WO2025088308A1 - Products and methods for treating autoimmune diseases - Google Patents
Products and methods for treating autoimmune diseases Download PDFInfo
- Publication number
- WO2025088308A1 WO2025088308A1 PCT/GB2024/052698 GB2024052698W WO2025088308A1 WO 2025088308 A1 WO2025088308 A1 WO 2025088308A1 GB 2024052698 W GB2024052698 W GB 2024052698W WO 2025088308 A1 WO2025088308 A1 WO 2025088308A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- car
- cells
- cell
- seq
- sle
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K40/00—Cellular immunotherapy
- A61K40/40—Cellular immunotherapy characterised by antigens that are targeted or presented by cells of the immune system
- A61K40/41—Vertebrate antigens
- A61K40/42—Cancer antigens
- A61K40/4202—Receptors, cell surface antigens or cell surface determinants
- A61K40/421—Immunoglobulin superfamily
- A61K40/4211—CD19 or B4
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K40/00—Cellular immunotherapy
- A61K40/10—Cellular immunotherapy characterised by the cell type used
- A61K40/11—T-cells, e.g. tumour infiltrating lymphocytes [TIL] or regulatory T [Treg] cells; Lymphokine-activated killer [LAK] cells
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K40/00—Cellular immunotherapy
- A61K40/30—Cellular immunotherapy characterised by the recombinant expression of specific molecules in the cells of the immune system
- A61K40/31—Chimeric antigen receptors [CAR]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K40/00—Cellular immunotherapy
- A61K40/40—Cellular immunotherapy characterised by antigens that are targeted or presented by cells of the immune system
- A61K40/41—Vertebrate antigens
- A61K40/416—Antigens related to auto-immune diseases; Preparations to induce self-tolerance
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2239/00—Indexing codes associated with cellular immunotherapy of group A61K40/00
- A61K2239/31—Indexing codes associated with cellular immunotherapy of group A61K40/00 characterized by the route of administration
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2239/00—Indexing codes associated with cellular immunotherapy of group A61K40/00
- A61K2239/38—Indexing codes associated with cellular immunotherapy of group A61K40/00 characterised by the dose, timing or administration schedule
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/60—Immunoglobulins specific features characterized by non-natural combinations of immunoglobulin fragments
- C07K2317/62—Immunoglobulins specific features characterized by non-natural combinations of immunoglobulin fragments comprising only variable region components
- C07K2317/622—Single chain antibody (scFv)
Definitions
- the present invention relates to a chimeric antigen receptor (CAR) which binds the B- lymphocyte antigen CD19 (Cluster of Differentiation 19) and to use of cytolytic cells expressing the CAR for the treatment of autoimmune diseases such as systemic lupus erythromatosus (SLE).
- CAR chimeric antigen receptor
- B cells along with T cells, form the core of the adaptive arm of the immune system and play a key role in regulating immune response in both normal physiological and pathological conditions.
- B cells are identified pharmacological targets in B cell malignancies and autoimmune diseases.
- autoimmune mechanisms Numerous diseases are believed to result from autoimmune mechanisms. Common among these are rheumatoid arthritis, systemic lupus erythematosus, inflammatory bowel disease, multiple sclerosis, Type I diabetes, Guillain-Barre syndrome, chronic inflammatory demyelinating polyneuropathy, psoriasis, Graves’ disease, Hashimoto’s thyroiditis, myasthenia gravis, scleroderma, vasculitis and pemphigus vulgaris. Autoimmune diseases affect millions of individuals world-wide and the cost of these diseases, in terms of actual treatment expenditures and lost productivity, is measured in billions of dollars annually.
- Systemic lupus erythematosus is an autoimmune disease characterised by the formation of autoantibodies and immune complex mediated inflammation and organ damage, including the skin, joints, central nervous system, heart, lung, and kidneys (Tsokos GC. (2011). Systemic lupus erythematosus. N Engl J Med 365(22):2110-21 ; Kaul A, Gordon C, Crow M, et al. (2016). Systemic lupus erythematosus. Nature Reviews 2:1-21 ; Mackensen A, Muller F, Mouglakakos D, et al. (2022). Anti-CD19 CAR T cell therapy for refractory lupus erythematosus.
- Anifrolumab a type I interferon (IFN) receptor antagonist, has also been approved in the US and EU and is indicated as an add-on for the treatment of adult patients with moderate to severe SLE who are receiving standard therapy (Saphnelo (anifrolumab- fnia) IISPI. 2022.
- IFN interferon
- voclosporin (Lupkynis USPI, 2021) was approved for lupus nephritis based on the AURORA1 trial (Rovin et al., Lancet 2021) (www.doi.org/10.1016/S0140-6736(21)00578-X). Voclosporin in combination with MMF and low-dose steroids led to a clinically and statistically superior complete renal response rate versus MMF and low-dose steroids alone, with a comparable safety profile. Despite these approvals, some patients have insufficient response, lack of response, or lack of sustained response and are at risk for further organ damage despite standard therapy. Hence, challenges remain with treatmentresistant disease.
- CD19-targeting chimeric antigen receptor (CAR) T cells Cappell KM, Kochenderfer JN. (2023) Longterm outcomes following CAR T cell therapy: what we know so far. Nat Review 20:359- 371).
- CD19-targeted CAR T cells are now approved for relapsed and/or refractory B cell lymphoma and B cell acute lymphoblastic leukemia (B-ALL) (axicabtagene ciloleucel, tisagenlecleucel, lisocabtagene maraleucel and brexucabtagene autoleucel).
- CD19-targeted CAR T cells are likely to be curative for a subset of patients with B cell lymphomas and possibly B-ALL (Cappell and Kochenderfer, supra; Westin J, Davis RE, Feng L, Hagemeister F, et al. (2023). Smart start: rituximab, lenalidomide, and ibrutinib in patients with newly diagnosed large B-cell lymphoma. J Clin Oncol 41 (4): 745-55).
- CD8 + T cells expressing CD19-targeted CARs [1 D3 CAR (Kochenderfer JN, Yu Z, Frasheri D, et al. (2010), Adoptive transfer of syngeneic T cells transduced with a chimeric antigen receptor that recognizes murine CD19 can eradicate lymphoma and normal B cells.
- Transferred anti-CD19 CAR T cells to MRL-lpr mice at onset of disease was assessed to determine their role in SLE prevention.
- the adoptive transfer of the constructed anti-CD 19 CAR-T cells showed a more sustained B-cell-depletion effect in MRL-lpr mice.
- the transfer of syngeneic anti-CD19 CAR-T cells not only prevented disease pathogenesis before the onset of disease symptoms but also displayed therapeutic benefits at a later stage after disease progression.
- CAR-T cells with the 4-1 BB costimulatory motif showed better therapeutic efficiency, without cell enrichment, compared to CAR-T cells with CD28 costimulatory motif.
- belimumab an anti-B cell activating factor (BAFF)/B lymphocyte stimulator (BlyS) monoclonal antibody
- BAFF anti-B cell activating factor
- BlyS B lymphocyte stimulator
- Tolerability and efficacy were initially tested in a single SLE patient treated with a CD19 CAR (FMC63 CAR) (Mougiakakos D, Krdnke G, Volkl S, Kretschmann S, et al. (2021). CD-19-targeted CAR T cells in refractory systemic lupus erythematosus. N Engl J Med 385(6): 567-9).
- CD19 CAR T cell treatment in another autoimmune disease has recently been shown in patients with refractory idiopathic inflammatory myopathy associated with antisynthetase syndrome (Muller F, Boeltz S, Knitza J, Aigner M, et al. (2023).
- CD19-targeted CAR T cells in refractory antisynthetase syndrome Lancet 401(10379):815-8; Percher AC, Hensen L, Klein R, et al. (2023).
- CAR Chimeric Antigen Receptor
- scFv single-chain variable fragments
- monoclonal antibodies which recognise a target antigen, fused via a spacer and a transmembrane domain to a signalling endodomain.
- Such molecules result in activation of the T cell in response to recognition by the scFv of its cognate target.
- T cells express such a CAR, they recognize and kill target cells that express the target antigen.
- CARs have been developed against tumour associated antigens, and adoptive transfer approaches using such CAR-expressing T cells are currently in clinical trial for the treatment of various cancers. To-date however, the main clinical exploration and potential application of CAR therapy is as treatment for B-cell malignancies.
- CD19 is a B-cell antigen which is expressed very early in B-cell differentiation and is only lost at terminal B-cell differentiation into plasma cells. Hence, CD19 is expressed on all B-cell malignancies apart from multiple myeloma. It is not expressed on other haematopoietic populations or non-haematopoietic cells and therefore targeting this antigen should not lead to toxicity to the bone marrow or non-haematopoietic organs. Loss of the normal B-cell compartment is considered an acceptable toxicity when treating lymphoid malignancies, because although effective CD 19 CAR T cell therapy will result in B cell aplasia, the consequent hypogammaglobulinaemia can be treated with pooled immunoglobulin.
- CD19 is therefore an attractive CAR target.
- the main clinical focus of the CAR field has been studies targeting CD19 on refractory B-cell cancers, as summarised in Table 1.
- Cytokine release syndrome encompasses a range of inflammatory symptoms ranging from mild to multi-organ failure with hypotension and respiratory failure.
- Some degree of CRS occurs commonly in patients treated with CD19 CAR T cells. Approximately 30% (21/73) patients treated in recent cohorts showed some degree of CRS (Davila et al ((2014). Sci. Transl. Med. 6, 224ra25; Lee et al (2014) Lancet. doi:10.1016/S0140-6736(14)61403-3; Kochenderfer et al. (2014) J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol., doi:10.1200/JCO.2014.56.2025).
- CRS has also been seen in patients treated with blinatumomab, a bi-specific recombinant single-chain antibody recognising both CD19 and CD3.
- CRS typically occurs 5-21 days after CAR T cell infusion.
- CRS can be life threatening and requires treatment in an intensive care setting.
- CRS is associated with elevated serum cytokine levels.
- the cytokines most significantly elevated are IL-6, IL-10 and interferon gamma (IFNy).
- IL-6 IL-6
- IFNy interferon gamma
- Clinical manifestations of severe CRS fever, hepatosplenomegaly, coagulopathy and hyperferritinaemia
- MAS macrophage activation syndrome
- MAS macrophage activation syndrome
- MAS macrophage activation syndrome
- a key initiating factor in MAS is release of copious Interferongamma (Lopez-Alvarez et al. (2009). Clin. Vaccine Immunol. CVI 16, 142-145).
- Durable responses appeared to correlate with higher peak levels of circulating CAR transduced T cells, as well as with the duration of B cell aplasia. With exception of patients relapsing with CD19- disease, relapse was generally associated with loss of circulating CAR T cells and recovery of normal B cells.
- T cell exhaustion is a state of T cell dysfunction that arises during many chronic infections and cancer. It is defined by poor effector function, sustained expression of inhibitory receptors and a transcriptional state distinct from that of functional effector or memory T cells. Exhaustion prevents optimal control of infection and tumors. Recently, a clearer picture of the functional and phenotypic profile of exhausted T cells has emerged with expression of inhibitory receptor programmed death 1 (PD-1 ; also known as PDCD1), a negative regulator of activated T cells, being a key feature (Day et al. (2006) Nature 443, 350-354).
- PD-1 inhibitory receptor programmed death 1
- the present disclosure provides methods of treatment of autoimmune diseases with CD19 CAR T cells.
- Treatment with CD19 CAR T cells is contemplated to more completely the B cell compartment than Rituximab, more effectively eliminating autoreactive B cells.
- CD19 as a target in autoimmune diseases is advantageous over CD20-directed therapies as CD19 is expressed more broadly on B cells, including very early B-progenitor cells as well as immature plasmablasts.
- the present invention provides methods of treating an autoimmune disease in a patient, where the patient is administered a cell comprising a chimeric antigen receptor (CAR) comprising a CD19-binding domain which comprises a) a heavy chain variable region (VH) having complementarity determining regions (CDRs) with the following sequences:
- CAR chimeric antigen receptor
- VH heavy chain variable region
- CDRs complementarity determining regions
- the autoimmune disease may be systemic lupus erythrematosus (SLE).
- SLE systemic lupus erythrematosus
- the autoimmune disease may be severe SLE or refractory SLE.
- the cell may be a T cell or NK cell.
- the cell may an autologous peripheral blood T cells transduced ex vivo to express the CAR.
- the cell may have been isolated from a peripheral blood mononuclear cell (PBMC) sample from the patient.
- PBMC peripheral blood mononuclear cell
- a dose of 50 x 10 6 ( ⁇ 20%) CD19 CAR-positive viable T cells may be administered to the patient.
- the dose may be 100 x 10 6 ( ⁇ 20%) CD 19 CAR-positive viable T cells or 30 x 10 6 ( ⁇ 20%) CD19 CAR-positive viable T cells.
- the administration may be an intravenous injection through a Hickman line or peripherally inserted central catheter.
- the CD19 binding domain may comprise the six CDRs (SEQ ID Nos. 1-6) grafted on to a human antibody framework.
- the CD19 binding domain may comprise a VH domain having the sequence shown as SEQ ID No. 7 and/or or a VL domain having the sequence shown as SEQ Id No. 8, or a variant of either having at least 95% sequence identity.
- the CD19 binding domain may comprise an scFv in the orientation VH-VL.
- the CD19 binding domain may comprise the sequence shown as SEQ ID No.. 9 or a variant thereof having at least 90% sequence identity.
- the CAR may comprise a spacer that is a CD8 stalk.
- the CAR may comprise an intracellular T cell signaling domain comprising the 41 BB endodomain and the CD3-Zeta endodomain.
- the invention provides a method of treating an autoimmune disease in a patient comprising administering to the patient a dose of 50 x 10 6 ( ⁇ 20%) CD19 CAR-positive viable T cells.
- the patient may be administered a dose of 30 x 10 6 ( ⁇ 20%) CD19 CAR-positive viable T cells.
- the patient may be administered a dose of 100 x 10 6 ( ⁇ 20%) CD19 CAR-positive viable T cells.
- the autoimmune disease according to the second aspect may be SLE.
- the autoimmune disease may be severe SLE or refractory SLE.
- the CAR may comprise the sequence shown as any of SEQ ID No. 10 to 15 or a variant thereof which has at least 80% sequence identity but retains the capacity to i) bind CD19 and ii) induce T cell signalling.
- the CAR may have advantageous properties compared to the fmc63-based CAR used in the UPENN studies.
- the CD19 (CAT) CAR herein has a lower affinity CD19 binder with a faster off-rate which mirrors more closely a physiological interaction and therefore, a more physiological T cell activation.
- the CAR when expressed by a T cell and used to target a CD19 expressing cell, may cause lower IFNy release by the CD19-expressing target cell than that caused by a T cell expressing a CAR comprising a CD19-binding domain which comprises: a) a heavy chain variable region (VH) having complementarity determining regions (CDRs) with the following sequences:CDR1 - GVSLPDY (SEQ ID No. 16); CDR2 - WGSET (SEQ ID No.
- CDR3 - HYYYGGSYAMDY SEQ ID No. 18
- VL light chain variable region
- CDR1 - RASQDISKYLN SEQ ID No. 19
- CDR2 - HTSRLHS SEQ ID No. 20
- CDR3 - QQGNTLPYT SEQ ID No. 21.
- the CDRs provided herein may be grafted on to a human or humanised framework. DESCRIPTION OF THE FIGURES
- FIG. 1 CAR-T cell expansion in first SLE patient treated with obe-cel.
- A) Flow cytometry of CD3+CAT19+ cells.
- Chimeric antigen receptors also known as chimeric T cell receptors, artificial T cell receptors and chimeric immunoreceptors, are engineered receptors, which graft an arbitrary specificity onto an immune effector cell.
- CARs also known as chimeric T cell receptors, artificial T cell receptors and chimeric immunoreceptors
- CAR-encoding nucleic acids may be transferred to T cells using, for example, retroviral vectors. In this way, a large number of CD19-specific T cells can be generated for adoptive cell transfer. Phase I clinical studies of this approach show efficacy.
- the target-antigen binding domain of a CAR is commonly fused via a spacer and transmembrane domain to an endodomain.
- the endodomain may comprise or associate with an intracellular T cell signaling domain.
- the human CD19 antigen is a 95 kd transmembrane glycoprotein belonging to the immunoglobulin superfamily.
- CD19 is classified as a type I transmembrane protein, with a single transmembrane domain, a cytoplasmic C-terminus, and extracellular N- terminus.
- CD19 is expressed very early in B-cell differentiation and is only lost at terminal B-cell differentiation into plasma cells.
- CD19 is a biomarker for normal B cells as well as follicular dendritic cells.
- CD19 primarily acts as a B cell co-receptor in conjunction with CD21 and CD81 . Upon activation, the cytoplasmic tail of CD19 becomes phosphorylated, which leads to binding by Src-family kinases and recruitment of PI-3 kinase.
- CD19 is also expressed on all B-cell malignancies but not multiple myeloma cells. It is not expressed on other haematopoietic populations or non-haematopoietic cells and therefore targeting this antigen should not lead to toxicity to the bone marrow or non-haematopoietic organs. Loss of the normal B-cell compartment is considered an acceptable toxicity when treating lymphoid malignancies, because although effective CD19 CAR T cell therapy will result in B cell aplasia, the consequent hypogammaglobulinaemia can be treated with pooled immunoglobulin.
- the antigen-binding domain of a CAR which binds to CD19 may be any domain which is capable of binding CD19.
- the antigen-binding domain may comprise a CD19 antigen-binding domain as described in Table 3.
- the gene encoding CD19 comprises ten exons: exons 1 to 4 encode the extracellular domain; exon 5 encodes the transmembrane domain; and exons 6 to 10 encode the cytoplasmic domain.
- the antigen-binding domain of a CD19 CAR herein may bind an epitope of CD19 encoded by exon 1 of the CD19 gene.
- the antigen-binding domain of a CD19 CAR herein may bind an epitope of CD19 encoded by exon 2 of the CD19 gene.
- the antigen-binding domain of a CD19 CAR herein may bind an epitope of CD19 encoded by exon 3 of the CD19 gene.
- the antigen-binding domain of a CD19 CAR herein may bind an epitope of CD19 encoded by exon 4 of the CD19 gene.
- a CD19-binding domain exemplified herein comprises variable regions with complementarity determining regions (CDRs) from an antibody referred to as CAT19, a) a heavy chain variable region (VH) having CAT19 CDRs with the following sequences:
- the CAT19 antibody is described in WO 2016/139487.
- Each CDR may, for example, have one, two or three amino acid mutations.
- the CDRs may be in the format of a single-chain variable fragment (scFv), which is a fusion protein of the heavy variable region (VH) and light chain variable region (VL) of an antibody, connected with a short linker peptide of ten to about 25 amino acids.
- scFv single-chain variable fragment
- VH heavy variable region
- VL light chain variable region
- the scFv may be in the orientation VH-VL, /.e., the VH is at the amino-terminus of the CAR molecule and the VL domain is linked to the spacer and, in turn the transmembrane domain and endodomain.
- the CDRs may be grafted on to the framework of a human antibody or scFv.
- the CAR may comprise a CD19-binding domain consisting or comprising one of the following sequences.
- the CD19 CAR may comprise the following VH sequence. SEQ ID No. 7 - VH sequence from CAT19 murine monoclonal antibody
- the CD19 CAR may comprise the following VL sequence.
- the CD19 CAR may comprise the following scFv sequence.
- the CAR may consist of or comprise one of the following sequences.
- “Campana” architecture refers to a CAR with a CD8a spacer and transmembrane domain, 4-1 BB endodomain and TCR CD3z endodomain.
- the CAR provided herein may comprise a variant of the polypeptide of SEQ ID Nos. 1-15 having at least 80, 85, 90, 95, 98 or 99% sequence identity, provided that the variant sequence retains the capacity to bind CD19 (when in conjunction with a complementary VL or VH domain, if appropriate).
- the percentage identity between two polypeptide sequences may be readily determined by programs such as BLAST which is freely available at blast.ncbi.nlm.nih.gov.
- the CD19 CAR exemplified herein (/.e., the CAT19CAR using “Campana” architecture, SEQ ID No. 10) has properties contemplated by the disclosure to result in lower toxicity and better efficacy in treated patients.
- the CAT19CAR exemplified herein effected killing of target cells expressing CD19 and proliferated in response to CD19 expressing targets, but Interferon-gamma release was less.
- a small animal model of an aggressive B-cell lymphoma showed equal efficacy and equal engraftment between the fmc63- and CAT 19-based CAR-T cells, but surprisingly, less of the CAT 19 CAR T cells were exhausted than fmc63 CAR T cells. See, Examples 2 and 3 of US Publication No.: 2018-0044417.
- the CAT19CAR provided herein may cause 25, 50, 70 or 90% lower IFNy release in a comparative assay involving bringing CAR T cells into contact with target cells.
- the CAT19CAR provided herein may result in a smaller proportion of CAR T cells becoming exhausted than fmc63 CAR T cells. T cell exhaustion may be assessed using methods known in the art, such as analysis of PD-1 expression.
- the CAR may cause 20, 30, 40, 50, 60 of 70% fewer CAR T cells to express PD-1 that fmc63 CAR T cells in a comparative assay involving bringing CAR T cells into contact with target cells.
- CD19 antigen-binding domain contemplated by the disclosure is based on the CD19 antigen-binding domain CD19ALAb (described in WO20 16/102965) and comprises: a) a heavy chain variable region (VH) having CDRs with the following sequences:
- Each CDR may, for example, have one, two or three amino acid mutations.
- the CAR may comprise one of the following amino acid sequences.
- SEQ ID No. 22 Murine CD19ALAb scFv sequence QVQLQQSGAELVRPGSSVKISCKASGYAFSSYWMNWVKQRPGQGLEWIGQIWPG DGDTNYNGKFKGKATLTADESSSTAYMQLSSLASEDSAVYFCARRETTTVGRYYYA MDYWGQGTTVTVSSDIQLTQSPASLAVSLGQRATISCKASQSVDYDGDSYLNWYQ QIPGQPPKLLIYDASNLVSGIPPRFSGSGSGTDFTLNIHPVEKVDAATYHCQQSTEDP
- the scFv may be in a VH-VL orientation (as shown in SEQ ID NO:s 9, 22, 23 and 24) or a VL-VH orientation.
- the CAR may comprise one of the following VH sequences:
- the CAR may comprise one of the following VL sequences:
- the CAR provided herein may comprise a variant of the sequence shown as any of SEQ ID Nos. 16-29 having at least 80, 85, 90, 95, 98 or 99% sequence identity, provided that the variant sequence retains the capacity to bind CD19 (when in conjunction with a complementary VL or VH domain, if appropriate).
- the CAR provided herein may comprise a variant of the sequence shown as any of SEQ ID Nos. 22-29 or 44-49 having at least 80, 85, 90, 95, 98 or 99% sequence identity, provided that the variant sequence retains the capacity to bind CD19 (when in conjunction with a complementary VL or VH domain, if appropriate).
- the percentage identity between two polypeptide sequences may be readily determined by programs such as BLAST which is freely available at blast.ncbi.nlm.nih.gov.
- the transmembrane domain is the domain of the CAR that spans the membrane.
- a transmembrane domain may be any protein structure which is thermodynamically stable in a membrane. This is typically an alpha helix comprising of several hydrophobic residues.
- the transmembrane domain of any transmembrane protein can be used to supply the transmembrane portion provided herein.
- the presence and span of a transmembrane domain of a protein can be determined by those skilled in the art using the TMHMM algorithm (http://www.cbs. dtu.dk/services/TMHMM-2.0/).
- transmembrane domain of a protein is a relatively simple structure, i.e, a polypeptide predicted to form a hydrophobic alpha helix of sufficient length to span the membrane
- an artificially designed transmembrane domain may also be used (US 7052906 B1 describes synthetic transmembrane components).
