WO2025087763A1 - Pyridine and pyrimidine derivatives as herbicides - Google Patents
Pyridine and pyrimidine derivatives as herbicides Download PDFInfo
- Publication number
- WO2025087763A1 WO2025087763A1 PCT/EP2024/079166 EP2024079166W WO2025087763A1 WO 2025087763 A1 WO2025087763 A1 WO 2025087763A1 EP 2024079166 W EP2024079166 W EP 2024079166W WO 2025087763 A1 WO2025087763 A1 WO 2025087763A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- mmol
- compounds
- methyl
- chloropyrimidin
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/28—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
- C07D239/32—One oxygen, sulfur or nitrogen atom
- C07D239/34—One oxygen atom
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01N—PRESERVATION OF BODIES OF HUMANS OR ANIMALS OR PLANTS OR PARTS THEREOF; BIOCIDES, e.g. AS DISINFECTANTS, AS PESTICIDES OR AS HERBICIDES; PEST REPELLANTS OR ATTRACTANTS; PLANT GROWTH REGULATORS
- A01N43/00—Biocides, pest repellants or attractants, or plant growth regulators containing heterocyclic compounds
- A01N43/34—Biocides, pest repellants or attractants, or plant growth regulators containing heterocyclic compounds having rings with one nitrogen atom as the only ring hetero atom
- A01N43/40—Biocides, pest repellants or attractants, or plant growth regulators containing heterocyclic compounds having rings with one nitrogen atom as the only ring hetero atom six-membered rings
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01N—PRESERVATION OF BODIES OF HUMANS OR ANIMALS OR PLANTS OR PARTS THEREOF; BIOCIDES, e.g. AS DISINFECTANTS, AS PESTICIDES OR AS HERBICIDES; PEST REPELLANTS OR ATTRACTANTS; PLANT GROWTH REGULATORS
- A01N43/00—Biocides, pest repellants or attractants, or plant growth regulators containing heterocyclic compounds
- A01N43/48—Biocides, pest repellants or attractants, or plant growth regulators containing heterocyclic compounds having rings with two nitrogen atoms as the only ring hetero atoms
- A01N43/54—1,3-Diazines; Hydrogenated 1,3-diazines
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01P—BIOCIDAL, PEST REPELLANT, PEST ATTRACTANT OR PLANT GROWTH REGULATORY ACTIVITY OF CHEMICAL COMPOUNDS OR PREPARATIONS
- A01P13/00—Herbicides; Algicides
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
Definitions
- the present invention relates to novel herbicidal compounds, to processes for their preparation, to herbicidal compositions which comprise the novel compounds, and to their use for controlling weeds, in particular in crops of useful plants, or for inhibiting plant growth.
- WO2010/064688 disclosed the use of pyrimidine derivatives as herbicides.
- WO2016/149315 discloses substituted pyrimidinyloxy pyridine derivatives as herbicides.
- W02020/002089 discloses substituted 2-heteroaryloxypyridines.
- W02021/204706 discloses herbicidal 5-haloalkoxy-pyrimidine compounds.
- GB2594931 teaches phenoxypyridine compounds.
- W02022/002838 discloses substituted heteroaryloxypyridines.
- WO2023/186691 discloses substituted 2-C- azines.
- the present invention relates to novel herbicidal compounds.
- X is O or CHR 8 ,
- Q is phenyl or a C-linked 6-membered heteroaryl wherein said phenyl or 6- membered heteroaryl is optionally substituted by one or more R 4 ;
- a 1 is CH or N
- a 2 is OR 2 or N, wherein at least one of A 1 and A 2 are N,
- a 3 is CR 5 or N; wherein if X is O then A 1 is not N when A 2 is CR 2 and A 3 is N;
- R 1 is selected from the group consisting of hydrogen, halogen, -CN, nitro, Ci- C4alkyl-, C2-C4alkenyl-, C2-C4alkynyl-, Ci-C4haloalkyl-, C1-C4 alkoxy-, C1- C4haloalkoxy-, -S(O) p Ci-C4alkyl and Cs-Cecycloalkyl-;
- R 2 is selected from the group consisting of hydrogen, halogen and Ci-Ce alkyl
- R 3 is selected from the group consisting of halogen, Ci-C4haloalkyl- and C1-C2 haloalkoxy-;
- R 4 is selected from the group consisting of halogen, C1-C4 alkyl, Ci- 04 haloalkyl, C1-C4 alkoxy, C1-C4 haloalkoxy, Ci-C4alkoxyCi-C3alkyl-, Ci- C4alkoxyCi-C3alkoxy-, Ci-C4alkoxyCi-C3alkoxyCi-C3alkyl-, -CN, NO2, C2- C4alkenyl, C2-C4alkynyl, -S(O) p Ci-C4alkyl, -S(O) p Ci-C4haloalkyl, -C(O)OCi- C 4 alkyl and -C(O)NR 6 R 7 ;
- R 5 is hydrogen or halogen
- R 6 is selected from the group consisting of hydrogen, C3-C4cycloalkyl, C1- C4alkyl and Ci-C4haloalkyl;
- R 7 is selected from the group consisting of hydrogen, C3-C4cycloalkyl, C1- C4alkyl and Ci-C4haloalkyl;
- R 8 is hydrogen or hydroxyl; and p is 0, 1 or 2; or an agriculturally acceptable salt thereof.
- Alkyl groups e.g Ci-Cealkyl
- Alkyl groups include, for example, methyl (Me, CH3), ethyl (Et, C2H5), n-propyl (n-Pr), isopropyl (/-Pr), n-butyl (n-Bu), isobutyl (/-Bu), sec-butyl (s-Bu) and tert- butyl (t-Bu).
- Alkenyl and alkynyl moieties can be in the form of straight or branched chains, and the alkenyl moieties, where appropriate, can be of either the (E)- or ( ⁇ -configuration. Examples are vinyl, allyl and propargyl. Alkenyl and alkynyl moieties can contain one or more double and/or triple bonds in any combination.
- Halogen encompasses fluorine, chlorine, bromine or iodine. The same correspondingly applies to halogen in the context of other definitions, such as haloalkyl.
- Haloalkyl groups are, for example, fluoromethyl, difluoromethyl, trifluoromethyl, chloromethyl, dichloromethyl, trichloromethyl, 2,2,2- trifluoroethyl, 2-fluoroethyl, 2-chloroethyl, pentafluoroethyl, 1 , 1 -difluoro-2,2,2- trichloroethyl, 2,2,3,3-tetrafluoroethyl and 2,2,2-trichloroethyl, heptafluoro-n-propyl and perfluoro-n-hexyl.
- Alkoxy groups are, for example, methoxy, ethoxy, propoxy, isopropoxy, n-butoxy, isobutoxy, sec-butoxy or tert-butoxy, preferably methoxy and ethoxy.
- Alkoxyalkyl groups (e.g Ci-C4alkoxy-Ci-C3alkyl-) includes, for example, methoxymethyl, methoxyethyl, ethoxymethyl, ethoxyethyl, n-propoxymethyl, n- propoxyethyl, isopropoxymethyl or isopropoxyethyl.
- Cycloalkyl groups include, for example cyclopropyl (c- propyl, c-Pr), cyclobutyl (c-butyl, c-Bu), cyclopentyl (c-pentyl) and cyclohexyl (c-hexyl) and may be substituted or unsubstituted as indicated.
- Ci-Cealkyl-S- (alkylthio) is, for example, methylthio, ethylthio, propylthio, isopropylthio, n-butylthio, isobutylthio, sec-butylthio or tert-butylthio, preferably methylthio or ethylthio.
- Ci-Cealkyl-S(O)- (alkylsulfinyl) is, for example, methylsulfinyl, ethylsulfinyl, propylsulfinyl, isopropylsulfinyl, n-butylsulfinyl, isobutylsulfinyl, sec-butylsulfinyl or tert-butylsulfinyl, preferably methylsulfinyl or ethylsulfinyl.
- Ci-Cealkyl-S(O)2- (alkylsulfonyl) is, for example, methylsulfonyl, ethylsulfonyl, propylsulfonyl, isopropylsulfonyl, n-butylsulfonyl, isobutylsulfonyl, sec-butylsulfonyl or tert-butylsulfonyl, preferably methylsulfonyl or ethylsulfonyl.
- X is CH2.
- R 2 is, for example, H or halo
- a compound of Formula (I) wherein A 1 is CH and A 2 is N there is provided a compound of Formula (I) wherein A 1 is N and A 2 is N.
- a 3 is N.
- a 3 is CR 5 .
- R 5 is hydrogen or halogen, for example chloro or fluoro, most preferably fluoro.
- a 1 is CH, A 2 is N and A 3 is N.
- a 1 is N, A 2 is N and A 3 is N.
- a compound of Formula (I) wherein A 1 is N, A 2 is CH and A 3 is CR 5 , preferably CH or CF. In another embodiment of the present invention, A 1 is CH, A 2 is N and A 3 is CR 5 , preferably CH or CF. In another embodiment of the present invention, A 1 is N, A 2 is N and A 3 is CR 5 , preferably CH or CF.
- R 1 is selected from the group consisting of hydrogen, halogen, Ci-C4alkyl- and Ci-C4haloalkyl-.
- R 1 is hydrogen, methyl or -CF3.
- R 3 is halogen (e.g bromo or chloro, most preferably chloro), Ci- C4haloalkyl- (e.g -CF3) or Ci-C4haloalkoxy- (e.g -OCF3).ln a more preferred embodiment R 3 is halogen (e.g bromo or chloro, most preferably chloro).
- a compound of Formula (I) wherein Q is selected from the group consisting of Q-1, Q-3 and Q-4 which are optionally substituted by one or two R 4 .
- Q is Q-1 or Q-4 which are optionally substituted by one or two R 4 .
- Q is Q-1 which is optionally substituted by one or two R 4 .
- Q is Q-1, R 3 is halogen or C1-C4 haloalkyl and n is 1 (wherein in a preferred embodiment Q is 4-CF3-phenyl-).
- Q is Q-1, R 3 is halogen and n is 2 (wherein in a preferred embodiment Q is 3,4-dihalophenyl- (e.g 3-F,4-CI-phenyl-, 3- CI,4-F-phenyl-, 3,4-diclorophenyl- or 3,4diflurophenyl-).
- Q is 3,4-dihalophenyl- (e.g 3-F,4-CI-phenyl-, 3- CI,4-F-phenyl-, 3,4-diclorophenyl- or 3,4diflurophenyl-).
- Q is Q-3 which is optionally substituted by one or two R 4 .
- Q is Q-4 which is optionally substituted by one or two R 4 .
- Q is Q-3, R 3 is halogen and n is 2 (wherein in a preferred embodiment Q is 6,5-dihalo-3-pyridyl- (e.g 6-chloro-5-fluoro-3-pyridyl or 6-fluoro-5- chloro-3-pyridyl).
- n is 0. In another embodiment, n is 1. In another embodiment, n is 2. In embodiments of the invention wherein n is 1 or 2, each R 4 is preferably selected from the group consisting of halogen (e.g fluoro or chloro) and C1-C4 haloalkyl (e.g -CF3).
- halogen e.g fluoro or chloro
- C1-C4 haloalkyl e.g -CF3
- the compounds of Formula (I) according to the invention can be used as herbicides by themselves, but they are generally formulated into herbicidal compositions using formulation adjuvants, such as carriers, solvents and surfaceactive agents (SAA).
- formulation adjuvants such as carriers, solvents and surfaceactive agents (SAA).
- the present invention further provides a herbicidal composition comprising a herbicidal compound according to the present invention and an agriculturally acceptable formulation adjuvant.
- the composition can be in the form of concentrates which are diluted prior to use, although ready-to-use compositions can also be made. The final dilution is usually made with water, but can be made instead of, or in addition to, water, with, for example, liquid fertilisers, micronutrients, biological organisms, oil or solvents.
- the compounds of the present invention may contain an asymmetric centre.
- the compound of the present invention may be present in the composition as a racemic mixture of the two enantiomers.
- the compound of the present invention may be present in an enantiomer enriched form.
- the herbicidal compositions generally comprise from 0.1 to 99 % by weight, especially from 0.1 to 95 % by weight, compounds of Formula I and from 1 to 99.9 % by weight of a formulation adjuvant which preferably includes from 0 to 25 % by weight of a surface-active substance.
- compositions can be chosen from a number of formulation types. These include an emulsion concentrate (EC), a suspension concentrate (SC), a suspo- emulsion (SE), a capsule suspension (CS), a water dispersible granule (WG), an emulsifiable granule (EG), an emulsion, water in oil (EG), an emulsion, oil in water (EW), a micro-emulsion (ME), an oil dispersion (OD), an oil miscible flowable (OF), an oil miscible liquid (OL), a soluble concentrate (SL), an ultra-low volume suspension (Sil), an ultra-low volume liquid (UL), a technical concentrate (TK), a dispersible concentrate (DC), a soluble powder (SP), a wettable powder (WP) and a soluble granule (SG).
- formulation type chosen in any instance will depend upon the particular purpose envisaged and the physical, chemical and biological properties of the compound of Formula (I).
- Soluble powders may be prepared by mixing a compound of Formula (I) with one or more water-soluble inorganic salts (such as sodium bicarbonate, sodium carbonate or magnesium sulphate) or one or more water-soluble organic solids (such as a polysaccharide) and, optionally, one or more wetting agents, one or more dispersing agents or a mixture of said agents to improve water dispersibility/solubility. The mixture is then ground to a fine powder. Similar compositions may also be granulated to form water soluble granules (SG).
- water-soluble inorganic salts such as sodium bicarbonate, sodium carbonate or magnesium sulphate
- water-soluble organic solids such as a polysaccharide
- WP Wettable powders
- WG Water dispersible granules
- Granules may be formed either by granulating a mixture of a compound of Formula (I) and one or more powdered solid diluents or carriers, or from preformed blank granules by absorbing a compound of Formula (I) (or a solution thereof, in a suitable agent) in a porous granular material (such as pumice, attapulgite clays, fuller's earth, kieselguhr, diatomaceous earths or ground corn cobs) or by adsorbing a compound of Formula (I) (or a solution thereof, in a suitable agent) on to a hard core material (such as sands, silicates, mineral carbonates, sulphates or phosphates) and drying if necessary.
- a hard core material such as sands, silicates, mineral carbonates, sulphates or phosphates
- Agents which are commonly used to aid absorption or adsorption include solvents (such as aliphatic and aromatic petroleum solvents, alcohols, ethers, ketones and esters) and sticking agents (such as polyvinyl acetates, polyvinyl alcohols, dextrins, sugars and vegetable oils).
- solvents such as aliphatic and aromatic petroleum solvents, alcohols, ethers, ketones and esters
- sticking agents such as polyvinyl acetates, polyvinyl alcohols, dextrins, sugars and vegetable oils.
- One or more other additives may also be included in granules (for example an emulsifying agent, wetting agent or dispersing agent).
- DC Dispersible Concentrates
- a compound of Formula (I) may be prepared by dissolving a compound of Formula (I) in water or an organic solvent, such as a ketone, alcohol or glycol ether.
- organic solvent such as a ketone, alcohol or glycol ether.
- surface-active agent for example to improve water dilution or prevent crystallisation in a spray tank.
- Emulsifiable concentrates or oil-in-water emulsions (EW) may be prepared by dissolving a compound of Formula (I) in an organic solvent (optionally containing one or more wetting agents, one or more emulsifying agents or a mixture of said agents).
- Suitable organic solvents for use in ECs include aromatic hydrocarbons (such as alkylbenzenes or alkylnaphthalenes, exemplified by SOLVESSO 100, SOLVESSO 150 and SOLVESSO 200; SOLVESSO is a Registered Trade Mark), ketones (such as cyclohexanone or methylcyclohexanone) and alcohols (such as benzyl alcohol, furfuryl alcohol or butanol), N-alkylpyrrolidones (such as N-methylpyrrolidone or N-octylpyrrolidone), dimethyl amides of fatty acids (such as Cs-C fatty acid dimethylamide) and chlorinated hydrocarbons.
- An EC product may spontaneously emulsify on addition to water, to produce an emulsion with sufficient stability to allow spray application through appropriate equipment.
- Preparation of an EW involves obtaining a compound of Formula (I) either as a liquid (if it is not a liquid at room temperature, it may be melted at a reasonable temperature, typically below 70°C) or in solution (by dissolving it in an appropriate solvent) and then emulsifying the resultant liquid or solution into water containing one or more SAAs, under high shear, to produce an emulsion.
- Suitable solvents for use in EWs include vegetable oils, chlorinated hydrocarbons (such as chlorobenzenes), aromatic solvents (such as alkylbenzenes or alkylnaphthalenes) and other appropriate organic solvents which have a low solubility in water.
- Microemulsions may be prepared by mixing water with a blend of one or more solvents with one or more SAAs, to produce spontaneously a thermodynamically stable isotropic liquid formulation.
- a compound of Formula (I) is present initially in either the water or the solvent/SAA blend.
- Suitable solvents for use in MEs include those hereinbefore described for use in in ECs or in EWs.
- An ME may be either an oil-in-water or a water-in-oil system (which system is present may be determined by conductivity measurements) and may be suitable for mixing water- soluble and oil-soluble pesticides in the same formulation.
- An ME is suitable for dilution into water, either remaining as a microemulsion or forming a conventional oil- in-water emulsion.
- SC Suspension concentrates
- SCs may comprise aqueous or non-aqueous suspensions of finely divided insoluble solid particles of a compound of Formula (I).
- SCs may be prepared by ball or bead milling the solid compound of Formula (I) in a suitable medium, optionally with one or more dispersing agents, to produce a fine particle suspension of the compound.
- One or more wetting agents may be included in the composition and a suspending agent may be included to reduce the rate at which the particles settle.
- a compound of Formula (I) may be dry milled and added to water, containing agents hereinbefore described, to produce the desired end product.
- Aerosol formulations comprise a compound of Formula (I) and a suitable propellant (for example n-butane).
- a compound of Formula (I) may also be dissolved or dispersed in a suitable medium (for example water or a water miscible liquid, such as n-propanol) to provide compositions for use in non-pressurised, hand-actuated spray pumps.
- Capsule suspensions may be prepared in a manner similar to the preparation of EW formulations but with an additional polymerisation stage such that an aqueous dispersion of oil droplets is obtained, in which each oil droplet is encapsulated by a polymeric shell and contains a compound of Formula (I) and, optionally, a carrier or diluent therefor.
- the polymeric shell may be produced by either an interfacial polycondensation reaction or by a coacervation procedure.
- the compositions may provide for controlled release of the compound of Formula (I) and they may be used for seed treatment.
- a compound of Formula (I) may also be formulated in a biodegradable polymeric matrix to provide a slow, controlled release of the compound.
- the composition may include one or more additives to improve the biological performance of the composition, for example by improving wetting, retention or distribution on surfaces; resistance to rain on treated surfaces; or uptake or mobility of a compound of Formula (I).
- additives include surface active agents (SAAs), spray additives based on oils, for example certain mineral oils or natural plant oils (such as soy bean and rape seed oil), modified plant oils such as methylated rape seed oil (MRSO), and blends of these with other bio-enhancing adjuvants (ingredients which may aid or modify the action of a compound of Formula (I).
- SAAs surface active agents
- spray additives based on oils for example certain mineral oils or natural plant oils (such as soy bean and rape seed oil), modified plant oils such as methylated rape seed oil (MRSO), and blends of these with other bio-enhancing adjuvants (ingredients which may aid or modify the action of a compound of Formula (I).
- SAAs of the cationic, anionic, ampho
- Suitable SAAs of the cationic type include quaternary ammonium compounds (for example cetyltri methyl ammonium bromide), imidazolines and amine salts.
- Suitable anionic SAAs include alkali metals salts of fatty acids, salts of aliphatic monoesters of sulphuric acid (for example sodium lauryl sulphate), salts of sulphonated aromatic compounds (for example sodium dodecylbenzenesulphonate, calcium dodecylbenzenesulphonate, butylnaphthalene sulphonate and mixtures of sodium di-/sopropyl- and tri-/sopropyl-naphthalene sulphonates), ether sulphates, alcohol ether sulphates (for example sodium laureth-3-sulphate), ether carboxylates (for example sodium laureth-3-carboxylate), phosphate esters (products from the reaction between one or more fatty alcohols and phosphoric acid (predominately mono-esters) or phosphorus pentoxide (predominately di-esters), for example the reaction between lauryl alcohol and tetraphosphoric acid
- Suitable SAAs of the amphoteric type include betaines, propionates and glycinates.
- Suitable SAAs of the non-ionic type include condensation products of alkylene oxides, such as ethylene oxide, propylene oxide, butylene oxide or mixtures thereof, with fatty alcohols (such as oleyl alcohol or cetyl alcohol) or with alkylphenols (such as octylphenol, nonylphenol or octylcresol); partial esters derived from long chain fatty acids or hexitol anhydrides; condensation products of said partial esters with ethylene oxide; block polymers (comprising ethylene oxide and propylene oxide); alkanolamides; simple esters (for example fatty acid polyethylene glycol esters); amine oxides (for example lauryl dimethyl amine oxide); lecithins and sorbitans and esters thereof, alkyl polyglycosides and tristyrylphenols.
- alkylene oxides such as ethylene oxide, propylene oxide, butylene oxide or mixtures thereof
- fatty alcohols such as oleyl
- Suitable suspending agents include hydrophilic colloids (such as polysaccharides, polyvinylpyrrolidone or sodium carboxymethylcellulose) and swelling clays (such as bentonite or attapulgite).
- hydrophilic colloids such as polysaccharides, polyvinylpyrrolidone or sodium carboxymethylcellulose
- swelling clays such as bentonite or attapulgite
- the compounds of present invention can also be used in mixture with one or more additional herbicides and/or plant growth regulators.
- additional herbicides or plant growth regulators include acetochlor, acifluorfen (including acifluorfen-sodium), aclonifen, ametryn, amicarbazone, aminopyralid, aminotriazole, atrazine, beflubutamid-M, benquitrione, bensulfuron (including bensulfuron-methyl), bentazone, bicyclopyrone, bilanafos, bipyrazone, bispyribac- sodium, bixlozone, bromacil, bromoxynil, butachlor, butafenacil, carfentrazone (including carfentrazone-ethyl), cloransulam (including cloransulam-methyl), chlorimuron (including chlorimuron-ethyl), chlorotoluron, chlorsulfuron, cinmethylin, cl
- 5-carboxylic acid ethyl ester 4-hydroxy-1-methoxy-5-methyl-3-[4-(trifluoromethyl)-2- pyridyl]imidazolidin-2-one, 4-hydroxy-1,5-dimethyl-3-[4-(trifluoromethyl)-2- pyridyl]imidazolidin-2-one, 5-ethoxy-4-hydroxy-1-methyl-3-[4-(trifluoromethyl)-2- pyridyl]imidazolidin-2-one, 4-hydroxy-1-methyl-3-[4-(trifluoromethyl)-2- pyridyl]imidazolidin-2-one, 4-hydroxy-1,5-dimethyl-3-[1-methyl-5-
- the mixing partners of the compound of Formula (I) may also be in the form of esters or salts, as mentioned e.g. in The Pesticide Manual, Sixteenth Edition, British Crop Protection Council, 2012.
- the compound of Formula (I) can also be used in mixtures with other agrochemicals such as fungicides, nematicides or insecticides, examples of which are given in The Pesticide Manual.
- the mixing ratio of the compound of Formula (I) to the mixing partner is preferably from 1 : 100 to 1000:1.
- mixtures can advantageously be used in the above-mentioned formulations (in which case "active ingredient” relates to the respective mixture of compound of Formula (I) with the mixing partner).
- the compounds or mixtures of the present invention can also be used in combination with one or more herbicide safeners.
- herbicide safeners include benoxacor, cloquintocet (including cloquintocet-mexyl), cyprosulfamide, dichlormid, fenchlorazole (including fenchlorazole-ethyl), fenclorim, fluxofenim, furilazole, isoxadifen (including isoxadifen-ethyl), mefenpyr (including mefenpyr-diethyl), metcamifen and oxabetrinil.
- the safeners of the compound of Formula (I) may also be in the form of esters or salts, as mentioned e.g. in The Pesticide Manual, 16 th Edition (BCPC), 2012.
- the reference to cloquintocet-mexyl also applies to a lithium, sodium, potassium, calcium, magnesium, aluminium, iron, ammonium, quaternary ammonium, sulfonium or phosphonium salt thereof as disclosed in WO 02/34048.
- the mixing ratio of compound of Formula (I) to safener is from 100: 1 to 1 : 10, especially from 20: 1 to 1 : 1 .
- Locus means the area in which the plants are growing or will grow.
- the application may be applied to the locus pre-emergence and/or postemergence of the crop plant.
- Some crop plants may be inherently tolerant to herbicidal effects of compounds of Formula (I).
- Preferred crop plants include maize, wheat, barley and rice.
- the rates of application of compounds of Formula I may vary within wide limits and depend on the nature of the soil, the method of application (pre- or postemergence; seed dressing; application to the seed furrow; no tillage application etc.), the crop plant, the weed(s) to be controlled, the prevailing climatic conditions, and other factors governed by the method of application, the time of application and the target crop.
- the compounds of Formula I according to the invention are generally applied at a rate of from 10 to 2500 g/ha, especially from 25 to 1000 g/ha, more especially from 25 to 250 g/ha.
- the application is generally made by spraying the composition, typically by tractor mounted sprayer for large areas, but other methods such as dusting (for powders), drip or drench can also be used.
- Crop plants are to be understood as also including those crop plants which have been rendered tolerant to other herbicides or classes of herbicides (e.g. ALS-, GS-, EPSPS-, PPO-, HPPD-, -PDS and ACCase-inhibitors) by conventional methods of breeding or by genetic engineering.
- herbicides or classes of herbicides e.g. ALS-, GS-, EPSPS-, PPO-, HPPD-, -PDS and ACCase-inhibitors
- An example of a crop that has been rendered tolerant to imidazolinones, e.g. imazamox, by conventional methods of breeding is Clearfield® summer rape (canola).
- crops that have been rendered tolerant to herbicides by genetic engineering methods include e.g. glyphosate- and glufosinate-resistant maize varieties commercially available under the trade names RoundupReady® and LibertyLink®.
- Crop plants are also to be understood as being those which have been rendered resistant to harmful insects by genetic engineering methods, for example Bt maize (resistant to European corn borer), Bt cotton (resistant to cotton boll weevil) and also Bt potatoes (resistant to Colorado beetle).
- Bt maize are the Bt 176 maize hybrids of NK® (Syngenta Seeds).
- the Bt toxin is a protein that is formed naturally by Bacillus thuringiensis soil bacteria.
- Examples of toxins, or transgenic plants able to synthesise such toxins are described in EP-A-451 878, EP-A-374 753, WO 93/07278, WO 95/34656, WO 03/052073 and EP-A-427 529.
- transgenic plants comprising one or more genes that code for an insecticidal resistance and express one or more toxins are KnockOut® (maize), Yield Gard® (maize), NuCOTIN33B® (cotton), Bollgard® (cotton), NewLeaf® (potatoes), NatureGard® and Protexcta®.
- Plant crops or seed material thereof can be both resistant to herbicides and, at the same time, resistant to insect feeding (“stacked” transgenic events).
- seed can have the ability to express an insecticidal Cry3 protein while at the same time being tolerant to glyphosate.
- Crop plants are also to be understood to include those which are obtained by conventional methods of breeding or genetic engineering and contain so-called output traits (e.g. improved storage stability, higher nutritional value and improved flavour).
- the compositions can be used to control unwanted plants (collectively, ‘weeds’).
- weeds to be controlled may be both monocotyledonous species, for example Agrostis, Alopecurus, Avena, Brachiaria, Bromus, Cenchrus, Cyperus, Digitaria, Echinochloa, Eleusine, Lolium, Monochoria, Rottboellia, Sagittaria, Scirpus, Setaria and Sorghum, and dicotyledonous species, for example Abutilon, Amaranthus, Ambrosia, Chenopodium, Chrysanthemum, Conyza, Galium, Ipomoea, Nasturtium, Sida, Sinapis, Solanum, Stellaria, Veronica, Viola and Xanthium.
- Agrostis Alopecurus
- Avena Brachiaria
- Bromus Cenchrus
- Cyperus Digitaria
- Echinochloa Eleusine
- Lolium Monochoria
- the compounds of the present invention can be prepared according to the following schemes.
- a compound of Formula 11 is compounds of Formula I wherein X is O and R 1 , A 1 , A 2 , Q, A 3 and R 3 are as defined in Formula I above.
- a compound of Formula 11 can be prepared via alkoxylation reaction which involves reacting compounds of formula III with compounds of formula II, wherein LG 1 is a halogen, preferably iodine, bromine or chlorine (or a pseudo-halogen leaving group, such as a (halo)alkyl or phenyl sulfonate ester, e.g. triflate), in the presence of a base, such as sodium hydride or an alkali earth metal hydride, carbonate (e.g.
- Compounds of formula IV can be prepared by Suzuki cross-coupling reactions which involves reacting compounds of formula V, wherein X 1 is halogen preferably bromide or iodide with compounds of formula Q-Yba, wherein Yba can be a boron-derived functional group, such as for example B(OH)2 or B(ORba)2 wherein Rba can be a Ci- C4alkyl group or the two groups ORba can form together with the boron atom a five membered ring, as for example a pinacol boronic ester.
- X 1 is halogen preferably bromide or iodide
- Q-Yba wherein Yba can be a boron-derived functional group, such as for example B(OH)2 or B(ORba)2 wherein Rba can be a Ci- C4alkyl group or the two groups ORba can form together with the boron atom a five membered ring, as for
- the reaction may be catalysed by a palladium based catalyst, for example tetrakis(triphenyl- phosphine)palladium(O), (1 ,1'bis(diphenylphosphino)ferrocene)dichloro-palladium- dichloromethane (1 :1 complex) or chloro(2-dicyclohexylphosphino-2',4',6'-triisopropyl- 1 ,1'-biphenyl)[2-(2'-amino-1 ,T-biphenyl)]palladium(ll) (XPhos palladacycle), in presence of a base, like sodium carbonate, tripotassium phosphate or cesium fluoride, in a solvent or a solvent mixture, like, for example dioxane, acetonitrile, N,N- dimethyl-formamide, a mixture of 1 ,2-dimethoxyethane and water or of dioxane
- Compounds of Formula 12 are compounds of formula I wherein X is CH2 and R1, A1, A2, A3, Q, and R3 are as defined in formula I above (scheme 2).
- Compounds of formula I2 can be prepared by decarboxylation of compounds of formula VI, wherein R 11 is Ci-Cealkyl or phenyl (scheme 2). Such decarboxylation reactions can be carried out under thermal conditions for example heating at 100 to 180 °C in solvent such as isopropanol/water or under basic conditions such as using sodium hydroxide or potassium hydroxide.
- Compounds of Formula VI can be prepared by reacting compounds of formula VII with compounds of Formula VIII, wherein LG 2 is a halogen, preferably iodine, bromine or chlorine (or a pseudo-halogen leaving group, such as a (halo)alkyl or phenyl sulfonate ester, e.g. triflate), in the presence of a base, such as sodium hydride or an alkali earth metal hydride, carbonate (e.g.
- sodium carbonate, potassium carbonate or cesium carbonate) or hydroxide optionally in the presence of potassium iodide in an inert solvent such as tetrahydrofuran, dioxane, water, N,N-dimethylformamide DMF, N,N-dimethylacetamide, sulfolane or acetonitrile and the like, at temperatures between 0 and 120°C.
- an inert solvent such as tetrahydrofuran, dioxane, water, N,N-dimethylformamide DMF, N,N-dimethylacetamide, sulfolane or acetonitrile and the like, at temperatures between 0 and 120°C.
- compounds of Formula 12 can be prepared from compounds of formula IX via deoxygenation or reduction of alcohol. Reduction of such alcohols are well described in literature and can be carried out using reducing agent such as LiAIF , DIBAL-H, or using triphenyl phosphine in the presence of iodine and imidazole or using triethyl silane in the presence of trifluoroacetic acid.
- reducing agent such as LiAIF , DIBAL-H, or using triphenyl phosphine in the presence of iodine and imidazole or using triethyl silane in the presence of trifluoroacetic acid.
- Compounds of formula IX can be prepared by reacting compounds of Formula XI, wherein X 11 is a halogen preferably bromine or iodine with an organometallic reagent such as BuLi or isopropylmagnesium chloride/LiCI complex amongst other metallating reagents to form an intermediate Xia, wherein M(Ln) p is a corresponding metal from the organometallic reagent such as lithium or magnesium and (Ln) p is its optionally substituted group like chloro and then subsequently reacting with compounds of Formula X.
- organometallic reagent such as BuLi or isopropylmagnesium chloride/LiCI complex amongst other metallating reagents
- M(Ln) p is a corresponding metal from the organometallic reagent such as lithium or magnesium
- (Ln) p is its optionally substituted group like chloro
- compounds of formula 12 can be prepared following scheme 4.
- compounds of formula 12 can be prepared by reacting compounds of formula IXab, wherein X 22 is a halogen, preferably iodine, bromine or chlorine (or a pseudo-halogen leaving group, such as a (halo)alkyl or phenyl sulfonate ester, e.g. triflate), and compounds of formula Va under Suzuki cross-coupling reaction under conditions analogous to procedure as described in scheme 1 for the conversion of compounds of formula V to compounds of formula IV.
- Compounds of formula IXab can be prepared from compounds of formula Xab via analogous procedure as described in scheme 3 for the conversion of compounds of formula X to compounds of formula 12.
- Compounds of formula 13 can be prepared by nucleophilic substitution reaction which involves reacting compounds of formula XVII with compounds of formula XVIIa or compounds of formula XVIIb wherein M is a metal such as sodium, lithium or potassium.
- Compounds of formula XVII can be prepared from compounds of formula XVI via oxidation reaction using oxidizing reagents such as hydrogen peroxide or meta-chloroperoxybenzoic acid.
- Compounds of formula XVI can be prepared by decarboxylation reaction from compounds of formula XV analogous to procedure as described in scheme 2 for synthesis of compounds of formula 12 from compounds of formula VI.
- Compounds of formula XV can be prepared by reaction of compounds of formula XIII, wherein R 15 is Ci-Cealklyl or phenyl and compounds of formula XIV, wherein LG 3 is a leaving group for e.g. Cl, Br, SO2CH3 or SChPh following procedure as described in scheme 2 for the synthesis of compounds of formula VI by the reaction of compounds of formula VII and VIII.
- Compounds of formula XIII can be prepared by the reaction of compounds of formula XII, wherein X 15 is halogen such as Cl or Br and compounds of formula Xlla, wherein Yb4 can be a boron-derived functional group, such as for example B(OH)2 or B(ORb4)2 wherein Rb4 can be a C1- C4alkyl group or the two groups ORb4 can form together with the boron atom a five membered ring, as for example a pinacol boronic ester following procedure as described in scheme 1 for the synthesis of compounds of formula IV from compounds of formula V and compounds of formula Va.
- X 15 is halogen such as Cl or Br
- Yb4 can be a boron-derived functional group, such as for example B(OH)2 or B(ORb4)2 wherein Rb4 can be a C1- C4alkyl group or the two groups ORb4 can form together with the boron atom a five membered
- compounds of formula 13 can be prepared by reacting compounds of formula XVIII, wherein X 12 is halogen such as Cl, Br or tosyl or mesyl functional group and compounds of formula Xlla following procedure as described in scheme 5 for the synthesis of compounds of formula XIII from compounds of formula XII and compounds of formula Xlla.
- Compounds of formula XVIII, wherein X 12 is halogen can be prepared from compounds of formula XIX by halogenation reaction using reagents such as POCh or POBra.
- compounds of formula XVIII, wherein X 12 is tosyl or mesyl can be prepared by the reaction of compounds of formula XIX with reagents such as p-toluenesulfonyl chloride or methanesulfonyl chloride respectively in the presence of base such as triethyl amine or pyridine and optionally reaction can be carried out in the presence of 4-dimethylaminopyridine.
- bases such as triethyl amine or pyridine
- 4-dimethylaminopyridine Such reactions are well known in the literature and known to those skilled in the state of art.
- Compounds of formula XIX can be prepared by the condensation reaction of amidine XXI and compounds of formula XX.
- Scheme 7 compounds of formula 12 can be prepared by reacting compounds of formula XXIV and compounds of formula Va wherein Yb 3 can be a boron-derived functional group, such as for example B(OH)2 or B(ORba)2 wherein Rb 3 can be a Ci- C4alkyl group or the two groups ORb 3 can form together with the boron atom a five membered ring, as for example a pinacol boronic ester following procedure as described in scheme 1 for the synthesis of compounds of formula IV from compounds of formula V and compounds of formula Va.
- Compounds of formula XXIV can be prepared from compounds of formula XXVI via two step procedure which involves base mediated arylation and decarboxylation reaction as described in scheme 2 for the conversion of compounds of formula VII to compounds of formula I2.
- reaction mixture was allowed to stir for another 15 min at room temperature.
- the reaction mixture was warmed to 40 °C and was allowed to stir for a further 1 h.
- the reaction mixture was allowed to cool to room temperature before being diluted with brine (20 mL) and extracted with ethyl acetate (2 x 15 mL). The combined organics were concentrated in vacuo. The residues were loaded onto celite and subjected to silica gel column chromatography using 0-40% ethyl acetate in cyclohexane.
- Step 2 Preparation of 2-(trifluoromethyl)-4-[4-(trifluoromethyl) phenyl]pyrimidin-5-ol (I-2)
- reaction mixture was allowed to cool to room temperature before being diluted with water (10 mL) and brine (10 mL) and extracted with tert-butylmethyl ether (2 x 15 mL). The combined organics were concentrated. The residues were loaded onto celite and subjected to silica gel column chromatography using 0-40% ethyl acetate in cyclohexane as eluent. The fractions forming the major peak of interest were combined and concentrated in vacuo, yielding 5-(5-chloropyrimidin-2-yl)oxy-2-(trifluoromethyl)-4-[4-(trifluoromethyl) phenyl]pyrimidine (1.007).
- 1 H NMR (400 MHz, CDCI 3 ) 5 8.85 (s, 1 H), 8.44 (s, 2H), 8.23 (d, 2H), 7.72 (d, 2H).
- Step 1 Preparation of (2-chloro-5-fluoro-3-pyridyl)-(5-chloropyrimidin-2- yljmethanol (I-3)
- Step 3 Preparation of 5-chloro-2-[[2-(3,4-difluorophenyl)-5-fluoro-3- pyridyl]methyl]pyrimidine (1.015)
- Step 1 Preparation of ethyl 2-[4-(3,4-difluorophenyl)-2-methylsulfanyl- pyrimidin-5-yl]acetate (I-5)
- Step 2 Preparation of ethyl 2-(5-chloropyrimidin-2-yl)-2-[4-(3,4-difluorophenyl)- 2-methylsulfanyl-pyrimidin-5-yl]acetate (I-6)
- Step 3 Preparation of 5-[(5-chloropyrimidin-2-yl)methyl]-4-(3,4-difluorophenyl)- 2-methylsulfanyl-pyrimidine (1.029)
- Step 4 Preparation of 5-[(5-chloropyrimidin-2-yl)methyl]-4-(3,4-difluorophenyl)- 2-methylsulfonyl-pyrimidine (1.028)
- Step 5 Preparation of 5-[(5-chloropyrimidin-2-yl)methyl]-6-(3,4-difluorophenyl)- 1H-pyrimidin-2-one (I-7)
- a solution of 5-[(5-chloropyrimidin-2-yl)methyl]-4-(3,4-difluorophenyl)-2- methylsulfonyl-pyrimidine 1.028 (760 mg, 1.82 mmol) in THF (9 mL) was treated with aqueous sodium hydroxide (2.0 M, 35 mL, 70 mmol) and was allowed to stir at room temperature for 2.5 hours.
- the mixture was acidified with 2M hydrochloric acid and extracted with ethyl acetate.
