WO2025089240A1 - Colloidal silica and method for producing colloidal silica - Google Patents
Colloidal silica and method for producing colloidal silica Download PDFInfo
- Publication number
- WO2025089240A1 WO2025089240A1 PCT/JP2024/037448 JP2024037448W WO2025089240A1 WO 2025089240 A1 WO2025089240 A1 WO 2025089240A1 JP 2024037448 W JP2024037448 W JP 2024037448W WO 2025089240 A1 WO2025089240 A1 WO 2025089240A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- colloidal silica
- mass
- concentration
- less
- reaction
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Images





Classifications
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B33/00—Silicon; Compounds thereof
- C01B33/113—Silicon oxides; Hydrates thereof
- C01B33/12—Silica; Hydrates thereof, e.g. lepidoic silicic acid
- C01B33/14—Colloidal silica, e.g. dispersions, gels, sols
- C01B33/141—Preparation of hydrosols or aqueous dispersions
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K3/00—Materials not provided for elsewhere
- C09K3/14—Anti-slip materials; Abrasives
Definitions
- This invention relates to colloidal silica and a method for producing colloidal silica.
- Methods that have been proposed and implemented as industrial methods for producing high-purity colloidal silica include ion-exchanging an aqueous solution of sodium silicate, thermal decomposition of silicon tetrachloride, and hydrolysis of organosilicate in a water-alcohol mixed solvent in the presence of an acid or alkali catalyst.
- the method of hydrolyzing organosilicate allows the use of high-purity organosilicate, catalyst, and solvent for the reaction, so that the amount of impurities derived from these raw materials is extremely small, and it is particularly suitable as a method for producing high-purity colloidal silica with low metal impurity content, and several methods have been proposed for hydrolyzing organosilicate.
- colloidal silica used for various purposes particularly colloidal silica used in the field of semiconductor wafer polishing, with the increasing integration of today's LSIs, various types of metal wiring and oxide films exist on a single wafer, and each semiconductor wafer requires its own appropriate polishing performance, so colloidal silica with various slightly different compositions and properties is required.
- colloidal silica used in applications where even the slightest alkali metal impurities are undesirable, such as binders for hard coatings and ceramics, chromate-based metal surface treatment agents, and ground improvement grouting agents, acidic colloidal silica is required, and several proposals for manufacturing such acidic colloidal silica are known.
- the applicant of the present application has investigated a method for easily producing colloidal silica having predetermined properties, such as spherical colloidal silica, which does not require special post-treatment such as acid treatment, ion exchange treatment, or further modification treatment, has an extremely low content of metal impurities, including alkali metals, and has an average particle size in the range of 5 to 500 nm, a standard deviation of 20 or less, and a polydispersity index of 0.15 or less, as determined by particle size distribution analysis using an electron microscope.
- spherical colloidal silica which does not require special post-treatment such as acid treatment, ion exchange treatment, or further modification treatment, has an extremely low content of metal impurities, including alkali metals, and has an average particle size in the range of 5 to 500 nm, a standard deviation of 20 or less, and a polydispersity index of 0.15 or less, as determined by particle size distribution analysis using an electron microscope.
- colloidal silica with a particle size smaller than that of conventional colloidal silica, that is, a particle size of a few nm, or a particle size close to that.
- the characteristics e.g., polishability, improvement of the number of defects on the polished object, etc.
- the inventors of the present application therefore conducted extensive research to develop colloidal silica with a small particle size of single nano size or close to that size, which does not substantially aggregate even when the solid concentration is relatively high. As a result, they discovered that this could be achieved by devising a raw material preparation process and a concentration adjustment process, etc., and by subjecting colloidal silica to a modification treatment at a predetermined timing, and thus completed the present invention.
- the object of the present invention is therefore to provide colloidal silica having a small particle size of single nano size or close to that size, which is substantially free of aggregation, and a method for producing the same.
- Patent Document 2 is related to modified colloidal silica that can improve the stability of polishing rate over time when used as abrasive grains with few microparticles, but does not specifically teach colloidal silica with a single nano size or a small particle size close to that, and does not disclose particle size in its examples.
- it is essential to provide a process of concentrating the colloidal silica so that the residual organic solvent concentration is 1 mass% or less (organic solvent distillation process) before the modification process of modifying the colloidal silica.Therefore, in such a manufacturing process, when trying to obtain colloidal silica with a small particle size close to that of single nano size, there is a risk of aggregation before the modification process is performed.
- Patent Document 3 relates to a chemical mechanical polishing composition capable of selectively polishing a silicon nitride film by sufficiently increasing the polishing rate of a silicon nitride film relative to the polishing rate of a silicon oxide film.
- a colloidal silica particle dispersion having a predetermined solid content concentration, primary particle size, and secondary particle size obtained by the method described in Example 8 or Example 1 of Patent Document 4 is mixed with 3-mercaptopropyltrimethoxysilane and heated under reflux to obtain a thiolated silica sol, and then hydrogen peroxide is added to the silica sol and heated under reflux to obtain a dispersion containing abrasive grains D or E having the predetermined solid content concentration, primary particle size, and secondary particle size.
- the primary particle size is 10 nm or less at the concentration (4% by mass) when obtained, the secondary particle size is not significantly different from the primary particle size, and the association ratio is about 1.2, so it is understood that there is little aggregation at that concentration.
- Reference Experimental Examples 1 and 2 described below it was found that these dispersions would likely aggregate and gel if the solids concentration was significantly greater than 10% by mass, and would not be stable over time.
- Patent Document 5 provides a chemical mechanical polishing composition capable of polishing a molybdenum film and a silicon oxide film at a stable polishing rate and suppressing the corrosion of the molybdenum film and the occurrence of defects in the silicon oxide film.
- a (3-triethoxysilyl)mercapto group-containing silane coupling agent is dropped into a colloidal silica dispersion and stirred, and then hydrogen peroxide is added and refluxed under normal pressure to obtain an aqueous dispersion J containing silica particles surface-modified with sulfo groups and having an average secondary particle diameter of 7.4 nm (see paragraph [0106]).
- the primary particle diameter of the silica particles was 7.0 nm.
- Patent Document 6 provides a method for converting the silica particles into nanoparticles having a diameter of 10 nm or less by mixing ordinary silica particles such as powder or granules, water, and a metal alkoxide compound having a chain group having an amino group to prepare a reaction mixture, and heating the reaction mixture at 75° C. or higher for a certain period of time.
- ordinary silica particles such as powder or granules, water, and a metal alkoxide compound having a chain group having an amino group
- Patent Document 6 provides a method for converting the silica particles into nanoparticles having a diameter of 10 nm or less by mixing ordinary silica particles such as powder or granules, water, and a metal alkoxide compound having a chain group having an amino group to prepare a reaction mixture, and heating the reaction mixture at 75° C. or higher for a certain period of time.
- the aqueous solution obtained after the reaction is a sol solution containing colloidal silica, and the particles observed with a tunneling electron microscope (TEM) are all nanoparticles of 10 nm or less, and are observed in a dispersed state without aggregation (see paragraphs [0057], [0061], etc.).
- TEM tunneling electron microscope
- the aqueous dispersion obtained in Patent Document 5 and the sol solution containing silica obtained in Patent Document 6 are likely to aggregate when the solid content concentration is increased due to their production methods, and are also highly likely to lack stability over time.
- an organic solvent-dispersed silica sol in which colloidal silica is dispersed in a dispersion medium containing an organic solvent is disclosed, in which a silane compound having at least one ether structure is bonded to the surface of colloidal silica particles.
- a silane compound having at least one ether structure is bonded to the surface of colloidal silica particles.
- the particle size measured by dynamic light scattering remains unchanged and the particle is relatively stable.
- the BET diameter is not necessarily small.
- the organic solvent-dispersed silica sol described in Patent Document 7 uses a large amount of benzyl alcohol in the dispersion solvent, exceeding 80% by mass, and has a different purpose from the colloidal silica of the present application.
- the gist of the present invention is as follows.
- the colloidal silica according to (3), wherein the anion-modification is performed with a sulfo group.
- the colloidal silica according to (1) having a solid content of 12% by mass or more.
- the colloidal silica according to (1) which has a solid content concentration of 12% by mass or more, and which exhibits, with respect to a cumulant average diameter measured by a dynamic light scattering method, a rate of change in the cumulant average diameter after being kept at a temperature of 60° C. for one week, compared to that before the keeping, of within 20%.
- a method for producing the colloidal silica according to any one of (1) to (7) comprising the steps of: a raw material preparation step of supplying and reacting a readily decomposable organosilicate to a reaction solution containing a hydrolysis catalyst made of an organic amine to prepare raw material colloidal silica having a BET diameter of 12 nm or less; a concentration adjusting step of adjusting the solid content concentration of the raw material colloidal silica to 13% by mass or less and adjusting the concentration of alcohols generated in the raw material preparation step to 1 to 25% by mass; a modification treatment step of modifying the raw colloidal silica having the adjusted concentration; and a concentrating step of concentrating the modified colloidal silica so that a residual organic solvent in the modified colloidal silica is 1 mass % or less.
- the method for producing colloidal silica according to (8) characterized in that the modification treatment step comprises a step of reacting a modifier having a functional group that can be converted into an anionic group with the colloidal silica after the concentration adjustment step, and a step of converting the functional group in the modifier after the reaction into an anionic group.
- the modification treatment step comprises a step of reacting a modifier having a functional group that can be converted into an anionic group with the colloidal silica after the concentration adjustment step, and a step of converting the functional group in the modifier after the reaction into an anionic group.
- the modifying agent has a mercapto group and/or a sulfide group, and the mercapto group and/or the sulfide group are converted to a sulfo group by treating with an oxidizing agent to obtain colloidal silica having sulfo groups on the surface.
- the modification treatment step comprises a step of reacting a modifying agent having a cationic group with the colloidal
- colloidal silica that is substantially free of aggregation, even when the particle size is single nano size or close to that size and the solid content concentration is relatively high.
- FIG. 1 shows an SEM image (magnification: 500,000) of the colloidal silica obtained in Example 1.
- FIG. 2 shows an SEM image (magnification: 500,000) of the colloidal silica obtained in Example 2.
- Figure 3 is a photograph showing the state of aggregated gel in Comparative Example 2.
- Fig. 3A is a photograph showing the state in which the gel was placed in a spherical glass container
- Fig. 3B is a photograph showing the state in which a part of the gel was taken out and placed on a petri dish
- Fig. 3C is a photograph showing the state in which the gel was crushed.
- the colloidal silica of the present invention is surface-modified, has a BET diameter of 12 nm or less, and is substantially free of aggregation.
- the colloidal silica used as the raw material is not limited as long as it has silanol groups on the surface, but considering that it does not contain metal impurities or corrosive ions such as chlorine, colloidal silica obtained by hydrolysis and condensation using hydrolyzable silicon compounds as raw materials (for example, easily hydrolyzable organosilicates or derivatives thereof, which will be described later) is preferred.
- the raw colloidal silica can be used alone or in a mixture of two or more types.
- the colloidal silica of the present invention has a small particle size of single nano size or close to single nano size, and has a BET diameter of 12 nm or less.
- small particle colloidal silica having a BET diameter of 12 nm or less is useful in that it can be easily prepared as an abrasive for semiconductor wafers and is less likely to be scratched during polishing.
- the BET diameter is preferably 12 nm or less, more preferably 10 nm or less, in terms of polishing efficiency. There is no lower limit to the BET diameter, but it can be set appropriately taking into account the application and characteristics. For example, to ensure dispersion stability, it is preferably 1 nm or more, more preferably 5 nm or more.
- the reason why the BET diameter is used to express a small particle diameter in this invention is that, for example, as will be explained later, it is possible for the colloidal silica to be not only monodispersed spherical products but also colloidal silica produced by two- or three-dimensional coalescence of multiple particles, and the production method of the present invention produces colloidal silica with an uneven surface and a relatively high specific surface area. In light of these circumstances, it is preferable to adopt the BET diameter assuming a spherical shape while also taking into account the specific surface area in judging the performance relative to the particle shape.
- the BET diameter is a particle diameter calculated from the BET specific surface area S (unit: m2 /g) of colloidal silica measured by the BET method and the true density ⁇ (unit: g/ cm3 ). Specifically, it can be calculated from the following formula (1).
- BET diameter (nm) 6000/(S x ⁇ ) ... (1)
- ⁇ is the typical true density of SiO2 , which is 2.2 g/ cm3 .
- the colloidal silica of the present invention preferably has a BET specific surface area of 227 m 2 /g or more. This BET specific surface area is suitable in terms of polishing efficiency.
- the preferred BET specific surface area is 230 m 2 /g or more, more preferably 300 m 2 /g or more. Although there is no upper limit for the BET specific surface area, it is preferably 500 m 2 /g or less, more preferably 400 m 2 /g or less, since this may cause concerns about stability.
- the colloidal silica of the present invention is obtained by subjecting raw colloidal silica to a modification treatment.
- the modification treatment in the present invention is not limited as long as it contributes to the improvement or modification of the properties such as the improvement of dispersion stability, the suppression of aggregation, and the affinity with the object to be polished, which are the objects of the present invention, and can be appropriately selected from the modification treatments of colloidal silica known in the art.
- nonionic modification, anionic modification, cationic modification, etc. can be mentioned.
- anionic or cationic modification is preferable, and although the reason why anionic modification is more preferable is not necessarily clear, it is presumed that the surface potential of colloidal silica is changed by modification, and the action of attracting with the object to be polished is expected, and the selectivity of the object to be polished is improved.
- the dispersion stability tends to be improved not only in alkaline conditions but also in acidic conditions where it is metastable and relatively prone to aggregation, so it is more preferable to perform anionic modification.
- the modification treatment can be appropriately selected from known methods, but as a preferred embodiment of the modification treatment, an anionic modification treatment and a cationic modification treatment are typically described below.
- an anionic modification treatment reference can be made to, for example, JP-A-2010-269985 and JP-A-2013-041992.
- a cationic modification treatment reference can be made to, for example, JP-A-2005-162533 and JP-A-2020-73445.
- Specific methods for the anion modification treatment are not limited, but include, for example, a method in which a modifier having an anionic group is chemically bonded to the surface of colloidal silica.
- Another method includes a method in which a modifier having a functional group that can be converted to an anionic group by a chemical method or the like is chemically bonded to the surface of colloidal silica, and then a treatment is performed to convert the functional group to an anionic group, thereby forming an anionic group on the surface of colloidal silica.
- a method using a modifier having a functional group that can be converted to an anionic group is preferable.
- the compound (modifier) having a functional group that can be converted into an anion group is preferably, but not limited to, a silane coupling agent having a functional group that can be converted into an anion group.
- the anion group is not limited to, but may be, for example, a sulfo group, a carboxy group, or a phosphate group. It may be ionic bonded with a cation to form a salt. Even with such an anion group, the cation is released in an aqueous solution and it functions as an anion group.
- Examples of cations that ionic bond with an anion group include alkali metal ions such as sodium ions and potassium ions, and alkaline earth metal ions such as calcium ions. From the viewpoint of improving the selectivity of the object to be polished, among the anion groups, the sulfo group and the carboxy group are preferable, and the sulfo group is more preferable.
- a sulfo group will be taken as an example of a preferred embodiment of the anion group.
- Modifiers having a functional group that can be converted to a sulfo group include silane coupling agents having a sulfonate ester group that can be converted to a sulfo group by hydrolysis, and silane coupling agents having a mercapto group and/or a sulfide group that can be converted to a sulfo group by oxidation.
- silane coupling agents having a mercapto group and/or a sulfide group that can be converted to a sulfo group by oxidation.
- a method using a silane coupling agent having a mercapto group and/or a sulfide group is more preferred because it is easy to modify the colloidal silica.
- only one type of modifier may be used, or two or more types may be used in combination.
- silane coupling agents having a mercapto group examples include 3-mercaptopropyltrimethoxysilane, 2-mercaptopropyltriethoxysilane, 2-mercaptoethyltrimethoxysilane, and 2-mercaptoethyltriethoxysilane.
- silane coupling agents having a sulfide group examples include bis(3-triethoxysilylpropyl)disulfide.
- the amount of the above-mentioned modifier used is preferably 0.1 to 10 parts by mass, more preferably 0.5 to 7 parts by mass, and even more preferably 0.8 to 6 parts by mass, relative to 100 parts by mass of the solid content of the raw colloidal silica.
- the modifier is preferably 0.1 to 10% by mass, more preferably 0.5 to 7% by mass, and even more preferably 0.8 to 6% by mass in the solid content of the colloidal silica after modification. If the amount used is within this range, the particle surface of the colloidal silica can be sufficiently anionized.
- colloidal silica that has been stably anion-modified without aggregation.
- a solvent water, a hydrophilic organic solvent, etc.
- dissolving the modifier may be used during the modification treatment.
- examples include a method in which a modifying agent having a cationic group is chemically bonded to the surface of colloidal silica, and a method in which a modifying agent having a functional group that can be converted to a cationic group by a chemical method or the like is chemically bonded to the surface of colloidal silica, and then a process is carried out to convert the functional group into a cationic group, thereby forming a cationic group on the surface of colloidal silica.
- a preferred method is to chemically bond a silane coupling agent having a cationic group to the surface of colloidal silica.
- Cation groups are not limited, but examples include primary amino groups, secondary amino groups, tertiary amino groups, quaternary ammonium groups, imino groups, and iminium groups. Primary amino groups, secondary amino groups, tertiary amino groups, and quaternary ammonium groups are preferred. Primary amino groups are more preferred.
- the cationic group may form an ionic bond with an anion to form a salt. Even with such cationic groups, the anion is released in the mixed liquid and the group functions as a cationic group.
- anions that form an ionic bond with a cationic group include fluoride ions, chloride ions, bromide ions, iodide ions, hydrochloride ions, acetate ions, sulfate ions, hydrofluoric acid ions, and carbonate ions.
- silane coupling agents having a cationic group examples include N-2-(aminoethyl)-3-aminopropylmethyldimethoxysilane, N-2-(aminoethyl)-3-aminopropyltrimethoxysilane, 3-aminopropyltrimethoxysilane, 3-aminopropyltriethoxysilane, 3-triethoxysilyl-N-(1,3-dimethyl-butylidene)propylamine, and N-phenyl-3-aminopropyltrimethoxysilane.
- the amount of the modifying agent having a cationic group used is preferably 0.1 to 1.5 parts by mass, more preferably 0.5 to 1.2 parts by mass, and even more preferably 0.6 to 1 part by mass, relative to 100 parts by mass of the solid content of the raw colloidal silica.
- the amount of the modifying agent having a cationic group in the solid content of the colloidal silica after cation modification is preferably 0.1 to 1.5% by mass, more preferably 0.5 to 1.2% by mass, and even more preferably 0.6 to 1% by mass.
- the particle surface of the colloidal silica can be sufficiently cationized. Furthermore, if the amount used is such, it is possible to produce a stably cation-modified colloidal silica without aggregation.
- a solvent water, a hydrophilic organic solvent, etc.
- dissolving the modifying agent may be used during the modification treatment.
- modified colloidal silica of the present invention obtained by the above-mentioned modification treatment (hereinafter, sometimes referred to as "modified colloidal silica”) has excellent dispersion stability and does not substantially aggregate even when the solid concentration is relatively high, and is suitable for various applications.
- the solid concentration is usually about 6% by mass due to the manufacturing process, etc., but it is preferably 12% by mass or more, more preferably 15% by mass or more, even more preferably 18% by mass or more, and particularly preferably 19% by mass or more, because it can improve polishing performance and reduce transportation costs.
- the solid concentration can be appropriately set depending on the application, etc.
- the upper limit of the solid concentration is not limited, but it is preferably 50% by mass or less because there is a tendency for the handleability to decrease due to an increase in viscosity, etc., and for the dispersion stability to decrease.
- the modified colloidal silica of the present invention has a relatively small particle size as described above, and does not substantially aggregate even at a relatively high solids concentration as described above, and a practical colloidal silica of this kind has not yet been clearly identified.
- the colloidal silica of the present invention is substantially free of aggregation.
- the wording "substantially” is used for the following reason. That is, in general, methods for confirming whether colloidal silica is aggregated include a method of confirming with an electron microscope, measuring the increase in viscosity, and measuring the cumulant mean diameter by dynamic light scattering (DLS).
- methods for confirming whether colloidal silica is aggregated include a method of confirming with an electron microscope, measuring the increase in viscosity, and measuring the cumulant mean diameter by dynamic light scattering (DLS).
- DLS dynamic light scattering
- the colloidal silica of the present invention which is an extremely small particle with a BET diameter of 12 nm or less, it is difficult to completely grasp whether each and every particle is monodispersed.
- the above-mentioned “substantially not aggregated” is not present, it can be measured by the above-mentioned method, but since the particles are small, some parts may be difficult to grasp by an electron microscope. Therefore, it is preferable to use dynamic light scattering (DLS) and judge from the change in the cumulant mean diameter measured by the method. More specifically, it is preferable to confirm the change (or the lack of change) in the cumulant mean diameter by DLS measurement over time. For example, in the manufacturing method of the present invention, in the concentration step described below, the presence or absence of aggregation can be confirmed by confirming the DLS measurement results before and after concentration.
- DLS dynamic light scattering
- a more practical method for grasping aggregation is to measure the change in particle diameter by DLS measurement after holding for a certain time (period) under heated conditions.
- this method it is possible to confirm whether the colloidal silica has the characteristics of being prone to aggregation without causing an active change in the solid content concentration.
- the change in the cumulant mean diameter after holding for 7 days at at least 60°C, which is a temperature at which particles are prone to aggregation is confirmed, and if the rate of change is within 20%, it can be determined that there is no aggregation.
- the reason why the method of holding at 60°C or higher for 7 days is preferably applied is that it is known that the condition of 60°C for 7 days is approximately equivalent to a time-dependent change test in a state held at room temperature for one year, and this makes it possible to confirm the occurrence of aggregation in a time-dependent change test at least at room temperature for one year.
- the rate of change in the cumulant mean diameter by DLS measurement is within 10%, more preferably within 5%.
- the solid content concentration is preferably maintained or measured at 12% by mass or more, more specifically 18% by mass or more, even more specifically 19% by mass or more, and preferably 50% by mass or less, more preferably 40% by mass or less, and even more preferably 30% by mass or less.
- the solid content concentration at this time is not limited, and it is preferable that the DLS measurement is performed at a solid content concentration that matches the application and actual use.
- the colloidal silica of the present invention may have any shape or other properties depending on the application or purpose, so long as it has the above-mentioned properties of BET diameter, modification, and substantial absence of aggregation.
- the colloidal silica obtained by the present invention preferably has the above-mentioned BET specific surface area, a relatively high particle surface area, and a large number of irregular small protrusions, so to speak, a confetti-like shape as a whole particle.
- Such a shape has a large BET specific surface area compared to the large SEM average particle diameter measured by measuring the arithmetic mean of particle images observed by SEM, and also has a high particle density (true specific gravity) measured by the liquid phase displacement method, in other words, a high hardness, and has an excellent polishing rate, making it suitable for use as an abrasive for CMP.
- the shape of the colloidal silica particles of the present invention can be controlled by the feed composition, etc., to be monodisperse spheres (spherical products) or to have a shape in which the particles are bonded together and associated (associated products). For example, by adding a large amount of catalyst and relatively slowly introducing an organosilicate as a silica raw material to the reaction field, the organosilicate hydrolyzes quickly and uniformly and grows mildly, so that the seed particles grow gradually while maintaining their spherical shape, and a spherical product can be obtained.
- the organosilicate hydrolyzes non-uniformly and acts like an adhesive between the particles, resulting in an associated product in which the particles are associated.
- the viscosity of the colloidal silica of the present invention is preferably 1 to 100 mPa ⁇ s, more preferably 1 to 50 mPa ⁇ s, and even more preferably 1 to 20 mPa ⁇ s.
- the colloidal silica of the present invention may be a monodisperse spherical product, or it may be an aggregated product (cocoon-shaped, chain-like, branched, etc.) that has a shape that appears to be formed by multiple particles bonding together in two or three dimensions when observed under an electron microscope.
- the colloidal silica of the present invention may be adjusted to a pH that does not impair dispersion stability, depending on the above-mentioned modification treatment.
- the pH is preferably 1 to 5, and more preferably 2 to 3.
- the pH is preferably 8 to 11, and more preferably 8.5 to 10. Adjusting the pH to such a range is favorable in terms of the dispersion stability of the colloidal silica.
- the colloidal silica of the present invention preferably has a metal impurity content of 1 ppm or less, more preferably 0.01 ppm or less, and even more preferably 0.0001 ppm or less.
- a metal impurity content of 1 ppm or less, more preferably 0.01 ppm or less, and even more preferably 0.0001 ppm or less.
- such high purity colloidal silica can be achieved, for example, in the manufacturing method described below, by using silica source, hydrolysis catalyst, and water used as raw materials in the hydrolysis reaction to obtain the raw material colloidal silica, which satisfy the above metal impurity content.
- the method for producing colloidal silica of the present invention essentially includes the following steps.
- a colloidal silica having a BET diameter of 12 nm or less is prepared by supplying and reacting an easily decomposable organosilicate to a reaction solution containing a hydrolysis catalyst made of an organic amine.
- the colloidal silica prepared in step (a) is called raw colloidal silica.
- a hydrolysis method is used because it reduces metal impurities, can obtain colloidal silica with a relatively large surface area, and can produce particles with high uniformity.
- hydrolysis method a method is used in which a silica source is supplied to a reaction solution containing a hydrolysis catalyst and hydrolyzed, and a method is used in which a easily decomposable organosilicate is supplied as a silica source to a reaction solution containing a hydrolysis catalyst made of an organic amine and hydrolyzed because it is easy to control the particle diameter.
- the silica source preferably used in step (a) is an easily hydrolyzable organosilicate with a fast hydrolysis rate.
- the easily hydrolyzable organosilicate is preferably one which is hydrolyzed within 1 hour by stirring 10 g of organosilicate and 100 g of pure water with impurities of 0.1 ppb or less at 25°C.
- Specific examples of such easily hydrolyzable organosilicates include trimethyl silicate (hydrolysis reaction time until hydrolysis reaction is completed: about 3 minutes), tetramethyl silicate (hydrolysis reaction time: about 5 minutes), triethyl silicate (hydrolysis reaction time: about 5 minutes), and methyl trimethyl silicate (hydrolysis reaction time: about 7 minutes).
- Tetraethyl silicate and organosilicates with a larger carbon number than tetraethyl silicate have a slow hydrolysis rate and tend to gel easily (hydrolysis reaction time: 24 hours or more for both), so the above-mentioned easily decomposable organosilicates are preferably used.
- the organic amines used as the hydrolysis catalyst in step (a) are not limited, but may be one or a mixture of two or more selected from quaternary ammoniums, tertiary amines, secondary amines, primary amines, and their carbonates, bicarbonates, and silicates.
- quaternary ammoniums include tetramethylammonium hydroxide (TMAH), tetraethylammonium hydroxide (TEAH), trimethylethylammonium hydroxide, trimethylethanolammonium hydroxide (choline), triethylethanolammonium hydroxide, tetrapropylammonium hydroxide, butylammonium hydroxide, and other quaternary ammoniums, as well as their carbonates, bicarbonates, and silicates. Since a relatively high pH is desirable for the hydrolysis reaction, tetramethylammonium hydroxide (TMAH), choline, or tetraethylammonium hydroxide (TEAH) is preferred.
- TMAH tetramethylammonium hydroxide
- TEAH tetraethylammonium hydroxide
- the primary amines, secondary amines, and tertiary amines of the organic amines used as hydrolysis catalysts are not limited, but examples thereof include aminoalcohols, morpholines, piperazines, aliphatic amines, and aliphatic ether amines.
- aminoalcohols including ethanolamine derivatives can be used as aminoalcohols, but ethanolamine derivatives are preferred, such as monoethanolamine, diethanolamine, triethanolamine, N,N-dimethylethanolamine, N,N-diethylethanolamine, N,N-di-n-butylethanolamine, N-( ⁇ -aminoethyl)ethanolamine, N-methylethanolamine, N-methyldiethanolamine, N-ethylethanolamine, N-n-butylethanolamine, N-n-butyldiethanolamine, N-tert-butylethanolamine, and N-tert-butyldiethanolamine.
- ethanolamine derivatives are preferred, such as monoethanolamine, diethanolamine, triethanolamine, N,N-dimethylethanolamine, N,N-diethylethanolamine, N,N-di-n-butylethanolamine, N-( ⁇ -aminoethyl)ethanolamine, N-methylethanolamine, N-methyldiethanol
- morpholines of the organic amines used as the hydrolysis catalyst various morpholine derivatives can be used, with preferred examples including morpholine, N-methylmorpholine, and N-ethylmorpholine.
- piperazines of the organic amines used as the hydrolysis catalyst various piperazine derivatives can be used, with preferred examples including piperazine and hydroxyethylpiperazine.
- aliphatic amines and aliphatic etheramines of the organic amines used as the hydrolysis catalyst preferred examples include alkylamines having 1 to 8 carbon atoms, such as triethylamine, dipropylamine, pentylamine, hexylamine, heptylamine, and octylamine.
- Preferred examples of the aliphatic etheramines include aliphatic etheramines having 1 to 8 carbon atoms, such as 2-methoxyethylamine, 3-methoxypropylamine, 3-ethoxypropylamine, 3-propoxypropylamine, 3-isopropoxypropylamine, and 3-butoxypropylamine.
- the above organic amines used as hydrolysis catalysts can be used alone or, if necessary, in a mixture of two or more kinds.
- the reaction liquid must contain an easily decomposable organosilicate as a silica source and a hydrolysis catalyst, but other than that, water, alcohols, aldehydes, ketones, surfactants, etc. can also be used. It is preferable that the total content of the silica source, hydrolysis catalyst, and water is 90% by mass or more. More preferably, these are 95% by mass or more.
