WO2025082632A1 - Method and composition for oligonucleotide synthesis - Google Patents
Method and composition for oligonucleotide synthesis Download PDFInfo
- Publication number
- WO2025082632A1 WO2025082632A1 PCT/EP2024/060228 EP2024060228W WO2025082632A1 WO 2025082632 A1 WO2025082632 A1 WO 2025082632A1 EP 2024060228 W EP2024060228 W EP 2024060228W WO 2025082632 A1 WO2025082632 A1 WO 2025082632A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- oligonucleotide
- group
- cycle
- protecting group
- aqueous
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H1/00—Processes for the preparation of sugar derivatives
- C07H1/06—Separation; Purification
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/06—Pyrimidine radicals
- C07H19/067—Pyrimidine radicals with ribosyl as the saccharide radical
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/16—Purine radicals
- C07H19/167—Purine radicals with ribosyl as the saccharide radical
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H21/00—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids
- C07H21/02—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids with ribosyl as saccharide radical
Definitions
- the present invention generally pertains to the field of oligonucleotide synthesis at an industrial or laboratory scale. Improved methods for the synthesis of oligonucleotides are disclosed. Efficient methods for the manufacture of oligonucleotides at an industrial or laboratory scale as well as solvents for use in these methods are of significant pharmaceutical and commercial interest. The synthesis of oligonucleotides commonly relies on iterative coupling cycles, in which oligomeric or monomeric building blocks are added to a growing oligonucleotide chain.
- Solid-phase synthesis is characterized in that a growing oligonucleotide chain is assembled on a solid support, typically made of polymer resins or controlled pore glass.
- Solid-phase synthesis processes using the so-called phosphoramidite method have been optimized and automated. Thus, such solid-phase synthesis processes may be advantageous in terms of speed and are used most widely.
- the upscaling of solid-phase synthesis processes is difficult for several reasons, including excessive consumption of reagents and raw materials.
- a further disadvantage of solid-phase synthesis processes is that it is difficult to follow the progress of the reaction in real time and to analyze the structure of intermediates.
- oligonucleotide chain is bonded to a soluble support, also referred to as pseudo solid-phase protecting group or tag.
- pseudo solid-phase protecting groups are typically hydrophobic and alter the solubility of the oligonucleotides.
- oligonucleotides bonded to one or more such pseudo solid-phase protecting groups herein also referred to as conjugates of oligonucleotides and pseudo solid-phase protecting groups, may be readily soluble in the organic solvents commonly used for oligonucleotide synthesis.
- solid-liquid separation typically filtration
- Such precipitation is typically achieved by addition of one or more polar solvents (e.g. water, acetonitrile, propionitrile, methanol, ethanol, 1-propanol, 2-propanol, and the like) and/or by concentrating the respective solution (e.g. in vacuo) and/or by decreasing the temperature of the respective solution.
- polar solvents e.g. water, acetonitrile, propionitrile, methanol, ethanol, 1-propanol, 2-propanol, and the like
- precipitation and solid-liquid separation approaches to oligonucleotide synthesis using pseudo solid-phase protecting groups are e.g. disclosed in: US2015112053A1, EP3825300A1, EP2711370A1, EP3398955A1, US2018291056A1, EP3925964A1, EP2921499A1, EP3733680A1, EP3378869A1, WO2020227618A2, EP3263579A1, EP3950698A1, EP4006045A1, and EP3015467A1.
- liquid-phase oligonucleotide synthesis using pseudo solid-phase protecting groups may rely on one or more (liquid-liquid) extraction steps, typically aqueous extraction steps, to remove excess reagents and/or side- products and/or by-products, thereby avoiding the need for precipitation.
- This strategy typically exploits the fact that the pseudo solid-phase protecting groups are usually quite hydrophobic.
- the conjugates of such pseudo solid-phase protecting groups and the growing oligonucleotide chains are typically (mostly) retained in an organic phase during aqueous extraction(s).
- At least one solid-liquid separation, typically filtration, step is usually performed per coupling cycle, either after evaporation to dryness and re-dissolution to achieve further purification, or prior to evaporation to dryness, if a drying agent such as sodium sulfate has been added, e.g. to the organic phase obtained from aqueous extraction.
- a drying agent such as sodium sulfate
- An oligonucleotide synthesis process which does not comprise any solid-liquid separation during and in between at least two consecutive coupling cycles, may be referred to as one-pot process.
- Such one- pot processes may be preferred, but have been reported very scarcely, namely in US2013267697A1, US2015080565A1, EP2816053A1, and EP3296306A1.
- a typical coupling cycle comprises a deprotection step, in which a temporary protecting group is removed from a hydroxyl moiety of a first nucleoside or oligonucleotide, so as to obtain a free hydroxyl group.
- a coupling step in which said free hydroxyl group engages in a condensation reaction with a phosphorus moiety of a second nucleoside or oligonucleotide, so as to obtain an elongated oligonucleotide.
- the phosphorus moiety of said second nucleoside or oligonucleotide is a P(III) moiety, e.g., a phosphoramidite moiety
- the coupling reaction is typically followed by a step of converting P(III) atoms to P(V) atoms by incubation with an oxidizing or sulfurizing agent. This step is also referred to as “oxidation step” or “sulfurization step”.
- P(III) moieties such as H- phosphonate diester moieties, may also be converted to P(V) moieties in the final coupling cycle only.
- a step of blocking unreacted hydroxyl groups (also referred to as “capping step”) is oftentimes carried out either after the coupling step or after the oxidation or sulfurization step.
- the present invention specifically focusses on methods for liquid- phase oligonucleotide synthesis using pseudo solid-phase protecting groups comprising one or more aqueous extractions of a nucleoside or oligonucleotide having a free backbone hydroxyl group (i.e., after the deprotection step of the typical coupling cycle). So far, little to no attention has been paid to the pH-values of the aqueous phases of the aqueous extractions of nucleosides or oligonucleotides having a free backbone hydroxyl group (i.e., after the deprotection step) in liquid-phase oligonucleotide synthesis using pseudo solid-phase protecting groups.
- optimized conditions for the aqueous extraction of solutions comprising nucleosides or oligonucleotides bound to a pseudo solid-phase protecting group and having a free backbone hydroxyl group are desirable. Such extractions will be conducted after the deprotection step of the typical coupling cycle. Such optimized extraction conditions should ideally afford the target oligonucleotide of the respective synthesis with higher yield and/or higher purity.
- the present invention provides methods addressing the above needs.
- One aspect of the invention pertains to a method for the synthesis of a target oligonucleotide O T , said method comprising a step of subjecting a solution comprising a component (C-0) # selected from the group consisting of a nucleoside and an oligonucleotide to one or more aqueous extractions, wherein the organic phase comprises the component (C-0) # , and wherein - said component (C-0) # is covalently bonded to a pseudo solid-phase protecting group PG-s and is a compound of the following Formula I-C: (Formula I-C), wherein in Formula I-C: each oxygen atom O depicted within each nucleoside subunit x-0 to x-m represents the oxygen atom of a hydroxyl moiety of the respective nucleoside subunit; each of the nucleoside subunits x-0 to x-m may be the same or different; m is an integer equal to or larger than 0;
- aqueous extraction may, in the context of the aforementioned aspect of the invention, be understood in the broadest sense as any liquid-liquid extraction operation during which said solution comprising said component (C-0) # is extracted with water or an aqueous solution in general.
- an “aqueous extraction” is further characterized in that it results in a mixture comprising at least one aqueous phase (preferably an aqueous layer) and at least one organic phase (preferably an organic layer), wherein the at least one aqueous phase (preferably layer) is then separated from the at least one organic phase (preferably layer). It will be understood that such phase separation is usually not complete, meaning that some (traces) of the aqueous phase may typically remain in the organic phase or vice versa.
- an organic solvent S O is not particularly limited with regard to its chemical structure, except for that it is an organic solvent as defined herein.
- the one or more organic solvents S O are selected so that an organic phase and an aqueous phase are formed during the respective aqueous extraction.
- the skilled person can easily identify one or more organic solvents which fulfill this criterion by routine experimentation. For example, the skilled person can simply add one or more organic solvents until an organic phase and an aqueous phase are formed during the respective aqueous extraction.
- each organic solvent S O comprises in its chemical structure at least 4 carbon atoms or at least 5 carbon atoms.
- each organic solvent S O comprises in its chemical structure 4–30 carbon atoms (i.e., not less than 4 carbon atoms and not more than 30 carbon atoms) or 5–30 carbon atoms. In some embodiments of the method of the invention, each organic solvent S O comprises in its chemical structure 4–24 or 5–24 carbon atoms. In some embodiments of the method of the invention, each organic solvent S O comprises in its chemical structure 4–18 or 5–18 carbon atoms. In some embodiments of the method of the invention, each organic solvent S O comprises in its chemical structure 5–12 carbon atoms.
- each organic solvent S O is selected from the group consisting of an amide solvent, an ether solvent, an alkane solvent, a heteroalkane solvent, and an aromatic solvent.
- amide solvent has been defined in another section of this text. Particularly preferred examples of such amide solvents are amide solvents S A as defined herein. This is to say that amide solvents S A as defined herein are organic solvents S O as defined herein, unless indicated differently in specific embodiments.
- ether solvent has been defined in another section of this text. Particularly preferred examples of such ether solvents are ether solvents S E as defined herein.
- alkane solvent refers to any solvent, whose chemical structure consists only of atoms of the elements carbon (C) and hydrogen (H), with the proviso that the carbon atoms of an alkane may only be bonded to each other via direct single bonds.
- alkane solvents are pentanes, hexanes, and heptanes.
- Such alkane solvents may be linear, branched or cyclic, unless indicated differently in specific embodiments. n-Heptane is particularly preferred.
- heteroalkane solvent refers to any solvent, whose chemical structure differs from that of an alkane solvent only in that one or more hydrogen atoms or carbon atoms are replaced by an atom of an element other than carbon (C) and hydrogen (H) (i.e., a “heteroatom”), wherein each heteroatom bonds to at least one carbon atom via a single bond (e.g., the oxygen atom of an alcohol group) or a double bond (e.g., the oxygen atom of a carbonyl group) or a triple bond (e.g., the nitrogen atom of a nitrile group).
- a single bond e.g., the oxygen atom of an alcohol group
- a double bond e.g., the oxygen atom of a carbonyl group
- a triple bond e.g., the nitrogen atom of a nitrile group
- heteroatoms are oxygen (O) and nitrogen (N) such as, e.g., in alcohols or amines or anhydrides or esters or nitriles.
- Alternative heteroatoms are halide atoms, in particular fluorine (F) or chlorine (Cl) such as, e.g., in dichloromethane and chloroform.
- aromatic solvent refers to benzene or any solvent, in which one or more hydrogen residues of said benzene moiety are substituted.
- substituents comprise alkyl groups as defined herein, heteroalkyl groups as defined herein, a nitrile group, an ether group, an ester group, an amide group, and a keto group.
- each organic solvent S O is selected from the group consisting of an ether solvent, an alkane solvent, a heteroalkane solvent, and an aromatic solvent.
- each organic solvent S O is selected from the group consisting of an amide solvent S A , an ether solvent S E , an alkane solvent, and an aromatic solvent.
- each organic solvent S O is selected from the group consisting of an ether solvent S E , an alkane solvent, and an aromatic solvent. In some embodiments of the method of the invention, each organic solvent S O is selected from the group consisting of an amide solvent S A , an ether solvent S E , an alkane solvent, and benzonitrile. In some embodiments of the method of the invention, each organic solvent S O is selected from the group consisting of an ether solvent S E , an alkane solvent, and benzonitrile.
- each organic solvent S O is selected from the group consisting of an amide solvent S A , an ether solvent S E , a pentane, a hexane, a heptane, and benzonitrile. In some embodiments of the method of the invention, each organic solvent S O is selected from the group consisting of an ether solvent S E , a pentane, a hexane, a heptane, and benzonitrile.
- each organic solvent S O is selected from the group consisting of an amide solvent S A , 4-methyltetrahydropyran, cyclopentyl methyl ether, a pentane (linear, branched or cyclic), a hexane (linear, branched or cyclic), a heptane (linear, branched or cyclic), and benzonitrile.
- each organic solvent S O is selected from the group consisting of 4-methyltetrahydropyran, cyclopentyl methyl ether, a pentane (linear, branched or cyclic), a hexane (linear, branched or cyclic), a heptane (linear, branched or cyclic), and benzonitrile.
- each organic solvent S O is selected from the group consisting of N-octyl-2-pyrrolidone, N,N- dibutylformamide, N-cyclohexyl-2-pyrrolidone, N,N-diethyldodecaneamide, 4-methyltetrahydropyran, cyclopentyl methyl ether, a pentane (linear, branched or cyclic), a hexane (linear, branched or cyclic), a heptane (linear, branched or cyclic), and benzonitrile.
- each organic solvent S O is selected from the group consisting of 4-methyltetrahydropyran, cyclopentyl methyl ether, a pentane (linear, branched or cyclic), a hexane (linear, branched or cyclic), a heptane (linear, branched or cyclic), and benzonitrile.
- each organic solvent S O is selected from the group consisting of N-octyl-2-pyrrolidone, N,N-dibutylformamide, N- cyclohexyl-2-pyrrolidone, N,N-diethyldodecaneamide, 4-methyltetrahydropyran, cyclopentyl methyl ether, a hexane (linear, branched or cyclic), a heptane (linear, branched or cyclic), and benzonitrile.
- each organic solvent S O is selected from the group consisting of 4-methyltetrahydropyran, cyclopentyl methyl ether, a hexane (linear, branched or cyclic), a heptane (linear, branched or cyclic), and benzonitrile.
- each organic solvent S O is selected from the group consisting of N-octyl-2-pyrrolidone, N,N-dibutylformamide, N- cyclohexyl-2-pyrrolidone, N,N-diethyldodecaneamide, 4-methyltetrahydropyran, cyclopentyl methyl ether, a heptane (linear, branched or cyclic), and benzonitrile.
- each organic solvent S O is selected from the group consisting of 4-methyltetrahydropyran, cyclopentyl methyl ether, a heptane (linear, branched or cyclic), and benzonitrile.
- each organic solvent S O is selected from the group consisting of N-octyl-2-pyrrolidone, N,N-dibutylformamide, N-cyclohexyl-2- pyrrolidone, 4-methyltetrahydropyran, cyclopentyl methyl ether, n-heptane, and benzonitrile.
- each organic solvent S O is selected from the group consisting of 4-methyltetrahydropyran, cyclopentyl methyl ether, n-heptane, and benzonitrile. In some embodiments of the method of the invention, each organic solvent S O is selected from the group consisting of N-octyl-2-pyrrolidone, N,N-dibutylformamide, 4-methyltetrahydropyran, cyclopentyl methyl ether, n-heptane, and benzonitrile.
- each organic solvent S O is selected from the group consisting of N-octyl-2-pyrrolidone, N,N-dibutylformamide, 4-methyltetrahydropyran, n-heptane, and benzonitrile. In some embodiments of the method of the invention, each organic solvent S O is selected from the group consisting of 4-methyltetrahydropyran, n-heptane, and benzonitrile. In some embodiments of the method of the invention, each organic solvent S O is selected from the group consisting of N-octyl-2-pyrrolidone, 4-methyltetrahydropyran, n-heptane, and benzonitrile.
- the one or more aqueous extractions are not carried out in the presence of an amide solvent S A . In some embodiments of the method of the invention, the one or more aqueous extractions are not carried out in the presence of an amide solvent comprising equal to more than 6 carbon atoms. In some embodiments of the method of the invention, the one or more aqueous extractions are not carried out in the presence of an amide solvent comprising equal to more than 5 carbon atoms. In some embodiments of the method of the invention, the one or more aqueous extractions are not carried out in the presence of an amide solvent comprising equal to more than 4 carbon atoms.
- An aqueous solution may optionally also comprise one or more dissolved components.
- dissolved components comprise: - inorganic salts such as alkali halides, in particular sodium chloride (e.g., typically used to facilitate and/or accelerate phase separation), - water-soluble protic acids such as acetic acid, - water-soluble organic or inorganic bases such as N-methylmorpholine (NMM), pyridine, sodium or potassium or ammonium carbonate or hydrogen carbonate, and sodium or potassium or ammonium phosphate or hydrogen phosphate or dihydrogen phosphate, and - mixtures thereof.
- - inorganic salts such as alkali halides, in particular sodium chloride (e.g., typically used to facilitate and/or accelerate phase separation)
- - water-soluble protic acids such as acetic acid
- - water-soluble organic or inorganic bases such as N-methylmorpholine (NMM)
- NMM N-methylmorpholine
- pyridine sodium or potassium or ammonium carbon
- the organic phase comprises the component (C-0) #
- the organic phase comprises the component (C-0) #
- the organic phase may be understood in the broadest sense to mean that some of the molecules of said component (C-0) # are dissolved in the organic phase. It is preferred that most molecules (e.g., more than 50 %, 60 %, 70 %, 80 %, or 90 % of the molecules) of the said component (C-0) # are comprised in the organic phase (and thus not in the aqueous phase).
- Said “solution comprising a component (C-0) # ” may, for example, be a reaction mixture or it may, for example, be obtained from a reaction mixture by addition of one or more compounds and/or solvents.
- bases preferably bases having a pKa-value (determined in water, i.e., an aqueous solution of the base, at 25 °C) equal to or smaller than 9.0 or 8.0 or 7.0 or 6.5 or 6.0 or 5.5.
- Pyridine is an example of such a base. The protonated from of pyridine has a pKa- value smaller than 5.5. Pyridine is a weak base compared to, e.g., triethylamine, whose protonated from has a pKa-value larger than 9.0.
- solvents which may be added to a reaction mixture comprising the component (C-0) # are non-polar solvents.
- non-polar solvents may also be added during or in between the one or more aqueous extractions.
- non-polar solvents which may be added comprise: - ether solvents such as, e.g., alkylated derivatives of tetrahydropyran or tetrahydrofuran, in particular 4-methyltetrahydropyran (MTHP) and 2-methyltetrahydrofuran, cyclopentyl methyl ether (CPME), tert-butyl methyl ether, diethyl ether, and anisole; - aromatic solvents such as, e.g., benzene, benzonitrile, toluene, o- or m- or p- xylene, and mesitylene; - aliphatic hydrocarbon solvents such as, e.g., pentanes, hexanes, heptanes, octanes, nonanes, and cyclic derivative
- MTHP 4-Methyltetrahydropyran
- MTHP 4-Methyltetrahydropyran
- one or more amide solvents S A may be added prior to or during or in between the aqueous extraction(s).
- the one or more aqueous extractions are carried out in essentially halogen-free solvents.
- essentially halogen-free solvent refers to a solvent which contains in total equal to or less than 3.0 vol-%, 2.0 vol-%, 1.0 vol-%, 0.1 vol-%, 0.01 vol-%, or 0.001 vol-% of halogenated solvents.
- halogenated solvent refers to any solvent comprising in its chemical structure at least one halogen atom. Examples of halogenated solvents comprise dichloromethane, chloroform, 1,1-dichloroethane, and 1,2-dichloroethane.
- the one or more aqueous extractions are carried out in essentially halogen-free solvents.
- - the solution comprising the component (C-0) # comprises in total equal to or less than 3.0 vol-%, 2.0 vol-%, 1.0 vol-%, 0.1 vol-%, 0.01 vol-%, or 0.001 vol-% of halogenated solvents; and - only essentially halogen-free solvents may be added prior to, during or in between the one or more aqueous extractions.
- each organic phase and each aqueous phase of the one or more aqueous extractions comprises in total less than 3.0 vol-%, 2.0 vol-%, 1.0 vol-%, 0.1 vol-%, 0.01 vol-%, or 0.001 vol-% of halogenated solvents.
- the expression “in total less than 3.0 vol-%, 2.0 vol-%, 1.0 vol-%, 0.1 vol-%, 0.01 vol-%, or 0.001 vol-% of halogenated solvents” means that, if more than one halogenated solvents (e.g., dichloromethane and chloroform) are comprised, their vol-% are to be summed up to obtain the total vol- % which is to be less than 3.0 vol-%, 2.0 vol-%, 1.0 vol-%, 0.1 vol-%, 0.01 vol-%, or 0.001 vol-%.
- halogenated solvents e.g., dichloromethane and chloroform
- said one or more aqueous extractions comprise a first aqueous extraction comprising the following steps (Ex-1) to (Ex-4): (Ex-1) Combining the solution comprising the component (C-0) # with a first aqueous solution AS-1; (Ex-2) Agitating the mixture of step (Ex-1); (Ex-3) Allowing the phases to separate, so as to obtain a first organic phase OP-1 and a first aqueous phase AP-1, wherein the first organic phase OP-1 comprises the component (C-0) # ; and (Ex-4) Removing the first aqueous phase AP-1 from the first organic phase OP-1 or vice versa; wherein the solution comprising the component (C-0) # of step (Ex-1) comprises one or more organic solvents S O and/or one or more organic solvents S O are added during any one of steps (Ex-1) and (Ex-2) and/or in between these steps.
- said one or more aqueous extractions comprise a first aqueous extraction comprising the aforementioned steps (Ex-1) to (Ex-4), and a second aqueous extraction comprising the following steps (Ex-5) to (Ex-8): (Ex-5) Combining the first organic phase OP-1 with a second aqueous solution AS-2; (Ex-6) Agitating the mixture of step (Ex-5); (Ex-7) Allowing the phases to separate, so as to obtain a second organic phase OP-2 and a second aqueous phase AP-2, wherein the second organic phase OP-2 comprises the component (C-0) # ; and (Ex-8) Removing the second aqueous phase AP-2 from the second organic phase OP-2 or vice versa; wherein the first organic phase OP-1 comprises one or more organic solvents S O and, optionally, one or more organic solvents S O are added during any one of steps (Ex-5) and (Ex-6) and/
- said one or more aqueous extractions comprise a first aqueous extraction comprising the aforementioned steps (Ex-1) to (Ex-4), a second aqueous extraction comprising the aforementioned steps (Ex-5) to (Ex-8), and a third aqueous extraction comprising the following steps (Ex-9) to (Ex-12): (Ex-9) Combining the second organic phase OP-2 with a third aqueous solution AS-3; (Ex-10) Agitating the mixture of step (Ex-9); (Ex-11) Allowing the phases to separate, so as to obtain a third organic phase OP-3 and a third aqueous phase AP-3, wherein the third organic phase OP-3 comprises the component (C-0) # ; and (Ex-12) Removing the third aqueous phase AP-3 from the third organic phase OP-3 or vice versa; wherein the second organic phase OP-2 comprises one or more organic solvents S O and, optionally, one or
- Such further aqueous extractions preferably comprise the following steps (Ex-A) to (Ex-D): (Ex-A) Combining the organic phase obtained from the previous aqueous extraction with an aqueous solution; (Ex-B) Agitating the mixture of step (Ex-A); (Ex-C) Allowing the phases to separate, so as to obtain an organic phase and an aqueous phase, wherein the organic phase comprises the component (C- 0) # ; and (Ex-D) Removing the aqueous phase from the organic phase or vice versa; wherein the organic phase obtained from the previous aqueous extraction comprises one or more organic solvents S O and, optionally, one or more organic solvents S O are added during any one of steps (Ex-A) and (Ex-B) and/or in between these steps.
