid (1.3 g, 2.0 mmol, 1.0 equiv), BOP (1.0 g, 2.4 mmol, 1.2 equiv.) and DIEA (774.1 mg, 5.9 mmol, 1.0 mL, 3.0 equiv.) in DMF (13.0 mL) was treated with [3-(2,2,2-trifluoro-1,1-dimethyl- ethyl)-1H-1,2,4-triazol-5-yl]methanamine hydrochloride (976.9 mg, 3.9 mmol, 2.0 equiv.). The mixture was stirred at ambient temperature for 2 h, treated with water (10 mL) and extracted with ethyl acetate (20 mL). The combined organic portions were washed with brine (20 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by prep-HPLC (Phenomenex Luna C18250x100mm, 15µm; [water(0.02%FA)-ACN]; gradient:20%-50% B over 25.0 min ) to give 167. LCMS [M+1] =805.4; 1H NMR (400 MHz, DMSO-d6) δ 13.89 (br s, 1H), 10.41 (s, 1H), 8.80 (br s, 1H), 8.18 (s, 1H), 8.12 (s, 1H), 7.91 (dd, J = 1.2, 2.0 Hz, 1H), 7.53 (dd, J = 2.0, 13.2 Hz, 1H), 7.46 (s, 1H), 7.29 (d, J = 8.8 Hz, 1H), 7.20 (d, J = 2.8 Hz, 1H), 6.96 (dd, J = 2.8, 8.8 Hz, 1H), 6.77 (s, 1H), 4.61 - 4.47 (m, 3H), 3.84 (br d, J = 7.2 Hz, 4H), 3.75 (s, 3H), 3.71 (t, J = 6.8 Hz, 2H), 2.70 (t, J = 6.4 Hz, 2H), 2.02 – 1.87 (m, 4H), 1.77 - 1.50 (m, 10H). Example 168: Synthesis of Compound 168
2,4(1H,3H)-dione (168a) A mixture of 2-fluoro-5-iodopyridine (10.4 g, 46.9 mmol, 1.1 equiv.), 3-(4- methoxybenzyl)dihydropyrimidine-2,4(1H,3H)-dione (10 g, 42.6 mmol, 1.0 equiv.), K3PO4 (18.1 g, 85.3 mmol, 2.0 equiv.), CuI (4.0 g, 21.3 mmol, 0.5 equiv.) and (1S,2S)-N1,N2- dimethylcyclohexane-1,2-diamine (6.0 g, 42.6.mmol, 1.0 equiv.) in dioxane (100.0 mL) was degassed and purged threefold with nitrogen, and then the mixture was stirred at 60°C for 1 h under a nitrogen atmosphere. The residue was diluted with water (40 mL) and extracted with ethyl acetate (40 mL x 3). The combined organic layers were washed with brine (40 mL x 2),
dried, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether/ethyl acetate = 1/0 to 1/1) to give 168a. LCMS [M+1] = 330.1. Step B – Synthesis of 1-(6-fluoropyridin-3-yl)dihydropyrimidine-2,4(1H,3H)-dione trifluoroacetate salt (168b) To a solution of 1-(6-fluoropyridin-3-yl)-3-(4-methoxybenzyl)dihydropyrimidine- 2,4(1H,3H)-dione (5.0 g, 15.1 mmol, 1.0 equiv.) in TFA (30.0 mL) was added TfOH (6.0 mL). The mixture was stirred at 70°C for 30 min. The mixture was concentrated under reduced pressure. The residue was purified by prep-HPLC (Phenomenex Luna C18 (250x70mm, 10 µm); [water(FA)-ACN];gradient:2%-30% B over 20 min) to give 168b. LCMS [M+1] = 210.1. 1H NMR (400 MHz, DMSO-d6) δ 10.53 (s, 1H), 8.23 (d, J = 1.0 Hz, 1H), 7.98 (ddd, J = 2.8, 7.2, 8.8 Hz, 1H), 7.24 (dd, J = 3.2, 8.8 Hz, 1H), 3.82 (t, J = 6.8 Hz, 2H), 2.73 (t, J = 6.8 Hz, 2H). Step C – Synthesis of ethyl 1-(4-hydroxy-2-methylphenyl)-1H-pyrazole-4-carboxylate (168c) To a solution of ethyl 1H-pyrazole-4-carboxylate (25.3 g, 180.9 mmol, 1.1 equiv.), (4- hydroxy-2-methylphenyl)boronic acid (25.0 g, 164.5 mmol, 1.0 equiv.) in DMF (250.0 mL) was added Cu(OAc)2 (22.4 g, 123.3 mmol, 0.7 equiv.) and pyridine (26.5 mL). The mixture was stirred at 15°C for 12 h under O2. The mixture was partitioned between water (800 mL) and ethyl acetate (300 mLx3). The organic phase was separated, washed with brine (300mL x2), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, Petroleum ether/Ethyl acetate=1/0 to 1/1) to afford 168c. LCMS [M+1] = 247.1. Step D – Synthesis of tert-butyl 7-(4-(4-(ethoxycarbonyl)-1H-pyrazol-1-yl)-3-methylphenoxy)-2- azaspiro[3.5]nonane-2-carboxylate (168d) To a solution of ethyl 1-(4-hydroxy-2-methylphenyl)-1H-pyrazole-4-carboxylate (2.0 g, 8.12 mmol, 1.0 equiv.) in toluene (10.0 mL) was added 2-(tributylphosphanylidene)acetonitrile (4.9 g, 20.3 mmol, 2.5 equiv.) and tert-butyl 7-hydroxy-2-azaspiro[3.5]nonane-2-carboxylate (3.9 g, 16.2 mmol, 2.0 equiv.). The mixture was stirred at 100°C for 6 h. The mixture was
concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, Petroleum ether : Ethyl acetate= 100:1 to 3:1) to afford 168d. LCMS [M-56] = 414.1. Step E – Synthesis of 1-(4-((2-(tert-butoxycarbonyl)-2-azaspiro[3.5]nonan-7-yl)oxy)-2- methylphenyl)-1H-pyrazole-4-carboxylic acid (168e) To a solution of tert-butyl 7-(4-(4-(ethoxycarbonyl)-1H-pyrazol-1-yl)-3-methylphenoxy)- 2-azaspiro[3.5]nonane-2-carboxylate (5.0 g, 7.4 mmol, 1.0 equiv.) in THF (24.0 mL) and MeOH (6.0 mL) was added the solution of LiOH hydrate in water (12.0 mL) (938.2 mg, 28.3 mmol, 3.0 equiv.). The mixture was stirred at 50°C for 2 h. The mixture was concentrated under reduced pressure and poured into water (300 mL). Aqueous HCl (1M) was added until pH=4~5. The aqueous phase was extracted with ethyl acetate (100mLx3). The combined organic phase was washed with brine (80 mLx2), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure to give 168e. LCMS [M-55] = 386.2. Step F – Synthesis of tert-butyl 7-(3-methyl-4-(4-(((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H- 1,2,4-triazol-5-yl)methyl)carbamoyl)-1H-pyrazol-1-yl)phenoxy)-2-azaspiro[3.5]nonane-2- carboxylate (168f) To a solution of 1-(4-((2-(tert-butoxycarbonyl)-2-azaspiro[3.5]nonan-7-yl)oxy)-2- methylphenyl)-1H-pyrazole-4-carboxylic acid (9.0 g, 20.3 mmol, 1.0 equiv.) and [3-(2,2,2- trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methanamine hydrochloride (5.0 g, 24.4 mmol, 1.2 equiv.) in DMF (60.0 mL) was added NMI (8.3 g, 101.9 mmol, 8.1 mL, 5.0 equiv.). The mixture was treated with TCFH (8.5 g, 30.5 mmol, 1.5 equiv.), stirred at 25°C for 1 h and filtered. The filtrate was purified by prep-HPLC(Phenomenex Luna C18250x100mm, 10 µm; [water(FA)-ACN];gradient:40%-70% B over 24 min) to afford 168f. LCMS [M-100] = 532.2. Step G – Synthesis of 1-(4-((2-azaspiro[3.5]nonan-7-yl)oxy)-2-methylphenyl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide trifluoroacetate salt (168g) To a solution of tert-butyl 7-(3-methyl-4-(4-(((3-(1,1,1-trifluoro-2-methylpropan-2-yl)- 1H-1,2,4-triazol-5-yl)methyl)carbamoyl)-1H-pyrazol-1-yl)phenoxy)-2-azaspiro[3.5]nonane-2- carboxylate (4.3 g, 6.8 mmol, 1.0 equiv.) in dichloromethane (30.0 mL) and TFA (10.0 mL). The
mixture was stirred at 25°C for 1.0 h. The mixture was concentrated under reduced pressure. The residue was triturated with MTBE (10 mL) at 20oC for 30 min and filtered. The filtered cake was concentrated under reduce pressure to give 168g.LCMS [M+1] = 532.3. 1H NMR (400 MHz, DMSO-d6) δ 8.82 (s, 1H), 8.67 (s, 2H), 8.37 (s, 1H), 8.10 (s, 1H), 7.26 (d, J = 8.4 Hz, 1H), 6.95 (d, J = 2.4 Hz, 1H), 6.89 (dd, J = 2.4, 8.8 Hz, 1H), 4.53 (d, J = 5.6 Hz, 2H), 4.44 - 4.41 (m, 1H), 3.75 - 3.69 (m, 4H), 2.13 (s, 3H), 1.98 - 1.90 (m, 2H), 1.88 - 1.80 (m, 2H), 1.70 - 1.63 (m, 2H), 1.53 (s, 8H). Step H – Synthesis of 1-(4-((2-(5-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyridin-2-yl)-2- azaspiro[3.5]nonan-7-yl)oxy)-2-methylphenyl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H- 1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide (168) To a solution of 1-(4-((2-azaspiro[3.5]nonan-7-yl)oxy)-2-methylphenyl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide trifluoroacetate (2.0 g, 3.1 mmol, 1.0 equiv.), 1-(6-fluoropyridin-3-yl)dihydropyrimidine- 2,4(1H,3H)-dione (1.9 g, 6.2 mmol, 2.0 equiv., TFA) in DMSO (20.0 mL) was added DIEA (2.0 g, 15.4 mmol, 2.7 mL, 5.0 equiv.). The mixture was stirred at 100°C for 1 h. The mixture was concentrated under reduced pressure. The residue was purified by prep-HPLC (Phenomenex Luna C18 (250x70mm,10 µm); [water(FA)-ACN];gradient:10%-40% B over 20 min) to give 168. LCMS [M+1] = 721.4. 1H NMR (400 MHz, DMSO-d6) δ 13.90 (s, 1H), 10.33 (s, 1H), 8.82 (s, 1H), 8.38 (s, 1H), 8.10 (s, 1H), 8.01 (d, J = 2.8 Hz, 1H), 7.47 (dd, J =2.8 Hz, 1H), 7.26 (d, J = 2.4 Hz, 1H), 6.98 (d, J = 2.4 Hz, 1H), 6.92 (dd, J = 2.4, 8.8 Hz, 1H), 6.39 (d, J = 8.8 Hz, 1H), 4.59 - 4.42 (m, 3H), 3.76 - 3.62 (m, 6H), 2.69 (t, J = 6.8 Hz, 2H), 2.13 (s, 3H), 1.92 (s, 4H), 1.74 - 1.64 (m, 2H), 1.63 - 1.55 (m, 2H), 1.53 (s, 6H). Example 169: Synthesis of Compound 169
Step A A solution of 6-fluoro-5-methyl-pyridin-3-amine (30.0 g, 237.8 mmol, 1.0 equiv.) and acrylic acid (47.9 g, 665.9 mmol, 45.6 mL, 2.8 equiv.) in toluene (300.0 mL) was stirred at 120°C for 30 min. The mixture was cooled to room temperature, filtered and the filtrate was concentrated under reduced pressure to give 169a, which was used without purification. LCMS [M+1] = 198.9. Step B – Synthesis of 1-(6-fluoro-5-methyl-3-pyridyl) hexahydropyrimidine-2, 4-dione (169b) To a solution of 3-[(6-fluoro-5-methyl-3-pyridyl)amino]propanoic acid (32.0 g, 161.4 mmol, 1.0 equiv.) in acetic acid (300.0 mL) was added urea (62.0 g, 1.0 mol, 55.5 mL, 6.4 equiv.). The mixture was stirred at 100°C for 6 h. The mixture was concentrated under reduced pressure to remove the volatile portion. The residue was diluted with water (100 mL) and extracted with dichloromethane (100 mLx3). The combined organic portions were dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was triturated with water (200 mL) at 20oC for 30 min to afford 169b. LCMS [M+1] = 224.1; 1H NMR (400 MHz, DMSO-d6) δ 10.51 (s, 1H), 8.04 (s, 1H), 7.85 (dd, J = 2.0, 8.8 Hz, 1H), 3.80 (t, J = 6.4 Hz, 2H), 2.72 (t, J = 6.4 Hz, 2H), 2.25 (s, 3H).
Step C – Synthesis of 1-[4-[[2-[5-(2,4-dioxohexahydropyrimidin-1-yl)-3-methyl-2-pyridyl]-2- azaspiro[3.5]nonan-7-yl]oxy]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H- 1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide (169) To a solution of 1-(6-fluoro-5-methyl-3-pyridyl)hexahydropyrimidine-2,4-dione (800.0 mg, 3.5 mmol, 1.0 equiv.) in DMSO (8.0 mL) was added DIEA (2.3 g, 17.9 mmol, 3.1 mL, 5.0 equiv.) and 1-[4-(2-azaspiro[3.5]nonan-7-yloxy)-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide trifluoroacetate salt (1.8 g, 2.8 mmol, 0.8 equiv.). The mixture was stirred at 80°C for 5 d. The mixture was cooled to 25°C and filtered. The filtrate was purified by prep-HPLC (Phenomenex Luna C18 (250x70mm,10 µm); [water ( NH4HCO3)-ACN];gradient:10%-40% B over 20 min) to give 169. LCMS [M+1] = 735.5; 1H NMR (400 MHz, DMSO-d6) δ 13.89 (s, 1H), 10.33 (s, 1H), 8.81 (s, 1H), 8.37 (s, 1H), 8.10 (s, 1H), 7.90 (d, J = 2.4 Hz, 1H), 7.29 (d, J = 2.0 Hz, 1H), 7.26 (d, J = 8.8 Hz, 1H), 6.98 (d, J = 2.4 Hz, 1H), 6.91 (dd, J = 2.4, 8.8 Hz, 1H), 4.54 (d, J = 5.6 Hz, 2H), 4.50 - 4.41 (m, 1H), 3.81 (d, J = 8.4 Hz, 4H), 3.68 (t, J = 6.8 Hz, 2H), 2.69 (t, J = 6.8 Hz, 2H), 2.16 (s, 3H), 2.13 (s, 3H), 1.96 - 1.87 (m, 4H), 1.71 - 1.56 (m, 4H), 1.53 (s, 6H). Example 170: Synthesis of Intermediate 170
Step A – Synthesis of ethyl 1-(4-((1,4-dioxaspiro[4.5]decan-8-yl)oxy)-2-methylphenyl)-1H- pyrazole-4-carboxylate (170a) A solution of ethyl 1-(4-hydroxy-2-methylphenyl)-1H-pyrazole-4-carboxylate (8.0 g, 32.4 mmol, 1.0 equiv.) in toluene. (80.0 mL) was added CMBP (31.6 g, 129.9 mmol, 4.0 equiv.) and 1-oxaspiro[4.5]decan-8-ol (10.0 g, 64.97 mmol, 2.0 equiv.). The mixture was stirred at 100°C for 2 h under an atmosphere of nitrogen. The mixture was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, petroleum ether/ethyl acetate=1/0 to 9/1) to afford 170a. LCMS [M+1] = 387.2. Step B – Synthesis of ethyl 1-(2-methyl-4-((4-oxocyclohexyl)oxy)phenyl)-1H-pyrazole-4- carboxylate (170b) A solution of ethyl 1-(4-((1,4-dioxaspiro[4.5]decan-8-yl)oxy)-2-methylphenyl)-1H- pyrazole-4-carboxylate (5.5 g, 14.2 mmol, 1.0 equiv.) in formic acid (50.0 mL) was stirred at 40°C for 1 h. The mixture was quenched into water (100 mL) and directly lyophilization to afford 170b, which was used without further purification. LCMS [M+1] = 343.2. Step C – Synthesis of 1-(2-methyl-4-((4-oxocyclohexyl)oxy)phenyl)-1H-pyrazole-4-carboxylic acid (170c) To a solution of ethyl 1-(2-methyl-4-((4-oxocyclohexyl)oxy)phenyl)-1H-pyrazole-4- carboxylate (5.5 g, 16.0 mmol, 1.0 eq) in THF (40.0 mL) and MeOH (1.00 mL) was added the solution of LiOH•H2O (2.0 g, 48.1 mmol, 3.0 equiv.) in water (20.0 mL). The mixture was stirred at 50°C. The mixture was concentrated under reduced pressure and the remainder was poured into water (150 mL). Aqueous HCl (1M) was added until the pH of the mixture was 4 to 5 . The aqueous phase was extracted with ethyl acetate (150 mLx3). The combined organic phase was washed with brine (150 mLx2), dried with anhydrous sodium sulfate , filtered and the filtrate was concentrated under reduced pressure to afford 170c. LCMS [M+1] = 315.2. Step D – Synthesis of 1-(2-methyl-4-((4-oxocyclohexyl)oxy)phenyl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide (170)
To a solution of 1-(2-methyl-4-((4-oxocyclohexyl)oxy)phenyl)-1H-pyrazole-4-carboxylic acid (4.3 g, 13.6 mmol, 1.0 equiv.) and (3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol- 5-yl)methanamine hydrochloride (5.44 g, 13.6 mmol, 1.0 equiv.) in DMF (43.0 mL) was added NMI (5.6 g, 68.4 mmol, 5.4 mL, 5.0 equiv.). The mixture was stirred at 25°C for 5 min, treated with TCFH (5.7 g, 20.5 mmol, 1.5 equiv.), stirred at 20°C for 1 h and filtered. The filtrate was purified by prep-HPLC(Phenomenex Luna C18 (250x70 mm, 10 µm); [water(FA)-ACN]; gradient:23%-53% B over 30 min) to afford 170. LCMS [M+1] = 505.3. 1H NMR (400 MHz, DMSO-d6) δ 13.88 (s, 1H), 8.86 (s, 1H), 8.39 (s, 1H), 8.11 (s, 1H), 7.29 (d, J = 8.8 Hz, 1H), 7.07 (d, J = 2.4 Hz, 1H), 7.00 (dd, J = 2.8, 8.0 Hz, 1H), 4.88 - 4.85 (m, 1H), 4.54 (s, 2H), 2.48 - 2.31 (m, 4H), 2.19 - 2.01 (m, 7H), 1.53 (s, 6H). Example 171: Synthesis of Compounds 171-1 and 171-2 Step A –
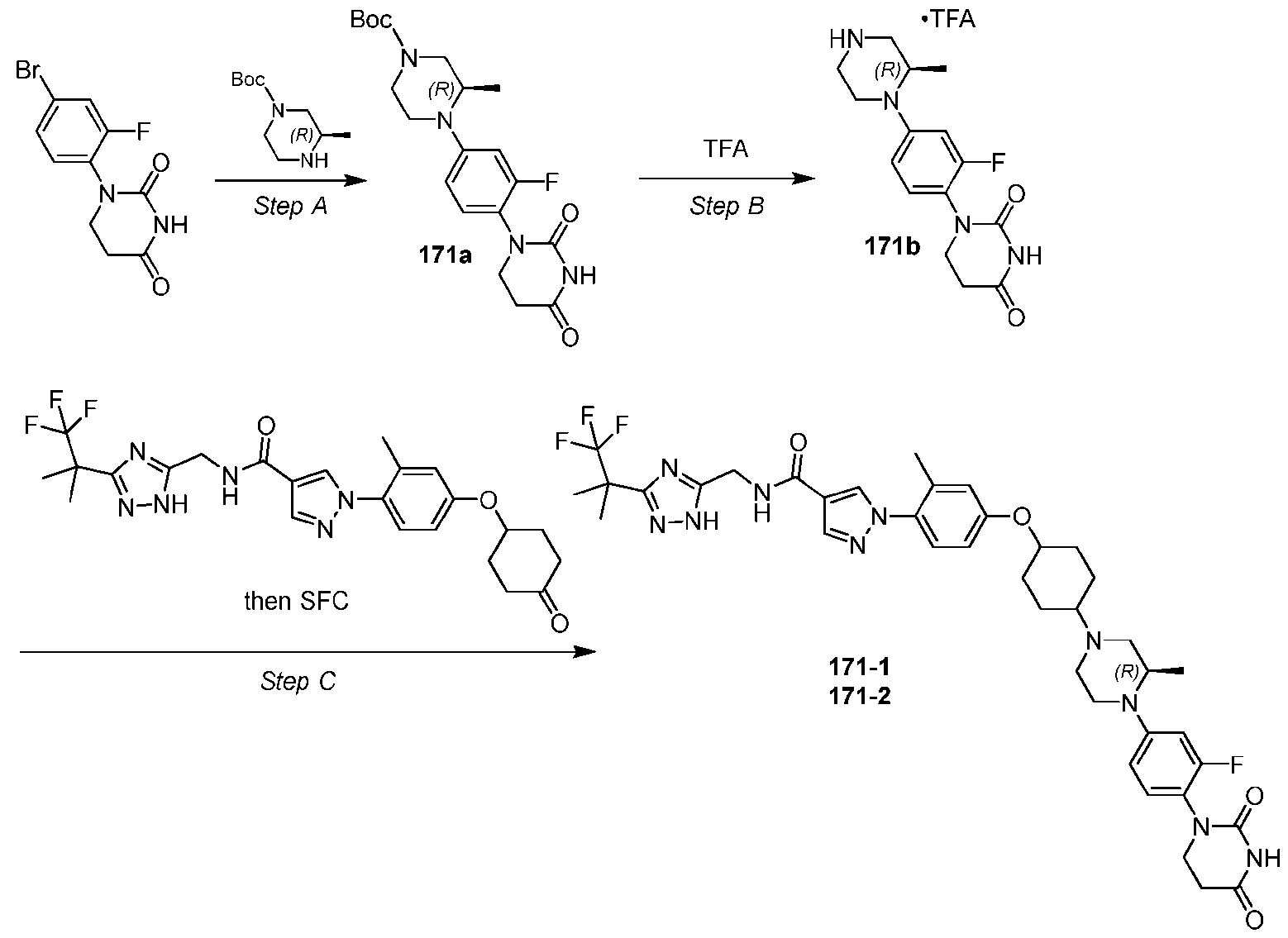 fluorophenyl)-3-methylpiperazine-1-carboxylate (171a) A mixture of 1-(4-bromo-2-fluorophenyl)dihydropyrimidine-2,4(1H,3H)-dione (1.0 g, 3.48 mmol, 1.0 equiv.), tert-butyl (R)-3-methylpiperazine-1-carboxylate (837.1 mg, 4.18 mmol, 1.2 equiv.), Pd-PEPPSI-ipentCl(o-picoline) (292.6 mg, 348.3 μmol, 0.03 equiv.) and cesium
carbonate (3.4 g, 10.4 mmol, 3.0 equiv.) in dioxane (15.0 mL) was degassed and purged threefold with nitrogen. The mixture was stirred at 100°C for 1 h under a nitrogen atmosphere, cooled to 25°C and partitioned between ethyl acetate (20 mL) and water (15 mL). The organic phase was separated, dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, Petroleum ether/Ethyl acetate=1/0 to 1/1) to afford 171a. 1H NMR (400 MHz, METHANOL-d4) δ 7.22 (t, J = 8.8 Hz, 1H), 6.80 - 6.73 (m, 2H), 4.03 (d, J = 4.0 Hz, 2H), 3.88 (d, J = 13.2 Hz, 1H), 3.74 (t, J = 6.8 Hz, 2H), 3.34 - 3.32 (m, 2H), 3.16 - 3.00 (m, 2H), 2.80 (t, J = 6.8 Hz, 2H), 1.49 (s, 9H), 1.03 (d, J = 6.4 Hz, 3H). Step B – Synthesis of (R)-1-(2-fluoro-4-(2-methylpiperazin-1-yl)phenyl)dihydropyrimidine- 2,4(1H,3H)-dione trifluoroacetate salt (171b) To a solution of tert-butyl (R)-4-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3- fluorophenyl)-3-methylpiperazine-1-carboxylate (1.4 g, 3.5 mmol, 1.0 equiv.) in dichloromethane (6.0 mL) was added TFA (3.0 mL), the mixture was stirred at 25°C for 5 min. The mixture was filtered and the filtrate was concentrated under reduced pressure to afford 171b. LCMS [M+1] = 307.0. Step C – Synthesis of (R)-1-(4-((4-(4-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3- fluorophenyl)-3-methylpiperazin-1-yl)cyclohexyl)oxy)-2-methylphenyl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide (171c-1 and 171c- 2) To a solution of 1-(2-methyl-4-((4-oxocyclohexyl)oxy)phenyl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide (1.0 g, 1.9 mmol, 1.0 equiv.) and (R)-1-(2-fluoro-4-(2-methylpiperazin-1-yl)phenyl)dihydropyrimidine-2,4(1H,3H)- dione trifluoroacetate salt (1.2 g, 2.9 mmol, 1.5 equiv.) in THF (20 mL) was added Ti(OEt)4 (4.5 g, 19.8 mmol, 4.1 mL, 10.0 equiv.), The mixture was stirred at 60°C for 30 min, and then NaBH(OAc)3 (1.2 g, 5.9 mmol, 3.0 equiv.) was added, the mixture was stirred at 60°C for 1 h. The mixture was filtered and the filtrate was concentrated under reduced pressure. The residue was purified by prep-HPLC (Phenomenex Luna C18150x40 mm; 10 µm; [water(FA)-ACN]; 0%- 30% B over 10 min). The diastereomers were separated by SFC (DAICEL CHIRALPAK AD
(250x30mm, 10 µm); [isopropanol-triethylamine];B%:52%, isocratic elution) to give 171-1 as the early eluting isomer and 171-2 as the late eluting isomer. 171-2: LCMS [M+1] = 795.2; 1H NMR (400 MHz, DMSO-d6) δ 13.92 (s, 1H), 10.37 (s, 1H), 8.83 (s, 1H), 8.37 (s, 1H), 8.23 (s, 1H), 8.11 (s, 1H), 7.25 (d, J = 8.4 Hz, 1H), 7.22 - 7.15 (m, 1H), 6.97 (d, J = 2.4 Hz, 1H), 6.90 (dd, J = 2.8, 8.8 Hz, 1H), 6.78 - 6.66 (m, 2H), 4.53 (d, J = 5.6 Hz, 2H), 4.43 - 4.31 (m, 1H), 4.05 (d, J = 5.6 Hz, 1H), 3.62 (t, J = 6.8 Hz, 2H), 2.96 - 2.88 (m, 2H), 2.76 (s, 1H), 2.69 (t, J = 6.8 Hz, 2H), 2.46 (s, 1H), 2.41 - 2.28 (m, 2H), 2.13 (s, 5H), 1.92 - 1.81 (m, 2H), 1.54 (s, 6H), 1.43 (d, J = 6.8 Hz, 4H), 1.24 (s, 1H), 1.05 (d, J = 6.4 Hz, 3H). Example 172: Synthesis of Compounds 172-1 and 172-2 Compounds
fluorophenyl)-3-methylpiperazine-1-carboxylate (171a) A mixture of 1-(4-bromo-2-fluorophenyl)dihydropyrimidine-2,4(1H,3H)-dione (1.0 g, 3.48 mmol, 1.0 equiv.), tert-butyl (R)-3-methylpiperazine-1-carboxylate (837.1 mg, 4.18 mmol, 1.2 equiv.), Pd-PEPPSI-ipentCl(o-picoline) (292.6 mg, 348.3 μmol, 0.03 equiv.) and cesium
carbonate (3.4 g, 10.4 mmol, 3.0 equiv.) in dioxane (15.0 mL) was degassed and purged threefold with nitrogen. The mixture was stirred at 100°C for 1 h under a nitrogen atmosphere, cooled to 25°C and partitioned between ethyl acetate (20 mL) and water (15 mL). The organic phase was separated, dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, Petroleum ether/Ethyl acetate=1/0 to 1/1) to afford 171a. 1H NMR (400 MHz, METHANOL-d4) δ 7.22 (t, J = 8.8 Hz, 1H), 6.80 - 6.73 (m, 2H), 4.03 (d, J = 4.0 Hz, 2H), 3.88 (d, J = 13.2 Hz, 1H), 3.74 (t, J = 6.8 Hz, 2H), 3.34 - 3.32 (m, 2H), 3.16 - 3.00 (m, 2H), 2.80 (t, J = 6.8 Hz, 2H), 1.49 (s, 9H), 1.03 (d, J = 6.4 Hz, 3H). Step B – Synthesis of (R)-1-(2-fluoro-4-(2-methylpiperazin-1-yl)phenyl)dihydropyrimidine- 2,4(1H,3H)-dione trifluoroacetate salt (171b) To a solution of tert-butyl (R)-4-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3- fluorophenyl)-3-methylpiperazine-1-carboxylate (1.4 g, 3.5 mmol, 1.0 equiv.) in dichloromethane (6.0 mL) was added TFA (3.0 mL), the mixture was stirred at 25°C for 5 min. The mixture was filtered and the filtrate was concentrated under reduced pressure to afford 171b. LCMS [M+1] = 307.0. Step C – Synthesis of (R)-1-(4-((4-(4-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3- fluorophenyl)-3-methylpiperazin-1-yl)cyclohexyl)oxy)-2-methylphenyl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide (171c-1 and 171c- 2) To a solution of 1-(2-methyl-4-((4-oxocyclohexyl)oxy)phenyl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide (1.0 g, 1.9 mmol, 1.0 equiv.) and (R)-1-(2-fluoro-4-(2-methylpiperazin-1-yl)phenyl)dihydropyrimidine-2,4(1H,3H)- dione trifluoroacetate salt (1.2 g, 2.9 mmol, 1.5 equiv.) in THF (20 mL) was added Ti(OEt)4 (4.5 g, 19.8 mmol, 4.1 mL, 10.0 equiv.), The mixture was stirred at 60°C for 30 min, and then NaBH(OAc)3 (1.2 g, 5.9 mmol, 3.0 equiv.) was added, the mixture was stirred at 60°C for 1 h. The mixture was filtered and the filtrate was concentrated under reduced pressure. The residue was purified by prep-HPLC (Phenomenex Luna C18150x40 mm; 10 µm; [water(FA)-ACN]; 0%- 30% B over 10 min). The diastereomers were separated by SFC (DAICEL CHIRALPAK AD
(250x30mm, 10 µm); [isopropanol-triethylamine];B%:52%, isocratic elution) to give 171-1 as the early eluting isomer and 171-2 as the late eluting isomer. 171-2: LCMS [M+1] = 795.2; 1H NMR (400 MHz, DMSO-d6) δ 13.92 (s, 1H), 10.37 (s, 1H), 8.83 (s, 1H), 8.37 (s, 1H), 8.23 (s, 1H), 8.11 (s, 1H), 7.25 (d, J = 8.4 Hz, 1H), 7.22 - 7.15 (m, 1H), 6.97 (d, J = 2.4 Hz, 1H), 6.90 (dd, J = 2.8, 8.8 Hz, 1H), 6.78 - 6.66 (m, 2H), 4.53 (d, J = 5.6 Hz, 2H), 4.43 - 4.31 (m, 1H), 4.05 (d, J = 5.6 Hz, 1H), 3.62 (t, J = 6.8 Hz, 2H), 2.96 - 2.88 (m, 2H), 2.76 (s, 1H), 2.69 (t, J = 6.8 Hz, 2H), 2.46 (s, 1H), 2.41 - 2.28 (m, 2H), 2.13 (s, 5H), 1.92 - 1.81 (m, 2H), 1.54 (s, 6H), 1.43 (d, J = 6.8 Hz, 4H), 1.24 (s, 1H), 1.05 (d, J = 6.4 Hz, 3H). Example 172: Synthesis of Compounds 172-1 and 172-2 Compounds
 1(2H)-yl)- 3-fluorophenyl)-3-methylpiperazin-1-yl)cyclohexyl)oxy)-2-methylphenyl)-N-((3-(1,1,1-trifluoro- 2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide, were prepared in a similar manner to Example 171 to afford, after separation by SFC (DAICEL CHIRALPAK AD(250x30mm,10µm); [isopropanol-triethylamine]; B%:55%, isocratic elution), 172-1 as the early eluting isomer and 172-2 as the late eluting isomer. 172-2: LCMS [M+1] = 795.2; 1H NMR (400 MHz, METHANOL-d4) δ 8.24 (s, 1H), 8.12 (s, 1H), 7.31 - 7.13 (m, 2H), 6.93 - 6.88 (m, 2H), 6.78 - 6.72 (m, 2H), 4.68 (s, 2H), 4.39 - 4.30 (m, 1H), 3.97 (dd, J = 2.8, 6.0 Hz, 1H), 3.74 (t, J = 6.8 Hz, 2H), 3.33 (s, 1H), 3.12 - 3.05 (m, 1H), 3.00 - 2.95 (m, 1H), 2.86 - 2.78 (m, 3H), 2.63 (dd, J = 3.2, 11.2 Hz, 1H), 2.50 (m, J = 1H), 2.41 (s, 1H), 2.23 (d, J = 2.4 Hz, 2H), 2.15 (s, 3H), 2.06 - 1.98 (m, 2H), 1.61 (s, 6H), 1.57 - 1.44 (m, 4H), 1.12 (d, J = 6.4 Hz, 3H). Example 173: Synthesis of Compound 173
1(2H)-yl)- 3-fluorophenyl)-3-methylpiperazin-1-yl)cyclohexyl)oxy)-2-methylphenyl)-N-((3-(1,1,1-trifluoro- 2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide, were prepared in a similar manner to Example 171 to afford, after separation by SFC (DAICEL CHIRALPAK AD(250x30mm,10µm); [isopropanol-triethylamine]; B%:55%, isocratic elution), 172-1 as the early eluting isomer and 172-2 as the late eluting isomer. 172-2: LCMS [M+1] = 795.2; 1H NMR (400 MHz, METHANOL-d4) δ 8.24 (s, 1H), 8.12 (s, 1H), 7.31 - 7.13 (m, 2H), 6.93 - 6.88 (m, 2H), 6.78 - 6.72 (m, 2H), 4.68 (s, 2H), 4.39 - 4.30 (m, 1H), 3.97 (dd, J = 2.8, 6.0 Hz, 1H), 3.74 (t, J = 6.8 Hz, 2H), 3.33 (s, 1H), 3.12 - 3.05 (m, 1H), 3.00 - 2.95 (m, 1H), 2.86 - 2.78 (m, 3H), 2.63 (dd, J = 3.2, 11.2 Hz, 1H), 2.50 (m, J = 1H), 2.41 (s, 1H), 2.23 (d, J = 2.4 Hz, 2H), 2.15 (s, 3H), 2.06 - 1.98 (m, 2H), 1.61 (s, 6H), 1.57 - 1.44 (m, 4H), 1.12 (d, J = 6.4 Hz, 3H). Example 173: Synthesis of Compound 173
Step A – Synthesis of tert-butyl 7-iodo-2-azaspiro[3.5]nonane-2-carboxylate (173a) To a solution of tert-butyl 7-hydroxy-2-azaspiro[3.5]nonane-2-carboxylate (4.0 g, 16.5 mmol, 1.0 equiv.) in toluene (40.0 mL) was added PPh3 (8.6 g, 33.1 mmol, 2.0 equiv.), imidazole (3.3 g, 49.7 mmol, 3.0 equiv.) and I2 (6.3 g, 24.8 mmol, 5.0 mL, 1.5 equiv.). The resulting mixture was stirred at 110°C for 2 h. The mixture was cooled to 25°C, washed with water (100 mL) and brine (100 mL). The organic layers were dried over anhydrous sodium sulfate and concentrated
under reduced pressure. The residue was purified by silica gel chromatography (0~10% Ethyl acetate/Petroleum ether) to give 173a. 1H NMR (400 MHz, CHLOROFORM-d) δ 4.30 (s, 1H), 3.64 (s, 2H), 3.53 (s, 2H), 2.01 - 1.78 (m, 5H), 1.73 - 1.54 (m, 3H), 1.44 (s, 9H). Step B – Synthesis of tert-butyl 7-(3-(4-(tert-butoxycarbonyl)-1H-pyrazol-1-yl)-5-fluoro-2- methylphenyl)-2-azaspiro[3.5]nonane-2-carboxylate (173b) A reaction vial equipped with a stir bar was charged with DME (14.0 mL), tert-butyl 1-(3- bromo-5-fluoro-2-methylphenyl)-1H-pyrazole-4-carboxylate (500.0 mg, 1.4 mmol, 1.0 equiv.), tert-butyl 7-iodo-2-azaspiro[3.5]nonane-2-carboxylate (642.7 mg, 1.8 mmol, 1.3 equiv.), Ir[dF(CF3)ppy]2(dtbpy)(PF6) (15.7 mg, 14.0 μmol, 0.01 equiv.), NiCl2·dtbbpy (21.1 μmol, 0.015 equiv.), TTMSS (350.0 mg, 1.4 mmol, 434.2 μL, 1.0 equiv.), and Na2CO3 (298.3 mg, 2.8 mmol, 2.0 equiv.) under an atmosphere of nitrogen. The reaction was stirred and irradiated with a 10 W blue LED lamp (3 cm distance from reaction vessel), with cooling water to maintain the reaction temperature at 25 °C for 14 h. The mixture was concentrated under reduced pressure. The residue was purified by silica gel chromatography (0~15% Ethyl acetate/Petroleum ether) to give 173b. LCMS [M-tBu+1] = 444.2. Step C – Synthesis of tert-butyl 1-(5-fluoro-2-methyl-3-(2-azaspiro[3.5]nonan-7-yl)phenyl)-1H- pyrazole-4-carboxylate (173c) To a solution of tert-butyl 7-(3-(4-(tert-butoxycarbonyl)-1H-pyrazol-1-yl)-5-fluoro-2- methylphenyl)-2-azaspiro[3.5]nonane-2-carboxylate (200.0 mg, 400.3 μmol, 1.0 equiv.) in t- BuOH (1.5 mL) was added aqueous HCl (12 M, 0.5 mL) and the mixture was stirred at 25°C for 4 h. The mixture pH was adjusted to 7 to 8 with NaHCO3 solution and extracted with ethyl acetate (10 mLx3). The combined organic layers were washed with brine (20 mL) and dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure to give 173c, which was used without purification. LCMS [M+1] = 400.3. Step D – Synthesis of tert-butyl 1-(3-(2-(5-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3- methylpyridin-2-yl)-2-azaspiro[3.5]nonan-7-yl)-5-fluoro-2-methylphenyl)-1H-pyrazole-4- carboxylate (173d)
To a solution of tert-butyl 1-(5-fluoro-2-methyl-3-(2-azaspiro[3.5]nonan-7-yl)phenyl)-1H- pyrazole-4-carboxylate (130.0 mg, 325.4 μmol, 1.0 equiv.) in DMSO (1.0 mL) was added 1-1-(6- fluoro-5-methylpyridin-3-yl)dihydropyrimidine-2,4(1H,3H)-dione (72.6 mg, 325.4 μmol, 1.0 equiv.) and DIEA (126.1 mg, 976.2 μmol, 170.0 μL, 3.0 equiv.), the mixture was stirred at 100°C for 8 h. The mixture was filtered and the filtrate purified by prep-HPLC (0.1% FA condition) to give 173d. LCMS [M+1] = 603.3. Step E – Synthesis of 1-(3-(2-(5-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-methylpyridin-2-yl)- 2-azaspiro[3.5]nonan-7-yl)-5-fluoro-2-methylphenyl)-1H-pyrazole-4-carboxylic acid trifluoroacetic acid salt (173e) A solution of tert-butyl 1-(3-(2-(5-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3- methylpyridin-2-yl)-2-azaspiro[3.5]nonan-7-yl)-5-fluoro-2-methylphenyl)-1H-pyrazole-4- carboxylate (50.0 mg, 82.9 μmol, 1.0 equiv.) in dichloromethane (0.6 mL) was added TFA (307.0 mg, 2.6 mmol, 0.2 mL, 32.4 equiv.), the mixture was stirred at 25°C for 3 h. The mixture was concentrated under reduced pressure. The residue was triturated with MTBE (5.0 mL) at 25°C for 5 min to afford 173e. LCMS [M+1] = 547.2. Step F – Synthesis of 1-(3-(2-(5-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-methylpyridin-2-yl)- 2-azaspiro[3.5]nonan-7-yl)-5-fluoro-2-methylphenyl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2- yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide (173) A solution of 1-(3-(2-(5-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-methylpyridin-2-yl)- 2-azaspiro[3.5]nonan-7-yl)-5-fluoro-2-methylphenyl)-1H-pyrazole-4-carboxylic acid trifluoroacetate salt (40.0 mg, 73.1 μmol, 1.0 equiv.) and (3-(1,1,1-trifluoro-2-methylpropan-2- yl)-1H-1,2,4-triazol-5-yl)methanamine methane sulfonic acid salt (58.5 mg, 146.3 μmol, 2.0 equiv) in DMF (0.5 mL) was added NMI (30.0 mg, 365.9 μmol, 29.1 μL, 5.0 equiv.). The mixture was stirred at 25°C for 1 h, treated with TCFH (30.8 mg, 109.7 μmol, 1.5 equiv.) and stirred at 25°C for 5 min. The mixture was filtered and the filtrate was purified by prep-HPLC (XPT PHS C18150x25mm, 10 µm; [water (FA)-ACN]; 16%-46% B over 10 min) to give 173. LCMS [M+1] = 737.2; 1H NMR (400 MHz, METHANOL-d4) δ 8.29 (s, 1H), 8.16 (s, 1H), 7.89 (d, J = 2.4 Hz, 1H), 7.46 (s, 1H), 7.24 (dd, J = 2.8, 10.2 Hz, 1H), 7.03 (dd, J = 2.8, 8.4 Hz, 1H), 4.69 (s, 2H), 4.11 (s, 2H), 3.98 - 3.93 (m, 2H), 3.80 (t, J = 6.8 Hz, 2H), 2.96 - 2.87 (m, 1H), 2.82
