WO2025096899A1 - Immunoassays and methods for diagnosing hepatitis d virus (hdv) infection - Google Patents
Immunoassays and methods for diagnosing hepatitis d virus (hdv) infection Download PDFInfo
- Publication number
- WO2025096899A1 WO2025096899A1 PCT/US2024/054061 US2024054061W WO2025096899A1 WO 2025096899 A1 WO2025096899 A1 WO 2025096899A1 US 2024054061 W US2024054061 W US 2024054061W WO 2025096899 A1 WO2025096899 A1 WO 2025096899A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- antigenic polypeptide
- seq
- sequence
- hdv
- polypeptide
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/53—Immunoassay; Biospecific binding assay; Materials therefor
- G01N33/576—Immunoassay; Biospecific binding assay; Materials therefor for hepatitis
- G01N33/5767—Immunoassay; Biospecific binding assay; Materials therefor for hepatitis non-A, non-B hepatitis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/005—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from viruses
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/08—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from viruses
- C07K16/081—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from viruses from DNA viruses
- C07K16/082—Hepadnaviridae, e.g. hepatitis B virus
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2760/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA ssRNA viruses negative-sense
- C12N2760/00011—Details
- C12N2760/10011—Arenaviridae
- C12N2760/10111—Deltavirus, e.g. hepatitis delta virus
- C12N2760/10122—New viral proteins or individual genes, new structural or functional aspects of known viral proteins or genes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2760/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA ssRNA viruses negative-sense
- C12N2760/00011—Details
- C12N2760/10011—Arenaviridae
- C12N2760/10111—Deltavirus, e.g. hepatitis delta virus
- C12N2760/10134—Use of virus or viral component as vaccine, e.g. live-attenuated or inactivated virus, VLP, viral protein
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N2469/00—Immunoassays for the detection of microorganisms
- G01N2469/20—Detection of antibodies in sample from host which are directed against antigens from microorganisms
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N2470/00—Immunochemical assays or immunoassays characterised by the reaction format or reaction type
- G01N2470/04—Sandwich assay format
Definitions
- HDV/HBV Co-infection of HDV/HBV leads to increased mortality over HBV mono- infection.
- Antibody (total Ig) to HDV is the serological marker of HDV infection and is recommended for screening alhepatitis B surface antigen positive (HBsAg +)patients with liver disease.
- HBsAg + alhepatitis B surface antigen positive
- a serology assay for detecting HDV Total Ig is not widely available and current state of the art assays stil use the less sensitive and less specific indirect assay format. Sensitive and accurate diagnostics are needed to identify people who have been infected with HDV and who might benefit from increased monitoring and treatment. New medications which wil ofer more opportunities for treatment of chronic HDV infection are in development and provide weight to the need to identify cases.
- compositions, kits, systems and methods for detecting antibodies against hepatitis D virusin a sample and for detecting hepatitis D virus infection in a subject provides an antigenic polypeptide comprising a sequence having at least 90% sequence identity to the sequence of SEQ ID NO: 8, wherein said antigenic polypeptide lacks a sequence comprising at least 90% sequence identity to a region of from about 12 to about 60 amino acids at the N-terminus region of SEQ ID NO: 1.
- the antigenic polypeptide lacks a sequence comprising at least 90% sequence identity with a sequence selected from the groupconsisting ofSEQ ID NO: 9, SEQ ID NO: 10, SEQ ID NO: 11; SEQ ID NO: 12; and SEQ ID NO: 13.
- the antigenic polypeptide comprises at least one amino acid mutation as compared to the sequence of amino acids 1 to 60 of SEQ ID NO: 1.
- the antigenic polypeptide comprises a sequence selected from the group consisting of SEQ ID NO: 2, SEQ ID NO: 4, SEQ ID NO: 5, SEQ ID NO: 6, SEQ ID NO: 7 and SEQ ID NO: 8.
- the antigenic polypeptide comprises the sequence of SEQ ID NO: 6 and lacks the sequence of SEQ ID NO: 11.
- the antigenic polypeptide comprises an amino acid sequence having a formula of: MSRSESKKNRGGX1EEIX2EQX3X4SGX5KKLEELERDLRKVKKKIKKLEDENP WLGNIKGILGKKDKDGEGAPPAKRARTDQMEVDSGPRKRPLRGGFTDKERQDHRRRK ALENKKKQLSGGGKNLSKEEEEELKRLTEEDERRERRVAGPPVGGVNPLEGGSRGAPG GGFVPSMQGVPESPFTRTGEGLDIRGNQGFPWDILFPADPPFSPQSCRPQ(SEQ ID NO:22), where: X1 is R or S; X2 is L or G; X3 is W or A; X4 is V or G; and X5 is R or S, provided that X1 if S if X2is L, X3is W, X4is V and X5is R.
- theantigenic polypeptide is capable of being bound by an antibody directed to HD protein.
- the antigenic polypeptide is fused to at least one heterologous peptide.
- the heterologous peptide comprises an afinity tag, an epitope tag, a fluorescentprotein, an enzyme or a carier protein.
- the presently disclosed subject mater provides an antigenic polypeptide comprising a sequence having at least 90% sequence identity to a truncated polypeptide derived from SEQ ID NO: 1, said truncated polypeptide lacking from 12 to 60 N- terminal amino acids of SEQ ID NO: 1.
- the truncated polypeptide comprises a sequence of at least 158 amino acids of SEQ ID NO: 1.
- the presently disclosed subject mater provides anantigenic polypeptide comprising an amino acid sequence having at least 90% sequence identity to any of the ful-length sequences of SEQ ID NO: 2 and SEQ ID NOs: 4-8, and a heterologous peptide.
- the heterologous peptide comprises an affinity tag, an epitope tag, a fluorescentprotein, an enzyme or a carrier protein.
- the presently disclosed subject mater provides a composition comprising any of the above-describedantigenic polypeptides.
- the composition further comprises a detectable label.
- the detectable label is conjugated to the antigenic polypeptide.
- thedetectable label is selected from the group consisting of a fluorescence label, a chemiluminescent label, an enzymatic label, and a particle label.
- thecomposition further comprises a second antigenic polypeptide that comprises the sequence of SEQ ID NO: 1.
- thesecond antigenic polypeptide comprises a means for binding to a solid support.
- the presently disclosed subject mater provides a kit for detecting antibodies against hepatitis D virus (HDV) in a biological sample, comprising any of the antigenic polypeptides described above or any of the compositions described above.
- HDV hepatitis D virus
- the presently disclosed subject mater provides a kitfor detecting antibodies against hepatitis D virus (HDV) in a biological sample, comprising any of the antigenic polypeptides described above and a second antigenic polypeptide that comprises the sequence of SEQ ID NO: 1.
- the presently disclosed subject mater provides a method for detecting antibodies specific for HD protein in a biological sample, wherein any of theantigenic polypeptides described aboveis used as a binding partner for said HD protein antibodies.
- the presently disclosed subject mater provides a method for detecting antibodies specific for HD protein in a biological sample, said method comprising: a) forming an immunoreaction mixture by mixing a biological sample with anantigenic polypeptide described above; b) maintaining said immunoreaction mixture for a time period sufficient for alowing antibodies against said antigenic polypeptide present in the biological sample to immunoreact with said antigenic polypeptide to form an immunoreaction product; and c) detecting the presence and/or the concentration of said immunoreaction product.
- the immunoreaction is caried out in a double antigen sandwich format comprising:a) adding to said biological sample a first antigenic polypeptide that comprises a means for binding to a solid support and a second antigenic polypeptide that caries a detectable label, said second antigenic polypeptide comprising anantigenic polypeptide described above, wherein said first antigenic polypeptide and said second antigenic polypeptide bind specificaly to the HD protein antibodies,b) forming an immunoreaction mixture
- the method further comprises a washing step prior to the detecting step.
- theimmunoreaction product is bound to the solid support and the washing step comprises removing or separating some oral of the first antigenic polypeptide, HD protein antibody and second antigenic polypeptidethat are not part of the immunoreaction product that is bound to the solid support.
- the solid support is selected from the group consisting of acolumn, bead, test tube, microtiter dish,multi-wel plate,microparticle, microsphere, a test stick, a test strip, microchip and membrane.
- the biological sample is a body fluid sample from a human subject.
- the biological sample is selected from the group consisting of blood, plasma andserum.
- the presently disclosed subject mater provides a polynucleotide encoding an antigenic polypeptide described above.
- the polynucleotide further comprises a heterologous regulatory element operably linked to a nucleic acid encoding the antigenic polypeptide.
- the heterologous regulatory element is a promoter.
- the presently disclosed subject mater provides a vector comprising the polynucleotide.
- the vector is a plasmid, naked nucleic acid, phage, viral vector, or virus.
- the presently disclosed subject mater provides a cel comprising apolynucleotidedescribed above.
- the presently disclosed subject mater provides a cel comprising a vector described above.
- the presently disclosed subject mater provides a cel expressing anantigenic polypeptidedescribed above.
- the presently disclosed subject mater provides a method for detecting antibodies against hepatitis D virus (HDV) in a biological sample, comprising performing a HDV serology assay using a direct assay format with a first antigenic polypeptide and a second antigenic polypeptide, wherein the first antigenic polypeptide comprises a ful- length HD protein and the second antigenic polypeptide comprises an N-terminaly truncated fragment of the ful-length HD protein.
- the assay is a double antigen sandwich assay.
- the second antigenic polypeptide lacks from 12 to 60 N- terminal amino acids of the HD protein.
- the second antigenic polypeptide lacks 21, 37 or 46 consecutive N-terminal amino acids of the HD protein.
- the ful-length HD protein comprises an amino acid sequence having at least 95% sequence identity to the ful-length sequence of SEQ ID NO: 1.
- the presently disclosed subject mater provides a method for detecting antibodies against hepatitis D virus (HDV) in a biological sample, comprising:a) contacting abiological sample, either simultaneously or sequentialy, in any order with:(1) a first antigenic polypeptide under conditions adequate to alow binding of anyHDV antibody present in the biological sample to the first antigenic polypeptide, wherein said first antigenic polypeptide is immobilized on a solid support, and (2) a second antigenic polypeptide under conditions adequate to alow binding of the second antigenic polypeptide to any HDV antibody present in the biological sample,such that a first antigenic polypeptide-HDV antibody-second antigenic polypeptide complex is formed and is bound to the solid support, and b) detecting the presence of said first antigenic polypeptide-HDV antibody-second antigenic polypeptide complex,wherein said first antigenic polypeptide comprises a sequence having at least 90% sequence identity to the ful-length sequence of SEQ ID NO: 1;wherein said second antigenic poly
- the method further comprises a washing step prior to the detecting step.
- the washing step comprises removingorseparating some or al of the first antigenic polypeptide, HDV antibody and second antigenic polypeptide that are not part of the first antigenic polypeptide-HDV antibody-second antigenic polypeptide complex that is bound to the solid support.
- the step of detecting comprises detecting the presence and/or the concentration of the first antigenic polypeptide-HDV antibody-second antigenic polypeptide complex that is formed and is bound to the solid support.
- the step of detecting comprises measuring an amount of saidfirst antigenic polypeptide-HDV antibody-second antigenic polypeptide complex that is formed and is bound to the solid support.
- the solid support is selected from the group consisting of acolumn, bead, test tube,microtiter dish,multi-wel plate, microparticle, microsphere, a test stick, a test strip, microchip and membrane.
- the biological sample is selected from the group consisting of blood, plasma and serum.
- the presently disclosed subject mater provides a method of detecting hepatitis D virus (HDV) infection in a subject comprising performing amethod described above, wherein the biological sample is a sample taken from the subject and wherein the step of detecting comprises detecting the presence of said first antigenic polypeptide-HDV antibody-second antigenic polypeptide complex that is formed and is bound to the solid support, thereby detecting the presence of past or present HDV infection in said subject.
- HDV hepatitis D virus
- the presently disclosed subject mater provides method of estimating HDV incidence in a population, the method comprising, a) providing a set of samples derived from a plurality of individuals within the population over a period of time;b) performing the method on each sample in the set of samples; andc) determining the percentage of recent HDV infections over the period of time;wherein the percentage of recent HDV infections over the period of time provides an estimate of HDV incidence in the population.
- the presently disclosed subject mater provides a system comprising:a) a sample receiving component configured to receive a sample from a subject, wherein the sample comprises or is suspected of comprising a hepatitis D virus (HDV) antibody; b) a first antigenic polypeptide and a second antigenic polypeptide configured to make contact with the sample to form a first antigenic polypeptide-HDV antibody-second antigenic polypeptide complex, wherein the first antigenic polypeptide comprises a ful-length HD protein and the second antigenic polypeptide comprises amutated and/or truncated HD protein, and wherein the second antigenic polypeptide caries a detectable label;c) a detection component configured to measure a signal generated by the detectable label in the first antigenic polypeptide-HDV antibody-second antigenic polypeptide complex; andd) an output component that indicates an amount of HDV antibody in the sample based on the signal.
- a sample receiving component configured to receive a sample from a subject, wherein the sample comprises or
- the second antigenic polypeptide comprises an N-terminaly truncated fragment of the ful-length HD protein.
- the ful-length HD protein comprises an amino acid sequence having at least 95% sequence identity the ful-length sequence of SEQ ID NO: 1.
- the second antigenic polypeptide lacks from 12 to 60 N-terminal amino acids of the ful-length HD protein. In some embodiments, the second antigenic polypeptide lacks 21, 37 or 46 consecutive N-terminal amino acids of the ful-length HD protein.
- the second antigenic polypeptide is selected from the group consisting of: i) a polypeptide that comprises the sequence of SEQ ID NO: 8 and lacks the sequence of SEQ ID NO: 13;i) a polypeptide that comprises the sequence of SEQ ID NO: 7and lacks the sequence of SEQ ID NO: 12;ii) a polypeptide that comprises the sequence of SEQ ID NO: 6 and lacks the sequence of SEQ ID NO: 11;iv) a polypeptide that comprises the sequence of SEQ ID NO: 5 and lacks the sequence of SEQ ID NO: 10;v) a polypeptide that comprises the sequence of SEQ ID NO: 4 and lacks the sequence of SEQ ID NO: 9; andvi) a polypeptide that comprises the sequence of SEQ ID NO: 2.
- the sample is selected from the group consisting of blood, plasma andserum.
- the detectable label is selected from the group consisting of a fluorescent label, a chemiluminescent marker, and
- FIG.3 An evaluation of the corelation between signal of HDV Antibody assays and HDV RNA viral load on 70HDV infected patient samples.
- the circle highlightsthe 8samples with high signal (193-1150 S/CO) and the rectangle showsthe 6samples with low signal (2.1-19.3 S/CO) in Architect HDV Ig within the same low viral load range (0.53-1.9 log10 IU/ml). Orange square indicatesthe 6 missed samples by both assays.
- FIG.4 Specificity and signal distribution of HDV antibody assays with 200 US normal blood donors.
- An antibody is a protein (or protein complex) that includes one or more polypeptides substantialy encoded by immunoglobulin genes or fragments of immunoglobulin genes.
- the recognized immunoglobulin genes include the kappa, lambda, alpha, gamma, delta, epsilon, and mu constant region genes, as wel as the myriad of immunoglobulinvariable region genes.
- Lightchains are classified as either kappa or lambda.
- Heavy chains are classified as gamma, mu,alpha, delta, or epsilon, which in turn define the immunoglobulinclasses, IgG, IgM, IgA, IgD and IgE, respectively.
- Antibodies are evoked in humans or other animals by a specificantigen (immunogen). Antibodies are characterized byreacting specificalywith the antigen in some demonstrable way,antibody and antigen each being defined in terms of theother. “Eliciting an antibody response”refers to the abilityof an antigen or other molecule to induce the production ofantibodies.
- the term“antigen” refers to a molecule, moiety, foreign particulate mater, or an Organenthat can bind to a specific antibody or T-cel receptor.An antigencan stimulate the production of antibodies or a T-cel response inan animal.
- the term “antigenic polypeptide” refers to a polypeptide that binds specificaly to antibodies that recognize the polypeptide.
- an “antigen-specific”antibody is an antibody that was elicited (produced and/oractivated) in response to a particular antigen.
- An “antigen-specific”antibody is capable ofbinding to the antigen,typicaly with high afinity.
- the “area under curve”or “AUC” refer to area under a ROC curve.
- AUC under a ROC curve is a measure of accuracy.
- An AUC of 1 represents a perfect test, whereas an AUC of 0.5 represents an insignificant test.
- a prefered AUC may be at least approximately 0.700, at least approximately 0.750, at least approximately 0.800, at least approximately 0.850, at least approximately 0.900, at least approximately 0.910, at least approximately 0.920 ⁇ at least approximately 0.930, at least approximately 0.940, at least approximately 0.950, at least approximately 0.960, at least approximately 0.970, at least approximately 0.980, at least approximately 0.990, or at least approximately 0.995.
- Bead”and “particle” are used herein interchangeably and refer to a substantialy spherical solid support.
- a bead or particle is a microparticle.
- Microparticles that can be used herein can be any type known in the art.
- the bead or particle can be a magnetic bead or magnetic particle.
- Magnetic beads/particles may be feromagnetic, ferimagnetic, paramagnetic, superparamagnetic or ferrofluidic.
- Exemplary feromagnetic materials include Fe, Co, Ni, Gd, Dy, CrO 2 , MnAs, MnBi, EuO, and NiO/Fe.
- Examples of ferimagnetic materials include NiFe 2 O 4 , CoFe 2 O 4 , Fe3O 4 (or FeO ⁇ Fe 2 O3).
- Beads can have a solid core portion that is magnetic and besurrounded by one or more non-magnetic layers. Alternately, the magnetic portion can be a layer around a non-magnetic core.
- the microparticles can be of any size that would work in the methods described herein, e.g.,from about 0.75 to about 5 nm, or from about 1 to about 5 nm, or from about 1 to about 3 nm.
- a “carier protein” is a protein that functions to facilitate expression from a host cel. A carier protein may facilitate soluble expression or promote the formation of inclusion bodies in the host cel.
- Non-limiting examples of carrier proteins include thioredoxin and GST(glutathione transferase).
- the carier/peptide junction contains an enzymatic or chemical cleavage site that enables the peptide to be released by the coresponding method.
- conjugated refersto two molecules that are bondedtogether, for example by covalent bonds.
- Control refers to a reagent whose purpose is to evaluate the performance of a measurementmethod orsystem in order toassure that it continues to produce results within permissible boundaries (e.g., boundaries ranging from measures appropriate for a research use assay on one end to analytic boundaries established by quality specifications for a commercial assay on the other end).
- a control should be indicative of patient results and optionaly should somehow assess theimpact of eror on the measurement (e.g., eror due to reagent stability, calibrator variability, instrument variability, and the like).
- detectable label refers to a moiety or compound thatcan be used to provide a detectable and/or quantifiable signal.
- the label can be atached, directly or indirectly, to a nucleic acid or protein.
- the detectablelabel generates a signal whichcan be measured and whose intensity is related to (e.g., proportional to) the amount of entitybound thereto.
- Suitable labels that can be atached to a nucleic acid or protein include, but are not limitedto, radioisotopes, fluorophores, chromophores, mass labels, electron dense particles, magnetic particles, spin labels, molecules that emit chemiluminescence, electrochemicaly active molecules, enzymes, cofactors, and enzyme substrates. Additional examples of suitable detectable labels are provided below.
- the term “derived from,”as used herein with reference to proteins and polypeptides (and sequences thereof) refers to various modifications, analogs, and products based upon an original protein or polypeptide (and sequence thereof) from which a derivative protein or polypeptide is derived, for example, based on the protein and polypeptide sequences disclosed herein.
- a polypeptide that is derived from an original protein or polypeptide (and sequence thereof) may be a truncated or augmented sequence, a sequence comprising at least one mutation relative to the original sequence, or some other modified sequence.
- the term “heterologous”in reference to an element refers to an element that is not in its natural environment.
- a heterologous element includes an element from one species introduced into another species.
- a heterologous element also includes an element native to an organism that has been altered in some way (e.g., mutated, added in multiple copies, linked to non-native regulatory sequences, etc.).
- Heterologous elements are distinguished from endogenous elements in that the heterologous element sequences are typicaly joined to sequences that are not found naturaly associated with the element sequences in the chromosome or are associated withportions of the chromosome not found in nature (e.g., genes expressed in loci where the gene is not normaly expressed).
- the term “host cel” means any cel that harbors or is susceptible to harboring foreign molecules, viruses, or microorganisms. It may also be a cel that has been introduced with or is susceptible to being introduced with a foreign (i.e., heterologous) nucleic acid molecule.
- a host cel is any cel type that is susceptible to transformation, transfection, transduction, or the like with a nucleic acid construct or expression vector of the present disclosure.
- An antibody that immunoreacts with a polypeptide e.g., antigenic polypeptide
- mutation refers to a change and/or alteration.
- mutations may be changes and/or alterations to proteins (including peptides and polypeptides) and/or nucleic acids (including polynucleic acids).
- mutations comprise changes and/or alterations to a protein and/or nucleic acid sequence.
- Such changes and/or alterations may comprise the addition, substitution and/or deletion of one or more amino acids (in the case of proteins and/or peptides) and/or nucleotides (in the case of nucleic acids and/or polynucleic acids).
- mutations comprise the addition and/or substitution of amino acids and/or nucleotides
- such additions and/or substitutions may comprise 1 or more amino acid and/or nucleotide residues and may include modified amino acids and/or nucleotides.
- the resulting construct, molecule or sequence of a mutation, change or alteration may be refered to herein as amutant.
- the “N-terminus region” refer to the region at or near the end of a polypeptide chain or protein where the first amino acid resides. Proteins are composed of linear chains of amino acids linked together by peptide bonds, forming a primary structure. The sequence of amino acids in this chain is read from the N-terminus to the C-terminus.
- the N-terminus (also known as the amino-terminus, NH2-terminus, N-terminal end or amine-terminus) is the start of a protein or polypeptide, refering to the free amine group (-NH2) located at the end of a polypeptide.
- the amine group is bonded to the carboxylic group of another amino acid, making it a chain. That leaves a free carboxylic group at one end of the peptide, caled the C-terminus, and a free amine group on the other end caled the N-terminus.
- the length of the N- terminus region is from at least one amino acid in length up to the length equal to 50% of the ful-length of the protein or polypeptide chain itself.
- polypeptide is a polymer in which themonomers are aminoacid residues which are joined together through amidebonds. When the amino acids are alpha-amino acids, eitherthe L-optical isomer or the D-optical isomer can be used.As used herein, the term “polypeptide”is used interchangeably with the terms“peptide”and “protein.” The terms “polypeptide”or “protein”as used herein areintended to encompass any amino acid sequence and include modified sequences such as glycoproteins. The term “polypeptide”is specificaly intended to cover naturaly occuring proteins, as wel as those which are recombinantly or syntheticaly produced.
- ROC discrimination threshold
- TPR is also known as sensitivity
- FPR is one minus the specificity or true negative rate.
- the ROC curve demonstrates the tradeof between sensitivity and specificity (any increase in sensitivity wil be accompanied by a decrease in specificity); the closer the curve folows the left-hand border and then the top border of the ROC space, the more accurate the test; the closer the curve comes to the 45-degree diagonal of the ROC space, the less accurate the test; the slope of the tangent line at a cutoff point gives the likelihood ratio (LR) for that value of the test; and the area under the curve is a measure of test accuracy.
- a “solid support” is any inert material having a rigid or semirigidsurface.
- thesolid support is capable of binding directly or indirectly to a polypeptide (e.g., an antigenic polypeptide that is a capture moiety).
- the solid support can have any shape, form or size (forexample, plate, sheet, tube, stick or particle).
- the solid support is a multi-wel plate(also refered to as a microtiter or microwel plate), membrane,glass, metal, bead, microsphere, test tube, test stick,test strip, porous matrix or resin.
- the solid support is amicroparticle.In some examples, the solid support includes polystyrene,polyethylene or polypropylene.
- nanoparticle refers to any particle having a diameter of less than 1000 nm.
- nanoparticles of the disclosure have a greatest dimension (e.g., diameter) of 500 nm or less.
- nanoparticles of the disclosure have a greatest dimension ranging between 25 nm and 200 nm.
- nanoparticles of the disclosure have a greatest dimension of 100 nm or less.
- nanoparticles of the disclosure have a greatest dimension ranging between 35 nm and 60 nm.
- sequence identity refers to the degree two polymer sequences (e.g., peptide, polypeptide, nucleic acid, etc.) have the same sequential compositionof monomer subunits.
- sequence identity can be determined with the aid of readily available sequence comparison programs. These available computer programs may calculate percent (%) sequence identity between two or more sequences and may also calculate the sequence identity shared by two or more amino acid or nucleic acid sequences.
- the “percent sequence identity” is calculated by: (1) comparing two optimaly aligned sequences over a window of comparison (e.g., the length of the longer sequence, the length of the shorter sequence, a specified window), (2) determining the number of positions containing identical (or similar) monomers (e.g., same amino acids occurs in both sequences) to yield the number of matched positions, (3) dividing the number of matched positions by the total number of positions in the comparison window (e.g., the length of the longer sequence, the length of the shorter sequence, a specified window), and (4) multiplying the result by 100 to yield the percent sequence identity.
- a window of comparison e.g., the length of the longer sequence, the length of the shorter sequence, a specified window
- peptides A and B are both 20 amino acids in length and have identical amino acids at al but 1 position, then peptide A and peptide B have 95% sequence identity.
- peptide C is 20 amino acids in length and peptide D is 15 amino acids in length, and 14 out of 15 amino acids in peptideD are identical to those of a portion of peptide C, then peptides C andD have 70% sequence identity, but peptideD has 93.3% sequence identity to an optimal comparison window of peptide C.
- any gaps in aligned sequences are treated as mismatches at that position.
- a window of comparison is not specified and a specific sequence identifier is indicated (i.e., an assigned SEQ ID NO is indicated)
- the sequence identity is calculated with respect to the ful-length sequence coresponding to that sequence identifier.
- the phrases“a sequence has at least 90% sequence identity to SEQ ID NO: 1”and “a sequence has at least 90% sequence identity to the sequence of SEQ ID NO: 1” arethe same as the phrase “a sequence has at least 90% sequence identity tothe ful-length sequence ofSEQ ID NO: 1.”
- the term “sample” is used in the broadest sense and generaly refers to a biological material being tested for and/or suspected of containing an analyte of interest, such as a target antibody described herein.
- Thesample may be derived from any biological source, such as, a physiological fluid, including, but not limited to, whole blood, serum, plasma, interstitial fluid, saliva, ocular lens fluid, cerebral spinal fluid, sweat, urine, milk, ascites fluid, mucous, nasal fluid, sputum, synovial fluid, peritoneal fluid, vaginal fluid, menses, amniotic fluid, semen and so forth.
- a physiological fluid including, but not limited to, whole blood, serum, plasma, interstitial fluid, saliva, ocular lens fluid, cerebral spinal fluid, sweat, urine, milk, ascites fluid, mucous, nasal fluid, sputum, synovial fluid, peritoneal fluid, vaginal fluid, menses, amniotic fluid, semen and so forth.
- the sample is a whole blood sample.
- the sample is a plasma sample.
- the sample is a serum sample.
- the test sample may be used directly as obtained from the biological source or folowing a pretreatment to modify the
- such pretreatment may include preparing plasma from blood, diluting viscous fluids and so forth. Methods of pretreatment may also involve filtration, precipitation, dilution, distilation, mixing, concentration, inactivation of interfering components, the addition of reagents, lysing, etc. Moreover, it may also be beneficial to modify a solid test sample to form a liquid medium or to release the analyte.
- Reference level refers to an assayorcutof value that is used to assess diagnostic(“diagnostic”cutof), prognostic, or therapeutic eficacy and that has been linked or is associated herein with various clinical parameters (e.g., presence of disease such as, for example, to rule a subject as having a disease (“rule in”) or rule a subject as not having a disease (“rule out”), stage of disease, severity of disease, progression, non-progression, or improvement of disease, etc.)
- reference levels may vary depending on the nature of the immunoassay (e.g.,such as, in an immunoassay, the antigens or antibodies employed, reaction conditions, sample purity, etc.) and that assays can be compared and standardized.
- a “reagent” refers broadly to any agent used in a reaction, other than the analyte (e.g., polypeptideor antibodybeing analyzed).
- Ilustrative reagents for enzyme reactions include, for example, substrates, cofactors, bufer, metal ions, inhibitors, and activators.
- the numbers 7 and 8 are contemplated in addition to 6 and 9, and for the range 6.0-7.0, the number 6.0, 6.1, 6.2, 6.3, 6.4, 6.5, 6.6, 6.7, 6.8, 6.9, and 7.0 are explicitly contemplated.
- “Sensitivity” refers to the proportion of subjects for whom the outcome is positive that are correctly identified as positive (e.g., correctly identifying those subjects with a disease or medical condition for which they are being tested). For example, this might include correctly identifying subjects as having an HDVinfection from those who do not have an infection.
- “Specificity”of an assay as used herein refers to the proportion of subjects for whom the outcome is negative that are correctly identified as negative (e.g., correctly identifying those subjects who do not have a disease or medical condition for which they are being tested). For example, this might include correctly identifying subjects not having an HDVinfection from those who do have an HDV infection.
- “Subject”and “patient”as used herein interchangeably refers to any vertebrate, including, but not limited to, a mammal and a human. In some embodiments, the subject may be a human or a non-human. The subject or patient may be undergoing forms of treatment.
- “Mammal”as used herein refers to any member of the class Mammalia, including, without limitation, humans and nonhuman primates such as chimpanzees and other apes and monkey species; farm animals such as catle, sheep, pigs, goats, lamas, camels,and horses; domestic mammals such as dogs and cats; laboratory animals including rodents such as mice, rats, rabbits, guinea pigs, and the like. The term does not denote a particular age or sex. Thus, adult and newborn subjects, as wel as fetuses, whether male or female, are intended to be included within the scope of this term.
- Treat,”“treating”or “treatment” are each used interchangeably herein to describe reversing, aleviating, or inhibiting the progress of a disease and/or injury, or one or more symptoms of such disease, to which such term applies.
- the term also refers to preventing a disease, and includes preventing the onset of a disease, or preventing the symptoms associated with a disease.
- a treatment may be either performed in an acute or chronic way.
- the term also refers to reducing the severity of a disease or symptoms associated with such disease prior to afliction with the disease.
- Such prevention or reduction of the severity of a disease prior to affliction refers to administration of a pharmaceutical composition to a subject that is not at the time of administration aflicted with the disease.
- Preventing also refers to preventing the recurrence of a disease or of one or more symptoms associated with such disease.
- a “truncated polypeptide” refers to a shortened molecule or sequence that has been shortened by removing a portion of it.
- scientific and technicalterms used in connection with the present disclosure shal havethe meanings that are commonly understood by those of ordinaryskil in the art.
- any nomenclatures used in connection with, andtechniques of, cel and tissue culture, molecular biology,immunology, microbiology, genetics and protein and nucleicacid chemistry and hybridization described herein are thosethat are wel known and commonly used in the art.
- Hepatitis Delta Virus The hepatitis delta virusesare negative-sense single-stranded RNA viruses (or virus- like particles) classified together as the genus Deltavirus. Hepatitisdelta virus(HDV) is one of five known hepatitis viruses: A, B, C, D, and E.HDV is considered a defective virus because it is unable to replicate on its own.
- HDV requires infection of the same cel with Hepatitis B virus (HBV), which contributes to virion assembly and infectivity. HDV only occurs in people who are also infected with the hepatitis B virus. Transmission of HDV can occur either via simultaneous infection with HBV (coinfection) or superimposed on chronic hepatitis B or hepatitis B carier state (superinfection).
- the HDV viral envelope contains host phospholipids, as wel as three proteins taken from the hepatitis B virus (the large, medium, and smal hepatitis B surface antigens).
- the viral envelope surounds an inner ribonucleoprotein (RNP) particle, which contains the genome surounded by about 200 molecules of hepatitis D antigen (HDAg) for each genome.
- Hepatitis D antigen (abbreviated herein as “HDAg”or “HD Ag”) is the only protein that is produced by the HDV virus.Throughout the present disclosure, Hepatitis D antigen may also be refered to as “HD protein”.
- Hepatitis D antigen comes in two forms: an approximately 27kDa HD large antigen (abbreviated herein as “large-HDAg”or “HD LAg”)and an approximately24kDa smal-HDAg.
- a consensus amino acid sequence of several HD large antigen sequences is used as a representative HD protein sequence and has the folowing sequence: MSRSESKKNRGGREEILEQWVSGRKKLEELERDLRKVKKKIKKLEDENPWLGNIKGILG KKDKDGEGAPPAKRARTDQMEVDSGPRKRPLRGGFTDKERQDHRRRKALENKKKQLS GGGKNLSKEEEEELKRLTEEDERRERRVAGPPVGGVNPLEGGSRGAPGGGFVPSMQGV PESPFTRTGEGLDIRGNQGFPWDILFPADPPFSPQSCRPQ (SEQ ID NO: 1).
- Hepatitis D can be an acute, short-term infection or become a long-term, chronic infection. Hepatitis D can cause severe symptoms and serious ilness that can lead to life-long liver damage and even death. People can become infected with both hepatitis B and hepatitis D viruses at the same time (known as “coinfection”) or get hepatitis D after first being infected with the hepatitis B virus (known as “superinfection”).Superinfections can occur when someone who already has chronic hepatitis B becomes infected with hepatitis D—thesetypes of infections are more common, and have a 70-90% chance of resulting in a chronic infection of both hepatitis B and D.
- Antibodies against the HDAg are the serological marker of HDV infection.Antibody testing is widely used as a primary screening test for HDV infection and recommended for universal screening of al HbsAg-positive individuals. Diagnosis of HDV can be established by detecting HDV antigen, HDV-specific IgM, or HDV-specific total antibodies (combined IgM and IgG) in the sera of infected patients with clinicaly evident acute or chronic hepatitis B. Anti-HDV IgM typicaly appears inserum at 2 to 3 weeks after onset of symptoms and disappears by 2 months after acute HDV infection, but it may persist up to 9 months in HDV superinfection.
- HDV IgG and HDV total antibodies persist in serum after resolution of acute HDV infection and in chronic coinfection.
- European and Asian-Pacific guidelines recommend universal screening for HDV in al HbsAg-positive patients.
- a recent study on the implementation of the universal screening in HbsAg-positive individuals found a 5-fold increase in diagnosis of HDV infection.
- While current guideline in the United States stil recommend risk-based screening for HDV, it has been suggested to expand the HDV screening structures to universal screening in al HbsAg-positive individuals based on updated prevalence data revealing a higher-than-expected prevalence. Screening for HDV requires testing for anti-HDV antibodies, which indicate past exposure to the virus or current infection.
- the first step is performing the HDV antibody total (anti-HDV total) test. People who have recovered from or are currently infected with hepatitis delta wil have antibodies.If the HDV antibody total test is positive, it should be folowed by the HDV RNA (Qualitative or Quantitative) test to confirm an active infection. Testing for HDV is indicated in those who are hepatitis B surface antigen positive (i.e., those who have had previous or active infection with hepatitis B). Ifliver fibrosis or cirhosis is suspected, a liver biopsy is usualy needed.
- Curent treatments for chronic hepatitis D include conventional or pegylated interferon alpha therapy.
- Evidence suggests that pegylated interferon alpha is effective in reducing the viral load and the effect of the disease during the time the drug is given, but the benefit generaly stops if the drug is discontinued. The eficiency of this treatment does not usualy exceed about 20%, and late relapse after therapy has been reported.
- New treatment options for HDV are on the horizon, including viral entry inhibitors, prenylation inhibitors and virion egress inhibitors.
- Emerging antiviral therapies include Hepcludex (bulevirtide) to treat hepatitis D.
- Bulevirtide binds and inactivates the sodium/bile acid cotransporter, blocking hepatitis D virus (as wel as hepatitis B virus) from entering hepatocytes.
- Bulevirtide may begiven along with pegylated interferon alpha.Other treatments for hepatitis D which are currently under development include pegylated interferon lambda ( ⁇ ), which binds to receptors on the hepatocyte surface leading to an intracellular signaling cascade via the JAK-STAT signaling pathway and activation of anti-viral cel mediated immunity.
- the prenylation inhibitor lonafarnib prevents hepatitis D viral particle assembly by inhibiting the farnesylation of the L-HDAg.
- REP2139-Ca is a nucleic acid polymer that prevents the release of hepatitis B surface antigen (which is required for assembly of hepatitis D viral particles). 3.Antigenic polypeptides In various embodiments, the present disclosure provides antigenic polypeptides.
- the present disclosure provides an antigenic polypeptide comprising a sequencederived from asequence of the Hepatitis D antigen(HD protein)and may be refered to as a “recombinant HD protein”or “engineered HD protein.”
- an antigenic polypeptide that is a recombinant HD protein comprises a sequence derived from the sequence of the HD large antigenand may be refered to as an “engineered HD large antigen”(engineered HD LAg).
- the sequence of the HD large antigen is derived from a consensus amino acid sequence of several HD large antigen sequences.
- the sequence of the HD large antigen of the present disclosure is SEQ ID NO: 1.
- the present disclosure provides antigenic polypeptidesthat are recombinant HD proteinshaving sequences derived from a single HD large antigen sequence or a consensus amino acid sequence of several HD large antigen sequences.