- the aqueous solutions of two or more aqueous extractions need not be identical, i.e., they may or may not comprise the same species/components/solvents and the amounts/volumes of said species/components/solvents may be the same or different, unless indicated differently in specific embodiments.
- the aqueous phase of each, of said one or more aqueous extractions has a pH-value equal to or smaller than 7, preferably in the range of 4–7 or 4–6.
- the pH-value of the aqueous phase of an aqueous extraction is to be determined from the respective aqueous phase after the aqueous extraction (i.e., after the phase separation, preferably after removing the aqueous phase from the organic phase or vice versa) and at a temperature in the range of 23–28 °C, preferably 25 °C.
- the term “aqueous phase” of an aqueous extraction is distinct from the aqueous solution, which is initially combined with the solution comprising the component (C-0) # , followed by extraction.
- the “aqueous phase” typically comprises species which have been extracted from the solution comprising the component (C-0) # , e.g., water-soluble species.
- the aqueous phase of an aqueous extraction has a certain pH-value is not the same as to say that the aqueous solution employed in the aqueous extraction has a certain pH-value.
- pure water or any other aqueous solution may be used for an aqueous extraction, and during the aqueous extraction one or more protic acids may partition from said solution comprising the component (C-0) # into the aqueous phase, so that the aqueous phase then has a pH-value which is lower than the pH-value of the water or other aqueous solution employed.
- said one or more aqueous extractions comprise a first aqueous extraction comprising the following steps (Ex-1) to (Ex-4): (Ex-1) Combining the solution comprising the component (C-0) # with a first aqueous solution AS-1; (Ex-2) Agitating the mixture of step (Ex-1); (Ex-3) Allowing the phases to separate, so as to obtain a first organic phase OP-1 and a first aqueous phase AP-1, wherein the first organic phase OP-1 comprises the component (C-0) # ; and (Ex-4) Removing the first aqueous phase AP-1 from the first organic phase OP-1 or vice versa; wherein - the solution comprising the component (C-0) # of step (Ex-1) comprises one or more organic solvents S O and/or one or more organic solvents S O are added during any one of steps (Ex-1) and (Ex-2) and/or in between these steps; and -
- said one or more aqueous extractions comprise a first aqueous extraction comprising the aforementioned steps (Ex-1) to (Ex-4), and a second aqueous extraction comprising the following steps (Ex-5) to (Ex-8): (Ex-5) Combining the first organic phase OP-1 with a second aqueous solution AS-2; (Ex-6) Agitating the mixture of step (Ex-5); (Ex-7) Allowing the phases to separate, so as to obtain a second organic phase OP-2 and a second aqueous phase AP-2, wherein the second organic phase OP-2 comprises the component (C-0) # ; and (Ex-8) Removing the second aqueous phase AP-2 from the second organic phase OP-2 or vice versa; wherein - the first organic phase OP-1 comprises one or more organic solvents S O and, optionally, one or more organic solvents S O are added during any one of steps (Ex-5) and (Ex-6)
- said one or more aqueous extractions comprise a first aqueous extraction comprising the aforementioned steps (Ex-1) to (Ex-4), a second aqueous extraction comprising the aforementioned steps (Ex-5) to (Ex-8), and a third aqueous extraction comprising the following steps (Ex-9) to (Ex-12): (Ex-9) Combining the second organic phase OP-2 with a third aqueous solution AS-3; (Ex-10) Agitating the mixture of step (Ex-9); (Ex-11) Allowing the phases to separate, so as to obtain a third organic phase OP- 3 and a third aqueous phase AP-3, wherein the third organic phase OP-3 comprises the component (C-0) # ; and (Ex-12) Removing the third aqueous phase AP-3 from the third organic phase OP-3 or vice versa; wherein - the second organic phase OP-2 comprises one or more organic solvents S O and, optionally
- Such further aqueous extractions preferably comprise the following steps (Ex-A) to (Ex-D): (Ex-A) Combining the organic phase obtained from the previous aqueous extraction with an aqueous solution; (Ex-B) Agitating the mixture of step (Ex-A); (Ex-C) Allowing the phases to separate, so as to obtain an organic phase and an aqueous phase, wherein the organic phase comprises the component (C- 0) # ; and (Ex-D) Removing the aqueous phase from the organic phase or vice versa; wherein - the organic phase obtained from the previous aqueous extraction comprises one or more organic solvents S O and, optionally, one or more organic solvents S O are added during any one of steps (Ex-A) and (Ex-B) and/or in between these steps; and - the aqueous phase obtained in each of steps (Ex-C) has a pH-value equal to or smaller than 7, preferably in the range of 4.0–7.0 or 4.0–6.0, in particular in the range
- the first aqueous solution AS-1 has a pH-value in the range of 4.0–8.0, 5.0–8.0, 4.0–7.5, 4.5–7.0, 5.0–7.0, 5.0–6.5 or 5.0–6.0.
- the first aqueous solution AS-1 does not comprise a compound having a pKa-value equal to or larger than 9.0, 8.5, 8.0, 7.5, 7.0, 6.5, 6.0 or 5.5.
- the pKa-value is to be determined in water (i.e., in an aqueous solution of the respective compound) at 25 °C.
- pyridine is a base and its protonated form has a pKa value of 5.2 (cf., e.g., A. Fischer et al., J. Chem. Soc.
- pyridine is not a compound having a pKa-value equal to or larger than 5.5.
- 4-aminopyridine is also a base and its protonated form has a pKa-value of 9.1 (cf., e.g., A. Fischer et al., J. Chem. Soc. 1964, 3591–3596; https://doi.org/10.1039/JR9640003591).
- 4-aminopyridine is a compound having a pKa-value equal to or larger than 9.0.
- each of the first aqueous solution AS-1 and the second aqueous solution AS-2 has a pH-value in the range of 5.0–8.0, 4.0–7.5, 4.5–7.0, 5.0–7.0, 5.0–6.5 or 5.0–6.0.
- none of the first aqueous solution AS-1 and the second aqueous solution AS-2 comprises a compound having a pKa-value equal to or larger than 9.0, 8.5, 8.0, 7.5, 7.0, 6.5, 6.0 or 5.5.
- the pKa-value is to be determined in water (i.e., in an aqueous solution of the respective compound) at 25 °C.
- each of the first aqueous solution AS-1, the second aqueous solution AS-2, and the third aqueous solution AS-3 has a pH-value in the range of 5.0–8.0, 4.0–7.5, 4.5–7.0, 5.0–7.0, 5.0–6.5 or 5.0–6.0.
- none of the first aqueous solution AS-1, the second aqueous solution AS-2, and the third aqueous solution AS-3 comprises a compound having a pKa-value equal to or larger than 9.0, 8.5, 8.0, 7.5, 7.0, 6.5, 6.0 or 5.5.
- Step (Ex-1) is: Combining the solution comprising the component (C-0) # with a first aqueous solution AS-1. This means that the solution comprising the component (C- 0) # and the first aqueous solution AS-1 are combined in the vessel or funnel or other receptacle, in which the agitation of step (Ex-2) is to be performed.
- Step (Ex-5) is: Combining the first organic phase OP-1 with a second aqueous solution AS-2.
- Step (Ex-9) is: Combining the second organic phase OP-2 with a third aqueous solution AS-3.
- Step (Ex-A) is: Combining the organic phase obtained from the previous aqueous extraction with an aqueous solution.
- step (Ex-B) the organic phase from the previous aqueous extraction and the aqueous solution are combined in the vessel or funnel or other receptacle, in which the agitation of step (Ex-B) is to be performed.
- the term “agitating” in steps (Ex-2), (Ex-6), (Ex-10), and (Ex-B) may be understood in the broadest sense to refer to any operation of inducing a movement of the respective mixture. This typically facilitates the extraction process.
- Suitable means of agitation are known to those skilled in the art and comprise, e.g., shaking, stirring, e.g., mechanical stirring, bubbling of an inert has such as nitrogen, and inversion of the vessel or funnel or other receptacle in which the one or more aqueous extractions may be carried out. More than one such means of agitation may be used in combination.
- a reaction vessel made of a suitable material, e.g., glass or stainless steel, equipped with a mechanical stirrer may preferably be used for the agitation step (Ex-2), (Ex-6), (Ex-10) or (Ex-B).
- step (Ex-2), (Ex-6), (Ex-10), and (Ex-B) the respective organic phase and the respective aqueous phase usually form a dispersion. Under such conditions, one phase may typically not be removed from the other. For this purpose, phase separation is usually required.
- - Step (Ex-3) comprises “Allowing the phases to separate, so as to obtain a first organic phase OP-1 and a first aqueous phase AP-1”
- - Step (Ex-7) comprises “Allowing the phases to separate, so as to obtain a second organic phase OP-2 and a second aqueous phase AP-2”
- - Step (Ex-11) comprises “Allowing the phases to separate, so as to obtain a third organic phase OP-3 and a third aqueous phase AP-3”
- - Step (Ex-C) comprises “Allowing the phases to separate, so as to obtain an organic phase and an aqueous phase.”
- the term “allowing the phases to separate so as to obtain an organic phase and an aqueous phase” may be understood in the broadest sense as establishing conditions under which the organic phase forms one or more, preferably one, layer (i.e., organic layer) and the aqueous phase forms one or more, preferably one, layer (i.e., aqueous layer).
- This definition likewise applies to the first, second, and third organic phase OP-1, OP-2, and OP-3 and the respective first, second, and third aqueous phase AP-1, AP-2, and AP-3.
- an organic layer formed from an organic phase may typically not be completely free of an aqueous phase and that an aqueous layer formed from an aqueous phase may typically not be completely free of an organic phase.
- an organic layer is mostly but not necessarily completely composed of an organic phase and an aqueous layer is mostly but not necessarily completely composed of an aqueous phase.
- “Establishing conditions under which the organic phase forms one or more, preferably one, layer (i.e., organic layer) and the aqueous phase forms one or more, preferably one, layer (i.e., aqueous layer)” may usually comprise stopping the agitation. This means that the shaking and/or stirring and/or inert gas bubbling and/or inversion of the vessel or funnel or other receptacle is stopped. It will be understood that some movement of the liquids may still occur.
- step (Ex-3) is: Stopping the agitation and allowing the phases to separate, so as to obtain a first organic phase OP-1 and a first aqueous phase AP-1, wherein the first organic phase OP-1 comprises the component (C-0) # .
- step (Ex-7) is: Stopping the agitation and allowing the phases to separate, so as to obtain a second organic phase OP-2 and a second aqueous phase AP-2, wherein the second organic phase OP-2 comprises the component (C-0) # .
- step (Ex- 11) is: Stopping the agitation and allowing the phases to separate, so as to obtain a third organic phase OP-3 and a third aqueous phase AP-3, wherein the third organic phase OP-3 comprises the component (C-0) # .
- step (Ex-C) is: Stopping the agitation and allowing the phases to separate, so as to obtain an organic phase and an aqueous phase, wherein the organic phase comprises the component (C-0) # . Typically, one may simply wait for the phases to separate.
- Phase separation may optionally be speeded up by addition of aqueous solutions comprising dissolved ions, e.g., by addition of brine (i.e., an aqueous solution of sodium chloride), as known to those skilled in the art.
- brine i.e., an aqueous solution of sodium chloride
- the respective aqueous phase is removed from the respective organic phase or vice versa. This means that at least one aqueous layer is physically removed from at least one organic layer or vice versa. This will be understood by the skilled artisan.
- This removal may typically comprise draining of one layer, typically the bottom layer, e.g., the aqueous layer from the vessel or funnel or other receptacle, in which the agitation has been carried out. It will be understood that this removal is not necessarily complete. In other words, “removing” in this context is not to mean “completely removing” but preferably means “mostly removing”. In this context, removing one phase from another phase may mean removing at least 51 vol-%, 60 vol-%, 70 vol-%, 80 vol- %, 90 vol-%, 95 vol-%, 96 vol-%, 97 vol-%, 98 vol-% or 99 vol-% of the respective phase to be removed.
- the component (C-0) # is a compound of the above- mentioned Formula I-C, which is a compound of Formula I below, wherein, PG-0 is absent, so that the hydroxyl moiety otherwise protected by PG-0 is a free hydroxyl group.
- any definitions and embodiments relating to the component C-0 of Formula I likewise apply to the component (C-0) # of the above- mentioned Formula I-C, with the proviso that the protecting group PG-0 of Formula I is absent in the component (C-0) # of Formula I-C, so that the hydroxyl moiety, which is protected by the protecting group PG-0 in Formula I is always a free hydroxyl group (OH) in the component (C-0) # of Formula I-C.
- said solution comprising the component (C-0) # further comprises one or more carbocation scavengers.
- the term “carbocation scavenger” relates to a nucleophilic compound, which may be used to bind a carbocation or to consume a carbocation by formal donation of a hydride anion, thereby preventing unwanted side reactions of the carbocation.
- Examples of carbocation scavengers are given below for the deprotection mixtures M-(b-1), M-(b-2), and M-(b-x) and likewise apply to the aforementioned embodiments.
- said solution comprising the component (C-0) # further comprises one or more carbocation scavengers selected from the group consisting of - compounds comprising one or more sulfhydryl groups and one or more carboxyl groups, and - compounds comprising an indole residue and one or more carboxyl groups.
- carbocation scavengers are extractable carbocation scavengers, meaning that they partly or mostly partition into the aqueous phase during an aqueous extraction. If a carbocation scavenger is used, it is preferred that it is such an extractable carbocation scavenger, so that the scavenger itself does (mostly) not remain in the organic phase.
- said solution comprising the component (C-0) # further comprises one or more carbocation scavengers selected from the group consisting of compounds comprising one or more sulfhydryl groups and one or more carboxyl groups.
- said solution comprising the component (C-0) # further comprises one or more carbocation scavengers selected from the group consisting of compounds comprising one sulfhydryl groups and one or two carboxyl groups. In some embodiments, said solution comprising the component (C-0) # further comprises one or more carbocation scavengers selected from the group consisting of glutathione, thiomalic acid, and 3-mercapropropionic acid. In some embodiments, said solution comprising the component (C-0) # further comprises one or more carbocation scavengers selected from the group consisting of glutathione and thiomalic acid.
- said solution comprising the component (C-0) # further comprises glutathione.
- the target oligonucleotide O T comprises a first cycle oligonucleotide O-1, and the method comprises the following step (a-1), and a first coupling cycle comprising the following steps: (a-1) providing a component C-0 selected from the group consisting of a nucleoside and an oligonucleotide, wherein the component C-0 is covalently bonded to a pseudo solid-phase protecting group PG-s and comprises a backbone hydroxyl moiety protected by a protecting group PG-0 removable under acidic conditions, wherein the component C-0 is a compound of the following Formula I: (Formula I), wherein in in Formula I: each oxygen atom O depicted within each nucleoside subunit x-0 to x-m represents the oxygen atom of a hydroxyl moiety of the respective nucleoside subunit; each
- no solid-liquid separation of the first cycle oligonucleotide O-1 or of any oligonucleotidic educts or intermediates involved in the synthesis of the first cycle oligonucleotide O-1 is performed during and in between steps (b-1) to (h-1) (as far as present) and steps (c-1) and (g-1) are carried out in the presence of one or more organic solvents S O .
- steps (c-1) and (g-1) (as far as present) are not carried out in the presence of an amide solvent S A .
- steps (c-1) and (g-1) (as far as present) are not carried out in the presence of an amide solvent comprising equal to more than 6 carbon atoms. In some embodiments of the method of the invention, steps (c-1) and (g-1) (as far as present) are not carried out in the presence of an amide solvent comprising equal to more than 5 carbon atoms. In some embodiments of the method of the invention, steps (c-1) and (g-1) (as far as present) are not carried out in the presence of an amide solvent comprising equal to more than 4 carbon atoms.
- the target oligonucleotide O T comprises a second cycle oligonucleotide O-2
- the method further comprises performing a second coupling cycle comprising the following steps: (b-2) incubating the first cycle oligonucleotide O-1 obtained in the first coupling cycle with a deprotection mixture M-(b-2), thereby cleaving the protecting group PG-1 from the first cycle oligonucleotide O-1, so as to obtain a first cycle oligonucleotide (O-1) # having a free backbone hydroxyl group; (c-2) subjecting a solution comprising the first cycle oligonucleotide (O-1) # to one or more aqueous extractions, wherein the organic phase comprises the first cycle oligonucleotide (O-1) # ; (d-2) optionally, reducing the water content of the organic phase comprising the first cycle oligonucleotide (O-1) # ;
- the target oligonucleotide O T comprises a second cycle oligonucleotide O-2
- the method further comprises performing a second coupling cycle comprising the above steps (b-2) to (h-2) (as far as present), wherein: no solid-liquid separation of the second cycle oligonucleotide O-2 or of any oligonucleotidic educts or intermediates involved in the synthesis of the second cycle oligonucleotide O-2 is performed during and in between steps (b-1) to (h-2) (as far as present) and steps (c-1), (g-1), (c-2), and (g-2) are carried out in the presence of one or more organic solvents S O .
- the first cycle oligonucleotide O-1 is an oligonucleotidic educt or intermediate involved in the in the synthesis of the second cycle oligonucleotide O- 2.
- the target oligonucleotide O T comprises a second cycle oligonucleotide O-2, and the method further comprises performing a second coupling cycle comprising the above steps (b-2) to (h-2) (as far as present), wherein steps (c-1), (g-1), (c-2), and (g-2) (as far as present) are not carried out in the presence of an amide solvent S A .
- the target oligonucleotide O T comprises a second cycle oligonucleotide O-2
- the method further comprises performing a second coupling cycle comprising the above steps (b-2) to (h-2) (as far as present), wherein steps (c-1), (g-1), (c-2), and (g-2) (as far as present) are not carried out in the presence of an amide solvent comprising equal to more than 6 carbon atoms.
- the target oligonucleotide O T comprises a second cycle oligonucleotide O-2
- the method further comprises performing a second coupling cycle comprising the above steps (b-2) to (h-2) (as far as present), wherein steps (c-1), (g-1), (c-2), and (g-2) (as far as present) are not carried out in the presence of an amide solvent comprising equal to more than 5 carbon atoms.
- the target oligonucleotide O T comprises a second cycle oligonucleotide O-2
- the method further comprises performing a second coupling cycle comprising the above steps (b-2) to (h-2) (as far as present), wherein steps (c-1), (g-1), (c-2), and (g-2) (as far as present) are not carried out in the presence of an amide solvent comprising equal to more than 4 carbon atoms.
- the target oligonucleotide O T comprises a n-th cycle oligonucleotide O-n
- the method further comprises performing performing (n ⁇ 2) iterations of a coupling cycle comprising the following steps (b-x) to (h-x) (as far as present), wherein n is an integer in the range of 3 to 99, which denotes the total number of coupling cycles performed to obtain the n-th cycle oligonucleotide O-n
- each individual coupling cycle comprising the following steps (b-x) to (h-x) (as far as present) is identified by a serial number x, which runs in steps of 1 from 3 to n: (b-x) incubating the (x ⁇ 1)-th cycle oligonucleotide O-(x ⁇ 1) obtained in the previous coupling cycle with a deprotection mixture M-(b-x), thereby cleaving the protecting group PG-(x ⁇ 1) from the (
- the target oligonucleotide O T comprises a n-th cycle oligonucleotide O-n
- the method further comprises performing (n ⁇ 2) iterations of a coupling cycle comprising the above steps (b-x) to (h-x) (as far as present), wherein n is an integer in the range of 3 to 99, which denotes the total number of coupling cycles performed to obtain to obtain the n-th cycle oligonucleotide O-n
- each individual coupling cycle comprising the above steps (b-x) to (h-x) (as far as present) is identified by a serial number x, which runs in steps of 1 from 3 to n, wherein: no solid-liquid separation of the n-th cycle oligonucleotide O-n or of any oligonucleotidic educts or intermediates involved in the synthesis of the n-th cycle oligonucleotide O-n is performed
- any previous cycle oligonucleotide is an oligonucleotidic educt or intermediate involved in the synthesis of the n-th cycle oligonucleotide O-n.
- the target oligonucleotide O T comprises a n-th cycle oligonucleotide O-n
- the method further comprises performing (n ⁇ 2) iterations of a coupling cycle comprising the above steps (b-x) to (h-x) (as far as present), wherein steps (c-1), (g-1), (c-2), and (g-2) (as far as present) as well as each iteration of steps (c-x) and (g-x) (as far as present) are not carried out in the presence of an amide solvent S A .
- the target oligonucleotide O T comprises a n-th cycle oligonucleotide O-n
- the method further comprises performing (n ⁇ 2) iterations of a coupling cycle comprising the above steps (b-x) to (h-x) (as far as present), wherein steps (c-1), (g-1), (c-2), and (g-2) (as far as present) as well as each iteration of steps (c-x) and (g-x) (as far as present) are not carried out in the presence of an amide solvent comprising equal to more than 6 carbon atoms.
- the target oligonucleotide O T comprises a n-th cycle oligonucleotide O-n
- the method further comprises performing (n ⁇ 2) iterations of a coupling cycle comprising the above steps (b-x) to (h-x) (as far as present), wherein steps (c-1), (g-1), (c-2), and (g-2) (as far as present) as well as each iteration of steps (c-x) and (g-x) (as far as present) are not carried out in the presence of an amide solvent comprising equal to more than 5 carbon atoms.
