WO2025090511A1 - Methods of preparing modulators of sodium channels and solid forms of the same for treating pain - Google Patents
Methods of preparing modulators of sodium channels and solid forms of the same for treating pain Download PDFInfo
- Publication number
- WO2025090511A1 WO2025090511A1 PCT/US2024/052419 US2024052419W WO2025090511A1 WO 2025090511 A1 WO2025090511 A1 WO 2025090511A1 US 2024052419 W US2024052419 W US 2024052419W WO 2025090511 A1 WO2025090511 A1 WO 2025090511A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- ppm
- compound
- theta
- degrees
- formula
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/61—Halogen atoms or nitro radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/62—Oxygen or sulfur atoms
- C07D213/63—One oxygen atom
- C07D213/64—One oxygen atom attached in position 2 or 6
- C07D213/643—2-Phenoxypyridines; Derivatives thereof
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F5/00—Compounds containing elements of Groups 3 or 13 of the Periodic Table
- C07F5/02—Boron compounds
- C07F5/025—Boronic and borinic acid compounds
Definitions
- Pain is a protective mechanism that allows healthy animals to avoid tissue damage and to prevent further damage to injured tissue. Nonetheless, there are many conditions where pain persists beyond its usefulness, or where patients would benefit from inhibition of pain. Acute and chronic pain are two common pain states and can be distinguished by the duration of the pain.
- Neuropathic pain is a form of chronic pain caused by an injury to the sensory nerves (Dieleman, J.P., et al., Incidence rates and treatment of neuropathic pain conditions in the general population. Pain, 2008.137(3): p.681-8). Neuropathic pain can be divided into two categories, pain caused by generalized metabolic damage to the nerve and pain caused by a discrete nerve injury.
- the metabolic neuropathies include post-herpetic neuropathy, diabetic neuropathy, and drug-induced neuropathy.
- Discrete nerve injury indications include post-amputation pain, post-surgical nerve injury pain, and nerve entrapment injuries like neuropathic back pain.
- Voltage-gated sodium channels Na V s are involved in pain signaling. Na V s are biological mediators of electrical signaling as they mediate the rapid upstroke of the action potential of many excitable cell types, for example, neurons, skeletal myocytes, cardiac myocytes (Hille, Bertil, Ion Channels of Excitable Membranes, Third ed. (Sinauer Associates, Inc., Sunderland, MA, 2001)).
- the local anesthetic drugs such as lidocaine block pain by inhibiting Na V channels
- other compounds such as carbamazepine, lamotrigine, and tricyclic antidepressants that have proven effective at reducing pain
- sodium channel inhibition Soderpalm, B., Anticonvulsants: aspects of their mechanisms of action. Eur. J. Pain 6 Suppl. A, p.3- 9 (2002); Wang, G. K., Mitchell, J., and Wang, S. Y., Block of persistent late Na + currents by antidepressant sertraline and paroxetine. J. Membr. Biol.222 (2), p.79-90 (2008)).
- the NaVs form a subfamily of the voltage-gated ion channel super-family and comprises 9 isoforms, designated Na V 1.1 – Na V 1.9.
- the tissue localizations of the nine isoforms vary.
- Na V 1.4 is the primary sodium channel of skeletal muscle
- Na V 1.5 is primary sodium channel of cardiac myocytes.
- Na V s 1.7, 1.8 and 1.9 are primarily localized to the peripheral nervous system, while Na V s 1.1, 1.2, 1.3, and 1.6 are neuronal channels found in both the central and peripheral nervous systems.
- the functional behaviors of the nine isoforms are similar but distinct in the specifics of their voltage- dependent and kinetic behavior (Catterall, W. A., Goldin, A.
- Na V 1.8 channels were identified as likely targets for analgesia (Akopian, A.N., L. Sivilotti, and J.N. Wood, A tetrodotoxin-resistant voltage-gated sodium channel expressed by sensory neurons. Nature, 1996.379(6562): p.257-62).
- Na V 1.8 has been shown to be a carrier of the sodium current that maintains action potential firing in small dorsal root ganglia (DRG) neurons (Blair, N.T. and B.P. Bean, Roles of tetrodotoxin (TTX)-sensitive Na + current, TTX- resistant Na + current, and Ca 2+ current in the action potentials of nociceptive sensory neurons. J. Neurosci., 2002.22(23): p.10277-90).
- DRG dorsal root ganglia
- TTX tetrodotoxin
- Na V 1.8 is involved in spontaneous firing in damaged neurons, like those that drive neuropathic pain (Roza, C., et al., The tetrodotoxin-resistant Na + channel Na V 1.8 is essential for the expression of spontaneous activity in damaged sensory axons of mice. J. Physiol., 2003.550(Pt 3): p.921-6; Jarvis, M.F., et al., A-803467, a potent and selective Na V 1.8 sodium channel blocker, attenuates neuropathic and inflammatory pain in the rat. Proc. Natl. Acad. Sci. U S A, 2007. 104(20): p.
- NaV1.8 is necessary for rapid repetitive action potentials in nociceptors, and for spontaneous activity of damaged neurons.
- Na V 1.8 Contribution of Na( V )1.8 sodium channels to action potential electrogenesis in DRG neurons. J. Neurophysiol., 2001.86(2): p. 629-40; Roza, C., et al., The tetrodotoxin-resistant Na + channel Na V 1.8 is essential for the expression of spontaneous activity in damaged sensory axons of mice. J. Physiol., 2003. 550(Pt 3): p. 921-6). In depolarized or damaged DRG neurons, Na V 1.8 appears to be a driver of hyper-excitablility (Rush, A.M., et al., A single sodium channel mutation produces hyper- or hypoexcitability in different types of neurons. Proc. Natl. Acad. Sci.
- Na V 1.8 mRNA expression levels have been shown to increase in the DRG (Sun, W., et al., Reduced conduction failure of the main axon of polymodal nociceptive C-fibers contributes to painful diabetic neuropathy in rats. Brain, 135(Pt 2): p.359-75; Strickland, I.T., et al., Changes in the expression of Na V 1.7, Na V 1.8 and Na V 1.9 in a distinct population of dorsal root ganglia innervating the rat knee joint in a model of chronic inflammatory joint pain. Eur. J. Pain, 2008. 12(5): p.
- Compound I 2-[2-(3,4-difluoro-2-methyl-phenoxy)-4-methyl-5-(trifluoromethyl)-3- pyridyl]-4-oxo-1H-1,6-naphthyridine-5-carboxamide, is a Na v inhibitor and thus useful in treating pain.
- Compound I has the following structure: [0011]
- Compound I is No. PCT/US2023/019469. Crystalline forms are of interest where the control of the crystalline form(s) of the active ingredient may be desirable or even required. Reproducible processes for producing a compound with a particular crystalline form in high purity may be desirable for compounds intended to be used in pharmaceuticals, as different crystalline forms may possess different properties.
- different crystalline forms may possess different chemical, physical, and/or pharmaceutical properties.
- one or more crystalline forms disclosed herein may exhibit a higher level of purity, chemical stability, and/or physical stability compared to the forms produced in PCT US/2023/019469.
- Certain crystalline forms e.g., crystalline free form, crystalline salt, crystalline salt solvate, and crystalline salt hydrate forms of Compound I (collectively referred to as “crystalline forms”)
- crystalline forms may exhibit lower hygroscopicity than any preexisting forms.
- the crystalline forms of this disclosure may provide advantages during drug substance manufacturing, storage, and handling over the amorphous forms produced in PCT US/2023/019469.
- compositions of Compound I may be particularly useful for the production of drugs for the treatment of pain.
- pharmaceutically acceptable crystalline forms of Compound I may be particularly useful for the production of drugs for the treatment of pain.
- the invention relates to a method of preparing a compound of formula I: I, or a solvate or a tautomer or a [0014]
- a method of preparing a compound of formula I: I or a solvate or a tautomer or a [0014]
- compounds of formulae A 1 , A 1-1 , A 1-2 , A 1-3 , A 1-4 , A 1-5 , A 2 , A 2-1 , A 3 , A 3-1 , A 3-2 , B-1, B-2, B-3, B-4, B-5, C-1, C-2, C-3, C-4, D-1, D-2, E-A 1 , E-A 2 , and E-A 3 to the compound of formula I or a solvate thereof or a tautomer or a pharmaceutically acceptable salt thereof following the reaction steps described herein.
- the invention relates to a compound selected from: , O NH O 2 , or a [0016] I, wherein the crystalline Compound I is selected from substantially pure compound I neat Form A, Compound I neat form B, Compound I neat form E, Compound I Acetone Solvate Hydrate Form A, Compound I Ethanol Solvate Form A, Compound I Hydrate Form A, and Compound I Dehydrated Hydrate Form A.
- the disclosure relates to substantially amorphous Compound I amorphous form.
- Figure 1 provides the X-ray powder diffraction (XRPD) pattern of Compound I amorphous form.
- Figure 2 provides the 13 C solid-state NMR (SSNMR) spectrum of Compound I amorphous form.
- Figure 3 provides an 19 F NMR spectrum of Compound I amorphous form.
- Figure 4 provides the XRPD pattern of crystalline Compound I neat Form A.
- Figure 5 provides the 13 C SSNMR spectrum of crystalline Compound I neat Form A.
- Figure 6 provides an 19 F NMR spectrum of crystalline Compound I neat Form A.
- Figure 7 provides the XRPD pattern of crystalline Compound I neat Form B.
- Figure 8 provides the 13 C SSNMR spectrum of crystalline Compound I neat Form B.
- Figure 9 provides an 19 F NMR spectrum of crystalline Compound I neat Form B.
- Figure 10 provides the XRPD pattern of crystalline Compound I neat Form E.
- Figure 11 provides the 13 C SSNMR spectrum of crystalline Compound I neat Form E.
- Figure 12 provides an 19 F NMR spectrum of crystalline Compound I neat Form E.
- Figure 13 provides the XRPD pattern of crystalline Compound I Acetone Solvate Hydrate Form A.
- Figure 14 provides the 13 C SSNMR spectrum of crystalline Compound I Acetone Solvate Hydrate Form A.
- Figure 15 provides an 19 F NMR spectrum of crystalline Compound I Acetone Solvate Hydrate Form A.
- Figure 16 provides the XRPD pattern of crystalline Compound I Ethanol Solvate Form A.
- Figure 17 provides the 13 C SSNMR spectrum of crystalline Compound I Ethanol Solvate Form A.
- Figure 18 provides an 19 F NMR spectrum of crystalline Compound I Ethanol Solvate Form A.
- Figure 19 provides the XRPD pattern of crystalline Compound I Hydrate Form A (wet form).
- Figure 20 provides the XRPD pattern of crystalline Compound I Dehydrated Hydrate Form A (dry form).
- Figure 21 provides the overlay of the XRPD patterns of crystalline Compound I Hydrate Form A with crystalline Compound I Dehydrated Hydrate Form A.
- Compound I refers to 2-[2-(3,4-difluoro-2-methyl- phenoxy)-4-methyl-5-(trifluoromethyl)-3-pyridyl]-4-oxo-1H-1,6-naphthyridine-5-carboxamide, and has the following structure: [0043] Compound I may be acceptable salt, solvate, and/or hydrate.
- the term “compound” includes such a collection of molecules in pure form, in a mixture (e.g., solution, suspension, colloid, or pharmaceutical composition, or dosage form) with one or more other substances, or in the form of a hydrate, solvate, or co-crystal.
- a mixture e.g., solution, suspension, colloid, or pharmaceutical composition, or dosage form
- any atom not specifically designated as a particular isotope in any compound of the invention is meant to represent any stable isotope of the specified element.
- H refers to hydrogen and includes any stable isotope of hydrogen, namely 1 H and D.
- the compounds described in the present application include each constituent atom at approximately the natural abundance isotopic composition of the specified element.
- the compounds described in the present application, and pharmaceutically acceptable salts thereof include one or more atoms having an atomic mass or mass number which differs from the atomic mass or mass number of the most abundant isotope of the specified element (“isotope-labeled” compounds and salts).
- isotope-labeled compounds and salts include without limitation isotopes of hydrogen, carbon, nitrogen, oxygen, and phosphorus, for example 2 H, 13 C, 15 N, 18 O, 17 O, and 31 P, respectively.
- “H” refers to hydrogen and includes any stable isotope of hydrogen, namely 1 H and D.
- the term “compound of formula” followed by a number (typically Roman number) and the term “compound” followed by the same number (Roman or otherwise) may interchangeably be used.
- the “compound of formula V” and “compound V” denote the same compound.
- reacting when referring to a chemical reaction, means to add or mix two or more reagents under appropriate conditions to produce the indicated and/or the desired product. It should be appreciated that the reaction which produces the indicated and/or the desired product may not necessarily result directly from the combination of two reagents which were initially added, i.e., there may be one or more intermediates which are produced in the mixture which ultimately leads to the formation of the indicated and/or the desired product.
- chromatographic purification refers to any method of purification based on differential retention by a stationary phase. Methods of chromatographic purification include flash chromatography, medium pressure liquid chromatography, preparative thin layer chromatography, and high performance liquid chromatography.
- the terms “converting” and “transforming” as used herein refer to a step of converting a first compound or salt to a second compound or salt, and refers to a process of transforming the first compound or salt to the second compound or salt in one or more chemical steps.
- the term “acid” refers to a chemical species having a pKa (in water) of less than 7.
- the term includes inorganic (mineral) acids, such as hydrochloric acid, hydrobromic acid, hydroiodic acid, sulfuric acid, phosphoric acid, nitric acid, and the like.
- the term also includes organic acids such as acetic acid, propionic acid, n-butyric acid, i-butyric acid, n-valeric acid, i-valeric acid, n-hexanoic acid, succinic acid, glutaric acid, adipic acid, aspartic acid, formic acid, citric acid, o-chlorobenzoic acid, chloroacetic acid, dichloroacetic acid, trichloroacetic acid, nicotinic acid, lactic acid, oxalic acid, picric acid, picolinic acid, fluoroacetic acid, difluoroacetic acid, trifluoroacetic acid, phthalic acid, isophthalic acid, terephthalic acid, maleic acid, malonic acid, and the like.
- organic acids such as acetic acid, propionic acid, n-butyric acid, i-butyric acid, n-valeric acid, i-valeric acid, n-
- base refers to a chemical species whose conjugate acid has a pKa (in water) of greater than 7.
- the term includes “inorganic bases,” such as sodium hydroxide, potassium hydroxide, sodium bicarbonate, potassium bicarbonate, sodium carbonate, potassium carbonate, cesium carbonate, potassium phosphate (mono-, di-, or tri-basic), sodium hydride, and potassium hydride.
- organic bases such as sodium hydroxide, potassium hydroxide, sodium bicarbonate, potassium bicarbonate, sodium carbonate, potassium carbonate, cesium carbonate, potassium phosphate (mono-, di-, or tri-basic), sodium hydride, and potassium hydride.
- anionic organic bases such as methyl lithium, butyl lithium, lithium diisopropyl amide, and sodium acetate.
- neutral organic bases such as trimethylamine, dimethylethylamine, diethylmethylamine, triethylamine, di-n-propylmethylamine, dimethylcyclohexylamine, diisopropylethylamine, tri-n-propylamine, diisopropylisobutylamine, dimethyl-n-nonylamine, tri-n-butylamine, di-n-hexylmethylamine, dimethyl-n-dodecylamine, tri-n-pentylamine, 1,4-diazabicyclo[2.2.2] octane (DABCO), dimethylaminopyridine (DMAP), 1,5-diazabicyclo[4.3.0] non-5-ene (DBN), 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), pyridine, 2,3-lutidine, 2,4-lutidine, 2,5-lutidine, 2,6-luti
- DABCO di
- alcohol protecting group refers to a chemical moiety suitable to protect an alcohol group against undesirable side reactions during synthetic procedures.
- Common alcohol protecting groups include methyl, ethyl, isopropyl, benzyl, 2-tetrahydropyranyl, acetyl, trifluoroacetyl, trialkylsilyl, aryldialkylsilyl, alkyldiarylsilyl, or triarylsilyl.
- Other alcohol protecting groups also are well known in the art. See, e.g., P.G.M. Wuts et al., Greene’s Protective Groups in Organic Synthesis (4th ed.2006).
- hydrogenation catalyst refers to any homogeneous or heterogeneous catalyst that catalyzes the hydrogenolysis of benzylic carbon-oxygen single bonds. Suitable hydrogenation catalysts are well-known in the art and include, for example, palladium on activated carbon, platinum oxide, and Raney Nickel.
- the term includes a direct reaction between the carboxylic acid and the amine, as well as a reaction between an activated derivative of the carboxylic acid (such as the derivative formed by the reaction between the carboxylic acid and a coupling reagent) and the amine.
- an activated derivative of the carboxylic acid such as the derivative formed by the reaction between the carboxylic acid and a coupling reagent
- the term “coupling reagent” refers to a reagent suitable to react with a carboxylic acid to activate the carboxylic acid for coupling with an amine to form an amide bond. Coupling reagents are well known in the art.
- Coupling reagents include, but are not limited to, thionyl chloride, oxalyl chloride, 1,1'-carbonylbis-(4,5-dicyanoimidazole) (CBDCI), 1,1'- carbonyldiimidazole (CDI), propylphosphonic anhydride (T3P), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI), N,N’-dicyclohexylcarbodiimide (DCC), 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5- b]pyridinium 3-oxid hexafluorophosphate (HATU), and 1-hydroxybenzotriazole (HOBt).
- CBDCI 1,1'-carbonylbis-(4,5-dicyanoimidazole)
- CDI 1,1'- carbonyldiimidazole
- T3P propylphosphonic anhydr
- the term “monovalent cation” refers to any cation with a charge of +1, such as alkali metal cations, NH 4 + , and tetraalkylammonium.
- alkali metal cation refers to a cation derived from a Group I metal atom, including without limitation lithium (Li + ), sodium (Na + ), potassium (K + ), rubidium (Rb + ), and cesium (Cs + ).
- substituted benzyl refers to a benzyl group that is substituted with 1-3 substituents selected from the group consisting of C 1 -C 3 alkyl, C 1 -C 3 alkoxy, halogen, and cyano.
- ketone solvent refers to a compound having the formula C n H 2n+1 C(O)C m H 2m+1 , wherein n and m are each independently an integer between 1 and 6.
- the C n H 2n+1 and C m H 2m+1 and groups may be linear or branched and each may be substituted with up to 3 halogens.
- Ketone solvents include without limitation acetone, methyl ethyl ketone, 3-pentanone, and methyl tert-butyl ketone.
- ethereal solvent refers to an organic solvent having at least one ether moiety. Ethereal solvents include without limitation tetrahydofuran, dimethoxyethane, dioxane, and dialkyl ethers such as diethyl ether and methyl isobutyl ether.
- esteer solvent refers to a compound having the formula C n H 2n+1 OC(O)C m H 2m+1 , wherein n and m are each independently an integer between 1 and 6.
- the C n H 2n+1 and C m H 2m+1 and groups may be linear or branched and each may be substituted with up to 3 halogens.
- Ester solvents include without limitation ethyl acetate, isopropyl acetate, butyl acetate, and ethylpropionate.
- halogenated solvent refers to a C 1 -C 6 alkane or C 2 -C 6 alkene substituted with up to six halogens.
- Halogenated solvents include without limitation dichloromethane, dichloroethane, chloroform, tetrachloroethylene, and carbon tetrachloride.
- aromatic solvent refers to a C 6-10 aromatic hydrocarbon.
- the aromatic hydrocarbon may be substituted with up to six halogens.
- Aromatic solvents include without limitation benzene, trifluoromethylbenzene, xylene, and toluene.
- the term “about” means that the stated number can vary from that value by ⁇ 10%. Where the term defines a temperature, the stated temperature can vary by ⁇ 10%. For example, about 80oC means between 72oC and 88oC. Where the term defines pressure, the term “about” means the pressure can vary by ⁇ 10%. Thus, about 100 bars means between 90 and 110 bars.
- the term means the quantity can vary by ⁇ 10%. For example, about 1 equivalent means between 0.9 and 1.1 equivalents.
- time the term means the stated time can vary by ⁇ 10%. For example, about 1 hour means between 0.9 and 1.1 hours.
- the term “leaving group” is a chemical group that is readily displaced by a desired incoming chemical moiety. Thus, the choice of the specific suitable leaving group is predicated upon its ability to be readily displaced by the incoming chemical moiety such as a CN group.
- cyanating agent refers to a reagent, such as trimethylsilyl cyanide (TMSCN), diethylaluminum cyanide, KCN, NaCN, TBACN, HCN and the like, useful in the synthesis of compounds disclosed herein, e.g., compound A 1 , A 2 , and A 3 .
- TMSCN trimethylsilyl cyanide
- KCN diethylaluminum cyanide
- KCN diethylaluminum cyanide
- KCN sodiumCN
- TBACN TBACN
- HCN trimethylsilyl cyanide
- the cyanating agent e.g., trimethylsilyl cyanide
- the cyanating agent may be combined with a Lewis acid.
- the Lewis acid is trifluoromethanesulfonic anhydride (Tf 2 O), boron trifluoride ethyl etherate (BF 3 OEt 2 ), TiCl 4 , InCl 3 , AgSbF 6 , iodine, ZnBr 2 , Al(OiPr) 3 , MgCl 2 , Mn(acac) 2 , MnCl 2 , TMSOTf, SnCl 4 , and the like.
- the Lewis acid is trifluoromethanesulfonic anhydride (Tf 2 O).
- the cyanation reaction may be conducted in an organic solvent, for example toluene, dichloromethane, 2-methyl THF, acetonitrile, methanol, 1,2-dichloroethane, nitromethane, and the like.
- an organic solvent for example toluene, dichloromethane, 2-methyl THF, acetonitrile, methanol, 1,2-dichloroethane, nitromethane, and the like.
- API active pharmaceutical ingredient
- therapeutic agent refers to a biologically active compound.
- patient and “subject” are used interchangeably and refer to an animal including humans.
- an effective dose and “effective amount” are used interchangeably herein and refer to that amount of a compound that produces the desired effect for which it is administered (e.g., improvement in pain or a symptom of pain, or lessening the severity of pain or a symptom of pain).
- the exact amount of an effective dose will depend on the purpose of the treatment and will be ascertainable by one skilled in the art using known techniques (see, e.g., Lloyd (1999) The Art, Science and Technology of Pharmaceutical Compounding).
- the terms “treatment,” “treating,” and the like generally mean the improvement of pain or one or more of its symptoms or lessening the severity of pain or one or more of its symptoms in a subject.
- Treatment includes, but is not limited to, the following: Chronic pain, gut pain, neuropathic pain, musculoskeletal pain, acute pain, inflammatory pain, cancer pain, idiopathic pain, postsurgical pain (e.g., bunionectomy pain, herniorrhaphy pain or abdominoplasty pain), visceral pain, multiple sclerosis, Charcot-Marie-Tooth syndrome, incontinence, pathological cough, or cardiac arrhythmia. Improvements in or lessening the severity of any of these symptoms can be readily assessed according to standard methods and techniques known in the art.
- the term “in combination with,” when referring to two or more compounds, agents, or additional active pharmaceutical ingredients, means the administration of two or more compounds, agents, or active pharmaceutical ingredients to the patient prior to, concurrently with, or subsequent to each other.
- “Selected from” and “chosen from” are used interchangeably herein.
- the term “ambient conditions” means room temperature, open air condition and uncontrolled humidity condition.
- the terms “room temperature” and “ambient temperature” mean 15 °C to 30 °C.
- the term “solvent” refers to any liquid in which the product is at least partially soluble (solubility of product > 1 g/L).
- stable refers to compounds or solid forms that are not substantially altered when subjected to conditions to allow for their production, detection, and preferably their recovery, purification, and use for one or more of the purposes disclosed herein.
- chemically stable means that the solid form of Compound I does not decompose into one or more different chemical compounds when subjected to specified conditions, e.g., 40 °C/75% relative humidity, for a specific period of time, e.g., 1 day, 2 days, 3 days, 1 week, 2 weeks, or longer. In some embodiments, less than 25% of the solid form of Compound I decomposes.
- the term "physically stable,” as used herein, means that the solid form of Compound I does not change into one or more different physical forms of Compound I (e.g., different solid forms as measured by XRPD, DSC, etc.) when subjected to specific conditions, e.g., 40 °C/75 % relative humidity, for a specific period of time, e.g, 1 day, 2 days, 3 days, 1 week, 2 weeks, or longer. In some embodiments, less than 25% of the solid form of Compound I changes into one or more different physical forms when subjected to specified conditions.
- specific conditions e.g. 40 °C/75 % relative humidity
- less than about 20%, less than about 15%, less than about 10%, less than about 5%, less than about 3%, less than about 1 %, less than about 0.5% of the solid form of Compound I changes into one or more different physical forms of Compound I when subjected to specified conditions. In some embodiments, no detectable amount of the solid form of Compound I changes into one or more physically different solid forms of Compound I.
- the term "hydrate" refers to any crystalline Compound I that contains water in its crystal lattice. The stoichiometry of a Compound I hydrate can vary.
- a hydrate of Compound I can be a quarter hydrate, hemihydrate, monohydrate, dihydrate, or a partially dehydrated form.
- pharmaceutically acceptable solid form refers to a solid form of Compound I of this disclosure wherein the solid form (e.g., crystalline free form, crystalline salt, crystalline salt solvate, crystalline salt hydrate, and amorphous form) of Compound I is nontoxic and suitable for use in pharmaceutical compositions.
- the term “amorphous” refers to a solid material having no long-range order in the position of its molecules. Amorphous solids are generally rather isotropic, i.e., exhibit similar properties in all directions.
- Amorphous solids do not have definite melting points.
- Amorphous solids are generally glasses or supercooled liquids in which the molecules are arranged in a random manner so that there is no well-defined arrangement, e.g., molecular packing, and no long-range order.
- an amorphous material is a solid material having no sharp characteristic crystalline peak(s) in its X-ray power diffraction (XRPD) pattern (i.e., is not crystalline as determined by XRPD). Instead, one or several broad peaks (e.g., halos) appear in its XRPD pattern. Broad peaks are characteristic of an amorphous solid.
- a solid material may comprise an amorphous compound, and the material may, for example, be characterized by a lack of sharp characteristic crystalline peak(s) in its XRPD spectrum (i.e., the material is not crystalline, but is amorphous, as determined by XRPD). Instead, one or several broad peaks (e.g., halos) may appear in the XRPD pattern of the material. See US 2004/0006237 for a comparison of XRPDs of an amorphous material and crystalline material.
- a solid material comprising an amorphous compound
- Other techniques such as, for example, solid state NMR may also be used to characterize crystalline or amorphous forms.
- crystal form As used herein, the terms "crystal form,” “crystalline form,” and “Form” interchangeably refer to a crystal structure (or polymorph) having a particular molecular packing arrangement in the crystal lattice.
- Crystalline forms can be identified and distinguished from each other by one or more characterization techniques including, for example, X-ray powder diffraction (XRPD), single crystal X-ray diffraction, and 13 C solid state nuclear magnetic resonance ( 13 C SSNMR).
- XRPD X-ray powder diffraction
- 13 C SSNMR 13 C solid state nuclear magnetic resonance
- the terms "crystalline Form [X] of Compound (I)” and “crystalline Form [C] potassium salt of Compound (I)” refer to unique crystalline forms that can be identified and distinguished from each other by one or more characterization techniques including, for example, XRPD, single crystal X-ray diffraction, and 13 C SSNMR
- the novel crystalline forms are characterized by an X-ray powder diffractogram having one or more signals at one or more specified degree two-theta values (°2 ⁇ ).
- the term “free form” refers to a non-ionized version of the compound in the solid state. Examples of free forms include free bases and free acids.
- the term “neat form” refers to an unsolvated and unhydrated free form version of a compound in the solid state.
- solvate refers to a crystal form comprising one or more molecules of a compound of the present disclosure and, incorporated into the crystal lattice, one or more molecules of a solvent or solvents in stoichiometric or nonstoichiometric amounts.
- hydrate refers to any crystalline Compound I that contains water in its crystal lattice.
- the stoichiometry of a Compound I hydrate can vary, i.e., Compound I can be a variable hydrate.
- a hydrate of Compound I can be a quarter hydrate, hemihydrate, monohydrate, dihydrate, or a partially dehydrated form.
- a solid material may comprise a mixture of crystalline solids and amorphous solids.
- a solid material comprising an amorphous compound may also, for example, contain up to 30% of a crystalline solid.
- a solid material prepared to comprise an amorphous compound may also, for example, contain up to 25%, 20%, 15%, 10%, 5%, or 2% of a crystalline solid.
- the characterizing data such as XRPD, may contain indicators of both crystalline and amorphous solids.
- a crystalline form of this disclosure may contain up to 30% amorphous compound.
- a crystalline preparation of Compound I may contain up to 25%, 20%, 15%, 10%, 5%, or 2% of an amorphous solid.
- substantially amorphous refers to a solid material having little or no long-range order in the position of its molecules.
- substantially amorphous materials have less than 15% crystallinity (e.g., less than 10% crystallinity, less than 5% crystallinity, or less than 2% crystallinity).
- substantially amorphous includes the descriptor, "amorphous,” which refers to materials having no (0%) crystallinity.
- substantially crystalline refers to a solid material having little or no amorphous molecules.
- substantially crystalline materials have less than 15% amorphous molecules (e.g., less than 10% amorphous molecules, less than 5% amorphous molecules, or less than 2% amorphous molecules). It is also noted that the term “substantially crystalline” includes the descriptor "crystalline,” which refers to materials that are 100% crystalline form. [0094] As used herein, a crystalline form is "substantially pure” when it accounts for an amount by weight equal to or greater than 90% of the sum of all solid form(s) in a sample as determined by a method in accordance with the art, such as quantitative XRPD.
- the solid form is “substantially pure” when it accounts for an amount by weight equal to or greater than 95% of the sum of all solid form(s) in a sample. In some embodiments, the solid form is “substantially pure” when it accounts for an amount by weight equal to or greater than 99% of the sum of all solid form(s) in a sample.
- the terms "X-ray powder diffractogram,” “X-ray powder diffraction pattern,” “XRPD pattern,” “XRPD spectrum” interchangeably refer to an experimentally obtained pattern plotting signal positions (on the abscissa) versus signal intensities (on the ordinate).
- a “signal” or “peak” as used herein refers to a point in the XRPD pattern where the intensity as measured in counts is at a local maximum.
- An XRPD peak is identified by its angular value as measured in degrees 2 ⁇ (°2 ⁇ ), depicted on the abscissa of an X-ray powder diffractogram, which may be expressed, for example, as "a signal at ... degrees two-theta,” “a signal at [a] two-theta value(s) of ... " and/or "a signal at at least ... two-theta value(s) selected from ....
- the repeatability of the measured angular values is in the range of ⁇ 0.2° 2 ⁇ , i.e., the angular value can be at the recited angular value +0.2 degrees two-theta, the angular value -0.2 degrees two-theta, or any value between those two end points (angular value +0.2 degrees two-theta and angular value -0.2 degrees two-theta).
- the angular value can be at the recited angular value +0.2 degrees two-theta, the angular value -0.2 degrees two-theta, or any value between those two end points (angular value +0.2 degrees two-theta and angular value -0.2 degrees two-theta).
- One of ordinary skill in the art would recognize that one or more signals (or peaks) in an XRPD pattern may overlap and may, for example, not be apparent to the naked eye.
- signal intensities and “peak intensities” interchangeably refer to relative signal intensities within a given X-ray powder diffractogram. Factors that can affect the relative signal or peak intensities include sample thickness and preferred orientation (e.g., the crystalline particles are not distributed randomly).
- an X-ray powder diffractogram is "substantially similar to that in [a particular] Figure" when at least 90%, such as at least 95%, at least 98%, or at least 99%, of the signals in the two diffractograms overlap.
- substantially similarity one of ordinary skill in the art will understand that there may be variation in the intensities and/or signal positions in XRPD diffractograms even for the same crystalline form.
- the signal maximum values in XRPD diffractograms in degrees two-theta generally mean that value is identified as ⁇ 0.2 degrees two-theta of the reported value, an art-recognized variance.
- the term “glass transition temperature” or “Tg” refers to the temperature above which a hard and brittle “glassy” amorphous solid becomes viscous or rubbery supercooled liquid.
- the term “melting temperature”, “melting point”, or “Tm” refers to the temperature at which a crystalline material is in equilibrium with liquid phase.
- the term “dispersion” refers to a disperse system in which one substance, the dispersed phase, is distributed, in discrete units, throughout a second substance (the continuous phase or vehicle). The size of the dispersed phase can vary considerably (e.g., colloidal particles of nanometer dimension, to multiple microns in size).
- the dispersed phases can be solids, liquids, or gases.
- the dispersed and continuous phases are both solids.
- a solid dispersion can include a crystalline drug (dispersed phase) in an amorphous polymer (continuous phase); or alternatively, an amorphous drug (dispersed phase) in an amorphous polymer (continuous phase).
- a solid dispersion includes the polymer constituting the dispersed phase, and the drug constitute the continuous phase or, a solid dispersion includes the drug constituting the dispersed phase, and the polymer constituting the continuous phase.
- One aspect of the disclosure provides a method of preparing a compound of formula I: I, or a solvate or a tautomer or a [0105]
- Another aspect of the disclosure provides solid forms of Compound I (e.g., crystalline forms, amorphous forms, solvates, hydrates, cocrystals), which can be used in the methods of treatment and pharmaceutical compositions described herein.
- the invention provides neat amorphous forms of Compound I.
- the invention provides neat crystalline forms of Compound I.
- the invention provides solvate crystalline forms of Compound I.
- the invention provides hydrate crystalline forms of Compound I. A.
- the method steps described herein may refer to conversion of a starting compound of formulae A 1 , A 1-1 , A 1-2 , A 1-3 , A 1-4 , A 1-5 , A 2 , A 2-1 , A 3 , A 3-1 , A 3-2 , B-1, B-2, B-3, B-4, B-5, C-1, C-2, C-3, C-4, D-1, D-2, E-A 1 , E-A 2 , E-A 3 , and F-A 2 to the compound of formula I.
- the skilled artisan would understand that such methods can also be used to prepare any intermediate between any starting compound and the compound of formula I.
- conversion of the compound of formula A 1-2 to the compound of formula I goes through intermediate compound E-A 1 .
- the skilled artisan would understand that the methods described for converting the compound of formula A 1-2 to the compound of formula I can be used to prepare intermediate compounds E-A1, A1, and D1 from the compounds of formula A 1-2 .
- conversion of the compound of formula A 3-2 to the compound of formula I goes through preparation of intermediate compounds A 3-1 , A 3 , D-1, and E-A 3 .
- the present application contemplates preparing intermediate compounds A 1 , A 1-1 , A 1-2 , A 1-3 , A 1-4 , A 1-5 , A 2 , A 2-1 , A 3 , A 3-1 , A 3-2 , B-1, B-2, B-3, B-4, B-5, C-1, C-2, C-3, C-4, D-1, D-2, E-A 1 , E-A 2 , E-A 3 , and F-A 2 starting with any intermediate or starting material that precedes the intermediate that is being prepared.
- Scheme 1 Scheme 1
- Scheme 9 comprising transforming a to the compound of formula (I); wherein are independently selected from halogen.
- X 1 and X 2 are the same.
- each of X 1 and X 2 are fluoro.
- each of X 1 and X 2 are chloro.
- each of X 1 and X 2 are bromo.
- each of X 1 and X 2 are iodo.
- X 1 and X 2 are different.
- the step of transforming the compound of formula (A 1 ) to the compound of formula (I) comprises transforming the compound of formula (A 1 ) to a compound of formula (E-A 1 ): X 2 CN N .
- the compound of formula (A 1 ) with a compound of formula (D-1) is presence a first palladium catalyst and a first base.
- the first palladium catalyst is a palladium-phosphine complex.
- the first palladium catalyst is PdCl2(dtbdpf).
- the step of contacting the compound of formula (A 1 ) with a compound of formula (D-1) is performed in the presence of a first palladium catalyst, a first ligand, and a first base.
- the first palladium catalyst is PdCl 2 .
- the first ligand is selected from 1,1′-Bis(di-tert- butylphosphino)ferrocene (dtbpf), 1,1′-Ferrocenediyl-bis(diphenylphosphine) (dppf), Bis[(2- diphenylphosphino)phenyl] ether (DPEPhos), 1,2-Bis(diphenylphosphino)ethane (dppe), triphenylphosphine (PPh 3 ), cyclohexyldiphenylphosphine (CyPh 2 P), tri(o-tolyl)phosphine (P(o-tol) 3 ), and Di(1-adamantyl)-n-butylphosphine (CataCXium A).
- the first ligand is 1,1′-Bis(di-tert-butylphosphino)ferrocene (dtbpf). In other embodiments, the first ligand is 1,1′-Ferrocenediyl-bis(diphenylphosphine) (dppf). In other embodiments, the first ligand is Bis[(2- diphenylphosphino)phenyl] ether (DPEPhos). In other embodiments, the first ligand is 1,2-Bis(diphenylphosphino)ethane (dppe). In other embodiments, the first ligand is triphenylphosphine (PPh 3 ).
- the first ligand is cyclohexyldiphenylphosphine (CyPh 2 P). In other embodiments, the first ligand is tri(o-tolyl)phosphine (P(o-tol) 3 ). In other embodiments, the first ligand is di(1-adamantyl)-n-butylphosphine (CataCXium A). [0126] In some embodiments, the first base is selected from potassium carbonate, potassium phosphate, and potassium fluoride.
- the first base is selected from potassium carbonate, potassium phosphate, potassium fluoride, potassium bicarbonate, tripotassium phosphate, dipotassium phosphate, potassium acetate, potassium pivalate, potassium hydroxide, potassium hexamethyldisilazide, 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU), and triethylamine.
- the first base is potassium carbonate.
- the first base is potassium phosphate.
- the first base is potassium fluoride.
- the first base is potassium bicarbonate.
- the first base is tripotassium phosphate.
- the first base is dipotassium phosphate. In other embodiments, the first base is potassium acetate. In other embodiments, the first base is potassium pivalate. In other embodiments, the first base is potassium hydroxide. In other embodiments, the first base is potassium hexamethyldisilazide. In other embodiments, the first base is 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU). In other embodiments, the first base is triethylamine. [0127] In some embodiments, the step of transforming the compound of formula (A1) to the compound of formula (I) further comprises transforming the compound of formula (E-A 1 ) to the compound of formula (I).
- the step of transforming the compound of formula (E-A 1 ) to the compound of formula (I) comprises treating the compound of formula (E-A 1 ) with a first acid.
- the first acid is selected from sulfuric acid, phosphoric acid, methanesulfonic acid, and trifluoroacetic acid. In some embodiments, the first acid is trifluoroacetic acid. In some embodiments, the first acid is sulfuric acid. In some embodiments, the first acid is methane sulfonic acid.
- the step of transforming the compound of formula (A 1 ) to the compound of formula (I) further comprises transforming a compound of formula (E-A 2 ): Cl CN N to the compound of formula (I).
- the step of transforming the compound of formula (E-A 2 ) to the compound of formula (I) comprises treating the compound of formula (E-A 2 ) with a first acid in the presence of water and heat to prepare a compound of formula (F-A 2 ): .
- the first acid formic acid, sulfuric acid, phosphoric acid, hydrochloric acid, toluenesulfonic acid, and trifluoroacetic acid.
- the first acid is acetic acid. In other embodiments, the first acid is formic acid. In other embodiments, the first acid is sulfuric acid. In other embodiments, the first acid is phosphoric acid. In other embodiments, the first acid is hydrochloric acid. In other embodiments, the first acid is methanesulfonic acid. In other embodiments, the first acid is oxalic acid. In other embodiments, the first acid is p-toluenesulfonic acid. In other embodiments, the first acid is trifluoroacetic acid.
- the step of treating the compound of formula (E-A 2 ) with a first acid in the presence of water and heat to prepare a compound of formula (F-A 2 ) is performed at a temperature of about 60 °C to about 100 °C. In other embodiments, the step of treating the compound of formula (E-A2) with a first acid in the presence of water and heat to prepare a compound of formula (F-A 2 ) is performed at a temperature of about 100 °C.
- the step of transforming the compound of formula (E-A 2 ) to the compound of formula (I) comprises treating the compound of formula (E-A 2 ) with a first lewis acid in the presence of acetic acid to prepare a compound of formula (F-A 2 ).
- the first lewis acid is selected from MoCl 2 O 2 , MoCl 5 , CuOTf 2 , VCl 3 , WOCl 4 , and WCl 6 .
- the first lewis acid is MoCl 2 O 2 .
- the first lewis acid is MoCl 5 .
- the first lewis acid is CuOTf 2 .
- the first lewis acid is VCl 3 . In other embodiments, the first lewis acid is WOCl 4 . In other embodiments, the first lewis acid is WCl 6 . [0132] In some embodiments, the step of transforming the compound of formula (E-A 2 ) to the compound of formula (I) comprises treating the compound of formula (E-A 2 ) with a first base in the presence of water and heat to prepare a compound of formula (F-A 2 ). In some embodiments, the first base is potassium acetate.
- the step of treating the compound of formula (E-A 2 ) with a first base in the presence of water and heat to prepare a compound of formula (F-A 2 ) is performed at a temperature of about 80 °C to about 100 °C.
- the step of transforming the compound of formula (A 1 ) to the compound of formula (I) further comprises transforming the compound of formula (F-A 2 ) to the compound of formula (I).
- the step of transforming the compound of formula (F-A 2 ) to the compound of formula (I) comprises treating the compound of formula (F-A 2 ) with an acid.
- the acid is selected from sulfuric acid, phosphoric acid, methanesulfonic acid, oxalic acid, and trifluoroacetic acid. In some embodiments, the acid is trifluoroacetic acid. In some embodiments, the acid is sulfuric acid. In some embodiments, the acid is methane sulfonic acid. In some embodiments, the acid is methanesulfonic acid. In some embodiments, the acid is phosphoric acid. In some embodiments, the first acid is oxalic acid. [0135] In some embodiments of preparing the compound of formula I, the process comprises transforming a compound of formula (A 1-1 ): to the compound of formula (A 1 ).
- the a compound of formula (A 1-1 ) to the compound of formula (A1) comprises the steps of: treating the compound of formula (A 1-1 ) with trifluoromethanesulfonic anhydride to form a triflyl intermediate; treating the triflyl intermediate with a cyanation reagent; treating the cyanated intermediate with a second base to form a cyanated triflyl intermediate; and treating the cyanated triflyl intermediate with an aqueous base to form the compound of formula (A 1 ).
- the cyanation reagent is trimethylsilyl cyanide, sodium cyanide, and potassium cyanide.
- the cyanation reagent is trimethylsilyl cyanide. In some embodiments, the cyanation reagent is sodium cyanide. In some embodiments, the cyanation reagent is potassium cyanide.
- the second base is selected from 4-methyl morpholine, trimethylamine, and Hünig’s base. In some embodiments, the second base is 4-methylmorpholine. In some embodiments, the second base is trimethylamine. In some embodiments, the second base is Hünig’s base.
- the aqueous base is aqueous sodium bicarbonate.
- the process for preparing a compound of formula I comprises transforming a compound of formula (A 1-2 ): to the compound of formula (A 1-1 ).
- the the compound of formula (A 1-2 ) to the compound of formula (A1-1) comprises treating the compound of formula (A1-2) with a halogenating reagent.
- the halogenating reagent is a chlorinating reagent.
- the chlorinating reagent is selected from HCl, oxalyl chloride, trichloroacetic anhydride/phosgene, triphenylphosphine dichloride, phenylphosphonic dichloride, phosphorous trichloride, phosphorous pentachloride, phosphorous oxychloride, thionyl chloride, p-toluenesulfonyl chloride, and methanesulfonyl chloride.
- the chlorinating reagent is phosphorous oxychloride.
- the chlorinating reagent is selected from Cl 2 (with/without oxidant), HCl/Cl - plus oxidant, N-chlorosuccinimide (NCS)/Dichlorodimethylhydantoin (DCDMH)/ Trichloroisocyanuric acid (TCCA) sodium dichloroiso-cyanurate NaDCC, NaOCl, SO 2 Cl 2 , and CCl 4 /C 2 Cl 6 .
- the chlorinating reagent is selected from phosphorus oxychloride and phosphorus pentachloride.
- the chlorinating reagent is phosphorus oxychloride.
- the process for preparing a compound of formula I comprises transforming a compound of formula (A 1-3 ): to the compound of formula (A 1-2 ).
- the step of transforming a compound of formula (A 1-3 ) to the compound of formula (A 1-3 ) comprises treating the compound of formula (A 1-3 ) with a second acid.
- the second acid is aqueous hydrochloric acid.
- the process of preparing a compound of formula I comprises transforming a compound of formula (A 1-4 ): to the compound of formula (A 1-3 ).
- the step of transforming a compound of formula (A 1-4 ) to the compound of formula (A 1-3 ) comprises treating the compound of formula (A 1-4 ) with diethyl malonate in the presence of a third base.
- the third base is an alkaline alkoxide.
- the third base is sodium ethoxide.
- the process of preparing a compound of formula I comprises transforming a compound of formula (A 1-5 ): to the compound of formula (A 1-4 ).
- the the compound of formula (A 1-5 ) to the compound of formula (A 1-4 ) comprises treating the compound of formula (A 1-5 ) with carbon monoxide in the presence of a fourth base, a second palladium catalyst, and a first suitable ligand.
- the fourth base is selected from triethylamine, Hünig’s Base, 4-methylmorpholine, 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU), and 1,4-Diazabicyclo[2.2.2]octane (DABCO).
- the fourth base is triethylamine.
- the second palladium catalyst is Pd(OAc) 2 .
- the first suitable ligand is selected from 1,1'-bis(diphenylphosphino)ferrocene (DPPF), 4,5-Bis(diphenylphosphino)-9,9-dimethylxanthene (XantPhos), triphenylphosphine (PPh3), and 2,2′-Bis(diphenylphosphino)-1,1′-binaphthalene (BINAP).
- the first suitable ligand is 1,1'-bis(diphenyl phosphino)ferrocene (DPPF).
- each of X 1 and X 2 are chloro.
- a compound of formula I comprises contacting the compound of formula (A 3 ) with a compound of formula (D-1) in the presence of a first palladium catalyst and a first base.
- the first palladium catalyst is PdCl2(dtbdpf).
- the first base is potassium phosphate.
- the step of contacting the compound of formula (A 3 ) with a compound of formula (D-1) is performed in the presence of a first palladium catalyst, a first ligand, and a first base.
- the first palladium catalyst is PdCl 2 .
- the first ligand is selected from 1,1′-Bis(di-tert- butylphosphino)ferrocene (dtbpf), 1,1′-Ferrocenediyl-bis(diphenylphosphine) (dppf), Bis[(2- diphenylphosphino)phenyl] ether (DPEPhos), 1,2-Bis(diphenylphosphino)ethane (dppe), triphenylphosphine (PPh 3 ), cyclohexyldiphenylphosphine (CyPh 2 P), tri(o-tolyl)phosphine (P(o-tol) 3 ), and Di(1-adamantyl)-n-butylphosphine (CataCXium A).
- the first ligand is 1,1′-Bis(di-tert-butylphosphino)ferrocene (dtbpf). In other embodiments, the first ligand is 1,1′-Ferrocenediyl-bis(diphenylphosphine) (dppf). In other embodiments, the first ligand is Bis[(2- diphenylphosphino)phenyl] ether (DPEPhos). In other embodiments, the first ligand is 1,2-Bis(diphenylphosphino)ethane (dppe). In other embodiments, the first ligand is triphenylphosphine (PPh 3 ).
- the first ligand is cyclohexyldiphenylphosphine (CyPh 2 P). In other embodiments, the first ligand is tri(o-tolyl)phosphine (P(o-tol) 3 ). In other embodiments, the first ligand is di(1-adamantyl)-n-butylphosphine (CataCXium A).
- the first base is selected from potassium carbonate, potassium phosphate, potassium fluoride, potassium bicarbonate, tripotassium phosphate, dipotassium phosphate, potassium acetate, potassium pivalate, potassium hydroxide, potassium hexamethyldisilazide, 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU), and triethylamine.
- the first base is potassium carbonate.
- the first base is potassium phosphate.
- the first base is potassium fluoride.
- the first base is potassium bicarbonate.
- the first base is tripotassium phosphate.
- the first base is dipotassium phosphate. In other embodiments, the first base is potassium acetate. In other embodiments, the first base is potassium pivalate. In other embodiments, the first base is potassium hydroxide. In other embodiments, the first base is potassium hexamethyldisilazide. In other embodiments, the first base is 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU). In other embodiments, the first base is triethylamine. [0159] In some embodiments, the step of transforming the compound of formula (A 3 ) to the compound of formula (I) further comprises transforming the compound of formula (E-A 3 ) to the compound of formula (I).
- the step of transforming the compound of formula (E-A 3 ) to the compound of formula (I) comprises treating the compound of formula (E-A 3 ) with a first acid.
- the first acid is trifluoroacetic acid, sulfuric acid, phosphoric acid, and methane sulfonic acid.
- the first acid is trifluoroacetic acid.
- the first acid is sulfuric acid.
- the first acid is methane sulfonic acid.
- the process of preparing a compound of formula I comprises transforming a compound of formula (A 3-1 ): to the compound of formula (A 3 ).
- the step of transforming a compound of formula (A 3-1 ) to the compound of formula (A 3 ) comprises treating the compound of formula (A 3-1 ) with a cyanation reagent and a second base.
- the cyanation reagent is trimethylsilyl cyanide, sodium cyanide, and potassium cyanide.
- the cyanation reagent is trimethylsilyl cyanide.
- the cyanation reagent is sodium cyanide.
- the cyanation reagent is potassium cyanide.
- the second base is triethylamine.
- the process of preparing a compound of formula I comprise transforming a compound of formula (A 3-2 ): to the compound of formula (A 3-1 ).
- a compound of formula (A 3-2 ) to the compound of formula (A 3-1 ) comprises treating the compound of formula (A 3-2 ) with benzyl alcohol in the presence of a third base.
- the third base is selected from carbonate base, phosphate base, sodium hexamethyldisilizane, alkyllythium reagent, hydride reagent, alkaline alkoxide, and alkaline hydroxide.
- the third base is an alkaline alkoxide.
- the third base is an alkaline methoxide. In some embodiments, the third base is selected from lithium methoxide, sodium methoxide, and potassium methoxide. In some embodiments, the third base is lithium methoxide. In some embodiments, the third base is sodium methoxide. In some embodiments, the third base is potassium methoxide. In some embodiments, the third base is an alkaline ethoxide. In some embodiments, the third base is selected from lithium ethoxide, sodium ethoxide, and potassium ethoxide. In some embodiments, the third base is lithium ethoxide. In some embodiments, the third base is sodium ethoxide.
- the third base is potassium ethoxide. In some embodiments, the third base is an alkaline tert-butoxide. In some embodiments, the third base is selected from lithium tert-butoxide, sodium tert-butoxide, and potassium tert-butoxide. In some embodiments, the third base is lithium tert-butoxide. In some embodiments, the third base is sodium tert-butoxide. In some embodiments, the third base is potassium tert-butoxide. In some embodiments, third base is an alkaline tert-pentoxide.
- the third base is selected from lithium tert-pentoxide, sodium tert-pentoxide, and potassium tert-pentoxide. In some embodiments, the third base is lithium tert- pentoxide. In some embodiments, the third base is sodium tert-pentoxide. In some embodiments, the third base is potassium tert-pentoxide. In some embodiments, the third base is selected from lithium bis(trimethylsilyl)amide, sodium bis(trimethylsilyl)amide, and potassium bis(trimethylsilyl)amide. In some embodiments, the third base is lithium bis(trimethylsilyl)amide. In some embodiments, the third base is sodium bis(trimethylsilyl)amide.
- the third base is potassium bis(trimethylsilyl) amide. In some embodiments, the third base is an alkaline hydride. In some embodiments, the third base is selected from lithium hydride, sodium hydride, and potassium hydride. In some embodiments, the third base is lithium hydride. In some embodiments, the third base is sodium hydride. In some embodiments, the third base is potassium hydride. [0168] In some embodiments, the process of preparing a compound of formula I comprises transforming a compound of formula (D-2): to the compound of formula (D-1).
- a compound of formula (D-2) to the compound of formula (D-1) comprises treating the compound of formula (D-2) with tetrahydroxydiboron (diboronic acid), in the presence of a third palladium catalyst, a second suitable ligand, and a fifth base.
- the third palladium catalyst is selected from Pd(OAc) 2 , Pd(dppf)Cl 2 , Pd(PCy 3 ) 2 , and Pd 2 dba 3 .
- the third palladium catalyst is Pd(OAc) 2 .
- the second suitable ligand is cyclohexyldiphenylphosphine.
- the step of transforming a compound of formula (B-1) to the compound of formula (D-2) comprises contacting the compound of formula (B-2) with a compound of formula (C-1) in the presence of a sixth base and a phase transfer catalyst [0175]
- the sixth base is potassium carbonate.
- the phase transfer catalyst is selected from tetrabutylammonium bromide (TBAB), tetrabutylammonium chloride (TBAC), tetrabutylphosphonium bromide (TBPB), tetrabutylammonium hydrogen sulfate (TBA HSO 4 ), and tetraoctylammonium chloride (TOAC).
- TBAB tetrabutylammonium bromide
- TBAC tetrabutylammonium chloride
- the phase transfer catalyst is tetrabutylphosphonium bromide (TBPB).
- the phase transfer catalyst is tetrabutylammonium hydrogen sulfate (TBA HSO 4 ). In other embodiments, the phase transfer catalyst is tetraoctylammonium chloride (TOAC).
- the process of preparing a compound of formula I comprises transforming a compound of formula (B-2): to the compound of formula (B-1).
- the step of transforming the compound of formula (B-2) to the compound of formula (B-1) comprises treating the compound of formula (B-2) with a chlorinating reagent.
- the chlorinating reagent is selected from HCl, oxalyl chloride, trichloroacetic anhydride/phosgene, triphenylphosphine dichloride, phenylphosphonic dichloride, phosphorous trichloride, phosphorous pentachloride, phosphorous oxychloride, thionyl chloride, p- toluenesulfonyl chloride, and methanesulfonyl chloride.
- the chlorinating reagent is phosphorous oxychloride.
- the chlorinating reagent is selected from Cl 2 (with/without oxidant), HCl/Cl - plus oxidant, N-chlorosuccinimide (NCS)/dichlorodimethyl- hydantoin (DCDMH)/trichloroisocyanuric acid (TCCA) sodium dichloroiso-cyanurate NaDCC, NaOCl, SO 2 Cl 2 , and CCl 4 /C 2 Cl 6 .
- the chlorinating reagent is selected from phosphorus oxychloride and phosphorus pentachloride.
- the chlorinating reagent is phosphorus oxychloride.
- the process of preparing a compound of formula I comprises transforming a compound of formula (B-3): to the compound of formula (B-2).
- the compound of formula (B-3) to the compound of formula (B-2) comprises treating the compound of formula (B-3) with a brominating reagent in the presence of a third acid.
- the brominating reagent is molecular bromine.
- the third acid is acetic acid.
- the process of preparing a compound of formula I comprises transforming a compound of formula (B-4): to the compound of formula (B-3).
- the a compound of formula (B-4) to the compound of formula (B-3) comprises treating the compound of formula (B-4) with a fourth acid in the presence of a first suitable solvent.
- the fourth acid is a mineral acid.
- the fourth acid is concentrated aqueous hydrochloric acid.
- the first suitable solvent comprises dioxane and water.
- the process of preparing a compound of formula I comprise transforming a compound of formula (B-5): to the compound of formula (B-4).
- the step of transforming a compound of formula (B-5) to the compound of formula (B-4) comprises treating the compound of formula (B-5) with a methylating reagent in the presence of a fourth palladium catalyst, and a seventh base.
- the first methylating reagent is methylboronic acid.
- the fourth palladium catalyst is Pd(dppf)Cl 2 •DCM.
- the seventh base is potassium carbonate.
- the process of preparing a compound of formula I comprises transforming a compound of formula (C-2): to the compound of formula (C-1), wherein protecting group.
- the a compound of formula (C-2) to the compound of formula (C-1) comprises deprotecting the compound of formula (C-1) to afford the compound of formula (C-2).
- PG is tetrahydropyranyl.
- the step of transforming the compound of formula (C-2) to the compound of formula (C-1) comprises treating the compound of formula (C-2) with a first acid catalyst.
- the first acid catalyst is pyridinium p-toluenesulfonate.
- the step of transforming a compound of formula (C-3) to the compound of formula (C-2), wherein protecting group.
- the PG is tetrahydropyranyl.
- the step of transforming a compound of formula (C-3) to the compound of formula (C-2) comprises treating the compound of formula (C-3) with a second methylating reagent in the presence of an eighth base.
- the second methylating reagent is methyl iodide.
- the eighth base is butyllithium.
- the eighth base is n-butyllithium.
- the process of preparing a compound of formula I comprises transforming a compound of formula (C-4): to the compound of formula (C-3).
- the a compound of formula (C-4) to the compound of C-3 comprises treating the compound of (C-4) with a suitable protecting group in the presence of a second acid catalyst.
- the suitable protecting group is 3,4-dihydropyran.
- the second acid catalyst is pyridinium p-toluenesulfonate.
- the process of preparing a compound of formula I comprises transforming the compound of formula (I) to a solvate of the compound of formula (I).
- the step of transforming the compound of formula (I) to the solvate of the compound of formula (I) comprises recrystallizing the compound of formula (I) from a suitable solvent.
- the suitable solvent is ethanol.
- the solvate of the compound of formula (I) is a compound of formula (F): O NH O 2 .
- In some of formula I comprises the step of transforming the solvate of the compound of formula (I) to Form A of the compound of formula (I).
- the invention relates to a compound selected from: , more compounds selected from: , , [0 und of formula (I) and a compound of formula (E-A 2 ): .
- the acid is trifluoroacetic acid.
- the invention relates to a composition
- a composition comprising a compound of formula (I) and a compound of formula (E-A 3 ): .
- the acid is [0207]
- a composition comprises a compound of formula (E-A 2 ), a compound of formula (A 2 ), and a compound of formula (D-1): A 3 ) a , a :
- a compound of formula (I): , or a pharmaceutically acceptable is prepared by the process of any one or more of the processes disclosed B.
- Compound I Amorphous Form [0210] In some embodiments, the disclosure provides a neat amorphous form of Compound I. In some embodiments, the disclosure provides Compound I amorphous form. FIG. 1 provides an X-ray powder diffractogram of Compound I amorphous form at room temperature. [0211] In some embodiments, Compound I amorphous form is substantially pure. In some embodiments, Compound I amorphous form is substantially amorphous.
- Compound I amorphous form is characterized by an X-ray powder diffractogram generated by an X-ray powder diffraction analysis with an incident beam of Cu K ⁇ radiation. [0212] In some embodiments, Compound I amorphous form is characterized by an X-ray powder diffractogram substantially similar to FIG.1. [0213] In some embodiments, Compound I amorphous form is characterized as having a 13 C SSNMR spectrum with a peak at 177.6 ⁇ 0.2 ppm. In some embodiments, Compound I amorphous form is characterized as having a 13 C SSNMR spectrum with a peak at 172.0 ⁇ 0.2 ppm.
- Compound I amorphous form is characterized as having a 13 C SSNMR spectrum with a peak at 162.6 ⁇ 0.2 ppm. In some embodiments, Compound I amorphous form is characterized as having a 13 C SSNMR spectrum with a peak at 155.2 ⁇ 0.2 ppm. In some embodiments, Compound I amorphous form is characterized as having a 13 C SSNMR spectrum with a peak at 149.9 ⁇ 0.2 ppm. In some embodiments, Compound I amorphous form is characterized as having a 13 C SSNMR spectrum with a peak at 147.0 ⁇ 0.2 ppm.
- Compound I amorphous form is characterized as having a 13 C SSNMR spectrum with a peak at 121.0 ⁇ 0.2 ppm. In some embodiments, Compound I amorphous form is characterized as having a 13 C SSNMR spectrum with a peak at 120.1 ⁇ 0.2 ppm. In some embodiments, Compound I amorphous form is characterized as having a 13 C SSNMR spectrum with a peak at 119.1 ⁇ 0.2 ppm. In some embodiments, Compound I amorphous form is characterized as having a 13 C SSNMR spectrum with a peak at 116.0 ⁇ 0.2 ppm.
- Compound I amorphous form is characterized as having a 13 C SSNMR spectrum with a peak at 114.6 ⁇ 0.2 ppm. In some embodiments, Compound I amorphous form is characterized as having a 13 C SSNMR spectrum with a peak at 113.8 ⁇ 0.2 ppm. In some embodiments, Compound I amorphous form is characterized as having a 13 C SSNMR spectrum with a peak at 15.5 ⁇ 0.2 ppm. In some embodiments, Compound I amorphous form is characterized as having a 13 C SSNMR spectrum with a peak at 7.7 ⁇ 0.2 ppm.
- Compound I amorphous form is characterized as having a 13 C SSNMR spectrum with one, two, three, four, five, six, seven, eight, nine, ten, or more peaks selected from 177.6 ⁇ 0.2 ppm, 172.0 ⁇ 0.2 ppm, 162.6 ⁇ 0.2 ppm, 155.2 ⁇ 0.2 ppm, 149.9 ⁇ 0.2 ppm, 147.0 ⁇ 0.2 ppm, 121.0 ⁇ 0.2 ppm, 120.1 ⁇ 0.2 ppm, 119.1 ⁇ 0.2 ppm, 116.0 ⁇ 0.2 ppm, 114.6 ⁇ 0.2 ppm, 113.8 ⁇ 0.2 ppm, 15.5 ⁇ 0.2 ppm, and 7.7 ⁇ 0.2 ppm.
- Compound I amorphous form is characterized as having a 13 C SSNMR spectrum with peaks at 177.6 ⁇ 0.2 ppm, 172.0 ⁇ 0.2 ppm, 162.6 ⁇ 0.2 ppm, 155.2 ⁇ 0.2 ppm, 149.9 ⁇ 0.2 ppm, 147.0 ⁇ 0.2 ppm, 121.0 ⁇ 0.2 ppm, 120.1 ⁇ 0.2 ppm, 119.1 ⁇ 0.2 ppm, 116.0 ⁇ 0.2 ppm, 114.6 ⁇ 0.2 ppm, 113.8 ⁇ 0.2 ppm, 15.5 ⁇ 0.2 ppm, and 7.7 ⁇ 0.2 ppm.
- Compound I amorphous form is characterized by a 19 F SSNMR spectrum substantially similar to FIG.3.
- Another aspect of the disclosure provides a method of making Compound I amorphous form.
- the method of making Compound I amorphous form comprises: comprising (i) dissolving Compound I, (ii) filtering and evaporating the solvent at 50 °C, 12 mbar, over 1 hour on a centrifugal evaporation, (iii) drying in a vacuum oven at 50 °C overnight to yield Compound I amorphous form.
- C. Crystalline Compound I Neat Form A [0220]
- the disclosure provides neat crystalline forms of Compound I.
- the disclosure provides crystalline Compound I neat Form A.
- FIG.4 provides an X-ray powder diffractogram of crystalline Compound I neat Form A.
- crystalline Compound I neat Form A is substantially pure.
- crystalline Compound I neat Form A is substantially crystalline.
- crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram generated by an X-ray powder diffraction analysis with an incident beam of Cu K ⁇ radiation.
- crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 9.5 ⁇ 0.2 degrees two-theta.
- crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 12.3 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 13.5 ⁇ 0.2 degrees two-theta. [0223] In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 7.4 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 8.3 ⁇ 0.2 degrees two-theta.
- crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 11.3 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 14.4 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 14.9 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 15.0 ⁇ 0.2 degrees two-theta.
- crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 15.9 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 16.6 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 17.6 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 18.0 ⁇ 0.2 degrees two-theta.
- crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 18.7 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 18.9 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 19.2 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 19.5 ⁇ 0.2 degrees two-theta.
- crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 19.8 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 20.5 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 21.4 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 21.9 ⁇ 0.2 degrees two-theta.
- crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 22.5 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 23.5 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 24.1 ⁇ 0.2 degrees two-theta.
- crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having signals at one or two of 9.5 ⁇ 0.2 degrees two theta, 12.3 ⁇ 0.2 degrees two theta, and 13.5 ⁇ 0.2 degrees two-theta.
- crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having (a) one or two signals selected from 9.5 ⁇ 0.2 degrees two-theta, 12.3 ⁇ 0.2 degrees two-theta, and 13.5 ⁇ 0.2 degrees two-theta, and (b) one or two signals selected from 11.3 ⁇ 0.2 degrees two-theta, 15.9 ⁇ 0.2 degrees two-theta, and 20.5 ⁇ 0.2 degrees two-theta.
- crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having two, three, four, five, or six signals selected from 9.5 ⁇ 0.2 degrees two- theta, 12.3 ⁇ 0.2 degrees two-theta, 13.5 ⁇ 0.2 degrees two-theta , 11.3 ⁇ 0.2 degrees two-theta, 15.9 ⁇ 0.2 degrees two-theta, 20.5 ⁇ 0.2 degrees two-theta, 14.9 ⁇ 0.2 degrees two-theta, 16.6 ⁇ 0.2 degrees two-theta, and 23.5 ⁇ 0.2 degrees two-theta.
- crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having one, two three, four five, six, seven, eight, nine, ten, eleven, twelve, thirteen, fourteen, fifteen, sixteen, seventeen, eighteen, nineteen, twenty, twenty-one, or twenty-two signals selected from 7.4 ⁇ 0.2 degrees two-theta; 8.3 ⁇ 0.2 degrees two-theta; 9.5 ⁇ 0.2 degrees two- theta; 11.3 ⁇ 0.2 degrees two-theta; 12.3 ⁇ 0.2 degrees two-theta; 13.5 ⁇ 0.2 degrees two-theta; 14.4 ⁇ 0.2 degrees two-theta; 14.9 ⁇ 0.2 degrees two-theta; 15.0 ⁇ 0.2 degrees two-theta; 15.9 ⁇ 0.2 degrees two-theta; 16.6 ⁇ 0.2 degrees two-theta; 17.6 ⁇ 0.2 degrees two-theta; 18.0 ⁇ 0.2 degrees two-theta; 18.
- crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having signals at 7.4 ⁇ 0.2 degrees two-theta, 8.3 ⁇ 0.2 degrees two-theta, 9.5 ⁇ 0.2 degrees two-theta, 11.3 ⁇ 0.2 degrees two-theta, 12.3 ⁇ 0.2 degrees two-theta, 13.5 ⁇ 0.2 degrees two-theta, 14.4 ⁇ 0.2 degrees two-theta, 14.9 ⁇ 0.2 degrees two-theta, 15.0 ⁇ 0.2 degrees two-theta, 15.9 ⁇ 0.2 degrees two-theta, 16.6 ⁇ 0.2 degrees two-theta, 17.6 ⁇ 0.2 degrees two-theta, 18.0 ⁇ 0.2 degrees two-theta, 18.7 ⁇ 0.2 degrees two-theta, 18.9 ⁇ 0.2 degrees two-theta, 19.2 ⁇ 0.2 degrees two-theta, 19.5 ⁇ 0.2 degrees two-theta,
- crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram substantially similar to FIG.4. [0230] In some embodiments, crystalline Compound I neat Form A is characterized as having a 13 C SSNMR spectrum with a peak at 170.4 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13 C SSNMR spectrum with a peak at 163.8 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13 C SSNMR spectrum with a peak at 162.4 ⁇ 0.2 ppm.
- crystalline Compound I neat Form A is characterized as having a 13 C SSNMR spectrum with a peak at 160.9 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13 C SSNMR spectrum with a peak at 158.6 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13 C SSNMR spectrum with a peak at 158.3 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13 C SSNMR spectrum with a peak at 154.6 ⁇ 0.2 ppm.
- crystalline Compound I neat Form A is characterized as having a 13 C SSNMR spectrum with a peak at 154.0 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13 C SSNMR spectrum with a peak at 148.3 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13 C SSNMR spectrum with a peak at 147.6 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13 C SSNMR spectrum with a peak at 146.9 ⁇ 0.2 ppm.
- crystalline Compound I neat Form A is characterized as having a 13 C SSNMR spectrum with a peak at 127.4 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13 C SSNMR spectrum with a peak at 125.3 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13 C SSNMR spectrum with a peak at 123.6 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13 C SSNMR spectrum with a peak at 122.2 ⁇ 0.2 ppm.
- crystalline Compound I neat Form A is characterized as having a 13 C SSNMR spectrum with a peak at 121.3 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13 C SSNMR spectrum with a peak at 120.7 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13 C SSNMR spectrum with a peak at 117.9 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13 C SSNMR spectrum with a peak at 117.3 ⁇ 0.2 ppm.
- crystalline Compound I neat Form A is characterized as having a 13 C SSNMR spectrum with a peak at 116.3 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13 C SSNMR spectrum with a peak at 115.8 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13 C SSNMR spectrum with a peak at 114.2 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13 C SSNMR spectrum with a peak at 113.6 ⁇ 0.2 ppm.
- crystalline Compound I neat Form A is characterized as having a 13 C SSNMR spectrum with a peak at 112.8 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13 C SSNMR spectrum with a peak at 15.4 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13 C SSNMR spectrum with a peak at 8.2 ⁇ 0.2 ppm.
- crystalline Compound I neat Form A is characterized as having a 13 C SSNMR spectrum with one, two, three, four, five, six, seven, eight, nine, ten, or more peaks selected from 170.4 ⁇ 0.2 ppm, 163.8 ⁇ 0.2 ppm, 162.4 ⁇ 0.2 ppm, 160.9 ⁇ 0.2 ppm, 158.6 ⁇ 0.2 ppm, 158.3 ⁇ 0.2 ppm, 154.6 ⁇ 0.2 ppm, 154.0 ⁇ 0.2 ppm, 148.3 ⁇ 0.2 ppm, 147.6 ⁇ 0.2 ppm, 146.9 ⁇ 0.2 ppm, 145.9 ⁇ 0.2 ppm, 143.5 ⁇ 0.2 ppm, 143.1 ⁇ 0.2 ppm, 141.9 ⁇ 0.2 ppm, 127.4 ⁇ 0.2 ppm, 125.3 ⁇ 0.2 ppm, 123.6 ⁇ 0.2 ppm, 122.2
- crystalline Compound I neat Form A is characterized as having a 13 C SSNMR spectrum with a 13 C SSNMR spectrum having peaks at 170.4 ⁇ 0.2 ppm, 163.8 ⁇ 0.2 ppm, 162.4 ⁇ 0.2 ppm, 160.9 ⁇ 0.2 ppm, 158.6 ⁇ 0.2 ppm, 158.3 ⁇ 0.2 ppm, 154.6 ⁇ 0.2 ppm, 154.0 ⁇ 0.2 ppm, 148.3 ⁇ 0.2 ppm, 147.6 ⁇ 0.2 ppm, 146.9 ⁇ 0.2 ppm, 145.9 ⁇ 0.2 ppm, 143.5 ⁇ 0.2 ppm, 143.1 ⁇ 0.2 ppm, 141.9 ⁇ 0.2 ppm, 127.4 ⁇ 0.2 ppm, 125.3 ⁇ 0.2 ppm, 123.6 ⁇ 0.2 ppm, 122.2 ⁇ 0.2 ppm, 121.3 ⁇ 0.2 ppm
- crystalline Compound I neat Form A is characterized by a 13 C SSNMR spectrum substantially similar to FIG.5.
- crystalline Compound I neat Form A is characterized as having a 19 F SSNMR spectrum with a peak at -61.2 ⁇ 0.2 ppm.
- crystalline Compound I neat Form A is characterized as having a 19 F SSNMR spectrum with a peak at -61.6 ⁇ 0.2 ppm.
- crystalline Compound I neat Form A is characterized as having a 19 F SSNMR spectrum with a peak at -138.6 ⁇ 0.2 ppm.
- crystalline Compound I neat Form A is characterized as having a 19 F SSNMR spectrum with one or two peaks selected from -61.2 ⁇ 0.2 ppm, -61.6 ⁇ 0.2 ppm, -138.6 ⁇ 0.2 ppm, -140.3 ⁇ 0.2 ppm, -142.0 ⁇ 0.2 ppm, and -146.0 ⁇ 0.2 ppm.
- crystalline Compound I neat Form A is characterized as having a 19 F SSNMR spectrum with peaks at -61.2 ⁇ 0.2 ppm, -61.6 ⁇ 0.2 ppm, -138.6 ⁇ 0.2 ppm, -140.3 ⁇ 0.2 ppm, -142.0 ⁇ 0.2 ppm, and -146.0 ⁇ 0.2 ppm.
- crystalline Compound I neat Form A is characterized by a 19 F SSNMR spectrum substantially similar to FIG.6.
- the diffractometer is a Bruker diffractometer.
- Another aspect of the invention provides a method of making crystalline Compound I neat Form A.
- the method of making crystalline Compound I neat Form A comprises: (i) combining Compound I and ethyl acetate, (ii) distilling under vacuum at 50 °C, (iii) adding ethyl acetate, (iv) distilling under vacuum at 50 °C, (v) heating to 75 °C, (vi) adding heptane, (vii) cooling to 40 °C, (viii) adding a seed of crystalline Compound I Form A, (ix) holding at 40 °C for 1.5 hours, (x) adding heptane over 5 hours, (xi) cooling the slurry to 20 °C over 5 hours, (xii) holding at 20 °C for 11 hours, (xiii) collecting the solids, (xiv) drying the solids in a vacuum oven at 45 °C with a slight nitrogen bleed for 46 hours to yield crystalline Compound I neat Form A.
- Crystalline Compound I Neat Form B [0239] In some embodiments, the disclosure provides crystalline Compound I neat Form B.
- FIG. 7 provides an X-ray powder diffractogram of crystalline Compound I neat Form B.
- crystalline Compound I neat Form B is substantially pure. In some embodiments, crystalline Compound I neat Form B is substantially crystalline. In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram generated by an X-ray powder diffraction analysis with an incident beam of Cu K ⁇ radiation.
- crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 4.8 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 13.3 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 18.5 ⁇ 0.2 degrees two-theta. [0242] In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 6.0 ⁇ 0.2 degrees two-theta.
- crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 7.0 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 8.8 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 10.5 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 12.1 ⁇ 0.2 degrees two-theta.
- crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 16.5 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 16.9 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 17.5 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 17.9 ⁇ 0.2 degrees two-theta.
- crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 18.9 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 19.2 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 19.5 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 20.9 ⁇ 0.2 degrees two-theta.
- crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 21.7 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 23.0 ⁇ 0.2 degrees two-theta. [0243] In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having signals at one or two 4.8 ⁇ 0.2 degrees two-theta, 13.3 ⁇ 0.2 degrees two-theta, and 18.5 ⁇ 0.2 degrees two-theta.
- crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having (a) one or two signals selected from from 4.8 ⁇ 0.2 degrees two-theta, 13.3 ⁇ 0.2 degrees two-theta, and 18.5 ⁇ 0.2 degrees two-theta, and (b) one or two signals selected from 6.0 ⁇ 0.2 degrees two-theta, 16.5 ⁇ 0.2 degrees two-theta, and 16.9 ⁇ 0.2 degrees two-theta.
- crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having two, three, four, five, or six signals selected from 4.8 ⁇ 0.2 degrees two-theta, 13.3 ⁇ 0.2 degrees two-theta, 18.5 ⁇ 0.2 degrees two-theta, 6.0 ⁇ 0.2 degrees two-theta, 16.5 ⁇ 0.2 degrees two-theta, 16.9 ⁇ 0.2 degrees two-theta, 14.1 ⁇ 0.2 degrees two-theta, 14.7 ⁇ 0.2 degrees two-theta, and 15.2 ⁇ 0.2 degrees two-theta.
- crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having one, two three, four five, six, seven, eight, nine, ten, eleven, twelve, thirteen, fourteen, fifteen, sixteen, seventeen, eighteen, nineteen, twenty, twenty-one, or twenty-two signals selected from 4.8 ⁇ 0.2 degrees two-theta; 6.0 ⁇ 0.2 degrees two-theta; 7.0 ⁇ 0.2 degrees two-theta; 8.8 ⁇ 0.2 degrees two-theta; 10.5 ⁇ 0.2 degrees two-theta; 12.1 ⁇ 0.2 degrees two-theta; 13.3 ⁇ 0.2 degrees two-theta; 13.8 ⁇ 0.2 degrees two-theta; 14.1 ⁇ 0.2 degrees two-theta; 14.7 ⁇ 0.2 degrees two-theta; 15.2 ⁇ 0.2 degrees two-theta; 16.5 ⁇ 0.2 degrees two-theta; 16.9 ⁇ 0.2 degrees two-theta; 1
- crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having signals at 4.8 ⁇ 0.2 degrees two-theta, 6.0 ⁇ 0.2 degrees two-theta, 7.0 ⁇ 0.2 degrees two-theta, 8.8 ⁇ 0.2 degrees two-theta, 10.5 ⁇ 0.2 degrees two-theta, 12.1 ⁇ 0.2 degrees two-theta, 13.3 ⁇ 0.2 degrees two-theta, 13.8 ⁇ 0.2 degrees two-theta, 14.1 ⁇ 0.2 degrees two-theta, 14.7 ⁇ 0.2 degrees two-theta, 15.2 ⁇ 0.2 degrees two-theta, 16.5 ⁇ 0.2 degrees two-theta, 16.9 ⁇ 0.2 degrees two-theta, 17.5 ⁇ 0.2 degrees two-theta, 17.9 ⁇ 0.2 degrees two-theta, 18.5 ⁇ 0.2 degrees two-theta, 18.9 ⁇ 0.2 degrees two-theta,
- crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram substantially similar to FIG.7. [0249] In some embodiments, crystalline Compound I neat Form B is characterized as having a 13 C SSNMR spectrum with a peak at 176.9 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13 C SSNMR spectrum with a peak at 171.7 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13 C SSNMR spectrum with a peak at 164.0 ⁇ 0.2 ppm.
- crystalline Compound I neat Form B is characterized as having a 13 C SSNMR spectrum with a peak at 161.4 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13 C SSNMR spectrum with a peak at 160.7 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13 C SSNMR spectrum with a peak at 155.3 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13 C SSNMR spectrum with a peak at 154.8 ⁇ 0.2 ppm.
- crystalline Compound I neat Form B is characterized as having a 13 C SSNMR spectrum with a peak at 148.6 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13 C SSNMR spectrum with a peak at 147.6 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13 C SSNMR spectrum with a peak at 146.4 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13 C SSNMR spectrum with a peak at 145.7 ⁇ 0.2 ppm.
- crystalline Compound I neat Form B is characterized as having a 13 C SSNMR spectrum with a peak at 144.8 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13 C SSNMR spectrum with a peak at 127.7 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13 C SSNMR spectrum with a peak at 125.4 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13 C SSNMR spectrum with a peak at 122.0 ⁇ 0.2 ppm.
- crystalline Compound I neat Form B is characterized as having a 13 C SSNMR spectrum with a peak at 121.5 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13 C SSNMR spectrum with a peak at 118.4 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13 C SSNMR spectrum with a peak at 116.3 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13 C SSNMR spectrum with a peak at 114.8 ⁇ 0.2 ppm.
- crystalline Compound I neat Form B is characterized as having a 13 C SSNMR spectrum with a peak at 113.8 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13 C SSNMR spectrum with a peak at 112.6 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13 C SSNMR spectrum with a peak at 17.8 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13 C SSNMR spectrum with a peak at 15.0 ⁇ 0.2 ppm.
- crystalline Compound I neat Form B is characterized as having a 13 C SSNMR spectrum with a peak at 10.7 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13 C SSNMR spectrum with a peak at 9.6 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13 C SSNMR spectrum with a peak at 8.8 ⁇ 0.2 ppm.
- crystalline Compound I neat Form B is characterized as having a 13 C SSNMR spectrum with one, two, three, four, five, six, seven, eight, nine, ten, or more peaks selected from 176.9 ⁇ 0.2 ppm, 171.7 ⁇ 0.2 ppm, 164.0 ⁇ 0.2 ppm, 161.4 ⁇ 0.2 ppm, 160.7 ⁇ 0.2 ppm, 155.3 ⁇ 0.2 ppm, 154.8 ⁇ 0.2 ppm, 148.6 ⁇ 0.2 ppm, 147.6 ⁇ 0.2 ppm, 146.4 ⁇ 0.2 ppm, 145.7 ⁇ 0.2 ppm, 144.8 ⁇ 0.2 ppm, 127.7 ⁇ 0.2 ppm, 125.4 ⁇ 0.2 ppm, 122.0 ⁇ 0.2 ppm, 121.5 ⁇ 0.2 ppm, 118.4 ⁇ 0.2 ppm, 116.3 ⁇ 0.2 ppm, 114.8 ⁇ 0.2 ppm, 116.3 ⁇
- crystalline Compound I neat Form B is characterized as having a 13 C SSNMR spectrum with peaks at 176.9 ⁇ 0.2 ppm, 171.7 ⁇ 0.2 ppm, 164.0 ⁇ 0.2 ppm, 161.4 ⁇ 0.2 ppm, 160.7 ⁇ 0.2 ppm, 155.3 ⁇ 0.2 ppm, 154.8 ⁇ 0.2 ppm, 148.6 ⁇ 0.2 ppm, 147.6 ⁇ 0.2 ppm, 146.4 ⁇ 0.2 ppm, 145.7 ⁇ 0.2 ppm, 144.8 ⁇ 0.2 ppm, 127.7 ⁇ 0.2 ppm, 125.4 ⁇ 0.2 ppm, 122.0 ⁇ 0.2 ppm, 121.5 ⁇ 0.2 ppm, 118.4 ⁇ 0.2 ppm, 116.3 ⁇ 0.2 ppm, 114.8 ⁇ 0.2 ppm, 113.8 ⁇ 0.2 ppm, 112.6 ⁇ 0.2 ppm, 17.
- crystalline Compound I neat Form B is characterized by a 13 C SSNMR spectrum substantially similar to FIG.8. [0253] In some embodiments, crystalline Compound I neat Form B is characterized as having a 19 F SSNMR spectrum with a peak at -61.6 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 19 F SSNMR spectrum with a peak at -138.0 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 19 F SSNMR spectrum with a peak at -139.7 ⁇ 0.2 ppm.
- crystalline Compound I neat Form B is characterized as having a 19 F SSNMR spectrum with a peak at -143.4 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 19 F SSNMR spectrum with a peak at -144.9 ⁇ 0.2 ppm. [0254] In some embodiments, crystalline Compound I neat Form B is characterized as having a 19 F SSNMR spectrum with one or two peaks selected from -61.6 ⁇ 0.2 ppm, -138.0 ⁇ 0.2 ppm, -139.7 ⁇ 0.2 ppm, -143.4 ⁇ 0.2 ppm, and -144.9 ⁇ 0.2 ppm.
- crystalline Compound I neat Form B is characterized as having a 19 F SSNMR spectrum with peaks at -61.6 ⁇ 0.2 ppm, -138.0 ⁇ 0.2 ppm, -139.7 ⁇ 0.2 ppm, -143.4 ⁇ 0.2 ppm, and -144.9 ⁇ 0.2 ppm.
- crystalline Compound I neat Form B is characterized by a 19 F SSNMR spectrum substantially similar to FIG.9.
- Another aspect of the disclosure provides a method of crystalline Compound I neat Form B.
- the method of making crystalline Compound I neat Form B comprises: (i) adding Compound I hydrate Form A to an oven set at 180 °C (ii) cooling under ambient conditions to yield crystalline Compound I neat Form B.
- the disclosure provides crystalline Compound I neat Form E.
- FIG. 10 provides an X-ray powder diffractogram of crystalline Compound I neat Form E.
- crystalline Compound I neat Form E is substantially pure.
- crystalline Compound I neat Form E is substantially crystalline.
- crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram generated by an X-ray powder diffraction analysis with an incident beam of Cu K ⁇ radiation. [0259] In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 8.6 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 10.1 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 12.6 ⁇ 0.2 degrees two-theta.
- crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 8.5 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 8.7 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 11.5 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 12.0 ⁇ 0.2 degrees two-theta.
- crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 14.1 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 14.2 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 15.1 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 16.4 ⁇ 0.2 degrees two-theta.
- crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 20.3 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 20.6 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 21.0 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 21.4 ⁇ 0.2 degrees two-theta.
- crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 22.1 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 22.5 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 23.0 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 23.2 ⁇ 0.2 degrees two-theta.
- crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 24.1 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 24.7 ⁇ 0.2 degrees two-theta. [0261] In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having signals at one or two selected from 8.6 ⁇ 0.2 degrees two-theta, 10.1 ⁇ 0.2 degrees two-theta, and 12.6 ⁇ 0.2 degrees two-theta.
- crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having (a) one or two signals selected from 8.6 ⁇ 0.2 degrees two-theta, 10.1 ⁇ 0.2 degrees two-theta, and 12.6 ⁇ 0.2 degrees two-theta and (b) one or two signals selected from 14.1 ⁇ 0.2 degrees two-theta, 20.3 ⁇ 0.2 degrees two-theta, and 23.2 ⁇ 0.2 degrees two-theta.
- crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having two, three, four, five, or six signals selected from 8.6 ⁇ 0.2 degrees two-theta, 10.1 ⁇ 0.2 degrees two-theta, 12.6 ⁇ 0.2 degrees two-theta, 14.1 ⁇ 0.2 degrees two-theta, 20.3 ⁇ 0.2 degrees two-theta, 23.2 ⁇ 0.2 degrees two-theta, 11.5 ⁇ 0.2 degrees two-theta, 14.2 ⁇ 0.2 degrees two-theta, and 15.1 ⁇ 0.2 degrees two-theta.
- crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having signals at 8.5 ⁇ 0.2 degrees two-theta, 8.6 ⁇ 0.2 degrees two-theta, 8.7 ⁇ 0.2 degrees two-theta, 10.1 ⁇ 0.2 degrees two-theta, 11.5 ⁇ 0.2 degrees two-theta, 12.0 ⁇ 0.2 degrees two-theta, 12.6 ⁇ 0.2 degrees two-theta, 14.1 ⁇ 0.2 degrees two-theta, 14.2 ⁇ 0.2 degrees two-theta, 15.1 ⁇ 0.2 degrees two-theta, 16.4 ⁇ 0.2 degrees two-theta, 20.3 ⁇ 0.2 degrees two-theta, 20.6 ⁇ 0.2 degrees two-theta, 21.0 ⁇ 0.2 degrees two-theta, 21.4 ⁇ 0.2 degrees two-theta, 22.1 ⁇ 0.2 degrees two-theta, 22.5 ⁇ 0.2 degrees two-theta,
- crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram substantially similar to FIG.10. [0267] In some embodiments, crystalline Compound I neat Form E is characterized as having a 13 C SSNMR spectrum with a peak at 170.0 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13 C SSNMR spectrum with a peak at 163.0 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13 C SSNMR spectrum with a peak at 162.3 ⁇ 0.2 ppm.
- crystalline Compound I neat Form E is characterized as having a 13 C SSNMR spectrum with a peak at 161.3 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13 C SSNMR spectrum with a peak at 159.3 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13 C SSNMR spectrum with a peak at 154.7 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13 C SSNMR spectrum with a peak at 150.6 ⁇ 0.2 ppm.
- crystalline Compound I neat Form E is characterized as having a 13 C SSNMR spectrum with a peak at 149.8 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13 C SSNMR spectrum with a peak at 148.6 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13 C SSNMR spectrum with a peak at 148.3 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13 C SSNMR spectrum with a peak at 147.1 ⁇ 0.2 ppm.
- crystalline Compound I neat Form E is characterized as having a 13 C SSNMR spectrum with a peak at 144.9 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13 C SSNMR spectrum with a peak at 143.9 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13 C SSNMR spectrum with a peak at 143.5 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13 C SSNMR spectrum with a peak at 126.9 ⁇ 0.2 ppm.
- crystalline Compound I neat Form E is characterized as having a 13 C SSNMR spectrum with a peak at 126.0 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13 C SSNMR spectrum with a peak at 124.1 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13 C SSNMR spectrum with a peak at 122.6 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13 C SSNMR spectrum with a peak at 120.8 ⁇ 0.2 ppm.
- crystalline Compound I neat Form E is characterized as having a 13 C SSNMR spectrum with a peak at 117.8 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13 C SSNMR spectrum with a peak at 116.7 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13 C SSNMR spectrum with a peak at 115.2 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13 C SSNMR spectrum with a peak at 114.1 ⁇ 0.2 ppm.
- crystalline Compound I neat Form E is characterized as having a 13 C SSNMR spectrum with a peak at 113.1 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13 C SSNMR spectrum with a peak at 15.9 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13 C SSNMR spectrum with a peak at 15.3 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13 C SSNMR spectrum with a peak at 8.4 ⁇ 0.2 ppm.
- crystalline Compound I neat Form E is characterized as having a 13 C SSNMR spectrum with one, two, three, four, five, six, seven, eight, nine, ten, or more peaks selected from 170.0 ⁇ 0.2 ppm, 163.0 ⁇ 0.2 ppm, 162.3 ⁇ 0.2 ppm, 161.3 ⁇ 0.2 ppm, 159.3 ⁇ 0.2 ppm, 154.7 ⁇ 0.2 ppm, 150.6 ⁇ 0.2 ppm, 149.8 ⁇ 0.2 ppm, 148.6 ⁇ 0.2 ppm, 148.3 ⁇ 0.2 ppm, 147.1 ⁇ 0.2 ppm, 144.9 ⁇ 0.2 ppm, 143.9 ⁇ 0.2 ppm, 143.5 ⁇ 0.2 ppm, 126.9 ⁇ 0.2 ppm, 126.0 ⁇ 0.2 ppm, 124.1 ⁇ 0.2 ppm, 122.6 ⁇ 0.2 ppm, 120.8 ⁇ 0.2
- crystalline Compound I neat Form E is characterized with peaks at 170.0 ⁇ 0.2 ppm, 163.0 ⁇ 0.2 ppm, 162.3 ⁇ 0.2 ppm, 161.3 ⁇ 0.2 ppm, 159.3 ⁇ 0.2 ppm, 154.7 ⁇ 0.2 ppm, 150.6 ⁇ 0.2 ppm, 149.8 ⁇ 0.2 ppm, 148.6 ⁇ 0.2 ppm, 148.3 ⁇ 0.2 ppm, 147.1 ⁇ 0.2 ppm, 144.9 ⁇ 0.2 ppm, 143.9 ⁇ 0.2 ppm, 143.5 ⁇ 0.2 ppm, 126.9 ⁇ 0.2 ppm, 126.0 ⁇ 0.2 ppm, 124.1 ⁇ 0.2 ppm, 122.6 ⁇ 0.2 ppm, 120.8 ⁇ 0.2 ppm, 117.8 ⁇ 0.2 ppm, 116.7 ⁇ 0.2 ppm, 115.2 ⁇ 0.2 ppm
- crystalline Compound I neat Form E is characterized by a 13 C SSNMR spectrum substantially similar to FIG.11. [0271] In some embodiments, crystalline Compound I neat Form E is characterized as having a 19 F SSNMR spectrum with a peak at -60.9 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 19 F SSNMR spectrum with a peak at -62.5 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 19 F SSNMR spectrum with a peak at -63.1 ⁇ 0.2 ppm.
- crystalline Compound I neat Form E is characterized as having a 19 F SSNMR spectrum with a peak at -135.1 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 19 F SSNMR spectrum with a peak at -137.1 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 19 F SSNMR spectrum with a peak at -140.7 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 19 F SSNMR spectrum with a peak at -141.9 ⁇ 0.2 ppm.
- crystalline Compound I neat Form E is characterized as having a 19 F SSNMR spectrum with one or two peaks selected from -60.9 ⁇ 0.2 ppm, -62.5 ⁇ 0.2 ppm, -63.1 ⁇ 0.2 ppm, -135.1 ⁇ 0.2 ppm, -137.1 ⁇ 0.2 ppm, -140.7 ⁇ 0.2 ppm, and -141.9 ⁇ 0.2 ppm.
- crystalline Compound I neat Form E is characterized as having a 19 F SSNMR spectrum with peaks at -60.9 ⁇ 0.2 ppm, -62.5 ⁇ 0.2 ppm, -63.1 ⁇ 0.2 ppm, -135.1 ⁇ 0.2 ppm, -137.1 ⁇ 0.2 ppm, -140.7 ⁇ 0.2 ppm, and -141.9 ⁇ 0.2 ppm.
- crystalline Compound I neat Form E is characterized by a 19 F SSNMR spectrum substantially similar to FIG.12.
- the diffractometer is a Rigaku diffractometer.
- the method of making crystalline Compound I neat Form E comprises: (i) combining Compound I hydrate Form A and trifluorotoluene, (ii) heating the slurry to 80 °C, (iii) cooling to room temperature to yield crystalline Compound I neat Form E.
- the disclosure provides crystalline Compound I Acetone Solvate Hydrate Form A.
- FIG.13 provides an X-ray powder diffractogram of crystalline Compound I Acetone Solvate Hydrate Form A.
- crystalline Compound I Acetone Solvate Hydrate Form A is substantially pure.
- crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 25.9 ⁇ 0.2 degrees two-theta. [0280] In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 7.2 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 8.1 ⁇ 0.2 degrees two-theta.
- crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 9.4 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 9.7 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 11.2 ⁇ 0.2 degrees two-theta.
- crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 12.9 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 13.1 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 13.9 ⁇ 0.2 degrees two-theta.
- crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 14.4 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 14.8 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 15.1 ⁇ 0.2 degrees two-theta.
- crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 15.6 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 16.2 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 16.5 ⁇ 0.2 degrees two-theta.
- crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 17.6 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 17.7 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 22.7 ⁇ 0.2 degrees two-theta.
- crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 24.8 ⁇ 0.2 degrees two-theta.
- crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having signals at one or two of 17.2 ⁇ 0.2 degrees two-theta, 18.1 ⁇ 0.2 degrees two-theta, and 25.9 ⁇ 0.2 degrees two-theta.
- crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having (a) one or two signals selected 17.2 ⁇ 0.2 degrees two-theta, 18.1 ⁇ 0.2 degrees two-theta, and 25.9 ⁇ 0.2 degrees two-theta, and (b) one or two signals selected from 12.9 ⁇ 0.2 degrees two-theta, 15.6 ⁇ 0.2 degrees two-theta, and 24.8 ⁇ 0.2 degrees two-theta.
- crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having two, three, four, five, or six signals selected from 9.5 ⁇ 0.2 degrees two-theta, 12.3 ⁇ 0.2 degrees two-theta, 13.5 ⁇ 0.2 degrees two-theta, 12.9 ⁇ 0.2 degrees two-theta, 15.6 ⁇ 0.2 degrees two-theta, 24.8 ⁇ 0.2 degrees two-theta, 8.1 ⁇ 0.2 degrees two-theta, 12.3 ⁇ 0.2 degrees two-theta, and 17.6 ⁇ 0.2 degrees two-theta.
- crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having one, two three, four five, six, seven, eight, nine, ten, eleven, twelve, thirteen, fourteen, fifteen, sixteen, seventeen, eighteen, nineteen, twenty, twenty-one, or twenty-two signals selected from 7.2 ⁇ 0.2 degrees two-theta; 8.1 ⁇ 0.2 degrees two-theta; 9.7 ⁇ 0.2 degrees two-theta; 11.2 ⁇ 0.2 degrees two-theta; 11.5 ⁇ 0.2 degrees two-theta; 12.3 ⁇ 0.2 degrees two-theta; 12.5 ⁇ 0.2 degrees two-theta; 12.9 ⁇ 0.2 degrees two-theta; 13.1 ⁇ 0.2 degrees two-theta; 13.9 ⁇ 0.2 degrees two-theta; 14.4 ⁇ 0.2 degrees two-theta; 14.8 ⁇ 0.2 degrees two-theta; 15.1 ⁇ 0.2 degrees two
- crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram substantially similar to FIG.13. [0287] In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 212.9 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 211.6 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 179.2 ⁇ 0.2 ppm.
- crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 178.9 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 178.4 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 174.7 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 174.1 ⁇ 0.2 ppm.
- crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 172.7 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 172.2 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 163.1 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 156.7 ⁇ 0.2 ppm.
- crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 155.6 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 152.3 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 150.1 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 148.7 ⁇ 0.2 ppm.
- crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 147.9 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 147.0 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 145.1 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 125.5 ⁇ 0.2 ppm.
- crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 124.5 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 122.5 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 121.6 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 121.1 ⁇ 0.2 ppm.
- crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 118.5 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 118.1 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 117.4 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 116.8 ⁇ 0.2 ppm.
- crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 115.7 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 115.3 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 113.9 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 113.2 ⁇ 0.2 ppm.
- crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 112.4 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 30.0 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 29.5 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 18.4 ⁇ 0.2 ppm.
- crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 17.6 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 8.8 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 8.1 ⁇ 0.2 ppm.
- crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13 C SSNMR spectrum with one, two, three, four, five, six, seven, eight, nine, ten, or more peaks selected from 212.9 ⁇ 0.2 ppm, 211.6 ⁇ 0.2 ppm, 179.2 ⁇ 0.2 ppm, 178.9 ⁇ 0.2 ppm, 178.4 ⁇ 0.2 ppm, 174.7 ⁇ 0.2 ppm, 174.1 ⁇ 0.2 ppm, 172.7 ⁇ 0.2 ppm, 172.2 ⁇ 0.2 ppm, 163.1 ⁇ 0.2 ppm, 156.7 ⁇ 0.2 ppm, 155.6 ⁇ 0.2 ppm, 152.3 ⁇ 0.2 ppm, 150.1 ⁇ 0.2 ppm, 148.7 ⁇ 0.2 ppm, 147.9 ⁇ 0.2 ppm, 147.0 ⁇ 0.2 ppm, 145.1 ⁇ 0.2 ppm, 12
- crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13 C SSNMR spectrum with peaks at 212.9 ⁇ 0.2 ppm, 211.6 ⁇ 0.2 ppm, 179.2 ⁇ 0.2 ppm, 178.9 ⁇ 0.2 ppm, 178.4 ⁇ 0.2 ppm, 174.7 ⁇ 0.2 ppm, 174.1 ⁇ 0.2 ppm, 172.7 ⁇ 0.2 ppm, 172.2 ⁇ 0.2 ppm, 163.1 ⁇ 0.2 ppm, 156.7 ⁇ 0.2 ppm, 155.6 ⁇ 0.2 ppm, 152.3 ⁇ 0.2 ppm, 150.1 ⁇ 0.2 ppm, 148.7 ⁇ 0.2 ppm, 147.9 ⁇ 0.2 ppm, 147.0 ⁇ 0.2 ppm, 145.1 ⁇ 0.2 ppm, 125.5 ⁇ 0.2 ppm, 124.5 ⁇ 0.2 ppm, 122.5 ⁇
- crystalline Compound I Acetone Solvate Hydrate Form A is characterized by a 13 C SSNMR spectrum substantially similar to FIG.14. [0291] In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 19 F SSNMR spectrum with a peak at -60.4 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 19 F SSNMR spectrum with a peak at -61.2 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 19 F SSNMR spectrum with a peak at -61.6 ⁇ 0.2 ppm.
- crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 19 F SSNMR spectrum with a peak at -62.7 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 19 F SSNMR spectrum with a peak at -135.9 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 19 F SSNMR spectrum with a peak at -136.1 ⁇ 0.2 ppm.
- crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 19 F SSNMR spectrum with a peak at -139.6 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 19 F SSNMR spectrum with a peak at -140.6 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 19 F SSNMR spectrum with a peak at -141.7 ⁇ 0.2 ppm.
- crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 19 F SSNMR spectrum with a peak at -145.2 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 19 F SSNMR spectrum with a peak at -145.7 ⁇ 0.2 ppm.
- crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 19 F SSNMR spectrum with one or two peaks selected from -60.4 ⁇ 0.2 ppm, -61.2 ⁇ 0.2 ppm, -61.6 ⁇ 0.2 ppm, -62.7 ⁇ 0.2 ppm, -135.9 ⁇ 0.2 ppm, -136.1 ⁇ 0.2 ppm, -139.6 ⁇ 0.2 ppm, -140.6 ⁇ 0.2 ppm, -141.7 ⁇ 0.2 ppm, -145.2 ⁇ 0.2 ppm, and -145.7 ⁇ 0.2 ppm.
- crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 19 F SSNMR spectrum with peaks at -60.4 ⁇ 0.2 ppm, -61.2 ⁇ 0.2 ppm, -61.6 ⁇ 0.2 ppm, -62.7 ⁇ 0.2 ppm, -135.9 ⁇ 0.2 ppm, -136.1 ⁇ 0.2 ppm, -139.6 ⁇ 0.2 ppm, -140.6 ⁇ 0.2 ppm, -141.7 ⁇ 0.2 ppm, -145.2 ⁇ 0.2 ppm, and -145.7 ⁇ 0.2 ppm.
- crystalline Compound I Acetone Solvate Hydrate Form A is characterized by a 19 F SSNMR spectrum substantially similar to FIG.15.
- the diffractometer is a Rigaku diffractometer.
- Another aspect of the disclosure provides a method of crystalline Compound I Acetone Solvate Hydrate Form A.
- the method of making crystalline Compound I Acetone Solvate Hydrate Form A comprises: (i) saturating acetone with Compound I, (ii) adding water dropwise to 20% of the final volume, (iii) holding overnight at room temperature to yield crystalline Compound I Acetone Solvate Hydrate Form A.
- G. Crystalline Compound Ethanol Solvate Form A [0296]
- the disclosure provides crystalline Compound I Ethanol Solvate Form A.
- FIG.16 provides an X-ray powder diffractogram of crystalline Compound I Ethanol Solvate Form A.
- crystalline Compound I Ethanol Solvate Form A is substantially pure. In some embodiments, crystalline Compound I Ethanol Solvate Form A is substantially crystalline. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram generated by an X-ray powder diffraction analysis with an incident beam of Cu K ⁇ radiation. [0298] In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having a signal at 9.4 ⁇ 0.2 degrees two-theta.
- crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having a signal at 16.1 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having a signal at 26.0 ⁇ 0.2 degrees two-theta. [0299] In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having a signal at 10.7 ⁇ 0.2 degrees two-theta.
- crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having a signal at 13.2 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having a signal at 13.6 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having a signal at 13.8 ⁇ 0.2 degrees two-theta.
- crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having a signal at 15.1 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having a signal at 15.9 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having a signal at 17.0 ⁇ 0.2 degrees two-theta.
- crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having a signal at 17.9 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having a signal at 18.3 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having a signal at 18.4 ⁇ 0.2 degrees two-theta.
- crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having a signal at 19.1 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having a signal at 19.9 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having a signal at 21.8 ⁇ 0.2 degrees two-theta.
- crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having a signal at 22.2 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having a signal at 22.4 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having a signal at 24.1 ⁇ 0.2 degrees two-theta.
- crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having signals at one or two of 9.4 ⁇ 0.2 degrees two-theta, 16.1 ⁇ 0.2 degrees two-theta, and 26.0 ⁇ 0.2 degrees two-theta.
- crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having (a) one or two signals selected from 9.4 ⁇ 0.2 degrees two-theta, 16.1 ⁇ 0.2 degrees two-theta, and 26.0 ⁇ 0.2 degrees two-theta, and (b) one or two signals selected from 15.1 ⁇ 0.2 degrees two-theta, 17.0 ⁇ 0.2 degrees two-theta, and 22.4 ⁇ 0.2 degrees two-theta.
- crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having two, three, four, five, or six signals selected from 9.4 ⁇ 0.2 degrees two-theta, 16.1 ⁇ 0.2 degrees two-theta, 26.0 ⁇ 0.2 degrees two-theta, 15.1 ⁇ 0.2 degrees two-theta, 17.0 ⁇ 0.2 degrees two-theta, 22.4 ⁇ 0.2 degrees two-theta, 13.2 ⁇ 0.2 degrees two-theta, 13.6 ⁇ 0.2 degrees two-theta, and 13.8 ⁇ 0.2 degrees two-theta.
- crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having one, two three, four five, six, seven, eight, nine, ten, eleven, twelve, thirteen, fourteen, fifteen, sixteen, seventeen, eighteen, nineteen, twenty, twenty-one, or twenty-two signals selected from 9.4 ⁇ 0.2 degrees two-theta; 10.7 ⁇ 0.2 degrees two-theta; 13.2 ⁇ 0.2 degrees two-theta; 13.6 ⁇ 0.2 degrees two-theta; 13.8 ⁇ 0.2 degrees two-theta; 15.1 ⁇ 0.2 degrees two-theta; 15.9 ⁇ 0.2 degrees two-theta; 16.1 ⁇ 0.2 degrees two-theta; 17.0 ⁇ 0.2 degrees two-theta; 17.9 ⁇ 0.2 degrees two-theta; 18.3 ⁇ 0.2 degrees two-theta; 18.4 ⁇ 0.2 degrees two-theta; 19.1 ⁇ 0.2 degrees two-theta
- crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having signals at 9.4 ⁇ 0.2 degrees two-theta, 10.7 ⁇ 0.2 degrees two-theta, 13.2 ⁇ 0.2 degrees two-theta, 13.6 ⁇ 0.2 degrees two-theta, 13.8 ⁇ 0.2 degrees two-theta, 15.1 ⁇ 0.2 degrees two-theta, 15.9 ⁇ 0.2 degrees two-theta, 16.1 ⁇ 0.2 degrees two-theta, 17.0 ⁇ 0.2 degrees two-theta, 17.9 ⁇ 0.2 degrees two-theta, 18.3 ⁇ 0.2 degrees two-theta, 18.4 ⁇ 0.2 degrees two-theta, 19.1 ⁇ 0.2 degrees two-theta, 19.9 ⁇ 0.2 degrees two-theta, 21.8 ⁇ 0.2 degrees two-theta, 22.2 ⁇ 0.2 degrees two-theta, 22.4 ⁇ 0.2 degrees
- crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram substantially similar to FIG.16. [0306] In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 178.2 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 175.7 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 163.9 ⁇ 0.2 ppm.
- crystalline Compound I Ethanol Solvate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 163.4 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 162.8 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 154.9 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 148.1 ⁇ 0.2 ppm.
- crystalline Compound I Ethanol Solvate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 146.0 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 145.2 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 125.9 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 123.1 ⁇ 0.2 ppm.
- crystalline Compound I Ethanol Solvate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 121.8 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 120.4 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 119.5 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 116.9 ⁇ 0.2 ppm.
- crystalline Compound I Ethanol Solvate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 116.3 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 114.8 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 113.8 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 56.5 ⁇ 0.2 ppm.
- crystalline Compound I Ethanol Solvate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 18.4 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 17.2 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 9.2 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 7.9 ⁇ 0.2 ppm.
- crystalline Compound I Ethanol Solvate Form A is characterized as having a 13 C SSNMR spectrum with one, two, three, four, five, six, seven, eight, nine, ten, or more peaks selected from 178.2 ⁇ 0.2 ppm, 175.7 ⁇ 0.2 ppm, 163.9 ⁇ 0.2 ppm, 163.4 ⁇ 0.2 ppm, 162.8 ⁇ 0.2 ppm, 154.9 ⁇ 0.2 ppm, 148.1 ⁇ 0.2 ppm, 146.0 ⁇ 0.2 ppm, 145.2 ⁇ 0.2 ppm, 125.9 ⁇ 0.2 ppm, 123.1 ⁇ 0.2 ppm, 121.8 ⁇ 0.2 ppm, 120.4 ⁇ 0.2 ppm, 119.5 ⁇ 0.2 ppm, 116.9 ⁇ 0.2 ppm, 116.3 ⁇ 0.2 ppm, 114.8 ⁇ 0.2 ppm, 113.8 ⁇ 0.2 ppm, 56.5 ⁇
- crystalline Compound I Ethanol Solvate Form A is characterized as having a 13 C SSNMR spectrum with peaks at 178.2 ⁇ 0.2 ppm, 175.7 ⁇ 0.2 ppm, 163.9 ⁇ 0.2 ppm, 163.4 ⁇ 0.2 ppm, 162.8 ⁇ 0.2 ppm, 154.9 ⁇ 0.2 ppm, 148.1 ⁇ 0.2 ppm, 146.0 ⁇ 0.2 ppm, 145.2 ⁇ 0.2 ppm, 125.9 ⁇ 0.2 ppm, 123.1 ⁇ 0.2 ppm, 121.8 ⁇ 0.2 ppm, 120.4 ⁇ 0.2 ppm, 119.5 ⁇ 0.2 ppm, 116.9 ⁇ 0.2 ppm, 116.3 ⁇ 0.2 ppm, 114.8 ⁇ 0.2 ppm, 113.8 ⁇ 0.2 ppm, 56.5 ⁇ 0.2 ppm, 18.4 ⁇ 0.2 ppm, 17.2 ⁇ 0.2 ppm, 18.4 ⁇ 0.2
- crystalline Compound I Ethanol Solvate Form A is characterized by a 13 C SSNMR spectrum substantially similar to FIG.17. [0310] In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 19 F SSNMR spectrum with a peak at -61.1 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 19 F SSNMR spectrum with a peak at -138.1 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 19 F SSNMR spectrum with a peak at -141.2 ⁇ 0.2 ppm.
- crystalline Compound I Ethanol Solvate Form A is characterized as having a 19 F SSNMR spectrum with one or two peaks selected from -61.1 ⁇ 0.2 ppm, -138.1 ⁇ 0.2 ppm, and -141.2 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 19 F SSNMR spectrum with peaks at -61.1 ⁇ 0.2 ppm, -138.1 ⁇ 0.2 ppm, and -141.2 ⁇ 0.2 ppm. [0312] In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by a 19 F SSNMR spectrum substantially similar to FIG.18.
- the diffractometer is a Bruker diffractometer.
- Another aspect of the disclosure provides a method of crystalline Compound I Ethanol Solvate Form A.
- the method of making crystalline Compound I Ethanol Solvate Form A comprises: (i) combining Compound I and ethyl acetate, (ii) heating the slurry to 50 °C, (iii) adding ethanol and distilling under vacuum at 50 °C, (iv) adding ethanol and distilling under vacuum at 50 °C, (v) adding ethanol and distilling under vacuum at 50 °C to a minimum volume, (vi) adding ethanol and distilling under vacuum at 50 °C, (vii) drying in a vacuum oven at 45 °C, with a slight nitrogen bleed for 42 hours to yield crystalline Compound I Ethanol Solvate Form A. H.
- Crystalline Compound I Hydrate Form A [0315] In some embodiments, the disclosure provides crystalline Compound I Hydrate Form A.
- FIG.19 provides an X-ray powder diffractogram of crystalline Compound I Hydrate Form A.
- crystalline Compound I Hydrate Form A is substantially pure. In some embodiments, crystalline Compound I Hydrate Form A is substantially crystalline. In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram generated by an X-ray powder diffraction analysis with an incident beam of Cu K ⁇ radiation.
- crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 8.7 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 12.4 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 18.8 ⁇ 0.2 degrees two-theta. [0318] In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 4.7 ⁇ 0.2 degrees two-theta.
- crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 9.5 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 11.0 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 12.7 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 13.1 ⁇ 0.2 degrees two-theta.
- crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 14.4 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 15.8 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 16.3 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 17.5 ⁇ 0.2 degrees two-theta.
- crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 19.8 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 20.2 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 21.1 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 21.4 ⁇ 0.2 degrees two-theta.
- crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 22.2 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 22.9 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 23.9 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 24.4 ⁇ 0.2 degrees two-theta.
- crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 24.7 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 25.4 ⁇ 0.2 degrees two-theta. [0319] In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having signals at one or two of 8.7 ⁇ 0.2 degrees two-theta, 12.4 ⁇ 0.2 degrees two-theta, and 18.8 ⁇ 0.2 degrees two-theta.
- crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram (a) one or two signals selected from 8.7 ⁇ 0.2 degrees two-theta, 12.4 ⁇ 0.2 degrees two-theta, and 18.8 ⁇ 0.2 degrees two-theta, and (b) one or two signals selected from 12.7 ⁇ 0.2 degrees two-theta, 17.5 ⁇ 0.2 degrees two-theta, and 19.8 ⁇ 0.2 degrees two-theta.
- crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having two, three, four, five, or six signals selected from 8.7 ⁇ 0.2 degrees two-theta, 12.4 ⁇ 0.2 degrees two-theta, 18.8 ⁇ 0.2 degrees two-theta, 12.7 ⁇ 0.2 degrees two-theta, 17.5 ⁇ 0.2 degrees two-theta, 19.8 ⁇ 0.2 degrees two-theta, 13.1 ⁇ 0.2 degrees two-theta, 16.3 ⁇ 0.2 degrees two-theta, and 21.1 ⁇ 0.2 degrees two-theta.
- crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having one, two three, four five, six, seven, eight, nine, ten, eleven, twelve, thirteen, fourteen, fifteen, sixteen, seventeen, eighteen, nineteen, twenty, twenty-one, or twenty-two signals selected from 8.7 ⁇ 0.2 degrees two-theta, 12.4 ⁇ 0.2 degrees two-theta, 18.8 ⁇ 0.2 degrees two-theta; 4.7 ⁇ 0.2 degrees two-theta; 9.5 ⁇ 0.2 degrees two-theta; 11.0 ⁇ 0.2 degrees two-theta; 12.7 ⁇ 0.2 degrees two-theta; 13.1 ⁇ 0.2 degrees two-theta; 14.4 ⁇ 0.2 degrees two-theta; 15.8 ⁇ 0.2 degrees two-theta; 16.3 ⁇ 0.2 degrees two-theta; 17.5 ⁇ 0.2 degrees two-theta; 19.8 ⁇ 0.2 degrees two-theta;
- crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having signals at 8.7 ⁇ 0.2 degrees two-theta, 12.4 ⁇ 0.2 degrees two-theta, 18.8 ⁇ 0.2 degrees two-theta; 4.7 ⁇ 0.2 degrees two-theta; 9.5 ⁇ 0.2 degrees two-theta; 11.0 ⁇ 0.2 degrees two-theta; 12.7 ⁇ 0.2 degrees two-theta; 13.1 ⁇ 0.2 degrees two-theta; 14.4 ⁇ 0.2 degrees two-theta; 15.8 ⁇ 0.2 degrees two-theta; 16.3 ⁇ 0.2 degrees two-theta; 17.5 ⁇ 0.2 degrees two-theta; 19.8 ⁇ 0.2 degrees two-theta; 20.2 ⁇ 0.2 degrees two-theta; 21.1 ⁇ 0.2 degrees two-theta; 21.4 ⁇ 0.2 degrees two-theta; 22.2 ⁇ 0.2 degrees two-theta
- crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram substantially similar to FIG.19. [0325] In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 177.3 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 172.8 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 170.5 ⁇ 0.2 ppm.
- crystalline Compound I Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 162.0 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 154.3 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 154.0 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 150.2 ⁇ 0.2 ppm.
- crystalline Compound I Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 148.7 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 147.6 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 146.6 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 145.5 ⁇ 0.2 ppm.
- crystalline Compound I Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 126.3 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 125.0 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 123.8 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 122.4 ⁇ 0.2 ppm.
- crystalline Compound I Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 121.6 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 120.6 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 118.4 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 117.9 ⁇ 0.2 ppm.
- crystalline Compound I Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 117.0 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 116.0 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 114.7 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 114.4 ⁇ 0.2 ppm.
- crystalline Compound I Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 112.8 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 17.7 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 17.1 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13 C SSNMR spectrum with a peak at 8.7 ⁇ 0.2 ppm.
- crystalline Compound I Hydrate Form A is characterized as having a 13 C SSNMR spectrum with one, two, three, four, five, six, seven, eight, nine, ten, or more peaks selected from 177.3 ⁇ 0.2 ppm, 172.8 ⁇ 0.2 ppm, 170.5 ⁇ 0.2 ppm, 162.0 ⁇ 0.2 ppm, 154.3 ⁇ 0.2 ppm, 154.0 ⁇ 0.2 ppm, 150.2 ⁇ 0.2 ppm, 148.7 ⁇ 0.2 ppm, 147.6 ⁇ 0.2 ppm, 146.6 ⁇ 0.2 ppm, 145.5 ⁇ 0.2 ppm, 126.3 ⁇ 0.2 ppm, 125.0 ⁇ 0.2 ppm, 123.8 ⁇ 0.2 ppm, 122.4 ⁇ 0.2 ppm, 121.6 ⁇ 0.2 ppm, 120.6 ⁇ 0.2 ppm, 118.4 ⁇ 0.2 ppm, 117.9 ⁇ 0.2 ppm
- crystalline Compound I Hydrate Form A is characterized as having a 13 C SSNMR spectrum with peaks at 177.3 ⁇ 0.2 ppm, 172.8 ⁇ 0.2 ppm, 170.5 ⁇ 0.2 ppm, 162.0 ⁇ 0.2 ppm, 154.3 ⁇ 0.2 ppm, 154.0 ⁇ 0.2 ppm, 150.2 ⁇ 0.2 ppm, 148.7 ⁇ 0.2 ppm, 147.6 ⁇ 0.2 ppm, 146.6 ⁇ 0.2 ppm, 145.5 ⁇ 0.2 ppm, 126.3 ⁇ 0.2 ppm, 125.0 ⁇ 0.2 ppm, 123.8 ⁇ 0.2 ppm, 122.4 ⁇ 0.2 ppm, 121.6 ⁇ 0.2 ppm, 120.6 ⁇ 0.2 ppm, 118.4 ⁇ 0.2 ppm, 117.9 ⁇ 0.2 ppm, 117.0 ⁇ 0.2 ppm, 116.0 ⁇ 0.2 ppm, 114.7
- crystalline Compound I Hydrate Form A is characterized by a 13 C SSNMR spectrum substantially similar to FIG.22. [0329] In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13 C SSNMR spectrum measured at 25 ⁇ 2 °C and 6% relative humidity (RH) with a peak at 171.5 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13 C SSNMR spectrum measured at 25 ⁇ 2 °C and 11% relative humidity (RH) with a peak at 171.6 ⁇ 0.2 ppm.
- crystalline Compound I Hydrate Form A is characterized as having a 13 C SSNMR spectrum measured at 25 ⁇ 2 °C and 33% relative humidity (RH) with a peak at 170.7 ⁇ 0.2 ppm. [0330] In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13 C SSNMR spectrum measured at 25 ⁇ 2 °C and 6% relative humidity (RH) with a peak at 155.0 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13 C SSNMR spectrum measured at 25 ⁇ 2 °C and 11% relative humidity (RH) with a peak at 155.0 ⁇ 0.2 ppm.
- crystalline Compound I Hydrate Form A is characterized as having a 13 C SSNMR spectrum measured at 25 ⁇ 2 °C and 22% relative humidity (RH) with a peak at 154.6 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13 C SSNMR spectrum measured at 25 ⁇ 2 °C and 33% relative humidity (RH) with a peak at 154.4 ⁇ 0.2 ppm. [0331] In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13 C SSNMR spectrum measured at 25 ⁇ 2 °C and 6% relative humidity (RH) with a peak at 149.5 ⁇ 0.2 ppm.
- crystalline Compound I Hydrate Form A is characterized as having a 13 C SSNMR spectrum measured at 25 ⁇ 2 °C and 11% relative humidity (RH) with a peak at 149.4 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13 C SSNMR spectrum measured at 25 ⁇ 2 °C and 22% relative humidity (RH) with a peak at 148.6 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13 C SSNMR spectrum measured at 25 ⁇ 2 °C and 33% relative humidity (RH) with a peak at 148.6 ⁇ 0.2 ppm.
- crystalline Compound I Hydrate Form A is characterized as having a 13 C SSNMR spectrum measured at 25 ⁇ 2 °C and 6% relative humidity (RH) with a peak at 145.8 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13 C SSNMR spectrum measured at 25 ⁇ 2 °C and 11% relative humidity (RH) with a peak at 145.8 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13 C SSNMR spectrum measured at 25 ⁇ 2 °C and 22% relative humidity (RH) with a peak at 145.6 ⁇ 0.2 ppm.
- crystalline Compound I Hydrate Form A is characterized as having a 13 C SSNMR spectrum measured at 25 ⁇ 2 °C and 33% relative humidity (RH) with a peak at 145.5 ⁇ 0.2 ppm.
- crystalline Compound I Hydrate Form A is characterized as having a 13 C SSNMR spectrum measured at 25 ⁇ 2 °C and 6% relative humidity (RH) with a peak at 8.5 ⁇ 0.2 ppm.
- crystalline Compound I Hydrate Form A is characterized as having a 13 C SSNMR spectrum measured at 25 ⁇ 2 °C and 11% relative humidity (RH) with a peak at 8.5 ⁇ 0.2 ppm.
- crystalline Compound I Hydrate Form A is characterized as having a 13 C SSNMR spectrum measured at 25 ⁇ 2 °C and 22% relative humidity (RH) with a peak at 8.6 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13 C SSNMR spectrum measured at 25 ⁇ 2 °C and 33% relative humidity (RH) with a peak at 8.6 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13 C SSNMR spectrum measured at 25 ⁇ 2 °C and 43% relative humidity (RH) with a peak at 8.6 ⁇ 0.2 ppm.
- crystalline Compound I Hydrate Form A is characterized as having a 19 F SSNMR spectrum with a peak at -59.4 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 19 F SSNMR spectrum with a peak at -60.2 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 19 F SSNMR spectrum with a peak at -135.0 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 19 F SSNMR spectrum with a peak at -138.4 ⁇ 0.2 ppm.
- crystalline Compound I Hydrate Form A is characterized as having a 19 F SSNMR spectrum with a peak at -141.7 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 19 F SSNMR spectrum with a peak at -144 ⁇ 0.2 ppm.
- crystalline Compound I Hydrate Form A is characterized as having a 19 F SSNMR spectrum with one or two peaks selected from -59.4 ⁇ 0.2 ppm, -60.2 ⁇ 0.2 ppm, -135.0 ⁇ 0.2 ppm, -138.4 ⁇ 0.2 ppm, -141.7 ⁇ 0.2 ppm, and -144.0 ⁇ 0.2 ppm.
- crystalline Compound I Hydrate Form A is characterized as having a 19 F SSNMR spectrum with peaks at -59.4 ⁇ 0.2 ppm, -60.2 ⁇ 0.2 ppm, -135.0 ⁇ 0.2 ppm, -138.4 ⁇ 0.2 ppm, -141.7 ⁇ 0.2 ppm, and -144.0 ⁇ 0.2 ppm.
- crystalline Compound I Hydrate Form A is characterized by a 19 F SSNMR spectrum substantially similar to FIG.23.
- crystalline Compound I Hydrate Form A is characterized as having a 19 F SSNMR spectrum measured at 25 ⁇ 2 °C and 11% relative humidity (RH) with a peak at -142.4 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 19 F SSNMR spectrum measured at 25 ⁇ 2 °C and 22% relative humidity (RH) with a peak at -141.9 ⁇ 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 19 F SSNMR spectrum measured at 25 ⁇ 2 °C and 33% relative humidity (RH) with a peak at -141.8 ⁇ 0.2 ppm.
- the diffractometer is a Bruker diffractometer.
- Another aspect of the disclosure provides a method of crystalline Compound I Hydrate Form A.
- the method of making crystalline Compound I Hydrate Form A comprises: (i) combining Compound I Ethanol Solvate Form A and acetone, (ii) stirring the slurry while heating 50 °C, (iii) adding water over 1 hour, (iv) adding a seed of crystalline Compound I hydrate Form A, (v) agitating for 30 minutes, (vi) adding water over 8 hours, (vii) stirring at 50 °C for 5 hours, (viii) cooling the slurry to 20 °C over 3 hours, (ix) agitating for 2 hours, and (x) collecting the solids and washing with 1:2 acetone:water to yield crystalline Compound I hydrate Form A.
- Crystalline Compound I Dehydrated Hydrate Form A [0340] In some embodiments, the disclosure provides crystalline Compound I Dehydrated Hydrate Form A.
- FIG. 20 provides an X-ray powder diffractogram of crystalline Compound I Dehydrated Hydrate Form A.
- crystalline Compound I Dehydrated Hydrate Form A is substantially pure. In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is substantially crystalline. In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram generated by an X-ray powder diffraction analysis with an incident beam of Cu K ⁇ radiation.
- crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 8.7 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 15.9 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 18.7 ⁇ 0.2 degrees two-theta.
- crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 4.7 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 9.5 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 10.9 ⁇ 0.2 degrees two-theta.
- crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 12.4 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 12.6 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 13.1 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 14.3 ⁇ 0.2 degrees two-theta.
- crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 16.3 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 17.3 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 19.8 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 20.3 ⁇ 0.2 degrees two-theta.
- crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 21.1 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 21.4 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 22.3 ⁇ 0.2 degrees two-theta.
- crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 23.0 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 23.9 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 24.3 ⁇ 0.2 degrees two-theta.
- crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 24.7 ⁇ 0.2 degrees two-theta. In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 25.3 ⁇ 0.2 degrees two-theta. [0344] In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having signals at one or two of 8.7 ⁇ 0.2 degrees two- theta, 15.9 ⁇ 0.2 degrees two-theta, and 18.7 ⁇ 0.2 degrees two-theta.
- crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram (a) one or two signals selected from 8.7 ⁇ 0.2 degrees two-theta, 15.9 ⁇ 0.2 degrees two-theta, and 18.7 ⁇ 0.2 degrees two-theta, and (b) one or two signals selected from 12.4 ⁇ 0.2 degrees two-theta, 12.6 ⁇ 0.2 degrees two-theta, 17.3 ⁇ 0.2 degrees two-theta, and 19.8 ⁇ 0.2 degrees two-theta.
- crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having two, three, four, five, or six signals selected 8.7 ⁇ 0.2 degrees two-theta, 15.9 ⁇ 0.2 degrees two-theta, 18.7 ⁇ 0.2 degrees two-theta, 12.4 ⁇ 0.2 degrees two-theta, 12.6 ⁇ 0.2 degrees two-theta, 17.3 ⁇ 0.2 degrees two-theta, 19.8 ⁇ 0.2 degrees two-theta, 13.1 ⁇ 0.2 degrees two-theta, 16.3 ⁇ 0.2 degrees two-theta, and 21.1 ⁇ 0.2 degrees two-theta.
- crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having one, two three, four five, six, seven, eight, nine, ten, eleven, twelve, thirteen, fourteen, fifteen, sixteen, seventeen, eighteen, nineteen, twenty, twenty-one, or twenty-two signals selected from 4.7 ⁇ 0.2 degrees two-theta; 8.7 ⁇ 0.2 degrees two-theta; 9.5 ⁇ 0.2 degrees two-theta; 10.9 ⁇ 0.2 degrees two-theta; 12.4 ⁇ 0.2 degrees two-theta; 12.6 ⁇ 0.2 degrees two-theta; 13.1 ⁇ 0.2 degrees two-theta; 14.3 ⁇ 0.2 degrees two-theta; 15.9 ⁇ 0.2 degrees two-theta; 16.3 ⁇ 0.2 degrees two-theta; 17.3 ⁇ 0.2 degrees two-theta; 18.7 ⁇ 0.2 degrees two-theta; 19.8 ⁇ 0.2 degrees two-theta;
- crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having signals at 4.7 ⁇ 0.2 degrees two-theta, 8.7 ⁇ 0.2 degrees two-theta, 9.5 ⁇ 0.2 degrees two-theta, 10.9 ⁇ 0.2 degrees two-theta, 12.4 ⁇ 0.2 degrees two-theta, 12.6 ⁇ 0.2 degrees two-theta, 13.1 ⁇ 0.2 degrees two-theta, 14.3 ⁇ 0.2 degrees two-theta, 15.9 ⁇ 0.2 degrees two-theta, 16.3 ⁇ 0.2 degrees two-theta, 17.3 ⁇ 0.2 degrees two-theta, 18.7 ⁇ 0.2 degrees two-theta, 19.8 ⁇ 0.2 degrees two-theta, 20.3 ⁇ 0.2 degrees two-theta, 21.1 ⁇ 0.2 degrees two-theta, 21.4 ⁇ 0.2 degrees two-theta, 22.3 ⁇ 0.2 degrees two-theta
- crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram substantially similar to FIG.20.
- the diffractometer is a Bruker diffractometer.
- the method of making crystalline Compound I Dehydrated Hydrate Form A comprises: (i) combining Compound I Ethanol Solvate Form A and acetone, (ii) stirring the slurry while heating 50 °C, (iii) adding water over 1 hour, (iv) adding a seed of crystalline Compound I Dehydrated Hydrate Form A, (v) agitating for 30 minutes, (vi) adding water over 8 hours, (vii) stirring at 50 °C for 5 hours, (viii) cooling the slurry to 20 °C over 3 hours, (ix) agitating for 2 hours, (x) collecting the solids and washing with 1:2 acetone:water, (xi) drying the solids in a vacuum over at 50 °C with a slight nitrogen bleed to yield crystalline Compound I Dehydrated Hydrate Form A.
- Compound I in any one of the pharmaceutically acceptable solid forms disclosed herein, act as a voltage-gated sodium channel inhibitor.
- the voltage-gated sodium channel is Na V 1.8.
- the disclosure features a method of inhibiting a voltage-gated sodium channel in a subject comprising administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof.
- the voltage-gated sodium channel is Na V 1.8.
- the invention features a method of inhibiting a voltage-gated sodium channel in a subject comprising administering to the subject a compound of formula I or a solvate thereof or a pharmaceutically acceptable salt thereof or a pharmaceutical composition thereof.
- the disclosure relates to a method of treating or lessening the severity of pain in a subject, comprising administering to the subject a compound of formula I, or a pharmaceutically acceptable salt thereof.
- the disclosure relates to a use of a compound of formula I, or a pharmaceutically acceptable salt thereof, in a method of treating or lessening the severity of pain in a subject, comprising administering to the subject a compound of formula I, or the pharmaceutically acceptable salt thereof [0355]
- the disclosure relates to a composition comprising a compound of formula I, or a solvate of formula I, or a pharmaceutically acceptable salt thereof, for use in a method of treating or lessening the severity of pain in a subject, wherein the composition is prepared for administration of a compound of formula I, or a solvate of formula I or the pharmaceutically acceptable salt thereof, to the subject.
- the disclosure features a method of treating or lessening the severity in a subject of chronic pain, gut pain, neuropathic pain, musculoskeletal pain, acute pain, inflammatory pain, cancer pain, idiopathic pain, postsurgical pain (e.g., bunionectomy pain, herniorrhaphy pain or abdominoplasty pain), visceral pain, multiple sclerosis, Charcot-Marie-Tooth syndrome, incontinence, pathological cough, or cardiac arrhythmia comprising administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e. g. crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof.
- the pharmaceutically acceptable solid e. g. crystalline or amorphous
- the disclosure features a method of treating or lessening the severity in a subject of chronic pain, gut pain, neuropathic pain, musculoskeletal pain, acute pain, inflammatory pain, cancer pain, idiopathic pain, postsurgical pain, herniorrhaphy pain, bunionectomy pain, multiple sclerosis, Charcot-Marie-Tooth syndrome, incontinence, or cardiac arrhythmia comprising administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof.
- the pharmaceutically acceptable solid e.g., crystalline or amorphous
- the disclosure features a method of treating or lessening the severity in a subject of gut pain, wherein gut pain comprises inflammatory bowel disease pain, Crohn’s disease pain, irritable bowel syndrome, endometriosis, polycystic ovarian disease, salpingitis, cervicitis or interstitial cystitis pain wherein said method comprises administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof.
- a pharmaceutically acceptable solid e.g., crystalline or amorphous
- the disclosure features a method of treating or lessening the severity in a subject of neuropathic pain comprising administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof.
- the neuropathic pain comprises post-herpetic neuralgia, small fiber neuropathy, diabetic neuropathy, or idiopathic small-fiber neuropathy.
- the neuropathic pain comprises diabetic neuropathy (e.g., diabetic peripheral neuropathy).
- the phrase “idiopathic small-fiber neuropathy” shall be understood to include any small fiber neuropathy.
- neuropathic pain comprises post-herpetic neuralgia, diabetic neuralgia, painful HIV-associated sensory neuropathy, trigeminal neuralgia, burning mouth syndrome, post-amputation pain, phantom pain, painful neuroma; traumatic neuroma; Morton’s neuroma; nerve entrapment injury, spinal stenosis, carpal tunnel syndrome, radicular pain, sciatica pain; nerve avulsion injury, brachial plexus avulsion injury; complex regional pain syndrome, drug therapy induced neuralgia, cancer chemotherapy induced neuralgia, anti-retroviral therapy induced neuralgia, HIV-induced neuropathy; post spinal cord injury pain, spinal stenosis pain, small fiber neuropathy, idiopathic small-fiber neuropathy, idiopathic sensory neuropathy or trigeminal autonomic cephalalgia where
- the disclosure features a method of treating or lessening the severity in a subject of musculoskeletal pain comprising administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof.
- the musculoskeletal pain comprises osteoarthritis pain.
- the disclosure features a method of treating or lessening the severity in a subject of musculoskeletal pain, wherein musculoskeletal pain comprises osteoarthritis pain, back pain, cold pain, burn pain or dental pain wherein said method comprises administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g. crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof.
- musculoskeletal pain comprises osteoarthritis pain, back pain, cold pain, burn pain or dental pain
- said method comprises administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g. crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof.
- the disclosure features a method of treating or lessening the severity in a subject of inflammatory pain, wherein inflammatory pain comprises rheumatoid arthritis pain, ankylosing spondylitis or vulvodynia wherein said method comprises administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof.
- inflammatory pain comprises rheumatoid arthritis pain, ankylosing spondylitis or vulvodynia
- said method comprises administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof.
- the disclosure features a method of treating or lessening the severity in a subject of inflammatory pain, wherein inflammatory pain comprises rheumatoid arthritis pain wherein said method comprises administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof of.
- inflammatory pain comprises rheumatoid arthritis pain
- said method comprises administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof of.
- the disclosure features a method of treating or lessening the severity in a subject of idiopathic pain, wherein idiopathic pain comprises fibromyalgia pain wherein said method comprises administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof.
- idiopathic pain comprises fibromyalgia pain
- said method comprises administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof.
- the disclosure features a method of treating or lessening the severity in a subject of idiopathic pain, wherein idiopathic pain comprises reflex sympathetic dystrophy pain, wherein said method comprises administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof.
- idiopathic pain comprises reflex sympathetic dystrophy pain
- said method comprises administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof.
- the disclosure features a method of treating or lessening the severity in a subject of acute pain comprising administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof.
- the acute pain comprises acute post-operative pain.
- the disclosure features a method of treating or lessening the severity in a subject of postsurgical pain (e.g., joint replacement pain, soft tissue surgery pain, post-thoracotomy pain, post-mastectomy pain, hemorrhoidectomy pain, herniorrhaphy pain, bunionectomy pain or abdominoplasty pain) comprising administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof.
- postsurgical pain e.g., joint replacement pain, soft tissue surgery pain, post-thoracotomy pain, post-mastectomy pain, hemorrhoidectomy pain, herniorrhaphy pain, bunionectomy pain or abdominoplasty pain
- the disclosure features a method of treating or lessening the severity in a subject of herniorrhaphy pain comprising administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof.
- the disclosure features a method of treating or lessening the severity in a subject of abdominoplasty pain comprising administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof.
- the disclosure features a method of treating or lessening the severity in a subject of visceral pain comprising administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof.
- the visceral pain comprises visceral pain from abdominoplasty.
- the disclosure features a method of treating or lessening the severity in a subject of a neurodegenerative disease comprising administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof.
- the neurodegenerative disease comprises multiple sclerosis.
- the neurodegenerative disease comprises Pitt Hopkins Syndrome (PTHS).
- PTHS Pitt Hopkins Syndrome
- the disclosure features a method wherein the subject is treated with one or more additional therapeutic agents administered concurrently with, prior to, or subsequent to treatment with an effective of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof.
- the additional therapeutic agent is a sodium channel inhibitor.
- the disclosure features a method of inhibiting a voltage-gated sodium channel in a biological sample comprising contacting the biological sample with an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof.
- the voltage-gated sodium channel is Na V 1.8.
- the disclosure features a method of treating or lessening the severity in a subject of acute pain, sub-acute and chronic pain, nociceptive pain, neuropathic pain, inflammatory pain, nociplastic pain, arthritis, migraine, cluster headaches, tension headaches, and all other forms of headaches, trigeminal neuralgia, herpetic neuralgia, general neuralgias, epilepsy, epilepsy conditions, neurodegenerative disorders, psychiatric disorders, anxiety, depression, bipolar disorder, myotonia, arrhythmia, movement disorders, neuroendocrine disorders, ataxia, central neuropathic pain of multiple sclerosis and irritable bowel syndrome, incontinence, pathological cough, visceral pain, osteoarthritis pain, postherpetic neuralgia, diabetic neuropathy, radicular pain, sciatica, back pain, unspecific chronic back pain, head pain, neck pain, moderate pain, severe pain, intractable pain, nociceptive pain, breakthrough pain, postsurgical
- the disclosure features a method of treating or lessening the severity in a subject of femur cancer pain; non-malignant chronic bone pain; rheumatoid arthritis; osteoarthritis; spinal stenosis; neuropathic low back pain; myofascial pain syndrome; fibromyalgia; temporomandibular joint pain; chronic visceral pain, abdominal pain; pancreatic pain; IBS pain; chronic and acute headache pain; migraine; tension headache; cluster headaches; chronic and acute neuropathic pain, post-herpetic neuralgia; diabetic neuropathy; HIV-associated neuropathy; trigeminal neuralgia; Charcot-Marie-Tooth neuropathy; hereditary sensory neuropathy; peripheral nerve injury; painful neuromas; ectopic proximal and distal discharges; radiculopathy; chemotherapy induced neuropathic pain; radiotherapy-induced neuropathic pain; persistent/chronic post-surgical pain (e.g., post amputation, post-t
- the disclosure features a method of treating or lessening the severity in a subject of trigeminal neuralgia, migraines treated with botox, cervical radiculopathy, occipital neuralgia, axillary neuropathy, radial neuropathy, ulnar neuropathy, brachial plexopathy, thoracic radiculopathy, intercostal neuralgia, lumbosacral radiculopathy, iliolingual neuralgia, pudendal neuralgia, femoral neuropathy, meralgia paresthetica, saphenous neuropathy, sciatic neuropathy, peroneal neuropathy, tibial neuropathy, lumbosacral plexopathy, traumatic neuroma stump pain or postamputation pain, comprising administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof.
- a pharmaceutically acceptable solid e.g.,
- the method of treating, or lessening the severity of, or symptomatically treating pain in a patient comprises administering to the patient an effective amount of Compound I in any one of the pharmaceutically acceptable crystalline forms disclosed herein.
- the pharmaceutically acceptable crystalline form of Compound I is Compound I neat Form A.
- the pharmaceutically acceptable crystalline form of Compound I is Compound I neat Form B.
- the pharmaceutically acceptable crystalline form of Compound I is Compound I neat Form E.
- the pharmaceutically acceptable crystalline form of Compound I is Compound I Acetone Solvate Hydrate Form A.
- the pharmaceutically acceptable crystalline form of Compound I is Compound I Ethanol Solvate Form A.
- the pharmaceutically acceptable crystalline form of Compound I is Compound I Hydrate Form A. In some embodiments the pharmaceutically acceptable crystalline form of Compound I is Compound I Hydrate Form A. In some embodiments the pharmaceutically acceptable crystalline form of Compound I is Compound I Dehydrated Hydrate Form A. [0382] In some embodiments, the method of treating, or lessening the severity of, or symptomatically treating pain in a patient comprises administering to the patient an effective amount of Compound I in a pharmaceutically acceptable amorphous form disclosed herein. In some embodiments the pharmaceutically acceptable form of Compound I is Compound I amorphous form.