- Step 6 Preparation of 5-[(5-chloropyrimidin-2-yl)methyl]-2-(difluoromethoxy)-4- (3,4-difluorophenyl)pyrimidine (1.021 )
- Step 1 Preparation of 5-[(5-chloropyrimidin-2-yl)methyl]-4-(3,4- difluorophenyl)pyrimidine-2-carbonitrile (1.025)
- Step 1 Preparation of 5-[(5-chloropyrimidin-2-yl)methyl]-2-cyclopropyl-4-(3,4- difluorophenyljpyrimidine (1.030)
- Step 1 Preparation of diethyl 2-(5-chloropyrimidin-2-yl)propanedioate (I-8)
- Ethyl 2-(5-chloropyrimidin-2-yl)-2-[2-chloro-6-(trifluoromethyl)-3-pyridyl]acetate 1-10 (310 mg, 0.81 mmol) was dissolved in 3.1 mL propanenitrile in a teflon coated vial. To this, 0.31 mL of water were added and the vial was placed inside a pressure reactor and heated at 175 °C for 8 h. After this time, the reaction mixture was cooled to room temperature and diluted with 5 mL of water.
- Step 1 Preparation of ethyl 4-(3,4-difluorophenyl)-2-
- Step 4 Preparation of (5-chloropyrimidin-2-yl)-[4-(3,4-difluorophenyl)-2- (trifluoromethyl)pyrimidin-5-yl]methanol (1.034)
- Step 5 Preparation of 5-[(5-chloropyrimidin-2-yl)methyl]-4-(3,4-difluorophenyl)- 2-(trifluoromethyl)pyrimidine (1.033)
- triphenylphosphine 230 mg, 0.86 mmol was added followed by [4,4’-bis(1,1-dimethylethyl)-2,2 ' -bipyridine-N1,N1]bis-[3,5-difluoro-2-[5- (trifluoromethyl)-2-pyridinyl-N]-phenyl-C]lridium(lll) hexafluorophosphate (3.9 mg, 0.0034 mmol) added and the reaction was stirred and irradiated with blue light (Penn photo-reactor, 450 nm) for 24 hours.
- diethyl propanedioate (11.79 g, 73.62 mmol) was dissolved in 100 mL dimethyl sulfoxide under nitrogen atmosphere.
- potassium phosphate 39.47 g, 184.1 mmol was added followed by 5-chloro-2- (chloromethyl)pyrimidine (10.00 g, 61.35 mmol) and the reaction mixture was stirred at room temperature for 12 h.
- 400 mL water were added, and the aqueous layer was extracted by ethyl acetate (3 X 150 mL).
- the combined organic was washed with brine (200 mL), dried over anhydrous sodium sulphate, filtered and evaporated to get the crude compound.
- the crude material was further purified by silica gel column chromatography using 30 % ethyl acetate in cyclohexane to get diethyl 2-[(5-chloropyrimidin-2-yl)methyl]propanedioate 1-15 (13.0 g, 66%) as yellowish liquid.
- the aqueous layer was extracted with ethyl acetate (3 x 75 mL) and the combined organic layer was dried over anhydrous sodium sulphate, filtered and concentrated to get the crude compound.
- the crude material was then purified by silica gel column chromatography using 40 % ethyl acetate in cyclohexane to afford ethyl 2-[(5-chloropyrimidin-2-yl)methyl]-3-oxo-propanoate 1-17 (2.1 g, 55%) yellowish liquid as a mixture of keto enol tautomer (1.6:1).
- Step 6 Preparation of 5-[(5-chloropyrimidin-2-yl)methyl]-4-(3,4-difluorophenyl)- 2-(methoxymethyl)pyrimidine (1-20)
- the reaction mixture was degassed by bubbling nitrogen for 10 min and then heated at 100 °C under microwave irradiation for 1 h.
- the reaction mixture was cooled to room temperature and 20 mL water was added.
- the aqueous layer was extracted with ethyl acetate (3 X 30 mL), dried over anhydrous sodium sulphate, filtered and concentrated.
- the crude product obtained after concentration was purified by silica gel column chromatography using 25-30% ethyl acetate in cyclohexane to get 5-[(5- chloropyrimidin-2-yl)methyl]-4-(3,4-difluorophenyl)-2-(methoxymethyl)pyrimidine I-20 (180 mg, 50%) as yellowish gummy material.
- Step 7 Preparation of [5-[(5-chloropyrimidin-2-yl)methyl]-4-(3,4- difluorophenyl)-pyrimidin-2-yl]methanol (1-21)
- the aqueous layer was extracted with ethyl acetate (3 X 20 mL), dried over anhydrous sodium sulphate, filtered and concentrated to get the crude compound.
- the crude was further purified by silica gel column chromatography using 50-60% ethyl acetate in cyclohexane to get [5-[(5-chloropyrimidin-2-yl)methyl]-4-(3,4- difluorophenyl)pyrimidin-2-yl]methanol 1-21 (65 mg, 38%) yellow liquid.
- Step 8 Preparation of 5-[(5-chloropyrimidin-2-yl)methyl]-4-(3,4-difluorophenyl)- pyrimidine-2-carbaldehyde (I-22)
- reaction mixture was then stirred at room temperature for 12 h and then quenched by adding 10 mL of aqueous sodium hydrogen carbonate solution followed by 10 mL aqueous sodium thiosulphate solution.
- the aqueous layer was extracted with ethyl acetate (3 X 20 mL) and the combined organic layer was dried over anhydrous sodium sulphate, filtered and evaporated to the crude compound.
- Step 9 Preparation of 5-[(5-chloropyrimidin-2-yl)methyl]-2-(difluoromethyl)-4- (3,4-difluorophenyl)pyrimidine (1.038)
- Step 1 Preparation of ethyl 2-[4-(6-chloro-5-fluoro-3-pyridyl)-2-methylsulfanyl- pyrimidin-5-yl]acetate (I-23)
- Step 2 Preparation of ethyl 2-[4-(6-chloro-5-fluoro-3-pyridyl)-2-methylsulfanyl- pyrimidin-5-yl]-2-(5-chloropyrimidin-2-yl)acetate (I-24)
- Step 3 Preparation of 4-(6-chloro-5-fluoro-3-pyridyl)-5-[(5-chloropyrimidin-2- yl)methyl]-2-methylsulfanyl-pyrimidine (1.036)
- Step 4 Preparation of 4-(6-chloro-5-fluoro-3-pyridyl)-5-[(5-chloropyrimidin-2- yl)methyl]-2-methylsulfonyl-pyrimidine (1.037)
- Step 5 Preparation of 4-(6-chloro-5-fluoro-3-pyridyl)-5-[(5-chloropyrimidin-2- yl)methyl]-2-(trifluoromethyl)pyrimidine (1.017)
- the plants After cultivation for one day (pre-emergence) or after 8 days cultivation (post-emergence) under controlled conditions in a glasshouse (at 24/16°C, day/night; 14 hours light; 65 % humidity), the plants are sprayed with an aqueous spray solution derived from the formulation of the technical active ingredient in acetone I water (50:50) solution containing 0.5% Tween 20 (polyoxyethelyene sorbitan monolaurate, CAS RN 9005- 64-5). Compounds are applied at 250 g/ha unless otherwise stated. The test plants are then grown in a glasshouse under controlled conditions in a glasshouse (at 24/16°C, day/night; 14 hours light; 65 % humidity) and watered twice daily.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Zoology (AREA)
- Pest Control & Pesticides (AREA)
- Plant Pathology (AREA)
- Engineering & Computer Science (AREA)
- Environmental Sciences (AREA)
- Wood Science & Technology (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Dentistry (AREA)
- Agronomy & Crop Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Agricultural Chemicals And Associated Chemicals (AREA)
Abstract
The present invention relates to compounds of Formula (I), wherein A1, A2, A3, R1, R3, Q and X are as defined herein. The invention further relates to herbicidal compositions which comprise a compound of Formula (I) and to the use of compounds of Formula (I) for controlling weeds, in particular in crops of useful plants.
Description
PYRIDINE AND PYRIMIDINE DERIVATIVES AS HERBICIDES
The present invention relates to novel herbicidal compounds, to processes for their preparation, to herbicidal compositions which comprise the novel compounds, and to their use for controlling weeds, in particular in crops of useful plants, or for inhibiting plant growth.
WO2010/064688 disclosed the use of pyrimidine derivatives as herbicides. WO2016/149315 discloses substituted pyrimidinyloxy pyridine derivatives as herbicides. W02020/002089 discloses substituted 2-heteroaryloxypyridines. W02021/204706 discloses herbicidal 5-haloalkoxy-pyrimidine compounds. GB2594931 teaches phenoxypyridine compounds. W02022/002838 discloses substituted heteroaryloxypyridines. WO2023/186691 discloses substituted 2-C- azines. The present invention relates to novel herbicidal compounds.
Thus, according to the present invention there is provided a compound of Formula (I):
 wherein
wherein
X is O or CHR8,
Q is phenyl or a C-linked 6-membered heteroaryl wherein said phenyl or 6- membered heteroaryl is optionally substituted by one or more R4;
A1 is CH or N
A2 is OR2 or N,
wherein at least one of A1 and A2 are N,
A3 is CR5 or N; wherein if X is O then A1 is not N when A2 is CR2 and A3 is N;
R1 is selected from the group consisting of hydrogen, halogen, -CN, nitro, Ci- C4alkyl-, C2-C4alkenyl-, C2-C4alkynyl-, Ci-C4haloalkyl-, C1-C4 alkoxy-, C1- C4haloalkoxy-, -S(O)pCi-C4alkyl and Cs-Cecycloalkyl-;
R2 is selected from the group consisting of hydrogen, halogen and Ci-Ce alkyl;
R3 is selected from the group consisting of halogen, Ci-C4haloalkyl- and C1-C2 haloalkoxy-;
R4 is selected from the group consisting of halogen, C1-C4 alkyl, Ci- 04 haloalkyl, C1-C4 alkoxy, C1-C4 haloalkoxy, Ci-C4alkoxyCi-C3alkyl-, Ci- C4alkoxyCi-C3alkoxy-, Ci-C4alkoxyCi-C3alkoxyCi-C3alkyl-, -CN, NO2, C2- C4alkenyl, C2-C4alkynyl, -S(O)pCi-C4alkyl, -S(O)pCi-C4haloalkyl, -C(O)OCi- C4alkyl and -C(O)NR6R7;
R5 is hydrogen or halogen;
R6 is selected from the group consisting of hydrogen, C3-C4cycloalkyl, C1- C4alkyl and Ci-C4haloalkyl;
R7 is selected from the group consisting of hydrogen, C3-C4cycloalkyl, C1- C4alkyl and Ci-C4haloalkyl;
R8 is hydrogen or hydroxyl; and p is 0, 1 or 2; or an agriculturally acceptable salt thereof.
Alkyl groups (e.g Ci-Cealkyl) include, for example, methyl (Me, CH3), ethyl (Et, C2H5), n-propyl (n-Pr), isopropyl (/-Pr), n-butyl (n-Bu), isobutyl (/-Bu), sec-butyl (s-Bu) and tert- butyl (t-Bu).
Alkenyl and alkynyl moieties can be in the form of straight or branched chains, and the alkenyl moieties, where appropriate, can be of either the (E)- or (^-configuration. Examples are vinyl, allyl and propargyl. Alkenyl and alkynyl moieties can contain one or more double and/or triple bonds in any combination.
Halogen (or halo) encompasses fluorine, chlorine, bromine or iodine. The same correspondingly applies to halogen in the context of other definitions, such as haloalkyl.
Haloalkyl groups (e.g Ci-Cehaloalkyl) are, for example, fluoromethyl, difluoromethyl, trifluoromethyl, chloromethyl, dichloromethyl, trichloromethyl, 2,2,2- trifluoroethyl, 2-fluoroethyl, 2-chloroethyl, pentafluoroethyl, 1 , 1 -difluoro-2,2,2- trichloroethyl, 2,2,3,3-tetrafluoroethyl and 2,2,2-trichloroethyl, heptafluoro-n-propyl and perfluoro-n-hexyl.
Alkoxy groups (e.g Ci-C4alkoxy-) are, for example, methoxy, ethoxy, propoxy, isopropoxy, n-butoxy, isobutoxy, sec-butoxy or tert-butoxy, preferably methoxy and ethoxy.
Alkoxyalkyl groups (e.g Ci-C4alkoxy-Ci-C3alkyl-) includes, for example, methoxymethyl, methoxyethyl, ethoxymethyl, ethoxyethyl, n-propoxymethyl, n- propoxyethyl, isopropoxymethyl or isopropoxyethyl.
Cycloalkyl groups (e.g Cs-Cecycloalkyl-) include, for example cyclopropyl (c- propyl, c-Pr), cyclobutyl (c-butyl, c-Bu), cyclopentyl (c-pentyl) and cyclohexyl (c-hexyl) and may be substituted or unsubstituted as indicated.
Ci-Cealkyl-S- (alkylthio) is, for example, methylthio, ethylthio, propylthio, isopropylthio, n-butylthio, isobutylthio, sec-butylthio or tert-butylthio, preferably methylthio or ethylthio.
Ci-Cealkyl-S(O)- (alkylsulfinyl) is, for example, methylsulfinyl, ethylsulfinyl, propylsulfinyl, isopropylsulfinyl, n-butylsulfinyl, isobutylsulfinyl, sec-butylsulfinyl or tert-butylsulfinyl, preferably methylsulfinyl or ethylsulfinyl.
Ci-Cealkyl-S(O)2- (alkylsulfonyl) is, for example, methylsulfonyl, ethylsulfonyl, propylsulfonyl, isopropylsulfonyl, n-butylsulfonyl, isobutylsulfonyl, sec-butylsulfonyl or tert-butylsulfonyl, preferably methylsulfonyl or ethylsulfonyl.
In a preferred embodiment of the present invention in the compound of Formula (I) X is CH2.
In one embodiment of the present invention there is provided a compound of Formula (I) wherein A1 is N and A2 is CR2 (wherein R2 is, for example, H or halo). In another embodiment of the present invention, there is provided a compound of Formula (I) wherein A1 is CH and A2 is N. In another embodiment of the present invention, there is provided a compound of Formula (I) wherein A1 is N and A2 is N.
In another embodiment of the present invention, there is provided a compound of Formula (I) wherein A3 is N. In another embodiment of the present invention A3 is CR5. In this embodiment it is preferred that R5 is hydrogen or halogen, for example chloro or fluoro, most preferably fluoro.
Thus, in one embodiment of the present invention, there is provided a compound of Formula (I) wherein A1 is N, A2 is CH and A3 is N (wherein X is CHR8). In another embodiment of the present invention, A1 is CH, A2 is N and A3 is N. In another embodiment of the present invention, A1 is N, A2 is N and A3 is N.
In another embodiment of the present invention, there is provided a compound of Formula (I) wherein A1 is N, A2 is CH and A3 is CR5, preferably CH or CF. In another embodiment of the present invention, A1 is CH, A2 is N and A3 is CR5, preferably CH or CF. In another embodiment of the present invention, A1 is N, A2 is N and A3 is CR5, preferably CH or CF.
In one embodiment of the present invention, there is provided a compound of Formula (I) wherein R1 is selected from the group consisting of hydrogen, halogen, Ci-C4alkyl- and Ci-C4haloalkyl-. In a more preferred embodiment of the present invention, R1 is hydrogen, methyl or -CF3.
In one embodiment of the present invention, there is provided a compound of Formula (I) wherein R3 is halogen (e.g bromo or chloro, most preferably chloro), Ci- C4haloalkyl- (e.g -CF3) or Ci-C4haloalkoxy- (e.g -OCF3).ln a more preferred embodiment R3 is halogen (e.g bromo or chloro, most preferably chloro).
In a further embodiment of the present invention, there is provided a compound of Formula (I) wherein Q is selected from the group consisting of:
 wherein n is 0, 1 or 2.
wherein n is 0, 1 or 2.
In a more preferred embodiment of the present invention, there is provided a compound of Formula (I) wherein Q is selected from the group consisting of Q-1, Q-3 and Q-4 which are optionally substituted by one or two R4. In one embodiment of the present invention, there is provided a compound of Formula (I) wherein Q is Q-1 or Q-4 which are optionally substituted by one or two R4. In another embodiment of the present invention, Q is Q-1 which is optionally substituted by one or two R4.
In a more preferred embodiment, Q is Q-1, R3 is halogen or C1-C4 haloalkyl and n is 1 (wherein in a preferred embodiment Q is 4-CF3-phenyl-). In another preferred embodiment of the present invention Q is Q-1, R3 is halogen and n is 2 (wherein in a preferred embodiment Q is 3,4-dihalophenyl- (e.g 3-F,4-CI-phenyl-, 3- CI,4-F-phenyl-, 3,4-diclorophenyl- or 3,4diflurophenyl-).
In another embodiment of the present invention, Q is Q-3 which is optionally substituted by one or two R4. In one embodiment of the present invention, Q is Q-4 which is optionally substituted by one or two R4. In a further preferred embodiment of the present invention Q is Q-3, R3 is halogen and n is 2 (wherein in a preferred embodiment Q is 6,5-dihalo-3-pyridyl- (e.g 6-chloro-5-fluoro-3-pyridyl or 6-fluoro-5- chloro-3-pyridyl).
In one embodiment of the present invention, n is 0. In another embodiment, n is 1. In another embodiment, n is 2. In embodiments of the invention wherein n is 1 or 2, each R4 is preferably selected from the group consisting of halogen (e.g fluoro or chloro) and C1-C4 haloalkyl (e.g -CF3).
The compounds of Formula (I) according to the invention can be used as herbicides by themselves, but they are generally formulated into herbicidal compositions using formulation adjuvants, such as carriers, solvents and surfaceactive agents (SAA). Thus, the present invention further provides a herbicidal composition comprising a herbicidal compound according to the present invention and an agriculturally acceptable formulation adjuvant. The composition can be in the form of concentrates which are diluted prior to use, although ready-to-use compositions can also be made. The final dilution is usually made with water, but can be made instead of, or in addition to, water, with, for example, liquid fertilisers, micronutrients, biological organisms, oil or solvents.
As outlined above, the compounds of the present invention may contain an asymmetric centre. As such, the compound of the present invention may be present in the composition as a racemic mixture of the two enantiomers. Alternatively, the compound of the present invention may be present in an enantiomer enriched form.
The herbicidal compositions generally comprise from 0.1 to 99 % by weight, especially from 0.1 to 95 % by weight, compounds of Formula I and from 1 to 99.9 %
by weight of a formulation adjuvant which preferably includes from 0 to 25 % by weight of a surface-active substance.
The compositions can be chosen from a number of formulation types. These include an emulsion concentrate (EC), a suspension concentrate (SC), a suspo- emulsion (SE), a capsule suspension (CS), a water dispersible granule (WG), an emulsifiable granule (EG), an emulsion, water in oil (EG), an emulsion, oil in water (EW), a micro-emulsion (ME), an oil dispersion (OD), an oil miscible flowable (OF), an oil miscible liquid (OL), a soluble concentrate (SL), an ultra-low volume suspension (Sil), an ultra-low volume liquid (UL), a technical concentrate (TK), a dispersible concentrate (DC), a soluble powder (SP), a wettable powder (WP) and a soluble granule (SG). The formulation type chosen in any instance will depend upon the particular purpose envisaged and the physical, chemical and biological properties of the compound of Formula (I).
Soluble powders (SP) may be prepared by mixing a compound of Formula (I) with one or more water-soluble inorganic salts (such as sodium bicarbonate, sodium carbonate or magnesium sulphate) or one or more water-soluble organic solids (such as a polysaccharide) and, optionally, one or more wetting agents, one or more dispersing agents or a mixture of said agents to improve water dispersibility/solubility. The mixture is then ground to a fine powder. Similar compositions may also be granulated to form water soluble granules (SG).
Wettable powders (WP) may be prepared by mixing a compound of Formula (I) with one or more solid diluents or carriers, one or more wetting agents and, preferably, one or more dispersing agents and, optionally, one or more suspending agents to facilitate the dispersion in liquids. The mixture is then ground to a fine powder. Similar compositions may also be granulated to form water dispersible granules (WG).
Granules (GR) may be formed either by granulating a mixture of a compound of Formula (I) and one or more powdered solid diluents or carriers, or from preformed blank granules by absorbing a compound of Formula (I) (or a solution thereof, in a suitable agent) in a porous granular material (such as pumice, attapulgite clays, fuller's earth, kieselguhr, diatomaceous earths or ground corn cobs) or by adsorbing a compound of Formula (I) (or a solution thereof, in a suitable agent) on to a hard core material (such as sands, silicates, mineral carbonates, sulphates or phosphates) and drying if necessary. Agents which are commonly used to aid absorption or adsorption include solvents (such as aliphatic and aromatic petroleum solvents,
alcohols, ethers, ketones and esters) and sticking agents (such as polyvinyl acetates, polyvinyl alcohols, dextrins, sugars and vegetable oils). One or more other additives may also be included in granules (for example an emulsifying agent, wetting agent or dispersing agent).
Dispersible Concentrates (DC) may be prepared by dissolving a compound of Formula (I) in water or an organic solvent, such as a ketone, alcohol or glycol ether. These solutions may contain a surface-active agent (for example to improve water dilution or prevent crystallisation in a spray tank).
Emulsifiable concentrates (EC) or oil-in-water emulsions (EW) may be prepared by dissolving a compound of Formula (I) in an organic solvent (optionally containing one or more wetting agents, one or more emulsifying agents or a mixture of said agents). Suitable organic solvents for use in ECs include aromatic hydrocarbons (such as alkylbenzenes or alkylnaphthalenes, exemplified by SOLVESSO 100, SOLVESSO 150 and SOLVESSO 200; SOLVESSO is a Registered Trade Mark), ketones (such as cyclohexanone or methylcyclohexanone) and alcohols (such as benzyl alcohol, furfuryl alcohol or butanol), N-alkylpyrrolidones (such as N-methylpyrrolidone or N-octylpyrrolidone), dimethyl amides of fatty acids (such as Cs-C fatty acid dimethylamide) and chlorinated hydrocarbons. An EC product may spontaneously emulsify on addition to water, to produce an emulsion with sufficient stability to allow spray application through appropriate equipment.
Preparation of an EW involves obtaining a compound of Formula (I) either as a liquid (if it is not a liquid at room temperature, it may be melted at a reasonable temperature, typically below 70°C) or in solution (by dissolving it in an appropriate solvent) and then emulsifying the resultant liquid or solution into water containing one or more SAAs, under high shear, to produce an emulsion. Suitable solvents for use in EWs include vegetable oils, chlorinated hydrocarbons (such as chlorobenzenes), aromatic solvents (such as alkylbenzenes or alkylnaphthalenes) and other appropriate organic solvents which have a low solubility in water.
Microemulsions (ME) may be prepared by mixing water with a blend of one or more solvents with one or more SAAs, to produce spontaneously a thermodynamically stable isotropic liquid formulation. A compound of Formula (I) is present initially in either the water or the solvent/SAA blend. Suitable solvents for use in MEs include those hereinbefore described for use in in ECs or in EWs. An ME may be either an oil-in-water or a water-in-oil system (which system is present may be determined by conductivity measurements) and may be suitable for mixing water-
soluble and oil-soluble pesticides in the same formulation. An ME is suitable for dilution into water, either remaining as a microemulsion or forming a conventional oil- in-water emulsion.
Suspension concentrates (SC) may comprise aqueous or non-aqueous suspensions of finely divided insoluble solid particles of a compound of Formula (I). SCs may be prepared by ball or bead milling the solid compound of Formula (I) in a suitable medium, optionally with one or more dispersing agents, to produce a fine particle suspension of the compound. One or more wetting agents may be included in the composition and a suspending agent may be included to reduce the rate at which the particles settle. Alternatively, a compound of Formula (I) may be dry milled and added to water, containing agents hereinbefore described, to produce the desired end product.
Aerosol formulations comprise a compound of Formula (I) and a suitable propellant (for example n-butane). A compound of Formula (I) may also be dissolved or dispersed in a suitable medium (for example water or a water miscible liquid, such as n-propanol) to provide compositions for use in non-pressurised, hand-actuated spray pumps.
Capsule suspensions (CS) may be prepared in a manner similar to the preparation of EW formulations but with an additional polymerisation stage such that an aqueous dispersion of oil droplets is obtained, in which each oil droplet is encapsulated by a polymeric shell and contains a compound of Formula (I) and, optionally, a carrier or diluent therefor. The polymeric shell may be produced by either an interfacial polycondensation reaction or by a coacervation procedure. The compositions may provide for controlled release of the compound of Formula (I) and they may be used for seed treatment. A compound of Formula (I) may also be formulated in a biodegradable polymeric matrix to provide a slow, controlled release of the compound.
The composition may include one or more additives to improve the biological performance of the composition, for example by improving wetting, retention or distribution on surfaces; resistance to rain on treated surfaces; or uptake or mobility of a compound of Formula (I). Such additives include surface active agents (SAAs), spray additives based on oils, for example certain mineral oils or natural plant oils (such as soy bean and rape seed oil), modified plant oils such as methylated rape seed oil (MRSO), and blends of these with other bio-enhancing adjuvants (ingredients which may aid or modify the action of a compound of Formula (I).
Wetting agents, dispersing agents and emulsifying agents may be SAAs of the cationic, anionic, amphoteric or non-ionic type.
Suitable SAAs of the cationic type include quaternary ammonium compounds (for example cetyltri methyl ammonium bromide), imidazolines and amine salts.
Suitable anionic SAAs include alkali metals salts of fatty acids, salts of aliphatic monoesters of sulphuric acid (for example sodium lauryl sulphate), salts of sulphonated aromatic compounds (for example sodium dodecylbenzenesulphonate, calcium dodecylbenzenesulphonate, butylnaphthalene sulphonate and mixtures of sodium di-/sopropyl- and tri-/sopropyl-naphthalene sulphonates), ether sulphates, alcohol ether sulphates (for example sodium laureth-3-sulphate), ether carboxylates (for example sodium laureth-3-carboxylate), phosphate esters (products from the reaction between one or more fatty alcohols and phosphoric acid (predominately mono-esters) or phosphorus pentoxide (predominately di-esters), for example the reaction between lauryl alcohol and tetraphosphoric acid; additionally these products may be ethoxylated), sulphosuccinamates, paraffin or olefine sulphonates, taurates, lignosulphonates and phosphates I sulphates of tristyrylphenols.
Suitable SAAs of the amphoteric type include betaines, propionates and glycinates.
Suitable SAAs of the non-ionic type include condensation products of alkylene oxides, such as ethylene oxide, propylene oxide, butylene oxide or mixtures thereof, with fatty alcohols (such as oleyl alcohol or cetyl alcohol) or with alkylphenols (such as octylphenol, nonylphenol or octylcresol); partial esters derived from long chain fatty acids or hexitol anhydrides; condensation products of said partial esters with ethylene oxide; block polymers (comprising ethylene oxide and propylene oxide); alkanolamides; simple esters (for example fatty acid polyethylene glycol esters); amine oxides (for example lauryl dimethyl amine oxide); lecithins and sorbitans and esters thereof, alkyl polyglycosides and tristyrylphenols.
Suitable suspending agents include hydrophilic colloids (such as polysaccharides, polyvinylpyrrolidone or sodium carboxymethylcellulose) and swelling clays (such as bentonite or attapulgite).
The compounds of present invention can also be used in mixture with one or more additional herbicides and/or plant growth regulators. Examples of such additional herbicides or plant growth regulators include acetochlor, acifluorfen (including acifluorfen-sodium), aclonifen, ametryn, amicarbazone, aminopyralid, aminotriazole, atrazine, beflubutamid-M, benquitrione, bensulfuron (including
bensulfuron-methyl), bentazone, bicyclopyrone, bilanafos, bipyrazone, bispyribac- sodium, bixlozone, bromacil, bromoxynil, butachlor, butafenacil, carfentrazone (including carfentrazone-ethyl), cloransulam (including cloransulam-methyl), chlorimuron (including chlorimuron-ethyl), chlorotoluron, chlorsulfuron, cinmethylin, clacyfos, clethodim, clodinafop (including clodinafop-propargyl), clomazone, clopyralid, cyclopyranil, cyclopyrimorate, cyclosulfamuron, cyhalofop (including cyhalofop-butyl), 2,4-D (including the choline salt and 2-ethylhexyl ester thereof), 2,4- DB, desmedipham, dicamba (including the aluminium, aminopropyl, bisaminopropylmethyl, choline, dichloroprop, diglycolamine, dimethylamine, dimethylammonium, potassium and sodium salts thereof) diclosulam, diflufenican, diflufenzopyr, dimethachlor, dimethenamid-P, dioxopyritrione, diquat dibromide, diuron, epyrifenacil, ethalfluralin, ethofumesate, fenoxaprop (including fenoxaprop-P- ethyl), fenoxasulfone, fenpyrazone, fenquinotrione, fentrazamide, flazasulfuron, florasulam, florpyrauxifen (including florpyrauxifen-benzyl), fluazifop (including fluazifop-P-butyl), flucarbazone (including flucarbazone-sodium), flufenacet, flumetsulam, flumioxazin, fluometuron, flupyrsulfuron (including flupyrsulfuron- methyl-sodium), fluroxypyr (including fluroxypyr-meptyl), fomesafen, foramsulfuron, glufosinate (including L-glufosinate and the ammonium salts of both), glyphosate (including the diammonium, isopropylammonium and potassium salts thereof), halauxifen (including halauxifen-methyl), haloxyfop (including haloxyfop-methyl), hexazinone, hydantocidin, imazamox (including R-imazamox), imazapic, imazapyr, imazethapyr, indaziflam, iodosulfuron (including iodosulfuron-methyl-sodium), iofensulfuron (including iofensulfuron-sodium), ioxynil, isoproturon, isoxaflutole, lancotrione, MCPA, MCPB, mecoprop-P, mesosulfuron (including mesosulfuron- methyl), mesotrione, metamitron, metazachlor, methiozolin, metolachlor, metosulam, metribuzin, metsulfuron, napropamide, nicosulfuron, norflurazon, oxadiazon, oxasulfuron, oxyfluorfen, paraquat dichloride, pendimethalin, penoxsulam, phenmedipham, picloram, pinoxaden, pretilachlor, primisulfuron-methyl, prometryne, propanil, propaquizafop, propyrisulfuron, propyzamide, prosulfocarb, prosulfuron, pyraclonil, pyraflufen (including pyraflufen-ethyl), pyrasulfotole, pyridate, pyriftalid, pyrimisulfan, pyroxasulfone, pyroxsulam, quinclorac, quinmerac, quizalofop (including quizalofop-P-ethyl and quizalofop-P-tefuryl), rimisoxafen, rimsulfuron, saflufenacil, sethoxydim, simazine, S-metalochlor, sulfentrazone, sulfosulfuron, tebuthiuron, tefuryltrione, tembotrione, terbuthylazine, terbutryn, tetflupyrolimet, thiencarbazone, thifensulfuron, tiafenacil, tolpyralate, topramezone, tralkoxydim, triafamone, triallate, triasulfuron, tribenuron (including tribenuron-methyl), triclopyr, trifloxysulfuron (including trifloxysulfuron-sodium), trifludimoxazin, trifluralin,
triflusulfuron, tripyrasulfone, 3-(2-chloro-4-fluoro-5-(3-methyl-2,6-dioxo-4- trifluoromethyl-3,6-dihydropyrimidin-1(2H)-yl)phenyl)-5-methyl-4,5-dihydroisoxazole-
5-carboxylic acid ethyl ester, 4-hydroxy-1-methoxy-5-methyl-3-[4-(trifluoromethyl)-2- pyridyl]imidazolidin-2-one, 4-hydroxy-1,5-dimethyl-3-[4-(trifluoromethyl)-2- pyridyl]imidazolidin-2-one, 5-ethoxy-4-hydroxy-1-methyl-3-[4-(trifluoromethyl)-2- pyridyl]imidazolidin-2-one, 4-hydroxy-1-methyl-3-[4-(trifluoromethyl)-2- pyridyl]imidazolidin-2-one, 4-hydroxy-1,5-dimethyl-3-[1-methyl-5-
(trifluoromethyl)pyrazol-3-yl]imidazolidin-2-one, (4R)1-(5-tert-butylisoxazol-3-yl)-4- ethoxy-5-hydroxy-3-methyl-imidazolidin-2-one, 4-amino-3-chloro-5-fluoro-6-(7-fluoro- 1H-indol-6-yl)pyridine-2-carboxylic acid (including agrochemically acceptable esters thereof, for example, methyl 4-amino-3-chloro-5-fluoro-6-(7-fluoro-1 H-indol-6- yl)pyridine-2-carboxylate, prop-2-ynyl 4-amino-3-chloro-5-fluoro-6-(7-fluoro-1 H-indol-
6-yl)pyridine-2-carboxylate and cyanomethyl 4-amino-3-chloro-5-fluoro-6-(7-fluoro- 1 H-indol-6-yl)pyridine-2-carboxylate), 3-ethylsulfanyl-N-(1 ,3,4-oxadiazol-2-yl)-5- (trifluoromethyl)-[1,2,4]triazolo[4,3-a]pyridine-8-carboxamide, 3-(isopropylsulfanyl methyl)-N-(5-methyl-1 ,3,4-oxadiazol-2-yl)-5-(trifluoromethyl)-[1,2,4]triazolo[4,3- a]pyridine-8-carboxamide, 3-(isopropylsulfonylmethyl)-N-(5-methyl-1,3,4-oxadiazol-2- yl)-5-(trifluoromethyl)-[1,2,4]triazolo[4,3-a]pyridine-8-carboxamide, 3-(ethylsulfonyl methyl)-N-(5-methyl-1 ,3,4-oxadiazol-2-yl)-5-(trifluoromethyl)-[1,2,4]triazolo[4,3-a] pyridine-8-carboxamide, ethyl-2-[[3-[[3-chloro-5-fluoro-6-[3-methyl-2,6-dioxo-4- (trifluoromethyl)pyrimidin-1-yl]-2-pyridyl]oxy]acetate,6-chloro-4-(2,7-dimethyl-1- naphthyl)-5-hydroxy-2-methyl-pyridazin-3-one, tetrahydrofuran-2-ylmethyl (2R)-2-[(4- amino-3,5-dichloro-6-fluoro-2-pyridyl)oxy]propanoate, tetrahydrofuran-2-ylmethyl (2R)-2-[(4-amino-3,5-dichloro-6-fluoro-2-pyridyl)oxy]propanoate, tetrahydrofuran-2- ylmethyl 2-[(4-amino-3,5-dichloro-6-fluoro-2-pyridyl)oxy]propanoate, 2-[(4-amino-3,5- dichloro-6-fluoro-2-pyridyl)oxy]propanoic acid, 2-fluoro-N-(5-methyl-1 ,3,4-oxadiazol- 2-yl)-3-[(R)-propylsulfinyl]-4-(trifluoromethyl)benzamide, 2-fluoro-N-(5-methyl-1,3,4- oxadiazol-2-yl)-3-propylsulfinyl-4-(trifluoromethyl)benzamide, (2-fluorophenyl)methyl 6-amino-5-chloro-2-(4-chloro-2-fluoro-3-methoxy-phenyl)pyrimidine-4-carboxylate and 6-amino-5-chloro-2-(4-chloro-2-fluoro-3-methoxy-phenyl)pyrimidine-4-carboxylic acid.
The mixing partners of the compound of Formula (I) may also be in the form of esters or salts, as mentioned e.g. in The Pesticide Manual, Sixteenth Edition, British Crop Protection Council, 2012.
The compound of Formula (I) can also be used in mixtures with other agrochemicals such as fungicides, nematicides or insecticides, examples of which are given in The Pesticide Manual.
The mixing ratio of the compound of Formula (I) to the mixing partner is preferably from 1 : 100 to 1000:1.
The mixtures can advantageously be used in the above-mentioned formulations (in which case "active ingredient" relates to the respective mixture of compound of Formula (I) with the mixing partner).
The compounds or mixtures of the present invention can also be used in combination with one or more herbicide safeners. Examples of such safeners include benoxacor, cloquintocet (including cloquintocet-mexyl), cyprosulfamide, dichlormid, fenchlorazole (including fenchlorazole-ethyl), fenclorim, fluxofenim, furilazole, isoxadifen (including isoxadifen-ethyl), mefenpyr (including mefenpyr-diethyl), metcamifen and oxabetrinil.
Particularly preferred are mixtures of a compound of Formula (I) with cyprosulfamide, isoxadifen-ethyl, cloquintocet-mexyl and/or N-(2-methoxybenzoyl)-4- [(methyl-aminocarbonyl)amino]benzenesulfonamide.
The safeners of the compound of Formula (I) may also be in the form of esters or salts, as mentioned e.g. in The Pesticide Manual, 16th Edition (BCPC), 2012. The reference to cloquintocet-mexyl also applies to a lithium, sodium, potassium, calcium, magnesium, aluminium, iron, ammonium, quaternary ammonium, sulfonium or phosphonium salt thereof as disclosed in WO 02/34048.
Preferably the mixing ratio of compound of Formula (I) to safener is from 100: 1 to 1 : 10, especially from 20: 1 to 1 : 1 .
The present invention still further provides a method of controlling weeds at a locus, said method comprising application to the locus of a weed controlling amount of a composition comprising a compound of Formula (I). Moreover, the present invention may further provide a method of selectively controlling weeds at a locus comprising crop plants and weeds, wherein the method comprises application to the locus of a weed controlling amount of a composition according to the present invention. ‘Controlling’ means killing, reducing or retarding growth or preventing or reducing germination. It is noted that the compounds of the present invention show a much-improved selectivity compared to know, structurally similar compounds. Generally the plants to be controlled are unwanted plants (weeds). ‘Locus’ means the area in which the plants are growing or will grow. The application may be applied to the locus pre-emergence and/or postemergence of the crop plant. Some crop plants may be inherently tolerant to herbicidal effects of compounds of Formula (I). Preferred crop plants include maize, wheat, barley and rice.
The rates of application of compounds of Formula I may vary within wide limits and depend on the nature of the soil, the method of application (pre- or postemergence; seed dressing; application to the seed furrow; no tillage application etc.), the crop plant, the weed(s) to be controlled, the prevailing climatic conditions, and other factors governed by the method of application, the time of application and the target crop. The compounds of Formula I according to the invention are generally applied at a rate of from 10 to 2500 g/ha, especially from 25 to 1000 g/ha, more especially from 25 to 250 g/ha.
The application is generally made by spraying the composition, typically by tractor mounted sprayer for large areas, but other methods such as dusting (for powders), drip or drench can also be used.
Crop plants are to be understood as also including those crop plants which have been rendered tolerant to other herbicides or classes of herbicides (e.g. ALS-, GS-, EPSPS-, PPO-, HPPD-, -PDS and ACCase-inhibitors) by conventional methods of breeding or by genetic engineering. An example of a crop that has been rendered tolerant to imidazolinones, e.g. imazamox, by conventional methods of breeding is Clearfield® summer rape (canola). Examples of crops that have been rendered tolerant to herbicides by genetic engineering methods include e.g. glyphosate- and glufosinate-resistant maize varieties commercially available under the trade names RoundupReady® and LibertyLink®.
Crop plants are also to be understood as being those which have been rendered resistant to harmful insects by genetic engineering methods, for example Bt maize (resistant to European corn borer), Bt cotton (resistant to cotton boll weevil) and also Bt potatoes (resistant to Colorado beetle). Examples of Bt maize are the Bt 176 maize hybrids of NK® (Syngenta Seeds). The Bt toxin is a protein that is formed naturally by Bacillus thuringiensis soil bacteria. Examples of toxins, or transgenic plants able to synthesise such toxins, are described in EP-A-451 878, EP-A-374 753, WO 93/07278, WO 95/34656, WO 03/052073 and EP-A-427 529. Examples of transgenic plants comprising one or more genes that code for an insecticidal resistance and express one or more toxins are KnockOut® (maize), Yield Gard® (maize), NuCOTIN33B® (cotton), Bollgard® (cotton), NewLeaf® (potatoes), NatureGard® and Protexcta®. Plant crops or seed material thereof can be both resistant to herbicides and, at the same time, resistant to insect feeding (“stacked” transgenic events). For example, seed can have the ability to express an insecticidal Cry3 protein while at the same time being tolerant to glyphosate.
Crop plants are also to be understood to include those which are obtained by conventional methods of breeding or genetic engineering and contain so-called output traits (e.g. improved storage stability, higher nutritional value and improved flavour).
The compositions can be used to control unwanted plants (collectively, ‘weeds’). The weeds to be controlled may be both monocotyledonous species, for example Agrostis, Alopecurus, Avena, Brachiaria, Bromus, Cenchrus, Cyperus, Digitaria, Echinochloa, Eleusine, Lolium, Monochoria, Rottboellia, Sagittaria, Scirpus, Setaria and Sorghum, and dicotyledonous species, for example Abutilon, Amaranthus, Ambrosia, Chenopodium, Chrysanthemum, Conyza, Galium, Ipomoea, Nasturtium, Sida, Sinapis, Solanum, Stellaria, Veronica, Viola and Xanthium.
In a further aspect of the present invention there is provided the use of a compound of Formula (I) as defined herein as a herbicide.
The compounds of the present invention can be prepared according to the following schemes.
Processes for the preparation of compounds of Formula (I)
Processes for preparation of compounds, e.g. a compound of Formula (I) (which optionally can be an agrochemically acceptable salt thereof), are now described, and form further aspects of the present invention.
Scheme 1:

Compounds of Formula 11 are compounds of Formula I wherein X is O and R1, A1, A2, Q, A3 and R3 are as defined in Formula I above. As shown in Scheme 1 , a compound of Formula 11 can be prepared via alkoxylation reaction which involves reacting compounds of formula III with compounds of formula II, wherein LG1 is a halogen, preferably iodine, bromine or chlorine (or a pseudo-halogen leaving group, such as a (halo)alkyl or phenyl sulfonate ester, e.g. triflate), in the presence of a base, such as sodium hydride or an alkali earth metal hydride, carbonate (e.g. sodium carbonate, potassium carbonate or cesium carbonate) or hydroxide, optionally in the presence of potassium iodide in an inert solvent such as tetrahydrofuran, dioxane, water, N,N- dimethylformamide DMF, N,N-dimethylacetamide, sulfolane or acetonitrile and the like, at temperatures between 0 and 120°C, by known procedures. Compounds of formula III can be prepared by deprotection reaction of compounds of formula IV, wherein PG is an alcohol protecting group like methyl, tert-butyl, amongst others. Such reactions are well known in the literature and known to those skilled in the art. Compounds of formula IV, can be prepared by Suzuki cross-coupling reactions which involves reacting compounds of formula V, wherein X1 is halogen preferably bromide or iodide with compounds of formula Q-Yba, wherein Yba can be a boron-derived functional group, such as for example B(OH)2 or B(ORba)2 wherein Rba can be a Ci- C4alkyl group or the two groups ORba can form together with the boron atom a five membered ring, as for example a pinacol boronic ester. The reaction may be catalysed by a palladium based catalyst, for example tetrakis(triphenyl- phosphine)palladium(O), (1 ,1'bis(diphenylphosphino)ferrocene)dichloro-palladium- dichloromethane (1 :1 complex) or chloro(2-dicyclohexylphosphino-2',4',6'-triisopropyl- 1 ,1'-biphenyl)[2-(2'-amino-1 ,T-biphenyl)]palladium(ll) (XPhos palladacycle), in presence of a base, like sodium carbonate, tripotassium phosphate or cesium
fluoride, in a solvent or a solvent mixture, like, for example dioxane, acetonitrile, N,N- dimethyl-formamide, a mixture of 1 ,2-dimethoxyethane and water or of dioxane/water, or of toluene/water, preferably under inert atmosphere. The reaction temperature can preferentially range from room temperature to the boiling point of the reaction mixture, or the reaction may be performed under microwave irradiation. Such Suzuki reactions are well known to those skilled in the art.