- reaction mixture In the mixture after the reaction of the silica source with the hydrolysis catalyst (hereinafter, this may be referred to as the "reaction mixture"), or in the reaction mixture after the solids concentration or alcohol concentration has been adjusted as described below, or after the treatment of dispersion stabilization with acid (hereinafter, this may be particularly referred to as the "reaction concentrate”), it is preferable to add the hydrolysis catalyst to the reaction system and carry out the hydrolysis reaction so that the ratio of the hydrolysis catalyst (A) to the silica (B) ⁇ catalyst remaining molar ratio (A/B) ⁇ is 0.012 or less, more preferably in the range of 0.00035 to 0.012, and even more preferably in the range of 0.0035 to 0.011. This is preferable because it is possible to optimize the pH of the reaction mixture or reaction concentrate, and to suppress thickening and gelation.
- the method for achieving such a catalyst remaining molar ratio is not particularly limited, but examples include a method of continuously or intermittently introducing a silica source calculated so that the final catalyst remaining molar ratio (A/B) falls within the above range into a reaction vessel charged with water and hydrolysis catalyst (A), a method of continuously or intermittently introducing a hydrolysis catalyst and a silica source calculated so that the final catalyst remaining molar ratio falls within the above range into a reaction vessel charged with only water, and a method of continuously or intermittently introducing a hydrolysis catalyst and a silica source calculated so that the final catalyst remaining molar ratio falls within the above range into a reaction vessel charged with water and a small amount of hydrolysis catalyst (A).
- colloidal silica seeds having particle growth properties may be charged into the reaction system for the hydrolysis reaction, and the silica source and hydrolysis catalyst may be gradually added to the reaction system so that the catalyst remaining molar ratio (A/B) falls within the above range. This is preferable because it allows the production of colloidal silica with uniform particles.
- the silica source, hydrolysis catalyst, and water used as raw materials for the hydrolysis reaction preferably have a metal impurity content of 1 ppm or less, and more preferably have a high purity of 0.01 ppm or less. This ensures that the raw colloidal silica obtained and the modified colloidal silica obtained after modification also satisfy the above-mentioned range of metal impurity content.
- step (a) the method for making the BET diameter of the raw colloidal silica 12 nm or less is not limited, but it is preferable to adjust the rate at which the silica source is dropped into the reaction solution (feed rate), the reaction temperature, and the reaction time.
- the supply rate is preferably less than 1.5 mass%/min of the total amount of readily decomposable organosilicate added, more preferably 1.3 mass% or less/min, and even more preferably 1.2 mass% or less/min. If the supply rate is 1.5 mass% or more/min, the particles tend not to be uniformly dispersed or to not be nearly spherical.
- the reason why the total amount is used as the standard is that the amount added varies depending on the production scale, etc., and it is preferable to consider completing the supply within the reaction time described below. In other words, it is preferable to make the supply rate as slow as possible, which makes it easier to form the desired small particle size. There is no lower limit to the supply rate, but if the supply rate is too slow, there is a risk that the target particle size will not be achieved, so it is preferable to make it 1.0 mass%/min or more of the total amount added.
- the reaction temperature is preferably 70°C or less, more preferably 65°C or less, and even more preferably 60°C or less. If the reaction temperature exceeds 70°C, the reaction liquid will volatilize more and the liquid composition will be more likely to change, which may make it difficult to control the particle size. On the other hand, the lower limit of the reaction temperature can be set as appropriate, but if the temperature is too low, the hydrolysis reaction tends to be slow and particle growth may be promoted, so it is preferably set to 20°C or more.
- the reaction time is preferably 6 hours or less, more preferably 3 hours or less, and even more preferably 2 hours or less. If the reaction time exceeds 6 hours, there is a risk that the target particle size will not be achieved. On the other hand, the lower limit of the reaction time can be set as appropriate, but if the reaction time is too short, the hydrolysis reaction may not be completed, and particle formation and particle growth may not occur sufficiently, so it is preferably 20 minutes or more.
- step (a) it is generally preferable that the solids concentration during the reaction is 3 to 13% by mass. If the solids concentration is less than 3% by mass, particle formation and growth may not occur sufficiently. On the other hand, if the solids concentration is too high, particles are likely to aggregate. In step (a), it is preferable that the solids concentration of the colloidal silica after the reaction is in the range of 3 to 10% by mass.
- step (b) the solid content concentration of the raw colloidal silica obtained in step (a) is adjusted to 13% by mass or less.
- the solid content concentration is 10% by mass or less, more preferably 8% by mass or less.
- aggregation gelation generally refers to a state in which the aggregation of colloidal silica has progressed further and has become a jelly-like mass when viewed with the naked eye (for example, see FIG. 3 in Comparative Example 2 described later).
- step (a) if the solids concentration of the raw colloidal silica obtained in step (a) is already 13% by mass or less, there is no need to actively adjust the solids concentration in step (b), and in that case, it may be treated as a step of confirming the solids concentration, or a step of maintaining the solids concentration as it is. Ensuring that the solids concentration is indeed 13% by mass or less in step (b) before the modification treatment in the next step (c) is a characteristic step in obtaining the final modified colloidal silica of the present invention that is substantially non-aggregated and has a relatively high solids concentration.
- the method for adjusting the solid content concentration in step (b) is not limited, but it is preferable to add the same solvent as that contained in the raw colloidal silica.
- the solvent to be added is preferably water and/or alcohols, more preferably water, methanol and/or ethanol, and even more preferably water and/or methanol.
- the solvent to be added may include water, alcohols, aldehydes, ketones, surfactants, etc.
- the concentration of the alcohols produced from the step (a) is adjusted to 1 to 25% by mass.
- the hydrolysis reaction produces alcohols according to the easily decomposable organosilicate used as the silica source.
- the easily decomposable organosilicate used has a methoxy group, such as trimethyl silicate or tetramethyl silicate, methanol is produced as the alcohol.
- the easily decomposable organosilicate used has an ethoxy group, such as triethyl silicate, ethanol is produced as the alcohol. The same applies to other cases.
- step (a) the concentration of the organic solvent in this range, the occurrence of aggregation due to the polarity of the solvent can be reduced even if the BET diameter is 12 nm or less, and the next step (c) (modification treatment step) can be performed without causing aggregation and gelation.
- the concentration of the generated alcohols can be appropriately adjusted by the amount of easily decomposable organosilicate used in step (a) or by replacing the alcohols with pure water by distillation, but the preferred lower limit is 5% by mass or more, and the more preferred lower limit is 12% by mass or more. On the other hand, the preferred upper limit is 20% by mass or less.
- step (b) As in the case of adjusting the solid content concentration (13% by mass or less) in this step (b), if the alcohol concentration of the raw colloidal silica obtained in step (a) is already 1-25% by mass, there is no need to actively adjust the alcohol concentration. In that case, it may be treated as a step of confirming the alcohol concentration, or as a step of maintaining the alcohol concentration produced in step (a) as it is.
- ensuring that the alcohol concentration is 1-25% by mass in this step (b) before the modification treatment in the next step (c) is a characteristic step in obtaining a substantially non-aggregated colloidal silica of the present invention with a relatively high solid content concentration as the final modified colloidal silica of the present invention.
- the alcohol may be part of the solvent in the modification treatment in the next step (c).
- an organic solvent containing the same organic solvent as the produced alcohols as the main component e.g., 95% by mass or more
- the organic solvent added here may include water, alcohols, aldehydes, ketones, surfactants, etc.
- an example of a method for adjusting to decrease the concentration of the alcohols is to volatilize the alcohols by heating them while distilling them from a distillation tube with a condenser, as is done in Comparative Example 1 described later. In this case, pure water may be added.
- step (c) the raw colloidal silica, the solid content concentration of which and the concentration of the generated alcohols have been adjusted through step (b), is modified.
- the modification is not limited and can be selected from known modification treatments, but anion modification or cation modification is preferred, and anion modification is more preferred.
- the anion modification or the cation modification is as exemplified above, and the anion modification is preferred, and a suitable method for the anion modification is a method in which a modifying agent having a functional group that can be converted to an anion group by a chemical method or the like is chemically bonded to the surface of the colloidal silica.
- the anion modifications the sulfo group exemplified as a preferred embodiment will be taken as an example.
- the primary amino group exemplified as a preferred embodiment will be taken as an example.
- a modifying agent having a functional group that can be converted to a sulfo group by chemical method or the like is chemically bonded to the surface of colloidal silica, and then the functional group is converted to a sulfo group, and a modifying agent having a functional group that can be converted to a sulfo group by oxidation is preferable.
- a preferred modifying agent is a silane coupling agent having a mercapto group and/or a sulfide group as described above, and a representative one will be described below.
- step c1 of reacting raw colloidal silica, the solid content of which has been adjusted through steps (a) and (b) in the presence of the silane coupling agent, and a step (step c2) of oxidizing the reaction product of step c1 to convert mercapto groups and/or sulfide groups to sulfo groups.
- steps c1 and c2 can also be performed with reference to the above-mentioned JP-A-2010-269985 and JP-A-2013-041992.
- steps c1 and c2 can also be performed with reference to the above-mentioned JP-A-2010-269985 and JP-A-2013-041992.
- steps may be included as appropriate as long as they do not impair the object of the present invention. Examples of other steps include adjusting the viscosity of the reaction solution and adjusting the pH.
- Step c1 the raw colloidal silica, the solid content of which has been adjusted through steps (a) and (b), is reacted in the presence of a silane coupling agent having a mercapto group and/or a sulfide group, so that the silane coupling agent is chemically bonded to the surface of the raw colloidal silica.
- the reaction in step c1 can be carried out within the range of temperatures suitable for use of the denaturant, and is not limited to a particular range, for example, 40°C or higher and below the boiling point of the reaction liquid (solvent). To improve reactivity, the reaction is preferably carried out at a temperature of 50°C or higher, more preferably 60°C or higher, and preferably below the boiling point of the reaction liquid (solvent), 100°C or lower.
- the reaction time is also not limited, but is preferably carried out for 10 minutes to 10 hours, and more preferably 1 to 8 hours.
- a solvent for improving the solubility of the modifier or silane coupling agent can be added within a range that does not impair the object of the present invention.
- a solvent can be a hydrophilic solvent, for example, alcohols such as methanol, ethanol, isopropanol, etc., but is not limited to these. It is more preferable to use the same alcohols as those produced by the hydrolysis reaction to obtain the raw colloidal silica.
- the amount of the modifier (silane coupling agent) used is preferably 0.1 to 10 parts by mass, more preferably 0.5 to 7 parts by mass, and even more preferably 0.8 to 5 parts by mass, per 100 parts by mass of the solid content of the raw colloidal silica.
- the reaction in step c1 can also be understood, for example, by analyzing the functional groups introduced into the colloidal silica.
- pulsed NMR TD-NMR
- TD-NMR pulsed NMR
- Step c2 the functional groups introduced onto the surface of the raw colloidal silica in step c1 are converted into anionic groups by a chemical method.
- a method for converting the functional groups introduced by a silane coupling agent having a mercapto group and/or a sulfide group into sulfo groups by oxidation treatment will be described.
- Methods for converting mercapto groups and/or sulfide groups to sulfo groups by oxidation treatment include, but are not limited to, the use of an oxidizing agent.
- oxidizing agents include nitric acid, hydrogen peroxide, oxygen, ozone, organic peracids (percarboxylic acids), bromine, hypochlorite, potassium permanganate, chromic acid, etc.
- hydrogen peroxide and organic peracids peracetic acid, perbenzoic acids
- the amount of oxidizing agent added may be in excess of the amount of the modifying agent (silane coupling agent), but it is preferable to minimize the amount of residual oxidizing agent, and it is more preferred to use 3 to 5 moles of oxidizing agent per mole of silane coupling agent in order to ensure a sufficient oxidation reaction.
- the oxidation reaction it can be carried out under suitable reaction conditions for the oxidizing agent used, as long as it does not impair the purpose of the present invention.
- step c2 colloidal silica having anionic groups (sulfo groups) on the surface can be obtained.
- step c2 may include other steps after the oxidation treatment. For example, it may include a step for removing the oxidizing agent, a step for adjusting the pH of the solution after the reaction, etc., and these can be appropriately selected and performed within the scope that does not impair the object of the present invention.
- a preferred method is to carry out a treatment in which a modifying agent having an amino group is chemically bonded to the surface of colloidal silica.
- a preferred modifying agent among these is a silane coupling agent having an amino group, which will be described below as a representative example.
- the raw colloidal silica the solid content concentration of which has been adjusted through steps (a) and (b) as described above, in the presence of the silane coupling agent.
- steps can also be performed with reference to the above-mentioned JP-A-2005-162533 and JP-A-2020-73445.
- Other steps may be included as appropriate as long as they do not impair the object of the present invention.
- Examples of other steps include adjusting the viscosity of the reaction solution and adjusting the pH, but for dispersion stability, it is preferable to adjust the pH before performing the cation modification treatment.
- the pH adjusted at this time is preferably 8 to 11, and more preferably 8.5 to 10.
- the pH can be adjusted by a method using a known compound (pH adjuster), but a preferred method is to use the same compound as the hydrolysis catalyst used in step (a) as the pH adjuster.
- the raw colloidal silica whose solid content concentration has been adjusted through steps (a) and (b), is reacted in the presence of a silane coupling agent having an amino group. This causes the silane coupling agent to chemically bond to the surface of the raw colloidal silica.
- This reaction can be carried out within the range of temperatures suitable for use with the denaturant, and is not limited, but can be carried out at a temperature range of, for example, 40°C or higher and below the boiling point of the reaction liquid (solvent). To improve reactivity, it is preferable to carry out the reaction at a temperature of 50°C or higher, more preferably 60°C or higher, and preferably below the boiling point of the reaction liquid (solvent), 100°C or lower. There is also no limit to the reaction time, but it is preferable to carry out the reaction for, for example, 10 minutes to 10 hours, and more preferably 1 to 8 hours.
- a solvent for improving the solubility of the modifier or silane coupling agent can be added within a range that does not impair the object of the present invention.
- a solvent can be a hydrophilic solvent, for example, alcohols such as methanol, ethanol, isopropanol, etc., but is not limited to these. It is more preferable to use the same alcohols as those produced by the hydrolysis reaction to obtain the raw colloidal silica.
- the amount of the modifier (silane coupling agent) used is preferably 0.1 to 1.5 parts by mass, more preferably 0.5 to 1.2 parts by mass, and even more preferably 0.6 to 1 part by mass, per 100 parts by mass of the solid content of the raw colloidal silica.
- This reaction can also be understood, for example, by analyzing the functional groups introduced into the colloidal silica.
- pulsed NMR TD-NMR
- TD-NMR pulsed NMR
- the pH adjusted in this case is preferably 8 to 11, and more preferably 8.5 to 10.
- the pH can be adjusted by a method using a known compound (pH adjuster), but a preferred method is to use the same compound as the hydrolysis catalyst used in step (a) as the pH adjuster.
- the residual organic solvent in the colloidal silica obtained after step (c) is concentrated to 1 mass % or less.
- the residual organic solvent is 0.1 mass % or less, more preferably 0.05 mass % or less.
- the colloidal silica obtained through the step (d) is dispersed in a dispersion medium having a residual organic solvent of 0.1 mass % or less.
- a dispersion medium is, for example, an aqueous solution having a residual organic solvent of 0.1 mass % or less, and is preferably substantially water, as in the embodiment described in the examples below.
- the dispersion medium contains trace amounts of alcohols, aldehydes, ketones, surfactants, etc., and these are also preferably 0.1 mass % or less.
- the solid content concentration can be adjusted or increased to a predetermined range, and in the case of a highly volatile residual organic solvent, it is preferable because the concentration of colloidal silica can be prevented from fluctuating due to the evaporation of the organic solvent.
- the colloidal silica of the present invention there is an advantage that it is not necessary to consider the resistance of the material used in the application to residual organic solvents.
- the method for removing and concentrating the residual organic solvent is not particularly limited, and any known method can be used.
- any known method can be used.
- the solid content concentration in this step (d) can be adjusted to a level suitable for use, taking into account the intended use and purpose, by concentrating the process of removing the residual organic solvent and water.
- modified colloidal silica is obtained through the above-mentioned steps (a) to (c), and even if the solid content concentration is relatively high in this step (d), colloidal silica that does not substantially aggregate can be obtained.
- the solid content concentration in the colloidal silica after the modification step (c) is preferably 12% by mass or more, more preferably 15% by mass or more, even more preferably 18% by mass or more, and particularly preferably 19% by mass or more.
- the upper limit of the solid content concentration is the same as described above, and is preferably 50% by mass or less, more preferably 40% by mass or less, and even more preferably 30% by mass or less.
- the colloidal silica of the present invention can be produced by carrying out the above steps (a) to (d), but other steps may be included as appropriate even after step (d) as long as they do not impair the object of the present invention.
- Publicly known treatments can be used for the dispersion stabilization treatment.
- Colloidal silica that has been subjected to dispersion stabilization treatment using the method of the present invention exhibits excellent dispersion stability for a period of at least one week, and even for a period of several years, and does not undergo two-layer separation.
- colloidal silica with a small particle size such as a BET diameter of 12 nm or less
- a method is also possible in which the obtained colloidal silica is used as seed particles, and a silica source is supplied to and reacted with a reaction liquid containing the seed particles and the hydrolysis catalyst to grow the particle size.
- Example 1 In a 5 liter (L) glass vessel equipped with a stirrer, a thermometer, a distillation tube with a condenser, and an organosilicate inlet tube, 3200 g of pure water having a metal impurity content of 0.1 ppb or less and 3.42 g of triethanolamine (boiling point (bp): 361°C) having a metal impurity content of 10 ppb or less were charged, and while the liquid temperature in the reaction vessel was kept at 60°C using a mantle heater, 706 g of tetramethylsilicate (manufactured by Tama Chemicals Co., Ltd.) having a metal impurity content of 10 ppb or less was continuously fed under stirring over a period of 90 minutes. The obtained raw colloidal silica was subjected to various analyses, and the results are shown in ⁇ 1> of Table 1.
- the temperature in the reaction vessel was temporarily lowered to 40°C, the pressure in the system was reduced with a vacuum pump, and then heating was resumed, the reaction mixture in the reaction vessel was further heated to 52 to 68°C, and the generated methanol was distilled from a distillation tube equipped with a condenser at a distillation temperature of 32 to 67°C, and 250 g of pure water was further added while distilling off the water and methanol, thereby obtaining colloidal silica with a solid concentration of about 20% by mass.
- the residual organic solvent in the obtained colloidal silica was 0.1% by mass.
- the obtained colloidal silica was subjected to various analyses, and the results are shown in ⁇ 2> of Table 1.
- the obtained colloidal silica was stored at 60° C. for 7 days, and various analyses of the obtained colloidal silica were performed. It was confirmed, particularly from the results of DLS measurement, that substantially no aggregation had occurred. The results are shown in ⁇ 3> of Table 1.
- Example 2 In a 5 L glass vessel equipped with a stirrer, a thermometer, a distillation tube with a condenser, and an organosilicate inlet tube, 3374 g of pure water containing 0.1 ppb or less of metal impurities and 3.608 g of triethanolamine (bp: 361° C.) containing 10 ppb or less of metal impurities were charged, and while the liquid temperature in the reaction vessel was kept at 70° C. using a mantle heater, 1122 g of tetramethylsilicate containing 10 ppb or less of metal impurities (manufactured by Tama Chemicals Co., Ltd.) was continuously fed with stirring over a period of 270 minutes. Various analyses of the obtained raw colloidal silica were carried out, and the results are shown in ⁇ 4> of Table 1.
- the resulting colloidal silica was stored at 60°C for 7 days, and various analyses were performed on the resulting colloidal silica, confirming that essentially no aggregation had occurred, particularly from the results of DLS measurements.
- the results are shown in ⁇ 6> of Table 1.
- Example 3 In a 5 L glass vessel equipped with a stirrer, a thermometer, a distillation tube with a condenser, and an organosilicate inlet tube, 3463 g of pure water containing 0.1 ppb or less of metal impurities and 0.336 g of triethanolamine (bp: 361°C) containing 10 ppb or less of metal impurities were charged, and while the liquid temperature in the reaction vessel was kept at 70°C using a mantle heater, 911.6 g of tetramethylsilicate (manufactured by Tama Chemicals Co., Ltd.) containing 10 ppb or less of metal impurities was continuously fed under stirring over a period of 90 minutes.
- Various analyses of the obtained raw colloidal silica were carried out, and the results are shown in ⁇ 7> of Table 1.
- the resulting colloidal silica was stored at 60°C for 7 days, and various analyses were performed on the resulting colloidal silica, confirming that no aggregation had occurred, particularly from the results of DLS measurements.
- the results are shown in ⁇ 9> of Table 1.
- the temperature in the reaction vessel was temporarily lowered to 40°C, the pressure in the system was reduced using a vacuum pump, and then heating was resumed.
- the reaction mixture in the reaction vessel was further heated to 52-68°C, and colloidal silica with a solid content of approximately 20% by mass was obtained.
- Various analyses of the obtained colloidal silica were carried out, and the results are shown in ⁇ 11> of Table 2. From the analysis results, particularly the DLS measurement results, the change in the cumulant mean diameter by DLS measurement exceeded 20%, confirming the presence of aggregation.
- the obtained colloidal silica was further stored at 60°C for 7 days, and various analyses of the obtained colloidal silica were carried out. The results are shown in ⁇ 12> of Table 2. Because the colloidal silica was aggregated at the time of result ⁇ 11>, the DLS result in this result ⁇ 12> was also similar to result ⁇ 11> and higher than result ⁇ 10>.
- Example 4 In a 5 L glass vessel equipped with a stirrer, a thermometer, a distillation tube with a condenser, and an organosilicate inlet tube, 3889 g of pure water containing 0.1 ppb or less of metal impurities and 3.42 g of triethanolamine (bp: 361°C) containing 10 ppb or less of metal impurities were charged, and while the temperature of the liquid in the reaction vessel was kept at 60°C using a mantle heater, 707 g of tetramethylsilicate containing 10 ppb or less of metal impurities (manufactured by Tama Chemicals Co., Ltd.) was continuously fed with stirring over a period of 90 minutes. The obtained raw colloidal silica was subjected to various analyses, and the results are shown in ⁇ 15> of Table 3.
- the temperature in the reaction vessel was temporarily lowered to 40° C., 20 g of triethanolamine (bp: 361° C.) having a metal impurity content of 10 ppb or less was added, the pressure in the system was reduced using a vacuum pump, and then heating was resumed.
- the reaction mixture in the reaction vessel was further heated to 52 to 68° C., and the produced methanol was distilled from the distillation tube equipped with a condenser at a distillation temperature of 32 to 67° C., and 670 g of pure water was further added while distilling off the water and methanol, thereby obtaining colloidal silica with a solid content concentration of about 20 mass%.
- the obtained colloidal silica was subjected to various analyses, and the results are shown in ⁇ 16> of Table 3. Furthermore, the obtained colloidal silica was stored at 60°C for 7 days, and various analyses of the obtained colloidal silica were performed. It was confirmed, particularly from the results of DLS measurement, that substantially no aggregation had occurred. The results are shown in ⁇ 17> of Table 3.
- Example 5 In a 5 L glass vessel equipped with a stirrer, a thermometer, a distillation tube with a condenser, and an organosilicate inlet tube, 3890 g of pure water containing 0.1 ppb or less of metal impurities and 3.42 g of triethanolamine (bp: 361°C) containing 10 ppb or less of metal impurities were charged, and while the temperature of the liquid in the reaction vessel was kept at 60°C using a mantle heater, 707 g of tetramethylsilicate containing 10 ppb or less of metal impurities (manufactured by Tama Chemicals Co., Ltd.) was continuously fed with stirring over a period of 90 minutes. The obtained raw colloidal silica was subjected to various analyses, and the results are shown in ⁇ 18> of Table 3.
- the temperature in the reaction vessel was lowered to 40° C.
- 10 g of triethanolamine (bp: 361° C.) having a metal impurity content of 10 ppb or less was added to 1,975 g of the reaction mixture
- the pressure in the system was reduced with a vacuum pump
- heating was then resumed
- the reaction mixture in the reaction vessel was further heated to 52 to 68° C.
- the produced methanol was distilled from a distillation tube equipped with a condenser at a distillation temperature of 32 to 67° C., and 340 g of pure water was added while distilling off the water and methanol, thereby obtaining colloidal silica with a solid content concentration of about 20 mass%.
- the obtained colloidal silica was subjected to various analyses, and the results are shown in ⁇ 19> of Table 3. Furthermore, the obtained colloidal silica was stored at 60°C for 7 days, and various analyses of the obtained colloidal silica were performed. It was confirmed, particularly from the results of DLS measurement, that substantially no aggregation had occurred. The results are shown in ⁇ 20> of Table 3.
- Example 6 In a 5 L glass vessel equipped with a stirrer, a thermometer, a distillation tube with a condenser, and an organosilicate inlet tube, 3890 g of pure water containing 0.1 ppb or less of metal impurities and 3.43 g of triethanolamine (bp: 361°C) containing 10 ppb or less of metal impurities were charged, and while the liquid temperature in the reaction vessel was kept at 60°C using a mantle heater, 707 g of tetramethylsilicate containing 10 ppb or less of metal impurities (manufactured by Tama Chemicals Co., Ltd.) was continuously fed with stirring over a period of 90 minutes. The obtained raw colloidal silica was subjected to various analyses, and the results are shown in ⁇ 21> of Table 3.
- the temperature in the reaction vessel was temporarily lowered to 40° C., 12 g of triethanolamine (bp: 361° C.) having a metal impurity content of 10 ppb or less was added, the pressure in the system was reduced using a vacuum pump, and then heating was resumed.
- the reaction mixture in the reaction vessel was further heated to 52 to 68° C., and the produced methanol was distilled from the distillation tube equipped with a condenser at a distillation temperature of 32 to 67° C., and 351 g of pure water was added while distilling off the water and methanol, thereby obtaining colloidal silica with a solid content concentration of about 20 mass%.
- the obtained colloidal silica was subjected to various analyses, and the results are shown in ⁇ 22> of Table 3. Furthermore, the obtained colloidal silica was stored at 60°C for 7 days, and various analyses of the obtained colloidal silica were performed. It was confirmed, particularly from the results of DLS measurement, that substantially no aggregation had occurred. The results are shown in ⁇ 23> of Table 3.
- FIG. 4 is a photograph of a portion of the colloidal silica particle dispersion concentrated to approximately 12% by mass taken and left to cool at room temperature in an inverted container, and shows that the colloidal silica particle dispersion has aggregated and gelled at the bottom of the container (i.e., the upper part of the photograph), and does not fall out.
- FIG. 5 is a photograph of a portion of the colloidal silica particle dispersion concentrated to approximately 11% by mass taken and left to cool at room temperature in an inverted container, and it can be seen that, like Figure 4, the colloidal silica particle dispersion has aggregated and gelled at the bottom of the container (i.e., the upper part of the photograph) and does not fall out.
- the physical properties of the resulting colloidal silica were evaluated by the following methods.
- BET specific surface area, BET diameter Measured using a NOVA4200e (manufactured by Anton Paar). The particle diameter is calculated by the formula 2727/S using the BET specific surface area S ( m2 /g) measured by the nitrogen adsorption method (BET method), the true density of SiO2 (2.2 g/ cm3 ), and the above formula (1).
- BET method nitrogen adsorption method
- SiO2 2.2 g/ cm3
- Cumulant mean diameter by dynamic scattering method Measured using an SZ-100 (manufactured by Horiba, Ltd.) When measuring, the silica content in the measurement sample was adjusted to 1.13 g with pure water and ammonium nitrate, and the adjusted liquid was measured.
- silica concentration Using an SMS-70 (manufactured by A&D Co., Ltd.) as an apparatus, the residue obtained after evaporating the water content was taken as the silica concentration.
- pH Measured at 25° C. using an instrument D-51 (manufactured by Horiba, Ltd.).
- Viscosity Measured at 25° C. using a VM-10A (manufactured by Sekonic Corporation).
- Methanol concentration Measured using a GC-2025 (Shimadzu Corporation).
- the colloidal silica of the present invention is suitable for applications such as abrasives (silicon wafers, hard disks, etc.), coating agents (eyeglasses, displays, building materials, paper, etc.), and binders (ceramics, catalysts, etc.).
- abrasives silicon wafers, hard disks, etc.
- coating agents eyeglasses, displays, building materials, paper, etc.
- binders ceramics, catalysts, etc.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Dispersion Chemistry (AREA)
- Inorganic Chemistry (AREA)
- Silicon Compounds (AREA)
Abstract
Description
この発明は、コロイダルシリカ及びコロイダルシリカの製造方法に関する。 This invention relates to colloidal silica and a method for producing colloidal silica.
高純度のコロイダルシリカを工業的に製造する方法として、珪酸ソーダ水溶液をイオン交換する方法、四塩化珪素の熱分解法、オルガノシリケートを酸触媒又はアルカリ触媒の存在下に水-アルコール混合溶媒中で加水分解する方法等が提案され実施されているが、オルガノシリケートを加水分解する方法は、反応に用いるオルガノシリケート、触媒及び溶媒等として高純度のものを使用することができるために、これら原料等に由来する不純物が極めて少なく、特に金属不純物含有量の少ない高純度コロイダルシリカを製造する方法として適しており、これまでにこのオルガノシリケートの加水分解法に関する幾つかの方法が提案されている。 Methods that have been proposed and implemented as industrial methods for producing high-purity colloidal silica include ion-exchanging an aqueous solution of sodium silicate, thermal decomposition of silicon tetrachloride, and hydrolysis of organosilicate in a water-alcohol mixed solvent in the presence of an acid or alkali catalyst. However, the method of hydrolyzing organosilicate allows the use of high-purity organosilicate, catalyst, and solvent for the reaction, so that the amount of impurities derived from these raw materials is extremely small, and it is particularly suitable as a method for producing high-purity colloidal silica with low metal impurity content, and several methods have been proposed for hydrolyzing organosilicate.
