WO2025099489A1 - Antibody-drug conjugate comprising bispecific antibody specifically binding to cd20 and cd22 and uses thereof - Google Patents
Antibody-drug conjugate comprising bispecific antibody specifically binding to cd20 and cd22 and uses thereof Download PDFInfo
- Publication number
- WO2025099489A1 WO2025099489A1 PCT/IB2024/000636 IB2024000636W WO2025099489A1 WO 2025099489 A1 WO2025099489 A1 WO 2025099489A1 IB 2024000636 W IB2024000636 W IB 2024000636W WO 2025099489 A1 WO2025099489 A1 WO 2025099489A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- amino acid
- seq
- acid sequence
- conjugate
- domain
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6801—Drug-antibody or immunoglobulin conjugates defined by the pharmacologically or therapeutically active agent
- A61K47/6803—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates
- A61K47/68031—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates the drug being an auristatin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6801—Drug-antibody or immunoglobulin conjugates defined by the pharmacologically or therapeutically active agent
- A61K47/6803—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates
- A61K47/68035—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates the drug being a pyrrolobenzodiazepine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6875—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody being a hybrid immunoglobulin
- A61K47/6879—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody being a hybrid immunoglobulin the immunoglobulin having two or more different antigen-binding sites, e.g. bispecific or multispecific immunoglobulin
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2887—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against CD20
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2896—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against molecules with a "CD"-designation, not provided for elsewhere
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/505—Medicinal preparations containing antigens or antibodies comprising antibodies
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/30—Immunoglobulins specific features characterized by aspects of specificity or valency
- C07K2317/31—Immunoglobulins specific features characterized by aspects of specificity or valency multispecific
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/73—Inducing cell death, e.g. apoptosis, necrosis or inhibition of cell proliferation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/77—Internalization into the cell
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/90—Immunoglobulins specific features characterized by (pharmaco)kinetic aspects or by stability of the immunoglobulin
Definitions
- Cancer refers to a disease caused by abnormally grown lumps due to autonomous overgrowth of body tissues, and is the result of uncontrolled cell growth in various tissues. Tumors in early stage can be removed by surgical and radio-therapeutic measures, and metastasized tumors are generally treated using chemotherapy.
- chemotherapeutic agents administered parenterally may induce unwanted side effects and even serious toxicity, as a result of systemic administration. Accordingly, the focus of development has been on developing treatments to achieve increased efficacy and/or reduced minimal toxicity/side effects, for example through the selective application of these chemotherapeutic agents in tumor cells or immediately adjacent tissues. There remains a demand for development of antibody-drug conjugates for effective therapeutic methods.
- the present disclosure provides, among other things, antibody-drug conjugates (ADCs) targeting both CD20 and CD22, active metabolites of such ADCs, methods for preparation of such ADCs, and compositions and uses of such ADCs for preventing or treating cancer or proliferative diseases. More particularly, the present invention provides ADCs comprising a bispecific antibody specifically binding to CD20 and CD22, and a pharmaceutical composition comprising the same. In some embodiments, the bispecific anti-CD20xCD22 antibody of the present disclosure has improved cell internalization as compared to antibodies known in the art (e.g., an anti-CD20 antibody).
- the present disclosure provides antibody-drug conjugates comprising a bispecific antibody specifically binding to CD20 and CD22.
- the antibody disclosed herein binds to CD20 and CD22 expressed in a tumor and may be used to deliver an active agent to the tumor.
- the antibody-drug conjugates disclosed herein have improved stability and/or cell internalization as compared to antibody-drug conjugates known in the art.
- Ab is a bispecific antibody comprising a first antigen-binding domain specifically binding to CD20 and a second antigen-binding domain specifically binding to CD22, each L is, independently, a linker, each B is, independently, an active agent, and
- 1 and m are each independently 1 to 20.
- Fig. 1 shows the binding affinity of an exemplified anti-CD20xCD22 bispecific antibody (anti-CD20xCD22 Bispecific Antibody 1) and exemplified anti-CD20, anti-CD22, and control IgG antibodies to cancer cell lines.
- Fig. 2 shows the internalization rate of an exemplified anti-CD20xCD22 bispecific antibody (anti-CD20xCD22 Bispecific Antibody 1) and exemplified anti-CD20 and control IgG antibodies in Ramos cells.
- Fig. 3 shows the internalization rate of exemplified anti-CD20xCD22 bispecific ADCs (ADC2, 6, and 7) and an exemplified anti-CD20 ADC (ADC1) in Ramos cells.
- Fig. 4 shows the plasma concentration of ADC2 at indicated time points in rats.
- Fig. 5 shows that in the SU-DHL-8 cell model, ADC2 exhibited better tumor growth inhibition than ADC 1 at the same dose (0.3 mg/kg, QDxl).
- Fig. 6 shows that in the Granta-519 cell model, ADC2 exhibited significant tumor growth inhibition compared to the control group.
- Fig. 7 shows that in the MINO cell model, ADC2 exhibited significant tumor growth inhibition compared to the control group.
- Fig. 8 shows that in Ramos cell model, ADC2 exhibited dose-dependent tumor growth inhibition. ADC2 exhibited better tumor growth inhibition than an exemplified anti-CD22 ADC (Inotuzumab Ozogamicin) at the same dose.
- Fig. 9 shows that in WSU-DLCL2 cell model, ADC2 exhibited dose-dependent tumor growth inhibition. ADC2 exhibited significant tumor growth inhibition than ADC 1 at the same dose (0.3 mg/kg, QDxl) or R-CHOP.
- An antibody-drug conjugate is a targeted technology for conjugating a toxin or a drug to an antibody that binds to an antigen, by which the toxin is released in a cell to cause death of cancer cells and the like.
- the ADC enables a drug to be accurately delivered to target cancer cells while minimally affecting healthy cells, and to be released only under specific conditions, and thus has excellent efficacy compared to antibody therapeutic agents themselves and can remarkably reduce the risk of side effects compared to existing anticancer agents.
- antibody-linker-small molecule drug toxin
- the linker plays a functional role in linking the antibody and the drug, but in some cases also ensures that the drug is released from the antibody at the appropriate time, for example after reaching target cells. That is, the stability of the linker can play a very important role in the efficacy and safety such as systemic toxicity of an antibody-drug conjugate (Discovery Medicine 2010, 10(53): 329-39).
- monoclonal antibodies for cancer treatment has had substantial success.
- monoclonal antibodies are suitable for target-directed addressing of tumor tissue and tumor cells.
- Antibody-drug conjugates have become a novel and powerful option for the treatment of lymphomas and solid cancers, and immunomodulatory antibodies also have recently had considerable success in clinical trials.
- the development of therapeutic antibodies is based on deep understanding of cancer serology, protein engineering technology and the action thereof, mechanisms of resistance, and interactions between immune systems and cancer cells.
- Antigens which are expressed on the surface of human cancer cells are defined as a broad range of targets which are over-expressed compared to normal tissues, mutated and selectively expressed.
- the key challenge is to identify antigens suitable for antibody-based therapies.
- These therapeutic agents mediate changes in antigen or receptor function (i.e., function as a stimulant or an antagonist), regulate the immune system through Fc and T cell activation, and exhibit efficacy through the delivery of specific drugs that bind to antibodies targeting specific antigens.
- Molecular techniques that can alter antibody pharmacokinetics, action function, size and immune stimulation are emerging as key factors in the development of novel antibody-based therapies.
- Evidence from clinical trials of therapeutic antibodies in cancer patients highlights the importance of approaches for selecting optimized antibodies, including affinity and binding of target antigens and antibodies, selection of an antibody structure, and therapeutic approaches (signaling blockade or immune function).
- a CD20 antigen (also referred to as a human B-lymphocyte-restricted differentiation antigen, Bp35) is a hydrophobic transmembrane protein with a molecular weight of approximately 35 kDa, which is located on pre-B and mature B lymphocytes. The antigen is also expressed in 90% or more of B-cell non-Hodgkin’s lymphomas (NHL), but may not be found in hematopoietic stem cells, pro-B cells, normal plasma cells, or other normal tissues.
- CD20 has been shown to regulate early step(s) in an activation process for cell cycle initiation and differentiation, and possibly act as a calcium ion channel.
- CD22 is a 135-kDa B cell-restricted sialoglycoprotein that is expressed on the B-cell surface only during a mature stage of differentiation.
- the main form of CD22 in humans is CD22 beta, which contains seven immunoglobulin superfamily domains in the extracellular domain.
- CD22 alpha contains seven immunoglobulin superfamily domains 3 and 4.
- Ligands that bind to human CD22 have been presented to associate with immunoglobulin superfamily domains 1 and 2.
- CD22 expression ranges from 91 % to 99% in the aggressive and indolent populations, respectively.
- CD22 may function as both a component of a B-cell activation complex and an adhesion molecule. After binding to natural ligands or antibodies thereof, CD22 is rapidly internalized to provide potent co-stimulatory signals to primary B cells and proapoptotic signals to neonatal B cells.
- compositions containing antibodies are useful for the treatment of diseases (e.g., cancers).
- ADCs antibody-drug conjugates
- the present disclosure provides antibody-drug conjugates comprising a bispecific antibody that specifically binds to CD20 and CD22.
- the antibody disclosed herein binds to CD20 and CD22 expressed in a tumor and may be used to deliver a drug to the tumor.
- the antibody-drug conjugates disclosed herein have improved stability and/or cell internalization as compared to antibody-drug conjugates known in the art.
- the antibody-drug conjugates disclosed herein enable a drug and/or toxin to be easily, specifically, and efficiently released in cancer cells to maximize efficacy, and enable a drug and/or toxin to stably reach a target cell.
- conjugates having a structure represented by General Formula I or a pharmaceutically acceptable salt thereof:
- Ab is a bispecific antibody comprising a first antigen-binding domain specifically binding to CD20 and a second antigen-binding domain specifically binding to CD22, each L is, independently, a linker, each B is, independently, an active agent, and 1 and m are each independently 1 to 20.
- L is a cleavable linker
- a cleavable linker refers to a linker or linker component that links two moieties by covalent linkage, but is degraded to cleave the covalent linkage between the moieties under physiologically relevant conditions.
- the cleavable linker is cleaved in vivo more rapidly in an intracellular environment than outside the cell to allow the release of a payload (e.g., an active agent) to occur preferentially inside a targeted cell.
- the cleavage may be enzymatic or non-enzymatic.
- the cleavage may release the payload from the antibody without degrading the antibody.
- the cleavage may allow a part of the linker or linker component to be attached to the payload, or may release the payload without any residue of the linker.
- L is a protease cleavable linker, an acid-cleavable linker, a disulfide linker, or a glycosidase cleavable linker. In some embodiments, L is a protease cleavable linker. In some embodiments, the protease is selected from one or more of a cysteine protease, a metalloprotease, a serine protease, a threonine protease, and an aspartic protease.
- a cysteine protease also known as a thiol protease, is a protease that shares a common catalytic mechanism including a nucleophilic cysteine thiol in a catalytic triad or dyad.
- a metalloprotease is a protease of which the catalytic mechanism contains metal. Most metalloproteases require zinc, but some use cobalt. The metal ion is coordinated to a protein via three ligands.
- the serine protease is an enzyme that cleaves peptide bonds in a protein. Serine acts as a nucleophilic amino acid at an active site of the enzyme.
- the serine protease is divided into two broad categories of chymotrypsin-like (trypsin-like) or subtilisin-like according to a structure.
- the threonine protease is a protease family that contains a threonine (Thr) residue in the active site.
- the prototypical member of the enzyme in the family is a catalytic subunit of the proteasome, but acyltransferases have convergently evolved the same active site geometry and mechanism.
- the aspartic protease is a catalytic type of protease enzyme that uses activated water molecules bound to one or more aspartate residues to catalyze a peptide substrate.
- two highly conserved aspartate salts exist in the active site and are optimally activated at acidic pH. Almost all known aspartic proteases are inhibited by pepstatin.
- the protease is a cathepsin.
- L comprises 2 or more amino acid residues, e.g., defining the recognition site of the protease. In some embodiments, L comprises 2 to 4 amino acid residues, e.g., defining the recognition site of the protease.
- L comprises valine-alanine (Vai-Ala), valine-citrulline (Val- Cit), valine-lysine (Val-Lys), valine-arginine (Val-Arg), phenylalanine-lysine-glycine-proline- leucine -glycine (Phe-Lys-Gly-Pro-Leu-Gly), or alanine-alanine-proline-valine (Ala-Ala-Pro- Val).
- L comprises Val-Cit, Vai-Ala, or Gly-Gly-Phe-Gly.
- L is a glycosidase cleavable linker.
- the glycosidase is P-glucuronidase or p-galactosidase.
- L comprises a P- glucuronosyl or P-galactosyl moiety.
- L comprises any one of the following components: 6- maleimidocaproyl (MC), maleimido propanoyl (MP), valine-citrulline (Val-Cit), alaninephenylalanine (Ala-Phe), lysine -phenylalanine (Lys-Phe), p-aminobenzyloxycarbonyl (PAB), 4-thio-pentanoate (SPP), 4-thio-butyrate (SPDB), 4-(N-maleimidomethyl)cyclo-hexane-l- carboxylate (MCC), maleimidoethyl (ME), 4-thio-2-hydroxysulfonyl-butyrate (2-Sulfo- SPDB), aryl-thiol (PySS), (4-acetyl)aminobenzoate (SIAB), oxylbenzylthio, aminobenzylthio, dioxylbenzylthio, diaminobenzylthio, amino
- L is covalently bonded to Ab by a thioether, thioester, disulfide, hydrazone, ester, carbamate, carbonate, alkoxy, or amide.
- L comprises a peptide further comprising at least one hydrophilic amino acid.
- the peptide comprises an amino acid having a side chain having a moiety that bears a charge at neutral pH in aqueous solution (e.g., an amine, guanidine, or carboxyl moiety).
- the peptide comprises an amino acid selected from alanine, histidine, arginine, aspartate, asparagine, glutamate, glutamine, glycine, lysine, ornithine, proline, serine, and threonine. In some embodiments, the peptide comprises an amino acid selected from alanine, aspartate, asparagine, glutamate, glutamine, glycine, lysine, ornithine, proline, serine, and threonine.
- L comprises a functional group capable of reacting with a terminal moiety of a particular molecule.
- functional groups capable of reacting with a terminal amine include N-hydroxysuccinimide ester, p-nitrophenyl ester, dinitrophenyl ester, pentafluorophenyl ester, carboxylic acid chloride, and carboxylic anhydride.
- functional groups capable of reacting with a terminal thiol include pyridyl disulfide, nitropyridyl disulfide, maleimide, haloacetate, methylsulfonphenyl oxadiazole (ODA), carboxylic acid chloride, and carboxylic acid anhydride.
- Examples of functional groups capable of reacting with a terminal ketone or aldehyde include amines, alkoxyamines, hydrazines, acyloxyamines, and hydrazides.
- Examples of functional groups capable of reacting with a terminal azide include alkynes.
- L comprises an oxime. In some embodiments, L comprises at least one isoprenyl group. In some embodiments, L comprises at least one succinimide group.
- L is covalently bonded to Ab by a thioether bond and the thioether bond comprises a sulfur atom of a cysteine of Ab.
- Ab at the C-terminus comprises an amino acid motif that is recognized by an isoprenoid transferase.
- the isoprenoid transferase is famesyl protein transferase (FTase) or geranylgeranyl transferase (GGTase).
- L is covalently bonded to Ab by a thioether bond and the thioether bond comprises a sulfur atom of a cysteine of the amino acid motif.
- the amino acid motif comprises a CY iY iX sequence, wherein: C is cysteine; each Y 1 is independently an aliphatic amino acid; and X is selected from glutamine, glutamate, serine, cysteine, methionine, alanine, and leucine. In some embodiments, each Yi is independently selected from alanine, isoleucine, leucine, methionine, and valine. In some embodiments, the amino acid motif comprises a CVIM (SEQ ID NO: 84) or CVLL (SEQ ID NO: 85). In some embodiments, at least one of the 1 to 20 amino acids preceding the amino acid motif is glycine. In some embodiments, the amino acid motif has the sequence GGGGGGGCVIM (SEQ ID NO: 77).
- L comprises a self-immolative moiety.
- the self- immolative moiety includes, without limitation, 2 -aminoimidazole -5 -methanol derivatives, heterocyclic PAB analogs, beta-glucuronides, and aromatic compounds electronically similar to a para-aminobenzylcarbamoyl (PAB) group, such as a group of ortho or paraaminobenzylacetals.
- PAB para-aminobenzylcarbamoyl
- the conjugate has a structure represented by the General Formula II or a pharmaceutically acceptable salt thereof:
- G represents a sugar moiety or a glucuronic acid moiety
- R 1 and R 2 are each independently hydrogen, C1-8 alkyl, or C3-8 cycloalkyl; or R 1 and R 2 combine to complete a (CYC's) cycloalkyl ring; W is -*C(O)-, -*C(O)N(R’)-, -*N(R’)C(O)-, -*(CH 2 )tN(R’)C(O)-, -*C(O)O-, -*S(O 2 )N(R’)-, -*P(O)(R”)N(R’)-, -*S(O)N(R’)-, or -*P(O 2 )N(R’)-, wherein the C(O), N, CH 2 , S, or P marked with an * is directly bonded to the phenyl ring of General Formula II,
- R’ and R” are each independently hydrogen, C1-8 alkyl, C3-8 cycloalkyl, C1-8 alkoxy, C1-8 alkylthio, mono- or di-C1-8 alkylamino, Cs- 2 o heteroaryl or Ce- 2 o aryl;
- Z is each independently C1-8 alkyl, halogen, cyano, or nitro; nl and n2 are each independently 1 to 20; n3 is 0 to 3; and
- Y is absent, C1-C50 alkylene, C1-C50 alkenylene, 1-50 atom heterocyclylene, or 1-50 atom heteroalkylene.
- Y comprises a C1-50 alkylene or C1-50 heteroalkylene and further comprises at least one of:
- a heterocyclylene or a heteroarylene e.g., a heteroarylene in the alkylene or heteroalkylene chain
- R 1 and R 2 are each independently hydrogen. In some embodiments, n3 is 0.
- the G has a structure represented by the General Formula IV :
- R3 is hydrogen, alkyl, CH2OR3A, or CO2R3B;
- R3A is hydrogen or a hydroxyl protecting group
- RSB is hydrogen or a carboxyl protecting group; and each R4 is independently a hydrogen or a hydroxyl protecting group.
- R3 is-COOH.
- each R4 is independently hydrogen.
- the W is -*C(O)NR’-.
- W is -*C(O)NR’-.
- Y is covalently bonded to Ab by a thioether, thioester, disulfide, hydrazone, ester, carbamate, carbonate, alkoxy, or amide.
- Y comprises a peptide further comprising at least one hydrophilic amino acid.
- the peptide comprises an amino acid having a side chain having a moiety that bears a charge at neutral pH in aqueous solution (e.g., an amine, guanidine, or carboxyl moiety).
- the peptide comprises an amino acid selected from alanine, histidine, arginine, aspartate, asparagine, glutamate, glutamine, glycine, lysine, ornithine, proline, serine, and threonine. In some embodiments, the peptide comprises an amino acid selected from alanine, aspartate, asparagine, glutamate, glutamine, glycine, lysine, ornithine, proline, serine, and threonine.
- Y comprises an oxime.
- the oxygen atom of the oxime is on the side of Y linked to W and the carbon atom of the oxime is on the side of
- the carbon atom of the oxime is on the side of Y linked to W and the oxygen atom of the oxime is on the side of Y linked to Ab.
- Y is a C1-50 heteroalkylene comprising an oxime.
- the oxygen atom of the oxime is on the side of Y linked to W, and the carbon atom of the oxime is on the side of Y linked to Ab.
- the carbon atom of the oxime is on the side of Y linked to W, and the oxygen atom of the oxime is on the side of
- Y linked to Ab.
- Y comprises an oxime, and at least one isoprenyl unit covalently bonds the oxime to Ab (e.g., the at least one isoprenyl unit directly or indirectly bonds the oxime to Ab).
- Y comprises at least one isoprenyl group.
- Y is covalently bonded to Ab by a thioether bond and the thioether bond comprises a sulfur atom of a cysteine of Ab.
- the Ab at the C-terminus comprises an amino acid motif that is recognized by an isoprenoid transferase.
- the isoprenoid transferase is famesyl protein transferase (FTase) or geranylgeranyl transferase (GGTase).
- Y is covalently bonded to Ab by a thioether bond and the thioether bond comprises a sulfur atom of a cysteine of the amino acid motif.
- the amino acid motif comprises a CY iY iX sequence, wherein C is cysteine; each Y 1 is independently an aliphatic amino acid; and X is selected from glutamine, glutamate, serine, cysteine, methionine, alanine, and leucine. In some embodiments, each Yi is independently selected from alanine, isoleucine, leucine, methionine, and valine. In some embodiments, the amino acid motif comprises a CVIM (SEQ ID NO: 84) or CVLL (SEQ ID NO: 85). In some embodiments, at least one of the 1 to 20 amino acids preceding the amino acid motif is glycine. In some embodiments, the amino acid motif has the sequence GGGGGGGCVIM (SEQ ID NO: 77).
- the Y comprises a connecting unit represented by the General Formula V or General Formula VI:
- V is a single bond, -O-, -S-, -NR 5 -, -C(O)NR 6 -, -NR 7 C(O)-, -NR 8 SO 2 -, or -SO2NR 9 -;
- X is -O-, C1-8 alkylene or -NR 5 -;
- R 5 to R 9 are each independently hydrogen, C1-6 alkyl, C1-6 alkyl C6-20 aryl, or
- q is 1 to 10.
- r is 1 or 2.
- p is 1 or 2.
- V is -O-.
- q is 1 to 10; r and p are each 1 or 2; and V is -O-.
- X is -O-.
- X is -O-; and w is 1 to 10.
- L comprises at least one polyethylene glycol monomer represented by In some embodiments, L comprises a polyethylene glycol oligomer represented by wherein n40 is 2 to 12.
- Y comprises an oxime, and the at least one polyethylene glycol unit covalently bonds the oxime to W.
- Y further comprises a binding unit formed by a reaction between an alkyne and an azide or between an aldehyde or ketone and hydrazine or hydroxylamine.
- Y further comprises a binding unit represented by General Formula Vila, Vllb, Vile, Vlld or Vile:
- Li is each independently a single bond or C1-30 alkylene; and R11 is hydrogen or C1-10 alkyl.
- the conjugate comprises
- the conjugate comprises a structure represented by:
- nl2 is 0 to 30; and the wavy bond represents a connection to Ab.
- Y is branched.
- Y comprises: i) a branching unit covalently bonded to Ab by a primary linker; ii) a first which couples a first B to the branching unit; and iiia) a second branch which couples a second B to the branching unit; or iiib) a second branch, in which an alkyl or heteroalkyl (e.g., a polyethylene glycol monomer or a polyethylene glycol oligomer) is covalently coupled to the branching unit.
- an alkyl or heteroalkyl e.g., a polyethylene glycol monomer or a polyethylene glycol oligomer
- Y comprises a second branch which couples a second B, via a cleavage group, to the branching unit.
- Y comprises a second branch, in which an alkyl or heteroalkyl (e.g., a polyethylene glycol monomer or a polyethylene glycol oligomer) is covalently coupled to the branching unit.
- an alkyl or heteroalkyl e.g., a polyethylene glycol monomer or a polyethylene glycol oligomer
- the branching unit has a structure represented by wherein
- R 30 is hydrogen or C1-30 alkyl
- R 40 is hydrogen or L 5 -COOR 50 ;
- R 50 is hydrogen or C1-30 alkyl
- L 2 , L 3 , and L 4 are each independently a bond or -C n 'H2n'-; and n' is 1 to 10.
- At least one branched linker is covalently coupled to Ab; and at least two B are covalently coupled to the branched linker.
- the conjugate comprises 1, 2, 3, or 4 branched linkers and each branched linker comprises two B.
- the branching unit comprises a lysine residue.
- the conjugate comprises a structure represented by: or a pharmaceutically acceptable salt thereof; wherein each B is, independently, an active agent; nl 1, n22, and n33 are each independently 0 to 30; and
- AA is a peptide comprising at least two amino acid residues; and the wavy bond represents a connection to Ab.
- the present disclosure provides, among other things, a bispecific antibody comprising a first antigen-binding domain specifically binding to CD20 and a second antigen-binding domain specifically binding to CD22, i.e., an anti-CD20xCD22 bispecific antibody (“Ab”).
- a bispecific antibody comprising a first antigen-binding domain specifically binding to CD20 and a second antigen-binding domain specifically binding to CD22, i.e., an anti-CD20xCD22 bispecific antibody (“Ab”).
- the present disclosure provides antibody-drug conjugates comprising Ab.
- the Ab disclosed herein binds to CD20 and CD22 expressed in a tumor and may be used to deliver a drug to the tumor.
- the antibodydrug conjugates and the Ab disclosed herein have improved stability and/or cell internalization as compared to antibody-drug conjugates or antibodies known in the art.
- the bispecific antibody according to the present disclosure is a polypeptide comprising one or more complementarity-determining areas or regions (CDRs).
- the CDR is included in a "framework" region, and the framework orients the CDR(s) so that the CDR(s) can have appropriate antigen-binding properties.
- the antibody according to the present disclosure may consist of a polypeptide of only light chains or only heavy chains including the variable regions shown in Tables 4-9 and 16.
- CDR sequences that may be included in the heavy and light chain variable regions of the antibody or antigen-binding fragment thereof according to an embodiment of the present disclosure are shown in Tables 4-9 and 16.
- an antibody according to the present disclosure shares certain regions or sequences with other antibodies disclosed herein.
- the constant region of the antibody or antigen-binding fragment thereof may be shared.
- Fc regions may be shared.
- the frame of a variable region may be shared.
- the heavy chain variable region and the light chain variable region according to the present disclosure may be linked to at least a part of a human constant region.
- the selection of a constant region may be determined partially by whether or not antibody-dependent cell- mediated cytotoxicity, antibody-dependent cellular phagocytosis, and/or complement- dependent cytotoxicity is required.
- human isotypes IgGl and IgG3 have complement-dependent cytotoxicity
- human isotypes IgG2 and IgG4 do not have such cytotoxicity.
- human IgGl and IgG3 induce a cell-mediated effector function stronger than that of human IgG2 and IgG4.
- the light chain constant region may be lambda or kappa.
- a variable region of an immunoglobulin chain generally has the same overall structure and includes a comparatively conserved framework region (FR) linked by three hypervariable regions called "complementarity determining areas or regions or domains" or complementarity determining regions (CDRs).
- the CDRs of a variable region derived from each chain including a heavy chain/light chain pair are typically aligned by a framework region to form a structure specifically binding to a specific epitope of a target protein.
- These factors of naturally occurring light chain and heavy chain variable regions are typically included from the N-terminus to the C-terminus in the following order: FR1, CDR1, FR2, CDR2, FR3, CDR3, and FR4.
- the position of amino acid sequences corresponding to each variable region may be determined by Kabat (Kabat et al., (1983) U.S. Dept, of Health and Human Services, "Sequences of Proteins of Immunological Interest"), Chothia (Chothia and Lesk, J. Mol. Biol. 196:901-917 (1987)) or in a manner related to the OPAL library (Hye Young Yang et. al., 2009 Mol. Cells 27: 225).
- the CDRs determined by each definition, when compared to each other, may be subsets which overlap or where one includes another. Those of ordinary skill in the art will be readily able to easily select CDR sequences according to the definitions above, given a variable region sequence of an antibody.
- amino acid sequences of CDRs are defined according to Kabat definition.
- CDRs of an antibody can be defined in the art according to a variety of methods, such as Chothia definition based on the location of a structural loop region (Al-Lazikani, B et al., J Mol Biol 273: 927-48 (1997)), and Kabat definition based on sequence variability (Kabat et al., “Sequences of proteins of immunological interest”, fifth edition, National Institutes of Health, Bethesda, MD. (1991)).
- amino acid residues in variable region sequences may also be determined using a Combined definition that incorporates both Kabat definition and Chothia definition.
- the Combined definition refers to the combination of the ranges of Kabat definition and Chothia definition. It should be understood by those skilled in the art that unless otherwise specified, the terms “CDR” and “complementarity determining region” of a given antibody or region thereof (e.g., a variable region) should be understood to encompass the complementarity determining region as defined according to any of the embodiments described in the present disclosure.
- the antibody according to the present disclosure is a humanized antibody.
- a humanized antibody refers to any antibody in which the constant region of a nonhuman antibody is completely substituted with a human form of the constant region, and at least a portion of the variable region of a non-human antibody, except for the three loops of an amino acid sequence outside each variable region that binds to a target structure, is completely or partially substituted with the corresponding portion of a human antibody.
- the antibody according to the present disclosure is a human antibody.
- Certain mutations may be introduced to the framework region to enhance the stability of antibodies while maintaining their antigen binding activity. Stabilization of therapeutic antibodies can result in improved serum half-life, lower dosage requirements, reduced sideeffects, improved shelf-life and reduced shipping and storage costs.
- the present disclosure discloses one or more amino acid sequences having substantial sequence identity to one or more amino acid sequences disclosed herein. Substantial identity means that the effects disclosed herein are maintained in the presence of sequence variations.
- the amino acid sequence has about 90% identity, about 95% identity, or about 99% identity to the heavy chain variable regions shown in Tables 4-9 and 16.
- the amino acid sequence has about 90% identity, about 95% identity, or about 99% identity to the light chain variable regions shown in Tables 4-9 and 16.
- any mutation occurs in the framework of the variable region rather than the CDRs.
- a nucleic acid encoding the antibody or fragment thereof according to the present disclosure is a nucleic acid encoding a full-length antibody including the CDRs disclosed herein, the variable region including the CDRs, and the variable region, and the constant region.
- Ab comprises a first constant region linked to the first antigenbinding domain and a second constant region linked to the second antigen-binding domain, and wherein the first constant region and the second constant region each comprises a light chain constant region CL domain and heavy chain constant region CHI, CH2, and CH3 domains.
- Ab is a chimeric antibody, a humanized antibody, or a human antibody.
- Ab is of IgA, IgG, IgM, IgE, or IgD isotype. In some embodiments, Ab is of IgG isotype (e.g., IgGl, IgG2, IgG3, and IgG4).
- Ab comprises LALA mutations in the first and/or the second heavy chain constant regions.
- the LALA mutations comprise L234A and L235A according to EU numbering convention.
- a bispecific antibody of the present disclosure is produced by the methods described in U.S. Patent No. 9,637,557B2, the entirety of which is incorporated herein by reference.
- a bispecific antibody according to some embodiments may be a heteromultimer.
- heteromultimer refers to a protein multimer consisting of a plurality types of polypeptides, wherein the polypeptides may associate with each other. More specifically, the “heteromultimer” is a molecule having at least a first polypeptide and a second polypeptide, wherein the second polypeptide has at least one amino acid residue different from the first polypeptide in the amino acid sequence.
- the heteromultimer may include at least one amino acid substitution (e.g., to form a knob-in-hole structure) within each domain (e.g., a CH3 domain) of the first constant region and the second constant region.
- the interaction surface of the two CH3 domains is modified so that the heterodimerization of all heavy chains containing the two CH3 domains is increased.
- One of the two CH3 domains (of the two heavy chains) may be a “knob” domain and the other may be a “hole” domain.
- the introduction of a disulfide bridge stabilizes the heterodimer (Merchant, A..M., et al, Nature Biotech 16 (1998) 677-681; Atwell, S., Ridgway, J.B., Wells, J.A., Carter, P., J Mol Biol 270 (1997) 26-35) and increases the yield.
- the bispecific antibody of the present disclosure may be characterized in that the CH3 domain of one heavy chain and the CH3 domain of the other heavy chain form an interface beyond the original interface between the antibody CH3 domains; wherein the interface is modified to facilitate the formation of a bivalent, bispecific antibody, and the modification is characterized as follows: a) the CH3 domain of one heavy chain (e.g., of the first constant region) is modified such that, within the original interface of the CH3 domain of one heavy chain that contacts the original interface of the CH3 domain of the other heavy chain (e.g., of the second constant region) in the bispecific antibody, an amino acid residue is replaced with an amino acid residue having a larger side chain volume to generate a ridge (“knob”) within the interface of the CH3 domain of one heavy chain that is positionable in a space within the interface of the CH3 domain of the other heavy chain, and b) the CH3 domain of the other heavy chain (e.g., of the second constant region) is modified such that, within the original interface of the CH3 domain of the other
- the amino acid residue having the larger side chain volume is selected from arginine (R), phenylalanine (F), tyrosine (Y), and tryptophan (W).
- the amino acid residue having the smaller side chain volume is selected from alanine (A), serine (S), threonine (T), and valine (V).
- both the CH3 domains may be further modified to introduce cysteine (C) as an amino acid into a corresponding position in each CH3 domain, thereby forming a disulfide bridge between the two CH3 domains.
- C cysteine
- the bispecific antibody includes, according to the EU index of Kabat, a T366W mutation in the CH3 domain of the “knob chain” (e.g., the CH3 domain of the first constant region or the second constant region) and T366S, L368A, and/or Y407V mutations in the CH3 domain of the “hole chain” (e.g., the CH3 domain of the first constant region or the second constant region).
- additional interchain disulfide bridges between the CH3 domains may also be used by introducing a Y349C mutation in the CH3 domain of the “knob chain” and an E356C mutation or S354C mutation in the CH3 domain of the “hole chain” (Merchant, A.M., et al., Nature Biotech. 16 (1998) 677-681).
- the bispecific antibody includes Y349C and T366W mutations in one of the two CH3 domains and E356C, T366S, L368A, and Y407V mutations in the other of the two CH3 domains, or the bispecific antibody includes Y349C and T366W mutations in one of the two CH3 domains and S354C, T366S, L368A, and Y407V mutations in the other of the two CH3 domains (wherein an additional Y349C mutation in one CH3 domain and an additional E356C or S354C mutation in the other CH3 domain form an interchain disulfide bridge).
- the bispecific antibody includes a T366W mutation in the CH3 domain of the “knob chain” and T366S, L368A, and Y407V mutations in the CH3 domain of the “hole chain”, and further includes a R409D; K370E mutation in the CH3 domain of the “knob chain” and a D399K; E357K mutation in the CH3 domain of the “hole chain”.
- the bispecific antibody includes Y349C and T366W mutations in one of the two CH3 domains and S354C, T366S, L368A, and Y407V mutations in the other of the two CH3 domains, or the bispecific antibody includes Y349C and T366W mutations in one of the two CH3 domains and S354C, T366S, L368A, and Y407V mutations in the other of the two CH3 domains and further includes a R409D; K370E mutation in the CH3 domain of the “knob chain” and a D399K; E357K mutation in the CH3 domain of the “hole chain”.
- the bispecific antibody of the present disclosure may have a knob-in-hole structure formed by substituting an amino acid residue in the CH3 domain of the first constant region with an amino acid residue having a larger side chain size, and substituting an amino acid residue in the CH3 domain of the second constant region with an amino acid residue having a smaller side chain size; or substituting an amino acid residue in the CH3 domain of the second constant region with an amino acid residue having a larger side chain size, and substituting an amino acid residue in the CH3 domain of the first constant region with an amino acid residue having a smaller side chain size.
- the bispecific antibody of the present disclosure may include T366S, L368A, and Y407V according to the Kabat EU index in the CH3 domain of the first constant region, and T366W according to the Kabat EU index in the CH3 domain of the second constant region; or include T366S, L368A, and Y407V according to the Kabat EU index in the CH3 domain of the second constant region, and T366W according to the Kabat EU index in the CH3 domain of the first constant region.
- At least one amino acid residue in the CH3 domain of the first heavy chain constant region is substituted with an amino acid residue having a larger side chain size, and at least one amino acid residue in the CH3 domain of the second heavy chain constant region is substituted with an amino acid residue having a smaller side chain size; or at least one amino acid residue in the CH3 domain of the second heavy chain constant region is substituted with an amino acid residue having a larger side chain size, and at least one amino acid residue in the CH3 domain of the first heavy chain constant region is substituted with an amino acid residue having a smaller side chain size.
- the amino acid residue having a larger side chain size is selected from arginine, phenylalanine, tyrosine, and tryptophan; and the amino acid residue having a smaller side chain size is selected from alanine, serine, threonine, and valine.
- the CH3 domain of the first heavy chain constant region comprises amino acid substitutions at positions T366, L368, and Y407 according to EU numbering
- the CH3 domain of the second heavy chain constant region comprises amino acid substitutions at position T366 according to EU numbering
- the CH3 domain of the second heavy chain constant region comprises amino acid substitutions at positions T366, L368, and Y407 according to EU numbering
- the CH3 domain of the first heavy chain constant region comprises amino acid substitutions at position T366 according to EU numbering.
- the CH3 domain of the first heavy chain constant region comprises T366S, L368A, and Y407V according to EU numbering
- the CH3 domain of the second heavy chain constant region comprises T366W according to EU numbering
- the CH3 domain of the second heavy chain constant region comprises T366S, L368A, and Y407V according to EU numbering
- the CH3 domain of the first heavy chain constant region comprises T366W according to EU numbering.
- the CH3 domain of each of the first and the second heavy chain constant regions further comprises at least one substitution of an amino acid residue to a cysteine residue.
- the CH3 domain of the first heavy chain constant region comprises E356C, T366S, L368A, and Y407V according to EU numbering
- the CH3 domain of the second heavy chain constant region comprises Y349C and T366W according to EU numbering
- the CH3 domain of the second heavy chain constant region comprises E356C, T366S, L368A, and Y407V according to EU numbering
- the CH3 domain of the first heavy chain constant region comprises Y349C and T366W according to EU numbering
- the CH3 domain of the first heavy chain constant region comprises S354C, T366S, L368A, and Y407V according to EU numbering
- the CH3 domain of the second heavy chain constant region comprises Y349C and T366W according to EU numbering
- the CH3 domain of the second heavy chain constant region comprises S354C, T366S, L368A, and Y407V according to EU numbering
- the CH3 domain of the first heavy chain constant region comprises E356C, E357K, T366S, L368A, D399K, and Y407V according to EU numbering
- the CH3 domain of the second heavy chain constant region comprises Y349C, T366W, K370E, and R409D according to EU numbering
- the CH3 domain of the second heavy chain constant region comprises E356C, E357K, T366S, L368A, D399K, and Y407V according to EU numbering
- the CH3 domain of the first heavy chain constant region comprises Y349C, T366W, K370E, and R409D according to EU numbering
- the CH3 domain of the first heavy chain constant region comprises S354C, E357K, T366S, L368A, D399K, and Y407V according to EU numbering
- the CH3 domain of the second heavy chain constant region comprises Y349C, T366W, K370E
- a bispecific antibody of the present disclosure is produced by the methods described in U.S. Patent No. 11,498,977B2, the entirety of which is incorporated herein by reference.
- the bispecific antibody may include one or more amino acid substitutions within the CH3 domains of each of the first constant region and the second constant region.
- the first constant region and the second constant region may include at least 5 amino acid substitutions at the following positions according to the Kabat EU index within the CH3 domain: 1) positions 366 and 399 within the first constant region (or the second constant region) and positions 351, 407, and 409 within the second constant region (or the first constant region); or 2) positions 366 and 409 within the first constant region (or the second constant region) and positions 351, 399, and 407 within the second constant region (or the first constant region).
- the bispecific antibody may include amino acid substitutions at positions T366 and D399 according to the Kabat EU index within the CH3 domain of the first constant region, and amino acid substitutions at positions L351, Y407, and K409 according to the Kabat EU index within the CH3 domain of the second constant region; or include amino acid substitutions at positions T366 and D399 according to the Kabat EU index in the CH3 domain of the second constant region, and amino acid substitutions at positions L351, Y407 and K409 according to the Kabat EU index in the CH3 domain of the first constant region.
- the first constant region and the second constant region include at least one of the following substitutions according to the Kabat EU index in the CH3 domain: a) glycine, tyrosine, valine, proline, aspartic acid, glutamic acid, lysine, or tryptophan at L351 in the second constant region (or the first constant region); b) leucine, proline, tryptophan, or valine at T366 in the first constant region (or the second constant region); c) cysteine, asparagine, isoleucine, glycine, arginine, threonine, or alanine at D399 in the first constant region and/or the second constant region; d) leucine, alanine, proline, phenylalanine, threonine, or histidine at Y407 in the second constant region (or the first constant region); and e) cysteine, proline, serine, phenylalanine, valine,
- the bispecific antibody includes T366L and D399R according to the Kabat EU index in the CH3 domain of the first constant region, and L35 IE, Y407L and K409Y according to the Kabat EU index in the CH3 domain of the second constant region; or may include T366L and D399R according to the Kabat EU index in the CH3 domain of the second constant region, and L351E, Y407L and K409Y according to the Kabat EU index in the CH3 domain of the first constant region, but is not limited thereto.
- the CH3 domain of the first heavy chain constant region comprises amino acid substitutions at positions T366 and D399 according to EU numbering
- the CH3 domain of the second heavy chain constant region comprises amino acid substitutions at positions L351, Y407, and K409 according to EU numbering
- the CH3 domain of the second heavy chain constant region comprises amino acid substitutions at positions T366 and D399 according to EU numbering
- the CH3 domain of the first heavy chain constant region comprises amino acid substitutions at positions L351, Y407, and K409 according to EU numbering
- the CH3 domain of the first heavy chain constant region comprises amino acid substitutions at positions T366 and K409 according to EU numbering
- the CH3 domain of the second heavy chain constant region comprises amino acid substitutions at positions L351, D399, and Y407 according to EU numbering
- the CH3 domain of the second heavy chain constant region comprises amino acid substitutions at positions T366 and K409 according to EU numbering
- the substituted amino acid at T366 is selected from leucine, proline, tryptophan, and valine;
- the substituted amino acid at D399 is selected from cysteine, asparagine, isoleucine, glycine, arginine, threonine, and alanine;
- the substituted amino acid at L351 is selected from glycine, tyrosine, valine, proline, aspartic acid, glutamic acid, lysine, and tryptophan;
- the substituted amino acid at Y407 is selected from leucine, alanine, proline, phenylalanine, threonine, and histidine;
- the substituted amino acid at K409 is selected from cysteine, proline, serine, phenylalanine, valine, glutamic acid, and arginine.
- the CH3 domain of the first heavy chain constant region comprises amino acid substitutions T366L and D399R according to EU numbering
- the CH3 domain of the second heavy chain constant region comprises amino acid substitutions L351E, Y407L and K409V according to EU numbering
- the CH3 domain of the second heavy chain constant region comprises amino acid substitutions T366L and D399R according to EU numbering
- the CH3 domain of the first heavy chain constant region comprises amino acid substitutions L351E, Y407L and K409V according to EU numbering
- the CH3 domain of the first heavy chain constant region comprises amino acid substitutions T366L and D399C according to EU numbering
- the CH3 domain of the second heavy chain constant region comprises amino acid substitutions L351G, Y407L and K409C according to EU numbering
- the CH3 domain of the second heavy chain constant region comprises amino acid substitutions T366L and D399C according to EU numbering
- the CH3 domain of the first heavy chain constant region comprises amino acid substitutions T366L and D399R
- the CH3 domain of the second heavy chain constant region comprises amino acid substitutions L351E, Y407L and K409Y
- the CH3 domain of the second heavy chain constant region comprises amino acid substitutions T366L and D399R
- the CH3 domain of the first heavy chain constant region comprises amino acid substitutions L35 IE, Y407L and K409Y.
- a bispecific antibody of the present disclosure is produced by the methods described in U.S. Patent No. 8,242,247B2, the entirety of which is incorporated herein by reference.
- the bispecific antibody includes a first constant region linked to a first antigen-binding domain; and a second constant region linked to a second antigen-binding domain, wherein the first constant region and the second constant region may each include a light chain constant domain and CHI, CH2, and CH3 domains of a heavy chain constant region of an antibody.
- variable heavy chain domain VH of the first constant region may be replaced with the variable light chain domain VL.
- variable heavy chain domain VH and the heavy chain constant domain CHI of the first constant region (or the second constant region) may be replaced with light chain domains.
- the structure described above may provide additional advantages in terms of the preparation and efficacy of the anti-CD20xCD22 bispecific antibody-drug conjugate of the present disclosure.
- (a) CL and CHI domains from the first constant region are replaced by each other; or CL and CHI domains from the second constant region are replaced by each other; or (b) VH and VL domains from the first antigen-binding domain are replaced by each other; or VH and VL domains from the second antigen-binding domain are replaced by each other; or (c) VH and CHI domains and VL and CL domains from the first constant region and the first antigen-binding domain are replaced by each other; or VH and CHI domains and VL and CL domains from the second constant region and the second antigenbinding domain are replaced by each other.
- CL and CHI domains from the first constant region are replaced by each other; or CL and CHI domains from the second constant region are replaced by each other.
- a bispecific antibody of the present disclosure is produced by the methods described in EP Patent Application No. 4286408A1, the entirety of which is incorporated herein by reference.
- S-DUALTM may effectively separate and analyze the bispecific antibody as impurities from the improper pairings form different sizes. It also applies “knob-in-hole” technique to avoid mispairing between two heavy chains and light chains.
- S- DUALTM has a unique asymmetrical structure that ensures high binding affinity among chains to produce high titer and purity for optimized manufacturability.
- the bispecific antibody of the present disclosure may have an asymmetrical structure between the first polypeptide and the second polypeptide.
- a CH3 dimer may be further included between the first antigen-binding domain and the CHI domain or the light chain constant domain of the first constant region, or a CH3 dimer may be further included between the second antigen-binding domain and the CHI domain or the light chain constant domain of the second constant region.
- the bispecific antibody may further include the knob-in-hole structure between the first antigen-binding domain and the CHI domain of the first constant region; or further include the knob-in-hole structure between the second antigen-binding domain and the CHI domain of the second constant region.
- the first constant region and the second constant region have an asymmetrical structure to each other.
- a knob-in-hole structure is further included between the first antigen-binding domain and the CHI domain of the first constant region; or a knob-in-hole structure is further included between the second antigen-binding domain and the CHI domain of the second constant region.
- the knobin-hole structure comprises a dimer of CH3 domains.
- the structures of the bispecific antibody disclosed herein may be combined with each other within a compatible aspect.
- the structures prepared by the Knob-and-Hole or the Pentambody methods may be combined with the structures prepared by the CrossMab or S-DUALTM methods.
- the combination of the structures may provide additional advantages in terms of the preparation and efficacy of the anti-CD20xCD22 bispecific antibody-drug conjugate of the present disclosure. Antibody and Antigen-Binding Fragment Thereof
- the first antigen-binding domain specifically binding to CD20 comprises a first heavy chain variable region and a first light chain variable region
- the second antigen-binding domain specifically binding to CD22 comprises a second heavy chain variable region and a second light chain variable region
- the first heavy chain variable region comprises a heavy chain CDR1 comprising the amino acid sequence of SEQ ID NO: 2, a heavy chain CDR2 comprising the amino acid sequence of SEQ ID NO: 4, and a heavy chain CDR3 comprising the amino acid sequence of SEQ ID NO: 6;
- the first light chain variable region comprises a light chain CDR1 comprising the amino acid sequence of SEQ ID NO: 9, a light chain CDR2 comprising the amino acid sequence of SEQ ID NO: 11, and a light chain CDR3 comprising the amino acid sequence of SEQ ID NO: 13;
- the second heavy chain variable region comprises a heavy chain CDR1 comprising the amino acid sequence of SEQ ID NO: 40, a heavy chain CDR2 comprising the amino acid sequence of SEQ ID NO: 42, and a heavy chain CDR3 comprising the amino acid sequence of SEQ ID NO: 44; and
- the second light chain variable region comprises a light chain CDR1 comprising the amino acid sequence of SEQ ID NO: 47, a light chain CDR2 comprising the
- the first heavy chain variable region comprises a heavy chain CDR1 comprising the amino acid sequence of SEQ ID NO: 2, a heavy chain CDR2 comprising the amino acid sequence of SEQ ID NO: 4, and a heavy chain CDR3 comprising the amino acid sequence of SEQ ID NO: 6;
- the first light chain variable region comprises a light chain CDR1 comprising the amino acid sequence of SEQ ID NO: 9, a light chain CDR2 comprising the amino acid sequence of SEQ ID NO: 11, and a light chain CDR3 comprising the amino acid sequence of SEQ ID NO: 13;
- the second heavy chain variable region comprises a heavy chain CDR1 comprising the amino acid sequence of SEQ ID NO: 58, a heavy chain CDR2 comprising the amino acid sequence of SEQ ID NO: 60, and a heavy chain CDR3 comprising the amino acid sequence of SEQ ID NO: 62; and
- the second light chain variable region comprises a light chain CDR1 comprising the amino acid sequence of SEQ ID NO: 65, a light chain CDR2 compris
- the first heavy chain variable region comprises a heavy chain CDR1 comprising the amino acid sequence of SEQ ID NO: 22, a heavy chain CDR2 comprising the amino acid sequence of SEQ ID NO: 24, and a heavy chain CDR3 comprising the amino acid sequence of SEQ ID NO: 26;
- the first light chain variable region comprises a light chain CDR1 comprising the amino acid sequence of SEQ ID NO: 29, a light chain CDR2 comprising the amino acid sequence of SEQ ID NO: 31, and a light chain CDR3 comprising the amino acid sequence of SEQ ID NO: 33;
- the second heavy chain variable region comprises a heavy chain CDR1 comprising the amino acid sequence of SEQ ID NO: 40, a heavy chain CDR2 comprising the amino acid sequence of SEQ ID NO: 42, and a heavy chain CDR3 comprising the amino acid sequence of SEQ ID NO: 44; and
- the second light chain variable region comprises a light chain CDR1 comprising the amino acid sequence of SEQ ID NO: 47, a light chain CDR2 comprising the
- the first heavy chain variable region comprises a heavy chain CDR1 comprising the amino acid sequence of SEQ ID NO: 22, a heavy chain CDR2 comprising the amino acid sequence of SEQ ID NO: 24, and a heavy chain CDR3 comprising the amino acid sequence of SEQ ID NO: 26;
- the first light chain variable region comprises a light chain CDR1 comprising the amino acid sequence of SEQ ID NO: 29, a light chain CDR2 comprising the amino acid sequence of SEQ ID NO: 31, and a light chain CDR3 comprising the amino acid sequence of SEQ ID NO: 33;
- the second heavy chain variable region comprises a heavy chain CDR1 comprising the amino acid sequence of SEQ ID NO: 58, a heavy chain CDR2 comprising the amino acid sequence of SEQ ID NO: 60, and a heavy chain CDR3 comprising the amino acid sequence of SEQ ID NO: 62; and the second light chain variable region comprises a light chain CDR1 comprising the amino acid sequence of SEQ ID NO: 65, a light chain CDR2 comprising the
- the VH domain of the first antigen-binding domain comprises: the amino acid sequence of SEQ ID NO: 15; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 15; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 15;
- the VL domain of the first antigen-binding domain comprises: the amino acid sequence of SEQ ID NO: 16; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 16; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 16;
- the VH domain of the first antigen-binding domain comprises: the amino acid sequence of SEQ ID NO: 15; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 15; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 15;
- the VL domain of the first antigen-binding domain comprises: the amino acid sequence of SEQ ID NO: 16; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 16; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 16;
- the VH domain of the second antigen-binding domain comprises: the amino acid sequence of SEQ ID NO: 71; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 71; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 71; and
- the VL domain of the second antigen-binding domain comprises: the amino acid sequence of SEQ ID NO: 72; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 72; or at least 9
- the VH domain of the first antigen-binding domain comprises: the amino acid sequence of SEQ ID NO: 35; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 35; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 35;
- the VL domain of the first antigen-binding domain comprises: the amino acid sequence of SEQ ID NO: 36; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 36; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 36;
- the VH domain of the first antigen-binding domain comprises: the amino acid sequence of SEQ ID NO: 35; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 35; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 35;
- the VL domain of the first antigen-binding domain comprises: the amino acid sequence of SEQ ID NO: 36; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 36; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 36;
- the VH domain of the second antigen-binding domain comprises: the amino acid sequence of SEQ ID NO: 71; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 71; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 71;
- the VL domain of the second antigen-binding domain comprises: the amino acid sequence of SEQ ID NO: 72; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 72; or at least 95%
- the VH domain of the first antigen-binding domain comprises the amino acid sequence of SEQ ID NO: 15; the VL domain of the first antigenbinding domain comprises the amino acid sequence of SEQ ID NO: 16; the VH domain of the second antigen-binding domain comprises the amino acid sequence of SEQ ID NO: 53; and the VL domain of the second antigen-binding domain comprises the amino acid sequence of SEQ ID NO: 54;
- the VH domain of the first antigen-binding domain comprises the amino acid sequence of SEQ ID NO: 15; the VL domain of the first antigen-binding domain comprises the amino acid sequence of SEQ ID NO: 16; the VH domain of the second antigen-binding domain comprises the amino acid sequence of SEQ ID NO: 71; and the VL domain of the second antigen-binding domain comprises the amino acid sequence of SEQ ID NO: 72;
- the VH domain of the first antigen-binding domain comprises the amino acid sequence of SEQ ID NO: 35; the VL domain of the first antigen-binding domain comprises the amino acid sequence of SEQ ID NO: 36; the VH domain of the second antigen-binding domain comprises the amino acid sequence of SEQ ID NO: 53; and the VL domain of the second antigen-binding domain comprises the amino acid sequence of SEQ ID NO: 54; or
- the VH domain of the first antigen-binding domain comprises the amino acid sequence of SEQ ID NO: 35; the VL domain of the first antigen-binding domain comprises the amino acid sequence of SEQ ID NO: 36; the VH domain of the second antigen-binding domain comprises the amino acid sequence of SEQ ID NO: 71; and the VL domain of the second antigen-binding domain comprises the amino acid sequence of SEQ ID NO: 72.
- Ab comprises a first heavy chain and a first light chain comprising the first antigen-binding domain specifically binding to CD20 and a second heavy chain and a second light chain comprising the second antigen-binding domain specifically binding to CD22
- the first heavy chain comprises: the amino acid sequence of SEQ ID NO: 20; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 20; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 20
- the first light chain comprises: the amino acid sequence of SEQ ID NO: 18; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 18; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 18
- the second heavy chain comprises: the amino acid sequence of SEQ ID NO: 55; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 55; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 55
- the second light chain comprises: the amino acid sequence of SEQ ID NO: 56; at least
- the first heavy chain comprises: the amino acid sequence of SEQ ID NO: 20; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 20; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 20;
- the first light chain comprises: the amino acid sequence of SEQ ID NO: 18; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 18; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 18;
- the second heavy chain comprises: the amino acid sequence of SEQ ID NO: 73; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 73; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 73; and the second light chain comprises: the amino acid sequence of SEQ ID NO: 74; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 74; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 74;
- the first heavy chain comprises: the amino acid sequence of SEQ ID NO: 37; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 37; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 37;
- the first light chain comprises: the amino acid sequence of SEQ ID NO: 38; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 38; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 38;
- the second heavy chain comprises: the amino acid sequence of SEQ ID NO: 55; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 55; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 55; and the second light chain comprises: the amino acid sequence of SEQ ID NO: 56; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 56; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 56;
- the first heavy chain comprises: the amino acid sequence of SEQ ID NO: 37; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 37; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 37;
- the first light chain comprises: the amino acid sequence of SEQ ID NO: 38; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 38; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 38;
- the second heavy chain comprises: the amino acid sequence of SEQ ID NO: 73; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 73; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 73; and the second light chain comprises: the amino acid sequence of SEQ ID NO: 74; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 74; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 74;
- the first heavy chain comprises: the amino acid sequence of SEQ ID NO: 75; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 75; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 75;
- the first light chain comprises: the amino acid sequence of SEQ ID NO: 18; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 18; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 18;
- the second heavy chain comprises: the amino acid sequence of SEQ ID NO: 76; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 76; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 76; and
- the second light chain comprises: the amino acid sequence of SEQ ID NO: 56; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 56; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 56; or
- the first heavy chain comprises: the amino acid sequence of SEQ ID NO: 20; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 20; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 20;
- the first light chain comprises: the amino acid sequence of SEQ ID NO: 18; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 18; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 18;
- the second heavy chain comprises: the amino acid sequence of SEQ ID NO: 81; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 81; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 81; and the second light chain comprises: the amino acid sequence of SEQ ID NO: 82; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 82; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 82.
- the first heavy chain comprises the amino acid sequence of SEQ ID NO: 20; the first light chain comprises the amino acid sequence of SEQ ID NO: 18; the second heavy chain comprises the amino acid sequence of SEQ ID NO: 55; and the second light chain comprises the amino acid sequence of SEQ ID NO: 56;
- the first heavy chain comprises the amino acid sequence of SEQ ID NO: 20; the first light chain comprises the amino acid sequence of SEQ ID NO: 18; the second heavy chain comprises the amino acid sequence of SEQ ID NO: 73; and the second light chain comprises the amino acid sequence of SEQ ID NO: 74;
- the first heavy chain comprises the amino acid sequence of SEQ ID NO: 37; the first light chain comprises the amino acid sequence of SEQ ID NO: 38; the second heavy chain comprises the amino acid sequence of SEQ ID NO: 55; and the second light chain comprises the amino acid sequence of SEQ ID NO: 56;
- the first heavy chain comprises the amino acid sequence of SEQ ID NO: 37; the first light chain comprises the amino acid sequence of SEQ ID NO: the first light chain comprises the amino
- Ab is a full-length antibody.
- Ab is an IgG-scFv, a trifiinctional antibody (triomab), a knobs into holes (KIH)-IgG, a K/.-body. a crossmab, an ortho-Fab IgG, a dual variable domain immunoglobulin (DVD-Ig), or a 2 in 1- IgG (dual action antibody), or a combination thereof.
- Ab is an antigen binding fragment.
- Ab is a scFv2-Fc, a bi-nanobody, a bispecific T cell engager (BiTE), a tandem diabody (tandAb), a dual affinity retargeting (DART) antibody, a DART-Fc, a scFv-human serum albumin (HSA)-scFv, a dock-and-lock (DNL)-Fab3, a minibody, a Fab2 fragment (bispecific), a Fab3 fragment (trispecific), a Bis-scFv fragment (bispecific), a sdAb fragment (VH/VHH), a tetrabody, a triabody, or a diabody, or a combination thereof.
- the bispecific antibodies of the present disclosure may be produced by any means known in the art, such as, without limitation, any chemical, biological, genetic or enzymatic technique, either alone or in combination, including recombinant expression, chemical synthesis, and enzymatic digestion of a full-length monoclonal antibody.
- the recombinant expression may take place in any suitable host cell known in the art, including without limitation to, mammalian host cells, bacterial host cells, yeast host cells, and insect host cells, or in a cell-free system (e.g., Sutro’s Xpress CF platform, World Wide Web at sutrobio.com/technology/) .
- antibodies or polypeptides may readily produce said antibodies or polypeptides, by standard techniques for production of polypeptides. For instance, they may be synthesized using well-known solid phase method, preferably using a commercially available peptide synthesis apparatus (such as that made by Applied Biosystems, Foster City, Calif.) and following the manufacturer's instructions. Alternatively, antibodies and other polypeptides may be synthesized by recombinant DNA techniques as is well-known in the art.
- these fragments may be obtained as DNA expression products after incorporation of DNA sequences encoding the desired (poly)peptide into expression vectors and introduction of such vectors into suitable eukaryotic or prokaryotic hosts that will express the desired polypeptide, from which they may be later isolated using well-known techniques.
- an antibody or a polypeptide which method comprises the steps consisting of: (i) culturing a transformed host cell under conditions suitable to allow expression of said antibody or polypeptide; and (ii) recovering the expressed antibody or polypeptide.
- Antibodies and other polypeptides are suitably separated from the culture medium by conventional immunoglobulin purification procedures such as, for example, protein A- Sepharose, hydroxylapatite chromatography, gel electrophoresis, dialysis, affinity chromatography, ammonium sulfate or ethanol precipitation, acid extraction, anion or cation exchange chromatography, phosphocellulose chromatography, hydrophobic interaction chromatography, hydroxylapatite chromatography and lectin chromatography.
- High performance liquid chromatography (“HPLC”) may also be employed for purification.
- the present disclosure also provides an expression vector and a host cell for producing any of the bispecific antibody described herein.
- Various expression vectors may be used to express a polynucleotide encoding the bispecific antibody.
- Both viral-based and nonviral expression vectors may be used to produce antibodies in mammalian host cells.
- Nonviral vectors and systems include plasmids, episomal vectors (typically, containing an expression cassette for protein or RNA expression), and human artificial chromosomes (e.g., see [Harrington et al., Nat Genet. 15:345, 1997]).
- Useful viral vectors include without limitation to, retrovirus vectors, adenovirus vectors, adeno-associated virus vectors, herpes virus-based vector, SV40 vectors, papilloma virus vectors, HBP Epstein-Barr virus vectors, vaccinia virus vectors, and Semliki Forest virus (SFV)-based vectors.
- the expression vector varies based on an intended host cell in which the vector is to be expressed.
- the expression vector contains a promoter and other regulatory sequences (e.g., enhancer) operably linked to a polynucleotide encoding the bispecific antibody or antibody fragment (e.g., antigen-binding fragment).
- an inducible promoter is used to prevent expression of inserted sequences except under inducing conditions.
- the inducible promoter includes, for example, arabinose, lacZ, metallothionein promoter, or heat shock promoter.
- a transformed organism culture may be proliferated under non-inducing conditions without biasing a population for coding sequences of which expression products are better tolerated by the host cell.
- the expression vector may also provide a secretion signal sequence location to form a fusion protein with a polypeptide encoded by the inserted bispecific antibody or antibody fragment (e.g., antigen-binding fragment) sequence. More often, the inserted bispecific antibody or antibody fragment (e.g., antigen-binding fragment) sequence is included in the vector after being linked to a signal sequence.
- a vector used to contain a sequence encoding the light and heavy chain variable domains of the bispecific antibody or antibody fragment (e.g., antigen-binding fragment) sometimes also encodes a constant region or a portion thereof. Such a vector allows the variable region to be expressed as a fusion protein with the constant region to induce production of a complete antibody or fragment thereof.
- mammalian host cells are used to express and produce the bispecific antibody or antibody fragment (e.g., antigen-binding fragment) polypeptide of the present disclosure.
- these mammalian host cells may be hybridoma cell lines expressing endogenous immunoglobulin genes (e.g., myeloma hybridoma clones as described in Examples), or mammalian cell lines harboring exogenous expression vectors (e.g., SP2/0 myeloma cells to be exemplified below).
- the mammalian host cells include any normal apoptotic or normal or abnormal immortalized animal or human cells.
- a plurality of suitable host cell lines capable of secreting intact immunoglobulins have been developed, including CHO cell lines, various COS cell lines, HeLa cells, myeloma cell lines, transformed B cells, and hybridomas.
- Expression vectors for mammalian host cells may include expression control sequences, such as an origin of replication, a promoter, and an enhancer, and necessary processing information sites, such as a ribosome binding site, an RNA splice site, a polyadenylation site, and a transcription terminator sequence. These expression vectors generally contain promoters derived from mammalian genes or mammalian viruses.
- Suitable promoters may be constitutive, cell type -specific, step-specific, and/or controllable or regulatable promoters.
- Useful promoters include a metallothionein promoter, a constitutive adenovirus major late promoter, a dexamethasone-inducible MMTV promoter, a SV40 promoter, a MRP polIII promoter, a constitutive MPSV promoter, a tetracycline -inducible CMV promoter (e.g., a human immediate early CMV promoter), a constitutive CMV promoter, and promoter-enhancer combinations known in the art, but are not limited thereto.
- a non-human antibody may be derived from, for example, any antibody-producing animal, for example, a mouse, a rat, a rabbit, a goat, a donkey, or non-human primates (e.g., monkeys such as cynomolgus or rhesus monkey) or apes (e.g., chimpanzees).
- a non-human antibody may be produced by immunizing an animal by using a method known in the art.
- Chimeric antibodies may be produced by obtaining nucleic sequences encoding VL and VH domains as previously described, constructing a human chimeric antibody expression vector by inserting them into an expression vector for animal cell having genes encoding human antibody CH and human antibody CL, and expressing the coding sequence by introducing the expression vector into an animal cell.
- the CH domain of a human chimeric antibody may be any region which belongs to human immunoglobulin, such as the IgG class or a subclass thereof, such as IgGl, IgG2, IgG3 and IgG4.
- the CL of a human chimeric antibody may be any region which belongs to Ig, such as the kappa class or lambda class
- chimeric and humanized monoclonal antibodies comprising both human and non-human portions may be made using standard recombinant DNA techniques.
- Such chimeric and humanized monoclonal antibodies may be produced by recombinant DNA techniques known in the art, for example using methods described in Robinson et al. International Patent Publication PCT/US86/02269; Akira et al. European Patent Application 184,187; Taniguchi, M. European Patent Application 171,496; Morrison et al. European Patent Application 173,494; Neuberger et al.
- humanized antibodies may be made according to standard protocols such as those disclosed in U.S. Patent 5,565,332.
- antibody chains or specific binding pair members may be produced by recombination between vectors comprising nucleic acid molecules encoding a fusion of a polypeptide chain of a specific binding pair member and a component of a replicable generic display package and vectors containing nucleic acid molecules encoding a second polypeptide chain of a single binding pair member using techniques known in the art, e.g., as described in U.S. Patents 5,565,332, 5,871,907, or 5,733,743.
- Humanized antibodies may be produced by obtaining nucleic acid sequences encoding CDR domains, as previously described, constructing a humanized antibody expression vector by inserting them into an expression vector for animal cell having genes encoding (i) a heavy chain constant region identical to that of a human antibody and (ii) a light chain constant region identical to that of a human antibody, and expressing the genes by introducing the expression vector into an animal cell.
- the humanized antibody expression vector may be either of a type in which a gene encoding an antibody heavy chain and a gene encoding an antibody light chain exists on separate vectors or of a type in which both genes exist on the same vector (tandem type).
- Antibodies may be humanized using a variety of techniques known in the art including, for example, CDR-grafting (EP 239,400; PCT publication WO91/09967; U.S. Pat. Nos. 5,225,539; 5,530,101; and 5,585,089), veneering or resurfacing (EP 592,106; EP 519,596; Padlan EA (1991); Studnicka G M et al. (1994); Roguska M A. et al. (1994)), and chain shuffling (U.S. Pat. No. 5,565,332).
- the general recombinant DNA technology for preparation of such antibodies is also known (see European Patent Application EP 125023 and International Patent Application WO 96/02576).
- a fully human antibody may be produced by administering an antigen to a transformed animal including a human immunoglobulin gene locus, or by treating a phage display library expressing a human antibody repertory with an antigen, and then selecting the target antibody.
- the antibody may be polyclonal or monoclonal, or may be synthesized within a cell host through the expression of recombinant DNA.
- a monoclonal antibody (mAb) may be produced using a conventional monoclonal antibody method, for example, a standard somatic hybridization technique in the literature (Kohler and Milstein, 1975, Nature 256:495).
- bispecific or multispecific antibodies described herein may be made according to standard procedures.
- triomas and hybrid hybridomas are two examples of cell lines that may secrete bispecific or multispecific antibodies.
- Examples of bispecific and multispecific antibodies produced by a hybrid hybridoma or a trioma are disclosed in U.S. Patent 4,474,893.
- Such antibodies may also be constructed by chemical means (Staerz et al. (1985) Nature 314:628, and Perez et al. (1985) Nature 316:354) and hybridoma technology (Staerz and Bevan (1986) Proc. Natl. Acad. Set. USA, 83: 1453, and Staerz and Bevan (1986) Immunol. Today 7:241).
- such antibodies may also be generated by making heterohybridomas by fusing hybridomas or other cells making different antibodies, followed by identification of clones producing and co-assembling the desired antibodies. They may also be generated by chemical or genetic conjugation of complete immunoglobulin chains or portions thereof such as Fab and Fv sequences.
- the present disclosure provides, among other things, active agents and antibody-drug conjugates that comprise the active agents.
- the anti-CD20xCD22 bispecific antibody (“Ab”) disclosed herein may be conjugated to many identical or different active agents using methods known in the art.
- the active agents of the present disclosure should not be construed as being limited to chemical therapeutic agents.
- active agents of the present disclosure may be proteins, peptides, or polypeptides having a desired biological activity.
- the active agent is a chemotherapeutic agent or a toxin.
- the active agent is selected from a chemotherapeutic compound, a cytotoxic compound, an immunomodulatory compound, an anticancer agent, an antiviral agent, an antibacterial agent, an antifungal agent, an antiparasitic agent, and a combination thereof.
- the cytotoxic compound is selected from a mitotic inhibitor, a DNA alkylating agent, a topoisomerase inhibitor, and a combination thereof.
- the cytotoxic compound is selected from auristatin, maytansinoid, tubulisin, calicheamicin, duocarmycin, pyrrolobenzodiazepine and derivatives thereof, camptothecin and derivatives thereof, and combinations thereof.
- the auristatin is MMAE (monomethyl auristatin E) or MMAF (monomethyl auristatin F).
- the active agent is a pyrrolobenzodiazepine dimer; position N10 of the pyrrolobenzodiazepine dimer is substituted with X or position N’ 10 is substituted with X’, wherein X or X' links the pyrrolobenzodiazepine dimer to the linker;
- X and X' are each independently -C(O)O-* or -C(O)-* ; and * refers to a binding site between the pyrrolobenzodiazepine dimer and the linker.
- the active agent is a pyrrolobenzodiazepine dimer and the pyrrolobenzodiazepine dimer has a structure represented by Formula VIII:
- R m is selected from R m , C0 2 R m , C0R m , CHO, CO 2 H. and halo;
- R m is selected from substituted or unsubstituted C1-12 alkyl, substituted or unsubstituted C2-12 alkenyl, substituted or unsubstituted C2-12 alkynyl, substituted or unsubstituted Cs- 2 o aryl, substituted or unsubstituted C3-6 heteroaryl, substituted or unsubstituted C3-6 cycloalkyl, substituted or unsubstituted 3- to 7-membered heterocyclyl, substituted or unsubstituted 3- to 7-membered heterocycloalkyl, and substituted or unsubstituted 5- to 7-membered heteroaryl, wherein when the C1-12 alkyl, C2-12 alkenyl, C2-12 alkynyl, Cs- 2 o aryl, Cs- 2 o heteroaryl, C3-6 cycloalkyl, 3- to 7-membered heterocyclyl, 3- to 7- membered heterocycloal
- R2, R3, Rs, R2, R3 , and Rs are each independently selected from H, R m , OH, OR m , SH, SR m , NH 2 , NHR m , NR m R m , NO2, Me 3 Sn, and halo;
- R4 and R4 are each independently selected from H, R m , OH, OR m , SH, SR m , NH2, NHR m , NR m R m , NO2, McsSn. halo, substituted or unsubstituted C1-6 alkyl, substituted or unsubstituted C1-6 alkoxy, substituted or unsubstituted C2-6 alkenyl, substituted or unsubstituted C2-6 alkynyl, substituted or unsubstituted C3-6 cycloalkyl, substituted or unsubstituted 3- to 7-membered heterocycloalkyl, substituted or unsubstituted C5-12 aryl, substituted or unsubstituted 5- to 7-membered heteroaryl, -CN, -NCO, -OR n , - OC(O)R n , -OC(O)NR n R n , -OS(O)R
- R n , R n , R°, R° R p , R p , and R q are each independently selected from H, C1-7 alkyl, C2-7 alkenyl, C2-7 alkynyl, C3-13 cycloalkyl, 3- to 7-membered heterocycloalkyl, C6-10 aryl, and 5- to 7-membered heteroaryl;
- X is selected from -C(O)O-, -S(O)O-, -C(O)-, -C(O)NR-, -S(O) 2 NR-, -P(O)R'NR-, -S(O)NR-, and -PO2NR-;
- Xa is a bond or substituted or unsubstituted C1-6 alkylene, wherein C1-6 alkylene is substituted with C1-8 alkyl, or C3-8 cycloalkyl when substituted;
- R and R' each independently denote H, OH, NH2, ONH2, NHNH2, substituted or unsubstituted Ci-s alkyl, substituted or unsubstituted C3-8 cycloalkyl, substituted or unsubstituted C1-8 alkoxy, substituted or unsubstituted C1-8 alkylthio, substituted or unsubstituted C3-20 heteroaryl, substituted or unsubstituted C5-20 aryl, or mono- or di- C1-8 alkylamino, wherein the C1-8 alkyl, C3-8 cycloalkyl, C1-8 alkoxy, C1-8 alkylthio, C3-20 heteroaryl, and C5-20 aryl are substituted with a substituent
- Y and Y' are each independently selected from O, S, and N(H);
- Re is a substituted or unsubstituted saturated or unsaturated C3-12 hydrocarbon chain, wherein the chain may be interrupted by one or more heteroatoms, NMe, or a substituted or unsubstituted aromatic ring, the chain or aromatic ring may be substituted with -NH, -NR m , -NHC(O)R m , -NHC(O)CH 2 -[OCH 2 CH 2 ]n-R, or-[CH 2 CH 2 O] n -R at any one or more positions of hydrogen atoms on the chain or aromatic ring or unsubstituted, wherein R m and R are each as defined for R m and R above, and n is 1 to 12; and
- R7 and R7 are each independently H, substituted or unsubstituted C1-6 alkyl, substituted or unsubstituted C2-6 alkenyl, substituted or unsubstituted C2-6 alkynyl, substituted or unsubstituted C3-6 cycloalkyl, substituted or unsubstituted 3- to 7-membered heterocycloalkyl, substituted or unsubstituted C6-10 aryl, substituted or unsubstituted 5- to 7-membered heteroaryl, -OR r , -OC(O)R r , -OC(O)NR r R r , -OS(O)R r , -OS(O)2R r , - SR r , -S(O)R r , -S(O) 2 R r , -S(O)NR r R r , -S(O) 2 NR r R r ',
- R r , R r , R s , R s , R ⁇ R f , R u , and R ul are each independently selected from H, C1-7 alkyl, C2-7 alkenyl, C2-7 alkynyl, C3-13 cycloalkyl, 3- to 7-membered heterocycloalkyl, C5-10 aryl, and 5- to 7-membered heteroaryl;
- G is a glucuronide group or a galactoside group; each Z is selected from H, C1-8 alkyl, halo, NO2, CN,
- R9, Rio, and Ris are each independently selected from H, C1-8 alkyl, C2-6 alkenyl, C1-6 alkoxy, and alkyloxyalkyl; and n30 is 0 to 3.
- Y is O. In some embodiments, is Y’ is O. In some embodiments, a dotted line represents presence of a double bond between the carbons bearing Ri and R? or R1 and R7 .
- Ri is selected from substituted or unsubstituted Ci-6 alkyl, substituted or unsubstituted C2-6 alkenyl, substituted or unsubstituted C5-7 aryl, and substituted or unsubstituted C3-6 heteroaryl.
- R2, R3, and Rs are each independently H or OH.
- R4 is C1-6 alkoxy. In some embodiments, R4 is methoxy, ethoxy, or butoxy.
- X is selected from -C(O)O-, -C(O)-, and -C(O)NR-. In some embodiments, X is -C(O)NR-.
- Rs is a substituted or unsubstituted saturated or unsaturated C3-8 hydrocarbon chain, wherein one or more of the carbon atoms of the hydrocarbon chain is replaced by a heteroatom or a substituted or unsubstituted aromatic ring, wherein the heteroatom is O, S, or N(H) and the aromatic ring is benzene, pyridine, imidazole, or pyrazole, and the chain or aromatic ring may be substituted with -NHC(O)CH2-[OCH2CH2] n -R or -[CH2CH2O] n -R at any one or more positions of hydrogen atoms on the chain or aromatic ring; and n is 1 to 6. In some embodiments, n is 1 to 6. In some embodiments, n is 1 to 6. In some
- Z is H, , and , wherein R9, R10, and Rie are each independently selected from H, C1-3 alkyl, C1-3 alkoxy, and alkyloxyalkyl .
- R9 is methyloxyalkyl.
- Rio is methyloxyalkyl.
- Rie is methyloxyalkyl.
- R9, Rio, or Rie is -(CH2CH2O) m - (CH2)m2CH3, further wherein m is 1-6 and m2 is 0-2.
- m is 1.
- m2 is 0.
- R2 is H.
- R3 is H.
- R7 is H.
- R4 is C1-6 alkoxy (e.g., methoxy).
- Rs is OH.
- Ri CH2, CH?. or phenyl, optionally substituted with methoxy.
- Y is O.
- R2’ is H.
- R3’ is H.
- R7’ is H.
- R4’ is C1-6 alkoxy (e.g., methoxy).
- Rs’ is OH.
- Y’ is O.
- X is -C(O)O-.
- Xa is CH2.
- G is a glucuronide group.
- n30 is 1.
- Z is . In some embodiments, R9 is
- R16 is alkyloxyalkyl (e.g., methoxyethyl).
- Z i s i n some embodiments, Rio is alkyl (e.g, methyl).
- Re is C3-12 alkyl (e.g., pentyl).
- the pyrrolobenzodiazepine dimer is selected from:
- the conjugate comprises:
- MMAE is monomethyl auristatin E, and MMAF is monomethyl auristatin F; and the dotted line represents a connection to Ab.
- MMAE is monomethyl auristatin E, and MMAF is monomethyl auristatin F; and the dotted line represents a connection to Ab.
- the active agent is selected from:
- affinity ligand where the affinity ligand is a substrate, inhibitor, active agent, neurotransmitter, radioactive isotope, or mixtures thereof;
- radioactive label 32P, 35S, fluorescent dye, electron dense reagent, enzyme, biotin, streptavidin, digoxigenin, hapten, immunogenic protein, nucleic acid molecule with a sequence complementary to a target, or mixtures thereof;
- immunomodulatory compound anti-cancer agent, anti-viral agent, anti-bacterial agent, anti-fungal agent, anti-parasitic agent, or mixtures thereof;
- CD20 and CD22 in cancer are associated with unfavorable prognosis of certain cancers (e.g., hematological cancers and B-cell malignancies) and is known to also affect cancer metastasis.
- the anti-CD20xCD22 bispecific antibody may, as described herein, be used in a form linked to various active agents via a linker to remove CD20 and CD22 overexpressing cancer cells.
- a bispecific antibody binding to CD20 and CD22 may be used in a form bonded to an active agent, and thus can be used as a targeted therapeutic agent for directing to CD20 and CD22 expressing cells.