(t, J = 6.8 Hz, 2H), 2.32 (s, 3H), 2.16 (d, J = 12.8 Hz, 2H), 2.09 (s, 3H), 1.87 - 1.73 (m, 4H), 1.61 (s, 6H), 1.59 - 1.53 (m, 2H). Step G – Synthesis of tert-butyl 1- (3-bromo-5-fluoro-2-methyl-phenyl) pyrazole-4-carboxylate (173g) To a solution of tert-butyl 1H-pyrazole-4-carboxylate (10 g, 59.46 mmol, 1.0 equiv.), 1,3- dibromo-5-fluoro-2-methyl-benzene (14.34 g, 53.51 mmol, 0.9 equiv.) in DMSO (100 mL) was added CuI (5.66 g, 29.73 mmol, 0.5 equiv.), (1S,2S) -N1, N2-dimethylcyclohexane-1,2-diamine (8.46 g, 59.46 mmol, 1 equiv.) and K3PO4 (25.24 g, 118.91 mmol, 2 equiv.). The mixture was stirred at 60°C for 1 h under a nitrogen atmosphere. The mixture was diluted with water (200 mL) and extracted with ethyl acetate (150 mLx3). The combined organic portions were washed with brine (100mL x2), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, Petroleum ether/Ethyl acetate = 1/0 to 20/1) to give 173g. LCMS [M+1] = 354.9, 356.9; 1H NMR (400 MHz, CHLOROFORM-d) δ 8.05 (s, 1H), 8.00 (s, 1H), 7.46 (dd, J = 2.4, 7.6 Hz, 1H), 7.08 (dd, J = 2.4, 8.4 Hz, 1H), 2.24 (s, 3H), 1.58 (s, 9H). Example 174: Synthesis of Compound 174
To a solution of 1-iodo-4-methylbenzene (5.0 g, 22.9 mmol, 1.0 equiv.) in DMA (300.0 mL) was added NiBr2∙dtbpy (2.2 g, 4.5 mmol, 0.2 equiv.), 1-(1,3-dioxoisoindolin-2-yl) 3-methyl bicyclo[1.1.1]pentane-1,3-dicarboxylate ((10.8 g, 34.4 mmol, 1.5 equiv.), NaHCO3 (7.7 g, 91.7 mmol, 3.5 mL, 4.0 equiv.) and diethyl 2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate (11.6 g, 45.8 mmol, 2.0 equiv.). The vial was flushed with nitrogen and sealed. The reaction was stirred and irradiated with a blue LED lamp (10 W, 395 nM, 3 cm distance from the reaction flask), with cooling water to keep the reaction temperature at 25°C for 14 h. The mixture was poured into H2O (800 mL). The aqueous phase was extracted with ethyl acetate (300 mLx3). The combined organic portion was washed with brine (400 mLx3), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was
purified by column chromatography (silica gel, Petroleum ether/Ethyl acetate=1/0 to 1/1) to give 174a. LCMS [M+1] = 217.1 Step B – Synthesis of (3-(p-tolyl)bicyclo[1.1.1]pentan-1-yl)methanol (174b) A solution of methyl 3-(p-tolyl)bicyclo[1.1.1]pentane-1-carboxylate (1.8 g, 8.5 mmol, 1.0 equiv.) in THF (20.0 mL) was treated with LiAlH4 (2.5 M in THF, 10.2 mL, 1.5 equiv.) under a nitrogen atmosphere at 0°C and then stirred for 1 h. The mixture was treated with water (10.2 mL) and 15% NaOH aqueous solution (10.2 mL) and stirred for 30 min. The mixture was filtered and the filtrate was concentrated under reduced pressure. The residue was diluted with water (30 mL) and extracted with ethyl acetate (30 mLx3). The combined organic layers were washed with brine (50 mL), dried, filtered the filtrate was concentrated under reduced pressure to afford 174b. 1H NMR (400 MHz, CHLOROFORM-d) δ 7.15 - 7.11 (m, 4H), 2.33 (s, 3H), 1.98 (s, 6H). Step C – Synthesis of 3-(p-tolyl)bicyclo[1.1.1]pentane-1-carbaldehyde (174c) To a solution of (3-(p-tolyl)bicyclo[1.1.1]pentan-1-yl)methanol (1.2 g, 6.6 mmol, 1.0 equiv.) in dichloromethane (15.0 mL) was added PCC (2.1 g, 10.0 mmol, 1.5 equiv.). The mixture was stirred at 25°C for 2 h. The mixture was poured into petroleum ether (20 mL) and filtered and the filtrate was concentrated under reduced pressure to give 174c. Step D – Synthesis of 3-(p-tolyl)bicyclo[1.1.1]pentane-1-carbaldehyde oxime (174d) To a solution of 3-(p-tolyl)bicyclo[1.1.1]pentane-1-carbaldehyde (1.2 g, 6.7 mmol, 1.0 equiv.) in MeOH (12.5 mL) and H2O (12.5 mL) was added NaHCO3 (338.3 mg, 4.0 mmol, 156.6 μL, 0.6 equiv.) and hydroxylamine (932.7 mg, 13.4 mmol, 2.0 equiv., HCl). The mixture was stirred at 25°C for 1 h. The mixture was poured into water (40 mL) and extracted with ethyl acetate (20 mL x 3). The combined organic portion was washed with brine (50 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure to afford 174d. LCMS [M+1] = 202.1 Step E – Synthesis of tert-butyl 3-(3-(p-tolyl)bicyclo[1.1.1]pentan-1-yl)isoxazole-5-carboxylate (174e)
To a solution of 3-(p-tolyl)bicyclo[1.1.1]pentane-1-carbaldehyde oxime (1.1 g, 5.4 mmol, 1.0 equiv.) and tert-butyl propiolate (1.7 g, 13.6 mmol, 1.8 mL, 2.5 equiv.) in water (22.0 mL) was added KCl (407.4 mg, 5.4 mmol, 1.0 equiv.) and oxone (1.2 g, 8.2 mmol, 1.5 equiv.). The mixture was stirred at 25°C for 2 h. The aqueous was extracted with ethyl acetate (10 mL x 3). The combined organic phase was washed with brine (30 mL), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by prep-HPLC (Phenomenex Luna C18 (250x70mm,10 µm);mobile phase: [water(FA)- ACN];gradient:55%-85% B over 20 min) to give 174e. LCMS [M-55, M+1] = 270.1, 326.1. Step F – Synthesis of tert-butyl 3-(3-(4-(bromomethyl)phenyl)bicyclo[1.1.1]pentan-1- yl)isoxazole-5-carboxylate (174f) To a solution of tert-butyl 3-(3-(p-tolyl)bicyclo[1.1.1]pentan-1-yl)isoxazole-5-carboxylate (500.0 mg, 1.5 mmol, 1.0 equiv.) in CCl4 (17.5 mL) was added AIBN (25.2 mg, 153.6 μmol, 0.1 equiv.) and NBS (273.4 mg, 1.5 mmol, 1.0 equiv.). The mixture was stirred at 90°C for 1 h, treated with water (50 mL), and extracted with dichloromethane (30 mLx3). The combined organic portions were washed with brine (50 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure to give 174f. LCMS [M+1] = 404.0, 406.0. Step G– Synthesis of tert-butyl 3-(3-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3- fluorophenyl)-2,6-diazaspiro[3.3]heptan-2-yl)methyl)phenyl)bicyclo[1.1.1]pentan-1-yl)isoxazole- 5-carboxylate (174g) To a solution of tert-butyl 3-(3-(4-(bromomethyl)phenyl)bicyclo[1.1.1]pentan-1- yl)isoxazole-5-carboxylate (500.0 mg, 1.2 mmol, 1.0 equiv.) and 1-(2-fluoro-4-(2,6- diazaspiro[3.3]heptan-2-yl)phenyl)dihydropyrimidine-2,4(1H,3H)-dione trifluoroacetate (517.3 mg, 1.2 mmol, 1.0 equiv.) in DMF (10.0 mL) was added DIEA (479.5 mg, 3.7 mmol, 646.2 μL, 3.0 equiv.). The mixture was stirred at 80°C for 1 h, cooled to 25°C and poured into water (5 mL). The aqueous phase was extracted with ethyl acetate (5 mLx3). The combined organic phase was washed with brine (5 mLx3), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by column
chromatography (silica gel, Ethyl acetate: Methanol=1/0 to 10/1) to afford 174g. LCMS [M+1] = 628.3. Step H– Synthesis of 3-(3-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2,6- diazaspiro[3.3]heptan-2-yl)methyl)phenyl)bicyclo[1.1.1]pentan-1-yl)isoxazole-5-carboxylic acid trifluoroacetate salt (174h) To a solution of tert-butyl 3-(3-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3- fluorophenyl)-2,6-diazaspiro[3.3]heptan-2-yl)methyl)phenyl)bicyclo[1.1.1]pentan-1-yl)isoxazole- 5-carboxylate (300.0 mg, 477.9 μmol, 1.0 equiv.) in dichloromethane (3.0 mL) was added TFA (1.5 g, 13.4 mmol, 1.0 mL, 28.1 equiv.). The mixture was stirred at 25°C for 2 h. The mixture was concentrated under reduced pressure. The residue was triturated with MTBE (5 mL) at 25oC for 5 min, then filtered to afford 174h. LCMS [M+1] = 572.2. Step I– Synthesis of 3-(3-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2,6- diazaspiro[3.3]heptan-2-yl)methyl)phenyl)bicyclo[1.1.1]pentan-1-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (174) To a solution of 3-(3-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)- 2,6-diazaspiro[3.3]heptan-2-yl)methyl)phenyl)bicyclo[1.1.1]pentan-1-yl)isoxazole-5-carboxylic acid trifluoroacetate salt (220.0 mg, 384.8 μmol, 1.0 equiv) and (3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methanamine hydrochloride (216.4 mg, 769.7 μmol, 2.0 equiv) in DMF (3.0 mL) was added NMI (158.0 mg, 1.9 mmol, 153.4 μL, 5.0 equiv.) and TCFH (161.9 mg, 577.3 μmol, 1.5 equiv.). The mixture was stirred at 25°C for 30 min. The mixture was poured into H2O (10 mL). The aqueous phase was extracted with ethyl acetate (5 mLx3). The combined organic portion was washed with brine (5 mLx3), dried with anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by prep-HPLC (Phenomenex Luna C18150x25mm, 10 µm; [water(FA)- ACN];gradient:14%-44% B over 10 min) to give 174. LCMS [M+1] = 762.4; 1H NMR (400 MHz, DMSO-d6) δ 13.92 (s, 1H), 10.34 (s, 1H), 9.51 (t, J = 4.4 Hz, 1H), 8.14 (s, 1H), 7.28 - 7.24 (m, 4H), 7.16 (t, J = 8.8 Hz, 1H), 7.12 (s, 1H), 6.32 (dd, J = 2.4, 12.8 Hz, 1H), 6.24 (dd, J = 2.4, 8.8 Hz, 1H), 4.54 (d, J = 6.0 Hz, 2H), 3.91 (s, 4H), 3.72 - 3.62 (m, 2H), 3.58 (t, J = 6.8 Hz, 2H), 3.52 - 3.42 (m, 4H), 2.67 (t, J = 6.8 Hz, 2H), 2.39 (s, 6H), 1.53 (s, 6H).
Example 175: Synthesis of Compound 175 Step
 yl)methyl)carbamate (175a) A solution of 2-methyl-2-methylsulfanyl-propanoic acid (900.0 mg, 6.7 mmol, 1.0 equiv.) in DMF (4.5 mL) was added CDI (1.1 g, 6.7 mmol, 1.0 equiv.). The mixture was stirred at 20°C for 1 h. The tert-butyl N-(2-amino-2-imino-ethyl)carbamate hydrochloride (1.4 g, 6.7 mmol, 1.0 equiv) and triethylamine (3.7 mL, 4.0 equiv.) in DMF (4.5 mL) was added to the mixture at 0°C. The reaction was stirred at 0°C for 15 min. The mixture was used without purification. LCMS [M+1] = 290.1. To a solution of tert-butyl (2-amino-2-((2-methyl-2- (methylthio)propanoyl)imino)ethyl)carbamate (1.9 g, 6.5 mmol, 1.0 equiv.) in acetic acid (3.9 g, 65.6 mmol, 3.7 mL, 10.0 equiv. ) was added hydrazine hydrate (328.6 mg, 6.5 mmol, 318.4 μL, 1.0 equiv.). The mixture was stirred at 0°C for 15 min and then poured into saturated aqueous NaHCO3 (20 mL) and extracted with EtOAc (4 × 20 mL). The combined organic portion was washed with brine (20 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated. The residue was purified by column chromatography (silica gel, petroleum ether/ethyl acetate=1/0 to 1/1) to afford 175a. LCMS [M+1] = 287.1.
Step B – Synthesis of (3-(2-(methylthio)propan-2-yl)-1H-1,2,4-triazol-5-yl)methanamine hydrochloride (175b) A mixture of tert-butyl ((3-(2-(methylthio)propan-2-yl)-1H-1,2,4-triazol-5- yl)methyl)carbamate (700.0 mg, 2.4 mmol, 1.0 equiv.) in HCl (2 M in EtOAc, 14.0 mL, 11.4 equiv.) was stirred at 20°C for 30 min. The mixture was concentrated and triturated with MTBE (20.0 mL) at 0oC for 30 min. The solids were collected by vacuum filtration to afford 175b. LCMS [M+1] = 187.1. Step C – Synthesis of 3-(5-(4-(1-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)- 2-azaspiro[3.3]heptan-2-yl)ethyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(2- (methylthio)propan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (175-1 and 175- 2) A solution of 155c-1 or 155c-2 (200.0 mg) and (3-(2-(methylthio)propan-2-yl)-1H-1,2,4- triazol-5-yl)methanamine (120.4 mg) in DMSO (3.0 mL) was added triethylamine (221 μL) and BOP (281 mg). The mixture was stirred at 20°C for 30 min. The mixture was filtered and the filtrate was purified by prep-HPLC (Welch Ultimate C18150 x 25mm, 7µm; [water(FA)- ACN];gradient:1%-31% B over 15 min) to afford 175-1 or 175-2. LCMS [M+1] = 797.2; 1H NMR (400 MHz, METHANOL-d4) δ = 8.75 (s, 1H), 8.50 - 8.43 (m, 1H), 7.68 (s, 1H), 7.55 - 7.49 (m, 1H), 7.45 - 7.37 (m, 2H), 7.34 (t, J = 8.0 Hz, 1H), 7.15 - 7.09 (m, 2H), 4.72 (s, 2H), 3.93 - 3.85 (m, 1H), 3.79 (t, J = 6.8 Hz, 3H), 3.73 - 3.66 (m, 1H), 3.61 (d, J = 9.2 Hz, 1H), 3.54 - 3.45 (m, 2H), 2.83 (t, J = 6.8 Hz, 2H), 2.71 - 2.62 (m, 2H), 2.56 (s, 3H), 2.40 - 2.31 (m, 2H), 1.95 (s, 3H), 1.74 (s, 6H), 1.44 (d, J = 6.4 Hz, 3H). Example 176: Synthesis of Compound 176-1 and 176-2
yl)methyl)carbamate (175a) A solution of 2-methyl-2-methylsulfanyl-propanoic acid (900.0 mg, 6.7 mmol, 1.0 equiv.) in DMF (4.5 mL) was added CDI (1.1 g, 6.7 mmol, 1.0 equiv.). The mixture was stirred at 20°C for 1 h. The tert-butyl N-(2-amino-2-imino-ethyl)carbamate hydrochloride (1.4 g, 6.7 mmol, 1.0 equiv) and triethylamine (3.7 mL, 4.0 equiv.) in DMF (4.5 mL) was added to the mixture at 0°C. The reaction was stirred at 0°C for 15 min. The mixture was used without purification. LCMS [M+1] = 290.1. To a solution of tert-butyl (2-amino-2-((2-methyl-2- (methylthio)propanoyl)imino)ethyl)carbamate (1.9 g, 6.5 mmol, 1.0 equiv.) in acetic acid (3.9 g, 65.6 mmol, 3.7 mL, 10.0 equiv. ) was added hydrazine hydrate (328.6 mg, 6.5 mmol, 318.4 μL, 1.0 equiv.). The mixture was stirred at 0°C for 15 min and then poured into saturated aqueous NaHCO3 (20 mL) and extracted with EtOAc (4 × 20 mL). The combined organic portion was washed with brine (20 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated. The residue was purified by column chromatography (silica gel, petroleum ether/ethyl acetate=1/0 to 1/1) to afford 175a. LCMS [M+1] = 287.1.
Step B – Synthesis of (3-(2-(methylthio)propan-2-yl)-1H-1,2,4-triazol-5-yl)methanamine hydrochloride (175b) A mixture of tert-butyl ((3-(2-(methylthio)propan-2-yl)-1H-1,2,4-triazol-5- yl)methyl)carbamate (700.0 mg, 2.4 mmol, 1.0 equiv.) in HCl (2 M in EtOAc, 14.0 mL, 11.4 equiv.) was stirred at 20°C for 30 min. The mixture was concentrated and triturated with MTBE (20.0 mL) at 0oC for 30 min. The solids were collected by vacuum filtration to afford 175b. LCMS [M+1] = 187.1. Step C – Synthesis of 3-(5-(4-(1-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)- 2-azaspiro[3.3]heptan-2-yl)ethyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(2- (methylthio)propan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (175-1 and 175- 2) A solution of 155c-1 or 155c-2 (200.0 mg) and (3-(2-(methylthio)propan-2-yl)-1H-1,2,4- triazol-5-yl)methanamine (120.4 mg) in DMSO (3.0 mL) was added triethylamine (221 μL) and BOP (281 mg). The mixture was stirred at 20°C for 30 min. The mixture was filtered and the filtrate was purified by prep-HPLC (Welch Ultimate C18150 x 25mm, 7µm; [water(FA)- ACN];gradient:1%-31% B over 15 min) to afford 175-1 or 175-2. LCMS [M+1] = 797.2; 1H NMR (400 MHz, METHANOL-d4) δ = 8.75 (s, 1H), 8.50 - 8.43 (m, 1H), 7.68 (s, 1H), 7.55 - 7.49 (m, 1H), 7.45 - 7.37 (m, 2H), 7.34 (t, J = 8.0 Hz, 1H), 7.15 - 7.09 (m, 2H), 4.72 (s, 2H), 3.93 - 3.85 (m, 1H), 3.79 (t, J = 6.8 Hz, 3H), 3.73 - 3.66 (m, 1H), 3.61 (d, J = 9.2 Hz, 1H), 3.54 - 3.45 (m, 2H), 2.83 (t, J = 6.8 Hz, 2H), 2.71 - 2.62 (m, 2H), 2.56 (s, 3H), 2.40 - 2.31 (m, 2H), 1.95 (s, 3H), 1.74 (s, 6H), 1.44 (d, J = 6.4 Hz, 3H). Example 176: Synthesis of Compound 176-1 and 176-2
Step 2,6- diazaspiro[3.3]heptane-2-carboxylate (176a) To a solution of 3-(4-bromo-2-fluorophenyl)-3-methylpiperidine-2,6-dione (300.0 mg, 999.5 μmol, 1.0 equiv.) and tert-butyl 2,6-diazaspiro[3.3]heptane-2-carboxylate (297.2 mg, 1.5 mmol, 1.5 equiv.) in DMSO (3.0 mL) was added cesium carbonate (651.3 mg, 2.0 mmol, 2.0 equiv.) and RuPhosPdG3 (83.6 mg, 99.9 μmol, 0.1 equiv.). The mixture was stirred at 90°C for 1 h. The mixture was cooled to 25 °C and diluted with water (5 mL) and extracted with EtOAc (5 mLx3). The combined organic layers were washed with brine (10 mL), dried over anhydrous sodium sulfate, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by chromatography column on silica gel (PE/EA=5/1 to 1/1) to give 176a. LCMS [M+1] = 418.2. 1H NMR (400 MHz, DMSO-d6) δ 10.80 (s, 1H), 7.08 (t, J = 8.0 Hz, 1H), 6.29 - 6.16 (m, 2H), 4.02 (s, 4H), 3.92 (s, 4H), 2.70 - 2.58 (m, 1H), 2.36 - 2.16 (m, 2H), 1.84 - 1.73 (m, 1H), 1.51 (s, 3H), 1.37 (s, 9H). Step B – Synthesis of 3-(2-fluoro-4-(2,6-diazaspiro[3.3]heptan-2-yl)phenyl)-3-methylpiperidine- 2,6-dione trifluoroacetate salt (176b)
A mixture of tert-butyl 6-(3-fluoro-4-(3-methyl-2,6-dioxopiperidin-3-yl)phenyl)-2,6- diazaspiro[3.3]heptane-2-carboxylate (250.0 mg, 599 μmol, 1.0 equiv.) in TFA (1.0 mL) and dichloromethane (3.0 mL) was stirred at 25°C for 1 h. The mixture was concentrated under pressure to give 176b, which was used without purification. LCMS [M+1] = 318.1. Step C – Synthesis of 3-(5-(2-fluoro-4-((6-(3-fluoro-4-(3-methyl-2,6-dioxopiperidin-3-yl)phenyl)- 2,6-diazaspiro[3.3]heptan-2-yl)methyl)phenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (176-1 and 176-2) To a solution of 3-(2-fluoro-4-(2,6-diazaspiro[3.3]heptan-2-yl)phenyl)-3- methylpiperidine-2,6-dione trifluoroacetate salt (220.0 mg, 693.2 μmol, 1.2 equiv) and 3-(5-(2- fluoro-4-formylphenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)isoxazole-5-carboxamide (290.8 mg, 577.6 μmol, 1.0 equiv.) in DMSO (2.0 mL) was treated with triethylamine (161 μL, 2.0 equiv.), stirred for 30 min and treated with NaBH(OAc)3 (367.3 mg, 1.7 mmol, 3.0 equiv.). The mixture was stirred at 25°C for 1 h. The mixture was purified with prep-HPLC (Waters Xbridge BEH C18150x25mm, 10µm; [water( NH4HCO3)-ACN];gradient:30%-60% B over 10 min) to afford the product as the racemate. LCMS [M+1] =805.6. The stereoisomers were separated by SFC (DAICEL CHIRALPAK IK (250x30mm,10um); [CO2-isopropanol/ACN];B%:70%, isocratic elution) to afford 176-1 as the early eluting isomer and 176-2 as the late eluting isomer. LCMS [M+1] = 805.5; 1H NMR (400 MHz, DMSO-d6) δ 13.99 (s, 1H), 10.79 (s, 1H), 9.73 (s, 1H), 9.23 (s, 2H), 7.79 - 7.69 (m, 2H), 7.39 - 7.25 (m, 2H), 7.07 (t, J = 8.0 Hz, 1H), 6.30 - 6.13 (m, 2H), 4.61 (d, J = 4.0 Hz, 2H), 3.91 (s, 4H), 3.65 (s, 2H), 3.37 (s, 4H), 2.69 - 2.59 (m, 1H), 2.30 - 2.18 (m, 2H), 1.83 - 1.72 (m, 1H), 1.56 - 1.50 (m, 9H) The following examples were prepared as either the free form or the formic acid salt using the methods outlined above. Example 177: Synthesis of Compound 177
F F O F N N N N H N NH O 3-(5-(4-((4-(4- piperazin-1-
 yl)methyl)piperidin-1- - 2-yl)-1H-1,2,4- triazol-5-yl)methyl)isoxazole-5-carboxamide (177) was prepared according to methods described above. LCMS [M+1] = 787.2; 1H NMR (400 MHz, METHANOL-d4) δ 8.57 (s, 2H), 8.29 (s, 1H), 7.52 (s, 1H), 6.68 (d, J = 11.6 Hz, 2H), 4.74 (s, 2H), 4.04 (d, J = 12.8 Hz, 2H), 3.74 (t, J = 6.8 Hz, 2H), 3.36 (s, 4H), 3.00 (t, J = 12.8 Hz, 2H), 2.83 (t, J = 6.8 Hz, 2H), 2.80 - 2.74 (m, 4H), 2.49 (d, J = 6.4 Hz, 2H), 2.03 - 1.94 (m, 3H), 1.63 (s, 6H), 1.46 - 1.33 (m, 2H). Example 178: Synthesis of Compound 178 3-(5-(5-((6-
yl)methyl)piperidin-1- - 2-yl)-1H-1,2,4- triazol-5-yl)methyl)isoxazole-5-carboxamide (177) was prepared according to methods described above. LCMS [M+1] = 787.2; 1H NMR (400 MHz, METHANOL-d4) δ 8.57 (s, 2H), 8.29 (s, 1H), 7.52 (s, 1H), 6.68 (d, J = 11.6 Hz, 2H), 4.74 (s, 2H), 4.04 (d, J = 12.8 Hz, 2H), 3.74 (t, J = 6.8 Hz, 2H), 3.36 (s, 4H), 3.00 (t, J = 12.8 Hz, 2H), 2.83 (t, J = 6.8 Hz, 2H), 2.80 - 2.74 (m, 4H), 2.49 (d, J = 6.4 Hz, 2H), 2.03 - 1.94 (m, 3H), 1.63 (s, 6H), 1.46 - 1.33 (m, 2H). Example 178: Synthesis of Compound 178 3-(5-(5-((6-
 -2- azaspiro[3.3]heptan-2-yl)methyl)-3-fluoropyridin-2-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (178) was prepared according to methods described above. LCMS [M+1] = 792.5; 1H NMR (400 MHz, DMSO-d6) δ 14.00 (m, 1H), 10.46 (s, 1H), 9.75 (s, 1H), 9.49 (s, 2H), 8.58 (s, 1H), 8.14 (s, 1H), 7.87 (d, J = 11.6 Hz, 1H), 7.75 (s, 1H), 7.33 (t, J = 8.4 Hz, 1H), 7.15 (d, J = 11.6 Hz, 1H), 7.08 (d, J = 8.4 Hz, 1H), 4.61 (d, J = 5.2 Hz, 2H), 3.76 - 3.63 (m, 4H), 3.47 - 3.36 (m, 5H), 3.19 (s, 2H), 2.70 (t, J = 6.8 Hz, 2H), 2.25 - 2.15 (m, 2H), 1.54 (s, 7H). Examples 179-232 were prepared in an analagous fashion to the previous examples.
Example 179: Synthesis of Compound 179 3-(5-(4-((6- -2-
-2- azaspiro[3.3]heptan-2-yl)methyl)-3-fluoropyridin-2-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (178) was prepared according to methods described above. LCMS [M+1] = 792.5; 1H NMR (400 MHz, DMSO-d6) δ 14.00 (m, 1H), 10.46 (s, 1H), 9.75 (s, 1H), 9.49 (s, 2H), 8.58 (s, 1H), 8.14 (s, 1H), 7.87 (d, J = 11.6 Hz, 1H), 7.75 (s, 1H), 7.33 (t, J = 8.4 Hz, 1H), 7.15 (d, J = 11.6 Hz, 1H), 7.08 (d, J = 8.4 Hz, 1H), 4.61 (d, J = 5.2 Hz, 2H), 3.76 - 3.63 (m, 4H), 3.47 - 3.36 (m, 5H), 3.19 (s, 2H), 2.70 (t, J = 6.8 Hz, 2H), 2.25 - 2.15 (m, 2H), 1.54 (s, 7H). Examples 179-232 were prepared in an analagous fashion to the previous examples.
Example 179: Synthesis of Compound 179 3-(5-(4-((6- -2-
 azaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (179) was prepared according to methods described above. LCMS [M+1] = 809.4; 1H NMR (400 MHz, DMSO-d6) δ 13.99 (s, 1H), 10.53 (s, 1H), 9.73 (t, J = 5.2 Hz, 1H), 9.24 (d, J = 1.2 Hz, 2H), 8.14 (s, 1H), 7.79 - 7.70 (m, 2H), 7.40 - 7.32 (m, 2H), 7.26 - 7.15 (m, 2H), 4.61 (d, J = 5.6 Hz, 2H), 3.77 - 3.69 (m, 4H), 3.63 - 3.56 (m, 1H), 3.49 (s, 2H), 3.26 (s, 2H), 2.72 (t, J = 6.4 Hz, 2H), 2.59 - 2.53 (m, 2H), 2.35 - 2.26 (m, 2H), 1.54 (s, 6H). Example 180: Synthesis of Compound 180 3-(5-(4-((6-(4-
azaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (179) was prepared according to methods described above. LCMS [M+1] = 809.4; 1H NMR (400 MHz, DMSO-d6) δ 13.99 (s, 1H), 10.53 (s, 1H), 9.73 (t, J = 5.2 Hz, 1H), 9.24 (d, J = 1.2 Hz, 2H), 8.14 (s, 1H), 7.79 - 7.70 (m, 2H), 7.40 - 7.32 (m, 2H), 7.26 - 7.15 (m, 2H), 4.61 (d, J = 5.6 Hz, 2H), 3.77 - 3.69 (m, 4H), 3.63 - 3.56 (m, 1H), 3.49 (s, 2H), 3.26 (s, 2H), 2.72 (t, J = 6.4 Hz, 2H), 2.59 - 2.53 (m, 2H), 2.35 - 2.26 (m, 2H), 1.54 (s, 6H). Example 180: Synthesis of Compound 180 3-(5-(4-((6-(4-
 -2- azaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (180) was prepared according to methods described above. LCMS [M+1] = 809.5; 1H NMR (400 MHz, DMSO-d6) δ
13.98 (s, 1H), 10.51 (s, 1H), 9.73 (d, J = 2.0 Hz, 1H), 9.24 (d, J = 1.2 Hz, 2H), 7.77 - 7.70 (m, 2H), 7.35 - 7.26 (m, 4H), 4.61 (s, 2H), 3.69 (t, J = 6.4 Hz, 2H), 3.63 (s, 2H), 3.53 (t, J = 8.8 Hz, 1H), 3.35 (s, 4H), 3.13 (s, 2H), 2.70 (t, J = 6.8 Hz, 2H), 2.32 - 2.23 (m, 2H), 1.54 (s, 6H). Example 181: Synthesis of Compound 181 3-(5-(4-((6-
-2- azaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (180) was prepared according to methods described above. LCMS [M+1] = 809.5; 1H NMR (400 MHz, DMSO-d6) δ
13.98 (s, 1H), 10.51 (s, 1H), 9.73 (d, J = 2.0 Hz, 1H), 9.24 (d, J = 1.2 Hz, 2H), 7.77 - 7.70 (m, 2H), 7.35 - 7.26 (m, 4H), 4.61 (s, 2H), 3.69 (t, J = 6.4 Hz, 2H), 3.63 (s, 2H), 3.53 (t, J = 8.8 Hz, 1H), 3.35 (s, 4H), 3.13 (s, 2H), 2.70 (t, J = 6.8 Hz, 2H), 2.32 - 2.23 (m, 2H), 1.54 (s, 6H). Example 181: Synthesis of Compound 181 3-(5-(4-((6-
 methylphenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (181) was prepared according to methods described above. LCMS [M+1] = 805.5; 1H NMR (400 MHz, DMSO-d6) δ = 14.02 (br s, 1H), 10.36 (s, 1H), 9.74 (t, J = 8.0 Hz, 1H), 9.24 (s, 2H), 8.15 (s, 1H), 7.77 (t, J = 8.0 Hz, 1H), 7.72 (s, 1H), 7.36 (t, J = 8.0 Hz, 2H), 7.20 (d, J = 8.4 Hz, 1H), 7.11 (d, J = 11.2 Hz, 1H), 4.61 (d, J = 6.0 Hz, 2H), 3.78 - 3.71 (m, 3H), 3.53 - 3.46 (m, 4H), 3.31 (s, 2H), 2.80 - 2.62 (m, 2H), 2.57 - 2.53 (m, 2H), 2.29 (t, J = 9.6 Hz, 2H), 2.14 (s, 3H), 1.54 (s, 6H). Example 182: Synthesis of Compound 182
methylphenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (181) was prepared according to methods described above. LCMS [M+1] = 805.5; 1H NMR (400 MHz, DMSO-d6) δ = 14.02 (br s, 1H), 10.36 (s, 1H), 9.74 (t, J = 8.0 Hz, 1H), 9.24 (s, 2H), 8.15 (s, 1H), 7.77 (t, J = 8.0 Hz, 1H), 7.72 (s, 1H), 7.36 (t, J = 8.0 Hz, 2H), 7.20 (d, J = 8.4 Hz, 1H), 7.11 (d, J = 11.2 Hz, 1H), 4.61 (d, J = 6.0 Hz, 2H), 3.78 - 3.71 (m, 3H), 3.53 - 3.46 (m, 4H), 3.31 (s, 2H), 2.80 - 2.62 (m, 2H), 2.57 - 2.53 (m, 2H), 2.29 (t, J = 9.6 Hz, 2H), 2.14 (s, 3H), 1.54 (s, 6H). Example 182: Synthesis of Compound 182
3-(5-(4-((6-(5-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluoro-6-methylpyridin-2-yl)-2- azaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (182) was prepared according to methods described above. LCMS [M+1] = 806.3; 1H NMR (400 MHz, METHANOL-d4) δ 9.18 (d, J = 0.4 Hz, 2H), 8.46 (br s, 1H), 7.86 - 7.72 (m, 1H), 7.68 (s, 1H), 7.50 (d, J = 9.6 Hz, 1H), 7.47 - 7.35 (m, 2H), 4.74 (s, 2H), 4.12 (br s, 2H), 3.94 (br s, 2H), 3.89 - 3.75 (m, 4H), 3.68 (m, 1H), 2.95 - 2.78 (m, 2H), 2.65 (br d, J = 8.0 Hz, 4H), 2.44 (s, 3H), 1.62 (s, 6H). Example 183: Synthesis of Compound 183 3-(5-(5-((6-
 -2- azaspiro[3.3]heptan-2-yl)methyl)pyridin-2-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (183) was prepared according to methods described above. LCMS [M +1] = 792.7; 1H NMR (400 MHz, DMSO-d6) δ 13.99 (br s, 1H), 10.59 (s, 1H), 9.73 (t, J = 5.4 Hz, 1H), 9.64 (s, 2H), 8.68 (d, J = 1.6 Hz, 1H), 8.20 (d, J = 8.4 Hz, 1H), 7.90 (dd, J = 2.0 Hz, 8.4 Hz, 1H), 7.74 (s, 1H), 7.09 (d, J = 9.2 Hz, 2H), 4.61 (d, J = 5.6 Hz, 2H), 3.67 - 3.64 (m, 4H), 3.44 - 3.39 (m, 1H), 3.36 (s, 2H), 3.16 (s, 2H), 2.70 (t, J = 6.8 Hz, 2H), 2.47 ( d, J = 2.8 Hz, 2H), 2.25 - 2.20 (m, 2H), 1.54 (s, 6H). Example 184: Synthesis of Compound 184
3-(5-(5-((4-( 5-a]pyrazin-6- yl)piperazin-1-yl)methyl)-3-fluoropyridin-2-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (184) was prepared according to methods described above. LCMS [M+1] = 804.0; 1H NMR (400 MHz, MeOH-d4) δ 9.56 (s, 2H), 8.88 (s, 1H), 8.67 (s, 1H), 7.94 (s, 1H), 7.91 (s, 1H), 7.88 (s, 2H), 7.72 (s, 1H), 4.77 (s, 2H), 3.99 (t, J = 6.8 Hz, 2H), 3.80 (s, 2H), 3.46 (d, J = 4.8 Hz, 4H), 2.91 (t, J = 6.8 Hz, 2H), 2.76 - 2.73 (m, 4H), 1.64 (s, 6H). Example 185: Synthesis of Compound 185 3-(5-(5-((6-
-2- azaspiro[3.3]heptan-2-yl)methyl)pyridin-2-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (183) was prepared according to methods described above. LCMS [M +1] = 792.7; 1H NMR (400 MHz, DMSO-d6) δ 13.99 (br s, 1H), 10.59 (s, 1H), 9.73 (t, J = 5.4 Hz, 1H), 9.64 (s, 2H), 8.68 (d, J = 1.6 Hz, 1H), 8.20 (d, J = 8.4 Hz, 1H), 7.90 (dd, J = 2.0 Hz, 8.4 Hz, 1H), 7.74 (s, 1H), 7.09 (d, J = 9.2 Hz, 2H), 4.61 (d, J = 5.6 Hz, 2H), 3.67 - 3.64 (m, 4H), 3.44 - 3.39 (m, 1H), 3.36 (s, 2H), 3.16 (s, 2H), 2.70 (t, J = 6.8 Hz, 2H), 2.47 ( d, J = 2.8 Hz, 2H), 2.25 - 2.20 (m, 2H), 1.54 (s, 6H). Example 184: Synthesis of Compound 184
3-(5-(5-((4-( 5-a]pyrazin-6- yl)piperazin-1-yl)methyl)-3-fluoropyridin-2-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (184) was prepared according to methods described above. LCMS [M+1] = 804.0; 1H NMR (400 MHz, MeOH-d4) δ 9.56 (s, 2H), 8.88 (s, 1H), 8.67 (s, 1H), 7.94 (s, 1H), 7.91 (s, 1H), 7.88 (s, 2H), 7.72 (s, 1H), 4.77 (s, 2H), 3.99 (t, J = 6.8 Hz, 2H), 3.80 (s, 2H), 3.46 (d, J = 4.8 Hz, 4H), 2.91 (t, J = 6.8 Hz, 2H), 2.76 - 2.73 (m, 4H), 1.64 (s, 6H). Example 185: Synthesis of Compound 185 3-(5-(5-((6-
 -2- azaspiro[3.3]heptan-2-yl)methyl)pyrazin-2-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (185) was prepared according to methods described above. LCMS [M+1] = 775.8; 1H NMR (400 MHz, DMSO-d6) δ 13.99 (br s, 1H), 10.45 (s, 1H), 9.74 (br s, 1H), 9.70 (s, 2H), 9.43 (s, 1H), 8.81 (s, 1H), 8.13 (s, 1H), 7.76 (s, 1H), 7.33 (t, J = 8.0 Hz, 1H), 7.18 - 7.08 (m, 2H), 4.61 (d, J = 5.2 Hz, 2H), 4.01 (s, 2H), 3.68 (t, J = 6.4 Hz, 4H), 3.42 (td, J = 8.4, 16.8 Hz, 3H), 2.70 (t, J = 6.4 Hz, 2H), 2.57 - 2.54 (m, 2H), 2.28 - 2.23 (m, 2H), 1.55 (s, 6H).
Example 186: Synthesis of Compound 186 (R)-3-(5-(4-( [1,5-a]pyrazin-6-yl)-
-2- azaspiro[3.3]heptan-2-yl)methyl)pyrazin-2-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (185) was prepared according to methods described above. LCMS [M+1] = 775.8; 1H NMR (400 MHz, DMSO-d6) δ 13.99 (br s, 1H), 10.45 (s, 1H), 9.74 (br s, 1H), 9.70 (s, 2H), 9.43 (s, 1H), 8.81 (s, 1H), 8.13 (s, 1H), 7.76 (s, 1H), 7.33 (t, J = 8.0 Hz, 1H), 7.18 - 7.08 (m, 2H), 4.61 (d, J = 5.2 Hz, 2H), 4.01 (s, 2H), 3.68 (t, J = 6.4 Hz, 4H), 3.42 (td, J = 8.4, 16.8 Hz, 3H), 2.70 (t, J = 6.4 Hz, 2H), 2.57 - 2.54 (m, 2H), 2.28 - 2.23 (m, 2H), 1.55 (s, 6H).