ここで、種々の用途に用いられるコロイダルシリカ、特に例えば半導体ウエハの研磨剤の分野で用いられるコロイダルシリカについては、今日のLSIの高集積化に伴って様々な種類の金属の配線や酸化膜等が1枚のウエハ上に存在し、また、各々の半導体ウエハについてそれぞれに適した研磨性能が要求されることから、微妙に異なる様々な組成や性状のコロイダルシリカが要求されている。 Here, regarding colloidal silica used for various purposes, particularly colloidal silica used in the field of semiconductor wafer polishing, with the increasing integration of today's LSIs, various types of metal wiring and oxide films exist on a single wafer, and each semiconductor wafer requires its own appropriate polishing performance, so colloidal silica with various slightly different compositions and properties is required.
また、僅かなアルカリ金属不純物の含有も嫌う例えばハードコート剤用途やセラミックス用途等のバインダー、クロム酸系の金属表面処理剤、地盤改良注入剤等の用途に用いるコロイダルシリカについては、酸性のコロイダルシリカが要求され、このような酸性コロイダルシリカの製造方法についても幾つかの提案が知られている。 In addition, for colloidal silica used in applications where even the slightest alkali metal impurities are undesirable, such as binders for hard coatings and ceramics, chromate-based metal surface treatment agents, and ground improvement grouting agents, acidic colloidal silica is required, and several proposals for manufacturing such acidic colloidal silica are known.
例えば、本願の出願人は、酸処理やイオン交換処理、更には変性処理等の特別な後処理をする必要がなく、また、アルカリ金属を始めとして金属不純物含有量が極めて少なく、しかも、例えば電子顕微鏡による粒度分布分析で求められる平均粒子径が5~500nmの範囲で、標準偏差が20以下で多分散度指数が0.15以下である球状コロイダルシリカ等の所定の性状を有するコロイダルシリカを容易に製造することができる方法を検討し、その結果、加水分解速度の速い易加水分解性オルガノシリケートを用い、また、加水分解触媒として特定の加水分解触媒を用い、この加水分解触媒を、少なくとも反応終了時の反応混合物中におけるシリカ(B)に対する加水分解触媒(A)の割合{触媒残存モル比(A/B)}が所定の値以下となるように、添加して反応させることにより、酸処理やイオン交換処理等の特別な後処理を行うことなく、容易にpH5~8の中性コロイダルシリカを製造できることを提案している(特許文献1)。 For example, the applicant of the present application has investigated a method for easily producing colloidal silica having predetermined properties, such as spherical colloidal silica, which does not require special post-treatment such as acid treatment, ion exchange treatment, or further modification treatment, has an extremely low content of metal impurities, including alkali metals, and has an average particle size in the range of 5 to 500 nm, a standard deviation of 20 or less, and a polydispersity index of 0.15 or less, as determined by particle size distribution analysis using an electron microscope. As a result, the applicant has proposed that neutral colloidal silica with a pH of 5 to 8 can be easily produced without special post-treatment such as acid treatment, ion exchange treatment, or the like, by using an easily hydrolyzable organosilicate with a fast hydrolysis rate and a specific hydrolysis catalyst as the hydrolysis catalyst, and adding and reacting this hydrolysis catalyst so that at least the ratio of hydrolysis catalyst (A) to silica (B) in the reaction mixture at the end of the reaction {catalyst remaining molar ratio (A/B)} is a predetermined value or less (Patent Document 1).
ところで、例えば半導体ウエハの研磨剤の分野においても、近年は様々な要求に応える必要性があり、従来に製造されてきた数十ナノメートル(nm)のコロイダルシリカであると、研磨する際の微調整が難しく、表面が削れ過ぎたり、きれいな表面を形成できなかったりするといった問題が生じる。また、半導体の微細化が進んでいるといった事情もある。 Incidentally, even in the field of abrasives for semiconductor wafers, for example, there has been a need to meet various demands in recent years, and with the colloidal silica of several tens of nanometers (nm) that has been conventionally produced, it is difficult to make fine adjustments during polishing, leading to problems such as over-removal of the surface or failure to form a clean surface. Another factor is the ongoing miniaturization of semiconductors.
そのため、従来のコロイダルシリカよりも、さらに粒子径が小さく、数nmといった所謂シングルナノサイズやそれに近い粒子径を有するものが求められるようになってきている。ところが、そのような粒子径になると、コロイダルシリカの固形分に起因して粒子の凝集が起こる傾向が高まり、せっかくシングルナノサイズに近い粒子径であっても、その特性(例えば、研磨性、研磨対象物への欠陥数の改善など)を十分に生かしきれないといったことが懸念される。この点、本願の発明者らの検討によれば、とくにコロイダルシリカの固形分濃度が高まるにつれて、凝集が顕著になることが把握された。
すなわち、シングルナノサイズやそれに近い粒子径であって、固形分濃度が比較的高い場合であっても凝集が起こらずに安定した分散状態を有するコロイダルシリカはこれまで見出されてこなかった。
Therefore, there is a demand for colloidal silica with a particle size smaller than that of conventional colloidal silica, that is, a particle size of a few nm, or a particle size close to that.However, when the particle size is such, the tendency of particles to aggregate due to the solid content of colloidal silica increases, and even if the particle size is close to single nano size, there is a concern that the characteristics (e.g., polishability, improvement of the number of defects on the polished object, etc.) cannot be fully utilized.In this regard, according to the study by the inventors of the present application, it has been found that aggregation becomes more prominent, especially as the solid content concentration of colloidal silica increases.
That is, colloidal silica that has a particle size of single nano size or close to single nano size and has a stable dispersed state without causing aggregation even when the solid content concentration is relatively high has not been found so far.
そこで、本願の発明者らは、このようなシングルナノサイズやそれに近い領域の小粒子径のコロイダルシリカであって、固形分濃度が比較的高い場合であっても実質的に凝集が起こらないコロイダルシリカを開発するために鋭意検討した結果、原料の準備工程や濃度調整工程等を工夫するとともに、所定のタイミングでコロイダルシリカに変性処理を施すことにより達成できることを見出して、本発明を完成させた。 The inventors of the present application therefore conducted extensive research to develop colloidal silica with a small particle size of single nano size or close to that size, which does not substantially aggregate even when the solid concentration is relatively high. As a result, they discovered that this could be achieved by devising a raw material preparation process and a concentration adjustment process, etc., and by subjecting colloidal silica to a modification treatment at a predetermined timing, and thus completed the present invention.
したがって、本発明の目的は、シングルナノサイズやそれに近い小粒子径であって実質的に凝集が起こらないコロイダルシリカ及びその製造方法を提供することである。 The object of the present invention is therefore to provide colloidal silica having a small particle size of single nano size or close to that size, which is substantially free of aggregation, and a method for producing the same.
なお、コロイダルシリカに対して変性処理を施す技術についてはいくつか報告されている。
ここで、特許文献2は、微小粒子が少なく砥粒として用いられたときに研磨レートの経時的な安定性を向上できる変性コロイダルシリカに関するものであり、シングルナノサイズやそれに近い小粒子径のコロイダルシリカを具体的に教示しておらず、その実施例においても粒子径の開示は無い。また、この特許文献2に記載の製造方法においては、コロイダルシリカを変性させる変性工程よりも前に、残留有機溶媒濃度が1質量%以下になるように濃縮する工程(有機溶媒留去工程)を設けることが必須である。そのため、このような製造工程では、シングルナノサイズやそれに近い小粒子径のコロイダルシリカを得ようとすると、変性工程を行うよりも前に凝集してしまうおそれがあった。
Several techniques for modifying colloidal silica have been reported.
Here, Patent Document 2 is related to modified colloidal silica that can improve the stability of polishing rate over time when used as abrasive grains with few microparticles, but does not specifically teach colloidal silica with a single nano size or a small particle size close to that, and does not disclose particle size in its examples.In addition, in the manufacturing method described in Patent Document 2, it is essential to provide a process of concentrating the colloidal silica so that the residual organic solvent concentration is 1 mass% or less (organic solvent distillation process) before the modification process of modifying the colloidal silica.Therefore, in such a manufacturing process, when trying to obtain colloidal silica with a small particle size close to that of single nano size, there is a risk of aggregation before the modification process is performed.
また、特許文献3は、シリコン酸化膜の研磨速度に対しシリコン窒化膜の研磨速度を十分に大きくできてシリコン窒化膜を選択的に研磨することができる化学機械研磨用組成物に関するものである。この特許文献3の実施例においては、特許文献4に記載の実施例8や実施例1に記載された方法で得られた所定の固形分濃度、一次粒子径及び二次粒子径を有するコロイダルシリカ粒子分散体に対して、3-メルカプトプロピルトリメトキシシランを混合し、加熱還流することによりチオール化シリカゾルを得た後に、そのシリカゾルに、過酸化水素を加えて加熱還流することにより、シリカ粒子の表面にスルホ基が固定化されてなり、前記所定の固形分濃度、一次粒子径及び二次粒子径を有する砥粒D又は砥粒Eを含有する分散体を得たことが示されている。これらの砥粒D又は砥粒Eを含有する分散体は、いずれも得られた際の濃度(4質量%)においては、一次粒子径が10nm以下であって、二次粒子径も一次粒子径と大きく相違せず、会合比も1.2程度であって、当該濃度においては凝集が少ないものであると解されるが、本願の発明者らが検証したところ、後述の参考実験例1及び2に示したように、これらの分散体は固形分濃度を大きく10質量%を超える程度とすると凝集ゲル化してしまい、経時の安定性も具備しない蓋然性が高いものであることが分かった。 Patent Document 3 relates to a chemical mechanical polishing composition capable of selectively polishing a silicon nitride film by sufficiently increasing the polishing rate of a silicon nitride film relative to the polishing rate of a silicon oxide film. In the examples of Patent Document 3, it is shown that a colloidal silica particle dispersion having a predetermined solid content concentration, primary particle size, and secondary particle size obtained by the method described in Example 8 or Example 1 of Patent Document 4 is mixed with 3-mercaptopropyltrimethoxysilane and heated under reflux to obtain a thiolated silica sol, and then hydrogen peroxide is added to the silica sol and heated under reflux to obtain a dispersion containing abrasive grains D or E having the predetermined solid content concentration, primary particle size, and secondary particle size. In the dispersions containing abrasive grains D or E, the primary particle size is 10 nm or less at the concentration (4% by mass) when obtained, the secondary particle size is not significantly different from the primary particle size, and the association ratio is about 1.2, so it is understood that there is little aggregation at that concentration. However, when the inventors of the present application verified this, as shown in Reference Experimental Examples 1 and 2 described below, it was found that these dispersions would likely aggregate and gel if the solids concentration was significantly greater than 10% by mass, and would not be stable over time.
また、特許文献5は、モリブデン膜およびシリコン酸化膜を安定した研磨速度で研磨でき、かつ、モリブデン膜の腐食およびシリコン酸化膜の欠陥の発生を抑制することができる化学機械研磨用組成物を提供するものである。この特許文献5の実施例においては、コロイダルシリカ分散液に、(3-トリエトキシシリル)メルカプト基含有シランカップリング剤を滴下して撹拌し、その後、過酸化水素を添加して常圧下で還流することで、平均二次粒子径7.4nmのスルホ基で表面修飾されたシリカ粒子を含む水分散体Jを得たことが示されている(段落[0106]参照)。また、このシリカ粒子の一次粒子径は7.0nmであった。また、特許文献6は、粉末や顆粒状といった通常のシリカ粒子と、水と、アミノ基を有する鎖状基を備えた金属アルコキシド化合物とを混合して反応混合物を調製し、この反応混合物を75℃以上で一定時間加熱することにより、上記のシリカ粒子を直径10nm以下のナノ粒子に変換する方法を提供するものである。この特許文献6の実施例においては、還流装置を装着した反応容器に、粉末状シリカゲル、3-アミノプロピルトリメトキシシラン、及び蒸留水を仕込み、加熱及び撹拌する方法が示されている。そして、反応終了後に得られた水溶液は、コロイド状となったシリカを含むゾル溶液であり、トンネル電子顕微鏡(TEM)で観察した粒子はいずれも10nm以下のナノ粒子であり、凝集のない分散状態として観察されたことが示されている(段落[0057]、[0061]などを参照)。
しかしながら、これら特許文献5で得られた水分散体や、特許文献6で得られたシリカを含むゾル溶液は、それらの製造方法に起因して、固形分濃度を大きくすると凝集するおそれがあり、また、経時の安定性も具備しない蓋然性が高いものであった。
Patent Document 5 provides a chemical mechanical polishing composition capable of polishing a molybdenum film and a silicon oxide film at a stable polishing rate and suppressing the corrosion of the molybdenum film and the occurrence of defects in the silicon oxide film. In the examples of Patent Document 5, a (3-triethoxysilyl)mercapto group-containing silane coupling agent is dropped into a colloidal silica dispersion and stirred, and then hydrogen peroxide is added and refluxed under normal pressure to obtain an aqueous dispersion J containing silica particles surface-modified with sulfo groups and having an average secondary particle diameter of 7.4 nm (see paragraph [0106]). The primary particle diameter of the silica particles was 7.0 nm. Patent Document 6 provides a method for converting the silica particles into nanoparticles having a diameter of 10 nm or less by mixing ordinary silica particles such as powder or granules, water, and a metal alkoxide compound having a chain group having an amino group to prepare a reaction mixture, and heating the reaction mixture at 75° C. or higher for a certain period of time. In the examples of this patent document 6, powdered silica gel, 3-aminopropyltrimethoxysilane, and distilled water are charged into a reaction vessel equipped with a reflux device, and the mixture is heated and stirred. The aqueous solution obtained after the reaction is a sol solution containing colloidal silica, and the particles observed with a tunneling electron microscope (TEM) are all nanoparticles of 10 nm or less, and are observed in a dispersed state without aggregation (see paragraphs [0057], [0061], etc.).
However, the aqueous dispersion obtained in Patent Document 5 and the sol solution containing silica obtained in Patent Document 6 are likely to aggregate when the solid content concentration is increased due to their production methods, and are also highly likely to lack stability over time.
なお、特許文献7のように、有機溶媒を含む分散媒中にコロイダルシリカが分散した有機溶媒分散シリカゾルとして、コロイダルシリカ粒子の表面に、少なくとも1個のエーテル構造を有するシラン化合物を結合したものが示されており、その実施例で確認されているように50℃の恒温槽内で1ヶ月保持した後でも動的光散乱法粒子径に変化がなく比較的安定なものであることが示されているが、BET径が必ずしも小さいとは言えず、また、この特許文献7に記載された有機溶媒分散シリカゾルは分散溶媒中のベンジルアルコール含有量が80質量%を超えて多量に用いられており、本願のコロイダルシリカとは目的が異なるものであった。 As shown in Patent Document 7, an organic solvent-dispersed silica sol in which colloidal silica is dispersed in a dispersion medium containing an organic solvent is disclosed, in which a silane compound having at least one ether structure is bonded to the surface of colloidal silica particles. As confirmed in the examples, even after being kept in a thermostatic chamber at 50°C for one month, the particle size measured by dynamic light scattering remains unchanged and the particle is relatively stable. However, the BET diameter is not necessarily small. Furthermore, the organic solvent-dispersed silica sol described in Patent Document 7 uses a large amount of benzyl alcohol in the dispersion solvent, exceeding 80% by mass, and has a different purpose from the colloidal silica of the present application.
すなわち、本発明の要旨は以下のとおりである。
(1)表面が変性されてなり、BET径が12nm以下であり、実質的に凝集していないことを特徴とするコロイダルシリカ。
(2)BET比表面積が227m2/g以上であることを特徴とする(1)に記載のコロイダルシリカ。
(3)前記変性がアニオン変性又はカチオン変性であることを特徴とする(1)に記載のコロイダルシリカ。
(4)前記アニオン変性がスルホ基によることを特徴とする(3)に記載のコロイダルシリカ。
(5)前記カチオン変性が1級アミノ基によることを特徴とする(3)に記載のコロイダルシリカ。
(6)固形分濃度が12質量%以上であることを特徴とする(1)に記載のコロイダルシリカ。
(7)固形分濃度が12質量%以上であり、また、動的光散乱法により測定されるキュムラント平均径について、温度60℃の条件下で1週間保持した後における当該キュムラント平均径の変化率が、前記保持前と比較して20%以内であることを特徴とする(1)に記載のコロイダルシリカ。
(8)(1)~(7)のいずれかに記載のコロイダルシリカを製造する方法であって、
有機アミンからなる加水分解触媒を含む反応液に、易分解性オルガノシリケートを供給及び反応させて、BET径が12nm以下である原料コロイダルシリカを準備する原料準備工程と、
原料コロイダルシリカの固形分濃度を13質量%以下とすると共に、前記原料準備工程に由来して生成するアルコール類の濃度を1~25質量%とする濃度調整工程と、
前記濃度調整された原料コロイダルシリカを変性処理する変性処理工程と、
前記変性処理されたコロイダルシリカ中の残留有機溶媒が1質量%以下となるように濃縮する濃縮工程と
を含むことを特徴とするコロイダルシリカの製造方法。
(9)前記原料準備工程では、前記易加水分解性オルガノシリケートの供給速度が該易加水分解性オルガノシリケートの総投入量の1.5質量%未満/分、反応時間が6時間以内、及び反応温度が70℃以下の条件下において反応を行うことを特徴とする(8)に記載のコロイダルシリカの製造方法。
(10)前記濃縮工程では、変性処理工程後のコロイダルシリカ中の固形分濃度を12質量%以上となるように濃縮することを特徴とする(8)に記載のコロイダルシリカの製造方法。
(11)前記変性処理工程では、アニオン基に変換できる官能基を有する変性剤と前記濃度調整工程後のコロイダルシリカとを反応させる工程と、当該反応後の変性剤における前記官能基をアニオン基に変換させる工程とを有することを特徴とする(8)に記載のコロイダルシリカの製造方法。
(12)前記変性剤がメルカプト基及び/又はスルフィド基を有し、酸化剤で処理することによって当該メルカプト基及び/又はスルフィド基をスルホ基に変換させて、表面にスルホ基を有するコロイダルシリカを得ることを特徴とする(11)に記載のコロイダルシリカの製造方法。
(13)前記変性処理工程では、カチオン基を有する変性剤と前記濃度調整工程後のコロイダルシリカとを反応させる工程を有することを特徴とする(8)に記載のコロイダルシリカの製造方法。
That is, the gist of the present invention is as follows.
(1) A colloidal silica having a modified surface, a BET diameter of 12 nm or less, and being substantially free of aggregation.
(2) The colloidal silica according to (1), which has a BET specific surface area of 227 m 2 /g or more.
(3) The colloidal silica according to (1), wherein the modification is anion-modified or cation-modified.
(4) The colloidal silica according to (3), wherein the anion-modification is performed with a sulfo group.
(5) The colloidal silica according to (3), wherein the cation modification is performed by a primary amino group.
(6) The colloidal silica according to (1), having a solid content of 12% by mass or more.
(7) The colloidal silica according to (1), which has a solid content concentration of 12% by mass or more, and which exhibits, with respect to a cumulant average diameter measured by a dynamic light scattering method, a rate of change in the cumulant average diameter after being kept at a temperature of 60° C. for one week, compared to that before the keeping, of within 20%.
(8) A method for producing the colloidal silica according to any one of (1) to (7), comprising the steps of:
a raw material preparation step of supplying and reacting a readily decomposable organosilicate to a reaction solution containing a hydrolysis catalyst made of an organic amine to prepare raw material colloidal silica having a BET diameter of 12 nm or less;
a concentration adjusting step of adjusting the solid content concentration of the raw material colloidal silica to 13% by mass or less and adjusting the concentration of alcohols generated in the raw material preparation step to 1 to 25% by mass;
a modification treatment step of modifying the raw colloidal silica having the adjusted concentration;
and a concentrating step of concentrating the modified colloidal silica so that a residual organic solvent in the modified colloidal silica is 1 mass % or less.
(9) The method for producing colloidal silica according to (8), wherein in the raw material preparing step, the reaction is carried out under conditions in which the feed rate of the easily hydrolyzable organosilicate is less than 1.5 mass%/min of the total amount of the easily hydrolyzable organosilicate fed, the reaction time is 6 hours or less, and the reaction temperature is 70° C. or lower.
(10) The method for producing colloidal silica according to (8), wherein in the concentrating step, the colloidal silica after the modification treatment step is concentrated so that the solid content concentration in the colloidal silica is 12 mass% or more.
(11) The method for producing colloidal silica according to (8), characterized in that the modification treatment step comprises a step of reacting a modifier having a functional group that can be converted into an anionic group with the colloidal silica after the concentration adjustment step, and a step of converting the functional group in the modifier after the reaction into an anionic group.
(12) The method for producing colloidal silica according to (11), wherein the modifying agent has a mercapto group and/or a sulfide group, and the mercapto group and/or the sulfide group are converted to a sulfo group by treating with an oxidizing agent to obtain colloidal silica having sulfo groups on the surface.
(13) The method for producing colloidal silica according to (8), wherein the modification treatment step comprises a step of reacting a modifying agent having a cationic group with the colloidal silica after the concentration adjustment step.
本発明によれば、シングルナノサイズやそれに近い小粒子径であって、比較的固形分濃度が高い場合であっても、実質的に凝集が起こらないコロイダルシリカを得ることができる。 According to the present invention, it is possible to obtain colloidal silica that is substantially free of aggregation, even when the particle size is single nano size or close to that size and the solid content concentration is relatively high.
<コロイダルシリカ>
本発明のコロイダルシリカは、表面が変性されてなり、BET径が12nm以下であり、実質的に凝集していないものである。
<Colloidal Silica>
The colloidal silica of the present invention is surface-modified, has a BET diameter of 12 nm or less, and is substantially free of aggregation.
原料となるコロイダルシリカは、表面にシラノール基を有するものであれば限定されないが、金属不純物や塩素等の腐食性イオンを含まないことを考慮すると、加水分解可能なケイ素化合物(例えば、後述の易加水分解性オルガノシリケート又はその誘導体など)を原料とし、加水分解・縮合により得られるコロイダルシリカが好ましい。原料となるコロイダルシリカは、1種又は2種以上を混合して使用できる。 The colloidal silica used as the raw material is not limited as long as it has silanol groups on the surface, but considering that it does not contain metal impurities or corrosive ions such as chlorine, colloidal silica obtained by hydrolysis and condensation using hydrolyzable silicon compounds as raw materials (for example, easily hydrolyzable organosilicates or derivatives thereof, which will be described later) is preferred. The raw colloidal silica can be used alone or in a mixture of two or more types.
先ず、BET径について説明する。
前述のとおり、本発明のコロイダルシリカは、シングルナノサイズやそれに近い小粒子径であって、BET径は12nm以下である。このようにBET径が12nm以下である小粒子のコロイダルシリカは、後述する産業上の利用可能性でも例示されるように、例えば、半導体ウエハの研磨剤として調整を行い易いことや、研磨時の傷が付きにくいなどの点で有用である。
First, the BET diameter will be described.
As described above, the colloidal silica of the present invention has a small particle size of single nano size or close to single nano size, and has a BET diameter of 12 nm or less. As exemplified in the industrial applicability described below, small particle colloidal silica having a BET diameter of 12 nm or less is useful in that it can be easily prepared as an abrasive for semiconductor wafers and is less likely to be scratched during polishing.
前記BET径については、研磨の効率性の面などで、好ましくは12nm以下であり、より好ましくは10nm以下である。BET径の下限については制限されないが、用途や特性などを勘案して適宜設定することが可能である。例えば、分散安定性の確保のために、好ましくは1nm以上であり、より好ましくは5nm以上である。 The BET diameter is preferably 12 nm or less, more preferably 10 nm or less, in terms of polishing efficiency. There is no lower limit to the BET diameter, but it can be set appropriately taking into account the application and characteristics. For example, to ensure dispersion stability, it is preferably 1 nm or more, more preferably 5 nm or more.
ここで、本発明において、BET径を用いて小粒子径であることを表現している意義としては、例えば後述でも説明するように、単分散性の球状品のみならず複数の粒子が2次元または3次元に合着し製造されたコロイダルシリカであったりしてもよいことや、本発明の製造方法によれば表面に凹凸があり、比表面積の比較的高いコロイダルシリカであったりすること等の実情を踏まえて、比表面積も考慮しつつ球形とみなしたBET径を採用することが粒形に対する性能を判断する上で好適であるからである。 The reason why the BET diameter is used to express a small particle diameter in this invention is that, for example, as will be explained later, it is possible for the colloidal silica to be not only monodispersed spherical products but also colloidal silica produced by two- or three-dimensional coalescence of multiple particles, and the production method of the present invention produces colloidal silica with an uneven surface and a relatively high specific surface area. In light of these circumstances, it is preferable to adopt the BET diameter assuming a spherical shape while also taking into account the specific surface area in judging the performance relative to the particle shape.
すなわち、本発明においてBET径とは、BET法により測定されるコロイダルシリカのBET比表面積S(単位:m2/g)と真密度ρ(単位:g/cm3)とから算出される粒子径である。具体的には、次の式(1)から求めることができる。
BET径(nm)=6000/(S×ρ) ・・・(1)
ここで、ρはSiO2の一般的な真密度であって、2.2g/cm3である。
That is, in the present invention, the BET diameter is a particle diameter calculated from the BET specific surface area S (unit: m2 /g) of colloidal silica measured by the BET method and the true density ρ (unit: g/ cm3 ). Specifically, it can be calculated from the following formula (1).
BET diameter (nm) = 6000/(S x ρ) ... (1)
Here, ρ is the typical true density of SiO2 , which is 2.2 g/ cm3 .
本発明のコロイダルシリカについて、BET比表面積は227m2/g以上であることが好ましい。このBET比表面積を有することにより、研磨の効率性の面で好適である。好ましいBET比表面積は230m2/g以上であり、より好ましくは300m2/g以上である。BET比表面積の上限値は制限されないものの、安定性に不安が出ることから、500m2/g以下であることが好ましく、より好ましくは400m2/g以下である。 The colloidal silica of the present invention preferably has a BET specific surface area of 227 m 2 /g or more. This BET specific surface area is suitable in terms of polishing efficiency. The preferred BET specific surface area is 230 m 2 /g or more, more preferably 300 m 2 /g or more. Although there is no upper limit for the BET specific surface area, it is preferably 500 m 2 /g or less, more preferably 400 m 2 /g or less, since this may cause concerns about stability.
次に、変性について説明する。
本発明のコロイダルシリカは、原料コロイダルシリカに対して変性処理が施されることにより得られる。ここで、本発明において変性処理は、本発明の目的である分散安定性の向上や凝集抑制やそのほか研磨対象物との親和性などの特性の向上や改質に寄与するものであれば制限されず、当業界で知られる従来公知のコロイダルシリカの変性処理から適宜選択することができる。例えば、ノニオン変性、アニオン変性、カチオン変性などを挙げることができる。具体的には、コロイダルシリカをアニオン変性又はカチオン変性させることが好ましく、より好ましくはアニオン変性である。このような変性処理として、アニオン変性又はカチオン変性が好ましく、アニオン変性がより好ましい理由は必ずしも明確ではないものの、変性させることによりコロイダルシリカの表面電位が変化し、研磨対象物と引き合うことなどの作用が見込まれ、研磨対象物の選択性が向上すると推測されるからである。とくに、変性処理を施していないコロイダルシリカと比べて、アルカリ性のみならず、準安定であって比較的凝集しやすい酸性側においても分散安定性が向上する傾向にあるために、アニオン変性を行うことがより好ましい。
Next, the modification will be described.
The colloidal silica of the present invention is obtained by subjecting raw colloidal silica to a modification treatment. Here, the modification treatment in the present invention is not limited as long as it contributes to the improvement or modification of the properties such as the improvement of dispersion stability, the suppression of aggregation, and the affinity with the object to be polished, which are the objects of the present invention, and can be appropriately selected from the modification treatments of colloidal silica known in the art. For example, nonionic modification, anionic modification, cationic modification, etc. can be mentioned. Specifically, it is preferable to subject colloidal silica to anionic or cationic modification, and more preferably anionic modification. As such a modification treatment, anionic or cationic modification is preferable, and although the reason why anionic modification is more preferable is not necessarily clear, it is presumed that the surface potential of colloidal silica is changed by modification, and the action of attracting with the object to be polished is expected, and the selectivity of the object to be polished is improved. In particular, compared with colloidal silica that has not been modified, the dispersion stability tends to be improved not only in alkaline conditions but also in acidic conditions where it is metastable and relatively prone to aggregation, so it is more preferable to perform anionic modification.
前述のとおり、変性処理は公知の方法から適宜選択して採用することができるが、変性処理の好ましい一実施形態として、代表的にアニオン変性処理及びカチオン変性処理について以下で説明する。アニオン変性処理については、例えば、特開2010-269985号公報や特開2013-041992号公報などを参照することができる。また、カチオン変性処理については、例えば、特開2005-162533号公報や特開2020-73445号公報などを参照することができる。 As mentioned above, the modification treatment can be appropriately selected from known methods, but as a preferred embodiment of the modification treatment, an anionic modification treatment and a cationic modification treatment are typically described below. For an anionic modification treatment, reference can be made to, for example, JP-A-2010-269985 and JP-A-2013-041992. For a cationic modification treatment, reference can be made to, for example, JP-A-2005-162533 and JP-A-2020-73445.
