WO2025106469A1 - Engineered heavy chain variable domains and uses thereof - Google Patents
Engineered heavy chain variable domains and uses thereof Download PDFInfo
- Publication number
- WO2025106469A1 WO2025106469A1 PCT/US2024/055618 US2024055618W WO2025106469A1 WO 2025106469 A1 WO2025106469 A1 WO 2025106469A1 US 2024055618 W US2024055618 W US 2024055618W WO 2025106469 A1 WO2025106469 A1 WO 2025106469A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- antigen
- binding molecule
- binding
- domain
- amino acid
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2803—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily
- C07K16/2809—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily against the T-cell receptor (TcR)-CD3 complex
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/30—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants from tumour cells
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/30—Immunoglobulins specific features characterized by aspects of specificity or valency
- C07K2317/31—Immunoglobulins specific features characterized by aspects of specificity or valency multispecific
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/40—Immunoglobulins specific features characterized by post-translational modification
- C07K2317/41—Glycosylation, sialylation, or fucosylation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/56—Immunoglobulins specific features characterized by immunoglobulin fragments variable (Fv) region, i.e. VH and/or VL
- C07K2317/567—Framework region [FR]
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/60—Immunoglobulins specific features characterized by non-natural combinations of immunoglobulin fragments
- C07K2317/64—Immunoglobulins specific features characterized by non-natural combinations of immunoglobulin fragments comprising a combination of variable region and constant region components
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/73—Inducing cell death, e.g. apoptosis, necrosis or inhibition of cell proliferation
Definitions
- Various biological therapeutics have been developed for the prevention and treatment of various diseases, including proliferative diseases (e.g., cancers), chronic inflammatory diseases (e.g., Crohn’s disease), and rheumatologic diseases (e.g., rheumatoid arthritis).
- proliferative diseases e.g., cancers
- chronic inflammatory diseases e.g., Crohn’s disease
- rheumatologic diseases e.g., rheumatoid arthritis
- antibodies and related antigen-binding molecules have proven effective in clinical practice. Yet, immunogenicity of these molecules can challenge their usefulness and hinder the development of new molecules for clinical application.
- ADAs anti-drug antibodies
- ADAs anti-drug antibodies
- the present disclosure further provides polypeptides comprising the VHs of the disclosure, having an N-linked glycosylation site near their C-termini.
- the polypeptides are antigen-binding molecules.
- the engineered VHs can be advantageously used in the context of a variety of antigen-binding molecules and components thereof, e.g., scFvs, Fabs, antibodies, antibody fragments, and other antigen-binding molecules, including multivalent and/or multispecific antigen-binding molecules.
- an antigen-binding molecule can include one or more additional target binding domains (e.g., one or more Fab moieties, one or more scFv moieties, or a combination thereof) and/or one or more linkers separating one or more moieties in the antigen-binding molecule.
- additional target binding domains e.g., one or more Fab moieties, one or more scFv moieties, or a combination thereof
- linkers separating one or more moieties in the antigen-binding molecule.
- the disclosure further provides nucleic acids encoding the VHs and antigen-binding molecules of the disclosure.
- the nucleic acids encoding the antigen-binding molecules can be in the form of a single nucleic acid (e.g., a vector encoding two or more polypeptide chains) or a plurality of nucleic acids (e.g., two or more vectors encoding different polypeptide chains).
- the disclosure further provides host cells and cell lines engineered to express the nucleic acids, VHs and antigen-binding molecules of the disclosure. Exemplary nucleic acids, host cells, and cell lines, are described in Section 6.5 and numbered embodiments 108 to 110. [0011] Methods of producing the antigen-binding molecules, methods of using the engineered VHs of the disclosure to decrease binging of an antigen-binding molecule to anti-drug antibodies, and methods of reducing antigenicity of antigen-binding molecules are described in Section 6.7 and numbered embodiments 111, and 118 to 150. [0012] The disclosure further provides pharmaceutical compositions comprising the antigen- binding molecules of the disclosure. Exemplary compositions are described in Section 6.6 and numbered embodiment 107.
- Exemplary administration methods, including treatment methods, are described in Section 6.7 and numbered embodiments 112 to 114. 5.
- FIGS.1A-1C illustrate exemplary antigen-binding molecules of the disclosure comprising a VH comprising at its C-terminus the amino acid sequence X 1 X 2 X 3 X 4 X 5 NX 6 X 7 X 8 X 9 X 10 X 11 X 12 (SEQ ID NO:1), wherein (a) X 1 , X 2 , X 3 , X 4 , and X 5 are each independently selected from any amino acid; (b) X 6 is selected from any amino acid, optionally wherein the amino acid is not proline; (c) X 7 is S or T, and (d) X 8 , X 9 , X 10 ,X 11 , and X 12 are each independently selected from any amino acid and absent.
- FIG.1A shows an scFv comprising a VH comprising the amino acid sequence of SEQ ID NO:1 at its C-terminus.
- FIG.1B shows a bivalent antigen-binding molecule comprising two scFv molecules linked by a linker, where at least one of the VHs comprises the sequence X 1 X 2 X 3 X 4 X 5 NX 6 [S/T]X 8 X 9 X 10 X 11 X 12 at its C- terminus.
- FIG.1C shows an exemplary multispecific antigen-binding molecule comprising a first polypeptide, which comprises from N-terminus to C-terminus: a first targeting moiety (e.g., a Fab or scFv), a first dimerization moiety (e.g., an Fc domain), an optional linker, and a first scFv; and a second polypeptide which comprises from N-terminus to C-terminus: a second targeting moiety (e.g., a Fab of scFv), a second dimerization moiety, an optional linker, and a second scFv, where each scFv comprises a VH comprising the sequence X 1 X 2 X 3 X 4 X 5 NX 6 [S/T]X 8 X 9 X 10 X 11 X 12 at its C-terminus.
- a first targeting moiety e.g., a Fab or scFv
- FIGS.2A-2G illustrate certain multispecific antigen-binding molecules of the disclosure, each comprising a first polypeptide, which comprises from N-terminus to C-terminus: a first targeting moiety (e.g., a Fab or scFv), a first dimerization moiety (e.g., an Fc domain), an optional linker, and a second targeting moiety; and a second polypeptide which comprises from N-terminus to C-terminus: a target antigen-binding moiety, such as a tumor-associated antigen targeting moiety (e.g., a Fab or scFv that specifically binds to a tumor-associated antigen), a second dimerization moiety, an optional linker and a second target antigen-binding moiety.
- a first targeting moiety e.g., a Fab or scFv
- a first dimerization moiety e.g., an Fc domain
- an optional linker e.g.,
- FIG. 2A shows a multispecific antigen-binding molecule, the scFv C-termini of which comprise the amino acid sequence VTVSS (SEQ ID NO:62).
- FIGs 2B-2G show multispecific antigen-binding molecules comprising engineered scFv C-terminal sequences, wherein modifications are presented in bold.
- FIG.2B shows a construct with an engineered scFv C-terminal amino acid sequence VTVSSPP (SEQ ID NO:63).
- FIG.2C shows a construct with an engineered scFv C- terminal amino acid sequence VTVKPGG (SEQ ID NO:64).
- FIG.2D shows a construct with an engineered scFv C-terminal amino acid sequence VTVKPGG (SEQ ID NO:64) and a V11K mutation.
- FIG.2E shows a construct with an engineered scFv C-terminal amino acid sequence VTVSSGGGGS (SEQ ID NO:65).
- FIG.2F shows a construct with an engineered scFv C- terminal amino acid sequence VTVNSS (SEQ ID NO:50).
- FIG.2G shows a construct with an engineered scFv C-terminal amino acid sequence VTVNST (SEQ ID NO:51).
- FIG.3A-3C show that the amino acid modifications to the C-termini of the antigen- binding molecule, REGN9930, do not affect binding affinity to target cell surface antigens expressed on Raji and Jurkat cells.
- FIG.3A shows binding of antigen-binding molecules to Raji MAGEA4 (230-239) cells.
- FIG.3B shows binding of antigen-binding molecules to Raji MAGEA4 (286-294).
- FIG.3C shows binding the antigen-binding molecules to Jurkat cells.
- FIG.4 shows that the C-terminal amino acid modifications do not affect cytotoxic potency of antigen-binding molecule, REGN9930.
- FIGs.5A-5E show SEC-MS analysis of limited-LysC-digested antigen-binding molecule, REGN9930-VNSS (C-terminal sequence VTVNSS; SEQ ID NO:50).
- FIG.5A shows the native SEC-UV/MS analysis whereas boxes labeled (i)-(iv) indicate the fragments shown in mass analyses in FIGs 5B-5E.
- FIGs.6A-6D show SEC-MS analysis of limited-LysC-digested antigen-binding molecule, REGN9930-VNST (C-terminal sequence VTVNST; SEQ ID NO:51).
- FIG.6A shows the native SEC-UV/MS analysis, whereas boxes labeled (i)-(iii) indicate the fragments shown in mass analyses in FIGs 6B-6D.
- FIG.7 demonstrates the reduced preexisting ADA reactivity of antigen-binding molecules with C-terminal amino acid modifications relative to REGN9930. 6. DETAILED DESCRIPTION 6.1.
- an “or” conjunction is intended to be used in its correct sense as a Boolean logical operator, encompassing both the selection of features in the alternative (A or B, where the selection of A is mutually exclusive from B) and the selection of features in conjunction (A or B, where both A and B are selected).
- the term “and/or” is used for the same purpose, which shall not be construed to imply that “or” is used with reference to mutually exclusive alternatives.
- Anti-drug Antibodies or ADAs The terms “Anti-drug antibodies” or “ADAs” refer to antibodies that bind specifically to any region of a drug.
- the “drug” is or comprises an antibody (as defined below).
- an anti- drug antibody may be an antibody or fragment thereof that specifically binds to a region of a drug antibody, e.g., the variable domain, the constant domains, or the glycostructure of the antibody.
- Such anti-drug antibodies may occur during drug therapy as an immunogenic reaction of a patient.
- An ADA may be one of any human immunoglobulin isotype (e.g., IgM, IgE, IgA, IgG, IgD) or IgG subclass (IgG1, 2, 3, and 4).
- ADAs include ADAs from any animal source, including, for example, human or non-human animal (e.g. veterinary) sources.
- Antibody refers to a polypeptide (or set of polypeptides) of the immunoglobulin family that is capable of binding an antigen non-covalently, reversibly and specifically.
- a naturally occurring “antibody” of the IgG type is a tetramer comprising at least two heavy (H) chains and two light (L) chains inter-connected by disulfide bonds.
- Each heavy chain is comprised of a heavy chain variable region (abbreviated herein as VH) and a heavy chain constant region.
- VH heavy chain variable region
- the heavy chain constant region is comprised of three domains, CH1, CH2 and CH3.
- Each light chain is comprised of a light chain variable region (abbreviated herein as VL) and a light chain constant region.
- the light chain constant region is comprised of one domain (abbreviated herein as CL).
- CL light chain constant region
- the VH and VL regions can be further subdivided into regions of hypervariability, termed complementarity determining regions (CDR), interspersed with regions that are more conserved, termed framework regions (FR).
- CDR complementarity determining regions
- FR framework regions
- Each VH and VL is composed of three CDRs and four FRs arranged from amino-terminus to carboxy-terminus in the following order: FR1, CDR1, FR2, CDR2, FR3, CDR3, FR4.
- the variable regions of the heavy and light chains contain a binding domain that interacts with an antigen.
- the constant regions of the antibodies may mediate the binding of the immunoglobulin to host tissues or factors, including various cells of the immune system (e.g., effector cells) and the first component (Clq) of the classical complement system.
- the term “antibody” includes, but is not limited to, monoclonal antibodies, human antibodies, humanized antibodies, camelized antibodies, chimeric antibodies, bispecific or multispecific antibodies and anti-idiotypic (anti-id) antibodies.
- the antibodies can be of any isotype/class (e.g., IgG, IgE, IgM, IgD, IgA and IgY) or subclass (e.g., lgG1, lgG2, lgG3, lgG4, lgA1 and lgA2).
- IgG isotype/class
- IgM immunoglobulin-like compound
- IgD immunoglobulin
- IgA and IgY subclass
- subclass e.g., lgG1, lgG2, lgG3, lgG4, lgA1 and lgA2
- Both the light and heavy chains are divided into regions of structural and functional homology.
- the terms “constant” and “variable” are used functionally.
- the variable domains of both the light (VL) and heavy (VH) chain portions determine antigen recognition and specificity.
- the constant domains of the light chain (CL) and the heavy chain (CH1, CH2 or CH3) confer important biological properties such as secretion, transplacental mobility, Fc receptor binding, complement binding, and the like.
- CL light chain
- CH2 or CH3 heavy chain
- the numbering of the constant region domains increases as they become more distal from the antigen-binding domain or amino-terminus of the antibody.
- the N-terminus is a variable region and at the C- terminus is a constant region; the CH3 and CL domains represent the carboxy-terminus of the heavy and light chain, respectively, of natural antibodies.
- Antigen-binding Domain refers to a portion of a binding molecule (e.g., a multispecific binding molecule, an antibody, or an antibody fragment) that has the ability to bind to a target molecule (e.g., an antigen) non- covalently, reversibly and specifically.
- an antibody fragment that can comprise an ABD include, but are not limited to, a single-chain Fv (scFv), a Fab fragment, a monovalent fragment consisting of the VL, VH, CL and CH1 domains; a F(ab)2 fragment, a bivalent fragment comprising two Fab fragments linked by a disulfide bridge at the hinge region; a Fd fragment consisting of the VH and CH1 domains; a Fv fragment consisting of the VL and VH domains of a single arm of an antibody; a dAb fragment (Ward et al., 1989, Nature 341:544- 546), which consists of a VH domain; and an isolated complementarity determining region (CDR).
- scFv single-chain Fv
- Fab fragment a monovalent fragment consisting of the VL, VH, CL and CH1 domains
- F(ab)2 fragment a bivalent fragment comprising two Fab fragments linked by a disulfide bridge
- antibody fragment encompasses both proteolytic fragments of antibodies (e.g., Fab and F(ab) 2 fragments) and engineered proteins comprising one or more portions of an antibody (e.g., an scFv).
- Antibody fragments can also be incorporated into single domain antibodies, maxibodies, minibodies, intrabodies, diabodies, triabodies, tetrabodies, v- NAR and bis-scFv (see, e.g., Hollinger and Hudson, 2005, Nature Biotechnology 23: 1126- 1136).
- association in the context of an antigen-binding molecule refers to a functional relationship between two or more polypeptide chains or portions of a polypeptide chain.
- association means that two or more polypeptides are associated with one another, e.g., non-covalently through molecular interactions or covalently through one or more disulfide bridges or chemical cross-linkages, so as to produce a functional antigen- binding molecule.
- association examples include (but are not limited to) associations between homodimeric or heterodimeric Fc domains in an Fc region, associations between VH and VL regions in a Fab or scFv, associations between CH1 and CL in a Fab, and associations between CH3 and CH3 in a domain substituted Fab.
- Bivalent The term “bivalent” as used herein in reference to an antigen-binding molecule means an antigen-binding molecule that has two antigen-binding sites. In some embodiments, the two antigen-binding sites bind to the same epitope of the same target.
- cancer antigen refers to a molecule (typically a protein, carbohydrate, lipid or some combination thereof) that is expressed on the surface of a cancer cell, either entirely or as a fragment (e.g., MHC/peptide), and which is useful for the preferential targeting of a pharmacological agent to the cancer cell.
- a cancer antigen is a marker expressed by both normal cells and cancer cells, e.g., a lineage marker, e.g., CD19 on B cells.
- a cancer antigen is a cell surface molecule that is overexpressed in a cancer cell in comparison to a normal cell, for instance, 1-fold over expression, 2-fold overexpression, 3-fold overexpression or more in comparison to a normal cell.
- a cancer antigen is a cell surface molecule that is inappropriately synthesized in the cancer cell, for instance, a molecule that contains deletions, additions or mutations in comparison to the molecule expressed on a normal cell.
- a cancer antigen will be expressed exclusively on the cell surface of a cancer cell, entirely or as a fragment (e.g., MHC/peptide), and not synthesized or expressed on the surface of a normal cell. Accordingly, the term “cancer antigen” encompasses antigens that are specific to cancer cells, sometimes known in the art as tumor-specific antigens (“TSAs”).
- TSAs tumor-specific antigens
- CDR Complementarity Determining Region or CDR: The terms “complementarity determining region” or “CDR,” as used herein, refer to the sequences of amino acids within antibody variable regions which confer antigen specificity and binding affinity.
- CDR-H1, CDR-H2, CDR-H3 there are three CDRs in each heavy chain variable region and three CDRs in each light chain variable region (CDR1-L1, CDR-L2, CDR-L3).
- Exemplary conventions that can be used to identify the boundaries of CDRs include, e.g., the Kabat definition, the Chothia definition, the ABD definition and the IMGT definition. See, e.g., Kabat, 1991, “Sequences of Proteins of Immunological Interest,” National Institutes of Health, Bethesda, Md. (Kabat numbering scheme); Al-Lazikani et al., 1997, J. Mol.
- CDR amino acid residues in the heavy chain variable domain (VH) are numbered 31-35 (CDR-H1), 50-65 (CDR- H2), and 95- 102 (CDR-H3); and the CDR amino acid residues in the light chain variable domain (VL) are numbered 24-34 (CDR-L1), 50-56 (CDR-L2), and 89-97 (CDR-L3).
- the CDRs consist of amino acid residues 26-35 (CDR-H1), 50-65 (CDR-H2), and 95-102 (CDR-H3) in human VH and amino acid residues 24-34 (CDR-L1), 50-56 (CDR-L2), and 89-97 (CDR-L3) in human VL.
- the CDR amino acid residues in the VH are numbered approximately 26-35 (CDR- H1), 51-57 (CDR-H2) and 93-102 (CDR-H3), and the CDR amino acid residues in the VL are numbered approximately 27-32 (CDR-L1), 50-52 (CDR-L2), and 89-97 (CDR-L3) (numbering according to “Kabat”).
- CDR-L1 the CDR amino acid residues in the VL
- CDR-L3 are numbered approximately 27-32 (CDR-L1), 50-52 (CDR-L2), and 89-97 (CDR-L3) (numbering according to “Kabat”).
- the CDR regions of an antibody can be determined using the program IMGT/DomainGap Align. Public databases are available for identifying CDR sequences within an antibody.
- the Fc region can be homodimeric or heterodimeric.
- the dimerization moiety is an Fc domain or an amino acid sequence of 1 to about 200 amino acids in length containing at least one cysteine residue.
- the dimerization moiety is a cysteine residue or a short cysteine-containing peptide.
- dimerization moieties include peptides or polypeptides comprising or consisting of a leucine zipper, a helix-loop motif, or a coiled-coil motif.
- EC50 refers to the half maximal effective concentration of a molecule, such as an antigen-binding molecule, which induces a response halfway between the baseline and maximum after a specified exposure time.
- the EC50 essentially represents the concentration of a molecule where 50% of its maximal effect is observed. Thus, reduced or weaker binding is observed with an increased EC50, or half maximal effective concentration value.
- Epitope An epitope, or antigenic determinant, is a portion of an antigen recognized by an antibody or other antigen-binding moiety as described herein. An epitope can be linear or conformational.
- the Fabs of the disclosure can be arranged according to the native orientation or include domain substitutions or swaps that facilitate correct VH and VL pairings. For example, it is possible to replace the CH1 and CL domain pair in a Fab with a CH3-domain pair to facilitate correct modified Fab-chain pairing in heterodimeric molecules. It is also possible to reverse CH1 and CL, so that the CH1 is attached to VL and CL is attached to the VH, a configuration generally known as Crossmab (a type of “domain exchanged” arrangement). Alternatively, or in addition to, the use of substituted or swapped constant domains, correct chain pairing can be achieved by the use of universal light chains that can pair with both variable regions of a heterodimeric antigen-binding molecule of the disclosure.
- Fc Domain and Fc Region refers to a portion of the heavy chain that pairs with the corresponding portion of another heavy chain.
- Fc region refers to the region of antibody-based binding molecules formed by association of two heavy chain Fc domains. The two Fc domains within the Fc region may be the same or different from one another. In a native antibody the Fc domains are typically identical, but one or both Fc domains might advantageously be modified to allow for heterodimerization, e.g., via a knob-in-hole interaction and/or for purification, e.g., via star mutations.
- Half Antibody refers to a molecule that comprises at least one Fc domain and can associate with another molecule comprising an Fc through, e.g., a disulfide bridge or molecular interactions.
- a half antibody can be composed of one polypeptide chain or more than one polypeptide chains (e.g., the two polypeptide chains of a Fab).
- a host cell can be a cell line of mammalian origin or mammalian-like characteristics, such as monkey kidney cells (COS, e.g., COS-1 , COS- 7), HEK293, baby hamster kidney (BHK, e.g., BHK21), Chinese hamster ovary (CHO), NSO, PerC6, BSC-1 , human hepatocellular carcinoma cells (e.g., Hep G2), SP2/0, HeLa, Madin- Darby bovine kidney (MDBK), myeloma and lymphoma cells, or derivatives and/or engineered variants thereof.
- COS monkey kidney cells
- BHK baby hamster kidney
- CHO Chinese hamster ovary
- NSO PerC6, BSC-1
- human hepatocellular carcinoma cells e.g., Hep G2
- SP2/0 heLa
- HeLa Madin- Darby bovine
- multispecific antigen-binding molecule (also “multispecific antigen-binding molecule” or “multispecific binding molecule”) as used herein refers to a molecule (e.g., assembly of multiple polypeptide chains) comprising two half antibodies and which specifically bind to at least two different epitopes (and in some instances three, four, or more different epitopes).
- a multispecific antigen-binding molecule of the disclosure may be bivalent, trivalent, tetravelant, or otherwise multivalent, and may be monospecific, bispecific, or otherwise multispecific.
- a multispecific antigen-binding molecule of the disclosure may specifically bind to epitopes on one, two, three, four, or more different antigens.
- Multivalent refers to an antigen-binding molecule comprising two or more ABDs, on one, two or more polypeptide chains.
- N-linked Glycosylation Site refers to an asparagine (Asn; N) residue of a polypeptide which is capable of being glycosylated.
- N-linked glycosylation sites contain an Asn residue of a polypeptide within a three amino acid sequence Asn-Xaa-Ser or Asn-Xaa-Thr, where Xaa is any amino acid, typically other than proline. Such a three amino acid consensus sequence is often referred to herein as “NX[S/T]”.
- NX[S/T] Such a three amino acid consensus sequence is often referred to herein as “NX[S/T]”.
- the term “N-linked glycosylation site” is used to describe such an asparagine residue whether or not an oligosaccharide is attached to the residue.
- a polypeptide described herein as comprising an N-linked glycosylation site at a particular location means that the Asn of the N-linked glycosylation site is at that particular location.
- an antigen- binding molecule or a domain thereof comprising an N-linked glycosylation site “within 10 amino acids of the C-terminus” describes an antigen-binding molecule or domain thereof having the amino acid sequence NX[S/T] such that the Asn residue is within 10 amino acids of the C-terminus of the antigen-binding molecule or domain (e.g., VH domain).
- Operably Linked refers to a functional relationship between two or more regions of a polypeptide chain in which the two or more regions are linked so as to produce a functional polypeptide, or two or more nucleic acid sequences, e.g., to produce an in-frame fusion of two polypeptide components or to link a regulatory sequence to a coding sequence.
- operably linked means that two or more amino acid segments are linked so as to produce a functional polypeptide.
- operably linked means that the two nucleic acids are joined such that the amino acid sequences encoded by the two nucleic acids remain in-frame.
- transcriptional regulation the term refers to the functional relationship of a transcriptional regulatory sequence to a transcribed sequence.
- a promoter or enhancer sequence is operably linked to a coding sequence if it stimulates or modulates the transcription of the coding sequence in an appropriate host cell or other expression system.
- Polypeptide, Peptide and Protein The terms “polypeptide”, “peptide” and “protein” are used interchangeably herein to refer to a polymer of amino acid residues.
- Single Chain Fab or scFab The term “single chain Fab” or “scFab” as used herein refers an ABD comprising a VH domain, a CH1 domain, a VL domain, a CL domain and a linker.
- the foregoing domains and linker are arranged in one of the following orders in a N-terminal to C-terminal orientation: (a) VH-CH1-linker-VL-CL, (b) VL-CL-linker-VH- CH1, (c) VH-CL-linker-VL-CH1 or (d) VL-CH1-linker-VH-CL.
- Linkers are suitably noncleavable linkers of at least 30 amino acids, preferably between 32 and 50 amino acids.
- Single chain Fab fragments are typically stabilized via the natural disulfide bond between the CL domain and the CH1 domain.
- Single Chain Fv or scFv refers to a polypeptide chain comprising the VH and VL domains of antibody, where these domains are present in a single polypeptide chain.
- the Fv polypeptide further comprises a polypeptide linker between the VH and VL domains which enables the scFv to form the desired structure for antigen-binding.
- Subject includes human and non-human animals.
- Non-human animals include all vertebrates, e.g., mammals and non-mammals, such as non-human primates, sheep, dog, cow, chickens, amphibians, and reptiles.
- the subject is human.
- the terms “patient” or “subject” are used herein interchangeably.
- Tetravalent refers to refers to an antigen-binding molecule that has four antigen-binding sites. In certain embodiments, all four of the antigen- binding sites bind to the same epitope. In some embodiments, two of the antigen-binding sites bind to the same epitope and the other two antigen-binding sites bind to a different epitope, whether of the same target molecule or different target molecules. In other embodiments, two of the antigen-binding sites bind to the same epitope, a third antigen-binding site binds to a different epitope, and a fourth antigen-binding site binds to yet a different epitope.
- a tetravalent antigen-binding molecule can be monospecific, bispecific, trispecific, or tetraspecific.
- Trivalent refers to refers to an antigen-binding molecule that has three antigen-binding sites. In certain embodiments, all three of the antigen- binding sites bind to the same epitope. In some embodiments, two of the antigen-binding sites bind to the same epitope and the other antigen-binding site binds to a different epitope, whether of the same target molecule or different target molecules.
- TAA Tumor-associated Antigen
- TAAs include cancer antigens as well as molecules expressed by or present on non- tumor cells which are useful in the treatment of a tumor, including, for example, extracellular matrix (“ECM”) proteins, cell surface molecules of tumor or viral lymphocytes, T-cell antigens (“TCAs”), and immune checkpoint molecules.
- ECM extracellular matrix
- TAAs T-cell antigens
- immune checkpoint molecules include, for example, extracellular matrix (“ECM”) proteins, cell surface molecules of tumor or viral lymphocytes, T-cell antigens (“TCAs”), and immune checkpoint molecules.
- ECM extracellular matrix
- TCAs T-cell antigens
- immune checkpoint molecules include, for example, extracellular matrix (“ECM”) proteins, cell surface molecules of tumor or viral lymphocytes, T-cell antigens (“TCAs”), and immune checkpoint molecules.
- ULC The term “universal light chain” or “ULC” as used herein in the context of an antigen-biding domain refers to a light chain polypeptide capable of pairing
- VH refers to the variable region of an immunoglobulin heavy chain of an antibody, including the heavy chain of an Fv, scFv, dsFv or Fab.
- VL refers to the variable region of an immunoglobulin light chain, including the light chain of an Fv, scFv, dsFv or Fab.
- VHs heavy chain variable domains
- ABSDs antigen-binding domains
- the ABDs can be in any format, including formats such as scFvs (e.g., as described in Section 6.3.1, infra) and Fabs (e.g., as described in Section 6.3.2, infra), and can have various specificities, including TAA ABDs (e.g., as described in Section 6.4.2, infra) and T-cell engager (TCE) ABDs (e.g., as described in Section 6.4.3, infra).
- TAA ABDs e.g., as described in Section 6.4.2, infra
- TCE T-cell engager
- An exemplary scFv comprising an engineered VH of the disclosure is depicted in FIG.1A.
- An exemplary bivalent antigen-binding molecule comprising two scFvs comprising an engineered VH of the disclosure is depicted in FIG.1B.
- the engineered heavy chain variable domains (VHs; also “VH regions” or “VH domains”) of the present disclosure generally comprise (1) an N-linked glycosylation site within 20, 19, 18, 17, 16, 15, 14, 13, 12, 11, 10, 9, 8, 7, 6, 5, 4, or 3 amino acids of its C-terminus, or (2) the amino acid sequence X 1 X 2 X 3 X 4 X 5 NX 6 X 7 X 8 X 9 X 10 X 11 X 12 (SEQ ID NO:1) at the C-terminus, where (a) X 1 , X 2 , X 3 , X 4 , and X 5 are each independently selected from any amino acid; (b) X 6 is selected from any amino acid, optionally wherein the amino acid is not proline; (c) X 7 is S or T, and (d) X 8 , X 9 , X 10 ,
- VHs are often referred to herein as “engineered VHs” or “engineered VH domains” for convenience.
- modification of a VH in an antigen-binding domain for example via amino acid substitution, insertion, or deletion, to incorporate an N- linked glycosylation site near the C-terminus of the VH (for example modification of the VH such that it comprises the sequence X 1 X 2 X 3 X 4 X 5 NX 6 [S/T]X 8 X 9 X 10 X 11 X 12 at its C-terminus) results in reduced binding of the antigen-binding molecule to anti-drug antibodies (see, e.g., Section [0219] (Example 4)).
- VHs comprising such engineered VHs.
- Any antigen-binding molecule comprising a VH may be modified such that the VH has an N-linked glycosylation site near its C-terminus (e.g., the sequence X 1 X 2 X 3 X 4 X 5 NX 6 [S/T]X 8 X 9 X 10 X 11 X 12 at its C-terminus).
- VHs can be further subdivided into regions of hypervariability, termed complementarity determining regions (CDR), interspersed with regions that are more conserved, termed framework regions (FR).
- CDR complementarity determining regions
- FR framework regions
- Each VH is composed of three CDRs and four FRs arranged from amino-terminus to carboxy-terminus in the following order: FR1, CDR1, FR2, CDR2, FR3, CDR3, FR4.
- an engineered VH of the disclosure comprises an N-linked glycosylation site within the FR4 region. In other embodiments, an engineered VH of the disclosure comprises an N-linked glycosylation site within the CDR3 region.
- a VH of the disclosure comprises, at its C-terminus, the amino acid sequence X 1 X 2 X 3 X 4 X 5 NX 6 X 7 X 8 X 9 X 10 X 11 X 12 (SEQ ID NO:1), wherein (a) X 1 , X 2 , X 3 , X 4 , and X 5 are each independently selected from any amino acid; (b) X 6 is selected from any amino acid, optionally wherein the amino acid is not proline; (c) X 7 is S or T, and (d) X 8 , X 9 , X 10 ,X 11 , and X 12 are each independently selected from any amino acid and absent.
- Such a VH may comprise the sequence X 1 X 2 X 3 X 4 X 5 NX 6 [S/T]X 8 X 9 X 10 X 11 X 12 by virtue of such a sequence being inserted at the C-terminus of the VH or due to one or more amino acid modifications made to the original VH sequence resulting in the sequence X 1 X 2 X 3 X 4 X 5 NX 6 [S/T]X 8 X 9 X 10 X 11 X 12 .
- X 1 is A, C, D, E, F, G, H, I, K, L, M, N, P, Q, R, S, T, V, W, or Y.
- X 2 is A, C, D, E, F, G, H, I, K, L, M, N, P, Q, R, S, T, V, W, or Y.
- X 3 is A, C, D, E, F, G, H, I, K, L, M, N, P, Q, R, S, T, V, W, or Y.
- X 4 is A, C, D, E, F, G, H, I, K, L, M, N, P, Q, R, S, T, V, W, or Y.
- X 5 is A, C, D, E, F, G, H, I, K, L, M, N, P, Q, R, S, T, V, W, or Y.
- X 6 is (i) A, C, D, E, F, G, H, I, K, L, M, N, P, Q, R, S, T, V, W, or Y or (ii) A, C, D, E, F, G, H, I, K, L, M, N, Q, R, S, T, V, W, or Y.
- X 7 is S or T.
- X 8 is A, C, D, E, F, G, H, I, K, L, M, N, Q, R, S, T, V, W, Y, or absent.
- X 9 is A, C, D, E, F, G, H, I, K, L, M, N, Q, R, S, T, V, W, Y, or absent.
- X 10 is A, C, D, E, F, G, H, I, K, L, M, N, Q, R, S, T, V, W, Y, or absent.
- X 11 is A, C, D, E, F, G, H, I, K, L, M, N, Q, R, S, T, V, W, Y, or absent.
- X 12 is A, C, D, E, F, G, H, I, K, L, M, N, Q, R, S, T, V, W, Y, or absent.
- one of X 8 , X 9 , X 10 , X 11 , and X 12 is absent.
- two of X 8 , X 9 , X 10 , X 11 , and X 12 are absent.
- X 8 , X 9 , X 10 , X 11 , and X 12 are absent.
- four of X 8 , X 9 , X 10 , X 11 , and X 12 are absent.
- all five of X 8 , X 9 , X 10 , X 11 , and X 12 are absent.
- Table 1 provides a complete list of possible amino acids at the C-terminal region of an engineered VH of the present disclosure, where the VH comprises at its C-terminus the amino acid sequence X 1 X 2 X 3 X 4 X 5 NX 6 X 7 X 8 X 9 X 10 X 11 X 12 (SEQ ID NO:1).
- the amino acid sequence X 1 X 2 X 3 X 4 X 5 NX 6 X 7 X 8 X 9 X 10 X 11 X 12 at the C-terminus of an engineered VH of the present disclosure may comprise any combination of amino acids at each position as outlined in Table 1.
- X 6 is selected from any amino acid except proline.
- the VH comprises one of the following sequences at its C- terminus (where X 1 , X 2 , X 3 , X 4 , and X 5 are each independently selected from any amino acid): X 1 X 2 X 3 X 4 X 5 NSS (SEQ ID NO:2), X 1 X 2 X 3 X 4 X 5 NST (SEQ ID NO:3), X 1 X 2 X 3 X 4 VNSS (SEQ ID NO:4), X 1 X 2 X 3 X 4 VNST (SEQ ID NO:5), X 1 X 2 X 3 TVNSS (SEQ ID NO:6), X 1 X 2 X 3 TVNST (SEQ ID NO:7), X 1 X 2 X 3 TVNST (SEQ ID NO:7), X 1 X 2 X 3 TVNST (SEQ ID NO:7), X 1 X 2 X 3 TVN
- the VH comprises the sequence VTVNSS (SEQ ID NO:50) at its C-terminus. In some embodiments, the VH comprises the sequence VTVNST (SEQ ID NO:51) at its C-terminus. In some embodiments, the VH comprises the sequence VTVNSSGGGG (SEQ ID NO:52) at its C-terminus. In some embodiments, the VH comprises the sequence VTVNSTGGGG (SEQ ID NO:53) at its C-terminus. In some embodiments, the VH comprises the sequence VTVNSSKPGG (SEQ ID NO:54) at its C-terminus. In some embodiments, the VH comprises the sequence VTVNSTKPGG (SEQ ID NO:55) at its C-terminus.
- the VH comprises the sequence VTVNSSPP (SEQ ID NO:56) at its C-terminus. In some embodiments, the VH comprises the sequence VTVNSTPP (SEQ ID NO:57) at its C- terminus.
- a human VH is understood to commonly comprise, at its C-terminus, the sequence VTVSS (SEQ ID NO:62). Accordingly, aspects of the present disclosure comprise modifying a VH comprising the sequence VTVSS (SEQ ID NO:62) so as to comprise an N-linked glycosylation site at the C-terminus by virtue of insertion and/or substitution of one or two amino acids within this sequence.
- an engineered VH of the disclosure is generated by inserting an N residue between the third and fourth positions of this sequence, generating the sequence VTVNSS (SEQ ID NO:50) at the C-terminus.
- an engineered VH of the disclosure is generated by inserting an N residue between the third and fourth positions of this sequence and also substituting a T residue at the fifth position, generating the sequence VTVNST (SEQ ID NO:51).
- a sequence at the C-terminus of the VH may be further followed by any number of additional sequences, for example in situations where the VH is not at the C-terminus of the antigen-binding molecule.
- a VH of the present disclosure may optionally further comprise one or more additional amino acid modifications or sequences designed to reduce binding of anti-drug antibodies.
- additional modifications include, for example, the sequence PP at the C-terminus, the sequence KPGG (SEQ ID NO: 66) at the C-terminus, a V11K substitution (EU numbering), and a polyglycine sequence (e.g., G 4 S (SEQ ID NO: 67)) at the C-terminus.
- a VH of the present disclosure may comprise any one or more of these additional modifications, including any combination thereof.
- the engineered VH domains of the disclosure can be incorporated into antigen-binding domains of antigen-binding molecules.
- Antigen-binding molecules comprising an engineered VH of the disclosure are described in, inter alia, Section 6.4 and include, for example antibodies of various formats such as multivalent and/or multispecific antigen-binding molecules (e.g., as described in Sections 6.4.1.1 and 6.4.1.2, infra), antibody fragments, scFvs, and chimeric antigen receptors. 6.3.
- an engineered VH domain can be incorporated into an ABD, for example an ABD that is incorporated into an antigen-binding molecule as described herein.
- the ABD is an immunoglobulin molecule or fragment thereof, particularly an IgG class immunoglobulin molecule, more particularly an IgG1 or IgG4 immunoglobulin molecule.
- Antibody fragments include, but are not limited to, VH fragments, VL fragments, Fab fragments, F(ab')2 fragments, scFv fragments, Fv fragments, minibodies, diabodies, triabodies, and tetrabodies. 6.3.1.
- an engineered VH of the disclosure is incorporated into an scFv.
- a VH of an scFv described herein can be, in some embodiments, an engineered VH.
- An example scFv comprising an engineered VH is depicted in FIG.1A.
- Single chain Fv or “scFv” antibody fragments comprise the VH and VL domains of an antibody in a single polypeptide chain, are capable of being expressed as a single chain polypeptide, and retain the specificity of the intact antibodies from which they are derived.
- the scFv polypeptide further comprises a polypeptide linker between the VH and VL domain that enables the scFv to form the desired structure for target binding.
- linkers suitable for connecting the VH and VL chains of an scFV are the linkers identified in Section 6.4.4.
- an scFv may have the VL and VH variable regions in either order, e.g., with respect to the N-terminal and C-terminal ends of the polypeptide, the scFv may comprise VL-linker-VH or may comprise VH-linker-VL.
- an scFv of the disclosure comprises an engineered VH comprising an N-linked glycosylation site (e.g., as a component of the amino acid sequence of SEQ ID NO:1) within 3 or more (e.g., 3, 4, 5, 6, 7, 8, 9, 10, 15, or 20) amino acids of the C-terminus of the VH
- N-linked glycosylation site may be within 3 or more amino acids of the C-terminus of the scFv itself (e.g., where the scFv comprises VL-linker-VH) or simply within 3 or more amino acids of the C-terminus of the VH but a further distance from the C-terminus of the scFv (e.g., where the scFv comprises VH- linker-VL).
- the scFv can comprise VH and VL sequences from any suitable species, such as murine, human or humanized VH and VL sequences.
- the VH and VL-encoding DNA fragments are operably linked to another fragment encoding a linker, e.g., encoding any of the linkers described in Section 6.4.4, for example a repeat of a sequence containing the amino acids glycine and serine, such that the VH and VL sequences can be expressed as a contiguous single-chain protein with the VL and VH regions joined by the flexible linker (see, e.g., Bird et al., 1988, Science 242:423-426; Huston et al., 1988, Proc.
- an engineered VH of the disclosure is incorporated into a Fab.
- Fab domains were traditionally produced by proteolytic cleavage of immunoglobulin molecules using enzymes such as papain.
- the Fab domains can comprise constant domain and variable region sequences from any suitable species, and thus can be murine, chimeric, human or humanized.
- Fab domains typically comprise a CH1 domain attached to a VH domain which pairs with a CL domain attached to a VL domain.
- the VH domain is paired with the VL domain to constitute the Fv region
- the CH1 domain is paired with the CL domain to further stabilize the binding site.
- a disulfide bond between the two constant domains can further stabilize the Fab domain.
- correct association between the two polypeptides of a Fab is promoted by exchanging the VL and VH domains of the Fab for each other or exchanging the CH1 and CL domains for each other, e.g., as described in WO 2009/080251.
- Correct Fab pairing can also be promoted by introducing one or more amino acid modifications in the CH1 domain and one or more amino acid modifications in the CL domain of the Fab and/or one or more amino acid modifications in the VH domain and one or more amino acid modifications in the VL domain.
- the amino acids that are modified are typically part of the VH:VL and CH1:CL interface such that the Fab components preferentially pair with each other rather than with components of other Fabs.
- the one or more amino acid modifications are limited to the conserved framework residues of the variable (VH, VL) and constant (CH1, CL) domains as indicated by the Kabat numbering of residues.
- VH, VL variable
- CH1, CL constant domains
- the modifications introduced in the VH and CH1 and/or VL and CL domains are complementary to each other.
- Complementarity at the heavy and light chain interface can be achieved on the basis of steric and hydrophobic contacts, electrostatic/charge interactions or a combination of the variety of interactions.
- the complementarity between protein surfaces is broadly described in the literature in terms of lock and key fit, knob into hole, protrusion and cavity, donor and acceptor etc., all implying the nature of structural and chemical match between the two interacting surfaces.
- the one or more introduced modifications introduce a new hydrogen bond across the interface of the Fab components.
- the one or more introduced modifications introduce a new salt bridge across the interface of the Fab components. Exemplary substitutions are described in WO 2014/150973 and WO 2014/082179, the contents of which are hereby incorporated by reference.
- the Fab domain comprises a 192E substitution in the CH1 domain and 114A and 137K substitutions in the CL domain, which introduces a salt-bridge between the CH1 and CL domains (see, e.g., Golay et al., 2016, J Immunol 196:3199-211).
- the Fab domain comprises a 143Q and 188V substitutions in the CH1 domain and 113T and 176V substitutions in the CL domain, which serves to swap hydrophobic and polar regions of contact between the CH1 and CL domain (see, e.g., Golay et al., 2016, J Immunol 196:3199-211).
- the Fab domain can comprise modifications in some or all of the VH, CH1, VL, CL domains to introduce orthogonal Fab interfaces which promote correct assembly of Fab domains (Lewis et al., 2014 Nature Biotechnology 32:191-198).
- 39K, 62E modifications are introduced in the VH domain
- H172A, F174G modifications are introduced in the CH1 domain
- 1 R, 38D, (36F) modifications are introduced in the VL domain
- L135Y, S176W modifications are introduced in the CL domain.
- a 39Y modification is introduced in the VH domain and a 38R modification is introduced in the VL domain.
- Fab domains can also be modified to replace the native CH1:CL disulfide bond with an engineered disulfide bond, thereby increasing the efficiency of Fab component pairing.
- an engineered disulfide bond can be introduced by introducing a 126C in the CH1 domain and a 121 C in the CL domain (see, e.g., Mazor et al., 2015, MAbs 7:377-89).
- Fab domains can also be modified by replacing the CH1 domain and CL domain with alternative domains that promote correct assembly.
- VL of common light chain (also referred to as a universal light chain) can be used for each unique ABD in the antigen-binding molecules of the disclosure.
- employing a common light chain as described herein reduces the number of inappropriate species in the antigen-binding molecules as compared to employing original cognate VLs.
- the VL domains of ABDs are identified from monospecific antibodies comprising a common light chain.
- the VH regions of the ABDs in the antigen-binding molecules comprise human heavy chain variable gene segments that are rearranged in vivo within mouse B cells that have been previously engineered to express a limited human light chain repertoire, or a single human light chain, cognate with human heavy chains and, in response to exposure with an antigen of interest, generate an antibody repertoire containing a plurality of human VHs that are cognate with one or one of two possible human VLs, wherein the antibody repertoire specific for the antigen of interest.
- Common light chains are those derived from a rearranged human V ⁇ 1-39J ⁇ 5 sequence or a rearranged human V ⁇ 3-20J ⁇ 1 sequence, and include somatically mutated (e.g., affinity matured) versions.
- the present disclosure provides antigen-binding molecules comprising an engineered VH of the disclosure.
- the antigen-binding molecule is an antibody.
- the antibody can be any type of engineered antibody, including chimeric, humanized, veneered, or human antibodies.
- the antibody can be a monoclonal antibody or a genetically engineered polyclonal antibody.
- Antibodies contemplated herein include both traditional antibodies as well as antibody- like molecules known in the art, including but not limited to nanobodies, diabodies, minibodies, antibody fragments, and other “alternative format” antibodies (e.g., as described in Spiess et al., 2015, Mol Immunol, 67(2 Pt A):95-106, incorporated herein by reference).
- An antibody may include an engineered VH of the disclosure as a component of one or more ABDs, including, for example, as a component of a Fab or as a component of an scFv.
- an antibody comprising an engineered VH, the engineered VH comprising (1) an N-linked glycosylation site within 20, 19, 18, 17, 16, 15, 14, 13, 12, 11, 10, 9, 8, 7, 6, 5, 4, or 3 amino acids of its C-terminus, or (2) the amino acid sequence X 1 X 2 X 3 X 4 X 5 NX 6 X 7 X 8 X 9 X 10 X 11 X 12 (SEQ ID NO:1) at its C-terminus, where (a) X 1 , X 2 , X 3 , X 4 , and X 5 are each independently selected from any amino acid; (b) X 6 is selected from any amino acid, optionally wherein the amino acid is not proline; (c) X 7 is S or T, and (d) X 8 , X 9 , X 10 ,X 11 , and X 12 are each independently selected from any amino acid and absent.
- an antibody of the present disclosure comprises a polypeptide comprising: (a) a VH comprising (1) an N-linked glycosylation site within 20, 19, 18, 17, 16, 15, 14, 13, 12, 11, 10, 9, 8, 7, 6, 5, 4, or 3 amino acids of its C-terminus, or (2) the amino acid sequence X 1 X 2 X 3 X 4 X 5 NX 6 X 7 X 8 X 9 X 10 X 11 X 12 (SEQ ID NO:1) at its C-terminus; and (b) an Fc domain.
- the antibody may further comprise an additional polypeptide comprising an additional VH and an additional Fc domain.
- the additional VH comprises (1) an N- linked glycosylation site within 20, 19, 18, 17, 16, 15, 14, 13, 12, 11, 10, 9, 8, 7, 6, 5, 4, or 3 amino acids of its C-terminus, or (2) the amino acid sequence X 1 X 2 X 3 X 4 X 5 NX 6 X 7 X 8 X 9 X 10 X 11 X 12 (SEQ ID NO:1) at its C-terminus.
- An antibody of the present disclosure may comprise any number of VHs (e.g., 1, 2, 3, 4, or more VHs), where at least one VH is an engineered VH described herein and where any number of the additional VHs may or may not be an engineered VH as described herein. 6.4.1.1.
- Antigen-binding molecules of the present disclosure include multivalent molecules having one or more engineered VH domains.
- Various multivalent antigen-binding molecule formats are recognized in the art and contemplated herein, including bivalent, trivalent, and tetravalent formats. Certain example multivalent antigen-binding molecules are described in more detail below.
- the antigen-binding molecule comprising an engineered VH is a multivalent antigen-binding molecule comprising two or more scFvs connected via a linker, where at least one of the scFvs comprises an engineered VH (as described in, e.g., Section 6.2 or as defined in numbered embodiments 1 to 34).
- an antigen-binding molecule comprising: (a) a first scFv, (b) a linker, and (c) a second scFv, where the first and/or the second scFv comprise an engineered VH.
- the first scFv comprises the engineered VH.
- the second scFv comprises the engineered VH.
- both the first and second scFvs comprise an engineered VH.
- the antigen-binding molecule further comprises a third scFv, which optionally comprises an engineered VH. An example of such a multivalent antigen-binding molecule comprising two scFvs is depicted in FIG.1B.
- the antigen-binding molecule comprising an engineered VH is a multivalent antigen-binding molecule comprising three or more antigen-binding domains, where at least one of the antigen-binding domains comprises an engineered VH.
- an antigen-binding molecule comprising: (a) a first polypeptide chain comprising, from N-terminus to C-terminus (i) a first antigen-binding domain (e.g., a Fab), (ii) a first dimerization moiety, and (iii) a second antigen-binding domain (e.g., an scFv), the second antigen-binding domain comprising an engineered VH; and (b) a second polypeptide chain comprising, from N-terminus to C-terminus (i) a third antigen-binding domain (e.g., a Fab), and (ii) a second dimerization moiety.
- a first antigen-binding domain e.g., a Fab
- a first dimerization moiety e.g., an scFv
- a second antigen-binding domain e.g., an scFv
- the second polypeptide further comprises a fourth antigen-binding domain (e.g., an scFv) C-terminal to the second dimerization moiety, where the fourth antigen-binding domain comprises an engineered VH.
- a fourth antigen-binding domain e.g., an scFv
- Multispecific Antigen-Binding Molecules [0096]
- Antigen-binding molecules of the present disclosure include multispecific molecules having one or more engineered VH domains.
- Various multispecific antigen-binding molecules are recognized in the art and contemplated herein, including bispecific, trispecific, and tetraspecific antigen-binding molecules.
- an antigen-binding molecule comprising an engineered VH of the disclosure is a bispecific T-cell engager.
- Bispecific T-cell engager describes a molecule comprising two scFvs, connected via a linker, where the first scFv comprises a first ABD that binds to a T cell antigen (e.g., a TCE ABD as described in Section 6.4.3) and the second scFv comprises a second ABD that binds to a tumor-associated antigen (e.g., a TAA ABD as described in Section 6.4.2).
- the second TCE ABD or TAA ABD is an scFv.
- the second polypeptide further comprises a TCE ABD or TAA ABD C-terminal to the second dimerization moiety, where the fourth TCE ABD or TAA ABD comprises an engineered VH.
- the fourth TCE ABD or TAA ABD is an scFv.
- the first ABD is a TCE ABD (e.g., as described in Section 6.4.3).
- the first ABD is a TAA ABD (e.g., as described in Section 6.4.2).
- the second ABD is a TCE ABD (e.g., as described in Section 6.4.3). In some embodiments, the second ABD is a TAA ABD (e.g., as described in Section 6.4.2). In some embodiments, the third ABD is a TCE ABD (e.g., as described in Section 6.4.3). In some embodiments, the third ABD is a TAA ABD (e.g., as described in Section 6.4.2). In some embodiments, the fourth ABD is a TCE ABD (e.g., as described in Section 6.4.3). In some embodiments, the fourth ABD is a TAA ABD (e.g., as described in Section 6.4.2). 6.4.2.
- the antigen-binding molecules of the disclosure comprise at least one ABD that binds specifically to a tumor-associated antigen (TAA), referred to herein as a “TAA ABD”.
- TAAs include cancer antigens, extracellular matrix (“ECM”) proteins, tumor reactive lymphocyte antigens, cell surface molecules of tumor or viral lymphocytes, T-cell antigens (“TCAs”), and immune checkpoint molecules.
- ECM extracellular matrix
- TAAs tumor reactive lymphocyte antigens
- TCAs T-cell antigens
- immune checkpoint molecules immune checkpoint molecules.
- the TAA is a human antigen.
- the antigen may or may not be present on normal cells.
- Certain aspects are directed to antigen-binding molecules comprising at least one ABD that binds specifically to a TAA.
- the TAA is preferentially expressed or upregulated on tumor cells as compared to normal cells.
- the TAA is a lineage marker. [0101] It is anticipated that any type of tumor and any type of TAA may be targeted by the antigen-binding molecules of the disclosure.
- Exemplary types of cancers that may be targeted include acute lymphoblastic leukemia, acute myelogenous leukemia, biliary cancer, B-cell leukemia, B-cell lymphoma, biliary cancer, bone cancer, brain cancer, breast cancer, triple- negative breast cancer, cervical cancer, Burkitt lymphoma, chronic lymphocytic leukemia, chronic myelogenous leukemia, colorectal cancer, endometrial cancer, esophageal cancer, gall bladder cancer, gastric cancer, gastrointestinal tract cancer, glioma, hairy cell leukemia, head and neck cancer, Hodgkin’s lymphoma, liver cancer, lung cancer, medullary thyroid cancer, melanoma, multiple myeloma, ovarian cancer, non-Hodgkin’s lymphoma, pancreatic cancer, prostate cancer, pulmonary tract cancer, renal cancer, sarcoma, skin cancer, testicular cancer, urothelial cancer, and other urinary bladder cancers
- Non-limiting examples of ECM antigens include syndecan, 27enzalkoni, integrins, osteopontin, link, cadherins, laminin, laminin type EGF, lectin, fibronectin, notch, nectin (e.g., nectin-4), tenascin, collagen (e.g., collagen type X) and matrixin.
- target molecules are cell surface molecules of tumor or viral lymphocytes, for example T-cell co-stimulatory proteins such as CD27, CD28, 4-1BB (CD137), OX40, CD30, CD40, ICOS, lymphocyte function-associated antigen-1 (LFA-1), CD2, CD7, LIGHT, NKG2C, and B7-H3.
- the target molecules are immune checkpoint molecules, for example CTLA-4, PD1, PDL1, PDL2, B7-H3, B7-H4, BTLA, HVEM, TIM3, GAL9, LAG3, VISTA, KIR, 2B4, CD160, CGEN-15049, CHK1, CHK2.
- the target molecule is PD1.
- WO2015/112800A1 SEQ ID Nos: 16/17 of US Patent No.11,034,765 B2; SEQ ID Nos.164/178, 165/179, 166/180, 167/181, 168/182, 169/183, 170/184, 171/185, 172/186, 173/187, 174/188, 175/189, 176/190 and 177/190 of US Patent No.10,294,299 B2.
- Examples of non-blocking or poorly-blocking anti-LAG3 antibodies includes antibodies having VH/VL amino acid sequences of SEQ ID Nos 23/24, 3 ⁇ 4 and 11/12 of US Pub. US2022/0056126A1.