- the present disclosure provides antigenic polypeptides (recombinant HD proteins) having sequences derived from SEQ ID NO: 1.
- an antigenic polypeptide of the present disclosure comprises a sequence that is an N-terminal truncation ofan HD large antigen sequence, such asSEQ ID NO: 1. In some embodiments from 1 to 60 N-terminal amino acids are truncated from the HD large antigen sequence (e.g., SEQ ID NO:1).
- from 1 to 56 N-terminal amino acids are truncated from the HD large antigen sequence (e.g., SEQ ID NO:1).
- from about 10 to about 60 N-terminal amino acids are truncated fromthe HD large antigen sequence (e.g.,SEQ ID NO: 1)to generate an antigenic polypeptide of the present disclosure.
- about 10, about 20, about 30, about 40, about 50 or about 60 amino acids are truncated from the N-terminus of the HD large antigen sequence (e.g., SEQ ID NO: 1)to generate an antigenic polypeptide of the present disclosure.
- 21 amino acids are truncated from the N-terminus of SEQ ID NO: 1 to generate an antigenic polypeptide.
- 24 amino acids are truncated from the N-terminus of SEQ ID NO: 1 to generate an antigenic polypeptide.
- 37 amino acids are truncated from the N-terminus of SEQ ID NO: 1 to generate an antigenic polypeptide.
- 46 amino acids are truncated from the N-terminus of SEQ ID NO: 1 to generate an antigenic polypeptide.
- an antigenic polypeptide of the present disclosure comprises a sequence that has between 1 to 10 amino acid mutations at the N-terminusregionas compared to the sequence of an HD large antigen (e.g., SEQ ID NO: 1). In some embodiments, an antigenic polypeptide of the present disclosure comprises 5 amino acid mutations at the N-terminusregionas compared to the sequence of an HD large antigen (e.g., SEQ ID NO: 1).
- an antigenic polypeptide of the present disclosure is designed based onthe sequence of the HD large antigen, such asSEQ ID NO: 1,by optimizing the length of an N-terminal truncation of the HD large antigen (e.g., SEQ ID NO: 1).
- optimization includes lowering assay background.
- an antigenic polypeptide can be optimized by testing background using a negative control, such as a sample that lacksatarget antibodythat is specific for the antigenic polypeptide.
- optimization includes optimizing both sensitivity and specificityof each antigenic polypeptide.
- optimization includes optimizing both sensitivity and specificity of combinationsoftwo antigenic polypeptides, one functioning as a capture moiety and a second one (which can be different from the first) functioning as a detection moiety.
- the present disclosure provides an antigenic polypeptide comprising a sequence having at least 90% sequence identity to the sequence of SEQ ID NO: 8, wherein said antigenic polypeptide lacks a sequence comprising at least 90% sequence identity to a region of from about 12 to about 60 amino acids at the N-terminus region of SEQ ID NO: 1.
- the antigenic polypeptide comprises a sequence having at least 95%, 96%, 97%, 98% or 99% sequence identity to the sequence of SEQ ID NO: 8.
- the antigenic polypeptide lacks a sequence comprising at least 95%, 96%, 97%, 98% or 99% sequence identity to a region of from about 12 to about 60 amino acids at the N- terminus region of SEQ ID NO: 1. In some embodiments, the antigenic polypeptide lacks from 12 to 60 N-terminal amino acids of SEQ ID NO: 1. In some embodiments, the antigenic polypeptide lacks a sequence comprising at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98% or 99% sequence identity with a sequence selected from the groupconsisting ofSEQ ID NO: 9, SEQ ID NO: 10, SEQ ID NO: 11; SEQ ID NO: 12; and SEQ ID NO: 13.
- the antigenic polypeptide lacks a sequence comprising at least 90% sequence identity with a sequence selected from the groupconsisting ofSEQ ID NO: 9, SEQ ID NO: 10, SEQ ID NO: 11; SEQ ID NO: 12; and SEQ ID NO: 13.
- the antigenic polypeptide lacks a sequence selected from the groupconsisting ofSEQ ID NO: 9, SEQ ID NO: 10, SEQ ID NO: 11; SEQ ID NO: 12; and SEQ ID NO: 13.
- the antigenic polypeptide comprises, consists essentialy of, or consists of a sequence selected from the group consisting of SEQ ID NO: 2, SEQ ID NO: 4, SEQ ID NO: 5, SEQ ID NO: 6, SEQ ID NO: 7 and SEQ ID NO: 8.
- the antigenic polypeptide comprises the sequence of SEQ ID NO: 6 and lacks the sequence of SEQ ID NO: 11.
- the present disclosure provides an antigenic polypeptide comprising a sequence having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% or 100% sequence identity to a truncated polypeptide derived from SEQ ID NO: 1, said truncated polypeptide lacking from 12 to 60 N-terminal amino acids of SEQ ID NO: 1.
- the antigenic polypeptide comprises a sequence having at least 90% sequence identity to a truncated polypeptide derived from SEQ ID NO: 1.
- the truncated polypeptide comprises a sequence of at least 158 amino acids of SEQ ID NO: 1.
- the truncated polypeptide comprises a sequence of at least 158 consecutive amino acids of SEQ ID NO: 1, such as the amino acids at positions 57 to 214 of SEQ ID NO: 1.
- the present disclosure provides an antigenic polypeptide comprising an amino acid sequence having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% or 100% sequence identity to any of the ful-length sequences of SEQ ID NO: 2 and SEQ ID NOs: 4-8, and a heterologous peptide.
- the antigenic polypeptide comprises a sequence having at least 90% sequenceidentity to any of the ful-length sequences of SEQ ID NO: 2 and SEQ ID NOs: 4-8, and a heterologous peptide.
- the heterologous peptide comprises an afinity tag, an epitope tag, a fluorescentprotein, an enzyme or a carier protein.
- the afinity tag is a His-tag.
- an antigenic polypeptide of the present disclosure is capable of being bound by an antibody directed to HD protein.As used herein, an antibody is “directed to” a particular entity (e.g., HD protein) when the antibody is capable of binding to that entity, typicaly with high afinity. Such an antibody may also be refered to herein as an antigen- specific antibody.
- the antibody isan antibody present in a subject after infection with HDV. In some embodiments, the antibody isan antibodyelicited by infection with HDV.
- the antibody is an antibody against the large-HDAg. In some embodiments, the antibody is an antibody againsta large-HDAgcomprising a sequence having at least 90% sequence identity to the ful-length sequence of SEQ ID NO: 1. In some embodiments, the antibody is an antibody against the smal-HDAg.In some embodiments, the antibody is an antibody againsta smal-HDAgcomprising a sequence having at least 90% sequence identity to the ful-length sequence of SEQ ID NO: 3. In some embodiments, an antigenic polypeptide of the present disclosure is fused to at least one heterologous peptide.
- the heterologous peptide comprises an afinity tag, an epitope tag, a fluorescentprotein, an enzyme or a carier protein.
- the present disclosure provides a composition comprising any of the antigenic polypeptides described above.
- the composition comprises a detectable label.
- the detectable label is conjugated to the antigenic polypeptide.
- the detectable label is selected from the group consisting of a fluorescence label, a chemiluminescent label, an enzymatic label, and a particle label.
- the detectable label is a chemiluminescent label, such as, acridinium (e.g., acridium esters, acridinium SPSP (N10-(3-sulfopropyl)-N-(3-sulfopropyl, etc.), luminol, isoluminol, thioesters, sulfonamides, phenanthridinium esters, etc.
- acridinium e.g., acridium esters, acridinium SPSP (N10-(3-sulfopropyl)-N-(3-sulfopropyl, etc.
- luminol isoluminol, thioesters, sulfonamides, phenanthridinium esters, etc.
- the detectable label is an enzymatic label, such as horseradish peroxidase, alkaline phosphatase, glucose 6-phosphate dehydrogenase, etc.
- the detectable label is a gold nanoparticle.
- the detectable label is a latex bead or a latex nanoparticle.
- the detectable label is a magnetic nanoparticle.
- a composition of the present disclosure further comprises a second antigenic polypeptide that comprises a sequencehaving at least 70%, 80%, 90%, 95%, 96%, 97%, 98%, or 99%sequence identity to the ful-length sequence of SEQ ID NO: 1.
- the second antigenic polypeptide comprises the sequence of SEQ ID NO: 1In some embodiments, the second antigenic polypeptide comprises a means for binding to a solid support.
- an antigenic polypeptide (recombinant HD protein) of the present disclosure is expressed from a plasmid construct. In some embodiments, the antigenic polypeptide is expressed from a plasmid construct in a host cel.
- a non-limiting example of a host cel is a bacterial cel, such as an E. coli cel.
- an antigenic polypeptide (recombinant HD protein) of the present disclosure comprisesa moiety that enables isolation/purification of the antigenic polypeptide.
- the antigenic polypeptide is prepared such that an N or C-terminal moiety (such as, but not limited to a histidine (His)-tag) isfused to the open reading frame (ORF) for theantigenic polypeptideto alow purification.
- an N or C-terminal moiety such as, but not limited to a histidine (His)-tag
- ORF open reading frame
- embodiments of the present disclosure include polynucleotides encoding the antigenic polypeptides of the disclosure.
- the present disclosure provides a polynucleotide encoding an antigenic polypeptideof the disclosure.
- the polynucleotide is codon optimized.
- the polynucleotide further comprises a heterologous regulatory element operably linked to a nucleic acid encoding the antigenic polypeptide.
- the heterologous regulatory element is a promoter.
- the heterologous regulatory element is a terminator.
- Polynucleotide sequences of the disclosure encompass DNA, RNA, DNA-RNA hybrids, peptide nucleic acid (PNA) or any other DNA-like or RNA-like material.
- the present disclosure provides isolated or recombinant nucleic acid molecules comprising nucleic acid sequences encoding the antigenic polypeptides.
- nucleic acid molecule refers to DNA molecules (e.g., recombinant DNA, cDNA, genomic DNA, plastid DNA, mitochondrial DNA) and RNA molecules (e.g., mRNA) and analogs of the DNA or RNA generated using nucleotide analogs.
- the nucleic acid molecule can be single-stranded or double-stranded, but preferably is double-stranded DNA.
- An “isolated”nucleic acid molecule (or DNA) is used herein to refer to a nucleic acid sequence (or DNA) that is no longer in its natural environment, for example in vitro.
- a “recombinant”nucleic acid molecule (or DNA) is used herein to refer to a nucleic acid sequence (or DNA) that is made by combining genetic material from multiple sources.
- a recombinant nucleic acid molecule (or DNA) is made in a recombinant cel, such as a recombinant host cel.
- an “isolated”or “recombinant”nucleic acid is free of sequences (preferably protein encoding sequences) that naturaly flank the nucleic acid (i.e., sequences located at the 5′ and 3′ ends of the nucleic acid) in the genomic DNA of the organism from which the nucleic acid is derived.
- Nucleic acid sequences of the disclosure can be used in DNA constructs or expression cassetes for transformation and expression in organisms, including microorganisms and plants.
- the nucleotide or amino acid sequences may be synthetic sequences that have been designed for expression in an organism including, but not limited to, a microorganism or a plant.
- the scope of the disclosure further encompasses any nucleic acid construct which codes for an antigenic polypeptidedescribed herein.
- Polynucleotide constructs of the disclosure may comprise single-stranded or double- stranded polynucleotides and may represent the sense or the antisense strand.
- the present disclosure provides a polynucleotide comprising, consisting of, or consisting essentialy of any of the ful-length sequences of SEQ ID NOs: 14- 21.
- the present disclosure provides a recombinant protein comprising an amino acid sequence encoded by the nucleotide sequence of any of SEQ ID NOs: 14-21.
- the present disclosure provides a recombinant protein comprising an amino acid sequence encoded by the nucleotide sequence of SEQ ID NO: 14, SEQ ID NO: 15, SEQ ID NO: 16, SEQ ID NO: 17, SEQ ID NO: 18, SEQ ID NO: 19, SEQ ID NO: 20, or SEQ ID NO: 21.
- the present disclosure also encompasses constructs comprising sequences which are derivatives of polynucleotide sequences encoding antigenic polypeptides described herein.
- derivative refers to complementary sequences, degenerate sequences, truncated or augmented sequences, modified sequences, and other polynucleotides based upon an original sequence from which a derivative sequence is derived.
- polynucleotide derivative contemplated within the scope of this disclosure is a polynucleotide comprising nucleotide substitutions.
- substitutions may be made within a given polynucleotide sequence that result in a codon which codes for the identical amino acid as coded for in the original sequence, and which such change does not alter the composition of the polypeptide coded by a polynucleotide.
- Such “silent” substitutions may be selected by one of skil in the art.
- nucleotide substitutions are contemplated which result in an amino acid substitution, wherein the amino acid is of similar polarity, charge, size, aromaticity, etc., such that the resulting polypeptide is of identical or substantialy similar structure and function as a polypeptide resulting from an unmodified sequence.
- nucleotide substitutions which result in amino acid substitutions which create a polypeptide derivative.
- nucleotide analogs, modified nucleotides, and other compositions may be substituted for the nucleotides of the sequences encoding for antigenic polypeptides, for example modified or non-naturaly occuring nucleotides such as 5-propynyl pyrimidines (i.e., 5-propynyl-dTTP and 5-propynyl-dTCP), 7- deaza purines (i.e., 7-deaza-dATP and 7-deaza-dGTP).
- Nucleotide analogs include base analogs and comprise modified forms of deoxyribonucleotides as wel as ribonucleotides. Additionaly, substitutions in a polynucleotide sequence may be made which enable the translation of polypeptides from the polynucleotide sequence within a specific expression system. For example, it is contemplated that the polynucleotide sequences may bemodified as necessary to enable or optimize expression of proteins in eukaryotic, yeast, bacterial, insect, plant, mammalian, or in other expression systems such as cel-free and chemical systems. The selection of proper substitutions for proper expression within a given expression system is within the skil of one in the art of molecular biology.
- Polynucleotide derivatives of the disclosure also comprise augmented or chimeric sequences, wherein a polynucleotide sequence has been modified to include additional nucleotides.
- a polynucleotide sequence, or subsequences thereof may be ligated with additional sequences which enhance expression (for example, promoter sequences), or which alter the properties of the resulting polypeptide, such as sequences which enhance secretion, enable isolation (e.g. sequences which code for display/affinitytags, His-Tags or like moieties), enable immobilization, or other useful sequences as known in the art.
- the scope of the disclosure additionaly includes vectors comprising the polynucleotide constructs of the disclosure integrated into the vectors.
- Exemplary vectors include plasmids, phages, and viral constructs which promote maintenance, amplification, and transcription of the polynucleotide sequences in an expression system.
- the nucleic acid constructs may comprise sequences integrated into the genome of an organism by transduction techniques known in the art.
- the present disclosure provides a vector comprising any of the polynucleotides of the disclosure.
- the vector is a plasmid, naked nucleic acid, phage, viral vector, or virus.
- the vector is capable of genome integration.
- the vector is an RNA.
- the present disclosure provides a vectoror construct comprising the nucleotide sequence of any of SEQ ID NOs: 14-21. In some embodiments, the present disclosure provides a vector or construct comprising thenucleotide sequence of SEQ ID NO: 15, SEQ ID NO: 17, SEQ ID NO: 18, SEQ ID NO: 19, SEQ ID NO: 20, or SEQ ID NO: 21.
- the present disclosure also provides a cel comprising any of the polynucleotides of the disclosure.
- the present disclosure also provides a cel comprising any of the vectors of the disclosure.
- the present disclosure also provides a cel comprising any of theconstructsof the disclosure.
- the present disclosure also provides a cel expressing any of the antigenic polypeptidesof the disclosure.
- the cel is a microorganism.
- the present disclosure also provides host cels that are engineered to express one or more antigenic polypeptidesof the disclosure.
- Suitable host cels include cels of any microorganism (e.g., cels of a bacterium, a protist, an alga, a fungus (e.g., a yeast or filamentous fungus), or other microbe), and are preferably cels of a bacterium.
- the disclosure further provides a recombinant host cel that is engineered to express one or more, two or more, three or more, four or more, or five or more antigenic polypeptides.
- immunoassays In some embodiments, provided herein are immunoassays.In some embodiments, the immunoassays provided herein are serologic assays. The terms “serologic assay,”“serologic” and “serology assay”as used herein refers to an assay that detects antibodies or antibody fragments in a sample. In some embodiments, provided herein are immunoassays for detecting a target antibody in a sample obtained from a subject.
- immunoassays for detecting antibodies elicited by infection with hepatitis D virus (HDV) in a sample obtained from a subject.
- the assays providedherein detect total antibodies (IgG and IgM) against HDV.
- An example of animmunoassay format for detecting a target antibody is an assay that comprises a capture moiety that binds to the antibody of interest (target antibody) and a detection moiety that detects presence of the target antibody.
- target antibody a capture moiety that binds to the antibody of interest
- detection moiety that detects presence of the target antibody.
- Such an assay can be either an indirect assay or a direct assay.
- An example of an indirect assay format for detecting a target antibody is an assay that comprises an antigen (capture moiety) that is recognized by the target antibody and an antibody (detection moiety) that recognizes the captured antibody and comprises a detectable label.
- an assay may be illustrated by the shorthand: Ag-Ab-Ab*, in which “Ag”represents an antigen that is the capture moiety, “Ab”represents the target antibody, and “Ab*”represents adetection moiety that is an antibody with a detectable label.
- the indirect assay format (Ag-Ab-Ab*) is known tohavelimitationsin sensitivity and specificity due to nonspecific binding of anti-human IgG and IgM conjugates (Ab*), which limits sample volume and conjugate concentration used in the indirect assay format.
- An example schematic of an indirect assay format is shown in Figure 2A.
- An example of a direct assay format for detecting a target antibody is an assay that comprises a first antigen (capture moiety) that is recognized by the target antibody and a second antigen (detection moiety) that is recognized by the target antibody and comprises a detectable label.
- Such an assay may be illustrated by the shorthand: (Ag-Ab-Ag*),in which “Ag” represents a first antigen that is the capture moiety, “Ab”represents the target antibody, and “Ag*”represents thedetection moiety that is a second antigen with a detectable label.
- the direct assay format has been used in many serology assays for detection of HIV, HTLV, HBV and HCV infections.
- the present disclosure providesdirect immunoassays.
- an assay of the present disclosure is refered to as a “double antigen sandwich assay”, or alternatively as a “double antigen bridging assay (DABA)”.
- a double antigen sandwich assay uses an antigen sandwichto detect target antibodiesand can be represented by the format Ag-Ab-Ag*.
- the double antigen sandwich format two antigens are bridged by an antibody analyte.
- the two antigens are diferent from each other, for example, one antigen may be boundto a solid support while the other antigen includes a detectable label.
- the amino acid sequences of the two antigens aredifferent.
- Double antigen sandwich assays are total antibody assays, meaning that they detect al immunoglobulin types and classes.
- an assay of the present disclosure comprises the format: Ag- Ab-Ag*.
- the assay usesrecombinant HD proteins as capture antigen (Ag) and detectionantigen(Ag*), which are bridged onlyby an HDV specific antibody (Ab)to generate positive signal.
- Ag capture antigen
- Ag* detectionantigen
- Abs HDV specific antibody
- an assay of the present disclosure utilizes a capture antigen on a solid phase/support (Ag/solid) and a detection antigen having a detectable label (Ag*, also refered to as a “detection conjugate”)to form a double antigen sandwich with ananti-HDV antibody (Ag/solid-Ab-Ag*).
- sample, capture antigen and detection antigen are combined.
- Anti-HDV antibodies present in the sample simultaneously form a double antigen sandwich immunoproduct (Ag/solid-Ab-Ag*) captured on the solid phase/support.
- the solid phase/support is washed at least once to remove or separate unbound antibodies and conjugates. After the washing, signal detection utilizing the detectable label is caried out.
- the measured signal is proportional to the amount of anti-HDV antibody in the sample.
- An example schematic of such a direct assay format is shown in Figure 2B.
- the present disclosure provides a direct format HDV total antibody assay with enhanced sensitivity and specificity.
- an assay of the present disclosure such as a double antigen sandwich assay utilizing recombinant HD proteins as capture antigen (Ag) and detection antigen (Ag*), provides an improvement to existing assays for detection of HDV specificantibodies in a sample.
- the improvement is compared to an HDV indirect assay.
- the improvement is improved specificity.
- the improvement is measurable as improved positive and/or negative agreement with an HDV RNA test.
- the improvement is enhanced dynamic range of assay signal.
- the assay provides utility to assess disease progressionofHDV-HBV coinfection and superinfection.
- a high antibody response with low viral load is indicative ofspontaneously clearing HDV RNA based on typical evolution of serological and virological markers in HDV infection.
- Initial atempts to develop a directformat HDV total antibody assay using the ful- length HD protein were not successful due to extremely high assay background. Analysis of the 3D structure of HD protein suggested the background wasdriven by aggregation between the capture and detection HD antigens as theN-terminal (12-60amino acids) tends to form a helix bundle octamer structure.
- immunoassays comprise contacting a sample (e.g., a sample obtained from a subject) with a capture moiety that binds to a target antibody in the sample.
- the immunoassays provided herein also comprise a separate detection moiety which is detectably labeled.
- capture moiety refers to a component of an immunoassay that binds and retains the target antibody.
- the capture moiety is an antigenic polypeptide.
- the antigenic polypeptide is a recombinant large- HDAg polypeptide.
- the antigenic polypeptide is a recombinant smal- HDAg polypeptide.
- an antigenic polypeptide that is a capture moiety is also refered to herein as a “capture antigen.”
- a capture antigen of the disclosure is acapture HD antigen.
- a capture moiety is a ful-length HD protein (HD large antigen or HD smal antigen).
- the ful-length HD protein sequence is SEQ ID NO: 1.
- the ful-length HD protein sequence is SEQ ID NO: 3.
- a capture moiety comprises, consists essentialy of, or consists of the sequence of SEQ ID NO: 1.
- a capture moiety comprises, consists essentialy of, or consists of the sequence of SEQ ID NO: 3.
- the term “detectionmoiety” refers to a component of an immunoassay that detects presence of the target antibody.
- the detection moiety is an antigenic polypeptide.
- the detection moiety is an antigenic polypeptide that is diferent from the antigenic polypeptide that functions as a capture moiety.
- the antigenic polypeptide is a recombinant large-HDAg polypeptide.
- the antigenic polypeptide is a recombinant smal-HDAg polypeptide.
- an antigenic polypeptide that is a detection moiety is also refered to herein as a “detection antigen.”
- a detection antigen of the disclosure is a detection HD antigen.
- a detection moiety is an antigenic polypeptide derived from the ful-length HD protein (HD large antigen or HD smal antigen) and comprisesa mutation (e.g., truncation(s), substitution(s), etc.) relative to the ful-length HD protein.
- the ful-length HD protein sequence is SEQ ID NO: 1.
- the ful-length HD protein sequence is SEQ ID NO: 3.
- the detection moiety may be any of the antigenic polypeptides comprising truncations and/or mutations describedabove in the “Antigenic polypeptides”section.
- an antigenic polypeptide that is a detection moiety comprises, consists essentialy of, or consists of a sequence selected from SEQ ID NO: 2, SEQ ID NO: 4, SEQ ID NO: 5, SEQ ID NO: 6, SEQ ID NO: 7 and SEQ ID NO: 8.
- an antigenic polypeptide that is a detection moiety comprises a sequence having at least 90% sequence identity to the sequence of SEQ ID NO: 8, whereinsaid antigenic polypeptide lacks a sequence comprising at least 90% sequence identity to a region of from about 12 to about 60 amino acids at the N-terminus region of SEQ ID NO: 1.
- an antigenic polypeptide that is a detection moiety comprises, consists essentialy of, or consists of a polypeptide selected from the group consisting of: i) a polypeptide that comprises the sequence of SEQ ID NO: 8 and lacks the sequence of SEQ ID NO: 13; i) a polypeptide that comprises the sequence of SEQ ID NO: 7 and lacks the sequence of SEQ ID NO: 12; ii) a polypeptide that comprises the sequence of SEQ ID NO: 6 and lacks the sequence of SEQ ID NO: 11; iv) a polypeptide that comprises the sequence of SEQ ID NO: 5 and lacks the sequence of SEQ ID NO: 10; v) a polypeptide that comprises the sequence of SEQ ID NO: 4 and lacks the sequence of SEQ ID NO: 9; and vi) a polypeptide that comprises the sequence of SEQ ID NO: 2.
- a detection moiety comprises, consists essentialy of, or consists of a sequence selected from SEQ ID NO:
- the detection moiety is detectably labeled, andbinding of the detection moiety to an antibody of interest (i.e. a target antibody) produces a detectable signal.
- an antibody of interest i.e. a target antibody
- a direct immunoassay comprising contacting asample with a capture antigen that binds to an antibody against a polypeptide comprising a sequence having at least 70% sequence identity to SEQ ID NO: 1 and a detection antigen that binds to the antibody, wherein the detection antigen is detectably labeled.
- the antibody is directed to a polypeptide comprising SEQ ID NO: 1.
- a direct immunoassay involving the use of separate capture and detection moieties, wherein the capture moiety binds to a targetantibodyof interest forming a capture moiety-target antibody complex, and the detection moietybinds to the targetantibodyof interest.
- the detection moiety comprises a detectable label.
- the detection moiety binds to the target antibody in the capture moiety-target antibody complex.
- the capture moiety is a capture antigen and the detection moiety is a detection antigen, such that a capture antigen-target antibody- detection antigen complexis formed in the assay.
- the immunoassays provided herein comprise contacting a sample with an antigenic polypeptide(e.g. a capture antigenand/or detection antigen) that binds to an antibody againsthepatitis D antigen (HDAg) of hepatitis D virus (HDV).
- an antigenic polypeptide e.g. a capture antigenand/or detection antigen
- HDAg hepatitis D antigen
- HDV hepatitis D virus
- the antibody bound by the antigenic polypeptide is an antibody against the large- HDAg and/or the smal-HDAg.
- the antibody is an antibody against the large-HDAg.
- the antigenic polypeptide is a recombinant large-HDAg polypeptide.
- the antigenic polypeptide binds to an antibody againstthe polypeptideofSEQ ID NO: 1.
- the antibody is an antibody against the smal-HDAg.
- the antigenic polypeptide is a recombinant smal-HDAg polypeptide.
- the antigenic polypeptide binds to an antibody againstthe polypeptide of SEQ ID NO: 3.
- the antigenic polypeptide may be any of the antigenic polypeptides described throughout the present disclosure.
- an antigen used in an immunoassay of the disclosure is a liquid phase antigen or a solid phase antigen.
- liquid phase antigen refers to an antigen in solution, which comprises one or more epitopes that bind to a target antibody also freely mobile within a solution.
- a “solid phase antigen” is defined as an antigen that is atached to a solid phase, which comprises one or more epitopes that can bind to a target antibody in solution.
- a “solid phase” may be a porous or non-porous material, a latex particle, a magnetic particle, a microparticle, a bead, a membrane, and a microtiter wel or a plastic tube.
- thecapture moiety e.g. capture antigen
- thecapture moiety isbound to a solid support or solid phase.
- the solid support or solid phase facilitates separation of the capture moiety-target antibody complex or the capture moiety-target antibody- detection moietycomplex from the test sample.
- Any solid support known in the art can be used, including but not limited to, solid supports made out of polymeric materials in the forms of wels of a reaction tray, test tubes or beads (for example, polystyrene beads, magnetic beads), nitrocelluloseose strips, membranes, microparticles (for example, latex particles).
- the solid phase also can comprise any suitable porous material with adequate porosity andsurface afinity.Microporous structures are generaly used, but materials with gel structure in the hydrated state may be used as wel.
- Such useful solid supports include, but are not limited to, nitrocellularose and nylon.
- Such porous solid supports are in the form of sheets of thickness from about 0.01 to 0.5 mm, including about 0.1 mm.
- the pore size may vary within wide limits, and can be from about 0.025 to about 15 microns, especialy from about 0.15 to about 15 microns.
- the surface of such supports may be activated by chemical processes which cause covalent linkage of the capture moietyto the support.
- the capture moiety e.g. capture antigen
- the capture moiety can be bound to the solid support or solid phasedirectly or indirectly.
- the capture moiety e.g.
- capture antigen can be bound to the solid support or solid phase by adsorption, by covalent bonding using a chemical coupling agent or by other means known in the art, provided that such binding does not interfere with the ability of the capture moietyto bind to the target antibody.
- the capture moiety can be bound to microparticles that have previouslybeencoated with streptavidin or biotinwith biotinylated moieties using means known in the art.
- the capture moiety can be bound using microparticles that have been previously coated with anti-species specific monoclonal antibodies.
- the solid support can be derivatized to alow reactivity with various functional groups on the capture moiety.
- Such derivatization requires the use of certain coupling agents such as, but not limited to, maleic anhydride, N-hydroxysuccinimide and 1- ethyl-3-(3-dimethylaminopropyl)carbodimide.
- the mixture is incubated in order to alow for the formation of acapturemoiety-target antibodycomplex, detectionmoiety-target antibody complex, and/or capture moiety-target antibody-detection moietycomplex.
- the incubation can be carried out at a pH of from about 4.5 to about 10.0, at a temperature of from about 2°C to about 45°C, and for a period from at least about one (1) minute to about eighteen (18) hours, including from about 1 to 20 minutes, also including from about 2-6 minutes.
- the immunoassay described herein can be conducted in one step (meaning the test sample, at least one capture moietyand at least one detection moiety are al added sequentialy or simultaneously to a reaction vessel) or in more than one step, such as two steps, three steps, etc.
- the sample is first contacted with the capture moiety.
- the complex is then contacted with at least one detection moiety under conditions such that the detection moiety binds to the capture moiety-target antibody complex,thereby forming a capture moiety-target antibody-detection moiety complex.
- multiple detection moieties are used. If the capture moiety-target antibodycomplexis contacted with more than one detection moiety, then multiplecapture moiety-target antibody-detection moietycomplexes areformed.
- thedetectionmoiety e.g. the detectionantigen
- the detectable label is bound to the detectionmoiety prior to, simultaneously with, or after formation of a complexbetween the detection moiety and the target antibody.
- Any detectable label known in the art can be usedas a detectable label of the present disclosure.
- the detectable label can be a radioactive label, such as, 3H, 125I, 35S, 14C, 32P, 33P, an enzymatic label, such as horseradish peroxidase, alkaline phosphatase, glucose 6- phosphate dehydrogenase, etc., a chemiluminescent label, such as, acridinium (e.g., acridium esters, acridinium SPSP (N10-(3-sulfopropyl)-N-(3-sulfopropyl, etc.), luminol, isoluminol, thioesters, sulfonamides, phenanthridinium esters, etc.
- acridinium
- a fluorescence label such as, fluorescein (5-fluorescein, 6-carboxyfluorescein, 3’6-carboxyfluorescein, 5(6)-carboxyfluorescein, 6- hexachloro-fluorescein, 6-tetrachlorofluorescein, fluorescein isothiocyanate, etc.), rhodamine, phycobiliproteins, R-phycoerythrin, quantum dots (zinc sulfide-capped cadmium selenide), a thermometric label or an immuno-polymerase chain reaction label.
- fluorescein 5-fluorescein, 6-carboxyfluorescein, 3’6-carboxyfluorescein, 5(6)-carboxyfluorescein, 6- hexachloro-fluorescein, 6-tetrachlorofluorescein, fluorescein isothiocyanate, etc.
- rhodamine rhodamine
- the detectable label is a particle label.
- a particle label produces a colored readout which requires no development process for visualization.
- the detectable label is a nanoparticle.
- the detectable label is a gold nanoparticle.
- the detectable label is a latex bead or a latex nanoparticle.
- the detectable label is a magnetic nanoparticle.
- any suitable detectable label that can be conjugated or linked to an antigenic polypeptidein order to detect binding of the antigenic polypeptideto a target antibody can be used,many of which are known in the art.
- the detectable label can be bound to the detection moiety either directly or through a coupling agent.
- An example of a coupling agent that can be used is EDAC (1-ethyl-3-(3- dimethylaminopropyl) carbodimide, hydrochloride) that is commercialy available from Sigma- Aldrich, St. Louis, MO. Other coupling agents that can be used are known in the art. Methods for binding a detectable label to a polypeptideare known in the art.
- detectable labels can be purchased or synthesized that already contain end groups that facilitate the coupling of the detectable label to the detection moiety, such as, N10-(3-sulfopropyl)-N-(3- carboxypropyl)-acridinium-9-carboxamide, otherwise known as CPSP-Acridinium Ester or N10- (3-sulfopropyl)-N-(3-sulfopropyl)-acridinium-9-carboxamide, otherwise known as SPSP- Acridinium Ester.
- the capture moiety-target antibody-detection moiety complex can be,but does not have to be, separated from the remainder of the test sample prior to quantification of the label.
- the capture moiety e.g. capture antigen
- a solid support or solid phase such as, but not limited to, a wel of a reaction tray, a bead or a microparticle
- separation can be accomplished by removing the fluid (of the test sample) from contact with the solid support.
- the capture moiety e.g. capture antigen
- the capture moiety is bound to a solid support it can be simultaneously contacted with the sample and the detection moiety, folowed by removal of the fluid (test sample) from contact with the solid support.
- the capture moiety-target antibody complex or the capture moiety-target antibody-detection moiety complexdoes not have to be removed from the test sample for quantification of the amount of the label.
- the amount of label in the complex is quantified using techniques known in the art. For example, if an enzymatic label is used, the labeled complex is reacted with a substrate for the label that gives a quantifiable reaction such as the development of color. If the label is a radioactive label, the label is quantified using a scintilation counter.
- the label is quantified by stimulating the label with a light of one color (which is known as the “excitation wavelength”) and detecting another color (which is known as the “emission wavelength”) that is emited by the label in response to the stimulation.
- the label is a chemiluminescent label
- the label is quantified detecting the light emited either visualy or by using luminometers, x-ray film, high speed photographic film, a CCD camera, etc.
- the concentration of the target antibodyin the test sample can be determined by use of a standard curve that has been generated using serial dilutions of the marker of known concentration.
- the standard curve can be generated gravimetricaly, by mass spectroscopy and by other techniques known in the art.
- the presence of HDV antibodyin asample is determined by means of a cutof value that alows for the semi-quantitative detection of theHDV antibody.
- negativeresults indicate the absence of HDV infection.
- equivocalresults indicate borderline level of anti-HDV total antibodies.
- the provided assays are employed for diagnosis of HDV infection and/or to identify patients for antiviral treatments.
- the assay is employed in the diagnosis of HDV infection.
- the provided assays are employed to determine prevalence of HDV infection for epidemiology studies.
- the assay is a rapid test assay.
- the assay is employed on a sample from a subject that is hepatitis B surface antigen positive (HBsAgpositive).
- a positive result from the assay indicates a subsequentHDV RNA assay.
- a positive result from the assay combined with a positive result from an HDV RNA assay indicates a need for treating the subject for HDV infection.
- Thisassay design wil provide a significant performance advantage versus competitors on the market today.
- Themethods and kits described elsewhere in the present disclosure encompass other reagents and methods for carying out the immunoassay.
- an automated or semi-automated system includes the substrate to which the capture moietyis atached (which can impact sandwich formation and analyte reactivity), and the length and timing of the capture, detection and/or any wash steps.
- a non-automated system e.g., ELISA
- a non-automated format such as an ELISA may include a relatively longer incubation time with sample and capture reagent (e.g., about 2 hours)
- an automated or semi-automated format e.g., ARCHITECT®
- sample and capture moiety e.g., approximately 18 minutes for ARCHITECT®
- an automated or semi-automated format may have a relatively shorter incubation time (e.g.,approximately 18minutes for the ARCHITECT®).
- methods of the present disclosure comprise incubation with sample and capture moiety along with detection moiety.
- incubation of sample, capture moiety and detection moiety is for a relatively shorter incubation time (e.g., approximately 18 minutes) as compared with a non-automated format.
- methods of the present disclosure comprise incubation of sample, capture moiety and detection moiety for approximately 15 minutes, 16 minutes, 17 minutes, 18 minutes, 19 minutes, 20 minutes, 21 minutes, 22 minutes, 23 minutes, 24 minutes, or 25 minutes. In some embodiments, methods of the present disclosure comprise incubation of sample, capture moiety and detection moiety for 30 minutes or less.
- animmunoassay provided herein is a chemiluminescent microparticle immunoassay(CMIA).
- the assay is used in a fuly automated and high-throughput system (e.g., 200 tests per hour), such as the ARCHITECT® system.
- a provided assay of the disclosure comprises the folowing steps: first, sample (serum or plasma) is combined with recombinant HDAg (capture moiety) coated microparticles and acridinium-labeled conjugates(detection moiety), HDV antibodies present in the sample simultaneously form a double antigen sandwich with the capture HDAg on microparticles and the detection HDAg conjugate in solution; folowing a wash cycle, alkaline hydrogen peroxide solution is then added to release acridinium chemiluminescent signal; and the intensity of the chemiluminescence, measured as relative light units (RLU), is proportional to the concentration of HDV antibodies in the sample.
- methods for detecting antibodies specific for HD protein in a biological sample comprise performing an immunoassayas describedherein, including an immunoassay as describedabove.
- the immunoassay comprises determining the presence or amount of atarget antibody in asample based upon the detectable signal generated upon formation of the capture moiety-target antibody-detection moiety complex (e.g., a capture antigen-target antibody- detection antigencomplex).