Compounds of Formula 12 are compounds of formula I wherein X is CH2 and R1, A1, A2, A3, Q, and R3 are as defined in formula I above (scheme 2). Compounds of formula I2 can be prepared by decarboxylation of compounds of formula VI, wherein R11 is Ci-Cealkyl or phenyl (scheme 2). Such decarboxylation reactions can be carried out under thermal conditions for example heating at 100 to 180 °C in solvent such as isopropanol/water or under basic conditions such as using sodium hydroxide or potassium hydroxide.
Compounds of Formula VI, can be prepared by reacting compounds of formula VII with compounds of Formula VIII, wherein LG2 is a halogen, preferably iodine, bromine or chlorine (or a pseudo-halogen leaving group, such as a (halo)alkyl or phenyl sulfonate ester, e.g. triflate), in the presence of a base, such as sodium hydride or an alkali earth metal hydride, carbonate (e.g. sodium carbonate, potassium carbonate or cesium carbonate) or hydroxide, optionally in the presence of potassium iodide in an inert solvent such as tetrahydrofuran, dioxane, water, N,N-dimethylformamide DMF, N,N-dimethylacetamide, sulfolane or acetonitrile and the like, at temperatures between 0 and 120°C.
Scheme 3:

Alternatively compounds of Formula 12 can be prepared from compounds of formula IX via deoxygenation or reduction of alcohol. Reduction of such alcohols are well described in literature and can be carried out using reducing agent such as LiAIF , DIBAL-H, or using triphenyl phosphine in the presence of iodine and imidazole or using triethyl silane in the presence of trifluoroacetic acid. Compounds of formula IX can be prepared by reacting compounds of Formula XI, wherein X11 is a halogen preferably bromine or iodine with an organometallic reagent such as BuLi or isopropylmagnesium chloride/LiCI complex amongst other metallating reagents to form an intermediate Xia, wherein M(Ln)p is a corresponding metal from the organometallic reagent such as lithium or magnesium and (Ln)p is its optionally substituted group like chloro and then subsequently reacting with compounds of Formula X.
 Alternatively compounds of formula 12 can be prepared following scheme 4. In scheme 4 compounds of formula 12 can be prepared by reacting compounds of formula IXab, wherein X22 is a halogen, preferably iodine, bromine or chlorine (or a pseudo-halogen leaving group, such as a (halo)alkyl or phenyl sulfonate ester, e.g. triflate), and compounds of formula Va under Suzuki cross-coupling reaction under conditions analogous to procedure as described in scheme 1 for the conversion of compounds of formula V to compounds of formula IV. Compounds of formula IXab can be prepared from compounds of formula Xab via analogous procedure as described in scheme 3 for the conversion of compounds of formula X to compounds of formula 12.
Alternatively compounds of formula 12 can be prepared following scheme 4. In scheme 4 compounds of formula 12 can be prepared by reacting compounds of formula IXab, wherein X22 is a halogen, preferably iodine, bromine or chlorine (or a pseudo-halogen leaving group, such as a (halo)alkyl or phenyl sulfonate ester, e.g. triflate), and compounds of formula Va under Suzuki cross-coupling reaction under conditions analogous to procedure as described in scheme 1 for the conversion of compounds of formula V to compounds of formula IV. Compounds of formula IXab can be prepared from compounds of formula Xab via analogous procedure as described in scheme 3 for the conversion of compounds of formula X to compounds of formula 12.
Alternatively compounds of formula I wherein A1 and A2 are nitrogen atoms can be prepared following scheme 5. Compounds of formula I3 are compounds of formula I wherein A1 and A2 are nitrogen atoms.
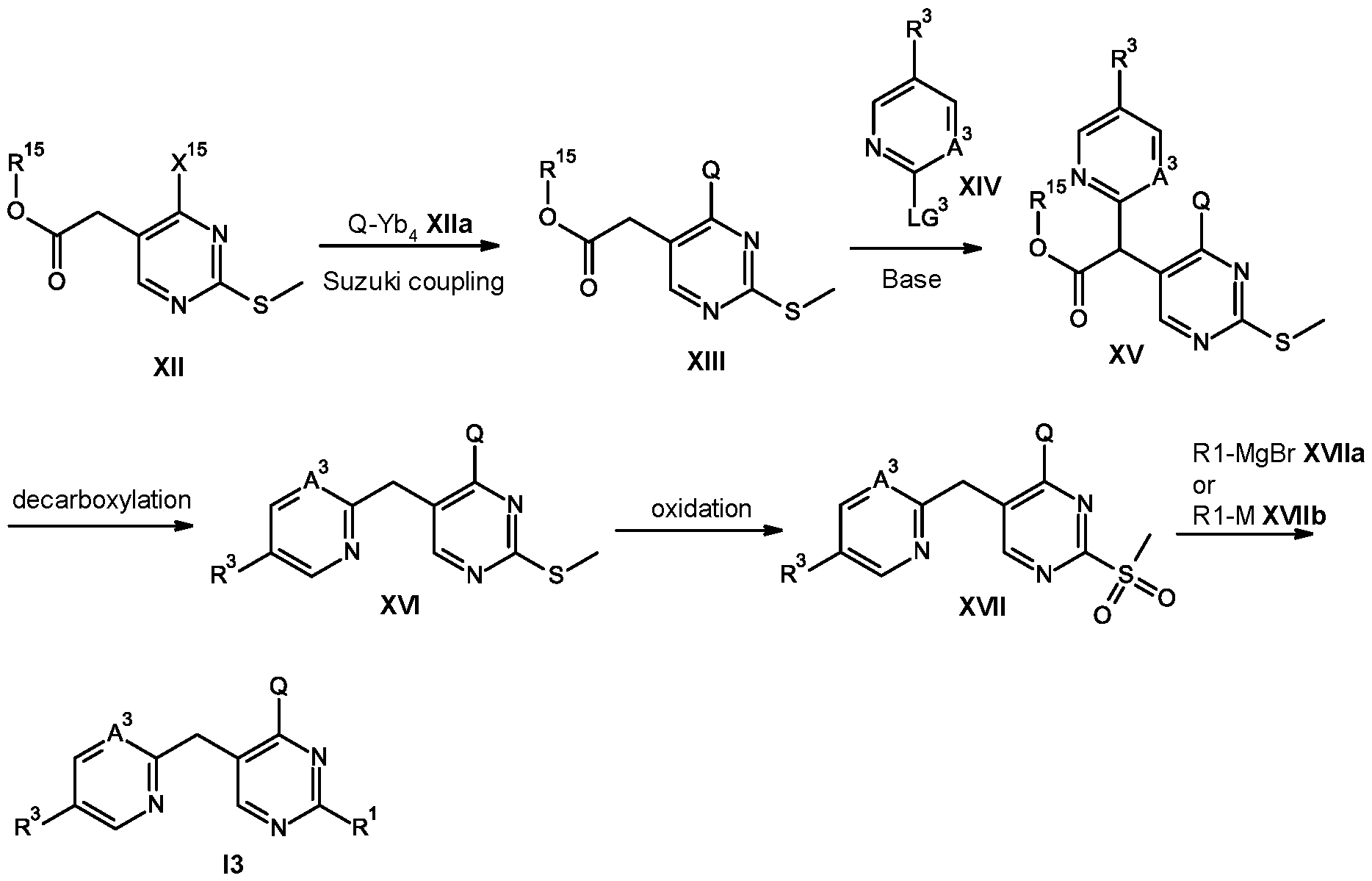
Compounds of formula 13 can be prepared by nucleophilic substitution reaction which involves reacting compounds of formula XVII with compounds of formula XVIIa or compounds of formula XVIIb wherein M is a metal such as sodium, lithium or potassium. Compounds of formula XVII can be prepared from compounds of formula XVI via oxidation reaction using oxidizing reagents such as hydrogen peroxide or meta-chloroperoxybenzoic acid. Compounds of formula XVI can be prepared by decarboxylation reaction from compounds of formula XV analogous to procedure as described in scheme 2 for synthesis of compounds of formula 12 from compounds of formula VI. Compounds of formula XV can be prepared by reaction of compounds of formula XIII, wherein R15 is Ci-Cealklyl or phenyl and compounds of formula XIV, wherein LG3 is a leaving group for e.g. Cl, Br, SO2CH3 or SChPh following procedure as described in scheme 2 for the synthesis of compounds of formula VI by the reaction of compounds of formula VII and VIII. Compounds of formula XIII can be prepared by the reaction of compounds of formula XII, wherein X15 is halogen such as Cl or Br and compounds of formula Xlla, wherein Yb4 can be a boron-derived functional group, such as for example B(OH)2 or B(ORb4)2 wherein Rb4 can be a C1- C4alkyl group or the two groups ORb4 can form together with the boron atom a five membered ring, as for example a pinacol boronic ester following procedure as
described in scheme 1 for the synthesis of compounds of formula IV from compounds of formula V and compounds of formula Va.
Alternatively compounds of formula I3 can be prepared following scheme 6.
Scheme 6:
 halogenation or Q-Yb4 Xlla tosylation or mesylation
halogenation or Q-Yb4 Xlla tosylation or mesylation
 Suzuki coupling
Suzuki coupling