- the method of treating, lessening the severity of, or symptomatically treating pain in a patient comprises administering to the patient an effective amount of Compound I as any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein in combination with at least one additional active pharmaceutical ingredient.
- an effective amount of Compound I as any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein in combination with at least one additional active pharmaceutical ingredient.
- the method of treating, lessening the severity of, or symptomatically treating pain in a patient comprises administering to the patient an effective amount of Compound I as a solid form selected from Compound I neat Form A, Compound I neat Form B, Compound I neat Form E, Compound I Acetone Solvate Hydrate Form A, Compound I Ethanol Solvate Form A, Compound I Hydrate Form A, Compound I Dehydrated Hydrate Form A, and Compound I amorphous form, in combination with at least one additional active pharmaceutical ingredient.
- the disclosure features Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof for use in any of the foregoing methods.
- the disclosure features the use of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof for the manufacture of a medicament for use in any of the foregoing methods.
- the disclosure features Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof for use as a medicament.
- the compounds, salts, and pharmaceutically acceptable compositions of the invention can be employed in combination therapies, that is, the compounds, salts, and pharmaceutically acceptable compositions can be administered concurrently with, prior to, or subsequent to, one or more other desired therapeutics or medical procedures.
- the particular combination of therapies (therapeutics or procedures) to employ in a combination regimen will take into account compatibility of the desired therapeutics and/or procedures and the desired therapeutic effect to be achieved.
- the therapies employed may achieve a desired effect for the same disorder (for example, an inventive compound may be administered concurrently with another agent used to treat the same disorder), or they may achieve different effects (e.g., control of any adverse effects).
- additional therapeutic agents that are normally administered to treat or prevent a particular disease, or condition, are known as “appropriate for the disease, or condition, being treated.”
- exemplary additional therapeutic agents include, but are not limited to: non-opioid analgesics (indoles such as Etodolac, Indomethacin, Sulindac, Tolmetin; naphthylalkanones such as Nabumetone; oxicams such as Piroxicam; para-aminophenol derivatives, such as Acetaminophen; propionic acids such as Fenoprofen, Flurbiprofen, Ibuprofen, Ketoprofen, Naproxen, Naproxen sodium, Oxaprozin; salicylates such as Aspirin, Choline magnesium trisalicylate, Diflunisal; fenamates such as meclofenamic acid, Mefenamic acid; and pyrazoles such as Phenylbutazone); or opioid (narcotic)
- nondrug analgesic approaches may be utilized in conjunction with administration of one or more compounds of the invention.
- anesthesiologic intraspinal infusion, neural blockade
- neurosurgical neurolysis of CNS pathways
- neurostimulatory transcutaneous electrical nerve stimulation, dorsal column stimulation
- physiatric physical therapy, orthotic devices, diathermy
- psychologic psychologic
- additional appropriate therapeutic agents are selected from the following: [0389] (1) an opioid analgesic, e.g.
- NSAID nonsteroidal antiinflammatory drug
- dextromethorphan (+)-3-hydroxy-N- methylmorphinan) or its metabolite dextrorphan ((+)-3-hydroxy-N-methylmorphinan), ketamine, memantine, pyrroloquinoline quinine, cis-4-(phosphonomethyl)-2- piperidinecarboxylic acid, budipine, EN-3231 (MorphiDex®), a combination formulation of morphine and dextromethorphan), topiramate, neramexane or perzinfotel including an NR2B antagonist, e.g.
- doxazosin tamsulosin, clonidine, guanfacine, dexmedetomidine, modafinil, or 4-amino-6,7-dimethoxy-2-(5-methane-sulfonamido-l, 2,3,4- tetrahydroisoquinolin-2-yl)-5-(2-pyridyl) quinazoline;
- a tricyclic antidepressant e.g. desipramine, imipramine, amitriptyline or nortriptyline
- an anticonvulsant e.g.
- a tachykinin (NK) antagonist particularly an NK-3, NK-2 or NK-1 antagonist, e.g.
- a coal-tar analgesic in particular paracetamol
- a neuroleptic such as droperidol, chlorpromazine, haloperidol, perphenazine, thioridazine, mesoridazine, trifluoperazine, fluphenazine, clozapine, olanzapine, risperidone, ziprasidone, quetiapine, sertindole, aripiprazole, sonepiprazole, blonanserin, iloperidone, perospirone, raclopride, zotepine, bifeprunox, asenapine, lurasidone, amisulpride, balaperidone, palindore, epli
- a Na V 1.8 blocker such as PF-04531083, PF-06372865 and such as those disclosed in WO2008/135826 (US2009048306), WO2006/011050 (US2008312235), WO2013/061205 (US2014296313), US20130303535, WO2013131018, US8466188, WO2013114250 (US2013274243), WO2014/120808 (US2014213616), WO2014/120815 (US2014228371) WO2014/120820 (US2014221435), WO2015/010065 (US20160152561), WO2015/089361 (US20150166589), WO2019/014352 (US20190016671), WO2018/213426, WO2020/146682, WO2020/146612, WO2020/014243, WO2020/014246, WO2020/092187, WO2020/092667 (US2020140411), WO2020/
- the additional appropriate therapeutic agents are selected from V- 116517, Pregabalin, controlled release Pregabalin, Ezogabine (Potiga®). Ketamine/amitriptyline topical cream (Amiket®), AVP-923, Perampanel (E-2007), Ralfinamide, transdermal bupivacaine (Eladur®), CNV1014802, JNJ-10234094 (Carisbamate), BMS-954561 or ARC-4558.
- the additional appropriate therapeutic agents are selected from N- (6-amino-5-(2,3,5-trichlorophenyl)pyridin-2-yl)acetamide; N-(6-amino-5-(2-chloro-5- methoxyphenyl)pyridin-2-yl)-1-methyl-1H-pyrazole-5-carboxamide; or 3-((4-(4- (trifluoromethoxy)phenyl)-1H-imidazol-2-yl)methyl)oxetan-3-amine.
- the additional therapeutic agent is selected from a GlyT2/5HT2 inhibitor, such as Operanserin (VVZ149), a TRPV modulator such as CA008, CMX-020, NEO6860, FTABS, CNTX4975, MCP101, MDR16523, or MDR652, a EGR1 inhibitor such as Brivoglide (AYX1), an NGF inhibitor such as Tanezumab, Fasinumab, ASP6294, MEDI7352, a Mu opioid agonist such as Cebranopadol, NKTR181 (oxycodegol), a CB-1 agonist such as NEO1940 (AZN1940), an imidazoline 12 agonist such as CR4056 or a p75NTR-Fc modulator such as LEVI-04.
- a GlyT2/5HT2 inhibitor such as Operanserin (VZ149), a TRPV modulator such as CA008, CMX-020, NEO6860, FTABS,
- the additional therapeutic agent is oliceridine or ropivacaine (TLC590).
- the additional therapeutic agent is a Na V 1.7 blocker such as ST- 2427, ST-2578 and those disclosed in WO2010/129864, WO2015/157559, WO2017/059385, WO2018/183781, WO2018/183782, WO2020/072835, and/or WO2022/036297 the entire contents of each application hereby incorporated by reference.
- the additional therapeutic agent is selected from ASP18071, CC- 8464, ANP-230, ANP-231, NOC-100, NTX-1175, ASN008, NW3509, AM-6120, AM-8145, AM- 0422, BL-017881, NTM-006, Opiranserin (Unafra TM ), brivoligide, SR419, NRD.E1, LX9211, LY3016859, ISC-17536, NFX-88, LAT-8881, AP-235, NYX 2925, CNTX-6016, S-600918, S- 637880, RQ-00434739, KLS-2031, MEDI 7352, and XT-150.
- the additional therapeutic agent is selected from Olinvyk, Zynrelef, Seglentis, Neumentum, Nevakar, HTX-034, CPL-01, ACP-044, HRS-4800, Tarlige, BAY2395840, LY3526318, Eliapixant, TRV045, RTA901, NRD1355-E1, MT-8554, LY3556050, AP-325, tetrodotoxin, Otenaproxesul, CFTX-1554, Funapide, iN1011-N17, JMKX000623/ODM- 111, ETX-801, OLP-1002, ANP-230/DSP-2230, iN1011-N17, DSP-3905 and ACD440.
- the additional therapeutic agent is selected from HRS4800, ODM-111/JMKX000623, LX9211, LY3556050, LY3857210, CFTX01554/CFTX-1554, MEDI7352, MEDI0618, BAY3178275, BAY2395840, GSK3858279, STC-004, HALNEURON, OLP-1002, ATX01, ANP230, CC-8464, iN1011-N17, ST-2427, MSD199, FZ008, VYNAV-01, BL-017881, Profervia (Cilnidipine), LS-04, vixotrigine, FX301/PCRX-301, PF-04531083, PF-01247324, and DSP-3905.
- the additional therapeutic agent is a sodium channel inhibitor (also known as a sodium channel blocker), such as the NaV1.7 and NaV1.8 blockers identified above.
- the amount of additional therapeutic agent present in the compositions of this invention may be no more than the amount that would normally be administered in a composition comprising that therapeutic agent as the only active agent.
- the amount of additional therapeutic agent in the presently disclosed compositions may range from about 10% to 100% of the amount normally present in a composition comprising that agent as the only therapeutically active agent.
- the compounds and salts of this invention or pharmaceutically acceptable compositions thereof may also be incorporated into compositions for coating an implantable medical device, such as prostheses, artificial valves, vascular grafts, stents and catheters.
- the invention in another aspect, includes a composition for coating an implantable device comprising a compound or salt of the invention as described generally above, and in classes and subclasses herein, and a carrier suitable for coating said implantable device.
- the invention includes an implantable device coated with a composition comprising a compound or salt of the invention as described generally above, and in classes and subclasses herein, and a carrier suitable for coating said implantable device. Suitable coatings and the general preparation of coated implantable devices are described in US Patents 6,099,562; 5,886,026; and 5,304,121.
- the coatings are typically biocompatible polymeric materials such as a hydrogel polymer, polymethyldisiloxane, polycaprolactone, polyethylene glycol, polylactic acid, ethylene vinyl acetate, and mixtures thereof.
- the coatings may optionally be further covered by a suitable topcoat of fluorosilicone, polysaccharides, polyethylene glycol, phospholipids or combinations thereof to impart controlled release characteristics in the composition.
- Another aspect of the invention relates to inhibiting Na V 1.8 activity in a biological sample or a subject, which method comprises administering to the subject, or contacting said biological sample with a compound of the invention, a pharmaceutically acceptable salt thereof, or a pharmaceutical composition thereof.
- biological sample includes, without limitation, cell cultures or extracts thereof; biopsied material obtained from a mammal or extracts thereof; and blood, saliva, urine, feces, semen, tears, or other body fluids or extracts thereof.
- Inhibition of Na V 1.8 activity in a biological sample is useful for a variety of purposes that are known to one of skill in the art. Examples of such purposes include, but are not limited to, the study of sodium channels in biological and pathological phenomena; and the comparative evaluation of new sodium channel inhibitors.
- K. Pharmaceutical Compositions [0454] Another aspect of the invention provides pharmaceutical compositions comprising Compound I in any one of the pharmaceutically acceptable solid (e.g. crystalline or amorphous) forms disclosed herein.
- the pharmaceutical composition comprises Compound I in a solid form selected from Compound I neat Form A, Compound I neat Form B, Compound I neat Form E, Compound I Acetone Solvate Hydrate Form A, Compound I Ethanol Solvate Form A, Compound I Hydrate Form A, and Compound I Dehydrated Hydrate Form A.
- the pharmaceutical composition comprises Compound I in a solid amorphous form that is Compound I amorphous form.
- the invention provides a pharmaceutical composition comprising (a) Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein, and (b) at least one pharmaceutically acceptable carrier.
- the invention provides pharmaceutical compositions comprising (a) Compound I in a solid form selected from Compound I neat Form A, Compound I neat Form B, Compound I neat Form E, Compound I Acetone Solvate Hydrate Form A, Compound I Ethanol Solvate Form A, Compound I Hydrate Form A, and Compound I Dehydrated Hydrate Form A and (b) at least one pharmaceutically acceptable carrier.
- the pharmaceutically acceptable compositions of the invention additionally comprise a pharmaceutically acceptable carrier, adjuvant, or vehicle, which, as used herein, includes any and all solvents, diluents, or other liquid vehicle, dispersion or suspension aids, surface active agents, isotonic agents, thickening or emulsifying agents, preservatives, solid binders, lubricants and the like, as suited to the particular dosage form desired.
- a pharmaceutically acceptable carrier, adjuvant, or vehicle which, as used herein, includes any and all solvents, diluents, or other liquid vehicle, dispersion or suspension aids, surface active agents, isotonic agents, thickening or emulsifying agents, preservatives, solid binders, lubricants and the like, as suited to the particular dosage form desired.
- Remington s Pharmaceutical Sciences, Sixteenth Edition, E. W. Martin (Mack Publishing Co., Easton, Pa., 1980) discloses various carriers used in formulating pharmaceutically acceptable compositions and known
- any conventional carrier medium is incompatible with the compounds of the invention, such as by producing any undesirable biological effect or otherwise interacting in a deleterious manner with any other component(s) of the pharmaceutically acceptable composition, its use is contemplated to be within the scope of this invention.
- materials which can serve as pharmaceutically acceptable carriers include, but are not limited to, ion exchangers, alumina, aluminum stearate, lecithin, serum proteins, such as human serum albumin, buffer substances such as phosphates, glycine, sorbic acid, or potassium sorbate, partial glyceride mixtures of saturated vegetable fatty acids, water, salts or electrolytes, such as protamine sulfate, disodium hydrogen phosphate, potassium hydrogen phosphate, sodium chloride, zinc salts, colloidal silica, magnesium trisilicate, polyvinyl pyrrolidone, polyacrylates, waxes, polyethylene-polyoxypropylene- block polymers, wool fat, sugars such as lactose, glucose and sucrose; starches such as corn starch and potato starch; cellulose and its derivatives such as sodium carboxymethyl cellulose, ethyl cellulose and cellulose acetate; powdered tragacanth; malt; gelatin; talc
- the compounds of the invention can be prepared from known materials by the methods described in the Examples, other similar methods, and other methods known to one skilled in the art.
- the functional groups of the intermediate compounds may need to be protected by suitable protecting groups.
- Protecting groups may be added or removed in accordance with standard techniques, which are well-known to those skilled in the art. The use of protecting groups is described in detail in T.G.M. Wuts et al., Greene’s Protective Groups in Organic Synthesis (4th ed.2006).
- M. Radiolabeled Analogs of the Compounds [0459] In another aspect, the invention relates to radiolabeled analogs of the compounds of the invention.
- the term “radiolabeled analogs of the compounds of the invention” refers to compounds that are identical to the compounds of the invention, as described herein including all embodiments thereof, except that one or more atoms has been replaced with a radioisotope of the atom present in the compounds of the invention.
- the term “radioisotope” refers to an isotope of an element that is known to undergo spontaneous radioactive decay. Examples of radioisotopes include 3 H, 14 C, 32 P, 35 S, 18 F, 36 Cl, and the like, as well as the isotopes for which a decay mode is identified in V.S. Shirley & C.M.
- the radiolabeled analogs can be used in a number of beneficial ways, including in various types of assays, such as substrate tissue distribution assays.
- assays such as substrate tissue distribution assays.
- tritium ( 3 H)- and/or carbon- 14 ( 14 C)-labeled compounds may be useful for various types of assays, such as substrate tissue distribution assays, due to relatively simple preparation and excellent detectability.
- the invention relates to pharmaceutically acceptable salts of the radiolabeled analogs, in accordance with any of the embodiments described herein in connection with the compounds of the invention.
- the invention relates to pharmaceutical compositions comprising the radiolabeled analogs, or pharmaceutically acceptable salts thereof, and a pharmaceutically acceptable carrier, adjuvant or vehicle, in accordance with any of the embodiments described herein in connection with the compounds of the invention.
- the invention relates to methods of inhibiting voltage-gated sodium channels and methods of treating or lessening the severity of various diseases and disorders, including pain, in a subject comprising administering an effective amount of the radiolabeled analogs, pharmaceutically acceptable salts thereof, and pharmaceutical compositions thereof, in accordance with any of the embodiments described herein in connection with the compounds of the invention.
- the invention relates to radiolabeled analogs, pharmaceutically acceptable salts thereof, and pharmaceutical compositions thereof, for use, in accordance with any of the embodiments described herein in connection with the compounds of the invention.
- the invention relates to the use of the radiolabeled analogs, or pharmaceutically acceptable salts thereof, and pharmaceutical compositions thereof, for the manufacture of medicaments, in accordance with any of the embodiments described herein in connection with the compounds of the invention.
- the radiolabeled analogs, pharmaceutically acceptable salts thereof, and pharmaceutical compositions thereof can be employed in combination therapies, in accordance with any of the embodiments described herein in connection with the compounds of the invention. N.
- Embodiments Further embodiments of the disclosure are set out in the following numbered Embodiments: 1. Compound I I) as substantially amorphous Compound I amorphous form (i.e., wherein less than 15% of Compound I is in crystalline form, wherein less than 10% of Compound I is in crystalline form, wherein less than 5% of Compound I is in crystalline form). 2. The substantially amorphous Compound I amorphous form according to Embodiment 1, wherein Compound I is 100% amorphous. 3. The substantially amorphous Compound I amorphous form according to Embodiment 1 or Embodiment 2, characterized by an X-ray powder diffractogram substantially similar to FIG.1. 4.
- the substantially amorphous Compound I amorphous form according to any one of Embodiments 1-3, characterized by a 13 C SSNMR spectrum having one, two, three, four, five, six, seven, eight, nine, ten, or more peaks selected from 177.6 ⁇ 0.2 ppm, 172.0 ⁇ 0.2 ppm, 162.6 ⁇ 0.2 ppm, 155.2 ⁇ 0.2 ppm, 149.9 ⁇ 0.2 ppm, 147.0 ⁇ 0.2 ppm, 121.0 ⁇ 0.2 ppm, 120.1 ⁇ 0.2 ppm, 119.1 ⁇ 0.2 ppm, 116.0 ⁇ 0.2 ppm, 114.6 ⁇ 0.2 ppm, 113.8 ⁇ 0.2 ppm, 15.5 ⁇ 0.2 ppm, and 7.7 ⁇ 0.2 ppm.
- the substantially amorphous Compound I amorphous form according to any one of Embodiments 1-5 characterized by a 13 C SSNMR spectrum substantially similar to FIG.2. 7.
- the substantially amorphous Compound I amorphous form according to any one of Embodiments 1-6 characterized by a 19 F SSNMR spectrum having one or two peaks selected from -61.9 ⁇ 0.2 ppm and -142.1 ⁇ 0.2 ppm.
- Substantially crystalline Compound I neat Form A (i.e., wherein less than 15% of Compound I is in amorphous form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous form). 10.
- the substantially crystalline Compound I neat Form A according to Embodiment 9 or Embodiment 10 characterized by an X-ray powder diffractogram having one or two signals selected from 9.5 ⁇ 0.2 degrees two-theta, 12.3 ⁇ 0.2 degrees two-theta, and 13.5 ⁇ 0.2 degrees two-theta. 12.
- the substantially crystalline Compound I neat Form A according to any one of Embodiments 9-11, characterized by an X-ray powder diffractogram having (a) one or two signals selected from 9.5 ⁇ 0.2 degrees two-theta, 12.3 ⁇ 0.2 degrees two-theta, and 13.5 ⁇ 0.2 degrees two-theta, and (b) one or two signals selected from 11.3 ⁇ 0.2 degrees two-theta, 15.9 ⁇ 0.2 degrees two-theta, and 20.5 ⁇ 0.2 degrees two-theta. 13.
- the substantially crystalline Compound I neat Form A according to any one of Embodiments 9-12, characterized by an X-ray powder diffractogram having two, three, four, five, or six signals selected from 9.5 ⁇ 0.2 degrees two-theta, 12.3 ⁇ 0.2 degrees two-theta, 13.5 ⁇ 0.2 degrees two-theta, 11.3 ⁇ 0.2 degrees two-theta, 15.9 ⁇ 0.2 degrees two-theta, 20.5 ⁇ 0.2 degrees two-theta, 14.6 ⁇ 0.2 degrees two-theta, 16.6 ⁇ 0.2 degrees two-theta, and 23.5 ⁇ 0.2 degrees two-theta. 14.
- the substantially crystalline Compound I neat Form A according to any one of Embodiments 9-16 characterized by a 13 C SSNMR spectrum substantially similar to FIG.5. 18.
- the substantially crystalline Compound I neat Form A according to any one of Embodiments 9-17 characterized as having a 19 F SSNMR spectrum with one or two peaks selected from -61.2 ⁇ 0.2 ppm, -61.6 ⁇ 0.2 ppm, -138.6 ⁇ 0.2 ppm, -140.3 ⁇ 0.2 ppm, -142.0 ⁇ 0.2 ppm, and-146.0 ⁇ 0.2 ppm. 19.
- Substantially crystalline Compound I neat Form B (i.e., wherein less than 15% of Compound I is in amorphous form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous form).
- the substantially crystalline Compound I neat Form B according to any one of Embodiments 22-24, characterized by an X-ray powder diffractogram having (a) one or two signals selected from 4.8 ⁇ 0.2 degrees two-theta, 13.3 ⁇ 0.2 degrees two-theta, and 18.5 ⁇ 0.2 degrees two-theta, and (b) one or two signals selected from 6.0 ⁇ 0.2 degrees two-theta, 16.5 ⁇ 0.2 degrees two-theta, and 16.9 ⁇ 0.2 degrees two-theta. 26.
- the substantially crystalline Compound I neat Form B according to any one of Embodiments 22-25, characterized by an X-ray powder diffractogram having two, three, four, five, or six signals selected from 4.8 ⁇ 0.2 degrees two-theta, 13.3 ⁇ 0.2 degrees two-theta, 18.5 ⁇ 0.2 degrees two-theta, 6.0 ⁇ 0.2 degrees two-theta, 16.5 ⁇ 0.2 degrees two-theta, 16.9 ⁇ 0.2 degrees two-theta, 14.1 ⁇ 0.2 degrees two-theta, 14.7 ⁇ 0.2 degrees two-theta, and 15.2 ⁇ 0.2 degrees two-theta. 27.
- the substantially crystalline Compound I neat Form B according to any one of Embodiments 22-29 characterized by a 13 C SSNMR spectrum substantially similar to FIG.8. 31.
- the substantially crystalline Compound I neat Form B according to any one of Embodiments 22-30 characterized as having a 19 F SSNMR spectrum with one or two peaks selected from -61.6 ⁇ 0.2 ppm, -138 ⁇ 0.2 ppm, -139.7 ⁇ 0.2 ppm, -143.4 ⁇ 0.2 ppm, and -144.9 ⁇ 0.2 ppm. 32.
- the substantially crystalline Compound I neat Form B according to any one of Embodiments 22-31 characterized as having a 19 F SSNMR spectrum having peaks at -61.6 ⁇ 0.2 ppm, -138 ⁇ 0.2 ppm, -139.7 ⁇ 0.2 ppm, -143.4 ⁇ 0.2 ppm, and -144.9 ⁇ 0.2 ppm. 33.
- the substantially crystalline Compound I neat Form B according to any one of Embodiments 22-32 characterized by a 19 F SSNMR spectrum substantially similar to FIG.9. 34.
- Substantially crystalline Compound I neat Form E (i.e., wherein less than 15% of Compound I is in amorphous form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous form).
- 36. The substantially crystalline Compound I neat Form E according to Embodiment 34 or Embodiment 35 characterized by an X-ray powder diffractogram having one or two signals selected from 8.6 ⁇ 0.2 degrees two-theta, 10.1 ⁇ 0.2 degrees two-theta, and 12.6 ⁇ 0.2 degrees two-theta. 37.
- the substantially crystalline Compound I neat Form E according to any one of Embodiments 34-36, characterized by an X-ray powder diffractogram having (a) one or two signals selected from 8.6 ⁇ 0.2 degrees two-theta, 10.1 ⁇ 0.2 degrees two-theta, and 12.6 ⁇ 0.2 degrees two-theta, and (b) one or two signals selected from 14.1 ⁇ 0.2 degrees two-theta, 20.3 ⁇ 0.2 degrees two-theta, and 23.2 ⁇ 0.2 degrees two-theta. 38.
- the substantially crystalline Compound I neat Form E according to any one of Embodiments 34-37, characterized by an X-ray powder diffractogram having two, three, four, five, or six signals selected from 8.6 ⁇ 0.2 degrees two-theta, 10.1 ⁇ 0.2 degrees two-theta, 12.6 ⁇ 0.2 degrees two-theta, 14.1 ⁇ 0.2 degrees two-theta, 20.3 ⁇ 0.2 degrees two-theta, 23.2 ⁇ 0.2 degrees two-theta, 11.5 ⁇ 0.2 degrees two-theta, 14.2 ⁇ 0.2 degrees two-theta, and 15.1 ⁇ 0.2 degrees two-theta. 39.
- the substantially crystalline Compound I neat Form E according to any one of Embodiments 34-41 characterized by a 13 C SSNMR spectrum substantially similar to FIG.11.
- 43. The substantially crystalline Compound I neat Form E according to any one of Embodiments 34-42, characterized as having a 19 F SSNMR spectrum with one or two peaks selected from -60.9 ⁇ 0.2 ppm, -62.5 ⁇ 0.2 ppm, -63.1 ⁇ 0.2 ppm, -135.1 ⁇ 0.2 ppm, -137.1 ⁇ 0.2 ppm, -140.7 ⁇ 0.2 ppm, and -141.9 ⁇ 0.2 ppm. 44.
- the substantially crystalline Compound I neat Form E according to any one of Embodiments 34-43 characterized as having a 19 F SSNMR spectrum having peaks at -60.9 ⁇ 0.2 ppm, -62.5 ⁇ 0.2 ppm, -63.1 ⁇ 0.2 ppm, -135.1 ⁇ 0.2 ppm, -137.1 ⁇ 0.2 ppm, -140.7 ⁇ 0.2 ppm, and -141.9 ⁇ 0.2 ppm. 45.
- the substantially crystalline Compound I neat Form E according to any one of Embodiments 34-44 characterized by a 19 F SSNMR spectrum substantially similar to FIG.12. 46.
- Substantially crystalline Compound I Acetone Solvate Hydrate Form A (i.e., wherein less than 15% of Compound I is in amorphous form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous form). 49.
- the substantially crystalline Compound I Acetone Solvate Hydrate Form A according to any one of Embodiments 48-50, characterized by an X-ray powder diffractogram having (a) one or two signals selected from 17.2 ⁇ 0.2 degrees two-theta, 18.1 ⁇ 0.2 degrees two-theta, and 25.9 ⁇ 0.2 degrees two-theta, and (b) one or two signals selected from 12.9 ⁇ 0.2 degrees two-theta, 15.6 ⁇ 0.2 degrees two-theta, and 24.8 ⁇ 0.2 degrees two-theta. 52.
- the substantially crystalline Compound I Acetone Solvate Hydrate Form A according to any one of Embodiments 48-51, characterized by an X-ray powder diffractogram having two, three, four, five, or six signals selected from 9.5 ⁇ 0.2 degrees two-theta, 12.3 ⁇ 0.2 degrees two-theta, 13.5 ⁇ 0.2 degrees two-theta, 12.9 ⁇ 0.2 degrees two-theta, 15.6 ⁇ 0.2 degrees two-theta, 24.8 ⁇ 0.2 degrees two-theta, 8.1 ⁇ 0.2 degrees two-theta, 12.3 ⁇ 0.2 degrees two-theta, and 17.6 ⁇ 0.2 degrees two-theta. 53.
- the substantially crystalline Compound I Acetone Solvate Hydrate Form According to any one of Embodiments 48-55, characterized by a 13 C SSNMR spectrum substantially similar to FIG.14. 57.
- the substantially crystalline Compound I Acetone Solvate Hydrate Form According to any one of Embodiments 48-58, characterized by a 19 F SSNMR spectrum substantially similar to FIG.15. 60.
- Substantially crystalline Compound I Ethanol Solvate Form A i.e., wherein less than 15% of Compound I is in amorphous form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous form.
- the substantially crystalline Compound I Ethanol Solvate Form A according to any one of Embodiments 61-63, characterized by an X-ray powder diffractogram having (a) one or two signals selected from 9.4 ⁇ 0.2 degrees two-theta, 16.1 ⁇ 0.2 degrees two-theta, and 26.0 ⁇ 0.2 degrees two-theta, and (b) one or two signals selected from 15.1 ⁇ 0.2 degrees two-theta, 17.0 ⁇ 0.2 degrees two-theta, and 22.4 ⁇ 0.2 degrees two-theta. 65.
- the substantially crystalline Compound I Ethanol Solvate Form A according to any one of Embodiments 61-64, characterized by an X-ray powder diffractogram having two, three, four, five, or six signals selected from 9.4 ⁇ 0.2 degrees two-theta, 16.1 ⁇ 0.2 degrees two-theta, 26.0 ⁇ 0.2 degrees two-theta, 15.1 ⁇ 0.2 degrees two-theta, 17.0 ⁇ 0.2 degrees two-theta, 22.4 ⁇ 0.2 degrees two-theta, 13.2 ⁇ 0.2 degrees two-theta, 13.6 ⁇ 0.2 degrees two-theta, and 13.8 ⁇ 0.2 degrees two-theta. 66.
- Substantially crystalline Compound I Hydrate Form A i.e., wherein less than 15% of Compound I is in amorphous form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous form.
- the substantially crystalline Compound I Hydrate Form A according to any one of Embodiments 74-76, characterized by an X-ray powder diffractogram having (a) one or two signals selected from 8.7 ⁇ 0.2 degrees two-theta, 12.4 ⁇ 0.2 degrees two-theta, and 18.8 ⁇ 0.2 degrees two-theta, and (b) one or two signals selected from 12.7 ⁇ 0.2 degrees two-theta, 17.5 ⁇ 0.2 degrees two-theta, and 19.8 ⁇ 0.2 degrees two-theta. 78.
- the substantially crystalline Compound I Hydrate Form A according to any one of Embodiments 74-77, characterized by an X-ray powder diffractogram having two, three, four, five, or six signals selected from 8.7 ⁇ 0.2 degrees two-theta, 12.4 ⁇ 0.2 degrees two-theta, 18.8 ⁇ 0.2 degrees two-theta, 12.7 ⁇ 0.2 degrees two-theta, 17.5 ⁇ 0.2 degrees two-theta, 19.8 ⁇ 0.2 degrees two-theta, 13.1 ⁇ 0.2 degrees two-theta, 16.3 ⁇ 0.2 degrees two-theta, and 21.1 ⁇ 0.2 degrees two-theta. 79.
- the substantially crystalline Compound I Hydrate Form A according to any one of Embodiments 74-78, characterized by an X-ray powder diffractogram substantially similar to FIG.19. 80.
- Substantially crystalline Compound I Dehydrated Hydrate Form A i.e., wherein less than 15% of Compound I is in amorphous form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous form).
- the substantially crystalline Compound I Dehydrated Hydrate Form A according to Embodiment 81 or Embodiment 82, characterized by an X-ray powder diffractogram having one or two signals selected from 8.7 ⁇ 0.2 degrees two-theta, 15.9 ⁇ 0.2 degrees two-theta, and 18.7 ⁇ 0.2 degrees two-theta. 83.
- the substantially crystalline Compound I Dehydrated Hydrate Form A according to any one of Embodiments 81-83, characterized by an X-ray powder diffractogram having (a) one or two signals selected from 8.7 ⁇ 0.2 degrees two-theta, 15.9 ⁇ 0.2 degrees two-theta, and 18.7 ⁇ 0.2 degrees two-theta, and (b) one or two signals selected from 12.4 ⁇ 0.2 degrees two-theta, 12.6 ⁇ 0.2 degrees two-theta, 17.3 ⁇ 0.2 degrees two-theta, and 19.8 ⁇ 0.2 degrees two-theta. 84.
- the substantially crystalline Compound I Dehydrated Hydrate Form A according to any one of Embodiments 81-84, characterized by an X-ray powder diffractogram having two, three, four, five, or six signals selected from 8.7 ⁇ 0.2 degrees two-theta, 15.9 ⁇ 0.2 degrees two-theta, 18.7 ⁇ 0.2 degrees two-theta, 12.4 ⁇ 0.2 degrees two-theta, 12.6 ⁇ 0.2 degrees two-theta, 17.3 ⁇ 0.2 degrees two-theta, 19.8 ⁇ 0.2 degrees two-theta, 13.1 ⁇ 0.2 degrees two-theta, 16.3 ⁇ 0.2 degrees two-theta, and 21.1 ⁇ 0.2 degrees two-theta. 85.
- the substantially crystalline Compound I Dehydrated Hydrate Form A according to any one of Embodiments 81-85, characterized by an X-ray powder diffractogram substantially similar to FIG.20. 86.
- the substantially crystalline Compound I Hydrate Form A according to any one of Embodiments 74-90, characterized as having a 19 F SSNMR spectrum having peaks at -59.4 ⁇ 0.2 ppm, -60.2 ⁇ 0.2 ppm, -135.0 ⁇ 0.2 ppm, -138.4 ⁇ 0.2 ppm, -141.7 ⁇ 0.2 ppm, and -144.0 ⁇ 0.2 ppm. 91.
- the substantially crystalline Compound I Hydrate Form A according to any one of Embodiments 74-91 characterized by a 19 F SSNMR spectrum substantially similar to FIG.23. 92.
- a pharmaceutical composition comprising Compound I according to any one of Embodiments 1-93, and optionally further comprising one or more additional therapeutic agents. 95.
- the pharmaceutical composition according to Embodiment 94 wherein the pharmaceutical composition comprises one or more additional pain modulating compounds.
- 96. The Compound I according to any one of Embodiments 1-93, or the pharmaceutical composition according to any one of Embodiments 94-95, for use in the treatment of pain.
- 97. Use of the Compound I according to any one of Embodiments 1-93, or the pharmaceutical composition according to any one of Embodiments 94-95, in the manufacture of a medicament for the treatment of pain.
- a method of treating pain comprising administering the Compound I according to any one of Embodiments 1-93, or the pharmaceutical composition according to any one of Embodiments 94-95, to a subject in need thereof. 99.
- Embodiment 96 The compound or composition for use of Embodiment 96, the use of Embodiment 97, or the method of Embodiment 98, wherein the Compound I according to any one of Embodiments 1-93 or the composition according to any one of Embodiments 94-95 is administered in combination with one or more additional therapeutic agents.
- a method of making crystalline Compound I neat Form A comprising (i) combining Compound I and ethyl acetate, (ii) distilling under vacuum at 50 °C, (iii) adding ethyl acetate, (iv) distilling under vacuum at 50 °C, (v) heating to 75 °C, (vi) adding heptane, (vii) cooling to 40 °C, (viii) adding a seed of crystalline Compound I Form A, (ix) holding at 40 °C for 1.5 hours, (x) adding heptane over 5 hours, (xi) cooling the slurry to 20 °C over 5 hours, (xii) holding at 20 °C for 11 hours, (xiii) collecting the solids, (xiv) drying the solids in a vacuum oven at 45 °C with a slight nitrogen bleed for 46 hours to yield crystalline Compound I neat Form A.
- a method of making crystalline Compound I neat Form B comprising (i) adding Compound I hydrate Form A to an oven set at 180 °C (ii) cooling under ambient conditions to yield crystalline Compound I neat Form B.
- a method of making crystalline Compound I neat Form E comprising (i) combining Compound I hydrate Form A and trifluorotoluene, (ii) heating the slurry to 80 °C, (iii) cooling to room temperature to yield crystalline Compound I neat Form E. 104.
- a method of making crystalline Compound I Acetone Solvate Hydrate Form A comprising (i) saturating acetone with Compound I, (ii) adding water dropwise to 20% of the final volume, (iii) holding overnight at room temperature to yield crystalline Compound I acetone solvate hydrate Form A. 105.
- a method of making crystalline Compound I Ethanol Solvate Form A comprising (i) combining Compound I and ethyl acetate, (ii) heating the slurry to 50 °C, (iii) adding ethanol and distilling under vacuum at 50 °C, (iv) adding ethanol and distilling under vacuum at 50 °C, (v) adding ethanol and distilling under vacuum at 50 °C to a minimum volume, (vi) adding ethanol and distilling under vacuum at 50 °C, (vii) drying in a vacuum oven at 45 °C, with a slight nitrogen bleed for 42 hours to yield crystalline Compound I ethanol solvate Form A. 106.
- a method of making crystalline Compound I Dehydrated Hydrate Form A comprising (i) combining Compound I Ethanol Solvate Form A and acetone, (ii) stirring the slurry while heating 50 °C, (iii) adding water over 1 hour, (iv) adding a seed of crystalline Compound I hydrate Form A, (v) agitating for 30 minutes, (vi) adding water over 8 hours, (vii) stirring at 50 °C for 5 hours, (viii) cooling the slurry to 20 °C over 3 hours, (ix) agitating for 2 hours, (x) collecting the solids and washing with 1:2 acetone:water, (xi) drying the solids in a vacuum over at 50 °C with a slight nitrogen bleed to yield crystalline Compound I hydrate Form A.
- a method of making Compound I amorphous form comprising (i) dissolving Compound I in acetone, (ii) filtering and evaporating the solvent at 50 °C, 12 mbar, over 1 hour on a centrifugal evaporation, (iii) drying in a vacuum oven at 50 °C overnight to yield Compound I amorphous form.
- Additional embodiments of the disclosure are set out in the following numbered clauses: 1.
- a process for preparing a compound of formula (I): comprising transforming a to the compound of formula (I); wherein are independently selected from halogen.
- transforming a compound of formula (A 1-1 ) to the compound of formula (A 1 ) comprises the steps of: treating the compound of formula (A 1-1 ) with trifluoromethanesulfonic anhydride to form a triflyl intermediate; treating the triflyl intermediate with a cyanation reagent; treating the cyanated intermediate with a second base to form a cyanated triflyl intermediate; and treating the cyanated triflyl intermediate with an aqueous base to form the compound of formula (A 1 ).
- the cyanation reagent is trimethylsilyl cyanide, sodium cyanide, and potassium cyanide.
- the process of Clause 33, of formula (A 3 ) to the compound of formula (E-A 3 ) comprises contacting the compound of formula (A 3 ) with a compound of formula (D-1): . 35.
- the process of Clause 34, of formula (A 3 ) with a compound of formula (D-1) is performed in the presence of a first palladium catalyst and a first base.
- the first palladium catalyst is PdCl 2 (dtbdpf).
- 37. The process of any one of Clauses 35 to 36, wherein the first base is potassium phosphate. 38.
- the process of Clause 57, wherein transforming the compound of formula (B-2) to the compound of formula (B-1) comprises treating the compound of formula (B-2) with a chlorinating reagent.
- the process of Clause 60, wherein transforming the compound of formula (B-3) to the compound of formula (B-2) comprises treating the compound of formula (B-3) with a brominating reagent in the presence of a third acid. 62.
- a composition comprising a compound of formula (I) and a compound of formula (E-A 3 ): : . 100.
- the composition of Clause 99 further comprising an acid.
- 101. The composition of Clause 100, wherein the acid is trifluoroacetic acid.
- 102. A composition comprising a compound of formula (E-A 2 ), a compound of formula (A 2 ), and a compound of formula (D-1): . 1) , and a compound of formula (D-1): . 1)
- the substantially crystalline Compound I according to Clause 105 wherein 100% of Compound I is crystalline.
- 110. Substantially amorphous Compound I amorphous form.
- 111. The substantially amorphous Compound I according to Clause 110, wherein less than 15% of Compound I is in crystalline form.
- 1112. The substantially crystalline Compound I according to Clause 110, wherein less than 10% of Compound I is in crystalline form.
- 113. The substantially crystalline Compound I according to Clause 110, wherein less than 5% of Compound I is in crystalline form.
- 114. The substantially crystalline Compound I according to Clause 110, wherein 100% of Compound I is amorphous.
- a pharmaceutical composition comprising the Compound I according to any one of Clauses 105 to 117.
- the pharmaceutical composition according to Clause 119 wherein the pharmaceutical composition comprises one or more additional pain modulating compounds.
- the pharmaceutical composition according to Clause 120 wherein the pharmaceutical composition comprises one or more combination compounds and pharmaceutically acceptable salts and deuterated derivatives thereof.
- 122. The Compound I according to any one of Clauses 105 to 117, or the pharmaceutical composition according to any one of Clauses 118 to 121, for use in the treatment of pain. 123.
- the compound or composition according to Clause 122 wherein the pain comprises chronic pain, gut pain, neuropathic pain, musculoskeletal pain, acute pain, inflammatory pain, cancer pain, idiopathic pain, postsurgical pain, visceral pain, multiple sclerosis, Charcot-Marie-Tooth syndrome, incontinence, pathological cough, or cardiac arrhythmia.
- the pain comprises chronic pain, gut pain, neuropathic pain, musculoskeletal pain, acute pain, inflammatory pain, cancer pain, idiopathic pain, postsurgical pain, visceral pain, multiple sclerosis, Charcot-Marie-Tooth syndrome, incontinence, pathological cough, or cardiac arrhythmia.
- a method of treating pain comprising administering the Compound I according to any one of Clauses 105 to 117, or the pharmaceutical composition according to any one of Clauses 118 to 121, to a subject in need thereof.
- 126. The compound or composition for use of Clause 122, the use of Clause 124, or the method of Clause 125, wherein the Compound I according to any one of Clauses 105 to 117 or the composition according to any one of Clauses 118 to 120 is administered in combination with one or more additional therapeutic agents.
- Mobile phase B 0.1% (v/v) Phosphoric Acid in Acetonitrile.
- Exemplary LC Method #2 HPLC analysis was conducted using an Agilent 1260 HPLC utilizing an Agilent Poroshell EC-C18 column (4.6 x 150 mm, 2.7 ⁇ m particle) guard column (pn: 693975-902), and a dual gradient run from 5-95% mobile phase B over 15 min.
- Mobile phase A 0.1% (v/v) phosphoric acid in water.
- Mobile phase B 0.1% (v/v) Phosphoric Acid in Acetonitrile.
- X-ray Powder Diffraction [0475] X-ray powder diffraction (XRPD) spectra were recorded at room temperature in reflection mode using a PANalytical Empyrean system equipped with a sealed tube source and a PIXcel 1D Medipix-2 detector (Malvern PANalytical Inc, Westborough, Massachusetts).
- the X-Ray generator operated at a voltage of 45 kV and a current of 40 mA with copper radiation (1.54060 ⁇ ).
- the powder sample was placed in a back filled sample holder and loaded into the instrument.
- the sample was scanned over the range of about 3° to about 40° 2 ⁇ with a step size of 0.0131303° and 49.725s per step.
- the slurry was distilled under vacuum at 50 °C until 15 g solvent remained in the flask, followed by additional ethanol (20 mL) being charged to the flask.
- the slurry was distilled under vacuum at 50 °C until 12 g solvent remained in the flask, followed by ethanol (30 mL) being charged to the flask.
- the slurry was distilled under vacuum at 50 °C to minimum volume. Ethanol (25 mL) was then charged to the flask.
- the slurry was distilled under vacuum at 50 °C until only 8 g solvent remained.
- Step 2 Synthesis of ethyl 4-hydroxy-2-oxo-1,2-dihydro-1,6-naphthyridine-3-carboxylate (A 1-3 )
- Methyl 4-aminonicotinate (A 1-4 ) 42 kg, 276.3 mol, 1 eq
- diethyl malonate 53.1 kg, 331.5 mol, 1.2 eq.
- EtOH 294 L
- EtONa 24.4 kg, 359.2 mol, 1.3 eq.
- the mixture was stirred at 90 °C. After the reaction was completed, as determined by HPLC analysis, the reaction mixture was acidified with HCl (1.5 M) to pH 3 ⁇ 4.
- Step 3 Synthesis of 4-hydroxy-1,6-naphthyridin-2(1H)-one (A 1-2 ) [0481] Crude ethyl 4-hydroxy-2-oxo-1,2-dihydro-1,6-naphthyridine-3-carboxylate (A 1-3 ) (64.8 kg) and toluene (32 L) were charged to a reactor followed by HCl (453 L, 27% w/w% in H 2 O). The mixture was stirred at 90 °C. After the reaction was completed, the mixture was cooled to 25 °C and the layers were separated. The aqueous layer was cooled to 0 °C and basified with 30% NaOH to pH 5-6.
- Step 4 Synthesis of 2,4-dichloro-1,6-naphthyridine (A 2-1 )
- 4-Hydroxy-1,6-naphthyridin-2(1H)-one (A 1-2 ) (33.5 kg, 206.6 mol, 1.0 eq.) and toluene (67 L) were charged to a reactor followed by POCl 3 (88 kg, 574.3 mol, 2.78 eq.). The mixture was stirred at 110 °C. After the reaction was completed, as determined by HPLC analysis, the mixture was concentrated in vacuo. The residue was diluted with CH 2 Cl 2 (5 V), quenched into ice water (5 V), and then the resulting mixture was poured into sat.
- Step 5 Synthesis of 2,4-dichloro-1,6-naphthyridine-5-carbonitrile (A2) [0483] 2,4-Dichloro-1,6-naphthyridine (A 2-1 ) (33.6 kg, 168.8 mol, 1 eq.) and CH 2 Cl 2 (336 L) were charged to a reactor. The solution was cooled to -10 °C. Tf 2 O (50 kg, 177.2 mol, 1.05 eq.) was charged. After complete addition, TMSCN (17.5 kg, 177.2 mol, 1.05 eq.) was charged followed by Et 3 N (22.2 kg, 219.4 mol, 1.3 eq.).
- reaction mixture was diluted with CH 2 Cl 2 (7.5 V) and basified to pH 7 with sat. NaHCO 3 .
- the layers were separated, and the aqueous layer was extracted with CH 2 Cl 2 (2 x 5 V).
- the combined organic layers were washed with water (2 x 5 L) and brine and dried over with anhydrous Na 2 SO 4 .
- the organic solution was concentrated under vacuum and the residue was triturated with MeOH (4 V).
- the slurry was filtered, and the wet cake was dried under vacuum to afford 2,4-dichloro-1,6- naphthyridine-5-carbonitrile (A 2 ) as a yellow solid (31.21 kg).
- the organic phase was washed with 20 wt% NH 4 Cl (3 Vol), water (3 Vol), and 2% aq. NaCl (3 Vol).
- the organic layer was treated with Si-Thiol (5% wt eq.).
- the resin was filtered, and the filtrate was concentrated to dryness.
- the concentrate was then diluted with DCM (6 Vol) and the solution was re- concentrated.
- the concentrate was slurried in 1:1 DCM:heptane (6 Vol) and filtered.
- the mixture was filtered through a pad of celite.
- the celite was washed with acetonitrile (3 vol).
- DCM (20 vol) and H 2 O (8 vol) were added to the reactor, along with the filter cake and additional celite.
- the layers were separated, and the isolated organic layer was filtered through a pad of celite.
- the celite was washed with DCM and the filtrate was washed with H 2 O (8 vol).
- the organic layer was treated with MP-TMT (0.32 w/w).
- the resin was filtered and the filtrate was concentrated.
- EtOH (6 vol) was added to the resulting residue and that solution was concentrated.
- the slurry was heated 40 °C and the solids were collected by filtration after cooling to 20 °C.
- the organic solution was washed with saturated aqueous NaHCO 3 (6 vol) followed an aqueous 2% w/w NaCl solution (4 vol). The organic solution was then filtered through a silica plug. The filtrate was then concentrated and heated to 70 °C.
- the solution was seeded (0.01 equiv) with 2-(2-(3,4-difluoro-2- methylphenoxy)-4-methyl-5-(trifluoromethyl)pyridin-3-yl)-4-oxo-1,4-dihydro-1,6-naphthyridine-5- carboxamide ethanol solvate, and ethanol (6 vol) was charged to the reactor. The slurry was cooled to 20 °C and filtered.
- the mixture was poured into a stirring mixture of 0.1 M aqueous HCl (100 mL) and 2-MeTHF (100 mL). The layers were separated and the aqueous layer extracted with additional 2-MeTHF (2 x 100 mL). The organic layers were combined and washed with water (2 x 50 mL), 1:1 water/brine (50 mL) and brine (50 mL). The solution was dried over sodium sulfate, filtered, and concentrated under reduced pressure.
- Step 2 2-chloro-4-[(4-methoxyphenyl)methoxy]-6-oxido-1,6-naphthyridin-6-ium [0493] A solution (5 g, 16.6 mmol) in DCM (100 acid (4.7 g, 21 mmol). The reaction was warmed to room temperature and stirred for 4 h. The mixture was quenched with saturated aqueous sodium bicarbonate (100 mL) and the layers were separated. The aqueous layer was extracted with additional DCM (3 x 25 mL).
- Step 3 2-chloro-4-[(4-methoxyphenyl)methoxy]-1,6-naphthyridine-5-carbonitrile
- a 1,6-naphthyridin-6- ium 6.1 g, 19.3 mmol
- trimethylsilyl cyanide 6.8 mL, 51 mmol
- TEA 8 mL, 57.4 mmol
- the mixture was stirred for 20 h at room temperature.
- the reaction was quenched with water and the aqueous layer was extracted with DCM (3x). The combined organic layers were dried over magnesium sulfate, filtered and concentrated.
- Example 2 Compound 1 Neat Amorphous Form Method for the Synthesis of Compound of Formula (I) using Intermediates A and Intermediate B Synthesis of Intermediate A-1
- Step 1 4-benzyloxy-2-chloro-1,6-naphthyridine
- the reaction mixture was stirred at 0 °C for 1 h, followed gradual warming to room temperature and stirring for 2 h.
- the reaction mixture was poured into a stirring mixture of 0.1 M aqueous HCl (50 mL) and 2-MeTHF (50 mL).
- the layers were separated, and the aqueous layer extracted with 2-MeTHF (2 x 100 mL).
- the combined organic layers were washed with water (2 x 50 mL), 1:1 water/brine (50 mL) and brine (50 mL), dried over sodium sulfate, filtered and evaporated under reduced pressure.
- Step 2 4-benzyloxy-2-chloro-6-oxido-1,6-naphthyridin-6-ium
- mCPBA 1.6 g, 7.14 mmol
- Step 1 2-chloro-4-[(4-methoxyphenyl)methoxy]-1,6-naphthyridine
- Step 2 2-chloro-4-[(4-methoxyphenyl)methoxy]-1,6-naphthyridine
- the mixture was stirred at 0 °C for 1 h, then gradually warmed to room temperature and stirred for 19.5 h.
- the mixture was poured into a stirring mixture of 0.1 M aqueous HCl (100 mL) and 2-MeTHF (100 mL).
- the layers were separated and the aqueous layer extracted with additional 2-MeTHF (2 x 100 mL).
- the organic layers were combined and washed with water (2 x 50 mL), 1:1 water/brine (50 mL) and brine (50 mL). The solution was dried over sodium sulfate, filtered, and concentrated under reduced pressure.
- Step 2 2-chloro-4-[(4-methoxyphenyl)methoxy]-6-oxido-1,6-naphthyridin-6-ium
- a solution of 2-chloro-4-[(4-methoxyphenoxy)methyl]-1,6-naphthyridine (5.0 g, 16.6 mmol) in DCM (100 mL) was cooled to 0 °C and treated with solid 3-chlorobenzenecarboperoxoic acid (4.7 g, 21 mmol). The reaction was warmed to room temperature and stirred for 4 h. The mixture was quenched with saturated aqueous sodium bicarbonate (100 mL) and the layers were separated.
- Step 3 2-chloro-4-[(4-methoxyphenyl)methoxy]-1,6-naphthyridine-5-carbonitrile
- 2-chloro-4-[(4-methoxyphenyl)methoxy]-6-oxido-1,6-naphthyridin-6- ium 6.1 g, 19.3 mmol
- DCM DCM
- trimethylsilyl cyanide 6.8 mL, 51 mmol
- TEA 8 mL, 57.4 mmol
- Step 2 [2-(3,4-difluoro-2-methyl-phenoxy)-4-methyl-5-(trifluoromethyl)-3- pyridyl]boronic acid
- 3-bromo-2-(3,4-difluoro-2-methyl-phenoxy)-4-methyl-5- (trifluoromethyl)pyridine 2.0 g, 5.2 mmol
- diethyl ether 20 mL
- n-BuLi 2.5 mL of 2.5 M in hexanes, 6.25 mmol
- the material was purified by reverse phase chromatography (C18, 5-95% acetonitrile/water containing 0.1 % formic acid) and the product-containing fractions concentrated to remove the acetonitrile.
- the resulting aqueous solution was extracted with ethyl acetate (3 x 100 mL).
- the combined organic layers were dried over sodium sulfate, filtered and concentrated under reduced pressure to provide [2- (3,4-difluoro-2-methyl-phenoxy)-4-methyl-5-(trifluoromethyl)-3-pyridyl]boronic acid (1.64 g, 90%) as a white solid.
- Step 1 A mixture of Intermediate A (i.e., Intermediate A-1 or Intermediate A-2, 1 eq), Intermediate B (e.g., Intermediate B-1, 1 - 2 eq), palladium catalyst (1-5 mol%), e.g. PdCl 2 (dppf) or PdCl 2 (dtbpf), base (2-3 eq, eg. potassium phosphate) in organic solvent (e.g.dioxane, DMSO, toluene) and water is degassed with nitrogen bubbling and stirred under inert atmosphere at a temperature ranging from room temperature to 120 °C.
- organic solvent e.g.dioxane, DMSO, toluene
- Step 2 A solution of the protected intermediate I in the appropriate solvent (e.g. toluene, dioxane) is treated with acid (TFA or HCl) and stirred at either room temperature or elevated temperature (e.g.70 oC) to convert the nitrile functionality to the carboxamide.
- the reaction mixture is neutralized and purified via silica gel column chromatography or reverse phase column chromatography to provide the desired product I.
- Preparation of Compound I amorphous form To a 20 ml glass vial, Compound I (0.5 g) was added.
- the resulting solution was distilled to 6 mL under vacuum at 50 °C.
- the resulting material was transferred to a 50 mL reactor with overhead stirrer.
- Ethyl acetate (10 mL) was used to rinse the flask and transfer to reactor.
- the solution was heated to 75 °C.
- heptane (12 mL) was charged.
- the batch was cool to 40 °C.
- the batch was seeded with Compound I Form A (70.1 mg) and held at 40 °C for 1.5 hours.
- heptane (30.6 mL) was charged over 5 hours.
- the slurry was cool to 20 °C over 5 hours and held at 20 °C for 11 hours.
- the resulting solids were collected by vacuum filtration.
- Table 14 Single Crystal Elucidation of Compound I Neat Form E at 298 K Crystal System Triclinic ⁇ (°) 97.140(2) ⁇ (°) 98.521(2) ⁇ 3
- Example 6 Compound I A Preparation of Compound I Acetone Solvate Hydrate Form A [0551] To a saturated Compound I acetone solution, water was added dropwise until up to 20% of the final volume. The crystals of Compound I Acetone Solvate Hydrate Form A formed overnight.
- the slurry was distilled under vacuum at 50 °C until 15 g solvent remained in the flask, followed by additional ethanol (20 mL) being charged to the flask.
- the slurry was distilled under vacuum at 50 °C until 12 g solvent remained in the flask, followed by ethanol (30 mL) being charged to the flask.
- the slurry was distilled under vacuum at 50 °C to minimum volume. Ethanol (25 mL) was then charged to the flask.
- the slurry was distilled under vacuum at 50 °C until only 8 g solvent remained.
- Table 20 13 C SSNMR Signals for Compound I Ethanol Solvate Form A Peak # Chemical Shift [ppm] Intensity [rel] 1 178.2 5.01 Solid-State 19 F NMR [0567] The 19 F SSNMR of Compound I Ethanol Solvate Form A was acquired using the procedure described in the General SSNMR Method. The 19 F SSNMR spectrum for Compound I Ethanol Solvate Form A is provided in FIG.18, and the data are summarized below in Table 21.
- Table 22 Single Crystal Elucidation of Compound I Ethanol Solvate Form A Crystal System monoclinic Space Group P2 1 /c
- Example 8 Compound I H Preparation of Compound I Hydrate Form A
- Compound I Ethanol Solvate Form A (5 g) and acetone (30 mL) were added to a reactor. The slurry was stirred and heated to 50 °C to obtain a clear solution. To the clear solution was added water (20 mL) evenly over 1 hour. The clear solution was then seeded with 5 wt% Compound I Hydrate Form A seeds and agitated for 30 minutes.
- the filtered solids were dried at 50 °C in a vacuum oven overnight.
- X-Ray Powder Diffraction [0573] The XRPD pattern of crystalline Compound I Hydrate Form A was recorded using the procedure described in the General XRPD Method. [0574] The XRPD diffractogram for crystalline Compound I Hydrate Form A (wet) is provided in FIG. 19, and the XRPD data are summarized below in Table 23. The XRPD diffractogram for crystalline Compound I Dehydrated Hydrate Form A is provided in FIG. 20, and the XRPD data are summarized below in Table 24. An overlay of the dry and dry crystalline Compound I Hydrate Form A XRPD diffractograms is provided in FIG.21.
- Table 23 XRPD Signals for Crystalline Compound I Hydrate Form A No. Pos. [°2 ⁇ ] Area [cts*°2 ⁇ ] Rel. Int. [%] No. Pos. [°2 ⁇ ] Area [cts*°2 ⁇ ] Rel. Int. [%] 2 8.7 1573.0 56 [0576] Table ydrate Form A No. Pos. [°2 ⁇ ] Area [cts*°2 ⁇ ] Rel. Int. [%] 1 47 3764 20 No. Pos. [°2 ⁇ ] Area [cts*°2 ⁇ ] Rel. Int.
- the 19 F SSNMR spectrum for Compound I Hydrate Form A is provided in FIG.23, and the data are summarized below in Table 26.
- Table 26 19 F SSNMR Signals for Compound I Hydrate Form A Peak # Chemical Shift [ppm] Intensity [rel] Peak # Chemical Shift [ppm] Intensity [rel] 2 -60.2 10 3 1350 11 Solid-State 13 C NMR [0581] The 13 C SSNMR of Compound I Hydrate Form A was acquired using the procedure described in the General SSNMR Method and the 19 F I Hydrate Form A was acquired using the procedure described in the General SSNMR Method.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Pain & Pain Management (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Saccharide Compounds (AREA)
Abstract
Processes and methods of preparing Compound (I) are disclosed. Crystalline forms of Compound (I), pharmaceutically acceptable salts, solvates, hydrates, and cocrystals thereof, pharmaceutical compositions comprising the same, methods of pain using the same, and methods for making the same are also disclosed.
Description
METHODS OF PREPARING MODULATORS OF SODIUM CHANNELS AND SOLID FORMS OF THE SAME FOR TREATING PAIN CROSS-REFERENCE TO RELATED APPLICATIONS [0001] This application claims the benefit of U.S. Provisional Application No. 63/592,530, filed October 23, 2023, and U.S. Provisional Application No. 63/592,494, filed October 23, 2023, each of which is incorporated by reference herein in its entirety. TECHNICAL FIELD [0002] Disclosed herein are processes and methods of making compounds that inhibit a voltage- gated sodium channel (NaV). [0003] Also disclosed herein are processes and methods of inhibiting a voltage-gated sodium channel (NaV) and crystalline and amorphous solid forms of (Nav) inhibitors, pharmaceutical compositions thereof, methods of treating pain with any of the foregoing, and processes for making the crystalline and amorphous forms. BACKGROUND [0004] Pain is a protective mechanism that allows healthy animals to avoid tissue damage and to prevent further damage to injured tissue. Nonetheless, there are many conditions where pain persists beyond its usefulness, or where patients would benefit from inhibition of pain. Acute and chronic pain are two common pain states and can be distinguished by the duration of the pain. Acute pain can arise for many reasons (e.g., a hospital procedure) and treatment options are generally limited by poor efficacy and/or adverse events. Like acute pain, chronic pain can arise for many reasons and treatment options are limited by poor efficacy and/or adverse events. [0005] Neuropathic pain is a form of chronic pain caused by an injury to the sensory nerves (Dieleman, J.P., et al., Incidence rates and treatment of neuropathic pain conditions in the general population. Pain, 2008.137(3): p.681-8). Neuropathic pain can be divided into two categories, pain caused by generalized metabolic damage to the nerve and pain caused by a discrete nerve injury. The metabolic neuropathies include post-herpetic neuropathy, diabetic neuropathy, and drug-induced neuropathy. Discrete nerve injury indications include post-amputation pain, post-surgical nerve injury pain, and nerve entrapment injuries like neuropathic back pain. [0006] Voltage-gated sodium channels (NaVs) are involved in pain signaling. NaVs are biological mediators of electrical signaling as they mediate the rapid upstroke of the action potential of many excitable cell types, for example, neurons, skeletal myocytes, cardiac myocytes (Hille, Bertil, Ion Channels of Excitable Membranes, Third ed. (Sinauer Associates, Inc., Sunderland, MA, 2001)). The evidence for the role of these channels in normal physiology, the pathological states arising from mutations in sodium channel genes, preclinical work in animal models, and the clinical pharmacology
of known sodium channel modulating agents all point to the central role of NaVs in pain sensation (Rush, A.M. and T.R. Cummins, Painful Research: Identification of a Small-Molecule Inhibitor that Selectively Targets NaV1.8 Sodium Channels. Mol. Interv., 2007.7(4): p.192-5); England, S., Voltage- gated sodium channels: the search for subtype-selective analgesics. Expert Opin. Investig. Drugs 17 (12), p. 1849-64 (2008); Krafte, D. S. and Bannon, A. W., Sodium channels and nociception: recent concepts and therapeutic opportunities. Curr. Opin. Pharmacol.8 (1), p.50-56 (2008)). Because of the role NaVs play in the initiation and propagation of neuronal signals, antagonists that reduce NaV currents can prevent or reduce neural signaling and NaV channels have been considered likely targets to reduce pain in conditions where hyper-excitability is observed (Chahine, M., Chatelier, A., Babich, O., and Krupp, J. J., Voltage-gated sodium channels in neurological disorders. CNS Neurol. Disord. Drug Targets 7 (2), p.144-58 (2008)). Several clinically useful analgesics have been identified as inhibitors of NaV channels. The local anesthetic drugs such as lidocaine block pain by inhibiting NaV channels, and other compounds, such as carbamazepine, lamotrigine, and tricyclic antidepressants that have proven effective at reducing pain have also been suggested to act by sodium channel inhibition (Soderpalm, B., Anticonvulsants: aspects of their mechanisms of action. Eur. J. Pain 6 Suppl. A, p.3- 9 (2002); Wang, G. K., Mitchell, J., and Wang, S. Y., Block of persistent late Na+ currents by antidepressant sertraline and paroxetine. J. Membr. Biol.222 (2), p.79-90 (2008)). [0007] The NaVs form a subfamily of the voltage-gated ion channel super-family and comprises 9 isoforms, designated NaV1.1 – NaV1.9. The tissue localizations of the nine isoforms vary. NaV1.4 is the primary sodium channel of skeletal muscle, and NaV1.5 is primary sodium channel of cardiac myocytes. NaVs 1.7, 1.8 and 1.9 are primarily localized to the peripheral nervous system, while NaVs 1.1, 1.2, 1.3, and 1.6 are neuronal channels found in both the central and peripheral nervous systems. The functional behaviors of the nine isoforms are similar but distinct in the specifics of their voltage- dependent and kinetic behavior (Catterall, W. A., Goldin, A. L., and Waxman, S. G., International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol. Rev.57 (4), p.397 (2005)). [0008] Upon their discovery, NaV1.8 channels were identified as likely targets for analgesia (Akopian, A.N., L. Sivilotti, and J.N. Wood, A tetrodotoxin-resistant voltage-gated sodium channel expressed by sensory neurons. Nature, 1996.379(6562): p.257-62). Since then, NaV1.8 has been shown to be a carrier of the sodium current that maintains action potential firing in small dorsal root ganglia (DRG) neurons (Blair, N.T. and B.P. Bean, Roles of tetrodotoxin (TTX)-sensitive Na+ current, TTX- resistant Na+ current, and Ca2+ current in the action potentials of nociceptive sensory neurons. J. Neurosci., 2002.22(23): p.10277-90). NaV1.8 is involved in spontaneous firing in damaged neurons, like those that drive neuropathic pain (Roza, C., et al., The tetrodotoxin-resistant Na+ channel NaV1.8 is essential for the expression of spontaneous activity in damaged sensory axons of mice. J. Physiol., 2003.550(Pt 3): p.921-6; Jarvis, M.F., et al., A-803467, a potent and selective NaV1.8 sodium channel blocker, attenuates neuropathic and inflammatory pain in the rat. Proc. Natl. Acad. Sci. U S A, 2007.
104(20): p. 8520-5; Joshi, S.K., et al., Involvement of the TTX-resistant sodium channel NaV1.8 in inflammatory and neuropathic, but not post-operative, pain states. Pain, 2006.123(1-2): pp.75-82; Lai, J., et al., Inhibition of neuropathic pain by decreased expression of the tetrodotoxin-resistant sodium channel, NaV1.8. Pain, 2002.95(1-2): p.143-52; Dong, X.W., et al., Small interfering RNA-mediated selective knockdown of NaV1.8 tetrodotoxin-resistant sodium channel reverses mechanical allodynia in neuropathic rats. Neuroscience, 2007. 146(2): p. 812-21; Huang, H.L., et al., Proteomic profiling of neuromas reveals alterations in protein composition and local protein synthesis in hyper-excitable nerves. Mol. Pain, 2008.4: p. 33; Black, J.A., et al., Multiple sodium channel isoforms and mitogen- activated protein kinases are present in painful human neuromas. Ann. Neurol., 2008.64(6): p.644-53; Coward, K., et al., Immunolocalization of SNS/PN3 and NaN/SNS2 sodium channels in human pain states. Pain, 2000. 85(1-2): p. 41-50; Yiangou, Y., et al., SNS/PN3 and SNS2/NaN sodium channel- like immunoreactivity in human adult and neonate injured sensory nerves. FEBS Lett., 2000.467(2-3): p. 249-52; Ruangsri, S., et al., Relationship of axonal voltage-gated sodium channel 1.8 (NaV1.8) mRNA accumulation to sciatic nerve injury-induced painful neuropathy in rats. J. Biol. Chem.286(46): p. 39836-47). The small DRG neurons where NaV1.8 is expressed include the nociceptors involved (Blair, N.T. and B.P. Bean, Roles of tetrodotoxin (TTX)-sensitive Na+ current, TTX-resistant Na+ current, and Ca2+ current in the action potentials of nociceptive sensory neurons. J. Neurosci., 2002. 22(23): p.10277-90). NaV1.8 is necessary for rapid repetitive action potentials in nociceptors, and for spontaneous activity of damaged neurons. (Choi, J.S. and S.G. Waxman, Physiological interactions between NaV1.7 and NaV1.8 sodium channels: a computer simulation study. J. Neurophysiol. 106(6): p. 3173-84; Renganathan, M., T.R. Cummins, and S.G. Waxman, Contribution of Na(V)1.8 sodium channels to action potential electrogenesis in DRG neurons. J. Neurophysiol., 2001.86(2): p. 629-40; Roza, C., et al., The tetrodotoxin-resistant Na+ channel NaV1.8 is essential for the expression of spontaneous activity in damaged sensory axons of mice. J. Physiol., 2003. 550(Pt 3): p. 921-6). In depolarized or damaged DRG neurons, NaV1.8 appears to be a driver of hyper-excitablility (Rush, A.M., et al., A single sodium channel mutation produces hyper- or hypoexcitability in different types of neurons. Proc. Natl. Acad. Sci. USA, 2006.103(21): p.8245-50). In some animal pain models, NaV1.8 mRNA expression levels have been shown to increase in the DRG (Sun, W., et al., Reduced conduction failure of the main axon of polymodal nociceptive C-fibers contributes to painful diabetic neuropathy in rats. Brain, 135(Pt 2): p.359-75; Strickland, I.T., et al., Changes in the expression of NaV1.7, NaV1.8 and NaV1.9 in a distinct population of dorsal root ganglia innervating the rat knee joint in a model of chronic inflammatory joint pain. Eur. J. Pain, 2008. 12(5): p. 564-72; Qiu, F., et al., Increased expression of tetrodotoxin-resistant sodium channels NaV1.8 and NaV1.9 within dorsal root ganglia in a rat model of bone cancer pain. Neurosci. Lett., 512(2): p.61-6). [0009] The inventors have discovered that some voltage-gated sodium channel inhibitors have limitations as therapeutic agents due to, for example, a poor therapeutic window (e.g., due to a lack of NaV isoform selectivity, low potency, and/or other reasons). Accordingly, there remains a need to
develop selective voltage-gated sodium channel inhibitors, such as selective NaV1.8 inhibitors, and methods for making the same. [0010] Compound I, 2-[2-(3,4-difluoro-2-methyl-phenoxy)-4-methyl-5-(trifluoromethyl)-3- pyridyl]-4-oxo-1H-1,6-naphthyridine-5-carboxamide, is a Nav inhibitor and thus useful in treating pain. Compound I has the following structure: [0011] Compound I is No. PCT/US2023/019469.
 Crystalline forms are of interest where the control of the crystalline form(s) of the active ingredient may be desirable or even required. Reproducible processes for producing a compound with a particular crystalline form in high purity may be desirable for compounds intended to be used in pharmaceuticals, as different crystalline forms may possess different properties. For example, different crystalline forms may possess different chemical, physical, and/or pharmaceutical properties. In some embodiments, one or more crystalline forms disclosed herein may exhibit a higher level of purity, chemical stability, and/or physical stability compared to the forms produced in PCT US/2023/019469. Certain crystalline forms (e.g., crystalline free form, crystalline salt, crystalline salt solvate, and crystalline salt hydrate forms of Compound I (collectively referred to as “crystalline forms”)) may exhibit lower hygroscopicity than any preexisting forms. Thus, the crystalline forms of this disclosure may provide advantages during drug substance manufacturing, storage, and handling over the amorphous forms produced in PCT US/2023/019469. Thus, pharmaceutically acceptable crystalline forms of Compound I may be particularly useful for the production of drugs for the treatment of pain. [0012] Moreover, there also remains a need for more efficient processes for the synthesis of Compound I that delivers this compound, or pharmaceutically acceptable salts thereof, for example, in higher yield, with higher selectivity, and/or with higher purity relative to known processes.
SUMMARY [0013] In one aspect, the invention relates to a method of preparing a compound of formula I: I, or a solvate or a tautomer or a [0014] In some
Crystalline forms are of interest where the control of the crystalline form(s) of the active ingredient may be desirable or even required. Reproducible processes for producing a compound with a particular crystalline form in high purity may be desirable for compounds intended to be used in pharmaceuticals, as different crystalline forms may possess different properties. For example, different crystalline forms may possess different chemical, physical, and/or pharmaceutical properties. In some embodiments, one or more crystalline forms disclosed herein may exhibit a higher level of purity, chemical stability, and/or physical stability compared to the forms produced in PCT US/2023/019469. Certain crystalline forms (e.g., crystalline free form, crystalline salt, crystalline salt solvate, and crystalline salt hydrate forms of Compound I (collectively referred to as “crystalline forms”)) may exhibit lower hygroscopicity than any preexisting forms. Thus, the crystalline forms of this disclosure may provide advantages during drug substance manufacturing, storage, and handling over the amorphous forms produced in PCT US/2023/019469. Thus, pharmaceutically acceptable crystalline forms of Compound I may be particularly useful for the production of drugs for the treatment of pain. [0012] Moreover, there also remains a need for more efficient processes for the synthesis of Compound I that delivers this compound, or pharmaceutically acceptable salts thereof, for example, in higher yield, with higher selectivity, and/or with higher purity relative to known processes.
SUMMARY [0013] In one aspect, the invention relates to a method of preparing a compound of formula I: I, or a solvate or a tautomer or a [0014] In some
 any of compounds of formulae A1, A1-1, A1-2, A1-3, A1-4, A1-5, A2, A2-1, A3, A3-1, A3-2, B-1, B-2, B-3, B-4, B-5, C-1, C-2, C-3, C-4, D-1, D-2, E-A1, E-A2, and E-A3 to the compound of formula I or a solvate thereof or a tautomer or a pharmaceutically acceptable salt thereof following the reaction steps described herein. [0015] In another aspect, the invention relates to a compound selected from: ,
O NH O 2 , or a [0016]
any of compounds of formulae A1, A1-1, A1-2, A1-3, A1-4, A1-5, A2, A2-1, A3, A3-1, A3-2, B-1, B-2, B-3, B-4, B-5, C-1, C-2, C-3, C-4, D-1, D-2, E-A1, E-A2, and E-A3 to the compound of formula I or a solvate thereof or a tautomer or a pharmaceutically acceptable salt thereof following the reaction steps described herein. [0015] In another aspect, the invention relates to a compound selected from: ,
O NH O 2 , or a [0016]
 I, wherein the crystalline Compound I is selected from substantially pure compound I neat Form A, Compound I neat form B, Compound I neat form E, Compound I Acetone Solvate Hydrate Form A, Compound I Ethanol Solvate Form A, Compound I Hydrate Form A, and Compound I Dehydrated Hydrate Form A. [0017] In some embodiments, the disclosure relates to substantially amorphous Compound I amorphous form. BRIEF DESCRIPTION OF THE FIGURES [0018] Figure 1 provides the X-ray powder diffraction (XRPD) pattern of Compound I amorphous form. [0019] Figure 2 provides the 13C solid-state NMR (SSNMR) spectrum of Compound I amorphous form. [0020] Figure 3 provides an 19F NMR spectrum of Compound I amorphous form. [0021] Figure 4 provides the XRPD pattern of crystalline Compound I neat Form A. [0022] Figure 5 provides the 13C SSNMR spectrum of crystalline Compound I neat Form A. [0023] Figure 6 provides an 19F NMR spectrum of crystalline Compound I neat Form A. [0024] Figure 7 provides the XRPD pattern of crystalline Compound I neat Form B. [0025] Figure 8 provides the 13C SSNMR spectrum of crystalline Compound I neat Form B. [0026] Figure 9 provides an 19F NMR spectrum of crystalline Compound I neat Form B. [0027] Figure 10 provides the XRPD pattern of crystalline Compound I neat Form E. [0028] Figure 11 provides the 13C SSNMR spectrum of crystalline Compound I neat Form E. [0029] Figure 12 provides an 19F NMR spectrum of crystalline Compound I neat Form E. [0030] Figure 13 provides the XRPD pattern of crystalline Compound I Acetone Solvate Hydrate Form A. [0031] Figure 14 provides the 13C SSNMR spectrum of crystalline Compound I Acetone Solvate Hydrate Form A. [0032] Figure 15 provides an 19F NMR spectrum of crystalline Compound I Acetone Solvate Hydrate Form A.
[0033] Figure 16 provides the XRPD pattern of crystalline Compound I Ethanol Solvate Form A. [0034] Figure 17 provides the 13C SSNMR spectrum of crystalline Compound I Ethanol Solvate Form A. [0035] Figure 18 provides an 19F NMR spectrum of crystalline Compound I Ethanol Solvate Form A. [0036] Figure 19 provides the XRPD pattern of crystalline Compound I Hydrate Form A (wet form). [0037] Figure 20 provides the XRPD pattern of crystalline Compound I Dehydrated Hydrate Form A (dry form). [0038] Figure 21 provides the overlay of the XRPD patterns of crystalline Compound I Hydrate Form A with crystalline Compound I Dehydrated Hydrate Form A. The dry form is shown as a dashed line. [0039] Figure 22 provides the 13C SSNMR spectrum of crystalline Compound I Hydrate Form A. [0040] Figure 23 provides an 19F NMR spectrum of crystalline Compound I Hydrate Form A. DETAILED DESCRIPTION Definitions [0041] The following definitions are meant to clarify, but not limit, the terms defined. If a particular term used herein is not specifically defined, such term should not be considered indefinite. Rather, terms are used within their accepted meanings. [0042] “Compound I” as used throughout this disclosure refers to 2-[2-(3,4-difluoro-2-methyl- phenoxy)-4-methyl-5-(trifluoromethyl)-3-pyridyl]-4-oxo-1H-1,6-naphthyridine-5-carboxamide, and has the following structure: [0043] Compound I may be
I, wherein the crystalline Compound I is selected from substantially pure compound I neat Form A, Compound I neat form B, Compound I neat form E, Compound I Acetone Solvate Hydrate Form A, Compound I Ethanol Solvate Form A, Compound I Hydrate Form A, and Compound I Dehydrated Hydrate Form A. [0017] In some embodiments, the disclosure relates to substantially amorphous Compound I amorphous form. BRIEF DESCRIPTION OF THE FIGURES [0018] Figure 1 provides the X-ray powder diffraction (XRPD) pattern of Compound I amorphous form. [0019] Figure 2 provides the 13C solid-state NMR (SSNMR) spectrum of Compound I amorphous form. [0020] Figure 3 provides an 19F NMR spectrum of Compound I amorphous form. [0021] Figure 4 provides the XRPD pattern of crystalline Compound I neat Form A. [0022] Figure 5 provides the 13C SSNMR spectrum of crystalline Compound I neat Form A. [0023] Figure 6 provides an 19F NMR spectrum of crystalline Compound I neat Form A. [0024] Figure 7 provides the XRPD pattern of crystalline Compound I neat Form B. [0025] Figure 8 provides the 13C SSNMR spectrum of crystalline Compound I neat Form B. [0026] Figure 9 provides an 19F NMR spectrum of crystalline Compound I neat Form B. [0027] Figure 10 provides the XRPD pattern of crystalline Compound I neat Form E. [0028] Figure 11 provides the 13C SSNMR spectrum of crystalline Compound I neat Form E. [0029] Figure 12 provides an 19F NMR spectrum of crystalline Compound I neat Form E. [0030] Figure 13 provides the XRPD pattern of crystalline Compound I Acetone Solvate Hydrate Form A. [0031] Figure 14 provides the 13C SSNMR spectrum of crystalline Compound I Acetone Solvate Hydrate Form A. [0032] Figure 15 provides an 19F NMR spectrum of crystalline Compound I Acetone Solvate Hydrate Form A.
[0033] Figure 16 provides the XRPD pattern of crystalline Compound I Ethanol Solvate Form A. [0034] Figure 17 provides the 13C SSNMR spectrum of crystalline Compound I Ethanol Solvate Form A. [0035] Figure 18 provides an 19F NMR spectrum of crystalline Compound I Ethanol Solvate Form A. [0036] Figure 19 provides the XRPD pattern of crystalline Compound I Hydrate Form A (wet form). [0037] Figure 20 provides the XRPD pattern of crystalline Compound I Dehydrated Hydrate Form A (dry form). [0038] Figure 21 provides the overlay of the XRPD patterns of crystalline Compound I Hydrate Form A with crystalline Compound I Dehydrated Hydrate Form A. The dry form is shown as a dashed line. [0039] Figure 22 provides the 13C SSNMR spectrum of crystalline Compound I Hydrate Form A. [0040] Figure 23 provides an 19F NMR spectrum of crystalline Compound I Hydrate Form A. DETAILED DESCRIPTION Definitions [0041] The following definitions are meant to clarify, but not limit, the terms defined. If a particular term used herein is not specifically defined, such term should not be considered indefinite. Rather, terms are used within their accepted meanings. [0042] “Compound I” as used throughout this disclosure refers to 2-[2-(3,4-difluoro-2-methyl- phenoxy)-4-methyl-5-(trifluoromethyl)-3-pyridyl]-4-oxo-1H-1,6-naphthyridine-5-carboxamide, and has the following structure: [0043] Compound I may be
 acceptable salt, solvate, and/or hydrate. Compound I and methods for making and using Compound I, tautomers of Compound I, stereoisomers of Compound I, deuterated derivatives of Compound I and its tautomers and stereoisomers, and pharmaceutically acceptable salts of any of the foregoing are disclosed in PCT/US2023/019469, incorporated herein by reference.
[0044] As used herein, the term “compound,” when referring to the compounds described in this application, refers to a collection of molecules having identical chemical structures, except that there may be isotopic variation among the constituent atoms of the molecules. The term “compound” includes such a collection of molecules without regard to the purity of a given sample containing the collection of molecules. Thus, the term “compound” includes such a collection of molecules in pure form, in a mixture (e.g., solution, suspension, colloid, or pharmaceutical composition, or dosage form) with one or more other substances, or in the form of a hydrate, solvate, or co-crystal. [0045] In the specification and claims, unless otherwise specified, any atom not specifically designated as a particular isotope in any compound of the invention is meant to represent any stable isotope of the specified element. In the Examples, where an atom is not specifically designated as a particular isotope in any compound of the invention, no effort was made to enrich that atom in a particular isotope, and therefore a person of ordinary skill in the art would understand that such atom likely was present at approximately the natural abundance isotopic composition of the specified element. [0046] As used herein in the specification and claims, “H” refers to hydrogen and includes any stable isotope of hydrogen, namely 1H and D. [0047] In some embodiments, the compounds described in the present application include each constituent atom at approximately the natural abundance isotopic composition of the specified element. [0048] In some embodiments, the compounds described in the present application, and pharmaceutically acceptable salts thereof, include one or more atoms having an atomic mass or mass number which differs from the atomic mass or mass number of the most abundant isotope of the specified element (“isotope-labeled” compounds and salts). Examples of stable isotopes which are commercially available and suitable for the invention include without limitation isotopes of hydrogen, carbon, nitrogen, oxygen, and phosphorus, for example 2H, 13C, 15N, 18O, 17O, and 31P, respectively. For example, as used in the specification and claims, “H” refers to hydrogen and includes any stable isotope of hydrogen, namely 1H and D. [0049] The term the “compound of formula” followed by a number (typically Roman number) and the term “compound” followed by the same number (Roman or otherwise) may interchangeably be used. For example, the “compound of formula V” and “compound V” denote the same compound. [0050] The term “reacting,” when referring to a chemical reaction, means to add or mix two or more reagents under appropriate conditions to produce the indicated and/or the desired product. It should be appreciated that the reaction which produces the indicated and/or the desired product may not necessarily result directly from the combination of two reagents which were initially added, i.e., there may be one or more intermediates which are produced in the mixture which ultimately leads to the formation of the indicated and/or the desired product.
[0051] The term “conducted in a solvent,” when referring to a reaction, means that the substrate(s) and reagent(s) are dissolved or suspended in the specified solvent or in a mixture of solvents comprising the specified solvent. [0052] The term “chromatographic purification” refers to any method of purification based on differential retention by a stationary phase. Methods of chromatographic purification include flash chromatography, medium pressure liquid chromatography, preparative thin layer chromatography, and high performance liquid chromatography. [0053] The terms “converting” and “transforming” as used herein refer to a step of converting a first compound or salt to a second compound or salt, and refers to a process of transforming the first compound or salt to the second compound or salt in one or more chemical steps. [0054] The term “acid” refers to a chemical species having a pKa (in water) of less than 7. The term includes inorganic (mineral) acids, such as hydrochloric acid, hydrobromic acid, hydroiodic acid, sulfuric acid, phosphoric acid, nitric acid, and the like. The term also includes organic acids such as acetic acid, propionic acid, n-butyric acid, i-butyric acid, n-valeric acid, i-valeric acid, n-hexanoic acid, succinic acid, glutaric acid, adipic acid, aspartic acid, formic acid, citric acid, o-chlorobenzoic acid, chloroacetic acid, dichloroacetic acid, trichloroacetic acid, nicotinic acid, lactic acid, oxalic acid, picric acid, picolinic acid, fluoroacetic acid, difluoroacetic acid, trifluoroacetic acid, phthalic acid, isophthalic acid, terephthalic acid, maleic acid, malonic acid, and the like. [0055] The term “base” refers to a chemical species whose conjugate acid has a pKa (in water) of greater than 7. The term includes “inorganic bases,” such as sodium hydroxide, potassium hydroxide, sodium bicarbonate, potassium bicarbonate, sodium carbonate, potassium carbonate, cesium carbonate, potassium phosphate (mono-, di-, or tri-basic), sodium hydride, and potassium hydride. The term also includes “anionic organic bases,” such as methyl lithium, butyl lithium, lithium diisopropyl amide, and sodium acetate. The term also includes “neutral organic bases,” such as trimethylamine, dimethylethylamine, diethylmethylamine, triethylamine, di-n-propylmethylamine, dimethylcyclohexylamine, diisopropylethylamine, tri-n-propylamine, diisopropylisobutylamine, dimethyl-n-nonylamine, tri-n-butylamine, di-n-hexylmethylamine, dimethyl-n-dodecylamine, tri-n-pentylamine, 1,4-diazabicyclo[2.2.2] octane (DABCO), dimethylaminopyridine (DMAP), 1,5-diazabicyclo[4.3.0] non-5-ene (DBN), 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), pyridine, 2,3-lutidine, 2,4-lutidine, 2,5-lutidine, 2,6-lutidine, 3,4-lutidine, 3,5-lutidine, 2,3,4-collidine, 2,4,5-collidine, 2,5,6-collidine, 2,4,6-collidine, 3,4,5-collidine, and 3,5,6-collidine. [0056] The term “alcohol protecting group” refers to a chemical moiety suitable to protect an alcohol group against undesirable side reactions during synthetic procedures. Common alcohol protecting groups include methyl, ethyl, isopropyl, benzyl, 2-tetrahydropyranyl, acetyl, trifluoroacetyl, trialkylsilyl, aryldialkylsilyl, alkyldiarylsilyl, or triarylsilyl. Other alcohol protecting groups also are well known in the art. See, e.g., P.G.M. Wuts et al., Greene’s Protective Groups in Organic Synthesis (4th ed.2006).
[0057] The term “deprotecting” refers to a step of reacting a compound or salt containing a protecting group, such as an alcohol protecting group, under conditions suitable to remove the protecting group and reveal the protected moiety. For example, where a compound or salt contains an alcohol protecting group, the term “deprotecting” refers to reacting the compound or salt under conditions suitable to remove the alcohol protecting group and reveal the alcohol. Conditions for removing various protecting groups are well known in the art. See, e.g., P.G.M. Wuts et al., Greene’s Protective Groups in Organic Synthesis (4th ed.2006). [0058] The term “hydrogenation catalyst” refers to any homogeneous or heterogeneous catalyst that catalyzes the hydrogenolysis of benzylic carbon-oxygen single bonds. Suitable hydrogenation catalysts are well-known in the art and include, for example, palladium on activated carbon, platinum oxide, and Raney Nickel. [0059] The term “coupling,” when referring to a reaction between a carboxylic acid or acid halide and an amine, refers to a net transformation linking the carboxylic acid or acid halide and the amine to form an amide. The term includes a direct reaction between the carboxylic acid and the amine, as well as a reaction between an activated derivative of the carboxylic acid (such as the derivative formed by the reaction between the carboxylic acid and a coupling reagent) and the amine. [0060] The term “coupling reagent” refers to a reagent suitable to react with a carboxylic acid to activate the carboxylic acid for coupling with an amine to form an amide bond. Coupling reagents are well known in the art. Coupling reagents include, but are not limited to, thionyl chloride, oxalyl chloride, 1,1'-carbonylbis-(4,5-dicyanoimidazole) (CBDCI), 1,1'- carbonyldiimidazole (CDI), propylphosphonic anhydride (T3P), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI), N,N’-dicyclohexylcarbodiimide (DCC), 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5- b]pyridinium 3-oxid hexafluorophosphate (HATU), and 1-hydroxybenzotriazole (HOBt). [0061] The term “monovalent cation” refers to any cation with a charge of +1, such as alkali metal cations, NH4 +, and tetraalkylammonium. [0062] The term “alkali metal cation” refers to a cation derived from a Group I metal atom, including without limitation lithium (Li+), sodium (Na+), potassium (K+), rubidium (Rb+), and cesium (Cs+). [0063] The term “substituted benzyl” refers to a benzyl group that is substituted with 1-3 substituents selected from the group consisting of C1-C3 alkyl, C1-C3 alkoxy, halogen, and cyano. [0064] The term “ketone solvent” refers to a compound having the formula CnH2n+1C(O)CmH2m+1, wherein n and m are each independently an integer between 1 and 6. The CnH2n+1 and CmH2m+1 and groups may be linear or branched and each may be substituted with up to 3 halogens. Ketone solvents include without limitation acetone, methyl ethyl ketone, 3-pentanone, and methyl tert-butyl ketone. [0065] The term “ethereal solvent” refers to an organic solvent having at least one ether moiety. Ethereal solvents include without limitation tetrahydofuran, dimethoxyethane, dioxane, and dialkyl ethers such as diethyl ether and methyl isobutyl ether.
[0066] The term “ester solvent” refers to a compound having the formula CnH2n+1OC(O)CmH2m+1, wherein n and m are each independently an integer between 1 and 6. The CnH2n+1 and CmH2m+1 and groups may be linear or branched and each may be substituted with up to 3 halogens. Ester solvents include without limitation ethyl acetate, isopropyl acetate, butyl acetate, and ethylpropionate. [0067] The term “halogenated solvent” refers to a C1-C6 alkane or C2-C6 alkene substituted with up to six halogens. Halogenated solvents include without limitation dichloromethane, dichloroethane, chloroform, tetrachloroethylene, and carbon tetrachloride. [0068] The term “aromatic solvent” refers to a C6-10 aromatic hydrocarbon. The aromatic hydrocarbon may be substituted with up to six halogens. Aromatic solvents include without limitation benzene, trifluoromethylbenzene, xylene, and toluene. [0069] The term “about” means that the stated number can vary from that value by ±10%. Where the term defines a temperature, the stated temperature can vary by ±10%. For example, about 80ºC means between 72ºC and 88ºC. Where the term defines pressure, the term “about” means the pressure can vary by ±10%. Thus, about 100 bars means between 90 and 110 bars. Where the term defines quantity (such as equivalents or weight), the term means the quantity can vary by ±10%. For example, about 1 equivalent means between 0.9 and 1.1 equivalents. Where the term defines time, the term means the stated time can vary by ±10%. For example, about 1 hour means between 0.9 and 1.1 hours. [0070] The term “leaving group” is a chemical group that is readily displaced by a desired incoming chemical moiety. Thus, the choice of the specific suitable leaving group is predicated upon its ability to be readily displaced by the incoming chemical moiety such as a CN group. Suitable leaving groups are well known in the art, e.g., see, “Advanced Organic Chemistry,” Jerry March, 5.sup.th Ed., pp.351-357, John Wiley and Sons, N.Y. [0071] As used here, the term “cyanating agent” refers to a reagent, such as trimethylsilyl cyanide (TMSCN), diethylaluminum cyanide, KCN, NaCN, TBACN, HCN and the like, useful in the synthesis of compounds disclosed herein, e.g., compound A1, A2, and A3. In one embodiment, the cyanating agent (e.g., trimethylsilyl cyanide) may be combined with a Lewis acid. In some embodiments, the Lewis acid is trifluoromethanesulfonic anhydride (Tf2O), boron trifluoride ethyl etherate (BF3OEt2), TiCl4, InCl3, AgSbF6, iodine, ZnBr2, Al(OiPr)3, MgCl2, Mn(acac)2, MnCl2, TMSOTf, SnCl4, and the like. In further embodiments, the Lewis acid is trifluoromethanesulfonic anhydride (Tf2O). The cyanation reaction may be conducted in an organic solvent, for example toluene, dichloromethane, 2-methyl THF, acetonitrile, methanol, 1,2-dichloroethane, nitromethane, and the like. [0072] As used herein, the term "active pharmaceutical ingredient" ("API") or "therapeutic agent" refers to a biologically active compound. [0073] The terms "patient" and "subject" are used interchangeably and refer to an animal including humans. [0074] The terms "effective dose" and "effective amount" are used interchangeably herein and refer to that amount of a compound that produces the desired effect for which it is administered (e.g.,
improvement in pain or a symptom of pain, or lessening the severity of pain or a symptom of pain). The exact amount of an effective dose will depend on the purpose of the treatment and will be ascertainable by one skilled in the art using known techniques (see, e.g., Lloyd (1999) The Art, Science and Technology of Pharmaceutical Compounding). [0075] As used herein, the terms "treatment," "treating," and the like generally mean the improvement of pain or one or more of its symptoms or lessening the severity of pain or one or more of its symptoms in a subject. "Treatment," as used herein, includes, but is not limited to, the following: Chronic pain, gut pain, neuropathic pain, musculoskeletal pain, acute pain, inflammatory pain, cancer pain, idiopathic pain, postsurgical pain (e.g., bunionectomy pain, herniorrhaphy pain or abdominoplasty pain), visceral pain, multiple sclerosis, Charcot-Marie-Tooth syndrome, incontinence, pathological cough, or cardiac arrhythmia. Improvements in or lessening the severity of any of these symptoms can be readily assessed according to standard methods and techniques known in the art. [0076] As used herein, the term "in combination with," when referring to two or more compounds, agents, or additional active pharmaceutical ingredients, means the administration of two or more compounds, agents, or active pharmaceutical ingredients to the patient prior to, concurrently with, or subsequent to each other. [0077] "Selected from" and "chosen from" are used interchangeably herein. [0078] As used herein, the term "ambient conditions" means room temperature, open air condition and uncontrolled humidity condition. As used herein, the terms "room temperature" and "ambient temperature" mean 15 °C to 30 °C. [0079] As used herein, the term "solvent" refers to any liquid in which the product is at least partially soluble (solubility of product > 1 g/L). [0080] The term "stable," as used herein, refers to compounds or solid forms that are not substantially altered when subjected to conditions to allow for their production, detection, and preferably their recovery, purification, and use for one or more of the purposes disclosed herein. [0081] The term "chemically stable," as used herein, means that the solid form of Compound I does not decompose into one or more different chemical compounds when subjected to specified conditions, e.g., 40 °C/75% relative humidity, for a specific period of time, e.g., 1 day, 2 days, 3 days, 1 week, 2 weeks, or longer. In some embodiments, less than 25% of the solid form of Compound I decomposes. In some embodiments, less than about 20%, less than about 15%, less than about 10%, less than about 5%, less than about 3%, less than about 1 %, less than about 0.5% of the form of Compound I decomposes under the conditions specified. In some embodiments, no detectable amount of the solid form of Compound I decomposes. [0082] The term "physically stable," as used herein, means that the solid form of Compound I does not change into one or more different physical forms of Compound I (e.g., different solid forms as measured by XRPD, DSC, etc.) when subjected to specific conditions, e.g., 40 °C/75 % relative humidity, for a specific period of time, e.g, 1 day, 2 days, 3 days, 1 week, 2 weeks, or longer. In some
embodiments, less than 25% of the solid form of Compound I changes into one or more different physical forms when subjected to specified conditions. In some embodiments, less than about 20%, less than about 15%, less than about 10%, less than about 5%, less than about 3%, less than about 1 %, less than about 0.5% of the solid form of Compound I changes into one or more different physical forms of Compound I when subjected to specified conditions. In some embodiments, no detectable amount of the solid form of Compound I changes into one or more physically different solid forms of Compound I. [0083] As used herein, the term "hydrate" refers to any crystalline Compound I that contains water in its crystal lattice. The stoichiometry of a Compound I hydrate can vary. For example, a hydrate of Compound I can be a quarter hydrate, hemihydrate, monohydrate, dihydrate, or a partially dehydrated form. [0084] As used herein, the term "pharmaceutically acceptable solid form" refers to a solid form of Compound I of this disclosure wherein the solid form (e.g., crystalline free form, crystalline salt, crystalline salt solvate, crystalline salt hydrate, and amorphous form) of Compound I is nontoxic and suitable for use in pharmaceutical compositions. [0085] As used herein, the term "amorphous" refers to a solid material having no long-range order in the position of its molecules. Amorphous solids are generally rather isotropic, i.e., exhibit similar properties in all directions. Amorphous solids do not have definite melting points. Amorphous solids are generally glasses or supercooled liquids in which the molecules are arranged in a random manner so that there is no well-defined arrangement, e.g., molecular packing, and no long-range order. For example, an amorphous material is a solid material having no sharp characteristic crystalline peak(s) in its X-ray power diffraction (XRPD) pattern (i.e., is not crystalline as determined by XRPD). Instead, one or several broad peaks (e.g., halos) appear in its XRPD pattern. Broad peaks are characteristic of an amorphous solid. See US 2004/0006237 for a comparison of XRPDs of an amorphous material and crystalline material. In some embodiments, a solid material may comprise an amorphous compound, and the material may, for example, be characterized by a lack of sharp characteristic crystalline peak(s) in its XRPD spectrum (i.e., the material is not crystalline, but is amorphous, as determined by XRPD). Instead, one or several broad peaks (e.g., halos) may appear in the XRPD pattern of the material. See US 2004/0006237 for a comparison of XRPDs of an amorphous material and crystalline material. A solid material, comprising an amorphous compound, may be characterized by, for example, a wider temperature range for the melting of the solid material, as compared to the range for the melting of a pure crystalline solid. Other techniques, such as, for example, solid state NMR may also be used to characterize crystalline or amorphous forms. [0086] As used herein, the terms "crystal form," "crystalline form," and "Form" interchangeably refer to a crystal structure (or polymorph) having a particular molecular packing arrangement in the crystal lattice. Crystalline forms can be identified and distinguished from each other by one or more characterization techniques including, for example, X-ray powder diffraction (XRPD), single crystal
X-ray diffraction, and 13C solid state nuclear magnetic resonance (13C SSNMR). Accordingly, as used herein, the terms "crystalline Form [X] of Compound (I)" and "crystalline Form [C] potassium salt of Compound (I)" refer to unique crystalline forms that can be identified and distinguished from each other by one or more characterization techniques including, for example, XRPD, single crystal X-ray diffraction, and 13C SSNMR In some embodiments, the novel crystalline forms are characterized by an X-ray powder diffractogram having one or more signals at one or more specified degree two-theta values (°2θ). [0087] As used herein, the term "free form" refers to a non-ionized version of the compound in the solid state. Examples of free forms include free bases and free acids. [0088] As used herein, the term "neat form" refers to an unsolvated and unhydrated free form version of a compound in the solid state. [0089] As used herein, the term "solvate" refers to a crystal form comprising one or more molecules of a compound of the present disclosure and, incorporated into the crystal lattice, one or more molecules of a solvent or solvents in stoichiometric or nonstoichiometric amounts. When the solvent is water, the solvate is referred to as a "hydrate." [0090] As used herein, the term "hydrate" refers to any crystalline Compound I that contains water in its crystal lattice. The stoichiometry of a Compound I hydrate can vary, i.e., Compound I can be a variable hydrate. For example, a hydrate of Compound I can be a quarter hydrate, hemihydrate, monohydrate, dihydrate, or a partially dehydrated form. [0091] In some embodiments, a solid material may comprise a mixture of crystalline solids and amorphous solids. A solid material comprising an amorphous compound may also, for example, contain up to 30% of a crystalline solid. In some embodiments, a solid material prepared to comprise an amorphous compound may also, for example, contain up to 25%, 20%, 15%, 10%, 5%, or 2% of a crystalline solid. In embodiments wherein the solid material contains a mixture of crystalline solids and amorphous solids, the characterizing data, such as XRPD, may contain indicators of both crystalline and amorphous solids. In some embodiments, a crystalline form of this disclosure may contain up to 30% amorphous compound. In some embodiments, a crystalline preparation of Compound I may contain up to 25%, 20%, 15%, 10%, 5%, or 2% of an amorphous solid. [0092] As used herein, the term "substantially amorphous" refers to a solid material having little or no long-range order in the position of its molecules. For example, substantially amorphous materials have less than 15% crystallinity (e.g., less than 10% crystallinity, less than 5% crystallinity, or less than 2% crystallinity). It is also noted that the term "substantially amorphous" includes the descriptor, "amorphous," which refers to materials having no (0%) crystallinity. [0093] As used herein, the term "substantially crystalline" refers to a solid material having little or no amorphous molecules. For example, substantially crystalline materials have less than 15% amorphous molecules (e.g., less than 10% amorphous molecules, less than 5% amorphous molecules,
or less than 2% amorphous molecules). It is also noted that the term "substantially crystalline" includes the descriptor "crystalline," which refers to materials that are 100% crystalline form. [0094] As used herein, a crystalline form is "substantially pure" when it accounts for an amount by weight equal to or greater than 90% of the sum of all solid form(s) in a sample as determined by a method in accordance with the art, such as quantitative XRPD. In some embodiments, the solid form is "substantially pure" when it accounts for an amount by weight equal to or greater than 95% of the sum of all solid form(s) in a sample. In some embodiments, the solid form is "substantially pure" when it accounts for an amount by weight equal to or greater than 99% of the sum of all solid form(s) in a sample. [0095] As used herein, the terms "X-ray powder diffractogram," "X-ray powder diffraction pattern," "XRPD pattern," "XRPD spectrum" interchangeably refer to an experimentally obtained pattern plotting signal positions (on the abscissa) versus signal intensities (on the ordinate). [0096] A "signal" or "peak" as used herein refers to a point in the XRPD pattern where the intensity as measured in counts is at a local maximum. An XRPD peak is identified by its angular value as measured in degrees 2θ (°2θ), depicted on the abscissa of an X-ray powder diffractogram, which may be expressed, for example, as "a signal at ... degrees two-theta," "a signal at [a] two-theta value(s) of ... " and/or "a signal at at least ... two-theta value(s) selected from .... " [0097] The repeatability of the measured angular values is in the range of ±0.2° 2θ, i.e., the angular value can be at the recited angular value +0.2 degrees two-theta, the angular value -0.2 degrees two-theta, or any value between those two end points (angular value +0.2 degrees two-theta and angular value -0.2 degrees two-theta). [0098] One of ordinary skill in the art would recognize that one or more signals (or peaks) in an XRPD pattern may overlap and may, for example, not be apparent to the naked eye. Indeed, one of ordinary skill in the art would recognize that some art-recognized methods are capable of and suitable for determining whether a signal exists in a pattern, such as Rietveld refinement. [0099] The terms "signal intensities" and "peak intensities" interchangeably refer to relative signal intensities within a given X-ray powder diffractogram. Factors that can affect the relative signal or peak intensities include sample thickness and preferred orientation (e.g., the crystalline particles are not distributed randomly). [0100] As used herein, an X-ray powder diffractogram is "substantially similar to that in [a particular] Figure" when at least 90%, such as at least 95%, at least 98%, or at least 99%, of the signals in the two diffractograms overlap. In determining "substantial similarity," one of ordinary skill in the art will understand that there may be variation in the intensities and/or signal positions in XRPD diffractograms even for the same crystalline form. Thus, those of ordinary skill in the art will understand that the signal maximum values in XRPD diffractograms (in degrees two-theta) generally mean that value is identified as ±0.2 degrees two-theta of the reported value, an art-recognized variance.
[0101] As used herein, the term "glass transition temperature" or "Tg" refers to the temperature above which a hard and brittle "glassy" amorphous solid becomes viscous or rubbery supercooled liquid. [0102] As used herein, the term "melting temperature", "melting point", or "Tm" refers to the temperature at which a crystalline material is in equilibrium with liquid phase. [0103] As used herein, the term "dispersion" refers to a disperse system in which one substance, the dispersed phase, is distributed, in discrete units, throughout a second substance (the continuous phase or vehicle). The size of the dispersed phase can vary considerably (e.g., colloidal particles of nanometer dimension, to multiple microns in size). In general, the dispersed phases can be solids, liquids, or gases. In the case of a solid dispersion, the dispersed and continuous phases are both solids. In pharmaceutical applications, a solid dispersion can include a crystalline drug (dispersed phase) in an amorphous polymer (continuous phase); or alternatively, an amorphous drug (dispersed phase) in an amorphous polymer (continuous phase). In some embodiments, a solid dispersion includes the polymer constituting the dispersed phase, and the drug constitute the continuous phase or, a solid dispersion includes the drug constituting the dispersed phase, and the polymer constituting the continuous phase. Detailed Description of Embodiments [0104] One aspect of the disclosure provides a method of preparing a compound of formula I: I, or a solvate or a tautomer or a
acceptable salt, solvate, and/or hydrate. Compound I and methods for making and using Compound I, tautomers of Compound I, stereoisomers of Compound I, deuterated derivatives of Compound I and its tautomers and stereoisomers, and pharmaceutically acceptable salts of any of the foregoing are disclosed in PCT/US2023/019469, incorporated herein by reference.
[0044] As used herein, the term “compound,” when referring to the compounds described in this application, refers to a collection of molecules having identical chemical structures, except that there may be isotopic variation among the constituent atoms of the molecules. The term “compound” includes such a collection of molecules without regard to the purity of a given sample containing the collection of molecules. Thus, the term “compound” includes such a collection of molecules in pure form, in a mixture (e.g., solution, suspension, colloid, or pharmaceutical composition, or dosage form) with one or more other substances, or in the form of a hydrate, solvate, or co-crystal. [0045] In the specification and claims, unless otherwise specified, any atom not specifically designated as a particular isotope in any compound of the invention is meant to represent any stable isotope of the specified element. In the Examples, where an atom is not specifically designated as a particular isotope in any compound of the invention, no effort was made to enrich that atom in a particular isotope, and therefore a person of ordinary skill in the art would understand that such atom likely was present at approximately the natural abundance isotopic composition of the specified element. [0046] As used herein in the specification and claims, “H” refers to hydrogen and includes any stable isotope of hydrogen, namely 1H and D. [0047] In some embodiments, the compounds described in the present application include each constituent atom at approximately the natural abundance isotopic composition of the specified element. [0048] In some embodiments, the compounds described in the present application, and pharmaceutically acceptable salts thereof, include one or more atoms having an atomic mass or mass number which differs from the atomic mass or mass number of the most abundant isotope of the specified element (“isotope-labeled” compounds and salts). Examples of stable isotopes which are commercially available and suitable for the invention include without limitation isotopes of hydrogen, carbon, nitrogen, oxygen, and phosphorus, for example 2H, 13C, 15N, 18O, 17O, and 31P, respectively. For example, as used in the specification and claims, “H” refers to hydrogen and includes any stable isotope of hydrogen, namely 1H and D. [0049] The term the “compound of formula” followed by a number (typically Roman number) and the term “compound” followed by the same number (Roman or otherwise) may interchangeably be used. For example, the “compound of formula V” and “compound V” denote the same compound. [0050] The term “reacting,” when referring to a chemical reaction, means to add or mix two or more reagents under appropriate conditions to produce the indicated and/or the desired product. It should be appreciated that the reaction which produces the indicated and/or the desired product may not necessarily result directly from the combination of two reagents which were initially added, i.e., there may be one or more intermediates which are produced in the mixture which ultimately leads to the formation of the indicated and/or the desired product.
[0051] The term “conducted in a solvent,” when referring to a reaction, means that the substrate(s) and reagent(s) are dissolved or suspended in the specified solvent or in a mixture of solvents comprising the specified solvent. [0052] The term “chromatographic purification” refers to any method of purification based on differential retention by a stationary phase. Methods of chromatographic purification include flash chromatography, medium pressure liquid chromatography, preparative thin layer chromatography, and high performance liquid chromatography. [0053] The terms “converting” and “transforming” as used herein refer to a step of converting a first compound or salt to a second compound or salt, and refers to a process of transforming the first compound or salt to the second compound or salt in one or more chemical steps. [0054] The term “acid” refers to a chemical species having a pKa (in water) of less than 7. The term includes inorganic (mineral) acids, such as hydrochloric acid, hydrobromic acid, hydroiodic acid, sulfuric acid, phosphoric acid, nitric acid, and the like. The term also includes organic acids such as acetic acid, propionic acid, n-butyric acid, i-butyric acid, n-valeric acid, i-valeric acid, n-hexanoic acid, succinic acid, glutaric acid, adipic acid, aspartic acid, formic acid, citric acid, o-chlorobenzoic acid, chloroacetic acid, dichloroacetic acid, trichloroacetic acid, nicotinic acid, lactic acid, oxalic acid, picric acid, picolinic acid, fluoroacetic acid, difluoroacetic acid, trifluoroacetic acid, phthalic acid, isophthalic acid, terephthalic acid, maleic acid, malonic acid, and the like. [0055] The term “base” refers to a chemical species whose conjugate acid has a pKa (in water) of greater than 7. The term includes “inorganic bases,” such as sodium hydroxide, potassium hydroxide, sodium bicarbonate, potassium bicarbonate, sodium carbonate, potassium carbonate, cesium carbonate, potassium phosphate (mono-, di-, or tri-basic), sodium hydride, and potassium hydride. The term also includes “anionic organic bases,” such as methyl lithium, butyl lithium, lithium diisopropyl amide, and sodium acetate. The term also includes “neutral organic bases,” such as trimethylamine, dimethylethylamine, diethylmethylamine, triethylamine, di-n-propylmethylamine, dimethylcyclohexylamine, diisopropylethylamine, tri-n-propylamine, diisopropylisobutylamine, dimethyl-n-nonylamine, tri-n-butylamine, di-n-hexylmethylamine, dimethyl-n-dodecylamine, tri-n-pentylamine, 1,4-diazabicyclo[2.2.2] octane (DABCO), dimethylaminopyridine (DMAP), 1,5-diazabicyclo[4.3.0] non-5-ene (DBN), 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), pyridine, 2,3-lutidine, 2,4-lutidine, 2,5-lutidine, 2,6-lutidine, 3,4-lutidine, 3,5-lutidine, 2,3,4-collidine, 2,4,5-collidine, 2,5,6-collidine, 2,4,6-collidine, 3,4,5-collidine, and 3,5,6-collidine. [0056] The term “alcohol protecting group” refers to a chemical moiety suitable to protect an alcohol group against undesirable side reactions during synthetic procedures. Common alcohol protecting groups include methyl, ethyl, isopropyl, benzyl, 2-tetrahydropyranyl, acetyl, trifluoroacetyl, trialkylsilyl, aryldialkylsilyl, alkyldiarylsilyl, or triarylsilyl. Other alcohol protecting groups also are well known in the art. See, e.g., P.G.M. Wuts et al., Greene’s Protective Groups in Organic Synthesis (4th ed.2006).
[0057] The term “deprotecting” refers to a step of reacting a compound or salt containing a protecting group, such as an alcohol protecting group, under conditions suitable to remove the protecting group and reveal the protected moiety. For example, where a compound or salt contains an alcohol protecting group, the term “deprotecting” refers to reacting the compound or salt under conditions suitable to remove the alcohol protecting group and reveal the alcohol. Conditions for removing various protecting groups are well known in the art. See, e.g., P.G.M. Wuts et al., Greene’s Protective Groups in Organic Synthesis (4th ed.2006). [0058] The term “hydrogenation catalyst” refers to any homogeneous or heterogeneous catalyst that catalyzes the hydrogenolysis of benzylic carbon-oxygen single bonds. Suitable hydrogenation catalysts are well-known in the art and include, for example, palladium on activated carbon, platinum oxide, and Raney Nickel. [0059] The term “coupling,” when referring to a reaction between a carboxylic acid or acid halide and an amine, refers to a net transformation linking the carboxylic acid or acid halide and the amine to form an amide. The term includes a direct reaction between the carboxylic acid and the amine, as well as a reaction between an activated derivative of the carboxylic acid (such as the derivative formed by the reaction between the carboxylic acid and a coupling reagent) and the amine. [0060] The term “coupling reagent” refers to a reagent suitable to react with a carboxylic acid to activate the carboxylic acid for coupling with an amine to form an amide bond. Coupling reagents are well known in the art. Coupling reagents include, but are not limited to, thionyl chloride, oxalyl chloride, 1,1'-carbonylbis-(4,5-dicyanoimidazole) (CBDCI), 1,1'- carbonyldiimidazole (CDI), propylphosphonic anhydride (T3P), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI), N,N’-dicyclohexylcarbodiimide (DCC), 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5- b]pyridinium 3-oxid hexafluorophosphate (HATU), and 1-hydroxybenzotriazole (HOBt). [0061] The term “monovalent cation” refers to any cation with a charge of +1, such as alkali metal cations, NH4 +, and tetraalkylammonium. [0062] The term “alkali metal cation” refers to a cation derived from a Group I metal atom, including without limitation lithium (Li+), sodium (Na+), potassium (K+), rubidium (Rb+), and cesium (Cs+). [0063] The term “substituted benzyl” refers to a benzyl group that is substituted with 1-3 substituents selected from the group consisting of C1-C3 alkyl, C1-C3 alkoxy, halogen, and cyano. [0064] The term “ketone solvent” refers to a compound having the formula CnH2n+1C(O)CmH2m+1, wherein n and m are each independently an integer between 1 and 6. The CnH2n+1 and CmH2m+1 and groups may be linear or branched and each may be substituted with up to 3 halogens. Ketone solvents include without limitation acetone, methyl ethyl ketone, 3-pentanone, and methyl tert-butyl ketone. [0065] The term “ethereal solvent” refers to an organic solvent having at least one ether moiety. Ethereal solvents include without limitation tetrahydofuran, dimethoxyethane, dioxane, and dialkyl ethers such as diethyl ether and methyl isobutyl ether.
[0066] The term “ester solvent” refers to a compound having the formula CnH2n+1OC(O)CmH2m+1, wherein n and m are each independently an integer between 1 and 6. The CnH2n+1 and CmH2m+1 and groups may be linear or branched and each may be substituted with up to 3 halogens. Ester solvents include without limitation ethyl acetate, isopropyl acetate, butyl acetate, and ethylpropionate. [0067] The term “halogenated solvent” refers to a C1-C6 alkane or C2-C6 alkene substituted with up to six halogens. Halogenated solvents include without limitation dichloromethane, dichloroethane, chloroform, tetrachloroethylene, and carbon tetrachloride. [0068] The term “aromatic solvent” refers to a C6-10 aromatic hydrocarbon. The aromatic hydrocarbon may be substituted with up to six halogens. Aromatic solvents include without limitation benzene, trifluoromethylbenzene, xylene, and toluene. [0069] The term “about” means that the stated number can vary from that value by ±10%. Where the term defines a temperature, the stated temperature can vary by ±10%. For example, about 80ºC means between 72ºC and 88ºC. Where the term defines pressure, the term “about” means the pressure can vary by ±10%. Thus, about 100 bars means between 90 and 110 bars. Where the term defines quantity (such as equivalents or weight), the term means the quantity can vary by ±10%. For example, about 1 equivalent means between 0.9 and 1.1 equivalents. Where the term defines time, the term means the stated time can vary by ±10%. For example, about 1 hour means between 0.9 and 1.1 hours. [0070] The term “leaving group” is a chemical group that is readily displaced by a desired incoming chemical moiety. Thus, the choice of the specific suitable leaving group is predicated upon its ability to be readily displaced by the incoming chemical moiety such as a CN group. Suitable leaving groups are well known in the art, e.g., see, “Advanced Organic Chemistry,” Jerry March, 5.sup.th Ed., pp.351-357, John Wiley and Sons, N.Y. [0071] As used here, the term “cyanating agent” refers to a reagent, such as trimethylsilyl cyanide (TMSCN), diethylaluminum cyanide, KCN, NaCN, TBACN, HCN and the like, useful in the synthesis of compounds disclosed herein, e.g., compound A1, A2, and A3. In one embodiment, the cyanating agent (e.g., trimethylsilyl cyanide) may be combined with a Lewis acid. In some embodiments, the Lewis acid is trifluoromethanesulfonic anhydride (Tf2O), boron trifluoride ethyl etherate (BF3OEt2), TiCl4, InCl3, AgSbF6, iodine, ZnBr2, Al(OiPr)3, MgCl2, Mn(acac)2, MnCl2, TMSOTf, SnCl4, and the like. In further embodiments, the Lewis acid is trifluoromethanesulfonic anhydride (Tf2O). The cyanation reaction may be conducted in an organic solvent, for example toluene, dichloromethane, 2-methyl THF, acetonitrile, methanol, 1,2-dichloroethane, nitromethane, and the like. [0072] As used herein, the term "active pharmaceutical ingredient" ("API") or "therapeutic agent" refers to a biologically active compound. [0073] The terms "patient" and "subject" are used interchangeably and refer to an animal including humans. [0074] The terms "effective dose" and "effective amount" are used interchangeably herein and refer to that amount of a compound that produces the desired effect for which it is administered (e.g.,
improvement in pain or a symptom of pain, or lessening the severity of pain or a symptom of pain). The exact amount of an effective dose will depend on the purpose of the treatment and will be ascertainable by one skilled in the art using known techniques (see, e.g., Lloyd (1999) The Art, Science and Technology of Pharmaceutical Compounding). [0075] As used herein, the terms "treatment," "treating," and the like generally mean the improvement of pain or one or more of its symptoms or lessening the severity of pain or one or more of its symptoms in a subject. "Treatment," as used herein, includes, but is not limited to, the following: Chronic pain, gut pain, neuropathic pain, musculoskeletal pain, acute pain, inflammatory pain, cancer pain, idiopathic pain, postsurgical pain (e.g., bunionectomy pain, herniorrhaphy pain or abdominoplasty pain), visceral pain, multiple sclerosis, Charcot-Marie-Tooth syndrome, incontinence, pathological cough, or cardiac arrhythmia. Improvements in or lessening the severity of any of these symptoms can be readily assessed according to standard methods and techniques known in the art. [0076] As used herein, the term "in combination with," when referring to two or more compounds, agents, or additional active pharmaceutical ingredients, means the administration of two or more compounds, agents, or active pharmaceutical ingredients to the patient prior to, concurrently with, or subsequent to each other. [0077] "Selected from" and "chosen from" are used interchangeably herein. [0078] As used herein, the term "ambient conditions" means room temperature, open air condition and uncontrolled humidity condition. As used herein, the terms "room temperature" and "ambient temperature" mean 15 °C to 30 °C. [0079] As used herein, the term "solvent" refers to any liquid in which the product is at least partially soluble (solubility of product > 1 g/L). [0080] The term "stable," as used herein, refers to compounds or solid forms that are not substantially altered when subjected to conditions to allow for their production, detection, and preferably their recovery, purification, and use for one or more of the purposes disclosed herein. [0081] The term "chemically stable," as used herein, means that the solid form of Compound I does not decompose into one or more different chemical compounds when subjected to specified conditions, e.g., 40 °C/75% relative humidity, for a specific period of time, e.g., 1 day, 2 days, 3 days, 1 week, 2 weeks, or longer. In some embodiments, less than 25% of the solid form of Compound I decomposes. In some embodiments, less than about 20%, less than about 15%, less than about 10%, less than about 5%, less than about 3%, less than about 1 %, less than about 0.5% of the form of Compound I decomposes under the conditions specified. In some embodiments, no detectable amount of the solid form of Compound I decomposes. [0082] The term "physically stable," as used herein, means that the solid form of Compound I does not change into one or more different physical forms of Compound I (e.g., different solid forms as measured by XRPD, DSC, etc.) when subjected to specific conditions, e.g., 40 °C/75 % relative humidity, for a specific period of time, e.g, 1 day, 2 days, 3 days, 1 week, 2 weeks, or longer. In some
embodiments, less than 25% of the solid form of Compound I changes into one or more different physical forms when subjected to specified conditions. In some embodiments, less than about 20%, less than about 15%, less than about 10%, less than about 5%, less than about 3%, less than about 1 %, less than about 0.5% of the solid form of Compound I changes into one or more different physical forms of Compound I when subjected to specified conditions. In some embodiments, no detectable amount of the solid form of Compound I changes into one or more physically different solid forms of Compound I. [0083] As used herein, the term "hydrate" refers to any crystalline Compound I that contains water in its crystal lattice. The stoichiometry of a Compound I hydrate can vary. For example, a hydrate of Compound I can be a quarter hydrate, hemihydrate, monohydrate, dihydrate, or a partially dehydrated form. [0084] As used herein, the term "pharmaceutically acceptable solid form" refers to a solid form of Compound I of this disclosure wherein the solid form (e.g., crystalline free form, crystalline salt, crystalline salt solvate, crystalline salt hydrate, and amorphous form) of Compound I is nontoxic and suitable for use in pharmaceutical compositions. [0085] As used herein, the term "amorphous" refers to a solid material having no long-range order in the position of its molecules. Amorphous solids are generally rather isotropic, i.e., exhibit similar properties in all directions. Amorphous solids do not have definite melting points. Amorphous solids are generally glasses or supercooled liquids in which the molecules are arranged in a random manner so that there is no well-defined arrangement, e.g., molecular packing, and no long-range order. For example, an amorphous material is a solid material having no sharp characteristic crystalline peak(s) in its X-ray power diffraction (XRPD) pattern (i.e., is not crystalline as determined by XRPD). Instead, one or several broad peaks (e.g., halos) appear in its XRPD pattern. Broad peaks are characteristic of an amorphous solid. See US 2004/0006237 for a comparison of XRPDs of an amorphous material and crystalline material. In some embodiments, a solid material may comprise an amorphous compound, and the material may, for example, be characterized by a lack of sharp characteristic crystalline peak(s) in its XRPD spectrum (i.e., the material is not crystalline, but is amorphous, as determined by XRPD). Instead, one or several broad peaks (e.g., halos) may appear in the XRPD pattern of the material. See US 2004/0006237 for a comparison of XRPDs of an amorphous material and crystalline material. A solid material, comprising an amorphous compound, may be characterized by, for example, a wider temperature range for the melting of the solid material, as compared to the range for the melting of a pure crystalline solid. Other techniques, such as, for example, solid state NMR may also be used to characterize crystalline or amorphous forms. [0086] As used herein, the terms "crystal form," "crystalline form," and "Form" interchangeably refer to a crystal structure (or polymorph) having a particular molecular packing arrangement in the crystal lattice. Crystalline forms can be identified and distinguished from each other by one or more characterization techniques including, for example, X-ray powder diffraction (XRPD), single crystal
X-ray diffraction, and 13C solid state nuclear magnetic resonance (13C SSNMR). Accordingly, as used herein, the terms "crystalline Form [X] of Compound (I)" and "crystalline Form [C] potassium salt of Compound (I)" refer to unique crystalline forms that can be identified and distinguished from each other by one or more characterization techniques including, for example, XRPD, single crystal X-ray diffraction, and 13C SSNMR In some embodiments, the novel crystalline forms are characterized by an X-ray powder diffractogram having one or more signals at one or more specified degree two-theta values (°2θ). [0087] As used herein, the term "free form" refers to a non-ionized version of the compound in the solid state. Examples of free forms include free bases and free acids. [0088] As used herein, the term "neat form" refers to an unsolvated and unhydrated free form version of a compound in the solid state. [0089] As used herein, the term "solvate" refers to a crystal form comprising one or more molecules of a compound of the present disclosure and, incorporated into the crystal lattice, one or more molecules of a solvent or solvents in stoichiometric or nonstoichiometric amounts. When the solvent is water, the solvate is referred to as a "hydrate." [0090] As used herein, the term "hydrate" refers to any crystalline Compound I that contains water in its crystal lattice. The stoichiometry of a Compound I hydrate can vary, i.e., Compound I can be a variable hydrate. For example, a hydrate of Compound I can be a quarter hydrate, hemihydrate, monohydrate, dihydrate, or a partially dehydrated form. [0091] In some embodiments, a solid material may comprise a mixture of crystalline solids and amorphous solids. A solid material comprising an amorphous compound may also, for example, contain up to 30% of a crystalline solid. In some embodiments, a solid material prepared to comprise an amorphous compound may also, for example, contain up to 25%, 20%, 15%, 10%, 5%, or 2% of a crystalline solid. In embodiments wherein the solid material contains a mixture of crystalline solids and amorphous solids, the characterizing data, such as XRPD, may contain indicators of both crystalline and amorphous solids. In some embodiments, a crystalline form of this disclosure may contain up to 30% amorphous compound. In some embodiments, a crystalline preparation of Compound I may contain up to 25%, 20%, 15%, 10%, 5%, or 2% of an amorphous solid. [0092] As used herein, the term "substantially amorphous" refers to a solid material having little or no long-range order in the position of its molecules. For example, substantially amorphous materials have less than 15% crystallinity (e.g., less than 10% crystallinity, less than 5% crystallinity, or less than 2% crystallinity). It is also noted that the term "substantially amorphous" includes the descriptor, "amorphous," which refers to materials having no (0%) crystallinity. [0093] As used herein, the term "substantially crystalline" refers to a solid material having little or no amorphous molecules. For example, substantially crystalline materials have less than 15% amorphous molecules (e.g., less than 10% amorphous molecules, less than 5% amorphous molecules,
or less than 2% amorphous molecules). It is also noted that the term "substantially crystalline" includes the descriptor "crystalline," which refers to materials that are 100% crystalline form. [0094] As used herein, a crystalline form is "substantially pure" when it accounts for an amount by weight equal to or greater than 90% of the sum of all solid form(s) in a sample as determined by a method in accordance with the art, such as quantitative XRPD. In some embodiments, the solid form is "substantially pure" when it accounts for an amount by weight equal to or greater than 95% of the sum of all solid form(s) in a sample. In some embodiments, the solid form is "substantially pure" when it accounts for an amount by weight equal to or greater than 99% of the sum of all solid form(s) in a sample. [0095] As used herein, the terms "X-ray powder diffractogram," "X-ray powder diffraction pattern," "XRPD pattern," "XRPD spectrum" interchangeably refer to an experimentally obtained pattern plotting signal positions (on the abscissa) versus signal intensities (on the ordinate). [0096] A "signal" or "peak" as used herein refers to a point in the XRPD pattern where the intensity as measured in counts is at a local maximum. An XRPD peak is identified by its angular value as measured in degrees 2θ (°2θ), depicted on the abscissa of an X-ray powder diffractogram, which may be expressed, for example, as "a signal at ... degrees two-theta," "a signal at [a] two-theta value(s) of ... " and/or "a signal at at least ... two-theta value(s) selected from .... " [0097] The repeatability of the measured angular values is in the range of ±0.2° 2θ, i.e., the angular value can be at the recited angular value +0.2 degrees two-theta, the angular value -0.2 degrees two-theta, or any value between those two end points (angular value +0.2 degrees two-theta and angular value -0.2 degrees two-theta). [0098] One of ordinary skill in the art would recognize that one or more signals (or peaks) in an XRPD pattern may overlap and may, for example, not be apparent to the naked eye. Indeed, one of ordinary skill in the art would recognize that some art-recognized methods are capable of and suitable for determining whether a signal exists in a pattern, such as Rietveld refinement. [0099] The terms "signal intensities" and "peak intensities" interchangeably refer to relative signal intensities within a given X-ray powder diffractogram. Factors that can affect the relative signal or peak intensities include sample thickness and preferred orientation (e.g., the crystalline particles are not distributed randomly). [0100] As used herein, an X-ray powder diffractogram is "substantially similar to that in [a particular] Figure" when at least 90%, such as at least 95%, at least 98%, or at least 99%, of the signals in the two diffractograms overlap. In determining "substantial similarity," one of ordinary skill in the art will understand that there may be variation in the intensities and/or signal positions in XRPD diffractograms even for the same crystalline form. Thus, those of ordinary skill in the art will understand that the signal maximum values in XRPD diffractograms (in degrees two-theta) generally mean that value is identified as ±0.2 degrees two-theta of the reported value, an art-recognized variance.
[0101] As used herein, the term "glass transition temperature" or "Tg" refers to the temperature above which a hard and brittle "glassy" amorphous solid becomes viscous or rubbery supercooled liquid. [0102] As used herein, the term "melting temperature", "melting point", or "Tm" refers to the temperature at which a crystalline material is in equilibrium with liquid phase. [0103] As used herein, the term "dispersion" refers to a disperse system in which one substance, the dispersed phase, is distributed, in discrete units, throughout a second substance (the continuous phase or vehicle). The size of the dispersed phase can vary considerably (e.g., colloidal particles of nanometer dimension, to multiple microns in size). In general, the dispersed phases can be solids, liquids, or gases. In the case of a solid dispersion, the dispersed and continuous phases are both solids. In pharmaceutical applications, a solid dispersion can include a crystalline drug (dispersed phase) in an amorphous polymer (continuous phase); or alternatively, an amorphous drug (dispersed phase) in an amorphous polymer (continuous phase). In some embodiments, a solid dispersion includes the polymer constituting the dispersed phase, and the drug constitute the continuous phase or, a solid dispersion includes the drug constituting the dispersed phase, and the polymer constituting the continuous phase. Detailed Description of Embodiments [0104] One aspect of the disclosure provides a method of preparing a compound of formula I: I, or a solvate or a tautomer or a
 [0105] Another aspect of the disclosure provides solid forms of Compound I (e.g., crystalline forms, amorphous forms, solvates, hydrates, cocrystals), which can be used in the methods of treatment and pharmaceutical compositions described herein. In some embodiments, the invention provides neat amorphous forms of Compound I. In some embodiments, the invention provides neat crystalline forms of Compound I. In some embodiments, the invention provides solvate crystalline forms of Compound I. In some embodiments, the invention provides hydrate crystalline forms of Compound I. A. Methods of Preparing Compound I [0106] In one embodiment, the skilled artisan could start with any compounds of formulae A1, A1-1, A1-2, A1-3, A1-4, A1-5, A2, A2-1, A3, A3-1, A3-2, B-1, B-2, B-3, B-4, B-5, C-1, C-2, C-3, C-4, D-1, D-2,
E-A1, E-A2, and E-A3 to prepare the compound of formula I or any of the intermediate compounds of formulae A1, A1-1, A1-2, A1-3, A1-4, A1-5, A2, A2-1, A3, A3-1, A3-2, B-1, B-2, B-3, B-4, B-5, C-1, C-2, C-3, C-4, D-1, D-2, E-A1, E-A2, and E-A3 by following the reactions illustrated in Schemes 1-9. [0107] The method steps described herein may refer to conversion of a starting compound of formulae A1, A1-1, A1-2, A1-3, A1-4, A1-5, A2, A2-1, A3, A3-1, A3-2, B-1, B-2, B-3, B-4, B-5, C-1, C-2, C-3, C-4, D-1, D-2, E-A1, E-A2, E-A3, and F-A2 to the compound of formula I. The skilled artisan would understand that such methods can also be used to prepare any intermediate between any starting compound and the compound of formula I. For example, conversion of the compound of formula A1-2 to the compound of formula I goes through intermediate compound E-A1. As such, the skilled artisan would understand that the methods described for converting the compound of formula A1-2 to the compound of formula I can be used to prepare intermediate compounds E-A1, A1, and D1 from the compounds of formula A1-2. [0108] Similarly, conversion of the compound of formula A3-2 to the compound of formula I goes through preparation of intermediate compounds A3-1, A3, D-1, and E-A3. As such, the skilled artisan would understand that the methods described for converting the compound of formula A3-2 to the compound of formula I can be used to prepare any of intermediate compounds A3-1, A3, and D-1, and E-A3 starting with the compound of formula A3-2 or any intermediate compound can be converted to the desired intermediate compound using the methods described herein. Thus, the present application contemplates preparing intermediate compounds A1, A1-1, A1-2, A1-3, A1-4, A1-5, A2, A2-1, A3, A3-1, A3-2, B-1, B-2, B-3, B-4, B-5, C-1, C-2, C-3, C-4, D-1, D-2, E-A1, E-A2, E-A3, and F-A2 starting with any intermediate or starting material that precedes the intermediate that is being prepared. [0109] Scheme 1
[0105] Another aspect of the disclosure provides solid forms of Compound I (e.g., crystalline forms, amorphous forms, solvates, hydrates, cocrystals), which can be used in the methods of treatment and pharmaceutical compositions described herein. In some embodiments, the invention provides neat amorphous forms of Compound I. In some embodiments, the invention provides neat crystalline forms of Compound I. In some embodiments, the invention provides solvate crystalline forms of Compound I. In some embodiments, the invention provides hydrate crystalline forms of Compound I. A. Methods of Preparing Compound I [0106] In one embodiment, the skilled artisan could start with any compounds of formulae A1, A1-1, A1-2, A1-3, A1-4, A1-5, A2, A2-1, A3, A3-1, A3-2, B-1, B-2, B-3, B-4, B-5, C-1, C-2, C-3, C-4, D-1, D-2,
E-A1, E-A2, and E-A3 to prepare the compound of formula I or any of the intermediate compounds of formulae A1, A1-1, A1-2, A1-3, A1-4, A1-5, A2, A2-1, A3, A3-1, A3-2, B-1, B-2, B-3, B-4, B-5, C-1, C-2, C-3, C-4, D-1, D-2, E-A1, E-A2, and E-A3 by following the reactions illustrated in Schemes 1-9. [0107] The method steps described herein may refer to conversion of a starting compound of formulae A1, A1-1, A1-2, A1-3, A1-4, A1-5, A2, A2-1, A3, A3-1, A3-2, B-1, B-2, B-3, B-4, B-5, C-1, C-2, C-3, C-4, D-1, D-2, E-A1, E-A2, E-A3, and F-A2 to the compound of formula I. The skilled artisan would understand that such methods can also be used to prepare any intermediate between any starting compound and the compound of formula I. For example, conversion of the compound of formula A1-2 to the compound of formula I goes through intermediate compound E-A1. As such, the skilled artisan would understand that the methods described for converting the compound of formula A1-2 to the compound of formula I can be used to prepare intermediate compounds E-A1, A1, and D1 from the compounds of formula A1-2. [0108] Similarly, conversion of the compound of formula A3-2 to the compound of formula I goes through preparation of intermediate compounds A3-1, A3, D-1, and E-A3. As such, the skilled artisan would understand that the methods described for converting the compound of formula A3-2 to the compound of formula I can be used to prepare any of intermediate compounds A3-1, A3, and D-1, and E-A3 starting with the compound of formula A3-2 or any intermediate compound can be converted to the desired intermediate compound using the methods described herein. Thus, the present application contemplates preparing intermediate compounds A1, A1-1, A1-2, A1-3, A1-4, A1-5, A2, A2-1, A3, A3-1, A3-2, B-1, B-2, B-3, B-4, B-5, C-1, C-2, C-3, C-4, D-1, D-2, E-A1, E-A2, E-A3, and F-A2 starting with any intermediate or starting material that precedes the intermediate that is being prepared. [0109] Scheme 1
[0110] Scheme 2
[0114] Scheme 6
[0117] Scheme 9
 , comprising transforming a
, comprising transforming a
 to the compound of formula (I); wherein
to the compound of formula (I); wherein
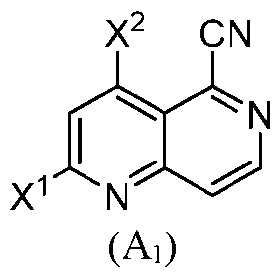 are independently selected from halogen. [0119] In some embodiments, X1 and X2 are the same. In some embodiments, each of X1 and X2 are fluoro. In some embodiments, each of X1 and X2 are chloro. In some embodiments, each of X1 and X2 are bromo. In some embodiments, each of X1 and X2 are iodo. [0120] In some embodiments, X1 and X2 are different.
[0121] In some embodiments, the step of transforming the compound of formula (A1) to the compound of formula (I) comprises transforming the compound of formula (A1) to a compound of formula (E-A1): X2 CN N . [0122] In some embodiments, the compound of formula (A1) to the compound of formula (E-A1)
are independently selected from halogen. [0119] In some embodiments, X1 and X2 are the same. In some embodiments, each of X1 and X2 are fluoro. In some embodiments, each of X1 and X2 are chloro. In some embodiments, each of X1 and X2 are bromo. In some embodiments, each of X1 and X2 are iodo. [0120] In some embodiments, X1 and X2 are different.
[0121] In some embodiments, the step of transforming the compound of formula (A1) to the compound of formula (I) comprises transforming the compound of formula (A1) to a compound of formula (E-A1): X2 CN N . [0122] In some embodiments, the compound of formula (A1) to the compound of formula (E-A1)
 of formula (A1) with a compound of formula (D-1): . [0123] In some embodiments, the compound of formula (A1) with a
of formula (A1) with a compound of formula (D-1): . [0123] In some embodiments, the compound of formula (A1) with a
 compound of formula (D-1) is presence a first palladium catalyst and a first base. In some embodiments, the first palladium catalyst is a palladium-phosphine complex. In some embodiments, the first palladium catalyst is PdCl2(dtbdpf). [0124] In some embodiments, the step of contacting the compound of formula (A1) with a compound of formula (D-1) is performed in the presence of a first palladium catalyst, a first ligand, and a first base. In some embodiments, the first palladium catalyst is PdCl2. [0125] In some embodiments, the first ligand is selected from 1,1′-Bis(di-tert- butylphosphino)ferrocene (dtbpf), 1,1′-Ferrocenediyl-bis(diphenylphosphine) (dppf), Bis[(2- diphenylphosphino)phenyl] ether (DPEPhos), 1,2-Bis(diphenylphosphino)ethane (dppe), triphenylphosphine (PPh3), cyclohexyldiphenylphosphine (CyPh2P), tri(o-tolyl)phosphine (P(o-tol)3), and Di(1-adamantyl)-n-butylphosphine (CataCXium A). In other embodiments, the first ligand is 1,1′-Bis(di-tert-butylphosphino)ferrocene (dtbpf). In other embodiments, the first ligand is 1,1′-Ferrocenediyl-bis(diphenylphosphine) (dppf). In other embodiments, the first ligand is Bis[(2- diphenylphosphino)phenyl] ether (DPEPhos). In other embodiments, the first ligand is