- the method comprises determining the presence or amount ofatarget antibody in asample based upon adetectable signalafterseparating orremoving the fluid (of the test sample) from contact with the solid support.
- adetectable signal afterseparating orremoving the fluid (of the test sample) from contact with the solid support.
- methods for detecting hepatitis D virus (HDV) infection in a subject comprising determining that the subject is infected with hepatitis D virus (HDV)when the amount of the target antibody (e.g. the level of the detectable signal, which is indicative of the amount of target antibody) is equal to or above a reference value.
- the method comprises determining that the subject is not infected with HDV when the amount of the target antibody (e.g. the level of the detectable signal, which is indicative of the amount of target antibody) is below a reference value.
- the methodsprovided herein comprise performing an immunoassay as describedherein.
- the immunoassay comprises contacting a sample obtained from a subject with a capture moiety (e.g., a capture antigen).
- thecapture moiety is an antigenic polypeptide describedherein, including an antigenic polypeptide as describedabove.
- anantigenic polypeptide that is a capture moiety is a recombinant large-HDAg polypeptide.
- anantigenic polypeptide that is a capture moiety is a recombinant smal-HDAg polypeptide.
- a capture moiety is a ful-length HD protein (HD large antigen or HD smal antigen).
- the ful-length HD protein sequence is SEQ ID NO: 1.
- the ful-length HD protein sequence is SEQ ID NO: 3.
- a capture moiety comprises, consists essentialy of, or consists of the sequence of SEQ ID NO: 1.
- a capture moiety comprises, consists essentialy of, or consists of the sequence of SEQ ID NO: 3.
- the immunoassay further comprises contacting the sample with a detection moiety.
- thedetection moiety is an antigenic polypeptide describedherein, including an antigenic polypeptideas describedabove.
- the detection moiety is an antigenic polypeptide that is diferent from the antigenic polypeptide that functions as a capture moiety.
- the antigenic polypeptide that is a detection moiety is a recombinant large-HDAg polypeptide.
- the antigenic polypeptide that is a detection moiety is a recombinant smal-HDAg polypeptide.
- a detection moiety is an antigenic polypeptide derived from the ful-length HD protein (HD large antigen or HD smal antigen) and comprisesa mutation relative to the ful- length HD protein.
- the ful-length HD protein sequence is SEQ ID NO: 1.
- the ful-length HD protein sequence is SEQ ID NO: 3.
- the detection moiety may be any of the antigenic polypeptides comprising truncations and/or mutations described above in the “Antigenic polypeptides”section.
- an antigenic polypeptide that is a detection moiety comprises, consists essentialy of, or consists of a sequence selected from the group consisting of SEQ ID NO: 2, SEQ ID NO: 4, SEQ ID NO: 5, SEQ ID NO: 6, SEQ ID NO: 7 and SEQ ID NO: 8.
- an antigenic polypeptide that is a detection moiety comprises a sequence having at least 90% sequence identity to the sequence of SEQ ID NO: 8, wherein said antigenic polypeptide lacks a sequence comprising at least 90% sequence identity to a region of from about 12 to about 60 amino acids at the N-terminus region of SEQ ID NO: 1.
- an antigenic polypeptide that is a detection moiety comprises, consists essentialy of, or consists of a polypeptide selected from the group consisting of: i) a polypeptide that comprises the sequence of SEQ ID NO: 8 and lacks the sequence of SEQ ID NO: 13; i) a polypeptide that comprises the sequence of SEQ ID NO: 7 and lacks the sequence of SEQ ID NO: 12; ii) a polypeptide that comprises the sequence of SEQ ID NO: 6 and lacks the sequence of SEQ ID NO: 11; iv) a polypeptide that comprises the sequence of SEQ ID NO: 5 and lacks the sequence of SEQ ID NO: 10; v) a polypeptide that comprises the sequence of SEQ ID NO: 4 and lacks the sequence of SEQ ID NO: 9;and vi) a polypeptide that comprises the sequence of SEQ ID NO: 2.
- a detection moiety comprises, consists essentialy of, or consists of a sequence selected from SEQ ID NO:
- themethodfurther comprises determining the presence or amount of the target antibody (e.g. the antibody against HDV) in the sample based upon the detectable signal. In some embodiments, the method further comprises determining that the subject is infected with HDVwhen the amount of the target antibody (e.g. the level of the detectable signal, which is indicative of the amount of target antibody) is equal to or above a reference value. In some embodiments, the method further comprises determining that the subject is not infected with HDV when the amount of the target antibody (e.g.
- the present disclosure provides a method for detecting antibodies specific for HD protein in a biological sample, wherein an antigenic polypeptideof the present disclosure is used as a binding partner for said HD protein antibodies.
- the present disclosure provides a method for detecting antibodies specific for HD protein in a biological sample, said method comprising:a) forming an immunoreaction mixture by mixing a biological sample withanantigenic polypeptide of the present disclosure; b) maintaining said immunoreaction mixture for a time period sufficient for alowing antibodies against said antigenic polypeptide present in the biological sample to immunoreact with said antigenic polypeptide to form an immunoreaction product; and c) detecting the presence and/or the concentration of said immunoreaction product.
- the immunoreaction is caried out in a double antigen sandwich format comprising:a) adding to said biological sample a first antigenic polypeptide that comprises a means for binding to a solid support and a second antigenic polypeptide that caries a detectable label, wherein said first antigenic polypeptide and said second antigenic polypeptide bind specificaly to the HD protein antibodies, b) forming an immunoreaction mixture comprising the first antigenic polypeptide, the biological sample antibody and the second antigenic polypeptide, wherein the solid support is added before, during or after forming the immunoreaction mixture, c) maintaining said immunoreaction mixture for a time period sufficient for HD protein antibodies in the biological sample to immunoreact with said first and second antigenic polypeptides to form an immunoreaction product, andd) detecting the presence of any of said immunoreaction product.
- the first antigenic polypeptide comprises a sequence having at least 90% sequence identity to the ful-length sequence of SEQ ID NO: 1.
- the method further comprises a washing step prior to the detecting step.
- the immunoreaction product is bound to the solid support and the washing step comprises removingorseparating someor al of the first antigenic polypeptide, HD protein antibody and second antigenic polypeptide that are not part of the immunoreaction product that is bound to the solid support.
- the solid support is selected from the group consisting of acolumn, bead, test tube, microtiter dish,multi- wel plate,microparticle, microsphere, a test stick, a test strip, microchip and membrane.
- the biological sample is a body fluid sample from a human subject.
- the biological sample is selected from the group consisting of blood, plasma and serum.
- methods for detecting antibodies against hepatitis D virus (HDV) in a biological sample comprising performing a HDV serology assay using a direct assay format with a first antigenic polypeptide and a second antigenic polypeptide, wherein the first antigenic polypeptide comprises a ful-length HD protein and the second antigenic polypeptide comprises an N-terminaly truncated fragment of the ful-length HD protein.
- HDV hepatitis D virus
- an “N-terminaly truncated fragment of theful-lengthHD protein” refers to a shortened HD protein (and sequence thereof) that has been shortened by removing a portion of the protein located at or near the N-terminus.
- the assay is a double antigen sandwich assay.
- theful-length HD protein comprises an amino acid sequence having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% or 100% sequence identity to the ful-length sequence of SEQ ID NO: 1.
- the ful-length HD protein comprises an amino acid sequence having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% or 100% sequence identity to the ful-length sequence of SEQ ID NO: 3.
- the second antigenic polypeptide lacks from 12 to 60 N- terminal amino acids of the HD protein.
- the second antigenic polypeptide lacks from 12 to 60 N-terminal amino acidsofthe ful-length HD protein of SEQ ID NO: 1.
- the assay is a double antigen sandwich assay.
- the second antigenic polypeptide lacks from 12 to 60 consecutive N-terminal amino acids of the HD protein.
- the second antigenic polypeptide lacks 21, 37 or 46 consecutive N- terminal amino acids of the HD protein.
- hepatitis D virus in a biological sample, comprising: a) contacting abiological sample, either simultaneously or sequentialy, in any order with:(1) a first antigenic polypeptide under conditions adequate to alow binding of anyHDV antibody present in the biological sample to the first antigenic polypeptide, wherein said first antigenic polypeptide is immobilized on a solid support, and (2) a second antigenic polypeptide under conditions adequate to alow binding of the second antigenic polypeptide to any HDV antibody present in the biological sample,such that a first antigenic polypeptide-HDV antibody-second antigenic polypeptide complex is formed and is bound to the solid support, andb) detecting the presence of said first antigenic polypeptide-HDV antibody-second antigenic polypeptide complex, wherein said first antigenic polypeptide comprises a sequence having at least 90% sequence identity to the ful-length sequence of SEQ ID NO: 1;wherein said second antigenic polypeptide comprises a sequence having
- the step of detecting comprises measuring an amount of saidfirst antigenic polypeptide-HDV antibody- second antigenic polypeptide complex that is formed and is bound to the solid support.
- the solid support is selected from the group consisting of acolumn, bead, test tube,microtiter dish,multi-wel plate,microparticle, microsphere, a test stick, a test strip, microchip and membrane. Incertainprefered embodiments, the solid support is a microparticle.
- thebiological sample is selected from the group consisting of blood, plasma andserum.
- the first antigenic polypeptide comprises a sequence having at least 95%, 96%, 97%, 98%, 99% or 100%sequence identity to the ful-length sequence of SEQ ID NO: 1.
- the second antigenic polypeptide is selected from the group consisting of:i) a polypeptide that comprises the sequence of SEQ ID NO: 8 and lacks the sequence of SEQ ID NO: 13;i) a polypeptide that comprises the sequence of SEQ ID NO: 7and lacks the sequence of SEQ ID NO: 12;ii) a polypeptide that comprises the sequence of SEQ ID NO: 6 and lacks the sequence of SEQ ID NO: 11;iv) a polypeptide that comprises the sequence of SEQ ID NO: 5 and lacks the sequence of SEQ ID NO: 10;v) a polypeptide that comprises the sequence of SEQ ID NO: 4 and lacks the sequence of SEQ ID NO: 9; andvi) a polypeptide that comprises the sequence of SEQ ID NO: 2.
- hepatitis D virus (HDV) infection in a subject comprising performing theimmunoassays orimmunoassay-based methods, such as the serology assays,described above.
- thesample is a sample taken from the subject and the step of detecting comprises detecting the presence of a first antigenic polypeptide-HDV antibody-second antigenic polypeptide complex that is formed and is bound to a solid support, thereby detecting the presence of past or present HDV infection in said subject.
- kits for estimating HDV incidence in a population comprising, a) providing a set of samples derived from a plurality of individuals within the population over a period of time;b) performing on each sample in the set of samples the immunoassays or immunoassay-based methods, such as the serology assays, described above; and c) determining the percentage of recent HDV infections over the period of time;wherein the percentage of recent HDV infections over the period of time provides an estimate of HDV incidence in the population.
- an immunoassay ofthe disclosure displays at least 75% sensitivity(e.g.
- the immunoassay displays at least 85% sensitivity. In some embodiments, the immunoassaydisplays at least 90% sensitivity. In some embodiments, the immunoassaydisplays at least 95% sensitivity. In some embodiments, the immunoassay displays at least 99% sensitivity. In some embodiments, the immunoassay displays at least 75% specificity(e.g. specificity in accurately diferentiating subjects not having HDVinfection from those having HDVinfection). In someembodiments, the immunoassay displays at least 85%specificity. In some embodiments, the immunoassay displays at least 90% specificity.
- the immunoassay displays at least 95% specificity.In some embodiments, the immunoassay displays at least 99% specificity. In some embodiments, the immunoassay displays at least 75% sensitivityand at least 75% specificity. In some embodiments, the immunoassay displays atleast 85% sensitivity and at least 75% specificity.In some embodiments, the immunoassay displays at least 95% sensitivity and at least 75% specificity. In some embodiments, the immunoassay displays at least 99% sensitivity and at least 75% specificity. In some embodiments, the immunoassay displays at least 85% sensitivity and at least 85% specificity. In some embodiments, the immunoassay displays at least 90% sensitivity and at least 85% specificity.
- the immunoassay displays at least 95% sensitivity and at least 85% specificity. In some embodiments, the immunoassay displays at least 99% sensitivity and at least 85%specificity. In some embodiments, the immunoassay displays at least 90% sensitivity and at least 90% specificity. In some embodiments, the immunoassay displays at least 95% sensitivity and at least 90% specificity. In some embodiments, the immunoassay displays at least 99% sensitivity and at least 90% specificity. In some embodiments, the immunoassay displays at least 95% sensitivity and at least 95% specificity.
- the immunoassay displays at least 99% sensitivity and at least 95% specificity. In some embodiments, the immunoassay displays at least 99% sensitivity and at least 99% specificity.
- the power of a diagnostic test to correctly predict status is commonly measured as the sensitivity of the assay, the specificity of the assay or the area under a receiver operated characteristic (“ROC”) curve.
- Sensitivity is the percentage of true positives that are predicted by a test to be positive, while specificity is the percentage of true negatives that are predicted by a test to be negative.
- a ROC curve provides the sensitivity of a test as a function of 1-specificity. The greater the area under the ROC curve, the more powerful the predictive value of the test.
- Positive predictive value is the percentage of people who test positive that are actualy positive.
- Negative predictive value is the percentage of people who test negative that are actualy negative.
- the immunoassays described herein may show an ROC of at least 0.6, at least about 0.7, at least about 0.8, or at least about 0.9.
- the methods provided herein further comprisetesting for active HDV infection by measuring hepatitis D RNA levels in a sample.
- methods of the present disclosure provide for a first line screening test for HDV.
- a negative result from an immunoassay of the disclosure provides for time and/or cost saving benefits by reducing the number of unnecessary HDV RNA testing.
- the methods provided herein further comprise treating the subject for an HDVinfection. Suitable treatments for HDVinfection include, for example, antiviral medications.
- treating a subject determined to have an HDV infection according to the presently disclosed methods comprises administering antiviral therapies.
- treating a subject according to the disclosure comprises administering one or more of pegylated interferon alpha therapyand bulevirtide.
- treating a subject according to the disclosure comprises administering one or more of pegylated interferon alpha therapy, bulevirtide, pegylated interferon lambda ( ⁇ ), lonafarnib, and REP2139-Ca.
- treating a subject according to the disclosure comprises treating the subject for both an HBV and an HDV infection. 7.Samples As used herein, “sample”, “test sample”, and “biological sample”refer to a sample that contains, is suspected of containing, or is to be tested for whether or not it contains a target antibody (e.g. an antibody produced during HDV infection).
- a target antibody e.g. an antibody produced during HDV infection
- the “sample”, “test sample”, and “biological sample” is a sample that is positive for HBV infection and/or obtained from a subject known to have an HBV infection.
- the sample is from a subject with clinicaly evident acute or chronic hepatitis B.
- the sample is from a subject that has a previous or active infection with hepatitis B.
- the sample is from a subject that is hepatitis B surface antigen positive.
- the sample may be processed prior to performing a method (e.g. an immunoassay)described herein. For example, the sample may be separated or purified from its source prior to analysis.
- an unprocessed sample canbe assayed directly.
- the sample is a fluid sample.
- the sample is a human bodily substance (e.g.,bodily fluid, blood such as whole blood, serum, plasma, urine, saliva, sweat, sputum, semen, mucus, lacrimal fluid, lymph fluid, amniotic fluid, interstitial fluid, lung lavage, cerebrospinal fluid, feces, tissue, organ, or the like).
- the sample is a whole blood sample, a serum sample, or a plasma sample. A wide range of volumes of the fluid sample may be analyzed.
- the sample volume may be about 0.5 nL, about 1 nL, about 3 nL, about 0.01 ⁇ L, about 0.1 ⁇ L, about 1 ⁇ L, about 5 ⁇ L, about 10 ⁇ L, about 100 ⁇ L, about 1 mL, about 5 mL, about 10 mL, or the like.
- the volume of the fluid sample is between about 0.01 ⁇ L and about 10 mL, between about 0.01 ⁇ L and about 1 mL, between about 0.01 ⁇ L and about 100 ⁇ L, or between about 0.1 ⁇ L and about 10 ⁇ L.
- the fluid sample may be diluted prior to use in an assaydescribed herein.
- the sample isdiluted with an appropriate solvent (e.g., a buffer such as PBS bufer).
- an appropriate solvent e.g., a buffer such as PBS bufer.
- a fluid sample may be diluted about 1-fold, about 2-fold, about 3-fold, about 4-fold, about 5-fold, about 6-fold, about 10-fold, about 100-fold, or greater, prior to use.
- the fluid sample is not diluted prior to use in an assay.
- the sample may undergo pre-analytical processing. Pre-analytical processing may offer additional functionality such as nonspecific protein removal and/or effete yet economicalyimplementable mixing functionality.
- the fluid sample may be concentrated prior to use in an assay.
- the sample maybe concentrated by precipitation, evaporation, filtration, centrifugation, or a combination thereof.
- a fluid sample may be concentrated about 1-fold, about 2-fold, about 3-fold, about 4-fold, about 5-fold, about 6-fold, about 10-fold, about 100-fold, or greater, prior to use. It may be desirable to include a calibrator and/or a controlin the methods described herein.
- the calibrator and/or control may be analyzed concurently with the sample from the subject as described above.
- the results obtained from the subject sample can be compared to the results obtained from the calibrator and/or control sample.
- Standard curves may be provided, with which assay results for the sample may be compared.
- Such standard curves present levels of marker (e.g., antibody) as a function of assay units, e.g.,fluorescent signal intensity, if a fluorescent label is used.
- standard curves can be provided for reference levels of a target antibody. 8.Kits and Systems
- the present disclosure further provides kitsand systems for performing an immunoassay described herein.
- kitsand systemsfor detecting antibodies elicited by infection with HDV in a sample obtained from a subject find use in multiplex and/or automated analysis methods.
- kits for detecting antibodies against hepatitis D virus (HDV) in a biological sample comprises an antigenic polypeptide of the present disclosure.
- the kit comprises at least two antigenic polypeptides of the present disclosure.
- the kit comprises a composition of the present disclosure, such as a composition comprising any of theantigenic polypeptides described above.
- the kit comprises a first and a second antigenic polypeptide of the disclosure, wherein either the first or second antigenic polypeptide comprises the sequence of SEQ ID NO: 1.
- either the first or second antigenic polypeptide comprises a sequence having at least 90% sequence identity to the ful- length sequence of SEQ ID NO: 1.
- Exemplary reagentsof the provided kits include, but are not limited to, colorimetric reagents, enzymes, buffers, etc.
- the kit can also contain at least one calibrator or control. Any calibrator or control can be included in the kit.
- the kit comprises a capture moiety and/or detection moiety described herein.
- the assays, kits and kit components of the disclosure are optimized for use on commercial platforms (e.g., immunoassays on the ARCHITECT® and Alinity®platforms of Abbot Laboratories, Abbot Park, IL, as wel as other commercial and/or in vitrodiagnostic assays).
- the assays, kits and kit components can be employed in other formats, for example, on electrochemical or other hand-held or point-of-care assay systems.
- the present disclosure is, for example, applicable to the commercial Abbot Point of Care (i-STAT®, Abbot Laboratories, Abbot Park, IL) electrochemical and rapid Lateral flow immunoassay systems.
- kits include quality control reagents (for example, sensitivity panels, calibrators, negative controls, and/orpositive controls). Preparation of quality control reagents is wel known in the art, and is described, e.g., on a variety of immunodiagnostic or nucleic acid product insert sheets.
- kits can optionaly include other reagents required to conduct a diagnostic assay or facilitate quality control evaluations, such as buffers, salts, enzymes, enzyme co-factors, substrates, detection reagents, and the like.
- Other components such as buffers and solutions for the isolation and/or treatment of a test sample (e.g., pretreatment reagents), may also be included in the kit.
- the kit may additionaly include one or more other controls.
- One or more of the components of the kit may be lyophilized and the kit may further comprise reagents suitable for the reconstitution of the lyophilized components.
- the “component,”“components,”or “at least one component,”of a kit refer generaly to a capture moiety, a detection moiety, a calibrator, a control, a sensitivitypanel, a container, a bufer, a diluent, a salt, an enzyme, a co-factor for an enzyme, a detection reagent, a pretreatment reagent/solution, a substrate (e.g.,as a solution), a stop solution, and the like that can be included in a kit for assay of a test sample, such as a patient urine, whole blood, serum or plasma sample, in accordance with the methods described herein and other methods known in the art.
- a test sample such as a patient urine, whole blood, serum or plasma sample
- kits for holding or storing a sample (e.g., a container or cartridge for a blood or urine sample).
- a sample e.g., a container or cartridge for a blood or urine sample.
- the kit may also optionaly contain reaction vessels, mixing vessels and other components that facilitate the preparation of reagents or the test sample.
- the kit may also include one or more instruments for assisting with obtaining a test sample, such as a syringe, pipete, forceps, measured spoon, or the like.
- the kit further can optionaly include instructions for use, which may be provided in paper form or in computer-readable form, such as a disc, CD, DVD or the like.
- the disclosure as described herein also can be adapted for use in a variety of automated and semi-automated systems (including those wherein the solid phase comprises a microparticle), as described, e.g., in U.S. Patent Nos.5,089,424 and 5,006,309, and as, e.g., commercialy marketed by Abbot Laboratories (Abbot Park, IL) including but not limited to Abbot’s ARCHITECT®and Alinity®instruments, as wel as other platforms.
- the disclosure optionaly is adaptable for the Abbot Laboratories commercial Point of Care (i- STATTM) electrochemical and rapid Lateral flow immunoassay systemsfor performing sandwich immunoassays.
- Immunosensors, and their methods of manufacture and operation in single-use test devices are described, for example in, U.S. Patent No.5,063,081, U.S. Patent Application 2003/0170881, U.S. Patent Application 2005/0054078, and U.S. Patent Application 2006/0160164, which are incorporated in their entireties by reference for their teachings regarding same.
- the present disclosure further provides a system comprising: a) a sample receiving component configured to receive a sample from a subject, wherein the sample comprises or is suspected of comprising a hepatitis D virus (HDV) antibody;b) a first antigenic polypeptide and a second antigenic polypeptide configured to make contact with the sample to form a first antigenic polypeptide-HDV antibody-second antigenic polypeptide complex, wherein the first antigenic polypeptide comprises a ful-length HD protein and the second antigenic polypeptide comprises amutated and/or truncatedHD protein, and wherein the second antigenic polypeptide carries a detectable label; c) a detection component configured to measure a signal generated by the detectable label in the first antigenic polypeptide-HDV antibody- second antigenic polypeptide complex; and d) an output component that indicates an amount of HDV antibody in the sample based on the signal.
- the second antigenic polypeptide comprises an N-terminaly trun
- an “N-terminaly truncated fragment of the ful-length HD protein” refers to a shortened HD protein (and sequence thereof) that has been shortened by removing a portion of the protein located at or near the N-terminus.
- the ful-lengthHD protein comprises an amino acid sequence having at least 95% sequence identity to the ful-length sequence of SEQ ID NO: 1.
- theful-length HD protein comprises an amino acid sequence having at least 95% sequence identity to the ful-length sequence of SEQ ID NO: 3.
- thesecond antigenic polypeptide lacks from 12 to 60 N-terminal amino acids of the ful-length HD protein.
- thesecond antigenic polypeptide lacks21, 37 or 46 consecutive N-terminal amino acids of the ful-length HD protein.
- thesecond antigenic polypeptide is selected from the group consisting of:i) a polypeptide that comprises the sequence of SEQ ID NO: 8 and lacks the sequence of SEQ ID NO: 13;i) a polypeptide that comprises the sequence of SEQ ID NO: 7and lacks the sequence of SEQ ID NO: 12;ii) a polypeptide that comprises the sequence of SEQ ID NO: 6 and lacks the sequence of SEQ ID NO: 11;iv) a polypeptide that comprises the sequence of SEQ ID NO: 5 and lacks the sequence of SEQ ID NO: 10;v) a polypeptide that comprises the sequence of SEQ ID NO: 4 and lacks the sequence of SEQ ID NO: 9; andvi) a polypeptide that comprises the sequence of SEQ ID NO: 2.
- thesample is selected from the group consisting of blood, plasma andser
- thedetectable label is selected from the group consisting of a fluorescent label, a chemiluminescent marker, and a chromophore.
- FIG.1 illustrates sequence alignment of the HD LAg, 5 N-terminal truncated HD Ags and HD LAg mutant as wel as the smal HD Ag.
- Example2 Design, expression, and purification of recombinant HD proteins
- the folowing is a general protocol to design, prepare and purify HD recombinant proteins expressed from plasmid constructs in E. coli.
- An N or C-terminal Histidine (His)-tag was fused to the ORF for each protein to alow purification using His-Bind Nickel Afinity Chromatography. Codon optimization and cloning of the DNA sequence was performed by a third-party vendor, ATUM (Newark, CA).
- Al proteins were expressed by transforming BL21(DE3) Chemicaly Competent E. coli cels by heat shock transformation method. Growth and Induction of E.
- E. coli harboring a pD454-SR-based construct was prepared by inoculating a single colony from an LB agar plate into a 125 ml Erlenmeyer flask (Corning Inc., Corning, NY) containing 500 ml LB Broth (Sigma-Aldrich) supplemented with 100 ⁇ g/ml ampicilin. The flask was placed in a shaking orbital incubator and incubated overnight ( ⁇ 16 hours) at 37oC.
- the pooled recombinant protein from the S-200 column was loaded onto a His-Bind nickel column (Novex-Life Technologies) equilibrated with binding bufer containing 20mM sodium Phosphate, pH 7.8, 0.5 M NaCl, 20 mM imidazole and 6 M urea.
- the bound protein was washed with 10 column volumes of binding bufer folowed by 6 column volumes of wash bufer (20mM sodium Phosphate, pH 7.8, 0.5 M NaCl, 20 mM imidazole and 6 M urea).
- the bound protein was eluted at 1 ml/min with a 0–500 mM linear imidazole gradient. Fractions containing the purified protein as assessed by Coomassie Blue staining were pooled and dialyzed against 1 L of 20mM sodium Phosphate, pH 7.8, 0.5 M NaCl and 6 M urea bufer over a 24 h period at 4°C. The dialyzed solution containing the purified protein was aliquoted and stored at –70 oC for future use.
- Example3 ARCHITECT® Chemiluminescent Immunoassay for Detection of Antibodies to HDV (indirect or anti-human assay format)
- HDV IgG prototype chemiluminescent microparticle immunoassays (CMIA) were developed on the automated ARCHITECT® instrument system (Abbot Laboratories, Abbot Park, IL).
- the indirect ARCHITECT® HDV IgG was a reference test for comparison to the direct HDV Total Ig assay (Example 4), it is a two-step immunoassay which utilizes an indirect anti-human assay format as illustrated in Figure 2A.
- the first step combines sample, assay diluent and paramagnetic microparticles.
- Anti-HDV antibodies (Ab) present in the sample are captured on paramagnetic particles coated with the recombinant HD protein (Ag/Solid-Ab). The microparticles are washed to remove unbound Anti-HDV antibodies.
- anti-HDV antibodies captured by the microparticles (Ag/Solid-Ab) are incubated with acridinium-labeled anti-human IgG monoclonal conjugate (Ab* to form immunoproduct(Ag/Solid-Ab-Ab*). Folowing an additional wash cycle, alkaline hydrogen peroxide solution is added to release acridinium chemiluminescence signal.
- the intensity of the chemiluminescence is proportional to the amount of specific antibody captured by the recombinant HD protein.
- the ARCHITECT® HDV IgG results are reported as the ratio of the sample RLU to the cutof RLU (S/CO) for each specimen. Specimens with S/CO values ⁇ 1.00 are considered non-reactive; specimens with S/CO values ⁇ 1.00 are considered reactive.
- Example 4 ARCHITECT® Chemiluminescent Immunoassay for Detection of Antibodies to HDV (Direct or Sandwich Assay Format)
- a direct double antigen sandwich assay (Ag/solid-Ab-Ag*) was developed on the automated ARCHITECT® instrument system (Abbot Laboratories, Abbot Park, IL).
- sample such as serum or plasma
- paramagnetic microparticles and antigen conjugate are combined.
- Anti-HDV antibodies present in the sample simultaneously form a double antigen sandwich immunoproduct (Ag/solid-Ab-Ag*) captured on paramagnetic particles coated with the Ful-length HD recombinant protein.
- the microparticles are washed to remove unbound antibodies and conjugates. Folowing an additional wash cycle, alkaline hydrogen peroxide solution is added to release acridinium chemiluminescence signal. The intensity of the chemiluminescence, measured as relative light unit (RLU), is proportional to the amount of anti- HDV antibody in the sample.
- RLU relative light unit
- the ARCHITECT® HDV Total Ig results are reported as the ratio of the sample RLU to the cutof RLU (S/CO) for each specimen. Specimens with S/CO values ⁇ 1.00 are considered non-reactive; specimens with S/CO values ⁇ 1.00 are considered reactive.
- D37 and D46 HD Ags were identified as potential candidates as detection antigen for the direct ARCHITECT® HDV Total Ig Assay.
- Table 1 Evaluation of engineered HD proteins as detection antigens paired with the full-length HD capture antigen in direct format HDV Total Ig prototype assay.
- Example 6 Performance of the direct ARCHITECT® HDV Total Ig Assay and Agreement with HDV RNA test Performance of the direct ARCHITECT® HDV Total Ig assay with the FL HD LAg as capture antigen and D37 HD LAg as detection antigen was further evaluated using an expanded HDV panel containing 132samples with previously determined HBV and HCV status as listed in Table 2.
- the panel included101 samples with and 31samples without HBV infection.17of the 101 HBV infected samples were coinfected with HCV. Among the 31HBV negative samples, 21samples had HCV mono-infection while 10samples were not infected with either HBV or HCV.
- the direct ARCHITECT® HDV Total Ig assay detected 70/101 HBV+ and HBV+/HCV+ samples, it also detected 2/21HBV-/HCV+ but 0/10HBV- /HCV-samples.
- the indirect LIAISON®XL Anti-HDV assay also referred to herein as DiaSorinanti-HDV IgG+
- DiaSorinanti-HDV IgG+ detected 79/101 HBV+ and HBV+/HCV+ samples and 9/21 HBV-/HCV+
- 6/10HBV-/HCV-samples indicating poor specificity of the assay (Table 2).Only 62of the 101 HBV positive samples had detectable HDV RNA viral load (0.34- 7.82 log10 IU/ml) and RNA was not detected in the 31HBV negative samples.
- Example 7 Potential utility of ARCHITECT HDV Total Ig for assessing disease progression of HDV coinfection and superinfection
- Another significant advantage of the direct ARCHITECT HDV Total Ig is its enhanced dynamic range of assay signal, which may provide potential utility to assess disease progressionof HDV-HBV coinfection and superinfection.
- Signal ploting of the 62HDV RNA positivesamples in ARCHITECT HDV Total Ig and LIAISON Anti-HDV assays against HDV RNA viral load revealed up to 80-fold higher signal of the direct ARCHITECT HDV Ig than LIAISON Anti-HDV for most RNA positive samples (Fig.3).
- the enhanced dynamic range enabled the assay to generate much higher signal for 8samples (193-1150 S/CO), separating them from the 6samples (2.1-19.3 S/CO) within the same low viral load range (0.53-1.9 log10 IU/ml, Fig.3).
- This reactivity patern high antibody response with low viral load
- This reactivity patern is halmark of spontaneously clearing HDV RNA based on typical evolution of serological and virological markers in HDV infection(Hughes SA, Wedemeyer H, Harison PM. Hepatitis delta virus. Lancet.2011 Jul 2;378(9785):73-85. doi: 10.1016/S0140-6736(10)61931-9).
- the direct ARCHITECT HDV Ig test may provide additionalinformation useful for assessing disease progression of HDV coinfection and superinfection.
- Both serology assays were able to detect samples with RNA viral load ranged from 0.51-7.82 log10 IU/ml except 6samples.Notably, there were 6RNA positive samples that were missed by LIAISON Anti-HDV and/or ARCHITECT HDV Total Ig, al had low viral load (2.2-34 IU/ml), suggesting they were in the acute HDV infection resulted seronegative window period.
- Clause 1 An antigenic polypeptide comprising a sequence having at least 90% sequence identity to the sequence of SEQ ID NO: 8, wherein said antigenic polypeptide lacks a sequence comprising at least 90% sequence identity to a region of from about 12 to about 60 amino acids at the N-terminus region of SEQ ID NO: 1.
- Clause 2 The antigenic polypeptide of clause 1, wherein said antigenic polypeptide lacks a sequence comprising at least 90% sequence identity with a sequence selected from the groupconsisting ofSEQ ID NO: 9, SEQ ID NO: 10, SEQ ID NO: 11; SEQ ID NO: 12; and SEQ ID NO: 13.
- Clause 3 The antigenic polypeptide of clause1, wherein said antigenic polypeptide comprises at least one amino acid mutation as compared to the sequence of amino acids 1 to 60 of SEQ ID NO: 1.
- Clause 4 The antigenic polypeptide of any one of clauses1-3, wherein said antigenic polypeptide comprises a sequence selected from the group consisting of SEQ ID NO: 2, SEQ ID NO: 4, SEQ ID NO: 5, SEQ ID NO: 6, SEQ ID NO: 7 and SEQ ID NO: 8.
- Clause 5 The antigenic polypeptide of clause1, wherein said antigenic polypeptide comprises the sequence of SEQ ID NO: 6 and lacks the sequence of SEQ ID NO: 11.
- Clause 6 The antigenic polypeptide of any one of clauses1-5, wherein the antigenic polypeptide is capable of being bound by an antibody directed to HD protein.
- Clause 7 The antigenic polypeptide of any one of clauses1-6, wherein said antigenic polypeptide is fused to at least one heterologous peptide.
- Clause 8 The antigenic polypeptide of clause7, wherein the heterologous peptide comprises an afinity tag, an epitope tag, a fluorescentprotein, an enzyme or a carier protein.
- Clause 9 An antigenic polypeptide comprising a sequence having at least 90% sequence identity to a truncated polypeptide derived from SEQ ID NO: 1, said truncated polypeptide lacking from 12 to 60 N-terminal amino acids of SEQ ID NO: 1.
- Clause 10 The antigenic polypeptide of clause9, wherein the truncated polypeptide comprises a sequence of at least 158 amino acids of SEQ ID NO: 1.
- Clause 11 An antigenic polypeptide comprising an amino acid sequence having at least 90% sequence identity to any of the ful-length sequences of SEQ ID NO: 2 and SEQ ID NOs: 4- 8, and a heterologous peptide.
- Clause 12 The antigenic polypeptide of clause11, wherein the heterologous peptide comprises an afinity tag, an epitope tag, a fluorescentprotein, an enzyme or a carier protein.
- Clause 13 A composition comprising the antigenic polypeptide of any one ofclauses 1-12.
- Clause 14 The composition of clause13, further comprising a detectable label.
- Clause 15 The composition of clause 14, wherein the detectable label is conjugated to the antigenic polypeptide.
- Clause 16 The composition of clause14 or 15, wherein the detectable label is selected from the group consisting of a fluorescence label, a chemiluminescent label, an enzymatic label, and a particle label.
- Clause 17 The composition of any one of clauses13-16, further comprising a second antigenic polypeptide that comprises the sequence of SEQ ID NO: 1.
- Clause 18 The composition of clause 17,wherein the second antigenic polypeptide comprises a means for binding to a solid support.
- Clause 19 A kit for detecting antibodies against hepatitis D virus (HDV) in a biological sample, comprising the antigenic polypeptide of any one of clauses1-12or the composition of any one of clauses 13-18.
- Clause 20 A kit for detecting antibodies against hepatitis D virus (HDV) in a biological sample, comprising the antigenic polypeptide of any one of clauses1-12 and a second antigenic polypeptide that comprises the sequence of SEQ ID NO: 1.
- Clause 21 A method for detecting antibodies specific for HD protein in a biological sample, wherein an antigenic polypeptide according to any one of clauses 1-12 is used as a binding partner for said HD protein antibodies.
- Clause 22 A method for detecting antibodies specific for HD protein in a biological sample, said method comprising: a) forming an immunoreaction mixture by mixing a biological sample with the antigenic polypeptide according toany one ofclauses 1-12;b) maintaining said immunoreaction mixture for a time period sufficient for alowing antibodies against said antigenic polypeptide present in the biological sample to immunoreact with said antigenic polypeptide to form an immunoreaction product; andc) detecting the presence and/or the concentration of said immunoreaction product.
- Clause 23 The method of clause 22,wherein said immunoreaction is carried out in a double antigen sandwich format comprising:a) adding to said biological sample a first antigenic polypeptide that comprises a means for binding to a solid support and a second antigenic polypeptide that caries a detectable label, said second antigenic polypeptide comprising the antigenic polypeptide of any one of clauses 1-12, wherein said first antigenic polypeptide and said second antigenic polypeptide bind specificaly to the HD protein antibodies, b) forming an immunoreaction mixture comprising the first antigenic polypeptide, the biological sample antibody and the second antigenic polypeptide, wherein the solid support is added before, during or after forming the immunoreaction mixture,c) maintaining said immunoreaction mixture for a time period adequate for HD protein antibodies in the biological sample to immunoreact with said first and second antigenic polypeptides to form an immunoreaction product, andd) detecting the presence of any of said immunoreaction product.