- the transmembrane domain may be derived from CD28, which gives good receptor stability.
- the CD28 transmembrane domain is shown as SEQ ID No. 50. SEQ ID No. 50
- the transmembrane domain may be derived from human Tyrp-1 .
- the tyrp-1 transmembrane domain sequence is shown as SEQ ID No. 51.
- the transmembrane domain may be derived from CD8A.
- the CD8A transmembrane domain sequence is shown as SEQ ID NO: 52.
- the endodomain is the signal-transmission portion of the CAR. After antigen recognition, receptors cluster and a signal is transmitted to the cell.
- the most commonly used endodomain component is that of CD3-zeta which contains 3 ITAMs. This transmits an activation signal to the T cell after antigen is bound.
- CD3-zeta may not provide a fully competent activation signal and additional co-stimulatory signaling may be needed.
- endodomains from CD28, or 0X40 or 41 BB can be used with CD3-Zeta to transmit a proliferative I survival signal, or all three can be used together.
- the endodomain of the CAR of the present invention may comprise combinations of one or more of the CD3-Zeta endodomain, the 41 BB endodomain, the 0X40 endodomain or the CD28 endodomain.
- the intracellular T cell signalling domain (endodomain) of the CAR of the present invention may comprise the sequence shown as SEQ ID Nos. 53, 54, 55, 56, 57, 30, 31 or 32 or a variant thereof having at least 80% sequence identity.
- Examples of combinations of such endodomains include 41 BB-Z, QX40-Z, CD28-Z and CD28-QX40-Zeta.
- a variant sequence may have at least 80%, 85%, 90%, 95%, 98% or 99% sequence identity to SEQ ID Nos. 25, 26, 27, 28, 29, 30, 31 or 32 provided that the sequence provides an effective transmembrane domain/intracellular T cell signaling domain.
- the CAR of the present invention may comprise a signal peptide so that when the CAR is expressed inside a cell, such as a T cell, the nascent protein is directed to the endoplasmic reticulum and subsequently to the cell surface, where it is expressed.
- the core of the signal peptide may contain a long stretch of hydrophobic amino acids that has a tendency to form a single alpha-helix.
- the signal peptide may begin with a short positively charged stretch of amino acids, which helps to enforce proper topology of the polypeptide during translocation.
- At the end of the signal peptide there is typically a stretch of amino acids that is recognized and cleaved by signal peptidase.
- Signal peptidase may cleave either during or after completion of translocation to generate a free signal peptide and a mature protein.
- the free signal peptides are then digested by specific proteases.
- the signal peptide may be at the amino terminus of the molecule.
- the CAR of the invention may have the general formula:
- Signal peptide - CD19-binding domain - spacer domain - transmembrane domain/intracellular T cell signaling domain may comprise a variant having 5, 4, 3, 2 or 1 amino acid mutations (insertions, substitutions, or additions) provided that the signal peptide still functions to cause cell surface expression of the CAR.
- the signal peptide of SEQ ID No. 33 is compact and highly efficient. It is predicted to give about 95% cleavage after the terminal glycine, giving efficient removal by signal peptidase.
- the signal peptide of SEQ ID No. 34 is compact and highly efficient. It is predicted to give about 95% cleavage after the terminal glycine, giving efficient removal by signal peptidase.
- the signal peptide of SEQ ID NO: 35 is derived from lgG1.
- the signal peptide of SEQ ID NO: 36 is derived from CD8.
- the CAR of the present invention may comprise a spacer sequence to connect the CD19-binding domain with the transmembrane domain and spatially separate the CD19-binding domain from the endodomain.
- a flexible spacer allows to the CD19- binding domain to orient in different directions to enable CD19 binding.
- the spacer sequence may, for example, comprise an lgG1 Fc region, an lgG1 hinge or a CD8 stalk, or a combination thereof.
- the spacer may alternatively comprise an alternative sequence which has similar length and/or domain spacing properties as an IgG 1 Fc region, an lgG1 hinge or a CD8 stalk.
- a human I gG 1 spacer may be altered to remove Fc binding motifs. Examples of amino acid sequences for these spacers are given below:
- SEQ ID No. 39 (human lgG1 hinge):
- SEQ ID No. 41 (lgG1 Hinge - Fc modified to remove Fc receptor recognition motifs)
- Modified residues are underlined; * denotes a deletion.
- SEQ ID NO: 42 (CD2 ectodomain)
- SEQ ID NO: 43 (CD34 ectodomain) SLDNNGTATPELPTQGTFSNVSTNVSYQETTTPSTLGSTSLHPVSQHGNEATTNITE TTVKFTSTSVITSVYGNTNSSVQSQTSVISTVFTTPANVSTPETTLKPSLSPGNVSDL STTSTSLATSPTKPYTSSSPILSDIKAEIKCSGIREVKLTQGICLEQNKTSSCAEFKKD RGEGLARVLCGEEQADADAGAQVCSLLLAQSEVRPQCLLLVLANRTEISSKLQLMK KHQSDLKKLGI LDFTEQDVASHQSYSQKT
- CD19 CAR based on the CAT19 scFv has properties which may result in lower toxicity and better efficacy.
- a small animal model of an aggressive B-cell lymphoma showed equal efficacy and equal engraftment between the fmc63 and CAT19 based CARs, but surprisingly, less of the CAT19 CAR T cells were exhausted than fmc63 CAR T cells.
- the CAR of the invention may cause 25, 50, 70 or 90% lower IFNy release in a comparative assay involving bringing CAR T cells into contact with target cells.
- the CAR of the invention may result in a smaller proportion of CAR T cells becoming exhausted than fmc63 CAR T cells. T cell exhaustion may be assessed using methods known in the art, such as analysis of PD-1 expression.
- the CAR of the present invention may cause 20, 30, 40, 50, 60 of 70% fewer CAR T cells to express PD-1 that fmc63 CAR T cells in a comparative assay involving bringing CAR T cells into contact with target cells.
- the second aspect of the invention relates to a nucleic acid sequence which codes for a CAR of the methods herein.
- the nucleic acid sequence may be capable of encoding a CAR having the amino acid sequence shown as any of SEQ ID Nos. 10-15.
- polynucleotide As used herein, the terms “polynucleotide”, “nucleotide”, and “nucleic acid” are intended to be synonymous with each other.
- the nucleic acid may be, for example, an RNA, a DNA or a cDNA.
- Nucleic acids may comprise DNA or RNA. They may be single-stranded or double-stranded. They may also be polynucleotides which include within them synthetic or modified nucleotides.
- oligonucleotides A number of different types of modification to oligonucleotides are known in the art. These include methylphosphonate and phosphorothioate backbones, addition of acridine or polylysine chains at the 3' and/or 5' ends of the molecule.
- polynucleotides may be modified by any method available in the art. Such modifications may be carried out in order to enhance the in vivo activity or life span of polynucleotides of interest.
- Alternative codons may be used in regions of sequence encoding the same or similar amino acid sequences, in order to avoid homologous recombination when both CARs are encoded by the same vector.
- codons “ccg” and “cca” both encode the amino acid proline, so using “ccg” may be exchanged for “cca” without affecting the amino acid in this position in the sequence of the translated protein.
- RNA codons which may be used to encode each amino acid are summarized in Table 4.
- Alternative codons may be used in the portions of nucleic acid which encode the spacer of the first CAR and the spacer of the second CAR, especially if the same or similar spacers are used in the first and second CARs.
- Alternative codons may be used in the portions of nucleic acid which encode the transmembrane domain of the first CAR and the transmembrane of the second CAR, especially if the same or similar transmembrane domains are used in the first and second CARs.
- Alternative codons may be used in one or more nucleic acids which encode costimulatory domains, such as the CD28 endodomain.
- Alternative codons may be used in one or more domains which transmit survival signals, such as 0X40 and 41 BB endodomains.
- Alternative codons may be used in the portions of nucleic acid encoding a CD3zeta endodomain and/or the portions of nucleic acid encoding one or more costimulatory domain(s) and/or the portions of nucleic acid encoding one or more domain(s) which transmit survival signals.
- the present invention also provides a vector which comprises a nucleic acid sequence according to the present invention.
- a vector may be used to introduce the nucleic acid sequence into a host cell so that it expresses and produces a molecule according to the first aspect of the invention.
- the vector may, for example, be a plasmid or a viral vector, such as a retroviral vector or a lentiviral vector.
- the vector may be capable of transfecting or transducing a cell, such as a T cell.
- the invention also provides a cell which comprises a nucleic acid according to the invention.
- the invention provides a cell which expresses a CAR provided herein at the cell surface.
- the cell may be any eukaryotic cell capable of expressing a CAR at the cell surface, such as an immunological cell.
- the cell may be an immune effector cell such as a T cell or a natural killer (NK) cell.
- an immune effector cell such as a T cell or a natural killer (NK) cell.
- T cells or T lymphocytes are a type of lymphocyte that play a central role in cell- mediated immunity. They can be distinguished from other lymphocytes, such as B cells and natural killer cells (NK cells), by the presence of a T cell receptor (TCR) on the cell surface.
- TCR T cell receptor
- Helper T helper cells assist other white blood cells in immunologic processes, including maturation of B cells into plasma cells and memory B cells, and activation of cytotoxic T cells and macrophages.
- TH cells express CD4 on their surface.
- TH cells become activated when they are presented with peptide antigens by MHC class II molecules on the surface of antigen presenting cells (APCs).
- APCs antigen presenting cells
- TH1 , TH2, TH3, TH 17, Th9, or TFH which secrete different cytokines to facilitate different types of immune responses.
- Cytotoxic T cells TC cells, or CTLs
- CTLs destroy vi rally infected cells and tumor cells, and are also implicated in transplant rejection.
- CTLs express the CD8 at their surface.
- CD8+ cells recognize their targets by binding to antigen associated with MHC class I, which is present on the surface of all nucleated cells.
- MHC class I MHC class I
- IL-10 adenosine and other molecules secreted by regulatory T cells, the CD8+ cells can be inactivated to an anergic state, which prevent autoimmune diseases such as experimental autoimmune encephalomyelitis.
- Memory T cells are a subset of antigen-specific T cells that persist long-term after an infection has resolved. They quickly expand to large numbers of effector T cells upon re-exposure to their cognate antigen, thus providing the immune system with "memory" against past infections.
- Memory T cells comprise three subtypes: central memory T cells (TCM cells) and two types of effector memory T cells (TEM cells and TEMRA cells). Memory cells may be either CD4+ or CD8+. Memory T cells typically express the cell surface protein CD45RO.
- Treg cells Regulatory T cells
- suppressor T cells are crucial for the maintenance of immunological tolerance. Their major role is to shut down T cell- mediated immunity toward the end of an immune reaction and to suppress auto- reactive T cells that escaped the process of negative selection in the thymus.
- Treg cells Two major classes of CD4+ Treg cells have been described — naturally occurring Treg cells and adaptive Treg cells.
- Naturally occurring Treg cells arise in the thymus and have been linked to interactions between developing T cells with both myeloid (CD11c+) and plasmacytoid (CD123+) dendritic cells that have been activated with TSLP.
- Naturally occurring Treg cells can be distinguished from other T cells by the presence of an intracellular molecule called FoxP3. Mutations of the FOXP3 gene can prevent regulatory T cell development, causing the fatal autoimmune disease IPEX.
- Adaptive Treg cells may originate during a normal immune response.
- the T cell provided herein may be any of the T cell types mentioned above, in particular a CTL.
- Natural killer (NK) cells are a type of cytolytic cell which forms part of the innate immune system. NK cells provide rapid responses to innate signals from virally infected cells in an MHC independent manner.
- NK cells (belonging to the group of innate lymphoid cells) are defined as large granular lymphocytes (LGL) and constitute the third kind of cells differentiated from the common lymphoid progenitor generating B and T lymphocytes. NK cells are known to differentiate and mature in the bone marrow, lymph node, spleen, tonsils and thymus where they then enter into the circulation.
- LGL large granular lymphocytes
- the CAR-expressing cells provided herein may be any of the cell types mentioned above.
- T or NK cells provided herein may be derived from ex vivo differentiation of inducible progenitor cells or embryonic progenitor cells to T or NK cells.
- an immortalized T cell line which retains its lytic function and could act as a therapeutic may be used.
- Binding kinetics of the CAT scFv (used in obe-cel) and FMC63 scFv (used in axicabtagene ciloleucel and tisagenlecleucel) with recombinant CD19 were determined using surface plasmon resonance.
- Equilibrium dissociation constants (KD) of 14.4 nM for CAT and 0.328 nM for FMC63 were determined, when the data were fitted to a 1 :1 Langmuir binding model.
- CD19 (CAT) CAR T cell proliferation was greater than CD19 (FMC63) CAR T cells when co-cultured with Raji and NALM-6 cells.
- the enhanced proliferation was not a result of increased IL-2 production indicating an IL-2 independent mechanism.
- CAT CD19
- FMC63 CD19
- Raji cells a Burkitt’s lymphoma cell line that expresses CD19
- pro-inflammatory cytokines were analysed in the supernatant at 48 hours.
- T cells transduced to express CD19 (CAT) CAR or CD19 (FMC63) CAR were incubated with CD19-negative and CD19 expressing targets.
- Cell killing was significantly greater for CD 19 (CAT) CAR T cells than for CD19 (FMC63) CAR T cells, particularly at low effectortarget ratios.
- CD19 (CAT) CAR T cells and CD19 (FMC63) CAR T cells were assessed in a NALM-6 NSG-mouse xenograft model and compared. In control mice receiving non-transduced T cells, rapid, disseminated tumour infiltration was observed. CD19 (FMC63) CAR T cells slowed but did not prevent growth of the tumour. In contrast, equivalent numbers of CD19 (CAT) CAR T cells resulted in tumour regression.
- the CAT scFv binding domain used in obe-cel has a > 40-fold lower affinity to recombinant CD19 in vitro than the FMC63 scFv binding domain used in other CAR T products.
- CD19 (CAT) CAR enables transduced T cells to proliferate, secrete cytokines in response to CD19-positive targets and specifically lyse CD19-positive cell lines in vitro with greater cytotoxicity compared to CD19 (FMC63) CAR T cells.
- CD19 (CAT) CAR T cells also have better anti-tumour efficacy and engraftment versus CD19 (FMC63) CAR T cells in an NSG NALM-6 mouse model of leukaemia.
- ALLCAR19 (EudraCT 2016-004027-22, NCT02935257 2017) (Roddie C, Dias J, O'Reiilly MA, et al (2021). Durable Responses and Low Toxicity After Fast Off-Rate CD19 Chimeric Antigen Receptor-T Therapy in Adults With Relapsed or Refractory B- Cell Acute Lymphoblastic Leukemia. J Clin Oncol; 39(30):3352-63; Roddie C, Dias J, O’Reilly M, Mitsikakou M, et al. (2022a).
- the Phase II portion of the FELIX study is the pivotal study to achieve the marketing authorization of obe-cel (Roddie C, Sandhu KS, Tholouli E, Shaughnessy P, et al. (2023a).
- Safety and effiacy of obecabtagene autoleucel (obe-cel, AUTO1), a fast-off rate CD19 CAR, in relapsed/refractory adult B-cell acute lymphoblastic leukemia (r/r IB- ALL): top line result of the pivotal FELIX study.
- r/r IB- ALL relapsed/refractory adult B-cell acute lymphoblastic leukemia
- Severe neurotoxicity was limited with 1 patient in the CARPALL study (5%); 3 patients in the ALLCAR19 study (15%) and 9 patients in the FELIX study (7.1 %) reporting >grade 3 neurological AE/ICANS.
- Preliminary results obtained in indolent B NHL look promising with all 10 patients treated with obe-cel achieving complete molecular response (data cut-off 18-Nov- 2021).
- the safety profile remains favourable in this small sample size with four patients with grade 1 , two patients with grade 2 CRS and no ICANS of any grade reported.
- Cells such as T cells or NK cells expressing a CAR molecule provided herein may be used for the treatment of an autoimmune disease, in particular an autoimmune disease mediated by CD19-positive B lymphocytes or B cells.
- the present disclosure provides a method for treating an autoimmune disease mediated by CD19-positive B lymphocytes in a patient comprising administering to the patient an CD19 CAR T cell or a population of CD19 CAR T cells.
- the present disclosure provides a CD19 CAR T cell or a population of CD19 CAR T cells for use in treating an autoimmune disease mediated by CD19-positive B lymphocytes.
- the present disclosure provides the use of a CD19 CAR T cell or a population of CD19 CAR T cells in the manufacture of a medicament for treating an autoimmune disease mediated by CD19-positive B lymphocytes.
- autoimmune diseases include, but are not limited to, systemic lupus erythematosus (SLE), rheumatoid arthritis, idiopathic inflammatory myopathy (IIM, myositis), systemic sclerosis, ANCA-associated vasculitis, inflammatory bowel disease (IBD), multiple sclerosis (MS), Type I diabetes, Guillain-Barre syndrome, chronic inflammatory demyelinating polyneuropathy (CIDP), Graves’ disease, Hashimoto’s thyroiditis or Hashimoto’s disease, myasthenia gravis, neuromyelitis optica, A/-methyl- D-aspartate receptor (NMDAR) encephalitis, Lambert-Eaton syndrome, scleroderma, vasculitis, pemphigus vulgaris, pemphigus foliaceus, epidermolysis bullosa acquisita, bullous pemphigoid, lupus nephritis, membranous l
- the autoimmune disease may be SLE.
- the autoimmune disease may be severe SLE.
- the autoimmune disease may be refractory SLE.
- the autoimmune disease may be severe, refractory SLE.
- the Lupus Nephritis may be severe.
- the Lupus Nephritis may be active.
- the SLE may present with active, severe Lupus Nephritis.
- Methods for treating autoimmune disease relate to the therapeutic use of a cell or population of cells provided herein.
- the cells may be administered to a subject having an existing disease or condition in order to lessen, reduce or improve at least one symptom associated with the disease and/or to slow down, reduce or block the progression of the disease.
- the method may cause or promote cell mediated killing of CD19-expressing cells, such as B cells.
- CD19 CAR T cell product for example, obe-cel
- infusion of a patient with a CD19 CAR T cell product will eliminate the malfunctioning autoreactive B cells and subsequently, normal, non-autoreactive B cells are produced.
- Patients to be treated include, but are not limited to, patients with severe, refractory SLE who are predicted to have a poor outcome if treated with standard of care therapy due to disease severity, one or more areas of SLE-related organ system involvement, and prior exposure to multiple SLE treatment strategies.
- Patients to be treated may have a diagnosis of SLE fulfilling the 2019 European League against Rheumatism (EULAR)/American College of Rheumatology (ACR) Classification Criteria for Systemic Lupus Erythematosus; may be positive for at least one of the following autoantibodies: antinuclear antibodies (ANA) at a titer of >1 :80, or anti-dsDNA (>30 IIJ/mL) or anti-Sm (> ULN), anti-histone or anti-chromatin (> ULN); may have severe SLE defined as a) a Systemic Lupus Erythematosus Disease Activity Index 2000 (SLEDAI-2K) score of > 8 points (of which 4 are non-laboratory items and with the exclusion of points associated with neurological findings [SLEDAI 2K items 1-7]) and b) at least one of the following significant SLE-related organ involvements: i) renal (ongoing active, biopsy-proven lupus nephritis), ii
- Patients may be subject to a pre-conditioning chemotherapy such as lymphodepletion chemotherapy prior to CD19 CAR treatment.
- the lymphodepletion chemotherapy may comprise treatment with fludarabine and cyclophosphamide.
- the patients may receive three doses of fludarabine 25 mg/m 2 /d intravenous administration on Day -5, Day -4, and Day -3 relative to CD 19 CAR treatment and one dose of cyclophosphamide 1000 mg/m 2 intravenous administration on Day -3 relative to CD 19 CAR treatment.
- the patients may be administered a single dose of 50 x 10 6 ( ⁇ 20%) CD19 CAR-positive viable T cells.
- Alternative doses contemplated are an escalated (100 x 10 6 [ ⁇ 20%]) or a de-escalated (30 x 10 6 [ ⁇ 20%]) CAR-positive T cell dose.
- the administration may be, for example, an intravenous injection through a Hickman line or peripherally inserted central catheter (PICC line).
- the starting material leukapheresate
- manufacturing of the CD19 CAR-positive T cells may lead to dose variability, i.e. variability in the amount of CD19 CARpositive T cells that is administered to the patient. It is contemplated herein that the dose may have a variability of ⁇ 5%, ⁇ 10%, ⁇ 15%, ⁇ 20%, ⁇ 25%, or ⁇ 30%.
- LDAS Lupus Low Disease Activity State
- improvements may include SLEDAI-2K ⁇ 4 (no activity in major organ systems (renal, central nervous system, cardiopulmonary, vasculitis, fever and no hemolytic anemia or gastrointestinal activity), no new features of lupus disease activity compared to the previous assessment, PGA ⁇ 1 on a visual analog scale from 0-3, current prednisolone (or equivalent) dose ⁇ 7.5 mg daily, and/or well-tolerated standard maintenance doses of immunosuppressive drugs and approved biologic agents, excluding investigational drugs;
- SLEDAI-2K Systemic Lupus Erythematosus Disease Activity Index-2000
- Physician Physician’s global assessment (PGA);
- HAQ-DI Health Assessment Questionnaire - Disability Index
- Autoantibody panel anti-double stranded DNA [anti- dsDNA], anti-Smith, anti-RNA binding protein [anti-RBP]);
- Antiphospholipid profile (lupus anticoagulant, anti-cardiolipin antibodies and beta-2 glycoprotein 1);
- T and B cell phenotype Determination of T and B cell phenotype over time as assessed by flow cytometry in the peripheral blood;
- Obe-cel obecabtagene autoleucel also referred to as ALITO1
- CAR T cell-based therapies such as obe-cel provide further advantages over antibodies because CAR T cells require only a single administration, expand, and have the potential to persist in vivo, migrate to multiple lymphoid tissues and organs in the recipient and develop into both effector and memory cell population. Longer persistency of obe-cel with its CAT CD19 binder, when indirectly compared with the approved FMC63 CD19 CAR T cells, provides another advantage.
- obe-cel is prepared from enriched autologous T cells that are transduced with a lentiviral vector to express a novel, second generation CAR targeting CD19 with a 4-1 BB and CD3- endodomain.
- the CAR of the obe-cel CAR T cells comprises a single chain variable fragment (scFv) called CAT with a lower affinity for CD19 and a faster off rate compared with that of the FMC63 scFv used in other approved CD19 CAR T therapies.
- This fast-off binding kinetics is contemplated to confer an opportunity for physiological T cell activation, reduced immune-toxicity and potential for improved persistence. See, WO20 16/139487.
- Obe-cel is classified as an advanced therapy investigational medicinal product (ATIMP). It is an autologous cellular therapy product where the active component is the patient’s own T cells transduced with a lentiviral vector to express a novel second generation CAR targeting CD19 (sometimes referred to herein as “CD19 CAT CAR”). Obe-cel also contains non-transduced autologous T cells and non-T cells.
- ATIMP advanced therapy investigational medicinal product
- CAR T cells Once administered to patients, obe-cel CAR T cells circulate throughout the body.
- the CAR T cells recognize CD19 surface antigens on all B cells and B-progenitor cells, resulting in activation and proliferation of the CAR T cells. Activated CAR T cells will then in turn cause destruction of these cells via several physiological effector mechanisms.
- the patients treated are patients with severe, refractory SLE who are predicted to have a poor outcome if treated with standard of care therapy due to disease severity, one or more areas of SLE-related organ system involvement, and prior exposure to multiple SLE treatment strategies.