Example 186: Synthesis of Compound 186 (R)-3-(5-(4-( [1,5-a]pyrazin-6-yl)-
 3-methylpiperazin-1-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (186) was prepared according to methods described above. LCMS [M+1] = 817.3; 1H NMR (400 MHz, DMSO-d6) δ 13.98 (s, 1H), 10.52 (s, 1H), 9.73 (s, 1H), 9.26 (d, J = 1.2 Hz, 2H), 8.89 (d, J = 1.2 Hz, 1H), 7.94 - 7.93 (m, 2H), 7.79 (t, J = 8.4 Hz, 1H), 7.73 (s, 1H), 7.45 - 7.42 (m, 2H), 4.61 (s, 2H), 4.49 - 4.48 (m, 1H), 3.87 (t, J = 6.8 Hz, 2H), 3.72 - 3.68 (m, 1H), 3.61 - 3.52 (m, 2H), 3.09 - 3.03 (m, 1H), 2.94 (d, J = 10.4 Hz, 1H), 2.78 (t, J = 6.8 Hz, 3H), 2.38 (dd, J = 3.2, 10.8 Hz, 1H), 2.29 - 2.24 (m, 1H), 1.54 (s, 6H), 1.12 (d, J = 6.4 Hz, 3H). Example 187: Synthesis of Compound 187 3-(5-(4-((4-
3-methylpiperazin-1-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (186) was prepared according to methods described above. LCMS [M+1] = 817.3; 1H NMR (400 MHz, DMSO-d6) δ 13.98 (s, 1H), 10.52 (s, 1H), 9.73 (s, 1H), 9.26 (d, J = 1.2 Hz, 2H), 8.89 (d, J = 1.2 Hz, 1H), 7.94 - 7.93 (m, 2H), 7.79 (t, J = 8.4 Hz, 1H), 7.73 (s, 1H), 7.45 - 7.42 (m, 2H), 4.61 (s, 2H), 4.49 - 4.48 (m, 1H), 3.87 (t, J = 6.8 Hz, 2H), 3.72 - 3.68 (m, 1H), 3.61 - 3.52 (m, 2H), 3.09 - 3.03 (m, 1H), 2.94 (d, J = 10.4 Hz, 1H), 2.78 (t, J = 6.8 Hz, 3H), 2.38 (dd, J = 3.2, 10.8 Hz, 1H), 2.29 - 2.24 (m, 1H), 1.54 (s, 6H), 1.12 (d, J = 6.4 Hz, 3H). Example 187: Synthesis of Compound 187 3-(5-(4-((4-
 a]pyridin-6- yl)piperazin-1-yl)methyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (187) was prepared according to methods described above. LCMS [M+1] = 816.1; 1H NMR (400 MHz, MeOH-d6) δ = 8.76 (s, 1H), 7.98 (s, 1H), 7.90 (s, 1H), 7.69 (s, 1H), 7.49 (t, J = 9.6 Hz, 2H), 7.45 - 7.39 (m, 2H), 7.30 (d, J = 9.6 Hz, 1H), 4.76 (s, 2H), 3.90 (t, J = 6.8 Hz, 2H), 3.74 (s, 2H), 3.22 (s, 4H), 2.89 (t, J = 6.8 Hz, 2H), 2.75 (s, 4H), 2.57 (s, 3H), 1.64 (s, 6H).
Example 188: Synthesis of Compound 188 3-(5-(4-((4-(3-(2,4- 6-yl)piperidin-1-
a]pyridin-6- yl)piperazin-1-yl)methyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (187) was prepared according to methods described above. LCMS [M+1] = 816.1; 1H NMR (400 MHz, MeOH-d6) δ = 8.76 (s, 1H), 7.98 (s, 1H), 7.90 (s, 1H), 7.69 (s, 1H), 7.49 (t, J = 9.6 Hz, 2H), 7.45 - 7.39 (m, 2H), 7.30 (d, J = 9.6 Hz, 1H), 4.76 (s, 2H), 3.90 (t, J = 6.8 Hz, 2H), 3.74 (s, 2H), 3.22 (s, 4H), 2.89 (t, J = 6.8 Hz, 2H), 2.75 (s, 4H), 2.57 (s, 3H), 1.64 (s, 6H).
Example 188: Synthesis of Compound 188 3-(5-(4-((4-(3-(2,4- 6-yl)piperidin-1-
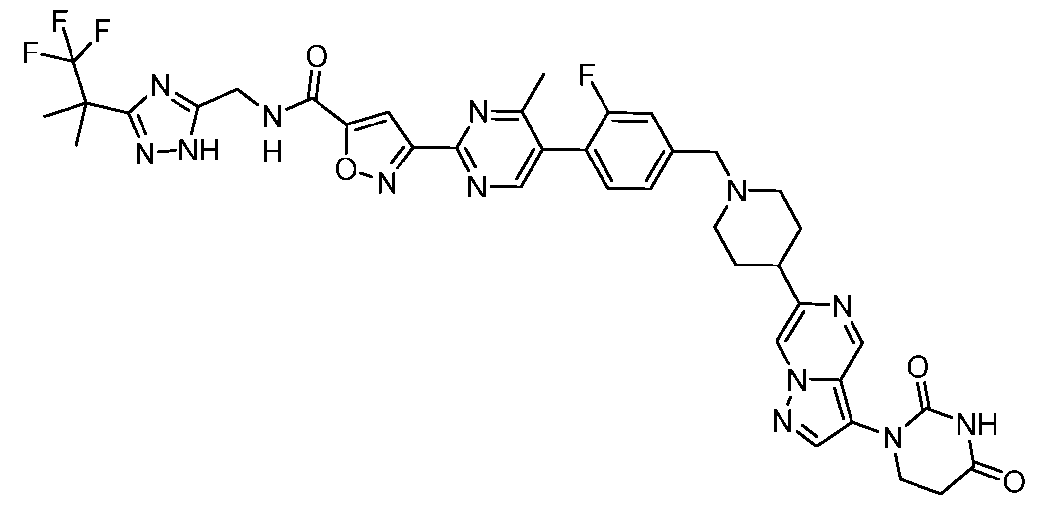 yl)methyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)- 1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (188) was prepared according to methods described above. LCMS [M+1] = 816.3. 1HNMR (400 MHz, METHANOL-d4) δ 9.06 (d, J = 1.2 Hz, 1H), 8.73 (s, 1H), 8.55 (s, 1H), 8.38 (s, 1H), 8.08 (s, 1H), 7.66 (s, 1H), 7.46 - 7.42 (m, 1H), 7.41 - 7.37 (m, 2H), 4.70 (s, 2H), 3.98 (d, J = 5.6 Hz, 2H), 3.70 (s, 2H), 3.14 - 3.08 (m, 2H), 2.93 - 2.88 (m, 1H), 2.84 - 2.74 (m, 1H), 2.54 (s, 4H), 2.32 - 2.24 (m, 2H), 2.05 - 1.99 (m, 3H), 1.98 - 1.94 (m, 1H), 1.59 (s, 6H). Example 189: Synthesis of Compounds 189-1 and 189-2 3-(5-(4-(1-(4-
yl)methyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)- 1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (188) was prepared according to methods described above. LCMS [M+1] = 816.3. 1HNMR (400 MHz, METHANOL-d4) δ 9.06 (d, J = 1.2 Hz, 1H), 8.73 (s, 1H), 8.55 (s, 1H), 8.38 (s, 1H), 8.08 (s, 1H), 7.66 (s, 1H), 7.46 - 7.42 (m, 1H), 7.41 - 7.37 (m, 2H), 4.70 (s, 2H), 3.98 (d, J = 5.6 Hz, 2H), 3.70 (s, 2H), 3.14 - 3.08 (m, 2H), 2.93 - 2.88 (m, 1H), 2.84 - 2.74 (m, 1H), 2.54 (s, 4H), 2.32 - 2.24 (m, 2H), 2.05 - 1.99 (m, 3H), 1.98 - 1.94 (m, 1H), 1.59 (s, 6H). Example 189: Synthesis of Compounds 189-1 and 189-2 3-(5-(4-(1-(4-
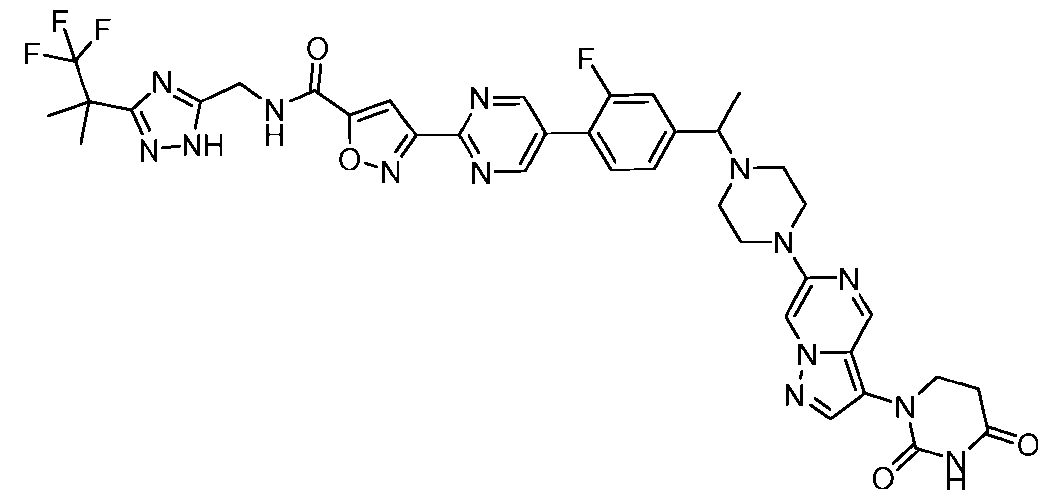 [1,5-a]pyrazin-6- yl)piperazin-1-yl)ethyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2- yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (189) was prepared according to methods described above. Stereoisomers were separated by SFC (DAICEL CHIRALPAK IK (250 x 50 mm, 10µm); [HEXANE-IPA/ACN(4:1)]; B%:93%, isocratic elution) to afford the early eluting isomer 189-1 and the late eluting isomer 189-2. LCMS [M+1] = 817.0; 1H NMR (400 MHz, METHANOL-d4) δ 9.18 (d, J = 1.2 Hz, 2H), 8.85 (d, J = 1.2 Hz, 1H), 7.91 (s, 1H), 7.84 (d,
J = 1.2 Hz, 1H), 7.72 - 7.67 (m, 2H), 7.44 - 7.38 (m, 2H), 4.74 (s, 2H), 3.96 (t, J = 6.8 Hz, 2H), 3.60 (q, J = 6.4 Hz, 1H), 3.40 (t, J = 4.8 Hz, 4H), 2.89 (t, J = 6.8 Hz, 2H), 2.80 - 2.73 (m, 2H), 2.66 - 2.59 (m, 2H), 1.62 (s, 6H), 1.48 (d, J = 6.8 Hz, 3H). Example 190: Synthesis of Compound 190 3-(5-(4-((4-
[1,5-a]pyrazin-6- yl)piperazin-1-yl)ethyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2- yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (189) was prepared according to methods described above. Stereoisomers were separated by SFC (DAICEL CHIRALPAK IK (250 x 50 mm, 10µm); [HEXANE-IPA/ACN(4:1)]; B%:93%, isocratic elution) to afford the early eluting isomer 189-1 and the late eluting isomer 189-2. LCMS [M+1] = 817.0; 1H NMR (400 MHz, METHANOL-d4) δ 9.18 (d, J = 1.2 Hz, 2H), 8.85 (d, J = 1.2 Hz, 1H), 7.91 (s, 1H), 7.84 (d,
J = 1.2 Hz, 1H), 7.72 - 7.67 (m, 2H), 7.44 - 7.38 (m, 2H), 4.74 (s, 2H), 3.96 (t, J = 6.8 Hz, 2H), 3.60 (q, J = 6.4 Hz, 1H), 3.40 (t, J = 4.8 Hz, 4H), 2.89 (t, J = 6.8 Hz, 2H), 2.80 - 2.73 (m, 2H), 2.66 - 2.59 (m, 2H), 1.62 (s, 6H), 1.48 (d, J = 6.8 Hz, 3H). Example 190: Synthesis of Compound 190 3-(5-(4-((4-
 a]pyrazin-6- yl)piperazin-1-yl)methyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (190) was prepared according to methods described above. LCMS [M+1] = 817.3; 1H NMR (400 MHz, METHANOL-d4) δ 8.87 (d, J = 0.8 Hz, 1H), 8.75 (s, 1H), 7.92 (s, 1H), 7.87 (s, 1H), 7.67 (s, 1H), 7.48 - 7.37 (m, 3H), 4.75 (s, 2H), 3.99 (t, J = 6.8 Hz, 2H), 3.73 (s, 2H), 3.51 - 3.40 (m, 4H), 2.89 (t, J = 6.8 Hz, 2H), 2.78 - 2.66 (m, 4H), 2.58 - 2.53 (m, 3H), 1.62 (s, 6H). Example 191: Synthesis of Compounds 191-1 and 191-2 3-(5-(4-(1-(4-
a]pyrazin-6- yl)piperazin-1-yl)methyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (190) was prepared according to methods described above. LCMS [M+1] = 817.3; 1H NMR (400 MHz, METHANOL-d4) δ 8.87 (d, J = 0.8 Hz, 1H), 8.75 (s, 1H), 7.92 (s, 1H), 7.87 (s, 1H), 7.67 (s, 1H), 7.48 - 7.37 (m, 3H), 4.75 (s, 2H), 3.99 (t, J = 6.8 Hz, 2H), 3.73 (s, 2H), 3.51 - 3.40 (m, 4H), 2.89 (t, J = 6.8 Hz, 2H), 2.78 - 2.66 (m, 4H), 2.58 - 2.53 (m, 3H), 1.62 (s, 6H). Example 191: Synthesis of Compounds 191-1 and 191-2 3-(5-(4-(1-(4-
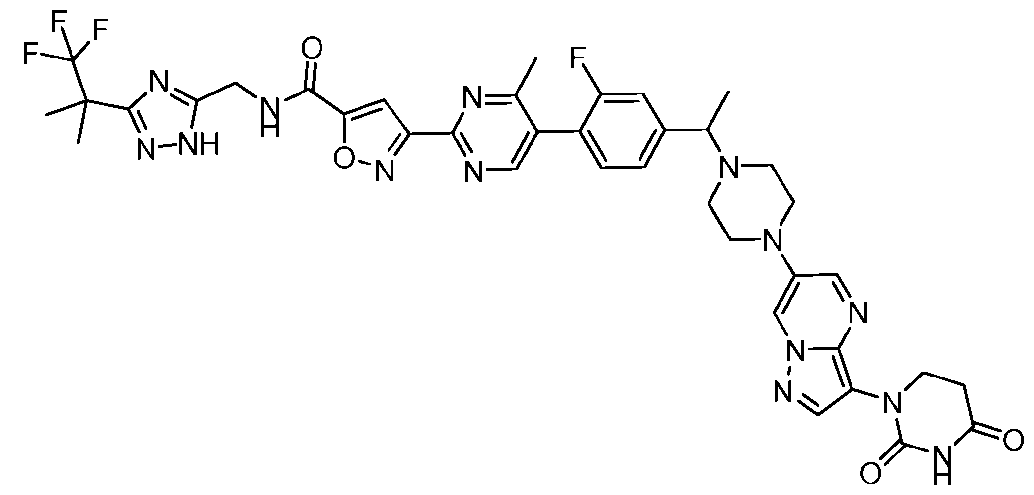 [1,5-a]pyrimidin-6- yl)piperazin-1-yl)ethyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (191) was prepared
according to methods described above. Stereoisomers were separated by SFC (DAICEL CHIRALPAK AS (250 x 30 mm, 10 µm); [CO2-i-PrOH (0.1%NH3•H2O)]; B%:50%, isocratic elution) to afford the early eluting isomer 191-1 and the late eluting isomer 192-2. LCMS [M+1] = 831.3. 1H NMR (400 MHz, DMSO-d6) δ 14.03 (s, 1H), 10.45 (s, 1H), 9.74 (t, J = 5.6 Hz, 1H), 8.88 (s, 1H), 8.66 (d, J = 2.5 Hz, 1H), 8.46 (d, J = 2.5 Hz, 1H), 8.22 (s, 1H), 8.12 (s, 1H), 7.73 (s, 1H), 7.54 (t, J = 7.9 Hz, 1H), 7.46 - 7.35 (m, 2H), 4.60 (d, J = 5.8 Hz, 2H), 3.88 (t, J = 6.8 Hz, 2H), 3.71 - 3.63 (m, 2H), 3.16 (s, 4H), 2.74 (t, J = 6.8 Hz, 2H), 2.69 - 2.62 (m, 2H), 2.61 - 2.51 (m, 4H), 1.54 (s, 6H), 1.41 (d, J = 6.6 Hz, 3H). Example 192: Synthesis of Compounds 192-1 and 192-2 3-(5-(5-(1-
[1,5-a]pyrimidin-6- yl)piperazin-1-yl)ethyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (191) was prepared
according to methods described above. Stereoisomers were separated by SFC (DAICEL CHIRALPAK AS (250 x 30 mm, 10 µm); [CO2-i-PrOH (0.1%NH3•H2O)]; B%:50%, isocratic elution) to afford the early eluting isomer 191-1 and the late eluting isomer 192-2. LCMS [M+1] = 831.3. 1H NMR (400 MHz, DMSO-d6) δ 14.03 (s, 1H), 10.45 (s, 1H), 9.74 (t, J = 5.6 Hz, 1H), 8.88 (s, 1H), 8.66 (d, J = 2.5 Hz, 1H), 8.46 (d, J = 2.5 Hz, 1H), 8.22 (s, 1H), 8.12 (s, 1H), 7.73 (s, 1H), 7.54 (t, J = 7.9 Hz, 1H), 7.46 - 7.35 (m, 2H), 4.60 (d, J = 5.8 Hz, 2H), 3.88 (t, J = 6.8 Hz, 2H), 3.71 - 3.63 (m, 2H), 3.16 (s, 4H), 2.74 (t, J = 6.8 Hz, 2H), 2.69 - 2.62 (m, 2H), 2.61 - 2.51 (m, 4H), 1.54 (s, 6H), 1.41 (d, J = 6.6 Hz, 3H). Example 192: Synthesis of Compounds 192-1 and 192-2 3-(5-(5-(1-
 -2- azaspiro[3.3]heptan-2-yl)ethyl)-3-fluoropyridin-2-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (192) was prepared according to methods described above. Stereoisomers were separated as the intermediate 3-(5-(5- (1-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3,5-difluorophenyl)-2-azaspiro[3.3]heptan-2- yl)ethyl)-3-fluoropyridin-2-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H- 1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide by SFC (DAICEL CHIRALPAK AD (250 x 30 mm, 10 µm); [CO2 - EtOH (0.1% NH3H2O)]; B%: 50%, isocratic elution) to afford the early eluting isomer that was processed to afford 192-1. The late eluting isomer was processed to afford 192-2. LCMS [M+1] = 824.3; 1H NMR (400MHz, DMSO-d6+ D2O) δ 9.44 (d, J = 0.4 Hz, 2H), 8.57 (s, 1H), 7.82 (dd, J = 0.8 Hz, 11.6 Hz, 1H), 7.70 (s, 1H), 7.03 (d, J = 8.8 Hz, 2H), 4.58 (s, 2H), 3.62 (t, J = 6.8 Hz, 2H), 3.52 - 3.48 (m, 1H), 3.42 - 3.34 (m, 1H), 3.29 (d, J = 6.8 Hz,
1H), 3.21 (d, J = 6.8 Hz, 1H), 3.10 (d, J = 7.2 Hz, 1H), 3.01 (d, J = 7.6 Hz, 1H), 2.69 (t, J = 6.8 Hz, 2H), 2.46 - 2.43 (m, 2H), 2.16 (t, J = 10.6 Hz, 2H), 1.51 (s, 6H), 1.18 (d, J = 6.0 Hz, 3H). Example 193: Synthesis of Compounds 193-1 and 193-2 3-(5-(3-(6- -2-
-2- azaspiro[3.3]heptan-2-yl)ethyl)-3-fluoropyridin-2-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (192) was prepared according to methods described above. Stereoisomers were separated as the intermediate 3-(5-(5- (1-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3,5-difluorophenyl)-2-azaspiro[3.3]heptan-2- yl)ethyl)-3-fluoropyridin-2-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H- 1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide by SFC (DAICEL CHIRALPAK AD (250 x 30 mm, 10 µm); [CO2 - EtOH (0.1% NH3H2O)]; B%: 50%, isocratic elution) to afford the early eluting isomer that was processed to afford 192-1. The late eluting isomer was processed to afford 192-2. LCMS [M+1] = 824.3; 1H NMR (400MHz, DMSO-d6+ D2O) δ 9.44 (d, J = 0.4 Hz, 2H), 8.57 (s, 1H), 7.82 (dd, J = 0.8 Hz, 11.6 Hz, 1H), 7.70 (s, 1H), 7.03 (d, J = 8.8 Hz, 2H), 4.58 (s, 2H), 3.62 (t, J = 6.8 Hz, 2H), 3.52 - 3.48 (m, 1H), 3.42 - 3.34 (m, 1H), 3.29 (d, J = 6.8 Hz,
1H), 3.21 (d, J = 6.8 Hz, 1H), 3.10 (d, J = 7.2 Hz, 1H), 3.01 (d, J = 7.6 Hz, 1H), 2.69 (t, J = 6.8 Hz, 2H), 2.46 - 2.43 (m, 2H), 2.16 (t, J = 10.6 Hz, 2H), 1.51 (s, 6H), 1.18 (d, J = 6.0 Hz, 3H). Example 193: Synthesis of Compounds 193-1 and 193-2 3-(5-(3-(6- -2-
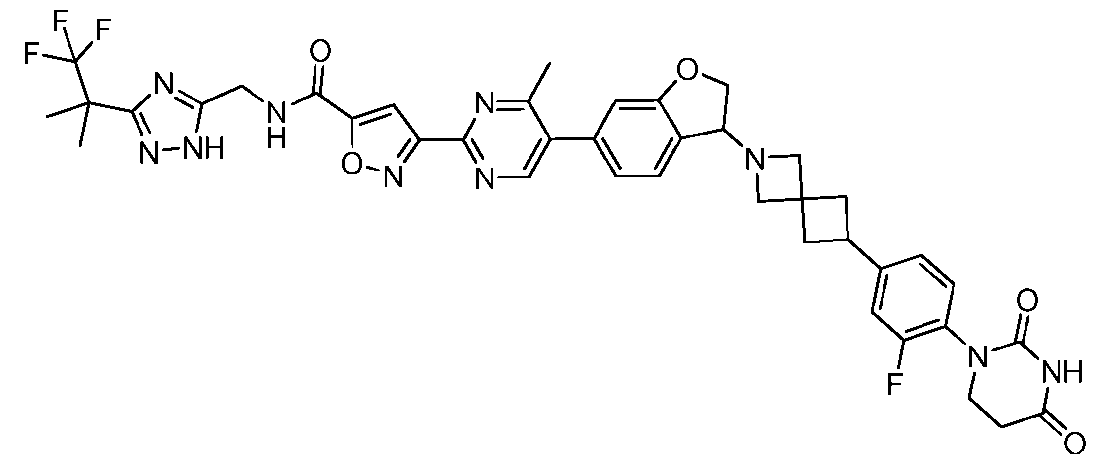 azaspiro[3.3]heptan-2-yl)-2,3-dihydrobenzofuran-6-yl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (193) was prepared according to the methods described above. Stereoisomers were separated as the intermediate 3-(5-(3-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)-2,3-dihydrobenzofuran-6-yl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide by SFC (DAICEL CHIRALPAK AD (250 x 30 mm, 10 µm); [CO2-i-PrOH/ACN]; B%:60%, isocratic elution) to afford the early eluting isomer that was processed to afford 193-1. The late eluting isomer was processed to afford 193-2. LCMS [M+1] =815.2; 1H NMR (400 MHz, DMSO-d6) δ 14.24 - 13.76 (s, 1H), 10.45 (s, 1H), 9.72 (t, J = 5.6 Hz, 1H), 8.79 (s, 1H), 8.18 (s, 1H), 7.72 (s, 1H), 7.48 (d, J = 7.6 Hz, 1H), 7.34 (t, J = 8.0 Hz, 1H), 7.16 (dd, J = 1.6, 11.6 Hz, 1H), 7.11 - 7.06 (m, 1H), 7.05 - 7.00 (m, 2H), 4.61 (d, J = 5.6 Hz, 2H), 4.47 - 4.35 (m, 2H), 4.11 (dd, J = 2.4, 6.8 Hz, 1H), 3.68 (t, J = 6.8 Hz, 2H), 3.46 -3.42 (m, 2H), 3.26 -3.22 (m, 2H), 3.12 (d, J = 7.2 Hz, 1H), 2.71 (t, J = 6.8 Hz, 2H), 2.57 (s, 3H), 2.49 - 2.43 (m, 2H), 2.20 (t, J = 9.6 Hz, 2H), 1.55 (s, 6H). Example 194: Synthesis of Compounds 194-1 and 194-2
azaspiro[3.3]heptan-2-yl)-2,3-dihydrobenzofuran-6-yl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (193) was prepared according to the methods described above. Stereoisomers were separated as the intermediate 3-(5-(3-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)-2,3-dihydrobenzofuran-6-yl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide by SFC (DAICEL CHIRALPAK AD (250 x 30 mm, 10 µm); [CO2-i-PrOH/ACN]; B%:60%, isocratic elution) to afford the early eluting isomer that was processed to afford 193-1. The late eluting isomer was processed to afford 193-2. LCMS [M+1] =815.2; 1H NMR (400 MHz, DMSO-d6) δ 14.24 - 13.76 (s, 1H), 10.45 (s, 1H), 9.72 (t, J = 5.6 Hz, 1H), 8.79 (s, 1H), 8.18 (s, 1H), 7.72 (s, 1H), 7.48 (d, J = 7.6 Hz, 1H), 7.34 (t, J = 8.0 Hz, 1H), 7.16 (dd, J = 1.6, 11.6 Hz, 1H), 7.11 - 7.06 (m, 1H), 7.05 - 7.00 (m, 2H), 4.61 (d, J = 5.6 Hz, 2H), 4.47 - 4.35 (m, 2H), 4.11 (dd, J = 2.4, 6.8 Hz, 1H), 3.68 (t, J = 6.8 Hz, 2H), 3.46 -3.42 (m, 2H), 3.26 -3.22 (m, 2H), 3.12 (d, J = 7.2 Hz, 1H), 2.71 (t, J = 6.8 Hz, 2H), 2.57 (s, 3H), 2.49 - 2.43 (m, 2H), 2.20 (t, J = 9.6 Hz, 2H), 1.55 (s, 6H). Example 194: Synthesis of Compounds 194-1 and 194-2
3-(5-(5-(1-(6-( ophenyl)-2- azaspiro[3.3]heptan-2-yl)ethyl)pyridin-2-yl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (194) was prepared according to the methods described above. Stereoisomers were separated as the intermediate tert- butyl 3-(5-(5-(1-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)ethyl)pyridin-2-yl)-4-methylpyrimidin-2-yl)isoxazole-5-carboxylate by SFC (ChiralPak IH, 250 x 30 mm, 10 µm; [CO2-EtOH(0.1% isopropylamine)]; B%:47%, isocratic elution) to afford the early eluting isomer that was processed to afford 194-1. The late eluting isomer was processed to afford 194-2. LCMS [M+H] = 802.5; 1H NMR (400 MHz, DMSO-d6) δ 8.97 (s, 1H), 8.67 (d, J = 1.6 Hz, 1H), 7.91 (dd, J = 1.8, 8.0 Hz, 1H), 7.75 (d, J = 8.0 Hz, 1H), 7.70 (s, 1H), 7.30 (t, J = 8.0 Hz, 1H), 7.11 (d, J = 11.6 Hz, 1H), 7.06 (d, J = 8.4 Hz, 1H), 4.58 (s, 2H), 3.65 (t, J = 6.6 Hz, 2H), 3.49 (q, J = 6.0 Hz, 1H), 3.42 - 3.32 (m, 2H), 3.24 (d, J = 7.2 Hz, 1H), 3.15 (d, J = 7.6 Hz, 1H), 3.04 (d, J = 7.6 Hz, 1H), 2.69 (t, J = 6.8 Hz, 2H), 2.66 (s, 3H), 2.47 - 2.45 (m, 2H), 2.16 (t, J = 10.2 Hz, 2H), 1.52 (s, 6H), 1.20 (d, J = 6.4 Hz, 3H). Example 195: Synthesis of Compounds 195-1 and 195-2
N-((3-(3,3-difluoro-2-methylbutan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-3-(5-(4-(1-(6-(4- (2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2-azaspiro[3.3]heptan-2-yl)propyl)-2- fluorophenyl)-4-methylpyrimidin-2-yl)isoxazole-5-carboxamide (195) was prepared according to the methods described above. Stereoisomers were separated as the intermediate tert-butyl 3-(5-(4- (1-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2-azaspiro[3.3]heptan-2- yl)propyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)isoxazole-5-carboxylate by SFC (ChiralPak IH, 250 x 50 mm, 10µm; [HEXANE-IPA/ACN(0.1% isopropylamine)]; B%:40%, isocratic elution) to afford the early eluting isomer that was processed to afford 195-1. The late eluting isomer was processed to afford 195-2. LCMS [M+1] = 829.2; 1H NMR (400 MHz, METHANOL- d4) δ 8.75 (s, 1H), 8.43 (s, 1H), 7.66 (s, 1H), 7.58 - 7.53 (m, 1H), 7.42 - 7.36 (m, 2H), 7.33 (t, J = 7.6 Hz, 1H), 7.13 - 7.06 (m, 2H), 4.71 (s, 2H), 4.64 - 4.55 (m, 1H), 3.96 (d, J = 8.4 Hz, 1H), 3.87 - 3.81 (m, 2H), 3.77 (t, J = 6.8 Hz, 2H), 3.61 (d, J = 8.8 Hz, 1H), 3.48 (t, J = 8.8 Hz, 1H), 2.81 (t, J = 6.8 Hz, 2H), 2.70 - 2.63 (m, 2H), 2.54 (s, 3H), 2.40 - 2.32 (m, 2H), 2.05 - 1.94 (m, 1H), 1.77 - 1.69 (m, 1H), 1.61 - 1.52 (m, 9H), 0.84 (t, J = 7.2 Hz, 3H) . Example 196: Synthesis of Compounds 196-1 and 196-2 3-(5-(1-
 -2- azaspiro[3.3]heptan-2-yl)-2,3-dihydro-1H-inden-5-yl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (196) was prepared according to the methods described above. Stereoisomers were separated as the intermediate tert-butyl 3-(5-(1-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)-2,3-dihydro-1H-inden-5-yl)-4-methylpyrimidin-2-yl)isoxazole-5- carboxylate by SFC (DAICEL CHIRALPAK IA (250 x 30 mm,10 µm); [hexane- isopropanol/ACN(0.1% isopropylamine)]; B%:60%, isocratic elution) to afford the early eluting isomer that was processed to afford 196-1. The late eluting isomer was processed to afford 196-2.
LCMS [M+1] = 813.2; 1H NMR (400 MHz, DMSO-d6) δ 8.71 (s, 1H), 8.49 (s, 1H), 7.69 - 7.65 (m, 2H), 7.50 (s, 1H), 7.42 (d, J = 7.6 Hz, 1H), 7.36 (t, J = 8.0 Hz, 1H), 7.16 - 7.10 (m, 2H), 4.76 (s, 2H), 4.67 - 4.64 (m, 1H), 4.29 - 4.11 (m, 2H), 4.05 - 3.89 (m, 2H), 3.80 (t, J = 6.8 Hz, 2H), 3.59 - 3.48 (m, 1H), 3.28 - 3.19 (m, 1H), 3.11 - 3.01 (m, 1H), 2.84 (t, J = 6.8 Hz, 2H), 2.75 - 2.67 (m, 2H), 2.61 (s, 3H), 2.55 - 2.46 (m, 1H), 2.45 - 2.37 (m, 2H), 2.19 - 2.10 (m, 1H), 1.64 (s, 6H). Example 197: Synthesis of Compounds 197-1 and 197-2 3-(5-(4-(1-
-2- azaspiro[3.3]heptan-2-yl)-2,3-dihydro-1H-inden-5-yl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (196) was prepared according to the methods described above. Stereoisomers were separated as the intermediate tert-butyl 3-(5-(1-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)-2,3-dihydro-1H-inden-5-yl)-4-methylpyrimidin-2-yl)isoxazole-5- carboxylate by SFC (DAICEL CHIRALPAK IA (250 x 30 mm,10 µm); [hexane- isopropanol/ACN(0.1% isopropylamine)]; B%:60%, isocratic elution) to afford the early eluting isomer that was processed to afford 196-1. The late eluting isomer was processed to afford 196-2.