- Clause 24 The method of clause 23, wherein the first antigenic polypeptide comprises a sequence having at least 90% sequence identity to the ful-length sequence of SEQ ID NO: 1.
- Clause 25 The method of clause 23 or 24, further comprising a washing step prior to the detecting step.
- Clause 26 The method of clause 25, wherein the immunoreaction product is bound to the solid support and the washing step comprises removing or separatingsome or al ofthe first antigenic polypeptide, HD protein antibody and second antigenic polypeptide that are not part of the immunoreaction product that is bound to the solid support.
- Clause 27 The method of any one of clauses 23-26, wherein the solid support is selected from the group consisting of acolumn, bead, test tube, microtiter dish,multi-wel plate, microparticle, microsphere, a test stick, a test strip, microchip and membrane.
- Clause 28 The method of any one of clauses 22-27, wherein the biological sample is a body fluid sample from a human subject.
- Clause 29 The method of clause 28, wherein the biological sample is selected from the group consisting of blood, plasma andserum.
- Clause 30 A polynucleotide encoding the antigenic polypeptide of any one of clauses1 to12.
- Clause 31 The polynucleotide of clause 30, further comprising a heterologous regulatory element operably linked to a nucleic acid encoding the antigenic polypeptide.
- Clause 32 The polynucleotide of clause 31, wherein the heterologous regulatory element is a promoter.
- Clause 33 A vector comprising the polynucleotide of any one ofclauses 30-32.
- Clause 34 The vector of clause 33, wherein the vector is a plasmid, naked nucleic acid, phage, viral vector, or virus.
- Clause 35 A cel comprising the polynucleotide of any one of clauses 30 to 32 or the vector of clause33 or 34, or expressing the antigenic polypeptide of any one of clauses1 to 12.
- Clause 36 A method for detecting antibodies against hepatitis D virus (HDV) in a biological sample, comprising performing a HDV serology assay using a direct assay format with a first antigenic polypeptide and a second antigenic polypeptide, wherein the first antigenic polypeptide comprises a ful-length HD protein and the second antigenic polypeptide comprises an N-terminaly truncated fragment of the ful-length HD protein.
- Clause 37 The method of clause 36, wherein the assay is a double antigen sandwich assay.
- Clause 38 The method of clause36 or 37, wherein the second antigenic polypeptide lacks from 12 to 60 N-terminal amino acids of the HD protein.
- Clause 39 The method of clause 38, wherein the second antigenic polypeptide lacks 21, 37 or 46 consecutive N-terminal amino acids of the HD protein.
- Clause 40 The method of any one of clauses 36-39, wherein the ful-length HD protein comprises an amino acid sequence having at least 95% sequence identity to the ful-length sequence of SEQ ID NO: 1.
- Clause 41 A method for detecting antibodies against hepatitis D virus (HDV) in a biological sample, comprising:a) contacting abiological sample, either simultaneously or sequentialy, in any order with:(1) a first antigenic polypeptide under conditions adequate to alow binding of anyHDV antibody present in the biological sample to the first antigenic polypeptide, wherein said first antigenic polypeptide is immobilized on a solid support, and (2) a second antigenic polypeptide under conditions adequate to alow binding of the second antigenic polypeptide to any HDV antibody present in the biological sample, such that a first antigenic polypeptide-HDV antibody-second antigenic polypeptide complex is formed and is bound to the solid support, and b) detecting the presence of said first antigenic polypeptide-HDV antibody- second antigenic polypeptide complex, wherein said first antigenic polypeptide comprises a sequence having at least 90% sequence identity to the ful-length sequence of SEQ ID NO: 1; wherein said second antigenic polypeptide comprises a sequence having at least 90%
- Clause 42 The method of clause 41, wherein the second antigenic polypeptide is selected from the group consisting of:i) a polypeptide that comprises the sequence of SEQ ID NO: 8and lacks the sequence of SEQ ID NO:13;i) a polypeptide that comprises the sequence of SEQ ID NO: 7and lacks the sequence of SEQ ID NO: 12;ii) a polypeptide that comprises the sequence of SEQ ID NO: 6and lacks the sequence of SEQ ID NO:11;iv) a polypeptide that comprises the sequence of SEQ ID NO: 5and lacks the sequenceof SEQ ID NO:10;v) a polypeptide that comprises the sequence of SEQ ID NO: 4and lacks the sequence of SEQ ID NO:9;and vi) a polypeptide that comprises the sequence of SEQ ID NO: 2.
- Clause 43 The method of clause 41 or 42, further comprising a washing step prior to the detecting step.
- Clause 44 The method of clause 43, wherein the washing step comprises removing some or al of the first antigenic polypeptide, HDV antibody and second antigenic polypeptide that are not part of the first antigenic polypeptide-HDV antibody-second antigenic polypeptide complex that is boundto the solid support.
- Clause 45 The method of any one of clauses 41-44, wherein the step of detecting comprises detecting the presence and/or the concentration of the first antigenic polypeptide-HDV antibody-second antigenic polypeptide complex that is formed and is bound to the solid support.
- Clause 46 The method of any one of clauses 41-44, wherein the step of detecting comprises measuring an amount of saidfirst antigenic polypeptide-HDV antibody-second antigenic polypeptide complex that is formed and is bound to the solid support.
- Clause 47 The method of any one of clauses 41-46, wherein the solid support is selected from the group consisting of acolumn, bead, test tube,microtiter dish,multi-wel plate, microparticle, microsphere, a test stick, a test strip, microchip and membrane.
- Clause 48 The method of any one of clauses 41-47, wherein the biological sample is selected from the group consisting of blood, plasma andserum.
- Clause 49 A method of detecting hepatitis D virus (HDV) infection in a subject comprising performing the method of any one of clauses 41-48, wherein the biological sample is a sample taken from the subject and wherein the step of detecting comprises detecting the presence of said first antigenic polypeptide-HDV antibody-second antigenic polypeptide complex that is formed and is bound to the solid support, thereby detecting the presence of past or present HDV infection in said subject.
- the biological sample is a sample taken from the subject and wherein the step of detecting comprises detecting the presence of said first antigenic polypeptide-HDV antibody-second antigenic polypeptide complex that is formed and is bound to the solid support, thereby detecting the presence of past or present HDV infection in said subject.
- Clause 50 A method of estimating HDV incidence in a population, the method comprising, a) providing a set of samples derived from a plurality of individuals within the population over a period of time;b) performing the method of clause 49 on each sample in the set of samples; andc) determining the percentage of recent HDV infections over the period of time; wherein the percentage of recent HDV infections over the period of time provides an estimate of HDV incidence in the population.
- Clause 52 The system of clause 51, wherein thesecond antigenic polypeptide comprises an N-terminaly truncated fragment of a ful-length HD protein.
- Clause 53 The system of clause 51or 52, wherein the ful-length HD protein comprises an amino acid sequence having at least 95% sequence identity the ful-length sequence of SEQ ID NO: 1.
- Clause 54 The system ofany one ofclauses51-53, wherein the second antigenic polypeptide lacks from 12 to 60 N-terminal amino acids of the ful-length HD protein.
- Clause 55 The system of clause 54, wherein the second antigenic polypeptide lacks 21, 37 or 46 consecutive N-terminal amino acids of the ful-length HD protein.
- Clause 56 The systemof clause 52 or 53, wherein the second antigenic polypeptide is selected from the group consisting of: i) a polypeptide that comprises the sequence of SEQ ID NO: 8and lacks the sequence of SEQ ID NO:13; i) a polypeptide that comprises the sequence of SEQ ID NO: 7and lacks the sequence of SEQ ID NO: 12;ii) a polypeptide that comprises the sequence of SEQ ID NO: 6and lacks the sequence of SEQ ID NO:11;iv) a polypeptide that comprises the sequence of SEQ ID NO: 5and lacks the sequence of SEQ ID NO:10;v) a polypeptide that comprises the sequence of SEQ ID NO: 4and lacks the sequence of SEQ ID NO:9;and vi) a polypeptide that comprises the sequence of SEQ ID NO: 2.
- Clause 57 The system of any one of clauses 51-56, wherein the sample is selected from the group consisting of blood, plasma andserum.
- Clause 58 The system of any one of clauses 51-57, wherein the detectable label is selected from the group consisting of a fluorescent label, a chemiluminescent marker, and a chromophore.
- An antigenic polypeptide comprising an amino acid sequence having a formula of: MSRSESKKNRGGX1EEIX2EQX3X4SGX5KKLEELERDLRKVKKKIKKLEDENP WLGNIKGILGKKDKDGEGAPPAKRARTDQMEVDSGPRKRPLRGGFTDKERQDHRRRK ALENKKKQLSGGGKNLSKEEEEELKRLTEEDERRERRVAGPPVGGVNPLEGGSRGAPG GGFVPSMQGVPESPFTRTGEGLDIRGNQGFPWDILFPADPPFSPQSCRPQ(SEQ ID NO:22), wherein X 1 is R or S; X 2 is L or G; X 3 is W or A; X 4 is V or G; and X 5 is R or S, provided that X 1 if S if X 2 is L, X 3 is W, X 4 is V and X 5 is R.
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Immunology (AREA)
- Organic Chemistry (AREA)
- Molecular Biology (AREA)
- Medicinal Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Biochemistry (AREA)
- General Health & Medical Sciences (AREA)
- Urology & Nephrology (AREA)
- Hematology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Biophysics (AREA)
- Communicable Diseases (AREA)
- Virology (AREA)
- Biomedical Technology (AREA)
- Genetics & Genomics (AREA)
- Cell Biology (AREA)
- Biotechnology (AREA)
- Microbiology (AREA)
- Gastroenterology & Hepatology (AREA)
- Food Science & Technology (AREA)
- Physics & Mathematics (AREA)
- Analytical Chemistry (AREA)
- General Physics & Mathematics (AREA)
- Pathology (AREA)
- Peptides Or Proteins (AREA)
Abstract
Provided herein are compositions, kits, systems, and methods for detecting antibodies against hepatitis D virus in a sample and for detecting hepatitis D virus infection in a subject.
Description
IMMUNOASSAYS AND METHODS FOR DIAGNOSING HEPATITIS D VIRUS (HDV) INFECTION RELATED APPLICATION INFORMATION This application claims priority to U.S. Application No.63/590,476filed on November 2, 2023, the contents of which are herein incorporated by reference. FIELD The present disclosure provides compositions, kits, systems,and methods for detecting antibodies against hepatitis D virusin a sample and for detecting hepatitis D virus infection in a subject. BACKGROUND Hepatitis Delta Virus(HDV)infection is a major cause of morbidity and mortality worldwide. HDVis classified as a defective RNA virus requiring Hepatitis B virus (HBV) help to be infectious. Co-infection of HDV/HBV leads to increased mortality over HBV mono- infection. Antibody (total Ig) to HDV is the serological marker of HDV infection and is recommended for screening alhepatitis B surface antigen positive (HBsAg +)patients with liver disease. However, a serology assay for detecting HDV Total Ig is not widely available and curent state of the art assays stil use the less sensitive and less specific indirect assay format. Sensitive and accurate diagnostics are needed to identify people who have been infected with HDV and who might benefit from increased monitoring and treatment. New medications which wil ofer more opportunities for treatment of chronic HDV infection are in development and provide weight to the need to identify cases. Understanding infection status can alow people to take steps to control transmission while understanding prevalence of infection in a population can inform a Ministry of Health or other public health agency to make informed decisions around healthcare.
SUMMARY The present disclosure provides compositions, kits, systems and methods for detecting antibodies against hepatitis D virusin a sample and for detecting hepatitis D virus infection in a subject. In some embodiments, the presently disclosed subject mater providesan antigenic polypeptide comprising a sequence having at least 90% sequence identity to the sequence of SEQ ID NO: 8, wherein said antigenic polypeptide lacks a sequence comprising at least 90% sequence identity to a region of from about 12 to about 60 amino acids at the N-terminus region of SEQ ID NO: 1. In some embodiments, the antigenic polypeptide lacks a sequence comprising at least 90% sequence identity with a sequence selected from the groupconsisting ofSEQ ID NO: 9, SEQ ID NO: 10, SEQ ID NO: 11; SEQ ID NO: 12; and SEQ ID NO: 13. In some embodiments, the antigenic polypeptide comprises at least one amino acid mutation as compared to the sequence of amino acids 1 to 60 of SEQ ID NO: 1.In some embodiments, the antigenic polypeptide comprises a sequence selected from the group consisting of SEQ ID NO: 2, SEQ ID NO: 4, SEQ ID NO: 5, SEQ ID NO: 6, SEQ ID NO: 7 and SEQ ID NO: 8.In some embodiments, the antigenic polypeptidecomprises the sequence of SEQ ID NO: 6 and lacks the sequence of SEQ ID NO: 11. In some embodiments, the antigenic polypeptide comprises an amino acid sequence having a formula of: MSRSESKKNRGGX1EEIX2EQX3X4SGX5KKLEELERDLRKVKKKIKKLEDENP WLGNIKGILGKKDKDGEGAPPAKRARTDQMEVDSGPRKRPLRGGFTDKERQDHRRRK ALENKKKQLSGGGKNLSKEEEEELKRLTEEDERRERRVAGPPVGGVNPLEGGSRGAPG GGFVPSMQGVPESPFTRTGEGLDIRGNQGFPWDILFPADPPFSPQSCRPQ(SEQ ID NO:22), where: X1 is R or S; X2 is L or G; X3 is W or A; X4 is V or G; and X5 is R or S, provided that X1 if S if X2is L, X3is W, X4is V and X5is R. In some aspects, X1is S, X2is G, X3is A, X4is G and X5is S.In some embodiments, theantigenic polypeptide is capable of being bound by an antibody directed to HD protein. In some embodiments, the
antigenic polypeptide is fused to at least one heterologous peptide.In some embodiments, the heterologous peptide comprises an afinity tag, an epitope tag, a fluorescentprotein, an enzyme or a carier protein. In some embodiments, the presently disclosed subject mater provides an antigenic polypeptide comprising a sequence having at least 90% sequence identity to a truncated polypeptide derived from SEQ ID NO: 1, said truncated polypeptide lacking from 12 to 60 N- terminal amino acids of SEQ ID NO: 1.In some embodiments, the truncated polypeptide comprises a sequence of at least 158 amino acids of SEQ ID NO: 1. In some embodiments, the presently disclosed subject mater provides anantigenic polypeptide comprising an amino acid sequence having at least 90% sequence identity to any of the ful-length sequences of SEQ ID NO: 2 and SEQ ID NOs: 4-8, and a heterologous peptide.In some embodiments, the heterologous peptide comprises an affinity tag, an epitope tag, a fluorescentprotein, an enzyme or a carrier protein. In some embodiments, the presently disclosed subject mater provides a composition comprising any of the above-describedantigenic polypeptides. In some embodiments, the composition further comprises a detectable label. In some embodiments, the detectable label is conjugated to the antigenic polypeptide. In some embodiments, thedetectable label is selected from the group consisting of a fluorescence label, a chemiluminescent label, an enzymatic label, and a particle label. In some embodiments, thecomposition further comprises a second antigenic polypeptide that comprises the sequence of SEQ ID NO: 1. In some embodiments, thesecond antigenic polypeptide comprises a means for binding to a solid support. In some embodiments, the presently disclosed subject mater provides a kit for detecting antibodies against hepatitis D virus (HDV) in a biological sample, comprising any of the antigenic polypeptides described above or any of the compositions described above. In some embodiments, the presently disclosed subject mater provides a kitfor detecting antibodies against hepatitis D virus (HDV) in a biological sample, comprising any of the antigenic polypeptides described above and a second antigenic polypeptide that comprises the sequence of SEQ ID NO: 1. In some embodiments, the presently disclosed subject mater provides a method for detecting antibodies specific for HD protein in a biological sample, wherein any of theantigenic polypeptides described aboveis used as a binding partner for said HD protein antibodies.
In some embodiments, the presently disclosed subject mater provides a method for detecting antibodies specific for HD protein in a biological sample, said method comprising: a) forming an immunoreaction mixture by mixing a biological sample with anantigenic polypeptide described above; b) maintaining said immunoreaction mixture for a time period suficient for alowing antibodies against said antigenic polypeptide present in the biological sample to immunoreact with said antigenic polypeptide to form an immunoreaction product; and c) detecting the presence and/or the concentration of said immunoreaction product.In some embodiments, the immunoreaction is caried out in a double antigen sandwich format comprising:a) adding to said biological sample a first antigenic polypeptide that comprises a means for binding to a solid support and a second antigenic polypeptide that caries a detectable label, said second antigenic polypeptide comprising anantigenic polypeptide described above, wherein said first antigenic polypeptide and said second antigenic polypeptide bind specificaly to the HD protein antibodies,b) forming an immunoreaction mixture comprising the first antigenic polypeptide, the biological sample antibody and the second antigenic polypeptide, wherein the solid support is added before, during or after forming the immunoreaction mixture, c) maintaining said immunoreaction mixture for a time period suficient for HD protein antibodies in the biological sample to immunoreact with said first and secondantigenic polypeptides to form an immunoreaction product, andd) detecting the presence of any of said immunoreaction product.In some embodiments, the first antigenic polypeptide comprises a sequence having at least 90% sequence identity to the ful-length sequence of SEQ ID NO: 1. In some embodiments, the method further comprises a washing step prior to the detecting step. In some embodiments, theimmunoreaction product is bound to the solid support and the washing step comprises removing or separating some oral of the first antigenic polypeptide, HD protein antibody and second antigenic polypeptidethat are not part of the immunoreaction product that is bound to the solid support.In some embodiments, the solid support is selected from the group consisting of acolumn, bead, test tube, microtiter dish,multi-wel plate,microparticle, microsphere, a test stick, a test strip, microchip and membrane. In some embodiments, the biological sample is a body fluid sample from a human subject.In some embodiments, the biological sample is selected from the group consisting of blood, plasma andserum. In some embodiments, the presently disclosed subject mater providesa polynucleotide encoding an antigenic polypeptide described above. In some embodiments, the
polynucleotide further comprises a heterologous regulatory element operably linked to a nucleic acid encoding the antigenic polypeptide. In some embodiments, the heterologous regulatory element is a promoter.In some embodiments, the presently disclosed subject mater provides a vector comprising the polynucleotide. In some embodiments, the vector is a plasmid, naked nucleic acid, phage, viral vector, or virus. In some embodiments, the presently disclosed subject mater provides a cel comprising apolynucleotidedescribed above. In some embodiments, the presently disclosed subject mater provides a cel comprising a vector described above. In some embodiments, the presently disclosed subject mater provides a cel expressing anantigenic polypeptidedescribed above. In some embodiments, the presently disclosed subject mater provides a method for detecting antibodies against hepatitis D virus (HDV) in a biological sample, comprising performing a HDV serology assay using a direct assay format with a first antigenic polypeptide and a second antigenic polypeptide, wherein the first antigenic polypeptide comprises a ful- length HD protein and the second antigenic polypeptide comprises an N-terminaly truncated fragment of the ful-length HD protein.In some embodiments, the assay is a double antigen sandwich assay. In some embodiments, the second antigenic polypeptide lacks from 12 to 60 N- terminal amino acids of the HD protein. In some embodiments, the second antigenic polypeptide lacks 21, 37 or 46 consecutive N-terminal amino acids of the HD protein. In some embodiments, the ful-length HD protein comprises an amino acid sequence having at least 95% sequence identity to the ful-length sequence of SEQ ID NO: 1. In some embodiments, the presently disclosed subject mater provides a method for detecting antibodies against hepatitis D virus (HDV) in a biological sample, comprising:a) contacting abiological sample, either simultaneously or sequentialy, in any order with:(1) a first antigenic polypeptide under conditions suficient to alow binding of anyHDV antibody present in the biological sample to the first antigenic polypeptide, wherein said first antigenic polypeptide is immobilized on a solid support, and (2) a second antigenic polypeptide under conditions suficient to alow binding of the second antigenic polypeptide to any HDV antibody present in the biological sample,such that a first antigenic polypeptide-HDV antibody-second antigenic polypeptide complex is formed and is bound to the solid support, and b) detecting the
presence of said first antigenic polypeptide-HDV antibody-second antigenic polypeptide complex,wherein said first antigenic polypeptide comprises a sequence having at least 90% sequence identity to the ful-length sequence of SEQ ID NO: 1;wherein said second antigenic polypeptide comprises a sequence having at least 90% sequence identity to a truncated polypeptide derived from SEQ ID NO: 1, said truncated polypeptide lacking from 12 to 60 N- terminal amino acids of SEQ ID NO: 1; andwherein said second antigenic polypeptide carries a detectable label.In some embodiments, the second antigenic polypeptide is selected from the group consisting of:i) a polypeptide that comprises the sequence of SEQ ID NO: 8 and lacks the sequence of SEQ ID NO: 13;i) a polypeptide that comprises the sequence of SEQ ID NO: 7and lacks the sequence of SEQ ID NO: 12;ii) a polypeptide that comprises the sequence of SEQ ID NO: 6 and lacks the sequence of SEQ ID NO: 11;iv) a polypeptide that comprises the sequence of SEQ ID NO: 5 and lacks the sequence of SEQ ID NO: 10;v) a polypeptide that comprises the sequence of SEQ ID NO: 4 and lacks the sequence of SEQ ID NO: 9; andvi) a polypeptide that comprises the sequence of SEQ ID NO: 2. In some embodiments, the method further comprises a washing step prior to the detecting step. In some embodiments, the washing step comprises removingorseparating some or al of the first antigenic polypeptide, HDV antibody and second antigenic polypeptide that are not part of the first antigenic polypeptide-HDV antibody-second antigenic polypeptide complex that is bound to the solid support.In some embodiments, the step of detecting comprises detecting the presence and/or the concentration of the first antigenic polypeptide-HDV antibody-second antigenic polypeptide complex that is formed and is bound to the solid support. In some embodiments, the step of detecting comprises measuring an amount of saidfirst antigenic polypeptide-HDV antibody-second antigenic polypeptide complex that is formed and is bound to the solid support. In some embodiments, the solid support is selected from the group consisting of acolumn, bead, test tube,microtiter dish,multi-wel plate, microparticle, microsphere, a test stick, a test strip, microchip and membrane.In some embodiments, the biological sample is selected from the group consisting of blood, plasma and serum. In some embodiments, the presently disclosed subject mater provides a method of detecting hepatitis D virus (HDV) infection in a subject comprising performing amethod described above, wherein the biological sample is a sample taken from the subject and wherein the step of detecting comprises detecting the presence of said first antigenic polypeptide-HDV
antibody-second antigenic polypeptide complex that is formed and is bound to the solid support, thereby detecting the presence of past or present HDV infection in said subject. In some embodiments, the presently disclosed subject mater provides method of estimating HDV incidence in a population, the method comprising, a) providing a set of samples derived from a plurality of individuals within the population over a period of time;b) performing the method on each sample in the set of samples; andc) determining the percentage of recent HDV infections over the period of time;wherein the percentage of recent HDV infections over the period of time provides an estimate of HDV incidence in the population. In some embodiments, the presently disclosed subject mater provides a system comprising:a) a sample receiving component configured to receive a sample from a subject, wherein the sample comprises or is suspected of comprising a hepatitis D virus (HDV) antibody; b) a first antigenic polypeptide and a second antigenic polypeptide configured to make contact with the sample to form a first antigenic polypeptide-HDV antibody-second antigenic polypeptide complex, wherein the first antigenic polypeptide comprises a ful-length HD protein and the second antigenic polypeptide comprises amutated and/or truncated HD protein, and wherein the second antigenic polypeptide caries a detectable label;c) a detection component configured to measure a signal generated by the detectable label in the first antigenic polypeptide-HDV antibody-second antigenic polypeptide complex; andd) an output component that indicates an amount of HDV antibody in the sample based on the signal. In some embodiments, the second antigenic polypeptide comprises an N-terminaly truncated fragment of the ful-length HD protein. In some embodiments, the ful-length HD protein comprises an amino acid sequence having at least 95% sequence identity the ful-length sequence of SEQ ID NO: 1. In some embodiments, the second antigenic polypeptide lacks from 12 to 60 N-terminal amino acids of the ful-length HD protein.In some embodiments, the second antigenic polypeptide lacks 21, 37 or 46 consecutive N-terminal amino acids of the ful-length HD protein. In some embodiments, the second antigenic polypeptide is selected from the group consisting of: i) a polypeptide that comprises the sequence of SEQ ID NO: 8 and lacks the sequence of SEQ ID NO: 13;i) a polypeptide that comprises the sequence of SEQ ID NO: 7and lacks the sequence of SEQ ID NO: 12;ii) a polypeptide that comprises the sequence of SEQ ID NO: 6 and lacks the sequence of SEQ ID NO: 11;iv) a polypeptide that comprises the sequence of SEQ ID NO: 5 and lacks the sequence of SEQ ID NO: 10;v) a polypeptide that comprises the sequence of SEQ
ID NO: 4 and lacks the sequence of SEQ ID NO: 9; andvi) a polypeptide that comprises the sequence of SEQ ID NO: 2.In some embodiments, the sample is selected from the group consisting of blood, plasma andserum.In some embodiments, the detectable label is selected from the group consisting of a fluorescent label, a chemiluminescent marker, and a chromophore. Section headings as used in this section and the entire disclosure herein are merely for organizational purposes and are not intended to be limiting. BRIEF DESCRIPTION OF THE DRAWINGS FIG.1: Representative schematic diagram ilustratingprotein sequence alignment of ful-length HD Large Antigen (HD LAg), SmalAntigen (HD SAg), HD LAg mutant (HD LAg M) and 5 truncated HD Ags (D21-D56). FIG.2: Representative schematic diagrams of ARCHITECT Indirect HDV IgG (FIG. 2A) and Direct HDV Total Ig (FIG.2B) assay formats. hv = chemiluminescent signal; HD Ag or HDAg= recombinant HD antigen. FIG.3: An evaluation of the corelation between signal of HDV Antibody assays and HDV RNA viral load on 70HDV infected patient samples. R2 = 0.35 for Architect HDV and R2 = 0.26 for LIAISON Anti-HDV.The circle highlightsthe 8samples with high signal (193-1150 S/CO) and the rectangle showsthe 6samples with low signal (2.1-19.3 S/CO) in Architect HDV Ig within the same low viral load range (0.53-1.9 log10 IU/ml). Orange square indicatesthe 6 missed samples by both assays. FIG.4: Specificity and signal distribution of HDV antibody assays with 200 US normal blood donors. DETAILED DESCRIPTION 1.Definitions Unless otherwise defined, al technical and scientific terms used herein have the same meaning as commonly understood by one of ordinary skil in the art. In case of conflict, the present document, including definitions, wil control. Various embodiments of themethods and materials are described below, although methods and materials similar or equivalent to those described herein can be used in practice or testing of the present disclosure. Al publications, patent applications, patents and other references mentioned herein are incorporated by reference in their entirety. The materials, methods, and examples disclosed herein are ilustrative only and not intended to be limiting.
As used herein, the term “antibody”refers to an immunoglobulin molecule or immunologicaly active portion thereof, namely, an antigen-binding portion. An antibodyis a protein (or protein complex) that includes one or more polypeptides substantialy encoded by immunoglobulin genes or fragments of immunoglobulin genes. The recognized immunoglobulin genes include the kappa, lambda, alpha, gamma, delta, epsilon, and mu constant region genes, as wel as the myriad of immunoglobulinvariable region genes. Lightchains are classified as either kappa or lambda. Heavy chains are classified as gamma, mu,alpha, delta, or epsilon, which in turn define the immunoglobulinclasses, IgG, IgM, IgA, IgD and IgE, respectively.Antibodies are evoked in humans or other animals by a specificantigen (immunogen). Antibodies are characterized byreacting specificalywith the antigen in some demonstrable way,antibody and antigen each being defined in terms of theother. “Eliciting an antibody response”refers to the abilityof an antigen or other molecule to induce the production ofantibodies. As used herein, the term“antigen”refers to a molecule, moiety, foreign particulate mater, or an alergenthat can bind to a specific antibody or T-cel receptor.An antigencan stimulate the production of antibodies or a T-cel response inan animal. The term “antigenic polypeptide”refers to a polypeptide that binds specificaly to antibodies that recognize the polypeptide. As used herein, an “antigen-specific”antibody is an antibody that was elicited (produced and/oractivated) in response to a particular antigen. An “antigen-specific”antibody is capable ofbinding to the antigen,typicaly with high afinity. The “area under curve”or “AUC”refers to area under a ROC curve. AUC under a ROC curve is a measure of accuracy. An AUC of 1 represents a perfect test, whereas an AUC of 0.5 represents an insignificant test. A prefered AUC may be at least approximately 0.700, at least approximately 0.750, at least approximately 0.800, at least approximately 0.850, at least approximately 0.900, at least approximately 0.910, at least approximately 0.920̧ at least approximately 0.930, at least approximately 0.940, at least approximately 0.950, at least approximately 0.960, at least approximately 0.970, at least approximately 0.980, at least approximately 0.990, or at least approximately 0.995. “Bead”and “particle”are used herein interchangeably and refer to a substantialy spherical solid support. One example of a bead or particle is a microparticle. Microparticles that can be used herein can be any type known in the art. For example, the bead or particle can be a
magnetic bead or magnetic particle. Magnetic beads/particles may be feromagnetic, ferimagnetic, paramagnetic, superparamagnetic or ferrofluidic. Exemplary feromagnetic materials include Fe, Co, Ni, Gd, Dy, CrO2, MnAs, MnBi, EuO, and NiO/Fe. Examples of ferimagnetic materials include NiFe2O4, CoFe2O4, Fe3O4(or FeO·Fe2O3). Beads can have a solid core portion that is magnetic and besurrounded by one or more non-magnetic layers. Alternately, the magnetic portion can be a layer around a non-magnetic core. The microparticles can be of any size that would work in the methods described herein, e.g.,from about 0.75 to about 5 nm, or from about 1 to about 5 nm, or from about 1 to about 3 nm. As used herein, a “carier protein”is a protein that functions to facilitate expression from a host cel. A carier protein may facilitate soluble expression or promote the formation of inclusion bodies in the host cel. Non-limiting examples of carrier proteins include thioredoxin and GST(glutathione transferase). In some embodiments, the carier/peptide junction contains an enzymatic or chemical cleavage site that enables the peptide to be released by the coresponding method. The term “conjugated”refersto two molecules that are bondedtogether, for example by covalent bonds. “Control”as used herein generaly refers to a reagent whose purpose is to evaluate the performance of a measurementmethod orsystem in order toassure that it continues to produce results within permissible boundaries (e.g., boundaries ranging from measures appropriate for a research use assay on one end to analytic boundaries established by quality specifications for a commercial assay on the other end). To accomplish this, a control should be indicative of patient results and optionaly should somehow assess theimpact of eror on the measurement (e.g., eror due to reagent stability, calibrator variability, instrument variability, and the like). The term “detectable label,”as used herein, refers to a moiety or compound thatcan be used to provide a detectable and/or quantifiable signal. In some cases, the label can be atached, directly or indirectly, to a nucleic acid or protein.In some embodiments, the detectablelabel generates a signal whichcan be measured and whose intensity is related to (e.g., proportional to) the amount of entitybound thereto. Suitable labels that can be atached to a nucleic acid or protein include, but are not limitedto, radioisotopes, fluorophores, chromophores, mass labels, electron dense particles, magnetic particles, spin labels, molecules that emit chemiluminescence,
electrochemicaly active molecules, enzymes, cofactors, and enzyme substrates. Additional examples of suitable detectable labels are provided below. The term “derived from,”as used herein with reference to proteins and polypeptides (and sequences thereof) refers to various modifications, analogs, and products based upon an original protein or polypeptide (and sequence thereof) from which a derivative protein or polypeptide is derived, for example, based on the protein and polypeptide sequences disclosed herein. A polypeptide that is derived from an original protein or polypeptide (and sequence thereof) may be a truncated or augmented sequence, a sequence comprising at least one mutation relative to the original sequence, or some other modified sequence. As used herein, the term “heterologous”in reference to an element (e.g., a sequence element, signal sequence, regulatory element, polypeptide, gene, etc.) refers to an element that is not in its natural environment. For example, a heterologous element includes an element from one species introduced into another species. A heterologous element also includes an element native to an organism that has been altered in some way (e.g., mutated, added in multiple copies, linked to non-native regulatory sequences, etc.). Heterologous elements are distinguished from endogenous elements in that the heterologous element sequences are typicaly joined to sequences that are not found naturaly associated with the element sequences in the chromosome or are associated withportions of the chromosome not found in nature (e.g., genes expressed in loci where the gene is not normaly expressed). The term “host cel”means any cel that harbors or is susceptible to harboring foreign molecules, viruses, or microorganisms. It may also be a cel that has been introduced with or is susceptible to being introduced with a foreign (i.e., heterologous) nucleic acid molecule. In some embodiments, a host cel is any cel type that is susceptible to transformation, transfection, transduction, or the like with a nucleic acid construct or expression vector of the present disclosure. The term “host cel”encompasses any progeny of a parent cel that is not identical to the parent cel due to mutations that occur during replication. The term “immunoreact”is used herein to describe the reaction of an antibody with a polypeptide (e.g., antigen, antigenic polypeptide) recognized by the antibody. An antibody that immunoreacts with a polypeptide (e.g., antigenic polypeptide) specificaly binds to said polypeptide.