1,2-Bis(diphenylphosphino)ethane (dppe). In other embodiments, the first ligand is triphenylphosphine (PPh3). In other embodiments, the first ligand is cyclohexyldiphenylphosphine (CyPh2P). In other embodiments, the first ligand is tri(o-tolyl)phosphine (P(o-tol)3). In other embodiments, the first ligand is di(1-adamantyl)-n-butylphosphine (CataCXium A). [0126] In some embodiments, the first base is selected from potassium carbonate, potassium phosphate, and potassium fluoride. In some embodiments, the first base is selected from potassium carbonate, potassium phosphate, potassium fluoride, potassium bicarbonate, tripotassium phosphate, dipotassium phosphate, potassium acetate, potassium pivalate, potassium hydroxide, potassium hexamethyldisilazide, 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU), and triethylamine. In other embodiments, the first base is potassium carbonate. In other embodiments, the first base is potassium phosphate. In other embodiments, the first base is potassium fluoride. In other embodiments, the first base is potassium bicarbonate. In other embodiments, the first base is tripotassium phosphate. In other embodiments, the first base is dipotassium phosphate. In other embodiments, the first base is potassium acetate. In other embodiments, the first base is potassium pivalate. In other embodiments, the first base is potassium hydroxide. In other embodiments, the first base is potassium hexamethyldisilazide. In other embodiments, the first base is 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU). In other embodiments, the first base is triethylamine. [0127] In some embodiments, the step of transforming the compound of formula (A1) to the compound of formula (I) further comprises transforming the compound of formula (E-A1) to the compound of formula (I). [0128] In some embodiments, the step of transforming the compound of formula (E-A1) to the compound of formula (I) comprises treating the compound of formula (E-A1) with a first acid. In some embodiments, the first acid is selected from sulfuric acid, phosphoric acid, methanesulfonic acid, and trifluoroacetic acid. In some embodiments, the first acid is trifluoroacetic acid. In some embodiments, the first acid is sulfuric acid. In some embodiments, the first acid is methane sulfonic acid. [0129] In some embodiments, the step of transforming the compound of formula (A1) to the compound of formula (I) further comprises transforming a compound of formula (E-A2): Cl CN N to the compound of formula (I).
compound of formula (D-1) is presence a first palladium catalyst and a first base. In some embodiments, the first palladium catalyst is a palladium-phosphine complex. In some embodiments, the first palladium catalyst is PdCl2(dtbdpf). [0124] In some embodiments, the step of contacting the compound of formula (A1) with a compound of formula (D-1) is performed in the presence of a first palladium catalyst, a first ligand, and a first base. In some embodiments, the first palladium catalyst is PdCl2. [0125] In some embodiments, the first ligand is selected from 1,1′-Bis(di-tert- butylphosphino)ferrocene (dtbpf), 1,1′-Ferrocenediyl-bis(diphenylphosphine) (dppf), Bis[(2- diphenylphosphino)phenyl] ether (DPEPhos), 1,2-Bis(diphenylphosphino)ethane (dppe), triphenylphosphine (PPh3), cyclohexyldiphenylphosphine (CyPh2P), tri(o-tolyl)phosphine (P(o-tol)3), and Di(1-adamantyl)-n-butylphosphine (CataCXium A). In other embodiments, the first ligand is 1,1′-Bis(di-tert-butylphosphino)ferrocene (dtbpf). In other embodiments, the first ligand is 1,1′-Ferrocenediyl-bis(diphenylphosphine) (dppf). In other embodiments, the first ligand is Bis[(2- diphenylphosphino)phenyl] ether (DPEPhos). In other embodiments, the first ligand is
1,2-Bis(diphenylphosphino)ethane (dppe). In other embodiments, the first ligand is triphenylphosphine (PPh3). In other embodiments, the first ligand is cyclohexyldiphenylphosphine (CyPh2P). In other embodiments, the first ligand is tri(o-tolyl)phosphine (P(o-tol)3). In other embodiments, the first ligand is di(1-adamantyl)-n-butylphosphine (CataCXium A). [0126] In some embodiments, the first base is selected from potassium carbonate, potassium phosphate, and potassium fluoride. In some embodiments, the first base is selected from potassium carbonate, potassium phosphate, potassium fluoride, potassium bicarbonate, tripotassium phosphate, dipotassium phosphate, potassium acetate, potassium pivalate, potassium hydroxide, potassium hexamethyldisilazide, 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU), and triethylamine. In other embodiments, the first base is potassium carbonate. In other embodiments, the first base is potassium phosphate. In other embodiments, the first base is potassium fluoride. In other embodiments, the first base is potassium bicarbonate. In other embodiments, the first base is tripotassium phosphate. In other embodiments, the first base is dipotassium phosphate. In other embodiments, the first base is potassium acetate. In other embodiments, the first base is potassium pivalate. In other embodiments, the first base is potassium hydroxide. In other embodiments, the first base is potassium hexamethyldisilazide. In other embodiments, the first base is 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU). In other embodiments, the first base is triethylamine. [0127] In some embodiments, the step of transforming the compound of formula (A1) to the compound of formula (I) further comprises transforming the compound of formula (E-A1) to the compound of formula (I). [0128] In some embodiments, the step of transforming the compound of formula (E-A1) to the compound of formula (I) comprises treating the compound of formula (E-A1) with a first acid. In some embodiments, the first acid is selected from sulfuric acid, phosphoric acid, methanesulfonic acid, and trifluoroacetic acid. In some embodiments, the first acid is trifluoroacetic acid. In some embodiments, the first acid is sulfuric acid. In some embodiments, the first acid is methane sulfonic acid. [0129] In some embodiments, the step of transforming the compound of formula (A1) to the compound of formula (I) further comprises transforming a compound of formula (E-A2): Cl CN N to the compound of formula (I).
 [0130] In some embodiments, the step of transforming the compound of formula (E-A2) to the compound of formula (I) comprises treating the compound of formula (E-A2) with a first acid in the presence of water and heat to prepare a compound of formula (F-A2): . In some embodiments, the first acid formic acid, sulfuric acid, phosphoric acid, hydrochloric acid,
[0130] In some embodiments, the step of transforming the compound of formula (E-A2) to the compound of formula (I) comprises treating the compound of formula (E-A2) with a first acid in the presence of water and heat to prepare a compound of formula (F-A2): . In some embodiments, the first acid formic acid, sulfuric acid, phosphoric acid, hydrochloric acid,
 toluenesulfonic acid, and trifluoroacetic acid. In other embodiments, the first acid is acetic acid. In other embodiments, the first acid is formic acid. In other embodiments, the first acid is sulfuric acid. In other embodiments, the first acid is phosphoric acid. In other embodiments, the first acid is hydrochloric acid. In other embodiments, the first acid is methanesulfonic acid. In other embodiments, the first acid is oxalic acid. In other embodiments, the first acid is p-toluenesulfonic acid. In other embodiments, the first acid is trifluoroacetic acid. In some embodiments, the step of treating the compound of formula (E-A2) with a first acid in the presence of water and heat to prepare a compound of formula (F-A2) is performed at a temperature of about 60 °C to about 100 °C. In other embodiments, the step of treating the compound of formula (E-A2) with a first acid in the presence of water and heat to prepare a compound of formula (F-A2) is performed at a temperature of about 100 °C. [0131] In some embodiments, the step of transforming the compound of formula (E-A2) to the compound of formula (I) comprises treating the compound of formula (E-A2) with a first lewis acid in the presence of acetic acid to prepare a compound of formula (F-A2). In some embodiments, the first lewis acid is selected from MoCl2O2, MoCl5, CuOTf2, VCl3, WOCl4, and WCl6. In other embodiments, the first lewis acid is MoCl2O2. In other embodiments, the first lewis acid is MoCl5. In other embodiments, the first lewis acid is CuOTf2. In other embodiments, the first lewis acid is VCl3. In other embodiments, the first lewis acid is WOCl4. In other embodiments, the first lewis acid is WCl6. [0132] In some embodiments, the step of transforming the compound of formula (E-A2) to the compound of formula (I) comprises treating the compound of formula (E-A2) with a first base in the presence of water and heat to prepare a compound of formula (F-A2). In some embodiments, the first base is potassium acetate. In some embodiments, the step of treating the compound of formula (E-A2)
with a first base in the presence of water and heat to prepare a compound of formula (F-A2) is performed at a temperature of about 80 °C to about 100 °C. [0133] In some embodiments, the step of transforming the compound of formula (A1) to the compound of formula (I) further comprises transforming the compound of formula (F-A2) to the compound of formula (I). [0134] In some embodiments, the step of transforming the compound of formula (F-A2) to the compound of formula (I) comprises treating the compound of formula (F-A2) with an acid. In some embodiments, the acid is selected from sulfuric acid, phosphoric acid, methanesulfonic acid, oxalic acid, and trifluoroacetic acid. In some embodiments, the acid is trifluoroacetic acid. In some embodiments, the acid is sulfuric acid. In some embodiments, the acid is methane sulfonic acid. In some embodiments, the acid is methanesulfonic acid. In some embodiments, the acid is phosphoric acid. In some embodiments, the first acid is oxalic acid. [0135] In some embodiments of preparing the compound of formula I, the process comprises transforming a compound of formula (A1-1): to the compound of formula (A1).
toluenesulfonic acid, and trifluoroacetic acid. In other embodiments, the first acid is acetic acid. In other embodiments, the first acid is formic acid. In other embodiments, the first acid is sulfuric acid. In other embodiments, the first acid is phosphoric acid. In other embodiments, the first acid is hydrochloric acid. In other embodiments, the first acid is methanesulfonic acid. In other embodiments, the first acid is oxalic acid. In other embodiments, the first acid is p-toluenesulfonic acid. In other embodiments, the first acid is trifluoroacetic acid. In some embodiments, the step of treating the compound of formula (E-A2) with a first acid in the presence of water and heat to prepare a compound of formula (F-A2) is performed at a temperature of about 60 °C to about 100 °C. In other embodiments, the step of treating the compound of formula (E-A2) with a first acid in the presence of water and heat to prepare a compound of formula (F-A2) is performed at a temperature of about 100 °C. [0131] In some embodiments, the step of transforming the compound of formula (E-A2) to the compound of formula (I) comprises treating the compound of formula (E-A2) with a first lewis acid in the presence of acetic acid to prepare a compound of formula (F-A2). In some embodiments, the first lewis acid is selected from MoCl2O2, MoCl5, CuOTf2, VCl3, WOCl4, and WCl6. In other embodiments, the first lewis acid is MoCl2O2. In other embodiments, the first lewis acid is MoCl5. In other embodiments, the first lewis acid is CuOTf2. In other embodiments, the first lewis acid is VCl3. In other embodiments, the first lewis acid is WOCl4. In other embodiments, the first lewis acid is WCl6. [0132] In some embodiments, the step of transforming the compound of formula (E-A2) to the compound of formula (I) comprises treating the compound of formula (E-A2) with a first base in the presence of water and heat to prepare a compound of formula (F-A2). In some embodiments, the first base is potassium acetate. In some embodiments, the step of treating the compound of formula (E-A2)
with a first base in the presence of water and heat to prepare a compound of formula (F-A2) is performed at a temperature of about 80 °C to about 100 °C. [0133] In some embodiments, the step of transforming the compound of formula (A1) to the compound of formula (I) further comprises transforming the compound of formula (F-A2) to the compound of formula (I). [0134] In some embodiments, the step of transforming the compound of formula (F-A2) to the compound of formula (I) comprises treating the compound of formula (F-A2) with an acid. In some embodiments, the acid is selected from sulfuric acid, phosphoric acid, methanesulfonic acid, oxalic acid, and trifluoroacetic acid. In some embodiments, the acid is trifluoroacetic acid. In some embodiments, the acid is sulfuric acid. In some embodiments, the acid is methane sulfonic acid. In some embodiments, the acid is methanesulfonic acid. In some embodiments, the acid is phosphoric acid. In some embodiments, the first acid is oxalic acid. [0135] In some embodiments of preparing the compound of formula I, the process comprises transforming a compound of formula (A1-1): to the compound of formula (A1).
 [0136] In some embodiments, the a compound of formula (A1-1) to the compound of formula (A1) comprises the steps of: treating the compound of formula (A1-1) with trifluoromethanesulfonic anhydride to form a triflyl intermediate; treating the triflyl intermediate with a cyanation reagent; treating the cyanated intermediate with a second base to form a cyanated triflyl intermediate; and treating the cyanated triflyl intermediate with an aqueous base to form the compound of formula (A1). [0137] In some embodiments, the cyanation reagent is trimethylsilyl cyanide, sodium cyanide, and potassium cyanide. In some embodiments, the cyanation reagent is trimethylsilyl cyanide. In some embodiments, the cyanation reagent is sodium cyanide. In some embodiments, the cyanation reagent is potassium cyanide. [0138] In some embodiments, the second base is selected from 4-methyl morpholine, trimethylamine, and Hünig’s base. In some embodiments, the second base is 4-methylmorpholine. In some embodiments, the second base is trimethylamine. In some embodiments, the second base is Hünig’s base. [0139] In some embodiments, the aqueous base is aqueous sodium bicarbonate.
[0140] In some embodiments, the process for preparing a compound of formula I comprises transforming a compound of formula (A1-2): to the compound of formula (A1-1). [0141] In some embodiments, the
[0136] In some embodiments, the a compound of formula (A1-1) to the compound of formula (A1) comprises the steps of: treating the compound of formula (A1-1) with trifluoromethanesulfonic anhydride to form a triflyl intermediate; treating the triflyl intermediate with a cyanation reagent; treating the cyanated intermediate with a second base to form a cyanated triflyl intermediate; and treating the cyanated triflyl intermediate with an aqueous base to form the compound of formula (A1). [0137] In some embodiments, the cyanation reagent is trimethylsilyl cyanide, sodium cyanide, and potassium cyanide. In some embodiments, the cyanation reagent is trimethylsilyl cyanide. In some embodiments, the cyanation reagent is sodium cyanide. In some embodiments, the cyanation reagent is potassium cyanide. [0138] In some embodiments, the second base is selected from 4-methyl morpholine, trimethylamine, and Hünig’s base. In some embodiments, the second base is 4-methylmorpholine. In some embodiments, the second base is trimethylamine. In some embodiments, the second base is Hünig’s base. [0139] In some embodiments, the aqueous base is aqueous sodium bicarbonate.
[0140] In some embodiments, the process for preparing a compound of formula I comprises transforming a compound of formula (A1-2): to the compound of formula (A1-1). [0141] In some embodiments, the
 the compound of formula (A1-2) to the compound of formula (A1-1) comprises treating the compound of formula (A1-2) with a halogenating reagent. In some embodiments, the halogenating reagent is a chlorinating reagent. In some embodiments, the chlorinating reagent is selected from HCl, oxalyl chloride, trichloroacetic anhydride/phosgene, triphenylphosphine dichloride, phenylphosphonic dichloride, phosphorous trichloride, phosphorous pentachloride, phosphorous oxychloride, thionyl chloride, p-toluenesulfonyl chloride, and methanesulfonyl chloride. In some embodiments, the chlorinating reagent is phosphorous oxychloride. In some embodiments, the chlorinating reagent is selected from Cl2 (with/without oxidant), HCl/Cl - plus oxidant, N-chlorosuccinimide (NCS)/Dichlorodimethylhydantoin (DCDMH)/ Trichloroisocyanuric acid (TCCA) sodium dichloroiso-cyanurate NaDCC, NaOCl, SO2Cl2, and CCl4/C2Cl6. In some embodiments, the chlorinating reagent is selected from phosphorus oxychloride and phosphorus pentachloride. In some embodiments, the chlorinating reagent is phosphorus oxychloride. [0142] In some embodiments, the process for preparing a compound of formula I comprises transforming a compound of formula (A1-3): to the compound of formula (A1-2).
the compound of formula (A1-2) to the compound of formula (A1-1) comprises treating the compound of formula (A1-2) with a halogenating reagent. In some embodiments, the halogenating reagent is a chlorinating reagent. In some embodiments, the chlorinating reagent is selected from HCl, oxalyl chloride, trichloroacetic anhydride/phosgene, triphenylphosphine dichloride, phenylphosphonic dichloride, phosphorous trichloride, phosphorous pentachloride, phosphorous oxychloride, thionyl chloride, p-toluenesulfonyl chloride, and methanesulfonyl chloride. In some embodiments, the chlorinating reagent is phosphorous oxychloride. In some embodiments, the chlorinating reagent is selected from Cl2 (with/without oxidant), HCl/Cl - plus oxidant, N-chlorosuccinimide (NCS)/Dichlorodimethylhydantoin (DCDMH)/ Trichloroisocyanuric acid (TCCA) sodium dichloroiso-cyanurate NaDCC, NaOCl, SO2Cl2, and CCl4/C2Cl6. In some embodiments, the chlorinating reagent is selected from phosphorus oxychloride and phosphorus pentachloride. In some embodiments, the chlorinating reagent is phosphorus oxychloride. [0142] In some embodiments, the process for preparing a compound of formula I comprises transforming a compound of formula (A1-3): to the compound of formula (A1-2).
 [0143] In some embodiments, the step of transforming a compound of formula (A1-3) to the compound of formula (A1-3) comprises treating the compound of formula (A1-3) with a second acid. In some embodiments, the second acid is aqueous hydrochloric acid. [0144] In some embodiments, the process of preparing a compound of formula I comprises transforming a compound of formula (A1-4):
to the compound of formula (A1-3). [0145] In some embodiments, the step of transforming a compound of formula (A1-4) to the compound of formula (A1-3) comprises treating the compound of formula (A1-4) with diethyl malonate in the presence of a third base. In some embodiments, the third base is an alkaline alkoxide. In some embodiments, the third base is sodium ethoxide. [0146] In some embodiments, the process of preparing a compound of formula I comprises transforming a compound of formula (A1-5): to the compound of formula (A1-4). [0147] In some embodiments, the
[0143] In some embodiments, the step of transforming a compound of formula (A1-3) to the compound of formula (A1-3) comprises treating the compound of formula (A1-3) with a second acid. In some embodiments, the second acid is aqueous hydrochloric acid. [0144] In some embodiments, the process of preparing a compound of formula I comprises transforming a compound of formula (A1-4):
to the compound of formula (A1-3). [0145] In some embodiments, the step of transforming a compound of formula (A1-4) to the compound of formula (A1-3) comprises treating the compound of formula (A1-4) with diethyl malonate in the presence of a third base. In some embodiments, the third base is an alkaline alkoxide. In some embodiments, the third base is sodium ethoxide. [0146] In some embodiments, the process of preparing a compound of formula I comprises transforming a compound of formula (A1-5): to the compound of formula (A1-4). [0147] In some embodiments, the
 the compound of formula (A1-5) to the compound of formula (A1-4) comprises treating the compound of formula (A1-5) with carbon monoxide in the presence of a fourth base, a second palladium catalyst, and a first suitable ligand. [0148] In some embodiments, the fourth base is selected from triethylamine, Hünig’s Base, 4-methylmorpholine, 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU), and 1,4-Diazabicyclo[2.2.2]octane (DABCO). In some embodiments, the fourth base is triethylamine. [0149] In some embodiments, the second palladium catalyst is Pd(OAc)2. [0150] In some embodiments, the first suitable ligand is selected from 1,1'-bis(diphenylphosphino)ferrocene (DPPF), 4,5-Bis(diphenylphosphino)-9,9-dimethylxanthene (XantPhos), triphenylphosphine (PPh3), and 2,2′-Bis(diphenylphosphino)-1,1′-binaphthalene (BINAP). In some embodiments, the first suitable ligand is 1,1'-bis(diphenyl phosphino)ferrocene (DPPF). [0151] In some embodiments, each of X1 and X2 are chloro. [0152] In some embodiments, a process for preparing a compound of formula (I):
the compound of formula (A1-5) to the compound of formula (A1-4) comprises treating the compound of formula (A1-5) with carbon monoxide in the presence of a fourth base, a second palladium catalyst, and a first suitable ligand. [0148] In some embodiments, the fourth base is selected from triethylamine, Hünig’s Base, 4-methylmorpholine, 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU), and 1,4-Diazabicyclo[2.2.2]octane (DABCO). In some embodiments, the fourth base is triethylamine. [0149] In some embodiments, the second palladium catalyst is Pd(OAc)2. [0150] In some embodiments, the first suitable ligand is selected from 1,1'-bis(diphenylphosphino)ferrocene (DPPF), 4,5-Bis(diphenylphosphino)-9,9-dimethylxanthene (XantPhos), triphenylphosphine (PPh3), and 2,2′-Bis(diphenylphosphino)-1,1′-binaphthalene (BINAP). In some embodiments, the first suitable ligand is 1,1'-bis(diphenyl phosphino)ferrocene (DPPF). [0151] In some embodiments, each of X1 and X2 are chloro. [0152] In some embodiments, a process for preparing a compound of formula (I):
comprises transforming a compound of formula (A3): to the compound of formula (I). [0153] In some embodiments, the
 the compound of formula (A3) to the compound of formula (I) comprises transforming the compound of formula (A3) to a compound of formula (E-A3): . [0154] In some embodiments, the compound of formula (A3) to the
the compound of formula (A3) to the compound of formula (I) comprises transforming the compound of formula (A3) to a compound of formula (E-A3): . [0154] In some embodiments, the compound of formula (A3) to the
 compound of formula (E-A3) of formula (A3) with a compound of formula (D-1): . [0155] In some embodiments,
compound of formula (E-A3) of formula (A3) with a compound of formula (D-1): . [0155] In some embodiments,
 a compound of formula I comprises contacting the compound of formula (A3) with a compound of formula (D-1) in the presence of a first palladium catalyst and a first base. In some embodiments, the first palladium catalyst is PdCl2(dtbdpf). In some embodiments, the first base is potassium phosphate. [0156] In some embodiments, the step of contacting the compound of formula (A3) with a compound of formula (D-1) is performed in the presence of a first palladium catalyst, a first ligand, and a first base. In some embodiments, the first palladium catalyst is PdCl2. [0157] In some embodimtns, the first ligand is selected from 1,1′-Bis(di-tert- butylphosphino)ferrocene (dtbpf), 1,1′-Ferrocenediyl-bis(diphenylphosphine) (dppf), Bis[(2-
diphenylphosphino)phenyl] ether (DPEPhos), 1,2-Bis(diphenylphosphino)ethane (dppe), triphenylphosphine (PPh3), cyclohexyldiphenylphosphine (CyPh2P), tri(o-tolyl)phosphine (P(o-tol)3), and Di(1-adamantyl)-n-butylphosphine (CataCXium A). In other embodiments, the first ligand is 1,1′-Bis(di-tert-butylphosphino)ferrocene (dtbpf). In other embodiments, the first ligand is 1,1′-Ferrocenediyl-bis(diphenylphosphine) (dppf). In other embodiments, the first ligand is Bis[(2- diphenylphosphino)phenyl] ether (DPEPhos). In other embodiments, the first ligand is 1,2-Bis(diphenylphosphino)ethane (dppe). In other embodiments, the first ligand is triphenylphosphine (PPh3). In other embodiments, the first ligand is cyclohexyldiphenylphosphine (CyPh2P). In other embodiments, the first ligand is tri(o-tolyl)phosphine (P(o-tol)3). In other embodiments, the first ligand is di(1-adamantyl)-n-butylphosphine (CataCXium A). [0158] In some embodiments, the first base is selected from potassium carbonate, potassium phosphate, potassium fluoride, potassium bicarbonate, tripotassium phosphate, dipotassium phosphate, potassium acetate, potassium pivalate, potassium hydroxide, potassium hexamethyldisilazide, 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU), and triethylamine. In other embodiments, the first base is potassium carbonate. In other embodiments, the first base is potassium phosphate. In other embodiments, the first base is potassium fluoride. In other embodiments, the first base is potassium bicarbonate. In other embodiments, the first base is tripotassium phosphate. In other embodiments, the first base is dipotassium phosphate. In other embodiments, the first base is potassium acetate. In other embodiments, the first base is potassium pivalate. In other embodiments, the first base is potassium hydroxide. In other embodiments, the first base is potassium hexamethyldisilazide. In other embodiments, the first base is 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU). In other embodiments, the first base is triethylamine. [0159] In some embodiments, the step of transforming the compound of formula (A3) to the compound of formula (I) further comprises transforming the compound of formula (E-A3) to the compound of formula (I). [0160] In some embodiments, the step of transforming the compound of formula (E-A3) to the compound of formula (I) comprises treating the compound of formula (E-A3) with a first acid. In some embodiments, the first acid is trifluoroacetic acid, sulfuric acid, phosphoric acid, and methane sulfonic acid. In some embodiments, the first acid is trifluoroacetic acid. In some embodiments, the first acid is sulfuric acid. In some embodiments, the first acid is methane sulfonic acid. [0161] In some embodiments, the process of preparing a compound of formula I comprises transforming a compound of formula (A3-1): to the compound of formula (A3).
a compound of formula I comprises contacting the compound of formula (A3) with a compound of formula (D-1) in the presence of a first palladium catalyst and a first base. In some embodiments, the first palladium catalyst is PdCl2(dtbdpf). In some embodiments, the first base is potassium phosphate. [0156] In some embodiments, the step of contacting the compound of formula (A3) with a compound of formula (D-1) is performed in the presence of a first palladium catalyst, a first ligand, and a first base. In some embodiments, the first palladium catalyst is PdCl2. [0157] In some embodimtns, the first ligand is selected from 1,1′-Bis(di-tert- butylphosphino)ferrocene (dtbpf), 1,1′-Ferrocenediyl-bis(diphenylphosphine) (dppf), Bis[(2-
diphenylphosphino)phenyl] ether (DPEPhos), 1,2-Bis(diphenylphosphino)ethane (dppe), triphenylphosphine (PPh3), cyclohexyldiphenylphosphine (CyPh2P), tri(o-tolyl)phosphine (P(o-tol)3), and Di(1-adamantyl)-n-butylphosphine (CataCXium A). In other embodiments, the first ligand is 1,1′-Bis(di-tert-butylphosphino)ferrocene (dtbpf). In other embodiments, the first ligand is 1,1′-Ferrocenediyl-bis(diphenylphosphine) (dppf). In other embodiments, the first ligand is Bis[(2- diphenylphosphino)phenyl] ether (DPEPhos). In other embodiments, the first ligand is 1,2-Bis(diphenylphosphino)ethane (dppe). In other embodiments, the first ligand is triphenylphosphine (PPh3). In other embodiments, the first ligand is cyclohexyldiphenylphosphine (CyPh2P). In other embodiments, the first ligand is tri(o-tolyl)phosphine (P(o-tol)3). In other embodiments, the first ligand is di(1-adamantyl)-n-butylphosphine (CataCXium A). [0158] In some embodiments, the first base is selected from potassium carbonate, potassium phosphate, potassium fluoride, potassium bicarbonate, tripotassium phosphate, dipotassium phosphate, potassium acetate, potassium pivalate, potassium hydroxide, potassium hexamethyldisilazide, 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU), and triethylamine. In other embodiments, the first base is potassium carbonate. In other embodiments, the first base is potassium phosphate. In other embodiments, the first base is potassium fluoride. In other embodiments, the first base is potassium bicarbonate. In other embodiments, the first base is tripotassium phosphate. In other embodiments, the first base is dipotassium phosphate. In other embodiments, the first base is potassium acetate. In other embodiments, the first base is potassium pivalate. In other embodiments, the first base is potassium hydroxide. In other embodiments, the first base is potassium hexamethyldisilazide. In other embodiments, the first base is 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU). In other embodiments, the first base is triethylamine. [0159] In some embodiments, the step of transforming the compound of formula (A3) to the compound of formula (I) further comprises transforming the compound of formula (E-A3) to the compound of formula (I). [0160] In some embodiments, the step of transforming the compound of formula (E-A3) to the compound of formula (I) comprises treating the compound of formula (E-A3) with a first acid. In some embodiments, the first acid is trifluoroacetic acid, sulfuric acid, phosphoric acid, and methane sulfonic acid. In some embodiments, the first acid is trifluoroacetic acid. In some embodiments, the first acid is sulfuric acid. In some embodiments, the first acid is methane sulfonic acid. [0161] In some embodiments, the process of preparing a compound of formula I comprises transforming a compound of formula (A3-1): to the compound of formula (A3).
 [0162] In some embodiments, the step of transforming a compound of formula (A3-1) to the compound of formula (A3) comprises treating the compound of formula (A3-1) with a cyanation reagent and a second base. [0163] In some embodiments, the cyanation reagent is trimethylsilyl cyanide, sodium cyanide, and potassium cyanide. In some embodiments, the cyanation reagent is trimethylsilyl cyanide. In some embodiments, the cyanation reagent is sodium cyanide. In some embodiments, the cyanation reagent is potassium cyanide. [0164] In some embodiments, the second base is triethylamine. [0165] In some embodiments, the process of preparing a compound of formula I comprise transforming a compound of formula (A3-2): to the compound of formula (A3-1). [0166] In some embodiments,
[0162] In some embodiments, the step of transforming a compound of formula (A3-1) to the compound of formula (A3) comprises treating the compound of formula (A3-1) with a cyanation reagent and a second base. [0163] In some embodiments, the cyanation reagent is trimethylsilyl cyanide, sodium cyanide, and potassium cyanide. In some embodiments, the cyanation reagent is trimethylsilyl cyanide. In some embodiments, the cyanation reagent is sodium cyanide. In some embodiments, the cyanation reagent is potassium cyanide. [0164] In some embodiments, the second base is triethylamine. [0165] In some embodiments, the process of preparing a compound of formula I comprise transforming a compound of formula (A3-2): to the compound of formula (A3-1). [0166] In some embodiments,
 a compound of formula (A3-2) to the compound of formula (A3-1) comprises treating the compound of formula (A3-2) with benzyl alcohol in the presence of a third base. [0167] In some embodiments, the third base is selected from carbonate base, phosphate base, sodium hexamethyldisilizane, alkyllythium reagent, hydride reagent, alkaline alkoxide, and alkaline hydroxide. In some embodiments, the third base is an alkaline alkoxide. In some embodiments, the third base is an alkaline methoxide. In some embodiments, the third base is selected from lithium methoxide, sodium methoxide, and potassium methoxide. In some embodiments, the third base is lithium methoxide. In some embodiments, the third base is sodium methoxide. In some embodiments, the third base is potassium methoxide. In some embodiments, the third base is an alkaline ethoxide. In some embodiments, the third base is selected from lithium ethoxide, sodium ethoxide, and potassium ethoxide. In some embodiments, the third base is lithium ethoxide. In some embodiments, the third base is sodium ethoxide. In some embodiments, the third base is potassium ethoxide. In some embodiments, the third base is an alkaline tert-butoxide. In some embodiments, the third base is selected from lithium tert-butoxide, sodium tert-butoxide, and potassium tert-butoxide. In some embodiments, the third base is lithium tert-butoxide. In some embodiments, the third base is sodium tert-butoxide. In some embodiments, the third base is potassium tert-butoxide. In some embodiments, third base is an alkaline tert-pentoxide. In some embodiments, the third base is selected from lithium tert-pentoxide, sodium tert-pentoxide, and potassium tert-pentoxide. In some embodiments, the third base is lithium tert- pentoxide. In some embodiments, the third base is sodium tert-pentoxide. In some embodiments, the third base is potassium tert-pentoxide. In some embodiments, the third base is selected from lithium bis(trimethylsilyl)amide, sodium bis(trimethylsilyl)amide, and potassium bis(trimethylsilyl)amide. In
some embodiments, the third base is lithium bis(trimethylsilyl)amide. In some embodiments, the third base is sodium bis(trimethylsilyl)amide. In some embodiments, the third base is potassium bis(trimethylsilyl) amide. In some embodiments, the third base is an alkaline hydride. In some embodiments, the third base is selected from lithium hydride, sodium hydride, and potassium hydride. In some embodiments, the third base is lithium hydride. In some embodiments, the third base is sodium hydride. In some embodiments, the third base is potassium hydride. [0168] In some embodiments, the process of preparing a compound of formula I comprises transforming a compound of formula (D-2): to the compound of formula (D-1). [0169] In some embodiments,
a compound of formula (A3-2) to the compound of formula (A3-1) comprises treating the compound of formula (A3-2) with benzyl alcohol in the presence of a third base. [0167] In some embodiments, the third base is selected from carbonate base, phosphate base, sodium hexamethyldisilizane, alkyllythium reagent, hydride reagent, alkaline alkoxide, and alkaline hydroxide. In some embodiments, the third base is an alkaline alkoxide. In some embodiments, the third base is an alkaline methoxide. In some embodiments, the third base is selected from lithium methoxide, sodium methoxide, and potassium methoxide. In some embodiments, the third base is lithium methoxide. In some embodiments, the third base is sodium methoxide. In some embodiments, the third base is potassium methoxide. In some embodiments, the third base is an alkaline ethoxide. In some embodiments, the third base is selected from lithium ethoxide, sodium ethoxide, and potassium ethoxide. In some embodiments, the third base is lithium ethoxide. In some embodiments, the third base is sodium ethoxide. In some embodiments, the third base is potassium ethoxide. In some embodiments, the third base is an alkaline tert-butoxide. In some embodiments, the third base is selected from lithium tert-butoxide, sodium tert-butoxide, and potassium tert-butoxide. In some embodiments, the third base is lithium tert-butoxide. In some embodiments, the third base is sodium tert-butoxide. In some embodiments, the third base is potassium tert-butoxide. In some embodiments, third base is an alkaline tert-pentoxide. In some embodiments, the third base is selected from lithium tert-pentoxide, sodium tert-pentoxide, and potassium tert-pentoxide. In some embodiments, the third base is lithium tert- pentoxide. In some embodiments, the third base is sodium tert-pentoxide. In some embodiments, the third base is potassium tert-pentoxide. In some embodiments, the third base is selected from lithium bis(trimethylsilyl)amide, sodium bis(trimethylsilyl)amide, and potassium bis(trimethylsilyl)amide. In
some embodiments, the third base is lithium bis(trimethylsilyl)amide. In some embodiments, the third base is sodium bis(trimethylsilyl)amide. In some embodiments, the third base is potassium bis(trimethylsilyl) amide. In some embodiments, the third base is an alkaline hydride. In some embodiments, the third base is selected from lithium hydride, sodium hydride, and potassium hydride. In some embodiments, the third base is lithium hydride. In some embodiments, the third base is sodium hydride. In some embodiments, the third base is potassium hydride. [0168] In some embodiments, the process of preparing a compound of formula I comprises transforming a compound of formula (D-2): to the compound of formula (D-1). [0169] In some embodiments,
 a compound of formula (D-2) to the compound of formula (D-1) comprises treating the compound of formula (D-2) with tetrahydroxydiboron (diboronic acid), in the presence of a third palladium catalyst, a second suitable ligand, and a fifth base. [0170] In some embodiments, the third palladium catalyst is selected from Pd(OAc)2, Pd(dppf)Cl2, Pd(PCy3)2, and Pd2dba3. In some embodiments, the third palladium catalyst is Pd(OAc)2. In some embodiments, the second suitable ligand is cyclohexyldiphenylphosphine. [0171] In some embodiments, the fifth base is selected from KOPh, triethylamine, Hünig’s Base, KOEt, and potassium acetate (KOAc). In some embodiments, the fifth base is potassium acetate. [0172] In some embodiments, the process of preparing a compound of formula I comprises transforming a compound of formula (B-1): to the compound of formula (D-2).
a compound of formula (D-2) to the compound of formula (D-1) comprises treating the compound of formula (D-2) with tetrahydroxydiboron (diboronic acid), in the presence of a third palladium catalyst, a second suitable ligand, and a fifth base. [0170] In some embodiments, the third palladium catalyst is selected from Pd(OAc)2, Pd(dppf)Cl2, Pd(PCy3)2, and Pd2dba3. In some embodiments, the third palladium catalyst is Pd(OAc)2. In some embodiments, the second suitable ligand is cyclohexyldiphenylphosphine. [0171] In some embodiments, the fifth base is selected from KOPh, triethylamine, Hünig’s Base, KOEt, and potassium acetate (KOAc). In some embodiments, the fifth base is potassium acetate. [0172] In some embodiments, the process of preparing a compound of formula I comprises transforming a compound of formula (B-1): to the compound of formula (D-2).
 [0173] In some embodiments, the step of transforming a compound of formula (B-1) to the compound of formula (D-2) comprises contacting the compound of formula (B-1) with a compound of formula (C-1): (C-1) in the presence of a sixth base. [0174] In some embodiments, the step of transforming a compound of formula (B-1) to the compound of formula (D-2) comprises contacting the compound of formula (B-2) with a compound of formula (C-1) in the presence of a sixth base and a phase transfer catalyst [0175] In some embodiments, the sixth base is potassium carbonate. [0176] In some embodiments, the phase transfer catalyst is selected from tetrabutylammonium bromide (TBAB), tetrabutylammonium chloride (TBAC), tetrabutylphosphonium bromide (TBPB), tetrabutylammonium hydrogen sulfate (TBA HSO4), and tetraoctylammonium chloride (TOAC). In other embodiments, the phase transfer catalyst is tetrabutylammonium bromide (TBAB). In other embodiments, the phase transfer catalyst is tetrabutylammonium chloride (TBAC). In other embodiments, the phase transfer catalyst is tetrabutylphosphonium bromide (TBPB). In other embodiments, the phase transfer catalyst is tetrabutylammonium hydrogen sulfate (TBA HSO4). In other embodiments, the phase transfer catalyst is tetraoctylammonium chloride (TOAC). [0177] In some embodiments, the process of preparing a compound of formula I comprises transforming a compound of formula (B-2): to the compound of formula (B-1).
[0173] In some embodiments, the step of transforming a compound of formula (B-1) to the compound of formula (D-2) comprises contacting the compound of formula (B-1) with a compound of formula (C-1): (C-1) in the presence of a sixth base. [0174] In some embodiments, the step of transforming a compound of formula (B-1) to the compound of formula (D-2) comprises contacting the compound of formula (B-2) with a compound of formula (C-1) in the presence of a sixth base and a phase transfer catalyst [0175] In some embodiments, the sixth base is potassium carbonate. [0176] In some embodiments, the phase transfer catalyst is selected from tetrabutylammonium bromide (TBAB), tetrabutylammonium chloride (TBAC), tetrabutylphosphonium bromide (TBPB), tetrabutylammonium hydrogen sulfate (TBA HSO4), and tetraoctylammonium chloride (TOAC). In other embodiments, the phase transfer catalyst is tetrabutylammonium bromide (TBAB). In other embodiments, the phase transfer catalyst is tetrabutylammonium chloride (TBAC). In other embodiments, the phase transfer catalyst is tetrabutylphosphonium bromide (TBPB). In other embodiments, the phase transfer catalyst is tetrabutylammonium hydrogen sulfate (TBA HSO4). In other embodiments, the phase transfer catalyst is tetraoctylammonium chloride (TOAC). [0177] In some embodiments, the process of preparing a compound of formula I comprises transforming a compound of formula (B-2): to the compound of formula (B-1).
 [0178] In some embodiments, the step of transforming the compound of formula (B-2) to the compound of formula (B-1) comprises treating the compound of formula (B-2) with a chlorinating reagent. In some embodiments, the chlorinating reagent is selected from HCl, oxalyl chloride, trichloroacetic anhydride/phosgene, triphenylphosphine dichloride, phenylphosphonic dichloride, phosphorous trichloride, phosphorous pentachloride, phosphorous oxychloride, thionyl chloride, p- toluenesulfonyl chloride, and methanesulfonyl chloride. In some embodiments, the chlorinating reagent is phosphorous oxychloride. In some embodiments, the chlorinating reagent is selected from Cl2 (with/without oxidant), HCl/Cl - plus oxidant, N-chlorosuccinimide (NCS)/dichlorodimethyl- hydantoin (DCDMH)/trichloroisocyanuric acid (TCCA) sodium dichloroiso-cyanurate NaDCC,
NaOCl, SO2Cl2, and CCl4/C2Cl6. In some embodiments, the chlorinating reagent is selected from phosphorus oxychloride and phosphorus pentachloride. In some embodiments, the chlorinating reagent is phosphorus oxychloride. [0179] In some embodiments, the process of preparing a compound of formula I comprises transforming a compound of formula (B-3): to the compound of formula (B-2). [0180] In some embodiments,
[0178] In some embodiments, the step of transforming the compound of formula (B-2) to the compound of formula (B-1) comprises treating the compound of formula (B-2) with a chlorinating reagent. In some embodiments, the chlorinating reagent is selected from HCl, oxalyl chloride, trichloroacetic anhydride/phosgene, triphenylphosphine dichloride, phenylphosphonic dichloride, phosphorous trichloride, phosphorous pentachloride, phosphorous oxychloride, thionyl chloride, p- toluenesulfonyl chloride, and methanesulfonyl chloride. In some embodiments, the chlorinating reagent is phosphorous oxychloride. In some embodiments, the chlorinating reagent is selected from Cl2 (with/without oxidant), HCl/Cl - plus oxidant, N-chlorosuccinimide (NCS)/dichlorodimethyl- hydantoin (DCDMH)/trichloroisocyanuric acid (TCCA) sodium dichloroiso-cyanurate NaDCC,
NaOCl, SO2Cl2, and CCl4/C2Cl6. In some embodiments, the chlorinating reagent is selected from phosphorus oxychloride and phosphorus pentachloride. In some embodiments, the chlorinating reagent is phosphorus oxychloride. [0179] In some embodiments, the process of preparing a compound of formula I comprises transforming a compound of formula (B-3): to the compound of formula (B-2). [0180] In some embodiments,
 the compound of formula (B-3) to the compound of formula (B-2) comprises treating the compound of formula (B-3) with a brominating reagent in the presence of a third acid. [0181] In some embodiments, the brominating reagent is molecular bromine. In some embodiments, the third acid is acetic acid. [0182] In some embodiments, the process of preparing a compound of formula I comprises transforming a compound of formula (B-4): to the compound of formula (B-3).
the compound of formula (B-3) to the compound of formula (B-2) comprises treating the compound of formula (B-3) with a brominating reagent in the presence of a third acid. [0181] In some embodiments, the brominating reagent is molecular bromine. In some embodiments, the third acid is acetic acid. [0182] In some embodiments, the process of preparing a compound of formula I comprises transforming a compound of formula (B-4): to the compound of formula (B-3).
 [0183] In some embodiments, the a compound of formula (B-4) to the compound of formula (B-3) comprises treating the compound of formula (B-4) with a fourth acid in the presence of a first suitable solvent. [0184] In some embodiments, the fourth acid is a mineral acid. In some embodiments, the fourth acid is concentrated aqueous hydrochloric acid. In some embodiments, the first suitable solvent comprises dioxane and water. [0185] In some embodiments, the process of preparing a compound of formula I comprise transforming a compound of formula (B-5): to the compound of formula (B-4).
[0183] In some embodiments, the a compound of formula (B-4) to the compound of formula (B-3) comprises treating the compound of formula (B-4) with a fourth acid in the presence of a first suitable solvent. [0184] In some embodiments, the fourth acid is a mineral acid. In some embodiments, the fourth acid is concentrated aqueous hydrochloric acid. In some embodiments, the first suitable solvent comprises dioxane and water. [0185] In some embodiments, the process of preparing a compound of formula I comprise transforming a compound of formula (B-5): to the compound of formula (B-4).
 [0186] In some embodiments, the step of transforming a compound of formula (B-5) to the compound of formula (B-4) comprises treating the compound of formula (B-5) with a methylating reagent in the presence of a fourth palladium catalyst, and a seventh base.
[0187] In some embodiments, the first methylating reagent is methylboronic acid. In some embodiments, the fourth palladium catalyst is Pd(dppf)Cl2•DCM. In some embodiments, the seventh base is potassium carbonate. [0188] In some embodiments, the process of preparing a compound of formula I comprises transforming a compound of formula (C-2): to the compound of formula (C-1), wherein protecting group. [0189] In some embodiments, the
[0186] In some embodiments, the step of transforming a compound of formula (B-5) to the compound of formula (B-4) comprises treating the compound of formula (B-5) with a methylating reagent in the presence of a fourth palladium catalyst, and a seventh base.
[0187] In some embodiments, the first methylating reagent is methylboronic acid. In some embodiments, the fourth palladium catalyst is Pd(dppf)Cl2•DCM. In some embodiments, the seventh base is potassium carbonate. [0188] In some embodiments, the process of preparing a compound of formula I comprises transforming a compound of formula (C-2): to the compound of formula (C-1), wherein protecting group. [0189] In some embodiments, the
 a compound of formula (C-2) to the compound of formula (C-1) comprises deprotecting the compound of formula (C-1) to afford the compound of formula (C-2). [0190] In some embodiments, PG is tetrahydropyranyl. [0191] In some embodiments, the step of transforming the compound of formula (C-2) to the compound of formula (C-1) comprises treating the compound of formula (C-2) with a first acid catalyst. In some embodiments, the first acid catalyst is pyridinium p-toluenesulfonate. [0192] In some embodiments, the step of transforming a compound of formula (C-3): to the compound of formula (C-2), wherein protecting group. In some embodiments,
a compound of formula (C-2) to the compound of formula (C-1) comprises deprotecting the compound of formula (C-1) to afford the compound of formula (C-2). [0190] In some embodiments, PG is tetrahydropyranyl. [0191] In some embodiments, the step of transforming the compound of formula (C-2) to the compound of formula (C-1) comprises treating the compound of formula (C-2) with a first acid catalyst. In some embodiments, the first acid catalyst is pyridinium p-toluenesulfonate. [0192] In some embodiments, the step of transforming a compound of formula (C-3): to the compound of formula (C-2), wherein protecting group. In some embodiments,
 the PG is tetrahydropyranyl. [0193] In some embodiments, the step of transforming a compound of formula (C-3) to the compound of formula (C-2) comprises treating the compound of formula (C-3) with a second methylating reagent in the presence of an eighth base. [0194] In some embodiments, the second methylating reagent is methyl iodide. In some embodiments, the eighth base is butyllithium. In some embodiments, the eighth base is n-butyllithium.
[0195] In some embodiments, the process of preparing a compound of formula I comprises transforming a compound of formula (C-4): to the compound of formula (C-3). [0196] In some embodiments, the
the PG is tetrahydropyranyl. [0193] In some embodiments, the step of transforming a compound of formula (C-3) to the compound of formula (C-2) comprises treating the compound of formula (C-3) with a second methylating reagent in the presence of an eighth base. [0194] In some embodiments, the second methylating reagent is methyl iodide. In some embodiments, the eighth base is butyllithium. In some embodiments, the eighth base is n-butyllithium.
[0195] In some embodiments, the process of preparing a compound of formula I comprises transforming a compound of formula (C-4): to the compound of formula (C-3). [0196] In some embodiments, the
 a compound of formula (C-4) to the compound of C-3 comprises treating the compound of (C-4) with a suitable protecting group in the presence of a second acid catalyst. In some embodiments, the suitable protecting group is 3,4-dihydropyran. In some embodiments, the second acid catalyst is pyridinium p-toluenesulfonate. [0197] In some embodiments, the process of preparing a compound of formula I comprises transforming the compound of formula (I) to a solvate of the compound of formula (I). [0198] In some embodiments, the step of transforming the compound of formula (I) to the solvate of the compound of formula (I) comprises recrystallizing the compound of formula (I) from a suitable solvent. In some embodiments, the suitable solvent is ethanol. [0199] In some embodiments, the solvate of the compound of formula (I) is a compound of formula (F): O NH O 2 . [0200] In some
a compound of formula (C-4) to the compound of C-3 comprises treating the compound of (C-4) with a suitable protecting group in the presence of a second acid catalyst. In some embodiments, the suitable protecting group is 3,4-dihydropyran. In some embodiments, the second acid catalyst is pyridinium p-toluenesulfonate. [0197] In some embodiments, the process of preparing a compound of formula I comprises transforming the compound of formula (I) to a solvate of the compound of formula (I). [0198] In some embodiments, the step of transforming the compound of formula (I) to the solvate of the compound of formula (I) comprises recrystallizing the compound of formula (I) from a suitable solvent. In some embodiments, the suitable solvent is ethanol. [0199] In some embodiments, the solvate of the compound of formula (I) is a compound of formula (F): O NH O 2 . [0200] In some
 of formula I comprises the step of transforming the solvate of the compound of formula (I) to Form A of the compound of formula (I).
of formula I comprises the step of transforming the solvate of the compound of formula (I) to Form A of the compound of formula (I).
[0201] In some embodiments, the invention relates to a compound selected from: , more
 compounds selected from: ,
, [0 und of formula (I) and a compound of formula (E-A2): . [0204] In
compounds selected from: ,
, [0 und of formula (I) and a compound of formula (E-A2): . [0204] In
 In some embodiments, the acid is trifluoroacetic acid.
In some embodiments, the acid is trifluoroacetic acid.
[0205] In some embodiments, the invention relates to a composition comprising a compound of formula (I) and a compound of formula (E-A3): . [0206] In embodiments, the acid is
 [0207] In some embodiments, a composition comprises a compound of formula (E-A2), a compound of formula (A2), and a compound of formula (D-1): A3) a
[0207] In some embodiments, a composition comprises a compound of formula (E-A2), a compound of formula (A2), and a compound of formula (D-1): A3) a
 , a :
, a :
[0209] In some embodiments, A compound of formula (I): , or a pharmaceutically acceptable is prepared by the process of any one or more of the processes disclosed
 B. Compound I Amorphous Form [0210] In some embodiments, the disclosure provides a neat amorphous form of Compound I. In some embodiments, the disclosure provides Compound I amorphous form. FIG. 1 provides an X-ray powder diffractogram of Compound I amorphous form at room temperature. [0211] In some embodiments, Compound I amorphous form is substantially pure. In some embodiments, Compound I amorphous form is substantially amorphous. In some embodiments, Compound I amorphous form is characterized by an X-ray powder diffractogram generated by an X-ray powder diffraction analysis with an incident beam of Cu Kα radiation. [0212] In some embodiments, Compound I amorphous form is characterized by an X-ray powder diffractogram substantially similar to FIG.1. [0213] In some embodiments, Compound I amorphous form is characterized as having a 13C SSNMR spectrum with a peak at 177.6 ± 0.2 ppm. In some embodiments, Compound I amorphous form is characterized as having a 13C SSNMR spectrum with a peak at 172.0 ± 0.2 ppm. In some embodiments, Compound I amorphous form is characterized as having a 13C SSNMR spectrum with a peak at 162.6 ± 0.2 ppm. In some embodiments, Compound I amorphous form is characterized as having a 13C SSNMR spectrum with a peak at 155.2 ± 0.2 ppm. In some embodiments, Compound I amorphous form is characterized as having a 13C SSNMR spectrum with a peak at 149.9 ± 0.2 ppm. In some embodiments, Compound I amorphous form is characterized as having a 13C SSNMR spectrum with a peak at 147.0 ± 0.2 ppm. In some embodiments, Compound I amorphous form is characterized as having a 13C SSNMR spectrum with a peak at 121.0 ± 0.2 ppm. In some embodiments, Compound I amorphous form is characterized as having a 13C SSNMR spectrum with a peak at 120.1 ± 0.2 ppm. In some embodiments, Compound I amorphous form is characterized as having a 13C SSNMR spectrum with a peak at 119.1 ± 0.2 ppm. In some embodiments, Compound I amorphous form is characterized as having a 13C SSNMR spectrum with a peak at 116.0 ± 0.2 ppm. In some embodiments, Compound I amorphous
form is characterized as having a 13C SSNMR spectrum with a peak at 114.6 ± 0.2 ppm. In some embodiments, Compound I amorphous form is characterized as having a 13C SSNMR spectrum with a peak at 113.8 ± 0.2 ppm. In some embodiments, Compound I amorphous form is characterized as having a 13C SSNMR spectrum with a peak at 15.5 ± 0.2 ppm. In some embodiments, Compound I amorphous form is characterized as having a 13C SSNMR spectrum with a peak at 7.7 ± 0.2 ppm. [0214] In some embodiments, Compound I amorphous form is characterized as having a 13C SSNMR spectrum with one, two, three, four, five, six, seven, eight, nine, ten, or more peaks selected from 177.6 ± 0.2 ppm, 172.0 ± 0.2 ppm, 162.6 ± 0.2 ppm, 155.2 ± 0.2 ppm, 149.9 ± 0.2 ppm, 147.0 ± 0.2 ppm, 121.0 ± 0.2 ppm, 120.1 ± 0.2 ppm, 119.1 ± 0.2 ppm, 116.0 ± 0.2 ppm, 114.6 ± 0.2 ppm, 113.8 ± 0.2 ppm, 15.5 ± 0.2 ppm, and 7.7 ± 0.2 ppm. In some embodiments, Compound I amorphous form is characterized as having a 13C SSNMR spectrum with peaks at 177.6 ± 0.2 ppm, 172.0 ± 0.2 ppm, 162.6 ± 0.2 ppm, 155.2 ± 0.2 ppm, 149.9 ± 0.2 ppm, 147.0 ± 0.2 ppm, 121.0 ± 0.2 ppm, 120.1 ± 0.2 ppm, 119.1 ± 0.2 ppm, 116.0 ± 0.2 ppm, 114.6 ± 0.2 ppm, 113.8 ± 0.2 ppm, 15.5 ± 0.2 ppm, and 7.7 ± 0.2 ppm. [0215] In some embodiments, Compound I amorphous form is characterized by a 13C SSNMR spectrum substantially similar to FIG.2. [0216] In some embodiments, Compound I amorphous form characterized as having a 19F SSNMR spectrum with a peak at -61.9 ± 0.2 ppm. In some embodiments, Compound I amorphous form characterized as having a 19F SSNMR spectrum with a peak at -142.1 ± 0.2 ppm. [0217] In some embodiments, Compound I amorphous form is characterized as having a 19F SSNMR spectrum with one or two peaks selected from -61.9 ± 0.2 ppm and -142.1 ± 0.2 ppm. [0218] In some embodiments, Compound I amorphous form is characterized by a 19F SSNMR spectrum substantially similar to FIG.3. [0219] Another aspect of the disclosure provides a method of making Compound I amorphous form. In some embodiments, the method of making Compound I amorphous form comprises: comprising (i) dissolving Compound I, (ii) filtering and evaporating the solvent at 50 °C, 12 mbar, over 1 hour on a centrifugal evaporation, (iii) drying in a vacuum oven at 50 °C overnight to yield Compound I amorphous form. C. Crystalline Compound I Neat Form A [0220] In some embodiments, the disclosure provides neat crystalline forms of Compound I. In some embodiments, the disclosure provides crystalline Compound I neat Form A. FIG.4 provides an X-ray powder diffractogram of crystalline Compound I neat Form A. [0221] In some embodiments, crystalline Compound I neat Form A is substantially pure. In some embodiments, crystalline Compound I neat Form A is substantially crystalline. In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram generated by an X-ray powder diffraction analysis with an incident beam of Cu Kα radiation.
[0222] In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 9.5 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 12.3 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 13.5 ± 0.2 degrees two-theta. [0223] In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 7.4 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 8.3 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 11.3 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 14.4 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 14.9 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 15.0 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 15.9 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 16.6 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 17.6 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 18.0 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 18.7 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 18.9 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 19.2 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 19.5 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 19.8 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 20.5 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 21.4 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 21.9 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 22.5 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form A is
characterized by an X-ray powder diffractogram having a signal at 23.5 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 24.1 ± 0.2 degrees two-theta. [0224] In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having signals at one or two of 9.5 ± 0.2 degrees two theta, 12.3 ± 0.2 degrees two theta, and 13.5 ± 0.2 degrees two-theta. [0225] In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having (a) one or two signals selected from 9.5 ± 0.2 degrees two-theta, 12.3 ± 0.2 degrees two-theta, and 13.5 ± 0.2 degrees two-theta, and (b) one or two signals selected from 11.3 ± 0.2 degrees two-theta, 15.9 ± 0.2 degrees two-theta, and 20.5 ± 0.2 degrees two-theta. [0226] In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having two, three, four, five, or six signals selected from 9.5 ± 0.2 degrees two- theta, 12.3 ± 0.2 degrees two-theta, 13.5 ± 0.2 degrees two-theta , 11.3 ± 0.2 degrees two-theta, 15.9 ± 0.2 degrees two-theta, 20.5 ± 0.2 degrees two-theta, 14.9 ± 0.2 degrees two-theta, 16.6 ± 0.2 degrees two-theta, and 23.5 ± 0.2 degrees two-theta. [0227] In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having one, two three, four five, six, seven, eight, nine, ten, eleven, twelve, thirteen, fourteen, fifteen, sixteen, seventeen, eighteen, nineteen, twenty, twenty-one, or twenty-two signals selected from 7.4 ± 0.2 degrees two-theta; 8.3 ± 0.2 degrees two-theta; 9.5 ± 0.2 degrees two- theta; 11.3 ± 0.2 degrees two-theta; 12.3 ± 0.2 degrees two-theta; 13.5 ± 0.2 degrees two-theta; 14.4 ± 0.2 degrees two-theta; 14.9 ± 0.2 degrees two-theta; 15.0 ± 0.2 degrees two-theta; 15.9 ± 0.2 degrees two-theta; 16.6 ± 0.2 degrees two-theta; 17.6 ± 0.2 degrees two-theta; 18.0 ± 0.2 degrees two-theta; 18.7 ± 0.2 degrees two-theta; 18.9 ± 0.2 degrees two-theta; 19.2 ± 0.2 degrees two-theta; 19.5 ± 0.2 degrees two-theta; 19.8 ± 0.2 degrees two-theta; 20.5 ± 0.2 degrees two-theta; 21.4 ± 0.2 degrees two-theta; 21.9 ± 0.2 degrees two-theta; 22.5 ± 0.2 degrees two-theta; 23.5 ± 0.2 degrees two-theta; and 24.1 ± 0.2 degrees two-theta. [0228] In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having signals at 7.4 ± 0.2 degrees two-theta, 8.3 ± 0.2 degrees two-theta, 9.5 ± 0.2 degrees two-theta, 11.3 ± 0.2 degrees two-theta, 12.3 ± 0.2 degrees two-theta, 13.5 ± 0.2 degrees two-theta, 14.4 ± 0.2 degrees two-theta, 14.9 ± 0.2 degrees two-theta, 15.0 ± 0.2 degrees two-theta, 15.9 ± 0.2 degrees two-theta, 16.6 ± 0.2 degrees two-theta, 17.6 ± 0.2 degrees two-theta, 18.0 ± 0.2 degrees two-theta, 18.7 ± 0.2 degrees two-theta, 18.9 ± 0.2 degrees two-theta, 19.2 ± 0.2 degrees two-theta, 19.5 ± 0.2 degrees two-theta, 19.8 ± 0.2 degrees two-theta, 20.5 ± 0.2 degrees two-theta, 21.4 ± 0.2 degrees two-theta, 21.9 ± 0.2 degrees two-theta, 22.5 ± 0.2 degrees two-theta, 23.5 ± 0.2 degrees two-theta, and 24.1 ± 0.2 degrees two-theta. [0229] In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram substantially similar to FIG.4.
[0230] In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a peak at 170.4 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a peak at 163.8 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a peak at 162.4 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a peak at 160.9 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a peak at 158.6 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a peak at 158.3 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a peak at 154.6 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a peak at 154.0 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a peak at 148.3 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a peak at 147.6 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a peak at 146.9 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a peak at 145.9 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a peak at 143.5 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a peak at 143.1 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a peak at 141.9 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a peak at 127.4 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a peak at 125.3 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a peak at 123.6 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a peak at 122.2 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a peak at 121.3 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a peak at 120.7 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a peak at 117.9 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a peak at 117.3 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a peak at 116.3 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a peak at 115.8 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a peak at
114.2 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a peak at 113.6 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a peak at 112.8 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a peak at 15.4 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a peak at 8.2 ± 0.2 ppm. [0231] In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with one, two, three, four, five, six, seven, eight, nine, ten, or more peaks selected from 170.4 ± 0.2 ppm, 163.8 ± 0.2 ppm, 162.4 ± 0.2 ppm, 160.9 ± 0.2 ppm, 158.6 ± 0.2 ppm, 158.3 ± 0.2 ppm, 154.6 ± 0.2 ppm, 154.0 ± 0.2 ppm, 148.3 ± 0.2 ppm, 147.6 ± 0.2 ppm, 146.9 ± 0.2 ppm, 145.9 ± 0.2 ppm, 143.5 ± 0.2 ppm, 143.1 ± 0.2 ppm, 141.9 ± 0.2 ppm, 127.4 ± 0.2 ppm, 125.3 ± 0.2 ppm, 123.6 ± 0.2 ppm, 122.2 ± 0.2 ppm, 121.3 ± 0.2 ppm, 120.7 ± 0.2 ppm, 117.9 ± 0.2 ppm, 117.3 ± 0.2 ppm, 116.3 ± 0.2 ppm, 115.8 ± 0.2 ppm, 114.2 ± 0.2 ppm, 113.6 ± 0.2 ppm, 112.8 ± 0.2 ppm, 15.4 ± 0.2 ppm, and 8.2 ± 0.2 ppm. [0232] In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a 13C SSNMR spectrum having peaks at 170.4 ± 0.2 ppm, 163.8 ± 0.2 ppm, 162.4 ± 0.2 ppm, 160.9 ± 0.2 ppm, 158.6 ± 0.2 ppm, 158.3 ± 0.2 ppm, 154.6 ± 0.2 ppm, 154.0 ± 0.2 ppm, 148.3 ± 0.2 ppm, 147.6 ± 0.2 ppm, 146.9 ± 0.2 ppm, 145.9 ± 0.2 ppm, 143.5 ± 0.2 ppm, 143.1 ± 0.2 ppm, 141.9 ± 0.2 ppm, 127.4 ± 0.2 ppm, 125.3 ± 0.2 ppm, 123.6 ± 0.2 ppm, 122.2 ± 0.2 ppm, 121.3 ± 0.2 ppm, 120.7 ± 0.2 ppm, 117.9 ± 0.2 ppm, 117.3 ± 0.2 ppm, 116.3 ± 0.2 ppm, 115.8 ± 0.2 ppm, 114.2 ± 0.2 ppm, 113.6 ± 0.2 ppm, 112.8 ± 0.2 ppm, 15.4 ± 0.2 ppm, and 8.2 ± 0.2 ppm. [0233] In some embodiments, crystalline Compound I neat Form A is characterized by a 13C SSNMR spectrum substantially similar to FIG.5. [0234] In some embodiments, crystalline Compound I neat Form A is characterized as having a 19F SSNMR spectrum with a peak at -61.2 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 19F SSNMR spectrum with a peak at -61.6 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 19F SSNMR spectrum with a peak at -138.6 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 19F SSNMR spectrum with a peak at -140.3 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 19F SSNMR spectrum with a peak at -142.0 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 19F SSNMR spectrum with a peak at -146.0 ± 0.2 ppm. [0235] In some embodiments, crystalline Compound I neat Form A is characterized as having a 19F SSNMR spectrum with one or two peaks selected from -61.2 ± 0.2 ppm, -61.6 ± 0.2 ppm, -138.6 ± 0.2 ppm, -140.3 ± 0.2 ppm, -142.0 ± 0.2 ppm, and -146.0 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 19F SSNMR spectrum with peaks at
-61.2 ± 0.2 ppm, -61.6 ± 0.2 ppm, -138.6 ± 0.2 ppm, -140.3 ± 0.2 ppm, -142.0 ± 0.2 ppm, and -146.0 ± 0.2 ppm. [0236] In some embodiments, crystalline Compound I neat Form A is characterized by a 19F SSNMR spectrum substantially similar to FIG.6. [0237] In some embodiments, crystalline Compound I neat Form A is characterized by a triclinic crystal system, P-1 space group and the following unit cell dimensions measured at 100 K on a diffractometer utilizing Cu Kα radiation (λ=1.54178 Å): a 9.6 ± 0.1 Å α 94.3 ± 0.1° b 12.6 ± 0.1 Å β 97.3 ± 0.1° c 18.7 ± 0.1 Å γ 107.3 ± 0.1°. In some embodiments, the diffractometer is a Bruker diffractometer. [0238] Another aspect of the invention provides a method of making crystalline Compound I neat Form A. In some embodiments, the method of making crystalline Compound I neat Form A comprises: (i) combining Compound I and ethyl acetate, (ii) distilling under vacuum at 50 °C, (iii) adding ethyl acetate, (iv) distilling under vacuum at 50 °C, (v) heating to 75 °C, (vi) adding heptane, (vii) cooling to 40 °C, (viii) adding a seed of crystalline Compound I Form A, (ix) holding at 40 °C for 1.5 hours, (x) adding heptane over 5 hours, (xi) cooling the slurry to 20 °C over 5 hours, (xii) holding at 20 °C for 11 hours, (xiii) collecting the solids, (xiv) drying the solids in a vacuum oven at 45 °C with a slight nitrogen bleed for 46 hours to yield crystalline Compound I neat Form A. D. Crystalline Compound I Neat Form B [0239] In some embodiments, the disclosure provides crystalline Compound I neat Form B. FIG. 7 provides an X-ray powder diffractogram of crystalline Compound I neat Form B. [0240] In some embodiments, crystalline Compound I neat Form B is substantially pure. In some embodiments, crystalline Compound I neat Form B is substantially crystalline. In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram generated by an X-ray powder diffraction analysis with an incident beam of Cu Kα radiation. [0241] In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 4.8 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 13.3 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 18.5 ± 0.2 degrees two-theta. [0242] In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 6.0 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 7.0 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 8.8 ± 0.2 degrees two-theta. In some embodiments,
crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 10.5 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 12.1 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 13.8 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 14.1 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 14.7 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 15.2 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 16.5 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 16.9 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 17.5 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 17.9 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 18.9 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 19.2 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 19.5 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 20.9 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 21.7 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 23.0 ± 0.2 degrees two-theta. [0243] In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having signals at one or two 4.8 ± 0.2 degrees two-theta, 13.3 ± 0.2 degrees two-theta, and 18.5 ± 0.2 degrees two-theta. [0244] In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having (a) one or two signals selected from from 4.8 ± 0.2 degrees two-theta, 13.3 ± 0.2 degrees two-theta, and 18.5 ± 0.2 degrees two-theta, and (b) one or two signals selected from 6.0 ± 0.2 degrees two-theta, 16.5 ± 0.2 degrees two-theta, and 16.9 ± 0.2 degrees two-theta. [0245] In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having two, three, four, five, or six signals selected from 4.8 ± 0.2 degrees two-theta, 13.3 ± 0.2 degrees two-theta, 18.5 ± 0.2 degrees two-theta, 6.0 ± 0.2 degrees two-theta,
16.5 ± 0.2 degrees two-theta, 16.9 ± 0.2 degrees two-theta, 14.1 ± 0.2 degrees two-theta, 14.7 ± 0.2 degrees two-theta, and 15.2 ± 0.2 degrees two-theta. [0246] In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having one, two three, four five, six, seven, eight, nine, ten, eleven, twelve, thirteen, fourteen, fifteen, sixteen, seventeen, eighteen, nineteen, twenty, twenty-one, or twenty-two signals selected from 4.8 ± 0.2 degrees two-theta; 6.0 ± 0.2 degrees two-theta; 7.0 ± 0.2 degrees two-theta; 8.8 ± 0.2 degrees two-theta; 10.5 ± 0.2 degrees two-theta; 12.1 ± 0.2 degrees two-theta; 13.3 ± 0.2 degrees two-theta; 13.8 ± 0.2 degrees two-theta; 14.1 ± 0.2 degrees two-theta; 14.7 ± 0.2 degrees two-theta; 15.2 ± 0.2 degrees two-theta; 16.5 ± 0.2 degrees two-theta; 16.9 ± 0.2 degrees two-theta; 17.5 ± 0.2 degrees two-theta; 17.9 ± 0.2 degrees two-theta; 18.5 ± 0.2 degrees two-theta; 18.9 ± 0.2 degrees two-theta; 19.2 ± 0.2 degrees two-theta; 19.5 ± 0.2 degrees two-theta; 20.9 ± 0.2 degrees two-theta; 21.7 ± 0.2 degrees two-theta; and 23.0 ± 0.2 degrees two-theta. [0247] In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having signals at 4.8 ± 0.2 degrees two-theta, 6.0 ± 0.2 degrees two-theta, 7.0 ± 0.2 degrees two-theta, 8.8 ± 0.2 degrees two-theta, 10.5 ± 0.2 degrees two-theta, 12.1 ± 0.2 degrees two-theta, 13.3 ± 0.2 degrees two-theta, 13.8 ± 0.2 degrees two-theta, 14.1 ± 0.2 degrees two-theta, 14.7 ± 0.2 degrees two-theta, 15.2 ± 0.2 degrees two-theta, 16.5 ± 0.2 degrees two-theta, 16.9 ± 0.2 degrees two-theta, 17.5 ± 0.2 degrees two-theta, 17.9 ± 0.2 degrees two-theta, 18.5 ± 0.2 degrees two-theta, 18.9 ± 0.2 degrees two-theta, 19.2 ± 0.2 degrees two-theta, 19.5 ± 0.2 degrees two-theta, 20.9 ± 0.2 degrees two-theta, 21.7 ± 0.2 degrees two-theta, and 23.0 ± 0.2 degrees two-theta. [0248] In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram substantially similar to FIG.7. [0249] In some embodiments, crystalline Compound I neat Form B is characterized as having a 13C SSNMR spectrum with a peak at 176.9 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13C SSNMR spectrum with a peak at 171.7 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13C SSNMR spectrum with a peak at 164.0 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13C SSNMR spectrum with a peak at 161.4 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13C SSNMR spectrum with a peak at 160.7 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13C SSNMR spectrum with a peak at 155.3 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13C SSNMR spectrum with a peak at 154.8 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13C SSNMR spectrum with a peak at 148.6 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13C SSNMR spectrum with a peak at 147.6 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13C SSNMR spectrum with a peak at 146.4 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is
characterized as having a 13C SSNMR spectrum with a peak at 145.7 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13C SSNMR spectrum with a peak at 144.8 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13C SSNMR spectrum with a peak at 127.7 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13C SSNMR spectrum with a peak at 125.4 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13C SSNMR spectrum with a peak at 122.0 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13C SSNMR spectrum with a peak at 121.5 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13C SSNMR spectrum with a peak at 118.4 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13C SSNMR spectrum with a peak at 116.3 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13C SSNMR spectrum with a peak at 114.8 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13C SSNMR spectrum with a peak at 113.8 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13C SSNMR spectrum with a peak at 112.6 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13C SSNMR spectrum with a peak at 17.8 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13C SSNMR spectrum with a peak at 15.0 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13C SSNMR spectrum with a peak at 10.7 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13C SSNMR spectrum with a peak at 9.6 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13C SSNMR spectrum with a peak at 8.8 ± 0.2 ppm. [0250] In some embodiments, crystalline Compound I neat Form B is characterized as having a 13C SSNMR spectrum with one, two, three, four, five, six, seven, eight, nine, ten, or more peaks selected from 176.9 ± 0.2 ppm, 171.7 ± 0.2 ppm, 164.0 ± 0.2 ppm, 161.4 ± 0.2 ppm, 160.7 ± 0.2 ppm, 155.3 ± 0.2 ppm, 154.8 ± 0.2 ppm, 148.6 ± 0.2 ppm, 147.6 ± 0.2 ppm, 146.4 ± 0.2 ppm, 145.7 ± 0.2 ppm, 144.8 ± 0.2 ppm, 127.7 ± 0.2 ppm, 125.4 ± 0.2 ppm, 122.0 ± 0.2 ppm, 121.5 ± 0.2 ppm, 118.4 ± 0.2 ppm, 116.3 ± 0.2 ppm, 114.8 ± 0.2 ppm, 113.8 ± 0.2 ppm, 112.6 ± 0.2 ppm, 17.8 ± 0.2 ppm, 15.0 ± 0.2 ppm, 10.7 ± 0.2 ppm, 9.6 ± 0.2 ppm, and 8.8 ± 0.2 ppm. [0251] In some embodiments, crystalline Compound I neat Form B is characterized as having a 13C SSNMR spectrum with peaks at 176.9 ± 0.2 ppm, 171.7 ± 0.2 ppm, 164.0 ± 0.2 ppm, 161.4 ± 0.2 ppm, 160.7 ± 0.2 ppm, 155.3 ± 0.2 ppm, 154.8 ± 0.2 ppm, 148.6 ± 0.2 ppm, 147.6 ± 0.2 ppm, 146.4 ± 0.2 ppm, 145.7 ± 0.2 ppm, 144.8 ± 0.2 ppm, 127.7 ± 0.2 ppm, 125.4 ± 0.2 ppm, 122.0 ± 0.2 ppm, 121.5 ± 0.2 ppm, 118.4 ± 0.2 ppm, 116.3 ± 0.2 ppm, 114.8 ± 0.2 ppm, 113.8 ± 0.2 ppm, 112.6 ± 0.2 ppm, 17.8 ± 0.2 ppm, 15.0 ± 0.2 ppm, 10.7 ± 0.2 ppm, 9.6 ± 0.2 ppm, and 8.8 ± 0.2 ppm. [0252] In some embodiments, crystalline Compound I neat Form B is characterized by a 13C SSNMR spectrum substantially similar to FIG.8.
[0253] In some embodiments, crystalline Compound I neat Form B is characterized as having a 19F SSNMR spectrum with a peak at -61.6 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 19F SSNMR spectrum with a peak at -138.0 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 19F SSNMR spectrum with a peak at -139.7 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 19F SSNMR spectrum with a peak at -143.4 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 19F SSNMR spectrum with a peak at -144.9 ± 0.2 ppm. [0254] In some embodiments, crystalline Compound I neat Form B is characterized as having a 19F SSNMR spectrum with one or two peaks selected from -61.6 ± 0.2 ppm, -138.0 ± 0.2 ppm, -139.7 ± 0.2 ppm, -143.4 ± 0.2 ppm, and -144.9 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 19F SSNMR spectrum with peaks at -61.6 ± 0.2 ppm, -138.0 ± 0.2 ppm, -139.7 ± 0.2 ppm, -143.4 ± 0.2 ppm, and -144.9 ± 0.2 ppm. [0255] In some embodiments, crystalline Compound I neat Form B is characterized by a 19F SSNMR spectrum substantially similar to FIG.9. [0256] Another aspect of the disclosure provides a method of crystalline Compound I neat Form B. In some embodiments, the method of making crystalline Compound I neat Form B, comprises: (i) adding Compound I hydrate Form A to an oven set at 180 °C (ii) cooling under ambient conditions to yield crystalline Compound I neat Form B. E. Crystalline Compound I Neat Form E [0257] In some embodiments, the disclosure provides crystalline Compound I neat Form E. FIG. 10 provides an X-ray powder diffractogram of crystalline Compound I neat Form E. [0258] In some embodiments, crystalline Compound I neat Form E is substantially pure. In some embodiments, crystalline Compound I neat Form E is substantially crystalline. In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram generated by an X-ray powder diffraction analysis with an incident beam of Cu Kα radiation. [0259] In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 8.6 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 10.1 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 12.6 ± 0.2 degrees two-theta. [0260] In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 8.5 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 8.7 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 11.5 ± 0.2 degrees two-theta. In some embodiments,
crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 12.0 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 14.1 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 14.2 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 15.1 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 16.4 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 20.3 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 20.6 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 21.0 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 21.4 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 22.1 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 22.5 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 23.0 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 23.2 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 24.1 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 24.7 ± 0.2 degrees two-theta. [0261] In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having signals at one or two selected from 8.6 ± 0.2 degrees two-theta, 10.1 ± 0.2 degrees two-theta, and 12.6 ± 0.2 degrees two-theta. [0262] In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having (a) one or two signals selected from 8.6 ± 0.2 degrees two-theta, 10.1 ± 0.2 degrees two-theta, and 12.6 ± 0.2 degrees two-theta and (b) one or two signals selected from 14.1 ± 0.2 degrees two-theta, 20.3 ± 0.2 degrees two-theta, and 23.2 ± 0.2 degrees two-theta. [0263] In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having two, three, four, five, or six signals selected from 8.6 ± 0.2 degrees two-theta, 10.1 ± 0.2 degrees two-theta, 12.6 ± 0.2 degrees two-theta, 14.1 ± 0.2 degrees two-theta, 20.3 ± 0.2 degrees two-theta, 23.2 ± 0.2 degrees two-theta, 11.5 ± 0.2 degrees two-theta, 14.2 ± 0.2 degrees two-theta, and 15.1 ± 0.2 degrees two-theta.
[0264] In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having one, two three, four five, six, seven, eight, nine, ten, eleven, twelve, thirteen, fourteen, fifteen, sixteen, seventeen, eighteen, nineteen, twenty, twenty-one, or twenty-two signals selected from 8.5 ± 0.2 degrees two-theta; 8.6 ± 0.2 degrees two-theta; 8.7 ± 0.2 degrees two-theta; 10.1 ± 0.2 degrees two-theta; 11.5 ± 0.2 degrees two-theta; 12.0 ± 0.2 degrees two-theta; 12.6 ± 0.2 degrees two-theta; 14.1 ± 0.2 degrees two-theta; 14.2 ± 0.2 degrees two-theta; 15.1 ± 0.2 degrees two-theta; 16.4 ± 0.2 degrees two-theta; 20.3 ± 0.2 degrees two-theta; 20.6 ± 0.2 degrees two-theta; 21.0 ± 0.2 degrees two-theta; 21.4 ± 0.2 degrees two-theta; 22.1 ± 0.2 degrees two-theta; 22.5 ± 0.2 degrees two-theta; 23.0 ± 0.2 degrees two-theta; 23.2 ± 0.2 degrees two-theta; 24.1 ± 0.2 degrees two-theta; and 24.7 ± 0.2 degrees two-theta. [0265] In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having signals at 8.5 ± 0.2 degrees two-theta, 8.6 ± 0.2 degrees two-theta, 8.7 ± 0.2 degrees two-theta, 10.1 ± 0.2 degrees two-theta, 11.5 ± 0.2 degrees two-theta, 12.0 ± 0.2 degrees two-theta, 12.6 ± 0.2 degrees two-theta, 14.1 ± 0.2 degrees two-theta, 14.2 ± 0.2 degrees two-theta, 15.1 ± 0.2 degrees two-theta, 16.4 ± 0.2 degrees two-theta, 20.3 ± 0.2 degrees two-theta, 20.6 ± 0.2 degrees two-theta, 21.0 ± 0.2 degrees two-theta, 21.4 ± 0.2 degrees two-theta, 22.1 ± 0.2 degrees two-theta, 22.5 ± 0.2 degrees two-theta, 23.0 ± 0.2 degrees two-theta, 23.2 ± 0.2 degrees two-theta, 24.1 ± 0.2 degrees two-theta, and 24.7 ± 0.2 degrees two-theta. [0266] In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram substantially similar to FIG.10. [0267] In some embodiments, crystalline Compound I neat Form E is characterized as having a 13C SSNMR spectrum with a peak at 170.0 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13C SSNMR spectrum with a peak at 163.0 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13C SSNMR spectrum with a peak at 162.3 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13C SSNMR spectrum with a peak at 161.3 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13C SSNMR spectrum with a peak at 159.3 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13C SSNMR spectrum with a peak at 154.7 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13C SSNMR spectrum with a peak at 150.6 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13C SSNMR spectrum with a peak at 149.8 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13C SSNMR spectrum with a peak at 148.6 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13C SSNMR spectrum with a peak at 148.3 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13C SSNMR spectrum with a peak at 147.1 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13C SSNMR spectrum with a peak at
144.9 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13C SSNMR spectrum with a peak at 143.9 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13C SSNMR spectrum with a peak at 143.5 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13C SSNMR spectrum with a peak at 126.9 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13C SSNMR spectrum with a peak at 126.0 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13C SSNMR spectrum with a peak at 124.1 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13C SSNMR spectrum with a peak at 122.6 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13C SSNMR spectrum with a peak at 120.8 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13C SSNMR spectrum with a peak at 117.8 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13C SSNMR spectrum with a peak at 116.7 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13C SSNMR spectrum with a peak at 115.2 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13C SSNMR spectrum with a peak at 114.1 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13C SSNMR spectrum with a peak at 113.1 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13C SSNMR spectrum with a peak at 15.9 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13C SSNMR spectrum with a peak at 15.3 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13C SSNMR spectrum with a peak at 8.4 ± 0.2 ppm. [0268] In some embodiments, crystalline Compound I neat Form E is characterized as having a 13C SSNMR spectrum with one, two, three, four, five, six, seven, eight, nine, ten, or more peaks selected from 170.0 ± 0.2 ppm, 163.0 ± 0.2 ppm, 162.3 ± 0.2 ppm, 161.3 ± 0.2 ppm, 159.3 ± 0.2 ppm, 154.7 ± 0.2 ppm, 150.6 ± 0.2 ppm, 149.8 ± 0.2 ppm, 148.6 ± 0.2 ppm, 148.3 ± 0.2 ppm, 147.1 ± 0.2 ppm, 144.9 ± 0.2 ppm, 143.9 ± 0.2 ppm, 143.5 ± 0.2 ppm, 126.9 ± 0.2 ppm, 126.0 ± 0.2 ppm, 124.1 ± 0.2 ppm, 122.6 ± 0.2 ppm, 120.8 ± 0.2 ppm, 117.8 ± 0.2 ppm, 116.7 ± 0.2 ppm, 115.2 ± 0.2 ppm, 114.1 ± 0.2 ppm, 113.1 ± 0.2 ppm, 15.9 ± 0.2 ppm, 15.3 ± 0.2 ppm, and 8.4 ± 0.2 ppm. [0269] In some embodiments, crystalline Compound I neat Form E is characterized with peaks at 170.0 ± 0.2 ppm, 163.0 ± 0.2 ppm, 162.3 ± 0.2 ppm, 161.3 ± 0.2 ppm, 159.3 ± 0.2 ppm, 154.7 ± 0.2 ppm, 150.6 ± 0.2 ppm, 149.8 ± 0.2 ppm, 148.6 ± 0.2 ppm, 148.3 ± 0.2 ppm, 147.1 ± 0.2 ppm, 144.9 ± 0.2 ppm, 143.9 ± 0.2 ppm, 143.5 ± 0.2 ppm, 126.9 ± 0.2 ppm, 126.0 ± 0.2 ppm, 124.1 ± 0.2 ppm, 122.6 ± 0.2 ppm, 120.8 ± 0.2 ppm, 117.8 ± 0.2 ppm, 116.7 ± 0.2 ppm, 115.2 ± 0.2 ppm, 114.1 ± 0.2 ppm, 113.1 ± 0.2 ppm, 15.9 ± 0.2 ppm, 15.3 ± 0.2 ppm, and 8.4 ± 0.2 ppm. [0270] In some embodiments, crystalline Compound I neat Form E is characterized by a 13C SSNMR spectrum substantially similar to FIG.11.
[0271] In some embodiments, crystalline Compound I neat Form E is characterized as having a 19F SSNMR spectrum with a peak at -60.9 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 19F SSNMR spectrum with a peak at -62.5 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 19F SSNMR spectrum with a peak at -63.1 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 19F SSNMR spectrum with a peak at -135.1 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 19F SSNMR spectrum with a peak at -137.1 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 19F SSNMR spectrum with a peak at -140.7 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 19F SSNMR spectrum with a peak at -141.9 ± 0.2 ppm. [0272] In some embodiments, crystalline Compound I neat Form E is characterized as having a 19F SSNMR spectrum with one or two peaks selected from -60.9 ± 0.2 ppm, -62.5 ± 0.2 ppm, -63.1 ± 0.2 ppm, -135.1 ± 0.2 ppm, -137.1 ± 0.2 ppm, -140.7 ± 0.2 ppm, and -141.9 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 19F SSNMR spectrum with peaks at -60.9 ± 0.2 ppm, -62.5 ± 0.2 ppm, -63.1 ± 0.2 ppm, -135.1 ± 0.2 ppm, -137.1 ± 0.2 ppm, -140.7 ± 0.2 ppm, and -141.9 ± 0.2 ppm. [0273] In some embodiments, crystalline Compound I neat Form E is characterized by a 19F SSNMR spectrum substantially similar to FIG.12. [0274] In some embodiments, crystalline Compound I neat Form E is characterized by a triclinic crystal system, P-1 space group and the following unit cell dimensions measured at 100 K on a diffractometer utilizing Cu Kα radiation (λ=1.54178 Å): a 8.7 ± 0.1 Å α 92.7 ± 0.1° b 12.2 ± 0.1 Å β 96.3 ± 0.1° c 20.9 ± 0.1 Å γ 98.6 ± 0.1°. In some embodiments, the diffractometer is a Rigaku diffractometer. [0275] In some embodiments, crystalline Compound I neat Form E is characterized by a triclinic crystal system, P-1 space group and the following unit cell dimensions measured at 298 K on a diffractometer utilizing Cu Kα radiation (λ=1.54178 Å): a 8.9 ± 0.1 Å α 92.6 ± 0.1° b 12.3 ± 0.1 Å β 97.1 ± 0.1° c 21.1 ± 0.1 Å γ 98.5 ± 0.1°. In some embodiments, the diffractometer is a Rigaku diffractometer. [0276] Another aspect of the disclosure provides a method of crystalline Compound I neat Form E. In some embodiments, the method of making crystalline Compound I neat Form E, comprises: (i) combining Compound I hydrate Form A and trifluorotoluene, (ii) heating the slurry to 80 °C, (iii) cooling to room temperature to yield crystalline Compound I neat Form E.
F. Crystalline Compound I Acetone Solvate Hydrate Form A [0277] In some embodiments, the disclosure provides crystalline Compound I Acetone Solvate Hydrate Form A. FIG.13 provides an X-ray powder diffractogram of crystalline Compound I Acetone Solvate Hydrate Form A. [0278] In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is substantially pure. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is substantially crystalline. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram generated by an X-ray powder diffraction analysis with an incident beam of Cu Kα radiation. [0279] In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 17.2 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 18.1 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 25.9 ± 0.2 degrees two-theta. [0280] In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 7.2 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 8.1 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 9.4 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 9.7 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 11.2 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 11.5 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 12.3 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 12.5 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 12.9 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 13.1 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 13.9 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 14.4 ± 0.2 degrees two-theta. In some embodiments, crystalline
Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 14.8 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 15.1 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 15.6 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 16.2 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 16.5 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 17.6 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 17.7 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 22.7 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 24.8 ± 0.2 degrees two-theta. [0281] In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having signals at one or two of 17.2 ± 0.2 degrees two-theta, 18.1 ± 0.2 degrees two-theta, and 25.9 ± 0.2 degrees two-theta. [0282] In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having (a) one or two signals selected 17.2 ± 0.2 degrees two-theta, 18.1 ± 0.2 degrees two-theta, and 25.9 ± 0.2 degrees two-theta, and (b) one or two signals selected from 12.9 ± 0.2 degrees two-theta, 15.6 ± 0.2 degrees two-theta, and 24.8 ± 0.2 degrees two-theta. [0283] In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having two, three, four, five, or six signals selected from 9.5 ± 0.2 degrees two-theta, 12.3 ± 0.2 degrees two-theta, 13.5 ± 0.2 degrees two-theta, 12.9 ± 0.2 degrees two-theta, 15.6 ± 0.2 degrees two-theta, 24.8 ± 0.2 degrees two-theta, 8.1 ± 0.2 degrees two-theta, 12.3 ± 0.2 degrees two-theta, and 17.6 ± 0.2 degrees two-theta. [0284] In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having one, two three, four five, six, seven, eight, nine, ten, eleven, twelve, thirteen, fourteen, fifteen, sixteen, seventeen, eighteen, nineteen, twenty, twenty-one, or twenty-two signals selected from 7.2 ± 0.2 degrees two-theta; 8.1 ± 0.2 degrees two-theta; 9.7 ± 0.2 degrees two-theta; 11.2 ± 0.2 degrees two-theta; 11.5 ± 0.2 degrees two-theta; 12.3 ± 0.2 degrees two-theta; 12.5 ± 0.2 degrees two-theta; 12.9 ± 0.2 degrees two-theta; 13.1 ± 0.2 degrees two-theta; 13.9 ± 0.2 degrees two-theta; 14.4 ± 0.2 degrees two-theta; 14.8 ± 0.2 degrees two-theta; 15.1 ± 0.2 degrees two-theta; 15.5 ± 0.2 degrees two-theta; 16.2 ± 0.2 degrees two-theta;
16.5 ± 0.2 degrees two-theta; 17.2 ± 0.2 degrees two-theta; 17.6 ± 0.2 degrees two-theta; 17.7 ± 0.2 degrees two-theta; 18.1 ± 0.2 degrees two-theta; 22.7 ± 0.2 degrees two-theta; 24.8 ± 0.2 degrees two-theta; and 25.9 ± 0.2 degrees two-theta. [0285] In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having signals at 7.2 ± 0.2 degrees two-theta, 8.1 ± 0.2 degrees two-theta, 9.7 ± 0.2 degrees two-theta, 11.2 ± 0.2 degrees two-theta, 11.5 ± 0.2 degrees two-theta, 12.3 ± 0.2 degrees two-theta, 12.5 ± 0.2 degrees two-theta, 12.9 ± 0.2 degrees two-theta, 13.1 ± 0.2 degrees two-theta, 13.9 ± 0.2 degrees two-theta, 14.4 ± 0.2 degrees two-theta, 14.8 ± 0.2 degrees two-theta, 15.1 ± 0.2 degrees two-theta, 15.5 ± 0.2 degrees two-theta, 16.2 ± 0.2 degrees two-theta, 16.5 ± 0.2 degrees two-theta, 17.2 ± 0.2 degrees two-theta, 17.6 ± 0.2 degrees two-theta, 17.7 ± 0.2 degrees two-theta, 18.1 ± 0.2 degrees two-theta, 22.7 ± 0.2 degrees two-theta, 24.8 ± 0.2 degrees two-theta, and 25.9 ± 0.2 degrees two-theta. [0286] In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram substantially similar to FIG.13. [0287] In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 212.9 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 211.6 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 179.2 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 178.9 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 178.4 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 174.7 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 174.1 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 172.7 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 172.2 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 163.1 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 156.7 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 155.6 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 152.3 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 150.1 ± 0.2 ppm. In some
embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 148.7 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 147.9 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 147.0 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 145.1 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 125.5 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 124.5 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 122.5 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 121.6 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 121.1 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 118.5 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 118.1 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 117.4 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 116.8 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 115.7 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 115.3 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 113.9 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 113.2 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 112.4 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 30.0 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 29.5 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 18.4 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 17.6 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with
a peak at 8.8 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 8.1 ± 0.2 ppm. [0288] In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with one, two, three, four, five, six, seven, eight, nine, ten, or more peaks selected from 212.9 ± 0.2 ppm, 211.6 ± 0.2 ppm, 179.2 ± 0.2 ppm, 178.9 ± 0.2 ppm, 178.4 ± 0.2 ppm, 174.7 ± 0.2 ppm, 174.1 ± 0.2 ppm, 172.7 ± 0.2 ppm, 172.2 ± 0.2 ppm, 163.1 ± 0.2 ppm, 156.7 ± 0.2 ppm, 155.6 ± 0.2 ppm, 152.3 ± 0.2 ppm, 150.1 ± 0.2 ppm, 148.7 ± 0.2 ppm, 147.9 ± 0.2 ppm, 147.0 ± 0.2 ppm, 145.1 ± 0.2 ppm, 125.5 ± 0.2 ppm, 124.5 ± 0.2 ppm, 122.5 ± 0.2 ppm, 121.6 ± 0.2 ppm, 121.1 ± 0.2 ppm, 118.5 ± 0.2 ppm, 118.1 ± 0.2 ppm, 117.4 ± 0.2 ppm, 116.8 ± 0.2 ppm, 115.7 ± 0.2 ppm, 115.3 ± 0.2 ppm, 113.9 ± 0.2 ppm, 113.2 ± 0.2 ppm, 112.4 ± 0.2 ppm, 30.0 ± 0.2 ppm, 29.5 ± 0.2 ppm, 18.4 ± 0.2 ppm, 17.6 ± 0.2 ppm, 8.8 ± 0.2 ppm, and 8.1 ± 0.2 ppm. [0289] In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with peaks at 212.9 ± 0.2 ppm, 211.6 ± 0.2 ppm, 179.2 ± 0.2 ppm, 178.9 ± 0.2 ppm, 178.4 ± 0.2 ppm, 174.7 ± 0.2 ppm, 174.1 ± 0.2 ppm, 172.7 ± 0.2 ppm, 172.2 ± 0.2 ppm, 163.1 ± 0.2 ppm, 156.7 ± 0.2 ppm, 155.6 ± 0.2 ppm, 152.3 ± 0.2 ppm, 150.1 ± 0.2 ppm, 148.7 ± 0.2 ppm, 147.9 ± 0.2 ppm, 147.0 ± 0.2 ppm, 145.1 ± 0.2 ppm, 125.5 ± 0.2 ppm, 124.5 ± 0.2 ppm, 122.5 ± 0.2 ppm, 121.6 ± 0.2 ppm, 121.1 ± 0.2 ppm, 118.5 ± 0.2 ppm, 118.1 ± 0.2 ppm, 117.4 ± 0.2 ppm, 116.8 ± 0.2 ppm, 115.7 ± 0.2 ppm, 115.3 ± 0.2 ppm, 113.9 ± 0.2 ppm, 113.2 ± 0.2 ppm, 112.4 ± 0.2 ppm, 30.0 ± 0.2 ppm, 29.5 ± 0.2 ppm, 18.4 ± 0.2 ppm, 17.6 ± 0.2 ppm, 8.8 ± 0.2 ppm, and 8.1 ± 0.2 ppm. [0290] In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by a 13C SSNMR spectrum substantially similar to FIG.14. [0291] In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 19F SSNMR spectrum with a peak at -60.4 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 19F SSNMR spectrum with a peak at -61.2 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 19F SSNMR spectrum with a peak at -61.6 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 19F SSNMR spectrum with a peak at -62.7 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 19F SSNMR spectrum with a peak at -135.9 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 19F SSNMR spectrum with a peak at -136.1 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 19F SSNMR spectrum with a peak at -139.6 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 19F SSNMR spectrum with a peak at -140.6 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 19F SSNMR spectrum with a peak at -141.7 ± 0.2 ppm. In some embodiments,
crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 19F SSNMR spectrum with a peak at -145.2 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 19F SSNMR spectrum with a peak at -145.7 ± 0.2 ppm. [0292] In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 19F SSNMR spectrum with one or two peaks selected from -60.4 ± 0.2 ppm, -61.2 ± 0.2 ppm, -61.6 ± 0.2 ppm, -62.7 ± 0.2 ppm, -135.9 ± 0.2 ppm, -136.1 ± 0.2 ppm, -139.6 ± 0.2 ppm, -140.6 ± 0.2 ppm, -141.7 ± 0.2 ppm, -145.2 ± 0.2 ppm, and -145.7 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 19F SSNMR spectrum with peaks at -60.4 ± 0.2 ppm, -61.2 ± 0.2 ppm, -61.6 ± 0.2 ppm, -62.7 ± 0.2 ppm, -135.9 ± 0.2 ppm, -136.1 ± 0.2 ppm, -139.6 ± 0.2 ppm, -140.6 ± 0.2 ppm, -141.7 ± 0.2 ppm, -145.2 ± 0.2 ppm, and -145.7 ± 0.2 ppm. [0293] In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by a 19F SSNMR spectrum substantially similar to FIG.15. [0294] In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by a monoclinic crystal system, P21 space group and the following unit cell dimensions measured at 100 K on a diffractometer utilizing Cu Kα radiation (λ=1.54178 Å): a 9.2 ± 0.1 Å α 90° b 39.8 ± 0.1 Å β 96.3 ± 0.1° c 12.9 ± 0.1 Å γ 90°. In some embodiments, the diffractometer is a Rigaku diffractometer. [0295] Another aspect of the disclosure provides a method of crystalline Compound I Acetone Solvate Hydrate Form A. In some embodiments, the method of making crystalline Compound I Acetone Solvate Hydrate Form A, comprises: (i) saturating acetone with Compound I, (ii) adding water dropwise to 20% of the final volume, (iii) holding overnight at room temperature to yield crystalline Compound I Acetone Solvate Hydrate Form A. G. Crystalline Compound Ethanol Solvate Form A [0296] In some embodiments, the disclosure provides crystalline Compound I Ethanol Solvate Form A. FIG.16 provides an X-ray powder diffractogram of crystalline Compound I Ethanol Solvate Form A. [0297] In some embodiments, crystalline Compound I Ethanol Solvate Form A is substantially pure. In some embodiments, crystalline Compound I Ethanol Solvate Form A is substantially crystalline. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram generated by an X-ray powder diffraction analysis with an incident beam of Cu Kα radiation.
[0298] In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having a signal at 9.4 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having a signal at 16.1 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having a signal at 26.0 ± 0.2 degrees two-theta. [0299] In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having a signal at 10.7 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having a signal at 13.2 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having a signal at 13.6 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having a signal at 13.8 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having a signal at 15.1 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having a signal at 15.9 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having a signal at 17.0 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having a signal at 17.9 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having a signal at 18.3 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having a signal at 18.4 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having a signal at 19.1 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having a signal at 19.9 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having a signal at 21.8 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having a signal at 22.2 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having a signal at 22.4 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having a signal at 24.1 ± 0.2 degrees two-theta. [0300] In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having signals at one or two of 9.4 ± 0.2 degrees two-theta, 16.1 ± 0.2 degrees two-theta, and 26.0 ± 0.2 degrees two-theta.
[0301] In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having (a) one or two signals selected from 9.4 ± 0.2 degrees two-theta, 16.1 ± 0.2 degrees two-theta, and 26.0 ± 0.2 degrees two-theta, and (b) one or two signals selected from 15.1 ± 0.2 degrees two-theta, 17.0 ± 0.2 degrees two-theta, and 22.4 ± 0.2 degrees two-theta. [0302] In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having two, three, four, five, or six signals selected from 9.4 ± 0.2 degrees two-theta, 16.1 ± 0.2 degrees two-theta, 26.0 ± 0.2 degrees two-theta, 15.1 ± 0.2 degrees two-theta, 17.0 ± 0.2 degrees two-theta, 22.4 ± 0.2 degrees two-theta, 13.2 ± 0.2 degrees two-theta, 13.6 ± 0.2 degrees two-theta, and 13.8 ± 0.2 degrees two-theta. [0303] In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having one, two three, four five, six, seven, eight, nine, ten, eleven, twelve, thirteen, fourteen, fifteen, sixteen, seventeen, eighteen, nineteen, twenty, twenty-one, or twenty-two signals selected from 9.4 ± 0.2 degrees two-theta; 10.7 ± 0.2 degrees two-theta; 13.2 ± 0.2 degrees two-theta; 13.6 ± 0.2 degrees two-theta; 13.8 ± 0.2 degrees two-theta; 15.1 ± 0.2 degrees two-theta; 15.9 ± 0.2 degrees two-theta; 16.1 ± 0.2 degrees two-theta; 17.0 ± 0.2 degrees two-theta; 17.9 ± 0.2 degrees two-theta; 18.3 ± 0.2 degrees two-theta; 18.4 ± 0.2 degrees two-theta; 19.1 ± 0.2 degrees two-theta; 19.9 ± 0.2 degrees two-theta; 21.8 ± 0.2 degrees two-theta; 22.2 ± 0.2 degrees two-theta; 22.4 ± 0.2 degrees two-theta; 24.1 ± 0.2 degrees two-theta; and 26.0 ± 0.2 degrees two-theta. [0304] In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having signals at 9.4 ± 0.2 degrees two-theta, 10.7 ± 0.2 degrees two-theta, 13.2 ± 0.2 degrees two-theta, 13.6 ± 0.2 degrees two-theta, 13.8 ± 0.2 degrees two-theta, 15.1 ± 0.2 degrees two-theta, 15.9 ± 0.2 degrees two-theta, 16.1 ± 0.2 degrees two-theta, 17.0 ± 0.2 degrees two-theta, 17.9 ± 0.2 degrees two-theta, 18.3 ± 0.2 degrees two-theta, 18.4 ± 0.2 degrees two-theta, 19.1 ± 0.2 degrees two-theta, 19.9 ± 0.2 degrees two-theta, 21.8 ± 0.2 degrees two-theta, 22.2 ± 0.2 degrees two-theta, 22.4 ± 0.2 degrees two-theta, 24.1 ± 0.2 degrees two-theta, and 26.0 ± 0.2 degrees two-theta. [0305] In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram substantially similar to FIG.16. [0306] In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13C SSNMR spectrum with a peak at 178.2 ± 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13C SSNMR spectrum with a peak at 175.7 ± 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13C SSNMR spectrum with a peak at 163.9 ± 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13C SSNMR spectrum with a peak at 163.4 ± 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13C SSNMR spectrum with a peak at 162.8 ± 0.2 ppm. In some
embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13C SSNMR spectrum with a peak at 154.9 ± 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13C SSNMR spectrum with a peak at 148.1 ± 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13C SSNMR spectrum with a peak at 146.0 ± 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13C SSNMR spectrum with a peak at 145.2 ± 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13C SSNMR spectrum with a peak at 125.9 ± 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13C SSNMR spectrum with a peak at 123.1 ± 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13C SSNMR spectrum with a peak at 121.8 ± 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13C SSNMR spectrum with a peak at 120.4 ± 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13C SSNMR spectrum with a peak at 119.5 ± 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13C SSNMR spectrum with a peak at 116.9 ± 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13C SSNMR spectrum with a peak at 116.3 ± 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13C SSNMR spectrum with a peak at 114.8 ± 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13C SSNMR spectrum with a peak at 113.8 ± 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13C SSNMR spectrum with a peak at 56.5 ± 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13C SSNMR spectrum with a peak at 18.4 ± 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13C SSNMR spectrum with a peak at 17.2 ± 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13C SSNMR spectrum with a peak at 9.2 ± 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13C SSNMR spectrum with a peak at 7.9 ± 0.2 ppm. [0307] In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13C SSNMR spectrum with one, two, three, four, five, six, seven, eight, nine, ten, or more peaks selected from 178.2 ± 0.2 ppm, 175.7 ± 0.2 ppm, 163.9 ± 0.2 ppm, 163.4 ± 0.2 ppm, 162.8 ± 0.2 ppm, 154.9 ± 0.2 ppm, 148.1 ± 0.2 ppm, 146.0 ± 0.2 ppm, 145.2 ± 0.2 ppm, 125.9 ± 0.2 ppm, 123.1 ± 0.2 ppm, 121.8 ± 0.2 ppm, 120.4 ± 0.2 ppm, 119.5 ± 0.2 ppm, 116.9 ± 0.2 ppm, 116.3 ± 0.2 ppm, 114.8 ± 0.2 ppm, 113.8 ± 0.2 ppm, 56.5 ± 0.2 ppm, 18.4 ± 0.2 ppm, 17.2 ± 0.2 ppm, 9.2 ± 0.2 ppm, and 7.9 ± 0.2 ppm. [0308] In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13C SSNMR spectrum with peaks at 178.2 ± 0.2 ppm, 175.7 ± 0.2 ppm, 163.9 ± 0.2 ppm,
163.4 ± 0.2 ppm, 162.8 ± 0.2 ppm, 154.9 ± 0.2 ppm, 148.1 ± 0.2 ppm, 146.0 ± 0.2 ppm, 145.2 ± 0.2 ppm, 125.9 ± 0.2 ppm, 123.1 ± 0.2 ppm, 121.8 ± 0.2 ppm, 120.4 ± 0.2 ppm, 119.5 ± 0.2 ppm, 116.9 ± 0.2 ppm, 116.3 ± 0.2 ppm, 114.8 ± 0.2 ppm, 113.8 ± 0.2 ppm, 56.5 ± 0.2 ppm, 18.4 ± 0.2 ppm, 17.2 ± 0.2 ppm, 9.2 ± 0.2 ppm, and 7.9 ± 0.2 ppm. [0309] In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by a 13C SSNMR spectrum substantially similar to FIG.17. [0310] In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 19F SSNMR spectrum with a peak at -61.1 ± 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 19F SSNMR spectrum with a peak at -138.1 ± 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 19F SSNMR spectrum with a peak at -141.2 ± 0.2 ppm. [0311] In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 19F SSNMR spectrum with one or two peaks selected from -61.1 ± 0.2 ppm, -138.1 ± 0.2 ppm, and -141.2 ± 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 19F SSNMR spectrum with peaks at -61.1 ± 0.2 ppm, -138.1 ± 0.2 ppm, and -141.2 ± 0.2 ppm. [0312] In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by a 19F SSNMR spectrum substantially similar to FIG.18. [0313] In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by a monoclinic crystal system, P21/c space group and the following unit cell dimensions measured at 100 K on a diffractometer utilizing Cu Kα radiation (λ=1.54178 Å): a 17.0 ± 0.1 Å α 90° b 11.1 ± 0.1 Å β 105.9 ± 0.1° c 13.2 ± 0.1 Å γ 90°. In some embodiments, the diffractometer is a Bruker diffractometer. [0314] Another aspect of the disclosure provides a method of crystalline Compound I Ethanol Solvate Form A. In some embodiments, the method of making crystalline Compound I Ethanol Solvate Form A comprises: (i) combining Compound I and ethyl acetate, (ii) heating the slurry to 50 °C, (iii) adding ethanol and distilling under vacuum at 50 °C, (iv) adding ethanol and distilling under vacuum at 50 °C, (v) adding ethanol and distilling under vacuum at 50 °C to a minimum volume, (vi) adding ethanol and distilling under vacuum at 50 °C, (vii) drying in a vacuum oven at 45 °C, with a slight nitrogen bleed for 42 hours to yield crystalline Compound I Ethanol Solvate Form A. H. Crystalline Compound I Hydrate Form A [0315] In some embodiments, the disclosure provides crystalline Compound I Hydrate Form A. FIG.19 provides an X-ray powder diffractogram of crystalline Compound I Hydrate Form A.
[0316] In some embodiments, crystalline Compound I Hydrate Form A is substantially pure. In some embodiments, crystalline Compound I Hydrate Form A is substantially crystalline. In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram generated by an X-ray powder diffraction analysis with an incident beam of Cu Kα radiation. [0317] In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 8.7 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 12.4 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 18.8 ± 0.2 degrees two-theta. [0318] In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 4.7 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 9.5 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 11.0 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 12.7 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 13.1 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 14.4 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 15.8 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 16.3 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 17.5 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 19.8 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 20.2 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 21.1 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 21.4 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 22.2 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 22.9 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 23.9 ± 0.2 degrees two-theta. In some embodiments, crystalline
Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 24.4 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 24.7 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 25.4 ± 0.2 degrees two-theta. [0319] In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having signals at one or two of 8.7 ± 0.2 degrees two-theta, 12.4 ± 0.2 degrees two-theta, and 18.8 ± 0.2 degrees two-theta. [0320] In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram (a) one or two signals selected from 8.7 ± 0.2 degrees two-theta, 12.4 ± 0.2 degrees two-theta, and 18.8 ± 0.2 degrees two-theta, and (b) one or two signals selected from 12.7 ± 0.2 degrees two-theta, 17.5 ± 0.2 degrees two-theta, and 19.8 ± 0.2 degrees two-theta. [0321] In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having two, three, four, five, or six signals selected from 8.7 ± 0.2 degrees two-theta, 12.4 ± 0.2 degrees two-theta, 18.8 ± 0.2 degrees two-theta, 12.7 ± 0.2 degrees two-theta, 17.5 ± 0.2 degrees two-theta, 19.8 ± 0.2 degrees two-theta, 13.1 ± 0.2 degrees two-theta, 16.3 ± 0.2 degrees two-theta, and 21.1 ± 0.2 degrees two-theta. [0322] In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having one, two three, four five, six, seven, eight, nine, ten, eleven, twelve, thirteen, fourteen, fifteen, sixteen, seventeen, eighteen, nineteen, twenty, twenty-one, or twenty-two signals selected from 8.7 ± 0.2 degrees two-theta, 12.4 ± 0.2 degrees two-theta, 18.8 ± 0.2 degrees two-theta; 4.7 ± 0.2 degrees two-theta; 9.5 ± 0.2 degrees two-theta; 11.0 ± 0.2 degrees two-theta; 12.7 ± 0.2 degrees two-theta; 13.1 ± 0.2 degrees two-theta; 14.4 ± 0.2 degrees two-theta; 15.8 ± 0.2 degrees two-theta; 16.3 ± 0.2 degrees two-theta; 17.5 ± 0.2 degrees two-theta; 19.8 ± 0.2 degrees two-theta; 20.2 ± 0.2 degrees two-theta; 21.1 ± 0.2 degrees two-theta; 21.4 ± 0.2 degrees two-theta; 22.2 ± 0.2 degrees two-theta; 22.9 ± 0.2 degrees two-theta; 23.9 ± 0.2 degrees two-theta; 24.4 ± 0.2 degrees two-theta; 24.7 ± 0.2 degrees two-theta; and 25.4 ± 0.2 degrees two-theta. [0323] In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having signals at 8.7 ± 0.2 degrees two-theta, 12.4 ± 0.2 degrees two-theta, 18.8 ± 0.2 degrees two-theta; 4.7 ± 0.2 degrees two-theta; 9.5 ± 0.2 degrees two-theta; 11.0 ± 0.2 degrees two-theta; 12.7 ± 0.2 degrees two-theta; 13.1 ± 0.2 degrees two-theta; 14.4 ± 0.2 degrees two-theta; 15.8 ± 0.2 degrees two-theta; 16.3 ± 0.2 degrees two-theta; 17.5 ± 0.2 degrees two-theta; 19.8 ± 0.2 degrees two-theta; 20.2 ± 0.2 degrees two-theta; 21.1 ± 0.2 degrees two-theta; 21.4 ± 0.2 degrees two-theta; 22.2 ± 0.2 degrees two-theta; 22.9 ± 0.2 degrees two-theta; 23.9 ± 0.2 degrees two-theta; 24.4 ± 0.2 degrees two-theta; 24.7 ± 0.2 degrees two-theta; and 25.4 ± 0.2 degrees two-theta. [0324] In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram substantially similar to FIG.19.
[0325] In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 177.3 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 172.8 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 170.5 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 162.0 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 154.3 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 154.0 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 150.2 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 148.7 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 147.6 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 146.6 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 145.5 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 126.3 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 125.0 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 123.8 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 122.4 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 121.6 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 120.6 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 118.4 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 117.9 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 117.0 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 116.0 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 114.7 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 114.4 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 112.8 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR
spectrum with a peak at 17.7 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 17.1 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 8.7 ± 0.2 ppm. [0326] In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum with one, two, three, four, five, six, seven, eight, nine, ten, or more peaks selected from 177.3 ± 0.2 ppm, 172.8 ± 0.2 ppm, 170.5 ± 0.2 ppm, 162.0 ± 0.2 ppm, 154.3 ± 0.2 ppm, 154.0 ± 0.2 ppm, 150.2 ± 0.2 ppm, 148.7 ± 0.2 ppm, 147.6 ± 0.2 ppm, 146.6 ± 0.2 ppm, 145.5 ± 0.2 ppm, 126.3 ± 0.2 ppm, 125.0 ± 0.2 ppm, 123.8 ± 0.2 ppm, 122.4 ± 0.2 ppm, 121.6 ± 0.2 ppm, 120.6 ± 0.2 ppm, 118.4 ± 0.2 ppm, 117.9 ± 0.2 ppm, 117.0 ± 0.2 ppm, 116.0 ± 0.2 ppm, 114.7 ± 0.2 ppm, 114.4 ± 0.2 ppm, 112.8 ± 0.2 ppm, 17.7 ± 0.2 ppm, 17.1 ± 0.2 ppm, and 8.7 ± 0.2 ppm. [0327] In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum with peaks at 177.3 ± 0.2 ppm, 172.8 ± 0.2 ppm, 170.5 ± 0.2 ppm, 162.0 ± 0.2 ppm, 154.3 ± 0.2 ppm, 154.0 ± 0.2 ppm, 150.2 ± 0.2 ppm, 148.7 ± 0.2 ppm, 147.6 ± 0.2 ppm, 146.6 ± 0.2 ppm, 145.5 ± 0.2 ppm, 126.3 ± 0.2 ppm, 125.0 ± 0.2 ppm, 123.8 ± 0.2 ppm, 122.4 ± 0.2 ppm, 121.6 ± 0.2 ppm, 120.6 ± 0.2 ppm, 118.4 ± 0.2 ppm, 117.9 ± 0.2 ppm, 117.0 ± 0.2 ppm, 116.0 ± 0.2 ppm, 114.7 ± 0.2 ppm, 114.4 ± 0.2 ppm, 112.8 ± 0.2 ppm, 17.7 ± 0.2 ppm, 17.1 ± 0.2 ppm, and 8.7 ± 0.2 ppm. [0328] In some embodiments, crystalline Compound I Hydrate Form A is characterized by a 13C SSNMR spectrum substantially similar to FIG.22. [0329] In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum measured at 25 ± 2 °C and 6% relative humidity (RH) with a peak at 171.5 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum measured at 25 ± 2 °C and 11% relative humidity (RH) with a peak at 171.6 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum measured at 25 ± 2 °C and 33% relative humidity (RH) with a peak at 170.7 ± 0.2 ppm. [0330] In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum measured at 25 ± 2 °C and 6% relative humidity (RH) with a peak at 155.0 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum measured at 25 ± 2 °C and 11% relative humidity (RH) with a peak at 155.0 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum measured at 25 ± 2 °C and 22% relative humidity (RH) with a peak at 154.6 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum measured at 25 ± 2 °C and 33% relative humidity (RH) with a peak at 154.4 ± 0.2 ppm. [0331] In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum measured at 25 ± 2 °C and 6% relative humidity (RH) with a peak at 149.5 ± 0.2
ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum measured at 25 ± 2 °C and 11% relative humidity (RH) with a peak at 149.4 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum measured at 25 ± 2 °C and 22% relative humidity (RH) with a peak at 148.6 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum measured at 25 ± 2 °C and 33% relative humidity (RH) with a peak at 148.6 ± 0.2 ppm. [0332] In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum measured at 25 ± 2 °C and 6% relative humidity (RH) with a peak at 145.8 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum measured at 25 ± 2 °C and 11% relative humidity (RH) with a peak at 145.8 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum measured at 25 ± 2 °C and 22% relative humidity (RH) with a peak at 145.6 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum measured at 25 ± 2 °C and 33% relative humidity (RH) with a peak at 145.5 ± 0.2 ppm. [0333] In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum measured at 25 ± 2 °C and 6% relative humidity (RH) with a peak at 8.5 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum measured at 25 ± 2 °C and 11% relative humidity (RH) with a peak at 8.5 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum measured at 25 ± 2 °C and 22% relative humidity (RH) with a peak at 8.6 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum measured at 25 ± 2 °C and 33% relative humidity (RH) with a peak at 8.6 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum measured at 25 ± 2 °C and 43% relative humidity (RH) with a peak at 8.6 ± 0.2 ppm. [0334] In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 19F SSNMR spectrum with a peak at -59.4 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 19F SSNMR spectrum with a peak at -60.2 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 19F SSNMR spectrum with a peak at -135.0 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 19F SSNMR spectrum with a peak at -138.4 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 19F SSNMR spectrum with a peak at -141.7 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 19F SSNMR spectrum with a peak at -144 ± 0.2 ppm.
[0335] In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 19F SSNMR spectrum with one or two peaks selected from -59.4 ± 0.2 ppm, -60.2 ± 0.2 ppm, -135.0 ± 0.2 ppm, -138.4 ± 0.2 ppm, -141.7 ± 0.2 ppm, and -144.0 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 19F SSNMR spectrum with peaks at -59.4 ± 0.2 ppm, -60.2 ± 0.2 ppm, -135.0 ± 0.2 ppm, -138.4 ± 0.2 ppm, -141.7 ± 0.2 ppm, and -144.0 ± 0.2 ppm. [0336] In some embodiments, crystalline Compound I Hydrate Form A is characterized by a 19F SSNMR spectrum substantially similar to FIG.23. [0337] In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 19F SSNMR spectrum measured at 25 ± 2 °C and 11% relative humidity (RH) with a peak at -142.4 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 19F SSNMR spectrum measured at 25 ± 2 °C and 22% relative humidity (RH) with a peak at -141.9 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 19F SSNMR spectrum measured at 25 ± 2 °C and 33% relative humidity (RH) with a peak at -141.8 ± 0.2 ppm. [0338] In some embodiments, crystalline Compound I Hydrate Form A is characterized by a triclinic crystal system, P-1 space group and the following unit cell dimensions measured at 100 K on a diffractometer utilizing Cu Kα radiation (λ=1.54178 Å): a 10.5 ± 0.1 Å α 91.3 ± 0.1° b 11.2 ± 0.1 Å β 103.9 ± 0.1° c 19.2 ± 0.1 Å γ 98.4 ± 0.1°. In some embodiments, the diffractometer is a Bruker diffractometer. [0339] Another aspect of the disclosure provides a method of crystalline Compound I Hydrate Form A. In some embodiments, the method of making crystalline Compound I Hydrate Form A, comprises: (i) combining Compound I Ethanol Solvate Form A and acetone, (ii) stirring the slurry while heating 50 °C, (iii) adding water over 1 hour, (iv) adding a seed of crystalline Compound I hydrate Form A, (v) agitating for 30 minutes, (vi) adding water over 8 hours, (vii) stirring at 50 °C for 5 hours, (viii) cooling the slurry to 20 °C over 3 hours, (ix) agitating for 2 hours, and (x) collecting the solids and washing with 1:2 acetone:water to yield crystalline Compound I hydrate Form A. I. Crystalline Compound I Dehydrated Hydrate Form A [0340] In some embodiments, the disclosure provides crystalline Compound I Dehydrated Hydrate Form A. FIG. 20 provides an X-ray powder diffractogram of crystalline Compound I Dehydrated Hydrate Form A. [0341] In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is substantially pure. In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is substantially crystalline. In some embodiments, crystalline Compound I Dehydrated Hydrate Form A
is characterized by an X-ray powder diffractogram generated by an X-ray powder diffraction analysis with an incident beam of Cu Kα radiation. [0342] In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 8.7 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 15.9 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 18.7 ± 0.2 degrees two-theta. [0343] In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 4.7 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 9.5 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 10.9 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 12.4 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 12.6 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 13.1 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 14.3 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 16.3 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 17.3 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 19.8 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 20.3 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 21.1 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 21.4 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 22.3 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 23.0 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 23.9 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized
by an X-ray powder diffractogram having a signal at 24.3 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 24.7 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 25.3 ± 0.2 degrees two-theta. [0344] In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having signals at one or two of 8.7 ± 0.2 degrees two- theta, 15.9 ± 0.2 degrees two-theta, and 18.7 ± 0.2 degrees two-theta. [0345] In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram (a) one or two signals selected from 8.7 ± 0.2 degrees two-theta, 15.9 ± 0.2 degrees two-theta, and 18.7 ± 0.2 degrees two-theta, and (b) one or two signals selected from 12.4 ± 0.2 degrees two-theta, 12.6 ± 0.2 degrees two-theta, 17.3 ± 0.2 degrees two-theta, and 19.8 ± 0.2 degrees two-theta. [0346] In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having two, three, four, five, or six signals selected 8.7 ± 0.2 degrees two-theta, 15.9 ± 0.2 degrees two-theta, 18.7 ± 0.2 degrees two-theta, 12.4 ± 0.2 degrees two-theta, 12.6 ± 0.2 degrees two-theta, 17.3 ± 0.2 degrees two-theta, 19.8 ± 0.2 degrees two-theta, 13.1 ± 0.2 degrees two-theta, 16.3 ± 0.2 degrees two-theta, and 21.1 ± 0.2 degrees two-theta. [0347] In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having one, two three, four five, six, seven, eight, nine, ten, eleven, twelve, thirteen, fourteen, fifteen, sixteen, seventeen, eighteen, nineteen, twenty, twenty-one, or twenty-two signals selected from 4.7 ± 0.2 degrees two-theta; 8.7 ± 0.2 degrees two-theta; 9.5 ± 0.2 degrees two-theta; 10.9 ± 0.2 degrees two-theta; 12.4 ± 0.2 degrees two-theta; 12.6 ± 0.2 degrees two-theta; 13.1 ± 0.2 degrees two-theta; 14.3 ± 0.2 degrees two-theta; 15.9 ± 0.2 degrees two-theta; 16.3 ± 0.2 degrees two-theta; 17.3 ± 0.2 degrees two-theta; 18.7 ± 0.2 degrees two-theta; 19.8 ± 0.2 degrees two-theta; 20.3 ± 0.2 degrees two-theta; 21.1 ± 0.2 degrees two-theta; 21.4 ± 0.2 degrees two-theta; 22.3 ± 0.2 degrees two-theta; 23.0 ± 0.2 degrees two-theta; 23.9 ± 0.2 degrees two-theta; 24.3 ± 0.2 degrees two-theta; 24.7 ± 0.2 degrees two-theta; and 25.3 ± 0.2 degrees two-theta. [0348] In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having signals at 4.7 ± 0.2 degrees two-theta, 8.7 ± 0.2 degrees two-theta, 9.5 ± 0.2 degrees two-theta, 10.9 ± 0.2 degrees two-theta, 12.4 ± 0.2 degrees two-theta, 12.6 ± 0.2 degrees two-theta, 13.1 ± 0.2 degrees two-theta, 14.3 ± 0.2 degrees two-theta, 15.9 ± 0.2 degrees two-theta, 16.3 ± 0.2 degrees two-theta, 17.3 ± 0.2 degrees two-theta, 18.7 ± 0.2 degrees two-theta, 19.8 ± 0.2 degrees two-theta, 20.3 ± 0.2 degrees two-theta, 21.1 ± 0.2 degrees two-theta, 21.4 ± 0.2 degrees two-theta, 22.3 ± 0.2 degrees two-theta, 23.0 ± 0.2 degrees two-theta, 23.9 ± 0.2 degrees two-theta, 24.3 ± 0.2 degrees two-theta, 24.7 ± 0.2 degrees two-theta, and 25.3 ± 0.2 degrees two-theta.
[0349] In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram substantially similar to FIG.20. [0350] In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by a triclinic crystal system, P-1 space group and the following unit cell dimensions measured at 100 K on a diffractometer utilizing Cu Kα radiation (λ=1.54178 Å): a 10.4 ± 0.1 Å α 90.7 ± 0.1° b 11.2 ± 0.1 Å β 104.7 ± 0.1° c 19.3 ± 0.1 Å γ 98.0 ± 0.1°. In some embodiments, the diffractometer is a Bruker diffractometer. [0351] Another aspect of the disclosure provides a method of crystalline Compound I Dehydrated Hydrate Form A. In some embodiments, the method of making crystalline Compound I Dehydrated Hydrate Form A, comprises: (i) combining Compound I Ethanol Solvate Form A and acetone, (ii) stirring the slurry while heating 50 °C, (iii) adding water over 1 hour, (iv) adding a seed of crystalline Compound I Dehydrated Hydrate Form A, (v) agitating for 30 minutes, (vi) adding water over 8 hours, (vii) stirring at 50 °C for 5 hours, (viii) cooling the slurry to 20 °C over 3 hours, (ix) agitating for 2 hours, (x) collecting the solids and washing with 1:2 acetone:water, (xi) drying the solids in a vacuum over at 50 °C with a slight nitrogen bleed to yield crystalline Compound I Dehydrated Hydrate Form A. J. Methods of Treatment [0352] Compound I, in any one of the pharmaceutically acceptable solid forms disclosed herein, act as a voltage-gated sodium channel inhibitor. In some aspects, the voltage-gated sodium channel is NaV1.8. Accordingly, in another aspect, the disclosure features a method of inhibiting a voltage-gated sodium channel in a subject comprising administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof. In another aspect, the voltage-gated sodium channel is NaV1.8. In another aspect, the invention features a method of inhibiting a voltage-gated sodium channel in a subject comprising administering to the subject a compound of formula I or a solvate thereof or a pharmaceutically acceptable salt thereof or a pharmaceutical composition thereof. [0353] In one aspect, the disclosure relates to a method of treating or lessening the severity of pain in a subject, comprising administering to the subject a compound of formula I, or a pharmaceutically acceptable salt thereof. [0354] In another aspect, the disclosure relates to a use of a compound of formula I, or a pharmaceutically acceptable salt thereof, in a method of treating or lessening the severity of pain in a subject, comprising administering to the subject a compound of formula I, or the pharmaceutically acceptable salt thereof
[0355] In another aspect, the disclosure relates to a composition comprising a compound of formula I, or a solvate of formula I, or a pharmaceutically acceptable salt thereof, for use in a method of treating or lessening the severity of pain in a subject, wherein the composition is prepared for administration of a compound of formula I, or a solvate of formula I or the pharmaceutically acceptable salt thereof, to the subject. [0356] In yet another aspect, the disclosure features a method of treating or lessening the severity in a subject of chronic pain, gut pain, neuropathic pain, musculoskeletal pain, acute pain, inflammatory pain, cancer pain, idiopathic pain, postsurgical pain (e.g., bunionectomy pain, herniorrhaphy pain or abdominoplasty pain), visceral pain, multiple sclerosis, Charcot-Marie-Tooth syndrome, incontinence, pathological cough, or cardiac arrhythmia comprising administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e. g. crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof. [0357] In yet another aspect, the disclosure features a method of treating or lessening the severity in a subject of chronic pain, gut pain, neuropathic pain, musculoskeletal pain, acute pain, inflammatory pain, cancer pain, idiopathic pain, postsurgical pain, herniorrhaphy pain, bunionectomy pain, multiple sclerosis, Charcot-Marie-Tooth syndrome, incontinence, or cardiac arrhythmia comprising administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof. [0358] In yet another aspect, the disclosure features a method of treating or lessening the severity in a subject of gut pain, wherein gut pain comprises inflammatory bowel disease pain, Crohn’s disease pain, irritable bowel syndrome, endometriosis, polycystic ovarian disease, salpingitis, cervicitis or interstitial cystitis pain wherein said method comprises administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof. [0359] In yet another aspect, the disclosure features a method of treating or lessening the severity in a subject of neuropathic pain comprising administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof. In some aspects, the neuropathic pain comprises post-herpetic neuralgia, small fiber neuropathy, diabetic neuropathy, or idiopathic small-fiber neuropathy. In some aspects, the neuropathic pain comprises diabetic neuropathy (e.g., diabetic peripheral neuropathy). As used herein, the phrase “idiopathic small-fiber neuropathy” shall be understood to include any small fiber neuropathy. [0360] In yet another aspect, the disclosure features a method of treating or lessening the severity in a subject of neuropathic pain, wherein neuropathic pain comprises post-herpetic neuralgia, diabetic neuralgia, painful HIV-associated sensory neuropathy, trigeminal neuralgia, burning mouth syndrome, post-amputation pain, phantom pain, painful neuroma; traumatic neuroma; Morton’s neuroma; nerve entrapment injury, spinal stenosis, carpal tunnel syndrome, radicular pain, sciatica pain; nerve avulsion
injury, brachial plexus avulsion injury; complex regional pain syndrome, drug therapy induced neuralgia, cancer chemotherapy induced neuralgia, anti-retroviral therapy induced neuralgia, HIV-induced neuropathy; post spinal cord injury pain, spinal stenosis pain, small fiber neuropathy, idiopathic small-fiber neuropathy, idiopathic sensory neuropathy or trigeminal autonomic cephalalgia wherein said method comprises administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g. crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof. [0361] In yet another aspect, the disclosure features a method of treating or lessening the severity in a subject of musculoskeletal pain comprising administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof. In some aspects, the musculoskeletal pain comprises osteoarthritis pain. [0362] In yet another aspect, the disclosure features a method of treating or lessening the severity in a subject of musculoskeletal pain, wherein musculoskeletal pain comprises osteoarthritis pain, back pain, cold pain, burn pain or dental pain wherein said method comprises administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g. crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof. [0363] In yet another aspect, the disclosure features a method of treating or lessening the severity in a subject of inflammatory pain, wherein inflammatory pain comprises rheumatoid arthritis pain, ankylosing spondylitis or vulvodynia wherein said method comprises administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof. [0364] In yet another aspect, the disclosure features a method of treating or lessening the severity in a subject of inflammatory pain, wherein inflammatory pain comprises rheumatoid arthritis pain wherein said method comprises administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof of. [0365] In yet another aspect, the disclosure features a method of treating or lessening the severity in a subject of idiopathic pain, wherein idiopathic pain comprises fibromyalgia pain wherein said method comprises administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof. [0366] In yet another aspect, the disclosure features a method of treating or lessening the severity in a subject of idiopathic pain, wherein idiopathic pain comprises reflex sympathetic dystrophy pain, wherein said method comprises administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof.
[0367] In yet another aspect, the disclosure features a method of treating or lessening the severity in a subject of pathological cough wherein said method comprises administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof. [0368] In yet another aspect, the disclosure features a method of treating or lessening the severity in a subject of acute pain comprising administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof. In some aspects, the acute pain comprises acute post-operative pain. [0369] In yet another aspect, the disclosure features a method of treating or lessening the severity in a subject of postsurgical pain (e.g., joint replacement pain, soft tissue surgery pain, post-thoracotomy pain, post-mastectomy pain, hemorrhoidectomy pain, herniorrhaphy pain, bunionectomy pain or abdominoplasty pain) comprising administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof. [0370] In yet another aspect, the disclosure features a method of treating or lessening the severity in a subject of bunionectomy pain comprising administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof. [0371] In yet another aspect, the disclosure features a method of treating or lessening the severity in a subject of shoulder arthroplasty pain or shoulder arthroscopy pain comprising administering an effective amount of a compound of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof. [0372] In yet another aspect, the disclosure features a method of treating or lessening the severity in a subject of herniorrhaphy pain comprising administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof. [0373] In yet another aspect, the disclosure features a method of treating or lessening the severity in a subject of abdominoplasty pain comprising administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof. [0374] In yet another aspect, the disclosure features a method of treating or lessening the severity in a subject of visceral pain comprising administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof. In some aspects, the visceral pain comprises visceral pain from abdominoplasty.
[0375] In yet another aspect, the disclosure features a method of treating or lessening the severity in a subject of a neurodegenerative disease comprising administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof. In some aspects, the neurodegenerative disease comprises multiple sclerosis. In some aspects, the neurodegenerative disease comprises Pitt Hopkins Syndrome (PTHS). [0376] In yet another aspect, the disclosure features a method wherein the subject is treated with one or more additional therapeutic agents administered concurrently with, prior to, or subsequent to treatment with an effective of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof. In some embodiments, the additional therapeutic agent is a sodium channel inhibitor. [0377] In another aspect, the disclosure features a method of inhibiting a voltage-gated sodium channel in a biological sample comprising contacting the biological sample with an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof. In another aspect, the voltage-gated sodium channel is NaV1.8. [0378] In another aspect, the disclosure features a method of treating or lessening the severity in a subject of acute pain, sub-acute and chronic pain, nociceptive pain, neuropathic pain, inflammatory pain, nociplastic pain, arthritis, migraine, cluster headaches, tension headaches, and all other forms of headaches, trigeminal neuralgia, herpetic neuralgia, general neuralgias, epilepsy, epilepsy conditions, neurodegenerative disorders, psychiatric disorders, anxiety, depression, bipolar disorder, myotonia, arrhythmia, movement disorders, neuroendocrine disorders, ataxia, central neuropathic pain of multiple sclerosis and irritable bowel syndrome, incontinence, pathological cough, visceral pain, osteoarthritis pain, postherpetic neuralgia, diabetic neuropathy, radicular pain, sciatica, back pain, unspecific chronic back pain, head pain, neck pain, moderate pain, severe pain, intractable pain, nociceptive pain, breakthrough pain, postsurgical pain (e.g., joint replacement pain, soft tissue surgery pain, post- thoracotomy pain, post-mastectomy pain, herniorrhaphy pain, bunionectomy pain or abdominoplasty pain), cancer pain including chronic cancer pain and breakthrough cancer pain, stroke (e.g., post stroke central neuropathic pain), whiplash associated disorders, fragility fractures, spinal fractures, ankylosing spondylitis, pemphigus, Raynaud’s Disease, scleroderma, systemic lupus erythematosus, Epidermolysis bullosa, gout, juvenile idiopathic arthritis, melorheostosis, polymyalgia reumatica, pyoderma gangrenosum, chronic widespread pain, diffuse idiopathic skeletal hyperostosis, disc degeneration/herniation pain, radiculopathy, facet joint syndrome, failed back surgery syndrome, burns, carpal tunnel syndrome, Paget’s disease pain, spinal canal stenosis, spondylodyscitis, transverse myelitis, Ehlers-Danlos syndrome, Fabry’s disease, mastocytocytosis, neurofibromatosis, ocular neuropathic pain, sarcoidosis, spondylolysis, spondylolisthesis, chemotherapy induced oral mucositis, Charcot neuropathic osteoarhropathy, temporo-mandibular joint disorder, painful joint arthroplasties,