LCMS [M+1] = 813.2; 1H NMR (400 MHz, DMSO-d6) δ 8.71 (s, 1H), 8.49 (s, 1H), 7.69 - 7.65 (m, 2H), 7.50 (s, 1H), 7.42 (d, J = 7.6 Hz, 1H), 7.36 (t, J = 8.0 Hz, 1H), 7.16 - 7.10 (m, 2H), 4.76 (s, 2H), 4.67 - 4.64 (m, 1H), 4.29 - 4.11 (m, 2H), 4.05 - 3.89 (m, 2H), 3.80 (t, J = 6.8 Hz, 2H), 3.59 - 3.48 (m, 1H), 3.28 - 3.19 (m, 1H), 3.11 - 3.01 (m, 1H), 2.84 (t, J = 6.8 Hz, 2H), 2.75 - 2.67 (m, 2H), 2.61 (s, 3H), 2.55 - 2.46 (m, 1H), 2.45 - 2.37 (m, 2H), 2.19 - 2.10 (m, 1H), 1.64 (s, 6H). Example 197: Synthesis of Compounds 197-1 and 197-2 3-(5-(4-(1-
 -2- azaspiro[3.3]heptan-2-yl)propyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro- 2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (197) was prepared according to the methods described above. Stereoisomers were separated as the intermediate tert- butyl 3-(5-(4-(1-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)propyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)isoxazole-5- carboxylate by SFC (ChiralPak IH, 250 x 50 mm, 10 µm; [hexane-isopropanol/ACN(0.1% isopropylamine)]; B%:40%, isocratic elution) to afford the early eluting isomer that was processed to afford 197-1. The late eluting isomer was processed to afford 197-2. LCMS [M+1] = 833.2; 1H NMR (400 MHz, METHANOL-d4) δ 8.77 (s, 1H), 7.69 (s, 1H), 7.50 - 7.43 (m, 1H), 7.37 - 7.30 (m, 3H), 7.11 (s, 1H), 7.09 (s, 1H), 4.77 (s, 2H), 3.79 (t, J = 6.8 Hz, 2H), 3.51 - 3.44 (m, 2H), 3.29 - 3.26 (m, 2H), 3.15 (d, J = 8.4 Hz, 2H), 2.83 (t, J = 6.8 Hz, 2H), 2.64 - 2.58 (m, 2H), 2.56 (s, 3H), 2.32 - 2.25 (m, 2H), 1.87 (m, 1H), 1.64 (s, 6H), 1.60 - 1.54 (m, 1H), 0.79 (t, J = 7.6 Hz, 3H). Example 198: Synthesis of Compounds 198-1 and 198-2
3-(5-(5-((2 yl)-6- azaspiro[3.4]octan-6-yl)methyl)-3-fluoropyridin-2-yl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (198) was prepared according to the methods described above. Stereoisomers were separated as the intermediate tert-butyl 3-(5-(5-((2-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-6- azaspiro[3.4]octan-6-yl)methyl)-3-fluoropyridin-2-yl)-4-methylpyrimidin-2-yl)isoxazole-5- carboxylate by SFC (DAICEL CHIRALPAK AD (250 x 30mm, 10 µm); [CO2-EtOH: acetonitrile = 1: 1 (0.1% isopropylamine)]; B%: 62%, isocratic elution) to afford the early eluting isomer that was processed to afford 198-1. The late eluting isomer was processed to afford 198-2. 198-1: LCMS [M+1] = 820.4; 1H NMR (400 MHz, METHANOL-d4) δ 8.91 (s, 1H), 8.59 (s, 1H), 7.84 (d, J = 10.4 Hz, 1H), 7.68 (d, J = 1.6 Hz, 1H), 7.31 (t, J = 8.0 Hz, 1H), 7.09 (d, J = 9.2 Hz, 2H), 4.75 (s, 2H), 3.83 - 3.73 (m, 4H), 3.59 - 3.50 (m, 1H), 2.85 - 2.74 (m, 4H), 2.70 (s, 2H), 2.60 (s, 3H), 2.47 - 2.38 (m, 2H), 2.26 - 2.14 (m, 4H), 1.62 (s, 6H). 198-2: LCMS [M+1] = 820.4; 1H NMR (400 MHz, METHANOL-d4) δ 8.92 (s, 1H), 8.61 (s, 1H), 7.87 (dd, J = 1.2, 10.4 Hz, 1H), 7.69 (s, 1H), 7.31 (t, J = 8.0 Hz, 1H), 7.09 (d, J = 9.6 Hz, 2H), 4.75 (s, 2H), 3.85 (s, 2H), 3.77 (t, J = 6.8 Hz, 2H), 3.50 - 3.41 (m, 1H), 2.88 (s, 2H), 2.81 (t, J = 6.8 Hz, 2H), 2.69 (t, J = 7.2 Hz, 2H), 2.62 (d, J = 0.8 Hz, 3H), 2.52 - 2.47 (m, 2H), 2.23 - 2.13 (m, 2H), 1.95 (t, J = 7.2 Hz, 2H), 1.62 (s, 6H). Example 199: Synthesis of Compounds 199-1 and 199-2
-2- azaspiro[3.3]heptan-2-yl)propyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro- 2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (197) was prepared according to the methods described above. Stereoisomers were separated as the intermediate tert- butyl 3-(5-(4-(1-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)propyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)isoxazole-5- carboxylate by SFC (ChiralPak IH, 250 x 50 mm, 10 µm; [hexane-isopropanol/ACN(0.1% isopropylamine)]; B%:40%, isocratic elution) to afford the early eluting isomer that was processed to afford 197-1. The late eluting isomer was processed to afford 197-2. LCMS [M+1] = 833.2; 1H NMR (400 MHz, METHANOL-d4) δ 8.77 (s, 1H), 7.69 (s, 1H), 7.50 - 7.43 (m, 1H), 7.37 - 7.30 (m, 3H), 7.11 (s, 1H), 7.09 (s, 1H), 4.77 (s, 2H), 3.79 (t, J = 6.8 Hz, 2H), 3.51 - 3.44 (m, 2H), 3.29 - 3.26 (m, 2H), 3.15 (d, J = 8.4 Hz, 2H), 2.83 (t, J = 6.8 Hz, 2H), 2.64 - 2.58 (m, 2H), 2.56 (s, 3H), 2.32 - 2.25 (m, 2H), 1.87 (m, 1H), 1.64 (s, 6H), 1.60 - 1.54 (m, 1H), 0.79 (t, J = 7.6 Hz, 3H). Example 198: Synthesis of Compounds 198-1 and 198-2
3-(5-(5-((2 yl)-6- azaspiro[3.4]octan-6-yl)methyl)-3-fluoropyridin-2-yl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (198) was prepared according to the methods described above. Stereoisomers were separated as the intermediate tert-butyl 3-(5-(5-((2-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-6- azaspiro[3.4]octan-6-yl)methyl)-3-fluoropyridin-2-yl)-4-methylpyrimidin-2-yl)isoxazole-5- carboxylate by SFC (DAICEL CHIRALPAK AD (250 x 30mm, 10 µm); [CO2-EtOH: acetonitrile = 1: 1 (0.1% isopropylamine)]; B%: 62%, isocratic elution) to afford the early eluting isomer that was processed to afford 198-1. The late eluting isomer was processed to afford 198-2. 198-1: LCMS [M+1] = 820.4; 1H NMR (400 MHz, METHANOL-d4) δ 8.91 (s, 1H), 8.59 (s, 1H), 7.84 (d, J = 10.4 Hz, 1H), 7.68 (d, J = 1.6 Hz, 1H), 7.31 (t, J = 8.0 Hz, 1H), 7.09 (d, J = 9.2 Hz, 2H), 4.75 (s, 2H), 3.83 - 3.73 (m, 4H), 3.59 - 3.50 (m, 1H), 2.85 - 2.74 (m, 4H), 2.70 (s, 2H), 2.60 (s, 3H), 2.47 - 2.38 (m, 2H), 2.26 - 2.14 (m, 4H), 1.62 (s, 6H). 198-2: LCMS [M+1] = 820.4; 1H NMR (400 MHz, METHANOL-d4) δ 8.92 (s, 1H), 8.61 (s, 1H), 7.87 (dd, J = 1.2, 10.4 Hz, 1H), 7.69 (s, 1H), 7.31 (t, J = 8.0 Hz, 1H), 7.09 (d, J = 9.6 Hz, 2H), 4.75 (s, 2H), 3.85 (s, 2H), 3.77 (t, J = 6.8 Hz, 2H), 3.50 - 3.41 (m, 1H), 2.88 (s, 2H), 2.81 (t, J = 6.8 Hz, 2H), 2.69 (t, J = 7.2 Hz, 2H), 2.62 (d, J = 0.8 Hz, 3H), 2.52 - 2.47 (m, 2H), 2.23 - 2.13 (m, 2H), 1.95 (t, J = 7.2 Hz, 2H), 1.62 (s, 6H). Example 199: Synthesis of Compounds 199-1 and 199-2
3-(5-(1-(6-(4-(2 henyl)-2- azaspiro[3.3]heptan-2-yl)ethyl)-4'-methyl-[2,5'-bipyrimidin]-2'-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (199) was prepared according to the methods described above. Stereoisomers were separated as the intermediate tert- butyl 3-(5-(1-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)ethyl)-4'-methyl-[2,5'-bipyrimidin]-2'-yl)isoxazole-5-carboxylate by SFC (DAICEL CHIRALPAK AD (250 x 30 mm, 10 µm); [CO2 - IPA (0.1% isopropylamine)]; B%: 60%, isocratic elution) to afford the early eluting isomer that was processed to afford 199-1. The late eluting isomer was processed to afford 199-2. LCMS [M+1] = 803.5. 1H NMR (400 MHz, CHLOROFORM-d) δ 9.42 (s, 1H), 8.85 (s, 2H), 7.73 (s, 1H), 7.51 (br s, 1H), 7.45 (t, J = 5.8 Hz, 1H), 7.25 - 7.23 (m, 1H), 7.01 (s, 1H), 6.98 (d, J = 1.6 Hz, 1H), 4.80 (d, J = 6.0 Hz, 2H), 3.78 (t, J = 6.6 Hz, 2H), 3.46 - 3.34 (m, 4H), 3.22 (d, J = 7.6 Hz, 1H), 3.14 (d, J = 7.2 Hz, 1H), 2.98 (s, 3H), 2.85 (t, J = 6.6 Hz, 2H), 2.62 - 2.57 (m, 2H), 2.29 - 2.22 (m, 2H), 1.64 (s, 6H), 1.34 (d, J = 6.4 Hz, 3H). Example 200: Synthesis of Compounds 200-1 and 200-2
N-((3-(3,3 l)-3-(5-(1-(6-(4-(2,4- dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2-azaspiro[3.3]heptan-2-yl)ethyl)-4'-methyl- [2,5'-bipyrimidin]-2'-yl)isoxazole-5-carboxamide (200) was prepared according to the methods described above. Stereoisomers were separated as the intermediate tert-butyl 3-(5-(1-(6-(4-(2,4- dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2-azaspiro[3.3]heptan-2-yl)ethyl)-4'-methyl- [2,5'-bipyrimidin]-2'-yl)isoxazole-5-carboxylate by SFC (DAICEL CHIRALPAK AD (250 x 30 mm, 10 µm); [CO2 - IPA (0.1% isopropylamine)]; B%: 60%, isocratic elution) to afford the early eluting isomer that was processed to afford 200-1. The late eluting isomer was processed to afford 200-2. LCMS [M+1] = 799.5; 1H NMR (400 MHz, CHLOROFORM-d) δ 9.41 (s, 1H), 8.85 (s, 2H), 7.71 (s, 1H), 7.54 (s, 1H), 7.42 (t, J = 5.6 Hz, 1H), 7.25 - 7.23 (m, 1H), 7.01 (s, 1H), 6.98 (d, J = 2.0 Hz, 1H), 4.80 (d, J = 5.6 Hz, 2H), 3.78 (t, J = 6.6 Hz, 2H), 3.46 - 3.34 (m, 4H), 3.24 (d, J = 7.6 Hz, 1H), 3.15 (d, J = 7.6 Hz, 1H), 2.97 (s, 3H), 2.85 (t, J = 6.6 Hz, 2H), 2.62 - 2.57 (m, 2H), 2.29 - 2.22 (m, 2H), 1.71 - 1.61 (m, 3H), 1.56 (s, 6H), 1.34 (d, J = 6.4 Hz, 3H). Example 201: Synthesis of Compounds 201-1 and 201-2
3-(5-(4-(1-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)butyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro- 2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (201) was prepared according to the methods described above. Stereoisomers were separated as the intermediate tert- butyl 3-(5-(4-(1-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)butyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)isoxazole-5-carboxylate by SFC (ChiralPak IH, 250 x 50 mm, 10 µm; [hexane-IPA/acetonitrile (0.1%isopropylamine)]; B%:40%, isocratic elution) to afford the early eluting isomer that was processed to afford 201-1. The late eluting isomer was processed to afford 201-2. LCMS [M+1] = 847.2; 1H NMR (400 MHz, METHANOL-d4) δ 8.76 (s, 1H), 8.42 - 8.28 (m, 1H), 7.67 (s, 1H), 7.64 - 7.56 (m, 1H), 7.48 - 7.38 (m, 2H), 7.37 - 7.29 (m, 1H), 7.15 - 7.05 (m, 2H), 4.74 (s, 2H), 4.25 - 4.12 (m, 2H), 4.07 - 3.92 (m, 2H), 3.86 - 3.72 (m, 3H), 3.50 (q, J = 8.8 Hz, 1H), 2.81 (t, J = 6.8 Hz, 2H), 2.75 - 2.63 (m, 2H), 2.54 (s, 3H), 2.40 (t, J = 10.4 Hz, 2H), 1.97 - 1.93 (m, 1H), 1.85 - 1.74 (m, 1H), 1.62 (s, 6H), 1.34 - 1.14 (m, 2H), 0.96 (t, J = 7.2 Hz, 3H). Example 202: Synthesis of Compounds 202-1 and 202-2 F F F O N F O NH O 3-(5-(4-(1-(6-
 -2- azaspiro[3.3]heptan-2-yl)-2-methylpropyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3- (1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (202) was prepared according to the methods described above. Stereoisomers were separated as the intermediate tert-butyl 3-(5-(4-(1-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3- fluorophenyl)-2-azaspiro[3.3]heptan-2-yl)-2-methylpropyl)-2-fluorophenyl)-4-methylpyrimidin- 2-yl)isoxazole-5-carboxylate by SFC (ChiralPak IH, 250 x 50 mm, 10 µm; [hexane-EtOH/
acetonitrile(4:1)]; B%:20%, isocratic elution) to afford the early eluting isomer that was processed to afford 202-1. The late eluting isomer was processed to afford 202-2. LCMS [M+1] = 847.4; 1H NMR (400 MHz, METHANOL-d4) δ 8.74 (s, 1H), 7.66 (s, 1H), 7.41 (t, J = 7.6 Hz, 1H), 7.35 - 7.21 (m, 3H), 7.07 (d, J = 9.6 Hz, 2H), 4.74 (s, 2H), 3.77 (t, J = 6.8 Hz, 2H), 3.47 - 3.41 (m, 1H), 3.40 - 3.33 (m, 2H), 3.24 (d, J = 4.8 Hz, 1H), 3.16 (q, J = 7.6 Hz, 2H), 2.81 (t, J = 6.8 Hz, 2H), 2.65 - 2.49 (m, 5H), 2.31 - 2.20 (m, 2H), 2.08 - 1.98 (m, 1H), 1.62 (s, 6H), 0.91 (d, J = 6.8 Hz, 3H), 0.78 (d, J = 6.8 Hz, 3H). Example 203: Synthesis of Compounds 203-1 and 203-2 3-(5-(4-(1-(6-
-2- azaspiro[3.3]heptan-2-yl)-2-methylpropyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3- (1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (202) was prepared according to the methods described above. Stereoisomers were separated as the intermediate tert-butyl 3-(5-(4-(1-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3- fluorophenyl)-2-azaspiro[3.3]heptan-2-yl)-2-methylpropyl)-2-fluorophenyl)-4-methylpyrimidin- 2-yl)isoxazole-5-carboxylate by SFC (ChiralPak IH, 250 x 50 mm, 10 µm; [hexane-EtOH/
acetonitrile(4:1)]; B%:20%, isocratic elution) to afford the early eluting isomer that was processed to afford 202-1. The late eluting isomer was processed to afford 202-2. LCMS [M+1] = 847.4; 1H NMR (400 MHz, METHANOL-d4) δ 8.74 (s, 1H), 7.66 (s, 1H), 7.41 (t, J = 7.6 Hz, 1H), 7.35 - 7.21 (m, 3H), 7.07 (d, J = 9.6 Hz, 2H), 4.74 (s, 2H), 3.77 (t, J = 6.8 Hz, 2H), 3.47 - 3.41 (m, 1H), 3.40 - 3.33 (m, 2H), 3.24 (d, J = 4.8 Hz, 1H), 3.16 (q, J = 7.6 Hz, 2H), 2.81 (t, J = 6.8 Hz, 2H), 2.65 - 2.49 (m, 5H), 2.31 - 2.20 (m, 2H), 2.08 - 1.98 (m, 1H), 1.62 (s, 6H), 0.91 (d, J = 6.8 Hz, 3H), 0.78 (d, J = 6.8 Hz, 3H). Example 203: Synthesis of Compounds 203-1 and 203-2 3-(5-(4-(1-(6-
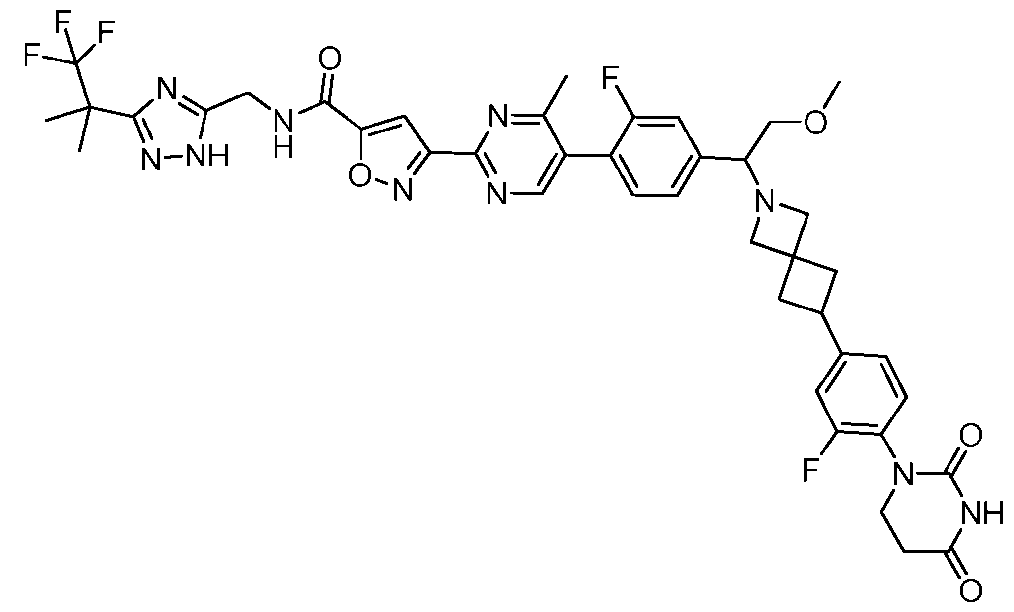 -2- azaspiro[3.3]heptan-2-yl)-2-methoxyethyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3- (1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (203) was prepared according to the methods described above. Stereoisomers were separated as the intermediate tert-butyl 3-(5-(4-(1-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3- fluorophenyl)-2-azaspiro[3.3]heptan-2-yl)-2-methoxyethyl)-2-fluorophenyl)-4-methylpyrimidin- 2-yl)isoxazole-5-carboxylate by SFC (DAICEL CHIRALPAK IA (250 x 30 mm, 10 µm); [hexane-EtOH/ acetonitrile (4:1)]; B%:30%, isocratic elution) to afford the early eluting isomer that was processed to afford 203-1. The late eluting isomer was processed to afford 203-2. LCMS [M+1] = 849.1; 1H NMR (400 MHz, METHANOL-d4) δ 8.74 (s, 1H), 8.37 (s, 1H), 7.67 (s, 1H), 7.55 - 7.48 (m, 1H), 7.44 - 7.38 (m, 2H), 7.32 (t, J = 8.2 Hz, 1H), 7.12 - 7.04 (m, 2H), 4.74 (s, 2H), 4.13 (d, J = 3.6 Hz, 1H), 3.94 - 3.81 (m, 2H), 3.77 (t, J = 6.8 Hz, 2H), 3.69 (d, J = 4.8 Hz, 3H), 3.63 (s, 1H), 3.51 - 3.45 (m, 1H), 3.40 (s, 3H), 2.81 (t, J = 6.8 Hz, 2H), 2.69 - 2.63 (m, 2H), 2.54 (d, J = 0.8 Hz, 3H), 2.39 - 2.31 (m, 2H), 1.62 (s, 6H).
Example 204: Synthesis of Compounds 204-1 and 204-2 3-(5-(7-(4-
-2- azaspiro[3.3]heptan-2-yl)-2-methoxyethyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3- (1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (203) was prepared according to the methods described above. Stereoisomers were separated as the intermediate tert-butyl 3-(5-(4-(1-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3- fluorophenyl)-2-azaspiro[3.3]heptan-2-yl)-2-methoxyethyl)-2-fluorophenyl)-4-methylpyrimidin- 2-yl)isoxazole-5-carboxylate by SFC (DAICEL CHIRALPAK IA (250 x 30 mm, 10 µm); [hexane-EtOH/ acetonitrile (4:1)]; B%:30%, isocratic elution) to afford the early eluting isomer that was processed to afford 203-1. The late eluting isomer was processed to afford 203-2. LCMS [M+1] = 849.1; 1H NMR (400 MHz, METHANOL-d4) δ 8.74 (s, 1H), 8.37 (s, 1H), 7.67 (s, 1H), 7.55 - 7.48 (m, 1H), 7.44 - 7.38 (m, 2H), 7.32 (t, J = 8.2 Hz, 1H), 7.12 - 7.04 (m, 2H), 4.74 (s, 2H), 4.13 (d, J = 3.6 Hz, 1H), 3.94 - 3.81 (m, 2H), 3.77 (t, J = 6.8 Hz, 2H), 3.69 (d, J = 4.8 Hz, 3H), 3.63 (s, 1H), 3.51 - 3.45 (m, 1H), 3.40 (s, 3H), 2.81 (t, J = 6.8 Hz, 2H), 2.69 - 2.63 (m, 2H), 2.54 (d, J = 0.8 Hz, 3H), 2.39 - 2.31 (m, 2H), 1.62 (s, 6H).
Example 204: Synthesis of Compounds 204-1 and 204-2 3-(5-(7-(4-
 a]pyrazin-6- yl)piperazin-1-yl)-5-oxa-2-azaspiro[3.4]octan-2-yl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (204) was prepared according to the methods described above.Stereoisomers were separated as the intermediate tert-butyl 3-(5-(7-(4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5- a]pyrazin-6-yl)piperazin-1-yl)-5-oxa-2-azaspiro[3.4]octan-2-yl)-4-methylpyrimidin-2- yl)isoxazole-5-carboxylate by SFC (DAICEL CHIRALCEL OJ (250 x 30 mm,10 µm); [CO2-i- PrOH/ acetonitrile]; B%:50%, isocratic elution) to afford the early eluting isomer that was processed to afford 204-1. The late eluting isomer was processed to afford 204-2. LCMS [M+1] = 820.1; 1H NMR (400 MHz, METHANOL-d4) δ 8.89 (s, 1H), 7.96 (d, J = 10.0 Hz, 2H), 7.89 (s, 1H), 7.52 (s, 1H), 4.74 (s, 2H), 4.33 (d, J= 8.4 Hz, 1H), 4.26 - 4.19 (m, 2H), 4.15 - 4.09 (m, 2H), 3.99 (t, J = 6.8 Hz, 2H), 3.85 (t, J = 8.0 Hz, 1H), 3.44 (t, J = 4.4 Hz, 4H), 3.23 - 3.14 (m, 1H), 2.91 (t, J = 6.8 Hz, 2H), 2.82 - 2.75 (m, 2H), 2.71 - 2.58 (m, 3H), 2.55 (s, 3H), 2.25 (dd, J = 8.0, 12.8 Hz, 1H), 1.63 (s, 6H). Example 205: Synthesis of Compound 205
a]pyrazin-6- yl)piperazin-1-yl)-5-oxa-2-azaspiro[3.4]octan-2-yl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (204) was prepared according to the methods described above.Stereoisomers were separated as the intermediate tert-butyl 3-(5-(7-(4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5- a]pyrazin-6-yl)piperazin-1-yl)-5-oxa-2-azaspiro[3.4]octan-2-yl)-4-methylpyrimidin-2- yl)isoxazole-5-carboxylate by SFC (DAICEL CHIRALCEL OJ (250 x 30 mm,10 µm); [CO2-i- PrOH/ acetonitrile]; B%:50%, isocratic elution) to afford the early eluting isomer that was processed to afford 204-1. The late eluting isomer was processed to afford 204-2. LCMS [M+1] = 820.1; 1H NMR (400 MHz, METHANOL-d4) δ 8.89 (s, 1H), 7.96 (d, J = 10.0 Hz, 2H), 7.89 (s, 1H), 7.52 (s, 1H), 4.74 (s, 2H), 4.33 (d, J= 8.4 Hz, 1H), 4.26 - 4.19 (m, 2H), 4.15 - 4.09 (m, 2H), 3.99 (t, J = 6.8 Hz, 2H), 3.85 (t, J = 8.0 Hz, 1H), 3.44 (t, J = 4.4 Hz, 4H), 3.23 - 3.14 (m, 1H), 2.91 (t, J = 6.8 Hz, 2H), 2.82 - 2.75 (m, 2H), 2.71 - 2.58 (m, 3H), 2.55 (s, 3H), 2.25 (dd, J = 8.0, 12.8 Hz, 1H), 1.63 (s, 6H). Example 205: Synthesis of Compound 205
N-((3-(tert-butyl)-1H-1,2,4-triazol-5-yl)methyl)-3-(5-(4-((6-(4-(2,4- dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2-azaspiro[3.3]heptan-2-yl)methyl)-2- fluorophenyl)-4-methylpyrimidin-2-yl)isoxazole-5-carboxamide (205) was prepared according to the methods described above. LCMS [M +1] = 751.5; 1H NMR (400 MHz, METHANOL-d4) δ = 8.73 (s, 1H), 7.66 (s, 1H), 7.51 - 7.47 (m, 1H), 7.38 - 7.31 (m, 3H), 7.12 - 7.09 (m, 2H), 4.68 (s, 2H), 3.96 (s, 2H), 3.79 - 3.76 (m, 4H), 3.57 (br s, 2H), 3.49 (br t, J = 9.2 Hz, 1H), 2.82 (t, J = 6.8 Hz, 2H), 2.68 - 2.62 (m, 2H), 2.53 (d, J = 0.8 Hz, 3H), 2.37 - 2.31 (m, 2H), 1.39 (s, 9H). Example 206: Synthesis of Compounds 206-1 and 206-2 N-((3-(3,3-
 -3-(5-(5-(1-(6-(4- (2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2-azaspiro[3.3]heptan-2-yl)ethyl)-3- fluoropyridin-2-yl)-4-methylpyrimidin-2-yl)isoxazole-5-carboxamide (206) was prepared according to the methods described above. Stereoisomers were separated as the intermediate tert- butyl 3-(5-(4-(1-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)ethyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)isoxazole-5-carboxylate by SFC (ChiralPak IH, 250 x 50mm, 10 µm; [hexane-EtOH: acetonitrile = 4:1(0.1% isopropylamine)]; B%:28%, isocratic elution) to afford the early eluting isomer that was processed to afford 206-1. The late eluting isomer was processed to afford 206-2. LCMS [M+1] = 816.4; 1H NMR (400 MHz, METHANOL-d4) δ 8.91 (s, 1H), 8.61 (s, 1H), 7.85 (dd, J = 1.2, 10.4 Hz, 1H), 7.68 (s, 1H), 7.32 (t, J = 7.6 Hz, 1H), 7.14 - 7.03 (m, 2H), 4.72 (s, 2H), 3.82 (d, J = 6.0 Hz, 1H), 3.77 (t, J = 6.8 Hz, 2H), 3.67 ( d, J = 7.6 Hz, 1H), 3.56 ( d, J = 8.0 Hz, 1H), 3.53 -
3.42 (m, 2H), 3.35 ( d, J = 9.2 Hz, 1H), 2.81 (t, J = 6.8 Hz, 2H), 2.70 - 2.54 (m, 6H), 2.32 (t, J = 10.4 Hz, 2H), 1.62 - 1.48 (m, 9H), 1.41 (d, J = 6.8 Hz, 3H). Example 207: Synthesis of Compounds 207-1 and 207-2 3-(5-(5-
-3-(5-(5-(1-(6-(4- (2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2-azaspiro[3.3]heptan-2-yl)ethyl)-3- fluoropyridin-2-yl)-4-methylpyrimidin-2-yl)isoxazole-5-carboxamide (206) was prepared according to the methods described above. Stereoisomers were separated as the intermediate tert- butyl 3-(5-(4-(1-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)ethyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)isoxazole-5-carboxylate by SFC (ChiralPak IH, 250 x 50mm, 10 µm; [hexane-EtOH: acetonitrile = 4:1(0.1% isopropylamine)]; B%:28%, isocratic elution) to afford the early eluting isomer that was processed to afford 206-1. The late eluting isomer was processed to afford 206-2. LCMS [M+1] = 816.4; 1H NMR (400 MHz, METHANOL-d4) δ 8.91 (s, 1H), 8.61 (s, 1H), 7.85 (dd, J = 1.2, 10.4 Hz, 1H), 7.68 (s, 1H), 7.32 (t, J = 7.6 Hz, 1H), 7.14 - 7.03 (m, 2H), 4.72 (s, 2H), 3.82 (d, J = 6.0 Hz, 1H), 3.77 (t, J = 6.8 Hz, 2H), 3.67 ( d, J = 7.6 Hz, 1H), 3.56 ( d, J = 8.0 Hz, 1H), 3.53 -
3.42 (m, 2H), 3.35 ( d, J = 9.2 Hz, 1H), 2.81 (t, J = 6.8 Hz, 2H), 2.70 - 2.54 (m, 6H), 2.32 (t, J = 10.4 Hz, 2H), 1.62 - 1.48 (m, 9H), 1.41 (d, J = 6.8 Hz, 3H). Example 207: Synthesis of Compounds 207-1 and 207-2 3-(5-(5-
 -2- azaspiro[3.3]heptan-2-yl)ethyl)-3-fluoropyridin-2-yl)-4-methylpyrimidin-2-yl)-N-((3-(3,3- difluoro-2-methylbutan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (207) was prepared according to the methods described above. Stereoisomers were separated as the intermediate tert-butyl 3-(5-(5-(1-(6-(2-chloro-3-(2,4-dioxotetrahydropyrimidin-1(2H)- yl)phenyl)-2-azaspiro[3.3]heptan-2-yl)ethyl)-3-fluoropyridin-2-yl)-4-methylpyrimidin-2- yl)isoxazole-5-carboxylate by SFC (DAICEL CHIRALPAK AD (250 x 30 mm, 10 µm); [CO2- EtOH (0.1% isopropylamine)]; B%: 56%, isocratic elution) to afford the early eluting isomer that was processed to afford 207-1. The late eluting isomer was processed to afford 207-2. LCMS [M+1] = 832.4; 1H NMR (400 MHz, DMSO-d6) δ 13.73 (br s, 1H), 10.43 (s, 1H), 9.66 (br s, 1H), 9.00 (d, J = 0.8 Hz, 1H), 8.60 (s, 1H), 7.88 (dd, J = 1.6, 10.8 Hz, 1H), 7.74 (s, 1H), 7.40 - 7.36 (m, 2H), 7.35 - 7.32 (m, 1H), 4.56 (d, J = 3.2 Hz, 2H), 3.71 - 3.60 (m, 2H), 3.59 - 3.48 (m, 2H), 3.35 (s, 1H), 3.28 (d, J = 6.8 Hz, 1H), 3.13 (d, J = 7.2 Hz, 1H), 3.04 (dd, J = 2.4, 7.2 Hz, 1H), 2.72 (t, J = 7.0 Hz, 2H), 2.61 - 2.56 (m, 2H), 2.54 (d, J = 0.8 Hz, 3H), 2.27 - 2.19 (m, 2H), 1.58 (t, J = 19.6 Hz, 3H), 1.44 (s, 6H), 1.22 (d, J = 6.4 Hz, 3H). Example 208: Synthesis of Compounds 208-1 and 208-2
3-(5-(1-(6-(2-chloro-3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)phenyl)-2- azaspiro[3.3]heptan-2-yl)ethyl)-4'-methyl-[2,5'-bipyrimidin]-2'-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (208) was prepared according to the methods described above. Stereoisomers were separated as the intermediate tert- butyl 3-(5-(1-(6-(2-chloro-3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)phenyl)-2- azaspiro[3.3]heptan-2-yl)ethyl)-4'-methyl-[2,5'-bipyrimidin]-2'-yl)isoxazole-5-carboxylate by SFC (DAICEL CHIRALPAK AD (250 x 30 mm, 10 µm); [CO2-IPA (0.1% isopropylamine)]; B%: 56%, isocratic elution) to afford the early eluting isomer that was process to afford 208-1. The late eluting isomer was processed to afford 208-2. LCMS [M+1] = 819.4; 1H NMR (400 MHz, DMSO-d6) δ 13.96 (br s, 1H), 10.43 (br s, 1H), 9.72 (br s, 1H), 9.38 (s, 1H), 8.97 (s, 2H), 7.75 (s, 1H), 7.42 - 7.28 (m, 3H), 4.60 (s, 2H), 3.72 - 3.52 (m, 3H), 3.48 (q, J = 6.4 Hz, 1H), 3.37 - 3.34 (m, 1H), 3.27 (d, J = 6.4 Hz, 1H), 3.14 (d, J = 7.2 Hz, 1H), 3.05 - 3.02 (m, 1H), 2.89 (s, 3H), 2.72 (t, J = 6.8 Hz, 2H), 2.61 - 2.52 (m, 2H), 2.26 - 2.19 (m, 2H), 1.54 (s, 6H), 1.25 (d, J = 6.8 Hz, 3H). Example 209: Synthesis of Compound 209
-2- azaspiro[3.3]heptan-2-yl)ethyl)-3-fluoropyridin-2-yl)-4-methylpyrimidin-2-yl)-N-((3-(3,3- difluoro-2-methylbutan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (207) was prepared according to the methods described above. Stereoisomers were separated as the intermediate tert-butyl 3-(5-(5-(1-(6-(2-chloro-3-(2,4-dioxotetrahydropyrimidin-1(2H)- yl)phenyl)-2-azaspiro[3.3]heptan-2-yl)ethyl)-3-fluoropyridin-2-yl)-4-methylpyrimidin-2- yl)isoxazole-5-carboxylate by SFC (DAICEL CHIRALPAK AD (250 x 30 mm, 10 µm); [CO2- EtOH (0.1% isopropylamine)]; B%: 56%, isocratic elution) to afford the early eluting isomer that was processed to afford 207-1. The late eluting isomer was processed to afford 207-2. LCMS [M+1] = 832.4; 1H NMR (400 MHz, DMSO-d6) δ 13.73 (br s, 1H), 10.43 (s, 1H), 9.66 (br s, 1H), 9.00 (d, J = 0.8 Hz, 1H), 8.60 (s, 1H), 7.88 (dd, J = 1.6, 10.8 Hz, 1H), 7.74 (s, 1H), 7.40 - 7.36 (m, 2H), 7.35 - 7.32 (m, 1H), 4.56 (d, J = 3.2 Hz, 2H), 3.71 - 3.60 (m, 2H), 3.59 - 3.48 (m, 2H), 3.35 (s, 1H), 3.28 (d, J = 6.8 Hz, 1H), 3.13 (d, J = 7.2 Hz, 1H), 3.04 (dd, J = 2.4, 7.2 Hz, 1H), 2.72 (t, J = 7.0 Hz, 2H), 2.61 - 2.56 (m, 2H), 2.54 (d, J = 0.8 Hz, 3H), 2.27 - 2.19 (m, 2H), 1.58 (t, J = 19.6 Hz, 3H), 1.44 (s, 6H), 1.22 (d, J = 6.4 Hz, 3H). Example 208: Synthesis of Compounds 208-1 and 208-2
3-(5-(1-(6-(2-chloro-3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)phenyl)-2- azaspiro[3.3]heptan-2-yl)ethyl)-4'-methyl-[2,5'-bipyrimidin]-2'-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (208) was prepared according to the methods described above. Stereoisomers were separated as the intermediate tert- butyl 3-(5-(1-(6-(2-chloro-3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)phenyl)-2- azaspiro[3.3]heptan-2-yl)ethyl)-4'-methyl-[2,5'-bipyrimidin]-2'-yl)isoxazole-5-carboxylate by SFC (DAICEL CHIRALPAK AD (250 x 30 mm, 10 µm); [CO2-IPA (0.1% isopropylamine)]; B%: 56%, isocratic elution) to afford the early eluting isomer that was process to afford 208-1. The late eluting isomer was processed to afford 208-2. LCMS [M+1] = 819.4; 1H NMR (400 MHz, DMSO-d6) δ 13.96 (br s, 1H), 10.43 (br s, 1H), 9.72 (br s, 1H), 9.38 (s, 1H), 8.97 (s, 2H), 7.75 (s, 1H), 7.42 - 7.28 (m, 3H), 4.60 (s, 2H), 3.72 - 3.52 (m, 3H), 3.48 (q, J = 6.4 Hz, 1H), 3.37 - 3.34 (m, 1H), 3.27 (d, J = 6.4 Hz, 1H), 3.14 (d, J = 7.2 Hz, 1H), 3.05 - 3.02 (m, 1H), 2.89 (s, 3H), 2.72 (t, J = 6.8 Hz, 2H), 2.61 - 2.52 (m, 2H), 2.26 - 2.19 (m, 2H), 1.54 (s, 6H), 1.25 (d, J = 6.8 Hz, 3H). Example 209: Synthesis of Compound 209
 1-(4-((4-(4-(5- - - 2-yl)piperazin-1- yl)cyclohexyl)oxy)-2-methylphenyl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide (209) was prepared according to the methods described above. LCMS [M+1] = 778.5. 1H NMR (400 MHz, DMSO-d6) δ 13.89 (br s, 1H), 10.33 (br s, 1H), 8.83 (br s, 1H), 8.37 (s, 1H), 8.10 (s, 1H), 7.96 (s, 1H), 7.25 (d, J = 8.8 Hz, 1H), 6.96 (d, J = 2.8 Hz, 1H), 6.91 - 6.88 (m, 1H), 6.75 (s, 1H), 4.55 (d, J = 4.4 Hz, 2H), 4.37 - 4.32
(m, 1H), 3.69 - 3.64 (m, 1H), 3.55 - 3.48 (m, 5H), 2.73 - 2.66 (m, 5H), 2.13 (d, J = 4.4 Hz, 8H), 1.91 (d, J = 9.6 Hz, 2H), 1.53 (s, 6H), 1.47 - 1.39 (m, 4H). Example 210: Synthesis of Compound 210 3-(5-(5-((6-
1-(4-((4-(4-(5- - - 2-yl)piperazin-1- yl)cyclohexyl)oxy)-2-methylphenyl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide (209) was prepared according to the methods described above. LCMS [M+1] = 778.5. 1H NMR (400 MHz, DMSO-d6) δ 13.89 (br s, 1H), 10.33 (br s, 1H), 8.83 (br s, 1H), 8.37 (s, 1H), 8.10 (s, 1H), 7.96 (s, 1H), 7.25 (d, J = 8.8 Hz, 1H), 6.96 (d, J = 2.8 Hz, 1H), 6.91 - 6.88 (m, 1H), 6.75 (s, 1H), 4.55 (d, J = 4.4 Hz, 2H), 4.37 - 4.32
(m, 1H), 3.69 - 3.64 (m, 1H), 3.55 - 3.48 (m, 5H), 2.73 - 2.66 (m, 5H), 2.13 (d, J = 4.4 Hz, 8H), 1.91 (d, J = 9.6 Hz, 2H), 1.53 (s, 6H), 1.47 - 1.39 (m, 4H). Example 210: Synthesis of Compound 210 3-(5-(5-((6-
 -2- azaspiro[3.3]heptan-2-yl)methyl)pyridin-2-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (210) was prepared according to the methods described above. LCMS [M+1] = 774.3; 1H NMR (400 MHz, DMSO- d6) δ 13.96 (br s, 1H), 10.44 (s, 1H), 9.73 (t, J = 5.2 Hz, 1H), 9.63 (s, 2H), 8.67 (d, J = 1.6 Hz, 1H), 8.20 (d, J = 8.0 Hz, 1H), 7.89 (dd, J = 2.0 Hz, 8.0 Hz, 1H), 7.74 (s, 1H), 7.32 (t, J = 8.2 Hz, 1H), 7.16 - 7.06 (m, 2H), 4.61 (d, J = 5.2 Hz, 2H), 3.69 - 3.64 (m, 4H), 3.44 - 3.37 (m, 1H), 3.34 (s, 2H), 3.13 (s, 2H), 2.70 (t, J = 6.8 Hz, 2H), 2.54 (s, 1H), 2.47 - 2.46 (m, 1H), 2.23 - 2.17 (m, 2H), 1.54 (s, 6H). Example 211: Synthesis of Compounds 211-1 and 211-2
-2- azaspiro[3.3]heptan-2-yl)methyl)pyridin-2-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (210) was prepared according to the methods described above. LCMS [M+1] = 774.3; 1H NMR (400 MHz, DMSO- d6) δ 13.96 (br s, 1H), 10.44 (s, 1H), 9.73 (t, J = 5.2 Hz, 1H), 9.63 (s, 2H), 8.67 (d, J = 1.6 Hz, 1H), 8.20 (d, J = 8.0 Hz, 1H), 7.89 (dd, J = 2.0 Hz, 8.0 Hz, 1H), 7.74 (s, 1H), 7.32 (t, J = 8.2 Hz, 1H), 7.16 - 7.06 (m, 2H), 4.61 (d, J = 5.2 Hz, 2H), 3.69 - 3.64 (m, 4H), 3.44 - 3.37 (m, 1H), 3.34 (s, 2H), 3.13 (s, 2H), 2.70 (t, J = 6.8 Hz, 2H), 2.54 (s, 1H), 2.47 - 2.46 (m, 1H), 2.23 - 2.17 (m, 2H), 1.54 (s, 6H). Example 211: Synthesis of Compounds 211-1 and 211-2
 3-(5-(4- - -2- azaspiro[3.3]heptan-2-yl)ethyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-
2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (211) was prepared according to the methods described above.Stereoisomers were separated as the intermediate tert- butyl 3-(5-(4-(1-(6-(2-chloro-3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)phenyl)-2- azaspiro[3.3]heptan-2-yl)ethyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)isoxazole-5-carboxylate by SFC (DAICEL CHIRALPAK IG (250 x 30mm, 10 µm); [CO2-EtOH: acetonitrile =1:1 (0.1% isopropylamine)]; B%:65%, isocratic elution) to afford the early eluting isomer that was processed to afford 211-1. The late eluting isomer was processed to afford 211-2. LCMS [M+1] = 835.3; 1H NMR (400 MHz, DMSO-d6) δ 13.97 (s, 1H), 10.43 (s, 1H), 9.72 (br t, J = 5.2 Hz, 1H), 8.85 (s, 1H), 7.72 (s, 1H), 7.48 (t, J = 8.0 Hz, 1H), 7.41 - 7.30 (m, 5H), 4.60 (br d, J = 5.6 Hz, 2H), 3.72 - 3.51 (m, 3H), 3.42 - 3.35 (m, 1H), 3.30 - 3.23 (m, 2H), 3.12 - 3.00 (m, 2H), 2.72 (t, J = 6.4 Hz, 2H), 2.58 - 2.52 (m, 2H), 2.47 (d, J = 0.8 Hz, 3H), 2.27 - 2.15 (m, 2H), 1.54 (s, 6H), 1.17 (d, J = 6.4 Hz, 3H). Example 212: Synthesis of Compound 212 3-(5-(4-((6-
3-(5-(4- - -2- azaspiro[3.3]heptan-2-yl)ethyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-
2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (211) was prepared according to the methods described above.Stereoisomers were separated as the intermediate tert- butyl 3-(5-(4-(1-(6-(2-chloro-3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)phenyl)-2- azaspiro[3.3]heptan-2-yl)ethyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)isoxazole-5-carboxylate by SFC (DAICEL CHIRALPAK IG (250 x 30mm, 10 µm); [CO2-EtOH: acetonitrile =1:1 (0.1% isopropylamine)]; B%:65%, isocratic elution) to afford the early eluting isomer that was processed to afford 211-1. The late eluting isomer was processed to afford 211-2. LCMS [M+1] = 835.3; 1H NMR (400 MHz, DMSO-d6) δ 13.97 (s, 1H), 10.43 (s, 1H), 9.72 (br t, J = 5.2 Hz, 1H), 8.85 (s, 1H), 7.72 (s, 1H), 7.48 (t, J = 8.0 Hz, 1H), 7.41 - 7.30 (m, 5H), 4.60 (br d, J = 5.6 Hz, 2H), 3.72 - 3.51 (m, 3H), 3.42 - 3.35 (m, 1H), 3.30 - 3.23 (m, 2H), 3.12 - 3.00 (m, 2H), 2.72 (t, J = 6.4 Hz, 2H), 2.58 - 2.52 (m, 2H), 2.47 (d, J = 0.8 Hz, 3H), 2.27 - 2.15 (m, 2H), 1.54 (s, 6H), 1.17 (d, J = 6.4 Hz, 3H). Example 212: Synthesis of Compound 212 3-(5-(4-((6-
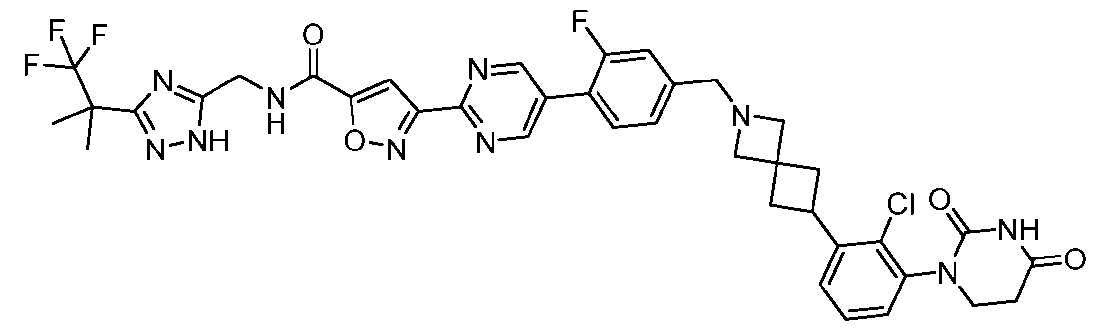 -2- azaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (212) was prepared according to the methods described above. LCMS [M+1] =807.3; 1H NMR (400 MHz, MEOD- d4) δ 9.18 (d, J = 0.8 Hz, 2H), 8.44 (s, 1H), 7.77 (t, J = 8.2 Hz, 1H), 7.68 (s, 1H), 7.45 - 7.36 (m, 4H), 7.34 - 7.30 (m, 1H), 4.74 (s, 2H), 4.11 (s, 2H), 3.99 (s, 2H), 3.82 - 3.67 (m, 5H), 2.92 - 2.81 (m, 2H), 2.80 - 2.71 (m, 2H), 2.46 - 2.36 (m, 2H), 1.62 (s, 6H). Example 213: Synthesis of Compounds 213-1 and 213-2
-2- azaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (212) was prepared according to the methods described above. LCMS [M+1] =807.3; 1H NMR (400 MHz, MEOD- d4) δ 9.18 (d, J = 0.8 Hz, 2H), 8.44 (s, 1H), 7.77 (t, J = 8.2 Hz, 1H), 7.68 (s, 1H), 7.45 - 7.36 (m, 4H), 7.34 - 7.30 (m, 1H), 4.74 (s, 2H), 4.11 (s, 2H), 3.99 (s, 2H), 3.82 - 3.67 (m, 5H), 2.92 - 2.81 (m, 2H), 2.80 - 2.71 (m, 2H), 2.46 - 2.36 (m, 2H), 1.62 (s, 6H). Example 213: Synthesis of Compounds 213-1 and 213-2
3-(5-(4-(1-(6-(4- orophenyl)-2- azaspiro[3.3]heptan-2-yl)ethyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (213) was prepared according to the methods described above. Stereoisomers were separated by SFC (DAICEL CHIRALPAK AS (250 x 30 mm, 10µm); [CO2- acetonitrile /i-PrOH(0.1% NH3H2O)]; B%:45%, isocratic elution) to afford 213-1 as the early eluting isomer and 213-2 as the late eluting isomer. LCMS [M+1] = 805.2; 1H NMR (400 MHz, DMSO-d6) δ 13.99 (s, 1H), 10.45 (s, 1H), 9.73 (s, 1H), 9.25 (s, 2H), 8.13 (s, 1H), 7.91 - 7.75 (m, 1H), 7.72 (s, 1H), 7.53 - 7.38 (m, 2H), 7.33 (t, J = 8.0 Hz, 1H), 7.17 (d, J = 11.6 Hz, 1H), 7.08 (d, J = 8.4 Hz, 1H), 4.61 (d, J = 4.0 Hz, 2H), 3.67 (t, J = 6.8 Hz, 2H), 3.48 - 3.34 (m, 4H), 2.70 (t, J = 6.8 Hz, 2H), 2.63 - 2.51 (m, 4H), 2.38 - 2.13 (m, 2H), 1.54 (s, 6H), 1.48 - 1.08 (m, 3H). Example 214: Synthesis of Compound 214 3-(5-(5-((6-(4-
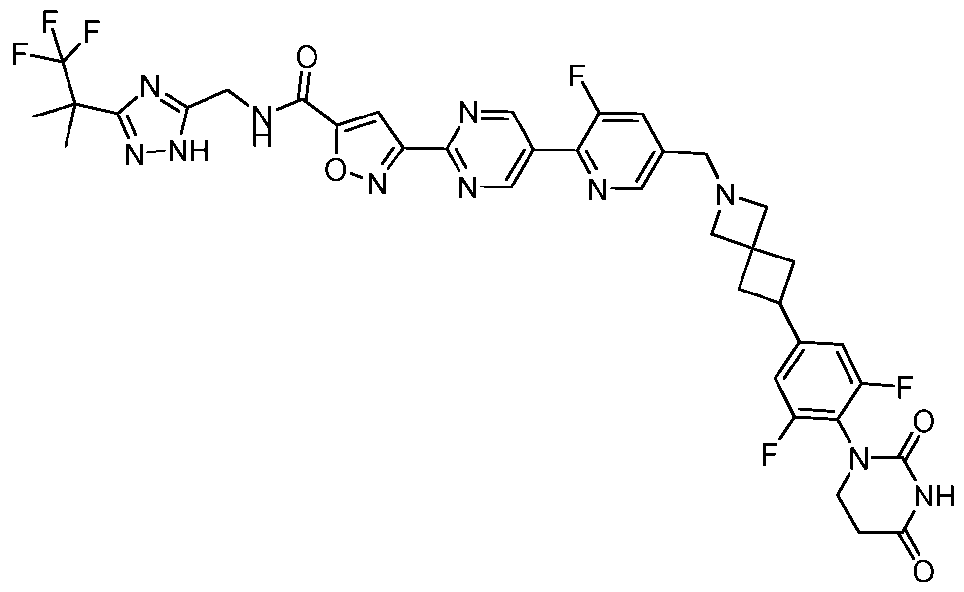 -2- azaspiro[3.3]heptan-2-yl)methyl)-3-fluoropyridin-2-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (214) was prepared according to the methods described above. LCMS [M+1] = 810.6; 1H NMR (400 MHz, DMSO-
d6) δ 13.97 ( s, 1H), 10.61 (s, 1H), 10.22 ( d, J = 5.2 Hz, 1H), 9.77 (s, 1H), 9.53 (d, J = 0.8 Hz, 2H), 8.76 (s, 1H), 8.11 ( d, J = 11.6 Hz, 1H), 7.75 (s, 1H), 7.15 (d, J = 9.6 Hz, 2H), 4.63 (s, 2H), 4.54 ( d, J = 5.2 Hz, 2H), 4.42 - 4.35 (m, 1H), 4.32 - 4.20 (m, 2H), 4.05 (s, 1H), 3.66 (t, J = 6.8 Hz, 2H), 3.45 (t, J = 9.2 Hz, 1H), 2.71 (t, J = 6.8 Hz, 3H), 2.61 - 2.58 (m, 1H), 2.45 - 2.33 (m, 2H), 1.54 (s, 6H). Example 215: Synthesis of Compound 215 3-(5-(4-((((3-
-2- azaspiro[3.3]heptan-2-yl)methyl)-3-fluoropyridin-2-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (214) was prepared according to the methods described above. LCMS [M+1] = 810.6; 1H NMR (400 MHz, DMSO-
d6) δ 13.97 ( s, 1H), 10.61 (s, 1H), 10.22 ( d, J = 5.2 Hz, 1H), 9.77 (s, 1H), 9.53 (d, J = 0.8 Hz, 2H), 8.76 (s, 1H), 8.11 ( d, J = 11.6 Hz, 1H), 7.75 (s, 1H), 7.15 (d, J = 9.6 Hz, 2H), 4.63 (s, 2H), 4.54 ( d, J = 5.2 Hz, 2H), 4.42 - 4.35 (m, 1H), 4.32 - 4.20 (m, 2H), 4.05 (s, 1H), 3.66 (t, J = 6.8 Hz, 2H), 3.45 (t, J = 9.2 Hz, 1H), 2.71 (t, J = 6.8 Hz, 3H), 2.61 - 2.58 (m, 1H), 2.45 - 2.33 (m, 2H), 1.54 (s, 6H). Example 215: Synthesis of Compound 215 3-(5-(4-((((3-
 difluorophenyl)cyclobutyl)methyl)amino)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (215) was prepared according to the methods described above.The stereoisomers were separated by SFC (DAICEL CHIRALPAK IE (50 x 250 mm, 10 µm), [HEXANE-IPA/ acetonitrile (0.1%isopropylamine)]; B%:50%, isocratic elution mode). The early eluting isomer was purified further with prep-HPLC (Welch Ultimate C18150 x 25 mm, 7 µm; [water (FA)- acetonitrile]; gradient: 13%-43% B over 10 min) and prep-HPLC (Phenomenex Luna C18150 x 25 mm, 10 µm; [water (FA)- acetonitrile]; gradient:10%-40% B over 10 min) to afford 215. LCMS [M+1] = 797.3; 1H NMR (400 MHz, DMSO-d6) δ 14.02 (s, 1H), 10.60 (s, 1H), 9.85 - 9.71 (m, 1H), 9.24 (s, 2H), 7.81 - 7.70 (m, 2H), 7.49 - 7.35 (m, 2H), 7.10 (d, J = 9.2 Hz, 2H), 4.61 (d, J = 3.6 Hz, 2H), 4.06 - 3.74 (m, 2H), 3.67 (t, J = 6.4 Hz, 2H), 3.58 - 3.54 (m, 1H), 2.75 - 2.66 (m, 3H), 2.58 (d, J = 12.8 Hz, 2H), 2.47 - 2.37 (m, 3H), 1.81 (d, J = 4.4 Hz, 2H), 1.55 (s, 6H). Example 216: Synthesis of Compound 216
3-(5-(4-( difluorophenyl)cyclobutyl)amino)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro- 2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (216) was prepared according to the methods described above. Stereoisomers were purified by preparative HPLC(Welch Utimate C18150 x 25 mm, 7 µm; [water(FA)- acetonitrile];gradient:14%-44% B over 10 min) and preparative HPLC (Waters Xbidge BEH C18150 x 25 mm, 10 µm; [water( NH4HCO3)- acetonitrile];gradient:21%-51% B over 10 min) to give afford 216 as the early eluting isomer. LCMS [M+1] = 783.0.1H NMR (400 MHz, DMSO-d6) δ 9.14 (s, 2H), 8.31 (s, 1H), 7.68 (t, J = 7.6 Hz, 1H), 7.64 (s, 1H), 7.42 - 7.34 (m, 2H), 7.08 (d, J = 9.2 Hz, 2H), 4.57 (s, 2H), 3.82 (s, 2H), 3.65 - 3.56 (m, 3H), 3.55 - 3.43 (m, 1H), 2.71 (t, J = 6.4 Hz, 2H), 2.43 - 2.24 (m, 4H), 1.49 (s, 6H). Example 217: Synthesis of Compound 217 3-(5-(4-((6-
difluorophenyl)cyclobutyl)methyl)amino)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (215) was prepared according to the methods described above.The stereoisomers were separated by SFC (DAICEL CHIRALPAK IE (50 x 250 mm, 10 µm), [HEXANE-IPA/ acetonitrile (0.1%isopropylamine)]; B%:50%, isocratic elution mode). The early eluting isomer was purified further with prep-HPLC (Welch Ultimate C18150 x 25 mm, 7 µm; [water (FA)- acetonitrile]; gradient: 13%-43% B over 10 min) and prep-HPLC (Phenomenex Luna C18150 x 25 mm, 10 µm; [water (FA)- acetonitrile]; gradient:10%-40% B over 10 min) to afford 215. LCMS [M+1] = 797.3; 1H NMR (400 MHz, DMSO-d6) δ 14.02 (s, 1H), 10.60 (s, 1H), 9.85 - 9.71 (m, 1H), 9.24 (s, 2H), 7.81 - 7.70 (m, 2H), 7.49 - 7.35 (m, 2H), 7.10 (d, J = 9.2 Hz, 2H), 4.61 (d, J = 3.6 Hz, 2H), 4.06 - 3.74 (m, 2H), 3.67 (t, J = 6.4 Hz, 2H), 3.58 - 3.54 (m, 1H), 2.75 - 2.66 (m, 3H), 2.58 (d, J = 12.8 Hz, 2H), 2.47 - 2.37 (m, 3H), 1.81 (d, J = 4.4 Hz, 2H), 1.55 (s, 6H). Example 216: Synthesis of Compound 216
3-(5-(4-( difluorophenyl)cyclobutyl)amino)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro- 2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (216) was prepared according to the methods described above. Stereoisomers were purified by preparative HPLC(Welch Utimate C18150 x 25 mm, 7 µm; [water(FA)- acetonitrile];gradient:14%-44% B over 10 min) and preparative HPLC (Waters Xbidge BEH C18150 x 25 mm, 10 µm; [water( NH4HCO3)- acetonitrile];gradient:21%-51% B over 10 min) to give afford 216 as the early eluting isomer. LCMS [M+1] = 783.0.1H NMR (400 MHz, DMSO-d6) δ 9.14 (s, 2H), 8.31 (s, 1H), 7.68 (t, J = 7.6 Hz, 1H), 7.64 (s, 1H), 7.42 - 7.34 (m, 2H), 7.08 (d, J = 9.2 Hz, 2H), 4.57 (s, 2H), 3.82 (s, 2H), 3.65 - 3.56 (m, 3H), 3.55 - 3.43 (m, 1H), 2.71 (t, J = 6.4 Hz, 2H), 2.43 - 2.24 (m, 4H), 1.49 (s, 6H). Example 217: Synthesis of Compound 217 3-(5-(4-((6-
 -2,6- diazaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (217) was prepared according to the methods described above. LCMS [M+1] = 810.5; 1H NMR (400 MHz, DMSO- d6) δ 9.72 (br s, 1H), 9.23 (d, J = 1.3 Hz, 2H), 7.79 - 7.65 (m, 2H), 7.40 - 7.24 (m, 2H), 6.21 (d, J = 10.0 Hz, 2H), 4.60 (s, 2H), 3.96 (s, 4H), 3.65 (s, 2H), 3.58 (t, J = 6.4 Hz, 2H), 3.37 (s, 4H), 2.67 (t, J = 6.8 Hz, 2H), 1.54 (s, 6H).