- the target oligonucleotide O T comprises a n-th cycle oligonucleotide O-n
- the method further comprises performing (n ⁇ 2) iterations of a coupling cycle comprising the above steps (b-x) to (h-x) (as far as present), wherein steps (c-1), (g-1), (c-2), and (g-2) (as far as present) as well as each iteration of steps (c-x) and (g-x) (as far as present) are not carried out in the presence of an amide solvent comprising equal to more than 4 carbon atoms.
- target oligonucleotide O T may be understood synonymously with “target oligonucleotide (O T )”. This means “O T “ and “(O T )” may be understood as reference marks, which do not imply any further limitation, unless indicated differently.
- step (b-1) is referred to as deprotection mixture M-(b-1).
- the component C-0 is a specific component (a nucleoside or oligonucleotide), which may be used as starting material in the method of the invention. Since it is provided in step (a-1), which is performed prior to the first coupling cycle, a "0" has been added to its reference mark. This is not to be construed to be limiting in any kind.
- the component C is also a specific component (a nucleoside or oligonucleotide).
- the component (C-0) # differs from the component C-0 in that the protecting group PG-0 is absent, so that (C-0) # comprises a free backbone hydroxyl group.
- target oligonucleotide O T refers to any specific oligonucleotide which is to be synthesized by the method of the invention.
- target oligonucleotide O T is generally the final oligonucleotide product of the method of the invention.
- the “target oligonucleotide O T may have the same sequence as the n-th cycle oligonucleotide O-n, or may comprise one or more further nucleoside moieties, and/or may be obtained by conjugating the n-th cycle oligonucleotide O-n to another compound.
- oligonucleotide is used in a most general way to relate to any oligomers comprising at least two nucleoside subunits interconnected via an internucleosidic linkage group of any one of Formulae A and B: (Formula A), wherein in Formula A: X 1 is selected from the group consisting of O and S; X 2 is selected from the group consisting of O-R 1 , S-R 1 , and H; and R 1 may be any conceivable residue, and is preferably selected from the group consisting of H and a protecting group (preferably a protecting group removable under alkaline conditions, in particular the 2-cyanoethyl group, i.e.
- internucleosidic linkage groups of Formulae A or B may be protonated (e.g. at a carbonyl or thiocarbonyl group) or deprotonated (e.g. hydroxyl or sulfhydryl groups may be deprotonated), without this being indicated specifically in Formulae A and B.
- Formulae A and B will be understood to embrace any salts, stereoisomeric and tautomeric forms of the respective internucleosidic linkage groups, without this being indicated specifically in Formulae A and B.
- Internucleosidic linkage groups also referred to as internucleoside linkage groups or linkage groups, may be classified depending on the oxidation state of the respective phosphorus (P) atom.
- Internucleosidic linkage groups typically comprise a P (III) atom or a P (V) atom.
- P (III) or phosphorus (III) atom are used interchangeably to denote a P atom of a certain oxidation state, namely with the oxidation state III (i.e. +3).
- P (V) or phosphorus (V) atom are used interchangeably to denote a P atom of a certain oxidation state, namely with the oxidation state V (i.e. +5).
- Internucleosidic linkage groups whose P atom is a P (III) atom are herein referred to as P (III) or phosphorus (III) linkage groups.
- Internucleosidic linkage groups whose P atom is a P (V) atom are herein referred to as P (V) or phosphorus (V) linkage groups.
- Examples of P (V) linkage groups are: - phosphate diester (i.e. a phosphodiester) groups (i.e.
- P (III) linkage groups are: - phosphite triester groups (i.e. groups of Formula B with X 3 being O), - thiophosphite triester groups (i.e.
- internucleosidic linkage groups may be the same or different (i.e. have the same or different chemical structures).
- An oligonucleotide may also comprise one or more P(III) linkage groups and one or more P(V) linkage groups.
- internucleosidic linkage groups may be modified in the course of oligonucleotide synthesis.
- P(III) linkage groups may be converted to P (V) linkage groups by incubation with an oxidizing or sulfurizing agent, and/or protecting groups may be removed.
- substituted may be understood in the broadest sense and may denote any chemical residue or moiety.
- substituted may be used interchangeably.
- group is also used to denote certain substituents (i.e. residues or moieties).
- Said di(C1 ⁇ C6-alkyl)amino group may also be a cyclic amino group in which formally two alkyl residues are bonded to each other to form a cyclic structure.
- Examples of such cyclic amino groups comprise a pyrrolidine group and a piperidine group.
- the term amine moiety additionally encompasses an amine residue, which results from a reaction of an amine group with another chemical group, e.g. an amine residue involved in a bond to a protecting group.
- the term “optionally substituted” may be understood in the broadest sense to mean that in the respective structure, which is optionally substituted, one or more hydrogen residues may optionally and independently of each other be substituted by another residue, also referred to as substituent. If a “substituent” (i.e. residue or moiety) is not further specified it may be any conceivable stable atom or atom group and may preferably be selected from the group consisting of an alkyl residue, O-alkyl, a halogen residue (F, Cl, Br, I), a cyano (i.e.
- CN CN residue, a heteroalkyl residue, an alkenyl residue, a heteroalkenyl residue, an alkynyl residue, a heteroalkynyl residue, an aryl residue, a heteroaryl residue, a ketone residue (in particular C(O)alkyl), an aldehyde residue (CHO), a carboxylic acid residue or ester (in particular C(O)O-alkyl) or amide thereof, an amine group or moiety, a boryl residue (i.e. a substituent bonded via a B atom), a silyl residue (i.e.
- alkyl may be understood in the broadest sense and may be used to denote any aliphatic group (in other words: residue, moiety or substituent) which is composed of atoms of the chemical elements carbon (C) and hydrogen (H), but does not comprise any heteroatoms.
- an alkyl group as defined herein may be characterized in that it comprises at least one carbon atom and in that the one or more carbon atoms may only be bonded to each other via direct single bonds.
- an alkyl group may comprise in total 1 to 40 carbon atoms (i.e. in total not less than 1 and not more than 40 carbon atoms).
- the term “alkyl” may generally embrace unbranched, branched, and cyclic groups.
- Non-limiting examples of alkyl groups comprise methyl, ethyl, n-propyl (propane-1-yl), isopropyl (propane-2-yl), n-butyl (butan-1-yl), sec-butyl (butan-2-yl), tert-butyl (2-methylpropan-2-yl), isobutyl (2-methylpropan-1-yl), cyclohexyl, cyclopentyl, and longer alkyl chains, in particular in pseudo solid-phase protecting groups, e.g. the pseudo solid-phase protecting group PG-s and amide solvents S A .
- pseudo solid-phase protecting groups e.g. the pseudo solid-phase protecting group PG-s and amide solvents S A .
- a heteroalkyl group may comprise in total 1 to 40, 1 to 19, 1 to 6, or 1 to 5 carbon atoms.
- the term “heteroalkyl” may generally embrace unbranched, branched, and cyclic groups.
- Non-limiting examples of heteroalkyl groups comprise halogenated alkyl groups such as trifluoromethyl groups, oxygenated alkyl groups (i.e. alkyloxy groups), aminated alkyl groups (i.e. alkylamine groups, e.g. dimethylamine groups), and cyclic groups such as e.g. piperidine groups, pyrrolidine groups or morpholine groups.
- a heteroalkyl-substituent may typically be bonded via one or more of its carbon atoms.
- alkenyl and “heteroalkenyl” generally embrace both, the (E)- and the (Z)-isomers, and mixtures thereof.
- alkynyl may be understood in the broadest sense and may be used to denote any aliphatic group which may be derived from an alkyl group by introducing one or more C ⁇ C-triple bonds between carbon atoms.
- heteroalkynyl may be understood in the broadest sense and may be used to denote any aliphatic group which may be derived from a heteroalkyl group by introducing one or more C ⁇ C- triple bonds between carbon atoms.
- an alkenyl group, a heteroalkenyl group, an alkynyl group, and a heteroalkynyl group comprise at least two carbon atoms.
- aryl may be understood in the broadest sense and may be used to denote any mono- or polycyclic aromatic group, with the proviso that all aromatic ring atoms are carbon atoms. Unless indicted differently in the context of specific embodiments, an aryl group may preferably comprise in total 6 to 30, 6 to 20, or 6 to 15, in particular 6, aromatic carbon atoms.
- Benzene i.e. the phenyl group, Ph
- Ph may be a preferred example of a monocyclic aromatic (i.e. aryl) group.
- polycyclic in this regard may refer to condensed aromatic ring systems in which two or more aromatic rings are fused together. This is to say that the condensed aromatic rings share at least one bond between aromatic carbon atoms, so that this shared bond as well as the carbon atoms forming the bond are part of two or more monocyclic aromatic groups which build up the polycyclic aromatic group.
- polycyclic aromatic (i.e. aryl) groups are naphthalene (i.e. napthyl), anthracene (i.e. anthracenyl), and phenanthrene (i.e. phenanthryl).
- heteroaryl may be understood in the broadest sense and may be used to denote any mono- or polycyclic heteroaromatic group, which differs from an aromatic (i.e. aryl) group in that at least one, preferably 1 to 5 or 1 to 3, aromatic ring atoms are heteroatoms.
- a heteroatom in this context is any atom of a chemical element other than carbon and hydrogen, and may, unless indicated differently in the context of specific embodiments, preferably be an atom which is at each occurrence independently selected from the group consisting of N, O, and S.
- Non-limiting examples of monocyclic heteroaromatic (i.e. heteroaryl) groups comprise pyridine (i.e.
- Non-limiting examples of polycyclic heteroaromatic (i.e. heteroaryl) groups comprise quinoline (i.e. quinolinyl), quinazoline (i.e. quinazolinyl), quinoxaline (i.e.
- aliphatic may be used to denote any residues or compounds which are not aromatic, i.e. which do not comprise an aromatic or heteroaromatic ring system.
- an aliphatic cyclic amine moiety may, e.g., be a piperidine moiety, a pyrrolidine moiety or a morpholine moiety, but may not be a pyridine moiety, which instead is a heteroaromatic amine moiety or, more general, a heteroaryl moiety or heteroaromatic moiety.
- Non-limiting examples of aliphatic moieties are aliphatic hydrocarbon groups.
- aliphatic hydrocarbon may be understood in the broadest sense and may be used to denote any aliphatic residue which is composed of atoms of the chemical elements carbon (C) and hydrogen (H), but does not comprise any heteroatoms.
- the term “aliphatic hydrocarbon” embraces alkyl groups, alkenyl groups, and alkynyl groups as defined herein, unless indicated differently in the context of specific embodiments.
- the names of substituents i.e. residues or moieties and also groups
- this wording may be used interchangeably with the chemical name of the respective chemical atom or atom group as if it was not a substituent, with common abbreviations, and with the element symbol(s) themselves.
- a CH3-substituent may also be referred to as methyl or Me
- a Cl-substituent may also be referred to as chlorine or simply Cl
- a phenyl substituent may also be denoted as Ph
- a cyano group may also be referred to as CN.
- a C 1 ⁇ C 24 -alkyl group comprises in total 1 to 24 carbon atoms, but not less than 1 or more than 24 carbon atoms.
- a C6 ⁇ C20- aryl group comprises in total 6 to 20 aromatic ring carbon atoms, but not less than 6 or more than 20 aromatic ring carbon atoms.
- oligonucleotide when referring to an oligonucleotide, said oligonucleotide may generally be present as a mixture of isomers, in particular as a mixture of stereoisomers.
- a molecule of an oligonucleotide may (typically) only be present in a single (stereo)isomeric form at a certain point in time.
- an oligonucleotide when referring to an oligonucleotide as optionally being present as a mixture of (stereo)isomers, this may refer to a population of oligonucleotide molecules having essentially the same nucleoside sequence, wherein the molecules of said population may be present in different (stereo)isomeric forms.
- said population of oligonucleotide molecules may be a mixture of numerous discrete stereoisomers, or may be enriched in one specific stereoisomeric form, or may essentially consist of molecules of a specific stereoisomeric form. It will be understood that certain groups, for example OH and SH groups within an oligonucleotide may be deprotonated.
- an oligonucleotide as defined herein may optionally bear any counter ions known in the art, in particular cations such as sodium cations, potassium cations, magnesium cations, ammonium cations as well as in general cations derived from amines such as trimethylamine (TEA), diisopropylamine (DIPEA) or derived from heteroaromatics such as pyridine or collidine and/or anions such as chloride anions, bromide anions, acetate anions, trifluoroacetate anions, carbonate anions, hydrocarbonate anions, phosphate anions, hydrogen phosphate anions, dihydrogen phosphate anions, perchlorate anions, and combinations thereof.
- cations such as sodium cations, potassium cations, magnesium cations, ammonium cations
- amines such as trimethylamine (TEA), diisopropylamine (DIPEA) or derived from heteroaromatics such as pyridine or
- oligonucleotide as well as the (non-covalent) attachment of one or more cations and/or anions may for example occur during the synthesis, isolation, and purification of an oligonucleotide.
- an oligonucleotide as defined herein may be non-covalently bonded or associated to/with species with whom the oligonucleotide was contacted in the course of its synthesis, isolation or purification, for example cations and/or anions or residuals of protecting groups as well as traces of one or more cation scavengers, such as, e.g., thiols or silanes.
- oligonucleotides of the invention are 2 ⁇ -deoxynucleic acids (DNA), ribonucleic acids (RNA), locked nucleic acids (LNA), constrained ethyl nucleic acid analogs (cET), bridged nucleic acids (BNA), tricycloDNA, unlocked nucleic acids (UNA), small interfering RNA (siRNA), microRNA, antisense oligonucleotides (ASO), gapmers, glycerol nucleic acids, phosphorothioate oligonucleotides, phosphorodithioate oligonucleotides, as well as derivatives and analogs thereof, all of which are known to those skilled in the art.
- DNA 2 ⁇ -deoxynucleic acids
- RNA ribonucleic acids
- LNA locked nucleic acids
- cET constrained ethyl nucleic acid analogs
- BNA bridged nucleic acids
- UPA unlocked nucleic
- An oligonucleotide preferably is a linear sequence of nucleoside subunits, wherein any two adjacent nucleoside subunits are interconnected by an internucleosidic linkage group as defined above. Such a linear sequence may also be referred to as an oligonucleotide strand.
- the number of nucleoside subunits in an oligonucleotide may be denoted by a number (an integer equal to or larger than 2) followed by the syllable “mer” or by using a suitable prefix (e.g. di for 2, tri for 3, and the like).
- an oligonucleotide comprising exactly two nucleoside subunits may be denoted as a 2mer or 2mer oligonucleotide or a dinucleotide and an oligonucleotide comprising exactly 20 nucleoside subunits may be denoted as a 20mer or a 20mer oligonucleotide.
- Such an oligonucleotide strand will comprise a first terminal nucleoside subunit and a second terminal nucleoside subunit, both of which only have exactly one adjacent nucleoside subunit.
- a dinucleotide i.e.
- oligonucleotide comprising more than two nucleoside subunits, will comprise exactly two terminal nucleoside subunits and one or more non-terminal nucleoside subunits, wherein non-terminal nucleoside subunits are characterized in that they have exactly two adjacent (i.e. one antecedent and one following) nucleoside subunits.
- nucleoside subunits of an oligonucleotide refers to any two nucleoside subunits which are interconnected by an internucleosidic linkage group. It is understood that a nucleoside moiety which forms part of an oligonucleotide is herein referred to as a nucleoside subunit of said oligonucleotide.
- nucleoside moiety embraces both, nucleosides as such and nucleoside subunits of an oligonucleotide as defined herein.
- nucleoside (without the addition “moiety” or “subunit”) is however only used to denote a mononucleoside, i.e. a nucleoside moiety which is not a nucleoside subunit of an oligonucleotide.
- mononucleoside i.e. a nucleoside moiety which is not a nucleoside subunit of an oligonucleotide.
- nucleoside moieties are known to those skilled in the art.
- a nucleoside moiety may be composed of: - a carbohydrate moiety (in other words: a sugar moiety), preferably a monosaccharide moiety, more preferably a pentose moiety, in particular a ribose moiety (such as typically present in RNA) or a 2 ⁇ -deoxyribose moiety (such as typically present DNA); and - a nucleobase, wherein said carbohydrate moiety and said nucleobase may typically be covalently bonded to each other via a direct covalent bond, typically an N-glycosidic bond.
- nucleoside moiety may comprise naturally occurring nucleoside moieties as well as non-natural nucleoside moieties, in which the carbohydrate moiety and/or the nucleobase have been chemically modified or in which the nucleobase may even be absent (i.e. an abasic site).
- Non-limiting examples of naturally occurring nucleoside moieties may comprise adenosine, 2 ⁇ -deoxyadenosine, guanosine, 2 ⁇ -deoxyguanosine, cytidine, 2 ⁇ -deoxycytidine, uridine, (2 ⁇ -deoxy)thymidine, ribothymidine, inosine, and methylated derivatives thereof, all of which are known to those skilled in the art. Further examples may comprise queuosine, archaeosine, wybutosine, lysidine, and N 6 -threonylcarbamoyladenosine.
- derivative as used herein may be understood in the broadest sense and may refer to a compound obtainable from a first compound (i.e. a parent compound) by means of one or more, preferably one, chemical reaction.
- a derivative may differ from the first (parent) compound for example with regards to the substitutional pattern or with regards to the presence or absence of one or more atom, atom groups, functional groups, or protecting groups.
- chemical modifications of (non-natural) nucleoside moieties may comprise modifications of the carbohydrate, in particular of the ribose or 2 ⁇ -deoxyribose moiety.
- Such carbohydrate-modifications may exemplarily be selected from the group consisting of: - introducing an O ⁇ CH3 (i.e. O ⁇ methyl or OMe) group, an O ⁇ CH2 ⁇ CH2 ⁇ O ⁇ CH3 (i.e. O ⁇ methoxyethyl or O ⁇ MOE) group or an F-substituent (i.e.
- nucleobase encompasses both non-natural nucleobases as well as naturally occurring nucleobases such as adenine, guanine, cytosine, thymine, and uracil.
- Non-natural nucleobases may preferably be derivatives of purine or pyrimidine, which are capable of a specific interaction with another nucleobase.
- Non-natural nucleoside moieties may for example be formed from a naturally occurring carbohydrate and a non-natural nucleobase or from a non-natural carbohydrate and a naturally occurring nucleobase or from a non-natural carbohydrate and a non-natural nucleobase.
- nucleoside moieties are nucleoside moieties of Formula C: (Formula C), wherein in Formula C: B N is a nucleobase which may carry one or more protecting groups; Q 1 is selected from the group consisting of OR 3 (if the nucleoside moiety is the 3 ⁇ -terminal nucleoside moiety of an oligonucleotide or if the nucleoside moiety is a nucleoside) and an oxygen atom covalently bonded to an internucleosidic linkage group (if the respective nucleoside moiety is part of an oligonucleotide and not the 3 ⁇ -terminal nucleoside moiety thereof); R 3 is selected from the group consisting of H, a protecting group, and a conjugated moiety which is not a nucleoside, nucleotide or oligonucleotide; Q 2 is selected from the group consisting of OR 4 (if the nucleoside moiety is the 5 ⁇ -terminal nucle
- an oligonucleotide may herein generally be present as a mixture of isomers. No stereochemical information may be deduced from Formula C.
- a nucleoside moiety of Formula C is a nucleoside moiety of the following Formula C-a: (Formula C-a; carbon atoms labelled from 1 ⁇ to 5 ⁇ ), wherein in Formula C-a, the carbon atoms have been numbered (in line with common practice) from 1 ⁇ to 5 ⁇ , which merely serves illustrative purposes and should not be construed to be limiting in any kind.
- Q 1 , Q 2 , B N , R I , R II , R III , R IV , R V , R 3 , R 4 , and R 5 are defined as for Formula C. It is understood that the carbon atom numbers will be identical in any ribose or 2 ⁇ -deoxyribose based nucleoside moieties. From what has been laid out above, it will be understood that any nucleoside subunits comprising a ribose or 2 ⁇ -deoxyribose moiety, e.g.
- nucleoside moieties of Formula C and/or C-a will preferably all be incorporated into an oligonucleotide in 3 ⁇ ⁇ 5 ⁇ direction or 5 ⁇ ⁇ 3 ⁇ direction (all in the same direction).
- Such nucleoside subunits will bond to one (if it is a terminal nucleoside subunit) or two (if it is a non- terminal nucleoside subunit) internucleosidic linkage groups, wherein, preferably, bonding to said internucleosidic linkage group(s) occurs through the 3 ⁇ -hydroxyl moiety (i.e. the hydroxyl moiety bonded to the 3 ⁇ -carbon atom) and/or the 5 ⁇ -hydroxyl moiety (i.e.
- any internucleosidic linkage groups between nucleoside subunits comprising a ribose or 2 ⁇ -deoxyribose moiety are preferably bonded to the 5 ⁇ -hydroxyl moiety of one nucleoside subunit and the 3 ⁇ -hydroxyl moiety of another nucleoside subunit.
- the 3 ⁇ -hydroxyl moiety of the 3 ⁇ -terminal nucleoside subunit does not engage in bonding to an internucleosidic linkage group.
- an oligonucleotide comprises two or more nucleoside subunits (nucleoside moieties). It will further be understood that these two or more nucleoside subunits of an oligonucleotide may be the same or different (i.e. have the same or different chemical structures).
- nucleotide may be understood in the broadest sense and may preferably refer to a conjugate of a nucleoside moiety and a phosphate group or derivative thereof, wherein a hydroxyl moiety of said nucleoside moiety bonds via its oxygen atom to the phosphorus atom of the phosphate moiety or derivative thereof.
- internucleosidic linkage groups and any carbohydrate moieties e.g. ribose- or 2 ⁇ -deoxyribose moieties, are herein referred to as the “backbone” of an oligonucleotide.
- the term “backbone” of an oligonucleotide excludes the nucleobases. It will also be understood that in a (mono-)nucleoside, the backbone is the carbohydrate moiety, e.g. the ribose- or 2 ⁇ -deoxyribose moiety, since a nucleoside does not contain any internucleosidic linkage groups. It forms part of the common knowledge of those skilled in the art that oligonucleotides may be conjugated to additional moieties, which are not a nucleoside, nucleotide or oligonucleotide as defined herein, for various purposes. In particular, free OH-groups (e.g.