XVIII

In Scheme 6 compounds of formula 13 can be prepared by reacting compounds of formula XVIII, wherein X12 is halogen such as Cl, Br or tosyl or mesyl functional group and compounds of formula Xlla following procedure as described in scheme 5 for the synthesis of compounds of formula XIII from compounds of formula XII and compounds of formula Xlla. Compounds of formula XVIII, wherein X12 is halogen can be prepared from compounds of formula XIX by halogenation reaction using reagents such as POCh or POBra. Alternatively compounds of formula XVIII, wherein X12 is tosyl or mesyl can be prepared by the reaction of compounds of formula XIX with reagents such as p-toluenesulfonyl chloride or methanesulfonyl chloride respectively in the presence of base such as triethyl amine or pyridine and optionally reaction can be carried out in the presence of 4-dimethylaminopyridine. Such reactions are well known in the literature and known to those skilled in the state of art. Compounds of formula XIX can be prepared by the condensation reaction of amidine XXI and compounds of formula XX. Such reactions are known in the literature and for example described in Tetrahedron 2017, 73, 27-28, 3939-3948. Compounds of formula XX can be prepared by the reaction of compounds of formula XXII with compounds of formula XXIII, wherein R41 is C1-C4alkyl in the presence of base such as sodium hydride, potassium hydride amongst others. Such reactions are known in the literature and described for example in Tetrahedron 2017, 73, 27-28, 3939-3948.
Compounds of formula I2 can also be alternatively prepared following scheme 7.
Scheme 7:
 In scheme 7 compounds of formula 12 can be prepared by reacting compounds of formula XXIV and compounds of formula Va wherein Yb3 can be a boron-derived functional group, such as for example B(OH)2 or B(ORba)2 wherein Rb3 can be a Ci- C4alkyl group or the two groups ORb3 can form together with the boron atom a five membered ring, as for example a pinacol boronic ester following procedure as described in scheme 1 for the synthesis of compounds of formula IV from compounds of formula V and compounds of formula Va. Compounds of formula XXIV can be prepared from compounds of formula XXVI via two step procedure which involves base mediated arylation and decarboxylation reaction as described in scheme 2 for the conversion of compounds of formula VII to compounds of formula I2.
In scheme 7 compounds of formula 12 can be prepared by reacting compounds of formula XXIV and compounds of formula Va wherein Yb3 can be a boron-derived functional group, such as for example B(OH)2 or B(ORba)2 wherein Rb3 can be a Ci- C4alkyl group or the two groups ORb3 can form together with the boron atom a five membered ring, as for example a pinacol boronic ester following procedure as described in scheme 1 for the synthesis of compounds of formula IV from compounds of formula V and compounds of formula Va. Compounds of formula XXIV can be prepared from compounds of formula XXVI via two step procedure which involves base mediated arylation and decarboxylation reaction as described in scheme 2 for the conversion of compounds of formula VII to compounds of formula I2.
The following non-limiting examples provide specific synthesis methods for representative compounds of the present invention, as referred to in the Table below.
Example 1 : 5-(5-chloropyrimidin-2-yl)oxy-2-(trifluoromethyl)-4-[4-
(trifluoromethyl)phenyl]-pyrimidine (1.007)

Step 1 : Preparation of 5-methoxy-2-(trifluoromethyl)-4-[4-
(trifluoromethyl)phenyl]-pyrimidine (1-1)

A mixture of 4-chloro-5-methoxy-2-(trifluoromethyl)pyrimidine (155 mg, 0.72 mmol), 4-(trifluoromethyl)phenylboronic acid (253 mg, 1.27 mmol) and (2- Dicyclohexylphosphino-2',4',6'-triisopropyl-1 , 1'-biphenyl)[2-(2'-amino-1 , 1'-biphenyl)] palladium^ I) methanesulfonate (30 mg, 0.034 mmol) in a 50 mL round bottomed flask was placed under an atmosphere of nitrogen and treated with acetonitrile (2.8 mL) and de-gassed potassium phosphate tribasic (1.0 M in water) (1.4 mL). The reaction mixture was allowed to stir for another 15 min at room temperature. The reaction mixture was warmed to 40 °C and was allowed to stir for a further 1 h. The reaction mixture was allowed to cool to room temperature before being diluted with brine (20 mL) and extracted with ethyl acetate (2 x 15 mL). The combined organics were concentrated in vacuo. The residues were loaded onto celite and subjected to
silica gel column chromatography using 0-40% ethyl acetate in cyclohexane. The fractions forming the major peak of interest were combined and concentrated in vacuo, yielding 5-methoxy-2-(trifluoromethyl)-4-[4-(trifluoromethyl)phenyl]-pyrimidine 1-1 (237 mg, 95%) as a beige solid. 1H NMR (400 MHz, CDCh) 6 = 8.59 (s, 1H), 8.29 (d, 2H), 7.76 (d, 2H), 4.11 (s, 3H)
Step 2: Preparation of 2-(trifluoromethyl)-4-[4-(trifluoromethyl) phenyl]pyrimidin-5-ol (I-2)
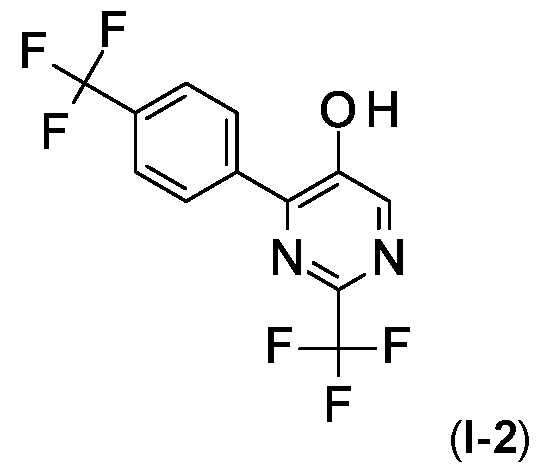
A solution of 5-methoxy-2-(trifluoromethyl)-4-[4-(trifluoromethyl)phenyl]pyrimidine 1-1 (61 mg, 0.18 mmol) in ZV-Methyl-2-pyrrolidone (0.5 mL) in a 50 mL round bottomed flask was treated with 1 -dodecanethiol (110 pL, 0.450 mmol) and sodium hydroxide (50 mass%) in water (25 pL, 0.485 mmol). Precipitate was observed on addition of the base. The reaction mixture was warmed to 100 °C and was allowed to stir for 30 minutes. The reaction mixture was allowed to cool to room temperature. The mixture was quenched with aqueous ammonium chloride (10 mL), diluted with water (10 mL) and was extracted with ethyl acetate (2 x 10 mL). The combined organics were washed with brine (10 mL) and concentrated in vacuo. The residues were loaded onto celite and subjected to column chromatography using 0-100% ethyl acetate in cyclohexane as eluent. The fractions forming the major peak of interest were combined and concentrated in vacuo, yielding 2-(trifluoromethyl)-4-[4- (trifluoromethyl)phenyl]pyrimidin-5-ol I-2 (54 mg, 87%) as an off-white solid. 1H NMR (400 MHz, CDCI3) 6 = 8.57 (s, 1 H), 8.29 (d, 2H), 7.80 (d, 2H), 6.49 (br s, 1 H).
Step 3: Preparation of 5-(5-chloropyrimidin-2-yl)oxy-2-(trifluoromethyl)-4-[4- (trifluoromethyl)phenyl]-pyrimidine (1.007)

(1.007)
A mixture of 2-(trifluoromethyl)-4-[4-(trifluoromethyl)phenyl]pyrimidin-5-ol 1-2 (54 mg, 0.157 mmol), 2,5-dichloropyrimidine (41 mg, 0.27 mmol) and potassium carbonate (44 mg, 0.31 mmol) in a 50 mL round bottomed flask was treated with acetonitrile (1.0 mL). The reaction mixture was warmed to 90 °C and was allowed to stir for 30 minutes. The reaction mixture was treated with sulfolane (1.0 mL) and was allowed to stir for an additional 4 h. The reaction mixture was allowed to cool to room temperature before being diluted with water (10 mL) and brine (10 mL) and extracted with tert-butylmethyl ether (2 x 15 mL). The combined organics were concentrated. The residues were loaded onto celite and subjected to silica gel column chromatography using 0-40% ethyl acetate in cyclohexane as eluent. The fractions forming the major peak of interest were combined and concentrated in vacuo, yielding 5-(5-chloropyrimidin-2-yl)oxy-2-(trifluoromethyl)-4-[4-(trifluoromethyl) phenyl]pyrimidine (1.007). 1H NMR (400 MHz, CDCI3) 5 = 8.85 (s, 1 H), 8.44 (s, 2H), 8.23 (d, 2H), 7.72 (d, 2H).
Example 2: 5-chloro-2-[[2-(3,4-difluorophenyl)-5-fluoro-3- pyridyl]methyl]pyrimidine (1.015)

(1.015)
Step 1 : Preparation of (2-chloro-5-fluoro-3-pyridyl)-(5-chloropyrimidin-2- yljmethanol (I-3)

(1-3)
A solution of 5-chloro-2-iodopyrimidine (2.525 g, 10.50 mmol) in toluene (35 mL) in a 250 mL round bottomed flask was placed under an atmosphere of nitrogen, cooled to -78°C and was treated with n-butyllithium (2.5 M in hexanes, 4.5 mL, 11 mmol). The resulting mixture was stirred for 10 minutes before being treated with a solution of 2- chloro-5-fluoronicotinaldehyde (1.126 g, 7.058 mmol) in toluene (23 mL). The resulting mixture was stirred for a further 30 minutes, quenched with 0.5 M hydrochloric acid and extracted with ethyl acetate. The combined organics were concentrated and subjected to silica gel column chromatography using 0-100% ethyl acetate in cyclohexane. The fractions forming the major peak of interest were combined and concentrated in vacuo, yielding (2-chloro-5-fluoro-3-pyridyl)-(5- chloropyrimidin-2-yl)methanol I-3 (1.062 g, 52%) as a yellow solid. 1H NMR (400 MHz, CDCI3) 6 = 8.71 (s, 2H), 8.22 (d, 1 H), 7.56 (dd, 1 H), 6.20 (d, 1 H), 4.52 (d, 1 H).
Step 2: Preparation of 5-chloro-2-[(2-chloro-5-fluoro-3- pyridyl)methyl]pyrimidine (I-4)

(I-4)
A solution of (2-chloro-5-fluoro-3-pyridyl)-(5-chloropyrimidin-2-yl)methanol I-3 (1.062 g, 3.68 mmol), imidazole (389 mg, 5.71 mmol) and triphenylphosphine (1.968 g, 7.35 mmol) in tetra hydrofuran (21 mL) in a 50 mL round bottomed flask was placed under an atmosphere of nitrogen and treated dropwise with a solution of iodine (1.217 g, 4.80 mmol) in tetra hydrofuran (5.5 mL). The resulting mixture was allowed to stir for 5
minutes, quenched with an aqueous sodium thiosulphate solution and extracted with ethyl acetate. The combined organics were concentrated and subjected to silica gel column chromatography using 0-40% ethyl acetate in cyclohexane. The fractions forming the major peak of interest were combined and concentrated in vacuo, yielding 5-chloro-2-[(2-chloro-5-fluoro-3-pyridyl)methyl]pyrimidine 1-4 (918 mg, 87%) as a light brown oil. 1H NMR (400 MHz, CDCI3) 5 = 8.64 (s, 2H), 8.20 (d, 1H), 7.44 (dd, 1H), 4.41 (s, 2H).
Step 3: Preparation of 5-chloro-2-[[2-(3,4-difluorophenyl)-5-fluoro-3- pyridyl]methyl]pyrimidine (1.015)

(1.015)
A mixture of 5-chloro-2-[(2-chloro-5-fluoro-3-pyridyl)methyl]pyrimidine I-4 (154 mg, 0.537 mmol), 3,4-difluorophenylboronic acid (185 mg, 1.11 mmol) and 1,1'- bis(diphenylphosphino)ferrocene-palladium(ll)dichloride dichloromethane complex (47 mg, 0.056 mmol) in a 2-5 mL microwave vial was placed under an atmosphere of nitrogen and treated with 2-methyltetrahydrofuran (2.0 mL) and potassium phosphate tribasic (1.0 M in water, 1.3 mL, 1.3 mmol). The resulting mixture was irradiated under microwave radiation to 100°C for 30 minutes, diluted with water and extracted with ethyl acetate. The combined organics were concentrated and subjected to silica gel column chromatography using 0-30% ethyl acetate in cyclohexane. The fractions forming the major peak of interest were combined and concentrated in vacuo, yielding 5-chloro-2-[[2-(3,4-difluorophenyl)-5-fluoro-3-pyridyl]methyl]pyrimidine 1.015. 1H NMR (400 MHz, CDCI3) 6 = 8.63 (s, 2H), 8.44 (d, 1 H), 7.50 - 7.40 (m, 2H), 7.32 - 7.27 (m, 1 H), 7.25 - 7.17 (m, 1 H), 4.31 (s, 2H).
Example 3: 5-[(5-chloropyrimidin-2-yl)methyl]-2-(difluoromethoxy)-4-(3,4- difluorophenyljpyrimidine (1.021)

(1.021)
Step 1: Preparation of ethyl 2-[4-(3,4-difluorophenyl)-2-methylsulfanyl- pyrimidin-5-yl]acetate (I-5)

(I-5) A mixture of ethyl 2-(4-chloro-2-methylsulfanyl-pyrimidin-5-yl)acetate (4.012 g, 16.26 mmol), 3,4-difluorophenylboronic acid (3.810 g, 24.13 mmol) and XPhos Pd G3 (422 mg, 0.499 mmol) was treated with 2-methyltetrahydrofuran (32 mL) and a solution of potassium phosphate in water (1.0 M, 24 mL, 24.0 mmol). The resulting mixture was stirred at 80°C for 2.5 hours, charged with additional XPhos Pd G3 (423 mg, 0.500 mmol) and was stirred for an additional hour. The mixture was cooled, diluted with water and extracted with ethyl acetate. The combined organics were concentrated and subjected to silica gel column chromatography using 0-30% ethyl acetate in cyclohexane. The fractions forming the major peak of interest were combined and concentrated in vacuo, yielding ethyl 2-[4-(3,4-difluorophenyl)-2-methylsulfanyl- pyrimidin-5-yl]acetate 1-5 (3.617 g, 69%) as a light yellow solid. 1H NMR (400 MHz, CDCI3) 6 = 8.51 (s, 1 H), 7.50 - 7.43 (m, 1 H), 7.36 - 7.30 (m, 1H), 7.30 - 7.22 (m, 1H), 4.15 (q, 2H), 3.60 (s, 2H), 2.59 (s, 3H), 1.24 (t, 3H).
Step 2: Preparation of ethyl 2-(5-chloropyrimidin-2-yl)-2-[4-(3,4-difluorophenyl)- 2-methylsulfanyl-pyrimidin-5-yl]acetate (I-6)

A solution of ethyl 2-[4-(3,4-difluorophenyl)-2-methylsulfanyl-pyrimidin-5-yl]acetate I-5 (3.617 g, 11.15 mmol) and 2,5-dichloropyrimidine (5.034 g, 33.79 mmol) in DMSO (20 mL) was treated with potassium phosphate (9.533 g, 44.91 mmol) and the resulting mixture was stirred at 70 °C for an hour. The mixture was cooled, treated with 1.0 M hydrochloric acid and extracted with terf-butyl methyl ether. The combined organics were concentrated and subjected to silica gel column chromatography using 0-40% ethyl acetate in cyclohexane. The fractions forming the major peak of interest were combined and concentrated in vacuo, yielding ethyl 2-(5-chloropyrimidin-2-yl)-2- [4-(3,4-difluorophenyl)-2-methylsulfanyl-pyrimidin-5-yl]acetate I-6 (4.682 g, 87%) as an orange oil. 1H NMR (400 MHz, CDCI3) 6 = 8.86 (s, 1 H), 8.67 (s, 2H), 7.58 - 7.52 (m, 1 H), 7.43 - 7.38 (m, 1H), 7.32 - 7.23 (m, 1H), 5.47 (s, 1 H), 4.26 - 4.18 (m, 2H), 2.58 (s, 3H), 1.23 (t, 3H).
Step 3: Preparation of 5-[(5-chloropyrimidin-2-yl)methyl]-4-(3,4-difluorophenyl)- 2-methylsulfanyl-pyrimidine (1.029)
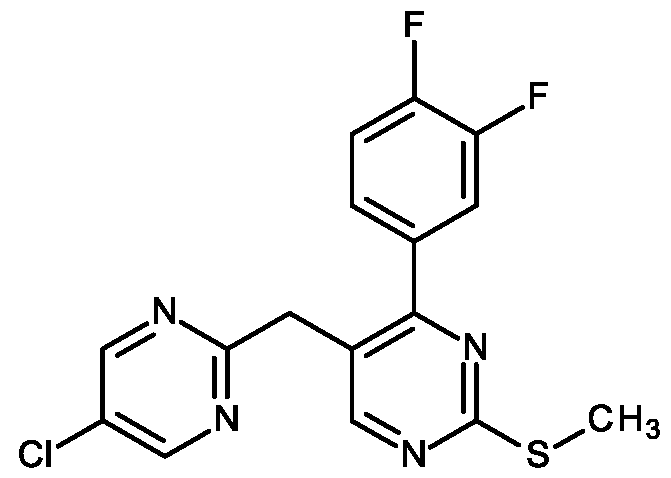
(1.029)
A solution of ethyl 2-(5-chloropyrimidin-2-yl)-2-[4-(3,4-difluorophenyl)-2- methylsulfanyl-pyrimidin-5-yl]acetate I-6 (4.682 g, 9.65 mmol) in methanol (48 mL) was treated with aqueous sodium hydroxide (2.0 M, 35 mL, 70 mmol) and was allowed to stir at room temperature for 1.5 hours. The mixture was acidified with 2M hydrochloric acid and extracted with ethyl acetate. The combined organics were dried and concentrated in vacuo, yielding 5-[(5-chloropyrimidin-2-yl)methyl]-4-(3,4- difluorophenyl)-2-methylsulfanyl-pynmidine 1.029 (3.453 g, 93%) as a light yellow
solid. 1H NMR (400 MHz, CDCh) 6 = 8.61 (s, 2H), 8.56 (s, 1 H), 7.60 - 7.54 (m, 1 H), 7.44 - 7.39 (m, 1 H), 7.26 - 7.18 (m, 1 H), 4.28 (s, 2H), 2.59 (s, 3H).
Step 4: Preparation of 5-[(5-chloropyrimidin-2-yl)methyl]-4-(3,4-difluorophenyl)- 2-methylsulfonyl-pyrimidine (1.028)