- the present disclosure provides a pharmaceutical composition comprising a conjugate disclosed herein and a pharmaceutically acceptable excipient.
- the present disclosure provides a pharmaceutical composition for use in preventing or treating a proliferative disease, the composition comprising a conjugate disclosed herein.
- the proliferative disease is cancer.
- the cancer is selected from liver cancer, thyroid cancer, ovarian cancer, brain cancer, multiple myeloma, colon cancer, head and neck cancer, lymphoma, leukemia, bladder cancer, kidney cancer, stomach cancer, breast cancer, uterine cancer, prostate cancer, pancreatic cancer, lung cancer, sarcoma, neuroendocrine tumor, melanoma, and combinations thereof.
- the lymphoma or leukemia is selected from Hodgkin lymphoma, non-Hodgkin lymphoma (NHL), T cell lymphoma, B cell lymphoma, natural killer cell lymphoma, diffuse large B cell lymphoma (DLBCL), mantle cell lymphoma (MCL), primary central nervous system (CNS) lymphoma, lymphoblastic lymphoma, enteropathy-type intestinal lymphoma, anaplastic large cell lymphoma, angioimmunoblastic T cell lymphoma, anaplastic large cell lymphoma, peripheral T cell lymphoma, marginal zone lymphoma, chronic lymphocytic leukemia (CLL), acute lymphoblastic leukemia (ALL), chronic myelogenous leukemia (CML), acute myelogenous leukemia (AML), and combinations thereof.
- the acute lymphoblastic leukemia is B-cell acute lymphoblastic leukemia (B-ALL).
- the present disclosure also provides a treatment method using an antibody-drug conjugate or a pharmaceutically acceptable salt or solvate thereof.
- the antibody-drug conjugate or pharmaceutically acceptable salt or solvate thereof is provided to a patient.
- the antibody-drug conjugate or pharmaceutically acceptable salt or solvate thereof inhibits cancer cell progression by binding to CD20 and CD22 expressed on the surface of cancer cells.
- the antibody binds to CD20 and CD22 expressed on the surface of cancer cells in a form bonded to an active agent disclosed herein, thereby specifically delivering the active agent bonded to the antibody to cancer cells, to induce the death of the cancer cells.
- the antibody binds to CD20 and CD22 expressed on the surface of cancer cells in the form of an antibody specific to the same target or another target, thereby increasing specificity of multiple antibodies for cancer cells or inducing connections between cancer cells and other types of cells such as immune cells, to induce the death of the cancer cells.
- the present disclosure provides a method of treating or preventing a proliferative disease in a subject in need thereof comprising administering a conjugate disclosed herein or a pharmaceutically acceptable salt thereof to the subject.
- the proliferative disease is cancer.
- the cancer is selected from liver cancer, thyroid cancer, ovarian cancer, brain cancer, multiple myeloma, colon cancer, head and neck cancer, lymphoma, leukemia, bladder cancer, kidney cancer, stomach cancer, breast cancer, uterine cancer, prostate cancer, pancreatic cancer, lung cancer, sarcoma, neuroendocrine tumor, melanoma, and combinations thereof.
- the lymphoma or leukemia is selected from Hodgkin lymphoma, non-Hodgkin lymphoma (NHL), T cell lymphoma, B cell lymphoma, natural killer cell lymphoma, diffuse large B cell lymphoma (DLBCL), mantle cell lymphoma (MCL), primary central nervous system (CNS) lymphoma, lymphoblastic lymphoma, enteropathy-type intestinal lymphoma, anaplastic large cell lymphoma, angioimmunoblastic T cell lymphoma, anaplastic large cell lymphoma, peripheral T cell lymphoma, marginal zone lymphoma, chronic lymphocytic leukemia (CLL), acute lymphoblastic leukemia (ALL), chronic myelogenous leukemia (CML), acute myelogenous leukemia (AML), and combinations thereof.
- the acute lymphoblastic leukemia is B-cell acute lymphoblastic leukemia (B-ALL).
- the present disclosure provides a pharmaceutical composition
- a pharmaceutical composition comprising a therapeutically effective amount of the antibody-drug conjugate or a pharmaceutically acceptable salt or solvate thereof, and a pharmaceutically acceptable diluent, a carrier, a solubilizer, an emulsifier, a preservative, and/or an adjuvant.
- a method of treating a cancer patient by administering such a pharmaceutical composition is provided.
- patient includes human patients.
- the pharmaceutical composition may include a pharmaceutically acceptable carrier.
- the carrier is used as a meaning including an excipient, a diluent, or an adjuvant.
- the carrier may be selected from the group consisting of, for example, lactose, dextrose, sucrose, sorbitol, mannitol, xylitol, erythritol, maltitol, starch, acacia rubber, alginate, gelatin, calcium phosphate, calcium silicate, cellulose, methyl cellulose, polyvinyl pyrrolidone, water, saline, buffer such as PBS, methylhydroxy benzoate, propylhydroxy benzoate, talc, magnesium stearate, and mineral oil.
- the composition may include a filler, an anticoagulant, a lubricant, a wetting agent, a flavoring agent, an emulsifier, a preservative, or a combination thereof.
- the pharmaceutical composition may be prepared as any formulation according to general methods.
- the composition may be formulated into formulations for oral administration (e.g., powders, tablets, capsules, syrups, pills or granules) or for parenteral administration (for example, injections).
- the composition may be prepared as a systemic or local formulation.
- the pharmaceutical composition may include an effective amount of the antibody or antigen-binding fragment thereof, an anticancer agent, or a combination thereof.
- the term "effective amount" refers to an amount sufficient to exhibit preventive or therapeutic effects when administered to an individual requiring prevention or treatment.
- the effective amount may be appropriately selected depending on a cell or individual that is selected by those or ordinary skill in the art.
- the effective amount may be determined according to factors including the severity of the disease, the age, body weight, health and gender of a patient, sensitivity of a patient to the drug, administration time, administration routes, excretion rate, treatment period, and drugs used in combination or simultaneously with the used composition, and other factors well known in the medical field.
- the dosage of the pharmaceutical composition may range, for example, from 10 pg/kg to about 30 mg/kg, optionally from 0.1 mg/kg to about 30 mg/kg, or alternatively from 0.3 mg/kg to about 20 mg/kg per adult.
- the pharmaceutical composition may be administered once a day, multiple times a day, once every 1 to 4 weeks, or once to 12 times a year.
- agent is used herein to denote a chemical compound (such as an organic or inorganic compound, a mixture of chemical compounds), a biological macromolecule (such as a nucleic acid, an antibody, including parts thereof as well as humanized, chimeric and human antibodies and monoclonal antibodies, a protein or portion thereof, e.g., a peptide, a lipid, a carbohydrate), or an extract made from biological materials such as bacteria, plants, fungi, or animal (particularly mammalian) cells or tissues.
- Agents include, for example, agents whose structure is known, and those whose structure is not known.
- a “patient,” “subject,” or “individual” are used interchangeably and refer to either a human or a non-human animal. These terms include mammals, such as humans, primates, livestock animals (including bovines, porcines, etc.), companion animals (e.g., canines, felines, etc.) and rodents (e.g., mice and rats).
- Treating” a condition or patient refers to taking steps to obtain beneficial or desired results, including clinical results.
- Beneficial or desired clinical results can include, but are not limited to, alleviation or amelioration of one or more symptoms or conditions, diminishment of extent of disease, stabilized (i.e. not worsening) state of disease, preventing spread of disease, delay or slowing of disease progression, amelioration or palliation of the disease state, and remission (whether partial or total), whether detectable or undetectable.
- Treatment can also mean prolonging survival as compared to expected survival if not receiving treatment.
- preventing is art-recognized, and when used in relation to a condition, such as a local recurrence (e.g., pain), a disease such as cancer, a syndrome complex such as heart failure or any other medical condition, is well understood in the art, and includes administration of a composition which reduces the frequency of, or delays the onset of, symptoms of a medical condition in a subject relative to a subject which does not receive the composition.
- a condition such as a local recurrence (e.g., pain)
- a disease such as cancer
- a syndrome complex such as heart failure or any other medical condition
- prevention of cancer includes, for example, reducing the number of detectable cancerous growths in a population of patients receiving a prophylactic treatment relative to an untreated control population, and/or delaying the appearance of detectable cancerous growths in a treated population versus an untreated control population, e.g., by a statistically and/or clinically significant amount.
- administering or “administration of’ a substance, a compound or an agent to a subject can be carried out using one of a variety of methods known to those skilled in the art.
- a compound or an agent can be administered, intravenously, arterially, intradermally, intramuscularly, intraperitoneally, subcutaneously, ocularly, sublingually, orally (by ingestion), intranasally (by inhalation), intraspinally, intracerebrally, and transdermally (by absorption, e.g., through a skin duct).
- a compound or agent can also appropriately be introduced by rechargeable or biodegradable polymeric devices or other devices, e.g., patches and pumps, or formulations, which provide for the extended, slow or controlled release of the compound or agent.
- Administering can also be performed, for example, once, a plurality of times, and/or over one or more extended periods.
- a compound or an agent is administered orally, e.g., to a subject by ingestion.
- the orally administered compound or agent is in an extended release or slow release formulation, or administered using a device for such slow or extended release.
- the phrase “conjoint administration” refers to any form of administration of two or more different therapeutic agents such that the second agent is administered while the previously administered therapeutic agent is still effective in the body (e.g., the two agents are simultaneously effective in the patient, which may include synergistic effects of the two agents).
- the different therapeutic compounds can be administered either in the same formulation or in separate formulations, either concomitantly or sequentially.
- an individual who receives such treatment can benefit from a combined effect of different therapeutic agents.
- a “therapeutically effective amount” or a “therapeutically effective dose” of a drug or agent is an amount of a drug or an agent that, when administered to a subject will have the intended therapeutic effect.
- the full therapeutic effect does not necessarily occur by administration of one dose, and may occur only after administration of a series of doses.
- a therapeutically effective amount may be administered in one or more administrations.
- the precise effective amount needed for a subject will depend upon, for example, the subject’s size, health and age, and the nature and extent of the condition being treated, such as cancer or MDS. The skilled worker can readily determine the effective amount for a given situation by routine experimentation.
- the terms “optional” or “optionally” mean that the subsequently described event or circumstance may occur or may not occur, and that the description includes instances where the event or circumstance occurs as well as instances in which it does not.
- “optionally substituted alkyl” refers to the alkyl may be substituted as well as where the alkyl is not substituted.
- substituent and substitution patterns on the compounds of the present disclosure can be selected by one of ordinary skilled person in the art to result chemically stable compounds which can be readily synthesized by techniques known in the art, as well as those methods set forth below, from readily available starting materials. If a substituent is itself substituted with more than one group, it is understood that these multiple groups may be on the same carbon or on different carbons, so long as a stable structure results.
- the term “optionally substituted” refers to the replacement of one to six hydrogen radicals in a given structure with the radical of a specified substituent including, but not limited to: hydroxyl, hydroxyalkyl, alkoxy, halogen, alkyl, nitro, silyl, acyl, acyloxy, aryl, cycloalkyl, heterocyclyl, amino, aminoalkyl, cyano, haloalkyl, haloalkoxy, -OCO-CH2-O- alkyl, -OP(O)(O-alkyl)2 or -CH2-OP(O)(O-alkyl)2.
- “optionally substituted” refers to the replacement of one to four hydrogen radicals in a given structure with the substituents mentioned above. More preferably, one to three hydrogen radicals are replaced by the substituents as mentioned above. It is understood that the substituent can be further substituted.
- conjugates refers to cell binding agents that are covalently bonded to one or more molecules of a cytotoxic compound.
- cell binding agent is a molecule having affinity for a biological target, and may be, for example, an antibody, particularly a monoclonal antibody, or an antibody fragment, and the binding agent functions to direct a biologically active compound to a biological target.
- the conjugate may be designed to target tumor cells through cell surface antigens.
- the antigen may be a cell surface antigen that is overexpressed or expressed in an abnormal cell type.
- the target antigen may be expressed only on proliferative cells (e.g., tumor cells).
- the target antigen may be selected on the basis of different expression, usually between proliferative tissues and normal tissues.
- the antibody is bonded to the linker.
- a "variant" of a polypeptide for example, an antigen-binding fragment, a protein, or an antibody, is a polypeptide in which insertion, deletion, addition, and/or substitution have occurred at one or more amino acid residues compared to other polypeptide sequences, and includes fusion polypeptides. Protein variants also include those modified by protein enzymatic cleavage, phosphorylation or other post-translational modifications, but retaining the biological activity of the antibody disclosed herein, such as binding and specificity to CD20 and/or CD22.
- Variants may have about 99% identity, about 98% identity, about 97% identity, about 96% identity, about 95% identity, about 94% identity, about 93% identity, about 92% identity, about 91% identity, about 90% identity, about 89% identity, about 88% identity, about 87% identity, about 86% identity, about 85% identity, about 84% identity, about 83% identity, about 82% identity, about 81% identity, or about 80% identity to the sequence of the antibody or antigen-binding fragment thereof according to the present disclosure. Percent identity (%) or homology may be calculated by methods known in the art.
- the percent homology or identity can be calculated by 100X[(same position)/min(TGA, TGB)], wherein TGA and TGB are the sum of the number of residues and internal gap positions in sequences A and B to be compared (Russell et al., J. Mol Biol., 244: 332-350 (1994).
- derivatives of a polypeptide refers to a polypeptide that has chemical modification at one or more residues through conjugation with other chemical moi eties, different from insertion, deletion, addition or substitution variants.
- percent sequence identity or “percent identity” between two polynucleotide or polypeptide sequences refers to the number of identical matched positions shared by the sequences over a comparison window, taking into account additions or deletions (i.e., gaps) that must be introduced for optimal alignment of the two sequences.
- a matched position is any position where an identical nucleotide or amino acid is presented in both the target and reference sequence. Gaps presented in the target sequence are not counted since gaps are not nucleotides or amino acids. Likewise, gaps presented in the reference sequence are not counted since target sequence nucleotides or amino acids are counted, not nucleotides or amino acids from the reference sequence.
- the percentage of sequence identity is calculated by determining the number of positions at which the identical amino-acid residue or nucleic acid base occurs in both sequences to yield the number of matched positions, dividing the number of matched positions by the total number of positions in the window of comparison and multiplying the result by 100 to yield the percentage of sequence identity.
- the comparison of sequences and determination of percent sequence identity between two sequences can be accomplished using readily available software programs. Suitable software programs are available from various sources, and for alignment of both protein and nucleotide sequences. One suitable program to determine percent sequence identity is bl2seq, part of the BLAST suite of program available from the U.S.
- B12seq performs a comparison between two sequences using either the BLASTN or BLASTP algorithm.
- BLASTN is used to compare nucleic acid sequences
- BLASTP is used to compare amino acid sequences.
- Other suitable programs are, e.g., Needle, Stretcher, Water, or Matcher, part of the EMBOSS suite of bioinformatics programs and also available from the European Bioinformatics Institute (EBI) at world wide web at ebi.ac.uk/Tools/psa.
- homology with respect to a peptide, polypeptide or antibody sequence refers to the percentage of amino acid residues in a candidate sequence that are identical with the amino acid residues in the specific peptide or polypeptide sequence, after aligning the sequences and introducing gaps, if necessary, to achieve the maximum percent sequence identity, and not considering any conservative substitutions as part of the sequence identity. Alignment for purposes of determining percent amino acid sequence identity can be achieved in various ways that are within the skill in the art, for instance, using publicly available computer software such as BLAST, BLAST-2, ALIGN or MEGALIGNTM (DNASTAR) software. Those skilled in the art can determine appropriate parameters for measuring alignment, including any algorithms known in the art needed to achieve maximal alignment over the full length of the sequences being compared.
- affinity refers to the strength of interaction between an antibody or its antigen-binding fragment and an antigen, determined by characteristics of the antigen such as size, shape, and/or charge, and the CDR sequences of the antibody or antigen-binding fragment. Methods for determining such affinities are known in the art and may also be referenced herein.
- Antibodies or their antigen-binding fragments used in the present invention are said to “specifically bind” to the target, such as the antigen, when the dissociation constant (KD) is ⁇ 10' 6 M.
- KD dissociation constant
- the “antigen-binding fragment” of a chain (heavy chain or light chain) of an antibody or immunoglobulin includes a part of an antibody which lacks some amino acids compared to a full-length chain, but can specifically bind to an antigen (e.g., CD22 and/or CD20).
- This fragment can be considered as having biological activity, in that the fragment can specifically bind to a target antigen, or can compete with other antibodies or antigen binding fragments thereof to bind to a specific epitope.
- such a fragment includes at least one CDR present in a full-length light chain or heavy chain, and in some embodiments, includes a short-chain heavy chain and/or light chain, or part thereof.
- This biological active fragment may be produced by a recombinant DNA technique or may be produced, for example, by cleaving an intact antibody enzymatically or chemically.
- An immunologically functional immunoglobulin fragment includes, but is not limited to, Fab, Fab', scFab, dsFv, Fv, scFV, scFV-Fc, 83cab, and dAb, scFv2-Fc, bi-nanobody, bispecific T cell engager (BiTE), tandem diabody (tandAb), dual affinity retargeting (DART) antibody, DART-Fc, scFv-human serum albumin (HSA)-scFv, dock-and-lock (DNL)-Fab3, minibody, Fab2 fragment (bispecific), Fab3 fragment (trispecific), Bis-scFv fragment (bispecific), sdAb fragment (VH/VHH), tetrabody, triabody, or a diabody,
- the “Fc” region includes two heavy chain fragments including CH2 and CH3 domains of an antibody. These two heavy chain fragments are linked to each other by hydrophobic interaction of two or more of disulfide bonds and a CH3 domain.
- the “Fab fragment” consists of one light chain and one heavy chain including a variable region and CHI only.
- the heavy chain of a Fab molecule cannot form a disulfide bond with another heavy chain molecule.
- two molecules of Fab are linked by a flexible linker.
- the “Fab' fragment” includes a Fab fragment and additionally a region between CHI and CH2 domains of a heavy chain. A disulfide bond may form between two heavy chains of Fab' fragments of two molecules, forming a F(ab')2 molecule.
- the “F(ab')2 fragment” includes two light chains and two heavy chains including a variable region CHI and part of a constant region between the CHI and CH2 domains, with an inter-chain disulfide bond formed between the two heavy chains. Accordingly, a F(ab')2 fragment consists of two Fab' fragments, and the two Fab' fragments are joined to each other by the disulfide bond therebetween.
- the “Fv region” is a fragment of an antibody which includes each variable region of a heavy chain and a light chain, but does not include constant regions.
- a heavy chain and a light chain are linked by a disulfide bond.
- the Fv is linked by a flexible linker.
- a scFv-Fc a Fc is linked to a scFV.
- CH3 is linked to a scFV.
- a diabody includes the scFVs of two molecules.
- the “single chain Fv” or “scFv” antibody fragment includes the VH and VL domains of an antibody, and these domains are present within a single polypeptide chain.
- An Fv polypeptide may additionally include a polypeptide linker between a Vh domain which enables the scFv to form the target structure for antigen binding, and a VL domain.
- the “short-chain antibody (84cab)” is a single polypeptide chain including one constant region of a heavy chain or a light chain constant region in which heavy chain and light chain variable regions are linked by a flexible linker.
- U.S. Pat. No. 5,260,203 may be referred to, and short-chain antibody is disclosed herein by reference.
- the “domain antibody (dAb)” is an immunologically functional immunoglobulin fragment including only a variable region of a heavy chain or a variable region of a light chain.
- two or more VH regions are linked by a covalent bond via a peptide linker, to form a bivalent domain antibody. Two VH regions of this bivalent domain antibody may target the same or different antigens.
- CDR complementarity determining region
- Each variable region typically has three CDR domains, identified as CDR1, CDR2, and CDR3.
- the “framework region” (FR) is a variable region residue other than the CDR residues.
- Each variable region typically has four FRs, identified as FR1, FR2, FR3, and FR4.
- the “bivalent antigen-binding protein” or “bivalent antibody” includes two antigen-binding sites.
- the two antigen-binding sites included in a bivalent antibody may have the same antigen specificity, or may be a bispecific antibody where the antigen-biding sites bind to different antigens.
- the “bispecific antibody” refers to an antibody or an antigenbinding fragment thereof capable of binding to two different epitopes on a single antigen or two different antigens.
- the bispecific antibody of the present disclosure may be bivalent, trivalent, or tetravalent.
- “-valent”, “-valent(s)” or other grammatical variations thereof, refer to the number of antigen-binding sites within an antibody molecule.
- multispecific antigen-binding protein or “multispecific antibody” targets two or more antigens or epitopes.
- a “chimeric antibody” refers to an antibody (immunoglobulin) in which a portion of the heavy and/or light chain is identical with or homologous to corresponding sequences in antibodies derived from a particular species or belonging to a particular antibody class or subclass, while the remainder of the chain(s) is(are) identical with or homologous to corresponding sequences in antibodies derived from another species or belonging to another antibody class or subclass, as well as fragments of such antibodies, so long as they exhibit the desired biological activity (U.S. Patent No. 4,816,567; Morrison et al., Proc. Nat’l Acad. Sci. USA, 81:6851-55 (1984)).
- Chimeric antibodies of interest herein include PRIMATIZED® antibodies wherein the antigen-binding region of the antibody is derived from an antibody produced by, e.g., immunizing macaque monkeys with an antigen of interest.
- “Humanized” forms of non-human (e.g., murine) antibodies are chimeric antibodies that contain minimal sequence derived from non-human immunoglobulin.
- a humanized antibody is a human immunoglobulin (recipient antibody) in which residues from an CDR of the recipient are replaced by residues from an CDR of a non-human species (donor antibody) such as mouse, rat, rabbit or non-human primate having the desired specificity, affinity, and/or capacity.
- donor antibody such as mouse, rat, rabbit or non-human primate having the desired specificity, affinity, and/or capacity.
- FR residues of the human immunoglobulin are replaced by corresponding non-human residues.
- humanized antibodies may comprise residues that are not found in the recipient antibody or in the donor antibody. These modifications may be made to further refine antibody performance, such as binding affinity.
- a humanized antibody will comprise substantially all of at least one, and typically two, variable domains, in which all or substantially all of the hypervariable loops correspond to those of a non-human immunoglobulin sequence, and all or substantially all of the FR regions are those of a human immunoglobulin sequence, although the FR regions may include one or more individual FR residue substitutions that improve antibody performance, such as binding affinity, isomerization, immunogenicity, and the like.
- the number of these amino acid substitutions in the FR is typically no more than 6 in the H chain, and in the L chain, no more than 3.
- the humanized antibody optionally will also comprise at least a portion of an immunoglobulin constant region (Fc), typically that of a human immunoglobulin.
- Fc immunoglobulin constant region
- a “human antibody” is one that possesses an amino-acid sequence corresponding to that of an antibody, produced by a human and/or has been made using any of the techniques for making human antibodies as disclosed herein. This definition of a human antibody specifically excludes a humanized antibody comprising non-human antigen-binding residues.
- Human antibodies can be produced using various techniques known in the art, including phagedisplay libraries. Hoogenboom and Winter, J. Mol. Biol., 227:381 (1991); Marks et al., J. Mol. Biol., 222:581 (1991). Also available forthe preparation of human monoclonal antibodies are methods described in Cole et al., Monoclonal Antibodies and Cancer Therapy, Alan R. Liss, p.
- Human antibodies can be prepared by administering the antigen to a transgenic animal that has been modified to produce such antibodies in response to antigenic challenge, but whose endogenous loci have been disabled, e.g., immunized xenomice (see, e.g., U.S. Patent Nos. 6,075,181 and 6,150,584 regarding XENOMOUSETM technology). See also, for example, Li et al., Proc. Nat’l Acad. Sci.
- a “human consensus framework” is a framework that represents the most commonly occurring amino acid residues in a selection of human immunoglobulin VL or VH framework sequences. Generally, the selection of human immunoglobulin VL or VH sequences is from a subgroup of variable domain sequences. Generally, the subgroup of sequences is a subgroup as in Kabat et al., Sequences of Proteins of Immunological Interest, 5th Ed. Public Health Service, National Institutes of Health, Bethesda, MD (1991).
- the subgroup may be subgroup kappa I, kappa II, kappa III or kappa IV as in Kabat et al., supra. Additionally, for the VH, the subgroup may be subgroup I, subgroup II, or subgroup III as in Kabat et al., supra.
- an “affinity-matured” antibody is one with one or more alterations in one or more CDRs thereof that result in an improvement in the affinity of the antibody for antigen, compared to a parent antibody that does not possess those alteration(s).
- an affinity-matured antibody has nanomolar or even picomolar affinities for the target antigen.
- Affinity-matured antibodies are produced by procedures known in the art. For example, Marks et al., Bio/Technology 10:779-783 (1992) describes affinity maturation by VH- and VL- domain shuffling. Random mutagenesis of CDR and/or framework residues is described by, for example: Barbas et al. Proc Nat. Acad. Sci.
- linker refers to a compound which covalently bonds a cytotoxic compound to an antibody.
- unsubstituted or substituted refers to a parent group which may be unsubstituted or substituted
- substituted refers to a parent group having at least one substituent
- a substituent refers to a chemical moiety covalently bonded to or fused with a parent group.
- halo refers to fluorine, chlorine, bromine, iodine, and the like.
- alkyl refers to saturated aliphatic groups, including but not limited to Ci-Cio straight-chain alkyl groups or Ci-Cio branched-chain alkyl groups.
- the “alkyl” group refers to Ci-Cg straight-chain alkyl groups or Ci-Cg branched- chain alkyl groups.
- the “alkyl” group refers to C1-C4 straight-chain alkyl groups or C1-C4 branched-chain alkyl groups.
- alkyl examples include, but are not limited to, methyl, ethyl, 1-propyl, 2-propyl, n-butyl, sec-butyl, tert-butyl, 1-pentyl, 2-pentyl, 3-pentyl, neo-pentyl, 1-hexyl, 2-hexyl, 3-hexyl, 1-heptyl, 2-heptyl, 3-heptyl, 4-heptyl, 1-octyl, 2-octyl, 3-octyl or 4-octyl and the like.
- the “alkyl” group may be optionally substituted.
- acyl is art-recognized and refers to a group represented by the general formula hydrocarbylC(O)-, preferably alkylC(O)-.
- acylamino is art-recognized and refers to an amino group substituted with an acyl group and may be represented, for example, by the formula hydrocarbylC(O)NH-.
- acyloxy is art-recognized and refers to a group represented by the general formula hydrocarbylC(O)O-, preferably alkylC(O)O-.
- alkoxy refers to an alkyl group having an oxygen attached thereto.
- Representative alkoxy groups include methoxy, ethoxy, propoxy, tert-butoxy and the like.
- alkoxyalkyl refers to an alkyl group substituted with an alkoxy group and may be represented by the general formula alkyl-O-alkyl.
- alkyl refers to saturated aliphatic groups, including straight-chain alkyl groups, branched-chain alkyl groups, cycloalkyl (alicyclic) groups, alkyl-substituted cycloalkyl groups, and cycloalkyl-substituted alkyl groups.
- a straight chain or branched chain alkyl has 30 or fewer carbon atoms in its backbone (e.g., Ci- 30 for straight chains, C3-30 for branched chains), and more preferably 20 or fewer.
- alkyl as used throughout the specification, examples, and claims is intended to include both unsubstituted and substituted alkyl groups, the latter of which refers to alkyl moieties having substituents replacing a hydrogen on one or more carbons of the hydrocarbon backbone, including haloalkyl groups such as trifluoromethyl and 2,2,2- trifluoroethyl, etc.
- C x-y or “C x -C y ”, when used in conjunction with a chemical moiety, such as, acyl, acyloxy, alkyl, alkenyl, alkynyl, or alkoxy is meant to include groups that contain from x to y carbons in the chain.
- Coalkyl indicates a hydrogen where the group is in a terminal position, a bond if internal.
- a Ci-ealkyl group for example, contains from one to six carbon atoms in the chain.
- alkylamino refers to an amino group substituted with at least one alkyl group.
- alkylthio refers to a thiol group substituted with an alkyl group and may be represented by the general formula alkylS-.
- alkylS- refers to a group wherein R 9 and R 10 each independently represent a hydrogen or hydrocarbyl group, or R 9 and R 10 taken together with the N atom to which they are attached complete a heterocycle having from 4 to 8 atoms in the ring structure.
- amine and “amino” are art-recognized and refer to both unsubstituted and substituted amines and salts thereof, e.g., a moiety that can be represented by wherein R 9 , R 10 , and R 10 ’ each independently represent a hydrogen or a hydrocarbyl group, or R 9 and R 10 taken together with the N atom to which they are attached complete a heterocycle having from 4 to 8 atoms in the ring structure.
- aminoalkyl refers to an alkyl group substituted with an amino group.
- aralkyl refers to an alkyl group substituted with an aryl group.
- aryl as used herein include substituted or unsubstituted single-ring aromatic groups in which each atom of the ring is carbon.
- the ring is a 5- to 7-membered ring, more preferably a 6-membered ring.
- aryl also includes polycyclic ring systems having two or more cyclic rings in which two or more carbons are common to two adjoining rings wherein at least one of the rings is aromatic, e.g., the other cyclic rings can be cycloalkyls, cycloalkenyls, cycloalkynyls, aryls, heteroaryls, and/or heterocyclyls.
- Aryl groups include benzene, naphthalene, phenanthrene, phenol, aniline, and the like.
- carboxylate is art-recognized and refers to a group wherein R 9 and R 10 independently represent hydrogen or a hydrocarbyl group.
- Carbocyclylalkyl refers to an alkyl group substituted with a carbocycle group.
- the term “carbocycle” includes 5-7 membered monocyclic and 8-12 membered bicyclic rings. Each ring of a bicyclic carbocycle may be selected from saturated, unsaturated and aromatic rings. Carbocycle includes bicyclic molecules in which one, two or three or more atoms are shared between the two rings.
- the term “fused carbocycle” refers to a bicyclic carbocycle in which each of the rings shares two adjacent atoms with the other ring. Each ring of a fused carbocycle may be selected from saturated, unsaturated and aromatic rings.
- an aromatic ring e.g., phenyl
- a saturated or unsaturated ring e.g., cyclohexane, cyclopentane, or cyclohexene.
- Exemplary “carbocycles” include cyclopentane, cyclohexane, bicyclo[2.2.1]heptane, 1,5-cyclooctadiene, 1,2,3,4-tetrahydronaphthalene, bicyclo[4.2.0]oct- 3-ene, naphthalene and adamantane.
- Exemplary fused carbocycles include decalin, naphthalene, 1,2,3,4-tetrahydronaphthalene, bicyclo[4.2.0]octane, 4,5,6,7-tetrahydro-lH- indene and bicyclo [4.1.0]hept-3-ene.
- Carbocycles may be substituted at any one or more positions capable of bearing a hydrogen atom.
- Carbocyclylalkyl refers to an alkyl group substituted with a carbocycle group.
- carbonate is art-recognized and refers to a group -OCO2-.
- cycloalkyl includes substituted or unsubstituted non-aromatic single ring structures, preferably 4- to 8-membered rings, more preferably 4- to 6-membered rings.
- cycloalkyl also includes polycyclic ring systems having two or more cyclic rings in which two or more carbons are common to two adjoining rings wherein at least one of the rings is cycloalkyl and the substituent (e.g., R 100 ) is attached to the cycloalkyl ring, e.g., the other cyclic rings can be cycloalkyls, cycloalkenyls, cycloalkynyls, aryls, heteroaryls, and/or heterocyclyls.
- Heteroaryl groups include, for example, pyrrole, furan, thiophene, imidazole, oxazole, thiazole, pyrazole, pyridine, pyrazine, pyridazine, pyrimidine, denzodioxane, tetrahydroquinoline, and the like.
- esters refers to a group -C(O)OR 9 wherein R 9 represents a hydrocarbyl group.
- ether refers to a hydrocarbyl group linked through an oxygen to another hydrocarbyl group . Accordingly, an ether substituent of a hydrocarbyl group may be hydrocarbyl-O-. Ethers may be either symmetrical or unsymmetrical. Examples of ethers include, but are not limited to, heterocycle-O-heterocycle and aryl-0 -heterocycle. Ethers include “alkoxyalkyl” groups, which may be represented by the general formula alkyl-O-alkyl.
- halo and “halogen” as used herein means halogen and includes chloro, fluoro, bromo, and iodo.
- heteroalkyl and “heteroaralkyl”, as used herein, refers to an alkyl group substituted with a hetaryl group.
- heteroaryl and “hetaryl” include substituted or unsubstituted aromatic single ring structures, preferably 5- to 7-membered rings, more preferably 5- to 6-membered rings, whose ring structures include at least one heteroatom, preferably one to four heteroatoms, more preferably one or two heteroatoms.
- heteroaryl and “hetaryl” also include polycyclic ring systems having two or more cyclic rings in which two or more carbons are common to two adjoining rings wherein at least one of the rings is heteroaromatic, e.g., the other cyclic rings can be cycloalkyls, cycloalkenyls, cycloalkynyls, aryls, heteroaryls, and/or heterocyclyls.
- Heteroaryl groups include, for example, pyrrole, furan, thiophene, imidazole, oxazole, thiazole, pyrazole, pyridine, pyrazine, pyridazine, and pyrimidine, and the like.
- heteroatom as used herein means an atom of any element other than carbon or hydrogen. Preferred heteroatoms are nitrogen, oxygen, and sulfur.
- heterocyclylalkyl refers to an alkyl group substituted with a heterocycle group.
- heterocyclyl refers to substituted or unsubstituted non-aromatic ring structures, preferably 3- to 10-membered rings, more preferably 3- to 7-membered rings, whose ring structures include at least one heteroatom, preferably one to four heteroatoms, more preferably one or two heteroatoms.
- heterocyclyl and “heterocyclic” also include polycyclic ring systems having two or more cyclic rings in which two or more carbons are common to two adjoining rings wherein at least one of the rings is heterocyclic, e.g., the other cyclic rings can be cycloalkyls, cycloalkenyls, cycloalkynyls, aryls, heteroaryls, and/or heterocyclyls.
- Heterocyclyl groups include, for example, piperidine, piperazine, pyrrolidine, morpholine, lactones, lactams, and the like.
- Hydrocarbyl groups include, but are not limited to aryl, heteroaryl, carbocycle, heterocycle, alkyl, alkenyl, alkynyl, and combinations thereof.
- hydroxyalkyl refers to an alkyl group substituted with a hydroxy group.
- lower when used in conjunction with a chemical moiety, such as, acyl, acyloxy, alkyl, alkenyl, alkynyl, or alkoxy is meant to include groups where there are ten or fewer atoms in the substituent, preferably six or fewer.
- acyl, acyloxy, alkyl, alkenyl, alkynyl, or alkoxy substituents defined herein are respectively lower acyl, lower acyloxy, lower alkyl, lower alkenyl, lower alkynyl, or lower alkoxy, whether they appear alone or in combination with other substituents, such as in the recitations hydroxyalkyl and aralkyl (in which case, for example, the atoms within the aryl group are not counted when counting the carbon atoms in the alkyl substituent).
- polycyclyl refers to two or more rings (e.g., cycloalkyls, cycloalkenyls, cycloalkynyls, aryls, heteroaryls, and/or heterocyclyls) in which two or more atoms are common to two adjoining rings, e.g., the rings are “fused rings”.
- Each of the rings of the polycycle can be substituted or unsubstituted.
- each ring of the poly cycle contains from 3 to 10 atoms in the ring, preferably from 5 to 7.
- sulfate is art-recognized and refers to the group -OSO3H, or a pharmaceutically acceptable salt thereof.
- sulfonamide is art-recognized and refers to the group represented by the general formulae wherein R 9 and R 10 independently represents hydrogen or hydrocarbyl.
- sulfoxide is art-recognized and refers to the group-S(O)-.
- sulfonate is art-recognized and refers to the group SO3H, or a pharmaceutically acceptable salt thereof.
- substituted refers to moieties having substituents replacing a hydrogen on one or more carbons of the backbone. It will be understood that “substitution” or “substituted with” includes the implicit proviso that such substitution is in accordance with permitted valence of the substituted atom and the substituent, and that the substitution results in a stable compound, e.g., which does not spontaneously undergo transformation such as by rearrangement, cyclization, elimination, etc. As used herein, the term “substituted” is contemplated to include all permissible substituents of organic compounds.
- the permissible substituents include acyclic and cyclic, branched and unbranched, carbocyclic and heterocyclic, aromatic and non-aromatic substituents of organic compounds.
- the permissible substituents can be one or more and the same or different for appropriate organic compounds.
- the heteroatoms such as nitrogen may have hydrogen substituents and/or any permissible substituents of organic compounds described herein which satisfy the valences of the heteroatoms.
- Substituents can include any substituents described herein, for example, a halogen, a hydroxyl, a carbonyl (such as a carboxyl, an alkoxycarbonyl, a formyl, or an acyl), a thiocarbonyl (such as a thioester, a thioacetate, or a thioformate), an alkoxyl, a phosphoryl, a phosphate, a phosphonate, a phosphinate, an amino, an amido, an amidine, an imine, a cyano, a nitro, an azido, a sulfhydryl, an alkylthio, a sulfate, a sulfonate, a sulfamoyl, a sulfonamide, a sulfonyl, a heterocyclyl, an aralkyl, or an aromatic or heteroaromatic moiety.
- thioalkyl refers to an alkyl group substituted with a thiol group.
- thioester refers to a group -C(O)SR 9 or -SC(O)R 9 wherein R 9 represents a hydrocarbyl.
- thioether is equivalent to an ether, wherein the oxygen is replaced with a sulfur.
- urea is art-recognized and may be represented by the general formula wherein R 9 and R 10 independently represent hydrogen or a hydrocarbyl.
- DBCO dibenzocyclooctyne moiety
- sugar moiety refers to a naturally occurring sugar or a modified sugar which is part of a larger molecule and is connected to the remainder of the larger molecule through, for example, one of the hydroxyl groups present on the sugar.
- sugar is connected to the remainder of the larger molecule via the hydroxyl group on the anomeric carbon.
- sugar moieties include, but are not limited to, glucuronosyl and galactosyl.
- glycosidase is an enzyme that breaks down glycosidic bonds in carbohydrates, glycoproteins, and glycolipids.
- glycosidases are P-glucuronidase and P- galactosidase.
- P-glucuronidase is a type of glucuronidase that catalyzes hydrolysis of P-D- glucuronic acid or P-glucuronosyl residues.
- P-Galactosidase is a glycoside hydrolase enzyme that catalyzes hydrolysis of terminal non-reducing P-D-galactose or P-galactosyl residues in P- D-galactosides.
- modulate includes the inhibition or suppression of a function or activity (such as cell proliferation) as well as the enhancement of a function or activity.
- cleavage group refers to a chemical moiety which dissociates when subjected to a stimulus, such as acidic conditions, basic conditions, reducing conditions, oxidizing conditions, light, or heat, or an enzyme, such as an esterase.
- a first exemplified anti-CD20 antibody CD20 1 (Rituximab), that specifically binds to CD20, was produced by the methods described in U.S. Patent No. 5,736,137 A and U.S. Patent No. 7,422,739 B2, the entirety of which are incorporated herein by reference.
- the amino acid sequences of the antibody CD20 1 are shown in Table 1 below.
- the amino acid sequences of the CDRs below are defined according to the Kabat definition.
- an exemplified antibody clone CD20_l-CaaX was constructed by introducing a CaaX peptide moiety (GGGGGGGCVIM, SEQ ID NO: 77) to the C-terminus of the light chain of SEQ ID NO: 18 in Table 1 with the method disclosed in International Patent Application No. PCT/IB2012/001065, the entirety of which is incorporated herein by reference.
- the amino acid sequences of CD20_l-CaaX are shown in Table 2 below.
- Exemplified halfinerl was prepared by introducing Hole mutations (T370S, L372A, Y41 IV of SEQ ID NO: 17 in Table 1) in the heavy chain constant region of the half-antibody of CD20_l, and introducing LALA mutations (L238A/L239A of SEQ ID NO: 17 in Table 1) into the heavy chain constant region of a half-antibody of CD20 1.
- a second exemplified anti-CD20 antibody CD20 2 (Ocaratuzumab), that specifically binds to CD20, was produced by the methods described in U.S. Patent No. 8,153,125 B2, the entirety of which is incorporated herein by reference.
- Exemplified halfiner2 was prepared by introducing Hole mutations (T370S, L372A, Y411V according to SEQ ID NO: 37) in the heavy chain constant region of a half-antibody of CD20 2, and introducing LALA mutations (L238A/L239A according to SEQ ID NO: 37) into the heavy chain constant region of a halfantibody of CD20_2.
- a first exemplified anti-CD22 antibody CD22 1 (Inotuzumab) was produced by the methods described in U. S . Patent No. 8,747,857 B2, the entirety of which is incorporated herein by reference.
- Exemplified halfmer3 was prepared by introducing Knob mutation (T370W according to SEQ ID NO: 55) in the heavy chain constant region of a half-antibody of CD22 1, and introducing LALA mutations (L238A/L239A according to SEQ ID NO: 55) into the heavy chain constant region of a half-antibody of CD22 1.
- Exemplified anti-CD22 antibody CD22 2 (Epratuzumab) was produced by the methods described in U.S. Patent No. 6,187,287 B2, the entirety of which is incorporated herein by reference.
- Exemplified halfiner4 is prepared by introducing Knob mutation (T365W according to SEQ ID NO: 73) in the heavy chain constant region of a half-antibody of CD22 2, and introducing LALA mutations (L233A/L234A according to SEQ ID NO: 73) into the heavy chain constant region of a half-antibody of CD22_2.
- Exemplified halfiner5 was prepared by introducing Pentambody substitutions (L355E, Y411L, K413V of SEQ ID NO: 17 in Table 1) in the heavy chain constant region of a halfantibody of CD20_l, and introducing LALA mutations (L238A/L239A of SEQ ID NO: 17 in Table 1) into the heavy chain constant region of a half-antibody of CD20 1.
- Exemplified halfmer6 was prepared by introducing Pentambody substitutions (T370L and D403R according to SEQ ID NO: 76) in the heavy chain constant region of a half-antibody of CD22_1, and introducing LALA mutations (L238A/L239A according to SEQ ID NO: 76) into the heavy chain constant region of a half-antibody of CD22 1.
- CD20 (Hole) halfinerl CD20 (Hole) halfiner2
- CD22 (Knob) halfiner3 CD22 (Knob) halfiner4.
- CD20 U351E, Y407L, K409V according to EU numbering convention
- CD22 T366L, D399R according to EU numbering convention
- exemplified halfmer-CaaX constructs were prepared by introducing a CaaX peptide moiety (GGGGGGGCVIM, SEQ ID NO: 77) to the C-terminus of the light chain of each of Halfinerl (LC1; SEQ ID NO: 18) in Table 4, Halfmer5 (LC1; SEQ ID NO: 18) in Table 8, Halfmer2 (LC2; SEQ ID NO: 38) in Table 5, Halfiner3 (LC3; SEQ ID NO: 56) in Table 6, Halfmer6 (LC3; SEQ ID NO: 56) in Table 9, and Halfmer4 (LC4; SEQ ID NO: 74) in Table 7 with the method disclosed in International Patent Application No. PCT/IB2012/001065, the entirety of which is incorporated herein by reference.
- LC1; SEQ ID NO: 18 Halfmer5
- Halfmer2 LC2; SEQ ID NO: 38
- Halfiner3 LC3; SEQ ID NO: 56
- Halfmer6 LC3; SEQ ID NO: 56
- Pairs of exemplified halfmer-CaaX constructs 1 and 3, 1 and 4, 2 and 3, and 2 and 4 were reacted using the Knob-into-Hole method, described in U.S. Patent No. 9,637,557 B2, to form bispecific antibodies, which were then used for subsequent ADC synthesis.
- each halfmer-CaaX construct was buffer-exchanged with a buffer containing 50 mM Arginine (Sigma-Aldrich, A4474) and 200 mM Histidine (Sigma-Aldrich, H6034) using vivaspin 20 (Sartorius, VS2002).
- the hole halfmer-CaaX and knob halfmer-CaaX pairs were then mixed at a 1: 1 ratio to achieve a concentration of 2 mg/mL each.
- Reduced L-glutathione (Sigma- Aldrich, G4251) was added to a final concentration of 2.6 mM.
- the reaction was then carried out in a shaking incubator at 36°C, 160 rpm, for 6 hours.
- CD20xCD22 bispecific antibodyl which specifically binds to CD20 and CD22, are shown in Table 10 below.
- CD20xCD22 bispecific antibody2 The amino acid sequences of CD20xCD22 bispecific antibody2, which specifically binds to CD20 and CD22, are shown in Table 11 below. Table 11. Amino Acid Sequences of Exemplified CD20xCD22 Bispecific Antibody2
- CD20xCD22 bispecific antibody3 which specifically binds to CD20 and CD22, are shown in Table 12 below.
- CD20xCD22 bispecific antibody4 which specifically binds to CD20 and CD22, are shown in Table 13 below.
- CD20xCD22 bispecific antibody5 The amino acid sequences of CD20xCD22 bispecific antibody5, which specifically binds to CD20 and CD22, are shown in Table 14 below.
- bispecific antibodies that specifically bind to CD20 and CD22 were produced using the CrossMab method, described in U.S. Patent No. 8,242,247 B2, and the S-DUALTM method, described in EP Patent Application No. 4286408 Al, the entirety of which are incorporated herein by reference.
- the list of exemplified bispecific antibodies produced by CrossMab or S-DUALTM is shown in Table 15 below.
- halfmerl which specifically binds to CD20
- halfiner7 which specifically binds to CD22
- the amino acid sequences of the CD Rs below are defined according to the Kabat definition.
- Halfiner7 was prepared based on halfiner3 (Table 6) by replacing the heavy chain CHI and light chain CL regions of halfiner3 with each other.
- bispecific antibody6 was produced using halfmerl (Table 4) as is, and halfiner7.
- exemplified halfiner-CaaX constructs were produced by introducing a CaaX peptide moiety (GGGGGGGCVIM; SEQ ID NO: 77) to the C-terminus of each of the light chain of Halfinerl (LC1; SEQ ID NO: 18) in Table 4 and Halfiner7 (LC7; SEQ ID NO: 82) in Table 16 with the method disclosed in International Patent Application No. PCT/IB2012/001065, the entirety of which is incorporated herein by reference.
- An exemplified pair of halfiner-CaaX constructs 1 and 7 were reacted using the CrossMab method, described in U.S. Patent No. 8,242,247 B2, to form bispecific antibodies for subsequent ADC synthesis.
- CD20xCD22 bispecific antibody6 which specifically binds to CD20 and CD22, are shown in Table 17 below.
- An exemplified CD20xCD22 bispecific antibody? was produced using the exemplified anti-CD20 and anti-CD22 antibodies disclosed herein with the S-DUAL method, described in EP Patent Application No. 4286408 Al.
- Compound 1 was produced with the method disclosed in U.S. Patent No. 11,654,197 B2, the entirety of which is incorporated herein by reference.
- Compound 2 (MC-Val-Cit-PAB-MMAE) has the above structure and was purchased from MedChemExpress (Catalog No. HY-15575).
- Compound 3 (MC-Val-Ala-PBD) has the above structure and was purchased from GLPBIO (Catalog No. GC39403).
- LCB 14-0606 was produced with the method disclosed in U.S. Patent No. 9,669,107 B2, the entirety of which is incorporated herein by reference.
- the structural formula of LCB14-0606 is as follows:
- Step 1 Production of Prenylated Antibody with LCB14-0606
- Exemplified anti-CD20 mono or anti-CD20xCD22 bispecific antibodies listed in below Table 18 were prepared according to Preparation Examples 1-3.
- a mixture comprising each of the antibodies for the prenylation reaction was prepared and reacted at 30°C for 16 hours.
- the reaction mixture for each antibody contained a total of 24 pM antibody, 400 nM FTase (Calbiochem #344145), and 0.14 mM LCB 14-0606 in a buffer solution (50 mM Tris-HCl (pH 7.4), 5 mM MgCh, 10 pM ZnCh, 0.25 mM DTT).
- the prenylated antibodies were purified using a G25 Sepharose column (AKTA purifier, GE healthcare) equilibrated with PBS buffer.
- Step 2 Drug-conjugation method
- the oxime bond formation reaction between each of the prenylated antibodies and linker-drug was carried out by mixing 100 mM sodium acetate buffer (pH 5.2, Sigma- Aldrich, S7899), 10% DMSO (Sigma-Aldrich, D4540), 24 pM of the prenylated antibody, and 10 equivalents (240 pM) of the linker-drug (Compound 1 of Preparation Example 4). The mixture was stirred gently at 30°C for 6 hours. After the reaction, excess low-molecular-weight compounds were removed through a desalting process using a G25 Sepharose column, and the protein fractions were collected and concentrated.
- ADCs The preparation of ADCs was analyzed using hydrophobic interaction chromatography to measure the drug-antibody ratio and size exclusion chromatography for monomer analysis.
- hydrophobic interaction chromatography was performed using a phenyl column (7.5 mm X 75 mm, 10 pm, Tosoh Bioscience, 7573) and potassium phosphate buffer as the mobile phase.
- the analysis used a buffer A consisting of 50 mM potassium phosphate buffer (pH 7.0) with 0.5 M ammonium sulfate and a buffer B consisting of 50 mM potassium phosphate buffer (pH 7.0) with 30% acetonitrile.
- the initial conditions were stabilized at 90% A/10% B, and a linear gradient of 90% A/10% B to 10% A/90% B was applied over 30 minutes, followed by an additional elution with 10% A/90% B for 5 minutes.
- the flow rate and temperature were set to 1.0 mL/min and 25°C, respectively, with detection at both 254 nm and 280 nm.
- Buffer A contained 50 mM potassium phosphate (pH 7.0) with 1.5 M ammonium sulfate
- buffer B contained 50 mM potassium phosphate (pH 7.0) with 30% acetonitrile.
- the initial conditions were stabilized at 70% A/30% B, and a linear gradient from 70% A/30% B to 10% A/90% B was applied over 30 minutes, followed by elution with 10% A/90% B for 5 minutes.
- the flow rate and temperature were maintained at 1.0 mL/min and 25°C, respectively, with detection at both 254 nm and 280 nm.
- hydrophobic interaction chromatography was performed using a butyl column (4.6 mm X 35 mm, 2.5 pm, Tosoh Bioscience, 14947) and potassium phosphate buffer.
- Buffer A contained 50 mM potassium phosphate (pH 7.0) with 1.5 M ammonium sulfate
- buffer B contained 50 mM potassium phosphate (pH 7.0) with 30% isopropyl alcohol.
- the initial conditions were stabilized at 100% A/0% B, and a linear gradient from 100% A/0% B to 0% A/ 100% B was applied over 20 minutes, followed by elution with 0% A/ 100% B for 2 minutes.
- the flow rate and temperature were set to 1.0 mL/min and 25°C, respectively, with detection at both 254 nm and 280 nm.
- ADC1, ADC2, ADC3, ADC4, ADC5, ADC6, ADC7, and ADC8 a SEC column (7.8 mm X 30 cm, Tosoh Bioscience, 8541) was used with a mobile phase of 0.1 M sodium phosphate and 0.3 M sodium chloride containing 10% acetonitrile (pH 6.8). The analysis was conducted with a linear gradient of 100% buffer over 30 minutes.
- the CD20xCD22 bispecific antibody 1 was conjugated with Compound 3 of Preparation Example 4 or Compound 2 of Preparation Example 4.
- bispecific antibody For conjugation of the bispecific antibody to the linker-toxin, 74 pM of tris(2- carboxyethyljphosphine (Sigma- Aldrich, C4706) was added to a solution of 24 pM CD20xCD22 bispecific antibody 1 containing 4 mM EDTA (Enzynomics, EBE001-500) at pH 7.4, and the mixture was gently stirred at 25°C. After a reaction time of 2 hours, 216 pM of Compound 3 or Compound 2 were added, and the mixture was gently stirred for an additional 2 hours at 25°C.
- ADC9 and ADC 10 were analyzed using reverse phase chromatography to measure the average drug-antibody ratio and size exclusion chromatography for monomer analysis.
- Reverse phase chromatography utilized a PLRP-S 1000A column (2.1 mm * 50 mm, 5 pm, Agilent, PL1912-1502) with a mobile phase containing 0.1% trifluoroacetic acid (Sigma- Aldrich, T6508). Water containing 0.1% trifluoroacetic acid was used as solution A, and acetonitrile containing 0.1% trifluoroacetic acid was used as solution B. The system was stabilized with 75% A and 25% B for 3 minutes, followed by a linear gradient of 50% A and 50% B for 28 minutes, and then an additional linear gradient of 5% A and 95% B for 1 minute, with a further 2 minutes of elution.
- the flow rate and temperature were set to 1.0 mL/min and 80°C, respectively. Detection was performed at both 254 nm and 280 nm. Size exclusion chromatography utilized a SEC column (7.8 mm x 30 cm, Tosoh Bioscience, 8541) and a mobile phase containing sodium phosphate 0. IM and 0.3M sodium chloride in 10% acetonitrile buffer (pH 6.8). The analysis was performed with a linear gradient of 100% buffer for 30 minutes.
- the above cancer cell lines were cultured and divided into two groups: a control IgG antibody-treated group and an anti-CD20xCD22 bispecific antibody 1 -treated group.
- the cells were then incubated in phosphate-buffered saline (PBS) containing 2% fetal bovine serum (FBS) at 4°C for 1 hour to minimize non-specific antibody binding.
- PBS phosphate-buffered saline
- FBS fetal bovine serum
- the control antibody and anti- CD20xCD22 bispecific antibody 1 were incubated with the respective cell groups at 4°C for 1 hour to allow antibody binding to the CD20 or CD22 proteins on the cancer cell surface.
- Fig. 1 The experimental results (Fig. 1) showed that anti-CD20xCD22 bispecific antibody 1 exhibited a binding affinity 10.4 times higher than that of the IgG control for SUP-B15 cells, which have low CD20 expression and high CD22 expression.
- anti-CD20xCD22 bispecific antibody 1 showed 9.9 times stronger binding compared to the IgG control.
- Granta-519 cells which express both CD20 and CD22 at high levels
- binding affinity of anti-CD20xCD22 bispecific antibody 1 was 11.2 times higher than that of the IgG control.
- Ramos cells a B-cell non-Hodgkin lymphoma cell line known for CD20 expression, were used, with human IgG used as the control antibody.
- the anti-CD20 antibody CD20_l-CaaX construct and the anti-CD20xCD22 bispecific antibody 1 were first incubated with Ramos cells at 4°C for 1 hour, respectively.
- the experimental groups were then incubated at 37°C for either 2 or 4 hours to induce internalization, while a control group was maintained at 4°C.
- a phycoerythrin (PE)-conjugated secondary antibody (BD, 555787) specific to human IgG was used.
- Fig. 2 showed that, in Ramos cells, the anti-CD20 antibody CD20_l-CaaX construct exhibited internalization rate of 42% after 2-hours and 44% after 4-hours of incubation at 37°C. In contrast, the anti-CD20xCD22 bispecific antibodyl demonstrated an internalization rate of 93% after 2-hours and 95% after 4-hours.
- the anti-CD20xCD22 bispecific antibody showed a more than 50% improvement of internalization rate compared to the anti-CD20 antibody in Ramos cells.
- ADC 1 which comprises an anti-CD20 antibody
- ADC2 Kerbe-into-Hole
- ADC6 Purbody
- ADC7 Cell binding experiments were conducted using a flow cytometer (FACS).
- the anti-CD20 ADC ADC1
- the anti-CD20xCD22 bispecific ADCs ADC 2, 6, and 7
- a control group was maintained at 4°C
- treatment groups were incubated at 37°C for 1 or 3 hours to induce internalization.
- a PE-conjugated secondary antibody BD, 555787 specific to human IgG was used to quantify remaining antibodies on the cell surface.
- anti-CD20xCD22 bispecific ADCs demonstrated an over 50% higher internalization rate than the anti-CD20 ADC in Ramos cells, regardless of platform.
- the cytotoxicity activity of the ADCs against various cancer cell lines was measured.
- the cancer cell lines used were the commercially available human B-cell non-Hodgkin lymphoma cell lines (SU-DHL-8, WSU-DLCL2), and acute lymphoblastic leukemia cell lines (SUP-B15 and Reh).
- the ADCs used for measuring cytotoxicity activity were ADC1, ADC2, ADC6, and ADC7, which were prepared in Preparation Example 3.
- Each cancer cell line was seeded at 5,000 to 20,000 cells per well in a 96-well plate and treated with each ADC at concentrations ranging from 0.128 fM to 50 nM (five-fold serial dilution).
- the number of living cells was quantified using Cell Titer Gio (Promega, G7570) after 96 or 144 hours, and the results are shown in Table 23 below.
- the pyrrolobenzodiazepine-based anti-CD20xCD22 bispecific ADCs demonstrated improved efficacy ranging from 22-fold to 380-fold compared to the pyrrolobenzodiazepine-based anti-CD20 ADC (ADC1).
- the anti-CD20xCD22 bispecific ADCs (ADC2, ADC6, and ADC7) exhibited consistently stronger cytotoxicity effects against cancer cell lines compared to anti-CD20 ADC1.
- the anti-CD20xCD22 ADCs effectively demonstrated cytotoxicity against cancer cells compared to the anti-CD20 ADC regardless of the bispecific antibody platform used.
- the cytotoxicity activity of the ADCs against various cancer cell lines was measured.
- the commercially available human B-cell non-Hodgkin lymphoma cell lines (WSU-DLCL2, WSU-NHL, Mino, and Granta-519 were used for this analysis.
- Each cancer cell line was seeded at 5,000 cells per well in a 96-well plate and treated with each ADC at concentrations ranging from 0.128 fM to 50 nM (five-fold serial dilution). The number of living cells (cell viability) was quantified using Cell Titer Gio (Promega, G7570) after 96 hours of treatment.
- the cytotoxicity evaluation results of the cancer cell lines indicated that ADC2, produced with a two-cell system using the Knob-in-Hole method disclosed herein, and ADC8, produced with a one-cell system using the S-DUAL method disclosed herein, exhibited comparable efficacy across the tested cell lines (WSU-DLCL2, WSU-NHL, Mino, and Granta-519).
- the cytotoxicity activity of drugs (SG2057) and ADCs was measured against various cancer cell lines and normal cells.
- SG2057 has the above structure and was purchased from MedChemExpress (Catalog No. HY-101160).
- the cancer cell lines used were commercially available human B-cell nonHodgkin lymphoma cell lines (Daudi, Ramos, NAMALWA, WSU-NHL, WSU-DLCL2) and acute lymphoblastic leukemia cell lines (SUP-B15 and Reh).
- the normal cell lines used were PBMC (STEMCELL; Catalog No. 70002.2) and HSC (STEMCELL; Catalog No. 70025.2).
- the ADCs tested were ADC 1 and ADC2 prepared in Preparation Example 5.
- Each cancer cell line was seeded at 5,000 to 20,000 cells per well in a 96-well plate and treated with SG2057 at concentrations ranging from 5.12 fM to 2 nM (five-fold serial dilution) or each ADC at concentrations ranging from 0. 128 fM to 50 nM (five-fold serial dilution).
- cells were treated with SG2057 at concentrations of 512 fM to 2 nM (fivefold serial dilution), or ADCs at concentrations of 10 pM to 5,000 nM (five-fold serial dilution).
- cancer cell viability The number of living cells in the cancer cell lines (cancer cell viability) was quantified using Cell Titer Gio (Promega, G7570) after 96 or 144 hours.
- PBMC and HSC were quantified using Cell Titer Gio after 144 hours, and the results are shown in Table 25 and 26 below.
- ADC2 demonstrated 8- to 29-fold weaker cytotoxicity, indicating a reduction in toxicity towards blood cells where CD20 and CD22 are commonly expressed in vivo.
- the cytotoxicity activity of exemplified ADCs against cancer cell lines was measured to observe potential differences in efficacy based on antibody sequence variations.
- the cancer cell line used was the commercially available human B-cell non-Hodgkin lymphoma cell line Ramos.
- the ADCs tested (ADC2, ADC3, ADC4, and ADC5) were prepared in Preparation Example 5.
- Ramos cells were seeded at 5,000 cells per well in a 96-well plate and treated with each ADC at concentrations ranging from 0.128 fMto 50 nM (five-fold serial dilution). The number of living cells was quantified using Cell Titer Gio (Promega, G7570) after 96 hours, and the results are shown in Table 27 below.
- cytotoxicity evaluation results of the cancer cell line as shown in Table 27, indicate that all pyrrolobenzodiazepine-based anti-CD20xCD22bispecific ADCs (ADC2, ADC3, ADC4, and ADC5) showed strong efficacy, regardless of antibody sequences variation.
- the cytotoxicity activity of ADC9 and ADC10, prepared in Preparation Example 5, against cancer cell line was measured to observe potential differences in efficacy based on variations in linker-payload combinations.
- the cancer cell line used was the commercially available human B-cell non-Hodgkin lymphoma cell line Ramos.
- Ramos cells were seeded at 5,000 cells per well in a 96-well plate and treated with each ADC at concentrations ranging from 0.128 fM to 50 nM (five-fold serial dilution).
- the number of living cells was quantified using Cell Titer Gio (Promega, G7570) after 96 hours, and the results are shown in Table 28 below. Table 28.
- anti-CD20xCD22 bispecific ADC conjugated with MC-Val-Ala-PBD (Compound 3) demonstrated potent cytotoxicity regardless of the specific linker used, where the efficacy is comparable to those of ADC2, ADC3, ADC4, and ADC5.
- anti-CD20xCD22 bispecific ADC conjugated with MC-Val-Cit-PAB- MMAE (Compound 2) demonstrated strong efficacy, independent of the specific payload.
- ADC2 prepared in Preparation Example 5 in Sprague Dawley (SD) rats single doses of ADC2 were given at 1.0 mg/kg intravenously into male SD rats (Orientbio, South Korea) at 9 weeks of age. Pharmacokinetics were studied following injection of ADC2 into SD rats. Plasma samples were taken at various time points and stored frozen for analysis. The plasma concentrations of the ADC2 at the indicated time points were measured using a LC-MS/MS analysis method.
- ACN acetonitrile
- sample 250 pL of sample and 50 pL of plasma containing 10 nM Dextromethorphan (internal standard), and the solutions were mixed vigorously using a vortex mixer for 5 minutes. The samples were then spun down at 14,000 rpm, 4 °C for 5 minutes. 100 pL of supernatants were combined with 100 pL of mobile phase A (0.1 % formic acid in water with 5% ACN) and mixed thoroughly. The samples were measured for ADC2 using a LC-MS/MS method (Nexera LC40 (SHIMADZU) and QTRAP4500 (SCIEX)). The PK profiles of ADC2 are presented in Table 29. and Fig. 4. ADC2 showed stable pharmacokinetic profiles in rats.
- pyrrolobenzodiazepine-based anti-CD20xCD22 bispecific ADC2 As shown in Fig. 5 and Table 30, administration of the pyrrolobenzodiazepine-based anti-CD20xCD22 bispecific ADC2 at 0.3 mg/kg resulted in 45.1% tumor growth inhibition compared to the control group (administered with PBS), while the anti-CD20 ADC1 at the same dosage achieved 13.9% tumor growth inhibition. Therefore, ADC2 demonstrated more than a threefold improvement in tumor growth inhibition efficacy compared to ADC1 at the same dosage.
- the pyrrolobenzodiazepine-based anti-CD20xCD22 bispecific drug conjugate ADC2 exhibited dose-dependent tumor growth inhibition at dosages of 0.1, 0.3, and 0.5 mg/kg in Ramos cell xenografts.
- ADC2 at 0.3 mg/kg demonstrated a 49% improvement in tumor growth inhibition.
- ADC2 achieved 3.14 times stronger tumor growth inhibition than Inotuzumab Ozogamicin at 2.5 mg/kg.
- the pyrrolobenzodiazepine-based anti-CD20xCD22 bispecific drug conjugate ADC2 demonstrated dose-dependent tumor growth inhibition at 0.1, 0.3, and 0.5 mg/kg in the WSU-DLCL2 cell CDX model. At 0.3 mg/kg, ADC2 achieved a 51% greater tumor growth inhibition compared to ADC1 at the same dosage.
- ADC2 exhibited strong tumor growth inhibition across all tested doses when compared to the standard R-CHOP therapy, commonly used as the first-line treatment for diffuse large B-cell lymphoma. These results indicate that ADC2 is effective in showing tumor growth inhibition even in CDX models that are resistance to R-CHOP therapy.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Immunology (AREA)
- General Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Organic Chemistry (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- Epidemiology (AREA)
- Genetics & Genomics (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Biochemistry (AREA)
- Molecular Biology (AREA)
- Biophysics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Peptides Or Proteins (AREA)
- Medicinal Preparation (AREA)
Abstract
The present disclosure relates to an antibody-drug conjugate (ADC) comprising a bispecific antibody specifically binding to CD20 and CD22, and compositions and uses of the ADCs for producing a drug for the treatment and/or treatment of diseases, more particularly, hyperproliferative and/or angiogenic diseases, such as cancers (e.g., hematological cancers).
Description
ANTIBODY-DRUG CONJUGATE COMPRISING BISPECIFIC ANTIBODY
SPECIFICALLY BINDING TO CD20 AND CD22 AND USES THEREOF
Related Applications
This application claims the benefit of priority to Korean Patent Application No. KR 10-2023-0155908, filed November 10, 2023, the content of which is hereby incorporated by reference in its entirety.
Background
Cancer refers to a disease caused by abnormally grown lumps due to autonomous overgrowth of body tissues, and is the result of uncontrolled cell growth in various tissues. Tumors in early stage can be removed by surgical and radio-therapeutic measures, and metastasized tumors are generally treated using chemotherapy.
Most chemotherapeutic agents administered parenterally may induce unwanted side effects and even serious toxicity, as a result of systemic administration. Accordingly, the focus of development has been on developing treatments to achieve increased efficacy and/or reduced minimal toxicity/side effects, for example through the selective application of these chemotherapeutic agents in tumor cells or immediately adjacent tissues. There remains a demand for development of antibody-drug conjugates for effective therapeutic methods.
Summary
The present disclosure provides, among other things, antibody-drug conjugates (ADCs) targeting both CD20 and CD22, active metabolites of such ADCs, methods for preparation of such ADCs, and compositions and uses of such ADCs for preventing or treating cancer or proliferative diseases. More particularly, the present invention provides ADCs comprising a bispecific antibody specifically binding to CD20 and CD22, and a pharmaceutical composition comprising the same. In some embodiments, the bispecific anti-CD20xCD22 antibody of the present disclosure has improved cell internalization as compared to antibodies known in the art (e.g., an anti-CD20 antibody).
In certain aspects, the present disclosure provides antibody-drug conjugates comprising a bispecific antibody specifically binding to CD20 and CD22. In certain embodiments, the antibody disclosed herein binds to CD20 and CD22 expressed in a tumor
and may be used to deliver an active agent to the tumor. In certain embodiments, the antibody-drug conjugates disclosed herein have improved stability and/or cell internalization as compared to antibody-drug conjugates known in the art.
In certain aspects, the present disclosure provides conjugates having a structure represented by General Formula I or a pharmaceutically acceptable salt thereof:
[General Formula 1]
Ab-[L-(B)i]m wherein,
Ab is a bispecific antibody comprising a first antigen-binding domain specifically binding to CD20 and a second antigen-binding domain specifically binding to CD22, each L is, independently, a linker, each B is, independently, an active agent, and
1 and m are each independently 1 to 20.
Brief Description of the Drawings
Fig. 1 shows the binding affinity of an exemplified anti-CD20xCD22 bispecific antibody (anti-CD20xCD22 Bispecific Antibody 1) and exemplified anti-CD20, anti-CD22, and control IgG antibodies to cancer cell lines.
Fig. 2 shows the internalization rate of an exemplified anti-CD20xCD22 bispecific antibody (anti-CD20xCD22 Bispecific Antibody 1) and exemplified anti-CD20 and control IgG antibodies in Ramos cells.
Fig. 3 shows the internalization rate of exemplified anti-CD20xCD22 bispecific ADCs (ADC2, 6, and 7) and an exemplified anti-CD20 ADC (ADC1) in Ramos cells.
Fig. 4 shows the plasma concentration of ADC2 at indicated time points in rats.
Fig. 5 shows that in the SU-DHL-8 cell model, ADC2 exhibited better tumor growth inhibition than ADC 1 at the same dose (0.3 mg/kg, QDxl).
Fig. 6 shows that in the Granta-519 cell model, ADC2 exhibited significant tumor growth inhibition compared to the control group.
Fig. 7 shows that in the MINO cell model, ADC2 exhibited significant tumor growth
inhibition compared to the control group.
Fig. 8 shows that in Ramos cell model, ADC2 exhibited dose-dependent tumor growth inhibition. ADC2 exhibited better tumor growth inhibition than an exemplified anti-CD22 ADC (Inotuzumab Ozogamicin) at the same dose.
Fig. 9 shows that in WSU-DLCL2 cell model, ADC2 exhibited dose-dependent tumor growth inhibition. ADC2 exhibited significant tumor growth inhibition than ADC 1 at the same dose (0.3 mg/kg, QDxl) or R-CHOP.
Detailed Description
An antibody-drug conjugate (ADC) is a targeted technology for conjugating a toxin or a drug to an antibody that binds to an antigen, by which the toxin is released in a cell to cause death of cancer cells and the like. The ADC enables a drug to be accurately delivered to target cancer cells while minimally affecting healthy cells, and to be released only under specific conditions, and thus has excellent efficacy compared to antibody therapeutic agents themselves and can remarkably reduce the risk of side effects compared to existing anticancer agents.
The basic structure of these antibody-drug conjugates is “antibody-linker-small molecule drug (toxin)”. In this structure, the linker plays a functional role in linking the antibody and the drug, but in some cases also ensures that the drug is released from the antibody at the appropriate time, for example after reaching target cells. That is, the stability of the linker can play a very important role in the efficacy and safety such as systemic toxicity of an antibody-drug conjugate (Discovery Medicine 2010, 10(53): 329-39).
The use of monoclonal antibodies for cancer treatment has had substantial success. For example, monoclonal antibodies are suitable for target-directed addressing of tumor tissue and tumor cells. Antibody-drug conjugates have become a novel and powerful option for the treatment of lymphomas and solid cancers, and immunomodulatory antibodies also have recently had considerable success in clinical trials. The development of therapeutic antibodies is based on deep understanding of cancer serology, protein engineering technology and the action thereof, mechanisms of resistance, and interactions between immune systems and cancer cells.
Antigens which are expressed on the surface of human cancer cells are defined as a broad range of targets which are over-expressed compared to normal tissues, mutated and selectively expressed. The key challenge is to identify antigens suitable for antibody-based
therapies. These therapeutic agents mediate changes in antigen or receptor function (i.e., function as a stimulant or an antagonist), regulate the immune system through Fc and T cell activation, and exhibit efficacy through the delivery of specific drugs that bind to antibodies targeting specific antigens. Molecular techniques that can alter antibody pharmacokinetics, action function, size and immune stimulation are emerging as key factors in the development of novel antibody-based therapies. Evidence from clinical trials of therapeutic antibodies in cancer patients highlights the importance of approaches for selecting optimized antibodies, including affinity and binding of target antigens and antibodies, selection of an antibody structure, and therapeutic approaches (signaling blockade or immune function).
A CD20 antigen (also referred to as a human B-lymphocyte-restricted differentiation antigen, Bp35) is a hydrophobic transmembrane protein with a molecular weight of approximately 35 kDa, which is located on pre-B and mature B lymphocytes. The antigen is also expressed in 90% or more of B-cell non-Hodgkin’s lymphomas (NHL), but may not be found in hematopoietic stem cells, pro-B cells, normal plasma cells, or other normal tissues. CD20 has been shown to regulate early step(s) in an activation process for cell cycle initiation and differentiation, and possibly act as a calcium ion channel.
CD22 is a 135-kDa B cell-restricted sialoglycoprotein that is expressed on the B-cell surface only during a mature stage of differentiation. The main form of CD22 in humans is CD22 beta, which contains seven immunoglobulin superfamily domains in the extracellular domain. One variant form, CD22 alpha, lacks immunoglobulin superfamily domains 3 and 4. Ligands that bind to human CD22 have been presented to associate with immunoglobulin superfamily domains 1 and 2. In B-cell NHL, CD22 expression ranges from 91 % to 99% in the aggressive and indolent populations, respectively. CD22 may function as both a component of a B-cell activation complex and an adhesion molecule. After binding to natural ligands or antibodies thereof, CD22 is rapidly internalized to provide potent co-stimulatory signals to primary B cells and proapoptotic signals to neonatal B cells.
Without being bound by any particular scientific theory, the present disclosure includes the recognition that compositions containing antibodies (e.g., a bispecific anti-CD20xCD20 antibody), such as antibody-drug conjugates (ADCs), are useful for the treatment of diseases (e.g., cancers). In certain aspects, the present disclosure provides antibody-drug conjugates comprising a bispecific antibody that specifically binds to CD20 and CD22. In certain embodiments, the antibody disclosed herein binds to CD20 and CD22 expressed in a tumor
and may be used to deliver a drug to the tumor. In certain embodiments, the antibody-drug conjugates disclosed herein have improved stability and/or cell internalization as compared to antibody-drug conjugates known in the art. Without being bound by any particular scientific theory, the antibody-drug conjugates disclosed herein enable a drug and/or toxin to be easily, specifically, and efficiently released in cancer cells to maximize efficacy, and enable a drug and/or toxin to stably reach a target cell.
In certain aspects, the present disclosure provides conjugates having a structure represented by General Formula I or a pharmaceutically acceptable salt thereof:
[General Formula 1]
Ab-[L-(B)i]m wherein,
Ab is a bispecific antibody comprising a first antigen-binding domain specifically binding to CD20 and a second antigen-binding domain specifically binding to CD22, each L is, independently, a linker, each B is, independently, an active agent, and 1 and m are each independently 1 to 20.
In some embodiments, L is a cleavable linker.
In some embodiments, a cleavable linker refers to a linker or linker component that links two moieties by covalent linkage, but is degraded to cleave the covalent linkage between the moieties under physiologically relevant conditions. In some embodiments, the cleavable linker is cleaved in vivo more rapidly in an intracellular environment than outside the cell to allow the release of a payload (e.g., an active agent) to occur preferentially inside a targeted cell. The cleavage may be enzymatic or non-enzymatic. The cleavage may release the payload from the antibody without degrading the antibody. The cleavage may allow a part of the linker or linker component to be attached to the payload, or may release the payload without any residue of the linker.
In some embodiments, L is a protease cleavable linker, an acid-cleavable linker, a disulfide linker, or a glycosidase cleavable linker. In some embodiments, L is a protease cleavable linker. In some embodiments, the protease is selected from one or more of a cysteine protease, a metalloprotease, a serine protease, a threonine protease, and an aspartic protease.
A cysteine protease, also known as a thiol protease, is a protease that shares a common catalytic mechanism including a nucleophilic cysteine thiol in a catalytic triad or dyad. A metalloprotease is a protease of which the catalytic mechanism contains metal. Most metalloproteases require zinc, but some use cobalt. The metal ion is coordinated to a protein via three ligands. The serine protease is an enzyme that cleaves peptide bonds in a protein. Serine acts as a nucleophilic amino acid at an active site of the enzyme. The serine protease is divided into two broad categories of chymotrypsin-like (trypsin-like) or subtilisin-like according to a structure. The threonine protease is a protease family that contains a threonine (Thr) residue in the active site. The prototypical member of the enzyme in the family is a catalytic subunit of the proteasome, but acyltransferases have convergently evolved the same active site geometry and mechanism. The aspartic protease is a catalytic type of protease enzyme that uses activated water molecules bound to one or more aspartate residues to catalyze a peptide substrate. Generally, two highly conserved aspartate salts exist in the active site and are optimally activated at acidic pH. Almost all known aspartic proteases are inhibited by pepstatin.
In some embodiments, the protease is a cathepsin. In some embodiments, L comprises 2 or more amino acid residues, e.g., defining the recognition site of the protease. In some embodiments, L comprises 2 to 4 amino acid residues, e.g., defining the recognition site of the protease. In some embodiments, L comprises valine-alanine (Vai-Ala), valine-citrulline (Val- Cit), valine-lysine (Val-Lys), valine-arginine (Val-Arg), phenylalanine-lysine-glycine-proline- leucine -glycine (Phe-Lys-Gly-Pro-Leu-Gly), or alanine-alanine-proline-valine (Ala-Ala-Pro- Val). In some embodiments, L comprises Val-Cit, Vai-Ala, or Gly-Gly-Phe-Gly.
In some embodiments, L is a glycosidase cleavable linker. In some embodiments, the glycosidase is P-glucuronidase or p-galactosidase. In some embodiments, L comprises a P- glucuronosyl or P-galactosyl moiety.
In some embodiments, L comprises any one of the following components: 6- maleimidocaproyl (MC), maleimido propanoyl (MP), valine-citrulline (Val-Cit), alaninephenylalanine (Ala-Phe), lysine -phenylalanine (Lys-Phe), p-aminobenzyloxycarbonyl (PAB), 4-thio-pentanoate (SPP), 4-thio-butyrate (SPDB), 4-(N-maleimidomethyl)cyclo-hexane-l- carboxylate (MCC), maleimidoethyl (ME), 4-thio-2-hydroxysulfonyl-butyrate (2-Sulfo- SPDB), aryl-thiol (PySS), (4-acetyl)aminobenzoate (SIAB), oxylbenzylthio, aminobenzylthio, dioxylbenzylthio, diaminobenzylthio, aminooxylbenzylthio, alkoxy amino (AOA),
ethyleneoxy (EO), 4-methyl-4-dithio-pentanoic acid (MPDP), triazole, dithio, alkyl sulfonyl, alkyl sulfonamide, sulfone-bisamide, phosphodiamide, alkylphosphonamide, phosphinic acid, N-methylphosphonamic acid, N,N’-dimethylphosphon-amic acid, N,N’- dimethylphosphondiamide, hydrazine, acetimidamide, oxime, acetylacetohydrazide, aminoethyl-amine, aminoethyl-aminoethyl-amine, and L- or D-, natural or unnatural peptides containing 1 to 20 amino acids.
In some embodiments, L is covalently bonded to Ab by a thioether, thioester, disulfide, hydrazone, ester, carbamate, carbonate, alkoxy, or amide. In some embodiments, L comprises a peptide further comprising at least one hydrophilic amino acid. In some embodiments, the peptide comprises an amino acid having a side chain having a moiety that bears a charge at neutral pH in aqueous solution (e.g., an amine, guanidine, or carboxyl moiety). In some embodiments, the peptide comprises an amino acid selected from alanine, histidine, arginine, aspartate, asparagine, glutamate, glutamine, glycine, lysine, ornithine, proline, serine, and threonine. In some embodiments, the peptide comprises an amino acid selected from alanine, aspartate, asparagine, glutamate, glutamine, glycine, lysine, ornithine, proline, serine, and threonine.
In some embodiments, L comprises a functional group capable of reacting with a terminal moiety of a particular molecule. Examples of functional groups capable of reacting with a terminal amine include N-hydroxysuccinimide ester, p-nitrophenyl ester, dinitrophenyl ester, pentafluorophenyl ester, carboxylic acid chloride, and carboxylic anhydride. Examples of functional groups capable of reacting with a terminal thiol include pyridyl disulfide, nitropyridyl disulfide, maleimide, haloacetate, methylsulfonphenyl oxadiazole (ODA), carboxylic acid chloride, and carboxylic acid anhydride. Examples of functional groups capable of reacting with a terminal ketone or aldehyde include amines, alkoxyamines, hydrazines, acyloxyamines, and hydrazides. Examples of functional groups capable of reacting with a terminal azide include alkynes.
In some embodiments, L comprises an oxime. In some embodiments, L comprises at least one isoprenyl group. In some embodiments, L comprises at least one succinimide group.
In some embodiments, L is covalently bonded to Ab by a thioether bond and the thioether bond comprises a sulfur atom of a cysteine of Ab. In some embodiments, Ab at the C-terminus comprises an amino acid motif that is recognized by an isoprenoid transferase. In some embodiments, the isoprenoid transferase is famesyl protein transferase (FTase) or
geranylgeranyl transferase (GGTase). In some embodiments, L is covalently bonded to Ab by a thioether bond and the thioether bond comprises a sulfur atom of a cysteine of the amino acid motif. In some embodiments, the amino acid motif comprises a CY iY iX sequence, wherein: C is cysteine; each Y 1 is independently an aliphatic amino acid; and X is selected from glutamine, glutamate, serine, cysteine, methionine, alanine, and leucine. In some embodiments, each Yi is independently selected from alanine, isoleucine, leucine, methionine, and valine. In some embodiments, the amino acid motif comprises a CVIM (SEQ ID NO: 84) or CVLL (SEQ ID NO: 85). In some embodiments, at least one of the 1 to 20 amino acids preceding the amino acid motif is glycine. In some embodiments, the amino acid motif has the sequence GGGGGGGCVIM (SEQ ID NO: 77).
In some embodiments, L comprises a self-immolative moiety. Examples of the self- immolative moiety includes, without limitation, 2 -aminoimidazole -5 -methanol derivatives, heterocyclic PAB analogs, beta-glucuronides, and aromatic compounds electronically similar to a para-aminobenzylcarbamoyl (PAB) group, such as a group of ortho or paraaminobenzylacetals.
In some embodiments, the conjugate has a structure represented by the General Formula II or a pharmaceutically acceptable salt thereof:
[General Formula II]
 wherein,
wherein,
B is the active agent,
G represents a sugar moiety or a glucuronic acid moiety;
R1 and R2 are each independently hydrogen, C1-8 alkyl, or C3-8 cycloalkyl; or R1 and R2 combine to complete a (CYC's) cycloalkyl ring;
W is -*C(O)-, -*C(O)N(R’)-, -*N(R’)C(O)-, -*(CH2)tN(R’)C(O)-, -*C(O)O-, -*S(O2)N(R’)-, -*P(O)(R”)N(R’)-, -*S(O)N(R’)-, or -*P(O2)N(R’)-, wherein the C(O), N, CH2, S, or P marked with an * is directly bonded to the phenyl ring of General Formula II,
R’ and R” are each independently hydrogen, C1-8 alkyl, C3-8 cycloalkyl, C1-8 alkoxy, C1-8 alkylthio, mono- or di-C1-8 alkylamino, Cs-2o heteroaryl or Ce-2o aryl;
Z is each independently C1-8 alkyl, halogen, cyano, or nitro; nl and n2 are each independently 1 to 20; n3 is 0 to 3; and
Y is absent, C1-C50 alkylene, C1-C50 alkenylene, 1-50 atom heterocyclylene, or 1-50 atom heteroalkylene.
In some embodiments, Y comprises a C1-50 alkylene or C1-50 heteroalkylene and further comprises at least one of:
(i) one or more unsaturated bonds;
(ii) a heterocyclylene or a heteroarylene (e.g., a heteroarylene in the alkylene or heteroalkylene chain);
(iii) at least one Ci-2o alkyl; and
(iv) at least one isoprenyl group having a structure represented by General Formula III: [General Formula III]
 wherein n4 is an integer of 1 to 20.
wherein n4 is an integer of 1 to 20.
In some embodiments, R1 and R2 are each independently hydrogen. In some embodiments, n3 is 0.
In some embodiments, the G has a structure represented by the General Formula IV :
[General Formula IV]
 wherein,
wherein,
R3 is hydrogen, alkyl, CH2OR3A, or CO2R3B;
R3A is hydrogen or a hydroxyl protecting group;
RSB is hydrogen or a carboxyl protecting group; and each R4 is independently a hydrogen or a hydroxyl protecting group.
In some embodiments, R3 is-COOH. In some embodiments, each R4 is independently hydrogen. In some embodiments, the W is -*C(O)NR’-.