- the target oligonucleotide O T may be an oligonucleotide conjugate, unless indicated differently in the context of specific embodiments.
- the term “oligonucleotide conjugate” may refer to any oligonucleotide comprising at least one nucleoside-subunit which is covalently bonded to another moiety, which is not a nucleoside, nucleotide or oligonucleotide, e.g. to a moiety comprising a peptide, protein, lipid, carbohydrate, or a hydrocarbon moiety, among which carbohydrate moieties may be preferred.
- the expression “the target oligonucleotide O T comprises a n-th cycle oligonucleotide O-n” herein means that the target oligonucleotide O T comprises the nucleoside sequence of said n-th cycle oligonucleotide O-n.
- the term “nucleoside sequence” refers to an array of the nucleoside subunits within an oligonucleotide. For a linear oligonucleotide, the nucleoside sequence is usually given starting from a first terminal nucleoside subunit, optionally continuing with one or more non-terminal nucleoside subunits, and ending with a second terminal nucleoside subunit.
- nucleoside sequence of an oligonucleotide may be referred to as “sequence” of the oligonucleotide herein. Unless otherwise noted, the sequence of oligonucleotides with a phosphoribose backbone is written herein from the 5’ end (left) to the 3’ end (right). As used herein, the term “nucleoside sequence” does not specify one or more internucleosidic linkage groups and does not take into account the presence or absence of any protecting groups.
- two oligonucleotides comprising the same nucleoside subunits in the same order are herein considered to comprise the same nucleoside sequence, regardless of whether or not the internucleosidic linkage groups interconnecting these nucleoside subunits are the same or not (i.e. have the same chemical structure or not) and regardless of whether or not any atoms or functional groups within the carbohydrate moieties, the nucleobases, and the internucleosidic linkage groups are protected.
- a pseudo solid-phase protecting group is herein regarded a (permanent) protecting group and thus comprised in the general term “protecting group”, unless indicated differently in the context of specific embodiments.
- the internucleosidic linkage groups may differ with regards to the oxidation state of the phosphorus atom and/or the presence or absence of protecting groups such as the 2-cyanoethyl group.
- the exocyclic amino groups of nucleobases such as cytosine, 5-methylcytosine, guanine, and adenine may be protected or may not be protected, and a 5 ⁇ -terminal hydroxyl moiety may or may not be protected without having any impact on what is herein referred to as the nucleoside sequence.
- the target oligonucleotide O T may comprise more nucleoside subunits than the first cycle oligonucleotide O-1, for example, if the first coupling cycle comprising steps (b-1) to (h-1) (as far as present) is followed by further steps including further coupling cycles. It will be understood that, unless indicated differently in the context of specific embodiments, the target oligonucleotide O T may comprise more nucleoside subunits than the second cycle oligonucleotide O-2, for example, if the second coupling cycle comprising steps (b-2) to (h-2) (as far as present) is followed by further steps including further coupling cycles.
- the target oligonucleotide O T may comprise more nucleoside subunits than the n-th cycle oligonucleotide O-n, for example, if said (n ⁇ 2) iterations of the coupling cycle comprising steps (b-x) to (h-x) (as far as present) are followed by further steps including further coupling cycles.
- the term “further coupling cycles” embraces fragment coupling approaches, since a fragment coupling may be considered a coupling cycle in which two fragments are coupled.
- the target oligonucleotide O T may also be a conjugate as defined herein, i.e.
- the target oligonucleotide O T consists of the same nucleoside sequence as the first cycle oligonucleotide O-1. In such embodiments, the target oligonucleotide O T may only differ from the first cycle oligonucleotide O-1 with regards to the internucleosidic linkage groups and the absence or presence of protecting groups.
- the target oligonucleotide O T consists of the same nucleoside sequence as the second cycle oligonucleotide O-2. In such embodiments, the target oligonucleotide O T may only differ from the second cycle oligonucleotide O-2 with regards to the internucleosidic linkage groups and the absence or presence of protecting groups. In some embodiments of the method of the invention, the target oligonucleotide O T consists of the same nucleoside sequence as the n-th cycle oligonucleotide O-n.
- the target oligonucleotide O T may only differ from the n-th cycle oligonucleotide O-n with regards to the internucleosidic linkage groups and the absence or presence of protecting groups.
- the target oligonucleotide may preferably only comprise phosphorus (V) linkage groups while the first cycle oligonucleotide O-1, the second cycle oligonucleotide O-2, or the n-th cycle oligonucleotide O-n may comprise one or more phosphorus (III) linkage groups.
- the target oligonucleotide O T may preferably not comprise any protecting groups, while the first cycle oligonucleotide O-1, the second-cycle oligonucleotide O-2, and the n-th cycle oligonucleotide O-n typically comprise several protecting groups and are at least covalently linked to a pseudo solid-phase protecting group, which is herein considered a (permanent) protecting group.
- a pseudo solid-phase protecting group which is herein considered a (permanent) protecting group.
- the target oligonucleotide O T comprises 2 ⁇ 200, 2 ⁇ 150, 2 ⁇ 100, 2 ⁇ 90, 2 ⁇ 80, 2 ⁇ 70, 2 ⁇ 60, 2 ⁇ 50, 3 ⁇ 50, 4 ⁇ 50, 5 ⁇ 50, 5 ⁇ 40, 5 ⁇ 30, 5 ⁇ 25, 5 ⁇ 20, 6 ⁇ 20 or 6 ⁇ 18 nucleoside subunits.
- protecting group may be understood in the broadest sense as any group which is introduced into a molecule by means of chemical modification (i.e. through one or more, typically one, chemical reaction) of an atom or functional group (i.e.
- protecting and “carrying a protecting group” are herein used interchangeably to denote an atom or functional group covalently bonded to a protecting group.
- a protecting group used for protecting an amine moiety may herein be referred to as an amine protecting group or amino protecting group.
- a protecting group used for protecting a hydroxyl moiety may herein be referred to as a hydroxyl protecting group. It will be understood by those skilled in the art that a protecting group may substitute a hydrogen residue from the atom or functional group to be protected.
- hydroxyl group of a first nucleoside or oligonucleotide is reacted with a phosphorus moiety of second nucleoside or oligonucleotide, which typically comprises a protected backbone hydroxyl moiety. Protection of this backbone hydroxyl moiety is required to avoid double insertion of said second nucleoside or oligonucleotide and/or multimerization.
- this hydroxyl moiety Prior to the next coupling step, this hydroxyl moiety is typicall deprotected to then engage in the next condensation reaction with the phosphorus moiety of a third nucleoside or oligonucleotide, and so on.
- the (hydroxyl) protecting groups cleaved prior to each condensation reaction (i.e.
- a pseudo solid-phase protecting group preferably is a permanent protecting group, since it shall typically influence the solubility of the growing oligonucleotide strand until it has been fully assembled (i.e.
- the temporary and the permanent protecting groups are typically orthogonal to each other, meaning that one type can be removed under conditions, which do not affect the other type of protecting group.
- the permanent protecting groups are removable (i.e. cleavable) under alkaline conditions and the temporary protecting groups are removable (i.e. cleavable) under acidic conditions.
- the terms “removing a protecting group”, “cleaving a protecting group”, and “deprotecting” a functional group are used interchangeably to denote a process, preferably a chemical reaction, of removing a protecting group from an atom or functional group, e.g.
- a hydroxyl or amine moiety so that the latter is again available in free form, e.g. as (free) hydroxyl group or (free) amine group.
- free may be used.
- the process of removing a protecting group comprising an optionally substituted triarylmethyl residue, e.g. a DMT group may be referred to as “detritylation”.
- cleavage of a protecting group refers to a process, preferably a chemical reaction, of removing a protecting group from an atom or functional group.
- the term “pseudo solid-phase protecting group” refers to a protecting group, preferably a hydroxyl protecting group or an amine protecting group, in particular a hydroxyl protecting group, characterized in that it comprises one or more aliphatic hydrocarbon residues, with the proviso that all aliphatic hydrocarbon residues comprised in a pseudo solid-phase protecting group together comprise in total 18 ⁇ 200 carbon atoms.
- oligonucleotide synthesis using pseudo solid-phase protecting groups may be understood in the broadest sense to refer to any oligonucleotide synthesis characterized in that during at least one, preferably each, coupling cycle, a nucleoside or oligonucleotide to be elongated (i.e. which is coupled with another nucleoside or oligonucleotide) is covalently bonded to at least one pseudo solid-phase protecting group.
- the term “coupling cycle” may be understood in the broadest sense and may refer to a sequence of two or more process steps, including a deprotection step used to provide a free hydroxyl group at a nucleoside or oligonucleotide, and a subsequent coupling step (also referred to as condensation step), in which said free hydroxyl group engages in a bond forming reaction with a phosphorus moiety of another nucleoside or oligonucleotide.
- Several iterations (i.e. rounds of) coupling cycles may be carried out.
- an elongated oligonucleotide is obtained.
- a coupling cycle may be further characterized in aspects and embodiments of the method of the invention.
- the first coupling cycle comprises steps (b-1) to (h- 1) (as far as present), i.e. steps (b-1), (c-1), (d-1), (e-1), (f-1), (g-1), and (h-1), which may preferably be carried out in this order, wherein any one of steps (d-1), (f-1), (g-1), and (h-1) is optional (i.e. may or may not be carried out), unless indicated differently in the context of specific embodiments.
- the first coupling cycle comprises steps (b-1) to (h-1) (as far as present) does not alter the fact that any one of steps (d-1), (f-1), (g-1), and (h-1) is optional. It will be understood that stating that the first coupling cycle comprises steps (b-1) to (h-1) does not alter the fact that any one of steps (d-1), (f-1), (g-1), and (h-1) is optional.
- the first coupling cycle is followed by a second coupling cycle characterized in that it comprises the steps (b-2) to (h-2) (as far as present), i.e.
- steps (b-2), (c-2), (d-2), (e-2), (f-2), (g-2), and (h-2) as far as present, which may preferably be carried out in this order, wherein any one of steps (d-2), (f-2), (g-2), and (h-2) is optional (i.e. may or may not be carried out), unless indicated differently in the context of specific embodiments. It will be understood that stating that the second coupling cycle comprises steps (b-2) to (h-2) does not alter the fact that any one of steps (d-2), (f-2), (g-2), and (h-2) is optional.
- n is an integer in the range of 3 ⁇ 89, 3 ⁇ 79, 3 ⁇ 69, 3 ⁇ 59, 3 ⁇ 49, 3 ⁇ 39, 3 ⁇ 29, 3 ⁇ 19, 3 ⁇ 18, 3 ⁇ 17, 3 ⁇ 16, 3 ⁇ 15, 3 ⁇ 14, 3 ⁇ 13, 3 ⁇ 12, 3 ⁇ 11, 3 ⁇ 10, 3 ⁇ 9, 3 ⁇ 8, 3 ⁇ 7, 3 ⁇ 6, 3 ⁇ 5, or 3 ⁇ 4. It will be understood that to state that an integer is in the range of 3 ⁇ 5 means that said integer may be 3 or 4 or 5. Said integer n denotes the total number of coupling cycles performed to obtain the n-th cycle oligonucleotide O-n.
- This total number of performed coupling cycles denoted by the integer n includes the first coupling cycle comprising steps (b-1) to (h-1) (as far as present), the second coupling cycle comprising steps (b-2) to (h-2) (as far as present), as well as the (n ⁇ 2) iterations of the coupling cycle comprising steps (b-x) to (h-x) (as far as present), but does not include any coupling cycles optionally performed, e.g. to prepare component C-0 or any of the building blocks used.
- Each coupling cycle comprising steps (b-x) to (h-x) (as far as present) is herein identified by a serial number x, which runs in steps of 1 from 3 to n, with n being said integer representing the total number of coupling cycles performed to obtain the n-th cycle oligonucleotide O-n.
- the coupling cycle previous to any given coupling cycle x may be referred to as the coupling cycle (x ⁇ 1) (or (x ⁇ 1)-th cycle) and the oligonucleotide obtained in this previous coupling cycle may be referred to as the (x ⁇ 1)-th cycle oligonucleotide O-(x ⁇ 1).
- the x-th coupling cycle will start with removing the protecting group PG-(x ⁇ 1) from the (x ⁇ 1)-th-cycle oligonucleotide O-(x ⁇ 1) of the previous coupling cycle.
- the x-th cycle oligonucleotide O-x of the coupling cycle x will then be obtained by reacting the deprotected (x ⁇ 1)-th cycle oligonucleotide (O-(x ⁇ 1)) # with the building block B-x.
- the (x ⁇ 1)-th cycle oligonucleotide O-(x ⁇ 1) will be the second cycle oligonucleotide O-2 obtained in the second coupling cycle.
- the (x ⁇ 1)-th cycle oligonucleotide O-(x ⁇ 1) will be the third cycle oligonucleotide O-3 obtained in the third coupling cycle.
- Step (a-1) of some methods of the invention is not part of a coupling cycle and is carried out prior to the first coupling cycle comprising steps (b-1) to (h-1) (as far as present).
- Step (a-1) of some methods of the invention is: providing a component C-0 selected from the group consisting of a nucleoside and an oligonucleotide, wherein the component C-0 is covalently bonded to a pseudo solid-phase protecting group PG-s and comprises a backbone hydroxyl moiety protected by a protecting group PG-0 removable under acidic conditions.
- the component C-0 is a nucleoside. In some embodiments of the method of the invention, the component C-0 is an oligonucleotide.
- backbone hydroxyl moiety refers to a hydroxyl moiety which is part of the backbone of the component C-0 and will be understood based on the above explanations of the terms “hydroxyl moiety” and “backbone”.
- said hydroxyl moiety is a hydroxyl moiety of a carbohydrate moiety, in particular of an optionally substituted and/or protected ribose or 2 ⁇ -deoxyribose moiety.
- said backbone hydroxyl moiety is comprised in a terminal nucleoside subunit of the component C-0.
- the component C-0 comprises exactly one protecting group PG-0.
- the protecting group PG-0 is a protecting group “removable under acidic conditions”.
- a protecting group is “removable under acidic conditions”, if it can be cleaved by treatment with a protic acid.
- protic acid may be understood in the broadest sense and may refer to any Br ⁇ nsted acid, also referred to as Br ⁇ nsted-Lowry acid.
- protecting groups removable under acidic conditions are protecting groups comprising an optionally substituted triarylmethyl residue.
- protecting group comprising an optionally substituted triarylmethyl residue and “triarylmethyl type protecting group” may be understood in the broadest sense as any protecting group which covalently binds to the atom or functional group to be protected (e.g. to the oxygen atom of the hydroxyl moiety to be protected) via a carbon atom to which three optionally substituted aryl moieties are bonded.
- the basic triarylmethyl type protecting group is the triphenylmethyl group (i.e. the trityl group).
- substituents e.g. alkyl or alkoxy substituents, capable of stabilizing a triarylmethyl-cation formed during acid-catalyzed removal of a triarylmethyl type protecting group, may facilitate acid-catalyzed cleavage of the respective protecting group.
- triarylmethyl type protecting groups are the trityl group, the (p-methylphenyl)diphenylmethyl group (i.e. the 4-methyltrityl group), the di(p-methylphenyl)phenylmethyl group (i.e.
- the IDT group 4,4'-dimethoxy-3''-[N- (imidazolylethyl)carbamoyl]trityl group (i.e. the IET group), the bis(4-methoxyphenyl)-1'-pyrenylmethyl group (i.e. the Bmpm group), and the 4-(17- tetrabenzo[a,c,g,i]fluorenylmethyl)-4',4''-dimethoxytrityl group (i.e. the Tbf-DMT group).
- the trityl group, the MMT group, and the DMT group may be preferred.
- the DMT group is herein most preferred as temporary hydroxyl protecting group.
- said protecting group PG-0 is selected from the group consisting of the triphenylmethyl group, the (p-methoxyphenyl)diphenylmethyl group, and the di(p-methoxyphenyl)phenylmethyl group. In some embodiments of the method of the invention, said protecting group PG-0 is a di(p-methoxyphenyl)phenylmethyl group. In some preferred embodiments, the component C-0 comprises exactly one backbone hydroxyl moiety protected by a protecting group comprising an optionally substituted triarylmethyl residue, which is said protecting group PG-0.
- the component C-0 comprises exactly one protecting group comprising an optionally substituted triarylmethyl residue, which is said protecting group PG-0.
- the term “pseudo solid-phase protecting group” has been defined above and will be exemplified in more detail in a later section of this text.
- the expression “the component C-0 is covalently bonded to a pseudo solid-phase protecting group PG-s” means that there are one or more, preferably exactly one, covalent chemical bonds interconnecting the component C-0 and said pseudo solid-phase protecting group PG-s.
- the name “PG-s” has been assigned for illustrative purposes only and should not be construed to be limiting in any kind, unless indicated differently in the context of specific embodiments.
- the component C-0 is covalently bonded to exactly one pseudo solid-phase protecting group, which is said pseudo solid-phase protecting group PG-s.
- a pseudo solid-phase protecting group as defined herein is not limited with regard to the atom or functional group of the respective nucleoside moiety to which it is covalently bonded, unless indicated differently in the context of specific embodiments.
- a pseudo solid-phase protecting group may be covalently bonded to - a hydroxyl moiety of a carbohydrate moiety of a nucleoside moiety, preferably the 2 ⁇ - or 3 ⁇ -, in particular the 3 ⁇ -hydroxyl moiety, of a ribose- or 2 ⁇ -deoxyribose- type nucleoside moiety (e.g. of any one of Formulae C and C-a), or - an amine moiety of a nucleobase (e.g.
- the component C-0 is a compound of the aforementioned Formula I.
- the component C-0 of Formula I is a compound of the following Formula I-a: (Formula I-a), wherein in Formula I-a: m, Y 1 , Z 1 , R z-1 , PG-0, and PG-s are defined as for Formula I; B N is a nucleobase, which may be the same or different at each occurrence; R VI is at each occurrence independently selected from the group consisting of H, F, O-(C1 ⁇ C5-alkyl), O-(C1 ⁇ C5-alkyl)-O-(C1 ⁇ C5-alkyl), O-Si(C1 ⁇ C5-alkyl)3, and O-CH2-O-Si(C1 ⁇ C5-alkyl)3; R VIII is independently at each occurrence H or R VIII and R VI of the same nucleoside subunit (i.e.
- R VII , R IX , and R X are either all H or they are bonded together so that the respective nucleoside subunit has a structure of the following Formula I-a-tc: (Formula I-a-tc), wherein in Formula I-a-tc: the oxygen atom from which the dashed line originates represents the oxygen atom bonded to the 3 ⁇ -carbon atom, i.e.
- the carbon atom to which R VII is bonded in Formula I-a indicates the covalent chemical bond interconnecting the respective oxygen atom of the nucleoside subunit of Formula I-a-tc and the pseudo solid-phase protecting group PG-s or P(Y 1 )(Z 1 ) in Formula I-a; the oxygen atom from which the wavy line originates represents the oxygen atom bonded to the 5 ⁇ -carbon atom, i.e.
- the component C-0 of any one of Formulae I, and I-a is a compound of the following Formula I-b: (Formula I-b), wherein in Formula I-b: m, PG-0, PG-s, Y 1 , Z 1 , R z-1 , B N , R VI , and R VIII are defined as for Formula I-a, and for each nucleoside subunit independently, R VII , R IX , and R X are either all H or they are bonded together so that the respective nucleoside subunit has a structure of the following Formula I-b-tc: (Formula I-b-tc), wherein in Formula I-b-tc: the oxygen atom from which the dashed line originates represents the oxygen atom bonded to the 3 ⁇ -carbon atom, i.e.
- the carbon atom to which R VII is bonded in Formula I-b indicates the covalent chemical bond interconnecting the respective oxygen atom of the nucleoside subunit of Formula I-b-tc and the pseudo solid-phase protecting group PG-s or P(Y 1 )(Z 1 ) in Formula I-b; the oxygen atom from which the wavy line originates represents the oxygen atom bonded to the 5 ⁇ -carbon atom, i.e.
- R z-1 is a protecting group removable under alkaline conditions, wherein R z-1 may be the same or different at each occurrence.
- a protecting group is “removable under alkaline conditions”, if it can be cleaved by treatment with a base (wherein “a base” is not to be construed to mean any base, but specific bases adapted to the protecting group to be removed based on the common knowledge of those skilled in the art and routine experimentation).
- the term “base” may in this context be understood as proton acceptor in the sense of the Br ⁇ nsted-Lowry theory. Examples of such a base include ammonia, in particular an aqueous solution of ammonia (i.e.
- R z-1 is for each repetitive unit m independently a protecting group of the chemical structure CH 2 -CH 2 -EWG, where EWG is an electron withdrawing group, preferably a cyano group.
- Z 1 is selected independently for each repetitive unit m from the group consisting of O-R z-1 and S-R z-1 . In some embodiments, in the component C-0 of any one of Formulae I, I-a, and I-b, Z 1 is selected independently for each repetitive unit m from the group consisting of O-R z-1 and S-R z-1 , where R z-1 is for each repetitive unit m a 2-cyanoethyl group. In such embodiments, Z 1 is selected independently for each repetitive unit m from the group consisting of O-CH 2 -CH 2 -CN and S-CH 2 -CH 2 -CN.
- Z 1 is for each repetitive unit m O-R z-1 , where R z-1 is for each repetitive unit m a 2-cyanoethyl group. In such embodiments, Z 1 is for each repetitive unit m O-CH2-CH2-CN. In some embodiments, in the component C-0 of any one of Formulae I, I-a, and I-b, Y 1 is for each repetitive unit m O. In some embodiments, in the component C-0 of any one of Formulae I, I-a, and I-b, Y 1 is for each repetitive unit m S.
- R VI is selected independently at each occurrence from the group consisting of H, F, O-CH3 (i.e. methoxy), O-CH2-CH2-O-CH3 (i.e. 2-methoxyethyl-1-oxy), O-Si(CH3)3 (i.e. trimethylsilyloxy), O-Si(CH3)2(C(CH3)3) (i.e. tert-butyl(dimethyl)silyloxy), and O- CH2-O-Si(C(CH3)3)3 (i.e.