As used herein, the term “mutation”refers to a change and/or alteration. In some embodiments, mutations may be changes and/or alterations to proteins (including peptides and polypeptides) and/or nucleic acids (including polynucleic acids). In some embodiments, mutations comprise changes and/or alterations to a protein and/or nucleic acid sequence. Such changes and/or alterations may comprise the addition, substitution and/or deletion of one or more amino acids (in the case of proteins and/or peptides) and/or nucleotides (in the case of nucleic acids and/or polynucleic acids). In some embodiments, wherein mutations comprise the addition and/or substitution of amino acids and/or nucleotides, such additions and/or substitutions may comprise 1 or more amino acid and/or nucleotide residues and may include modified amino acids and/or nucleotides. The resulting construct, molecule or sequence of a mutation, change or alteration may be refered to herein as amutant. The “N-terminus region”refers to the region at or near the end of a polypeptide chain or protein where the first amino acid resides. Proteins are composed of linear chains of amino acids linked together by peptide bonds, forming a primary structure. The sequence of amino acids in this chain is read from the N-terminus to the C-terminus. The N-terminus (also known as the amino-terminus, NH2-terminus, N-terminal end or amine-terminus) is the start of a protein or polypeptide, refering to the free amine group (-NH2) located at the end of a polypeptide. Within a peptide, the amine group is bonded to the carboxylic group of another amino acid, making it a chain. That leaves a free carboxylic group at one end of the peptide, caled the C-terminus, and a free amine group on the other end caled the N-terminus. As used herein, the length of the N- terminus region is from at least one amino acid in length up to the length equal to 50% of the ful-length of the protein or polypeptide chain itself. In some embodiments,the amino acid length of the N-terminus region is up to the length equal to 40%, 30%, 20%,10% or 5% of the ful-length of the protein or polypeptide itself. In some embodiments, the region begins at the first amino acid in the polypeptide chain or protein. In some embodiments, the region begins somewhere between the first amino acid and the amino acid positioned at 50% of the length of the protein. In some embodiments, an N-terminus region of the present disclosure is the region of amino acids 1 to 60 of SEQ ID NO: 1 (i.e., from the 1stamino acid to the 60thamino acid of SEQ ID NO: 1, including the 1stand 60thamino acids). A “polypeptide”is a polymer in which themonomers are aminoacid residues which are joined together through amidebonds. When the amino acids are alpha-amino acids, eitherthe
L-optical isomer or the D-optical isomer can be used.As used herein, the term “polypeptide”is used interchangeably with the terms“peptide”and “protein.”The terms “polypeptide”or “protein”as used herein areintended to encompass any amino acid sequence and include modified sequences such as glycoproteins. The term “polypeptide”is specificaly intended to cover naturaly occuring proteins, as wel as those which are recombinantly or syntheticaly produced. The term “residue”or “amino acidresidue”includes reference to an amino acid that is incorporatedinto a protein, polypeptide, or peptide. A “receiver operating characteristic”curve or “ROC”curve refers to a graphical plot that ilustrates the performance of a binary classifier system as its discrimination threshold is varied. For example, an ROC curve can be a plot of the true positive rate against the false positive rate for the different possible cutof points of a diagnostic test. It is created by ploting the fraction of true positives out of the positives (TPR = true positive rate) vs. the fraction of false positives out of the negatives (FPR = false positive rate), at various threshold setings. TPR is also known as sensitivity, and FPR is one minus the specificity or true negative rate. The ROC curve demonstrates the tradeof between sensitivity and specificity (any increase in sensitivity wil be accompanied by a decrease in specificity); the closer the curve folows the left-hand border and then the top border of the ROC space, the more accurate the test; the closer the curve comes to the 45-degree diagonal of the ROC space, the less accurate the test; the slope of the tangent line at a cutoff point gives the likelihood ratio (LR) for that value of the test; and the area under the curve is a measure of test accuracy. A “solid support”is any inert material having a rigid or semirigidsurface. In the context of the present disclosure, thesolid support is capable of binding directly or indirectly toa polypeptide (e.g., an antigenic polypeptide that is a capture moiety).The solid support can have any shape, form or size (forexample, plate, sheet, tube, stick or particle). In someembodiments herein, the solid support is a multi-wel plate(also refered to as a microtiter or microwel plate), membrane,glass, metal, bead, microsphere, test tube, test stick,test strip, porous matrix or resin. In someembodiments herein, the solid support is amicroparticle.In some examples, the solid support includes polystyrene,polyethylene or polypropylene. In general, a “nanoparticle”refers to any particle having a diameter of less than 1000 nm. In certain prefered embodiments, nanoparticles of the disclosure have a greatest dimension (e.g., diameter) of 500 nm or less. In other prefered embodiments, nanoparticles of the
disclosure have a greatest dimension ranging between 25 nm and 200 nm. In other prefered embodiments, nanoparticles of the disclosure have a greatest dimension of 100 nm or less. In other prefered embodiments, nanoparticles of the disclosure have a greatest dimension ranging between 35 nm and 60 nm. As used herein, the phrase “operably linked”refers to a functional connection between two or more molecules, constructs, transcripts, entities, moieties or the like. As used herein, the term “sequence identity”refers to the degree two polymer sequences (e.g., peptide, polypeptide, nucleic acid, etc.) have the same sequential compositionof monomer subunits. The “percent sequence identity”can be determined with the aid of readily available sequence comparison programs. These available computer programs may calculate percent (%) sequence identity between two or more sequences and may also calculate the sequence identity shared by two or more amino acid or nucleic acid sequences. As an example, the “percent sequence identity”is calculated by: (1) comparing two optimaly aligned sequences over a window of comparison (e.g., the length of the longer sequence, the length of the shorter sequence, a specified window), (2) determining the number of positions containing identical (or similar) monomers (e.g., same amino acids occurs in both sequences) to yield the number of matched positions, (3) dividing the number of matched positions by the total number of positions in the comparison window (e.g., the length of the longer sequence, the length of the shorter sequence, a specified window), and (4) multiplying the result by 100 to yield the percent sequence identity. For example, if peptides A and B are both 20 amino acids in length and have identical amino acids at al but 1 position, then peptide A and peptide B have 95% sequence identity. As another example, if peptide C is 20 amino acids in length and peptide D is 15 amino acids in length, and 14 out of 15 amino acids in peptideD are identical to those of a portion of peptide C, then peptides C andD have 70% sequence identity, but peptideD has 93.3% sequence identity to an optimal comparison window of peptide C. For the purpose of calculating “percent sequence identity”herein, any gaps in aligned sequences are treated as mismatches at that position.In some embodiments, if a window of comparison is not specified and a specific sequence identifier is indicated (i.e., an assigned SEQ ID NO is indicated), then the sequence identity is calculated with respect to the ful-length sequence coresponding to that sequence identifier. For example, unlessotherwise indicated herein orclearly contradicted by context, the phrases“a sequence has at least 90% sequence identity to SEQ ID NO: 1”and “a sequence has at
least 90% sequence identity to the sequence of SEQ ID NO: 1” arethe same as the phrase “a sequence has at least 90% sequence identity tothe ful-length sequence ofSEQ ID NO: 1.” As used herein, the term “sample”is used in the broadest sense and generaly refers to a biological material being tested for and/or suspected of containing an analyte of interest, such as a target antibody described herein. Thesample may be derived from any biological source, such as, a physiological fluid, including, but not limited to, whole blood, serum, plasma, interstitial fluid, saliva, ocular lens fluid, cerebral spinal fluid, sweat, urine, milk, ascites fluid, mucous, nasal fluid, sputum, synovial fluid, peritoneal fluid, vaginal fluid, menses, amniotic fluid, semen and so forth. In some embodiments, the sample is a whole blood sample. In some embodiments, the sample is a plasma sample. In yet other embodiments, the sample is a serum sample. The test sample may be used directly as obtained from the biological source or folowing a pretreatment to modify the character of the sample. For example, such pretreatment may include preparing plasma from blood, diluting viscous fluids and so forth. Methods of pretreatment may also involve filtration, precipitation, dilution, distilation, mixing, concentration, inactivation of interfering components, the addition of reagents, lysing, etc. Moreover, it may also be beneficial to modify a solid test sample to form a liquid medium or to release the analyte. “Reference level”or “reference value”as used herein refers to an assayorcutof value that is used to assess diagnostic(“diagnostic”cutof), prognostic, or therapeutic eficacy and that has been linked or is associated herein with various clinical parameters (e.g., presence of disease such as, for example, to rule a subject as having a disease (“rule in”) or rule a subject as not having a disease (“rule out”), stage of disease, severity of disease, progression, non-progression, or improvement of disease, etc.) However, it is wel-known that reference levels may vary depending on the nature of the immunoassay (e.g.,such as, in an immunoassay, the antigens or antibodies employed, reaction conditions, sample purity, etc.) and that assays can be compared and standardized. It further is wel within the ordinary skil of one in the art to adapt the disclosure herein for other assays to obtain assay-specific reference levels for those other immunoassaysbased on the description provided by this disclosure. Whereas the precise value of the reference level may vary between assays, the findings as described herein should be generaly applicable and capable of being extrapolated to other assays.
A “reagent”refers broadly to any agent used in a reaction, other than the analyte (e.g., polypeptideor antibodybeing analyzed). Ilustrative reagents for enzyme reactions include, for example, substrates, cofactors, bufer, metal ions, inhibitors, and activators. Reagents for immunoassay include, for example, antibodies specific for a target marker, detection (e.g., labeled) antibodies, antigenic polypeptides, capture antigens, detection antigens, controls, bufers, and the like. The use of the terms “a”and “an”and “the”and “at least one”and similar references are tobe construed to cover both the singular and the plural, unless otherwise indicated herein or clearly contradicted by context. The use of the term “at least one”folowed by a list of one or more items (for example, “at least one of A and B”) is to be construed to mean one item selected from the listed items (A or B) or any combination of two or more of the listed items (A and B), unless otherwise indicated herein orclearly contradicted by context. The terms “comprise(s),”“include(s),”“having,”“has,”“can,”“contain(s),”and variants thereof, as used herein, are intended to be open-ended transitional phrases, terms, or words that do not preclude the possibility of additional acts or structures. The phrase “consisting essentialy of”also is construed to be an open-ended phrase meant to include steps or materials which do notmaterialy afect the basic and novel characteristics of a described product or method. Thephrase “consisting of”is construed to be a closed phrase which excludes any element, step, oringredient notexplicitly specified in the specification or claims. Recitation of ranges of valuesherein are merely intended to serve as a shorthand method of refering individualy to eachseparate value faling within the range, unless otherwise indicated herein, and each separatevalue is incorporated into the specification as if it were individualy recited herein. Al methodsdescribed herein can be performed in any suitable order unless otherwise indicated herein orotherwise clearly contradicted by context. The use of any and al examples, or exemplarylanguage (e.g., “such as”) provided herein, is intended merely to beter iluminate the inventionand does not pose a limitation on the scope of the invention unless otherwise claimed. Nolanguage in the specification should be construed as indicating any non-claimed element as essential to the practice of the invention. The present disclosure also contemplates other embodiments “comprising,” “consisting of”and “consisting essentialy of,”the embodiments or elements presentedherein, whether explicitly set forth or not.
For the recitation of numeric ranges herein, each intervening number there between with the same degree of precision is explicitly contemplated. For example, for the range of 6-9, the numbers 7 and 8 are contemplated in addition to 6 and 9, and for the range 6.0-7.0, the number 6.0, 6.1, 6.2, 6.3, 6.4, 6.5, 6.6, 6.7, 6.8, 6.9, and 7.0 are explicitly contemplated. “Sensitivity”refers to the proportion of subjects for whom the outcome is positive that are corectly identified as positive (e.g., corectly identifying those subjects with a disease or medical condition for which they are being tested). For example, this might include corectly identifying subjects as having an HDVinfection from those who do not have an infection. “Specificity”of an assay as used herein refers to the proportion of subjects for whom the outcome is negative that are corectly identified as negative (e.g., correctly identifying those subjects who do not have a disease or medical condition for which they are being tested). For example, this might include corectly identifying subjects not having an HDVinfection from those who do have an HDV infection. “Subject”and “patient”as used herein interchangeably refers to any vertebrate, including, but not limited to, a mammal and a human. In some embodiments, the subject may be a human or a non-human. The subject or patient may be undergoing forms of treatment. “Mammal”as used herein refers to any member of the class Mammalia, including, without limitation, humans and nonhuman primates such as chimpanzees and other apes and monkey species; farm animals such as catle, sheep, pigs, goats, lamas, camels,and horses; domestic mammals such as dogs and cats; laboratory animals including rodents such as mice, rats, rabbits, guinea pigs, and the like. The term does not denote a particular age or sex. Thus, adult and newborn subjects, as wel as fetuses, whether male or female, are intended to be included within the scope of this term. “Treat,”“treating”or “treatment”are each used interchangeably herein to describe reversing, aleviating, or inhibiting the progress of a disease and/or injury, or one or more symptoms of such disease, to which such term applies. Depending on the condition of the subject, the term also refers to preventing a disease, and includes preventing the onset of a disease, or preventing the symptoms associated with a disease. A treatment may be either performed in an acute or chronic way.The term also refers to reducing the severity of a disease or symptoms associated with such disease prior to afliction with the disease. Such prevention or reduction of the severity of a disease prior to affliction refers to administration of a
pharmaceutical composition to a subject that is not at the time of administration aflicted with the disease. “Preventing”also refers to preventing the recurrence of a disease or of one or more symptoms associated with such disease. As used herein, a “truncated polypeptide”refers to a shortened molecule or sequence that has been shortened by removing a portion of it. Unless otherwise defined herein, scientific and technicalterms used in connection with the present disclosure shal havethe meanings that are commonly understood by those of ordinaryskil in the art. For example, any nomenclatures used in connection with, andtechniques of, cel and tissue culture, molecular biology,immunology, microbiology, genetics and protein and nucleicacid chemistry and hybridization described herein are thosethat are wel known and commonly used in the art. The meaning and scope of the terms shouldbe clear; in the event, however of any latent ambiguity,definitions provided herein take precedent over any dictionaryor extrinsic definition. Further, unless otherwise requiredby context, singular terms shal include pluralities andplural terms shal include the singular. 2.Hepatitis Delta Virus (HDV) The hepatitis delta virusesare negative-sense single-stranded RNA viruses (or virus- like particles) classified together as the genus Deltavirus. Hepatitisdelta virus(HDV) is one of five known hepatitis viruses: A, B, C, D, and E.HDV is considered a defective virus because it is unable to replicate on its own. HDV requires infection of the same cel with Hepatitis B virus (HBV), which contributes to virion assembly and infectivity. HDV only occurs in people who are also infected with the hepatitis B virus. Transmission of HDV can occur either via simultaneous infection with HBV (coinfection) or superimposed on chronic hepatitis B or hepatitis B carier state (superinfection). The HDV viral envelope contains host phospholipids, as wel as three proteins taken from the hepatitis B virus (the large, medium, and smal hepatitis B surface antigens). The viral envelope surounds an inner ribonucleoprotein (RNP) particle, which contains the genome surounded by about 200 molecules of hepatitis D antigen (HDAg) for each genome. Hepatitis D antigen (abbreviated herein as “HDAg”or “HD Ag”) is the only protein that is produced by the HDV virus.Throughout the present disclosure, Hepatitis D antigen may also be refered to as “HD protein”. Hepatitis D antigencomes in two forms: an approximately 27kDa HD large antigen (abbreviated herein as “large-HDAg”or “HD LAg”)and an
approximately24kDa smal-HDAg. The N-terminals of the two forms are identical,butthey difer by19 more amino acids in the C-terminal of the large-HDAg.In some embodiments of the present disclosure, a consensus amino acid sequence of several HD large antigen sequences is used as a representative HD protein sequence and has the folowing sequence: MSRSESKKNRGGREEILEQWVSGRKKLEELERDLRKVKKKIKKLEDENPWLGNIKGILG KKDKDGEGAPPAKRARTDQMEVDSGPRKRPLRGGFTDKERQDHRRRKALENKKKQLS GGGKNLSKEEEEELKRLTEEDERRERRVAGPPVGGVNPLEGGSRGAPGGGFVPSMQGV PESPFTRTGEGLDIRGNQGFPWDILFPADPPFSPQSCRPQ (SEQ ID NO: 1). Hepatitis D can be an acute, short-term infection or become a long-term, chronic infection. Hepatitis D can cause severe symptoms and serious ilness that can lead to life-long liver damage and even death. People can become infected with both hepatitis B and hepatitis D viruses at the same time (known as “coinfection”) or get hepatitis D after first being infected with the hepatitis B virus (known as “superinfection”).Superinfections can occur when someone who already has chronic hepatitis B becomes infected with hepatitis D—thesetypes of infections are more common, and have a 70-90% chance of resulting in a chronic infection of both hepatitis B and D. Coinfections can occur when someone becomes infected with hepatitis B and delta at the same time, andhave less than a 5% chance of resulting in chronic infections.Persisting HDV infection leads tohigher risk of developing liver cirhosis and hepatocelular carcinoma than HBV infection alone. The global estimates of HDV infection vary between 12 and 74 milion. Such widely variable results highlight the chalenges of accurately estimating the global burden of HDV due to lack of epidemiological data in many countries, heterogeneous prevalence indiferent geographic areas and populations as wel as use of suboptimum diagnostic tests. Antibodies against the HDAg, including IgM and IgG, are the serological marker of HDV infection.Antibody testing is widely used as a primary screening test for HDV infection and recommended for universal screening of al HbsAg-positive individuals. Diagnosis of HDV can be established by detecting HDV antigen, HDV-specific IgM, or HDV-specific total antibodies (combined IgM and IgG) in the sera of infected patients with clinicaly evident acute or chronic hepatitis B. Anti-HDV IgM typicaly appears inserum at 2 to 3 weeks after onset of symptoms and disappears by 2 months after acute HDV infection, but it
may persist up to 9 months in HDV superinfection. HDV IgG and HDV total antibodies persist in serum after resolution of acute HDV infection and in chronic coinfection. European and Asian-Pacific guidelines recommend universal screening for HDV in al HbsAg-positive patients. A recent study on the implementation of the universal screening in HbsAg-positive individuals found a 5-fold increase in diagnosis of HDV infection. While curent guideline in the United States stil recommend risk-based screening for HDV, it has been suggested to expand the HDV screening eforts to universal screening in al HbsAg-positive individuals based on updated prevalence data revealing a higher-than-expected prevalence. Screening for HDV requires testing for anti-HDV antibodies, which indicate past exposure to the virus or curent infection. If anti-HDV antibodies are present, then active HDV infection is confirmed by measuring hepatitis D RNA levels. The first step is performing the HDV antibody total (anti-HDV total) test. People who have recovered from or are curently infected with hepatitis delta wil have antibodies.If the HDV antibody total test is positive, it should be folowed by the HDV RNA (Qualitative or Quantitative) test to confirm an active infection. Testing for HDV is indicated in those who are hepatitis B surface antigen positive (i.e., those who have had previous or active infection with hepatitis B). Ifliver fibrosis or cirhosis is suspected, a liver biopsy is usualy needed. Curent treatments for chronic hepatitis D include conventional or pegylated interferon alpha therapy. Evidence suggests that pegylated interferon alpha is effective in reducing the viral load and the effect of the disease during the time the drug is given, but the benefit generaly stops if the drug is discontinued. The eficiency of this treatment does not usualy exceed about 20%, and late relapse after therapy has been reported.New treatment options for HDV are on the horizon, including viral entry inhibitors, prenylation inhibitors and virion egress inhibitors. Emerging antiviral therapies include Hepcludex (bulevirtide) to treat hepatitis D. Bulevirtide binds and inactivates the sodium/bile acid cotransporter, blocking hepatitis D virus (as wel as hepatitis B virus) from entering hepatocytes. Bulevirtide may begiven along with pegylated interferon alpha.Other treatments for hepatitis D which are currently under development include pegylated interferon lambda (λ), which binds to receptors on the hepatocyte surface leading to an intracelular signaling cascade via the JAK-STAT signaling pathway and activation of anti-viral cel mediated immunity. The prenylation inhibitor lonafarnib prevents hepatitis D viral particle assembly by inhibiting the farnesylation of the L-HDAg. REP2139-Ca
is a nucleic acid polymer that prevents the release of hepatitis B surface antigen (which is required for assembly of hepatitis D viral particles). 3.Antigenic polypeptides In various embodiments, the present disclosure provides antigenic polypeptides. In some embodiments, the present disclosure provides an antigenic polypeptide comprising a sequencederived from asequence of the Hepatitis D antigen(HD protein)and may be refered to as a “recombinant HD protein”or “engineered HD protein.”In some embodiments, an antigenic polypeptide that is a recombinant HD protein comprises a sequence derived from the sequence of the HD large antigenand may be refered to as an “engineered HD large antigen”(engineered HD LAg).In some embodiments, the sequence of the HD large antigen is derived from a consensus amino acid sequence of several HD large antigen sequences. In some embodiments, the sequence of the HD large antigen of the present disclosure is SEQ ID NO: 1. In some embodiments, the present disclosure provides antigenic polypeptidesthat are recombinant HD proteinshaving sequences derived from a single HD large antigen sequence or a consensus amino acid sequence of several HD large antigen sequences. In some embodiments, the present disclosure provides antigenic polypeptides (recombinant HD proteins) having sequences derived from SEQ ID NO: 1. In some embodiments, an antigenic polypeptide of the present disclosure comprises a sequence that is an N-terminal truncation ofan HD large antigen sequence, such asSEQ ID NO: 1. In some embodiments from 1 to 60 N-terminal amino acids are truncated from the HD large antigen sequence (e.g., SEQ ID NO:1). In some embodiments, from 1 to 56 N-terminal amino acids are truncated from the HD large antigen sequence (e.g., SEQ ID NO:1). In yet other embodiments, from about 10 to about 60 N-terminal amino acids are truncated fromthe HD large antigen sequence (e.g.,SEQ ID NO: 1)to generate an antigenic polypeptide of the present disclosure. In some embodiments, about 10, about 20, about 30, about 40, about 50 or about 60 amino acids are truncated from the N-terminus of the HD large antigen sequence (e.g., SEQ ID NO: 1)to generate an antigenic polypeptide of the present disclosure. In some embodiments, 21 amino acids are truncated from the N-terminus of SEQ ID NO: 1 to generate an antigenic polypeptide. In some embodiments, 24 amino acids are truncated from the N-terminus of SEQ ID NO: 1 to generate an antigenic polypeptide. In some embodiments, 37 amino acids are truncated from the N-terminus of SEQ ID NO: 1 to generate an antigenic
polypeptide. In some embodiments, 46 amino acids are truncated from the N-terminus of SEQ ID NO: 1 to generate an antigenic polypeptide. In some embodiments, 56 amino acids are truncated from the N-terminus of SEQ ID NO: 1 to generate an antigenic polypeptide.In some embodiments, an antigenic polypeptide of the present disclosure comprises a sequence that has between 1 to 10 amino acid mutations at the N-terminusregionas compared to the sequence of an HD large antigen (e.g., SEQ ID NO: 1). In some embodiments, an antigenic polypeptide of the present disclosure comprises 5 amino acid mutations at the N-terminusregionas compared to the sequence of an HD large antigen (e.g., SEQ ID NO: 1). In some embodiments, an antigenic polypeptide of the present disclosure is designed based onthe sequence of the HD large antigen, such asSEQ ID NO: 1,by optimizing the length of an N-terminal truncation of the HD large antigen (e.g., SEQ ID NO: 1). In some embodiments, optimization includes lowering assay background. For example, an antigenic polypeptide can be optimized by testing background using a negative control, such as a sample that lacksatarget antibodythat is specific for the antigenic polypeptide. In some embodiments, optimization includes optimizing both sensitivity and specificityof each antigenic polypeptide. In some embodiments, optimization includes optimizing both sensitivity and specificity of combinationsoftwo antigenic polypeptides, one functioning as a capture moiety and a second one (which can be different from the first) functioning as a detection moiety. In some embodiments, the present disclosure provides an antigenic polypeptide comprising a sequence having at least 90% sequence identity to the sequence of SEQ ID NO: 8, wherein said antigenic polypeptide lacks a sequence comprising at least 90% sequence identity to a region of from about 12 to about 60 amino acids at the N-terminus region of SEQ ID NO: 1. In some embodiments, the antigenic polypeptide comprises a sequence having at least 95%, 96%, 97%, 98% or 99% sequence identity to the sequence of SEQ ID NO: 8. In some embodiments, the antigenic polypeptide lacks a sequence comprising at least 95%, 96%, 97%, 98% or 99% sequence identity to a region of from about 12 to about 60 amino acids at the N- terminus region of SEQ ID NO: 1.In some embodiments, the antigenic polypeptide lacks from 12 to 60 N-terminal amino acids of SEQ ID NO: 1. In some embodiments, the antigenic polypeptide lacks a sequence comprising at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98% or 99% sequence identity with a sequence selected from the groupconsisting ofSEQ ID NO: 9, SEQ ID NO: 10, SEQ ID NO: 11; SEQ ID NO: 12; and SEQ ID NO: 13. In some
embodiments, the antigenic polypeptide lacks a sequence comprising at least 90% sequence identity with a sequence selected from the groupconsisting ofSEQ ID NO: 9, SEQ ID NO: 10, SEQ ID NO: 11; SEQ ID NO: 12; and SEQ ID NO: 13. In some embodiments, the antigenic polypeptide lacks a sequence selected from the groupconsisting ofSEQ ID NO: 9, SEQ ID NO: 10, SEQ ID NO: 11; SEQ ID NO: 12; and SEQ ID NO: 13.In some embodiments, the antigenic polypeptidecomprises at least one amino acid mutation as compared to the sequence of amino acids 1 to 60 of SEQ ID NO: 1.In some embodiments, the antigenic polypeptidecomprises between 1 and 10 amino acid mutations as compared to the sequence of amino acids 1 to 60 of SEQ ID NO: 1. In some embodiments, the antigenic polypeptidecomprises between 1 and 5 amino acid mutations as compared to the sequence of amino acids 1 to 60 of SEQ ID NO: 1. In some embodiments, the antigenic polypeptidecomprises, consists essentialy of, or consists of a sequence selected from the group consisting of SEQ ID NO: 2, SEQ ID NO: 4, SEQ ID NO: 5, SEQ ID NO: 6, SEQ ID NO: 7 and SEQ ID NO: 8. In some embodiments, the antigenic polypeptide comprises the sequence of SEQ ID NO: 6 and lacks the sequence of SEQ ID NO: 11. In some embodiments, the present disclosure provides an antigenic polypeptide comprising a sequence having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% or 100% sequence identity to a truncated polypeptide derived from SEQ ID NO: 1, said truncated polypeptide lacking from 12 to 60 N-terminal amino acids of SEQ ID NO: 1. In some embodiments, the antigenic polypeptide comprises a sequence having at least 90% sequence identity to a truncated polypeptide derived from SEQ ID NO: 1. In some embodiments, the truncated polypeptide comprises a sequence of at least 158 amino acids of SEQ ID NO: 1.For example, the truncated polypeptide comprises a sequence of at least 158 consecutive amino acids of SEQ ID NO: 1, such as the amino acids at positions 57 to 214 of SEQ ID NO: 1. In some embodiments, the present disclosure provides an antigenic polypeptide comprising an amino acid sequence having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% or 100% sequence identity to any of the ful-length sequences of SEQ ID NO: 2 and SEQ ID NOs: 4-8, and a heterologous peptide.In some embodiments, the antigenic polypeptide comprises a sequence having at least 90% sequenceidentity to any of the ful-length sequences of SEQ ID NO: 2 and SEQ ID NOs: 4-8, and a heterologous peptide. In some embodiments, the
heterologous peptide comprises an afinity tag, an epitope tag, a fluorescentprotein, an enzyme or a carier protein.In some embodiments, the afinity tag is a His-tag. In some embodiments, an antigenic polypeptide of the present disclosure is capable of being bound by an antibody directed to HD protein.As used herein, an antibody is “directed to” a particular entity (e.g., HD protein) when the antibody is capable of binding to that entity, typicaly with high afinity. Such an antibody may also be refered to herein as an antigen- specific antibody.In some embodiments, the antibody isan antibody present in a subject after infection with HDV. In some embodiments, the antibody isan antibodyelicited by infection with HDV. In some embodiments, the antibody is an antibody against the large-HDAg.In some embodiments, the antibody is an antibody againsta large-HDAgcomprising a sequence having at least 90% sequence identity to the ful-length sequence of SEQ ID NO: 1.In some embodiments, the antibody is an antibody against the smal-HDAg.In some embodiments, the antibody is an antibody againsta smal-HDAgcomprising a sequence having at least 90% sequence identity to the ful-length sequence of SEQ ID NO: 3. In some embodiments, an antigenic polypeptide of the present disclosure is fused to at least one heterologous peptide. In some embodiments, the heterologous peptide comprises an afinity tag, an epitope tag, a fluorescentprotein, an enzyme or a carier protein. In some embodiments, the present disclosure provides a composition comprising any of the antigenic polypeptides described above. In some embodiments, the composition comprises a detectable label. In some embodiments, the detectable label is conjugated to the antigenic polypeptide. In some embodiments, the detectable label is selected from the group consisting of a fluorescence label, a chemiluminescent label, an enzymatic label, and a particle label.In some embodiments, the detectable label is a chemiluminescent label, such as, acridinium (e.g., acridium esters, acridinium SPSP (N10-(3-sulfopropyl)-N-(3-sulfopropyl, etc.), luminol, isoluminol, thioesters, sulfonamides, phenanthridinium esters, etc. In some embodiments, the detectable label is an enzymatic label, such as horseradish peroxidase, alkaline phosphatase, glucose 6-phosphate dehydrogenase, etc.In some embodiments, the detectable label is a gold nanoparticle. In some embodiments, the detectable label is a latex bead or a latex nanoparticle. In someembodiments, the detectable label is a magnetic nanoparticle. In some embodiments, a composition of the present disclosure further comprises a second antigenic polypeptide that comprises a sequencehaving at least 70%, 80%, 90%, 95%,
96%, 97%, 98%, or 99%sequence identity to the ful-length sequence of SEQ ID NO: 1. In some embodiments, the second antigenic polypeptide comprises the sequence of SEQ ID NO: 1In some embodiments, the second antigenic polypeptide comprises a means for binding to a solid support. In some embodiments, an antigenic polypeptide (recombinant HD protein) of the present disclosure is expressed from a plasmid construct. In some embodiments, the antigenic polypeptide is expressed from a plasmid construct in a host cel. A non-limiting example of a host cel is a bacterial cel, such as an E. coli cel. In some embodiments, an antigenic polypeptide (recombinant HD protein) of the present disclosurecomprisesa moiety that enables isolation/purification of the antigenic polypeptide. For example, the antigenic polypeptide is prepared such that an N or C-terminal moiety (such as, but not limited to a histidine (His)-tag) isfused to the open reading frame (ORF) for theantigenic polypeptideto alow purification. 4.Polynucleotides and host cels As described further herein, embodiments of the present disclosure include polynucleotides encoding the antigenic polypeptides of the disclosure. In some embodiments, the present disclosure provides a polynucleotide encoding an antigenic polypeptideof the disclosure. In some embodiments, the polynucleotide is codon optimized. In some embodiments, the polynucleotide further comprises a heterologous regulatory element operably linked to a nucleic acid encoding the antigenic polypeptide. In some embodiments, the heterologous regulatory element is a promoter. In some embodiments, the heterologous regulatory element is a terminator. Polynucleotide sequences of the disclosure encompass DNA, RNA, DNA-RNA hybrids, peptide nucleic acid (PNA) or any other DNA-like or RNA-like material. In some embodiments, the present disclosure provides isolated or recombinant nucleic acid molecules comprising nucleic acid sequences encoding the antigenic polypeptides. As used herein, the term “nucleic acid molecule”refers to DNA molecules (e.g., recombinant DNA, cDNA, genomic DNA, plastid DNA, mitochondrial DNA) and RNA molecules (e.g., mRNA) and analogs of the DNA or RNA generated using nucleotide analogs. The nucleic acid molecule can be single-stranded or double-stranded, but preferably is double-stranded DNA.