アニオン変性処理の具体的な方法は、制限されるものではないが、例えば、アニオン基を有する変性剤をコロイダルシリカの表面に化学的に結合させる方法が挙げられる。また、他の方法としては、化学的な方法などによりアニオン基に変換できる官能基を有する変性剤を、コロイダルシリカの表面に化学的に結合させた後に、該官能基をアニオン基に変換させる処理を行うことでコロイダルシリカの表面にアニオン基を形成する方法などを挙げることができる。変性処理の効率性やアニオン基を安定性よくコロイダルシリカに導入することができることや、アニオン基を有する変性剤が直接得られ難いといった理由などから、好ましくは、アニオン基に変換できる官能基を有する変性剤を用いた方法が好ましい。 Specific methods for the anion modification treatment are not limited, but include, for example, a method in which a modifier having an anionic group is chemically bonded to the surface of colloidal silica. Another method includes a method in which a modifier having a functional group that can be converted to an anionic group by a chemical method or the like is chemically bonded to the surface of colloidal silica, and then a treatment is performed to convert the functional group to an anionic group, thereby forming an anionic group on the surface of colloidal silica. For reasons such as the efficiency of the modification treatment, the ability to stably introduce anionic groups into colloidal silica, and the difficulty of directly obtaining a modifier having an anionic group, a method using a modifier having a functional group that can be converted to an anionic group is preferable.
アニオン基に変換できる官能基を有する化合物(変性剤)としては、限定されないものの、例えば、アニオン基に変換できる官能基を有するシランカップリング剤を好適に挙げることができる。ここで、アニオン基としては、限定されないが、例えば、スルホ基、カルボキシ基、リン酸基などを挙げることができる。陽イオンとイオン結合し、塩を形成したものでもよい。このようなアニオン基であっても、水溶液中では陽イオンが脱離し、アニオン基として機能する。アニオン基とイオン結合する陽イオンとしては、ナトリウムイオンやカリウムイオン等のアルカリ金属イオン、及びカルシウムイオン等のアルカリ土類金属イオン等を挙げることができる。研磨対象物の選択性の向上の観点から、アニオン基のうち、好ましくはスルホ基、カルボキシ基であり、より好ましくはスルホ基である。 The compound (modifier) having a functional group that can be converted into an anion group is preferably, but not limited to, a silane coupling agent having a functional group that can be converted into an anion group. Here, the anion group is not limited to, but may be, for example, a sulfo group, a carboxy group, or a phosphate group. It may be ionic bonded with a cation to form a salt. Even with such an anion group, the cation is released in an aqueous solution and it functions as an anion group. Examples of cations that ionic bond with an anion group include alkali metal ions such as sodium ions and potassium ions, and alkaline earth metal ions such as calcium ions. From the viewpoint of improving the selectivity of the object to be polished, among the anion groups, the sulfo group and the carboxy group are preferable, and the sulfo group is more preferable.
アニオン基として好ましい実施形態としてスルホ基を例にとって説明する。スルホ基に変換できる官能基を有する変性剤としては、加水分解によりスルホ基に変換できるスルホン酸エステル基を有するシランカップリング剤や、酸化によりスルホ基に変換できるメルカプト基及び/又はスルフィド基を有するシランカップリング剤を挙げることができる。このうち、コロイダルシリカへの修飾を行いやすいという理由から、メルカプト基及び/又はスルフィド基を有するシランカップリング剤を用いた方法がより好ましい。なお、本発明において、変性剤は1種のみでもよく、2種以上を併用することもできる。 A sulfo group will be taken as an example of a preferred embodiment of the anion group. Modifiers having a functional group that can be converted to a sulfo group include silane coupling agents having a sulfonate ester group that can be converted to a sulfo group by hydrolysis, and silane coupling agents having a mercapto group and/or a sulfide group that can be converted to a sulfo group by oxidation. Of these, a method using a silane coupling agent having a mercapto group and/or a sulfide group is more preferred because it is easy to modify the colloidal silica. In the present invention, only one type of modifier may be used, or two or more types may be used in combination.
メルカプト基を有するシランカップリング剤としては、例えば、3-メルカプトプロピルトリメトキシシラン、2-メルカプトプロピルトリエトキシシラン、2-メルカプトエチルトリメトキシシラン、2-メルカプトエチルトリエトキシシラン等を挙げることができる。また、スルフィド基を有するシランカップリング剤としては、例えば、ビス(3-トリエトキシシリルプロピル)ジスルフィドを挙げることができる。 Examples of silane coupling agents having a mercapto group include 3-mercaptopropyltrimethoxysilane, 2-mercaptopropyltriethoxysilane, 2-mercaptoethyltrimethoxysilane, and 2-mercaptoethyltriethoxysilane. Examples of silane coupling agents having a sulfide group include bis(3-triethoxysilylpropyl)disulfide.
アニオン変性処理のより具体的な方法については、後述の製造方法の説明において詳述するが、上記の変性剤の使用量は、原料コロイダルシリカにおける固形分100質量部に対して、好ましくは0.1~10質量部であり、より好ましくは0.5~7質量部であり、さらに好ましくは0.8~6質量部である。すなわち、変性後のコロイダルシリカの固形分中に、変性剤は0.1~10質量%であることが好ましく、0.5~7質量%がより好ましく、0.8~6質量%がさらに好ましい。この範囲の使用量であれば、コロイダルシリカの粒子表面を十分にアニオン化させることができる。また、このような使用量であれば、凝集することなく安定的にアニオン変性処理されたコロイダルシリカを製造することができる。なお、変性処理に際しては、変性剤を溶解させるための溶媒(水、親水性の有機溶媒など)が用いられてもよい。 A more specific method for the anion modification treatment will be described in detail in the explanation of the manufacturing method described later, but the amount of the above-mentioned modifier used is preferably 0.1 to 10 parts by mass, more preferably 0.5 to 7 parts by mass, and even more preferably 0.8 to 6 parts by mass, relative to 100 parts by mass of the solid content of the raw colloidal silica. In other words, the modifier is preferably 0.1 to 10% by mass, more preferably 0.5 to 7% by mass, and even more preferably 0.8 to 6% by mass in the solid content of the colloidal silica after modification. If the amount used is within this range, the particle surface of the colloidal silica can be sufficiently anionized. Furthermore, if the amount used is such, it is possible to produce colloidal silica that has been stably anion-modified without aggregation. Note that a solvent (water, a hydrophilic organic solvent, etc.) for dissolving the modifier may be used during the modification treatment.
なお、カチオン変性についても、例えば、カチオン基を有する変性剤をコロイダルシリカの表面に化学的に結合させる方法や、化学的な方法などによりカチオン基に変換できる官能基を有する変性剤を、コロイダルシリカの表面に化学的に結合させた後に、該官能基をカチオン基に変換させる処理を行うことでコロイダルシリカの表面にカチオン基を形成する方法などを挙げることができる。好ましくは、カチオン基を有するシランカップリング剤をコロイダルシリカの表面に化学的に結合させる方法が挙げられる。 As for cationic modification, examples include a method in which a modifying agent having a cationic group is chemically bonded to the surface of colloidal silica, and a method in which a modifying agent having a functional group that can be converted to a cationic group by a chemical method or the like is chemically bonded to the surface of colloidal silica, and then a process is carried out to convert the functional group into a cationic group, thereby forming a cationic group on the surface of colloidal silica. A preferred method is to chemically bond a silane coupling agent having a cationic group to the surface of colloidal silica.
カチオン基としては、限定されないが、例えば、1級アミノ基、2級アミノ基、3級アミノ基、4級アンモニウム基、イミノ基及びイミニウム基が挙げられる。好ましくは、1級アミノ基、2級アミノ基、3級アミノ基、4級アンモニウム基である。より好ましくは、1級アミノ基である。カチオン基は、陰イオンとイオン結合し、塩を形成してもよい。このようなカチオン基であっても、混合液中では陰イオンが脱離し、カチオン基として機能する。カチオン基とイオン結合する陰イオンとしては、フッ化物イオン、塩化物イオン、臭化物イオン、ヨウ化物イオン、塩酸イオン、酢酸イオン、硫酸イオン、フッ化水素酸イオン、及び炭酸イオンを挙げることができる。 Cation groups are not limited, but examples include primary amino groups, secondary amino groups, tertiary amino groups, quaternary ammonium groups, imino groups, and iminium groups. Primary amino groups, secondary amino groups, tertiary amino groups, and quaternary ammonium groups are preferred. Primary amino groups are more preferred. The cationic group may form an ionic bond with an anion to form a salt. Even with such cationic groups, the anion is released in the mixed liquid and the group functions as a cationic group. Examples of anions that form an ionic bond with a cationic group include fluoride ions, chloride ions, bromide ions, iodide ions, hydrochloride ions, acetate ions, sulfate ions, hydrofluoric acid ions, and carbonate ions.
カチオン基を有するシランカップリング剤としては、例えば、N-2-(アミノエチル)-3-アミノプロピルメチルジメトキシシラン、N-2-(アミノエチル)-3-アミノプロピルトリメトキシシラン、3-アミノプロピルトリメトキシシラン、3-アミノプロピルトリエトキシシラン、3-トリエトキシシリル-N-(1,3-ジメチル-ブチリデン)プロピルアミン、N-フェニル-3-アミノプロピルトリメトキシシランが挙げられる。 Examples of silane coupling agents having a cationic group include N-2-(aminoethyl)-3-aminopropylmethyldimethoxysilane, N-2-(aminoethyl)-3-aminopropyltrimethoxysilane, 3-aminopropyltrimethoxysilane, 3-aminopropyltriethoxysilane, 3-triethoxysilyl-N-(1,3-dimethyl-butylidene)propylamine, and N-phenyl-3-aminopropyltrimethoxysilane.
カチオン変性処理のより具体的な方法については、後述の製造方法の説明において詳述するが、前記したようにカチオン基を有する変性剤を用いる方法であることが好ましい。カチオン基を有する変性剤の使用量は、原料コロイダルシリカにおける固形分100質量部に対して、好ましくは0.1~1.5質量部であり、より好ましくは0.5~1.2質量部であり、さらに好ましくは0.6~1質量部である。すなわち、カチオン変性後のコロイダルシリカの固形分中に、カチオン基を有する変性剤は0.1~1.5質量%であることが好ましく、0.5~1.2質量%がより好ましく、0.6~1質量%がさらに好ましい。この範囲の使用量であれば、コロイダルシリカの粒子表面を十分にカチオン化させることができる。また、このような使用量であれば、凝集することなく安定的にカチオン変性処理されたコロイダルシリカを製造することができる。なお、変性処理に際しては、変性剤を溶解させるための溶媒(水、親水性の有機溶媒など)が用いられてもよい。 A more specific method for the cation modification treatment will be described in detail in the explanation of the manufacturing method below, but as described above, it is preferable to use a method using a modifying agent having a cationic group. The amount of the modifying agent having a cationic group used is preferably 0.1 to 1.5 parts by mass, more preferably 0.5 to 1.2 parts by mass, and even more preferably 0.6 to 1 part by mass, relative to 100 parts by mass of the solid content of the raw colloidal silica. In other words, the amount of the modifying agent having a cationic group in the solid content of the colloidal silica after cation modification is preferably 0.1 to 1.5% by mass, more preferably 0.5 to 1.2% by mass, and even more preferably 0.6 to 1% by mass. If the amount used is within this range, the particle surface of the colloidal silica can be sufficiently cationized. Furthermore, if the amount used is such, it is possible to produce a stably cation-modified colloidal silica without aggregation. In addition, a solvent (water, a hydrophilic organic solvent, etc.) for dissolving the modifying agent may be used during the modification treatment.
前記の変性処理により得られる本発明の変性後のコロイダルシリカ(以下、「変性コロイダルシリカ」という場合がある。)は、固形分濃度が比較的少ない場合はもちろんのこと、固形分濃度が比較的高い場合でも分散安定性に優れ、実質的な凝集は見られず、様々な用途に好適である。固形分濃度としては、通常、製造過程などに起因して6質量%程度となることが多いが、研磨性能の向上や、運送上のコストを下げることが出来るという理由から、好ましくは12質量%以上、より好ましくは15質量%以上、さらに好ましくは18質量%以上であり、とくに好ましくは19質量%以上である。固形分濃度は用途などに応じて適宜設定することができる。他方、固形分濃度の上限値は限定されないが、粘度上昇などによる取り扱い性の低下や、分散安定性が低下する傾向があることから、好ましくは50質量%以下である。 The modified colloidal silica of the present invention obtained by the above-mentioned modification treatment (hereinafter, sometimes referred to as "modified colloidal silica") has excellent dispersion stability and does not substantially aggregate even when the solid concentration is relatively high, and is suitable for various applications. The solid concentration is usually about 6% by mass due to the manufacturing process, etc., but it is preferably 12% by mass or more, more preferably 15% by mass or more, even more preferably 18% by mass or more, and particularly preferably 19% by mass or more, because it can improve polishing performance and reduce transportation costs. The solid concentration can be appropriately set depending on the application, etc. On the other hand, the upper limit of the solid concentration is not limited, but it is preferably 50% by mass or less because there is a tendency for the handleability to decrease due to an increase in viscosity, etc., and for the dispersion stability to decrease.
一般的に固形分濃度が上昇するにつれて、粒子間の距離が短くなることから、コロイダルシリカには凝集が生じやすくなることが知られている。
ところが、本発明の変性コロイダルシリカは、前述のとおりの比較的小さな粒子径を有するとともに、前記のように比較的高い固形分濃度においても、実質的に凝集が生じないものであって、このようなコロイダルシリカとして実用的なものは未だ明確に確認されていなかった。
It is known that, in general, as the solid content increases, the distance between particles decreases, and therefore, colloidal silica becomes more susceptible to aggregation.
However, the modified colloidal silica of the present invention has a relatively small particle size as described above, and does not substantially aggregate even at a relatively high solids concentration as described above, and a practical colloidal silica of this kind has not yet been clearly identified.
また、前述のとおり、本発明のコロイダルシリカは、実質的に凝集していないものである。ここで、「実質的に」としたのは、次の理由からである。すなわち、一般的にコロイダルシリカが凝集していることを確認するには、電子顕微鏡により確認する方法や、粘度上昇を測定することや、動的光散乱法(DLS)によるキュムラント平均径の測定などによる手法が挙げられるが、BET径が12nm以下といった非常に小粒子である本発明のコロイダルシリカについて、その全ての粒子の一粒一粒が単分散しているかどうかを完全に把握することが困難である。前記のような手法で把握できるのは、ある一定程度の確からしさで粒子の分散状態を把握ないし理解できるに留まると考えられることから、実用上問題ない程度の実質的に凝集の発生が無いものであれば、コロイダルシリカの機能の発揮や、用途上で問題が生じないからである。 As mentioned above, the colloidal silica of the present invention is substantially free of aggregation. Here, the wording "substantially" is used for the following reason. That is, in general, methods for confirming whether colloidal silica is aggregated include a method of confirming with an electron microscope, measuring the increase in viscosity, and measuring the cumulant mean diameter by dynamic light scattering (DLS). However, for the colloidal silica of the present invention, which is an extremely small particle with a BET diameter of 12 nm or less, it is difficult to completely grasp whether each and every particle is monodispersed. Since it is considered that the above-mentioned methods can only grasp or understand the dispersion state of the particles with a certain degree of certainty, as long as there is substantially no aggregation to the extent that it does not cause practical problems, the colloidal silica will not exhibit its functions or cause problems in use.
そして、前記した「実質的に凝集していない」ことをより具体的に把握するためには、前記した手法により測定することができるが、小粒子であるために電子顕微鏡では一部把握困難な部分が生じる可能性がある。そのため、好ましくは、動的光散乱法(DLS)を用いて、それにより測定されるキュムラント平均径の変化により判断することが好適である。より具体的には、経時のDLS測定により、キュムラント平均径の変化(或いは、変化していないこと)を確認することがよく、例えば、本発明の製造方法においては、後述する濃縮工程において、濃縮前と濃縮後のDLS測定結果をそれぞれ確認することで凝集発生の有無を確認することができる。すなわち、固形分濃度上昇に伴う凝集の有無を把握することで、コロイダルシリカとして凝集が起こりやすい特性を有するものかどうかを確認することができるようになる。これは前述のとおり、固形分濃度が比較的低い場合は凝集の発生が少なく、固形分濃度が高くなるにつれて凝集しやすくなるといった実情があるからである。このような傾向は、通常、固形分濃度が10質量%以上、より具体的には12質量%以上の場合に顕著になる傾向がある。 Then, in order to grasp more specifically that the above-mentioned "substantially not aggregated" is not present, it can be measured by the above-mentioned method, but since the particles are small, some parts may be difficult to grasp by an electron microscope. Therefore, it is preferable to use dynamic light scattering (DLS) and judge from the change in the cumulant mean diameter measured by the method. More specifically, it is preferable to confirm the change (or the lack of change) in the cumulant mean diameter by DLS measurement over time. For example, in the manufacturing method of the present invention, in the concentration step described below, the presence or absence of aggregation can be confirmed by confirming the DLS measurement results before and after concentration. In other words, by grasping the presence or absence of aggregation with an increase in solid concentration, it becomes possible to confirm whether the colloidal silica has a property that is likely to cause aggregation. This is because, as mentioned above, when the solid concentration is relatively low, aggregation does not occur much, and as the solid concentration increases, aggregation becomes more likely. This tendency usually tends to be noticeable when the solid concentration is 10% by mass or more, more specifically, 12% by mass or more.
DLS測定による方法のうちでも、実用上、より好ましい凝集の把握の方法としては、加温した条件下において一定時間(期間)保持した後におけるDLS測定による粒子径の変化を測定することが挙げられる。この方法では、固形分濃度の積極的な変化を起こすことなく、コロイダルシリカとして凝集が起こりやすい特性を有するものかどうかを確認することができる。具体的には、後述の実施例でも行っているとおり、粒子が凝集し易い温度として少なくとも60℃以上で7日間保持した後におけるキュムラント平均径の変化を確認し、その変化率が20%以内である場合に凝集が無いと判断することができる。ここで、60℃以上で7日間保持する方法を好ましく適用する理由としては、60℃で7日間の条件とすることで、おおよそ常温で1年間保持した状態の経時変化試験と同等となることが分かっているからであり、これにより、少なくとも常温で1年間の経時変化試験において凝集の発生の有無を確認することができるからである。そして、好ましくは、DLS測定によるキュムラント平均径の変化率が10%以内、より好ましくは5%以内である。ここで、このようなDLS測定に際しては、前述の理由から、固形分濃度が12質量%以上、より具体的には18質量%以上、さらに具体的には、19質量%以上であって、好ましくは50質量%以下、より好ましくは40質量%以下、さらに好ましくは30質量%以下で保持ないし測定されることがよい。この時の固形分濃度は制限されず、用途や実使用に合わせた固形分濃度で前記DLS測定が行われことが好ましい。 Among the DLS measurement methods, a more practical method for grasping aggregation is to measure the change in particle diameter by DLS measurement after holding for a certain time (period) under heated conditions. With this method, it is possible to confirm whether the colloidal silica has the characteristics of being prone to aggregation without causing an active change in the solid content concentration. Specifically, as will be described in the examples below, the change in the cumulant mean diameter after holding for 7 days at at least 60°C, which is a temperature at which particles are prone to aggregation, is confirmed, and if the rate of change is within 20%, it can be determined that there is no aggregation. Here, the reason why the method of holding at 60°C or higher for 7 days is preferably applied is that it is known that the condition of 60°C for 7 days is approximately equivalent to a time-dependent change test in a state held at room temperature for one year, and this makes it possible to confirm the occurrence of aggregation in a time-dependent change test at least at room temperature for one year. And, preferably, the rate of change in the cumulant mean diameter by DLS measurement is within 10%, more preferably within 5%. Here, for the reasons described above, when performing such DLS measurement, the solid content concentration is preferably maintained or measured at 12% by mass or more, more specifically 18% by mass or more, even more specifically 19% by mass or more, and preferably 50% by mass or less, more preferably 40% by mass or less, and even more preferably 30% by mass or less. The solid content concentration at this time is not limited, and it is preferable that the DLS measurement is performed at a solid content concentration that matches the application and actual use.
また、本発明のコロイダルシリカは、前記のとおりのBET径、変性、実質的な凝集が無いといった特性を有するものであれば、用途や目的などによって任意の形状やその他特性を有するものであってよい。とりわけ、本発明で得られるコロイダルシリカについては、前述のとおりのBET比表面積を有するものであって、粒子の表面積が比較的高いものであることが好ましく、多数の不規則な小突起を有して粒子全体としていわば金平糖の如き形状を有しているものであることが好ましい。このような形状については、SEMにより観察した粒子像の算術平均を測定したSEM平均粒子径が大きい割にはBET比表面積が大きく、また、液相置換法にて測定された粒子密度(真比重)が高い、言い換えると硬度が高いという性質を有しており、優れた研磨速度を有してCMP用研磨剤には好適である。 The colloidal silica of the present invention may have any shape or other properties depending on the application or purpose, so long as it has the above-mentioned properties of BET diameter, modification, and substantial absence of aggregation. In particular, the colloidal silica obtained by the present invention preferably has the above-mentioned BET specific surface area, a relatively high particle surface area, and a large number of irregular small protrusions, so to speak, a confetti-like shape as a whole particle. Such a shape has a large BET specific surface area compared to the large SEM average particle diameter measured by measuring the arithmetic mean of particle images observed by SEM, and also has a high particle density (true specific gravity) measured by the liquid phase displacement method, in other words, a high hardness, and has an excellent polishing rate, making it suitable for use as an abrasive for CMP.
なお、本発明のコロイダルシリカの粒子の形状としては、仕込み組成等により、単分散の球状とすることや(球状品)、粒子どうしが合着して会合したような形状(会合品)などに制御することが可能である。例えば、触媒を多めに、かつ、シリカ原料としてのオルガノシリケートを反応場に比較的緩慢に投入することにより、オルガノシリケートが敏速に、均一に加水分解し、かつマイルドに成長するため、種粒子が球状を維持したまま徐々に成長し、球状品とすることができる。また、例えば、触媒を少なめに、かつ、シリカ原料としてのオルガノシリケートを反応場に比較的敏速に投入することにより、オルガノシリケートが不均一に加水分解するため、粒子同士の接着剤のようにふるまい、その結果、粒子が会合した会合品とすることも可能である。 The shape of the colloidal silica particles of the present invention can be controlled by the feed composition, etc., to be monodisperse spheres (spherical products) or to have a shape in which the particles are bonded together and associated (associated products). For example, by adding a large amount of catalyst and relatively slowly introducing an organosilicate as a silica raw material to the reaction field, the organosilicate hydrolyzes quickly and uniformly and grows mildly, so that the seed particles grow gradually while maintaining their spherical shape, and a spherical product can be obtained. Also, by adding a small amount of catalyst and relatively quickly introducing an organosilicate as a silica raw material to the reaction field, the organosilicate hydrolyzes non-uniformly and acts like an adhesive between the particles, resulting in an associated product in which the particles are associated.
また、本発明のコロイダルシリカは、その粘度が1~100mPa・sとすることが好ましく、より好ましくは1~50mPa・s、さらに好ましくは1~20mPa・sとすることがよい。 The viscosity of the colloidal silica of the present invention is preferably 1 to 100 mPa·s, more preferably 1 to 50 mPa·s, and even more preferably 1 to 20 mPa·s.
また、本発明のコロイダルシリカは、前述のとおり、単分散性の球状品であってもよく、又は、電子顕微鏡観察にて複数の粒子が2次元または3次元に合着して形成されて見える形状をもつ会合品(繭型、鎖状、分岐状など)でもよい。 As described above, the colloidal silica of the present invention may be a monodisperse spherical product, or it may be an aggregated product (cocoon-shaped, chain-like, branched, etc.) that has a shape that appears to be formed by multiple particles bonding together in two or three dimensions when observed under an electron microscope.
また、本発明のコロイダルシリカは、前記した変性処理などに応じて、分散安定性が損なわれないpHに調整されてもよい。例えば、前記アニオン変性処理されたコロイダルシリカの場合には、pHが1~5であることが好ましく、より好ましくはpHが2~3である。また、例えば、前記カチオン変性処理されたコロイダルシリカの場合には、pHが8~11であることが好ましく、より好ましくはpHが8.5~10である。pHがこのような範囲に調整されることにより、コロイダルシリカの分散安定性の面で好適である。 The colloidal silica of the present invention may be adjusted to a pH that does not impair dispersion stability, depending on the above-mentioned modification treatment. For example, in the case of the anion-modified colloidal silica, the pH is preferably 1 to 5, and more preferably 2 to 3. For example, in the case of the cation-modified colloidal silica, the pH is preferably 8 to 11, and more preferably 8.5 to 10. Adjusting the pH to such a range is favorable in terms of the dispersion stability of the colloidal silica.
更に、本発明のコロイダルシリカは、金属不純物含有量が1ppm以下であることが好ましく、より好ましくは0.01ppm以下、さらに好ましくは0.0001ppm以下である。このような高純度のコロイダルシリカは、限定されるものではないが、例えば、後述の製造方法において、原料であるコロイダルシリカを得る際の加水分解反応の原料に用いるシリカ源、加水分解触媒及び水として、前記金属不純物含有量を満足するものを使用することで達成することができる。 Furthermore, the colloidal silica of the present invention preferably has a metal impurity content of 1 ppm or less, more preferably 0.01 ppm or less, and even more preferably 0.0001 ppm or less. Although not limited thereto, such high purity colloidal silica can be achieved, for example, in the manufacturing method described below, by using silica source, hydrolysis catalyst, and water used as raw materials in the hydrolysis reaction to obtain the raw material colloidal silica, which satisfy the above metal impurity content.
<コロイダルシリカの製造方法>
本発明のコロイダルシリカの製造方法は、以下の工程を必須として含む。
(a)有機アミンからなる加水分解触媒を含む反応液に、易分解性オルガノシリケートを供給及び反応させて、BET径が12nm以下である原料コロイダルシリカを準備する原料準備工程。
(b)原料コロイダルシリカの固形分濃度を13質量%以下とすると共に、前記原料準備工程に由来して生成するアルコール類の濃度を1~25質量%とする濃度調整工程。
(c)前記工程(b)で濃度調整された原料コロイダルシリカを変性処理する変性処理工程。
(d)前記工程(c)で変性処理されたコロイダルシリカ中の残留有機溶媒が1質量%以下となるように濃縮する濃縮工程。
これらの工程について以下で説明する。
<Method of producing colloidal silica>
The method for producing colloidal silica of the present invention essentially includes the following steps.
(a) a raw material preparation step of supplying and reacting a readily decomposable organosilicate to a reaction liquid containing a hydrolysis catalyst made of an organic amine, thereby preparing raw material colloidal silica having a BET diameter of 12 nm or less.
(b) a concentration adjusting step of adjusting the solid content concentration of the raw material colloidal silica to 13% by mass or less and adjusting the concentration of alcohols generated in the raw material preparation step to 1 to 25% by mass.
(c) a modification treatment step of modifying the raw material colloidal silica having its concentration adjusted in the step (b).
(d) a concentration step of concentrating the colloidal silica modified in the step (c) so that the residual organic solvent in the colloidal silica is 1 mass % or less.
These steps are described below.
[工程(a)]
先ず、工程(a)において、有機アミンからなる加水分解触媒を含む反応液に、易分解性オルガノシリケートを供給及び反応させて、BET径が12nm以下であるコロイダルシリカを準備する。本発明においては、工程(a)において準備されるコロイダルシリカを原料コロイダルシリカと称する。原料コロイダルシリカには、市販品を用いることも可能であるが、金属不純物が少なくなることや、比較的表面積が大きいコロイダルシリカを得ることできることや、また、均一性の高い粒子を製造できることから、加水分解法を用いるようにする。加水分解法としては、加水分解触媒を含む反応液に、シリカ源を供給し、加水分解反応させる方法が用いられ、粒子径のコントロールが容易であるとの理由から、有機アミンからなる加水分解触媒を含む反応液に、シリカ源として易分解性オルガノシリケートを供給し、加水分解反応させる方法を用いる。
[Step (a)]
First, in step (a), a colloidal silica having a BET diameter of 12 nm or less is prepared by supplying and reacting an easily decomposable organosilicate to a reaction solution containing a hydrolysis catalyst made of an organic amine. In the present invention, the colloidal silica prepared in step (a) is called raw colloidal silica. Although a commercially available product can be used as the raw colloidal silica, a hydrolysis method is used because it reduces metal impurities, can obtain colloidal silica with a relatively large surface area, and can produce particles with high uniformity. As the hydrolysis method, a method is used in which a silica source is supplied to a reaction solution containing a hydrolysis catalyst and hydrolyzed, and a method is used in which a easily decomposable organosilicate is supplied as a silica source to a reaction solution containing a hydrolysis catalyst made of an organic amine and hydrolyzed because it is easy to control the particle diameter.
ここで、工程(a)において好ましく使用されるシリカ源は、加水分解速度の速い易加水分解性オルガノシリケートである。易加水分解性オルガノシリケートとは、オルガノシリケート10gと不純物0.1ppb以下の純水100gとを攪拌下に25℃で加水分解反応させ、1時間以内にこの加水分解反応が終了するものが好ましく用いられる。このような易加水分解性オルガノシリケートとしては、具体的には、トリメチルシリケート(加水分解反応が終了するまでの加水分解反応時間:3分程度)、テトラメチルシリケート(加水分解反応時間:5分程度)、トリエチルシリケート(加水分解反応時間:5分程度)、メチルトリメチルシリケート(加水分解反応時間:7分程度)等を挙げることができる。テトラエチルシリケート及びこれより炭素数の多いオルガノシリケートはその加水分解速度が遅くてゲル化し易い傾向があるため(何れも加水分解反応時間:24時間以上)、前記した易分解性オルガノシリケートが好ましく用いられる。 Here, the silica source preferably used in step (a) is an easily hydrolyzable organosilicate with a fast hydrolysis rate. The easily hydrolyzable organosilicate is preferably one which is hydrolyzed within 1 hour by stirring 10 g of organosilicate and 100 g of pure water with impurities of 0.1 ppb or less at 25°C. Specific examples of such easily hydrolyzable organosilicates include trimethyl silicate (hydrolysis reaction time until hydrolysis reaction is completed: about 3 minutes), tetramethyl silicate (hydrolysis reaction time: about 5 minutes), triethyl silicate (hydrolysis reaction time: about 5 minutes), and methyl trimethyl silicate (hydrolysis reaction time: about 7 minutes). Tetraethyl silicate and organosilicates with a larger carbon number than tetraethyl silicate have a slow hydrolysis rate and tend to gel easily (hydrolysis reaction time: 24 hours or more for both), so the above-mentioned easily decomposable organosilicates are preferably used.