W is -*C(O)NR’-.
In some embodiments, Y is covalently bonded to Ab by a thioether, thioester, disulfide, hydrazone, ester, carbamate, carbonate, alkoxy, or amide. In some embodiments, Y comprises a peptide further comprising at least one hydrophilic amino acid. In some embodiments, the peptide comprises an amino acid having a side chain having a moiety that bears a charge at neutral pH in aqueous solution (e.g., an amine, guanidine, or carboxyl moiety). In some embodiments, the peptide comprises an amino acid selected from alanine, histidine, arginine, aspartate, asparagine, glutamate, glutamine, glycine, lysine, ornithine, proline, serine, and threonine. In some embodiments, the peptide comprises an amino acid selected from alanine,
aspartate, asparagine, glutamate, glutamine, glycine, lysine, ornithine, proline, serine, and threonine.
In some embodiments, Y comprises an oxime. In some embodiments, the oxygen atom of the oxime is on the side of Y linked to W and the carbon atom of the oxime is on the side of
Y linked to Ab. In some embodiments, the carbon atom of the oxime is on the side of Y linked to W and the oxygen atom of the oxime is on the side of Y linked to Ab.
In some embodiments, Y is a C1-50 heteroalkylene comprising an oxime. In some embodiments, the oxygen atom of the oxime is on the side of Y linked to W, and the carbon atom of the oxime is on the side of Y linked to Ab. In some embodiments, the carbon atom of the oxime is on the side of Y linked to W, and the oxygen atom of the oxime is on the side of
Y linked to Ab. In some embodiments, Y comprises an oxime, and at least one isoprenyl unit covalently bonds the oxime to Ab (e.g., the at least one isoprenyl unit directly or indirectly bonds the oxime to Ab). In some embodiments, Y comprises at least one isoprenyl group.
In some embodiments, Y is covalently bonded to Ab by a thioether bond and the thioether bond comprises a sulfur atom of a cysteine of Ab. In some embodiments, the Ab at the C-terminus comprises an amino acid motif that is recognized by an isoprenoid transferase. In some embodiments, the isoprenoid transferase is famesyl protein transferase (FTase) or geranylgeranyl transferase (GGTase). In some embodiments, Y is covalently bonded to Ab by a thioether bond and the thioether bond comprises a sulfur atom of a cysteine of the amino acid motif. In some embodiments, the amino acid motif comprises a CY iY iX sequence, wherein C is cysteine; each Y 1 is independently an aliphatic amino acid; and X is selected from glutamine, glutamate, serine, cysteine, methionine, alanine, and leucine. In some embodiments, each Yi is independently selected from alanine, isoleucine, leucine, methionine, and valine. In some embodiments, the amino acid motif comprises a CVIM (SEQ ID NO: 84) or CVLL (SEQ ID NO: 85). In some embodiments, at least one of the 1 to 20 amino acids preceding the amino acid motif is glycine. In some embodiments, the amino acid motif has the sequence GGGGGGGCVIM (SEQ ID NO: 77).
In some embodiments, the Y comprises a connecting unit represented by the General Formula V or General Formula VI:
[General Formula V] -(CH2)r(V(CH2)P)q- [General Formula VI]
-(CH2CH2X)W- wherein,
V is a single bond, -O-, -S-, -NR5-, -C(O)NR6-, -NR7C(O)-, -NR8SO2-, or -SO2NR9-;
X is -O-, C1-8 alkylene or -NR5-;
R5 to R9 are each independently hydrogen, C1-6 alkyl, C1-6 alkyl C6-20 aryl, or
C1-6 alkyl-C’3-20 heteroaryl; r is 0 to 10; p is 0 to 10; q is 1 to 20; and w is 1 to 20.
In some embodiments, q is 1 to 10. In some embodiments, r is 1 or 2. In some embodiments, p is 1 or 2. In some embodiments, V is -O-. In some embodiments, q is 1 to 10; r and p are each 1 or 2; and V is -O-. In some embodiments, X is -O-. In some embodiments, X is -O-; and w is 1 to 10.
In some embodiments, L comprises at least one polyethylene glycol monomer represented by
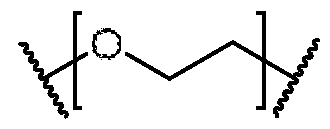 In some embodiments, L comprises a polyethylene glycol oligomer represented by
In some embodiments, L comprises a polyethylene glycol oligomer represented by
 wherein n40 is 2 to 12.
wherein n40 is 2 to 12.
In some embodiments, Y comprises an oxime, and the at least one polyethylene glycol unit covalently bonds the oxime to W.
In some embodiments, Y further comprises a binding unit formed by a reaction between an alkyne and an azide or between an aldehyde or ketone and hydrazine or hydroxylamine.
In some embodiments, Y further comprises a binding unit represented by General Formula Vila, Vllb, Vile, Vlld or Vile:
[General Formula Vila]