- antinuclear antibodies at a titer of >1:80, or anti-dsDNA (>30 IIJ/mL) or antiSmith (> ULN), anti-histone or anti-chromatin (> ULN).
- SLEDAI-2K A Systemic Lupus Erythematosus Disease Activity Index 2000 (SLEDAI-2K) score of > 8 points (of which 4 are non-laboratory items and with the exclusion of points associated with neurological findings [SLEDAI 2K items 1-7]) AND
- Severe hematologic manifestation e.g., severe thrombocytopenia or severe autoimmune hemolytic anemia
- Refractory SLE (defined as lack of response, insufficient response or lack of sustained response) or intolerance to: a. Hydroxychloroquine treatment in combination with corticosteroids AND b. s2 of the following treatment groups used for at least 3 months each or less if intolerant: i Immunosuppressive drugs (e.g., methotrexate, azathioprine, mycophenolate mofetil, mycophenolic acid, tacrolimus, leflunomide, cyclosporine, voclosporin or cyclophosphamide) ii. B cell-targeting agents (e.g., belimumab, anti-CD20 mAb) iii Cytokine inhibitors (e.g., anifrolumab)
- Immunosuppressive drugs e.g., methotrexate, azathioprine, mycophenolate mofetil, mycophenolic acid, tacrolimus, leflunomide, cyclosporine,
- non-SLE autoimmune disease e.g., dermatomyositis, polymyositis, scleroderma, rheumatoid arthritis
- other non-SLE autoimmune disease or overlap syndrome e.g., dermatomyositis, polymyositis, scleroderma, rheumatoid arthritis
- other non-SLE autoimmune disease or overlap syndrome e.g., dermatomyositis, polymyositis, scleroderma, rheumatoid arthritis
- CNS pathology such as epilepsy, paresis, aphasia, or stroke
- Prior treatment with anti-CD19 therapy including bispecifics, adoptive T cell therapy or any prior gene therapy product (e.g., CAR T cell therapy).
- indwelling line or drain e.g., percutaneous nephrostomy tube, indwelling Foley catheter, biliary drain, or pleural/peritoneal/pericardial catheter.
- indwelling line or drain e.g., percutaneous nephrostomy tube, indwelling Foley catheter, biliary drain, or pleural/peritoneal/pericardial catheter.
- Central venous access catheters such as a Port-a-Cath or Hickman catheter are permitted.
- patients may receive bridging therapy with permitted standard of care (SOC) medications to treat SLE.
- SOC standard of care
- Lymphodepletion chemotherapy with fludarabine and cyclophosphamide is recognized as standard of care before CAR T cell infusion.
- Clinical CAR T cell data collectively indicate that Fludarabine/Cyclophosphamide are required for CAR T cell engraftment, and CAR T cell engraftment is required for activity towards B cells
- This lymphodepletion regimen matches the one published by Mackensen et al, 2022.
- lymphodepletion therapy may be repeated.
- the starting material for generation of obe-cel is mononuclear cells from a nonmobilized leukapheresis collection from the patient.
- obe-cel is an autologous product and is therefore specific for each patient.
- the autologous T cells are enriched and transduced ex vivo with a lentiviral vector to express a novel CD19-targeting (i.e., anti-CD19) CAR referred to as “CD19 (CAT) CAR.”
- CD19 (CAT) CAR novel CD19-targeting CAR referred to as “CD19 (CAT) CAR.”
- the “CAT” binding domain was chosen as a binding domain as it showed a substantially lower affinity to CD19 (>40 fold) than FMC63 scFv, a binder used in already marketed CAR T cell therapies
- the CD19 (CAT) CAR construct is derived from a murine CAT13.1 E10 hybridoma, where the CAR comprises an anti-CD19 scFv, CD8-derived stalk and trans-membrane domain, and a compound endodomain fusion of 4-1 BB- CD3 endodomains.
- Obe-cel is generated by ex vivo transduction of activated peripheral blood mononuclear cells using an engineered HIV derived lentiviral vector containing the CD19 CAR expression cassette.
- the lentiviral vector is produced under Good Manufacturing Practice (GMP) conditions by four plasmid co transfection of HEK293T cells and subsequent harvest and purification of the culture supernatant.
- GMP Good Manufacturing Practice
- Cells from the leukapheresis starting material are enriched by positive selection for CD4+/CD8+ cells using magnetic beads on the Miltenyi CliniMACS® Prodigy bioreactor. Cells are then washed and stimulated with mitogenic ligands and cytokines. One day after activation, cells are transduced with the lentiviral vector. Posttransduction, cells are expanded (drug substance) to produce the desired dose. The cells are then washed and formulated with a phosphate-buffered saline (PBS)/ ethylenediaminetetraacetic acid (EDTA) I human serum albumin (HSA) I dimethyl sulfoxide (DMSO) buffer and filled into final packaging and cryopreserved (drug product). The obe-cel product comprises transduced and non-transduced T cells. The dose given to patients is expressed as the total number of CD19 CAR-positive T cells (the active substance).
- PBS phosphate-buffered saline
- Obe-cel is supplied as a cryopreserved cell suspension in a cryostorage-compatible infusion bag, to be thawed at 37°C.
- the selection and justification for the obe-cel dose is based in part on the prior use of obe-cel in patients with B-ALL and other B cell malignancies.
- a single dose of 50 x 10 6 ( ⁇ 20%) CD19 CAR-positive viable T cells is to be infused to patients on Day 1 after lymphodepletion chemotherapy.
- Other doses contemplated are an escalated (100 x 10 6 [ ⁇ 20%]) or a de-escalated (30 x 10 6 [ ⁇ 20%]) CAR-positive T cell dose.
- the starting dose of 50 x 10 6 ( ⁇ 20%) CD19 CAR-positive viable T cells is 8-fold lower than the total obe-cel target dose utilized in adult B-ALL patients, which have demonstrated a tolerable safety profile in the B-ALL patient population.
- the mean (standard deviation) body weight in two large, randomized phase 3 belimumab studies in a total of 865 SLE patients was 61 ⁇ 13 kg, the lowest body weight in these studies was 35 kg (US Food & Drug Administration. Arthritis advisory committee meeting briefing document BLA 125370. October 2010) suggesting that indeed a flat dose of 50 x 10 6 [ ⁇ 20%]) CARpositive T cells is contemplated to be safe and effective.
- Example 5 Obe-cel infusion and assessments of disease activity and response to treatment
- IV intravenously
- Obe-cel is infused on Day 1.
- obe-cel is thawed in a 37°C water bath under sterile conditions.
- Obe-cel is given as an IV infusion using a syringe or gravity-assisted infusion through a central or peripheral venous line through an 18-gauge blood set over a few minutes (maximum 30 minutes from obe-cel being thawed to preserve cell viability).
- Example 6 A single-arm, open-label, Phase I study to determine the safety, tolerability and preliminary efficacy of obecabtagene autoleucel (obe-cel) in patients with severe, refractory systemic lupus erythematosus (SLE)
- Participants are expected to receive a single infusion of obe-cel at a target dose of 50 x 10 6 ( ⁇ 25%) CD19 CAR-positive T cells.
- the primary endpoint is the proportion of participants achieving a CRR at Month 12 after obe-cel infusion as adjudicated by an Independent Response Review Committee (IRRC).
- IRRC Independent Response Review Committee
- the study may be stopped due to insufficient response, or excessive treatment toxicity.
- the study comprises 3 periods:
- Screening Period From Day -30 to Day of Enrollment a. Screening: After informed consent is obtained, participants are to undergo screening procedures to confirm they are eligible for the study and are able to undergo leukapheresis. b. Leukapheresis: Eligible participants are to proceed to leukapheresis to collect mononuclear cells to be used for manufacturing obe-cel. c. Enrollment: A participant is to be enrolled into the study at the point they meet all eligibility criteria and the participant’s leukapheresis material has been accepted for manufacturing.
- Treatment Period From Day of Enrollment to Day 1 to End of Treatment (EOT)/Day 1 a. Lymphodepletion (LD) Evaluation: Participants are to undergo an evaluation from Day -8 to Day -6 to confirm eligibility for start of LD. b. LD Chemotherapy: Participants are to receive intravenous (i.v.) fludarabine and cyclophosphamide for LD to enhance treatment efficacy and CD19 CAR T cell survival.
- EOT End of Treatment
- LD Lymphodepletion
- Fludarabine 25 mg/m 2 /day i.v. on Day -5, Day -4, and Day -3
- Cyclophosphamide 1 ,000 mg/m 2 i.v. on Day -3
- Day -1 the next step is the infusion, defined as Day 1.
- the EOT is defined as the day of infusion (Day 1) or the day of premature study discontinuation if prior to infusion.
- Efficacy and Safety follow-up: a. After the infusion, all participants are to be closely monitored for safety and efficacy. The first efficacy assessment is to be performed at Day 28 post-infusion. b. Thereafter, participants are to be followed with monthly or 3-monthly assessments of safety, tolerability, and efficacy until the EOS. c. Following the Month 24 visit, participants are to be followed every 3 months until the last participant, last visit (LPLV) is completed for the study (i.e., when the last enrolled participant reaches Month 24). The last 3-monthly follow-up assessment occurring prior to LPLV is to determine the EOS date for each individual participant. d. The EOS is defined as completion of the LPLV for the study.
- Severe, Active SLE defined as: o ASLEDAI-2K score of > 8 points (of which 4 are non-laboratory items and with the exclusion of points associated with neurological findings [SLEDAI-2K items 1-7]), and
- Severe active LN defined as: o SLE-related renal organ involvement. A biopsy must be performed in the 6 months prior to the screening visit or during the screening period to confirm ongoing, active, class III, IV or V (V only in combination with class III or IV) active or active/chronic LN in accordance with kidney biopsy evidence of International Society of Nephrology/Renal Pathology Society 2019.
- o UPGR > 1 mg/mg from a 24-hour urine collection, and eGFR of > 30 mL/min/1.73 m 2
- Refractory SLE (defined as lack of response, insufficient response, or lack of sustained response or intolerance related to such drugs not allowing their further use): a. hydroxychloroquine in combination with corticosteroids AND b. to > 2 of the following treatment groups used for at least 6 months: i. Immunosuppressive drugs (e.g., methotrexate, azathioprine, mycophenolate mofetil, mycophenolic acid, leflunomide, or cyclophosphamide). ii. Calcineurin inhibitors (e.g., cyclosporine, voclosporin, tacrolimus). iii. B cell-targeting agents (e.g., belimumab, anti-CD20 mAb) OR cytokine inhibitors (e.g., anifrolumab).
- Immunosuppressive drugs e.g., methotrexate, azathioprine, mycophenolate mofetil, mycophenolic acid
- LD lymphodepletion
- SLE systemic lupus erythematosus
- Example 7 A single-arm, open-label, Phase I study to determine the safety, tolerability and preliminary efficacy of obecabtagene autoleucel (obe-cel) in patients with severe, refractory systemic lupus erythematosus (SLE): Results of first treated patient
- a 32-year-old female SLE patient was treated with obe-cel. This patient was black or African American, with a SLE diagnosis in 2021, showing renal (LN class IVA/), musculoskeletal, dermal, immunologic organ system involvement and a medical history of hypertension. She had ANA, anti-dsDNA, anti-Smith, anti-RBP, anti-chromatin autoantibodies and a SLEDAI-2K score of 24 at screening. Prior SLE treatments included hydroxychloroquine, corticosteroids, mycophenolate, rituximab, and Obinutuzumab.
- this patient received a single dose of 50 x 10 6 ( ⁇ 20%) CD19 CAR-positive viable T cells on Day 1 after lymphodepletion chemotherapy. Methylprednisolone was used as bridging therapy.
- anti-dsDNA decreased from >300 at screening to 40.6;
- IgG levels decreased from 6.07 at screening to 2.39 at Day 28.
- IgA decreased from 1.84 to 0.46 and IgM from 0.53 to 0.12, respectively;
- SAE Grade 3 serious adverse effect
- the CD19 CAR-T cells (obe-cel) showed a good expansion with a peak at Day 10 post CAR-T cell infusion by flow cytometry and ddPCR, as shown by Figure 2a and 2b, respectively.
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Immunology (AREA)
- Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Organic Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicines Containing Material From Animals Or Micro-Organisms (AREA)
Abstract
The present invention relates to a chimeric antigen receptor (CAR) which binds the B- lymphocyte antigen CD19 (Cluster of Differentiation 19) and to use of T cells expressing such a CAR for the treatment of autoimmune diseases, such as systemic lupus erythematosus (SLE)
Description
PRODUCTS AND METHODS FOR TREATING AUTOIMMUNE DISEASES
FIELD OF THE INVENTION
The present invention relates to a chimeric antigen receptor (CAR) which binds the B- lymphocyte antigen CD19 (Cluster of Differentiation 19) and to use of cytolytic cells expressing the CAR for the treatment of autoimmune diseases such as systemic lupus erythromatosus (SLE).
BACKGROUND
B cells, along with T cells, form the core of the adaptive arm of the immune system and play a key role in regulating immune response in both normal physiological and pathological conditions. B cells are identified pharmacological targets in B cell malignancies and autoimmune diseases.
Autoimmune Diseases
Numerous diseases are believed to result from autoimmune mechanisms. Common among these are rheumatoid arthritis, systemic lupus erythematosus, inflammatory bowel disease, multiple sclerosis, Type I diabetes, Guillain-Barre syndrome, chronic inflammatory demyelinating polyneuropathy, psoriasis, Graves’ disease, Hashimoto’s thyroiditis, myasthenia gravis, scleroderma, vasculitis and pemphigus vulgaris. Autoimmune diseases affect millions of individuals world-wide and the cost of these diseases, in terms of actual treatment expenditures and lost productivity, is measured in billions of dollars annually.
Systemic Lupus Erythromatosus
Systemic lupus erythematosus (SLE) is an autoimmune disease characterised by the formation of autoantibodies and immune complex mediated inflammation and organ damage, including the skin, joints, central nervous system, heart, lung, and kidneys (Tsokos GC. (2011). Systemic lupus erythematosus. N Engl J Med 365(22):2110-21 ; Kaul A, Gordon C, Crow M, et al. (2016). Systemic lupus erythematosus. Nature Reviews 2:1-21 ; Mackensen A, Muller F, Mouglakakos D, et al. (2022). Anti-CD19 CAR T cell therapy for refractory lupus erythematosus. Nature Medicine 28:2124-2132). It is a genetically complex disease, exhibiting genetic heterogeneity (Kaul et al, supra). The
vast variation in clinical and serological manifestations makes SLE a very heterogeneous disease. Disease severity also changes over time with periods of no disease activity alternated by periods with disease flares/relapses. Besides this heterogeneity, SLE can be complicated by co-morbidities like infections, premature atherosclerosis, and chronic organ damage, as well as subjective manifestations including a reduced quality of life. The disease onset is generally between the ages of 20 and 40, and it affects predominantly young women. Patients with SLE have about a three-fold greater risk of mortality than the general population.
Currently available treatments aim to reduce inflammation, manage symptoms, and prevent organ damage but have significant side effects, raising safety concerns. Durable complete remissions without serological or clinical disease activity, and a glucocorticoid and immunosuppressant-free state are rarely achieved. Hence, there is an unmet medical need for treatments with potentially curative intent with a better safety profile and a convenient treatment-free interval, particularly for severe, refractory patients.
Autoreactive B cells with autoantibody formation play a key role in the pathogenesis of SLE (Mackensen et al., supra). However, B cell-depleting agents, such as the anti- CD20 antibody rituximab, did not improve clinical outcomes compared to placebo in randomized studies in SLE and Lupus Nephritis (LN) (Merrill J, Neuwelt CM, Wallace DJ, Shanahan JC, et al. (2010). Efficacy and safety of rituximab in moderately-to- severely active systemic lupus erythematosus: the randomized, double-blind, phase ll/lll systemic lupus erythematosus evaluation of rituximab trial. Arthritis Rheum 62(1):222-33; Rovin et al., (2012). Efficacy and safety of rituximab in patients with active proliferative lupus nephritis: the Lupus Nephritis Assessment with Rituximab study Arthritis Rheum 64(4): 1215-26) while two different biologies have recently been approved in SLE: Belimumab, an anti-BAFF/BLyS monoclonal antibody, has been approved as add-on therapy in adult patients with active, autoantibody-positive SLE with a high degree of disease activity despite standard therapy (Benlysta (belimumab) USPI. 2023. Available at: gskpro.com/content/dam/global/hcpportal/en_US/Prescribing_lnformation/Benlysta/pd f/BENLYSTA-PI-MG-IFU.PDF. Last accessed 25-Aug-2023; Benlysta (belimumab) SmPC. 2023. Available at: www.ema.europa.eu/en/documents/product- information/benlysta-epar-product-information_en.pdf. Last accessed 30-Aug-2023). Anifrolumab, a type I interferon (IFN) receptor antagonist, has also been approved in the US and EU and is indicated as an add-on for the treatment of adult patients with
moderate to severe SLE who are receiving standard therapy (Saphnelo (anifrolumab- fnia) IISPI. 2022. Available at: www.den8dhaj6zs0e.cloudfront.net/50fd68b9-106b- 4550-b5d0-12b045f8b184/44b6985c-8268-46b1-ba3e-2bb43bfd4d4c/44b6985c- 8268-46b1-ba3e-2bb43bfd4d4c_viewable_rendition v.pdf. Last accessed 25-Aug- 2023; Saphnelo (anifrolumab-fnia) SmPC. 2022. Available at: www.ema.europa.eu/en/documents/product-information/saphnelo-epar-product- information_en.pdf. Last accessed 25-Aug-2023). In addition, voclosporin (Lupkynis USPI, 2021) was approved for lupus nephritis based on the AURORA1 trial (Rovin et al., Lancet 2021) (www.doi.org/10.1016/S0140-6736(21)00578-X). Voclosporin in combination with MMF and low-dose steroids led to a clinically and statistically superior complete renal response rate versus MMF and low-dose steroids alone, with a comparable safety profile. Despite these approvals, some patients have insufficient response, lack of response, or lack of sustained response and are at risk for further organ damage despite standard therapy. Hence, challenges remain with treatmentresistant disease.
Another strategy to induce deeper depletion of the B cell compartment originates from the highly effective treatment of patients with B cell malignancies using CD19-targeting chimeric antigen receptor (CAR) T cells (Cappell KM, Kochenderfer JN. (2023) Longterm outcomes following CAR T cell therapy: what we know so far. Nat Review 20:359- 371). Several CD19-targeted CAR T cells are now approved for relapsed and/or refractory B cell lymphoma and B cell acute lymphoblastic leukemia (B-ALL) (axicabtagene ciloleucel, tisagenlecleucel, lisocabtagene maraleucel and brexucabtagene autoleucel). Long-term follow-up data indicate that CD19-targeted CAR T cells are likely to be curative for a subset of patients with B cell lymphomas and possibly B-ALL (Cappell and Kochenderfer, supra; Westin J, Davis RE, Feng L, Hagemeister F, et al. (2023). Smart start: rituximab, lenalidomide, and ibrutinib in patients with newly diagnosed large B-cell lymphoma. J Clin Oncol 41 (4): 745-55).
Two preclinical studies in lupus-prone mice support the efficacy of CD19 CAR T cells in SLE. Upon administration of CD19-targeting CAR T cells (1 D3 CAR and FMC63 CAR), B cells in these mice were depleted, autoantibody production was suspended, and glomerulonephritis and other organ manifestations were reversed.
Using autoimmune New Zealand mixed mice (NZB x NZW) F1 and Murphys Roths Large (MRL) fas/fas mouse models of lupus, CD8+ T cells expressing CD19-targeted CARs [1 D3 CAR (Kochenderfer JN, Yu Z, Frasheri D, et al. (2010), Adoptive transfer
of syngeneic T cells transduced with a chimeric antigen receptor that recognizes murine CD19 can eradicate lymphoma and normal B cells. Blood 116(19), 3875-3886] were shown to persistently deplete CD19+ B cells, eliminate autoantibody production, reverse disease manifestations in target organs, and extend life spans well beyond normal range in the autoimmune mouse models (Kansal R, Richardson N Neeli I, et al. (2019). Sustained B cell depletion by CD19-targeted CAR T cells is a highly effective treatment for murine lupus. Sci Transl Med 11(482): eaav1648-eaav1648). CAR T cells were present for one year in vivo and were significantly enriched in the CD44+CD62L+ central memory CD8+T cell subset, suggesting this population may harbor the CAR and contribute to the sustained B cell depletion. Kansel et al showed that adoptive transfer of splenic T cells from CAR T cell-treated mice depleted CD19+ B cells and reduced disease in naive autoimmune mice indicating that disease control was cell-mediated.
Using constructed murine anti-CD19 CARs [1 D3 CAR (Kochenderfer et al., 2010, supra] with either CD28 or 4-1 BB as the intracellular costimulatory motif, Jin et al (Jin X, Xu Q, Pu C, Zhu K, et al. (2021). Therapeutic efficacy of anti-CD19 CAR-T cells in a mouse model of systemic lupus erythematosus. Cell Mol Immunol 18(8): 1896-1903) evaluated the therapeutic function of the corresponding CAR-T cells by infusing them into lupus mouse model (MRL-lpr). Transferred anti-CD19 CAR T cells to MRL-lpr mice at onset of disease was assessed to determine their role in SLE prevention. Compared with antibody treatment, the adoptive transfer of the constructed anti-CD 19 CAR-T cells showed a more sustained B-cell-depletion effect in MRL-lpr mice. The transfer of syngeneic anti-CD19 CAR-T cells not only prevented disease pathogenesis before the onset of disease symptoms but also displayed therapeutic benefits at a later stage after disease progression. CAR-T cells with the 4-1 BB costimulatory motif showed better therapeutic efficiency, without cell enrichment, compared to CAR-T cells with CD28 costimulatory motif.
Taken together, the results from these two pre-clinical show that anti-CD19 CAR-T cell therapy was effective in prevention and treatment, albeit in murine models of SLE.
Recently, belimumab, an anti-B cell activating factor (BAFF)/B lymphocyte stimulator (BlyS) monoclonal antibody, has been approved as add-on therapy in adult patients with active, autoantibody-positive SLE who are receiving standard therapy (Benlysta USPI, supra).
Tolerability and efficacy were initially tested in a single SLE patient treated with a CD19 CAR (FMC63 CAR) (Mougiakakos D, Krdnke G, Volkl S, Kretschmann S, et al. (2021). CD-19-targeted CAR T cells in refractory systemic lupus erythematosus. N Engl J Med 385(6): 567-9). The same research group further assessed tolerability and efficacy of a CD19 CAR T cell therapy (FMC63 CAR) in a small series of seriously ill and treatmentresistant patients with SLE (Mackensen et al, supra). In this series, autologous T cells from five patients with SLE were transduced with a lentiviral anti-CD19 CAR vector, expanded and reinfused at a dose of 1 xio6 CAR T cells per kg body weight into the patients after lymphodepletion with fludarabine and cyclophosphamide. CAR T cells were expanded in vivo and led to deep depletion of B cells with improvement of clinical symptoms and normalization of laboratory parameters including seroconversion of anti- ds DNA antibodies. Remission of SLE according to DORIS criteria was achieved in all five patients after 3 months, and drug-free remission was maintained during longer follow-up after CAR T cell administration.
Additional anecdotal evidence of CD19 CAR T cell treatment (FMC63 CAR) in another autoimmune disease has recently been shown in patients with refractory idiopathic inflammatory myopathy associated with antisynthetase syndrome (Muller F, Boeltz S, Knitza J, Aigner M, et al. (2023). CD19-targeted CAR T cells in refractory antisynthetase syndrome. Lancet 401(10379):815-8; Percher AC, Hensen L, Klein R, et al. (2023). CD19-targeting CAR T cells for myositis and interstitial lung disease associated with antisynthetase syndrome. JAMA 329(24):2154-62).