Example 218: Synthesis of Compound 218 3-(5-(4-((4- piperazin-1-
-2,6- diazaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (217) was prepared according to the methods described above. LCMS [M+1] = 810.5; 1H NMR (400 MHz, DMSO- d6) δ 9.72 (br s, 1H), 9.23 (d, J = 1.3 Hz, 2H), 7.79 - 7.65 (m, 2H), 7.40 - 7.24 (m, 2H), 6.21 (d, J = 10.0 Hz, 2H), 4.60 (s, 2H), 3.96 (s, 4H), 3.65 (s, 2H), 3.58 (t, J = 6.4 Hz, 2H), 3.37 (s, 4H), 2.67 (t, J = 6.8 Hz, 2H), 1.54 (s, 6H).
Example 218: Synthesis of Compound 218 3-(5-(4-((4- piperazin-1-
 yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)isoxazole-5-carboxamide (218) was prepared according to the methods described above. LCMS [M+1] =798.3; 1H NMR (400 MHz, DMSO-d6) δ 13.99 (s, 1H), 10.51 (s, 1H), 9.74 (s, 1H), 9.25 (d, J = 1.2 Hz, 2H), 8.16 (s, 1H), 7.78 (t, J = 8.0 Hz, 1H), 7.73 (s, 1H), 7.50 - 7.34 (m, 2H), 6.74 (d, J = 11.6 Hz, 2H), 4.61 (d, J = 4.4 Hz, 2H), 3.67 - 3.57 (m, 4H), 3.27 (s, 4H), 2.69 (t, J = 6.4 Hz, 2H), 2.56 - 2.53 (m, 4H), 1.54 (s, 6H). Example 219: Synthesis of Compound 219 ’
yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)isoxazole-5-carboxamide (218) was prepared according to the methods described above. LCMS [M+1] =798.3; 1H NMR (400 MHz, DMSO-d6) δ 13.99 (s, 1H), 10.51 (s, 1H), 9.74 (s, 1H), 9.25 (d, J = 1.2 Hz, 2H), 8.16 (s, 1H), 7.78 (t, J = 8.0 Hz, 1H), 7.73 (s, 1H), 7.50 - 7.34 (m, 2H), 6.74 (d, J = 11.6 Hz, 2H), 4.61 (d, J = 4.4 Hz, 2H), 3.67 - 3.57 (m, 4H), 3.27 (s, 4H), 2.69 (t, J = 6.4 Hz, 2H), 2.56 - 2.53 (m, 4H), 1.54 (s, 6H). Example 219: Synthesis of Compound 219 ’
 3-(5- - - piperidin-1- yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)isoxazole-5-carboxamide (219) was prepared according to the methods described above. LCMS [M+1] = 813.4; 1H NMR (400 MHz, DMSO-d6) δ 13.99 (br s, 1H), 10.57 (s, 1H), 9.73 (br s, 1H), 9.25 (s, 2H), 7.82 - 7.66 (m, 2H), 7.47 - 7.30 (m, 2H), 6.90 (d, J = 10.0 Hz, 2H), 4.61 (d, J = 4.0 Hz, 2H), 4.55 - 4.45 (m, 1H), 3.68 - 3.57 (m, 4H), 2.77 - 2.65 (m, 4H), 2.33 (t, J = 9.6 Hz, 2H), 1.98 (d, J = 9.2 Hz, 2H), 1.73 - 1.61 (m, 2H), 1.54 (s, 6H).
Example 220: Synthesis of Compound 220 3-(5-(4-( azetidin-1-
3-(5- - - piperidin-1- yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)isoxazole-5-carboxamide (219) was prepared according to the methods described above. LCMS [M+1] = 813.4; 1H NMR (400 MHz, DMSO-d6) δ 13.99 (br s, 1H), 10.57 (s, 1H), 9.73 (br s, 1H), 9.25 (s, 2H), 7.82 - 7.66 (m, 2H), 7.47 - 7.30 (m, 2H), 6.90 (d, J = 10.0 Hz, 2H), 4.61 (d, J = 4.0 Hz, 2H), 4.55 - 4.45 (m, 1H), 3.68 - 3.57 (m, 4H), 2.77 - 2.65 (m, 4H), 2.33 (t, J = 9.6 Hz, 2H), 1.98 (d, J = 9.2 Hz, 2H), 1.73 - 1.61 (m, 2H), 1.54 (s, 6H).
Example 220: Synthesis of Compound 220 3-(5-(4-( azetidin-1-
 yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)isoxazole-5-carboxamide (220) was prepared according to the methods described above. LCMS [M+1] = 785.4; 1H NMR (400MHz, DMSO-d6) δ 13.99 (br s, 1H), 10.58 (s, 1H), 9.73 (br s, 1H), 9.23 (s, 2H), 7.77 - 7.72 (m, 2H), 7.38 - 7.33 (m, 2H), 6.81 (d, J = 9.2 Hz, 2H), 4.95 - 4.89 (m, 1H), 6.61 (d, J = 4.8 Hz, 2H), 3.82 (d, J = 6.8 Hz, 2H), 3.76 (s, 2H), 3.63 (t, J = 6.6 Hz, 2H), 3.14 - 3.11 (m, 2H), 2.70 (t, J = 6.6 Hz, 2H), 1.54 (s, 6H). Example 221: Synthesis of Compound 221 3-(5-(4-(
yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)isoxazole-5-carboxamide (220) was prepared according to the methods described above. LCMS [M+1] = 785.4; 1H NMR (400MHz, DMSO-d6) δ 13.99 (br s, 1H), 10.58 (s, 1H), 9.73 (br s, 1H), 9.23 (s, 2H), 7.77 - 7.72 (m, 2H), 7.38 - 7.33 (m, 2H), 6.81 (d, J = 9.2 Hz, 2H), 4.95 - 4.89 (m, 1H), 6.61 (d, J = 4.8 Hz, 2H), 3.82 (d, J = 6.8 Hz, 2H), 3.76 (s, 2H), 3.63 (t, J = 6.6 Hz, 2H), 3.14 - 3.11 (m, 2H), 2.70 (t, J = 6.6 Hz, 2H), 1.54 (s, 6H). Example 221: Synthesis of Compound 221 3-(5-(4-(
 -2- azaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro- 2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (221) was prepared according to the methods described above. LCMS [M+1] = 839.1; 1H NMR (400 MHz, DMSO- d6) δ 14.00 (s, 1H), 10.57 (s, 1H), 9.72 (s, 1H), 8.84 (s, 1H), 8.13 (s, 1H), 7.72 (s, 1H), 7.57 - 7.45 (m, 1H), 7.40 - 7.25 (m, 2H), 6.77 (d, J = 9.6 Hz, 2H), 4.72 - 4.64 (m, 1H), 4.60 (d, J = 5.6 Hz, 2H), 3.90 - 3.71 (m, 2H), 3.63 (t, J = 6.8 Hz, 2H), 2.77 - 2.71 (m, 2H), 2.71 - 2.67 (m, 2H), 2.54 (s, 4H), 2.47 (s, 3H), 2.25 - 2.14 (m, 2H), 1.54 (s, 6H). Example 222: Synthesis of Compound 222
3-(3-(4-((4-(3-( [1,5-a]pyrimidin-6- yl)piperazin-1-yl)methyl)phenyl)bicyclo[1.1.1]pentan-1-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (222) was prepared according to the methods described above. LCMS [M+1] = 773.1; 1H NMR (400 MHz, METHANOL-d4) δ 8.62 (d, J = 1.6 Hz, 1H), 8.31 (d, J = 1.6 Hz, 1H), 8.11 (s, 1H), 7.38 - 7.33 (m, 2H), 7.31 - 7.27 (m, 2H), 6.98 (s, 1H), 4.69 (s, 2H), 3.96 (t, J = 6.8 Hz, 2H), 3.65 (s, 2H), 3.25 - 3.20 (m, 4H), 2.87 (t, J = 6.8 Hz, 2H), 2.75 - 2.70 (m, 4H), 2.44 (s, 6H), 1.61 (s, 6H). Example 223: Synthesis of Compound 223 3-(3-(4-((4-(3-
-2- azaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro- 2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (221) was prepared according to the methods described above. LCMS [M+1] = 839.1; 1H NMR (400 MHz, DMSO- d6) δ 14.00 (s, 1H), 10.57 (s, 1H), 9.72 (s, 1H), 8.84 (s, 1H), 8.13 (s, 1H), 7.72 (s, 1H), 7.57 - 7.45 (m, 1H), 7.40 - 7.25 (m, 2H), 6.77 (d, J = 9.6 Hz, 2H), 4.72 - 4.64 (m, 1H), 4.60 (d, J = 5.6 Hz, 2H), 3.90 - 3.71 (m, 2H), 3.63 (t, J = 6.8 Hz, 2H), 2.77 - 2.71 (m, 2H), 2.71 - 2.67 (m, 2H), 2.54 (s, 4H), 2.47 (s, 3H), 2.25 - 2.14 (m, 2H), 1.54 (s, 6H). Example 222: Synthesis of Compound 222
3-(3-(4-((4-(3-( [1,5-a]pyrimidin-6- yl)piperazin-1-yl)methyl)phenyl)bicyclo[1.1.1]pentan-1-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (222) was prepared according to the methods described above. LCMS [M+1] = 773.1; 1H NMR (400 MHz, METHANOL-d4) δ 8.62 (d, J = 1.6 Hz, 1H), 8.31 (d, J = 1.6 Hz, 1H), 8.11 (s, 1H), 7.38 - 7.33 (m, 2H), 7.31 - 7.27 (m, 2H), 6.98 (s, 1H), 4.69 (s, 2H), 3.96 (t, J = 6.8 Hz, 2H), 3.65 (s, 2H), 3.25 - 3.20 (m, 4H), 2.87 (t, J = 6.8 Hz, 2H), 2.75 - 2.70 (m, 4H), 2.44 (s, 6H), 1.61 (s, 6H). Example 223: Synthesis of Compound 223 3-(3-(4-((4-(3-
 a]pyrazin-6- yl)piperazin-1-yl)methyl)phenyl)bicyclo[1.1.1]pentan-1-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (223) was prepared according to the methods described above. LCMS [M+1] = 773.3; 1H NMR (400 MHz, METHANOL-d4) δ 8.86 (d, J = 0.8 Hz, 1H), 7.92 (s, 1H), 7.87 (s, 1H), 7.39 - 7.30 (m, 4H), 6.97 (s, 1H), 4.69 (s, 2H), 3.96 (t, J = 6.8 Hz, 2H), 3.72 (s, 2H), 3.43 (s, 4H), 2.89 (t, J = 6.8 Hz, 2H), 2.76 (d, J = 4.4 Hz, 4H), 2.45 (s, 6H), 1.61 (s, 6H). Example 224: Synthesis of Compound 224
a]pyrazin-6- yl)piperazin-1-yl)methyl)phenyl)bicyclo[1.1.1]pentan-1-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (223) was prepared according to the methods described above. LCMS [M+1] = 773.3; 1H NMR (400 MHz, METHANOL-d4) δ 8.86 (d, J = 0.8 Hz, 1H), 7.92 (s, 1H), 7.87 (s, 1H), 7.39 - 7.30 (m, 4H), 6.97 (s, 1H), 4.69 (s, 2H), 3.96 (t, J = 6.8 Hz, 2H), 3.72 (s, 2H), 3.43 (s, 4H), 2.89 (t, J = 6.8 Hz, 2H), 2.76 (d, J = 4.4 Hz, 4H), 2.45 (s, 6H), 1.61 (s, 6H). Example 224: Synthesis of Compound 224
3-(5-(4-((6-(4-(2 ophenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)phenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2- yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (224) was prepared according to the methods described above. LCMS [M+1] = 773.5; 1H NMR (400 MHz, DMSO-d6) δ 14.01 (s, 1H), 10.45 (s, 1H), 9.74 (s, 1H), 9.35 (s, 2H), 8.17 (s, 1H), 7.86 (d, J = 8.0 Hz, 2H), 7.71 (s, 1H), 7.47 (d, J = 8.0 Hz, 2H), 7.33 (t, J = 8.0 Hz, 1H), 7.19 - 7.02 (m, 2H), 4.61 (d, J = 5.6 Hz, 2H), 3.67 (t, J = 6.8 Hz, 2H), 3.63 (s, 2H), 3.41 (d, J = 8.4 Hz, 2H), 3.13 (s, 2H), 2.75 - 2.61 (m, 3H), 2.56 - 2.53 (m, 2H), 2.28 - 2.14 (m, 2H), 1.54 (s, 6H). Example 225: Synthesis of Compound 225 3-(5-(4-((4-
 - - piperidin-1- yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)isoxazole-5-carboxamide (225) was prepared according to the methods described above. LCMS [M+1] =775.5; 1H NMR (400 MHz, DMSO-d6) δ 14.01 (bs, 1H), 10.31 (s, 1H), 9.75 (t, J = 5.6 Hz, 1H), 9.25 (d, J = 1.2 Hz, 2H), 8.20 (s, 1H), 7.77 (t, J = 8.0 Hz, 1H), 7.73 (s, 1H), 7.47 - 7.34 (m, 2H), 7.30 - 7.17 (m, 2H), 7.11 (d, J = 6.4 Hz, 1H), 4.61 (d, J = 5.6 Hz, 2H), 3.76 – 3.69 (m, 1H), 3.63 (s, 2H), 3.51 - 3.44 (m, 2H), 2.98 (s, 2H), 2.82 - 2.72 (m, 2H), 2.70 - 2.63 (m, 1H), 2.17 (s, 1H), 2.14 (s, 3H), 1.71 (d, J = 4.0 Hz, 4H), 1.54 (s, 6H).
Example 226: Synthesis of Compound 226 3-(5-(4-((4-
- - piperidin-1- yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)isoxazole-5-carboxamide (225) was prepared according to the methods described above. LCMS [M+1] =775.5; 1H NMR (400 MHz, DMSO-d6) δ 14.01 (bs, 1H), 10.31 (s, 1H), 9.75 (t, J = 5.6 Hz, 1H), 9.25 (d, J = 1.2 Hz, 2H), 8.20 (s, 1H), 7.77 (t, J = 8.0 Hz, 1H), 7.73 (s, 1H), 7.47 - 7.34 (m, 2H), 7.30 - 7.17 (m, 2H), 7.11 (d, J = 6.4 Hz, 1H), 4.61 (d, J = 5.6 Hz, 2H), 3.76 – 3.69 (m, 1H), 3.63 (s, 2H), 3.51 - 3.44 (m, 2H), 2.98 (s, 2H), 2.82 - 2.72 (m, 2H), 2.70 - 2.63 (m, 1H), 2.17 (s, 1H), 2.14 (s, 3H), 1.71 (d, J = 4.0 Hz, 4H), 1.54 (s, 6H).
Example 226: Synthesis of Compound 226 3-(5-(4-((4-
 piperidin-1- yl)methyl)piperidin-1-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)isoxazole-5-carboxamide (226) was prepared according to the methods described above. LCMS [M+1] = 764.3; 1H NMR (400 MHz, METHANOL-d4) δ 8.60 (s, 2H), 7.52 (s, 1H), 7.37 - 7.27 (m, 2H), 7.21 (dd, J = 2.0, 6.8 Hz, 1H), 4.75 (s, 2H), 4.09 (d, J = 12.8 Hz, 2H), 3.85 (ddd, J = 6.0, 9.2, 12.8 Hz, 1H), 3.77 (d, J = 12.4 Hz, 2H), 3.64 (td, J = 6.4, 12.4 Hz, 1H), 3.27 - 3.11 (m, 5H), 3.05 (t, J = 12 Hz, 2H), 2.95 - 2.77 (m, 2H), 2.34 - 2.20 (m, 4H), 2.15 - 1.94 (m, 6H), 1.62 (s, 6H), 1.58 - 1.44 (m, 2H). Example 227: Synthesis of Compound 227 3-(5-(4-(
piperidin-1- yl)methyl)piperidin-1-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)isoxazole-5-carboxamide (226) was prepared according to the methods described above. LCMS [M+1] = 764.3; 1H NMR (400 MHz, METHANOL-d4) δ 8.60 (s, 2H), 7.52 (s, 1H), 7.37 - 7.27 (m, 2H), 7.21 (dd, J = 2.0, 6.8 Hz, 1H), 4.75 (s, 2H), 4.09 (d, J = 12.8 Hz, 2H), 3.85 (ddd, J = 6.0, 9.2, 12.8 Hz, 1H), 3.77 (d, J = 12.4 Hz, 2H), 3.64 (td, J = 6.4, 12.4 Hz, 1H), 3.27 - 3.11 (m, 5H), 3.05 (t, J = 12 Hz, 2H), 2.95 - 2.77 (m, 2H), 2.34 - 2.20 (m, 4H), 2.15 - 1.94 (m, 6H), 1.62 (s, 6H), 1.58 - 1.44 (m, 2H). Example 227: Synthesis of Compound 227 3-(5-(4-(
 piperidin-1- yl)methyl)piperidin-1-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)isoxazole-5-carboxamide (227) was prepared according to the methods described above. LCMS [M+1] = 784.5. 1H NMR (400 MHz, DMSO-d6) δ 13.76 (br s, 1H), 10.45 (br s, 1H), 9.63 (t, J = 5.2 Hz, 1H), 8.63 (s, 2H), 7.53 (s, 1H), 7.43 - 7.34 (m, 3H), 4.58 (d, J = 5.2 Hz, 2H), 4.01 (d, J = 12.4 Hz, 2H), 3.74 - 3.66 (m, 1H), 3.62 - 3.56 (m, 1H), 2.99 - 2.88 (m, 5H), 2.75 - 2.72 (m, 2H), 2.22 (d, J = 6.8 Hz, 2H), 2.03 (t, J = 11.2 Hz, 2H), 1.86 - 1.64 (m, 7H), 1.54 (s, 6H), 1.26 - 1.18 (m, 2H). Example 228: Synthesis of Compounds 228-1 and 228-2
piperidin-1- yl)methyl)piperidin-1-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)isoxazole-5-carboxamide (227) was prepared according to the methods described above. LCMS [M+1] = 784.5. 1H NMR (400 MHz, DMSO-d6) δ 13.76 (br s, 1H), 10.45 (br s, 1H), 9.63 (t, J = 5.2 Hz, 1H), 8.63 (s, 2H), 7.53 (s, 1H), 7.43 - 7.34 (m, 3H), 4.58 (d, J = 5.2 Hz, 2H), 4.01 (d, J = 12.4 Hz, 2H), 3.74 - 3.66 (m, 1H), 3.62 - 3.56 (m, 1H), 2.99 - 2.88 (m, 5H), 2.75 - 2.72 (m, 2H), 2.22 (d, J = 6.8 Hz, 2H), 2.03 (t, J = 11.2 Hz, 2H), 1.86 - 1.64 (m, 7H), 1.54 (s, 6H), 1.26 - 1.18 (m, 2H). Example 228: Synthesis of Compounds 228-1 and 228-2
3-(5-(4-(((3aS*,6 H)-yl)-3- fluorophenyl)hexahydropyrrolo[3,2-b]pyrrol-1(2H)-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)- N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5- carboxamide (228) was prepared according to the methods described above. Stereoisomers were separated by SFC ( (S,S)-WHELK-O1 (250 x 30 mm, 10 µm); [CO2-i-PrOH/ acetonitrile]; B%:63%, isocratic elution) to afford 228-1 as the early eluting isomer and 228-2 as the late eluting isomer. LCMS [M+1] = 806.2; 1H NMR (400 MHz, DMSO-d6) δ 13.99 (s, 1H), 10.33 (s, 1H), 9.73 (s, 1H), 9.24 (d, J = 1.2 Hz, 2H), 7.78 - 7.72 (m, 2H), 7.41 - 7.39 (m, 2H), 7.15 (t, J = 8.8 Hz, 1H), 6.43 - 6.37 (m, 2H), 4.61 (d, J = 4.0 Hz, 2H), 4.28 - 4.23 (m, 1H), 4.06 (d, J = 14.0 Hz, 1H), 3.60 (t, J = 6.8 Hz, 2H), 3.48 (d, J = 13.6 Hz, 1H), 3.39 - 3.35 (m, 2H), 2.84 (t, J = 6.8 Hz, 1H), 2.68 (t, J = 6.8 Hz, 2H), 2.33 - 2.25 (m, 2H), 2.06 - 2.03 (m, 1H), 1.89 - 1.80 (m, 1H), 1.54 (s, 6H), 1.41 - 1.35 (m, 1H), 1.26 - 1.17 (m, 1H). Example 229: Synthesis of Compound 229 3-(5-(4-((6-
 -2- azaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-
2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (229) was prepared according to the methods described above. LCMS [M+1] = 823.4; 1HNMR (400 MHz, DMSO- d6) δ 14.34 (bs, 1H), 10.59 (s, 1H), 9.73 (t, J = 5.6 Hz, 1H), 8.85 (s, 1H), 8.18 (s, 1H), 7.72 (s, 1H), 7.49 (t, J = 7.2 Hz, 1H), 7.34 - 7.26 (m, 2H), 7.09 (d, J = 9.2 Hz, 2H), 4.60 (d, J = 5.6 Hz, 2H), 3.68 - 3.64 (m, 4H), 3.44 (s, 1H), 3.39 - 3.37 (m, 2H), 3.16 (s, 2H), 2.70 (t, J = 6.8 Hz, 2H), 2.55 - 2.52 (m, 2H), 2.48 - 2.46 (m, 3H), 2.27 - 2.20 (m, 2H), 1.54 (s, 6H). Example 230: Synthesis of Compound 230 3-(5-(5-((6-(4-
-2- azaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-
2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (229) was prepared according to the methods described above. LCMS [M+1] = 823.4; 1HNMR (400 MHz, DMSO- d6) δ 14.34 (bs, 1H), 10.59 (s, 1H), 9.73 (t, J = 5.6 Hz, 1H), 8.85 (s, 1H), 8.18 (s, 1H), 7.72 (s, 1H), 7.49 (t, J = 7.2 Hz, 1H), 7.34 - 7.26 (m, 2H), 7.09 (d, J = 9.2 Hz, 2H), 4.60 (d, J = 5.6 Hz, 2H), 3.68 - 3.64 (m, 4H), 3.44 (s, 1H), 3.39 - 3.37 (m, 2H), 3.16 (s, 2H), 2.70 (t, J = 6.8 Hz, 2H), 2.55 - 2.52 (m, 2H), 2.48 - 2.46 (m, 3H), 2.27 - 2.20 (m, 2H), 1.54 (s, 6H). Example 230: Synthesis of Compound 230 3-(5-(5-((6-(4-
 -2- azaspiro[3.3]heptan-2-yl)methyl)-3-fluoropyridin-2-yl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (230) was prepared according to the methods described above. LCMS [M+1] = 806.2; 1H NMR (400 MHz, DMSO-d6) δ 13.98 (bs, 1H), 10.46 (s, 1H), 9..73 (s, 1H), 9.01 (d, J = 0.8 Hz, 1H), 8.56 (s, 1H), 7.85 (dd, J = 0.8, 10.8 Hz, 1H), 7.75 (s, 1H), 7.34 (t, J = 8.4 Hz, 1H), 7.16 (dd, J = 1.2, 11.8 Hz, 1H), 7.09 (d, J = 8.0 Hz, 1H), 4.60 (s, 2H), 3.71 (s, 2H), 3.70 (t, J = 6.8 Hz, 2H), 3.44 (s, 1H), 3.39 (s, 2H), 3.19 (s, 2H), 2.71 (t, J = 6.8 Hz, 2H), 2.55 (s, 5H), 2.26 - 2.19 (m, 2H), 1.55 (s, 6H). Example 231: Synthesis of Compound 231
-2- azaspiro[3.3]heptan-2-yl)methyl)-3-fluoropyridin-2-yl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (230) was prepared according to the methods described above. LCMS [M+1] = 806.2; 1H NMR (400 MHz, DMSO-d6) δ 13.98 (bs, 1H), 10.46 (s, 1H), 9..73 (s, 1H), 9.01 (d, J = 0.8 Hz, 1H), 8.56 (s, 1H), 7.85 (dd, J = 0.8, 10.8 Hz, 1H), 7.75 (s, 1H), 7.34 (t, J = 8.4 Hz, 1H), 7.16 (dd, J = 1.2, 11.8 Hz, 1H), 7.09 (d, J = 8.0 Hz, 1H), 4.60 (s, 2H), 3.71 (s, 2H), 3.70 (t, J = 6.8 Hz, 2H), 3.44 (s, 1H), 3.39 (s, 2H), 3.19 (s, 2H), 2.71 (t, J = 6.8 Hz, 2H), 2.55 (s, 5H), 2.26 - 2.19 (m, 2H), 1.55 (s, 6H). Example 231: Synthesis of Compound 231
3-(5-((6-(4-(2, nyl)-2- azaspiro[3.3]heptan-2-yl)methyl)-[2,5'-bipyrimidin]-2'-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan- 2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (231) was prepared according to the methods described above. LCMS [M+1] = 775.4; 1H NMR (400 MHz, DMSO-d6) δ 13.99 (br s, 1H), 10.45 (s, 1H), 9.79 (s, 2H), 9.74 (br s, 1H), 8.95 (s, 2H), 7.77 (s, 1H), 7.33 (t, J = 8.0 Hz, 1H), 7.15 (dd, J = 1.6, 11.6 Hz, 1H), 7.08 (dd, J = 1.6, 8.4 Hz, 1H), 4.61 (br d, J = 5.6 Hz, 2H), 3.82 - 3.71 (m, 2H), 3.67 (t, J = 6.4 Hz, 2H), 3.50 - 3.37 (m, 3H), 3.24 (br s, 2H), 2.73 - 2.67 (m, 2H), 2.53 (br s, 2H), 2.28 - 2.16 (m, 2H), 1.54 (s, 6H). Example 232: Synthesis of Compound 232 3-(5-(4-((6-(4-
 -2,6- diazaspiro[3.3]heptan-2-yl)methyl)phenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan- 2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (232) was prepared according to the methods described above. LCMS [M+1] = 774.3; 1H NMR (400 MHz, METHANOL-d4) δ 9.22 (s, 2H), 7.80 (d, J = 8.4 Hz, 2H), 7.66 (s, 1H), 7.52 (d, J = 8.4 Hz, 2H), 7.15 (t, J = 8.4 Hz, 1H),
6.27 (d, J = 10.0 Hz, 2H), 4.74 (s, 2H), 3.96 (s, 4H), 3.75 - 3.67 (m, 4H), 3.51 (s, 4H), 2.78 (t, J = 6.8 Hz, 2H), 1.62 (s, 6H). Example A: Androgen Receptor Degradation HiBiT Reporter Assay LNCaP clone FGC prostate cancer cells were genetically modified by genetically engineering a HiBiT epitope tag into the endogenous locus for the androgen receptor (AR) gene to generate a stable cell line expressing HiBit-AR fusion protein (LNCaP.7-HiBiT-AR). LNCaP.7-HiBiT-AR and LNCaP clone FGC cells were treated with a dose-titration of AR modulators in duplicate in the presence of 100 nM dihydrotestosterone in 384-well plates and incubated for 24 hours in a 37 °C incubator. Multiple replicates of vehicle (DMSO) and a positive control compound were included for data normalization. After incubation with compounds, the relative abundance of the HiBiT-AR fusion protein in LNCaP.7-HiBiT-AR was evaluated using Nano-Glo® HiBiT Lytic Detection System (Promega) with luminescence detection being performed using an EnVision (Perkin Elmer) multimode plate reader. Cell health and non-specifc toxicity was evaluated at the same time point in LNCaP clone FGC using CellTiter-Glo Luminescent Cell Viability Assay with luminescence detection being performed using an EnVision multimode plate reader. The percent degradation was calculated by normalizing the luminescence units to DMSO (0% inhibition) and positive control (100% inhibition). Curve fitting was performed using 4-parameter PKI Logistic Regression in Spotfire (Tibco) for calculation of DC50, and maximal degradation efficacy (Dmax) for AR degradation. Data are shown below in Table 1. Example B: PSA Expression HiBiT Reporter Assay LNCaP clone FGC prostate cancer cells were genetically modified by genetically engineering a HiBiT epitope tag into the endogenous locus of KLK3 (kallikrein related peptidase 3) gene which encodes prostate specific antigen (PSA) protein to generate a stable cell line expressing HiBit-PSA fusion protein (LNCaP.18-HiBiT-PSA). LNCaP.18-HiBiT-PSA and LNCaP clone FGC cells were treated with a dose-titration of AR modulators in duplicate in the presence of 100 nM dihydrotestosterone in 384-well plates and incubated for 24 hours in a 37 °C incubator. Multiple replicates of vehicle (DMSO) and a positive control compound were included for data normalization. After incubation with compounds, the
relative abundance of the HiBiT-PSA fusion protein in LNCaP.18-HiBiT-PSA was evaluated using Nano-Glo® HiBiT Lytic Detection System (Promega) with luminescence detection being performed using an EnVision (Perkin Elmer) multimode plate reader. Cell health and non-specifc toxicity was evaluated at the same time point in LNCaP clone FGC using CellTiter-Glo Luminescent Cell Viability Assay with luminescence detection being performed using an EnVision multimode plate reader. The percent decrease in HiBiT-PSA was calculated by first normalizing the luminescence units to DMSO (0% inhibition) and positive control (100% inhibition) followed by curve fitting using 4-parameter PKI Logistic Regression in Spotfire (Tibco). EC50, and maximal efficacy (Emax) for reduction in PSA protein are reported. Data are shown below in Table 1. Table 1. Data (Examples A and B) Compound AR DC50 AR DMax PSA EC50 PSA Emax
-2,6- diazaspiro[3.3]heptan-2-yl)methyl)phenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan- 2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide (232) was prepared according to the methods described above. LCMS [M+1] = 774.3; 1H NMR (400 MHz, METHANOL-d4) δ 9.22 (s, 2H), 7.80 (d, J = 8.4 Hz, 2H), 7.66 (s, 1H), 7.52 (d, J = 8.4 Hz, 2H), 7.15 (t, J = 8.4 Hz, 1H),
6.27 (d, J = 10.0 Hz, 2H), 4.74 (s, 2H), 3.96 (s, 4H), 3.75 - 3.67 (m, 4H), 3.51 (s, 4H), 2.78 (t, J = 6.8 Hz, 2H), 1.62 (s, 6H). Example A: Androgen Receptor Degradation HiBiT Reporter Assay LNCaP clone FGC prostate cancer cells were genetically modified by genetically engineering a HiBiT epitope tag into the endogenous locus for the androgen receptor (AR) gene to generate a stable cell line expressing HiBit-AR fusion protein (LNCaP.7-HiBiT-AR). LNCaP.7-HiBiT-AR and LNCaP clone FGC cells were treated with a dose-titration of AR modulators in duplicate in the presence of 100 nM dihydrotestosterone in 384-well plates and incubated for 24 hours in a 37 °C incubator. Multiple replicates of vehicle (DMSO) and a positive control compound were included for data normalization. After incubation with compounds, the relative abundance of the HiBiT-AR fusion protein in LNCaP.7-HiBiT-AR was evaluated using Nano-Glo® HiBiT Lytic Detection System (Promega) with luminescence detection being performed using an EnVision (Perkin Elmer) multimode plate reader. Cell health and non-specifc toxicity was evaluated at the same time point in LNCaP clone FGC using CellTiter-Glo Luminescent Cell Viability Assay with luminescence detection being performed using an EnVision multimode plate reader. The percent degradation was calculated by normalizing the luminescence units to DMSO (0% inhibition) and positive control (100% inhibition). Curve fitting was performed using 4-parameter PKI Logistic Regression in Spotfire (Tibco) for calculation of DC50, and maximal degradation efficacy (Dmax) for AR degradation. Data are shown below in Table 1. Example B: PSA Expression HiBiT Reporter Assay LNCaP clone FGC prostate cancer cells were genetically modified by genetically engineering a HiBiT epitope tag into the endogenous locus of KLK3 (kallikrein related peptidase 3) gene which encodes prostate specific antigen (PSA) protein to generate a stable cell line expressing HiBit-PSA fusion protein (LNCaP.18-HiBiT-PSA). LNCaP.18-HiBiT-PSA and LNCaP clone FGC cells were treated with a dose-titration of AR modulators in duplicate in the presence of 100 nM dihydrotestosterone in 384-well plates and incubated for 24 hours in a 37 °C incubator. Multiple replicates of vehicle (DMSO) and a positive control compound were included for data normalization. After incubation with compounds, the
relative abundance of the HiBiT-PSA fusion protein in LNCaP.18-HiBiT-PSA was evaluated using Nano-Glo® HiBiT Lytic Detection System (Promega) with luminescence detection being performed using an EnVision (Perkin Elmer) multimode plate reader. Cell health and non-specifc toxicity was evaluated at the same time point in LNCaP clone FGC using CellTiter-Glo Luminescent Cell Viability Assay with luminescence detection being performed using an EnVision multimode plate reader. The percent decrease in HiBiT-PSA was calculated by first normalizing the luminescence units to DMSO (0% inhibition) and positive control (100% inhibition) followed by curve fitting using 4-parameter PKI Logistic Regression in Spotfire (Tibco). EC50, and maximal efficacy (Emax) for reduction in PSA protein are reported. Data are shown below in Table 1. Table 1. Data (Examples A and B) Compound AR DC50 AR DMax PSA EC50 PSA Emax
 24 1.14 77 1.64 66 25 2.22 59 1.05 80
24 1.14 77 1.64 66 25 2.22 59 1.05 80
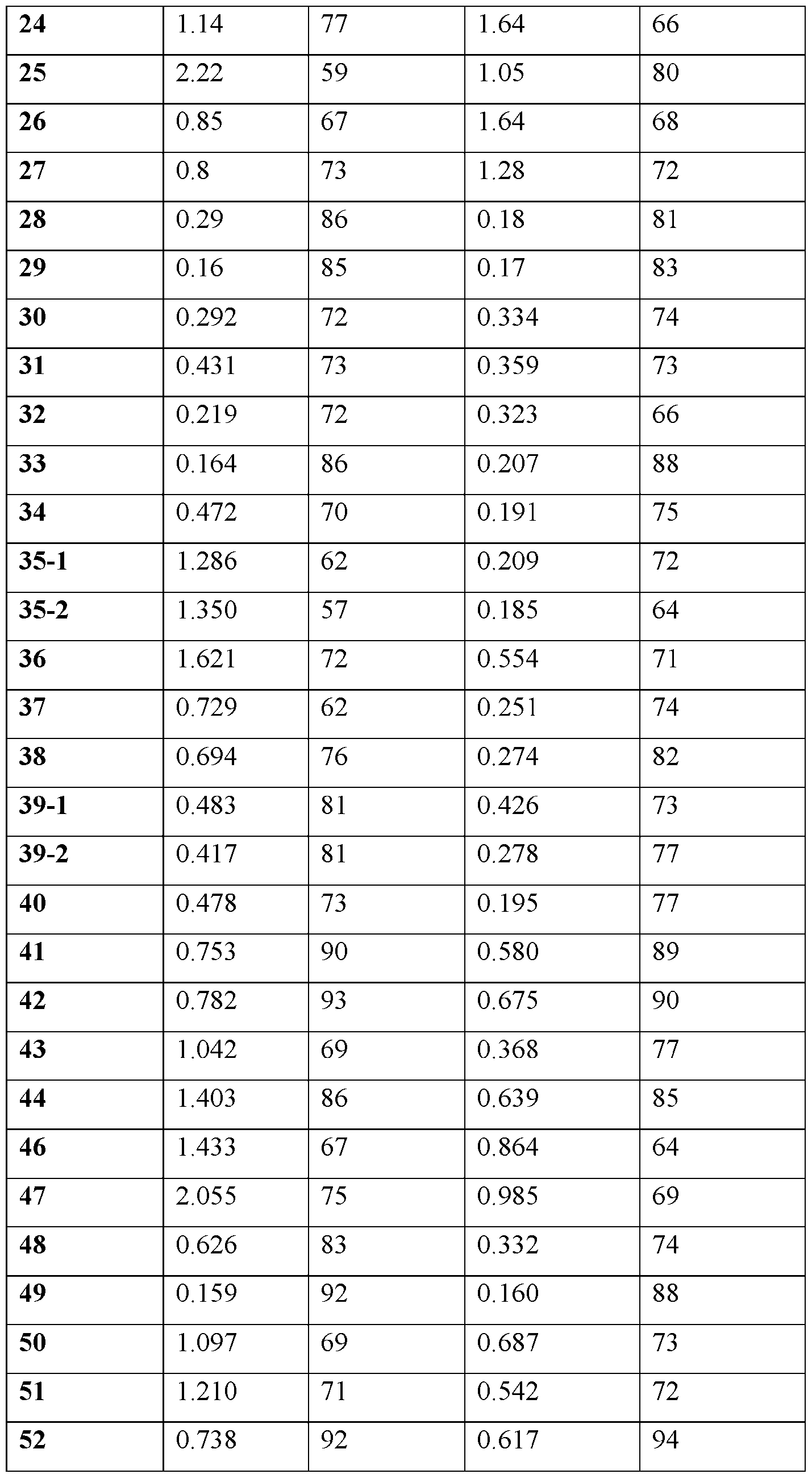 53 0.783 79 0.420 74 54 0.909 75 0.360 75
53 0.783 79 0.420 74 54 0.909 75 0.360 75
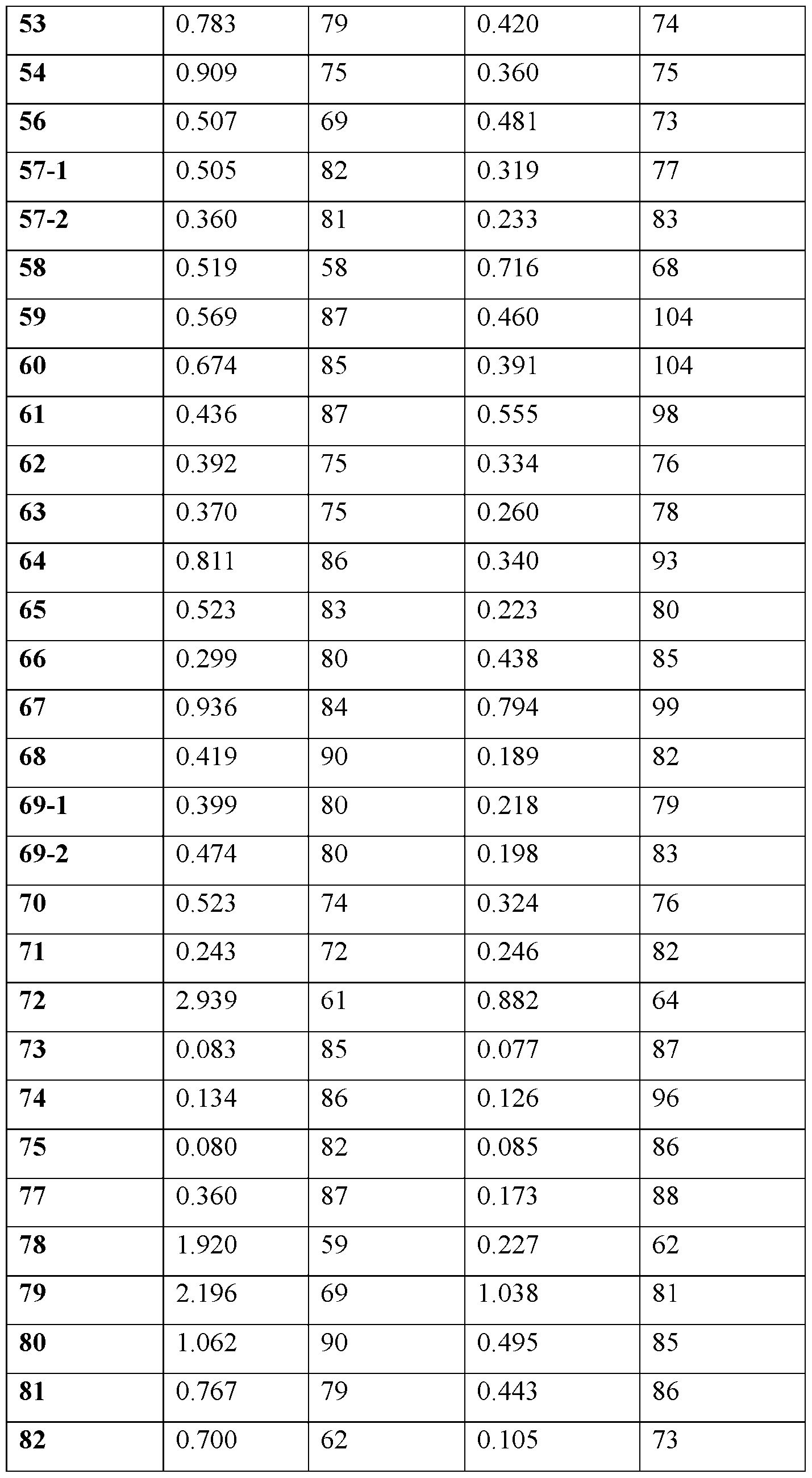 83 0.244 67 0.201 70 84 1.425 59 0.545 81
83 0.244 67 0.201 70 84 1.425 59 0.545 81
 125 0.079 84 0.031 95 127 0.860 73 0.901 79
125 0.079 84 0.031 95 127 0.860 73 0.901 79