- R VIII is independently at each occurrence H or R VIII and R VI of the same nucleoside subunit (i.e. bonded to the 4 ⁇ - and 2 ⁇ -C atom of the same carbohydrate moiety) together form a structure +–CH 2 -O ⁇ ++, +–CH(CH 3 )-O ⁇ ++, or +–CH 2 -CH 2 -O ⁇ ++, where + is the point of attachment to the 4 ⁇ -carbon atom (i.e. the carbon atom to which R VIII is bonded) and ++ is the point of attachment to the 2 ⁇ -carbon (i.e.
- R VI is selected independently at each occurrence from the group consisting of H, F, O-CH3 (i.e.
- the integer m is an integer in the range of 0 ⁇ 49, 0 ⁇ 39, 0 ⁇ 29, 0 ⁇ 24, 0 ⁇ 19, 0 ⁇ 18, 0 ⁇ 17, 0 ⁇ 16, 0 ⁇ 15, 0 ⁇ 14, 0 ⁇ 13, 0 ⁇ 12, 0 ⁇ 11, 0 ⁇ 10, 0 ⁇ 9, 0 ⁇ 8, 0 ⁇ 7, 0 ⁇ 6, 0 ⁇ 5 or 0 ⁇ 4.
- the integer m is an integer in the range of 4 ⁇ 49, 4 ⁇ 39, 4 ⁇ 29, 4 ⁇ 24, 4 ⁇ 19, 4 ⁇ 18, 4 ⁇ 17, 4 ⁇ 16, 4 ⁇ 15, 4 ⁇ 14, 4 ⁇ 13, 4 ⁇ 12, 4 ⁇ 11, 4 ⁇ 10, or 4 ⁇ 9.
- the integer m is an integer equal to or larger than 5, in particular an integer in the range of 5–18 or 5–17. In some embodiments, in the component C-0 of any one of Formulae I, I-a, and I-b, the integer m is 0.
- any kind of nucleobase B N may be present in the component C-0 of any one of Formulae I, I-a, and I-b. Reference is e.g. made to the explanations relating to the nucleobase given above.
- B N is a nucleobase and at each occurrence independently selected from the group consisting of adenine, guanine, cytosine, 5-methylcytosine, thymine, and uracil. It will be understood by those skilled in the art, that any nucleobase B N in any one of Formulae I, I-a, and I-b may optionally be protected, i.e. carry one or more protecting groups, without this being indicated specifically.
- nucleobases in protected form and in free form (i.e. with and without any protecting groups).
- nucleobases in general. The skilled artisan is familiar with nucleobase protecting groups and knows how to select, introduce, and remove them.
- the exocyclic amino groups in nucleobases such as in adenine, guanine, cytosine, and 5-methylcytosine may be protected.
- Non-limiting examples of protecting groups for exocyclic amino groups of nucleobases comprise the acetyl group, the benzoyl group, the isobutyryl group, the pivaloyl group, the pivaloyloxymethyl group, the trifluoroacetyl group, the phenoxyacetyl group, the 4-isopropylphenoxyacetyl group, the 4-tert- butylphenoxyacetyl group, and the dimethylformamidinyl group.
- Carbonyl groups present in nucleobases such as in thymine, uracil, and guanine may also be protected, for example, by reaction with phenol, 2,5-dichlorophenol, 3-chlorophenol, 3,5-dichlorophenol, 2-formylphenol, 2-naphthol, 4-methoxyphenol, 4-chlorophenol, 2-nitrophenol, 4-nitrophenol, 4-acetylaminophenol, pentafluorophenol, 4-pivaloyloxybenzyl alcohol, 4-nitrophenethyl alcohol, 2-(methylsulfonyl)ethanol, 2-(phenylsulfonyl)ethanol, 2-cyanoethanol, 2-(trimethylsilyl)ethanol, dimethylcarbamoyl chloride, diethylcarbamoyl chloride, ethylphenylcarbamoyl chloride, 1-pyrrolidinecarbonyl chloride, 4-morpholinecarbonyl chloride, diphenylcarbamoyl chloride,
- each nucleobase of the component C-0 in particular each nucleobase B N of the component C-0 of any one of Formulae I, I-a, and I-b is independently selected from the group consisting of - adenine, in which the exocyclic amino group is protected;
- m is an integer in the range of 0 ⁇ 49, 0 ⁇ 39, 0 ⁇ 29, 0 ⁇ 24, 0 ⁇ 19, 0 ⁇ 18, 0 ⁇ 17, 0 ⁇ 16, 0 ⁇ 15, 0 ⁇ 14, 0 ⁇ 13, 0 ⁇ 12, 0 ⁇ 11, 0 ⁇ 10, 0 ⁇ 9, 0 ⁇ 8, 0 ⁇ 7, 0 ⁇ 6, 0 ⁇ 5 or 0 ⁇ 4 or in the range of 4 ⁇ 49, 4 ⁇ 39, 4 ⁇ 29, 4 ⁇ 24, 4 ⁇ 19, 4 ⁇ 18, 4 ⁇ 17, 4 ⁇ 16, 4 ⁇ 15, 4 ⁇ 14, 4 ⁇ 13, 4 ⁇ 12, 4 ⁇ 11, 4 ⁇ 10, or 4 ⁇ 9; PG-0 is a protecting group comprising an optionally substituted triarylmethyl residue, preferably a di(p-methoxyphenyl)phenylmethyl (DMT) protecting group; Y 1 is selected independently for each repetitive unit m from the group consisting of O and
- m is an integer in the range of 0 ⁇ 49, 0 ⁇ 39, 0 ⁇ 29, 0 ⁇ 24, 0 ⁇ 19, 0 ⁇ 18, 0 ⁇ 17, 0 ⁇ 16, 0 ⁇ 15, 0 ⁇ 14, 0 ⁇ 13, 0 ⁇ 12, 0 ⁇ 11, 0 ⁇ 10, 0 ⁇ 9, 0 ⁇ 8, 0 ⁇ 7, 0 ⁇ 6, 0 ⁇ 5 or 0 ⁇ 4 or in the range of 4 ⁇ 49, 4 ⁇ 39, 4 ⁇ 29, 4 ⁇ 24, 4 ⁇ 19, 4 ⁇ 18, 4 ⁇ 17, 4 ⁇ 16, 4 ⁇ 15, 4 ⁇ 14, 4 ⁇ 13, 4 ⁇ 12, 4 ⁇ 11, 4 ⁇ 10, or 4 ⁇ 9; PG-0 is a protecting group comprising an optionally substituted triarylmethyl residue, preferably a di(p-methoxyphenyl)phenylmethyl (DMT) protecting group; Y 1 is selected independently for each repetitive unit m from the group consisting of O and
- m is an integer in the range of 0 ⁇ 49, 0 ⁇ 39, 0 ⁇ 29, 0 ⁇ 24, 0 ⁇ 19, 0 ⁇ 18, 0 ⁇ 17, 0 ⁇ 16, 0 ⁇ 15, 0 ⁇ 14, 0 ⁇ 13, 0 ⁇ 12, 0 ⁇ 11, 0 ⁇ 10, 0 ⁇ 9, 0 ⁇ 8, 0 ⁇ 7, 0 ⁇ 6, 0 ⁇ 5 or 0 ⁇ 4 or in the range of 4 ⁇ 49, 4 ⁇ 39, 4 ⁇ 29, 4 ⁇ 24, 4 ⁇ 19, 4 ⁇ 18, 4 ⁇ 17, 4 ⁇ 16, 4 ⁇ 15, 4 ⁇ 14, 4 ⁇ 13, 4 ⁇ 12, 4 ⁇ 11, 4 ⁇ 10, or 4 ⁇ 9; PG-0 is a protecting group comprising an optionally substituted triarylmethyl residue, preferably a di(p-methoxyphenyl)phenylmethyl (DMT) protecting group; Y 1 is selected independently for each repetitive unit m from the group consisting of O and
- m is an integer in the range of 0 ⁇ 49, 0 ⁇ 39, 0 ⁇ 29, 0 ⁇ 24, 0 ⁇ 19, 0 ⁇ 18, 0 ⁇ 17, 0 ⁇ 16, 0 ⁇ 15, 0 ⁇ 14, 0 ⁇ 13, 0 ⁇ 12, 0 ⁇ 11, 0 ⁇ 10, 0 ⁇ 9, 0 ⁇ 8, 0 ⁇ 7, 0 ⁇ 6, 0 ⁇ 5 or 0 ⁇ 4 or in the range of 4 ⁇ 49, 4 ⁇ 39, 4 ⁇ 29, 4 ⁇ 24, 4 ⁇ 19, 4 ⁇ 18, 4 ⁇ 17, 4 ⁇ 16, 4 ⁇ 15, 4 ⁇ 14, 4 ⁇ 13, 4 ⁇ 12, 4 ⁇ 11, 4 ⁇ 10, or 4 ⁇ 9; PG-0 is a protecting group comprising an optionally substituted triarylmethyl residue, preferably a di(p-methoxyphenyl)phenylmethyl (DMT) protecting group; Y 1 is selected independently for each repetitive unit
- R VII , R IX , and R X are either all H or they are bonded together so that the respective nucleoside subunit has a structure of the aforementioned Formula I-a-tc (in a component C-0 of Fomula I-a) or the aforementioned Formula I-b-tc (in a component C-0 of Fomula I-b).
- m is an integer in the range of 0 ⁇ 49, 0 ⁇ 39, 0 ⁇ 29, 0 ⁇ 24, 0 ⁇ 19, 0 ⁇ 18, 0 ⁇ 17, 0 ⁇ 16, 0 ⁇ 15, 0 ⁇ 14, 0 ⁇ 13, 0 ⁇ 12, 0 ⁇ 11, 0 ⁇ 10, 0 ⁇ 9, 0 ⁇ 8, 0 ⁇ 7, 0 ⁇ 6, 0 ⁇ 5 or 0 ⁇ 4 or in the range of 4 ⁇ 49, 4 ⁇ 39, 4 ⁇ 29, 4 ⁇ 24, 4 ⁇ 19, 4 ⁇ 18, 4 ⁇ 17, 4 ⁇ 16, 4 ⁇ 15, 4 ⁇ 14, 4 ⁇ 13, 4 ⁇ 12, 4 ⁇ 11, 4 ⁇ 10, or 4 ⁇ 9; PG-0 is a protecting group comprising an optionally substituted triarylmethyl residue, preferably a di(p-methoxyphenyl)phenylmethyl (DMT) protecting group; Y 1 is selected independently for each repetitive unit
- R VII , R IX , and R X are either all H or they are bonded together so that the respective nucleoside subunit has a structure of the aforementioned Formula I-a-tc (in a component C-0 of Fomula I-a) or the aforementioned Formula I-b-tc (in a component C-0 of Fomula I-b).
- m is an integer in the range of 0 ⁇ 49, 0 ⁇ 39, 0 ⁇ 29, 0 ⁇ 24, 0 ⁇ 19, 0 ⁇ 18, 0 ⁇ 17, 0 ⁇ 16, 0 ⁇ 15, 0 ⁇ 14, 0 ⁇ 13, 0 ⁇ 12, 0 ⁇ 11, 0 ⁇ 10, 0 ⁇ 9, 0 ⁇ 8, 0 ⁇ 7, 0 ⁇ 6, 0 ⁇ 5 or 0 ⁇ 4 or in the range of 4 ⁇ 49, 4 ⁇ 39, 4 ⁇ 29, 4 ⁇ 24, 4 ⁇ 19, 4 ⁇ 18, 4 ⁇ 17, 4 ⁇ 16, 4 ⁇ 15, 4 ⁇ 14, 4 ⁇ 13, 4 ⁇ 12, 4 ⁇ 11, 4 ⁇ 10, or 4 ⁇ 9; PG-0 is a protecting group comprising an optionally substituted triarylmethyl residue, preferably a di(p-methoxyphenyl)phenylmethyl (DMT) protecting group; Y 1 is selected independently for each repetitive unit
- any definitions and embodiments laid out herein for the component C-0 of Formula I likewise apply to the component (C-0) # of Formula I-C, with the proviso that the protecting PG-0 is absent and the hydroxyl moiety, to which the protecting group PG-0 was bonded, is a free hydroxyl group (OH).
- the component (C-0) # of Formula I-C is a compound of Formula I-a, with the proviso that the protecting group PG-0 is absent and the hydroxyl moiety, to which the protecting group PG-0 was bonded, is a free hydroxyl group (OH).
- any definitions and embodiments laid out herein for the component C-0 of Formula I-a are likewise applicable to the component (C-0) # of Formula I-C, with the proviso that the protecting group PG-0 is absent and the hydroxyl moiety, to which the protecting group PG-0 was bonded, is a free hydroxyl group (OH).
- the component (C-0) # of Formula I-C is a compound of Formula I-b, with the proviso that the protecting group PG-0 is absent and the hydroxyl moiety, to which the protecting group PG-0 was bonded, is a free hydroxyl group (OH).
- any definitions and embodiments laid out herein for the component C-0 of Formula I-b are likewise applicable to the component (C-0) # of Formula I-C, with the proviso that the protecting group PG-0 is absent and the hydroxyl moiety, to which the protecting group PG-0 was bonded, is a free hydroxyl group (OH).
- the term “providing a component C-0” in step (a-1) may be understood in the broadest sense to refer to any means of obtaining a component C-0.
- the component C-0 may, for example, be obtained commercially, in particular, if it is a nucleoside. Alternatively, the component C-0 may be provided by means of chemical synthesis.
- Such means are well known to the skilled artisan and comprise, for example, solid-phase and liquid-phase oligonucleotide synthesis.
- the following documents disclose means of providing components C-0 for use in the method of the invention: PCT/EP2022/059528, US2015112053A1, , EP3825300A1, EP2711370A1, EP3398955A1, US2018291056A1, EP3925964A1, EP2921499A1, EP3733680A1, EP3378869A1, WO2020227618A2, EP3263579A1, EP3950698A1, EP3015467A1, M.C. de Koning et al., Organic Process Research & Development 2006, 10, 1238–1245; A.
- Step (b-1) of some methods of the invention is: incubating the component C-0 of step (a-1) with a deprotection mixture M-(b-1), thereby cleaving the protecting group PG-0 from the component C-0, so as to obtain a component (C-0) # having a free backbone hydroxyl group.
- Step (b-2) of some methods of the invention is: incubating the first cycle oligonucleotide O-1 obtained in the first coupling cycle with a deprotection mixture M-(b-2), thereby cleaving the protecting group PG-1 from the first cycle oligonucleotide O-1, so as to obtain a first cycle oligonucleotide (O-1) # having a free backbone hydroxyl group.
- Step (b-x) of some methods of the invention is: incubating the (x ⁇ 1)-th cycle oligonucleotide O-(x ⁇ 1) obtained in the previous coupling cycle with a deprotection mixture M-(b-x), thereby cleaving the protecting group PG-(x ⁇ 1) from the (x ⁇ 1)-th cycle oligonucleotide O-(x ⁇ 1), so as to obtain a (x ⁇ 1)-th cycle oligonucleotide (O- (x ⁇ 1)) # having a free backbone hydroxyl group.
- the component C-0 and the protecting group PG-0 have been defined.
- the first cycle oligonucleotide O-1 is obtained in the first coupling cycle.
- the building block B-1 used to prepare said first cycle oligonucleotide O-1 comprises a backbone hydroxyl moiety protected by a protecting group PG-1 removable under acidic conditions
- the first cycle oligonucleotide O-1 also comprises a backbone hydroxyl moiety protected by a protecting group PG-1 removable under acidic conditions.
- the term (x ⁇ 1)-th cycle oligonucleotide O-(x ⁇ 1) refers to the x-th cycle oligonucleotide obtained in the previous coupling cycle, and, in the case of the first coupling cycle comprising steps (b-x) to (h-x) (as far as present) (i.e.
- the second cycle oligonucleotide O-2 refers to the second cycle oligonucleotide O-2. It will be understood that, since the building block B-2 used to prepare the second cycle oligonucleotide O-2 comprises a backbone hydroxyl moiety protected by a protecting group PG-2 removable under acidic conditions, the second cycle oligonucleotide O-2 also comprises a backbone hydroxyl moiety protected by a protecting group PG-2 removable under acidic conditions.
- each building block B-x comprises a backbone hydroxyl moiety protected by a protecting group PG-x removable under acidic conditions
- each (x ⁇ 1)-th cycle oligonucleotide O-(x ⁇ 1) also comprises a backbone hydroxyl moiety protected by a protecting group PG-(x ⁇ 1) removable under acidic conditions.
- the term “removable under acidic conditions” has been defined above in the context of the protecting group PG-0 and likewise applies to all protecting groups PG-1, PG-2, PG-x, PG-(x ⁇ 1), and PG-n.
- each of the protecting groups PG-1, PG-2, PG-x, PG-(x ⁇ 1), and PG-n is independently of each other a protecting group comprising an optionally substituted triarylmethyl residue.
- each of the protecting groups PG-1, PG-2, PG-x, PG-(x ⁇ 1), and PG-n is independently of each other selected from the group consisting of the triphenylmethyl group (i.e. the trityl group), the (p-methylphenyl)diphenylmethyl group (i.e. the 4-methyltrityl group), the di(p-methylphenyl)phenylmethyl group (i.e.
- the 4,4'-dimethyltrityl group the tri(p-methylphenyl)methyl group (i.e. the 4,4',4"-trimethyltrityl group), the (p-methoxyphenyl)diphenylmethyl group (i.e. the MMT group), and the di(p-methoxyphenyl)phenylmethyl group (i.e. the DMT group).
- each of the protecting groups PG-1, PG-2, PG-x, PG-(x ⁇ 1), and PG-n is independently of each other selected from the group consisting of the triphenylmethyl group, the (p-methoxyphenyl)diphenylmethyl group, and the di(p-methoxyphenyl)phenylmethyl group.
- each of the protecting groups PG-1, PG-2, PG-x, PG- (x ⁇ 1), and PG-n is a di(p-methoxyphenyl)phenylmethyl group.
- the first cycle oligonucleotide O-1 comprises exactly one backbone hydroxyl moiety protected by a protecting group comprising an optionally substituted triarylmethyl residue, which is said protecting group PG-1. In some preferred embodiments, the first cycle oligonucleotide O-1 comprises exactly one protecting group comprising an optionally substituted triarylmethyl residue, which is said protecting group PG-1. In some preferred embodiments, the second cycle oligonucleotide O-2 comprises exactly one backbone hydroxyl moiety protected by a protecting group comprising an optionally substituted triarylmethyl residue, which is said protecting group PG-2.
- the second cycle oligonucleotide O-2 comprises exactly one protecting group comprising an optionally substituted triarylmethyl residue, which is said protecting group PG-2.
- each (x ⁇ 1)-th cycle oligonucleotide O-(x ⁇ 1) comprises exactly one backbone hydroxyl moiety protected by a protecting group comprising an optionally substituted triarylmethyl residue, which is said protecting group PG-(x ⁇ 1).
- each (x ⁇ 1)-th cycle oligonucleotide O-(x ⁇ 1) comprises exactly one protecting group comprising an optionally substituted triarylmethyl residue, which is said protecting group PG-(x ⁇ 1).
- the component (C-0) # differs from the component C-0 only in that the backbone hydroxyl moiety protected by the protecting group PG-0 in the component C-0 is a free backbone hydroxyl group in the component (C-0) # ;
- the first cycle oligonucleotide (O-1) # differs from the first cycle oligonucleotide O-1 only in that the backbone hydroxyl moiety protected by the protecting group PG-1 in the first cycle oligonucleotide O-1 is a free backbone hydroxyl group in the first cycle oligonucleotide (O-1) # ;
- - the second cycle oligonucleotide (O-2) # differs from the second cycle oligonucleotide O-2 only in that the backbone hydroxyl moiety protected by the protecting group PG-2 in the second cycle oligonucleotide O-2 is a free backbone hydroxyl group in the second cycle
- free backbone hydroxyl group refers to a free hydroxyl group, which is part of the backbone of the respective nucleoside or oligonucleotide.
- deprotection mixture M-(b-1) refers to any mixture which may be used to effect cleavage of the protecting group PG-0 from the component C-0.
- deprotection mixture M-(b-2) refers to any mixture which may be used to effect cleavage of the protecting group PG-1 from the first cycle oligonucleotide O-1.
- deprotection mixture M-(b-3) refers to any mixture which may be used to effect cleavage of the protecting group PG-2 from the second cycle oligonucleotide O-2.
- deprotection mixture M-(b-x) refers to any mixture which may be used to effect cleavage of the protecting group PG-(x ⁇ 1) from the (x ⁇ 1)-th cycle oligonucleotide O-(x ⁇ 1). It will be understood that said deprotection mixture M-(b-x) may be the same or different (i.e. comprise the same or different components in the same or different ratios and/or concentrations) between different iterations of step (b-x).
- each of the protecting groups PG-0, PG-1, PG-2, PG-(x ⁇ 1), PG-x, and PG-n is a protecting group “removable under acidic conditions”, which has been explained above to mean that each of these protecting groups can be cleaved (i.e. removed) by treatment with a protic acid, also referred to as Br ⁇ nsted-Lowry acid.
- a protic acid also referred to as Br ⁇ nsted-Lowry acid.
- the acid strength simply speaking, the tendency of a Br ⁇ nsted-Lowry acid to donate a proton, may be expressed in terms of a pKa value, wherein a strong acid has a lower (i.e. smaller) pKa value than a weak acid.
- pKa value as such and the means of determining the pKa value of a protic acid form part of the common knowledge of the skilled artisan. Additionally, pKa values for commonly used Br ⁇ nsted-Lowry acids (and many more) are available in tabulated form from the literature. As used herein, the pKa value is such determinable in water (i.e. aqueous solution) at 25 °C, unless indicated differently.
- the deprotection mixture M-(b-1), the deprotection mixture M-(b-2), and each deprotection mixture M-(b-x) comprise a protic acid.
- the deprotection mixture M-(b-1), the deprotection mixture M-(b-2), and each deprotection mixture M-(b-x) comprise a protic acid having a pKa equal to or smaller than 5, equal to or smaller than 4, equal to or smaller than 3.5, equal to or smaller than 3, equal to or smaller than 2.5, or equal to or smaller than 2.