Chimeric Antigen Receptors
Traditionally, antigen-specific T cells have been generated by selective expansion of peripheral blood T cells natively specific for the target antigen. However, it is difficult and quite often impossible to select and expand large numbers of T cells specific for most cancer antigens. Gene-therapy with integrating vectors affords us a solution to this problem: transgenic expression of Chimeric Antigen Receptor (CAR) allows large numbers of T cells specific to any surface antigen to be easily generated by ex vivo viral vector transduction of a bulk population of peripheral blood T cells.
The most common forms of these molecules are fusions of single-chain variable fragments (scFv) derived from monoclonal antibodies which recognise a target antigen, fused via a spacer and a transmembrane domain to a signalling endodomain. Such molecules result in activation of the T cell in response to recognition by the scFv of its
cognate target. When T cells express such a CAR, they recognize and kill target cells that express the target antigen. Several CARs have been developed against tumour associated antigens, and adoptive transfer approaches using such CAR-expressing T cells are currently in clinical trial for the treatment of various cancers. To-date however, the main clinical exploration and potential application of CAR therapy is as treatment for B-cell malignancies.
CA s directed against CD19
CD19 is a B-cell antigen which is expressed very early in B-cell differentiation and is only lost at terminal B-cell differentiation into plasma cells. Hence, CD19 is expressed on all B-cell malignancies apart from multiple myeloma. It is not expressed on other haematopoietic populations or non-haematopoietic cells and therefore targeting this antigen should not lead to toxicity to the bone marrow or non-haematopoietic organs. Loss of the normal B-cell compartment is considered an acceptable toxicity when treating lymphoid malignancies, because although effective CD 19 CAR T cell therapy will result in B cell aplasia, the consequent hypogammaglobulinaemia can be treated with pooled immunoglobulin.
CD19 is therefore an attractive CAR target. To date, the main clinical focus of the CAR field has been studies targeting CD19 on refractory B-cell cancers, as summarised in Table 1.
Different designs of CARs have been tested against CD19 in different centres, as outlined in Table 1.
Table 1. Summary of CAR experience targeting CD 19

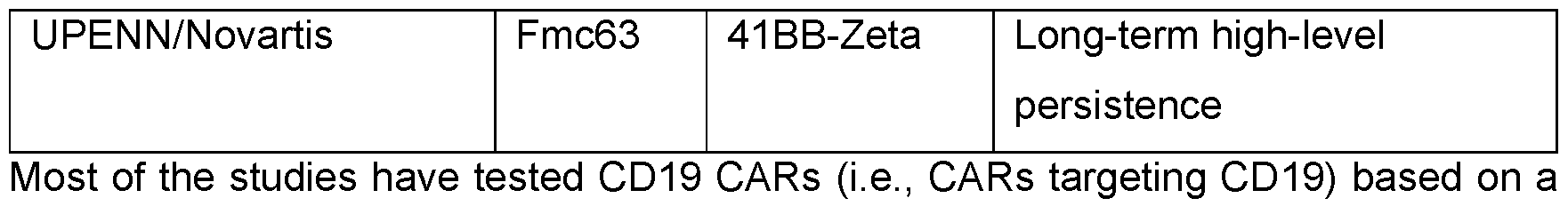 scFv derived from the hybridoma fmc63. The most promising have been in the treatment of Acute Lymphoblastic Leukaemia (ALL).
scFv derived from the hybridoma fmc63. The most promising have been in the treatment of Acute Lymphoblastic Leukaemia (ALL).
Immune toxicity of CD19 CAR therapy
Cytokine release syndrome (CRS) encompasses a range of inflammatory symptoms ranging from mild to multi-organ failure with hypotension and respiratory failure. Some degree of CRS occurs commonly in patients treated with CD19 CAR T cells. Approximately 30% (21/73) patients treated in recent cohorts showed some degree of CRS (Davila et al ((2014). Sci. Transl. Med. 6, 224ra25; Lee et al (2014) Lancet. doi:10.1016/S0140-6736(14)61403-3; Kochenderfer et al. (2014) J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol., doi:10.1200/JCO.2014.56.2025). CRS has also been seen in patients treated with blinatumomab, a bi-specific recombinant single-chain antibody recognising both CD19 and CD3. CRS typically occurs 5-21 days after CAR T cell infusion.
CRS can be life threatening and requires treatment in an intensive care setting. CRS is associated with elevated serum cytokine levels. The cytokines most significantly elevated are IL-6, IL-10 and interferon gamma (IFNy). Clinical manifestations of severe CRS (fever, hepatosplenomegaly, coagulopathy and hyperferritinaemia) resemble macrophage activation syndrome (MAS) found for instance in patients with congenital defects in T cells. This suggests that common immunopathological processes are involved. At present it is not clear which cell type (CAR T cells, dying tumour cells, or locally-activated macrophages) are responsible for production of the key cytokines, particularly IL-6. However, a key initiating factor in MAS is release of copious Interferongamma (Lopez-Alvarez et al. (2009). Clin. Vaccine Immunol. CVI 16, 142-145).
Neurotoxicity
A number of patients in CD19 CAR studies across institutions have developed transient neurotoxicity with a spectrum of severity from aphasia to obtundation, delirium and seizures (Davilia et al, supra). This appears to be restricted to patients with ALL and a similar syndrome has been documented after blinatumomab therapy. Brain imaging
appears normal. Neurotoxicity may reflect high levels of systemic cytokines crossing the blood-brain barrier.
Persistence, relapse and T cell exhaustion
Durable responses appeared to correlate with higher peak levels of circulating CAR transduced T cells, as well as with the duration of B cell aplasia. With exception of patients relapsing with CD19- disease, relapse was generally associated with loss of circulating CAR T cells and recovery of normal B cells.
T cell exhaustion is a state of T cell dysfunction that arises during many chronic infections and cancer. It is defined by poor effector function, sustained expression of inhibitory receptors and a transcriptional state distinct from that of functional effector or memory T cells. Exhaustion prevents optimal control of infection and tumors. Recently, a clearer picture of the functional and phenotypic profile of exhausted T cells has emerged with expression of inhibitory receptor programmed death 1 (PD-1 ; also known as PDCD1), a negative regulator of activated T cells, being a key feature (Day et al. (2006) Nature 443, 350-354).
Responses in CD19 CAR studies suggest that persistence of T cells for a protracted period at high levels seems to be important in effecting durable responses. A CD19 CAR which reduces T cell exhaustion may result in improved clinical responses.
There is thus a need in treatment methods for autoimmune disease for an alternative CAR directed against CD19 which is not associated with the above disadvantages.
BRIEF SUMMARY
The present disclosure provides methods of treatment of autoimmune diseases with CD19 CAR T cells. Treatment with CD19 CAR T cells is contemplated to more completely the B cell compartment than Rituximab, more effectively eliminating autoreactive B cells. CD19 as a target in autoimmune diseases is advantageous over CD20-directed therapies as CD19 is expressed more broadly on B cells, including very early B-progenitor cells as well as immature plasmablasts.
Thus, in a first aspect the present invention provides methods of treating an autoimmune disease in a patient, where the patient is administered a cell comprising a
chimeric antigen receptor (CAR) comprising a CD19-binding domain which comprises a) a heavy chain variable region (VH) having complementarity determining regions (CDRs) with the following sequences:
CDR1 - GYAFSSS (SEQ ID No. 1);
CDR2 - YPGDED (SEQ ID No. 2)
CDR3 - SLLYGDYLDY (SEQ ID No. 3); and b) a light chain variable region (VL) having CDRs with the following sequences:
CDR1 - SASSSVSYMH (SEQ ID No. 4);
CDR2 - DTSKLAS (SEQ ID No. 5)
CDR3 - QQWNINPLT (SEQ ID No. 6).
The autoimmune disease may be systemic lupus erythrematosus (SLE). The autoimmune disease may be severe SLE or refractory SLE.
The cell may be a T cell or NK cell. The cell may an autologous peripheral blood T cells transduced ex vivo to express the CAR. The cell may have been isolated from a peripheral blood mononuclear cell (PBMC) sample from the patient.
A dose of 50 x 106 (±20%) CD19 CAR-positive viable T cells may be administered to the patient. Alternatively, the dose may be 100 x 106 (±20%) CD 19 CAR-positive viable T cells or 30 x 106 (±20%) CD19 CAR-positive viable T cells.
The administration may be an intravenous injection through a Hickman line or peripherally inserted central catheter.
The CD19 binding domain may comprise the six CDRs (SEQ ID Nos. 1-6) grafted on to a human antibody framework.
The CD19 binding domain may comprise a VH domain having the sequence shown as SEQ ID No. 7 and/or or a VL domain having the sequence shown as SEQ Id No. 8, or a variant of either having at least 95% sequence identity.
The CD19 binding domain may comprise an scFv in the orientation VH-VL.
The CD19 binding domain may comprise the sequence shown as SEQ ID No.. 9 or a variant thereof having at least 90% sequence identity.
The CAR may comprise a spacer that is a CD8 stalk.
The CAR may comprise an intracellular T cell signaling domain comprising the 41 BB endodomain and the CD3-Zeta endodomain.
In a second aspect, the invention provides a method of treating an autoimmune disease in a patient comprising administering to the patient a dose of 50 x 106 (±20%) CD19 CAR-positive viable T cells. Alternatively, the patient may be administered a dose of 30 x 106 (±20%) CD19 CAR-positive viable T cells. Alternatively, the patient may be administered a dose of 100 x 106 (±20%) CD19 CAR-positive viable T cells.
The autoimmune disease according to the second aspect may be SLE. The autoimmune disease may be severe SLE or refractory SLE.
In the first or second aspect, the CAR may comprise the sequence shown as any of SEQ ID No. 10 to 15 or a variant thereof which has at least 80% sequence identity but retains the capacity to i) bind CD19 and ii) induce T cell signalling. The CAR may have advantageous properties compared to the fmc63-based CAR used in the UPENN studies. For example, the CD19 (CAT) CAR herein has a lower affinity CD19 binder with a faster off-rate which mirrors more closely a physiological interaction and therefore, a more physiological T cell activation. This is contemplated herein to reduce toxicity of cells expressing the CAR and to render those cells less prone to T cell exhaustion, in turn enhancing persistence and improving the ability of the programmed T cells to engage in serial killing of target cancer cells. As another example, the CAR, when expressed by a T cell and used to target a CD19 expressing cell, may cause lower IFNy release by the CD19-expressing target cell than that caused by a T cell expressing a CAR comprising a CD19-binding domain which comprises: a) a heavy chain variable region (VH) having complementarity determining regions (CDRs) with the following sequences:CDR1 - GVSLPDY (SEQ ID No. 16); CDR2 - WGSET (SEQ ID No. 17); CDR3 - HYYYGGSYAMDY (SEQ ID No. 18); and b) a light chain variable region (VL) having CDRs with the following sequences:CDR1 - RASQDISKYLN (SEQ ID No. 19); CDR2 - HTSRLHS (SEQ ID No. 20) CDR3 - QQGNTLPYT (SEQ ID No. 21). The CDRs provided herein may be grafted on to a human or humanised framework.
DESCRIPTION OF THE FIGURES
Figure 1. SLEDAI-2K score of first SLE patient treated with obe-cel.
Figure 2. CAR-T cell expansion in first SLE patient treated with obe-cel. A) Flow cytometry of CD3+CAT19+ cells. B) ddPCR.
DETAILED DESCRIPTION
CHIMERIC ANTIGEN RECEPTORS (CARS)
Chimeric antigen receptors (CARs), also known as chimeric T cell receptors, artificial T cell receptors and chimeric immunoreceptors, are engineered receptors, which graft an arbitrary specificity onto an immune effector cell. In a classical CAR, the specificity of a monoclonal antibody is grafted on to a T cell. CAR-encoding nucleic acids may be transferred to T cells using, for example, retroviral vectors. In this way, a large number of CD19-specific T cells can be generated for adoptive cell transfer. Phase I clinical studies of this approach show efficacy.
The target-antigen binding domain of a CAR is commonly fused via a spacer and transmembrane domain to an endodomain. The endodomain may comprise or associate with an intracellular T cell signaling domain. When the CAR binds the targetantigen, this results in the transmission of an activating signal to the T cell it is expressed on.
BINDING DOMAINS SPECIFIC FOR CD19 TARGET ANTIGEN
The human CD19 antigen is a 95 kd transmembrane glycoprotein belonging to the immunoglobulin superfamily. CD19 is classified as a type I transmembrane protein, with a single transmembrane domain, a cytoplasmic C-terminus, and extracellular N- terminus. CD19 is expressed very early in B-cell differentiation and is only lost at terminal B-cell differentiation into plasma cells. CD19 is a biomarker for normal B cells as well as follicular dendritic cells. CD19 primarily acts as a B cell co-receptor in conjunction with CD21 and CD81 . Upon activation, the cytoplasmic tail of CD19 becomes phosphorylated, which leads to binding by Src-family kinases and recruitment of PI-3 kinase.
CD19 is also expressed on all B-cell malignancies but not multiple myeloma cells. It is not expressed on other haematopoietic populations or non-haematopoietic cells
and therefore targeting this antigen should not lead to toxicity to the bone marrow or non-haematopoietic organs. Loss of the normal B-cell compartment is considered an acceptable toxicity when treating lymphoid malignancies, because although effective CD19 CAR T cell therapy will result in B cell aplasia, the consequent hypogammaglobulinaemia can be treated with pooled immunoglobulin.
Different designs of CARs have been tested against CD19 in various clinical trials, as outlined in the following Table 2.
Table 2
 As shown above, most of the studies conducted to date have used an scFv derived from the hybridoma fmc63 as part of the binding domain to recognize CD19.
As shown above, most of the studies conducted to date have used an scFv derived from the hybridoma fmc63 as part of the binding domain to recognize CD19.
The antigen-binding domain of a CAR which binds to CD19 (referred to as a CD19 CAR herein) may be any domain which is capable of binding CD19.
For example, the antigen-binding domain may comprise a CD19 antigen-binding domain as described in Table 3.
Table 3
 The gene encoding CD19 comprises ten exons: exons 1 to 4 encode the extracellular domain; exon 5 encodes the transmembrane domain; and exons 6 to 10 encode the cytoplasmic domain. The antigen-binding domain of a CD19 CAR herein may bind an epitope of CD19 encoded by exon 1 of the CD19 gene. The antigen-binding domain of a CD19 CAR herein may bind an epitope of CD19 encoded by exon 2 of the CD19 gene. The antigen-binding domain of a CD19 CAR herein may bind an epitope of CD19 encoded by exon 3 of the CD19 gene. The antigen-binding domain of a CD19 CAR herein may bind an epitope of CD19 encoded by exon 4 of the CD19 gene.
The gene encoding CD19 comprises ten exons: exons 1 to 4 encode the extracellular domain; exon 5 encodes the transmembrane domain; and exons 6 to 10 encode the cytoplasmic domain. The antigen-binding domain of a CD19 CAR herein may bind an epitope of CD19 encoded by exon 1 of the CD19 gene. The antigen-binding domain of a CD19 CAR herein may bind an epitope of CD19 encoded by exon 2 of the CD19 gene. The antigen-binding domain of a CD19 CAR herein may bind an epitope of CD19 encoded by exon 3 of the CD19 gene. The antigen-binding domain of a CD19 CAR herein may bind an epitope of CD19 encoded by exon 4 of the CD19 gene.
A CD19-binding domain exemplified herein comprises variable regions with complementarity determining regions (CDRs) from an antibody referred to as CAT19, a) a heavy chain variable region (VH) having CAT19 CDRs with the following sequences:
CDR1 - GYAFSSS (SEQ ID No. 1),
CDR2 - YPGDED (SEQ ID No. 2) and
CDR3 - SLLYGDYLDY (SEQ ID No. 3), and b) a light chain variable region (VL) having CAT 19 CDRs with the following sequences:
CDR1 - SASSSVSYMH (SEQ ID No. 4), CDR2 - DTSKLAS (SEQ ID No. 5) and CDR3 - QQWNINPLT (SEQ ID No. 6).
The CAT19 antibody is described in WO 2016/139487.
It is contemplated that one or more mutations (substitutions, additions or deletions) can be introduced into one or more CDRs without negatively affecting CD19-binding activity. Each CDR may, for example, have one, two or three amino acid mutations. The CDRs may be in the format of a single-chain variable fragment (scFv), which is a fusion protein of the heavy variable region (VH) and light chain variable region (VL) of an antibody, connected with a short linker peptide of ten to about 25 amino acids. The scFv may be in the orientation VH-VL, /.e., the VH is at the amino-terminus of the CAR molecule and the VL domain is linked to the spacer and, in turn the transmembrane domain and endodomain.
The CDRs may be grafted on to the framework of a human antibody or scFv. For example, the CAR may comprise a CD19-binding domain consisting or comprising one of the following sequences.
The CD19 CAR may comprise the following VH sequence.
SEQ ID No. 7 - VH sequence from CAT19 murine monoclonal antibody
QVQLQQSGPELVKPGASVKISCKASGYAFSSSWMNWVKQRPGKGLEWIGRIYPGD EDTNYSGKFKDKATLTADKSSTTAYMQLSSLTSEDSAVYFCARSLLYGDYLDYWGQ GTTLTVSS
The CD19 CAR may comprise the following VL sequence.
SEQ ID No. 8 - VL sequence from CAT19 murine monoclonal antibody
QIVLTQSPAIMSASPGEKVTMTCSASSSVSYMHWYQQKSGTSPKRWIYDTSKLASG VPDRFSGSGSGTSYFLTINNMEAEDAATYYCQQWNINPLTFGAGTKLELKR
The CD19 CAR may comprise the following scFv sequence.
SEQ ID No. 9 - VH-VL scFv sequence from murine monoclonal antibody
QVQLQQSGPELVKPGASVKISCKASGYAFSSSWMNWVKQRPGKGLEWIGRIYPGD
EDTNYSGKFKDKATLTADKSSTTAYMQLSSLTSEDSAVYFCARSLLYGDYLDYWGQ
GTTLTVSSGGGGSGGGGSGGGGSQIVLTQSPAIMSASPGEKVTMTCSASSSVSYM HWYQQKSGTSPKRWIYDTSKLASGVPDRFSGSGSGTSYFLTINNMEAEDAATYYC QQWNINPLTFGAGTKLELKR
The CAR may consist of or comprise one of the following sequences.
SEQ ID No. 10 - CAT19 CAR using “Campana” architecture
MGTSLLCWMALCLLGADHADAQVQLQQSGPELVKPGASVKISCKASGYAFSSSWM
NWVKQRPGKGLEWIGRIYPGDEDTNYSGKFKDKATLTADKSSTTAYMQLSSLTSED
SAVYFCARSLLYGDYLDYWGQGTTLTVSSGGGGSGGGGSGGGGSQIVLTQSPAIM
SASPGEKVTMTCSASSSVSYMHWYQQKSGTSPKRWIYDTSKLASGVPDRFSGSGS
GTSYFLTINNMEAEDAATYYCQQWNINPLTFGAGTKLELKRSDPTTTPAPRPPTPAP TIASQPLSLRPEACRPAAGGAVHTRGLDFACDIYIWAPLAGTCGVLLLSLVITLYCKR GRKKLLYIFKQPFMRPVQTTQEEDGCSCRFPEEEEGGCELRVKFSRSADAPAYQQ
GQNQLYNELNLGRREEYDVLDKRRGRDPEMGGKPRRKNPQEGLYNELQKDKMAE AYSEIGMKGERRRGKGHDGLYQGLSTATKDTYDALHMQALPPR
“Campana” architecture refers to a CAR with a CD8a spacer and transmembrane domain, 4-1 BB endodomain and TCR CD3z endodomain.
SEQ ID No. 11 - CAT19 CAR with an QX40-Zeta endodomain
MGTSLLCWMALCLLGADHADAQVQLQQSGPELVKPGASVKISCKASGYAFSSSWM NWVKQRPGKGLEWIGRIYPGDEDTNYSGKFKDKATLTADKSSTTAYMQLSSLTSED SAVYFCARSLLYGDYLDYWGQGTTLTVSSGGGGSGGGGSGGGGSQIVLTQSPAIM SASPGEKVTMTCSASSSVSYMHWYQQKSGTSPKRWIYDTSKLASGVPDRFSGSGS GTSYFLTINNMEAEDAATYYCQQWNINPLTFGAGTKLELKRSDPTTTPAPRPPTPAP TIASQPLSLRPEACRPAAGGAVHTRGLDFACDIYIWAPLAGTCGVLLLSLVITLYCRR DQRLPPDAHKPPGGGSFRTPIQEEQADAHSTLAKIRVKFSRSADAPAYQQGQNQLY NELNLGRREEYDVLDKRRGRDPEMGGKPRRKNPQEGLYNELQKDKMAEAYSEIGM KGERRRGKGHDGLYQGLSTATKDTYDALHMQALPPR
SEQ ID No. 12 - CAT19 CAR with a CD28-Zeta endodomain
MGTSLLCWMALCLLGADHADAQVQLQQSGPELVKPGASVKISCKASGYAFSSSWM NWVKQRPGKGLEWIGRIYPGDEDTNYSGKFKDKATLTADKSSTTAYMQLSSLTSED SAVYFCARSLLYGDYLDYWGQGTTLTVSSGGGGSGGGGSGGGGSQIVLTQSPAIM SASPGEKVTMTCSASSSVSYMHWYQQKSGTSPKRWIYDTSKLASGVPDRFSGSGS GTSYFLTINNMEAEDAATYYCQQWNINPLTFGAGTKLELKRSDPTTTPAPRPPTPAP TIASQPLSLRPEACRPAAGGAVHTRGLDFACDIYIWAPLAGTCGVLLLSLVITLYCRS KRSRLLHSDYMNMTPRRPGPTRKHYQPYAPPRDFAAYRSRVKFSRSADAPAYQQG QNQLYNELNLGRREEYDVLDKRRGRDPEMGGKPRRKNPQEGLYNELQKDKMAEA YSEIGMKGERRRGKGHDGLYQGLSTATKDTYDALHMQALPPR
SEQ ID No. 13 - Third generation CD19 CAR
MGTSLLCWMALCLLGADHADAQVQLQQSGPELVKPGASVKISCKASGYAFSSSWM NWVKQRPGKGLEWIGRIYPGDEDTNYSGKFKDKATLTADKSSTTAYMQLSSLTSED SAVYFCARSLLYGDYLDYWGQGTTLTVSSGGGGSGGGGSGGGGSQIVLTQSPAIM SASPGEKVTMTCSASSSVSYMHWYQQKSGTSPKRWIYDTSKLASGVPDRFSGSGS GTSYFLTINNMEAEDAATYYCQQWNINPLTFGAGTKLELKRSDPTTTPAPRPPTPAP Tl ASQPLSLRPEACRPAAGGAVHTRGLDFACDI FWVLWVGGVLACYSLLVTVAFI I F WVRSKRSRLLHSDYMNMTPRRPGPTRKHYQPYAPPRDFAAYRSRDQRLPPDAHK
PPGGGSFRTPIQEEQADAHSTLAKIRVKFSRSADAPAYQQGQNQLYNELNLGRREE YDVLDKRRGRDPEMGGKPRRKNPQEGLYNELQKDKMAEAYSEIGMKGERRRGKG HDGLYQGLSTATKDTYDALHMQALPPR
SEQ ID No. 14 - CD19 CAR with lgG1 hinge spacer
MGTSLLCWMALCLLGADHADAQVQLQQSGPELVKPGASVKISCKASGYAFSSSWM NWVKQRPGKGLEWIGRIYPGDEDTNYSGKFKDKATLTADKSSTTAYMQLSSLTSED SAVYFCARSLLYGDYLDYWGQGTTLTVSSGGGGSGGGGSGGGGSQIVLTQSPAIM
SASPGEKVTMTCSASSSVSYMHWYQQKSGTSPKRWIYDTSKLASGVPDRFSGSGS GTSYFLTINNMEAEDAATYYCQQWNINPLTFGAGTKLELKRSDPAEPKSPDKTHTCP PCPKDPKFWVLVWGGVLACYSLLVTVAFIIFWVRSKRSRLLHSDYMNMTPRRPGP TRKHYQPYAPPRDFAAYRSRVKFSRSADAPAYQQGQNQLYNELNLGRREEYDVLD KRRGRDPEMGGKPRRKNPQEGLYNELQKDKMAEAYSEIGMKGERRRGKGHDGLY QGLSTATKDTYDALHMQALPPR
SEQ ID No. 15 - CD19 CAR with hinge-CH2-CH3 of human lgG1 with FcR binding sites mutated out MGTSLLCWMALCLLGADHADAQVQLQQSGPELVKPGASVKISCKASGYAFSSSWM NWVKQRPGKGLEWIGRIYPGDEDTNYSGKFKDKATLTADKSSTTAYMQLSSLTSED SAVYFCARSLLYGDYLDYWGQGTTLTVSSGGGGSGGGGSGGGGSQIVLTQSPAIM SASPGEKVTMTCSASSSVSYMHWYQQKSGTSPKRWIYDTSKLASGVPDRFSGSGS GTSYFLTINNMEAEDAATYYCQQWNINPLTFGAGTKLELKRSDPAEPKSPDKTHTCP PCPAPPVAGPSVFLFPPKPKDTLMIARTPEVTCWVDVSHEDPEVKFNWYVDGVEV HNAKTKPREEQYNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIEKTISKAKG QPREPQVYTLPPSRDELTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPV LDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGKKDPKF WVLVWGGVLACYSLLVTVAFIIFWVRSKRSRLLHSDYMNMTPRRPGPTRKHYQPY APPRDFAAYRSRVKFSRSADAPAYQQGQNQLYNELNLGRREEYDVLDKRRGRDPE MGGKPRRKNPQEGLYNELQKDKMAEAYSEIGMKGERRRGKGHDGLYQGLSTATK DTYDALHMQALPPR
The CAR provided herein may comprise a variant of the polypeptide of SEQ ID Nos. 1-15 having at least 80, 85, 90, 95, 98 or 99% sequence identity, provided that the variant sequence retains the capacity to bind CD19 (when in conjunction with a complementary VL or VH domain, if appropriate). The percentage identity between two polypeptide sequences may be readily determined by programs such as BLAST which is freely available at blast.ncbi.nlm.nih.gov.