[General Formula Vllb]

[General Formula Vile] ]

[General Formula Vile]
 wherein
wherein
Li is each independently a single bond or C1-30 alkylene; and R11 is hydrogen or C1-10 alkyl.
In some embodiments, the conjugate comprises

In some embodiments, the conjugate comprises a structure represented by:

In some embodiments, Y is branched. In some embodiments, Y comprises: i) a branching unit covalently bonded to Ab by a primary linker; ii) a first which couples a first B to the branching unit; and iiia) a second branch which couples a second B to the branching unit; or iiib) a second branch, in which an alkyl or heteroalkyl (e.g., a polyethylene glycol monomer or a polyethylene glycol oligomer) is covalently coupled to the branching unit.
In some embodiments, Y comprises a second branch which couples a second B, via a cleavage group, to the branching unit. In some embodiments, Y comprises a second branch, in which an alkyl or heteroalkyl (e.g., a polyethylene glycol monomer or a polyethylene glycol oligomer) is covalently coupled to the branching unit.

In some embodiments, the branching unit has a structure represented by

 wherein
wherein

R30 is hydrogen or C1-30 alkyl;
R40 is hydrogen or L5-COOR50;
R50 is hydrogen or C1-30 alkyl; and
L2, L3, and L4 are each independently a bond or -Cn'H2n'-; and n' is 1 to 10.
In some embodiments, at least one branched linker is covalently coupled to Ab; and at least two B are covalently coupled to the branched linker.
In some embodiments, the conjugate comprises 1, 2, 3, or 4 branched linkers and each branched linker comprises two B. In some embodiments, the branching unit comprises a lysine residue.
In some embodiments, the conjugate comprises a structure represented by:
 or a pharmaceutically acceptable salt thereof; wherein each B is, independently, an active agent;
nl 1, n22, and n33 are each independently 0 to 30; and
or a pharmaceutically acceptable salt thereof; wherein each B is, independently, an active agent;
nl 1, n22, and n33 are each independently 0 to 30; and
AA is a peptide comprising at least two amino acid residues; and the wavy bond represents a connection to Ab.
Bi-specific Antibody or Antigen-Binding Fragment thereof
The present disclosure provides, among other things, a bispecific antibody comprising a first antigen-binding domain specifically binding to CD20 and a second antigen-binding domain specifically binding to CD22, i.e., an anti-CD20xCD22 bispecific antibody (“Ab”).
In certain aspects, the present disclosure provides antibody-drug conjugates comprising Ab. In certain embodiments, the Ab disclosed herein binds to CD20 and CD22 expressed in a tumor and may be used to deliver a drug to the tumor. In certain embodiments, the antibodydrug conjugates and the Ab disclosed herein have improved stability and/or cell internalization as compared to antibody-drug conjugates or antibodies known in the art.
As described herein, the bispecific antibody according to the present disclosure is a polypeptide comprising one or more complementarity-determining areas or regions (CDRs).
In some embodiments, the CDR is included in a "framework" region, and the framework orients the CDR(s) so that the CDR(s) can have appropriate antigen-binding properties.
In certain embodiments, the antibody according to the present disclosure may consist of a polypeptide of only light chains or only heavy chains including the variable regions shown in Tables 4-9 and 16.
CDR sequences that may be included in the heavy and light chain variable regions of the antibody or antigen-binding fragment thereof according to an embodiment of the present disclosure are shown in Tables 4-9 and 16.
An antibody according to the present disclosure shares certain regions or sequences with other antibodies disclosed herein. In certain embodiments, the constant region of the antibody or antigen-binding fragment thereof may be shared. In certain embodiments, Fc regions may be shared. In certain embodiments, the frame of a variable region may be shared.
The heavy chain variable region and the light chain variable region according to the present disclosure may be linked to at least a part of a human constant region. The selection of a constant region may be determined partially by whether or not antibody-dependent cell- mediated cytotoxicity, antibody-dependent cellular phagocytosis, and/or complement-
dependent cytotoxicity is required. For example, human isotypes IgGl and IgG3 have complement-dependent cytotoxicity, and human isotypes IgG2 and IgG4 do not have such cytotoxicity. In addition, human IgGl and IgG3 induce a cell-mediated effector function stronger than that of human IgG2 and IgG4. The light chain constant region may be lambda or kappa.
A variable region of an immunoglobulin chain generally has the same overall structure and includes a comparatively conserved framework region (FR) linked by three hypervariable regions called "complementarity determining areas or regions or domains" or complementarity determining regions (CDRs). The CDRs of a variable region derived from each chain including a heavy chain/light chain pair are typically aligned by a framework region to form a structure specifically binding to a specific epitope of a target protein. These factors of naturally occurring light chain and heavy chain variable regions are typically included from the N-terminus to the C-terminus in the following order: FR1, CDR1, FR2, CDR2, FR3, CDR3, and FR4. The position of amino acid sequences corresponding to each variable region may be determined by Kabat (Kabat et al., (1983) U.S. Dept, of Health and Human Services, "Sequences of Proteins of Immunological Interest"), Chothia (Chothia and Lesk, J. Mol. Biol. 196:901-917 (1987)) or in a manner related to the OPAL library (Hye Young Yang et. al., 2009 Mol. Cells 27: 225). The CDRs determined by each definition, when compared to each other, may be subsets which overlap or where one includes another. Those of ordinary skill in the art will be readily able to easily select CDR sequences according to the definitions above, given a variable region sequence of an antibody.
In certain embodiments, amino acid sequences of CDRs are defined according to Kabat definition. However, it is well known to those skilled in the art that CDRs of an antibody can be defined in the art according to a variety of methods, such as Chothia definition based on the location of a structural loop region (Al-Lazikani, B et al., J Mol Biol 273: 927-48 (1997)), and Kabat definition based on sequence variability (Kabat et al., “Sequences of proteins of immunological interest”, fifth edition, National Institutes of Health, Bethesda, MD. (1991)).
In certain embodiments, amino acid residues in variable region sequences may also be determined using a Combined definition that incorporates both Kabat definition and Chothia definition. The Combined definition refers to the combination of the ranges of Kabat definition and Chothia definition. It should be understood by those skilled in the art that unless otherwise specified, the terms “CDR” and “complementarity determining region” of a given antibody or
region thereof (e.g., a variable region) should be understood to encompass the complementarity determining region as defined according to any of the embodiments described in the present disclosure.
Although the scope of protection claimed in the claims of the present disclosure is based on the sequences defined according to Kabat definition, amino acid sequences defined according to other CDRs definitions should also fall within the scope of protection of the present disclosure.
In certain embodiments, the antibody according to the present disclosure is a humanized antibody. A humanized antibody refers to any antibody in which the constant region of a nonhuman antibody is completely substituted with a human form of the constant region, and at least a portion of the variable region of a non-human antibody, except for the three loops of an amino acid sequence outside each variable region that binds to a target structure, is completely or partially substituted with the corresponding portion of a human antibody. In certain embodiments, the antibody according to the present disclosure is a human antibody.
Certain mutations may be introduced to the framework region to enhance the stability of antibodies while maintaining their antigen binding activity. Stabilization of therapeutic antibodies can result in improved serum half-life, lower dosage requirements, reduced sideeffects, improved shelf-life and reduced shipping and storage costs.
In certain embodiments, the present disclosure discloses one or more amino acid sequences having substantial sequence identity to one or more amino acid sequences disclosed herein. Substantial identity means that the effects disclosed herein are maintained in the presence of sequence variations. In certain embodiments, the amino acid sequence has about 90% identity, about 95% identity, or about 99% identity to the heavy chain variable regions shown in Tables 4-9 and 16. In another embodiment, the amino acid sequence has about 90% identity, about 95% identity, or about 99% identity to the light chain variable regions shown in Tables 4-9 and 16. For example, in the case of variants exhibiting 90% identity, 95% identity, or 99% identity to the sequence of the antibody or antigen-binding fragment thereof according to the present disclosure, any mutation occurs in the framework of the variable region rather than the CDRs.
In certain embodiments, a nucleic acid encoding the antibody or fragment thereof according to the present disclosure is a nucleic acid encoding a full-length antibody including the CDRs disclosed herein, the variable region including the CDRs, and the variable region,
and the constant region. Once the amino acid sequence is determined, the nucleic acid sequence may be easily determined in consideration of a known reverse transcription program, codon usage, and the like.
In some embodiments, Ab comprises a first constant region linked to the first antigenbinding domain and a second constant region linked to the second antigen-binding domain, and wherein the first constant region and the second constant region each comprises a light chain constant region CL domain and heavy chain constant region CHI, CH2, and CH3 domains.
In some embodiments, Ab is a chimeric antibody, a humanized antibody, or a human antibody.
In some embodiments, Ab is of IgA, IgG, IgM, IgE, or IgD isotype. In some embodiments, Ab is of IgG isotype (e.g., IgGl, IgG2, IgG3, and IgG4).
In some embodiments, Ab comprises LALA mutations in the first and/or the second heavy chain constant regions. In some embodiments, the LALA mutations comprise L234A and L235A according to EU numbering convention.
Knob-in-Hole
In some embodiments, a bispecific antibody of the present disclosure is produced by the methods described in U.S. Patent No. 9,637,557B2, the entirety of which is incorporated herein by reference.
A bispecific antibody according to some embodiments may be a heteromultimer.
As used herein, the term “heteromultimer” refers to a protein multimer consisting of a plurality types of polypeptides, wherein the polypeptides may associate with each other. More specifically, the “heteromultimer” is a molecule having at least a first polypeptide and a second polypeptide, wherein the second polypeptide has at least one amino acid residue different from the first polypeptide in the amino acid sequence.
In some embodiments, the heteromultimer may include at least one amino acid substitution (e.g., to form a knob-in-hole structure) within each domain (e.g., a CH3 domain) of the first constant region and the second constant region.
In this method, the interaction surface of the two CH3 domains is modified so that the heterodimerization of all heavy chains containing the two CH3 domains is increased. One of the two CH3 domains (of the two heavy chains) may be a “knob” domain and the other may be a “hole” domain. The introduction of a disulfide bridge stabilizes the heterodimer
(Merchant, A..M., et al, Nature Biotech 16 (1998) 677-681; Atwell, S., Ridgway, J.B., Wells, J.A., Carter, P., J Mol Biol 270 (1997) 26-35) and increases the yield.
The bispecific antibody of the present disclosure may be characterized in that the CH3 domain of one heavy chain and the CH3 domain of the other heavy chain form an interface beyond the original interface between the antibody CH3 domains; wherein the interface is modified to facilitate the formation of a bivalent, bispecific antibody, and the modification is characterized as follows: a) the CH3 domain of one heavy chain (e.g., of the first constant region) is modified such that, within the original interface of the CH3 domain of one heavy chain that contacts the original interface of the CH3 domain of the other heavy chain (e.g., of the second constant region) in the bispecific antibody, an amino acid residue is replaced with an amino acid residue having a larger side chain volume to generate a ridge (“knob”) within the interface of the CH3 domain of one heavy chain that is positionable in a space within the interface of the CH3 domain of the other heavy chain, and b) the CH3 domain of the other heavy chain (e.g., of the second constant region) is modified such that, within the original interface of the CH3 domain of the other heavy chain that contacts the original interface of the CH3 domain of one heavy chain (e.g., the first constant region) in the bispecific antibody, an amino acid residue is replaced with an amino acid residue having a smaller side chain volume to generate a space (“hole”) within the interface of the CH3 domain of the other heavy chain that is positionable in a ridge within the interface of the CH3 domain of one heavy chain.
Preferably, the amino acid residue having the larger side chain volume is selected from arginine (R), phenylalanine (F), tyrosine (Y), and tryptophan (W).
Preferably, the amino acid residue having the smaller side chain volume is selected from alanine (A), serine (S), threonine (T), and valine (V).
In some embodiments, both the CH3 domains may be further modified to introduce cysteine (C) as an amino acid into a corresponding position in each CH3 domain, thereby forming a disulfide bridge between the two CH3 domains.
In some preferred embodiments, the bispecific antibody includes, according to the EU index of Kabat, a T366W mutation in the CH3 domain of the “knob chain” (e.g., the CH3 domain of the first constant region or the second constant region) and T366S, L368A, and/or Y407V mutations in the CH3 domain of the “hole chain” (e.g., the CH3 domain of the first
constant region or the second constant region). For example, additional interchain disulfide bridges between the CH3 domains may also be used by introducing a Y349C mutation in the CH3 domain of the “knob chain” and an E356C mutation or S354C mutation in the CH3 domain of the “hole chain” (Merchant, A.M., et al., Nature Biotech. 16 (1998) 677-681). Therefore, in other preferred embodiments, the bispecific antibody includes Y349C and T366W mutations in one of the two CH3 domains and E356C, T366S, L368A, and Y407V mutations in the other of the two CH3 domains, or the bispecific antibody includes Y349C and T366W mutations in one of the two CH3 domains and S354C, T366S, L368A, and Y407V mutations in the other of the two CH3 domains (wherein an additional Y349C mutation in one CH3 domain and an additional E356C or S354C mutation in the other CH3 domain form an interchain disulfide bridge).
In some embodiments, the bispecific antibody includes a T366W mutation in the CH3 domain of the “knob chain” and T366S, L368A, and Y407V mutations in the CH3 domain of the “hole chain”, and further includes a R409D; K370E mutation in the CH3 domain of the “knob chain” and a D399K; E357K mutation in the CH3 domain of the “hole chain”.
In other preferred embodiments, the bispecific antibody includes Y349C and T366W mutations in one of the two CH3 domains and S354C, T366S, L368A, and Y407V mutations in the other of the two CH3 domains, or the bispecific antibody includes Y349C and T366W mutations in one of the two CH3 domains and S354C, T366S, L368A, and Y407V mutations in the other of the two CH3 domains and further includes a R409D; K370E mutation in the CH3 domain of the “knob chain” and a D399K; E357K mutation in the CH3 domain of the “hole chain”.
Therefore, in some embodiments, the bispecific antibody of the present disclosure may have a knob-in-hole structure formed by substituting an amino acid residue in the CH3 domain of the first constant region with an amino acid residue having a larger side chain size, and substituting an amino acid residue in the CH3 domain of the second constant region with an amino acid residue having a smaller side chain size; or substituting an amino acid residue in the CH3 domain of the second constant region with an amino acid residue having a larger side chain size, and substituting an amino acid residue in the CH3 domain of the first constant region with an amino acid residue having a smaller side chain size.
In some embodiments, the bispecific antibody of the present disclosure may include T366S, L368A, and Y407V according to the Kabat EU index in the CH3 domain of the first
constant region, and T366W according to the Kabat EU index in the CH3 domain of the second constant region; or include T366S, L368A, and Y407V according to the Kabat EU index in the CH3 domain of the second constant region, and T366W according to the Kabat EU index in the CH3 domain of the first constant region.
In some embodiments, at least one amino acid residue in the CH3 domain of the first heavy chain constant region is substituted with an amino acid residue having a larger side chain size, and at least one amino acid residue in the CH3 domain of the second heavy chain constant region is substituted with an amino acid residue having a smaller side chain size; or at least one amino acid residue in the CH3 domain of the second heavy chain constant region is substituted with an amino acid residue having a larger side chain size, and at least one amino acid residue in the CH3 domain of the first heavy chain constant region is substituted with an amino acid residue having a smaller side chain size.
In some embodiments, the amino acid residue having a larger side chain size is selected from arginine, phenylalanine, tyrosine, and tryptophan; and the amino acid residue having a smaller side chain size is selected from alanine, serine, threonine, and valine.
In some embodiments, the CH3 domain of the first heavy chain constant region comprises amino acid substitutions at positions T366, L368, and Y407 according to EU numbering, and the CH3 domain of the second heavy chain constant region comprises amino acid substitutions at position T366 according to EU numbering; or the CH3 domain of the second heavy chain constant region comprises amino acid substitutions at positions T366, L368, and Y407 according to EU numbering, and the CH3 domain of the first heavy chain constant region comprises amino acid substitutions at position T366 according to EU numbering.
In some embodiments, the CH3 domain of the first heavy chain constant region comprises T366S, L368A, and Y407V according to EU numbering, and the CH3 domain of the second heavy chain constant region comprises T366W according to EU numbering; or the CH3 domain of the second heavy chain constant region comprises T366S, L368A, and Y407V according to EU numbering, and the CH3 domain of the first heavy chain constant region comprises T366W according to EU numbering.
In some embodiments, the CH3 domain of each of the first and the second heavy chain constant regions further comprises at least one substitution of an amino acid residue to a cysteine residue.
In some embodiments, (a) the CH3 domain of the first heavy chain constant region comprises E356C, T366S, L368A, and Y407V according to EU numbering, and the CH3 domain of the second heavy chain constant region comprises Y349C and T366W according to EU numbering; or the CH3 domain of the second heavy chain constant region comprises E356C, T366S, L368A, and Y407V according to EU numbering, and the CH3 domain of the first heavy chain constant region comprises Y349C and T366W according to EU numbering; or (b) the CH3 domain of the first heavy chain constant region comprises S354C, T366S, L368A, and Y407V according to EU numbering, and the CH3 domain of the second heavy chain constant region comprises Y349C and T366W according to EU numbering; or the CH3 domain of the second heavy chain constant region comprises S354C, T366S, L368A, and Y407V according to EU numbering, and the CH3 domain of the first heavy chain constant region comprises Y349C and T366W according to EU numbering.
In some embodiments, (a) the CH3 domain of the first heavy chain constant region comprises E356C, E357K, T366S, L368A, D399K, and Y407V according to EU numbering, and the CH3 domain of the second heavy chain constant region comprises Y349C, T366W, K370E, and R409D according to EU numbering; or the CH3 domain of the second heavy chain constant region comprises E356C, E357K, T366S, L368A, D399K, and Y407V according to EU numbering, and the CH3 domain of the first heavy chain constant region comprises Y349C, T366W, K370E, and R409D according to EU numbering; or (b) the CH3 domain of the first heavy chain constant region comprises S354C, E357K, T366S, L368A, D399K, and Y407V according to EU numbering, and the CH3 domain of the second heavy chain constant region comprises Y349C, T366W, K370E, and R409D according to EU numbering; or the CH3 domain of the second heavy chain constant region comprises S354C, E357K, T366S, L368A, D399K, and Y407V according to EU numbering, and the CH3 domain of the first heavy chain constant region comprises Y349C, T366W, K370E, and R409D according to EU numbering.
Pentambody
In some embodiments, a bispecific antibody of the present disclosure is produced by the methods described in U.S. Patent No. 11,498,977B2, the entirety of which is incorporated herein by reference.
In some embodiments, the bispecific antibody may include one or more amino acid substitutions within the CH3 domains of each of the first constant region and the second constant region. The first constant region and the second constant region may include at least 5 amino acid substitutions at the following positions according to the Kabat EU index within the CH3 domain: 1) positions 366 and 399 within the first constant region (or the second constant region) and positions 351, 407, and 409 within the second constant region (or the first constant region); or 2) positions 366 and 409 within the first constant region (or the second constant region) and positions 351, 399, and 407 within the second constant region (or the first constant region).
Specifically, the bispecific antibody may include amino acid substitutions at positions T366 and D399 according to the Kabat EU index within the CH3 domain of the first constant region, and amino acid substitutions at positions L351, Y407, and K409 according to the Kabat EU index within the CH3 domain of the second constant region; or include amino acid substitutions at positions T366 and D399 according to the Kabat EU index in the CH3 domain of the second constant region, and amino acid substitutions at positions L351, Y407 and K409 according to the Kabat EU index in the CH3 domain of the first constant region.
In some embodiments, the first constant region and the second constant region include at least one of the following substitutions according to the Kabat EU index in the CH3 domain: a) glycine, tyrosine, valine, proline, aspartic acid, glutamic acid, lysine, or tryptophan at L351 in the second constant region (or the first constant region); b) leucine, proline, tryptophan, or valine at T366 in the first constant region (or the second constant region); c) cysteine, asparagine, isoleucine, glycine, arginine, threonine, or alanine at D399 in the first constant region and/or the second constant region; d) leucine, alanine, proline, phenylalanine, threonine, or histidine at Y407 in the second constant region (or the first constant region); and e) cysteine, proline, serine, phenylalanine, valine, glutamic acid, or arginine at K409 in the first constant region and/or the second constant region.
The combinations of the amino acid substitutions are all possible within the mentioned substitutions, and specifically, the bispecific antibody includes T366L and D399R according to the Kabat EU index in the CH3 domain of the first constant region, and L35 IE, Y407L and K409Y according to the Kabat EU index in the CH3 domain of the second constant region; or may include T366L and D399R according to the Kabat EU index in the CH3 domain of the second constant region, and L351E, Y407L and K409Y according to the Kabat EU index in the CH3 domain of the first constant region, but is not limited thereto.
In some embodiments, (a) the CH3 domain of the first heavy chain constant region comprises amino acid substitutions at positions T366 and D399 according to EU numbering, and the CH3 domain of the second heavy chain constant region comprises amino acid substitutions at positions L351, Y407, and K409 according to EU numbering; or the CH3 domain of the second heavy chain constant region comprises amino acid substitutions at positions T366 and D399 according to EU numbering, and the CH3 domain of the first heavy chain constant region comprises amino acid substitutions at positions L351, Y407, and K409 according to EU numbering; or (b) the CH3 domain of the first heavy chain constant region comprises amino acid substitutions at positions T366 and K409 according to EU numbering, and the CH3 domain of the second heavy chain constant region comprises amino acid substitutions at positions L351, D399, and Y407 according to EU numbering; or the CH3 domain of the second heavy chain constant region comprises amino acid substitutions at positions T366 and K409 according to EU numbering, and the CH3 domain of the first heavy chain constant region comprises amino acid substitutions at positions L351, D399, and Y407 according to EU numbering.
In some embodiments, the substituted amino acid at T366 is selected from leucine, proline, tryptophan, and valine; the substituted amino acid at D399 is selected from cysteine, asparagine, isoleucine, glycine, arginine, threonine, and alanine; the substituted amino acid at L351 is selected from glycine, tyrosine, valine, proline, aspartic acid, glutamic acid, lysine, and tryptophan; the substituted amino acid at Y407 is selected from leucine, alanine, proline, phenylalanine, threonine, and histidine; and the substituted amino acid at K409 is selected from cysteine, proline, serine, phenylalanine, valine, glutamic acid, and arginine.
In some embodiments, (a) the CH3 domain of the first heavy chain constant region comprises amino acid substitutions T366L and D399R according to EU numbering, and the CH3 domain of the second heavy chain constant region comprises amino acid substitutions
L351E, Y407L and K409V according to EU numbering; or the CH3 domain of the second heavy chain constant region comprises amino acid substitutions T366L and D399R according to EU numbering, and the CH3 domain of the first heavy chain constant region comprises amino acid substitutions L351E, Y407L and K409V according to EU numbering; or (b) the CH3 domain of the first heavy chain constant region comprises amino acid substitutions T366L and D399C according to EU numbering, and the CH3 domain of the second heavy chain constant region comprises amino acid substitutions L351G, Y407L and K409C according to EU numbering; or the CH3 domain of the second heavy chain constant region comprises amino acid substitutions T366L and D399C according to EU numbering, and the CH3 domain of the first heavy chain constant region comprises amino acid substitutions L351G, Y407L and K409C according to EU numbering; or (c) the CH3 domain of the first heavy chain constant region comprises amino acid substitutions T366L and D399C according to EU numbering, and the CH3 domain of the second heavy chain constant region comprises amino acid substitutions L351Y, Y407A and K409P according to EU numbering; or the CH3 domain of the second heavy chain constant region comprises amino acid substitutions T366L and D399C according to EU numbering, and the CH3 domain of the first heavy chain constant region comprises amino acid substitutions L351Y, Y407A and K409P according to EU numbering; or (d) the CH3 domain of the first heavy chain constant region comprises amino acid substitutions T366P and D399N according to EU numbering, and the CH3 domain of the second heavy chain constant region comprises amino acid substitutions L351V, Y407P and K409S according to EU numbering; or the CH3 domain of the second heavy chain constant region comprises amino acid substitutions T366P and D399N according to EU numbering, and the CH3 domain of the first heavy chain constant region comprises amino acid substitutions L351V, Y407P and K409S according to EU numbering; or (e) the CH3 domain of the first heavy chain constant region comprises amino acid substitutions T366W and D399G according to EU numbering, and the CH3 domain of the second heavy chain constant region comprises amino acid substitutions L351D, Y407P and K409S according to EU numbering; or the CH3 domain of the second heavy chain constant region comprises amino acid substitutions T366W and D399G according to EU numbering, and the CH3 domain of the first heavy chain constant region comprises amino acid substitutions L35 ID, Y407P and K409S according to EU numbering; or (f) the CH3 domain of the first heavy chain constant region comprises amino acid substitutions T366P and D399I according to EU numbering, and the CH3 domain of the second heavy chain
constant region comprises amino acid substitutions L35 IP, Y407F and K409F according to EU numbering; or the CH3 domain of the second heavy chain constant region comprises amino acid substitutions T366P and D399I according to EU numbering, and the CH3 domain of the first heavy chain constant region comprises amino acid substitutions L35 IP, Y407F and K409F according to EU numbering; or (g) the CH3 domain of the first heavy chain constant region comprises amino acid substitutions T366V and D399T according to EU numbering, and the CH3 domain of the second heavy chain constant region comprises amino acid substitutions L351K, Y407T and K409Q according to EU numbering; or the CH3 domain of the second heavy chain constant region comprises amino acid substitutions T366V and D399T according to EU numbering, and the CH3 domain of the first heavy chain constant region comprises amino acid substitutions L351K, Y407T and K409Q according to EU numbering; or (h) the CH3 domain of the first heavy chain constant region comprises amino acid substitutions T366L and D399A according to EU numbering, and the CH3 domain of the second heavy chain constant region comprises amino acid substitutions L351W, Y407H and K409R according to EU numbering; or the CH3 domain of the second heavy chain constant region comprises amino acid substitutions T366L and D399A according to EU numbering, and the CH3 domain of the first heavy chain constant region comprises amino acid substitutions L351W, Y407H and K409R according to EU numbering.
In some embodiments, the CH3 domain of the first heavy chain constant region comprises amino acid substitutions T366L and D399R, and the CH3 domain of the second heavy chain constant region comprises amino acid substitutions L351E, Y407L and K409Y; or the CH3 domain of the second heavy chain constant region comprises amino acid substitutions T366L and D399R, and the CH3 domain of the first heavy chain constant region comprises amino acid substitutions L35 IE, Y407L and K409Y.
CrossMab
In some embodiments, a bispecific antibody of the present disclosure is produced by the methods described in U.S. Patent No. 8,242,247B2, the entirety of which is incorporated herein by reference.
In some embodiments, the bispecific antibody includes a first constant region linked to a first antigen-binding domain; and a second constant region linked to a second antigen-binding domain, wherein the first constant region and the second constant region may each include a
light chain constant domain and CHI, CH2, and CH3 domains of a heavy chain constant region of an antibody.
The bispecific antibody of the present disclosure may be improved by replacing a specific domain in a pair of heavy and light chains (HC/LC). For example, the light chain constant domain and the CHI domain of the first constant region may be replaced with each other, and/or the light chain constant domain and the CH 1 domain of the second constant region may be replaced with each other.
In some embodiments, the variable heavy chain domain VH of the first constant region (or the second constant region) may be replaced with the variable light chain domain VL.
In some embodiments, the variable heavy chain domain VH and the heavy chain constant domain CHI of the first constant region (or the second constant region) may be replaced with light chain domains.
The structure described above may provide additional advantages in terms of the preparation and efficacy of the anti-CD20xCD22 bispecific antibody-drug conjugate of the present disclosure.
In some embodiments, (a) CL and CHI domains from the first constant region are replaced by each other; or CL and CHI domains from the second constant region are replaced by each other; or (b) VH and VL domains from the first antigen-binding domain are replaced by each other; or VH and VL domains from the second antigen-binding domain are replaced by each other; or (c) VH and CHI domains and VL and CL domains from the first constant region and the first antigen-binding domain are replaced by each other; or VH and CHI domains and VL and CL domains from the second constant region and the second antigenbinding domain are replaced by each other.
In some embodiments, CL and CHI domains from the first constant region are replaced by each other; or CL and CHI domains from the second constant region are replaced by each other.
S-DUAL™
In some embodiments, a bispecific antibody of the present disclosure is produced by the methods described in EP Patent Application No. 4286408A1, the entirety of which is incorporated herein by reference.
With a unique asymmetric design, S-DUAL™may effectively separate and analyze the bispecific antibody as impurities from the improper pairings form different sizes. It also applies “knob-in-hole” technique to avoid mispairing between two heavy chains and light chains. S- DUAL™ has a unique asymmetrical structure that ensures high binding affinity among chains to produce high titer and purity for optimized manufacturability.
In some embodiments, the bispecific antibody of the present disclosure may have an asymmetrical structure between the first polypeptide and the second polypeptide. Specifically, a CH3 dimer may be further included between the first antigen-binding domain and the CHI domain or the light chain constant domain of the first constant region, or a CH3 dimer may be further included between the second antigen-binding domain and the CHI domain or the light chain constant domain of the second constant region. By further introducing the CH3 dimer into one of the two Fab regions, the two arms of the Fab region may form an asymmetrical shape.
Therefore, the bispecific antibody according to some embodiments may further include the knob-in-hole structure between the first antigen-binding domain and the CHI domain of the first constant region; or further include the knob-in-hole structure between the second antigen-binding domain and the CHI domain of the second constant region.
In some embodiments, the first constant region and the second constant region have an asymmetrical structure to each other. In some embodiments, a knob-in-hole structure is further included between the first antigen-binding domain and the CHI domain of the first constant region; or a knob-in-hole structure is further included between the second antigen-binding domain and the CHI domain of the second constant region. In some embodiments, the knobin-hole structure comprises a dimer of CH3 domains.
The structures of the bispecific antibody disclosed herein may be combined with each other within a compatible aspect. For example, in some preferred embodiments, the structures prepared by the Knob-and-Hole or the Pentambody methods may be combined with the structures prepared by the CrossMab or S-DUAL™ methods. In some embodiments, the combination of the structures may provide additional advantages in terms of the preparation and efficacy of the anti-CD20xCD22 bispecific antibody-drug conjugate of the present disclosure.
Antibody and Antigen-Binding Fragment Thereof
In some embodiments, the first antigen-binding domain specifically binding to CD20 comprises a first heavy chain variable region and a first light chain variable region, and the second antigen-binding domain specifically binding to CD22 comprises a second heavy chain variable region and a second light chain variable region, wherein
(a) the first heavy chain variable region comprises a heavy chain CDR1 comprising the amino acid sequence of SEQ ID NO: 2, a heavy chain CDR2 comprising the amino acid sequence of SEQ ID NO: 4, and a heavy chain CDR3 comprising the amino acid sequence of SEQ ID NO: 6; the first light chain variable region comprises a light chain CDR1 comprising the amino acid sequence of SEQ ID NO: 9, a light chain CDR2 comprising the amino acid sequence of SEQ ID NO: 11, and a light chain CDR3 comprising the amino acid sequence of SEQ ID NO: 13; the second heavy chain variable region comprises a heavy chain CDR1 comprising the amino acid sequence of SEQ ID NO: 40, a heavy chain CDR2 comprising the amino acid sequence of SEQ ID NO: 42, and a heavy chain CDR3 comprising the amino acid sequence of SEQ ID NO: 44; and the second light chain variable region comprises a light chain CDR1 comprising the amino acid sequence of SEQ ID NO: 47, a light chain CDR2 comprising the amino acid sequence of SEQ ID NO: 49, and a light chain CDR3 comprising the amino acid sequence of SEQ ID NO: 51;
(b) the first heavy chain variable region comprises a heavy chain CDR1 comprising the amino acid sequence of SEQ ID NO: 2, a heavy chain CDR2 comprising the amino acid sequence of SEQ ID NO: 4, and a heavy chain CDR3 comprising the amino acid sequence of SEQ ID NO: 6; the first light chain variable region comprises a light chain CDR1 comprising the amino acid sequence of SEQ ID NO: 9, a light chain CDR2 comprising the amino acid sequence of SEQ ID NO: 11, and a light chain CDR3 comprising the amino acid sequence of SEQ ID NO: 13; the second heavy chain variable region comprises a heavy chain CDR1 comprising the amino acid sequence of SEQ ID NO: 58, a heavy chain CDR2 comprising the amino acid sequence of SEQ ID NO: 60, and a heavy chain CDR3 comprising the amino acid sequence of SEQ ID NO: 62; and the second light chain variable region comprises a light chain CDR1 comprising the amino acid sequence of SEQ ID NO: 65, a light
chain CDR2 comprising the amino acid sequence of SEQ ID NO: 67, and a light chain CDR3 comprising the amino acid sequence of SEQ ID NO: 69;
(c) the first heavy chain variable region comprises a heavy chain CDR1 comprising the amino acid sequence of SEQ ID NO: 22, a heavy chain CDR2 comprising the amino acid sequence of SEQ ID NO: 24, and a heavy chain CDR3 comprising the amino acid sequence of SEQ ID NO: 26; the first light chain variable region comprises a light chain CDR1 comprising the amino acid sequence of SEQ ID NO: 29, a light chain CDR2 comprising the amino acid sequence of SEQ ID NO: 31, and a light chain CDR3 comprising the amino acid sequence of SEQ ID NO: 33; the second heavy chain variable region comprises a heavy chain CDR1 comprising the amino acid sequence of SEQ ID NO: 40, a heavy chain CDR2 comprising the amino acid sequence of SEQ ID NO: 42, and a heavy chain CDR3 comprising the amino acid sequence of SEQ ID NO: 44; and the second light chain variable region comprises a light chain CDR1 comprising the amino acid sequence of SEQ ID NO: 47, a light chain CDR2 comprising the amino acid sequence of SEQ ID NO: 49, and a light chain CDR3 comprising the amino acid sequence of SEQ ID NO: 51; or
(d) the first heavy chain variable region comprises a heavy chain CDR1 comprising the amino acid sequence of SEQ ID NO: 22, a heavy chain CDR2 comprising the amino acid sequence of SEQ ID NO: 24, and a heavy chain CDR3 comprising the amino acid sequence of SEQ ID NO: 26; the first light chain variable region comprises a light chain CDR1 comprising the amino acid sequence of SEQ ID NO: 29, a light chain CDR2 comprising the amino acid sequence of SEQ ID NO: 31, and a light chain CDR3 comprising the amino acid sequence of SEQ ID NO: 33; the second heavy chain variable region comprises a heavy chain CDR1 comprising the amino acid sequence of SEQ ID NO: 58, a heavy chain CDR2 comprising the amino acid sequence of SEQ ID NO: 60, and a heavy chain CDR3 comprising the amino acid sequence of SEQ ID NO: 62; and the second light chain variable region comprises a light chain CDR1 comprising the amino acid sequence of SEQ ID NO: 65, a light chain CDR2 comprising the amino acid sequence of SEQ ID NO: 67, and a light chain CDR3 comprising the amino acid sequence of SEQ ID NO: 69.
In some embodiments, (a) the VH domain of the first antigen-binding domain comprises: the amino acid sequence of SEQ ID NO: 15; at least 90% sequence identity to the
amino acid sequence of SEQ ID NO: 15; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 15; the VL domain of the first antigen-binding domain comprises: the amino acid sequence of SEQ ID NO: 16; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 16; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 16; the VH domain of the second antigen-binding domain comprises: the amino acid sequence of SEQ ID NO: 53; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 53; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 53; and the VL domain of the second antigen-binding domain comprises: the amino acid sequence of SEQ ID NO: 54; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 54; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 54;
(b) the VH domain of the first antigen-binding domain comprises: the amino acid sequence of SEQ ID NO: 15; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 15; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 15; the VL domain of the first antigen-binding domain comprises: the amino acid sequence of SEQ ID NO: 16; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 16; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 16; the VH domain of the second antigen-binding domain comprises: the amino acid sequence of SEQ ID NO: 71; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 71; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 71; and the VL domain of the second antigen-binding domain comprises: the amino acid sequence of SEQ ID NO: 72; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 72; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 72;
(c) the VH domain of the first antigen-binding domain comprises: the amino acid sequence of SEQ ID NO: 35; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 35; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 35; the VL domain of the first antigen-binding domain comprises: the amino acid sequence of SEQ ID NO: 36; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 36; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 36; the VH domain of the second antigen-binding domain comprises: the amino acid sequence of SEQ ID NO: 53; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 53; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 53; and the VL domain of the second antigen-binding domain comprises: the amino acid sequence of SEQ ID NO: 54; at least
90% sequence identity to the amino acid sequence of SEQ ID NO: 54; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 54; or
(d) the VH domain of the first antigen-binding domain comprises: the amino acid sequence of SEQ ID NO: 35; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 35; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 35; the VL domain of the first antigen-binding domain comprises: the amino acid sequence of SEQ ID NO: 36; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 36; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 36; the VH domain of the second antigen-binding domain comprises: the amino acid sequence of SEQ ID NO: 71; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 71; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 71; and the VL domain of the second antigen-binding domain comprises: the amino acid sequence of SEQ ID NO: 72; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 72; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 72.
In some embodiments, (a) the VH domain of the first antigen-binding domain comprises the amino acid sequence of SEQ ID NO: 15; the VL domain of the first antigenbinding domain comprises the amino acid sequence of SEQ ID NO: 16; the VH domain of the second antigen-binding domain comprises the amino acid sequence of SEQ ID NO: 53; and the VL domain of the second antigen-binding domain comprises the amino acid sequence of SEQ ID NO: 54;
(b) the VH domain of the first antigen-binding domain comprises the amino acid sequence of SEQ ID NO: 15; the VL domain of the first antigen-binding domain comprises the amino acid sequence of SEQ ID NO: 16; the VH domain of the second antigen-binding domain comprises the amino acid sequence of SEQ ID NO: 71; and the VL domain of the second antigen-binding domain comprises the amino acid sequence of SEQ ID NO: 72;
(c) the VH domain of the first antigen-binding domain comprises the amino acid sequence of SEQ ID NO: 35; the VL domain of the first antigen-binding domain comprises the amino acid sequence of SEQ ID NO: 36; the VH domain of the second antigen-binding domain comprises the amino acid sequence of SEQ ID NO: 53; and the VL domain of the second antigen-binding domain comprises the amino acid sequence of SEQ ID NO: 54; or
(d) the VH domain of the first antigen-binding domain comprises the amino acid sequence of SEQ ID NO: 35; the VL domain of the first antigen-binding domain comprises the
amino acid sequence of SEQ ID NO: 36; the VH domain of the second antigen-binding domain comprises the amino acid sequence of SEQ ID NO: 71; and the VL domain of the second antigen-binding domain comprises the amino acid sequence of SEQ ID NO: 72.
In some embodiments, Ab comprises a first heavy chain and a first light chain comprising the first antigen-binding domain specifically binding to CD20 and a second heavy chain and a second light chain comprising the second antigen-binding domain specifically binding to CD22, wherein (a) the first heavy chain comprises: the amino acid sequence of SEQ ID NO: 20; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 20; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 20; the first light chain comprises: the amino acid sequence of SEQ ID NO: 18; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 18; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 18; the second heavy chain comprises: the amino acid sequence of SEQ ID NO: 55; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 55; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 55; and the second light chain comprises: the amino acid sequence of SEQ ID NO: 56; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 56; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 56;
(b) the first heavy chain comprises: the amino acid sequence of SEQ ID NO: 20; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 20; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 20; the first light chain comprises: the amino acid sequence of SEQ ID NO: 18; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 18; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 18; the second heavy chain comprises: the amino acid sequence of SEQ ID NO: 73; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 73; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 73; and the second light chain comprises: the amino acid sequence of SEQ ID NO: 74; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 74; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 74;
(c) the first heavy chain comprises: the amino acid sequence of SEQ ID NO: 37; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 37; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 37; the first light chain comprises: the amino acid sequence of SEQ ID NO: 38; at least 90% sequence identity to the amino acid
sequence of SEQ ID NO: 38; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 38; the second heavy chain comprises: the amino acid sequence of SEQ ID NO: 55; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 55; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 55; and the second light chain comprises: the amino acid sequence of SEQ ID NO: 56; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 56; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 56;
(d) the first heavy chain comprises: the amino acid sequence of SEQ ID NO: 37; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 37; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 37; the first light chain comprises: the amino acid sequence of SEQ ID NO: 38; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 38; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 38; the second heavy chain comprises: the amino acid sequence of SEQ ID NO: 73; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 73; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 73; and the second light chain comprises: the amino acid sequence of SEQ ID NO: 74; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 74; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 74;
(e) the first heavy chain comprises: the amino acid sequence of SEQ ID NO: 75; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 75; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 75; the first light chain comprises: the amino acid sequence of SEQ ID NO: 18; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 18; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 18; the second heavy chain comprises: the amino acid sequence of SEQ ID NO: 76; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 76; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 76; and the second light chain comprises: the amino acid sequence of SEQ ID NO: 56; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 56; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 56; or
(f) the first heavy chain comprises: the amino acid sequence of SEQ ID NO: 20; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 20; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 20; the first light chain comprises: the
amino acid sequence of SEQ ID NO: 18; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 18; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 18; the second heavy chain comprises: the amino acid sequence of SEQ ID NO: 81; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 81; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 81; and the second light chain comprises: the amino acid sequence of SEQ ID NO: 82; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 82; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 82.
In some embodiments, (a) the first heavy chain comprises the amino acid sequence of SEQ ID NO: 20; the first light chain comprises the amino acid sequence of SEQ ID NO: 18; the second heavy chain comprises the amino acid sequence of SEQ ID NO: 55; and the second light chain comprises the amino acid sequence of SEQ ID NO: 56; (b) the first heavy chain comprises the amino acid sequence of SEQ ID NO : 20; the first light chain comprises the amino acid sequence of SEQ ID NO: 18; the second heavy chain comprises the amino acid sequence of SEQ ID NO: 73; and the second light chain comprises the amino acid sequence of SEQ ID NO: 74; (c) the first heavy chain comprises the amino acid sequence of SEQ ID NO: 37; the first light chain comprises the amino acid sequence of SEQ ID NO: 38; the second heavy chain comprises the amino acid sequence of SEQ ID NO: 55; and the second light chain comprises the amino acid sequence of SEQ ID NO: 56; (d) the first heavy chain comprises the amino acid sequence of SEQ ID NO: 37; the first light chain comprises the amino acid sequence of SEQ ID NO: 38; the second heavy chain comprises the amino acid sequence of SEQ ID NO: 73; and the second light chain comprises the amino acid sequence of SEQ ID NO: 74; (e) the first heavy chain comprises the amino acid sequence of SEQ ID NO: 75; the first light chain comprises the amino acid sequence of SEQ ID NO: 18; the second heavy chain comprises the amino acid sequence of SEQ ID NO: 76; and the second light chain comprises the amino acid sequence of SEQ ID NO: 56; or (f) the first heavy chain comprises the amino acid sequence of SEQ ID NO: 20; the first light chain comprises the amino acid sequence of SEQ ID NO: 18; the second heavy chain comprises the amino acid sequence of SEQ ID NO: 81 ; and the second light chain comprises the amino acid sequence of SEQ ID NO: 82.
In some embodiments, Ab is a full-length antibody. In some embodiments, Ab is an IgG-scFv, a trifiinctional antibody (triomab), a knobs into holes (KIH)-IgG, a K/.-body. a
crossmab, an ortho-Fab IgG, a dual variable domain immunoglobulin (DVD-Ig), or a 2 in 1- IgG (dual action antibody), or a combination thereof.
In some embodiments, Ab is an antigen binding fragment. In some embodiments, Ab is a scFv2-Fc, a bi-nanobody, a bispecific T cell engager (BiTE), a tandem diabody (tandAb), a dual affinity retargeting (DART) antibody, a DART-Fc, a scFv-human serum albumin (HSA)-scFv, a dock-and-lock (DNL)-Fab3, a minibody, a Fab2 fragment (bispecific), a Fab3 fragment (trispecific), a Bis-scFv fragment (bispecific), a sdAb fragment (VH/VHH), a tetrabody, a triabody, or a diabody, or a combination thereof.
Production of Antibody
The bispecific antibodies of the present disclosure may be produced by any means known in the art, such as, without limitation, any chemical, biological, genetic or enzymatic technique, either alone or in combination, including recombinant expression, chemical synthesis, and enzymatic digestion of a full-length monoclonal antibody. The recombinant expression may take place in any suitable host cell known in the art, including without limitation to, mammalian host cells, bacterial host cells, yeast host cells, and insect host cells, or in a cell-free system (e.g., Sutro’s Xpress CF platform, World Wide Web at sutrobio.com/technology/) .
Knowing the amino acid sequence of the desired sequence, one skilled in the art may readily produce said antibodies or polypeptides, by standard techniques for production of polypeptides. For instance, they may be synthesized using well-known solid phase method, preferably using a commercially available peptide synthesis apparatus (such as that made by Applied Biosystems, Foster City, Calif.) and following the manufacturer's instructions. Alternatively, antibodies and other polypeptides may be synthesized by recombinant DNA techniques as is well-known in the art. For example, these fragments may be obtained as DNA expression products after incorporation of DNA sequences encoding the desired (poly)peptide into expression vectors and introduction of such vectors into suitable eukaryotic or prokaryotic hosts that will express the desired polypeptide, from which they may be later isolated using well-known techniques.
In particular, described herein are methods of producing an antibody or a polypeptide, which method comprises the steps consisting of: (i) culturing a transformed host cell under
conditions suitable to allow expression of said antibody or polypeptide; and (ii) recovering the expressed antibody or polypeptide.
Antibodies and other polypeptides are suitably separated from the culture medium by conventional immunoglobulin purification procedures such as, for example, protein A- Sepharose, hydroxylapatite chromatography, gel electrophoresis, dialysis, affinity chromatography, ammonium sulfate or ethanol precipitation, acid extraction, anion or cation exchange chromatography, phosphocellulose chromatography, hydrophobic interaction chromatography, hydroxylapatite chromatography and lectin chromatography. High performance liquid chromatography (“HPLC”) may also be employed for purification. See, e.g., Colligan, Current Protocols in Immunology, or Current Protocols in Protein Science, John Wiley & Sons, NY, N.Y., (1997-2001), e.g., Chapters 1, 4, 6, 8, 9, 10, each entirely incorporated herein by reference.
The present disclosure also provides an expression vector and a host cell for producing any of the bispecific antibody described herein. Various expression vectors may be used to express a polynucleotide encoding the bispecific antibody. Both viral-based and nonviral expression vectors may be used to produce antibodies in mammalian host cells. Nonviral vectors and systems include plasmids, episomal vectors (typically, containing an expression cassette for protein or RNA expression), and human artificial chromosomes (e.g., see [Harrington et al., Nat Genet. 15:345, 1997]). Useful viral vectors include without limitation to, retrovirus vectors, adenovirus vectors, adeno-associated virus vectors, herpes virus-based vector, SV40 vectors, papilloma virus vectors, HBP Epstein-Barr virus vectors, vaccinia virus vectors, and Semliki Forest virus (SFV)-based vectors.
The selection of the expression vector varies based on an intended host cell in which the vector is to be expressed. Typically, the expression vector contains a promoter and other regulatory sequences (e.g., enhancer) operably linked to a polynucleotide encoding the bispecific antibody or antibody fragment (e.g., antigen-binding fragment). In some embodiments, an inducible promoter is used to prevent expression of inserted sequences except under inducing conditions. The inducible promoter includes, for example, arabinose, lacZ, metallothionein promoter, or heat shock promoter. A transformed organism culture may be proliferated under non-inducing conditions without biasing a population for coding sequences of which expression products are better tolerated by the host cell.
The expression vector may also provide a secretion signal sequence location to form a fusion protein with a polypeptide encoded by the inserted bispecific antibody or antibody fragment (e.g., antigen-binding fragment) sequence. More often, the inserted bispecific antibody or antibody fragment (e.g., antigen-binding fragment) sequence is included in the vector after being linked to a signal sequence. A vector used to contain a sequence encoding the light and heavy chain variable domains of the bispecific antibody or antibody fragment (e.g., antigen-binding fragment) sometimes also encodes a constant region or a portion thereof. Such a vector allows the variable region to be expressed as a fusion protein with the constant region to induce production of a complete antibody or fragment thereof.
In some embodiments, mammalian host cells are used to express and produce the bispecific antibody or antibody fragment (e.g., antigen-binding fragment) polypeptide of the present disclosure. For example, these mammalian host cells may be hybridoma cell lines expressing endogenous immunoglobulin genes (e.g., myeloma hybridoma clones as described in Examples), or mammalian cell lines harboring exogenous expression vectors (e.g., SP2/0 myeloma cells to be exemplified below). The mammalian host cells include any normal apoptotic or normal or abnormal immortalized animal or human cells. For example, a plurality of suitable host cell lines capable of secreting intact immunoglobulins have been developed, including CHO cell lines, various COS cell lines, HeLa cells, myeloma cell lines, transformed B cells, and hybridomas. Expression vectors for mammalian host cells may include expression control sequences, such as an origin of replication, a promoter, and an enhancer, and necessary processing information sites, such as a ribosome binding site, an RNA splice site, a polyadenylation site, and a transcription terminator sequence. These expression vectors generally contain promoters derived from mammalian genes or mammalian viruses. Suitable promoters may be constitutive, cell type -specific, step-specific, and/or controllable or regulatable promoters. Useful promoters include a metallothionein promoter, a constitutive adenovirus major late promoter, a dexamethasone-inducible MMTV promoter, a SV40 promoter, a MRP polIII promoter, a constitutive MPSV promoter, a tetracycline -inducible CMV promoter (e.g., a human immediate early CMV promoter), a constitutive CMV promoter, and promoter-enhancer combinations known in the art, but are not limited thereto.
A method of introducing an expression vector including a polynucleotide sequence of interest varies depending on a type of host cell. For example, calcium chloride transfection is
commonly used for prokaryotic cells, whereas calcium phosphate treatment or electroporation may be used for other host cells.
In the present disclosure, a non-human antibody may be derived from, for example, any antibody-producing animal, for example, a mouse, a rat, a rabbit, a goat, a donkey, or non-human primates (e.g., monkeys such as cynomolgus or rhesus monkey) or apes (e.g., chimpanzees). A non-human antibody may be produced by immunizing an animal by using a method known in the art.
Chimeric antibodies (e.g, mouse-human chimeras) may be produced by obtaining nucleic sequences encoding VL and VH domains as previously described, constructing a human chimeric antibody expression vector by inserting them into an expression vector for animal cell having genes encoding human antibody CH and human antibody CL, and expressing the coding sequence by introducing the expression vector into an animal cell. The CH domain of a human chimeric antibody may be any region which belongs to human immunoglobulin, such as the IgG class or a subclass thereof, such as IgGl, IgG2, IgG3 and IgG4. Similarly, the CL of a human chimeric antibody may be any region which belongs to Ig, such as the kappa class or lambda class, chimeric and humanized monoclonal antibodies, comprising both human and non-human portions may be made using standard recombinant DNA techniques. Such chimeric and humanized monoclonal antibodies may be produced by recombinant DNA techniques known in the art, for example using methods described in Robinson et al. International Patent Publication PCT/US86/02269; Akira et al. European Patent Application 184,187; Taniguchi, M. European Patent Application 171,496; Morrison et al. European Patent Application 173,494; Neuberger et al. PCT Application WO 86/01533; Cabilly e/ a/. U.S. Patent No. 4,816,567; Cabilly et al. European Patent Application 125,023; Better et al. (1988) Science 240: 1041-1043; Liu et al. (1987) Proc. Natl. Acad. Sci. USA 84:3439-3443; Liu et al. (1987) J. Immunol. 139:3521-3526; Sun et al. (1987) Proc. Natl. Acad. Sci. 84:214-218; Nishimura et al. (1987) Cancer Res. 47:999-1005; Wood et a/. (1985) Nature 314:446-449; Shaw etal. (1988) J. Natl. Cancer Inst. 80: 1553-1559); Morrison, S. L. (1985) Science 229: 1202-1207; Oi et al. (1986) Biotechniques 4:214; Winter U.S. Patent 5,225,539; Jones et al. (1986) Nature 321:552-525; Verhoeyan et al. (1988) Science 239: 1534; and Beidler et al. (1988) J. Immunol. 141:4053-4060.
In addition, humanized antibodies may be made according to standard protocols such as those disclosed in U.S. Patent 5,565,332. In another embodiment, antibody chains or
specific binding pair members may be produced by recombination between vectors comprising nucleic acid molecules encoding a fusion of a polypeptide chain of a specific binding pair member and a component of a replicable generic display package and vectors containing nucleic acid molecules encoding a second polypeptide chain of a single binding pair member using techniques known in the art, e.g., as described in U.S. Patents 5,565,332, 5,871,907, or 5,733,743. Humanized antibodies may be produced by obtaining nucleic acid sequences encoding CDR domains, as previously described, constructing a humanized antibody expression vector by inserting them into an expression vector for animal cell having genes encoding (i) a heavy chain constant region identical to that of a human antibody and (ii) a light chain constant region identical to that of a human antibody, and expressing the genes by introducing the expression vector into an animal cell.
The humanized antibody expression vector may be either of a type in which a gene encoding an antibody heavy chain and a gene encoding an antibody light chain exists on separate vectors or of a type in which both genes exist on the same vector (tandem type).
Methods for producing humanized antibodies based on conventional recombinant DNA and gene transfection techniques are well known in the art (See, e.g., Riechmann L. et al. 1988; Neuberger M S. et al. 1985). Antibodies may be humanized using a variety of techniques known in the art including, for example, CDR-grafting (EP 239,400; PCT publication WO91/09967; U.S. Pat. Nos. 5,225,539; 5,530,101; and 5,585,089), veneering or resurfacing (EP 592,106; EP 519,596; Padlan EA (1991); Studnicka G M et al. (1994); Roguska M A. et al. (1994)), and chain shuffling (U.S. Pat. No. 5,565,332). The general recombinant DNA technology for preparation of such antibodies is also known (see European Patent Application EP 125023 and International Patent Application WO 96/02576).
A fully human antibody may be produced by administering an antigen to a transformed animal including a human immunoglobulin gene locus, or by treating a phage display library expressing a human antibody repertory with an antigen, and then selecting the target antibody. The antibody may be polyclonal or monoclonal, or may be synthesized within a cell host through the expression of recombinant DNA. A monoclonal antibody (mAb) may be produced using a conventional monoclonal antibody method, for example, a standard somatic hybridization technique in the literature (Kohler and Milstein, 1975, Nature 256:495).
Bispecific or multispecific antibodies described herein may be made according to standard procedures. For example, triomas and hybrid hybridomas are two examples of cell
lines that may secrete bispecific or multispecific antibodies. Examples of bispecific and multispecific antibodies produced by a hybrid hybridoma or a trioma are disclosed in U.S. Patent 4,474,893. Such antibodies may also be constructed by chemical means (Staerz et al. (1985) Nature 314:628, and Perez et al. (1985) Nature 316:354) and hybridoma technology (Staerz and Bevan (1986) Proc. Natl. Acad. Set. USA, 83: 1453, and Staerz and Bevan (1986) Immunol. Today 7:241). Alternatively, such antibodiesmay also be generated by making heterohybridomas by fusing hybridomas or other cells making different antibodies, followed by identification of clones producing and co-assembling the desired antibodies. They may also be generated by chemical or genetic conjugation of complete immunoglobulin chains or portions thereof such as Fab and Fv sequences.
Active Agent and Payload
The present disclosure provides, among other things, active agents and antibody-drug conjugates that comprise the active agents. The anti-CD20xCD22 bispecific antibody (“Ab”) disclosed herein may be conjugated to many identical or different active agents using methods known in the art. The active agents of the present disclosure should not be construed as being limited to chemical therapeutic agents. For example, active agents of the present disclosure may be proteins, peptides, or polypeptides having a desired biological activity.
In some embodiments, the active agent is a chemotherapeutic agent or a toxin. In some embodiments, the active agent is selected from a chemotherapeutic compound, a cytotoxic compound, an immunomodulatory compound, an anticancer agent, an antiviral agent, an antibacterial agent, an antifungal agent, an antiparasitic agent, and a combination thereof. In some embodiments, the cytotoxic compound is selected from a mitotic inhibitor, a DNA alkylating agent, a topoisomerase inhibitor, and a combination thereof. In some embodiments, the cytotoxic compound is selected from auristatin, maytansinoid, tubulisin, calicheamicin, duocarmycin, pyrrolobenzodiazepine and derivatives thereof, camptothecin and derivatives thereof, and combinations thereof. In some embodiments, the auristatin is MMAE (monomethyl auristatin E) or MMAF (monomethyl auristatin F).
In some embodiments, the active agent is a pyrrolobenzodiazepine dimer; position N10 of the pyrrolobenzodiazepine dimer is substituted with X or position N’ 10 is substituted with X’, wherein X or X' links the pyrrolobenzodiazepine dimer to the linker;
X and X' are each independently -C(O)O-* or -C(O)-* ; and
* refers to a binding site between the pyrrolobenzodiazepine dimer and the linker.
In some embodiments, the active agent is a pyrrolobenzodiazepine dimer and the pyrrolobenzodiazepine dimer has a structure represented by Formula VIII:

[Formula VIII] wherein the wavy line indicates a connection point to the linker; a dotted line represents an optional double bond;
Ri and Ri are each independently selected from H, OH, =0, =CH2, CN, Rm, 0Rm, =CH-Rm' =C(Rm)2, O-SO2-Rm, C02Rm, C0Rm, halo, and dihalo;
Rm is selected from Rm, C02Rm, C0Rm, CHO, CO2H. and halo;
Rm is selected from substituted or unsubstituted C1-12 alkyl, substituted or unsubstituted C2-12 alkenyl, substituted or unsubstituted C2-12 alkynyl, substituted or unsubstituted Cs-2o aryl, substituted or unsubstituted C3-6 heteroaryl, substituted or unsubstituted C3-6 cycloalkyl, substituted or unsubstituted 3- to 7-membered heterocyclyl, substituted or unsubstituted 3- to 7-membered heterocycloalkyl, and substituted or unsubstituted 5- to 7-membered heteroaryl, wherein when the C1-12 alkyl, C2-12 alkenyl, C2-12 alkynyl, Cs-2o aryl, Cs-2o heteroaryl, C3-6 cycloalkyl, 3- to 7-membered heterocyclyl, 3- to 7- membered heterocycloalkyl, or 5- to 7-membered heteroaryl is substituted, the respective hydrogen atoms in the C1-12 alkyl, C2-12 alkenyl, C2-12 alkynyl, Cs-2o aryl, Cs-2o heteroaryl, C3-6 cycloalkyl, 3- to 7-membered heterocyclyl, 3- to 7-membered heterocycloalkyl, or 5- to 7-membered heteroaryl may each be independently replaced with methoxy, C1-12 alkyl, C2-12 alkenyl, C2-12 alkynyl, Cs-2o aryl, Cs-2o heteroaryl, C3-6
cycloalkyl, 3- to 7-membered heterocyclyl, 3- to 7-membered heterocycloalkyl, and 5- to 7-membered heteroaryl;
R2, R3, Rs, R2, R3 , and Rs are each independently selected from H, Rm, OH, ORm, SH, SRm, NH2, NHRm, NRmRm, NO2, Me3Sn, and halo;
R4 and R4 are each independently selected from H, Rm, OH, ORm, SH, SRm, NH2, NHRm, NRmRm, NO2, McsSn. halo, substituted or unsubstituted C1-6 alkyl, substituted or unsubstituted C1-6 alkoxy, substituted or unsubstituted C2-6 alkenyl, substituted or unsubstituted C2-6 alkynyl, substituted or unsubstituted C3-6 cycloalkyl, substituted or unsubstituted 3- to 7-membered heterocycloalkyl, substituted or unsubstituted C5-12 aryl, substituted or unsubstituted 5- to 7-membered heteroaryl, -CN, -NCO, -ORn, - OC(O)Rn, -OC(O)NRnRn, -OS(O)Rn, -OS(O)2Rn, -SRn, -S(O)Rn, -S(O)2Rn, - S(O)NRnRn, -S(O)2NRnRn , -OS(O)NRnRn', -OS(O)2NRnRn', -NRnRn, -NRnC(O)R°, - NRnC(O)OR°, -NRnC(O)NR°R° , -NRnS(O)R°, -NRnS(O)2R°, -NRnS(O)NR°R° , - NRnS(O)2NR°R° , -C(O)Rn, -C(O)ORn, and -C(O)NRnRn , wherein the hydrogen atoms in the C1-6 alkyl, C1-6 alkoxy, C2-6 alkenyl, C2-6 alkynyl, C3-6 cycloalkyl, 3- to 7- membered heterocycloalkyl, C5-12 aryl, and 5- to 7-membered heteroaryl may each be independently replaced with C1-6 alkyl, C1-6 alkoxy, C2-6 alkenyl, C2-6 alkynyl, C3-6 cycloalkyl, 3- to 7-membered heterocycloalkyl, C5-12 aryl, 5- to 7-membered heteroaryl, -ORP, -OC(O)RP, -OC(O)NRPRP', -OS(O)RP, -OS(O)2RP, -SRP, -S(O)RP, - S(O)2Rp, -S(O)NRpRp ', -S(O)2NRPRP , -OS(O)NRPRP', -OS(O)2NRPRP', -NRpRp ', - NRpC(O)Rq, -NRpC(O)ORq, -NRpC(O)NRqH, -NRpS(O)Rq, -NRpS(O)2Rq, - NRpS(O)NRqH, -NRpS(O)2NRqH, -C(O)RP, -C(O)ORP, or -C(O)NRPRP when the C1-6 alkyl, C1-6 alkoxy, C2-6 alkenyl, C2-6 alkynyl, C3-6 cycloalkyl, 3- to 7-membered heterocycloalkyl, C5-12 aryl, and 5- to 7-membered heteroaryl;
Rn, Rn, R°, R° Rp, Rp , and Rq are each independently selected from H, C1-7 alkyl, C2-7 alkenyl, C2-7 alkynyl, C3-13 cycloalkyl, 3- to 7-membered heterocycloalkyl, C6-10 aryl, and 5- to 7-membered heteroaryl;
X is selected from -C(O)O-, -S(O)O-, -C(O)-, -C(O)NR-, -S(O)2NR-, -P(O)R'NR-, -S(O)NR-, and -PO2NR-;
Xa is a bond or substituted or unsubstituted C1-6 alkylene, wherein C1-6 alkylene is substituted with C1-8 alkyl, or C3-8 cycloalkyl when substituted;
R and R' each independently denote H, OH, NH2, ONH2, NHNH2, substituted or unsubstituted Ci-s alkyl, substituted or unsubstituted C3-8 cycloalkyl, substituted or unsubstituted C1-8 alkoxy, substituted or unsubstituted C1-8 alkylthio, substituted or unsubstituted C3-20 heteroaryl, substituted or unsubstituted C5-20 aryl, or mono- or di- C1-8 alkylamino, wherein the C1-8 alkyl, C3-8 cycloalkyl, C1-8 alkoxy, C1-8 alkylthio, C3-20 heteroaryl, and C5-20 aryl are substituted with a substituent selected from OH, N3, CN, NO2, SH, NH2, ONH2, NHNH2, halo, C1-6 alkyl, C1-6 alkoxy, and Cg-12 aryl when substituted;
Y and Y' are each independently selected from O, S, and N(H);
Re is a substituted or unsubstituted saturated or unsaturated C3-12 hydrocarbon chain, wherein the chain may be interrupted by one or more heteroatoms, NMe, or a substituted or unsubstituted aromatic ring, the chain or aromatic ring may be substituted with -NH, -NRm, -NHC(O)Rm, -NHC(O)CH2-[OCH2CH2]n-R, or-[CH2CH2O]n-R at any one or more positions of hydrogen atoms on the chain or aromatic ring or unsubstituted, wherein Rm and R are each as defined for Rm and R above, and n is 1 to 12; and
R7 and R7 are each independently H, substituted or unsubstituted C1-6 alkyl, substituted or unsubstituted C2-6 alkenyl, substituted or unsubstituted C2-6 alkynyl, substituted or unsubstituted C3-6 cycloalkyl, substituted or unsubstituted 3- to 7-membered heterocycloalkyl, substituted or unsubstituted C6-10 aryl, substituted or unsubstituted 5- to 7-membered heteroaryl, -ORr, -OC(O)Rr, -OC(O)NRrRr , -OS(O)Rr, -OS(O)2Rr, - SRr, -S(O)Rr, -S(O)2Rr, -S(O)NRrRr , -S(O)2NRrRr', -OS(O)NRrRr', -OS(O)2NRrRr', - NRrRr , -NRrC(O)Rs, -NRrC(O)ORs, -NRrC(O)NRsRs', -NRrS(O)Rs, -NRrS(O)2Rs, - NRrS(O)NRsRs , -NRrS(O)2NRsRs, -C(O)Rr, -C(O)ORS, or -C(O)NRRr ', wherein the hydrogen atoms in the C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-6 cycloalkyl, 3- to 7- membered heterocycloalkyl, C6-10 aryl, and 5- to 7-membered heteroaryl may each be independently replaced with C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-6 cycloalkyl, 3- to 7-membered heterocycloalkyl, Ce-io aryl, 5- to 7-membered heteroaryl, -OR1, - OC(O)R‘, -OC(O)NRtRt, -OS(O)Rl, -OS(O)2Rl, -SR‘, -S(O)Rl, -S(O)2Rl, -S(O)NRtRt, -S(O)2NRtRt, -OS(O)NRtRt , -OS(O)2NRtRt', -NFfR1'. -NRlC(O)Ru, -NRlC(O)ORu, - NRlC(O)NRuRu , -NRlS(O)Ru, -NRtS(O)2Ru,-NRtS(O)NRuRu', -NRtS(O)2NRuRu', - C(O)Rl, -C(O)ORl, or -C(O)NRtRt when the C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-6
cycloalkyl, 3- to 7-membered heterocycloalkyl, Ce-io aryl, and 5- to 7-membered heteroaryl;
Rr, Rr , Rs, Rs , R\ Rf, Ru, and Rul are each independently selected from H, C1-7 alkyl, C2-7 alkenyl, C2-7 alkynyl, C3-13 cycloalkyl, 3- to 7-membered heterocycloalkyl, C5-10 aryl, and 5- to 7-membered heteroaryl;
G is a glucuronide group or a galactoside group; each Z is selected from H, C1-8 alkyl, halo, NO2, CN,

R9, Rio, and Ris are each independently selected from H, C1-8 alkyl, C2-6 alkenyl, C1-6 alkoxy, and alkyloxyalkyl; and n30 is 0 to 3.
In some embodiments, Y is O. In some embodiments, is Y’ is O. In some embodiments, a dotted line represents presence of a double bond between the carbons bearing Ri and R? or R1 and R7 . In some embodiments, Ri is selected from substituted or unsubstituted Ci-6 alkyl, substituted or unsubstituted C2-6 alkenyl, substituted or unsubstituted C5-7 aryl, and substituted or unsubstituted C3-6 heteroaryl. In some embodiments, R2, R3, and Rs are each independently H or OH. In some embodiments, R4 is C1-6 alkoxy. In some embodiments, R4 is methoxy, ethoxy, or butoxy. In some embodiments, X is selected from -C(O)O-, -C(O)-, and -C(O)NR-. In some embodiments, X is -C(O)NR-. In some embodiments, Rs is a substituted or unsubstituted saturated or unsaturated C3-8 hydrocarbon chain, wherein one or more of the carbon atoms of the hydrocarbon chain is replaced by a heteroatom or a substituted or unsubstituted aromatic ring, wherein the heteroatom is O, S, or N(H) and the aromatic ring is benzene, pyridine, imidazole, or pyrazole, and the chain or aromatic ring may be substituted with -NHC(O)CH2-[OCH2CH2]n-R or -[CH2CH2O]n-R at any one or more positions of hydrogen atoms on the chain or aromatic ring; and n is 1 to 6. In some embodiments, n is 1 to 6. In some embodiments, Xa is a bond or C1-3 alkylene.
0 vSi/Rl6
In some embodiments, Z is H, , and
 , wherein R9, R10, and Rie are each independently selected from H, C1-3 alkyl, C1-3 alkoxy, and alkyloxyalkyl . In some embodiments, R9 is methyloxyalkyl. In some embodiments, Rio is methyloxyalkyl. In some
embodiments, Rie is methyloxyalkyl. In some embodiments, R9, Rio, or Rie is -(CH2CH2O)m- (CH2)m2CH3, further wherein m is 1-6 and m2 is 0-2. In some embodiments, m is 1. In some embodiments, m2 is 0. In some embodiments, R2is H. In some embodiments, R3 is H. In some embodiments, R7 is H. In some embodiments, R4 is C1-6 alkoxy (e.g., methoxy). In some embodiments, Rs is OH. In some embodiments, Ri is =CH2, CH?. or phenyl, optionally substituted with methoxy. In some embodiments, Y is O.
, wherein R9, R10, and Rie are each independently selected from H, C1-3 alkyl, C1-3 alkoxy, and alkyloxyalkyl . In some embodiments, R9 is methyloxyalkyl. In some embodiments, Rio is methyloxyalkyl. In some
embodiments, Rie is methyloxyalkyl. In some embodiments, R9, Rio, or Rie is -(CH2CH2O)m- (CH2)m2CH3, further wherein m is 1-6 and m2 is 0-2. In some embodiments, m is 1. In some embodiments, m2 is 0. In some embodiments, R2is H. In some embodiments, R3 is H. In some embodiments, R7 is H. In some embodiments, R4 is C1-6 alkoxy (e.g., methoxy). In some embodiments, Rs is OH. In some embodiments, Ri is =CH2, CH?. or phenyl, optionally substituted with methoxy. In some embodiments, Y is O.
In some embodiments, R2’ is H. In some embodiments, R3’ is H. In some embodiments,
R7’ is H. In some embodiments, R4’ is C1-6 alkoxy (e.g., methoxy). In some embodiments, Rs’ is OH. In some embodiments, Ri’ is =CH2, CH3, or phenyl, optionally substituted with methoxy. In some embodiments, Y’ is O. In some embodiments, X is -C(O)O-. In some embodiments, Xa is CH2. In some embodiments, G is a glucuronide group. In some embodiments,
 some embodiments, n30 is 1.
some embodiments, n30 is 1.
In some embodiments, Z is
 . In some embodiments, R9 is
. In some embodiments, R9 is
H. In some embodiments, R16 is alkyloxyalkyl (e.g., methoxyethyl). In some embodiments, Z is
 in some embodiments, Rio is alkyl (e.g, methyl). In some embodiments, Re is C3-12 alkyl (e.g., pentyl).
in some embodiments, Rio is alkyl (e.g, methyl). In some embodiments, Re is C3-12 alkyl (e.g., pentyl).
In some embodiments, the pyrrolobenzodiazepine dimer is selected from:


OMe or -NH2.
In certain embodiments, the conjugate comprises:






MMAE is monomethyl auristatin E, and MMAF is monomethyl auristatin F; and the dotted line represents a connection to Ab.