- the target molecules are TAAs.
- t e TAA ABD competes wt an antbody set ort n Tabe A or binding to the TAA.
- the TAA ABD comprises CDRs having CDR sequences of an anti-TAA antibody set forth in Table A.
- the TAA ABD comprises all 6 CDR sequences of an anti-TAA antibody set forth in Table A.
- the TAA ABD comprises at least the heavy chain CDR sequences (CDR-H1, CDR-H2, CDR-H3) of an anti-TAA antibody set forth in Table A and the light chain CDR sequences of a universal light chain.
- a TAA ABD comprises a VH comprising the amino acid sequence of the VH of an anti-TAA antibody set forth in Table A.
- the TAA ABD further comprises a VL comprising the amino acid sequence of the VL of the anti-TAA antibody set forth in Table A.
- the TAA ABD further comprises a universal light chain VL sequence.
- TAAs that can be targeted by the antigen-binding molecules are disclosed in, e.g., Hafeez et al., 2020, Molecules 25:4764, doi:10.3390/molecules25204764, particularly in Table 1. Table 1 of Hafeez et al. is incorporated by reference in its entirety here.
- TAAs include Fibroblast Activation Protein (FAP), the A1 domain of Tenascin-C (TNC A1), the A2 domain of Tenascin-C (TNC A2), the Extra Domain B of Fibronectin (EDB), the Melanoma-associated Chondroitin Sulfate Proteoglycan (MCSP), MART-1/Melan-A, gp100, Dipeptidyl peptidase IV (DPPIV), adenosine deaminase-binding protein (ADAbp), cyclophilin b, colorectal associated antigen (CRC)-C017-1A/GA733, Carcinoembryonic Antigen (CEA) and its immunogenic epitopes CAP-1 and CAP-2, etv6, aml1, Prostate Specific Antigen (PSA) and its immunogenic epitopes PSA-1, PSA-2, and PSA-3, prostate-specific membrane antigen (PSMA), T-
- FAP Fibroblast
- the antigen-binding molecules of the disclosure comprise at least a T-cell engaging ABD that binds specifically to a T-cell receptor complex component, referred to herein as “TCE ABD”. In other embodiments, the antigen-binding molecules of the disclosure do not comprise a TCE ABD.
- Exemplary targets for the TCE ABD are CD3 and the T-cell receptor (e.g., TCR ⁇ or TCR ⁇ ).
- the TCE ABD target is a human T-cell receptor complex component.
- the epitope of the TCE ABD can be an individual polypeptide (e.g., CD3 epsilon) or a multimeric component of the T-cell receptor complex (e.g., the TCR ⁇ dimer or the TCR ⁇ dimer).
- CD3 and TCR antibodies or antibody sequences are set forth in Table T below, upon which the TCE ABD can be based.
- the TCE ABD competes with a T-cell engaging (TCE) antibody set forth in Table T for binding to the TCE antibody’s target (e.g., CD3 or a T-cell receptor).
- TCE T-cell engaging
- the TCE ABD comprises CDRs having CDR sequences of a TCE antibody set forth in Table T.
- the TCE ABD comprises all 6 CDR sequences of a TCE antibody set forth in Table T. In other embodiments, the TCE ABD comprises at least the heavy chain CDR sequences (CDR-H1, CDR-H2, CDR-H3) of a TCE antibody set forth in Table T and the light chain CDR sequences of a universal light chain. In further aspects, a TCE ABD comprises a VH comprising the amino acid sequence of the VH of a TCE antibody set forth in Table T. In some embodiments, the TCE ABD further comprises a VL comprising the amino acid sequence of the VL of the TCE antibody set forth in Table T. In other embodiments, the TCE ABD further comprises a universal light chain VL sequence. 6.4.4.
- linkers can be used to connect (a) a target binding domain and a constant domain; (b) a first target binding domain and a second target binding domain (e.g. a first scFv and a second scFv); or (c) different domains within a target binding domain (e.g., the VH and VL domains in an scFv).
- a peptide linker can range from 2 amino acids to 60 or more amino acids, and in certain aspects a peptide linker ranges from 3 amino acids to 50 amino acids, from 4 to 30 amino acids, from 5 to 25 amino acids, from 10 to 25 amino acids, 10 amino acids to 60 amino acids, from 12 amino acids to 20 amino acids, from 20 amino acids to 50 amino acids, or from 25 amino acids to 35 amino acids in length.
- a peptide linker is at least 5 amino acids, at least 6 amino acids or at least 7 amino acids in length and optionally is up to 30 amino acids, up to 40 amino acids, up to 50 amino acids or up to 60 amino acids in length.
- the linker ranges from 5 amino acids to 50 amino acids in length, e.g., ranges from 5 to 50, from 5 to 45, from 5 to 40, from 5 to 35, from 5 to 30, from 5 to 25, or from 5 to 20 amino acids in length.
- the linker ranges from 6 amino acids to 50 amino acids in length, e.g., ranges from 6 to 50, from 6 to 45, from 6 to 40, from 6 to 35, from 6 to 30, from 6 to 25, or from 6 to 20 amino acids in length.
- the linker ranges from 7 amino acids to 50 amino acids in length, e.g., ranges from 7 to 50, from 7 to 45, from 7 to 40, from 7 to 35, from 7 to 30, from 7 to 25, or from 7 to 20 amino acids in length.
- Charged (e.g., charged hydrophilic linkers) and/or flexible linkers are particularly preferred.
- Examples of flexible linkers that can be used in the recombinant polypeptides of the disclosure include those disclosed by Chen et al., 2013, Adv Drug Deliv Rev.65(10): 1357-1369 and Klein et al., 2014, Protein Engineering, Design & Selection 27(10): 325-330.
- Particularly useful flexible linkers are or comprise repeats of glycines and serines, e.g., a monomer or multimer of GnS (SEQ ID NO: 68) or SGn (SEQ ID NO: 69), where n is an integer from 1 to 10, e.g., 12, 3, 4, 5, 6, 7, 8, 9 or 10.
- the linker is or comprises a monomer or multimer of repeat of G 4 S (SEQ ID NO: 67) e.g., (GGGGS) n, where n is an integer from 1 to 10 (SEQ ID NO: 70), e.g., 12, 3, 4, 5, 6, 7, 8, 9 or 10.
- the linker is GGGGS (SEQ ID NO: 67), GGGGSGGGGS (SEQ ID NO: 71), GGGGSGGGGSGGGGS (SEQ ID NO: 72), GGGGSGGGGSGGGGSGGGGS (SEQ ID NO: 73), or GGGGSGGGGSGGGGSGGGGSGGGGS (SEQ ID NO: 74).
- Polyglycine linkers can suitably be used in the recombinant polypeptides of the disclosure.
- a peptide linker comprises two consecutive glycines (2Gly), three consecutive glycines (3Gly), four consecutive glycines (4Gly), five consecutive glycines (5Gly), six consecutive glycines (6Gly), seven consecutive glycines (7Gly), eight consecutive glycines (8Gly) or nine consecutive glycines (9Gly). 6.4.5.
- Fc Regions [0119]
- the antigen-binding molecules of the disclosure comprise a pair of Fc domains that associate to form an Fc region. In native antibodies, Fc regions comprise hinge regions at their N-termini to form a constant domain.
- an Fc domain encompasses an Fc domain with a hinge domain at its N-terminus unless specified otherwise.
- the Fc domains can be derived from any suitable species operably linked to an ABD or component thereof.
- the Fc domain is derived from a human Fc domain.
- an antigen-binding domain of an antigen-binding molecule of the disclosure is fused to an IgG Fc molecule.
- An antigen-binding domain may be fused to the N- terminus or the C-terminus of the IgG Fc domain or both.
- the Fc domains can be derived from any suitable class of antibody, including IgA (including subclasses lgA1 and lgA2), IgD, IgE, IgG (including subclasses lgG1, lgG2, lgG3 and lgG4), and IgM.
- the Fc domain is derived from lgG1, lgG2, lgG3 or lgG4.
- the Fc domain is derived from lgG1.
- the Fc domain is derived from lgG4.
- the two Fc domains within the Fc region can be the same or different from one another.
- the Fc domains are typically identical, but for the purpose of producing multispecific binding molecules, e.g., antigen-binding molecules described herein, the Fc domains might advantageously be different to allow for heterodimerization, as described in Section 6.4.5.2 below. In other embodiments, the two Fc domains of antigen-binding molecules disclosed herein are the same.
- the heavy chain Fc domain of IgA, IgD and IgG is composed of two heavy chain constant domains (CH2 and CH3) and that of IgE and IgM is composed of three heavy chain constant domains (CH2, CH3 and CH4). These dimerize to create an Fc region.
- the Fc region, and / or the Fc domains within it can comprise heavy chain constant domains from one or more different classes of antibody, for example one, two or three different classes.
- the Fc region comprises CH2 and CH3 domains derived from lgG1.
- the Fc region comprises CH2 and CH3 domains derived from lgG2.
- the Fc region comprises CH2 and CH3 domains derived from lgG3.
- the Fc region comprises CH2 and CH3 domains derived from lgG4.
- the Fc region comprises a CH4 domain from IgM.
- the IgM CH4 domain is typically located at the C-terminus of the CH3 domain.
- the Fc region comprises CH2 and CH3 domains derived from IgG and a CH4 domain derived from IgM.
- the heavy chain constant domains for use in producing an Fc region for antigen-binding molecules of the present disclosure may include variants of the naturally occurring constant domains described above. Such variants may comprise one or more amino acid variations compared to wild type constant domains.
- the Fc region of the present disclosure comprises at least one constant domain that varies in sequence from the wild type constant domain. It will be appreciated that the variant constant domains may be longer or shorter than the wild-type constant domain.
- the variant constant domains are at least 60% identical or similar to a wild-type constant domain. In another example the variant constant domains are at least 70% identical or similar. In another example the variant constant domains are at least 80% identical or similar. In another example the variant constant domains are at least 90% identical or similar. In another example the variant constant domains are at least 95% identical or similar. [0132] IgM and IgA occur naturally in humans as covalent multimers of the common H2L2 antibody unit. IgM occurs as a pentamer when it has incorporated a J-chain, or as a hexamer when it lacks a J-chain. IgA occurs as monomer and dimer forms.
- the heavy chains of IgM and IgA possess an 18 amino acid extension to the C-terminal constant domain, known as a tailpiece.
- the tailpiece includes a cysteine residue that forms a disulfide bond between heavy chains in the polymer, and is believed to have an important role in polymerization.
- the tailpiece also contains a glycosylation site.
- the antigen-binding molecules of the present disclosure do not comprise a tailpiece.
- the Fc domains that are incorporated into the antigen-binding molecules of the present disclosure may comprise one or more modifications that alter the functional properties of the proteins, for example, binding to Fc-receptors such as FcRn or leukocyte receptors, binding to complement, modified disulfide bond architecture, or altered glycosylation patterns. Exemplary Fc modifications that alter effector function are described in Section 6.4.5.1.
- the Fc domains can also be altered to include modifications that improve manufacturability of asymmetric antigen-binding molecules, for example by allowing heterodimerization, which is the preferential pairing of non-identical Fc domains over identical Fc domains.
- Heterodimerization permits the production of antigen-binding molecules in which different polypeptide components are connected to one another by an Fc region containing Fc domains that differ in sequence. Examples of heterodimerization strategies are exemplified in Section 6.4.5.2. [0135] It will be appreciated that any of the modifications mentioned above can be combined in any suitable manner to achieve the desired functional properties and/or combined with other modifications to alter the properties of the antigen-binding molecules.
- an Fc domain comprises an amino acid sequence having at least about 90%, at least about 91%, at least about 92%, about at least 93%, at least about 94%, at eat least about 95%, at least about 96%, at least about 97%, at least about 98%, at least about 99%, or 100% sequence identity to SEQ ID NO: 58.
- an Fc domain may also comprise one or more amino acid substitutions described herein, for example one or more substitutions that reduce effector function (e.g., as described in Section 6.4.5.1) and/or one or more substitutions that promote Fc heterodimerization (e.g., as described in Section 6.4.5.2).
- an Fc domain comprises an amino acid sequence having at least about 90%, at least about 91%, at least about 92%, about at least 93%, at least about 94%, at least about 95%, at least about 96%, at least about 97%, at least about 98%, at least about 99%, or 100% sequence identity to SEQ ID NO: 59.
- an Fc domain comprises an amino acid sequence having at least about 90%, at least about 91%, at least about 92%, about at least 93%, at least about 94%, at eat least about 95%, at least about 96%, at least about 97%, at least about 98%, at least about 99%, or 100% sequence identity to SEQ ID NO: 60.
- an Fc domain may also comprise one or more amino acid substitutions described herein, for example one or more substitutions that reduce effector function (e.g., as described in Section 6.4.5.1) and/or one or more substitutions that promote Fc heterodimerization (e.g., as described in Section 6.4.5.2).
- an Fc domain comprises an amino acid sequence having at least about 90%, at least about 91%, at least about 92%, about at least 93%, at least about 94%, at eat least about 95%, at least about 96%, at least about 97%, at least about 98%, at least about 99%, or 100% sequence identity to SEQ ID NO: 61.
- an Fc domain may also comprise one or more amino acid substitutions described herein, for example one or more substitutions that reduce effector function (e.g., as described in Section 6.4.5.1) and/or one or more substitutions that promote Fc heterodimerization (e.g., as described in Section 6.4.5.2).
- 6.4.5.1. Fc Domains with Altered Effector Function [0140]
- the Fc domain comprises one or more amino acid substitutions that reduces binding to an Fc receptor and/or effector function.
- the Fc receptor is an Fc ⁇ receptor.
- the Fc receptor is a human Fc receptor.
- the Fc receptor is an activating Fc receptor.
- the Fc receptor is an activating human Fc ⁇ receptor, more specifically human Fc ⁇ RIIIa, Fc ⁇ RI or Fc ⁇ Rlla, most specifically human Fc ⁇ Rllla.
- the effector function is one or more selected from the group of complement dependent cytotoxicity (CDC), antibody-dependent cell-mediated cytotoxicity (ADCC), antibody- dependent cellular phagocytosis (ADCP), and cytokine secretion.
- the effector function is ADCC.
- the Fc domain (e.g., an Fc domain of an antigen-binding molecule) or the Fc region (e.g., one or both Fc domains of an antigen-binding molecule that can associate to form an Fc region) comprises an amino acid substitution at a position selected from the group of E233, L234, L235, N297, P331 and P329 (numberings according to Kabat EU index).
- the Fc domain or the Fc region comprises an amino acid substitution at a position selected from the group of L234, L235 and P329 (numberings according to Kabat EU index).
- the Fc domain or the Fc region comprises the amino acid substitutions L234A and L235A (numberings according to Kabat EU index).
- the Fc domain or region is an Igd Fc domain or region, particularly a human Igd Fc domain or region.
- the Fc domain or the Fc region comprises an amino acid substitution at position P329.
- the amino acid substitution is P329A or P329G, particularly P329G (numberings according to Kabat EU index).
- the Fc domain or the Fc region comprises an amino acid substitution at position P329 and a further amino acid substitution at a position selected from E233, L234, L235, N297 and P331 (numberings according to Kabat EU index).
- the further amino acid substitution is E233P, L234A, L235A, L235E, N297A, N297D or P331S.
- the Fc domain or the Fc region comprises amino acid substitutions at positions P329, L234 and L235 (numberings according to Kabat EU index).
- the Fc domain comprises the amino acid mutations L234A, L235A and P329G (“P329G LALA”, “PGLALA” or “LALAPG”). [0143] Typically, the same one or more amino acid substitution is present in each of the two Fc domains of an Fc region.
- each Fc domain of the Fc region comprises the amino acid substitutions L234A, L235A and P329G (Kabat EU index numbering), i.e.
- the Fc domain is an IgG1 Fc domain, particularly a human IgG1 Fc domain.
- the IgG1 Fc domain is a variant IgG1 comprising D265A, N297A mutations (EU numbering) to reduce effector function.
- the Fc domain is an IgG4 Fc domain with reduced binding to Fc receptors.
- Exemplary IgG4 Fc domains with reduced binding to Fc receptors may comprise an amino acid sequence selected from Table F-2 below.
- the Fc domain includes only the bolded portion of the sequences shown below: TABLE F-2 Fc Domain Sequence SEQ ID NO TABLE F-2 Fc Domain Sequence SEQ ID NO TABLE F-2 Fc Domain Sequence SEQ ID NO TABLE F-2 Fc Domain Sequence SEQ ID NO TABLE F-2 Fc Domain Sequence SEQ ID NO [0146]
- the IgG4 with reduced effector function comprises the bolded portion of the amino acid sequence of SEQ ID NO:31 of WO2014/121087, sometimes referred to herein as IgG4s or hIgG4s.
- an Fc region comprising an Fc domain comprising the amino acid sequence of SEQ ID NO:30 of WO2014/121087 (or the bolded portion thereof) and an Fc domain comprising the amino acid sequence of SEQ ID NO:37 of WO2014/121087 (or the bolded portion thereof) or an Fc region comprising an Fc domain comprising the amino acid sequence of SEQ ID NO:31 of WO2014/121087 (or the bolded portion thereof) and an Fc domain comprising the amino acid sequence of SEQ ID NO:38 of WO2014/121087 (or the bolded portion thereof).
- an Fc region comprising an Fc domain comprising the amino acid sequence of SEQ ID NO:30 of WO2014/121087 (or the bolded portion thereof) and an Fc domain comprising the amino acid sequence of SEQ ID NO:37 of WO2014/121087 (or the bolded portion thereof)
- an Fc region comprising an Fc domain comprising the amino acid sequence of SEQ ID NO:31 of WO2014/121087
- Certain antigen-binding molecules entail dimerization between two Fc domains that, unlike a native immunoglobulin, are operably linked to non-identical N-terminal or C-terminal regions. Inadequate heterodimerization of two Fc domains to form an Fc region has can be an obstacle for increasing the yield of desired heterodimeric molecules and represents challenges for purification.
- a variety of approaches available in the art can be used in for enhancing dimerization of Fc domains that might be present in the antigen-binding molecules of the disclosure, for example as disclosed in EP 1870459A1; U.S. Patent No.5,582,996; U.S. Patent No.5,731,168; U.S.
- the present disclosure provides antigen-binding molecules comprising Fc heterodimers, i.e., Fc regions comprising heterologous, non-identical Fc domains.
- Fc heterodimers i.e., Fc regions comprising heterologous, non-identical Fc domains.
- each Fc domain in the Fc heterodimer comprises a CH3 domain of an antibody.
- the CH3 domains are derived from the constant region of an antibody of any isotype, class or subclass, and preferably of IgG (lgG1, lgG2, lgG3 and lgG4) class, as described in the preceding section.
- said modification promoting the formation of Fc heterodimers is a so-called “knob-into-hole” or “knob-in-hole” modification, comprising a “knob” modification in one of the Fc domains and a “hole” modification in the other Fc domain.
- the knob-into-hole technology is described e.g., in U.S.
- the method involves introducing a protuberance (“knob”) at the interface of a first polypeptide and a corresponding cavity (“hole”) in the interface of a second polypeptide, such that the protuberance can be positioned in the cavity so as to promote heterodimer formation and hinder homodimer formation.
- Protuberances are constructed by replacing small amino acid side chains from the interface of the first polypeptide with larger side chains (e.g., tyrosine or tryptophan).
- Compensatory cavities of identical or similar size to the protuberances are created in the interface of the second polypeptide by replacing large amino acid side chains with smaller ones (e.g., alanine or threonine).
- an amino acid residue in the CH3 domain of the first subunit of the Fc domain is replaced with an amino acid residue having a larger side chain volume, thereby generating a protuberance within the CH3 domain of the first subunit which is positionable in a cavity within the CH3 domain of the second subunit, and an amino acid residue in the CH3 domain of the second subunit of the Fc domain is replaced with an amino acid residue having a smaller side chain volume, thereby generating a cavity within the CH3 domain of the second subunit within which the protuberance within the CH3 domain of the first subunit is positionable.
- said amino acid residue having a larger side chain volume is selected from the group consisting of arginine I, phenylalanine (F), tyrosine (Y), and tryptophan (W).
- said amino acid residue having a smaller side chain volume is selected from the group consisting of alanine (A), serine (S), threonine (T), and valine (V).
- the protuberance and cavity can be made by altering the nucleic acid encoding the polypeptides, e.g., by site-specific mutagenesis, or by peptide synthesis.
- An exemplary substitution is Y470T.
- the threonine residue at position 366 is replaced with a tryptophan residue (T366W), and in the Fc domain the tyrosine residue at position 407 is replaced with a valine residue (Y407V) and optionally the threonine residue at position 366 is replaced with a serine residue (T366S) and the leucine residue at position 368 is replaced with an alanine residue (L368A) (numbering according to Kabat EU index).
- the serine residue at position 354 is replaced with a cysteine residue (S354C) or the glutamic acid residue at position 356 is replaced with a cysteine residue (E356C) (particularly the serine residue at position 354 is replaced with a cysteine residue), and in the second Fc domain additionally the tyrosine residue at position 349 is replaced by a cysteine residue (Y349C) (numbering according to Kabat EU index).
- the first Fc domain comprises the amino acid substitutions S354C and T366W
- the second Fc domain comprises the amino acid substitutions Y349C, T366S, L368A and Y407V (numbering according to Kabat EU index).
- electrostatic steering e.g., as described in Gunasekaran et al., 2010, J Biol Chem 285(25): 19637-46) can be used to promote the association of the first and the second Fc domains of the Fc region.
- an Fc domain can be modified to allow a purification strategy that enables selections of Fc heterodimers.
- one polypeptide comprises a modified Fc domain that abrogates its binding to Protein A, thus enabling a purification method that yields a heterodimeric protein. See, for example, U.S. Patent No.8,586,713.
- the antigen- binding molecules comprise a first CH3 domain and a second Ig CH3 domain, wherein the first and second Ig CH3 domains differ from one another by at least one amino acid, and wherein at least one amino acid difference reduces binding of the antigen-binding molecules to Protein A as compared to a corresponding antigen-binding molecule lacking the amino acid difference.
- the first CH3 domain binds Protein A and the second CH3 domain contains a mutation/modification that reduces or abolishes Protein A binding such as an H95R modification (by IMGT exon numbering; H435R by EU numbering).
- the second CH3 may further comprise a Y96F modification (by IMGT; Y436F by EU).
- the Fc can contain one or more mutations (e.g., knob and hole mutations) to facilitate heterodimerization as well as star mutations to facilitate purification.
- the antigen-binding molecules of the disclosure can comprise an Fc domain comprising a hinge domain at its N-terminus.
- the hinge region can be a native or a modified hinge region. Hinge regions are typically found at the N-termini of Fc regions.
- hinge domain refers to a naturally or non-naturally occurring hinge sequence that in the context of a single or monomeric polypeptide chain is a monomeric hinge domain and in the context of a dimeric polypeptide (e.g., a homodimeric or heterodimeric antigen-binding molecule formed by the association of two Fc domains) can comprise two associated hinge sequences on separate polypeptide chains. Sometimes, the two associated hinge sequences are referred to as a “hinge region”. In certain embodiments of antigen-binding molecules of the disclosure, additional iterations of hinge regions may be incorporated into the polypeptide sequence.
- a native hinge region is the hinge region that would normally be found between Fab and Fc domains in a naturally occurring antibody.
- a modified hinge region is any hinge that differs in length and/or composition from the native hinge region. Such hinges can include hinge regions from other species, such as human, mouse, rat, rabbit, shark, pig, hamster, camel, llama or goat hinge regions. Other modified hinge regions may comprise a complete hinge region derived from an antibody of a different class or subclass from that of the heavy chain Fc domain or Fc region. Alternatively, the modified hinge region may comprise part of a natural hinge or a repeating unit in which each unit in the repeat is derived from a natural hinge region.
- the natural hinge region may be altered by converting one or more cysteine or other residues into neutral residues, such as serine or alanine, or by converting suitably placed residues into cysteine residues. By such means the number of cysteine residues in the hinge region may be increased or decreased.
- Other modified hinge regions may be entirely synthetic and may be designed to possess desired properties such as length, cysteine composition and flexibility. [0158] A number of modified hinge regions have already been described for example, in U.S. Patent No.5,677,425, WO 99/15549, WO 2005/003170, WO 2005/003169, WO 2005/003170, WO 98/25971 and WO 2005/003171 and these are incorporated herein by reference.
- an antigen-binding molecule of the disclosure comprises an Fc region in which one or both Fc domains possesses an intact hinge domain at its N-terminus.
- positions 233-236 within a hinge region may be G, G, G and unoccupied; G, G, unoccupied, and unoccupied; G, unoccupied, unoccupied, and unoccupied; or all unoccupied, with positions numbered by EU numbering.
- the antigen-binding molecules of the disclosure comprise a modified hinge region that reduces binding affinity for an Fc ⁇ receptor relative to a wild-type hinge region of the same isotype (e.g., human IgG1 or human IgG4).
- the serine residue present in the lgG4 sequence leads to increased flexibility in this region, and therefore a proportion of molecules form disulfide bonds within the same protein chain (an intrachain disulfide) rather than bridging to the other heavy chain in the IgG molecule to form the interchain disulfide.
- an intrachain disulfide an intrachain disulfide
- Changing the serine residue to a proline to give the same core sequence as lgG1 allows complete formation of inter-chain disulfides in the lgG4 hinge region, thus reducing heterogeneity in the purified product. This altered isotype is termed lgG4P. 6.4.5.3.1.
- the hinge domain can be a chimeric hinge domain (also “chimeric hinge region”).
- a chimeric hinge domain may comprise an “upper hinge” sequence, derived from a human IgG1, a human IgG2 or a human IgG4 hinge region, combined with a “lower hinge” sequence, derived from a human IgG1, a human IgG2 or a human IgG4 hinge region.
- a chimeric hinge region comprises the amino acid sequence EPKSCDKTHTCPPCPAPPVA (SEQ ID NO: 89) (previously disclosed as SEQ ID NO:8 of WO2014/121087, which is incorporated by reference in its entirety herein) or ESKYGPPCPPCPAPPVA (SEQ ID NO: 90) (previously disclosed as SEQ ID NO:9 of WO2014/121087).
- EPKSCDKTHTCPPCPAPPVA amino acid sequence EPKSCDKTHTCPPCPAPPVA
- ESKYGPPCPPCPAPPVA SEQ ID NO: 90
- Such chimeric hinge sequences can be suitably linked to an IgG4 CH2 region (for example by incorporation into an IgG4 Fc domain, for example a human or murine Fc domain, which can be further modified in the CH2 and/or CH3 domain to reduce effector function, for example as described in Section 6.4.5.1).
- the hinge region can be modified to reduce effector function, for example as described in WO2016161010A2, which is incorporated by reference in its entirety herein.
- the positions 233-236 of the modified hinge region are G, G, G and unoccupied; G, G, unoccupied, and unoccupied; G, unoccupied, unoccupied, and unoccupied; or all unoccupied, with positions numbered by EU numbering (as shown in FIG.1 of WO2016161010A2). These segments can be represented as GGG-, GG--, G--- or ---- with “-“ representing an unoccupied position.
- Position 236 is unoccupied in canonical human IgG2 but is occupied by in other canonical human IgG isotypes. Positions 233-235 are occupied by residues other than G in all four human isotypes (as shown in FIG.1 of WO2016161010A2). [0168] The hinge modification within positions 233-236 can be combined with position 228 being occupied by P. Position 228 is naturally occupied by P in human IgG1 and IgG2 but is occupied by S in human IgG4 and R in human IgG3. An S228P mutation in an IgG4 antibody is advantageous in stabilizing an IgG4 antibody and reducing exchange of heavy chain light chain pairs between exogenous and endogenous antibodies.
- positions 226-229 are occupied by C, P, P and C respectively.
- Exemplary hinge regions have residues 226-236, sometimes referred to as middle (or core) and lower hinge, occupied by the modified hinge sequences designated GGG-(233-236), GG—(233-236), G---(233-236) and no G(233-236).
- the hinge domain amino acid sequence comprises CPPCPAPGGG-GPSVF (SEQ ID NO: 91) (previously disclosed as SEQ ID NO:1 of WO2016161010A2), CPPCPAPGG—GPSVF (SEQ ID NO: 92) (previously disclosed as SEQ ID NO:2 of WO2016161010A2), CPPCPAPG---GPSVF (SEQ ID NO: 93) (previously disclosed as SEQ ID NO:3 of WO2016161010A2), or CPPCPAP----GPSVF (SEQ ID NO: 94) (previously disclosed as SEQ ID NO:4 of WO2016161010A2).
- the modified hinge regions described above can be incorporated into a heavy chain constant region, which typically include CH2 and CH3 domains, and which may have an additional hinge segment (e.g., an upper hinge) flanking the designated region.
- additional constant region segments present are typically of the same isotype, preferably a human isotype, although can be hybrids of different isotypes.
- the isotype of such additional human constant regions segments is preferably human IgG4 but can also be human IgG1, IgG2, or IgG3 or hybrids thereof in which domains are of different isotypes. Exemplary sequences of human IgG1, IgG2 and IgG4 are shown in FIGS.2-4 of WO2016161010A2.
- the modified hinge sequences can be linked to an IgG4 CH2 region (for example by incorporation into an IgG4 Fc domain, for example a human or murine Fc domain, which can be further modified in the CH2 and/or CH3 domain to reduce effector function, for example as described in Section 6.4.5.1).
- Nucleic Acids and Host Cells [0172]
- the disclosure provides nucleic acids encoding the antigen-binding molecules of the disclosure.
- the antigen-binding molecules are encoded by a single nucleic acid.
- the antigen-binding molecules can be encoded by a plurality (e.g., two, three, four or more) nucleic acids.
- a single nucleic acid can encode an antigen-binding molecule that comprises a single polypeptide chain, an antigen-binding molecule that comprises two or more polypeptide chains, or a portion of an antigen-binding molecule that comprises more than two polypeptide chains (for example, a single nucleic acid can encode two polypeptide chains of an antigen-binding molecule comprising three, four or more polypeptide chains, or three polypeptide chains of an antigen-binding molecule comprising four or more polypeptide chains).
- the open reading frames encoding two or more polypeptide chains can be under the control of separate transcriptional regulatory elements (e.g., promoters and/or enhancers).
- an antigen-binding molecule comprising two or more polypeptide chains is encoded by two or more nucleic acids.
- the number of nucleic acids encoding an antigen-binding molecule can be equal to or less than the number of polypeptide chains in the antigen-binding molecule (for example, when more than one polypeptide chains are encoded by a single nucleic acid).
- the nucleic acids of the disclosure can be DNA (e.g., plasmid) or RNA (e.g., mRNA).
- the disclosure provides host cells and vectors containing the nucleic acids of the disclosure.
- the nucleic acids may be present in a single vector or separate vectors present in the same host cell or separate host cell, as described in more detail herein below. 6.6.
- Pharmaceutical Compositions [0177]
- the antigen-binding molecules of the disclosure may be in the form of compositions comprising the antigen-binding molecule and one or more carriers, excipients and/or diluents.
- the compositions may be formulated for specific uses, such as for veterinary uses or pharmaceutical uses in humans.
- compositions may be supplied as part of a sterile, pharmaceutical composition that includes a pharmaceutically acceptable carrier.
- This composition can be in any suitable form (depending upon the desired method of administering it to a patient).
- the pharmaceutical composition can be administered to a patient by a variety of routes such as orally, transdermally, subcutaneously, intranasally, intravenously, intramuscularly, intratumorally, intrathecally, topically, or locally.
- Preservatives may be added to retard microbial growth, and can be added in amounts ranging from about 0.2%-1 % (w/v).
- Non-ionic surfactants may be present in a range of about 0.05 mg/mL to about 1.0 mg/mL, for example about 0.07 mg/mL to about 0.2 mg/mL.
- Additional miscellaneous excipients include bulking agents (e.g., starch), chelating agents (e.g., EDTA), antioxidants (e.g., ascorbic acid, methionine, vitamin E), and cosolvents. 6.6.1.
- Non-limiting examples of adenovirus-based or AAV-based therapeutics for use in the methods, uses or compositions herein include, but are not limited to: rAd-p53, which is a recombinant adenoviral vector encoding the wild-type human tumor suppressor protein p53, for example, for the use in treating a cancer (also known as Gendicine®, Genkaxin®, Qi et al., 2006, Modern Oncology, 14:1295-1297); Ad5_d11520, which is an adenovirus lacking the E1B gene for inactivating host p53 (also called H101 or ONYX-015; see, e.g., Russell et al., 2012, Nature Biotechnology 30:658-670); AD5-D24-GM-CSF, an adenovirus containing the cytokine GM-CSF, for example, for the use in treating a cancer (Cerullo et al., 2010, Cancer Res.70:42
- rAd- TNF ⁇ a replication-deficient adenoviral vector expressing human tumor necrosis factor alpha (TNF ⁇ ) under the control of the chemoradiation-inducible EGR-1 promoter, for example, for the treatment of cancer (TNFeradeTM, GenVec; Rasmussen et al., 2002, Cancer Gene Ther.9:951- 7;
- Ad-IFN ⁇ an adenovirus serotype 5 vector from which the E1 and E3 genes have been deleted expressing the human interferon-beta gene under the direction of the cytomegalovirus (CMV) immediate-early promoter, for example for treating cancers (BG00001 and H5.110CMVhIFN- ⁇ , Biogen; Sterman et al., 2010, Mol.
- CMV cytomegalovirus
- exemplary non-limiting nucleic acid complexes for use as a delivery vector include lipoplexes, polymersomes, polypexes, dendrimers, inorganic nanoparticles (e.g., polynucleotide coated gold, silica, iron oxide, calcium phosphate, etc.).
- a delivery vector as described herein comprises a combination of a viral vector, naked nucleic acids, and nucleic acid complexes.
- the delivery vector is a virus, including a retrovirus, adenovirus, herpes simplex virus, pox virus, vaccinia virus, lentivirus, or an adeno-associated virus.
- the delivery vector is an adeno-associated virus (AAV), including serotypes AAV1, AAV2, AAV3, AAV4, AAV5, AAV6, AAV7, AAV8, AAV9, AAV10, and AAV11, or engineered or naturally selected variants thereof.
- AAV adeno-associated virus
- a nucleic acid encoding an antigen-binding molecule (or component thereof) also contains adeno-associated virus (AAV) nucleic acid sequence.
- the vector is a chimeric adeno-associated virus containing genetic elements from two or more serotypes.
- an AAV vector with rep genes from AAV1 and cap genes from AAV2 may be used as a delivery vector to deliver an antigen-binding molecule expressing nucleic acid to a cell or a cell of a patient in need.
- the delivery vector is an AAV1/2, AAV1/3, AAV1/4, AAV1/5, AAV1/6, AAV1/7, AAV1/8, AAV1/9, AAV1/10, AAV1/11, AAV2/1, AAV2/3, AAV2/4, AAV2/5, AAV2/6, AAV2/7, AAV2/8, AAV2/9, AAV2/10, AAV2/11, AAV3/1, AAV3/2, AAV3/4, AAV3/5, AAV3/6, AAV3/7, AAV3/8, AAV3/9, AAV3/10, AAV3/10, AAV4/1, AAV4/2, AAV4/3, AAV4/5, AAV4/6, AAV4/7, AAV4/8, AAV4/9, AAV4/10, AAV4/11, AAV5/1, AAV5/2, AAV5/3, AAV5/4, AAV5/6, AAV5/7, AAV5/8, AAV5/9, AAV4/10, AAV4/11, AAV5/1, AAV5/2, AAV5/3
- the present disclosure includes methods for producing an engineered VH.
- protein engineering methods comprising generating an engineered VH having an N-linked glycosylation site within 10 amino acids of its C-terminus.
- methods for producing an antigen-binding molecule comprising such an engineered VH.
- the disclosed methods comprise inserting an N-linked glycosylation site within 10 (e.g., 10, 9, 8, 7, 6, 5, 4, or 3) amino acids of the C-terminus of a VH.
- the methods comprise inserting the amino acid sequence X 1 X 2 X 3 X 4 X 5 NX 6 X 7 X 8 X 9 X 10 X 11 X 12 (SEQ ID NO:1), or a portion thereof, into the VH, where (a) X 1 , X 2 , X 3 , X 4 , and X 5 are each independently selected from any amino acid; (b) X 6 is selected from any amino acid, where the amino acid is optionally not proline; (c) X 7 is S or T, and (d) X 8 , X 9 , X 10 ,X 11 , and X 12 are each independently selected from any amino acid and absent.
- the methods comprise inserting the full amino acid sequence X 1 X 2 X 3 X 4 X 5 NX 6 X 7 X 8 X 9 X 10 X 11 X 12 into the VH. In other embodiments, the methods comprise inserting a portion of the amino acid sequence X 1 X 2 X 3 X 4 X 5 NX 6 X 7 X 8 X 9 X 10 X 11 X 12 into the VH such that the resultant engineered VH comprises the sequence NX 6 X 7 .
- the sequence NX 6 X 7 may be inserted within 10 amino acids of the C-terminus of the VH, thereby generating an engineered VH.
- the disclosed methods comprise inserting, deleting, and/or substituting one or more amino acids within the sequence of the VH such that the VH comprises an N-linked glycosylation site within 10 (e.g., 10, 9, 8, 7, 6, 5, 4, or 3) amino acids of the C- terminus of the VH.
- the methods comprise inserting, deleting, and/or substituting one or more amino acids within the sequence of the VH such that the VH comprises the amino acid sequence X 1 X 2 X 3 X 4 X 5 NX 6 X 7 X 8 X 9 X 10 X 11 X 12 (SEQ ID NO:1) within 10 amino acids of its C-terminus, where (a) X 1 , X 2 , X 3 , X 4 , and X 5 are each independently selected from any amino acid; (b) X 6 is selected from any amino acid, where the amino acid is optionally not proline; (c) X 7 is S or T, and (d) X 8 , X 9 , X 10 ,X 11 , and X 12 are each independently selected from any amino acid and absent.
- the methods may comprise inserting, deleting, and/or substituting 1, 2, 3, 4, 5, or more amino acids such that the VH comprises the N-linked glycosylation site.
- a single amino acid is inserted such that the engineered VH comprises the sequence NX 6 X 7 , for example insertion of an asparagine residue prior to the sequence SS or ST.
- a single amino acid residue is substituted such that that the engineered VH comprises the sequence NX 6 X 7 , for example substitution of an asparagine residue for X in the sequence XSS or XST to generate the sequence NSS or NST.
- the methods comprise inserting an asparagine residue between the third and fourth positions of the sequence VTVSS (SEQ ID NO: 62) of a VH, generating the sequence VTVNSS (SEQ ID NO: 50) at the C-terminus of the engineered VH.
- two or more amino acid residues are inserted, deleted, and/or substituted such that that the engineered VH comprises the sequence NX 6 X 7 , for example both insertion of an asparagine residue prior to the sequence SX and also substitution of a serine or threonine residue for X, generating the sequence NSS or NST.
- the methods comprise inserting an N residue between the third and fourth positions of the sequence VTVSS (SEQ ID NO: 62) of a VH and also substituting a T residue at the fifth position of this sequence, generating the sequence VTVNST (SEQ ID NO: 51) at the C-terminus of the engineered VH.
- engineered VHs of the disclosure are useful for decreasing binding of anti-drug antibodies to an antigen-binding molecule. Accordingly, disclosed are methods for reducing antigenicity of an antigen-binding molecule comprising producing an antigen-binding molecule comprising an engineered VH as disclosed herein.
- antigenicity describes the degree to which an antigen-binding molecule binds to anti-drug antibodies from a subject.
- references herein to “modifying,” “inserting an amino acid,” “deleting an amino acid,” “substituting an amino acid,” and the like, in reference to generating an engineered VH, are used purely for convenience and do not require that the engineered VH be derived or obtained directly from a particular VH. Such references describe an engineered VH that is related in sequence and has one or more sequence differences relative to the particular reference VH.
- a method for generating an engineered VH comprises “substituting one or more amino acids” of a VH
- such a description includes generation of the engineered VH through mutagenesis of a coding sequence for the VH and also includes direct synthesis of an engineered VH having the one or more amino acid differences (e.g., insertions, deletions or substitutions) relative to the VH.
- the reference VH is referred to as a parental VH and an antigen-binding molecule that incorporates the reference VH instead of the engineered VH but is otherwise identical is referred to as a counterpart antigen-binding molecule. 6.8.
- Therapeutic Methods comprising administering an antigen-binding molecule comprising an engineered VH of the present disclosure.
- engineered VHs having an N-glycosylation site within 10 amino acids of their C-terminus have reduced binding to anti-drug antibodies relative to a corresponding non-engineered VH.
- the disclosed therapeutic methods which involve administration of an antigen-binding molecule comprising an engineered VH of the disclosure, are understood to have reduced adverse immune response and/or increased durability relative to treatment methods involving administration of a counterpart antigen-binding molecule in which the VH is not engineered.
- the present disclosure provides methods for reducing an adverse immune response associated with an antigen-binding molecule therapeutic.
- the methods typically comprise administering to a subject an antigen-binding molecule comprising an engineered VH having an N-glycosylation site within 10 amino acids (e.g.,10, 9, 8, 7, 6, 5, 4, or 3 amino acids) of its C-terminus, such as an engineered VH as described in Section 6.3.
- administering reduces an adverse immune response associated with an antigen-binding molecule (comprising a non-engineered VH) by at least 10%, at least 20%, at least 30%, at least 40%, at least 50%, at least 60%, at least 70%, at least 80%, at least 90%, at least 95%, at least 99%, or more.
- the subject may be administered an antigen-binding molecule comprising an engineered VH following an adverse reaction to a corresponding antigen-binding molecule comprising a non-engineered VH.
- the present disclosure provides methods for increasing treatment durability of an antigen-binding molecule therapeutic.
- the methods typically comprise administering to a subject an antigen-binding molecule comprising an engineered VH having an N-glycosylation site within 10 amino acids (e.g.,10, 9, 8, 7, 6, 5, 4, or 3 amino acids) of its C- terminus, such as an engineered VH as described in Section 6.3.
- administration of an antigen-binding molecule comprising an engineered VH described herein increases treatment durability of an antigen-binding molecule (comprising a non-engineered VH) by at least 10%, at least 20%, at least 30%, at least 40%, at least 50%, at least 60%, at least 70%, at least 80%, at least 90%, at least 100%, at least 200%, at least 300%, or more.
- antigen-binding molecules of the disclosure can, in some embodiments, be used in the treatment of a proliferative disorder (e.g., cancer) that expresses a tumor-associated antigen.
- a proliferative disorder e.g., cancer
- the cancer is acute lymphoblastic leukemia (ALL), acute myeloid leukemia (AML), adrenocortical carcinoma, anal cancer, appendix cancer, astrocytoma, basal cell carcinoma, brain tumor, bile duct cancer, bladder cancer, bone cancer, breast cancer, bronchial tumor, Burkitt Lymphoma, carcinoma of unknown primary origin, cardiac tumor, cervical cancer, chordoma, chronic lymphocytic leukemia (CLL), chronic myelogenous leukemia (CML), chronic myeloproliferative neoplasm, colon cancer, colorectal cancer, craniopharyngioma, cutaneous T-cell lymphoma, ductal carcinoma, embryonal tumor, endometrial cancer, ependymoma, esophageal cancer, esthesioneuroblastoma, fibrous histiocytoma, Ewing sarcoma, eye cancer, germ cell tumor, gallbladder cancer, gas
- Table I below shows exemplary indications for which antigen-binding molecules targeting particular TAAs can be used.
- Additional tumor-associated antigens and corresponding indications are disclosed in, e.g., Hafeez et al., 2020, Molecules 25:4764, doi:10.3390/molecules25204764, particularly in Table 1.
- an additional therapeutic agent is an immunomodulatory agent, a cytostatic agent, an inhibitor of cell adhesion, a cytotoxic agent, an activator of cell apoptosis, or an agent that increases the sensitivity of cells to apoptotic inducers.
- the additional therapeutic agent is an anti-cancer agent, for example a microtubule disruptor, an antimetabolite, a topoisomerase inhibitor, a DNA intercalator, an alkylating agent, a hormonal therapy, a kinase inhibitor, a receptor antagonist, an activator of tumor cell apoptosis, or an antiangiogenic agent.
- Such other agents are suitably present in combination in amounts that are effective for the purpose intended.
- the effective amount of such other agents depends on the amount of antigen-binding molecule used, the type of disorder or treatment, and other factors discussed above.
- the antigen-binding molecules are generally used in the same dosages and with administration routes as described herein, or about from 1 to 99% of the dosages described herein, or in any dosage and by any route that is empirically/clinically determined to be appropriate.
- Such combination therapies noted above encompass combined administration (where two or more therapeutic agents are included in the same or separate compositions), and separate administration, in which case, administration of the antigen-binding molecule of the disclosure can occur prior to, simultaneously, and/or following, administration of the additional therapeutic agent and/or adjuvant. 7.
- SEQUENCES [0205] Certain sequences of the disclosure are provided in Table S below.
- An antigen-binding molecule comprising a heavy chain variable domain (VH), said VH comprising an N-linked glycosylation site within 10 amino acids of the C-terminus of the VH.
- VH heavy chain variable domain
- An antigen-binding molecule comprising a heavy chain variable domain (VH), optionally wherein the antigen-binding molecule is an antigen-binding molecule according to any one of embodiments 1 to 6, wherein the VH comprises at its C-terminus the amino acid sequence X 1 X 2 X 3 X 4 X 5 NX 6 X 7 X 8 X 9 X 10 X 11 X 12 (SEQ ID NO:1), wherein (a) X 1 , X 2 , X 3 , X 4 , and X 5 are each independently selected from any amino acid; (b) X 6 is selected from any amino acid, optionally wherein the amino acid is not proline; (c) X 7 is S or T; and (d) X 8 , X 9 , X 10 ,X 11 , and X 12 are each independently selected from any amino acid and absent.
- VH heavy chain variable domain
- the antigen-binding molecule of any one of embodiments 7 to 18, wherein X 8 is K. 22.
- the antigen-binding molecule of any one of embodiments 7 to 18, 21, and 22 wherein X 10 is G. 24.
- the antigen-binding molecule of any one of embodiments 7 to 18, wherein X 8 is G. 26.
- the antigen-binding molecule of any one of embodiments 7 to 18, wherein the VH is at least 100 amino acids in length.
- 48. The antigen-binding molecule of any one of embodiments 45 to 47, wherein the Fc domain and the VH are separated by a linker.
- 49. The antigen-binding molecule of embodiment 48, wherein the linker is a glycine- serine linker. 50.
- the antigen-binding molecule of embodiment 88 wherein the TAA ABD is capable of binding to syndecan, heparanase, integrins, osteopontin, link, cadherins, laminin, laminin type EGF, lectin, fibronectin, notch, nectin (e.g., nectin-4), tenascin, collagen (e.g., collagen type X), or matrixin.
- the antigen-binding molecule of embodiment 94, wherein the VH comprising the N-linked glycosylation site is a component of the TCE ABD.
- the antigen-binding molecule of embodiment 96, wherein the component of the TCR complex is CD3.
- the antigen-binding molecule of embodiment 96, wherein the component of the TCR complex is TCR ⁇ .
- the antigen-binding molecule of embodiment 96, wherein the component of the TCR complex is TCR ⁇ . 100.
- the antigen-binding molecule of any one of embodiments 95 to 99, wherein the TCE ABD (a) comprises the (i) CDR or (ii) VH and VL sequences of antibody set forth in Table T or (b) competes with the antibody set forth in Table T for binding to its target.
- TAA ABD tumor-associated antigen
- the antigen-binding molecule of embodiment 102 wherein the TAA ABD is capable of binding to AFP, ALK, a BAGE protein, BIRC5 (survivin), BIRC7, ⁇ -catenin, brc-abl, BRCA1, BORIS, CA9, carbonic anhydrase IX, caspase-8, CALR, CEACAM5 (also known as carcinoembryonic antigen or CEA), CCR5, CD19, CD20 (MS4A1), CD22, CD30, CD40, CDK4, CEA, CTLA4, cyclin-B1, CYP1B1, EGFR, EGFRvIII, ErbB2/Her2, ErbB3, ErbB4, ETV6-AML, EpCAM, EphA2, Fra-1, FOLR1, a GAGE protein (e.g., GAGE-1 or -2), GD2, GD3, GloboH, glypican-3, GM3, gp100, Her2, HLA/B-raf, HLA
- the antigen-binding molecule of embodiment 102, wherein the TAA ABD is capable of binding to CTLA-4, PD1, PDL1, PDL2, B7-H3, B7-H4, BTLA, HVEM, TIM3, GAL9, LAG3, VISTA, KIR, 2B4, CD160, CGEN-15049, CHK1, or CHK2.
- 105. The antigen-binding molecule of embodiment 102, wherein the TAA ABD is capable of binding to CD27, CD28, 4-1BB (CD137), OX40, CD30, CD40, ICOS, lymphocyte function-associated antigen-1 (LFA-1), CD2, CD7, LIGHT, NKG2C, or B7-H3.
- 106 The antigen-binding molecule of embodiment 102, wherein the TAA ABD is capable of binding to CD27, CD28, 4-1BB (CD137), OX40, CD30, CD40, ICOS, lymphocyte function-associated antigen-1 (LFA-1), CD2, CD7
- the antigen-binding molecule of embodiment 102 wherein the TAA ABD is capable of binding to syndecan, heparanase, integrins, osteopontin, link, cadherins, laminin, laminin type EGF, lectin, fibronectin, notch, nectin (e.g., nectin-4), tenascin, collagen (e.g., collagen type X), or matrixin.
- a pharmaceutical composition comprising the antigen-binding molecule of any one of embodiments 1 to 106.
- a method of producing the antigen-binding molecule of any one of embodiments 1 to 106 comprising culturing the host cell of embodiment 109 or 110 and recovering the antigen-binding molecule expressed thereby.
- a method comprising administering the antigen-binding molecule of any one of embodiments 1 to 106 to a subject. 113.
- a method of treating cancer comprising administering to a subject in need thereof the antigen-binding molecule of any one of embodiments 1 to 106 or the pharmaceutical composition of embodiment 107.
- the antigen-binding molecule is a bispecific antigen-binding molecule comprising a TCE ABD and a TAA ABD and the cancer is associated with the expression of the tumor-associated antigen, e.g., as set forth in Table A.
- a polypeptide comprising an engineered VH as defined in any one of embodiments 1 to 34.
- a method of producing the polypeptide of embodiment 115 comprising culturing the host cell of embodiment 117.
- a protein engineering method comprising generating an antigen-binding molecule comprising a VH having an N-linked glycosylation site within 10 amino acids of its C- terminus.
- a protein engineering method comprising generating a VH comprising an N- linked glycosylation site within 10 amino acids of its C-terminus.
- a method of reducing antigenicity of an antigen-binding molecule comprising a VH comprising incorporating an N-linked glycosylation site within 10 amino acids of the C-terminus of the VH. 122.
- the method embodiment 121, wherein the antigen-binding molecule further comprises an additional VH.
- the method of embodiment 122, wherein the method further comprises incorporating an N-linked glycosylation site within 10 amino acids of the C-terminus of the additional VH. 124.
- any one of embodiments 119 to 123 wherein the method comprises inserting the amino acid sequence X 1 X 2 X 3 X 4 X 5 NX 6 X 7 X 8 X 9 X 10 X 11 X 12 (SEQ ID NO:1), or a portion thereof, into the VH, wherein (a) X 1 , X 2 , X 3 , X 4 , and X 5 are each independently selected from any amino acid; (b) X 6 is selected from any amino acid, optionally wherein the amino acid is not proline; (c) X 7 is S or T, and (d) X 8 , X 9 , X 10 ,X 11 , and X 12 are each independently selected from any amino acid and absent.
- any one of embodiments 119 to 123, wherein the method comprises substituting one or more amino acids within the sequence of the VH such that the VH comprises the amino acid sequence X 1 X 2 X 3 X 4 X 5 NX 6 X 7 X 8 X 9 X 10 X 11 X 12 (SEQ ID NO:1) within 10 amino acids of its C-terminus, wherein (a) X 1 , X 2 , X 3 , X 4 , and X 5 are each independently selected from any amino acid; (b) X 6 is selected from any amino acid, optionally wherein the amino acid is not proline; (c) X 7 is S or T, and (d) X 8 , X 9 , X 10 ,X 11 , and X 12 are each independently selected from any amino acid and absent.
- X 4 is S. 134.
- the method of any one of embodiments 124 to 128 and 132 to 134, wherein X 2 is T. 136.
- the method of any one of embodiments 124 to 136, wherein X 8 is P. 138.
- Antigen-binding molecules were designed to comprise two polypeptides connected to one another via their Fc domains.
- the first polypeptide was designed to comprise from N- terminus to C-terminus: a CD3-targeting Fab, a first Fc domain, a linker, and a CD3-targeting scFv.
- the second polypeptide was designed to comprise from N-terminus to C-terminus: a TAA- targeting Fab, a second Fc domain, a linker, and a TAA-targeting scFv.
- Antigen-binding molecules were engineered by introducing amino acid modifications at or near the C-terminus of the scFvs.
- FIG.2A shows a parental antigen-binding molecule
- FIGs.2B-2E show variants with C-terminal amino acid modifications that do not incorporate an N-glycosylation consensus sequence
- FIGs.2F and 2G show variants that incorporate an N-glycosylation consensus sequence at the C-terminus of the scFvs.
- APC-conjugated goat anti-human IgG (Jackson Immuno Research, 109-607-003, 1:400) was added to stain the cells for 30 min at 2–8°C along with LIVE/DEAD Fixable Violet Dead Cell Stain Kit (Thermo Fisher Scientific). Following washing, cells were fixed in 2% paraformaldehyde for 30 min at 2–8°C. After two washes, stained cells were analyzed using BD FACSCantoII instrument. The results were analyzed by FlowJo. FSC/SSC gates were used to select single cells and BV421 negative gating was used to select live cells. 9.1.3.