- the deprotection mixture M-(b-1), the deprotection mixture M-(b-2), and each deprotection mixture M-(b-x) comprise a protic acid having a pKa in the range of 0 ⁇ 5, 0 ⁇ 4, 0 ⁇ 3.5, 0 ⁇ 3, 0 ⁇ 2.5, or 0 ⁇ 2.
- the protic acid is not particularly limited in terms of its chemical structure.
- the protic acid may be a carboxylic acid, a sulfonic acid, a mineral acid or mixtures thereof.
- carboxylic acids comprise trifluoroacetic acid (TFA), dichloroacetic acid (DCA), trichloroacetic acid (TCA), and acetic acid (AcOH).
- a carboxylic acid such as acetic acid may, for example, also be employed in combination with trimethylsilyl chloride (TMSCl).
- TMSCl trimethylsilyl chloride
- sulfonic acids comprise methanesulfonic acid and p-toluenesulfonic acid.
- mineral acids comprise hydrochloric acid and sulfuric acid. It will be understood that the molar amount of the protic acid in each deprotection mixture M-(b-1), M-(b-2), and M-(b-x) is preferably higher than the molar amount of the respective component or oligonucleotide from which the respective protecting group is to be cleaved.
- the total molar amount of the protic acid comprised in the respective deprotection mixture may be in the range of 1 ⁇ 100 mol, 1 ⁇ 90 mol, 1 ⁇ 80 mol, 1 ⁇ 70 mol, 1 ⁇ 60 mol, 1 ⁇ 50 mol, 1 ⁇ 40 mol, or 1 ⁇ 30 mol per 1 mol of the respective component or oligonucleotide from which the respective protecting group is to be cleaved.
- each of the deprotection mixtures M-(b-1), M-(b-2), and M-(b-x) may further comprise one or more carbocation scavengers.
- the term “carbocation scavenger” relates to a nucleophilic compound, which may be used to bind a carbocation or to consume a carbocation by formal donation of a hydride anion, thereby preventing unwanted side reactions of the carbocation.
- Typical examples of such carbocations are carbocations formed during the cleavage of protecting groups comprising an optionally substituted triarylmethyl residue.
- the cleavage of a di(p-methoxyphenyl)phenylmethyl (DMT) protecting group may result in a DMT cation (i.e. a di(p-methoxyphenyl)phenylmethyl cation).
- Non-limiting examples of carbocation scavengers comprise: - compounds comprising one or more sulfhydryl groups (SH) and one or more carboxyl groups (C(O)OH or C(O)O ⁇ ) such as, e.g., glutathione, thiomalic acid, 3-mercaptopropionic acid, cysteine, cysteinyl-glutamic acid, and cysteinyl-aspartic acid, wherein the N-terminal amino group in any one of cysteine, cysteinyl-glutamic acid, and cysteinyl aspartic acid may optionally be protected by a protecting group removable under alkaline conditions such as, e.g., the 9-fluorenylmethyloxycarbonyl (Fmoc) protecting group, - compounds comprising an indole residue and one or more carboxyl groups (C(O)OH or C(O)O ⁇ ), such as, e.g., 1-(1H-indol-5-y
- the above-mentioned compounds comprising one or more sulfhydryl groups (SH) and one or more carboxyl groups (C(O)OH or C(O)O ⁇ ) as well as the above-mentioned compounds comprising an indole residue and one or more carboxyl groups (C(O)OH or C(O)O ⁇ ) may be preferred carbocation scavengers, and glutathione, thiomalic acid, and 3-mercaptopropionic acid may be a particularly preferred carbocation scavenger. Glutathione is most preferred.
- the molar amount of the optional carbocation scavenger in each deprotection mixture M-(b-1), M-(b-2), and M-(b-x) is preferably higher than the molar amount of the respective component or oligonucleotide from which the respective protecting group is to be cleaved.
- the total molar amount of the carbocation scavenger optionally comprised in each deprotection mixture M-(b-1), M-(b-2), and M-(b-x) may be in the range of 1 ⁇ 60 mol, 1 ⁇ 50 mol, 1 ⁇ 40 mol, 1 ⁇ 35 mol, 1 ⁇ 30 mol, 5 ⁇ 30 mol, 10 ⁇ 30 mol, or 15 ⁇ 30 mol, per 1 mol of of the respective component or oligonucleotide from which the respective protecting group is to be cleaved.
- glutathione as carbocation scavenger 1–10 mol or 2–9 mol or 3–8 mol or 3–7 mol or 4–6 mor or 5 mol of glutathione per 1 mol of the respective component or oligonucleotide from which the respective protecting group is to be cleaved may preferably be used.
- TFA as protic acid may preferably be used alongside glutathione as carbocation scavenger.
- oligonucleotide synthesis it may be unpractical to determine the molar amount of the component or oligonucleotide from which the protecting group is to be cleaved in each coupling cycle.
- the molar amount of the respective component or oligonucleotide from which the respective protecting group is to be cleaved is essentially constant throughout the synthesis and equal to the molar amount of the component C-0 as provided in step (a-1).
- each deprotection mixture M-(b-1), M-(b-2), and M-(b-x) comprises: - a protic acid, preferably a protic acid having a pKa equal to or smaller than 5, 4, 3.5, 3, 2.5 or 2, and - one or more carbocation scavengers (as defined herein).
- each deprotection mixture M-(b-1), M-(b-2), and M-(b-x) comprises: - a protic acid, preferably a protic acid having a pKa equal to or smaller than 5, 4, 3.5, 3, 2.5 or 2, and - one or more carbocation scavengers selected from the group consisting of compounds comprising one or more sulfhydryl groups (SH) and one or more carboxyl groups (C(O)OH or C(O)O ⁇ ) and compounds comprising one indole residue and one or more carboxyl groups (C(O)OH or C(O)O ⁇ ).
- a protic acid preferably a protic acid having a pKa equal to or smaller than 5, 4, 3.5, 3, 2.5 or 2
- one or more carbocation scavengers selected from the group consisting of compounds comprising one or more sulfhydryl groups (SH) and one or more carboxyl groups (C(O)OH or C(O)O
- each deprotection mixture M-(b-1), M-(b-2), and M-(b-x) comprises: - a protic acid selected from the group consisting of a carboxylic acid, a sulfonic acid, a mineral acid, and mixtures thereof, and - one or more carbocation scavengers, each of which is a compound comprising one sulfhydryl group (SH) and one or two carboxyl groups (C(O)OH or C(O)O ⁇ ).
- each deprotection mixture M-(b-1), M-(b-2), and M-(b-x) comprises: - a protic acid having a pKa equal to or smaller than 5, 4, 3.5, 3, 2.5 or 2 and being selected from the group consisting of a carboxylic acid, a sulfonic acid, a mineral acid, and mixtures thereof, and - one or more carbocation scavengers, each of which is a compound comprising one sulfhydryl group (SH) and one or two carboxyl groups (C(O)OH or C(O)O ⁇ ).
- each deprotection mixture M-(b-1), M-(b-2), and M-(b-x) comprises: - a protic acid selected from the group consisting of trifluoroacetic acid, dichloroacetic acid, trichloroacetic acid, acetic acid, hydrochloric acid, sulfuric acid, methanesulfonic acid, benzenesulfonic acid, p-toluenesulfonic acid, and mixtures thereof, and - one or more carbocation scavengers, each of which is a compound comprising one sulfhydryl group (SH) and one or two carboxyl groups (C(O)OH or C(O)O ⁇ ).
- a protic acid selected from the group consisting of trifluoroacetic acid, dichloroacetic acid, trichloroacetic acid, acetic acid, hydrochloric acid, sulfuric acid, methanesulfonic acid, benzenesulfonic acid, p
- each deprotection mixture M-(b-1), M-(b-2), and M-(b-x) comprises: - a protic acid selected from the group consisting of trifluoroacetic acid, dichloroacetic acid, trichloroacetic acid, hydrochloric acid, sulfuric acid, methanesulfonic acid, benzenesulfonic acid, p-toluenesulfonic acid, and mixtures thereof, and - one or more carbocation scavengers selected from the group consisting of glutathione, thiomalic acid, 3-mercaptopropionic acid, cysteine, cysteinyl- glutamic acid, and cysteinyl-aspartic acid, wherein the N-terminal amino group in any one of cysteine, cysteinyl-glutamic acid, and cysteinyl aspartic acid may optionally be protected by a protecting group removable under alkaline conditions such as, e.g.,
- each deprotection mixture M-(b-1), M-(b-2), and M-(b-x) comprises: - a protic acid selected from the group consisting of trifluoroacetic acid, dichloroacetic acid, and trichloroacetic acid, and - a carbocation scavenger selected from the group consisting of glutathione, thiomalic acid, and 3-mercaptopropionic acid.
- each deprotection mixture M-(b-1), M-(b-2), and M-(b-x) comprises trifluoroacetic acid and glutathione.
- Each deprotection mixture M-(b-1), M-(b-2), and M-(b-x) may be a solution, i.e. may further comprise a solvent or mixed solvent capable of dissolving said protic acid, and, if present, also said one or more carbocation scavengers and/or said one or more further additives.
- Said solvent or mixed solvent may preferably comprise one or more non-polar solvents.
- Non-limiting examples of non-polar solvents comprise: - ether solvents such as, e.g., alkylated derivatives of tetrahydropyran or tetrahydrofuran, in particular 4-methyltetrahydropyran (MTHP) and 2-methyltetrahydrofuran, cyclopentyl methyl ether, tert-butyl methyl ether, diethyl ether, and anisole; - aromatic solvents such as, e.g., benzene, toluene, o- or m- or p-xylene, and mesitylene; - aliphatic hydrocarbon solvents such as, e.g., pentanes, hexanes, heptanes, octanes, nonanes, and cyclic derivatives thereof such as cyclohexane; - ester solvents such as, e.g., ethyl acetate and isopropyl acetate
- said one or more non-polar solvents may be mixed with one or more polar solvents.
- polar solvents comprise: - nitrile solvents such as, e.g., acetonitrile and propionitrile; - amide solvents such as, e.g., N-N-dimethylformamide, N-N-dimethylacetamine, N-methyl-2-piperidone, and N-methyl-2-pyrrolidone; - ketone solvents such as, e.g., acetone and 2-butanone; - polar ether solvents such as, e.g., 1,4-dioxane and tetrahydrofuran; - sulfoxide solvents such as, e.g., dimethylsulfoxide; and - mixtures thereof.
- any one of the deprotection mixtures M-(b-1), M-(b-2), and M-(b-x) may be formed by adding said protic acid, and optionally the one or more carbocation scavengers and/or further additives directly into the respective reaction mixture or organic phase of the preceding step of the coupling cycle.
- the term “incubating” in steps (b-1), (b-2), and (b-x) may be understood in the broadest sense to refer to any process of contacting the respective component or oligonucleotide from which the respective protecting group is to be cleaved and the respective deprotection mixture, preferably inside the same reaction vessel or reactor.
- the respective component or oligonucleotide from which the respective protecting group is to be cleaved may already be contained in a reaction vessel or reactor, to which the respective deprotection mixture is then added.
- the components of the respective deprotection mixture may be added individually to the reaction vessel or reactor containing the respective component or oligonucleotide from which the respective protecting group is to be cleaved, so that the respective deprotection mixture may be formed directly within said reaction vessel or reactor containing the respective component or oligonucleotide from which the respective protecting group is to be cleaved.
- the term “incubating” does not imply that a preformed deprotection mixture is employed.
- the respective deprotection mixture may already be contained in a reaction vessel or reactor, to which the respective component or oligonucleotide from which the respective protecting group is to be cleaved is then added.
- Any one of steps (b-1), (b-2), and (b-x) may, for example, be performed at a temperature in the range of ⁇ 15 to 90 °C, ⁇ 10 to 90 °C, 0 to 90 °C, 5 to 90 °C, 10 to 70 °C, 15 to 60 °C, or 15 to 50 °C.
- any one of steps (b-1), (b-2), and (b-x) may simply be performed at room temperature. Increased temperatures may result in shorter reaction times.
- the reaction time may also depend on the chemical structure of the reactants and will routinely be selected by a skilled person, for example based on reaction monitoring using, e.g., thin-layer chromatography and/or high performance liquid chromatography (HPLC), optionally coupled to mass spectrometry.
- a base may be added to the reaction mixture to completely or partly neutralize any excess of the protic acid. Such neutralization may typically be achieved by addition of one or more bases or a solution thereof directly into the reaction mixture of the respective step (b-1), (b-2), and (b-x).
- Non-limiting examples of bases which may be used for such neutralization comprise pyridine, benzimidazole, 1,2,4-triazole, N- phenylimidazole, 2-amino-4,6-dimethylpyrimidine, 1,10-phenanthroline, imidazole, N-methylimidazole, 2-chlorobenzimidazole, 2-bromobenzimidazole, 2-methylimidazole, 2-phenylbenzimidazole, N-phenylbenzimidazole, and 5-nitrobenzimidazole, among which pyridine may be preferred.
- Step (c-1) of some methods of the invention is: subjecting a solution comprising the component (C-0) # to one or more aqueous extractions, wherein the organic phase comprises the component (C-0) # .
- Step (c-2) of some methods of the invention is: subjecting a solution comprising the first cycle oligonucleotide (O-1) # to one or more aqueous extractions, wherein the organic phase comprises the first cycle oligonucleotide (O-1) # .
- Step (c-x) of some methods of the invention is: subjecting a solution comprising the (x ⁇ 1)-th cycle oligonucleotide (O-(x ⁇ 1)) # to one or more aqueous extractions, wherein the organic phase comprises the (x ⁇ 1)-th cycle oligonucleotide (O-(x ⁇ 1)) # .
- Said solution comprising the component (C-0) # of step (c-1) may, for example, be the reaction mixture obtained from carrying step (b-1).
- Said solution comprising the first cycle oligonucleotide (O-1) # of step (c-2) may, for example, be the reaction mixture obtained from carrying step (b-2).
- Said solution comprising the (x ⁇ 1)-th cycle oligonucleotide (O-(x ⁇ 1)) # of step (c-x) may, for example, be the reaction mixture obtained from carrying step (b-x) of the same coupling cycle.
- said solutions comprising the component (C-0) # or the first cycle oligonucleotide (O-1) # or the (x ⁇ 1)-th cycle oligonucleotide (O-(x ⁇ 1)) # may be obtained from the respective reaction mixture by addition of one or more non-polar solvents, for example in order to facilitate the phase separation.
- non-polar solvents may also be added during or in between the one or more aqueous extractions of any one of steps (c-1), (c-2), and (c-x).
- non-polar solvents which may be added comprise: - ether solvents such as, e.g., alkylated derivatives of tetrahydropyran or tetrahydrofuran, in particular 4-methyltetrahydropyran (MTHP) and 2-methyltetrahydrofuran, cyclopentyl methyl ether, tert-butyl methyl ether, diethyl ether, and anisole; - aromatic solvents such as, e.g., benzene, benzonitrile, toluene, o- or m- or p- xylene, and mesitylene; - aliphatic hydrocarbon solvents such as, e.g., pentanes, hexanes, heptanes,
- 4-Methyltetrahydropyran may be a preferred non-polar ether solvent which may be added prior to or during or in between the aqueous extraction(s) of any one of steps (c-1), (c-2), and (c-x). Additionally, or alternatively, one or more amide solvents S A (as defined herein below) may be added prior to or during or in between the aqueous extraction(s) of any one of steps (c-1), (c-2), and (c-x).
- aqueous extraction in steps (c-1), (c-2), and (c-x) of the method of the invention may be understood in the broadest sense as any liquid-liquid extraction operation during which the respective solution comprising the component (C-0) # or the first cycle oligonucleotide (O-1) # or the (x ⁇ 1)-th cycle oligonucleotide (O-(x ⁇ 1)) # is extracted with water or an aqueous solution.
- an aqueous solution is a solution comprising water, preferably at least 10 vol-%, 20 vol-%, 30 vol-%, 40 vol-%, 50 vol-%, or 60 vol-% of water.
- water is herein also considered a specific aqueous solution, since it comprises more than 10 vol-%, 20 vol-%, 30 vol- %, 40 vol-%, 50 vol-%, or 60 vol-% of water.
- An aqueous solution may optionally comprise one or more water-miscible organic solvents.
- Non-limiting examples of such water-miscible organic solvents comprise: - alcohol solvents such as methanol, ethanol, and isopropyl alcohol (propan-2-ol, IPA), - nitrile solvents such as acetonitrile and propionitrile, - ketone solvents such as acetone and 2-butanone, - polar ether solvents such as tetrahydrofuran and 1,4-dioxane, - polar amide solvents such as N,N-dimethylformamide, N,N-dimethylacetamide, N-methyl-2-pyrrolidone, and N-methyl-2-piperidone, - sulfoxide solvents such as dimethyl sulfoxide, and - mixtures thereof.
- alcohol solvents such as methanol, ethanol, and isopropyl alcohol (propan-2-ol, IPA)
- - nitrile solvents such as acetonitrile and propionitrile
- An aqueous solution may optionally also comprise one or more dissolved components.
- dissolved components comprise: - inorganic salts such as alkali halides, in particular sodium chloride (e.g. typically used to facilitate and/or accelerate phase separation), - water-soluble protic acids (as defined herein) such as acetic acid, - water-soluble organic or inorganic bases such as N-methylmorpholine (NMM), pyridine, sodium or potassium or ammonium carbonate or hydrogen carbonate, and sodium or potassium or ammonium phosphate or hydrogen phosphate or dihydrogen phosphate, and - mixtures thereof.
- - inorganic salts such as alkali halides, in particular sodium chloride (e.g. typically used to facilitate and/or accelerate phase separation)
- - water-soluble protic acids as acetic acid
- - water-soluble organic or inorganic bases such as N-methylmorpholine (NMM)
- pyridine sodium or potassium or ammonium carbonate or hydrogen carbonate
- an “aqueous extraction” is further characterized in that it results in a mixture comprising at least one aqueous phase (preferably an aqueous layer) and at least one organic phase (preferably an organic layer), wherein the at least one aqueous phase (preferably layer) is then separated from the at least one organic phase (preferably layer).
- the organic phase comprises the component (C-0) # ” in step (c-1) may be understood in the broadest sense to mean that some of the molecules of said component (C-0) # are dissolved in the organic phase.
- the organic phase comprises the first cycle oligonucleotide (O-1) # ” in step (c-2) may be understood in the broadest sense to mean that some of the molecules of said first cycle oligonucleotide (O-1) # are dissolved in the organic phase.
- the expression “the organic phase comprises the (x ⁇ 1)-th cycle oligonucleotide (O-(x ⁇ 1)) # ” in step (c-x) may be understood in the broadest sense to mean that some of the molecules of said (x ⁇ 1)-th cycle oligonucleotide (O-(x ⁇ 1)) # are dissolved in the organic phase. It is preferred that most molecules (e.g.
- the pH-value of the aqueous phase of an aqueous extraction is to be determined from the respective aqueous phase after the aqueous extraction (i.e., after the phase separation, preferably after removing the aqueous phase from the organic phase or vice versa) and at a temperature in the range of 23–28 °C, preferably 25 °C.
- aqueous phase of an aqueous extraction is distinct from the aqueous solution, which is initially combined with the solution comprising the component (C-0) # (in step (c-1)) or the first cycle oligonucleotide (O-1) # (in step (c- 2)) or the (x ⁇ 1)-th cycle oligonucleotide (O-(x ⁇ 1)) # (in a step (c-x)), followed by extraction.
- the “aqueous phase” typically comprises species which have been extracted from the solution comprising the component (C-0) # (in step (c-1)) or the first cycle oligonucleotide (O-1) # (in step (c- 2)) or the (x ⁇ 1)-th cycle oligonucleotide (O-(x ⁇ 1)) # (in a step (c-x)), e.g., water- soluble species.
- species which have been extracted from the solution comprising the component (C-0) # (in step (c-1)) or the first cycle oligonucleotide (O-1) # (in step (c- 2)) or the (x ⁇ 1)-th cycle oligonucleotide (O-(x ⁇ 1)) # (in a step (c-x) e.g., water- soluble species.
- pure water or any other aqueous solution may be used for an aqueous extraction, and during the aqueous extraction one or more protic acids may partition from said solution comprising the component (C-0) # (in step (c-1)) or the first cycle oligonucleotide (O- 1) # (in step (c-2)) or the (x ⁇ 1)-th cycle oligonucleotide (O-(x ⁇ 1)) # (in a step (c-x)) into the aqueous phase, so that the aqueous phase then has a pH-value which is lower than the pH-value of the water or other aqueous solution employed.
- said one or more aqueous extractions of steps (c-1), (c-2), and (c-x) comprise a first aqueous extraction comprising the following steps (Ex-I) to (Ex-IV): (Ex-I) Combining the solution comprising the component (C-0) # (in step (c-1)) or the first cycle oligonucleotide (O-1) # (in step (c-2)) or the (x ⁇ 1)-th cycle oligonucleotide (O-(x ⁇ 1)) # (in a step (c-x)) with a first aqueous solution AS-I; (Ex-II) Agitating the mixture of step (Ex-I); (Ex-III) Allowing the phases to separate, so as to obtain a first organic phase OP-I and a first aqueous phase AP-I, wherein the first organic phase OP-I comprises the component (C-0) # (in step (c-1)) or the first cycle oli
- the first aqueous phase AP-I preferably has a pH-value equal to or smaller than 7, preferably in the range of 1.0– 7.0, 2.0–7.0, 3.0–7.0, 3.5–7.0, 4.0–7.0, 4.0–6.0, 4.5–7.0, 4.5–6.5, 4.5–6.0, or 4.5 ⁇ 5.5.