IS















MMAE is monomethyl auristatin E, and MMAF is monomethyl auristatin F; and the dotted line represents a connection to Ab.
In some embodiments, the active agent is selected from:
(a) erlotinib, bortezomib, fulvestrant, sunitinib, letrozole, imatinib mesylate, PTK787/ZK 222584, oxaliplatin, 5 -fluorouracil, leucovorin, rapamycin, lapatinib, lonafamib, sorafenib, gefitinib, AG1478, AG1571, thiotepa, cyclophosphamide, busulfan, improsulfan, piposulfan, benzodopa, carboquone, meturedopa, uredopa, ethylenimine, altretamine, triethylenemelamine, triethylenephosphoramide, triethylenethiophosphoramide, trimethylolomelamine, bullatacin, bullatacinone,
camptothecin, topotecan, bryostatin, callystatin, CC-1065, adozelesin, carzelesin, bizelesin, cryptophycin 1, cryptophycin 8, dolastatin, duocarmycin, KW-2189, CB1- TM1, eleutherobin, pancrati statin, sarcodictyin, spongistatin, chlorambucil, chlomaphazine, cholophosphamide, estramustine, ifosfamide, mechlorethamine, melphalan, novembichin, phenesterine, prednimustine, trofosfamide, uracil mustard, carmustine, chlorozotoxin, fotemustine, lomustine, nimustine, ranimustine, calicheamicin, calicheamicin gamma 1, calicheamicin omega 1, dynemicin, dynemicin A, clodronate, esperamicin, neocarzino statin chromophore, aclacinomysins, actinomycin, anthramycin, azaserine, bleomycins, catcinomycin, carabicin, caminomycin, carzinophilin, chromomycins, dactinomycin, daunorubicin, 6-diazo-5-oxo-L-norleucine, doxorubicin, morpholino-doxorubicin, cyanomorpholino-doxorubicin, 2-pyrrolino-doxorubucin, liposomal doxorubicin, deoxydoxorubicin, epirubicin, esorubicin, marcellomycin, mitomycin C, mycophenolic acid, nogalamycin, olivomycins, peplomycin, potfiromycin, puromycin, quelamycin, rodorubicin, streptomigrin, streptozocin, tubercidin, ubenimex, zinostatin, zorubicin, denopterin, methotrexate, pteropterin, trimetrexate, fludarabine, 6-mercaptopurine, thiamiprine, thioguanine, ancitabine, azacytidine, 6- azauridine, carmofur, cytarabine, dideoxyuridine, doxifluridine, enocitabine, floxuridine, calusterone, dromostanolone, propionate, epitiostanol, mepitiostane, testolactone, aminoglutethimide, mitotane, trilostane, folinic acid, aceglatone, aldophosphamide glycoside, aminolevulinic acid, eniluracil, amsacrine, bestrabucil, bisantrene, edatrexate, democolcine, diaziquone, elfomithine, elliptinium acetate, etoglucid, gallium nitrate, hydroxyurea, lentinan, lonidainine, maytansine, ansamitocins, mitoguazone, mitoxantrone, mopidanmol, pentostatin, phenamet, pirarubicin, losoxantrone, 2-ethylhydrazide, procarbazine, polysaccharide-k, razoxane, rhizoxin, sizofiran, spirogermanium, tenuazonic acid, triaziquone, 2,2’, 2”- trichlorotriethylamine, T-2 toxin, verracurin A, roridin A, anguidine, urethane, vindesine, dacarbazine, mannomustine, mitobronitol, mitolactol, pipobroman, arabinoside, cyclophosphamide, paclitaxel, albumin-engineered nanoparticle formulation of paclitaxel, docetaxel, chlorambucil, gemcitabine, 6-thioguanine, mercaptopurine, cisplatin, carboplatin, vinblastine, platinum, etoposide, vincristine, vinorelbine, novantrone, teniposide, edatrexate, daunomycin, aminopterin,
ibandronate, CPT-11, topoisomerase inhibitor RFS 2000, difluoromethylomithine, retinoic acid, capecitabine, or pharmaceutically acceptable salts, solvates or acids thereof;
(b) monokine, lymphokine, traditional polypeptide hormone, parathyroid hormone, thyroxine, relaxin, prorelaxin, glycoprotein hormone, follicle stimulating hormone, thyroid stimulating hormone, luteinizing hormone, hepatic growth factor, fibroblast growth factor, prolactin, placental lactogen, tumor necrosis factor, tumor necrosis factor-a, tumor necrosis factor-p, Mullerian inhibiting substance, mouse gonadotropin associated peptide, inhibin, activin, vascular endothelial growth factor, thrombopoietin, erythropoietin, osteoinductive factor, interferon, interferon-a, interferon-P, interferon- y, colony stimulating factor (CSF), macrophage-CSF, granulocyte-macrophage-CSF), granulocyte-macrophage-CSF, granulocyte-CSF, interleukin (IL), IL-1, IL-1, IL-2, IL-3, IL-4, IL-5, IL-6, IL-7, IL-8, IL-9, IL-10, IL- 11, IL- 12, polypeptide factor, LIF, kit ligand, or mixtures thereof;
(c) diphtheria toxin, botulinum toxin, tetanus toxin, dysentery toxin, cholera toxin, amanitin, a-amatinin, pyrrolobenzodiazepine, pyrrolobenzodiazepine derivative, indolinobenzodiazepine, pyridobenzodiazepine, tetrodotoxin, brevetoxin, ciguatoxin, ricin, AM toxin, auristatin, tubulysin, geldanamycin, maytansinoid, calicheamicin, daunomycin, doxorubicin, methotrexate, vindesine, SG2285, dolastatin, dolastatin analog, cryptophycin, camptothecin, rhizoxin, rhizoxin derivatives, CC-1065, CC- 1065 analogs or derivatives, duocarmycin, enediyne antibiotic, esperamicin, epothilone, toxoid, or mixtures thereof;
(d) affinity ligand, where the affinity ligand is a substrate, inhibitor, active agent, neurotransmitter, radioactive isotope, or mixtures thereof;
(e) radioactive label, 32P, 35S, fluorescent dye, electron dense reagent, enzyme, biotin, streptavidin, digoxigenin, hapten, immunogenic protein, nucleic acid molecule with a sequence complementary to a target, or mixtures thereof;
(f) immunomodulatory compound, anti-cancer agent, anti-viral agent, anti-bacterial agent, anti-fungal agent, anti-parasitic agent, or mixtures thereof;
(g) tamoxifen, raloxifene, droloxifene, 4-hydroxytamoxifen, trioxifene, LY117018, onapristone or toremifene;
(h) 4(5)-imidazole, aminoglutethimide, megestrol acetate, exemestane, letrozole or anastrozole
(i) flutamide, nilutamide, bicalutamide, leuprolide, goserelin, ortroxacitabine;
(j) aromatase inhibitor;
(k) protein kinase inhibitor;
(l) lipid kinase inhibitor;
(m) antisense oligonucleotide;
(n) ribozyme;
(o) vaccine; and
(p) anti-angiogenic agent.
Use of Anti-Human anti-CD20xCD22 Bispecific Antibody Drug-Conjugates for Therapeutic Purposes
The expression of CD20 and CD22 in cancer is associated with unfavorable prognosis of certain cancers (e.g., hematological cancers and B-cell malignancies) and is known to also affect cancer metastasis. For anticancer antibody treatment, for example, the anti-CD20xCD22 bispecific antibody may, as described herein, be used in a form linked to various active agents via a linker to remove CD20 and CD22 overexpressing cancer cells. Accordingly, a bispecific antibody binding to CD20 and CD22 may be used in a form bonded to an active agent, and thus can be used as a targeted therapeutic agent for directing to CD20 and CD22 expressing cells.
In some aspects, the present disclosure provides a pharmaceutical composition comprising a conjugate disclosed herein and a pharmaceutically acceptable excipient.
In some aspects, the present disclosure provides a pharmaceutical composition for use in preventing or treating a proliferative disease, the composition comprising a conjugate disclosed herein. In some embodiments, the proliferative disease is cancer. In some embodiments, the cancer is selected from liver cancer, thyroid cancer, ovarian cancer, brain cancer, multiple myeloma, colon cancer, head and neck cancer, lymphoma, leukemia, bladder cancer, kidney cancer, stomach cancer, breast cancer, uterine cancer, prostate cancer, pancreatic cancer, lung cancer, sarcoma, neuroendocrine tumor, melanoma, and combinations thereof. In some embodiments, the lymphoma or leukemia is selected from Hodgkin lymphoma, non-Hodgkin lymphoma (NHL), T cell lymphoma, B cell lymphoma, natural killer
cell lymphoma, diffuse large B cell lymphoma (DLBCL), mantle cell lymphoma (MCL), primary central nervous system (CNS) lymphoma, lymphoblastic lymphoma, enteropathy-type intestinal lymphoma, anaplastic large cell lymphoma, angioimmunoblastic T cell lymphoma, anaplastic large cell lymphoma, peripheral T cell lymphoma, marginal zone lymphoma, chronic lymphocytic leukemia (CLL), acute lymphoblastic leukemia (ALL), chronic myelogenous leukemia (CML), acute myelogenous leukemia (AML), and combinations thereof. In some embodiments, the acute lymphoblastic leukemia is B-cell acute lymphoblastic leukemia (B-ALL).
Treatment Method: Pharmaceutical Formulation and Administration Route
In some embodiments, the present disclosure also provides a treatment method using an antibody-drug conjugate or a pharmaceutically acceptable salt or solvate thereof. In some embodiments, the antibody-drug conjugate or pharmaceutically acceptable salt or solvate thereof is provided to a patient. The antibody-drug conjugate or pharmaceutically acceptable salt or solvate thereof inhibits cancer cell progression by binding to CD20 and CD22 expressed on the surface of cancer cells. In some embodiments, the antibody binds to CD20 and CD22 expressed on the surface of cancer cells in a form bonded to an active agent disclosed herein, thereby specifically delivering the active agent bonded to the antibody to cancer cells, to induce the death of the cancer cells. In some embodiments, the antibody binds to CD20 and CD22 expressed on the surface of cancer cells in the form of an antibody specific to the same target or another target, thereby increasing specificity of multiple antibodies for cancer cells or inducing connections between cancer cells and other types of cells such as immune cells, to induce the death of the cancer cells.
In some aspects, the present disclosure provides a method of treating or preventing a proliferative disease in a subject in need thereof comprising administering a conjugate disclosed herein or a pharmaceutically acceptable salt thereof to the subject. In some embodiments, the proliferative disease is cancer. In some embodiments, the cancer is selected from liver cancer, thyroid cancer, ovarian cancer, brain cancer, multiple myeloma, colon cancer, head and neck cancer, lymphoma, leukemia, bladder cancer, kidney cancer, stomach cancer, breast cancer, uterine cancer, prostate cancer, pancreatic cancer, lung cancer, sarcoma, neuroendocrine tumor, melanoma, and combinations thereof. In some embodiments, the lymphoma or leukemia is selected from Hodgkin lymphoma, non-Hodgkin lymphoma (NHL),
T cell lymphoma, B cell lymphoma, natural killer cell lymphoma, diffuse large B cell lymphoma (DLBCL), mantle cell lymphoma (MCL), primary central nervous system (CNS) lymphoma, lymphoblastic lymphoma, enteropathy-type intestinal lymphoma, anaplastic large cell lymphoma, angioimmunoblastic T cell lymphoma, anaplastic large cell lymphoma, peripheral T cell lymphoma, marginal zone lymphoma, chronic lymphocytic leukemia (CLL), acute lymphoblastic leukemia (ALL), chronic myelogenous leukemia (CML), acute myelogenous leukemia (AML), and combinations thereof. In some embodiments, the acute lymphoblastic leukemia is B-cell acute lymphoblastic leukemia (B-ALL).
Pharmaceutical compositions
The present disclosure, among other things, provides a pharmaceutical composition comprising a therapeutically effective amount of the antibody-drug conjugate or a pharmaceutically acceptable salt or solvate thereof, and a pharmaceutically acceptable diluent, a carrier, a solubilizer, an emulsifier, a preservative, and/or an adjuvant. Also, for example, a method of treating a cancer patient by administering such a pharmaceutical composition is provided. The term "patient" includes human patients.
The pharmaceutical composition may include a pharmaceutically acceptable carrier. The carrier is used as a meaning including an excipient, a diluent, or an adjuvant. The carrier may be selected from the group consisting of, for example, lactose, dextrose, sucrose, sorbitol, mannitol, xylitol, erythritol, maltitol, starch, acacia rubber, alginate, gelatin, calcium phosphate, calcium silicate, cellulose, methyl cellulose, polyvinyl pyrrolidone, water, saline, buffer such as PBS, methylhydroxy benzoate, propylhydroxy benzoate, talc, magnesium stearate, and mineral oil. The composition may include a filler, an anticoagulant, a lubricant, a wetting agent, a flavoring agent, an emulsifier, a preservative, or a combination thereof.
The pharmaceutical composition may be prepared as any formulation according to general methods. The composition may be formulated into formulations for oral administration (e.g., powders, tablets, capsules, syrups, pills or granules) or for parenteral administration (for example, injections). In addition, the composition may be prepared as a systemic or local formulation.
The pharmaceutical composition may include an effective amount of the antibody or antigen-binding fragment thereof, an anticancer agent, or a combination thereof. The term "effective amount" refers to an amount sufficient to exhibit preventive or therapeutic effects
when administered to an individual requiring prevention or treatment. The effective amount may be appropriately selected depending on a cell or individual that is selected by those or ordinary skill in the art. The effective amount may be determined according to factors including the severity of the disease, the age, body weight, health and gender of a patient, sensitivity of a patient to the drug, administration time, administration routes, excretion rate, treatment period, and drugs used in combination or simultaneously with the used composition, and other factors well known in the medical field.
The dosage of the pharmaceutical composition may range, for example, from 10 pg/kg to about 30 mg/kg, optionally from 0.1 mg/kg to about 30 mg/kg, or alternatively from 0.3 mg/kg to about 20 mg/kg per adult. The pharmaceutical composition may be administered once a day, multiple times a day, once every 1 to 4 weeks, or once to 12 times a year.
Hereinafter, the present disclosure will be described in more detail with reference to examples and experimental examples.
The following examples and experimental Examples are intended to aid in understanding of the present disclosure and are not intended to limit the scope of the present disclosure.
Definitions
Unless otherwise defined herein, scientific and technical terms used in this application shall have the meanings that are commonly understood by those of ordinary skill in the art. Generally, nomenclature used in connection with, and techniques of, chemistry, cell and tissue culture, molecular biology, cell and cancer biology, neurobiology, neurochemistry, virology, immunology, microbiology, pharmacology, genetics and protein and nucleic acid chemistry, described herein, are those well-known and commonly used in the art.
The methods and techniques of the present disclosure are generally performed, unless otherwise indicated, according to conventional methods well known in the art and as described in various general and more specific references that are cited and discussed throughout this specification. See, e.g. “Principles ofNeural Science”, McGraw-Hill Medical, New York, N.Y. (2000); Motulsky, “Intuitive Biostatistics”, Oxford University Press, Inc. (1995); Eodish et al., “Molecular Cell Biology, 4th ed ”, W. H. Freeman & Co., New York (2000); Griffiths et al., “Introduction to Genetic Analysis, 7th ed.”, W. H. Freeman & Co., N.Y. (1999); and Gilbert et al., “Developmental Biology, 6th ed.”, Sinauer Associates, Inc., Sunderland, MA (2000).
Chemistry terms used herein, unless otherwise defined herein, are used according to conventional usage in the art, as exemplified by “The McGraw-Hill Dictionary of Chemical Terms”, Parker S., Ed., McGraw-Hill, San Francisco, C.A. (1985).
All of the above, and any other publications, patents and published patent applications referred to in this application are specifically incorporated by reference herein. In case of conflict, the present specification, including its specific definitions, will control.
The term “agent” is used herein to denote a chemical compound (such as an organic or inorganic compound, a mixture of chemical compounds), a biological macromolecule (such as a nucleic acid, an antibody, including parts thereof as well as humanized, chimeric and human antibodies and monoclonal antibodies, a protein or portion thereof, e.g., a peptide, a lipid, a carbohydrate), or an extract made from biological materials such as bacteria, plants, fungi, or animal (particularly mammalian) cells or tissues. Agents include, for example, agents whose structure is known, and those whose structure is not known.
A “patient,” “subject,” or “individual” are used interchangeably and refer to either a human or a non-human animal. These terms include mammals, such as humans, primates, livestock animals (including bovines, porcines, etc.), companion animals (e.g., canines, felines, etc.) and rodents (e.g., mice and rats).
“Treating” a condition or patient refers to taking steps to obtain beneficial or desired results, including clinical results. Beneficial or desired clinical results can include, but are not limited to, alleviation or amelioration of one or more symptoms or conditions, diminishment of extent of disease, stabilized (i.e. not worsening) state of disease, preventing spread of disease, delay or slowing of disease progression, amelioration or palliation of the disease state, and remission (whether partial or total), whether detectable or undetectable. “Treatment” can also mean prolonging survival as compared to expected survival if not receiving treatment.
The term “preventing” is art-recognized, and when used in relation to a condition, such as a local recurrence (e.g., pain), a disease such as cancer, a syndrome complex such as heart failure or any other medical condition, is well understood in the art, and includes administration of a composition which reduces the frequency of, or delays the onset of, symptoms of a medical condition in a subject relative to a subject which does not receive the composition. Thus, prevention of cancer includes, for example, reducing the number of detectable cancerous growths in a population of patients receiving a prophylactic treatment relative to an untreated control population, and/or delaying the appearance of detectable cancerous growths in a treated
population versus an untreated control population, e.g., by a statistically and/or clinically significant amount.
“Administering” or “administration of’ a substance, a compound or an agent to a subject can be carried out using one of a variety of methods known to those skilled in the art. For example, a compound or an agent can be administered, intravenously, arterially, intradermally, intramuscularly, intraperitoneally, subcutaneously, ocularly, sublingually, orally (by ingestion), intranasally (by inhalation), intraspinally, intracerebrally, and transdermally (by absorption, e.g., through a skin duct). A compound or agent can also appropriately be introduced by rechargeable or biodegradable polymeric devices or other devices, e.g., patches and pumps, or formulations, which provide for the extended, slow or controlled release of the compound or agent. Administering can also be performed, for example, once, a plurality of times, and/or over one or more extended periods.
Appropriate methods of administering a substance, a compound or an agent to a subject will also depend, for example, on the age and/or the physical condition of the subject and the chemical and biological properties of the compound or agent (e.g., solubility, digestibility, bioavailability, stability and toxicity). In some embodiments, a compound or an agent is administered orally, e.g., to a subject by ingestion. In some embodiments, the orally administered compound or agent is in an extended release or slow release formulation, or administered using a device for such slow or extended release.
As used herein, the phrase “conjoint administration” refers to any form of administration of two or more different therapeutic agents such that the second agent is administered while the previously administered therapeutic agent is still effective in the body (e.g., the two agents are simultaneously effective in the patient, which may include synergistic effects of the two agents). For example, the different therapeutic compounds can be administered either in the same formulation or in separate formulations, either concomitantly or sequentially. Thus, an individual who receives such treatment can benefit from a combined effect of different therapeutic agents.
A “therapeutically effective amount” or a “therapeutically effective dose” of a drug or agent is an amount of a drug or an agent that, when administered to a subject will have the intended therapeutic effect. The full therapeutic effect does not necessarily occur by administration of one dose, and may occur only after administration of a series of doses. Thus, a therapeutically effective amount may be administered in one or more administrations. The
precise effective amount needed for a subject will depend upon, for example, the subject’s size, health and age, and the nature and extent of the condition being treated, such as cancer or MDS. The skilled worker can readily determine the effective amount for a given situation by routine experimentation.
As used herein, the terms “optional” or “optionally” mean that the subsequently described event or circumstance may occur or may not occur, and that the description includes instances where the event or circumstance occurs as well as instances in which it does not. For example, “optionally substituted alkyl” refers to the alkyl may be substituted as well as where the alkyl is not substituted.
It is understood that substituent and substitution patterns on the compounds of the present disclosure can be selected by one of ordinary skilled person in the art to result chemically stable compounds which can be readily synthesized by techniques known in the art, as well as those methods set forth below, from readily available starting materials. If a substituent is itself substituted with more than one group, it is understood that these multiple groups may be on the same carbon or on different carbons, so long as a stable structure results.
As used herein, the term “optionally substituted” refers to the replacement of one to six hydrogen radicals in a given structure with the radical of a specified substituent including, but not limited to: hydroxyl, hydroxyalkyl, alkoxy, halogen, alkyl, nitro, silyl, acyl, acyloxy, aryl, cycloalkyl, heterocyclyl, amino, aminoalkyl, cyano, haloalkyl, haloalkoxy, -OCO-CH2-O- alkyl, -OP(O)(O-alkyl)2 or -CH2-OP(O)(O-alkyl)2. Preferably, “optionally substituted” refers to the replacement of one to four hydrogen radicals in a given structure with the substituents mentioned above. More preferably, one to three hydrogen radicals are replaced by the substituents as mentioned above. It is understood that the substituent can be further substituted.
The term “conjugates” as used herein refers to cell binding agents that are covalently bonded to one or more molecules of a cytotoxic compound. In this regard, "cell binding agent" is a molecule having affinity for a biological target, and may be, for example, an antibody, particularly a monoclonal antibody, or an antibody fragment, and the binding agent functions to direct a biologically active compound to a biological target. In certain embodiments, the conjugate may be designed to target tumor cells through cell surface antigens. The antigen may be a cell surface antigen that is overexpressed or expressed in an abnormal cell type. Specifically, the target antigen may be expressed only on proliferative cells (e.g., tumor cells). The target antigen may be selected on the basis of different expression, usually between
proliferative tissues and normal tissues. In the present disclosure, the antibody is bonded to the linker.
In the present disclosure, a "variant" of a polypeptide, for example, an antigen-binding fragment, a protein, or an antibody, is a polypeptide in which insertion, deletion, addition, and/or substitution have occurred at one or more amino acid residues compared to other polypeptide sequences, and includes fusion polypeptides. Protein variants also include those modified by protein enzymatic cleavage, phosphorylation or other post-translational modifications, but retaining the biological activity of the antibody disclosed herein, such as binding and specificity to CD20 and/or CD22. Variants may have about 99% identity, about 98% identity, about 97% identity, about 96% identity, about 95% identity, about 94% identity, about 93% identity, about 92% identity, about 91% identity, about 90% identity, about 89% identity, about 88% identity, about 87% identity, about 86% identity, about 85% identity, about 84% identity, about 83% identity, about 82% identity, about 81% identity, or about 80% identity to the sequence of the antibody or antigen-binding fragment thereof according to the present disclosure. Percent identity (%) or homology may be calculated by methods known in the art.
In certain embodiments, the percent homology or identity can be calculated by 100X[(same position)/min(TGA, TGB)], wherein TGA and TGB are the sum of the number of residues and internal gap positions in sequences A and B to be compared (Russell et al., J. Mol Biol., 244: 332-350 (1994).
The term "derivative" of a polypeptide as used herein refers to a polypeptide that has chemical modification at one or more residues through conjugation with other chemical moi eties, different from insertion, deletion, addition or substitution variants.
The term "percent sequence identity" or "percent identity" between two polynucleotide or polypeptide sequences refers to the number of identical matched positions shared by the sequences over a comparison window, taking into account additions or deletions (i.e., gaps) that must be introduced for optimal alignment of the two sequences. A matched position is any position where an identical nucleotide or amino acid is presented in both the target and reference sequence. Gaps presented in the target sequence are not counted since gaps are not nucleotides or amino acids. Likewise, gaps presented in the reference sequence are not counted since target sequence nucleotides or amino acids are counted, not nucleotides or amino acids from the reference sequence. The percentage of sequence identity is calculated by determining
the number of positions at which the identical amino-acid residue or nucleic acid base occurs in both sequences to yield the number of matched positions, dividing the number of matched positions by the total number of positions in the window of comparison and multiplying the result by 100 to yield the percentage of sequence identity. The comparison of sequences and determination of percent sequence identity between two sequences can be accomplished using readily available software programs. Suitable software programs are available from various sources, and for alignment of both protein and nucleotide sequences. One suitable program to determine percent sequence identity is bl2seq, part of the BLAST suite of program available from the U.S. government's National Center for Biotechnology Information BLAST web site (at world wide web at blast.ncbi.nlm.nih.gov). B12seq performs a comparison between two sequences using either the BLASTN or BLASTP algorithm. BLASTN is used to compare nucleic acid sequences, while BLASTP is used to compare amino acid sequences. Other suitable programs are, e.g., Needle, Stretcher, Water, or Matcher, part of the EMBOSS suite of bioinformatics programs and also available from the European Bioinformatics Institute (EBI) at world wide web at ebi.ac.uk/Tools/psa.
As used herein, “homology” with respect to a peptide, polypeptide or antibody sequence refers to the percentage of amino acid residues in a candidate sequence that are identical with the amino acid residues in the specific peptide or polypeptide sequence, after aligning the sequences and introducing gaps, if necessary, to achieve the maximum percent sequence identity, and not considering any conservative substitutions as part of the sequence identity. Alignment for purposes of determining percent amino acid sequence identity can be achieved in various ways that are within the skill in the art, for instance, using publicly available computer software such as BLAST, BLAST-2, ALIGN or MEGALIGN™ (DNASTAR) software. Those skilled in the art can determine appropriate parameters for measuring alignment, including any algorithms known in the art needed to achieve maximal alignment over the full length of the sequences being compared.
The term “affinity” or “avidity” refers to the strength of interaction between an antibody or its antigen-binding fragment and an antigen, determined by characteristics of the antigen such as size, shape, and/or charge, and the CDR sequences of the antibody or antigen-binding fragment. Methods for determining such affinities are known in the art and may also be referenced herein.
Antibodies or their antigen-binding fragments used in the present invention are said to “specifically bind” to the target, such as the antigen, when the dissociation constant (KD) is <10'6 M. Antibodies bind “with high affinity” to the target when KD is <lx 10'8 M.
As used in the present disclosure, the “antigen-binding fragment” of a chain (heavy chain or light chain) of an antibody or immunoglobulin includes a part of an antibody which lacks some amino acids compared to a full-length chain, but can specifically bind to an antigen (e.g., CD22 and/or CD20). This fragment can be considered as having biological activity, in that the fragment can specifically bind to a target antigen, or can compete with other antibodies or antigen binding fragments thereof to bind to a specific epitope. In certain embodiments, such a fragment includes at least one CDR present in a full-length light chain or heavy chain, and in some embodiments, includes a short-chain heavy chain and/or light chain, or part thereof. This biological active fragment may be produced by a recombinant DNA technique or may be produced, for example, by cleaving an intact antibody enzymatically or chemically. An immunologically functional immunoglobulin fragment includes, but is not limited to, Fab, Fab', scFab, dsFv, Fv, scFV, scFV-Fc, 83cab, and dAb, scFv2-Fc, bi-nanobody, bispecific T cell engager (BiTE), tandem diabody (tandAb), dual affinity retargeting (DART) antibody, DART-Fc, scFv-human serum albumin (HSA)-scFv, dock-and-lock (DNL)-Fab3, minibody, Fab2 fragment (bispecific), Fab3 fragment (trispecific), Bis-scFv fragment (bispecific), sdAb fragment (VH/VHH), tetrabody, triabody, or a diabody, and may be derived from any mammal, including, but not being limited to, a human, a mouse, a rat, a camelid, or a rabbit. The functional parts of antibodies such as the one or more CDRs disclosed in the present disclosure may be linked with a secondary protein or a small compound by a covalent bond, and thereby used as a targeted therapeutic agent for a specific target.
In the present disclosure, the “Fc” region includes two heavy chain fragments including CH2 and CH3 domains of an antibody. These two heavy chain fragments are linked to each other by hydrophobic interaction of two or more of disulfide bonds and a CH3 domain.
In the present disclosure, the “Fab fragment” consists of one light chain and one heavy chain including a variable region and CHI only. The heavy chain of a Fab molecule cannot form a disulfide bond with another heavy chain molecule. In an scFab, two molecules of Fab are linked by a flexible linker.
In the present disclosure, the “Fab' fragment” includes a Fab fragment and additionally a region between CHI and CH2 domains of a heavy chain. A disulfide bond may form between two heavy chains of Fab' fragments of two molecules, forming a F(ab')2 molecule.
In the present disclosure, as described above, the “F(ab')2 fragment” includes two light chains and two heavy chains including a variable region CHI and part of a constant region between the CHI and CH2 domains, with an inter-chain disulfide bond formed between the two heavy chains. Accordingly, a F(ab')2 fragment consists of two Fab' fragments, and the two Fab' fragments are joined to each other by the disulfide bond therebetween.
In the present disclosure, the “Fv region” is a fragment of an antibody which includes each variable region of a heavy chain and a light chain, but does not include constant regions. In an sdFV, a heavy chain and a light chain are linked by a disulfide bond. In an scFc, the Fv is linked by a flexible linker. In a scFv-Fc, a Fc is linked to a scFV. In a minibody, CH3 is linked to a scFV. A diabody includes the scFVs of two molecules.
In the present disclosure, the “single chain Fv” or “scFv” antibody fragment includes the VH and VL domains of an antibody, and these domains are present within a single polypeptide chain. An Fv polypeptide may additionally include a polypeptide linker between a Vh domain which enables the scFv to form the target structure for antigen binding, and a VL domain.
In the present disclosure, the “short-chain antibody (84cab)” is a single polypeptide chain including one constant region of a heavy chain or a light chain constant region in which heavy chain and light chain variable regions are linked by a flexible linker. For a short-chain antibody, U.S. Pat. No. 5,260,203 may be referred to, and short-chain antibody is disclosed herein by reference.
In the present disclosure, the “domain antibody (dAb)” is an immunologically functional immunoglobulin fragment including only a variable region of a heavy chain or a variable region of a light chain. In certain embodiments, two or more VH regions are linked by a covalent bond via a peptide linker, to form a bivalent domain antibody. Two VH regions of this bivalent domain antibody may target the same or different antigens.
In the present disclosure, “complementarity determining region” (CDR; that is, CDR1, CDR2, and CDR3) denotes amino acid residues of the variable region of an antibody, which are necessary for binding to antigen. Each variable region typically has three CDR domains, identified as CDR1, CDR2, and CDR3.
In the present disclosure, the “framework region” (FR) is a variable region residue other than the CDR residues. Each variable region typically has four FRs, identified as FR1, FR2, FR3, and FR4.
In the present disclosure, the “bivalent antigen-binding protein” or “bivalent antibody” includes two antigen-binding sites. The two antigen-binding sites included in a bivalent antibody may have the same antigen specificity, or may be a bispecific antibody where the antigen-biding sites bind to different antigens.
In the present disclosure, the “bispecific antibody” refers to an antibody or an antigenbinding fragment thereof capable of binding to two different epitopes on a single antigen or two different antigens. The bispecific antibody of the present disclosure may be bivalent, trivalent, or tetravalent. As used herein, “-valent”, “-valent(s)” or other grammatical variations thereof, refer to the number of antigen-binding sites within an antibody molecule.
In the present disclosure, the “multispecific antigen-binding protein” or “multispecific antibody” targets two or more antigens or epitopes.
As used herein, a “chimeric antibody” refers to an antibody (immunoglobulin) in which a portion of the heavy and/or light chain is identical with or homologous to corresponding sequences in antibodies derived from a particular species or belonging to a particular antibody class or subclass, while the remainder of the chain(s) is(are) identical with or homologous to corresponding sequences in antibodies derived from another species or belonging to another antibody class or subclass, as well as fragments of such antibodies, so long as they exhibit the desired biological activity (U.S. Patent No. 4,816,567; Morrison et al., Proc. Nat’l Acad. Sci. USA, 81:6851-55 (1984)). Chimeric antibodies of interest herein include PRIMATIZED® antibodies wherein the antigen-binding region of the antibody is derived from an antibody produced by, e.g., immunizing macaque monkeys with an antigen of interest.
“Humanized” forms of non-human (e.g., murine) antibodies, are chimeric antibodies that contain minimal sequence derived from non-human immunoglobulin. In one embodiment, a humanized antibody is a human immunoglobulin (recipient antibody) in which residues from an CDR of the recipient are replaced by residues from an CDR of a non-human species (donor antibody) such as mouse, rat, rabbit or non-human primate having the desired specificity, affinity, and/or capacity. In some instances, FR residues of the human immunoglobulin are replaced by corresponding non-human residues. Furthermore, humanized antibodies may comprise residues that are not found in the recipient antibody or in the donor antibody. These
modifications may be made to further refine antibody performance, such as binding affinity. In general, a humanized antibody will comprise substantially all of at least one, and typically two, variable domains, in which all or substantially all of the hypervariable loops correspond to those of a non-human immunoglobulin sequence, and all or substantially all of the FR regions are those of a human immunoglobulin sequence, although the FR regions may include one or more individual FR residue substitutions that improve antibody performance, such as binding affinity, isomerization, immunogenicity, and the like. The number of these amino acid substitutions in the FR is typically no more than 6 in the H chain, and in the L chain, no more than 3. The humanized antibody optionally will also comprise at least a portion of an immunoglobulin constant region (Fc), typically that of a human immunoglobulin. For further details, see, e.g., Jones et al., Nature 321:522-525 (1986); Riechmann et al., Nature 332:323- 329 (1988); and Presta, Curr. Op. Struct. Biol. 2:593-596 (1992). See also, for example, Vaswani and Hamilton, Ann. Allergy, Asthma & Immunol. 1: 105-115 (1998); Harris, Biochem. Soc. Transactions 23: 1035-1038 (1995); Hurle and Gross, Curr. Op. Biotech. 5:428- 433 (1994); and U.S. Patent Nos. 6,982,321 and 7,087,409.
A “human antibody” is one that possesses an amino-acid sequence corresponding to that of an antibody, produced by a human and/or has been made using any of the techniques for making human antibodies as disclosed herein. This definition of a human antibody specifically excludes a humanized antibody comprising non-human antigen-binding residues. Human antibodies can be produced using various techniques known in the art, including phagedisplay libraries. Hoogenboom and Winter, J. Mol. Biol., 227:381 (1991); Marks et al., J. Mol. Biol., 222:581 (1991). Also available forthe preparation of human monoclonal antibodies are methods described in Cole et al., Monoclonal Antibodies and Cancer Therapy, Alan R. Liss, p. 77 (1985); Boemer et al., J. Immunol., 147(l):86-95 (1991). See also van Dijk and van de Winkel, Curr. Opin. Pharmacol. 5:368-74 (2001). Human antibodies can be prepared by administering the antigen to a transgenic animal that has been modified to produce such antibodies in response to antigenic challenge, but whose endogenous loci have been disabled, e.g., immunized xenomice (see, e.g., U.S. Patent Nos. 6,075,181 and 6,150,584 regarding XENOMOUSETM technology). See also, for example, Li et al., Proc. Nat’l Acad. Sci. USA, 103:3557-3562 (2006) regarding human antibodies generated via a human B-cell hybridoma technology.
A “human consensus framework” is a framework that represents the most commonly occurring amino acid residues in a selection of human immunoglobulin VL or VH framework sequences. Generally, the selection of human immunoglobulin VL or VH sequences is from a subgroup of variable domain sequences. Generally, the subgroup of sequences is a subgroup as in Kabat et al., Sequences of Proteins of Immunological Interest, 5th Ed. Public Health Service, National Institutes of Health, Bethesda, MD (1991). Examples include for the VL, the subgroup may be subgroup kappa I, kappa II, kappa III or kappa IV as in Kabat et al., supra. Additionally, for the VH, the subgroup may be subgroup I, subgroup II, or subgroup III as in Kabat et al., supra.
An “affinity-matured” antibody is one with one or more alterations in one or more CDRs thereof that result in an improvement in the affinity of the antibody for antigen, compared to a parent antibody that does not possess those alteration(s). In one embodiment, an affinity-matured antibody has nanomolar or even picomolar affinities for the target antigen. Affinity-matured antibodies are produced by procedures known in the art. For example, Marks et al., Bio/Technology 10:779-783 (1992) describes affinity maturation by VH- and VL- domain shuffling. Random mutagenesis of CDR and/or framework residues is described by, for example: Barbas et al. Proc Nat. Acad. Sci. USA 91:3809-3813 (1994); Schier et al. Gene 169: 147- 155 (1995); Yelton et al. J. Immunol. 155: 1994-2004 (1995); Jackson et al., J. Immunol. 154(7):3310-9 (1995); and Hawkins et al, J. Mol. Biol. 226:889-896 (1992).
In the present disclosure, “linker” refers to a compound which covalently bonds a cytotoxic compound to an antibody.
In the present disclosure, "unsubstituted or substituted" is used to refer to a parent group which may be unsubstituted or substituted, "substituted" refers to a parent group having at least one substituent, and a substituent refers to a chemical moiety covalently bonded to or fused with a parent group.
In the present disclosure, “halo” refers to fluorine, chlorine, bromine, iodine, and the like.
As used herein, the term “alkyl” refers to saturated aliphatic groups, including but not limited to Ci-Cio straight-chain alkyl groups or Ci-Cio branched-chain alkyl groups. Preferably, the “alkyl” group refers to Ci-Cg straight-chain alkyl groups or Ci-Cg branched- chain alkyl groups. Most preferably, the “alkyl” group refers to C1-C4 straight-chain alkyl groups or C1-C4 branched-chain alkyl groups. Examples of “alkyl” include, but are not limited
to, methyl, ethyl, 1-propyl, 2-propyl, n-butyl, sec-butyl, tert-butyl, 1-pentyl, 2-pentyl, 3-pentyl, neo-pentyl, 1-hexyl, 2-hexyl, 3-hexyl, 1-heptyl, 2-heptyl, 3-heptyl, 4-heptyl, 1-octyl, 2-octyl, 3-octyl or 4-octyl and the like. The “alkyl” group may be optionally substituted.
The term “acyl” is art-recognized and refers to a group represented by the general formula hydrocarbylC(O)-, preferably alkylC(O)-.
The term “acylamino” is art-recognized and refers to an amino group substituted with an acyl group and may be represented, for example, by the formula hydrocarbylC(O)NH-.
The term “acyloxy” is art-recognized and refers to a group represented by the general formula hydrocarbylC(O)O-, preferably alkylC(O)O-.
The term “alkoxy” refers to an alkyl group having an oxygen attached thereto. Representative alkoxy groups include methoxy, ethoxy, propoxy, tert-butoxy and the like.
The term “alkoxyalkyl” refers to an alkyl group substituted with an alkoxy group and may be represented by the general formula alkyl-O-alkyl.
The term “alkyl” refers to saturated aliphatic groups, including straight-chain alkyl groups, branched-chain alkyl groups, cycloalkyl (alicyclic) groups, alkyl-substituted cycloalkyl groups, and cycloalkyl-substituted alkyl groups. In preferred embodiments, a straight chain or branched chain alkyl has 30 or fewer carbon atoms in its backbone (e.g., Ci- 30 for straight chains, C3-30 for branched chains), and more preferably 20 or fewer.
Moreover, the term “alkyl” as used throughout the specification, examples, and claims is intended to include both unsubstituted and substituted alkyl groups, the latter of which refers to alkyl moieties having substituents replacing a hydrogen on one or more carbons of the hydrocarbon backbone, including haloalkyl groups such as trifluoromethyl and 2,2,2- trifluoroethyl, etc.
The term “Cx-y” or “Cx-Cy”, when used in conjunction with a chemical moiety, such as, acyl, acyloxy, alkyl, alkenyl, alkynyl, or alkoxy is meant to include groups that contain from x to y carbons in the chain. Coalkyl indicates a hydrogen where the group is in a terminal position, a bond if internal. A Ci-ealkyl group, for example, contains from one to six carbon atoms in the chain.
The term “alkylamino”, as used herein, refers to an amino group substituted with at least one alkyl group.
The term “alkylthio”, as used herein, refers to a thiol group substituted with an alkyl group and may be represented by the general formula alkylS-.
The term “amido”, as used herein, refers to a group
 wherein R9 and R10 each independently represent a hydrogen or hydrocarbyl group, or R9 and R10 taken together with the N atom to which they are attached complete a heterocycle having from 4 to 8 atoms in the ring structure.
wherein R9 and R10 each independently represent a hydrogen or hydrocarbyl group, or R9 and R10 taken together with the N atom to which they are attached complete a heterocycle having from 4 to 8 atoms in the ring structure.
The terms “amine” and “amino” are art-recognized and refer to both unsubstituted and substituted amines and salts thereof, e.g., a moiety that can be represented by
 wherein R9, R10, and R10’ each independently represent a hydrogen or a hydrocarbyl group, or R9 and R10 taken together with the N atom to which they are attached complete a heterocycle having from 4 to 8 atoms in the ring structure.
wherein R9, R10, and R10’ each independently represent a hydrogen or a hydrocarbyl group, or R9 and R10 taken together with the N atom to which they are attached complete a heterocycle having from 4 to 8 atoms in the ring structure.
The term “aminoalkyl”, as used herein, refers to an alkyl group substituted with an amino group.
The term “aralkyl”, as used herein, refers to an alkyl group substituted with an aryl group.
The term “aryl” as used herein include substituted or unsubstituted single-ring aromatic groups in which each atom of the ring is carbon. Preferably the ring is a 5- to 7-membered ring, more preferably a 6-membered ring. The term “aryl” also includes polycyclic ring systems having two or more cyclic rings in which two or more carbons are common to two adjoining rings wherein at least one of the rings is aromatic, e.g., the other cyclic rings can be cycloalkyls, cycloalkenyls, cycloalkynyls, aryls, heteroaryls, and/or heterocyclyls. Aryl groups include benzene, naphthalene, phenanthrene, phenol, aniline, and the like.
The term “carbamate” is art-recognized and refers to a group
 wherein R9 and R10 independently represent hydrogen or a hydrocarbyl group.
wherein R9 and R10 independently represent hydrogen or a hydrocarbyl group.
The term “carbocyclylalkyl”, as used herein, refers to an alkyl group substituted with a carbocycle group.
The term “carbocycle” includes 5-7 membered monocyclic and 8-12 membered bicyclic rings. Each ring of a bicyclic carbocycle may be selected from saturated, unsaturated and aromatic rings. Carbocycle includes bicyclic molecules in which one, two or three or more atoms are shared between the two rings. The term “fused carbocycle” refers to a bicyclic carbocycle in which each of the rings shares two adjacent atoms with the other ring. Each ring of a fused carbocycle may be selected from saturated, unsaturated and aromatic rings. In an exemplary embodiment, an aromatic ring, e.g., phenyl, may be fused to a saturated or unsaturated ring, e.g., cyclohexane, cyclopentane, or cyclohexene. Any combination of saturated, unsaturated and aromatic bicyclic rings, as valence permits, is included in the definition of carbocyclic. Exemplary “carbocycles” include cyclopentane, cyclohexane, bicyclo[2.2.1]heptane, 1,5-cyclooctadiene, 1,2,3,4-tetrahydronaphthalene, bicyclo[4.2.0]oct- 3-ene, naphthalene and adamantane. Exemplary fused carbocycles include decalin, naphthalene, 1,2,3,4-tetrahydronaphthalene, bicyclo[4.2.0]octane, 4,5,6,7-tetrahydro-lH- indene and bicyclo [4.1.0]hept-3-ene. “Carbocycles” may be substituted at any one or more positions capable of bearing a hydrogen atom.
The term “carbocyclylalkyl”, as used herein, refers to an alkyl group substituted with a carbocycle group.
The term “carbonate” is art-recognized and refers to a group -OCO2-.
The term “carboxy”, as used herein, refers to a group represented by the formula -CO2H.
The term “cycloalkyl” includes substituted or unsubstituted non-aromatic single ring structures, preferably 4- to 8-membered rings, more preferably 4- to 6-membered rings. The term “cycloalkyl” also includes polycyclic ring systems having two or more cyclic rings in which two or more carbons are common to two adjoining rings wherein at least one of the rings is cycloalkyl and the substituent (e.g., R100) is attached to the cycloalkyl ring, e.g., the other cyclic rings can be cycloalkyls, cycloalkenyls, cycloalkynyls, aryls, heteroaryls, and/or heterocyclyls. Heteroaryl groups include, for example, pyrrole, furan, thiophene, imidazole, oxazole, thiazole, pyrazole, pyridine, pyrazine, pyridazine, pyrimidine, denzodioxane, tetrahydroquinoline, and the like.
The term “ester”, as used herein, refers to a group -C(O)OR9 wherein R9 represents a hydrocarbyl group.
The term “ether”, as used herein, refers to a hydrocarbyl group linked through an oxygen to another hydrocarbyl group . Accordingly, an ether substituent of a hydrocarbyl group may be hydrocarbyl-O-. Ethers may be either symmetrical or unsymmetrical. Examples of ethers include, but are not limited to, heterocycle-O-heterocycle and aryl-0 -heterocycle. Ethers include “alkoxyalkyl” groups, which may be represented by the general formula alkyl-O-alkyl.
The terms “halo” and “halogen” as used herein means halogen and includes chloro, fluoro, bromo, and iodo.
The terms “hetaralkyl” and “heteroaralkyl”, as used herein, refers to an alkyl group substituted with a hetaryl group.
The terms “heteroaryl” and “hetaryl” include substituted or unsubstituted aromatic single ring structures, preferably 5- to 7-membered rings, more preferably 5- to 6-membered rings, whose ring structures include at least one heteroatom, preferably one to four heteroatoms, more preferably one or two heteroatoms. The terms “heteroaryl” and “hetaryl” also include polycyclic ring systems having two or more cyclic rings in which two or more carbons are common to two adjoining rings wherein at least one of the rings is heteroaromatic, e.g., the other cyclic rings can be cycloalkyls, cycloalkenyls, cycloalkynyls, aryls, heteroaryls, and/or heterocyclyls. Heteroaryl groups include, for example, pyrrole, furan, thiophene, imidazole, oxazole, thiazole, pyrazole, pyridine, pyrazine, pyridazine, and pyrimidine, and the like.
The term “heteroatom” as used herein means an atom of any element other than carbon or hydrogen. Preferred heteroatoms are nitrogen, oxygen, and sulfur.
The term “heterocyclylalkyl”, as used herein, refers to an alkyl group substituted with a heterocycle group.
The terms “heterocyclyl”, “heterocycle”, and “heterocyclic” refer to substituted or unsubstituted non-aromatic ring structures, preferably 3- to 10-membered rings, more preferably 3- to 7-membered rings, whose ring structures include at least one heteroatom, preferably one to four heteroatoms, more preferably one or two heteroatoms. The terms “heterocyclyl” and “heterocyclic” also include polycyclic ring systems having two or more cyclic rings in which two or more carbons are common to two adjoining rings wherein at least one of the rings is heterocyclic, e.g., the other cyclic rings can be cycloalkyls, cycloalkenyls, cycloalkynyls, aryls, heteroaryls, and/or heterocyclyls. Heterocyclyl groups include, for example, piperidine, piperazine, pyrrolidine, morpholine, lactones, lactams, and the like.
The term “hydrocarbyl”, as used herein, refers to a group that is bonded through a carbon atom that does not have a =0 or =S substituent, and typically has at least one carbonhydrogen bond and a primarily carbon backbone, but may optionally include heteroatoms. Thus, groups like methyl, ethoxyethyl, 2-pyridyl, and even trifluoromethyl are considered to be hydrocarbyl for the purposes of this application, but substituents such as acetyl (which has a =0 substituent on the linking carbon) and ethoxy (which is linked through oxygen, not carbon) are not. Hydrocarbyl groups include, but are not limited to aryl, heteroaryl, carbocycle, heterocycle, alkyl, alkenyl, alkynyl, and combinations thereof.
The term “hydroxyalkyl”, as used herein, refers to an alkyl group substituted with a hydroxy group.
The term “lower” when used in conjunction with a chemical moiety, such as, acyl, acyloxy, alkyl, alkenyl, alkynyl, or alkoxy is meant to include groups where there are ten or fewer atoms in the substituent, preferably six or fewer. A “lower alkyl”, for example, refers to an alkyl group that contains ten or fewer carbon atoms, preferably six or fewer. In certain embodiments, acyl, acyloxy, alkyl, alkenyl, alkynyl, or alkoxy substituents defined herein are respectively lower acyl, lower acyloxy, lower alkyl, lower alkenyl, lower alkynyl, or lower alkoxy, whether they appear alone or in combination with other substituents, such as in the recitations hydroxyalkyl and aralkyl (in which case, for example, the atoms within the aryl group are not counted when counting the carbon atoms in the alkyl substituent).
The terms “polycyclyl”, “polycycle”, and “polycyclic” refer to two or more rings (e.g., cycloalkyls, cycloalkenyls, cycloalkynyls, aryls, heteroaryls, and/or heterocyclyls) in which two or more atoms are common to two adjoining rings, e.g., the rings are “fused rings”. Each of the rings of the polycycle can be substituted or unsubstituted. In certain embodiments, each ring of the poly cycle contains from 3 to 10 atoms in the ring, preferably from 5 to 7.
The term “sulfate” is art-recognized and refers to the group -OSO3H, or a pharmaceutically acceptable salt thereof.
The term “sulfonamide” is art-recognized and refers to the group represented by the general formulae
 wherein R9 and R10 independently represents hydrogen or hydrocarbyl.
The term “sulfoxide” is art-recognized and refers to the group-S(O)-.
wherein R9 and R10 independently represents hydrogen or hydrocarbyl.
The term “sulfoxide” is art-recognized and refers to the group-S(O)-.
The term “sulfonate” is art-recognized and refers to the group SO3H, or a pharmaceutically acceptable salt thereof.
The term “sulfone” is art-recognized and refers to the group -S(O)2-.
The term “substituted” refers to moieties having substituents replacing a hydrogen on one or more carbons of the backbone. It will be understood that “substitution” or “substituted with” includes the implicit proviso that such substitution is in accordance with permitted valence of the substituted atom and the substituent, and that the substitution results in a stable compound, e.g., which does not spontaneously undergo transformation such as by rearrangement, cyclization, elimination, etc. As used herein, the term “substituted” is contemplated to include all permissible substituents of organic compounds. In a broad aspect, the permissible substituents include acyclic and cyclic, branched and unbranched, carbocyclic and heterocyclic, aromatic and non-aromatic substituents of organic compounds. The permissible substituents can be one or more and the same or different for appropriate organic compounds. For purposes of this invention, the heteroatoms such as nitrogen may have hydrogen substituents and/or any permissible substituents of organic compounds described herein which satisfy the valences of the heteroatoms. Substituents can include any substituents described herein, for example, a halogen, a hydroxyl, a carbonyl (such as a carboxyl, an alkoxycarbonyl, a formyl, or an acyl), a thiocarbonyl (such as a thioester, a thioacetate, or a thioformate), an alkoxyl, a phosphoryl, a phosphate, a phosphonate, a phosphinate, an amino, an amido, an amidine, an imine, a cyano, a nitro, an azido, a sulfhydryl, an alkylthio, a sulfate, a sulfonate, a sulfamoyl, a sulfonamide, a sulfonyl, a heterocyclyl, an aralkyl, or an aromatic or heteroaromatic moiety. It will be understood by those skilled in the art that the moieties substituted on the hydrocarbon chain can themselves be substituted, if appropriate.
The term “thioalkyl”, as used herein, refers to an alkyl group substituted with a thiol group.
The term “thioester”, as used herein, refers to a group -C(O)SR9 or -SC(O)R9 wherein R9 represents a hydrocarbyl.
The term “thioether”, as used herein, is equivalent to an ether, wherein the oxygen is replaced with a sulfur.
The term “urea” is art-recognized and may be represented by the general formula
 wherein R9 and R10 independently represent hydrogen or a hydrocarbyl.
wherein R9 and R10 independently represent hydrogen or a hydrocarbyl.
The term “DBCO” used herein refers to an optionally substituted dibenzocyclooctyne moiety, e.g., the following structure:

The term “sugar moiety” used herein refers to a naturally occurring sugar or a modified sugar which is part of a larger molecule and is connected to the remainder of the larger molecule through, for example, one of the hydroxyl groups present on the sugar. For example, in some embodiments the sugar is connected to the remainder of the larger molecule via the hydroxyl group on the anomeric carbon. Examples of sugar moieties include, but are not limited to, glucuronosyl and galactosyl.
A “glycosidase” is an enzyme that breaks down glycosidic bonds in carbohydrates, glycoproteins, and glycolipids. Examples of glycosidases are P-glucuronidase and P- galactosidase. P-glucuronidase is a type of glucuronidase that catalyzes hydrolysis of P-D- glucuronic acid or P-glucuronosyl residues. P-Galactosidase is a glycoside hydrolase enzyme that catalyzes hydrolysis of terminal non-reducing P-D-galactose or P-galactosyl residues in P- D-galactosides.
The term “modulate” as used herein includes the inhibition or suppression of a function or activity (such as cell proliferation) as well as the enhancement of a function or activity.
The term “cleavage group” refers to a chemical moiety which dissociates when subjected to a stimulus, such as acidic conditions, basic conditions, reducing conditions, oxidizing conditions, light, or heat, or an enzyme, such as an esterase.
Examples
The invention now being generally described, it will be more readily understood by reference to the following examples which are included merely for purposes of illustration of certain aspects and embodiments of the present invention and are not intended to limit the invention.
Preparation Example 1: Preparation of Exemplified CD20-Specific Antibody
A first exemplified anti-CD20 antibody CD20 1 (Rituximab), that specifically binds to CD20, was produced by the methods described in U.S. Patent No. 5,736,137 A and U.S. Patent No. 7,422,739 B2, the entirety of which are incorporated herein by reference. The amino acid sequences of the antibody CD20 1 are shown in Table 1 below. The amino acid sequences of the CDRs below are defined according to the Kabat definition.
Table 1. Amino Acid Sequences of Exemplified CD20 Antibody CD20 1 (Rituximab)


For ADC synthesis, an exemplified antibody clone CD20_l-CaaX was constructed by introducing a CaaX peptide moiety (GGGGGGGCVIM, SEQ ID NO: 77) to the C-terminus of the light chain of SEQ ID NO: 18 in Table 1 with the method disclosed in International Patent Application No. PCT/IB2012/001065, the entirety of which is incorporated herein by reference. The amino acid sequences of CD20_l-CaaX are shown in Table 2 below.
Table 2. Amino Acid Sequences of CD20_l-CaaX


Preparation Example 2: Preparation of Exemplified Halfmers of CD20- or CD22- Specific Antibodies
Six exemplified halfmers of antibodies specifically binding to CD20 or CD22 were produced using the Knob-into-Hole method, described in U.S. Patent No. 9,637,557 B2, and the Pentambody method, described in U.S. Patent No. 11,498,977 B2, the entirety of which are incorporated herein by reference. The list of exemplified halfmers is shown in Table 3 below.
Table 3. Exemplified Halfmers

* amino acid positions according to EU numbering convention
The amino acid sequences of Halfinerl and Halfmer2, which specifically bind to CD20, are shown in Tables 4 and 5 below, respectively. The amino acid sequences of the CDRs below are defined according to the Kabat definition.
Exemplified halfinerl was prepared by introducing Hole mutations (T370S, L372A, Y41 IV of SEQ ID NO: 17 in Table 1) in the heavy chain constant region of the half-antibody
of CD20_l, and introducing LALA mutations (L238A/L239A of SEQ ID NO: 17 in Table 1) into the heavy chain constant region of a half-antibody of CD20 1.
Table 4. Amino Acid Sequences of Exemplified Halfmerl


A second exemplified anti-CD20 antibody CD20 2 (Ocaratuzumab), that specifically binds to CD20, was produced by the methods described in U.S. Patent No. 8,153,125 B2, the entirety of which is incorporated herein by reference. Exemplified halfiner2 was prepared by introducing Hole mutations (T370S, L372A, Y411V according to SEQ ID NO: 37) in the heavy chain constant region of a half-antibody of CD20 2, and introducing LALA mutations (L238A/L239A according to SEQ ID NO: 37) into the heavy chain constant region of a halfantibody of CD20_2.
Table 5. Amino Acid Sequences of Exemplified Halfmer2


The amino acid sequences of Halfiner3 and Halfmer4, which specifically bind to CD22, are shown in Tables 6 and 7 below, respectively. The amino acid sequences of the CDRs below are defined according to the Kabat definition.
A first exemplified anti-CD22 antibody CD22 1 (Inotuzumab) was produced by the methods described in U. S . Patent No. 8,747,857 B2, the entirety of which is incorporated herein by reference. Exemplified halfmer3 was prepared by introducing Knob mutation (T370W according to SEQ ID NO: 55) in the heavy chain constant region of a half-antibody of CD22 1, and introducing LALA mutations (L238A/L239A according to SEQ ID NO: 55) into the heavy
chain constant region of a half-antibody of CD22 1.
Table 6. Amino Acid Sequences of Exemplified Halfmer3


A second exemplified anti-CD22 antibody CD22 2 (Epratuzumab) was produced by the methods described in U.S. Patent No. 6,187,287 B2, the entirety of which is incorporated herein by reference. Exemplified halfiner4 is prepared by introducing Knob mutation (T365W according to SEQ ID NO: 73) in the heavy chain constant region of a half-antibody of CD22 2, and introducing LALA mutations (L233A/L234A according to SEQ ID NO: 73) into the heavy chain constant region of a half-antibody of CD22_2.
Table 7. Amino Acid Sequences of Exemplified Halfmer4


The amino acid sequences of Halfmer5, which specifically binds to CD20, and Halfiner6, which specifically binds to CD22, are shown in Tables 8 and 9 below, respectively. The amino acid sequences of the CDRs below are defined according to the Kabat definition.
Exemplified halfiner5 was prepared by introducing Pentambody substitutions (L355E, Y411L, K413V of SEQ ID NO: 17 in Table 1) in the heavy chain constant region of a halfantibody of CD20_l, and introducing LALA mutations (L238A/L239A of SEQ ID NO: 17 in
Table 1) into the heavy chain constant region of a half-antibody of CD20 1.
Table 8. Amino Acid Sequences of Exemplified Halfmer5


Exemplified halfmer6 was prepared by introducing Pentambody substitutions (T370L and D403R according to SEQ ID NO: 76) in the heavy chain constant region of a half-antibody of CD22_1, and introducing LALA mutations (L238A/L239A according to SEQ ID NO: 76) into the heavy chain constant region of a half-antibody of CD22 1.
Table 9. Amino Acid Sequences of Exemplified Halfmer6