- PBMC Peripheral blood mononuclear cells
- the PBMC suspension was mixed with human IgG1 at a final concentration of 5 mg/mL to mimic human serum, and PBMC at 2 ⁇ 10 6 cells/mL.
- PBMC were added to a final amount of 50,000 cells/well.
- Multispecific antigen binding molecules were diluted with a ratio of 1:10 and added to the assay plates with a final starting concentration of 6.7 x 10 -08 M in R10 media.
- the plates were incubated in a cell culture incubator for 72 hours or longer. The supernatant was then removed and spun down at 300 x g for 4 minutes to pellet all cells in suspension.
- the tumor cells were washed and harvested with trypsin, and combined with the suspension cell pellet.
- the final cell pellet was washed with PBS and stained with LIVE/DEAD Fixable Near-IR Dead Cell Stain Kit (Thermo Fisher Scientific). After 2 washes with PBS, cells were resuspended in FACS wash and analyzed on a cytometer such as the BD FACSCelesta instrument or BD FACSCantoII instrument. Results were analyzed by FlowJo: CellTrace positive (BV421 channel) cells were gated to identify tumor cells, FSC/SSC gates were used to select single cells, and APC-Cy7 negative gating was used to select live cells. All appropriate compensation samples and fluorescence minus one (FMO) controls were included. 9.1.4.
- Multispecific antigen-binding molecules REGN9930-VNST and REGN9930-VNSS were subjected to limited LysC digestion to generate full-length scFv fragments (arising from preferential cleavage at Lys site N-terminal to GS-linkers). Such fragments were well separated from the mAbs or Fabs and thus could be discerned from N-linked glycans in the scFv region based on the predicted mass. 9.1.5. ADA Reactivity Assay [0212] In order to assess antigenicity, antibodies were tested in an ADA reactivity assay using an Electrochemiluminescent Immunoassay (ECL) or a SMCxPro Immunogenicity method for measuring binding to anti-drug antibodies.
- ECL Electrochemiluminescent Immunoassay
- SMCxPro Immunogenicity method for measuring binding to anti-drug antibodies.
- Example 3 Native SEC-MS Analysis of Limited LysC Digested REGN9930- VNSS and REGN9930-VNST [0218] To determine whether C-terminal modifications of constructs REGN9930-VNSS and REGN9930-VNST have incorporated N-glycans, these two constructs were subjected to limited LysC digestion and evaluated with native SEC-UV/MS as described in Section 9.1.4. [0219] Limited LysC digestion freed the scFv domains attached to Fc C-termini of both polypeptide chains of REGN9930-VNSS and yielded full-length scFv fragments.
- Example 4 C-Terminal Modifications Reduce ADA Reactivity of REGN9930
- REGN9930 has pre-existing ADA reactivity.
- REGN8503 was used as a negative control and displayed no preexisting reactivity.
- REGN9930 was associated with high levels of preexisting reactivity.
- All C-terminal engineered variants of REGN9930 exhibited greatly reduced ADA binding (FIG.6).
Landscapes
- Health & Medical Sciences (AREA)
- Immunology (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Biophysics (AREA)
- Biochemistry (AREA)
- General Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Medicinal Chemistry (AREA)
- Molecular Biology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Cell Biology (AREA)
- Peptides Or Proteins (AREA)
Abstract
The present disclosure relates to engineered heavy chain variable domains. Also disclosed are antigen-binding molecules comprising such engineered heavy chain variable domains, including multivalent and multispecific antigen-binding molecules. Further disclosed are methods of producing the antigen-binding molecules, pharmaceutical compositions comprising the antigen-binding molecules, nucleic acids expressing the antigen-binding molecules, and methods for use of the antigen-binding molecules.
Description
ENGINEERED HEAVY CHAIN VARIABLE DOMAINS AND USES THEREOF 1. CROSS-REFERENCE TO RELATED APPLICATIONS [0001] This application claims the priority benefit of U.S. provisional application no.63/598,776, filed on November 14, 2023, the contents of which are incorporated herein in their entirety by reference thereto. 2. SEQUENCE LISTING [0002] The instant application contains a Sequence Listing which has been submitted electronically and is hereby incorporated by reference in its entirety. Said copy, created on November 5, 2024, is named RGN-031WO_SL.xml and is 126,612 bytes in size. 3. BACKGROUND [0003] Various biological therapeutics have been developed for the prevention and treatment of various diseases, including proliferative diseases (e.g., cancers), chronic inflammatory diseases (e.g., Crohn’s disease), and rheumatologic diseases (e.g., rheumatoid arthritis). [0004] Among biological therapeutics, antibodies and related antigen-binding molecules have proven effective in clinical practice. Yet, immunogenicity of these molecules can challenge their usefulness and hinder the development of new molecules for clinical application. For instance, even fully human therapeutic monoclonal antibodies have the potential to trigger the production of anti-drug antibodies (ADAs) that can cause undesired effects, such as suboptimal targeting, loss of efficacy, formation of highly immunogenic complexes, and other adverse events (Koren et al., 2002, Curr Pharm Biotechnol, 3(4):349-60; Schellekens, 2002, Clin Ther, 24(11):1720-40; van Schie et al., 2015, Ann Rheum Dis.74(1):311–314; Hansel, 2010, Nat Rev Drug Discov. 9(4):325–338), which may limit the clinical applicability and use of antigen-binding molecules and other biologicals. [0005] Therefore, there is need in the art to mitigate production and/or binding of ADAs associated with antigen-binding molecules and other biologicals. 4. SUMMARY [0006] The present disclosure relates to engineered heavy chain variable domains (VHs) and antigen-binding molecules comprising the engineered VHs.
[0007] In particular, the present disclosure provides VHs that are engineered to incorporate an N-linked glycosylation site near their C-termini. Without being bound by theory, it is believed that incorporation of an N-linked glycosylation site reduces binding to anti-drug antibodies and thus reduces the antigenicity of antigen-binding molecules that incorporate the engineered VHs as compared to a non-engineered VH. [0008] The present disclosure further provides polypeptides comprising the VHs of the disclosure, having an N-linked glycosylation site near their C-termini. In some embodiments, the polypeptides are antigen-binding molecules. The engineered VHs can be advantageously used in the context of a variety of antigen-binding molecules and components thereof, e.g., scFvs, Fabs, antibodies, antibody fragments, and other antigen-binding molecules, including multivalent and/or multispecific antigen-binding molecules. In addition to an engineered VH of the disclosure, an antigen-binding molecule can include one or more additional target binding domains (e.g., one or more Fab moieties, one or more scFv moieties, or a combination thereof) and/or one or more linkers separating one or more moieties in the antigen-binding molecule. [0009] Exemplary engineered VHs are disclosed in Section 6.2. Exemplary antigen-binding molecules comprising engineered VHs are disclosed in Section 6.3, and numbered embodiments 1 to 106. Exemplary target binding domains for incorporation in an antigen- binding molecule are disclosed in Sections 6.4.2 and 6.4.3 and exemplary linkers useful for connecting constant domains to the target binding domains or different components of the target binding domains are described in Section 6.4.4. [0010] The disclosure further provides nucleic acids encoding the VHs and antigen-binding molecules of the disclosure. The nucleic acids encoding the antigen-binding molecules can be in the form of a single nucleic acid (e.g., a vector encoding two or more polypeptide chains) or a plurality of nucleic acids (e.g., two or more vectors encoding different polypeptide chains). The disclosure further provides host cells and cell lines engineered to express the nucleic acids, VHs and antigen-binding molecules of the disclosure. Exemplary nucleic acids, host cells, and cell lines, are described in Section 6.5 and numbered embodiments 108 to 110. [0011] Methods of producing the antigen-binding molecules, methods of using the engineered VHs of the disclosure to decrease binging of an antigen-binding molecule to anti-drug antibodies, and methods of reducing antigenicity of antigen-binding molecules are described in Section 6.7 and numbered embodiments 111, and 118 to 150.
[0012] The disclosure further provides pharmaceutical compositions comprising the antigen- binding molecules of the disclosure. Exemplary compositions are described in Section 6.6 and numbered embodiment 107. [0013] Also disclosed are methods of administration of the antigen-binding molecules of the disclosure, including methods of treatment comprising administering the antigen-binding molecules. Exemplary administration methods, including treatment methods, are described in Section 6.7 and numbered embodiments 112 to 114. 5. BRIEF DESCRIPTION OF THE FIGURES [0014] FIGS.1A-1C illustrate exemplary antigen-binding molecules of the disclosure comprising a VH comprising at its C-terminus the amino acid sequence X1X2X3X4X5NX6X7X8X9X10X11X12 (SEQ ID NO:1), wherein (a) X1, X2, X3, X4, and X5 are each independently selected from any amino acid; (b) X6 is selected from any amino acid, optionally wherein the amino acid is not proline; (c) X7 is S or T, and (d) X8, X9, X10,X11, and X12 are each independently selected from any amino acid and absent. This sequence is represented as “X1X2X3X4X5NX6[S/T]X8X9X10X11X12” in FIGs.1A-1C. FIG.1A shows an scFv comprising a VH comprising the amino acid sequence of SEQ ID NO:1 at its C-terminus. FIG.1B shows a bivalent antigen-binding molecule comprising two scFv molecules linked by a linker, where at least one of the VHs comprises the sequence X1X2X3X4X5NX6[S/T]X8X9X10X11X12 at its C- terminus. FIG.1C shows an exemplary multispecific antigen-binding molecule comprising a first polypeptide, which comprises from N-terminus to C-terminus: a first targeting moiety (e.g., a Fab or scFv), a first dimerization moiety (e.g., an Fc domain), an optional linker, and a first scFv; and a second polypeptide which comprises from N-terminus to C-terminus: a second targeting moiety (e.g., a Fab of scFv), a second dimerization moiety, an optional linker, and a second scFv, where each scFv comprises a VH comprising the sequence X1X2X3X4X5NX6[S/T]X8X9X10X11X12 at its C-terminus. [0015] FIGS.2A-2G illustrate certain multispecific antigen-binding molecules of the disclosure, each comprising a first polypeptide, which comprises from N-terminus to C-terminus: a first targeting moiety (e.g., a Fab or scFv), a first dimerization moiety (e.g., an Fc domain), an optional linker, and a second targeting moiety; and a second polypeptide which comprises from N-terminus to C-terminus: a target antigen-binding moiety, such as a tumor-associated antigen targeting moiety (e.g., a Fab or scFv that specifically binds to a tumor-associated antigen), a second dimerization moiety, an optional linker and a second target antigen-binding moiety. FIG.
2A shows a multispecific antigen-binding molecule, the scFv C-termini of which comprise the amino acid sequence VTVSS (SEQ ID NO:62). FIGs 2B-2G show multispecific antigen-binding molecules comprising engineered scFv C-terminal sequences, wherein modifications are presented in bold. FIG.2B shows a construct with an engineered scFv C-terminal amino acid sequence VTVSSPP (SEQ ID NO:63). FIG.2C shows a construct with an engineered scFv C- terminal amino acid sequence VTVKPGG (SEQ ID NO:64). FIG.2D shows a construct with an engineered scFv C-terminal amino acid sequence VTVKPGG (SEQ ID NO:64) and a V11K mutation. FIG.2E shows a construct with an engineered scFv C-terminal amino acid sequence VTVSSGGGGS (SEQ ID NO:65). FIG.2F shows a construct with an engineered scFv C- terminal amino acid sequence VTVNSS (SEQ ID NO:50). FIG.2G shows a construct with an engineered scFv C-terminal amino acid sequence VTVNST (SEQ ID NO:51). Although the two targeting moieties in each polypeptide in FIGs.2A-2G are depicted to comprise identical VH and VL domains, they may comprise distinct VH and VL domains, which may recognize the same epitope, different epitopes of the same antigen, or different antigens. [0016] FIG.3A-3C show that the amino acid modifications to the C-termini of the antigen- binding molecule, REGN9930, do not affect binding affinity to target cell surface antigens expressed on Raji and Jurkat cells. FIG.3A shows binding of antigen-binding molecules to Raji MAGEA4 (230-239) cells. FIG.3B shows binding of antigen-binding molecules to Raji MAGEA4 (286-294). FIG.3C shows binding the antigen-binding molecules to Jurkat cells. [0017] FIG.4 shows that the C-terminal amino acid modifications do not affect cytotoxic potency of antigen-binding molecule, REGN9930. [0018] FIGs.5A-5E show SEC-MS analysis of limited-LysC-digested antigen-binding molecule, REGN9930-VNSS (C-terminal sequence VTVNSS; SEQ ID NO:50). FIG.5A shows the native SEC-UV/MS analysis whereas boxes labeled (i)-(iv) indicate the fragments shown in mass analyses in FIGs 5B-5E. [0019] FIGs.6A-6D show SEC-MS analysis of limited-LysC-digested antigen-binding molecule, REGN9930-VNST (C-terminal sequence VTVNST; SEQ ID NO:51). FIG.6A shows the native SEC-UV/MS analysis, whereas boxes labeled (i)-(iii) indicate the fragments shown in mass analyses in FIGs 6B-6D. [0020] FIG.7 demonstrates the reduced preexisting ADA reactivity of antigen-binding molecules with C-terminal amino acid modifications relative to REGN9930.
6. DETAILED DESCRIPTION 6.1. Definitions [0021] About, Approximately: The terms “about”, “approximately” and the like are used throughout the specification in front of a number to show that the number is not necessarily exact (e.g., to account for fractions, variations in measurement accuracy and/or precision, timing, etc.). It should be understood that a disclosure of “about X” or “approximately X” where X is a number is also a disclosure of “X.” Thus, for example, a disclosure of an embodiment in which one sequence has “about X% sequence identity” to another sequence is also a disclosure of an embodiment in which the sequence has “X% sequence identity” to the other sequence. [0022] And, or: Unless indicated otherwise, an “or” conjunction is intended to be used in its correct sense as a Boolean logical operator, encompassing both the selection of features in the alternative (A or B, where the selection of A is mutually exclusive from B) and the selection of features in conjunction (A or B, where both A and B are selected). In some places in the text, the term “and/or” is used for the same purpose, which shall not be construed to imply that “or” is used with reference to mutually exclusive alternatives. [0023] Anti-drug Antibodies or ADAs: The terms “Anti-drug antibodies” or “ADAs” refer to antibodies that bind specifically to any region of a drug. In some embodiments, the “drug” is or comprises an antibody (as defined below). When the drug is or comprises an antibody, an anti- drug antibody may be an antibody or fragment thereof that specifically binds to a region of a drug antibody, e.g., the variable domain, the constant domains, or the glycostructure of the antibody. Such anti-drug antibodies may occur during drug therapy as an immunogenic reaction of a patient. An ADA may be one of any human immunoglobulin isotype (e.g., IgM, IgE, IgA, IgG, IgD) or IgG subclass (IgG1, 2, 3, and 4). ADAs include ADAs from any animal source, including, for example, human or non-human animal (e.g. veterinary) sources. [0024] Antibody: The term “antibody” as used herein refers to a polypeptide (or set of polypeptides) of the immunoglobulin family that is capable of binding an antigen non-covalently, reversibly and specifically. For example, a naturally occurring “antibody” of the IgG type is a tetramer comprising at least two heavy (H) chains and two light (L) chains inter-connected by disulfide bonds. Each heavy chain is comprised of a heavy chain variable region (abbreviated herein as VH) and a heavy chain constant region. The heavy chain constant region is comprised of three domains, CH1, CH2 and CH3. Each light chain is comprised of a light chain variable region (abbreviated herein as VL) and a light chain constant region. The light chain constant region is comprised of one domain (abbreviated herein as CL). The VH and VL regions can be
further subdivided into regions of hypervariability, termed complementarity determining regions (CDR), interspersed with regions that are more conserved, termed framework regions (FR). Each VH and VL is composed of three CDRs and four FRs arranged from amino-terminus to carboxy-terminus in the following order: FR1, CDR1, FR2, CDR2, FR3, CDR3, FR4. The variable regions of the heavy and light chains contain a binding domain that interacts with an antigen. The constant regions of the antibodies may mediate the binding of the immunoglobulin to host tissues or factors, including various cells of the immune system (e.g., effector cells) and the first component (Clq) of the classical complement system. The term “antibody” includes, but is not limited to, monoclonal antibodies, human antibodies, humanized antibodies, camelized antibodies, chimeric antibodies, bispecific or multispecific antibodies and anti-idiotypic (anti-id) antibodies. The antibodies can be of any isotype/class (e.g., IgG, IgE, IgM, IgD, IgA and IgY) or subclass (e.g., lgG1, lgG2, lgG3, lgG4, lgA1 and lgA2). Both the light and heavy chains are divided into regions of structural and functional homology. The terms “constant” and “variable” are used functionally. In this regard, it will be appreciated that the variable domains of both the light (VL) and heavy (VH) chain portions determine antigen recognition and specificity. Conversely, the constant domains of the light chain (CL) and the heavy chain (CH1, CH2 or CH3) confer important biological properties such as secretion, transplacental mobility, Fc receptor binding, complement binding, and the like. By convention the numbering of the constant region domains increases as they become more distal from the antigen-binding domain or amino-terminus of the antibody. The N-terminus is a variable region and at the C- terminus is a constant region; the CH3 and CL domains represent the carboxy-terminus of the heavy and light chain, respectively, of natural antibodies. For convenience, and unless the context dictates otherwise, the reference to an antibody also refers to antibody fragments as well as engineered antibodies that include non-naturally occurring antigen-binding domains and/or antigen-binding domains having non-native configurations. [0025] Antigen-binding Domain: The term “antigen-binding domain” or “ABD” as used herein refers to a portion of a binding molecule (e.g., a multispecific binding molecule, an antibody, or an antibody fragment) that has the ability to bind to a target molecule (e.g., an antigen) non- covalently, reversibly and specifically. Examples of an antibody fragment that can comprise an ABD include, but are not limited to, a single-chain Fv (scFv), a Fab fragment, a monovalent fragment consisting of the VL, VH, CL and CH1 domains; a F(ab)2 fragment, a bivalent fragment comprising two Fab fragments linked by a disulfide bridge at the hinge region; a Fd fragment consisting of the VH and CH1 domains; a Fv fragment consisting of the VL and VH domains of a single arm of an antibody; a dAb fragment (Ward et al., 1989, Nature 341:544-
546), which consists of a VH domain; and an isolated complementarity determining region (CDR). Thus, the term “antibody fragment” encompasses both proteolytic fragments of antibodies (e.g., Fab and F(ab)2 fragments) and engineered proteins comprising one or more portions of an antibody (e.g., an scFv). Antibody fragments can also be incorporated into single domain antibodies, maxibodies, minibodies, intrabodies, diabodies, triabodies, tetrabodies, v- NAR and bis-scFv (see, e.g., Hollinger and Hudson, 2005, Nature Biotechnology 23: 1126- 1136). [0026] Associated: The term “associated” in the context of an antigen-binding molecule refers to a functional relationship between two or more polypeptide chains or portions of a polypeptide chain. In particular, the term “associated” means that two or more polypeptides are associated with one another, e.g., non-covalently through molecular interactions or covalently through one or more disulfide bridges or chemical cross-linkages, so as to produce a functional antigen- binding molecule. Examples of associations that might be present in an antigen-binding molecule of the disclosure include (but are not limited to) associations between homodimeric or heterodimeric Fc domains in an Fc region, associations between VH and VL regions in a Fab or scFv, associations between CH1 and CL in a Fab, and associations between CH3 and CH3 in a domain substituted Fab. [0027] Bivalent: The term “bivalent” as used herein in reference to an antigen-binding molecule means an antigen-binding molecule that has two antigen-binding sites. In some embodiments, the two antigen-binding sites bind to the same epitope of the same target. In other embodiments, the two antigen-binding sites specifically bind to different epitopes of the same target molecule. In other embodiments, the two antigen-binding sites specifically bind to different epitopes of two different target molecules. Accordingly, a bivalent antigen-binding molecule can be monospecific or bispecific. [0028] Cancer Antigen: The term “cancer antigen” refers to a molecule (typically a protein, carbohydrate, lipid or some combination thereof) that is expressed on the surface of a cancer cell, either entirely or as a fragment (e.g., MHC/peptide), and which is useful for the preferential targeting of a pharmacological agent to the cancer cell. In some embodiments, a cancer antigen is a marker expressed by both normal cells and cancer cells, e.g., a lineage marker, e.g., CD19 on B cells. In some embodiments, a cancer antigen is a cell surface molecule that is overexpressed in a cancer cell in comparison to a normal cell, for instance, 1-fold over expression, 2-fold overexpression, 3-fold overexpression or more in comparison to a normal cell. In some embodiments, a cancer antigen is a cell surface molecule that is inappropriately
synthesized in the cancer cell, for instance, a molecule that contains deletions, additions or mutations in comparison to the molecule expressed on a normal cell. In some embodiments, a cancer antigen will be expressed exclusively on the cell surface of a cancer cell, entirely or as a fragment (e.g., MHC/peptide), and not synthesized or expressed on the surface of a normal cell. Accordingly, the term “cancer antigen” encompasses antigens that are specific to cancer cells, sometimes known in the art as tumor-specific antigens (“TSAs”). [0029] Complementarity Determining Region or CDR: The terms “complementarity determining region” or “CDR,” as used herein, refer to the sequences of amino acids within antibody variable regions which confer antigen specificity and binding affinity. In general, there are three CDRs in each heavy chain variable region (CDR-H1, CDR-H2, CDR-H3) and three CDRs in each light chain variable region (CDR1-L1, CDR-L2, CDR-L3). Exemplary conventions that can be used to identify the boundaries of CDRs include, e.g., the Kabat definition, the Chothia definition, the ABD definition and the IMGT definition. See, e.g., Kabat, 1991, “Sequences of Proteins of Immunological Interest,” National Institutes of Health, Bethesda, Md. (Kabat numbering scheme); Al-Lazikani et al., 1997, J. Mol. Biol.273:927-948 (Chothia numbering scheme); Martin et al., 1989, Proc. Natl. Acad. Sci. USA 86:9268-9272 (ABD numbering scheme); and Lefranc et al., 2003, Dev. Comp. Immunol.27:55-77 (IMGT numbering scheme). For example, for classic formats, under Kabat, the CDR amino acid residues in the heavy chain variable domain (VH) are numbered 31-35 (CDR-H1), 50-65 (CDR- H2), and 95- 102 (CDR-H3); and the CDR amino acid residues in the light chain variable domain (VL) are numbered 24-34 (CDR-L1), 50-56 (CDR-L2), and 89-97 (CDR-L3). Under Chothia, the CDR amino acids in the VH are numbered 26-32 (CDR-H1), 52-56 (CDR-H2), and 95-102 (CDR-H3); and the amino acid residues in VL are numbered 26-32 (CDR-L1), 50-52 (CDR-L2), and 91-96 (CDR-L3). By combining the CDR definitions of both Kabat and Chothia, the CDRs consist of amino acid residues 26-35 (CDR-H1), 50-65 (CDR-H2), and 95-102 (CDR-H3) in human VH and amino acid residues 24-34 (CDR-L1), 50-56 (CDR-L2), and 89-97 (CDR-L3) in human VL. Under IMGT the CDR amino acid residues in the VH are numbered approximately 26-35 (CDR- H1), 51-57 (CDR-H2) and 93-102 (CDR-H3), and the CDR amino acid residues in the VL are numbered approximately 27-32 (CDR-L1), 50-52 (CDR-L2), and 89-97 (CDR-L3) (numbering according to “Kabat”). Under IMGT, the CDR regions of an antibody can be determined using the program IMGT/DomainGap Align. Public databases are available for identifying CDR sequences within an antibody.
[0030] Dimerization Moiety: The term “dimerization moiety” refers to a polypeptide chain or an amino acid sequence capable of facilitating an association between two polypeptide chains to form a dimer. A first dimerization moiety can associate with an identical second dimerization moiety, or can associate with a second dimerization moiety that is different from the first. In some embodiments, a dimerization moiety is an Fc domain (e.g., an Fc domain of an IgG selected from the isotypes lgG1 , lgG2, lgG3, and lgG4, as well as any allotype within each isotype group), with the association of two Fc domains forming an Fc region. Thus, the Fc region can be homodimeric or heterodimeric. In certain embodiments, the dimerization moiety is an Fc domain or an amino acid sequence of 1 to about 200 amino acids in length containing at least one cysteine residue. In other embodiments, the dimerization moiety is a cysteine residue or a short cysteine-containing peptide. Other dimerization moieties include peptides or polypeptides comprising or consisting of a leucine zipper, a helix-loop motif, or a coiled-coil motif. [0031] EC50: The term “EC50” refers to the half maximal effective concentration of a molecule, such as an antigen-binding molecule, which induces a response halfway between the baseline and maximum after a specified exposure time. The EC50 essentially represents the concentration of a molecule where 50% of its maximal effect is observed. Thus, reduced or weaker binding is observed with an increased EC50, or half maximal effective concentration value. [0032] Epitope: An epitope, or antigenic determinant, is a portion of an antigen recognized by an antibody or other antigen-binding moiety as described herein. An epitope can be linear or conformational. As described herein, an epitope may be described as “comprising a sequence” of a particular region (e.g., protein domain) of an antigen. Such description includes both linear epitopes and conformational epitopes, and describes epitopes comprising at least one amino acid present in the particular region of the antigen. Such epitopes may or may not comprise additional amino acids which are not present in the particular region. [0033] Fab: The term “Fab” in the context of an antigen-binding molecule of the disclosure of the disclosure refers to a pair of polypeptide chains, the first comprising a variable heavy (VH) domain of an antibody N-terminal to a first constant domain (referred to herein as C1), and the second comprising variable light (VL) domain of an antibody N-terminal to a second constant domain (referred to herein as C2) capable of pairing with the first constant domain. In a native antibody, the VH is N-terminal to the first constant domain (CH1) of the heavy chain and the VL is N-terminal to the constant domain of the light chain (CL). The Fabs of the disclosure can be
arranged according to the native orientation or include domain substitutions or swaps that facilitate correct VH and VL pairings. For example, it is possible to replace the CH1 and CL domain pair in a Fab with a CH3-domain pair to facilitate correct modified Fab-chain pairing in heterodimeric molecules. It is also possible to reverse CH1 and CL, so that the CH1 is attached to VL and CL is attached to the VH, a configuration generally known as Crossmab (a type of “domain exchanged” arrangement). Alternatively, or in addition to, the use of substituted or swapped constant domains, correct chain pairing can be achieved by the use of universal light chains that can pair with both variable regions of a heterodimeric antigen-binding molecule of the disclosure. The term “Fab” encompasses single chain Fabs. [0034] Fc Domain and Fc Region: The term “Fc domain” refers to a portion of the heavy chain that pairs with the corresponding portion of another heavy chain. The term “Fc region” refers to the region of antibody-based binding molecules formed by association of two heavy chain Fc domains. The two Fc domains within the Fc region may be the same or different from one another. In a native antibody the Fc domains are typically identical, but one or both Fc domains might advantageously be modified to allow for heterodimerization, e.g., via a knob-in-hole interaction and/or for purification, e.g., via star mutations. [0035] Fv: The term “Fv” refers to the minimum antibody fragment derivable from an immunoglobulin that contains a complete target recognition and binding site. This region consists of a dimer of one heavy and one light chain variable domain in a tight, noncovalent association (VH-VL dimer). It is in this configuration that the three CDRs of each variable domain interact to define a target binding site on the surface of the VH-VL dimer. Often, the six CDRs confer target binding specificity to the antibody. However, in some instances even a single variable domain (or half of an Fv comprising only three CDRs specific for a target) can have the ability to recognize and bind target. The reference to a VH-VL dimer herein is not intended to convey any particular configuration. When present on a single polypeptide chain (e.g., a scFv), the VH and be N- terminal or C-terminal to the VL. [0036] Half Antibody: The term “half antibody” refers to a molecule that comprises at least one Fc domain and can associate with another molecule comprising an Fc through, e.g., a disulfide bridge or molecular interactions. A half antibody can be composed of one polypeptide chain or more than one polypeptide chains (e.g., the two polypeptide chains of a Fab). An example of a half antibody is a molecule comprising a heavy and light chain of an antibody (e.g., an IgG antibody). Another example of a half antibody is a molecule comprising a first polypeptide chain comprising a VL domain and a CL domain, and a second polypeptide chain comprising a VH
domain, a CH1 domain, a hinge domain, a CH2 domain, and a CH3 domain, wherein said VL and VH domains form an ABD. Yet another example of a half antibody is a polypeptide comprising an scFv domain, a CH2 domain and a CH3 domain. The term “half antibody” is intended for descriptive purposes only and does not connote a particular configuration or method of production. Descriptions of a half antibody as a “first” half antibody, a “second” half antibody, a “left” half antibody, a “right” half antibody or the like are merely for convenience and descriptive purposes. [0037] Host Cell or Recombinant Host Cell: The terms “host cell” and “recombinant host cell” as used herein refer to a cell that has been genetically engineered, e.g., through introduction of a heterologous nucleic acid. It should be understood that such terms are intended to refer not only to the particular subject cell but to the progeny of such a cell. Because certain modifications can occur in succeeding generations due to either mutation or environmental influences, such progeny may not, in fact, be identical to the parent cell, but are still included within the scope of the term “host cell” as used herein. A host cell can carry the heterologous nucleic acid transiently, e.g., on an extrachromosomal heterologous expression vector, or stably, e.g., through integration of the heterologous nucleic acid into the host cell genome. For purposes of expressing an antigen-binding molecule, a host cell can be a cell line of mammalian origin or mammalian-like characteristics, such as monkey kidney cells (COS, e.g., COS-1 , COS- 7), HEK293, baby hamster kidney (BHK, e.g., BHK21), Chinese hamster ovary (CHO), NSO, PerC6, BSC-1 , human hepatocellular carcinoma cells (e.g., Hep G2), SP2/0, HeLa, Madin- Darby bovine kidney (MDBK), myeloma and lymphoma cells, or derivatives and/or engineered variants thereof. The engineered variants include, e.g., glycan profile modified and/or site- specific integration site derivatives. [0038] Multispecific Antigen-binding Molecule: The term “multispecific antigen-binding molecule” (also “multispecific antigen-binding molecule” or “multispecific binding molecule”) as used herein refers to a molecule (e.g., assembly of multiple polypeptide chains) comprising two half antibodies and which specifically bind to at least two different epitopes (and in some instances three, four, or more different epitopes). A multispecific antigen-binding molecule of the disclosure may be bivalent, trivalent, tetravelant, or otherwise multivalent, and may be monospecific, bispecific, or otherwise multispecific. A multispecific antigen-binding molecule of the disclosure may specifically bind to epitopes on one, two, three, four, or more different antigens.
[0039] Multivalent: The term “multivalent” as used herein refers to an antigen-binding molecule comprising two or more ABDs, on one, two or more polypeptide chains. [0040] N-linked Glycosylation Site: The term “N-linked glycosylation site” as used herein refers to an asparagine (Asn; N) residue of a polypeptide which is capable of being glycosylated. Canonical “N-linked glycosylation sites” contain an Asn residue of a polypeptide within a three amino acid sequence Asn-Xaa-Ser or Asn-Xaa-Thr, where Xaa is any amino acid, typically other than proline. Such a three amino acid consensus sequence is often referred to herein as “NX[S/T]”. The term “N-linked glycosylation site” is used to describe such an asparagine residue whether or not an oligosaccharide is attached to the residue. A polypeptide described herein as comprising an N-linked glycosylation site at a particular location means that the Asn of the N-linked glycosylation site is at that particular location. For example, an antigen- binding molecule or a domain thereof (e.g., a VH domain) comprising an N-linked glycosylation site “within 10 amino acids of the C-terminus” describes an antigen-binding molecule or domain thereof having the amino acid sequence NX[S/T] such that the Asn residue is within 10 amino acids of the C-terminus of the antigen-binding molecule or domain (e.g., VH domain). [0041] Operably Linked: The term “operably linked” as used herein refers to a functional relationship between two or more regions of a polypeptide chain in which the two or more regions are linked so as to produce a functional polypeptide, or two or more nucleic acid sequences, e.g., to produce an in-frame fusion of two polypeptide components or to link a regulatory sequence to a coding sequence. In the context of a fusion protein or other polypeptide, the term “operably linked” means that two or more amino acid segments are linked so as to produce a functional polypeptide. In the context of a nucleic acid encoding a fusion protein, such as an antigen-binding molecule of the disclosure, “operably linked” means that the two nucleic acids are joined such that the amino acid sequences encoded by the two nucleic acids remain in-frame. In the context of transcriptional regulation, the term refers to the functional relationship of a transcriptional regulatory sequence to a transcribed sequence. For example, a promoter or enhancer sequence is operably linked to a coding sequence if it stimulates or modulates the transcription of the coding sequence in an appropriate host cell or other expression system. [0042] Polypeptide, Peptide and Protein: The terms “polypeptide”, “peptide” and “protein” are used interchangeably herein to refer to a polymer of amino acid residues. [0043] Single Chain Fab or scFab: The term “single chain Fab” or “scFab” as used herein refers an ABD comprising a VH domain, a CH1 domain, a VL domain, a CL domain and a linker.
In some embodiments, the foregoing domains and linker are arranged in one of the following orders in a N-terminal to C-terminal orientation: (a) VH-CH1-linker-VL-CL, (b) VL-CL-linker-VH- CH1, (c) VH-CL-linker-VL-CH1 or (d) VL-CH1-linker-VH-CL. Linkers are suitably noncleavable linkers of at least 30 amino acids, preferably between 32 and 50 amino acids. Single chain Fab fragments are typically stabilized via the natural disulfide bond between the CL domain and the CH1 domain. In addition, these single chain Fab molecules might be further stabilized by generation of interchain disulfide bonds via insertion of cysteine residues (e.g., at position 44 in the VH domain and position 100 in the VL domain according to Kabat numbering). [0044] Single Chain Fv or scFv: The term “single chain Fv” or “scFv” as used herein refers to a polypeptide chain comprising the VH and VL domains of antibody, where these domains are present in a single polypeptide chain. Preferably, the Fv polypeptide further comprises a polypeptide linker between the VH and VL domains which enables the scFv to form the desired structure for antigen-binding. For a review of scFv see Pluckthun in The Pharmacology of Monoclonal Antibodies, vol.113, Rosenburg and Moore eds. (1994), Springer-Verlag, New York, pp.269-315. [0045] Subject: The term “subject” includes human and non-human animals. Non-human animals include all vertebrates, e.g., mammals and non-mammals, such as non-human primates, sheep, dog, cow, chickens, amphibians, and reptiles. In certain embodiments, the subject is human. Except when noted, the terms “patient” or “subject” are used herein interchangeably. [0046] Tetravalent: The term “tetravalent” as used herein refers to refers to an antigen-binding molecule that has four antigen-binding sites. In certain embodiments, all four of the antigen- binding sites bind to the same epitope. In some embodiments, two of the antigen-binding sites bind to the same epitope and the other two antigen-binding sites bind to a different epitope, whether of the same target molecule or different target molecules. In other embodiments, two of the antigen-binding sites bind to the same epitope, a third antigen-binding site binds to a different epitope, and a fourth antigen-binding site binds to yet a different epitope. In still other embodiments, all four epitopes bind to different epitopes, whether of the same target molecule or different target molecules. Accordingly, a tetravalent antigen-binding molecule can be monospecific, bispecific, trispecific, or tetraspecific. [0047] Trivalent: The term “trivalent” as used herein refers to refers to an antigen-binding molecule that has three antigen-binding sites. In certain embodiments, all three of the antigen- binding sites bind to the same epitope. In some embodiments, two of the antigen-binding sites
bind to the same epitope and the other antigen-binding site binds to a different epitope, whether of the same target molecule or different target molecules. In other embodiments, all three of the antigen-binding sites bind to different epitopes, whether on the same target molecule or on any combination of two or more different target molecules. Accordingly, an antigen-binding molecule may be monospecific, bispecific, or trispecific. [0048] Tumor-associated Antigen, TAA: The term “tumor-associated antigen” or “TAA” refers to a molecule which, when targeted by a pharmacologic agent, is useful in the treatment of a tumor. TAAs include cancer antigens as well as molecules expressed by or present on non- tumor cells which are useful in the treatment of a tumor, including, for example, extracellular matrix (“ECM”) proteins, cell surface molecules of tumor or viral lymphocytes, T-cell antigens (“TCAs”), and immune checkpoint molecules. [0049] Universal Light Chain, ULC: The term “universal light chain” or “ULC” as used herein in the context of an antigen-biding domain refers to a light chain polypeptide capable of pairing with the heavy chain region of the antigen-biding domain and also capable of pairing with other heavy chain regions. Universal light chains are also known as “common light chains.” [0050] VH: The term “VH” refers to the variable region of an immunoglobulin heavy chain of an antibody, including the heavy chain of an Fv, scFv, dsFv or Fab. [0051] VL: The term “VL” refers to the variable region of an immunoglobulin light chain, including the light chain of an Fv, scFv, dsFv or Fab. 6.2. Engineered Heavy Chain Variable Domains [0052] The present disclosure provides heavy chain variable domains (VHs) whose primary amino acid sequences are modified to include an N-linked glycosylation site near the C- terminus (e.g., within 20, within 10, within 5, or within 3 amino acids of the C-terminus). [0053] The engineered VH domains can be incorporated into antigen-binding domains (ABDs) of antigen-binding molecules. The ABDs can be in any format, including formats such as scFvs (e.g., as described in Section 6.3.1, infra) and Fabs (e.g., as described in Section 6.3.2, infra), and can have various specificities, including TAA ABDs (e.g., as described in Section 6.4.2, infra) and T-cell engager (TCE) ABDs (e.g., as described in Section 6.4.3, infra). An exemplary scFv comprising an engineered VH of the disclosure is depicted in FIG.1A. An exemplary bivalent antigen-binding molecule comprising two scFvs comprising an engineered VH of the disclosure is depicted in FIG.1B. An exemplary multivalent antigen-binding molecule comprising an engineered VH of the disclosure is depicted in FIG.1C.
[0054] The engineered heavy chain variable domains (VHs; also “VH regions” or “VH domains”) of the present disclosure generally comprise (1) an N-linked glycosylation site within 20, 19, 18, 17, 16, 15, 14, 13, 12, 11, 10, 9, 8, 7, 6, 5, 4, or 3 amino acids of its C-terminus, or (2) the amino acid sequence X1X2X3X4X5NX6X7X8X9X10X11X12 (SEQ ID NO:1) at the C-terminus, where (a) X1, X2, X3, X4, and X5 are each independently selected from any amino acid; (b) X6 is selected from any amino acid, optionally wherein the amino acid is not proline; (c) X7 is S or T, and (d) X8, X9, X10,X11, and X12 are each independently selected from any amino acid and absent. Such VHs are often referred to herein as “engineered VHs” or “engineered VH domains” for convenience. [0055] As described in herein, it was found that modification of a VH in an antigen-binding domain, for example via amino acid substitution, insertion, or deletion, to incorporate an N- linked glycosylation site near the C-terminus of the VH (for example modification of the VH such that it comprises the sequence X1X2X3X4X5NX6[S/T]X8X9X10X11X12 at its C-terminus) results in reduced binding of the antigen-binding molecule to anti-drug antibodies (see, e.g., Section [0219] (Example 4)). Accordingly, the present disclosure provides antigen-binding molecules comprising such engineered VHs. Any antigen-binding molecule comprising a VH may be modified such that the VH has an N-linked glycosylation site near its C-terminus (e.g., the sequence X1X2X3X4X5NX6[S/T]X8X9X10X11X12 at its C-terminus). [0056] VHs can be further subdivided into regions of hypervariability, termed complementarity determining regions (CDR), interspersed with regions that are more conserved, termed framework regions (FR). Each VH is composed of three CDRs and four FRs arranged from amino-terminus to carboxy-terminus in the following order: FR1, CDR1, FR2, CDR2, FR3, CDR3, FR4. In some embodiments, an engineered VH of the disclosure comprises an N-linked glycosylation site within the FR4 region. In other embodiments, an engineered VH of the disclosure comprises an N-linked glycosylation site within the CDR3 region. [0057] In some embodiments, a VH of the disclosure comprises, at its C-terminus, the amino acid sequence X1X2X3X4X5NX6X7X8X9X10X11X12 (SEQ ID NO:1), wherein (a) X1, X2, X3, X4, and X5 are each independently selected from any amino acid; (b) X6 is selected from any amino acid, optionally wherein the amino acid is not proline; (c) X7 is S or T, and (d) X8, X9, X10,X11, and X12 are each independently selected from any amino acid and absent. Such a VH may comprise the sequence X1X2X3X4X5NX6[S/T]X8X9X10X11X12 by virtue of such a sequence being inserted at the C-terminus of the VH or due to one or more amino acid modifications made to the original VH sequence resulting in the sequence X1X2X3X4X5NX6[S/T]X8X9X10X11X12.