- said one or more aqueous extractions of steps (c-1), (c-2), and (c-x) comprise a first aqueous extraction comprising the aforementioned steps (Ex-I) to (Ex-IV), and a second aqueous extraction comprising the following steps (Ex-V) to (Ex-VIII): (Ex-V) Combining the first organic phase OP-I with a second aqueous solution AS-II; (Ex-VI) Agitating the mixture of step (Ex-V); (Ex-VII) Allowing the phases to separate, so as to obtain a second organic phase OP-II and a second aqueous phase AP-II, wherein the second organic phase OP-II comprises the component (C-0) # (in step (c-1)) or the first cycle oligonucleotide (O-1) # (in step (c-2)) or the (x ⁇ 1)-th cycle oligonucleotide (O-(x ⁇
- the second aqueous phase AP-II preferably has a pH-value in the range of 4.0–7.0, 4.5–7.0, 5.0–7.0, 5.0–6.5, or 5.0–6.0.
- said one or more aqueous extractions of steps (c-1), (c-2), and (c-x) comprise a first aqueous extraction comprising the aforementioned steps (Ex-I) to (Ex-IV), a second aqueous extraction comprising the aforementioned steps (Ex-V) to (Ex-VIII), and a third aqueous extraction comprising the following steps (Ex-IX) to (Ex-XII): (Ex-IX) Combining the second organic phase OP-II with a third aqueous solution AS-III; (Ex-X) Agitating the mixture of step (Ex-IX); (Ex-XI) Allowing the phases to separate, so as to obtain a third organic phase OP-III and a third a
- the third aqueous phase AP-III preferably has a pH-value in the range of 5.0–7.0, 5.0– 6.5, or 5.0–6.0.
- steps (Ex-I) to (Ex-IV) or steps (Ex-V) to (Ex-VIII) or steps (Ex-IX) to (Ex-XII), respectively may be carried out in addition in any one of steps (c-1), (c-2), and (c-x).
- Such further aqueous extractions preferably comprise the following steps (Ex-A ⁇ ) to (Ex-D ⁇ ): (Ex-A ⁇ ) Combining the organic phase obtained from the previous aqueous extraction with an aqueous solution; (Ex-B ⁇ ) Agitating the mixture of step (Ex-A ⁇ ); (Ex-C ⁇ ) Allowing the phases to separate, so as to obtain an organic phase and an aqueous phase, wherein the organic phase comprises the component (C- 0) # (in step (c-1)) or the first cycle oligonucleotide (O-1) # (in step (c-2)) or the (x ⁇ 1)-th cycle oligonucleotide (O-(x ⁇ 1)) # (in a step (c-x)); and (Ex-D ⁇ ) Removing the aqueous phase from the organic phase or vice versa; wherein the organic phase obtained from the previous aqueous extraction comprises one or more organic solvents S O and, optionally, one or more organic solvents
- the aqueous phase obtained in each step (Ex-C ⁇ ) preferably has a pH-value equal to or smaller than 7, preferably in the range of 4.0–7.0 or 4.0–6.0, in particular in the range of 5.0–7.0, 5.0–6.5, or 5.0–6.0
- the aqueous solutions of two or more aqueous extractions e.g., the aqueous solutions AS-I, AS-I, and AS-III, need not be identical, i.e., they may or may not comprise the same species/components/solvents and the amounts/volumes of said species/components/solvents may be the same or different, unless indicated differently in specific embodiments.
- the first aqueous solution AS-I has a pH-value in the range of 5.0–8.0, 4.0–7.5, 4.5–7.0, 5.0–7.0, 5.0–6.5 or 5.0– 6.0.
- the first aqueous solution AS- I does not comprise a compound having a pKa-value equal to or larger than 9.0, 8.5, 8.0, 7.5, 7.0, 6.5, 6.0 or 5.5.
- the pKa-value is to be determined in water (i.e., in an aqueous solution of the respective compound) at 25 °C.
- each of the first aqueous solution AS-I and the second aqueous solution AS-II has a pH-value in the range of 5.0–8.0, 4.0–7.5, 4.5–7.0, 5.0–7.0, 5.0–6.5 or 5.0–6.0.
- none of the first aqueous solution AS-I and the second aqueous solution AS-II comprises a compound having a pKa-value equal to or larger than 9.0, 8.5, 8.0, 7.5, 7.0, 6.5, 6.0 or 5.5.
- the pKa-value is to be determined in water (i.e., in an aqueous solution of the respective compound) at 25 °C.
- each of the first aqueous solution AS-I, the second aqueous solution AS-II, and the third aqueous solution AS-III has a pH-value in the range of 5.0–8.0, 4.0–7.5, 4.5–7.0, 5.0–7.0, 5.0–6.5 or 5.0–6.0.
- none of the first aqueous solution AS-I, the second aqueous solution AS-II, and the third aqueous solution AS-III comprises a compound having a pKa-value equal to or larger than 9.0, 8.5, 8.0, 7.5, 7.0, 6.5, 6.0 or 5.5.
- Step (Ex-I) is: Combining the solution comprising the component (C-0) # (in step (c- 1)) or the first cycle oligonucleotide (O-1) # (in step (c-2)) or the (x ⁇ 1)-th cycle oligonucleotide (O-(x ⁇ 1)) # (in a step (c-x)) with a first aqueous solution AS-I.
- Step (Ex-V) is: Combining the first organic phase OP-I with a second aqueous solution AS-II.
- Step (Ex-IX) is: Combining the second organic phase OP-II with a third aqueous solution AS-III. This means that the second organic phase OP-II and the third aqueous solution AS-III are combined in the vessel or funnel or other receptacle, in which the agitation of step (Ex-X) is to be performed.
- Step (Ex-A ⁇ ) is: Combining the organic phase obtained from the previous aqueous extraction with an aqueous solution.
- step (Ex-B ⁇ ) the organic phase from the previous aqueous extraction and the aqueous solution are combined in the vessel or funnel or other receptacle, in which the agitation of step (Ex-B ⁇ ) is to be performed.
- the term “agitating” in steps (Ex-II), (Ex-VI), (Ex-X), and (Ex-B ⁇ ) may be understood in the broadest sense to refer to any operation of inducing a movement of the respective mixture. This typically facilitates the extraction process.
- Suitable means of agitation are known to those skilled in the art and comprise, e.g., shaking, stirring, e.g., mechanical stirring, bubbling of an inert has such as nitrogen, and inversion of the vessel or funnel or other receptacle in which the one or more aqueous extractions may be carried out. More than one such means of agitation may be used in combination.
- a reaction vessel made of a suitable material e.g., stainless steel, equipped with a mechanical stirrer may preferably be used for the agitation step (Ex-2), (Ex-6), (Ex-X) or (Ex-B ⁇ ). In practice, it is usually most convenient, to carry out all steps of an aqueous extraction in the same vessel.
- step (Ex-II), (Ex-VI), (Ex-X), and (Ex-B ⁇ ) the respective organic phase and the respective aqueous phase usually form a dispersion. Under such conditions, one phase may typically not be removed from the other. For this purpose, phase separation is usually required.
- - Step (Ex-III) comprises “Allowing the phases to separate, so as to obtain a first organic phase OP-I and a first aqueous phase AP-I”
- - Step (Ex-VII) comprises “Allowing the phases to separate, so as to obtain a second organic phase OP-II and a second aqueous phase AP-II”
- - Step (Ex-XI) comprises “Allowing the phases to separate, so as to obtain a third organic phase OP-III and a third aqueous phase AP-III”
- - Step (Ex-C ⁇ ) comprises “Allowing the phases to separate, so as to obtain an organic phase and an aqueous phase.”
- the term “allowing the phases to separate so as to obtain an organic phase and an aqueous phase” may be understood in the broadest sense as establishing conditions under which the organic phase forms one or more, preferably one, layer (i.e., organic layer) and the aqueous phase forms one or more, preferably
- an organic layer formed from an organic phase may typically not be completely free of an aqueous phase and that an aqueous layer formed from an aqueous phase may typically not be completely free of an organic phase.
- an organic layer is mostly but not necessarily completely composed of an organic phase and an aqueous layer is mostly but not necessarily completely composed of an aqueous phase.
- “Establishing conditions under which the organic phase forms one or more, preferably one, layer (i.e., organic layer) and the aqueous phase forms one or more, preferably one, layer (i.e., aqueous layer)” may usually comprise stopping the agitation.
- step (Ex-III) is: Stopping the agitation and allowing the phases to separate, so as to obtain a first organic phase OP-I and a first aqueous phase AP-I, wherein the first organic phase OP-I comprises the component (C-0) # (in step (c-1)) or the first cycle oligonucleotide (O-1) # (in step (c- 2)) or the (x ⁇ 1)-th cycle oligonucleotide (O-(x ⁇ 1)) # (in a step (c-x)).
- step (Ex-VII) is: Stopping the agitation and allowing the phases to separate, so as to obtain a second organic phase OP-II and a second aqueous phase AP-II, wherein the second organic phase OP-II comprises the component (C- 0) # (in step (c-1)) or the first cycle oligonucleotide (O-1) # (in step (c-2)) or the (x ⁇ 1)- th cycle oligonucleotide (O-(x ⁇ 1)) # (in a step (c-x)).
- step (Ex-XI) is: Stopping the agitation and allowing the phases to separate, so as to obtain a third organic phase OP-III and a third aqueous phase AP-III, wherein the third organic phase OP-III comprises the component (C-0) # (in step (c-1)) or the first cycle oligonucleotide (O-1) # (in step (c-2)) or the (x ⁇ 1)-th cycle oligonucleotide (O-(x ⁇ 1)) # (in a step (c-x)).
- step (Ex-C ⁇ ) is: Stopping the agitation and allowing the phases to separate, so as to obtain an organic phase and an aqueous phase, wherein the organic phase comprises the component (C-0) # (in step (c-1)) or the first cycle oligonucleotide (O-1) # (in step (c- 2)) or the (x ⁇ 1)-th cycle oligonucleotide (O-(x ⁇ 1)) # (in a step (c-x)). Typically, one may simply wait for the phases to separate.
- Phase separation may optionally be speeded up by addition of aqueous solutions comprising dissolved ions, e.g., by addition of brine (i.e., an aqueous solution of sodium chloride), as known to those skilled in the art.
- brine i.e., an aqueous solution of sodium chloride
- the respective aqueous phase is removed from the respective organic phase or vice versa.
- This means that at least one aqueous layer is physically removed from at least one organic layer or vice versa.
- This removal may typically comprise draining of one layer, typically the bottom layer, e.g., the aqueous layer. It will be understood that this removal is not necessarily complete.
- removing in this context is not to mean “completely removing” but preferably means “mostly removing”.
- removing one phase from another phase may mean removing at least 51 vol-%, 60 vol-%, 70 vol-%, 80 vol-%, 90 vol-%, 95 vol-%, 96 vol-%, 97 vol-%, 98 vol-% or 99 vol-% of the respective phase to be removed.
- Step (d-1) of some methods of the invention is: optionally, reducing the water content of the organic phase comprising the component (C-0) # .
- Step (d-2) of some of the methods of the invention is: optionally, reducing the water content of the organic phase comprising the first cycle oligonucleotide (O-1) # .
- Step (d-x) of some of the methods of the invention is: optionally, reducing the water content of the organic phase comprising the (x ⁇ 1)-th cycle oligonucleotide (O-(x ⁇ 1)) # .
- steps (d-1), (d-2), and (d-x) denotes that the respective step may or may not be carried out in a given coupling cycle, unless indicated differently in the context of specific embodiments.
- step (d-1) is carried out (i.e. is not optional).
- step (d-2) is carried out (i.e. is not optional).
- step (d-x) is carried out (i.e. is not optional).
- the organic phase in steps (d-1), (d-2), and (d-x) refers to “the organic phase” of the respective step (c-1) or (c-2) or (c-x) of the same coupling cycle.
- the term “reducing the water content” may be understood in the broadest sense as any operation during which the concentration of water, preferably measured via standard Karl Fischer titration at 25 °C, is reduced.
- the set up may, for example, be as follows:
- the titration cell may be filled with HYDRANAL TM -Coulomat Oil for anolyte reagent, and the inner burette may be filled with HYDRANAL TM -Coulomat CG for catholyte reagent.
- sample 100 ⁇ L
- sample 100 ⁇ L
- the water content of solutions is herein reported in parts per million (ppm). Said reduction of the water content may be characterized by its end point. Preferably, the water content is reduced so that (i.e.
- 2000 ppm 1900 ppm, 1800 ppm, 1700 ppm, 1600 ppm, 1500 ppm, 1400 ppm, 1300 ppm, 1200 ppm, 1100 ppm, 1000 ppm, 900 ppm, 800 ppm, 700 ppm, 600 ppm, 500 ppm, 400 ppm, 300 ppm, 250 ppm, 200 ppm, 150 ppm, or 100 ppm.
- step (d-1) is: optionally, reducing the water content of the organic phase comprising the component (C-0) # , wherein the water content is adjusted to be equal to or less than 2000 ppm, 1900 ppm, 1800 ppm, 1700 ppm, 1600 ppm, 1500 ppm, 1400 ppm, 1300 ppm, 1200 ppm, 1100 ppm, 1000 ppm, 900 ppm, 800 ppm, 700 ppm, 600 ppm, 500 ppm, 400 ppm, 300 ppm, 250 ppm, 200 ppm, 150 ppm, or 100 ppm.
- step (d-2) is: optionally, reducing the water content of the organic phase comprising the first cycle oligonucleotide (O-1) # , wherein the water content is adjusted to be equal to or less than 2000 ppm, 1900 ppm, 1800 ppm, 1700 ppm, 1600 ppm, 1500 ppm, 1400 ppm, 1300 ppm, 1200 ppm, 1100 ppm, 1000 ppm, 900 ppm, 800 ppm, 700 ppm, 600 ppm, 500 ppm, 400 ppm, 300 ppm, 250 ppm, 200 ppm, 150 ppm, or 100 ppm.
- step (d-x) is: optionally, reducing the water content of the organic phase comprising the (x ⁇ 1)-th cycle oligonucleotide (O-(x ⁇ 1)) # , wherein the water content is adjusted to be equal to or less than 2000 ppm, 1900 ppm, 1800 ppm, 1700 ppm, 1600 ppm, 1500 ppm, 1400 ppm, 1300 ppm, 1200 ppm, 1100 ppm, 1000 ppm, 900 ppm, 800 ppm, 700 ppm, 600 ppm, 500 ppm, 400 ppm, 300 ppm, 250 ppm, 200 ppm, 150 ppm, or 100 ppm.
- - step (d-1) is carried out (i.e. is not optional), if the organic phase comprising the component (C-0) # of step (c-1) comprises more than 2000 ppm, 1900 ppm, 1800 ppm, 1700 ppm, 1600 ppm, 1500 ppm, 1400 ppm, 1300 ppm, 1200 ppm, 1100 ppm, 1000 ppm, 900 ppm, 800 ppm, 700 ppm, 600 ppm, 500 ppm, 400 ppm, 300 ppm, 250 ppm, 200 ppm, 150 ppm, or 100 ppm of water; - step (d-2) is carried out (i.e.
- the organic phase comprising the first cycle oligonucleotide (O-1) # of step (c-2) comprises more than 2000 ppm, 1900 ppm, 1800 ppm, 1700 ppm, 1600 ppm, 1500 ppm, 1400 ppm, 1300 ppm, 1200 ppm, 1100 ppm, 1000 ppm, 900 ppm, 800 ppm, 700 ppm, 600 ppm, 500 ppm, 400 ppm, 300 ppm, 250 ppm, 200 ppm, 150 ppm, or 100 ppm of water; and - step (d-x) is carried out (i.e.
- the organic phase comprising the (x ⁇ 1)-th cycle oligonucleotide (O-(x ⁇ 1)) # of step (c-x) of the same coupling cycle comprises more than 2000 ppm, 1900 ppm, 1800 ppm, 1700 ppm, 1600 ppm, 1500 ppm, 1400 ppm, 1300 ppm, 1200 ppm, 1100 ppm, 1000 ppm, 900 ppm, 800 ppm, 700 ppm, 600 ppm, 500 ppm, 400 ppm, 300 ppm, 250 ppm, 200 ppm, 150 ppm, or 100 ppm of water.
- any means of reducing the water content may be employed, preferred examples of which comprise azeotropic distillation and the use of a drying agent.
- reducing the water content is achieved by means of azeotropic distillation (i.e.
- reducing the water content is achieved by means of azeotropic distillation.
- reducing the water content is achieved by means of contacting said organic phase comprising the component (C-0) # or the first cycle oligonucleotide (O-1) # or the (x ⁇ 1)-th cycle oligonucleotide (O-(x ⁇ 1)) # with a drying agent.
- any one of steps (d-1), (d-2), and (d-x) one or more solvents and/or further components may be added to said organic phase comprising the component (C-0) # or the first cycle oligonucleotide (O-1) # or the (x ⁇ 1)-th cycle oligonucleotide (O-(x ⁇ 1)) # prior to, during, and/or after reduction of the water content.
- azeotropic distillation may be understood in the broadest sense to refer to a distillation, in which an azeotrope of water and one or more organic solvents is distilled off.
- Said one or more organic solvents may already be comprised in the organic phase and/or may be added to the organic phase prior to and/or during azeotropic distillation.
- said one or more organic solvents are selected so that the azeotrope with water has a lower boiling point than water itself and any one of the one or more organic solvents used to form said azeotrope.
- Preferred examples of said one or more organic solvents comprise toluene, benzene, and ether solvents, e.g. ether solvents S E as defined herein (vide infra), in particular 4-methyltetrahydropyran (MTHP).
- drying agent may be understood in the broadest sense to refer to any salt or compound or macroscopic structure used to remove water from one or more organic solvents or solutions comprising one or more organic solvents. Any drying agent used for reducing the water content of organic solvents or solutions comprising organic solvents may be used in any one of steps (d-1), (d-2), and (d-x) of the method of the invention.
- Non-limiting examples of suitable drying agents comprise molecular sieves, in particular 3 ⁇ (i.e.3 angstrom) or 4 ⁇ molecular sieves, sulfuric acid, and inorganic salts such as sodium sulfate, magnesium sulfate, and calcium chloride.
- the drying agent may be added to the organic phase, the water content of which is to be reduced.
- the organic phase, the water content of which is to be reduced may be passed through a compartment, e.g. a column, packed with a drying agent, e.g. molecular sieves, as, for example, disclosed in X.
- Step (e-1) of some methods of the invention is: reacting the component (C-0) # with a building block B-1, wherein said building block B-1 is selected from the group consisting of a nucleoside and an oligonucleotide and comprises - a backbone hydroxyl moiety protected by a protecting group PG-1 removable under acidic conditions, and - a phosphorus moiety covalently bonded via its phosphorus atom to an oxygen atom of the backbone of the building block B-1, under conditions suitable to form a covalent bond between said free backbone hydroxyl group of the component (C-0) # and the phosphorus atom of said phosphorus moiety of the building block B-1, thereby obtaining a first cycle oligonucleotide O-1.
- Step (e-2) of some of the methods of the invention is: reacting the first cycle oligonucleotide (O-1) # with a building block B-2, wherein said building block B-2 is selected from the group consisting of a nucleoside and an oligonucleotide and comprises - a backbone hydroxyl moiety protected by a protecting group PG-2 removable under acidic conditions, and - a phosphorus moiety covalently bonded via its phosphorus atom to an oxygen atom of the backbone of the building block B-2, under conditions suitable to form a covalent bond between said free backbone hydroxyl group of the first cycle oligonucleotide (O-1) # and the phosphorus atom of said phosphorus moiety of the building block B-2, thereby obtaining a second cycle oligonucleotide O-2.
- said building block B-2 is selected from the group consisting of a nucleoside and an oligonucleotide and comprises - a backbone
- Step (e-x) of some of the methods of the invention is: reacting the (x ⁇ 1)-th cycle oligonucleotide (O-(x ⁇ 1)) # with a building block B-x, wherein said building block B-x is selected from the group consisting of a nucleoside and an oligonucleotide and comprises - a backbone hydroxyl moiety protected by a protecting group PG-x removable under acidic conditions, and - a phosphorus moiety covalently bonded via its phosphorus atom to an oxygen atom of the backbone of the building block B-x, under conditions suitable to form a covalent bond between said free backbone hydroxyl group of the (x ⁇ 1)-th cycle oligonucleotide (O-(x ⁇ 1)) # and the phosphorus atom of said phosphorus moiety of the building block B-x, thereby obtaining a x-th cycle oligonucleotide O-x.
- building block B-x is selected from the group
- each of the building blocks B-1, B-2, and B-x is selected from the group consisting of a nucleoside and an oligonucleotide comprising not more than 50, 40, 30, 25, 20, 15, 10, or 5 nucleoside subunits.
- each of the building blocks B-1, B-2, and B-x is a nucleoside.
- each of the building blocks B-1, B-2, and B-x is an oligonucleotide.
- each of the building blocks B-1, B-2, and B-x is an oligonucleotide comprising not more than 50, 40, 30, 25, 20, 15, 10, or 5 nucleoside subunits.
- backbone hydroxyl moiety refers to a hydroxyl moiety which is part of the backbone of the respective building block B-1, B-2 or B-x and will be understood based on the above explanations of the terms “hydroxyl moiety” and “backbone”.
- the terms “protecting group” and “removable under acidic conditions” have been defined.
- the protecting groups PG-1, PG-2, and PG-x have been explained. It will be understood that said protecting group PG-x may be the same or different (i.e.
- each of the building blocks B-1, B-2, and B-x further comprises a phosphorus moiety covalently bonded via its phosphorus atom to an oxygen atom of the backbone of the respective building block B-1, B-2, and B-x.
- an oxygen atom of the backbone refers to an oxygen atom which is part of the backbone of the respective building block B-1, B-2 or B-x and will be understood based on the above explanation of the term “backbone”.
- each of the building blocks B-1, B-2, and B-x comprises: - exactly one backbone hydroxyl moiety protected by a protecting group PG-1 or PG-2 or PG-x removable under acidic conditions, and - exactly one phosphorus moiety covalently bonded via its phosphorus atom to an oxygen atom of the backbone of the respective building block B-1 or B-2 or B-x.
- each of the building blocks B-1, B-2, and B-x comprises: - exactly one protecting group removable under acidic conditions, which is said protecting group PG-1 or PG-2 or PG-x, and - exactly one phosphorus moiety covalently bonded via its phosphorus atom to an oxygen atom of the backbone of the respective building block B-1 or B-2 or B-x.