 158 0.022 100 0.019 100 159 0.035 100 0.028 99
158 0.022 100 0.019 100 159 0.035 100 0.028 99
 185 0.090 96 0.087 98 186 0.041 97 0.034 98
185 0.090 96 0.087 98 186 0.041 97 0.034 98
 202-2 0.063 93 0.045 96 203-1 0.086 96 0.062 97
202-2 0.063 93 0.045 96 203-1 0.086 96 0.062 97
 225 0.038 81 0.042 80 226 0.099 85 0.089 87 Vario
225 0.038 81 0.042 80 226 0.099 85 0.089 87 Vario
 us o ca o s o e e o , a o o ose esc e een, will be apparent to those skilled in the art from the foregoing description. Such modifications are also intended to fall within the scope of the appended claims. Each reference, including all patent, patent applications, and publications, cited in the present application is incorporated herein by reference in its entirety.
us o ca o s o e e o , a o o ose esc e een, will be apparent to those skilled in the art from the foregoing description. Such modifications are also intended to fall within the scope of the appended claims. Each reference, including all patent, patent applications, and publications, cited in the present application is incorporated herein by reference in its entirety.
Claims
What is claimed is: 1. A compound of Formula I:
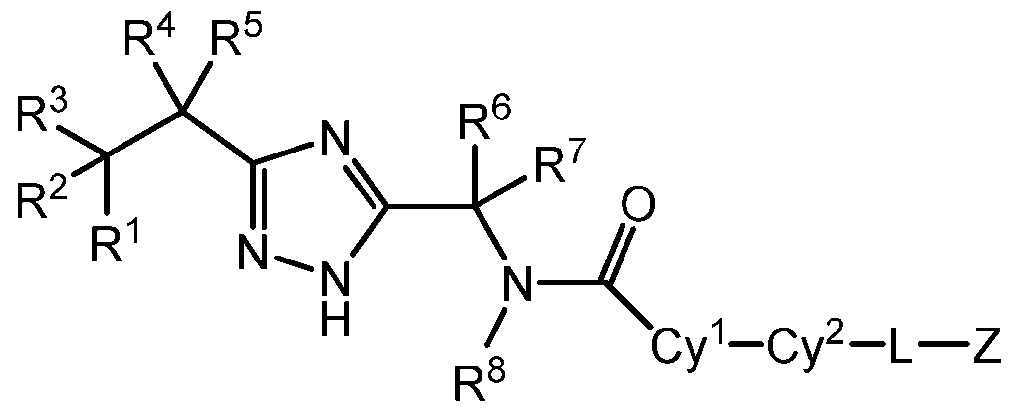 or a pharmaceutically acceptable salt thereof, wherein: R1, R2, and R3 are each independently selected from H, halo, SRA, ORA, C1-4 alkyl, C2-4 alkenyl, and C1-4 haloalkyl, wherein the C2-4 alkenyl is optionally substituted with 1, 2, or 3 substituents independently selected from halo, and wherein RA is H, C1-4 alkyl, or C1-4 haloalkyl; R4 and R5 are each independently selected from H, halo, SRA, ORA, C1-4 alkyl, C2-4 alkenyl, and C1-4 haloalkyl, wherein the C2-4 alkenyl is optionally substituted with 1, 2, or 3 substituents independently selected from halo, and wherein RA is H, C1-4 alkyl, or C1-4 haloalkyl; or R4 and R5 together with the carbon atom to which they are attached form a C3-6 cycloalkyl group or a 3-6 membered heterocycloalkyl group, each optionally substituted with 1, 2, or 3 substituents independently selected from H, halo, ORA, C1-4 alkyl, and C1-4 haloalkyl; R6 and R7 are each independently selected from H, halo, C1-4 alkyl, and C1-4 haloalkyl; R8 is H or C1-4 alkyl; Cy1 is selected from C6-10 aryl, C3-14 cycloalkyl, 5-14 membered heteroaryl, 4-14 membered heterocycloalkyl, C3-7 cycloalkyl fused with phenyl to form a bicyclic ring, C4-7 cycloalkyl fused with 5-6 membered heteroaryl to form a bicyclic ring, 4-7 membered heterocycloalkyl fused with phenyl to form a bicyclic ring, and 4-7 membered heterocycloalkyl fused with 5-6 membered heteroaryl to form a bicyclic ring, each optionally substituted by 1, 2, 3, or 4 substituents independently selected from halo, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C1-6 haloalkyl, CN, NO2, ORa, SRa, C1-6 alkyl-NRcRd, C(O)Rb, C(O)NRcRd, C(O)ORa, OC(O)Rb, OC(O)NRcRd, C(=NRe)NRcRd, NRcC(=NRe)NRcRd, NRcRd, NRcC(O)Rb, NRcC(O)ORa,
Cy2 is absent or selected from C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, and 4- 7 membered heterocycloalkyl, each optionally substituted by 1, 2, 3, or 4 substituents independently selected from 5-6 membered heteroaryl, halo, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C1-6 haloalkyl, CN, NO2, ORa1, SRa1, C(O)Rb1, C(O)NRc1Rd1, C(O)ORa1, OC(O)Rb1, OC(O)NRc1Rd1, C(=NRe1)NRc1Rd1, NRc1C(=NRe1)NRc1Rd1, NRc1Rd1, NRc1C(O)Rb1, NRc1C(O)ORa1, NRc1C(O)NRc1Rd1, NRc1S(O)Rb1, NRc1S(O)2Rb1, NRc1S(O)2NRc1Rd1, S(O)Rb1, S(O)NRc1Rd1, S(O)2Rb1, and S(O)2NRc1Rd1, wherein said 5-6 membered heteroaryl substituent of Cy2 is optionally substituted with 1 or 2 substituents independently selected from halo and C1-6 alkyl; L is a linker; and Z is a binder of cereblon E3 ligase; each Ra, Rb, Rc, Rd, Ra1, Rb1, Rc1, and Rd1 is independently selected from H, C1-6 alkyl, C1-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl-C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl, wherein said C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl-C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl of Ra, Rb, Rc, Rd, Ra1, Rb1, Rc1, and Rd1 is optionally substituted with 1, 2, 3, 4, or 5 substituents independently selected from halo, C1-4 alkyl, C1-4 haloalkyl, C1-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, CN, ORa3, SRa3, C(O)Rb3, C(O)NRc3Rd3, C(O)ORa3, OC(O)Rb3, OC(O)NRc3Rd3, NRc3Rd3, NRc3C(O)Rb3, NRc3C(O)NRc3Rd3,
or a pharmaceutically acceptable salt thereof, wherein: R1, R2, and R3 are each independently selected from H, halo, SRA, ORA, C1-4 alkyl, C2-4 alkenyl, and C1-4 haloalkyl, wherein the C2-4 alkenyl is optionally substituted with 1, 2, or 3 substituents independently selected from halo, and wherein RA is H, C1-4 alkyl, or C1-4 haloalkyl; R4 and R5 are each independently selected from H, halo, SRA, ORA, C1-4 alkyl, C2-4 alkenyl, and C1-4 haloalkyl, wherein the C2-4 alkenyl is optionally substituted with 1, 2, or 3 substituents independently selected from halo, and wherein RA is H, C1-4 alkyl, or C1-4 haloalkyl; or R4 and R5 together with the carbon atom to which they are attached form a C3-6 cycloalkyl group or a 3-6 membered heterocycloalkyl group, each optionally substituted with 1, 2, or 3 substituents independently selected from H, halo, ORA, C1-4 alkyl, and C1-4 haloalkyl; R6 and R7 are each independently selected from H, halo, C1-4 alkyl, and C1-4 haloalkyl; R8 is H or C1-4 alkyl; Cy1 is selected from C6-10 aryl, C3-14 cycloalkyl, 5-14 membered heteroaryl, 4-14 membered heterocycloalkyl, C3-7 cycloalkyl fused with phenyl to form a bicyclic ring, C4-7 cycloalkyl fused with 5-6 membered heteroaryl to form a bicyclic ring, 4-7 membered heterocycloalkyl fused with phenyl to form a bicyclic ring, and 4-7 membered heterocycloalkyl fused with 5-6 membered heteroaryl to form a bicyclic ring, each optionally substituted by 1, 2, 3, or 4 substituents independently selected from halo, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C1-6 haloalkyl, CN, NO2, ORa, SRa, C1-6 alkyl-NRcRd, C(O)Rb, C(O)NRcRd, C(O)ORa, OC(O)Rb, OC(O)NRcRd, C(=NRe)NRcRd, NRcC(=NRe)NRcRd, NRcRd, NRcC(O)Rb, NRcC(O)ORa,
Cy2 is absent or selected from C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, and 4- 7 membered heterocycloalkyl, each optionally substituted by 1, 2, 3, or 4 substituents independently selected from 5-6 membered heteroaryl, halo, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C1-6 haloalkyl, CN, NO2, ORa1, SRa1, C(O)Rb1, C(O)NRc1Rd1, C(O)ORa1, OC(O)Rb1, OC(O)NRc1Rd1, C(=NRe1)NRc1Rd1, NRc1C(=NRe1)NRc1Rd1, NRc1Rd1, NRc1C(O)Rb1, NRc1C(O)ORa1, NRc1C(O)NRc1Rd1, NRc1S(O)Rb1, NRc1S(O)2Rb1, NRc1S(O)2NRc1Rd1, S(O)Rb1, S(O)NRc1Rd1, S(O)2Rb1, and S(O)2NRc1Rd1, wherein said 5-6 membered heteroaryl substituent of Cy2 is optionally substituted with 1 or 2 substituents independently selected from halo and C1-6 alkyl; L is a linker; and Z is a binder of cereblon E3 ligase; each Ra, Rb, Rc, Rd, Ra1, Rb1, Rc1, and Rd1 is independently selected from H, C1-6 alkyl, C1-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl-C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl, wherein said C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl-C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl of Ra, Rb, Rc, Rd, Ra1, Rb1, Rc1, and Rd1 is optionally substituted with 1, 2, 3, 4, or 5 substituents independently selected from halo, C1-4 alkyl, C1-4 haloalkyl, C1-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, CN, ORa3, SRa3, C(O)Rb3, C(O)NRc3Rd3, C(O)ORa3, OC(O)Rb3, OC(O)NRc3Rd3, NRc3Rd3, NRc3C(O)Rb3, NRc3C(O)NRc3Rd3,
 heterocycloalkyl group optionally substituted with 1, 2, or 3 substituents independently selected from halo, C1-4 alkyl, C1-4 haloalkyl, CN, ORa3, SRa3, C(O)Rb3, C(O)NRc3Rd3, C(O)ORa3, OC(O)Rb3, OC(O)NRc3Rd3, NRc3Rd3, NRc3C(O)Rb3, NRc3C(O)NRc3Rd3, NRc3C(O)ORa3,
heterocycloalkyl group optionally substituted with 1, 2, or 3 substituents independently selected from halo, C1-4 alkyl, C1-4 haloalkyl, CN, ORa3, SRa3, C(O)Rb3, C(O)NRc3Rd3, C(O)ORa3, OC(O)Rb3, OC(O)NRc3Rd3, NRc3Rd3, NRc3C(O)Rb3, NRc3C(O)NRc3Rd3, NRc3C(O)ORa3,
 heterocycloalkyl group optionally substituted with 1, 2, or 3 substituents independently selected
from halo, C1-4 alkyl, C1-4 haloalkyl, CN, ORa3, SRa3, C(O)Rb3, C(O)NRc3Rd3, C(O)ORa3, OC(O)Rb3, OC(O)NRc3Rd3, NRc3Rd3, NRc3C(O)Rb3, NRc3C(O)NRc3Rd3, NRc3C(O)ORa3, C2-
heterocycloalkyl group optionally substituted with 1, 2, or 3 substituents independently selected
from halo, C1-4 alkyl, C1-4 haloalkyl, CN, ORa3, SRa3, C(O)Rb3, C(O)NRc3Rd3, C(O)ORa3, OC(O)Rb3, OC(O)NRc3Rd3, NRc3Rd3, NRc3C(O)Rb3, NRc3C(O)NRc3Rd3, NRc3C(O)ORa3, C2-
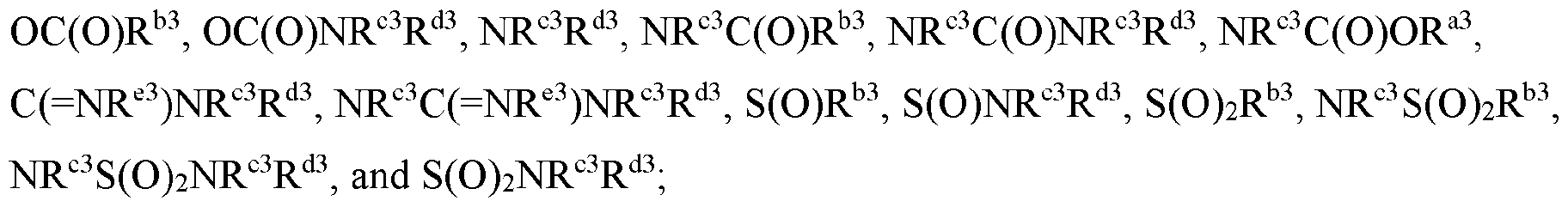 heterocycloalkyl, C6-10 aryl-C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl, wherein said C1-6 alkyl, C1-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl-C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl are each optionally substituted with 1, 2, or 3 substituents independently selected from OH, CN, amino, halo, C1-6 alkyl, C1-6 alkylamino, di(C1-6 alkyl)amino, C1-6 alkoxy, C1-6 haloalkyl, and C1-6 haloalkoxy; and each Re, Re1, Re2, and Re3 is independently selected from H, C1-4 alkyl, and CN. 2. A compound of Formula I: or a pharmaceutically
heterocycloalkyl, C6-10 aryl-C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl, wherein said C1-6 alkyl, C1-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl-C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl are each optionally substituted with 1, 2, or 3 substituents independently selected from OH, CN, amino, halo, C1-6 alkyl, C1-6 alkylamino, di(C1-6 alkyl)amino, C1-6 alkoxy, C1-6 haloalkyl, and C1-6 haloalkoxy; and each Re, Re1, Re2, and Re3 is independently selected from H, C1-4 alkyl, and CN. 2. A compound of Formula I: or a pharmaceutically
 R1, R2, and R3 are each independently selected from H, halo, C1-4 alkyl, and C1-4 haloalkyl; R4 and R5 are each independently selected from H, halo, ORA, C1-4 alkyl, and C1-4 haloalkyl, wherein RA is H, C1-4 alkyl, or C1-4 haloalkyl; or R4 and R5 together with the carbon atom to which they are attached form a C3-6 cycloalkyl group or a 3-6 membered heterocycloalkyl group, each optionally substituted with 1, 2, or 3 substituents independently selected from H, halo, ORA, C1-4 alkyl, and C1-4 haloalkyl; R6 and R7 are each independently selected from H, halo, C1-4 alkyl, and C1-4 haloalkyl; R8 is H or C1-4 alkyl;
Cy1 is selected from C6-10 aryl, C3-14 cycloalkyl, 5-14 membered heteroaryl, 4-14 membered heterocycloalkyl, C3-7 cycloalkyl fused with phenyl to form a bicyclic ring, C4-7 cycloalkyl fused with 5-6 membered heteroaryl to form a bicyclic ring, 4-7 membered heterocycloalkyl fused with phenyl to form a bicyclic ring, and 4-7 membered heterocycloalkyl fused with 5-6 membered heteroaryl to form a bicyclic ring, each optionally substituted by 1, 2, 3, or 4 substituents independently selected from halo, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C1-6 haloalkyl, CN, NO2, ORa, SRa, C1-6 alkyl-NRcRd, C(O)Rb, C(O)NRcRd, C(O)ORa, OC(O)Rb, OC(O)NRcRd, C(=NRe)NRcRd, NRcC(=NRe)NRcRd, NRcRd, NRcC(O)Rb, NRcC(O)ORa, NRcC(O)NRcRd, NRcS(O)Rb, NRcS(O)2Rb, NRcS(O)2NRcRd, S(O)Rb, S(O)NRcRd, S(O)2Rb, and S(O)2NRcRd; Cy2 is absent or selected from C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, and 4- 7 membered heterocycloalkyl, each optionally substituted by 1, 2, 3, or 4 substituents independently selected from halo, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C1-6 haloalkyl, CN, NO2, ORa1, SRa1, C(O)Rb1, C(O)NRc1Rd1, C(O)ORa1, OC(O)Rb1, OC(O)NRc1Rd1, C(=NRe1)NRc1Rd1, NRc1C(=NRe1)NRc1Rd1, NRc1Rd1, NRc1C(O)Rb1, NRc1C(O)ORa1, NRc1C(O)NRc1Rd1, NRc1S(O)Rb1, NRc1S(O)2Rb1, NRc1S(O)2NRc1Rd1, S(O)Rb1, S(O)NRc1Rd1, S(O)2Rb1, and S(O)2NRc1Rd1; L is a linker; and Z is a binder of cereblon E3 ligase; each Ra, Rb, Rc, Rd, Ra1, Rb1, Rc1, and Rd1 is independently selected from H, C1-6 alkyl, C1-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl-C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl, wherein said C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl-C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl of Ra, Rb, Rc, Rd, Ra1, Rb1, Rc1, and Rd1 is optionally substituted with 1, 2, 3, 4, or 5 substituents independently selected from halo, C1-4 alkyl, C1-4 haloalkyl, C1-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, CN, ORa3, SRa3, C(O)Rb3, C(O)NRc3Rd3, C(O)ORa3, OC(O)Rb3, OC(O)NRc3Rd3, NRc3Rd3, NRc3C(O)Rb3, NRc3C(O)NRc3Rd3,
or Rc and Rd together with the N atom to which they are attached form a 4-7 membered heterocycloalkyl group optionally substituted with 1, 2, or 3 substituents independently selected from halo, C1-4 alkyl, C1-4 haloalkyl, CN, ORa3, SRa3, C(O)Rb3, C(O)NRc3Rd3, C(O)ORa3, OC(O)Rb3, OC(O)NRc3Rd3, NRc3Rd3, NRc3C(O)Rb3, NRc3C(O)NRc3Rd3, NRc3C(O)ORa3, C(=NRe3)NRc3Rd3, NRc3C(=NRe3)NRc3Rd3, S(O)Rb3, S(O)NRc3Rd3, S(O)2Rb3, NRc3S(O)2Rb3, NRc3S(O)2NRc3Rd3, and S(O)2NRc3Rd3; or Rc1 and Rd1 together with the N atom to which they are attached form a 4-7 membered heterocycloalkyl group optionally substituted with 1, 2, or 3 substituents independently selected from halo, C1-4 alkyl, C1-4 haloalkyl, CN, ORa3, SRa3, C(O)Rb3, C(O)NRc3Rd3, C(O)ORa3, OC(O)Rb3, OC(O)NRc3Rd3, NRc3Rd3, NRc3C(O)Rb3, NRc3C(O)NRc3Rd3, NRc3C(O)ORa3, C(=NRe3)NRc3Rd3, NRc3C(=NRe3)NRc3Rd3, S(O)Rb3, S(O)NRc3Rd3, S(O)2Rb3, NRc3S(O)2Rb3, NRc3S(O)2NRc3Rd3, and S(O)2NRc3Rd3; each Ra3, Rb3, Rc3, and Rd3 is independently selected from H, C1-6 alkyl, C1-6 haloalkyl, C2- 6 alkenyl, C2-6 alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl-C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl, wherein said C1-6 alkyl, C1-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl-C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl are each optionally substituted with 1, 2, or 3 substituents independently selected from OH, CN, amino, halo, C1-6 alkyl, C1-6 alkylamino, di(C1-6 alkyl)amino, C1-6 alkoxy, C1-6 haloalkyl, and C1-6 haloalkoxy; and each Re, Re1, Re2, and Re3 is independently selected from H, C1-4 alkyl, and CN. 3. The compound of claim 1 or 2, or a pharmaceutically acceptable salt thereof, wherein L is a linker having Formula II: II wherein CyA, CyB, CyC, and CyD are each independently absent or independently selected from C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, and 4-10 membered heterocycloalkyl, each optionally substituted by 1, 2, 3, or 4 substituents independently selected from halo, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C1-6 haloalkyl, CN, NO2, ORa11, SRa11, C(O)Rb11, C(O)NRc11Rd11,
C(O)ORa11, OC(O)Rb11, OC(O)NRc11Rd11, C(=NRe11)NRc11Rd11, NRc11C(=NRe11)NRc11Rd11, NRc11Rd11, NRc11C(O)Rb11, NRc11C(O)ORa11, NRc11C(O)NRc11Rd11, NRc11S(O)Rb11, NRc11S(O)2Rb11, NRc11S(O)2NRc11Rd11, S(O)Rb11, S(O)NRc11Rd11, S(O)2Rb11, and S(O)2NRc11Rd11; and LA, LB, and LC are each independently absent or independently selected from C1-6 alkylene, C2-6 alkenylene, C2-6 alkynylene, -C(O)-, -O-, -O-(C1-6 alkylene)-, -NRc23-, -S-(C1-6 alkylene)-, -C(O)-(C1-6 alkylene)-, -C(O)NRc23-(C1-6 alkylene)-, -C(O)O-, -C(O)O-(C1-6 alkylene)- , -OC(O)-(C1-6 alkylene)-, -OC(O)NRc23-(C1-6 alkylene)-, -NRc23-(C1-6 alkylene)-, -N-(C1-6 alkylene)-C(O)-, -NRc23C(O)-(C1-6 alkylene)-, -NRc23C(O)NRc23-(C1-6 alkylene)-, -NRc23C(O)O- (C1-6 alkylene)-, -C(=NRe23)NRc23-(C1-6 alkylene)-, -NRc23C(=NRe23)NRc23-(C1-6 alkylene)-, - S(O)-(C1-6 alkylene)-, -S(O)NRc23-(C1-6 alkylene)-, -S(O)2-(C1-6 alkylene)-, -NRc23S(O)2-(C1-6 alkylene)-, -NRc23S(O)2NRc23-(C1-6 alkylene)-, and -S(O)2NRc23-(C1-6 alkylene)-, wherein the C1-6 alkylene, C2-6 alkenylene, or C2-6 alkynylene is optionally substituted by 1, 2, or 3 substituents independently selected from C1-4 alkyl, C1-4 haloalkyl, C1-4 alkoxy, halo, and OH; each Ra11, Rb11, Rc11, and Rd11 is independently selected from H, C1-6 alkyl, C1-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl-C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl, wherein said C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl-C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl of Ra11, Rb11, Rc11, and Rd11 is optionally substituted with 1,
R1, R2, and R3 are each independently selected from H, halo, C1-4 alkyl, and C1-4 haloalkyl; R4 and R5 are each independently selected from H, halo, ORA, C1-4 alkyl, and C1-4 haloalkyl, wherein RA is H, C1-4 alkyl, or C1-4 haloalkyl; or R4 and R5 together with the carbon atom to which they are attached form a C3-6 cycloalkyl group or a 3-6 membered heterocycloalkyl group, each optionally substituted with 1, 2, or 3 substituents independently selected from H, halo, ORA, C1-4 alkyl, and C1-4 haloalkyl; R6 and R7 are each independently selected from H, halo, C1-4 alkyl, and C1-4 haloalkyl; R8 is H or C1-4 alkyl;
Cy1 is selected from C6-10 aryl, C3-14 cycloalkyl, 5-14 membered heteroaryl, 4-14 membered heterocycloalkyl, C3-7 cycloalkyl fused with phenyl to form a bicyclic ring, C4-7 cycloalkyl fused with 5-6 membered heteroaryl to form a bicyclic ring, 4-7 membered heterocycloalkyl fused with phenyl to form a bicyclic ring, and 4-7 membered heterocycloalkyl fused with 5-6 membered heteroaryl to form a bicyclic ring, each optionally substituted by 1, 2, 3, or 4 substituents independently selected from halo, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C1-6 haloalkyl, CN, NO2, ORa, SRa, C1-6 alkyl-NRcRd, C(O)Rb, C(O)NRcRd, C(O)ORa, OC(O)Rb, OC(O)NRcRd, C(=NRe)NRcRd, NRcC(=NRe)NRcRd, NRcRd, NRcC(O)Rb, NRcC(O)ORa, NRcC(O)NRcRd, NRcS(O)Rb, NRcS(O)2Rb, NRcS(O)2NRcRd, S(O)Rb, S(O)NRcRd, S(O)2Rb, and S(O)2NRcRd; Cy2 is absent or selected from C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, and 4- 7 membered heterocycloalkyl, each optionally substituted by 1, 2, 3, or 4 substituents independently selected from halo, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C1-6 haloalkyl, CN, NO2, ORa1, SRa1, C(O)Rb1, C(O)NRc1Rd1, C(O)ORa1, OC(O)Rb1, OC(O)NRc1Rd1, C(=NRe1)NRc1Rd1, NRc1C(=NRe1)NRc1Rd1, NRc1Rd1, NRc1C(O)Rb1, NRc1C(O)ORa1, NRc1C(O)NRc1Rd1, NRc1S(O)Rb1, NRc1S(O)2Rb1, NRc1S(O)2NRc1Rd1, S(O)Rb1, S(O)NRc1Rd1, S(O)2Rb1, and S(O)2NRc1Rd1; L is a linker; and Z is a binder of cereblon E3 ligase; each Ra, Rb, Rc, Rd, Ra1, Rb1, Rc1, and Rd1 is independently selected from H, C1-6 alkyl, C1-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl-C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl, wherein said C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl-C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl of Ra, Rb, Rc, Rd, Ra1, Rb1, Rc1, and Rd1 is optionally substituted with 1, 2, 3, 4, or 5 substituents independently selected from halo, C1-4 alkyl, C1-4 haloalkyl, C1-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, CN, ORa3, SRa3, C(O)Rb3, C(O)NRc3Rd3, C(O)ORa3, OC(O)Rb3, OC(O)NRc3Rd3, NRc3Rd3, NRc3C(O)Rb3, NRc3C(O)NRc3Rd3,
or Rc and Rd together with the N atom to which they are attached form a 4-7 membered heterocycloalkyl group optionally substituted with 1, 2, or 3 substituents independently selected from halo, C1-4 alkyl, C1-4 haloalkyl, CN, ORa3, SRa3, C(O)Rb3, C(O)NRc3Rd3, C(O)ORa3, OC(O)Rb3, OC(O)NRc3Rd3, NRc3Rd3, NRc3C(O)Rb3, NRc3C(O)NRc3Rd3, NRc3C(O)ORa3, C(=NRe3)NRc3Rd3, NRc3C(=NRe3)NRc3Rd3, S(O)Rb3, S(O)NRc3Rd3, S(O)2Rb3, NRc3S(O)2Rb3, NRc3S(O)2NRc3Rd3, and S(O)2NRc3Rd3; or Rc1 and Rd1 together with the N atom to which they are attached form a 4-7 membered heterocycloalkyl group optionally substituted with 1, 2, or 3 substituents independently selected from halo, C1-4 alkyl, C1-4 haloalkyl, CN, ORa3, SRa3, C(O)Rb3, C(O)NRc3Rd3, C(O)ORa3, OC(O)Rb3, OC(O)NRc3Rd3, NRc3Rd3, NRc3C(O)Rb3, NRc3C(O)NRc3Rd3, NRc3C(O)ORa3, C(=NRe3)NRc3Rd3, NRc3C(=NRe3)NRc3Rd3, S(O)Rb3, S(O)NRc3Rd3, S(O)2Rb3, NRc3S(O)2Rb3, NRc3S(O)2NRc3Rd3, and S(O)2NRc3Rd3; each Ra3, Rb3, Rc3, and Rd3 is independently selected from H, C1-6 alkyl, C1-6 haloalkyl, C2- 6 alkenyl, C2-6 alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl-C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl, wherein said C1-6 alkyl, C1-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl-C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl are each optionally substituted with 1, 2, or 3 substituents independently selected from OH, CN, amino, halo, C1-6 alkyl, C1-6 alkylamino, di(C1-6 alkyl)amino, C1-6 alkoxy, C1-6 haloalkyl, and C1-6 haloalkoxy; and each Re, Re1, Re2, and Re3 is independently selected from H, C1-4 alkyl, and CN. 3. The compound of claim 1 or 2, or a pharmaceutically acceptable salt thereof, wherein L is a linker having Formula II: II wherein CyA, CyB, CyC, and CyD are each independently absent or independently selected from C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, and 4-10 membered heterocycloalkyl, each optionally substituted by 1, 2, 3, or 4 substituents independently selected from halo, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C1-6 haloalkyl, CN, NO2, ORa11, SRa11, C(O)Rb11, C(O)NRc11Rd11,
C(O)ORa11, OC(O)Rb11, OC(O)NRc11Rd11, C(=NRe11)NRc11Rd11, NRc11C(=NRe11)NRc11Rd11, NRc11Rd11, NRc11C(O)Rb11, NRc11C(O)ORa11, NRc11C(O)NRc11Rd11, NRc11S(O)Rb11, NRc11S(O)2Rb11, NRc11S(O)2NRc11Rd11, S(O)Rb11, S(O)NRc11Rd11, S(O)2Rb11, and S(O)2NRc11Rd11; and LA, LB, and LC are each independently absent or independently selected from C1-6 alkylene, C2-6 alkenylene, C2-6 alkynylene, -C(O)-, -O-, -O-(C1-6 alkylene)-, -NRc23-, -S-(C1-6 alkylene)-, -C(O)-(C1-6 alkylene)-, -C(O)NRc23-(C1-6 alkylene)-, -C(O)O-, -C(O)O-(C1-6 alkylene)- , -OC(O)-(C1-6 alkylene)-, -OC(O)NRc23-(C1-6 alkylene)-, -NRc23-(C1-6 alkylene)-, -N-(C1-6 alkylene)-C(O)-, -NRc23C(O)-(C1-6 alkylene)-, -NRc23C(O)NRc23-(C1-6 alkylene)-, -NRc23C(O)O- (C1-6 alkylene)-, -C(=NRe23)NRc23-(C1-6 alkylene)-, -NRc23C(=NRe23)NRc23-(C1-6 alkylene)-, - S(O)-(C1-6 alkylene)-, -S(O)NRc23-(C1-6 alkylene)-, -S(O)2-(C1-6 alkylene)-, -NRc23S(O)2-(C1-6 alkylene)-, -NRc23S(O)2NRc23-(C1-6 alkylene)-, and -S(O)2NRc23-(C1-6 alkylene)-, wherein the C1-6 alkylene, C2-6 alkenylene, or C2-6 alkynylene is optionally substituted by 1, 2, or 3 substituents independently selected from C1-4 alkyl, C1-4 haloalkyl, C1-4 alkoxy, halo, and OH; each Ra11, Rb11, Rc11, and Rd11 is independently selected from H, C1-6 alkyl, C1-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl-C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl, wherein said C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl-C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl of Ra11, Rb11, Rc11, and Rd11 is optionally substituted with 1,
2,
3, 4, or 5 substituents independently selected from halo, C1-4 alkyl, C1-4 haloalkyl, C1-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, CN, ORa3, SRa3, C(O)Rb3, C(O)NRc3Rd3, C(O)ORa3, OC(O)Rb3, OC(O)NRc3Rd3, NRc3Rd3, NRc3C(O)Rb3, NRc3C(O)NRc3Rd3, NRc3C(O)ORa3,
 alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl- C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl, wherein said C1-6 alkyl, C1-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl-C1-4
alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl are each optionally substituted with 1, 2, or 3 substituents independently selected from OH, CN, amino, halo, C1-6 alkyl, C1-6 alkylamino, di(C1-6 alkyl)amino, C1-6 alkoxy, C1-6 haloalkyl, and C1-6 haloalkoxy; and Re23 is independently selected from H, C1-4 alkyl, and CN; wherein at least one of LA, LB, LC, CyA, CyB, CyC, and CyD is not absent.
alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl- C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl, wherein said C1-6 alkyl, C1-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl-C1-4
alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl are each optionally substituted with 1, 2, or 3 substituents independently selected from OH, CN, amino, halo, C1-6 alkyl, C1-6 alkylamino, di(C1-6 alkyl)amino, C1-6 alkoxy, C1-6 haloalkyl, and C1-6 haloalkoxy; and Re23 is independently selected from H, C1-4 alkyl, and CN; wherein at least one of LA, LB, LC, CyA, CyB, CyC, and CyD is not absent.