[0058] In some embodiments, X1 is A, C, D, E, F, G, H, I, K, L, M, N, P, Q, R, S, T, V, W, or Y. In some embodiments, X2 is A, C, D, E, F, G, H, I, K, L, M, N, P, Q, R, S, T, V, W, or Y. In some embodiments, X3 is A, C, D, E, F, G, H, I, K, L, M, N, P, Q, R, S, T, V, W, or Y. In some embodiments, X4 is A, C, D, E, F, G, H, I, K, L, M, N, P, Q, R, S, T, V, W, or Y. In some embodiments, X5 is A, C, D, E, F, G, H, I, K, L, M, N, P, Q, R, S, T, V, W, or Y. In some embodiments, X6 is (i) A, C, D, E, F, G, H, I, K, L, M, N, P, Q, R, S, T, V, W, or Y or (ii) A, C, D, E, F, G, H, I, K, L, M, N, Q, R, S, T, V, W, or Y. In some embodiments, X7 is S or T. In some embodiments, X8 is A, C, D, E, F, G, H, I, K, L, M, N, Q, R, S, T, V, W, Y, or absent. In some embodiments, X9 is A, C, D, E, F, G, H, I, K, L, M, N, Q, R, S, T, V, W, Y, or absent. In some embodiments, X10 is A, C, D, E, F, G, H, I, K, L, M, N, Q, R, S, T, V, W, Y, or absent. In some embodiments, X11 is A, C, D, E, F, G, H, I, K, L, M, N, Q, R, S, T, V, W, Y, or absent. In some embodiments, X12 is A, C, D, E, F, G, H, I, K, L, M, N, Q, R, S, T, V, W, Y, or absent. [0059] In certain aspects, one of X8, X9, X10, X11, and X12 is absent. In other aspects, two of X8, X9, X10, X11, and X12 are absent. In further aspects, three of X8, X9, X10, X11, and X12 are absent. In yet further aspects, four of X8, X9, X10, X11, and X12 are absent. In yet further aspects, all five of X8, X9, X10, X11, and X12 are absent. [0060] Table 1 provides a complete list of possible amino acids at the C-terminal region of an engineered VH of the present disclosure, where the VH comprises at its C-terminus the amino acid sequence X1X2X3X4X5NX6X7X8X9X10X11X12 (SEQ ID NO:1). The amino acid sequence X1X2X3X4X5NX6X7X8X9X10X11X12 at the C-terminus of an engineered VH of the present disclosure may comprise any combination of amino acids at each position as outlined in Table 1. Table 1
 Table 1 X X X X X X X X X X X X
Table 1 X X X X X X X X X X X X
 nt
nt
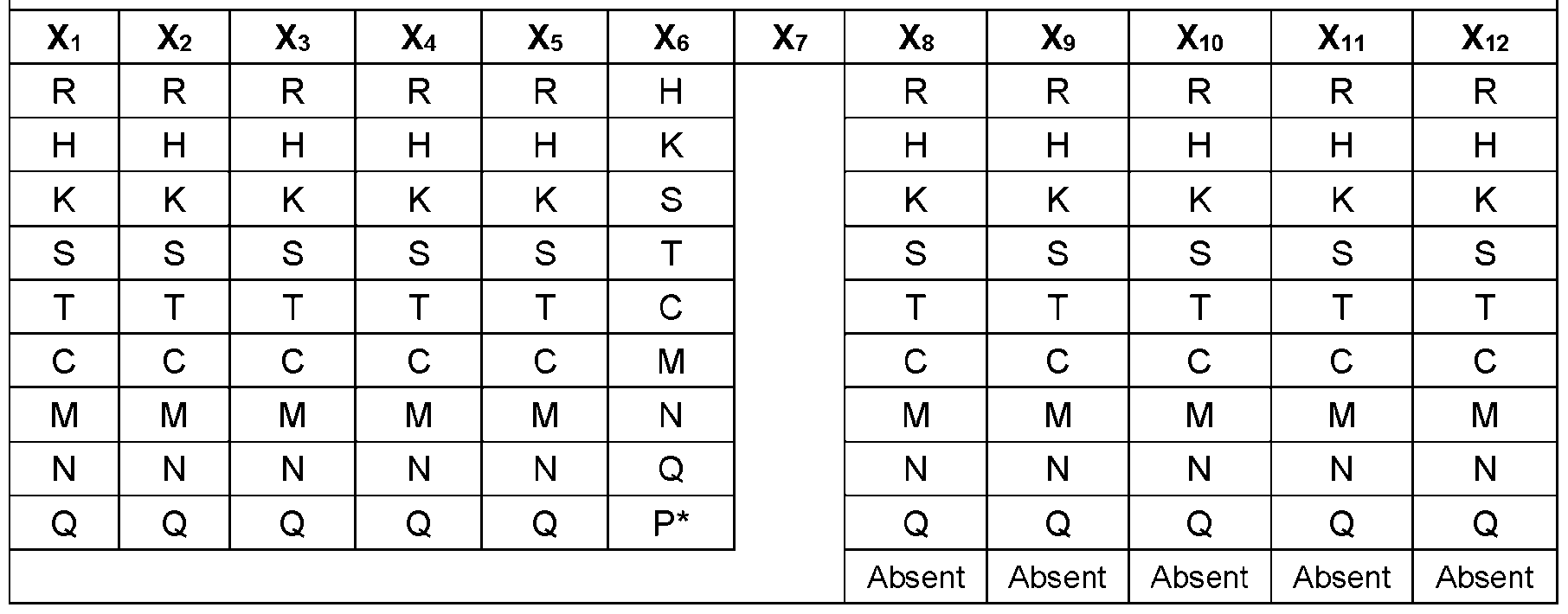 [0061] In certain embodiments, X6 is selected from any amino acid except proline. [0062] In particular embodiments, the VH comprises one of the following sequences at its C- terminus (where X1, X2, X3, X4, and X5 are each independently selected from any amino acid): X1X2X3X4X5NSS (SEQ ID NO:2), X1X2X3X4X5NST (SEQ ID NO:3), X1X2X3X4VNSS (SEQ ID NO:4), X1X2X3X4VNST (SEQ ID NO:5), X1X2X3TVNSS (SEQ ID NO:6), X1X2X3TVNST (SEQ ID NO:7), X1X2VTVNSS (SEQ ID NO:8), X1X2VTVNST (SEQ ID NO:9), X1TVTVNSS (SEQ ID NO:10), X1TVTVNST (SEQ ID NO:11), TTVTVNSS (SEQ ID NO:12), TTVTVNST (SEQ ID NO:13), X1X2X3X4X5NSSGGGG (SEQ ID NO:14), X1X2X3X4X5NSTGGGG (SEQ ID NO:15), X1X2X3X4VNSSGGGG (SEQ ID NO:16), X1X2X3X4VNSTGGGG (SEQ ID NO:17), X1X2X3TVNSSGGGG (SEQ ID NO:18), X1X2X3TVNSTGGGG (SEQ ID NO:19), X1X2VTVNSSGGGG (SEQ ID NO:20), X1X2VTVNSTGGGG (SEQ ID NO:21), X1TVTVNSSGGGG (SEQ ID NO:22), X1TVTVNSTGGGG (SEQ ID NO:23), TTVTVNSSGGGG (SEQ ID NO:24), TTVTVNSTGGGG (SEQ ID NO:25), X1X2X3X4X5NSSKPGG (SEQ ID NO:26), X1X2X3X4X5NSTKPGG (SEQ ID NO:27), X1X2X3X4VNSSKPGG (SEQ ID NO:28), X1X2X3X4VNSTKPGG (SEQ ID NO:29), X1X2X3VTNSSKPGG (SEQ ID NO:30), X1X2X3VTNSTKPGG (SEQ ID NO:31), X1X2VTVNSSKPGG (SEQ ID NO:32), X1X2VTVNSTKPGG (SEQ ID NO:33), X1TVTVNSSKPGG (SEQ ID NO:34), X1TVTVNSTKPGG (SEQ ID NO:35), TTVTVNSSKPGG (SEQ ID NO:36), TTVTVNSTKPGG (SEQ ID NO:37), X1X2X3X4X5NSSPP (SEQ ID NO:38), X1X2X3X4X5NSTPP (SEQ ID NO:39), X1X2X3X4VNSSPP (SEQ ID NO:40), X1X2X3X4VNSTPP (SEQ ID NO:41), X1X2X3TVNSSPP (SEQ ID NO:42), X1X2X3TVNSTPP (SEQ ID NO:43), X1X2VTVNSSPP (SEQ ID NO:44), X1X2VTVNSTPP (SEQ ID
NO:45), X1TVTVNSSPP (SEQ ID NO:46), X1TVTVNSTPP (SEQ ID NO:47), TTVTVNSSPP (SEQ ID NO:48), or TTVTVNSTPP (SEQ ID NO:49). [0063] In some embodiments, the VH comprises the sequence VTVNSS (SEQ ID NO:50) at its C-terminus. In some embodiments, the VH comprises the sequence VTVNST (SEQ ID NO:51) at its C-terminus. In some embodiments, the VH comprises the sequence VTVNSSGGGG (SEQ ID NO:52) at its C-terminus. In some embodiments, the VH comprises the sequence VTVNSTGGGG (SEQ ID NO:53) at its C-terminus. In some embodiments, the VH comprises the sequence VTVNSSKPGG (SEQ ID NO:54) at its C-terminus. In some embodiments, the VH comprises the sequence VTVNSTKPGG (SEQ ID NO:55) at its C-terminus. In some embodiments, the VH comprises the sequence VTVNSSPP (SEQ ID NO:56) at its C-terminus. In some embodiments, the VH comprises the sequence VTVNSTPP (SEQ ID NO:57) at its C- terminus. [0064] Without being bound by theory, a human VH is understood to commonly comprise, at its C-terminus, the sequence VTVSS (SEQ ID NO:62). Accordingly, aspects of the present disclosure comprise modifying a VH comprising the sequence VTVSS (SEQ ID NO:62) so as to comprise an N-linked glycosylation site at the C-terminus by virtue of insertion and/or substitution of one or two amino acids within this sequence. For example, in one embodiment, an engineered VH of the disclosure is generated by inserting an N residue between the third and fourth positions of this sequence, generating the sequence VTVNSS (SEQ ID NO:50) at the C-terminus. In another embodiment, an engineered VH of the disclosure is generated by inserting an N residue between the third and fourth positions of this sequence and also substituting a T residue at the fifth position, generating the sequence VTVNST (SEQ ID NO:51). [0065] In the context of the complete antigen-binding molecule, a sequence at the C-terminus of the VH may be further followed by any number of additional sequences, for example in situations where the VH is not at the C-terminus of the antigen-binding molecule. Alternatively, in some embodiments, such a sequence at the C-terminus of the VH is also at the C-terminus of the antigen-binding molecule. [0066] In addition to an N-glycosylation site, a VH of the present disclosure may optionally further comprise one or more additional amino acid modifications or sequences designed to reduce binding of anti-drug antibodies. Such additional modifications include, for example, the sequence PP at the C-terminus, the sequence KPGG (SEQ ID NO: 66) at the C-terminus, a V11K substitution (EU numbering), and a polyglycine sequence (e.g., G4S (SEQ ID NO: 67)) at
the C-terminus. A VH of the present disclosure may comprise any one or more of these additional modifications, including any combination thereof. [0067] The engineered VH domains of the disclosure can be incorporated into antigen-binding domains of antigen-binding molecules. Antigen-binding molecules comprising an engineered VH of the disclosure are described in, inter alia, Section 6.4 and include, for example antibodies of various formats such as multivalent and/or multispecific antigen-binding molecules (e.g., as described in Sections 6.4.1.1 and 6.4.1.2, infra), antibody fragments, scFvs, and chimeric antigen receptors. 6.3. Antigen-Binding Domains Comprising Engineered VH Domains [0068] In certain aspects, an engineered VH domain can be incorporated into an ABD, for example an ABD that is incorporated into an antigen-binding molecule as described herein. In one embodiment the ABD is an immunoglobulin molecule or fragment thereof, particularly an IgG class immunoglobulin molecule, more particularly an IgG1 or IgG4 immunoglobulin molecule. Antibody fragments include, but are not limited to, VH fragments, VL fragments, Fab fragments, F(ab')2 fragments, scFv fragments, Fv fragments, minibodies, diabodies, triabodies, and tetrabodies. 6.3.1. scFv [0069] In some embodiments, an engineered VH of the disclosure is incorporated into an scFv. A VH of an scFv described herein can be, in some embodiments, an engineered VH. An example scFv comprising an engineered VH is depicted in FIG.1A. [0070] Single chain Fv or “scFv” antibody fragments comprise the VH and VL domains of an antibody in a single polypeptide chain, are capable of being expressed as a single chain polypeptide, and retain the specificity of the intact antibodies from which they are derived. Generally, the scFv polypeptide further comprises a polypeptide linker between the VH and VL domain that enables the scFv to form the desired structure for target binding. Examples of linkers suitable for connecting the VH and VL chains of an scFV are the linkers identified in Section 6.4.4. [0071] Unless specified, as used herein an scFv may have the VL and VH variable regions in either order, e.g., with respect to the N-terminal and C-terminal ends of the polypeptide, the scFv may comprise VL-linker-VH or may comprise VH-linker-VL. For avoidance of doubt, where an scFv of the disclosure comprises an engineered VH comprising an N-linked glycosylation site (e.g., as a component of the amino acid sequence of SEQ ID NO:1) within 3 or more (e.g., 3, 4,
5, 6, 7, 8, 9, 10, 15, or 20) amino acids of the C-terminus of the VH, such N-linked glycosylation site may be within 3 or more amino acids of the C-terminus of the scFv itself (e.g., where the scFv comprises VL-linker-VH) or simply within 3 or more amino acids of the C-terminus of the VH but a further distance from the C-terminus of the scFv (e.g., where the scFv comprises VH- linker-VL). [0072] The scFv can comprise VH and VL sequences from any suitable species, such as murine, human or humanized VH and VL sequences. [0073] To create an scFv-encoding nucleic acid, the VH and VL-encoding DNA fragments are operably linked to another fragment encoding a linker, e.g., encoding any of the linkers described in Section 6.4.4, for example a repeat of a sequence containing the amino acids glycine and serine, such that the VH and VL sequences can be expressed as a contiguous single-chain protein with the VL and VH regions joined by the flexible linker (see, e.g., Bird et al., 1988, Science 242:423-426; Huston et al., 1988, Proc. Natl. Acad. Sci. USA 85:5879-5883; McCafferty et al., 1990, Nature 348:552-554). 6.3.2. Fab [0074] In some embodiments, an engineered VH of the disclosure is incorporated into a Fab. [0075] Fab domains were traditionally produced by proteolytic cleavage of immunoglobulin molecules using enzymes such as papain. The Fab domains can comprise constant domain and variable region sequences from any suitable species, and thus can be murine, chimeric, human or humanized. [0076] Fab domains typically comprise a CH1 domain attached to a VH domain which pairs with a CL domain attached to a VL domain. In a wild-type immunoglobulin, the VH domain is paired with the VL domain to constitute the Fv region, and the CH1 domain is paired with the CL domain to further stabilize the binding site. A disulfide bond between the two constant domains can further stabilize the Fab domain. [0077] For the antigen-binding molecules of the disclosure, particularly when the light chains of the ABDs are not common or universal light chains, it is advantageous to use Fab heterodimerization strategies to permit the correct association of Fab domains belonging to the same ABD and minimize aberrant pairing of Fab domains belonging to different ABDs. For example, the Fab heterodimerization strategies shown in Table H below can be used:
TABLE H Fab Heterodimerization Strategies . . bs 4
[0061] In certain embodiments, X6 is selected from any amino acid except proline. [0062] In particular embodiments, the VH comprises one of the following sequences at its C- terminus (where X1, X2, X3, X4, and X5 are each independently selected from any amino acid): X1X2X3X4X5NSS (SEQ ID NO:2), X1X2X3X4X5NST (SEQ ID NO:3), X1X2X3X4VNSS (SEQ ID NO:4), X1X2X3X4VNST (SEQ ID NO:5), X1X2X3TVNSS (SEQ ID NO:6), X1X2X3TVNST (SEQ ID NO:7), X1X2VTVNSS (SEQ ID NO:8), X1X2VTVNST (SEQ ID NO:9), X1TVTVNSS (SEQ ID NO:10), X1TVTVNST (SEQ ID NO:11), TTVTVNSS (SEQ ID NO:12), TTVTVNST (SEQ ID NO:13), X1X2X3X4X5NSSGGGG (SEQ ID NO:14), X1X2X3X4X5NSTGGGG (SEQ ID NO:15), X1X2X3X4VNSSGGGG (SEQ ID NO:16), X1X2X3X4VNSTGGGG (SEQ ID NO:17), X1X2X3TVNSSGGGG (SEQ ID NO:18), X1X2X3TVNSTGGGG (SEQ ID NO:19), X1X2VTVNSSGGGG (SEQ ID NO:20), X1X2VTVNSTGGGG (SEQ ID NO:21), X1TVTVNSSGGGG (SEQ ID NO:22), X1TVTVNSTGGGG (SEQ ID NO:23), TTVTVNSSGGGG (SEQ ID NO:24), TTVTVNSTGGGG (SEQ ID NO:25), X1X2X3X4X5NSSKPGG (SEQ ID NO:26), X1X2X3X4X5NSTKPGG (SEQ ID NO:27), X1X2X3X4VNSSKPGG (SEQ ID NO:28), X1X2X3X4VNSTKPGG (SEQ ID NO:29), X1X2X3VTNSSKPGG (SEQ ID NO:30), X1X2X3VTNSTKPGG (SEQ ID NO:31), X1X2VTVNSSKPGG (SEQ ID NO:32), X1X2VTVNSTKPGG (SEQ ID NO:33), X1TVTVNSSKPGG (SEQ ID NO:34), X1TVTVNSTKPGG (SEQ ID NO:35), TTVTVNSSKPGG (SEQ ID NO:36), TTVTVNSTKPGG (SEQ ID NO:37), X1X2X3X4X5NSSPP (SEQ ID NO:38), X1X2X3X4X5NSTPP (SEQ ID NO:39), X1X2X3X4VNSSPP (SEQ ID NO:40), X1X2X3X4VNSTPP (SEQ ID NO:41), X1X2X3TVNSSPP (SEQ ID NO:42), X1X2X3TVNSTPP (SEQ ID NO:43), X1X2VTVNSSPP (SEQ ID NO:44), X1X2VTVNSTPP (SEQ ID
NO:45), X1TVTVNSSPP (SEQ ID NO:46), X1TVTVNSTPP (SEQ ID NO:47), TTVTVNSSPP (SEQ ID NO:48), or TTVTVNSTPP (SEQ ID NO:49). [0063] In some embodiments, the VH comprises the sequence VTVNSS (SEQ ID NO:50) at its C-terminus. In some embodiments, the VH comprises the sequence VTVNST (SEQ ID NO:51) at its C-terminus. In some embodiments, the VH comprises the sequence VTVNSSGGGG (SEQ ID NO:52) at its C-terminus. In some embodiments, the VH comprises the sequence VTVNSTGGGG (SEQ ID NO:53) at its C-terminus. In some embodiments, the VH comprises the sequence VTVNSSKPGG (SEQ ID NO:54) at its C-terminus. In some embodiments, the VH comprises the sequence VTVNSTKPGG (SEQ ID NO:55) at its C-terminus. In some embodiments, the VH comprises the sequence VTVNSSPP (SEQ ID NO:56) at its C-terminus. In some embodiments, the VH comprises the sequence VTVNSTPP (SEQ ID NO:57) at its C- terminus. [0064] Without being bound by theory, a human VH is understood to commonly comprise, at its C-terminus, the sequence VTVSS (SEQ ID NO:62). Accordingly, aspects of the present disclosure comprise modifying a VH comprising the sequence VTVSS (SEQ ID NO:62) so as to comprise an N-linked glycosylation site at the C-terminus by virtue of insertion and/or substitution of one or two amino acids within this sequence. For example, in one embodiment, an engineered VH of the disclosure is generated by inserting an N residue between the third and fourth positions of this sequence, generating the sequence VTVNSS (SEQ ID NO:50) at the C-terminus. In another embodiment, an engineered VH of the disclosure is generated by inserting an N residue between the third and fourth positions of this sequence and also substituting a T residue at the fifth position, generating the sequence VTVNST (SEQ ID NO:51). [0065] In the context of the complete antigen-binding molecule, a sequence at the C-terminus of the VH may be further followed by any number of additional sequences, for example in situations where the VH is not at the C-terminus of the antigen-binding molecule. Alternatively, in some embodiments, such a sequence at the C-terminus of the VH is also at the C-terminus of the antigen-binding molecule. [0066] In addition to an N-glycosylation site, a VH of the present disclosure may optionally further comprise one or more additional amino acid modifications or sequences designed to reduce binding of anti-drug antibodies. Such additional modifications include, for example, the sequence PP at the C-terminus, the sequence KPGG (SEQ ID NO: 66) at the C-terminus, a V11K substitution (EU numbering), and a polyglycine sequence (e.g., G4S (SEQ ID NO: 67)) at
the C-terminus. A VH of the present disclosure may comprise any one or more of these additional modifications, including any combination thereof. [0067] The engineered VH domains of the disclosure can be incorporated into antigen-binding domains of antigen-binding molecules. Antigen-binding molecules comprising an engineered VH of the disclosure are described in, inter alia, Section 6.4 and include, for example antibodies of various formats such as multivalent and/or multispecific antigen-binding molecules (e.g., as described in Sections 6.4.1.1 and 6.4.1.2, infra), antibody fragments, scFvs, and chimeric antigen receptors. 6.3. Antigen-Binding Domains Comprising Engineered VH Domains [0068] In certain aspects, an engineered VH domain can be incorporated into an ABD, for example an ABD that is incorporated into an antigen-binding molecule as described herein. In one embodiment the ABD is an immunoglobulin molecule or fragment thereof, particularly an IgG class immunoglobulin molecule, more particularly an IgG1 or IgG4 immunoglobulin molecule. Antibody fragments include, but are not limited to, VH fragments, VL fragments, Fab fragments, F(ab')2 fragments, scFv fragments, Fv fragments, minibodies, diabodies, triabodies, and tetrabodies. 6.3.1. scFv [0069] In some embodiments, an engineered VH of the disclosure is incorporated into an scFv. A VH of an scFv described herein can be, in some embodiments, an engineered VH. An example scFv comprising an engineered VH is depicted in FIG.1A. [0070] Single chain Fv or “scFv” antibody fragments comprise the VH and VL domains of an antibody in a single polypeptide chain, are capable of being expressed as a single chain polypeptide, and retain the specificity of the intact antibodies from which they are derived. Generally, the scFv polypeptide further comprises a polypeptide linker between the VH and VL domain that enables the scFv to form the desired structure for target binding. Examples of linkers suitable for connecting the VH and VL chains of an scFV are the linkers identified in Section 6.4.4. [0071] Unless specified, as used herein an scFv may have the VL and VH variable regions in either order, e.g., with respect to the N-terminal and C-terminal ends of the polypeptide, the scFv may comprise VL-linker-VH or may comprise VH-linker-VL. For avoidance of doubt, where an scFv of the disclosure comprises an engineered VH comprising an N-linked glycosylation site (e.g., as a component of the amino acid sequence of SEQ ID NO:1) within 3 or more (e.g., 3, 4,
5, 6, 7, 8, 9, 10, 15, or 20) amino acids of the C-terminus of the VH, such N-linked glycosylation site may be within 3 or more amino acids of the C-terminus of the scFv itself (e.g., where the scFv comprises VL-linker-VH) or simply within 3 or more amino acids of the C-terminus of the VH but a further distance from the C-terminus of the scFv (e.g., where the scFv comprises VH- linker-VL). [0072] The scFv can comprise VH and VL sequences from any suitable species, such as murine, human or humanized VH and VL sequences. [0073] To create an scFv-encoding nucleic acid, the VH and VL-encoding DNA fragments are operably linked to another fragment encoding a linker, e.g., encoding any of the linkers described in Section 6.4.4, for example a repeat of a sequence containing the amino acids glycine and serine, such that the VH and VL sequences can be expressed as a contiguous single-chain protein with the VL and VH regions joined by the flexible linker (see, e.g., Bird et al., 1988, Science 242:423-426; Huston et al., 1988, Proc. Natl. Acad. Sci. USA 85:5879-5883; McCafferty et al., 1990, Nature 348:552-554). 6.3.2. Fab [0074] In some embodiments, an engineered VH of the disclosure is incorporated into a Fab. [0075] Fab domains were traditionally produced by proteolytic cleavage of immunoglobulin molecules using enzymes such as papain. The Fab domains can comprise constant domain and variable region sequences from any suitable species, and thus can be murine, chimeric, human or humanized. [0076] Fab domains typically comprise a CH1 domain attached to a VH domain which pairs with a CL domain attached to a VL domain. In a wild-type immunoglobulin, the VH domain is paired with the VL domain to constitute the Fv region, and the CH1 domain is paired with the CL domain to further stabilize the binding site. A disulfide bond between the two constant domains can further stabilize the Fab domain. [0077] For the antigen-binding molecules of the disclosure, particularly when the light chains of the ABDs are not common or universal light chains, it is advantageous to use Fab heterodimerization strategies to permit the correct association of Fab domains belonging to the same ABD and minimize aberrant pairing of Fab domains belonging to different ABDs. For example, the Fab heterodimerization strategies shown in Table H below can be used:
TABLE H Fab Heterodimerization Strategies . . bs 4
 [0078] Accordingly, in certain embodiments, correct association between the two polypeptides of a Fab is promoted by exchanging the VL and VH domains of the Fab for each other or exchanging the CH1 and CL domains for each other, e.g., as described in WO 2009/080251. [0079] Correct Fab pairing can also be promoted by introducing one or more amino acid modifications in the CH1 domain and one or more amino acid modifications in the CL domain of the Fab and/or one or more amino acid modifications in the VH domain and one or more amino acid modifications in the VL domain. The amino acids that are modified are typically part of the VH:VL and CH1:CL interface such that the Fab components preferentially pair with each other rather than with components of other Fabs. [0080] In one embodiment, the one or more amino acid modifications are limited to the conserved framework residues of the variable (VH, VL) and constant (CH1, CL) domains as indicated by the Kabat numbering of residues. Almagro, 2008, Frontiers In Bioscience 13:1619-
1633 provides a definition of the framework residues on the basis of Kabat, Chothia, and IMGT numbering schemes. [0081] In one embodiment, the modifications introduced in the VH and CH1 and/or VL and CL domains are complementary to each other. Complementarity at the heavy and light chain interface can be achieved on the basis of steric and hydrophobic contacts, electrostatic/charge interactions or a combination of the variety of interactions. The complementarity between protein surfaces is broadly described in the literature in terms of lock and key fit, knob into hole, protrusion and cavity, donor and acceptor etc., all implying the nature of structural and chemical match between the two interacting surfaces. [0082] In one embodiment, the one or more introduced modifications introduce a new hydrogen bond across the interface of the Fab components. In one embodiment, the one or more introduced modifications introduce a new salt bridge across the interface of the Fab components. Exemplary substitutions are described in WO 2014/150973 and WO 2014/082179, the contents of which are hereby incorporated by reference. [0083] In some embodiments, the Fab domain comprises a 192E substitution in the CH1 domain and 114A and 137K substitutions in the CL domain, which introduces a salt-bridge between the CH1 and CL domains (see, e.g., Golay et al., 2016, J Immunol 196:3199-211). [0084] In some embodiments, the Fab domain comprises a 143Q and 188V substitutions in the CH1 domain and 113T and 176V substitutions in the CL domain, which serves to swap hydrophobic and polar regions of contact between the CH1 and CL domain (see, e.g., Golay et al., 2016, J Immunol 196:3199-211). [0085] In some embodiments, the Fab domain can comprise modifications in some or all of the VH, CH1, VL, CL domains to introduce orthogonal Fab interfaces which promote correct assembly of Fab domains (Lewis et al., 2014 Nature Biotechnology 32:191-198). In an embodiment, 39K, 62E modifications are introduced in the VH domain, H172A, F174G modifications are introduced in the CH1 domain, 1 R, 38D, (36F) modifications are introduced in the VL domain, and L135Y, S176W modifications are introduced in the CL domain. In another embodiment, a 39Y modification is introduced in the VH domain and a 38R modification is introduced in the VL domain. [0086] Fab domains can also be modified to replace the native CH1:CL disulfide bond with an engineered disulfide bond, thereby increasing the efficiency of Fab component pairing. For
example, an engineered disulfide bond can be introduced by introducing a 126C in the CH1 domain and a 121 C in the CL domain (see, e.g., Mazor et al., 2015, MAbs 7:377-89). [0087] Fab domains can also be modified by replacing the CH1 domain and CL domain with alternative domains that promote correct assembly. For example, Wu et al., 2015, MAbs 7:364- 76, describes substituting the CH1 domain with the constant domain of the T cell receptor and substituting the CL domain with the b domain of the T cell receptor, and pairing these domain replacements with an additional charge-charge interaction between the VL and VH domains by introducing a 38D modification in the VL domain and a 39K modification in the VH domain. [0088] In lieu of, or in addition to, the use of Fab heterodimerization strategies to promote correct VH-VL pairings, the VL of common light chain (also referred to as a universal light chain) can be used for each unique ABD in the antigen-binding molecules of the disclosure. In various embodiments, employing a common light chain as described herein reduces the number of inappropriate species in the antigen-binding molecules as compared to employing original cognate VLs. In various embodiments, the VL domains of ABDs are identified from monospecific antibodies comprising a common light chain. In various embodiments, the VH regions of the ABDs in the antigen-binding molecules comprise human heavy chain variable gene segments that are rearranged in vivo within mouse B cells that have been previously engineered to express a limited human light chain repertoire, or a single human light chain, cognate with human heavy chains and, in response to exposure with an antigen of interest, generate an antibody repertoire containing a plurality of human VHs that are cognate with one or one of two possible human VLs, wherein the antibody repertoire specific for the antigen of interest. Common light chains are those derived from a rearranged human Vκ1-39Jκ5 sequence or a rearranged human Vκ3-20Jκ1 sequence, and include somatically mutated (e.g., affinity matured) versions. See, for example, U.S. Patent No.10,412,940. 6.4. Antigen-Binding Molecules Comprising Engineered VH Domains [0089] The present disclosure provides antigen-binding molecules comprising an engineered VH of the disclosure. [0090] In some embodiments, the antigen-binding molecule is an antibody. The antibody can be any type of engineered antibody, including chimeric, humanized, veneered, or human antibodies. The antibody can be a monoclonal antibody or a genetically engineered polyclonal antibody. Antibodies contemplated herein include both traditional antibodies as well as antibody- like molecules known in the art, including but not limited to nanobodies, diabodies, minibodies,
antibody fragments, and other “alternative format” antibodies (e.g., as described in Spiess et al., 2015, Mol Immunol, 67(2 Pt A):95-106, incorporated herein by reference). An antibody may include an engineered VH of the disclosure as a component of one or more ABDs, including, for example, as a component of a Fab or as a component of an scFv. [0091] Accordingly, in some embodiments, disclosed herein is an antibody comprising an engineered VH, the engineered VH comprising (1) an N-linked glycosylation site within 20, 19, 18, 17, 16, 15, 14, 13, 12, 11, 10, 9, 8, 7, 6, 5, 4, or 3 amino acids of its C-terminus, or (2) the amino acid sequence X1X2X3X4X5NX6X7X8X9X10X11X12 (SEQ ID NO:1) at its C-terminus, where (a) X1, X2, X3, X4, and X5 are each independently selected from any amino acid; (b) X6 is selected from any amino acid, optionally wherein the amino acid is not proline; (c) X7 is S or T, and (d) X8, X9, X10,X11, and X12 are each independently selected from any amino acid and absent. [0092] In some embodiments, an antibody of the present disclosure comprises a polypeptide comprising: (a) a VH comprising (1) an N-linked glycosylation site within 20, 19, 18, 17, 16, 15, 14, 13, 12, 11, 10, 9, 8, 7, 6, 5, 4, or 3 amino acids of its C-terminus, or (2) the amino acid sequence X1X2X3X4X5NX6X7X8X9X10X11X12 (SEQ ID NO:1) at its C-terminus; and (b) an Fc domain. The antibody may further comprise an additional polypeptide comprising an additional VH and an additional Fc domain. In some embodiments, the additional VH comprises (1) an N- linked glycosylation site within 20, 19, 18, 17, 16, 15, 14, 13, 12, 11, 10, 9, 8, 7, 6, 5, 4, or 3 amino acids of its C-terminus, or (2) the amino acid sequence X1X2X3X4X5NX6X7X8X9X10X11X12 (SEQ ID NO:1) at its C-terminus. An antibody of the present disclosure may comprise any number of VHs (e.g., 1, 2, 3, 4, or more VHs), where at least one VH is an engineered VH described herein and where any number of the additional VHs may or may not be an engineered VH as described herein. 6.4.1.1. Multivalent Antigen-Binding Molecules [0093] Antigen-binding molecules of the present disclosure include multivalent molecules having one or more engineered VH domains. Various multivalent antigen-binding molecule formats are recognized in the art and contemplated herein, including bivalent, trivalent, and tetravalent formats. Certain example multivalent antigen-binding molecules are described in more detail below. [0094] In some embodiments, the antigen-binding molecule comprising an engineered VH is a multivalent antigen-binding molecule comprising two or more scFvs connected via a linker, where at least one of the scFvs comprises an engineered VH (as described in, e.g., Section 6.2
or as defined in numbered embodiments 1 to 34). Accordingly, disclosed herein, in some embodiments, is an antigen-binding molecule comprising: (a) a first scFv, (b) a linker, and (c) a second scFv, where the first and/or the second scFv comprise an engineered VH. In some embodiments, the first scFv comprises the engineered VH. In some embodiments, the second scFv comprises the engineered VH. In some embodiments, both the first and second scFvs comprise an engineered VH. In some embodiments, the antigen-binding molecule further comprises a third scFv, which optionally comprises an engineered VH. An example of such a multivalent antigen-binding molecule comprising two scFvs is depicted in FIG.1B. [0095] In some embodiments, the antigen-binding molecule comprising an engineered VH is a multivalent antigen-binding molecule comprising three or more antigen-binding domains, where at least one of the antigen-binding domains comprises an engineered VH. Accordingly, disclosed herein, in some embodiments, is an antigen-binding molecule comprising: (a) a first polypeptide chain comprising, from N-terminus to C-terminus (i) a first antigen-binding domain (e.g., a Fab), (ii) a first dimerization moiety, and (iii) a second antigen-binding domain (e.g., an scFv), the second antigen-binding domain comprising an engineered VH; and (b) a second polypeptide chain comprising, from N-terminus to C-terminus (i) a third antigen-binding domain (e.g., a Fab), and (ii) a second dimerization moiety. In some embodiments, the second polypeptide further comprises a fourth antigen-binding domain (e.g., an scFv) C-terminal to the second dimerization moiety, where the fourth antigen-binding domain comprises an engineered VH. Examples of such multivalent antigen-binding molecules are depicted in FIG.1C and FIGS. 2A-2G. 6.4.1.2. Multispecific Antigen-Binding Molecules [0096] Antigen-binding molecules of the present disclosure include multispecific molecules having one or more engineered VH domains. Various multispecific antigen-binding molecules are recognized in the art and contemplated herein, including bispecific, trispecific, and tetraspecific antigen-binding molecules. Certain example multispecific antigen-binding molecules are described in more detail below. [0097] In some embodiments, an antigen-binding molecule comprising an engineered VH of the disclosure is a bispecific T-cell engager. “Bispecific T-cell engager” describes a molecule comprising two scFvs, connected via a linker, where the first scFv comprises a first ABD that binds to a T cell antigen (e.g., a TCE ABD as described in Section 6.4.3) and the second scFv comprises a second ABD that binds to a tumor-associated antigen (e.g., a TAA ABD as described in Section 6.4.2). In some embodiments, the TCE ABD comprises the engineered VH.
In some embodiments, the TAA ABD comprises the engineered VH. In some embodiments, both the TCE ABD and the TAA ABD comprise an engineered VH. [0098] In some embodiments, an antigen-binding molecule comprising an engineered VH of the disclosure is a multivalent antigen-binding molecule comprising: (a) a first polypeptide chain comprising, from N-terminus to C-terminus (i) a first TCE ABD or TAA ABD, (ii) a first dimerization moiety, and (iii) a second TCE ABD or TAA ABD; and (b) a second polypeptide chain comprising, from N-terminus to C-terminus (i) a third TCE ABD or TAA ABD, and (ii) a second dimerization domain, where the second TCE ABD or TAA ABD comprises an engineered VH. In some embodiments, the second TCE ABD or TAA ABD is an scFv. In some embodiments, the second polypeptide further comprises a TCE ABD or TAA ABD C-terminal to the second dimerization moiety, where the fourth TCE ABD or TAA ABD comprises an engineered VH. In some embodiments, the fourth TCE ABD or TAA ABD is an scFv. [0099] In some embodiments, the first ABD is a TCE ABD (e.g., as described in Section 6.4.3). In some embodiments, the first ABD is a TAA ABD (e.g., as described in Section 6.4.2). In some embodiments, the second ABD is a TCE ABD (e.g., as described in Section 6.4.3). In some embodiments, the second ABD is a TAA ABD (e.g., as described in Section 6.4.2). In some embodiments, the third ABD is a TCE ABD (e.g., as described in Section 6.4.3). In some embodiments, the third ABD is a TAA ABD (e.g., as described in Section 6.4.2). In some embodiments, the fourth ABD is a TCE ABD (e.g., as described in Section 6.4.3). In some embodiments, the fourth ABD is a TAA ABD (e.g., as described in Section 6.4.2). 6.4.2. Tumor-associated Antigen ABDs [0100] In some embodiments, the antigen-binding molecules of the disclosure comprise at least one ABD that binds specifically to a tumor-associated antigen (TAA), referred to herein as a “TAA ABD”. Examples of TAAs include cancer antigens, extracellular matrix (“ECM”) proteins, tumor reactive lymphocyte antigens, cell surface molecules of tumor or viral lymphocytes, T-cell antigens (“TCAs”), and immune checkpoint molecules. The skilled artisan would recognize that the foregoing categories of target molecules are not mutually exclusive and thus a given target molecule may fall into more than one of the foregoing categories of target molecules. For example, some molecules may be considered both TCAs and immune checkpoint molecules. Preferably, the TAA is a human antigen. The antigen may or may not be present on normal cells. Certain aspects are directed to antigen-binding molecules comprising at least one ABD that binds specifically to a TAA. In certain embodiments, the TAA is preferentially expressed or
upregulated on tumor cells as compared to normal cells. In other embodiments, the TAA is a lineage marker. [0101] It is anticipated that any type of tumor and any type of TAA may be targeted by the antigen-binding molecules of the disclosure. Exemplary types of cancers that may be targeted include acute lymphoblastic leukemia, acute myelogenous leukemia, biliary cancer, B-cell leukemia, B-cell lymphoma, biliary cancer, bone cancer, brain cancer, breast cancer, triple- negative breast cancer, cervical cancer, Burkitt lymphoma, chronic lymphocytic leukemia, chronic myelogenous leukemia, colorectal cancer, endometrial cancer, esophageal cancer, gall bladder cancer, gastric cancer, gastrointestinal tract cancer, glioma, hairy cell leukemia, head and neck cancer, Hodgkin’s lymphoma, liver cancer, lung cancer, medullary thyroid cancer, melanoma, multiple myeloma, ovarian cancer, non-Hodgkin’s lymphoma, pancreatic cancer, prostate cancer, pulmonary tract cancer, renal cancer, sarcoma, skin cancer, testicular cancer, urothelial cancer, and other urinary bladder cancers. However, the skilled artisan will realize that TAAs are known for virtually any type of cancer. [0102] Non-limiting examples of ECM antigens include syndecan, 27enzalkoni, integrins, osteopontin, link, cadherins, laminin, laminin type EGF, lectin, fibronectin, notch, nectin (e.g., nectin-4), tenascin, collagen (e.g., collagen type X) and matrixin. [0103] Other target molecules are cell surface molecules of tumor or viral lymphocytes, for example T-cell co-stimulatory proteins such as CD27, CD28, 4-1BB (CD137), OX40, CD30, CD40, ICOS, lymphocyte function-associated antigen-1 (LFA-1), CD2, CD7, LIGHT, NKG2C, and B7-H3. [0104] In particular embodiments, the target molecules are immune checkpoint molecules, for example CTLA-4, PD1, PDL1, PDL2, B7-H3, B7-H4, BTLA, HVEM, TIM3, GAL9, LAG3, VISTA, KIR, 2B4, CD160, CGEN-15049, CHK1, CHK2. In particular embodiments, the target molecule is PD1. In other embodiments, the target molecule is LAG3. In some embodiments, where the target molecule is an immune checkpoint molecules, the TAA ABD is non-blocking or poorly- blocking of ligand-receptor binding. Examples of non-blocking or poorly-blocking anti-PD1 antibodies includes antibodies having VH/VL amino acid sequences of SEQ ID Nos: 2/10 of PCT Pub. No. WO2015/112800A1; SEQ ID Nos: 16/17 of US Patent No.11,034,765 B2; SEQ ID Nos.164/178, 165/179, 166/180, 167/181, 168/182, 169/183, 170/184, 171/185, 172/186, 173/187, 174/188, 175/189, 176/190 and 177/190 of US Patent No.10,294,299 B2. Examples of non-blocking or poorly-blocking anti-LAG3 antibodies includes antibodies having VH/VL amino acid sequences of SEQ ID Nos 23/24, ¾ and 11/12 of US Pub. US2022/0056126A1.
[0105] In certain embodiments, the target molecules are TAAs. Exemplary TAAs are set forth in Table A below, together with references to exemplary antibodies or antibody sequences upon which the TAA ABD can be based. TABLE A Exemplary Anti-TAAs . o. 6 r f
[0078] Accordingly, in certain embodiments, correct association between the two polypeptides of a Fab is promoted by exchanging the VL and VH domains of the Fab for each other or exchanging the CH1 and CL domains for each other, e.g., as described in WO 2009/080251. [0079] Correct Fab pairing can also be promoted by introducing one or more amino acid modifications in the CH1 domain and one or more amino acid modifications in the CL domain of the Fab and/or one or more amino acid modifications in the VH domain and one or more amino acid modifications in the VL domain. The amino acids that are modified are typically part of the VH:VL and CH1:CL interface such that the Fab components preferentially pair with each other rather than with components of other Fabs. [0080] In one embodiment, the one or more amino acid modifications are limited to the conserved framework residues of the variable (VH, VL) and constant (CH1, CL) domains as indicated by the Kabat numbering of residues. Almagro, 2008, Frontiers In Bioscience 13:1619-
1633 provides a definition of the framework residues on the basis of Kabat, Chothia, and IMGT numbering schemes. [0081] In one embodiment, the modifications introduced in the VH and CH1 and/or VL and CL domains are complementary to each other. Complementarity at the heavy and light chain interface can be achieved on the basis of steric and hydrophobic contacts, electrostatic/charge interactions or a combination of the variety of interactions. The complementarity between protein surfaces is broadly described in the literature in terms of lock and key fit, knob into hole, protrusion and cavity, donor and acceptor etc., all implying the nature of structural and chemical match between the two interacting surfaces. [0082] In one embodiment, the one or more introduced modifications introduce a new hydrogen bond across the interface of the Fab components. In one embodiment, the one or more introduced modifications introduce a new salt bridge across the interface of the Fab components. Exemplary substitutions are described in WO 2014/150973 and WO 2014/082179, the contents of which are hereby incorporated by reference. [0083] In some embodiments, the Fab domain comprises a 192E substitution in the CH1 domain and 114A and 137K substitutions in the CL domain, which introduces a salt-bridge between the CH1 and CL domains (see, e.g., Golay et al., 2016, J Immunol 196:3199-211). [0084] In some embodiments, the Fab domain comprises a 143Q and 188V substitutions in the CH1 domain and 113T and 176V substitutions in the CL domain, which serves to swap hydrophobic and polar regions of contact between the CH1 and CL domain (see, e.g., Golay et al., 2016, J Immunol 196:3199-211). [0085] In some embodiments, the Fab domain can comprise modifications in some or all of the VH, CH1, VL, CL domains to introduce orthogonal Fab interfaces which promote correct assembly of Fab domains (Lewis et al., 2014 Nature Biotechnology 32:191-198). In an embodiment, 39K, 62E modifications are introduced in the VH domain, H172A, F174G modifications are introduced in the CH1 domain, 1 R, 38D, (36F) modifications are introduced in the VL domain, and L135Y, S176W modifications are introduced in the CL domain. In another embodiment, a 39Y modification is introduced in the VH domain and a 38R modification is introduced in the VL domain. [0086] Fab domains can also be modified to replace the native CH1:CL disulfide bond with an engineered disulfide bond, thereby increasing the efficiency of Fab component pairing. For
example, an engineered disulfide bond can be introduced by introducing a 126C in the CH1 domain and a 121 C in the CL domain (see, e.g., Mazor et al., 2015, MAbs 7:377-89). [0087] Fab domains can also be modified by replacing the CH1 domain and CL domain with alternative domains that promote correct assembly. For example, Wu et al., 2015, MAbs 7:364- 76, describes substituting the CH1 domain with the constant domain of the T cell receptor and substituting the CL domain with the b domain of the T cell receptor, and pairing these domain replacements with an additional charge-charge interaction between the VL and VH domains by introducing a 38D modification in the VL domain and a 39K modification in the VH domain. [0088] In lieu of, or in addition to, the use of Fab heterodimerization strategies to promote correct VH-VL pairings, the VL of common light chain (also referred to as a universal light chain) can be used for each unique ABD in the antigen-binding molecules of the disclosure. In various embodiments, employing a common light chain as described herein reduces the number of inappropriate species in the antigen-binding molecules as compared to employing original cognate VLs. In various embodiments, the VL domains of ABDs are identified from monospecific antibodies comprising a common light chain. In various embodiments, the VH regions of the ABDs in the antigen-binding molecules comprise human heavy chain variable gene segments that are rearranged in vivo within mouse B cells that have been previously engineered to express a limited human light chain repertoire, or a single human light chain, cognate with human heavy chains and, in response to exposure with an antigen of interest, generate an antibody repertoire containing a plurality of human VHs that are cognate with one or one of two possible human VLs, wherein the antibody repertoire specific for the antigen of interest. Common light chains are those derived from a rearranged human Vκ1-39Jκ5 sequence or a rearranged human Vκ3-20Jκ1 sequence, and include somatically mutated (e.g., affinity matured) versions. See, for example, U.S. Patent No.10,412,940. 6.4. Antigen-Binding Molecules Comprising Engineered VH Domains [0089] The present disclosure provides antigen-binding molecules comprising an engineered VH of the disclosure. [0090] In some embodiments, the antigen-binding molecule is an antibody. The antibody can be any type of engineered antibody, including chimeric, humanized, veneered, or human antibodies. The antibody can be a monoclonal antibody or a genetically engineered polyclonal antibody. Antibodies contemplated herein include both traditional antibodies as well as antibody- like molecules known in the art, including but not limited to nanobodies, diabodies, minibodies,
antibody fragments, and other “alternative format” antibodies (e.g., as described in Spiess et al., 2015, Mol Immunol, 67(2 Pt A):95-106, incorporated herein by reference). An antibody may include an engineered VH of the disclosure as a component of one or more ABDs, including, for example, as a component of a Fab or as a component of an scFv. [0091] Accordingly, in some embodiments, disclosed herein is an antibody comprising an engineered VH, the engineered VH comprising (1) an N-linked glycosylation site within 20, 19, 18, 17, 16, 15, 14, 13, 12, 11, 10, 9, 8, 7, 6, 5, 4, or 3 amino acids of its C-terminus, or (2) the amino acid sequence X1X2X3X4X5NX6X7X8X9X10X11X12 (SEQ ID NO:1) at its C-terminus, where (a) X1, X2, X3, X4, and X5 are each independently selected from any amino acid; (b) X6 is selected from any amino acid, optionally wherein the amino acid is not proline; (c) X7 is S or T, and (d) X8, X9, X10,X11, and X12 are each independently selected from any amino acid and absent. [0092] In some embodiments, an antibody of the present disclosure comprises a polypeptide comprising: (a) a VH comprising (1) an N-linked glycosylation site within 20, 19, 18, 17, 16, 15, 14, 13, 12, 11, 10, 9, 8, 7, 6, 5, 4, or 3 amino acids of its C-terminus, or (2) the amino acid sequence X1X2X3X4X5NX6X7X8X9X10X11X12 (SEQ ID NO:1) at its C-terminus; and (b) an Fc domain. The antibody may further comprise an additional polypeptide comprising an additional VH and an additional Fc domain. In some embodiments, the additional VH comprises (1) an N- linked glycosylation site within 20, 19, 18, 17, 16, 15, 14, 13, 12, 11, 10, 9, 8, 7, 6, 5, 4, or 3 amino acids of its C-terminus, or (2) the amino acid sequence X1X2X3X4X5NX6X7X8X9X10X11X12 (SEQ ID NO:1) at its C-terminus. An antibody of the present disclosure may comprise any number of VHs (e.g., 1, 2, 3, 4, or more VHs), where at least one VH is an engineered VH described herein and where any number of the additional VHs may or may not be an engineered VH as described herein. 6.4.1.1. Multivalent Antigen-Binding Molecules [0093] Antigen-binding molecules of the present disclosure include multivalent molecules having one or more engineered VH domains. Various multivalent antigen-binding molecule formats are recognized in the art and contemplated herein, including bivalent, trivalent, and tetravalent formats. Certain example multivalent antigen-binding molecules are described in more detail below. [0094] In some embodiments, the antigen-binding molecule comprising an engineered VH is a multivalent antigen-binding molecule comprising two or more scFvs connected via a linker, where at least one of the scFvs comprises an engineered VH (as described in, e.g., Section 6.2
or as defined in numbered embodiments 1 to 34). Accordingly, disclosed herein, in some embodiments, is an antigen-binding molecule comprising: (a) a first scFv, (b) a linker, and (c) a second scFv, where the first and/or the second scFv comprise an engineered VH. In some embodiments, the first scFv comprises the engineered VH. In some embodiments, the second scFv comprises the engineered VH. In some embodiments, both the first and second scFvs comprise an engineered VH. In some embodiments, the antigen-binding molecule further comprises a third scFv, which optionally comprises an engineered VH. An example of such a multivalent antigen-binding molecule comprising two scFvs is depicted in FIG.1B. [0095] In some embodiments, the antigen-binding molecule comprising an engineered VH is a multivalent antigen-binding molecule comprising three or more antigen-binding domains, where at least one of the antigen-binding domains comprises an engineered VH. Accordingly, disclosed herein, in some embodiments, is an antigen-binding molecule comprising: (a) a first polypeptide chain comprising, from N-terminus to C-terminus (i) a first antigen-binding domain (e.g., a Fab), (ii) a first dimerization moiety, and (iii) a second antigen-binding domain (e.g., an scFv), the second antigen-binding domain comprising an engineered VH; and (b) a second polypeptide chain comprising, from N-terminus to C-terminus (i) a third antigen-binding domain (e.g., a Fab), and (ii) a second dimerization moiety. In some embodiments, the second polypeptide further comprises a fourth antigen-binding domain (e.g., an scFv) C-terminal to the second dimerization moiety, where the fourth antigen-binding domain comprises an engineered VH. Examples of such multivalent antigen-binding molecules are depicted in FIG.1C and FIGS. 2A-2G. 6.4.1.2. Multispecific Antigen-Binding Molecules [0096] Antigen-binding molecules of the present disclosure include multispecific molecules having one or more engineered VH domains. Various multispecific antigen-binding molecules are recognized in the art and contemplated herein, including bispecific, trispecific, and tetraspecific antigen-binding molecules. Certain example multispecific antigen-binding molecules are described in more detail below. [0097] In some embodiments, an antigen-binding molecule comprising an engineered VH of the disclosure is a bispecific T-cell engager. “Bispecific T-cell engager” describes a molecule comprising two scFvs, connected via a linker, where the first scFv comprises a first ABD that binds to a T cell antigen (e.g., a TCE ABD as described in Section 6.4.3) and the second scFv comprises a second ABD that binds to a tumor-associated antigen (e.g., a TAA ABD as described in Section 6.4.2). In some embodiments, the TCE ABD comprises the engineered VH.
In some embodiments, the TAA ABD comprises the engineered VH. In some embodiments, both the TCE ABD and the TAA ABD comprise an engineered VH. [0098] In some embodiments, an antigen-binding molecule comprising an engineered VH of the disclosure is a multivalent antigen-binding molecule comprising: (a) a first polypeptide chain comprising, from N-terminus to C-terminus (i) a first TCE ABD or TAA ABD, (ii) a first dimerization moiety, and (iii) a second TCE ABD or TAA ABD; and (b) a second polypeptide chain comprising, from N-terminus to C-terminus (i) a third TCE ABD or TAA ABD, and (ii) a second dimerization domain, where the second TCE ABD or TAA ABD comprises an engineered VH. In some embodiments, the second TCE ABD or TAA ABD is an scFv. In some embodiments, the second polypeptide further comprises a TCE ABD or TAA ABD C-terminal to the second dimerization moiety, where the fourth TCE ABD or TAA ABD comprises an engineered VH. In some embodiments, the fourth TCE ABD or TAA ABD is an scFv. [0099] In some embodiments, the first ABD is a TCE ABD (e.g., as described in Section 6.4.3). In some embodiments, the first ABD is a TAA ABD (e.g., as described in Section 6.4.2). In some embodiments, the second ABD is a TCE ABD (e.g., as described in Section 6.4.3). In some embodiments, the second ABD is a TAA ABD (e.g., as described in Section 6.4.2). In some embodiments, the third ABD is a TCE ABD (e.g., as described in Section 6.4.3). In some embodiments, the third ABD is a TAA ABD (e.g., as described in Section 6.4.2). In some embodiments, the fourth ABD is a TCE ABD (e.g., as described in Section 6.4.3). In some embodiments, the fourth ABD is a TAA ABD (e.g., as described in Section 6.4.2). 6.4.2. Tumor-associated Antigen ABDs [0100] In some embodiments, the antigen-binding molecules of the disclosure comprise at least one ABD that binds specifically to a tumor-associated antigen (TAA), referred to herein as a “TAA ABD”. Examples of TAAs include cancer antigens, extracellular matrix (“ECM”) proteins, tumor reactive lymphocyte antigens, cell surface molecules of tumor or viral lymphocytes, T-cell antigens (“TCAs”), and immune checkpoint molecules. The skilled artisan would recognize that the foregoing categories of target molecules are not mutually exclusive and thus a given target molecule may fall into more than one of the foregoing categories of target molecules. For example, some molecules may be considered both TCAs and immune checkpoint molecules. Preferably, the TAA is a human antigen. The antigen may or may not be present on normal cells. Certain aspects are directed to antigen-binding molecules comprising at least one ABD that binds specifically to a TAA. In certain embodiments, the TAA is preferentially expressed or
upregulated on tumor cells as compared to normal cells. In other embodiments, the TAA is a lineage marker. [0101] It is anticipated that any type of tumor and any type of TAA may be targeted by the antigen-binding molecules of the disclosure. Exemplary types of cancers that may be targeted include acute lymphoblastic leukemia, acute myelogenous leukemia, biliary cancer, B-cell leukemia, B-cell lymphoma, biliary cancer, bone cancer, brain cancer, breast cancer, triple- negative breast cancer, cervical cancer, Burkitt lymphoma, chronic lymphocytic leukemia, chronic myelogenous leukemia, colorectal cancer, endometrial cancer, esophageal cancer, gall bladder cancer, gastric cancer, gastrointestinal tract cancer, glioma, hairy cell leukemia, head and neck cancer, Hodgkin’s lymphoma, liver cancer, lung cancer, medullary thyroid cancer, melanoma, multiple myeloma, ovarian cancer, non-Hodgkin’s lymphoma, pancreatic cancer, prostate cancer, pulmonary tract cancer, renal cancer, sarcoma, skin cancer, testicular cancer, urothelial cancer, and other urinary bladder cancers. However, the skilled artisan will realize that TAAs are known for virtually any type of cancer. [0102] Non-limiting examples of ECM antigens include syndecan, 27enzalkoni, integrins, osteopontin, link, cadherins, laminin, laminin type EGF, lectin, fibronectin, notch, nectin (e.g., nectin-4), tenascin, collagen (e.g., collagen type X) and matrixin. [0103] Other target molecules are cell surface molecules of tumor or viral lymphocytes, for example T-cell co-stimulatory proteins such as CD27, CD28, 4-1BB (CD137), OX40, CD30, CD40, ICOS, lymphocyte function-associated antigen-1 (LFA-1), CD2, CD7, LIGHT, NKG2C, and B7-H3. [0104] In particular embodiments, the target molecules are immune checkpoint molecules, for example CTLA-4, PD1, PDL1, PDL2, B7-H3, B7-H4, BTLA, HVEM, TIM3, GAL9, LAG3, VISTA, KIR, 2B4, CD160, CGEN-15049, CHK1, CHK2. In particular embodiments, the target molecule is PD1. In other embodiments, the target molecule is LAG3. In some embodiments, where the target molecule is an immune checkpoint molecules, the TAA ABD is non-blocking or poorly- blocking of ligand-receptor binding. Examples of non-blocking or poorly-blocking anti-PD1 antibodies includes antibodies having VH/VL amino acid sequences of SEQ ID Nos: 2/10 of PCT Pub. No. WO2015/112800A1; SEQ ID Nos: 16/17 of US Patent No.11,034,765 B2; SEQ ID Nos.164/178, 165/179, 166/180, 167/181, 168/182, 169/183, 170/184, 171/185, 172/186, 173/187, 174/188, 175/189, 176/190 and 177/190 of US Patent No.10,294,299 B2. Examples of non-blocking or poorly-blocking anti-LAG3 antibodies includes antibodies having VH/VL amino acid sequences of SEQ ID Nos 23/24, ¾ and 11/12 of US Pub. US2022/0056126A1.
[0105] In certain embodiments, the target molecules are TAAs. Exemplary TAAs are set forth in Table A below, together with references to exemplary antibodies or antibody sequences upon which the TAA ABD can be based. TABLE A Exemplary Anti-TAAs . o. 6 r f
 TABLE A Exemplary Anti-TAAs T t A tib d N d Bi di S
TABLE A Exemplary Anti-TAAs T t A tib d N d Bi di S
 TABLE A Exemplary Anti-TAAs T t A tib d N d Bi di S 1- 4
TABLE A Exemplary Anti-TAAs T t A tib d N d Bi di S 1- 4
 TABLE A Exemplary Anti-TAAs T t A tib d N d Bi di S 6 6
TABLE A Exemplary Anti-TAAs T t A tib d N d Bi di S 6 6
 TABLE A Exemplary Anti-TAAs T t A tib d N d Bi di S g
TABLE A Exemplary Anti-TAAs T t A tib d N d Bi di S g
 TABLE A Exemplary Anti-TAAs T t A tib d N d Bi di S , 1, A A , , ,
TABLE A Exemplary Anti-TAAs T t A tib d N d Bi di S , 1, A A , , ,
 TABLE A Exemplary Anti-TAAs T t A tib d N d Bi di S , , f n m ) - -
TABLE A Exemplary Anti-TAAs T t A tib d N d Bi di S , , f n m ) - -
 TABLE A Exemplary Anti-TAAs T t A tib d N d Bi di S - - - - - 2 , 0, o. ; e ; ; 1
TABLE A Exemplary Anti-TAAs T t A tib d N d Bi di S - - - - - 2 , 0, o. ; e ; ; 1
 TABLE A Exemplary Anti-TAAs T t A tib d N d Bi di S
TABLE A Exemplary Anti-TAAs T t A tib d N d Bi di S
 [0106] In some aspects, t e TAA ABD competes wt an antbody set ort n Tabe A or binding to the TAA. In further aspects, the TAA ABD comprises CDRs having CDR sequences of an anti-TAA antibody set forth in Table A. In some embodiments, the TAA ABD comprises all 6 CDR sequences of an anti-TAA antibody set forth in Table A. In other embodiments, the TAA ABD comprises at least the heavy chain CDR sequences (CDR-H1, CDR-H2, CDR-H3) of an anti-TAA antibody set forth in Table A and the light chain CDR sequences of a universal light chain. In further aspects, a TAA ABD comprises a VH comprising the amino acid sequence of the VH of an anti-TAA antibody set forth in Table A. In some embodiments, the TAA ABD further comprises a VL comprising the amino acid sequence of the VL of the anti-TAA antibody set forth in Table A. In other embodiments, the TAA ABD further comprises a universal light chain VL sequence. [0107] Additional TAAs that can be targeted by the antigen-binding molecules are disclosed in, e.g., Hafeez et al., 2020, Molecules 25:4764, doi:10.3390/molecules25204764, particularly in Table 1. Table 1 of Hafeez et al. is incorporated by reference in its entirety here. [0108] Yet additional exemplary TAAs include Fibroblast Activation Protein (FAP), the A1 domain of Tenascin-C (TNC A1), the A2 domain of Tenascin-C (TNC A2), the Extra Domain B of Fibronectin (EDB), the Melanoma-associated Chondroitin Sulfate Proteoglycan (MCSP), MART-1/Melan-A, gp100, Dipeptidyl peptidase IV (DPPIV), adenosine deaminase-binding protein (ADAbp), cyclophilin b, colorectal associated antigen (CRC)-C017-1A/GA733, Carcinoembryonic Antigen (CEA) and its immunogenic epitopes CAP-1 and CAP-2, etv6, aml1, Prostate Specific Antigen (PSA) and its immunogenic epitopes PSA-1, PSA-2, and PSA-3, prostate-specific membrane antigen (PSMA), T-cell receptor/CD3-zeta chain, MAGE-family of tumor antigens (e.g., MAGE-A1, MAGE-A2, MAGE-A3, MAGE-A4, MAGE-A5, MAGE-A6, MAGE-A7, MAGE-A8, MAGE-A9, MAGE-A10, MAGE-A11, MAGE-A12, MAGE-Xp2 (MAGE- B2), MAGE-Xp3 (MAGE-B3), MAGE-Xp4 (MAGE-B4), MAGE-C1, MAGE-C2, MAGE-C3,
MAGE-C4, MAGE-C5), GAGE-family of tumor antigens (e.g., GAGE-1, GAGE-2, GAGE-3, GAGE-4, GAGE-5, GAGE-6, GAGE-7, GAGE-8, GAGE-9), BAGE, RAGE, LAGE-1, NAG, GnT- V, MUM-1, CDK4, tyrosinase, p53, MUC family, HER2/neu, p21ras, RCAS1, α-fetoprotein, E- cadherin, α-catenin, β-catenin and γ-catenin, p120ctn, gp100 Pmel117, PRAME, NY-ESO-1, cdc27, adenomatous polyposis coli protein (APC), fodrin, Connexin 37, Ig-idiotype, p15, gp75, GM2 and GD2 gangliosides, viral products such as human papilloma virus proteins, Smad family of tumor antigens, Imp-1, P1A, EBV-encoded nuclear antigen (EBNA)-1, brain glycogen phosphorylase, SSX-1, SSX-2 (HOM-MEL-40), SSX-1, SSX-4, SSX-5, SCP-1 and CT-7, c- erbB-2, Her2, EGFR, IGF-1R, CD2 (T-cell surface antigen), CD3 (heteromultimer associated with the TCR), CD22 (B-cell receptor), CD23 (low affinity IgE receptor), CD30 (cytokine receptor), CD33 (myeloid cell surface antigen), CD40 (tumor necrosis factor receptor), IL-6R- (IL6 receptor), CD20, MCSP, PDGFβR (β-platelet-derived growth factor receptor), ErbB2 epithelial cell adhesion molecule (EpCAM), EGFR variant III (EGFRvIII), CD19, disialoganglioside GD2, ductal-epithelial mucine, gp36, TAG-72, glioma-associated antigen, β- human chorionic gonadotropin, alphafetoprotein (AFP), lectin-reactive AFP, thyroglobulin, MN- CA IX, human telomerase reverse transcriptase, RU1, RU2 (AS), intestinal carboxyl esterase, mut hsp70-2, M-CSF, prostase, prostase specific antigen (PSA), PAP, LAGA-1a, p53, prostein, PSMA, surviving and telomerase, prostate-carcinoma tumor antigen-1 (PCTA-1), ELF2M, neutrophil elastase, ephrin B2, insulin growth factor (IGF1)-I, IGF-II, IGFI receptor, 5T4, ROR1, Nkp30, NKG2D, tumor stromal antigens, the extra domain A (EDA) and extra domain B (EDB) of fibronectin and the A1 domain of tenascin-C (TnC A1). 6.4.3. TCE ABDs [0109] In some embodiments, the antigen-binding molecules of the disclosure comprise at least a T-cell engaging ABD that binds specifically to a T-cell receptor complex component, referred to herein as “TCE ABD”. In other embodiments, the antigen-binding molecules of the disclosure do not comprise a TCE ABD. Exemplary targets for the TCE ABD are CD3 and the T-cell receptor (e.g., TCRαβ or TCRγδ). Preferably, the TCE ABD target is a human T-cell receptor complex component. The epitope of the TCE ABD can be an individual polypeptide (e.g., CD3 epsilon) or a multimeric component of the T-cell receptor complex (e.g., the TCRαβ dimer or the TCRγδ dimer). [0110] Exemplary CD3 and TCR antibodies or antibody sequences are set forth in Table T below, upon which the TCE ABD can be based.