Using CHO cell-based transient expression, the following halfiners were produced: CD20 (Hole) halfinerl, CD20 (Hole) halfiner2, CD22 (Knob) halfiner3, CD22 (Knob) halfiner4.
Using HEK293 cell-based transient expression, the following halfmers were produced: CD20 (U351E, Y407L, K409V according to EU numbering convention) halfiner5, and CD22 (T366L, D399R according to EU numbering convention) halfmer6.
Preparation Example 3: Preparation of Exemplified CD20- and CD22- Bispecific
Antibodies
For ADC synthesis, exemplified halfmer-CaaX constructs were prepared by introducing a CaaX peptide moiety (GGGGGGGCVIM, SEQ ID NO: 77) to the C-terminus of the light chain of each of Halfinerl (LC1; SEQ ID NO: 18) in Table 4, Halfmer5 (LC1; SEQ ID NO: 18) in Table 8, Halfmer2 (LC2; SEQ ID NO: 38) in Table 5, Halfiner3 (LC3; SEQ ID NO: 56) in Table 6, Halfmer6 (LC3; SEQ ID NO: 56) in Table 9, and Halfmer4 (LC4; SEQ ID NO: 74) in Table 7 with the method disclosed in International Patent Application No. PCT/IB2012/001065, the entirety of which is incorporated herein by reference.
Pairs of exemplified halfmer-CaaX constructs 1 and 3, 1 and 4, 2 and 3, and 2 and 4 were reacted using the Knob-into-Hole method, described in U.S. Patent No. 9,637,557 B2, to form bispecific antibodies, which were then used for subsequent ADC synthesis.
Specifically, for assembly of a bispecific antibody using the Knob-into-Hole method, each halfmer-CaaX construct was buffer-exchanged with a buffer containing 50 mM Arginine (Sigma-Aldrich, A4474) and 200 mM Histidine (Sigma-Aldrich, H6034) using vivaspin 20 (Sartorius, VS2002). The hole halfmer-CaaX and knob halfmer-CaaX pairs were then mixed at a 1: 1 ratio to achieve a concentration of 2 mg/mL each. Reduced L-glutathione (Sigma- Aldrich, G4251) was added to a final concentration of 2.6 mM. The reaction was then carried out in a shaking incubator at 36°C, 160 rpm, for 6 hours.
An exemplified pair of halfmer-CaaX constructs 5 and 6 was reacted using the Pentambody method, described in U.S. Patent No. 11,498,977 B2, to form bispecific antibodies for subsequent ADC synthesis.
The amino acid sequences of CD20xCD22 bispecific antibodyl, which specifically binds to CD20 and CD22, are shown in Table 10 below.
Table 10. Amino Acid Sequences of Exemplified CD20xCD22 Bispecific Antibodyl


The amino acid sequences of CD20xCD22 bispecific antibody2, which specifically binds to CD20 and CD22, are shown in Table 11 below.
Table 11. Amino Acid Sequences of Exemplified CD20xCD22 Bispecific Antibody2


The amino acid sequences of CD20xCD22 bispecific antibody3, which specifically binds to CD20 and CD22, are shown in Table 12 below.
Table 12. Amino Acid Sequences of Exemplified CD20xCD22 Bispecific Antibody3


The amino acid sequences of CD20xCD22 bispecific antibody4, which specifically binds to CD20 and CD22, are shown in Table 13 below.
Table 13. Amino Acid Sequences of Exemplified CD20xCD22 Bispecific Antibody4


The amino acid sequences of CD20xCD22 bispecific antibody5, which specifically binds to CD20 and CD22, are shown in Table 14 below.
Table 14. Amino Acid Sequences of Exemplified CD20xCD22 Bispecific Antibody5


Two additional types of bispecific antibodies that specifically bind to CD20 and CD22 were produced using the CrossMab method, described in U.S. Patent No. 8,242,247 B2, and the S-DUAL™ method, described in EP Patent Application No. 4286408 Al, the entirety of which are incorporated herein by reference. The list of exemplified bispecific antibodies produced by CrossMab or S-DUAL™ is shown in Table 15 below.
Table 15. Exemplified CD20xCD22 Bispecific Antibodies


* amino acid positions according to EU numbering convention
The amino acid sequences of halfmerl, which specifically binds to CD20, and halfiner7, which specifically binds to CD22, are shown in Tables 16 below. The amino acid sequences of the CD Rs below are defined according to the Kabat definition. Halfiner7 was prepared based on halfiner3 (Table 6) by replacing the heavy chain CHI and light chain CL regions of halfiner3 with each other.
An exemplified bispecific antibody6 was produced using halfmerl (Table 4) as is, and halfiner7.
Table 16. Amino Acid Sequences of Exemplified Halfmerl and Halfmer7



For ADC synthesis, exemplified halfiner-CaaX constructs were produced by introducing a CaaX peptide moiety (GGGGGGGCVIM; SEQ ID NO: 77) to the C-terminus of each of the light chain of Halfinerl (LC1; SEQ ID NO: 18) in Table 4 and Halfiner7 (LC7; SEQ ID NO: 82) in Table 16 with the method disclosed in International Patent Application No. PCT/IB2012/001065, the entirety of which is incorporated herein by reference.
An exemplified pair of halfiner-CaaX constructs 1 and 7 were reacted using the CrossMab method, described in U.S. Patent No. 8,242,247 B2, to form bispecific antibodies for subsequent ADC synthesis.
The amino acid sequences of CD20xCD22 bispecific antibody6, which specifically binds to CD20 and CD22, are shown in Table 17 below.
Table 17. Amino Acid Sequences of Exemplified CD20xCD22 Bispecific Antibody6


An exemplified CD20xCD22 bispecific antibody? was produced using the exemplified anti-CD20 and anti-CD22 antibodies disclosed herein with the S-DUAL method, described in EP Patent Application No. 4286408 Al.
Preparation Example 4: Synthesis of Exemplified Compounds of the Disclosure
Production of Compound 1

Compound 1 was produced with the method disclosed in U.S. Patent No. 11,654,197 B2, the entirety of which is incorporated herein by reference.
EI-MS m/z: [M+H]+ 1698.2, [ '/2M+H| 849.6.
Production of Compound 2

Compound 2 (MC-Val-Cit-PAB-MMAE) has the above structure and was purchased from MedChemExpress (Catalog No. HY-15575).
Production of Compound 3

Compound 3 (MC-Val-Ala-PBD) has the above structure and was purchased from GLPBIO (Catalog No. GC39403).
Preparation Example 5: Preparation of Exemplified ADCs by Site-specific Conjugation
Exemplified ADCs were produced with the below two steps. LCB 14-0606 was produced with the method disclosed in U.S. Patent No. 9,669,107 B2, the entirety of which is incorporated herein by reference. The structural formula of LCB14-0606 is as follows:

LCB 14-0606
Step 1: Production of Prenylated Antibody with LCB14-0606
Exemplified anti-CD20 mono or anti-CD20xCD22 bispecific antibodies listed in below Table 18 were prepared according to Preparation Examples 1-3. A mixture comprising each of the antibodies for the prenylation reaction was prepared and reacted at 30°C for 16 hours. The reaction mixture for each antibody contained a total of 24 pM antibody, 400 nM FTase (Calbiochem #344145), and 0.14 mM LCB 14-0606 in a buffer solution (50 mM Tris-HCl (pH 7.4), 5 mM MgCh, 10 pM ZnCh, 0.25 mM DTT). After the reaction was completed, the prenylated antibodies were purified using a G25 Sepharose column (AKTA purifier, GE healthcare) equilibrated with PBS buffer.
Step 2: Drug-conjugation method
Oxime Bond Reaction of ADC (conjugation by oxime bond formation)
The oxime bond formation reaction between each of the prenylated antibodies and linker-drug was carried out by mixing 100 mM sodium acetate buffer (pH 5.2, Sigma- Aldrich, S7899), 10% DMSO (Sigma-Aldrich, D4540), 24 pM of the prenylated antibody, and 10 equivalents (240 pM) of the linker-drug (Compound 1 of Preparation Example 4). The mixture was stirred gently at 30°C for 6 hours. After the reaction, excess low-molecular-weight compounds were removed through a desalting process using a G25 Sepharose column, and the protein fractions were collected and concentrated.
Exemplified ADCs produced by the above two steps are presented in Table 18 below.
Table 18. Exemplified ADCs by Site-specific Conjugation


The preparation of ADCs was analyzed using hydrophobic interaction chromatography to measure the drug-antibody ratio and size exclusion chromatography for monomer analysis.
For ADC1, ADC2, ADC3, ADC4, and ADC7, hydrophobic interaction chromatography was performed using a phenyl column (7.5 mm X 75 mm, 10 pm, Tosoh Bioscience, 7573) and potassium phosphate buffer as the mobile phase. The analysis used a buffer A consisting of 50 mM potassium phosphate buffer (pH 7.0) with 0.5 M ammonium sulfate and a buffer B consisting of 50 mM potassium phosphate buffer (pH 7.0) with 30% acetonitrile. The initial conditions were stabilized at 90% A/10% B, and a linear gradient of 90% A/10% B to 10% A/90% B was applied over 30 minutes, followed by an additional elution with 10% A/90% B for 5 minutes. The flow rate and temperature were set to 1.0 mL/min and 25°C, respectively, with detection at both 254 nm and 280 nm.
For ADC5 and ADC6, the same phenyl column (7.5 mm X 75 mm, 10 pm, Tosoh Bioscience, 7573) and potassium phosphate buffer were used. Buffer A contained 50 mM potassium phosphate (pH 7.0) with 1.5 M ammonium sulfate, and buffer B contained 50 mM potassium phosphate (pH 7.0) with 30% acetonitrile. The initial conditions were stabilized at
70% A/30% B, and a linear gradient from 70% A/30% B to 10% A/90% B was applied over 30 minutes, followed by elution with 10% A/90% B for 5 minutes. The flow rate and temperature were maintained at 1.0 mL/min and 25°C, respectively, with detection at both 254 nm and 280 nm.
For ADC8, hydrophobic interaction chromatography was performed using a butyl column (4.6 mm X 35 mm, 2.5 pm, Tosoh Bioscience, 14947) and potassium phosphate buffer. Buffer A contained 50 mM potassium phosphate (pH 7.0) with 1.5 M ammonium sulfate, and buffer B contained 50 mM potassium phosphate (pH 7.0) with 30% isopropyl alcohol. The initial conditions were stabilized at 100% A/0% B, and a linear gradient from 100% A/0% B to 0% A/ 100% B was applied over 20 minutes, followed by elution with 0% A/ 100% B for 2 minutes. The flow rate and temperature were set to 1.0 mL/min and 25°C, respectively, with detection at both 254 nm and 280 nm.
For size exclusion chromatography of ADC1, ADC2, ADC3, ADC4, ADC5, ADC6, ADC7, and ADC8, a SEC column (7.8 mm X 30 cm, Tosoh Bioscience, 8541) was used with a mobile phase of 0.1 M sodium phosphate and 0.3 M sodium chloride containing 10% acetonitrile (pH 6.8). The analysis was conducted with a linear gradient of 100% buffer over 30 minutes.
The results of ADCs preparation analysis are presented in Table 19 below.
Table 19. Exemplified ADCs Preparation Analysis Results
 Preparation Example 6: Preparation of Exemplified ADCs by Random Conjugation
Preparation Example 6: Preparation of Exemplified ADCs by Random Conjugation
To produce randomly conjugated ADCs, the CD20xCD22 bispecific antibody 1 was conjugated with Compound 3 of Preparation Example 4 or Compound 2 of Preparation Example 4.
For conjugation of the bispecific antibody to the linker-toxin, 74 pM of tris(2- carboxyethyljphosphine (Sigma- Aldrich, C4706) was added to a solution of 24 pM CD20xCD22 bispecific antibody 1 containing 4 mM EDTA (Enzynomics, EBE001-500) at pH 7.4, and the mixture was gently stirred at 25°C. After a reaction time of 2 hours, 216 pM of Compound 3 or Compound 2 were added, and the mixture was gently stirred for an additional 2 hours at 25°C. Subsequently, 1 mM of dehydroabietic acid (Sigma-Aldrich, 261556) was added, and the mixture was stirred gently for another 2 hours at 25 °C. The excess low- molecular-weight compounds were removed through a desalting process using a G25 Sepharose column, and the protein fractions were collected and concentrated.
Exemplified ADCs produced by random conjugation are presented in Table 20 below.
Table 20. Exemplified ADCs by Random conjugation

The preparation of ADC9 and ADC 10 was analyzed using reverse phase chromatography to measure the average drug-antibody ratio and size exclusion chromatography for monomer analysis.
Reverse phase chromatography utilized a PLRP-S 1000A column (2.1 mm * 50 mm, 5 pm, Agilent, PL1912-1502) with a mobile phase containing 0.1% trifluoroacetic acid (Sigma- Aldrich, T6508). Water containing 0.1% trifluoroacetic acid was used as solution A, and acetonitrile containing 0.1% trifluoroacetic acid was used as solution B. The system was stabilized with 75% A and 25% B for 3 minutes, followed by a linear gradient of 50% A and 50% B for 28 minutes, and then an additional linear gradient of 5% A and 95% B for 1 minute, with a further 2 minutes of elution. The flow rate and temperature were set to 1.0 mL/min and
80°C, respectively. Detection was performed at both 254 nm and 280 nm. Size exclusion chromatography utilized a SEC column (7.8 mm x 30 cm, Tosoh Bioscience, 8541) and a mobile phase containing sodium phosphate 0. IM and 0.3M sodium chloride in 10% acetonitrile buffer (pH 6.8). The analysis was performed with a linear gradient of 100% buffer for 30 minutes.
The results of preparation analysis of ADC9 and ADC10 are presented in Table 21 below.
Table 21. Exemplified ADCs Preparation Analysis Results

Experimental Example 1: In Vitro Evaluation of the Binding Affinity of the CD20xCD22 Bispecific Antibody to Cancer Cells
The cell binding affinity of anti-CD20xCD22 bispecific antibody 1, produced in Preparation Example 3, was evaluated in hematological cancer cell lines using flow cytometry (FACS). The cell lines tested included SUP-B15, characterized by low CD20 and high CD22 expression; Mino, characterized by low CD22 and high CD20 expression; and Granta-519, which expresses both CD20 and CD22 at high levels.
Specifically, the above cancer cell lines were cultured and divided into two groups: a control IgG antibody-treated group and an anti-CD20xCD22 bispecific antibody 1 -treated group. The cells were then incubated in phosphate-buffered saline (PBS) containing 2% fetal bovine serum (FBS) at 4°C for 1 hour to minimize non-specific antibody binding. Next, the control antibody and anti- CD20xCD22 bispecific antibody 1 were incubated with the respective cell groups at 4°C for 1 hour to allow antibody binding to the CD20 or CD22 proteins on the cancer cell surface.
After incubation, unbound antibodies were removed with PBS containing 2% FBS. The cells were then incubated with fluorescent-labeled secondary antibodies specific to the control IgG antibody and anti- CD20xCD22 bispecific antibody 1 at 4°C for 1 hour. Following this,
unbound secondary antibodies were washed off with PBS, and the fluorescence intensity of each cell group was measured using a flow cytometer. By comparing the fluorescence intensity of the anti- CD20xCD22 bispecific antibody 1 group with the control IgG group, the binding affinity of the antibodies to the cancer cell lines was confirmed. The results are presented in Table 22 below.
Table 22. Binding Affinity of Exemplified CD20xCD22 bispecific antibody

The experimental results (Fig. 1) showed that anti-CD20xCD22 bispecific antibody 1 exhibited a binding affinity 10.4 times higher than that of the IgG control for SUP-B15 cells, which have low CD20 expression and high CD22 expression. For Mino cells, characterized by low CD22 and high CD20 expression, anti-CD20xCD22 bispecific antibody 1 showed 9.9 times stronger binding compared to the IgG control. In Granta-519 cells, which express both CD20 and CD22 at high levels, binding affinity of anti-CD20xCD22 bispecific antibody 1 was 11.2 times higher than that of the IgG control. Overall, these findings confirm that anti-CD20xCD22 bispecific antibody 1 is capable of binding to cancer cells expressing either CD20 or CD22, as well as hematological cancer cells co-expressing both CD20 and CD22.
Experimental Example 2: In Vitro Evaluation of Internalization Rate of Exemplified Antibodies in Cancer Cells
To analyze the internalization rates of the anti-CD20 antibody CD20_l-CaaX construct prepared in Preparation Example 1 and the anti-CD20xCD22 bispecific antibody 1 prepared in Preparation Example 3, cell binding experiments were conducted using a flow cytometer (FACS).
Ramos cells, a B-cell non-Hodgkin lymphoma cell line known for CD20 expression, were used, with human IgG used as the control antibody.
To evaluate the internalization rates, the anti-CD20 antibody CD20_l-CaaX construct and the anti-CD20xCD22 bispecific antibody 1 were first incubated with Ramos cells at 4°C
for 1 hour, respectively. The experimental groups were then incubated at 37°C for either 2 or 4 hours to induce internalization, while a control group was maintained at 4°C. To quantify the remaining antibodies on the cell surface across groups, a phycoerythrin (PE)-conjugated secondary antibody (BD, 555787) specific to human IgG was used.
The results (Fig. 2) showed that, in Ramos cells, the anti-CD20 antibody CD20_l-CaaX construct exhibited internalization rate of 42% after 2-hours and 44% after 4-hours of incubation at 37°C. In contrast, the anti-CD20xCD22 bispecific antibodyl demonstrated an internalization rate of 93% after 2-hours and 95% after 4-hours.
Overall, the anti-CD20xCD22 bispecific antibody showed a more than 50% improvement of internalization rate compared to the anti-CD20 antibody in Ramos cells.
Experimental Example 3: In Vitro Evaluation of Internalization Rates of Exemplified Bispecific ADCs in Cancer Cell by Platform
To evaluate the internalization rates of ADC 1, which comprises an anti-CD20 antibody, and those of ADC2 (Knob-into-Hole), ADC6 (Pentambody), and ADC7 (CrossMab), which comprise anti-CD20xCD22 bispecific antibodies that share the same variable region as prepared in Preparation Example 3, cell binding experiments were conducted using a flow cytometer (FACS). Ramos cells, a CD20-expressing B-cell non-Hodgkin lymphoma cell line, were used, with human IgG as the control antibody.
For internalization rate analysis, the anti-CD20 ADC (ADC1) and the anti- CD20xCD22 bispecific ADCs (ADC 2, 6, and 7) were first incubated at 4°C for 1 hour, respectively. A control group was maintained at 4°C, while treatment groups were incubated at 37°C for 1 or 3 hours to induce internalization. A PE-conjugated secondary antibody (BD, 555787) specific to human IgG was used to quantify remaining antibodies on the cell surface.
The results (Fig. 3) showed that ADC1 exhibited internalization rate of 19% after 1- hour and 40% after 3 -hour in Ramos cells. In contrast, ADC2 (Knob-into-Hole platform) demonstrated an internalization rate of 94% after 1-hour incubation and 98% after 3-hour incubation. ADC6 (Pentambody platform) showed an internalization rate of 88% after 1-hour incubation and 85% after 3-hour incubation. ADC7 (CrossMab platform) exhibited an internalization rate of 87% after 1-hour incubation and 88% after 3-hour incubation.
Overall, the anti-CD20xCD22 bispecific ADCs demonstrated an over 50% higher internalization rate than the anti-CD20 ADC in Ramos cells, regardless of platform. These
findings confirm that anti-CD20xCD22 bispecific ADCs have an improved internalization rate compared to CD20-targeted ADCs, independent of the platform used.
Experimental Example 4: In vitro Cytotoxicity Evaluation of Exemplified Bispecific ADCs by Platform
The cytotoxicity activity of the ADCs against various cancer cell lines, as presented in Table 23 below, was measured. The cancer cell lines used were the commercially available human B-cell non-Hodgkin lymphoma cell lines (SU-DHL-8, WSU-DLCL2), and acute lymphoblastic leukemia cell lines (SUP-B15 and Reh). The ADCs used for measuring cytotoxicity activity were ADC1, ADC2, ADC6, and ADC7, which were prepared in Preparation Example 3.
Each cancer cell line was seeded at 5,000 to 20,000 cells per well in a 96-well plate and treated with each ADC at concentrations ranging from 0.128 fM to 50 nM (five-fold serial dilution). The number of living cells (cell viability) was quantified using Cell Titer Gio (Promega, G7570) after 96 or 144 hours, and the results are shown in Table 23 below.
Table 23. 50% Cytotoxicity Concentrations of Exemplified ADCs in Cancer Cell Lines

As shown in Table 23, in various cancer cell lines (SU-DHL-8, WSU-DLCL2, SUP- B15, Reh), the pyrrolobenzodiazepine-based anti-CD20xCD22 bispecific ADCs (ADC2, ADC6, ADC7) demonstrated improved efficacy ranging from 22-fold to 380-fold compared to the pyrrolobenzodiazepine-based anti-CD20 ADC (ADC1). The anti-CD20xCD22 bispecific ADCs (ADC2, ADC6, and ADC7) exhibited consistently stronger cytotoxicity effects against
cancer cell lines compared to anti-CD20 ADC1.
Therefore, the anti-CD20xCD22 ADCs effectively demonstrated cytotoxicity against cancer cells compared to the anti-CD20 ADC regardless of the bispecific antibody platform used.
Experimental Example 5: In vitro Cytotoxicity Evaluation of Exemplified Bispecific ADCs by Platform
The cytotoxicity activity of the ADCs against various cancer cell lines, as listed in Table 24 below, was measured. The commercially available human B-cell non-Hodgkin lymphoma cell lines (WSU-DLCL2, WSU-NHL, Mino, and Granta-519 were used for this analysis. ADC2 and ADC8, prepared in Preparation Example 5, were tested for their cytotoxicity.
Each cancer cell line was seeded at 5,000 cells per well in a 96-well plate and treated with each ADC at concentrations ranging from 0.128 fM to 50 nM (five-fold serial dilution). The number of living cells (cell viability) was quantified using Cell Titer Gio (Promega, G7570) after 96 hours of treatment.
Table 24. 50% Cytotoxicity Concentrations of Exemplified ADCs in Cancer Cell Lines

As shown in Table 24, the cytotoxicity evaluation results of the cancer cell lines indicated that ADC2, produced with a two-cell system using the Knob-in-Hole method disclosed herein, and ADC8, produced with a one-cell system using the S-DUAL method disclosed herein, exhibited comparable efficacy across the tested cell lines (WSU-DLCL2, WSU-NHL, Mino, and Granta-519).
These findings confirm that, regardless of the bispecific antibody production method, CD20xCD22 bispecific ADCs demonstrated consistent cytotoxicity.
Experimental Example 6: Comparison of In vitro Cytotoxicity Between ADCs Comprising Monoclonal Antibodies and Bispecific Antibodies
The cytotoxicity activity of drugs (SG2057) and ADCs was measured against various cancer cell lines and normal cells.

SG2057 has the above structure and was purchased from MedChemExpress (Catalog No. HY-101160). The cancer cell lines used were commercially available human B-cell nonHodgkin lymphoma cell lines (Daudi, Ramos, NAMALWA, WSU-NHL, WSU-DLCL2) and acute lymphoblastic leukemia cell lines (SUP-B15 and Reh). The normal cell lines used were PBMC (STEMCELL; Catalog No. 70002.2) and HSC (STEMCELL; Catalog No. 70025.2). The ADCs tested were ADC 1 and ADC2 prepared in Preparation Example 5.
Each cancer cell line was seeded at 5,000 to 20,000 cells per well in a 96-well plate and treated with SG2057 at concentrations ranging from 5.12 fM to 2 nM (five-fold serial dilution) or each ADC at concentrations ranging from 0. 128 fM to 50 nM (five-fold serial dilution). For normal cell lines, cells were treated with SG2057 at concentrations of 512 fM to 2 nM (fivefold serial dilution), or ADCs at concentrations of 10 pM to 5,000 nM (five-fold serial dilution). The number of living cells in the cancer cell lines (cancer cell viability) was quantified using Cell Titer Gio (Promega, G7570) after 96 or 144 hours. For normal cell lines, PBMC and HSC were quantified using Cell Titer Gio after 144 hours, and the results are shown in Table 25 and 26 below.
Table 25. 50% Cytotoxicity Concentrations of Exemplified Drug or ADCs in Cancer Cell
Lines


Table 26. 50% Cytotoxicity Concentrations of Exemplified Drug or ADCs in Normal Cell Lines

As shown in Table 25, the cytotoxicity evaluation results in cancer cell lines (Daudi, Ramos, NAMALWA, WSU-NHL, WSU-DLCL2, SUP-B15, Reh) showed that ADC2, a pyrrolobenzodiazepine-based anti-CD20xCD22bispecific ADC, demonstrated 4- to 75 -fold greater efficacy compared to ADC1, an anti-CD20 pyrrolobenzodiazepine-based ADC .
In normal cell lines PBMC and HSC, as shown in Table 26, ADC2 demonstrated 8- to 29-fold weaker cytotoxicity, indicating a reduction in toxicity towards blood cells where CD20 and CD22 are commonly expressed in vivo.
These findings confirm that ADC2’s cytotoxicity is selectively enhanced in cancer cells, with significantly reduced impact on normal cells, underscoring ADC2’s potential for targeted cytotoxicity against cancer cells over ADC1.
Experimental Example 7: In vitro Cytotoxicity Evaluation of Exemplified Bispecific ADCs with Different Antibody Sequences
The cytotoxicity activity of exemplified ADCs against cancer cell lines was measured
to observe potential differences in efficacy based on antibody sequence variations. The cancer cell line used was the commercially available human B-cell non-Hodgkin lymphoma cell line Ramos. The ADCs tested (ADC2, ADC3, ADC4, and ADC5) were prepared in Preparation Example 5.
Ramos cells were seeded at 5,000 cells per well in a 96-well plate and treated with each ADC at concentrations ranging from 0.128 fMto 50 nM (five-fold serial dilution). The number of living cells was quantified using Cell Titer Gio (Promega, G7570) after 96 hours, and the results are shown in Table 27 below.
Table 27. 50% Cytotoxicity Concentrations of Exemplified ADCs in Cancer Cell Line

The cytotoxicity evaluation results of the cancer cell line (Ramos), as shown in Table 27, indicate that all pyrrolobenzodiazepine-based anti-CD20xCD22bispecific ADCs (ADC2, ADC3, ADC4, and ADC5) showed strong efficacy, regardless of antibody sequences variation.
It was found that the anti-CD20xCD22 bispecific antibody combination ADC consistently exhibited potent, cancer cell-specific cytotoxicity, unaffected by variations in antibody sequences.
Experimental Example 8: In vitro Cytotoxicity Assessment of Exemplified Bispecific ADCs with Different Linker Payload Combination
The cytotoxicity activity of ADC9 and ADC10, prepared in Preparation Example 5, against cancer cell line was measured to observe potential differences in efficacy based on variations in linker-payload combinations.
The cancer cell line used was the commercially available human B-cell non-Hodgkin lymphoma cell line Ramos. Ramos cells were seeded at 5,000 cells per well in a 96-well plate and treated with each ADC at concentrations ranging from 0.128 fM to 50 nM (five-fold serial dilution). The number of living cells was quantified using Cell Titer Gio (Promega, G7570) after 96 hours, and the results are shown in Table 28 below.
Table 28. 50% Cytotoxicity Concentrations of Exemplified Bispecific ADCs in Cancer
Cell Lines

As shown in Table 28, anti-CD20xCD22 bispecific ADC (ADC9) conjugated with MC-Val-Ala-PBD (Compound 3) demonstrated potent cytotoxicity regardless of the specific linker used, where the efficacy is comparable to those of ADC2, ADC3, ADC4, and ADC5. Similarly, the anti-CD20xCD22 bispecific ADC (ADC 10) conjugated with MC-Val-Cit-PAB- MMAE (Compound 2) demonstrated strong efficacy, independent of the specific payload.
These findings suggest that the efficacy of anti-CD20xCD22 bispecific ADCs is not limited by linker or payload variations and consistently exhibits cancer cell specific cytotoxicity.
Experimental Example 9: Assessment of Pharmacokinetics (PK)
For in vivo evaluation of ADC2 prepared in Preparation Example 5 in Sprague Dawley (SD) rats, single doses of ADC2 were given at 1.0 mg/kg intravenously into male SD rats (Orientbio, South Korea) at 9 weeks of age. Pharmacokinetics were studied following injection of ADC2 into SD rats. Plasma samples were taken at various time points and stored frozen for analysis. The plasma concentrations of the ADC2 at the indicated time points were measured using a LC-MS/MS analysis method.
Briefly, 250 pL of acetonitrile (ACN) solution was combined with both 50 pL of sample and 50 pL of plasma containing 10 nM Dextromethorphan (internal standard), and the solutions were mixed vigorously using a vortex mixer for 5 minutes. The samples were then spun down at 14,000 rpm, 4 °C for 5 minutes. 100 pL of supernatants were combined with 100 pL of mobile phase A (0.1 % formic acid in water with 5% ACN) and mixed thoroughly. The samples were measured for ADC2 using a LC-MS/MS method (Nexera LC40 (SHIMADZU) and QTRAP4500 (SCIEX)).
The PK profiles of ADC2 are presented in Table 29. and Fig. 4. ADC2 showed stable pharmacokinetic profiles in rats.
Table 29. The PK Profiles of Exemplified ADC

Experimental Example 10: In vivo Efficacy Evaluation (SU-DHL-8)
After culturing the hematologic cancer cell line SU-DHL-8, 5,000,000 cells were mixed with 50 pl of Matrigel™ (Coming) in 50 pl of PBS and implanted into CB17/SCID mice. When tumor size reached 70 to 110 mm3, exemplified ADC1 or ADC2 was administered via intravenous injection. The results comparing the efficacy of the ADC samples are presented in Table 30 and Fig. 5.
Table 30. In vivo anti-cancer efficacy of Exemplified ADCs (SU-DHL-8)

As shown in Fig. 5 and Table 30, administration of the pyrrolobenzodiazepine-based anti-CD20xCD22 bispecific ADC2 at 0.3 mg/kg resulted in 45.1% tumor growth inhibition compared to the control group (administered with PBS), while the anti-CD20 ADC1 at the same dosage achieved 13.9% tumor growth inhibition. Therefore, ADC2 demonstrated more
than a threefold improvement in tumor growth inhibition efficacy compared to ADC1 at the same dosage.
Experimental Example 11: In vivo Efficacy Evaluation (Granta-519)
After culturing the hematologic cancer cell line Granta-519, 5,000,000 cells were mixed with 50 pl of Matrigel™ (Coming) in 50 pl of PBS and implanted into CB 17/SCID mice. When the tumor size reached 100 mm3, exemplified ADC2 was administered via intravenous injection at different doses, as shown in Table 31 below. The results verifying the efficacy of the ADC samples are presented in Table 31 and Fig. 6.
Table 31. In vivo anti-cancer efficacy of Exemplified ADCs (Granta-519)

As shown in Fig. 6 and Table 31, complete remission was achieved with the pyrrolobenzodiazepine-based anti-CD20xCD22 bispecific ADC2 at a dosage of 0. 1 mg/kg.
Experimental Example 12: In vivo Efficacy Evaluation (MINO)
After culturing the hematologic cancer cell line MINO, 10,000,000 cells were mixed with 100 pl of Matrigel™ (Coming) in 100 pl of PBS and implanted into CB 17/SCID mice. When the tumor size reached 126 mm3, exemplified ADC2 was administered via intravenous injection at different doses, as shown in Table 32 below. The results verifying the efficacy of the ADC samples are presented in Table 32 and Fig. 7.
Table 32. In vivo anti-cancer efficacy of Exemplified ADCs (MINO)


As shown in Fig. 7 and Table 32, complete remission was achieved with the pyrrolobenzodiazepine-based anti-CD20xCD22bispecific drug conjugate ADC2 at a dosage of 0.2 mg/kg.
Experimental Example 13: In vivo Efficacy Evaluation (Ramos)
After culturing the hematologic cancer cell line Ramos, 5,000,000 cells were mixed with 100 pl of Matrigel™ (Coming) in 100 pl of PBS and implanted into BALB/c mice. When the tumor size reached 70 mm3, exemplified ADC2 or Inotuzumab Ozogamicin (Besponsa™) was administered via intravenous injection at different doses, as shown in Table 33 below. The results verifying the efficacy of the ADC samples are presented in Table 33 and Fig. 8.
Table 33. In vivo anti-cancer efficacy of Exemplified ADC (Ramos)

As shown in Fig. 8 and Table 33, the pyrrolobenzodiazepine-based anti-CD20xCD22 bispecific drug conjugate ADC2 exhibited dose-dependent tumor growth inhibition at dosages of 0.1, 0.3, and 0.5 mg/kg in Ramos cell xenografts. When compared to the commercially available CD22 -targeting ADC, Inotuzumab Ozogamicin, ADC2 at 0.3 mg/kg demonstrated a 49% improvement in tumor growth inhibition. At 0.5 mg/kg, ADC2 achieved 3.14 times stronger tumor growth inhibition than Inotuzumab Ozogamicin at 2.5 mg/kg. These findings
confirm that ADC2 effectively inhibits tumor growth in cell line-derived xenograft (CDX) models that are resistant to Inotuzumab Ozogamicin.
Experimental Example 14: In vivo Efficacy Evaluation (WSU-DLCL2)
After culturing the hematologic cancer cell line WSU-DLCL2, 10,000,000 cells were mixed with 100 pl of Matrigel™ (Coming) in 100 pl of PBS and implanted into CB17 SCID mice. When the tumor size reached 150 mm3, exemplified ADC1 or ADC2 were administered via intravenous injection, as shown in Table 34 below. In Rituximab-CHOP treatment group, Rituximab and Cyclophosphamide were administered intravenously, while Doxorubicin, Vincristine, and Prednisone were given orally. The results verifying the efficacy of the ADC samples and R-CHOP are presented in Table 34 and Fig. 9.
Table 34. In vivo anti-cancer efficacy of Exemplified ADCs (WSU-DLCL2)

As shown in Fig. 9 and Table 34, the pyrrolobenzodiazepine-based anti-CD20xCD22 bispecific drug conjugate ADC2 demonstrated dose-dependent tumor growth inhibition at 0.1, 0.3, and 0.5 mg/kg in the WSU-DLCL2 cell CDX model. At 0.3 mg/kg, ADC2 achieved a 51% greater tumor growth inhibition compared to ADC1 at the same dosage. These results confirm that the anti-CD20xCD22 ADC, which addresses the low internalization challenge associated
with CD20-targeting ADCs, provides robust tumor growth inhibition.
Additionally, ADC2 exhibited strong tumor growth inhibition across all tested doses when compared to the standard R-CHOP therapy, commonly used as the first-line treatment for diffuse large B-cell lymphoma. These results indicate that ADC2 is effective in showing tumor growth inhibition even in CDX models that are resistance to R-CHOP therapy.
Incorporation by Reference
All publications and patents mentioned herein are hereby incorporated by reference in their entirety as if each individual publication or patent was specifically and individually indicated to be incorporated by reference. In case of conflict, the present application, including any definitions herein, will control.
Equivalents
While specific embodiments of the disclosure have been discussed, the above specification is illustrative and not restrictive. Many variations of the disclosure will become apparent to those skilled in the art upon review of this specification and the claims below. The full scope of the disclosure should be determined by reference to the claims, along with their full scope of equivalents, and the specification, along with such variations.
Claims
1. A conjugate having a structure represented by the following General Formula I or a pharmaceutically acceptable salt thereof:
[General Formula 1]
Ab-[L-(B)i]m wherein,
Ab is a bispecific antibody comprising a first antigen-binding domain specifically binding to CD20 and a second antigen-binding domain specifically binding to CD22, each L is, independently, a linker, each B is, independently, an active agent, and
1 and m are each independently 1 to 20.
2. The conjugate of claim 1, wherein L is a cleavable linker.
3. The conjugate of claim 1 or 2, wherein L is a protease cleavable linker, an acid- cleavable linker, a disulfide linker, or a glycosidase cleavable linker.
4. The conjugate of claim 1 or 2, wherein L is a protease cleavable linker.
5. The conjugate of claim 4, wherein the protease is a cathepsin.
6. The conjugate of claim 4 or 5, wherein L comprises 2 to 4 amino acid residues, e.g., defining the recognition site of the protease.
7. The conjugate of claim 6, wherein L comprises Val-Cit, Vai-Ala, or Gly-Gly-Phe- Gly.
8. The conjugate of claim 1 or 2, wherein L is a glycosidase cleavable linker.
9. The conjugate of claim 8, wherein the glycosidase is [3-glucuronidase or [3- galactosidase.
10. The conjugate of claim 9, wherein L comprises a P-glucuronosyl or P-galactosyl moiety.
11. The conjugate of any one of claims 1-10, wherein L is covalently bonded to Ab by a thioether, thioester, disulfide, hydrazone, ester, carbamate, carbonate, alkoxy, or amide.
12. The conjugate of any one of claims 1-11, wherein L comprises a peptide further comprising at least one hydrophilic amino acid.
13. The conjugate of claim 12, wherein the peptide comprises an amino acid having a side chain having a moiety that bears a charge at neutral pH in aqueous solution (e.g., an amine, guanidine, or carboxyl moiety).
14. The conjugate of any one of claims 1-13, wherein L comprises an oxime.
15. The conjugate of any one of claims 1-14, wherein L comprises at least one isoprenyl group.
16. The conjugate of any one of claims 1-15, wherein L comprises at least one succinimide group.
17. The conjugate of any one of claims 1-16, wherein L is covalently bonded to Ab by a thioether bond and the thioether bond comprises a sulfur atom of a cysteine of Ab.
18. The conjugate of any one of claims 1-17, wherein Ab at the C-terminus comprises an amino acid motif that is recognized by an isoprenoid transferase.
19. The conjugate of claim 18, wherein the isoprenoid transferase is famesyl protein transferase (FTase) or geranylgeranyl transferase (GGTase).
20. The conjugate of claim 18 or 19, wherein L is covalently bonded to Ab by a thioether bond and the thioether bond comprises a sulfur atom of a cysteine of the amino acid motif.
21. The conjugate of any one of claims 18-20, wherein the amino acid motif comprises a CY iY iX sequence, wherein:
C is cysteine; each Y i is independently an aliphatic amino acid; and
X is selected from glutamine, glutamate, serine, cysteine, methionine, alanine, and leucine.
22. The conjugate of claim 21, wherein each Y i is independently selected from alanine, isoleucine, leucine, methionine, and valine.
23. The conjugate of any one of claims 18-22, wherein the amino acid motif comprises a CVIM (SEQ ID NO: 84) or CVLL (SEQ ID NO: 85).
24. The conjugate of any one of claims 18-23, wherein at least one of the 1 to 20 amino acids preceding the amino acid motif is glycine.
25. The conjugate of any one of claims 18-24, wherein the amino acid motif has the sequence GGGGGGGCVIM (SEQ ID NO: 77).
26. The conjugate of any one of claims 1-25, wherein L comprises a self-immolative moiety.
27. The conjugate of any one of claims 1-10, wherein the conjugate has a structure represented by the General Formula II or a pharmaceutically acceptable salt thereof:
[General Formula II]
 wherein,
wherein,
B is the active agent,
G represents a sugar moiety or a glucuronic acid moiety;
R1 and R2 are each independently hydrogen, C1-8 alkyl, or C3-8 cycloalkyl; or R1 and R2 combine to complete a (CYC's) cycloalkyl ring;
W is -*C(O)-, -*C(O)N(R’)-, -*N(R’)C(O)-, -*(CH2)tN(R’)C(O)-, -*C(O)O-, -*S(O2)N(R’)-, -*P(O)(R”)N(R’)-, -*S(O)N(R’)-, or -*P(O2)N(R’)-, wherein the C(O), N, CH2, S, or P marked with an * is directly bonded to the phenyl ring of General Formula II,
R’ and R” are each independently hydrogen, C1-8 alkyl, C3-8 cycloalkyl, C1-8 alkoxy, C1-8 alkylthio, mono- or di-C1-8 alkylamino, Cs-2o heteroaryl or Ce-2o aryl;
Z is each independently C1-8 alkyl, halogen, cyano, or nitro; nl and n2 are each independently 1 to 20; n3 is 0 to 3; and
Y is absent, C1-C50 alkylene, C1-C50 alkenylene, 1-50 atom heterocyclylene, or 1-50 atom heteroalkylene.
28. The conjugate of claim 27, wherein Y comprises a C1-50 alkylene or C1-50 heteroalkylene and further comprises at least one of:
(i) one or more unsaturated bonds;
(ii) a heterocyclylene or a heteroarylene (e.g., a heteroarylene in the alkylene or heteroalkylene chain);
(iii) at least one Ci-2o alkyl; and
(iv) at least one isoprenyl group having a structure represented by General Formula III: [General Formula III]
 wherein n4 is an integer of 1 to 20.
wherein n4 is an integer of 1 to 20.
29. The conjugate of claim 27 or 28, wherein R1 and R2 are each independently hydrogen.
30. The conjugate of any one of claims 27-29, wherein n3 is 0.
31. The conjugate of any one of claims 27-30, wherein the G has a structure represented by the General Formula IV :
[General Formula IV]
 wherein,
wherein,
R3 is hydrogen, alkyl, CH2OR3A, or CO2R3B;
RSA is hydrogen or a hydroxyl protecting group;
R3B is hydrogen or a carboxyl protecting group; and each R4 is independently a hydrogen or a hydroxyl protecting group.
32. The conjugate of claim 31, wherein R3 is-COOH.
33. The conjugate of claim 31 or 32, wherein each R4 is independently hydrogen.
34. The conjugate of any one of claims 27-33, wherein the W is -*C(O)NR’-.
35. The conjugate of any one of claims 27-34, wherein

W is -*C(O)NR’-.
36. The conjugate of any one of claims 27-35, wherein Y is covalently bonded to Ab by a thioether, thioester, disulfide, hydrazone, ester, carbamate, carbonate, alkoxy, or amide.
37. The conjugate of claim 36, wherein Y comprises a peptide further comprising at least one hydrophilic amino acid.
38. The conjugate of claim 37, wherein the peptide comprises an amino acid having a side chain having a moiety that bears a charge at neutral pH in aqueous solution (e.g., an amine, guanidine, or carboxyl moiety).
39. The conjugate of claim 37 or 38, wherein the peptide comprises an amino acid selected from alanine, histidine, arginine, aspartate, asparagine, glutamate, glutamine, glycine, lysine, ornithine, proline, serine, and threonine.
40. The conjugate of claim 37 or 38, wherein the peptide comprises an amino acid selected from alanine, aspartate, asparagine, glutamate, glutamine, glycine, lysine, ornithine, proline, serine, and threonine.
41. The conjugate of any one of claims 27-40, wherein Y comprises an oxime.
42. The conjugate of claim 41, wherein the oxygen atom of the oxime is on the side of Y linked to W and the carbon atom of the oxime is on the side of Y linked to Ab.
43. The conjugate of claim 41, wherein the carbon atom of the oxime is on the side of Y linked to W and the oxygen atom of the oxime is on the side of Y linked to Ab.
44. The conjugate of any one of claims 27-43, wherein Y is a C1-50 heteroalkylene comprising an oxime.
45. The conjugate of claim 44, wherein the oxygen atom of the oxime is on the side of Y linked to W, and the carbon atom of the oxime is on the side of Y linked to Ab.
46. The conjugate of claim 44, wherein the carbon atom of the oxime is on the side of Y linked to W, and the oxygen atom of the oxime is on the side of Y linked to Ab.
47. The conjugate of any one of claims 44-46, wherein Y comprises an oxime, and at least one isoprenyl unit covalently bonds the oxime to Ab (e.g., the at least one isoprenyl unit directly or indirectly bonds the oxime to Ab).
48. The conjugate of any one of claims 27-46, wherein Y comprises at least one isoprenyl group.
49. The conjugate of any one of claims 27-48, wherein Y is covalently bonded to Ab by a thioether bond and the thioether bond comprises a sulfur atom of a cysteine of Ab.
50. The conjugate of any one of claims 27-49, wherein the Ab at the C-terminus comprises an amino acid motif that is recognized by an isoprenoid transferase.
51. The conjugate of claim 50, wherein the isoprenoid transferase is famesyl protein transferase (FTase) or geranylgeranyl transferase (GGTase).
52. The conjugate of claim 50 or 51, wherein Y is covalently bonded to Ab by a thioether bond and the thioether bond comprises a sulfur atom of a cysteine of the amino acid motif.
53. The conjugate of any one of claims 50-52, wherein the amino acid motif comprises a CY iY iX sequence, wherein
C is cysteine; each Y i is independently an aliphatic amino acid; and
X is selected from glutamine, glutamate, serine, cysteine, methionine, alanine, and leucine.
54. The conjugate of claim 53, wherein each Y i is independently selected from alanine, isoleucine, leucine, methionine, and valine.
55. The conjugate of any one of claims 50-54, wherein the amino acid motif comprises a CVIM (SEQ ID NO: 84) or CVLL (SEQ ID NO: 85).
56. The conjugate of any one of claims 50-55, wherein at least one of the 1 to 20 amino acids preceding the amino acid motif is glycine.
57. The conjugate of any one of claims 50-56, wherein the amino acid motif has the sequence GGGGGGGCVIM (SEQ ID NO: 77).
58. The conjugate of any one of claims 27-57, wherein the Y comprises a connecting unit represented by the General Formula V or General Formula VI:
[General Formula V]
-(CH2)r(V(CH2)P)q-
[General Formula VI]
-(CH2CH2X)W- wherein,
V is a single bond, -O-, -S-, -NR5-, -C(O)NR6-, -NR7C(O)-, -NR8SO2-, or -SO2NR9-;
X is -O-, C1-8 alkylene or -NR5-;
R5 to R9 are each independently hydrogen, Ci-6 alkyl, Ci-6 alkyl Cg-2o aryl, or
Ci-6 alkyl-C3-2o heteroaryl; r is 0 to 10; p is 0 to 10; q is 1 to 20; and
w is 1 to 20.
59. The conjugate of claim 58, wherein q is 1 to 10.
60. The conjugate of claim 58 or 59, wherein r is 1 or 2.
61. The conjugate of any one of claims 58-60, wherein p is 1 or 2.
62. The conjugate of any one of claims 58-61, wherein V is -O-.
63. The conjugate of claim 58, wherein: q is 1 to 10; r and p are each 1 or 2; and
V is -O-.
64. The conjugate of any one of claims 58-63, wherein X is -O-.
65. The conjugate of any one of claims 58-64, wherein the X is -O-; and w is 1 to 10.
66. The conjugate of any one of claims 58-65, wherein L comprises at least one polyethylene glycol monomer represented by

67. The conjugate of claim 66, wherein L comprises a polyethylene glycol oligomer represented by
 ? wherein n40 is 2 to 12.
? wherein n40 is 2 to 12.
68. The conjugate of claim 66 or 67, wherein Y comprises an oxime, and the at least one polyethylene glycol unit covalently bonds the oxime to W.
69. The conjugate of any one of claims 27-68, wherein Y further comprises a binding unit formed by a reaction between an alkyne and an azide or between an aldehyde or ketone and hydrazine or hydroxylamine.
70. The conjugate of any one of claims 27-69, wherein Y further comprises a binding unit represented by General Formula Vila, Vllb, Vile, Vlld or Vile:
[General Formula Vila]

[General Formula Vllb]

[General Formula Vile] ]

[General Formula Vile]

Li is each independently a single bond or C1-30 alkylene; and R11 is hydrogen or C1-10 alkyl.
71. The conjugate of any one of claims 27-70, wherein the conjugate comprises

72. The conjugate of any one of claims 1-71, wherein the conjugate comprises a structure represented by:
 or a pharmaceutically acceptable salt thereof; wherein nl2 is 0 to 30; and the wavy bond represents a connection to Ab.
or a pharmaceutically acceptable salt thereof; wherein nl2 is 0 to 30; and the wavy bond represents a connection to Ab.
73. The conjugate of any one of claims 27-72, wherein Y is branched.
74. The conjugate of claim 73, wherein Y comprises: i) a branching unit covalently bonded to Ab by a primary linker;
ii) a first which couples a first B to the branching unit; and iiia) a second branch which couples a second B to the branching unit; or iiib) a second branch, in which an alkyl or heteroalkyl (e.g., a polyethylene glycol monomer or a polyethylene glycol oligomer) is covalently coupled to the branching unit.
75. The conjugate of claim 74, wherein Y comprises a second branch which couples a second B, via a cleavage group, to the branching unit.
76. The conjugate of claim 74, wherein Y comprises a second branch, in which an alkyl or heteroalkyl (e.g., a polyethylene glycol monomer or a polyethylene glycol oligomer) is covalently coupled to the branching unit.
77. The conjugate of any one of claims 74-76, wherein the branching unit has a structure