An “isolated”nucleic acid molecule (or DNA) is used herein to refer to a nucleic acid sequence (or DNA) that is no longer in its natural environment, for example in vitro. A “recombinant”nucleic acid molecule (or DNA) is used herein to refer to a nucleic acid sequence (or DNA) that is made by combining genetic material from multiple sources. In some embodiments, a recombinant nucleic acid molecule (or DNA) is made in a recombinant cel, such as a recombinant host cel. In some embodiments, an “isolated”or “recombinant”nucleic acid is free of sequences (preferably protein encoding sequences) that naturaly flank the nucleic acid (i.e., sequences located at the 5′ and 3′ ends of the nucleic acid) in the genomic DNA of the organism from which the nucleic acid is derived. Nucleic acid sequences of the disclosure can be used in DNA constructs or expression cassetes for transformation and expression in organisms, including microorganisms and plants. The nucleotide or amino acid sequences may be synthetic sequences that have been designed for expression in an organism including, but not limited to, a microorganism or a plant. The scope of the disclosure further encompasses any nucleic acid construct which codes for an antigenic polypeptidedescribed herein. Polynucleotide constructs of the disclosure may comprise single-stranded or double- stranded polynucleotides and may represent the sense or the antisense strand. In some embodiments, the present disclosure provides a polynucleotide comprising, consisting of, or consisting essentialy of any of the ful-length sequences of SEQ ID NOs: 14- 21. In some embodiments, the present disclosure provides a recombinant protein comprising an amino acid sequence encoded by the nucleotide sequence of any of SEQ ID NOs: 14-21. In some embodiments, the present disclosure provides a recombinant protein comprising an amino acid sequence encoded by the nucleotide sequence of SEQ ID NO: 14, SEQ ID NO: 15, SEQ ID NO: 16, SEQ ID NO: 17, SEQ ID NO: 18, SEQ ID NO: 19, SEQ ID NO: 20, or SEQ ID NO: 21. The present disclosure also encompasses constructs comprising sequences which are derivatives of polynucleotide sequences encoding antigenic polypeptides described herein. As used herein, the term “derivative”refers to complementary sequences, degenerate sequences, truncated or augmented sequences, modified sequences, and other polynucleotides based upon an original sequence from which a derivative sequence is derived. One form of polynucleotide derivative contemplated within the scope of this disclosure is a polynucleotide comprising nucleotide substitutions. For example, utilizing the redundancy in the genetic code, various
substitutions may be made within a given polynucleotide sequence that result in a codon which codes for the identical amino acid as coded for in the original sequence, and which such change does not alter the composition of the polypeptide coded by a polynucleotide. Such “silent” substitutions may be selected by one of skil in the art. Likewise, nucleotide substitutions are contemplated which result in an amino acid substitution, wherein the amino acid is of similar polarity, charge, size, aromaticity, etc., such that the resulting polypeptide is of identical or substantialy similar structure and function as a polypeptide resulting from an unmodified sequence. Further, the disclosure also comprises nucleotide substitutions which result in amino acid substitutions which create a polypeptide derivative. It is also understood by one of skil in the art that various nucleotide analogs, modified nucleotides, and other compositions may be substituted for the nucleotides of the sequences encoding for antigenic polypeptides, for example modified or non-naturaly occuring nucleotides such as 5-propynyl pyrimidines (i.e., 5-propynyl-dTTP and 5-propynyl-dTCP), 7- deaza purines (i.e., 7-deaza-dATP and 7-deaza-dGTP). Nucleotide analogs include base analogs and comprise modified forms of deoxyribonucleotides as wel as ribonucleotides. Additionaly, substitutions in a polynucleotide sequence may be made which enable the translation of polypeptides from the polynucleotide sequence within a specific expression system. For example, it is contemplated that the polynucleotide sequences may bemodified as necessary to enable or optimize expression of proteins in eukaryotic, yeast, bacterial, insect, plant, mammalian, or in other expression systems such as cel-free and chemical systems. The selection of proper substitutions for proper expression within a given expression system is within the skil of one in the art of molecular biology. Polynucleotide derivatives of the disclosure also comprise augmented or chimeric sequences, wherein a polynucleotide sequence has been modified to include additional nucleotides. For example, a polynucleotide sequence, or subsequences thereof, may be ligated with additional sequences which enhance expression (for example, promoter sequences), or which alter the properties of the resulting polypeptide, such as sequences which enhance secretion, enable isolation (e.g. sequences which code for display/affinitytags, His-Tags or like moieties), enable immobilization, or other useful sequences as known in the art. The scope of the disclosure additionaly includes vectors comprising the polynucleotide constructs of the disclosure integrated into the vectors. Exemplary vectors include
plasmids, phages, and viral constructs which promote eficient maintenance, amplification, and transcription of the polynucleotide sequences in an expression system. The nucleic acid constructs may comprise sequences integrated into the genome of an organism by transduction techniques known in the art. In some embodiments, the present disclosure provides a vector comprising any of the polynucleotides of the disclosure. In some embodiments, the vector is a plasmid, naked nucleic acid, phage, viral vector, or virus. In some embodiments, the vector is capable of genome integration. In some embodiments, the vector is an RNA. In some embodiments, the present disclosure provides a vectoror construct comprising the nucleotide sequence of any of SEQ ID NOs: 14-21. In some embodiments, the present disclosure provides a vector or construct comprising thenucleotide sequence of SEQ ID NO: 15, SEQ ID NO: 17, SEQ ID NO: 18, SEQ ID NO: 19, SEQ ID NO: 20, or SEQ ID NO: 21. The present disclosure also provides a cel comprising any of the polynucleotides of the disclosure. The present disclosure also provides a cel comprising any of the vectors of the disclosure. The present disclosure also provides a cel comprising any of theconstructsof the disclosure. The present disclosure also provides a cel expressing any of the antigenic polypeptidesof the disclosure. In some embodiments, the cel is a microorganism. The present disclosure also provides host cels that are engineered to express one or more antigenic polypeptidesof the disclosure. Suitable host cels include cels of any microorganism (e.g., cels of a bacterium, a protist, an alga, a fungus (e.g., a yeast or filamentous fungus), or other microbe), and are preferably cels of a bacterium. The disclosure further provides a recombinant host cel that is engineered to express one or more, two or more, three or more, four or more, or five or more antigenic polypeptides. 5.Immunoassays In some embodiments, provided herein are immunoassays.In some embodiments, the immunoassays provided herein are serologic assays. The terms “serologic assay,”“serologic” and “serology assay”as used herein refers to an assay that detects antibodies or antibody fragments in a sample. In some embodiments, provided herein are immunoassays for detecting a target antibody in a sample obtained from a subject. In some embodiments, provided herein are immunoassaysfor detecting antibodies elicited by infection with hepatitis D virus (HDV) in a
sample obtained from a subject.In some embodiments, the assays providedherein detect total antibodies (IgG and IgM) against HDV. An example of animmunoassay format for detecting a target antibodyis an assay that comprises a capture moiety that binds to the antibody of interest (target antibody) and a detection moiety that detects presence of the target antibody. Such an assay can be either an indirect assay or a direct assay. An example of an indirect assay format for detecting a target antibodyis an assay that comprises an antigen (capture moiety) that is recognized by the target antibody and an antibody (detection moiety) that recognizes the captured antibody and comprises a detectable label. Such an assay may be ilustrated by the shorthand: Ag-Ab-Ab*, in which “Ag”represents an antigen that is the capture moiety, “Ab”represents the target antibody, and “Ab*”represents adetection moiety that is an antibody with a detectable label. The indirect assay format (Ag-Ab-Ab*) is known tohavelimitationsin sensitivity and specificity due to nonspecific binding of anti-human IgG and IgM conjugates (Ab*), which limits sample volume and conjugate concentration used in the indirect assay format. An example schematic of an indirect assay format is shown in Figure 2A. An example of a direct assay format for detecting a target antibodyis an assay that comprises a first antigen (capture moiety) that is recognized by the target antibody and a second antigen (detection moiety) that is recognized by the target antibody and comprises a detectable label. Such an assay may be ilustrated by the shorthand: (Ag-Ab-Ag*),in which “Ag” represents a first antigen that is the capture moiety, “Ab”represents the target antibody, and “Ag*”represents thedetection moiety that is a second antigen with a detectable label. The direct assay format has been used in many serology assays for detection of HIV, HTLV, HBV and HCV infections.In some embodiments, the present disclosure providesdirect immunoassays. In some embodiments, an assay of the present disclosure is refered to as a “double antigen sandwich assay”, or alternatively as a “double antigen bridging assay (DABA)”. A double antigen sandwich assay uses an antigen sandwichto detect target antibodiesand can be represented by the format Ag-Ab-Ag*. According to the double antigen sandwich format, two antigens are bridged by an antibody analyte. In various embodiments, the two antigens are diferent from each other, for example, one antigen may be boundto a solid support while the other antigen includes a detectable label. In some embodiments, the amino acid sequences of the
two antigens aredifferent. Double antigen sandwich assaysare total antibody assays, meaning that they detect al immunoglobulin types and classes. This immunoassay design poses several advantages to conventional ELISA formats, especialy whentrying to reduce the effects of cross- reactive antibodies.In some embodiments, the second part of the sandwich isan antigenhaving a detectable label. In some embodiments, an assay of the present disclosure comprises the format: Ag- Ab-Ag*. In some embodiments, the assay usesrecombinant HD proteins as capture antigen (Ag) and detectionantigen(Ag*), which are bridged onlyby an HDV specific antibody (Ab)to generate positive signal. Such double selection significantly improves assay specificity. In addition, the direct format alows the use of large sample volume and high conjugate concentration, which substantialy enhances sensitivity. In some embodiments, an assay of the present disclosureutilizes a capture antigen on a solid phase/support (Ag/solid) and a detection antigen having a detectable label (Ag*, also refered to as a “detection conjugate”)to form a double antigen sandwich with ananti-HDV antibody (Ag/solid-Ab-Ag*).In some embodiments, sample, capture antigen and detection antigen are combined. Anti-HDV antibodies present in the sample simultaneously form a double antigen sandwich immunoproduct (Ag/solid-Ab-Ag*) captured on the solid phase/support. The solid phase/support is washed at least once to remove or separate unbound antibodies and conjugates. After the washing, signal detection utilizing the detectable label is caried out. The measured signal is proportional to the amount of anti-HDV antibody in the sample. An example schematic of such a direct assay format is shown in Figure 2B. In some embodiments,the present disclosure provides a direct format HDV total antibody assay with enhanced sensitivity and specificity. In some embodiments, an assay of the present disclosure, such as a double antigen sandwich assay utilizing recombinant HD proteins as capture antigen (Ag) and detection antigen (Ag*), provides an improvement to existing assays for detection of HDV specificantibodies in a sample. In some embodiments, the improvement is compared to an HDV indirect assay. In some embodiments, the improvement is improved specificity. In some embodiments, the improvement is measurable as improved positive and/or negative agreement with an HDV RNA test. In some embodiments, the improvement is enhanced dynamic range of assay signal.In some embodiments, the assay provides utility to assess disease progressionofHDV-HBV coinfection
and superinfection.In some embodiments, a high antibody response with low viral load is indicative ofspontaneously clearing HDV RNA based on typical evolution of serological and virological markers in HDV infection. Initial atempts to develop a directformat HDV total antibody assay using the ful- length HD protein were not successful due to extremely high assay background. Analysis of the 3D structure of HD protein suggested the background wasdriven by aggregation between the capture and detection HD antigens as theN-terminal (12-60amino acids) tends to form a helix bundle octamer structure. Since the N-terminal helix region is notanimmunodominant region, the N-terminal helix region of the HD protein was truncated. Employing the truncated HD protein as detection antigen (Ag*)drasticaly reducedassay background and alowed successful creation of the direct format HDV total antibodyassay to achieve significant sensitivity and specificity improvement over current state of the art HDV antibodyassays. Broadly speaking, immunoassays provided herein comprise contacting a sample (e.g., a sample obtained from a subject) with a capture moiety that binds to a target antibody in the sample. The immunoassays provided herein also comprise a separate detection moiety which is detectably labeled. Both the capture moiety and detection moiety bind to thetarget antibody in the sample, forming a capture moiety-target antibody-detection moiety complex. In some embodiments, the capture moiety-target antibody-detection moiety complex produces a detectable signal. The sample can be contacted with the capture moietyand the detection moiety either simultaneously or sequentialy, in any order. As used herein, the term “capture moiety”refers to a component of an immunoassay that binds and retains the target antibody. In various embodiments, the capture moiety is an antigenic polypeptide. In some embodiments, the antigenic polypeptide is a recombinant large- HDAg polypeptide. In some embodiments, the antigenic polypeptide is a recombinant smal- HDAg polypeptide. In some embodiments, an antigenic polypeptide that is a capture moiety is also refered to herein as a “capture antigen.”In some embodiments, a capture antigen of the disclosure is acapture HD antigen. In some embodiments, a capture moiety is a ful-length HD protein (HD large antigen or HD smal antigen). In some embodiments, the ful-length HD protein sequence is SEQ ID NO: 1. In some embodiments, the ful-length HD protein sequence is SEQ ID NO: 3.In some embodiments, a capture moiety comprises, consists essentialy of, or
consists of the sequence of SEQ ID NO: 1. In some embodiments, a capture moiety comprises, consists essentialy of, or consists of the sequence of SEQ ID NO: 3. As used herein, the term “detectionmoiety”refers to a component of an immunoassay that detects presence of the target antibody. In various embodiments, the detection moiety is an antigenic polypeptide.In some embodiments, the detection moiety is an antigenic polypeptide that is diferent from the antigenic polypeptide that functions as a capture moiety. In some embodiments, the antigenic polypeptide is a recombinant large-HDAg polypeptide. In some embodiments, the antigenic polypeptide is a recombinant smal-HDAg polypeptide. In some embodiments, an antigenic polypeptide that is a detection moiety is also refered to herein as a “detection antigen.”In some embodiments, a detection antigen of the disclosure is a detection HD antigen.In some embodiments, a detection moiety is an antigenic polypeptide derived from the ful-length HD protein (HD large antigen or HD smal antigen) and comprisesa mutation (e.g., truncation(s), substitution(s), etc.) relative to the ful-length HD protein. In some embodiments, the ful-length HD protein sequence is SEQ ID NO: 1. In some embodiments, the ful-length HD protein sequence is SEQ ID NO: 3. For example, the detection moiety may be any of the antigenic polypeptides comprising truncations and/or mutations describedabove in the “Antigenic polypeptides”section. In some embodiments, an antigenic polypeptide that is a detection moiety comprises, consists essentialy of, or consists of a sequence selected from SEQ ID NO: 2, SEQ ID NO: 4, SEQ ID NO: 5, SEQ ID NO: 6, SEQ ID NO: 7 and SEQ ID NO: 8. In some embodiments, an antigenic polypeptide that is a detection moiety comprises a sequence having at least 90% sequence identity to the sequence of SEQ ID NO: 8, whereinsaid antigenic polypeptide lacks a sequence comprising at least 90% sequence identity to a region of from about 12 to about 60 amino acids at the N-terminus region of SEQ ID NO: 1. In some embodiments, an antigenic polypeptide that is a detection moiety comprises, consists essentialy of, or consists of a polypeptide selected from the group consisting of: i) a polypeptide that comprises the sequence of SEQ ID NO: 8 and lacks the sequence of SEQ ID NO: 13; i) a polypeptide that comprises the sequence of SEQ ID NO: 7 and lacks the sequence of SEQ ID NO: 12; ii) a polypeptide that comprises the sequence of SEQ ID NO: 6 and lacks the sequence of SEQ ID NO: 11; iv) a polypeptide that comprises the sequence of SEQ ID NO: 5 and lacks the sequence of SEQ ID NO: 10; v) a polypeptide that comprises the sequence of SEQ ID NO: 4 and lacks the sequence of SEQ ID NO: 9; and vi) a polypeptide that comprises the sequence of SEQ
ID NO: 2.In some embodiments, a detection moiety comprises, consists essentialy of, or consists of a sequence selected from SEQ ID NO: 2 and SEQID NOs: 4-8. In some embodiments, the detection moiety is detectably labeled, andbinding of the detection moiety to an antibody of interest (i.e. a target antibody) produces a detectable signal. In some embodiments, provided herein is a direct immunoassay comprising contacting asample with a capture antigen that binds to an antibody against a polypeptide comprising a sequence having at least 70% sequence identity to SEQ ID NO: 1 and a detection antigen that binds to the antibody, wherein the detection antigen is detectably labeled. In some embodiments, the antibody is directed to a polypeptide comprising SEQ ID NO: 1. In some embodiments, provided herein is a direct immunoassay involving the use of separate capture and detection moieties, wherein the capture moiety binds to a targetantibodyof interest forming a capture moiety-target antibody complex, and the detection moietybinds to the targetantibodyof interest. In some embodiments, the detection moiety comprises a detectable label. In some embodiments, the detection moiety binds to the target antibody in the capture moiety-target antibody complex.In some embodiments,the capture moiety is a capture antigen and the detection moiety is a detection antigen, such that a capture antigen-target antibody- detection antigen complexis formed in the assay. In some embodiments, the immunoassays provided herein comprise contacting a sample with an antigenic polypeptide(e.g. a capture antigenand/or detection antigen) that binds to an antibody againsthepatitis D antigen (HDAg) of hepatitis D virus (HDV). In some embodiments, the antibody bound by the antigenic polypeptideis an antibody against the large- HDAg and/or the smal-HDAg. In some embodiments, the antibody is an antibody against the large-HDAg. In some embodiments, the antigenic polypeptideis a recombinant large-HDAg polypeptide. In some embodiments, the antigenic polypeptidebinds to an antibody againstthe polypeptideofSEQ ID NO: 1.In some embodiments, the antibody is an antibody against the smal-HDAg.In some embodiments, the antigenic polypeptideis a recombinant smal-HDAg polypeptide.In some embodiments, the antigenic polypeptidebinds to an antibody againstthe polypeptide of SEQ ID NO: 3. The antigenic polypeptide may be any of the antigenic polypeptides described throughout the present disclosure. In some embodiments, an antigen used in an immunoassay of the disclosure is a liquid phase antigen or a solid phase antigen. As used herein, the term “liquid phase antigen”refers to
an antigen in solution, which comprises one or more epitopes that bind to a target antibody also freely mobile within a solution. In contrast, a “solid phase antigen”is defined as an antigen that is atached to a solid phase, which comprises one or more epitopes that can bind to a target antibody in solution. A “solid phase”may be a porous or non-porous material, a latex particle, a magnetic particle, a microparticle, a bead, a membrane, and a microtiter wel or a plastic tube. In some embodiments, thecapture moiety (e.g. capture antigen)isbound to a solid support or solid phase. In some embodiments, the solid support or solid phasefacilitates separation of the capture moiety-target antibody complex or the capture moiety-target antibody- detection moietycomplex from the test sample.Any solid support known in the art can be used, including but not limited to, solid supports made out of polymeric materials in the forms of wels of a reaction tray, test tubes or beads (for example, polystyrene beads, magnetic beads), nitrocelulose strips, membranes, microparticles (for example, latex particles). The solid phase also can comprise any suitable porous material with suficient porosity andsurface afinity.Microporous structures are generaly used, but materials with gel structure in the hydrated state may be used as wel. Such useful solid supports include, but are not limited to, nitrocelulose and nylon. Such porous solid supports are in the form of sheets of thickness from about 0.01 to 0.5 mm, including about 0.1 mm. The pore size may vary within wide limits, and can be from about 0.025 to about 15 microns, especialy from about 0.15 to about 15 microns. The surface of such supports may be activated by chemical processes which cause covalent linkage of the capture moietyto the support. The capture moiety (e.g. capture antigen) can be bound to the solid support or solid phasedirectly or indirectly.The capture moiety (e.g. capture antigen)can be bound to the solid support or solid phase by adsorption, by covalent bonding using a chemical coupling agent or by other means known in the art, provided that such binding does not interfere with the ability of the capture moietyto bind to the target antibody. Alternatively, the capture moiety can be bound to microparticles that have previouslybeencoated with streptavidin or biotinwith biotinylated moieties using means known in the art. Alternatively, the capture moietycan be bound using microparticles that have been previously coated with anti-species specific monoclonal antibodies. Moreover, if necessary, the solid support can be derivatized to alow reactivity with various functional groups on the capture moiety. Such derivatization requires the use of certain
coupling agents such as, but not limited to, maleic anhydride, N-hydroxysuccinimide and 1- ethyl-3-(3-dimethylaminopropyl)carbodimide. After the test sample being tested for and/or suspected of containing the target antibodyis brought into contact with thecapture moiety (e.g. the capture antigen)and/or detection moiety (e.g., detection antigen),the mixture is incubated in order to alow for the formation of acapturemoiety-target antibodycomplex, detectionmoiety-target antibody complex, and/or capture moiety-target antibody-detection moietycomplex. The incubation can be carried out at a pH of from about 4.5 to about 10.0, at a temperature of from about 2°C to about 45°C, and for a period from at least about one (1) minute to about eighteen (18) hours, including from about 1 to 20 minutes, also including from about 2-6 minutes. The immunoassay described herein can be conducted in one step (meaning the test sample, at least one capture moietyand at least one detection moiety are al added sequentialy or simultaneously to a reaction vessel) or in more than one step, such as two steps, three steps, etc. In some embodiments, the sample is first contacted with the capture moiety. In some embodiments, after formation of the capture moiety-target antibody complex,the complex is then contacted with at least one detection moiety under conditions such that the detection moiety binds to the capture moiety-target antibody complex,thereby forming a capture moiety-target antibody-detection moiety complex. In some embodiments, multiple detection moieties are used. If the capture moiety-target antibodycomplexis contacted with more than one detection moiety, then multiplecapture moiety-target antibody-detection moietycomplexes areformed. In some embodiments, thedetectionmoiety (e.g. the detectionantigen) contains a detectable label. In some embodiments, the detectable label is bound to the detectionmoiety prior to, simultaneously with, or after formation ofa complexbetween the detection moiety and the target antibody. Any detectable label known in the art can be usedas a detectable label of the present disclosure. For example, the detectable label can be a radioactive label, such as, 3H, 125I, 35S, 14C, 32P, 33P, an enzymatic label, such as horseradish peroxidase, alkaline phosphatase, glucose 6- phosphate dehydrogenase, etc., a chemiluminescent label, such as, acridinium (e.g., acridium esters, acridinium SPSP (N10-(3-sulfopropyl)-N-(3-sulfopropyl, etc.), luminol, isoluminol, thioesters, sulfonamides, phenanthridinium esters, etc. a fluorescence label, such as, fluorescein (5-fluorescein, 6-carboxyfluorescein, 3’6-carboxyfluorescein, 5(6)-carboxyfluorescein, 6-
hexachloro-fluorescein, 6-tetrachlorofluorescein, fluorescein isothiocyanate, etc.), rhodamine, phycobiliproteins, R-phycoerythrin, quantum dots (zinc sulfide-capped cadmium selenide), a thermometric label or an immuno-polymerase chain reaction label. An introduction to labels, labeling procedures and detection of labels is found in Polak and Van Noorden, Introduction to Immunocytochemistry, 2nded., Springer Verlag, N.Y. (1997) and in Haugland, Handbook of Fluorescent Probes and Research Chemicals(1996),which is a combined handbook and catalogue published by Molecular Probes, Inc., Eugene, Oregon. In some embodiments, the detectable label is a particle label. A particle label produces a colored readout which requires no development process for visualization. In some embodiments, the detectable label is a nanoparticle. In some embodiments, the detectable label is a gold nanoparticle. In some embodiments, the detectable label is a latex bead or a latex nanoparticle. In some embodiments, the detectable label is a magnetic nanoparticle. In some embodiments, any suitable detectable label that can be conjugated or linked to an antigenic polypeptidein order to detect binding of the antigenic polypeptideto a target antibodycan be used,many of which are known in the art. The detectable label can be bound to the detection moiety either directly or through a coupling agent. An example of a coupling agent that can be used is EDAC (1-ethyl-3-(3- dimethylaminopropyl) carbodimide, hydrochloride) that is commercialy available from Sigma- Aldrich, St. Louis, MO. Other coupling agents that can be used are known in the art. Methods for binding a detectable label to a polypeptideare known in the art. Additionaly, many detectable labels can be purchased or synthesized that already contain end groups that facilitate the coupling of the detectable label to the detection moiety, such as, N10-(3-sulfopropyl)-N-(3- carboxypropyl)-acridinium-9-carboxamide, otherwise known as CPSP-Acridinium Ester or N10- (3-sulfopropyl)-N-(3-sulfopropyl)-acridinium-9-carboxamide, otherwise known as SPSP- Acridinium Ester. The capture moiety-target antibody-detection moiety complex can be,but does not have to be, separated from the remainder of the test sample prior to quantification of the label. For example, if the capture moiety (e.g. capture antigen) is bound to a solid support or solid phase, such as, but not limited to, a wel of a reaction tray, a bead or a microparticle, separation can be accomplished by removing the fluid (of the test sample) from contact with the solid support. In some embodiments, if the capture moiety (e.g. capture antigen)is bound to a solid
support it can be simultaneously contacted with the sample and the detection moiety, folowed by removal of the fluid (test sample) from contact with the solid support. If the capture moietyis not bound to a solid support, then the capture moiety-target antibody complex or the capture moiety-target antibody-detection moiety complexdoes not have to be removed from the test sample for quantification of the amount of the label. After formation of the capture moiety-target antibody-detection moiety complex,the amount of label in the complex is quantified using techniques known in the art. For example, if an enzymatic label is used, the labeled complex is reacted with a substrate for the label that gives a quantifiable reaction such as the development of color. If the label is a radioactive label, the label is quantified using a scintilation counter. If the label is a fluorescent label, the label is quantified by stimulating the label with a light of one color (which is known as the “excitation wavelength”) and detecting another color (which is known as the “emission wavelength”) that is emited by the label in response to the stimulation. If the label is a chemiluminescent label, the label is quantified detecting the light emited either visualy or by using luminometers, x-ray film, high speed photographic film, a CCD camera, etc. Once the amount of the label in the complex has been quantified, the concentration of the target antibodyin the test sample can be determined by use of a standard curve that has been generated using serial dilutions of the marker of known concentration. Other than using serial dilutions of the marker, the standard curve can be generated gravimetricaly, by mass spectroscopy and by other techniques known in the art. In some embodiments, the presence of HDV antibodyin asample is determined by means of a cutof value that alows for the semi-quantitative detection of theHDV antibody. In some embodiments, negativeresults indicate the absence of HDV infection.In some embodiments, equivocalresults indicate borderline level of anti-HDV total antibodies. Repeat testing is recommended to determine the definitive HDV infection status.In some embodiments, positiveresults usualy indicate simultaneous acute or chronic coinfection with hepatitis B virus (HBV) and HDV; acute HDV infection in patients with known chronic HBV infection (i.e., HDV superinfection); or resolved HDV infection. Results should be corelated with medical history and clinical findings. In some embodiments,the provided assays are employed for diagnosis of HDV infection and/or to identify patients for antiviral treatments. In some embodiments,the assay is
employed in the diagnosis of HDV infection. In some embodiments,the provided assays are employed to determine prevalence of HDV infection for epidemiology studies. In some embodiments, the assay is a rapid test assay. In some embodiments, the assay is employed on a sample from a subject that is hepatitis B surface antigen positive (HBsAgpositive). In some embodiments, a positive result from the assay indicates a subsequentHDV RNA assay. In some embodiments, a positive result from the assay combined with a positive result from an HDV RNA assay indicates a need for treating the subject for HDV infection. Increasing the specificity while alsoincreasing sensitivity aforded by the presently disclosed assay design wil benefit every aspect of HDV diagnosis and control. Thisassay design wil provide a significant performance advantage versus competitors on the market today. Themethods and kits described elsewhere in the present disclosure encompass other reagents and methods for carying out the immunoassay. For instance, encompassedare various bufers such as are known in the art and/or which can be readily prepared or optimized to be employed, e.g., for washing. Furthermore, the methods and kits optionaly are adapted for use on an automated or semi-automated system. Some of the diferences between an automated or semi-automated system as compared to a non-automated system (e.g., ELISA) include the substrate to which the capture moietyis atached (which can impact sandwich formation and analyte reactivity), and the length and timing of the capture, detection and/or any wash steps. Whereas a non-automated format such as an ELISA may include a relatively longer incubation time with sample and capture reagent (e.g., about 2 hours) an automated or semi-automated format (e.g., ARCHITECT®) may havea relatively shorter incubation timewith sample and capture moiety (e.g., approximately 18 minutes for ARCHITECT®). Similarly, whereas a non-automated format such as an ELISA may incubate a detection moietyfor a relatively longer incubation time (e.g., about 2 hours), an automated or semi-automated format (e.g., ARCHITECT®) may have a relatively shorter incubation time (e.g.,approximately 18minutes for the ARCHITECT®). In some embodiments,methods of the present disclosure comprise incubation with sample and capture moiety along with detection moiety. In some embodiments,incubation of sample, capture moiety and detection moiety is for a relatively shorter incubation time (e.g., approximately 18 minutes) as compared with a non-automated format. In some embodiments,
methods of the present disclosure comprise incubation of sample, capture moiety and detection moiety for approximately 15 minutes, 16 minutes, 17 minutes, 18 minutes, 19 minutes, 20 minutes, 21 minutes, 22 minutes, 23 minutes, 24 minutes, or 25 minutes. In some embodiments, methods of the present disclosure comprise incubation of sample, capture moiety and detection moiety for 30 minutes or less. In some embodiments, animmunoassay provided hereinis a chemiluminescent microparticle immunoassay(CMIA). In some embodiments,the assay is used in a fuly automated and high-throughput system (e.g., 200 tests per hour), such as the ARCHITECT® system.As a non-limiting example, a provided assay of the disclosure comprises the folowing steps: first, sample (serum or plasma) is combined with recombinant HDAg (capture moiety) coated microparticles and acridinium-labeled conjugates(detection moiety), HDV antibodies present in the sample simultaneously form a double antigen sandwich with the capture HDAg on microparticles and the detection HDAg conjugate in solution; folowing a wash cycle, alkaline hydrogen peroxide solution is then added to release acridinium chemiluminescent signal; and the intensity of the chemiluminescence, measured as relative light units (RLU), is proportional to the concentration of HDV antibodies in the sample. 6.Methods In some embodiments, provided herein aremethods for detecting antibodies specific for HD protein in a biological sample. In some embodiments, the methods provided herein comprise performing an immunoassayas describedherein, including an immunoassay as describedabove. In some embodiments, the immunoassay comprises determining the presence or amount of atarget antibody in asample based upon the detectable signal generated upon formation of the capture moiety-target antibody-detection moiety complex (e.g., a capture antigen-target antibody- detection antigencomplex). In some embodiments,if the capture moiety is bound to a solid support or solid phase, the method comprises determining the presence or amount ofatarget antibody in asample based upon adetectable signalafterseparating orremoving the fluid (of the test sample) from contact with the solid support. In some embodiments, provided herein are methods for detecting hepatitis D virus (HDV) infection in a subject.In some embodiments, the methodcomprises determining that the subject is infected with hepatitis D virus (HDV)when the amount of the target antibody (e.g. the
level of the detectable signal, which is indicative of the amount of target antibody) is equal to or above a reference value. In some embodiments, the method comprises determining that the subject is not infected with HDV when the amount of the target antibody (e.g. the level of the detectable signal, which is indicative of the amount of target antibody) is below a reference value. In some embodiments, the methodsprovided herein comprise performing an immunoassay as describedherein. In some embodiments, the immunoassay comprises contacting a sample obtained from a subject with a capture moiety (e.g., a capture antigen). In some embodiments, thecapture moiety is an antigenic polypeptide describedherein, including an antigenic polypeptide as describedabove.In some embodiments, anantigenic polypeptide that is a capture moiety is a recombinant large-HDAg polypeptide. In some embodiments, anantigenic polypeptide that is a capture moiety is a recombinant smal-HDAg polypeptide. In some embodiments, a capture moiety is a ful-length HD protein (HD large antigen or HD smal antigen). In some embodiments, the ful-length HD protein sequence is SEQ ID NO: 1. In some embodiments, the ful-length HD protein sequence is SEQ ID NO: 3.In some embodiments, a capture moiety comprises, consists essentialy of, or consists of the sequence of SEQ ID NO: 1. In some embodiments, a capture moiety comprises, consists essentialy of, or consists of the sequence of SEQ ID NO: 3. In some embodiments, the immunoassay further comprises contacting the sample with a detection moiety. In some embodiments, thedetection moiety is an antigenic polypeptide describedherein, including an antigenic polypeptideas describedabove.In some embodiments, the detection moiety is an antigenic polypeptide that is diferent from the antigenic polypeptide that functions as a capture moiety. In some embodiments, the antigenic polypeptide that is a detection moiety is a recombinant large-HDAg polypeptide. In some embodiments, the antigenic polypeptide that is a detection moiety is a recombinant smal-HDAg polypeptide. In some embodiments, a detection moiety is an antigenic polypeptide derived from the ful-length HD protein (HD large antigen or HD smal antigen) and comprisesa mutation relative to the ful- length HD protein. In some embodiments, the ful-length HD protein sequence is SEQ ID NO: 1. In some embodiments, the ful-length HD protein sequence is SEQ ID NO: 3. For example, the detection moiety may be any of the antigenic polypeptides comprising truncations and/or mutations described above in the “Antigenic polypeptides”section. In some embodiments, an
antigenic polypeptide that is a detection moiety comprises, consists essentialy of, or consists of a sequence selected from the group consisting of SEQ ID NO: 2, SEQ ID NO: 4, SEQ ID NO: 5, SEQ ID NO: 6, SEQ ID NO: 7 and SEQ ID NO: 8. In some embodiments,an antigenic polypeptide that is a detection moiety comprises a sequence having at least 90% sequence identity to the sequence of SEQ ID NO: 8, wherein said antigenic polypeptide lacks a sequence comprising at least 90% sequence identity to a region of from about 12 to about 60 amino acids at the N-terminus region of SEQ ID NO: 1. In some embodiments, an antigenic polypeptide that is a detection moiety comprises, consists essentialy of, or consists of a polypeptide selected from the group consisting of: i) a polypeptide that comprises the sequence of SEQ ID NO: 8 and lacks the sequence of SEQ ID NO: 13; i) a polypeptide that comprises the sequence of SEQ ID NO: 7 and lacks the sequence of SEQ ID NO: 12; ii) a polypeptide that comprises the sequence of SEQ ID NO: 6 and lacks the sequence of SEQ ID NO: 11; iv) a polypeptide that comprises the sequence of SEQ ID NO: 5 and lacks the sequence of SEQ ID NO: 10; v) a polypeptide that comprises the sequence of SEQ ID NO: 4 and lacks the sequence of SEQ ID NO: 9;and vi) a polypeptide that comprises the sequence of SEQ ID NO: 2.In some embodiments, a detection moiety comprises, consists essentialy of, or consists of a sequence selected from SEQ ID NO: 2 and SEQID NOs: 4-8. In some embodiments, contacting the sample with the capture moietyand the detection moiety results in the formation of a capture moiety-target antibody-detection moiety complex, which complex produces a detectable signal. In some embodiments, themethodfurther comprises determining the presence or amount of the target antibody (e.g. the antibody against HDV) in the sample based upon the detectable signal. In some embodiments, the method further comprises determining that the subject is infected with HDVwhen the amount of the target antibody (e.g. the level of the detectable signal, which is indicative of the amount of target antibody) is equal to or above a reference value. In some embodiments, the method further comprises determining that the subject is not infected with HDV when the amount of the target antibody (e.g. the level of the detectable signal, which is indicative of the amount of target antibody) is below a reference value. In some embodiments, the present disclosure provides a method for detecting antibodies specific for HD protein in a biological sample, wherein an antigenic polypeptideof the present disclosure is used as a binding partner for said HD protein antibodies.
In some embodiments, the present disclosure provides a method for detecting antibodies specific for HD protein in a biological sample, said method comprising:a) forming an immunoreaction mixture by mixing a biological sample withanantigenic polypeptide of the present disclosure; b) maintaining said immunoreaction mixture for a time period suficient for alowing antibodies against said antigenic polypeptide present in the biological sample to immunoreact with said antigenic polypeptide to form an immunoreaction product; and c) detecting the presence and/or the concentration of said immunoreaction product. In some embodiments, the immunoreaction is caried out in a double antigen sandwich format comprising:a) adding to said biological sample a first antigenic polypeptide that comprises a means for binding to a solid support and a second antigenic polypeptide that caries a detectable label, wherein said first antigenic polypeptide and said second antigenic polypeptide bind specificaly to the HD protein antibodies, b) forming an immunoreaction mixture comprising the first antigenic polypeptide, the biological sample antibody and the second antigenic polypeptide, wherein the solid support is added before, during or after forming the immunoreaction mixture, c) maintaining said immunoreaction mixture for a time period suficient for HD protein antibodies in the biological sample to immunoreact with said first and second antigenic polypeptides to form an immunoreaction product, andd) detecting the presence of any of said immunoreaction product. In some embodiments of this method, the first antigenic polypeptide comprises a sequence having at least 90% sequence identity to the ful-length sequence of SEQ ID NO: 1. In some embodiments, the method further comprises a washing step prior to the detecting step. In some embodiments, the immunoreaction product is bound to the solid support and the washing step comprises removingorseparating someor al of the first antigenic polypeptide, HD protein antibody and second antigenic polypeptide that are not part of the immunoreaction product that is bound to the solid support. In some embodiments, the solid support is selected from the group consisting of acolumn, bead, test tube, microtiter dish,multi- wel plate,microparticle, microsphere, a test stick, a test strip, microchip and membrane. In some embodiments, the biological sample is a body fluid sample from a human subject.In some embodiments, the biological sample is selected from the group consisting of blood, plasma and serum. In some embodiments, provided herein are methodsfor detecting antibodies against hepatitis D virus (HDV) in a biological sample, comprising performing a HDV serology assay
using a direct assay format with a first antigenic polypeptide and a second antigenic polypeptide, wherein the first antigenic polypeptide comprises a ful-length HD protein and the second antigenic polypeptide comprises an N-terminaly truncated fragment of the ful-length HD protein. As used herein, an “N-terminaly truncated fragment of theful-lengthHD protein” refers to a shortened HD protein (and sequence thereof) that has been shortened by removing a portion of the protein located at or near the N-terminus. In some embodiments,the assay is a double antigen sandwich assay.In some embodiments,theful-length HD protein comprises an amino acid sequence having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% or 100% sequence identity to the ful-length sequence of SEQ ID NO: 1.In some embodiments,the ful-length HD protein comprises an amino acid sequence having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% or 100% sequence identity to the ful-length sequence of SEQ ID NO: 3.In some embodiments,the second antigenic polypeptide lacks from 12 to 60 N- terminal amino acids of the HD protein. For example, the second antigenic polypeptide lacks from 12 to 60 N-terminal amino acidsofthe ful-length HD protein of SEQ ID NO: 1. In some embodiments,the assay is a double antigen sandwich assay.In some embodiments,the second antigenic polypeptide lacks from 12 to 60 consecutive N-terminal amino acids of the HD protein. In some embodiments,the second antigenic polypeptide lacks 21, 37 or 46 consecutive N- terminal amino acids of the HD protein. In some embodiments, provided herein are methodsfordetecting antibodies against hepatitis D virus (HDV) in a biological sample, comprising: a) contacting abiological sample, either simultaneously or sequentialy, in any order with:(1) a first antigenic polypeptide under conditions suficient to alow binding of anyHDV antibody present in the biological sample to the first antigenic polypeptide, wherein said first antigenic polypeptide is immobilized on a solid support, and (2) a second antigenic polypeptide under conditions suficient to alow binding of the second antigenic polypeptide to any HDV antibody present in the biological sample,such that a first antigenic polypeptide-HDV antibody-second antigenic polypeptide complex is formed and is bound to the solid support, andb) detecting the presence of said first antigenic polypeptide-HDV antibody-second antigenic polypeptide complex,wherein said first antigenic polypeptide comprises a sequence having at least 90% sequence identity to the ful-length sequence of SEQ ID NO: 1;wherein said second antigenic polypeptide comprises a sequence having at least 90% sequence identity to a truncated polypeptide derived from SEQ ID NO: 1,
said truncated polypeptide lacking from 12 to 60 N-terminal amino acids of SEQ ID NO: 1; and wherein said second antigenic polypeptide caries a detectable label.In some embodiments,the method further comprisesa washing step prior to the detecting step.In some embodiments,the washing step comprises removing orseparating someor al of the first antigenic polypeptide, HDV antibody and second antigenic polypeptide that are not part of the first antigenic polypeptide-HDV antibody-second antigenic polypeptide complex that is bound to the solid support.In some embodiments,the step of detecting comprises detecting the presence and/or the concentration of the first antigenic polypeptide-HDV antibody-second antigenic polypeptide complex that is formed and is bound to the solid support. In some embodiments,the step of detecting comprises measuring an amount of saidfirst antigenic polypeptide-HDV antibody- second antigenic polypeptide complex that is formed and is bound to the solid support. In some embodiments,the solid support is selected from the group consisting of acolumn, bead, test tube,microtiter dish,multi-wel plate,microparticle, microsphere, a test stick, a test strip, microchip and membrane. Incertainprefered embodiments, the solid support is a microparticle. In someembodiments,thebiological sample is selected from the group consisting of blood, plasma andserum. In some embodiments,thefirst antigenic polypeptide comprises a sequence having at least 95%, 96%, 97%, 98%, 99% or 100%sequence identity to the ful-length sequence of SEQ ID NO: 1. In some embodiments,the second antigenic polypeptide is selected from the group consisting of:i) a polypeptide that comprises the sequence of SEQ ID NO: 8 and lacks the sequence of SEQ ID NO: 13;i) a polypeptide that comprises the sequence of SEQ ID NO: 7and lacks the sequence of SEQ ID NO: 12;ii) a polypeptide that comprises the sequence of SEQ ID NO: 6 and lacks the sequence of SEQ ID NO: 11;iv) a polypeptide that comprises the sequence of SEQ ID NO: 5 and lacks the sequence of SEQ ID NO: 10;v) a polypeptide that comprises the sequence of SEQ ID NO: 4 and lacks the sequence of SEQ ID NO: 9; andvi) a polypeptide that comprises the sequence of SEQ ID NO: 2. In some embodiments, provided herein are methodsof detecting hepatitis D virus (HDV) infection in a subject comprising performing theimmunoassays orimmunoassay-based methods, such as the serology assays,described above. In some embodiments, thesample is a sample taken from the subject and the step of detecting comprises detecting the presence of a first antigenic polypeptide-HDV antibody-second antigenic polypeptide complex that is formed
and is bound to a solid support, thereby detecting the presence of past or present HDV infection in said subject. In some embodiments, provided herein are methodsof estimating HDV incidence in a population, the method comprising, a) providing a set of samples derived from a plurality of individuals within the population over a period of time;b) performing on each sample in the set of samples the immunoassays or immunoassay-based methods, such as the serology assays, described above; and c) determining the percentage of recent HDV infections over the period of time;wherein the percentage of recent HDV infections over the period of time provides an estimate of HDV incidence in the population. In some embodiments, an immunoassay ofthe disclosuredisplays at least 75% sensitivity(e.g. sensitivity in accurately diagnosing HDV infection in a subject).In some embodiments, the immunoassaydisplays at least 85% sensitivity. In some embodiments, the immunoassaydisplays at least 90% sensitivity. In some embodiments, the immunoassaydisplays at least 95% sensitivity. In some embodiments, the immunoassay displays at least 99% sensitivity. In some embodiments, the immunoassay displays at least 75% specificity(e.g. specificity in accurately diferentiating subjects not having HDVinfection from those having HDVinfection). In someembodiments, the immunoassay displays at least 85%specificity. In some embodiments, the immunoassay displays at least 90% specificity. In some embodiments, the immunoassay displays at least 95% specificity.In some embodiments, the immunoassay displays at least 99% specificity. In some embodiments, the immunoassay displays at least 75% sensitivityand at least 75% specificity. In some embodiments, the immunoassay displays atleast 85% sensitivity and at least 75% specificity.In some embodiments, the immunoassay displays at least 95% sensitivity and at least 75% specificity. In some embodiments, the immunoassay displays at least 99% sensitivity and at least 75% specificity. In some embodiments, the immunoassay displays at least 85% sensitivity and at least 85% specificity. In some embodiments, the immunoassay displays at least 90% sensitivity and at least 85% specificity. In some embodiments, the immunoassay displays at least 95% sensitivity and at least 85% specificity. In some embodiments, the immunoassay displays at least 99% sensitivity and at least 85%specificity.