TABLE T Exemplary T-Cell Engaging Antibodies i i i
[0106] In some aspects, t e TAA ABD competes wt an antbody set ort n Tabe A or binding to the TAA. In further aspects, the TAA ABD comprises CDRs having CDR sequences of an anti-TAA antibody set forth in Table A. In some embodiments, the TAA ABD comprises all 6 CDR sequences of an anti-TAA antibody set forth in Table A. In other embodiments, the TAA ABD comprises at least the heavy chain CDR sequences (CDR-H1, CDR-H2, CDR-H3) of an anti-TAA antibody set forth in Table A and the light chain CDR sequences of a universal light chain. In further aspects, a TAA ABD comprises a VH comprising the amino acid sequence of the VH of an anti-TAA antibody set forth in Table A. In some embodiments, the TAA ABD further comprises a VL comprising the amino acid sequence of the VL of the anti-TAA antibody set forth in Table A. In other embodiments, the TAA ABD further comprises a universal light chain VL sequence. [0107] Additional TAAs that can be targeted by the antigen-binding molecules are disclosed in, e.g., Hafeez et al., 2020, Molecules 25:4764, doi:10.3390/molecules25204764, particularly in Table 1. Table 1 of Hafeez et al. is incorporated by reference in its entirety here. [0108] Yet additional exemplary TAAs include Fibroblast Activation Protein (FAP), the A1 domain of Tenascin-C (TNC A1), the A2 domain of Tenascin-C (TNC A2), the Extra Domain B of Fibronectin (EDB), the Melanoma-associated Chondroitin Sulfate Proteoglycan (MCSP), MART-1/Melan-A, gp100, Dipeptidyl peptidase IV (DPPIV), adenosine deaminase-binding protein (ADAbp), cyclophilin b, colorectal associated antigen (CRC)-C017-1A/GA733, Carcinoembryonic Antigen (CEA) and its immunogenic epitopes CAP-1 and CAP-2, etv6, aml1, Prostate Specific Antigen (PSA) and its immunogenic epitopes PSA-1, PSA-2, and PSA-3, prostate-specific membrane antigen (PSMA), T-cell receptor/CD3-zeta chain, MAGE-family of tumor antigens (e.g., MAGE-A1, MAGE-A2, MAGE-A3, MAGE-A4, MAGE-A5, MAGE-A6, MAGE-A7, MAGE-A8, MAGE-A9, MAGE-A10, MAGE-A11, MAGE-A12, MAGE-Xp2 (MAGE- B2), MAGE-Xp3 (MAGE-B3), MAGE-Xp4 (MAGE-B4), MAGE-C1, MAGE-C2, MAGE-C3,
MAGE-C4, MAGE-C5), GAGE-family of tumor antigens (e.g., GAGE-1, GAGE-2, GAGE-3, GAGE-4, GAGE-5, GAGE-6, GAGE-7, GAGE-8, GAGE-9), BAGE, RAGE, LAGE-1, NAG, GnT- V, MUM-1, CDK4, tyrosinase, p53, MUC family, HER2/neu, p21ras, RCAS1, α-fetoprotein, E- cadherin, α-catenin, β-catenin and γ-catenin, p120ctn, gp100 Pmel117, PRAME, NY-ESO-1, cdc27, adenomatous polyposis coli protein (APC), fodrin, Connexin 37, Ig-idiotype, p15, gp75, GM2 and GD2 gangliosides, viral products such as human papilloma virus proteins, Smad family of tumor antigens, Imp-1, P1A, EBV-encoded nuclear antigen (EBNA)-1, brain glycogen phosphorylase, SSX-1, SSX-2 (HOM-MEL-40), SSX-1, SSX-4, SSX-5, SCP-1 and CT-7, c- erbB-2, Her2, EGFR, IGF-1R, CD2 (T-cell surface antigen), CD3 (heteromultimer associated with the TCR), CD22 (B-cell receptor), CD23 (low affinity IgE receptor), CD30 (cytokine receptor), CD33 (myeloid cell surface antigen), CD40 (tumor necrosis factor receptor), IL-6R- (IL6 receptor), CD20, MCSP, PDGFβR (β-platelet-derived growth factor receptor), ErbB2 epithelial cell adhesion molecule (EpCAM), EGFR variant III (EGFRvIII), CD19, disialoganglioside GD2, ductal-epithelial mucine, gp36, TAG-72, glioma-associated antigen, β- human chorionic gonadotropin, alphafetoprotein (AFP), lectin-reactive AFP, thyroglobulin, MN- CA IX, human telomerase reverse transcriptase, RU1, RU2 (AS), intestinal carboxyl esterase, mut hsp70-2, M-CSF, prostase, prostase specific antigen (PSA), PAP, LAGA-1a, p53, prostein, PSMA, surviving and telomerase, prostate-carcinoma tumor antigen-1 (PCTA-1), ELF2M, neutrophil elastase, ephrin B2, insulin growth factor (IGF1)-I, IGF-II, IGFI receptor, 5T4, ROR1, Nkp30, NKG2D, tumor stromal antigens, the extra domain A (EDA) and extra domain B (EDB) of fibronectin and the A1 domain of tenascin-C (TnC A1). 6.4.3. TCE ABDs [0109] In some embodiments, the antigen-binding molecules of the disclosure comprise at least a T-cell engaging ABD that binds specifically to a T-cell receptor complex component, referred to herein as “TCE ABD”. In other embodiments, the antigen-binding molecules of the disclosure do not comprise a TCE ABD. Exemplary targets for the TCE ABD are CD3 and the T-cell receptor (e.g., TCRαβ or TCRγδ). Preferably, the TCE ABD target is a human T-cell receptor complex component. The epitope of the TCE ABD can be an individual polypeptide (e.g., CD3 epsilon) or a multimeric component of the T-cell receptor complex (e.g., the TCRαβ dimer or the TCRγδ dimer). [0110] Exemplary CD3 and TCR antibodies or antibody sequences are set forth in Table T below, upon which the TCE ABD can be based.
TABLE T Exemplary T-Cell Engaging Antibodies i i i
 TABLE T Exemplary T-Cell Engaging Antibodies i i i
TABLE T Exemplary T-Cell Engaging Antibodies i i i
 TABLE T Exemplary T-Cell Engaging Antibodies i i i 2
TABLE T Exemplary T-Cell Engaging Antibodies i i i 2
 TABLE T Exemplary T-Cell Engaging Antibodies i i i , , ,
TABLE T Exemplary T-Cell Engaging Antibodies i i i , , ,
 [0111] In some aspects, the TCE ABD competes with a T-cell engaging (TCE) antibody set forth in Table T for binding to the TCE antibody’s target (e.g., CD3 or a T-cell receptor). In further aspects, the TCE ABD comprises CDRs having CDR sequences of a TCE antibody set forth in Table T. In some embodiments, the TCE ABD comprises all 6 CDR sequences of a TCE antibody set forth in Table T. In other embodiments, the TCE ABD comprises at least the heavy chain CDR sequences (CDR-H1, CDR-H2, CDR-H3) of a TCE antibody set forth in Table T and the light chain CDR sequences of a universal light chain. In further aspects, a TCE ABD comprises a VH comprising the amino acid sequence of the VH of a TCE antibody set forth in Table T. In some embodiments, the TCE ABD further comprises a VL comprising the amino acid sequence of the VL of the TCE antibody set forth in Table T. In other embodiments, the TCE ABD further comprises a universal light chain VL sequence. 6.4.4. Linkers [0112] In certain aspects, the present disclosure provides recombinant polypeptides in which two or more components of the recombinant polypeptide are connected to one another by a linker (also “peptide linker”). By way of example and not limitation, linkers can be used to connect (a) a target binding domain and a constant domain; (b) a first target binding domain and a second target binding domain (e.g. a first scFv and a second scFv); or (c) different domains within a target binding domain (e.g., the VH and VL domains in an scFv). [0113] A peptide linker can range from 2 amino acids to 60 or more amino acids, and in certain aspects a peptide linker ranges from 3 amino acids to 50 amino acids, from 4 to 30 amino acids, from 5 to 25 amino acids, from 10 to 25 amino acids, 10 amino acids to 60 amino acids, from 12
amino acids to 20 amino acids, from 20 amino acids to 50 amino acids, or from 25 amino acids to 35 amino acids in length. [0114] In particular aspects, a peptide linker is at least 5 amino acids, at least 6 amino acids or at least 7 amino acids in length and optionally is up to 30 amino acids, up to 40 amino acids, up to 50 amino acids or up to 60 amino acids in length. [0115] In some embodiments of the foregoing, the linker ranges from 5 amino acids to 50 amino acids in length, e.g., ranges from 5 to 50, from 5 to 45, from 5 to 40, from 5 to 35, from 5 to 30, from 5 to 25, or from 5 to 20 amino acids in length. In other embodiments of the foregoing, the linker ranges from 6 amino acids to 50 amino acids in length, e.g., ranges from 6 to 50, from 6 to 45, from 6 to 40, from 6 to 35, from 6 to 30, from 6 to 25, or from 6 to 20 amino acids in length. In yet other embodiments of the foregoing, the linker ranges from 7 amino acids to 50 amino acids in length, e.g., ranges from 7 to 50, from 7 to 45, from 7 to 40, from 7 to 35, from 7 to 30, from 7 to 25, or from 7 to 20 amino acids in length. [0116] Charged (e.g., charged hydrophilic linkers) and/or flexible linkers are particularly preferred. [0117] Examples of flexible linkers that can be used in the recombinant polypeptides of the disclosure include those disclosed by Chen et al., 2013, Adv Drug Deliv Rev.65(10): 1357-1369 and Klein et al., 2014, Protein Engineering, Design & Selection 27(10): 325-330. Particularly useful flexible linkers are or comprise repeats of glycines and serines, e.g., a monomer or multimer of GnS (SEQ ID NO: 68) or SGn (SEQ ID NO: 69), where n is an integer from 1 to 10, e.g., 12, 3, 4, 5, 6, 7, 8, 9 or 10. In one embodiment, the linker is or comprises a monomer or multimer of repeat of G4S (SEQ ID NO: 67) e.g., (GGGGS)n, where n is an integer from 1 to 10 (SEQ ID NO: 70), e.g., 12, 3, 4, 5, 6, 7, 8, 9 or 10. For example, in some embodiments, the linker is GGGGS (SEQ ID NO: 67), GGGGSGGGGS (SEQ ID NO: 71), GGGGSGGGGSGGGGS (SEQ ID NO: 72), GGGGSGGGGSGGGGSGGGGS (SEQ ID NO: 73), or GGGGSGGGGSGGGGSGGGGSGGGGS (SEQ ID NO: 74). [0118] Polyglycine linkers can suitably be used in the recombinant polypeptides of the disclosure. In some embodiments, a peptide linker comprises two consecutive glycines (2Gly), three consecutive glycines (3Gly), four consecutive glycines (4Gly), five consecutive glycines (5Gly), six consecutive glycines (6Gly), seven consecutive glycines (7Gly), eight consecutive glycines (8Gly) or nine consecutive glycines (9Gly).
6.4.5. Fc Regions [0119] In certain aspects, the antigen-binding molecules of the disclosure comprise a pair of Fc domains that associate to form an Fc region. In native antibodies, Fc regions comprise hinge regions at their N-termini to form a constant domain. Throughout this disclosure, the reference to an Fc domain encompasses an Fc domain with a hinge domain at its N-terminus unless specified otherwise. [0120] The Fc domains can be derived from any suitable species operably linked to an ABD or component thereof. In one embodiment the Fc domain is derived from a human Fc domain. In preferred embodiments, an antigen-binding domain of an antigen-binding molecule of the disclosure is fused to an IgG Fc molecule. An antigen-binding domain may be fused to the N- terminus or the C-terminus of the IgG Fc domain or both. [0121] The Fc domains can be derived from any suitable class of antibody, including IgA (including subclasses lgA1 and lgA2), IgD, IgE, IgG (including subclasses lgG1, lgG2, lgG3 and lgG4), and IgM. In one embodiment, the Fc domain is derived from lgG1, lgG2, lgG3 or lgG4. In one embodiment the Fc domain is derived from lgG1. In one embodiment the Fc domain is derived from lgG4. [0122] The two Fc domains within the Fc region can be the same or different from one another. In a native antibody the Fc domains are typically identical, but for the purpose of producing multispecific binding molecules, e.g., antigen-binding molecules described herein, the Fc domains might advantageously be different to allow for heterodimerization, as described in Section 6.4.5.2 below. In other embodiments, the two Fc domains of antigen-binding molecules disclosed herein are the same. [0123] In native antibodies, the heavy chain Fc domain of IgA, IgD and IgG is composed of two heavy chain constant domains (CH2 and CH3) and that of IgE and IgM is composed of three heavy chain constant domains (CH2, CH3 and CH4). These dimerize to create an Fc region. [0124] In antigen-binding molecules of the present disclosure, the Fc region, and / or the Fc domains within it, can comprise heavy chain constant domains from one or more different classes of antibody, for example one, two or three different classes. [0125] In one embodiment the Fc region comprises CH2 and CH3 domains derived from lgG1. [0126] In one embodiment the Fc region comprises CH2 and CH3 domains derived from lgG2. [0127] In one embodiment the Fc region comprises CH2 and CH3 domains derived from lgG3.
[0128] In one embodiment the Fc region comprises CH2 and CH3 domains derived from lgG4. [0129] In one embodiment the Fc region comprises a CH4 domain from IgM. The IgM CH4 domain is typically located at the C-terminus of the CH3 domain. [0130] In one embodiment the Fc region comprises CH2 and CH3 domains derived from IgG and a CH4 domain derived from IgM. [0131] It will be appreciated that the heavy chain constant domains for use in producing an Fc region for antigen-binding molecules of the present disclosure may include variants of the naturally occurring constant domains described above. Such variants may comprise one or more amino acid variations compared to wild type constant domains. In one example the Fc region of the present disclosure comprises at least one constant domain that varies in sequence from the wild type constant domain. It will be appreciated that the variant constant domains may be longer or shorter than the wild-type constant domain. Preferably the variant constant domains are at least 60% identical or similar to a wild-type constant domain. In another example the variant constant domains are at least 70% identical or similar. In another example the variant constant domains are at least 80% identical or similar. In another example the variant constant domains are at least 90% identical or similar. In another example the variant constant domains are at least 95% identical or similar. [0132] IgM and IgA occur naturally in humans as covalent multimers of the common H2L2 antibody unit. IgM occurs as a pentamer when it has incorporated a J-chain, or as a hexamer when it lacks a J-chain. IgA occurs as monomer and dimer forms. The heavy chains of IgM and IgA possess an 18 amino acid extension to the C-terminal constant domain, known as a tailpiece. The tailpiece includes a cysteine residue that forms a disulfide bond between heavy chains in the polymer, and is believed to have an important role in polymerization. The tailpiece also contains a glycosylation site. In certain embodiments, the antigen-binding molecules of the present disclosure do not comprise a tailpiece. [0133] The Fc domains that are incorporated into the antigen-binding molecules of the present disclosure may comprise one or more modifications that alter the functional properties of the proteins, for example, binding to Fc-receptors such as FcRn or leukocyte receptors, binding to complement, modified disulfide bond architecture, or altered glycosylation patterns. Exemplary Fc modifications that alter effector function are described in Section 6.4.5.1. [0134] The Fc domains can also be altered to include modifications that improve manufacturability of asymmetric antigen-binding molecules, for example by allowing
heterodimerization, which is the preferential pairing of non-identical Fc domains over identical Fc domains. Heterodimerization permits the production of antigen-binding molecules in which different polypeptide components are connected to one another by an Fc region containing Fc domains that differ in sequence. Examples of heterodimerization strategies are exemplified in Section 6.4.5.2. [0135] It will be appreciated that any of the modifications mentioned above can be combined in any suitable manner to achieve the desired functional properties and/or combined with other modifications to alter the properties of the antigen-binding molecules. TABLE F-1 Fc Sequences Q O
[0111] In some aspects, the TCE ABD competes with a T-cell engaging (TCE) antibody set forth in Table T for binding to the TCE antibody’s target (e.g., CD3 or a T-cell receptor). In further aspects, the TCE ABD comprises CDRs having CDR sequences of a TCE antibody set forth in Table T. In some embodiments, the TCE ABD comprises all 6 CDR sequences of a TCE antibody set forth in Table T. In other embodiments, the TCE ABD comprises at least the heavy chain CDR sequences (CDR-H1, CDR-H2, CDR-H3) of a TCE antibody set forth in Table T and the light chain CDR sequences of a universal light chain. In further aspects, a TCE ABD comprises a VH comprising the amino acid sequence of the VH of a TCE antibody set forth in Table T. In some embodiments, the TCE ABD further comprises a VL comprising the amino acid sequence of the VL of the TCE antibody set forth in Table T. In other embodiments, the TCE ABD further comprises a universal light chain VL sequence. 6.4.4. Linkers [0112] In certain aspects, the present disclosure provides recombinant polypeptides in which two or more components of the recombinant polypeptide are connected to one another by a linker (also “peptide linker”). By way of example and not limitation, linkers can be used to connect (a) a target binding domain and a constant domain; (b) a first target binding domain and a second target binding domain (e.g. a first scFv and a second scFv); or (c) different domains within a target binding domain (e.g., the VH and VL domains in an scFv). [0113] A peptide linker can range from 2 amino acids to 60 or more amino acids, and in certain aspects a peptide linker ranges from 3 amino acids to 50 amino acids, from 4 to 30 amino acids, from 5 to 25 amino acids, from 10 to 25 amino acids, 10 amino acids to 60 amino acids, from 12
amino acids to 20 amino acids, from 20 amino acids to 50 amino acids, or from 25 amino acids to 35 amino acids in length. [0114] In particular aspects, a peptide linker is at least 5 amino acids, at least 6 amino acids or at least 7 amino acids in length and optionally is up to 30 amino acids, up to 40 amino acids, up to 50 amino acids or up to 60 amino acids in length. [0115] In some embodiments of the foregoing, the linker ranges from 5 amino acids to 50 amino acids in length, e.g., ranges from 5 to 50, from 5 to 45, from 5 to 40, from 5 to 35, from 5 to 30, from 5 to 25, or from 5 to 20 amino acids in length. In other embodiments of the foregoing, the linker ranges from 6 amino acids to 50 amino acids in length, e.g., ranges from 6 to 50, from 6 to 45, from 6 to 40, from 6 to 35, from 6 to 30, from 6 to 25, or from 6 to 20 amino acids in length. In yet other embodiments of the foregoing, the linker ranges from 7 amino acids to 50 amino acids in length, e.g., ranges from 7 to 50, from 7 to 45, from 7 to 40, from 7 to 35, from 7 to 30, from 7 to 25, or from 7 to 20 amino acids in length. [0116] Charged (e.g., charged hydrophilic linkers) and/or flexible linkers are particularly preferred. [0117] Examples of flexible linkers that can be used in the recombinant polypeptides of the disclosure include those disclosed by Chen et al., 2013, Adv Drug Deliv Rev.65(10): 1357-1369 and Klein et al., 2014, Protein Engineering, Design & Selection 27(10): 325-330. Particularly useful flexible linkers are or comprise repeats of glycines and serines, e.g., a monomer or multimer of GnS (SEQ ID NO: 68) or SGn (SEQ ID NO: 69), where n is an integer from 1 to 10, e.g., 12, 3, 4, 5, 6, 7, 8, 9 or 10. In one embodiment, the linker is or comprises a monomer or multimer of repeat of G4S (SEQ ID NO: 67) e.g., (GGGGS)n, where n is an integer from 1 to 10 (SEQ ID NO: 70), e.g., 12, 3, 4, 5, 6, 7, 8, 9 or 10. For example, in some embodiments, the linker is GGGGS (SEQ ID NO: 67), GGGGSGGGGS (SEQ ID NO: 71), GGGGSGGGGSGGGGS (SEQ ID NO: 72), GGGGSGGGGSGGGGSGGGGS (SEQ ID NO: 73), or GGGGSGGGGSGGGGSGGGGSGGGGS (SEQ ID NO: 74). [0118] Polyglycine linkers can suitably be used in the recombinant polypeptides of the disclosure. In some embodiments, a peptide linker comprises two consecutive glycines (2Gly), three consecutive glycines (3Gly), four consecutive glycines (4Gly), five consecutive glycines (5Gly), six consecutive glycines (6Gly), seven consecutive glycines (7Gly), eight consecutive glycines (8Gly) or nine consecutive glycines (9Gly).
6.4.5. Fc Regions [0119] In certain aspects, the antigen-binding molecules of the disclosure comprise a pair of Fc domains that associate to form an Fc region. In native antibodies, Fc regions comprise hinge regions at their N-termini to form a constant domain. Throughout this disclosure, the reference to an Fc domain encompasses an Fc domain with a hinge domain at its N-terminus unless specified otherwise. [0120] The Fc domains can be derived from any suitable species operably linked to an ABD or component thereof. In one embodiment the Fc domain is derived from a human Fc domain. In preferred embodiments, an antigen-binding domain of an antigen-binding molecule of the disclosure is fused to an IgG Fc molecule. An antigen-binding domain may be fused to the N- terminus or the C-terminus of the IgG Fc domain or both. [0121] The Fc domains can be derived from any suitable class of antibody, including IgA (including subclasses lgA1 and lgA2), IgD, IgE, IgG (including subclasses lgG1, lgG2, lgG3 and lgG4), and IgM. In one embodiment, the Fc domain is derived from lgG1, lgG2, lgG3 or lgG4. In one embodiment the Fc domain is derived from lgG1. In one embodiment the Fc domain is derived from lgG4. [0122] The two Fc domains within the Fc region can be the same or different from one another. In a native antibody the Fc domains are typically identical, but for the purpose of producing multispecific binding molecules, e.g., antigen-binding molecules described herein, the Fc domains might advantageously be different to allow for heterodimerization, as described in Section 6.4.5.2 below. In other embodiments, the two Fc domains of antigen-binding molecules disclosed herein are the same. [0123] In native antibodies, the heavy chain Fc domain of IgA, IgD and IgG is composed of two heavy chain constant domains (CH2 and CH3) and that of IgE and IgM is composed of three heavy chain constant domains (CH2, CH3 and CH4). These dimerize to create an Fc region. [0124] In antigen-binding molecules of the present disclosure, the Fc region, and / or the Fc domains within it, can comprise heavy chain constant domains from one or more different classes of antibody, for example one, two or three different classes. [0125] In one embodiment the Fc region comprises CH2 and CH3 domains derived from lgG1. [0126] In one embodiment the Fc region comprises CH2 and CH3 domains derived from lgG2. [0127] In one embodiment the Fc region comprises CH2 and CH3 domains derived from lgG3.
[0128] In one embodiment the Fc region comprises CH2 and CH3 domains derived from lgG4. [0129] In one embodiment the Fc region comprises a CH4 domain from IgM. The IgM CH4 domain is typically located at the C-terminus of the CH3 domain. [0130] In one embodiment the Fc region comprises CH2 and CH3 domains derived from IgG and a CH4 domain derived from IgM. [0131] It will be appreciated that the heavy chain constant domains for use in producing an Fc region for antigen-binding molecules of the present disclosure may include variants of the naturally occurring constant domains described above. Such variants may comprise one or more amino acid variations compared to wild type constant domains. In one example the Fc region of the present disclosure comprises at least one constant domain that varies in sequence from the wild type constant domain. It will be appreciated that the variant constant domains may be longer or shorter than the wild-type constant domain. Preferably the variant constant domains are at least 60% identical or similar to a wild-type constant domain. In another example the variant constant domains are at least 70% identical or similar. In another example the variant constant domains are at least 80% identical or similar. In another example the variant constant domains are at least 90% identical or similar. In another example the variant constant domains are at least 95% identical or similar. [0132] IgM and IgA occur naturally in humans as covalent multimers of the common H2L2 antibody unit. IgM occurs as a pentamer when it has incorporated a J-chain, or as a hexamer when it lacks a J-chain. IgA occurs as monomer and dimer forms. The heavy chains of IgM and IgA possess an 18 amino acid extension to the C-terminal constant domain, known as a tailpiece. The tailpiece includes a cysteine residue that forms a disulfide bond between heavy chains in the polymer, and is believed to have an important role in polymerization. The tailpiece also contains a glycosylation site. In certain embodiments, the antigen-binding molecules of the present disclosure do not comprise a tailpiece. [0133] The Fc domains that are incorporated into the antigen-binding molecules of the present disclosure may comprise one or more modifications that alter the functional properties of the proteins, for example, binding to Fc-receptors such as FcRn or leukocyte receptors, binding to complement, modified disulfide bond architecture, or altered glycosylation patterns. Exemplary Fc modifications that alter effector function are described in Section 6.4.5.1. [0134] The Fc domains can also be altered to include modifications that improve manufacturability of asymmetric antigen-binding molecules, for example by allowing
heterodimerization, which is the preferential pairing of non-identical Fc domains over identical Fc domains. Heterodimerization permits the production of antigen-binding molecules in which different polypeptide components are connected to one another by an Fc region containing Fc domains that differ in sequence. Examples of heterodimerization strategies are exemplified in Section 6.4.5.2. [0135] It will be appreciated that any of the modifications mentioned above can be combined in any suitable manner to achieve the desired functional properties and/or combined with other modifications to alter the properties of the antigen-binding molecules. TABLE F-1 Fc Sequences Q O
 [0136] In some aspects, an Fc domain comprises an amino acid sequence having at least about 90%, at least about 91%, at least about 92%, about at least 93%, at least about 94%, at
eat least about 95%, at least about 96%, at least about 97%, at least about 98%, at least about 99%, or 100% sequence identity to SEQ ID NO: 58. In cases where an Fc domain comprises at least 90% sequence identity and less than 100% sequence identity to SEQ ID NO: 58 (e.g., between 90% and 99% sequence identity to SEQ ID NO: 58), an Fc domain may also comprise one or more amino acid substitutions described herein, for example one or more substitutions that reduce effector function (e.g., as described in Section 6.4.5.1) and/or one or more substitutions that promote Fc heterodimerization (e.g., as described in Section 6.4.5.2). [0137] In some aspects, an Fc domain comprises an amino acid sequence having at least about 90%, at least about 91%, at least about 92%, about at least 93%, at least about 94%, at least about 95%, at least about 96%, at least about 97%, at least about 98%, at least about 99%, or 100% sequence identity to SEQ ID NO: 59. In cases where an Fc domain comprises at least 90% sequence identity and less than 100% sequence identity to SEQ ID NO: 59 (e.g., between 90% and 99% sequence identity to SEQ ID NO: 59), an Fc domain may also comprise one or more amino acid substitutions described herein, for example one or more substitutions that reduce effector function (e.g., as described in Section 6.4.5.1) and/or one or more substitutions that promote Fc heterodimerization (e.g., as described in Section 6.4.5.2). [0138] In some aspects, an Fc domain comprises an amino acid sequence having at least about 90%, at least about 91%, at least about 92%, about at least 93%, at least about 94%, at eat least about 95%, at least about 96%, at least about 97%, at least about 98%, at least about 99%, or 100% sequence identity to SEQ ID NO: 60. In cases where an Fc domain comprises at least 90% sequence identity and less than 100% sequence identity to SEQ ID NO: 60 (e.g., between 90% and 99% sequence identity to SEQ ID NO: 60), an Fc domain may also comprise one or more amino acid substitutions described herein, for example one or more substitutions that reduce effector function (e.g., as described in Section 6.4.5.1) and/or one or more substitutions that promote Fc heterodimerization (e.g., as described in Section 6.4.5.2). [0139] In some aspects, an Fc domain comprises an amino acid sequence having at least about 90%, at least about 91%, at least about 92%, about at least 93%, at least about 94%, at eat least about 95%, at least about 96%, at least about 97%, at least about 98%, at least about 99%, or 100% sequence identity to SEQ ID NO: 61. In cases where an Fc domain comprises at least 90% sequence identity and less than 100% sequence identity to SEQ ID NO: 61 (e.g., between 90% and 99% sequence identity to SEQ ID NO: 61), an Fc domain may also comprise one or more amino acid substitutions described herein, for example one or more substitutions
that reduce effector function (e.g., as described in Section 6.4.5.1) and/or one or more substitutions that promote Fc heterodimerization (e.g., as described in Section 6.4.5.2). 6.4.5.1. Fc Domains with Altered Effector Function [0140] In some embodiments, the Fc domain comprises one or more amino acid substitutions that reduces binding to an Fc receptor and/or effector function. [0141] In a particular embodiment the Fc receptor is an Fcγ receptor. In one embodiment the Fc receptor is a human Fc receptor. In one embodiment the Fc receptor is an activating Fc receptor. In a specific embodiment the Fc receptor is an activating human Fcγ receptor, more specifically human FcγRIIIa, FcγRI or FcγRlla, most specifically human FcγRllla. In one embodiment the effector function is one or more selected from the group of complement dependent cytotoxicity (CDC), antibody-dependent cell-mediated cytotoxicity (ADCC), antibody- dependent cellular phagocytosis (ADCP), and cytokine secretion. In a particular embodiment, the effector function is ADCC. [0142] In one embodiment, the Fc domain (e.g., an Fc domain of an antigen-binding molecule) or the Fc region (e.g., one or both Fc domains of an antigen-binding molecule that can associate to form an Fc region) comprises an amino acid substitution at a position selected from the group of E233, L234, L235, N297, P331 and P329 (numberings according to Kabat EU index). In a more specific embodiment, the Fc domain or the Fc region comprises an amino acid substitution at a position selected from the group of L234, L235 and P329 (numberings according to Kabat EU index). In some embodiments, the Fc domain or the Fc region comprises the amino acid substitutions L234A and L235A (numberings according to Kabat EU index). In one such embodiment, the Fc domain or region is an Igd Fc domain or region, particularly a human Igd Fc domain or region. In one embodiment, the Fc domain or the Fc region comprises an amino acid substitution at position P329. In a more specific embodiment, the amino acid substitution is P329A or P329G, particularly P329G (numberings according to Kabat EU index). In one embodiment, the Fc domain or the Fc region comprises an amino acid substitution at position P329 and a further amino acid substitution at a position selected from E233, L234, L235, N297 and P331 (numberings according to Kabat EU index). In a more specific embodiment, the further amino acid substitution is E233P, L234A, L235A, L235E, N297A, N297D or P331S. In particular embodiments, the Fc domain or the Fc region comprises amino acid substitutions at positions P329, L234 and L235 (numberings according to Kabat EU index). In more particular embodiments, the Fc domain comprises the amino acid mutations L234A, L235A and P329G (“P329G LALA”, “PGLALA” or “LALAPG”).
[0143] Typically, the same one or more amino acid substitution is present in each of the two Fc domains of an Fc region. Thus, in a particular embodiment, each Fc domain of the Fc region comprises the amino acid substitutions L234A, L235A and P329G (Kabat EU index numbering), i.e. in each of the first and the second Fc domains in the Fc region the leucine residue at position 234 is replaced with an alanine residue (L234A), the leucine residue at position 235 is replaced with an alanine residue (L235A) and the proline residue at position 329 is replaced by a glycine residue (P329G) (numbering according to Kabat EU index). [0144] In one embodiment, the Fc domain is an IgG1 Fc domain, particularly a human IgG1 Fc domain. In some embodiments, the IgG1 Fc domain is a variant IgG1 comprising D265A, N297A mutations (EU numbering) to reduce effector function. [0145] In another embodiment, the Fc domain is an IgG4 Fc domain with reduced binding to Fc receptors. Exemplary IgG4 Fc domains with reduced binding to Fc receptors may comprise an amino acid sequence selected from Table F-2 below. In some embodiments, the Fc domain includes only the bolded portion of the sequences shown below: TABLE F-2 Fc Domain Sequence SEQ ID NO
[0136] In some aspects, an Fc domain comprises an amino acid sequence having at least about 90%, at least about 91%, at least about 92%, about at least 93%, at least about 94%, at
eat least about 95%, at least about 96%, at least about 97%, at least about 98%, at least about 99%, or 100% sequence identity to SEQ ID NO: 58. In cases where an Fc domain comprises at least 90% sequence identity and less than 100% sequence identity to SEQ ID NO: 58 (e.g., between 90% and 99% sequence identity to SEQ ID NO: 58), an Fc domain may also comprise one or more amino acid substitutions described herein, for example one or more substitutions that reduce effector function (e.g., as described in Section 6.4.5.1) and/or one or more substitutions that promote Fc heterodimerization (e.g., as described in Section 6.4.5.2). [0137] In some aspects, an Fc domain comprises an amino acid sequence having at least about 90%, at least about 91%, at least about 92%, about at least 93%, at least about 94%, at least about 95%, at least about 96%, at least about 97%, at least about 98%, at least about 99%, or 100% sequence identity to SEQ ID NO: 59. In cases where an Fc domain comprises at least 90% sequence identity and less than 100% sequence identity to SEQ ID NO: 59 (e.g., between 90% and 99% sequence identity to SEQ ID NO: 59), an Fc domain may also comprise one or more amino acid substitutions described herein, for example one or more substitutions that reduce effector function (e.g., as described in Section 6.4.5.1) and/or one or more substitutions that promote Fc heterodimerization (e.g., as described in Section 6.4.5.2). [0138] In some aspects, an Fc domain comprises an amino acid sequence having at least about 90%, at least about 91%, at least about 92%, about at least 93%, at least about 94%, at eat least about 95%, at least about 96%, at least about 97%, at least about 98%, at least about 99%, or 100% sequence identity to SEQ ID NO: 60. In cases where an Fc domain comprises at least 90% sequence identity and less than 100% sequence identity to SEQ ID NO: 60 (e.g., between 90% and 99% sequence identity to SEQ ID NO: 60), an Fc domain may also comprise one or more amino acid substitutions described herein, for example one or more substitutions that reduce effector function (e.g., as described in Section 6.4.5.1) and/or one or more substitutions that promote Fc heterodimerization (e.g., as described in Section 6.4.5.2). [0139] In some aspects, an Fc domain comprises an amino acid sequence having at least about 90%, at least about 91%, at least about 92%, about at least 93%, at least about 94%, at eat least about 95%, at least about 96%, at least about 97%, at least about 98%, at least about 99%, or 100% sequence identity to SEQ ID NO: 61. In cases where an Fc domain comprises at least 90% sequence identity and less than 100% sequence identity to SEQ ID NO: 61 (e.g., between 90% and 99% sequence identity to SEQ ID NO: 61), an Fc domain may also comprise one or more amino acid substitutions described herein, for example one or more substitutions
that reduce effector function (e.g., as described in Section 6.4.5.1) and/or one or more substitutions that promote Fc heterodimerization (e.g., as described in Section 6.4.5.2). 6.4.5.1. Fc Domains with Altered Effector Function [0140] In some embodiments, the Fc domain comprises one or more amino acid substitutions that reduces binding to an Fc receptor and/or effector function. [0141] In a particular embodiment the Fc receptor is an Fcγ receptor. In one embodiment the Fc receptor is a human Fc receptor. In one embodiment the Fc receptor is an activating Fc receptor. In a specific embodiment the Fc receptor is an activating human Fcγ receptor, more specifically human FcγRIIIa, FcγRI or FcγRlla, most specifically human FcγRllla. In one embodiment the effector function is one or more selected from the group of complement dependent cytotoxicity (CDC), antibody-dependent cell-mediated cytotoxicity (ADCC), antibody- dependent cellular phagocytosis (ADCP), and cytokine secretion. In a particular embodiment, the effector function is ADCC. [0142] In one embodiment, the Fc domain (e.g., an Fc domain of an antigen-binding molecule) or the Fc region (e.g., one or both Fc domains of an antigen-binding molecule that can associate to form an Fc region) comprises an amino acid substitution at a position selected from the group of E233, L234, L235, N297, P331 and P329 (numberings according to Kabat EU index). In a more specific embodiment, the Fc domain or the Fc region comprises an amino acid substitution at a position selected from the group of L234, L235 and P329 (numberings according to Kabat EU index). In some embodiments, the Fc domain or the Fc region comprises the amino acid substitutions L234A and L235A (numberings according to Kabat EU index). In one such embodiment, the Fc domain or region is an Igd Fc domain or region, particularly a human Igd Fc domain or region. In one embodiment, the Fc domain or the Fc region comprises an amino acid substitution at position P329. In a more specific embodiment, the amino acid substitution is P329A or P329G, particularly P329G (numberings according to Kabat EU index). In one embodiment, the Fc domain or the Fc region comprises an amino acid substitution at position P329 and a further amino acid substitution at a position selected from E233, L234, L235, N297 and P331 (numberings according to Kabat EU index). In a more specific embodiment, the further amino acid substitution is E233P, L234A, L235A, L235E, N297A, N297D or P331S. In particular embodiments, the Fc domain or the Fc region comprises amino acid substitutions at positions P329, L234 and L235 (numberings according to Kabat EU index). In more particular embodiments, the Fc domain comprises the amino acid mutations L234A, L235A and P329G (“P329G LALA”, “PGLALA” or “LALAPG”).
[0143] Typically, the same one or more amino acid substitution is present in each of the two Fc domains of an Fc region. Thus, in a particular embodiment, each Fc domain of the Fc region comprises the amino acid substitutions L234A, L235A and P329G (Kabat EU index numbering), i.e. in each of the first and the second Fc domains in the Fc region the leucine residue at position 234 is replaced with an alanine residue (L234A), the leucine residue at position 235 is replaced with an alanine residue (L235A) and the proline residue at position 329 is replaced by a glycine residue (P329G) (numbering according to Kabat EU index). [0144] In one embodiment, the Fc domain is an IgG1 Fc domain, particularly a human IgG1 Fc domain. In some embodiments, the IgG1 Fc domain is a variant IgG1 comprising D265A, N297A mutations (EU numbering) to reduce effector function. [0145] In another embodiment, the Fc domain is an IgG4 Fc domain with reduced binding to Fc receptors. Exemplary IgG4 Fc domains with reduced binding to Fc receptors may comprise an amino acid sequence selected from Table F-2 below. In some embodiments, the Fc domain includes only the bolded portion of the sequences shown below: TABLE F-2 Fc Domain Sequence SEQ ID NO
 TABLE F-2 Fc Domain Sequence SEQ ID NO
TABLE F-2 Fc Domain Sequence SEQ ID NO
 TABLE F-2 Fc Domain Sequence SEQ ID NO
TABLE F-2 Fc Domain Sequence SEQ ID NO
 TABLE F-2 Fc Domain Sequence SEQ ID NO
TABLE F-2 Fc Domain Sequence SEQ ID NO
 [0146] In a particular embodiment, the IgG4 with reduced effector function comprises the bolded portion of the amino acid sequence of SEQ ID NO:31 of WO2014/121087, sometimes referred to herein as IgG4s or hIgG4s. [0147] For heterodimeric Fc regions, it is possible to incorporate a combination of the variant IgG4 Fc sequences set forth above, for example an Fc region comprising an Fc domain comprising the amino acid sequence of SEQ ID NO:30 of WO2014/121087 (or the bolded portion thereof) and an Fc domain comprising the amino acid sequence of SEQ ID NO:37 of WO2014/121087 (or the bolded portion thereof) or an Fc region comprising an Fc domain comprising the amino acid sequence of SEQ ID NO:31 of WO2014/121087 (or the bolded portion thereof) and an Fc domain comprising the amino acid sequence of SEQ ID NO:38 of WO2014/121087 (or the bolded portion thereof). 6.4.5.2. Fc Heterodimerization Variants [0148] Certain antigen-binding molecules entail dimerization between two Fc domains that, unlike a native immunoglobulin, are operably linked to non-identical N-terminal or C-terminal regions. Inadequate heterodimerization of two Fc domains to form an Fc region has can be an
obstacle for increasing the yield of desired heterodimeric molecules and represents challenges for purification. A variety of approaches available in the art can be used in for enhancing dimerization of Fc domains that might be present in the antigen-binding molecules of the disclosure, for example as disclosed in EP 1870459A1; U.S. Patent No.5,582,996; U.S. Patent No.5,731,168; U.S. Patent No.5,910,573; U.S. Patent No.5,932,448; U.S. Patent No. 6,833,441; U.S. Patent No.7,183,076; U.S. Patent Application Publication No.2006204493A1; and PCT Publication No. WO 2009/089004A1. [0149] In some embodiments, the present disclosure provides antigen-binding molecules comprising Fc heterodimers, i.e., Fc regions comprising heterologous, non-identical Fc domains. Typically, each Fc domain in the Fc heterodimer comprises a CH3 domain of an antibody. The CH3 domains are derived from the constant region of an antibody of any isotype, class or subclass, and preferably of IgG (lgG1, lgG2, lgG3 and lgG4) class, as described in the preceding section. [0150] In a specific embodiment said modification promoting the formation of Fc heterodimers is a so-called “knob-into-hole” or “knob-in-hole” modification, comprising a “knob” modification in one of the Fc domains and a “hole” modification in the other Fc domain. The knob-into-hole technology is described e.g., in U.S. Patent No.5,731,168; US 7,695,936; Ridgway et al., 1996, Prot Eng 9:617-621, and Carter, 2001, Immunol Meth 248:7-15. Generally, the method involves introducing a protuberance (“knob”) at the interface of a first polypeptide and a corresponding cavity (“hole”) in the interface of a second polypeptide, such that the protuberance can be positioned in the cavity so as to promote heterodimer formation and hinder homodimer formation. Protuberances are constructed by replacing small amino acid side chains from the interface of the first polypeptide with larger side chains (e.g., tyrosine or tryptophan). Compensatory cavities of identical or similar size to the protuberances are created in the interface of the second polypeptide by replacing large amino acid side chains with smaller ones (e.g., alanine or threonine). [0151] Accordingly, in some embodiments, an amino acid residue in the CH3 domain of the first subunit of the Fc domain is replaced with an amino acid residue having a larger side chain volume, thereby generating a protuberance within the CH3 domain of the first subunit which is positionable in a cavity within the CH3 domain of the second subunit, and an amino acid residue in the CH3 domain of the second subunit of the Fc domain is replaced with an amino acid residue having a smaller side chain volume, thereby generating a cavity within the CH3 domain of the second subunit within which the protuberance within the CH3 domain of the first subunit is
positionable. Preferably said amino acid residue having a larger side chain volume is selected from the group consisting of arginine I, phenylalanine (F), tyrosine (Y), and tryptophan (W). Preferably said amino acid residue having a smaller side chain volume is selected from the group consisting of alanine (A), serine (S), threonine (T), and valine (V). The protuberance and cavity can be made by altering the nucleic acid encoding the polypeptides, e.g., by site-specific mutagenesis, or by peptide synthesis. An exemplary substitution is Y470T. [0152] In a specific such embodiment, in the first Fc domain the threonine residue at position 366 is replaced with a tryptophan residue (T366W), and in the Fc domain the tyrosine residue at position 407 is replaced with a valine residue (Y407V) and optionally the threonine residue at position 366 is replaced with a serine residue (T366S) and the leucine residue at position 368 is replaced with an alanine residue (L368A) (numbering according to Kabat EU index). In a further embodiment, in the first Fc domain additionally the serine residue at position 354 is replaced with a cysteine residue (S354C) or the glutamic acid residue at position 356 is replaced with a cysteine residue (E356C) (particularly the serine residue at position 354 is replaced with a cysteine residue), and in the second Fc domain additionally the tyrosine residue at position 349 is replaced by a cysteine residue (Y349C) (numbering according to Kabat EU index). In a particular embodiment, the first Fc domain comprises the amino acid substitutions S354C and T366W, and the second Fc domain comprises the amino acid substitutions Y349C, T366S, L368A and Y407V (numbering according to Kabat EU index). [0153] In some embodiments, electrostatic steering (e.g., as described in Gunasekaran et al., 2010, J Biol Chem 285(25): 19637-46) can be used to promote the association of the first and the second Fc domains of the Fc region. [0154] As an alternative, or in addition, to the use of Fc domains that are modified to promote heterodimerization, an Fc domain can be modified to allow a purification strategy that enables selections of Fc heterodimers. In one such embodiment, one polypeptide comprises a modified Fc domain that abrogates its binding to Protein A, thus enabling a purification method that yields a heterodimeric protein. See, for example, U.S. Patent No.8,586,713. As such, the antigen- binding molecules comprise a first CH3 domain and a second Ig CH3 domain, wherein the first and second Ig CH3 domains differ from one another by at least one amino acid, and wherein at least one amino acid difference reduces binding of the antigen-binding molecules to Protein A as compared to a corresponding antigen-binding molecule lacking the amino acid difference. In one embodiment, the first CH3 domain binds Protein A and the second CH3 domain contains a mutation/modification that reduces or abolishes Protein A binding such as an H95R modification
(by IMGT exon numbering; H435R by EU numbering). The second CH3 may further comprise a Y96F modification (by IMGT; Y436F by EU). This class of modifications is referred to herein as “star” mutations. [0155] In some embodiments, the Fc can contain one or more mutations (e.g., knob and hole mutations) to facilitate heterodimerization as well as star mutations to facilitate purification. 6.4.5.3. Hinge Domains [0156] The antigen-binding molecules of the disclosure can comprise an Fc domain comprising a hinge domain at its N-terminus. The hinge region can be a native or a modified hinge region. Hinge regions are typically found at the N-termini of Fc regions. The term “hinge domain”, unless the context dictates otherwise, refers to a naturally or non-naturally occurring hinge sequence that in the context of a single or monomeric polypeptide chain is a monomeric hinge domain and in the context of a dimeric polypeptide (e.g., a homodimeric or heterodimeric antigen-binding molecule formed by the association of two Fc domains) can comprise two associated hinge sequences on separate polypeptide chains. Sometimes, the two associated hinge sequences are referred to as a “hinge region”. In certain embodiments of antigen-binding molecules of the disclosure, additional iterations of hinge regions may be incorporated into the polypeptide sequence. [0157] A native hinge region is the hinge region that would normally be found between Fab and Fc domains in a naturally occurring antibody. A modified hinge region is any hinge that differs in length and/or composition from the native hinge region. Such hinges can include hinge regions from other species, such as human, mouse, rat, rabbit, shark, pig, hamster, camel, llama or goat hinge regions. Other modified hinge regions may comprise a complete hinge region derived from an antibody of a different class or subclass from that of the heavy chain Fc domain or Fc region. Alternatively, the modified hinge region may comprise part of a natural hinge or a repeating unit in which each unit in the repeat is derived from a natural hinge region. In a further alternative, the natural hinge region may be altered by converting one or more cysteine or other residues into neutral residues, such as serine or alanine, or by converting suitably placed residues into cysteine residues. By such means the number of cysteine residues in the hinge region may be increased or decreased. Other modified hinge regions may be entirely synthetic and may be designed to possess desired properties such as length, cysteine composition and flexibility.
[0158] A number of modified hinge regions have already been described for example, in U.S. Patent No.5,677,425, WO 99/15549, WO 2005/003170, WO 2005/003169, WO 2005/003170, WO 98/25971 and WO 2005/003171 and these are incorporated herein by reference. [0159] In one embodiment, an antigen-binding molecule of the disclosure comprises an Fc region in which one or both Fc domains possesses an intact hinge domain at its N-terminus. [0160] In various embodiments, positions 233-236 within a hinge region may be G, G, G and unoccupied; G, G, unoccupied, and unoccupied; G, unoccupied, unoccupied, and unoccupied; or all unoccupied, with positions numbered by EU numbering. [0161] In some embodiments, the antigen-binding molecules of the disclosure comprise a modified hinge region that reduces binding affinity for an Fcγ receptor relative to a wild-type hinge region of the same isotype (e.g., human IgG1 or human IgG4). [0162] In one embodiment, the antigen-binding molecules of the disclosure comprise an Fc region in which each Fc domain possesses an intact hinge domain at its N-terminus, where each Fc domain and hinge domain is derived from lgG4 and each hinge domain comprises the modified sequence CPPC (SEQ ID NO: 87). The core hinge region of human lgG4 contains the sequence CPSC (SEQ ID NO: 88) compared to lgG1 that contains the sequence CPPC (SEQ ID NO: 87). The serine residue present in the lgG4 sequence leads to increased flexibility in this region, and therefore a proportion of molecules form disulfide bonds within the same protein chain (an intrachain disulfide) rather than bridging to the other heavy chain in the IgG molecule to form the interchain disulfide. (Angel et al., 1993, Mol Immunol 30(1):105-108). Changing the serine residue to a proline to give the same core sequence as lgG1 allows complete formation of inter-chain disulfides in the lgG4 hinge region, thus reducing heterogeneity in the purified product. This altered isotype is termed lgG4P. 6.4.5.3.1. Chimeric Hinge Sequences [0163] The hinge domain can be a chimeric hinge domain (also “chimeric hinge region”). [0164] For example, a chimeric hinge domain may comprise an “upper hinge” sequence, derived from a human IgG1, a human IgG2 or a human IgG4 hinge region, combined with a “lower hinge” sequence, derived from a human IgG1, a human IgG2 or a human IgG4 hinge region. [0165] In particular embodiments, a chimeric hinge region comprises the amino acid sequence EPKSCDKTHTCPPCPAPPVA (SEQ ID NO: 89) (previously disclosed as SEQ ID NO:8 of WO2014/121087, which is incorporated by reference in its entirety herein) or
ESKYGPPCPPCPAPPVA (SEQ ID NO: 90) (previously disclosed as SEQ ID NO:9 of WO2014/121087). Such chimeric hinge sequences can be suitably linked to an IgG4 CH2 region (for example by incorporation into an IgG4 Fc domain, for example a human or murine Fc domain, which can be further modified in the CH2 and/or CH3 domain to reduce effector function, for example as described in Section 6.4.5.1). 6.4.5.3.2. Hinge Sequences with Reduced Effector Function [0166] In further embodiments, the hinge region can be modified to reduce effector function, for example as described in WO2016161010A2, which is incorporated by reference in its entirety herein. In various embodiments, the positions 233-236 of the modified hinge region are G, G, G and unoccupied; G, G, unoccupied, and unoccupied; G, unoccupied, unoccupied, and unoccupied; or all unoccupied, with positions numbered by EU numbering (as shown in FIG.1 of WO2016161010A2). These segments can be represented as GGG-, GG--, G--- or ---- with “-“ representing an unoccupied position. [0167] Position 236 is unoccupied in canonical human IgG2 but is occupied by in other canonical human IgG isotypes. Positions 233-235 are occupied by residues other than G in all four human isotypes (as shown in FIG.1 of WO2016161010A2). [0168] The hinge modification within positions 233-236 can be combined with position 228 being occupied by P. Position 228 is naturally occupied by P in human IgG1 and IgG2 but is occupied by S in human IgG4 and R in human IgG3. An S228P mutation in an IgG4 antibody is advantageous in stabilizing an IgG4 antibody and reducing exchange of heavy chain light chain pairs between exogenous and endogenous antibodies. Preferably positions 226-229 are occupied by C, P, P and C respectively. [0169] Exemplary hinge regions have residues 226-236, sometimes referred to as middle (or core) and lower hinge, occupied by the modified hinge sequences designated GGG-(233-236), GG—(233-236), G---(233-236) and no G(233-236). Optionally, the hinge domain amino acid sequence comprises CPPCPAPGGG-GPSVF (SEQ ID NO: 91) (previously disclosed as SEQ ID NO:1 of WO2016161010A2), CPPCPAPGG—GPSVF (SEQ ID NO: 92) (previously disclosed as SEQ ID NO:2 of WO2016161010A2), CPPCPAPG---GPSVF (SEQ ID NO: 93) (previously disclosed as SEQ ID NO:3 of WO2016161010A2), or CPPCPAP----GPSVF (SEQ ID NO: 94) (previously disclosed as SEQ ID NO:4 of WO2016161010A2). [0170] The modified hinge regions described above can be incorporated into a heavy chain constant region, which typically include CH2 and CH3 domains, and which may have an
additional hinge segment (e.g., an upper hinge) flanking the designated region. Such additional constant region segments present are typically of the same isotype, preferably a human isotype, although can be hybrids of different isotypes. The isotype of such additional human constant regions segments is preferably human IgG4 but can also be human IgG1, IgG2, or IgG3 or hybrids thereof in which domains are of different isotypes. Exemplary sequences of human IgG1, IgG2 and IgG4 are shown in FIGS.2-4 of WO2016161010A2. [0171] In specific embodiments, the modified hinge sequences can be linked to an IgG4 CH2 region (for example by incorporation into an IgG4 Fc domain, for example a human or murine Fc domain, which can be further modified in the CH2 and/or CH3 domain to reduce effector function, for example as described in Section 6.4.5.1). 6.5. Nucleic Acids and Host Cells [0172] In another aspect, the disclosure provides nucleic acids encoding the antigen-binding molecules of the disclosure. In some embodiments, the antigen-binding molecules are encoded by a single nucleic acid. In other embodiments, for example in the case of a heterodimeric molecule or a molecule comprising a component composed of more than one polypeptide chain, the antigen-binding molecules can be encoded by a plurality (e.g., two, three, four or more) nucleic acids. [0173] A single nucleic acid can encode an antigen-binding molecule that comprises a single polypeptide chain, an antigen-binding molecule that comprises two or more polypeptide chains, or a portion of an antigen-binding molecule that comprises more than two polypeptide chains (for example, a single nucleic acid can encode two polypeptide chains of an antigen-binding molecule comprising three, four or more polypeptide chains, or three polypeptide chains of an antigen-binding molecule comprising four or more polypeptide chains). For separate control of expression, the open reading frames encoding two or more polypeptide chains can be under the control of separate transcriptional regulatory elements (e.g., promoters and/or enhancers). The open reading frames encoding two or more polypeptides can also be controlled by the same transcriptional regulatory elements, and separated by internal ribosome entry site (IRES) sequences allowing for translation into separate polypeptides. [0174] In some embodiments, an antigen-binding molecule comprising two or more polypeptide chains is encoded by two or more nucleic acids. The number of nucleic acids encoding an antigen-binding molecule can be equal to or less than the number of polypeptide chains in the
antigen-binding molecule (for example, when more than one polypeptide chains are encoded by a single nucleic acid). [0175] The nucleic acids of the disclosure can be DNA (e.g., plasmid) or RNA (e.g., mRNA). [0176] In another aspect, the disclosure provides host cells and vectors containing the nucleic acids of the disclosure. The nucleic acids may be present in a single vector or separate vectors present in the same host cell or separate host cell, as described in more detail herein below. 6.6. Pharmaceutical Compositions [0177] The antigen-binding molecules of the disclosure may be in the form of compositions comprising the antigen-binding molecule and one or more carriers, excipients and/or diluents. The compositions may be formulated for specific uses, such as for veterinary uses or pharmaceutical uses in humans. The form of the composition (e.g., dry powder, liquid formulation, etc.) and the excipients, diluents and/or carriers used will depend upon the intended uses of the antigen-binding molecule and, for therapeutic uses, the mode of administration. [0178] For therapeutic uses, the compositions may be supplied as part of a sterile, pharmaceutical composition that includes a pharmaceutically acceptable carrier. This composition can be in any suitable form (depending upon the desired method of administering it to a patient). The pharmaceutical composition can be administered to a patient by a variety of routes such as orally, transdermally, subcutaneously, intranasally, intravenously, intramuscularly, intratumorally, intrathecally, topically, or locally. The most suitable route for administration in any given case will depend on the particular antibody, the subject, and the nature and severity of the disease and the physical condition of the subject. Typically, the pharmaceutical composition will be administered intravenously or subcutaneously. [0179] Pharmaceutical compositions can be conveniently presented in unit dosage forms containing a predetermined amount of an antigen-binding molecule of the disclosure per dose. The quantity of antigen-binding molecule included in a unit dose will depend on the disease being treated, as well as other factors as are well known in the art. Such unit dosages may be in the form of a lyophilized dry powder containing an amount of antigen-binding molecule suitable for a single administration, or in the form of a liquid. Dry powder unit dosage forms may be packaged in a kit with a syringe, a suitable quantity of diluent and/or other components useful for administration. Unit dosages in liquid form may be conveniently supplied in the form of a syringe pre-filled with a quantity of antigen-binding molecule suitable for a single administration.