The CD19 CAR exemplified herein (/.e., the CAT19CAR using “Campana” architecture, SEQ ID No. 10) has properties contemplated by the disclosure to result in lower toxicity and better efficacy in treated patients. When compared with an fmc63-Campana CAR, the CAT19CAR exemplified herein effected killing of target cells expressing CD19 and proliferated in response to CD19 expressing targets, but Interferon-gamma release was less. Further, a small animal model of an aggressive B-cell lymphoma showed equal efficacy and equal engraftment between the fmc63-
and CAT 19-based CAR-T cells, but surprisingly, less of the CAT 19 CAR T cells were exhausted than fmc63 CAR T cells. See, Examples 2 and 3 of US Publication No.: 2018-0044417.
The CAT19CAR provided herein may cause 25, 50, 70 or 90% lower IFNy release in a comparative assay involving bringing CAR T cells into contact with target cells. The CAT19CAR provided herein may result in a smaller proportion of CAR T cells becoming exhausted than fmc63 CAR T cells. T cell exhaustion may be assessed using methods known in the art, such as analysis of PD-1 expression. The CAR may cause 20, 30, 40, 50, 60 of 70% fewer CAR T cells to express PD-1 that fmc63 CAR T cells in a comparative assay involving bringing CAR T cells into contact with target cells.
Another exemplary CD19 antigen-binding domain contemplated by the disclosure is based on the CD19 antigen-binding domain CD19ALAb (described in WO20 16/102965) and comprises: a) a heavy chain variable region (VH) having CDRs with the following sequences:
CDR1 - SYWMN (SEQ ID No. 44);
CDR2 - QIWPGDGDTNYNGKFK (SEQ ID No. 45)
CDR3 - RETTTVGRYYYAMDY (SEQ ID No. 46); and b) a light chain variable region (VL) having CDRs with the following sequences:
CDR1 - KASQSVDYDGDSYLN (SEQ ID No. 47);
CDR2 - DASNLVS (SEQ ID No. 48) CDR3 - QQSTEDPWT (SEQ ID No. 49).
It is contemplated that it is possible to introduce one or more mutations (substitutions, additions or deletions) into one or more CDRs without negatively affecting CD19- binding activity. Each CDR may, for example, have one, two or three amino acid mutations.
The CAR may comprise one of the following amino acid sequences.
SEQ ID No. 22 - Murine CD19ALAb scFv sequence QVQLQQSGAELVRPGSSVKISCKASGYAFSSYWMNWVKQRPGQGLEWIGQIWPG DGDTNYNGKFKGKATLTADESSSTAYMQLSSLASEDSAVYFCARRETTTVGRYYYA MDYWGQGTTVTVSSDIQLTQSPASLAVSLGQRATISCKASQSVDYDGDSYLNWYQ
QIPGQPPKLLIYDASNLVSGIPPRFSGSGSGTDFTLNIHPVEKVDAATYHCQQSTEDP
WTFGGGTKLEIK
SEQ ID No. 23 - Humanized CD19ALAb scFv sequence - Heavy 19, Kappa 16
QVQLVQSGAEVKKPGASVKLSCKASGYAFSSYWMNWVRQAPGQSLEWIGQIWPG
DGDTNYNGKFKGRATLTADESARTAYMELSSLRSGDTAVYFCARRETTTVGRYYYA
MDYWGKGTLVTVSSDIQLTQSPDSLAVSLGERATINCKASQSVDYDGDSYLNWYQ QKPGQPPKLLIYDASNLVSGVPDRFSGSGSGTDFTLTISSLQAADVAVYHCQQSTED PWTFGQGTKVEIKR
SEQ ID No. 24 (Humanized CD19ALAb scFv sequence - Heavy 19, Kappa 7)
QVQLVQSGAEVKKPGASVKLSCKASGYAFSSYWMNWVRQAPGQSLEWIGQIWPG
DGDTNYNGKFKGRATLTADESARTAYMELSSLRSGDTAVYFCARRETTTVGRYYYA
MDYWGKGTLVTVSSDIQLTQSPDSLAVSLGERATINCKASQSVDYDGDSYLNWYQ
QKPGQPPKVLIYDASNLVSGVPDRFSGSGSGTDFTLTISSLQAADVAVYYCQQSTE DPWTFGQGTKVEIKR
The scFv may be in a VH-VL orientation (as shown in SEQ ID NO:s 9, 22, 23 and 24) or a VL-VH orientation.
The CAR may comprise one of the following VH sequences:
SEQ ID No. 25 - Murine CD19ALAb VH sequence
QVQLQQSGAELVRPGSSVKISCKASGYAFSSYWMNWVKQRPGQGLEWIGQIWPG DGDTNYNGKFKGKATLTADESSSTAYMQLSSLASEDSAVYFCARRETTTVGRYYYA MDYWGQGTTVTVSS
SEQ ID No. 26 - Humanized CD19ALAb VH sequence
QVQLVQSGAEVKKPGASVKLSCKASGYAFSSYWMNWVRQAPGQSLEWIGQIWPG DGDTNYNGKFKGRATLTADESARTAYMELSSLRSGDTAVYFCARRETTTVGRYYYA MDYWGKGTLVTVSS
The CAR may comprise one of the following VL sequences:
SEQ ID No. 27 - Murine CD19ALAb VL sequence
DIQLTQSPASLAVSLGQRATISCKASQSVDYDGDSYLNWYQQIPGQPPKLLIYDASN
LVSGIPPRFSGSGSGTDFTLNIHPVEKVDAATYHCQQSTEDPWTFGGGTKLEIK
SEQ ID No. 28 (Humanized CD19ALAb VL sequence, Kappa 16)
DIQLTQSPDSLAVSLGERATINCKASQSVDYDGDSYLNWYQQKPGQPPKLLIYDASN
LVSGVPDRFSGSGSGTDFTLTISSLQAADVAVYHCQQSTEDPWTFGQGTKVEIKR
SEQ ID No. 29 - Humanized CD19ALAb VL sequence, Kappa 7 DIQLTQSPDSLAVSLGERATINCKASQSVDYDGDSYLNWYQQKPGQPPKVLIYDAS NLVSGVPDRFSGSGSGTDFTLTISSLQAADVAVYYCQQSTEDPWTFGQGTKVEIKR
The CAR provided herein may comprise a variant of the sequence shown as any of SEQ ID Nos. 16-29 having at least 80, 85, 90, 95, 98 or 99% sequence identity, provided that the variant sequence retains the capacity to bind CD19 (when in conjunction with a complementary VL or VH domain, if appropriate). The CAR provided herein may comprise a variant of the sequence shown as any of SEQ ID Nos. 22-29 or 44-49 having at least 80, 85, 90, 95, 98 or 99% sequence identity, provided that the variant sequence retains the capacity to bind CD19 (when in conjunction with a complementary VL or VH domain, if appropriate). The percentage identity between two polypeptide sequences may be readily determined by programs such as BLAST which is freely available at blast.ncbi.nlm.nih.gov.
TRANSMEMBRANE DOMAIN
The transmembrane domain is the domain of the CAR that spans the membrane.
A transmembrane domain may be any protein structure which is thermodynamically stable in a membrane. This is typically an alpha helix comprising of several hydrophobic residues. The transmembrane domain of any transmembrane protein can be used to supply the transmembrane portion provided herein. The presence and span of a transmembrane domain of a protein can be determined by those skilled in the art using the TMHMM algorithm (http://www.cbs. dtu.dk/services/TMHMM-2.0/). Further, given that the transmembrane domain of a protein is a relatively simple structure, i.e, a polypeptide predicted to form a hydrophobic alpha helix of sufficient length to span the membrane, an artificially designed transmembrane domain may also be used (US 7052906 B1 describes synthetic transmembrane components).
The transmembrane domain may be derived from CD28, which gives good receptor stability. The CD28 transmembrane domain is shown as SEQ ID No. 50.
SEQ ID No. 50
FWVLVWGGVLACYSLLVTVAFI I FWV
The transmembrane domain may be derived from human Tyrp-1 . The tyrp-1 transmembrane domain sequence is shown as SEQ ID No. 51.
SEQ ID No. 51 IIAIAVVGALLLVALIFGTASYLI
The transmembrane domain may be derived from CD8A. The CD8A transmembrane domain sequence is shown as SEQ ID NO: 52.
SEQ ID No. 52 IYIWAPLAGTCGVLLLSLVITLYC
INTRACELLULAR T CELL SIGNALING DOMAIN (ENDODOMAIN)
The endodomain is the signal-transmission portion of the CAR. After antigen recognition, receptors cluster and a signal is transmitted to the cell. The most commonly used endodomain component is that of CD3-zeta which contains 3 ITAMs. This transmits an activation signal to the T cell after antigen is bound. CD3-zeta may not provide a fully competent activation signal and additional co-stimulatory signaling may be needed. For example, endodomains from CD28, or 0X40 or 41 BB can be used with CD3-Zeta to transmit a proliferative I survival signal, or all three can be used together.
Early CAR designs had endodomains derived from the intracellular parts of either the y chain of the FCER1 or CD3^. Consequently, these first generation receptors transmitted immunological signal 1 , which was sufficient to trigger T cell killing of cognate target cells but failed to fully activate the T cell to proliferate and survive. To overcome this limitation, compound endodomains were constructed. Fusion of the intracellular part of a T cell co-stimulatory molecule to that of CD3^ resulted in second generation receptors which could transmit an activating and co-stimulatory signal simultaneously after antigen recognition. The co-stimulatory domain most commonly used was that of CD28. This supplies the most potent co-stimulatory signal, namely immunological signal 2, which triggers T cell proliferation. Some receptors were also described which included TNF receptor family endodomains such as 0X40 and 41 BB which transmit survival signals. Finally, even more potent third generation CARs were
described which had endodomains capable of transmitting activation, proliferation and survival signals.
The endodomain of the CAR of the present invention may comprise combinations of one or more of the CD3-Zeta endodomain, the 41 BB endodomain, the 0X40 endodomain or the CD28 endodomain.
The intracellular T cell signalling domain (endodomain) of the CAR of the present invention may comprise the sequence shown as SEQ ID Nos. 53, 54, 55, 56, 57, 30, 31 or 32 or a variant thereof having at least 80% sequence identity.
SEQ ID No. 53 (CD3 zeta endodomain) RSRVKFSRSADAPAYQQGQNQLYNELNLGRREEYDVLDKRRGRDPEMGGKPRRK NPQEGLYNELQKDKMAEAYSEIGMKGERRRGKGHDGLYQGLSTATKDTYDALHMQ ALPPR
SEQ ID No. 54 (41 BB endodomain)
KRGRKKLLYIFKQPFMRPVQTTQEEDGCSCRFPEEEEGGCEL
SEQ ID No. 55 (0X40 endodomain)
RRDQRLPPDAHKPPGGGSFRTPIQEEQADAHSTLAKI
SEQ ID No. 56 (CD28 endodomain)
KRSRLLHSDYMNMTPRRPGPTRKHYQPYAPPRDFAAY
Examples of combinations of such endodomains include 41 BB-Z, QX40-Z, CD28-Z and CD28-QX40-Zeta.
SEQ ID No. 57 (41 BB-Z endodomain fusion) KRGRKKLLYIFKQPFMRPVQTTQEEDGCSCRFPEEEEGGCELRVKFSRSADAPAYQ QGQNQLYNELNLGRREEYDVLDKRRGRDPEMGGKPRRKNPQEGLYNELQKDKMA EAYSEIGMKGERRRGKGHDGLYQGLSTATKDTYDALHMQALPPR
SEQ ID No. 30 (QX40-Z endodomain fusion) RRDQRLPPDAHKPPGGGSFRTPIQEEQADAHSTLAKIRVKFSRSADAPAYQQGQN QLYNELNLGRREEYDVLDKRRGRDPEMGGKPRRKNPQEGLYNELQKDKMAEAYS EIGMKGERRRGKGHDGLYQGLSTATKDTYDALHMQALPPR
SEQ ID No. 31 (CD28Z endodomain fusion)
KRSRLLHSDYMNMTPRRPGPTRKHYQPYAPPRDFAAYRSRVKFSRSADAPAYQQG
QNQLYNELNLGRREEYDVLDKRRGRDPEMGGKPRRKNPQEGLYNELQKDKMAEA
YSEIGMKGERRRGKGHDGLYQGLSTATKDTYDALHMQALPPR
SEQ ID No. 32 (CD28OXZ)
KRSRLLHSDYMNMTPRRPGPTRKHYQPYAPPRDFAAYRSRDQRLPPDAHKPPGG GSFRTPIQEEQADAHSTLAKIRVKFSRSADAPAYQQGQNQLYNELNLGRREEYDVL DKRRGRDPEMGGKPRRKNPQEGLYNELQKDKMAEAYSEIGMKGERRRGKGHDGL YQGLSTATKDTYDALHMQALPPR
A variant sequence may have at least 80%, 85%, 90%, 95%, 98% or 99% sequence identity to SEQ ID Nos. 25, 26, 27, 28, 29, 30, 31 or 32 provided that the sequence provides an effective transmembrane domain/intracellular T cell signaling domain.
SIGNAL PEPTIDE
The CAR of the present invention may comprise a signal peptide so that when the CAR is expressed inside a cell, such as a T cell, the nascent protein is directed to the endoplasmic reticulum and subsequently to the cell surface, where it is expressed.
The core of the signal peptide may contain a long stretch of hydrophobic amino acids that has a tendency to form a single alpha-helix. The signal peptide may begin with a short positively charged stretch of amino acids, which helps to enforce proper topology of the polypeptide during translocation. At the end of the signal peptide there is typically a stretch of amino acids that is recognized and cleaved by signal peptidase. Signal peptidase may cleave either during or after completion of translocation to generate a free signal peptide and a mature protein. The free signal peptides are then digested by specific proteases.
The signal peptide may be at the amino terminus of the molecule.
The CAR of the invention may have the general formula:
Signal peptide - CD19-binding domain - spacer domain - transmembrane domain/intracellular T cell signaling domain.
The signal peptide may comprise a variant having 5, 4, 3, 2 or 1 amino acid mutations (insertions, substitutions, or additions) provided that the signal peptide still functions to cause cell surface expression of the CAR.
The signal peptide of SEQ ID No. 33 is compact and highly efficient. It is predicted to give about 95% cleavage after the terminal glycine, giving efficient removal by signal peptidase.
SEQ ID No. 33: METDTLLLWVLLLWVPGSTG
The signal peptide of SEQ ID No. 34 is compact and highly efficient. It is predicted to give about 95% cleavage after the terminal glycine, giving efficient removal by signal peptidase.
SEQ ID No. 34 MGTSLLCWMALCLLGADHADA
The signal peptide of SEQ ID NO: 35 is derived from lgG1.
SEQ ID No. 35: MSLPVTALLLPLALLLHAARP
The signal peptide of SEQ ID NO: 36 is derived from CD8.
SEQ ID No. 36: MAVPTQVLGLLLLWLTDARC
SPACER
The CAR of the present invention may comprise a spacer sequence to connect the CD19-binding domain with the transmembrane domain and spatially separate the CD19-binding domain from the endodomain. A flexible spacer allows to the CD19- binding domain to orient in different directions to enable CD19 binding.
The spacer sequence may, for example, comprise an lgG1 Fc region, an lgG1 hinge or a CD8 stalk, or a combination thereof. The spacer may alternatively comprise an alternative sequence which has similar length and/or domain spacing properties as an IgG 1 Fc region, an lgG1 hinge or a CD8 stalk.
A human I gG 1 spacer may be altered to remove Fc binding motifs.
Examples of amino acid sequences for these spacers are given below:
SEQ ID No. 37 (hinge-CH2CH3 of human lgG1)
AEPKSPDKTHTCPPCPAPPVAGPSVFLFPPKPKDTLMIARTPEVTCWVDVSHEDPE
VKFNWYVDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKA LPAPIEKTISKAKGQPREPQVYTLPPSRDELTKNQVSLTCLVKGFYPSDIAVEWESN GQPENNYKTTPPVLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQK SLSLSPGKKD
SEQ ID No. 38 (human CD8 stalk):
TTTPAPRPPTPAPTIASQPLSLRPEACRPAAGGAVHTRGLDFACDI
SEQ ID No. 39 (human lgG1 hinge):
AEPKSPDKTHTCPPCPKDPK
SEQ ID No. 40 (lgG1 Hinge-Fc)
AEPKSPDKTHTCPPCPAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVWDVSHEDP EVKFNWYVDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDWLNGKEYKCKVSNK ALPAPIEKTISKAKGQPREPQVYTLPPSRDELTKNQVSLTCLVKGFYPSDIAVEWESN GQPENNYKTTPPVLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQK
SLSLSPGKKDPK
SEQ ID No. 41 (lgG1 Hinge - Fc modified to remove Fc receptor recognition motifs) AEPKSPDKTHTCPPCPAPPVA*GPSVFLFPPKPKDTLMIARTPEVTCWVDVSHEDP EVKFNWYVDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDWLNGKEYKCKVSNK ALPAPIEKTISKAKGQPREPQVYTLPPSRDELTKNQVSLTCLVKGFYPSDIAVEWESN
GQPENNYKTTPPVLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQK SLSLSPGKKDPK
Modified residues are underlined; * denotes a deletion.
SEQ ID NO: 42 (CD2 ectodomain)
KEITNALETWGALGQDI N LDI PSFQMSDDI DDI KWEKTSDKKKI AQFRKEKETFKEKD TYKLFKNGTLKIKHLKTDDQDIYKVSIYDTKGKNVLEKIFDLKIQERVSKPKISWTCINT TLTCEVMNGTDPELNLYQDGKHLKLSQRVITHKWTTSLSAKFKCTAGNKVSKESSV EPVSCPEKGLD
SEQ ID NO: 43 (CD34 ectodomain)
SLDNNGTATPELPTQGTFSNVSTNVSYQETTTPSTLGSTSLHPVSQHGNEATTNITE TTVKFTSTSVITSVYGNTNSSVQSQTSVISTVFTTPANVSTPETTLKPSLSPGNVSDL STTSTSLATSPTKPYTSSSPILSDIKAEIKCSGIREVKLTQGICLEQNKTSSCAEFKKD RGEGLARVLCGEEQADADAGAQVCSLLLAQSEVRPQCLLLVLANRTEISSKLQLMK KHQSDLKKLGI LDFTEQDVASHQSYSQKT
INTERFERON RELEASE AND CAR T CELL EXHAUSTION
The present inventors have found that a CD19 CAR based on the CAT19 scFv has properties which may result in lower toxicity and better efficacy.
Given that the main experience with CD19 CAR therapy has been with CARs based on the fmc63 scFv, and that the oldest, largest and perhaps most significant clinical data set is with the fmc63 based Campana CAR, the present inventors took this Campana CAR as the “gold-standard”. A comparison was hence made between the fmc63-Campana CAR and a similar CAR but with CAT19 scFv instead of fmc63. Surprisingly, the present inventors found that while CAT19 CAR T cells effected killing of target cell expressing CD19, and proliferated in response to CD19 expressing targets, Interferon-gamma release was less. Further, a small animal model of an aggressive B-cell lymphoma showed equal efficacy and equal engraftment between the fmc63 and CAT19 based CARs, but surprisingly, less of the CAT19 CAR T cells were exhausted than fmc63 CAR T cells.
The CAR of the invention may cause 25, 50, 70 or 90% lower IFNy release in a comparative assay involving bringing CAR T cells into contact with target cells.
The CAR of the invention may result in a smaller proportion of CAR T cells becoming exhausted than fmc63 CAR T cells. T cell exhaustion may be assessed using methods known in the art, such as analysis of PD-1 expression. The CAR of the present invention may cause 20, 30, 40, 50, 60 of 70% fewer CAR T cells to express PD-1 that fmc63 CAR T cells in a comparative assay involving bringing CAR T cells into contact with target cells.
NUCLEIC ACID SEQUENCE
The second aspect of the invention relates to a nucleic acid sequence which codes for a CAR of the methods herein.
The nucleic acid sequence may be capable of encoding a CAR having the amino acid sequence shown as any of SEQ ID Nos. 10-15.
As used herein, the terms “polynucleotide”, “nucleotide”, and “nucleic acid” are intended to be synonymous with each other.
The nucleic acid may be, for example, an RNA, a DNA or a cDNA. Nucleic acids may comprise DNA or RNA. They may be single-stranded or double-stranded. They may also be polynucleotides which include within them synthetic or modified nucleotides.
A number of different types of modification to oligonucleotides are known in the art. These include methylphosphonate and phosphorothioate backbones, addition of acridine or polylysine chains at the 3' and/or 5' ends of the molecule. For the purposes of the use as described herein, it is to be understood that the polynucleotides may be modified by any method available in the art. Such modifications may be carried out in order to enhance the in vivo activity or life span of polynucleotides of interest.
Alternative codons may be used in regions of sequence encoding the same or similar amino acid sequences, in order to avoid homologous recombination when both CARs are encoded by the same vector.
Due to the degeneracy of the genetic code, it is possible to use alternative codons which encode the same amino acid sequence. For example, the codons “ccg” and “cca” both encode the amino acid proline, so using “ccg” may be exchanged for “cca” without affecting the amino acid in this position in the sequence of the translated protein.