(1.028)
A solution of 5-[(5-chloropyrimidin-2-yl)methyl]-4-(3,4-difluorophenyl)-2- methylsulfanyl-pyrimidine 1.029 (3.453 g, 9.47 mmol) in ethyl acetate (28 mL) was cooled over ice and was treated with 3-chloroperoxybenzoic acid (77%, 4.667 g, 20.82 mmol). The resulting mixture was allowed to stir at 0°C for 2 hours. The mixture was diluted with ethyl acetate and quenched with aqueous sodium thiosulphate. The organics were washed with aqueous sodium bicarbonate and water before being concentrated in vacuo and subjected to silica gel column chromatography using 30- 100% ethyl acetate in cyclohexane. The fractions forming the major peak of interest were combined and concentrated in vacuo, yielding 5-[(5-chloropyrimidin-2- yl)methyl]-4-(3,4-difluorophenyl)-2-methylsulfonyl-pyrimidine 1.028 (3.295 g, 83%) as a cream coloured solid. 1H NMR (400 MHz, CDCI3) 6 = 8.96 (s, 1 H), 8.64 (s, 2H), 7.68 - 7.61 (m, 1 H), 7.52 - 7.45 (m, 1 H), 7.32 - 7.24 (m, 1 H), 4.47 (s, 2H), 3.39 (s, 3H).
Step 5: Preparation of 5-[(5-chloropyrimidin-2-yl)methyl]-6-(3,4-difluorophenyl)- 1H-pyrimidin-2-one (I-7)
 A solution of 5-[(5-chloropyrimidin-2-yl)methyl]-4-(3,4-difluorophenyl)-2- methylsulfonyl-pyrimidine 1.028 (760 mg, 1.82 mmol) in THF (9 mL) was treated with aqueous sodium hydroxide (2.0 M, 35 mL, 70 mmol) and was allowed to stir at room temperature for 2.5 hours. The mixture was acidified with 2M hydrochloric acid and extracted with ethyl acetate. The combined organics were dried and concentrated in vacuo. The resulting solids were triturated from tert-butyl methyl ether, yielding 5-[(5- chloropyrimidin-2-yl)methyl]-6-(3,4-difluorophenyl)-1H-pyrimidin-2-one I-7 (560 g, 89%) as a beige solid. 1H NMR (400 MHz, DMSO-d6) 5 = 12.10 (br s, 1H), 8.77 (s, 2H), 8.17 (br s, 1 H), 7.58 - 7.49 (m, 1 H), 7.48 - 7.38 (m, 1 H), 7.29 - 7.21 (m, 1H), 4.12 (s, 2H).
A solution of 5-[(5-chloropyrimidin-2-yl)methyl]-4-(3,4-difluorophenyl)-2- methylsulfonyl-pyrimidine 1.028 (760 mg, 1.82 mmol) in THF (9 mL) was treated with aqueous sodium hydroxide (2.0 M, 35 mL, 70 mmol) and was allowed to stir at room temperature for 2.5 hours. The mixture was acidified with 2M hydrochloric acid and extracted with ethyl acetate. The combined organics were dried and concentrated in vacuo. The resulting solids were triturated from tert-butyl methyl ether, yielding 5-[(5- chloropyrimidin-2-yl)methyl]-6-(3,4-difluorophenyl)-1H-pyrimidin-2-one I-7 (560 g, 89%) as a beige solid. 1H NMR (400 MHz, DMSO-d6) 5 = 12.10 (br s, 1H), 8.77 (s, 2H), 8.17 (br s, 1 H), 7.58 - 7.49 (m, 1 H), 7.48 - 7.38 (m, 1 H), 7.29 - 7.21 (m, 1H), 4.12 (s, 2H).
Step 6: Preparation of 5-[(5-chloropyrimidin-2-yl)methyl]-2-(difluoromethoxy)-4- (3,4-difluorophenyl)pyrimidine (1.021 )

A solution of 5-[(5-chloropyrimidin-2-yl)methyl]-4-(3,4-difluorophenyl)pyrimidin-2-ol I-7 (159 mg, 0.475 mmol) in DMF (2.4 mL) was treated with potassium carbonate (149 mg, 1.08 mmol) and sodium chlorodifluoroacetate (121 mg, 0.794 mmol). The resulting mixture was stirred at 70 °C for 45 minutes, charged with additional sodium chlorodifluoroacetate (114 mg, 0.748 mmol) and stirred for a further 90 minutes. The mixture was cooled, diluted with water and extracted with tert-butyl methyl ether. The combined organics were concentrated and subjected to silica gel column chromatography using 0-100% ethyl acetate in cyclohexane. The product-rich fractions were concentrated and subjected to reverse-phase chromatography (MeCN in water, both with 0.1% formic acid). The fractions forming the major peak of interest were combined and concentrated in vacuo, yielding 5-[(5-chloropyrimidin-2- yl)methyl]-2-(difluoromethoxy)-4-(3,4-difluorophenyl)pyrimidine 1.021. 1 H NMR (400 MHz, chloroform) 5 = 8.61 (s, 2H), 7.93 (s, 1H), 7.59 (t, 1H), 7.56 - 7.49 (m, 1H), 7.42 - 7.36 (m, 1 H), 7.25 - 7.17 (m, 1 H), 4.18 (s, 2H).
Example 4: 5-[(5-chloropyrimidin-2-yl)methyl]-4-(3,4-difluorophenyl)pyrimidine- 2-carbonitrile (1.025)

(1.025)
Step 1 : Preparation of 5-[(5-chloropyrimidin-2-yl)methyl]-4-(3,4- difluorophenyl)pyrimidine-2-carbonitrile (1.025)

(1.025) A solution of 5-[(5-chloropyrimidin-2-yl)methyl]-4-(3,4-difluorophenyl)-2- methylsulfonyl-pyrimidine 1.028 (53 mg, 0.127 mmol) and potassium cyanide (18 mg, 0.263 mmol) in MeCN (0.6 mL) and DMSO (10 pL) was stirred at room temperature for 90 hours. The mixture was diluted with aqueous sodium and extracted with ethyl acetate. The combined organics were concentrated and subjected to silica gel column chromatography using 0-80% ethyl acetate in cyclohexane. The fractions forming the major peak of interest were combined and concentrated in vacuo, yielding 5-[(5-chloropyrimidin-2-yl)methyl]-4-(3,4-difluorophenyl)pyrimidine-2- carbonitrile 1.025. 1H NMR (400 MHz, CDCI3) 6 = 8.88 (s, 1 H), 8.64 (s, 2H), 7.66 - 7.59 (m, 1 H), 7.49 - 7.43 (m, 1 H), 7.33 - 7.27 (m, 1 H), 4.44 (s, 2H). Example 5: 5-[(5-chloropyrimidin-2-yl)methyl]-2-cyclopropyl-4-(3,4- difluorophenyljpyrimidine (1.030)

(1.030)
Step 1 : Preparation of 5-[(5-chloropyrimidin-2-yl)methyl]-2-cyclopropyl-4-(3,4- difluorophenyljpyrimidine (1.030)

(1.030)
A solution of 5-[(5-chloropyrimidin-2-yl)methyl]-4-(3,4-difluorophenyl)-2- methylsulfonyl-pyrimidine 1.028 (47 mg, 0.113 mmol) in THF (1.1 mL) was cooled to 0°C and was treated dropwise with solution of cyclopropylmagnesium bromide in THF (0.5 M, 0.3 mL, 0.15 mmol). The mixture was stirred for 5 minutes, treated with further cyclopropylmagnesium bromide in THF (0.5 M, 0.08 mL, 0.04 mmol) and stirred for a further 45 minutes. The mixture was quenched with water and extracted with ethyl acetate. The combined organics were concentrated and subjected to reverse-phase column chromatography using MeCN in water, both with 0.1% formic acid. The fractions forming the major peak of interest were combined and concentrated in vacuo, yielding 5-[(5-chloropyrimidin-2-yl)methyl]-2-cyclopropyl-4- (3,4-difluorophenyl)pyrimidine 1.030. 1H NMR (400 MHz, CDCI3) 5 = 8.61 (s, 2H), 8.58 (s, 1 H), 7.57 - 7.50 (m, 1 H), 7.41 - 7.35 (m, 1H), 7.25 - 7.17 (m, 1H), 4.28 (s, 2H), 2.32 - 2.24 (m, 1 H), 1.20 - 1.04 (m, 4H).
Example 6: 5-chloro-2-[[2-(6-chloro-5-fluoro-3-pyridyl)-6-(trifluoromethyl)-3- pyridyl]methyl]pyrimidine (1.016)

(1.016)
Step 1 : Preparation of diethyl 2-(5-chloropyrimidin-2-yl)propanedioate (I-8)
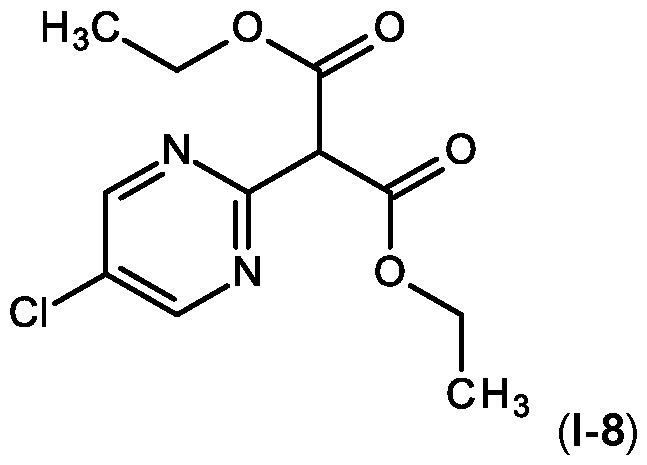
A mixture of 2,5-dichloropyrimidine (3.0 g, 20.20 mmol) and potassium phosphate tribasic (9.59 g, 44.3 mmol) in a 250 mL round bottomed flask was placed under an atmosphere of nitrogen and 33 mL dimethyl sulfoxide (DMSO) was added followed by diethyl malonate (5.1 g, 31.0 mmol). The resulting mixture was warmed to 80 °C and was allowed to stir for 12 h. The reaction mixture was allowed to cool to room temperature and diluted with water (150 mL) and extracted with ethyl acetate (3 X 100 mL). The combined organics were washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated in vacuo. The crude product was purified by silica gel column chromatography eluting with 0-15% ethyl acetate in cyclohexane.
Upon concentration of the fractions, diethyl 2-(5-chloropyrimidin-2-yl)propanedioate I- 8 was obtained as light yellow oil (5.2 g, 90%). 1H NMR (400 MHz, CDCh) 5 = 8.71 (s, 2 H), 5.09 (s, 1 H), 4.29 (q, 4 H), 1.29 (t, 6 H).
Step 2: Preparation of ethyl 2-(5-chloropyrimidin-2-yl)acetate (I-9)

To a solution of diethyl 2-(5-chloropyrimidin-2-yl)propanedioate (1-8, 5.2 g) in DMSO (52 mL) were added sodium chloride (1.4 g, 58.44 mmol) and water (1.0 mL) and the reaction mixture was heated at 150 °C for 3 h. The reaction mixture was allowed to cool to room temperature before being diluted with water (100 mL) and extracted with ethyl acetate (2 x 100 mL). The combined organics were concentrated in vacuo. The crude product was purified by silica gel column chromatography using 0-15% ethyl acetate in cyclohexane as eluent. Upon concentration, ethyl 2-(5-chloropyrimidin-2- yl)acetate I-9 was obtained as colourless oil. 1H NMR (400 MHz, CDCh) 6 = 8.68 (s, 2 H), 4.22 (q, 2 H), 4.02 (s, 2 H), 1.28 (t, 3 H).
Step 3: Preparation of ethyl 2-(5-chloropyrimidin-2-yl)-2-[2-chloro-6- (trifluoromethyl)-3-pyridyl]acetate (1-10)

(1-10)
To a solution of ethyl 2-(5-chloropyrimidin-2-yl)acetate (I-9, 2.1 g, 10.5 mmol) in DMSO (17.5 mL) were added 2-chloro-3-fluoro-6-(trifluoromethyl)pyridine (1.75 g, 8.77 mmol) followed by potassium phosphate tribasic (3.76 g, 17.54 mmol) and the reaction mixture was stirred at room temperature for 16 h. The reaction mixture was quenched by adding 5 mL of saturated ammonium chloride solution and diluted with 100 mL water. The aqueous phase was extracted with ethyl acetate (3 X 100 mL) and the combined organic layer was washed with brine and concentrated in vacuo. The crude product was purified by silica gel column chromatography using 0-20%
ethyl acetate in cyclohexane as eluent. Upon concentration, ethyl 2-(5- chloropyrimidin-2-yl)-2-[2-chloro-6-(trifluoromethyl)-3-pyridyl]acetate 1-10 was obtained as yellowish gummy mass (1.6 g, 43%). 1H NMR (400 MHz, CDCh) 6 = 8.70 (s, 2 H), 8.04 (d, 1 H), 7.64 (d, 1 H), 5.90 (s, 1 H), 4.28 (q, 2 H), 1.27 (t, 3 H)
Step 4: Preparation of 5-chloro-2-[[2-chloro-6-(trifluoromethyl)-3- pyridyl]methyl]pyrimidine (1-11)

(1-11)
Ethyl 2-(5-chloropyrimidin-2-yl)-2-[2-chloro-6-(trifluoromethyl)-3-pyridyl]acetate 1-10 (310 mg, 0.81 mmol) was dissolved in 3.1 mL propanenitrile in a teflon coated vial. To this, 0.31 mL of water were added and the vial was placed inside a pressure reactor and heated at 175 °C for 8 h. After this time, the reaction mixture was cooled to room temperature and diluted with 5 mL of water. The aqueous layer was extracted with ethyl acetate (3X15 mL) and the combined organic layer was evaporated in vacuo to get 5-chloro-2-[[2-chloro-6-(trifluoromethyl)-3- pyridyl]methyl]pyrimidine 1-11 as pale brown liquid (200 mg, 72%). 1H NMR (400 MHz, CDCh) 6 = 8.64 (s, 2 H), 7.86 (d, 1 H), 7.63 (d, 1 H), 4.50 (s, 2 H).
Step 5: Preparation of 5-chloro-2-[[2-(6-chloro-5-fluoro-3-pyridyl)-6-
(trifluoromethyl)-3-pyridyl]methyl]pyrimidine (1.016)

(1.016)
In a 25 mL two neck flask, 5-chloro-2-[[2-chloro-6-(trifluoromethyl)-3- pyridyl]methyl]pyrimidine 1-11 (240 mg, 0.78 mmol), (6-chloro-5-fluoro-3-
pyridyl)boronic acid (137 mg, 0.78 mmol) and potassium phosphate tribasic (334 mg, 1.55 mmol) were dissolved in 4.8 mL cyclopentyl methyl ether and the reaction mixture was degassed by bubbling nitrogen for 5 min. To this, 0.5 mL water was added followed by dichloro-[1,T-bis(diphenylphosphino)-ferrocene]palladium(ll) (60.0 mg, 0.078 mmol) and the reaction mixture was heated at 75 °C for 3 h. The reaction mixture was cooled to room temperature and quenched by adding 25 mL of water. The aqueous layer was extracted with ethyl acetate (3X 20 mL) and the combined organic layer was concentrated in vacuo. The crude product was purified by reverse phase chromatography using water/acetonitrile as eluent. Upon concentration of the fraction, 5-chloro-2-[[2-(6-chloro-5-fluoro-3-pyridyl)-6-(trifluoromethyl)-3- pyridyl]methyl]pyrimidine 1.016 was obtained as off white solid. 1H NMR (400 MHz, CDCI3) 6 = 8.65 (s, 2 H), 8.57 (br s, 1 H) 8.00 (br d, 1 H), 7.94 (br d, 1 H), 7.71 (br d, 1H), 4.40 (s, 2 H).
Example 7: 5-[(5-chloropyrimidin-2-yl)methyl]-4-(3,4-difluorophenyl)-2- (trifluoromethyl)pyrimidine (1.033)

(1.033)
Step 1 : Preparation of ethyl 4-(3,4-difluorophenyl)-2-
(trifluoromethyl)pyrimidine-5-carboxylate (1-12)
 To a 250 ml 2 neck flask, was added ethyl 4-chloro-2-(trifluoromethyl)pyrimidine-5- carboxylate (3.6 g, 14 mmol), (3,4-difluorophenyl)boronic acid (3.3 g, 21 mmol) and tetrahydrofuran (36 mL) followed by potassium fluoride (2.6 g, 42 mmol). The reaction mixture was degassed for 15 mins by bubbling nitrogen. To this solution, bis(tri-tert-butylphosphine)palladium(0) (0.38 g, 0.71 mmol) were added and the reaction was heated to 45 °C for 12 h. After the completion of the reaction, the reaction mixture was diluted with water (100 mL) and extracted with ethyl acetate (3 X 100 mL). The combined organic layer was washed with brine, dried over sodium sulphate, filtered and concentrated under reduced pressure to obtain the crude. The crude material was purified by silica gel column chromatography using 0-15% Ethyl acetate in cyclohexane as eluent. Upon concentration of the fraction, ethyl 4-(3,4- difluorophenyl)-2-(trifluoromethyl)pyrimidine-5-carboxylate 1-12 was obtained as white solid (4 g, 85%). 1H NMR (400 MHz, CDCI3) 5 = 9.20 (s, 1 H), 7.60 (m, 1 H), 7.42 - 7.47 (m, 1 H), 7.26 - 7.33 (m, 1 H), 4.35 (q, 2 H), 1.26 (t, 3 H).
To a 250 ml 2 neck flask, was added ethyl 4-chloro-2-(trifluoromethyl)pyrimidine-5- carboxylate (3.6 g, 14 mmol), (3,4-difluorophenyl)boronic acid (3.3 g, 21 mmol) and tetrahydrofuran (36 mL) followed by potassium fluoride (2.6 g, 42 mmol). The reaction mixture was degassed for 15 mins by bubbling nitrogen. To this solution, bis(tri-tert-butylphosphine)palladium(0) (0.38 g, 0.71 mmol) were added and the reaction was heated to 45 °C for 12 h. After the completion of the reaction, the reaction mixture was diluted with water (100 mL) and extracted with ethyl acetate (3 X 100 mL). The combined organic layer was washed with brine, dried over sodium sulphate, filtered and concentrated under reduced pressure to obtain the crude. The crude material was purified by silica gel column chromatography using 0-15% Ethyl acetate in cyclohexane as eluent. Upon concentration of the fraction, ethyl 4-(3,4- difluorophenyl)-2-(trifluoromethyl)pyrimidine-5-carboxylate 1-12 was obtained as white solid (4 g, 85%). 1H NMR (400 MHz, CDCI3) 5 = 9.20 (s, 1 H), 7.60 (m, 1 H), 7.42 - 7.47 (m, 1 H), 7.26 - 7.33 (m, 1 H), 4.35 (q, 2 H), 1.26 (t, 3 H).
Step 2: Preparation of [4-(3,4-difluorophenyl)-2-(trifluoromethyl)pyrimidin-5- yl]methanol (1-13)

(1-13)
To a 3-neck round bottom flask were added ethyl 4-(3,4-difluorophenyl)-2- (trifluoromethyl)pyrimidine-5-carboxylate 1-12 (4 g, 12.04 mmol) and tetrahydrofuran (120 mL). This was cooled to 0 °C using ice bath and diisobutylaluminium hydride (1.0 mol/L) in Toluene (25.2 mL, 25.2 mmol) was added slowly over a period of 5 min and stirred at 0 °C for 30 min. Reaction mixture was quenched with aqueous ammonium chloride solution (25 mL) and extracted with ethyl acetate (3 X 50 mL). The combined organic layer was dried over anhydrous sodium sulfate, filtered and evaporated to get the crude compound. The crude was
purified by silica gel column chromatography using 15-20% of ethyl acetate in cyclohexane. After concentration, [4-(3,4-difluorophenyl)-2-(trifluoromethyl)pyrimidin- 5-yl]methanol 1-13 was obtained as white solid (1.8 g, 52%). 1H NMR (400 MHz, CDCI3) 6 = 9.09 (s, 1 H), 7.70 - 7.76 (m, 1 H), 7.56 - 7.61 (m, 1 H), 7.28 - 7.37 (m, 1 H), 4.86 (s, 2 H).
Step 3: Preparation of 4-(3,4-difluorophenyl)-2-(trifluoromethyl)pyrimidine-5- carbaldehyde (1-14)

(1-14)
To a 2-neck round bottom flask, equipped with a nitrogen inlet, were added [4-(3,4- difluorophenyl)-2-(trifluoromethyl)pyrimidin-5-yl]methanol 1-13 (1.8 g, 6.2 mmol) in acetonitrile (18 mL) and cooled to 0 °C. To this solution, 1 ,1,1-Tris(acetyloxy)-1,1- dihydro-1,2-benziodoxol-3-(1H)-one (3.2 g, 7.4 mmol) was added portion wise and the reaction mixture was stirred at room temperature for 5 hr. The reaction mixture was quenched with 10% sodium thiosulfate solution (6 ml) and extracted with ethyl acetate (2X100 mL). The combined organic layer was washed with saturated sodium bicarbonate solution, dried over sodium sulphate, filtered and concentrated to get the crude product. This crude material was purified by silica gel column chromatography by eluting it with 15 to 20 % ethyl acetate in Cyclohexane to afford 4-(3,4- difluorophenyl)-2-(trifluoromethyl)pyrimidine-5-carbaldehyde 1-14 (1.5 g, 80%) as brown gummy mass. 1H NMR (400 MHz, CDCI3) 5 = 10.22 (s, 1 H), 9.37 (s, 1 H), 7.69 - 7.76 (m, 1 H), 7.41 - 7.48 (m, 2 H).
Step 4: Preparation of (5-chloropyrimidin-2-yl)-[4-(3,4-difluorophenyl)-2- (trifluoromethyl)pyrimidin-5-yl]methanol (1.034)

(1.034)
A solution of 5-chloro-2-iodopyrimidine (0.41 g, 1.71 mmol) in toluene (5 mL) in a 250 mL round bottomed flask was placed under an atmosphere of nitrogen, cooled to -78 °C and was treated with n-butyllithium (2.5 M in hexanes) (0.94 mL 2.34 mmol). The mixture turned red in colour and the mixture turned gel-like in consistency. The mixture was allowed to stir for 10 minutes before being treated with a solution of 4- (3,4-difluorophenyl)-2-(trifluoromethyl)pyrimidine-5-carbaldehyde 1-14 (500 mg, 1.56 mmol) in toluene (5 mL). The mixture was stirred for 30 minutes and let it warm slowly to 0 °C. The reaction mixture was quenched by adding 20 mL saturated ammonium chloride solution slowly at 0 °C. The reaction mixture was then extracted with ethyl acetate (3 X 50 mL) and the combined organic layer was dried over anhydrous sodium sulfate, filtered and evaporated to get the crude material. The crude product was purified by silica gel column chromatography using 40-50% of ethyl acetate in cyclohexane. Upon concentration of the fraction, (5-chloropyrimidin- 2-yl)-[4-(3,4-difluorophenyl)-2-(trifluoromethyl)pyrimidin-5-yl]methanol 1.034 (0.19 g, %) was obtained as yellow gummy mass. 1H NMR (400 MHz, CDCh) 6 = 8.78 (s, 2H), 8.75 (s, 1H), 7.94 (m, 1 H), 7.78 (m, 1H), 7.31 -7.38 (m, 1 H), 6.14 (s, 1H).
Step 5: Preparation of 5-[(5-chloropyrimidin-2-yl)methyl]-4-(3,4-difluorophenyl)- 2-(trifluoromethyl)pyrimidine (1.033)