[0180] The pharmaceutical compositions may also be supplied in bulk from containing quantities of antigen-binding molecule suitable for multiple administrations. [0181] Pharmaceutical compositions may be prepared for storage as lyophilized formulations or aqueous solutions by mixing an antigen-binding molecule having the desired degree of purity with optional pharmaceutically-acceptable carriers, excipients or stabilizers typically employed in the art (all of which are referred to herein as “carriers”), i.e., buffering agents, stabilizing agents, preservatives, isotonifiers, non-ionic detergents, antioxidants, and other miscellaneous additives. See, Remington’s Pharmaceutical Sciences, 16th edition (Osol, ed.1980). Such additives should be nontoxic to the recipients at the dosages and concentrations employed. [0182] Buffering agents help to maintain the pH in the range which approximates physiological conditions. They may be present at a wide variety of concentrations, but will typically be present in concentrations ranging from about 2 mM to about 50 mM. Suitable buffering agents for use with the present disclosure include both organic and inorganic acids and salts thereof such as citrate buffers (e.g., monosodium citrate-disodium citrate mixture, citric acid-trisodium citrate mixture, citric acid-monosodium citrate mixture, etc.), succinate buffers (e.g., succinic acid- monosodium succinate mixture, succinic acid-sodium hydroxide mixture, succinic acid-disodium succinate mixture, etc.), tartrate buffers (e.g., tartaric acid-sodium tartrate mixture, tartaric acid- potassium tartrate mixture, tartaric acid-sodium hydroxide mixture, etc.), fumarate buffers (e.g., fumaric acid-monosodium fumarate mixture, fumaric acid-disodium fumarate mixture, monosodium fumarate-disodium fumarate mixture, etc.), gluconate buffers (e.g., gluconic acid- sodium glyconate mixture, gluconic acid-sodium hydroxide mixture, gluconic acid-potassium glyuconate mixture, etc.), oxalate buffer (e.g., oxalic acid-sodium oxalate mixture, oxalic acid- sodium hydroxide mixture, oxalic acid-potassium oxalate mixture, etc.), lactate buffers (e.g., lactic acid-sodium lactate mixture, lactic acid-sodium hydroxide mixture, lactic acid-potassium lactate mixture, etc.) and acetate buffers (e.g., acetic acid-sodium acetate mixture, acetic acid- sodium hydroxide mixture, etc.). Additionally, phosphate buffers, histidine buffers and trimethylamine salts such as Tris can be used. [0183] Preservatives may be added to retard microbial growth, and can be added in amounts ranging from about 0.2%-1 % (w/v). Suitable preservatives for use with the present disclosure include phenol, benzyl alcohol, meta-cresol, methyl paraben, propyl paraben, octadecyldimethylbenzyl ammonium chloride, 59enzalkonium halides (e.g., chloride, bromide, and iodide), hexamethonium chloride, and alkyl parabens such as methyl or propyl paraben, catechol, resorcinol, cyclohexanol, and 3-pentanol. Isotonicifiers sometimes known as
“stabilizers” can be added to ensure isotonicity of liquid compositions of the present disclosure and include polyhydric sugar alcohols, for example trihydric or higher sugar alcohols, such as glycerin, erythritol, arabitol, xylitol, sorbitol and mannitol. Stabilizers refer to a broad category of excipients which can range in function from a bulking agent to an additive which solubilizes the therapeutic agent or helps to prevent denaturation or adherence to the container wall. Typical stabilizers can be polyhydric sugar alcohols (enumerated above); amino acids such as arginine, lysine, glycine, glutamine, asparagine, histidine, alanine, ornithine, L-leucine, 2-phenylalanine, glutamic acid, threonine, etc., organic sugars or sugar alcohols, such as lactose, trehalose, stachyose, mannitol, sorbitol, xylitol, ribitol, myoinisitol, galactitol, glycerol and the like, including cyclitols such as inositol; polyethylene glycol; amino acid polymers; sulfur containing reducing agents, such as urea, glutathione, thioctic acid, sodium thioglycolate, thioglycerol, a- monothioglycerol and sodium thio sulfate; low molecular weight polypeptides (e.g., peptides of 10 residues or fewer); proteins such as human serum albumin, bovine serum albumin, gelatin or immunoglobulins; hydrophylic polymers, such as polyvinylpyrrolidone monosaccharides, such as xylose, mannose, fructose, glucose; disaccharides such as lactose, maltose, sucrose and trehalose; and trisaccacharides such as raffinose; and polysaccharides such as dextran. Stabilizers may be present in amounts ranging from 0.5 to 10 wt % per wt of antigen-binding molecule. [0184] Non-ionic surfactants or detergents (also known as “wetting agents”) may be added to help solubilize the glycoprotein as well as to protect the glycoprotein against agitation-induced aggregation, which also permits the formulation to be exposed to shear surface stressed without causing denaturation of the protein. Suitable non-ionic surfactants include polysorbates (20, 80, etc.), polyoxamers (184, 188 etc.), and pluronic polyols. Non-ionic surfactants may be present in a range of about 0.05 mg/mL to about 1.0 mg/mL, for example about 0.07 mg/mL to about 0.2 mg/mL. [0185] Additional miscellaneous excipients include bulking agents (e.g., starch), chelating agents (e.g., EDTA), antioxidants (e.g., ascorbic acid, methionine, vitamin E), and cosolvents. 6.6.1. Pharmaceutical Compositions for Delivery of Antigen-Binding Molecule Encoding Nucleic Acids [0186] An antigen-binding molecule of the disclosure can be delivered by means of a nucleic acid encoding the antigen-binding molecule, for example as a plasmid, DNA, mRNA or through viral vectors encoding the antigen-binding molecule under the control of a suitable promoter.
[0187] Exemplary vectors include adenovirus- or AAV-based therapeutics. Non-limiting examples of adenovirus-based or AAV-based therapeutics for use in the methods, uses or compositions herein include, but are not limited to: rAd-p53, which is a recombinant adenoviral vector encoding the wild-type human tumor suppressor protein p53, for example, for the use in treating a cancer (also known as Gendicine®, Genkaxin®, Qi et al., 2006, Modern Oncology, 14:1295-1297); Ad5_d11520, which is an adenovirus lacking the E1B gene for inactivating host p53 (also called H101 or ONYX-015; see, e.g., Russell et al., 2012, Nature Biotechnology 30:658-670); AD5-D24-GM-CSF, an adenovirus containing the cytokine GM-CSF, for example, for the use in treating a cancer (Cerullo et al., 2010, Cancer Res.70:4297); rAd-HSVtk, a replication deficient adenovirus with HSV thymidine kinase gene, for example, for the treatment of cancer (developed as Cerepro®, Ark Therapeutics, see e.g. U.S. Pat. No.6,579,855; developed as ProstAtak™ by Advantagene; International PCT Appl. No. WO2005/049094); rAd- TNFα, a replication-deficient adenoviral vector expressing human tumor necrosis factor alpha (TNFα) under the control of the chemoradiation-inducible EGR-1 promoter, for example, for the treatment of cancer (TNFerade™, GenVec; Rasmussen et al., 2002, Cancer Gene Ther.9:951- 7; Ad-IFNβ, an adenovirus serotype 5 vector from which the E1 and E3 genes have been deleted expressing the human interferon-beta gene under the direction of the cytomegalovirus (CMV) immediate-early promoter, for example for treating cancers (BG00001 and H5.110CMVhIFN-β, Biogen; Sterman et al., 2010, Mol. Ther.18:852-860). Additional vectors are recognized in the art and include, for example, lentiviral vectors (e.g., VSV), retroviral vectors, and others. [0188] Any now-known or future-developed delivery vector, natural or engineered, can be used in the delivery of an antigen-binding molecule of the disclosure. In some embodiments, the delivery vector is a viral vector, e.g., comprises a virus, viral capsid, viral genome etc. In some embodiments, the delivery vector is a naked nucleic acid, e.g., an episome. In some embodiments, the delivery vector comprises a nucleic acid complex. Exemplary non-limiting nucleic acid complexes for use as a delivery vector include lipoplexes, polymersomes, polypexes, dendrimers, inorganic nanoparticles (e.g., polynucleotide coated gold, silica, iron oxide, calcium phosphate, etc.). In some embodiments, a delivery vector as described herein comprises a combination of a viral vector, naked nucleic acids, and nucleic acid complexes. [0189] In one embodiment, the delivery vector is a virus, including a retrovirus, adenovirus, herpes simplex virus, pox virus, vaccinia virus, lentivirus, or an adeno-associated virus. In one embodiment, the delivery vector is an adeno-associated virus (AAV), including serotypes AAV1,
AAV2, AAV3, AAV4, AAV5, AAV6, AAV7, AAV8, AAV9, AAV10, and AAV11, or engineered or naturally selected variants thereof. [0190] In one embodiment, a nucleic acid encoding an antigen-binding molecule (or component thereof) also contains adeno-associated virus (AAV) nucleic acid sequence. In one embodiment, the vector is a chimeric adeno-associated virus containing genetic elements from two or more serotypes. For example, an AAV vector with rep genes from AAV1 and cap genes from AAV2 (designated as AAV1/2 or AAV RC1/2) may be used as a delivery vector to deliver an antigen-binding molecule expressing nucleic acid to a cell or a cell of a patient in need. In one embodiment, the delivery vector is an AAV1/2, AAV1/3, AAV1/4, AAV1/5, AAV1/6, AAV1/7, AAV1/8, AAV1/9, AAV1/10, AAV1/11, AAV2/1, AAV2/3, AAV2/4, AAV2/5, AAV2/6, AAV2/7, AAV2/8, AAV2/9, AAV2/10, AAV2/11, AAV3/1, AAV3/2, AAV3/4, AAV3/5, AAV3/6, AAV3/7, AAV3/8, AAV3/9, AAV3/10, AAV3/10, AAV4/1, AAV4/2, AAV4/3, AAV4/5, AAV4/6, AAV4/7, AAV4/8, AAV4/9, AAV4/10, AAV4/11, AAV5/1, AAV5/2, AAV5/3, AAV5/4, AAV5/6, AAV5/7, AAV5/8, AAV5/9, AAV5/10, AAV5/11, AAV6/1, AAV6/2, AAV6/3, AAV6/4, AAV6/5, AAV6/7, AAV6/8, AAV6/9, AAV6/10, AAV6/10, AAV7/1, AAV7/2, AAV7/3, AAV7/4, AAV7/5, AAV7/6, AAV7/8, AAV7/9, AAV7/10, AAV7/11, AAV8/1, AAV8/2, AAV8/3, AAV8/4, AAV8/5, AAV8/6, AAV8/7, AAV8/9, AAV8/10, AAV8/11, AAV9/1, AAV9/2, AAV9/3, AAV9/4, AAV9/5, AAV9/6, AAV9/7, AAV9/8, AAV9/10, AAV9/11, AAV10/1, AAV10/2, AAV10/3, AAV10/4, AAV10/5, AAV10/6, AAV10/7, AAV10/8, AAV10/9, AAV10/11, AAV11/1, AAV11/2, AAV11/3, AAV11/4, AAV11/5, AAV11/6, AAV11/7, AAV11/8, AAV11/9, AAV11/10, chimeric viral vector or, derivative thereof. Gao et al., “Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy,” PNAS 99(18): 11854-11859, Sep.3, 2002, is incorporated herein by reference for AAV vectors and chimeric viral vectors useful as delivery vectors, and their construction and use. 6.7. Production Methods [0191] The present disclosure includes methods for producing an engineered VH. Disclosed are protein engineering methods comprising generating an engineered VH having an N-linked glycosylation site within 10 amino acids of its C-terminus. Also disclosed are methods for producing an antigen-binding molecule comprising such an engineered VH. [0192] In some embodiments, the disclosed methods comprise inserting an N-linked glycosylation site within 10 (e.g., 10, 9, 8, 7, 6, 5, 4, or 3) amino acids of the C-terminus of a VH. In some embodiments, the methods comprise inserting the amino acid sequence X1X2X3X4X5NX6X7X8X9X10X11X12 (SEQ ID NO:1), or a portion thereof, into the VH, where (a) X1,
X2, X3, X4, and X5 are each independently selected from any amino acid; (b) X6 is selected from any amino acid, where the amino acid is optionally not proline; (c) X7 is S or T, and (d) X8, X9, X10,X11, and X12 are each independently selected from any amino acid and absent. In some embodiments, the methods comprise inserting the full amino acid sequence X1X2X3X4X5NX6X7X8X9X10X11X12 into the VH. In other embodiments, the methods comprise inserting a portion of the amino acid sequence X1X2X3X4X5NX6X7X8X9X10X11X12 into the VH such that the resultant engineered VH comprises the sequence NX6X7. For example, the sequence NX6X7 may be inserted within 10 amino acids of the C-terminus of the VH, thereby generating an engineered VH. [0193] In other embodiments, the disclosed methods comprise inserting, deleting, and/or substituting one or more amino acids within the sequence of the VH such that the VH comprises an N-linked glycosylation site within 10 (e.g., 10, 9, 8, 7, 6, 5, 4, or 3) amino acids of the C- terminus of the VH. In some embodiments, the methods comprise inserting, deleting, and/or substituting one or more amino acids within the sequence of the VH such that the VH comprises the amino acid sequence X1X2X3X4X5NX6X7X8X9X10X11X12 (SEQ ID NO:1) within 10 amino acids of its C-terminus, where (a) X1, X2, X3, X4, and X5 are each independently selected from any amino acid; (b) X6 is selected from any amino acid, where the amino acid is optionally not proline; (c) X7 is S or T, and (d) X8, X9, X10,X11, and X12 are each independently selected from any amino acid and absent. The methods may comprise inserting, deleting, and/or substituting 1, 2, 3, 4, 5, or more amino acids such that the VH comprises the N-linked glycosylation site. In some embodiments, a single amino acid is inserted such that the engineered VH comprises the sequence NX6X7, for example insertion of an asparagine residue prior to the sequence SS or ST. In some embodiments, a single amino acid residue is substituted such that that the engineered VH comprises the sequence NX6X7, for example substitution of an asparagine residue for X in the sequence XSS or XST to generate the sequence NSS or NST. In one embodiment, the methods comprise inserting an asparagine residue between the third and fourth positions of the sequence VTVSS (SEQ ID NO: 62) of a VH, generating the sequence VTVNSS (SEQ ID NO: 50) at the C-terminus of the engineered VH. In some embodiments, two or more amino acid residues are inserted, deleted, and/or substituted such that that the engineered VH comprises the sequence NX6X7, for example both insertion of an asparagine residue prior to the sequence SX and also substitution of a serine or threonine residue for X, generating the sequence NSS or NST. In one embodiment, the methods comprise inserting an N residue between the third and fourth positions of the sequence VTVSS (SEQ ID NO: 62) of a
VH and also substituting a T residue at the fifth position of this sequence, generating the sequence VTVNST (SEQ ID NO: 51) at the C-terminus of the engineered VH. [0194] As disclosed herein, engineered VHs of the disclosure are useful for decreasing binding of anti-drug antibodies to an antigen-binding molecule. Accordingly, disclosed are methods for reducing antigenicity of an antigen-binding molecule comprising producing an antigen-binding molecule comprising an engineered VH as disclosed herein. As used herein, “antigenicity” describes the degree to which an antigen-binding molecule binds to anti-drug antibodies from a subject. In some embodiments, disclosed are methods for reducing antigenicity of an antigen- binding molecule by at least 10%, at least 20%, at least 30%, at least 40%, at least 50%, at least 60%, at least 70%, at least 80%, at least 90%, at least 95%, at least 99%, or more (e.g., as measured by an ADA reactivity assay as described in Section 9.1.5), comprising incorporating an N-linked glycosylation site within 10 amino acids of the C-terminus of one or more VHs of the antigen-binding molecule. [0195] References herein to “modifying,” “inserting an amino acid,” “deleting an amino acid,” “substituting an amino acid,” and the like, in reference to generating an engineered VH, are used purely for convenience and do not require that the engineered VH be derived or obtained directly from a particular VH. Such references describe an engineered VH that is related in sequence and has one or more sequence differences relative to the particular reference VH. For example, where a method for generating an engineered VH comprises “substituting one or more amino acids” of a VH, such a description includes generation of the engineered VH through mutagenesis of a coding sequence for the VH and also includes direct synthesis of an engineered VH having the one or more amino acid differences (e.g., insertions, deletions or substitutions) relative to the VH. Sometimes, for convenience only, the reference VH is referred to as a parental VH and an antigen-binding molecule that incorporates the reference VH instead of the engineered VH but is otherwise identical is referred to as a counterpart antigen-binding molecule. 6.8. Therapeutic Methods [0196] Disclosed herein, in some aspects, are therapeutic methods comprising administering an antigen-binding molecule comprising an engineered VH of the present disclosure. As described herein, engineered VHs having an N-glycosylation site within 10 amino acids of their C-terminus have reduced binding to anti-drug antibodies relative to a corresponding non-engineered VH. Accordingly, without being bound by theory, the disclosed therapeutic methods, which involve administration of an antigen-binding molecule comprising an engineered VH of the disclosure,
are understood to have reduced adverse immune response and/or increased durability relative to treatment methods involving administration of a counterpart antigen-binding molecule in which the VH is not engineered. [0197] In some embodiments, the present disclosure provides methods for reducing an adverse immune response associated with an antigen-binding molecule therapeutic. The methods typically comprise administering to a subject an antigen-binding molecule comprising an engineered VH having an N-glycosylation site within 10 amino acids (e.g.,10, 9, 8, 7, 6, 5, 4, or 3 amino acids) of its C-terminus, such as an engineered VH as described in Section 6.3. In particular embodiments, administration of an antigen-binding molecule comprising an engineered VH described herein reduces an adverse immune response associated with an antigen-binding molecule (comprising a non-engineered VH) by at least 10%, at least 20%, at least 30%, at least 40%, at least 50%, at least 60%, at least 70%, at least 80%, at least 90%, at least 95%, at least 99%, or more. The subject may be administered an antigen-binding molecule comprising an engineered VH following an adverse reaction to a corresponding antigen-binding molecule comprising a non-engineered VH. [0198] In some embodiments, the present disclosure provides methods for increasing treatment durability of an antigen-binding molecule therapeutic. The methods typically comprise administering to a subject an antigen-binding molecule comprising an engineered VH having an N-glycosylation site within 10 amino acids (e.g.,10, 9, 8, 7, 6, 5, 4, or 3 amino acids) of its C- terminus, such as an engineered VH as described in Section 6.3. In particular embodiments, administration of an antigen-binding molecule comprising an engineered VH described herein increases treatment durability of an antigen-binding molecule (comprising a non-engineered VH) by at least 10%, at least 20%, at least 30%, at least 40%, at least 50%, at least 60%, at least 70%, at least 80%, at least 90%, at least 100%, at least 200%, at least 300%, or more. The subject may be administered an antigen-binding molecule comprising an engineered VH following an adverse reaction to a corresponding antigen-binding molecule comprising a non- engineered VH. [0199] Certain antigen-binding molecules of the disclosure can, in some embodiments, be used in the treatment of a proliferative disorder (e.g., cancer) that expresses a tumor-associated antigen. In particular embodiments, the cancer is acute lymphoblastic leukemia (ALL), acute myeloid leukemia (AML), adrenocortical carcinoma, anal cancer, appendix cancer, astrocytoma, basal cell carcinoma, brain tumor, bile duct cancer, bladder cancer, bone cancer, breast cancer, bronchial tumor, Burkitt Lymphoma, carcinoma of unknown primary origin, cardiac tumor,
cervical cancer, chordoma, chronic lymphocytic leukemia (CLL), chronic myelogenous leukemia (CML), chronic myeloproliferative neoplasm, colon cancer, colorectal cancer, craniopharyngioma, cutaneous T-cell lymphoma, ductal carcinoma, embryonal tumor, endometrial cancer, ependymoma, esophageal cancer, esthesioneuroblastoma, fibrous histiocytoma, Ewing sarcoma, eye cancer, germ cell tumor, gallbladder cancer, gastric cancer, gastrointestinal carcinoid tumor, gastrointestinal stromal tumor, gestational trophoblastic disease, glioma, head and neck cancer, hairy cell leukemia, hepatocellular cancer, histiocytosis, Hodgkin lymphoma, hypopharyngeal cancer, intraocular melanoma, islet cell tumor, Kaposi sarcoma, kidney cancer, Langerhans cell histiocytosis, laryngeal cancer, leukemia, lip and oral cavity cancer, liver cancer, lobular carcinoma in situ, lung cancer, lymphoma, macroglobulinemia, malignant fibrous histiocytoma, melanoma, Merkel cell carcinoma, mesothelioma, metastatic squamous neck cancer with occult primary, midline tract carcinoma involving NUT gene, mouth cancer, multiple endocrine neoplasia syndrome, multiple myeloma, mycosis fungoides, myelodysplastic syndrome, myelodysplastic/myeloproliferative neoplasm, nasal cavity and para-nasal sinus cancer, nasopharyngeal cancer, neuroblastoma, non-Hodgkin lymphoma, non-small cell lung cancer, oropharyngeal cancer, osteosarcoma, ovarian cancer, pancreatic cancer, papillomatosis, paraganglioma, parathyroid cancer, penile cancer, pharyngeal cancer, pheochromocytomas, pituitary tumor, pleuropulmonary blastoma, primary central nervous system lymphoma, prostate cancer, rectal cancer, renal cell cancer, renal pelvis and ureter cancer, retinoblastoma, rhabdoid tumor, salivary gland cancer, Sezary syndrome, skin cancer, small cell lung cancer, small intestine cancer, soft tissue sarcoma, spinal cord tumor, stomach cancer, T-cell lymphoma, teratoid tumor, testicular cancer, throat cancer, thymoma and thymic carcinoma, thyroid cancer, urethral cancer, uterine cancer, vaginal cancer, vulvar cancer, or Wilms tumor. [0200] Table I below shows exemplary indications for which antigen-binding molecules targeting particular TAAs can be used. TABLE I E l f T A i t d A ti I di ti c,
[0146] In a particular embodiment, the IgG4 with reduced effector function comprises the bolded portion of the amino acid sequence of SEQ ID NO:31 of WO2014/121087, sometimes referred to herein as IgG4s or hIgG4s. [0147] For heterodimeric Fc regions, it is possible to incorporate a combination of the variant IgG4 Fc sequences set forth above, for example an Fc region comprising an Fc domain comprising the amino acid sequence of SEQ ID NO:30 of WO2014/121087 (or the bolded portion thereof) and an Fc domain comprising the amino acid sequence of SEQ ID NO:37 of WO2014/121087 (or the bolded portion thereof) or an Fc region comprising an Fc domain comprising the amino acid sequence of SEQ ID NO:31 of WO2014/121087 (or the bolded portion thereof) and an Fc domain comprising the amino acid sequence of SEQ ID NO:38 of WO2014/121087 (or the bolded portion thereof). 6.4.5.2. Fc Heterodimerization Variants [0148] Certain antigen-binding molecules entail dimerization between two Fc domains that, unlike a native immunoglobulin, are operably linked to non-identical N-terminal or C-terminal regions. Inadequate heterodimerization of two Fc domains to form an Fc region has can be an
obstacle for increasing the yield of desired heterodimeric molecules and represents challenges for purification. A variety of approaches available in the art can be used in for enhancing dimerization of Fc domains that might be present in the antigen-binding molecules of the disclosure, for example as disclosed in EP 1870459A1; U.S. Patent No.5,582,996; U.S. Patent No.5,731,168; U.S. Patent No.5,910,573; U.S. Patent No.5,932,448; U.S. Patent No. 6,833,441; U.S. Patent No.7,183,076; U.S. Patent Application Publication No.2006204493A1; and PCT Publication No. WO 2009/089004A1. [0149] In some embodiments, the present disclosure provides antigen-binding molecules comprising Fc heterodimers, i.e., Fc regions comprising heterologous, non-identical Fc domains. Typically, each Fc domain in the Fc heterodimer comprises a CH3 domain of an antibody. The CH3 domains are derived from the constant region of an antibody of any isotype, class or subclass, and preferably of IgG (lgG1, lgG2, lgG3 and lgG4) class, as described in the preceding section. [0150] In a specific embodiment said modification promoting the formation of Fc heterodimers is a so-called “knob-into-hole” or “knob-in-hole” modification, comprising a “knob” modification in one of the Fc domains and a “hole” modification in the other Fc domain. The knob-into-hole technology is described e.g., in U.S. Patent No.5,731,168; US 7,695,936; Ridgway et al., 1996, Prot Eng 9:617-621, and Carter, 2001, Immunol Meth 248:7-15. Generally, the method involves introducing a protuberance (“knob”) at the interface of a first polypeptide and a corresponding cavity (“hole”) in the interface of a second polypeptide, such that the protuberance can be positioned in the cavity so as to promote heterodimer formation and hinder homodimer formation. Protuberances are constructed by replacing small amino acid side chains from the interface of the first polypeptide with larger side chains (e.g., tyrosine or tryptophan). Compensatory cavities of identical or similar size to the protuberances are created in the interface of the second polypeptide by replacing large amino acid side chains with smaller ones (e.g., alanine or threonine). [0151] Accordingly, in some embodiments, an amino acid residue in the CH3 domain of the first subunit of the Fc domain is replaced with an amino acid residue having a larger side chain volume, thereby generating a protuberance within the CH3 domain of the first subunit which is positionable in a cavity within the CH3 domain of the second subunit, and an amino acid residue in the CH3 domain of the second subunit of the Fc domain is replaced with an amino acid residue having a smaller side chain volume, thereby generating a cavity within the CH3 domain of the second subunit within which the protuberance within the CH3 domain of the first subunit is
positionable. Preferably said amino acid residue having a larger side chain volume is selected from the group consisting of arginine I, phenylalanine (F), tyrosine (Y), and tryptophan (W). Preferably said amino acid residue having a smaller side chain volume is selected from the group consisting of alanine (A), serine (S), threonine (T), and valine (V). The protuberance and cavity can be made by altering the nucleic acid encoding the polypeptides, e.g., by site-specific mutagenesis, or by peptide synthesis. An exemplary substitution is Y470T. [0152] In a specific such embodiment, in the first Fc domain the threonine residue at position 366 is replaced with a tryptophan residue (T366W), and in the Fc domain the tyrosine residue at position 407 is replaced with a valine residue (Y407V) and optionally the threonine residue at position 366 is replaced with a serine residue (T366S) and the leucine residue at position 368 is replaced with an alanine residue (L368A) (numbering according to Kabat EU index). In a further embodiment, in the first Fc domain additionally the serine residue at position 354 is replaced with a cysteine residue (S354C) or the glutamic acid residue at position 356 is replaced with a cysteine residue (E356C) (particularly the serine residue at position 354 is replaced with a cysteine residue), and in the second Fc domain additionally the tyrosine residue at position 349 is replaced by a cysteine residue (Y349C) (numbering according to Kabat EU index). In a particular embodiment, the first Fc domain comprises the amino acid substitutions S354C and T366W, and the second Fc domain comprises the amino acid substitutions Y349C, T366S, L368A and Y407V (numbering according to Kabat EU index). [0153] In some embodiments, electrostatic steering (e.g., as described in Gunasekaran et al., 2010, J Biol Chem 285(25): 19637-46) can be used to promote the association of the first and the second Fc domains of the Fc region. [0154] As an alternative, or in addition, to the use of Fc domains that are modified to promote heterodimerization, an Fc domain can be modified to allow a purification strategy that enables selections of Fc heterodimers. In one such embodiment, one polypeptide comprises a modified Fc domain that abrogates its binding to Protein A, thus enabling a purification method that yields a heterodimeric protein. See, for example, U.S. Patent No.8,586,713. As such, the antigen- binding molecules comprise a first CH3 domain and a second Ig CH3 domain, wherein the first and second Ig CH3 domains differ from one another by at least one amino acid, and wherein at least one amino acid difference reduces binding of the antigen-binding molecules to Protein A as compared to a corresponding antigen-binding molecule lacking the amino acid difference. In one embodiment, the first CH3 domain binds Protein A and the second CH3 domain contains a mutation/modification that reduces or abolishes Protein A binding such as an H95R modification
(by IMGT exon numbering; H435R by EU numbering). The second CH3 may further comprise a Y96F modification (by IMGT; Y436F by EU). This class of modifications is referred to herein as “star” mutations. [0155] In some embodiments, the Fc can contain one or more mutations (e.g., knob and hole mutations) to facilitate heterodimerization as well as star mutations to facilitate purification. 6.4.5.3. Hinge Domains [0156] The antigen-binding molecules of the disclosure can comprise an Fc domain comprising a hinge domain at its N-terminus. The hinge region can be a native or a modified hinge region. Hinge regions are typically found at the N-termini of Fc regions. The term “hinge domain”, unless the context dictates otherwise, refers to a naturally or non-naturally occurring hinge sequence that in the context of a single or monomeric polypeptide chain is a monomeric hinge domain and in the context of a dimeric polypeptide (e.g., a homodimeric or heterodimeric antigen-binding molecule formed by the association of two Fc domains) can comprise two associated hinge sequences on separate polypeptide chains. Sometimes, the two associated hinge sequences are referred to as a “hinge region”. In certain embodiments of antigen-binding molecules of the disclosure, additional iterations of hinge regions may be incorporated into the polypeptide sequence. [0157] A native hinge region is the hinge region that would normally be found between Fab and Fc domains in a naturally occurring antibody. A modified hinge region is any hinge that differs in length and/or composition from the native hinge region. Such hinges can include hinge regions from other species, such as human, mouse, rat, rabbit, shark, pig, hamster, camel, llama or goat hinge regions. Other modified hinge regions may comprise a complete hinge region derived from an antibody of a different class or subclass from that of the heavy chain Fc domain or Fc region. Alternatively, the modified hinge region may comprise part of a natural hinge or a repeating unit in which each unit in the repeat is derived from a natural hinge region. In a further alternative, the natural hinge region may be altered by converting one or more cysteine or other residues into neutral residues, such as serine or alanine, or by converting suitably placed residues into cysteine residues. By such means the number of cysteine residues in the hinge region may be increased or decreased. Other modified hinge regions may be entirely synthetic and may be designed to possess desired properties such as length, cysteine composition and flexibility.
[0158] A number of modified hinge regions have already been described for example, in U.S. Patent No.5,677,425, WO 99/15549, WO 2005/003170, WO 2005/003169, WO 2005/003170, WO 98/25971 and WO 2005/003171 and these are incorporated herein by reference. [0159] In one embodiment, an antigen-binding molecule of the disclosure comprises an Fc region in which one or both Fc domains possesses an intact hinge domain at its N-terminus. [0160] In various embodiments, positions 233-236 within a hinge region may be G, G, G and unoccupied; G, G, unoccupied, and unoccupied; G, unoccupied, unoccupied, and unoccupied; or all unoccupied, with positions numbered by EU numbering. [0161] In some embodiments, the antigen-binding molecules of the disclosure comprise a modified hinge region that reduces binding affinity for an Fcγ receptor relative to a wild-type hinge region of the same isotype (e.g., human IgG1 or human IgG4). [0162] In one embodiment, the antigen-binding molecules of the disclosure comprise an Fc region in which each Fc domain possesses an intact hinge domain at its N-terminus, where each Fc domain and hinge domain is derived from lgG4 and each hinge domain comprises the modified sequence CPPC (SEQ ID NO: 87). The core hinge region of human lgG4 contains the sequence CPSC (SEQ ID NO: 88) compared to lgG1 that contains the sequence CPPC (SEQ ID NO: 87). The serine residue present in the lgG4 sequence leads to increased flexibility in this region, and therefore a proportion of molecules form disulfide bonds within the same protein chain (an intrachain disulfide) rather than bridging to the other heavy chain in the IgG molecule to form the interchain disulfide. (Angel et al., 1993, Mol Immunol 30(1):105-108). Changing the serine residue to a proline to give the same core sequence as lgG1 allows complete formation of inter-chain disulfides in the lgG4 hinge region, thus reducing heterogeneity in the purified product. This altered isotype is termed lgG4P. 6.4.5.3.1. Chimeric Hinge Sequences [0163] The hinge domain can be a chimeric hinge domain (also “chimeric hinge region”). [0164] For example, a chimeric hinge domain may comprise an “upper hinge” sequence, derived from a human IgG1, a human IgG2 or a human IgG4 hinge region, combined with a “lower hinge” sequence, derived from a human IgG1, a human IgG2 or a human IgG4 hinge region. [0165] In particular embodiments, a chimeric hinge region comprises the amino acid sequence EPKSCDKTHTCPPCPAPPVA (SEQ ID NO: 89) (previously disclosed as SEQ ID NO:8 of WO2014/121087, which is incorporated by reference in its entirety herein) or
ESKYGPPCPPCPAPPVA (SEQ ID NO: 90) (previously disclosed as SEQ ID NO:9 of WO2014/121087). Such chimeric hinge sequences can be suitably linked to an IgG4 CH2 region (for example by incorporation into an IgG4 Fc domain, for example a human or murine Fc domain, which can be further modified in the CH2 and/or CH3 domain to reduce effector function, for example as described in Section 6.4.5.1). 6.4.5.3.2. Hinge Sequences with Reduced Effector Function [0166] In further embodiments, the hinge region can be modified to reduce effector function, for example as described in WO2016161010A2, which is incorporated by reference in its entirety herein. In various embodiments, the positions 233-236 of the modified hinge region are G, G, G and unoccupied; G, G, unoccupied, and unoccupied; G, unoccupied, unoccupied, and unoccupied; or all unoccupied, with positions numbered by EU numbering (as shown in FIG.1 of WO2016161010A2). These segments can be represented as GGG-, GG--, G--- or ---- with “-“ representing an unoccupied position. [0167] Position 236 is unoccupied in canonical human IgG2 but is occupied by in other canonical human IgG isotypes. Positions 233-235 are occupied by residues other than G in all four human isotypes (as shown in FIG.1 of WO2016161010A2). [0168] The hinge modification within positions 233-236 can be combined with position 228 being occupied by P. Position 228 is naturally occupied by P in human IgG1 and IgG2 but is occupied by S in human IgG4 and R in human IgG3. An S228P mutation in an IgG4 antibody is advantageous in stabilizing an IgG4 antibody and reducing exchange of heavy chain light chain pairs between exogenous and endogenous antibodies. Preferably positions 226-229 are occupied by C, P, P and C respectively. [0169] Exemplary hinge regions have residues 226-236, sometimes referred to as middle (or core) and lower hinge, occupied by the modified hinge sequences designated GGG-(233-236), GG—(233-236), G---(233-236) and no G(233-236). Optionally, the hinge domain amino acid sequence comprises CPPCPAPGGG-GPSVF (SEQ ID NO: 91) (previously disclosed as SEQ ID NO:1 of WO2016161010A2), CPPCPAPGG—GPSVF (SEQ ID NO: 92) (previously disclosed as SEQ ID NO:2 of WO2016161010A2), CPPCPAPG---GPSVF (SEQ ID NO: 93) (previously disclosed as SEQ ID NO:3 of WO2016161010A2), or CPPCPAP----GPSVF (SEQ ID NO: 94) (previously disclosed as SEQ ID NO:4 of WO2016161010A2). [0170] The modified hinge regions described above can be incorporated into a heavy chain constant region, which typically include CH2 and CH3 domains, and which may have an
additional hinge segment (e.g., an upper hinge) flanking the designated region. Such additional constant region segments present are typically of the same isotype, preferably a human isotype, although can be hybrids of different isotypes. The isotype of such additional human constant regions segments is preferably human IgG4 but can also be human IgG1, IgG2, or IgG3 or hybrids thereof in which domains are of different isotypes. Exemplary sequences of human IgG1, IgG2 and IgG4 are shown in FIGS.2-4 of WO2016161010A2. [0171] In specific embodiments, the modified hinge sequences can be linked to an IgG4 CH2 region (for example by incorporation into an IgG4 Fc domain, for example a human or murine Fc domain, which can be further modified in the CH2 and/or CH3 domain to reduce effector function, for example as described in Section 6.4.5.1). 6.5. Nucleic Acids and Host Cells [0172] In another aspect, the disclosure provides nucleic acids encoding the antigen-binding molecules of the disclosure. In some embodiments, the antigen-binding molecules are encoded by a single nucleic acid. In other embodiments, for example in the case of a heterodimeric molecule or a molecule comprising a component composed of more than one polypeptide chain, the antigen-binding molecules can be encoded by a plurality (e.g., two, three, four or more) nucleic acids. [0173] A single nucleic acid can encode an antigen-binding molecule that comprises a single polypeptide chain, an antigen-binding molecule that comprises two or more polypeptide chains, or a portion of an antigen-binding molecule that comprises more than two polypeptide chains (for example, a single nucleic acid can encode two polypeptide chains of an antigen-binding molecule comprising three, four or more polypeptide chains, or three polypeptide chains of an antigen-binding molecule comprising four or more polypeptide chains). For separate control of expression, the open reading frames encoding two or more polypeptide chains can be under the control of separate transcriptional regulatory elements (e.g., promoters and/or enhancers). The open reading frames encoding two or more polypeptides can also be controlled by the same transcriptional regulatory elements, and separated by internal ribosome entry site (IRES) sequences allowing for translation into separate polypeptides. [0174] In some embodiments, an antigen-binding molecule comprising two or more polypeptide chains is encoded by two or more nucleic acids. The number of nucleic acids encoding an antigen-binding molecule can be equal to or less than the number of polypeptide chains in the
antigen-binding molecule (for example, when more than one polypeptide chains are encoded by a single nucleic acid). [0175] The nucleic acids of the disclosure can be DNA (e.g., plasmid) or RNA (e.g., mRNA). [0176] In another aspect, the disclosure provides host cells and vectors containing the nucleic acids of the disclosure. The nucleic acids may be present in a single vector or separate vectors present in the same host cell or separate host cell, as described in more detail herein below. 6.6. Pharmaceutical Compositions [0177] The antigen-binding molecules of the disclosure may be in the form of compositions comprising the antigen-binding molecule and one or more carriers, excipients and/or diluents. The compositions may be formulated for specific uses, such as for veterinary uses or pharmaceutical uses in humans. The form of the composition (e.g., dry powder, liquid formulation, etc.) and the excipients, diluents and/or carriers used will depend upon the intended uses of the antigen-binding molecule and, for therapeutic uses, the mode of administration. [0178] For therapeutic uses, the compositions may be supplied as part of a sterile, pharmaceutical composition that includes a pharmaceutically acceptable carrier. This composition can be in any suitable form (depending upon the desired method of administering it to a patient). The pharmaceutical composition can be administered to a patient by a variety of routes such as orally, transdermally, subcutaneously, intranasally, intravenously, intramuscularly, intratumorally, intrathecally, topically, or locally. The most suitable route for administration in any given case will depend on the particular antibody, the subject, and the nature and severity of the disease and the physical condition of the subject. Typically, the pharmaceutical composition will be administered intravenously or subcutaneously. [0179] Pharmaceutical compositions can be conveniently presented in unit dosage forms containing a predetermined amount of an antigen-binding molecule of the disclosure per dose. The quantity of antigen-binding molecule included in a unit dose will depend on the disease being treated, as well as other factors as are well known in the art. Such unit dosages may be in the form of a lyophilized dry powder containing an amount of antigen-binding molecule suitable for a single administration, or in the form of a liquid. Dry powder unit dosage forms may be packaged in a kit with a syringe, a suitable quantity of diluent and/or other components useful for administration. Unit dosages in liquid form may be conveniently supplied in the form of a syringe pre-filled with a quantity of antigen-binding molecule suitable for a single administration.
[0180] The pharmaceutical compositions may also be supplied in bulk from containing quantities of antigen-binding molecule suitable for multiple administrations. [0181] Pharmaceutical compositions may be prepared for storage as lyophilized formulations or aqueous solutions by mixing an antigen-binding molecule having the desired degree of purity with optional pharmaceutically-acceptable carriers, excipients or stabilizers typically employed in the art (all of which are referred to herein as “carriers”), i.e., buffering agents, stabilizing agents, preservatives, isotonifiers, non-ionic detergents, antioxidants, and other miscellaneous additives. See, Remington’s Pharmaceutical Sciences, 16th edition (Osol, ed.1980). Such additives should be nontoxic to the recipients at the dosages and concentrations employed. [0182] Buffering agents help to maintain the pH in the range which approximates physiological conditions. They may be present at a wide variety of concentrations, but will typically be present in concentrations ranging from about 2 mM to about 50 mM. Suitable buffering agents for use with the present disclosure include both organic and inorganic acids and salts thereof such as citrate buffers (e.g., monosodium citrate-disodium citrate mixture, citric acid-trisodium citrate mixture, citric acid-monosodium citrate mixture, etc.), succinate buffers (e.g., succinic acid- monosodium succinate mixture, succinic acid-sodium hydroxide mixture, succinic acid-disodium succinate mixture, etc.), tartrate buffers (e.g., tartaric acid-sodium tartrate mixture, tartaric acid- potassium tartrate mixture, tartaric acid-sodium hydroxide mixture, etc.), fumarate buffers (e.g., fumaric acid-monosodium fumarate mixture, fumaric acid-disodium fumarate mixture, monosodium fumarate-disodium fumarate mixture, etc.), gluconate buffers (e.g., gluconic acid- sodium glyconate mixture, gluconic acid-sodium hydroxide mixture, gluconic acid-potassium glyuconate mixture, etc.), oxalate buffer (e.g., oxalic acid-sodium oxalate mixture, oxalic acid- sodium hydroxide mixture, oxalic acid-potassium oxalate mixture, etc.), lactate buffers (e.g., lactic acid-sodium lactate mixture, lactic acid-sodium hydroxide mixture, lactic acid-potassium lactate mixture, etc.) and acetate buffers (e.g., acetic acid-sodium acetate mixture, acetic acid- sodium hydroxide mixture, etc.). Additionally, phosphate buffers, histidine buffers and trimethylamine salts such as Tris can be used. [0183] Preservatives may be added to retard microbial growth, and can be added in amounts ranging from about 0.2%-1 % (w/v). Suitable preservatives for use with the present disclosure include phenol, benzyl alcohol, meta-cresol, methyl paraben, propyl paraben, octadecyldimethylbenzyl ammonium chloride, 59enzalkonium halides (e.g., chloride, bromide, and iodide), hexamethonium chloride, and alkyl parabens such as methyl or propyl paraben, catechol, resorcinol, cyclohexanol, and 3-pentanol. Isotonicifiers sometimes known as
“stabilizers” can be added to ensure isotonicity of liquid compositions of the present disclosure and include polyhydric sugar alcohols, for example trihydric or higher sugar alcohols, such as glycerin, erythritol, arabitol, xylitol, sorbitol and mannitol. Stabilizers refer to a broad category of excipients which can range in function from a bulking agent to an additive which solubilizes the therapeutic agent or helps to prevent denaturation or adherence to the container wall. Typical stabilizers can be polyhydric sugar alcohols (enumerated above); amino acids such as arginine, lysine, glycine, glutamine, asparagine, histidine, alanine, ornithine, L-leucine, 2-phenylalanine, glutamic acid, threonine, etc., organic sugars or sugar alcohols, such as lactose, trehalose, stachyose, mannitol, sorbitol, xylitol, ribitol, myoinisitol, galactitol, glycerol and the like, including cyclitols such as inositol; polyethylene glycol; amino acid polymers; sulfur containing reducing agents, such as urea, glutathione, thioctic acid, sodium thioglycolate, thioglycerol, a- monothioglycerol and sodium thio sulfate; low molecular weight polypeptides (e.g., peptides of 10 residues or fewer); proteins such as human serum albumin, bovine serum albumin, gelatin or immunoglobulins; hydrophylic polymers, such as polyvinylpyrrolidone monosaccharides, such as xylose, mannose, fructose, glucose; disaccharides such as lactose, maltose, sucrose and trehalose; and trisaccacharides such as raffinose; and polysaccharides such as dextran. Stabilizers may be present in amounts ranging from 0.5 to 10 wt % per wt of antigen-binding molecule. [0184] Non-ionic surfactants or detergents (also known as “wetting agents”) may be added to help solubilize the glycoprotein as well as to protect the glycoprotein against agitation-induced aggregation, which also permits the formulation to be exposed to shear surface stressed without causing denaturation of the protein. Suitable non-ionic surfactants include polysorbates (20, 80, etc.), polyoxamers (184, 188 etc.), and pluronic polyols. Non-ionic surfactants may be present in a range of about 0.05 mg/mL to about 1.0 mg/mL, for example about 0.07 mg/mL to about 0.2 mg/mL. [0185] Additional miscellaneous excipients include bulking agents (e.g., starch), chelating agents (e.g., EDTA), antioxidants (e.g., ascorbic acid, methionine, vitamin E), and cosolvents. 6.6.1. Pharmaceutical Compositions for Delivery of Antigen-Binding Molecule Encoding Nucleic Acids [0186] An antigen-binding molecule of the disclosure can be delivered by means of a nucleic acid encoding the antigen-binding molecule, for example as a plasmid, DNA, mRNA or through viral vectors encoding the antigen-binding molecule under the control of a suitable promoter.