また、工程(a)において加水分解触媒として使用される有機アミン類については、制限されないが、第四級アンモニウム類、第三級アミン類、第二級アミン類及び第一級アミン類並びにこれらの炭酸塩、重炭酸塩及びケイ酸塩から選ばれた1種であるか又はそれらの2種以上の混合物を広く使用することができる。ここで、例えば、第四級アンモニウム類については、水酸化テトラメチルアンモニウム(TMAH)、水酸化テトラエチルアンモニウム(TEAH)、水酸化トリメチルエチルアンモニウム、水酸化トリメチルエタノールアンモニウム(コリン)、水酸化トリエチルエタノールアンモニウム、水酸化テトラプロピルアンモニウム、水酸化ブチルアンモニウム等の第四級アンモニウムや、これらの炭酸塩、重炭酸塩及びケイ酸塩を挙げることができる。加水分解反応には比較的高いpHが望ましいので、好ましくはテトラメチルアンモニウムヒドロキシド(TMAH)、コリン、又はテトラエチルアンモニウムヒドロキシド(TEAH)である。 The organic amines used as the hydrolysis catalyst in step (a) are not limited, but may be one or a mixture of two or more selected from quaternary ammoniums, tertiary amines, secondary amines, primary amines, and their carbonates, bicarbonates, and silicates. For example, quaternary ammoniums include tetramethylammonium hydroxide (TMAH), tetraethylammonium hydroxide (TEAH), trimethylethylammonium hydroxide, trimethylethanolammonium hydroxide (choline), triethylethanolammonium hydroxide, tetrapropylammonium hydroxide, butylammonium hydroxide, and other quaternary ammoniums, as well as their carbonates, bicarbonates, and silicates. Since a relatively high pH is desirable for the hydrolysis reaction, tetramethylammonium hydroxide (TMAH), choline, or tetraethylammonium hydroxide (TEAH) is preferred.
また、加水分解触媒として使用する有機アミン類の第一級アミン類、第二級アミン類、第三級アミン類としても制限されるものではないが、例えば、アミノアルコール類、モルホリン類、ピペラジン類、脂肪族アミン、脂肪族エーテルアミン等が挙げられる。ここで、アミノアルコール類については、エタノールアミン誘導体を始めとして種々のアミノアルコールを用いることができるが、好適にはエタノールアミン誘導体であり、例えばモノエタノールアミン、ジエタノールアミン、トリエタノールアミン、N,N-ジメチルエタノールアミン、N,N-ジエチルエタノールアミン、N,N-ジn-ブチルエタノールアミン、N-(β-アミノエチル)エタノールアミン、N-メチルエタノールアミン、N-メチルジエタノールアミン、N-エチルエタノールアミン、N-n-ブチルエタノールアミン、N-n-ブチルジエタノールアミン、N-tert-ブチルエタノールアミン、N-tert-ブチルジエタノールアミン等を挙げることができる。 Furthermore, the primary amines, secondary amines, and tertiary amines of the organic amines used as hydrolysis catalysts are not limited, but examples thereof include aminoalcohols, morpholines, piperazines, aliphatic amines, and aliphatic ether amines. Here, various aminoalcohols including ethanolamine derivatives can be used as aminoalcohols, but ethanolamine derivatives are preferred, such as monoethanolamine, diethanolamine, triethanolamine, N,N-dimethylethanolamine, N,N-diethylethanolamine, N,N-di-n-butylethanolamine, N-(β-aminoethyl)ethanolamine, N-methylethanolamine, N-methyldiethanolamine, N-ethylethanolamine, N-n-butylethanolamine, N-n-butyldiethanolamine, N-tert-butylethanolamine, and N-tert-butyldiethanolamine.
更に、加水分解触媒として使用する有機アミン類のモルホリン類についても、種々のモルホリン誘導体を用いることができるが、好ましくはモルホリン、N-メチルモルホリン、N-エチルモルホリン等を挙げることができる。更にまた、加水分解触媒として使用する有機アミン類のピペラジン類についても、種々のピペラジン誘導体を用いることができるが、好ましくはピペラジン、ヒドロキシエチルピペラジン等を挙げることができる。また、加水分解触媒として使用する有機アミン類の脂肪族アミン及び脂肪族エーテルアミンについては、脂肪族アミンとしてはトリエチルアミン、ジプロピルアミン、ペンチルアミン、ヘキシルアミン、ヘプチルアミン、オクチルアミンなどの炭素数1~8のアルキルアミンを好適に挙げることができる。また、脂肪族エーテルアミンとしては2-メトキシエチルアミン、3-メトキシプロピルアミン、3-エトキシプロピルアミン、3-プロポキシプロピルアミン、3-イソプロポキシプロピルアミン、3-ブトキシプロピルアミン等の炭素数1~8の脂肪族エーテルアミンを好適に挙げることができる。 Furthermore, for the morpholines of the organic amines used as the hydrolysis catalyst, various morpholine derivatives can be used, with preferred examples including morpholine, N-methylmorpholine, and N-ethylmorpholine. For the piperazines of the organic amines used as the hydrolysis catalyst, various piperazine derivatives can be used, with preferred examples including piperazine and hydroxyethylpiperazine. For the aliphatic amines and aliphatic etheramines of the organic amines used as the hydrolysis catalyst, preferred examples of the aliphatic amines include alkylamines having 1 to 8 carbon atoms, such as triethylamine, dipropylamine, pentylamine, hexylamine, heptylamine, and octylamine. Preferred examples of the aliphatic etheramines include aliphatic etheramines having 1 to 8 carbon atoms, such as 2-methoxyethylamine, 3-methoxypropylamine, 3-ethoxypropylamine, 3-propoxypropylamine, 3-isopropoxypropylamine, and 3-butoxypropylamine.
これら加水分解触媒として使用する前記有機アミン類は、その1種のみを単独で使用できるほか、必要により2種以上を混合物として使用することができる。 The above organic amines used as hydrolysis catalysts can be used alone or, if necessary, in a mixture of two or more kinds.
反応液については、前記のとおりシリカ源としての易分解性オルガノシリケートと、加水分解触媒とが必須で含まれるが、それ以外は、水、アルコール類、アルデヒド類、ケトン類、界面活性剤などを使用することができる。好ましくは、シリカ源と加水分解触媒と水とを合計で90質量%以上含むことが好ましい。より好ましくは、これらが95質量%以上である。 As described above, the reaction liquid must contain an easily decomposable organosilicate as a silica source and a hydrolysis catalyst, but other than that, water, alcohols, aldehydes, ketones, surfactants, etc. can also be used. It is preferable that the total content of the silica source, hydrolysis catalyst, and water is 90% by mass or more. More preferably, these are 95% by mass or more.
前記のシリカ源と加水分解触媒との反応後の混合物(以下、これを「反応混合物」ということがある。)や、或いはその後に、例えば後述するような固形分濃度やアルコール類の濃度の調整や、酸による分散安定化の処理を行った反応混合物(以下、これを特に「反応濃縮物」ということがある。)においては、シリカ(B)に対する加水分解触媒(A)の割合{触媒残存モル比(A/B)}が0.012以下となるようにすることが好ましく、より好ましくは0.00035~0.012の範囲内、さらに好ましくは0.0035~0.011の範囲内となるように、加水分解触媒を反応系内に添加して加水分解反応させることが良い。このようにすることにより、上記反応混合物や反応濃縮物のpHを適正化することができること、また、増粘やゲル化を抑えることができるため、好ましい。 In the mixture after the reaction of the silica source with the hydrolysis catalyst (hereinafter, this may be referred to as the "reaction mixture"), or in the reaction mixture after the solids concentration or alcohol concentration has been adjusted as described below, or after the treatment of dispersion stabilization with acid (hereinafter, this may be particularly referred to as the "reaction concentrate"), it is preferable to add the hydrolysis catalyst to the reaction system and carry out the hydrolysis reaction so that the ratio of the hydrolysis catalyst (A) to the silica (B) {catalyst remaining molar ratio (A/B)} is 0.012 or less, more preferably in the range of 0.00035 to 0.012, and even more preferably in the range of 0.0035 to 0.011. This is preferable because it is possible to optimize the pH of the reaction mixture or reaction concentrate, and to suppress thickening and gelation.
このような触媒残存モル比にする方法については、特に制限されるものではないが、例えば、水と加水分解触媒(A)とを仕込んだ反応容器内に最終的に触媒残存モル比(A/B)が前記の範囲内となるように計算されたシリカ源を連続的にあるいは間欠的に導入する方法や、水だけを仕込んだ反応容器内に上記の最終的な触媒残存モル比の範囲内となるように計算された加水分解触媒とシリカ源とを連続的にあるいは間欠的に導入する方法や、水と少量の加水分解触媒(A)とを仕込んだ反応容器内に上記の最終的な触媒残存モル比の範囲内となるように計算された加水分解触媒とシリカ源とを連続的にあるいは間欠的に導入する方法等を挙げることができる。 The method for achieving such a catalyst remaining molar ratio is not particularly limited, but examples include a method of continuously or intermittently introducing a silica source calculated so that the final catalyst remaining molar ratio (A/B) falls within the above range into a reaction vessel charged with water and hydrolysis catalyst (A), a method of continuously or intermittently introducing a hydrolysis catalyst and a silica source calculated so that the final catalyst remaining molar ratio falls within the above range into a reaction vessel charged with only water, and a method of continuously or intermittently introducing a hydrolysis catalyst and a silica source calculated so that the final catalyst remaining molar ratio falls within the above range into a reaction vessel charged with water and a small amount of hydrolysis catalyst (A).
また、加水分解反応の反応系内にはシリカ源の加水分解反応に先駆けて粒子成長性能を有するコロイダルシリカの種子を仕込み、この反応系内にシリカ源及び加水分解触媒を、触媒残存モル比(A/B)が上記の範囲内となるように、徐々に添加してもよく、これによって均一な粒子のコロイダルシリカを製造することができるため、好ましい。 In addition, prior to the hydrolysis reaction of the silica source, colloidal silica seeds having particle growth properties may be charged into the reaction system for the hydrolysis reaction, and the silica source and hydrolysis catalyst may be gradually added to the reaction system so that the catalyst remaining molar ratio (A/B) falls within the above range. This is preferable because it allows the production of colloidal silica with uniform particles.
なお、加水分解反応の原料に用いるシリカ源、加水分解触媒及び水としては、金属不純物含有量が1ppm以下であることが好ましく、より好ましくは0.01ppm以下の高純度のものを用いる。それにより、得られる原料コロイダルシリカ及び変性後の変性コロイダルシリカについても前述の金属不純物含有量の範囲が満足される。 The silica source, hydrolysis catalyst, and water used as raw materials for the hydrolysis reaction preferably have a metal impurity content of 1 ppm or less, and more preferably have a high purity of 0.01 ppm or less. This ensures that the raw colloidal silica obtained and the modified colloidal silica obtained after modification also satisfy the above-mentioned range of metal impurity content.
工程(a)において、原料コロイダルシリカのBET径を12nm以下とする方法は制限されないが、反応液にシリカ源を滴下する速度(供給速度)、反応温度及び反応時間を調整することが好ましい。 In step (a), the method for making the BET diameter of the raw colloidal silica 12 nm or less is not limited, but it is preferable to adjust the rate at which the silica source is dropped into the reaction solution (feed rate), the reaction temperature, and the reaction time.
シリカ源として易分解性オルガノシリケートを用いる場合、供給速度としては、易分解性オルガノシリケートの総投入量の1.5質量%未満/分とすることが好ましく、1.3質量%以下/分とすることがより好ましく、1.2質量%以下/分とすることがさらに好ましい。当該供給速度が1.5質量%以上/分である場合、均等に分散しないことや、真球に近い粒子にならない傾向がある。ここで、総投入量が基準とされる理由は、製造スケール等によって投入する量が変化するからであるとともに、後述の反応時間内に供給を完了することを考慮することが好ましいからである。すなわち、供給速度をなるべく遅くすることが好ましく、所望の小粒子径を形成しやすくなる。供給速度の下限は制限されないが、供給速度が遅過ぎると目標とする粒子のサイズにならないおそれがあることから、好ましくは総投入量の1.0質量%/分以上とすることが好ましい。 When a readily decomposable organosilicate is used as the silica source, the supply rate is preferably less than 1.5 mass%/min of the total amount of readily decomposable organosilicate added, more preferably 1.3 mass% or less/min, and even more preferably 1.2 mass% or less/min. If the supply rate is 1.5 mass% or more/min, the particles tend not to be uniformly dispersed or to not be nearly spherical. The reason why the total amount is used as the standard is that the amount added varies depending on the production scale, etc., and it is preferable to consider completing the supply within the reaction time described below. In other words, it is preferable to make the supply rate as slow as possible, which makes it easier to form the desired small particle size. There is no lower limit to the supply rate, but if the supply rate is too slow, there is a risk that the target particle size will not be achieved, so it is preferable to make it 1.0 mass%/min or more of the total amount added.
また、反応温度としては、70℃以下とすることが好ましく、より好ましくは65℃以下、さらに好ましくは60℃以下である。反応温度が70℃を超過すると、反応液の揮発が大きくなって液組成が変化しやすくなることから、粒子径の制御が困難となるおそれがある。他方、反応温度の下限は適宜設定することができるが、温度が低すぎると加水分解反応が遅くなる傾向があり、粒子成長が促進されるおそれがあることから、好ましくは20℃以上とすることが好ましい。 The reaction temperature is preferably 70°C or less, more preferably 65°C or less, and even more preferably 60°C or less. If the reaction temperature exceeds 70°C, the reaction liquid will volatilize more and the liquid composition will be more likely to change, which may make it difficult to control the particle size. On the other hand, the lower limit of the reaction temperature can be set as appropriate, but if the temperature is too low, the hydrolysis reaction tends to be slow and particle growth may be promoted, so it is preferably set to 20°C or more.
また、反応時間としては、6時間以内とすることが好ましく、より好ましくは3時間以内、さらに好ましくは2時間以内である。反応時間が6時間を超過すると、目標とする粒子のサイズにならないおそれがある。他方、反応時間の下限は適宜設定することができるが、反応時間が短か過ぎると加水分解反応が終了せずに、粒子形成や粒子成長が十分に行われない場合があることから、好ましくは20分以上である。 The reaction time is preferably 6 hours or less, more preferably 3 hours or less, and even more preferably 2 hours or less. If the reaction time exceeds 6 hours, there is a risk that the target particle size will not be achieved. On the other hand, the lower limit of the reaction time can be set as appropriate, but if the reaction time is too short, the hydrolysis reaction may not be completed, and particle formation and particle growth may not occur sufficiently, so it is preferably 20 minutes or more.
工程(a)においては、反応時の固形分濃度は、通常3~13質量%となるようにすることが好ましい。固形分濃度が3質量%未満であると、粒子形成や粒子成長が十分に行われない場合がある。他方、固形分濃度が高すぎる場合、粒子の凝集が起こりやすい。工程(a)において、反応後のコロイダルシリカの固形分濃度は3~10質量%の範囲内であることが好ましい。 In step (a), it is generally preferable that the solids concentration during the reaction is 3 to 13% by mass. If the solids concentration is less than 3% by mass, particle formation and growth may not occur sufficiently. On the other hand, if the solids concentration is too high, particles are likely to aggregate. In step (a), it is preferable that the solids concentration of the colloidal silica after the reaction is in the range of 3 to 10% by mass.
[工程(b)]
工程(b)では、工程(a)で得られた原料コロイダルシリカの固形分濃度を13質量%以下に濃度調整する。固形分濃度を13質量%以下に調整することにより、BET径が12nm以下であっても、凝集の発生を低下させることができ、凝集ゲル化までを起こすことなく、次の工程(c)(変性処理工程)を行うことができる。好ましくは、固形分濃度を10質量%以下、より好ましくは8質量%以下にすることがよい。ここで、「凝集ゲル化」とは、明確な定義や区分は難しいものの、一般的には、コロイダルシリカの凝集がさらに進み、目視ではゼリー状の塊になってしまった状態を表すことが多い(例えば、後述の比較例2における図3を参照)。
[Step (b)]
In step (b), the solid content concentration of the raw colloidal silica obtained in step (a) is adjusted to 13% by mass or less. By adjusting the solid content concentration to 13% by mass or less, even if the BET diameter is 12 nm or less, the occurrence of aggregation can be reduced, and the next step (c) (modification treatment step) can be performed without causing aggregation gelation. Preferably, the solid content concentration is 10% by mass or less, more preferably 8% by mass or less. Here, although it is difficult to clearly define or classify, the term "aggregation gelation" generally refers to a state in which the aggregation of colloidal silica has progressed further and has become a jelly-like mass when viewed with the naked eye (for example, see FIG. 3 in Comparative Example 2 described later).
ここで、工程(a)で得られた原料コロイダルシリカの固形分濃度が既に13質量%以下となっている場合には、工程(b)において積極的な固形分濃度の調整を行わなくてもよく、その場合は、固形分濃度を確認する工程であるとか、或いは、固形分濃度をそのまま維持する工程などのような取り扱いとしても構わない。次の工程(c)の変性処理の前に、この工程(b)において固形分濃度が確かに13質量%以下となっているようにすることは、最終的に変性された本発明のコロイダルシリカとして実質的に凝集していないものを、比較的固形分濃度を高いものとして得る上でも特徴的となる工程である。 Here, if the solids concentration of the raw colloidal silica obtained in step (a) is already 13% by mass or less, there is no need to actively adjust the solids concentration in step (b), and in that case, it may be treated as a step of confirming the solids concentration, or a step of maintaining the solids concentration as it is. Ensuring that the solids concentration is indeed 13% by mass or less in step (b) before the modification treatment in the next step (c) is a characteristic step in obtaining the final modified colloidal silica of the present invention that is substantially non-aggregated and has a relatively high solids concentration.
工程(b)における固形分濃度の調整方法は制限されないが、原料コロイダルシリカに含まれる溶媒と同じ溶媒を添加する方法により行うことが好ましい。添加する溶媒としては、水及び/又はアルコール類が好ましく、水、メタノール及び/又はエタノールがより好ましく、水及び/又はメタノールが更に好ましい。なお、添加する溶媒には、前記のとおり、水、アルコール類、アルデヒド類、ケトン類、界面活性剤などが含まれてもよい。 The method for adjusting the solid content concentration in step (b) is not limited, but it is preferable to add the same solvent as that contained in the raw colloidal silica. The solvent to be added is preferably water and/or alcohols, more preferably water, methanol and/or ethanol, and even more preferably water and/or methanol. As described above, the solvent to be added may include water, alcohols, aldehydes, ketones, surfactants, etc.
また、この工程(b)では、前記工程(a)に由来して生成するアルコール類の濃度を1~25質量%に濃度調整する。前記工程(a)においては、前記加水分解反応によって、使用されたシリカ源である易分解性オルガノシリケートに応じてアルコール類が生成する。例えば、使用される易分解性オルガノシリケートがトリメチルシリケートやテトラメチルシリケート等のメトキシ基を有するものである場合には、アルコール類としてメタノールが生成される。また、使用される易分解性オルガノシリケートがトリエチルシリケート等のエトキシ基を有するものである場合には、アルコール類としてエタノールが生成される。それ以外についても同様である。 In addition, in this step (b), the concentration of the alcohols produced from the step (a) is adjusted to 1 to 25% by mass. In the step (a), the hydrolysis reaction produces alcohols according to the easily decomposable organosilicate used as the silica source. For example, when the easily decomposable organosilicate used has a methoxy group, such as trimethyl silicate or tetramethyl silicate, methanol is produced as the alcohol. When the easily decomposable organosilicate used has an ethoxy group, such as triethyl silicate, ethanol is produced as the alcohol. The same applies to other cases.
ここで、このような、工程(a)における加水分解反応に由来して生成されるアルコール類の濃度を1~25質量%に調整する必要があることについては、詳細な機序は明らかではないものの、当該有機溶媒の濃度をこのような範囲に維持することにより、溶媒の極性に起因して、BET径が12nm以下であっても、凝集の発生を低下させることができ、凝集ゲル化までを起こすことなく、次の工程(c)(変性処理工程)を行うことができると推測される。当該生成されるアルコール類の濃度については、工程(a)で使用される易分解性オルガノシリケートの使用量や、蒸留によるアルコール類の純水への置換等により適宜調整され得るが、好ましい下限は5質量%以上、より好ましい下限は12質量%以上となるようにすることがよい。他方、好ましい上限については20質量%以下となるようにすることがよい。 Although the detailed mechanism behind the need to adjust the concentration of the alcohols resulting from the hydrolysis reaction in step (a) to 1-25% by mass is unclear, it is presumed that by maintaining the concentration of the organic solvent in this range, the occurrence of aggregation due to the polarity of the solvent can be reduced even if the BET diameter is 12 nm or less, and the next step (c) (modification treatment step) can be performed without causing aggregation and gelation. The concentration of the generated alcohols can be appropriately adjusted by the amount of easily decomposable organosilicate used in step (a) or by replacing the alcohols with pure water by distillation, but the preferred lower limit is 5% by mass or more, and the more preferred lower limit is 12% by mass or more. On the other hand, the preferred upper limit is 20% by mass or less.
なお、この工程(b)における前述の固形分濃度(13質量%以下)の調整の場合と同様に、工程(a)で得られた原料コロイダルシリカのアルコール類の濃度が既に1~25質量%となっている場合には、アルコール類の積極的な濃度調整を行わなくてもよく、その場合は、アルコール類の濃度を確認する工程であるとか、或いは、工程(a)で生成したアルコール類の濃度をそのまま維持する工程などのような取り扱いとしても構わない。次の工程(c)の変性処理の前に、この工程(b)において当該アルコール類の濃度が確かに1~25質量%となっているようにすることは、前述の固形分濃度(13質量%以下)の調整の場合と同様に、最終的に変性された本発明のコロイダルシリカとして実質的に凝集していないものを、比較的固形分濃度を高いものとして得る上でも特徴的となる工程である。なお、このアルコール類は、次の工程(c)の変性処理における溶媒の一部となり得る。 As in the case of adjusting the solid content concentration (13% by mass or less) in this step (b), if the alcohol concentration of the raw colloidal silica obtained in step (a) is already 1-25% by mass, there is no need to actively adjust the alcohol concentration. In that case, it may be treated as a step of confirming the alcohol concentration, or as a step of maintaining the alcohol concentration produced in step (a) as it is. As in the case of adjusting the solid content concentration (13% by mass or less) described above, ensuring that the alcohol concentration is 1-25% by mass in this step (b) before the modification treatment in the next step (c) is a characteristic step in obtaining a substantially non-aggregated colloidal silica of the present invention with a relatively high solid content concentration as the final modified colloidal silica of the present invention. The alcohol may be part of the solvent in the modification treatment in the next step (c).
この工程(b)における前記アルコール類の濃度の調整方法は制限されないが、当該アルコール類の濃度を高めるように調整する場合には、生成されたアルコール類と同じ有機溶媒を主成分(例えば、95質量%以上)としたものを添加する方法により行うことが好ましい。ここで添加される有機溶媒には、前記のとおり、水、アルコール類、アルデヒド類、ケトン類、界面活性剤などが含まれてもよい。他方、当該アルコール類の濃度を低めるように調整する方法としては、後述の比較例1において行っているように、例えば、加熱することで揮発させながらコンデンサー付留出管から留出させる方法を挙げることができる。この際、さらに純水を添加しながら行ってもよい。 There are no limitations on the method for adjusting the concentration of the alcohols in step (b), but when adjusting to increase the concentration of the alcohols, it is preferable to add an organic solvent containing the same organic solvent as the produced alcohols as the main component (e.g., 95% by mass or more). As described above, the organic solvent added here may include water, alcohols, aldehydes, ketones, surfactants, etc. On the other hand, an example of a method for adjusting to decrease the concentration of the alcohols is to volatilize the alcohols by heating them while distilling them from a distillation tube with a condenser, as is done in Comparative Example 1 described later. In this case, pure water may be added.
[工程(c)]
工程(c)では、工程(b)を経て固形分濃度や前記生成されたアルコール類の濃度が調整された原料コロイダルシリカを変性処理する。前述のとおり変性処理は制限されず、公知の変性処理から選択することができるが、アニオン変性又はカチオン変性が好ましく、アニオン変性がより好ましい。
ここで、アニオン変性又はカチオン変性についても前述の例示のとおりであり、好ましくは、アニオン変性であり、アニオン変性の好適な方法は、化学的な方法などによりアニオン基に変換できる官能基を有する変性剤をコロイダルシリカの表面に化学的に結合させる方法が挙げられる。アニオン変性のうち、好ましい実施形態として例示したスルホ基を例にとって説明する。また、カチオン変性のうち、好ましい実施形態として例示した1級アミノ基を例にとって説明する。
[Step (c)]
In step (c), the raw colloidal silica, the solid content concentration of which and the concentration of the generated alcohols have been adjusted through step (b), is modified. As described above, the modification is not limited and can be selected from known modification treatments, but anion modification or cation modification is preferred, and anion modification is more preferred.
Here, the anion modification or the cation modification is as exemplified above, and the anion modification is preferred, and a suitable method for the anion modification is a method in which a modifying agent having a functional group that can be converted to an anion group by a chemical method or the like is chemically bonded to the surface of the colloidal silica. Among the anion modifications, the sulfo group exemplified as a preferred embodiment will be taken as an example. Also, among the cation modifications, the primary amino group exemplified as a preferred embodiment will be taken as an example.
(スルホ基を例にとったアニオン変性処理の実施形態について)
スルホ基をコロイダルシリカの表面に形成させるためには、前述のとおり公知の方法で行うことができる。好ましくは、化学的な方法などによりスルホ基に変換できる官能基を有する変性剤をコロイダルシリカの表面に化学的に結合させた後に、該官能基をスルホ基に変換させる処理を行う方法を挙げることができ、酸化によりスルホ基に変換できる官能基を有する変性剤が好ましい。このうちの好ましい変性剤としては、前記したとおり、メルカプト基及び/又はスルフィド基を有するシランカップリング剤であり、代表的に以下で説明する。
(Embodiment of anion modification treatment using sulfo group as an example)
To form sulfo group on the surface of colloidal silica, it can be carried out by a known method as described above.Preferably, a method can be mentioned in which a modifying agent having a functional group that can be converted to a sulfo group by chemical method or the like is chemically bonded to the surface of colloidal silica, and then the functional group is converted to a sulfo group, and a modifying agent having a functional group that can be converted to a sulfo group by oxidation is preferable.Among these, a preferred modifying agent is a silane coupling agent having a mercapto group and/or a sulfide group as described above, and a representative one will be described below.
この方法において、当該シランカップリング剤の存在下で、前記のように工程(a)及び工程(b)を経て固形分濃度等が濃度調整された原料コロイダルシリカを反応させる工程(工程c1)と、工程c1の反応物を酸化処理して、メルカプト基及び/又はスルフィド基をスルホ基に変換させる工程(工程c2)とを含むようにすることが好ましい。工程c1及び工程c2についても前記した特開2010-269985号公報や特開2013-041992号公報などを参照して行うことができる。なお、本発明の目的を害しない限りは、それ以外の工程を適宜含ませてもよい。それ以外の工程としては、例えば、反応液の粘度を調整することや、pHの調整を行うことなどを挙げることができる。 In this method, it is preferable to include a step (step c1) of reacting raw colloidal silica, the solid content of which has been adjusted through steps (a) and (b) in the presence of the silane coupling agent, and a step (step c2) of oxidizing the reaction product of step c1 to convert mercapto groups and/or sulfide groups to sulfo groups. Steps c1 and c2 can also be performed with reference to the above-mentioned JP-A-2010-269985 and JP-A-2013-041992. Note that other steps may be included as appropriate as long as they do not impair the object of the present invention. Examples of other steps include adjusting the viscosity of the reaction solution and adjusting the pH.
(工程c1)
工程c1では、工程(a)及び工程(b)を経て固形分濃度等が濃度調整された原料コロイダルシリカを、メルカプト基及び/又はスルフィド基を有するシランカップリング剤の存在下で反応させる。これにより、該シランカップリング剤が原料コロイダルシリカの表面に化学的に結合される。
(Step c1)
In step c1, the raw colloidal silica, the solid content of which has been adjusted through steps (a) and (b), is reacted in the presence of a silane coupling agent having a mercapto group and/or a sulfide group, so that the silane coupling agent is chemically bonded to the surface of the raw colloidal silica.
工程c1の反応は、変性剤の使用適正温度の範囲内で行うことができ、制限されるものではないが、例えば、40℃以上であって、反応液(溶媒)の沸点以下の温度範囲で行うことができる。好ましくは、反応性の向上のために、温度50℃以上が好ましく、より好ましくは60℃以上であって、反応液(溶媒)の沸点以下としては好ましくは100℃以下で行われることがよい。反応時間も制限されないが、例えば、10分以上10時間以下で行われることが好ましく、1~8時間がより好ましい。 The reaction in step c1 can be carried out within the range of temperatures suitable for use of the denaturant, and is not limited to a particular range, for example, 40°C or higher and below the boiling point of the reaction liquid (solvent). To improve reactivity, the reaction is preferably carried out at a temperature of 50°C or higher, more preferably 60°C or higher, and preferably below the boiling point of the reaction liquid (solvent), 100°C or lower. The reaction time is also not limited, but is preferably carried out for 10 minutes to 10 hours, and more preferably 1 to 8 hours.