non-cardiac chest pain, pudendal neuralgia, renal colic, biliary tract diseases, vascular leg ulcers, pain in Parkinson’s disease, pain in Alzheimer’s disease, cerebral ischemia, traumatic brain injury, amyotrophic lateral sclerosis, stress induced angina, exercise induced angina, palpitations, hypertension, or abnormal gastro-intestinal motility, comprising administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof. [0379] In another aspect, the disclosure features a method of treating or lessening the severity in a subject of femur cancer pain; non-malignant chronic bone pain; rheumatoid arthritis; osteoarthritis; spinal stenosis; neuropathic low back pain; myofascial pain syndrome; fibromyalgia; temporomandibular joint pain; chronic visceral pain, abdominal pain; pancreatic pain; IBS pain; chronic and acute headache pain; migraine; tension headache; cluster headaches; chronic and acute neuropathic pain, post-herpetic neuralgia; diabetic neuropathy; HIV-associated neuropathy; trigeminal neuralgia; Charcot-Marie-Tooth neuropathy; hereditary sensory neuropathy; peripheral nerve injury; painful neuromas; ectopic proximal and distal discharges; radiculopathy; chemotherapy induced neuropathic pain; radiotherapy-induced neuropathic pain; persistent/chronic post-surgical pain (e.g., post amputation, post-thoracotomy, post-cardiac surgery), post-mastectomy pain; central pain; spinal cord injury pain; post-stroke pain; thalamic pain; phantom pain (e.g., following removal of lower extremity, upper extremity, breast); intractable pain; acute pain, acute post-operative pain; acute musculoskeletal pain; joint pain; mechanical low back pain; neck pain; tendonitis; injury pain; exercise pain; acute visceral pain; pyelonephritis; appendicitis; cholecystitis; intestinal obstruction; hernias; chest pain, cardiac pain; pelvic pain, renal colic pain, acute obstetric pain, labor pain; cesarean section pain; acute inflammatory pain, burn pain, trauma pain; acute intermittent pain, endometriosis; acute herpes zoster pain; sickle cell anemia; acute pancreatitis; breakthrough pain; orofacial pain; sinusitis pain; dental pain; multiple sclerosis (MS) pain; pain in depression; leprosy pain; Behcet's disease pain; adiposis dolorosa; phlebitic pain; Guillain-Barre pain; painful legs and moving toes; Haglund syndrome; erythromelalgia pain; Fabry's disease pain; bladder and urogenital disease; urinary incontinence, pathological cough; hyperactive bladder; painful bladder syndrome; interstitial cystitis (IC); prostatitis; complex regional pain syndrome (CRPS), type I, complex regional pain syndrome (CRPS) type II; widespread pain, paroxysmal extreme pain, pruritus, tinnitus, or angina-induced pain, comprising administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof. [0380] In another aspect, the disclosure features a method of treating or lessening the severity in a subject of trigeminal neuralgia, migraines treated with botox, cervical radiculopathy, occipital neuralgia, axillary neuropathy, radial neuropathy, ulnar neuropathy, brachial plexopathy, thoracic radiculopathy, intercostal neuralgia, lumbosacral radiculopathy, iliolingual neuralgia, pudendal neuralgia, femoral neuropathy, meralgia paresthetica, saphenous neuropathy, sciatic neuropathy, peroneal neuropathy, tibial neuropathy, lumbosacral plexopathy, traumatic neuroma stump pain or
postamputation pain, comprising administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof. [0381] In some embodiments, the method of treating, or lessening the severity of, or symptomatically treating pain in a patient comprises administering to the patient an effective amount of Compound I in any one of the pharmaceutically acceptable crystalline forms disclosed herein. In some embodiments the pharmaceutically acceptable crystalline form of Compound I is Compound I neat Form A. In some embodiments the pharmaceutically acceptable crystalline form of Compound I is Compound I neat Form B. In some embodiments the pharmaceutically acceptable crystalline form of Compound I is Compound I neat Form E. In some embodiments the pharmaceutically acceptable crystalline form of Compound I is Compound I Acetone Solvate Hydrate Form A. In some embodiments the pharmaceutically acceptable crystalline form of Compound I is Compound I Ethanol Solvate Form A. In some embodiments the pharmaceutically acceptable crystalline form of Compound I is Compound I Hydrate Form A. In some embodiments the pharmaceutically acceptable crystalline form of Compound I is Compound I Hydrate Form A. In some embodiments the pharmaceutically acceptable crystalline form of Compound I is Compound I Dehydrated Hydrate Form A. [0382] In some embodiments, the method of treating, or lessening the severity of, or symptomatically treating pain in a patient comprises administering to the patient an effective amount of Compound I in a pharmaceutically acceptable amorphous form disclosed herein. In some embodiments the pharmaceutically acceptable form of Compound I is Compound I amorphous form. [0383] In some embodiments, the method of treating, lessening the severity of, or symptomatically treating pain in a patient comprises administering to the patient an effective amount of Compound I as any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein in combination with at least one additional active pharmaceutical ingredient. In some embodiments, the method of treating, lessening the severity of, or symptomatically treating pain in a patient comprises administering to the patient an effective amount of Compound I as a solid form selected from Compound I neat Form A, Compound I neat Form B, Compound I neat Form E, Compound I Acetone Solvate Hydrate Form A, Compound I Ethanol Solvate Form A, Compound I Hydrate Form A, Compound I Dehydrated Hydrate Form A, and Compound I amorphous form, in combination with at least one additional active pharmaceutical ingredient. [0384] In some embodiments, the disclosure features Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof for use in any of the foregoing methods. [0385] In some embodiments, the disclosure features the use of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof for the manufacture of a medicament for use in any of the foregoing methods.
[0386] In some embodiments, the disclosure features Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof for use as a medicament. [0387] It will also be appreciated that the compounds, salts, and pharmaceutically acceptable compositions of the invention can be employed in combination therapies, that is, the compounds, salts, and pharmaceutically acceptable compositions can be administered concurrently with, prior to, or subsequent to, one or more other desired therapeutics or medical procedures. The particular combination of therapies (therapeutics or procedures) to employ in a combination regimen will take into account compatibility of the desired therapeutics and/or procedures and the desired therapeutic effect to be achieved. It will also be appreciated that the therapies employed may achieve a desired effect for the same disorder (for example, an inventive compound may be administered concurrently with another agent used to treat the same disorder), or they may achieve different effects (e.g., control of any adverse effects). As used herein, additional therapeutic agents that are normally administered to treat or prevent a particular disease, or condition, are known as “appropriate for the disease, or condition, being treated.” For example, exemplary additional therapeutic agents include, but are not limited to: non-opioid analgesics (indoles such as Etodolac, Indomethacin, Sulindac, Tolmetin; naphthylalkanones such as Nabumetone; oxicams such as Piroxicam; para-aminophenol derivatives, such as Acetaminophen; propionic acids such as Fenoprofen, Flurbiprofen, Ibuprofen, Ketoprofen, Naproxen, Naproxen sodium, Oxaprozin; salicylates such as Aspirin, Choline magnesium trisalicylate, Diflunisal; fenamates such as meclofenamic acid, Mefenamic acid; and pyrazoles such as Phenylbutazone); or opioid (narcotic) agonists (such as Codeine, Fentanyl, Hydromorphone, Levorphanol, Meperidine, Methadone, Morphine, Oxycodone, Oxymorphone, Propoxyphene, Buprenorphine, Butorphanol, Dezocine, Nalbuphine, and Pentazocine). Additionally, nondrug analgesic approaches may be utilized in conjunction with administration of one or more compounds of the invention. For example, anesthesiologic (intraspinal infusion, neural blockade), neurosurgical (neurolysis of CNS pathways), neurostimulatory (transcutaneous electrical nerve stimulation, dorsal column stimulation), physiatric (physical therapy, orthotic devices, diathermy), or psychologic (cognitive methods-hypnosis, biofeedback, or behavioral methods) approaches may also be utilized. Additional appropriate therapeutic agents or approaches are described generally in The Merck Manual, Nineteenth Edition, Ed. Robert S. Porter and Justin L. Kaplan, Merck Sharp &Dohme Corp., a subsidiary of Merck & Co., Inc., 2011, and the Food and Drug Administration website, www.fda.gov, the entire contents of which are hereby incorporated by reference. [0388] In another embodiment, additional appropriate therapeutic agents are selected from the following: [0389] (1) an opioid analgesic, e.g. morphine, heroin, hydromorphone, oxymorphone, levorphanol, levallorphan, methadone, meperidine, fentanyl, cocaine, codeine, dihydrocodeine,
oxycodone, hydrocodone, propoxyphene, nalmefene, nalorphine, naloxone, naltrexone, buprenorphine, butorphanol, nalbuphine, pentazocine, or difelikefalin; [0390] (2) a nonsteroidal antiinflammatory drug (NSAID), e.g. aspirin, diclofenac, diflunisal, etodolac, fenbufen, fenoprofen, flufenisal, flurbiprofen, ibuprofen (including without limitation intravenous ibuprofen (e.g., Caldolor®)), indomethacin, ketoprofen, ketorolac (including without limitation ketorolac tromethamine (e.g., Toradol®)), meclofenamic acid, mefenamic acid, meloxicam, IV meloxicam (e.g., Anjeso®), nabumetone, naproxen, nimesulide, nitroflurbiprofen, olsalazine, oxaprozin, phenylbutazone, piroxicam, sulfasalazine, sulindac, tolmetin or zomepirac; [0391] (3) a barbiturate sedative, e.g. amobarbital, aprobarbital, butabarbital, butalbital, mephobarbital, metharbital, methohexital, pentobarbital, phenobarbital, secobarbital, talbutal, thiamylal or thiopental; [0392] (4) a benzodiazepine having a sedative action, e.g. chlordiazepoxide, clorazepate, diazepam, flurazepam, lorazepam, oxazepam, temazepam or triazolam; [0393] (5) a histamine (H1) antagonist having a sedative action, e.g. diphenhydramine, pyrilamine, promethazine, chlorpheniramine or chlorcyclizine; [0394] (6) a sedative such as glutethimide, meprobamate, methaqualone or dichloralphenazone; [0395] (7) a skeletal muscle relaxant, e.g. baclofen, carisoprodol, chlorzoxazone, cyclobenzaprine, methocarbamol or orphenadrine; [0396] (8) an NMDA receptor antagonist, e.g. dextromethorphan ((+)-3-hydroxy-N- methylmorphinan) or its metabolite dextrorphan ((+)-3-hydroxy-N-methylmorphinan), ketamine, memantine, pyrroloquinoline quinine, cis-4-(phosphonomethyl)-2- piperidinecarboxylic acid, budipine, EN-3231 (MorphiDex®), a combination formulation of morphine and dextromethorphan), topiramate, neramexane or perzinfotel including an NR2B antagonist, e.g. ifenprodil, traxoprodil or (- )-(R)-6-{2-[4-(3-fluorophenyl)-4-hydroxy-l- piperidinyl]-l-hydroxyethyl-3,4-dihydro-2(lH)- quinolinone; [0397] (9) an alpha-adrenergic, e.g. doxazosin, tamsulosin, clonidine, guanfacine, dexmedetomidine, modafinil, or 4-amino-6,7-dimethoxy-2-(5-methane-sulfonamido-l, 2,3,4- tetrahydroisoquinolin-2-yl)-5-(2-pyridyl) quinazoline; [0398] (10) a tricyclic antidepressant, e.g. desipramine, imipramine, amitriptyline or nortriptyline; [0399] (11) an anticonvulsant, e.g. carbamazepine (Tegretol®), lamotrigine, topiramate, lacosamide (Vimpat®) or valproate; [0400] (12) a tachykinin (NK) antagonist, particularly an NK-3, NK-2 or NK-1 antagonist, e.g. (alphaR,9R)-7-[3,5-bis(trifluoromethyl)benzyl]-8,9,10,11 -tetrahydro-9-methyl-5-(4- methylphenyl)- 7H-[l,4]diazocino[2,l-g][l,7]-naphthyridine-6-13-dione (TAK-637), 5- [[(2R,3S)-2-[(lR)-l-[3,5- bis(trifluoromethyl)phenyl]ethoxy-3-(4-fluorophenyl)-4-morpholinyl]-methyl]-l,2-dihydro-3H-l,2,4-
triazol-3-one (MK-869), aprepitant, lanepitant, dapitant or 3-[[2-methoxy-5- (trifluoromethoxy)phenyl]-methylamino]-2-phenylpiperidine (2S,3S); [0401] (13) a muscarinic antagonist, e.g oxybutynin, tolterodine, propiverine, tropsium chloride, darifenacin, solifenacin, temiverine and ipratropium; [0402] (14) a COX-2 selective inhibitor, e.g. celecoxib, rofecoxib, parecoxib, valdecoxib, deracoxib, etoricoxib, or lumiracoxib; [0403] (15) a coal-tar analgesic, in particular paracetamol; [0404] (16) a neuroleptic such as droperidol, chlorpromazine, haloperidol, perphenazine, thioridazine, mesoridazine, trifluoperazine, fluphenazine, clozapine, olanzapine, risperidone, ziprasidone, quetiapine, sertindole, aripiprazole, sonepiprazole, blonanserin, iloperidone, perospirone, raclopride, zotepine, bifeprunox, asenapine, lurasidone, amisulpride, balaperidone, palindore, eplivanserin, osanetant, rimonabant, meclinertant, Miraxion® or sarizotan; [0405] (17) a vanilloid receptor agonist (e.g. resinferatoxin or civamide) or antagonist (e.g. capsazepine, GRC-15300); [0406] (18) a beta-adrenergic such as propranolol; [0407] (19) a local anesthetic such as mexiletine; [0408] (20) a corticosteroid such as dexamethasone; [0409] (21) a 5-HT receptor agonist or antagonist, particularly a 5-HT1B/1D agonist such as eletriptan, sumatriptan, naratriptan, zolmitriptan or rizatriptan; [0410] (22) a 5-HT2A receptor antagonist such as R(+)-alpha-(2,3-dimethoxy-phenyl)-l-[2-(4- fluorophenylethyl)]-4-piperidinemethanol (MDL-100907); [0411] (23) a cholinergic (nicotinic) analgesic, such as ispronicline (TC-1734), (E)-N-methyl-4- (3-pyridinyl)-3-buten-l-amine (RJR-2403), (R)-5-(2-azetidinylmethoxy)-2-chloropyridine (ABT-594) or nicotine; [0412] (24) Tramadol®, Tramadol ER (Ultram ER®), IV Tramadol, Tapentadol ER (Nucynta®); [0413] (25) a PDE5 inhibitor, such as 5-[2-ethoxy-5-(4-methyl-l-piperazinyl-sulphonyl)phenyl]- l-methyl-3-n-propyl-l,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one (sildenafil), (6R,12aR)- 2,3,6,7,12,12a-hexahydro-2-methyl-6-(3,4-methylenedioxyphenyl)-pyrazino[2',l':6,l]-pyrido[3,4- b]indole-l,4-dione (IC-351 or tadalafil), 2-[2-ethoxy-5-(4-ethyl-piperazin-l-yl-l-sulphonyl)-phenyl]-5- methyl-7-propyl-3H-imidazo[5,l-f][l,2,4]triazin-4-one (vardenafil), 5-(5-acetyl-2-butoxy-3-pyridinyl)- 3-ethyl-2-(l-ethyl-3-azetidinyl)-2,6-dihydro-7H- pyrazolo[4,3-d]pyrimidin-7-one, 5-(5-acetyl-2- propoxy-3-pyridinyl)-3-ethyl-2-(l-isopropyl-3-azetidinyl)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin- 7-one, 5-[2-ethoxy-5-(4-ethylpiperazin-l-ylsulphonyl)pyridin-3-yl]-3-ethyl-2-[2-methoxyethyl]-2,6- dihydro-7H- pyrazolo[4,3-d]pyrimidin-7-one, 4-[(3-chloro-4-methoxybenzyl)amino]-2-[(2S)-2- (hydroxymethyl)pyrrolidin-l-yl]-N-(pyrimidin-2-ylmethyl)pyrimidine-5-carboxamide, 3-(l- methyl-7- oxo-3-propyl-6,7-dihydro-lH-pyrazolo[4,3-d]pyrimidin-5-yl)-N-[2-(l-methylpyrrolidin-2-yl)ethyl]-4- propoxybenzenesulfonamide;
[0414] (26) an alpha-2-delta ligand such as gabapentin (Neurontin®), gabapentin GR (Gralise®), gabapentin, enacarbil (Horizant®), pregabalin (Lyrica®), 3-methyl gabapentin, (l[alpha],3[alpha],5[alpha])(3-amino-methyl-bicyclo[3.2.0]hept-3-yl)-acetic acid, (3S,5R)-3- aminomethyl-5-methyl-heptanoic acid, (3S,5R)-3-amino-5-methyl-heptanoic acid, (3S,5R)-3-amino- 5-methyl-octanoic acid, (2S,4S)-4-(3-chlorophenoxy)proline, (2S,4S)-4-(3-fluorobenzyl)-proline, [(lR,5R,6S)-6-(aminomethyl)bicyclo[3.2.0]hept-6-yl]acetic acid, 3-(l-aminomethyl- cyclohexylmethyl)-4H-[1,2,4]oxadiazol-5-one, C-[1-(1H-tetrazol-5-ylmethyl)-cycloheptyl]- methylamine, (3S,4S)-(l-aminomethyl-3,4-dimethyl-cyclopentyl)-acetic acid, (3S,5R)-3- aminomethyl-5-methyl-octanoic acid, (3S,5R)-3-amino-5-methyl-nonanoic acid, (3S,5R)-3-amino-5- methyl-octanoic acid, (3R,4R,5R)-3-amino-4,5-dimethyl-heptanoic acid and (3R,4R,5R)-3-amino- 4,5-dimethyl-octanoic acid; [0415] (27) a cannabinoid such as KHK-6188; [0416] (28) metabotropic glutamate subtype 1 receptor (mGluRl) antagonist; [0417] (29) a serotonin reuptake inhibitor such as sertraline, sertraline metabolite demethylsertraline, fluoxetine, norfluoxetine (fluoxetine desmethyl metabolite), fluvoxamine, paroxetine, citalopram, citalopram metabolite desmethylcitalopram, escitalopram, d,l-fenfluramine, femoxetine, ifoxetine, cyanodothiepin, litoxetine, dapoxetine, nefazodone, cericlamine and trazodone; [0418] (30) a noradrenaline (norepinephrine) reuptake inhibitor, such as maprotiline, lofepramine, mirtazepine, oxaprotiline, fezolamine, tomoxetine, mianserin, bupropion, bupropion metabolite hydroxybupropion, nomifensine and viloxazine (Vivalan®), especially a selective noradrenaline reuptake inhibitor such as reboxetine, in particular (S,S)-reboxetine; [0419] (31) a dual serotonin-noradrenaline reuptake inhibitor, such as venlafaxine, venlafaxine metabolite O-desmethylvenlafaxine, clomipramine, clomipramine metabolite desmethylclomipramine, duloxetine (Cymbalta®), milnacipran and imipramine; [0420] (32) an inducible nitric oxide synthase (iNOS) inhibitor such as S-[2-[(l- iminoethyl)amino]ethyl]-L-homocysteine, S-[2-[(l-iminoethyl)-amino]ethyl]-4,4-dioxo-L-cysteine, S- [2-[(l-iminoethyl)amino]ethyl]-2-methyl-L-cysteine, (2S,5Z)-2-amino-2-methyl-7-[(l- iminoethyl)amino]-5-heptenoic acid, 2-[[(lR,3S)-3-amino-4-hydroxy-l-(5-thiazolyl)-butyl]thio]-S- chloro-S-pyridinecarbonitrile; 2-[[(lR,3S)-3-amino-4-hydroxy-l-(5- thiazolyl)butyl]thio]-4- chlorobenzonitrile, (2S,4R)-2-amino-4-[[2-chloro-5- (trifluoromethyl)phenyl]thio]-5-thiazolebutanol, 2-[[(lR,3S)-3-amino-4-hydroxy-l-(5-thiazolyl) butyl]thio]-6-(trifluoromethyl)-3-pyridinecarbonitrile, 2-[[(lR,3S)-3-amino-4-hydroxy-1-(5-thiazolyl)butyl]thio]-5-chlorobenzonitrile, N-[4-[2-(3- chlorobenzylamino)ethyl]phenyl]thiophene-2-carboxamidine, NXN-462, or guanidinoethyldisulfide; [0421] (33) an acetylcholinesterase inhibitor such as donepezil; [0422] (34) a prostaglandin E2 subtype 4 (EP4) antagonist such as N-[({2-[4-(2-ethyl-4,6- dimethyl-lH-imidazo[4,5-c]pyridin-l-yl)phenyl]ethyl}amino)-carbonyl]-4- methylbenzenesulfonamide or 4-[(15)-l-({[5-chloro-2-(3-fluorophenoxy)pyridin-3- yl]carbonyl}amino)ethyl]benzoic acid;
[0423] (35) a leukotriene B4 antagonist; such as l-(3-biphenyl-4-ylmethyl-4-hydroxy-chroman-7- yl)-cyclopentanecarboxylic acid (CP- 105696), 5-[2-(2-Carboxyethyl)-3-[6-(4-methoxyphenyl)-5E- hexenyl]oxyphenoxy]-valeric acid (ONO-4057) or DPC-11870; [0424] (36) a 5-lipoxygenase inhibitor, such as zileuton, 6-[(3-fluoro-5-[4-methoxy-3,4,5,6- tetrahydro-2H-pyran-4-yl])phenoxy-methyl]-l-methyl-2-quinolone (ZD-2138), or 2,3,5- trimethyl-6- (3-pyridylmethyl)-l,4-benzoquinone (CV-6504); [0425] (37) a sodium channel blocker, such as lidocaine, lidocaine plus tetracaine cream (ZRS- 201) or eslicarbazepine acetate; [0426] (38) a NaV1.7 blocker, such as XEN-402, XEN403, TV-45070, PF-05089771, CNV1014802, GDC-0276, RG7893 BIIB-074 (Vixotrigine), BIIB-095, ASP-1807, DSP-3905, OLP- 1002, RQ-00432979, FX-301, DWP-1706, DWP-17061, IMB-110, IMB-111, IMB-112 and such as those disclosed in WO2011/140425 (US2011/306607); WO2012/106499 (US2012196869); WO2012/112743 (US2012245136); WO2012/125613 (US2012264749), WO2012/116440 (US2014187533), WO2011026240 (US2012220605), US8883840, US8466188, WO2013/109521 (US2015005304), CN111217776, WO2020/117626, WO2021/252822, WO2021/252818, WO2021/252820, WO2014/201173, WO2012/125973, WO2013/086229, WO2013/134518, WO2014/201206, or WO2016/141035 the entire contents of each application hereby incorporated by reference; [0427] (38a) a NaV1.7 blocker such as (2-benzylspiro[3,4-dihydropyrrolo[1,2-a]pyrazine-1,4'- piperidine]-1'-yl)-(4-isopropoxy-3-methyl-phenyl)methanone, 2,2,2-trifluoro-1-[1'-[3-methoxy-4-[2- (trifluoromethoxy)ethoxy]benzoyl]-2,4-dimethyl-spiro[3,4-dihydropyrrolo[1,2-a]pyrazine-1,4'- piperidine]-6-yl]ethanone, [8-fluoro-2-methyl-6-(trifluoromethyl)spiro[3,4-dihydropyrrolo[1,2- a]pyrazine-1,4'-piperidine]-1'-yl]-(4-isobutoxy-3-methoxy-phenyl)methanone, 1-(4- benzhydrylpiperazin-1-yl)-3-[2-(3,4-dimethylphenoxy)ethoxy]propan-2-ol, (4-butoxy-3-methoxy- phenyl)-[2-methyl-6-(trifluoromethyl)spiro[3,4-dihydropyrrolo[1,2-a]pyrazine-1,4'-piperidine]-1'- yl]methanone, [8-fluoro-2-methyl-6-(trifluoromethyl)spiro[3,4-dihydropyrrolo[1,2-a]pyrazine-1,4'- piperidine]-1'-yl]-(5-isopropoxy-6-methyl-2-pyridyl)methanone, (4-isopropoxy-3-methyl-phenyl)-[2- methyl-6-(1,1,2,2,2-pentafluoroethyl)spiro[3,4-dihydropyrrolo[1,2-a]pyrazine-1,4'-piperidine]-1'- yl]methanone, 5-[2-methyl-4-[2-methyl-6-(2,2,2-trifluoroacetyl)spiro[3,4-dihydropyrrolo[1,2- a]pyrazine-1,4'-piperidine]-1'-carbonyl]phenyl]pyridine-2-carbonitrile, (4-isopropoxy-3-methyl- phenyl)-[6-(trifluoromethyl)spiro[3,4-dihydro-2H-pyrrolo[1,2-a]pyrazine-1,4'-piperidine]-1'- yl]methanone, 2,2,2-trifluoro-1-[1'-[3-methoxy-4-[2-(trifluoromethoxy)ethoxy]benzoyl]-2-methyl- spiro[3,4-dihydropyrrolo[1,2-a]pyrazine-1,4'-piperidine]-6-yl]ethanone, 2,2,2-trifluoro-1-[1'-(5- isopropoxy-6-methyl-pyridine-2-carbonyl)-3,3-dimethyl-spiro[2,4-dihydropyrrolo[1,2-a]pyrazine- 1,4'-piperidine]-6-yl]ethanone, 2,2,2-trifluoro-1-[1'-(5-isopentyloxypyridine-2-carbonyl)-2-methyl- spiro[3,4-dihydropyrrolo[1,2-a]pyrazine-1,4'-piperidine]-6-yl]ethanone, (4-isopropoxy-3-methoxy- phenyl)-[2-methyl-6-(trifluoromethyl)spiro[3,4-dihydropyrrolo[1,2-a]pyrazine-1,4'-piperidine]-1'-
yl]methanone, 2,2,2-trifluoro-1-[1'-(5-isopentyloxypyridine-2-carbonyl)-2,4-dimethyl-spiro[3,4- dihydropyrrolo[1,2-a]pyrazine-1,4'-piperidine]-6-yl]ethanone, 1-[(3S)-2,3-dimethyl-1'-[4-(3,3,3- trifluoropropoxymethyl)benzoyl]spiro[3,4-dihydropyrrolo[1,2-a]pyrazine-1,4'-piperidine]-6-yl]-2,2,2- trifluoro-ethanone, [8-fluoro-2-methyl-6-(trifluoromethyl)spiro[3,4-dihydropyrrolo[1,2-a]pyrazine- 1,4'-piperidine]-1'-yl]-[3-methoxy-4-[(1R)-1-methylpropoxy]phenyl]methanone, 2,2,2-trifluoro-1-[1'- (5-isopropoxy-6-methyl-pyridine-2-carbonyl)-2,4-dimethyl-spiro[3,4-dihydropyrrolo[1,2-a]pyrazine- 1,4'-piperidine]-6-yl]ethanone, 1-[1'-[4-methoxy-3-(trifluoromethyl)benzoyl]-2-methyl-spiro
B. Compound I Amorphous Form [0210] In some embodiments, the disclosure provides a neat amorphous form of Compound I. In some embodiments, the disclosure provides Compound I amorphous form. FIG. 1 provides an X-ray powder diffractogram of Compound I amorphous form at room temperature. [0211] In some embodiments, Compound I amorphous form is substantially pure. In some embodiments, Compound I amorphous form is substantially amorphous. In some embodiments, Compound I amorphous form is characterized by an X-ray powder diffractogram generated by an X-ray powder diffraction analysis with an incident beam of Cu Kα radiation. [0212] In some embodiments, Compound I amorphous form is characterized by an X-ray powder diffractogram substantially similar to FIG.1. [0213] In some embodiments, Compound I amorphous form is characterized as having a 13C SSNMR spectrum with a peak at 177.6 ± 0.2 ppm. In some embodiments, Compound I amorphous form is characterized as having a 13C SSNMR spectrum with a peak at 172.0 ± 0.2 ppm. In some embodiments, Compound I amorphous form is characterized as having a 13C SSNMR spectrum with a peak at 162.6 ± 0.2 ppm. In some embodiments, Compound I amorphous form is characterized as having a 13C SSNMR spectrum with a peak at 155.2 ± 0.2 ppm. In some embodiments, Compound I amorphous form is characterized as having a 13C SSNMR spectrum with a peak at 149.9 ± 0.2 ppm. In some embodiments, Compound I amorphous form is characterized as having a 13C SSNMR spectrum with a peak at 147.0 ± 0.2 ppm. In some embodiments, Compound I amorphous form is characterized as having a 13C SSNMR spectrum with a peak at 121.0 ± 0.2 ppm. In some embodiments, Compound I amorphous form is characterized as having a 13C SSNMR spectrum with a peak at 120.1 ± 0.2 ppm. In some embodiments, Compound I amorphous form is characterized as having a 13C SSNMR spectrum with a peak at 119.1 ± 0.2 ppm. In some embodiments, Compound I amorphous form is characterized as having a 13C SSNMR spectrum with a peak at 116.0 ± 0.2 ppm. In some embodiments, Compound I amorphous
form is characterized as having a 13C SSNMR spectrum with a peak at 114.6 ± 0.2 ppm. In some embodiments, Compound I amorphous form is characterized as having a 13C SSNMR spectrum with a peak at 113.8 ± 0.2 ppm. In some embodiments, Compound I amorphous form is characterized as having a 13C SSNMR spectrum with a peak at 15.5 ± 0.2 ppm. In some embodiments, Compound I amorphous form is characterized as having a 13C SSNMR spectrum with a peak at 7.7 ± 0.2 ppm. [0214] In some embodiments, Compound I amorphous form is characterized as having a 13C SSNMR spectrum with one, two, three, four, five, six, seven, eight, nine, ten, or more peaks selected from 177.6 ± 0.2 ppm, 172.0 ± 0.2 ppm, 162.6 ± 0.2 ppm, 155.2 ± 0.2 ppm, 149.9 ± 0.2 ppm, 147.0 ± 0.2 ppm, 121.0 ± 0.2 ppm, 120.1 ± 0.2 ppm, 119.1 ± 0.2 ppm, 116.0 ± 0.2 ppm, 114.6 ± 0.2 ppm, 113.8 ± 0.2 ppm, 15.5 ± 0.2 ppm, and 7.7 ± 0.2 ppm. In some embodiments, Compound I amorphous form is characterized as having a 13C SSNMR spectrum with peaks at 177.6 ± 0.2 ppm, 172.0 ± 0.2 ppm, 162.6 ± 0.2 ppm, 155.2 ± 0.2 ppm, 149.9 ± 0.2 ppm, 147.0 ± 0.2 ppm, 121.0 ± 0.2 ppm, 120.1 ± 0.2 ppm, 119.1 ± 0.2 ppm, 116.0 ± 0.2 ppm, 114.6 ± 0.2 ppm, 113.8 ± 0.2 ppm, 15.5 ± 0.2 ppm, and 7.7 ± 0.2 ppm. [0215] In some embodiments, Compound I amorphous form is characterized by a 13C SSNMR spectrum substantially similar to FIG.2. [0216] In some embodiments, Compound I amorphous form characterized as having a 19F SSNMR spectrum with a peak at -61.9 ± 0.2 ppm. In some embodiments, Compound I amorphous form characterized as having a 19F SSNMR spectrum with a peak at -142.1 ± 0.2 ppm. [0217] In some embodiments, Compound I amorphous form is characterized as having a 19F SSNMR spectrum with one or two peaks selected from -61.9 ± 0.2 ppm and -142.1 ± 0.2 ppm. [0218] In some embodiments, Compound I amorphous form is characterized by a 19F SSNMR spectrum substantially similar to FIG.3. [0219] Another aspect of the disclosure provides a method of making Compound I amorphous form. In some embodiments, the method of making Compound I amorphous form comprises: comprising (i) dissolving Compound I, (ii) filtering and evaporating the solvent at 50 °C, 12 mbar, over 1 hour on a centrifugal evaporation, (iii) drying in a vacuum oven at 50 °C overnight to yield Compound I amorphous form. C. Crystalline Compound I Neat Form A [0220] In some embodiments, the disclosure provides neat crystalline forms of Compound I. In some embodiments, the disclosure provides crystalline Compound I neat Form A. FIG.4 provides an X-ray powder diffractogram of crystalline Compound I neat Form A. [0221] In some embodiments, crystalline Compound I neat Form A is substantially pure. In some embodiments, crystalline Compound I neat Form A is substantially crystalline. In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram generated by an X-ray powder diffraction analysis with an incident beam of Cu Kα radiation.
[0222] In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 9.5 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 12.3 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 13.5 ± 0.2 degrees two-theta. [0223] In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 7.4 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 8.3 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 11.3 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 14.4 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 14.9 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 15.0 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 15.9 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 16.6 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 17.6 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 18.0 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 18.7 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 18.9 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 19.2 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 19.5 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 19.8 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 20.5 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 21.4 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 21.9 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 22.5 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form A is
characterized by an X-ray powder diffractogram having a signal at 23.5 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having a signal at 24.1 ± 0.2 degrees two-theta. [0224] In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having signals at one or two of 9.5 ± 0.2 degrees two theta, 12.3 ± 0.2 degrees two theta, and 13.5 ± 0.2 degrees two-theta. [0225] In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having (a) one or two signals selected from 9.5 ± 0.2 degrees two-theta, 12.3 ± 0.2 degrees two-theta, and 13.5 ± 0.2 degrees two-theta, and (b) one or two signals selected from 11.3 ± 0.2 degrees two-theta, 15.9 ± 0.2 degrees two-theta, and 20.5 ± 0.2 degrees two-theta. [0226] In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having two, three, four, five, or six signals selected from 9.5 ± 0.2 degrees two- theta, 12.3 ± 0.2 degrees two-theta, 13.5 ± 0.2 degrees two-theta , 11.3 ± 0.2 degrees two-theta, 15.9 ± 0.2 degrees two-theta, 20.5 ± 0.2 degrees two-theta, 14.9 ± 0.2 degrees two-theta, 16.6 ± 0.2 degrees two-theta, and 23.5 ± 0.2 degrees two-theta. [0227] In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having one, two three, four five, six, seven, eight, nine, ten, eleven, twelve, thirteen, fourteen, fifteen, sixteen, seventeen, eighteen, nineteen, twenty, twenty-one, or twenty-two signals selected from 7.4 ± 0.2 degrees two-theta; 8.3 ± 0.2 degrees two-theta; 9.5 ± 0.2 degrees two- theta; 11.3 ± 0.2 degrees two-theta; 12.3 ± 0.2 degrees two-theta; 13.5 ± 0.2 degrees two-theta; 14.4 ± 0.2 degrees two-theta; 14.9 ± 0.2 degrees two-theta; 15.0 ± 0.2 degrees two-theta; 15.9 ± 0.2 degrees two-theta; 16.6 ± 0.2 degrees two-theta; 17.6 ± 0.2 degrees two-theta; 18.0 ± 0.2 degrees two-theta; 18.7 ± 0.2 degrees two-theta; 18.9 ± 0.2 degrees two-theta; 19.2 ± 0.2 degrees two-theta; 19.5 ± 0.2 degrees two-theta; 19.8 ± 0.2 degrees two-theta; 20.5 ± 0.2 degrees two-theta; 21.4 ± 0.2 degrees two-theta; 21.9 ± 0.2 degrees two-theta; 22.5 ± 0.2 degrees two-theta; 23.5 ± 0.2 degrees two-theta; and 24.1 ± 0.2 degrees two-theta. [0228] In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram having signals at 7.4 ± 0.2 degrees two-theta, 8.3 ± 0.2 degrees two-theta, 9.5 ± 0.2 degrees two-theta, 11.3 ± 0.2 degrees two-theta, 12.3 ± 0.2 degrees two-theta, 13.5 ± 0.2 degrees two-theta, 14.4 ± 0.2 degrees two-theta, 14.9 ± 0.2 degrees two-theta, 15.0 ± 0.2 degrees two-theta, 15.9 ± 0.2 degrees two-theta, 16.6 ± 0.2 degrees two-theta, 17.6 ± 0.2 degrees two-theta, 18.0 ± 0.2 degrees two-theta, 18.7 ± 0.2 degrees two-theta, 18.9 ± 0.2 degrees two-theta, 19.2 ± 0.2 degrees two-theta, 19.5 ± 0.2 degrees two-theta, 19.8 ± 0.2 degrees two-theta, 20.5 ± 0.2 degrees two-theta, 21.4 ± 0.2 degrees two-theta, 21.9 ± 0.2 degrees two-theta, 22.5 ± 0.2 degrees two-theta, 23.5 ± 0.2 degrees two-theta, and 24.1 ± 0.2 degrees two-theta. [0229] In some embodiments, crystalline Compound I neat Form A is characterized by an X-ray powder diffractogram substantially similar to FIG.4.
[0230] In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a peak at 170.4 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a peak at 163.8 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a peak at 162.4 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a peak at 160.9 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a peak at 158.6 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a peak at 158.3 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a peak at 154.6 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a peak at 154.0 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a peak at 148.3 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a peak at 147.6 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a peak at 146.9 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a peak at 145.9 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a peak at 143.5 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a peak at 143.1 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a peak at 141.9 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a peak at 127.4 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a peak at 125.3 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a peak at 123.6 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a peak at 122.2 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a peak at 121.3 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a peak at 120.7 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a peak at 117.9 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a peak at 117.3 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a peak at 116.3 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a peak at 115.8 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a peak at
114.2 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a peak at 113.6 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a peak at 112.8 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a peak at 15.4 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a peak at 8.2 ± 0.2 ppm. [0231] In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with one, two, three, four, five, six, seven, eight, nine, ten, or more peaks selected from 170.4 ± 0.2 ppm, 163.8 ± 0.2 ppm, 162.4 ± 0.2 ppm, 160.9 ± 0.2 ppm, 158.6 ± 0.2 ppm, 158.3 ± 0.2 ppm, 154.6 ± 0.2 ppm, 154.0 ± 0.2 ppm, 148.3 ± 0.2 ppm, 147.6 ± 0.2 ppm, 146.9 ± 0.2 ppm, 145.9 ± 0.2 ppm, 143.5 ± 0.2 ppm, 143.1 ± 0.2 ppm, 141.9 ± 0.2 ppm, 127.4 ± 0.2 ppm, 125.3 ± 0.2 ppm, 123.6 ± 0.2 ppm, 122.2 ± 0.2 ppm, 121.3 ± 0.2 ppm, 120.7 ± 0.2 ppm, 117.9 ± 0.2 ppm, 117.3 ± 0.2 ppm, 116.3 ± 0.2 ppm, 115.8 ± 0.2 ppm, 114.2 ± 0.2 ppm, 113.6 ± 0.2 ppm, 112.8 ± 0.2 ppm, 15.4 ± 0.2 ppm, and 8.2 ± 0.2 ppm. [0232] In some embodiments, crystalline Compound I neat Form A is characterized as having a 13C SSNMR spectrum with a 13C SSNMR spectrum having peaks at 170.4 ± 0.2 ppm, 163.8 ± 0.2 ppm, 162.4 ± 0.2 ppm, 160.9 ± 0.2 ppm, 158.6 ± 0.2 ppm, 158.3 ± 0.2 ppm, 154.6 ± 0.2 ppm, 154.0 ± 0.2 ppm, 148.3 ± 0.2 ppm, 147.6 ± 0.2 ppm, 146.9 ± 0.2 ppm, 145.9 ± 0.2 ppm, 143.5 ± 0.2 ppm, 143.1 ± 0.2 ppm, 141.9 ± 0.2 ppm, 127.4 ± 0.2 ppm, 125.3 ± 0.2 ppm, 123.6 ± 0.2 ppm, 122.2 ± 0.2 ppm, 121.3 ± 0.2 ppm, 120.7 ± 0.2 ppm, 117.9 ± 0.2 ppm, 117.3 ± 0.2 ppm, 116.3 ± 0.2 ppm, 115.8 ± 0.2 ppm, 114.2 ± 0.2 ppm, 113.6 ± 0.2 ppm, 112.8 ± 0.2 ppm, 15.4 ± 0.2 ppm, and 8.2 ± 0.2 ppm. [0233] In some embodiments, crystalline Compound I neat Form A is characterized by a 13C SSNMR spectrum substantially similar to FIG.5. [0234] In some embodiments, crystalline Compound I neat Form A is characterized as having a 19F SSNMR spectrum with a peak at -61.2 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 19F SSNMR spectrum with a peak at -61.6 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 19F SSNMR spectrum with a peak at -138.6 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 19F SSNMR spectrum with a peak at -140.3 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 19F SSNMR spectrum with a peak at -142.0 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 19F SSNMR spectrum with a peak at -146.0 ± 0.2 ppm. [0235] In some embodiments, crystalline Compound I neat Form A is characterized as having a 19F SSNMR spectrum with one or two peaks selected from -61.2 ± 0.2 ppm, -61.6 ± 0.2 ppm, -138.6 ± 0.2 ppm, -140.3 ± 0.2 ppm, -142.0 ± 0.2 ppm, and -146.0 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form A is characterized as having a 19F SSNMR spectrum with peaks at
-61.2 ± 0.2 ppm, -61.6 ± 0.2 ppm, -138.6 ± 0.2 ppm, -140.3 ± 0.2 ppm, -142.0 ± 0.2 ppm, and -146.0 ± 0.2 ppm. [0236] In some embodiments, crystalline Compound I neat Form A is characterized by a 19F SSNMR spectrum substantially similar to FIG.6. [0237] In some embodiments, crystalline Compound I neat Form A is characterized by a triclinic crystal system, P-1 space group and the following unit cell dimensions measured at 100 K on a diffractometer utilizing Cu Kα radiation (λ=1.54178 Å): a 9.6 ± 0.1 Å α 94.3 ± 0.1° b 12.6 ± 0.1 Å β 97.3 ± 0.1° c 18.7 ± 0.1 Å γ 107.3 ± 0.1°. In some embodiments, the diffractometer is a Bruker diffractometer. [0238] Another aspect of the invention provides a method of making crystalline Compound I neat Form A. In some embodiments, the method of making crystalline Compound I neat Form A comprises: (i) combining Compound I and ethyl acetate, (ii) distilling under vacuum at 50 °C, (iii) adding ethyl acetate, (iv) distilling under vacuum at 50 °C, (v) heating to 75 °C, (vi) adding heptane, (vii) cooling to 40 °C, (viii) adding a seed of crystalline Compound I Form A, (ix) holding at 40 °C for 1.5 hours, (x) adding heptane over 5 hours, (xi) cooling the slurry to 20 °C over 5 hours, (xii) holding at 20 °C for 11 hours, (xiii) collecting the solids, (xiv) drying the solids in a vacuum oven at 45 °C with a slight nitrogen bleed for 46 hours to yield crystalline Compound I neat Form A. D. Crystalline Compound I Neat Form B [0239] In some embodiments, the disclosure provides crystalline Compound I neat Form B. FIG. 7 provides an X-ray powder diffractogram of crystalline Compound I neat Form B. [0240] In some embodiments, crystalline Compound I neat Form B is substantially pure. In some embodiments, crystalline Compound I neat Form B is substantially crystalline. In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram generated by an X-ray powder diffraction analysis with an incident beam of Cu Kα radiation. [0241] In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 4.8 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 13.3 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 18.5 ± 0.2 degrees two-theta. [0242] In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 6.0 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 7.0 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 8.8 ± 0.2 degrees two-theta. In some embodiments,
crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 10.5 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 12.1 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 13.8 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 14.1 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 14.7 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 15.2 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 16.5 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 16.9 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 17.5 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 17.9 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 18.9 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 19.2 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 19.5 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 20.9 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 21.7 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having a signal at 23.0 ± 0.2 degrees two-theta. [0243] In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having signals at one or two 4.8 ± 0.2 degrees two-theta, 13.3 ± 0.2 degrees two-theta, and 18.5 ± 0.2 degrees two-theta. [0244] In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having (a) one or two signals selected from from 4.8 ± 0.2 degrees two-theta, 13.3 ± 0.2 degrees two-theta, and 18.5 ± 0.2 degrees two-theta, and (b) one or two signals selected from 6.0 ± 0.2 degrees two-theta, 16.5 ± 0.2 degrees two-theta, and 16.9 ± 0.2 degrees two-theta. [0245] In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having two, three, four, five, or six signals selected from 4.8 ± 0.2 degrees two-theta, 13.3 ± 0.2 degrees two-theta, 18.5 ± 0.2 degrees two-theta, 6.0 ± 0.2 degrees two-theta,
16.5 ± 0.2 degrees two-theta, 16.9 ± 0.2 degrees two-theta, 14.1 ± 0.2 degrees two-theta, 14.7 ± 0.2 degrees two-theta, and 15.2 ± 0.2 degrees two-theta. [0246] In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having one, two three, four five, six, seven, eight, nine, ten, eleven, twelve, thirteen, fourteen, fifteen, sixteen, seventeen, eighteen, nineteen, twenty, twenty-one, or twenty-two signals selected from 4.8 ± 0.2 degrees two-theta; 6.0 ± 0.2 degrees two-theta; 7.0 ± 0.2 degrees two-theta; 8.8 ± 0.2 degrees two-theta; 10.5 ± 0.2 degrees two-theta; 12.1 ± 0.2 degrees two-theta; 13.3 ± 0.2 degrees two-theta; 13.8 ± 0.2 degrees two-theta; 14.1 ± 0.2 degrees two-theta; 14.7 ± 0.2 degrees two-theta; 15.2 ± 0.2 degrees two-theta; 16.5 ± 0.2 degrees two-theta; 16.9 ± 0.2 degrees two-theta; 17.5 ± 0.2 degrees two-theta; 17.9 ± 0.2 degrees two-theta; 18.5 ± 0.2 degrees two-theta; 18.9 ± 0.2 degrees two-theta; 19.2 ± 0.2 degrees two-theta; 19.5 ± 0.2 degrees two-theta; 20.9 ± 0.2 degrees two-theta; 21.7 ± 0.2 degrees two-theta; and 23.0 ± 0.2 degrees two-theta. [0247] In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram having signals at 4.8 ± 0.2 degrees two-theta, 6.0 ± 0.2 degrees two-theta, 7.0 ± 0.2 degrees two-theta, 8.8 ± 0.2 degrees two-theta, 10.5 ± 0.2 degrees two-theta, 12.1 ± 0.2 degrees two-theta, 13.3 ± 0.2 degrees two-theta, 13.8 ± 0.2 degrees two-theta, 14.1 ± 0.2 degrees two-theta, 14.7 ± 0.2 degrees two-theta, 15.2 ± 0.2 degrees two-theta, 16.5 ± 0.2 degrees two-theta, 16.9 ± 0.2 degrees two-theta, 17.5 ± 0.2 degrees two-theta, 17.9 ± 0.2 degrees two-theta, 18.5 ± 0.2 degrees two-theta, 18.9 ± 0.2 degrees two-theta, 19.2 ± 0.2 degrees two-theta, 19.5 ± 0.2 degrees two-theta, 20.9 ± 0.2 degrees two-theta, 21.7 ± 0.2 degrees two-theta, and 23.0 ± 0.2 degrees two-theta. [0248] In some embodiments, crystalline Compound I neat Form B is characterized by an X-ray powder diffractogram substantially similar to FIG.7. [0249] In some embodiments, crystalline Compound I neat Form B is characterized as having a 13C SSNMR spectrum with a peak at 176.9 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13C SSNMR spectrum with a peak at 171.7 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13C SSNMR spectrum with a peak at 164.0 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13C SSNMR spectrum with a peak at 161.4 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13C SSNMR spectrum with a peak at 160.7 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13C SSNMR spectrum with a peak at 155.3 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13C SSNMR spectrum with a peak at 154.8 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13C SSNMR spectrum with a peak at 148.6 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13C SSNMR spectrum with a peak at 147.6 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13C SSNMR spectrum with a peak at 146.4 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is
characterized as having a 13C SSNMR spectrum with a peak at 145.7 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13C SSNMR spectrum with a peak at 144.8 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13C SSNMR spectrum with a peak at 127.7 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13C SSNMR spectrum with a peak at 125.4 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13C SSNMR spectrum with a peak at 122.0 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13C SSNMR spectrum with a peak at 121.5 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13C SSNMR spectrum with a peak at 118.4 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13C SSNMR spectrum with a peak at 116.3 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13C SSNMR spectrum with a peak at 114.8 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13C SSNMR spectrum with a peak at 113.8 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13C SSNMR spectrum with a peak at 112.6 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13C SSNMR spectrum with a peak at 17.8 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13C SSNMR spectrum with a peak at 15.0 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13C SSNMR spectrum with a peak at 10.7 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13C SSNMR spectrum with a peak at 9.6 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 13C SSNMR spectrum with a peak at 8.8 ± 0.2 ppm. [0250] In some embodiments, crystalline Compound I neat Form B is characterized as having a 13C SSNMR spectrum with one, two, three, four, five, six, seven, eight, nine, ten, or more peaks selected from 176.9 ± 0.2 ppm, 171.7 ± 0.2 ppm, 164.0 ± 0.2 ppm, 161.4 ± 0.2 ppm, 160.7 ± 0.2 ppm, 155.3 ± 0.2 ppm, 154.8 ± 0.2 ppm, 148.6 ± 0.2 ppm, 147.6 ± 0.2 ppm, 146.4 ± 0.2 ppm, 145.7 ± 0.2 ppm, 144.8 ± 0.2 ppm, 127.7 ± 0.2 ppm, 125.4 ± 0.2 ppm, 122.0 ± 0.2 ppm, 121.5 ± 0.2 ppm, 118.4 ± 0.2 ppm, 116.3 ± 0.2 ppm, 114.8 ± 0.2 ppm, 113.8 ± 0.2 ppm, 112.6 ± 0.2 ppm, 17.8 ± 0.2 ppm, 15.0 ± 0.2 ppm, 10.7 ± 0.2 ppm, 9.6 ± 0.2 ppm, and 8.8 ± 0.2 ppm. [0251] In some embodiments, crystalline Compound I neat Form B is characterized as having a 13C SSNMR spectrum with peaks at 176.9 ± 0.2 ppm, 171.7 ± 0.2 ppm, 164.0 ± 0.2 ppm, 161.4 ± 0.2 ppm, 160.7 ± 0.2 ppm, 155.3 ± 0.2 ppm, 154.8 ± 0.2 ppm, 148.6 ± 0.2 ppm, 147.6 ± 0.2 ppm, 146.4 ± 0.2 ppm, 145.7 ± 0.2 ppm, 144.8 ± 0.2 ppm, 127.7 ± 0.2 ppm, 125.4 ± 0.2 ppm, 122.0 ± 0.2 ppm, 121.5 ± 0.2 ppm, 118.4 ± 0.2 ppm, 116.3 ± 0.2 ppm, 114.8 ± 0.2 ppm, 113.8 ± 0.2 ppm, 112.6 ± 0.2 ppm, 17.8 ± 0.2 ppm, 15.0 ± 0.2 ppm, 10.7 ± 0.2 ppm, 9.6 ± 0.2 ppm, and 8.8 ± 0.2 ppm. [0252] In some embodiments, crystalline Compound I neat Form B is characterized by a 13C SSNMR spectrum substantially similar to FIG.8.
[0253] In some embodiments, crystalline Compound I neat Form B is characterized as having a 19F SSNMR spectrum with a peak at -61.6 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 19F SSNMR spectrum with a peak at -138.0 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 19F SSNMR spectrum with a peak at -139.7 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 19F SSNMR spectrum with a peak at -143.4 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 19F SSNMR spectrum with a peak at -144.9 ± 0.2 ppm. [0254] In some embodiments, crystalline Compound I neat Form B is characterized as having a 19F SSNMR spectrum with one or two peaks selected from -61.6 ± 0.2 ppm, -138.0 ± 0.2 ppm, -139.7 ± 0.2 ppm, -143.4 ± 0.2 ppm, and -144.9 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form B is characterized as having a 19F SSNMR spectrum with peaks at -61.6 ± 0.2 ppm, -138.0 ± 0.2 ppm, -139.7 ± 0.2 ppm, -143.4 ± 0.2 ppm, and -144.9 ± 0.2 ppm. [0255] In some embodiments, crystalline Compound I neat Form B is characterized by a 19F SSNMR spectrum substantially similar to FIG.9. [0256] Another aspect of the disclosure provides a method of crystalline Compound I neat Form B. In some embodiments, the method of making crystalline Compound I neat Form B, comprises: (i) adding Compound I hydrate Form A to an oven set at 180 °C (ii) cooling under ambient conditions to yield crystalline Compound I neat Form B. E. Crystalline Compound I Neat Form E [0257] In some embodiments, the disclosure provides crystalline Compound I neat Form E. FIG. 10 provides an X-ray powder diffractogram of crystalline Compound I neat Form E. [0258] In some embodiments, crystalline Compound I neat Form E is substantially pure. In some embodiments, crystalline Compound I neat Form E is substantially crystalline. In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram generated by an X-ray powder diffraction analysis with an incident beam of Cu Kα radiation. [0259] In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 8.6 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 10.1 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 12.6 ± 0.2 degrees two-theta. [0260] In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 8.5 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 8.7 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 11.5 ± 0.2 degrees two-theta. In some embodiments,
crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 12.0 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 14.1 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 14.2 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 15.1 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 16.4 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 20.3 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 20.6 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 21.0 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 21.4 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 22.1 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 22.5 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 23.0 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 23.2 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 24.1 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having a signal at 24.7 ± 0.2 degrees two-theta. [0261] In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having signals at one or two selected from 8.6 ± 0.2 degrees two-theta, 10.1 ± 0.2 degrees two-theta, and 12.6 ± 0.2 degrees two-theta. [0262] In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having (a) one or two signals selected from 8.6 ± 0.2 degrees two-theta, 10.1 ± 0.2 degrees two-theta, and 12.6 ± 0.2 degrees two-theta and (b) one or two signals selected from 14.1 ± 0.2 degrees two-theta, 20.3 ± 0.2 degrees two-theta, and 23.2 ± 0.2 degrees two-theta. [0263] In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having two, three, four, five, or six signals selected from 8.6 ± 0.2 degrees two-theta, 10.1 ± 0.2 degrees two-theta, 12.6 ± 0.2 degrees two-theta, 14.1 ± 0.2 degrees two-theta, 20.3 ± 0.2 degrees two-theta, 23.2 ± 0.2 degrees two-theta, 11.5 ± 0.2 degrees two-theta, 14.2 ± 0.2 degrees two-theta, and 15.1 ± 0.2 degrees two-theta.
[0264] In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having one, two three, four five, six, seven, eight, nine, ten, eleven, twelve, thirteen, fourteen, fifteen, sixteen, seventeen, eighteen, nineteen, twenty, twenty-one, or twenty-two signals selected from 8.5 ± 0.2 degrees two-theta; 8.6 ± 0.2 degrees two-theta; 8.7 ± 0.2 degrees two-theta; 10.1 ± 0.2 degrees two-theta; 11.5 ± 0.2 degrees two-theta; 12.0 ± 0.2 degrees two-theta; 12.6 ± 0.2 degrees two-theta; 14.1 ± 0.2 degrees two-theta; 14.2 ± 0.2 degrees two-theta; 15.1 ± 0.2 degrees two-theta; 16.4 ± 0.2 degrees two-theta; 20.3 ± 0.2 degrees two-theta; 20.6 ± 0.2 degrees two-theta; 21.0 ± 0.2 degrees two-theta; 21.4 ± 0.2 degrees two-theta; 22.1 ± 0.2 degrees two-theta; 22.5 ± 0.2 degrees two-theta; 23.0 ± 0.2 degrees two-theta; 23.2 ± 0.2 degrees two-theta; 24.1 ± 0.2 degrees two-theta; and 24.7 ± 0.2 degrees two-theta. [0265] In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram having signals at 8.5 ± 0.2 degrees two-theta, 8.6 ± 0.2 degrees two-theta, 8.7 ± 0.2 degrees two-theta, 10.1 ± 0.2 degrees two-theta, 11.5 ± 0.2 degrees two-theta, 12.0 ± 0.2 degrees two-theta, 12.6 ± 0.2 degrees two-theta, 14.1 ± 0.2 degrees two-theta, 14.2 ± 0.2 degrees two-theta, 15.1 ± 0.2 degrees two-theta, 16.4 ± 0.2 degrees two-theta, 20.3 ± 0.2 degrees two-theta, 20.6 ± 0.2 degrees two-theta, 21.0 ± 0.2 degrees two-theta, 21.4 ± 0.2 degrees two-theta, 22.1 ± 0.2 degrees two-theta, 22.5 ± 0.2 degrees two-theta, 23.0 ± 0.2 degrees two-theta, 23.2 ± 0.2 degrees two-theta, 24.1 ± 0.2 degrees two-theta, and 24.7 ± 0.2 degrees two-theta. [0266] In some embodiments, crystalline Compound I neat Form E is characterized by an X-ray powder diffractogram substantially similar to FIG.10. [0267] In some embodiments, crystalline Compound I neat Form E is characterized as having a 13C SSNMR spectrum with a peak at 170.0 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13C SSNMR spectrum with a peak at 163.0 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13C SSNMR spectrum with a peak at 162.3 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13C SSNMR spectrum with a peak at 161.3 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13C SSNMR spectrum with a peak at 159.3 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13C SSNMR spectrum with a peak at 154.7 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13C SSNMR spectrum with a peak at 150.6 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13C SSNMR spectrum with a peak at 149.8 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13C SSNMR spectrum with a peak at 148.6 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13C SSNMR spectrum with a peak at 148.3 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13C SSNMR spectrum with a peak at 147.1 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13C SSNMR spectrum with a peak at
144.9 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13C SSNMR spectrum with a peak at 143.9 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13C SSNMR spectrum with a peak at 143.5 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13C SSNMR spectrum with a peak at 126.9 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13C SSNMR spectrum with a peak at 126.0 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13C SSNMR spectrum with a peak at 124.1 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13C SSNMR spectrum with a peak at 122.6 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13C SSNMR spectrum with a peak at 120.8 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13C SSNMR spectrum with a peak at 117.8 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13C SSNMR spectrum with a peak at 116.7 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13C SSNMR spectrum with a peak at 115.2 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13C SSNMR spectrum with a peak at 114.1 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13C SSNMR spectrum with a peak at 113.1 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13C SSNMR spectrum with a peak at 15.9 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13C SSNMR spectrum with a peak at 15.3 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 13C SSNMR spectrum with a peak at 8.4 ± 0.2 ppm. [0268] In some embodiments, crystalline Compound I neat Form E is characterized as having a 13C SSNMR spectrum with one, two, three, four, five, six, seven, eight, nine, ten, or more peaks selected from 170.0 ± 0.2 ppm, 163.0 ± 0.2 ppm, 162.3 ± 0.2 ppm, 161.3 ± 0.2 ppm, 159.3 ± 0.2 ppm, 154.7 ± 0.2 ppm, 150.6 ± 0.2 ppm, 149.8 ± 0.2 ppm, 148.6 ± 0.2 ppm, 148.3 ± 0.2 ppm, 147.1 ± 0.2 ppm, 144.9 ± 0.2 ppm, 143.9 ± 0.2 ppm, 143.5 ± 0.2 ppm, 126.9 ± 0.2 ppm, 126.0 ± 0.2 ppm, 124.1 ± 0.2 ppm, 122.6 ± 0.2 ppm, 120.8 ± 0.2 ppm, 117.8 ± 0.2 ppm, 116.7 ± 0.2 ppm, 115.2 ± 0.2 ppm, 114.1 ± 0.2 ppm, 113.1 ± 0.2 ppm, 15.9 ± 0.2 ppm, 15.3 ± 0.2 ppm, and 8.4 ± 0.2 ppm. [0269] In some embodiments, crystalline Compound I neat Form E is characterized with peaks at 170.0 ± 0.2 ppm, 163.0 ± 0.2 ppm, 162.3 ± 0.2 ppm, 161.3 ± 0.2 ppm, 159.3 ± 0.2 ppm, 154.7 ± 0.2 ppm, 150.6 ± 0.2 ppm, 149.8 ± 0.2 ppm, 148.6 ± 0.2 ppm, 148.3 ± 0.2 ppm, 147.1 ± 0.2 ppm, 144.9 ± 0.2 ppm, 143.9 ± 0.2 ppm, 143.5 ± 0.2 ppm, 126.9 ± 0.2 ppm, 126.0 ± 0.2 ppm, 124.1 ± 0.2 ppm, 122.6 ± 0.2 ppm, 120.8 ± 0.2 ppm, 117.8 ± 0.2 ppm, 116.7 ± 0.2 ppm, 115.2 ± 0.2 ppm, 114.1 ± 0.2 ppm, 113.1 ± 0.2 ppm, 15.9 ± 0.2 ppm, 15.3 ± 0.2 ppm, and 8.4 ± 0.2 ppm. [0270] In some embodiments, crystalline Compound I neat Form E is characterized by a 13C SSNMR spectrum substantially similar to FIG.11.
[0271] In some embodiments, crystalline Compound I neat Form E is characterized as having a 19F SSNMR spectrum with a peak at -60.9 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 19F SSNMR spectrum with a peak at -62.5 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 19F SSNMR spectrum with a peak at -63.1 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 19F SSNMR spectrum with a peak at -135.1 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 19F SSNMR spectrum with a peak at -137.1 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 19F SSNMR spectrum with a peak at -140.7 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 19F SSNMR spectrum with a peak at -141.9 ± 0.2 ppm. [0272] In some embodiments, crystalline Compound I neat Form E is characterized as having a 19F SSNMR spectrum with one or two peaks selected from -60.9 ± 0.2 ppm, -62.5 ± 0.2 ppm, -63.1 ± 0.2 ppm, -135.1 ± 0.2 ppm, -137.1 ± 0.2 ppm, -140.7 ± 0.2 ppm, and -141.9 ± 0.2 ppm. In some embodiments, crystalline Compound I neat Form E is characterized as having a 19F SSNMR spectrum with peaks at -60.9 ± 0.2 ppm, -62.5 ± 0.2 ppm, -63.1 ± 0.2 ppm, -135.1 ± 0.2 ppm, -137.1 ± 0.2 ppm, -140.7 ± 0.2 ppm, and -141.9 ± 0.2 ppm. [0273] In some embodiments, crystalline Compound I neat Form E is characterized by a 19F SSNMR spectrum substantially similar to FIG.12. [0274] In some embodiments, crystalline Compound I neat Form E is characterized by a triclinic crystal system, P-1 space group and the following unit cell dimensions measured at 100 K on a diffractometer utilizing Cu Kα radiation (λ=1.54178 Å): a 8.7 ± 0.1 Å α 92.7 ± 0.1° b 12.2 ± 0.1 Å β 96.3 ± 0.1° c 20.9 ± 0.1 Å γ 98.6 ± 0.1°. In some embodiments, the diffractometer is a Rigaku diffractometer. [0275] In some embodiments, crystalline Compound I neat Form E is characterized by a triclinic crystal system, P-1 space group and the following unit cell dimensions measured at 298 K on a diffractometer utilizing Cu Kα radiation (λ=1.54178 Å): a 8.9 ± 0.1 Å α 92.6 ± 0.1° b 12.3 ± 0.1 Å β 97.1 ± 0.1° c 21.1 ± 0.1 Å γ 98.5 ± 0.1°. In some embodiments, the diffractometer is a Rigaku diffractometer. [0276] Another aspect of the disclosure provides a method of crystalline Compound I neat Form E. In some embodiments, the method of making crystalline Compound I neat Form E, comprises: (i) combining Compound I hydrate Form A and trifluorotoluene, (ii) heating the slurry to 80 °C, (iii) cooling to room temperature to yield crystalline Compound I neat Form E.
F. Crystalline Compound I Acetone Solvate Hydrate Form A [0277] In some embodiments, the disclosure provides crystalline Compound I Acetone Solvate Hydrate Form A. FIG.13 provides an X-ray powder diffractogram of crystalline Compound I Acetone Solvate Hydrate Form A. [0278] In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is substantially pure. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is substantially crystalline. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram generated by an X-ray powder diffraction analysis with an incident beam of Cu Kα radiation. [0279] In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 17.2 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 18.1 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 25.9 ± 0.2 degrees two-theta. [0280] In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 7.2 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 8.1 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 9.4 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 9.7 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 11.2 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 11.5 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 12.3 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 12.5 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 12.9 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 13.1 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 13.9 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 14.4 ± 0.2 degrees two-theta. In some embodiments, crystalline
Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 14.8 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 15.1 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 15.6 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 16.2 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 16.5 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 17.6 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 17.7 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 22.7 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 24.8 ± 0.2 degrees two-theta. [0281] In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having signals at one or two of 17.2 ± 0.2 degrees two-theta, 18.1 ± 0.2 degrees two-theta, and 25.9 ± 0.2 degrees two-theta. [0282] In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having (a) one or two signals selected 17.2 ± 0.2 degrees two-theta, 18.1 ± 0.2 degrees two-theta, and 25.9 ± 0.2 degrees two-theta, and (b) one or two signals selected from 12.9 ± 0.2 degrees two-theta, 15.6 ± 0.2 degrees two-theta, and 24.8 ± 0.2 degrees two-theta. [0283] In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having two, three, four, five, or six signals selected from 9.5 ± 0.2 degrees two-theta, 12.3 ± 0.2 degrees two-theta, 13.5 ± 0.2 degrees two-theta, 12.9 ± 0.2 degrees two-theta, 15.6 ± 0.2 degrees two-theta, 24.8 ± 0.2 degrees two-theta, 8.1 ± 0.2 degrees two-theta, 12.3 ± 0.2 degrees two-theta, and 17.6 ± 0.2 degrees two-theta. [0284] In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having one, two three, four five, six, seven, eight, nine, ten, eleven, twelve, thirteen, fourteen, fifteen, sixteen, seventeen, eighteen, nineteen, twenty, twenty-one, or twenty-two signals selected from 7.2 ± 0.2 degrees two-theta; 8.1 ± 0.2 degrees two-theta; 9.7 ± 0.2 degrees two-theta; 11.2 ± 0.2 degrees two-theta; 11.5 ± 0.2 degrees two-theta; 12.3 ± 0.2 degrees two-theta; 12.5 ± 0.2 degrees two-theta; 12.9 ± 0.2 degrees two-theta; 13.1 ± 0.2 degrees two-theta; 13.9 ± 0.2 degrees two-theta; 14.4 ± 0.2 degrees two-theta; 14.8 ± 0.2 degrees two-theta; 15.1 ± 0.2 degrees two-theta; 15.5 ± 0.2 degrees two-theta; 16.2 ± 0.2 degrees two-theta;
16.5 ± 0.2 degrees two-theta; 17.2 ± 0.2 degrees two-theta; 17.6 ± 0.2 degrees two-theta; 17.7 ± 0.2 degrees two-theta; 18.1 ± 0.2 degrees two-theta; 22.7 ± 0.2 degrees two-theta; 24.8 ± 0.2 degrees two-theta; and 25.9 ± 0.2 degrees two-theta. [0285] In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram having signals at 7.2 ± 0.2 degrees two-theta, 8.1 ± 0.2 degrees two-theta, 9.7 ± 0.2 degrees two-theta, 11.2 ± 0.2 degrees two-theta, 11.5 ± 0.2 degrees two-theta, 12.3 ± 0.2 degrees two-theta, 12.5 ± 0.2 degrees two-theta, 12.9 ± 0.2 degrees two-theta, 13.1 ± 0.2 degrees two-theta, 13.9 ± 0.2 degrees two-theta, 14.4 ± 0.2 degrees two-theta, 14.8 ± 0.2 degrees two-theta, 15.1 ± 0.2 degrees two-theta, 15.5 ± 0.2 degrees two-theta, 16.2 ± 0.2 degrees two-theta, 16.5 ± 0.2 degrees two-theta, 17.2 ± 0.2 degrees two-theta, 17.6 ± 0.2 degrees two-theta, 17.7 ± 0.2 degrees two-theta, 18.1 ± 0.2 degrees two-theta, 22.7 ± 0.2 degrees two-theta, 24.8 ± 0.2 degrees two-theta, and 25.9 ± 0.2 degrees two-theta. [0286] In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by an X-ray powder diffractogram substantially similar to FIG.13. [0287] In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 212.9 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 211.6 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 179.2 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 178.9 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 178.4 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 174.7 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 174.1 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 172.7 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 172.2 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 163.1 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 156.7 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 155.6 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 152.3 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 150.1 ± 0.2 ppm. In some
embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 148.7 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 147.9 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 147.0 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 145.1 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 125.5 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 124.5 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 122.5 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 121.6 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 121.1 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 118.5 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 118.1 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 117.4 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 116.8 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 115.7 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 115.3 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 113.9 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 113.2 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 112.4 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 30.0 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 29.5 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 18.4 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 17.6 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with
a peak at 8.8 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 8.1 ± 0.2 ppm. [0288] In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with one, two, three, four, five, six, seven, eight, nine, ten, or more peaks selected from 212.9 ± 0.2 ppm, 211.6 ± 0.2 ppm, 179.2 ± 0.2 ppm, 178.9 ± 0.2 ppm, 178.4 ± 0.2 ppm, 174.7 ± 0.2 ppm, 174.1 ± 0.2 ppm, 172.7 ± 0.2 ppm, 172.2 ± 0.2 ppm, 163.1 ± 0.2 ppm, 156.7 ± 0.2 ppm, 155.6 ± 0.2 ppm, 152.3 ± 0.2 ppm, 150.1 ± 0.2 ppm, 148.7 ± 0.2 ppm, 147.9 ± 0.2 ppm, 147.0 ± 0.2 ppm, 145.1 ± 0.2 ppm, 125.5 ± 0.2 ppm, 124.5 ± 0.2 ppm, 122.5 ± 0.2 ppm, 121.6 ± 0.2 ppm, 121.1 ± 0.2 ppm, 118.5 ± 0.2 ppm, 118.1 ± 0.2 ppm, 117.4 ± 0.2 ppm, 116.8 ± 0.2 ppm, 115.7 ± 0.2 ppm, 115.3 ± 0.2 ppm, 113.9 ± 0.2 ppm, 113.2 ± 0.2 ppm, 112.4 ± 0.2 ppm, 30.0 ± 0.2 ppm, 29.5 ± 0.2 ppm, 18.4 ± 0.2 ppm, 17.6 ± 0.2 ppm, 8.8 ± 0.2 ppm, and 8.1 ± 0.2 ppm. [0289] In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 13C SSNMR spectrum with peaks at 212.9 ± 0.2 ppm, 211.6 ± 0.2 ppm, 179.2 ± 0.2 ppm, 178.9 ± 0.2 ppm, 178.4 ± 0.2 ppm, 174.7 ± 0.2 ppm, 174.1 ± 0.2 ppm, 172.7 ± 0.2 ppm, 172.2 ± 0.2 ppm, 163.1 ± 0.2 ppm, 156.7 ± 0.2 ppm, 155.6 ± 0.2 ppm, 152.3 ± 0.2 ppm, 150.1 ± 0.2 ppm, 148.7 ± 0.2 ppm, 147.9 ± 0.2 ppm, 147.0 ± 0.2 ppm, 145.1 ± 0.2 ppm, 125.5 ± 0.2 ppm, 124.5 ± 0.2 ppm, 122.5 ± 0.2 ppm, 121.6 ± 0.2 ppm, 121.1 ± 0.2 ppm, 118.5 ± 0.2 ppm, 118.1 ± 0.2 ppm, 117.4 ± 0.2 ppm, 116.8 ± 0.2 ppm, 115.7 ± 0.2 ppm, 115.3 ± 0.2 ppm, 113.9 ± 0.2 ppm, 113.2 ± 0.2 ppm, 112.4 ± 0.2 ppm, 30.0 ± 0.2 ppm, 29.5 ± 0.2 ppm, 18.4 ± 0.2 ppm, 17.6 ± 0.2 ppm, 8.8 ± 0.2 ppm, and 8.1 ± 0.2 ppm. [0290] In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by a 13C SSNMR spectrum substantially similar to FIG.14. [0291] In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 19F SSNMR spectrum with a peak at -60.4 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 19F SSNMR spectrum with a peak at -61.2 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 19F SSNMR spectrum with a peak at -61.6 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 19F SSNMR spectrum with a peak at -62.7 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 19F SSNMR spectrum with a peak at -135.9 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 19F SSNMR spectrum with a peak at -136.1 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 19F SSNMR spectrum with a peak at -139.6 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 19F SSNMR spectrum with a peak at -140.6 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 19F SSNMR spectrum with a peak at -141.7 ± 0.2 ppm. In some embodiments,
crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 19F SSNMR spectrum with a peak at -145.2 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 19F SSNMR spectrum with a peak at -145.7 ± 0.2 ppm. [0292] In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 19F SSNMR spectrum with one or two peaks selected from -60.4 ± 0.2 ppm, -61.2 ± 0.2 ppm, -61.6 ± 0.2 ppm, -62.7 ± 0.2 ppm, -135.9 ± 0.2 ppm, -136.1 ± 0.2 ppm, -139.6 ± 0.2 ppm, -140.6 ± 0.2 ppm, -141.7 ± 0.2 ppm, -145.2 ± 0.2 ppm, and -145.7 ± 0.2 ppm. In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized as having a 19F SSNMR spectrum with peaks at -60.4 ± 0.2 ppm, -61.2 ± 0.2 ppm, -61.6 ± 0.2 ppm, -62.7 ± 0.2 ppm, -135.9 ± 0.2 ppm, -136.1 ± 0.2 ppm, -139.6 ± 0.2 ppm, -140.6 ± 0.2 ppm, -141.7 ± 0.2 ppm, -145.2 ± 0.2 ppm, and -145.7 ± 0.2 ppm. [0293] In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by a 19F SSNMR spectrum substantially similar to FIG.15. [0294] In some embodiments, crystalline Compound I Acetone Solvate Hydrate Form A is characterized by a monoclinic crystal system, P21 space group and the following unit cell dimensions measured at 100 K on a diffractometer utilizing Cu Kα radiation (λ=1.54178 Å): a 9.2 ± 0.1 Å α 90° b 39.8 ± 0.1 Å β 96.3 ± 0.1° c 12.9 ± 0.1 Å γ 90°. In some embodiments, the diffractometer is a Rigaku diffractometer. [0295] Another aspect of the disclosure provides a method of crystalline Compound I Acetone Solvate Hydrate Form A. In some embodiments, the method of making crystalline Compound I Acetone Solvate Hydrate Form A, comprises: (i) saturating acetone with Compound I, (ii) adding water dropwise to 20% of the final volume, (iii) holding overnight at room temperature to yield crystalline Compound I Acetone Solvate Hydrate Form A. G. Crystalline Compound Ethanol Solvate Form A [0296] In some embodiments, the disclosure provides crystalline Compound I Ethanol Solvate Form A. FIG.16 provides an X-ray powder diffractogram of crystalline Compound I Ethanol Solvate Form A. [0297] In some embodiments, crystalline Compound I Ethanol Solvate Form A is substantially pure. In some embodiments, crystalline Compound I Ethanol Solvate Form A is substantially crystalline. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram generated by an X-ray powder diffraction analysis with an incident beam of Cu Kα radiation.
[0298] In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having a signal at 9.4 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having a signal at 16.1 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having a signal at 26.0 ± 0.2 degrees two-theta. [0299] In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having a signal at 10.7 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having a signal at 13.2 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having a signal at 13.6 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having a signal at 13.8 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having a signal at 15.1 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having a signal at 15.9 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having a signal at 17.0 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having a signal at 17.9 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having a signal at 18.3 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having a signal at 18.4 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having a signal at 19.1 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having a signal at 19.9 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having a signal at 21.8 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having a signal at 22.2 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having a signal at 22.4 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having a signal at 24.1 ± 0.2 degrees two-theta. [0300] In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having signals at one or two of 9.4 ± 0.2 degrees two-theta, 16.1 ± 0.2 degrees two-theta, and 26.0 ± 0.2 degrees two-theta.
[0301] In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having (a) one or two signals selected from 9.4 ± 0.2 degrees two-theta, 16.1 ± 0.2 degrees two-theta, and 26.0 ± 0.2 degrees two-theta, and (b) one or two signals selected from 15.1 ± 0.2 degrees two-theta, 17.0 ± 0.2 degrees two-theta, and 22.4 ± 0.2 degrees two-theta. [0302] In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having two, three, four, five, or six signals selected from 9.4 ± 0.2 degrees two-theta, 16.1 ± 0.2 degrees two-theta, 26.0 ± 0.2 degrees two-theta, 15.1 ± 0.2 degrees two-theta, 17.0 ± 0.2 degrees two-theta, 22.4 ± 0.2 degrees two-theta, 13.2 ± 0.2 degrees two-theta, 13.6 ± 0.2 degrees two-theta, and 13.8 ± 0.2 degrees two-theta. [0303] In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having one, two three, four five, six, seven, eight, nine, ten, eleven, twelve, thirteen, fourteen, fifteen, sixteen, seventeen, eighteen, nineteen, twenty, twenty-one, or twenty-two signals selected from 9.4 ± 0.2 degrees two-theta; 10.7 ± 0.2 degrees two-theta; 13.2 ± 0.2 degrees two-theta; 13.6 ± 0.2 degrees two-theta; 13.8 ± 0.2 degrees two-theta; 15.1 ± 0.2 degrees two-theta; 15.9 ± 0.2 degrees two-theta; 16.1 ± 0.2 degrees two-theta; 17.0 ± 0.2 degrees two-theta; 17.9 ± 0.2 degrees two-theta; 18.3 ± 0.2 degrees two-theta; 18.4 ± 0.2 degrees two-theta; 19.1 ± 0.2 degrees two-theta; 19.9 ± 0.2 degrees two-theta; 21.8 ± 0.2 degrees two-theta; 22.2 ± 0.2 degrees two-theta; 22.4 ± 0.2 degrees two-theta; 24.1 ± 0.2 degrees two-theta; and 26.0 ± 0.2 degrees two-theta. [0304] In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram having signals at 9.4 ± 0.2 degrees two-theta, 10.7 ± 0.2 degrees two-theta, 13.2 ± 0.2 degrees two-theta, 13.6 ± 0.2 degrees two-theta, 13.8 ± 0.2 degrees two-theta, 15.1 ± 0.2 degrees two-theta, 15.9 ± 0.2 degrees two-theta, 16.1 ± 0.2 degrees two-theta, 17.0 ± 0.2 degrees two-theta, 17.9 ± 0.2 degrees two-theta, 18.3 ± 0.2 degrees two-theta, 18.4 ± 0.2 degrees two-theta, 19.1 ± 0.2 degrees two-theta, 19.9 ± 0.2 degrees two-theta, 21.8 ± 0.2 degrees two-theta, 22.2 ± 0.2 degrees two-theta, 22.4 ± 0.2 degrees two-theta, 24.1 ± 0.2 degrees two-theta, and 26.0 ± 0.2 degrees two-theta. [0305] In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by an X-ray powder diffractogram substantially similar to FIG.16. [0306] In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13C SSNMR spectrum with a peak at 178.2 ± 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13C SSNMR spectrum with a peak at 175.7 ± 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13C SSNMR spectrum with a peak at 163.9 ± 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13C SSNMR spectrum with a peak at 163.4 ± 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13C SSNMR spectrum with a peak at 162.8 ± 0.2 ppm. In some
embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13C SSNMR spectrum with a peak at 154.9 ± 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13C SSNMR spectrum with a peak at 148.1 ± 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13C SSNMR spectrum with a peak at 146.0 ± 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13C SSNMR spectrum with a peak at 145.2 ± 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13C SSNMR spectrum with a peak at 125.9 ± 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13C SSNMR spectrum with a peak at 123.1 ± 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13C SSNMR spectrum with a peak at 121.8 ± 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13C SSNMR spectrum with a peak at 120.4 ± 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13C SSNMR spectrum with a peak at 119.5 ± 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13C SSNMR spectrum with a peak at 116.9 ± 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13C SSNMR spectrum with a peak at 116.3 ± 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13C SSNMR spectrum with a peak at 114.8 ± 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13C SSNMR spectrum with a peak at 113.8 ± 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13C SSNMR spectrum with a peak at 56.5 ± 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13C SSNMR spectrum with a peak at 18.4 ± 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13C SSNMR spectrum with a peak at 17.2 ± 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13C SSNMR spectrum with a peak at 9.2 ± 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13C SSNMR spectrum with a peak at 7.9 ± 0.2 ppm. [0307] In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13C SSNMR spectrum with one, two, three, four, five, six, seven, eight, nine, ten, or more peaks selected from 178.2 ± 0.2 ppm, 175.7 ± 0.2 ppm, 163.9 ± 0.2 ppm, 163.4 ± 0.2 ppm, 162.8 ± 0.2 ppm, 154.9 ± 0.2 ppm, 148.1 ± 0.2 ppm, 146.0 ± 0.2 ppm, 145.2 ± 0.2 ppm, 125.9 ± 0.2 ppm, 123.1 ± 0.2 ppm, 121.8 ± 0.2 ppm, 120.4 ± 0.2 ppm, 119.5 ± 0.2 ppm, 116.9 ± 0.2 ppm, 116.3 ± 0.2 ppm, 114.8 ± 0.2 ppm, 113.8 ± 0.2 ppm, 56.5 ± 0.2 ppm, 18.4 ± 0.2 ppm, 17.2 ± 0.2 ppm, 9.2 ± 0.2 ppm, and 7.9 ± 0.2 ppm. [0308] In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 13C SSNMR spectrum with peaks at 178.2 ± 0.2 ppm, 175.7 ± 0.2 ppm, 163.9 ± 0.2 ppm,
163.4 ± 0.2 ppm, 162.8 ± 0.2 ppm, 154.9 ± 0.2 ppm, 148.1 ± 0.2 ppm, 146.0 ± 0.2 ppm, 145.2 ± 0.2 ppm, 125.9 ± 0.2 ppm, 123.1 ± 0.2 ppm, 121.8 ± 0.2 ppm, 120.4 ± 0.2 ppm, 119.5 ± 0.2 ppm, 116.9 ± 0.2 ppm, 116.3 ± 0.2 ppm, 114.8 ± 0.2 ppm, 113.8 ± 0.2 ppm, 56.5 ± 0.2 ppm, 18.4 ± 0.2 ppm, 17.2 ± 0.2 ppm, 9.2 ± 0.2 ppm, and 7.9 ± 0.2 ppm. [0309] In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by a 13C SSNMR spectrum substantially similar to FIG.17. [0310] In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 19F SSNMR spectrum with a peak at -61.1 ± 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 19F SSNMR spectrum with a peak at -138.1 ± 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 19F SSNMR spectrum with a peak at -141.2 ± 0.2 ppm. [0311] In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 19F SSNMR spectrum with one or two peaks selected from -61.1 ± 0.2 ppm, -138.1 ± 0.2 ppm, and -141.2 ± 0.2 ppm. In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized as having a 19F SSNMR spectrum with peaks at -61.1 ± 0.2 ppm, -138.1 ± 0.2 ppm, and -141.2 ± 0.2 ppm. [0312] In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by a 19F SSNMR spectrum substantially similar to FIG.18. [0313] In some embodiments, crystalline Compound I Ethanol Solvate Form A is characterized by a monoclinic crystal system, P21/c space group and the following unit cell dimensions measured at 100 K on a diffractometer utilizing Cu Kα radiation (λ=1.54178 Å): a 17.0 ± 0.1 Å α 90° b 11.1 ± 0.1 Å β 105.9 ± 0.1° c 13.2 ± 0.1 Å γ 90°. In some embodiments, the diffractometer is a Bruker diffractometer. [0314] Another aspect of the disclosure provides a method of crystalline Compound I Ethanol Solvate Form A. In some embodiments, the method of making crystalline Compound I Ethanol Solvate Form A comprises: (i) combining Compound I and ethyl acetate, (ii) heating the slurry to 50 °C, (iii) adding ethanol and distilling under vacuum at 50 °C, (iv) adding ethanol and distilling under vacuum at 50 °C, (v) adding ethanol and distilling under vacuum at 50 °C to a minimum volume, (vi) adding ethanol and distilling under vacuum at 50 °C, (vii) drying in a vacuum oven at 45 °C, with a slight nitrogen bleed for 42 hours to yield crystalline Compound I Ethanol Solvate Form A. H. Crystalline Compound I Hydrate Form A [0315] In some embodiments, the disclosure provides crystalline Compound I Hydrate Form A. FIG.19 provides an X-ray powder diffractogram of crystalline Compound I Hydrate Form A.
[0316] In some embodiments, crystalline Compound I Hydrate Form A is substantially pure. In some embodiments, crystalline Compound I Hydrate Form A is substantially crystalline. In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram generated by an X-ray powder diffraction analysis with an incident beam of Cu Kα radiation. [0317] In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 8.7 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 12.4 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 18.8 ± 0.2 degrees two-theta. [0318] In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 4.7 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 9.5 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 11.0 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 12.7 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 13.1 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 14.4 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 15.8 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 16.3 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 17.5 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 19.8 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 20.2 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 21.1 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 21.4 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 22.2 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 22.9 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 23.9 ± 0.2 degrees two-theta. In some embodiments, crystalline
Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 24.4 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 24.7 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 25.4 ± 0.2 degrees two-theta. [0319] In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having signals at one or two of 8.7 ± 0.2 degrees two-theta, 12.4 ± 0.2 degrees two-theta, and 18.8 ± 0.2 degrees two-theta. [0320] In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram (a) one or two signals selected from 8.7 ± 0.2 degrees two-theta, 12.4 ± 0.2 degrees two-theta, and 18.8 ± 0.2 degrees two-theta, and (b) one or two signals selected from 12.7 ± 0.2 degrees two-theta, 17.5 ± 0.2 degrees two-theta, and 19.8 ± 0.2 degrees two-theta. [0321] In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having two, three, four, five, or six signals selected from 8.7 ± 0.2 degrees two-theta, 12.4 ± 0.2 degrees two-theta, 18.8 ± 0.2 degrees two-theta, 12.7 ± 0.2 degrees two-theta, 17.5 ± 0.2 degrees two-theta, 19.8 ± 0.2 degrees two-theta, 13.1 ± 0.2 degrees two-theta, 16.3 ± 0.2 degrees two-theta, and 21.1 ± 0.2 degrees two-theta. [0322] In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having one, two three, four five, six, seven, eight, nine, ten, eleven, twelve, thirteen, fourteen, fifteen, sixteen, seventeen, eighteen, nineteen, twenty, twenty-one, or twenty-two signals selected from 8.7 ± 0.2 degrees two-theta, 12.4 ± 0.2 degrees two-theta, 18.8 ± 0.2 degrees two-theta; 4.7 ± 0.2 degrees two-theta; 9.5 ± 0.2 degrees two-theta; 11.0 ± 0.2 degrees two-theta; 12.7 ± 0.2 degrees two-theta; 13.1 ± 0.2 degrees two-theta; 14.4 ± 0.2 degrees two-theta; 15.8 ± 0.2 degrees two-theta; 16.3 ± 0.2 degrees two-theta; 17.5 ± 0.2 degrees two-theta; 19.8 ± 0.2 degrees two-theta; 20.2 ± 0.2 degrees two-theta; 21.1 ± 0.2 degrees two-theta; 21.4 ± 0.2 degrees two-theta; 22.2 ± 0.2 degrees two-theta; 22.9 ± 0.2 degrees two-theta; 23.9 ± 0.2 degrees two-theta; 24.4 ± 0.2 degrees two-theta; 24.7 ± 0.2 degrees two-theta; and 25.4 ± 0.2 degrees two-theta. [0323] In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having signals at 8.7 ± 0.2 degrees two-theta, 12.4 ± 0.2 degrees two-theta, 18.8 ± 0.2 degrees two-theta; 4.7 ± 0.2 degrees two-theta; 9.5 ± 0.2 degrees two-theta; 11.0 ± 0.2 degrees two-theta; 12.7 ± 0.2 degrees two-theta; 13.1 ± 0.2 degrees two-theta; 14.4 ± 0.2 degrees two-theta; 15.8 ± 0.2 degrees two-theta; 16.3 ± 0.2 degrees two-theta; 17.5 ± 0.2 degrees two-theta; 19.8 ± 0.2 degrees two-theta; 20.2 ± 0.2 degrees two-theta; 21.1 ± 0.2 degrees two-theta; 21.4 ± 0.2 degrees two-theta; 22.2 ± 0.2 degrees two-theta; 22.9 ± 0.2 degrees two-theta; 23.9 ± 0.2 degrees two-theta; 24.4 ± 0.2 degrees two-theta; 24.7 ± 0.2 degrees two-theta; and 25.4 ± 0.2 degrees two-theta. [0324] In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram substantially similar to FIG.19.
[0325] In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 177.3 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 172.8 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 170.5 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 162.0 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 154.3 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 154.0 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 150.2 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 148.7 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 147.6 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 146.6 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 145.5 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 126.3 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 125.0 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 123.8 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 122.4 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 121.6 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 120.6 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 118.4 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 117.9 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 117.0 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 116.0 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 114.7 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 114.4 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 112.8 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR
spectrum with a peak at 17.7 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 17.1 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum with a peak at 8.7 ± 0.2 ppm. [0326] In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum with one, two, three, four, five, six, seven, eight, nine, ten, or more peaks selected from 177.3 ± 0.2 ppm, 172.8 ± 0.2 ppm, 170.5 ± 0.2 ppm, 162.0 ± 0.2 ppm, 154.3 ± 0.2 ppm, 154.0 ± 0.2 ppm, 150.2 ± 0.2 ppm, 148.7 ± 0.2 ppm, 147.6 ± 0.2 ppm, 146.6 ± 0.2 ppm, 145.5 ± 0.2 ppm, 126.3 ± 0.2 ppm, 125.0 ± 0.2 ppm, 123.8 ± 0.2 ppm, 122.4 ± 0.2 ppm, 121.6 ± 0.2 ppm, 120.6 ± 0.2 ppm, 118.4 ± 0.2 ppm, 117.9 ± 0.2 ppm, 117.0 ± 0.2 ppm, 116.0 ± 0.2 ppm, 114.7 ± 0.2 ppm, 114.4 ± 0.2 ppm, 112.8 ± 0.2 ppm, 17.7 ± 0.2 ppm, 17.1 ± 0.2 ppm, and 8.7 ± 0.2 ppm. [0327] In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum with peaks at 177.3 ± 0.2 ppm, 172.8 ± 0.2 ppm, 170.5 ± 0.2 ppm, 162.0 ± 0.2 ppm, 154.3 ± 0.2 ppm, 154.0 ± 0.2 ppm, 150.2 ± 0.2 ppm, 148.7 ± 0.2 ppm, 147.6 ± 0.2 ppm, 146.6 ± 0.2 ppm, 145.5 ± 0.2 ppm, 126.3 ± 0.2 ppm, 125.0 ± 0.2 ppm, 123.8 ± 0.2 ppm, 122.4 ± 0.2 ppm, 121.6 ± 0.2 ppm, 120.6 ± 0.2 ppm, 118.4 ± 0.2 ppm, 117.9 ± 0.2 ppm, 117.0 ± 0.2 ppm, 116.0 ± 0.2 ppm, 114.7 ± 0.2 ppm, 114.4 ± 0.2 ppm, 112.8 ± 0.2 ppm, 17.7 ± 0.2 ppm, 17.1 ± 0.2 ppm, and 8.7 ± 0.2 ppm. [0328] In some embodiments, crystalline Compound I Hydrate Form A is characterized by a 13C SSNMR spectrum substantially similar to FIG.22. [0329] In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum measured at 25 ± 2 °C and 6% relative humidity (RH) with a peak at 171.5 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum measured at 25 ± 2 °C and 11% relative humidity (RH) with a peak at 171.6 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum measured at 25 ± 2 °C and 33% relative humidity (RH) with a peak at 170.7 ± 0.2 ppm. [0330] In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum measured at 25 ± 2 °C and 6% relative humidity (RH) with a peak at 155.0 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum measured at 25 ± 2 °C and 11% relative humidity (RH) with a peak at 155.0 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum measured at 25 ± 2 °C and 22% relative humidity (RH) with a peak at 154.6 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum measured at 25 ± 2 °C and 33% relative humidity (RH) with a peak at 154.4 ± 0.2 ppm. [0331] In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum measured at 25 ± 2 °C and 6% relative humidity (RH) with a peak at 149.5 ± 0.2
ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum measured at 25 ± 2 °C and 11% relative humidity (RH) with a peak at 149.4 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum measured at 25 ± 2 °C and 22% relative humidity (RH) with a peak at 148.6 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum measured at 25 ± 2 °C and 33% relative humidity (RH) with a peak at 148.6 ± 0.2 ppm. [0332] In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum measured at 25 ± 2 °C and 6% relative humidity (RH) with a peak at 145.8 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum measured at 25 ± 2 °C and 11% relative humidity (RH) with a peak at 145.8 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum measured at 25 ± 2 °C and 22% relative humidity (RH) with a peak at 145.6 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum measured at 25 ± 2 °C and 33% relative humidity (RH) with a peak at 145.5 ± 0.2 ppm. [0333] In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum measured at 25 ± 2 °C and 6% relative humidity (RH) with a peak at 8.5 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum measured at 25 ± 2 °C and 11% relative humidity (RH) with a peak at 8.5 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum measured at 25 ± 2 °C and 22% relative humidity (RH) with a peak at 8.6 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum measured at 25 ± 2 °C and 33% relative humidity (RH) with a peak at 8.6 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 13C SSNMR spectrum measured at 25 ± 2 °C and 43% relative humidity (RH) with a peak at 8.6 ± 0.2 ppm. [0334] In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 19F SSNMR spectrum with a peak at -59.4 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 19F SSNMR spectrum with a peak at -60.2 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 19F SSNMR spectrum with a peak at -135.0 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 19F SSNMR spectrum with a peak at -138.4 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 19F SSNMR spectrum with a peak at -141.7 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 19F SSNMR spectrum with a peak at -144 ± 0.2 ppm.
[0335] In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 19F SSNMR spectrum with one or two peaks selected from -59.4 ± 0.2 ppm, -60.2 ± 0.2 ppm, -135.0 ± 0.2 ppm, -138.4 ± 0.2 ppm, -141.7 ± 0.2 ppm, and -144.0 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 19F SSNMR spectrum with peaks at -59.4 ± 0.2 ppm, -60.2 ± 0.2 ppm, -135.0 ± 0.2 ppm, -138.4 ± 0.2 ppm, -141.7 ± 0.2 ppm, and -144.0 ± 0.2 ppm. [0336] In some embodiments, crystalline Compound I Hydrate Form A is characterized by a 19F SSNMR spectrum substantially similar to FIG.23. [0337] In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 19F SSNMR spectrum measured at 25 ± 2 °C and 11% relative humidity (RH) with a peak at -142.4 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 19F SSNMR spectrum measured at 25 ± 2 °C and 22% relative humidity (RH) with a peak at -141.9 ± 0.2 ppm. In some embodiments, crystalline Compound I Hydrate Form A is characterized as having a 19F SSNMR spectrum measured at 25 ± 2 °C and 33% relative humidity (RH) with a peak at -141.8 ± 0.2 ppm. [0338] In some embodiments, crystalline Compound I Hydrate Form A is characterized by a triclinic crystal system, P-1 space group and the following unit cell dimensions measured at 100 K on a diffractometer utilizing Cu Kα radiation (λ=1.54178 Å): a 10.5 ± 0.1 Å α 91.3 ± 0.1° b 11.2 ± 0.1 Å β 103.9 ± 0.1° c 19.2 ± 0.1 Å γ 98.4 ± 0.1°. In some embodiments, the diffractometer is a Bruker diffractometer. [0339] Another aspect of the disclosure provides a method of crystalline Compound I Hydrate Form A. In some embodiments, the method of making crystalline Compound I Hydrate Form A, comprises: (i) combining Compound I Ethanol Solvate Form A and acetone, (ii) stirring the slurry while heating 50 °C, (iii) adding water over 1 hour, (iv) adding a seed of crystalline Compound I hydrate Form A, (v) agitating for 30 minutes, (vi) adding water over 8 hours, (vii) stirring at 50 °C for 5 hours, (viii) cooling the slurry to 20 °C over 3 hours, (ix) agitating for 2 hours, and (x) collecting the solids and washing with 1:2 acetone:water to yield crystalline Compound I hydrate Form A. I. Crystalline Compound I Dehydrated Hydrate Form A [0340] In some embodiments, the disclosure provides crystalline Compound I Dehydrated Hydrate Form A. FIG. 20 provides an X-ray powder diffractogram of crystalline Compound I Dehydrated Hydrate Form A. [0341] In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is substantially pure. In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is substantially crystalline. In some embodiments, crystalline Compound I Dehydrated Hydrate Form A
is characterized by an X-ray powder diffractogram generated by an X-ray powder diffraction analysis with an incident beam of Cu Kα radiation. [0342] In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 8.7 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 15.9 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 18.7 ± 0.2 degrees two-theta. [0343] In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 4.7 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 9.5 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 10.9 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 12.4 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 12.6 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 13.1 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 14.3 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 16.3 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 17.3 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 19.8 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 20.3 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 21.1 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 21.4 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 22.3 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 23.0 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 23.9 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized
by an X-ray powder diffractogram having a signal at 24.3 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 24.7 ± 0.2 degrees two-theta. In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having a signal at 25.3 ± 0.2 degrees two-theta. [0344] In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having signals at one or two of 8.7 ± 0.2 degrees two- theta, 15.9 ± 0.2 degrees two-theta, and 18.7 ± 0.2 degrees two-theta. [0345] In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram (a) one or two signals selected from 8.7 ± 0.2 degrees two-theta, 15.9 ± 0.2 degrees two-theta, and 18.7 ± 0.2 degrees two-theta, and (b) one or two signals selected from 12.4 ± 0.2 degrees two-theta, 12.6 ± 0.2 degrees two-theta, 17.3 ± 0.2 degrees two-theta, and 19.8 ± 0.2 degrees two-theta. [0346] In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram having two, three, four, five, or six signals selected 8.7 ± 0.2 degrees two-theta, 15.9 ± 0.2 degrees two-theta, 18.7 ± 0.2 degrees two-theta, 12.4 ± 0.2 degrees two-theta, 12.6 ± 0.2 degrees two-theta, 17.3 ± 0.2 degrees two-theta, 19.8 ± 0.2 degrees two-theta, 13.1 ± 0.2 degrees two-theta, 16.3 ± 0.2 degrees two-theta, and 21.1 ± 0.2 degrees two-theta. [0347] In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having one, two three, four five, six, seven, eight, nine, ten, eleven, twelve, thirteen, fourteen, fifteen, sixteen, seventeen, eighteen, nineteen, twenty, twenty-one, or twenty-two signals selected from 4.7 ± 0.2 degrees two-theta; 8.7 ± 0.2 degrees two-theta; 9.5 ± 0.2 degrees two-theta; 10.9 ± 0.2 degrees two-theta; 12.4 ± 0.2 degrees two-theta; 12.6 ± 0.2 degrees two-theta; 13.1 ± 0.2 degrees two-theta; 14.3 ± 0.2 degrees two-theta; 15.9 ± 0.2 degrees two-theta; 16.3 ± 0.2 degrees two-theta; 17.3 ± 0.2 degrees two-theta; 18.7 ± 0.2 degrees two-theta; 19.8 ± 0.2 degrees two-theta; 20.3 ± 0.2 degrees two-theta; 21.1 ± 0.2 degrees two-theta; 21.4 ± 0.2 degrees two-theta; 22.3 ± 0.2 degrees two-theta; 23.0 ± 0.2 degrees two-theta; 23.9 ± 0.2 degrees two-theta; 24.3 ± 0.2 degrees two-theta; 24.7 ± 0.2 degrees two-theta; and 25.3 ± 0.2 degrees two-theta. [0348] In some embodiments, crystalline Compound I Hydrate Form A is characterized by an X-ray powder diffractogram having signals at 4.7 ± 0.2 degrees two-theta, 8.7 ± 0.2 degrees two-theta, 9.5 ± 0.2 degrees two-theta, 10.9 ± 0.2 degrees two-theta, 12.4 ± 0.2 degrees two-theta, 12.6 ± 0.2 degrees two-theta, 13.1 ± 0.2 degrees two-theta, 14.3 ± 0.2 degrees two-theta, 15.9 ± 0.2 degrees two-theta, 16.3 ± 0.2 degrees two-theta, 17.3 ± 0.2 degrees two-theta, 18.7 ± 0.2 degrees two-theta, 19.8 ± 0.2 degrees two-theta, 20.3 ± 0.2 degrees two-theta, 21.1 ± 0.2 degrees two-theta, 21.4 ± 0.2 degrees two-theta, 22.3 ± 0.2 degrees two-theta, 23.0 ± 0.2 degrees two-theta, 23.9 ± 0.2 degrees two-theta, 24.3 ± 0.2 degrees two-theta, 24.7 ± 0.2 degrees two-theta, and 25.3 ± 0.2 degrees two-theta.
[0349] In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by an X-ray powder diffractogram substantially similar to FIG.20. [0350] In some embodiments, crystalline Compound I Dehydrated Hydrate Form A is characterized by a triclinic crystal system, P-1 space group and the following unit cell dimensions measured at 100 K on a diffractometer utilizing Cu Kα radiation (λ=1.54178 Å): a 10.4 ± 0.1 Å α 90.7 ± 0.1° b 11.2 ± 0.1 Å β 104.7 ± 0.1° c 19.3 ± 0.1 Å γ 98.0 ± 0.1°. In some embodiments, the diffractometer is a Bruker diffractometer. [0351] Another aspect of the disclosure provides a method of crystalline Compound I Dehydrated Hydrate Form A. In some embodiments, the method of making crystalline Compound I Dehydrated Hydrate Form A, comprises: (i) combining Compound I Ethanol Solvate Form A and acetone, (ii) stirring the slurry while heating 50 °C, (iii) adding water over 1 hour, (iv) adding a seed of crystalline Compound I Dehydrated Hydrate Form A, (v) agitating for 30 minutes, (vi) adding water over 8 hours, (vii) stirring at 50 °C for 5 hours, (viii) cooling the slurry to 20 °C over 3 hours, (ix) agitating for 2 hours, (x) collecting the solids and washing with 1:2 acetone:water, (xi) drying the solids in a vacuum over at 50 °C with a slight nitrogen bleed to yield crystalline Compound I Dehydrated Hydrate Form A. J. Methods of Treatment [0352] Compound I, in any one of the pharmaceutically acceptable solid forms disclosed herein, act as a voltage-gated sodium channel inhibitor. In some aspects, the voltage-gated sodium channel is NaV1.8. Accordingly, in another aspect, the disclosure features a method of inhibiting a voltage-gated sodium channel in a subject comprising administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof. In another aspect, the voltage-gated sodium channel is NaV1.8. In another aspect, the invention features a method of inhibiting a voltage-gated sodium channel in a subject comprising administering to the subject a compound of formula I or a solvate thereof or a pharmaceutically acceptable salt thereof or a pharmaceutical composition thereof. [0353] In one aspect, the disclosure relates to a method of treating or lessening the severity of pain in a subject, comprising administering to the subject a compound of formula I, or a pharmaceutically acceptable salt thereof. [0354] In another aspect, the disclosure relates to a use of a compound of formula I, or a pharmaceutically acceptable salt thereof, in a method of treating or lessening the severity of pain in a subject, comprising administering to the subject a compound of formula I, or the pharmaceutically acceptable salt thereof
[0355] In another aspect, the disclosure relates to a composition comprising a compound of formula I, or a solvate of formula I, or a pharmaceutically acceptable salt thereof, for use in a method of treating or lessening the severity of pain in a subject, wherein the composition is prepared for administration of a compound of formula I, or a solvate of formula I or the pharmaceutically acceptable salt thereof, to the subject. [0356] In yet another aspect, the disclosure features a method of treating or lessening the severity in a subject of chronic pain, gut pain, neuropathic pain, musculoskeletal pain, acute pain, inflammatory pain, cancer pain, idiopathic pain, postsurgical pain (e.g., bunionectomy pain, herniorrhaphy pain or abdominoplasty pain), visceral pain, multiple sclerosis, Charcot-Marie-Tooth syndrome, incontinence, pathological cough, or cardiac arrhythmia comprising administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e. g. crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof. [0357] In yet another aspect, the disclosure features a method of treating or lessening the severity in a subject of chronic pain, gut pain, neuropathic pain, musculoskeletal pain, acute pain, inflammatory pain, cancer pain, idiopathic pain, postsurgical pain, herniorrhaphy pain, bunionectomy pain, multiple sclerosis, Charcot-Marie-Tooth syndrome, incontinence, or cardiac arrhythmia comprising administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof. [0358] In yet another aspect, the disclosure features a method of treating or lessening the severity in a subject of gut pain, wherein gut pain comprises inflammatory bowel disease pain, Crohn’s disease pain, irritable bowel syndrome, endometriosis, polycystic ovarian disease, salpingitis, cervicitis or interstitial cystitis pain wherein said method comprises administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof. [0359] In yet another aspect, the disclosure features a method of treating or lessening the severity in a subject of neuropathic pain comprising administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof. In some aspects, the neuropathic pain comprises post-herpetic neuralgia, small fiber neuropathy, diabetic neuropathy, or idiopathic small-fiber neuropathy. In some aspects, the neuropathic pain comprises diabetic neuropathy (e.g., diabetic peripheral neuropathy). As used herein, the phrase “idiopathic small-fiber neuropathy” shall be understood to include any small fiber neuropathy. [0360] In yet another aspect, the disclosure features a method of treating or lessening the severity in a subject of neuropathic pain, wherein neuropathic pain comprises post-herpetic neuralgia, diabetic neuralgia, painful HIV-associated sensory neuropathy, trigeminal neuralgia, burning mouth syndrome, post-amputation pain, phantom pain, painful neuroma; traumatic neuroma; Morton’s neuroma; nerve entrapment injury, spinal stenosis, carpal tunnel syndrome, radicular pain, sciatica pain; nerve avulsion
injury, brachial plexus avulsion injury; complex regional pain syndrome, drug therapy induced neuralgia, cancer chemotherapy induced neuralgia, anti-retroviral therapy induced neuralgia, HIV-induced neuropathy; post spinal cord injury pain, spinal stenosis pain, small fiber neuropathy, idiopathic small-fiber neuropathy, idiopathic sensory neuropathy or trigeminal autonomic cephalalgia wherein said method comprises administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g. crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof. [0361] In yet another aspect, the disclosure features a method of treating or lessening the severity in a subject of musculoskeletal pain comprising administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof. In some aspects, the musculoskeletal pain comprises osteoarthritis pain. [0362] In yet another aspect, the disclosure features a method of treating or lessening the severity in a subject of musculoskeletal pain, wherein musculoskeletal pain comprises osteoarthritis pain, back pain, cold pain, burn pain or dental pain wherein said method comprises administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g. crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof. [0363] In yet another aspect, the disclosure features a method of treating or lessening the severity in a subject of inflammatory pain, wherein inflammatory pain comprises rheumatoid arthritis pain, ankylosing spondylitis or vulvodynia wherein said method comprises administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof. [0364] In yet another aspect, the disclosure features a method of treating or lessening the severity in a subject of inflammatory pain, wherein inflammatory pain comprises rheumatoid arthritis pain wherein said method comprises administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof of. [0365] In yet another aspect, the disclosure features a method of treating or lessening the severity in a subject of idiopathic pain, wherein idiopathic pain comprises fibromyalgia pain wherein said method comprises administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof. [0366] In yet another aspect, the disclosure features a method of treating or lessening the severity in a subject of idiopathic pain, wherein idiopathic pain comprises reflex sympathetic dystrophy pain, wherein said method comprises administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof.
[0367] In yet another aspect, the disclosure features a method of treating or lessening the severity in a subject of pathological cough wherein said method comprises administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof. [0368] In yet another aspect, the disclosure features a method of treating or lessening the severity in a subject of acute pain comprising administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof. In some aspects, the acute pain comprises acute post-operative pain. [0369] In yet another aspect, the disclosure features a method of treating or lessening the severity in a subject of postsurgical pain (e.g., joint replacement pain, soft tissue surgery pain, post-thoracotomy pain, post-mastectomy pain, hemorrhoidectomy pain, herniorrhaphy pain, bunionectomy pain or abdominoplasty pain) comprising administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof. [0370] In yet another aspect, the disclosure features a method of treating or lessening the severity in a subject of bunionectomy pain comprising administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof. [0371] In yet another aspect, the disclosure features a method of treating or lessening the severity in a subject of shoulder arthroplasty pain or shoulder arthroscopy pain comprising administering an effective amount of a compound of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof. [0372] In yet another aspect, the disclosure features a method of treating or lessening the severity in a subject of herniorrhaphy pain comprising administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof. [0373] In yet another aspect, the disclosure features a method of treating or lessening the severity in a subject of abdominoplasty pain comprising administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof. [0374] In yet another aspect, the disclosure features a method of treating or lessening the severity in a subject of visceral pain comprising administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof. In some aspects, the visceral pain comprises visceral pain from abdominoplasty.
[0375] In yet another aspect, the disclosure features a method of treating or lessening the severity in a subject of a neurodegenerative disease comprising administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof. In some aspects, the neurodegenerative disease comprises multiple sclerosis. In some aspects, the neurodegenerative disease comprises Pitt Hopkins Syndrome (PTHS). [0376] In yet another aspect, the disclosure features a method wherein the subject is treated with one or more additional therapeutic agents administered concurrently with, prior to, or subsequent to treatment with an effective of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof. In some embodiments, the additional therapeutic agent is a sodium channel inhibitor. [0377] In another aspect, the disclosure features a method of inhibiting a voltage-gated sodium channel in a biological sample comprising contacting the biological sample with an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof. In another aspect, the voltage-gated sodium channel is NaV1.8. [0378] In another aspect, the disclosure features a method of treating or lessening the severity in a subject of acute pain, sub-acute and chronic pain, nociceptive pain, neuropathic pain, inflammatory pain, nociplastic pain, arthritis, migraine, cluster headaches, tension headaches, and all other forms of headaches, trigeminal neuralgia, herpetic neuralgia, general neuralgias, epilepsy, epilepsy conditions, neurodegenerative disorders, psychiatric disorders, anxiety, depression, bipolar disorder, myotonia, arrhythmia, movement disorders, neuroendocrine disorders, ataxia, central neuropathic pain of multiple sclerosis and irritable bowel syndrome, incontinence, pathological cough, visceral pain, osteoarthritis pain, postherpetic neuralgia, diabetic neuropathy, radicular pain, sciatica, back pain, unspecific chronic back pain, head pain, neck pain, moderate pain, severe pain, intractable pain, nociceptive pain, breakthrough pain, postsurgical pain (e.g., joint replacement pain, soft tissue surgery pain, post- thoracotomy pain, post-mastectomy pain, herniorrhaphy pain, bunionectomy pain or abdominoplasty pain), cancer pain including chronic cancer pain and breakthrough cancer pain, stroke (e.g., post stroke central neuropathic pain), whiplash associated disorders, fragility fractures, spinal fractures, ankylosing spondylitis, pemphigus, Raynaud’s Disease, scleroderma, systemic lupus erythematosus, Epidermolysis bullosa, gout, juvenile idiopathic arthritis, melorheostosis, polymyalgia reumatica, pyoderma gangrenosum, chronic widespread pain, diffuse idiopathic skeletal hyperostosis, disc degeneration/herniation pain, radiculopathy, facet joint syndrome, failed back surgery syndrome, burns, carpal tunnel syndrome, Paget’s disease pain, spinal canal stenosis, spondylodyscitis, transverse myelitis, Ehlers-Danlos syndrome, Fabry’s disease, mastocytocytosis, neurofibromatosis, ocular neuropathic pain, sarcoidosis, spondylolysis, spondylolisthesis, chemotherapy induced oral mucositis, Charcot neuropathic osteoarhropathy, temporo-mandibular joint disorder, painful joint arthroplasties,
non-cardiac chest pain, pudendal neuralgia, renal colic, biliary tract diseases, vascular leg ulcers, pain in Parkinson’s disease, pain in Alzheimer’s disease, cerebral ischemia, traumatic brain injury, amyotrophic lateral sclerosis, stress induced angina, exercise induced angina, palpitations, hypertension, or abnormal gastro-intestinal motility, comprising administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof. [0379] In another aspect, the disclosure features a method of treating or lessening the severity in a subject of femur cancer pain; non-malignant chronic bone pain; rheumatoid arthritis; osteoarthritis; spinal stenosis; neuropathic low back pain; myofascial pain syndrome; fibromyalgia; temporomandibular joint pain; chronic visceral pain, abdominal pain; pancreatic pain; IBS pain; chronic and acute headache pain; migraine; tension headache; cluster headaches; chronic and acute neuropathic pain, post-herpetic neuralgia; diabetic neuropathy; HIV-associated neuropathy; trigeminal neuralgia; Charcot-Marie-Tooth neuropathy; hereditary sensory neuropathy; peripheral nerve injury; painful neuromas; ectopic proximal and distal discharges; radiculopathy; chemotherapy induced neuropathic pain; radiotherapy-induced neuropathic pain; persistent/chronic post-surgical pain (e.g., post amputation, post-thoracotomy, post-cardiac surgery), post-mastectomy pain; central pain; spinal cord injury pain; post-stroke pain; thalamic pain; phantom pain (e.g., following removal of lower extremity, upper extremity, breast); intractable pain; acute pain, acute post-operative pain; acute musculoskeletal pain; joint pain; mechanical low back pain; neck pain; tendonitis; injury pain; exercise pain; acute visceral pain; pyelonephritis; appendicitis; cholecystitis; intestinal obstruction; hernias; chest pain, cardiac pain; pelvic pain, renal colic pain, acute obstetric pain, labor pain; cesarean section pain; acute inflammatory pain, burn pain, trauma pain; acute intermittent pain, endometriosis; acute herpes zoster pain; sickle cell anemia; acute pancreatitis; breakthrough pain; orofacial pain; sinusitis pain; dental pain; multiple sclerosis (MS) pain; pain in depression; leprosy pain; Behcet's disease pain; adiposis dolorosa; phlebitic pain; Guillain-Barre pain; painful legs and moving toes; Haglund syndrome; erythromelalgia pain; Fabry's disease pain; bladder and urogenital disease; urinary incontinence, pathological cough; hyperactive bladder; painful bladder syndrome; interstitial cystitis (IC); prostatitis; complex regional pain syndrome (CRPS), type I, complex regional pain syndrome (CRPS) type II; widespread pain, paroxysmal extreme pain, pruritus, tinnitus, or angina-induced pain, comprising administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof. [0380] In another aspect, the disclosure features a method of treating or lessening the severity in a subject of trigeminal neuralgia, migraines treated with botox, cervical radiculopathy, occipital neuralgia, axillary neuropathy, radial neuropathy, ulnar neuropathy, brachial plexopathy, thoracic radiculopathy, intercostal neuralgia, lumbosacral radiculopathy, iliolingual neuralgia, pudendal neuralgia, femoral neuropathy, meralgia paresthetica, saphenous neuropathy, sciatic neuropathy, peroneal neuropathy, tibial neuropathy, lumbosacral plexopathy, traumatic neuroma stump pain or
postamputation pain, comprising administering an effective amount of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof. [0381] In some embodiments, the method of treating, or lessening the severity of, or symptomatically treating pain in a patient comprises administering to the patient an effective amount of Compound I in any one of the pharmaceutically acceptable crystalline forms disclosed herein. In some embodiments the pharmaceutically acceptable crystalline form of Compound I is Compound I neat Form A. In some embodiments the pharmaceutically acceptable crystalline form of Compound I is Compound I neat Form B. In some embodiments the pharmaceutically acceptable crystalline form of Compound I is Compound I neat Form E. In some embodiments the pharmaceutically acceptable crystalline form of Compound I is Compound I Acetone Solvate Hydrate Form A. In some embodiments the pharmaceutically acceptable crystalline form of Compound I is Compound I Ethanol Solvate Form A. In some embodiments the pharmaceutically acceptable crystalline form of Compound I is Compound I Hydrate Form A. In some embodiments the pharmaceutically acceptable crystalline form of Compound I is Compound I Hydrate Form A. In some embodiments the pharmaceutically acceptable crystalline form of Compound I is Compound I Dehydrated Hydrate Form A. [0382] In some embodiments, the method of treating, or lessening the severity of, or symptomatically treating pain in a patient comprises administering to the patient an effective amount of Compound I in a pharmaceutically acceptable amorphous form disclosed herein. In some embodiments the pharmaceutically acceptable form of Compound I is Compound I amorphous form. [0383] In some embodiments, the method of treating, lessening the severity of, or symptomatically treating pain in a patient comprises administering to the patient an effective amount of Compound I as any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein in combination with at least one additional active pharmaceutical ingredient. In some embodiments, the method of treating, lessening the severity of, or symptomatically treating pain in a patient comprises administering to the patient an effective amount of Compound I as a solid form selected from Compound I neat Form A, Compound I neat Form B, Compound I neat Form E, Compound I Acetone Solvate Hydrate Form A, Compound I Ethanol Solvate Form A, Compound I Hydrate Form A, Compound I Dehydrated Hydrate Form A, and Compound I amorphous form, in combination with at least one additional active pharmaceutical ingredient. [0384] In some embodiments, the disclosure features Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof for use in any of the foregoing methods. [0385] In some embodiments, the disclosure features the use of Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof for the manufacture of a medicament for use in any of the foregoing methods.
[0386] In some embodiments, the disclosure features Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein or a pharmaceutical composition thereof for use as a medicament. [0387] It will also be appreciated that the compounds, salts, and pharmaceutically acceptable compositions of the invention can be employed in combination therapies, that is, the compounds, salts, and pharmaceutically acceptable compositions can be administered concurrently with, prior to, or subsequent to, one or more other desired therapeutics or medical procedures. The particular combination of therapies (therapeutics or procedures) to employ in a combination regimen will take into account compatibility of the desired therapeutics and/or procedures and the desired therapeutic effect to be achieved. It will also be appreciated that the therapies employed may achieve a desired effect for the same disorder (for example, an inventive compound may be administered concurrently with another agent used to treat the same disorder), or they may achieve different effects (e.g., control of any adverse effects). As used herein, additional therapeutic agents that are normally administered to treat or prevent a particular disease, or condition, are known as “appropriate for the disease, or condition, being treated.” For example, exemplary additional therapeutic agents include, but are not limited to: non-opioid analgesics (indoles such as Etodolac, Indomethacin, Sulindac, Tolmetin; naphthylalkanones such as Nabumetone; oxicams such as Piroxicam; para-aminophenol derivatives, such as Acetaminophen; propionic acids such as Fenoprofen, Flurbiprofen, Ibuprofen, Ketoprofen, Naproxen, Naproxen sodium, Oxaprozin; salicylates such as Aspirin, Choline magnesium trisalicylate, Diflunisal; fenamates such as meclofenamic acid, Mefenamic acid; and pyrazoles such as Phenylbutazone); or opioid (narcotic) agonists (such as Codeine, Fentanyl, Hydromorphone, Levorphanol, Meperidine, Methadone, Morphine, Oxycodone, Oxymorphone, Propoxyphene, Buprenorphine, Butorphanol, Dezocine, Nalbuphine, and Pentazocine). Additionally, nondrug analgesic approaches may be utilized in conjunction with administration of one or more compounds of the invention. For example, anesthesiologic (intraspinal infusion, neural blockade), neurosurgical (neurolysis of CNS pathways), neurostimulatory (transcutaneous electrical nerve stimulation, dorsal column stimulation), physiatric (physical therapy, orthotic devices, diathermy), or psychologic (cognitive methods-hypnosis, biofeedback, or behavioral methods) approaches may also be utilized. Additional appropriate therapeutic agents or approaches are described generally in The Merck Manual, Nineteenth Edition, Ed. Robert S. Porter and Justin L. Kaplan, Merck Sharp &Dohme Corp., a subsidiary of Merck & Co., Inc., 2011, and the Food and Drug Administration website, www.fda.gov, the entire contents of which are hereby incorporated by reference. [0388] In another embodiment, additional appropriate therapeutic agents are selected from the following: [0389] (1) an opioid analgesic, e.g. morphine, heroin, hydromorphone, oxymorphone, levorphanol, levallorphan, methadone, meperidine, fentanyl, cocaine, codeine, dihydrocodeine,
oxycodone, hydrocodone, propoxyphene, nalmefene, nalorphine, naloxone, naltrexone, buprenorphine, butorphanol, nalbuphine, pentazocine, or difelikefalin; [0390] (2) a nonsteroidal antiinflammatory drug (NSAID), e.g. aspirin, diclofenac, diflunisal, etodolac, fenbufen, fenoprofen, flufenisal, flurbiprofen, ibuprofen (including without limitation intravenous ibuprofen (e.g., Caldolor®)), indomethacin, ketoprofen, ketorolac (including without limitation ketorolac tromethamine (e.g., Toradol®)), meclofenamic acid, mefenamic acid, meloxicam, IV meloxicam (e.g., Anjeso®), nabumetone, naproxen, nimesulide, nitroflurbiprofen, olsalazine, oxaprozin, phenylbutazone, piroxicam, sulfasalazine, sulindac, tolmetin or zomepirac; [0391] (3) a barbiturate sedative, e.g. amobarbital, aprobarbital, butabarbital, butalbital, mephobarbital, metharbital, methohexital, pentobarbital, phenobarbital, secobarbital, talbutal, thiamylal or thiopental; [0392] (4) a benzodiazepine having a sedative action, e.g. chlordiazepoxide, clorazepate, diazepam, flurazepam, lorazepam, oxazepam, temazepam or triazolam; [0393] (5) a histamine (H1) antagonist having a sedative action, e.g. diphenhydramine, pyrilamine, promethazine, chlorpheniramine or chlorcyclizine; [0394] (6) a sedative such as glutethimide, meprobamate, methaqualone or dichloralphenazone; [0395] (7) a skeletal muscle relaxant, e.g. baclofen, carisoprodol, chlorzoxazone, cyclobenzaprine, methocarbamol or orphenadrine; [0396] (8) an NMDA receptor antagonist, e.g. dextromethorphan ((+)-3-hydroxy-N- methylmorphinan) or its metabolite dextrorphan ((+)-3-hydroxy-N-methylmorphinan), ketamine, memantine, pyrroloquinoline quinine, cis-4-(phosphonomethyl)-2- piperidinecarboxylic acid, budipine, EN-3231 (MorphiDex®), a combination formulation of morphine and dextromethorphan), topiramate, neramexane or perzinfotel including an NR2B antagonist, e.g. ifenprodil, traxoprodil or (- )-(R)-6-{2-[4-(3-fluorophenyl)-4-hydroxy-l- piperidinyl]-l-hydroxyethyl-3,4-dihydro-2(lH)- quinolinone; [0397] (9) an alpha-adrenergic, e.g. doxazosin, tamsulosin, clonidine, guanfacine, dexmedetomidine, modafinil, or 4-amino-6,7-dimethoxy-2-(5-methane-sulfonamido-l, 2,3,4- tetrahydroisoquinolin-2-yl)-5-(2-pyridyl) quinazoline; [0398] (10) a tricyclic antidepressant, e.g. desipramine, imipramine, amitriptyline or nortriptyline; [0399] (11) an anticonvulsant, e.g. carbamazepine (Tegretol®), lamotrigine, topiramate, lacosamide (Vimpat®) or valproate; [0400] (12) a tachykinin (NK) antagonist, particularly an NK-3, NK-2 or NK-1 antagonist, e.g. (alphaR,9R)-7-[3,5-bis(trifluoromethyl)benzyl]-8,9,10,11 -tetrahydro-9-methyl-5-(4- methylphenyl)- 7H-[l,4]diazocino[2,l-g][l,7]-naphthyridine-6-13-dione (TAK-637), 5- [[(2R,3S)-2-[(lR)-l-[3,5- bis(trifluoromethyl)phenyl]ethoxy-3-(4-fluorophenyl)-4-morpholinyl]-methyl]-l,2-dihydro-3H-l,2,4-
triazol-3-one (MK-869), aprepitant, lanepitant, dapitant or 3-[[2-methoxy-5- (trifluoromethoxy)phenyl]-methylamino]-2-phenylpiperidine (2S,3S); [0401] (13) a muscarinic antagonist, e.g oxybutynin, tolterodine, propiverine, tropsium chloride, darifenacin, solifenacin, temiverine and ipratropium; [0402] (14) a COX-2 selective inhibitor, e.g. celecoxib, rofecoxib, parecoxib, valdecoxib, deracoxib, etoricoxib, or lumiracoxib; [0403] (15) a coal-tar analgesic, in particular paracetamol; [0404] (16) a neuroleptic such as droperidol, chlorpromazine, haloperidol, perphenazine, thioridazine, mesoridazine, trifluoperazine, fluphenazine, clozapine, olanzapine, risperidone, ziprasidone, quetiapine, sertindole, aripiprazole, sonepiprazole, blonanserin, iloperidone, perospirone, raclopride, zotepine, bifeprunox, asenapine, lurasidone, amisulpride, balaperidone, palindore, eplivanserin, osanetant, rimonabant, meclinertant, Miraxion® or sarizotan; [0405] (17) a vanilloid receptor agonist (e.g. resinferatoxin or civamide) or antagonist (e.g. capsazepine, GRC-15300); [0406] (18) a beta-adrenergic such as propranolol; [0407] (19) a local anesthetic such as mexiletine; [0408] (20) a corticosteroid such as dexamethasone; [0409] (21) a 5-HT receptor agonist or antagonist, particularly a 5-HT1B/1D agonist such as eletriptan, sumatriptan, naratriptan, zolmitriptan or rizatriptan; [0410] (22) a 5-HT2A receptor antagonist such as R(+)-alpha-(2,3-dimethoxy-phenyl)-l-[2-(4- fluorophenylethyl)]-4-piperidinemethanol (MDL-100907); [0411] (23) a cholinergic (nicotinic) analgesic, such as ispronicline (TC-1734), (E)-N-methyl-4- (3-pyridinyl)-3-buten-l-amine (RJR-2403), (R)-5-(2-azetidinylmethoxy)-2-chloropyridine (ABT-594) or nicotine; [0412] (24) Tramadol®, Tramadol ER (Ultram ER®), IV Tramadol, Tapentadol ER (Nucynta®); [0413] (25) a PDE5 inhibitor, such as 5-[2-ethoxy-5-(4-methyl-l-piperazinyl-sulphonyl)phenyl]- l-methyl-3-n-propyl-l,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one (sildenafil), (6R,12aR)- 2,3,6,7,12,12a-hexahydro-2-methyl-6-(3,4-methylenedioxyphenyl)-pyrazino[2',l':6,l]-pyrido[3,4- b]indole-l,4-dione (IC-351 or tadalafil), 2-[2-ethoxy-5-(4-ethyl-piperazin-l-yl-l-sulphonyl)-phenyl]-5- methyl-7-propyl-3H-imidazo[5,l-f][l,2,4]triazin-4-one (vardenafil), 5-(5-acetyl-2-butoxy-3-pyridinyl)- 3-ethyl-2-(l-ethyl-3-azetidinyl)-2,6-dihydro-7H- pyrazolo[4,3-d]pyrimidin-7-one, 5-(5-acetyl-2- propoxy-3-pyridinyl)-3-ethyl-2-(l-isopropyl-3-azetidinyl)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin- 7-one, 5-[2-ethoxy-5-(4-ethylpiperazin-l-ylsulphonyl)pyridin-3-yl]-3-ethyl-2-[2-methoxyethyl]-2,6- dihydro-7H- pyrazolo[4,3-d]pyrimidin-7-one, 4-[(3-chloro-4-methoxybenzyl)amino]-2-[(2S)-2- (hydroxymethyl)pyrrolidin-l-yl]-N-(pyrimidin-2-ylmethyl)pyrimidine-5-carboxamide, 3-(l- methyl-7- oxo-3-propyl-6,7-dihydro-lH-pyrazolo[4,3-d]pyrimidin-5-yl)-N-[2-(l-methylpyrrolidin-2-yl)ethyl]-4- propoxybenzenesulfonamide;
[0414] (26) an alpha-2-delta ligand such as gabapentin (Neurontin®), gabapentin GR (Gralise®), gabapentin, enacarbil (Horizant®), pregabalin (Lyrica®), 3-methyl gabapentin, (l[alpha],3[alpha],5[alpha])(3-amino-methyl-bicyclo[3.2.0]hept-3-yl)-acetic acid, (3S,5R)-3- aminomethyl-5-methyl-heptanoic acid, (3S,5R)-3-amino-5-methyl-heptanoic acid, (3S,5R)-3-amino- 5-methyl-octanoic acid, (2S,4S)-4-(3-chlorophenoxy)proline, (2S,4S)-4-(3-fluorobenzyl)-proline, [(lR,5R,6S)-6-(aminomethyl)bicyclo[3.2.0]hept-6-yl]acetic acid, 3-(l-aminomethyl- cyclohexylmethyl)-4H-[1,2,4]oxadiazol-5-one, C-[1-(1H-tetrazol-5-ylmethyl)-cycloheptyl]- methylamine, (3S,4S)-(l-aminomethyl-3,4-dimethyl-cyclopentyl)-acetic acid, (3S,5R)-3- aminomethyl-5-methyl-octanoic acid, (3S,5R)-3-amino-5-methyl-nonanoic acid, (3S,5R)-3-amino-5- methyl-octanoic acid, (3R,4R,5R)-3-amino-4,5-dimethyl-heptanoic acid and (3R,4R,5R)-3-amino- 4,5-dimethyl-octanoic acid; [0415] (27) a cannabinoid such as KHK-6188; [0416] (28) metabotropic glutamate subtype 1 receptor (mGluRl) antagonist; [0417] (29) a serotonin reuptake inhibitor such as sertraline, sertraline metabolite demethylsertraline, fluoxetine, norfluoxetine (fluoxetine desmethyl metabolite), fluvoxamine, paroxetine, citalopram, citalopram metabolite desmethylcitalopram, escitalopram, d,l-fenfluramine, femoxetine, ifoxetine, cyanodothiepin, litoxetine, dapoxetine, nefazodone, cericlamine and trazodone; [0418] (30) a noradrenaline (norepinephrine) reuptake inhibitor, such as maprotiline, lofepramine, mirtazepine, oxaprotiline, fezolamine, tomoxetine, mianserin, bupropion, bupropion metabolite hydroxybupropion, nomifensine and viloxazine (Vivalan®), especially a selective noradrenaline reuptake inhibitor such as reboxetine, in particular (S,S)-reboxetine; [0419] (31) a dual serotonin-noradrenaline reuptake inhibitor, such as venlafaxine, venlafaxine metabolite O-desmethylvenlafaxine, clomipramine, clomipramine metabolite desmethylclomipramine, duloxetine (Cymbalta®), milnacipran and imipramine; [0420] (32) an inducible nitric oxide synthase (iNOS) inhibitor such as S-[2-[(l- iminoethyl)amino]ethyl]-L-homocysteine, S-[2-[(l-iminoethyl)-amino]ethyl]-4,4-dioxo-L-cysteine, S- [2-[(l-iminoethyl)amino]ethyl]-2-methyl-L-cysteine, (2S,5Z)-2-amino-2-methyl-7-[(l- iminoethyl)amino]-5-heptenoic acid, 2-[[(lR,3S)-3-amino-4-hydroxy-l-(5-thiazolyl)-butyl]thio]-S- chloro-S-pyridinecarbonitrile; 2-[[(lR,3S)-3-amino-4-hydroxy-l-(5- thiazolyl)butyl]thio]-4- chlorobenzonitrile, (2S,4R)-2-amino-4-[[2-chloro-5- (trifluoromethyl)phenyl]thio]-5-thiazolebutanol, 2-[[(lR,3S)-3-amino-4-hydroxy-l-(5-thiazolyl) butyl]thio]-6-(trifluoromethyl)-3-pyridinecarbonitrile, 2-[[(lR,3S)-3-amino-4-hydroxy-1-(5-thiazolyl)butyl]thio]-5-chlorobenzonitrile, N-[4-[2-(3- chlorobenzylamino)ethyl]phenyl]thiophene-2-carboxamidine, NXN-462, or guanidinoethyldisulfide; [0421] (33) an acetylcholinesterase inhibitor such as donepezil; [0422] (34) a prostaglandin E2 subtype 4 (EP4) antagonist such as N-[({2-[4-(2-ethyl-4,6- dimethyl-lH-imidazo[4,5-c]pyridin-l-yl)phenyl]ethyl}amino)-carbonyl]-4- methylbenzenesulfonamide or 4-[(15)-l-({[5-chloro-2-(3-fluorophenoxy)pyridin-3- yl]carbonyl}amino)ethyl]benzoic acid;
[0423] (35) a leukotriene B4 antagonist; such as l-(3-biphenyl-4-ylmethyl-4-hydroxy-chroman-7- yl)-cyclopentanecarboxylic acid (CP- 105696), 5-[2-(2-Carboxyethyl)-3-[6-(4-methoxyphenyl)-5E- hexenyl]oxyphenoxy]-valeric acid (ONO-4057) or DPC-11870; [0424] (36) a 5-lipoxygenase inhibitor, such as zileuton, 6-[(3-fluoro-5-[4-methoxy-3,4,5,6- tetrahydro-2H-pyran-4-yl])phenoxy-methyl]-l-methyl-2-quinolone (ZD-2138), or 2,3,5- trimethyl-6- (3-pyridylmethyl)-l,4-benzoquinone (CV-6504); [0425] (37) a sodium channel blocker, such as lidocaine, lidocaine plus tetracaine cream (ZRS- 201) or eslicarbazepine acetate; [0426] (38) a NaV1.7 blocker, such as XEN-402, XEN403, TV-45070, PF-05089771, CNV1014802, GDC-0276, RG7893 BIIB-074 (Vixotrigine), BIIB-095, ASP-1807, DSP-3905, OLP- 1002, RQ-00432979, FX-301, DWP-1706, DWP-17061, IMB-110, IMB-111, IMB-112 and such as those disclosed in WO2011/140425 (US2011/306607); WO2012/106499 (US2012196869); WO2012/112743 (US2012245136); WO2012/125613 (US2012264749), WO2012/116440 (US2014187533), WO2011026240 (US2012220605), US8883840, US8466188, WO2013/109521 (US2015005304), CN111217776, WO2020/117626, WO2021/252822, WO2021/252818, WO2021/252820, WO2014/201173, WO2012/125973, WO2013/086229, WO2013/134518, WO2014/201206, or WO2016/141035 the entire contents of each application hereby incorporated by reference; [0427] (38a) a NaV1.7 blocker such as (2-benzylspiro[3,4-dihydropyrrolo[1,2-a]pyrazine-1,4'- piperidine]-1'-yl)-(4-isopropoxy-3-methyl-phenyl)methanone, 2,2,2-trifluoro-1-[1'-[3-methoxy-4-[2- (trifluoromethoxy)ethoxy]benzoyl]-2,4-dimethyl-spiro[3,4-dihydropyrrolo[1,2-a]pyrazine-1,4'- piperidine]-6-yl]ethanone, [8-fluoro-2-methyl-6-(trifluoromethyl)spiro[3,4-dihydropyrrolo[1,2- a]pyrazine-1,4'-piperidine]-1'-yl]-(4-isobutoxy-3-methoxy-phenyl)methanone, 1-(4- benzhydrylpiperazin-1-yl)-3-[2-(3,4-dimethylphenoxy)ethoxy]propan-2-ol, (4-butoxy-3-methoxy- phenyl)-[2-methyl-6-(trifluoromethyl)spiro[3,4-dihydropyrrolo[1,2-a]pyrazine-1,4'-piperidine]-1'- yl]methanone, [8-fluoro-2-methyl-6-(trifluoromethyl)spiro[3,4-dihydropyrrolo[1,2-a]pyrazine-1,4'- piperidine]-1'-yl]-(5-isopropoxy-6-methyl-2-pyridyl)methanone, (4-isopropoxy-3-methyl-phenyl)-[2- methyl-6-(1,1,2,2,2-pentafluoroethyl)spiro[3,4-dihydropyrrolo[1,2-a]pyrazine-1,4'-piperidine]-1'- yl]methanone, 5-[2-methyl-4-[2-methyl-6-(2,2,2-trifluoroacetyl)spiro[3,4-dihydropyrrolo[1,2- a]pyrazine-1,4'-piperidine]-1'-carbonyl]phenyl]pyridine-2-carbonitrile, (4-isopropoxy-3-methyl- phenyl)-[6-(trifluoromethyl)spiro[3,4-dihydro-2H-pyrrolo[1,2-a]pyrazine-1,4'-piperidine]-1'- yl]methanone, 2,2,2-trifluoro-1-[1'-[3-methoxy-4-[2-(trifluoromethoxy)ethoxy]benzoyl]-2-methyl- spiro[3,4-dihydropyrrolo[1,2-a]pyrazine-1,4'-piperidine]-6-yl]ethanone, 2,2,2-trifluoro-1-[1'-(5- isopropoxy-6-methyl-pyridine-2-carbonyl)-3,3-dimethyl-spiro[2,4-dihydropyrrolo[1,2-a]pyrazine- 1,4'-piperidine]-6-yl]ethanone, 2,2,2-trifluoro-1-[1'-(5-isopentyloxypyridine-2-carbonyl)-2-methyl- spiro[3,4-dihydropyrrolo[1,2-a]pyrazine-1,4'-piperidine]-6-yl]ethanone, (4-isopropoxy-3-methoxy- phenyl)-[2-methyl-6-(trifluoromethyl)spiro[3,4-dihydropyrrolo[1,2-a]pyrazine-1,4'-piperidine]-1'-
yl]methanone, 2,2,2-trifluoro-1-[1'-(5-isopentyloxypyridine-2-carbonyl)-2,4-dimethyl-spiro[3,4- dihydropyrrolo[1,2-a]pyrazine-1,4'-piperidine]-6-yl]ethanone, 1-[(3S)-2,3-dimethyl-1'-[4-(3,3,3- trifluoropropoxymethyl)benzoyl]spiro[3,4-dihydropyrrolo[1,2-a]pyrazine-1,4'-piperidine]-6-yl]-2,2,2- trifluoro-ethanone, [8-fluoro-2-methyl-6-(trifluoromethyl)spiro[3,4-dihydropyrrolo[1,2-a]pyrazine- 1,4'-piperidine]-1'-yl]-[3-methoxy-4-[(1R)-1-methylpropoxy]phenyl]methanone, 2,2,2-trifluoro-1-[1'- (5-isopropoxy-6-methyl-pyridine-2-carbonyl)-2,4-dimethyl-spiro[3,4-dihydropyrrolo[1,2-a]pyrazine- 1,4'-piperidine]-6-yl]ethanone, 1-[1'-[4-methoxy-3-(trifluoromethyl)benzoyl]-2-methyl-spiro
 dihydropyrrolo[1,2-a]pyrazine-1,4'-piperidine]-6-yl]-2,2-dimethyl-propan-1-one, (4-isopropoxy-3- methyl-phenyl)-[2-methyl-6-(trifluoromethyl)spiro[3,4-dihydropyrrolo[1,2-a]pyrazine-1,4'- piperidine]-1'-yl]methanone, [2-methyl-6-(1-methylcyclopropanecarbonyl)spiro[3,4- dihydropyrrolo[1,2-a]pyrazine-1,4'-piperidine]-1'-yl]-[4-(3,3,3- trifluoropropoxymethyl)phenyl]methanone, 4-bromo-N-(4-bromophenyl)-3-[(1-methyl-2-oxo-4- piperidyl)sulfamoyl]benzamide or (3-chloro-4-isopropoxy-phenyl)-[2-methyl-6-(1,1,2,2,2- pentafluoroethyl)spiro[3,4-dihydropyrrolo[1,2-a]pyrazine-1,4'-piperidine]-1'-yl]methanone. [0428] (39) a NaV1.8 blocker, such as PF-04531083, PF-06372865 and such as those disclosed in WO2008/135826 (US2009048306), WO2006/011050 (US2008312235), WO2013/061205 (US2014296313), US20130303535, WO2013131018, US8466188, WO2013114250 (US2013274243), WO2014/120808 (US2014213616), WO2014/120815 (US2014228371) WO2014/120820 (US2014221435), WO2015/010065 (US20160152561), WO2015/089361 (US20150166589), WO2019/014352 (US20190016671), WO2018/213426, WO2020/146682, WO2020/146612, WO2020/014243, WO2020/014246, WO2020/092187, WO2020/092667 (US2020140411), WO2020/144375, WO2020/261114, WO2020/140959, WO2020/151728, WO2021/032074, WO2021/047622 (CN112479996), WO2021/257490, WO2021/257420, WO2021/257418, WO2022/263498, WO2022/235558, WO2022/235859, WO2023/138599, CN112390745, CN111808019, CN112225695, CN112457294, CN112300051, CN112300069, CN112441969, and CN114591293, the entire contents of each application hereby incorporated by reference; [0429] (39a) a NaV1.8 blocker such as 4,5-dichloro-2-(4-fluoro-2-methoxyphenoxy)-N-(2-oxo- 1,2-dihydropyridin-4-yl)benzamide, 2-(4-fluoro-2-methoxyphenoxy)-N-(2-oxo-1,2-dihydropyridin-4- yl)-4-(perfluoroethyl)benzamide, 4,5-dichloro-2-(4-fluorophenoxy)-N-(2-oxo-1,2-dihydropyridin-4- yl)benzamide, 4,5-dichloro-2-(3-fluoro-4-methoxyphenoxy)-N-(2-oxo-1,2-dihydropyridin-4- yl)benzamide, 2-(4-fluoro-2-methoxyphenoxy)-N-(2-oxo-1,2-dihydropyridin-4-yl)-5- (trifluoromethyl)benzamide, N-(2-oxo-1,2-dihydropyridin-4-yl)-2-(4-(trifluoromethoxy)phenoxy)-4- (trifluoromethyl)benzamide, 2-(4-fluorophenoxy)-N-(2-oxo-1,2-dihydropyridin-4-yl)-4- (perfluoroethyl)benzamide, 5-chloro-2-(4-fluoro-2-methoxyphenoxy)-N-(2-oxo-1,2-dihydropyridin-4- yl)benzamide, N-(2-oxo-1,2-dihydropyridin-4-yl)-2-(4-(trifluoromethoxy)phenoxy)-5- (trifluoromethyl)benzamide, 2-(4-fluoro-2-methylphenoxy)-N-(2-oxo-1,2-dihydropyridin-4-yl)-5-
(trifluoromethyl)benzamide, 2-(2-chloro-4-fluorophenoxy)-N-(2-oxo-1,2-dihydropyridin-4-yl)-5- (trifluoromethyl)benzamide, 5-chloro-2-(4-fluoro-2-methylphenoxy)-N-(2-oxo-1,2-dihydropyridin-4- yl)benzamide, 4-chloro-2-(4-fluoro-2-methylphenoxy)-N-(2-oxo-1,2-dihydropyridin-4-yl)benzamide, 5-chloro-2-(2-chloro-4-fluorophenoxy)-N-(2-oxo-1,2-dihydropyridin-4-yl)benzamide, 2-((5-fluoro-2- hydroxybenzyl)oxy)-N-(2-oxo-1,2-dihydropyridin-4-yl)-4-(trifluoromethyl)benzamide, N-(2-oxo-1,2- dihydropyridin-4-yl)-2-(o-tolyloxy)-5-(trifluoromethyl)benzamide, 2-(2,4-difluorophenoxy)-N-(2- oxo-1,2-dihydropyridin-4-yl)-4-(trifluoromethyl)benzamide, N-(2-oxo-1,2-dihydropyridin-4-yl)-2-(2- (trifluoromethoxy)phenoxy)-5-(trifluoromethyl)benzamide, 2-(4-fluorophenoxy)-N-(2-oxo-1,2- dihydropyridin-4-yl)-5-(trifluoromethyl)benzamide, 2-(4-fluoro-2-methyl-phenoxy)-N-(2-oxo-1H- pyridin-4-yl)-4-(trifluoromethyl)benzamide, [4-[[2-(4-fluoro-2-methyl-phenoxy)-4- (trifluoromethyl)benzoyl]amino]-2-oxo-1-pyridyl]methyl dihydrogen phosphate, 2-(4-fluoro-2- (methyl-d3)phenoxy)-N-(2-oxo-1,2-dihydropyridin-4-yl)-4-(trifluoromethyl)benzamide, (4-(2-(4- fluoro-2-(methyl-d3)phenoxy)-4-(trifluoromethyl)benzamido)-2-oxopyridin-1(2H)-yl)methyl dihydrogen phosphate, 3-(4-fluoro-2-methoxyphenoxy)-N-(3-(methylsulfonyl)phenyl)quinoxaline-2- carboxamide, 3-(2-chloro-4-fluorophenoxy)-N-(3-sulfamoylphenyl)quinoxaline-2-carboxamide, 3-(2- chloro-4-methoxyphenoxy)-N-(3-sulfamoylphenyl)quinoxaline-2-carboxamide, 3-(4-chloro-2- methoxyphenoxy)-N-(3-sulfamoylphenyl)quinoxaline-2-carboxamide, 4-(3-(4- (trifluoromethoxy)phenoxy)quinoxaline-2-carboxamido)picolinic acid, 2-(2,4-difluorophenoxy)-N-(3- sulfamoylphenyl)quinoline-3-carboxamide, 2-(4-fluoro-2-methoxyphenoxy)-N-(3- sulfamoylphenyl)quinoline-3-carboxamide, 3-(2,4-difluorophenoxy)-N-(3- sulfamoylphenyl)quinoxaline-2-carboxamide, N-(3-sulfamoylphenyl)-2-(4- (trifluoromethoxy)phenoxy)quinoline-3-carboxamide, N-(3-sulfamoylphenyl)-3-(4- (trifluoromethoxy)phenoxy)quinoxaline-2-carboxamide, 3-(4-chloro-2-methylphenoxy)-N-(3- sulfamoylphenyl)quinoxaline-2-carboxamide, 5-(3-(4-(trifluoromethoxy)phenoxy)quinoxaline-2- carboxamido)picolinic acid, 3-(4-fluoro-2-methoxyphenoxy)-N-(2-oxo-2,3-dihydro-1H- benzo[d]imidazol-5-yl)quinoxaline-2-carboxamide, 3-(4-fluoro-2-methoxyphenoxy)-N-(pyridin-4- yl)quinoxaline-2-carboxamide, 3-(4-fluorophenoxy)-N-(3-sulfamoylphenyl)quinoxaline-2- carboxamide, N-(3-cyanophenyl)-3-(4-fluoro-2-methoxyphenoxy)quinoxaline-2-carboxamide, N-(4- carbamoylphenyl)-3-(4-fluoro-2-methoxyphenoxy)quinoxaline-2-carboxamide, 4-(3-(4- (trifluoromethoxy)phenoxy)quinoxaline-2-carboxamido)benzoic acid, N-(4-cyanophenyl)-3-(4-fluoro- 2-methoxyphenoxy)quinoxaline-2-carboxamide, 5-(4,5-dichloro-2-(4-fluoro-2- methoxyphenoxy)benzamido)picolinic acid, 5-(2-(2,4-dimethoxyphenoxy)-4,6- bis(trifluoromethyl)benzamido)picolinic acid, 4-(4,5-dichloro-2-(4-fluoro-2- methoxyphenoxy)benzamido)benzoic acid, 5-(2-(4-fluoro-2-methoxyphenoxy)-4,6- bis(trifluoromethyl)benzamido)picolinic acid, 4-(2-(4-fluoro-2-methoxyphenoxy)-4- (perfluoroethyl)benzamido)benzoic acid, 5-(2-(4-fluoro-2-methoxyphenoxy)-4- (perfluoroethyl)benzamido)picolinic acid, 4-(2-(4-fluoro-2-methylphenoxy)-4-
(trifluoromethyl)benzamido)benzoic acid, 5-(4,5-dichloro-2-(4-fluoro-2- methoxyphenoxy)benzamido)picolinic acid, 4-(2-(2-chloro-4-fluorophenoxy)-4- (perfluoroethyl)benzamido)benzoic acid, 4-(2-(4-fluoro-2-methylphenoxy)-4- (perfluoroethyl)benzamido)benzoic acid, 4-(4,5-dichloro-2-(4- (trifluoromethoxy)phenoxy)benzamido)benzoic acid, 4-(4,5-dichloro-2-(4-chloro-2- methylphenoxy)benzamido)benzoic acid, 5-(4-(tert-butyl)-2-(4-fluoro-2- methoxyphenoxy)benzamido)picolinic acid, 5-(4,5-dichloro-2-(4- (trifluoromethoxy)phenoxy)benzamido)picolinic acid, 4-(4,5-dichloro-2-(4-fluoro-2- methylphenoxy)benzamido)benzoic acid, 5-(4,5-dichloro-2-(2,4- dimethoxyphenoxy)benzamido)picolinic acid, 5-(4,5-dichloro-2-(2-chloro-4- fluorophenoxy)benzamido)picolinic acid, 5-(4,5-dichloro-2-(4-fluoro-2- methylphenoxy)benzamido)picolinic acid, 4-(4,5-dichloro-2-(4-chloro-2- methoxyphenoxy)benzamido)benzoic acid, 5-(4,5-dichloro-2-(2,4- difluorophenoxy)benzamido)picolinic acid, 2-(4-fluorophenoxy)-N-(3-sulfamoylphenyl)-5- (trifluoromethyl)benzamide, 2-(4-fluorophenoxy)-N-(3-sulfamoylphenyl)-4- (trifluoromethyl)benzamide, 2-(2-chloro-4-fluorophenoxy)-N-(3-sulfamoylphenyl)-5- (trifluoromethyl)benzamide, 2-(4-fluorophenoxy)-N-(3-sulfamoylphenyl)-4- (trifluoromethyl)benzamide, 2-(2-chloro-4-fluorophenoxy)-N-(3-sulfamoylphenyl)-6- (trifluoromethyl)benzamide, 2-(2-chloro-4-fluorophenoxy)-5-(difluoromethyl)-N-(3- sulfamoylphenyl)benzamide, 2-(4-fluorophenoxy)-4-(perfluoroethyl)-N-(3- sulfamoylphenyl)benzamide, 2-(4-chloro-2-methoxyphenoxy)-4-(perfluoroethyl)-N-(3- sulfamoylphenyl)benzamide, 2-(4-fluoro-2-methoxyphenoxy)-N-(3-sulfamoylphenyl)-5- (trifluoromethyl)benzamide, 5-chloro-2-(4-fluoro-2-methylphenoxy)-N-(3- sulfamoylphenyl)benzamide, 4,5-dichloro-2-(4-fluoro-2-methoxyphenoxy)-N-(3- sulfamoylphenyl)benzamide, 2,4-dichloro-6-(4-chloro-2-methoxyphenoxy)-N-(3- sulfamoylphenyl)benzamide, 2,4-dichloro-6-(4-fluoro-2-methylphenoxy)-N-(3- sulfamoylphenyl)benzamide, 2-(4-fluoro-2-methoxyphenoxy)-N-(3-sulfamoylphenyl)-4,6- bis(trifluoromethyl)benzamide, 2-(4-fluoro-2-methylphenoxy)-N-(3-sulfamoylphenyl)-4,6- bis(trifluoromethyl)benzamide, 5-chloro-2-(2-chloro-4-fluorophenoxy)-N-(3- sulfamoylphenyl)benzamide, 2-(4-fluoro-2-methoxyphenoxy)-N-(3-sulfamoylphenyl)-4- (trifluoromethoxy)benzamide, 2-(4-fluoro-2-methoxyphenoxy)-N-(3-sulfamoylphenyl)-4- (trifluoromethyl)benzamide, 4,5-dichloro-2-(4-fluorophenoxy)-N-(3-sulfamoylphenyl)benzamide, 2- (4-fluoro-2-methoxyphenoxy)-4-(perfluoroethyl)-N-(3-sulfamoylphenyl)benzamide, 5-fluoro-2-(4- fluoro-2-methylphenoxy)-N-(3-sulfamoylphenyl)benzamide, 2-(2-chloro-4-fluorophenoxy)-4-cyano- N-(3-sulfamoylphenyl)benzamide, N-(3-sulfamoylphenyl)-2-(4-(trifluoromethoxy)phenoxy)-4- (trifluoromethyl)benzamide, N-(3-carbamoyl-4-fluoro-phenyl)-2-fluoro-6-[2-(trideuteriomethoxy)-4- (trifluoromethoxy)phenoxy]-3-(trifluoromethyl)benzamide, N-(3-carbamoyl-4-fluoro-phenyl)-2-