(1.033)
In a vial, (5-chloropyrimidin-2-yl)-[4-(3,4-difluorophenyl)-2-(trifluoromethyl)pyrimidin-5- yl]methanol 1.034 (70 mg, 0.17 mmol) and 2,4,6-trimethylpyridine (21 mg, 0.17 mmol) were dissolved in 1.7 mL acetonitrile. This solution was degassed for 10 min by bubbling with nitrogen. To this, triphenylphosphine (230 mg, 0.86 mmol) was added followed by [4,4’-bis(1,1-dimethylethyl)-2,2 ' -bipyridine-N1,N1]bis-[3,5-difluoro-2-[5- (trifluoromethyl)-2-pyridinyl-N]-phenyl-C]lridium(lll) hexafluorophosphate (3.9 mg, 0.0034 mmol) added and the reaction was stirred and irradiated with blue light (Penn photo-reactor, 450 nm) for 24 hours. Reaction mass quenched with water (5 mL) and extracted with ethyl acetate (3 X 10 mL). The combined organic layer was dried over sodium sulfate, filtered and concentrated to get the crude the crude product. This was purified by reverse-phase chromatography using 60-65% of acetonitrile in water to get 5-[(5-chloropyrimidin-2-yl)methyl]-4-(3,4-difluorophenyl)-2-
(trifluoromethyl)pyrimidine 1.033 as brown gummy mass. 1 H NMR (400 MHz, CDCh) 5 = 8.94 (s, 1 H), 8.64 (s, 2H), 7.68 - 7.60 (m, 1H), 7.51 - 7.45 (m, 1 H), 7.32 - 7.23 (m, 1H), 4.45 (s, 2H)
Example 8: 5-[(5-chloropyrimidin-2-yl)methyl]-2-(difluoromethyl)-4-(3,4- difluorophenyl)pyrimidine (1.038)

(1.038)
Step 1 : Preparation of diethyl 2-[(5-chloropyrimidin-2-yl)methyl]propanedioate (1-15)

In a 500 mL two neck flask, diethyl propanedioate (11.79 g, 73.62 mmol) was dissolved in 100 mL dimethyl sulfoxide under nitrogen atmosphere. To this, potassium phosphate (39.47 g, 184.1 mmol) was added followed by 5-chloro-2- (chloromethyl)pyrimidine (10.00 g, 61.35 mmol) and the reaction mixture was stirred at room temperature for 12 h. To this, 400 mL water were added, and the aqueous layer was extracted by ethyl acetate (3 X 150 mL). The combined organic was washed with brine (200 mL), dried over anhydrous sodium sulphate, filtered and evaporated to get the crude compound. The crude material was further purified by silica gel column chromatography using 30 % ethyl acetate in cyclohexane to get diethyl 2-[(5-chloropyrimidin-2-yl)methyl]propanedioate 1-15 (13.0 g, 66%) as yellowish liquid. 1H NMR (400 MHz, CDCh): (Mixture of Keto enol tautomer) 5 = 8.62 (s, 2H), 4.13-4.27 (m, 4H), 3.60 (d, 2H), 3.39 (s, 1H), 1.25-1.33 (m, 6H).
Step 2: Preparation of ethyl 3-(5-chloropyrimidin-2-yl)propanoate (1-16)

To a 150 mL Teflon vial, were added diethyl 2-[(5-chloropyrimidin-2- yl)methyl]propanedioate 1-15 (3.0 g, 10.0 mmol), dimethyl sulfoxide (30 mL), water (2 mL) and sodium chloride (0.6 g, 10 mmol) and the vial was placed in a pressure reactor and heated at 160 °C for 16 h. After cooling the reaction mixture to room temperature, 200 mL water was added to the reaction mixture and the aqueous layer
was extracted with ethyl acetate (3 X 100 mL). The combined organic layer was concentrated in vacuo and the crude obtained was purified by silica gel column chromatography using 20-25% ethyl acetate in cyclohexane. Upon concentration, ethyl 3-(5-chloropyrimidin-2-yl)propanoate was obtained 1-16 (1.5 g, 60.1%). 1H NMR (400 MHz, CDCI3) 6 = 8.61 (s, 2 H), 4.13 (q, 2 H), 3.29 (t, 2 H), 2.86 (t, 2 H), 1.23 (t, 3 H).
Step 3: Preparation of ethyl 2-[(5-chloropyrimidin-2-yl)methyl]-3-oxo- propanoate (1-17)
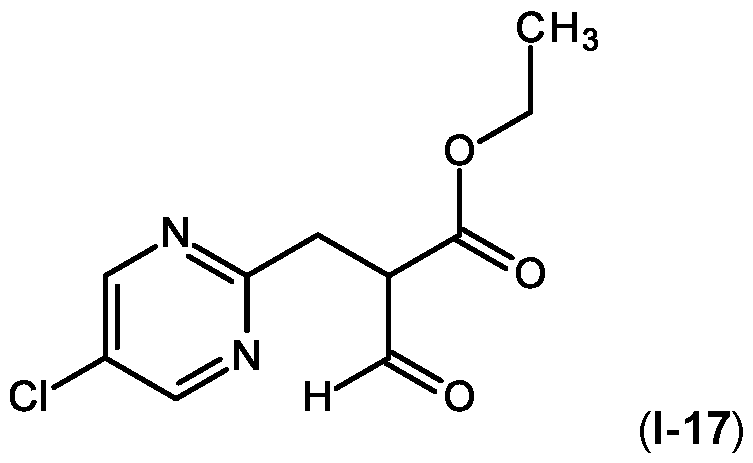
A mixture of ethyl 3-(5-chloropyrimidin-2-yl)propanoate 1-16 (3.5 g, 16.0 mmol) and ethyl formate (4.7 g, 64 mmol) were treated with 70 mL tetrahydrofuran and the mixture was cooled to 0 °C by an ice/water bath. To this solution, sodium hydride (2.6 g, 64 mmol, 60 wt%) were added portion wise. After the addition was over, the reaction mixture was stirred at room temperature for 12 h. The reaction mixture was cooled to 0 °C and quenched by slowly adding aqueous ammonium chloride (100 mL). The aqueous layer was extracted with ethyl acetate (3 x 75 mL) and the combined organic layer was dried over anhydrous sodium sulphate, filtered and concentrated to get the crude compound. The crude material was then purified by silica gel column chromatography using 40 % ethyl acetate in cyclohexane to afford ethyl 2-[(5-chloropyrimidin-2-yl)methyl]-3-oxo-propanoate 1-17 (2.1 g, 55%) yellowish liquid as a mixture of keto enol tautomer (1.6:1). 1H NMR (400 MHz, CDCI3) Keto form 5 = 10.04 (s, 1 H), 8.58 (s, 2 H), 4.25 (q, 2H), 4.01-404 (m, 1 H), 3.60-3.63 (m, 2H), 1.26 (t, 3H); Enol form 5 = 11.58 (d, 1 H), 8.62-8.65 (m, 2H), 7.21 (d, 1 H), 4.12- 4.17 (q, 2H), 3.74 (s, 2H), 1.24 (q, 3H).
Step 4: Preparation of 5-[(5-chloropyrimidin-2-yl)methyl]-2-
(methoxymethyl)pyrimidin-4-ol (1-18)

In a Teflon coated vial, ethyl 2-[(5-chloropyrimidin-2-yl)methyl]-3-oxo-propanoate 1-17 (2.1 g, 7.7 mmol) was dissolved in 63 mL methanol and to this, (2- methoxyethanimidoyl)ammonium chloride (2.46 g, 19.47 mmol) and potassium carbonate (6.5 g, 46.7 mmol) were added. The vial was placed inside a pressure reactor and heated at 80 °C for 5 h. The reaction mixture was cooled to room temperature and the solid was filtered off. The filtrate was evaporated and the solid obtained was washed with tert-butyl dimethyl ether (30 mL) and cyclohexane (70 mL). Th crude solid was then purified by reverse phase chromatography using water/acetonitrile as eluent. Upon concentration of the fraction, 5-[(5-chloropyrimidin- 2-yl)methyl]-2-(methoxymethyl)pyrimidin-4-ol 1-18 (1.25 g, 50%) was obtained as brown solid. 1 H NMR (400 MHz, DMSO-d6) 5 = 8.75 (s, 2H), 7.44 (s, 1 H), 7.26 (br s, 1H), 4.02 (s, 2H), 3.79 (s, 2H), 3.72 (s, 3H).
Step 5: Preparation of 5-[(5-chloropyrimidin-2-yl)methyl]-2-
(methoxymethyl)pyrimidin-4-ol (1-19)

A mixture of 5-[(5-chloropyrimidin-2-yl)methyl]-2-(methoxymethyl)pyrimidin-4-ol 1-18 (2.0 g, 3.0 mmol) in 40 mL dichloromethane was cooled to 0 °C, and N,N- dimethylaminopyridine (74 mg, 0.6 mmol) followed by triethyl amine (1.51 g, 15.0 mmol) were added to this mixture. The reaction mixture was stirred at 0 °C for 5 minutes and p-toluenesulfonyl chloride (1.71 g, 9.0 mmol) was added to it and stirred at room temperature for 12 h. After this time, the reaction was diluted with 100 mL water and the aqueous layer was extracted with ethyl acetate (3 X 100 mL). The combined organic layer was dried over anhydrous sodium sulphate, filtered and
evaporated to get the crude compound. The crude material was purified by silica gel column chromatography using 40% of ethyl acetate in cyclohexane to afford 5-[(5- chloropyrimidin-2-yl)methyl]-2-(methoxymethyl)pyrimidin-4-ol 1-19 (0.57 g, 21%) as yellowish gummy material. 1H-NMR (400 MHz, CDCI3) 6 = 8.66 (s, 1H), 8.60 (s, 2H), 8.00 (d, 2H), 7.37 (d, 2H), 4.61 (s, 2H), 4.31 (s, 2H), 3.53 (s, 3H), 2.49 (s, 3H).
Step 6: Preparation of 5-[(5-chloropyrimidin-2-yl)methyl]-4-(3,4-difluorophenyl)- 2-(methoxymethyl)pyrimidine (1-20)

In a microwave vial, [5-[(5-chloropyrimidin-2-yl)methyl]-2-(methoxymethyl)pyrimidin- 4-yl] 4-methylbenzenesulfonate 1-19 (250 mg, 0.59 mmol) was dissolved in 15 mL dioxane. To this solution were added (3,4-difluorophenyl)boronic acid (93.80 mg, 0.59 mmol), tetrakis(triphenylphosphine)palladium(0) (69.33 mg, 0.059 mmol), potassium carbonate (331.70 mg, 2.37 mmol) and water (1 mL). The reaction mixture was degassed by bubbling nitrogen for 10 min and then heated at 100 °C under microwave irradiation for 1 h. The reaction mixture was cooled to room temperature and 20 mL water was added. The aqueous layer was extracted with ethyl acetate (3 X 30 mL), dried over anhydrous sodium sulphate, filtered and concentrated. The crude product obtained after concentration was purified by silica gel column chromatography using 25-30% ethyl acetate in cyclohexane to get 5-[(5- chloropyrimidin-2-yl)methyl]-4-(3,4-difluorophenyl)-2-(methoxymethyl)pyrimidine I-20 (180 mg, 50%) as yellowish gummy material. 1H-NMR (400 MHz, CDCI3) 6 = 8.80 (s, 1H), 8.62 (s, 2H), 7.54-7.56 (m, 1H), 7.39-7.43 (m, 1 H), 7.21-7.27 (m, 1H), 4.74 (s, 2H), 4.35 (s, 2H), 3.58 (s, 3H).
Step 7: Preparation of [5-[(5-chloropyrimidin-2-yl)methyl]-4-(3,4- difluorophenyl)-pyrimidin-2-yl]methanol (1-21)

A solution of 5-[(5-chloropyrimidin-2-yl)methyl]-4-(3,4-difluorophenyl)-2- (methoxymethyl)-pyrimidine 1-20 (300 mg, 0.49 mmol) in dichloromethane (2 mL) was cooled to 0 °C and boron tribromide solution (1M) in dichloromethane (2.48 mL, 2.48 mmol) was added to the reaction mixture. The reaction was stirred at room temperature for 12 h. After this time, the reaction mixture was cooled to 0 °C and slowly quenched by adding 20 mL saturated aqueous sodium hydrogen carbonate solution. The aqueous layer was extracted with ethyl acetate (3 X 20 mL), dried over anhydrous sodium sulphate, filtered and concentrated to get the crude compound. The crude was further purified by silica gel column chromatography using 50-60% ethyl acetate in cyclohexane to get [5-[(5-chloropyrimidin-2-yl)methyl]-4-(3,4- difluorophenyl)pyrimidin-2-yl]methanol 1-21 (65 mg, 38%) yellow liquid. 1H NMR (400 MHz, CDCI3): 5 = 8.79 (s, 1H), 8.64 (s, 2H), 7.55-7.58 (m, 1 H), 7.41-7.45 (m, 1 H), 7.23-7.28 (m, 1H), 4.89 (s, 2H), 4.38 (s, 2H).
Step 8: Preparation of 5-[(5-chloropyrimidin-2-yl)methyl]-4-(3,4-difluorophenyl)- pyrimidine-2-carbaldehyde (I-22)

(I-22)
A solution of [5-[(5-chloropyrimidin-2-yl)methyl]-4-(3,4-difluorophenyl)pyrimidin-2- yl]methanol 1-21 (100 mg, 0.14 mmol) in acetonitrile (6 mL) was cooled to 0 °C and 1 , 1 , 1-T riacetoxy-1 , 1 -dihydro-1 ,2-benziodoxol-3(1 H)-one (Dess-Martin periodinane)
(121.62 mg, 0.28 mmol) was added portion wise. The reaction mixture was then stirred at room temperature for 12 h and then quenched by adding 10 mL of aqueous sodium hydrogen carbonate solution followed by 10 mL aqueous sodium thiosulphate solution. The aqueous layer was extracted with ethyl acetate (3 X 20 mL) and the combined organic layer was dried over anhydrous sodium sulphate, filtered and evaporated to the crude compound. Purification of the crude via silica gel column chromatography using 40 % ethyl acetate in cyclohexane afforded 5-[(5- chloropyrimidin-2-yl)methyl]-4-(3,4-difluorophenyl)pyrimidine-2-carbaldehyde I-22 (35 mg, 39%) yellowish gummy material. 1H NMR (400 MHz, CDCI3): 5 = 10.16 (s, 1H), 9.02 (s, 1H), 8.64 (s, 2H), 7.45 - 7.68 (m, 3H), 4.47 (s, 2H).
Step 9: Preparation of 5-[(5-chloropyrimidin-2-yl)methyl]-2-(difluoromethyl)-4- (3,4-difluorophenyl)pyrimidine (1.038)
(1.038)
A solution of 5-[(5-chloropyrimidin-2-yl)methyl]-4-(3,4-difluorophenyl)pyrimidine-2- carbaldehyde 1-22 (30 mg, 0.05 mmol) in dichloromethane (1.5 mL) was cooled to 0 °C and to this, diethylaminosulfur trifluoride (33.46 mg, 0.027 mL) was added dropwise. The reaction mixture was then stirred overnight at room temperature. The reaction mixture was then slowly poured into 20 mL of aqueous sodium hydrogen carbonate solution and the aqueous layer was extracted with ethyl acetate (3 X 20 mL). The combined organic layer was dried over anhydrous sodium sulphate, filtered and evaporated to get the crude compound. The crude was then purified by silica gel column chromatography using 25% ethyl acetate in cyclohexane to get 5-[(5- chloropyrimidin-2-yl)methyl]-2-(difluoromethyl)-4-(3,4-difluorophenyl)pyrimidine 1.038 as yellowish gummy material. 1H NMR (400 MHz, CDCh) 6 = 8.92 (s, 1H), 8.64 (s, 2H), 7.62 (br d, 1 H), 7.46 (br dd, 1 H), 7.22-7.32 (m, 1 H), 6.70 (t, 1 H), 4.43 (s, 2H).
Example 9: 4-(6-chloro-5-fluoro-3-pyridyl)-5-[(5-chloropyrimidin-2-yl)methyl]-2- (trifluoromethyl)pyrimidine (1.017)

(1.017)
Step 1 : Preparation of ethyl 2-[4-(6-chloro-5-fluoro-3-pyridyl)-2-methylsulfanyl- pyrimidin-5-yl]acetate (I-23)

(I-23)
A mixture of ethyl 2-(4-chloro-2-methylsulfanyl-pyrimidin-5-yl)acetate (703 mg, 2.85 mmol), (6-chloro-5-fluoro-3-pyridyl)boronic acid (322 mg, 1.84 mmol) and XPhos Pd G3 (79 mg, 0.093 mmol) was treated with 2-methyltetrahydrofuran (3.7 mL) and a solution of potassium phosphate in water (1.0 M, 2.7 mL, 2.7 mmol). The resulting mixture was stirred at 80°C for 1.5 hours. The mixture was cooled, diluted with water and extracted with ethyl acetate. The combined organics were concentrated and subjected to silica gel column chromatography using 0-50% ethyl acetate in cyclohexane. The product-rich fractions were concentrated and subjected to reversephase chromatography (acetonitrile in water, both with 0.1% formic acid). The fractions forming the major peak of interest were combined and concentrated in vacuo, yielding ethyl 2-[4-(6-chloro-5-fluoro-3-pyridyl)-2-methylsulfanyl-pyrimidin-5- yl]acetate I-23 (57 mg, 9%) as a colourless oil. 1H NMR (400 MHz, CDCh) 6 = 8.56 (s, 1H), 8.45 (d, 1 H), 7.83 (dd, 1 H), 4.17 (q, 2H), 3.62 (s, 2H), 2.59 (s, 3H), 1.25 (t, 3H).
Step 2: Preparation of ethyl 2-[4-(6-chloro-5-fluoro-3-pyridyl)-2-methylsulfanyl- pyrimidin-5-yl]-2-(5-chloropyrimidin-2-yl)acetate (I-24)

(1-24)
A solution of ethyl 2-[4-(6-chloro-5-fluoro-3-pyridyl)-2-methylsulfanyl-pyrimidin-5- yl]acetate 1-23 (57 mg, 0.167 mmol) and 2,5-dichloropyrimidine (78 mg, 0.524 mmol) in DMSO (0.8 mL) was treated with potassium phosphate (148 mg, 0.697 mmol) and the resulting mixture was stirred at 70 °C for 30 minutes. The mixture was cooled, treated with 0.5 M hydrochloric acid and extracted with tert-butyl methyl ether. The combined organics were concentrated and subjected to silica gel column chromatography using 0-30% ethyl acetate in cyclohexane. The fractions forming the major peak of interest were combined and concentrated in vacuo, yielding ethyl 2-[4- (6-chloro-5-fluoro-3-pyridyl)-2-methylsulfanyl-pyrimidin-5-yl]-2-(5-chloropyrimidin-2- yl)acetate I-24 (42 mg, 51%) as a yellow gum. 1H NMR (400 MHz, CDCI3) 5 = 8.94 (s, 1 H), 8.68 (s, 2H), 8.59 (d, 1H), 7.94 (dd, 1 H), 5.40 (s, 1 H), 4.28 - 4.18 (m, 2H), 2.58 (s, 3H), 1.23 (t, 3H).
Step 3: Preparation of 4-(6-chloro-5-fluoro-3-pyridyl)-5-[(5-chloropyrimidin-2- yl)methyl]-2-methylsulfanyl-pyrimidine (1.036)

(1.036)
A solution of ethyl 2-[4-(6-chloro-5-fluoro-3-pyridyl)-2-methylsulfanyl-pyrimidin-5-yl]-2- (5-chloropyrimidin-2-yl)acetate I-24 (42 mg, 0.0878 mmol) in acetonitrile (1 mL) and water (0.1 mL) heated to 180 °C for 30 minutes under microwave radiation. On completion, the mixture was concentrated in vacuo, yielding 4-(6-chloro-5-fluoro-3- pyridyl)-5-[(5-chloropyrimidin-2-yl)methyl]-2-methylsulfanyl-pyrimidine 1.036 (33 mg, 93%) as a beige solid. 1H NMR (400 MHz, CDCI3) 6 = 8.64 (s, 1 H), 8.63 (s, 2H), 8.60 (d, 1 H), 7.99 (dd, 1H), 4.27 (s, 2H), 2.58 (s, 3H).
Step 4: Preparation of 4-(6-chloro-5-fluoro-3-pyridyl)-5-[(5-chloropyrimidin-2- yl)methyl]-2-methylsulfonyl-pyrimidine (1.037)

(1.037)
A solution of 4-(6-chloro-5-fluoro-3-pyridyl)-5-[(5-chloropyrimidin-2-yl)methyl]-2- methylsulfanyl-pyrimidine 1.036 (42 mg, 0.0820 mmol) in ethyl acetate (1 mL) was treated with 3-chloroperoxybenzoic acid (77%, 42 mg, 0.187 mmol). The resulting mixture was allowed to stir for 2 hours. The mixture was diluted with ethyl acetate and quenched with aqueous sodium thiosulphate. The organics were washed with aqueous sodium bicarbonate and water before being concentrated in vacuo and subjected to silica gel column chromatography using 40-100% ethyl acetate in cyclohexane. The fractions forming the major peak of interest were combined and concentrated in vacuo, yielding 4-(6-chloro-5-fluoro-3-pyridyl)-5-[(5-chloropyrimidin-2- yl)methyl]-2-methylsulfonyl-pyrimidine 1.037 (25 mg, 70%) as a white solid. 1H NMR (400 MHz, CDCI3) 6 = 9.05 (s, 1H), 8.65 (s, 2H), 8.63 (d, 1H), 8.04 (dd, 1H), 4.47 (s, 2H), 3.40 (s, 3H).
Step 5: Preparation of 4-(6-chloro-5-fluoro-3-pyridyl)-5-[(5-chloropyrimidin-2- yl)methyl]-2-(trifluoromethyl)pyrimidine (1.017)
(1.017)
A solution of 4-(6-chloro-5-fluoro-3-pyridyl)-5-[(5-chloropyrimidin-2-yl)methyl]-2- methylsulfonyl-pyrimidine 1.037 (25 mg, 0.573 mmol) in THF (0.5 mL) was treated with potassium fluoride (19 mg, 0.327 mmol) and
trimethyl(trifluoromethyl)silane (50 pL, 0.338 mmol). The reaction mixture was stirred for 5 hours, treated with additional trimethyl(trifluoromethyl)silane (50 pL, 0.338 mmol) and was stirred for a further 16 hours. The reaction mixture was treated with additional trimethyl(trifluoromethyl)silane (50 pL, 0.338 mmol) and was stirred for a further 2.5 hours. The mixture was treated with aqueous sodium bicarbonate and extracted with ethyl acetate before being concentrated in vacuo and subjected to silica gel column chromatography using 0-50% ethyl acetate in cyclohexane. The fractions forming the major peak of interest were combined and concentrated in vacuo, yielding 4-(6-chloro-5-fluoro-3-pyridyl)-5-[(5-chloropyrimidin-2-yl)methyl]-2- (trifluoromethyl)pyrimidine 1.017. 1H NMR (400 MHz, CDCI3) 6 = 9.03 (s, 1 H), 8.67 - 8.64 (m, 3H), 8.04 (dd, 1H), 4.46 (s, 2H).
Table 1. Compounds of the present invention