4. The compound of any one of claims 1-3, or a pharmaceutically acceptable salt thereof wherein R1, R2, and R3 are each independently selected from H, F, methyl, and trifluoromethyl.
5. The compound of any one of claims 1-4, or a pharmaceutically acceptable salt thereof, wherein R1, R2, and R3 are each F.
6. The compound of any one of claims 1-4, or a pharmaceutically acceptable salt thereof, wherein R1, R2, and R3 are each H.
7. The compound of any one of claims 1-6, or a pharmaceutically acceptable salt thereof, wherein R4 and R5 are each independently selected from SRA, C1-4 alkyl, and C2-4 alkenyl, wherein the C2-4 alkenyl is optionally substituted with 1, 2, or 3 substituents independently selected from halo, and wherein RA is C1-4 alkyl.
8. The compound of any one of claims 1-6, or a pharmaceutically acceptable salt thereof, wherein R4 and R5 are each independently selected from OH, C1-4 alkyl, and C1-4 haloalkyl; or R4 and R5 together with the carbon atom to which they are attached form a C3-6 cycloalkyl group optionally substituted with 1, 2, or 3 substituents independently selected from halo, ORA, C1-4 alkyl, and C1-4 haloalkyl.
9. The compound of any one of claims 1-6, or a pharmaceutically acceptable salt thereof, wherein R4 and R5 are each C1-4 alkyl.
10. The compound of any one of claim 1-6, or a pharmaceutically acceptable salt thereof, wherein R4 and R5 are each methyl.
11. The compound of any one of claims 1-6, or a pharmaceutically acceptable salt thereof, wherein R4 and R5 together with the carbon atom to which they are attached form a C3-6 cycloalkyl group optionally substituted with 1 or 2 substituents independently selected from halo and C1-2 alkyl.
12. The compound of any one of claims 1-11, or a pharmaceutically acceptable salt thereof, wherein R6 and R7 are each H.
13. The compound of any one of claim 1-12, or a pharmaceutically acceptable salt thereof, wherein R8 is H.
14. The compound of any one of claims 1-13, or a pharmaceutically acceptable salt thereof, wherein Cy1 is selected from C3-14 cycloalkyl, 5-14 membered heteroaryl, C4-7 cycloalkyl fused with phenyl to form a bicyclic ring, and 4-7 membered heterocycloalkyl fused with phenyl to form a bicyclic ring, each optionally substituted by 1 or 2 substituents independently selected from halo, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C1-6 haloalkyl, CN, NO2, ORa, SRa, C1-6 alkyl- NRcRd, C(O)Rb, C(O)NRcRd, C(O)ORa, OC(O)Rb, OC(O)NRcRd, C(=NRe)NRcRd, NRcC(=NRe)NRcRd, NRcRd, NRcC(O)Rb, NRcC(O)ORa, NRcC(O)NRcRd, NRcS(O)Rb, NRcS(O)2Rb, NRcS(O)2NRcRd, S(O)Rb, S(O)NRcRd, S(O)2Rb, and S(O)2NRcRd.
15. The compound of any one of claims 1-13, or a pharmaceutically acceptable salt thereof, wherein Cy1 is selected from C3-6 cycloalkyl, 5-6 membered heteroaryl, C5-6 cycloalkyl fused with phenyl to form a bicyclic ring, and 5-6 membered heterocycloalkyl fused with phenyl to form a bicyclic ring, each optionally substituted by 1 or 2 substituents independently selected from halo, C1-6 alkyl, C1-6 haloalkyl, CN, C1-6 alkyl-NRcRd, C(O)NRcRd, and NRcRd.
16. The compound of any one of claims 1-13, or a pharmaceutically acceptable salt thereof, wherein Cy1 is C3-6 cycloalkyl.
17. The compound of any one of claims 1-13, or a pharmaceutically acceptable salt thereof, wherein Cy1 is cyclopropyl or cyclobutyl.
18. The compound of any one of claims 1-13, or a pharmaceutically acceptable salt thereof, wherein Cy1 is cyclopropyl.
19. The compound of any one of claims 1-13, or a pharmaceutically acceptable salt thereof, wherein Cy1 is 5-6 membered heteroaryl, optionally substituted by 1 or 2 substituents independently selected from halo, C1-6 alkyl, and C(O)NRcRd.
20. The compound of any one of claims 1-13, or a pharmaceutically acceptable salt thereof, wherein Cy1 is pyrazolyl, pyrrolyl, or isoxazolyl, each optionally substituted with C(O)NRcRd.
21. The compound of any one of claims 1-13, or a pharmaceutically acceptable salt thereof, wherein Cy1 is pyrazolyl.
22. The compound of any one of claims 1-13, or a pharmaceutically acceptable salt thereof, wherein Cy1 is C5-6 cycloalkyl fused with phenyl to form a bicyclic ring, optionally substituted by 1 or 2 substituents independently selected from halo, C1-6 alkyl, CN, C1-6 alkyl-NRcRd, and NRcRd.
23. The compound of any one of claims 1-13, or a pharmaceutically acceptable salt thereof, wherein Cy1 is cyclopentyl fused with phenyl to form a bicyclic ring, optionally substituted by methyl, CN, Cl, F, -CH2NHRd, and NH2.
24. The compound of any one of claims 1-13, or a pharmaceutically acceptable salt thereof, wherein Cy1 is 5-6 membered heterocycloalkyl fused with phenyl to form a bicyclic ring, optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl.
25. The compound of any one of claims 1-13, or a pharmaceutically acceptable salt thereof, wherein Cy1 is dihydrobenzofuran, optionally substituted by 1 or 2 substituents independently selected from chloro and methyl.
26. The compound of any one of claims 1-25, or a pharmaceutically acceptable salt thereof, wherein Cy2 is selected from C6-10 aryl and 5-10 membered heteroaryl, optionally substituted by 1, 2, or 3 substituents independently selected from halo, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C1-6 haloalkyl, CN, NO2, ORa1, SRa1, C(O)Rb1, C(O)NRc1Rd1, C(O)ORa1, OC(O)Rb1, OC(O)NRc1Rd1, C(=NRe1)NRc1Rd1, NRc1C(=NRe1)NRc1Rd1, NRc1Rd1, NRc1C(O)Rb1, NRc1C(O)ORa1, NRc1C(O)NRc1Rd1, NRc1S(O)Rb1, NRc1S(O)2Rb1, NRc1S(O)2NRc1Rd1, S(O)Rb1, S(O)NRc1Rd1, S(O)2Rb1, and S(O)2NRc1Rd1.
27. The compound of any one of claims 1-25, or a pharmaceutically acceptable salt thereof, wherein Cy2 is bicyclo[1.1.1]pentyl.
28. The compound of any one of claims 1-25, or a pharmaceutically acceptable salt thereof, wherein Cy2 is phenyl or 5-6 membered heteroaryl, optionally substituted by 1 or 2 substituents selected from halo and C1-6 alkyl.
29. The compound of any one of claims 1-25, or a pharmaceutically acceptable salt thereof, wherein Cy2 is phenyl optionally substituted with 1 or 2 substituents selected from methyl and chloro.
30. The compound of any one of claims 1-25, or a pharmaceutically acceptable salt thereof, wherein Cy2 is pyridinyl or pyrimidinyl, each optionally substituted with methyl.
31. The compound of any one of claims 3-30, or a pharmaceutically acceptable salt thereof, wherein CyA, CyB, CyC, and CyD are each independently absent or independently selected from C6-10 aryl, 5-10 membered heteroaryl, and 4-10 membered heterocycloalkyl, each optionally substituted by 1, 2, or 3 substituents independently selected from halo and C1-6 alkyl.
32. The compound of any one of claims 3-30, or a pharmaceutically acceptable salt thereof, wherein CyA, CyB, CyC, and CyD are each independently absent or independently selected from phenyl, 5-9 membered heteroaryl, and 4-7 membered heterocycloalkyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl.
33. The compound of any one of claims 3-32, or a pharmaceutically acceptable salt thereof, wherein CyA is absent or selected from phenyl, pyrazolyl, pyridinyl, pyrazinyl, pyrimidinyl, indazolyl, 2,3-dihydrobenzofuranyl, and piperidinyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl.
34. The compound of any one of claims 3-32, or a pharmaceutically acceptable salt thereof, wherein CyA is absent or selected from phenyl, pyrazolyl, pyridinyl, indazolyl, and piperidinyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl.
35. The compound of any one of claims 3-32, or a pharmaceutically acceptable salt thereof, z CyA is phenyl or pyridinyl, each optionally substituted by 1 or 2 substituents selected from halo and methyl.
36. The compound of any one of claims 3-35, or a pharmaceutically acceptable salt thereof, wherein CyB is absent or selected from phenyl, cyclohexyl, pyrazolyl, azetidinyl, pyrrolidinyl, piperazinyl, piperidinyl, azasprio[3.3]heptanyl, diazaspiro[3.5]nonanyl, 2,6- diazaspiro[3.3]heptanyl, 3,6-diazabicyclo[3.1.1]heptanyl, 2,5-diazabicyclo[2.2.1]heptanyl, 3,8- diazabicyclo[3.2.1]octanyl, 2-azaspiro[3.5]nonanyl, 7-azaspiro[3.5]nonanyl, 6- azaspiro[3.4]octanyl, and octahydropyrrolo[3,2-b]pyrrolyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl.
37. The compound of any one of claims 3-35, or a pharmaceutically acceptable salt thereof, wherein CyB is absent or selected from phenyl, cyclohexyl, pyrazolyl, azetidinyl, pyrrolidinyl, piperazinyl, piperidinyl, azasprio[3.3]heptanyl, diazaspiro[3.5]nonanyl, 2,6- diazaspiro[3.3]heptanyl, 3,6-diazabicyclo[3.1.1]heptanyl, 2,5-diazabicyclo[2.2.1]heptanyl, 3,8-
diazabicyclo[3.2.1]octanyl, and azaspiro[3.5]nonanyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl.
38. The compound of any one of claims 3-35, or a pharmaceutically acceptable salt thereof, wherein CyB is piperidinyl or piperazinyl.
39. The compound of any one of claims 3-35, or a pharmaceutically acceptable salt thereof, wherein CyB is 2-azaspiro[3.3]heptan-2-yl.
40. The compound of any one of claims 3-39, or a pharmaceutically acceptable salt thereof, wherein CyC is absent or selected from indazolyl, piperidinyl, piperazinyl, azetidinyl, and pyrrolidinyl, each optionally substituted by 1 or 2 substituents independently selected from halo and C1-6 alkyl.
41. The compound of any one of claims 3-39, or a pharmaceutically acceptable salt thereof, wherein CyC is piperidinyl, piperazinyl, or cyclobutyl.
42. The compound of any one of claims 3-41, or a pharmaceutically acceptable salt thereof, wherein CyD is absent or selected from piperazinyl and piperidinyl.
43. The compound of any one of claims 3-41, or a pharmaceutically acceptable salt thereof, wherein CyD is absent or piperazinyl.
44. The compound of any one of claims 3-43, or a pharmaceutically acceptable salt thereof, wherein LA, LB, and LC are each independently absent or independently selected from C1-6 alkylene, -C(O)-, -O-, -O-(C1-6 alkylene)-, -NRc23-, -C(O)NRc23-, -C(O)-(C1-6 alkylene)-, - C(O)NRc23-(C1-6 alkylene)-, and -NRc23C(O)-.
45. The compound of claim 44, or a pharmaceutically acceptable salt thereof, wherein Rc23 is selected from H and C1-6 alkyl.
46. The compound of any one of claims 3-43, or a pharmaceutically acceptable salt thereof, wherein LA, LB, and LC are each independently absent or independently selected from methylene, -C(O)-, -O-, -O-(C1-3 alkylene)-, -C(O)CH2-, -C(O)NH-(C1-3 alkylene).
47. The compound of any one of claims 3-43, or a pharmaceutically acceptable salt thereof, wherein LA is absent or selected from methylene, -C(O)-, -C(O)CH2-, -C(O)NH-(C1-3 alkylene)-, - NHC(O)-, -CH(CH3)-, -CH(CH(CH3)2)-, -CH(CH2CH2CH3)-, -CH(CH2OCH3)-, and -O-.
48. The compound of any one of claims 3-43, or a pharmaceutically acceptable salt thereof, wherein LA is absent or selected from methylene, -C(O)-, -C(O)CH2-, -C(O)NH-(C1-3 alkylene)-, and -NHC(O)-.
49. The compound of any one of claims 3-43, or a pharmaceutically acceptable salt thereof, wherein LA is absent or selected from methylene and -NHC(O)-.
50. The compound of any one of claims 3-49, or a pharmaceutically acceptable salt thereof, wherein LB is absent or selected from methylene, -C(O)-, -ORa3-, -C(O)CH2-, -C(O)NH-(C1-3 alkylene)-, -NHC(O)-, -O-, -O-(C1-6 alkylene)-, and -NRc23-(C1-6 alkylene)-.
51. The compound of any one of claims 3-49, or a pharmaceutically acceptable salt thereof, wherein LB is absent or selected from methylene, -C(O)-, -ORa3-, -C(O)CH2-, -C(O)NH-(C1-3 alkylene)-, -NHC(O)-, -O-, and -O-(C1-6 alkylene)-.
52. The compound of any one of claims 3-49, or a pharmaceutically acceptable salt thereof, wherein LB is absent or methylene.
53. The compound of any one of claim 3-52, or a pharmaceutically acceptable salt thereof, wherein LC is absent or selected from methylene, -O-, -C(O)CH2-, and -NHC(O)-.
54. The compound of any one of claims 3-52, or a pharmaceutically acceptable salt thereof, wherein LC is absent or methylene.
55. The compound of any one of claims 1-54, or a pharmaceutically acceptable salt thereof, wherein Z is a group having Formula III:
 wherein ring A is C6-10 aryl, 5-14 membered heteroaryl, C3-7 cycloalkyl fused with phenyl to form a bicyclic ring, 4-7 membered heterocycloalkyl fused with phenyl to form a bicyclic ring, each optionally substituted with 1, 2, or 3 substituents independently selected from halo, C1-6 alkyl, and C1-6 haloalkyl; L1 is absent, CH2, NH, C(O)NH, or O; and W is CH or N, wherein the wavy line represents the point of attachment to group L.
wherein ring A is C6-10 aryl, 5-14 membered heteroaryl, C3-7 cycloalkyl fused with phenyl to form a bicyclic ring, 4-7 membered heterocycloalkyl fused with phenyl to form a bicyclic ring, each optionally substituted with 1, 2, or 3 substituents independently selected from halo, C1-6 alkyl, and C1-6 haloalkyl; L1 is absent, CH2, NH, C(O)NH, or O; and W is CH or N, wherein the wavy line represents the point of attachment to group L.
56. The compound of any one of claims 1-55, or a pharmaceutically acceptable salt thereof, wherein Z is selected from:
; ch independently selected from CH, CF, CCl, C(CH3), and N; and each V is independently selected from CH, C(CH3), and N.
57. The compound of any one of claims 1-55, or a pharmaceutically acceptable salt thereof, wherein Z is selected from:
; from CH , CF, and N; and each V is independently selected from CH and N.
58. The compound of claim any one of claims 1-55, or a pharmaceutically acceptable salt thereof, wherein Z is selected from: O
 selected from CH , CF, and N; and each V is independently selected from CH and N.
selected from CH , CF, and N; and each V is independently selected from CH and N.
59. The compound of claim 3, wherein the compound has Formula IA: Z
 or a pharmaceutically acceptable salt thereof.
or a pharmaceutically acceptable salt thereof.
60. The compound of claim 3 or 59, or a pharmaceutically acceptable salt thereof, wherein: Cy1 is selected from C3-6 cycloalkyl, 5-6 membered heteroaryl, C5-6 cycloalkyl fused with phenyl to form a bicyclic ring, and 5-6 membered heterocycloalkyl fused with phenyl to form a bicyclic ring, each optionally substituted by 1 or 2 substituents independently selected from halo, C1-6 alkyl, C1-6 haloalkyl, CN, C1-6 alkyl-NRcRd, C(O)NRcRd, and NRcRd; Cy2 is selected from C6-10 aryl and 5-10 membered heteroaryl, optionally substituted by 1, 2, or 3 substituents independently selected from halo, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C1-6 haloalkyl, CN, NO2, ORa1, SRa1, C(O)Rb1, C(O)NRc1Rd1, C(O)ORa1, OC(O)Rb1, OC(O)NRc1Rd1, C(=NRe1)NRc1Rd1, NRc1C(=NRe1)NRc1Rd1, NRc1Rd1, NRc1C(O)Rb1, NRc1C(O)ORa1, NRc1C(O)NRc1Rd1, NRc1S(O)Rb1, NRc1S(O)2Rb1, NRc1S(O)2NRc1Rd1, S(O)Rb1, S(O)NRc1Rd1, S(O)2Rb1, and S(O)2NRc1Rd1; CyA, CyB, CyC, and CyD are each independently absent or independently selected from C6- 10 aryl, 5-10 membered heteroaryl, and 4-10 membered heterocycloalkyl, each optionally substituted by 1, 2, or 3 substituents independently selected from halo and C1-6 alkyl; and LA, LB, and LC are each independently absent or independently selected from C1-6 alkylene, -C(O)-, -O-, -O-(C1-6 alkylene)-, -C(O)-(C1-6 alkylene)-, -C(O)NRc23-(C1-6 alkylene)-, and -NRc23C(O)-.
61. The compound of claim 60, or a pharmaceutically acceptable salt thereof, wherein Z is selected from: O
62. The compound of claim 3 or 59-61, wherein the compound has Formula IB:
 wherein is selected from halo, C1-4 alkyl, and C1-4 haloalkyl; and p is 0, 1, or 2.
wherein is selected from halo, C1-4 alkyl, and C1-4 haloalkyl; and p is 0, 1, or 2.
63. The compound of claim 62, wherein the compound has Formula IB-1:
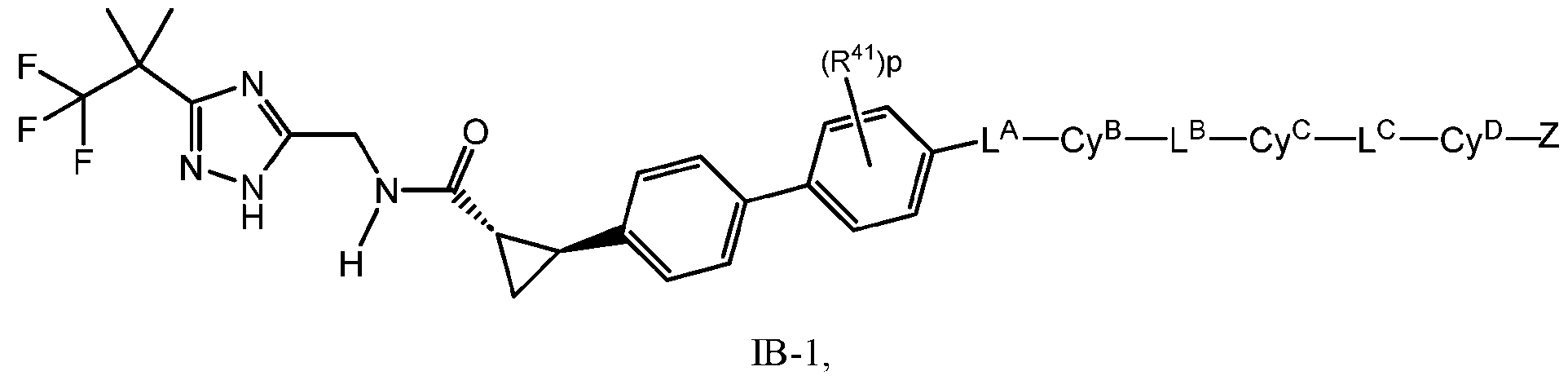
64. The compound of claim 3 or 59-61, wherein the compound has Formula IC: Z
 or a pharmaceutically acceptable salt thereof;
wherein X1 and X2 are each independently selected from CH and N; each R42 and R43 is independently selected from halo, C1-4 alkyl, and C1-4 haloalkyl; and q and r are each independently 0, 1, or 2.
or a pharmaceutically acceptable salt thereof;
wherein X1 and X2 are each independently selected from CH and N; each R42 and R43 is independently selected from halo, C1-4 alkyl, and C1-4 haloalkyl; and q and r are each independently 0, 1, or 2.
65. The compound of claim 64, or a pharmaceutically acceptable salt thereof, wherein R42 and R43 are methyl.
66. The compound of claim 3 or 59-61, wherein the compound has Formula ID:
 wherein X is CH or N; and R44 is selected from H, halo, C1-4 alkyl, and C1-4 haloalkyl.
wherein X is CH or N; and R44 is selected from H, halo, C1-4 alkyl, and C1-4 haloalkyl.
67. The compound of claim 3 or 59-61, wherein the compound has Formula IE:
 or a pharmaceutically acceptable salt thereof; wherein each R42 and R43 is independently selected from halo, C1-4 alkyl, and C1-4 haloalkyl; X1 and X2 are each independently selected from CH and N; and q and r are each independently 0, 1, or 2.
or a pharmaceutically acceptable salt thereof; wherein each R42 and R43 is independently selected from halo, C1-4 alkyl, and C1-4 haloalkyl; X1 and X2 are each independently selected from CH and N; and q and r are each independently 0, 1, or 2.
68. The compound of claims 3 or 59-61, wherein the compound has Formula IF: or a
 wherein each is independently selected from halo, C1-4 alkyl, and C1-4 haloalkyl; and q is 0, 1, or 2.
wherein each is independently selected from halo, C1-4 alkyl, and C1-4 haloalkyl; and q is 0, 1, or 2.
69. The compound of claim 3 or 59-61, wherein the compound has Formula IG: or a
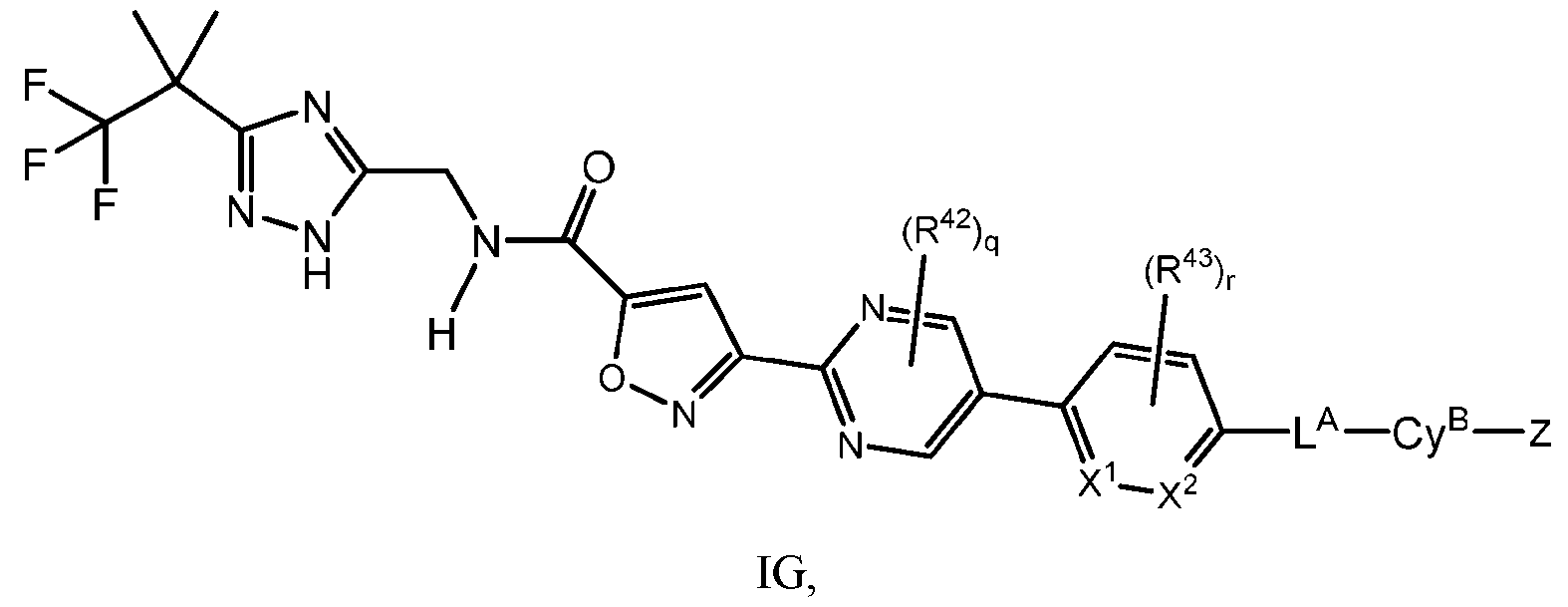 wherein each R42 and R43 is independently selected from halo, C1-4 alkyl, and C1-4 haloalkyl; X1 and X2 are each independently selected from CH and N; and q and r are each independently 0, 1, or 2.
wherein each R42 and R43 is independently selected from halo, C1-4 alkyl, and C1-4 haloalkyl; X1 and X2 are each independently selected from CH and N; and q and r are each independently 0, 1, or 2.
70. The compound of claim 1 or 2, wherein the compound is selected from: (2R)-5-chloro-6-(4-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperazin-1-yl)methyl)phenyl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5- yl)methyl)-2,3-dihydrobenzofuran-2-carboxamide;
(2R)-5-chloro-6-(4-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperazine-1-carbonyl)phenyl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5- yl)methyl)-2,3-dihydrobenzofuran-2-carboxamide; (2R)-5-chloro-6-(6-(4-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperazin-1-yl)methyl)piperidin-1-yl)pyridin-3-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2- yl)-1H-1,2,4-triazol-5-yl)methyl)-2,3-dihydrobenzofuran-2-carboxamide; (1S,2S)-2-(4'-(1-(2-(4-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperazin-1-yl)methyl)piperidin-1-yl)-2-oxoethyl)-1H-pyrazol-4-yl)-[1,1'-biphenyl]-4-yl)-N- ((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)cyclopropane-1- carboxamide; (1S,2S)-2-[4-[4-[1-[2-[4-[[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]piperazin-1-yl]methyl]-1-piperidyl]-2-oxo-ethyl]pyrazol-3-yl]phenyl]phenyl]-N-[[3-(2,2,2- trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]cyclopropanecarboxamide hydrochloride; 5-(4-((1-(2-(4-(3-chloro-4'-((1S,2S)-2-(3-(3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H- 1,2,4-triazol-5-yl)propanoyl)cyclopropyl)-[1,1'-biphenyl]-4-yl)-1H-pyrazol-1-yl)acetyl)piperidin- 4-yl)methyl)piperazin-1-yl)-2-(2,6-dioxopiperidin-3-yl)-6-fluoroisoindoline-1,3-dione; (1S,2S)-2-(2'-chloro-4'-(1-(2-(4-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3- dioxoisoindolin-5-yl)piperazin-1-yl)methyl)piperidin-1-yl)-2-oxoethyl)-1H-pyrazol-4-yl)-[1,1'- biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5- yl)methyl)cyclopropane-1-carboxamide; (1S,2S)-2-[4-[4-[1-[2-[3-[[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]piperazin-1-yl]methyl]azetidin-1-yl]-2-oxo-ethyl]pyrazol-4-yl]phenyl]phenyl]-N-[[3-(2,2,2- trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]cyclopropanecarboxamide; 3-chloro-N-(3-(2-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperazin-1-yl)ethoxy)propyl)-4'-((1S,2S)-2-(((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H- 1,2,4-triazol-5-yl)methyl)carbamoyl)cyclopropyl)-[1,1'-biphenyl]-4-carboxamide; 3-chloro-N-(2-(4-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperazin-1-yl)methyl)piperidin-1-yl)-2-oxoethyl)-4'-((1S,2S)-2-(((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)carbamoyl)cyclopropyl)-[1,1'-biphenyl]-4- carboxamide;
2-chloro-N-[1-[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]-4- piperidyl]-4-piperidyl]-4-[4-[(1S,2S)-2-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol- 5-yl]methylcarbamoyl]cyclopropyl]phenyl]benzamide; (1S,2S)-2-[4-[1-[2-[4-[[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]piperazin-1-yl]methyl]-1-piperidyl]-2-oxo-ethyl]-3-methyl-indazol-5-yl]phenyl]-N-[[3-(2,2,2- trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]cyclopropanecarboxamide; (1S,2S)-2-[4-[4-[1-[1-[2-[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]piperazin-1-yl]-2-oxo-ethyl]azetidin-3-yl]pyrazol-4-yl]phenyl]phenyl]-N-[[3-(2,2,2-trifluoro- 1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]cyclopropanecarboxamide; (1S,2S)-2-[4-[4-[1-[[1-[2-[4- [2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl] piperazin-1-yl]-2-oxo-ethyl] -4-piperidyl]methyl]pyrazol-4-yl]phenyl]phenyl]-N-[[3-(2,2,2- trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]cyclopropanecarboxamide; (1S,2S)-2-[4-[4-[1-[2-[(3S)-3-[[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo- isoindolin-5-yl]-4-piperidyl] oxy]pyrrolidin-1-yl]-2-oxo-ethyl]pyrazol-4-yl]phenyl]phenyl]-N-[[3- (2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]cyclopropanecarboxamide; (1S,2S)-2-[4-[1-[4-[4-[[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]piperazin-1-yl]methyl]piperidine-1-carbonyl]phenyl]pyrazol-4-yl]phenyl]-N-[[3-(2,2,2- trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]cyclopropanecarboxamide; (1S,2S)-2-(4'-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperazin-1-yl)methyl)-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H- 1,2,4-triazol-5-yl)methyl)cyclopropane-1-carboxamide; (1S,2S)-2-(4'-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperazin-1-yl)methyl)-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H- 1,2,4-triazol-5-yl)methyl)cyclopropane-1-carboxamide; 1-[3-[4-[[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]piperazin-1- yl]methyl]phenyl]-2,4-dimethyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4- triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-[4-[4-[[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]piperazin-1- yl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol- 5-yl]methyl]pyrazole-4-carboxamide;
1-[4-[3-[[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]piperazin-1- yl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol- 5-yl]methyl]pyrazole-4-carboxamide; 1-[4-[4-[[4-[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]piperazin-1- yl]-1-piperidyl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H- 1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-[4-[4-[[4-[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]-4- piperidyl]piperazin-1-yl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl- ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-(4'-((4-((1-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperidin-4- yl)methyl)piperazin-1-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-(4'-((4-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1- yl)methyl)piperidin-1-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-(4'-((4-((1-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperidin-4- yl)oxy)piperidin-1-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-(4'-((4-(1-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperidin-4- yl)-1,4-diazepan-1-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-(4'-((1'-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)-[4,4'- bipiperidin]-1-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2- yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-(4'-((3-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1- yl)pyrrolidin-1-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan- 2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-(4-(6-((4-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1- yl)piperidin-1-yl)methyl)pyridin-3-yl)-2-methylphenyl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2- yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide;
1-(2'-chloro-4'-((4-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperazin-1-yl)piperidin-1-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-(3'-((4-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1- yl)piperidin-1-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2- yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-(4'-(1-(4-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1- yl)piperidin-1-yl)ethyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2- yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-[4-[4-[[4-[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]piperazin-1- yl]-1-piperidyl]methyl]-3,5-difluoro-phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-(4-(1-(1'-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)-[1,4'- bipiperidin]-4-yl)-3-methyl-1H-indazol-5-yl)-2-methylphenyl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-[4-[6-[[1-[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]-4-piperidyl]- 4-piperidyl]oxy]-3-pyridyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H- 1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-[4-[4-[2-[4-[[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]piperazin- 1-yl]methyl]-1-piperidyl]ethoxy]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl- ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-(3'-(2-(4-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1- yl)methyl)piperidin-1-yl)ethoxy)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-[4-[4-[1-[4-[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]piperazin- 1-yl]-1-piperidyl]-1-methyl-ethyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl- ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-[4-[5-[[4-[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]piperazin-1- yl]-1-piperidyl]methyl]pyrazin-2-yl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide;
1-[4-[5-[[4-[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]piperazin-1- yl]-1-piperidyl]methyl]-2-pyridyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-(3'-chloro-4'-((4-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperazin-1-yl)piperidin-1-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-[4-[4-[[5-[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl] piperazin-1- yl]-2-pyridyl]oxy]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4- triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-[4-[4-[[4-[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]piperazin-1- yl]-2-oxo-1-piperidyl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl- ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-[4-[6-[4-[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]piperazin-1- yl]cyclohexoxy]-3-pyridyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H- 1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-[4-[1-[[1-[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]-4- piperidyl]-4-piperidyl]methyl]pyrazol-4-yl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-[4-[6-[[1-[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]-4-piperidyl]- 4-piperidyl]amino]-3-pyridyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H- 1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-[4-[4-[[2-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]-2,7- diazaspiro[3.5]nonan-7-yl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl- ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-(4'-((4-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1- yl)azepan-1-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2- yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-(4'-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)-1,4-diazepan-1- yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide;
1-[4-[4-[[8-[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]-4-piperidyl]- 3,8-diazabicyclo[3.2.1]octan-3-yl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-(4'-((3-(1-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperidin-4- yl)-3,6-diazabicyclo[3.1.1]heptan-6-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-(4'-(((1S,4S)-5-(1-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperidin-4-yl)-2,5-diazabicyclo[2.2.1]heptan-2-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N- ((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4- carboxamide; 1-(4'-(((1R,4R)-5-(1-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperidin-4-yl)-2,5-diazabicyclo[2.2.1]heptan-2-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N- ((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4- carboxamide; 1-(4'-((3-(1-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperidin-4- yl)-3,8-diazabicyclo[3.2.1]octan-8-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-(4'-((3-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1- yl)azetidin-1-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2- yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-[4-[4-[[2-[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]-4-piperidyl]- 2,6-diazaspiro[3.3]heptan-6-yl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-[4-[4-[[4-[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]piperazin-1- yl]-3,3-difluoro-1-piperidyl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-[4-[4-[[4-[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]piperazin-1- yl]-4-methyl-1-piperidyl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl- ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide;
1-[4-[4-[[4-[6-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]-2,6- diazaspiro[3.3]heptan-2-yl]-1-piperidyl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro- 1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-(4'-(((2S,4S)-4-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperazin-1-yl)-2-methylpiperidin-1-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-(4'-(((2S,4R)-4-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperazin-1-yl)-2-methylpiperidin-1-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-(4'-(((2R,4R)-4-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperazin-1-yl)-2-methylpiperidin-1-yl)methyl)-3-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-(4'-((4-(1-(2,6-dioxopiperidin-3-yl)-1H-indazol-5-yl)-[1,4'-bipiperidin]-1'-yl)methyl)-3- methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5- yl)methyl)-1H-pyrazole-4-carboxamide; 1-(4'-((4-(4-(3-(2,6-dioxopiperidin-3-yl)phenyl)piperazin-1-yl)piperidin-1-yl)methyl)-3- methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5- yl)methyl)-1H-pyrazole-4-carboxamide; 1-[4-[4-[[4-[4-[4-(2,6-dioxo-3-piperidyl)phenyl]piperazin-1-yl]-1- piperidyl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4- triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-[4-[4-[[4-[4-(2,6-dioxo-3-piperidyl)phenyl]piperazin-1-yl]methyl]phenyl]-2-methyl- phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4- carboxamide; 1-[4-[4-[[4-[4-[(2,6-dioxo-3-piperidyl)amino]-2-fluoro-phenyl]-1- piperidyl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4- triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-(2-chloro-4'-((4-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperazin-1-yl)piperidin-1-yl)methyl)-5-methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide;
1-[3-[4-[[(3S)-3-[[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5- yl]amino]pyrrolidin-1-yl]methyl]phenyl]-2,4-dimethyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-[3-[6-[[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]-4- piperidyl]oxy]-3-pyridyl]-2,4-dimethyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H- 1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-[3-[6-[4-[[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]-4- piperidyl]methyl]piperazin-1-yl]-3-pyridyl]-2,4-dimethyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-(3-(6-(4-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1- yl)methyl)piperidin-1-yl)pyridin-3-yl)-2,4-dimethylphenyl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-[3-[4-[[4-[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]piperazin-1- yl]-1-piperidyl]methyl]phenyl]-2,4-dimethyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)- 1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-[3-[4-[[4-[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]-4- piperidyl]piperazin-1-yl]methyl]phenyl]-2,4-dimethyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-[3-[2-[4-[[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]-4- piperidyl]methyl]piperazin-1-yl]-4-pyridyl]-2,4-dimethyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-(3-(2-(4-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5-yl)piperazin-1- yl)methyl)piperidin-1-yl)pyridin-4-yl)-2,4-dimethylphenyl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-[3-[6-[4-[[2-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]-2,6- diazaspiro[3.3]heptan-6-yl]methyl]-1-piperidyl]-3-pyridyl]-2,4-dimethyl-phenyl]-N-[[3-(2,2,2- trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-[3-[6-[6-[[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]-4- piperidyl]methyl]-2,6-diazaspiro[3.3]heptan-2-yl]-3-pyridyl]-2,4-dimethyl-phenyl]-N-[[3-(2,2,2- trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide;
1-[3-[2-[4-[[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]piperazin-1- yl]methyl]-1-piperidyl]pyrimidin-5-yl]-2,4-dimethyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl- ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; (2S)-5-chloro-6-[4-[[4-[4-(2,6-dioxo-3-piperidyl)phenyl]piperazin-1-yl]methyl]phenyl]-N- [[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl] -2,3-dihydrobenzofuran-2- carboxamide; (2R)-5-chloro-6-(4-((4-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperazin-1-yl)piperidin-1-yl)methyl)phenyl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H- 1,2,4-triazol-5-yl)methyl)-2,3-dihydrobenzofuran-2-carboxamide; (2R)-5-chloro-2-cyano-6-(6-(4-((4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3- dioxoisoindolin-5-yl)piperazin-1-yl)methyl)piperidin-1-yl)pyridin-3-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-2,3-dihydro-1H-indene-2-carboxamide; 5-chloro-2-cyano-6-(4-((4-(4-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperazin-1-yl)piperidin-1-yl)methyl)phenyl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H- 1,2,4-triazol-5-yl)methyl)-2,3-dihydro-1H-indene-2-carboxamide; (2R)-5-chloro-2-cyano-6-[4-[[4-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin- 5-yl]piperazin-1-yl]methyl]phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methyl]indane-2-carboxamide; (1S,2S)-2-[4-[6-[[1-[1-[[1-[2-(2,6-dioxo-3-piperidyl)-6-fluoro-1,3-dioxo-isoindolin-5-yl]- 4-piperidyl]methyl]-4-piperidyl]pyrazol-4-yl]amino]-3-pyridyl]phenyl]-N-[[3-(2,2,2-trifluoro-1,1- dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]cyclopropanecarboxamide; (1S,2S)-2-(4-(6-((1-(1-((1-(2-(2,6-dioxopiperidin-3-yl)-6-fluoro-1,3-dioxoisoindolin-5- yl)piperidin-4-yl)methyl)piperidin-4-yl)-1H-pyrazol-4-yl)oxy)pyridin-3-yl)phenyl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)cyclopropane-1-carboxamide; 1-[4-[4-[[4-[5-[(2,6-dioxo-3-piperidyl)amino]-3-fluoro-2-pyridyl]-1-piperidyl]methyl]- phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methyl]pyrazole-4-carboxamide; 1-[4-[4-[[4-[4-[(3S)-2,6-dioxo-3-piperidyl]phenyl]piperazin-1-yl]methyl]phenyl]-2- methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole- 4-carboxamide;
1-[4-[4-[[4-[5-(2,6-dioxo-3-piperidyl)-3-fluoro-2-pyridyl]piperazin-1-yl]methyl]phenyl]- 2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methyl]pyrazole-4-carboxamide; 1-(4'-((4-(4-(2,6-dioxopiperidin-3-yl)-2-fluorophenyl)piperazin-1-yl)methyl)-3-methyl- [1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)- 1H-pyrazole-4-carboxamide; 1-(4'-((4-(5-((2,6-dioxopiperidin-3-yl)amino)pyrazin-2-yl)piperidin-1-yl)methyl)-3- methyl-[1,1'-biphenyl]-4-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5- yl)methyl)-1H-pyrazole-4-carboxamide; 1-[4-[4-[[4-[5-[(2,6-dioxo-3-piperidyl)amino]-3-fluoro-2-pyridyl]piperazin-1- yl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol- 5-yl]methyl]pyrazole-4-carboxamide; 1-[4-[4-[[4-[5-(2,6-dioxo-3-piperidyl)-3-fluoro-2-pyridyl]-1-piperidyl]methyl]phenyl]-2- methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole- 4-carboxamide; 1-[4-[4-[[4-[5-(2,6-dioxo-3-piperidyl)-4-fluoro-2-pyridyl]piperazin-1-yl]methyl]phenyl]- 2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methyl]pyrazole-4-carboxamide; 1-[5-[4-[[4-[4-[(2,6-dioxo-3-piperidyl)amino]-2-fluoro-phenyl]-1- piperidyl]methyl]phenyl]pyrimidin-2-yl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4- triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-[5-[4-[[4-[5-(2,6-dioxo-3-piperidyl)-3-fluoro-2-pyridyl]piperazin-1- yl]methyl]phenyl]pyrimidin-2-yl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methyl]pyrazole-4-carboxamide; 1-[5-[4-[[4-[5-[(2,6-dioxo-3-piperidyl)amino]-3-fluoro-2-pyridyl]piperazin-1- yl]methyl]phenyl]pyrimidin-2-yl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methyl]pyrazole-4-carboxamide; 1-[5-[4-[[4-[4-[(2,4-dioxohexahydropyrimidin-1-yl)methyl]-2-fluoro-phenyl]piperazin-1- yl]methyl]phenyl]pyrimidin-2-yl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methyl]pyrazole-4-carboxamide;
1-[5-[4-[[4-[5-[(2,6-dioxo-3-piperidyl)amino]-3-fluoro-2-pyridyl]piperazin-1- yl]methyl]phenyl]-2-pyridyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methyl]pyrazole-4-carboxamide; 1-(5-(5-((4-(5-((2,6-dioxopiperidin-3-yl)amino)-3-fluoropyridin-2-yl)piperazin-1- yl)methyl)pyridin-2-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-(5-(5-((4-(5-((2,6-dioxopiperidin-3-yl)amino)-3-fluoropyridin-2-yl)piperazin-1- yl)methyl)-3-fluoropyridin-2-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H- 1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-[5-[4-[[4-[4-[[(3S)-2,6-dioxo-3-piperidyl]carbamoyl]-3-fluoro-phenyl]piperazin-1- yl]methyl]phenyl]pyrimidin-2-yl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methyl]pyrazole-4-carboxamide; 1-[4-[4-[[4-[4-[[(3S)-2,6-dioxo-3-piperidyl]carbamoyl]-3-fluoro-phenyl]piperazin-1- yl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol- 5-yl]methyl]pyrazole-4-carboxamide hydrochloride; 1-[4-[4-[[4-[4-[[(3S)-2,6-dioxo-3-piperidyl]carbamoyl]-3-fluoro-phenyl]-1- piperidyl]methyl]phenyl]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4- triazol-5-yl]methyl]pyrazole-4-carboxamide; (S)-N-((3-(1,1-difluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1-(5-(4-((4- (4-((2,6-dioxopiperidin-3-yl)carbamoyl)-3-fluorophenyl)piperidin-1-yl)methyl)phenyl)pyridin-2- yl)-1H-pyrazole-4-carboxamide; 1-[5-[4-[[4-[4-[[(3S)-2,6-dioxo-3-piperidyl]carbamoyl]-3-fluoro-phenyl]-1-piperidyl]- methyl]phenyl]pyrimidin-2-yl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methyl]pyrazole-4-carboxamide; 1-[5-[4-[[4-[5-(2,6-dioxo-3-piperidyl)-3-fluoro-2-pyridyl]-1-piperidyl]methyl]phenyl]- pyrimidin-2-yl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole- 4-carboxamide; 1-(4-((4-(5-(2,6-dioxopiperidin-3-yl)-3-fluoropyridin-2-yl)piperazin-1-yl)methyl)-2- methylphenyl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H- pyrazole-4-carboxamide;
1-[4-[[1-[5-(2,6-dioxo-3-piperidyl)-3-fluoro-2-pyridyl]-4-piperidyl]methyl]-2-methyl- phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole-4- carboxamide; 1-[4-[[2-[5-(2,6-dioxo-3-piperidyl)-3-fluoro-2-pyridyl]-2-azaspiro[3.5]nonan-7-yl]oxy]-2- methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]pyrazole- 4-carboxamide; and 3-[5-[4-[[4-[5-[(2,6-dioxo-3-piperidyl)amino]-3-fluoro-2-pyridyl]piperazin-1- yl]methyl]phenyl]pyrimidin-2-yl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methyl]isoxazole-5-carboxamide, or a pharmaceutically acceptable salt of any of the aforementioned.