工程c1の反応においては、本発明の目的を害さない範囲内において、変性剤やシランカップリング剤の溶解性を向上するための溶媒を添加することができる。このような溶媒としては、親水性溶媒を挙げることができ、例えば、メタノール、エタノール、イソプロパノールなどのアルコール類などを挙げることができるがこれらに限定されない。原料コロイダルシリカを得る加水分解反応により生成されるアルコールと同じアルコール類を用いることがより好ましい。 In the reaction of step c1, a solvent for improving the solubility of the modifier or silane coupling agent can be added within a range that does not impair the object of the present invention. Such a solvent can be a hydrophilic solvent, for example, alcohols such as methanol, ethanol, isopropanol, etc., but is not limited to these. It is more preferable to use the same alcohols as those produced by the hydrolysis reaction to obtain the raw colloidal silica.
上記の変性剤(シランカップリング剤)の使用量は、前述のとおり、原料コロイダルシリカにおける固形分100質量部に対して、好ましくは0.1~10質量部であり、より好ましくは0.5~7質量部であり、さらに好ましくは0.8~5質量部である。 As mentioned above, the amount of the modifier (silane coupling agent) used is preferably 0.1 to 10 parts by mass, more preferably 0.5 to 7 parts by mass, and even more preferably 0.8 to 5 parts by mass, per 100 parts by mass of the solid content of the raw colloidal silica.
工程c1の反応については、例えば、コロイダルシリカに導入された官能基を分析することにより把握することもできる、例えば、パルスNMR(TD-NMR)を用いて、官能基の定性及び/又は定量分析を行うことができる。 The reaction in step c1 can also be understood, for example, by analyzing the functional groups introduced into the colloidal silica. For example, pulsed NMR (TD-NMR) can be used to perform qualitative and/or quantitative analysis of the functional groups.
(工程c2)
工程c2では、工程c1により原料コロイダルシリカの表面に導入された官能基を化学的な方法によりアニオン基に変換を行う。ここでは、メルカプト基及び/又はスルフィド基を有するシランカップリング剤により導入された該官能基を、酸化処理でスルホ基に変換する方法について説明する。
(Step c2)
In step c2, the functional groups introduced onto the surface of the raw colloidal silica in step c1 are converted into anionic groups by a chemical method. Here, a method for converting the functional groups introduced by a silane coupling agent having a mercapto group and/or a sulfide group into sulfo groups by oxidation treatment will be described.
メルカプト基及び/又はスルフィド基を酸化処理によりスルホ基に変換する方法としては、制限されないが、酸化剤を用いることが挙げられる。酸化剤としては、例えば、硝酸、過酸化水素、酸素、オゾン、有機過酸(過カルボン酸)、臭素、次亜塩素酸塩、過マンガン酸カリウム、クロム酸等が挙げられる。これらの中でも過酸化水素および有機過酸(過酢酸、過安息香酸類)が取り扱いや反応性(酸化の収率)などの面から好ましい。なお、反応で副生する物質が少ないことから、過酸化水素を用いることが最も好ましい。酸化剤の添加量は、変性剤(シランカップリング剤)に対して過剰量であってもよいが、残留する酸化剤の量を極力少なくすることが好ましく、酸化反応を十分なものとするとの理由から、シランカップリング剤1モルに対して、酸化剤を3~5モル使用することがより好ましい。酸化反応については、制限されないが、本願発明の目的を害さない範囲において、使用される酸化剤の好適な反応条件において行うことができる。例えば、室温以上であって反応させる反応液に使用される溶媒の沸点以下(例えば、100℃以下)で、3~5時間の範囲内で行うことが好ましい。 Methods for converting mercapto groups and/or sulfide groups to sulfo groups by oxidation treatment include, but are not limited to, the use of an oxidizing agent. Examples of oxidizing agents include nitric acid, hydrogen peroxide, oxygen, ozone, organic peracids (percarboxylic acids), bromine, hypochlorite, potassium permanganate, chromic acid, etc. Among these, hydrogen peroxide and organic peracids (peracetic acid, perbenzoic acids) are preferred in terms of handling and reactivity (oxidation yield). It is most preferred to use hydrogen peroxide because it produces fewer by-products in the reaction. The amount of oxidizing agent added may be in excess of the amount of the modifying agent (silane coupling agent), but it is preferable to minimize the amount of residual oxidizing agent, and it is more preferred to use 3 to 5 moles of oxidizing agent per mole of silane coupling agent in order to ensure a sufficient oxidation reaction. There are no limitations on the oxidation reaction, but it can be carried out under suitable reaction conditions for the oxidizing agent used, as long as it does not impair the purpose of the present invention. For example, it is preferable to carry out the reaction at a temperature above room temperature and below the boiling point of the solvent used in the reaction solution (e.g., below 100°C) for a period of 3 to 5 hours.
この工程c2により、表面にアニオン基(スルホ基)を有するコロイダルシリカを得ることができる。なお、工程c2においては、酸化処理を行った後に、それ以外の工程を含んでもよい。例えば、酸化剤を除去するための工程や、反応後の溶液のpHを調整するための工程などを含んでもよく、本願発明の目的を害さない範囲において、適宜選択して行うことができる。 By this step c2, colloidal silica having anionic groups (sulfo groups) on the surface can be obtained. Note that step c2 may include other steps after the oxidation treatment. For example, it may include a step for removing the oxidizing agent, a step for adjusting the pH of the solution after the reaction, etc., and these can be appropriately selected and performed within the scope that does not impair the object of the present invention.
(アミノ基を例としたカチオン変性処理の実施形態について)
アミノ基をコロイダルシリカの表面に形成させるためには、前述のとおり公知の方法で行うことができる。好ましくは、アミノ基を有する変性剤をコロイダルシリカの表面に化学的に結合させる処理を行う方法を挙げることができる。このうちの好ましい変性剤としては、前記したとおり、アミノ基を有するシランカップリング剤であり、代表的に以下で説明する。
(Embodiment of cationic modification treatment using amino group as an example)
To form amino groups on the surface of colloidal silica, known methods can be used as described above. A preferred method is to carry out a treatment in which a modifying agent having an amino group is chemically bonded to the surface of colloidal silica. As described above, a preferred modifying agent among these is a silane coupling agent having an amino group, which will be described below as a representative example.
この方法において、当該シランカップリング剤の存在下で、前記のように工程(a)及び工程(b)を経て固形分濃度等が濃度調整された原料コロイダルシリカを反応させる工程を含むようにすることが好ましい。このような工程についても前記した特開2005-162533号公報や特開2020-73445号公報などを参照して行うことができる。なお、本発明の目的を害しない限りは、それ以外の工程を適宜含ませてもよい。それ以外の工程としては、例えば、反応液の粘度を調整することや、pHの調整を行うことなどを挙げることができるが、分散安定性のために、カチオン変性処理を行う前にpHの調整を行うことが好ましい。この際に調整されるpHについては、好ましくは8~11、より好ましくはpHが8.5~10である。pHの調整方法は公知の化合物(pH調整剤)を用いる方法により行うことができるが、例えば、工程(a)で使用された加水分解触媒と同じ化合物をpH調整剤として用いて行うことが好ましい方法として挙げられる。 In this method, it is preferable to include a step of reacting the raw colloidal silica, the solid content concentration of which has been adjusted through steps (a) and (b) as described above, in the presence of the silane coupling agent. Such steps can also be performed with reference to the above-mentioned JP-A-2005-162533 and JP-A-2020-73445. Other steps may be included as appropriate as long as they do not impair the object of the present invention. Examples of other steps include adjusting the viscosity of the reaction solution and adjusting the pH, but for dispersion stability, it is preferable to adjust the pH before performing the cation modification treatment. The pH adjusted at this time is preferably 8 to 11, and more preferably 8.5 to 10. The pH can be adjusted by a method using a known compound (pH adjuster), but a preferred method is to use the same compound as the hydrolysis catalyst used in step (a) as the pH adjuster.
すなわち、工程(a)及び工程(b)を経て固形分濃度等が濃度調整された原料コロイダルシリカを、アミノ基を有するシランカップリング剤の存在下で反応させる。これにより、該シランカップリング剤が原料コロイダルシリカの表面に化学的に結合される。 In other words, the raw colloidal silica, whose solid content concentration has been adjusted through steps (a) and (b), is reacted in the presence of a silane coupling agent having an amino group. This causes the silane coupling agent to chemically bond to the surface of the raw colloidal silica.
この反応は、変性剤の使用適正温度の範囲内で行うことができ、制限されるものではないが、例えば、40℃以上であって、反応液(溶媒)の沸点以下の温度範囲で行うことができる。好ましくは、反応性の向上のために、温度50℃以上が好ましく、より好ましくは60℃以上であって、反応液(溶媒)の沸点以下としては好ましくは100℃以下で行われることがよい。反応時間も制限されないが、例えば、10分以上10時間以下で行われることが好ましく、1~8時間がより好ましい。 This reaction can be carried out within the range of temperatures suitable for use with the denaturant, and is not limited, but can be carried out at a temperature range of, for example, 40°C or higher and below the boiling point of the reaction liquid (solvent). To improve reactivity, it is preferable to carry out the reaction at a temperature of 50°C or higher, more preferably 60°C or higher, and preferably below the boiling point of the reaction liquid (solvent), 100°C or lower. There is also no limit to the reaction time, but it is preferable to carry out the reaction for, for example, 10 minutes to 10 hours, and more preferably 1 to 8 hours.
この反応においては、本発明の目的を害さない範囲内において、変性剤やシランカップリング剤の溶解性を向上するための溶媒を添加することができる。このような溶媒としては、親水性溶媒を挙げることができ、例えば、メタノール、エタノール、イソプロパノールなどのアルコール類などを挙げることができるがこれらに限定されない。原料コロイダルシリカを得る加水分解反応により生成されるアルコールと同じアルコール類を用いることがより好ましい。 In this reaction, a solvent for improving the solubility of the modifier or silane coupling agent can be added within a range that does not impair the object of the present invention. Such a solvent can be a hydrophilic solvent, for example, alcohols such as methanol, ethanol, isopropanol, etc., but is not limited to these. It is more preferable to use the same alcohols as those produced by the hydrolysis reaction to obtain the raw colloidal silica.
上記の変性剤(シランカップリング剤)の使用量は、前述のとおり、原料コロイダルシリカにおける固形分100質量部に対して、好ましくは0.1~1.5質量部であり、より好ましくは0.5~1.2質量部であり、さらに好ましくは0.6~1質量部である。 As mentioned above, the amount of the modifier (silane coupling agent) used is preferably 0.1 to 1.5 parts by mass, more preferably 0.5 to 1.2 parts by mass, and even more preferably 0.6 to 1 part by mass, per 100 parts by mass of the solid content of the raw colloidal silica.
この反応については、例えば、コロイダルシリカに導入された官能基を分析することにより把握することもできる、例えば、パルスNMR(TD-NMR)を用いて、官能基の定性及び/又は定量分析を行うことができる。 This reaction can also be understood, for example, by analyzing the functional groups introduced into the colloidal silica. For example, pulsed NMR (TD-NMR) can be used to perform qualitative and/or quantitative analysis of the functional groups.
さらに、分散安定性のために、カチオン変性処理を行った後においてもpHの調整を行うことが好ましい。この際に調整されるpHについても、好ましくは8~11、より好ましくはpHが8.5~10である。pHの調整方法は、前述のとおり公知の化合物(pH調整剤)を用いる方法により行うことができるが、例えば、工程(a)で使用された加水分解触媒と同じ化合物をpH調整剤として用いて行うことが好ましい方法として挙げられる。 Furthermore, for the sake of dispersion stability, it is preferable to adjust the pH even after the cationic modification treatment. The pH adjusted in this case is preferably 8 to 11, and more preferably 8.5 to 10. As described above, the pH can be adjusted by a method using a known compound (pH adjuster), but a preferred method is to use the same compound as the hydrolysis catalyst used in step (a) as the pH adjuster.
[工程(d)]
前記工程(c)を行った後には、工程(d)として、工程(c)後に得られるコロイダルシリカ中の残留有機溶媒を1質量%以下となるように濃縮する。好ましくは、残留有機溶媒を0.1質量%以下、より好ましくは0.05質量%以下である。すなわち、当該工程(d)を経て得られたコロイダルシリカが、残留有機溶媒が0.1質量%以下である分散媒に分散されているようにする。このような分散媒は、例えば後述の実施例で挙げられる実施形態のように、残留有機溶媒が0.1質量%以下である水溶液であって、実質的には水であることが好ましい実施形態である。なお、分散媒には、極微量のアルコール類、アルデヒド類、ケトン類、界面活性剤などが含まれることは排除されず、これらも同様に0.1質量%以下であることが好ましい。そして、残留有機溶媒をこの範囲に低下させることで、固形分濃度を所定の範囲に調整ないし高めることができるとともに、揮発性が高い残留有機溶媒である場合には、有機溶媒の揮発に伴うコロイダルシリカの濃度の変動を防止することができるため好ましい。また、本発明のコロイダルシリカを使用する際に、その用途先で使用される材料において残留有機溶媒の耐性を考慮する必要が無くなること等の優位な点も得られる。
[Step (d)]
After carrying out the step (c), in step (d), the residual organic solvent in the colloidal silica obtained after step (c) is concentrated to 1 mass % or less. Preferably, the residual organic solvent is 0.1 mass % or less, more preferably 0.05 mass % or less. That is, the colloidal silica obtained through the step (d) is dispersed in a dispersion medium having a residual organic solvent of 0.1 mass % or less. Such a dispersion medium is, for example, an aqueous solution having a residual organic solvent of 0.1 mass % or less, and is preferably substantially water, as in the embodiment described in the examples below. It is not excluded that the dispersion medium contains trace amounts of alcohols, aldehydes, ketones, surfactants, etc., and these are also preferably 0.1 mass % or less. By reducing the residual organic solvent to this range, the solid content concentration can be adjusted or increased to a predetermined range, and in the case of a highly volatile residual organic solvent, it is preferable because the concentration of colloidal silica can be prevented from fluctuating due to the evaporation of the organic solvent. In addition, when the colloidal silica of the present invention is used, there is an advantage that it is not necessary to consider the resistance of the material used in the application to residual organic solvents.
ここで、残留有機溶媒を除去して濃縮する方法は特に制限されず、公知の方法を用いることができる。例えば、コンデンサー付留出管を備えた機器を用いて加熱により残留有機溶媒を留出させる方法を挙げることができる。 Here, the method for removing and concentrating the residual organic solvent is not particularly limited, and any known method can be used. For example, there is a method in which the residual organic solvent is distilled by heating using an apparatus equipped with a distillation tube with a condenser.
この工程(d)においては、残留有機溶媒や水を除去する過程の濃縮により、その用途や目的などを勘案して、使用される固形分濃度にすることが可能である。本発明においては、前記した工程(a)~工程(c)を経て変性処理されたコロイダルシリカを得ており、この工程(d)において固形分濃度を比較的高くしても、実質的に凝集しないコロイダルシリカを得ることができる。このような本発明の特徴や、コロイダルシリカの用途や目的などを勘案すると、前述もしたように、変性処理工程(c)後のコロイダルシリカ中の固形分濃度を12質量%以上とすることが好ましく、より好ましくは15質量%以上、さらに好ましくは18質量%以上であり、とくに好ましくは19質量%以上である。固形分濃度の上限についても前述と同様であり、好ましくは50質量%以下、より好ましくは40質量%以下、さらに好ましくは30質量%以下とすることがよい。 In this step (d), the solid content concentration can be adjusted to a level suitable for use, taking into account the intended use and purpose, by concentrating the process of removing the residual organic solvent and water. In the present invention, modified colloidal silica is obtained through the above-mentioned steps (a) to (c), and even if the solid content concentration is relatively high in this step (d), colloidal silica that does not substantially aggregate can be obtained. Considering such features of the present invention and the intended use and purpose of colloidal silica, as described above, the solid content concentration in the colloidal silica after the modification step (c) is preferably 12% by mass or more, more preferably 15% by mass or more, even more preferably 18% by mass or more, and particularly preferably 19% by mass or more. The upper limit of the solid content concentration is the same as described above, and is preferably 50% by mass or less, more preferably 40% by mass or less, and even more preferably 30% by mass or less.
前記の工程(a)~工程(d)を行うことで、本発明のコロイダルシリカを製造することができるが、工程(d)の後においても、本発明の目的を害さない限りは、それ以外の工程を適宜含ませてもよい。例えば、工程(d)後のコロイダルシリカに対して、分散安定化させることが好ましい。分散安定化処理は公知の処理を用いることができる。 The colloidal silica of the present invention can be produced by carrying out the above steps (a) to (d), but other steps may be included as appropriate even after step (d) as long as they do not impair the object of the present invention. For example, it is preferable to subject the colloidal silica after step (d) to dispersion stabilization. Publicly known treatments can be used for the dispersion stabilization treatment.
本発明の方法により分散安定化処理されたコロイダルシリカは、通常1数週間以上、更には数年に亘って優れた分散安定性が発揮され、二層分離現象が発生することがない。 Colloidal silica that has been subjected to dispersion stabilization treatment using the method of the present invention exhibits excellent dispersion stability for a period of at least one week, and even for a period of several years, and does not undergo two-layer separation.
本発明においては、前記のようにして、BET径が12nm以下であるような小粒子径のコロイダルシリカを得ることができるが、従来の方法でもあるとおり、得られたコロイダルシリカを種粒子として用い、この種粒子と前記加水分解触媒とを含む反応液に、シリカ源を供給及び反応させて、粒子径を成長させるような方法も排除はされない。 In the present invention, colloidal silica with a small particle size, such as a BET diameter of 12 nm or less, can be obtained as described above, but as in conventional methods, a method is also possible in which the obtained colloidal silica is used as seed particles, and a silica source is supplied to and reacted with a reaction liquid containing the seed particles and the hydrolysis catalyst to grow the particle size.
以下、実施例及び比較例に基づいて、本発明の好適な実施の形態を具体的に説明する。 Below, we will explain in detail the preferred embodiment of the present invention based on examples and comparative examples.
[実施例1]
攪拌機、温度計、コンデンサー付留出管及びオルガノシリケート導入管を備えた5リットル(L)のガラス容器中に、金属不純物含有量0.1ppb以下の純水3200gと、金属不純物含有量10ppb以下のトリエタノールアミン(沸点(bp):361℃)3.42gとを仕込み、マントルヒーターを用いて反応容器内液温を60℃に保ちながら、金属不純物含有量10ppb以下のテトラメチルシリケート(多摩化学工業株式会社製)706gを攪拌下に90分間かけて連続的に供給した。得られた原料コロイダルシリカについて各種分析を行い、結果を表1の<1>に示した。
[Example 1]
In a 5 liter (L) glass vessel equipped with a stirrer, a thermometer, a distillation tube with a condenser, and an organosilicate inlet tube, 3200 g of pure water having a metal impurity content of 0.1 ppb or less and 3.42 g of triethanolamine (boiling point (bp): 361°C) having a metal impurity content of 10 ppb or less were charged, and while the liquid temperature in the reaction vessel was kept at 60°C using a mantle heater, 706 g of tetramethylsilicate (manufactured by Tama Chemicals Co., Ltd.) having a metal impurity content of 10 ppb or less was continuously fed under stirring over a period of 90 minutes. The obtained raw colloidal silica was subjected to various analyses, and the results are shown in <1> of Table 1.
続いて、得られた反応物(コロイダルシリカ、固形分濃度6.45質量%、メタノール濃度15質量%)の3900gを5Lガラス製容器に加え、固形分濃度及びメタノール濃度を変更する操作は行わなかった。次いで、80℃に保ちながら、3-メルカプトプロピルトリメトキシシラン13.1gをメタノール117.9gに溶解させたものを6時間かけて連続的に供給した。供給した後、更に30質量%の過酸化水素水22.7gを、80℃を保ちながら、4時間かけて連続的に供給し、原料コロイダルシリカに変性処理を行った。 Next, 3900 g of the resulting reaction product (colloidal silica, solids concentration 6.45% by mass, methanol concentration 15% by mass) was added to a 5 L glass container, and no operations were performed to change the solids concentration or methanol concentration. Next, while maintaining the temperature at 80°C, 13.1 g of 3-mercaptopropyltrimethoxysilane dissolved in 117.9 g of methanol was continuously fed over a period of 6 hours. After feeding, 22.7 g of 30% by mass hydrogen peroxide solution was further continuously fed over a period of 4 hours while maintaining the temperature at 80°C, and the raw colloidal silica was modified.
過酸化水素水を供給した後、一旦、反応容器内の温度を40℃まで下げ、真空ポンプで系内を減圧にし、その後加熱を再開し、反応容器内の反応混合物を更に52~68℃に加熱し、生成したメタノールをコンデンサー付留出管から留出温度32~67℃で留出させ、更に純水250gを添加しながら、水とメタノールを留去して、固形分濃度が約20質量%のコロイダルシリカを得た。得られたコロイダルシリカの残留有機溶媒は0.1質量%であった。
得られたコロイダルシリカの各種分析を行い、結果は表1の<2>に示した。更に得られたコロイダルシリカを60℃で7日間保存し、得られたコロイダルシリカの各種分析を行うことにより、とくにDLS測定の結果から、実質的に凝集が発生していないことを確認した。結果を表1の<3>に示した。
After the hydrogen peroxide solution was supplied, the temperature in the reaction vessel was temporarily lowered to 40°C, the pressure in the system was reduced with a vacuum pump, and then heating was resumed, the reaction mixture in the reaction vessel was further heated to 52 to 68°C, and the generated methanol was distilled from a distillation tube equipped with a condenser at a distillation temperature of 32 to 67°C, and 250 g of pure water was further added while distilling off the water and methanol, thereby obtaining colloidal silica with a solid concentration of about 20% by mass. The residual organic solvent in the obtained colloidal silica was 0.1% by mass.
The obtained colloidal silica was subjected to various analyses, and the results are shown in <2> of Table 1. Furthermore, the obtained colloidal silica was stored at 60° C. for 7 days, and various analyses of the obtained colloidal silica were performed. It was confirmed, particularly from the results of DLS measurement, that substantially no aggregation had occurred. The results are shown in <3> of Table 1.
[実施例2]
攪拌機、温度計、コンデンサー付留出管及びオルガノシリケート導入管を備えた5Lのガラス容器中に、金属不純物含有量0.1ppb以下の純水3374gと、金属不純物含有量10ppb以下のトリエタノールアミン(bp:361℃)3.608gとを仕込み、マントルヒーターを用いて反応容器内液温を70℃に保ちながら、金属不純物含有量10ppb以下のテトラメチルシリケート(多摩化学工業株式会社製)1122gを攪拌下に270分間かけて連続的に供給した。得られた原料コロイダルシリカの各種分析を行い、結果は表1の<4>に示した。
[Example 2]
In a 5 L glass vessel equipped with a stirrer, a thermometer, a distillation tube with a condenser, and an organosilicate inlet tube, 3374 g of pure water containing 0.1 ppb or less of metal impurities and 3.608 g of triethanolamine (bp: 361° C.) containing 10 ppb or less of metal impurities were charged, and while the liquid temperature in the reaction vessel was kept at 70° C. using a mantle heater, 1122 g of tetramethylsilicate containing 10 ppb or less of metal impurities (manufactured by Tama Chemicals Co., Ltd.) was continuously fed with stirring over a period of 270 minutes. Various analyses of the obtained raw colloidal silica were carried out, and the results are shown in <4> of Table 1.
続いて、得られた反応物(コロイダルシリカ、固形分濃度10.38質量%、メタノール濃度20質量%)の2000gを2Lガラス製容器に加え、固形分濃度及びメタノール濃度を変更する操作は行わなかった。次いで、80℃に保ちながら、3-メルカプトプロピルトリメトキシシラン6.71gをメタノール60.35gに溶解させたものを6時間かけて連続的に供給した。供給した後、更に30質量%の過酸化水素水11.63gを、80℃を保ちながら、4時間かけて連続的に供給し、原料コロイダルシリカに変性処理を行った。 Next, 2000 g of the resulting reaction product (colloidal silica, solids concentration 10.38% by mass, methanol concentration 20% by mass) was added to a 2 L glass container, and no operations were performed to change the solids concentration or methanol concentration. Next, while maintaining the temperature at 80°C, 6.71 g of 3-mercaptopropyltrimethoxysilane dissolved in 60.35 g of methanol was continuously fed over a period of 6 hours. After feeding, 11.63 g of 30% by mass hydrogen peroxide solution was further continuously fed over a period of 4 hours while maintaining the temperature at 80°C, and the raw colloidal silica was modified.
過酸化水素水を供給した後、一旦、反応容器内の温度を40℃まで下げ、真空ポンプで系内を減圧にし、その後加熱を再開し、反応容器内の反応混合物を更に52~68℃に加熱し、生成したメタノールをコンデンサー付留出管から留出温度32~67℃で留出させ、更に純水320gを添加しながら、水とメタノールを留去して、固形分濃度が約20質量%のコロイダルシリカを得た。得られたコロイダルシリカの各種分析を行い、結果は表1の<5>に示した。更に、得られたコロイダルシリカを60℃で7日間保存し、得られたコロイダルシリカの各種分析を行うことにより、とくにDLS測定の結果から、実質的に凝集が発生していないことを確認した。結果は表1の<6>に示した。 After the hydrogen peroxide solution was supplied, the temperature in the reaction vessel was temporarily lowered to 40°C, and the pressure in the system was reduced using a vacuum pump. Heating was then resumed, and the reaction mixture in the reaction vessel was further heated to 52-68°C. The methanol produced was distilled from the distillation tube with a condenser at a distillation temperature of 32-67°C. 320 g of pure water was then added while distilling off the water and methanol, yielding colloidal silica with a solid concentration of approximately 20% by mass. Various analyses were performed on the resulting colloidal silica, and the results are shown in <5> of Table 1. Furthermore, the resulting colloidal silica was stored at 60°C for 7 days, and various analyses were performed on the resulting colloidal silica, confirming that essentially no aggregation had occurred, particularly from the results of DLS measurements. The results are shown in <6> of Table 1.
[実施例3]
攪拌機、温度計、コンデンサー付留出管及びオルガノシリケート導入管を備えた5Lのガラス容器中に、金属不純物含有量0.1ppb以下の純水3463gと、金属不純物含有量10ppb以下のトリエタノールアミン(bp:361℃)0.336gとを仕込み、マントルヒーターを用いて反応容器内液温を70℃に保ちながら、金属不純物含有量10ppb以下のテトラメチルシリケート(多摩化学工業株式会社製)911.6gを攪拌下に90分間かけて連続的に供給した。得られた原料コロイダルシリカの各種分析を行い、結果は表1の<7>に示した。
[Example 3]
In a 5 L glass vessel equipped with a stirrer, a thermometer, a distillation tube with a condenser, and an organosilicate inlet tube, 3463 g of pure water containing 0.1 ppb or less of metal impurities and 0.336 g of triethanolamine (bp: 361°C) containing 10 ppb or less of metal impurities were charged, and while the liquid temperature in the reaction vessel was kept at 70°C using a mantle heater, 911.6 g of tetramethylsilicate (manufactured by Tama Chemicals Co., Ltd.) containing 10 ppb or less of metal impurities was continuously fed under stirring over a period of 90 minutes. Various analyses of the obtained raw colloidal silica were carried out, and the results are shown in <7> of Table 1.
続いて、得られた反応物(コロイダルシリカ、固形分濃度8.82質量%、メタノール濃度17質量%)の4200gを5Lガラス製容器に加え、固形分濃度及びメタノール濃度を変更する操作は行わなかった。次いで、80℃に保ちながら、3-メルカプトプロピルトリメトキシシラン11.98gをメタノール107.8gに溶解させたものを6時間かけて連続的に供給した。供給した後、更に30質量%の過酸化水素水20.77gを、80℃を保ちながら、4時間かけて連続的に供給し、原料コロイダルシリカに変性処理を行った。 Next, 4,200 g of the resulting reaction product (colloidal silica, solids concentration 8.82% by mass, methanol concentration 17% by mass) was added to a 5 L glass container, and no operations were performed to change the solids concentration or methanol concentration. Next, while maintaining the temperature at 80°C, 11.98 g of 3-mercaptopropyltrimethoxysilane dissolved in 107.8 g of methanol was continuously fed over a period of 6 hours. After feeding, 20.77 g of 30% by mass hydrogen peroxide solution was further continuously fed over a period of 4 hours while maintaining the temperature at 80°C, and the raw colloidal silica was modified.
過酸化水素水を供給した後、一旦、反応容器内の温度を40℃まで下げ、真空ポンプで系内を減圧にし、その後加熱を再開し、反応容器内の反応混合物を更に52~68℃に加熱し、生成したメタノールをコンデンサー付留出管から留出温度32~67℃で留出させ、更に純水700gを添加しながら、水とメタノールを留去して、固形分濃度が約20質量%のコロイダルシリカを得た。得られたコロイダルシリカの各種分析を行い、結果は表1の<8>に示した。更に、得られたコロイダルシリカを60℃で7日間保存し、得られたコロイダルシリカの各種分析を行うことにより、とくにDLS測定の結果から、凝集が発生していないことを確認した。結果は表1の<9>に示した。 After the hydrogen peroxide solution was supplied, the temperature in the reaction vessel was temporarily lowered to 40°C, and the pressure in the system was reduced using a vacuum pump. Heating was then resumed, and the reaction mixture in the reaction vessel was further heated to 52-68°C. The resulting methanol was distilled from the condenser-equipped distillation tube at a distillation temperature of 32-67°C. 700 g of pure water was then added while distilling off the water and methanol, yielding colloidal silica with a solids concentration of approximately 20% by mass. Various analyses were performed on the resulting colloidal silica, and the results are shown in <8> of Table 1. Furthermore, the resulting colloidal silica was stored at 60°C for 7 days, and various analyses were performed on the resulting colloidal silica, confirming that no aggregation had occurred, particularly from the results of DLS measurements. The results are shown in <9> of Table 1.