[0187] Exemplary vectors include adenovirus- or AAV-based therapeutics. Non-limiting examples of adenovirus-based or AAV-based therapeutics for use in the methods, uses or compositions herein include, but are not limited to: rAd-p53, which is a recombinant adenoviral vector encoding the wild-type human tumor suppressor protein p53, for example, for the use in treating a cancer (also known as Gendicine®, Genkaxin®, Qi et al., 2006, Modern Oncology, 14:1295-1297); Ad5_d11520, which is an adenovirus lacking the E1B gene for inactivating host p53 (also called H101 or ONYX-015; see, e.g., Russell et al., 2012, Nature Biotechnology 30:658-670); AD5-D24-GM-CSF, an adenovirus containing the cytokine GM-CSF, for example, for the use in treating a cancer (Cerullo et al., 2010, Cancer Res.70:4297); rAd-HSVtk, a replication deficient adenovirus with HSV thymidine kinase gene, for example, for the treatment of cancer (developed as Cerepro®, Ark Therapeutics, see e.g. U.S. Pat. No.6,579,855; developed as ProstAtak™ by Advantagene; International PCT Appl. No. WO2005/049094); rAd- TNFα, a replication-deficient adenoviral vector expressing human tumor necrosis factor alpha (TNFα) under the control of the chemoradiation-inducible EGR-1 promoter, for example, for the treatment of cancer (TNFerade™, GenVec; Rasmussen et al., 2002, Cancer Gene Ther.9:951- 7; Ad-IFNβ, an adenovirus serotype 5 vector from which the E1 and E3 genes have been deleted expressing the human interferon-beta gene under the direction of the cytomegalovirus (CMV) immediate-early promoter, for example for treating cancers (BG00001 and H5.110CMVhIFN-β, Biogen; Sterman et al., 2010, Mol. Ther.18:852-860). Additional vectors are recognized in the art and include, for example, lentiviral vectors (e.g., VSV), retroviral vectors, and others. [0188] Any now-known or future-developed delivery vector, natural or engineered, can be used in the delivery of an antigen-binding molecule of the disclosure. In some embodiments, the delivery vector is a viral vector, e.g., comprises a virus, viral capsid, viral genome etc. In some embodiments, the delivery vector is a naked nucleic acid, e.g., an episome. In some embodiments, the delivery vector comprises a nucleic acid complex. Exemplary non-limiting nucleic acid complexes for use as a delivery vector include lipoplexes, polymersomes, polypexes, dendrimers, inorganic nanoparticles (e.g., polynucleotide coated gold, silica, iron oxide, calcium phosphate, etc.). In some embodiments, a delivery vector as described herein comprises a combination of a viral vector, naked nucleic acids, and nucleic acid complexes. [0189] In one embodiment, the delivery vector is a virus, including a retrovirus, adenovirus, herpes simplex virus, pox virus, vaccinia virus, lentivirus, or an adeno-associated virus. In one embodiment, the delivery vector is an adeno-associated virus (AAV), including serotypes AAV1,
AAV2, AAV3, AAV4, AAV5, AAV6, AAV7, AAV8, AAV9, AAV10, and AAV11, or engineered or naturally selected variants thereof. [0190] In one embodiment, a nucleic acid encoding an antigen-binding molecule (or component thereof) also contains adeno-associated virus (AAV) nucleic acid sequence. In one embodiment, the vector is a chimeric adeno-associated virus containing genetic elements from two or more serotypes. For example, an AAV vector with rep genes from AAV1 and cap genes from AAV2 (designated as AAV1/2 or AAV RC1/2) may be used as a delivery vector to deliver an antigen-binding molecule expressing nucleic acid to a cell or a cell of a patient in need. In one embodiment, the delivery vector is an AAV1/2, AAV1/3, AAV1/4, AAV1/5, AAV1/6, AAV1/7, AAV1/8, AAV1/9, AAV1/10, AAV1/11, AAV2/1, AAV2/3, AAV2/4, AAV2/5, AAV2/6, AAV2/7, AAV2/8, AAV2/9, AAV2/10, AAV2/11, AAV3/1, AAV3/2, AAV3/4, AAV3/5, AAV3/6, AAV3/7, AAV3/8, AAV3/9, AAV3/10, AAV3/10, AAV4/1, AAV4/2, AAV4/3, AAV4/5, AAV4/6, AAV4/7, AAV4/8, AAV4/9, AAV4/10, AAV4/11, AAV5/1, AAV5/2, AAV5/3, AAV5/4, AAV5/6, AAV5/7, AAV5/8, AAV5/9, AAV5/10, AAV5/11, AAV6/1, AAV6/2, AAV6/3, AAV6/4, AAV6/5, AAV6/7, AAV6/8, AAV6/9, AAV6/10, AAV6/10, AAV7/1, AAV7/2, AAV7/3, AAV7/4, AAV7/5, AAV7/6, AAV7/8, AAV7/9, AAV7/10, AAV7/11, AAV8/1, AAV8/2, AAV8/3, AAV8/4, AAV8/5, AAV8/6, AAV8/7, AAV8/9, AAV8/10, AAV8/11, AAV9/1, AAV9/2, AAV9/3, AAV9/4, AAV9/5, AAV9/6, AAV9/7, AAV9/8, AAV9/10, AAV9/11, AAV10/1, AAV10/2, AAV10/3, AAV10/4, AAV10/5, AAV10/6, AAV10/7, AAV10/8, AAV10/9, AAV10/11, AAV11/1, AAV11/2, AAV11/3, AAV11/4, AAV11/5, AAV11/6, AAV11/7, AAV11/8, AAV11/9, AAV11/10, chimeric viral vector or, derivative thereof. Gao et al., “Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy,” PNAS 99(18): 11854-11859, Sep.3, 2002, is incorporated herein by reference for AAV vectors and chimeric viral vectors useful as delivery vectors, and their construction and use. 6.7. Production Methods [0191] The present disclosure includes methods for producing an engineered VH. Disclosed are protein engineering methods comprising generating an engineered VH having an N-linked glycosylation site within 10 amino acids of its C-terminus. Also disclosed are methods for producing an antigen-binding molecule comprising such an engineered VH. [0192] In some embodiments, the disclosed methods comprise inserting an N-linked glycosylation site within 10 (e.g., 10, 9, 8, 7, 6, 5, 4, or 3) amino acids of the C-terminus of a VH. In some embodiments, the methods comprise inserting the amino acid sequence X1X2X3X4X5NX6X7X8X9X10X11X12 (SEQ ID NO:1), or a portion thereof, into the VH, where (a) X1,
X2, X3, X4, and X5 are each independently selected from any amino acid; (b) X6 is selected from any amino acid, where the amino acid is optionally not proline; (c) X7 is S or T, and (d) X8, X9, X10,X11, and X12 are each independently selected from any amino acid and absent. In some embodiments, the methods comprise inserting the full amino acid sequence X1X2X3X4X5NX6X7X8X9X10X11X12 into the VH. In other embodiments, the methods comprise inserting a portion of the amino acid sequence X1X2X3X4X5NX6X7X8X9X10X11X12 into the VH such that the resultant engineered VH comprises the sequence NX6X7. For example, the sequence NX6X7 may be inserted within 10 amino acids of the C-terminus of the VH, thereby generating an engineered VH. [0193] In other embodiments, the disclosed methods comprise inserting, deleting, and/or substituting one or more amino acids within the sequence of the VH such that the VH comprises an N-linked glycosylation site within 10 (e.g., 10, 9, 8, 7, 6, 5, 4, or 3) amino acids of the C- terminus of the VH. In some embodiments, the methods comprise inserting, deleting, and/or substituting one or more amino acids within the sequence of the VH such that the VH comprises the amino acid sequence X1X2X3X4X5NX6X7X8X9X10X11X12 (SEQ ID NO:1) within 10 amino acids of its C-terminus, where (a) X1, X2, X3, X4, and X5 are each independently selected from any amino acid; (b) X6 is selected from any amino acid, where the amino acid is optionally not proline; (c) X7 is S or T, and (d) X8, X9, X10,X11, and X12 are each independently selected from any amino acid and absent. The methods may comprise inserting, deleting, and/or substituting 1, 2, 3, 4, 5, or more amino acids such that the VH comprises the N-linked glycosylation site. In some embodiments, a single amino acid is inserted such that the engineered VH comprises the sequence NX6X7, for example insertion of an asparagine residue prior to the sequence SS or ST. In some embodiments, a single amino acid residue is substituted such that that the engineered VH comprises the sequence NX6X7, for example substitution of an asparagine residue for X in the sequence XSS or XST to generate the sequence NSS or NST. In one embodiment, the methods comprise inserting an asparagine residue between the third and fourth positions of the sequence VTVSS (SEQ ID NO: 62) of a VH, generating the sequence VTVNSS (SEQ ID NO: 50) at the C-terminus of the engineered VH. In some embodiments, two or more amino acid residues are inserted, deleted, and/or substituted such that that the engineered VH comprises the sequence NX6X7, for example both insertion of an asparagine residue prior to the sequence SX and also substitution of a serine or threonine residue for X, generating the sequence NSS or NST. In one embodiment, the methods comprise inserting an N residue between the third and fourth positions of the sequence VTVSS (SEQ ID NO: 62) of a
VH and also substituting a T residue at the fifth position of this sequence, generating the sequence VTVNST (SEQ ID NO: 51) at the C-terminus of the engineered VH. [0194] As disclosed herein, engineered VHs of the disclosure are useful for decreasing binding of anti-drug antibodies to an antigen-binding molecule. Accordingly, disclosed are methods for reducing antigenicity of an antigen-binding molecule comprising producing an antigen-binding molecule comprising an engineered VH as disclosed herein. As used herein, “antigenicity” describes the degree to which an antigen-binding molecule binds to anti-drug antibodies from a subject. In some embodiments, disclosed are methods for reducing antigenicity of an antigen- binding molecule by at least 10%, at least 20%, at least 30%, at least 40%, at least 50%, at least 60%, at least 70%, at least 80%, at least 90%, at least 95%, at least 99%, or more (e.g., as measured by an ADA reactivity assay as described in Section 9.1.5), comprising incorporating an N-linked glycosylation site within 10 amino acids of the C-terminus of one or more VHs of the antigen-binding molecule. [0195] References herein to “modifying,” “inserting an amino acid,” “deleting an amino acid,” “substituting an amino acid,” and the like, in reference to generating an engineered VH, are used purely for convenience and do not require that the engineered VH be derived or obtained directly from a particular VH. Such references describe an engineered VH that is related in sequence and has one or more sequence differences relative to the particular reference VH. For example, where a method for generating an engineered VH comprises “substituting one or more amino acids” of a VH, such a description includes generation of the engineered VH through mutagenesis of a coding sequence for the VH and also includes direct synthesis of an engineered VH having the one or more amino acid differences (e.g., insertions, deletions or substitutions) relative to the VH. Sometimes, for convenience only, the reference VH is referred to as a parental VH and an antigen-binding molecule that incorporates the reference VH instead of the engineered VH but is otherwise identical is referred to as a counterpart antigen-binding molecule. 6.8. Therapeutic Methods [0196] Disclosed herein, in some aspects, are therapeutic methods comprising administering an antigen-binding molecule comprising an engineered VH of the present disclosure. As described herein, engineered VHs having an N-glycosylation site within 10 amino acids of their C-terminus have reduced binding to anti-drug antibodies relative to a corresponding non-engineered VH. Accordingly, without being bound by theory, the disclosed therapeutic methods, which involve administration of an antigen-binding molecule comprising an engineered VH of the disclosure,
are understood to have reduced adverse immune response and/or increased durability relative to treatment methods involving administration of a counterpart antigen-binding molecule in which the VH is not engineered. [0197] In some embodiments, the present disclosure provides methods for reducing an adverse immune response associated with an antigen-binding molecule therapeutic. The methods typically comprise administering to a subject an antigen-binding molecule comprising an engineered VH having an N-glycosylation site within 10 amino acids (e.g.,10, 9, 8, 7, 6, 5, 4, or 3 amino acids) of its C-terminus, such as an engineered VH as described in Section 6.3. In particular embodiments, administration of an antigen-binding molecule comprising an engineered VH described herein reduces an adverse immune response associated with an antigen-binding molecule (comprising a non-engineered VH) by at least 10%, at least 20%, at least 30%, at least 40%, at least 50%, at least 60%, at least 70%, at least 80%, at least 90%, at least 95%, at least 99%, or more. The subject may be administered an antigen-binding molecule comprising an engineered VH following an adverse reaction to a corresponding antigen-binding molecule comprising a non-engineered VH. [0198] In some embodiments, the present disclosure provides methods for increasing treatment durability of an antigen-binding molecule therapeutic. The methods typically comprise administering to a subject an antigen-binding molecule comprising an engineered VH having an N-glycosylation site within 10 amino acids (e.g.,10, 9, 8, 7, 6, 5, 4, or 3 amino acids) of its C- terminus, such as an engineered VH as described in Section 6.3. In particular embodiments, administration of an antigen-binding molecule comprising an engineered VH described herein increases treatment durability of an antigen-binding molecule (comprising a non-engineered VH) by at least 10%, at least 20%, at least 30%, at least 40%, at least 50%, at least 60%, at least 70%, at least 80%, at least 90%, at least 100%, at least 200%, at least 300%, or more. The subject may be administered an antigen-binding molecule comprising an engineered VH following an adverse reaction to a corresponding antigen-binding molecule comprising a non- engineered VH. [0199] Certain antigen-binding molecules of the disclosure can, in some embodiments, be used in the treatment of a proliferative disorder (e.g., cancer) that expresses a tumor-associated antigen. In particular embodiments, the cancer is acute lymphoblastic leukemia (ALL), acute myeloid leukemia (AML), adrenocortical carcinoma, anal cancer, appendix cancer, astrocytoma, basal cell carcinoma, brain tumor, bile duct cancer, bladder cancer, bone cancer, breast cancer, bronchial tumor, Burkitt Lymphoma, carcinoma of unknown primary origin, cardiac tumor,
cervical cancer, chordoma, chronic lymphocytic leukemia (CLL), chronic myelogenous leukemia (CML), chronic myeloproliferative neoplasm, colon cancer, colorectal cancer, craniopharyngioma, cutaneous T-cell lymphoma, ductal carcinoma, embryonal tumor, endometrial cancer, ependymoma, esophageal cancer, esthesioneuroblastoma, fibrous histiocytoma, Ewing sarcoma, eye cancer, germ cell tumor, gallbladder cancer, gastric cancer, gastrointestinal carcinoid tumor, gastrointestinal stromal tumor, gestational trophoblastic disease, glioma, head and neck cancer, hairy cell leukemia, hepatocellular cancer, histiocytosis, Hodgkin lymphoma, hypopharyngeal cancer, intraocular melanoma, islet cell tumor, Kaposi sarcoma, kidney cancer, Langerhans cell histiocytosis, laryngeal cancer, leukemia, lip and oral cavity cancer, liver cancer, lobular carcinoma in situ, lung cancer, lymphoma, macroglobulinemia, malignant fibrous histiocytoma, melanoma, Merkel cell carcinoma, mesothelioma, metastatic squamous neck cancer with occult primary, midline tract carcinoma involving NUT gene, mouth cancer, multiple endocrine neoplasia syndrome, multiple myeloma, mycosis fungoides, myelodysplastic syndrome, myelodysplastic/myeloproliferative neoplasm, nasal cavity and para-nasal sinus cancer, nasopharyngeal cancer, neuroblastoma, non-Hodgkin lymphoma, non-small cell lung cancer, oropharyngeal cancer, osteosarcoma, ovarian cancer, pancreatic cancer, papillomatosis, paraganglioma, parathyroid cancer, penile cancer, pharyngeal cancer, pheochromocytomas, pituitary tumor, pleuropulmonary blastoma, primary central nervous system lymphoma, prostate cancer, rectal cancer, renal cell cancer, renal pelvis and ureter cancer, retinoblastoma, rhabdoid tumor, salivary gland cancer, Sezary syndrome, skin cancer, small cell lung cancer, small intestine cancer, soft tissue sarcoma, spinal cord tumor, stomach cancer, T-cell lymphoma, teratoid tumor, testicular cancer, throat cancer, thymoma and thymic carcinoma, thyroid cancer, urethral cancer, uterine cancer, vaginal cancer, vulvar cancer, or Wilms tumor. [0200] Table I below shows exemplary indications for which antigen-binding molecules targeting particular TAAs can be used. TABLE I E l f T A i t d A ti I di ti c,
 TABLE I Examples of Tumor-Associated Antigen Indications Tar et Exem lar Indication(s) ng n ma t, n t, n g uct
TABLE I Examples of Tumor-Associated Antigen Indications Tar et Exem lar Indication(s) ng n ma t, n t, n g uct
 TABLE I Examples of Tumor-Associated Antigen Indications Tar et Exem lar Indication(s) e , a,
TABLE I Examples of Tumor-Associated Antigen Indications Tar et Exem lar Indication(s) e , a,
 TABLE I Examples of Tumor-Associated Antigen Indications Tar et Exem lar Indication(s) s
TABLE I Examples of Tumor-Associated Antigen Indications Tar et Exem lar Indication(s) s
 [0201] Additional tumor-associated antigens and corresponding indications are disclosed in, e.g., Hafeez et al., 2020, Molecules 25:4764, doi:10.3390/molecules25204764, particularly in Table 1. Table 1 is incorporated by reference in its entirety here. 6.9. Combination Therapy [0202] The antigen-binding molecules according to the disclosure may be administered in combination with one or more other agents in therapy. For instance, an antigen-binding molecule of the disclosure may be co-administered with at least one additional therapeutic agent. The term “therapeutic agent” encompasses any agent administered to treat a symptom or disease in a subject in need of such treatment. Such additional therapeutic agent may comprise any active ingredients suitable for the particular indication being treated, preferably those with complementary activities that do not adversely affect each other. In certain
embodiments, an additional therapeutic agent is an immunomodulatory agent, a cytostatic agent, an inhibitor of cell adhesion, a cytotoxic agent, an activator of cell apoptosis, or an agent that increases the sensitivity of cells to apoptotic inducers. In a particular embodiment, the additional therapeutic agent is an anti-cancer agent, for example a microtubule disruptor, an antimetabolite, a topoisomerase inhibitor, a DNA intercalator, an alkylating agent, a hormonal therapy, a kinase inhibitor, a receptor antagonist, an activator of tumor cell apoptosis, or an antiangiogenic agent. [0203] Such other agents are suitably present in combination in amounts that are effective for the purpose intended. The effective amount of such other agents depends on the amount of antigen-binding molecule used, the type of disorder or treatment, and other factors discussed above. The antigen-binding molecules are generally used in the same dosages and with administration routes as described herein, or about from 1 to 99% of the dosages described herein, or in any dosage and by any route that is empirically/clinically determined to be appropriate. [0204] Such combination therapies noted above encompass combined administration (where two or more therapeutic agents are included in the same or separate compositions), and separate administration, in which case, administration of the antigen-binding molecule of the disclosure can occur prior to, simultaneously, and/or following, administration of the additional therapeutic agent and/or adjuvant. 7. SEQUENCES [0205] Certain sequences of the disclosure are provided in Table S below. TABLE S Sequences O
[0201] Additional tumor-associated antigens and corresponding indications are disclosed in, e.g., Hafeez et al., 2020, Molecules 25:4764, doi:10.3390/molecules25204764, particularly in Table 1. Table 1 is incorporated by reference in its entirety here. 6.9. Combination Therapy [0202] The antigen-binding molecules according to the disclosure may be administered in combination with one or more other agents in therapy. For instance, an antigen-binding molecule of the disclosure may be co-administered with at least one additional therapeutic agent. The term “therapeutic agent” encompasses any agent administered to treat a symptom or disease in a subject in need of such treatment. Such additional therapeutic agent may comprise any active ingredients suitable for the particular indication being treated, preferably those with complementary activities that do not adversely affect each other. In certain
embodiments, an additional therapeutic agent is an immunomodulatory agent, a cytostatic agent, an inhibitor of cell adhesion, a cytotoxic agent, an activator of cell apoptosis, or an agent that increases the sensitivity of cells to apoptotic inducers. In a particular embodiment, the additional therapeutic agent is an anti-cancer agent, for example a microtubule disruptor, an antimetabolite, a topoisomerase inhibitor, a DNA intercalator, an alkylating agent, a hormonal therapy, a kinase inhibitor, a receptor antagonist, an activator of tumor cell apoptosis, or an antiangiogenic agent. [0203] Such other agents are suitably present in combination in amounts that are effective for the purpose intended. The effective amount of such other agents depends on the amount of antigen-binding molecule used, the type of disorder or treatment, and other factors discussed above. The antigen-binding molecules are generally used in the same dosages and with administration routes as described herein, or about from 1 to 99% of the dosages described herein, or in any dosage and by any route that is empirically/clinically determined to be appropriate. [0204] Such combination therapies noted above encompass combined administration (where two or more therapeutic agents are included in the same or separate compositions), and separate administration, in which case, administration of the antigen-binding molecule of the disclosure can occur prior to, simultaneously, and/or following, administration of the additional therapeutic agent and/or adjuvant. 7. SEQUENCES [0205] Certain sequences of the disclosure are provided in Table S below. TABLE S Sequences O
 TABLE S Sequences SEQ O
TABLE S Sequences SEQ O
 TABLE S Sequences SEQ O
TABLE S Sequences SEQ O
 8. SPECIFIC EMBODIMENTS [0206] While various specific embodiments have been illustrated and described, it will be appreciated that various changes can be made without departing from the spirit and scope of the disclosure(s). The present disclosure is exemplified by the numbered embodiments set forth below. 1. An antigen-binding molecule comprising a heavy chain variable domain (VH), said VH comprising an N-linked glycosylation site within 10 amino acids of the C-terminus of the VH. 2. The antigen-binding molecule of embodiment 1, wherein the N-linked glycosylation site is within 8 amino acids of the C-terminus of the VH. 3. The antigen-binding molecule of embodiment 1, wherein the N-linked glycosylation site is within 5 amino acids of the C-terminus of the VH. 4. The antigen-binding molecule of embodiment 1, wherein the N-linked glycosylation site is within 4 amino acids of the C-terminus of the VH. 5. The antigen-binding molecule of embodiment 1, wherein N-linked glycosylation site is within 3 amino acids of the C-terminus of the VH. 6. The antigen-binding molecule of any one of embodiments 1 to 5, wherein the N- linked glycosylation site is defined by the sequence motif NX[S/T], wherein X is any amino acid, optionally wherein the amino acid is not proline. 7. An antigen-binding molecule comprising a heavy chain variable domain (VH), optionally wherein the antigen-binding molecule is an antigen-binding molecule according to any one of embodiments 1 to 6, wherein the VH comprises at its C-terminus the amino acid sequence X1X2X3X4X5NX6X7X8X9X10X11X12 (SEQ ID NO:1), wherein (a) X1, X2, X3, X4, and X5 are each independently selected from any amino acid; (b) X6 is selected from any amino acid,
optionally wherein the amino acid is not proline; (c) X7 is S or T; and (d) X8, X9, X10,X11, and X12 are each independently selected from any amino acid and absent. 8. The antigen-binding molecule of any one of embodiments 1 to 7, wherein X6 is S. 9. The antigen-binding molecule of any one of embodiments 1 to 8, wherein X7 is S. 10. The antigen-binding molecule of any one of embodiments 1 to 8, wherein X7 is T. 11. The antigen-binding molecule of any one of embodiments 7 to 10, wherein X5 is V. 12. The antigen-binding molecule of any one of embodiments 7 to 11, wherein X4 is T. 13. The antigen-binding molecule of any one of embodiments 7 to 12, wherein X3 is V. 14. The antigen-binding molecule of any one of embodiments 7 to 10, wherein X5 is S. 15. The antigen-binding molecule of any one of embodiments 7 to 10 and 14, wherein X4 is S. 16. The antigen-binding molecule of any one of embodiments 7 to 10 and 14 to 15, wherein X3 is V. 17. The antigen-binding molecule of any one of embodiments 7 to 10 and 14 to 16, wherein X2 is T. 18. The antigen-binding molecule of any one of embodiments 7 to 10 and 14 to 17, wherein X1 is V.
19. The antigen-binding molecule of any one of embodiments 7 to 18, wherein X8 is P. 20. The antigen-binding molecule of any one of embodiments 7 to 19, wherein X9 is P. 21. The antigen-binding molecule of any one of embodiments 7 to 18, wherein X8 is K. 22. The antigen-binding molecule of any one of embodiments 7 to 18 and 21, wherein X9 is P. 23. The antigen-binding molecule of any one of embodiments 7 to 18, 21, and 22 wherein X10 is G. 24. The antigen-binding molecule of any one of embodiments 7 to 18 and 21 to 23, wherein X11 is G. 25. The antigen-binding molecule of any one of embodiments 7 to 18, wherein X8 is G. 26. The antigen-binding molecule of any one of embodiments 7 to 18 and 25, wherein X9 is G. 27. The antigen-binding molecule of any one of embodiments 7 to 18, 25, and 26, wherein X10 is G. 28. The antigen-binding molecule of any one of embodiments 7 to 18 and 25 to 27, wherein X11 is G. 29. The antigen-binding molecule of any one of embodiments 7 to 18, wherein at least two of X8, X9, X10, X11, and X12 are absent. 30. The antigen-binding molecule of any one of embodiments 7 to 18, wherein at least three of X8, X9, X10, X11, and X12 are absent.
31. The antigen-binding molecule of any one of embodiments 7 to 18, wherein at least four of X8, X9, X10, X11, and X12 are absent. 32. The antigen-binding molecule of any one of embodiments 7 to 18, wherein all of X8, X9, X10, X11, and X12 are absent. 33. The antigen-binding molecule of any one of embodiments 7 to 18, wherein the VH is at least 100 amino acids in length. 34. The antigen-binding molecule of any one of embodiments 1 to 33, wherein the VH is at most 125 amino acids in length. 35. The antigen-binding molecule of any one of embodiments 1 to 34, which comprises a single chain Fv (scFv) comprising the VH. 36. The antigen-binding molecule of embodiment 35, which comprises a light chain variable domain (VL) N-terminal to the VH. 37. The antigen-binding molecule of embodiment 35, which comprises a light chain variable domain (VL) C-terminal to the VH. 38. The antigen-binding molecule of any one of embodiments 35 to 37, wherein the VH and VL are separated by a linker. 39. The antigen-binding molecule of any one of embodiments 35 to 38, wherein the scFv is at least 225 amino acids in length. 40. The antigen-binding molecule of any one of embodiments 35 to 39, wherein the scFv is at most 300 amino acids in length. 41. The antigen-binding molecule of any one of embodiments 1 to 34, further comprising a VL. 42. The antigen-binding molecule of any one of embodiments 1 to 34, which lacks a VL.
43. The antigen-binding molecule of any one of embodiments 1 to 42, wherein the antigen-binding molecule further comprises a dimerization moiety. 44. The antigen-binding molecule of embodiment 43, wherein the dimerization moiety is an Fc domain. 45. The antigen-binding molecule of embodiment 44, wherein the Fc domain is C- terminal to the VH. 46. The antigen-binding molecule of embodiment 44, wherein the Fc domain is N- terminal to the VH. 47. The antigen-binding molecule of embodiment 46, wherein the VH is at the C terminus of at least one polypeptide chain in the antigen-binding molecule. 48. The antigen-binding molecule of any one of embodiments 45 to 47, wherein the Fc domain and the VH are separated by a linker. 49. The antigen-binding molecule of embodiment 48, wherein the linker is a glycine- serine linker. 50. The antigen-binding molecule of any one of embodiments 1 to 49, further comprising an additional VH as defined in any one of embodiments 1 to 34. 51. The antigen-binding molecule of embodiment 50, wherein the antigen-binding molecule is a dimer. 52. The antigen-binding molecule of embodiment 51, wherein the antigen-binding molecule is a homodimer. 53. The antigen-binding molecule of embodiment 51, wherein the antigen-binding molecule is a heterodimer.
54. The antigen-binding molecule of any one of embodiments 1 to 53, wherein the VH comprises a glycan at the N-linked glycosylation site and/or the N residue of the X1X2X3X4X5NX6X7X8X9X10X11X12 (SEQ ID NO:1) sequence. 55. The antigen-binding molecule of embodiment 54, wherein the glycan is G1F, G2F, G1F and GlcNAc, G2S, G2F and GlycNac, or G2FS. 56. The antigen-binding molecule of any one of embodiments 1 to 55, wherein the antigen-binding molecule is a multispecific antigen-binding molecule. 57. An antigen-binding molecule, which is optionally an antigen-binding molecule of any one of embodiments 1 to 56, having the configuration illustrated in FIG.1A. 58. An antigen-binding molecule, which is optionally an antigen-binding molecule of any one of embodiments 1 to 56, having the configuration illustrated in FIG.1B. 59. An antigen-binding molecule, which is optionally an antigen-binding molecule of any one of embodiments 1 to 56, having the configuration illustrated in FIG.1C. 60. An antigen-binding molecule, which is optionally an antigen-binding molecule of any one of embodiments 1 to 56, comprising: (a) a first polypeptide chain comprising, from N-terminus to C-terminus (i) a first antigen-binding domain or component thereof, (ii) a first dimerization moiety, and (iii) a second antigen-binding domain, the second antigen-binding domain comprising a VH as defined in any one of embodiments 1 to 34; and (b) a second polypeptide chain comprising, from N-terminus to C-terminus (i) a third antigen-binding domain or component thereof, and (ii) a second dimerization moiety, wherein the first and the second dimerization moieties associate with one another to form the molecule. 61. The antigen-binding molecule of embodiment 60, wherein the second antigen- binding domain is an scFv, optionally wherein the scFv is as defined in any one of embodiments 35 to 40.
62. The antigen-binding molecule of embodiment 60 or 61, wherein the second polypeptide further comprises a fourth antigen-binding domain C-terminal to the second dimerization moiety. 63. The antigen-binding molecule of embodiment 62, wherein the fourth antigen- binding domain comprises a VH comprising an N-linked glycosylation site within 10 amino acids of the C-terminus of the VH. 64. The antigen-binding molecule of embodiment 62 or 63, wherein the fourth antigen-binding domain is an scFv. 65. The antigen-binding molecule of any one of embodiments 62 to 64, wherein the first antigen-binding domain specifically binds to a T cell antigen. 66. The antigen-binding molecule of any one of embodiments 62 to 65, wherein the second antigen-binding domain specifically binds to a T cell antigen. 67. The antigen-binding molecule of any one of embodiments 62 to 66, wherein the third antigen-binding domain specifically binds to a T cell antigen. 68. The antigen-binding molecule of any one of embodiments 62 to 67, wherein the fourth antigen-binding domain specifically binds to a T cell antigen. 69. The antigen-binding molecule of any one of embodiments 65 to 68, wherein the T cell antigen is CD3. 70. The antigen-binding molecule of any one of embodiments 65 to 68, wherein the T cell antigen is CD28. 71. The antigen-binding molecule of any one of embodiments 62 to 66, wherein the third antigen-binding domain specifically binds to a tumor-associated antigen. 72. The antigen-binding molecule of any one of embodiments 62 to 66, wherein the fourth antigen-binding domain specifically binds to a tumor-associated antigen.
73. The antigen-binding molecule of any one of embodiments 60 to 72, wherein the first and second dimerization moieties are Fc domains. 74. An antigen-binding molecule, which is optionally an antigen-binding molecule of any one of embodiments 1 to 56, comprising: (a) a first scFv; (b) a linker; and (c) a second scFv, wherein the first scFv or the second scFv comprises a VH as defined in any one of embodiments 1 to 34. 75. The antigen-binding molecule of embodiment 74, wherein the first scFv comprises a VH as defined in any one of embodiments 1 to 34. 76. The antigen-binding molecule of embodiment 75, wherein the first scFv comprises, from N- to C-terminal orientation, a VL, a linker, and a VH as defined in any one of embodiments 1 to 34. 77. The antigen-binding molecule of embodiment 75, wherein the first scFv comprises, from N- to C-terminal orientation, a VH as defined in any one of embodiments 1 to 34, a linker, and a VL. 78. The antigen-binding molecule of any one of embodiments 74 to 77, wherein the second scFv comprises a VH as defined in any one of embodiments 1 to 34. 79. The antigen-binding molecule of embodiment 78, wherein the second scFv comprises, from N- to C-terminal orientation, a VL, a linker, and a VH as defined in any one of embodiments 1 to 34. 80. The antigen-binding molecule of embodiment 78, wherein the second scFv comprises, from N- to C-terminal orientation, a VH as defined in any one of embodiments 1 to 34, a linker, and a VL.
81. The antigen-binding molecule of any one of embodiments 1 to 80, wherein the antigen-binding molecule has reduced binding to anti-drug antibodies, e.g., as measured by an ADA reactivity assay as presented in Section 7.1.5 relative to a control antigen-binding molecule. 82. The antigen-binding molecule of any one of embodiments 1 to 81, wherein the VH comprising the N-linked glycosylation site is a component of a T-cell engaging antigen- binding domain (“TCE ABD”). 83. The antigen-binding molecule of embodiment 82, wherein the TCE ABD is capable of binding to a component of the T-cell receptor (TCR) complex. 84. The antigen-binding molecule of embodiment 83, wherein the component of the TCR complex is CD3. 85. The antigen-binding molecule of embodiment 83, wherein the component of the TCR complex is TCRαβ. 86. The antigen-binding molecule of embodiment 83, wherein the component of the TCR complex is TCRγδ. 87. The antigen-binding molecule of any one of embodiments 82 to 86, wherein the TCE ABD (a) comprises the (i) CDR or (ii) VH and VL sequences of antibody set forth in Table T or (b) competes with the antibody set forth in Table T for binding to its target. 88. The antigen-binding molecule of any one of embodiments 1 to 81, wherein the VH comprising the N-linked glycosylation site is a component of an antigen-binding domain that is capable of binding to a tumor-associated antigen (“TAA ABD”). 89. The antigen-binding molecule of embodiment 88, wherein the TAA ABD is capable of binding to any tumor-associated antigen identified in Section 6.4.2. 90. The antigen-binding molecule of embodiment 88, wherein the TAA ABD is capable of binding to AFP, ALK, a BAGE protein, BIRC5 (survivin), BIRC7, β-catenin, brc-abl,
BRCA1, BORIS, CA9, carbonic anhydrase IX, caspase-8, CALR, CEACAM5 (also known as carcinoembryonic antigen or CEA), CCR5, CD19, CD20 (MS4A1), CD22, CD30, CD40, CDK4, CEA, CTLA4, cyclin-B1, CYP1B1, EGFR, EGFRvIII, ErbB2/Her2, ErbB3, ErbB4, ETV6-AML, EpCAM, EphA2, Fra-1, FOLR1, a GAGE protein (e.g., GAGE-1 or -2), GD2, GD3, GloboH, glypican-3, GM3, gp100, Her2, HLA/B-raf, HLA/k-ras, HLA/MAGE-A3, hTERT, LMP2, MAGE proteins (e.g., MAGE-1, -2, -3, -4, -6, and -12), MART-1, mesothelin, ML-IAP, Muc1, Muc2, Muc3, Muc4, Muc5, Muc16 (CA-125), MUM1, NA17, NY-BR1, NY-BR62, NY-BR85, NY-ESO1, OX40, p15, p53, PAP, PAX3, PAX5, PCTA-1, PLAC1, PRLR, PRAME, PSMA (FOLH1), RAGE proteins, Ras, RGS5, Rho, SART-1, SART-3, STEAP1, STEAP2, TAG-72, TGF-β, TMPRSS2, Thompson-nouvelle antigen (Tn), TRP-1, TRP-2, tyrosinase, or uroplakin-3. 91. The antigen-binding molecule of embodiment 88, wherein the TAA ABD is capable of binding to CTLA-4, PD1, PDL1, PDL2, B7-H3, B7-H4, BTLA, HVEM, TIM3, GAL9, LAG3, VISTA, KIR, 2B4, CD160, CGEN-15049, CHK1, or CHK2. 92. The antigen-binding molecule of embodiment 88, wherein the TAA ABD is capable of binding to CD27, CD28, 4-1BB (CD137), OX40, CD30, CD40, ICOS, lymphocyte function-associated antigen-1 (LFA-1), CD2, CD7, LIGHT, NKG2C, or B7-H3. 93. The antigen-binding molecule of embodiment 88, wherein the TAA ABD is capable of binding to syndecan, heparanase, integrins, osteopontin, link, cadherins, laminin, laminin type EGF, lectin, fibronectin, notch, nectin (e.g., nectin-4), tenascin, collagen (e.g., collagen type X), or matrixin. 94. The antigen-binding molecule of any one of embodiments 1 to 81, wherein the antigen-binding molecule comprises (1) a TCE ABD or component thereof; and (2) a TAA ABD or component thereof. 95. The antigen-binding molecule of embodiment 94, wherein the VH comprising the N-linked glycosylation site is a component of the TCE ABD. 96. The antigen-binding molecule of embodiment 95, wherein the TCE ABD is capable of binding to a component of the TCR complex.
97. The antigen-binding molecule of embodiment 96, wherein the component of the TCR complex is CD3. 98. The antigen-binding molecule of embodiment 96, wherein the component of the TCR complex is TCRαβ. 99. The antigen-binding molecule of embodiment 96, wherein the component of the TCR complex is TCRγδ. 100. The antigen-binding molecule of any one of embodiments 95 to 99, wherein the TCE ABD (a) comprises the (i) CDR or (ii) VH and VL sequences of antibody set forth in Table T or (b) competes with the antibody set forth in Table T for binding to its target. 101. The antigen-binding molecule of embodiment 94, wherein the VH comprising the N-linked glycosylation site is a component of an antigen-binding domain that is capable of binding to a tumor-associated antigen (“TAA ABD”). 102. The antigen-binding molecule of embodiment 101, wherein the TAA ABD is capable of binding to any tumor-associated antigen identified in Section 6.4.2. 103. The antigen-binding molecule of embodiment 102, wherein the TAA ABD is capable of binding to AFP, ALK, a BAGE protein, BIRC5 (survivin), BIRC7, β-catenin, brc-abl, BRCA1, BORIS, CA9, carbonic anhydrase IX, caspase-8, CALR, CEACAM5 (also known as carcinoembryonic antigen or CEA), CCR5, CD19, CD20 (MS4A1), CD22, CD30, CD40, CDK4, CEA, CTLA4, cyclin-B1, CYP1B1, EGFR, EGFRvIII, ErbB2/Her2, ErbB3, ErbB4, ETV6-AML, EpCAM, EphA2, Fra-1, FOLR1, a GAGE protein (e.g., GAGE-1 or -2), GD2, GD3, GloboH, glypican-3, GM3, gp100, Her2, HLA/B-raf, HLA/k-ras, HLA/MAGE-A3, hTERT, LMP2, MAGE proteins (e.g., MAGE-1, -2, -3, -4, -6, and -12), MART-1, mesothelin, ML-IAP, Muc1, Muc2, Muc3, Muc4, Muc5, Muc16 (CA-125), MUM1, NA17, NY-BR1, NY-BR62, NY-BR85, NY-ESO1, OX40, p15, p53, PAP, PAX3, PAX5, PCTA-1, PLAC1, PRLR, PRAME, PSMA (FOLH1), RAGE proteins, Ras, RGS5, Rho, SART-1, SART-3, STEAP1, STEAP2, TAG-72, TGF-β, TMPRSS2, Thompson-nouvelle antigen (Tn), TRP-1, TRP-2, tyrosinase, or uroplakin-3.
104. The antigen-binding molecule of embodiment 102, wherein the TAA ABD is capable of binding to CTLA-4, PD1, PDL1, PDL2, B7-H3, B7-H4, BTLA, HVEM, TIM3, GAL9, LAG3, VISTA, KIR, 2B4, CD160, CGEN-15049, CHK1, or CHK2. 105. The antigen-binding molecule of embodiment 102, wherein the TAA ABD is capable of binding to CD27, CD28, 4-1BB (CD137), OX40, CD30, CD40, ICOS, lymphocyte function-associated antigen-1 (LFA-1), CD2, CD7, LIGHT, NKG2C, or B7-H3. 106. The antigen-binding molecule of embodiment 102, wherein the TAA ABD is capable of binding to syndecan, heparanase, integrins, osteopontin, link, cadherins, laminin, laminin type EGF, lectin, fibronectin, notch, nectin (e.g., nectin-4), tenascin, collagen (e.g., collagen type X), or matrixin. 107. A pharmaceutical composition comprising the antigen-binding molecule of any one of embodiments 1 to 106. 108. A nucleic acid or plurality of nucleic acids encoding the antigen-binding molecule of any one of embodiments 1 to 106. 109. A host cell engineered to express the antigen-binding molecule of any one of embodiments 1 to 106. 110. A host cell transfected with one or more expression vectors comprising one or more nucleic acid sequences encoding the antigen-binding molecule of any one of embodiments 1 to 106 under the control of one or more promoters. 111. A method of producing the antigen-binding molecule of any one of embodiments 1 to 106 comprising culturing the host cell of embodiment 109 or 110 and recovering the antigen-binding molecule expressed thereby. 112. A method comprising administering the antigen-binding molecule of any one of embodiments 1 to 106 to a subject.
113. A method of treating cancer comprising administering to a subject in need thereof the antigen-binding molecule of any one of embodiments 1 to 106 or the pharmaceutical composition of embodiment 107. 114. The method of embodiment 113, wherein the antigen-binding molecule is a bispecific antigen-binding molecule comprising a TCE ABD and a TAA ABD and the cancer is associated with the expression of the tumor-associated antigen, e.g., as set forth in Table A. 115. A polypeptide comprising an engineered VH as defined in any one of embodiments 1 to 34. 116. A nucleic acid or plurality of nucleic acids encoding the polypeptide of embodiment 115. 117. A host cell engineered to express the polypeptide of embodiment 115 or the nucleic acid(s) of embodiment 116. 118. A method of producing the polypeptide of embodiment 115 comprising culturing the host cell of embodiment 117. 119. A protein engineering method comprising generating an antigen-binding molecule comprising a VH having an N-linked glycosylation site within 10 amino acids of its C- terminus. 120. A protein engineering method comprising generating a VH comprising an N- linked glycosylation site within 10 amino acids of its C-terminus. 121. A method of reducing antigenicity of an antigen-binding molecule comprising a VH, the method comprising incorporating an N-linked glycosylation site within 10 amino acids of the C-terminus of the VH. 122. The method embodiment 121, wherein the antigen-binding molecule further comprises an additional VH.
123. The method of embodiment 122, wherein the method further comprises incorporating an N-linked glycosylation site within 10 amino acids of the C-terminus of the additional VH. 124. The method of any one of embodiments 119 to 123, wherein the method comprises inserting the amino acid sequence X1X2X3X4X5NX6X7X8X9X10X11X12 (SEQ ID NO:1), or a portion thereof, into the VH, wherein (a) X1, X2, X3, X4, and X5 are each independently selected from any amino acid; (b) X6 is selected from any amino acid, optionally wherein the amino acid is not proline; (c) X7 is S or T, and (d) X8, X9, X10,X11, and X12 are each independently selected from any amino acid and absent. 125. The method of any one of embodiments 119 to 123, wherein the method comprises substituting one or more amino acids within the sequence of the VH such that the VH comprises the amino acid sequence X1X2X3X4X5NX6X7X8X9X10X11X12 (SEQ ID NO:1) within 10 amino acids of its C-terminus, wherein (a) X1, X2, X3, X4, and X5 are each independently selected from any amino acid; (b) X6 is selected from any amino acid, optionally wherein the amino acid is not proline; (c) X7 is S or T, and (d) X8, X9, X10,X11, and X12 are each independently selected from any amino acid and absent. 126. The method of embodiment 124 or 125, wherein X6 is S. 127. The method of any one of embodiments 124 to 126, wherein X7 is S. 128. The method of any one of embodiments 124 to 126, wherein X7 is T. 129. The method of any one of embodiments 124 to 128, wherein X5 is V. 130. The method of any one of embodiments 124 to 129, wherein X4 is T. 131. The method of any one of embodiments 124 to 130, wherein X3 is V. 132. The method of any one of embodiments 124 to 128, wherein X5 is S. 133. The method of any one of embodiments 124 to 128 and 132, wherein X4 is S.
134. The method of any one of embodiments 124 to 128 and 132 to 133, wherein X3 is V. 135. The method of any one of embodiments 124 to 128 and 132 to 134, wherein X2 is T. 136. The method of any one of embodiments 124 to 128 and 132 to 135, wherein X1 is V. 137. The method of any one of embodiments 124 to 136, wherein X8 is P. 138. The method of any one of embodiments 124 to 137, wherein X9 is P. 139. The method of any one of embodiments 124 to 136, wherein X8 is K. 140. The method of any one of embodiments 124 to 136 and 139, wherein X9 is P. 141. The method of any one of embodiments 124 to 136 and 139 to 140, wherein X10 is G. 142. The method of any one of embodiments 124 to 136 and 139 to 141, wherein X11 is G. 143. The method of any one of embodiments 124 to 136, wherein X8 is G. 144. The method of any one of embodiments 124 to 136 and 143, wherein X9 is G. 145. The method of any one of embodiments 124 to 136 and 143 to 144, wherein X10 is G. 146. The method of any one of embodiments 124 to 136 and 143 to 145, wherein X11 is G. 147. The method of any one of embodiments 124 to 136, wherein at least two of X8, X9, X10, X11, and X12 are absent.
148. The method of any one of embodiments 124 to 136, wherein at least three of X8, X9, X10, X11, and X12 are absent. 149. The method of any one of embodiments 124 to 136, wherein at least four of X8, X9, X10, X11, and X12 are absent. 150. The method of any one of embodiments 124 to 136, wherein all of X8, X9, X10, X11, and X12 are absent. 9. EXAMPLES 9.1. Materials and Methods 9.1.1. Design and Production of Antigen-binding Molecules [0207] Antigen-binding molecules were designed to comprise two polypeptides connected to one another via their Fc domains. The first polypeptide was designed to comprise from N- terminus to C-terminus: a CD3-targeting Fab, a first Fc domain, a linker, and a CD3-targeting scFv. The second polypeptide was designed to comprise from N-terminus to C-terminus: a TAA- targeting Fab, a second Fc domain, a linker, and a TAA-targeting scFv. Antigen-binding molecules were engineered by introducing amino acid modifications at or near the C-terminus of the scFvs. FIG.2A shows a parental antigen-binding molecule, FIGs.2B-2E show variants with C-terminal amino acid modifications that do not incorporate an N-glycosylation consensus sequence, and FIGs.2F and 2G show variants that incorporate an N-glycosylation consensus sequence at the C-terminus of the scFvs. [0001] All constructs were expressed in Expi293F™ cells by transient transfection (Thermo Fisher Scientific). Proteins in Expi293F supernatant were purified using the ProteinMaker system (Protein BioSolutions, Gaithersburg, MD) with HiTrap MabSelect SuRe Protein A columns (GE Healthcare). After single step elution with IgG Elution Buffer (Thermo Fisher Scientific), the proteins were neutralized, dialyzed into a final buffer of phosphate buffered saline (PBS) with 5% glycerol, aliquoted and stored at -80°C. [0208] Antigen-binding molecules and control constructs utilized in these examples are presented in Table 2. Table 2
8. SPECIFIC EMBODIMENTS [0206] While various specific embodiments have been illustrated and described, it will be appreciated that various changes can be made without departing from the spirit and scope of the disclosure(s). The present disclosure is exemplified by the numbered embodiments set forth below. 1. An antigen-binding molecule comprising a heavy chain variable domain (VH), said VH comprising an N-linked glycosylation site within 10 amino acids of the C-terminus of the VH. 2. The antigen-binding molecule of embodiment 1, wherein the N-linked glycosylation site is within 8 amino acids of the C-terminus of the VH. 3. The antigen-binding molecule of embodiment 1, wherein the N-linked glycosylation site is within 5 amino acids of the C-terminus of the VH. 4. The antigen-binding molecule of embodiment 1, wherein the N-linked glycosylation site is within 4 amino acids of the C-terminus of the VH. 5. The antigen-binding molecule of embodiment 1, wherein N-linked glycosylation site is within 3 amino acids of the C-terminus of the VH. 6. The antigen-binding molecule of any one of embodiments 1 to 5, wherein the N- linked glycosylation site is defined by the sequence motif NX[S/T], wherein X is any amino acid, optionally wherein the amino acid is not proline. 7. An antigen-binding molecule comprising a heavy chain variable domain (VH), optionally wherein the antigen-binding molecule is an antigen-binding molecule according to any one of embodiments 1 to 6, wherein the VH comprises at its C-terminus the amino acid sequence X1X2X3X4X5NX6X7X8X9X10X11X12 (SEQ ID NO:1), wherein (a) X1, X2, X3, X4, and X5 are each independently selected from any amino acid; (b) X6 is selected from any amino acid,
optionally wherein the amino acid is not proline; (c) X7 is S or T; and (d) X8, X9, X10,X11, and X12 are each independently selected from any amino acid and absent. 8. The antigen-binding molecule of any one of embodiments 1 to 7, wherein X6 is S. 9. The antigen-binding molecule of any one of embodiments 1 to 8, wherein X7 is S. 10. The antigen-binding molecule of any one of embodiments 1 to 8, wherein X7 is T. 11. The antigen-binding molecule of any one of embodiments 7 to 10, wherein X5 is V. 12. The antigen-binding molecule of any one of embodiments 7 to 11, wherein X4 is T. 13. The antigen-binding molecule of any one of embodiments 7 to 12, wherein X3 is V. 14. The antigen-binding molecule of any one of embodiments 7 to 10, wherein X5 is S. 15. The antigen-binding molecule of any one of embodiments 7 to 10 and 14, wherein X4 is S. 16. The antigen-binding molecule of any one of embodiments 7 to 10 and 14 to 15, wherein X3 is V. 17. The antigen-binding molecule of any one of embodiments 7 to 10 and 14 to 16, wherein X2 is T. 18. The antigen-binding molecule of any one of embodiments 7 to 10 and 14 to 17, wherein X1 is V.
19. The antigen-binding molecule of any one of embodiments 7 to 18, wherein X8 is P. 20. The antigen-binding molecule of any one of embodiments 7 to 19, wherein X9 is P. 21. The antigen-binding molecule of any one of embodiments 7 to 18, wherein X8 is K. 22. The antigen-binding molecule of any one of embodiments 7 to 18 and 21, wherein X9 is P. 23. The antigen-binding molecule of any one of embodiments 7 to 18, 21, and 22 wherein X10 is G. 24. The antigen-binding molecule of any one of embodiments 7 to 18 and 21 to 23, wherein X11 is G. 25. The antigen-binding molecule of any one of embodiments 7 to 18, wherein X8 is G. 26. The antigen-binding molecule of any one of embodiments 7 to 18 and 25, wherein X9 is G. 27. The antigen-binding molecule of any one of embodiments 7 to 18, 25, and 26, wherein X10 is G. 28. The antigen-binding molecule of any one of embodiments 7 to 18 and 25 to 27, wherein X11 is G. 29. The antigen-binding molecule of any one of embodiments 7 to 18, wherein at least two of X8, X9, X10, X11, and X12 are absent. 30. The antigen-binding molecule of any one of embodiments 7 to 18, wherein at least three of X8, X9, X10, X11, and X12 are absent.
31. The antigen-binding molecule of any one of embodiments 7 to 18, wherein at least four of X8, X9, X10, X11, and X12 are absent. 32. The antigen-binding molecule of any one of embodiments 7 to 18, wherein all of X8, X9, X10, X11, and X12 are absent. 33. The antigen-binding molecule of any one of embodiments 7 to 18, wherein the VH is at least 100 amino acids in length. 34. The antigen-binding molecule of any one of embodiments 1 to 33, wherein the VH is at most 125 amino acids in length. 35. The antigen-binding molecule of any one of embodiments 1 to 34, which comprises a single chain Fv (scFv) comprising the VH. 36. The antigen-binding molecule of embodiment 35, which comprises a light chain variable domain (VL) N-terminal to the VH. 37. The antigen-binding molecule of embodiment 35, which comprises a light chain variable domain (VL) C-terminal to the VH. 38. The antigen-binding molecule of any one of embodiments 35 to 37, wherein the VH and VL are separated by a linker. 39. The antigen-binding molecule of any one of embodiments 35 to 38, wherein the scFv is at least 225 amino acids in length. 40. The antigen-binding molecule of any one of embodiments 35 to 39, wherein the scFv is at most 300 amino acids in length. 41. The antigen-binding molecule of any one of embodiments 1 to 34, further comprising a VL. 42. The antigen-binding molecule of any one of embodiments 1 to 34, which lacks a VL.
43. The antigen-binding molecule of any one of embodiments 1 to 42, wherein the antigen-binding molecule further comprises a dimerization moiety. 44. The antigen-binding molecule of embodiment 43, wherein the dimerization moiety is an Fc domain. 45. The antigen-binding molecule of embodiment 44, wherein the Fc domain is C- terminal to the VH. 46. The antigen-binding molecule of embodiment 44, wherein the Fc domain is N- terminal to the VH. 47. The antigen-binding molecule of embodiment 46, wherein the VH is at the C terminus of at least one polypeptide chain in the antigen-binding molecule. 48. The antigen-binding molecule of any one of embodiments 45 to 47, wherein the Fc domain and the VH are separated by a linker. 49. The antigen-binding molecule of embodiment 48, wherein the linker is a glycine- serine linker. 50. The antigen-binding molecule of any one of embodiments 1 to 49, further comprising an additional VH as defined in any one of embodiments 1 to 34. 51. The antigen-binding molecule of embodiment 50, wherein the antigen-binding molecule is a dimer. 52. The antigen-binding molecule of embodiment 51, wherein the antigen-binding molecule is a homodimer. 53. The antigen-binding molecule of embodiment 51, wherein the antigen-binding molecule is a heterodimer.
54. The antigen-binding molecule of any one of embodiments 1 to 53, wherein the VH comprises a glycan at the N-linked glycosylation site and/or the N residue of the X1X2X3X4X5NX6X7X8X9X10X11X12 (SEQ ID NO:1) sequence. 55. The antigen-binding molecule of embodiment 54, wherein the glycan is G1F, G2F, G1F and GlcNAc, G2S, G2F and GlycNac, or G2FS. 56. The antigen-binding molecule of any one of embodiments 1 to 55, wherein the antigen-binding molecule is a multispecific antigen-binding molecule. 57. An antigen-binding molecule, which is optionally an antigen-binding molecule of any one of embodiments 1 to 56, having the configuration illustrated in FIG.1A. 58. An antigen-binding molecule, which is optionally an antigen-binding molecule of any one of embodiments 1 to 56, having the configuration illustrated in FIG.1B. 59. An antigen-binding molecule, which is optionally an antigen-binding molecule of any one of embodiments 1 to 56, having the configuration illustrated in FIG.1C. 60. An antigen-binding molecule, which is optionally an antigen-binding molecule of any one of embodiments 1 to 56, comprising: (a) a first polypeptide chain comprising, from N-terminus to C-terminus (i) a first antigen-binding domain or component thereof, (ii) a first dimerization moiety, and (iii) a second antigen-binding domain, the second antigen-binding domain comprising a VH as defined in any one of embodiments 1 to 34; and (b) a second polypeptide chain comprising, from N-terminus to C-terminus (i) a third antigen-binding domain or component thereof, and (ii) a second dimerization moiety, wherein the first and the second dimerization moieties associate with one another to form the molecule. 61. The antigen-binding molecule of embodiment 60, wherein the second antigen- binding domain is an scFv, optionally wherein the scFv is as defined in any one of embodiments 35 to 40.
62. The antigen-binding molecule of embodiment 60 or 61, wherein the second polypeptide further comprises a fourth antigen-binding domain C-terminal to the second dimerization moiety. 63. The antigen-binding molecule of embodiment 62, wherein the fourth antigen- binding domain comprises a VH comprising an N-linked glycosylation site within 10 amino acids of the C-terminus of the VH. 64. The antigen-binding molecule of embodiment 62 or 63, wherein the fourth antigen-binding domain is an scFv. 65. The antigen-binding molecule of any one of embodiments 62 to 64, wherein the first antigen-binding domain specifically binds to a T cell antigen. 66. The antigen-binding molecule of any one of embodiments 62 to 65, wherein the second antigen-binding domain specifically binds to a T cell antigen. 67. The antigen-binding molecule of any one of embodiments 62 to 66, wherein the third antigen-binding domain specifically binds to a T cell antigen. 68. The antigen-binding molecule of any one of embodiments 62 to 67, wherein the fourth antigen-binding domain specifically binds to a T cell antigen. 69. The antigen-binding molecule of any one of embodiments 65 to 68, wherein the T cell antigen is CD3. 70. The antigen-binding molecule of any one of embodiments 65 to 68, wherein the T cell antigen is CD28. 71. The antigen-binding molecule of any one of embodiments 62 to 66, wherein the third antigen-binding domain specifically binds to a tumor-associated antigen. 72. The antigen-binding molecule of any one of embodiments 62 to 66, wherein the fourth antigen-binding domain specifically binds to a tumor-associated antigen.
73. The antigen-binding molecule of any one of embodiments 60 to 72, wherein the first and second dimerization moieties are Fc domains. 74. An antigen-binding molecule, which is optionally an antigen-binding molecule of any one of embodiments 1 to 56, comprising: (a) a first scFv; (b) a linker; and (c) a second scFv, wherein the first scFv or the second scFv comprises a VH as defined in any one of embodiments 1 to 34. 75. The antigen-binding molecule of embodiment 74, wherein the first scFv comprises a VH as defined in any one of embodiments 1 to 34. 76. The antigen-binding molecule of embodiment 75, wherein the first scFv comprises, from N- to C-terminal orientation, a VL, a linker, and a VH as defined in any one of embodiments 1 to 34. 77. The antigen-binding molecule of embodiment 75, wherein the first scFv comprises, from N- to C-terminal orientation, a VH as defined in any one of embodiments 1 to 34, a linker, and a VL. 78. The antigen-binding molecule of any one of embodiments 74 to 77, wherein the second scFv comprises a VH as defined in any one of embodiments 1 to 34. 79. The antigen-binding molecule of embodiment 78, wherein the second scFv comprises, from N- to C-terminal orientation, a VL, a linker, and a VH as defined in any one of embodiments 1 to 34. 80. The antigen-binding molecule of embodiment 78, wherein the second scFv comprises, from N- to C-terminal orientation, a VH as defined in any one of embodiments 1 to 34, a linker, and a VL.
81. The antigen-binding molecule of any one of embodiments 1 to 80, wherein the antigen-binding molecule has reduced binding to anti-drug antibodies, e.g., as measured by an ADA reactivity assay as presented in Section 7.1.5 relative to a control antigen-binding molecule. 82. The antigen-binding molecule of any one of embodiments 1 to 81, wherein the VH comprising the N-linked glycosylation site is a component of a T-cell engaging antigen- binding domain (“TCE ABD”). 83. The antigen-binding molecule of embodiment 82, wherein the TCE ABD is capable of binding to a component of the T-cell receptor (TCR) complex. 84. The antigen-binding molecule of embodiment 83, wherein the component of the TCR complex is CD3. 85. The antigen-binding molecule of embodiment 83, wherein the component of the TCR complex is TCRαβ. 86. The antigen-binding molecule of embodiment 83, wherein the component of the TCR complex is TCRγδ. 87. The antigen-binding molecule of any one of embodiments 82 to 86, wherein the TCE ABD (a) comprises the (i) CDR or (ii) VH and VL sequences of antibody set forth in Table T or (b) competes with the antibody set forth in Table T for binding to its target. 88. The antigen-binding molecule of any one of embodiments 1 to 81, wherein the VH comprising the N-linked glycosylation site is a component of an antigen-binding domain that is capable of binding to a tumor-associated antigen (“TAA ABD”). 89. The antigen-binding molecule of embodiment 88, wherein the TAA ABD is capable of binding to any tumor-associated antigen identified in Section 6.4.2. 90. The antigen-binding molecule of embodiment 88, wherein the TAA ABD is capable of binding to AFP, ALK, a BAGE protein, BIRC5 (survivin), BIRC7, β-catenin, brc-abl,
BRCA1, BORIS, CA9, carbonic anhydrase IX, caspase-8, CALR, CEACAM5 (also known as carcinoembryonic antigen or CEA), CCR5, CD19, CD20 (MS4A1), CD22, CD30, CD40, CDK4, CEA, CTLA4, cyclin-B1, CYP1B1, EGFR, EGFRvIII, ErbB2/Her2, ErbB3, ErbB4, ETV6-AML, EpCAM, EphA2, Fra-1, FOLR1, a GAGE protein (e.g., GAGE-1 or -2), GD2, GD3, GloboH, glypican-3, GM3, gp100, Her2, HLA/B-raf, HLA/k-ras, HLA/MAGE-A3, hTERT, LMP2, MAGE proteins (e.g., MAGE-1, -2, -3, -4, -6, and -12), MART-1, mesothelin, ML-IAP, Muc1, Muc2, Muc3, Muc4, Muc5, Muc16 (CA-125), MUM1, NA17, NY-BR1, NY-BR62, NY-BR85, NY-ESO1, OX40, p15, p53, PAP, PAX3, PAX5, PCTA-1, PLAC1, PRLR, PRAME, PSMA (FOLH1), RAGE proteins, Ras, RGS5, Rho, SART-1, SART-3, STEAP1, STEAP2, TAG-72, TGF-β, TMPRSS2, Thompson-nouvelle antigen (Tn), TRP-1, TRP-2, tyrosinase, or uroplakin-3. 91. The antigen-binding molecule of embodiment 88, wherein the TAA ABD is capable of binding to CTLA-4, PD1, PDL1, PDL2, B7-H3, B7-H4, BTLA, HVEM, TIM3, GAL9, LAG3, VISTA, KIR, 2B4, CD160, CGEN-15049, CHK1, or CHK2. 92. The antigen-binding molecule of embodiment 88, wherein the TAA ABD is capable of binding to CD27, CD28, 4-1BB (CD137), OX40, CD30, CD40, ICOS, lymphocyte function-associated antigen-1 (LFA-1), CD2, CD7, LIGHT, NKG2C, or B7-H3. 93. The antigen-binding molecule of embodiment 88, wherein the TAA ABD is capable of binding to syndecan, heparanase, integrins, osteopontin, link, cadherins, laminin, laminin type EGF, lectin, fibronectin, notch, nectin (e.g., nectin-4), tenascin, collagen (e.g., collagen type X), or matrixin. 94. The antigen-binding molecule of any one of embodiments 1 to 81, wherein the antigen-binding molecule comprises (1) a TCE ABD or component thereof; and (2) a TAA ABD or component thereof. 95. The antigen-binding molecule of embodiment 94, wherein the VH comprising the N-linked glycosylation site is a component of the TCE ABD. 96. The antigen-binding molecule of embodiment 95, wherein the TCE ABD is capable of binding to a component of the TCR complex.