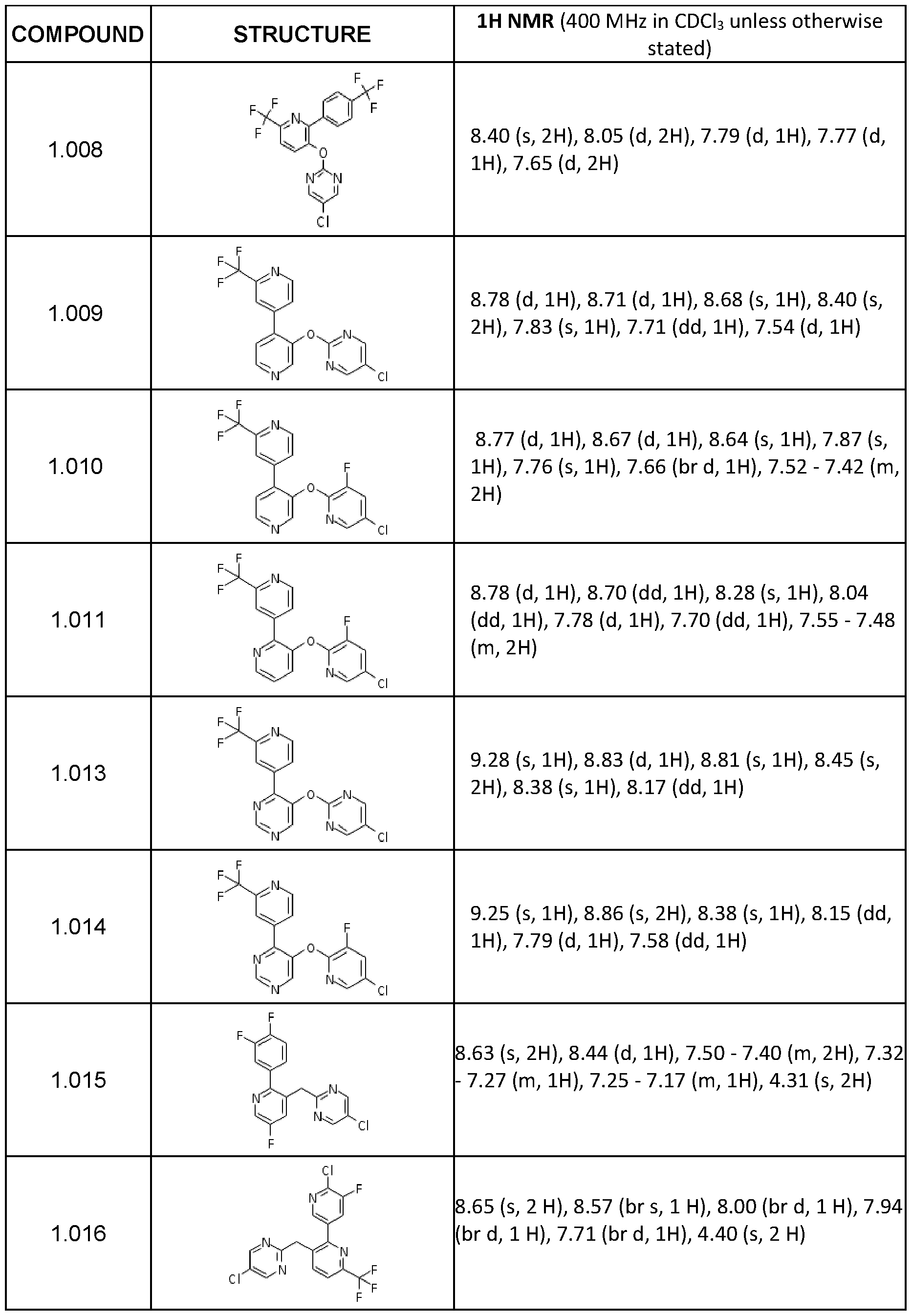
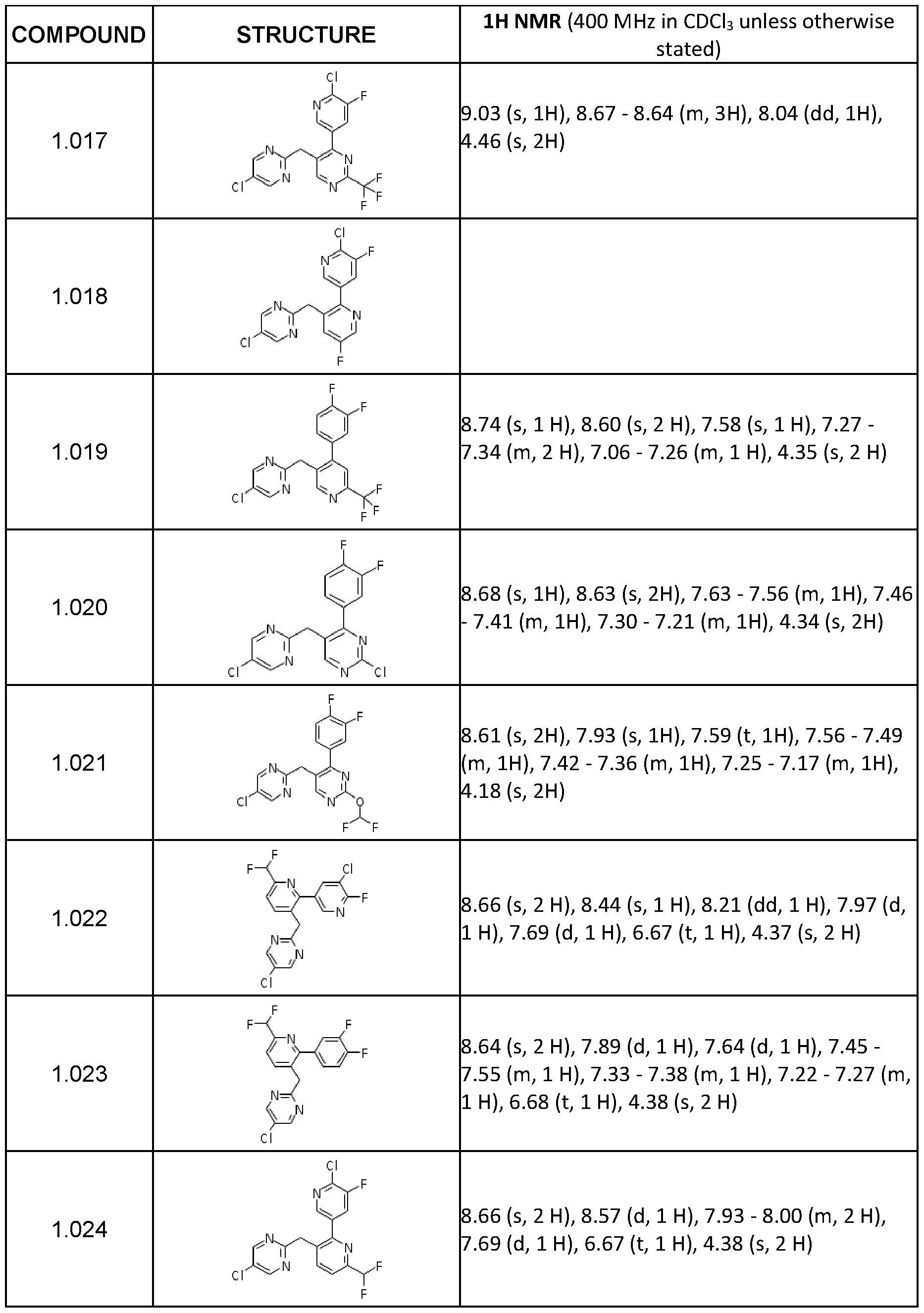

 Seeds of a variety of test species are sown in standard soil in pots: Amaranthus retoflexus (AMARE), Amaranthus palmeri (AMAPA), Setaria faberi (SETFA), Echinochloa crus-galli (ECHCG) and Ipomoea hederacea (IPOHE)). After cultivation for one day (pre-emergence) or after 8 days cultivation (post-emergence) under controlled conditions in a glasshouse (at 24/16°C, day/night; 14 hours light; 65 % humidity), the plants are sprayed with an aqueous spray solution derived from the formulation of the technical active ingredient in acetone I water (50:50) solution containing 0.5% Tween 20 (polyoxyethelyene sorbitan monolaurate, CAS RN 9005- 64-5). Compounds are applied at 250 g/ha unless otherwise stated. The test plants are then grown in a glasshouse under controlled conditions in a glasshouse (at 24/16°C, day/night; 14 hours light; 65 % humidity) and watered twice daily. After 13 days for pre and post-emergence, the test is evaluated for the percentage damage caused to the plant. The biological activities (% phytotoxicity) are shown in Tables B1 and B2 below using a five-point scale (5 = 81 to 100%; 4 = 61 to 80%; 3 = 41 to 60%; 2 = 21 to 40%; 7 = 0 to 20%). NT = Not tested.
Seeds of a variety of test species are sown in standard soil in pots: Amaranthus retoflexus (AMARE), Amaranthus palmeri (AMAPA), Setaria faberi (SETFA), Echinochloa crus-galli (ECHCG) and Ipomoea hederacea (IPOHE)). After cultivation for one day (pre-emergence) or after 8 days cultivation (post-emergence) under controlled conditions in a glasshouse (at 24/16°C, day/night; 14 hours light; 65 % humidity), the plants are sprayed with an aqueous spray solution derived from the formulation of the technical active ingredient in acetone I water (50:50) solution containing 0.5% Tween 20 (polyoxyethelyene sorbitan monolaurate, CAS RN 9005- 64-5). Compounds are applied at 250 g/ha unless otherwise stated. The test plants are then grown in a glasshouse under controlled conditions in a glasshouse (at 24/16°C, day/night; 14 hours light; 65 % humidity) and watered twice daily. After 13 days for pre and post-emergence, the test is evaluated for the percentage damage caused to the plant. The biological activities (% phytotoxicity) are shown in Tables B1 and B2 below using a five-point scale (5 = 81 to 100%; 4 = 61 to 80%; 3 = 41 to 60%; 2 = 21 to 40%; 7 = 0 to 20%). NT = Not tested.
TABLE B1. Post-emergence application.


TABLE B2. Pre-emergence application.

Claims
1. A compound of Formula (I):
 wherein
wherein
X is O or CHR8,
Q is phenyl or a C-linked 6-membered heteroaryl wherein said phenyl or 6- membered heteroaryl is optionally substituted by one or more R4;
A1 is CH or N
A2 is CR2 or N, wherein at least one of A1 and A2 are N,
A3 is CR5 or N; wherein if X is O then A1 is not N when A2 is CR2 and A3 is N;
R1 is selected from the group consisting of hydrogen, halogen, -CN, nitro, Ci- C4alkyl-, C2-C4alkenyl-, C2-C4alkynyl-, Ci-C4haloalkyl-, C1-C4 alkoxy-, C1- C4haloalkoxy-, -S(O)pCi-C4alkyl and Cs-Cecycloalkyl-;
R2 is selected from the group consisting of hydrogen, halogen and Ci-Ce alkyl;
R3 is selected from the group consisting of halogen, Ci-C4haloalkyl- and C1-C2 haloalkoxy-;
R4 is selected from the group consisting of halogen, C1-C4 alkyl, Ci- 04 haloalkyl, C1-C4 alkoxy, C1-C4 haloalkoxy, Ci-C4alkoxyCi-C3alkyl-, Ci- C4alkoxyCi-C3alkoxy-, Ci-C4alkoxyCi-C3alkoxyCi-C3alkyl-, -ON, NO2, C2- C4alkenyl, C2-C4alkynyl, -S(O)pCi-C4alkyl, -S(O)pCi-C4haloalkyl, -C(O)OCi- C4alkyl and -C(O)NR6R7;
R5 is hydrogen or halogen;
R6 is selected from the group consisting of hydrogen, C3-C4cycloalkyl, Ci- C4alkyl and Ci-C4haloalkyl;
R7 is selected from the group consisting of hydrogen, C3-C4cycloalkyl, Ci- C4alkyl and Ci-C4haloalkyl;
R8 is hydrogen or hydroxyl; and p is 0, 1 or 2; or an agriculturally acceptable salt thereof.
2. A compound according to claim 1 , wherein A1 is N and A2 is CH.
3. A compound according to claim 1 , wherein A1 is CH and A2 is N.
4. A compound according to claim 1 , wherein A1 is N and A2 is N.
5. A compound according to any one of the previous claims, wherein A3 is N or
CR5 wherein R5 is fluoro.
6. A compound according to any one of the previous claims, wherein R1 is selected from the group consisting of halogen, Ci-C4alkyl- and Ci-C4haloalkyl-.
7. A compound according to any one of the previous claims, wherein R3 is halogen or Ci-C4haloalkyl-.
8. A compound according to any one of the previous claims, wherein Q is selected from the group consisting of:
 wherein n is 0, 1 or 2.
wherein n is 0, 1 or 2.
9. A compound according to claim 8, wherein Q is selected from the group consisting of Q-1 , Q-3 and Q-4.
10. A compound according to claim 9, wherein n is 1 or 2 and R4 is independently selected from group consisting of halogen and C1-C4 haloalkyl.
11. A herbicidal composition comprising a compound according to any one of the previous claims and an agriculturally acceptable formulation adjuvant.
12. A herbicidal composition according to claim 11, further comprising at least one additional pesticide.
13. A herbicidal composition according to claim 12, wherein the additional pesticide is a herbicide or herbicide safener.
14. A method of controlling weeds at a locus comprising application to the locus of a weed controlling amount of a composition according to any one of claims
11 to 13.
15. Use of a compound of Formula (I) as defined in claim 1 as a herbicide.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP23205313.2 | 2023-10-23 | ||
| EP23205313 | 2023-10-23 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2025087763A1 true WO2025087763A1 (en) | 2025-05-01 |
Family
ID=88511210
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2024/079166 Pending WO2025087763A1 (en) | 2023-10-23 | 2024-10-16 | Pyridine and pyrimidine derivatives as herbicides |
Country Status (2)
| Country | Link |
|---|---|
| AR (1) | AR134159A1 (en) |
| WO (1) | WO2025087763A1 (en) |
Citations (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0374753A2 (en) | 1988-12-19 | 1990-06-27 | American Cyanamid Company | Insecticidal toxines, genes coding therefor, antibodies binding them, transgenic plant cells and plants expressing these toxines |
| EP0427529A1 (en) | 1989-11-07 | 1991-05-15 | Pioneer Hi-Bred International, Inc. | Larvicidal lectins and plant insect resistance based thereon |
| EP0451878A1 (en) | 1985-01-18 | 1991-10-16 | Plant Genetic Systems, N.V. | Modifying plants by genetic engineering to combat or control insects |
| WO1993007278A1 (en) | 1991-10-04 | 1993-04-15 | Ciba-Geigy Ag | Synthetic dna sequence having enhanced insecticidal activity in maize |
| WO1995034656A1 (en) | 1994-06-10 | 1995-12-21 | Ciba-Geigy Ag | Novel bacillus thuringiensis genes coding toxins active against lepidopteran pests |
| WO2002034048A1 (en) | 2000-10-23 | 2002-05-02 | Syngenta Participations Ag | Agrochemical compositions with quinoline safeners |
| WO2003052073A2 (en) | 2001-12-17 | 2003-06-26 | Syngenta Participations Ag | Novel corn event |
| WO2010064688A1 (en) | 2008-12-03 | 2010-06-10 | 日本農薬株式会社 | Pyrimidine derivatives, horticultural insecticides comprising said derivatives and method of use thereof |
| WO2016149315A1 (en) | 2015-03-18 | 2016-09-22 | E. I. Du Pont De Nemours And Company | Substituted pyrimidinyloxy pyridine derivatives as herbicides |
| WO2020002089A1 (en) | 2018-06-25 | 2020-01-02 | Bayer Aktiengesellschaft | Substituted 2-heteroaryloxypyridines and salts thereof and their use as herbicidal agents |
| WO2021204706A1 (en) | 2020-04-08 | 2021-10-14 | Syngenta Crop Protection Ag | 5-haloalkoxy-pyrimidine compounds as herbicides |
| GB2594931A (en) | 2020-05-06 | 2021-11-17 | Syngenta Crop Protection Ag | Improvements in or relating to organic compounds |
| WO2022002838A1 (en) | 2020-06-30 | 2022-01-06 | Bayer Aktiengesellschaft | Substituted heteroaryloxypyridines, the salts thereof and their use as herbicidal agents |
| WO2023186691A1 (en) | 2022-03-28 | 2023-10-05 | Bayer Aktiengesellschaft | Substituted 2-c-azines and salts thereof, and use thereof as herbicidal active substances |
-
2024
- 2024-10-16 WO PCT/EP2024/079166 patent/WO2025087763A1/en active Pending
- 2024-10-21 AR ARP240102841A patent/AR134159A1/en unknown
Patent Citations (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0451878A1 (en) | 1985-01-18 | 1991-10-16 | Plant Genetic Systems, N.V. | Modifying plants by genetic engineering to combat or control insects |
| EP0374753A2 (en) | 1988-12-19 | 1990-06-27 | American Cyanamid Company | Insecticidal toxines, genes coding therefor, antibodies binding them, transgenic plant cells and plants expressing these toxines |
| EP0427529A1 (en) | 1989-11-07 | 1991-05-15 | Pioneer Hi-Bred International, Inc. | Larvicidal lectins and plant insect resistance based thereon |
| WO1993007278A1 (en) | 1991-10-04 | 1993-04-15 | Ciba-Geigy Ag | Synthetic dna sequence having enhanced insecticidal activity in maize |
| WO1995034656A1 (en) | 1994-06-10 | 1995-12-21 | Ciba-Geigy Ag | Novel bacillus thuringiensis genes coding toxins active against lepidopteran pests |
| WO2002034048A1 (en) | 2000-10-23 | 2002-05-02 | Syngenta Participations Ag | Agrochemical compositions with quinoline safeners |
| WO2003052073A2 (en) | 2001-12-17 | 2003-06-26 | Syngenta Participations Ag | Novel corn event |
| WO2010064688A1 (en) | 2008-12-03 | 2010-06-10 | 日本農薬株式会社 | Pyrimidine derivatives, horticultural insecticides comprising said derivatives and method of use thereof |
| WO2016149315A1 (en) | 2015-03-18 | 2016-09-22 | E. I. Du Pont De Nemours And Company | Substituted pyrimidinyloxy pyridine derivatives as herbicides |
| WO2020002089A1 (en) | 2018-06-25 | 2020-01-02 | Bayer Aktiengesellschaft | Substituted 2-heteroaryloxypyridines and salts thereof and their use as herbicidal agents |
| WO2021204706A1 (en) | 2020-04-08 | 2021-10-14 | Syngenta Crop Protection Ag | 5-haloalkoxy-pyrimidine compounds as herbicides |
| GB2594931A (en) | 2020-05-06 | 2021-11-17 | Syngenta Crop Protection Ag | Improvements in or relating to organic compounds |
| WO2022002838A1 (en) | 2020-06-30 | 2022-01-06 | Bayer Aktiengesellschaft | Substituted heteroaryloxypyridines, the salts thereof and their use as herbicidal agents |
| WO2023186691A1 (en) | 2022-03-28 | 2023-10-05 | Bayer Aktiengesellschaft | Substituted 2-c-azines and salts thereof, and use thereof as herbicidal active substances |
Non-Patent Citations (2)
| Title |
|---|
| "CAS", Database accession no. 9005-64-5 |
| TETRAHEDRON, vol. 73, 2017, pages 3939 - 3948 |
Also Published As
| Publication number | Publication date |
|---|---|
| AR134159A1 (en) | 2025-12-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP4441041B1 (en) | Herbicidal pyrazole pyrimidine compounds | |
| EP4182309B1 (en) | Herbicidal compounds | |
| AU2022372260B9 (en) | Herbicidal imidazole compounds | |
| EP4210489A1 (en) | Herbicidal compounds | |
| US20230002357A1 (en) | 7-pyrimidine-2-yl-oxy-indazole derivatives and their use as herbicides | |
| EP4649075A1 (en) | Herbicidal imidazole compounds | |
| WO2024046890A1 (en) | Herbicidal pyrazole compounds | |
| WO2025087763A1 (en) | Pyridine and pyrimidine derivatives as herbicides | |
| WO2021028316A1 (en) | 2-phenoxy-pyrimidine derivatives as herbicidal compounds | |
| EP4058449B1 (en) | Herbicidal thiazole compounds | |
| EP4649076A1 (en) | Herbicidal pyrazole compounds | |
| WO2023165874A1 (en) | Pyrimidinyl-oxy-quinoline based herbicidal compounds | |
| WO2025098854A1 (en) | 5-membered heteroaryl and pyrazole herbicides | |
| WO2022207482A1 (en) | Herbicidal compounds | |
| WO2025021650A1 (en) | Herbicidal pyrazole compounds | |
| EP4543874A1 (en) | Herbicidal imidazole-containing compounds | |
| WO2025157695A1 (en) | Herbicidal pyrazole and triazole compounds | |
| WO2024013018A1 (en) | Herbicidal imidazole compounds |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 24790935 Country of ref document: EP Kind code of ref document: A1 |