In some embodiments, the immunoassay displays at least 90% sensitivity and at least 90% specificity. In some embodiments, the immunoassay displays at least 95% sensitivity and at least 90% specificity. In some embodiments, the immunoassay displays at least 99% sensitivity and at least 90% specificity. In some embodiments, the immunoassay displays at least 95% sensitivity and at least 95% specificity. In some embodiments, the immunoassay displays at least 99% sensitivity and at least 95% specificity.In some embodiments, the immunoassay displays at least 99% sensitivity and at least 99% specificity. The power of a diagnostic test to corectly predict status is commonly measured as the sensitivity of the assay, the specificity of the assay or the area under a receiver operated characteristic (“ROC”) curve. Sensitivity is the percentage of true positives that are predicted by a test to be positive, while specificity is the percentage of true negatives that are predicted by a test to be negative. A ROC curve provides the sensitivity of a test as a function of 1-specificity. The greater the area under the ROC curve, the more powerful the predictive value of the test. Other useful measures of the utility of a test are positive predictive value and negative predictive value. Positive predictive value is the percentage of people who test positive that are actualy positive. Negative predictive value is the percentage of people who test negative that are actualy negative.The immunoassays described herein may show an ROC of at least 0.6, at least about 0.7, at least about 0.8, or at least about 0.9. In some embodiments, the methods provided herein further comprisetesting for active HDV infection by measuring hepatitis D RNA levels in a sample. In some embodiments, methods of the present disclosureprovide for a first line screening test for HDV. In some embodiments, the methods provided hereinenable cost efective identification of patients with active HDV infection for therapeutic treatment. In some embodiments, a negative result from an immunoassay of the disclosure provides for time and/or cost saving benefits by reducing the number of unnecessary HDV RNA testing. In some embodiments, the methods provided herein further comprise treating the subject for an HDVinfection. Suitable treatments for HDVinfection include, for example, antiviral medications. In some embodiments, treating a subject determined to have an HDV infection according to the presently disclosed methods comprises administering antiviral therapies. In some embodiments, treating a subject according to the disclosure comprises
administering one or more of pegylated interferon alpha therapyand bulevirtide. In some embodiments, treating a subject according to the disclosure comprises administering one or more of pegylated interferon alpha therapy, bulevirtide, pegylated interferon lambda (λ), lonafarnib, and REP2139-Ca. In some embodiments, treating a subject according to the disclosure comprises treating the subject for both an HBV and an HDV infection. 7.Samples As used herein, “sample”, “test sample”, and “biological sample”refer to a sample that contains, is suspected of containing, or is to be tested for whether or not it contains a target antibody (e.g. an antibody produced during HDV infection). In some embodiments, the “sample”, “test sample”, and “biological sample”is a sample that is positive for HBV infection and/or obtained from a subject known to have an HBV infection. In some embodiments, the sample is from a subject with clinicaly evident acute or chronic hepatitis B.In some embodiments, the sample is from a subject that has a previous or active infection with hepatitis B. In some embodiments, the sample is from a subject that is hepatitis B surface antigen positive. In some cases, the sample may be processed prior to performing a method (e.g. an immunoassay)described herein. For example, the sample may be separated or purified from its source prior to analysis. In some embodiments, an unprocessed sample canbe assayed directly. In some embodiments, the sample is a fluid sample. In some embodiments, the sample is a human bodily substance (e.g.,bodily fluid, blood such as whole blood, serum, plasma, urine, saliva, sweat, sputum, semen, mucus, lacrimal fluid, lymph fluid, amniotic fluid, interstitial fluid, lung lavage, cerebrospinal fluid, feces, tissue, organ, or the like). In some embodiments, the sample is a whole blood sample, a serum sample, or a plasma sample. A wide range of volumes of the fluid sample may be analyzed. In a few exemplary embodiments, the sample volume may be about 0.5 nL, about 1 nL, about 3 nL, about 0.01 µL, about 0.1 µL, about 1 µL, about 5 µL, about 10 µL, about 100 µL, about 1 mL, about 5 mL, about 10 mL, or the like. In some cases, the volume of the fluid sample is between about 0.01 µL and about 10 mL, between about 0.01 µL and about 1 mL, between about 0.01 µL and about 100 µL, or between about 0.1 µL and about 10 µL. In some cases, the fluid sample may be diluted prior to use in an assaydescribed herein. For example, in some embodiments the sample isdiluted with an appropriate solvent
(e.g.,a buffer such as PBS bufer). A fluid sample may be diluted about 1-fold, about 2-fold, about 3-fold, about 4-fold, about 5-fold, about 6-fold, about 10-fold, about 100-fold, or greater, prior to use. In other cases, the fluid sample is not diluted prior to use in an assay. In some cases, the sample may undergo pre-analytical processing. Pre-analytical processing may offer additional functionality such as nonspecific protein removal and/or efective yet economicalyimplementable mixing functionality. General methods of pre- analytical processing may include the use of electrokinetic trapping, AC electrokinetics, surface acoustic waves, isotachophoresis, dielectrophoresis, electrophoresis, or other pre-concentration techniques known in the art. In some cases, the fluid sample may be concentrated prior to use in an assay. For example, in some embodiments the sample maybe concentrated by precipitation, evaporation, filtration, centrifugation, or a combination thereof. A fluid sample may be concentrated about 1-fold, about 2-fold, about 3-fold, about 4-fold, about 5-fold, about 6-fold, about 10-fold, about 100-fold, or greater, prior to use. It may be desirable to include a calibrator and/or a controlin the methods described herein. The calibrator and/or control may be analyzed concurently with the sample from the subject as described above. The results obtained from the subject sample can be compared to the results obtained from the calibrator and/or control sample. Standard curves may be provided, with which assay results for the sample may be compared. Such standard curves present levels of marker (e.g., antibody) as a function of assay units, e.g.,fluorescent signal intensity, if a fluorescent label is used. Using samples taken from multiple donors, standard curves can be provided for reference levels of a target antibody. 8.Kits and Systems In some embodiments, the present disclosure further provides kitsand systems for performing an immunoassay described herein. In some embodiments, the disclosure further provides kitsand systemsfor detecting antibodies elicited by infection with HDV in a sample obtained from a subject. In some embodiments, the kits or systems find use in multiplex and/or automated analysis methods. In some embodiments, provided herein is a kit for detecting antibodies against hepatitis D virus (HDV) in a biological sample. In some embodiments, the kit comprises an antigenic polypeptide of the present disclosure. In some embodiments, the kit comprises at least two antigenic polypeptides of the present disclosure. In some embodiments, the kit comprises a
composition of the present disclosure, such as a composition comprising any of theantigenic polypeptides described above. In some embodiments, the kit comprises a first and a second antigenic polypeptide of the disclosure, wherein either the first or second antigenic polypeptide comprises the sequence of SEQ ID NO: 1. In some embodiments, either the first or second antigenic polypeptide comprises a sequence having at least 90% sequence identity to the ful- length sequence of SEQ ID NO: 1. Exemplary reagentsof the provided kitsinclude, but are not limited to, colorimetric reagents, enzymes, buffers, etc. Optionaly, the kit can also contain at least one calibrator or control. Any calibrator or control can be included in the kit. In some embodiments, the kit comprises a capture moiety and/or detection moiety described herein. Optionaly the assays, kits and kit components of the disclosure are optimized for use on commercial platforms (e.g., immunoassays on the ARCHITECT® and Alinity®platforms of Abbot Laboratories, Abbot Park, IL, as wel as other commercial and/or in vitrodiagnostic assays). Additionaly, the assays, kits and kit components can be employed in other formats, for example, on electrochemical or other hand-held or point-of-care assay systems. The present disclosure is, for example, applicable to the commercial Abbot Point of Care (i-STAT®, Abbot Laboratories, Abbot Park, IL) electrochemical and rapid Lateral flow immunoassay systems. Immunosensors and methods of operating them in single-use test devices are described, for example, in U.S. Patent Applications 2003/0170881, 2005/0054078 and 2006/0160164 which are incorporated herein by reference. Additional background on the manufacture of electrochemical and other types of immunosensors is found in U.S. Patent 5,063,081 which is also incorporated by reference for its teachings regarding same. Optionaly the kits include quality control reagents (for example, sensitivity panels, calibrators, negative controls, and/orpositive controls). Preparation of quality control reagents is wel known in the art, and is described, e.g., on a variety of immunodiagnostic or nucleic acid product insert sheets. The kits can optionaly include other reagents required to conduct a diagnostic assay or facilitate quality control evaluations, such as buffers, salts, enzymes, enzyme co-factors, substrates, detection reagents, and the like. Other components, such as buffers and solutions for the isolation and/or treatment of a test sample (e.g., pretreatment reagents), may also be included
in the kit. The kit may additionaly include one or more other controls. One or more of the components of the kit may be lyophilized and the kit may further comprise reagents suitable for the reconstitution of the lyophilized components. The “component,”“components,”or “at least one component,”of a kit refer generaly to a capture moiety, a detection moiety, a calibrator, a control, a sensitivitypanel, a container, a bufer, a diluent, a salt, an enzyme, a co-factor for an enzyme, a detection reagent, a pretreatment reagent/solution, a substrate (e.g.,as a solution), a stop solution, and the like that can be included in a kit for assay of a test sample, such as a patient urine, whole blood, serum or plasma sample, in accordance with the methods described herein and other methods known in the art. Some components can be in solution or lyophilized for reconstitution for use in an assay. The various components of the kit optionaly are provided in suitable containers. As indicated above, one or more of the containers may be a microtiter plate. The kit further can include containers for holding or storing a sample (e.g., a container or cartridge for a blood or urine sample). Where appropriate, the kit may also optionaly contain reaction vessels, mixing vessels and other components that facilitate the preparation of reagents or the test sample. The kit may also include one or more instruments for assisting with obtaining a test sample, such as a syringe, pipete, forceps, measured spoon, or the like. The kit further can optionaly include instructions for use, which may be provided in paper form or in computer-readable form, such as a disc, CD, DVD or the like. The disclosure as described herein also can be adapted for use in a variety of automated and semi-automated systems (including those wherein the solid phase comprises a microparticle), as described, e.g., in U.S. Patent Nos.5,089,424 and 5,006,309, and as, e.g., commercialy marketed by Abbot Laboratories (Abbot Park, IL) including but not limited to Abbot’s ARCHITECT®and Alinity®instruments, as wel as other platforms. Moreover, the disclosure optionaly is adaptable for the Abbot Laboratories commercial Point of Care (i- STAT™) electrochemical and rapid Lateral flow immunoassay systemsfor performing sandwich immunoassays. Immunosensors, and their methods of manufacture and operation in single-use test devices are described, for example in, U.S. Patent No.5,063,081, U.S. Patent Application 2003/0170881, U.S. Patent Application 2005/0054078, and U.S. Patent Application 2006/0160164, which are incorporated in their entireties by reference for their teachings regarding same.
In some embodiments, the present disclosure further providesa system comprising: a) a sample receiving component configured to receive a sample from a subject, wherein the sample comprises or is suspected of comprising a hepatitis D virus (HDV) antibody;b) a first antigenic polypeptide and a second antigenic polypeptide configured to make contact with the sample to form a first antigenic polypeptide-HDV antibody-second antigenic polypeptide complex, wherein the first antigenic polypeptide comprises a ful-length HD protein and the second antigenic polypeptide comprises amutated and/or truncatedHD protein, and wherein the second antigenic polypeptide carries a detectable label; c) a detection component configured to measure a signal generated by the detectable label in the first antigenic polypeptide-HDV antibody- second antigenic polypeptide complex; and d) an output component that indicates an amount of HDV antibody in the sample based on the signal.In some embodiments, the second antigenic polypeptide comprises an N-terminaly truncated fragment of the ful-length HD protein. As used herein, an “N-terminaly truncated fragment of the ful-length HD protein”refers to a shortened HD protein (and sequence thereof) that has been shortened by removing a portion of the protein located at or near the N-terminus. In some embodiments, the ful-lengthHD protein comprises an amino acid sequence having at least 95% sequence identity to the ful-length sequence of SEQ ID NO: 1. In some embodiments, theful-length HD protein comprises an amino acid sequence having at least 95% sequence identity to the ful-length sequence of SEQ ID NO: 3. In some embodiments, thesecond antigenic polypeptide lacks from 12 to 60 N-terminal amino acids of the ful-length HD protein. In some embodiments, thesecond antigenic polypeptide lacks21, 37 or 46 consecutive N-terminal amino acids of the ful-length HD protein. In some embodiments, thesecond antigenic polypeptide is selected from the group consisting of:i) a polypeptide that comprises the sequence of SEQ ID NO: 8 and lacks the sequence of SEQ ID NO: 13;i) a polypeptide that comprises the sequence of SEQ ID NO: 7and lacks the sequence of SEQ ID NO: 12;ii) a polypeptide that comprises the sequence of SEQ ID NO: 6 and lacks the sequence of SEQ ID NO: 11;iv) a polypeptide that comprises the sequence of SEQ ID NO: 5 and lacks the sequence of SEQ ID NO: 10;v) a polypeptide that comprises the sequence of SEQ ID NO: 4 and lacks the sequence of SEQ ID NO: 9; andvi) a polypeptide that comprises the sequence of SEQ ID NO: 2.In some embodiments, thesample is selected from the group consisting of blood, plasma andserum. In some embodiments, thedetectable label is selected from the group consisting of a fluorescent label, a chemiluminescent marker, and a chromophore.
Al patents and publications mentionedin the specification are indicative of the levels of those skiled in the art to which the disclosure pertains. Al patents and publications are herein incorporated by reference to the same extent as if each individual publication was specificaly and individualy indicated to be incorporated by reference. The disclosure ilustratively described herein suitably may be practiced in the absence of any element or elements, limitation or limitations which is not specificaly disclosed herein. Thus, for example, in each instance herein any of the terms “comprising,”“consisting essentialy of”and “consisting of”may be replaced with either of the other two terms. The terms and expressions which have been employed are used as terms of description and not of limitation, and there is no intention that in the use of such terms and expressions of excluding any equivalents of the features shown and described or portions thereof, but it is recognized that various modifications are possible within the scope of the disclosure claimed. Thus, it should be understood that although the present disclosure includes various embodimentsand optional features, modification and variation of the concepts herein disclosed may be resorted to by those skiled in the art, and that such modifications and variations are considered to be within the scope of this disclosure as defined by the appended claims. EXAMPLES Example 1 Design and engineering HD Large antigen The ful-length HD Large antigen (HD LAg, SEQ ID NO:1) was derived from consensus amino acid sequence of 227 HD large antigen sequences. Al 227 HD LAg sequences were retrieved from NCBI database including 205 sequences from HDV Genotype 1, 7 HDV sequences from genotype 2 and 15 HDV sequences from genotype 3. Amino acid alignments of the 227 HD LAgs were manualy edited in BioEdit version 7.2.5 or higher to remove gaps to generate the consensus sequence. Subsequent 5 N terminal truncated (HD D21-D56) and one mutant (HD LAg M) proteins were designed based on the consensus sequence of HD LAg. FIG.1 ilustrates sequence alignment of the HD LAg, 5 N-terminal truncated HD Ags and HD LAg mutant as wel as the smal HD Ag.
Example2 Design, expression, and purification of recombinant HD proteins The folowing is a general protocol to design, prepare and purify HD recombinant proteins expressed from plasmid constructs in E. coli. An N or C-terminal Histidine (His)-tag was fused to the ORF for each protein to alow purification using His-Bind Nickel Afinity Chromatography. Codon optimization and cloning of the DNA sequence was performed by a third-party vendor, ATUM (Newark, CA). Al proteins were expressed by transforming BL21(DE3) Chemicaly Competent E. coli cels by heat shock transformation method. Growth and Induction of E. coli Strains with HD Recombinant Protein Constructs A culture of BL21(DE3) Chemicaly Competent E. coli harboring a pD454-SR-based construct was prepared by inoculating a single colony from an LB agar plate into a 125 ml Erlenmeyer flask (Corning Inc., Corning, NY) containing 500 ml LB Broth (Sigma-Aldrich) supplemented with 100 µg/ml ampicilin. The flask was placed in a shaking orbital incubator and incubated overnight (~16 hours) at 37ºC. Four ml of the overnight culture was transfered to a sterile 2-liter flask containing 400 ml of Terific Broth + 100 µg/ml ampicilin. The culture was incubated in a shaking orbital air incubator at 37ºC until reaching a cel density of OD600 = 0.6 – 0.8.Cels were then induced with a final concentration of 1 mM isopropyl β-D- thiogalactopyranoside (IPTG) at 37 ºC for 4 hours. After the induction period, cels were harvested by centrifugation and the Terific Broth supernatant was discarded. Cel pelets were stored at –70 ºC until further processing. Recombinant proteins produced as insoluble inclusion bodies within E. coli. Frozen cel pelets were thawed and resuspended thoroughly with lysis bufer containing 6 M guanidine-HCl, 30 mM NaCl, pH 7.8 and 50mM Sodium Phosphate using 10 ml of lysis buffer per gram of pelet. The solubilized recombinant proteins were then clarified by centrifugation. Purification of HD Recombinant Proteins by His-Bind Nickel Afinity Chromatography The pooled recombinant protein from the S-200 column was loaded onto a His-Bind nickel column (Novex-Life Technologies) equilibrated with binding bufer containing 20mM sodium Phosphate, pH 7.8, 0.5 M NaCl, 20 mM imidazole and 6 M urea. The bound protein was
washed with 10 column volumes of binding bufer folowed by 6 column volumes of wash bufer (20mM sodium Phosphate, pH 7.8, 0.5 M NaCl, 20 mM imidazole and 6 M urea). The bound protein was eluted at 1 ml/min with a 0–500 mM linear imidazole gradient. Fractions containing the purified protein as assessed by Coomassie Blue staining were pooled and dialyzed against 1 L of 20mM sodium Phosphate, pH 7.8, 0.5 M NaCl and 6 M urea bufer over a 24 h period at 4°C. The dialyzed solution containing the purified protein was aliquoted and stored at –70 ºC for future use. Example3 ARCHITECT® Chemiluminescent Immunoassay for Detection of Antibodies to HDV (indirect or anti-human assay format) Using the ful-length HD Large antigen (SEQ ID NO:1), HDV IgG prototype chemiluminescent microparticle immunoassays (CMIA) were developed on the automated ARCHITECT® instrument system (Abbot Laboratories, Abbot Park, IL).The indirect ARCHITECT® HDV IgG was a reference test for comparison to the direct HDV Total Ig assay (Example 4), it is a two-step immunoassay which utilizes an indirect anti-human assay format as ilustrated in Figure 2A. The first step combines sample, assay diluent and paramagnetic microparticles. Anti-HDV antibodies (Ab) present in the sample are captured on paramagnetic particles coated with the recombinant HD protein (Ag/Solid-Ab). The microparticles are washed to remove unbound Anti-HDV antibodies. In the second step, anti-HDV antibodies captured by the microparticles (Ag/Solid-Ab) are incubated with acridinium-labeled anti-human IgG monoclonal conjugate (Ab* to form immunoproduct(Ag/Solid-Ab-Ab*). Folowing an additional wash cycle, alkaline hydrogen peroxide solution is added to release acridinium chemiluminescence signal. The intensity of the chemiluminescence, measured as relative light unit (RLU), is proportional to the amount of specific antibody captured by the recombinant HD protein. The ARCHITECT® HDV IgG results are reported as the ratio of the sample RLU to the cutof RLU (S/CO) for each specimen. Specimens with S/CO values < 1.00 are considered non-reactive; specimens with S/CO values ≥1.00 are considered reactive. Example 4 ARCHITECT® Chemiluminescent Immunoassay for Detection of Antibodies to HDV (Direct or Sandwich Assay Format)
To improve HDV antibody assay performance, a direct double antigen sandwich assay (Ag/solid-Ab-Ag*) was developed on the automated ARCHITECT® instrument system (Abbot Laboratories, Abbot Park, IL).The direct ARCHITECT® HDV Total Ig is a One-step immunoassay that utilizes two HD antigens (i.e., the Ful-length HD = SEQ ID NO:1) as capture Ag on solid phase (Ag/solid), and Ful-length HD = SEQ ID NO:1 or the mutated/truncated HD Ags = SEQ ID NO: 2 and SEQ ID NOs: 4-8 as detection conjugate(Ag*) to formadouble antigen sandwich with the anti-HDV antibody (Ag/solid-Ab-Ag*). As ilustrated in Figure 2B, in the first step, sample(such as serum or plasma), paramagnetic microparticles and antigen conjugate are combined.Anti-HDV antibodies, present in the sample simultaneously form a double antigen sandwich immunoproduct (Ag/solid-Ab-Ag*) captured on paramagnetic particles coated with the Ful-length HD recombinant protein. In the second step, the microparticles are washed to remove unbound antibodies and conjugates. Folowing an additional wash cycle, alkaline hydrogen peroxide solution is added to release acridinium chemiluminescence signal. The intensity of the chemiluminescence, measured as relative light unit (RLU), is proportional to the amount of anti- HDV antibody in the sample. The ARCHITECT® HDV Total Ig results are reported as the ratio of the sample RLU to the cutof RLU (S/CO) for each specimen. Specimens with S/CO values < 1.00 are considered non-reactive; specimens with S/CO values ≥1.00 are considered reactive. Example 5 Identification of optimal engineered HD protein sequences for the direct ARCHITECT® HDV Total Ig Assay Performance of the direct ARCHITECT® HDV Total Ig Assay using the ful-length (FL) HD LAg (SEQ IDNO:1) as the capture antigen and the FL HD LAg (SEQ IDNO:1) or the 6 individual engineered HD LAgs (SEQ ID NO: 2 and SEQ ID NOs: 4-8) as detection antigen was first evaluated withnegativeand positive controls and a smal HDV panel including HDV RNA positive (n=12) and negative (n=4) samples. The indirect ARCHITECT® HDV IgG Assay was used as a reference test. Initial testing of the negative control revealed extremely high background, >100,000 RLU, in the direct ARCHITECT® HDV Total Ig Assay using the FL HD LAg as both capture and detection antigen, resulting in much lowersignal-to-noise ratios (S/Ns)than the indirect ARCHITECT® HDV IgG assay (Table 1). However, replacing the FL HD LAg conjugate with the N-terminal truncated HD LAgs (D21-D56) or the HD LAg mutant detection conjugate, remarkably reduced the assay background to 70 RLU. There is a clear trend, the longer amino acid
deletion, the lower assay background. However, sensitivity testing of HDV RNA positive samples, D37 and D46 HD Ags were identified as potential candidates as detection antigen for the direct ARCHITECT® HDV Total Ig Assay. Notably, the D37 detected 11/12 HDV RNA positive samples while D46 detected 10/12 of them. Also, both D37 and D46 had minimal reactivity with the 4 HDV RNA negative samples that were falsely reactive in the indirect ARCHITECT® HDV IgG Assay.
[0211 ] Table 1 : Evaluation of engineered HD proteins as detection antigens paired with the full-length HD capture antigen in direct format HDV Total Ig prototype assay.
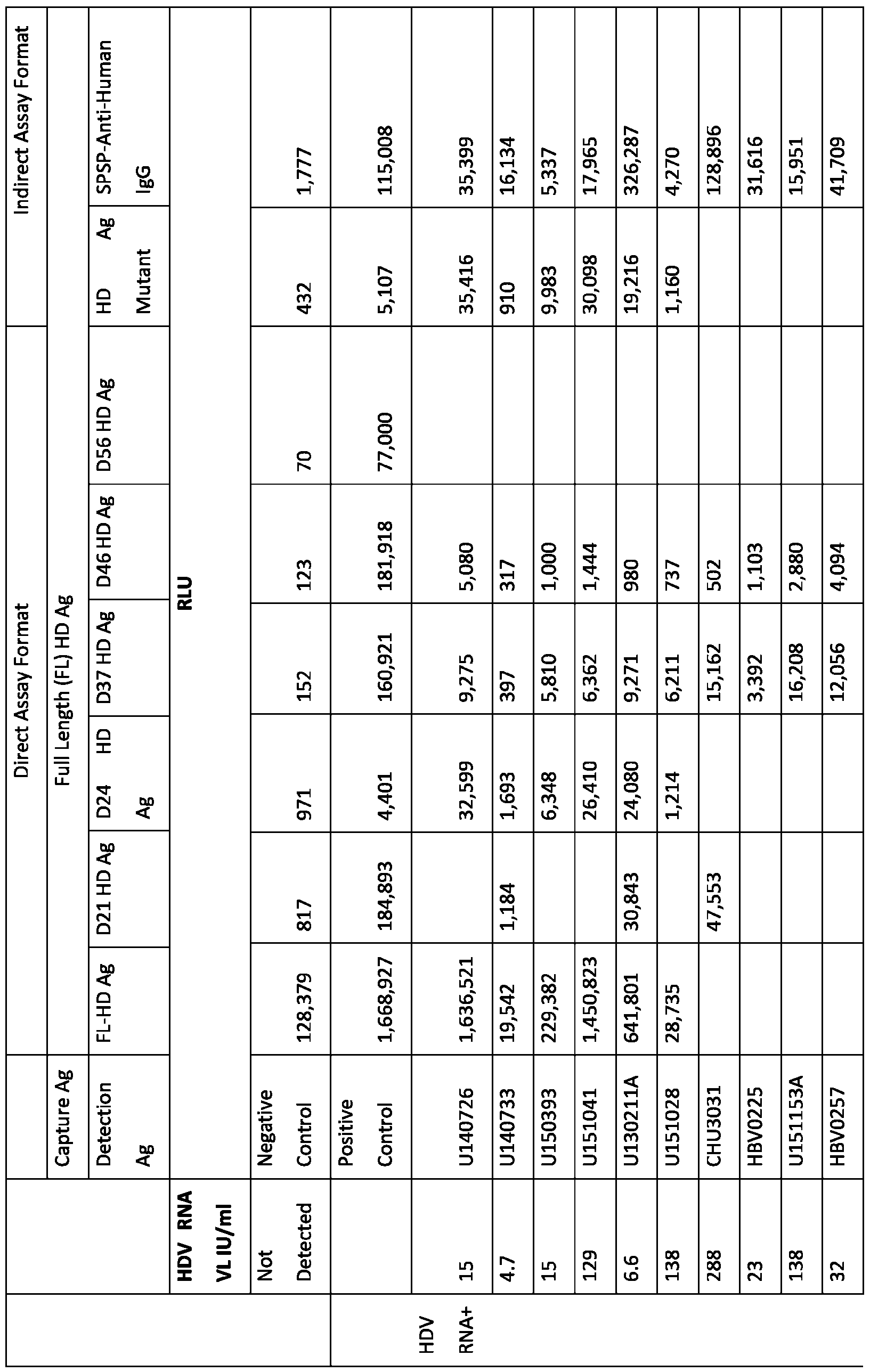

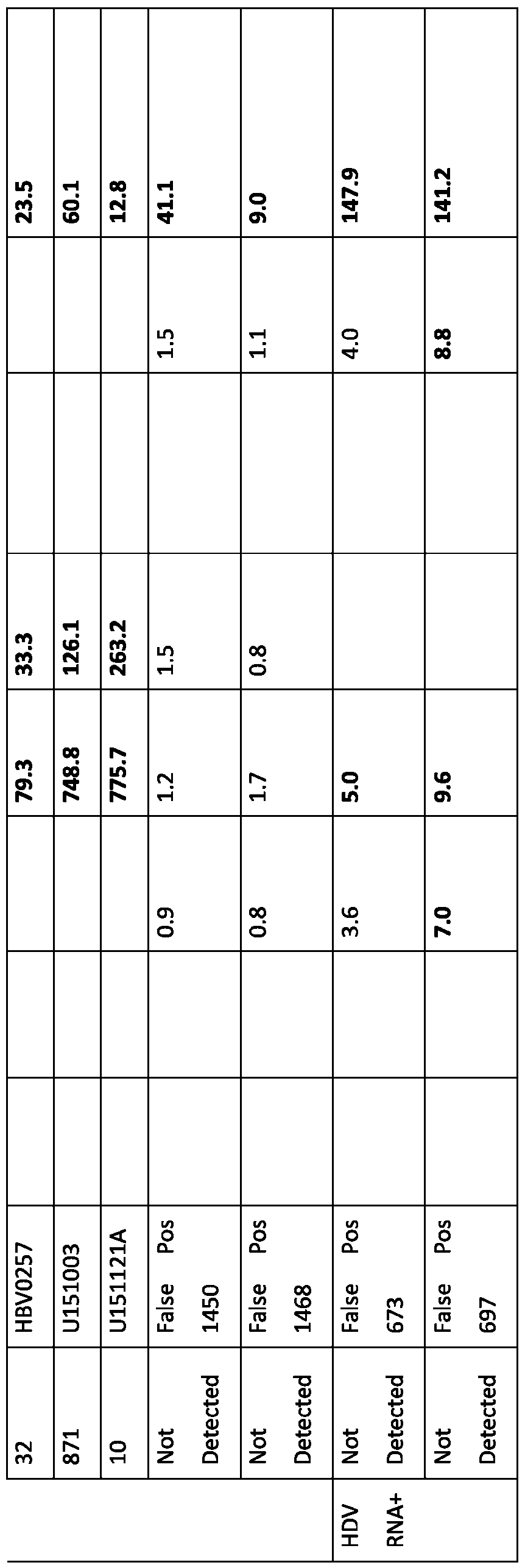
Example 6 Performance of the direct ARCHITECT® HDV Total Ig Assay and Agreement with HDV RNA test Performance of the direct ARCHITECT® HDV Total Ig assay with the FL HD LAg as capture antigen and D37 HD LAg as detection antigenwas further evaluated using an expanded HDV panel containing 132samples with previously determined HBV and HCV status as listed in Table 2. The panel included101 samples with and 31samples without HBV infection.17of the 101 HBV infected samples were coinfected with HCV. Among the 31HBV negative samples, 21samples had HCV mono-infection while 10samples were not infected with either HBV or HCV. Al samples were pre-screened using a research use only (RUO) HDV RNA test, m2000 RealTime HDV(Coler KE et al., Development and performance of prototype serologic and molecular tests for hepatitis delta infection. Sci Rep.2018 Feb 1;8(1):2095), 62samples were determined having detectable HDV RNA whereas 70samples without detectable HDV RNA.Also,a CE marked LIAISON®XL Anti-HDVassay(code 311260, DiaSorin, Saluggia, Italy) was used as acomparator assay folowing the manufacturers’ recommendations. The direct ARCHITECT® HDV Total Ig assay detected 70/101 HBV+ and HBV+/HCV+ samples, it also detected 2/21HBV-/HCV+ but 0/10HBV- /HCV-samples. Although the indirect LIAISON®XL Anti-HDV assay (also referred to herein as DiaSorinanti-HDV IgG+) detected 79/101 HBV+ and HBV+/HCV+ samples and 9/21 HBV-/HCV+, it detected 6/10HBV-/HCV-samples indicating poor specificity of the assay (Table 2).Only 62of the 101 HBV positive samples had detectable HDV RNA viral load (0.34- 7.82 log10 IU/ml) and RNA was not detected in the 31HBV negative samples. Analysis of agreement with molecular HDV RNA testshowed that the direct ARCHITECT HDV Total Ig detected 59/62RNA positive samples demonstrating 95.2% positive agreement and comparable sensitivity (p=0.2482) to the RNA test, whereas the positive agreement of LIAISON Anti-HDV was 90.3% (56/62), significantly lower sensitivity (p=0.0412) than the RNA test (Table 3). Although both serology assays had relatively low negative agreement with the RNA test on the 70RNA negative samples, the 81.4% (57/70) negative agreement of ARCHITECT HDV Ig was significantly greater (p=0.0001) than the 45.7% (32/70) negative agreement of LIAISON Anti-HDV. The results of the comparative study indicate beter agreement of the ARCHITECT HDV Ig assay with the HDV RNAtest than LIAISON Anti-HDVassay.The data clearly demonstrated significant beter positive and negative agreementof the ARCHITECT HDV Ig assay withthe HDV RNA test.
Table 2. Characteristics of HDV panel and performance of serology assays.
 Table 3.Percent Agreement of HDV Antibody assays with HDV RNA test on 126 samples.
Table 3.Percent Agreement of HDV Antibody assays with HDV RNA test on 126 samples.
 Table 3 lists p values compared to RNA test. Also, P=0.0001 comparing between 81.4% Negative Agreementof Architect HDV Total Ig and 45.7% Negative Agreement of LIAISON Anti-HDV, significant specificity difference. As shown in Table 3, 13/70of HDV RNA negative samples were reactive in ARCHITECT HDV Ig, resultingin81.4% negative agreement with the RNA test.Most likely, the 13 sero-reactive samples were acute HBV-HDV coinfection or superinfection with self-resolved outcome. Studies have shown, >90% coinfection and ~10% superinfection result in spontaneous clearance of HDV RNA, but the antibody responses persist for several years even after the clearance of RNA.In this regard, the highly specific ARCHITECT HDV Igassay provides an optimum test for universal screening of al HBsAg positive individuals to provide accurate prevalence data on past and active HDV infection. Example 7 Potential utility of ARCHITECT HDV Total Ig for assessing disease progression of HDV coinfection and superinfection Another significant advantage of the direct ARCHITECT HDV Total Ig is its enhanced dynamic range of assay signal, which may provide potential utility to assess disease
progressionof HDV-HBV coinfection and superinfection. Signal ploting of the 62HDV RNA positivesamples in ARCHITECT HDV Total Ig and LIAISON Anti-HDV assays against HDV RNA viral load revealed up to 80-fold higher signal of the direct ARCHITECT HDV Ig than LIAISON Anti-HDV for most RNA positive samples (Fig.3). The enhanced dynamic range enabled the assay to generate much higher signal for 8samples (193-1150 S/CO), separating them from the 6samples (2.1-19.3 S/CO) within the same low viral load range (0.53-1.9 log10 IU/ml, Fig.3). This reactivity patern (high antibody response with low viral load) is halmark of spontaneously clearing HDV RNA based on typical evolution of serological and virological markers in HDV infection(Hughes SA, Wedemeyer H, Harison PM. Hepatitis delta virus. Lancet.2011 Jul 2;378(9785):73-85. doi: 10.1016/S0140-6736(10)61931-9). Thus, the direct ARCHITECT HDV Ig test may provide additionalinformation useful for assessing disease progression of HDV coinfection and superinfection. Both serology assays were able to detect samples with RNA viral load ranged from 0.51-7.82 log10 IU/ml except 6samples.Notably, there were 6RNA positive samples that were missed by LIAISON Anti-HDV and/or ARCHITECT HDV Total Ig, al had low viral load (2.2-34 IU/ml), suggesting they were in the acute HDV infection resulted seronegative window period. Also, due to diferent infection types (coinfection or superinfection) and stages (acute or convalescence) of the samples as wel as the limit of high dynamic range, linear corelations with HDV RNA viral load were low, R2 =0.35 for Architect HDV Total Ig and R2 = 0.26 for LIAISON Anti-HDV. Example 8 Specificity evaluation of ARCHITECT HDV Total Ig Normal blood donors (100 serum and 100 plasma samples) pre-screened negative for HBV, HCV, HIV and HTLV from Gulf Coast Regional Blood Center (Houston, Texas) were used for specificity evaluation of ARCHITECT HDV Total Ig assayand LIAISON Anti- HDV assays. Only one sample was low reactive (2.3 S/CO)in ARCHITECT HDV Total Ig assay, resulting 99.5% (199/200, 95%CI=97.65-99.98) specificity, whereas 3 samples were reactive (1.0, 2.0 and 7.0 AU) in LIAISON Anti-HDV and specificity was 98.5% (197/200, 95%CI=96.1-99.6) (Fig.4). The data supportsthe observed beter specificity of ARCHITECT HDV Total Ig assayand high negative agreement with HDV RNA test than the LIAISON Anti- HDV.
9.Sequences Sequences relevant to the embodiments of the present disclosure are provided in the tables below. Table 5: Polypeptide Sequences
Table 3 lists p values compared to RNA test. Also, P=0.0001 comparing between 81.4% Negative Agreementof Architect HDV Total Ig and 45.7% Negative Agreement of LIAISON Anti-HDV, significant specificity difference. As shown in Table 3, 13/70of HDV RNA negative samples were reactive in ARCHITECT HDV Ig, resultingin81.4% negative agreement with the RNA test.Most likely, the 13 sero-reactive samples were acute HBV-HDV coinfection or superinfection with self-resolved outcome. Studies have shown, >90% coinfection and ~10% superinfection result in spontaneous clearance of HDV RNA, but the antibody responses persist for several years even after the clearance of RNA.In this regard, the highly specific ARCHITECT HDV Igassay provides an optimum test for universal screening of al HBsAg positive individuals to provide accurate prevalence data on past and active HDV infection. Example 7 Potential utility of ARCHITECT HDV Total Ig for assessing disease progression of HDV coinfection and superinfection Another significant advantage of the direct ARCHITECT HDV Total Ig is its enhanced dynamic range of assay signal, which may provide potential utility to assess disease
progressionof HDV-HBV coinfection and superinfection. Signal ploting of the 62HDV RNA positivesamples in ARCHITECT HDV Total Ig and LIAISON Anti-HDV assays against HDV RNA viral load revealed up to 80-fold higher signal of the direct ARCHITECT HDV Ig than LIAISON Anti-HDV for most RNA positive samples (Fig.3). The enhanced dynamic range enabled the assay to generate much higher signal for 8samples (193-1150 S/CO), separating them from the 6samples (2.1-19.3 S/CO) within the same low viral load range (0.53-1.9 log10 IU/ml, Fig.3). This reactivity patern (high antibody response with low viral load) is halmark of spontaneously clearing HDV RNA based on typical evolution of serological and virological markers in HDV infection(Hughes SA, Wedemeyer H, Harison PM. Hepatitis delta virus. Lancet.2011 Jul 2;378(9785):73-85. doi: 10.1016/S0140-6736(10)61931-9). Thus, the direct ARCHITECT HDV Ig test may provide additionalinformation useful for assessing disease progression of HDV coinfection and superinfection. Both serology assays were able to detect samples with RNA viral load ranged from 0.51-7.82 log10 IU/ml except 6samples.Notably, there were 6RNA positive samples that were missed by LIAISON Anti-HDV and/or ARCHITECT HDV Total Ig, al had low viral load (2.2-34 IU/ml), suggesting they were in the acute HDV infection resulted seronegative window period. Also, due to diferent infection types (coinfection or superinfection) and stages (acute or convalescence) of the samples as wel as the limit of high dynamic range, linear corelations with HDV RNA viral load were low, R2 =0.35 for Architect HDV Total Ig and R2 = 0.26 for LIAISON Anti-HDV. Example 8 Specificity evaluation of ARCHITECT HDV Total Ig Normal blood donors (100 serum and 100 plasma samples) pre-screened negative for HBV, HCV, HIV and HTLV from Gulf Coast Regional Blood Center (Houston, Texas) were used for specificity evaluation of ARCHITECT HDV Total Ig assayand LIAISON Anti- HDV assays. Only one sample was low reactive (2.3 S/CO)in ARCHITECT HDV Total Ig assay, resulting 99.5% (199/200, 95%CI=97.65-99.98) specificity, whereas 3 samples were reactive (1.0, 2.0 and 7.0 AU) in LIAISON Anti-HDV and specificity was 98.5% (197/200, 95%CI=96.1-99.6) (Fig.4). The data supportsthe observed beter specificity of ARCHITECT HDV Total Ig assayand high negative agreement with HDV RNA test than the LIAISON Anti- HDV.