R30 is hydrogen or C1-30 alkyl;
R40 is hydrogen or L5-COOR50;
R50 is hydrogen or C1-30 alkyl; and
L2, L3, and L4 are each independently a bond or -Cn'H2n'-; and n' is 1 to 10.
78. The conjugate of any one of claims 74-77, wherein: at least one branched linker is covalently coupled to Ab; and at least two B are covalently coupled to the branched linker.
79. The conjugate of claim 78, wherein the conjugate comprises 1, 2, 3, or 4 branched linkers and each branched linker comprises two B.
80. The conjugate of any one of claims 74-79, wherein the branching unit comprises a lysine residue.
81. The conjugate of any one of claims 74-80, wherein the conjugate comprises a structure represented by:
 or a pharmaceutically acceptable salt thereof; wherein each B is, independently, an active agent; nl 1, n22, and n33 are each independently 0 to 30; and
or a pharmaceutically acceptable salt thereof; wherein each B is, independently, an active agent; nl 1, n22, and n33 are each independently 0 to 30; and
AA is a peptide comprising at least two amino acid residues; and the wavy bond represents a connection to Ab.
82. The conjugate of any one of claims 1-81, wherein Ab comprises a first constant region linked to the first antigen-binding domain and a second constant region linked to the second antigen-binding domain, and wherein the first constant region and the second constant region each comprises a light chain constant region CL domain and heavy chain constant region CHI, CH2, and CH3 domains.
83. The conjugate of any one of claims 1-82, wherein Ab is a chimeric antibody, a humanized antibody, or a human antibody.
84. The conjugate of any one of claims 1-82, wherein Ab is of IgA, IgG, IgM, IgE, or IgD isotype.
85. The conjugate of claim 84, wherein Ab is of IgG isotype (e.g., IgGl, IgG2, IgG3, and IgG4).
86. The conjugate of any one of claims 1-85, wherein Ab comprises LALA mutations in the first and/or the second heavy chain constant regions.
87. The conjugate of claim 86, wherein the LALA mutations comprise L234A and L235A according to EU numbering convention.
88. The conjugate of any one of claims 83-87, wherein at least one amino acid residue in the CH3 domain of the first heavy chain constant region is substituted with an amino acid residue having a larger side chain size, and at least one amino acid residue in the CH3 domain of the second heavy chain constant region is substituted with an amino acid residue having a smaller side chain size; or at least one amino acid residue in the CH3 domain of the second heavy chain constant region is substituted with an amino acid residue having a larger side chain size, and at least one amino acid residue in the CH3 domain of the first heavy chain constant region is substituted with an amino acid residue having a smaller side chain size.
89. The conjugate of claim 88, wherein
the amino acid residue having a larger side chain size is selected from arginine, phenylalanine, tyrosine, and tryptophan; and the amino acid residue having a smaller side chain size is selected from alanine, serine, threonine, and valine.
90. The conjugate of claim 88 or 89, wherein the CH3 domain of the first heavy chain constant region comprises amino acid substitutions at positions T366, L368, and Y407 according to EU numbering, and the CH3 domain of the second heavy chain constant region comprises amino acid substitutions at position T366 according to EU numbering; or the CH3 domain of the second heavy chain constant region comprises amino acid substitutions at positions T366, L368, and Y407 according to EU numbering, and the CH3 domain of the first heavy chain constant region comprises amino acid substitutions at position T366 according to EU numbering.
91. The conjugate of claim 90, wherein the CH3 domain of the first heavy chain constant region comprises T366S, L368A, and Y407V according to EU numbering, and the CH3 domain of the second heavy chain constant region comprises T366W according to EU numbering; or the CH3 domain of the second heavy chain constant region comprises T366S, L368A, and Y407V according to EU numbering, and the CH3 domain of the first heavy chain constant region comprises T366W according to EU numbering.
92. The conjugate of any one of claims 88-91, wherein the CH3 domain of each of the first and the second heavy chain constant regions further comprises at least one substitution of an amino acid residue to a cysteine residue.
93. The conjugate of claim 88 or 89, wherein
(a) the CH3 domain of the first heavy chain constant region comprises E356C, T366S, L368A, and Y407V according to EU numbering, and the CH3 domain of the second heavy chain constant region comprises Y349C and T366W according to EU numbering; or
the CH3 domain of the second heavy chain constant region comprises E356C, T366S, L368A, and Y407V according to EU numbering, and the CH3 domain of the first heavy chain constant region comprises Y349C and T366W according to EU numbering; or
(b) the CH3 domain of the first heavy chain constant region comprises S354C, T366S, L368A, and Y407V according to EU numbering, and the CH3 domain of the second heavy chain constant region comprises Y349C and T366W according to EU numbering; or the CH3 domain of the second heavy chain constant region comprises S354C, T366S, L368A, and Y407V according to EU numbering, and the CH3 domain of the first heavy chain constant region comprises Y349C and T366W according to EU numbering.
94. The conjugate of claim 88 or 89, wherein
(a) the CH3 domain of the first heavy chain constant region comprises E356C, E357K, T366S, L368A, D399K, and Y407V according to EU numbering, and the CH3 domain of the second heavy chain constant region comprises Y349C, T366W, K370E, and R409D according to EU numbering; or the CH3 domain of the second heavy chain constant region comprises E356C, E357K, T366S, L368A, D399K, and Y407V according to EU numbering, and the CH3 domain of the first heavy chain constant region comprises Y349C, T366W, K370E, and R409D according to EU numbering; or
(b) the CH3 domain of the first heavy chain constant region comprises S354C, E357K, T366S, L368A, D399K, and Y407V according to EU numbering, and the CH3 domain of the second heavy chain constant region comprises Y349C, T366W, K370E, and R409D according to EU numbering; or the CH3 domain of the second heavy chain constant region comprises S354C, E357K, T366S, L368A, D399K, and Y407V according to EU numbering, and the CH3 domain of the first heavy chain constant region comprises Y349C, T366W, K370E, and R409D according to EU numbering.
95. The conjugate of any one of claims 83-87, wherein
(a) the CH3 domain of the first heavy chain constant region comprises amino acid substitutions at positions T366 and D399 according to EU numbering, and the CH3 domain of the second heavy chain constant region comprises amino acid substitutions at positions L351, Y407, and K409 according to EU numbering; or the CH3 domain of the second heavy chain constant region comprises amino acid substitutions at positions T366 and D399 according to EU numbering, and the CH3 domain of the first heavy chain constant region comprises amino acid substitutions at positions L351, Y407, and K409 according to EU numbering; or
(b) the CH3 domain of the first heavy chain constant region comprises amino acid substitutions at positions T366 and K409 according to EU numbering, and the CH3 domain of the second heavy chain constant region comprises amino acid substitutions at positions L351, D399, and Y407 according to EU numbering; or the CH3 domain of the second heavy chain constant region comprises amino acid substitutions at positions T366 and K409 according to EU numbering, and the CH3 domain of the first heavy chain constant region comprises amino acid substitutions at positions L351, D399, and Y407 according to EU numbering.
96. The conjugate of claim 95, wherein the substituted amino acid at T366 is selected from leucine, proline, tryptophan, and valine; the substituted amino acid at D399 is selected from cysteine, asparagine, isoleucine, glycine, arginine, threonine, and alanine; the substituted amino acid at L351 is selected from glycine, tyrosine, valine, proline, aspartic acid, glutamic acid, lysine, and tryptophan; the substituted amino acid at Y407 is selected from leucine, alanine, proline, phenylalanine, threonine, and histidine; and the substituted amino acid at K409 is selected from cysteine, proline, serine, phenylalanine, valine, glutamic acid, and arginine.
97. The conjugate of claim 95, wherein
(a) the CH3 domain of the first heavy chain constant region comprises amino acid substitutions T366L and D399R according to EU numbering, and the CH3 domain of
the second heavy chain constant region comprises amino acid substitutions L35 IE, Y407L and K409V according to EU numbering; or the CH3 domain of the second heavy chain constant region comprises amino acid substitutions T366L and D399R according to EU numbering, and the CH3 domain of the first heavy chain constant region comprises amino acid substitutions L351E, Y407L and K409V according to EU numbering; or
(b) the CH3 domain of the first heavy chain constant region comprises amino acid substitutions T366L and D399C according to EU numbering, and the CH3 domain of the second heavy chain constant region comprises amino acid substitutions L351G, Y407L and K409C according to EU numbering; or the CH3 domain of the second heavy chain constant region comprises amino acid substitutions T366L and D399C according to EU numbering, and the CH3 domain of the first heavy chain constant region comprises amino acid substitutions L351G, Y407L and K409C according to EU numbering; or
(c) the CH3 domain of the first heavy chain constant region comprises amino acid substitutions T366L and D399C according to EU numbering, and the CH3 domain of the second heavy chain constant region comprises amino acid substitutions L351Y, Y407A and K409P according to EU numbering; or the CH3 domain of the second heavy chain constant region comprises amino acid substitutions T366L and D399C according to EU numbering, and the CH3 domain of the first heavy chain constant region comprises amino acid substitutions L351Y, Y407A and K409P according to EU numbering; or
(d) the CH3 domain of the first heavy chain constant region comprises amino acid substitutions T366P and D399N according to EU numbering, and the CH3 domain of the second heavy chain constant region comprises amino acid substitutions L35 IV, Y407P and K409S according to EU numbering; or the CH3 domain of the second heavy chain constant region comprises amino acid substitutions T366P and D399N according to EU numbering, and the CH3 domain of the first heavy chain constant region comprises amino acid substitutions L351V, Y407P and K409S according to EU numbering; or
(e) the CH3 domain of the first heavy chain constant region comprises amino acid substitutions T366W and D399G according to EU numbering, and the CH3 domain of
the second heavy chain constant region comprises amino acid substitutions L35 ID, Y407P and K409S according to EU numbering; or the CH3 domain of the second heavy chain constant region comprises amino acid substitutions T366W and D399G according to EU numbering, and the CH3 domain of the first heavy chain constant region comprises amino acid substitutions L351D, Y407P and K409S according to EU numbering; or
(f) the CH3 domain of the first heavy chain constant region comprises amino acid substitutions T366P and D399I according to EU numbering, and the CH3 domain of the second heavy chain constant region comprises amino acid substitutions L35 IP, Y407F and K409F according to EU numbering; or the CH3 domain of the second heavy chain constant region comprises amino acid substitutions T366P and D399I according to EU numbering, and the CH3 domain of the first heavy chain constant region comprises amino acid substitutions L351P, Y407F and K409F according to EU numbering; or
(g) the CH3 domain of the first heavy chain constant region comprises amino acid substitutions T366V and D399T according to EU numbering, and the CH3 domain of the second heavy chain constant region comprises amino acid substitutions L35 IK, Y407T and K409Q according to EU numbering; or the CH3 domain of the second heavy chain constant region comprises amino acid substitutions T366V and D399T according to EU numbering, and the CH3 domain of the first heavy chain constant region comprises amino acid substitutions L351K, Y407T and K409Q according to EU numbering; or
(h) the CH3 domain of the first heavy chain constant region comprises amino acid substitutions T366L and D399A according to EU numbering, and the CH3 domain of the second heavy chain constant region comprises amino acid substitutions L351W, Y407H and K409R according to EU numbering; or the CH3 domain of the second heavy chain constant region comprises amino acid substitutions T366L and D399A according to EU numbering, and the CH3 domain of the first heavy chain constant region comprises amino acid substitutions L351W, Y407H and K409R according to EU numbering.
98. The conjugate of claim 95, wherein
the CH3 domain of the first heavy chain constant region comprises amino acid substitutions T366L and D399R, and the CH3 domain of the second heavy chain constant region comprises amino acid substitutions L351E, Y407L and K409Y; or the CH3 domain of the second heavy chain constant region comprises amino acid substitutions T366L and D399R, and the CH3 domain of the first heavy chain constant region comprises amino acid substitutions L351E, Y407L and K409Y.
99. The conjugate of any one of claims 83-98, wherein
(a) CL and CHI domains from the first constant region are replaced by each other; or CL and CHI domains from the second constant region are replaced by each other; or
(b) VH and VL domains from the first antigen-binding domain are replaced by each other; or
VH and VL domains from the second antigen-binding domain are replaced by each other; or
(c) VH and CHI domains and VL and CL domains from the first constant region and the first antigen-binding domain are replaced by each other; or
VH and CHI domains and VL and CL domains from the second constant region and the second antigen-binding domain are replaced by each other.
100. The conjugate of claim 99, wherein
CL and CHI domains from the first constant region are replaced by each other; or CL and CHI domains from the second constant region are replaced by each other.
101. The conjugate of any one of claims 83-98, wherein the first constant region and the second constant region have an asymmetrical structure to each other.
102. The conjugate of claim 101, wherein a knob-in-hole structure is further included between the first antigen-binding domain and the CHI domain of the first constant region; or a knob-in-hole structure is further included between the second antigen-binding domain and the CHI domain of the second constant region.
103. The conjugate of claim 102, wherein the knob-in -hole structure comprises a dimer of CH3 domains.
104. The conjugate of any one of claims 1-102, wherein the first antigen-binding domain specifically binding to CD20 comprises a first heavy chain variable region and a first light chain variable region, and the second antigen-binding domain specifically binding to CD22 comprises a second heavy chain variable region and a second light chain variable region, wherein
(a) the first heavy chain variable region comprises a heavy chain CDR1 comprising the amino acid sequence of SEQ ID NO: 2, a heavy chain CDR2 comprising the amino acid sequence of SEQ ID NO: 4, and a heavy chain CDR3 comprising the amino acid sequence of SEQ ID NO: 6, the first light chain variable region comprises a light chain CDR1 comprising the amino acid sequence of SEQ ID NO: 9, a light chain CDR2 comprising the amino acid sequence of SEQ ID NO: 11, and a light chain CDR3 comprising the amino acid sequence of SEQ ID NO: 13, the second heavy chain variable region comprises a heavy chain CDR1 comprising the amino acid sequence of SEQ ID NO: 40, a heavy chain CDR2 comprising the amino acid sequence of SEQ ID NO: 42, and a heavy chain CDR3 comprising the amino acid sequence of SEQ ID NO: 44, and the second light chain variable region comprises a light chain CDR1 comprising the amino acid sequence of SEQ ID NO: 47, a light chain CDR2 comprising the amino acid sequence of SEQ ID NO: 49, and a light chain CDR3 comprising the amino acid sequence of SEQ ID NO: 51;
(b) the first heavy chain variable region comprises a heavy chain CDR1 comprising the amino acid sequence of SEQ ID NO: 2, a heavy chain CDR2 comprising the amino acid sequence of SEQ ID NO: 4, and a heavy chain CDR3 comprising the amino acid sequence of SEQ ID NO: 6, the first light chain variable region comprises a light chain CDR1 comprising the amino acid sequence of SEQ ID NO: 9, a light chain CDR2 comprising the amino acid sequence of SEQ ID NO: 11, and a light chain CDR3 comprising the amino acid sequence of SEQ ID NO: 13,
the second heavy chain variable region comprises a heavy chain CDR1 comprising the amino acid sequence of SEQ ID NO: 58, a heavy chain CDR2 comprising the amino acid sequence of SEQ ID NO: 60, and a heavy chain CDR3 comprising the amino acid sequence of SEQ ID NO: 62, and the second light chain variable region comprises a light chain CDR1 comprising the amino acid sequence of SEQ ID NO: 65, a light chain CDR2 comprising the amino acid sequence of SEQ ID NO: 67, and a light chain CDR3 comprising the amino acid sequence of SEQ ID NO: 69;
(c) the first heavy chain variable region comprises a heavy chain CDR1 comprising the amino acid sequence of SEQ ID NO: 22, a heavy chain CDR2 comprising the amino acid sequence of SEQ ID NO: 24, and a heavy chain CDR3 comprising the amino acid sequence of SEQ ID NO: 26, the first light chain variable region comprises a light chain CDR1 comprising the amino acid sequence of SEQ ID NO: 29, a light chain CDR2 comprising the amino acid sequence of SEQ ID NO: 31, and a light chain CDR3 comprising the amino acid sequence of SEQ ID NO: 33, the second heavy chain variable region comprises a heavy chain CDR1 comprising the amino acid sequence of SEQ ID NO: 40, a heavy chain CDR2 comprising the amino acid sequence of SEQ ID NO: 42, and a heavy chain CDR3 comprising the amino acid sequence of SEQ ID NO: 44, and the second light chain variable region comprises a light chain CDR1 comprising the amino acid sequence of SEQ ID NO: 47, a light chain CDR2 comprising the amino acid sequence of SEQ ID NO: 49, and a light chain CDR3 comprising the amino acid sequence of SEQ ID NO: 51; or
(d) the first heavy chain variable region comprises a heavy chain CDR1 comprising the amino acid sequence of SEQ ID NO: 22, a heavy chain CDR2 comprising the amino acid sequence of SEQ ID NO: 24, and a heavy chain CDR3 comprising the amino acid sequence of SEQ ID NO: 26, the first light chain variable region comprises a light chain CDR1 comprising the amino acid sequence of SEQ ID NO: 29, a light chain CDR2 comprising the amino acid sequence of SEQ ID NO: 31, and a light chain CDR3 comprising the amino acid sequence of SEQ ID NO: 33,
the second heavy chain variable region comprises a heavy chain CDR1 comprising the amino acid sequence of SEQ ID NO: 58, a heavy chain CDR2 comprising the amino acid sequence of SEQ ID NO: 60, and a heavy chain CDR3 comprising the amino acid sequence of SEQ ID NO: 62, and the second light chain variable region comprises a light chain CDR1 comprising the amino acid sequence of SEQ ID NO: 65, a light chain CDR2 comprising the amino acid sequence of SEQ ID NO: 67, and a light chain CDR3 comprising the amino acid sequence of SEQ ID NO: 69.
105. The conjugate of claim 104, wherein
(a) the VH domain of the first antigen-binding domain comprises: the amino acid sequence of SEQ ID NO: 15; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 15; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 15; the VL domain of the first antigen-binding domain comprises: the amino acid sequence of SEQ ID NO: 16; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 16; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 16; the VH domain of the second antigen-binding domain comprises: the amino acid sequence of SEQ ID NO: 53; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 53; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 53; and the VL domain of the second antigen-binding domain comprises: the amino acid sequence of SEQ ID NO: 54; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 54; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 54;
(b) the VH domain of the first antigen-binding domain comprises: the amino acid sequence of SEQ ID NO: 15; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 15; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 15; the VL domain of the first antigen-binding domain comprises: the amino acid sequence of SEQ ID NO: 16;
at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 16; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 16; the VH domain of the second antigen-binding domain comprises: the amino acid sequence of SEQ ID NO: 71; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 71; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 71; and the VL domain of the second antigen-binding domain comprises: the amino acid sequence of SEQ ID NO: 72; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 72; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 72;
(c) the VH domain of the first antigen-binding domain comprises: the amino acid sequence of SEQ ID NO: 35; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 35; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 35; the VL domain of the first antigen-binding domain comprises: the amino acid sequence of SEQ ID NO: 36; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 36; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 36; the VH domain of the second antigen-binding domain comprises: the amino acid sequence of SEQ ID NO: 53; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 53; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 53; and the VL domain of the second antigen-binding domain comprises: the amino acid sequence of SEQ ID NO: 54; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 54; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 54; or
(d) the VH domain of the first antigen-binding domain comprises: the amino acid sequence of SEQ ID NO: 35; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 35; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 35; the VL domain of the first antigen-binding domain comprises: the amino acid sequence of SEQ ID NO: 36;
at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 36; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 36; the VH domain of the second antigen-binding domain comprises: the amino acid sequence of SEQ ID NO: 71; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 71; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 71; and the VL domain of the second antigen-binding domain comprises: the amino acid sequence of SEQ ID NO: 72; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 72; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 72.
106. The conjugate of claim 104, wherein
(a) the VH domain of the first antigen-binding domain comprises: the amino acid sequence of SEQ ID NO: 15; the VL domain of the first antigen-binding domain comprises: the amino acid sequence of SEQ ID NO: 16; the VH domain of the second antigen-binding domain comprises: the amino acid sequence of SEQ ID NO: 53; and the VL domain of the second antigen-binding domain comprises: the amino acid sequence of SEQ ID NO: 54;
(b) the VH domain of the first antigen-binding domain comprises: the amino acid sequence of SEQ ID NO: 15; the VL domain of the first antigen-binding domain comprises: the amino acid sequence of SEQ ID NO: 16; the VH domain of the second antigen-binding domain comprises: the amino acid sequence of SEQ ID NO: 71; and the VL domain of the second antigen-binding domain comprises: the amino acid sequence of SEQ ID NO: 72;
(c) the VH domain of the first antigen-binding domain comprises: the amino acid sequence of SEQ ID NO: 35; the VL domain of the first antigen-binding domain comprises: the amino acid sequence of SEQ ID NO: 36;
the VH domain of the second antigen-binding domain comprises: the amino acid sequence of SEQ ID NO: 53; and the VL domain of the second antigen-binding domain comprises: the amino acid sequence of SEQ ID NO: 54; or
(d) the VH domain of the first antigen-binding domain comprises: the amino acid sequence of SEQ ID NO: 35; the VL domain of the first antigen-binding domain comprises: the amino acid sequence of SEQ ID NO: 36; the VH domain of the second antigen-binding domain comprises: the amino acid sequence of SEQ ID NO: 71; and the VL domain of the second antigen-binding domain comprises: the amino acid sequence of SEQ ID NO: 72.
107. The conjugate of any one of claims 1-87, wherein Ab comprises a first heavy chain and a first light chain comprising the first antigen-binding domain specifically binding to CD20 and a second heavy chain and a second light chain comprising the second antigenbinding domain specifically binding to CD22, wherein
(a) the first heavy chain comprises: the amino acid sequence of SEQ ID NO: 20; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 20; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 20; the first light chain comprises: the amino acid sequence of SEQ ID NO: 18; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 18; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 18; the second heavy chain comprises: the amino acid sequence of SEQ ID NO: 55; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 55; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 55; and the second light chain comprises: the amino acid sequence of SEQ ID NO: 56; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 56; or
at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 56;
(b) the first heavy chain comprises: the amino acid sequence of SEQ ID NO: 20; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 20; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 20; the first light chain comprises: the amino acid sequence of SEQ ID NO: 18; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 18; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 18; the second heavy chain comprises: the amino acid sequence of SEQ ID NO: 73; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 73; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 73; and the second light chain comprises: the amino acid sequence of SEQ ID NO: 74; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 74; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 74;
(c) the first heavy chain comprises: the amino acid sequence of SEQ ID NO: 37; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 37; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 37; the first light chain comprises: the amino acid sequence of SEQ ID NO: 38; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 38; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 38; the second heavy chain comprises: the amino acid sequence of SEQ ID NO: 55; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 55; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 55; and the second light chain comprises: the amino acid sequence of SEQ ID NO: 56; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 56; or
at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 56;
(d) the first heavy chain comprises: the amino acid sequence of SEQ ID NO: 37; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 37; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 37; the first light chain comprises: the amino acid sequence of SEQ ID NO: 38; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 38; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 38; the second heavy chain comprises: the amino acid sequence of SEQ ID NO: 73; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 73; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 73; and the second light chain comprises: the amino acid sequence of SEQ ID NO: 74; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 74; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 74;
(e) the first heavy chain comprises: the amino acid sequence of SEQ ID NO: 75; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 75; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 75; the first light chain comprises: the amino acid sequence of SEQ ID NO: 18; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 18; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 18; the second heavy chain comprises: the amino acid sequence of SEQ ID NO: 76; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 76; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 76; and the second light chain comprises: the amino acid sequence of SEQ ID NO: 56; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 56; or
at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 56; or
(f) the first heavy chain comprises: the amino acid sequence of SEQ ID NO: 20; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 20; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 20; the first light chain comprises: the amino acid sequence of SEQ ID NO: 18; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 18; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 18; the second heavy chain comprises: the amino acid sequence of SEQ ID NO: 81; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 81; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 81; and the second light chain comprises: the amino acid sequence of SEQ ID NO: 82; at least 90% sequence identity to the amino acid sequence of SEQ ID NO: 82; or at least 95% sequence identity to the amino acid sequence of SEQ ID NO: 82.
108. The conjugate of claim 107, wherein
(a) the first heavy chain comprises the amino acid sequence of SEQ ID NO: 20; the first light chain comprises the amino acid sequence of SEQ ID NO: 18; the second heavy chain comprises the amino acid sequence of SEQ ID NO: 55; and the second light chain comprises the amino acid sequence of SEQ ID NO: 56;
(b) the first heavy chain comprises the amino acid sequence of SEQ ID NO: 20; the first light chain comprises the amino acid sequence of SEQ ID NO: 18; the second heavy chain comprises the amino acid sequence of SEQ ID NO: 73; and the second light chain comprises the amino acid sequence of SEQ ID NO: 74;
(c) the first heavy chain comprises the amino acid sequence of SEQ ID NO: 37; the first light chain comprises the amino acid sequence of SEQ ID NO: 38; the second heavy chain comprises the amino acid sequence of SEQ ID NO: 55; and the second light chain comprises the amino acid sequence of SEQ ID NO: 56;
(d) the first heavy chain comprises the amino acid sequence of SEQ ID NO: 37;
the first light chain comprises the amino acid sequence of SEQ ID NO: 38; the second heavy chain comprises the amino acid sequence of SEQ ID NO: 73; and the second light chain comprises the amino acid sequence of SEQ ID NO: 74;
(e) the first heavy chain comprises the amino acid sequence of SEQ ID NO: 75; the first light chain comprises the amino acid sequence of SEQ ID NO: 18; the second heavy chain comprises the amino acid sequence of SEQ ID NO: 76; and the second light chain comprises the amino acid sequence of SEQ ID NO: 56; or
(f) the first heavy chain comprises the amino acid sequence of SEQ ID NO: 20; the first light chain comprises the amino acid sequence of SEQ ID NO: 18; the second heavy chain comprises the amino acid sequence of SEQ ID NO: 81; and the second light chain comprises the amino acid sequence of SEQ ID NO: 82.
109. The conjugate of any one of claims 1-108, wherein Ab is a full-length antibody.
110. The conjugate of claim 109, wherein Ab is an IgG-scFv, a trifunctional antibody (triomab), a knobs into holes (KIH)-IgG, a K/.-body. a crossmab, an ortho-Fab IgG, a dual variable domain immunoglobulin (DVD-Ig), or a 2 in 1-IgG (dual action antibody), or a combination thereof.
111. The conjugate of any one of claims 1-106, wherein Ab is an antigen binding fragment.
112. The conjugate of claim 111, wherein Ab is a scFv2-Fc, a bi-nanobody, a bispecific T cell engager (BiTE), a tandem diabody (tandAb), a dual affinity retargeting (DART) antibody, a DART-Fc, a scFv-human serum albumin (HSA)-scFv, a dock-and-lock (DNL)- Fab3, a minibody, a Fab2 fragment (bispecific), a Fab3 fragment (trispecific), a Bis-scFv fragment (bispecific), a sdAb fragment (VH/VHH). a tetrabody, a triabody, or a diabody, or a combination thereof.
113. The conjugate of any one of claims 1-112, wherein the active agent is a chemotherapeutic agent or a toxin.
114. The conjugate of any one of claims 1-112, wherein the active agent is selected from a chemotherapeutic compound, a cytotoxic compound, an immunomodulatory compound, an anticancer agent, an antiviral agent, an antibacterial agent, an antifungal agent, an antiparasitic agent, and a combination thereof.
115. The conjugate of claim 114, wherein the cytotoxic compound is selected from a mitotic inhibitor, a DNA alkylating agent, a topoisomerase inhibitor, and a combination thereof.
116. The conjugate of claim 114, wherein the cytotoxic compound is selected from auristatin, maytansinoid, tubulisin, calicheamicin, duocarmycin, pyrrolobenzodiazepine and derivatives thereof, camptothecin and derivatives thereof, and combinations thereof.
117. The conjugate of claim 116, wherein the auristatin is MMAE (monomethyl auristatin E) or MMAF (monomethyl auristatin F).
118. The conjugate of any one of claims 1-112, wherein: the active agent is a pyrrolobenzodiazepine dimer; position N10 of the pyrrolobenzodiazepine dimer is substituted with X or position N’ 10 is substituted with X’, wherein X or X' links the pyrrolobenzodiazepine dimer to the linker;
X and X' are each independently -C(O)O-* or -C(O)-* ; and
* refers to a binding site between the pyrrolobenzodiazepine dimer and the linker.
119. The conjugate of any one of claims 1-112, wherein the active agent is a pyrrolobenzodiazepine dimer and the pyrrolobenzodiazepine dimer has a structure represented by Formula VIII:

[Formula VIII] wherein the wavy line indicates a connection point to the linker; a dotted line represents an optional double bond;
Ri and Ri are each independently selected from H, OH, =0, =CH2, CN, Rm, 0Rm, =CH-Rm' =C(Rm)2, O-SO2-Rm, C02Rm, C0Rm, halo, and dihalo;
Rm is selected from Rm, C02Rm, C0Rm, CHO, CO2H. and halo;
Rm is selected from substituted or unsubstituted C1-12 alkyl, substituted or unsubstituted C2-12 alkenyl, substituted or unsubstituted C2-12 alkynyl, substituted or unsubstituted Cs-2o aryl, substituted or unsubstituted C3-6 heteroaryl, substituted or unsubstituted C3-6 cycloalkyl, substituted or unsubstituted 3- to 7-membered heterocyclyl, substituted or unsubstituted 3- to 7-membered heterocycloalkyl, and substituted or unsubstituted 5- to 7-membered heteroaryl, wherein when the C1-12 alkyl, C2-12 alkenyl, C2-12 alkynyl, Cs-2o aryl, Cs-2o heteroaryl, C3-6 cycloalkyl, 3- to 7-membered heterocyclyl, 3- to 7- membered heterocycloalkyl, or 5- to 7-membered heteroaryl is substituted, the respective hydrogen atoms in the C1-12 alkyl, C2-12 alkenyl, C2-12 alkynyl, Cs-2o aryl, Cs-2o heteroaryl, C3-6 cycloalkyl, 3- to 7-membered heterocyclyl, 3- to 7-membered heterocycloalkyl, or 5- to 7-membered heteroaryl may each be independently replaced with methoxy, C1-12 alkyl, C2-12 alkenyl, C2-12 alkynyl, Cs-2o aryl, Cs-2o heteroaryl, C3-6 cycloalkyl, 3- to 7-membered heterocyclyl, 3- to 7-membered heterocycloalkyl, and 5- to 7-membered heteroaryl;
R.2, R3, Rs, R2, R3 , and Rs are each independently selected from H, Rm, OH, ORm, SH, SRm, NH2, NHRm, NRmRm, NO2, Me3Sn, and halo;
R4 and R4 are each independently selected from H, Rm, OH, ORm, SH, SRm, NH2, NHRm, NRmRm , NO2, McsSn. halo, substituted or unsubstituted C1-6 alkyl, substituted or unsubstituted C1-6 alkoxy, substituted or unsubstituted C2-6 alkenyl, substituted or unsubstituted C2-6 alkynyl, substituted or unsubstituted C3-6 cycloalkyl, substituted or unsubstituted 3- to 7-membered heterocycloalkyl, substituted or unsubstituted C5-12 aryl, substituted or unsubstituted 5- to 7-membered heteroaryl, -CN, -NCO, -ORn, - OC(O)Rn, -OC(O)NRnRn, -OS(O)Rn, -OS(O)2Rn, -SRn, -S(O)Rn, -S(O)2Rn, - S(O)NRnRn, -S(O)2NRnRn , -OS(O)NRnRn', -OS(O)2NRnRn', -NRnRn, -NRnC(O)R°, - NRnC(O)OR°, -NRnC(O)NR°R° , -NRnS(O)R°, -NRnS(O)2R°, -NRnS(O)NR°R° , - NRnS(O)2NR°R° , -C(O)Rn, -C(O)ORn, and -C(O)NRnRn , wherein the hydrogen atoms in the C1-6 alkyl, C1-6 alkoxy, C2-6 alkenyl, C2-6 alkynyl, C3-6 cycloalkyl, 3- to 7- membered heterocycloalkyl, C5-12 aryl, and 5- to 7-membered heteroaryl may each be independently replaced with C1-6 alkyl, C1-6 alkoxy, C2-6 alkenyl, C2-6 alkynyl, C3-6 cycloalkyl, 3- to 7-membered heterocycloalkyl, C5-12 aryl, 5- to 7-membered heteroaryl, -ORP, -OC(O)RP, -OC(O)NRPRP', -OS(O)RP, -OS(O)2RP, -SRP, -S(O)RP, - S(O)2Rp, -S(O)NRpRp ', -S(O)2NRPRP , -OS(O)NRPRP', -OS(O)2NRPRP', -NRpRp ', - NRpC(O)Rq, -NRpC(O)ORq, -NRpC(0)NRqH, -NRpS(O)Rq, -NRpS(O)2Rq, - NRpS(O)NRqH, -NRpS(O)2NRqH, -C(O)RP, -C(O)ORP, or -C(O)NRPRP when the Ci-6 alkyl, C1-6 alkoxy, C2-6 alkenyl, C2-6 alkynyl, C3-6 cycloalkyl, 3- to 7-membered heterocycloalkyl, C5-12 aryl, and 5- to 7-membered heteroaryl;
Rn, Rn, R°, R° Rp, Rp , and Rq are each independently selected from H, C1-7 alkyl, C2-7 alkenyl, C2-7 alkynyl, C3-13 cycloalkyl, 3- to 7-membered heterocycloalkyl, C6-10 aryl, and 5- to 7-membered heteroaryl;
X is selected from -C(O)O-, -S(O)O-, -C(O)-, -C(O)NR-, -S(O)2NR-, -P(O)R'NR-, -S(O)NR-, and -PO2NR-;
Xa is a bond or substituted or unsubstituted C1-6 alkylene, wherein C1-6 alkylene is substituted with C1-8 alkyl, or C3-8 cycloalkyl when substituted;
R and R each independently denote H, OH, NH2, ONH2, NHNH2, substituted or unsubstituted C1-8 alkyl, substituted or unsubstituted C3-8 cycloalkyl, substituted or unsubstituted C1-8 alkoxy, substituted or unsubstituted C1-8 alkylthio, substituted or
unsubstituted C3-20 heteroaryl, substituted or unsubstituted C5-20 aryl, or mono- or di- C1-8 alkylamino, wherein the C1-8 alkyl, C3-8 cycloalkyl, C1-8 alkoxy, C1-8 alkylthio, C3-20 heteroaryl, and C5-20 aryl are substituted with a substituent selected from OH, N3, CN, NO2, SH, NH2, ONH2, NHNH2, halo, C1-6 alkyl, C1-6 alkoxy, and Ce-12 aryl when substituted;
Y and Y' are each independently selected from O, S, and N(H);
Rs is a substituted or unsubstituted saturated or unsaturated C3-12 hydrocarbon chain, wherein the chain may be interrupted by one or more heteroatoms, NMe, or a substituted or unsubstituted aromatic ring, the chain or aromatic ring may be substituted with -NH, -NRm, -NHC(O)Rm, -NHC(O)CH2-[OCH2CH2]n-R, or-[CH2CH2O]n-R at any one or more positions of hydrogen atoms on the chain or aromatic ring or unsubstituted, wherein Rm and R are each as defined for Rm and R above, and n is 1 to 12; and
R7 and R7 are each independently H, substituted or unsubstituted C1-6 alkyl, substituted or unsubstituted C2-6 alkenyl, substituted or unsubstituted C2-6 alkynyl, substituted or unsubstituted C3-6 cycloalkyl, substituted or unsubstituted 3- to 7-membered heterocycloalkyl, substituted or unsubstituted Ce-io aryl, substituted or unsubstituted 5- to 7-membered heteroaryl, -ORr, -OC(O)Rr, -OC(O)NRrRr , -OS(O)Rr, -OS(O)2Rr, - SRr, -S(O)Rr, -S(O)2Rr, -S(O)NRrRr , -S(O)2NRrRr', -OS(O)NRrRr', -OS(O)2NRrRr', - NRrRr , -NRrC(O)Rs, -NRrC(O)ORs, -NRrC(O)NRsRs', -NRrS(O)Rs, -NRrS(O)2Rs, - NRrS(O)NRsRs , -NRrS(O)2NRsRs, -C(O)Rr, -C(O)ORS, or -C(O)NRRr ', wherein the hydrogen atoms in the C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-6 cycloalkyl, 3- to 7- membered heterocycloalkyl, Ce-io aryl, and 5- to 7-membered heteroaryl may each be independently replaced with C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-6 cycloalkyl, 3- to 7-membered heterocycloalkyl, C6-10 aryl, 5- to 7-membered heteroaryl, -OR1, - OC(O)R, -OC(O)NRtRt, -OS(O)R, -OS(O)2Rl, -SR‘, -S(O)R, -S(O)2Rl, -S(O)NRtRt, -S(O)2NRtRt, -OS(O)NRtRt , -OS(O)2NRtRt', -NRR. -NRC(O)RU, -NRC(O)ORU, - NRlC(O)NRuRu , -NRS(O)RU, -NRtS(O)2Ru,-NRtS(O)NRuRu', -NRtS(O)2NRuRu', - C(O)Rl, -C(O)ORl, or -C(O)NRtRt when the C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-6 cycloalkyl, 3- to 7-membered heterocycloalkyl, C6-10 aryl, and 5- to 7-membered heteroaryl;
Rr, Rr, Rs, Rs, R\ Rv, Ru, and RUI are each independently selected from H, C1-7 alkyl, C2-7 alkenyl, C2-7 alkynyl, C3-13 cycloalkyl, 3- to 7-membered heterocycloalkyl, C5-10 aryl, and 5- to 7-membered heteroaryl;
G is a glucuronide group or a galactoside group;
O each Z is selected from H, Ci-s alkyl, halo, NO2, CN,
 ;
;
R9, Rio, and Ris are each independently selected from H, Ci-s alkyl, C2-6 alkenyl, C1-6 alkoxy, and alkyloxyalkyl; and n30 is 0 to 3.
120. The conjugate claim 119, wherein Y is O.
121. The conjugate claim 119 or 120, wherein is Y’ is O.
122. The conjugate of any one of claims 119-121, wherein a dotted line represents presence of a double bond between the carbons bearing Ri and R7 or Ri and R7 .
123. The conjugate of any one of claims 119-122, wherein Ri is selected from substituted or unsubstituted C1-6 alkyl, substituted or unsubstituted C2-6 alkenyl, substituted or unsubstituted C5-7 aryl, and substituted or unsubstituted C3-6 heteroaryl.
124. The conjugate of any one of claims 119-123, wherein R2, R3, and Rs are each independently H or OH.
125. The conjugate of any one of claims 119-124, wherein R4 is C1-6 alkoxy.
126. The conjugate of any one of claims 119-124, wherein R4 is methoxy, ethoxy, or butoxy.
127. The conjugate of any one of claims 119-126, wherein
X is selected from -C(O)O-, -C(O)-, and -C(O)NR-.
128. The conjugate of any one of claims 119-127, wherein X is -C(O)NR-.
129. The conjugate of any one of claims 119-128, wherein Rs is a substituted or unsubstituted saturated or unsaturated C3-8 hydrocarbon chain, wherein one or more of the carbon atoms of the hydrocarbon chain is replaced by a heteroatom or a substituted or unsubstituted aromatic ring, wherein the heteroatom is O, S, or N(H) and the aromatic ring is benzene, pyridine, imidazole, or pyrazole, and the chain or aromatic ring may be substituted with -NHC(O)CH2-[OCH2CH2]n-R or-fCTbCTbOjn- R at any one or more positions of hydrogen atoms on the chain or aromatic ring; and n is 1 to 6.
130. The conjugate of any one of claims 119-129, wherein n is 1 to 6.
131. The conjugate of any one of claims 119-130, wherein Xa is a bond or C 1-3 alkylene .
O
132. The conjugate of any one of claims 119-131, wherein Z is H,
 , and
, and
 , wherein R9, Rio, and Rie are each independently selected from H, C1-3 alkyl, C1-3 alkoxy, and alkyloxyalkyl .
, wherein R9, Rio, and Rie are each independently selected from H, C1-3 alkyl, C1-3 alkoxy, and alkyloxyalkyl .
133. The conjugate of claim 132, wherein R9 is methyloxyalkyl.
134. The conjugate of claim 132 or 133, wherein Rio is methyl oxyalkyl.
135. The conjugate of any one of claims 132-134, wherein Rig is methyloxyalkyl.
136. The conjugate of any one of claims 132-135, wherein R9, Rio, or Ris is -
(CH2CH2O)m-(CH2)m2CH3, further wherein m is 1-6 and m2 is 0-2.
137. The conjugate of claim 136, wherein m is 1.
138. The conjugate of claim 136 or 137, wherein m2 is 0.
139. The conjugate of claim 119, wherein R2 is H.
140. The conjugate of claim 139, wherein R3 is H.
141. The conjugate of claim 139 or 140, wherein R7 is H.
142. The conjugate of any one of claims 139-141, wherein R4 is C1-6 alkoxy (e.g., methoxy).
143. The conjugate of any one of claims 139-142, wherein R5 is OH.
144. The conjugate of any one of claims 139-143, wherein Ri is =CH2, CH3, or phenyl, optionally substituted with methoxy.
145. The conjugate of any one of claims 139-144, wherein Y is O.
146. The conjugate of any one of claims 139-145, wherein R2’ is H.
147. The conjugate of any one of claims 139-146, wherein R3’ is H.
148. The conjugate of any one of claims 139-147, wherein R7’ is H.
149. The conjugate of any one of claims 139-148, wherein R4’ is C1-6 alkoxy (e.g., methoxy).
150. The conjugate of any one of claims 139-149, wherein R5’ is OH.
151. The conjugate of any one of claims 139-150, wherein Rf is =CH2, CH3, or phenyl, optionally substituted with methoxy.
152. The conjugate of any one of claims 139-151, wherein Y’ is O.
153. The conjugate of any one of claims 139-152, wherein X is -C(O)O-.
154. The conjugate of any one of claims 139-153, wherein Xa is CH2.
155. The conjugtae of any one of claims 139-154, wherein G is a glucuronide group.
156. The conjugate of calim 155, wherein

157. The conjugate of any one of calims 139-156, wherein n30 is 1.
O
158. The conjugate of any one of calims 139-157, wherein Z is
 or
or

159. The conjugate of claim 158, wherein R9 is H.
160. The conjugate of claim 158 or 159, wherein Ri6 is alkyloxyalkyl (e.g., methoxy ethyl).
161. The conjugate of any one of calims 139-157, wherein Z is

162. The conjugate of claim 161, wherein Rio is alkyl (e.g., methyl).
163. The conjugate of claim 161, wherein R$ is C3-12 alkyl (e.g., pentyl).
164. The conjugate of claim 118, wherein the pyrrolobenzodiazepine dimer is selected from:

OMe or -NH2.
165. A pharmaceutical composition comprising the conjugate according to any one of claims 1-164 and a pharmaceutically acceptable excipient.
166. A pharmaceutical composition for use in preventing or treating a proliferative disease, the composition comprising the conjugate according to any one of claims 1-164.
167. The pharmaceutical composition for use of claim 166, wherein the proliferative disease is cancer.
168. The pharmaceutical composition for use of claim 167, wherein the cancer is selected from liver cancer, thyroid cancer, ovarian cancer, brain cancer, multiple myeloma, colon cancer, head and neck cancer, lymphoma, leukemia, bladder cancer, kidney cancer, stomach cancer, breast cancer, uterine cancer, prostate cancer, pancreatic cancer, lung cancer, sarcoma, neuroendocrine tumor, melanoma, and combinations thereof.
169. The pharmaceutical composition for use of claim 168, wherein the lymphoma or leukemia is selected from Hodgkin lymphoma, non-Hodgkin lymphoma (NHL), T cell
lymphoma, B cell lymphoma, natural killer cell lymphoma, diffuse large B cell lymphoma (DLBCL), mantle cell lymphoma (MCL), primary central nervous system (CNS) lymphoma, lymphoblastic lymphoma, enteropathy-type intestinal lymphoma, anaplastic large cell lymphoma, angioimmunoblastic T cell lymphoma, anaplastic large cell lymphoma, peripheral T cell lymphoma, marginal zone lymphoma, chronic lymphocytic leukemia (CLL), acute lymphoblastic leukemia (ALL), chronic myelogenous leukemia (CML), acute myelogenous leukemia (AML), and combinations thereof.
170. The pharmaceutical composition for use of claim 169, wherein the acute lymphoblastic leukemia (ALL) is B-cell acute lymphoblastic leukemia (B-ALL).
171. A method of treating or preventing a proliferative disease in a subj ect in need thereof comprising administering a conjugate of any one of claims 1-164 or a pharmaceutically acceptable salt thereof to the subject.
172. The method of claim 171, wherein the proliferative disease is cancer.
173. The method of claim 172, wherein the cancer is selected from liver cancer, thyroid cancer, ovarian cancer, brain cancer, multiple myeloma, colon cancer, head and neck cancer, lymphoma, leukemia, bladder cancer, kidney cancer, stomach cancer, breast cancer, uterine cancer, prostate cancer, pancreatic cancer, lung cancer, sarcoma, neuroendocrine tumor, melanoma, and combinations thereof.
174. The method of claim 173, wherein the lymphoma or leukemia is selected from Hodgkin lymphoma, non-Hodgkin lymphoma (NHL), T cell lymphoma, B cell lymphoma, natural killer cell lymphoma, diffuse large B cell lymphoma (DLBCL), mantle cell lymphoma (MCL), primary central nervous system (CNS) lymphoma, lymphoblastic lymphoma, enteropathy-type intestinal lymphoma, anaplastic large cell lymphoma, angioimmunoblastic T cell lymphoma, anaplastic large cell lymphoma, peripheral T cell lymphoma, marginal zone lymphoma, chronic lymphocytic leukemia (CLL), acute lymphoblastic leukemia (ALL), chronic myelogenous leukemia (CML), acute myelogenous leukemia (AML), and combinations thereof.
175. The method of claim 174, wherein the acute lymphoblastic leukemia (ALL) is B-cell acute lymphoblastic leukemia (B-ALL).
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KRKR10-2023-0155908 | 2023-11-10 | ||
| KR20230155908 | 2023-11-10 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2025099489A1 true WO2025099489A1 (en) | 2025-05-15 |
Family
ID=95695087
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2024/000636 Pending WO2025099489A1 (en) | 2023-11-10 | 2024-11-08 | Antibody-drug conjugate comprising bispecific antibody specifically binding to cd20 and cd22 and uses thereof |
Country Status (2)
| Country | Link |
|---|---|
| TW (1) | TW202535946A (en) |
| WO (1) | WO2025099489A1 (en) |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20180193477A1 (en) * | 2015-07-15 | 2018-07-12 | Zymeworks Inc. | Drug-conjugated bi-specific antigen-binding constructs |
| US20220380480A1 (en) * | 2021-04-30 | 2022-12-01 | Hoffmann-La Roche Inc. | Dosing for combination treatment with anti-cd20/anti-cd3 bispecific antibody and anti-cd79b antibody drug conjugate |
-
2024
- 2024-11-08 WO PCT/IB2024/000636 patent/WO2025099489A1/en active Pending
- 2024-11-11 TW TW113143188A patent/TW202535946A/en unknown
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20180193477A1 (en) * | 2015-07-15 | 2018-07-12 | Zymeworks Inc. | Drug-conjugated bi-specific antigen-binding constructs |
| US20220380480A1 (en) * | 2021-04-30 | 2022-12-01 | Hoffmann-La Roche Inc. | Dosing for combination treatment with anti-cd20/anti-cd3 bispecific antibody and anti-cd79b antibody drug conjugate |
Non-Patent Citations (3)
| Title |
|---|
| DE GOEIJ ET AL.: "Efficient Payload Delivery by a Bispecific Antibody-Drug Conjugate Targeting HER2 and CD 6 3", MOL. CANCER THER, vol. 15, no. 11, 2016, pages 2688 - 2697, XP055358969, DOI: 10.1158/1535-7163.MCT-16-0364 * |
| QU, Z. ET AL.: "Bispecific anti- CD 20/22 antibodies inhibit B- cell lymphoma proliferation by a unique mechanism of action", BLOOD, vol. 111, no. 4, 2008, pages 2211 - 2219, XP086509619, DOI: 10.1182/blood-2007-08-110072 * |
| TUSCANO, J. M. ET AL.: "The Bs20x22 anti- CD 20- CD 22 bispecific antibody has more lymphomacidal activity than do the parent antibodies alone", CANCER IMMUNOL. IMMUNOTHER, vol. 60, 2011, pages 771 - 780, XP019906892, DOI: 10.1007/s00262-011-0978-6 * |
Also Published As
| Publication number | Publication date |
|---|---|
| TW202535946A (en) | 2025-09-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP2024102229A (en) | Anti-EGFR antibodies and antibody-drug conjugates | |
| KR20210028544A (en) | Antibody-drug conjugate comprising antibody binding to antibody against human ROR1 and its use | |
| CA3075087A1 (en) | Anti- folate receptor alpha antibody conjugates and their uses | |
| US12144869B2 (en) | Anti-ROR1 antibody conjugates, compositions comprising anti ROR1 antibody conjugates, and methods of making and using anti-ROR1 antibody conjugates | |
| KR20220136267A (en) | Antibody-drug conjugate comprising antibody binding to antibody against human CLDN18.2 and its use | |
| KR20220158181A (en) | ROR1 and B7-H3 binding antibody-drug conjugate and use thereof | |
| US12419963B2 (en) | Diels-Alder conjugation methods | |
| WO2024189428A1 (en) | Antibody-drug conjugate comprising antibodies against human l1cam and uses thereof | |
| WO2025099489A1 (en) | Antibody-drug conjugate comprising bispecific antibody specifically binding to cd20 and cd22 and uses thereof | |
| JP2022500454A (en) | Combination therapy with antifolate receptor antibody conjugate | |
| WO2022232488A1 (en) | Combination therapies using an anti-bcma antibody drug conjugate (adc) in combination with a gamma secretase inhibitor (gsi) | |
| KR20210028556A (en) | Antibody-drug conjugate comprising antibody binding to antibody against human ROR1 and its use | |
| US12540197B2 (en) | Anti-tissue factor antibodies and antibody conjugates, compositions comprising anti-tissue factor antibodies or antibody conjugates, and methods of making and using anti-tissue factor antibodies and antibody conjugates | |
| US20250242043A1 (en) | Antibody-drug conjugate comprising antibody against human trop2 and use thereof | |
| RU2847195C1 (en) | Diels alder conjugation methods | |
| WO2025233682A1 (en) | Antibody drug conjugates comprising sting agonists and uses thereof | |
| WO2025250825A1 (en) | Anti-trop2 antibodies, compositions comprising anti-trop2 antibodies and methods of making and using anti-trop2 antibodies | |
| WO2025037120A1 (en) | A compound comprising a first and second antibody or antigen-binding fragment and methods of conjugating the same |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 24888152 Country of ref document: EP Kind code of ref document: A1 |