97. The antigen-binding molecule of embodiment 96, wherein the component of the TCR complex is CD3. 98. The antigen-binding molecule of embodiment 96, wherein the component of the TCR complex is TCRαβ. 99. The antigen-binding molecule of embodiment 96, wherein the component of the TCR complex is TCRγδ. 100. The antigen-binding molecule of any one of embodiments 95 to 99, wherein the TCE ABD (a) comprises the (i) CDR or (ii) VH and VL sequences of antibody set forth in Table T or (b) competes with the antibody set forth in Table T for binding to its target. 101. The antigen-binding molecule of embodiment 94, wherein the VH comprising the N-linked glycosylation site is a component of an antigen-binding domain that is capable of binding to a tumor-associated antigen (“TAA ABD”). 102. The antigen-binding molecule of embodiment 101, wherein the TAA ABD is capable of binding to any tumor-associated antigen identified in Section 6.4.2. 103. The antigen-binding molecule of embodiment 102, wherein the TAA ABD is capable of binding to AFP, ALK, a BAGE protein, BIRC5 (survivin), BIRC7, β-catenin, brc-abl, BRCA1, BORIS, CA9, carbonic anhydrase IX, caspase-8, CALR, CEACAM5 (also known as carcinoembryonic antigen or CEA), CCR5, CD19, CD20 (MS4A1), CD22, CD30, CD40, CDK4, CEA, CTLA4, cyclin-B1, CYP1B1, EGFR, EGFRvIII, ErbB2/Her2, ErbB3, ErbB4, ETV6-AML, EpCAM, EphA2, Fra-1, FOLR1, a GAGE protein (e.g., GAGE-1 or -2), GD2, GD3, GloboH, glypican-3, GM3, gp100, Her2, HLA/B-raf, HLA/k-ras, HLA/MAGE-A3, hTERT, LMP2, MAGE proteins (e.g., MAGE-1, -2, -3, -4, -6, and -12), MART-1, mesothelin, ML-IAP, Muc1, Muc2, Muc3, Muc4, Muc5, Muc16 (CA-125), MUM1, NA17, NY-BR1, NY-BR62, NY-BR85, NY-ESO1, OX40, p15, p53, PAP, PAX3, PAX5, PCTA-1, PLAC1, PRLR, PRAME, PSMA (FOLH1), RAGE proteins, Ras, RGS5, Rho, SART-1, SART-3, STEAP1, STEAP2, TAG-72, TGF-β, TMPRSS2, Thompson-nouvelle antigen (Tn), TRP-1, TRP-2, tyrosinase, or uroplakin-3.
104. The antigen-binding molecule of embodiment 102, wherein the TAA ABD is capable of binding to CTLA-4, PD1, PDL1, PDL2, B7-H3, B7-H4, BTLA, HVEM, TIM3, GAL9, LAG3, VISTA, KIR, 2B4, CD160, CGEN-15049, CHK1, or CHK2. 105. The antigen-binding molecule of embodiment 102, wherein the TAA ABD is capable of binding to CD27, CD28, 4-1BB (CD137), OX40, CD30, CD40, ICOS, lymphocyte function-associated antigen-1 (LFA-1), CD2, CD7, LIGHT, NKG2C, or B7-H3. 106. The antigen-binding molecule of embodiment 102, wherein the TAA ABD is capable of binding to syndecan, heparanase, integrins, osteopontin, link, cadherins, laminin, laminin type EGF, lectin, fibronectin, notch, nectin (e.g., nectin-4), tenascin, collagen (e.g., collagen type X), or matrixin. 107. A pharmaceutical composition comprising the antigen-binding molecule of any one of embodiments 1 to 106. 108. A nucleic acid or plurality of nucleic acids encoding the antigen-binding molecule of any one of embodiments 1 to 106. 109. A host cell engineered to express the antigen-binding molecule of any one of embodiments 1 to 106. 110. A host cell transfected with one or more expression vectors comprising one or more nucleic acid sequences encoding the antigen-binding molecule of any one of embodiments 1 to 106 under the control of one or more promoters. 111. A method of producing the antigen-binding molecule of any one of embodiments 1 to 106 comprising culturing the host cell of embodiment 109 or 110 and recovering the antigen-binding molecule expressed thereby. 112. A method comprising administering the antigen-binding molecule of any one of embodiments 1 to 106 to a subject.
113. A method of treating cancer comprising administering to a subject in need thereof the antigen-binding molecule of any one of embodiments 1 to 106 or the pharmaceutical composition of embodiment 107. 114. The method of embodiment 113, wherein the antigen-binding molecule is a bispecific antigen-binding molecule comprising a TCE ABD and a TAA ABD and the cancer is associated with the expression of the tumor-associated antigen, e.g., as set forth in Table A. 115. A polypeptide comprising an engineered VH as defined in any one of embodiments 1 to 34. 116. A nucleic acid or plurality of nucleic acids encoding the polypeptide of embodiment 115. 117. A host cell engineered to express the polypeptide of embodiment 115 or the nucleic acid(s) of embodiment 116. 118. A method of producing the polypeptide of embodiment 115 comprising culturing the host cell of embodiment 117. 119. A protein engineering method comprising generating an antigen-binding molecule comprising a VH having an N-linked glycosylation site within 10 amino acids of its C- terminus. 120. A protein engineering method comprising generating a VH comprising an N- linked glycosylation site within 10 amino acids of its C-terminus. 121. A method of reducing antigenicity of an antigen-binding molecule comprising a VH, the method comprising incorporating an N-linked glycosylation site within 10 amino acids of the C-terminus of the VH. 122. The method embodiment 121, wherein the antigen-binding molecule further comprises an additional VH.
123. The method of embodiment 122, wherein the method further comprises incorporating an N-linked glycosylation site within 10 amino acids of the C-terminus of the additional VH. 124. The method of any one of embodiments 119 to 123, wherein the method comprises inserting the amino acid sequence X1X2X3X4X5NX6X7X8X9X10X11X12 (SEQ ID NO:1), or a portion thereof, into the VH, wherein (a) X1, X2, X3, X4, and X5 are each independently selected from any amino acid; (b) X6 is selected from any amino acid, optionally wherein the amino acid is not proline; (c) X7 is S or T, and (d) X8, X9, X10,X11, and X12 are each independently selected from any amino acid and absent. 125. The method of any one of embodiments 119 to 123, wherein the method comprises substituting one or more amino acids within the sequence of the VH such that the VH comprises the amino acid sequence X1X2X3X4X5NX6X7X8X9X10X11X12 (SEQ ID NO:1) within 10 amino acids of its C-terminus, wherein (a) X1, X2, X3, X4, and X5 are each independently selected from any amino acid; (b) X6 is selected from any amino acid, optionally wherein the amino acid is not proline; (c) X7 is S or T, and (d) X8, X9, X10,X11, and X12 are each independently selected from any amino acid and absent. 126. The method of embodiment 124 or 125, wherein X6 is S. 127. The method of any one of embodiments 124 to 126, wherein X7 is S. 128. The method of any one of embodiments 124 to 126, wherein X7 is T. 129. The method of any one of embodiments 124 to 128, wherein X5 is V. 130. The method of any one of embodiments 124 to 129, wherein X4 is T. 131. The method of any one of embodiments 124 to 130, wherein X3 is V. 132. The method of any one of embodiments 124 to 128, wherein X5 is S. 133. The method of any one of embodiments 124 to 128 and 132, wherein X4 is S.
134. The method of any one of embodiments 124 to 128 and 132 to 133, wherein X3 is V. 135. The method of any one of embodiments 124 to 128 and 132 to 134, wherein X2 is T. 136. The method of any one of embodiments 124 to 128 and 132 to 135, wherein X1 is V. 137. The method of any one of embodiments 124 to 136, wherein X8 is P. 138. The method of any one of embodiments 124 to 137, wherein X9 is P. 139. The method of any one of embodiments 124 to 136, wherein X8 is K. 140. The method of any one of embodiments 124 to 136 and 139, wherein X9 is P. 141. The method of any one of embodiments 124 to 136 and 139 to 140, wherein X10 is G. 142. The method of any one of embodiments 124 to 136 and 139 to 141, wherein X11 is G. 143. The method of any one of embodiments 124 to 136, wherein X8 is G. 144. The method of any one of embodiments 124 to 136 and 143, wherein X9 is G. 145. The method of any one of embodiments 124 to 136 and 143 to 144, wherein X10 is G. 146. The method of any one of embodiments 124 to 136 and 143 to 145, wherein X11 is G. 147. The method of any one of embodiments 124 to 136, wherein at least two of X8, X9, X10, X11, and X12 are absent.
148. The method of any one of embodiments 124 to 136, wherein at least three of X8, X9, X10, X11, and X12 are absent. 149. The method of any one of embodiments 124 to 136, wherein at least four of X8, X9, X10, X11, and X12 are absent. 150. The method of any one of embodiments 124 to 136, wherein all of X8, X9, X10, X11, and X12 are absent. 9. EXAMPLES 9.1. Materials and Methods 9.1.1. Design and Production of Antigen-binding Molecules [0207] Antigen-binding molecules were designed to comprise two polypeptides connected to one another via their Fc domains. The first polypeptide was designed to comprise from N- terminus to C-terminus: a CD3-targeting Fab, a first Fc domain, a linker, and a CD3-targeting scFv. The second polypeptide was designed to comprise from N-terminus to C-terminus: a TAA- targeting Fab, a second Fc domain, a linker, and a TAA-targeting scFv. Antigen-binding molecules were engineered by introducing amino acid modifications at or near the C-terminus of the scFvs. FIG.2A shows a parental antigen-binding molecule, FIGs.2B-2E show variants with C-terminal amino acid modifications that do not incorporate an N-glycosylation consensus sequence, and FIGs.2F and 2G show variants that incorporate an N-glycosylation consensus sequence at the C-terminus of the scFvs. [0001] All constructs were expressed in Expi293F™ cells by transient transfection (Thermo Fisher Scientific). Proteins in Expi293F supernatant were purified using the ProteinMaker system (Protein BioSolutions, Gaithersburg, MD) with HiTrap MabSelect SuRe Protein A columns (GE Healthcare). After single step elution with IgG Elution Buffer (Thermo Fisher Scientific), the proteins were neutralized, dialyzed into a final buffer of phosphate buffered saline (PBS) with 5% glycerol, aliquoted and stored at -80°C. [0208] Antigen-binding molecules and control constructs utilized in these examples are presented in Table 2. Table 2
 Construct Antigen-binding Domains C-Terminal Structure Modification(s) n
Construct Antigen-binding Domains C-Terminal Structure Modification(s) n
 9.1.2. Flow Cytometry Binding Assay [0209] Cells were resuspended in FACS wash (PBS with 1% FBS and 0.5mM EDTA) at 1×106 cells/mL. The staining was performed in 1 ×105 cells per well. The antibodies were diluted with a ratio of 1:5 from a starting concentration of 1.3 x 10-07 M. The diluted antibodies were then added into the wells containing cells. Cells were stained for 30 min at 2-8°C and washed twice with FACS wash buffer. APC-conjugated goat anti-human IgG (Jackson Immuno Research, 109-607-003, 1:400) was added to stain the cells for 30 min at 2–8°C along with LIVE/DEAD Fixable Violet Dead Cell Stain Kit (Thermo Fisher Scientific). Following washing, cells were fixed in 2% paraformaldehyde for 30 min at 2–8°C. After two washes, stained cells were analyzed using BD FACSCantoII instrument. The results were analyzed by FlowJo. FSC/SSC gates were used to select single cells and BV421 negative gating was used to select live cells. 9.1.3. Cytotoxicity Assay [0210] Tumor cell lines were resuspended in PBS and stained with CellTrace Violet (Thermo Fisher Scientific) based on manufacturer protocols. Cells were resuspended in RPMI media containing 10% FBS and penicillin-streptomycin-glutamine supplementation (R10 media) at a density of 100,000 cells/mL and 50μL was plated in a 96-well plate to provide 5,000 cells per well. Peripheral blood mononuclear cells (PBMC) were thawed and resuspended in R10 media at a density of 1×106 cells/mL and allowed to rest overnight in a cell culture incubator. The following day, the PBMC suspension was mixed with human IgG1 at a final concentration of 5 mg/mL to mimic human serum, and PBMC at 2 ×106 cells/mL. PBMC were added to a final amount of 50,000 cells/well. Multispecific antigen binding molecules were diluted with a ratio of 1:10 and added to the assay plates with a final starting concentration of 6.7 x 10-08 M in R10 media. The plates were incubated in a cell culture incubator for 72 hours or longer. The supernatant was then removed and spun down at 300 x g for 4 minutes to pellet all cells in suspension. The tumor cells were washed and harvested with trypsin, and combined with the suspension cell pellet. The final cell pellet was washed with PBS and stained with LIVE/DEAD Fixable Near-IR Dead Cell Stain Kit (Thermo Fisher Scientific). After 2 washes with PBS, cells were resuspended in FACS wash and analyzed on a cytometer such as the BD FACSCelesta instrument or BD FACSCantoII instrument. Results were analyzed by FlowJo: CellTrace positive (BV421 channel) cells were gated to identify tumor cells, FSC/SSC gates were used to select single cells, and APC-Cy7 negative gating was used to select live cells. All appropriate compensation samples and fluorescence minus one (FMO) controls were included.
9.1.4. Native SEC-UV/MS [0211] Intact mass measurement using native SEC-MS analysis of mAbs was performed to confirm that the molecular weight of non-reduced mAb matched the predicted molecular weight of a given mAb based on the cDNA-derived amino acid sequence. In addition, this method was also able to identify and characterize any size variants (due to SEC separation) that may be present within the samples. The native ESI mass spectrum of each peak from the SEC total ion chromatograms (TICs) were averaged and deconvoluted using Intact Mass software from Protein Metrics and the resulting mass spectra were output. Multispecific antigen-binding molecules REGN9930-VNST and REGN9930-VNSS were subjected to limited LysC digestion to generate full-length scFv fragments (arising from preferential cleavage at Lys site N-terminal to GS-linkers). Such fragments were well separated from the mAbs or Fabs and thus could be discerned from N-linked glycans in the scFv region based on the predicted mass. 9.1.5. ADA Reactivity Assay [0212] In order to assess antigenicity, antibodies were tested in an ADA reactivity assay using an Electrochemiluminescent Immunoassay (ECL) or a SMCxPro Immunogenicity method for measuring binding to anti-drug antibodies. For the ECL method, antibodies were labeled with biotin or Ruthenium. Labelled antibodies were co-incubated with human or monkey serum. Bridged complexes were captured by streptavidin-coated plates, and an electrochemiluminescent signal produced by an electric current activating Ruthenium-labeled antibodies was recorded. [0213] For the SMCxPro Immunogenicity method, antibodies were labeled with biotin and Alexa647 according to the Millipore SMC Immunogenicity Plate Based Assay Development Kit protocol (Catalog #03-0189-00). Biotinylated-antibody at 1μg/mL, Alexa647-antibody at 0.063ug/mL, and human or monkey serum diluted 1:10 were combined 1:1:1 and incubated overnight shaking at 4°C. After incubation, the bridged complex was transferred to a pre- blocked streptavidin-coated assay plate and incubated for 1 hour shaking at RT. The plate was washed 6 times in 1X Wash Buffer. Elution Buffer B was added to the assay plate and incubated for 10 minutes shaking at RT. After 10 minutes the reaction was quenched with Buffer C. The neutralized solution was transferred to an SMCxPro Aurora plate, sealed, and read on the SMCxPro. [0214] Samples with an average response 2 fold or higher than the monkey serum background were identified as positive for pre-existing ADA. This background was determined for each antibody individually.
9.2. Example 1: C-Terminal Modifications Do Not Affect Target Binding Affinity of REGN9930 [0215] Antigen-binding molecule REGN9930 and C-terminal modification constructs derived from REGN9930 (Table 2) were generated as described in Section 9.1.1. Binding affinity of antigen-binding molecules to target cell surface antigens was assessed using Raji and Jurkat cells via flow cytometry as described in in Section 9.1.2. [0216] Binding affinities of the parental construct REGN9930 and C-terminal engineered constructs to MAGE-A4 peptides (MAGE-A4 (230-239) and MAGE-A4 (286-294)) overexpressed on Raji cells, as well as to CD3 expressed on Jurkat cells, were assessed. All constructs with C-terminal modifications displayed binding affinities to Raji MAGE-A4 (230-239) cells that were comparable to the binding affinity of REGN9930 (FIG.2A). Binding affinities of all constructs with C-terminal modifications to Raji MAGE-A4 (286-294) cells were also comparable to the binding affinity of REGN9930 (FIG.2B). Similarly, all constructs displayed similar binding affinities to Jurkat cells. These results indicate that none of the C-terminal modifications evaluated in this example affected the target binding affinity observed with the parental construct, REGN9930. 9.3. Example 2: C-Terminal Modifications Do Not Affect Cytotoxic Potency of REGN9930 [0217] Cytotoxic potency of the parental construct REGN9930 and C-terminal engineered constructs was assessed as described in Section 9.1.3. Percent viability of A375 tumor cells was comparable with REGN9930 and all C-terminal engineered constructs (FIG.3), indicating that none of the C-terminal modifications affected the cytotoxic potency observed with the parental construct, REGN9930. 9.4. Example 3: Native SEC-MS Analysis of Limited LysC Digested REGN9930- VNSS and REGN9930-VNST [0218] To determine whether C-terminal modifications of constructs REGN9930-VNSS and REGN9930-VNST have incorporated N-glycans, these two constructs were subjected to limited LysC digestion and evaluated with native SEC-UV/MS as described in Section 9.1.4. [0219] Limited LysC digestion freed the scFv domains attached to Fc C-termini of both polypeptide chains of REGN9930-VNSS and yielded full-length scFv fragments. Although intact mass analysis showed the incorporation of the C-terminal modification, which changed the C- terminal amino acid sequence VTVSS (SEQ ID NO: 62) to VTVNSS (SEQ ID NO: 50), N-glycan occupancy at this site was not detected (FIGs.5A-5E). Similarly, when REGN9930-VNST was
evaluated, intact mass analysis showed that the C-terminal modification, which changed the C- terminal amino acid sequence VTVSS (SEQ ID NO: 62) to VTVNST (SEQ ID NO: 51), was incorporated. However, as was the case with REGN9930-VNSS, there was no evidence of N- glycan occupancy at the modified amino acid sequence (FIGs.6A-6D). 9.5. Example 4: C-Terminal Modifications Reduce ADA Reactivity of REGN9930 [0220] REGN9930 has pre-existing ADA reactivity. To evaluate the impact of C-terminal VH engineering on REGN9930 reactivity, ADA binding of the six REGN C-terminal variants (Table 2) along with REGN9930 and REGN8503, were assessed using a subset of 24 human naïve serum samples as described in Section 9.1.5. [0221] REGN8503 was used as a negative control and displayed no preexisting reactivity. In contrast, REGN9930 was associated with high levels of preexisting reactivity. All C-terminal engineered variants of REGN9930 exhibited greatly reduced ADA binding (FIG.6). These results indicated that the C-terminal VH engineered variants of REGN9930 were effective in reducing the reactivity observed with REGN9930.
9.1.2. Flow Cytometry Binding Assay [0209] Cells were resuspended in FACS wash (PBS with 1% FBS and 0.5mM EDTA) at 1×106 cells/mL. The staining was performed in 1 ×105 cells per well. The antibodies were diluted with a ratio of 1:5 from a starting concentration of 1.3 x 10-07 M. The diluted antibodies were then added into the wells containing cells. Cells were stained for 30 min at 2-8°C and washed twice with FACS wash buffer. APC-conjugated goat anti-human IgG (Jackson Immuno Research, 109-607-003, 1:400) was added to stain the cells for 30 min at 2–8°C along with LIVE/DEAD Fixable Violet Dead Cell Stain Kit (Thermo Fisher Scientific). Following washing, cells were fixed in 2% paraformaldehyde for 30 min at 2–8°C. After two washes, stained cells were analyzed using BD FACSCantoII instrument. The results were analyzed by FlowJo. FSC/SSC gates were used to select single cells and BV421 negative gating was used to select live cells. 9.1.3. Cytotoxicity Assay [0210] Tumor cell lines were resuspended in PBS and stained with CellTrace Violet (Thermo Fisher Scientific) based on manufacturer protocols. Cells were resuspended in RPMI media containing 10% FBS and penicillin-streptomycin-glutamine supplementation (R10 media) at a density of 100,000 cells/mL and 50μL was plated in a 96-well plate to provide 5,000 cells per well. Peripheral blood mononuclear cells (PBMC) were thawed and resuspended in R10 media at a density of 1×106 cells/mL and allowed to rest overnight in a cell culture incubator. The following day, the PBMC suspension was mixed with human IgG1 at a final concentration of 5 mg/mL to mimic human serum, and PBMC at 2 ×106 cells/mL. PBMC were added to a final amount of 50,000 cells/well. Multispecific antigen binding molecules were diluted with a ratio of 1:10 and added to the assay plates with a final starting concentration of 6.7 x 10-08 M in R10 media. The plates were incubated in a cell culture incubator for 72 hours or longer. The supernatant was then removed and spun down at 300 x g for 4 minutes to pellet all cells in suspension. The tumor cells were washed and harvested with trypsin, and combined with the suspension cell pellet. The final cell pellet was washed with PBS and stained with LIVE/DEAD Fixable Near-IR Dead Cell Stain Kit (Thermo Fisher Scientific). After 2 washes with PBS, cells were resuspended in FACS wash and analyzed on a cytometer such as the BD FACSCelesta instrument or BD FACSCantoII instrument. Results were analyzed by FlowJo: CellTrace positive (BV421 channel) cells were gated to identify tumor cells, FSC/SSC gates were used to select single cells, and APC-Cy7 negative gating was used to select live cells. All appropriate compensation samples and fluorescence minus one (FMO) controls were included.
9.1.4. Native SEC-UV/MS [0211] Intact mass measurement using native SEC-MS analysis of mAbs was performed to confirm that the molecular weight of non-reduced mAb matched the predicted molecular weight of a given mAb based on the cDNA-derived amino acid sequence. In addition, this method was also able to identify and characterize any size variants (due to SEC separation) that may be present within the samples. The native ESI mass spectrum of each peak from the SEC total ion chromatograms (TICs) were averaged and deconvoluted using Intact Mass software from Protein Metrics and the resulting mass spectra were output. Multispecific antigen-binding molecules REGN9930-VNST and REGN9930-VNSS were subjected to limited LysC digestion to generate full-length scFv fragments (arising from preferential cleavage at Lys site N-terminal to GS-linkers). Such fragments were well separated from the mAbs or Fabs and thus could be discerned from N-linked glycans in the scFv region based on the predicted mass. 9.1.5. ADA Reactivity Assay [0212] In order to assess antigenicity, antibodies were tested in an ADA reactivity assay using an Electrochemiluminescent Immunoassay (ECL) or a SMCxPro Immunogenicity method for measuring binding to anti-drug antibodies. For the ECL method, antibodies were labeled with biotin or Ruthenium. Labelled antibodies were co-incubated with human or monkey serum. Bridged complexes were captured by streptavidin-coated plates, and an electrochemiluminescent signal produced by an electric current activating Ruthenium-labeled antibodies was recorded. [0213] For the SMCxPro Immunogenicity method, antibodies were labeled with biotin and Alexa647 according to the Millipore SMC Immunogenicity Plate Based Assay Development Kit protocol (Catalog #03-0189-00). Biotinylated-antibody at 1μg/mL, Alexa647-antibody at 0.063ug/mL, and human or monkey serum diluted 1:10 were combined 1:1:1 and incubated overnight shaking at 4°C. After incubation, the bridged complex was transferred to a pre- blocked streptavidin-coated assay plate and incubated for 1 hour shaking at RT. The plate was washed 6 times in 1X Wash Buffer. Elution Buffer B was added to the assay plate and incubated for 10 minutes shaking at RT. After 10 minutes the reaction was quenched with Buffer C. The neutralized solution was transferred to an SMCxPro Aurora plate, sealed, and read on the SMCxPro. [0214] Samples with an average response 2 fold or higher than the monkey serum background were identified as positive for pre-existing ADA. This background was determined for each antibody individually.
9.2. Example 1: C-Terminal Modifications Do Not Affect Target Binding Affinity of REGN9930 [0215] Antigen-binding molecule REGN9930 and C-terminal modification constructs derived from REGN9930 (Table 2) were generated as described in Section 9.1.1. Binding affinity of antigen-binding molecules to target cell surface antigens was assessed using Raji and Jurkat cells via flow cytometry as described in in Section 9.1.2. [0216] Binding affinities of the parental construct REGN9930 and C-terminal engineered constructs to MAGE-A4 peptides (MAGE-A4 (230-239) and MAGE-A4 (286-294)) overexpressed on Raji cells, as well as to CD3 expressed on Jurkat cells, were assessed. All constructs with C-terminal modifications displayed binding affinities to Raji MAGE-A4 (230-239) cells that were comparable to the binding affinity of REGN9930 (FIG.2A). Binding affinities of all constructs with C-terminal modifications to Raji MAGE-A4 (286-294) cells were also comparable to the binding affinity of REGN9930 (FIG.2B). Similarly, all constructs displayed similar binding affinities to Jurkat cells. These results indicate that none of the C-terminal modifications evaluated in this example affected the target binding affinity observed with the parental construct, REGN9930. 9.3. Example 2: C-Terminal Modifications Do Not Affect Cytotoxic Potency of REGN9930 [0217] Cytotoxic potency of the parental construct REGN9930 and C-terminal engineered constructs was assessed as described in Section 9.1.3. Percent viability of A375 tumor cells was comparable with REGN9930 and all C-terminal engineered constructs (FIG.3), indicating that none of the C-terminal modifications affected the cytotoxic potency observed with the parental construct, REGN9930. 9.4. Example 3: Native SEC-MS Analysis of Limited LysC Digested REGN9930- VNSS and REGN9930-VNST [0218] To determine whether C-terminal modifications of constructs REGN9930-VNSS and REGN9930-VNST have incorporated N-glycans, these two constructs were subjected to limited LysC digestion and evaluated with native SEC-UV/MS as described in Section 9.1.4. [0219] Limited LysC digestion freed the scFv domains attached to Fc C-termini of both polypeptide chains of REGN9930-VNSS and yielded full-length scFv fragments. Although intact mass analysis showed the incorporation of the C-terminal modification, which changed the C- terminal amino acid sequence VTVSS (SEQ ID NO: 62) to VTVNSS (SEQ ID NO: 50), N-glycan occupancy at this site was not detected (FIGs.5A-5E). Similarly, when REGN9930-VNST was
evaluated, intact mass analysis showed that the C-terminal modification, which changed the C- terminal amino acid sequence VTVSS (SEQ ID NO: 62) to VTVNST (SEQ ID NO: 51), was incorporated. However, as was the case with REGN9930-VNSS, there was no evidence of N- glycan occupancy at the modified amino acid sequence (FIGs.6A-6D). 9.5. Example 4: C-Terminal Modifications Reduce ADA Reactivity of REGN9930 [0220] REGN9930 has pre-existing ADA reactivity. To evaluate the impact of C-terminal VH engineering on REGN9930 reactivity, ADA binding of the six REGN C-terminal variants (Table 2) along with REGN9930 and REGN8503, were assessed using a subset of 24 human naïve serum samples as described in Section 9.1.5. [0221] REGN8503 was used as a negative control and displayed no preexisting reactivity. In contrast, REGN9930 was associated with high levels of preexisting reactivity. All C-terminal engineered variants of REGN9930 exhibited greatly reduced ADA binding (FIG.6). These results indicated that the C-terminal VH engineered variants of REGN9930 were effective in reducing the reactivity observed with REGN9930.
Claims
WHAT IS CLAIMED IS 1. An antigen-binding molecule comprising a heavy chain variable domain (VH), said VH comprising an N-linked glycosylation site within 10 amino acids of the C-terminus of the VH.
2. The antigen-binding molecule of claim 1, wherein the N-linked glycosylation site is within 8 amino acids, within 5 amino acids, within 4 amino acids, or within 3 amino acids of the C-terminus of the VH.
3. The antigen-binding molecule of claim 1 or 2, wherein the N-linked glycosylation site is defined by the sequence motif NX[S/T], wherein X is any amino acid, optionally wherein the amino acid is not proline.
4. An antigen-binding molecule comprising a heavy chain variable domain (VH), optionally wherein the antigen-binding molecule is an antigen-binding molecule according to any one of claims 1 to 3, wherein the VH comprises at its C-terminus the amino acid sequence X1X2X3X4X5NX6X7X8X9X10X11X12, wherein (a) X1, X2, X3, X4, and X5 are each independently selected from any amino acid; (b) X6 is selected from any amino acid, optionally wherein the amino acid is not proline; (c) X7 is S or T; and (d) X8, X9, X10,X11, and X12 are each independently selected from any amino acid and absent.
5. The antigen-binding molecule of claim 4, wherein X6 is S.
6. The antigen-binding molecule of claim 4 or claim 5, wherein X7 is S.
7. The antigen-binding molecule of claim 4 or claim 5, wherein X7 is T.
8. The antigen-binding molecule of any one of claims 4 to 7, wherein X5 is V.
9. The antigen-binding molecule of any one of claims 4 to 8, wherein X4 is T.
10. The antigen-binding molecule of any one of claims 4 to 9, wherein X3 is V.
11. The antigen-binding molecule of any one of claims 4 to 7, wherein X5 is S.
12. The antigen-binding molecule of any one of claims 4 to 7 and 11, wherein X4 is S.
13. The antigen-binding molecule of any one of claims 4 to 7 and 11 to 12, wherein X3 is V.
14. The antigen-binding molecule of any one of claims 4 to 7 and 11 to 13, wherein X2 is T.
15. The antigen-binding molecule of any one of claims 4 to 7 and 11 to 14, wherein X1 is V.
16. The antigen-binding molecule of any one of claims 4 to 15, wherein X8 is P.
17. The antigen-binding molecule of any one of claims 4 to 16, wherein X9 is P.
18. The antigen-binding molecule of any one of claims 4 to 15, wherein X8 is K.
19. The antigen-binding molecule of any one of claims 4 to 15 and 18, wherein X9 is P.
20. The antigen-binding molecule of any one of claims 4 to 15, 18, and 19, wherein X10 is G.
21. The antigen-binding molecule of any one of claims 4 to 15 and 18 to 20, wherein X11 is G.
22. The antigen-binding molecule of any one of claims 4 to 15, wherein X8 is G.
23. The antigen-binding molecule of any one of claims 4 to 15 and 22, wherein X9 is G.
24. The antigen-binding molecule of any one of claims 4 to 15, 22, and 23, wherein X10 is G.
25. The antigen-binding molecule of any one of claims 4 to 15 and 22 to 24, wherein X11 is G.
26. The antigen-binding molecule of any one of claims 4 to 15, wherein at least two, at least three, at least four, or all of X8, X9, X10, X11, and X12 are absent.
27. The antigen-binding molecule of any one of claims 1 to 26, which comprises a single chain Fv (scFv) comprising the VH.
28. The antigen-binding molecule of any one of claims 1 to 27, wherein the antigen- binding molecule further comprises an Fc domain.
29. The antigen-binding molecule of claim 28, wherein the Fc domain is N-terminal to the VH.
30. The antigen-binding molecule of claim 29, wherein the VH is at the C terminus of at least one polypeptide chain in the antigen-binding molecule.
31. The antigen-binding molecule of any one of claims 1 to 30, wherein the VH comprises a glycan at the N-linked glycosylation site and/or the, when dependent from any one of claims 4 to 30, the N residue of the X1X2X3X4X5NX6X7X8X9X10X11X12 sequence.
32. An antigen-binding molecule, which is optionally an antigen-binding molecule of any one of claims 1 to 31, comprising: (a) a first polypeptide chain comprising, from N-terminus to C-terminus (i) a first antigen-binding domain or component thereof, (ii) a first dimerization moiety, and (iii) a second antigen-binding domain, the second antigen-binding domain comprising a VH as defined in any one of claims 1 to 26; and (b) a second polypeptide chain comprising, from N-terminus to C-terminus (i) a third antigen-binding domain or component thereof, and (ii) a second dimerization moiety, wherein the first and the second dimerization moieties associate with one another to form the molecule.
33. The antigen-binding molecule of claim 32, wherein the second antigen-binding domain is an scFv, optionally wherein the scFv is as defined in claim 27.
34. An antigen-binding molecule, which is optionally an antigen-binding molecule of any one of claims 1 to 31, comprising: (a) a first scFv; (b) a linker; and (c) a second scFv, wherein the first scFv or the second scFv comprises a VH as defined in any one of claims 1 to 26.
35. The antigen-binding molecule of any one of claims 1 to 34, wherein the antigen- binding molecule has reduced binding to anti-drug antibodies.
36. The antigen-binding molecule of any one of claims 1 to 35, wherein the VH comprising the N-linked glycosylation site is a component of a T-cell engaging antigen-binding domain (“TCE ABD”).
37. The antigen-binding molecule of any one of claims 1 to 35, wherein the VH comprising the N-linked glycosylation site is a component of an antigen-binding domain that is capable of binding to a tumor-associated antigen (“TAA ABD”).
38. A pharmaceutical composition comprising the antigen-binding molecule of any one of claims 1 to 37.
39. A nucleic acid or plurality of nucleic acids encoding the antigen-binding molecule of any one of claims 1 to 37.
40. A host cell transfected with one or more expression vectors comprising one or more nucleic acid sequences encoding the antigen-binding molecule of any one of claims 1 to 37 under the control of one or more promoters.
41. A method of producing the antigen-binding molecule of any one of claims 1 to 37 comprising culturing the host cell of claim 40 and recovering the antigen-binding molecule expressed thereby.
42. A method comprising administering the antigen-binding molecule of any one of claims 1 to 37 to a subject.
43. A method of treating cancer comprising administering to a subject in need thereof the antigen-binding molecule of any one of claims 1 to 37 or the pharmaceutical composition of claim 38, wherein the antigen-binding molecule is a bispecific antigen-binding molecule comprising a TCE ABD and a TAA ABD and the cancer is associated with the expression of the tumor-associated antigen, e.g., as set forth in Table A.
44. A polypeptide comprising an engineered VH as defined in any one of claims 1 to 37.
45. A nucleic acid or plurality of nucleic acids encoding the polypeptide of claim 44.
46. A host cell engineered to express the polypeptide of claim 44 or the nucleic acid(s) of claim 45.
47. A method of producing the polypeptide of claim 44 comprising culturing the host cell of claim 46.
48. A protein engineering method comprising generating a VH comprising an N- linked glycosylation site within 10 amino acids of its C-terminus.
49. A method of reducing antigenicity of an antigen-binding molecule comprising a VH, the method comprising incorporating an N-linked glycosylation site within 10 amino acids of the C-terminus of the VH.
50. The method of claim 49, wherein the method comprises substituting one or more amino acids within the sequence of the VH such that the VH comprises the amino acid sequence X1X2X3X4X5NX6X7X8X9X10X11X12 within 10 amino acids of its C-terminus, wherein (a)
X1, X2, X3, X4, and X5 are each independently selected from any amino acid; (b) X6 is selected from any amino acid, optionally wherein the amino acid is not proline; (c) X7 is S or T, and (d) X8, X9, X10,X11, and X12 are each independently selected from any amino acid and absent, optionally wherein the amino acid sequence X1X2X3X4X5NX6X7X8X9X10X11X12 is as defined in any one of claims 5 to 26.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US202363598776P | 2023-11-14 | 2023-11-14 | |
| US63/598,776 | 2023-11-14 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2025106469A1 true WO2025106469A1 (en) | 2025-05-22 |
Family
ID=93962490
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2024/055618 Pending WO2025106469A1 (en) | 2023-11-14 | 2024-11-13 | Engineered heavy chain variable domains and uses thereof |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2025106469A1 (en) |
Citations (34)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5582996A (en) | 1990-12-04 | 1996-12-10 | The Wistar Institute Of Anatomy & Biology | Bifunctional antibodies and method of preparing same |
| US5677425A (en) | 1987-09-04 | 1997-10-14 | Celltech Therapeutics Limited | Recombinant antibody |
| US5731168A (en) | 1995-03-01 | 1998-03-24 | Genentech, Inc. | Method for making heteromultimeric polypeptides |
| WO1998025971A1 (en) | 1996-12-10 | 1998-06-18 | Celltech Therapeutics Limited | Monovalent antibody fragments |
| WO1999015549A2 (en) | 1997-09-19 | 1999-04-01 | Celltech Therapeutics Limited | Peptide sequences as hinge regions in proteins like immunoglobulin fragments and their use in medicine |
| US5910573A (en) | 1992-01-23 | 1999-06-08 | Merck Patent Gesellschaft Mit Beschrankter Haftung | Monomeric and dimeric antibody-fragment fusion proteins |
| US5932448A (en) | 1991-11-29 | 1999-08-03 | Protein Design Labs., Inc. | Bispecific antibody heterodimers |
| US6579855B1 (en) | 1998-11-06 | 2003-06-17 | Ark Therapeutics, Ltd. | Adenovirus-mediated gene therapy |
| US6833441B2 (en) | 2001-08-01 | 2004-12-21 | Abmaxis, Inc. | Compositions and methods for generating chimeric heteromultimers |
| WO2005003171A2 (en) | 2003-07-01 | 2005-01-13 | Celltech R & D Limited | Modified antibody fragments |
| WO2005003169A2 (en) | 2003-07-01 | 2005-01-13 | Celltech R & D Limited | Modified antibody fab fragments |
| WO2005003170A2 (en) | 2003-07-01 | 2005-01-13 | Celltech R & D Limited | Modified antibody fragments |
| WO2005049094A1 (en) | 2003-11-13 | 2005-06-02 | Advantagene, Inc. | A mixed complementary viral vector for gene therapy |
| US20060204493A1 (en) | 2004-09-02 | 2006-09-14 | Genentech, Inc. | Heteromultimeric molecules |
| US7183076B2 (en) | 1997-05-02 | 2007-02-27 | Genentech, Inc. | Method for making multispecific antibodies having heteromultimeric and common components |
| EP1870459A1 (en) | 2005-03-31 | 2007-12-26 | Chugai Seiyaku Kabushiki Kaisha | Methods for producing polypeptides by regulating polypeptide association |
| US20080008713A1 (en) * | 2002-06-28 | 2008-01-10 | Domantis Limited | Single domain antibodies against tnfr1 and methods of use therefor |
| WO2009080251A1 (en) | 2007-12-21 | 2009-07-02 | F. Hoffmann-La Roche Ag | Bivalent, bispecific antibodies |
| WO2009089004A1 (en) | 2008-01-07 | 2009-07-16 | Amgen Inc. | Method for making antibody fc-heterodimeric molecules using electrostatic steering effects |
| US8586713B2 (en) | 2009-06-26 | 2013-11-19 | Regeneron Pharmaceuticals, Inc. | Readily isolated bispecific antibodies with native immunoglobulin format |
| WO2014082179A1 (en) | 2012-11-28 | 2014-06-05 | Zymeworks Inc. | Engineered immunoglobulin heavy chain-light chain pairs and uses thereof |
| WO2014121087A1 (en) | 2013-02-01 | 2014-08-07 | Regeneron Pharmaceuticals, Inc. | Antibodies comprising chimeric constant domains |
| WO2014150973A1 (en) | 2013-03-15 | 2014-09-25 | Eli Lilly And Company | Methods for producing fabs and bi-specific antibodies |
| WO2015112800A1 (en) | 2014-01-23 | 2015-07-30 | Regeneron Pharmaceuticals, Inc. | Human antibodies to pd-1 |
| WO2015179918A1 (en) * | 2014-05-27 | 2015-12-03 | The University Of Queensland | Modulation of cellular stress |
| WO2016161010A2 (en) | 2015-03-30 | 2016-10-06 | Regeneron Pharmaceuticals, Inc. | Heavy chain constant regions with reduced binding to fc gamma receptors |
| US20180371088A1 (en) * | 2017-06-22 | 2018-12-27 | Development Center For Biotechnology | TARGET CELL-DEPENDENT T CELL ENGAGING AND ACTIVATION ASYMMETRIC HETERODIMERIC Fc-ScFv FUSION ANTIBODY FORMAT AND USES THEREOF IN CANCER THERAPY |
| US10294299B2 (en) | 2016-01-22 | 2019-05-21 | MabQuest SA | Immunological reagents |
| US10412940B2 (en) | 2010-02-08 | 2019-09-17 | Regeneron Pharmaceuticals, Inc. | Mice expressing a limited immunoglobulin light chain repertoire |
| US11034765B2 (en) | 2015-10-02 | 2021-06-15 | Symphogen A/S | Anti-PD-1 antibodies and compositions |
| US20220056126A1 (en) | 2016-10-13 | 2022-02-24 | Symphogen A/S | Anti-lag-3 antibodies and compositions |
| US11306139B2 (en) * | 2015-03-20 | 2022-04-19 | Ablynx N.V. | Glycosylated immunoglobulin single variable domains |
| WO2023180346A1 (en) * | 2022-03-22 | 2023-09-28 | Morphosys Ag | Deimmunized antibodies specific for cd3 |
| WO2023215839A1 (en) * | 2022-05-06 | 2023-11-09 | 76Bio, Inc. | Bifunctional fusion proteins for ubiquitin-mediated degradation |
-
2024
- 2024-11-13 WO PCT/US2024/055618 patent/WO2025106469A1/en active Pending
Patent Citations (35)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5677425A (en) | 1987-09-04 | 1997-10-14 | Celltech Therapeutics Limited | Recombinant antibody |
| US5582996A (en) | 1990-12-04 | 1996-12-10 | The Wistar Institute Of Anatomy & Biology | Bifunctional antibodies and method of preparing same |
| US5932448A (en) | 1991-11-29 | 1999-08-03 | Protein Design Labs., Inc. | Bispecific antibody heterodimers |
| US5910573A (en) | 1992-01-23 | 1999-06-08 | Merck Patent Gesellschaft Mit Beschrankter Haftung | Monomeric and dimeric antibody-fragment fusion proteins |
| US5731168A (en) | 1995-03-01 | 1998-03-24 | Genentech, Inc. | Method for making heteromultimeric polypeptides |
| US7695936B2 (en) | 1995-03-01 | 2010-04-13 | Genentech, Inc. | Knobs and holes heteromeric polypeptides |
| WO1998025971A1 (en) | 1996-12-10 | 1998-06-18 | Celltech Therapeutics Limited | Monovalent antibody fragments |
| US7183076B2 (en) | 1997-05-02 | 2007-02-27 | Genentech, Inc. | Method for making multispecific antibodies having heteromultimeric and common components |
| WO1999015549A2 (en) | 1997-09-19 | 1999-04-01 | Celltech Therapeutics Limited | Peptide sequences as hinge regions in proteins like immunoglobulin fragments and their use in medicine |
| US6579855B1 (en) | 1998-11-06 | 2003-06-17 | Ark Therapeutics, Ltd. | Adenovirus-mediated gene therapy |
| US6833441B2 (en) | 2001-08-01 | 2004-12-21 | Abmaxis, Inc. | Compositions and methods for generating chimeric heteromultimers |
| US20080008713A1 (en) * | 2002-06-28 | 2008-01-10 | Domantis Limited | Single domain antibodies against tnfr1 and methods of use therefor |
| WO2005003170A2 (en) | 2003-07-01 | 2005-01-13 | Celltech R & D Limited | Modified antibody fragments |
| WO2005003169A2 (en) | 2003-07-01 | 2005-01-13 | Celltech R & D Limited | Modified antibody fab fragments |
| WO2005003171A2 (en) | 2003-07-01 | 2005-01-13 | Celltech R & D Limited | Modified antibody fragments |
| WO2005049094A1 (en) | 2003-11-13 | 2005-06-02 | Advantagene, Inc. | A mixed complementary viral vector for gene therapy |
| US20060204493A1 (en) | 2004-09-02 | 2006-09-14 | Genentech, Inc. | Heteromultimeric molecules |
| EP1870459A1 (en) | 2005-03-31 | 2007-12-26 | Chugai Seiyaku Kabushiki Kaisha | Methods for producing polypeptides by regulating polypeptide association |
| WO2009080251A1 (en) | 2007-12-21 | 2009-07-02 | F. Hoffmann-La Roche Ag | Bivalent, bispecific antibodies |
| WO2009089004A1 (en) | 2008-01-07 | 2009-07-16 | Amgen Inc. | Method for making antibody fc-heterodimeric molecules using electrostatic steering effects |
| US8586713B2 (en) | 2009-06-26 | 2013-11-19 | Regeneron Pharmaceuticals, Inc. | Readily isolated bispecific antibodies with native immunoglobulin format |
| US10412940B2 (en) | 2010-02-08 | 2019-09-17 | Regeneron Pharmaceuticals, Inc. | Mice expressing a limited immunoglobulin light chain repertoire |
| WO2014082179A1 (en) | 2012-11-28 | 2014-06-05 | Zymeworks Inc. | Engineered immunoglobulin heavy chain-light chain pairs and uses thereof |
| WO2014121087A1 (en) | 2013-02-01 | 2014-08-07 | Regeneron Pharmaceuticals, Inc. | Antibodies comprising chimeric constant domains |
| WO2014150973A1 (en) | 2013-03-15 | 2014-09-25 | Eli Lilly And Company | Methods for producing fabs and bi-specific antibodies |
| WO2015112800A1 (en) | 2014-01-23 | 2015-07-30 | Regeneron Pharmaceuticals, Inc. | Human antibodies to pd-1 |
| WO2015179918A1 (en) * | 2014-05-27 | 2015-12-03 | The University Of Queensland | Modulation of cellular stress |
| US11306139B2 (en) * | 2015-03-20 | 2022-04-19 | Ablynx N.V. | Glycosylated immunoglobulin single variable domains |
| WO2016161010A2 (en) | 2015-03-30 | 2016-10-06 | Regeneron Pharmaceuticals, Inc. | Heavy chain constant regions with reduced binding to fc gamma receptors |
| US11034765B2 (en) | 2015-10-02 | 2021-06-15 | Symphogen A/S | Anti-PD-1 antibodies and compositions |
| US10294299B2 (en) | 2016-01-22 | 2019-05-21 | MabQuest SA | Immunological reagents |
| US20220056126A1 (en) | 2016-10-13 | 2022-02-24 | Symphogen A/S | Anti-lag-3 antibodies and compositions |
| US20180371088A1 (en) * | 2017-06-22 | 2018-12-27 | Development Center For Biotechnology | TARGET CELL-DEPENDENT T CELL ENGAGING AND ACTIVATION ASYMMETRIC HETERODIMERIC Fc-ScFv FUSION ANTIBODY FORMAT AND USES THEREOF IN CANCER THERAPY |
| WO2023180346A1 (en) * | 2022-03-22 | 2023-09-28 | Morphosys Ag | Deimmunized antibodies specific for cd3 |
| WO2023215839A1 (en) * | 2022-05-06 | 2023-11-09 | 76Bio, Inc. | Bifunctional fusion proteins for ubiquitin-mediated degradation |
Non-Patent Citations (35)
| Title |
|---|
| "Remington's Pharmaceutical Sciences", 1980 |
| AL-LAZIKANI ET AL., J. MOL. BIOL., vol. 273, 1997, pages 927 - 948 |
| ALMAGRO, FRONTIERS IN BIOSCIENCE, vol. 13, 2008, pages 1619 - 1633 |
| ANGEL ET AL., MOL IMMUNOL, vol. 30, no. 1, 1993, pages 105 - 108 |
| BIRD ET AL., SCIENCE, vol. 242, 1988, pages 423 - 426 |
| CARTER, IMMUNOL METH, vol. 248, 2001, pages 7 - 15 |
| CERULLO ET AL., CANCER RES., vol. 70, 2010, pages 4297 |
| CHEN ET AL., ADV DRUG DELIV REV., vol. 65, no. 10, 2013, pages 1357 - 1369 |
| GAO ET AL.: "Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy", PNAS, vol. 99, no. 18, 3 September 2002 (2002-09-03), pages 11854 - 11859 |
| GOLAY ET AL., J IMMUNOL, vol. 196, 2016, pages 3199 - 211 |
| GUNASEKARAN ET AL., J BIOL CHEM, vol. 285, no. 25, 2010, pages 19637 - 46 |
| HAFEEZ ET AL., MOLECULES, vol. 25, 2020, pages 4764 |
| HANSEL, NAT REV DRUG DISCOV, vol. 9, no. 4, 2010, pages 325 - 338 |
| HOLLINGERHUDSON, NATURE BIOTECHNOLOGY, vol. 23, 2005, pages 1126 - 1136 |
| HUSTON ET AL., PROC. NATL. ACAD. SCI. USA, vol. 85, 1988, pages 5879 - 5883 |
| KABAT: "Sequences of Proteins of Immunological Interest", 1991, NATIONAL INSTITUTES OF HEALTH |
| KLEIN ET AL., PROTEIN ENGINEERING, DESIGN & SELECTION, vol. 27, no. 10, 2014, pages 325 - 330 |
| KOREN ET AL., CURR PHARM BIOTECHNOL, vol. 3, no. 4, 2002, pages 349 - 60 |
| KUMAR VINAY ET AL: "TNFR1 Contributes to Activation-Induced Cell Death of Pathological CD4+ T Lymphocytes During Ischemic Heart Failure", JACC: BASIC TO TRANSLATIONAL SCIENCE, vol. 7, no. 10, 1 October 2022 (2022-10-01), pages 1038 - 1049, XP093256782, ISSN: 2452-302X, DOI: 10.1016/j.jacbts.2022.05.005 * |
| LEFRANC ET AL., DEV. COMP. IMMUNOL., vol. 27, 2003, pages 55 - 77 |
| LEWIS ET AL., NATURE BIOTECHNOLOGY, vol. 32, 2014, pages 191 - 198 |
| MARTIN ET AL., PROC. NATL. ACAD. SCI. USA, vol. 86, 1989, pages 9268 - 9272 |
| MAZOR ET AL., MABS, vol. 7, 2015, pages 364 - 76 |
| MCCAFFERTY ET AL., NATURE, vol. 348, 1990, pages 552 - 554 |
| PLUCKTHUN: "The Pharmacology of Monoclonal Antibodies", vol. 113, 1994, SPRINGER-VERLAG, pages: 269 - 315 |
| QI ET AL., MODERN ONCOLOGY, vol. 14, 2006, pages 1295 - 1297 |
| RASMUSSEN ET AL., CANCER GENE THER, vol. 9, 2002, pages 951 - 7 |
| RIDGWAY ET AL., PROT ENG, vol. 9, 1996, pages 617 - 621 |
| RUSSELL ET AL., NATURE BIOTECHNOLOGY, vol. 30, 2012, pages 658 - 670 |
| SCHELLEKENS, CLIN THER, vol. 24, no. 11, 2002, pages 1720 - 40 |
| SPIESS ET AL., MOL IMMUNOL, vol. 67, 2015, pages 95 - 106 |
| STERMAN ET AL., MOL. THER., vol. 18, 2010, pages 852 - 860 |
| VAN SCHIE ET AL., ANN RHEUM DIS, vol. 74, no. 1, 2015, pages 311 - 314 |
| WARD ET AL., NATURE, vol. 341, 1989, pages 544 - 546 |
| ZHAO YILING ET AL: "miR-29a suppresses MCF-7 cell growth by downregulating tumor necrosis factor receptor 1", TUMOR BIOLOGY, vol. 39, no. 2, 1 February 2017 (2017-02-01), CH, XP093256780, ISSN: 1010-4283, DOI: 10.1177/1010428317692264 * |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US12522638B2 (en) | IL12 receptor agonists and methods of use thereof | |
| CA3229369A1 (en) | Novel il27 receptor agonists and methods of use thereof | |
| WO2024040249A1 (en) | Interferon receptor agonists and uses thereof | |
| AU2023325400A1 (en) | Interferon proproteins and uses thereof | |
| AU2023269792A1 (en) | Multispecific binding molecule proproteins and uses thereof | |
| WO2025106469A1 (en) | Engineered heavy chain variable domains and uses thereof | |
| WO2025199243A1 (en) | Trivalent multispecific binding molecules and methods of use thereof | |
| US20240270836A1 (en) | Il12 receptor agonists and methods of use thereof | |
| JP2025520175A (en) | Interleukin-2 proprotein and uses thereof | |
| WO2024182540A2 (en) | T cell activators and methods of use thereof | |
| KR20250030976A (en) | Interleukin-2 proprotein and uses thereof | |
| JP2026504072A (en) | IL12 receptor agonists and methods of use thereof | |
| HK40123893A (en) | Interleukin-2 proproteins and uses thereof | |
| WO2025245494A1 (en) | Tumor-targeted split il12 receptor agonists | |
| HK40116914A (en) | Multispecific binding molecule proproteins and uses thereof | |
| HK40126891A (en) | Interferon proproteins and uses thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 24827970 Country of ref document: EP Kind code of ref document: A1 |