fluoro-6-[2-methoxy-4-(trifluoromethoxy)phenoxy]-3-(trifluoromethyl)benzamide, N-(3-carbamoyl- 4-fluoro-phenyl)-2-fluoro-6-[2-(trideuteriomethoxy)-4-(trifluoromethoxy)phenoxy]-3- (trifluoromethoxy)benzamide, 4-[[2-fluoro-6-[2-methoxy-4-(trifluoromethoxy)phenoxy]-3- (trifluoromethyl)benzoyl]amino]pyridine-2-carboxamide, 4-[[3-chloro-2-fluoro-6-[2-methoxy-4- (trifluoromethoxy)phenoxy]benzoyl]amino]pyridine-2-carboxamide, 4-[[2-fluoro-6-[2- (trideuteriomethoxy)-4-(trifluoromethoxy)phenoxy]-3-(trifluoromethyl)benzoyl]amino]pyridine-2- carboxamide, N-(3-carbamoyl-4-fluoro-phenyl)-3-(difluoromethyl)-2-fluoro-6-[2-methoxy-4- (trifluoromethoxy)phenoxy]benzamide, 4-[[2-fluoro-6-[2-(trideuteriomethoxy)-4- (trifluoromethoxy)phenoxy]-3-(trifluoromethoxy)benzoyl]amino]pyridine-2-carboxamide, N-(3- carbamoyl-4-fluoro-phenyl)-6-[2-chloro-4-(trifluoromethoxy)phenoxy]-2-fluoro-3- (trifluoromethyl)benzamide, N-(3-carbamoyl-4-fluoro-phenyl)-2-fluoro-6-[2-methyl-4- (trifluoromethoxy)phenoxy]-3-(trifluoromethyl)benzamide, N-(3-carbamoyl-4-fluoro-phenyl)-2,3,4- trifluoro-6-[2-methoxy-4-(trifluoromethoxy)phenoxy]benzamide, N-(2-carbamoyl-4-pyridyl)-3- fluoro-5-[2-methoxy-4-(trifluoromethoxy)phenoxy]-2-(trifluoromethyl)pyridine-4-carboxamide, 4- [[6-[2-(difluoromethoxy)-4-(trifluoromethoxy)phenoxy]-2-fluoro-3- (trifluoromethyl)benzoyl]amino]pyridine-2-carboxamide, N-(3-carbamoyl-4-fluoro-phenyl)-6-[3- chloro-4-(trifluoromethoxy)phenoxy]-2-fluoro-3-(trifluoromethyl)benzamide, N-(3-carbamoyl-4- fluoro-phenyl)-2-fluoro-6-[4-(trifluoromethoxy)phenoxy]-3-(trifluoromethyl)benzamide, N-(4- carbamoyl-3-fluoro-phenyl)-2-fluoro-6-[2-methoxy-4-(trifluoromethoxy)phenoxy]-3- (trifluoromethyl)benzamide, 4-[[2-fluoro-6-[2-(trideuteriomethoxy)-4-(trifluoromethoxy)phenoxy]-4- (trifluoromethyl)benzoyl]amino]pyridine-2-carboxamide, N-(3-carbamoyl-4-fluoro-phenyl)-2-fluoro- 6-[3-fluoro-4-(trifluoromethoxy)phenoxy]-3-(trifluoromethyl)benzamide, N-(3-carbamoyl-4-fluoro- phenyl)-2-[2-methoxy-4-(trifluoromethoxy)phenoxy]-5-(1,1,2,2,2-pentafluoroethyl)benzamide, 4-[[4- (difluoromethoxy)-2-fluoro-6-[2-methoxy-4-(trifluoromethoxy)phenoxy]benzoyl]amino]pyridine-2- carboxamide, N-(3-carbamoyl-4-fluoro-phenyl)-2-fluoro-6-[2-fluoro-4-(trifluoromethoxy)phenoxy]- 3-(trifluoromethyl)benzamide, 4-[[4-cyclopropyl-2-fluoro-6-[2-methoxy-4- (trifluoromethoxy)phenoxy]benzoyl]amino]pyridine-2-carboxamide, N-(3-carbamoyl-4-fluoro- phenyl)-5-fluoro-2-[2-methoxy-4-(trifluoromethoxy)phenoxy]-4-(trifluoromethyl)benzamide, 5-[[2- fluoro-6-[2-(trideuteriomethoxy)-4-(trifluoromethoxy)phenoxy]-3- (trifluoromethyl)benzoyl]amino]pyridine-2-carboxamide, N-(3-carbamoyl-4-fluoro-phenyl)-2-fluoro- 6-(4-fluorophenoxy)-3-(trifluoromethyl)benzamide, or 4-[[2-fluoro-6-[3-fluoro-2-methoxy-4- (trifluoromethoxy)phenoxy]-3-(trifluoromethyl)benzoyl]amino]pyridine-2-carboxamide; [0430] (40) a combined NaV1.7 and NaV1.8 blocker, such as DSP-2230, Lohocla201 or BL-1021; [0431] (41) a 5-HT3 antagonist, such as ondansetron; [0432] (42) a TPRV 1 receptor agonist, such as capsaicin (NeurogesX®, Qutenza®); and the pharmaceutically acceptable salts and solvates thereof; [0433] (43) a nicotinic receptor antagonist, such as varenicline;
[0434] (44) an N-type calcium channel antagonist, such as Z-160; [0435] (45) a nerve growth factor antagonist, such as tanezumab; [0436] (46) an endopeptidase stimulant, such as senrebotase; [0437] (47) an angiotensin II antagonist, such as EMA-401; [0438] (48) acetaminophen (including without limitation intravenous acetaminophen (e.g., Ofirmev®)); [0439] (49) bupivacaine (including without limitation bupivacaine liposome injectable suspension (e.g., Exparel®) bupivacaine ER (Posimir), bupivacaine collagen (Xaracoll) and transdermal bupivacaine (Eladur®)); and [0440] (50) bupivacaine and meloxicam combination (e.g., HTX-011). [0441] In one embodiment, the additional appropriate therapeutic agents are selected from V- 116517, Pregabalin, controlled release Pregabalin, Ezogabine (Potiga®). Ketamine/amitriptyline topical cream (Amiket®), AVP-923, Perampanel (E-2007), Ralfinamide, transdermal bupivacaine (Eladur®), CNV1014802, JNJ-10234094 (Carisbamate), BMS-954561 or ARC-4558. [0442] In another embodiment, the additional appropriate therapeutic agents are selected from N- (6-amino-5-(2,3,5-trichlorophenyl)pyridin-2-yl)acetamide; N-(6-amino-5-(2-chloro-5- methoxyphenyl)pyridin-2-yl)-1-methyl-1H-pyrazole-5-carboxamide; or 3-((4-(4- (trifluoromethoxy)phenyl)-1H-imidazol-2-yl)methyl)oxetan-3-amine. [0443] In another embodiment, the additional therapeutic agent is selected from a GlyT2/5HT2 inhibitor, such as Operanserin (VVZ149), a TRPV modulator such as CA008, CMX-020, NEO6860, FTABS, CNTX4975, MCP101, MDR16523, or MDR652, a EGR1 inhibitor such as Brivoglide (AYX1), an NGF inhibitor such as Tanezumab, Fasinumab, ASP6294, MEDI7352, a Mu opioid agonist such as Cebranopadol, NKTR181 (oxycodegol), a CB-1 agonist such as NEO1940 (AZN1940), an imidazoline 12 agonist such as CR4056 or a p75NTR-Fc modulator such as LEVI-04. [0444] In another embodiment, the additional therapeutic agent is oliceridine or ropivacaine (TLC590). [0445] In another embodiment, the additional therapeutic agent is a NaV1.7 blocker such as ST- 2427, ST-2578 and those disclosed in WO2010/129864, WO2015/157559, WO2017/059385, WO2018/183781, WO2018/183782, WO2020/072835, and/or WO2022/036297 the entire contents of each application hereby incorporated by reference. [0446] In another embodiment, the additional therapeutic agent is selected from ASP18071, CC- 8464, ANP-230, ANP-231, NOC-100, NTX-1175, ASN008, NW3509, AM-6120, AM-8145, AM- 0422, BL-017881, NTM-006, Opiranserin (UnafraTM), brivoligide, SR419, NRD.E1, LX9211, LY3016859, ISC-17536, NFX-88, LAT-8881, AP-235, NYX 2925, CNTX-6016, S-600918, S- 637880, RQ-00434739, KLS-2031, MEDI 7352, and XT-150. [0447] In another embodiment, the additional therapeutic agent is selected from Olinvyk, Zynrelef, Seglentis, Neumentum, Nevakar, HTX-034, CPL-01, ACP-044, HRS-4800, Tarlige,
BAY2395840, LY3526318, Eliapixant, TRV045, RTA901, NRD1355-E1, MT-8554, LY3556050, AP-325, tetrodotoxin, Otenaproxesul, CFTX-1554, Funapide, iN1011-N17, JMKX000623/ODM- 111, ETX-801, OLP-1002, ANP-230/DSP-2230, iN1011-N17, DSP-3905 and ACD440. [0448] In another embodiment, the additional therapeutic agent is selected from HRS4800, ODM-111/JMKX000623, LX9211, LY3556050, LY3857210, CFTX01554/CFTX-1554, MEDI7352, MEDI0618, BAY3178275, BAY2395840, GSK3858279, STC-004, HALNEURON, OLP-1002, ATX01, ANP230, CC-8464, iN1011-N17, ST-2427, MSD199, FZ008, VYNAV-01, BL-017881, Profervia (Cilnidipine), LS-04, vixotrigine, FX301/PCRX-301, PF-04531083, PF-01247324, and DSP-3905. [0449] In another embodiment, the additional therapeutic agent is a sodium channel inhibitor (also known as a sodium channel blocker), such as the NaV1.7 and NaV1.8 blockers identified above. [0450] The amount of additional therapeutic agent present in the compositions of this invention may be no more than the amount that would normally be administered in a composition comprising that therapeutic agent as the only active agent. The amount of additional therapeutic agent in the presently disclosed compositions may range from about 10% to 100% of the amount normally present in a composition comprising that agent as the only therapeutically active agent. [0451] The compounds and salts of this invention or pharmaceutically acceptable compositions thereof may also be incorporated into compositions for coating an implantable medical device, such as prostheses, artificial valves, vascular grafts, stents and catheters. Accordingly, the invention, in another aspect, includes a composition for coating an implantable device comprising a compound or salt of the invention as described generally above, and in classes and subclasses herein, and a carrier suitable for coating said implantable device. In still another aspect, the invention includes an implantable device coated with a composition comprising a compound or salt of the invention as described generally above, and in classes and subclasses herein, and a carrier suitable for coating said implantable device. Suitable coatings and the general preparation of coated implantable devices are described in US Patents 6,099,562; 5,886,026; and 5,304,121. The coatings are typically biocompatible polymeric materials such as a hydrogel polymer, polymethyldisiloxane, polycaprolactone, polyethylene glycol, polylactic acid, ethylene vinyl acetate, and mixtures thereof. The coatings may optionally be further covered by a suitable topcoat of fluorosilicone, polysaccharides, polyethylene glycol, phospholipids or combinations thereof to impart controlled release characteristics in the composition. [0452] Another aspect of the invention relates to inhibiting NaV1.8 activity in a biological sample or a subject, which method comprises administering to the subject, or contacting said biological sample with a compound of the invention, a pharmaceutically acceptable salt thereof, or a pharmaceutical composition thereof. The term “biological sample,” as used herein, includes, without limitation, cell cultures or extracts thereof; biopsied material obtained from a mammal or extracts thereof; and blood, saliva, urine, feces, semen, tears, or other body fluids or extracts thereof.
[0453] Inhibition of NaV1.8 activity in a biological sample is useful for a variety of purposes that are known to one of skill in the art. Examples of such purposes include, but are not limited to, the study of sodium channels in biological and pathological phenomena; and the comparative evaluation of new sodium channel inhibitors. K. Pharmaceutical Compositions [0454] Another aspect of the invention provides pharmaceutical compositions comprising Compound I in any one of the pharmaceutically acceptable solid (e.g. crystalline or amorphous) forms disclosed herein. In some embodiments, the pharmaceutical composition comprises Compound I in a solid form selected from Compound I neat Form A, Compound I neat Form B, Compound I neat Form E, Compound I Acetone Solvate Hydrate Form A, Compound I Ethanol Solvate Form A, Compound I Hydrate Form A, and Compound I Dehydrated Hydrate Form A. In some embodiments, the pharmaceutical composition comprises Compound I in a solid amorphous form that is Compound I amorphous form. [0455] In some embodiments, the invention provides a pharmaceutical composition comprising (a) Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein, and (b) at least one pharmaceutically acceptable carrier. [0456] In some embodiments, the invention provides pharmaceutical compositions comprising (a) Compound I in a solid form selected from Compound I neat Form A, Compound I neat Form B, Compound I neat Form E, Compound I Acetone Solvate Hydrate Form A, Compound I Ethanol Solvate Form A, Compound I Hydrate Form A, and Compound I Dehydrated Hydrate Form A and (b) at least one pharmaceutically acceptable carrier. [0457] As described herein, the pharmaceutically acceptable compositions of the invention additionally comprise a pharmaceutically acceptable carrier, adjuvant, or vehicle, which, as used herein, includes any and all solvents, diluents, or other liquid vehicle, dispersion or suspension aids, surface active agents, isotonic agents, thickening or emulsifying agents, preservatives, solid binders, lubricants and the like, as suited to the particular dosage form desired. Remington’s Pharmaceutical Sciences, Sixteenth Edition, E. W. Martin (Mack Publishing Co., Easton, Pa., 1980) discloses various carriers used in formulating pharmaceutically acceptable compositions and known techniques for the preparation thereof. Except insofar as any conventional carrier medium is incompatible with the compounds of the invention, such as by producing any undesirable biological effect or otherwise interacting in a deleterious manner with any other component(s) of the pharmaceutically acceptable composition, its use is contemplated to be within the scope of this invention. Some examples of materials which can serve as pharmaceutically acceptable carriers include, but are not limited to, ion exchangers, alumina, aluminum stearate, lecithin, serum proteins, such as human serum albumin, buffer substances such as phosphates, glycine, sorbic acid, or potassium sorbate, partial glyceride mixtures of saturated vegetable fatty acids, water, salts or electrolytes, such as protamine sulfate, disodium
hydrogen phosphate, potassium hydrogen phosphate, sodium chloride, zinc salts, colloidal silica, magnesium trisilicate, polyvinyl pyrrolidone, polyacrylates, waxes, polyethylene-polyoxypropylene- block polymers, wool fat, sugars such as lactose, glucose and sucrose; starches such as corn starch and potato starch; cellulose and its derivatives such as sodium carboxymethyl cellulose, ethyl cellulose and cellulose acetate; powdered tragacanth; malt; gelatin; talc; excipients such as cocoa butter and suppository waxes; oils such as peanut oil, cottonseed oil; safflower oil; sesame oil; olive oil; corn oil and soybean oil; glycols; such a propylene glycol or polyethylene glycol; esters such as ethyl oleate and ethyl laurate; agar; buffering agents such as magnesium hydroxide and aluminum hydroxide; alginic acid; pyrogen-free water; isotonic saline; Ringer’s solution; ethyl alcohol, and phosphate buffer solutions, as well as other non-toxic compatible lubricants such as sodium lauryl sulfate and magnesium stearate, as well as coloring agents, releasing agents, coating agents, sweetening, flavoring and perfuming agents, preservatives and antioxidants can also be present in the composition, according to the judgment of the formulator. L. Synthesis of the Compounds [0458] The compounds of the invention can be prepared from known materials by the methods described in the Examples, other similar methods, and other methods known to one skilled in the art. As one skilled in the art would appreciate, the functional groups of the intermediate compounds may need to be protected by suitable protecting groups. Protecting groups may be added or removed in accordance with standard techniques, which are well-known to those skilled in the art. The use of protecting groups is described in detail in T.G.M. Wuts et al., Greene’s Protective Groups in Organic Synthesis (4th ed.2006). M. Radiolabeled Analogs of the Compounds [0459] In another aspect, the invention relates to radiolabeled analogs of the compounds of the invention. As used herein, the term “radiolabeled analogs of the compounds of the invention” refers to compounds that are identical to the compounds of the invention, as described herein including all embodiments thereof, except that one or more atoms has been replaced with a radioisotope of the atom present in the compounds of the invention. [0460] As used herein, the term “radioisotope” refers to an isotope of an element that is known to undergo spontaneous radioactive decay. Examples of radioisotopes include 3H, 14C, 32P, 35S, 18F, 36Cl, and the like, as well as the isotopes for which a decay mode is identified in V.S. Shirley & C.M. Lederer, Isotopes Project, Nuclear Science Division, Lawrence Berkeley Laboratory, Table of Nuclides (January 1980). [0461] The radiolabeled analogs can be used in a number of beneficial ways, including in various types of assays, such as substrate tissue distribution assays. For example, tritium (3H)- and/or carbon-
14 (14C)-labeled compounds may be useful for various types of assays, such as substrate tissue distribution assays, due to relatively simple preparation and excellent detectability. [0462] In another aspect, the invention relates to pharmaceutically acceptable salts of the radiolabeled analogs, in accordance with any of the embodiments described herein in connection with the compounds of the invention. [0463] In another aspect, the invention relates to pharmaceutical compositions comprising the radiolabeled analogs, or pharmaceutically acceptable salts thereof, and a pharmaceutically acceptable carrier, adjuvant or vehicle, in accordance with any of the embodiments described herein in connection with the compounds of the invention. [0464] In another aspect, the invention relates to methods of inhibiting voltage-gated sodium channels and methods of treating or lessening the severity of various diseases and disorders, including pain, in a subject comprising administering an effective amount of the radiolabeled analogs, pharmaceutically acceptable salts thereof, and pharmaceutical compositions thereof, in accordance with any of the embodiments described herein in connection with the compounds of the invention. [0465] In another aspect, the invention relates to radiolabeled analogs, pharmaceutically acceptable salts thereof, and pharmaceutical compositions thereof, for use, in accordance with any of the embodiments described herein in connection with the compounds of the invention. [0466] In another aspect, the invention relates to the use of the radiolabeled analogs, or pharmaceutically acceptable salts thereof, and pharmaceutical compositions thereof, for the manufacture of medicaments, in accordance with any of the embodiments described herein in connection with the compounds of the invention. [0467] In another aspect, the radiolabeled analogs, pharmaceutically acceptable salts thereof, and pharmaceutical compositions thereof, can be employed in combination therapies, in accordance with any of the embodiments described herein in connection with the compounds of the invention. N. Non-limiting Exemplary Embodiments [0468] Further embodiments of the disclosure are set out in the following numbered Embodiments: 1. Compound I I)
as substantially amorphous Compound I amorphous form (i.e., wherein less than 15% of Compound I is in crystalline form, wherein less than 10% of Compound I is in crystalline form, wherein less than 5% of Compound I is in crystalline form). 2. The substantially amorphous Compound I amorphous form according to Embodiment 1, wherein Compound I is 100% amorphous. 3. The substantially amorphous Compound I amorphous form according to Embodiment 1 or Embodiment 2, characterized by an X-ray powder diffractogram substantially similar to FIG.1. 4. The substantially amorphous Compound I amorphous form according to any one of Embodiments 1-3, characterized by a 13C SSNMR spectrum having one, two, three, four, five, six, seven, eight, nine, ten, or more peaks selected from 177.6 ± 0.2 ppm, 172.0 ± 0.2 ppm, 162.6 ± 0.2 ppm, 155.2 ± 0.2 ppm, 149.9 ± 0.2 ppm, 147.0 ± 0.2 ppm, 121.0 ± 0.2 ppm, 120.1 ± 0.2 ppm, 119.1 ± 0.2 ppm, 116.0 ± 0.2 ppm, 114.6 ± 0.2 ppm, 113.8 ± 0.2 ppm, 15.5 ± 0.2 ppm, and 7.7 ± 0.2 ppm. 5. The substantially amorphous Compound I amorphous form according to any one of Embodiments 1-4, characterized by a 13C SSNMR spectrum having peaks at 177.6 ± 0.2 ppm, 172.0 ± 0.2 ppm, 162.6 ± 0.2 ppm, 155.2 ± 0.2 ppm, 149.9 ± 0.2 ppm, 147.0 ± 0.2 ppm, 121.0 ± 0.2 ppm, 120.1 ± 0.2 ppm, 119.1 ± 0.2 ppm, 116.0 ± 0.2 ppm, 114.6 ± 0.2 ppm, 113.8 ± 0.2 ppm, 15.5 ± 0.2 ppm, and 7.7 ± 0.2 ppm. 6. The substantially amorphous Compound I amorphous form according to any one of Embodiments 1-5, characterized by a 13C SSNMR spectrum substantially similar to FIG.2. 7. The substantially amorphous Compound I amorphous form according to any one of Embodiments 1-6, characterized by a 19F SSNMR spectrum having one or two peaks selected from -61.9 ± 0.2 ppm and -142.1 ± 0.2 ppm. 8. The substantially amorphous Compound I amorphous form according to any one of Embodiments 1-7, characterized by a 19F SSNMR spectrum substantially similar to FIG.3. 9. Substantially crystalline Compound I neat Form A (i.e., wherein less than 15% of Compound I is in amorphous form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous form). 10. The substantially crystalline Compound I neat Form A according to Embodiment 9, wherein Compound I neat Form A is 100% crystalline.
11. The substantially crystalline Compound I neat Form A according to Embodiment 9 or Embodiment 10, characterized by an X-ray powder diffractogram having one or two signals selected from 9.5 ± 0.2 degrees two-theta, 12.3 ± 0.2 degrees two-theta, and 13.5 ± 0.2 degrees two-theta. 12. The substantially crystalline Compound I neat Form A according to any one of Embodiments 9-11, characterized by an X-ray powder diffractogram having (a) one or two signals selected from 9.5 ± 0.2 degrees two-theta, 12.3 ± 0.2 degrees two-theta, and 13.5 ± 0.2 degrees two-theta, and (b) one or two signals selected from 11.3 ± 0.2 degrees two-theta, 15.9 ± 0.2 degrees two-theta, and 20.5 ± 0.2 degrees two-theta. 13. The substantially crystalline Compound I neat Form A according to any one of Embodiments 9-12, characterized by an X-ray powder diffractogram having two, three, four, five, or six signals selected from 9.5 ± 0.2 degrees two-theta, 12.3 ± 0.2 degrees two-theta, 13.5 ± 0.2 degrees two-theta, 11.3 ± 0.2 degrees two-theta, 15.9 ± 0.2 degrees two-theta, 20.5 ± 0.2 degrees two-theta, 14.6 ± 0.2 degrees two-theta, 16.6 ± 0.2 degrees two-theta, and 23.5 ± 0.2 degrees two-theta. 14. The substantially crystalline Compound I neat Form A according to any one of Embodiments 9-13, characterized by an X-ray powder diffractogram substantially similar to FIG.4. 15. The substantially crystalline Compound I neat Form A according to any one of Embodiments 9-14, characterized by a 13C SSNMR spectrum having one, two, three, four, five, six, seven, eight, nine, ten, or more peaks selected from 170.4 ± 0.2 ppm, 163.8 ± 0.2 ppm, 162.4 ± 0.2 ppm, 160.9 ± 0.2 ppm, 158.6 ± 0.2 ppm, 158.3 ± 0.2 ppm, 154.6 ± 0.2 ppm, 154.0 ± 0.2 ppm, 148.3 ± 0.2 ppm, 147.6 ± 0.2 ppm, 146.9 ± 0.2 ppm, 145.9 ± 0.2 ppm, 143.5 ± 0.2 ppm, 143.1 ± 0.2 ppm, 141.9 ± 0.2 ppm, 127.4 ± 0.2 ppm, 125.3 ± 0.2 ppm, 123.6 ± 0.2 ppm, 122.2 ± 0.2 ppm, 121.3 ± 0.2 ppm, 120.7 ± 0.2 ppm, 117.9 ± 0.2 ppm, 117.3 ± 0.2 ppm, 116.3 ± 0.2 ppm, 115.8 ± 0.2 ppm, 114.2 ± 0.2 ppm, 113.6 ± 0.2 ppm, 112.8 ± 0.2 ppm, 15.4 ± 0.2 ppm, and 8.2 ± 0.2 ppm. 16. The substantially crystalline Compound I neat Form A according to any one of Embodiments 9-15, characterized by a 13C SSNMR spectrum having peaks at 170.4 ± 0.2 ppm, 163.8 ± 0.2 ppm, 162.4 ± 0.2 ppm, 160.9 ± 0.2 ppm, 158.6 ± 0.2 ppm, 158.3 ± 0.2 ppm, 154.6 ± 0.2 ppm, 154.0 ± 0.2 ppm, 148.3 ± 0.2 ppm, 147.6 ± 0.2 ppm, 146.9 ± 0.2 ppm, 145.9 ± 0.2 ppm, 143.5 ± 0.2 ppm, 143.1 ± 0.2 ppm, 141.9 ± 0.2 ppm, 127.4 ± 0.2 ppm, 125.3 ± 0.2 ppm, 123.6 ± 0.2 ppm, 122.2 ± 0.2 ppm, 121.3 ± 0.2 ppm, 120.7 ± 0.2 ppm, 117.9 ± 0.2 ppm, 117.3 ± 0.2 ppm, 116.3 ± 0.2 ppm, 115.8 ± 0.2 ppm, 114.2 ± 0.2 ppm, 113.6 ± 0.2 ppm, 112.8 ± 0.2 ppm, 15.4 ± 0.2 ppm, and 8.2 ± 0.2 ppm. 17. The substantially crystalline Compound I neat Form A according to any one of Embodiments 9-16, characterized by a 13C SSNMR spectrum substantially similar to FIG.5.
18. The substantially crystalline Compound I neat Form A according to any one of Embodiments 9-17, characterized as having a 19F SSNMR spectrum with one or two peaks selected from -61.2 ± 0.2 ppm, -61.6 ± 0.2 ppm, -138.6 ± 0.2 ppm, -140.3 ± 0.2 ppm, -142.0 ± 0.2 ppm, and-146.0 ± 0.2 ppm. 19. The substantially crystalline Compound I neat Form A according to any one of Embodiments 9-18, characterized as having a 19F SSNMR spectrum having peaks at -61.2 ± 0.2 ppm, -61.6 ± 0.2 ppm, -138.6 ± 0.2 ppm, -140.3 ± 0.2 ppm, -142.0 ± 0.2 ppm, and-146.0 ± 0.2 ppm. 20. The substantially crystalline Compound I neat Form A according to any one of Embodiments 9-19, characterized by a 19F SSNMR spectrum substantially similar to FIG.6. 21. The substantially crystalline Compound I neat Form A according to any one of Embodiments 9-20, characterized by a triclinic crystal system, P-1 space group, and unit cell dimensions measured at 100 K on a Bruker diffractometer utilizing Cu Kα radiation (λ=1.54178 Å) of: a 9.6 ± 0.1 Å α 94.3 ± 0.1° b 12.6 ± 0.1 Å β 97.3 ± 0.1° c 18.7 ± 0.1 Å γ 107.3 ± 0.1°. 22. Substantially crystalline Compound I neat Form B (i.e., wherein less than 15% of Compound I is in amorphous form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous form). 23. The substantially crystalline Compound I neat Form B according to Embodiment 22, wherein Compound I neat Form B is 100% crystalline. 24. The substantially crystalline Compound I neat Form B according to Embodiment 22 or Embodiment 23, characterized by an X-ray powder diffractogram having one or two signals selected from 4.8 ± 0.2 degrees two-theta, 13.3 ± 0.2 degrees two-theta, and 18.5 ± 0.2 degrees two-theta. 25. The substantially crystalline Compound I neat Form B according to any one of Embodiments 22-24, characterized by an X-ray powder diffractogram having (a) one or two signals selected from 4.8 ± 0.2 degrees two-theta, 13.3 ± 0.2 degrees two-theta, and 18.5 ± 0.2 degrees two-theta, and (b) one or two signals selected from 6.0 ± 0.2 degrees two-theta, 16.5 ± 0.2 degrees two-theta, and 16.9 ± 0.2 degrees two-theta. 26. The substantially crystalline Compound I neat Form B according to any one of Embodiments 22-25, characterized by an X-ray powder diffractogram having two, three, four, five, or six signals selected from 4.8 ± 0.2 degrees two-theta, 13.3 ± 0.2 degrees two-theta, 18.5 ± 0.2 degrees two-theta, 6.0 ± 0.2 degrees two-theta, 16.5 ± 0.2 degrees two-theta, 16.9 ± 0.2 degrees two-theta, 14.1 ± 0.2 degrees two-theta, 14.7 ± 0.2 degrees two-theta, and 15.2 ± 0.2 degrees two-theta.
27. The substantially crystalline Compound I neat Form B according to any one of Embodiments 22-26, characterized by an X-ray powder diffractogram substantially similar to FIG.7. 28. The substantially crystalline Compound I neat Form B according to any one of Embodiments 22-27, characterized by a 13C SSNMR spectrum having one, two, three, four, five, six, seven, eight, nine, ten, or more peaks selected from 176.9 ± 0.2 ppm, 171.7 ± 0.2 ppm, 164.0 ± 0.2 ppm, 161.4 ± 0.2 ppm, 160.7 ± 0.2 ppm, 155.3 ± 0.2 ppm, 154.8 ± 0.2 ppm, 148.6 ± 0.2 ppm, 147.6 ± 0.2 ppm, 146.4 ± 0.2 ppm, 145.7 ± 0.2 ppm, 144.8 ± 0.2 ppm, 127.7 ± 0.2 ppm, 125.4 ± 0.2 ppm, 122.0 ± 0.2 ppm, 121.5 ± 0.2 ppm, 118.4 ± 0.2 ppm, 116.3 ± 0.2 ppm, 114.8 ± 0.2 ppm, 113.8 ± 0.2 ppm, 112.6 ± 0.2 ppm, 17.8 ± 0.2 ppm, 15.0 ± 0.2 ppm, 10.7 ± 0.2 ppm, 9.6 ± 0.2 ppm, and 8.8 ± 0.2 ppm. 29. The substantially crystalline Compound I neat Form B according to any one of Embodiments 22-28, characterized by a 13C SSNMR spectrum having peaks at 176.9 ± 0.2 ppm, 171.7 ± 0.2 ppm, 164 ± 0.2 ppm, 161.4 ± 0.2 ppm, 160.7 ± 0.2 ppm, 155.3 ± 0.2 ppm, 154.8 ± 0.2 ppm, 148.6 ± 0.2 ppm, 147.6 ± 0.2 ppm, 146.4 ± 0.2 ppm, 145.7 ± 0.2 ppm, 144.8 ± 0.2 ppm, 127.7 ± 0.2 ppm, 125.4 ± 0.2 ppm, 122.0 ± 0.2 ppm, 121.5 ± 0.2 ppm, 118.4 ± 0.2 ppm, 116.3 ± 0.2 ppm, 114.8 ± 0.2 ppm, 113.8 ± 0.2 ppm, 112.6 ± 0.2 ppm, 17.8 ± 0.2 ppm, 15.0 ± 0.2 ppm, 10.7 ± 0.2 ppm, 9.6 ± 0.2 ppm, and 8.8 ± 0.2 ppm. 30. The substantially crystalline Compound I neat Form B according to any one of Embodiments 22-29, characterized by a 13C SSNMR spectrum substantially similar to FIG.8. 31. The substantially crystalline Compound I neat Form B according to any one of Embodiments 22-30, characterized as having a 19F SSNMR spectrum with one or two peaks selected from -61.6 ± 0.2 ppm, -138 ± 0.2 ppm, -139.7 ± 0.2 ppm, -143.4 ± 0.2 ppm, and -144.9 ± 0.2 ppm. 32. The substantially crystalline Compound I neat Form B according to any one of Embodiments 22-31, characterized as having a 19F SSNMR spectrum having peaks at -61.6 ± 0.2 ppm, -138 ± 0.2 ppm, -139.7 ± 0.2 ppm, -143.4 ± 0.2 ppm, and -144.9 ± 0.2 ppm. 33. The substantially crystalline Compound I neat Form B according to any one of Embodiments 22-32, characterized by a 19F SSNMR spectrum substantially similar to FIG.9. 34. Substantially crystalline Compound I neat Form E (i.e., wherein less than 15% of Compound I is in amorphous form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous form). 35. The substantially crystalline Compound I neat Form E according to Embodiment 34, wherein Compound I neat Form E is 100% crystalline.
36. The substantially crystalline Compound I neat Form E according to Embodiment 34 or Embodiment 35, characterized by an X-ray powder diffractogram having one or two signals selected from 8.6 ± 0.2 degrees two-theta, 10.1 ± 0.2 degrees two-theta, and 12.6 ± 0.2 degrees two-theta. 37. The substantially crystalline Compound I neat Form E according to any one of Embodiments 34-36, characterized by an X-ray powder diffractogram having (a) one or two signals selected from 8.6 ± 0.2 degrees two-theta, 10.1 ± 0.2 degrees two-theta, and 12.6 ± 0.2 degrees two-theta, and (b) one or two signals selected from 14.1 ± 0.2 degrees two-theta, 20.3 ± 0.2 degrees two-theta, and 23.2 ± 0.2 degrees two-theta. 38. The substantially crystalline Compound I neat Form E according to any one of Embodiments 34-37, characterized by an X-ray powder diffractogram having two, three, four, five, or six signals selected from 8.6 ± 0.2 degrees two-theta, 10.1 ± 0.2 degrees two-theta, 12.6 ± 0.2 degrees two-theta, 14.1 ± 0.2 degrees two-theta, 20.3 ± 0.2 degrees two-theta, 23.2 ± 0.2 degrees two-theta, 11.5 ± 0.2 degrees two-theta, 14.2 ± 0.2 degrees two-theta, and 15.1 ± 0.2 degrees two-theta. 39. The substantially crystalline Compound I neat Form E according to any one of Embodiments 34-38, characterized by an X-ray powder diffractogram substantially similar to FIG.10. 40. The substantially crystalline Compound I neat Form E according to any one of Embodiments 34-39, characterized by a 13C SSNMR spectrum having one, two, three, four, five, six, seven, eight, nine, ten, or more peaks selected from 170.0 ± 0.2 ppm, 163.0 ± 0.2 ppm, 162.3 ± 0.2 ppm, 161.3 ± 0.2 ppm, 159.3 ± 0.2 ppm, 154.7 ± 0.2 ppm, 150.6 ± 0.2 ppm, 149.8 ± 0.2 ppm, 148.6 ± 0.2 ppm, 148.3 ± 0.2 ppm, 147.1 ± 0.2 ppm, 144.9 ± 0.2 ppm, 143.9 ± 0.2 ppm, 143.5 ± 0.2 ppm, 126.9 ± 0.2 ppm, 126.0 ± 0.2 ppm, 124.1 ± 0.2 ppm, 122.6 ± 0.2 ppm, 120.8 ± 0.2 ppm, 117.8 ± 0.2 ppm, 116.7 ± 0.2 ppm, 115.2 ± 0.2 ppm, 114.1 ± 0.2 ppm, 113.1 ± 0.2 ppm, 15.9 ± 0.2 ppm, 15.3 ± 0.2 ppm, and 8.4 ± 0.2 ppm. 41. The substantially crystalline Compound I neat Form E according to any one of Embodiments 34-40, characterized by a 13C SSNMR spectrum having peaks at 170.0 ± 0.2 ppm, 163.0 ± 0.2 ppm, 162.3 ± 0.2 ppm, 161.3 ± 0.2 ppm, 159.3 ± 0.2 ppm, 154.7 ± 0.2 ppm, 150.6 ± 0.2 ppm, 149.8 ± 0.2 ppm, 148.6 ± 0.2 ppm, 148.3 ± 0.2 ppm, 147.1 ± 0.2 ppm, 144.9 ± 0.2 ppm, 143.9 ± 0.2 ppm, 143.5 ± 0.2 ppm, 126.9 ± 0.2 ppm, 126.0 ± 0.2 ppm, 124.1 ± 0.2 ppm, 122.6 ± 0.2 ppm, 120.8 ± 0.2 ppm, 117.8 ± 0.2 ppm, 116.7 ± 0.2 ppm, 115.2 ± 0.2 ppm, 114.1 ± 0.2 ppm, 113.1 ± 0.2 ppm, 15.9 ± 0.2 ppm, 15.3 ± 0.2 ppm, and 8.4 ± 0.2 ppm. 42. The substantially crystalline Compound I neat Form E according to any one of Embodiments 34-41, characterized by a 13C SSNMR spectrum substantially similar to FIG.11.
43. The substantially crystalline Compound I neat Form E according to any one of Embodiments 34-42, characterized as having a 19F SSNMR spectrum with one or two peaks selected from -60.9 ± 0.2 ppm, -62.5 ± 0.2 ppm, -63.1 ± 0.2 ppm, -135.1 ± 0.2 ppm, -137.1 ± 0.2 ppm, -140.7 ± 0.2 ppm, and -141.9 ± 0.2 ppm. 44. The substantially crystalline Compound I neat Form E according to any one of Embodiments 34-43, characterized as having a 19F SSNMR spectrum having peaks at -60.9 ± 0.2 ppm, -62.5 ± 0.2 ppm, -63.1 ± 0.2 ppm, -135.1 ± 0.2 ppm, -137.1 ± 0.2 ppm, -140.7 ± 0.2 ppm, and -141.9 ± 0.2 ppm. 45. The substantially crystalline Compound I neat Form E according to any one of Embodiments 34-44, characterized by a 19F SSNMR spectrum substantially similar to FIG.12. 46. The substantially crystalline Compound I neat Form E according to any one of Embodiments 34-45, characterized by a triclinic crystal system, P-1 space group, and unit cell dimensions measured at 100 K on a Rigaku diffractometer utilizing Cu Kα radiation (λ=1.54178 Å) of: a 8.7 ± 0.1 Å α 92.7 ± 0.1° b 12.2 ± 0.1 Å β 96.3 ± 0.1° c 20.9 ± 0.1 Å γ 98.6 ± 0.1°. 47. The substantially crystalline Compound I neat Form E according to any one of Embodiments 34-46, characterized by a triclinic crystal system, P-1 space group, and unit cell dimensions measured at 298 K on a Rigaku diffractometer utilizing Cu Kα radiation (λ=1.54178 Å) of: a 8.9 ± 0.1 Å α 92.6 ± 0.1° b 12.3 ± 0.1 Å β 97.1 ± 0.1° c 21.1 ± 0.1 Å γ 98.5 ± 0.1°. 48. Substantially crystalline Compound I Acetone Solvate Hydrate Form A (i.e., wherein less than 15% of Compound I is in amorphous form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous form). 49. The substantially crystalline Compound I Acetone Solvate Hydrate Form A according to Embodiment 48, wherein Compound I Acetone Solvate Hydrate is 100% crystalline. 50. The substantially crystalline Compound I Acetone Solvate Hydrate Form A according to Embodiment 48 or Embodiment 49, characterized by an X-ray powder diffractogram having one or two signals selected from 17.2 ± 0.2 degrees two-theta, 18.1 ± 0.2 degrees two-theta, and 25.9 ± 0.2 degrees two-theta. 51. The substantially crystalline Compound I Acetone Solvate Hydrate Form A according to any one of Embodiments 48-50, characterized by an X-ray powder diffractogram having (a) one or two
signals selected from 17.2 ± 0.2 degrees two-theta, 18.1 ± 0.2 degrees two-theta, and 25.9 ± 0.2 degrees two-theta, and (b) one or two signals selected from 12.9 ± 0.2 degrees two-theta, 15.6 ± 0.2 degrees two-theta, and 24.8 ± 0.2 degrees two-theta. 52. The substantially crystalline Compound I Acetone Solvate Hydrate Form A according to any one of Embodiments 48-51, characterized by an X-ray powder diffractogram having two, three, four, five, or six signals selected from 9.5 ± 0.2 degrees two-theta, 12.3 ± 0.2 degrees two-theta, 13.5 ± 0.2 degrees two-theta, 12.9 ± 0.2 degrees two-theta, 15.6 ± 0.2 degrees two-theta, 24.8 ± 0.2 degrees two-theta, 8.1 ± 0.2 degrees two-theta, 12.3 ± 0.2 degrees two-theta, and 17.6 ± 0.2 degrees two-theta. 53. The substantially crystalline Compound I Acetone Solvate Hydrate Form A according to any one of Embodiments 48-52, characterized by an X-ray powder diffractogram substantially similar to FIG.13. 54. The substantially crystalline Compound I Acetone Solvate Hydrate Form A according to any one of Embodiments 48-53, characterized by a 13C SSNMR spectrum having one, two, three, four, five, six, seven, eight, nine, ten, or more peaks selected from 212.9 ± 0.2 ppm, 211.6 ± 0.2 ppm, 179.2 ± 0.2 ppm, 178.9 ± 0.2 ppm, 178.4 ± 0.2 ppm, 174.7 ± 0.2 ppm, 174.1 ± 0.2 ppm, 172.7 ± 0.2 ppm, 172.2 ± 0.2 ppm, 163.1 ± 0.2 ppm, 156.7 ± 0.2 ppm, 155.6 ± 0.2 ppm, 152.3 ± 0.2 ppm, 150.1 ± 0.2 ppm, 148.7 ± 0.2 ppm, 147.9 ± 0.2 ppm, 147.0 ± 0.2 ppm, 145.1 ± 0.2 ppm, 125.5 ± 0.2 ppm, 124.5 ± 0.2 ppm, 122.5 ± 0.2 ppm, 121.6 ± 0.2 ppm, 121.1 ± 0.2 ppm, 118.5 ± 0.2 ppm, 118.1 ± 0.2 ppm, 117.4 ± 0.2 ppm, 116.8 ± 0.2 ppm, 115.7 ± 0.2 ppm, 115.3 ± 0.2 ppm, 113.9 ± 0.2 ppm, 113.2 ± 0.2 ppm, 112.4 ± 0.2 ppm, 30 ± 0.2 ppm, 29.5 ± 0.2 ppm, 18.4 ± 0.2 ppm, 17.6 ± 0.2 ppm, 8.8 ± 0.2 ppm, and 8.1 ± 0.2 ppm. 55. The substantially crystalline Compound I Acetone Solvate Hydrate Form A according to any one of Embodiments 48-54, characterized by a 13C SSNMR spectrum having peaks at 212.9 ± 0.2 ppm, 211.6 ± 0.2 ppm, 179.2 ± 0.2 ppm, 178.9 ± 0.2 ppm, 178.4 ± 0.2 ppm, 174.7 ± 0.2 ppm, 174.1 ± 0.2 ppm, 172.7 ± 0.2 ppm, 172.2 ± 0.2 ppm, 163.1 ± 0.2 ppm, 156.7 ± 0.2 ppm, 155.6 ± 0.2 ppm, 152.3 ± 0.2 ppm, 150.1 ± 0.2 ppm, 148.7 ± 0.2 ppm, 147.9 ± 0.2 ppm, 147.0 ± 0.2 ppm, 145.1 ± 0.2 ppm, 125.5 ± 0.2 ppm, 124.5 ± 0.2 ppm, 122.5 ± 0.2 ppm, 121.6 ± 0.2 ppm, 121.1 ± 0.2 ppm, 118.5 ± 0.2 ppm, 118.1 ± 0.2 ppm, 117.4 ± 0.2 ppm, 116.8 ± 0.2 ppm, 115.7 ± 0.2 ppm, 115.3 ± 0.2 ppm, 113.9 ± 0.2 ppm, 113.2 ± 0.2 ppm, 112.4 ± 0.2 ppm, 30 ± 0.2 ppm, 29.5 ± 0.2 ppm, 18.4 ± 0.2 ppm, 17.6 ± 0.2 ppm, 8.8 ± 0.2 ppm, and 8.1 ± 0.2 ppm. 56. The substantially crystalline Compound I Acetone Solvate Hydrate Form According to any one of Embodiments 48-55, characterized by a 13C SSNMR spectrum substantially similar to FIG.14. 57. The substantially crystalline Compound I Acetone Solvate Hydrate Form A according to any one of Embodiments 48-56, characterized as having a 19F SSNMR spectrum with one or two peaks
selected from -60.4 ± 0.2 ppm, -61.2 ± 0.2 ppm, -61.6 ± 0.2 ppm, -62.7 ± 0.2 ppm, -135.9 ± 0.2 ppm, -136.1 ± 0.2 ppm, -139.6 ± 0.2 ppm, -140.6 ± 0.2 ppm, -141.7 ± 0.2 ppm, -145.2 ± 0.2 ppm, and -145.7 ± 0.2 ppm. 58. The substantially crystalline Compound I Acetone Solvate Hydrate Form A according to any one of Embodiments 48-57, characterized as having a 19F SSNMR spectrum having peaks at -60.4 ± 0.2 ppm, -61.2 ± 0.2 ppm, -61.6 ± 0.2 ppm, -62.7 ± 0.2 ppm, -135.9 ± 0.2 ppm, -136.1 ± 0.2 ppm, -139.6 ± 0.2 ppm, -140.6 ± 0.2 ppm, -141.7 ± 0.2 ppm, -145.2 ± 0.2 ppm, and -145.7 ± 0.2 ppm. 59. The substantially crystalline Compound I Acetone Solvate Hydrate Form According to any one of Embodiments 48-58, characterized by a 19F SSNMR spectrum substantially similar to FIG.15. 60. The substantially crystalline Compound I Acetone Solvate Hydrate Form According to any one of Embodiments 48-59, characterized by a monoclinic crystal system, P21 space group, and unit cell dimensions measured at 100 K on a Rigaku diffractometer utilizing Cu Kα radiation (λ=1.54178 Å) of: a 9.2 ± 0.1 Å α 90° b 39.8 ± 0.1 Å β 96.3 ± 0.1° c 12.9 ± 0.1 Å γ 90°. 61. Substantially crystalline Compound I Ethanol Solvate Form A (i.e., wherein less than 15% of Compound I is in amorphous form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous form). 62. The substantially crystalline Compound I Ethanol Solvate Form A according to Embodiment 61, wherein Compound I Ethanol Solvate Hydrate is 100% crystalline. 63. The substantially crystalline Compound I Ethanol Solvate Form A according to Embodiment 61 or Embodiment 62, characterized by an X-ray powder diffractogram having one or two signals selected from 9.4 ± 0.2 degrees two-theta, 16.1 ± 0.2 degrees two-theta, and 26.0 ± 0.2 degrees two-theta. 64. The substantially crystalline Compound I Ethanol Solvate Form A according to any one of Embodiments 61-63, characterized by an X-ray powder diffractogram having (a) one or two signals selected from 9.4 ± 0.2 degrees two-theta, 16.1 ± 0.2 degrees two-theta, and 26.0 ± 0.2 degrees two-theta, and (b) one or two signals selected from 15.1 ± 0.2 degrees two-theta, 17.0 ± 0.2 degrees two-theta, and 22.4 ± 0.2 degrees two-theta. 65. The substantially crystalline Compound I Ethanol Solvate Form A according to any one of Embodiments 61-64, characterized by an X-ray powder diffractogram having two, three, four, five, or six signals selected from 9.4 ± 0.2 degrees two-theta, 16.1 ± 0.2 degrees two-theta, 26.0 ± 0.2 degrees
two-theta, 15.1 ± 0.2 degrees two-theta, 17.0 ± 0.2 degrees two-theta, 22.4 ± 0.2 degrees two-theta, 13.2 ± 0.2 degrees two-theta, 13.6 ± 0.2 degrees two-theta, and 13.8 ± 0.2 degrees two-theta. 66. The substantially crystalline Compound I Ethanol Solvate Form A according to any one of Embodiments 61-65, characterized by an X-ray powder diffractogram substantially similar to FIG.16. 67. The substantially crystalline Compound I Ethanol Solvate Form A according to any one of Embodiments 61-66, characterized by a 13C SSNMR spectrum having one, two, three, four, five, six, seven, eight, nine, ten, or more peaks selected from 178.2 ± 0.2 ppm, 175.7 ± 0.2 ppm, 163.9 ± 0.2 ppm, 163.4 ± 0.2 ppm, 162.8 ± 0.2 ppm, 154.9 ± 0.2 ppm, 148.1 ± 0.2 ppm, 146.0 ± 0.2 ppm, 145.2 ± 0.2 ppm, 125.9 ± 0.2 ppm, 123.1 ± 0.2 ppm, 121.8 ± 0.2 ppm, 120.4 ± 0.2 ppm, 119.5 ± 0.2 ppm, 116.9 ± 0.2 ppm, 116.3 ± 0.2 ppm, 114.8 ± 0.2 ppm, 113.8 ± 0.2 ppm, 56.5 ± 0.2 ppm, 18.4 ± 0.2 ppm, 17.2 ± 0.2 ppm, 9.2 ± 0.2 ppm, and 7.9 ± 0.2 ppm. 68. The substantially crystalline Compound I Ethanol Solvate Form A according to any one of Embodiments 61-67, characterized by a 13C SSNMR spectrum having peaks at 178.2 ± 0.2 ppm, 175.7 ± 0.2 ppm, 163.9 ± 0.2 ppm, 163.4 ± 0.2 ppm, 162.8 ± 0.2 ppm, 154.9 ± 0.2 ppm, 148.1 ± 0.2 ppm, 146.0 ± 0.2 ppm, 145.2 ± 0.2 ppm, 125.9 ± 0.2 ppm, 123.1 ± 0.2 ppm, 121.8 ± 0.2 ppm, 120.4 ± 0.2 ppm, 119.5 ± 0.2 ppm, 116.9 ± 0.2 ppm, 116.3 ± 0.2 ppm, 114.8 ± 0.2 ppm, 113.8 ± 0.2 ppm, 56.5 ± 0.2 ppm, 18.4 ± 0.2 ppm, 17.2 ± 0.2 ppm, 9.2 ± 0.2 ppm, and 7.9 ± 0.2 ppm. 69. The substantially crystalline Compound I Ethanol Solvate Form A according to any one of Embodiments 61-68, characterized by a 13C SSNMR spectrum substantially similar to FIG.17. 70. The substantially crystalline Compound I Ethanol Solvate Form A according to any one of Embodiments 61-69, characterized as having a 19F SSNMR spectrum with one or two peaks selected from -61.1 ± 0.2 ppm, -138.1 ± 0.2 ppm, and -141.2 ± 0.2 ppm. 71. The substantially crystalline Compound I Ethanol Solvate Form A according to any one of Embodiments 61-70, characterized as having a 19F SSNMR spectrum having peaks at -61.1 ± 0.2 ppm, -138.1 ± 0.2 ppm, and -141.2 ± 0.2 ppm. 72. The substantially crystalline Compound I Ethanol Solvate Form A according to any one of Embodiments 61-71, characterized by a 19F SSNMR spectrum substantially similar to FIG.18. 73. The substantially crystalline Compound I Ethanol Solvate Form A according to any one of Embodiments 61-72, characterized by a monoclinic crystal system, P21/c space group, and unit cell dimensions measured at 100 K on a Bruker diffractometer utilizing Cu Kα radiation (λ=1.54178 Å) of: a 17.0 ± 0.1 Å α 90° b 11.1 ± 0.1 Å β 105.9 ± 0.1°
c 13.2 ± 0.1 Å γ 90°. 74. Substantially crystalline Compound I Hydrate Form A (i.e., wherein less than 15% of Compound I is in amorphous form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous form). 75. The substantially crystalline Compound I Hydrate Form A according to Embodiment 74, wherein Compound I Dehydrated Hydrate Form A is 100% crystalline. 76. The substantially crystalline Compound I Hydrate Form A according to Embodiment 74 or Embodiment 75, characterized by an X-ray powder diffractogram having one or two signals selected from 8.7 ± 0.2 degrees two-theta, 12.4 ± 0.2 degrees two-theta, and 18.8 ± 0.2 degrees two-theta. 77. The substantially crystalline Compound I Hydrate Form A according to any one of Embodiments 74-76, characterized by an X-ray powder diffractogram having (a) one or two signals selected from 8.7 ± 0.2 degrees two-theta, 12.4 ± 0.2 degrees two-theta, and 18.8 ± 0.2 degrees two-theta, and (b) one or two signals selected from 12.7 ± 0.2 degrees two-theta, 17.5 ± 0.2 degrees two-theta, and 19.8 ± 0.2 degrees two-theta. 78. The substantially crystalline Compound I Hydrate Form A according to any one of Embodiments 74-77, characterized by an X-ray powder diffractogram having two, three, four, five, or six signals selected from 8.7 ± 0.2 degrees two-theta, 12.4 ± 0.2 degrees two-theta, 18.8 ± 0.2 degrees two-theta, 12.7 ± 0.2 degrees two-theta, 17.5 ± 0.2 degrees two-theta, 19.8 ± 0.2 degrees two-theta, 13.1 ± 0.2 degrees two-theta, 16.3 ± 0.2 degrees two-theta, and 21.1 ± 0.2 degrees two-theta. 79. The substantially crystalline Compound I Hydrate Form A according to any one of Embodiments 74-78, characterized by an X-ray powder diffractogram substantially similar to FIG.19. 80. Substantially crystalline Compound I Dehydrated Hydrate Form A (i.e., wherein less than 15% of Compound I is in amorphous form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous form). 81. The substantially crystalline Compound I Dehydrated Hydrate Form A according to Embodiment 81, wherein Compound I Dehydrated Hydrate Form A is 100% crystalline. 82. The substantially crystalline Compound I Dehydrated Hydrate Form A according to Embodiment 81 or Embodiment 82, characterized by an X-ray powder diffractogram having one or two signals selected from 8.7 ± 0.2 degrees two-theta, 15.9 ± 0.2 degrees two-theta, and 18.7 ± 0.2 degrees two-theta.
83. The substantially crystalline Compound I Dehydrated Hydrate Form A according to any one of Embodiments 81-83, characterized by an X-ray powder diffractogram having (a) one or two signals selected from 8.7 ± 0.2 degrees two-theta, 15.9 ± 0.2 degrees two-theta, and 18.7 ± 0.2 degrees two-theta, and (b) one or two signals selected from 12.4 ± 0.2 degrees two-theta, 12.6 ± 0.2 degrees two-theta, 17.3 ± 0.2 degrees two-theta, and 19.8 ± 0.2 degrees two-theta. 84. The substantially crystalline Compound I Dehydrated Hydrate Form A according to any one of Embodiments 81-84, characterized by an X-ray powder diffractogram having two, three, four, five, or six signals selected from 8.7 ± 0.2 degrees two-theta, 15.9 ± 0.2 degrees two-theta, 18.7 ± 0.2 degrees two-theta, 12.4 ± 0.2 degrees two-theta, 12.6 ± 0.2 degrees two-theta, 17.3 ± 0.2 degrees two-theta, 19.8 ± 0.2 degrees two-theta, 13.1 ± 0.2 degrees two-theta, 16.3 ± 0.2 degrees two-theta, and 21.1 ± 0.2 degrees two-theta. 85. The substantially crystalline Compound I Dehydrated Hydrate Form A according to any one of Embodiments 81-85, characterized by an X-ray powder diffractogram substantially similar to FIG.20. 86. The substantially crystalline Compound I Hydrate Form A according to any one of Embodiments 74-86, characterized by a 13C SSNMR spectrum having one, two, three, four, five, six, seven, eight, nine, ten, or more peaks selected from 177.3 ± 0.2 ppm, 172.8 ± 0.2 ppm, 170.5 ± 0.2 ppm, 162.0 ± 0.2 ppm, 154.3 ± 0.2 ppm, 154.0 ± 0.2 ppm, 150.2 ± 0.2 ppm, 148.7 ± 0.2 ppm, 147.6 ± 0.2 ppm, 146.6 ± 0.2 ppm, 145.5 ± 0.2 ppm, 126.3 ± 0.2 ppm, 125.0 ± 0.2 ppm, 123.8 ± 0.2 ppm, 122.4 ± 0.2 ppm, 121.6 ± 0.2 ppm, 120.6 ± 0.2 ppm, 118.4 ± 0.2 ppm, 117.9 ± 0.2 ppm, 117.0 ± 0.2 ppm, 116.0 ± 0.2 ppm, 114.7 ± 0.2 ppm, 114.4 ± 0.2 ppm, 112.8 ± 0.2 ppm, 17.7 ± 0.2 ppm, 17.1 ± 0.2 ppm, and 8.7 ± 0.2 ppm. 87. The substantially crystalline Compound I Hydrate Form A according to any one of Embodiments 74-87, characterized by a 13C SSNMR spectrum having peaks at 177.3 ± 0.2 ppm, 172.8 ± 0.2 ppm, 170.5 ± 0.2 ppm, 162.0 ± 0.2 ppm, 154.3 ± 0.2 ppm, 154.0 ± 0.2 ppm, 150.2 ± 0.2 ppm, 148.7 ± 0.2 ppm, 147.6 ± 0.2 ppm, 146.6 ± 0.2 ppm, 145.5 ± 0.2 ppm, 126.3 ± 0.2 ppm, 125.0 ± 0.2 ppm, 123.8 ± 0.2 ppm, 122.4 ± 0.2 ppm, 121.6 ± 0.2 ppm, 120.6 ± 0.2 ppm, 118.4 ± 0.2 ppm, 117.9 ± 0.2 ppm, 117.0 ± 0.2 ppm, 116.0 ± 0.2 ppm, 114.7 ± 0.2 ppm, 114.4 ± 0.2 ppm, 112.8 ± 0.2 ppm, 17.7 ± 0.2 ppm, 17.1 ± 0.2 ppm, and 8.7 ± 0.2 ppm. 88. The substantially crystalline Compound I Hydrate Form A according to any one of Embodiments 74-88, characterized by a 13C SSNMR spectrum substantially similar to FIG.22. 89. The substantially crystalline Compound I Hydrate Form A according to any one of Embodiments 74-89, characterized as having a 19F SSNMR spectrum with one or two peaks selected
from -59.4 ± 0.2 ppm, -60.2 ± 0.2 ppm, -135.0 ± 0.2 ppm, -138.4 ± 0.2 ppm, -141.7 ± 0.2 ppm, and -144.0 ± 0.2 ppm. 90. The substantially crystalline Compound I Hydrate Form A according to any one of Embodiments 74-90, characterized as having a 19F SSNMR spectrum having peaks at -59.4 ± 0.2 ppm, -60.2 ± 0.2 ppm, -135.0 ± 0.2 ppm, -138.4 ± 0.2 ppm, -141.7 ± 0.2 ppm, and -144.0 ± 0.2 ppm. 91. The substantially crystalline Compound I Hydrate Form A according to any one of Embodiments 74-91, characterized by a 19F SSNMR spectrum substantially similar to FIG.23. 92. The substantially crystalline Compound I Hydrate Form A according to any one of Embodiments 74-92, characterized by a triclinic crystal system, P-1 space group, and unit cell dimensions measured at 100 K on a Bruker diffractometer utilizing Cu Kα radiation (λ=1.54178 Å) of: a 10.5 ± 0.1 Å α 91.3 ± 0.1° b 11.2 ± 0.1 Å β 103.9 ± 0.1° c 19.2 ± 0.1 Å γ 98.4 ± 0.1°. 93. The substantially crystalline Compound I Dehydrated Hydrate Form A according to any one of Embodiments 81-92, characterized by a triclinic crystal system, P-1 space group, and unit cell dimensions measured at 100 K on a Bruker diffractometer utilizing Cu Kα radiation (λ=1.54178 Å) of: a 10.4 ± 0.1 Å α 90.7 ± 0.1° b 11.2 ± 0.1 Å β 104.7 ± 0.1° c 19.3 ± 0.1 Å γ 98.0 ± 0.1°. 94. A pharmaceutical composition comprising Compound I according to any one of Embodiments 1-93, and optionally further comprising one or more additional therapeutic agents. 95. The pharmaceutical composition according to Embodiment 94, wherein the pharmaceutical composition comprises one or more additional pain modulating compounds. 96. The Compound I according to any one of Embodiments 1-93, or the pharmaceutical composition according to any one of Embodiments 94-95, for use in the treatment of pain. 97. Use of the Compound I according to any one of Embodiments 1-93, or the pharmaceutical composition according to any one of Embodiments 94-95, in the manufacture of a medicament for the treatment of pain.
98. A method of treating pain comprising administering the Compound I according to any one of Embodiments 1-93, or the pharmaceutical composition according to any one of Embodiments 94-95, to a subject in need thereof. 99. The compound or composition for use of Embodiment 96, the use of Embodiment 97, or the method of Embodiment 98, wherein the Compound I according to any one of Embodiments 1-93 or the composition according to any one of Embodiments 94-95 is administered in combination with one or more additional therapeutic agents. 100. The compound or composition for use, the use, or the method of Embodiment 99, wherein the one or more additional therapeutic agents comprises one or more additional pain modulating compounds. 101. A method of making crystalline Compound I neat Form A, comprising (i) combining Compound I and ethyl acetate, (ii) distilling under vacuum at 50 °C, (iii) adding ethyl acetate, (iv) distilling under vacuum at 50 °C, (v) heating to 75 °C, (vi) adding heptane, (vii) cooling to 40 °C, (viii) adding a seed of crystalline Compound I Form A, (ix) holding at 40 °C for 1.5 hours, (x) adding heptane over 5 hours, (xi) cooling the slurry to 20 °C over 5 hours, (xii) holding at 20 °C for 11 hours, (xiii) collecting the solids, (xiv) drying the solids in a vacuum oven at 45 °C with a slight nitrogen bleed for 46 hours to yield crystalline Compound I neat Form A. 102. A method of making crystalline Compound I neat Form B, comprising (i) adding Compound I hydrate Form A to an oven set at 180 °C (ii) cooling under ambient conditions to yield crystalline Compound I neat Form B. 103. A method of making crystalline Compound I neat Form E, comprising (i) combining Compound I hydrate Form A and trifluorotoluene, (ii) heating the slurry to 80 °C, (iii) cooling to room temperature to yield crystalline Compound I neat Form E. 104. A method of making crystalline Compound I Acetone Solvate Hydrate Form A, comprising (i) saturating acetone with Compound I, (ii) adding water dropwise to 20% of the final volume, (iii) holding overnight at room temperature to yield crystalline Compound I acetone solvate hydrate Form A. 105. A method of making crystalline Compound I Ethanol Solvate Form A, comprising (i) combining Compound I and ethyl acetate, (ii) heating the slurry to 50 °C, (iii) adding ethanol and distilling under vacuum at 50 °C, (iv) adding ethanol and distilling under vacuum at 50 °C, (v) adding ethanol and distilling under vacuum at 50 °C to a minimum volume, (vi) adding ethanol and distilling under vacuum at 50 °C, (vii) drying in a vacuum oven at 45 °C, with a slight nitrogen bleed for 42 hours to yield crystalline Compound I ethanol solvate Form A.
106. A method of making crystalline Compound I Dehydrated Hydrate Form A, comprising (i) combining Compound I Ethanol Solvate Form A and acetone, (ii) stirring the slurry while heating 50 °C, (iii) adding water over 1 hour, (iv) adding a seed of crystalline Compound I hydrate Form A, (v) agitating for 30 minutes, (vi) adding water over 8 hours, (vii) stirring at 50 °C for 5 hours, (viii) cooling the slurry to 20 °C over 3 hours, (ix) agitating for 2 hours, (x) collecting the solids and washing with 1:2 acetone:water, (xi) drying the solids in a vacuum over at 50 °C with a slight nitrogen bleed to yield crystalline Compound I hydrate Form A. 107. A method of making Compound I amorphous form, comprising (i) dissolving Compound I in acetone, (ii) filtering and evaporating the solvent at 50 °C, 12 mbar, over 1 hour on a centrifugal evaporation, (iii) drying in a vacuum oven at 50 °C overnight to yield Compound I amorphous form. [0469] Additional embodiments of the disclosure are set out in the following numbered clauses: 1. A process for preparing a compound of formula (I): , comprising transforming a
dihydropyrrolo[1,2-a]pyrazine-1,4'-piperidine]-6-yl]-2,2-dimethyl-propan-1-one, (4-isopropoxy-3- methyl-phenyl)-[2-methyl-6-(trifluoromethyl)spiro[3,4-dihydropyrrolo[1,2-a]pyrazine-1,4'- piperidine]-1'-yl]methanone, [2-methyl-6-(1-methylcyclopropanecarbonyl)spiro[3,4- dihydropyrrolo[1,2-a]pyrazine-1,4'-piperidine]-1'-yl]-[4-(3,3,3- trifluoropropoxymethyl)phenyl]methanone, 4-bromo-N-(4-bromophenyl)-3-[(1-methyl-2-oxo-4- piperidyl)sulfamoyl]benzamide or (3-chloro-4-isopropoxy-phenyl)-[2-methyl-6-(1,1,2,2,2- pentafluoroethyl)spiro[3,4-dihydropyrrolo[1,2-a]pyrazine-1,4'-piperidine]-1'-yl]methanone. [0428] (39) a NaV1.8 blocker, such as PF-04531083, PF-06372865 and such as those disclosed in WO2008/135826 (US2009048306), WO2006/011050 (US2008312235), WO2013/061205 (US2014296313), US20130303535, WO2013131018, US8466188, WO2013114250 (US2013274243), WO2014/120808 (US2014213616), WO2014/120815 (US2014228371) WO2014/120820 (US2014221435), WO2015/010065 (US20160152561), WO2015/089361 (US20150166589), WO2019/014352 (US20190016671), WO2018/213426, WO2020/146682, WO2020/146612, WO2020/014243, WO2020/014246, WO2020/092187, WO2020/092667 (US2020140411), WO2020/144375, WO2020/261114, WO2020/140959, WO2020/151728, WO2021/032074, WO2021/047622 (CN112479996), WO2021/257490, WO2021/257420, WO2021/257418, WO2022/263498, WO2022/235558, WO2022/235859, WO2023/138599, CN112390745, CN111808019, CN112225695, CN112457294, CN112300051, CN112300069, CN112441969, and CN114591293, the entire contents of each application hereby incorporated by reference; [0429] (39a) a NaV1.8 blocker such as 4,5-dichloro-2-(4-fluoro-2-methoxyphenoxy)-N-(2-oxo- 1,2-dihydropyridin-4-yl)benzamide, 2-(4-fluoro-2-methoxyphenoxy)-N-(2-oxo-1,2-dihydropyridin-4- yl)-4-(perfluoroethyl)benzamide, 4,5-dichloro-2-(4-fluorophenoxy)-N-(2-oxo-1,2-dihydropyridin-4- yl)benzamide, 4,5-dichloro-2-(3-fluoro-4-methoxyphenoxy)-N-(2-oxo-1,2-dihydropyridin-4- yl)benzamide, 2-(4-fluoro-2-methoxyphenoxy)-N-(2-oxo-1,2-dihydropyridin-4-yl)-5- (trifluoromethyl)benzamide, N-(2-oxo-1,2-dihydropyridin-4-yl)-2-(4-(trifluoromethoxy)phenoxy)-4- (trifluoromethyl)benzamide, 2-(4-fluorophenoxy)-N-(2-oxo-1,2-dihydropyridin-4-yl)-4- (perfluoroethyl)benzamide, 5-chloro-2-(4-fluoro-2-methoxyphenoxy)-N-(2-oxo-1,2-dihydropyridin-4- yl)benzamide, N-(2-oxo-1,2-dihydropyridin-4-yl)-2-(4-(trifluoromethoxy)phenoxy)-5- (trifluoromethyl)benzamide, 2-(4-fluoro-2-methylphenoxy)-N-(2-oxo-1,2-dihydropyridin-4-yl)-5-
(trifluoromethyl)benzamide, 2-(2-chloro-4-fluorophenoxy)-N-(2-oxo-1,2-dihydropyridin-4-yl)-5- (trifluoromethyl)benzamide, 5-chloro-2-(4-fluoro-2-methylphenoxy)-N-(2-oxo-1,2-dihydropyridin-4- yl)benzamide, 4-chloro-2-(4-fluoro-2-methylphenoxy)-N-(2-oxo-1,2-dihydropyridin-4-yl)benzamide, 5-chloro-2-(2-chloro-4-fluorophenoxy)-N-(2-oxo-1,2-dihydropyridin-4-yl)benzamide, 2-((5-fluoro-2- hydroxybenzyl)oxy)-N-(2-oxo-1,2-dihydropyridin-4-yl)-4-(trifluoromethyl)benzamide, N-(2-oxo-1,2- dihydropyridin-4-yl)-2-(o-tolyloxy)-5-(trifluoromethyl)benzamide, 2-(2,4-difluorophenoxy)-N-(2- oxo-1,2-dihydropyridin-4-yl)-4-(trifluoromethyl)benzamide, N-(2-oxo-1,2-dihydropyridin-4-yl)-2-(2- (trifluoromethoxy)phenoxy)-5-(trifluoromethyl)benzamide, 2-(4-fluorophenoxy)-N-(2-oxo-1,2- dihydropyridin-4-yl)-5-(trifluoromethyl)benzamide, 2-(4-fluoro-2-methyl-phenoxy)-N-(2-oxo-1H- pyridin-4-yl)-4-(trifluoromethyl)benzamide, [4-[[2-(4-fluoro-2-methyl-phenoxy)-4- (trifluoromethyl)benzoyl]amino]-2-oxo-1-pyridyl]methyl dihydrogen phosphate, 2-(4-fluoro-2- (methyl-d3)phenoxy)-N-(2-oxo-1,2-dihydropyridin-4-yl)-4-(trifluoromethyl)benzamide, (4-(2-(4- fluoro-2-(methyl-d3)phenoxy)-4-(trifluoromethyl)benzamido)-2-oxopyridin-1(2H)-yl)methyl dihydrogen phosphate, 3-(4-fluoro-2-methoxyphenoxy)-N-(3-(methylsulfonyl)phenyl)quinoxaline-2- carboxamide, 3-(2-chloro-4-fluorophenoxy)-N-(3-sulfamoylphenyl)quinoxaline-2-carboxamide, 3-(2- chloro-4-methoxyphenoxy)-N-(3-sulfamoylphenyl)quinoxaline-2-carboxamide, 3-(4-chloro-2- methoxyphenoxy)-N-(3-sulfamoylphenyl)quinoxaline-2-carboxamide, 4-(3-(4- (trifluoromethoxy)phenoxy)quinoxaline-2-carboxamido)picolinic acid, 2-(2,4-difluorophenoxy)-N-(3- sulfamoylphenyl)quinoline-3-carboxamide, 2-(4-fluoro-2-methoxyphenoxy)-N-(3- sulfamoylphenyl)quinoline-3-carboxamide, 3-(2,4-difluorophenoxy)-N-(3- sulfamoylphenyl)quinoxaline-2-carboxamide, N-(3-sulfamoylphenyl)-2-(4- (trifluoromethoxy)phenoxy)quinoline-3-carboxamide, N-(3-sulfamoylphenyl)-3-(4- (trifluoromethoxy)phenoxy)quinoxaline-2-carboxamide, 3-(4-chloro-2-methylphenoxy)-N-(3- sulfamoylphenyl)quinoxaline-2-carboxamide, 5-(3-(4-(trifluoromethoxy)phenoxy)quinoxaline-2- carboxamido)picolinic acid, 3-(4-fluoro-2-methoxyphenoxy)-N-(2-oxo-2,3-dihydro-1H- benzo[d]imidazol-5-yl)quinoxaline-2-carboxamide, 3-(4-fluoro-2-methoxyphenoxy)-N-(pyridin-4- yl)quinoxaline-2-carboxamide, 3-(4-fluorophenoxy)-N-(3-sulfamoylphenyl)quinoxaline-2- carboxamide, N-(3-cyanophenyl)-3-(4-fluoro-2-methoxyphenoxy)quinoxaline-2-carboxamide, N-(4- carbamoylphenyl)-3-(4-fluoro-2-methoxyphenoxy)quinoxaline-2-carboxamide, 4-(3-(4- (trifluoromethoxy)phenoxy)quinoxaline-2-carboxamido)benzoic acid, N-(4-cyanophenyl)-3-(4-fluoro- 2-methoxyphenoxy)quinoxaline-2-carboxamide, 5-(4,5-dichloro-2-(4-fluoro-2- methoxyphenoxy)benzamido)picolinic acid, 5-(2-(2,4-dimethoxyphenoxy)-4,6- bis(trifluoromethyl)benzamido)picolinic acid, 4-(4,5-dichloro-2-(4-fluoro-2- methoxyphenoxy)benzamido)benzoic acid, 5-(2-(4-fluoro-2-methoxyphenoxy)-4,6- bis(trifluoromethyl)benzamido)picolinic acid, 4-(2-(4-fluoro-2-methoxyphenoxy)-4- (perfluoroethyl)benzamido)benzoic acid, 5-(2-(4-fluoro-2-methoxyphenoxy)-4- (perfluoroethyl)benzamido)picolinic acid, 4-(2-(4-fluoro-2-methylphenoxy)-4-
(trifluoromethyl)benzamido)benzoic acid, 5-(4,5-dichloro-2-(4-fluoro-2- methoxyphenoxy)benzamido)picolinic acid, 4-(2-(2-chloro-4-fluorophenoxy)-4- (perfluoroethyl)benzamido)benzoic acid, 4-(2-(4-fluoro-2-methylphenoxy)-4- (perfluoroethyl)benzamido)benzoic acid, 4-(4,5-dichloro-2-(4- (trifluoromethoxy)phenoxy)benzamido)benzoic acid, 4-(4,5-dichloro-2-(4-chloro-2- methylphenoxy)benzamido)benzoic acid, 5-(4-(tert-butyl)-2-(4-fluoro-2- methoxyphenoxy)benzamido)picolinic acid, 5-(4,5-dichloro-2-(4- (trifluoromethoxy)phenoxy)benzamido)picolinic acid, 4-(4,5-dichloro-2-(4-fluoro-2- methylphenoxy)benzamido)benzoic acid, 5-(4,5-dichloro-2-(2,4- dimethoxyphenoxy)benzamido)picolinic acid, 5-(4,5-dichloro-2-(2-chloro-4- fluorophenoxy)benzamido)picolinic acid, 5-(4,5-dichloro-2-(4-fluoro-2- methylphenoxy)benzamido)picolinic acid, 4-(4,5-dichloro-2-(4-chloro-2- methoxyphenoxy)benzamido)benzoic acid, 5-(4,5-dichloro-2-(2,4- difluorophenoxy)benzamido)picolinic acid, 2-(4-fluorophenoxy)-N-(3-sulfamoylphenyl)-5- (trifluoromethyl)benzamide, 2-(4-fluorophenoxy)-N-(3-sulfamoylphenyl)-4- (trifluoromethyl)benzamide, 2-(2-chloro-4-fluorophenoxy)-N-(3-sulfamoylphenyl)-5- (trifluoromethyl)benzamide, 2-(4-fluorophenoxy)-N-(3-sulfamoylphenyl)-4- (trifluoromethyl)benzamide, 2-(2-chloro-4-fluorophenoxy)-N-(3-sulfamoylphenyl)-6- (trifluoromethyl)benzamide, 2-(2-chloro-4-fluorophenoxy)-5-(difluoromethyl)-N-(3- sulfamoylphenyl)benzamide, 2-(4-fluorophenoxy)-4-(perfluoroethyl)-N-(3- sulfamoylphenyl)benzamide, 2-(4-chloro-2-methoxyphenoxy)-4-(perfluoroethyl)-N-(3- sulfamoylphenyl)benzamide, 2-(4-fluoro-2-methoxyphenoxy)-N-(3-sulfamoylphenyl)-5- (trifluoromethyl)benzamide, 5-chloro-2-(4-fluoro-2-methylphenoxy)-N-(3- sulfamoylphenyl)benzamide, 4,5-dichloro-2-(4-fluoro-2-methoxyphenoxy)-N-(3- sulfamoylphenyl)benzamide, 2,4-dichloro-6-(4-chloro-2-methoxyphenoxy)-N-(3- sulfamoylphenyl)benzamide, 2,4-dichloro-6-(4-fluoro-2-methylphenoxy)-N-(3- sulfamoylphenyl)benzamide, 2-(4-fluoro-2-methoxyphenoxy)-N-(3-sulfamoylphenyl)-4,6- bis(trifluoromethyl)benzamide, 2-(4-fluoro-2-methylphenoxy)-N-(3-sulfamoylphenyl)-4,6- bis(trifluoromethyl)benzamide, 5-chloro-2-(2-chloro-4-fluorophenoxy)-N-(3- sulfamoylphenyl)benzamide, 2-(4-fluoro-2-methoxyphenoxy)-N-(3-sulfamoylphenyl)-4- (trifluoromethoxy)benzamide, 2-(4-fluoro-2-methoxyphenoxy)-N-(3-sulfamoylphenyl)-4- (trifluoromethyl)benzamide, 4,5-dichloro-2-(4-fluorophenoxy)-N-(3-sulfamoylphenyl)benzamide, 2- (4-fluoro-2-methoxyphenoxy)-4-(perfluoroethyl)-N-(3-sulfamoylphenyl)benzamide, 5-fluoro-2-(4- fluoro-2-methylphenoxy)-N-(3-sulfamoylphenyl)benzamide, 2-(2-chloro-4-fluorophenoxy)-4-cyano- N-(3-sulfamoylphenyl)benzamide, N-(3-sulfamoylphenyl)-2-(4-(trifluoromethoxy)phenoxy)-4- (trifluoromethyl)benzamide, N-(3-carbamoyl-4-fluoro-phenyl)-2-fluoro-6-[2-(trideuteriomethoxy)-4- (trifluoromethoxy)phenoxy]-3-(trifluoromethyl)benzamide, N-(3-carbamoyl-4-fluoro-phenyl)-2-
fluoro-6-[2-methoxy-4-(trifluoromethoxy)phenoxy]-3-(trifluoromethyl)benzamide, N-(3-carbamoyl- 4-fluoro-phenyl)-2-fluoro-6-[2-(trideuteriomethoxy)-4-(trifluoromethoxy)phenoxy]-3- (trifluoromethoxy)benzamide, 4-[[2-fluoro-6-[2-methoxy-4-(trifluoromethoxy)phenoxy]-3- (trifluoromethyl)benzoyl]amino]pyridine-2-carboxamide, 4-[[3-chloro-2-fluoro-6-[2-methoxy-4- (trifluoromethoxy)phenoxy]benzoyl]amino]pyridine-2-carboxamide, 4-[[2-fluoro-6-[2- (trideuteriomethoxy)-4-(trifluoromethoxy)phenoxy]-3-(trifluoromethyl)benzoyl]amino]pyridine-2- carboxamide, N-(3-carbamoyl-4-fluoro-phenyl)-3-(difluoromethyl)-2-fluoro-6-[2-methoxy-4- (trifluoromethoxy)phenoxy]benzamide, 4-[[2-fluoro-6-[2-(trideuteriomethoxy)-4- (trifluoromethoxy)phenoxy]-3-(trifluoromethoxy)benzoyl]amino]pyridine-2-carboxamide, N-(3- carbamoyl-4-fluoro-phenyl)-6-[2-chloro-4-(trifluoromethoxy)phenoxy]-2-fluoro-3- (trifluoromethyl)benzamide, N-(3-carbamoyl-4-fluoro-phenyl)-2-fluoro-6-[2-methyl-4- (trifluoromethoxy)phenoxy]-3-(trifluoromethyl)benzamide, N-(3-carbamoyl-4-fluoro-phenyl)-2,3,4- trifluoro-6-[2-methoxy-4-(trifluoromethoxy)phenoxy]benzamide, N-(2-carbamoyl-4-pyridyl)-3- fluoro-5-[2-methoxy-4-(trifluoromethoxy)phenoxy]-2-(trifluoromethyl)pyridine-4-carboxamide, 4- [[6-[2-(difluoromethoxy)-4-(trifluoromethoxy)phenoxy]-2-fluoro-3- (trifluoromethyl)benzoyl]amino]pyridine-2-carboxamide, N-(3-carbamoyl-4-fluoro-phenyl)-6-[3- chloro-4-(trifluoromethoxy)phenoxy]-2-fluoro-3-(trifluoromethyl)benzamide, N-(3-carbamoyl-4- fluoro-phenyl)-2-fluoro-6-[4-(trifluoromethoxy)phenoxy]-3-(trifluoromethyl)benzamide, N-(4- carbamoyl-3-fluoro-phenyl)-2-fluoro-6-[2-methoxy-4-(trifluoromethoxy)phenoxy]-3- (trifluoromethyl)benzamide, 4-[[2-fluoro-6-[2-(trideuteriomethoxy)-4-(trifluoromethoxy)phenoxy]-4- (trifluoromethyl)benzoyl]amino]pyridine-2-carboxamide, N-(3-carbamoyl-4-fluoro-phenyl)-2-fluoro- 6-[3-fluoro-4-(trifluoromethoxy)phenoxy]-3-(trifluoromethyl)benzamide, N-(3-carbamoyl-4-fluoro- phenyl)-2-[2-methoxy-4-(trifluoromethoxy)phenoxy]-5-(1,1,2,2,2-pentafluoroethyl)benzamide, 4-[[4- (difluoromethoxy)-2-fluoro-6-[2-methoxy-4-(trifluoromethoxy)phenoxy]benzoyl]amino]pyridine-2- carboxamide, N-(3-carbamoyl-4-fluoro-phenyl)-2-fluoro-6-[2-fluoro-4-(trifluoromethoxy)phenoxy]- 3-(trifluoromethyl)benzamide, 4-[[4-cyclopropyl-2-fluoro-6-[2-methoxy-4- (trifluoromethoxy)phenoxy]benzoyl]amino]pyridine-2-carboxamide, N-(3-carbamoyl-4-fluoro- phenyl)-5-fluoro-2-[2-methoxy-4-(trifluoromethoxy)phenoxy]-4-(trifluoromethyl)benzamide, 5-[[2- fluoro-6-[2-(trideuteriomethoxy)-4-(trifluoromethoxy)phenoxy]-3- (trifluoromethyl)benzoyl]amino]pyridine-2-carboxamide, N-(3-carbamoyl-4-fluoro-phenyl)-2-fluoro- 6-(4-fluorophenoxy)-3-(trifluoromethyl)benzamide, or 4-[[2-fluoro-6-[3-fluoro-2-methoxy-4- (trifluoromethoxy)phenoxy]-3-(trifluoromethyl)benzoyl]amino]pyridine-2-carboxamide; [0430] (40) a combined NaV1.7 and NaV1.8 blocker, such as DSP-2230, Lohocla201 or BL-1021; [0431] (41) a 5-HT3 antagonist, such as ondansetron; [0432] (42) a TPRV 1 receptor agonist, such as capsaicin (NeurogesX®, Qutenza®); and the pharmaceutically acceptable salts and solvates thereof; [0433] (43) a nicotinic receptor antagonist, such as varenicline;
[0434] (44) an N-type calcium channel antagonist, such as Z-160; [0435] (45) a nerve growth factor antagonist, such as tanezumab; [0436] (46) an endopeptidase stimulant, such as senrebotase; [0437] (47) an angiotensin II antagonist, such as EMA-401; [0438] (48) acetaminophen (including without limitation intravenous acetaminophen (e.g., Ofirmev®)); [0439] (49) bupivacaine (including without limitation bupivacaine liposome injectable suspension (e.g., Exparel®) bupivacaine ER (Posimir), bupivacaine collagen (Xaracoll) and transdermal bupivacaine (Eladur®)); and [0440] (50) bupivacaine and meloxicam combination (e.g., HTX-011). [0441] In one embodiment, the additional appropriate therapeutic agents are selected from V- 116517, Pregabalin, controlled release Pregabalin, Ezogabine (Potiga®). Ketamine/amitriptyline topical cream (Amiket®), AVP-923, Perampanel (E-2007), Ralfinamide, transdermal bupivacaine (Eladur®), CNV1014802, JNJ-10234094 (Carisbamate), BMS-954561 or ARC-4558. [0442] In another embodiment, the additional appropriate therapeutic agents are selected from N- (6-amino-5-(2,3,5-trichlorophenyl)pyridin-2-yl)acetamide; N-(6-amino-5-(2-chloro-5- methoxyphenyl)pyridin-2-yl)-1-methyl-1H-pyrazole-5-carboxamide; or 3-((4-(4- (trifluoromethoxy)phenyl)-1H-imidazol-2-yl)methyl)oxetan-3-amine. [0443] In another embodiment, the additional therapeutic agent is selected from a GlyT2/5HT2 inhibitor, such as Operanserin (VVZ149), a TRPV modulator such as CA008, CMX-020, NEO6860, FTABS, CNTX4975, MCP101, MDR16523, or MDR652, a EGR1 inhibitor such as Brivoglide (AYX1), an NGF inhibitor such as Tanezumab, Fasinumab, ASP6294, MEDI7352, a Mu opioid agonist such as Cebranopadol, NKTR181 (oxycodegol), a CB-1 agonist such as NEO1940 (AZN1940), an imidazoline 12 agonist such as CR4056 or a p75NTR-Fc modulator such as LEVI-04. [0444] In another embodiment, the additional therapeutic agent is oliceridine or ropivacaine (TLC590). [0445] In another embodiment, the additional therapeutic agent is a NaV1.7 blocker such as ST- 2427, ST-2578 and those disclosed in WO2010/129864, WO2015/157559, WO2017/059385, WO2018/183781, WO2018/183782, WO2020/072835, and/or WO2022/036297 the entire contents of each application hereby incorporated by reference. [0446] In another embodiment, the additional therapeutic agent is selected from ASP18071, CC- 8464, ANP-230, ANP-231, NOC-100, NTX-1175, ASN008, NW3509, AM-6120, AM-8145, AM- 0422, BL-017881, NTM-006, Opiranserin (UnafraTM), brivoligide, SR419, NRD.E1, LX9211, LY3016859, ISC-17536, NFX-88, LAT-8881, AP-235, NYX 2925, CNTX-6016, S-600918, S- 637880, RQ-00434739, KLS-2031, MEDI 7352, and XT-150. [0447] In another embodiment, the additional therapeutic agent is selected from Olinvyk, Zynrelef, Seglentis, Neumentum, Nevakar, HTX-034, CPL-01, ACP-044, HRS-4800, Tarlige,
BAY2395840, LY3526318, Eliapixant, TRV045, RTA901, NRD1355-E1, MT-8554, LY3556050, AP-325, tetrodotoxin, Otenaproxesul, CFTX-1554, Funapide, iN1011-N17, JMKX000623/ODM- 111, ETX-801, OLP-1002, ANP-230/DSP-2230, iN1011-N17, DSP-3905 and ACD440. [0448] In another embodiment, the additional therapeutic agent is selected from HRS4800, ODM-111/JMKX000623, LX9211, LY3556050, LY3857210, CFTX01554/CFTX-1554, MEDI7352, MEDI0618, BAY3178275, BAY2395840, GSK3858279, STC-004, HALNEURON, OLP-1002, ATX01, ANP230, CC-8464, iN1011-N17, ST-2427, MSD199, FZ008, VYNAV-01, BL-017881, Profervia (Cilnidipine), LS-04, vixotrigine, FX301/PCRX-301, PF-04531083, PF-01247324, and DSP-3905. [0449] In another embodiment, the additional therapeutic agent is a sodium channel inhibitor (also known as a sodium channel blocker), such as the NaV1.7 and NaV1.8 blockers identified above. [0450] The amount of additional therapeutic agent present in the compositions of this invention may be no more than the amount that would normally be administered in a composition comprising that therapeutic agent as the only active agent. The amount of additional therapeutic agent in the presently disclosed compositions may range from about 10% to 100% of the amount normally present in a composition comprising that agent as the only therapeutically active agent. [0451] The compounds and salts of this invention or pharmaceutically acceptable compositions thereof may also be incorporated into compositions for coating an implantable medical device, such as prostheses, artificial valves, vascular grafts, stents and catheters. Accordingly, the invention, in another aspect, includes a composition for coating an implantable device comprising a compound or salt of the invention as described generally above, and in classes and subclasses herein, and a carrier suitable for coating said implantable device. In still another aspect, the invention includes an implantable device coated with a composition comprising a compound or salt of the invention as described generally above, and in classes and subclasses herein, and a carrier suitable for coating said implantable device. Suitable coatings and the general preparation of coated implantable devices are described in US Patents 6,099,562; 5,886,026; and 5,304,121. The coatings are typically biocompatible polymeric materials such as a hydrogel polymer, polymethyldisiloxane, polycaprolactone, polyethylene glycol, polylactic acid, ethylene vinyl acetate, and mixtures thereof. The coatings may optionally be further covered by a suitable topcoat of fluorosilicone, polysaccharides, polyethylene glycol, phospholipids or combinations thereof to impart controlled release characteristics in the composition. [0452] Another aspect of the invention relates to inhibiting NaV1.8 activity in a biological sample or a subject, which method comprises administering to the subject, or contacting said biological sample with a compound of the invention, a pharmaceutically acceptable salt thereof, or a pharmaceutical composition thereof. The term “biological sample,” as used herein, includes, without limitation, cell cultures or extracts thereof; biopsied material obtained from a mammal or extracts thereof; and blood, saliva, urine, feces, semen, tears, or other body fluids or extracts thereof.
[0453] Inhibition of NaV1.8 activity in a biological sample is useful for a variety of purposes that are known to one of skill in the art. Examples of such purposes include, but are not limited to, the study of sodium channels in biological and pathological phenomena; and the comparative evaluation of new sodium channel inhibitors. K. Pharmaceutical Compositions [0454] Another aspect of the invention provides pharmaceutical compositions comprising Compound I in any one of the pharmaceutically acceptable solid (e.g. crystalline or amorphous) forms disclosed herein. In some embodiments, the pharmaceutical composition comprises Compound I in a solid form selected from Compound I neat Form A, Compound I neat Form B, Compound I neat Form E, Compound I Acetone Solvate Hydrate Form A, Compound I Ethanol Solvate Form A, Compound I Hydrate Form A, and Compound I Dehydrated Hydrate Form A. In some embodiments, the pharmaceutical composition comprises Compound I in a solid amorphous form that is Compound I amorphous form. [0455] In some embodiments, the invention provides a pharmaceutical composition comprising (a) Compound I in any one of the pharmaceutically acceptable solid (e.g., crystalline or amorphous) forms disclosed herein, and (b) at least one pharmaceutically acceptable carrier. [0456] In some embodiments, the invention provides pharmaceutical compositions comprising (a) Compound I in a solid form selected from Compound I neat Form A, Compound I neat Form B, Compound I neat Form E, Compound I Acetone Solvate Hydrate Form A, Compound I Ethanol Solvate Form A, Compound I Hydrate Form A, and Compound I Dehydrated Hydrate Form A and (b) at least one pharmaceutically acceptable carrier. [0457] As described herein, the pharmaceutically acceptable compositions of the invention additionally comprise a pharmaceutically acceptable carrier, adjuvant, or vehicle, which, as used herein, includes any and all solvents, diluents, or other liquid vehicle, dispersion or suspension aids, surface active agents, isotonic agents, thickening or emulsifying agents, preservatives, solid binders, lubricants and the like, as suited to the particular dosage form desired. Remington’s Pharmaceutical Sciences, Sixteenth Edition, E. W. Martin (Mack Publishing Co., Easton, Pa., 1980) discloses various carriers used in formulating pharmaceutically acceptable compositions and known techniques for the preparation thereof. Except insofar as any conventional carrier medium is incompatible with the compounds of the invention, such as by producing any undesirable biological effect or otherwise interacting in a deleterious manner with any other component(s) of the pharmaceutically acceptable composition, its use is contemplated to be within the scope of this invention. Some examples of materials which can serve as pharmaceutically acceptable carriers include, but are not limited to, ion exchangers, alumina, aluminum stearate, lecithin, serum proteins, such as human serum albumin, buffer substances such as phosphates, glycine, sorbic acid, or potassium sorbate, partial glyceride mixtures of saturated vegetable fatty acids, water, salts or electrolytes, such as protamine sulfate, disodium
hydrogen phosphate, potassium hydrogen phosphate, sodium chloride, zinc salts, colloidal silica, magnesium trisilicate, polyvinyl pyrrolidone, polyacrylates, waxes, polyethylene-polyoxypropylene- block polymers, wool fat, sugars such as lactose, glucose and sucrose; starches such as corn starch and potato starch; cellulose and its derivatives such as sodium carboxymethyl cellulose, ethyl cellulose and cellulose acetate; powdered tragacanth; malt; gelatin; talc; excipients such as cocoa butter and suppository waxes; oils such as peanut oil, cottonseed oil; safflower oil; sesame oil; olive oil; corn oil and soybean oil; glycols; such a propylene glycol or polyethylene glycol; esters such as ethyl oleate and ethyl laurate; agar; buffering agents such as magnesium hydroxide and aluminum hydroxide; alginic acid; pyrogen-free water; isotonic saline; Ringer’s solution; ethyl alcohol, and phosphate buffer solutions, as well as other non-toxic compatible lubricants such as sodium lauryl sulfate and magnesium stearate, as well as coloring agents, releasing agents, coating agents, sweetening, flavoring and perfuming agents, preservatives and antioxidants can also be present in the composition, according to the judgment of the formulator. L. Synthesis of the Compounds [0458] The compounds of the invention can be prepared from known materials by the methods described in the Examples, other similar methods, and other methods known to one skilled in the art. As one skilled in the art would appreciate, the functional groups of the intermediate compounds may need to be protected by suitable protecting groups. Protecting groups may be added or removed in accordance with standard techniques, which are well-known to those skilled in the art. The use of protecting groups is described in detail in T.G.M. Wuts et al., Greene’s Protective Groups in Organic Synthesis (4th ed.2006). M. Radiolabeled Analogs of the Compounds [0459] In another aspect, the invention relates to radiolabeled analogs of the compounds of the invention. As used herein, the term “radiolabeled analogs of the compounds of the invention” refers to compounds that are identical to the compounds of the invention, as described herein including all embodiments thereof, except that one or more atoms has been replaced with a radioisotope of the atom present in the compounds of the invention. [0460] As used herein, the term “radioisotope” refers to an isotope of an element that is known to undergo spontaneous radioactive decay. Examples of radioisotopes include 3H, 14C, 32P, 35S, 18F, 36Cl, and the like, as well as the isotopes for which a decay mode is identified in V.S. Shirley & C.M. Lederer, Isotopes Project, Nuclear Science Division, Lawrence Berkeley Laboratory, Table of Nuclides (January 1980). [0461] The radiolabeled analogs can be used in a number of beneficial ways, including in various types of assays, such as substrate tissue distribution assays. For example, tritium (3H)- and/or carbon-
14 (14C)-labeled compounds may be useful for various types of assays, such as substrate tissue distribution assays, due to relatively simple preparation and excellent detectability. [0462] In another aspect, the invention relates to pharmaceutically acceptable salts of the radiolabeled analogs, in accordance with any of the embodiments described herein in connection with the compounds of the invention. [0463] In another aspect, the invention relates to pharmaceutical compositions comprising the radiolabeled analogs, or pharmaceutically acceptable salts thereof, and a pharmaceutically acceptable carrier, adjuvant or vehicle, in accordance with any of the embodiments described herein in connection with the compounds of the invention. [0464] In another aspect, the invention relates to methods of inhibiting voltage-gated sodium channels and methods of treating or lessening the severity of various diseases and disorders, including pain, in a subject comprising administering an effective amount of the radiolabeled analogs, pharmaceutically acceptable salts thereof, and pharmaceutical compositions thereof, in accordance with any of the embodiments described herein in connection with the compounds of the invention. [0465] In another aspect, the invention relates to radiolabeled analogs, pharmaceutically acceptable salts thereof, and pharmaceutical compositions thereof, for use, in accordance with any of the embodiments described herein in connection with the compounds of the invention. [0466] In another aspect, the invention relates to the use of the radiolabeled analogs, or pharmaceutically acceptable salts thereof, and pharmaceutical compositions thereof, for the manufacture of medicaments, in accordance with any of the embodiments described herein in connection with the compounds of the invention. [0467] In another aspect, the radiolabeled analogs, pharmaceutically acceptable salts thereof, and pharmaceutical compositions thereof, can be employed in combination therapies, in accordance with any of the embodiments described herein in connection with the compounds of the invention. N. Non-limiting Exemplary Embodiments [0468] Further embodiments of the disclosure are set out in the following numbered Embodiments: 1. Compound I I)
as substantially amorphous Compound I amorphous form (i.e., wherein less than 15% of Compound I is in crystalline form, wherein less than 10% of Compound I is in crystalline form, wherein less than 5% of Compound I is in crystalline form). 2. The substantially amorphous Compound I amorphous form according to Embodiment 1, wherein Compound I is 100% amorphous. 3. The substantially amorphous Compound I amorphous form according to Embodiment 1 or Embodiment 2, characterized by an X-ray powder diffractogram substantially similar to FIG.1. 4. The substantially amorphous Compound I amorphous form according to any one of Embodiments 1-3, characterized by a 13C SSNMR spectrum having one, two, three, four, five, six, seven, eight, nine, ten, or more peaks selected from 177.6 ± 0.2 ppm, 172.0 ± 0.2 ppm, 162.6 ± 0.2 ppm, 155.2 ± 0.2 ppm, 149.9 ± 0.2 ppm, 147.0 ± 0.2 ppm, 121.0 ± 0.2 ppm, 120.1 ± 0.2 ppm, 119.1 ± 0.2 ppm, 116.0 ± 0.2 ppm, 114.6 ± 0.2 ppm, 113.8 ± 0.2 ppm, 15.5 ± 0.2 ppm, and 7.7 ± 0.2 ppm. 5. The substantially amorphous Compound I amorphous form according to any one of Embodiments 1-4, characterized by a 13C SSNMR spectrum having peaks at 177.6 ± 0.2 ppm, 172.0 ± 0.2 ppm, 162.6 ± 0.2 ppm, 155.2 ± 0.2 ppm, 149.9 ± 0.2 ppm, 147.0 ± 0.2 ppm, 121.0 ± 0.2 ppm, 120.1 ± 0.2 ppm, 119.1 ± 0.2 ppm, 116.0 ± 0.2 ppm, 114.6 ± 0.2 ppm, 113.8 ± 0.2 ppm, 15.5 ± 0.2 ppm, and 7.7 ± 0.2 ppm. 6. The substantially amorphous Compound I amorphous form according to any one of Embodiments 1-5, characterized by a 13C SSNMR spectrum substantially similar to FIG.2. 7. The substantially amorphous Compound I amorphous form according to any one of Embodiments 1-6, characterized by a 19F SSNMR spectrum having one or two peaks selected from -61.9 ± 0.2 ppm and -142.1 ± 0.2 ppm. 8. The substantially amorphous Compound I amorphous form according to any one of Embodiments 1-7, characterized by a 19F SSNMR spectrum substantially similar to FIG.3. 9. Substantially crystalline Compound I neat Form A (i.e., wherein less than 15% of Compound I is in amorphous form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous form). 10. The substantially crystalline Compound I neat Form A according to Embodiment 9, wherein Compound I neat Form A is 100% crystalline.
11. The substantially crystalline Compound I neat Form A according to Embodiment 9 or Embodiment 10, characterized by an X-ray powder diffractogram having one or two signals selected from 9.5 ± 0.2 degrees two-theta, 12.3 ± 0.2 degrees two-theta, and 13.5 ± 0.2 degrees two-theta. 12. The substantially crystalline Compound I neat Form A according to any one of Embodiments 9-11, characterized by an X-ray powder diffractogram having (a) one or two signals selected from 9.5 ± 0.2 degrees two-theta, 12.3 ± 0.2 degrees two-theta, and 13.5 ± 0.2 degrees two-theta, and (b) one or two signals selected from 11.3 ± 0.2 degrees two-theta, 15.9 ± 0.2 degrees two-theta, and 20.5 ± 0.2 degrees two-theta. 13. The substantially crystalline Compound I neat Form A according to any one of Embodiments 9-12, characterized by an X-ray powder diffractogram having two, three, four, five, or six signals selected from 9.5 ± 0.2 degrees two-theta, 12.3 ± 0.2 degrees two-theta, 13.5 ± 0.2 degrees two-theta, 11.3 ± 0.2 degrees two-theta, 15.9 ± 0.2 degrees two-theta, 20.5 ± 0.2 degrees two-theta, 14.6 ± 0.2 degrees two-theta, 16.6 ± 0.2 degrees two-theta, and 23.5 ± 0.2 degrees two-theta. 14. The substantially crystalline Compound I neat Form A according to any one of Embodiments 9-13, characterized by an X-ray powder diffractogram substantially similar to FIG.4. 15. The substantially crystalline Compound I neat Form A according to any one of Embodiments 9-14, characterized by a 13C SSNMR spectrum having one, two, three, four, five, six, seven, eight, nine, ten, or more peaks selected from 170.4 ± 0.2 ppm, 163.8 ± 0.2 ppm, 162.4 ± 0.2 ppm, 160.9 ± 0.2 ppm, 158.6 ± 0.2 ppm, 158.3 ± 0.2 ppm, 154.6 ± 0.2 ppm, 154.0 ± 0.2 ppm, 148.3 ± 0.2 ppm, 147.6 ± 0.2 ppm, 146.9 ± 0.2 ppm, 145.9 ± 0.2 ppm, 143.5 ± 0.2 ppm, 143.1 ± 0.2 ppm, 141.9 ± 0.2 ppm, 127.4 ± 0.2 ppm, 125.3 ± 0.2 ppm, 123.6 ± 0.2 ppm, 122.2 ± 0.2 ppm, 121.3 ± 0.2 ppm, 120.7 ± 0.2 ppm, 117.9 ± 0.2 ppm, 117.3 ± 0.2 ppm, 116.3 ± 0.2 ppm, 115.8 ± 0.2 ppm, 114.2 ± 0.2 ppm, 113.6 ± 0.2 ppm, 112.8 ± 0.2 ppm, 15.4 ± 0.2 ppm, and 8.2 ± 0.2 ppm. 16. The substantially crystalline Compound I neat Form A according to any one of Embodiments 9-15, characterized by a 13C SSNMR spectrum having peaks at 170.4 ± 0.2 ppm, 163.8 ± 0.2 ppm, 162.4 ± 0.2 ppm, 160.9 ± 0.2 ppm, 158.6 ± 0.2 ppm, 158.3 ± 0.2 ppm, 154.6 ± 0.2 ppm, 154.0 ± 0.2 ppm, 148.3 ± 0.2 ppm, 147.6 ± 0.2 ppm, 146.9 ± 0.2 ppm, 145.9 ± 0.2 ppm, 143.5 ± 0.2 ppm, 143.1 ± 0.2 ppm, 141.9 ± 0.2 ppm, 127.4 ± 0.2 ppm, 125.3 ± 0.2 ppm, 123.6 ± 0.2 ppm, 122.2 ± 0.2 ppm, 121.3 ± 0.2 ppm, 120.7 ± 0.2 ppm, 117.9 ± 0.2 ppm, 117.3 ± 0.2 ppm, 116.3 ± 0.2 ppm, 115.8 ± 0.2 ppm, 114.2 ± 0.2 ppm, 113.6 ± 0.2 ppm, 112.8 ± 0.2 ppm, 15.4 ± 0.2 ppm, and 8.2 ± 0.2 ppm. 17. The substantially crystalline Compound I neat Form A according to any one of Embodiments 9-16, characterized by a 13C SSNMR spectrum substantially similar to FIG.5.
18. The substantially crystalline Compound I neat Form A according to any one of Embodiments 9-17, characterized as having a 19F SSNMR spectrum with one or two peaks selected from -61.2 ± 0.2 ppm, -61.6 ± 0.2 ppm, -138.6 ± 0.2 ppm, -140.3 ± 0.2 ppm, -142.0 ± 0.2 ppm, and-146.0 ± 0.2 ppm. 19. The substantially crystalline Compound I neat Form A according to any one of Embodiments 9-18, characterized as having a 19F SSNMR spectrum having peaks at -61.2 ± 0.2 ppm, -61.6 ± 0.2 ppm, -138.6 ± 0.2 ppm, -140.3 ± 0.2 ppm, -142.0 ± 0.2 ppm, and-146.0 ± 0.2 ppm. 20. The substantially crystalline Compound I neat Form A according to any one of Embodiments 9-19, characterized by a 19F SSNMR spectrum substantially similar to FIG.6. 21. The substantially crystalline Compound I neat Form A according to any one of Embodiments 9-20, characterized by a triclinic crystal system, P-1 space group, and unit cell dimensions measured at 100 K on a Bruker diffractometer utilizing Cu Kα radiation (λ=1.54178 Å) of: a 9.6 ± 0.1 Å α 94.3 ± 0.1° b 12.6 ± 0.1 Å β 97.3 ± 0.1° c 18.7 ± 0.1 Å γ 107.3 ± 0.1°. 22. Substantially crystalline Compound I neat Form B (i.e., wherein less than 15% of Compound I is in amorphous form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous form). 23. The substantially crystalline Compound I neat Form B according to Embodiment 22, wherein Compound I neat Form B is 100% crystalline. 24. The substantially crystalline Compound I neat Form B according to Embodiment 22 or Embodiment 23, characterized by an X-ray powder diffractogram having one or two signals selected from 4.8 ± 0.2 degrees two-theta, 13.3 ± 0.2 degrees two-theta, and 18.5 ± 0.2 degrees two-theta. 25. The substantially crystalline Compound I neat Form B according to any one of Embodiments 22-24, characterized by an X-ray powder diffractogram having (a) one or two signals selected from 4.8 ± 0.2 degrees two-theta, 13.3 ± 0.2 degrees two-theta, and 18.5 ± 0.2 degrees two-theta, and (b) one or two signals selected from 6.0 ± 0.2 degrees two-theta, 16.5 ± 0.2 degrees two-theta, and 16.9 ± 0.2 degrees two-theta. 26. The substantially crystalline Compound I neat Form B according to any one of Embodiments 22-25, characterized by an X-ray powder diffractogram having two, three, four, five, or six signals selected from 4.8 ± 0.2 degrees two-theta, 13.3 ± 0.2 degrees two-theta, 18.5 ± 0.2 degrees two-theta, 6.0 ± 0.2 degrees two-theta, 16.5 ± 0.2 degrees two-theta, 16.9 ± 0.2 degrees two-theta, 14.1 ± 0.2 degrees two-theta, 14.7 ± 0.2 degrees two-theta, and 15.2 ± 0.2 degrees two-theta.
27. The substantially crystalline Compound I neat Form B according to any one of Embodiments 22-26, characterized by an X-ray powder diffractogram substantially similar to FIG.7. 28. The substantially crystalline Compound I neat Form B according to any one of Embodiments 22-27, characterized by a 13C SSNMR spectrum having one, two, three, four, five, six, seven, eight, nine, ten, or more peaks selected from 176.9 ± 0.2 ppm, 171.7 ± 0.2 ppm, 164.0 ± 0.2 ppm, 161.4 ± 0.2 ppm, 160.7 ± 0.2 ppm, 155.3 ± 0.2 ppm, 154.8 ± 0.2 ppm, 148.6 ± 0.2 ppm, 147.6 ± 0.2 ppm, 146.4 ± 0.2 ppm, 145.7 ± 0.2 ppm, 144.8 ± 0.2 ppm, 127.7 ± 0.2 ppm, 125.4 ± 0.2 ppm, 122.0 ± 0.2 ppm, 121.5 ± 0.2 ppm, 118.4 ± 0.2 ppm, 116.3 ± 0.2 ppm, 114.8 ± 0.2 ppm, 113.8 ± 0.2 ppm, 112.6 ± 0.2 ppm, 17.8 ± 0.2 ppm, 15.0 ± 0.2 ppm, 10.7 ± 0.2 ppm, 9.6 ± 0.2 ppm, and 8.8 ± 0.2 ppm. 29. The substantially crystalline Compound I neat Form B according to any one of Embodiments 22-28, characterized by a 13C SSNMR spectrum having peaks at 176.9 ± 0.2 ppm, 171.7 ± 0.2 ppm, 164 ± 0.2 ppm, 161.4 ± 0.2 ppm, 160.7 ± 0.2 ppm, 155.3 ± 0.2 ppm, 154.8 ± 0.2 ppm, 148.6 ± 0.2 ppm, 147.6 ± 0.2 ppm, 146.4 ± 0.2 ppm, 145.7 ± 0.2 ppm, 144.8 ± 0.2 ppm, 127.7 ± 0.2 ppm, 125.4 ± 0.2 ppm, 122.0 ± 0.2 ppm, 121.5 ± 0.2 ppm, 118.4 ± 0.2 ppm, 116.3 ± 0.2 ppm, 114.8 ± 0.2 ppm, 113.8 ± 0.2 ppm, 112.6 ± 0.2 ppm, 17.8 ± 0.2 ppm, 15.0 ± 0.2 ppm, 10.7 ± 0.2 ppm, 9.6 ± 0.2 ppm, and 8.8 ± 0.2 ppm. 30. The substantially crystalline Compound I neat Form B according to any one of Embodiments 22-29, characterized by a 13C SSNMR spectrum substantially similar to FIG.8. 31. The substantially crystalline Compound I neat Form B according to any one of Embodiments 22-30, characterized as having a 19F SSNMR spectrum with one or two peaks selected from -61.6 ± 0.2 ppm, -138 ± 0.2 ppm, -139.7 ± 0.2 ppm, -143.4 ± 0.2 ppm, and -144.9 ± 0.2 ppm. 32. The substantially crystalline Compound I neat Form B according to any one of Embodiments 22-31, characterized as having a 19F SSNMR spectrum having peaks at -61.6 ± 0.2 ppm, -138 ± 0.2 ppm, -139.7 ± 0.2 ppm, -143.4 ± 0.2 ppm, and -144.9 ± 0.2 ppm. 33. The substantially crystalline Compound I neat Form B according to any one of Embodiments 22-32, characterized by a 19F SSNMR spectrum substantially similar to FIG.9. 34. Substantially crystalline Compound I neat Form E (i.e., wherein less than 15% of Compound I is in amorphous form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous form). 35. The substantially crystalline Compound I neat Form E according to Embodiment 34, wherein Compound I neat Form E is 100% crystalline.
36. The substantially crystalline Compound I neat Form E according to Embodiment 34 or Embodiment 35, characterized by an X-ray powder diffractogram having one or two signals selected from 8.6 ± 0.2 degrees two-theta, 10.1 ± 0.2 degrees two-theta, and 12.6 ± 0.2 degrees two-theta. 37. The substantially crystalline Compound I neat Form E according to any one of Embodiments 34-36, characterized by an X-ray powder diffractogram having (a) one or two signals selected from 8.6 ± 0.2 degrees two-theta, 10.1 ± 0.2 degrees two-theta, and 12.6 ± 0.2 degrees two-theta, and (b) one or two signals selected from 14.1 ± 0.2 degrees two-theta, 20.3 ± 0.2 degrees two-theta, and 23.2 ± 0.2 degrees two-theta. 38. The substantially crystalline Compound I neat Form E according to any one of Embodiments 34-37, characterized by an X-ray powder diffractogram having two, three, four, five, or six signals selected from 8.6 ± 0.2 degrees two-theta, 10.1 ± 0.2 degrees two-theta, 12.6 ± 0.2 degrees two-theta, 14.1 ± 0.2 degrees two-theta, 20.3 ± 0.2 degrees two-theta, 23.2 ± 0.2 degrees two-theta, 11.5 ± 0.2 degrees two-theta, 14.2 ± 0.2 degrees two-theta, and 15.1 ± 0.2 degrees two-theta. 39. The substantially crystalline Compound I neat Form E according to any one of Embodiments 34-38, characterized by an X-ray powder diffractogram substantially similar to FIG.10. 40. The substantially crystalline Compound I neat Form E according to any one of Embodiments 34-39, characterized by a 13C SSNMR spectrum having one, two, three, four, five, six, seven, eight, nine, ten, or more peaks selected from 170.0 ± 0.2 ppm, 163.0 ± 0.2 ppm, 162.3 ± 0.2 ppm, 161.3 ± 0.2 ppm, 159.3 ± 0.2 ppm, 154.7 ± 0.2 ppm, 150.6 ± 0.2 ppm, 149.8 ± 0.2 ppm, 148.6 ± 0.2 ppm, 148.3 ± 0.2 ppm, 147.1 ± 0.2 ppm, 144.9 ± 0.2 ppm, 143.9 ± 0.2 ppm, 143.5 ± 0.2 ppm, 126.9 ± 0.2 ppm, 126.0 ± 0.2 ppm, 124.1 ± 0.2 ppm, 122.6 ± 0.2 ppm, 120.8 ± 0.2 ppm, 117.8 ± 0.2 ppm, 116.7 ± 0.2 ppm, 115.2 ± 0.2 ppm, 114.1 ± 0.2 ppm, 113.1 ± 0.2 ppm, 15.9 ± 0.2 ppm, 15.3 ± 0.2 ppm, and 8.4 ± 0.2 ppm. 41. The substantially crystalline Compound I neat Form E according to any one of Embodiments 34-40, characterized by a 13C SSNMR spectrum having peaks at 170.0 ± 0.2 ppm, 163.0 ± 0.2 ppm, 162.3 ± 0.2 ppm, 161.3 ± 0.2 ppm, 159.3 ± 0.2 ppm, 154.7 ± 0.2 ppm, 150.6 ± 0.2 ppm, 149.8 ± 0.2 ppm, 148.6 ± 0.2 ppm, 148.3 ± 0.2 ppm, 147.1 ± 0.2 ppm, 144.9 ± 0.2 ppm, 143.9 ± 0.2 ppm, 143.5 ± 0.2 ppm, 126.9 ± 0.2 ppm, 126.0 ± 0.2 ppm, 124.1 ± 0.2 ppm, 122.6 ± 0.2 ppm, 120.8 ± 0.2 ppm, 117.8 ± 0.2 ppm, 116.7 ± 0.2 ppm, 115.2 ± 0.2 ppm, 114.1 ± 0.2 ppm, 113.1 ± 0.2 ppm, 15.9 ± 0.2 ppm, 15.3 ± 0.2 ppm, and 8.4 ± 0.2 ppm. 42. The substantially crystalline Compound I neat Form E according to any one of Embodiments 34-41, characterized by a 13C SSNMR spectrum substantially similar to FIG.11.
43. The substantially crystalline Compound I neat Form E according to any one of Embodiments 34-42, characterized as having a 19F SSNMR spectrum with one or two peaks selected from -60.9 ± 0.2 ppm, -62.5 ± 0.2 ppm, -63.1 ± 0.2 ppm, -135.1 ± 0.2 ppm, -137.1 ± 0.2 ppm, -140.7 ± 0.2 ppm, and -141.9 ± 0.2 ppm. 44. The substantially crystalline Compound I neat Form E according to any one of Embodiments 34-43, characterized as having a 19F SSNMR spectrum having peaks at -60.9 ± 0.2 ppm, -62.5 ± 0.2 ppm, -63.1 ± 0.2 ppm, -135.1 ± 0.2 ppm, -137.1 ± 0.2 ppm, -140.7 ± 0.2 ppm, and -141.9 ± 0.2 ppm. 45. The substantially crystalline Compound I neat Form E according to any one of Embodiments 34-44, characterized by a 19F SSNMR spectrum substantially similar to FIG.12. 46. The substantially crystalline Compound I neat Form E according to any one of Embodiments 34-45, characterized by a triclinic crystal system, P-1 space group, and unit cell dimensions measured at 100 K on a Rigaku diffractometer utilizing Cu Kα radiation (λ=1.54178 Å) of: a 8.7 ± 0.1 Å α 92.7 ± 0.1° b 12.2 ± 0.1 Å β 96.3 ± 0.1° c 20.9 ± 0.1 Å γ 98.6 ± 0.1°. 47. The substantially crystalline Compound I neat Form E according to any one of Embodiments 34-46, characterized by a triclinic crystal system, P-1 space group, and unit cell dimensions measured at 298 K on a Rigaku diffractometer utilizing Cu Kα radiation (λ=1.54178 Å) of: a 8.9 ± 0.1 Å α 92.6 ± 0.1° b 12.3 ± 0.1 Å β 97.1 ± 0.1° c 21.1 ± 0.1 Å γ 98.5 ± 0.1°. 48. Substantially crystalline Compound I Acetone Solvate Hydrate Form A (i.e., wherein less than 15% of Compound I is in amorphous form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous form). 49. The substantially crystalline Compound I Acetone Solvate Hydrate Form A according to Embodiment 48, wherein Compound I Acetone Solvate Hydrate is 100% crystalline. 50. The substantially crystalline Compound I Acetone Solvate Hydrate Form A according to Embodiment 48 or Embodiment 49, characterized by an X-ray powder diffractogram having one or two signals selected from 17.2 ± 0.2 degrees two-theta, 18.1 ± 0.2 degrees two-theta, and 25.9 ± 0.2 degrees two-theta. 51. The substantially crystalline Compound I Acetone Solvate Hydrate Form A according to any one of Embodiments 48-50, characterized by an X-ray powder diffractogram having (a) one or two
signals selected from 17.2 ± 0.2 degrees two-theta, 18.1 ± 0.2 degrees two-theta, and 25.9 ± 0.2 degrees two-theta, and (b) one or two signals selected from 12.9 ± 0.2 degrees two-theta, 15.6 ± 0.2 degrees two-theta, and 24.8 ± 0.2 degrees two-theta. 52. The substantially crystalline Compound I Acetone Solvate Hydrate Form A according to any one of Embodiments 48-51, characterized by an X-ray powder diffractogram having two, three, four, five, or six signals selected from 9.5 ± 0.2 degrees two-theta, 12.3 ± 0.2 degrees two-theta, 13.5 ± 0.2 degrees two-theta, 12.9 ± 0.2 degrees two-theta, 15.6 ± 0.2 degrees two-theta, 24.8 ± 0.2 degrees two-theta, 8.1 ± 0.2 degrees two-theta, 12.3 ± 0.2 degrees two-theta, and 17.6 ± 0.2 degrees two-theta. 53. The substantially crystalline Compound I Acetone Solvate Hydrate Form A according to any one of Embodiments 48-52, characterized by an X-ray powder diffractogram substantially similar to FIG.13. 54. The substantially crystalline Compound I Acetone Solvate Hydrate Form A according to any one of Embodiments 48-53, characterized by a 13C SSNMR spectrum having one, two, three, four, five, six, seven, eight, nine, ten, or more peaks selected from 212.9 ± 0.2 ppm, 211.6 ± 0.2 ppm, 179.2 ± 0.2 ppm, 178.9 ± 0.2 ppm, 178.4 ± 0.2 ppm, 174.7 ± 0.2 ppm, 174.1 ± 0.2 ppm, 172.7 ± 0.2 ppm, 172.2 ± 0.2 ppm, 163.1 ± 0.2 ppm, 156.7 ± 0.2 ppm, 155.6 ± 0.2 ppm, 152.3 ± 0.2 ppm, 150.1 ± 0.2 ppm, 148.7 ± 0.2 ppm, 147.9 ± 0.2 ppm, 147.0 ± 0.2 ppm, 145.1 ± 0.2 ppm, 125.5 ± 0.2 ppm, 124.5 ± 0.2 ppm, 122.5 ± 0.2 ppm, 121.6 ± 0.2 ppm, 121.1 ± 0.2 ppm, 118.5 ± 0.2 ppm, 118.1 ± 0.2 ppm, 117.4 ± 0.2 ppm, 116.8 ± 0.2 ppm, 115.7 ± 0.2 ppm, 115.3 ± 0.2 ppm, 113.9 ± 0.2 ppm, 113.2 ± 0.2 ppm, 112.4 ± 0.2 ppm, 30 ± 0.2 ppm, 29.5 ± 0.2 ppm, 18.4 ± 0.2 ppm, 17.6 ± 0.2 ppm, 8.8 ± 0.2 ppm, and 8.1 ± 0.2 ppm. 55. The substantially crystalline Compound I Acetone Solvate Hydrate Form A according to any one of Embodiments 48-54, characterized by a 13C SSNMR spectrum having peaks at 212.9 ± 0.2 ppm, 211.6 ± 0.2 ppm, 179.2 ± 0.2 ppm, 178.9 ± 0.2 ppm, 178.4 ± 0.2 ppm, 174.7 ± 0.2 ppm, 174.1 ± 0.2 ppm, 172.7 ± 0.2 ppm, 172.2 ± 0.2 ppm, 163.1 ± 0.2 ppm, 156.7 ± 0.2 ppm, 155.6 ± 0.2 ppm, 152.3 ± 0.2 ppm, 150.1 ± 0.2 ppm, 148.7 ± 0.2 ppm, 147.9 ± 0.2 ppm, 147.0 ± 0.2 ppm, 145.1 ± 0.2 ppm, 125.5 ± 0.2 ppm, 124.5 ± 0.2 ppm, 122.5 ± 0.2 ppm, 121.6 ± 0.2 ppm, 121.1 ± 0.2 ppm, 118.5 ± 0.2 ppm, 118.1 ± 0.2 ppm, 117.4 ± 0.2 ppm, 116.8 ± 0.2 ppm, 115.7 ± 0.2 ppm, 115.3 ± 0.2 ppm, 113.9 ± 0.2 ppm, 113.2 ± 0.2 ppm, 112.4 ± 0.2 ppm, 30 ± 0.2 ppm, 29.5 ± 0.2 ppm, 18.4 ± 0.2 ppm, 17.6 ± 0.2 ppm, 8.8 ± 0.2 ppm, and 8.1 ± 0.2 ppm. 56. The substantially crystalline Compound I Acetone Solvate Hydrate Form According to any one of Embodiments 48-55, characterized by a 13C SSNMR spectrum substantially similar to FIG.14. 57. The substantially crystalline Compound I Acetone Solvate Hydrate Form A according to any one of Embodiments 48-56, characterized as having a 19F SSNMR spectrum with one or two peaks
selected from -60.4 ± 0.2 ppm, -61.2 ± 0.2 ppm, -61.6 ± 0.2 ppm, -62.7 ± 0.2 ppm, -135.9 ± 0.2 ppm, -136.1 ± 0.2 ppm, -139.6 ± 0.2 ppm, -140.6 ± 0.2 ppm, -141.7 ± 0.2 ppm, -145.2 ± 0.2 ppm, and -145.7 ± 0.2 ppm. 58. The substantially crystalline Compound I Acetone Solvate Hydrate Form A according to any one of Embodiments 48-57, characterized as having a 19F SSNMR spectrum having peaks at -60.4 ± 0.2 ppm, -61.2 ± 0.2 ppm, -61.6 ± 0.2 ppm, -62.7 ± 0.2 ppm, -135.9 ± 0.2 ppm, -136.1 ± 0.2 ppm, -139.6 ± 0.2 ppm, -140.6 ± 0.2 ppm, -141.7 ± 0.2 ppm, -145.2 ± 0.2 ppm, and -145.7 ± 0.2 ppm. 59. The substantially crystalline Compound I Acetone Solvate Hydrate Form According to any one of Embodiments 48-58, characterized by a 19F SSNMR spectrum substantially similar to FIG.15. 60. The substantially crystalline Compound I Acetone Solvate Hydrate Form According to any one of Embodiments 48-59, characterized by a monoclinic crystal system, P21 space group, and unit cell dimensions measured at 100 K on a Rigaku diffractometer utilizing Cu Kα radiation (λ=1.54178 Å) of: a 9.2 ± 0.1 Å α 90° b 39.8 ± 0.1 Å β 96.3 ± 0.1° c 12.9 ± 0.1 Å γ 90°. 61. Substantially crystalline Compound I Ethanol Solvate Form A (i.e., wherein less than 15% of Compound I is in amorphous form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous form). 62. The substantially crystalline Compound I Ethanol Solvate Form A according to Embodiment 61, wherein Compound I Ethanol Solvate Hydrate is 100% crystalline. 63. The substantially crystalline Compound I Ethanol Solvate Form A according to Embodiment 61 or Embodiment 62, characterized by an X-ray powder diffractogram having one or two signals selected from 9.4 ± 0.2 degrees two-theta, 16.1 ± 0.2 degrees two-theta, and 26.0 ± 0.2 degrees two-theta. 64. The substantially crystalline Compound I Ethanol Solvate Form A according to any one of Embodiments 61-63, characterized by an X-ray powder diffractogram having (a) one or two signals selected from 9.4 ± 0.2 degrees two-theta, 16.1 ± 0.2 degrees two-theta, and 26.0 ± 0.2 degrees two-theta, and (b) one or two signals selected from 15.1 ± 0.2 degrees two-theta, 17.0 ± 0.2 degrees two-theta, and 22.4 ± 0.2 degrees two-theta. 65. The substantially crystalline Compound I Ethanol Solvate Form A according to any one of Embodiments 61-64, characterized by an X-ray powder diffractogram having two, three, four, five, or six signals selected from 9.4 ± 0.2 degrees two-theta, 16.1 ± 0.2 degrees two-theta, 26.0 ± 0.2 degrees
two-theta, 15.1 ± 0.2 degrees two-theta, 17.0 ± 0.2 degrees two-theta, 22.4 ± 0.2 degrees two-theta, 13.2 ± 0.2 degrees two-theta, 13.6 ± 0.2 degrees two-theta, and 13.8 ± 0.2 degrees two-theta. 66. The substantially crystalline Compound I Ethanol Solvate Form A according to any one of Embodiments 61-65, characterized by an X-ray powder diffractogram substantially similar to FIG.16. 67. The substantially crystalline Compound I Ethanol Solvate Form A according to any one of Embodiments 61-66, characterized by a 13C SSNMR spectrum having one, two, three, four, five, six, seven, eight, nine, ten, or more peaks selected from 178.2 ± 0.2 ppm, 175.7 ± 0.2 ppm, 163.9 ± 0.2 ppm, 163.4 ± 0.2 ppm, 162.8 ± 0.2 ppm, 154.9 ± 0.2 ppm, 148.1 ± 0.2 ppm, 146.0 ± 0.2 ppm, 145.2 ± 0.2 ppm, 125.9 ± 0.2 ppm, 123.1 ± 0.2 ppm, 121.8 ± 0.2 ppm, 120.4 ± 0.2 ppm, 119.5 ± 0.2 ppm, 116.9 ± 0.2 ppm, 116.3 ± 0.2 ppm, 114.8 ± 0.2 ppm, 113.8 ± 0.2 ppm, 56.5 ± 0.2 ppm, 18.4 ± 0.2 ppm, 17.2 ± 0.2 ppm, 9.2 ± 0.2 ppm, and 7.9 ± 0.2 ppm. 68. The substantially crystalline Compound I Ethanol Solvate Form A according to any one of Embodiments 61-67, characterized by a 13C SSNMR spectrum having peaks at 178.2 ± 0.2 ppm, 175.7 ± 0.2 ppm, 163.9 ± 0.2 ppm, 163.4 ± 0.2 ppm, 162.8 ± 0.2 ppm, 154.9 ± 0.2 ppm, 148.1 ± 0.2 ppm, 146.0 ± 0.2 ppm, 145.2 ± 0.2 ppm, 125.9 ± 0.2 ppm, 123.1 ± 0.2 ppm, 121.8 ± 0.2 ppm, 120.4 ± 0.2 ppm, 119.5 ± 0.2 ppm, 116.9 ± 0.2 ppm, 116.3 ± 0.2 ppm, 114.8 ± 0.2 ppm, 113.8 ± 0.2 ppm, 56.5 ± 0.2 ppm, 18.4 ± 0.2 ppm, 17.2 ± 0.2 ppm, 9.2 ± 0.2 ppm, and 7.9 ± 0.2 ppm. 69. The substantially crystalline Compound I Ethanol Solvate Form A according to any one of Embodiments 61-68, characterized by a 13C SSNMR spectrum substantially similar to FIG.17. 70. The substantially crystalline Compound I Ethanol Solvate Form A according to any one of Embodiments 61-69, characterized as having a 19F SSNMR spectrum with one or two peaks selected from -61.1 ± 0.2 ppm, -138.1 ± 0.2 ppm, and -141.2 ± 0.2 ppm. 71. The substantially crystalline Compound I Ethanol Solvate Form A according to any one of Embodiments 61-70, characterized as having a 19F SSNMR spectrum having peaks at -61.1 ± 0.2 ppm, -138.1 ± 0.2 ppm, and -141.2 ± 0.2 ppm. 72. The substantially crystalline Compound I Ethanol Solvate Form A according to any one of Embodiments 61-71, characterized by a 19F SSNMR spectrum substantially similar to FIG.18. 73. The substantially crystalline Compound I Ethanol Solvate Form A according to any one of Embodiments 61-72, characterized by a monoclinic crystal system, P21/c space group, and unit cell dimensions measured at 100 K on a Bruker diffractometer utilizing Cu Kα radiation (λ=1.54178 Å) of: a 17.0 ± 0.1 Å α 90° b 11.1 ± 0.1 Å β 105.9 ± 0.1°
c 13.2 ± 0.1 Å γ 90°. 74. Substantially crystalline Compound I Hydrate Form A (i.e., wherein less than 15% of Compound I is in amorphous form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous form). 75. The substantially crystalline Compound I Hydrate Form A according to Embodiment 74, wherein Compound I Dehydrated Hydrate Form A is 100% crystalline. 76. The substantially crystalline Compound I Hydrate Form A according to Embodiment 74 or Embodiment 75, characterized by an X-ray powder diffractogram having one or two signals selected from 8.7 ± 0.2 degrees two-theta, 12.4 ± 0.2 degrees two-theta, and 18.8 ± 0.2 degrees two-theta. 77. The substantially crystalline Compound I Hydrate Form A according to any one of Embodiments 74-76, characterized by an X-ray powder diffractogram having (a) one or two signals selected from 8.7 ± 0.2 degrees two-theta, 12.4 ± 0.2 degrees two-theta, and 18.8 ± 0.2 degrees two-theta, and (b) one or two signals selected from 12.7 ± 0.2 degrees two-theta, 17.5 ± 0.2 degrees two-theta, and 19.8 ± 0.2 degrees two-theta. 78. The substantially crystalline Compound I Hydrate Form A according to any one of Embodiments 74-77, characterized by an X-ray powder diffractogram having two, three, four, five, or six signals selected from 8.7 ± 0.2 degrees two-theta, 12.4 ± 0.2 degrees two-theta, 18.8 ± 0.2 degrees two-theta, 12.7 ± 0.2 degrees two-theta, 17.5 ± 0.2 degrees two-theta, 19.8 ± 0.2 degrees two-theta, 13.1 ± 0.2 degrees two-theta, 16.3 ± 0.2 degrees two-theta, and 21.1 ± 0.2 degrees two-theta. 79. The substantially crystalline Compound I Hydrate Form A according to any one of Embodiments 74-78, characterized by an X-ray powder diffractogram substantially similar to FIG.19. 80. Substantially crystalline Compound I Dehydrated Hydrate Form A (i.e., wherein less than 15% of Compound I is in amorphous form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous form). 81. The substantially crystalline Compound I Dehydrated Hydrate Form A according to Embodiment 81, wherein Compound I Dehydrated Hydrate Form A is 100% crystalline. 82. The substantially crystalline Compound I Dehydrated Hydrate Form A according to Embodiment 81 or Embodiment 82, characterized by an X-ray powder diffractogram having one or two signals selected from 8.7 ± 0.2 degrees two-theta, 15.9 ± 0.2 degrees two-theta, and 18.7 ± 0.2 degrees two-theta.
83. The substantially crystalline Compound I Dehydrated Hydrate Form A according to any one of Embodiments 81-83, characterized by an X-ray powder diffractogram having (a) one or two signals selected from 8.7 ± 0.2 degrees two-theta, 15.9 ± 0.2 degrees two-theta, and 18.7 ± 0.2 degrees two-theta, and (b) one or two signals selected from 12.4 ± 0.2 degrees two-theta, 12.6 ± 0.2 degrees two-theta, 17.3 ± 0.2 degrees two-theta, and 19.8 ± 0.2 degrees two-theta. 84. The substantially crystalline Compound I Dehydrated Hydrate Form A according to any one of Embodiments 81-84, characterized by an X-ray powder diffractogram having two, three, four, five, or six signals selected from 8.7 ± 0.2 degrees two-theta, 15.9 ± 0.2 degrees two-theta, 18.7 ± 0.2 degrees two-theta, 12.4 ± 0.2 degrees two-theta, 12.6 ± 0.2 degrees two-theta, 17.3 ± 0.2 degrees two-theta, 19.8 ± 0.2 degrees two-theta, 13.1 ± 0.2 degrees two-theta, 16.3 ± 0.2 degrees two-theta, and 21.1 ± 0.2 degrees two-theta. 85. The substantially crystalline Compound I Dehydrated Hydrate Form A according to any one of Embodiments 81-85, characterized by an X-ray powder diffractogram substantially similar to FIG.20. 86. The substantially crystalline Compound I Hydrate Form A according to any one of Embodiments 74-86, characterized by a 13C SSNMR spectrum having one, two, three, four, five, six, seven, eight, nine, ten, or more peaks selected from 177.3 ± 0.2 ppm, 172.8 ± 0.2 ppm, 170.5 ± 0.2 ppm, 162.0 ± 0.2 ppm, 154.3 ± 0.2 ppm, 154.0 ± 0.2 ppm, 150.2 ± 0.2 ppm, 148.7 ± 0.2 ppm, 147.6 ± 0.2 ppm, 146.6 ± 0.2 ppm, 145.5 ± 0.2 ppm, 126.3 ± 0.2 ppm, 125.0 ± 0.2 ppm, 123.8 ± 0.2 ppm, 122.4 ± 0.2 ppm, 121.6 ± 0.2 ppm, 120.6 ± 0.2 ppm, 118.4 ± 0.2 ppm, 117.9 ± 0.2 ppm, 117.0 ± 0.2 ppm, 116.0 ± 0.2 ppm, 114.7 ± 0.2 ppm, 114.4 ± 0.2 ppm, 112.8 ± 0.2 ppm, 17.7 ± 0.2 ppm, 17.1 ± 0.2 ppm, and 8.7 ± 0.2 ppm. 87. The substantially crystalline Compound I Hydrate Form A according to any one of Embodiments 74-87, characterized by a 13C SSNMR spectrum having peaks at 177.3 ± 0.2 ppm, 172.8 ± 0.2 ppm, 170.5 ± 0.2 ppm, 162.0 ± 0.2 ppm, 154.3 ± 0.2 ppm, 154.0 ± 0.2 ppm, 150.2 ± 0.2 ppm, 148.7 ± 0.2 ppm, 147.6 ± 0.2 ppm, 146.6 ± 0.2 ppm, 145.5 ± 0.2 ppm, 126.3 ± 0.2 ppm, 125.0 ± 0.2 ppm, 123.8 ± 0.2 ppm, 122.4 ± 0.2 ppm, 121.6 ± 0.2 ppm, 120.6 ± 0.2 ppm, 118.4 ± 0.2 ppm, 117.9 ± 0.2 ppm, 117.0 ± 0.2 ppm, 116.0 ± 0.2 ppm, 114.7 ± 0.2 ppm, 114.4 ± 0.2 ppm, 112.8 ± 0.2 ppm, 17.7 ± 0.2 ppm, 17.1 ± 0.2 ppm, and 8.7 ± 0.2 ppm. 88. The substantially crystalline Compound I Hydrate Form A according to any one of Embodiments 74-88, characterized by a 13C SSNMR spectrum substantially similar to FIG.22. 89. The substantially crystalline Compound I Hydrate Form A according to any one of Embodiments 74-89, characterized as having a 19F SSNMR spectrum with one or two peaks selected
from -59.4 ± 0.2 ppm, -60.2 ± 0.2 ppm, -135.0 ± 0.2 ppm, -138.4 ± 0.2 ppm, -141.7 ± 0.2 ppm, and -144.0 ± 0.2 ppm. 90. The substantially crystalline Compound I Hydrate Form A according to any one of Embodiments 74-90, characterized as having a 19F SSNMR spectrum having peaks at -59.4 ± 0.2 ppm, -60.2 ± 0.2 ppm, -135.0 ± 0.2 ppm, -138.4 ± 0.2 ppm, -141.7 ± 0.2 ppm, and -144.0 ± 0.2 ppm. 91. The substantially crystalline Compound I Hydrate Form A according to any one of Embodiments 74-91, characterized by a 19F SSNMR spectrum substantially similar to FIG.23. 92. The substantially crystalline Compound I Hydrate Form A according to any one of Embodiments 74-92, characterized by a triclinic crystal system, P-1 space group, and unit cell dimensions measured at 100 K on a Bruker diffractometer utilizing Cu Kα radiation (λ=1.54178 Å) of: a 10.5 ± 0.1 Å α 91.3 ± 0.1° b 11.2 ± 0.1 Å β 103.9 ± 0.1° c 19.2 ± 0.1 Å γ 98.4 ± 0.1°. 93. The substantially crystalline Compound I Dehydrated Hydrate Form A according to any one of Embodiments 81-92, characterized by a triclinic crystal system, P-1 space group, and unit cell dimensions measured at 100 K on a Bruker diffractometer utilizing Cu Kα radiation (λ=1.54178 Å) of: a 10.4 ± 0.1 Å α 90.7 ± 0.1° b 11.2 ± 0.1 Å β 104.7 ± 0.1° c 19.3 ± 0.1 Å γ 98.0 ± 0.1°. 94. A pharmaceutical composition comprising Compound I according to any one of Embodiments 1-93, and optionally further comprising one or more additional therapeutic agents. 95. The pharmaceutical composition according to Embodiment 94, wherein the pharmaceutical composition comprises one or more additional pain modulating compounds. 96. The Compound I according to any one of Embodiments 1-93, or the pharmaceutical composition according to any one of Embodiments 94-95, for use in the treatment of pain. 97. Use of the Compound I according to any one of Embodiments 1-93, or the pharmaceutical composition according to any one of Embodiments 94-95, in the manufacture of a medicament for the treatment of pain.
98. A method of treating pain comprising administering the Compound I according to any one of Embodiments 1-93, or the pharmaceutical composition according to any one of Embodiments 94-95, to a subject in need thereof. 99. The compound or composition for use of Embodiment 96, the use of Embodiment 97, or the method of Embodiment 98, wherein the Compound I according to any one of Embodiments 1-93 or the composition according to any one of Embodiments 94-95 is administered in combination with one or more additional therapeutic agents. 100. The compound or composition for use, the use, or the method of Embodiment 99, wherein the one or more additional therapeutic agents comprises one or more additional pain modulating compounds. 101. A method of making crystalline Compound I neat Form A, comprising (i) combining Compound I and ethyl acetate, (ii) distilling under vacuum at 50 °C, (iii) adding ethyl acetate, (iv) distilling under vacuum at 50 °C, (v) heating to 75 °C, (vi) adding heptane, (vii) cooling to 40 °C, (viii) adding a seed of crystalline Compound I Form A, (ix) holding at 40 °C for 1.5 hours, (x) adding heptane over 5 hours, (xi) cooling the slurry to 20 °C over 5 hours, (xii) holding at 20 °C for 11 hours, (xiii) collecting the solids, (xiv) drying the solids in a vacuum oven at 45 °C with a slight nitrogen bleed for 46 hours to yield crystalline Compound I neat Form A. 102. A method of making crystalline Compound I neat Form B, comprising (i) adding Compound I hydrate Form A to an oven set at 180 °C (ii) cooling under ambient conditions to yield crystalline Compound I neat Form B. 103. A method of making crystalline Compound I neat Form E, comprising (i) combining Compound I hydrate Form A and trifluorotoluene, (ii) heating the slurry to 80 °C, (iii) cooling to room temperature to yield crystalline Compound I neat Form E. 104. A method of making crystalline Compound I Acetone Solvate Hydrate Form A, comprising (i) saturating acetone with Compound I, (ii) adding water dropwise to 20% of the final volume, (iii) holding overnight at room temperature to yield crystalline Compound I acetone solvate hydrate Form A. 105. A method of making crystalline Compound I Ethanol Solvate Form A, comprising (i) combining Compound I and ethyl acetate, (ii) heating the slurry to 50 °C, (iii) adding ethanol and distilling under vacuum at 50 °C, (iv) adding ethanol and distilling under vacuum at 50 °C, (v) adding ethanol and distilling under vacuum at 50 °C to a minimum volume, (vi) adding ethanol and distilling under vacuum at 50 °C, (vii) drying in a vacuum oven at 45 °C, with a slight nitrogen bleed for 42 hours to yield crystalline Compound I ethanol solvate Form A.
106. A method of making crystalline Compound I Dehydrated Hydrate Form A, comprising (i) combining Compound I Ethanol Solvate Form A and acetone, (ii) stirring the slurry while heating 50 °C, (iii) adding water over 1 hour, (iv) adding a seed of crystalline Compound I hydrate Form A, (v) agitating for 30 minutes, (vi) adding water over 8 hours, (vii) stirring at 50 °C for 5 hours, (viii) cooling the slurry to 20 °C over 3 hours, (ix) agitating for 2 hours, (x) collecting the solids and washing with 1:2 acetone:water, (xi) drying the solids in a vacuum over at 50 °C with a slight nitrogen bleed to yield crystalline Compound I hydrate Form A. 107. A method of making Compound I amorphous form, comprising (i) dissolving Compound I in acetone, (ii) filtering and evaporating the solvent at 50 °C, 12 mbar, over 1 hour on a centrifugal evaporation, (iii) drying in a vacuum oven at 50 °C overnight to yield Compound I amorphous form. [0469] Additional embodiments of the disclosure are set out in the following numbered clauses: 1. A process for preparing a compound of formula (I): , comprising transforming a
 to the compound of formula (I); wherein
to the compound of formula (I); wherein
 are independently selected from halogen.
are independently selected from halogen.
2. The process of Clause 1, wherein transforming the compound of formula (A1) to the compound of formula (I) comprises transforming the compound of formula (A1) to a compound of formula (E-A1): X2 CN N . 3. The process of Clause 2,
 of formula (A1) to the compound of formula (E-A1) comprises contacting the compound of formula (A1) with a compound of formula (D-1): . 4. The process of Clause 3,
of formula (A1) to the compound of formula (E-A1) comprises contacting the compound of formula (A1) with a compound of formula (D-1): . 4. The process of Clause 3,
 of formula (A1) with a compound of formula (D-1) is performed in the presence of a first palladium catalyst and a first base. 5. The process of Clause 4, wherein the first palladium catalyst is a palladium-phosphine complex. 6. The process of any one of Clauses 4 and 5, wherein the first palladium catalyst is PdCl2(dtbdpf). 7. The process of any one of Clauses Clause 4-6, wherein the first base is selected from potassium carbonate, potassium phosphate, and potassium fluoride. 8. The process of any one of Clauses 1 to 7, wherein transforming the compound of formula (A1) to the compound of formula (I) further comprises transforming the compound of formula (E-A1) to the compound of formula (I). 9. The process of Clause 8, wherein transforming the compound of formula (E-A1) to the compound of formula (I) comprises treating the compound of formula (E-A1) with a first acid.
10. The process of Clause 9, wherein transforming the first acid is selected from sulfuric acid, methanesulfonic acid, and trifluoroacetic acid. 11. The process of any one of Clauses 1 to 10, further comprising transforming a compound of formula (A1-1): to the compound of formula (A1).
of formula (A1) with a compound of formula (D-1) is performed in the presence of a first palladium catalyst and a first base. 5. The process of Clause 4, wherein the first palladium catalyst is a palladium-phosphine complex. 6. The process of any one of Clauses 4 and 5, wherein the first palladium catalyst is PdCl2(dtbdpf). 7. The process of any one of Clauses Clause 4-6, wherein the first base is selected from potassium carbonate, potassium phosphate, and potassium fluoride. 8. The process of any one of Clauses 1 to 7, wherein transforming the compound of formula (A1) to the compound of formula (I) further comprises transforming the compound of formula (E-A1) to the compound of formula (I). 9. The process of Clause 8, wherein transforming the compound of formula (E-A1) to the compound of formula (I) comprises treating the compound of formula (E-A1) with a first acid.
10. The process of Clause 9, wherein transforming the first acid is selected from sulfuric acid, methanesulfonic acid, and trifluoroacetic acid. 11. The process of any one of Clauses 1 to 10, further comprising transforming a compound of formula (A1-1): to the compound of formula (A1).
 12. The process of Clause 11, wherein transforming a compound of formula (A1-1) to the compound of formula (A1) comprises the steps of: treating the compound of formula (A1-1) with trifluoromethanesulfonic anhydride to form a triflyl intermediate; treating the triflyl intermediate with a cyanation reagent; treating the cyanated intermediate with a second base to form a cyanated triflyl intermediate; and treating the cyanated triflyl intermediate with an aqueous base to form the compound of formula (A1). 13. The process of Clause 12, wherein the cyanation reagent is trimethylsilyl cyanide, sodium cyanide, and potassium cyanide. 14. The process of any of Clauses 12 and 13, wherein the second base is selected from 4-methyl morpholine, trimethylamine, and Hunig’s base. 15. The process of any one of Clauses 12 to 14, wherein the aqueous base is aqueous sodium bicarbonate. 16. The process of any one of Clauses 11 to 15, further comprising transforming a compound of formula (A1-2): to the compound of formula (A1-1).
12. The process of Clause 11, wherein transforming a compound of formula (A1-1) to the compound of formula (A1) comprises the steps of: treating the compound of formula (A1-1) with trifluoromethanesulfonic anhydride to form a triflyl intermediate; treating the triflyl intermediate with a cyanation reagent; treating the cyanated intermediate with a second base to form a cyanated triflyl intermediate; and treating the cyanated triflyl intermediate with an aqueous base to form the compound of formula (A1). 13. The process of Clause 12, wherein the cyanation reagent is trimethylsilyl cyanide, sodium cyanide, and potassium cyanide. 14. The process of any of Clauses 12 and 13, wherein the second base is selected from 4-methyl morpholine, trimethylamine, and Hunig’s base. 15. The process of any one of Clauses 12 to 14, wherein the aqueous base is aqueous sodium bicarbonate. 16. The process of any one of Clauses 11 to 15, further comprising transforming a compound of formula (A1-2): to the compound of formula (A1-1).
 17. The process of Clause 16, wherein transforming the compound of formula (A1-2) to the compound of formula (A1-1) comprises treating the compound of formula (A1-2) with a halogenating reagent. 18. The process of Clause 17, wherein the halogenating reagent is a chlorinating reagent selected from phosphorous oxychloride. 19. The process of any one of Clauses 16 to 18, further comprising transforming a compound of formula (A1-3): to the compound of formula (A1-2).
17. The process of Clause 16, wherein transforming the compound of formula (A1-2) to the compound of formula (A1-1) comprises treating the compound of formula (A1-2) with a halogenating reagent. 18. The process of Clause 17, wherein the halogenating reagent is a chlorinating reagent selected from phosphorous oxychloride. 19. The process of any one of Clauses 16 to 18, further comprising transforming a compound of formula (A1-3): to the compound of formula (A1-2).
 20. The process of Clause 19, wherein transforming a compound of formula (A1-3) to the compound of formula (A1-3) comprises treating the compound of formula (A1-3) with a second acid. 21. The process of Clause 20, wherein the second acid is aqueous hydrochloric acid. 22. The process of any one of Clauses 19 to 21, further comprising transforming a compound of formula (A1-4): to the compound of formula (A1-3).
20. The process of Clause 19, wherein transforming a compound of formula (A1-3) to the compound of formula (A1-3) comprises treating the compound of formula (A1-3) with a second acid. 21. The process of Clause 20, wherein the second acid is aqueous hydrochloric acid. 22. The process of any one of Clauses 19 to 21, further comprising transforming a compound of formula (A1-4): to the compound of formula (A1-3).
 23. The process of Clause 22, wherein transforming a compound of formula (A1-4) to the compound of formula (A1-3) comprises treating the compound of formula (A1-4) with diethyl malonate in the presence of a third base. 24. The process of Clause 23, wherein the third base is an alkaline alkoxide. 25. The process of any one of Clauses 23 to 24, wherein the third base is sodium ethoxide.
26. The process of any one of Clauses 23 to 25, further comprising transforming a compound of formula (A1-5): to the compound of formula (A1-4).
23. The process of Clause 22, wherein transforming a compound of formula (A1-4) to the compound of formula (A1-3) comprises treating the compound of formula (A1-4) with diethyl malonate in the presence of a third base. 24. The process of Clause 23, wherein the third base is an alkaline alkoxide. 25. The process of any one of Clauses 23 to 24, wherein the third base is sodium ethoxide.
26. The process of any one of Clauses 23 to 25, further comprising transforming a compound of formula (A1-5): to the compound of formula (A1-4).
 27. The process of Clause 26, wherein transforming the compound of formula (A1-5) to the compound of formula (A1-4) comprises treating the compound of formula (A1-5) with carbon monoxide in the presence of a fourth base, a second palladium catalyst, and a first suitable ligand. 28. The process of Clause 27, wherein the fourth base is triethylamine. 29. The process of Clause 27 or 28, wherein the second palladium catalyst is Pd(OAc)2. 30. The process of any one of Clauses 27 to 29, wherein the first suitable ligand is 1,1'-bis(diphenyl phosphino)ferrocene (DPPF). 31. The process of any one of Clauses 1 to 30, wherein each of X1 and X2 are chloro. 32. A process for preparing a compound of formula (I): comprising transforming a
27. The process of Clause 26, wherein transforming the compound of formula (A1-5) to the compound of formula (A1-4) comprises treating the compound of formula (A1-5) with carbon monoxide in the presence of a fourth base, a second palladium catalyst, and a first suitable ligand. 28. The process of Clause 27, wherein the fourth base is triethylamine. 29. The process of Clause 27 or 28, wherein the second palladium catalyst is Pd(OAc)2. 30. The process of any one of Clauses 27 to 29, wherein the first suitable ligand is 1,1'-bis(diphenyl phosphino)ferrocene (DPPF). 31. The process of any one of Clauses 1 to 30, wherein each of X1 and X2 are chloro. 32. A process for preparing a compound of formula (I): comprising transforming a
 to the compound of formula (I).
to the compound of formula (I).
 33. The process of Clause 32, wherein transforming the compound of formula (A3) to the compound of formula (I) comprises transforming the compound of formula (A3) to a compound of formula (E-A3): . 34. The process of Clause 33,
33. The process of Clause 32, wherein transforming the compound of formula (A3) to the compound of formula (I) comprises transforming the compound of formula (A3) to a compound of formula (E-A3): . 34. The process of Clause 33,
 of formula (A3) to the compound of formula (E-A3) comprises contacting the compound of formula (A3) with a compound of formula (D-1): . 35. The process of Clause 34,
of formula (A3) to the compound of formula (E-A3) comprises contacting the compound of formula (A3) with a compound of formula (D-1): . 35. The process of Clause 34,
 of formula (A3) with a compound of formula (D-1) is performed in the presence of a first palladium catalyst and a first base. 36. The process of Clause 35, wherein the first palladium catalyst is PdCl2(dtbdpf). 37. The process of any one of Clauses 35 to 36, wherein the first base is potassium phosphate. 38. The process of any one of Clauses 32 to 37, wherein transforming the compound of formula (A3) to the compound of formula (I) further comprises transforming the compound of formula (E-A3) to the compound of formula (I). 39. The process of Clause 38, wherein transforming the compound of formula (E-A3) to the compound of formula (I) comprises treating the compound of formula (E-A3) with a first acid. 40. The process of Clause 39, wherein the first acid is trifluoroacetic acid, sulfuric acid, and methane sulfonic acid.
41. The process of any one of Clauses 32 to 40, further comprising transforming a compound of formula (A3-1): to the compound of formula (A3).
of formula (A3) with a compound of formula (D-1) is performed in the presence of a first palladium catalyst and a first base. 36. The process of Clause 35, wherein the first palladium catalyst is PdCl2(dtbdpf). 37. The process of any one of Clauses 35 to 36, wherein the first base is potassium phosphate. 38. The process of any one of Clauses 32 to 37, wherein transforming the compound of formula (A3) to the compound of formula (I) further comprises transforming the compound of formula (E-A3) to the compound of formula (I). 39. The process of Clause 38, wherein transforming the compound of formula (E-A3) to the compound of formula (I) comprises treating the compound of formula (E-A3) with a first acid. 40. The process of Clause 39, wherein the first acid is trifluoroacetic acid, sulfuric acid, and methane sulfonic acid.
41. The process of any one of Clauses 32 to 40, further comprising transforming a compound of formula (A3-1): to the compound of formula (A3).
 42. The process of Clause 41, wherein transforming a compound of formula (A3-1) to the compound of formula (A3) comprises treating the compound of formula (A3-1) with a cyanation reagent and a second base. 43. The process of Clause 42, wherein the cyanation reagent is trimethylsilyl cyanide, sodium cyanide, and potassium cyanide. 44. The process of Clause 42 or 43, wherein the second base is triethylamine. 45. The process of any one of Clauses 41 to 44, further comprising transforming a compound of formula (A3-2): to the compound of formula (A3-1).
42. The process of Clause 41, wherein transforming a compound of formula (A3-1) to the compound of formula (A3) comprises treating the compound of formula (A3-1) with a cyanation reagent and a second base. 43. The process of Clause 42, wherein the cyanation reagent is trimethylsilyl cyanide, sodium cyanide, and potassium cyanide. 44. The process of Clause 42 or 43, wherein the second base is triethylamine. 45. The process of any one of Clauses 41 to 44, further comprising transforming a compound of formula (A3-2): to the compound of formula (A3-1).
 46. The process of Clause 45, wherein transforming a compound of formula (A3-2) to the compound of formula (A3-1) comprises treating the compound of formula (A3-2) with benzyl alcohol in the presence of a third base. 47. The process of Clause 46, wherein the third base is an alkaline alkoxide. 48. The process of Clause 46 or 47, wherein the third base is lithium tert-butoxide.
46. The process of Clause 45, wherein transforming a compound of formula (A3-2) to the compound of formula (A3-1) comprises treating the compound of formula (A3-2) with benzyl alcohol in the presence of a third base. 47. The process of Clause 46, wherein the third base is an alkaline alkoxide. 48. The process of Clause 46 or 47, wherein the third base is lithium tert-butoxide.
49. The process of any one of Clauses 3 to 31 or 34 to 48, further comprising transforming a compound of formula (D-2): to the compound of formula (D-1).
 50. The process of Clause 49, wherein transforming a compound of formula (D-2) to the compound of formula (D-1) comprises treating the compound of formula (D-2) with tetrahydroxydiboron, in the presence of a third palladium catalyst, a second suitable ligand, and a fifth base. 51. The process of Clause 50, wherein the third palladium catalyst is Pd(OAc)2. 52. The process of Clause 50 or 51, wherein the second suitable ligand is cyclohexyldiphenylphosphine. 53. The process of any one of Clauses 50 to 52, wherein the fifth base is potassium acetate. 54. The process of any one of Clauses 50 to 53, further comprising transforming a compound of formula (B-1): to the compound of formula (D-2).
50. The process of Clause 49, wherein transforming a compound of formula (D-2) to the compound of formula (D-1) comprises treating the compound of formula (D-2) with tetrahydroxydiboron, in the presence of a third palladium catalyst, a second suitable ligand, and a fifth base. 51. The process of Clause 50, wherein the third palladium catalyst is Pd(OAc)2. 52. The process of Clause 50 or 51, wherein the second suitable ligand is cyclohexyldiphenylphosphine. 53. The process of any one of Clauses 50 to 52, wherein the fifth base is potassium acetate. 54. The process of any one of Clauses 50 to 53, further comprising transforming a compound of formula (B-1): to the compound of formula (D-2).
 55. The process of Clause 54, wherein transforming a compound of formula (B-2) to the compound of formula (B-1) comprises contacting the compound of formula (B-2) with a compound of formula (C-1): in the presence of a sixth base.
55. The process of Clause 54, wherein transforming a compound of formula (B-2) to the compound of formula (B-1) comprises contacting the compound of formula (B-2) with a compound of formula (C-1): in the presence of a sixth base.
 56. The process of Clause 55, wherein the sixth base is potassium carbonate. 57. The process of any one of Clauses 54 to 56, further comprising transforming a compound of formula (B-2): to the compound of formula (B-1).
56. The process of Clause 55, wherein the sixth base is potassium carbonate. 57. The process of any one of Clauses 54 to 56, further comprising transforming a compound of formula (B-2): to the compound of formula (B-1).
 58. The process of Clause 57, wherein transforming the compound of formula (B-2) to the compound of formula (B-1) comprises treating the compound of formula (B-2) with a chlorinating reagent. 59. The process of Clause 58, wherein the chlorinating reagent is phosphorous oxychloride. 60. The process of any one of Clauses 57 to 59, further comprising transforming a compound of formula (B-3): to the compound of formula (B-2).
58. The process of Clause 57, wherein transforming the compound of formula (B-2) to the compound of formula (B-1) comprises treating the compound of formula (B-2) with a chlorinating reagent. 59. The process of Clause 58, wherein the chlorinating reagent is phosphorous oxychloride. 60. The process of any one of Clauses 57 to 59, further comprising transforming a compound of formula (B-3): to the compound of formula (B-2).
 61. The process of Clause 60, wherein transforming the compound of formula (B-3) to the compound of formula (B-2) comprises treating the compound of formula (B-3) with a brominating reagent in the presence of a third acid. 62. The process of Clause 61, wherein the brominating reagent is molecular bromine. 63. The process of Clause 61 or 62, wherein the third acid is acetic acid. 64. The process of any one of Clauses 60 to 62, further comprising transforming a compound of formula (B-4): to the compound of formula (B-3).
61. The process of Clause 60, wherein transforming the compound of formula (B-3) to the compound of formula (B-2) comprises treating the compound of formula (B-3) with a brominating reagent in the presence of a third acid. 62. The process of Clause 61, wherein the brominating reagent is molecular bromine. 63. The process of Clause 61 or 62, wherein the third acid is acetic acid. 64. The process of any one of Clauses 60 to 62, further comprising transforming a compound of formula (B-4): to the compound of formula (B-3).
 65. The process of Clause 64, wherein transforming a compound of formula (B-4) to the compound of formula (B-3) comprises treating the compound of formula (B-4) with a fourth acid in the presence of a first suitable solvent. 66. The process of Clause 65, wherein the fourth acid is a mineral acid. 67. The process of Clause 65 or 66, wherein the fourth acid is concentrated aqueous hydrochloric acid. 68. The process of any one of Clauses 65-67, wherein the first suitable solvent comprises dioxane and water. 69. The process of any one of Clauses 64 to 68, further comprising transforming a compound of formula (B-5): to the compound of formula (B-4).
65. The process of Clause 64, wherein transforming a compound of formula (B-4) to the compound of formula (B-3) comprises treating the compound of formula (B-4) with a fourth acid in the presence of a first suitable solvent. 66. The process of Clause 65, wherein the fourth acid is a mineral acid. 67. The process of Clause 65 or 66, wherein the fourth acid is concentrated aqueous hydrochloric acid. 68. The process of any one of Clauses 65-67, wherein the first suitable solvent comprises dioxane and water. 69. The process of any one of Clauses 64 to 68, further comprising transforming a compound of formula (B-5): to the compound of formula (B-4).
 70. The process of Clause 69, wherein transforming a compound of formula (B-5) to the compound of formula (B-4) comprises treating the compound of formula (B-5) with a methylating reagent in the presence of a fourth palladium catalyst, and a seventh base. 71. The process of Clause 70, wherein the first methylating reagent is methylboronic acid. 72. The process of Clause 70 or 71, wherein the fourth palladium catalyst is Pd(dppf)Cl2•DCM. 73. The process of any one of Clauses 70 to 72, wherein the seventh base is potassium carbonate. 74. The process of any one of Clauses 55 to 73, further comprising transforming a compound of formula (C-2): to the compound of formula (C-1), wherein
70. The process of Clause 69, wherein transforming a compound of formula (B-5) to the compound of formula (B-4) comprises treating the compound of formula (B-5) with a methylating reagent in the presence of a fourth palladium catalyst, and a seventh base. 71. The process of Clause 70, wherein the first methylating reagent is methylboronic acid. 72. The process of Clause 70 or 71, wherein the fourth palladium catalyst is Pd(dppf)Cl2•DCM. 73. The process of any one of Clauses 70 to 72, wherein the seventh base is potassium carbonate. 74. The process of any one of Clauses 55 to 73, further comprising transforming a compound of formula (C-2): to the compound of formula (C-1), wherein
 protecting group.
75. The process of Clause 74, wherein transforming a compound of formula (C-2) to the compound of formula (C-1) comprises deprotecting the compound of formula (C-1) to afford the compound of formula (C-2). 76. The process of Clause 74 or 75, wherein PG is tetrahydropyranyl. 77. The process of any one of Clauses 74 to 76, wherein transforming the compound of formula (C-2) to the compound of formula (C-1) comprises treating the compound of formula (C-2) with a first acid catalyst. 78. The process of Clause 77, wherein the first acid catalyst is pyridinium p-toluenesulfonate. 79. The process of any one of Clauses 74 to 78, further comprising transforming a compound of formula (C-3): to the compound of formula (C-2), wherein protecting group.
protecting group.
75. The process of Clause 74, wherein transforming a compound of formula (C-2) to the compound of formula (C-1) comprises deprotecting the compound of formula (C-1) to afford the compound of formula (C-2). 76. The process of Clause 74 or 75, wherein PG is tetrahydropyranyl. 77. The process of any one of Clauses 74 to 76, wherein transforming the compound of formula (C-2) to the compound of formula (C-1) comprises treating the compound of formula (C-2) with a first acid catalyst. 78. The process of Clause 77, wherein the first acid catalyst is pyridinium p-toluenesulfonate. 79. The process of any one of Clauses 74 to 78, further comprising transforming a compound of formula (C-3): to the compound of formula (C-2), wherein protecting group.
 80. The process of Clause 79, wherein PG is tetrahydropyranyl. 81. The process of Clause 79 or 80, wherein transforming a compound of formula (C-3) to the compound of formula (C-2) comprises treating the compound of formula (C-3) with a second methylating reagent in the presence of an eighth base. 82. The process of Clause 81, wherein the second methylating reagent is methyl iodide. 83. The process of Clause 81 or 82, wherein the eighth base is butyllithium. 84. The process of any one of Clauses 81 to 83, wherein the eighth base is n-butyllithium. 85. The process of any one of Clauses 79 to 84, further comprising transforming a compound of formula (C-4):
80. The process of Clause 79, wherein PG is tetrahydropyranyl. 81. The process of Clause 79 or 80, wherein transforming a compound of formula (C-3) to the compound of formula (C-2) comprises treating the compound of formula (C-3) with a second methylating reagent in the presence of an eighth base. 82. The process of Clause 81, wherein the second methylating reagent is methyl iodide. 83. The process of Clause 81 or 82, wherein the eighth base is butyllithium. 84. The process of any one of Clauses 81 to 83, wherein the eighth base is n-butyllithium. 85. The process of any one of Clauses 79 to 84, further comprising transforming a compound of formula (C-4):
 to the compound of formula (C-3). 86. The process of Clause 85, wherein transforming a compound of formula (C-4) to the compound of C-3 comprises treating the compound of (C-4) with a suitable protecting group in the presence of a second acid catalyst. 87. The process of Clause 86, wherein the suitable protecting group is 3,4-dihydropyran. 88. The process of Clause 86 or 87, wherein the second acid catalyst is pyridinium p-toluenesulfonate. 89. The process of any one of Clauses 1 to 88, further comprising transforming the compound of formula (I) to a solvate of the compound of formula (I). 90. The process of Clause 89, wherein transforming the compound of formula (I) to the solvate of the compound of formula (I) comprises recrystallizing the compound of formula (I) from a suitable solvent. 91. The process of Clause 90, wherein the suitable solvent is ethanol. 92. The process of any one of Clauses 89 to 91, wherein the solvate of the compound of formula (I) is a compound of formula (F): O NH O 2 . 93. The process of any one
to the compound of formula (C-3). 86. The process of Clause 85, wherein transforming a compound of formula (C-4) to the compound of C-3 comprises treating the compound of (C-4) with a suitable protecting group in the presence of a second acid catalyst. 87. The process of Clause 86, wherein the suitable protecting group is 3,4-dihydropyran. 88. The process of Clause 86 or 87, wherein the second acid catalyst is pyridinium p-toluenesulfonate. 89. The process of any one of Clauses 1 to 88, further comprising transforming the compound of formula (I) to a solvate of the compound of formula (I). 90. The process of Clause 89, wherein transforming the compound of formula (I) to the solvate of the compound of formula (I) comprises recrystallizing the compound of formula (I) from a suitable solvent. 91. The process of Clause 90, wherein the suitable solvent is ethanol. 92. The process of any one of Clauses 89 to 91, wherein the solvate of the compound of formula (I) is a compound of formula (F): O NH O 2 . 93. The process of any one
 transforming the solvate of the compound of formula (I) to Form A of the compound of formula (I).
94. A compound selected from: ,
transforming the solvate of the compound of formula (I) to Form A of the compound of formula (I).
94. A compound selected from: ,
 ,
, 2): )
,
, 2): )