The alternative RNA codons which may be used to encode each amino acid are summarized in Table 4.

Alternative codons may be used in the portions of nucleic acid which encode the spacer of the first CAR and the spacer of the second CAR, especially if the same or similar spacers are used in the first and second CARs.
Alternative codons may be used in the portions of nucleic acid which encode the transmembrane domain of the first CAR and the transmembrane of the second CAR, especially if the same or similar transmembrane domains are used in the first and second CARs.
Alternative codons may be used in one or more nucleic acids which encode costimulatory domains, such as the CD28 endodomain.
Alternative codons may be used in one or more domains which transmit survival signals, such as 0X40 and 41 BB endodomains.
Alternative codons may be used in the portions of nucleic acid encoding a CD3zeta endodomain and/or the portions of nucleic acid encoding one or more costimulatory domain(s) and/or the portions of nucleic acid encoding one or more domain(s) which transmit survival signals.
VECTOR
The present invention also provides a vector which comprises a nucleic acid sequence according to the present invention. Such a vector may be used to introduce the nucleic acid sequence into a host cell so that it expresses and produces a molecule according to the first aspect of the invention.
The vector may, for example, be a plasmid or a viral vector, such as a retroviral vector or a lentiviral vector.
The vector may be capable of transfecting or transducing a cell, such as a T cell.
CELLS
The invention also provides a cell which comprises a nucleic acid according to the invention. The invention provides a cell which expresses a CAR provided herein at the cell surface.
The cell may be any eukaryotic cell capable of expressing a CAR at the cell surface, such as an immunological cell.
In particular, the cell may be an immune effector cell such as a T cell or a natural killer (NK) cell.
T cells or T lymphocytes are a type of lymphocyte that play a central role in cell- mediated immunity. They can be distinguished from other lymphocytes, such as B cells and natural killer cells (NK cells), by the presence of a T cell receptor (TCR) on the cell surface. There are various types of T cell, as summarized below.
Helper T helper cells (TH cells) assist other white blood cells in immunologic processes, including maturation of B cells into plasma cells and memory B cells, and activation of cytotoxic T cells and macrophages. TH cells express CD4 on their surface. TH cells become activated when they are presented with peptide antigens by MHC class II molecules on the surface of antigen presenting cells (APCs). These cells can differentiate into one of several subtypes, including TH1 , TH2, TH3, TH 17, Th9, or TFH, which secrete different cytokines to facilitate different types of immune responses.
Cytotoxic T cells (TC cells, or CTLs) destroy vi rally infected cells and tumor cells, and are also implicated in transplant rejection. CTLs express the CD8 at their surface. These cells recognize their targets by binding to antigen associated with MHC class I, which is present on the surface of all nucleated cells. Through IL-10, adenosine and other molecules secreted by regulatory T cells, the CD8+ cells can be inactivated to an anergic state, which prevent autoimmune diseases such as experimental autoimmune encephalomyelitis.
Memory T cells are a subset of antigen-specific T cells that persist long-term after an infection has resolved. They quickly expand to large numbers of effector T cells upon re-exposure to their cognate antigen, thus providing the immune system with "memory" against past infections. Memory T cells comprise three subtypes: central memory T cells (TCM cells) and two types of effector memory T cells (TEM cells and TEMRA cells). Memory cells may be either CD4+ or CD8+. Memory T cells typically express the cell surface protein CD45RO.
Regulatory T cells (Treg cells), formerly known as suppressor T cells, are crucial for the maintenance of immunological tolerance. Their major role is to shut down T cell- mediated immunity toward the end of an immune reaction and to suppress auto- reactive T cells that escaped the process of negative selection in the thymus.
Two major classes of CD4+ Treg cells have been described — naturally occurring Treg cells and adaptive Treg cells.
Naturally occurring Treg cells (also known as CD4+CD25+FoxP3+ Treg cells) arise in the thymus and have been linked to interactions between developing T cells with both myeloid (CD11c+) and plasmacytoid (CD123+) dendritic cells that have been activated with TSLP. Naturally occurring Treg cells can be distinguished from other T cells by the presence of an intracellular molecule called FoxP3. Mutations of the FOXP3 gene can prevent regulatory T cell development, causing the fatal autoimmune disease IPEX.
Adaptive Treg cells (also known as Tr1 cells or Th3 cells) may originate during a normal immune response.
The T cell provided herein may be any of the T cell types mentioned above, in particular a CTL.
Natural killer (NK) cells are a type of cytolytic cell which forms part of the innate immune system. NK cells provide rapid responses to innate signals from virally infected cells in an MHC independent manner.
NK cells (belonging to the group of innate lymphoid cells) are defined as large granular lymphocytes (LGL) and constitute the third kind of cells differentiated from the common lymphoid progenitor generating B and T lymphocytes. NK cells are known to differentiate and mature in the bone marrow, lymph node, spleen, tonsils and thymus where they then enter into the circulation.
The CAR-expressing cells provided herein may be any of the cell types mentioned above.
The CAR-expressing cell of the invention may be generated ex vivo. The cell may be from a cell sample, such as a peripheral blood mononuclear cell (PBMC) sample from the patient or a donor. Cells may be activated and/or expanded prior to being transduced with CAR-encoding nucleic acid, for example by treatment with an anti-CD3 monoclonal antibody.
Alternatively, T or NK cells provided herein may be derived from ex vivo differentiation of inducible progenitor cells or embryonic progenitor cells to T or NK cells.
Alternatively, an immortalized T cell line which retains its lytic function and could act as a therapeutic may be used.
Obecabtagene autoleucel (referred to herein as “obe-cel”) consists of autologous peripheral blood T cells transduced ex vivo with a lentiviral vector which encodes the CD19 CAR “CD19 (CAT) CAR” disclosed herein, which has a lower affinity and faster disengagement from CD19 compared to CD19 (FMC63) CAR used in the approved CAR T cell therapies Kymriah® (tisagenlecleucel),Yescarta® (axicabtagene ciloleucel) and Tecartus® (brexucabtagene autoleucel).
Binding kinetics of the CAT scFv (used in obe-cel) and FMC63 scFv (used in axicabtagene ciloleucel and tisagenlecleucel) with recombinant CD19 were determined using surface plasmon resonance. Equilibrium dissociation constants (KD) of 14.4 nM for CAT and 0.328 nM for FMC63 were determined, when the data were fitted to a 1 :1 Langmuir binding model. A greater than 40-fold lower KD of the CAT scFv was the
result of a much faster off-rate (CAT: 3.1 x 10-3 s_1 versus FMC63: 6.8 x 10-5 s'1), while the on-rate was equivalent (CAT: 2.2 x 105 M'1S'1 versus FMC63: 2.1 x 105 M'1S'1).
CD19 (CAT) CAR T cell proliferation was greater than CD19 (FMC63) CAR T cells when co-cultured with Raji and NALM-6 cells. The enhanced proliferation was not a result of increased IL-2 production indicating an IL-2 independent mechanism.
Normal donor T cells were transduced to express CD19 (CAT) CAR or CD19 (FMC63) CAR, challenged with Raji cells (a Burkitt’s lymphoma cell line that expresses CD19), and pro-inflammatory cytokines were analysed in the supernatant at 48 hours. The cytokine release profiles were similar for both CAR expressing T cell lines with the exception of a higher tumour necrosis factor a secretion from the CD19 (CAT) CAR T cells than from the CD19 (FMC63) CAR T cells (mean 750.7 ± 103.3 pg/mL, and 292.1 ± 36.51 pg/mL, respectively; n=4, p<0.01).
In a standard 51Cr release assay, T cells transduced to express CD19 (CAT) CAR or CD19 (FMC63) CAR were incubated with CD19-negative and CD19 expressing targets. Cell killing was significantly greater for CD 19 (CAT) CAR T cells than for CD19 (FMC63) CAR T cells, particularly at low effectortarget ratios.
Anti-tumour efficacies of CD19 (CAT) CAR T cells and CD19 (FMC63) CAR T cells were assessed in a NALM-6 NSG-mouse xenograft model and compared. In control mice receiving non-transduced T cells, rapid, disseminated tumour infiltration was observed. CD19 (FMC63) CAR T cells slowed but did not prevent growth of the tumour. In contrast, equivalent numbers of CD19 (CAT) CAR T cells resulted in tumour regression. On Day 12 post-T cell injection, substantial differences were seen in tumour burden; mean CD19 (CAT) CAR T cells: 1.1 x 108 to 9.3 x 107 photons/second/cm2/steradian; CD19 (FMC63) CAR T cells 3.2 x 109 to 7.7 x 108 photons/second/cm2/steradian, n=18, p<0.001. Two weeks after the infusion of NALM-6 NSG mice with CAR T cells, blood and bone marrow (BM) were analysed for residual tumour cells and persisting CAR T cells. A significantly higher number of residual NALM-6 tumour cells were observed in the BM of the CD19 (FMC63) CAR T cell cohort compared with the CD19 (CAT) CAR T cell cohort (mean 2.8 x 105 versus 3 x 102 NALM-6 cells/mL, p<0.0001). Conversely, a significantly greater absolute number of CD19 (CAT) CAR T cells than CD19 (FMC63) CAR T cells persisted in the BM of NALM-6 NSG mice (mean 3.4 x 104 CD19 (CAT) CAR T cells/mL versus 1.3 x 104 CD19 (FMC63) CAR T cells/mL, n=18, p<0.05) and in the peripheral blood
(mean: 1.9 xio4 CD19 (CAT) CAR T cells/mL versus 2.8 x 103 CD19 (FMC63) CAR T cells/mL, n=9, p<0.001).
In summary, the CAT scFv binding domain used in obe-cel has a > 40-fold lower affinity to recombinant CD19 in vitro than the FMC63 scFv binding domain used in other CAR T products. CD19 (CAT) CAR enables transduced T cells to proliferate, secrete cytokines in response to CD19-positive targets and specifically lyse CD19-positive cell lines in vitro with greater cytotoxicity compared to CD19 (FMC63) CAR T cells. CD19 (CAT) CAR T cells also have better anti-tumour efficacy and engraftment versus CD19 (FMC63) CAR T cells in an NSG NALM-6 mouse model of leukaemia.
Clinical experience with obe-cel CAR T cells includes clinical studies in various hematological malignancies. There are currently four ongoing studies evaluating obe- cel in pediatric and adult patients with B cell acute lymphoblastic leukemia (B ALL) and other B cell malignancies: 3 Investigator-initiated Phase I studies, CARPALL (EudraCT 2015-001144-10 2019, NCT02443831 2016) (Ghorashian S, Kramer AM, Onuoha S, Wright G, et al. (2019). Enhanced CAR T cell expansion and prolonged persistence in pediatric patients with ALL treated with a low-affinity CD19 CAR. Nat Med. 25(9):1408- 14), ALLCAR19 (EudraCT 2016-004027-22, NCT02935257 2017) (Roddie C, Dias J, O'Reiilly MA, et al (2021). Durable Responses and Low Toxicity After Fast Off-Rate CD19 Chimeric Antigen Receptor-T Therapy in Adults With Relapsed or Refractory B- Cell Acute Lymphoblastic Leukemia. J Clin Oncol; 39(30):3352-63; Roddie C, Dias J, O’Reilly M, Mitsikakou M, et al. (2022a). Safety, efficacy and long-term follow-up of AUTO1 , a fost-off rate CD19 CAR in relapsed/refractory B-cell acute lymphoblastic leukaemia and other B-cell malignancies. American Society of Hematology Annual Meeting Poster 3318; Roddie C, Dias J, O’Reilly M, Mitsikakou M, et al. (2023b). Longterm follow-up of AUTO1 , a fast-off rate CD 19 CAR, in relapsed/refractory B-cell acute lymphoblastic leukemia and factors associated with durable response. American Society for Transplantation and Cellular Therapy Annual Meeting Poster 277), CAROUSEL (NCT04443829) (Roddie C, Dias J, O’Reilly M, Green L. (2022c). Safety and efficacy findings of AUTO1 , a fast-off rate CD19 CAR, in relapsed/refractory primary CNS lymphoma. European Hematology Association Annual Meeting Poster P1460) and 1 Autolus sponsored Phase Ib/ll AUTO1-AL1 (FELIX) study in adult patients with relapsed/refractory B-ALL (EudraCT 2019-001937-16, NCT04404660). The Phase II portion of the FELIX study is the pivotal study to achieve the marketing authorization of obe-cel (Roddie C, Sandhu KS, Tholouli E, Shaughnessy P, et al. (2023a). Safety and effiacy of obecabtagene autoleucel (obe-cel, AUTO1), a fast-off
rate CD19 CAR, in relapsed/refractory adult B-cell acute lymphoblastic leukemia (r/r IB- ALL): top line result of the pivotal FELIX study. American Society of Clinical Oncology Annual Meeting Oral abstract #7000). In total, as of 01-Aug-2023, 201 patients have been treated with obe-cel across different B cell malignancies in these 4 studies. Based on the novel mode of action of the CAT CAR construct with a more physiologic T cell activation and a reduced cytokine release, the safety and tolerability profile of obe-cel has been shown to be favorable with very low rates of cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome (ICANS) across different indications in both pediatric and adult patients (Ghorashian et al, supra; Roddie et al (2021), supra; Roddie et al (2022a), supra; Roddie et al (2023a), supra).
No clinical studies with obe-cel have been performed in patients with autoimmune disease.
Table 5 Ongoing Clinical Studies with Obe-cel



Abbreviations: AE=adverse event; ATIMP=advanced therapy medicinal product; B ALL=B-cell acute lymphoblastic leukaemia; B CLL=B-cell chronic lymphocytic leukaemia; B NHL=B-cell Non-Hodgkin’s lymphoma; BM =bone marrow; B SLL=B-cell small lymphocytic lymphoma ;CAR=chimeric antigen receptor; CD=cluster of differentiation; CNS=central nervous system; CR=complete remission; CRi=complete remission with incomplete recovery of blood counts; DLBCL=diffuse large B-cell lymphoma; EudraCT= European Union Drug Regulating Authorities Clinical Trials; ICANS=immune effector cell- associated neurotoxicity syndrome; IRRC=lndependent Response Review Committee; IV=intravenous; min=minimal; MRD=minimal residual disease; NCT=National clinical trial; NGS=next generation sequencing; ORR=overall response rate; PCNSL=primary central nervous system lymphoma; r/r=relapsed or refractory; qPCR=quantitative polymerase chain reaction; PD=Progressive disease; SAE=serious adverse event; SD=Stable disease
The available clinical data for obe-cel in the treatment of patients with B ALL from the Phase I studies CARPALL and ALLCAR19 and the Phase Ib/ll FELIX study support the non-clinical data and demonstrate promising efficacy with manageable toxicity in patients with r/r B ALL.
• Ten of 13 paediatric or young adult patients treated with obe-cel manufactured using an open process (Cohort 1) in CARPALL achieved MRDnegative CR resulting in an ORR of 77% following obe-cel infusion. All 7 patients treated with obe-cel manufactured using a closed process (Cohort 2) were in MRD-negative CR after obe-cel infusion (ORR=100%).
• Seventeen of 20 evaluable adult patients in Study ALLCAR19 achieved MRDnegative CR resulting in an ORR of 85% post obe-cel infusion.
• As of 16-Mar-2023, of the 94 patients who had > 5% blasts in the bone marrow (BM) at screening and were infused with obe-cel in FELIX, 71 patients achieved CR + CRi resulting in an ORR of 75.5%.
• CRS of any grade occurred in 95% of paediatric and young adult B ALL patients in CARPALL, in 55% of adult B ALL patients in ALLCAR19, and 69% of total infused adult patients with B ALL (N=126) in FELIX. Three patients (2.4%) experienced CRS > grade 3. Severe neurotoxicity was limited with 1 patient in the CARPALL study (5%); 3 patients in the ALLCAR19 study (15%) and 9 patients in the FELIX study (7.1 %) reporting >grade 3 neurological AE/ICANS.
Preliminary results obtained in indolent B NHL look promising with all 10 patients treated with obe-cel achieving complete molecular response (data cut-off 18-Nov- 2021). The safety profile remains favourable in this small sample size with four patients with grade 1 , two patients with grade 2 CRS and no ICANS of any grade reported.
PHARMACEUTICAL COMPOSITION
The present invention also relates to a pharmaceutical composition containing a CAR- expressing cell, or plurality of cells, of the invention together with a pharmaceutically acceptable carrier, diluent or excipient, and optionally one or more further pharmaceutically active polypeptides and/or compounds. Such a formulation may, for example, be in a form suitable for intravenous infusion.
METHOD OF TREATMENT
Cells such as T cells or NK cells expressing a CAR molecule provided herein may be used for the treatment of an autoimmune disease, in particular an autoimmune disease mediated by CD19-positive B lymphocytes or B cells.
The present disclosure provides a method for treating an autoimmune disease mediated by CD19-positive B lymphocytes in a patient comprising administering to the patient an CD19 CAR T cell or a population of CD19 CAR T cells.
The present disclosure provides a CD19 CAR T cell or a population of CD19 CAR T cells for use in treating an autoimmune disease mediated by CD19-positive B lymphocytes.
The present disclosure provides the use of a CD19 CAR T cell or a population of CD19 CAR T cells in the manufacture of a medicament for treating an autoimmune disease mediated by CD19-positive B lymphocytes.
Examples of autoimmune diseases include, but are not limited to, systemic lupus erythematosus (SLE), rheumatoid arthritis, idiopathic inflammatory myopathy (IIM, myositis), systemic sclerosis, ANCA-associated vasculitis, inflammatory bowel disease (IBD), multiple sclerosis (MS), Type I diabetes, Guillain-Barre syndrome, chronic inflammatory demyelinating polyneuropathy (CIDP), Graves’ disease, Hashimoto’s
thyroiditis or Hashimoto’s disease, myasthenia gravis, neuromyelitis optica, A/-methyl- D-aspartate receptor (NMDAR) encephalitis, Lambert-Eaton syndrome, scleroderma, vasculitis, pemphigus vulgaris, pemphigus foliaceus, epidermolysis bullosa acquisita, bullous pemphigoid, lupus nephritis, membranous nephropathy, Goodpasture’s syndrome, immune thrombocytopenic purpura, thrombotic thrombocytopenic purpura, antiphospholipid syndrome, autoimmune haemolytic anaemia, and acquired haemophilia.
The autoimmune disease may be SLE.
The autoimmune disease may be severe SLE.
The autoimmune disease may be refractory SLE.
The autoimmune disease may be severe, refractory SLE.
The SLE may present with Lupus Nephritis.
The Lupus Nephritis may be severe.
The Lupus Nephritis may be active.
The SLE may present with active, severe Lupus Nephritis.
Methods for treating autoimmune disease provided herein relate to the therapeutic use of a cell or population of cells provided herein. In this respect, the cells may be administered to a subject having an existing disease or condition in order to lessen, reduce or improve at least one symptom associated with the disease and/or to slow down, reduce or block the progression of the disease. The method may cause or promote cell mediated killing of CD19-expressing cells, such as B cells.
TREATMENT OF SLE
Considering the important role of B cells in SLE disease pathogenesis and the evidence of safety and activity of CD19 CAR T cell therapy, it is contemplated herein that infusion of a patient with a CD19 CAR T cell product (for example, obe-cel) will eliminate the
malfunctioning autoreactive B cells and subsequently, normal, non-autoreactive B cells are produced.
Patients to be treated include, but are not limited to, patients with severe, refractory SLE who are predicted to have a poor outcome if treated with standard of care therapy due to disease severity, one or more areas of SLE-related organ system involvement, and prior exposure to multiple SLE treatment strategies.
Patients to be treated: may have a diagnosis of SLE fulfilling the 2019 European League Against Rheumatism (EULAR)/American College of Rheumatology (ACR) Classification Criteria for Systemic Lupus Erythematosus; may be positive for at least one of the following autoantibodies: antinuclear antibodies (ANA) at a titer of >1 :80, or anti-dsDNA (>30 IIJ/mL) or anti-Sm (> ULN), anti-histone or anti-chromatin (> ULN); may have severe SLE defined as a) a Systemic Lupus Erythematosus Disease Activity Index 2000 (SLEDAI-2K) score of > 8 points (of which 4 are non-laboratory items and with the exclusion of points associated with neurological findings [SLEDAI 2K items 1-7]) and b) at least one of the following significant SLE-related organ involvements: i) renal (ongoing active, biopsy-proven lupus nephritis), ii) moderate or severe pericarditis/myocarditis, iii) moderate or severe pleuritis or other lung involvement, iv) vasculitis, and v) severe hematologic manifestation (e.g. severe thrombocytopenia or severe autoimmune hemolytic anemia); and/or may have refractory SLE defined as lack of response, insufficient response or lack of sustained response or intolerance to: a) hydroxychloroquine treatment in combination with corticosteroids, and b) >2 of the following treatment groups used for at least 3 months each or less if intolerant:
i) Immunosuppressive drugs (e.g., methotrexate, azathioprine, mycophenolate mofetil, mycophenolic acid, tacrolimus, leflunomide, cyclosporine, voclosporin or cyclophosphamide), ii) B cell-targeting agents (e.g., belimumab, anti-CD20 mAb), and iii) cytokine inhibitors (e.g., anifrolumab).
Patients may be subject to a pre-conditioning chemotherapy such as lymphodepletion chemotherapy prior to CD19 CAR treatment. The lymphodepletion chemotherapy may comprise treatment with fludarabine and cyclophosphamide. The patients may receive three doses of fludarabine 25 mg/m2/d intravenous administration on Day -5, Day -4, and Day -3 relative to CD 19 CAR treatment and one dose of cyclophosphamide 1000 mg/m2 intravenous administration on Day -3 relative to CD 19 CAR treatment.
In the methods of treatment provided herein for an autoimmune disease, such as SLE, the patients may be administered a single dose of 50 x 106 (±20%) CD19 CAR-positive viable T cells. Alternative doses contemplated are an escalated (100 x 106 [±20%]) or a de-escalated (30 x 106 [±20%]) CAR-positive T cell dose. The administration may be, for example, an intravenous injection through a Hickman line or peripherally inserted central catheter (PICC line).
It will be understood that there may be differences in e.g. the starting material (leukapheresate) and/or manufacturing of the CD19 CAR-positive T cells. These differences may lead to dose variability, i.e. variability in the amount of CD19 CARpositive T cells that is administered to the patient. It is contemplated herein that the dose may have a variability of ±5%, ±10%, ±15%, ±20%, ±25%, or ±30%.
Treated patients will exhibit improvement in one or more of the following assessments: Definition of Remission in SLE (Doris), improvements may include SLEDAI = 0 and/or PGA < 0.5 on a visual analog scale from 0-3;
Lupus Low Disease Activity State (LLDAS), improvements may include SLEDAI-2K < 4 (no activity in major organ systems (renal, central nervous system, cardiopulmonary, vasculitis, fever and no hemolytic anemia or gastrointestinal activity), no new features of lupus disease activity compared to the previous assessment, PGA < 1 on a visual analog scale from 0-3, current prednisolone (or equivalent) dose < 7.5 mg daily, and/or well-tolerated standard maintenance doses of immunosuppressive drugs and approved biologic agents, excluding investigational drugs;
Systemic Lupus Erythematosus Disease Activity Index-2000 (SLEDAI-2K) score;
Physician’s global assessment (PGA);
Health Assessment Questionnaire - Disability Index (HAQ-DI);
Functional Assessment of Chronic Illness Therapy - Fatigue Scale (FACIT-Fatigue) score;
Safety of Estrogens in Lupus Erythematosus National Assessment-SLEDAI (SELENA- SLEDAI) Flare Index (SFI) score;
Urinary protein creatinine ratio (UPGR);
Autoantibody panel (antinuclear antibody [ANA], anti-double stranded DNA [anti- dsDNA], anti-Smith, anti-RNA binding protein [anti-RBP]);
Antiphospholipid profile (lupus anticoagulant, anti-cardiolipin antibodies and beta-2 glycoprotein 1);
Complement panel (CH50, C3, C4);
Duration of B cell aplasia;
Determination of T and B cell phenotype over time as assessed by flow cytometry in the peripheral blood;
Determination of BlyS/BAFF concentration dynamics in the peripheral blood; and Determination of cytokine concentration dynamics in the peripheral blood.