[比較例1]
攪拌機、温度計、コンデンサー付留出管及びオルガノシリケート導入管を備えた5Lのガラス容器中に、金属不純物含有量0.1ppb以下の純水3200gと金属不純物含有量10ppb以下のトリエタノールアミン(bp:361℃)3.42gとを仕込み、マントルヒーターを用いて反応容器内液温を60℃に保ちながら、金属不純物含有量10ppb以下のテトラメチルシリケート(多摩化学工業株式会社製)706gを攪拌下に90分間かけて連続的に供給した。得られた原料コロイダルシリカの各種分析を行い、結果は表2の<10>に示した。
[Comparative Example 1]
In a 5 L glass vessel equipped with a stirrer, a thermometer, a distillation tube with a condenser, and an organosilicate inlet tube, 3200 g of pure water containing 0.1 ppb or less of metal impurities and 3.42 g of triethanolamine (bp: 361° C.) containing 10 ppb or less of metal impurities were charged, and while maintaining the liquid temperature in the reaction vessel at 60° C. using a mantle heater, 706 g of tetramethylsilicate (manufactured by Tama Chemicals Co., Ltd.) containing 10 ppb or less of metal impurities was continuously fed with stirring over a period of 90 minutes. Various analyses of the obtained raw colloidal silica were carried out, and the results are shown in <10> of Table 2.
続いて、得られた反応物(コロイダルシリカ)の3900gを5Lガラス製容器に加え、上記実施例1~3とは異なり、この時点で52℃~72℃に加熱し、生成したメタノールをコンデンサー付留出管から留出温度32~67℃で留出させ、更に純水1800gを添加しながら、水とメタノールを留去させた。 Next, 3,900 g of the resulting reaction product (colloidal silica) was added to a 5 L glass container, and unlike in Examples 1 to 3 above, it was heated to 52°C to 72°C at this point, and the resulting methanol was distilled from a distillation tube with a condenser at a distillation temperature of 32 to 67°C. Furthermore, 1,800 g of pure water was added while distilling off the water and methanol.
水とメタノールを留去させた反応物(コロイダルシリカ、固形分濃度6.45質量%、メタノール濃度0.1質量%)3900gを5Lガラス製容器に加え、固形分濃度を変更する操作は行わなかった。次いで、100℃に保ちながら、3-メルカプトプロピルトリメトキシシラン13.7gをメタノール123.6gに溶解させたものを6時間かけて連続的に供給した。供給した後、更に30質量%の過酸化水素水23.8gを、100℃に保ちながら4時間かけて連続的に供給し、原料コロイダルシリカに変性処理を行った。 3,900 g of the reaction product from which water and methanol had been distilled off (colloidal silica, solids concentration 6.45% by mass, methanol concentration 0.1% by mass) was added to a 5 L glass vessel, and no operation was performed to change the solids concentration. Next, while maintaining the temperature at 100°C, 13.7 g of 3-mercaptopropyltrimethoxysilane dissolved in 123.6 g of methanol was continuously fed over a period of 6 hours. After feeding, 23.8 g of 30% by mass hydrogen peroxide was further continuously fed over a period of 4 hours while maintaining the temperature at 100°C, and the raw colloidal silica was modified.
過酸化水素水を供給した後、一旦、反応容器内の温度を40℃まで下げ、真空ポンプで系内を減圧にし、その後加熱を再開し、反応容器内の反応混合物を更に52~68℃に加熱し、固形分濃度が約20質量%のコロイダルシリカを得た。得られたコロイダルシリカの各種分析を行い、結果は表2の<11>に示した。分析結果より、とくにDLS測定の結果から、DLS測定によるキュムラント平均径の変化が20%超過であって、凝集していることが確認された。更に得られたコロイダルシリカを60℃で7日間保存し、得られたコロイダルシリカの各種分析を行った。結果は表2の<12>に示した。結果<11>の時点でコロイダルシリカが凝集しているため、この結果<12>においても、DLSの結果は結果<11>と同程度であり、結果<10>よりも高値であった。 After the hydrogen peroxide solution was supplied, the temperature in the reaction vessel was temporarily lowered to 40°C, the pressure in the system was reduced using a vacuum pump, and then heating was resumed. The reaction mixture in the reaction vessel was further heated to 52-68°C, and colloidal silica with a solid content of approximately 20% by mass was obtained. Various analyses of the obtained colloidal silica were carried out, and the results are shown in <11> of Table 2. From the analysis results, particularly the DLS measurement results, the change in the cumulant mean diameter by DLS measurement exceeded 20%, confirming the presence of aggregation. The obtained colloidal silica was further stored at 60°C for 7 days, and various analyses of the obtained colloidal silica were carried out. The results are shown in <12> of Table 2. Because the colloidal silica was aggregated at the time of result <11>, the DLS result in this result <12> was also similar to result <11> and higher than result <10>.
[比較例2]
攪拌機、温度計、コンデンサー付留出管及びオルガノシリケート導入管を備えた5Lのガラス容器中に、金属不純物含有量0.1ppb以下の純水3200gと金属不純物含有量10ppb以下のトリエタノールアミン(bp:361℃)3.42gとを仕込み、マントルヒーターを用いて反応容器内液温を60℃に保ちながら、金属不純物含有量10ppb以下のテトラメチルシリケート(多摩化学工業株式会社製)706gを攪拌下に90分間かけて連続的に供給した。得られたコロイダルシリカの各種分析を行い、結果は表2の<13>に示した。
[Comparative Example 2]
In a 5 L glass vessel equipped with a stirrer, a thermometer, a distillation tube with a condenser, and an organosilicate inlet tube, 3200 g of pure water containing 0.1 ppb or less of metal impurities and 3.42 g of triethanolamine (bp: 361° C.) containing 10 ppb or less of metal impurities were charged, and while keeping the liquid temperature in the reaction vessel at 60° C. using a mantle heater, 706 g of tetramethylsilicate containing 10 ppb or less of metal impurities (manufactured by Tama Chemicals Co., Ltd.) was continuously fed with stirring over a period of 90 minutes. Various analyses of the obtained colloidal silica were carried out, and the results are shown in <13> of Table 2.
続いて、得られた反応物(コロイダルシリカ、固形分濃度6.01質量%、メタノール濃度15質量%)の4500gを3Lガラス製容器に加え、固形分濃度及びメタノール濃度を変更する操作は行わなかった。そして、変性処理工程を行うことなく、52℃~72℃で加熱し、生成したメタノールをコンデンサー付留出管から留出温度32~67℃で留出させ、更に純水を添加しながら、水とメタノールを留去させたところ、固形分濃度が16質量%に到達した時点で凝集ゲル化した。凝集ゲル化の様子については、図3に示した。 Next, 4,500 g of the resulting reaction product (colloidal silica, solids concentration 6.01% by mass, methanol concentration 15% by mass) was added to a 3 L glass container, and no operations were performed to change the solids concentration or methanol concentration. Then, without carrying out a modification treatment process, it was heated at 52°C to 72°C, and the generated methanol was distilled from a distillation tube with a condenser at a distillation temperature of 32°C to 67°C. Pure water was further added while distilling off the water and methanol, and when the solids concentration reached 16% by mass, coagulation and gelation occurred. The state of coagulation and gelation is shown in Figure 3.
[比較例3]
攪拌機、温度計、コンデンサー付留出管及びオルガノシリケート導入管を備えた5Lのガラス容器中に、金属不純物含有量0.1ppb以下の純水3463gと金属不純物含有量10ppb以下のトリエタノールアミン(bp:361℃)0.336gとを仕込み、マントルヒーターを用いて反応容器内液温を80℃に保ちながら、金属不純物含有量10ppb以下のテトラメチルシリケート(多摩化学工業株式会社製)1136gを攪拌下に480分間かけて連続的に供給した。得られたコロイダルシリカの各種分析を行い、結果は表2の<14>に示した。この比較例3では、シリカ源の添加時の温度に起因して、BET径が12nm超過であった。
[Comparative Example 3]
In a 5L glass vessel equipped with a stirrer, a thermometer, a distillation tube with a condenser, and an organosilicate inlet tube, 3463 g of pure water containing 0.1 ppb or less of metal impurities and 0.336 g of triethanolamine (bp: 361°C) containing 10 ppb or less of metal impurities were charged, and while the liquid temperature in the reaction vessel was kept at 80°C using a mantle heater, 1136 g of tetramethylsilicate (manufactured by Tama Chemicals Co., Ltd.) containing 10 ppb or less of metal impurities was continuously fed under stirring for 480 minutes. Various analyses of the obtained colloidal silica were carried out, and the results are shown in <14> of Table 2. In this Comparative Example 3, the BET diameter exceeded 12 nm due to the temperature at the time of adding the silica source.
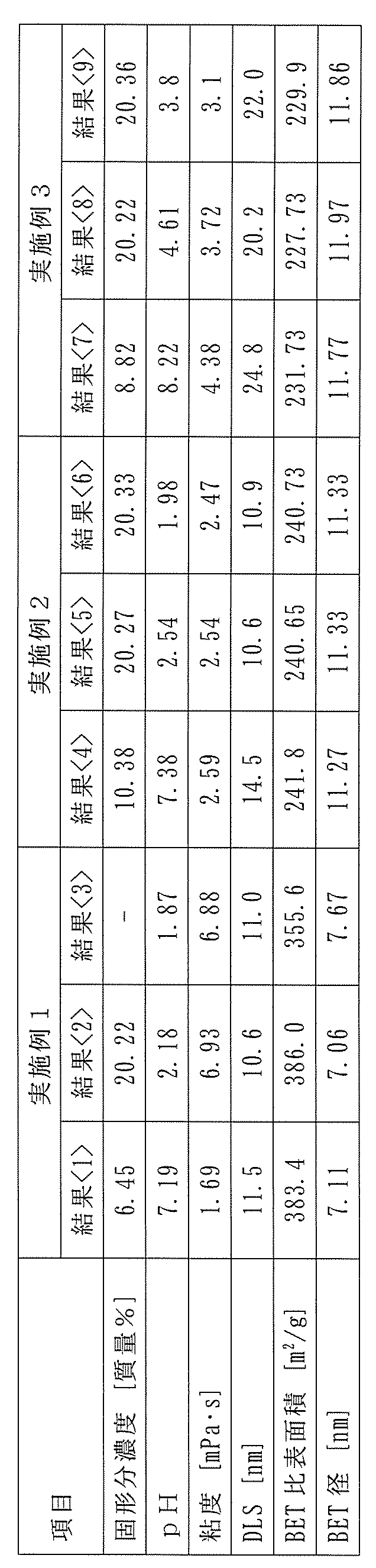
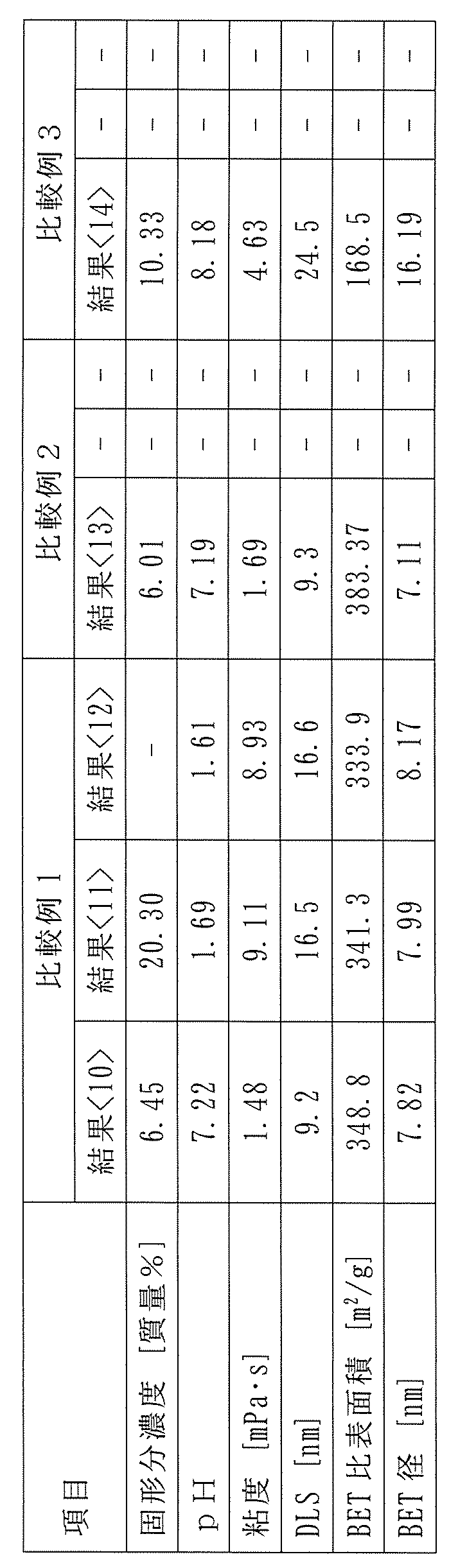
[実施例4]
撹拌機、温度計、コンデンサー付留出管及びオルガノシリケート導入管を備えた5Lガラス容器中に、金属不純物含有量0.1ppb以下の純水3889gと、金属不純物含有量10ppb以下のトリエタノールアミン(bp:361℃)3.42gとを仕込み、マントルヒーターを用いて反応容器内液温を60℃に保ちながら、金属不純物含有量10ppb以下のテトラメチルシリケート(多摩化学工業株式会社製)707gを撹拌下に90分間かけて連続的に供給した。得られた原料コロイダルシリカについて各種分析を行い、結果を表3の<15>に示した。
[Example 4]
In a 5 L glass vessel equipped with a stirrer, a thermometer, a distillation tube with a condenser, and an organosilicate inlet tube, 3889 g of pure water containing 0.1 ppb or less of metal impurities and 3.42 g of triethanolamine (bp: 361°C) containing 10 ppb or less of metal impurities were charged, and while the temperature of the liquid in the reaction vessel was kept at 60°C using a mantle heater, 707 g of tetramethylsilicate containing 10 ppb or less of metal impurities (manufactured by Tama Chemicals Co., Ltd.) was continuously fed with stirring over a period of 90 minutes. The obtained raw colloidal silica was subjected to various analyses, and the results are shown in <15> of Table 3.
続いて、得られた反応物(コロイダルシリカ、固形分濃度6.39質量%、メタノール濃度13質量%)の3800gと金属不純物含有量10ppb以下のトリエタノールアミン(bp:361℃)20gとを5Lガラス製容器に加え、固形分濃度及びメタノール濃度を変更する操作は行わなかった。このときのコロイダルシリカのpHは8.9であった。次いで、80℃に保ちながら、変性剤として、3-アミノプロピルトリメトキシシラン1.71gをメタノール50gに溶解させたものを6時間かけて連続的に供給し、原料コロイダルシリカに変性処理を行った。 Next, 3,800 g of the resulting reaction product (colloidal silica, solids concentration 6.39% by mass, methanol concentration 13% by mass) and 20 g of triethanolamine (bp: 361°C) with a metal impurity content of 10 ppb or less were added to a 5 L glass container, and no operation was performed to change the solids concentration or methanol concentration. The pH of the colloidal silica at this time was 8.9. Next, while maintaining the temperature at 80°C, a modifying agent obtained by dissolving 1.71 g of 3-aminopropyltrimethoxysilane in 50 g of methanol was continuously supplied over a period of 6 hours to modify the raw colloidal silica.
変性剤を供給した後、一旦、反応容器内の温度を40℃まで下げ、金属不純物含有量10ppb以下のトリエタノールアミン(bp:361℃)20gを加え、真空ポンプで系内を減圧にし、その後加熱を再開し、反応容器内の反応混合物を更に52~68℃に加熱し、生成したメタノールをコンデンサー付留出管から留出温度32~67℃で留出させ、さらに純水670gを添加しながら、水とメタノールを留去して、固形分濃度が約20質量%のコロイダルシリカを得た。
得られたコロイダルシリカの各種分析を行い、結果は表3の<16>に示した。さらに得られたコロイダルシリカを60℃で7日間保存し、得られたコロイダルシリカの各種分析を行うことにより、とくにDLS測定の結果から、実質的に凝集が発生していないことを確認した。結果を表3の<17>に示した。
After the modifier was supplied, the temperature in the reaction vessel was temporarily lowered to 40° C., 20 g of triethanolamine (bp: 361° C.) having a metal impurity content of 10 ppb or less was added, the pressure in the system was reduced using a vacuum pump, and then heating was resumed. The reaction mixture in the reaction vessel was further heated to 52 to 68° C., and the produced methanol was distilled from the distillation tube equipped with a condenser at a distillation temperature of 32 to 67° C., and 670 g of pure water was further added while distilling off the water and methanol, thereby obtaining colloidal silica with a solid content concentration of about 20 mass%.
The obtained colloidal silica was subjected to various analyses, and the results are shown in <16> of Table 3. Furthermore, the obtained colloidal silica was stored at 60°C for 7 days, and various analyses of the obtained colloidal silica were performed. It was confirmed, particularly from the results of DLS measurement, that substantially no aggregation had occurred. The results are shown in <17> of Table 3.
[実施例5]
撹拌機、温度計、コンデンサー付留出管及びオルガノシリケート導入管を備えた5Lガラス容器中に、金属不純物含有量0.1ppb以下の純水3890gと、金属不純物含有量10ppb以下のトリエタノールアミン(bp:361℃)3.42gとを仕込み、マントルヒーターを用いて反応容器内液温を60℃に保ちながら、金属不純物含有量10ppb以下のテトラメチルシリケート(多摩化学工業株式会社製)707gを撹拌下に90分間かけて連続的に供給した。得られた原料コロイダルシリカについて各種分析を行い、結果を表3の<18>に示した。
[Example 5]
In a 5 L glass vessel equipped with a stirrer, a thermometer, a distillation tube with a condenser, and an organosilicate inlet tube, 3890 g of pure water containing 0.1 ppb or less of metal impurities and 3.42 g of triethanolamine (bp: 361°C) containing 10 ppb or less of metal impurities were charged, and while the temperature of the liquid in the reaction vessel was kept at 60°C using a mantle heater, 707 g of tetramethylsilicate containing 10 ppb or less of metal impurities (manufactured by Tama Chemicals Co., Ltd.) was continuously fed with stirring over a period of 90 minutes. The obtained raw colloidal silica was subjected to various analyses, and the results are shown in <18> of Table 3.
続いて、得られた反応物(コロイダルシリカ、固形分濃度6.41質量%、メタノール濃度13質量%)の4001gと金属不純物含有量10ppb以下のトリエタノールアミン(bp:361℃)20gとを5Lガラス製容器に加え、固形分濃度及びメタノール濃度を変更する操作は行わなかった。このときのコロイダルシリカのpHは9.0であった。次いで、80℃に保ちながら、変性剤として、3-アミノプロピルトリメトキシシラン1.83gをメタノール54gに溶解させたものを6時間かけて連続的に供給し、原料コロイダルシリカに変性処理を行った。 Next, 4001 g of the resulting reaction product (colloidal silica, solids concentration 6.41% by mass, methanol concentration 13% by mass) and 20 g of triethanolamine (bp: 361°C) with a metal impurity content of 10 ppb or less were added to a 5 L glass container, and no operation was performed to change the solids concentration or methanol concentration. The pH of the colloidal silica at this time was 9.0. Next, while maintaining the temperature at 80°C, a modifying agent obtained by dissolving 1.83 g of 3-aminopropyltrimethoxysilane in 54 g of methanol was continuously supplied over a period of 6 hours to modify the raw colloidal silica.
変性剤を供給した後、反応混合物うちの1975gに対し、反応容器内の温度を40℃まで下げ、金属不純物含有量10ppb以下のトリエタノールアミン(bp:361℃)10gを加え、真空ポンプで系内を減圧にし、その後加熱を再開し、反応容器内の反応混合物を更に52~68℃に加熱し、生成したメタノールをコンデンサー付留出管から留出温度32~67℃で留出させ、さらに純水340gを添加しながら、水とメタノールを留去して、固形分濃度が約20質量%のコロイダルシリカを得た。
得られたコロイダルシリカの各種分析を行い、結果は表3の<19>に示した。さらに得られたコロイダルシリカを60℃で7日間保存し、得られたコロイダルシリカの各種分析を行うことにより、とくにDLS測定の結果から、実質的に凝集が発生していないことを確認した。結果を表3の<20>に示した。
After the modifier was supplied, the temperature in the reaction vessel was lowered to 40° C., 10 g of triethanolamine (bp: 361° C.) having a metal impurity content of 10 ppb or less was added to 1,975 g of the reaction mixture, the pressure in the system was reduced with a vacuum pump, heating was then resumed, and the reaction mixture in the reaction vessel was further heated to 52 to 68° C., and the produced methanol was distilled from a distillation tube equipped with a condenser at a distillation temperature of 32 to 67° C., and 340 g of pure water was added while distilling off the water and methanol, thereby obtaining colloidal silica with a solid content concentration of about 20 mass%.
The obtained colloidal silica was subjected to various analyses, and the results are shown in <19> of Table 3. Furthermore, the obtained colloidal silica was stored at 60°C for 7 days, and various analyses of the obtained colloidal silica were performed. It was confirmed, particularly from the results of DLS measurement, that substantially no aggregation had occurred. The results are shown in <20> of Table 3.
[実施例6]
撹拌機、温度計、コンデンサー付留出管及びオルガノシリケート導入管を備えた5Lガラス容器中に、金属不純物含有量0.1ppb以下の純水3890gと、金属不純物含有量10ppb以下のトリエタノールアミン(bp:361℃)3.43gとを仕込み、マントルヒーターを用いて反応容器内液温を60℃に保ちながら、金属不純物含有量10ppb以下のテトラメチルシリケート(多摩化学工業株式会社製)707gを撹拌下に90分間かけて連続的に供給した。得られた原料コロイダルシリカについて各種分析を行い、結果を表3の<21>に示した。
[Example 6]
In a 5 L glass vessel equipped with a stirrer, a thermometer, a distillation tube with a condenser, and an organosilicate inlet tube, 3890 g of pure water containing 0.1 ppb or less of metal impurities and 3.43 g of triethanolamine (bp: 361°C) containing 10 ppb or less of metal impurities were charged, and while the liquid temperature in the reaction vessel was kept at 60°C using a mantle heater, 707 g of tetramethylsilicate containing 10 ppb or less of metal impurities (manufactured by Tama Chemicals Co., Ltd.) was continuously fed with stirring over a period of 90 minutes. The obtained raw colloidal silica was subjected to various analyses, and the results are shown in <21> of Table 3.
続いて、得られた反応物(コロイダルシリカ、固形分濃度6.47質量%、メタノール濃度13質量%)の2350gと金属不純物含有量10ppb以下のトリエタノールアミン(bp:361℃)12gとを5Lガラス製容器に加え、固形分濃度及びメタノール濃度を変更する操作は行わなかった。このときのコロイダルシリカのpHは8.9であった。次いで、80℃に保ちながら、変性剤として、3-アミノプロピルトリメトキシシラン1.08gをメタノール30gに溶解させたものを6時間かけて連続的に供給し、原料コロイダルシリカに変性処理を行った。 Next, 2350 g of the resulting reaction product (colloidal silica, solids concentration 6.47% by mass, methanol concentration 13% by mass) and 12 g of triethanolamine (bp: 361°C) with a metal impurity content of 10 ppb or less were added to a 5 L glass container, and no operation was performed to change the solids concentration or methanol concentration. The pH of the colloidal silica at this time was 8.9. Next, while maintaining the temperature at 80°C, a modifying agent obtained by dissolving 1.08 g of 3-aminopropyltrimethoxysilane in 30 g of methanol was continuously supplied over a period of 6 hours to modify the raw colloidal silica.
変性剤を供給した後、一旦、反応容器内の温度を40℃まで下げ、金属不純物含有量10ppb以下のトリエタノールアミン(bp:361℃)12gを加え、真空ポンプで系内を減圧にし、その後加熱を再開し、反応容器内の反応混合物を更に52~68℃に加熱し、生成したメタノールをコンデンサー付留出管から留出温度32~67℃で留出させ、さらに純水351gを添加しながら、水とメタノールを留去して、固形分濃度が約20質量%のコロイダルシリカを得た。
得られたコロイダルシリカの各種分析を行い、結果は表3の<22>に示した。さらに得られたコロイダルシリカを60℃で7日間保存し、得られたコロイダルシリカの各種分析を行うことにより、とくにDLS測定の結果から、実質的に凝集が発生していないことを確認した。結果を表3の<23>に示した。
After the modifying agent was supplied, the temperature in the reaction vessel was temporarily lowered to 40° C., 12 g of triethanolamine (bp: 361° C.) having a metal impurity content of 10 ppb or less was added, the pressure in the system was reduced using a vacuum pump, and then heating was resumed. The reaction mixture in the reaction vessel was further heated to 52 to 68° C., and the produced methanol was distilled from the distillation tube equipped with a condenser at a distillation temperature of 32 to 67° C., and 351 g of pure water was added while distilling off the water and methanol, thereby obtaining colloidal silica with a solid content concentration of about 20 mass%.
The obtained colloidal silica was subjected to various analyses, and the results are shown in <22> of Table 3. Furthermore, the obtained colloidal silica was stored at 60°C for 7 days, and various analyses of the obtained colloidal silica were performed. It was confirmed, particularly from the results of DLS measurement, that substantially no aggregation had occurred. The results are shown in <23> of Table 3.
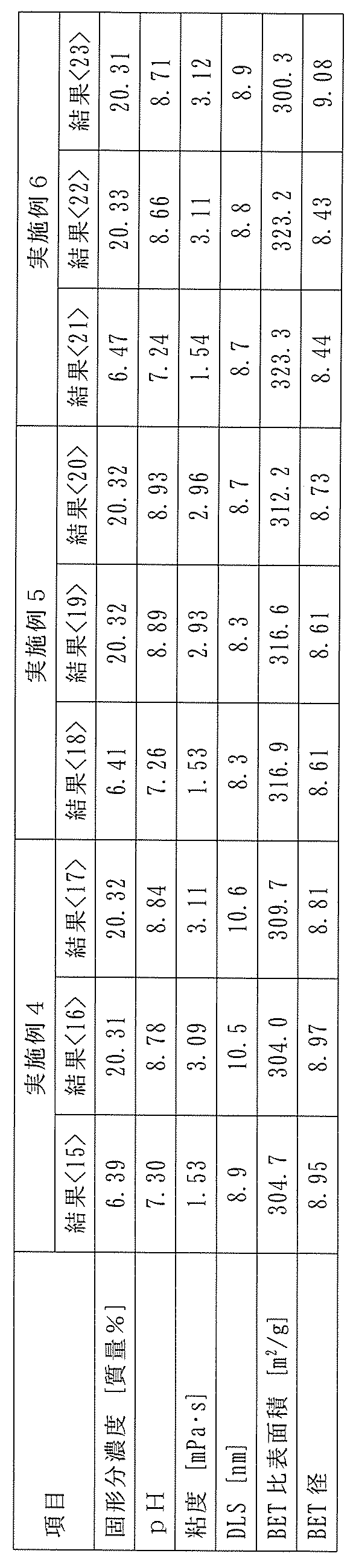
次に、前述のとおり、特許文献4に記載の実施例8や実施例1に記載された方法で得られた所定の固形分濃度、一次粒子径及び二次粒子径を有するコロイダルシリカ粒子分散体を用い、特許文献3に記載の砥粒D又は砥粒Eを含有する分散体を以下の手順で調製して、その評価を行った。
すなわち、以下に示した[参考実験例1]及び[参考実験例2]は、それぞれ、特許文献3の段落0075及びそれに記載された特許文献4の実施例8(段落0053)と、特許文献3の段落0076及びそれに記載された特許文献4の実施例1(段落0045)とに記載された手順と殆ど同じ手順によって、実際に、砥粒D及び砥粒Eに相当するコロイダルシリカ粒子分散体を調製し、その評価結果を示したものである。
Next, as described above, using a colloidal silica particle dispersion having a predetermined solid content concentration, primary particle size, and secondary particle size obtained by the method described in Example 8 or Example 1 described in Patent Document 4, a dispersion containing Abrasive D or Abrasive E described in Patent Document 3 was prepared in the following manner, and the dispersion was evaluated.
That is, the following [Reference Experimental Example 1] and [Reference Experimental Example 2] show the evaluation results of colloidal silica particle dispersions corresponding to Abrasives D and E prepared by procedures almost identical to those described in paragraph 0075 of Patent Document 3 and Example 8 (paragraph 0053) of Patent Document 4 described therein, and in paragraph 0076 of Patent Document 3 and Example 1 (paragraph 0045) of Patent Document 4 described therein, respectively.