99. A composition comprising a compound of formula (I) and a compound of formula (E-A3): :
 . 100. The composition of Clause 99, further comprising an acid. 101. The composition of Clause 100, wherein the acid is trifluoroacetic acid. 102. A composition comprising a compound of formula (E-A2), a compound of formula (A2), and a compound of formula (D-1): . 1)
. 100. The composition of Clause 99, further comprising an acid. 101. The composition of Clause 100, wherein the acid is trifluoroacetic acid. 102. A composition comprising a compound of formula (E-A2), a compound of formula (A2), and a compound of formula (D-1): . 1)
 , and a compound of formula (D-1): . 1)
104. A compound of formula (I): , or a pharmaceutically acceptable prepared by the process of any one of Clauses 1 to 93.
, and a compound of formula (D-1): . 1)
104. A compound of formula (I): , or a pharmaceutically acceptable prepared by the process of any one of Clauses 1 to 93.
 105. A substantially crystalline Compound I I. wherein the crystalline pure compound I neat Form
105. A substantially crystalline Compound I I. wherein the crystalline pure compound I neat Form
 A, Compound I neat form B, Compound I neat form E, Compound I Acetone Solvate Hydrate Form A, Compound I Ethanol Solvate Form A, Compound I Hydrate Form A, and Compound I Dehydrated Hydrate Form A. 106. The substantially crystalline Compound I according to Clause 105, wherein less than 15% of Compound I is in amorphous form. 107. The substantially crystalline Compound I according to Clause 105, wherein less than 10% of Compound I is in amorphous form. 108. The substantially crystalline Compound I according to Clause 105, wherein less than 5% of Compound I is in amorphous form. 109. The substantially crystalline Compound I according to Clause 105, wherein 100% of Compound I is crystalline.
110. Substantially amorphous Compound I amorphous form. 111. The substantially amorphous Compound I according to Clause 110, wherein less than 15% of Compound I is in crystalline form. 112. The substantially crystalline Compound I according to Clause 110, wherein less than 10% of Compound I is in crystalline form. 113. The substantially crystalline Compound I according to Clause 110, wherein less than 5% of Compound I is in crystalline form. 114. The substantially crystalline Compound I according to Clause 110, wherein 100% of Compound I is amorphous. 115. The substantially crystalline Compound I according to any one of Clauses 105 to 109, characterized by crystal lattice parameters. 116. The substantially crystalline Compound I according to any one of Clauses 105 to 115, characterized by an X-ray powder diffractogram (XRPD). 117. The substantially crystalline Compound I according to any one of Clauses 105 to 116., characterized by 13C solid-state nuclear magnetic resonance (13C SSNMR) spectrum. 118. A pharmaceutical composition comprising the Compound I according to any one of Clauses 105 to 117. 119. The pharmaceutical composition according to Clause 118, further comprising one or more additional therapeutic agents. 120. The pharmaceutical composition according to Clause 119, wherein the pharmaceutical composition comprises one or more additional pain modulating compounds. 121. The pharmaceutical composition according to Clause 120, wherein the pharmaceutical composition comprises one or more combination compounds and pharmaceutically acceptable salts and deuterated derivatives thereof. 122. The Compound I according to any one of Clauses 105 to 117, or the pharmaceutical composition according to any one of Clauses 118 to 121, for use in the treatment of pain. 123. The compound or composition according to Clause 122, wherein the pain comprises chronic pain, gut pain, neuropathic pain, musculoskeletal pain, acute pain, inflammatory pain, cancer pain,
idiopathic pain, postsurgical pain, visceral pain, multiple sclerosis, Charcot-Marie-Tooth syndrome, incontinence, pathological cough, or cardiac arrhythmia. 124. Use of the Compound I according to any one of Clauses 105 to 117, or the pharmaceutical composition according to any one of Clauses 118 to 121, in the manufacture of a medicament for the treatment of pain. 125. A method of treating pain comprising administering the Compound I according to any one of Clauses 105 to 117, or the pharmaceutical composition according to any one of Clauses 118 to 121, to a subject in need thereof. 126. The compound or composition for use of Clause 122, the use of Clause 124, or the method of Clause 125, wherein the Compound I according to any one of Clauses 105 to 117 or the composition according to any one of Clauses 118 to 120 is administered in combination with one or more additional therapeutic agents. 127. The compound or composition for use, the use, or the method of Clause 126, wherein the one or more additional therapeutic agents comprises one or more additional pain modulating compounds. 128. The compound or composition for use, the use, or the method of Clause 126 or Clause 127, wherein the one or more additional therapeutic agents comprises one or more combination compounds selected and pharmaceutically acceptable salts and deuterated derivatives thereof. 129. A method of preparing the Compound I according to any one of Clauses 105 to 117, comprising: (a) (i) combining Compound I and ethyl acetate, (ii) distilling under vacuum at 50 °C, (iii) adding ethyl acetate, (iv) distilling under vacuum at 50 °C, (v) heating to 75 °C, (vi) adding heptane, (vii) cooling to 40 °C, (viii) adding a seed of crystalline Compound I Form A, (ix) holding at 40 °C for 1.5 hours, (x) adding heptane over 5 hours, (xi) cooling the slurry to 20 °C over 5 hours, (xii) holding at 20 °C for 11 hours, (xiii) collecting the solids, (xiv) drying the solids in a vacuum oven at 45 °C with a slight nitrogen bleed for 46 hours to yield crystalline Compound I neat Form A; (b) (i) adding Compound I hydrate Form A to an oven set at 180 °C (ii) cooling under ambient conditions to yield crystalline Compound I neat Form B; (c) (i) combining Compound I hydrate Form A and trifluorotoluene, (ii) heating the slurry to 80 °C, (iii) cooling to room temperature to yield crystalline Compound I neat Form E; (d) (i) saturating acetone with Compound I, (ii) adding water dropwise to 20% of the final volume, (iii) holding overnight at room temperature to yield crystalline Compound I Acetone Solvate Hydrate Form A; (e) (i) combining Compound I Ethanol Solvate Form A and acetone, (ii) stirring the slurry while heating 50 °C, (iii) adding water over 1 hour, (iv) adding a seed of crystalline Compound I hydrate
Form A, (v) agitating for 30 minutes, (vi) adding water over 8 hours, (vii) stirring at 50 °C for 5 hours, (viii) cooling the slurry to 20 °C over 3 hours, (ix) agitating for 2 hours, (x) collecting the solids and washing with 1:2 acetone:water, (xi) drying the solids in a vacuum over at 50 °C with a slight nitrogen bleed to yield crystalline Compound I Hydrate Form A; (f) (i) combining Compound I and ethyl acetate, (ii) heating the slurry to 50 °C, (iii) adding ethanol and distilling under vacuum at 50 °C, (iv) adding ethanol and distilling under vacuum at 50 °C, (v) adding ethanol and distilling under vacuum at 50 °C to a minimum volume, (vi) adding ethanol and distilling under vacuum at 50 °C, (vii) drying in a vacuum oven at 45 °C, with a slight nitrogen bleed for 42 hours to yield crystalline Compound I Ethanol Solvate Form A; (g) (i) dissolving Compound I in acetone, (ii) filtering and evaporating the solvent at 50 °C, 12 mbar, over 1 hour on a centrifugal evaporation, (iii) drying in a vacuum oven at 50 °C overnight to yield Compound I Amorphous Form. EXAMPLES General Experimental Details [0470] General methods. [0471] 1H NMR (400 MHz) and 19F NMR (376 MHz) spectra were obtained as solutions in an appropriate deuterated solvent such as dimethyl sulfoxide-d6 (DMSO-d6). [0472] Exemplary LC Method #1: HPLC analysis was conducted using an Agilent 1260 HPLC utilizing an Agilent Poroshell EC-C18 column (4.6 x 150 mm, 2.7μm particle) guard column (pn: 693975-902), and a dual gradient run from 5-95% mobile phase B over 45 min. Mobile phase A = 0.1% (v/v) phosphoric acid in water. Mobile phase B = 0.1% (v/v) Phosphoric Acid in Acetonitrile. Flow rate = 1.0 mL/min, injection volume = 5 μL, and column temperature = 35 °C. [0473] Exemplary LC Method #2: HPLC analysis was conducted using an Agilent 1260 HPLC utilizing an Agilent Poroshell EC-C18 column (4.6 x 150 mm, 2.7μm particle) guard column (pn: 693975-902), and a dual gradient run from 5-95% mobile phase B over 15 min. Mobile phase A = 0.1% (v/v) phosphoric acid in water. Mobile phase B = 0.1% (v/v) Phosphoric Acid in Acetonitrile. Flow rate = 1.0 mL/min, injection volume = 5 μL, and column temperature = 35 °C. Solid State NMR [0474] Bruker-Biospin 400 MHz wide-bore spectrometer equipped with Bruker-Biospin 4 mm HFX probe was used. Samples were packed into 4 mm ZrO2 rotors and spun under Magic Angle Spinning (MAS) condition with spinning speed typically set to 12.5 kHz. The proton relaxation time was measured using 1H MAS T1 saturation recovery relaxation experiment in order to set up proper recycle delay of the 13C cross-polarization (CP) MAS experiments. The fluorine relaxation time was measured using 19F MAS T1 saturation recovery relaxation experiment in order to set up proper recycle delay of the 19F MAS experiment. The CP contact time of carbon CPMAS experiments was set to 2 ms.
A CP proton pulse with linear ramp (from 50% to 100%) was employed. The carbon Hartmann-Hahn match was optimized on external reference sample (glycine). All carbon and fluorine spectra were recorded with proton decoupling using TPPM15 or SPINAL64 decoupling sequence with the field strength of approximately 100 kHz. X-ray Powder Diffraction [0475] X-ray powder diffraction (XRPD) spectra were recorded at room temperature in reflection mode using a PANalytical Empyrean system equipped with a sealed tube source and a PIXcel 1D Medipix-2 detector (Malvern PANalytical Inc, Westborough, Massachusetts). The X-Ray generator operated at a voltage of 45 kV and a current of 40 mA with copper radiation (1.54060 Å). The powder sample was placed in a back filled sample holder and loaded into the instrument. The sample was scanned over the range of about 3° to about 40° 2θ with a step size of 0.0131303° and 49.725s per step. Abbreviations [0476] Unless otherwise noted, or where the context dictates otherwise, the following abbreviations shall be understood to have the following meanings: Abbreviation Meaning NMR Nuclear magnetic resonance LC/MS Liquid chromatography-mass spectrometry UPLC Ultra performance liquid chromatography HPLC/MS/MS High performance liquid chromatography/tandem mass spectrometry IS Internal standard HPLC High performance liquid chromatography SFC Supercritical fluid chromatography MDAP Mass directed auto purification g grams mg milligrams L Liter(s) mL Milliliters μL Microliters mmol millimole h hours min Minutes MHz Megahertz Hz Hertz N Normal (concentration) M Molar (concentration)
Abbreviation Meaning mM Millimolar (concentration) tBuOH tert-butyl alcohol nBuAc n-butyl acetate CPME Cyclopentyl methyl ether DCM Dichloromethane DMF N,N-Dimethylformamide DMSO Dimethyl sulfoxide DRG Dorsal root ganglia EtOH Ethanol EtOAc Ethyl acetate HATU 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate EDCI 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide IPA Isopropyl alcohol T3P Propylphosphonic anhydride, i.e., 2,4,6-tripropyl-1,3,5,2,4,6- trioxatriphosphinane 2,4,6-trioxide LC Liquid chromatography MeOH Methanol MTBE Methyl tert-butyl ether MSA methane sulfonic acid THF Tetrahydrofuran Mn(acac)2 manganese(II) acetylacetonate TEA triethylamine RB(F) Round bottom (flask) RT Room temperature TFT α,α,α-Trifluorotoluene TMSOTf trimethylsilyl trifluoromethanesulfonate DIPEA N,N-diisopropylethyl amine NMM N-methylmorpholine TBD Triazabicyclodecene DBU 1,8-diazabicyclo[5.4.0]undec-7-ene DABCO 1,4-diazabicyclo[2.2.2] octane MTBD 7-Methyl-1,5,7-triazabicyclo [4.4.0]dec-5-ene LiHMDS Lithium hexamethyldisilazide (LiN(SiMe3)2)
Example 1: Preparation of Compound I 2-[2-(3,4-difluoro-2-methyl-phenoxy)-4-methyl-5-(trifluoromethyl)-3-pyridyl]-4-oxo-1H-1,6- naphthyridine-5-carboxamide (Compound I) [0477] A -3- pyridyl]
A, Compound I neat form B, Compound I neat form E, Compound I Acetone Solvate Hydrate Form A, Compound I Ethanol Solvate Form A, Compound I Hydrate Form A, and Compound I Dehydrated Hydrate Form A. 106. The substantially crystalline Compound I according to Clause 105, wherein less than 15% of Compound I is in amorphous form. 107. The substantially crystalline Compound I according to Clause 105, wherein less than 10% of Compound I is in amorphous form. 108. The substantially crystalline Compound I according to Clause 105, wherein less than 5% of Compound I is in amorphous form. 109. The substantially crystalline Compound I according to Clause 105, wherein 100% of Compound I is crystalline.
110. Substantially amorphous Compound I amorphous form. 111. The substantially amorphous Compound I according to Clause 110, wherein less than 15% of Compound I is in crystalline form. 112. The substantially crystalline Compound I according to Clause 110, wherein less than 10% of Compound I is in crystalline form. 113. The substantially crystalline Compound I according to Clause 110, wherein less than 5% of Compound I is in crystalline form. 114. The substantially crystalline Compound I according to Clause 110, wherein 100% of Compound I is amorphous. 115. The substantially crystalline Compound I according to any one of Clauses 105 to 109, characterized by crystal lattice parameters. 116. The substantially crystalline Compound I according to any one of Clauses 105 to 115, characterized by an X-ray powder diffractogram (XRPD). 117. The substantially crystalline Compound I according to any one of Clauses 105 to 116., characterized by 13C solid-state nuclear magnetic resonance (13C SSNMR) spectrum. 118. A pharmaceutical composition comprising the Compound I according to any one of Clauses 105 to 117. 119. The pharmaceutical composition according to Clause 118, further comprising one or more additional therapeutic agents. 120. The pharmaceutical composition according to Clause 119, wherein the pharmaceutical composition comprises one or more additional pain modulating compounds. 121. The pharmaceutical composition according to Clause 120, wherein the pharmaceutical composition comprises one or more combination compounds and pharmaceutically acceptable salts and deuterated derivatives thereof. 122. The Compound I according to any one of Clauses 105 to 117, or the pharmaceutical composition according to any one of Clauses 118 to 121, for use in the treatment of pain. 123. The compound or composition according to Clause 122, wherein the pain comprises chronic pain, gut pain, neuropathic pain, musculoskeletal pain, acute pain, inflammatory pain, cancer pain,
idiopathic pain, postsurgical pain, visceral pain, multiple sclerosis, Charcot-Marie-Tooth syndrome, incontinence, pathological cough, or cardiac arrhythmia. 124. Use of the Compound I according to any one of Clauses 105 to 117, or the pharmaceutical composition according to any one of Clauses 118 to 121, in the manufacture of a medicament for the treatment of pain. 125. A method of treating pain comprising administering the Compound I according to any one of Clauses 105 to 117, or the pharmaceutical composition according to any one of Clauses 118 to 121, to a subject in need thereof. 126. The compound or composition for use of Clause 122, the use of Clause 124, or the method of Clause 125, wherein the Compound I according to any one of Clauses 105 to 117 or the composition according to any one of Clauses 118 to 120 is administered in combination with one or more additional therapeutic agents. 127. The compound or composition for use, the use, or the method of Clause 126, wherein the one or more additional therapeutic agents comprises one or more additional pain modulating compounds. 128. The compound or composition for use, the use, or the method of Clause 126 or Clause 127, wherein the one or more additional therapeutic agents comprises one or more combination compounds selected and pharmaceutically acceptable salts and deuterated derivatives thereof. 129. A method of preparing the Compound I according to any one of Clauses 105 to 117, comprising: (a) (i) combining Compound I and ethyl acetate, (ii) distilling under vacuum at 50 °C, (iii) adding ethyl acetate, (iv) distilling under vacuum at 50 °C, (v) heating to 75 °C, (vi) adding heptane, (vii) cooling to 40 °C, (viii) adding a seed of crystalline Compound I Form A, (ix) holding at 40 °C for 1.5 hours, (x) adding heptane over 5 hours, (xi) cooling the slurry to 20 °C over 5 hours, (xii) holding at 20 °C for 11 hours, (xiii) collecting the solids, (xiv) drying the solids in a vacuum oven at 45 °C with a slight nitrogen bleed for 46 hours to yield crystalline Compound I neat Form A; (b) (i) adding Compound I hydrate Form A to an oven set at 180 °C (ii) cooling under ambient conditions to yield crystalline Compound I neat Form B; (c) (i) combining Compound I hydrate Form A and trifluorotoluene, (ii) heating the slurry to 80 °C, (iii) cooling to room temperature to yield crystalline Compound I neat Form E; (d) (i) saturating acetone with Compound I, (ii) adding water dropwise to 20% of the final volume, (iii) holding overnight at room temperature to yield crystalline Compound I Acetone Solvate Hydrate Form A; (e) (i) combining Compound I Ethanol Solvate Form A and acetone, (ii) stirring the slurry while heating 50 °C, (iii) adding water over 1 hour, (iv) adding a seed of crystalline Compound I hydrate
Form A, (v) agitating for 30 minutes, (vi) adding water over 8 hours, (vii) stirring at 50 °C for 5 hours, (viii) cooling the slurry to 20 °C over 3 hours, (ix) agitating for 2 hours, (x) collecting the solids and washing with 1:2 acetone:water, (xi) drying the solids in a vacuum over at 50 °C with a slight nitrogen bleed to yield crystalline Compound I Hydrate Form A; (f) (i) combining Compound I and ethyl acetate, (ii) heating the slurry to 50 °C, (iii) adding ethanol and distilling under vacuum at 50 °C, (iv) adding ethanol and distilling under vacuum at 50 °C, (v) adding ethanol and distilling under vacuum at 50 °C to a minimum volume, (vi) adding ethanol and distilling under vacuum at 50 °C, (vii) drying in a vacuum oven at 45 °C, with a slight nitrogen bleed for 42 hours to yield crystalline Compound I Ethanol Solvate Form A; (g) (i) dissolving Compound I in acetone, (ii) filtering and evaporating the solvent at 50 °C, 12 mbar, over 1 hour on a centrifugal evaporation, (iii) drying in a vacuum oven at 50 °C overnight to yield Compound I Amorphous Form. EXAMPLES General Experimental Details [0470] General methods. [0471] 1H NMR (400 MHz) and 19F NMR (376 MHz) spectra were obtained as solutions in an appropriate deuterated solvent such as dimethyl sulfoxide-d6 (DMSO-d6). [0472] Exemplary LC Method #1: HPLC analysis was conducted using an Agilent 1260 HPLC utilizing an Agilent Poroshell EC-C18 column (4.6 x 150 mm, 2.7μm particle) guard column (pn: 693975-902), and a dual gradient run from 5-95% mobile phase B over 45 min. Mobile phase A = 0.1% (v/v) phosphoric acid in water. Mobile phase B = 0.1% (v/v) Phosphoric Acid in Acetonitrile. Flow rate = 1.0 mL/min, injection volume = 5 μL, and column temperature = 35 °C. [0473] Exemplary LC Method #2: HPLC analysis was conducted using an Agilent 1260 HPLC utilizing an Agilent Poroshell EC-C18 column (4.6 x 150 mm, 2.7μm particle) guard column (pn: 693975-902), and a dual gradient run from 5-95% mobile phase B over 15 min. Mobile phase A = 0.1% (v/v) phosphoric acid in water. Mobile phase B = 0.1% (v/v) Phosphoric Acid in Acetonitrile. Flow rate = 1.0 mL/min, injection volume = 5 μL, and column temperature = 35 °C. Solid State NMR [0474] Bruker-Biospin 400 MHz wide-bore spectrometer equipped with Bruker-Biospin 4 mm HFX probe was used. Samples were packed into 4 mm ZrO2 rotors and spun under Magic Angle Spinning (MAS) condition with spinning speed typically set to 12.5 kHz. The proton relaxation time was measured using 1H MAS T1 saturation recovery relaxation experiment in order to set up proper recycle delay of the 13C cross-polarization (CP) MAS experiments. The fluorine relaxation time was measured using 19F MAS T1 saturation recovery relaxation experiment in order to set up proper recycle delay of the 19F MAS experiment. The CP contact time of carbon CPMAS experiments was set to 2 ms.
A CP proton pulse with linear ramp (from 50% to 100%) was employed. The carbon Hartmann-Hahn match was optimized on external reference sample (glycine). All carbon and fluorine spectra were recorded with proton decoupling using TPPM15 or SPINAL64 decoupling sequence with the field strength of approximately 100 kHz. X-ray Powder Diffraction [0475] X-ray powder diffraction (XRPD) spectra were recorded at room temperature in reflection mode using a PANalytical Empyrean system equipped with a sealed tube source and a PIXcel 1D Medipix-2 detector (Malvern PANalytical Inc, Westborough, Massachusetts). The X-Ray generator operated at a voltage of 45 kV and a current of 40 mA with copper radiation (1.54060 Å). The powder sample was placed in a back filled sample holder and loaded into the instrument. The sample was scanned over the range of about 3° to about 40° 2θ with a step size of 0.0131303° and 49.725s per step. Abbreviations [0476] Unless otherwise noted, or where the context dictates otherwise, the following abbreviations shall be understood to have the following meanings: Abbreviation Meaning NMR Nuclear magnetic resonance LC/MS Liquid chromatography-mass spectrometry UPLC Ultra performance liquid chromatography HPLC/MS/MS High performance liquid chromatography/tandem mass spectrometry IS Internal standard HPLC High performance liquid chromatography SFC Supercritical fluid chromatography MDAP Mass directed auto purification g grams mg milligrams L Liter(s) mL Milliliters μL Microliters mmol millimole h hours min Minutes MHz Megahertz Hz Hertz N Normal (concentration) M Molar (concentration)
Abbreviation Meaning mM Millimolar (concentration) tBuOH tert-butyl alcohol nBuAc n-butyl acetate CPME Cyclopentyl methyl ether DCM Dichloromethane DMF N,N-Dimethylformamide DMSO Dimethyl sulfoxide DRG Dorsal root ganglia EtOH Ethanol EtOAc Ethyl acetate HATU 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate EDCI 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide IPA Isopropyl alcohol T3P Propylphosphonic anhydride, i.e., 2,4,6-tripropyl-1,3,5,2,4,6- trioxatriphosphinane 2,4,6-trioxide LC Liquid chromatography MeOH Methanol MTBE Methyl tert-butyl ether MSA methane sulfonic acid THF Tetrahydrofuran Mn(acac)2 manganese(II) acetylacetonate TEA triethylamine RB(F) Round bottom (flask) RT Room temperature TFT α,α,α-Trifluorotoluene TMSOTf trimethylsilyl trifluoromethanesulfonate DIPEA N,N-diisopropylethyl amine NMM N-methylmorpholine TBD Triazabicyclodecene DBU 1,8-diazabicyclo[5.4.0]undec-7-ene DABCO 1,4-diazabicyclo[2.2.2] octane MTBD 7-Methyl-1,5,7-triazabicyclo [4.4.0]dec-5-ene LiHMDS Lithium hexamethyldisilazide (LiN(SiMe3)2)
Example 1: Preparation of Compound I 2-[2-(3,4-difluoro-2-methyl-phenoxy)-4-methyl-5-(trifluoromethyl)-3-pyridyl]-4-oxo-1H-1,6- naphthyridine-5-carboxamide (Compound I) [0477] A -3- pyridyl]
 , -1,6- naphthyridine-5-carbonitrile (35 mg, 0.11 mmol) and potassium phosphate (41 mg, 0.19 mmol) in dioxane (720 µL) and water (240 µL) was flushed with nitrogen for 30 seconds then SPhos Pd G3 (11 mg, 0.014 mmol) added. The resulting mixture was flushed with nitrogen for 30 seconds, capped and stirred at room temperature for 16 h. The mixture was concentrated under reduced pressure and purified by silica gel chromatography (4 g silica, 0-70% ethyl acetate/hexanes to provide 2-[2-(3,4- difluoro-2-methyl-phenoxy)-4-methyl-5-(trifluoromethyl)-3-pyridyl]-4-[(4-methoxyphenyl)methoxy]- 1,6-naphthyridine-5-carbonitrile. ESI-MS m/z calc.592.15, found 593.3 (M+1)+. The PMB-protected intermediate was dissolved in toluene (1 mL) and treated with TFA (500 µL, 6.49 mmol). The reaction mixture was stirred at 60 °C for 3 days. The mixture was concentrated under reduced pressure and purified by reverse phase HPLC (C18 column, 1-99% acetonitrile/5 mM HCl) to provide 2-[2-(3,4-difluoro-2-methyl-phenoxy)-4-methyl-5-(trifluoromethyl)-3-pyridyl]-4-oxo-1H-1,6- naphthyridine-5-carboxamide (Compound I) (42, 21.9 mg, 44%) as an off-white solid. ESI-MS m/z calc.490.11, found 491.2 (M+1)+.1H NMR (400 MHz, DMSO-d6) δ 12.48 (s,1H), 8.61 (s, 1H), 8.55 (d, J = 5.8 Hz, 1H), 7.63 (s, 1H), 7.50 (d, J = 5.9 Hz, 1H), 7.44 (s, 1H), 7.35 (q, J = 9.4 Hz, 1H), 7.12 - 7.03 (m, 1H), 6.45 (s, 1H), 2.42 (s, 3H), 2.02 (d, J = 2.0 Hz, 3H).19F NMR (376 MHz, DMSO-d6) δ -59.29, -139.15 (d, J = 22.4 Hz), -141.51 (d, J = 22.2 Hz). [0478] 2-[2-(3,4-difluoro-2-methyl-phenoxy)-4-methyl-5-(trifluoromethyl)-3-pyridyl]-4-oxo-1H- 1,6-naphthyridine-5-carboxamide (Compound I) (10.00 g) was charge with ethyl acetate (30 mL) in a 100 mL round bottom flask. The resulting slurry was heated to 50 °C, followed by ethanol (20 mL) being charged to the flask. The slurry was distilled under vacuum at 50 °C until 15 g solvent remained in the flask, followed by additional ethanol (20 mL) being charged to the flask. The slurry was distilled under vacuum at 50 °C until 12 g solvent remained in the flask, followed by ethanol (30 mL) being charged to the flask. The slurry was distilled under vacuum at 50 °C to minimum volume. Ethanol (25 mL) was then charged to the flask. The slurry was distilled under vacuum at 50 °C until
only 8 g solvent remained. The resulting material was transferred to a vacuum oven at 45 °C with a slight nitrogen bleed for 42 hours to yield Compound I Ethanol Solvate (11.00 g) (Formula F). Synthesis of 2,4-dichloro-1,6-naphthyridine-5-carbonitrile (A2) O diethyl malonate Br N Et3N, dppf, Pd(OAc)2 NaOEt MeO N N oC
, -1,6- naphthyridine-5-carbonitrile (35 mg, 0.11 mmol) and potassium phosphate (41 mg, 0.19 mmol) in dioxane (720 µL) and water (240 µL) was flushed with nitrogen for 30 seconds then SPhos Pd G3 (11 mg, 0.014 mmol) added. The resulting mixture was flushed with nitrogen for 30 seconds, capped and stirred at room temperature for 16 h. The mixture was concentrated under reduced pressure and purified by silica gel chromatography (4 g silica, 0-70% ethyl acetate/hexanes to provide 2-[2-(3,4- difluoro-2-methyl-phenoxy)-4-methyl-5-(trifluoromethyl)-3-pyridyl]-4-[(4-methoxyphenyl)methoxy]- 1,6-naphthyridine-5-carbonitrile. ESI-MS m/z calc.592.15, found 593.3 (M+1)+. The PMB-protected intermediate was dissolved in toluene (1 mL) and treated with TFA (500 µL, 6.49 mmol). The reaction mixture was stirred at 60 °C for 3 days. The mixture was concentrated under reduced pressure and purified by reverse phase HPLC (C18 column, 1-99% acetonitrile/5 mM HCl) to provide 2-[2-(3,4-difluoro-2-methyl-phenoxy)-4-methyl-5-(trifluoromethyl)-3-pyridyl]-4-oxo-1H-1,6- naphthyridine-5-carboxamide (Compound I) (42, 21.9 mg, 44%) as an off-white solid. ESI-MS m/z calc.490.11, found 491.2 (M+1)+.1H NMR (400 MHz, DMSO-d6) δ 12.48 (s,1H), 8.61 (s, 1H), 8.55 (d, J = 5.8 Hz, 1H), 7.63 (s, 1H), 7.50 (d, J = 5.9 Hz, 1H), 7.44 (s, 1H), 7.35 (q, J = 9.4 Hz, 1H), 7.12 - 7.03 (m, 1H), 6.45 (s, 1H), 2.42 (s, 3H), 2.02 (d, J = 2.0 Hz, 3H).19F NMR (376 MHz, DMSO-d6) δ -59.29, -139.15 (d, J = 22.4 Hz), -141.51 (d, J = 22.2 Hz). [0478] 2-[2-(3,4-difluoro-2-methyl-phenoxy)-4-methyl-5-(trifluoromethyl)-3-pyridyl]-4-oxo-1H- 1,6-naphthyridine-5-carboxamide (Compound I) (10.00 g) was charge with ethyl acetate (30 mL) in a 100 mL round bottom flask. The resulting slurry was heated to 50 °C, followed by ethanol (20 mL) being charged to the flask. The slurry was distilled under vacuum at 50 °C until 15 g solvent remained in the flask, followed by additional ethanol (20 mL) being charged to the flask. The slurry was distilled under vacuum at 50 °C until 12 g solvent remained in the flask, followed by ethanol (30 mL) being charged to the flask. The slurry was distilled under vacuum at 50 °C to minimum volume. Ethanol (25 mL) was then charged to the flask. The slurry was distilled under vacuum at 50 °C until
only 8 g solvent remained. The resulting material was transferred to a vacuum oven at 45 °C with a slight nitrogen bleed for 42 hours to yield Compound I Ethanol Solvate (11.00 g) (Formula F). Synthesis of 2,4-dichloro-1,6-naphthyridine-5-carbonitrile (A2) O diethyl malonate Br N Et3N, dppf, Pd(OAc)2 NaOEt MeO N N oC