OTHER TERMINOLOGY AND DISCLOSURE
As used herein and in the appended claims, the singular forms "a," "and," and "the" include plural referents unless the context clearly dictates otherwise. It is further noted that the claims may be drafted to exclude any element, e.g., any optional element. As such, this statement is intended to serve as antecedent basis for use of such exclusive terminology as "solely," "only" and the like in connection with the recitation of claim elements, or use of a "negative" limitation.
When a range of values is provided herein, it is understood that each intervening value, to the tenth of the unit of the lower limit unless the context clearly dictates otherwise, between the upper and lower limit of that range and any other stated or intervening value in that stated range, is encompassed within the disclosure. The upper and lower limits of these smaller ranges may independently be included in the smaller ranges, and are also encompassed within the disclosure, subject to any specifically excluded limit in the stated range. Where the stated range includes one or both of the limits, ranges excluding either or both of those included limits are also included in the disclosure.
Unless defined otherwise, all technical and scientific terms used herein have the same meaning as commonly understood by one of ordinary skill in the art to which this disclosure belongs. Any methods and materials similar or equivalent to those described herein can also be used in the practice or testing of the present disclosure.
All publications mentioned herein are incorporated herein by reference to disclose and describe the methods and/or materials for the purpose for which the publications are cited.
As will be apparent to those of skill in the art upon reading this disclosure, each of the individual embodiments described and illustrated herein has discrete components and features which may be readily separated from or combined with the features of any of the other several embodiments without departing from the scope or spirit of the present disclosure. Any recited method can be carried out in the order of events recited or in any other order which is logically possible. This disclosure is intended to provide support for all such combinations.
As used herein, “may,” “may comprise,” “may be,” “can,” “can comprise” and “can be” all indicate something envisaged by the inventors that is functional and available as part of the subject matter provided.
Various abbreviations follow.
Abbreviation Term
ACR American College of Rheumatology
AE adverse Event
AESI adverse event of special interest
ALC absolute lymphocyte count
ALT alanine aminotransferase
ANA antinuclear antibodies
Anti-dsDN Anti-double stranded DNA
ATI MP Advanced Therapy Investigational Medicinal Product
B-ALL B cell acute lymphoblastic leukemia
BAFF B cell activating factor
BlyS B lymphocyte stimulator
BOIN Bayesian Optimal Interval
CAR Chimeric Antigen Receptor
CCG Case Report Form Completing Guidelines
Abbreviation Term
CD Cluster of differentiation
CG Cockcroft Gault
CIOMS Council for International Organizations of Medical Sciences
CNS central nervous system
CRF case report form
CRS cytokine release syndrome
CSF cerebrospinal fluid
CTCAE Common Terminology Criteria for Adverse Events
DIC disseminated intravascular coagulation
DDS Dose-Determining Set
DLT dose-limiting toxicity
DORIS Definition of Remission in Systemic Lupus Erythematosus
DRE disease-related events
ECG Electrocardiogram
EDC electronic data capture
ELISA enzyme-linked immunosorbent assay
EU European Union
FACIT- Functional Assessment of Chronic Illness Therapy - Fatigue Scale
Fatigue
GCP Good Clinical Practice
GMP Good Manufacturing Practice
HLH hemophagocytic lymphohistiocytosis
IB Investigator’s Brochure
ICANS immune effector cell-associated neurotoxicity syndrome
ICE immune effector cell-associated encephalopathy
ICF informed consent form
ICH International Council on Harmonisation
IDMC Independent Data Monitoring Committee
IEC Independent Ethics Committee
IUD intrauterine device
KM Kaplan-Meier
LLDAS Lupus Low Disease Activity State
LN lupus nephritis
LPLV last patient last visit
MAS macrophage activation syndrome
MTD maximum tolerated dose
Abbreviation Term
NCI National Cancer Institute
NCCN National Comprehensive Cancer Network
Obe-cel obecabtagene autoleucel (also referred to as ALITO1)
PBMC peripheral blood mononuclear cell
PCR polymerase chain reaction
PCP Pneumocystis carinii
PGA Physician Global Assessment
PI Principal Investigator
PJP Pneumocystis jirovecii
PK Pharmacokinetic
RCL replication-competent lentivirus
SAE serious adverse event
SAP Statistical Analysis Plan
SFI Safety of Estrogens in Lupus Erythematosus National Assessment-
SLEDAI (SELENA-SLEDAI) Flare Index
SLE systemic lupus erythematosus
SLEDAI Systemic Lupus Erythematosus Disease Activity Index
SoA Schedule of Activities
SRC Safety Review Committee
SRI Systemic Lupus Erythematosus Responder Index
SUSAR suspected unexpected serious adverse reaction
UK United Kingdom
ULN upper limit of normal
US United States
VAS visual analogue scale
The invention will now be further described by way of Examples, which are meant to serve to assist one of ordinary skill in the art in carrying out the invention and are not intended in any way to limit the scope of the invention.
EXAMPLES
The examples describe a study designed to demonstrate the safety, tolerability, and preliminary efficacy of obe-cel CAR T cells in patients with severe, refractory SLE. Conceptually, CAR T cell-based therapies such as obe-cel provide further advantages over antibodies because CAR T cells require only a single administration, expand, and
have the potential to persist in vivo, migrate to multiple lymphoid tissues and organs in the recipient and develop into both effector and memory cell population. Longer persistency of obe-cel with its CAT CD19 binder, when indirectly compared with the approved FMC63 CD19 CAR T cells, provides another advantage.
As mentioned above, obe-cel is prepared from enriched autologous T cells that are transduced with a lentiviral vector to express a novel, second generation CAR targeting CD19 with a 4-1 BB and CD3- endodomain.
The CAR of the obe-cel CAR T cells comprises a single chain variable fragment (scFv) called CAT with a lower affinity for CD19 and a faster off rate compared with that of the FMC63 scFv used in other approved CD19 CAR T therapies. This fast-off binding kinetics is contemplated to confer an opportunity for physiological T cell activation, reduced immune-toxicity and potential for improved persistence. See, WO20 16/139487.
Obe-cel is classified as an advanced therapy investigational medicinal product (ATIMP). It is an autologous cellular therapy product where the active component is the patient’s own T cells transduced with a lentiviral vector to express a novel second generation CAR targeting CD19 (sometimes referred to herein as “CD19 CAT CAR”). Obe-cel also contains non-transduced autologous T cells and non-T cells.
Once administered to patients, obe-cel CAR T cells circulate throughout the body. The CAR T cells recognize CD19 surface antigens on all B cells and B-progenitor cells, resulting in activation and proliferation of the CAR T cells. Activated CAR T cells will then in turn cause destruction of these cells via several physiological effector mechanisms.
Example 1 - Eligibility Criteria
The patients treated are patients with severe, refractory SLE who are predicted to have a poor outcome if treated with standard of care therapy due to disease severity, one or more areas of SLE-related organ system involvement, and prior exposure to multiple SLE treatment strategies.
Patients must meet all eligibility criteria.
Key Inclusion Criteria
• Diagnosis of SLE fulfilling the 2019 European League Against Rheumatism (EULAR)/American College of Rheumatology (ACR) Classification Criteria for Systemic Lupus Erythematosus.
• Positive for at least one of the following autoantibodies: antinuclear antibodies (ANA) at a titer of >1:80, or anti-dsDNA (>30 IIJ/mL) or antiSmith (> ULN), anti-histone or anti-chromatin (> ULN).
• Severe SLE defined as
- A Systemic Lupus Erythematosus Disease Activity Index 2000 (SLEDAI-2K) score of > 8 points (of which 4 are non-laboratory items and with the exclusion of points associated with neurological findings [SLEDAI 2K items 1-7]) AND
- At least one of the following significant SLE-related organ involvements:
■ Renal (ongoing active, biopsy-proven lupus nephritis)
NOTE: A biopsy should be performed (unless contra-indicated) in the 12 months prior to the screening visit or during the screening period to confirm ongoing active, biopsy-proven lupus nephritis. The histopathological report needs to be available as a source.
■ Moderate or severe pericarditis/myocarditis
■ Moderate or severe pleuritis or other lung involvement
■ Vasculitis
■ Severe hematologic manifestation (e.g., severe thrombocytopenia or severe autoimmune hemolytic anemia)
• Refractory SLE (defined as lack of response, insufficient response or lack of sustained response) or intolerance to: a. Hydroxychloroquine treatment in combination with corticosteroids AND b. s2 of the following treatment groups used for at least 3 months each or less if intolerant: i Immunosuppressive drugs (e.g., methotrexate, azathioprine, mycophenolate mofetil, mycophenolic acid, tacrolimus, leflunomide, cyclosporine, voclosporin or cyclophosphamide)
ii. B cell-targeting agents (e.g., belimumab, anti-CD20 mAb) iii Cytokine inhibitors (e.g., anifrolumab)
Key Exclusion Criteria
SLE and Autoimmunity
• Recurrent neuropsychiatric lupus at any point prior to the screening visit or active, severe, or unstable neuropsychiatric lupus disease within 2 years.
• Diagnosis of drug-induced SLE rather than idiopathic SLE.
• Any acute, severe lupus-related flare during screening that needs immediate treatment and/or makes the immunosuppressive washout impossible; thus, making the patient ineligible for CD19 CAR T therapy as judged by the Investigator or Sponsor.
• Significant, likely irreversible organ damage related to SLE (e.g., endstage renal disease) that in the opinion of the Investigator renders CD19 CAR T cell therapy unlikely to benefit the patient.
• Diagnosis of primary antiphospholipid syndrome.
• Diagnosis of clinically significant uveitis.
• Diagnosis of other non-SLE autoimmune disease (e.g., dermatomyositis, polymyositis, scleroderma, rheumatoid arthritis) or other non-SLE autoimmune disease or overlap syndrome.
Medical History
1. History or presence of a. Within 3 months before screening visit
■ clinically relevant CNS pathology such as epilepsy, paresis, aphasia, or stroke
■ evidence of deep venous thrombosis or pulmonary embolism b. Any time before screening visit: severe brain injuries, dementia, Parkinson’s disease, cerebellar disease, organic brain syndrome, uncontrolled mental illness, or psychosis.
2. Clinically significant, uncontrolled heart disease not due to SLE (New York Heart Association Class III or IV heart failure, uncontrolled angina, severe
uncontrolled cardiac arrhythmia, or electrocardiographic evidence of acute ischemia or Grade 3 conduction system abnormalities unless the patient has a pacemaker) or a recent (within 12 months of screening) cardiac event.
3. Active or uncontrolled fungal, bacterial, viral (including COVID-19), or other infection requiring systemic antimicrobials for management.
4. Active or latent Hepatitis B or active Hepatitis C.
5. Human Immunodeficiency Virus, HTLV-1 , HTLV-2 or syphilis positive test at screening.
6. History of malignant neoplasms unless disease free for at least 24 months (basal cell or squamous cell carcinoma in situ, or in situ breast cancer on hormonal therapy allowed).
7. History of heart, lung, kidney, liver transplant or hematopoietic stem cell transplant.
8. Pregnancy and lactating.
Laboratory and Organ Function
9. Patients are not eligible if the below laboratory criteria are met. a. Neutrophil count < 1 ,000/pL b. Platelet count < 50,000/pL c. Hemoglobin < 7 g/dL for SLE-related hemolytic anemia or < 8 g/dL for all other patients d. B-cell aplasia e. Serum alanine aminotransferase/aspartate aminotransferase > 2.5 x ULN f. Total bilirubin >1.5 x ULN for patients without Gilbert’s syndrome or direct bilirubin > 1 x ULN in patients with Gilbert’s syndrome g. Creatinine clearance (as estimated by Cockcroft Gault) < 30 mL/min/1.73m2 h. INR and PTT > 1.5 ULN.
10. Left ventricular ejection fraction < 45% (or < institutes lower limit of normal) confirmed by ECHO.
11 . SpO2 < 90% in the absence of oxygen support.
Medications
• Prior treatment with anti-CD19 therapy (including bispecifics), adoptive T cell therapy or any prior gene therapy product (e.g., CAR T cell therapy).
• Within 2 months of leukapheresis: use of anti-CD20 therapy.
• Immunization with a live or attenuated vaccine within 2 months of leukapheresis.
• The following are excluded within 14 days before leukapheresis: a. Use of standard antimalarials, immunosuppressive therapy or immunomodulatory therapy including biologies. b. Systemic corticosteroids greater than 10 mg daily of prednisone or its equivalent
• Within 2 months or 5 half-lives of leukapheresis: any investigational drug, whichever is longer.
NOTE: If the half-life is not known, please consider a 2 months washout is required.
General
• Presence of any indwelling line or drain (e.g., percutaneous nephrostomy tube, indwelling Foley catheter, biliary drain, or pleural/peritoneal/pericardial catheter). NOTE: Central venous access catheters such as a Port-a-Cath or Hickman catheter are permitted.
• Inability to tolerate leukapheresis.
• Inability to understand or comply with the safety monitoring requirements of the study or unlikely to complete all protocol-required study visits or procedures, including follow-up visits.
• Presence of any contraindications to cyclophosphamide or fludarabine.
Example 2 - Leukapheresis and Lymphodepletion Therapy
Leukapheresis
Before leukapheresis, from Day -6 to Day -8, patients will undergo clinical and laboratory assessments to confirm the patient meet all eligibility criteria.
Adequate washout and tapering of SLE medications is ensured.
After completing the required wash-out, patients will undergo unstimulated leukapheresis for the generation of obe-cel. This may require insertion of a central venous access device and is a day case procedure to collect PBMCs only.
Inadequate Leukapheresis
Should there be any issues with the procedure (i.e. , inadequate cell collection for dose manufacture, contamination of the leukapheresis material), then the patient may undergo a repeat leukapheresis if clinically appropriate.
Bridging Therapies
If necessary to control the disease/symptoms, patients may receive bridging therapy with permitted standard of care (SOC) medications to treat SLE.
Lymphodepletion Chemotherapy
Lymphodepletion chemotherapy with fludarabine and cyclophosphamide is recognized as standard of care before CAR T cell infusion. Clinical CAR T cell data collectively indicate that Fludarabine/Cyclophosphamide are required for CAR T cell engraftment, and CAR T cell engraftment is required for activity towards B cells
Patients will receive three doses of fludarabine and one dose of cyclophosphamide as follows:
Fludarabine: 25 mg/m2/d IV administration on Day -5, Day -4, and Day -3 Cyclophosphamide: 1000 mg/m2 IV administration on Day -3
This lymphodepletion regimen matches the one published by Mackensen et al, 2022.
If the obe-cel infusion cannot occur within 10 days from lymphodepletion, the lymphodepletion therapy may be repeated.
Example 3 - Obe-cel
The starting material for generation of obe-cel is mononuclear cells from a nonmobilized leukapheresis collection from the patient. Thus, obe-cel is an autologous product and is therefore specific for each patient.
The autologous T cells are enriched and transduced ex vivo with a lentiviral vector to express a novel CD19-targeting (i.e., anti-CD19) CAR referred to as “CD19 (CAT)
CAR.” The “CAT” binding domain was chosen as a binding domain as it showed a substantially lower affinity to CD19 (>40 fold) than FMC63 scFv, a binder used in already marketed CAR T cell therapies
More specifically, The CD19 (CAT) CAR construct is derived from a murine CAT13.1 E10 hybridoma, where the CAR comprises an anti-CD19 scFv, CD8-derived stalk and trans-membrane domain, and a compound endodomain fusion of 4-1 BB- CD3 endodomains.
Obe-cel is generated by ex vivo transduction of activated peripheral blood mononuclear cells using an engineered HIV derived lentiviral vector containing the CD19 CAR expression cassette. The lentiviral vector is produced under Good Manufacturing Practice (GMP) conditions by four plasmid co transfection of HEK293T cells and subsequent harvest and purification of the culture supernatant.
Cells from the leukapheresis starting material are enriched by positive selection for CD4+/CD8+ cells using magnetic beads on the Miltenyi CliniMACS® Prodigy bioreactor. Cells are then washed and stimulated with mitogenic ligands and cytokines. One day after activation, cells are transduced with the lentiviral vector. Posttransduction, cells are expanded (drug substance) to produce the desired dose. The cells are then washed and formulated with a phosphate-buffered saline (PBS)/ ethylenediaminetetraacetic acid (EDTA) I human serum albumin (HSA) I dimethyl sulfoxide (DMSO) buffer and filled into final packaging and cryopreserved (drug product). The obe-cel product comprises transduced and non-transduced T cells. The dose given to patients is expressed as the total number of CD19 CAR-positive T cells (the active substance).
Obe-cel is supplied as a cryopreserved cell suspension in a cryostorage-compatible infusion bag, to be thawed at 37°C.
Obe-cel dose
The selection and justification for the obe-cel dose is based in part on the prior use of obe-cel in patients with B-ALL and other B cell malignancies.
To date obe-cel was administered to over 200 patients with B-cell malignancies. Different dosing strategies were evaluated.
In the pivotal FELIX study in patients with relapsed/refractory CD19+ B-ALL, a planned total target dose of 410 x 106 CAR T cells was administered as a split dose on Day 1 and Day 10 based on the percentage of blast cells in bone marrow prior to the start of lymphodepletion chemotherapy (Roddie et al (2021), supra; Roddie et al (2023a), supra). This dosing regimen was safe and effective in these difficult to treat patients with relapsed/refractory B-ALL. Importantly, in B-ALL patients in complete remission (<5% bone marrow blasts at the start of lymphodepletion chemotherapy), no events of Grade 3 or higher CRS and/or ICANS were observed. Any grade CRS or ICANS were observed in 48% and 7%, respectively.
It is contemplated herein though, that in contrast to patients with relapsed/refractory B- ALL who harbor a much higher number of CD19 positive B lymphoblasts, the overall number of B cells including autoreactive B cells in patients with refractory SLE is limited. Therefore, it is contemplated herein that a much lower dose of obe-cel is needed to deplete and reset the B cell compartment in patients with refractory SLE.
In the first clinical evaluation of CD19 CAR T treatment in a limited number of SLE patients (Mackensen et al, supra), a single dose of 1 x 106 CAR T cells per kg body weight was administered, well tolerated, and demonstrated promising signs of initial efficacy.
Herein a single dose of 50 x 106 (±20%) CD19 CAR-positive viable T cells is to be infused to patients on Day 1 after lymphodepletion chemotherapy. Other doses contemplated are an escalated (100 x 106 [±20%]) or a de-escalated (30 x 106 [±20%]) CAR-positive T cell dose.
Across the currently evaluated dosing strategies, obe-cel demonstrated a tolerable safety profile, and profound CAR T expansion across a wide-variety of patients body weights. Obe-cel has a different mode of action, a propensity for greater AUC, and improved expansion when compared with the FMC63 CAR T construct utilized by Mackensen et al, 2022. Based on the above product characteristics and taking in considerations the effects of CAR T expansion, effects on safety and efficacy due to patient body weight or body surface area variations are expected to be minimal. The starting dose of 50 x 106 (±20%) CD19 CAR-positive viable T cells is 8-fold lower than the total obe-cel target dose utilized in adult B-ALL patients, which have demonstrated a tolerable safety profile in the B-ALL patient population. The mean (standard deviation) body weight in two large, randomized phase 3 belimumab studies in a total of 865 SLE
patients was 61 ±13 kg, the lowest body weight in these studies was 35 kg (US Food & Drug Administration. Arthritis advisory committee meeting briefing document BLA 125370. October 2010) suggesting that indeed a flat dose of 50 x 106 [±20%]) CARpositive T cells is contemplated to be safe and effective.
Example 4 - Exclusion criteria for obe-cel infusion
The exclusion criteria for obe-cel infusion follow.
1. Use of any systemic corticosteroid greater than 10 mg daily of prednisone or its equivalent within 72 hours before obe-cel infusion.
2. Body temperature > 38 °C not related to SLE.
3. Severe intercurrent infection at the time of obe-cel infusion requiring systemic antimicrobials. (If severe intercurrent infection is considered controlled, continuation of systemic antimicrobials to complete a therapeutic course does not constitute a criterion for withholding obe-cel infusion.)
4. SpO2 < 90% in the absence of oxygen support at the time of scheduled obe- cel infusion.
5. Ongoing non-hematological AEs that did not recover to baseline or < Grade 2 due to any cause.
6. Ongoing therapeutic anticoagulation.
7. Uncontrolled cardiac arrhythmia.
Example 5 - Obe-cel infusion and assessments of disease activity and response to treatment
Patients receive a single dose of obe-cel intravenously (IV) after a resting period of 2 days after lymphodepletion (Day -2 and Day -1).
Obe-cel is infused on Day 1. In brief, obe-cel is thawed in a 37°C water bath under sterile conditions. Obe-cel is given as an IV infusion using a syringe or gravity-assisted infusion through a central or peripheral venous line through an 18-gauge blood set over a few minutes (maximum 30 minutes from obe-cel being thawed to preserve cell viability).
Assessments of disease activity and response to treatment include the following.
Table 6 Endpoints




Table 7 Additional Endpoints


Example 6 - A single-arm, open-label, Phase I study to determine the safety, tolerability and preliminary efficacy of obecabtagene autoleucel (obe-cel) in patients with severe, refractory systemic lupus erythematosus (SLE)
Study design/description
Participants are expected to receive a single infusion of obe-cel at a target dose of 50 x 106 (± 25%) CD19 CAR-positive T cells.
The primary endpoint is the proportion of participants achieving a CRR at Month 12 after obe-cel infusion as adjudicated by an Independent Response Review Committee (IRRC).
Following Day 1 (infusion day), at least a 10-day post-infusion safety surveillance is required with special focus on identifying signs and symptoms indicating CAR T cellspecific potential AEs of special interest with contact from the site’s clinical team every day and on-site visits as per the SoA. Once the 10-day post-infusion safety surveillance period is over, the participant is to be followed up with contact from the site’s clinical study team every 1 to 3 days and on-site outpatient visits on Days 15, 22, 25 and 28. In case of fever > 38°C or a change in neurological behavior, the Investigator must be called promptly, and the participant may be re-admitted to hospital.
Based on emerging data, the study may be stopped due to insufficient response, or excessive treatment toxicity.
The study comprises 3 periods:
1. Screening Period: From Day -30 to Day of Enrollment a. Screening: After informed consent is obtained, participants are to undergo screening procedures to confirm they are eligible for the study and are able to undergo leukapheresis. b. Leukapheresis: Eligible participants are to proceed to leukapheresis to collect mononuclear cells to be used for manufacturing obe-cel.
c. Enrollment: A participant is to be enrolled into the study at the point they meet all eligibility criteria and the participant’s leukapheresis material has been accepted for manufacturing.
2. Treatment Period: From Day of Enrollment to Day 1 to End of Treatment (EOT)/Day 1 a. Lymphodepletion (LD) Evaluation: Participants are to undergo an evaluation from Day -8 to Day -6 to confirm eligibility for start of LD. b. LD Chemotherapy: Participants are to receive intravenous (i.v.) fludarabine and cyclophosphamide for LD to enhance treatment efficacy and CD19 CAR T cell survival.