71. The compound of claim 1 or 2, wherein the compound is selected from: 3-(5-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2,6- diazaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-1-methyl-1H-indazol-6- yl)piperidin-1-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan- 2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5-a]pyridin-6- yl)piperazin-1-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan- 2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5-a]pyrimidin-6- yl)piperazin-1-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan- 2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5-a]pyrazin-6- yl)piperazin-1-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan- 2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide;
3-(5-(4-((4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5-a]pyrazin-6- yl)piperidin-1-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan- 2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(2-fluoro-4-((6-(3-fluoro-4-(3-methyl-2,6-dioxopiperidin-3-yl)phenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)phenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2- yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5-a]pyridin-6- yl)piperidin-1-yl)methyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-(1-(4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5-a]pyridin-6- yl)piperidin-1-yl)ethyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-5-fluorobenzo[d]isoxazol-6- yl)piperidin-1-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan- 2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-(1-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)ethyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro- 2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; N-((3-(3,3-difluoro-2-methylbutan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-3-(5-(4-(1-(6-(4- (2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2-azaspiro[3.3]heptan-2-yl)ethyl)-2- fluorophenyl)-4-methylpyrimidin-2-yl)isoxazole-5-carboxamide; 3-(5-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro- 2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2-azaspiro- [3.3]heptan-2-yl)methyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(3-fluoro-2-methylbut- 3-en-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; N-((3-(1-(1,1-difluoroethyl)cyclopropyl)-1H-1,2,4-triazol-5-yl)methyl)-3-(5-(4-((6-(4- (2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2-azaspiro[3.3]heptan-2-yl)methyl)-2- fluorophenyl)-4-methylpyrimidin-2-yl)isoxazole-5-carboxamide;
3-(5-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1- (trifluoromethyl)cyclobutyl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-[5-[4-[1-[6-[4-(2,4-dioxohexahydropyrimidin-1-yl)-3-fluoro-phenyl]-2- azaspiro[3.3]heptan-2-yl]ethyl]-2-fluoro-phenyl]-4,6-dimethyl-pyrimidin-2-yl]-N-[[3-(2,2,2- trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]isoxazole-5-carboxamide; 3-[5-[5-[1-[6-[4- (2,4-dioxohexahydropyrimidin-1-yl)-3-fluoro-phenyl]-2- azaspiro[3.3]heptan-2-yl]ethyl]-3-fluoro-2-pyridyl]-4-methyl-pyrimidin-2-yl]-N-[[3-(2,2,2- trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]isoxazole-5-carboxamide; 3-(5-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)piperidin-1-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((4-(4-((2,4-dioxotetrahydropyrimidin-1(2H)-yl)methyl)-2,5- difluorophenyl)piperidin-1-yl)methyl)phenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((6-(2-chloro-3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)phenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)piperidin-1-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(2-(4-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)piperazin-1-yl)-7- azaspiro[3.5]nonan-7-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)isoxazole-5-carboxamide; 1-(4-((2-(5-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluoropyridin-2-yl)-2- azaspiro[3.5]nonan-7-yl)oxy)-2-(1-methyl-1H-pyrazol-4-yl)phenyl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-(4-((2-(5-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyridin-2-yl)-2-azaspiro[3.5]nonan-7- yl)oxy)-2-methylphenyl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5- yl)methyl)-1H-pyrazole-4-carboxamide; 1-[4-[[2-[5-(2,4-dioxohexahydropyrimidin-1-yl)-3-methyl-2-pyridyl]-2- azaspiro[3.5]nonan-7-yl]oxy]-2-methyl-phenyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H- 1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide;
(R)-1-(4-((4-(4-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-3- methylpiperazin-1-yl)cyclohexyl)oxy)-2-methylphenyl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2- yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; (S)-1-(4-((4-(4-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-3- methylpiperazin-1-yl)cyclohexyl)oxy)-2-methylphenyl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2- yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 1-(3-(2-(5-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-methylpyridin-2-yl)-2- azaspiro[3.5]nonan-7-yl)-5-fluoro-2-methylphenyl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)- 1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 3-(3-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2,6- diazaspiro[3.3]heptan-2-yl)methyl)phenyl)bicyclo[1.1.1]pentan-1-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-(1-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)ethyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(2- (methylthio)propan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(2-fluoro-4-((6-(3-fluoro-4-(3-methyl-2,6-dioxopiperidin-3-yl)phenyl)-2,6- diazaspiro[3.3]heptan-2-yl)methyl)phenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan- 2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((4-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3,5-difluorophenyl)piperazin-1- yl)methyl)piperidin-1-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(5-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)-3-fluoropyridin-2-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-2,3-difluorophenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-2,5-difluorophenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide;
3-(5-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-2-fluoro-5-methylphenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((6-(5-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluoro-6-methylpyridin-2-yl)-2- azaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(5-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3,5-difluorophenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)pyridin-2-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(5-((4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5-a]pyrazin-6- yl)piperazin-1-yl)methyl)-3-fluoropyridin-2-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(5-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)pyrazin-2-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; (R)-3-(5-(4-((4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5-a]pyrazin-6-yl)- 3-methylpiperazin-1-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5-a]pyridin-6- yl)piperazin-1-yl)methyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5-a]pyrazin-6- yl)piperidin-1-yl)methyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-(1-(4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5-a]pyrazin-6- yl)piperazin-1-yl)ethyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2- yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5-a]pyrazin-6- yl)piperazin-1-yl)methyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide;
3-(5-(4-(1-(4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5-a]pyrimidin-6- yl)piperazin-1-yl)ethyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(5-(1-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3,5-difluorophenyl)-2- azaspiro[3.3]heptan-2-yl)ethyl)-3-fluoropyridin-2-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(3-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)-2,3-dihydrobenzofuran-6-yl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(5-(1-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)ethyl)pyridin-2-yl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; N-((3-(3,3-difluoro-2-methylbutan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-3-(5-(4-(1-(6-(4- (2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2-azaspiro[3.3]heptan-2-yl)propyl)-2- fluorophenyl)-4-methylpyrimidin-2-yl)isoxazole-5-carboxamide; 3-(5-(1-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)-2,3-dihydro-1H-inden-5-yl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-(1-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)propyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro- 2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(5-((2-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-6- azaspiro[3.4]octan-6-yl)methyl)-3-fluoropyridin-2-yl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(1-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)ethyl)-4'-methyl-[2,5'-bipyrimidin]-2'-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; N-((3-(3,3-difluoro-2-methylbutan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-3-(5-(1-(6-(4-(2,4- dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2-azaspiro[3.3]heptan-2-yl)ethyl)-4'-methyl- [2,5'-bipyrimidin]-2'-yl)isoxazole-5-carboxamide;
3-(5-(4-(1-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)butyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro- 2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-(1-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)-2-methylpropyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3- (1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-(1-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)-2-methoxyethyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3- (1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(7-(4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5-a]pyrazin-6- yl)piperazin-1-yl)-5-oxa-2-azaspiro[3.4]octan-2-yl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; N-((3-(tert-butyl)-1H-1,2,4-triazol-5-yl)methyl)-3-(5-(4-((6-(4-(2,4- dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2-azaspiro[3.3]heptan-2-yl)methyl)-2- fluorophenyl)-4-methylpyrimidin-2-yl)isoxazole-5-carboxamide; N-((3-(3,3-difluoro-2-methylbutan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-3-(5-(5-(1-(6-(4- (2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2-azaspiro[3.3]heptan-2-yl)ethyl)-3- fluoropyridin-2-yl)-4-methylpyrimidin-2-yl)isoxazole-5-carboxamide; 3-(5-(5-(1-(6-(2-chloro-3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)phenyl)-2- azaspiro[3.3]heptan-2-yl)ethyl)-3-fluoropyridin-2-yl)-4-methylpyrimidin-2-yl)-N-((3-(3,3- difluoro-2-methylbutan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(1-(6-(2-chloro-3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)phenyl)-2- azaspiro[3.3]heptan-2-yl)ethyl)-4'-methyl-[2,5'-bipyrimidin]-2'-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 1-(4-((4-(4-(5-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-4-methylpyridin-2-yl)piperazin-1- yl)cyclohexyl)oxy)-2-methylphenyl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide; 3-(5-(5-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)pyridin-2-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide;
3-(5-(4-(1-(6-(2-chloro-3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)phenyl)-2- azaspiro[3.3]heptan-2-yl)ethyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro- 2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((6-(2-chloro-3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)phenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-(1-(6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)ethyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(5-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3,5-difluorophenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)-3-fluoropyridin-2-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((((3-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3,5- difluorophenyl)cyclobutyl)methyl)amino)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-(((3-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3,5- difluorophenyl)cyclobutyl)amino)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro- 2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3,5-difluorophenyl)-2,6- diazaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((4-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3,5-difluorophenyl)piperazin-1- yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((4-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3,5-difluorophenoxy)piperidin-1- yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((3-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3,5-difluorophenoxy)azetidin-1- yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)isoxazole-5-carboxamide;
3-(5-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3,5-difluorophenoxy)-2- azaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro- 2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(3-(4-((4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5-a]pyrimidin-6- yl)piperazin-1-yl)methyl)phenyl)bicyclo[1.1.1]pentan-1-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(3-(4-((4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)pyrazolo[1,5-a]pyrazin-6- yl)piperazin-1-yl)methyl)phenyl)bicyclo[1.1.1]pentan-1-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)phenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2- yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-2-methylphenyl)piperidin-1- yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((4-(3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-2-methylphenyl)piperidin-1- yl)methyl)piperidin-1-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((4-(2-chloro-3-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)phenyl)piperidin-1- yl)methyl)piperidin-1-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4- triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-(((3aS*,6aS*)-4-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3- fluorophenyl)hexahydropyrrolo[3,2-b]pyrrol-1(2H)-yl)methyl)-2-fluorophenyl)pyrimidin-2-yl)- N-((3-(1,1,1-trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5- carboxamide; 3-(5-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3,5-difluorophenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)-2-fluorophenyl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1-trifluoro- 2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(5-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)-3-fluoropyridin-2-yl)-4-methylpyrimidin-2-yl)-N-((3-(1,1,1- trifluoro-2-methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide;
3-(5-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2- azaspiro[3.3]heptan-2-yl)methyl)-[2,5'-bipyrimidin]-2'-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan- 2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; 3-(5-(4-((6-(4-(2,4-dioxotetrahydropyrimidin-1(2H)-yl)-3-fluorophenyl)-2,6- diazaspiro[3.3]heptan-2-yl)methyl)phenyl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2-methylpropan- 2-yl)-1H-1,2,4-triazol-5-yl)methyl)isoxazole-5-carboxamide; or a pharmaceutically acceptable salt of any of the aforementioned.
72. A pharmaceutical composition comprising a compound of any one of claims 1-71, or a pharmaceutically acceptable salt thereof, and at least one pharmaceutically acceptable carrier.
73. A method of treating cancer in a patient in need of treatment comprising administering to said patient a therapeutically effective amount of a compound of any one of claims 1-71, or a pharmaceutically acceptable salt thereof.
74. The method of claim 73, wherein the cancer is selected from prostate cancer, breast cancer, glioblastoma, bladder cancer, renal cell carcinoma, salivary gland cancer, colorectal cancer, esophageal cancer, pancreatic cancer, and stomach cancer.
75. A compound of Formula (A1):
 or a pharmaceutically acceptable salt thereof, wherein: R1, R2, and R3 are each independently selected from H, halo, C1-4 alkyl, and C1-4 haloalkyl; R4 and R5 are each independently selected from H, halo, ORA, C1-4 alkyl, and C1-4 haloalkyl, wherein RA is H, C1-4 alkyl, or C1-4 haloalkyl; or
R4 and R5 together with the carbon atom to which they are attached form a C3-6 cycloalkyl group or a 3-6 membered heterocycloalkyl group, each optionally substituted with 1, 2, or 3 substituents independently selected from H, halo, ORA, C1-4 alkyl, and C1-4 haloalkyl; R6 and R7 are each independently selected from H, halo, C1-4 alkyl, and C1-4 haloalkyl; R8 is H or C1-4 alkyl; Cy1 is selected from C6-10 aryl, C3-14 cycloalkyl, 5-14 membered heteroaryl, 4-14 membered heterocycloalkyl, C3-7 cycloalkyl fused with phenyl to form a bicyclic ring, C4-7 cycloalkyl fused with 5-6 membered heteroaryl to form a bicyclic ring, 4-7 membered heterocycloalkyl fused with phenyl to form a bicyclic ring, and 4-7 membered heterocycloalkyl fused with 5-6 membered heteroaryl to form a bicyclic ring, each optionally substituted by 1, 2, 3, or 4 substituents independently selected from halo, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C1-6 haloalkyl, CN, NO2, ORa, SRa, C1-6 alkyl-NRcRd, C(O)Rb, C(O)NRcRd, C(O)ORa, OC(O)Rb, OC(O)NRcRd, C(=NRe)NRcRd, NRcC(=NRe)NRcRd, NRcRd, NRcC(O)Rb, NRcC(O)ORa, NRcC(O)NRcRd, NRcS(O)Rb, NRcS(O)2Rb, NRcS(O)2NRcRd, S(O)Rb, S(O)NRcRd, S(O)2Rb, and S(O)2NRcRd; A is selected from H, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, and 4-7 membered heterocycloalkyl, each optionally substituted by 1, 2, 3, or 4 substituents independently selected from halo, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C1-6 haloalkyl, CN, NO2, ORa1, SRa1, C(O)Rb1, C(O)NRc1Rd1, C(O)ORa1, OC(O)Rb1, OC(O)NRc1Rd1, C(=NRe1)NRc1Rd1, NRc1C(=NRe1)NRc1Rd1, NRc1Rd1, NRc1C(O)Rb1, NRc1C(O)ORa1, NRc1C(O)NRc1Rd1, NRc1S(O)Rb1, NRc1S(O)2Rb1, NRc1S(O)2NRc1Rd1, S(O)Rb1, S(O)NRc1Rd1, S(O)2Rb1, and S(O)2NRc1Rd1; each Ra, Rb, Rc, Rd, Ra1, Rb1, Rc1, and Rd1 is independently selected from H, C1-6 alkyl, C1-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl-C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl, wherein said C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl-C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl of Ra, Rb, Rc, Rd, Ra1, Rb1, Rc1, and Rd1 is optionally substituted with 1, 2, 3, 4, or 5 substituents independently selected from halo, C1-4 alkyl, C1-4 haloalkyl, C1-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, CN, ORa3, SRa3, C(O)Rb3,
C(O)NRc3Rd3, C(O)ORa3, OC(O)Rb3, OC(O)NRc3Rd3, NRc3Rd3, NRc3C(O)Rb3, NRc3C(O)NRc3Rd3, NRc3C C NRc3C S S S
or a pharmaceutically acceptable salt thereof, wherein: R1, R2, and R3 are each independently selected from H, halo, C1-4 alkyl, and C1-4 haloalkyl; R4 and R5 are each independently selected from H, halo, ORA, C1-4 alkyl, and C1-4 haloalkyl, wherein RA is H, C1-4 alkyl, or C1-4 haloalkyl; or
R4 and R5 together with the carbon atom to which they are attached form a C3-6 cycloalkyl group or a 3-6 membered heterocycloalkyl group, each optionally substituted with 1, 2, or 3 substituents independently selected from H, halo, ORA, C1-4 alkyl, and C1-4 haloalkyl; R6 and R7 are each independently selected from H, halo, C1-4 alkyl, and C1-4 haloalkyl; R8 is H or C1-4 alkyl; Cy1 is selected from C6-10 aryl, C3-14 cycloalkyl, 5-14 membered heteroaryl, 4-14 membered heterocycloalkyl, C3-7 cycloalkyl fused with phenyl to form a bicyclic ring, C4-7 cycloalkyl fused with 5-6 membered heteroaryl to form a bicyclic ring, 4-7 membered heterocycloalkyl fused with phenyl to form a bicyclic ring, and 4-7 membered heterocycloalkyl fused with 5-6 membered heteroaryl to form a bicyclic ring, each optionally substituted by 1, 2, 3, or 4 substituents independently selected from halo, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C1-6 haloalkyl, CN, NO2, ORa, SRa, C1-6 alkyl-NRcRd, C(O)Rb, C(O)NRcRd, C(O)ORa, OC(O)Rb, OC(O)NRcRd, C(=NRe)NRcRd, NRcC(=NRe)NRcRd, NRcRd, NRcC(O)Rb, NRcC(O)ORa, NRcC(O)NRcRd, NRcS(O)Rb, NRcS(O)2Rb, NRcS(O)2NRcRd, S(O)Rb, S(O)NRcRd, S(O)2Rb, and S(O)2NRcRd; A is selected from H, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, and 4-7 membered heterocycloalkyl, each optionally substituted by 1, 2, 3, or 4 substituents independently selected from halo, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C1-6 haloalkyl, CN, NO2, ORa1, SRa1, C(O)Rb1, C(O)NRc1Rd1, C(O)ORa1, OC(O)Rb1, OC(O)NRc1Rd1, C(=NRe1)NRc1Rd1, NRc1C(=NRe1)NRc1Rd1, NRc1Rd1, NRc1C(O)Rb1, NRc1C(O)ORa1, NRc1C(O)NRc1Rd1, NRc1S(O)Rb1, NRc1S(O)2Rb1, NRc1S(O)2NRc1Rd1, S(O)Rb1, S(O)NRc1Rd1, S(O)2Rb1, and S(O)2NRc1Rd1; each Ra, Rb, Rc, Rd, Ra1, Rb1, Rc1, and Rd1 is independently selected from H, C1-6 alkyl, C1-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl-C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl, wherein said C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl-C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl of Ra, Rb, Rc, Rd, Ra1, Rb1, Rc1, and Rd1 is optionally substituted with 1, 2, 3, 4, or 5 substituents independently selected from halo, C1-4 alkyl, C1-4 haloalkyl, C1-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, CN, ORa3, SRa3, C(O)Rb3,
C(O)NRc3Rd3, C(O)ORa3, OC(O)Rb3, OC(O)NRc3Rd3, NRc3Rd3, NRc3C(O)Rb3, NRc3C(O)NRc3Rd3, NRc3C C NRc3C S S S
 from halo, C1-4 alkyl, C1-4 haloalkyl, CN, ORa3, SRa3, C(O)Rb3, C(O)NRc3Rd3, C(O)ORa3, OC(O)Rb3, OC(O)NRc3Rd3, NRc3Rd3, NRc3C(O)Rb3, NRc3C(O)NRc3Rd3, NRc3C(O)ORa3, C(=NRe3)NRc3Rd3, NRc3C(=NRe3)NRc3Rd3, S(O)Rb3, S(O)NRc3Rd3, S(O)2Rb3, NRc3S(O)2Rb3, NRc3S(O)2NRc3Rd3, and S(O)2NRc3Rd3; or Rc1 and Rd1 together with the N atom to which they are attached form a 4-7 membered heterocycloalkyl group optionally substituted with 1, 2, or 3 substituents independently selected from halo, C1-4 alkyl, C1-4 haloalkyl, CN, ORa3, SRa3, C(O)Rb3, C(O)NRc3Rd3, C(O)ORa3, OC(O)Rb3, OC(O)NRc3Rd3, NRc3Rd3, NRc3C(O)Rb3, NRc3C(O)NRc3Rd3, NRc3C(O)ORa3, C2-
from halo, C1-4 alkyl, C1-4 haloalkyl, CN, ORa3, SRa3, C(O)Rb3, C(O)NRc3Rd3, C(O)ORa3, OC(O)Rb3, OC(O)NRc3Rd3, NRc3Rd3, NRc3C(O)Rb3, NRc3C(O)NRc3Rd3, NRc3C(O)ORa3, C(=NRe3)NRc3Rd3, NRc3C(=NRe3)NRc3Rd3, S(O)Rb3, S(O)NRc3Rd3, S(O)2Rb3, NRc3S(O)2Rb3, NRc3S(O)2NRc3Rd3, and S(O)2NRc3Rd3; or Rc1 and Rd1 together with the N atom to which they are attached form a 4-7 membered heterocycloalkyl group optionally substituted with 1, 2, or 3 substituents independently selected from halo, C1-4 alkyl, C1-4 haloalkyl, CN, ORa3, SRa3, C(O)Rb3, C(O)NRc3Rd3, C(O)ORa3, OC(O)Rb3, OC(O)NRc3Rd3, NRc3Rd3, NRc3C(O)Rb3, NRc3C(O)NRc3Rd3, NRc3C(O)ORa3, C2-
 6 alkenyl, C2-6 alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl-C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl, wherein said C1-6 alkyl, C1-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl-C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl are each optionally substituted with 1, 2, or 3 substituents independently selected from OH, CN, amino, halo, C1-6 alkyl, C1-6 alkylamino, di(C1-6 alkyl)amino, C1-6 alkoxy, C1-6 haloalkyl, and C1-6 haloalkoxy; and each Re, Re1, Re2, and Re3 is independently selected from H, C1-4 alkyl, and CN.
6 alkenyl, C2-6 alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl-C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl, wherein said C1-6 alkyl, C1-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, C6-10 aryl, C3-7 cycloalkyl, 5-10 membered heteroaryl, 4-10 membered heterocycloalkyl, C6-10 aryl-C1-4 alkyl, C3-7 cycloalkyl-C1-4 alkyl, 5-10 membered heteroaryl-C1-4 alkyl, and 4-10 membered heterocycloalkyl-C1-4 alkyl are each optionally substituted with 1, 2, or 3 substituents independently selected from OH, CN, amino, halo, C1-6 alkyl, C1-6 alkylamino, di(C1-6 alkyl)amino, C1-6 alkoxy, C1-6 haloalkyl, and C1-6 haloalkoxy; and each Re, Re1, Re2, and Re3 is independently selected from H, C1-4 alkyl, and CN.
76. The compound of claim 75, or a pharmaceutically acceptable salt thereof wherein R1, R2, and R3 are each independently selected from H, F, methyl, and trifluoromethyl.
77. The compound of claim 75 or 76, or a pharmaceutically acceptable salt thereof, wherein R1, R2, and R3 are each F.
78. The compound of any one of claims 75-77, or a pharmaceutically acceptable salt thereof, wherein R4 and R5 are each C1-4 alkyl.
79. The compound of any one of claim 75-77, or a pharmaceutically acceptable salt thereof, wherein R4 and R5 are each methyl.
80. The compound of any one of claims 75-79, or a pharmaceutically acceptable salt thereof, wherein R6 and R7 are each H.
81. The compound of any one of claim 75-80, or a pharmaceutically acceptable salt thereof, wherein R8 is H.
82. The compound of any one of claims 75-81, or a pharmaceutically acceptable salt thereof, wherein Cy1 is selected from C3-6 cycloalkyl, 5-6 membered heteroaryl, C5-6 cycloalkyl fused with phenyl to form a bicyclic ring, and 5-6 membered heterocycloalkyl fused with phenyl to form a bicyclic ring, each optionally substituted by 1 or 2 substituents independently selected from halo, C1-6 alkyl, C1-6 haloalkyl, CN, C1-6 alkyl-NRcRd, C(O)NRcRd, and NRcRd.
83. The compound of any one of claims 75-81, or a pharmaceutically acceptable salt thereof, wherein Cy1 is cyclopropyl, pyrazolyl, or isoxazolyl.
84. The compound of any one of claims 75-81, or a pharmaceutically acceptable salt thereof, wherein Cy1 is C5-6 cycloalkyl fused with phenyl to form a bicyclic ring, optionally substituted by 1 or 2 substituents independently selected from halo, C1-6 alkyl, CN, C1-6 alkyl-NRcRd, and NRcRd.
85. The compound of any one of claims 75-81, or a pharmaceutically acceptable salt thereof, wherein Cy1 is cyclopentyl fused with phenyl to form a bicyclic ring, optionally substituted by CN, Cl, and F.
86. The compound of any one of claims 75-81, or a pharmaceutically acceptable salt thereof, wherein Cy1 is 5-6 membered heterocycloalkyl fused with phenyl to form a bicyclic ring, optionally substituted by 1, 2, or 3 substituents independently selected from halo and C1-6 alkyl.
87. The compound of any one of claims 75-81, or a pharmaceutically acceptable salt thereof, wherein Cy1 is dihydrobenzofuran, optionally substituted by 1, 2, or 3 substituents independently selected from Cl and I.
88. The compound of any one of claims 75-87, or a pharmaceutically acceptable salt thereof, wherein A is selected from H, C6-10 aryl and 5-10 membered heteroaryl, optionally substituted by 1, 2, or 3 substituents independently selected from halo, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C1-6 haloalkyl, CN, NO2, ORa1, SRa1, C(O)Rb1, C(O)NRc1Rd1, C(O)ORa1, OC(O)Rb1, OC(O)NRc1Rd1, C(=NRe1)NRc1Rd1, NRc1C(=NRe1)NRc1Rd1, NRc1Rd1, NRc1C(O)Rb1, NRc1C(O)ORa1, NRc1C(O)NRc1Rd1, NRc1S(O)Rb1, NRc1S(O)2Rb1, NRc1S(O)2NRc1Rd1, S(O)Rb1, S(O)NRc1Rd1, S(O)2Rb1, and S(O)2NRc1Rd1.
89. The compound of any one of claims 75-87, or a pharmaceutically acceptable salt thereof, wherein A is phenyl or 5-6 membered heteroaryl, optionally substituted by 1, 2, or 3 substituents selected from halo and C1-6 alkyl.
90. The compound of any one of claims 75-87, or a pharmaceutically acceptable salt thereof, wherein A is phenyl optionally substituted by 1, 2, or 3 substituents selected from methyl, Br, and Cl.
91. The compound of any one of claims 75-87, or a pharmaceutically acceptable salt thereof, wherein A is H.
92. The compound of claim 75, wherein the compound is selected from: 5-chloro-6-iodo-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5-yl]methyl]- 2,3-dihydrobenzofuran-2-carboxamide;
(1S,2S)-2-(4-bromophenyl)-N-[[5-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-3- yl]methyl]cyclopropanecarboxamide; 1-(4-bromo-2-methyl-phenyl)-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol- 5-yl]methyl]pyrazole-4-carboxamide; 1-(3-bromo-2,4-dimethyl-phenyl)-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4- triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-(4-bromo-5-chloro-2-methyl-phenyl)-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H- 1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 5,6-dichloro-2-cyano-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methyl]indane-2-carboxamide; 1-[5-(4-formylphenyl)pyrimidin-2-yl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H- 1,2,4-triazol-5-yl]methyl]pyrazole-4-carboxamide; 1-(5-bromo-2-pyridyl)-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4-triazol-5- yl]methyl]pyrazole-4-carboxamide; 1-[5-(4-formylphenyl)-2-pyridyl]-N-[[3-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-1H-1,2,4- triazol-5-yl]methyl]pyrazole-4-carboxamide; and 1-(5-(3-fluoro-5-formylpyridin-2-yl)pyrimidin-2-yl)-N-((3-(1,1,1-trifluoro-2- methylpropan-2-yl)-1H-1,2,4-triazol-5-yl)methyl)-1H-pyrazole-4-carboxamide, or a pharmaceutically acceptable salt of any of the aforementioned.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US202363600190P | 2023-11-17 | 2023-11-17 | |
| US63/600,190 | 2023-11-17 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2025106910A1 true WO2025106910A1 (en) | 2025-05-22 |
Family
ID=93843421
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2024/056259 Pending WO2025106910A1 (en) | 2023-11-17 | 2024-11-15 | Degrader molecules targeting androgen receptor |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2025106910A1 (en) |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017197056A1 (en) | 2016-05-10 | 2017-11-16 | C4 Therapeutics, Inc. | Bromodomain targeting degronimers for target protein degradation |
| WO2019099926A1 (en) | 2017-11-17 | 2019-05-23 | Arvinas, Inc. | Compounds and methods for the targeted degradation of interleukin-1 receptor-associated kinase 4 polypeptides |
| WO2020023851A1 (en) | 2018-07-26 | 2020-01-30 | Yale University | Bifunctional substitued pyrimidines as modulators of fak proteolyse |
-
2024
- 2024-11-15 WO PCT/US2024/056259 patent/WO2025106910A1/en active Pending
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017197056A1 (en) | 2016-05-10 | 2017-11-16 | C4 Therapeutics, Inc. | Bromodomain targeting degronimers for target protein degradation |
| WO2019099926A1 (en) | 2017-11-17 | 2019-05-23 | Arvinas, Inc. | Compounds and methods for the targeted degradation of interleukin-1 receptor-associated kinase 4 polypeptides |
| WO2020023851A1 (en) | 2018-07-26 | 2020-01-30 | Yale University | Bifunctional substitued pyrimidines as modulators of fak proteolyse |
Non-Patent Citations (15)
| Title |
|---|
| "Physicians' Desk Reference", MEDICAL ECONOMICS COMPANY |
| "Remington's Pharmaceutical Sciences", 1985, MACK PUBLISHING COMPANY, pages: 1418 |
| AN S ET AL., EBIOMEDICINE, vol. 36, 2018, pages 553 - 562 |
| BONDESON, D. P. ET AL., NAT CHEM BIOL, vol. 11, no. 8, 2015, pages 611 - 617 |
| CYRUS K ET AL: "Impact of linker length on the activity of PROTACs", MOLECULAR BIOSYSTEMS, ROYAL SOCIETY OF CHEMISTRY, GB, vol. 7, no. 2, 1 February 2011 (2011-02-01), pages 359 - 364, XP002721196, ISSN: 1742-206X, [retrieved on 20100401], DOI: 10.1039/C0MB00074D * |
| FUJII SHINYA ET AL: "Androgen receptor modulators: a review of recent patents and reports (2012-2018)", EXPERT OPINION ON THERAPEUTIC PATENTS, vol. 29, no. 6, 19 May 2019 (2019-05-19), GB, pages 439 - 453, XP055853428, ISSN: 1354-3776, DOI: 10.1080/13543776.2019.1618831 * |
| HA SI ET AL: "A Comprehensive Overview of Small-Molecule Androgen Receptor Degraders: Recent Progress and Future Perspectives", JOURNAL OF MEDICINAL CHEMISTRY, vol. 65, no. 24, 2 December 2022 (2022-12-02), US, pages 16128 - 16154, XP093083336, ISSN: 0022-2623, Retrieved from the Internet <URL:https://pubs.acs.org/doi/pdf/10.1021/acs.jmedchem.2c01487> DOI: 10.1021/acs.jmedchem.2c01487 * |
| HU, C., GENOMICS, vol. 112, no. 2, 2020, pages 1926 - 1940 |
| JOURNAL OF PHARMACEUTICAL SCIENCE, vol. 66, 1977, pages 2 |
| MICHAEL L MOHLER ET AL: "Androgen receptor antagonists: a patent review (2008 - 2011)", EXPERT OPINION ON THERAPEUTIC PATENTS, vol. 22, no. 5, 1 May 2012 (2012-05-01), pages 541 - 565, XP055111543, ISSN: 1354-3776, DOI: 10.1517/13543776.2012.682571 * |
| PAIVA S-L. ET AL., CURR. OP. IN CHEM. BIO., vol. 50, 2010, pages 111 - 119 |
| SCHWEIZER, M. T., CANCERS, vol. 9, 2017, pages 1 - 19 |
| STEINEBACH CHRISTIAN ET AL: "A MedChem toolbox for cereblon-directed PROTACs", MEDCHEMCOMM, vol. 10, no. 6, 19 June 2019 (2019-06-19), United Kingdom, pages 1037 - 1041, XP055857543, ISSN: 2040-2503, DOI: 10.1039/C9MD00185A * |
| T.W. GREENEP.G.M. WUTS: "Protective Groups in Organic Synthesis", 1999, WILEY & SONS, INC. |
| TROUP ROBERT I. ET AL: "Current strategies for the design of PROTAC linkers: a critical review", EXPLORATION OF TARGETED ANTI-TUMOR THERAPY, vol. 1, no. 5, 30 October 2020 (2020-10-30), XP055828975, Retrieved from the Internet <URL:https://www.explorationpub.com/uploads/Article/A100218/100218.pdf> DOI: 10.37349/etat.2020.00018 * |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP3573987B1 (en) | Tyrosine amide derivatives as rho- kinase inhibitors | |
| CN104470925B (en) | Imidazo-triazine formonitrile HCN as kinase inhibitor | |
| CN112262139B (en) | Aminopyrrolotriazines as Kinase Inhibitors | |
| TW201938555A (en) | KRAS G12C inhibitor | |
| KR101535954B1 (en) | Piperidine Derivatives for GPR119 agonist | |
| KR102454129B1 (en) | Aminopyridine derivatives and their use as selective alk-2 inhibitors | |
| WO2024028169A1 (en) | Novel specifically substituted thiophenolic compounds | |
| TW202237573A (en) | Aryl derivatives for treating trpm3 mediated disorders | |
| AU2023330130A1 (en) | Compounds and methods for modulating her2 | |
| CA3176957A1 (en) | Aminopyrimidine derivatives and their use as aryl hydrocarbon receptor modulators | |
| JP2023547892A (en) | Pyrimidine derivatives as modulators of 5-HT2A serotonin receptors useful for treating disorders associated with 5-HT2A serotonin receptors | |
| JP2023527961A (en) | Antagonist of gpr39 protein | |
| KR101700906B1 (en) | Amide Derivatives for GPR119 agonist | |
| WO2025106910A1 (en) | Degrader molecules targeting androgen receptor | |
| TW202448898A (en) | Compounds for the degradation of egfr kinase | |
| TW202346281A (en) | Compounds and their use in treating cancer | |
| JP2024528614A (en) | Nitrile sumo inhibitors and uses thereof | |
| CN115279757A (en) | Pyrimidine bicyclic compounds as antiviral agents for the treatment and prevention of HIV infection | |
| RU2780103C1 (en) | Antiviral remedies for treatment and prevention of hiv infection | |
| WO2024112692A1 (en) | Casitas b-lineage lymphoma protooncogene b (cbl-b) degrading compounds and associated methods of use | |
| CA3246917A1 (en) | C-myc mrna translation modulators and uses thereof in the treatment of cancer | |
| WO2025262297A1 (en) | Protac degraders of mllt1 and/or mllt3 | |
| EA049662B1 (en) | FGFR3 INHIBITOR COMPOUNDS | |
| JPWO2021222858A5 (en) |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 24821214 Country of ref document: EP Kind code of ref document: A1 |