 1) Tf2O OH POCl3 2) TMSCN Cl CN Cl 27% HCl N
1) Tf2O OH POCl3 2) TMSCN Cl CN Cl 27% HCl N
 , eq.), DPPF (317.2 g, 0.57 mol, 0.00165 eq.) and MeOH (225 L) were charged to an autoclave and agitation was started. Pd(OAc)2 (85.6 g, 0.38 mol, 0.0011 eq) was then charged and the mixture was stirred under CO (100 psi) at 80 °C. After the reaction was completed, as determined by HPLC analysis, the mixture was concentrated in vacuo to give the crude product. H2O (3.5 V) was charged, and the resulting mixture was stirred at 20 °C. The resulting suspension was filtered, and the wet cake was dried under reduced pressure to give methyl 4-aminonicotinate (A1-4) (42 kg, 276.3 mol, 79.6% yield) as an off-white solid. m/z = [M+H]+ 153.1; 1H NMR (400 MHz, DMSO-d6) δ 8.66 (s, 1H), 8.07 (s, 1H), 7.27 (s, 2H), 6.68 (d, J = 4.0 Hz, 1H), 3.81 (s, 3H). Step 2: Synthesis of ethyl 4-hydroxy-2-oxo-1,2-dihydro-1,6-naphthyridine-3-carboxylate (A1-3) [0480] Methyl 4-aminonicotinate (A1-4) (42 kg, 276.3 mol, 1 eq) and diethyl malonate (53.1 kg, 331.5 mol, 1.2 eq.) were charged to a reactor at 25 °C followed by EtOH (294 L) and EtONa (24.4 kg, 359.2 mol, 1.3 eq.). The mixture was stirred at 90 °C. After the reaction was completed, as determined by HPLC analysis, the reaction mixture was acidified with HCl (1.5 M) to pH 3~4. The mixture was then concentrated in vacuo. The slurry was cooled to 25 oC and filtered. The filter cake was dried under vacuum to afford crude ethyl 4-hydroxy-2-oxo-1,2-dihydro-1,6-naphthyridine-3-carboxylate (A1-3) (~65 kg), which was used directly in the next step without further purification. m/z = [M-OEt]+ 189.0; 1H NMR (400 MHz, DMSO-d6): δ 10.02 (s, 1H), 8.78 (s, 1H), 8.20 (d, J = 5.4 Hz, 1H), 6.87 (d, J = 6.1 Hz, 1H), 4.03 (q, J = 7.1 Hz, 2H), 1.19 (t, J = 7.0 Hz, 3H).
Step 3: Synthesis of 4-hydroxy-1,6-naphthyridin-2(1H)-one (A1-2) [0481] Crude ethyl 4-hydroxy-2-oxo-1,2-dihydro-1,6-naphthyridine-3-carboxylate (A1-3) (64.8 kg) and toluene (32 L) were charged to a reactor followed by HCl (453 L, 27% w/w% in H2O). The mixture was stirred at 90 °C. After the reaction was completed, the mixture was cooled to 25 °C and the layers were separated. The aqueous layer was cooled to 0 °C and basified with 30% NaOH to pH 5-6. The resulting slurry was filtered and the filter cake was triturated with MeCN (5 V). The wet cake was dried under vacuum to afford 4-hydroxy-1,6-naphthyridin-2(1H)-one (A1-2) (33.5 kg) as an off-white solid. m/z = [M+H]+ 163.0; 1H NMR (400 MHz, DMSO-d6) δ 11.97 (s, 1H), 8.97 (s, 1H), 8.53 (d, J = 8.0 Hz, 1H), 7.36 (d, J = 8.0 Hz, 1H), 5.91 (s, 1H). Step 4: Synthesis of 2,4-dichloro-1,6-naphthyridine (A2-1) [0482] 4-Hydroxy-1,6-naphthyridin-2(1H)-one (A1-2) (33.5 kg, 206.6 mol, 1.0 eq.) and toluene (67 L) were charged to a reactor followed by POCl3 (88 kg, 574.3 mol, 2.78 eq.). The mixture was stirred at 110 °C. After the reaction was completed, as determined by HPLC analysis, the mixture was concentrated in vacuo. The residue was diluted with CH2Cl2 (5 V), quenched into ice water (5 V), and then the resulting mixture was poured into sat. NaHCO3. The resulting mixture was filtered, and the layers of the bi-phasic filtrate were separated. The aqueous phase was extracted with EtOAc. All organic phases were combined, washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated under vacuum. The residue was triturated with MTBE (2 V) to afford 2,4-dichloro-1,6- naphthyridine (A2-1) (33.6 kg, 168.8 mol, 81.7% yield) as a yellow solid. m/z = [M35Cl35Cl+H]+ 199.0; [M35Cl37Cl+H]+ 201.0; [M37Cl37Cl+H]+ 203.0 (9:6:1 ratio); 1H NMR (400 MHz, DMSO-d6) δ 9.57 (s, 1H), 8.91 (d, J = 4.0 Hz, 1H), 8.12 (s, 1H), 7.94 (d, J = 4.0 Hz, 1H). Step 5: Synthesis of 2,4-dichloro-1,6-naphthyridine-5-carbonitrile (A2) [0483] 2,4-Dichloro-1,6-naphthyridine (A2-1) (33.6 kg, 168.8 mol, 1 eq.) and CH2Cl2 (336 L) were charged to a reactor. The solution was cooled to -10 °C. Tf2O (50 kg, 177.2 mol, 1.05 eq.) was charged. After complete addition, TMSCN (17.5 kg, 177.2 mol, 1.05 eq.) was charged followed by Et3N (22.2 kg, 219.4 mol, 1.3 eq.). After reaction completion, as determined by HPLC analysis, the reaction mixture was diluted with CH2Cl2 (7.5 V) and basified to pH 7 with sat. NaHCO3. The layers were separated, and the aqueous layer was extracted with CH2Cl2 (2 x 5 V). The combined organic layers were washed with water (2 x 5 L) and brine and dried over with anhydrous Na2SO4. The organic solution was concentrated under vacuum and the residue was triturated with MeOH (4 V). The slurry was filtered, and the wet cake was dried under vacuum to afford 2,4-dichloro-1,6- naphthyridine-5-carbonitrile (A2) as a yellow solid (31.21 kg). m/z = [M+H]+ 223.9; 1H NMR (400 MHz, DMSO-d6) δ 9.01 (d, J = 5.6 Hz, 1H), 8.32 (s, 1H), 8.29 (d, J = 5.6 Hz, 1H).
Synthesis of 3-bromo-2-(3,4-difluoro-2-methylphenoxy)-4-methyl-5-(trifluoromethyl)pyridine (D-2) [0484] was stirred
, eq.), DPPF (317.2 g, 0.57 mol, 0.00165 eq.) and MeOH (225 L) were charged to an autoclave and agitation was started. Pd(OAc)2 (85.6 g, 0.38 mol, 0.0011 eq) was then charged and the mixture was stirred under CO (100 psi) at 80 °C. After the reaction was completed, as determined by HPLC analysis, the mixture was concentrated in vacuo to give the crude product. H2O (3.5 V) was charged, and the resulting mixture was stirred at 20 °C. The resulting suspension was filtered, and the wet cake was dried under reduced pressure to give methyl 4-aminonicotinate (A1-4) (42 kg, 276.3 mol, 79.6% yield) as an off-white solid. m/z = [M+H]+ 153.1; 1H NMR (400 MHz, DMSO-d6) δ 8.66 (s, 1H), 8.07 (s, 1H), 7.27 (s, 2H), 6.68 (d, J = 4.0 Hz, 1H), 3.81 (s, 3H). Step 2: Synthesis of ethyl 4-hydroxy-2-oxo-1,2-dihydro-1,6-naphthyridine-3-carboxylate (A1-3) [0480] Methyl 4-aminonicotinate (A1-4) (42 kg, 276.3 mol, 1 eq) and diethyl malonate (53.1 kg, 331.5 mol, 1.2 eq.) were charged to a reactor at 25 °C followed by EtOH (294 L) and EtONa (24.4 kg, 359.2 mol, 1.3 eq.). The mixture was stirred at 90 °C. After the reaction was completed, as determined by HPLC analysis, the reaction mixture was acidified with HCl (1.5 M) to pH 3~4. The mixture was then concentrated in vacuo. The slurry was cooled to 25 oC and filtered. The filter cake was dried under vacuum to afford crude ethyl 4-hydroxy-2-oxo-1,2-dihydro-1,6-naphthyridine-3-carboxylate (A1-3) (~65 kg), which was used directly in the next step without further purification. m/z = [M-OEt]+ 189.0; 1H NMR (400 MHz, DMSO-d6): δ 10.02 (s, 1H), 8.78 (s, 1H), 8.20 (d, J = 5.4 Hz, 1H), 6.87 (d, J = 6.1 Hz, 1H), 4.03 (q, J = 7.1 Hz, 2H), 1.19 (t, J = 7.0 Hz, 3H).
Step 3: Synthesis of 4-hydroxy-1,6-naphthyridin-2(1H)-one (A1-2) [0481] Crude ethyl 4-hydroxy-2-oxo-1,2-dihydro-1,6-naphthyridine-3-carboxylate (A1-3) (64.8 kg) and toluene (32 L) were charged to a reactor followed by HCl (453 L, 27% w/w% in H2O). The mixture was stirred at 90 °C. After the reaction was completed, the mixture was cooled to 25 °C and the layers were separated. The aqueous layer was cooled to 0 °C and basified with 30% NaOH to pH 5-6. The resulting slurry was filtered and the filter cake was triturated with MeCN (5 V). The wet cake was dried under vacuum to afford 4-hydroxy-1,6-naphthyridin-2(1H)-one (A1-2) (33.5 kg) as an off-white solid. m/z = [M+H]+ 163.0; 1H NMR (400 MHz, DMSO-d6) δ 11.97 (s, 1H), 8.97 (s, 1H), 8.53 (d, J = 8.0 Hz, 1H), 7.36 (d, J = 8.0 Hz, 1H), 5.91 (s, 1H). Step 4: Synthesis of 2,4-dichloro-1,6-naphthyridine (A2-1) [0482] 4-Hydroxy-1,6-naphthyridin-2(1H)-one (A1-2) (33.5 kg, 206.6 mol, 1.0 eq.) and toluene (67 L) were charged to a reactor followed by POCl3 (88 kg, 574.3 mol, 2.78 eq.). The mixture was stirred at 110 °C. After the reaction was completed, as determined by HPLC analysis, the mixture was concentrated in vacuo. The residue was diluted with CH2Cl2 (5 V), quenched into ice water (5 V), and then the resulting mixture was poured into sat. NaHCO3. The resulting mixture was filtered, and the layers of the bi-phasic filtrate were separated. The aqueous phase was extracted with EtOAc. All organic phases were combined, washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated under vacuum. The residue was triturated with MTBE (2 V) to afford 2,4-dichloro-1,6- naphthyridine (A2-1) (33.6 kg, 168.8 mol, 81.7% yield) as a yellow solid. m/z = [M35Cl35Cl+H]+ 199.0; [M35Cl37Cl+H]+ 201.0; [M37Cl37Cl+H]+ 203.0 (9:6:1 ratio); 1H NMR (400 MHz, DMSO-d6) δ 9.57 (s, 1H), 8.91 (d, J = 4.0 Hz, 1H), 8.12 (s, 1H), 7.94 (d, J = 4.0 Hz, 1H). Step 5: Synthesis of 2,4-dichloro-1,6-naphthyridine-5-carbonitrile (A2) [0483] 2,4-Dichloro-1,6-naphthyridine (A2-1) (33.6 kg, 168.8 mol, 1 eq.) and CH2Cl2 (336 L) were charged to a reactor. The solution was cooled to -10 °C. Tf2O (50 kg, 177.2 mol, 1.05 eq.) was charged. After complete addition, TMSCN (17.5 kg, 177.2 mol, 1.05 eq.) was charged followed by Et3N (22.2 kg, 219.4 mol, 1.3 eq.). After reaction completion, as determined by HPLC analysis, the reaction mixture was diluted with CH2Cl2 (7.5 V) and basified to pH 7 with sat. NaHCO3. The layers were separated, and the aqueous layer was extracted with CH2Cl2 (2 x 5 V). The combined organic layers were washed with water (2 x 5 L) and brine and dried over with anhydrous Na2SO4. The organic solution was concentrated under vacuum and the residue was triturated with MeOH (4 V). The slurry was filtered, and the wet cake was dried under vacuum to afford 2,4-dichloro-1,6- naphthyridine-5-carbonitrile (A2) as a yellow solid (31.21 kg). m/z = [M+H]+ 223.9; 1H NMR (400 MHz, DMSO-d6) δ 9.01 (d, J = 5.6 Hz, 1H), 8.32 (s, 1H), 8.29 (d, J = 5.6 Hz, 1H).
Synthesis of 3-bromo-2-(3,4-difluoro-2-methylphenoxy)-4-methyl-5-(trifluoromethyl)pyridine (D-2) [0484] was stirred
 at 20 °C. to 40 °C. A solution of 3-bromo-2-chloro-4-methyl-5-(trifluoromethyl)pyridine (B-1, 1.0 equiv.) in DMSO (2.0 Vol) was added. After reaction completion, as determined by HPLC, the reaction was cooled, MTBE (8 Vol) and water (12 Vol, 5-10°C) were added, and the phases were separated. The aqueous layer was back extracted with MTBE (8 Vol). The combined organic phases were washed with 0.25 M NaOH (3 x 4 Vol), followed by 2% brine (4 Vol). The organic solution was filtered through a bed of silica. The filtrate was concentrated to dryness.3-bromo-2-(3,4-difluoro-2-methylphenoxy)-4-methyl- 5-(trifluoromethyl) pyridine (D-2) was isolated as an oil (3434 g; 98.6% yield).1H NMR (400 MHz, DMSO-d6) δ 8.39 (s, 1H), 7.37 (q, J = 9.4 Hz, 1H), 7.09 (ddd, J = 9.3, 4.4, 1.9 Hz, 1H), 2.58 (s, 3H), 2.02 (s, 3H).19F NMR (376 MHz, DMSO-d6) δ -58.43 – -60.75 (m), -139.10 – -139.23 (m), -141.40 (ddd, J = 22.2, 10.1, 4.4 Hz). MS ESI+: 382.1 m/z. Synthesis of (2-(3,4-difluoro-2-methylphenoxy)-4-methyl-5-(trifluoromethyl)pyridin-3- yl)boronic acid (D-1)
at 20 °C. to 40 °C. A solution of 3-bromo-2-chloro-4-methyl-5-(trifluoromethyl)pyridine (B-1, 1.0 equiv.) in DMSO (2.0 Vol) was added. After reaction completion, as determined by HPLC, the reaction was cooled, MTBE (8 Vol) and water (12 Vol, 5-10°C) were added, and the phases were separated. The aqueous layer was back extracted with MTBE (8 Vol). The combined organic phases were washed with 0.25 M NaOH (3 x 4 Vol), followed by 2% brine (4 Vol). The organic solution was filtered through a bed of silica. The filtrate was concentrated to dryness.3-bromo-2-(3,4-difluoro-2-methylphenoxy)-4-methyl- 5-(trifluoromethyl) pyridine (D-2) was isolated as an oil (3434 g; 98.6% yield).1H NMR (400 MHz, DMSO-d6) δ 8.39 (s, 1H), 7.37 (q, J = 9.4 Hz, 1H), 7.09 (ddd, J = 9.3, 4.4, 1.9 Hz, 1H), 2.58 (s, 3H), 2.02 (s, 3H).19F NMR (376 MHz, DMSO-d6) δ -58.43 – -60.75 (m), -139.10 – -139.23 (m), -141.40 (ddd, J = 22.2, 10.1, 4.4 Hz). MS ESI+: 382.1 m/z. Synthesis of (2-(3,4-difluoro-2-methylphenoxy)-4-methyl-5-(trifluoromethyl)pyridin-3- yl)boronic acid (D-1)
 [0485] Under an inert atmosphere, PPh2Cy (0.044 equiv.) and MeOH (0.5 Vol) were charged to a reactor. The suspension was stirred, degassed and sparged with N2. Pd(OAc)2 (0.025 equiv.) was charged, and the suspension warmed at about 50 – 55 °C. In a separate reactor, 3-bromo-2-(3,4- difluoro-2-methylphenoxy)-4-methyl-5-(trifluoromethyl)pyridine (D-2, 1.0 equiv.), KOAc (2.5 equiv.), diboronic acid (1.5 equiv.), and MeOH (7.5 Vol) were stirred at 20 °C and the mixture was degassed sparged with nitrogen. The catalyst-containing solution was transferred to the other reactor. The combined mixture was then heated to 40 °C. Upon reaction completion, as determined by HPLC
analysis, the mixture was cooled to RT and filtered through a pad of charcoal and celite. The filtrate was concentrated, and the residue was partitioned between MTBE (10 Vol) and water (6 Vol). The organic phase was washed with 20 wt% NH4Cl (3 Vol), water (3 Vol), and 2% aq. NaCl (3 Vol). The organic layer was treated with Si-Thiol (5% wt eq.). The resin was filtered, and the filtrate was concentrated to dryness. The concentrate was then diluted with DCM (6 Vol) and the solution was re- concentrated. The concentrate was slurried in 1:1 DCM:heptane (6 Vol) and filtered. The isolated solid was air-dried with suction and dried further under vacuum (40 °C) to afford (2-(3,4-difluoro-2- methylphenoxy)-4-methyl-5-(trifluoromethyl)pyridin-3-yl)boronic acid (D-1) as a white to off-white solid (2617 g; 85% yield). 1H NMR (400 MHz, DMSO-d6) δ 8.71 (s, 1H), 8.33 (s, 1H), 7.33 (q, J = 9.4 Hz, 1H), 6.98 (ddd, J = 9.2, 4.3, 1.9 Hz, 1H), 2.44 (s, 2H), 2.06 (s, 2H).19F NMR (376 MHz, DMSO-d6) δ -58.90 – -59.95 (m), -139.32 – -139.72 (m), -142.33 (ddd, J = 22.6, 10.3, 4.6 Hz). MS ESI+: 348.4 m/z. Synthesis of 4-chloro-2-(2-(3,4-difluoro-2-methylphenoxy)-4-methyl-5-(trifluoromethyl)pyridin- 3-yl)-1,6-naphthyridine-5-carbonitrile (E-A2) [0486]
[0485] Under an inert atmosphere, PPh2Cy (0.044 equiv.) and MeOH (0.5 Vol) were charged to a reactor. The suspension was stirred, degassed and sparged with N2. Pd(OAc)2 (0.025 equiv.) was charged, and the suspension warmed at about 50 – 55 °C. In a separate reactor, 3-bromo-2-(3,4- difluoro-2-methylphenoxy)-4-methyl-5-(trifluoromethyl)pyridine (D-2, 1.0 equiv.), KOAc (2.5 equiv.), diboronic acid (1.5 equiv.), and MeOH (7.5 Vol) were stirred at 20 °C and the mixture was degassed sparged with nitrogen. The catalyst-containing solution was transferred to the other reactor. The combined mixture was then heated to 40 °C. Upon reaction completion, as determined by HPLC
analysis, the mixture was cooled to RT and filtered through a pad of charcoal and celite. The filtrate was concentrated, and the residue was partitioned between MTBE (10 Vol) and water (6 Vol). The organic phase was washed with 20 wt% NH4Cl (3 Vol), water (3 Vol), and 2% aq. NaCl (3 Vol). The organic layer was treated with Si-Thiol (5% wt eq.). The resin was filtered, and the filtrate was concentrated to dryness. The concentrate was then diluted with DCM (6 Vol) and the solution was re- concentrated. The concentrate was slurried in 1:1 DCM:heptane (6 Vol) and filtered. The isolated solid was air-dried with suction and dried further under vacuum (40 °C) to afford (2-(3,4-difluoro-2- methylphenoxy)-4-methyl-5-(trifluoromethyl)pyridin-3-yl)boronic acid (D-1) as a white to off-white solid (2617 g; 85% yield). 1H NMR (400 MHz, DMSO-d6) δ 8.71 (s, 1H), 8.33 (s, 1H), 7.33 (q, J = 9.4 Hz, 1H), 6.98 (ddd, J = 9.2, 4.3, 1.9 Hz, 1H), 2.44 (s, 2H), 2.06 (s, 2H).19F NMR (376 MHz, DMSO-d6) δ -58.90 – -59.95 (m), -139.32 – -139.72 (m), -142.33 (ddd, J = 22.6, 10.3, 4.6 Hz). MS ESI+: 348.4 m/z. Synthesis of 4-chloro-2-(2-(3,4-difluoro-2-methylphenoxy)-4-methyl-5-(trifluoromethyl)pyridin- 3-yl)-1,6-naphthyridine-5-carbonitrile (E-A2) [0486]
 2- methylphenoxy)-4-methyl-5-(trifluoromethyl)pyridin-3-yl)boronic acid (D-1, 1.1 eq), and potassium fluoride (2.2 equiv) were mixed in acetonitrile (10 vol). The mixture was sparged with nitrogen and PdCl2(dtbpf) (0.05 equiv) was added to the reactor. After reaction completion, as determined by HPLC analysis, the mixture was filtered through a pad of celite. The celite was washed with acetonitrile (3 vol). DCM (20 vol) and H2O (8 vol) were added to the reactor, along with the filter cake and additional celite. The layers were separated, and the isolated organic layer was filtered through a pad of celite. The celite was washed with DCM and the filtrate was washed with H2O (8 vol). The organic layer was treated with MP-TMT (0.32 w/w). The resin was filtered and the filtrate was concentrated. EtOH (6 vol) was added to the resulting residue and that solution was concentrated. The slurry was heated 40 °C and the solids were collected by filtration after cooling to 20 °C. The product was dried at 20 °C to afford 4-chloro-2-(2-(3,4-difluoro-2-methylphenoxy)-4-methyl-5- (trifluoromethyl)pyridin-3-yl)-1,6-naphthyridine-5-carbonitrile (E-A2) as an off-white solid (91 g, 84% yield).1H NMR (400 MHz, CDCl3) δ 8.99 (d, J = 5.7 Hz, 1H), 8.22 (d, J = 5.7 Hz, 1H), 7.89 (s, 1H), 7.02 (q, J = 9.0 Hz, 1H), 6.77 (ddd, J = 9.1, 4.1, 2.1 Hz, 1H), 2.34 (d, J = 1.6 Hz, 3H), 2.05 (d, J
= 2.1 Hz, 3H) ppm.19F NMR (376 MHz, CDCl3) δ -60.67, -137.63, -137.68, -140.19, -140.24 ppm. MS ESI+: 491.5 m/z. Synthesis of 2-(2-(3,4-difluoro-2-methylphenoxy)-4-methyl-5-(trifluoromethyl)pyridin-3-yl)-4- oxo-1,4-dihydro-1,6-naphthyridine-5-carboxamide ethanol solvate (Compound I • ethanol solvate; Formula F) [0487] 4- pyridin-3-yl)-
2- methylphenoxy)-4-methyl-5-(trifluoromethyl)pyridin-3-yl)boronic acid (D-1, 1.1 eq), and potassium fluoride (2.2 equiv) were mixed in acetonitrile (10 vol). The mixture was sparged with nitrogen and PdCl2(dtbpf) (0.05 equiv) was added to the reactor. After reaction completion, as determined by HPLC analysis, the mixture was filtered through a pad of celite. The celite was washed with acetonitrile (3 vol). DCM (20 vol) and H2O (8 vol) were added to the reactor, along with the filter cake and additional celite. The layers were separated, and the isolated organic layer was filtered through a pad of celite. The celite was washed with DCM and the filtrate was washed with H2O (8 vol). The organic layer was treated with MP-TMT (0.32 w/w). The resin was filtered and the filtrate was concentrated. EtOH (6 vol) was added to the resulting residue and that solution was concentrated. The slurry was heated 40 °C and the solids were collected by filtration after cooling to 20 °C. The product was dried at 20 °C to afford 4-chloro-2-(2-(3,4-difluoro-2-methylphenoxy)-4-methyl-5- (trifluoromethyl)pyridin-3-yl)-1,6-naphthyridine-5-carbonitrile (E-A2) as an off-white solid (91 g, 84% yield).1H NMR (400 MHz, CDCl3) δ 8.99 (d, J = 5.7 Hz, 1H), 8.22 (d, J = 5.7 Hz, 1H), 7.89 (s, 1H), 7.02 (q, J = 9.0 Hz, 1H), 6.77 (ddd, J = 9.1, 4.1, 2.1 Hz, 1H), 2.34 (d, J = 1.6 Hz, 3H), 2.05 (d, J
= 2.1 Hz, 3H) ppm.19F NMR (376 MHz, CDCl3) δ -60.67, -137.63, -137.68, -140.19, -140.24 ppm. MS ESI+: 491.5 m/z. Synthesis of 2-(2-(3,4-difluoro-2-methylphenoxy)-4-methyl-5-(trifluoromethyl)pyridin-3-yl)-4- oxo-1,4-dihydro-1,6-naphthyridine-5-carboxamide ethanol solvate (Compound I • ethanol solvate; Formula F) [0487] 4- pyridin-3-yl)-
 1,6-naphthyridine- were to a reactor, followed by addition of TFA (7 vol). The mixture was heated to 70 °C. Upon reaction completion, as determined by HPLC analysis, the mixture was concentrated, and toluene was charged (2 vol) to the mixture. The mixture was concentrated again and then EtOAc (10 vol) was added. The organic solution was washed with saturated aqueous NaHCO3 (6 vol) followed an aqueous 2% w/w NaCl solution (4 vol). The organic solution was then filtered through a silica plug. The filtrate was then concentrated and heated to 70 °C. The solution was seeded (0.01 equiv) with 2-(2-(3,4-difluoro-2- methylphenoxy)-4-methyl-5-(trifluoromethyl)pyridin-3-yl)-4-oxo-1,4-dihydro-1,6-naphthyridine-5- carboxamide ethanol solvate, and ethanol (6 vol) was charged to the reactor. The slurry was cooled to 20 °C and filtered. The wet cake was washed with ethanol and dried at 40 °C to obtain 2-(2-(3,4- difluoro-2-methylphenoxy)-4-methyl-5-(trifluoromethyl)pyridin-3-yl)-4-oxo-1,4-dihydro-1,6- naphthyridine-5-carboxamide ethanol solvate (I • EtOH) as an off-white solid (3280 g, 92% yield).1H NMR (400 MHz, DMSO-d6) δ 12.28 (s, 1H), 8.61 (s, 1H), 8.53 (d, J = 5.8 Hz, 1H), 7.53 (s, 1H), 7.42 (d, J = 5.8 Hz, 1H), 7.40 – 7.29 (m, 2H), 7.06 (ddd, J = 9.3, 4.3, 1.9 Hz, 1H), 6.40 (s, 1H), 4.34 (t, J = 5.1 Hz, 1H), 3.44 (qd, J = 7.0, 5.0 Hz, 2H), 2.42 (d, J = 1.7 Hz, 3H), 2.02 (d, J = 2.0 Hz, 3H), 1.06 (t, J = 7.0 Hz, 3H) ppm.19F NMR (376 MHz, DMSO-d6) δ -58.89 – -59.90 (m), -139.14 (dd, J = 22.4, 9.0 Hz), -141.51 (ddd, J = 22.2, 10.2, 4.3 Hz). MS ESI+: 491.5 m/z.
Synthesis of 2-(2-(3,4-difluoro-2-methylphenoxy)-4-methyl-5-(trifluoromethyl)pyridin-3-yl)-4- oxo-1,4-dihydro-1,6-naphthyridine-5-carboxamide monohydrate (Compound I • H2O) [0488] 3-yl)-4-oxo- 1,4-
1,6-naphthyridine- were to a reactor, followed by addition of TFA (7 vol). The mixture was heated to 70 °C. Upon reaction completion, as determined by HPLC analysis, the mixture was concentrated, and toluene was charged (2 vol) to the mixture. The mixture was concentrated again and then EtOAc (10 vol) was added. The organic solution was washed with saturated aqueous NaHCO3 (6 vol) followed an aqueous 2% w/w NaCl solution (4 vol). The organic solution was then filtered through a silica plug. The filtrate was then concentrated and heated to 70 °C. The solution was seeded (0.01 equiv) with 2-(2-(3,4-difluoro-2- methylphenoxy)-4-methyl-5-(trifluoromethyl)pyridin-3-yl)-4-oxo-1,4-dihydro-1,6-naphthyridine-5- carboxamide ethanol solvate, and ethanol (6 vol) was charged to the reactor. The slurry was cooled to 20 °C and filtered. The wet cake was washed with ethanol and dried at 40 °C to obtain 2-(2-(3,4- difluoro-2-methylphenoxy)-4-methyl-5-(trifluoromethyl)pyridin-3-yl)-4-oxo-1,4-dihydro-1,6- naphthyridine-5-carboxamide ethanol solvate (I • EtOH) as an off-white solid (3280 g, 92% yield).1H NMR (400 MHz, DMSO-d6) δ 12.28 (s, 1H), 8.61 (s, 1H), 8.53 (d, J = 5.8 Hz, 1H), 7.53 (s, 1H), 7.42 (d, J = 5.8 Hz, 1H), 7.40 – 7.29 (m, 2H), 7.06 (ddd, J = 9.3, 4.3, 1.9 Hz, 1H), 6.40 (s, 1H), 4.34 (t, J = 5.1 Hz, 1H), 3.44 (qd, J = 7.0, 5.0 Hz, 2H), 2.42 (d, J = 1.7 Hz, 3H), 2.02 (d, J = 2.0 Hz, 3H), 1.06 (t, J = 7.0 Hz, 3H) ppm.19F NMR (376 MHz, DMSO-d6) δ -58.89 – -59.90 (m), -139.14 (dd, J = 22.4, 9.0 Hz), -141.51 (ddd, J = 22.2, 10.2, 4.3 Hz). MS ESI+: 491.5 m/z.
Synthesis of 2-(2-(3,4-difluoro-2-methylphenoxy)-4-methyl-5-(trifluoromethyl)pyridin-3-yl)-4- oxo-1,4-dihydro-1,6-naphthyridine-5-carboxamide monohydrate (Compound I • H2O) [0488] 3-yl)-4-oxo- 1,4-
 and acetone (41 L, 6 vol) were charged to a reactor. The slurry was heated to 50 °C and then water (27 L, 4 vol) was charged at 50 °C. The solution was seeded with 2-(2-(3,4-difluoro-2-methylphenoxy)-4-methyl- 5-(trifluoromethyl)pyridine-3-yl)-4-oxo-1,4-dihydro-1,6-naphthyridine-5-carboxamide monohydrate I (322 g, 0.005 eq). Water (54 L, 8 vol) was charged over time and held at 50 °C for 5 hours before cooling down to 20 °C. The slurry was filtered, washed with acetone/ water mixture and dried at 50 °C to afford 2-(2-(3,4-difluoro-2-methylphenoxy)-4-methyl-5-(trifluoromethyl)pyridin-3-yl)-4-oxo- 1,4-dihydro-1,6-naphthyridine-5-carboxamide monohydrate (I • H2O) as an off-white solid (6311 g, 93%). 1H NMR (400 MHz, DMSO-d6) δ 12.28 (s, 1H), 8.61 (s, 1H), 8.53 (d, J = 5.8 Hz, 1 H), 7.54 (s, 1H), 7.42 (d, J = 5.8 Hz, 1H), 7.39 – 7.24 (m, 2H), 7.06 (ddd, J = 9.2, 4.3, 1.9 Hz, 1H), 6.40 (s, 1H), 2.47 – 2.35 (m, 3H), 2.02 (d, J = 2.1 Hz, 3H) ppm.19F NMR (376 MHz, DMSO-d6) δ -59.30, - 139.11, -139.17, -141.48, -141.54 ppm. MS ESI+: 491.5 m/z. Alternative Synthesis of Compound I from Compound E-A2 5-(trifluoromethyl)pyridin-3-yl)-4-
and acetone (41 L, 6 vol) were charged to a reactor. The slurry was heated to 50 °C and then water (27 L, 4 vol) was charged at 50 °C. The solution was seeded with 2-(2-(3,4-difluoro-2-methylphenoxy)-4-methyl- 5-(trifluoromethyl)pyridine-3-yl)-4-oxo-1,4-dihydro-1,6-naphthyridine-5-carboxamide monohydrate I (322 g, 0.005 eq). Water (54 L, 8 vol) was charged over time and held at 50 °C for 5 hours before cooling down to 20 °C. The slurry was filtered, washed with acetone/ water mixture and dried at 50 °C to afford 2-(2-(3,4-difluoro-2-methylphenoxy)-4-methyl-5-(trifluoromethyl)pyridin-3-yl)-4-oxo- 1,4-dihydro-1,6-naphthyridine-5-carboxamide monohydrate (I • H2O) as an off-white solid (6311 g, 93%). 1H NMR (400 MHz, DMSO-d6) δ 12.28 (s, 1H), 8.61 (s, 1H), 8.53 (d, J = 5.8 Hz, 1 H), 7.54 (s, 1H), 7.42 (d, J = 5.8 Hz, 1H), 7.39 – 7.24 (m, 2H), 7.06 (ddd, J = 9.2, 4.3, 1.9 Hz, 1H), 6.40 (s, 1H), 2.47 – 2.35 (m, 3H), 2.02 (d, J = 2.1 Hz, 3H) ppm.19F NMR (376 MHz, DMSO-d6) δ -59.30, - 139.11, -139.17, -141.48, -141.54 ppm. MS ESI+: 491.5 m/z. Alternative Synthesis of Compound I from Compound E-A2 5-(trifluoromethyl)pyridin-3-yl)-4-
 5-carbonitrile (F-A2)
5-carbonitrile (F-A2)
 [0489] 4-chloro-2-(2-(3,4-difluoro-2-methylphenoxy)-4-methyl-5-(trifluoromethyl)pyridin-3-yl)- 1,6-naphthyridine-5-carbonitrile (E-A2, 1.0 equiv) and acetic acid (7 vol) were charged to a reactor. The mixture was heated to 65 °C. Upon complete consumption of E-A2, as determined by HPLC analysis, the mixture was cooled to 20 °C and filtered, and the resulting solids were washed with
water. The wet cake was dried at 45 °C to obtain 2-(2-(3,4-difluoro-2-methylphenoxy)-4-methyl-5- (trifluoromethyl)pyridin-3-yl)-4-oxo-1,4-dihydro-1,6-naphthyridine-5-carbonitrile (F-A2) as an off- white solid (0.942 g, 98% yield).1H NMR (400 MHz, DMSO) δ 12.72 – 12.67 (m, 1H), 8.75 (d, J = 5.8 Hz, 1H), 8.62 (s, 1H), 7.35 (q, J = 9.4 Hz, 1H), 7.07 (ddd, J = 9.2, 4.3, 2.0 Hz, 1H), 6.57 (d, J = 1.4 Hz, 1H), 2.42 (d, J = 1.7 Hz, 3H), 2.02 (d, J = 2.0 Hz, 3H).19F NMR (376 MHz, DMSO) δ -59.31, -139.18 (dd, J = 22.3, 8.8 Hz), -141.44 (ddd, J = 22.2, 10.2, 4.2 Hz). MS ESI-: 471.1 m/z. Synthesis of Compound I Ethanol Solvate from F-A2 [0490] F-A2 by addition of
[0489] 4-chloro-2-(2-(3,4-difluoro-2-methylphenoxy)-4-methyl-5-(trifluoromethyl)pyridin-3-yl)- 1,6-naphthyridine-5-carbonitrile (E-A2, 1.0 equiv) and acetic acid (7 vol) were charged to a reactor. The mixture was heated to 65 °C. Upon complete consumption of E-A2, as determined by HPLC analysis, the mixture was cooled to 20 °C and filtered, and the resulting solids were washed with
water. The wet cake was dried at 45 °C to obtain 2-(2-(3,4-difluoro-2-methylphenoxy)-4-methyl-5- (trifluoromethyl)pyridin-3-yl)-4-oxo-1,4-dihydro-1,6-naphthyridine-5-carbonitrile (F-A2) as an off- white solid (0.942 g, 98% yield).1H NMR (400 MHz, DMSO) δ 12.72 – 12.67 (m, 1H), 8.75 (d, J = 5.8 Hz, 1H), 8.62 (s, 1H), 7.35 (q, J = 9.4 Hz, 1H), 7.07 (ddd, J = 9.2, 4.3, 2.0 Hz, 1H), 6.57 (d, J = 1.4 Hz, 1H), 2.42 (d, J = 1.7 Hz, 3H), 2.02 (d, J = 2.0 Hz, 3H).19F NMR (376 MHz, DMSO) δ -59.31, -139.18 (dd, J = 22.3, 8.8 Hz), -141.44 (ddd, J = 22.2, 10.2, 4.2 Hz). MS ESI-: 471.1 m/z. Synthesis of Compound I Ethanol Solvate from F-A2 [0490] F-A2 by addition of
 TFA (7 vol). The was to 70 °C. Upon starting material, as determined by HPLC analysis, the mixture was concentrated, and toluene was charged (2 vol) to the mixture. The mixture was concentrated again and then EtOAc (10 vol) was added. The organic solution was washed with saturated aqueous NaHCO3 (6 vol) followed an aqueous 2% w/w NaCl solution (4 vol). The organic solution was then filtered through a silica plug. The filtrate was then concentrated and heated to 70 °C. The solution was seeded (0.01 equiv) with 2-(2-(3,4-difluoro-2- methylphenoxy)-4-methyl-5-(trifluoromethyl)pyridin-3-yl)-4-oxo-1,4-dihydro-1,6-naphthyridine-5- carboxamide (Compound I) ethanol solvate, and ethanol (6 vol) was charged to the reactor. The slurry was cooled to 20 °C and filtered. The wet cake was washed with ethanol and dried at 40 °C to obtain Compound I Ethanol Solvate as an off-white solid (3280 g, 92% yield).1H NMR (400 MHz, DMSO- d6) δ 12.28 (s, 1H), 8.61 (s, 1H), 8.53 (d, J = 5.8 Hz, 1H), 7.53 (s, 1H), 7.42 (d, J = 5.8 Hz, 1H), 7.40 – 7.29 (m, 2H), 7.06 (ddd, J = 9.3, 4.3, 1.9 Hz, 1H), 6.40 (s, 1H), 4.34 (t, J = 5.1 Hz, 1H), 3.44 (qd, J = 7.0, 5.0 Hz, 2H), 2.42 (d, J = 1.7 Hz, 3H), 2.02 (d, J = 2.0 Hz, 3H), 1.06 (t, J = 7.0 Hz, 3H) ppm.19F NMR (376 MHz, DMSO-d6) δ -58.89 – -59.90 (m), -139.14 (dd, J = 22.4, 9.0 Hz), -141.51 (ddd, J = 22.2, 10.2, 4.3 Hz). MS ESI+: 491.5 m/z.
Alternative Synthesis of Compound I from E-A2 [0491] E-A2
TFA (7 vol). The was to 70 °C. Upon starting material, as determined by HPLC analysis, the mixture was concentrated, and toluene was charged (2 vol) to the mixture. The mixture was concentrated again and then EtOAc (10 vol) was added. The organic solution was washed with saturated aqueous NaHCO3 (6 vol) followed an aqueous 2% w/w NaCl solution (4 vol). The organic solution was then filtered through a silica plug. The filtrate was then concentrated and heated to 70 °C. The solution was seeded (0.01 equiv) with 2-(2-(3,4-difluoro-2- methylphenoxy)-4-methyl-5-(trifluoromethyl)pyridin-3-yl)-4-oxo-1,4-dihydro-1,6-naphthyridine-5- carboxamide (Compound I) ethanol solvate, and ethanol (6 vol) was charged to the reactor. The slurry was cooled to 20 °C and filtered. The wet cake was washed with ethanol and dried at 40 °C to obtain Compound I Ethanol Solvate as an off-white solid (3280 g, 92% yield).1H NMR (400 MHz, DMSO- d6) δ 12.28 (s, 1H), 8.61 (s, 1H), 8.53 (d, J = 5.8 Hz, 1H), 7.53 (s, 1H), 7.42 (d, J = 5.8 Hz, 1H), 7.40 – 7.29 (m, 2H), 7.06 (ddd, J = 9.3, 4.3, 1.9 Hz, 1H), 6.40 (s, 1H), 4.34 (t, J = 5.1 Hz, 1H), 3.44 (qd, J = 7.0, 5.0 Hz, 2H), 2.42 (d, J = 1.7 Hz, 3H), 2.02 (d, J = 2.0 Hz, 3H), 1.06 (t, J = 7.0 Hz, 3H) ppm.19F NMR (376 MHz, DMSO-d6) δ -58.89 – -59.90 (m), -139.14 (dd, J = 22.4, 9.0 Hz), -141.51 (ddd, J = 22.2, 10.2, 4.3 Hz). MS ESI+: 491.5 m/z.
Alternative Synthesis of Compound I from E-A2 [0491] E-A2
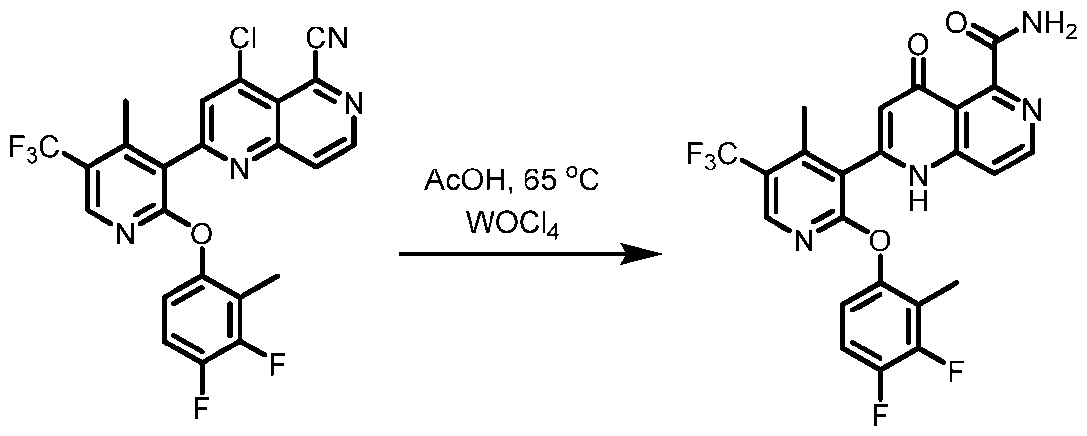 , (7 Vol) were charge to a reactor. The mixture was heated to 65 °C. After 21 hours, the mixture was cooled to room temperature. HPLC analysis showed complete consumption of E-A2 and formation of Compound I. Alternative Preparation of Starting Materials 2-chloro-4-[(4-methoxyphenyl)methoxy]-1,6-naphthyridine-5-carbonitrile Step 1: 2-chloro-4-[(4-methoxyphenyl)methoxy]-1,6-naphthyridine [0492] To a mixture g, 16.5 mmol) and (4-
, (7 Vol) were charge to a reactor. The mixture was heated to 65 °C. After 21 hours, the mixture was cooled to room temperature. HPLC analysis showed complete consumption of E-A2 and formation of Compound I. Alternative Preparation of Starting Materials 2-chloro-4-[(4-methoxyphenyl)methoxy]-1,6-naphthyridine-5-carbonitrile Step 1: 2-chloro-4-[(4-methoxyphenyl)methoxy]-1,6-naphthyridine [0492] To a mixture g, 16.5 mmol) and (4-
 methoxyphenyl)methanol g, 2.05 mL, 16.47 DMF and 2-MeTHF (33 mL) was added sodium hydride (715 mg, 60% in mineral oil, 17.88 mmol) portionwise. The mixture was stirred at 0 °C for 1 h, then gradually warmed to room temperature and stirred for 19.5 h. The mixture was poured into a stirring mixture of 0.1 M aqueous HCl (100 mL) and 2-MeTHF (100 mL). The layers were separated and the aqueous layer extracted with additional 2-MeTHF (2 x 100 mL). The organic layers were combined and washed with water (2 x 50 mL), 1:1 water/brine (50 mL) and brine (50 mL). The solution was dried over sodium sulfate, filtered, and concentrated under reduced pressure. Purification by silica gel chromatography (5-100% ethyl acetate/heptanes) provided 2-chloro-4-[(4-methoxyphenyl)methoxy]-1,6-naphthyridine (1.95 g, 39%) as a pale yellow solid. ESI- MS m/z calc.300.06, found 301.2 (M+1)+.1H NMR (400 MHz, CDCl3) δ (ppm) 9.44 (s, 1H), 8.74 (d, J= 5.9 Hz, 1H), 7.72 - 7.67 (m, 1H), 7.48 - 7.42 (m, 2H), 7.09 (s, 1H), 6.97 - 6.88 (m, 2H), 5.50 (s, 2H), 3.83 (s, 3H). ESI-MS m/z calc.300.06, found 301.2 (M+1)+.
Step 2: 2-chloro-4-[(4-methoxyphenyl)methoxy]-6-oxido-1,6-naphthyridin-6-ium [0493] A solution (5 g, 16.6 mmol) in DCM (100
methoxyphenyl)methanol g, 2.05 mL, 16.47 DMF and 2-MeTHF (33 mL) was added sodium hydride (715 mg, 60% in mineral oil, 17.88 mmol) portionwise. The mixture was stirred at 0 °C for 1 h, then gradually warmed to room temperature and stirred for 19.5 h. The mixture was poured into a stirring mixture of 0.1 M aqueous HCl (100 mL) and 2-MeTHF (100 mL). The layers were separated and the aqueous layer extracted with additional 2-MeTHF (2 x 100 mL). The organic layers were combined and washed with water (2 x 50 mL), 1:1 water/brine (50 mL) and brine (50 mL). The solution was dried over sodium sulfate, filtered, and concentrated under reduced pressure. Purification by silica gel chromatography (5-100% ethyl acetate/heptanes) provided 2-chloro-4-[(4-methoxyphenyl)methoxy]-1,6-naphthyridine (1.95 g, 39%) as a pale yellow solid. ESI- MS m/z calc.300.06, found 301.2 (M+1)+.1H NMR (400 MHz, CDCl3) δ (ppm) 9.44 (s, 1H), 8.74 (d, J= 5.9 Hz, 1H), 7.72 - 7.67 (m, 1H), 7.48 - 7.42 (m, 2H), 7.09 (s, 1H), 6.97 - 6.88 (m, 2H), 5.50 (s, 2H), 3.83 (s, 3H). ESI-MS m/z calc.300.06, found 301.2 (M+1)+.
Step 2: 2-chloro-4-[(4-methoxyphenyl)methoxy]-6-oxido-1,6-naphthyridin-6-ium [0493] A solution (5 g, 16.6 mmol) in DCM (100
 acid (4.7 g, 21 mmol). The reaction was warmed to room temperature and stirred for 4 h. The mixture was quenched with saturated aqueous sodium bicarbonate (100 mL) and the layers were separated. The aqueous layer was extracted with additional DCM (3 x 25 mL). The combined organic layers were washed with brine (20 mL x 2), dried with anhydrous magnesium sulfate, filtered, and concentrated in vacuo to obtain 2-chloro-4-[(4-methoxyphenyl)methoxy]-6-oxido-1,6-naphthyridin-6- ium (5.1 g, 97%). ESI-MS m/z calc.316.06, found 317.2 (M+1)+.1H NMR (400 MHz, DMSO-d6) δ (ppm) 8.66 (d, J= 2.1 Hz, 1H), 8.40 (dd, J= 7.3, 2.1 Hz, 1H), 7.86 (d, J= 7.3 Hz, 1H), 7.53 - 7.47 (m, 2H), 7.41 (s, 1H), 7.05 - 6.95 (m, 2H), 5.39 (s, 2H), 3.78 (s, 3H). Step 3: 2-chloro-4-[(4-methoxyphenyl)methoxy]-1,6-naphthyridine-5-carbonitrile [0494] To a 1,6-naphthyridin-6-
acid (4.7 g, 21 mmol). The reaction was warmed to room temperature and stirred for 4 h. The mixture was quenched with saturated aqueous sodium bicarbonate (100 mL) and the layers were separated. The aqueous layer was extracted with additional DCM (3 x 25 mL). The combined organic layers were washed with brine (20 mL x 2), dried with anhydrous magnesium sulfate, filtered, and concentrated in vacuo to obtain 2-chloro-4-[(4-methoxyphenyl)methoxy]-6-oxido-1,6-naphthyridin-6- ium (5.1 g, 97%). ESI-MS m/z calc.316.06, found 317.2 (M+1)+.1H NMR (400 MHz, DMSO-d6) δ (ppm) 8.66 (d, J= 2.1 Hz, 1H), 8.40 (dd, J= 7.3, 2.1 Hz, 1H), 7.86 (d, J= 7.3 Hz, 1H), 7.53 - 7.47 (m, 2H), 7.41 (s, 1H), 7.05 - 6.95 (m, 2H), 5.39 (s, 2H), 3.78 (s, 3H). Step 3: 2-chloro-4-[(4-methoxyphenyl)methoxy]-1,6-naphthyridine-5-carbonitrile [0494] To a 1,6-naphthyridin-6-
 ium (6.1 g, 19.3 mmol) DCM an was added trimethylsilyl cyanide (6.8 mL, 51 mmol), followed by the addition of TEA (8 mL, 57.4 mmol). The mixture was stirred for 20 h at room temperature. The reaction was quenched with water and the aqueous layer was extracted with DCM (3x). The combined organic layers were dried over magnesium sulfate, filtered and concentrated. Purification by silica gel chromatography (0-100% ethyl acetate/DCM) provided 2- chloro-4-[(4-methoxyphenyl)methoxy]-1,6-naphthyridine-5-carbonitrile (5.5 g, 88%).1H NMR (400 MHz, DMSO-d6) δ 8.90 (d, J= 5.8 Hz, 1H), 8.11 (d, J= 5.7 Hz, 1H), 7.61 (s, 1H), 7.60 - 7.52 (m, 2H), 7.03 - 6.95 (m, 2H), 5.52 (s, 2H), 3.77 (s, 3H). Example 2: Compound 1 Neat Amorphous Form Method for the Synthesis of Compound of Formula (I) using Intermediates A and Intermediate B Synthesis of Intermediate A-1
[0495] Step 1: 4-benzyloxy-2-chloro-1,6-naphthyridine [0496] To a mixture of 2,4-dichloro-1,6-naphthyridine (1.0 g, 4.9 mmol) and benzyl alcohol (0.5 mL, 4.8 mmol) in DMF (10 mL) and 2-MeTHF (10 mL) at 0 °C was added sodium hydride (60% in mineral oil, 208 mg, 5.20 mmol) portionwise. The reaction mixture was stirred at 0 °C for 1 h, followed gradual warming to room temperature and stirring for 2 h. The reaction mixture was poured into a stirring mixture of 0.1 M aqueous HCl (50 mL) and 2-MeTHF (50 mL). The layers were separated, and the aqueous layer extracted with 2-MeTHF (2 x 100 mL). The combined organic layers were washed with water (2 x 50 mL), 1:1 water/brine (50 mL) and brine (50 mL), dried over sodium sulfate, filtered and evaporated under reduced pressure. Purification by silica gel chromatography (0-35% ethyl acetate/heptanes) afforded 4-benzyloxy-2-chloro-1,6-naphthyridine (528 mg, 38%) as an off-white solid. ESI-MS m/z calc.270.06, found 271.1 (M+1)+.1H NMR (400 MHz, CDCl3) δ 9.58 (s, 1H), 8.78 (d, J = 5.9 Hz, 1H), 7.75 (dd, J = 5.9, 0.6 Hz, 1H), 7.54 - 7.37 (m, 5H), 6.92 (s, 1H), 5.34 (s, 2H). [0497] Step 2: 4-benzyloxy-2-chloro-6-oxido-1,6-naphthyridin-6-ium [0498] To a solution of 4-benzyloxy-2-chloro-1,6-naphthyridine (1.75 g, 6.46 mmol) in DCM (14 mL) at 0 °C was added mCPBA (1.6 g, 7.14 mmol). The resulting mixture was stirred at room temperature for 18 h, then diluted with 2 M aqueous sodium carbonate (60 mL) and water (90 mL). The aqueous layer was extracted with additional DCM (3 x 100 mL), and the combined organic layers dried over sodium sulfate, filtered and concentrated under reduced pressure to give 4-benzyloxy-2-chloro-6- oxido-1,6-naphthyridin-6-ium (1.796 g, 97%) as an off-white solid. ESI-MS m/z calc. 286.05, found 287.1 (M+1)+.1H NMR (400 MHz, CDCl3) δ 9.02 - 8.94 (m, 1H), 8.30 (dd, J = 7.3, 2.1 Hz, 1H), 7.79 - 7.72 (m, 1H), 7.44 (d, J = 2.6 Hz, 5H), 6.93 (s, 1H), 5.29 (s, 2H). [0499] Step 3: 4-benzyloxy-2-chloro-1,6-naphthyridine-5-carbonitrile [0500] To a solution of 4-benzyloxy-2-chloro-6-oxido-1,6-naphthyridin-6-ium (1.0 g, 3.5 mmol) in DCM (10 mL) under an atmosphere of nitrogen was added trimethylsilylformonitrile (1.3 mL, 9.8 mmol), followed by the addition of TEA (1.25 mL, 8.97 mmol). The reaction mixture was stirred at room temperature for 20 h. The mixture was quenched with water and the aqueous layer was extracted with DCM (3x). The combined organic layers were dried over magnesium sulfate, filtered and concentrated. Purification by silica gel chromatography (0-30% ethyl acetate/DCM) provided 4- benzyloxy-2-chloro-1,6-naphthyridine-5-carbonitrile (840 mg, 81%). ESI-MS m/z calc.295.05, found 296.1 (M+1)+.1H NMR (400 MHz, DMSO-d6) δ 8.91 (d, J = 5.7 Hz, 1H), 8.12 (d, J = 5.7 Hz, 1H), 7.67 - 7.58 (m, 3H), 7.50 - 7.34 (m, 3H), 5.61 (s, 2H). Synthesis of Intermediate A-2
[0501] Step 1: 2-chloro-4-[(4-methoxyphenyl)methoxy]-1,6-naphthyridine [0502] To a mixture at 0 °C of 2,4-dichloro-1,6-naphthyridine (3.29 g, 16.5 mmol) and (4- methoxyphenyl)methanol (2.28 g, 2.05 mL, 16.47 mmol) in DMF (33 mL) and 2-MeTHF (33 mL) was added sodium hydride (715 mg, 60% in mineral oil, 17.88 mmol) portionwise. The mixture was stirred at 0 °C for 1 h, then gradually warmed to room temperature and stirred for 19.5 h. The mixture was poured into a stirring mixture of 0.1 M aqueous HCl (100 mL) and 2-MeTHF (100 mL). The layers were separated and the aqueous layer extracted with additional 2-MeTHF (2 x 100 mL). The organic layers were combined and washed with water (2 x 50 mL), 1:1 water/brine (50 mL) and brine (50 mL). The solution was dried over sodium sulfate, filtered, and concentrated under reduced pressure. Purification by silica gel chromatography (5-100% ethyl acetate/heptanes) provided 2-chloro- 4-[(4-methoxyphenoxy)methoxy]-1,6-naphthyridine (1.95 g, 39%) as a pale yellow solid. ESI-MS m/z calc.300.06, found 301.2 (M+1)+.1H NMR (400 MHz, CDCl3) δ 9.44 (s, 1H), 8.74 (d, J = 5.9 Hz, 1H), 7.72 - 7.67 (m, 1H), 7.48 - 7.42 (m, 2H), 7.09 (s, 1H), 6.97 - 6.88 (m, 2H), 5.50 (s, 2H), 3.83 (s, 3H). ESI-MS m/z calc.300.06, found 301.2 (M+1)+. [0503] Step 2: 2-chloro-4-[(4-methoxyphenyl)methoxy]-6-oxido-1,6-naphthyridin-6-ium [0504] A solution of 2-chloro-4-[(4-methoxyphenoxy)methyl]-1,6-naphthyridine (5.0 g, 16.6 mmol) in DCM (100 mL) was cooled to 0 °C and treated with solid 3-chlorobenzenecarboperoxoic acid (4.7 g, 21 mmol). The reaction was warmed to room temperature and stirred for 4 h. The mixture was quenched with saturated aqueous sodium bicarbonate (100 mL) and the layers were separated. The aqueous layer was extracted with additional DCM (3 x 25 mL). The combined organic layers were washed with brine (20 mL x 2), dried with anhydrous magnesium sulfate, filtered, and concentrated in vacuo to obtain 2-chloro-4-[(4-methoxyphenyl)methoxy]-6-oxido-1,6-naphthyridin-6-ium (5.1 g, 97%). ESI-MS m/z calc. 316.06, found 317.2 (M+1)+. 1H NMR (400 MHz, DMSO-d6) δ 8.66 (d, J = 2.1 Hz, 1H), 8.40 (dd, J = 7.3, 2.1 Hz, 1H), 7.86 (d, J = 7.3 Hz, 1H), 7.53 - 7.47 (m, 2H), 7.41 (s, 1H), 7.05 - 6.95 (m, 2H), 5.39 (s, 2H), 3.78 (s, 3H). [0505] Step 3: 2-chloro-4-[(4-methoxyphenyl)methoxy]-1,6-naphthyridine-5-carbonitrile [0506] To a solution of 2-chloro-4-[(4-methoxyphenyl)methoxy]-6-oxido-1,6-naphthyridin-6- ium (6.1 g, 19.3 mmol) in DCM (50 mL) under an atmosphere of nitrogen was added trimethylsilyl cyanide (6.8 mL, 51 mmol), followed by the addition of TEA (8 mL, 57.4 mmol). The mixture was stirred for 20 h at room temperature. The reaction was quenched with water and the aqueous layer was extracted with DCM (3x). The combined organic layers were dried over magnesium sulfate, filtered and concentrated. Purification by silica gel chromatography (0-100% ethyl acetate/DCM) provided 2- chloro-4-[(4-methoxyphenyl)methoxy]-1,6-naphthyridine-5-carbonitrile (5.5 g, 88%).1H NMR (400 MHz, DMSO-d6) δ 8.90 (d, J = 5.8 Hz, 1H), 8.11 (d, J = 5.7 Hz, 1H), 7.61 (s, 1H), 7.60 - 7.52 (m, 2H), 7.03 - 6.95 (m, 2H), 5.52 (s, 2H), 3.77 (s, 3H).
Synthesis of Intermediate B-1 [0507]
ium (6.1 g, 19.3 mmol) DCM an was added trimethylsilyl cyanide (6.8 mL, 51 mmol), followed by the addition of TEA (8 mL, 57.4 mmol). The mixture was stirred for 20 h at room temperature. The reaction was quenched with water and the aqueous layer was extracted with DCM (3x). The combined organic layers were dried over magnesium sulfate, filtered and concentrated. Purification by silica gel chromatography (0-100% ethyl acetate/DCM) provided 2- chloro-4-[(4-methoxyphenyl)methoxy]-1,6-naphthyridine-5-carbonitrile (5.5 g, 88%).1H NMR (400 MHz, DMSO-d6) δ 8.90 (d, J= 5.8 Hz, 1H), 8.11 (d, J= 5.7 Hz, 1H), 7.61 (s, 1H), 7.60 - 7.52 (m, 2H), 7.03 - 6.95 (m, 2H), 5.52 (s, 2H), 3.77 (s, 3H). Example 2: Compound 1 Neat Amorphous Form Method for the Synthesis of Compound of Formula (I) using Intermediates A and Intermediate B Synthesis of Intermediate A-1
[0495] Step 1: 4-benzyloxy-2-chloro-1,6-naphthyridine [0496] To a mixture of 2,4-dichloro-1,6-naphthyridine (1.0 g, 4.9 mmol) and benzyl alcohol (0.5 mL, 4.8 mmol) in DMF (10 mL) and 2-MeTHF (10 mL) at 0 °C was added sodium hydride (60% in mineral oil, 208 mg, 5.20 mmol) portionwise. The reaction mixture was stirred at 0 °C for 1 h, followed gradual warming to room temperature and stirring for 2 h. The reaction mixture was poured into a stirring mixture of 0.1 M aqueous HCl (50 mL) and 2-MeTHF (50 mL). The layers were separated, and the aqueous layer extracted with 2-MeTHF (2 x 100 mL). The combined organic layers were washed with water (2 x 50 mL), 1:1 water/brine (50 mL) and brine (50 mL), dried over sodium sulfate, filtered and evaporated under reduced pressure. Purification by silica gel chromatography (0-35% ethyl acetate/heptanes) afforded 4-benzyloxy-2-chloro-1,6-naphthyridine (528 mg, 38%) as an off-white solid. ESI-MS m/z calc.270.06, found 271.1 (M+1)+.1H NMR (400 MHz, CDCl3) δ 9.58 (s, 1H), 8.78 (d, J = 5.9 Hz, 1H), 7.75 (dd, J = 5.9, 0.6 Hz, 1H), 7.54 - 7.37 (m, 5H), 6.92 (s, 1H), 5.34 (s, 2H). [0497] Step 2: 4-benzyloxy-2-chloro-6-oxido-1,6-naphthyridin-6-ium [0498] To a solution of 4-benzyloxy-2-chloro-1,6-naphthyridine (1.75 g, 6.46 mmol) in DCM (14 mL) at 0 °C was added mCPBA (1.6 g, 7.14 mmol). The resulting mixture was stirred at room temperature for 18 h, then diluted with 2 M aqueous sodium carbonate (60 mL) and water (90 mL). The aqueous layer was extracted with additional DCM (3 x 100 mL), and the combined organic layers dried over sodium sulfate, filtered and concentrated under reduced pressure to give 4-benzyloxy-2-chloro-6- oxido-1,6-naphthyridin-6-ium (1.796 g, 97%) as an off-white solid. ESI-MS m/z calc. 286.05, found 287.1 (M+1)+.1H NMR (400 MHz, CDCl3) δ 9.02 - 8.94 (m, 1H), 8.30 (dd, J = 7.3, 2.1 Hz, 1H), 7.79 - 7.72 (m, 1H), 7.44 (d, J = 2.6 Hz, 5H), 6.93 (s, 1H), 5.29 (s, 2H). [0499] Step 3: 4-benzyloxy-2-chloro-1,6-naphthyridine-5-carbonitrile [0500] To a solution of 4-benzyloxy-2-chloro-6-oxido-1,6-naphthyridin-6-ium (1.0 g, 3.5 mmol) in DCM (10 mL) under an atmosphere of nitrogen was added trimethylsilylformonitrile (1.3 mL, 9.8 mmol), followed by the addition of TEA (1.25 mL, 8.97 mmol). The reaction mixture was stirred at room temperature for 20 h. The mixture was quenched with water and the aqueous layer was extracted with DCM (3x). The combined organic layers were dried over magnesium sulfate, filtered and concentrated. Purification by silica gel chromatography (0-30% ethyl acetate/DCM) provided 4- benzyloxy-2-chloro-1,6-naphthyridine-5-carbonitrile (840 mg, 81%). ESI-MS m/z calc.295.05, found 296.1 (M+1)+.1H NMR (400 MHz, DMSO-d6) δ 8.91 (d, J = 5.7 Hz, 1H), 8.12 (d, J = 5.7 Hz, 1H), 7.67 - 7.58 (m, 3H), 7.50 - 7.34 (m, 3H), 5.61 (s, 2H). Synthesis of Intermediate A-2
[0501] Step 1: 2-chloro-4-[(4-methoxyphenyl)methoxy]-1,6-naphthyridine [0502] To a mixture at 0 °C of 2,4-dichloro-1,6-naphthyridine (3.29 g, 16.5 mmol) and (4- methoxyphenyl)methanol (2.28 g, 2.05 mL, 16.47 mmol) in DMF (33 mL) and 2-MeTHF (33 mL) was added sodium hydride (715 mg, 60% in mineral oil, 17.88 mmol) portionwise. The mixture was stirred at 0 °C for 1 h, then gradually warmed to room temperature and stirred for 19.5 h. The mixture was poured into a stirring mixture of 0.1 M aqueous HCl (100 mL) and 2-MeTHF (100 mL). The layers were separated and the aqueous layer extracted with additional 2-MeTHF (2 x 100 mL). The organic layers were combined and washed with water (2 x 50 mL), 1:1 water/brine (50 mL) and brine (50 mL). The solution was dried over sodium sulfate, filtered, and concentrated under reduced pressure. Purification by silica gel chromatography (5-100% ethyl acetate/heptanes) provided 2-chloro- 4-[(4-methoxyphenoxy)methoxy]-1,6-naphthyridine (1.95 g, 39%) as a pale yellow solid. ESI-MS m/z calc.300.06, found 301.2 (M+1)+.1H NMR (400 MHz, CDCl3) δ 9.44 (s, 1H), 8.74 (d, J = 5.9 Hz, 1H), 7.72 - 7.67 (m, 1H), 7.48 - 7.42 (m, 2H), 7.09 (s, 1H), 6.97 - 6.88 (m, 2H), 5.50 (s, 2H), 3.83 (s, 3H). ESI-MS m/z calc.300.06, found 301.2 (M+1)+. [0503] Step 2: 2-chloro-4-[(4-methoxyphenyl)methoxy]-6-oxido-1,6-naphthyridin-6-ium [0504] A solution of 2-chloro-4-[(4-methoxyphenoxy)methyl]-1,6-naphthyridine (5.0 g, 16.6 mmol) in DCM (100 mL) was cooled to 0 °C and treated with solid 3-chlorobenzenecarboperoxoic acid (4.7 g, 21 mmol). The reaction was warmed to room temperature and stirred for 4 h. The mixture was quenched with saturated aqueous sodium bicarbonate (100 mL) and the layers were separated. The aqueous layer was extracted with additional DCM (3 x 25 mL). The combined organic layers were washed with brine (20 mL x 2), dried with anhydrous magnesium sulfate, filtered, and concentrated in vacuo to obtain 2-chloro-4-[(4-methoxyphenyl)methoxy]-6-oxido-1,6-naphthyridin-6-ium (5.1 g, 97%). ESI-MS m/z calc. 316.06, found 317.2 (M+1)+. 1H NMR (400 MHz, DMSO-d6) δ 8.66 (d, J = 2.1 Hz, 1H), 8.40 (dd, J = 7.3, 2.1 Hz, 1H), 7.86 (d, J = 7.3 Hz, 1H), 7.53 - 7.47 (m, 2H), 7.41 (s, 1H), 7.05 - 6.95 (m, 2H), 5.39 (s, 2H), 3.78 (s, 3H). [0505] Step 3: 2-chloro-4-[(4-methoxyphenyl)methoxy]-1,6-naphthyridine-5-carbonitrile [0506] To a solution of 2-chloro-4-[(4-methoxyphenyl)methoxy]-6-oxido-1,6-naphthyridin-6- ium (6.1 g, 19.3 mmol) in DCM (50 mL) under an atmosphere of nitrogen was added trimethylsilyl cyanide (6.8 mL, 51 mmol), followed by the addition of TEA (8 mL, 57.4 mmol). The mixture was stirred for 20 h at room temperature. The reaction was quenched with water and the aqueous layer was extracted with DCM (3x). The combined organic layers were dried over magnesium sulfate, filtered and concentrated. Purification by silica gel chromatography (0-100% ethyl acetate/DCM) provided 2- chloro-4-[(4-methoxyphenyl)methoxy]-1,6-naphthyridine-5-carbonitrile (5.5 g, 88%).1H NMR (400 MHz, DMSO-d6) δ 8.90 (d, J = 5.8 Hz, 1H), 8.11 (d, J = 5.7 Hz, 1H), 7.61 (s, 1H), 7.60 - 7.52 (m, 2H), 7.03 - 6.95 (m, 2H), 5.52 (s, 2H), 3.77 (s, 3H).
Synthesis of Intermediate B-1 [0507]
 [0508] A mixture of 3-bromo-2-chloro-4-methyl-5-(trifluoromethyl)pyridine (2.63 g, 9.58 mmol) and 3,4-difluoro-2-methyl-phenol (2.6 g, 18 mmol) was dissolved in DMSO (26 mL). To this solution was added cesium carbonate (7.73 g, 23.7 mmol) and the mixture stirred at 90 °C for 2.5 h. The mixture was allowed to cool to room temperature then diluted with ethyl acetate. The organic solution was washed with water and brine. dried over sodium sulfate, filtered and concentrated under reduced pressure. Purification using silica gel chromatography (120 g silica, 0-20% ethyl acetate/hexanes) provided 3-bromo-2-(3,4-difluoro-2-methyl-phenoxy)-4-methyl-5-(trifluoromethyl)pyridine (3.340 g, 91%) as light orange solid. ESI-MS m/z calc.380.98, found 382.0 (M+1)+.1H NMR (400 MHz, DMSO-d6) δ 8.41 (s, 1H), 7.38 (q, J = 9.4 Hz, 1H), 7.10 (ddd, J = 9.2, 4.4, 2.1 Hz, 1H), 2.59 (d, J = 1.4 Hz, 3H), 2.03 (d, J = 2.2 Hz, 3H). [0509] Step 2: [2-(3,4-difluoro-2-methyl-phenoxy)-4-methyl-5-(trifluoromethyl)-3- pyridyl]boronic acid [0510] To a stirring solution of 3-bromo-2-(3,4-difluoro-2-methyl-phenoxy)-4-methyl-5- (trifluoromethyl)pyridine (2.0 g, 5.2 mmol) in diethyl ether (20 mL) at -78 °C was slowly added a solution of n-BuLi (2.5 mL of 2.5 M in hexanes, 6.25 mmol) under nitrogen atmosphere. The mixture was stirred at this temperature for 20 min after which a solution of trimethyl borate (1 mL, 9 mmol) in diethyl ether (6 mL) was added dropwise. The resulting mixture was allowed to warm to room temperature and stirred for 90 min. The mixture was quenched with saturated aqueous ammonium chloride (150 mL) and extracted with ethyl acetate (3 x 100 mL). The combine extracts were washed with brine (100 mL), dried over sodium sulfate, filtered and concentrated under reduced pressure. The material was purified by reverse phase chromatography (C18, 5-95% acetonitrile/water containing 0.1 % formic acid) and the product-containing fractions concentrated to remove the acetonitrile. The resulting aqueous solution was extracted with ethyl acetate (3 x 100 mL). The combined organic layers were dried over sodium sulfate, filtered and concentrated under reduced pressure to provide [2- (3,4-difluoro-2-methyl-phenoxy)-4-methyl-5-(trifluoromethyl)-3-pyridyl]boronic acid (1.64 g, 90%) as a white solid.1H NMR (400 MHz, CDCl3) δ 8.32 (s, 1H), 7.06 (q, J = 9.0 Hz, 1H), 6.85 - 6.80 (m, 1H), 5.45 (s, 2H), 2.71 - 2.67 (m, 3H), 2.10 (d, J = 2.0 Hz, 3H).19F NMR (377 MHz, CDCl3) δ -60.66 (s, 3F), -137.75 - -137.88 (m, 1F), -140.42 - -140.54 (m, 1F). ESI-MS m/z calc.347.08, found 348.2 (M+1)+.
Synthesis of Compound (I)
[0508] A mixture of 3-bromo-2-chloro-4-methyl-5-(trifluoromethyl)pyridine (2.63 g, 9.58 mmol) and 3,4-difluoro-2-methyl-phenol (2.6 g, 18 mmol) was dissolved in DMSO (26 mL). To this solution was added cesium carbonate (7.73 g, 23.7 mmol) and the mixture stirred at 90 °C for 2.5 h. The mixture was allowed to cool to room temperature then diluted with ethyl acetate. The organic solution was washed with water and brine. dried over sodium sulfate, filtered and concentrated under reduced pressure. Purification using silica gel chromatography (120 g silica, 0-20% ethyl acetate/hexanes) provided 3-bromo-2-(3,4-difluoro-2-methyl-phenoxy)-4-methyl-5-(trifluoromethyl)pyridine (3.340 g, 91%) as light orange solid. ESI-MS m/z calc.380.98, found 382.0 (M+1)+.1H NMR (400 MHz, DMSO-d6) δ 8.41 (s, 1H), 7.38 (q, J = 9.4 Hz, 1H), 7.10 (ddd, J = 9.2, 4.4, 2.1 Hz, 1H), 2.59 (d, J = 1.4 Hz, 3H), 2.03 (d, J = 2.2 Hz, 3H). [0509] Step 2: [2-(3,4-difluoro-2-methyl-phenoxy)-4-methyl-5-(trifluoromethyl)-3- pyridyl]boronic acid [0510] To a stirring solution of 3-bromo-2-(3,4-difluoro-2-methyl-phenoxy)-4-methyl-5- (trifluoromethyl)pyridine (2.0 g, 5.2 mmol) in diethyl ether (20 mL) at -78 °C was slowly added a solution of n-BuLi (2.5 mL of 2.5 M in hexanes, 6.25 mmol) under nitrogen atmosphere. The mixture was stirred at this temperature for 20 min after which a solution of trimethyl borate (1 mL, 9 mmol) in diethyl ether (6 mL) was added dropwise. The resulting mixture was allowed to warm to room temperature and stirred for 90 min. The mixture was quenched with saturated aqueous ammonium chloride (150 mL) and extracted with ethyl acetate (3 x 100 mL). The combine extracts were washed with brine (100 mL), dried over sodium sulfate, filtered and concentrated under reduced pressure. The material was purified by reverse phase chromatography (C18, 5-95% acetonitrile/water containing 0.1 % formic acid) and the product-containing fractions concentrated to remove the acetonitrile. The resulting aqueous solution was extracted with ethyl acetate (3 x 100 mL). The combined organic layers were dried over sodium sulfate, filtered and concentrated under reduced pressure to provide [2- (3,4-difluoro-2-methyl-phenoxy)-4-methyl-5-(trifluoromethyl)-3-pyridyl]boronic acid (1.64 g, 90%) as a white solid.1H NMR (400 MHz, CDCl3) δ 8.32 (s, 1H), 7.06 (q, J = 9.0 Hz, 1H), 6.85 - 6.80 (m, 1H), 5.45 (s, 2H), 2.71 - 2.67 (m, 3H), 2.10 (d, J = 2.0 Hz, 3H).19F NMR (377 MHz, CDCl3) δ -60.66 (s, 3F), -137.75 - -137.88 (m, 1F), -140.42 - -140.54 (m, 1F). ESI-MS m/z calc.347.08, found 348.2 (M+1)+.
Synthesis of Compound (I)
 [0511] Step 1: A mixture of Intermediate A (i.e., Intermediate A-1 or Intermediate A-2, 1 eq), Intermediate B (e.g., Intermediate B-1, 1 - 2 eq), palladium catalyst (1-5 mol%), e.g. PdCl2(dppf) or PdCl2(dtbpf), base (2-3 eq, eg. potassium phosphate) in organic solvent (e.g.dioxane, DMSO, toluene) and water is degassed with nitrogen bubbling and stirred under inert atmosphere at a temperature ranging from room temperature to 120 °C. The reaction mixture is filtered and purified via silica gel column chromatography or reverse phase HPLC to obtain protected Intermediate I. [0512] Step 2: A solution of the protected intermediate I in the appropriate solvent (e.g. toluene, dioxane) is treated with acid (TFA or HCl) and stirred at either room temperature or elevated temperature (e.g.70 ºC) to convert the nitrile functionality to the carboxamide. The reaction mixture is neutralized and purified via silica gel column chromatography or reverse phase column chromatography to provide the desired product I. Preparation of Compound I amorphous form [0513] To a 20 ml glass vial, Compound I (0.5 g) was added. Then, Acetone (3 ml) was added. The solid was fully dissolved. The solution was filtered, and then solvent was evaporated at 50 °C, 12 mbar, over 1 hour, on a Genevac centrifugal evaporator. Amorphous solid was obtained after the evaporation. [0514] The solid was further dried in vacuum oven at 50 °C overnight. X-Ray Powder Diffraction [0515] The XRPD pattern for Compound I amorphous form was recording using the procedure described in the General XRPD Method. [0516] The XRPD diffractogram for Compound I amorphous form is provided in FIG.1.
Solid-State 13C NMR [0517] The 13C SSNMR of Compound I amorphous form was acquired using the procedure described in the General SSNMR Method. The 13C SSNMR spectrum for Compound I amorphous form is provided in FIG.2, and the data are summarized below in Table 1. [0518] Table 1: 13C SSNMR Signals for Compound I amorphous form Peak # Chemical Shift [ppm] Intensity [rel] 1 177.6 1.8
[0511] Step 1: A mixture of Intermediate A (i.e., Intermediate A-1 or Intermediate A-2, 1 eq), Intermediate B (e.g., Intermediate B-1, 1 - 2 eq), palladium catalyst (1-5 mol%), e.g. PdCl2(dppf) or PdCl2(dtbpf), base (2-3 eq, eg. potassium phosphate) in organic solvent (e.g.dioxane, DMSO, toluene) and water is degassed with nitrogen bubbling and stirred under inert atmosphere at a temperature ranging from room temperature to 120 °C. The reaction mixture is filtered and purified via silica gel column chromatography or reverse phase HPLC to obtain protected Intermediate I. [0512] Step 2: A solution of the protected intermediate I in the appropriate solvent (e.g. toluene, dioxane) is treated with acid (TFA or HCl) and stirred at either room temperature or elevated temperature (e.g.70 ºC) to convert the nitrile functionality to the carboxamide. The reaction mixture is neutralized and purified via silica gel column chromatography or reverse phase column chromatography to provide the desired product I. Preparation of Compound I amorphous form [0513] To a 20 ml glass vial, Compound I (0.5 g) was added. Then, Acetone (3 ml) was added. The solid was fully dissolved. The solution was filtered, and then solvent was evaporated at 50 °C, 12 mbar, over 1 hour, on a Genevac centrifugal evaporator. Amorphous solid was obtained after the evaporation. [0514] The solid was further dried in vacuum oven at 50 °C overnight. X-Ray Powder Diffraction [0515] The XRPD pattern for Compound I amorphous form was recording using the procedure described in the General XRPD Method. [0516] The XRPD diffractogram for Compound I amorphous form is provided in FIG.1.
Solid-State 13C NMR [0517] The 13C SSNMR of Compound I amorphous form was acquired using the procedure described in the General SSNMR Method. The 13C SSNMR spectrum for Compound I amorphous form is provided in FIG.2, and the data are summarized below in Table 1. [0518] Table 1: 13C SSNMR Signals for Compound I amorphous form Peak # Chemical Shift [ppm] Intensity [rel] 1 177.6 1.8
 Solid-State 19F NMR [0519] The 19F SSNMR of Compound I amorphous form was acquired using the procedure described in the General SSNMR Method. The 19F SSNMR spectrum for Compound I amorphous form is provided in FIG.3, and the data are summarized below in Table 2. [0520] Table 2: 19F SSNMR Signals for Compound I amorphous form Peak # Chemical Shift [ppm] Intensity [rel] 1 -619 10
Solid-State 19F NMR [0519] The 19F SSNMR of Compound I amorphous form was acquired using the procedure described in the General SSNMR Method. The 19F SSNMR spectrum for Compound I amorphous form is provided in FIG.3, and the data are summarized below in Table 2. [0520] Table 2: 19F SSNMR Signals for Compound I amorphous form Peak # Chemical Shift [ppm] Intensity [rel] 1 -619 10
 Example 3: Compound I Neat Form A Preparation of Compound I Neat Form A [0521] Compound I (3.00 g) was charged with ethyl acetate (33 mL) in a 100 mL round bottom flask. The resulting solution was distilled to 9 mL under vacuum at 50 °C. Then ethyl acetate (15 mL) was charged to the flask. The resulting solution was distilled to 6 mL under vacuum at 50 °C. The resulting material was transferred to a 50 mL reactor with overhead stirrer. Ethyl acetate (10 mL) was used to rinse the flask and transfer to reactor. The solution was heated to 75 °C. Then heptane (12 mL) was charged. The batch was cool to 40 °C. Then the batch was seeded with Compound I Form A (70.1 mg) and held at 40 °C for 1.5 hours. Then heptane (30.6 mL) was charged over 5 hours. The slurry was cool to 20 °C over 5 hours and held at 20 °C for 11 hours. The resulting solids were collected by vacuum
filtration. The dry cake was transferred to a vacuum oven at 45 °C with a slight nitrogen bleed for 46 hours to yield 2.27 g of product, Compound I Neat Form A. X-Ray Powder Diffraction [0522] The XRPD pattern of crystalline Compound I neat Form A was recorded using the procedure described in the General XRPD Method. [0523] The XRPD diffractogram for crystalline Compound I neat Form A is provided in FIG.4, and the XRPD data are summarized below in Table 3 [0524] Table 3: XRPD Signals for Crystalline Compound I Neat Form A No. Pos. [°2θ] Area [cts*°2θ] Rel. Int.[%] 1 7.4 11.7 6 Solid-State 13C N
Example 3: Compound I Neat Form A Preparation of Compound I Neat Form A [0521] Compound I (3.00 g) was charged with ethyl acetate (33 mL) in a 100 mL round bottom flask. The resulting solution was distilled to 9 mL under vacuum at 50 °C. Then ethyl acetate (15 mL) was charged to the flask. The resulting solution was distilled to 6 mL under vacuum at 50 °C. The resulting material was transferred to a 50 mL reactor with overhead stirrer. Ethyl acetate (10 mL) was used to rinse the flask and transfer to reactor. The solution was heated to 75 °C. Then heptane (12 mL) was charged. The batch was cool to 40 °C. Then the batch was seeded with Compound I Form A (70.1 mg) and held at 40 °C for 1.5 hours. Then heptane (30.6 mL) was charged over 5 hours. The slurry was cool to 20 °C over 5 hours and held at 20 °C for 11 hours. The resulting solids were collected by vacuum
filtration. The dry cake was transferred to a vacuum oven at 45 °C with a slight nitrogen bleed for 46 hours to yield 2.27 g of product, Compound I Neat Form A. X-Ray Powder Diffraction [0522] The XRPD pattern of crystalline Compound I neat Form A was recorded using the procedure described in the General XRPD Method. [0523] The XRPD diffractogram for crystalline Compound I neat Form A is provided in FIG.4, and the XRPD data are summarized below in Table 3 [0524] Table 3: XRPD Signals for Crystalline Compound I Neat Form A No. Pos. [°2θ] Area [cts*°2θ] Rel. Int.[%] 1 7.4 11.7 6 Solid-State 13C N
 [0525] The 13C SSNMR of Compound I neat Form A was acquired using the procedure described in the General SSNMR Method. The 13C SSNMR spectrum for Compound I neat Form A is provided in FIG.5 and the data are summarized below in Table 4. [0526] Table 4: 13C SSNMR Signals for Compound I Neat Form A Peak # Chemical Shift [ppm] Intensity [rel]
[0525] The 13C SSNMR of Compound I neat Form A was acquired using the procedure described in the General SSNMR Method. The 13C SSNMR spectrum for Compound I neat Form A is provided in FIG.5 and the data are summarized below in Table 4. [0526] Table 4: 13C SSNMR Signals for Compound I Neat Form A Peak # Chemical Shift [ppm] Intensity [rel]
 Peak # Chemical Shift [ppm] Intensity [rel] 2 163.8 4.64 3 1624 372 19
Peak # Chemical Shift [ppm] Intensity [rel] 2 163.8 4.64 3 1624 372 19
 Solid-State F NMR [0527] The 19F SSNMR of Compound I neat Form A was acquired using the procedure described in the General SSNMR Method. The 19F SSNMR spectrum for Compound I neat Form A is provided in FIG.6, and the data are summarized below in Table 5. [0528] Table 5: 19F SSNMR Signals for Compound I Neat Form A Peak # Chemical Shift [ppm] Intensity [rel] 1 612 10 Single Crystal Elucidat
Solid-State F NMR [0527] The 19F SSNMR of Compound I neat Form A was acquired using the procedure described in the General SSNMR Method. The 19F SSNMR spectrum for Compound I neat Form A is provided in FIG.6, and the data are summarized below in Table 5. [0528] Table 5: 19F SSNMR Signals for Compound I Neat Form A Peak # Chemical Shift [ppm] Intensity [rel] 1 612 10 Single Crystal Elucidat
 p [0529] Single crystals having the Compound I Neat Form A structure were grown by refluxing Compound I ethanol solvate form A in heptane. X-ray diffraction data were acquired at 100K on a
Bruker diffractometer equipped with Cu Kα radiation (λ=1.54178 Å) and a CPAD detector. The structure was solved and refined using SHELX programs (Sheldrick, G.M., Acta Cryst., (2008) A64, 112-122) and results are summarized in the Table 6 below. [0530] Table 6: Single Crystal Elucidation of Compound I Neat Form A Crystal System Triclinic Space Group P-1 Example 4: Compound I N
p [0529] Single crystals having the Compound I Neat Form A structure were grown by refluxing Compound I ethanol solvate form A in heptane. X-ray diffraction data were acquired at 100K on a
Bruker diffractometer equipped with Cu Kα radiation (λ=1.54178 Å) and a CPAD detector. The structure was solved and refined using SHELX programs (Sheldrick, G.M., Acta Cryst., (2008) A64, 112-122) and results are summarized in the Table 6 below. [0530] Table 6: Single Crystal Elucidation of Compound I Neat Form A Crystal System Triclinic Space Group P-1 Example 4: Compound I N
 Preparation of Compound I Neat Form B [0531] Compound I (50 g) Hydrate Form A was added to a glass tray. The glass tray was placed in an oven of temperature set at 180 °C. The tray was kept in the oven at 180 °C for 10 minutes, and then cooled down to ambient condition. X-Ray Powder Diffraction [0532] The XRPD pattern of crystalline Compound I neat Form B was recorded using the procedure described in the General XRPD Method. [0533] The XRPD diffractogram for crystalline Compound I neat Form B is provided in FIG.7, and the XRPD data are summarized below in Table 7. [0534] Table 7: XRPD Signals for Crystalline Compound I Neat Form B No. Pos. [°2θ] Area [cts*°2θ] Rel. Int. [%] 1 4 2 12
Preparation of Compound I Neat Form B [0531] Compound I (50 g) Hydrate Form A was added to a glass tray. The glass tray was placed in an oven of temperature set at 180 °C. The tray was kept in the oven at 180 °C for 10 minutes, and then cooled down to ambient condition. X-Ray Powder Diffraction [0532] The XRPD pattern of crystalline Compound I neat Form B was recorded using the procedure described in the General XRPD Method. [0533] The XRPD diffractogram for crystalline Compound I neat Form B is provided in FIG.7, and the XRPD data are summarized below in Table 7. [0534] Table 7: XRPD Signals for Crystalline Compound I Neat Form B No. Pos. [°2θ] Area [cts*°2θ] Rel. Int. [%] 1 4 2 12
 No. Pos. [°2θ] Area [cts*°2θ] Rel. Int. [%] 13 16.9 85.8 28 Solid-State 13C N 13
No. Pos. [°2θ] Area [cts*°2θ] Rel. Int. [%] 13 16.9 85.8 28 Solid-State 13C N 13
 [0535] The C SSNMR of Compound I neat Form B was acquired using the procedure described in the General SSNMR Method. The 13C SSNMR spectrum for Compound I neat Form B is provided in FIG.8 and the data are summarized below in Table 8. [0536] Table 8: 13C SSNMR Signals for Compound I Neat Form B Peak # Chemical Shift [ppm] Intensity [rel] 1 176.9 3.12
[0535] The C SSNMR of Compound I neat Form B was acquired using the procedure described in the General SSNMR Method. The 13C SSNMR spectrum for Compound I neat Form B is provided in FIG.8 and the data are summarized below in Table 8. [0536] Table 8: 13C SSNMR Signals for Compound I Neat Form B Peak # Chemical Shift [ppm] Intensity [rel] 1 176.9 3.12
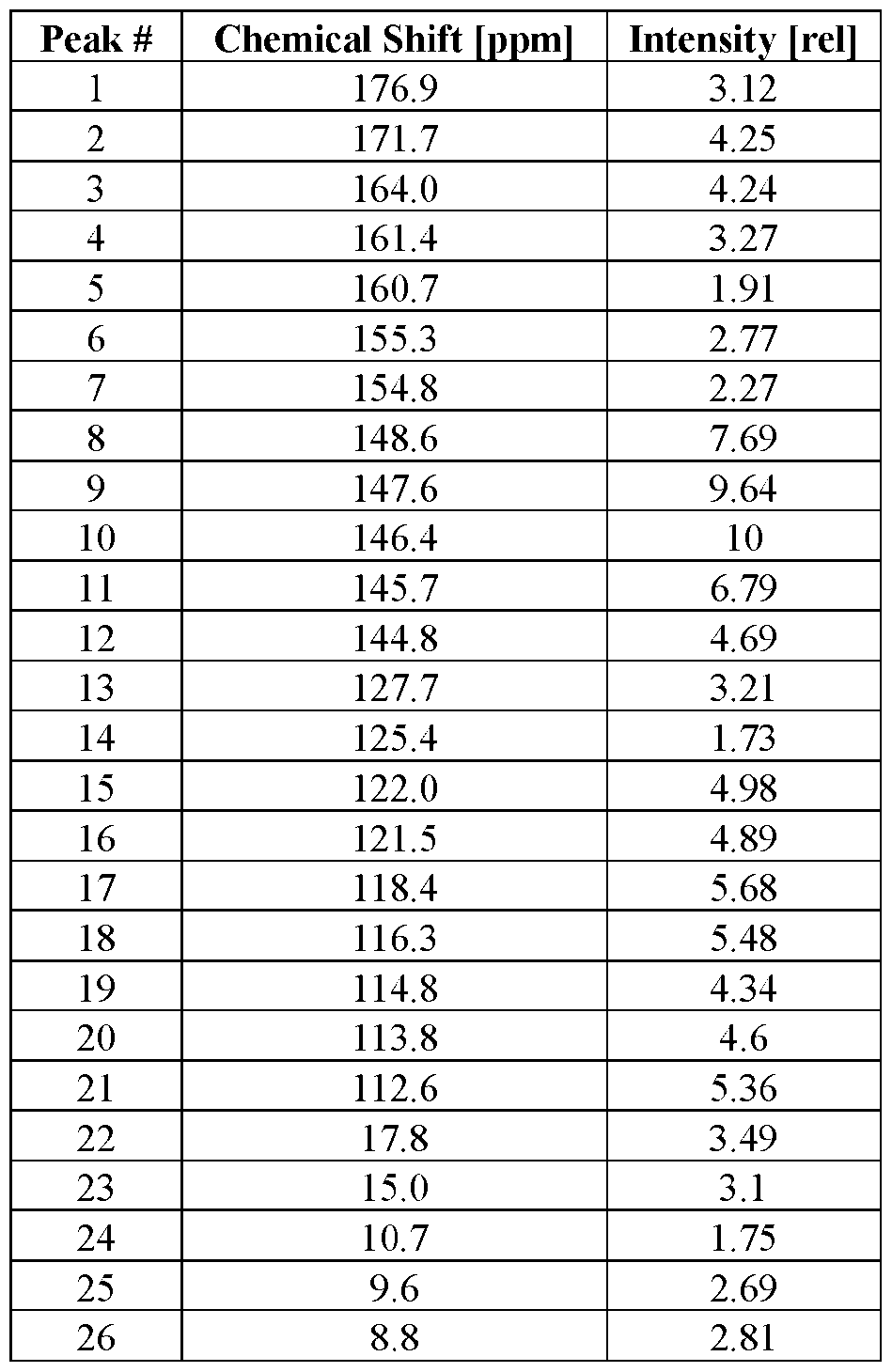 Solid-State 19F NMR [0537] The 19F SSNMR of Compound I neat Form B was acquired using the procedure described in the General SSNMR Method. The 19F SSNMR spectrum for Compound I neat Form B is provided in FIG.9, and the data are summarized below in Table 9. [0538] Table 9: 19F SSNMR Signals for Compound I Neat Form B Peak # Chemical Shift [ppm] Intensity [rel] 1 -61.6 10 Example 5: Compoun
Solid-State 19F NMR [0537] The 19F SSNMR of Compound I neat Form B was acquired using the procedure described in the General SSNMR Method. The 19F SSNMR spectrum for Compound I neat Form B is provided in FIG.9, and the data are summarized below in Table 9. [0538] Table 9: 19F SSNMR Signals for Compound I Neat Form B Peak # Chemical Shift [ppm] Intensity [rel] 1 -61.6 10 Example 5: Compoun
 Preparation of Compound I Neat Form E [0539] Trifluorotoluene (TFT) (2 mL) was added to Compound I Hydrate Form A (200 mg). The slurry was heated to 80 °C until all solids were dissolved. The solution was cooled down to room temperature, and light-yellow crystals of Compound 1 Neat Form E grew. The crystals were isolated by filtration, and dried in vacuum oven at 50 °C overnight. X-Ray Powder Diffraction [0540] The XRPD pattern of crystalline Compound I neat Form E was recorded using the procedure described in the General XRPD Method. [0541] The XRPD diffractogram for crystalline Compound I neat Form E is provided in FIG.10, and the XRPD data are summarized below in Table 10. [0542] Table 10: XRPD Signals for Crystalline Compound I Neat Form E No. Pos. [°2θ] Area [cts*°2θ] Rel. Int. [%] 1 85 770 8
Preparation of Compound I Neat Form E [0539] Trifluorotoluene (TFT) (2 mL) was added to Compound I Hydrate Form A (200 mg). The slurry was heated to 80 °C until all solids were dissolved. The solution was cooled down to room temperature, and light-yellow crystals of Compound 1 Neat Form E grew. The crystals were isolated by filtration, and dried in vacuum oven at 50 °C overnight. X-Ray Powder Diffraction [0540] The XRPD pattern of crystalline Compound I neat Form E was recorded using the procedure described in the General XRPD Method. [0541] The XRPD diffractogram for crystalline Compound I neat Form E is provided in FIG.10, and the XRPD data are summarized below in Table 10. [0542] Table 10: XRPD Signals for Crystalline Compound I Neat Form E No. Pos. [°2θ] Area [cts*°2θ] Rel. Int. [%] 1 85 770 8
 No. Pos. [°2θ] Area [cts*°2θ] Rel. Int. [%] 16 22.1 63.1 6 Solid-State 13C NM 13
No. Pos. [°2θ] Area [cts*°2θ] Rel. Int. [%] 16 22.1 63.1 6 Solid-State 13C NM 13
 [0543] The C SSNMR of Compound I neat Form E was acquired using the procedure described in the General SSNMR Method. The 13C SSNMR spectrum for Compound I neat Form E is provided in FIG.11 and the data are summarized below in Table 11. [0544] Table 11: 13C SSNMR Signals for Compound I Neat Form E Peak # Chemical Shift [ppm] Intensity [rel] 1 170.0 5.86 Solid-State 19F NMR
[0543] The C SSNMR of Compound I neat Form E was acquired using the procedure described in the General SSNMR Method. The 13C SSNMR spectrum for Compound I neat Form E is provided in FIG.11 and the data are summarized below in Table 11. [0544] Table 11: 13C SSNMR Signals for Compound I Neat Form E Peak # Chemical Shift [ppm] Intensity [rel] 1 170.0 5.86 Solid-State 19F NMR
 [0545] The 19F SSNMR of Compound I neat Form E was acquired using the procedure described in the General SSNMR Method. The 19F SSNMR spectrum for Compound I neat Form E is provided in FIG.12, and the data are summarized below in Table 12.
[0546] Table 12: 19F SSNMR Signals for Compound I Neat Form E Peak # Chemical Shift [ppm] Intensity [rel] 1 -60.9 1.09 2 625 10 Single Crystal Elucid
[0545] The 19F SSNMR of Compound I neat Form E was acquired using the procedure described in the General SSNMR Method. The 19F SSNMR spectrum for Compound I neat Form E is provided in FIG.12, and the data are summarized below in Table 12.
[0546] Table 12: 19F SSNMR Signals for Compound I Neat Form E Peak # Chemical Shift [ppm] Intensity [rel] 1 -60.9 1.09 2 625 10 Single Crystal Elucid
 [0547] Single crystals having the Compound I Neat Form E structure were grown by slowly cooling a trifluorotoluene solution of Compound I. X-ray diffraction data were acquired at 100K on a Rigaku diffractometer equipped with Cu Kα radiation (λ=1.54178 Å) and a hybrid pixel array detector. The structure was solved and refined using SHELX programs (Sheldrick, G.M., Acta Cryst., (2008) A64, 112-122) and results are summarized in Table 13 below. [0548] Table 13: Single Crystal Elucidation of Compound I Neat Form E at 100 K Crystal System Triclinic Space Group P-1 Single Crystal Elucidation o
[0547] Single crystals having the Compound I Neat Form E structure were grown by slowly cooling a trifluorotoluene solution of Compound I. X-ray diffraction data were acquired at 100K on a Rigaku diffractometer equipped with Cu Kα radiation (λ=1.54178 Å) and a hybrid pixel array detector. The structure was solved and refined using SHELX programs (Sheldrick, G.M., Acta Cryst., (2008) A64, 112-122) and results are summarized in Table 13 below. [0548] Table 13: Single Crystal Elucidation of Compound I Neat Form E at 100 K Crystal System Triclinic Space Group P-1 Single Crystal Elucidation o
 [0549] Single crystals having the Compound I Neat Form E structure were grown by slowly cooling a trifluorotoluene solution of Compound I. X-ray diffraction data were acquired at 298 K on a Rigaku diffractometer equipped with Cu Kα radiation (λ=1.54178 Å) and a hybrid pixel array detector. The structure was solved and refined using SHELX programs (Sheldrick, G.M., Acta Cryst., (2008) A64, 112-122) and results are summarized in Table 14 below. [0550] Table 14: Single Crystal Elucidation of Compound I Neat Form E at 298 K Crystal System Triclinic
[0549] Single crystals having the Compound I Neat Form E structure were grown by slowly cooling a trifluorotoluene solution of Compound I. X-ray diffraction data were acquired at 298 K on a Rigaku diffractometer equipped with Cu Kα radiation (λ=1.54178 Å) and a hybrid pixel array detector. The structure was solved and refined using SHELX programs (Sheldrick, G.M., Acta Cryst., (2008) A64, 112-122) and results are summarized in Table 14 below. [0550] Table 14: Single Crystal Elucidation of Compound I Neat Form E at 298 K Crystal System Triclinic
 β (°) 97.140(2) γ (°) 98.521(2) Å3 Example 6: Compound I A
β (°) 97.140(2) γ (°) 98.521(2) Å3 Example 6: Compound I A
 Preparation of Compound I Acetone Solvate Hydrate Form A [0551] To a saturated Compound I acetone solution, water was added dropwise until up to 20% of the final volume. The crystals of Compound I Acetone Solvate Hydrate Form A formed overnight. X-Ray Powder Diffraction [0552] The XRPD pattern of crystalline Compound I Acetone Solvate Hydrate Form A was recorded using the procedure described in the General XRPD Method. [0553] The XRPD diffractogram for crystalline Compound I Acetone Solvate Hydrate Form A is provided in FIG.13, and the XRPD data are summarized below in Table 15. [0554] Table 15: XRPD Signals for Crystalline Compound I Acetone Solvate Hydrate Form A No. Pos. [°2θ] Area [cts*°2θ] Rel. Int. [%] 1 7.2 80.42 23.24 3 4 3 7 4 6 8 3 3 6 2 7 2 7 2 3 6 8 3 0 6 5 5
Preparation of Compound I Acetone Solvate Hydrate Form A [0551] To a saturated Compound I acetone solution, water was added dropwise until up to 20% of the final volume. The crystals of Compound I Acetone Solvate Hydrate Form A formed overnight. X-Ray Powder Diffraction [0552] The XRPD pattern of crystalline Compound I Acetone Solvate Hydrate Form A was recorded using the procedure described in the General XRPD Method. [0553] The XRPD diffractogram for crystalline Compound I Acetone Solvate Hydrate Form A is provided in FIG.13, and the XRPD data are summarized below in Table 15. [0554] Table 15: XRPD Signals for Crystalline Compound I Acetone Solvate Hydrate Form A No. Pos. [°2θ] Area [cts*°2θ] Rel. Int. [%] 1 7.2 80.42 23.24 3 4 3 7 4 6 8 3 3 6 2 7 2 7 2 3 6 8 3 0 6 5 5
 Solid-State 13C NMR [0555] The 13C SSNMR of Compound I Acetone Solvate Hydrate Form A was acquired using the procedure described in the General SSNMR Method. The 13C SSNMR spectrum for Compound I Acetone Solvate Hydrate Form A is provided in FIG.14 and the data are summarized below in Table 16. [0556] Table 16: 13C SSNMR Signals for Compound I Acetone Solvate Hydrate Form A Peak # Chemical Shift [ppm] Intensity [rel] 1 212.9 0.66
Solid-State 13C NMR [0555] The 13C SSNMR of Compound I Acetone Solvate Hydrate Form A was acquired using the procedure described in the General SSNMR Method. The 13C SSNMR spectrum for Compound I Acetone Solvate Hydrate Form A is provided in FIG.14 and the data are summarized below in Table 16. [0556] Table 16: 13C SSNMR Signals for Compound I Acetone Solvate Hydrate Form A Peak # Chemical Shift [ppm] Intensity [rel] 1 212.9 0.66
 Solid-State 19F NMR [0557] The 19F SSNMR of Compound I Acetone Solvate Hydrate Form A was acquired using the procedure described in the General SSNMR Method. The 19F SSNMR spectrum for Compound I Acetone Solvate Hydrate Form A is provided in FIG.15, and the data are summarized below in Table 17. [0558] Table 17: 19F SSNMR Signals for Compound I Acetone Solvate Hydrate Form A Peak # Chemical Shift [ppm] Intensity [rel] 1 -60.4 6.46
Solid-State 19F NMR [0557] The 19F SSNMR of Compound I Acetone Solvate Hydrate Form A was acquired using the procedure described in the General SSNMR Method. The 19F SSNMR spectrum for Compound I Acetone Solvate Hydrate Form A is provided in FIG.15, and the data are summarized below in Table 17. [0558] Table 17: 19F SSNMR Signals for Compound I Acetone Solvate Hydrate Form A Peak # Chemical Shift [ppm] Intensity [rel] 1 -60.4 6.46
 Single Crystal Elucidation of Compound I Acetone Solvate Hydrate Form A [0559] Single crystals having the Compound I Acetone Solvate Hydrate Form A structure were grown by slurrying a mixture of Compound I Hydrate Form A and Compound I Acetone Solvate Hydrate in an acetone/water mixture at 50 °C. X-ray diffraction data were acquired at 100 K on a Rigaku diffractometer equipped with Cu Kα radiation (λ=1.54178 Å) and a hybrid pixel array detector. The structure was solved and refined using SHELX programs (Sheldrick, G.M., Acta Cryst., (2008) A64, 112-122) and results are summarized in Table 18 below. [0560] Table 18: Single Crystal Elucidation of Compound I Acetone Solvate Hydrate Form A Crystal System monoclinic S ace Grou P2
Single Crystal Elucidation of Compound I Acetone Solvate Hydrate Form A [0559] Single crystals having the Compound I Acetone Solvate Hydrate Form A structure were grown by slurrying a mixture of Compound I Hydrate Form A and Compound I Acetone Solvate Hydrate in an acetone/water mixture at 50 °C. X-ray diffraction data were acquired at 100 K on a Rigaku diffractometer equipped with Cu Kα radiation (λ=1.54178 Å) and a hybrid pixel array detector. The structure was solved and refined using SHELX programs (Sheldrick, G.M., Acta Cryst., (2008) A64, 112-122) and results are summarized in Table 18 below. [0560] Table 18: Single Crystal Elucidation of Compound I Acetone Solvate Hydrate Form A Crystal System monoclinic S ace Grou P2
 Example 7: Compound I Ethanol Solvate Form A Preparation of Compound I Ethanol Solvate Form A [0561] Compound I (10.00 g) was charge with ethyl acetate (30 mL) in a 100 mL round bottom flask. The resulting slurry was heated to 50 °C, followed by ethanol (20 mL) being charged to the flask. The slurry was distilled under vacuum at 50 °C until 15 g solvent remained in the flask, followed by additional ethanol (20 mL) being charged to the flask. The slurry was distilled under vacuum at 50 °C until 12 g solvent remained in the flask, followed by ethanol (30 mL) being charged to the flask. The slurry was distilled under vacuum at 50 °C to minimum volume. Ethanol (25 mL) was then charged to the flask. The slurry was distilled under vacuum at 50 °C until only 8 g solvent remained. The resulting material was transferred to a vacuum oven at 45 °C with a slight nitrogen bleed for 42 hours to yield Compound I Ethanol Solvate Form A (11.00 g). X-Ray Powder Diffraction [0562] The XRPD pattern of crystalline Compound I Ethanol Solvate Form A was recorded using the procedure described in the General XRPD Method. [0563] The XRPD diffractogram for crystalline Compound I Ethanol Solvate Form A is provided in FIG.16, and the XRPD data are summarized below in Table 19. [0564] Table 19: XRPD Signals for Crystalline Compound I Ethanol Solvate Form A No. Pos. [°2θ] Area [cts*°2θ] Rel. Int. [%] 1 9.4 195.5 24
Example 7: Compound I Ethanol Solvate Form A Preparation of Compound I Ethanol Solvate Form A [0561] Compound I (10.00 g) was charge with ethyl acetate (30 mL) in a 100 mL round bottom flask. The resulting slurry was heated to 50 °C, followed by ethanol (20 mL) being charged to the flask. The slurry was distilled under vacuum at 50 °C until 15 g solvent remained in the flask, followed by additional ethanol (20 mL) being charged to the flask. The slurry was distilled under vacuum at 50 °C until 12 g solvent remained in the flask, followed by ethanol (30 mL) being charged to the flask. The slurry was distilled under vacuum at 50 °C to minimum volume. Ethanol (25 mL) was then charged to the flask. The slurry was distilled under vacuum at 50 °C until only 8 g solvent remained. The resulting material was transferred to a vacuum oven at 45 °C with a slight nitrogen bleed for 42 hours to yield Compound I Ethanol Solvate Form A (11.00 g). X-Ray Powder Diffraction [0562] The XRPD pattern of crystalline Compound I Ethanol Solvate Form A was recorded using the procedure described in the General XRPD Method. [0563] The XRPD diffractogram for crystalline Compound I Ethanol Solvate Form A is provided in FIG.16, and the XRPD data are summarized below in Table 19. [0564] Table 19: XRPD Signals for Crystalline Compound I Ethanol Solvate Form A No. Pos. [°2θ] Area [cts*°2θ] Rel. Int. [%] 1 9.4 195.5 24
 Solid-State 13C NMR [0565] The 13C SSNMR of Compound I Ethanol Solvate Form A was acquired using the procedure described in the General SSNMR Method. The 13C SSNMR spectrum for Compound I Ethanol Solvate Form A is provided in FIG.17 and the data are summarized below in Table 20. [0566] Table 20: 13C SSNMR Signals for Compound I Ethanol Solvate Form A Peak # Chemical Shift [ppm] Intensity [rel] 1 178.2 5.01 Solid-State 19F NMR
Solid-State 13C NMR [0565] The 13C SSNMR of Compound I Ethanol Solvate Form A was acquired using the procedure described in the General SSNMR Method. The 13C SSNMR spectrum for Compound I Ethanol Solvate Form A is provided in FIG.17 and the data are summarized below in Table 20. [0566] Table 20: 13C SSNMR Signals for Compound I Ethanol Solvate Form A Peak # Chemical Shift [ppm] Intensity [rel] 1 178.2 5.01 Solid-State 19F NMR
 [0567] The 19F SSNMR of Compound I Ethanol Solvate Form A was acquired using the procedure described in the General SSNMR Method. The 19F SSNMR spectrum for Compound I Ethanol Solvate Form A is provided in FIG.18, and the data are summarized below in Table 21. [0568] Table 21: 19F SSNMR Signals for Compound I Ethanol Solvate Form A Peak # Chemical Shift [ppm] Intensity [rel] 1 11 1 Single Crystal Elucida
[0567] The 19F SSNMR of Compound I Ethanol Solvate Form A was acquired using the procedure described in the General SSNMR Method. The 19F SSNMR spectrum for Compound I Ethanol Solvate Form A is provided in FIG.18, and the data are summarized below in Table 21. [0568] Table 21: 19F SSNMR Signals for Compound I Ethanol Solvate Form A Peak # Chemical Shift [ppm] Intensity [rel] 1 11 1 Single Crystal Elucida
 [0569] Single crystals of Compound I Ethanol Solvate Form A were grown by slowly cooling an ethanol solution of Compound I. X-ray diffraction data were acquired at 100 K on a Bruker diffractometer equipped with Cu Kα radiation (λ=1.54178 Å) and a CPAD detector. The structure was
solved and refined using SHELX programs (Sheldrick, G.M., Acta Cryst., (2008) A64, 112-122) and results are summarized in the Table 22 below. [0570] Table 22: Single Crystal Elucidation of Compound I Ethanol Solvate Form A Crystal System monoclinic Space Group P21/c Example 8: Compound I H
[0569] Single crystals of Compound I Ethanol Solvate Form A were grown by slowly cooling an ethanol solution of Compound I. X-ray diffraction data were acquired at 100 K on a Bruker diffractometer equipped with Cu Kα radiation (λ=1.54178 Å) and a CPAD detector. The structure was
solved and refined using SHELX programs (Sheldrick, G.M., Acta Cryst., (2008) A64, 112-122) and results are summarized in the Table 22 below. [0570] Table 22: Single Crystal Elucidation of Compound I Ethanol Solvate Form A Crystal System monoclinic Space Group P21/c Example 8: Compound I H
 Preparation of Compound I Hydrate Form A [0571] Compound I Ethanol Solvate Form A (5 g) and acetone (30 mL) were added to a reactor. The slurry was stirred and heated to 50 °C to obtain a clear solution. To the clear solution was added water (20 mL) evenly over 1 hour. The clear solution was then seeded with 5 wt% Compound I Hydrate Form A seeds and agitated for 30 minutes. To the slurry, water (40 mL) was added over 8 hours, and the resulting slurry was stirred at 50 °C for an additional 5 hours. The slurry was then cooled to 20 °C over 3 hours, agitated for another 2 hours, and filtered. The filtered solids were washed with a 15 mL mixture of 1:2 acetone:water and then dried at 50 °C in a vacuum oven with a nitrogen bleed. [0572] Compound I Ethanol Solvate Form A (50 mg) and water (2 mL) were added to a 8 ml vial. The resulting slurry was stirred at 80 °C for 4 hours. The slurry was then cooled to 20 °C and filtered. The filtered solids were dried at 50 °C in a vacuum oven overnight. X-Ray Powder Diffraction [0573] The XRPD pattern of crystalline Compound I Hydrate Form A was recorded using the procedure described in the General XRPD Method. [0574] The XRPD diffractogram for crystalline Compound I Hydrate Form A (wet) is provided in FIG. 19, and the XRPD data are summarized below in Table 23. The XRPD diffractogram for crystalline Compound I Dehydrated Hydrate Form A is provided in FIG. 20, and the XRPD data are summarized below in Table 24. An overlay of the dry and dry crystalline Compound I Hydrate Form A XRPD diffractograms is provided in FIG.21. [0575] Table 23: XRPD Signals for Crystalline Compound I Hydrate Form A No. Pos. [°2θ] Area [cts*°2θ] Rel. Int. [%]
Preparation of Compound I Hydrate Form A [0571] Compound I Ethanol Solvate Form A (5 g) and acetone (30 mL) were added to a reactor. The slurry was stirred and heated to 50 °C to obtain a clear solution. To the clear solution was added water (20 mL) evenly over 1 hour. The clear solution was then seeded with 5 wt% Compound I Hydrate Form A seeds and agitated for 30 minutes. To the slurry, water (40 mL) was added over 8 hours, and the resulting slurry was stirred at 50 °C for an additional 5 hours. The slurry was then cooled to 20 °C over 3 hours, agitated for another 2 hours, and filtered. The filtered solids were washed with a 15 mL mixture of 1:2 acetone:water and then dried at 50 °C in a vacuum oven with a nitrogen bleed. [0572] Compound I Ethanol Solvate Form A (50 mg) and water (2 mL) were added to a 8 ml vial. The resulting slurry was stirred at 80 °C for 4 hours. The slurry was then cooled to 20 °C and filtered. The filtered solids were dried at 50 °C in a vacuum oven overnight. X-Ray Powder Diffraction [0573] The XRPD pattern of crystalline Compound I Hydrate Form A was recorded using the procedure described in the General XRPD Method. [0574] The XRPD diffractogram for crystalline Compound I Hydrate Form A (wet) is provided in FIG. 19, and the XRPD data are summarized below in Table 23. The XRPD diffractogram for crystalline Compound I Dehydrated Hydrate Form A is provided in FIG. 20, and the XRPD data are summarized below in Table 24. An overlay of the dry and dry crystalline Compound I Hydrate Form A XRPD diffractograms is provided in FIG.21. [0575] Table 23: XRPD Signals for Crystalline Compound I Hydrate Form A No. Pos. [°2θ] Area [cts*°2θ] Rel. Int. [%]
 No. Pos. [°2θ] Area [cts*°2θ] Rel. Int. [%] 2 8.7 1573.0 56 [0576] Table
No. Pos. [°2θ] Area [cts*°2θ] Rel. Int. [%] 2 8.7 1573.0 56 [0576] Table
 ydrate Form A No. Pos. [°2θ] Area [cts*°2θ] Rel. Int. [%] 1 47 3764 20
ydrate Form A No. Pos. [°2θ] Area [cts*°2θ] Rel. Int. [%] 1 47 3764 20
 No. Pos. [°2θ] Area [cts*°2θ] Rel. Int. [%] 20 24.3 570.3 17 Solid-State 13C N [0577] The 1
No. Pos. [°2θ] Area [cts*°2θ] Rel. Int. [%] 20 24.3 570.3 17 Solid-State 13C N [0577] The 1
 C SSNMR of Compound I Hydrate Form A was acquired using the procedure described in the General SSNMR Method. The 13C SSNMR spectrum for Compound I Hydrate Form A is provided in FIG.22 and the data are summarized below in Table 25. [0578] Table 25: 13C SSNMR Signals for Compound I Hydrate Form A Peak # Chemical Shift [ppm] Intensity [rel] 1 177.3 4 Solid-State 19F NMR
C SSNMR of Compound I Hydrate Form A was acquired using the procedure described in the General SSNMR Method. The 13C SSNMR spectrum for Compound I Hydrate Form A is provided in FIG.22 and the data are summarized below in Table 25. [0578] Table 25: 13C SSNMR Signals for Compound I Hydrate Form A Peak # Chemical Shift [ppm] Intensity [rel] 1 177.3 4 Solid-State 19F NMR
 [0579] The 19F SSNMR of Compound I Hydrate Form A was acquired using the procedure described in the General SSNMR Method. The 19F SSNMR spectrum for Compound I Hydrate Form A is provided in FIG.23, and the data are summarized below in Table 26. [0580] Table 26: 19F SSNMR Signals for Compound I Hydrate Form A Peak # Chemical Shift [ppm] Intensity [rel]
[0579] The 19F SSNMR of Compound I Hydrate Form A was acquired using the procedure described in the General SSNMR Method. The 19F SSNMR spectrum for Compound I Hydrate Form A is provided in FIG.23, and the data are summarized below in Table 26. [0580] Table 26: 19F SSNMR Signals for Compound I Hydrate Form A Peak # Chemical Shift [ppm] Intensity [rel]
 Peak # Chemical Shift [ppm] Intensity [rel] 2 -60.2 10 3 1350 11 Solid-State 13C NMR
Peak # Chemical Shift [ppm] Intensity [rel] 2 -60.2 10 3 1350 11 Solid-State 13C NMR
 [0581] The 13C SSNMR of Compound I Hydrate Form A was acquired using the procedure described in the General SSNMR Method and the 19F
[0581] The 13C SSNMR of Compound I Hydrate Form A was acquired using the procedure described in the General SSNMR Method and the 19F
 I Hydrate Form A was acquired using the procedure described in the General SSNMR Method. [0582] The 13C SSNMR spectrum for Compound I Hydrate Form A taken with variation in humidity is summarized below in Table 26. The 19F SSNMR spectrum for Compound I Hydrate Form A taken with variation in humidity is summarized below in Table 27. [0583] Table 26: 13C SSNMR Signals as a Function of Relative Humidity (%) for Compound I Hydrate Form A Chemical Shift [ppm] at Relative Humidity Peak # 6% 11% 22% 33% 43%
I Hydrate Form A was acquired using the procedure described in the General SSNMR Method. [0582] The 13C SSNMR spectrum for Compound I Hydrate Form A taken with variation in humidity is summarized below in Table 26. The 19F SSNMR spectrum for Compound I Hydrate Form A taken with variation in humidity is summarized below in Table 27. [0583] Table 26: 13C SSNMR Signals as a Function of Relative Humidity (%) for Compound I Hydrate Form A Chemical Shift [ppm] at Relative Humidity Peak # 6% 11% 22% 33% 43%
 [0584] Table 27: 19F SSNMR Signals as a Function of Relative Humidity (%) for Compound I Hydrate Form A Chemical Shift [ppm] at Relative Humidity Peak # 6% 11% 22% 33% 43% 1 4 4 Single Crystal El
[0584] Table 27: 19F SSNMR Signals as a Function of Relative Humidity (%) for Compound I Hydrate Form A Chemical Shift [ppm] at Relative Humidity Peak # 6% 11% 22% 33% 43% 1 4 4 Single Crystal El
 [0585] Single crystals having the Compound I Hydrate Form A structure were grown in a water slurry of Compound I. X-ray diffraction data were acquired at 100 K on a Bruker diffractometer equipped with Cu Kα radiation (λ=1.54178 Å) and a CPAD detector. The structure was solved and refined using SHELX programs (Sheldrick, G.M., Acta Cryst., (2008) A64, 112-122) and results are summarized in Table 28 below. [0586] Table 28: Single Crystal Elucidation of Compound I Hydrate Form A Crystal System Triclinic Space Group P-1 Single Crystal Elucidation o
[0585] Single crystals having the Compound I Hydrate Form A structure were grown in a water slurry of Compound I. X-ray diffraction data were acquired at 100 K on a Bruker diffractometer equipped with Cu Kα radiation (λ=1.54178 Å) and a CPAD detector. The structure was solved and refined using SHELX programs (Sheldrick, G.M., Acta Cryst., (2008) A64, 112-122) and results are summarized in Table 28 below. [0586] Table 28: Single Crystal Elucidation of Compound I Hydrate Form A Crystal System Triclinic Space Group P-1 Single Crystal Elucidation o
 A [0587] Single crystals having the Compound I Dehydrated Hydrate Form A structure were grown by drying a single crystal of Compound I Hydrate Form A in a stream of dry nitrogen held at 373(2) K for 1 hour. X-ray diffraction data were acquired at 100 K on a Bruker diffractometer equipped with Cu Kα radiation (λ=1.54178 Å) and a CPAD detector. The structure was solved and refined using SHELX programs (Sheldrick, G.M., Acta Cryst., (2008) A64, 112-122) and results are summarized in Table 29 below. [0588] Table 29: Single Crystal Elucidation of Compound I Dehydrated Hydrate Form A Crystal System Triclinic
A [0587] Single crystals having the Compound I Dehydrated Hydrate Form A structure were grown by drying a single crystal of Compound I Hydrate Form A in a stream of dry nitrogen held at 373(2) K for 1 hour. X-ray diffraction data were acquired at 100 K on a Bruker diffractometer equipped with Cu Kα radiation (λ=1.54178 Å) and a CPAD detector. The structure was solved and refined using SHELX programs (Sheldrick, G.M., Acta Cryst., (2008) A64, 112-122) and results are summarized in Table 29 below. [0588] Table 29: Single Crystal Elucidation of Compound I Dehydrated Hydrate Form A Crystal System Triclinic
 c (Å) 19.2707(7) α (°) 90.698(2) ° Other Embodiments
c (Å) 19.2707(7) α (°) 90.698(2) ° Other Embodiments
 [0589] The present disclosure enables one of skill in the relevant art to make and use the inventions provided herein in accordance with multiple and varied embodiments. Various alterations, modifications, and improvements of the present disclosure that readily occur to those skilled in the art, including certain alterations, modifications, substitutions, and improvements are also part of this disclosure. Accordingly, the foregoing description are by way of example to illustrate the discoveries provided herein. Furthermore, the foregoing Description and Examples are exemplary of the present invention and not limiting thereof. The scope of the invention is therefore set out in the appended claims. [0590] Although specific embodiments of the present disclosure are herein illustrated and described in detail, the disclosure is not limited thereto. The above detailed descriptions are provided as exemplary of the present disclosure and should not be construed as constituting any limitation of the disclosure. Modifications will be obvious to those skilled in the art, and all modifications that do not depart from the spirit of the disclosure are intended to be included with the scope of the appended claims.
[0589] The present disclosure enables one of skill in the relevant art to make and use the inventions provided herein in accordance with multiple and varied embodiments. Various alterations, modifications, and improvements of the present disclosure that readily occur to those skilled in the art, including certain alterations, modifications, substitutions, and improvements are also part of this disclosure. Accordingly, the foregoing description are by way of example to illustrate the discoveries provided herein. Furthermore, the foregoing Description and Examples are exemplary of the present invention and not limiting thereof. The scope of the invention is therefore set out in the appended claims. [0590] Although specific embodiments of the present disclosure are herein illustrated and described in detail, the disclosure is not limited thereto. The above detailed descriptions are provided as exemplary of the present disclosure and should not be construed as constituting any limitation of the disclosure. Modifications will be obvious to those skilled in the art, and all modifications that do not depart from the spirit of the disclosure are intended to be included with the scope of the appended claims.
Claims
CLAIMS 1. A substantially crystalline Compound I: I, wherein the crystalline Compound I I neat Form A.
 2. The substantially crystalline Compound I of claim 1, wherein the Compound I neat form A is characterized by: (i) an X-ray powder diffractogram having two, three, four, five, or six signals selected from 9.5 ± 0.2 degrees two-theta, 12.3 ± 0.2 degrees two-theta, 13.5 ± 0.2 degrees two-theta, 11.3 ± 0.2 degrees two-theta, 15.9 ± 0.2 degrees two-theta, 20.5 ± 0.2 degrees two-theta, 14.6 ± 0.2 degrees two-theta, 16.6 ± 0.2 degrees two-theta, and 23.5 ± 0.2 degrees two-theta; and/or (ii) an X-ray powder diffractogram substantially similar to FIG.4; and/or (iii) a 13C SSNMR spectrum having one, two, three, four, five, six, seven, eight, nine, ten, or more peaks selected from 170.4 ± 0.2 ppm, 163.8 ± 0.2 ppm, 162.4 ± 0.2 ppm, 160.9 ± 0.2 ppm, 158.6 ± 0.2 ppm, 158.3 ± 0.2 ppm, 154.6 ± 0.2 ppm, 154.0 ± 0.2 ppm, 148.3 ± 0.2 ppm, 147.6 ± 0.2 ppm, 146.9 ± 0.2 ppm, 145.9 ± 0.2 ppm, 143.5 ± 0.2 ppm, 143.1 ± 0.2 ppm, 141.9 ± 0.2 ppm, 127.4 ± 0.2 ppm, 125.3 ± 0.2 ppm, 123.6 ± 0.2 ppm, 122.2 ± 0.2 ppm, 121.3 ± 0.2 ppm, 120.7 ± 0.2 ppm, 117.9 ± 0.2 ppm, 117.3 ± 0.2 ppm, 116.3 ± 0.2 ppm, 115.8 ± 0.2 ppm, 114.2 ± 0.2 ppm, 113.6 ± 0.2 ppm, 112.8 ± 0.2 ppm, 15.4 ± 0.2 ppm, and 8.2 ± 0.2 ppm; and/or (iv) a 13C SSNMR spectrum substantially similar to FIG.5; and/or (v) a 19F SSNMR spectrum with one, two, three, four, five, or six peaks selected from -61.2 ± 0.2 ppm, -61.6 ± 0.2 ppm, -138.6 ± 0.2 ppm, -140.3 ± 0.2 ppm, -142.0 ± 0.2 ppm, and -146.0 ± 0.
2. The substantially crystalline Compound I of claim 1, wherein the Compound I neat form A is characterized by: (i) an X-ray powder diffractogram having two, three, four, five, or six signals selected from 9.5 ± 0.2 degrees two-theta, 12.3 ± 0.2 degrees two-theta, 13.5 ± 0.2 degrees two-theta, 11.3 ± 0.2 degrees two-theta, 15.9 ± 0.2 degrees two-theta, 20.5 ± 0.2 degrees two-theta, 14.6 ± 0.2 degrees two-theta, 16.6 ± 0.2 degrees two-theta, and 23.5 ± 0.2 degrees two-theta; and/or (ii) an X-ray powder diffractogram substantially similar to FIG.4; and/or (iii) a 13C SSNMR spectrum having one, two, three, four, five, six, seven, eight, nine, ten, or more peaks selected from 170.4 ± 0.2 ppm, 163.8 ± 0.2 ppm, 162.4 ± 0.2 ppm, 160.9 ± 0.2 ppm, 158.6 ± 0.2 ppm, 158.3 ± 0.2 ppm, 154.6 ± 0.2 ppm, 154.0 ± 0.2 ppm, 148.3 ± 0.2 ppm, 147.6 ± 0.2 ppm, 146.9 ± 0.2 ppm, 145.9 ± 0.2 ppm, 143.5 ± 0.2 ppm, 143.1 ± 0.2 ppm, 141.9 ± 0.2 ppm, 127.4 ± 0.2 ppm, 125.3 ± 0.2 ppm, 123.6 ± 0.2 ppm, 122.2 ± 0.2 ppm, 121.3 ± 0.2 ppm, 120.7 ± 0.2 ppm, 117.9 ± 0.2 ppm, 117.3 ± 0.2 ppm, 116.3 ± 0.2 ppm, 115.8 ± 0.2 ppm, 114.2 ± 0.2 ppm, 113.6 ± 0.2 ppm, 112.8 ± 0.2 ppm, 15.4 ± 0.2 ppm, and 8.2 ± 0.2 ppm; and/or (iv) a 13C SSNMR spectrum substantially similar to FIG.5; and/or (v) a 19F SSNMR spectrum with one, two, three, four, five, or six peaks selected from -61.2 ± 0.2 ppm, -61.6 ± 0.2 ppm, -138.6 ± 0.2 ppm, -140.3 ± 0.2 ppm, -142.0 ± 0.2 ppm, and -146.0 ± 0.
2 ppm; and/or (vi) a 19F SSNMR spectrum substantially similar to FIG.6; and/or (vii) a triclinic crystal system, P-1 space group, and unit cell dimensions measured at 100 K on a Bruker diffractometer utilizing Cu Kα radiation (λ=1.54178 Å) of: a 9.6 ± 0.1 Å α 94.3 ± 0.1° b 12.6 ± 0.1 Å β 97.3 ± 0.1° c 18.7 ± 0.1 Å γ 107.3 ± 0.1°.
3. A substantially crystalline Compound I: I. wherein the crystalline Compound I
 I neat form B. 4. The substantially crystalline Compound I of claim 1, wherein the Compound I neat form B is characterized by: (i) an X-ray powder diffractogram having two, three, four, five, six, seven, eight, or nine signals selected from 4.8 ± 0.2 degrees two-theta, 13.3 ± 0.2 degrees two-theta, 18.5 ± 0.2 degrees two-theta, 6.0 ± 0.2 degrees two-theta, 16.5 ± 0.2 degrees two-theta, 16.9 ± 0.2 degrees two-theta, 14.1 ± 0.2 degrees two-theta, 14.7 ± 0.2 degrees two-theta, and 15.2 ± 0.2 degrees two-theta; and/or (ii) an X-ray powder diffractogram substantially similar to FIG.7; and/or (iii) a 13C SSNMR spectrum having one, two, three, four, five, six, seven, eight, nine, ten, or more peaks selected from 176.9 ± 0.2 ppm, 171.7 ± 0.2 ppm, 164.0 ± 0.2 ppm, 161.4 ± 0.2 ppm, 160.7 ± 0.2 ppm, 155.3 ± 0.2 ppm, 154.8 ± 0.2 ppm, 148.6 ± 0.2 ppm, 147.6 ± 0.2 ppm, 146.4 ± 0.2 ppm, 145.7 ± 0.2 ppm, 144.8 ± 0.2 ppm, 127.7 ± 0.2 ppm, 125.4 ± 0.2 ppm, 122.0 ± 0.2 ppm, 121.5 ± 0.2 ppm, 118.4 ± 0.2 ppm, 116.3 ± 0.2 ppm, 114.8 ± 0.2 ppm, 113.8 ± 0.2 ppm, 112.6 ± 0.2 ppm, 17.8 ± 0.2 ppm, 15.0 ± 0.2 ppm, 10.7 ± 0.2 ppm, 9.6 ± 0.2 ppm, and 8.8 ± 0.2 ppm; and/or (iv) a 13C SSNMR spectrum substantially similar to FIG.8; and/or (v) a 19F SSNMR spectrum with one, two, three, four, or five peaks selected from -61.6 ± 0.2 ppm, -138 ± 0.2 ppm, -139.7 ± 0.2 ppm, -143.
I neat form B. 4. The substantially crystalline Compound I of claim 1, wherein the Compound I neat form B is characterized by: (i) an X-ray powder diffractogram having two, three, four, five, six, seven, eight, or nine signals selected from 4.8 ± 0.2 degrees two-theta, 13.3 ± 0.2 degrees two-theta, 18.5 ± 0.2 degrees two-theta, 6.0 ± 0.2 degrees two-theta, 16.5 ± 0.2 degrees two-theta, 16.9 ± 0.2 degrees two-theta, 14.1 ± 0.2 degrees two-theta, 14.7 ± 0.2 degrees two-theta, and 15.2 ± 0.2 degrees two-theta; and/or (ii) an X-ray powder diffractogram substantially similar to FIG.7; and/or (iii) a 13C SSNMR spectrum having one, two, three, four, five, six, seven, eight, nine, ten, or more peaks selected from 176.9 ± 0.2 ppm, 171.7 ± 0.2 ppm, 164.0 ± 0.2 ppm, 161.4 ± 0.2 ppm, 160.7 ± 0.2 ppm, 155.3 ± 0.2 ppm, 154.8 ± 0.2 ppm, 148.6 ± 0.2 ppm, 147.6 ± 0.2 ppm, 146.4 ± 0.2 ppm, 145.7 ± 0.2 ppm, 144.8 ± 0.2 ppm, 127.7 ± 0.2 ppm, 125.4 ± 0.2 ppm, 122.0 ± 0.2 ppm, 121.5 ± 0.2 ppm, 118.4 ± 0.2 ppm, 116.3 ± 0.2 ppm, 114.8 ± 0.2 ppm, 113.8 ± 0.2 ppm, 112.6 ± 0.2 ppm, 17.8 ± 0.2 ppm, 15.0 ± 0.2 ppm, 10.7 ± 0.2 ppm, 9.6 ± 0.2 ppm, and 8.8 ± 0.2 ppm; and/or (iv) a 13C SSNMR spectrum substantially similar to FIG.8; and/or (v) a 19F SSNMR spectrum with one, two, three, four, or five peaks selected from -61.6 ± 0.2 ppm, -138 ± 0.2 ppm, -139.7 ± 0.2 ppm, -143.
4 ± 0.2 ppm, and -144.9 ± 0.2 ppm; and/or (vi) a 19F SSNMR spectrum substantially similar to FIG.9.
5. A substantially crystalline Compound I: I. wherein the crystalline Compound I
 I neat form E. 6. The substantially crystalline Compound I of claim 1, wherein the Compound I neat form E is characterized by: (i) an X-ray powder diffractogram having two, three, four, five, six, seven, eight, or nine signals selected from 8.6 ± 0.2 degrees two-theta, 10.1 ± 0.2 degrees two-theta, 12.6 ± 0.2 degrees two-theta, 14.1 ± 0.2 degrees two-theta, 20.3 ± 0.2 degrees two-theta, 23.2 ± 0.2 degrees two-theta, 11.5 ± 0.2 degrees two-theta, 14.2 ± 0.2 degrees two-theta, and 15.1 ± 0.2 degrees two-theta; and/or (ii) an X-ray powder diffractogram substantially similar to FIG.10; and/or (iii) a 13C SSNMR spectrum having one, two, three, four, five, six, seven, eight, nine, ten, or more peaks selected from 170.0 ± 0.2 ppm, 163.0 ± 0.2 ppm, 162.3 ± 0.2 ppm, 161.3 ± 0.2 ppm, 159.3 ± 0.2 ppm, 154.7 ± 0.2 ppm, 150.6 ± 0.2 ppm, 149.8 ± 0.2 ppm, 148.6 ± 0.2 ppm, 148.3 ± 0.2 ppm, 147.1 ± 0.2 ppm, 144.9 ± 0.2 ppm, 143.9 ± 0.2 ppm, 143.5 ± 0.2 ppm, 126.9 ± 0.2 ppm, 126.0 ± 0.2 ppm, 124.1 ± 0.2 ppm, 122.6 ± 0.2 ppm, 120.8 ± 0.2 ppm, 117.8 ± 0.2 ppm, 116.7 ± 0.2 ppm, 115.2 ± 0.2 ppm, 114.1 ± 0.2 ppm, 113.1 ± 0.2 ppm, 15.9 ± 0.2 ppm, 15.3 ± 0.2 ppm, and 8.4 ± 0.2 ppm; and/or (iv) a 13C SSNMR spectrum substantially similar to FIG.11; and/or (v) a 19F SSNMR spectrum with one, two, three, four, five, six, or seven peaks selected from -60.9 ± 0.2 ppm, -62.5 ± 0.2 ppm, -63.1 ± 0.2 ppm, -135.1 ± 0.2 ppm, -137.1 ± 0.2 ppm, -140.7 ± 0.2 ppm, and -141.9 ± 0.2 ppm; and/or (vi) a 19F SSNMR spectrum substantially similar to FIG.12; and/or (vii) a triclinic crystal system, P-1 space group, and unit cell dimensions measured at 100 K on a Rigaku diffractometer utilizing Cu Kα radiation (λ=1.54178 Å) of: a 8.7 ± 0.1 Å α 92.7 ± 0.1° b 12.2 ± 0.1 Å β 96.3 ± 0.1° c 20.9 ± 0.1 Å γ 98.6 ± 0.1°; and/or
(viii) a triclinic crystal system, P-1 space group, and unit cell dimensions measured at 298 K on a Rigaku diffractometer utilizing Cu Kα radiation (λ=1.54178 Å) of: a 8.9 ± 0.1 Å α 92.6 ± 0.1° b 12.3 ± 0.1 Å β 97.1 ± 0.1° c 21.1 ± 0.1 Å γ 98.5 ± 0.1°. 7. A substantially crystalline Compound I: I. wherein the crystalline Compound I
I neat form E. 6. The substantially crystalline Compound I of claim 1, wherein the Compound I neat form E is characterized by: (i) an X-ray powder diffractogram having two, three, four, five, six, seven, eight, or nine signals selected from 8.6 ± 0.2 degrees two-theta, 10.1 ± 0.2 degrees two-theta, 12.6 ± 0.2 degrees two-theta, 14.1 ± 0.2 degrees two-theta, 20.3 ± 0.2 degrees two-theta, 23.2 ± 0.2 degrees two-theta, 11.5 ± 0.2 degrees two-theta, 14.2 ± 0.2 degrees two-theta, and 15.1 ± 0.2 degrees two-theta; and/or (ii) an X-ray powder diffractogram substantially similar to FIG.10; and/or (iii) a 13C SSNMR spectrum having one, two, three, four, five, six, seven, eight, nine, ten, or more peaks selected from 170.0 ± 0.2 ppm, 163.0 ± 0.2 ppm, 162.3 ± 0.2 ppm, 161.3 ± 0.2 ppm, 159.3 ± 0.2 ppm, 154.7 ± 0.2 ppm, 150.6 ± 0.2 ppm, 149.8 ± 0.2 ppm, 148.6 ± 0.2 ppm, 148.3 ± 0.2 ppm, 147.1 ± 0.2 ppm, 144.9 ± 0.2 ppm, 143.9 ± 0.2 ppm, 143.5 ± 0.2 ppm, 126.9 ± 0.2 ppm, 126.0 ± 0.2 ppm, 124.1 ± 0.2 ppm, 122.6 ± 0.2 ppm, 120.8 ± 0.2 ppm, 117.8 ± 0.2 ppm, 116.7 ± 0.2 ppm, 115.2 ± 0.2 ppm, 114.1 ± 0.2 ppm, 113.1 ± 0.2 ppm, 15.9 ± 0.2 ppm, 15.3 ± 0.2 ppm, and 8.4 ± 0.2 ppm; and/or (iv) a 13C SSNMR spectrum substantially similar to FIG.11; and/or (v) a 19F SSNMR spectrum with one, two, three, four, five, six, or seven peaks selected from -60.9 ± 0.2 ppm, -62.5 ± 0.2 ppm, -63.1 ± 0.2 ppm, -135.1 ± 0.2 ppm, -137.1 ± 0.2 ppm, -140.7 ± 0.2 ppm, and -141.9 ± 0.2 ppm; and/or (vi) a 19F SSNMR spectrum substantially similar to FIG.12; and/or (vii) a triclinic crystal system, P-1 space group, and unit cell dimensions measured at 100 K on a Rigaku diffractometer utilizing Cu Kα radiation (λ=1.54178 Å) of: a 8.7 ± 0.1 Å α 92.7 ± 0.1° b 12.2 ± 0.1 Å β 96.3 ± 0.1° c 20.9 ± 0.1 Å γ 98.6 ± 0.1°; and/or
(viii) a triclinic crystal system, P-1 space group, and unit cell dimensions measured at 298 K on a Rigaku diffractometer utilizing Cu Kα radiation (λ=1.54178 Å) of: a 8.9 ± 0.1 Å α 92.6 ± 0.1° b 12.3 ± 0.1 Å β 97.1 ± 0.1° c 21.1 ± 0.1 Å γ 98.5 ± 0.1°. 7. A substantially crystalline Compound I: I. wherein the crystalline Compound I
 I Acetone Solvate Hydrate Form A. 8. The substantially crystalline Compound I of claim 1, wherein the Compound I Acetone Solvate Hydrate Form A is characterized by: (i) an X-ray powder diffractogram having two, three, four, five, six, seven, eight, or nine signals selected from 9.5 ± 0.2 degrees two-theta, 12.3 ± 0.2 degrees two-theta, 13.5 ± 0.2 degrees two-theta, 12.9 ± 0.2 degrees two-theta, 15.6 ± 0.2 degrees two-theta, 24.8 ± 0.2 degrees two-theta, 8.1 ± 0.2 degrees two-theta, 12.3 ± 0.2 degrees two-theta, and 17.6 ± 0.2 degrees two-theta; and/or (ii) an X-ray powder diffractogram substantially similar to FIG.13; and/or (iii) a 13C SSNMR spectrum having one, two, three, four, five, six, seven, eight, nine, ten, or more peaks selected from 212.9 ± 0.2 ppm, 211.6 ± 0.2 ppm, 179.2 ± 0.2 ppm, 178.9 ± 0.2 ppm, 178.4 ± 0.2 ppm, 174.7 ± 0.2 ppm, 174.1 ± 0.2 ppm, 172.7 ± 0.2 ppm, 172.2 ± 0.2 ppm, 163.1 ± 0.2 ppm, 156.7 ± 0.2 ppm, 155.6 ± 0.2 ppm, 152.3 ± 0.2 ppm, 150.1 ± 0.2 ppm, 148.7 ± 0.2 ppm, 147.9 ± 0.2 ppm, 147.0 ± 0.2 ppm, 145.1 ± 0.2 ppm, 125.5 ± 0.2 ppm, 124.5 ± 0.2 ppm, 122.5 ± 0.2 ppm, 121.6 ± 0.2 ppm, 121.1 ± 0.2 ppm, 118.5 ± 0.2 ppm, 118.1 ± 0.2 ppm, 117.4 ± 0.2 ppm, 116.8 ± 0.2 ppm, 115.7 ± 0.2 ppm, 115.3 ± 0.2 ppm, 113.9 ± 0.2 ppm, 113.2 ± 0.2 ppm, 112.4 ± 0.2 ppm, 30 ± 0.2 ppm, 29.5 ± 0.2 ppm, 18.4 ± 0.2 ppm, 17.6 ± 0.2 ppm, 8.8 ± 0.2 ppm, and 8.1 ± 0.2 ppm; and/or (iv) a 13C SSNMR spectrum substantially similar to FIG.14; and/or
(v) a 19F SSNMR spectrum with one or two peaks selected from -60.4 ± 0.2 ppm, -61.2 ± 0.2 ppm, -61.6 ± 0.2 ppm, -62.7 ± 0.2 ppm, -135.9 ± 0.2 ppm, -136.1 ± 0.2 ppm, -139.6 ± 0.2 ppm, -140.
I Acetone Solvate Hydrate Form A. 8. The substantially crystalline Compound I of claim 1, wherein the Compound I Acetone Solvate Hydrate Form A is characterized by: (i) an X-ray powder diffractogram having two, three, four, five, six, seven, eight, or nine signals selected from 9.5 ± 0.2 degrees two-theta, 12.3 ± 0.2 degrees two-theta, 13.5 ± 0.2 degrees two-theta, 12.9 ± 0.2 degrees two-theta, 15.6 ± 0.2 degrees two-theta, 24.8 ± 0.2 degrees two-theta, 8.1 ± 0.2 degrees two-theta, 12.3 ± 0.2 degrees two-theta, and 17.6 ± 0.2 degrees two-theta; and/or (ii) an X-ray powder diffractogram substantially similar to FIG.13; and/or (iii) a 13C SSNMR spectrum having one, two, three, four, five, six, seven, eight, nine, ten, or more peaks selected from 212.9 ± 0.2 ppm, 211.6 ± 0.2 ppm, 179.2 ± 0.2 ppm, 178.9 ± 0.2 ppm, 178.4 ± 0.2 ppm, 174.7 ± 0.2 ppm, 174.1 ± 0.2 ppm, 172.7 ± 0.2 ppm, 172.2 ± 0.2 ppm, 163.1 ± 0.2 ppm, 156.7 ± 0.2 ppm, 155.6 ± 0.2 ppm, 152.3 ± 0.2 ppm, 150.1 ± 0.2 ppm, 148.7 ± 0.2 ppm, 147.9 ± 0.2 ppm, 147.0 ± 0.2 ppm, 145.1 ± 0.2 ppm, 125.5 ± 0.2 ppm, 124.5 ± 0.2 ppm, 122.5 ± 0.2 ppm, 121.6 ± 0.2 ppm, 121.1 ± 0.2 ppm, 118.5 ± 0.2 ppm, 118.1 ± 0.2 ppm, 117.4 ± 0.2 ppm, 116.8 ± 0.2 ppm, 115.7 ± 0.2 ppm, 115.3 ± 0.2 ppm, 113.9 ± 0.2 ppm, 113.2 ± 0.2 ppm, 112.4 ± 0.2 ppm, 30 ± 0.2 ppm, 29.5 ± 0.2 ppm, 18.4 ± 0.2 ppm, 17.6 ± 0.2 ppm, 8.8 ± 0.2 ppm, and 8.1 ± 0.2 ppm; and/or (iv) a 13C SSNMR spectrum substantially similar to FIG.14; and/or
(v) a 19F SSNMR spectrum with one or two peaks selected from -60.4 ± 0.2 ppm, -61.2 ± 0.2 ppm, -61.6 ± 0.2 ppm, -62.7 ± 0.2 ppm, -135.9 ± 0.2 ppm, -136.1 ± 0.2 ppm, -139.6 ± 0.2 ppm, -140.
6 ± 0.2 ppm, -141.7 ± 0.2 ppm, -145.2 ± 0.2 ppm, and -145.
7 ± 0.2 ppm; and/or (vi) a 19F SSNMR spectrum substantially similar to FIG.15; and/or (vii) a monoclinic crystal system, P21 space group, and unit cell dimensions measured at 100 K on a Rigaku diffractometer utilizing Cu Kα radiation (λ=1.54178 Å) of: a 9.2 ± 0.1 Å α 90° b 39.
8 ± 0.1 Å β 96.3 ± 0.1° c 12.9 ± 0.1 Å γ 90°.
9. A substantially crystalline Compound I: I. wherein the crystalline Compound I
 I Ethanol Solvate Form A.
I Ethanol Solvate Form A.
10. The substantially crystalline Compound I of claim 1, wherein the Compound I Ethanol Solvate Form A is characterized by: (i) an X-ray powder diffractogram having two, three, four, five, six, seven, eight, or nine signals selected from 9.4 ± 0.2 degrees two-theta, 16.1 ± 0.2 degrees two-theta, 26.0 ± 0.2 degrees two-theta, 15.1 ± 0.2 degrees two-theta, 17.0 ± 0.2 degrees two-theta, 22.4 ± 0.2 degrees two-theta, 13.2 ± 0.2 degrees two-theta, 13.6 ± 0.2 degrees two-theta, and 13.8 ± 0.2 degrees two-theta; and/or (ii) an X-ray powder diffractogram substantially similar to FIG.16; and/or (iii) a 13C SSNMR spectrum having one, two, three, four, five, six, seven, eight, nine, ten, or more peaks selected from 178.2 ± 0.2 ppm, 175.7 ± 0.2 ppm, 163.9 ± 0.2 ppm, 163.4 ± 0.2 ppm, 162.8 ± 0.2 ppm, 154.9 ± 0.2 ppm, 148.1 ± 0.2 ppm, 146.0 ± 0.2 ppm, 145.2 ± 0.2 ppm, 125.9 ± 0.2 ppm, 123.1 ± 0.2 ppm, 121.8 ± 0.2 ppm, 120.4 ± 0.2 ppm, 119.5 ± 0.2 ppm, 116.9 ± 0.2 ppm, 116.3 ± 0.2 ppm, 114.8 ± 0.2 ppm, 113.8 ± 0.2 ppm, 56.5 ± 0.2 ppm, 18.4 ± 0.2 ppm, 17.2 ± 0.2 ppm, 9.2 ± 0.2 ppm, and 7.9 ± 0.2 ppm; and/or (iv) a 13C SSNMR spectrum substantially similar to FIG.17; and/or (v) a 19F SSNMR spectrum with one, two, or three peaks selected from -61.1 ± 0.2 ppm, -138.1 ± 0.2 ppm, and -141.2 ± 0.2 ppm;
(vi) a 19F SSNMR spectrum substantially similar to FIG.18; and/or (vii) a monoclinic crystal system, P21/c space group, and unit cell dimensions measured at 100 K on a Bruker diffractometer utilizing Cu Kα radiation (λ=1.54178 Å) of: a 17.0 ± 0.1 Å α 90° b 11.1 ± 0.1 Å β 105.9 ± 0.1° c 13.2 ± 0.1 Å γ 90°.
11. A substantially crystalline Compound I: I. wherein the crystalline Compound I
 I Hydrate Form A. 12. The substantially crystalline Compound I of claim 1, wherein the Compound I Hydrate Form A is characterized by: (i) an X-ray powder diffractogram having two, three, four, five, six, seven, eight, or nine signals selected from 8.7 ± 0.2 degrees two-theta, 12.4 ± 0.2 degrees two-theta, 18.8 ± 0.2 degrees two-theta,
I Hydrate Form A. 12. The substantially crystalline Compound I of claim 1, wherein the Compound I Hydrate Form A is characterized by: (i) an X-ray powder diffractogram having two, three, four, five, six, seven, eight, or nine signals selected from 8.7 ± 0.2 degrees two-theta, 12.4 ± 0.2 degrees two-theta, 18.8 ± 0.2 degrees two-theta,
12.7 ± 0.2 degrees two-theta, 17.5 ± 0.2 degrees two-theta, 19.8 ± 0.2 degrees two-theta, 13.1 ± 0.2 degrees two-theta, 16.3 ± 0.2 degrees two-theta, and 21.1 ± 0.2 degrees two-theta; and/or (ii) an X-ray powder diffractogram substantially similar to FIG.19; and/or (iii) a 13C SSNMR spectrum having one, two, three, four, five, six, seven, eight, nine, ten, or more peaks selected from 177.3 ± 0.2 ppm, 172.8 ± 0.2 ppm, 170.5 ± 0.2 ppm, 162.0 ± 0.2 ppm, 154.3 ± 0.2 ppm, 154.0 ± 0.2 ppm, 150.2 ± 0.2 ppm, 148.7 ± 0.2 ppm, 147.6 ± 0.2 ppm, 146.6 ± 0.2 ppm, 145.5 ± 0.2 ppm, 126.3 ± 0.2 ppm, 125.0 ± 0.2 ppm, 123.8 ± 0.2 ppm, 122.4 ± 0.2 ppm, 121.6 ± 0.2 ppm, 120.6 ± 0.2 ppm, 118.4 ± 0.2 ppm, 117.9 ± 0.2 ppm, 117.0 ± 0.2 ppm, 116.0 ± 0.2 ppm, 114.7 ± 0.2 ppm, 114.4 ± 0.2 ppm, 112.8 ± 0.2 ppm, 17.7 ± 0.2 ppm, 17.1 ± 0.2 ppm, and 8.7 ± 0.2 ppm; and/or (iv) a 13C SSNMR spectrum substantially similar to FIG.22; and/or (v) a 19F SSNMR spectrum with one, two, three, four, five, or six peaks selected from -59.4 ± 0.2 ppm, -60.2 ± 0.2 ppm, -135.0 ± 0.2 ppm, -138.4 ± 0.2 ppm, -141.7 ± 0.2 ppm, and -144.0 ± 0.2 ppm; and/or (vi) a 19F SSNMR spectrum substantially similar to FIG.23; and/or
(vii) a triclinic crystal system, P-1 space group, and unit cell dimensions measured at 100 K on a Bruker diffractometer utilizing Cu Kα radiation (λ=1.54178 Å) of: a 10.5 ± 0.1 Å α 91.3 ± 0.1° b 11.2 ± 0.1 Å β 103.9 ± 0.1° c 19.2 ± 0.1 Å γ 98.4 ± 0.1°.
13. A substantially crystalline Compound I: I. wherein the crystalline Compound I
 I Dehydrated Hydrate Form A.
I Dehydrated Hydrate Form A.
14. The substantially crystalline Compound I of claim 1, wherein the Compound I Dehydrated Hydrate Form A is characterized by: (i) characterized by an X-ray powder diffractogram having two, three, four, five, six, seven, eight, nine, or ten signals selected from 8.7 ± 0.2 degrees two-theta, 15.9 ± 0.2 degrees two-theta, 18.7 ± 0.2 degrees two-theta, 12.4 ± 0.2 degrees two-theta, 12.6 ± 0.2 degrees two-theta, 17.3 ± 0.2 degrees two-theta, 19.8 ± 0.2 degrees two-theta, 13.1 ± 0.2 degrees two-theta, 16.3 ± 0.2 degrees two-theta, and 21.1 ± 0.2 degrees two-theta; and/or (ii) an X-ray powder diffractogram substantially similar to FIG.20; and/or (iii) a triclinic crystal system, P-1 space group, and unit cell dimensions measured at 100 K on a Bruker diffractometer utilizing Cu Kα radiation (λ=1.54178 Å) of: a 10.4 ± 0.1 Å α 90.7 ± 0.1° b 11.2 ± 0.1 Å β 104.7 ± 0.1° c 19.3 ± 0.1 Å γ 98.0 ± 0.1°.
15. A substantially amorphous Compound I: I. wherein the Compound I is in an
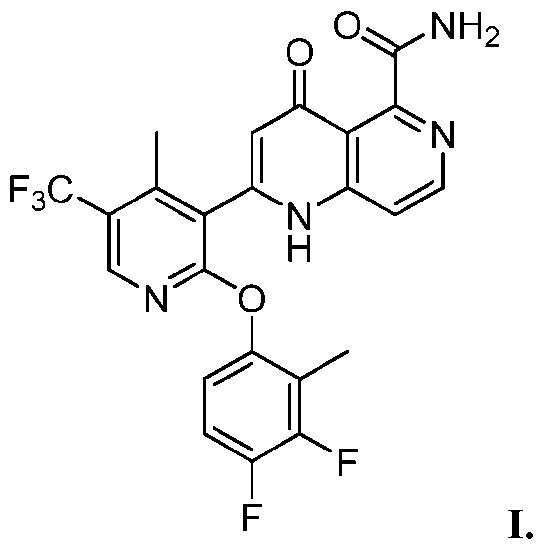
16. The substantially amorphous Compound I of claim 15, wherein the Compound I amorphous form is characterized by: (i) an X-ray powder diffractogram substantially similar to FIG.1; and/or (ii) a 13C SSNMR spectrum having one, two, three, four, five, six, seven, eight, nine, ten, or more peaks selected from 177.6 ± 0.2 ppm, 172.0 ± 0.2 ppm, 162.6 ± 0.2 ppm, 155.2 ± 0.2 ppm, 149.9 ± 0.2 ppm, 147.0 ± 0.2 ppm, 121.0 ± 0.2 ppm, 120.1 ± 0.2 ppm, 119.1 ± 0.2 ppm, 116.0 ± 0.2 ppm, 114.6 ± 0.2 ppm, 113.8 ± 0.2 ppm, 15.5 ± 0.2 ppm, and 7.7 ± 0.2 ppm; and/or (iii) a 13C SSNMR spectrum substantially similar to FIG.2; and/or (iv) a 19F SSNMR spectrum having one or two peaks selected from -61.9 ± 0.2 ppm and -142.1 ± 0.2 ppm; and/or (v) a 19F SSNMR spectrum substantially similar to FIG.3.
17. A pharmaceutical composition comprising the Compound I according to any one of claims 1 to 16.
18. The pharmaceutical composition according to claim 17, further comprising one or more additional therapeutic agents, optionally wherein the one or more additional therapeutic agents comprise one or more additional pain modulating compounds.
19. The Compound I according to any one of claims 1 to 16, or the pharmaceutical composition according to claim 17 or 18, for use in the treatment of pain, optionally wherein the pain comprises chronic pain, gut pain, neuropathic pain, musculoskeletal pain, acute pain, inflammatory pain, cancer pain, idiopathic pain, postsurgical pain, visceral pain, multiple sclerosis, Charcot-Marie-Tooth syndrome, incontinence, pathological cough, or cardiac arrhythmia.
20. Use of the Compound I according to any one of claims 1 to 16, or the pharmaceutical composition according to claim 17 or 18, in the manufacture of a medicament for the treatment of pain, optionally wherein the pain comprises chronic pain, gut pain, neuropathic pain, musculoskeletal pain,
acute pain, inflammatory pain, cancer pain, idiopathic pain, postsurgical pain, visceral pain, multiple sclerosis, Charcot-Marie-Tooth syndrome, incontinence, pathological cough, or cardiac arrhythmia.
21. A method of treating pain comprising administering the Compound I according to any one of claims 1 to 16, or the pharmaceutical composition according to claim 17 or 18, to a subject in need thereof, optionally wherein the pain comprises chronic pain, gut pain, neuropathic pain, musculoskeletal pain, acute pain, inflammatory pain, cancer pain, idiopathic pain, postsurgical pain, visceral pain, multiple sclerosis, Charcot-Marie-Tooth syndrome, incontinence, pathological cough, or cardiac arrhythmia.
22. The compound or composition for use of claim 19, the use of claim 20, or the method of claim 21, wherein the Compound I according to any one of claims 1 to 16 or the composition according to claim 17 or 18 is administered in combination with one or more additional therapeutic agents, optionally wherein the one or more additional therapeutic agents comprises one or more additional pain modulating compounds.
23. Use of the Compound I according to any one of claims 1 to 16, or the pharmaceutical composition according to claim 17 or 18, as a medicament.
24. A process for preparing a compound of formula (I): , comprising transforming a
 to the compound of formula (I); wherein
to the compound of formula (I); wherein
 are independently selected from halogen.
are independently selected from halogen.
25. The process of claim 24, wherein transforming the compound of formula (A1) to the compound of formula (I) comprises transforming the compound of formula (A1) to a compound of formula (E-A1): X2 CN N .
26. The process of claim 25,
 of formula (A1) to the compound of formula (E-A1) comprises contacting the compound of formula (A1) with a compound of formula (D-1): .
of formula (A1) to the compound of formula (E-A1) comprises contacting the compound of formula (A1) with a compound of formula (D-1): .
27. The process of claim 25 or
 the compound of formula (A1) to the compound of formula (I) further comprises transforming the compound of formula (E-A1) to the compound of formula (I).
the compound of formula (A1) to the compound of formula (I) further comprises transforming the compound of formula (E-A1) to the compound of formula (I).
28. The process of any one of claims 24 to 27, further comprising transforming a compound of formula (A1-1): to the compound of formula (A1).