. Fludarabine: 25 mg/m2/day i.v. on Day -5, Day -4, and Day -3
. Cyclophosphamide: 1 ,000 mg/m2 i.v. on Day -3
. Day -2, Day -1 = washout for 48 hours. NOTE: There is no Day 0. After Day -1 , the next step is the infusion, defined as Day 1. c. Administration: If the participant’s eligibility for receiving the infusion is confirmed, the participant is to receive a single infusion on Day 1. NOTE: The EOT is defined as the day of infusion (Day 1) or the day of premature study discontinuation if prior to infusion.
3. Post-treatment Period: From Day 1 to End of Study (EOS)
Efficacy and Safety Follow-up: a. After the infusion, all participants are to be closely monitored for safety and efficacy. The first efficacy assessment is to be performed at Day 28 post-infusion. b. Thereafter, participants are to be followed with monthly or 3-monthly assessments of safety, tolerability, and efficacy until the EOS. c. Following the Month 24 visit, participants are to be followed every 3 months until the last participant, last visit (LPLV) is completed for the study (i.e., when the last enrolled participant reaches Month 24). The last 3-monthly follow-up assessment occurring prior to LPLV is to determine the EOS date for each individual participant. d. The EOS is defined as completion of the LPLV for the study.
Key Inclusion Criteria
• Participants must be 16 to 65 years of age inclusive at the time of signing the informed consent.
• Diagnosis of SLE fulfilling the 2019 European League Against Rheumatism /American College of Rheumatology Classification Criteria for SLE.
• Positive for at least one of the following autoantibodies: ANA at a titer of > 1 :80, or anti-dsDNA (> 30 ILI/mL) or anti-Smith (> upper limit of normal [ULN]), anti-histone or anti-chromatin (> ULN).
• Severe, Active SLE defined as: o ASLEDAI-2K score of > 8 points (of which 4 are non-laboratory items and with the exclusion of points associated with neurological findings [SLEDAI-2K items 1-7]), and
• Severe active LN, defined as: o SLE-related renal organ involvement. A biopsy must be performed in the 6 months prior to the screening visit or during the screening period to confirm ongoing, active, class III, IV or V (V only in combination with class III or IV) active or active/chronic LN in accordance with kidney biopsy evidence of International Society of Nephrology/Renal Pathology Society 2019. o UPGR > 1 mg/mg from a 24-hour urine collection, and eGFR of > 30 mL/min/1.73 m2
• Refractory SLE (defined as lack of response, insufficient response, or lack of sustained response or intolerance related to such drugs not allowing their further use): a. hydroxychloroquine in combination with corticosteroids AND b. to > 2 of the following treatment groups used for at least 6 months: i. Immunosuppressive drugs (e.g., methotrexate, azathioprine, mycophenolate mofetil, mycophenolic acid, leflunomide, or cyclophosphamide). ii. Calcineurin inhibitors (e.g., cyclosporine, voclosporin, tacrolimus). iii. B cell-targeting agents (e.g., belimumab, anti-CD20 mAb) OR cytokine inhibitors (e.g., anifrolumab).
Key Exclusion Criteria
• Recurrent neuropsychiatric lupus at any point prior to screening, or active, severe, or unstable neuropsychiatric lupus within 1 year from screening.
• More than one acute, severe lupus-related flare during screening that needs immediate treatment and/or makes the immunosuppressive washout impossible; thus, making the participant ineligible for CD19 CAR T therapy. (One treatment of flare is allowed and participant must be fully rescreened; such cases should be discussed with the Medical Monitor).
• Significant, likely irreversible organ damage related to SLE (e.g., ESRD) that in the opinion of the Investigator renders CD19 CAR T-cell-based gene therapy unlikely to benefit the participant.
• Diagnosis of clinically significant uveitis
• Participants are not eligible if any of the following laboratory criteria are met. NOTE: If one or more laboratory parameters does not satisfy eligibility, it may be repeated once within the 30-day screening period.
- Neutrophil count < 1,000/pL.
- Platelet count < 50,000/pL. Hemoglobin < 7 g/dL for SLE-related hemolytic anemia or < 8 g/dL for all other participants.
- B cell aplasia.
- Serum alanine aminotransferase/aspartate aminotransferase > 2.5 x ULN.
- Total bilirubin > 1.5 x ULN for participants without Gilbert’s syndrome or direct bilirubin > 1 x ULN in participants with Gilbert’s syndrome.
- International normalized ratio and activated partial thromboplastin clotting time > 1.5 ULN.
The medications that are prohibited and permitted are listed in Table 8.
Table 8: Prohibited/ Permitted Medications


Abbreviations: LD = lymphodepletion; SLE = systemic lupus erythematosus
Table 9: Schedule of assessments.



Example 7 - A single-arm, open-label, Phase I study to determine the safety, tolerability and preliminary efficacy of obecabtagene autoleucel (obe-cel) in patients with severe, refractory systemic lupus erythematosus (SLE): Results of first treated patient
A 32-year-old female SLE patient was treated with obe-cel. This patient was black or African American, with a SLE diagnosis in 2021, showing renal (LN class IVA/), musculoskeletal, dermal, immunologic organ system involvement and a medical history of hypertension. She had ANA, anti-dsDNA, anti-Smith, anti-RBP, anti-chromatin autoantibodies and a SLEDAI-2K score of 24 at screening. Prior SLE treatments included hydroxychloroquine, corticosteroids, mycophenolate, rituximab, and
Obinutuzumab. After tapering the immunosuppressive drugs, this patient received a single dose of 50 x 106 (±20%) CD19 CAR-positive viable T cells on Day 1 after lymphodepletion chemotherapy. Methylprednisolone was used as bridging therapy.
Efficacy
At 2-month follow-up, this patient displayed clear signs of the treatment efficacy with obe-cel:
• anti-dsDNA: decreased from >300 at screening to 40.6;
• proteinuria improved;
• complement was back to normal;
• serum creatinine was 4.6 mg/dL;
• IgG levels decreased from 6.07 at screening to 2.39 at Day 28. IgA decreased from 1.84 to 0.46 and IgM from 0.53 to 0.12, respectively;
• the SLEDAI-2K score decreased from 24 at screening to less than 2 at Month 2 (Figure 1); and
• B cell aplasia.
Safety
Prior to infusion, the patient suffered a Grade 3 serious adverse effect (SAE), i.e. infective abscess of left sternocleidomastoid.
After infusion, no CRS or ICANS were observed.
Post obe-cel infusion, the patient showed the following SAEs:
• Grade 2 CMV reactivation, which was treated with valganciclovir;
• Grade 3 malignant hypertension;
• Grade 3 neutropenia, suspected secondary to valganciclovir. New medications administered were acyclovir and G-CSF.
CAR-T cell expansion
The CD19 CAR-T cells (obe-cel) showed a good expansion with a peak at Day 10 post CAR-T cell infusion by flow cytometry and ddPCR, as shown by Figure 2a and 2b, respectively.
This patent application claims the benefit of United Kingdom application No. 2316183.9 filed 23rd Oct 2023, United Kingdom application No. 2404412.5 filed 27th Mar 2024, United Kingdom application No. 2412689.8 filed 29 Aug 2024, and United Kingdom application No. 2414148.3 filed 26th September 2024. These applications are incorporated herein by reference in their entirety.
All publications mentioned in the above specification are herein incorporated by reference. Various modifications and variations of the described methods and system of the invention will be apparent to those skilled in the art without departing from the scope and spirit of the invention. Although the invention has been described in connection with specific preferred embodiments, it should be understood that the invention as claimed should not be unduly limited to such specific embodiments. Indeed, various modifications of the described modes for carrying out the invention which are obvious to those skilled in molecular biology, CAR technology or related fields are intended to be within the scope of the following claims.
Claims
1. A method of treating an autoimmune disease in a patient comprising administering to the patient a cell comprising a chimeric antigen receptor (CAR), wherein the CAR comprises a CD19-binding domain which comprises: a) a heavy chain variable region (VH) having complementarity determining regions (CDRs) with the following sequences:
CDR1 - GYAFSSS (SEQ ID NO: 1),
CDR2 - YPGDED (SEQ ID NO: 2) and
CDR3 - SLLYGDYLDY (SEQ ID NO: 3), and b) a light chain variable region (VL) having CDRs with the following sequences:
CDR1 - SASSSVSYMH (SEQ ID NO: 4),
CDR2 - DTSKLAS (SEQ ID NO: 5) and
CDR3 - QQWNINPLT (SEQ ID NO: 6).
2. The method of claim 1 wherein the autoimmune disease is systemic lupus erythematosus (SLE).
3. The method of claim 2 wherein the autoimmune disease is severe SLE.
4. The method of claim 2 wherein the autoimmune disease is refractory SLE.
5. The method of any preceding claim wherein the cells are autologous peripheral blood T cells transduced ex vivo to express the CAR.
6. The method of any preceding claim wherein a dose of 50 x 106 (±20%) CD19 CAR-positive viable T cells is administered to the patient.
7. The method of any preceding claim wherein a dose of 100 x 106 (±20%) CD19 CAR-positive viable T cells is administered to the patient.
8. The method of any preceding claim wherein a dose of 30 x 106 (±20%) CD19 CAR-positive viable T cells is administered to the patient.
9. The method of any preceding claim wherein the administration is an intravenous injection.
10. The method of any preceding claim wherein the CD19-binding domain comprises a VH domain having the sequence shown as SEQ ID NO: 7 and/or or a VL domain having the sequence shown as SEQ ID NO: 8, or a variant of either having at least 95% sequence identity.
11. The method of claim 10, wherein the CD19-binding domain comprises an scFv in the orientation VH-VL.
12. The method of claim 11 , wherein the CD19-binding domain comprises the scFv sequence shown as SEQ ID NO: 9 or a variant thereof having at least 90% sequence identity.
13. The method of any preceding claim, wherein the CAR comprises a CD8 stalk spacer.
14. The method of any preceding claim wherein CAR comprising an intracellular T cell signaling domain comprising the 41 BB endodomain and the CD3-Zeta endodomain.
15. A method of treating an autoimmune disease in a patient comprising administering to the patient a dose of 50 x 106 (±20%) CD19 CAR-positive viable T cells.
16. A method of treating an autoimmune disease in a patient comprising administering to the patient a dose of 30 x 106 (±20%) CD19 CAR-positive viable T cells.
17. A method of treating an autoimmune disease in a patient comprising administering to the patient a dose of 100 x 106 (±20%) CD19 CAR-positive viable T cells.
18. The method of claims 15 to 17 wherein the autoimmune disease is SLE.
19. The method of claim 18 wherein the autoimmune disease is severe SLE.
20. The method of claim 18 wherein the autoimmune disease is refractory SLE.
21. The method of claim 18 to 20 wherein the SLE presents with active Lupus Nephritis.
Applications Claiming Priority (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB2316183.9 | 2023-10-23 | ||
| GBGB2316183.9A GB202316183D0 (en) | 2023-10-23 | 2023-10-23 | Products and methods for treating autoimmune diseases |
| GBGB2404412.5A GB202404412D0 (en) | 2024-03-27 | 2024-03-27 | Products and methods for treating autoimmune diseases |
| GB2404412.5 | 2024-03-27 | ||
| GB2412689.8 | 2024-08-29 | ||
| GBGB2412689.8A GB202412689D0 (en) | 2024-08-29 | 2024-08-29 | Products and methods for treating autoimmine disease |
| GBGB2414148.3A GB202414148D0 (en) | 2024-09-26 | 2024-09-26 | Products and method for treating autoimmune diseases |
| GB2414148.3 | 2024-09-26 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2025088308A1 true WO2025088308A1 (en) | 2025-05-01 |
Family
ID=93335206
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/GB2024/052698 Pending WO2025088308A1 (en) | 2023-10-23 | 2024-10-22 | Products and methods for treating autoimmune diseases |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2025088308A1 (en) |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7052906B1 (en) | 1999-04-16 | 2006-05-30 | Celltech R & D Limited | Synthetic transmembrane components |
| WO2016102965A1 (en) | 2014-12-24 | 2016-06-30 | Ucl Business Plc | Cell |
| WO2016139487A1 (en) | 2015-03-05 | 2016-09-09 | Ucl Business Plc | Chimeric antigen receptor (car) comprising a cd19-binding domain |
| US20230210900A1 (en) * | 2022-01-04 | 2023-07-06 | Kyverna Therapeutics, Inc. | Methods for treating autoimmune diseases |
| US20240285762A1 (en) * | 2023-02-28 | 2024-08-29 | Juno Therapeutics, Inc. | Cell therapy for treating systemic autoimmune diseases |
| WO2024211554A1 (en) * | 2023-04-07 | 2024-10-10 | Nkarta, Inc. | Methods for treatment of autoimmune diseases |
-
2024
- 2024-10-22 WO PCT/GB2024/052698 patent/WO2025088308A1/en active Pending
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7052906B1 (en) | 1999-04-16 | 2006-05-30 | Celltech R & D Limited | Synthetic transmembrane components |
| WO2016102965A1 (en) | 2014-12-24 | 2016-06-30 | Ucl Business Plc | Cell |
| WO2016139487A1 (en) | 2015-03-05 | 2016-09-09 | Ucl Business Plc | Chimeric antigen receptor (car) comprising a cd19-binding domain |
| US20180044417A1 (en) | 2015-03-05 | 2018-02-15 | Ucl Business Plc | Chimeric Antigen Receptor (CAR) Comprising a CD19-Binding Domain |
| US20230220077A1 (en) * | 2015-03-05 | 2023-07-13 | Autolus Limited | Chimeric Antigen Receptor (CAR) Comprising A CD19-Binding Domain |
| US20230210900A1 (en) * | 2022-01-04 | 2023-07-06 | Kyverna Therapeutics, Inc. | Methods for treating autoimmune diseases |
| US20240285762A1 (en) * | 2023-02-28 | 2024-08-29 | Juno Therapeutics, Inc. | Cell therapy for treating systemic autoimmune diseases |
| WO2024211554A1 (en) * | 2023-04-07 | 2024-10-10 | Nkarta, Inc. | Methods for treatment of autoimmune diseases |
Non-Patent Citations (26)
| Title |
|---|
| CAPPELL KMKOCHENDERFER JN: "Long-term outcomes following CAR T cell therapy: what we know so fa", NAT REVIEW, vol. 20, 2023, pages 359 - 371 |
| CORTÉS HERNÁNDEZ JOSEFINA ET AL: "An Open-label, Multicenter, Phase 1/2 Study to Assess Safety, E cacy and Cellular Kinetics of YTB323, a Rapid Manufacturing CAR-T Cell Therapy Targeting CD19 on B Cells, for Severe Refractory Systemic Lupus Erythematosus: Preliminary Results", ACR CONVERGENCE 2023, 24 October 2023 (2023-10-24), XP093239863, Retrieved from the Internet <URL:https://acrabstracts.org/abstract/an-open-label-multicenter-phase-1-2-study-to-assess-safety-efficacy-and-cellular-kinetics-of-ytb323-a-rapid-manufacturing-car-t-cell-therapy-targeting-cd19-on-b-cells-for-severe-refractory-system/> * |
| DAVILA ET AL., SCI. TRANSL. MED., vol. 6, 2014, pages 224 - 25 |
| DAY ET AL., NATURE, vol. 443, 2006, pages 350 - 354 |
| GHORASHIAN SKRAMER AMONUOHA SWRIGHT G ET AL.: "Enhanced CAR T cell expansion and prolonged persistence in pediatric patients with ALL treated with a low-affinity CD19 CAR", NAT MED., vol. 25, no. 9, 2019, pages 1408 - 14, XP036881211, DOI: 10.1038/s41591-019-0549-5 |
| JIN XXU QPU CZHU K ET AL.: "Therapeutic efficacy of anti-CD19 CAR-T cells in a mouse model of systemic lupus erythematosus", CELL MOL IMMUNOL, vol. 18, no. 8, 2021, pages 1896 - 1903, XP037522670, DOI: 10.1038/s41423-020-0472-1 |
| KANSAL R, RICHARDSON N NEELI I: "Sustained B cell depletion by CD19-targeted CAR T cells is a highly effective treatment for murine lupus", SCI TRANSL MED, vol. 11, no. 482, 2019, pages 1648 - 1648, XP093037267, DOI: 10.1126/scitranslmed.aav1648 |
| KAUL AGORDON CCROW M ET AL.: "Systemic lupus erythematosus.", NATURE REVIEWS, vol. 2, 2016, pages 1 - 21, XP037922555, DOI: 10.1038/nrdp.2016.39 |
| KOCHENDERFER ET AL., J. CLIN. ONCOL. OFF. J. AM. SOC. CLIN. ONCOL., 2014 |
| KOCHENDERFER JNYU ZFRASHERI D ET AL.: "Adoptive transfer of syngeneic T cells transduced with a chimeric antigen receptor that recognizes murine CD19 can eradicate lymphoma and normal B cells", BLOOD, vol. 116, no. 19, 2010, pages 3875 - 3886, XP055204922, DOI: 10.1182/blood-2010-01-265041 |
| LEE ET AL., LANCET, 2014 |
| LOPEZ-ALVAREZ ET AL., CLIN. VACCINE IMMUNOL. CVI, vol. 16, 2009, pages 142 - 145 |
| MACKENSEN AMULLER FMOUGLAKAKOS D ET AL.: "Anti-CD19 CAR T cell therapy for refractory lupus erythematosus", NATURE MEDICINE, vol. 28, 2022, pages 2124 - 2132, XP093037276, DOI: 10.1038/s41591-022-02017-5 |
| MACKENSEN ANDREAS ET AL: "Anti-CD19 CAR T cell therapy for refractory systemic lupus erythematosus", NATURE MEDICINE, vol. 28, no. 10, 15 September 2022 (2022-09-15), New York, pages 2124 - 2132, XP093037276, ISSN: 1078-8956, Retrieved from the Internet <URL:https://www.nature.com/articles/s41591-022-02017-5> DOI: 10.1038/s41591-022-02017-5 * |
| MERRILL JNEUWELT CMWALLACE DJSHANAHAN JC ET AL.: "Efficacy and safety of rituximab in moderately-to-severely active systemic lupus erythematosus: the randomized, double-blind, phase II/III systemic lupus erythematosus evaluation of rituximab trial", ARTHRITIS RHEUM, vol. 62, no. 1, 2010, pages 222 - 33 |
| MOUGIAKAKOS D, KRONKE G, VÖLKL S, KRETSCHMANN S: "CD-19-targeted CAR T cells in refractory systemic lupus erythematosus", N ENGL J MED, vol. 385, no. 6, 2021, pages 567 - 9, XP093037292, DOI: 10.1056/NEJMc2107725 |
| MOUGIAKAKOS DIMITRIOS ET AL: "CD19-Targeted CAR T Cells in Refractory Systemic Lupus Erythematosus", NEW ENGL. J. MED., vol. 385, no. 6, 5 August 2021 (2021-08-05), pages 567 - 569, XP093037292, DOI: 10.1056/NEJMc2107725 * |
| MULLER FBOELTZ SKNITZA JAIGNER M ET AL.: "CD19-targeted CAR T cells in refractory antisynthetase syndrome", LANCET, vol. 401, no. 10379, 2023, pages 815 - 8, XP093219245 |
| PERCHER ACHENSEN LKLEIN R ET AL.: "CD19-targeting CAR T cells for myositis and interstitial lung disease associated with antisynthetase syndrome", JAMA, vol. 329, no. 24, 2023, pages 2154 - 62 |
| RODDIE C, SANDHU KS, THOLOULI E, SHAUGHNESSY P: "Safety and effiacy of obecabtagene autoleucel (obe-cel, AUTO1), a fast-off rate CD19 CAR, in relapsed/refractory adult B-cell acute lymphoblastic leukemia (r/r B-ALL): top line result of the pivotal FELIX study", AMERICAN SOCIETY OF CLINICAL ONCOLOGY ANNUAL MEETING ORAL ABSTRACT #, vol. 7000, 2023 |
| RODDIE CDIAS JO'REIILLY MA ET AL.: "Durable Responses and Low Toxicity After Fast Off-Rate CD19 Chimeric Antigen Receptor-T Therapy in Adults With Relapsed or Refractory B-Cell Acute Lymphoblastic Leukemia", J CLIN ONCOL;, vol. 39, no. 30, 2021, pages 3352 - 63 |
| RODDIE CDIAS JO'REILLY MMITSIKAKOU M ET AL.: "Long-term follow-up of AUTO1, a fast-off rate CD 19 CAR, in relapsed/refractory B-cell acute lymphoblastic leukemia and factors associated with durable response", AMERICAN SOCIETY FOR TRANSPLANTATION AND CELLULAR THERAPY ANNUAL MEETING POSTER, vol. 277, 2023 |
| RODDIE CDIAS JO'REILLY MMITSIKAKOU M ET AL.: "Safety, efficacy and long-term follow-up of AUTO1, a fost-off rate CD19 CAR in relapsed/refractory B-cell acute lymphoblastic leukaemia and other B-cell malignancies", AMERICAN SOCIETY OF HEMATOLOGY ANNUAL MEETING POSTER, vol. 3318, 2022 |
| ROVIN ET AL.: "Efficacy and safety of rituximab in patients with active proliferative lupus nephritis: the Lupus Nephritis Assessment with Rituximab study", ARTHRITIS RHEUM, vol. 64, no. 4, 2012, pages 1215 - 26, XP055287443, DOI: 10.1002/art.34359 |
| TSOKOS GC.: "Systemic lupus erythematosus.", N ENGL J MED, vol. 365, no. 22, 2011, pages 2110 - 21 |
| WESTIN JDAVIS REFENG LHAGEMEISTER F ET AL.: "Smart start: rituximab, lenalidomide, and ibrutinib in patients with newly diagnosed large B-cell lymphoma", J CLIN ONCOL, vol. 41, no. 4, 2023, pages 745 - 55 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7379561B2 (en) | Chimeric antigens and T cell receptors and methods of use | |
| JP7673272B2 (en) | Chimeric receptors and methods of use thereof | |
| US20230013642A1 (en) | Toxicity Management for Anti-Tumor Activity of CARs | |
| US20250011387A1 (en) | Articles of manufacture and methods for treatment using adoptive cell therapy | |
| ES3003809T3 (en) | Chimeric antigen receptors specific for b-cell maturation antigen and encoding polynucleotides | |
| JP6574848B2 (en) | Chimeric antigen receptor (CAR) comprising a CD19 binding domain | |
| TW202323273A (en) | Bcma binding molecules and uses thereof | |
| AU2019314452B2 (en) | Chimeric antigen receptor therapy T cell expansion kinetics and uses thereof | |
| US20220023344A1 (en) | Allogeneic cell therapy of acute lymphoblastic leukemia using genetically engineered t cells targeting cd19 | |
| KR20240117530A (en) | CAR-T cell therapy targeting BCMA for multiple myeloma | |
| IL324284A (en) | Anti-CD19 CAR T cells for the treatment of B-cell malignancies and autoimmune diseases | |
| CN117693528A (en) | anti-CLL 1 antibodies and constructs thereof | |
| US20240000843A1 (en) | Compositions and methods for inhibiting or screening for cd8 and methods and assays for detecting cd8 in cells | |
| WO2025088308A1 (en) | Products and methods for treating autoimmune diseases | |
| JP2025516531A (en) | CD19/22 CAR T cell treatment of high-risk or relapsed pediatric acute lymphoblastic leukemia | |
| WO2025215360A1 (en) | Method | |
| HK40126667A (en) | Chimeric antigen and t cell receptors and methods of use | |
| WO2025250591A1 (en) | Abc and gate car-t cell therapy for autoimmune disease | |
| CN121378503A (en) | Chimeric antigen receptor targeting CD19 and/or CD20 and application thereof | |
| HK40069741A (en) | Toxicity management for anti-tumor activity of cars | |
| HK40039113A (en) | Toxicity management for anti-tumor activity of cars | |
| HK1253365B (en) | Toxicity management for anti-tumor activity of cars |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 24799649 Country of ref document: EP Kind code of ref document: A1 |