[参考実験例1]
(手順)
反応容器にメタノール250部、金属不純物10ppb以下の純水10部及び29%アンモニア水2部の溶液を調製し、温度63℃に保持した後、撹拌条件下に金属不純物10ppb以下のテトラメトキシシラン(多摩化学工業株式会社製)27部を1cm3/min・Lの滴下速度で滴下し、加水分解、重縮合反応を行った。ここで、滴下速度の単位は特許文献4の段落0029に記載の定義に従った。反応終了後、1時間の熟成を実施したのちに、単蒸留操作により80部を留出させ、200部の超純水を添加したところ、特許文献4に記載の通り、わずかにコロイド溶液が白色を帯びたので、29%アンモニア水を5部添加し、透明状態に戻した。
[Reference Experimental Example 1]
(procedure)
A solution of 250 parts of methanol, 10 parts of pure water containing 10 ppb or less of metal impurities, and 2 parts of 29% aqueous ammonia was prepared in a reaction vessel, and after maintaining the temperature at 63° C., 27 parts of tetramethoxysilane (manufactured by Tama Chemicals Co., Ltd.) containing 10 ppb or less of metal impurities was added dropwise at a rate of 1 cm 3 /min·L under stirring conditions to carry out hydrolysis and polycondensation reactions. Here, the unit of the dropping rate was defined as described in paragraph 0029 of Patent Document 4. After the reaction was completed and aging was carried out for 1 hour, 80 parts were distilled out by simple distillation, and 200 parts of ultrapure water were added. As described in Patent Document 4, the colloidal solution became slightly white, so 5 parts of 29% aqueous ammonia were added to return it to a transparent state.
以上のようにして得られた、シリカ濃度3.8質量%、一次粒子径9.0nm、二次粒子径10.1nmのコロイダルシリカ粒子分散体5kgと、3-メルカプトプロピルトリメトキシシラン2gとを混合し、2時間加熱還流することにより、チオール化シリカゾルを得た。そのシリカゾルに、過酸化水素を加えて8時間加熱還流することにより、シリカ粒子の表面にスルホ基が固定化された、一次粒子9.0nm、二次粒子10.1nmの砥粒を3.7質量%含有する分散体を得た。 5 kg of the colloidal silica particle dispersion obtained above, with a silica concentration of 3.8% by mass, a primary particle diameter of 9.0 nm, and a secondary particle diameter of 10.1 nm, was mixed with 2 g of 3-mercaptopropyltrimethoxysilane and heated under reflux for 2 hours to obtain a thiolated silica sol. Hydrogen peroxide was added to the silica sol and heated under reflux for 8 hours to obtain a dispersion containing 3.7% by mass of abrasive grains with primary particles of 9.0 nm and secondary particles of 10.1 nm, with sulfo groups fixed to the surfaces of the silica particles.
このコロイダルシリカ粒子分散体を前述の実施例1に記載の方法と同様に、単蒸留操作により、コンデンサー付留出管から留出温度32~67℃で留出させ、12質量%まで濃縮したところ、図4に示すように凝集しゲル化が生じた。図4は、約12質量%まで濃縮したコロイダルシリカ粒子分散体の一部を採取して室温下で放冷した後、容器の上下を逆さにした状態の写真であり、容器の底部側(すなわち、写真の上部側)において、コロイダルシリカ粒子分散体が凝集ゲル化して落ちてこない状態が示されていることが分かる。 This colloidal silica particle dispersion was distilled at a distillation temperature of 32-67°C from a distillation tube equipped with a condenser by simple distillation in the same manner as in Example 1 above, and concentrated to 12% by mass, resulting in aggregation and gelation, as shown in Figure 4. Figure 4 is a photograph of a portion of the colloidal silica particle dispersion concentrated to approximately 12% by mass taken and left to cool at room temperature in an inverted container, and shows that the colloidal silica particle dispersion has aggregated and gelled at the bottom of the container (i.e., the upper part of the photograph), and does not fall out.
[参考実験例2]
(手順)
反応容器にメタノール310部、金属不純物10ppb以下の純水16部及び29%アンモニア水3部の溶液を調製し、温度60℃に保持した後、撹拌条件下に金属不純物10ppb以下のテトラメトキシシラン(多摩化学工業株式会社製)38部とメタノール18部との混合希釈溶液を1cm3/min・Lの滴下速度で滴下し、加水分解、重縮合反応を行い、固形分濃度が約3.8質量%のコロイダルシリカ粒子分散体を得た。ここで、滴下速度の単位は特許文献4の段落0029に記載の定義に従った。
[Reference Experimental Example 2]
(procedure)
A solution of 310 parts of methanol, 16 parts of pure water containing less than 10 ppb of metal impurities, and 3 parts of 29% aqueous ammonia was prepared in a reaction vessel, and after maintaining the temperature at 60° C., a mixed diluted solution of 38 parts of tetramethoxysilane (manufactured by Tama Chemicals Co., Ltd.) containing less than 10 ppb of metal impurities and 18 parts of methanol was dropped at a dropping rate of 1 cm 3 /min·L under stirring conditions to carry out hydrolysis and polycondensation reactions, thereby obtaining a colloidal silica particle dispersion with a solid content concentration of about 3.8 mass %. Here, the unit of the dropping rate was defined according to the definition described in paragraph 0029 of Patent Document 4.
以上のようにして得られた、シリカ濃度3.8質量%、一次粒子径5.9nm、二次粒子径7.3nmのコロイダルシリカ粒子分散体5kgと、3-メルカプトプロピルトリメトキシシラン2gとを混合し、2時間加熱還流することにより、チオール化シリカゾルを得た。そのシリカゾルに、過酸化水素を加えて8時間加熱還流することにより、シリカ粒子の表面にスルホ基が固定化された、一次粒子5.7nm、二次粒子7.0nmの砥粒を3.8質量%含有する分散体を得た。 5 kg of the colloidal silica particle dispersion obtained above, with a silica concentration of 3.8% by mass, a primary particle diameter of 5.9 nm, and a secondary particle diameter of 7.3 nm, was mixed with 2 g of 3-mercaptopropyltrimethoxysilane and heated under reflux for 2 hours to obtain a thiolated silica sol. Hydrogen peroxide was added to the silica sol and heated under reflux for 8 hours to obtain a dispersion containing 3.8% by mass of abrasive grains with primary particles of 5.7 nm and secondary particles of 7.0 nm, with sulfo groups fixed to the surfaces of the silica particles.
このコロイダルシリカ粒子分散体を前述の実施例1に記載の方法と同様に、単蒸留操作により、コンデンサー付留出管から留出温度32~67℃で留出させ、11質量%まで濃縮したところ、図5に示すように凝集しゲル化が生じた。図5は約11質量%まで濃縮したコロイダルシリカ粒子分散体の一部を採取して室温下で放冷した後、容器の上下を逆さにした状態の写真であり、図4と同様に、容器の底部側(すなわち、写真の上部側)において、コロイダルシリカ粒子分散体が凝集ゲル化して落ちてこない状態が示されていることが分かる。 This colloidal silica particle dispersion was distilled at a distillation temperature of 32-67°C from a distillation tube equipped with a condenser by simple distillation in the same manner as in Example 1 above, and concentrated to 11% by mass, resulting in aggregation and gelation, as shown in Figure 5. Figure 5 is a photograph of a portion of the colloidal silica particle dispersion concentrated to approximately 11% by mass taken and left to cool at room temperature in an inverted container, and it can be seen that, like Figure 4, the colloidal silica particle dispersion has aggregated and gelled at the bottom of the container (i.e., the upper part of the photograph) and does not fall out.
[結果と考察について]
前記のとおり、特許文献3に記載された砥粒D又は砥粒Eに相当する砥粒を含有する各コロイダルシリカ粒子分散体は、いずれも、少なくとも12質量%の濃度まで濃縮すると、当該コロイダルシリカ粒子分散体がいずれも凝集ゲル化することから、前述の本願の実施形態として示したBET径が12nm以下であり固形分濃度が12質量%以上のコロイダルシリカとして得られず、ひいては、本願の実施形態として示した経時安定性と同等の経時安定性は具備されないと言える。
[Results and Discussion]
As described above, when each of the colloidal silica particle dispersions containing abrasive grains corresponding to Abrasive grain D or Abrasive grain E described in Patent Document 3 is concentrated to a concentration of at least 12 mass %, the colloidal silica particle dispersions all aggregate and gel, and therefore the colloidal silica having a BET diameter of 12 nm or less and a solids concentration of 12 mass % or more shown in the above-mentioned embodiment of the present application is not obtained, and therefore it can be said that the same stability over time as that shown in the embodiment of the present application is not provided.
(考察)
ここで、特許文献3に示された方法によって作製されたコロイダルシリカ粒子分散体が12質量%を超過できない理由としては、次の(i)及び(ii)の点、すなわち、(i)チオール化反応(変性処理工程)時のシリカ粒子分散体のアルコール(メタノール)含有濃度と、(ii)シランカップリング剤(3-メルカプトプロピルトリメトキシシラン)に対するメタノール希釈の有無の二点が影響していると考えられる。この(i)について、前述の比較例1のように、変性処理工程前にシリカコロイド粒子分散体中に含まれるアルコールを純水へ置換した場合、前述の比較例1における説明や表2に示すように、DLS測定によるキュムラント平均径の変化が変性処理工程前後で20%を超過し、凝集傾向が見られた。特許文献4の実施例8ではシリカ粒子作成後に単蒸留操作を実施していることから、そのシリカ粒子分散体を用いた特許文献3の砥粒Dは、変性処理を行っても12質量%までは濃縮できなかったと考えられる。
(Discussion)
Here, the reason why the colloidal silica particle dispersion prepared by the method shown in Patent Document 3 cannot exceed 12% by mass is considered to be influenced by the following two points (i) and (ii), that is, (i) the alcohol (methanol) content concentration of the silica particle dispersion during the thiolation reaction (modification treatment process) and (ii) the presence or absence of methanol dilution of the silane coupling agent (3-mercaptopropyltrimethoxysilane). Regarding this (i), when the alcohol contained in the silica colloid particle dispersion is replaced with pure water before the modification treatment process as in the above-mentioned Comparative Example 1, as explained in the above-mentioned Comparative Example 1 and shown in Table 2, the change in the cumulant average diameter by DLS measurement exceeded 20% before and after the modification treatment process, and a tendency to aggregate was observed. Since a simple distillation operation is performed after the preparation of the silica particles in Example 8 of Patent Document 4, it is considered that the abrasive D of Patent Document 3 using the silica particle dispersion could not be concentrated to 12% by mass even after the modification treatment.
また、シリカ粒子作成後に単蒸留操作を行わず、変性処理工程を実施している特許文献3の砥粒E(特許文献4の実施例1を利用)でも同様にゲル化することが確認されたが、これは、上記の理由(ii)が原因だと考えられる。前述の実施例1では変性処理工程時にシランカップリング剤をアルコールで希釈してから6時間かけて連続的に供給(つまり、滴下して添加)している一方で、特許文献3では希釈せずに加熱還流前に混合されている。このことについては、例えば、カップリング剤による改質の動向を教示した前掲の非特許文献1(青木恂次郎,日本ゴム協会誌,Vol.59,No.11,1986,610-619.)の第611ページの「3.1 シラン」において、『構造的にはRSi(OCH3)3で,Rは有機官能基,極性ポリマーを選び組合せる.メタノール水に溶け加水分解しシラントリオール,縮合してシロキサノールオリゴマーをつくり,特に,シリカ系粉体,繊維に化学吸着する.(図省略) 溶剤を揮発,弱く加熱すると,ポリマーと化学結合する有機官能基をもつ薄膜となる.加水分解と化学反応を伴い,界面で化学吸着と橋かけ・重合する.』と記載されている。つまり、3-メルカプトプロピルトリメトキシシランを含む、一般的なシランカップリング剤の反応において、シランカップリング剤のアルコキシ基が加水分解した後、シランカップリング剤同士の自己縮合反応(結果的に凝集物の生成)とシリカ粒子への修飾反応が競合することが知られているが、変性処理工程においては、前述の実施例1のように、シランカップリング剤をメタノールで希釈して連続的に供給することで、シリカ粒子への効率的な修飾反応が進行することが確認される。 In addition, gelation was also confirmed in the abrasive grain E of Patent Document 3 (using Example 1 of Patent Document 4), which was subjected to a modification treatment process without performing a simple distillation operation after the preparation of silica particles, and this is believed to be due to the above reason (ii). In the above-mentioned Example 1, the silane coupling agent is diluted with alcohol and then continuously supplied (i.e., added dropwise) for 6 hours during the modification treatment process, while in Patent Document 3, it is mixed without dilution before heating and refluxing. Regarding this, for example, in the above-mentioned Non-Patent Document 1 (Aoki Shinjiro, Journal of the Japan Rubber Association, Vol. 59, No. 11, 1986, 610-619.), which teaches the trend of modification using coupling agents, it is described in "3.1 Silane" on page 611 that "Structurally, RSi(OCH 3 ) 3 , R is an organic functional group, and a polar polymer is selected and combined. It dissolves in methanol and water, hydrolyzes, and condenses to form silane triol, which is chemically adsorbed to silica-based powders and fibers. (Diagram omitted) When the solvent is evaporated and weakly heated, it becomes a thin film with organic functional groups that chemically bond with the polymer. It undergoes hydrolysis and chemical reaction, and chemical adsorption and crosslinking/polymerization at the interface. ' It is known that in the reaction of general silane coupling agents, including 3-mercaptopropyltrimethoxysilane, after the alkoxy group of the silane coupling agent is hydrolyzed, the self-condensation reaction between the silane coupling agents (resulting in the formation of aggregates) and the modification reaction to the silica particles compete with each other, but in the modification treatment process, as in the above-mentioned Example 1, it is confirmed that an efficient modification reaction to the silica particles proceeds by diluting the silane coupling agent with methanol and continuously supplying it.
そして、このような修飾反応において濃度や温度が影響することは、例えば、前掲の非特許文献2(M.-C. B. Salon, et al., Colloids and Surfaces A: Physicochem. Eng. Aspects, 2008, 312, 83-91.)に示されている。この文献は、「Kinetics of hydrolysis and self condensation reactions of silanes by NMR spectroscopy」(和訳:NMR分光法によるシランの加水分解および自己縮合反応の速度論)と題する論文であり、89ページの「3.4.1. Silane concentration」では、反応におけるシランカップリング剤の濃度の影響が調査されている。アルコール溶媒に対して、シランカップリング剤を10%と4%で比較している。10%に対する、4%の結果として、『The hydrolysis rate decreased (complete hydrolysis within 10 h). The silanol formation T0 H was also slowed down (only 80% of such species within 6 h reaction).』(和訳:加水分解速度は低下した(10時間以内に完全に加水分解)。また、T0 Hシラノール生成も遅くなった(6時間の反応で80%しか生成しなかった)。)と記載されている。さらに、別のシランカップリング剤を用いて、20%に濃度を上げた実験では、『However, the maximum of formed silanol units (T0 H) was reduced to 50% probably because of the increase of self condensation reactions; the T1 and T2formations were accelerated, as presented in Figure 9b.』(和訳:しかし、形成されたシラノール単位(T0 H)の最大値は、おそらく自己縮合反応の増加のため50%に減少した。)と記載されており、上記の濃度検討に関する結論として、『These data show that a concentration of 10% gave optimal experimental conditions to enhance the maximum silanol formation and to lower self condensation reactions.』(和訳:これらのデータは、10%の濃度が最大シラノール形成を促進し、自己縮合反応を低下させる最適な実験条件であることを示している。)と結ばれている。 The effect of concentration and temperature on such modification reactions is shown, for example, in the above-mentioned non-patent document 2 (M.-C. B. Salon, et al., Colloids and Surfaces A: Physicochem. Eng. Aspects, 2008, 312, 83-91.). This document is a paper titled "Kinetics of hydrolysis and self condensation reactions of silanes by NMR spectroscopy" and, in "3.4.1. Silane concentration" on page 89, the effect of the concentration of the silane coupling agent on the reaction is investigated. The silane coupling agent is compared at 10% and 4% in the alcohol solvent. As a result of 4% compared to 10%, it is stated that "The hydrolysis rate decreased (complete hydrolysis within 10 h). The silanol formation T 0 H was also slowed down (only 80% of such species within 6 h reaction)." Furthermore, in an experiment in which a different silane coupling agent was used and the concentration was increased to 20%, it was stated that "However, the maximum of formed silanol units (T 0 H ) was reduced to 50% probably because of the increase of self condensation reactions; the T 1 and T 2 formations were accelerated, as presented in Figure 9b." The conclusion regarding the above concentration study was concluded as "These data show that a concentration of 10% gave optimal experimental conditions to enhance the maximum silanol formation and to lower self condensation reactions."
さらに、その90ページの「3.4.2. Effect of the temperature」において、反応における温度の影響が調査されている。すなわち、『The hydrolysis reaction was started at room temperature (25℃) for 2h. Then the temperature was kept to 70℃ (corresponding to the boiling temperature of the solvent mixture), as displayed in Fig. 9c.』(和訳:加水分解反応は室温(25℃)で2時間行った。その後、温度を70℃(混合溶媒の沸騰温度に相当)に保ち、Fig. 9cに示した。)と記載されているように、Fig. 9cでは、70℃に加温した場合のシランカップリング剤の加水分解と自己縮合の継時変化が示されている。加水分解速度(T0 Hの減少)は昇温の影響を受けていないが、T1の急激な増加とその後に続くT2の緩やかな増大から、自己縮合反応は大きく促進されていることが分かる。したがって、筆者らによれば、例えば無機材料にシランカップリング剤を修飾する場合、自己縮合を抑制したいのであれば温度はなるべく低く保つ必要があると報告されている。 Furthermore, in "3.4.2. Effect of the temperature" on page 90, the effect of temperature on the reaction is investigated. That is, as described in "The hydrolysis reaction was started at room temperature (25℃) for 2h. Then the temperature was kept to 70℃ (corresponding to the boiling temperature of the solvent mixture), as displayed in Fig. 9c.", Fig. 9c shows the time course of hydrolysis and self-condensation of the silane coupling agent when heated to 70℃. The hydrolysis rate (decrease in T 0 H ) is not affected by the temperature rise, but the rapid increase in T 1 followed by the gradual increase in T 2 indicates that the self-condensation reaction is greatly accelerated. Therefore, the authors report that, for example, when modifying an inorganic material with a silane coupling agent, the temperature must be kept as low as possible if self-condensation is to be suppressed.
そして、その91ページの「4.Conclusion」において、『The optimal concentration of silanes hydrolysis was found to be around 10% (w/w) with respect to the solvents and the temperature raise enhanced drastically the self condensation reactions.』(和訳:シラン加水分解の最適濃度は、溶媒に対して約10%(w/w)であり、温度を上げると自己縮合反応が急激に促進されることが分かった。)と示されている。
したがって、特許文献3の砥粒Eは、変性処理時のシランカップリング剤の投入方法が原因で12質量%までは濃縮できなかったと考えられる。
Then, on page 91, in "4. Conclusion," it is stated that "The optimal concentration of silanes hydrolysis was found to be around 10% (w/w) with respect to the solvents and the temperature raise enhanced drastically the self condensation reactions."
Therefore, it is believed that the abrasive grain E of Patent Document 3 could not be concentrated to 12 mass % due to the method of adding the silane coupling agent during the modification treatment.
なお、得られたコロイダルシリカの物性などについては、以下の方法で評価した。
(1)BET比表面積、BET径:機器としてNOVA4200e(Anton Paar社製)を用いて測定した。窒素吸着法(BET法)により測定されたBET比表面積S(m2/g)と、SiO2の真密度2.2g/cm3)と、前記の式(1)とから、2727/Sの式によって導いた粒子径をBET径としている。
(2)動的散乱法によるキュムラント平均径:機器としてSZ-100(株式会社堀場製作所製)を用いて測定した。測定時は、測定サンプル中のシリカ分を1.13gとなるように純水・硝酸アンモニウムで調整し、調整した液を測定した。
(3)シリカ固形分濃度:機器としてSMS-70(株式会社エー・アンド・デイ社製)を用い、含有水分を蒸発させた残渣分をシリカ濃度とした。
(4)pH:機器としてD-51(株式会社堀場製作所社製)にて、25℃で測定した。
(5)粘度:機器としてVM-10A(株式会社セコニック社製)にて、25℃で測定した。
(6)メタノール濃度:機器としてGC-2025(株式会社島津製作所)を用いて測定した。
The physical properties of the resulting colloidal silica were evaluated by the following methods.
(1) BET specific surface area, BET diameter: Measured using a NOVA4200e (manufactured by Anton Paar). The particle diameter is calculated by the formula 2727/S using the BET specific surface area S ( m2 /g) measured by the nitrogen adsorption method (BET method), the true density of SiO2 (2.2 g/ cm3 ), and the above formula (1).
(2) Cumulant mean diameter by dynamic scattering method: Measured using an SZ-100 (manufactured by Horiba, Ltd.) When measuring, the silica content in the measurement sample was adjusted to 1.13 g with pure water and ammonium nitrate, and the adjusted liquid was measured.
(3) Silica solids concentration: Using an SMS-70 (manufactured by A&D Co., Ltd.) as an apparatus, the residue obtained after evaporating the water content was taken as the silica concentration.
(4) pH: Measured at 25° C. using an instrument D-51 (manufactured by Horiba, Ltd.).
(5) Viscosity: Measured at 25° C. using a VM-10A (manufactured by Sekonic Corporation).
(6) Methanol concentration: Measured using a GC-2025 (Shimadzu Corporation).
本発明のコロイダルシリカは、例えば、研磨剤(シリコンウエーハ、ハードディスクなど)、コーティング剤(眼鏡、ディスプレー、建材、紙など)、バインダー(セラミックス、触媒など)などの用途に好適である。
The colloidal silica of the present invention is suitable for applications such as abrasives (silicon wafers, hard disks, etc.), coating agents (eyeglasses, displays, building materials, paper, etc.), and binders (ceramics, catalysts, etc.).
Claims (13)
有機アミンからなる加水分解触媒を含む反応液に、易分解性オルガノシリケートを供給及び反応させて、BET径が12nm以下である原料コロイダルシリカを準備する原料準備工程と、
原料コロイダルシリカの固形分濃度を13質量%以下とすると共に、前記原料準備工程に由来して生成するアルコール類の濃度を1~25質量%とする濃度調整工程と、
前記濃度調整された原料コロイダルシリカを変性処理する変性処理工程と、
前記変性処理されたコロイダルシリカ中の残留有機溶媒が1質量%以下となるように濃縮する濃縮工程と
を含むことを特徴とするコロイダルシリカの製造方法。 A method for producing the colloidal silica according to any one of claims 1 to 7, comprising the steps of:
a raw material preparation step of supplying and reacting a readily decomposable organosilicate to a reaction solution containing a hydrolysis catalyst made of an organic amine to prepare raw material colloidal silica having a BET diameter of 12 nm or less;
a concentration adjusting step of adjusting the solid content concentration of the raw material colloidal silica to 13% by mass or less and adjusting the concentration of alcohols generated in the raw material preparation step to 1 to 25% by mass;
a modification treatment step of modifying the raw colloidal silica having the adjusted concentration;
and a concentrating step of concentrating the modified colloidal silica so that a residual organic solvent in the modified colloidal silica is 1 mass % or less.
9. The method for producing colloidal silica according to claim 8, wherein the modification treatment step comprises a step of reacting a modifying agent having a cationic group with the colloidal silica after the concentration adjustment step.
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2023-183082 | 2023-10-25 | ||
| JP2023183082 | 2023-10-25 | ||
| JP2024-048265 | 2024-03-25 | ||
| JP2024048265A JP7668924B1 (en) | 2023-10-25 | 2024-03-25 | Colloidal silica and method for producing colloidal silica |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2025089240A1 true WO2025089240A1 (en) | 2025-05-01 |
Family
ID=95450326
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2024/037448 Pending WO2025089240A1 (en) | 2023-10-25 | 2024-10-21 | Colloidal silica and method for producing colloidal silica |
Country Status (3)
| Country | Link |
|---|---|
| JP (1) | JP7668924B1 (en) |
| TW (1) | TW202525712A (en) |
| WO (1) | WO2025089240A1 (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP7771475B1 (en) * | 2025-05-30 | 2025-11-17 | 扶桑化学工業株式会社 | Amino-modified colloidal silica and method for producing amino-modified colloidal silica |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2021246137A1 (en) | 2020-06-04 | 2021-12-09 | Phcホールディングス株式会社 | Binary refrigeration device |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2003535804A (en) * | 2000-06-22 | 2003-12-02 | アクゾ ノーベル エヌ.ブイ. | Silica sol mixture |
| WO2020179555A1 (en) * | 2019-03-06 | 2020-09-10 | 扶桑化学工業株式会社 | Colloidal silica and production method therefor |
| JP2022152370A (en) * | 2021-03-29 | 2022-10-12 | 多摩化学工業株式会社 | Method of producing colloidal silica, and colloidal silica |
| WO2023007938A1 (en) * | 2021-07-30 | 2023-02-02 | Jsr株式会社 | Composition for chemical mechanical polishing and polishing method |
| JP2023146033A (en) * | 2022-03-29 | 2023-10-12 | 株式会社フジミインコーポレーテッド | Sulfonic acid modified colloidal silica |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2004315300A (en) * | 2003-04-17 | 2004-11-11 | Nippon Steel Chem Co Ltd | Silica fine particles, silica colloid in which silica particles are dispersed, and method for producing the same |
| JPWO2012161157A1 (en) * | 2011-05-24 | 2014-07-31 | 日産化学工業株式会社 | Organic solvent-dispersed silica sol |
| JP6757969B2 (en) * | 2016-07-07 | 2020-09-23 | 学校法人神奈川大学 | Method for Producing Silica Nanoparticles, Silica Nanoparticles, and Fluorescent Agent |
| TW202323189A (en) * | 2021-08-31 | 2023-06-16 | 日商日產化學股份有限公司 | Surface-treated inorganic oxide particle dispersion containing silicon oxide and its production method |
-
2024
- 2024-03-25 JP JP2024048265A patent/JP7668924B1/en active Active
- 2024-10-21 WO PCT/JP2024/037448 patent/WO2025089240A1/en active Pending
- 2024-10-24 TW TW113140545A patent/TW202525712A/en unknown
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2003535804A (en) * | 2000-06-22 | 2003-12-02 | アクゾ ノーベル エヌ.ブイ. | Silica sol mixture |
| WO2020179555A1 (en) * | 2019-03-06 | 2020-09-10 | 扶桑化学工業株式会社 | Colloidal silica and production method therefor |
| JP2022152370A (en) * | 2021-03-29 | 2022-10-12 | 多摩化学工業株式会社 | Method of producing colloidal silica, and colloidal silica |
| WO2023007938A1 (en) * | 2021-07-30 | 2023-02-02 | Jsr株式会社 | Composition for chemical mechanical polishing and polishing method |
| JP2023146033A (en) * | 2022-03-29 | 2023-10-12 | 株式会社フジミインコーポレーテッド | Sulfonic acid modified colloidal silica |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP7771475B1 (en) * | 2025-05-30 | 2025-11-17 | 扶桑化学工業株式会社 | Amino-modified colloidal silica and method for producing amino-modified colloidal silica |
Also Published As
| Publication number | Publication date |
|---|---|
| TW202525712A (en) | 2025-07-01 |
| JP7668924B1 (en) | 2025-04-25 |
| JP2025073046A (en) | 2025-05-12 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5080061B2 (en) | Method for producing neutral colloidal silica | |
| WO2025089240A1 (en) | Colloidal silica and method for producing colloidal silica | |
| JP7206695B2 (en) | Silica sol, polishing composition, method for polishing silicon wafer, method for producing silicon wafer, chemical-mechanical polishing composition, and method for producing semiconductor device | |
| JP5127452B2 (en) | Method for producing deformed silica sol | |
| JP5221517B2 (en) | Aluminum modified colloidal silica and method for producing the same | |
| JP5905767B2 (en) | Dispersion stabilization method of neutral colloidal silica dispersion and neutral colloidal silica dispersion excellent in dispersion stability | |
| WO2018124117A1 (en) | Silica particle dispersion and method for producing same | |
| JP2021147267A (en) | Method for producing silica particle, method for producing silica sol, polishing process, method for producing semiconductor wafer, and method for producing semiconductor device | |
| JP2025133817A (en) | Method for producing silica particles, method for producing silica sol, polishing method, method for producing semiconductor wafer, and method for producing semiconductor device | |
| JP2025036640A (en) | Silica particles, silica sol, polishing composition, polishing method, method for manufacturing semiconductor wafer, and method for manufacturing semiconductor device | |
| JP2023115394A (en) | Method for producing silica sol and method for suppressing intermediate products in silica sol | |
| JP7782174B2 (en) | Silica sol manufacturing method, polishing method, semiconductor wafer manufacturing method, and semiconductor device manufacturing method | |
| JP7618362B2 (en) | Method for producing water-dispersed surface-treated colloidal silica | |
| JP2926915B2 (en) | Elongated silica sol and method for producing the same | |
| WO2020171134A1 (en) | Silica particles and production method therefor, silica sol, polishing composition, polishing method, semiconductor wafer production method, and semiconductor device production method | |
| JP7588019B2 (en) | Method for producing colloidal silica and colloidal silica | |
| JP7782173B2 (en) | Silica sol manufacturing method, polishing method, semiconductor wafer manufacturing method, and semiconductor device manufacturing method | |
| JP2021147266A (en) | Method for producing silica particle, method for producing silica sol, polishing process, method for producing semiconductor wafer, and method for producing semiconductor device | |
| JP7331437B2 (en) | Silica particles, silica sol, polishing composition, polishing method, semiconductor wafer manufacturing method, and semiconductor device manufacturing method | |
| JP7464201B2 (en) | Silica particles and their manufacturing method, silica sol, polishing composition, polishing method, manufacturing method for semiconductor wafer, and manufacturing method for semiconductor device | |
| WO2020171140A1 (en) | Silica particles and production method therefor, silica sol, polishing composition, polishing method, semiconductor wafer production method, and semiconductor device production method | |
| JP7731017B1 (en) | Method for producing colloidal silica | |
| WO2023219004A1 (en) | Colloidal silica and production method therefor | |
| JP7225898B2 (en) | Method for producing silica particles, method for producing silica sol, and method for polishing | |
| JP2022167826A (en) | Chemical mechanical polishing composition and method |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 24882353 Country of ref document: EP Kind code of ref document: A1 |