9.Sequences Sequences relevant to the embodiments of the present disclosure are provided in the tables below. Table 5: Polypeptide Sequences






For reasons of completeness, various aspects of the disclosureare set out in the folowing numbered clauses: Clause 1: An antigenic polypeptide comprising a sequence having at least 90% sequence identity to the sequence of SEQ ID NO: 8, wherein said antigenic polypeptide lacks a sequence comprising at least 90% sequence identity to a region of from about 12 to about 60 amino acids at the N-terminus region of SEQ ID NO: 1. Clause 2:The antigenic polypeptide of clause 1, wherein said antigenic polypeptide lacks a sequence comprising at least 90% sequence identity with a sequence selected from the groupconsisting ofSEQ ID NO: 9, SEQ ID NO: 10, SEQ ID NO: 11; SEQ ID NO: 12; and SEQ ID NO: 13. Clause 3:The antigenic polypeptide of clause1, wherein said antigenic polypeptide comprises at least one amino acid mutation as compared to the sequence of amino acids 1 to 60 of SEQ ID NO: 1. Clause 4:The antigenic polypeptide of any one of clauses1-3, wherein said antigenic polypeptide comprises a sequence selected from the group consisting of SEQ ID NO: 2, SEQ ID NO: 4, SEQ ID NO: 5, SEQ ID NO: 6, SEQ ID NO: 7 and SEQ ID NO: 8. Clause 5: The antigenic polypeptide of clause1, wherein said antigenic polypeptide comprises the sequence of SEQ ID NO: 6 and lacks the sequence of SEQ ID NO: 11. Clause 6:The antigenic polypeptide of any one of clauses1-5, wherein the antigenic polypeptide is capable of being bound by an antibody directed to HD protein. Clause 7:The antigenic polypeptide of any one of clauses1-6, wherein said antigenic polypeptide is fused to at least one heterologous peptide. Clause 8:The antigenic polypeptide of clause7, wherein the heterologous peptide comprises an afinity tag, an epitope tag, a fluorescentprotein, an enzyme or a carier protein. Clause 9: An antigenic polypeptide comprising a sequence having at least 90% sequence identity to a truncated polypeptide derived from SEQ ID NO: 1, said truncated polypeptide lacking from 12 to 60 N-terminal amino acids of SEQ ID NO: 1. Clause 10:The antigenic polypeptide of clause9, wherein the truncated polypeptide comprises a sequence of at least 158 amino acids of SEQ ID NO: 1.
Clause 11:An antigenic polypeptide comprising an amino acid sequence having at least 90% sequence identity to any of the ful-length sequences of SEQ ID NO: 2 and SEQ ID NOs: 4- 8, and a heterologous peptide. Clause 12:The antigenic polypeptide of clause11, wherein the heterologous peptide comprises an afinity tag, an epitope tag, a fluorescentprotein, an enzyme or a carier protein. Clause 13: A composition comprising the antigenic polypeptide of any one ofclauses 1-12. Clause 14:The composition of clause13, further comprising a detectable label. Clause 15:The composition of clause 14, wherein the detectable label is conjugated to the antigenic polypeptide. Clause 16:The composition of clause14 or 15, wherein the detectable label is selected from the group consisting of a fluorescence label, a chemiluminescent label, an enzymatic label, and a particle label. Clause 17: The composition of any one of clauses13-16, further comprising a second antigenic polypeptide that comprises the sequence of SEQ ID NO: 1. Clause 18:The composition of clause 17,wherein the second antigenic polypeptide comprises a means for binding to a solid support. Clause 19:A kit for detecting antibodies against hepatitis D virus (HDV) in a biological sample, comprising the antigenic polypeptide of any one of clauses1-12or the composition of any one of clauses 13-18. Clause 20:A kit for detecting antibodies against hepatitis D virus (HDV) in a biological sample, comprising the antigenic polypeptide of any one of clauses1-12 and a second antigenic polypeptide that comprises the sequence of SEQ ID NO: 1. Clause 21: A method for detecting antibodies specific for HD protein in a biological sample, wherein an antigenic polypeptide according to any one of clauses 1-12 is used as a binding partner for said HD protein antibodies. Clause 22:A method for detecting antibodies specific for HD protein in a biological sample, said method comprising: a) forming an immunoreaction mixture by mixing a biological sample with the antigenic polypeptide according toany one ofclauses 1-12;b) maintaining said immunoreaction mixture for a time period suficient for alowing antibodies against said antigenic polypeptide present in the biological sample to immunoreact with said antigenic polypeptide to
form an immunoreaction product; andc) detecting the presence and/or the concentration of said immunoreaction product. Clause 23:The method of clause 22,wherein said immunoreaction is carried out in a double antigen sandwich format comprising:a) adding to said biological sample a first antigenic polypeptide that comprises a means for binding to a solid support and a second antigenic polypeptide that caries a detectable label, said second antigenic polypeptide comprising the antigenic polypeptide of any one of clauses 1-12, wherein said first antigenic polypeptide and said second antigenic polypeptide bind specificaly to the HD protein antibodies, b) forming an immunoreaction mixture comprising the first antigenic polypeptide, the biological sample antibody and the second antigenic polypeptide, wherein the solid support is added before, during or after forming the immunoreaction mixture,c) maintaining said immunoreaction mixture for a time period suficient for HD protein antibodies in the biological sample to immunoreact with said first and second antigenic polypeptides to form an immunoreaction product, andd) detecting the presence of any of said immunoreaction product. Clause 24:The method of clause 23, wherein the first antigenic polypeptide comprises a sequence having at least 90% sequence identity to the ful-length sequence of SEQ ID NO: 1. Clause 25:The method of clause 23 or 24, further comprising a washing step prior to the detecting step. Clause 26: The method of clause 25, wherein the immunoreaction product is bound to the solid support and the washing step comprises removing or separatingsome or al ofthe first antigenic polypeptide, HD protein antibody and second antigenic polypeptide that are not part of the immunoreaction product that is bound to the solid support. Clause 27:The method of any one of clauses 23-26, wherein the solid support is selected from the group consisting of acolumn, bead, test tube, microtiter dish,multi-wel plate, microparticle, microsphere, a test stick, a test strip, microchip and membrane. Clause 28:The method of any one of clauses 22-27, wherein the biological sample is a body fluid sample from a human subject. Clause 29:The method of clause 28, wherein the biological sample is selected from the group consisting of blood, plasma andserum. Clause 30: A polynucleotide encoding the antigenic polypeptide of any one of clauses1 to12.
Clause 31:The polynucleotide of clause 30, further comprising a heterologous regulatory element operably linked to a nucleic acid encoding the antigenic polypeptide. Clause 32:The polynucleotide of clause 31, wherein the heterologous regulatory element is a promoter. Clause 33:A vector comprising the polynucleotide of any one ofclauses 30-32. Clause 34: The vector of clause 33, wherein the vector is a plasmid, naked nucleic acid, phage, viral vector, or virus. Clause 35:A cel comprising the polynucleotide of any one of clauses 30 to 32 or the vector of clause33 or 34, or expressing the antigenic polypeptide of any one of clauses1 to 12. Clause 36:A method for detecting antibodies against hepatitis D virus (HDV) in a biological sample, comprising performing a HDV serology assay using a direct assay format with a first antigenic polypeptide and a second antigenic polypeptide, wherein the first antigenic polypeptide comprises a ful-length HD protein and the second antigenic polypeptide comprises an N-terminaly truncated fragment of the ful-length HD protein. Clause 37:The method of clause 36, wherein the assay is a double antigen sandwich assay. Clause 38:The method of clause36 or 37, wherein the second antigenic polypeptide lacks from 12 to 60 N-terminal amino acids of the HD protein. Clause 39: The method of clause 38, wherein the second antigenic polypeptide lacks 21, 37 or 46 consecutive N-terminal amino acids of the HD protein. Clause 40:The method of any one of clauses 36-39, wherein the ful-length HD protein comprises an amino acid sequence having at least 95% sequence identity to the ful-length sequence of SEQ ID NO: 1. Clause 41:A method for detecting antibodies against hepatitis D virus (HDV) in a biological sample, comprising:a) contacting abiological sample, either simultaneously or sequentialy, in any order with:(1) a first antigenic polypeptide under conditions suficient to alow binding of anyHDV antibody present in the biological sample to the first antigenic polypeptide, wherein said first antigenic polypeptide is immobilized on a solid support, and (2) a second antigenic polypeptide under conditions suficient to alow binding of the second antigenic polypeptide to any HDV antibody present in the biological sample, such that a first antigenic polypeptide-HDV antibody-second antigenic polypeptide complex is formed and is bound to the
solid support, and b) detecting the presence of said first antigenic polypeptide-HDV antibody- second antigenic polypeptide complex, wherein said first antigenic polypeptide comprises a sequence having at least 90% sequence identity to the ful-length sequence of SEQ ID NO: 1; wherein said second antigenic polypeptide comprises a sequence having at least 90% sequence identity to a truncated polypeptide derived from SEQ ID NO: 1, said truncated polypeptide lacking from 12 to 60 N-terminal amino acids of SEQ ID NO: 1; and wherein said second antigenic polypeptide carries a detectable label. Clause 42:The method of clause 41, wherein the second antigenic polypeptide is selected from the group consisting of:i) a polypeptide that comprises the sequence of SEQ ID NO: 8and lacks the sequence of SEQ ID NO:13;i) a polypeptide that comprises the sequence of SEQ ID NO: 7and lacks the sequence of SEQ ID NO: 12;ii) a polypeptide that comprises the sequence of SEQ ID NO: 6and lacks the sequence of SEQ ID NO:11;iv) a polypeptide that comprises the sequence of SEQ ID NO: 5and lacks the sequenceof SEQ ID NO:10;v) a polypeptide that comprises the sequence of SEQ ID NO: 4and lacks the sequence of SEQ ID NO:9;and vi) a polypeptide that comprises the sequence of SEQ ID NO: 2. Clause 43: The method of clause 41 or 42, further comprising a washing step prior to the detecting step. Clause 44:The method of clause 43, wherein the washing step comprises removing some or al of the first antigenic polypeptide, HDV antibody and second antigenic polypeptide that are not part of the first antigenic polypeptide-HDV antibody-second antigenic polypeptide complex that is boundto the solid support. Clause 45:The method of any one of clauses 41-44, wherein the step of detecting comprises detecting the presence and/or the concentration of the first antigenic polypeptide-HDV antibody-second antigenic polypeptide complex that is formed and is bound to the solid support. Clause 46:The method of any one of clauses 41-44, wherein the step of detecting comprises measuring an amount of saidfirst antigenic polypeptide-HDV antibody-second antigenic polypeptide complex that is formed and is bound to the solid support. Clause 47:The method of any one of clauses 41-46, wherein the solid support is selected from the group consisting of acolumn, bead, test tube,microtiter dish,multi-wel plate, microparticle, microsphere, a test stick, a test strip, microchip and membrane.
Clause 48:The method of any one of clauses 41-47, wherein the biological sample is selected from the group consisting of blood, plasma andserum. Clause 49:A method of detecting hepatitis D virus (HDV) infection in a subject comprising performing the method of any one of clauses 41-48, wherein the biological sample is a sample taken from the subject and wherein the step of detecting comprises detecting the presence of said first antigenic polypeptide-HDV antibody-second antigenic polypeptide complex that is formed and is bound to the solid support, thereby detecting the presence of past or present HDV infection in said subject. Clause 50:A method of estimating HDV incidence in a population, the method comprising, a) providing a set of samples derived from a plurality of individuals within the population over a period of time;b) performing the method of clause 49 on each sample in the set of samples; andc) determining the percentage of recent HDV infections over the period of time; wherein the percentage of recent HDV infections over the period of time provides an estimate of HDV incidence in the population. Clause 51:A system comprising: a) a sample receiving component configured to receive a sample from a subject, wherein the sample comprises or is suspected of comprising a hepatitis D virus (HDV) antibody; b) a first antigenic polypeptide and a second antigenic polypeptide configured to make contact with the sample to form a first antigenic polypeptide-HDV antibody- second antigenic polypeptide complex, wherein the first antigenic polypeptide comprises a ful- length HD protein and the second antigenic polypeptide comprises a mutatedHD protein, and wherein the second antigenic polypeptide caries a detectable label; c) a detection component configured to measure a signal generated by the detectable label in the first antigenic polypeptide- HDV antibody-second antigenic polypeptide complex; and d) an output component that indicates an amount of HDV antibody in the sample based on the signal. Clause 52.The system of clause 51, wherein thesecond antigenic polypeptide comprises an N-terminaly truncated fragment of a ful-length HD protein. Clause 53:The system of clause 51or 52, wherein the ful-length HD protein comprises an amino acid sequence having at least 95% sequence identity the ful-length sequence of SEQ ID NO: 1. Clause 54: The system ofany one ofclauses51-53, wherein the second antigenic polypeptide lacks from 12 to 60 N-terminal amino acids of the ful-length HD protein.
Clause 55: The system of clause 54, wherein the second antigenic polypeptide lacks 21, 37 or 46 consecutive N-terminal amino acids of the ful-length HD protein. Clause 56:The systemof clause 52 or 53, wherein the second antigenic polypeptide is selected from the group consisting of: i) a polypeptide that comprises the sequence of SEQ ID NO: 8and lacks the sequence of SEQ ID NO:13; i) a polypeptide that comprises the sequence of SEQ ID NO: 7and lacks the sequence of SEQ ID NO: 12;ii) a polypeptide that comprises the sequence of SEQ ID NO: 6and lacks the sequence of SEQ ID NO:11;iv) a polypeptide that comprises the sequence of SEQ ID NO: 5and lacks the sequence of SEQ ID NO:10;v) a polypeptide that comprises the sequence of SEQ ID NO: 4and lacks the sequence of SEQ ID NO:9;and vi) a polypeptide that comprises the sequence of SEQ ID NO: 2. Clause 57:The system of any one of clauses 51-56, wherein the sample is selected from the group consisting of blood, plasma andserum. Clause 58: The system of any one of clauses 51-57, wherein the detectable label is selected from the group consisting of a fluorescent label, a chemiluminescent marker, and a chromophore. Clause 59: An antigenic polypeptide comprising an amino acid sequence having a formula of: MSRSESKKNRGGX1EEIX2EQX3X4SGX5KKLEELERDLRKVKKKIKKLEDENP WLGNIKGILGKKDKDGEGAPPAKRARTDQMEVDSGPRKRPLRGGFTDKERQDHRRRK ALENKKKQLSGGGKNLSKEEEEELKRLTEEDERRERRVAGPPVGGVNPLEGGSRGAPG GGFVPSMQGVPESPFTRTGEGLDIRGNQGFPWDILFPADPPFSPQSCRPQ(SEQ ID NO:22), wherein X1 is R or S; X2 is L or G; X3 is W or A; X4 is V or G; and X5 is R or S, provided that X1 if S if X2 is L, X3 is W, X4 is V and X5 is R. Clause 60. The antigenic polypeptide of claim 59, wherein when X1is S, X2is G, X3 is A, X4 is G and X5 is S.
Claims
CLAIMS What is claimed is: 1. An antigenic polypeptide comprising a sequence having at least 90% sequence identity to the sequence of SEQ ID NO: 8, wherein said antigenic polypeptide lacks a sequence comprising at least 90% sequence identity to a region of from about 12 to about 60 amino acids at the N- terminus region of SEQ ID NO: 1.
2. The antigenic polypeptide of claim 1, wherein said antigenic polypeptide lacks a sequence comprising at least 90% sequence identity with a sequence selected from the group consisting of SEQ ID NO: 9, SEQ ID NO: 10, SEQ ID NO: 11; SEQ ID NO: 12; and SEQ ID NO: 13.
3. The antigenic polypeptide of claim 1, wherein said antigenic polypeptide comprises at least one amino acid mutation as compared to the sequence of amino acids 1 to 60 of SEQ ID NO: 1.
4. The antigenic polypeptide of any one of claims 1-3, wherein said antigenic polypeptide comprises a sequence selected from the group consisting of SEQ ID NO: 2, SEQ ID NO: 4, SEQ ID NO: 5, SEQ ID NO: 6, SEQ ID NO: 7 and SEQ ID NO: 8.
5. The antigenic polypeptide of claim 1, wherein said antigenic polypeptide comprises the sequence of SEQ ID NO: 6 and lacks the sequence of SEQ ID NO: 11.
6. The antigenic polypeptide of any one of claims 1-5, wherein the antigenic polypeptide is capable of being bound by an antibody directed to HD protein.
7. The antigenic polypeptide of any one of claims 1-6, wherein said antigenic polypeptide is fused to at least one heterologous peptide.
8. The antigenic polypeptide of claim 7, wherein the heterologous peptide comprises an afinity tag, an epitope tag, a fluorescent protein, an enzyme or a carier protein.
9. An antigenic polypeptide comprising a sequence having at least 90% sequence identity to a truncated polypeptide derived from SEQ ID NO: 1, said truncated polypeptide lacking from 12 to 60 N-terminal amino acids of SEQ ID NO: 1.
10. The antigenic polypeptide of claim 9, wherein the truncated polypeptide comprises a sequence of at least 158 amino acids of SEQ ID NO: 1.
11. An antigenic polypeptide comprising an amino acid sequence having at least 90% sequence identity to any of the ful-length sequences of SEQ ID NO: 2 and SEQ ID NOs: 4-8, and a heterologous peptide.
12. The antigenic polypeptide of claim 11, wherein the heterologous peptide comprises an afinity tag, an epitope tag, a fluorescent protein, an enzyme or a carier protein.
13. A composition comprising the antigenic polypeptide of any one of claims 1-12.
14. The composition of claim 13, further comprising a detectable label.
15. The composition of claim 14, wherein the detectable label is conjugated to the antigenic polypeptide.
16. The composition of claim 14 or 15, wherein the detectable label is selected from the group consisting of a fluorescence label, a chemiluminescent label, an enzymatic label, and a particle label.
17. The composition of any one of claims 13-16, further comprising a second antigenic polypeptide that comprises the sequence of SEQ ID NO: 1.
18. The composition of claim 17, wherein the second antigenic polypeptide comprises a means for binding to a solid support.
19. A kit for detecting antibodies against hepatitis D virus (HDV) in a biological sample, comprising the antigenic polypeptide of any one of claims 1-12 or the composition of any one of claims 13-18.
20. A kit for detecting antibodies against hepatitis D virus (HDV) in a biological sample, comprising the antigenic polypeptide of any one of claims 1-12 and a second antigenic polypeptide that comprises the sequence of SEQ ID NO: 1.
21. A method for detecting antibodies specific for HD protein in a biological sample, wherein an antigenic polypeptide according to any one of claims 1-12 is used as a binding partner for said HD protein antibodies.
22. A method for detecting antibodies specific for HD protein in a biological sample, said method comprising: a) forming an immunoreaction mixture by mixing a biological sample with the antigenic polypeptide according to any one of claims 1-12; b) maintaining said immunoreaction mixture for a time period suficient for alowing antibodies against said antigenic polypeptide present in the biological sample to immunoreact with said antigenic polypeptide to form an immunoreaction product; and c) detecting the presence and/or the concentration of said immunoreaction product.
23. The method of claim 22, wherein said immunoreaction is caried out in a double antigen sandwich format comprising: a) adding to said biological sample a first antigenic polypeptide that comprises a means for binding to a solid support and a second antigenic polypeptide that carries a detectable label, said second antigenic polypeptide comprising the antigenic polypeptide of any one of claims 1-12, wherein said first antigenic polypeptide and said second antigenic polypeptide bind specificaly to the HD protein antibodies,
b) forming an immunoreaction mixture comprising the first antigenic polypeptide, the biological sample antibody and the second antigenic polypeptide, wherein the solid support is added before, during or after forming the immunoreaction mixture, c) maintaining said immunoreaction mixture for a time period suficient for HD protein antibodies in the biological sample to immunoreact with said first and second antigenic polypeptides to form an immunoreaction product, and d) detecting the presence of any of said immunoreaction product.
24. The method of claim 23, wherein the first antigenic polypeptide comprises a sequence having at least 90% sequence identity to the ful-length sequence of SEQ ID NO: 1.
25. The method of claim 23 or 24, further comprising a washing step prior to the detecting step.
26. The method of claim 25, wherein the immunoreaction product is bound to the solid support and the washing step comprises removing or separating some or al of the first antigenic polypeptide, HD protein antibody and second antigenic polypeptide that are not part of the immunoreaction product that is bound to the solid support.
27. The method of any one of claims 23-26, wherein the solid support is selected from the group consisting of a column, bead, test tube, microtiter dish, multi-wel plate, microparticle, microsphere, a test stick, a test strip, microchip and membrane.
28. The method of any one of claims 22-27, wherein the biological sample is a body fluid sample from a human subject.
29. The method of claim 28, wherein the biological sample is selected from the group consisting of blood, plasma and serum.
30. A polynucleotide encoding the antigenic polypeptide of any one of claims 1 to 12.
31. The polynucleotide of claim 30, further comprising a heterologous regulatory element operably linked to a nucleic acid encoding the antigenic polypeptide.
32. The polynucleotide of claim 31, wherein the heterologous regulatory element is a promoter.
33. A vector comprising the polynucleotide of any one of claims 30-32.
34. The vector of claim 33, wherein the vector is a plasmid, naked nucleic acid, phage, viral vector, or virus.
35. A cel comprising the polynucleotide of any one of claims 30 to 32 or the vector of claim 33 or 34, or expressing the antigenic polypeptide of any one of claims 1 to 12.
36. A method for detecting antibodies against hepatitis D virus (HDV) in a biological sample, comprising performing a HDV serology assay using a direct assay format with a first antigenic polypeptide and a second antigenic polypeptide, wherein the first antigenic polypeptide comprises a ful-length HD protein and the second antigenic polypeptide comprises an N- terminaly truncated fragment of the ful-length HD protein.
37. The method of claim 36, wherein the assay is a double antigen sandwich assay.
38. The method of claim 36 or 37, wherein the second antigenic polypeptide lacks from 12 to 60 N-terminal amino acids of the HD protein.
39. The method of claim 38, wherein the second antigenic polypeptide lacks 21, 37 or 46 consecutive N-terminal amino acids of the HD protein.
40. The method of any one of claims 36-39, wherein the ful-length HD protein comprises an amino acid sequence having at least 95% sequence identity to the ful-length sequence of SEQ ID NO: 1.
41. A method for detecting antibodies against hepatitis D virus (HDV) in a biological sample, comprising: a) contacting a biological sample, either simultaneously or sequentialy, in any order with: (1) a first antigenic polypeptide under conditions suficient to alow binding of any HDV antibody present in the biological sample to the first antigenic polypeptide, wherein said first antigenic polypeptide is immobilized on a solid support, and (2) a second antigenic polypeptide under conditions suficient to alow binding of the second antigenic polypeptide to any HDV antibody present in the biological sample, such that a first antigenic polypeptide-HDV antibody-second antigenic polypeptide complex is formed and is bound to the solid support, and b) detecting the presence of said first antigenic polypeptide-HDV antibody-second antigenic polypeptide complex, wherein said first antigenic polypeptide comprises a sequence having at least 90% sequence identity to the ful-length sequence of SEQ ID NO: 1; wherein said second antigenic polypeptide comprises a sequence having at least 90% sequence identity to a truncated polypeptide derived from SEQ ID NO: 1, said truncated polypeptide lacking from 12 to 60 N-terminal amino acids of SEQ ID NO: 1; and wherein said second antigenic polypeptide caries a detectable label.
42. The method of claim 41, wherein the second antigenic polypeptide is selected from the group consisting of: i) a polypeptide that comprises the sequence of SEQ ID NO: 8 and lacks the sequence of SEQ ID NO: 13; i) a polypeptide that comprises the sequence of SEQ ID NO: 7 and lacks the sequence of SEQ ID NO: 12; ii) a polypeptide that comprises the sequence of SEQ ID NO: 6 and lacks the sequence of SEQ ID NO: 11; iv) a polypeptide that comprises the sequence of SEQ ID NO: 5 and lacks the sequence of SEQ ID NO: 10;
v) a polypeptide that comprises the sequence of SEQ ID NO: 4 and lacks the sequence of SEQ ID NO: 9; and vi) a polypeptide that comprises the sequence of SEQ ID NO: 2.
43. The method of claim 41 or 42, further comprising a washing step prior to the detecting step.
44. The method of claim 43, wherein the washing step comprises removing or separating some or al of the first antigenic polypeptide, HDV antibody and second antigenic polypeptide that are not part of the first antigenic polypeptide-HDV antibody-second antigenic polypeptide complex that is bound to the solid support.
45. The method of any one of claims 41-44, wherein the step of detecting comprises detecting the presence and/or the concentration of the first antigenic polypeptide-HDV antibody- second antigenic polypeptide complex that is formed and is bound to the solid support.
46. The method of any one of claims 41-44, wherein the step of detecting comprises measuring an amount of said first antigenic polypeptide-HDV antibody-second antigenic polypeptide complex that is formed and is bound to the solid support.
47. The method of any one of claims 41-46, wherein the solid support is selected from the group consisting of a column, bead, test tube, microtiter dish, multi-wel plate, microparticle, microsphere, a test stick, a test strip, microchip and membrane.
48. The method of any one of claims 41-47, wherein the biological sample is selected from the group consisting of blood, plasma and serum.
49. A method of detecting hepatitis D virus (HDV) infection in a subject comprising performing the method of any one of claims 41-48, wherein the biological sample is a sample taken from the subject and wherein the step of detecting comprises detecting the presence of said first antigenic polypeptide-HDV antibody-second antigenic polypeptide complex that is formed
and is bound to the solid support, thereby detecting the presence of past or present HDV infection in said subject.
50. A method of estimating HDV incidence in a population, the method comprising, a) providing a set of samples derived from a plurality of individuals within the population over a period of time; b) performing the method of claim 49 on each sample in the set of samples; and c) determining the percentage of recent HDV infections over the period of time; wherein the percentage of recent HDV infections over the period of time provides an estimate of HDV incidence in the population.
51. A system comprising: a) a sample receiving component configured to receive a sample from a subject, wherein the sample comprises or is suspected of comprising a hepatitis D virus (HDV) antibody; b) a first antigenic polypeptide and a second antigenic polypeptide configured to make contact with the sample to form a first antigenic polypeptide-HDV antibody-second antigenic polypeptide complex, wherein the first antigenic polypeptide comprises a ful-length HD protein and the second antigenic polypeptide comprises a mutated HD protein, and wherein the second antigenic polypeptide carries a detectable label; c) a detection component configured to measure a signal generated by the detectable label in the first antigenic polypeptide-HDV antibody-second antigenic polypeptide complex; and d) an output component that indicates an amount of HDV antibody in the sample based on the signal.
52. The system of claim 51, wherein the second antigenic polypeptide comprises an N- terminaly truncated fragment of a ful-length HD protein.
53. The system of claim 51 or 52, wherein the ful-length HD protein comprises an amino acid sequence having at least 95% sequence identity to the ful-length sequence of SEQ ID NO: 1.
54. The system of any one of claims 51-53, wherein the second antigenic polypeptide lacks from 12 to 60 N-terminal amino acids of the ful-length HD protein.
55. The system of claim 54, wherein the second antigenic polypeptide lacks 21, 37 or 46 consecutive N-terminal amino acids of the ful-length HD protein.
56. The system of claim 52 or 53, wherein the second antigenic polypeptide is selected from the group consisting of: i) a polypeptide that comprises the sequence of SEQ ID NO: 8 and lacks the sequence of SEQ ID NO: 13; i) a polypeptide that comprises the sequence of SEQ ID NO: 7 and lacks the sequence of SEQ ID NO: 12; ii) a polypeptide that comprises the sequence of SEQ ID NO: 6 and lacks the sequence of SEQ ID NO: 11; iv) a polypeptide that comprises the sequence of SEQ ID NO: 5 and lacks the sequence of SEQ ID NO: 10; v) a polypeptide that comprises the sequence of SEQ ID NO: 4 and lacks the sequence of SEQ ID NO: 9; and vi) a polypeptide that comprises the sequence of SEQ ID NO: 2.
57. The system of any one of claims 51-56, wherein the sample is selected from the group consisting of blood, plasma and serum.
58. The system of any one of claims 51-57, wherein the detectable label is selected from the group consisting of a fluorescent label, a chemiluminescent marker, and a chromophore.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US202363547068P | 2023-11-02 | 2023-11-02 | |
| US63/547,068 | 2023-11-02 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2025096899A1 true WO2025096899A1 (en) | 2025-05-08 |
Family
ID=95581615
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2024/054061 Pending WO2025096899A1 (en) | 2023-11-02 | 2024-11-01 | Immunoassays and methods for diagnosing hepatitis d virus (hdv) infection |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2025096899A1 (en) |
Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0251575A1 (en) * | 1986-06-17 | 1988-01-07 | Chiron Corporation | Hepatitis delta diagnostics and vaccines, their preparation and use |
| US5006309A (en) | 1988-04-22 | 1991-04-09 | Abbott Laboratories | Immunoassay device with liquid transfer between wells by washing |
| US5063081A (en) | 1988-11-14 | 1991-11-05 | I-Stat Corporation | Method of manufacturing a plurality of uniform microfabricated sensing devices having an immobilized ligand receptor |
| US5089424A (en) | 1988-06-14 | 1992-02-18 | Abbott Laboratories | Method and apparatus for heterogeneous chemiluminescence assay |
| US20030170881A1 (en) | 2002-03-05 | 2003-09-11 | I-Stat Corporation | Apparatus and methods for analyte measurement and immuno assay |
| US20050054078A1 (en) | 2003-09-10 | 2005-03-10 | Miller Cary James | Immunoassay device with improved sample closure |
| US20060160164A1 (en) | 2003-09-10 | 2006-07-20 | Miller Cary J | Immunoassay device with immuno-reference electrode |
-
2024
- 2024-11-01 WO PCT/US2024/054061 patent/WO2025096899A1/en active Pending
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0251575A1 (en) * | 1986-06-17 | 1988-01-07 | Chiron Corporation | Hepatitis delta diagnostics and vaccines, their preparation and use |
| US5006309A (en) | 1988-04-22 | 1991-04-09 | Abbott Laboratories | Immunoassay device with liquid transfer between wells by washing |
| US5089424A (en) | 1988-06-14 | 1992-02-18 | Abbott Laboratories | Method and apparatus for heterogeneous chemiluminescence assay |
| US5063081A (en) | 1988-11-14 | 1991-11-05 | I-Stat Corporation | Method of manufacturing a plurality of uniform microfabricated sensing devices having an immobilized ligand receptor |
| US20030170881A1 (en) | 2002-03-05 | 2003-09-11 | I-Stat Corporation | Apparatus and methods for analyte measurement and immuno assay |
| US20050054078A1 (en) | 2003-09-10 | 2005-03-10 | Miller Cary James | Immunoassay device with improved sample closure |
| US20060160164A1 (en) | 2003-09-10 | 2006-07-20 | Miller Cary J | Immunoassay device with immuno-reference electrode |
Non-Patent Citations (8)
| Title |
|---|
| ALVES CAROLINA ET AL: "Structural and nucleic acid binding properties of hepatitis delta virus small antigen", WORLD JOURNAL OF VIROLOGY, 12 May 2017 (2017-05-12), US, XP093238952, ISSN: 2220-3249, Retrieved from the Internet <URL:https://pmc.ncbi.nlm.nih.gov/articles/PMC5437381/pdf/WJV-6-26.pdf> [retrieved on 20250114] * |
| COLLER KE ET AL.: "Development and performance of prototype serologic and molecular tests for hepatitis delta infection", SCI REP., vol. 8, no. 1, 1 February 2018 (2018-02-01), pages 2095 |
| HUGHES SAWEDEMEYER HHARRISON PM: "Hepatitis delta virus", LANCET, vol. 378, no. 9785, 2 July 2011 (2011-07-02), pages 73 - 85 |
| LEE C Z ET AL: "Histone H1e interacts with small hepatitis delta antigen and affects hepatitis delta virus replication", VIROLOGY, ELSEVIER, AMSTERDAM, NL, vol. 375, no. 1, 25 May 2008 (2008-05-25), pages 197 - 204, XP022638394, ISSN: 0042-6822, [retrieved on 20080307], DOI: 10.1016/J.VIROL.2008.02.003 * |
| QIU XIAOXING ET AL: "Evaluation of a fully automated high-throughput serology assay for detection of Hepatitis D virus antibodies", JOURNAL OF CLINICAL VIROLOGY, vol. 173, 1 August 2024 (2024-08-01), NL, pages 105689, XP093238801, ISSN: 1386-6532, DOI: 10.1016/j.jcv.2024.105689 * |
| SHIH KO-NIEN ET AL: "The HDV Large-Delta Antigen Fused with GFP Remains Functional and Provides for Studying Its Dynamic Distribution", VIROLOGY, vol. 285, no. 1, 1 June 2001 (2001-06-01), AMSTERDAM, NL, pages 138 - 152, XP093238735, ISSN: 0042-6822, Retrieved from the Internet <URL:https://pdf.sciencedirectassets.com/272412/1-s2.0-S0042682200X00165/1-s2.0-S0042682201908451/main.pdf?hash=9c5f1b8a277ef0a4d7dc6ec01c4f73914596facec0fcf20ebbc08fc81f510dd1&host=68042c943591013ac2b2430a89b270f6af2c76d8dfd086a07176afe7c76c2c61&pii=S0042682201908451&tid=spdf-f78405c8-4b6b-4ad8-9a2f-8e1> [retrieved on 20250113], DOI: 10.1006/viro.2001.0845 * |
| UNKNOWN: "Q812E6 Q912E6_HDV", 12 January 2001 (2001-01-12), XP093238646, Retrieved from the Internet <URL:https://www.uniprot.org/uniparc/UPI00000EF56F/entry?facets=dbTypes%3AEMBL> [retrieved on 20250113] * |
| VALERIA IVANIUSHINA ET AL: "Hepatitis delta virus genotypes I and II cocirculate in an endemic area of Yakutia, Russia", JOURNAL OF GENERAL VIROLOGY, SOCIETY FOR GENERAL MICROBIOLOGY, vol. 82, no. Pt. 11, 1 November 2001 (2001-11-01), pages 2709 - 2718, XP002607359, ISSN: 0022-1317 * |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Zhang et al. | Evaluation of recombinant nucleocapsid and spike proteins for serological diagnosis of novel coronavirus disease 2019 (COVID-19) | |
| US9841427B2 (en) | HCV antigen-antibody combination assay and methods and compositions for use therein | |
| JP5946218B2 (en) | Assay for JC virus antibody | |
| US8846391B2 (en) | Methods and compositions for detecting herpes simplex virus type 2 | |
| CN103698511B (en) | Use of mycobacterium tuberculosis protein in preparing products for diagnosing latent tuberculosis infection | |
| CN114152748A (en) | Double-antibody sandwich ELISA diagnostic kit for detecting African swine fever virus and method thereof | |
| WO2025096899A1 (en) | Immunoassays and methods for diagnosing hepatitis d virus (hdv) infection | |
| CN104569425A (en) | Antigen protein specifically bound with tyrosine phosphatase antibody | |
| CN107110868B (en) | Subject Anti-HCV Antibody Detection Assay Using NS3 Capture Peptide | |
| CN102590505A (en) | Reagent kit for detecting immunoglobulin m (IgM) AlphaLISA for resisting enterovirus71 capsid protein1 | |
| CN101523216B (en) | Method for Determination of Human Immunodeficiency Virus Series Protein Antibody | |
| JP7489228B2 (en) | SARS-CoV-2 derived nucleocapsid fragment and method and kit for detecting anti-SARS-CoV-2 antibodies using said fragment | |
| JP7523129B2 (en) | Method and kit for identifying Helicobacter pylori strains | |
| CN117209571A (en) | VP1 antigen, kit and method for detecting BKV virus VP1 antibody | |
| CN118344492B (en) | Application of nano luciferase fusion antigen in African swine fever virus antibody detection | |
| CN115925827B (en) | Recombinant M protein applied to detection of canine coronavirus antibody and preparation method and application thereof | |
| CN110456052A (en) | A rapid detection method for Treponema pallidum antibody in serum and its application | |
| JP2003043044A (en) | Antibody immunoassay by antibody capture method | |
| Savevska et al. | COMPARISON OF TWO METHODS FOR SARS-COV-2 ANTIBODY TESTING | |
| JP2023128698A (en) | Anti-sars-cov-2 antibody against sars-cov-2 antigen in body fluid, comprising mutant strain, method for detecting sars-cov-2 using the antibody, and kit comprising the antibody | |
| Talha et al. | Europium Nanoparticle-Based High Performing Immunoassay for the Screening of | |
| Лебедин et al. | THE IMPORTANCE OF DETERMINING SARS-COV-2 N-Ag SERODIAGNOSTICS FOR THE MANAGEMENT OF COVID-19 PNEUMONIA IN HOSPITAL SETTINGS |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 24809124 Country of ref document: EP Kind code of ref document: A1 |