29. The process of claim 28, further comprising transforming a compound of formula (A1-2): to the compound of formula (A1-1).

30. The process of claim 29, further comprising transforming a compound of formula (A1-3): to the compound of formula (A1-2).

31. The process of claim 30, further comprising transforming a compound of formula (A1-4): to the compound of formula (A1-3).

32. The process of claim 31, further comprising transforming a compound of formula (A1-5): to the compound of formula (A1-4).

33. The process of any one of claims 24 to 32, wherein each of X1 and X2 are chloro.
34. A process for preparing a compound of formula (I): comprising transforming a
 to the compound of formula (I).
to the compound of formula (I).

35. The process of claim 34, wherein transforming the compound of formula (A3) to the compound of formula (I) comprises transforming the compound of formula (A3) to a compound of formula (E-A3): .
36. The process of claim 35,
 of formula (A3) to the compound of formula (E-A3) comprises contacting the compound of formula (A3) with a compound of formula (D-1): .
of formula (A3) to the compound of formula (E-A3) comprises contacting the compound of formula (A3) with a compound of formula (D-1): .
37. The process of claim 35 or 36, wherein transforming the compound of formula (A3) to the compound of formula (I) further comprises transforming the compound of formula (E-A3) to the compound of formula (I).
38. The process of any one of claims 34 to 38, further comprising transforming a compound of formula (A3-1): to the compound of formula (A3).

39. The process of claim 38, further comprising transforming a compound of formula (A3-2): to the compound of formula (A3-1).
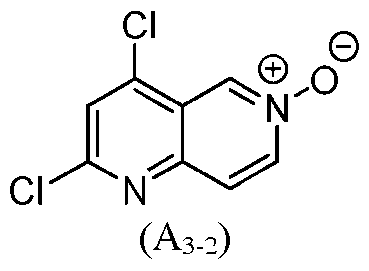
40. The process of any one of claims 26 to 33 or 36 to 39, further comprising transforming a compound of formula (D-2): to the compound of formula (D-1).

41. The process of claim 40, further comprising transforming a compound of formula (B-1): to the compound of formula (D-2).

42. The process of claim 41, wherein transforming a compound of formula (B-1) to the compound of formula (D-2) comprises contacting the compound of formula (B-1) with a compound of formula (C-1): in the presence of a base, optionally wherein is potassium carbonate.

43. The process of claim 42, further comprising transforming a compound of formula (B-2): to the compound of formula (B-1).

44. The process of claim 43, further comprising transforming a compound of formula (B-3): to the compound of formula (B-2).

45. The process of claim 44, further comprising transforming a compound of formula (B-4): to the compound of formula (B-3).

46. The process of claim 45, further comprising transforming a compound of formula (B-5): to the compound of formula (B-4).

47. The process of any one of claims 42 to 46, further comprising transforming a compound of formula (C-2): to the compound of formula (C-1), wherein protecting group.

48. The process of claim 47, further comprising transforming a compound of formula (C-3): to the compound of formula (C-2), wherein protecting group.

49. The process of claim 48, further comprising transforming a compound of formula (C-4): to the compound of formula (C-3).

50. The process of any one of claims 24 to 49, further comprising transforming the compound of formula (I) to a solvate of the compound of formula (I), optionally wherein the solvate of the compound of formula (I) is a compound of formula (F): O NH O 2 .
51. The process of claim 50, further comprising transforming the solvate of the compound of formula (I) to Form A of the compound of formula (I).
52. A compound selected from: ,

53. A compound of formula (I): , or a pharmaceutically acceptable prepared by the process of any one of claims 24 to 51.

Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US202363592494P | 2023-10-23 | 2023-10-23 | |
| US202363592530P | 2023-10-23 | 2023-10-23 | |
| US63/592,494 | 2023-10-23 | ||
| US63/592,530 | 2023-10-23 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2025090511A1 true WO2025090511A1 (en) | 2025-05-01 |
Family
ID=93430839
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2024/052419 Pending WO2025090511A1 (en) | 2023-10-23 | 2024-10-22 | Methods of preparing modulators of sodium channels and solid forms of the same for treating pain |
Country Status (2)
| Country | Link |
|---|---|
| TW (1) | TW202535867A (en) |
| WO (1) | WO2025090511A1 (en) |
Citations (71)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5304121A (en) | 1990-12-28 | 1994-04-19 | Boston Scientific Corporation | Drug delivery system making use of a hydrogel polymer coating |
| US5886026A (en) | 1993-07-19 | 1999-03-23 | Angiotech Pharmaceuticals Inc. | Anti-angiogenic compositions and methods of use |
| US6099562A (en) | 1996-06-13 | 2000-08-08 | Schneider (Usa) Inc. | Drug coating with topcoat |
| US20040006237A1 (en) | 2001-11-14 | 2004-01-08 | Teva Pharmaceutical Industries Ltd. | Amorphous and crystalline forms of losartan potassium and process for their preparation |
| WO2006011050A2 (en) | 2004-07-23 | 2006-02-02 | Pfizer Limited | Pyridine derivatives |
| WO2008135826A2 (en) | 2007-05-03 | 2008-11-13 | Pfizer Limited | 2 -pyridine carboxamide derivatives as sodium channel modulators |
| WO2010129864A2 (en) | 2009-05-07 | 2010-11-11 | The Board Of Trustees Of The Leland Stanford Junior University | Methods and compositions for studying, imaging, and treating pain |
| WO2011026240A1 (en) | 2009-09-04 | 2011-03-10 | Zalicus Pharmaceuticals Ltd. | Oxopiperazine derivatives for the treatment of pain and epilepsy |
| WO2011140425A1 (en) | 2010-05-06 | 2011-11-10 | Vertex Pharmaceuticals Incorporated | Heterocyclic chromene-spirocyclic piperidine amides as modulators of ion channels |
| US20120196869A1 (en) | 2011-02-02 | 2012-08-02 | Sara Sabina Hadida Ruah | Pyrrolopyrazine-spirocyclic piperidine amides as modulators of ion channels |
| WO2012112743A1 (en) | 2011-02-18 | 2012-08-23 | Vertex Pharmaceuticals Incorporated | Chroman - spirocyclic piperidine amides as modulators of ion channels |
| WO2012116440A1 (en) | 2011-03-03 | 2012-09-07 | Zalicus Pharmaceuticals Ltd. | Benzimidazole inhibitors of the sodium channel |
| WO2012125613A1 (en) | 2011-03-14 | 2012-09-20 | Vertex Pharmaceuticals Incorporated | Morpholine-spirocyclic piperidine amides as modulators of ion channels |
| WO2012125973A2 (en) | 2011-03-16 | 2012-09-20 | Amgen Inc. | Potent and selective inhibitors of nav1.3 and nav1.7 |
| WO2013061205A2 (en) | 2011-10-26 | 2013-05-02 | Pfizer Limited | Chemical compounds |
| WO2013086229A1 (en) | 2011-12-07 | 2013-06-13 | Amgen Inc. | Bicyclic aryl and heteroaryl sodium channel inhibitors |
| US8466188B2 (en) | 2006-10-12 | 2013-06-18 | Xenon Pharmaceuticals Inc. | Use of spiro-oxindole compounds as therapeutic agents |
| WO2013109521A1 (en) | 2012-01-16 | 2013-07-25 | Vertex Pharmaceuticals Incorporated | Pyran-spirocyclic piperidine amides as modulators of ion channels |
| WO2013114250A1 (en) | 2012-02-03 | 2013-08-08 | Pfizer Inc. | Benziimidazole and imidazopyridine derivatives as sodium channel modulators |
| WO2013131018A1 (en) | 2012-03-02 | 2013-09-06 | Zalicus Pharmaceuticals Ltd. | Biaryl inhibitors of the sodium channel |
| WO2013134518A1 (en) | 2012-03-09 | 2013-09-12 | Amgen Inc. | Sulfamide sodium channel inhibitors |
| US20130303535A1 (en) | 2008-12-26 | 2013-11-14 | Dainippon Sumitomo Pharma Co., Ltd. | Novel bicyclic heterocyclic compound |
| US20140213616A1 (en) | 2013-01-31 | 2014-07-31 | Vertex Pharmaceuticals Incorporated | Pyridone amides as modulators of sodium channels |
| WO2014120820A1 (en) | 2013-01-31 | 2014-08-07 | Vertex Pharmaceuticals Incorporated | Amides as modulators of sodium channels |
| WO2014120815A1 (en) | 2013-01-31 | 2014-08-07 | Vertex Pharmaceuticals Incorporated | Quinoline and quinazoline amides as modulators of sodium channels |
| US8883840B2 (en) | 2009-06-29 | 2014-11-11 | Xenon Pharmaceuticals Inc. | Enantiomers of spiro-oxindole compounds and their uses as therapeutic agents |
| WO2014201206A1 (en) | 2013-06-12 | 2014-12-18 | Amgen Inc. | Bicyclic sulfonamide compounds as sodium channel inhibitors |
| WO2015010065A1 (en) | 2013-07-19 | 2015-01-22 | Vertex Pharmaceuticals Incorporated | Sulfonamides as modulators of sodium channels |
| US20150166589A1 (en) | 2013-12-13 | 2015-06-18 | Vertex Pharmaceuticals Incorporated | Prodrugs of pyridone amides useful as modulators of sodium channels |
| WO2015157559A2 (en) | 2014-04-09 | 2015-10-15 | Siteone Therapeutics, Inc. | 10',11'-modified saxitoxins for the treatment of pain |
| WO2016141035A1 (en) | 2015-03-02 | 2016-09-09 | Amgen Inc. | Bicyclic ketone sulfonamide compounds |
| WO2017059385A1 (en) | 2015-09-30 | 2017-04-06 | Siteone Therapeutics, Inc. | 11,13-modified saxitoxins for the treatment of pain |
| WO2018183782A1 (en) | 2017-03-29 | 2018-10-04 | Siteone Therapeutics, Inc. | 11,13-modified saxitoxins for the treatment of pain |
| WO2018183781A1 (en) | 2017-03-29 | 2018-10-04 | Siteone Therapeutics, Inc. | 11,13-modified saxitoxins for the treatment of pain |
| WO2018213426A1 (en) | 2017-05-16 | 2018-11-22 | Vertex Pharmaceuticals Incorporated | Deuterated pyridone amides and prodrugs thereof as modulators of sodium channels |
| WO2019014352A1 (en) | 2017-07-11 | 2019-01-17 | Vertex Pharmaceuticals Incorporated | Carboxamides as modulators of sodium channels |
| WO2020014246A1 (en) | 2018-07-09 | 2020-01-16 | Lieber Institute, Inc. | Pyridine carboxamide compounds for inhibiting nav1.8 |
| WO2020014243A1 (en) | 2018-07-09 | 2020-01-16 | Lieber Institute, Inc. | PYRIDAZINE COMPOUNDS FOR INHIBITING Nav1.8 |
| WO2020072835A1 (en) | 2018-10-03 | 2020-04-09 | Siteone Therapeutics, Inc. | 11,13-modified saxitoxins for the treatment of pain |
| WO2020092667A1 (en) | 2018-11-02 | 2020-05-07 | Merck Sharp & Dohme Corp. | 2-amino-n-heteroaryl-nicotinamides as nav1.8 inhibitors |
| WO2020092187A1 (en) | 2018-11-02 | 2020-05-07 | Merck Sharp & Dohme Corp. | 2-amino-n-phenyl-nicotinamides as nav1.8 inhibitors |
| CN111217776A (en) | 2020-01-19 | 2020-06-02 | 中国人民解放军军事科学院军事医学研究院 | Amide derivative containing benzo heterocyclic structure, composition and application |
| WO2020117626A1 (en) | 2018-12-05 | 2020-06-11 | Merck Sharp & Dohme Corp. | 4-amino or 4-alkoxy-substituted aryl sulfonamide compounds with selective activity in voltage-gated sodium channels |
| WO2020140959A1 (en) | 2019-01-04 | 2020-07-09 | 江苏恒瑞医药股份有限公司 | 6-oxo-1,6-dihydropyridazine derivative, preparation method therefor and medical use thereof |
| WO2020144375A1 (en) | 2019-01-11 | 2020-07-16 | Grünenthal GmbH | Substituted pyrrolidine amides iii |
| WO2020146682A1 (en) | 2019-01-10 | 2020-07-16 | Vertex Pharmaceuticals Incorporated | Carboxamides as modulators of sodium channels |
| WO2020146612A1 (en) | 2019-01-10 | 2020-07-16 | Vertex Pharmaceuticals Incorporated | Esters and carbamates as modulators of sodium channels |
| WO2020151728A1 (en) | 2019-01-25 | 2020-07-30 | 江苏恒瑞医药股份有限公司 | 2-oxo-1,2-dihydropyridin derivative, preparation method therefor and medical uses thereof |
| CN111808019A (en) | 2020-09-08 | 2020-10-23 | 上海济煜医药科技有限公司 | A kind of cyclic compound and its application |
| WO2020261114A1 (en) | 2019-06-27 | 2020-12-30 | Glaxosmithkline Intellectual Property Development Limited | 2,3-dihydroquinazolin compounds as nav1.8 inhibitors |
| CN112225695A (en) | 2020-12-15 | 2021-01-15 | 上海济煜医药科技有限公司 | Oxynitride and preparation method and application thereof |
| CN112300069A (en) | 2019-07-31 | 2021-02-02 | 明慧医药(上海)有限公司 | A kind of selective sodium channel regulator and its preparation and application |
| CN112300051A (en) | 2019-07-31 | 2021-02-02 | 明慧医药(上海)有限公司 | Selective sodium channel regulator and preparation and application thereof |
| CN112390745A (en) | 2019-08-19 | 2021-02-23 | 江苏恒瑞医药股份有限公司 | Pyridine nicotinamide derivatives, preparation method and medical application thereof |
| WO2021032074A1 (en) | 2019-08-19 | 2021-02-25 | 江苏恒瑞医药股份有限公司 | Benzamide fused aromatic ring derivative, preparation method therefor and application thereof in medicine |
| CN112441969A (en) | 2019-08-30 | 2021-03-05 | 明慧医药(上海)有限公司 | Selective sodium channel regulator and preparation and application thereof |
| CN112457294A (en) | 2021-01-27 | 2021-03-09 | 上海济煜医药科技有限公司 | Compound serving as NaV1.8 retarder and preparation method and application thereof |
| CN112479996A (en) | 2019-09-12 | 2021-03-12 | 上海济煜医药科技有限公司 | Pyridine oxynitride and preparation method and application thereof |
| WO2021252822A1 (en) | 2020-06-10 | 2021-12-16 | Amgen Inc. | Heteroalkyl dihydroquinoline sulfonamide compounds |
| WO2021252820A1 (en) | 2020-06-10 | 2021-12-16 | Amgen Inc. | Cyclobutyl dihydroquinoline sulfonamide compounds |
| WO2021252818A1 (en) | 2020-06-10 | 2021-12-16 | Amgen Inc. | Cyclopropyl dihydroquinoline sulfonamide compounds |
| WO2021257490A1 (en) | 2020-06-17 | 2021-12-23 | Merck Sharp & Dohme Corp. | 2-oxoimidazolidine-4-carboxamides as nav1.8 inhibitors |
| WO2021257418A1 (en) | 2020-06-17 | 2021-12-23 | Merck Sharp & Dohme Corp. | 2-oxo-oxazolidine-5-carboxamides as nav1.8 inhibitors |
| WO2021257420A1 (en) | 2020-06-17 | 2021-12-23 | Merck Sharp & Dohme Corp. | 5-oxopyrrolidine-3-carboxamides as nav1.8 inhibitors |
| WO2022036297A1 (en) | 2020-08-14 | 2022-02-17 | Siteone Therapeutics, Inc. | Non-hydrated ketone inhibitors of nav1.7 for the treatment of pain |
| CN114591293A (en) | 2020-12-07 | 2022-06-07 | 成都康弘药业集团股份有限公司 | Conjunctive compounds as Nav1.8 inhibitors and their uses |
| WO2022235859A1 (en) | 2021-05-07 | 2022-11-10 | Merck Sharp & Dohme Llc | Cycloalkyl 3-oxopiperazine carboxamides and cycloheteroalkyl 3-oxopiperazine carboxamides as nav1.8 inhibitors |
| WO2022235558A1 (en) | 2021-05-07 | 2022-11-10 | Merck Sharp & Dohme Llc | Aryl 3-oxopiperazine carboxamides and heteroaryl 3-oxopiperazine carboxamides as nav1.8 inhibitors |
| WO2022263498A1 (en) | 2021-06-15 | 2022-12-22 | Grünenthal GmbH | Substituted pyrazole amides |
| WO2023138599A1 (en) | 2022-01-18 | 2023-07-27 | 成都康弘药业集团股份有限公司 | Aromatic fused ring nav1.8 inhibitor, and use thereof |
| WO2023205463A1 (en) * | 2022-04-22 | 2023-10-26 | Vertex Pharmaceuticals Incorporated | Heteroaryl compounds for the treatment of pain |
-
2024
- 2024-10-22 WO PCT/US2024/052419 patent/WO2025090511A1/en active Pending
- 2024-10-22 TW TW113140119A patent/TW202535867A/en unknown
Patent Citations (91)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5304121A (en) | 1990-12-28 | 1994-04-19 | Boston Scientific Corporation | Drug delivery system making use of a hydrogel polymer coating |
| US5886026A (en) | 1993-07-19 | 1999-03-23 | Angiotech Pharmaceuticals Inc. | Anti-angiogenic compositions and methods of use |
| US6099562A (en) | 1996-06-13 | 2000-08-08 | Schneider (Usa) Inc. | Drug coating with topcoat |
| US20040006237A1 (en) | 2001-11-14 | 2004-01-08 | Teva Pharmaceutical Industries Ltd. | Amorphous and crystalline forms of losartan potassium and process for their preparation |
| WO2006011050A2 (en) | 2004-07-23 | 2006-02-02 | Pfizer Limited | Pyridine derivatives |
| US20080312235A1 (en) | 2004-07-23 | 2008-12-18 | Charlotte Alice Louise Lane | Pyridine Derivatives |
| US8466188B2 (en) | 2006-10-12 | 2013-06-18 | Xenon Pharmaceuticals Inc. | Use of spiro-oxindole compounds as therapeutic agents |
| WO2008135826A2 (en) | 2007-05-03 | 2008-11-13 | Pfizer Limited | 2 -pyridine carboxamide derivatives as sodium channel modulators |
| US20090048306A1 (en) | 2007-05-03 | 2009-02-19 | Pfizer, Inc. | Pyridine derivatives |
| US20130303535A1 (en) | 2008-12-26 | 2013-11-14 | Dainippon Sumitomo Pharma Co., Ltd. | Novel bicyclic heterocyclic compound |
| WO2010129864A2 (en) | 2009-05-07 | 2010-11-11 | The Board Of Trustees Of The Leland Stanford Junior University | Methods and compositions for studying, imaging, and treating pain |
| US8883840B2 (en) | 2009-06-29 | 2014-11-11 | Xenon Pharmaceuticals Inc. | Enantiomers of spiro-oxindole compounds and their uses as therapeutic agents |
| WO2011026240A1 (en) | 2009-09-04 | 2011-03-10 | Zalicus Pharmaceuticals Ltd. | Oxopiperazine derivatives for the treatment of pain and epilepsy |
| US20120220605A1 (en) | 2009-09-04 | 2012-08-30 | Zalicus Pharmaceuticals Ltd. | Oxopiperazine derivatives for the treatment of pain and epilepsy |
| WO2011140425A1 (en) | 2010-05-06 | 2011-11-10 | Vertex Pharmaceuticals Incorporated | Heterocyclic chromene-spirocyclic piperidine amides as modulators of ion channels |
| US20110306607A1 (en) | 2010-05-06 | 2011-12-15 | Vertex Pharmaceuticals Incorporated | Heterocyclic chromene-spirocyclic piperidine amides as modulators of ion channels |
| US20120196869A1 (en) | 2011-02-02 | 2012-08-02 | Sara Sabina Hadida Ruah | Pyrrolopyrazine-spirocyclic piperidine amides as modulators of ion channels |
| WO2012106499A1 (en) | 2011-02-02 | 2012-08-09 | Vertex Pharmaceuticals Incorporated | Pyrrolopyrazine-spirocyclic piperidine amides as modulators of ion channels |
| US20120245136A1 (en) | 2011-02-18 | 2012-09-27 | Sara Sabina Hadida-Ruah | Chroman-spirocyclic piperidine amides as modulators of ion channels |
| WO2012112743A1 (en) | 2011-02-18 | 2012-08-23 | Vertex Pharmaceuticals Incorporated | Chroman - spirocyclic piperidine amides as modulators of ion channels |
| WO2012116440A1 (en) | 2011-03-03 | 2012-09-07 | Zalicus Pharmaceuticals Ltd. | Benzimidazole inhibitors of the sodium channel |
| US20140187533A1 (en) | 2011-03-03 | 2014-07-03 | Zalicus Pharmaceuticals Ltd. | Benzimidazole inhibitors of the sodium channel |
| WO2012125613A1 (en) | 2011-03-14 | 2012-09-20 | Vertex Pharmaceuticals Incorporated | Morpholine-spirocyclic piperidine amides as modulators of ion channels |
| US20120264749A1 (en) | 2011-03-14 | 2012-10-18 | Sara Sabina Hadida-Ruah | Morpholine-spirocyclic piperidine amides as modulators of ion channels |
| WO2012125973A2 (en) | 2011-03-16 | 2012-09-20 | Amgen Inc. | Potent and selective inhibitors of nav1.3 and nav1.7 |
| WO2013061205A2 (en) | 2011-10-26 | 2013-05-02 | Pfizer Limited | Chemical compounds |
| US20140296313A1 (en) | 2011-10-26 | 2014-10-02 | Pfizer Limited | (4-Phenylimidazol-2-yl) Ethylamine Derivatives Useful As Sodium Channel Modulators |
| WO2013086229A1 (en) | 2011-12-07 | 2013-06-13 | Amgen Inc. | Bicyclic aryl and heteroaryl sodium channel inhibitors |
| US20150005304A1 (en) | 2012-01-16 | 2015-01-01 | Vertex Pharmaceuticals Incorporated | Pyran-Spirocyclic Piperidine Amides as Modulators of Ion Channels |
| WO2013109521A1 (en) | 2012-01-16 | 2013-07-25 | Vertex Pharmaceuticals Incorporated | Pyran-spirocyclic piperidine amides as modulators of ion channels |
| US20130274243A1 (en) | 2012-02-03 | 2013-10-17 | Pfizer Limited | Chemical Compounds |
| WO2013114250A1 (en) | 2012-02-03 | 2013-08-08 | Pfizer Inc. | Benziimidazole and imidazopyridine derivatives as sodium channel modulators |
| WO2013131018A1 (en) | 2012-03-02 | 2013-09-06 | Zalicus Pharmaceuticals Ltd. | Biaryl inhibitors of the sodium channel |
| WO2013134518A1 (en) | 2012-03-09 | 2013-09-12 | Amgen Inc. | Sulfamide sodium channel inhibitors |
| US20140228371A1 (en) | 2013-01-31 | 2014-08-14 | Vertex Pharmaceuticals Incorporated | Quinoline and quinazoline amides as modulators of sodium channels |
| WO2014120808A1 (en) | 2013-01-31 | 2014-08-07 | Vertex Parmaceuticals Incorporated | Pyridone amides as modulators of sodium channels |
| US20140221435A1 (en) | 2013-01-31 | 2014-08-07 | Vertex Pharmaceuticals Incorporated | Amides as modulators of sodium channels |
| WO2014120815A1 (en) | 2013-01-31 | 2014-08-07 | Vertex Pharmaceuticals Incorporated | Quinoline and quinazoline amides as modulators of sodium channels |
| WO2014120820A1 (en) | 2013-01-31 | 2014-08-07 | Vertex Pharmaceuticals Incorporated | Amides as modulators of sodium channels |
| US20140213616A1 (en) | 2013-01-31 | 2014-07-31 | Vertex Pharmaceuticals Incorporated | Pyridone amides as modulators of sodium channels |
| WO2014201206A1 (en) | 2013-06-12 | 2014-12-18 | Amgen Inc. | Bicyclic sulfonamide compounds as sodium channel inhibitors |
| WO2014201173A1 (en) | 2013-06-12 | 2014-12-18 | Amgen Inc. | Bicyclic sulfonamide compounds as sodium channel inhibitors |
| WO2015010065A1 (en) | 2013-07-19 | 2015-01-22 | Vertex Pharmaceuticals Incorporated | Sulfonamides as modulators of sodium channels |
| US20160152561A1 (en) | 2013-07-19 | 2016-06-02 | Vertex Pharmaceuticals Incorporated | Sulfonamides as modulators of sodium channels |
| WO2015089361A1 (en) | 2013-12-13 | 2015-06-18 | Vertex Pharmaceuticals Incorporated | Prodrugs of pyridone amides useful as modulators of sodium channels |
| US20150166589A1 (en) | 2013-12-13 | 2015-06-18 | Vertex Pharmaceuticals Incorporated | Prodrugs of pyridone amides useful as modulators of sodium channels |
| WO2015157559A2 (en) | 2014-04-09 | 2015-10-15 | Siteone Therapeutics, Inc. | 10',11'-modified saxitoxins for the treatment of pain |
| WO2016141035A1 (en) | 2015-03-02 | 2016-09-09 | Amgen Inc. | Bicyclic ketone sulfonamide compounds |
| WO2017059385A1 (en) | 2015-09-30 | 2017-04-06 | Siteone Therapeutics, Inc. | 11,13-modified saxitoxins for the treatment of pain |
| WO2018183782A1 (en) | 2017-03-29 | 2018-10-04 | Siteone Therapeutics, Inc. | 11,13-modified saxitoxins for the treatment of pain |
| WO2018183781A1 (en) | 2017-03-29 | 2018-10-04 | Siteone Therapeutics, Inc. | 11,13-modified saxitoxins for the treatment of pain |
| WO2018213426A1 (en) | 2017-05-16 | 2018-11-22 | Vertex Pharmaceuticals Incorporated | Deuterated pyridone amides and prodrugs thereof as modulators of sodium channels |
| WO2019014352A1 (en) | 2017-07-11 | 2019-01-17 | Vertex Pharmaceuticals Incorporated | Carboxamides as modulators of sodium channels |
| US20190016671A1 (en) | 2017-07-11 | 2019-01-17 | Vertex Pharmaceuticals Incorporated | Carboxamides as modulators of sodium channels |
| WO2020014246A1 (en) | 2018-07-09 | 2020-01-16 | Lieber Institute, Inc. | Pyridine carboxamide compounds for inhibiting nav1.8 |
| WO2020014243A1 (en) | 2018-07-09 | 2020-01-16 | Lieber Institute, Inc. | PYRIDAZINE COMPOUNDS FOR INHIBITING Nav1.8 |
| WO2020072835A1 (en) | 2018-10-03 | 2020-04-09 | Siteone Therapeutics, Inc. | 11,13-modified saxitoxins for the treatment of pain |
| WO2020092187A1 (en) | 2018-11-02 | 2020-05-07 | Merck Sharp & Dohme Corp. | 2-amino-n-phenyl-nicotinamides as nav1.8 inhibitors |
| WO2020092667A1 (en) | 2018-11-02 | 2020-05-07 | Merck Sharp & Dohme Corp. | 2-amino-n-heteroaryl-nicotinamides as nav1.8 inhibitors |
| US20200140411A1 (en) | 2018-11-02 | 2020-05-07 | Merck Sharp & Dohme Corp. | 2-amino-n-heteroaryl-nicotinamides as nav1.8 inhibitors |
| WO2020117626A1 (en) | 2018-12-05 | 2020-06-11 | Merck Sharp & Dohme Corp. | 4-amino or 4-alkoxy-substituted aryl sulfonamide compounds with selective activity in voltage-gated sodium channels |
| WO2020140959A1 (en) | 2019-01-04 | 2020-07-09 | 江苏恒瑞医药股份有限公司 | 6-oxo-1,6-dihydropyridazine derivative, preparation method therefor and medical use thereof |
| WO2020146612A1 (en) | 2019-01-10 | 2020-07-16 | Vertex Pharmaceuticals Incorporated | Esters and carbamates as modulators of sodium channels |
| WO2020146682A1 (en) | 2019-01-10 | 2020-07-16 | Vertex Pharmaceuticals Incorporated | Carboxamides as modulators of sodium channels |
| WO2020144375A1 (en) | 2019-01-11 | 2020-07-16 | Grünenthal GmbH | Substituted pyrrolidine amides iii |
| WO2020151728A1 (en) | 2019-01-25 | 2020-07-30 | 江苏恒瑞医药股份有限公司 | 2-oxo-1,2-dihydropyridin derivative, preparation method therefor and medical uses thereof |
| WO2020261114A1 (en) | 2019-06-27 | 2020-12-30 | Glaxosmithkline Intellectual Property Development Limited | 2,3-dihydroquinazolin compounds as nav1.8 inhibitors |
| CN112300069A (en) | 2019-07-31 | 2021-02-02 | 明慧医药(上海)有限公司 | A kind of selective sodium channel regulator and its preparation and application |
| CN112300051A (en) | 2019-07-31 | 2021-02-02 | 明慧医药(上海)有限公司 | Selective sodium channel regulator and preparation and application thereof |
| CN112390745A (en) | 2019-08-19 | 2021-02-23 | 江苏恒瑞医药股份有限公司 | Pyridine nicotinamide derivatives, preparation method and medical application thereof |
| WO2021032074A1 (en) | 2019-08-19 | 2021-02-25 | 江苏恒瑞医药股份有限公司 | Benzamide fused aromatic ring derivative, preparation method therefor and application thereof in medicine |
| CN112441969A (en) | 2019-08-30 | 2021-03-05 | 明慧医药(上海)有限公司 | Selective sodium channel regulator and preparation and application thereof |
| CN112479996A (en) | 2019-09-12 | 2021-03-12 | 上海济煜医药科技有限公司 | Pyridine oxynitride and preparation method and application thereof |
| WO2021047622A1 (en) | 2019-09-12 | 2021-03-18 | 上海济煜医药科技有限公司 | Pyridine oxynitride, preparation method therefor and use thereof |
| CN111217776A (en) | 2020-01-19 | 2020-06-02 | 中国人民解放军军事科学院军事医学研究院 | Amide derivative containing benzo heterocyclic structure, composition and application |
| WO2021252822A1 (en) | 2020-06-10 | 2021-12-16 | Amgen Inc. | Heteroalkyl dihydroquinoline sulfonamide compounds |
| WO2021252820A1 (en) | 2020-06-10 | 2021-12-16 | Amgen Inc. | Cyclobutyl dihydroquinoline sulfonamide compounds |
| WO2021252818A1 (en) | 2020-06-10 | 2021-12-16 | Amgen Inc. | Cyclopropyl dihydroquinoline sulfonamide compounds |
| WO2021257418A1 (en) | 2020-06-17 | 2021-12-23 | Merck Sharp & Dohme Corp. | 2-oxo-oxazolidine-5-carboxamides as nav1.8 inhibitors |
| WO2021257490A1 (en) | 2020-06-17 | 2021-12-23 | Merck Sharp & Dohme Corp. | 2-oxoimidazolidine-4-carboxamides as nav1.8 inhibitors |
| WO2021257420A1 (en) | 2020-06-17 | 2021-12-23 | Merck Sharp & Dohme Corp. | 5-oxopyrrolidine-3-carboxamides as nav1.8 inhibitors |
| WO2022036297A1 (en) | 2020-08-14 | 2022-02-17 | Siteone Therapeutics, Inc. | Non-hydrated ketone inhibitors of nav1.7 for the treatment of pain |
| CN111808019A (en) | 2020-09-08 | 2020-10-23 | 上海济煜医药科技有限公司 | A kind of cyclic compound and its application |
| CN114591293A (en) | 2020-12-07 | 2022-06-07 | 成都康弘药业集团股份有限公司 | Conjunctive compounds as Nav1.8 inhibitors and their uses |
| CN112225695A (en) | 2020-12-15 | 2021-01-15 | 上海济煜医药科技有限公司 | Oxynitride and preparation method and application thereof |
| CN112457294A (en) | 2021-01-27 | 2021-03-09 | 上海济煜医药科技有限公司 | Compound serving as NaV1.8 retarder and preparation method and application thereof |
| WO2022235859A1 (en) | 2021-05-07 | 2022-11-10 | Merck Sharp & Dohme Llc | Cycloalkyl 3-oxopiperazine carboxamides and cycloheteroalkyl 3-oxopiperazine carboxamides as nav1.8 inhibitors |
| WO2022235558A1 (en) | 2021-05-07 | 2022-11-10 | Merck Sharp & Dohme Llc | Aryl 3-oxopiperazine carboxamides and heteroaryl 3-oxopiperazine carboxamides as nav1.8 inhibitors |
| WO2022263498A1 (en) | 2021-06-15 | 2022-12-22 | Grünenthal GmbH | Substituted pyrazole amides |
| WO2023138599A1 (en) | 2022-01-18 | 2023-07-27 | 成都康弘药业集团股份有限公司 | Aromatic fused ring nav1.8 inhibitor, and use thereof |
| WO2023205463A1 (en) * | 2022-04-22 | 2023-10-26 | Vertex Pharmaceuticals Incorporated | Heteroaryl compounds for the treatment of pain |
Non-Patent Citations (32)
Also Published As
| Publication number | Publication date |
|---|---|
| TW202535867A (en) | 2025-09-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| TWI891687B (en) | Substituted tetrahydrofurans as modulators of sodium channels | |
| JP7277431B2 (en) | Carboxamides as modulators of sodium channels | |
| JP2024520643A (en) | Hydroxy and (halo)alkoxy substituted tetrahydrofurans as modulators of sodium channels | |
| JP2024522292A (en) | N-(hydroxyalkyl(hetero)aryl)tetrahydrofurancarboxamides as sodium channel modulators | |
| WO2020146612A1 (en) | Esters and carbamates as modulators of sodium channels | |
| IL321269A (en) | Process for the synthesis of modified tetrahydrofuran-based sodium channel modulators | |
| TW202523313A (en) | Heteroaryl compounds for the treatment of pain | |
| EP4511117A1 (en) | Heteroaryl compounds for the treatment of pain | |
| WO2025090511A1 (en) | Methods of preparing modulators of sodium channels and solid forms of the same for treating pain | |
| WO2025090516A1 (en) | Methods of preparing compounds for treating pain and solid forms thereof | |
| WO2025090480A1 (en) | Heteroaryl compounds for the treatment of pain | |
| HK40121637A (en) | Substituted tetrahydrofurans as modulators of sodium channels | |
| HK40080163A (en) | Substituted tetrahydrofurans as modulators of sodium channels | |
| HK40080163B (en) | Substituted tetrahydrofurans as modulators of sodium channels | |
| OA21241A (en) | Substituted tetrahydrofurans as modulators of sodium channels. |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 24802100 Country of ref document: EP Kind code of ref document: A1 |