- each of the building blocks B- 1, B-2, and B-x comprises: - exactly one protecting group comprising an optionally substituted triarylmethyl residue, which is said protecting group PG-1 or PG-2 or PG-x, and - exactly one phosphorus moiety covalently bonded via its phosphorus atom to an oxygen atom of the backbone of the respective building block B-1 or B-2 or B-x.
- each of the building blocks B-1, B-2, and B-x is a compound of the following Formula II: wherein in Formula II: each oxygen atom (O) depicted within each nucleoside subunit y-0 to y-q represents the oxygen atom of a hydroxyl moiety of the respective nucleoside subunit; each nucleoside subunit y-0 to y-q may be the same or different (i.e.
- PM is a phosphorus moiety
- PG is the protecting group PG-1 (for building block B-1) or PG-2 (for building block B-2) or PG-x (for a building block B-x) and is a protecting group removable under acidic conditions
- q is an integer equal to or larger than 0
- Y 2 is selected independently for each repetitive unit q from the group consisting of O and S
- Z 2 is selected independently for each repetitive unit q from the group consisting of O-R z-2 , and S-R z-2
- R z-2 is a protecting group, which may be the same or different for each repetitive unit q.
- each of the building blocks B-1, B-2, and B-x is a compound of the following Formula II-a: (Formula II-a), wherein in Formula II-a: q, PG, Y 2 , Z 2 , R Z-2 , and PM are defined as for Formula II; B N is a nucleobase, which may be the same or different (i.e.
- R XI is at each occurrence independently selected from the group consisting of H, F, O-(C 1 ⁇ C 5 -alkyl), O-(C 1 ⁇ C 5 -alkyl)-O-(C 1 -C 5 -alkyl), O-Si(C 1 ⁇ C 5 -alkyl) 3 , and O-CH 2 -O-Si(C 1 ⁇ C 5 -alkyl) 3 ;
- R XIII is independently at each occurrence H or R XIII and R XI of the same nucleoside subunit (i.e.
- R XII , R XIV , and R XV are either all H or they are bonded together so that the respective nucleoside subunit has a structure of the following Formula II-a-tc: (Formula II-a-tc), wherein in Formula II-a-tc: the oxygen atom from which the dashed line originates represents the oxygen atom bonded to the 3 ⁇ -carbon atom, i.e.
- the carbon atom to which R XII is bonded in Formula II-a indicates the covalent chemical bond interconnecting the respective oxygen atom of the nucleoside subunit of Formula II-a-tc and the phosphorus moiety PM or P(Y 2 )(Z 2 ) in Formula II-a; the oxygen atom from which the wavy line originates represents the oxygen atom bonded to the 5 ⁇ -carbon atom, i.e.
- each of the building blocks B-1, B-2, and B-x is a compound of the following Formula II-b: (Formula II-b), wherein in Formula II-b: q, PG, Y 2 , Z 2 , R z-2 , PM, B N , R XI , and R XIII are defined as for Formula II-a, and for each nucleoside subunit independently, R XII , R XIV , and R XV are either all H or they are bonded together so that the respective nucleoside subunit has a structure of the following Formula II-b-tc: (Formula II-b-tc), wherein in Formula II-b-tc: the oxygen atom from which the dashed line originates represents the oxygen atom bonded to the 3 ⁇ -carbon atom, i.e.
- the carbon atom to which R XII is bonded in Formula II-b indicates the covalent chemical bond interconnecting the respective oxygen atom of the nucleoside subunit of Formula II-b-tc and either the phosphorus moiety PM or P(Y 2 )(Z 2 ) in Formula II-b; the oxygen atom from which the wavy line originates represents the oxygen atom bonded to the 5 ⁇ -carbon atom, i.e.
- phosphorus moiety may be understood in the broadest sense and may refer to any atom group comprising at least one, preferably exactly one, phosphorus atom, wherein the term “atom group” may refer to two or more atoms.
- each atom is covalently bonded to each further atom of said atom group, but that each atom is covalently bonded to at least one further atom of said atom group.
- said phosphorus moiety covalently bonded via its phosphorus atom to an oxygen atom of the backbone of the respective building block B-1 or B-2 or B-x e.g. the phosphorus moiety PM of any one of Formulae II, II-a, and II-b, is a phosphorus moiety capable of engaging in a bond forming (condensation) reaction with a free hydroxyl group.
- phosphorus moieties which fulfill this requirement and is able to select a suitable phosphorus moiety without undue experimentation. It is further known to those skilled in the art, that the phosphorus moiety which engages in such a bond forming reaction will typically form an internucleosidic linkage group of the elongated oligonucleotide formed via said bond forming reaction. Said phosphorus moiety covalently bonded via its phosphorus atom to an oxygen atom of the backbone of the respective building block B-1 or B- 2 or B-x, e.g.
- the phosphorus moiety PM of any one of Formulae II, II-a, and II-b may be a phosphorus (III) moiety, also referred to as P (III) moiety, i.e. a phosphorus moiety comprising a P (III) atom as defined herein.
- P (III) moieties are phosphoramidite moieties, as e.g. disclosed in X. Wei et al., Tetrahedron 2013, 69, 3615–3637, and H-phosphonate monoester moieties, as e.g. disclosed in J. Stawinski and R.
- the phosphorus moiety PM of any one of Formulae II, II-a, and II-b may be a phosphorus (V) moiety, also referred to as P (V) moiety, i.e. a phosphorus moiety comprising a P (V) atom as defined herein.
- P (V) moieties comprise classical aryl phosphate diester moieties as e.g. exemplified by H. Lönneberg (Beilstein Journal of Organic Chemistry, 2017, 13, 1368–1387) and the P (V) moieties disclosed by Baran et al. (Science 2018, 361, 12341238 and ACS Central Science 2021, 7, 1473–1485).
- each of the building blocks B-1, B-2, and B-x is a compound of the following Formula II-1:
- each oxygen atom (O) depicted within each nucleoside subunit y-0 to y-q represents the oxygen atom of a hydroxyl moiety of the respective nucleoside subunit; each nucleoside subunit y-0 to y-q may be the same or different;
- PG is the protecting group PG-1 (for the building block B-1) or PG-2 (for the building block B-2) or PG-x (for a building block B-x) and is a protecting group removable under acidic conditions;
- q is an integer equal to or larger than 0;
- Y 2 is selected independently for each repetitive unit q from the group consisting of O and S;
- Z 2 is selected independently for each repetitive unit q from the group consisting of O-R z-2 and S-R z-2 ;
- R z-2 is a protecting group, which may be the same or different for each repetitive unit q;
- Z 3 is selected from the group consisting of O and S; and
- R z-3 is
- each of the building blocks B-1, B-2, and B-x is a compound of the following Formula II-1-a: (Formula II-1-a), wherein in Formula II-1-a: q, PG, Y 2 , Z 2 , R z-2 , Z 3 , R z-3 , R a , and R b are defined as for Formula II-1; and B N , R XI , R XII , R XIII , R XIV , and R XV are defined as for Formula II-a, and wherein step (f-1) or (f-2) or (f-x) is carried out in each coupling cycle.
- each of the building blocks B-1, B-2, and B-x is a compound of the following Formula II-1-b:
- the integer q is an integer in the range of 0 ⁇ 50, 0 ⁇ 35, 0 ⁇ 30, 0 ⁇ 25, 0 ⁇ 20, 0 ⁇ 15, 0 ⁇ 10, 0 ⁇ 5, 0 ⁇ 3, 0 ⁇ 2, 0 ⁇ 1 or q is 0. In some embodiments, in the building block B-1 or B-2 or B-x of any one of Formulae II, II-a, II-b, II-1, II-1-a, and II-1-b, the integer q is 0.
- the protecting group PG is selected from the group consisting of the triphenylmethyl group (i.e. the trityl group), the (p-methylphenyl)diphenylmethyl group (i.e. the 4-methyltrityl group), the di(p-methylphenyl)phenylmethyl group (i.e. the 4,4'-dimethyltrityl group), the tri(p-methylphenyl)methyl group (i.e.
- the protecting group PG is selected from the group consisting of the triphenylmethyl group, the (p-methoxyphenyl)diphenylmethyl group, and the di(p-methoxyphenyl)phenylmethyl group.
- the protecting group PG is the di(p-methoxyphenyl)phenylmethyl group.
- Y 2 is at each occurrence O.
- Y 2 is at each occurrence S.
- R z-2 is a protecting group removable under alkaline conditions, wherein R z-2 may be the same or different at each occurrence.
- the term “removable under alkaline conditions” has been explained above.
- R z-2 is for each repetitive unit q independently a protecting group of the chemical structure CH2-CH2-EWG, where EWG is an electron withdrawing group, preferably a cyano group.
- the electron withdrawing group may for example be selected from the group consisting of a cyano group, a halogen atom such as a chlorine, fluorine, or bromine atom, a formyl group, a keto group, a carboxyester group, or a carboxamide group.
- R z-2 is for each repetitive unit q a 2-cyanoethyl group (i.e. CH 2 -CH 2 -CN).
- Z 2 is selected independently for each repetitive unit q from the group consisting of O-R z-2 and S-R z-2 .
- Z 2 is selected independently for each repetitive unit q from the group consisting of O-R z-2 and S-R z-2 , where R z-2 is for each repetitive unit q a 2-cyanoethyl group (i.e.
- Z 2 is selected independently for each repetitive unit q from the group consisting of O-CH2-CH2-CN and S-CH2- CH2-CN.
- Z 2 in the building block B-1 or B-2 or B-x of any one of Formulae II, II-a, II-b, II-1, II-1-a, and II-1-b, Z 2 is for each repetitive unit q O-R z-2 , where R z-2 is for each repetitive unit q a 2-cyanoethyl group (i.e. CH2-CH2-CN).
- Z 2 is for each repetitive unit q O-CH2-CH2-CN.
- Z 2 is for each repetitive unit q H.
- Y 2 is for each repetitive unit q O and Z 2 is for each repetitive unit q H.
- nucleobase B N may be present in a building block B-1 or B-2 or B-x of any one of Formulae II-a, II-b, II-1-a, and II- 1-b.
- B N is a nucleobase and at each occurrence independently selected from the group consisting of adenine, guanine, cytosine, 5- methylcytosine, thymine, and uracil. It will be understood by those skilled in the art, that any nucleobase B N in any one of Formulae II-a, II-b, II-1-a, and II-1-b may optionally be protected, i.e. carry one or more protecting groups, without this being indicated specifically.
- nucleobase protecting groups are known to those skilled in the art and have also been explained above, including, for example, the preferred nucleobase protecting groups compiled in Table T-1.
- each nucleobase of each building block B-1, B-2 and B-x in particular each nucleobase B N of each building block B-1, B-2 and B-x of any one of Formulae II-a, II-b, II-1-a, and II-1-b is independently selected from the group consisting of - adenine, in which the exocyclic amino group is protected; - guanine, in which the exocyclic amino group is protected; - cytosine, in which the exocyclic amino group is protected; - 5-methylcytosine, in which the exocyclic amino group is protected; - thymine; and - uracil.
- each nucleobase of each building block B-1, B-2 and B-x in particular each nucleobase B N of each building block B-1, B-2 and B-x of any one of Formulae II-a, II-b, II-1-a, and II-1-b is independently selected from the group consisting of - adenine, in which the exocyclic amino group is protected by a benzoyl group, an isobutyryl group or a phenoxyacetyl group; - guanine, in which the exocyclic amino group is protected by an isobutyryl group, a 4-isopropylphenoxyacetyl group or a dimethylformamidino group; - cytosine, in which the exocyclic amino group is protected by an acetyl group or a benzoyl group; - 5-methylcytosine, in which the exocyclic amino group is protected by an acetyl group or a benzoyl group; - thymine
- R XI is at each occurrence independently selected from the group consisting of H, F, O-(C1 ⁇ C5-alkyl), O-(C1 ⁇ C5-alkyl)-O-(C1 ⁇ C5-alkyl), O-Si(C1 ⁇ C5-alkyl)3, and O-CH2-O-Si(C1 ⁇ C5-alkyl)3;
- R XIII is independently at each occurrence H or R XIII and R XI of the same nucleoside subunit (i.e.
- R XII , R XIV , and R XV are either all H or they are bonded together so that the respective nucleoside subunit has a structure of the aforementioned Formula II-a-tc (in a building block B-1 or B-2 or B-x of any one of Formulae II-a, II-1-a) or Formula II-b-tc (in a building block B-1 or B-2 or B-x of any one of Formula II-b, II-1-b).
- R XI is at each occurrence independently selected from the group consisting of H, F, O-CH 3 (i.e. methoxy), O-CH 2 -CH 2 -O-CH 3 (i.e. 2-methoxyethyl-1-oxy), O-Si(CH3)3 (i.e. trimethylsilyloxy), O-Si(CH3)2(C(CH3)3) (i.e. tert- butyl(dimethyl)silyloxy), and O-CH2-O-Si(C(CH3)3)3 (i.e.
- R XIII is independently at each occurrence H or R XIII and R XI of the same nucleoside subunit (i.e. bonded to the 4 ⁇ - and 2 ⁇ -C atom of the same carbohydrate moiety) together form a structure +–CH2-O ⁇ ++, +–CH(CH3)-O ⁇ ++, or +–CH2-CH2-O ⁇ ++, where + is the point of attachment to the 4 ⁇ -carbon atom (i.e. the carbon atom to which R XIII is bonded) and ++ is the point of attachment to the 2 ⁇ -carbon (i.e.
- R XII , R XIV , and R XV are either all H or they are bonded together so that the respective nucleoside subunit has a structure of the aforementioned Formula II-a-tc (in a building block B-1 or B-2 or B-x of any one of Formulae II-a, II-1-a) or Formula II-b-tc (in a building block B-1 or B-2 or B-x of any one of Formula II-b, II-1-b).
- R XI is selected independently at each occurrence from the group consisting of H, F, O-CH3 (i.e. methoxy), and O-CH2-CH2-O-CH3 (i.e.2-methoxyethyl-1-oxy); and each of R XII , R XIII , R XIV , and R XV is H at each occurrence.
- Z 3 is O.
- Z 3 is S.
- R z-3 is a protecting group removable under alkaline conditions. The term “removable under alkaline conditions” has been explained above.
- R z-3 is a protecting group of the chemical structure CH 2 -CH 2 -EWG, where EWG is an electron withdrawing group, preferably a cyano group.
- EWG is an electron withdrawing group, preferably a cyano group.
- the electron withdrawing group may for example be selected from the group consisting of a cyano group, a halogen atom such as a chlorine, fluorine, or bromine atom, a formyl group, a keto group, a carboxyester group, or a carboxamide group.
- R z-3 is a 2-cyanoethyl group (i.e. CH 2 -CH 2 -CN).
- each of R a and R b is a C1–C6-alkyl group, wherein R a and R b may be the same or different, and wherein R a and R b may not bond to each other.
- each of R a and R b is an isopropyl group (i.e. CH(CH3)2).
- PM is a phosphorus moiety, preferably a phosphorus moiety selected from the group consisting of a phosphoramidite moiety and a H-phosphonate monoester moiety
- PG is the protecting group PG-1 (for building block B-1) or PG-2 (for building block B-2) or PG-x (for a building block B-x) and is a di(p-methoxyphenyl)phenylmethyl (DMT) protecting group
- q is an integer in the range of 0 ⁇ 50, 0 ⁇ 35, 0 ⁇ 30, 0 ⁇ 25, 0 ⁇ 20, 0 ⁇ 15, 0 ⁇ 10, or 0 ⁇ 5, or q is 0
- Y 2 is selected independently for each repetitive unit q from the group consisting of O and S
- Z 2 is selected independently for each repetitive unit q from the group consisting of O-R z-2 , S-R z-2 , and H
- PM is a phosphoramidite moiety
- PG is the protecting group PG-1 (for building block B-1) or PG-2 (for building block B-2) or PG-x (for a building block B-x) and is a di(p-methoxyphenyl)phenylmethyl (DMT) protecting group
- q is an integer in the range of 0 ⁇ 50, 0 ⁇ 35, 0 ⁇ 30, 0 ⁇ 25, 0 ⁇ 20, 0 ⁇ 15, 0 ⁇ 10, or 0 ⁇ 5, or q is 0
- Y 2 is selected independently for each repetitive unit q from the group consisting of O and S
- Z 2 is selected independently for each repetitive unit q from the group consisting of O-R z-2 and S-R z-2
- R z-2 is at each occurrence a 2-cyanoethyl group.
- PM is a phosphoramidite moiety
- PG is the protecting group PG-1 (for building block B-1) or PG-2 (for building block B-2) or PG-x (for a building block B-x) and is a di(p-methoxyphenyl)phenylmethyl (DMT) protecting group
- q is an integer in the range of 0 ⁇ 50, 0 ⁇ 35, 0 ⁇ 30, 0 ⁇ 25, 0 ⁇ 20, 0 ⁇ 15, 0 ⁇ 10, or 0 ⁇ 5, or q is 0
- Y 2 is selected independently for each repetitive unit q from the group consisting of O and S
- Z 2 is for each repetitive unit q O-R z-2
- R z-2 is at each occurrence a 2-cyanoethyl group.
- PM is a H-phosphonate monoester moiety
- PG is the protecting group PG-1 (for building block B-1) or PG-2 (for building block B-2) or PG-x (for a building block B-x) and is a di(p-methoxyphenyl)phenylmethyl (DMT) protecting group
- q is an integer in the range of 0 ⁇ 50, 0 ⁇ 35, 0 ⁇ 30, 0 ⁇ 25, 0 ⁇ 20, 0 ⁇ 15, 0 ⁇ 10, or 0 ⁇ 5, or q is 0
- Y 2 is selected independently for each repetitive unit q from the group consisting of O and S, and preferably is O
- Z 2 is for each repetitive unit q H.
- PM is a phosphorus moiety, preferably a phosphorus moiety selected from the group consisting of a phosphoramidite moiety and a H-phosphonate monoester moiety
- PG is the protecting group PG-1 (for building block B-1) or PG-2 (for building block B-2) or PG-x (for a building block B-x) and is a di(p-methoxyphenyl)phenylmethyl (DMT) protecting group
- q is an integer in the range of 0 ⁇ 50, 0 ⁇ 35, 0 ⁇ 30, 0 ⁇ 25, 0 ⁇ 20, 0 ⁇ 15, 0 ⁇ 10, or 0 ⁇ 5, or q is 0
- Y 2 is selected independently for each repetitive unit q from the group consisting of O and S
- Z 2 is selected independently for each repetitive unit q from the group consisting of O-R z-2 , S-
- R XII , R XIV , and R XV are either all H or they are bonded together so that the respective nucleoside subunit has a structure of the aforementioned Formula II-a-tc (in a building block B-1, B-2 or B-x of Formula II-a) or Formula II-b-tc (in a building block B-1, B-2 or B-x of Formula II-b).
- R XIII is independently at each occurrence H or R XIII and R XI of the same nucleoside subunit (i.e.
- R XII , R XIV , and R XV are either all H or they are bonded together so that the respective nucleoside subunit has a structure of the aforementioned Formula II-a-tc (in a building block B-1, B-2 or B-x of Formula II-a) or Formula II-b-tc (in a building block B-1, B-2 or B-x of Formula II-b).
- PM is a phosphoramidite moiety
- PG is the protecting group PG-1 (for building block B-1) or PG-2 (for building block B-2) or PG-x (for a building block B-x) and is a di(p-methoxyphenyl)phenylmethyl (DMT) protecting group
- q is an integer in the range of 0 ⁇ 50, 0 ⁇ 35, 0 ⁇ 30, 0 ⁇ 25, 0 ⁇ 20, 0 ⁇ 15, 0 ⁇ 10, or 0 ⁇ 5, or q is 0
- Y 2 is selected independently for each repetitive unit q from the group consisting of O and S
- Z 2 is for each repetitive unit q O-R z-2
- R z-2 is at each occurrence a 2-cyanoethyl group
- R XI is selected independently at each occurrence from the group consisting of H, F, O-CH3 (i
- R XII , R XIII , R XIV , and R XV is H at each occurrence.
- PG is the protecting group PG-1 (for building block B-1) or PG-2 (for building block B-2) or PG-x (for a building block B-x) and is a di(p-methoxyphenyl)phenylmethyl (DMT) protecting group;
- q is an integer in the range of 0 ⁇ 50, 0 ⁇ 35, 0 ⁇ 30, 0 ⁇ 25, 0 ⁇ 20, 0 ⁇ 15, 0 ⁇ 10, or 0 ⁇ 5, or q is 0;
- Y 2 is selected independently for each repetitive unit q from the group consisting of O and S;
- Z 2 is selected independently for each repetitive unit q from the group consisting of O-R z-2 and S-R z-2 , and Z 2 preferably is for each repetitive unit q O-R z-2 ;
- R z-2 is at each occurrence a 2-cyanoethyl group;
- Z 3 is selected from the group consisting of O and S
- PG is the protecting group PG-1 (for building block B-1) or PG-2 (for building block B-2) or PG-x (for a building block B-x) and is a di(p-methoxyphenyl)phenylmethyl (DMT) protecting group;
- q is an integer in the range of 0 ⁇ 50, 0 ⁇ 35, 0 ⁇ 30, 0 ⁇ 25, 0 ⁇ 20, 0 ⁇ 15, 0 ⁇ 10, or 0 ⁇ 5, or q is 0;
- Y 2 is selected independently for each repetitive unit q from the group consisting of O and S;
- Z 2 is for each repetitive unit q O-R z-2 ;
- R z-2 is at each occurrence a 2-cyanoethyl group;
- Z 3 is O;
- PG is the protecting group PG-1 (for building block B-1) or PG-2 (for building block B-2) or PG-x (for a building block B-x) and is a di(p-methoxyphenyl)phenylmethyl (DMT) protecting group;
- q is an integer in the range of 0 ⁇ 50, 0 ⁇ 35, 0 ⁇ 30, 0 ⁇ 25, 0 ⁇ 20, 0 ⁇ 15, 0 ⁇ 10, or 0 ⁇ 5, or q is 0;
- Y 2 is selected independently for each repetitive unit q from the group consisting of O and S;
- Z 2 is selected independently for each repetitive unit q from the group consisting of O-R z-2 and S-R z-2 , and Z 2 preferably is for each repetitive unit q O-R z-2 ;
- R z-2 is at each occurrence a 2-cyanoethyl group;
- Z 3 is
- R XII , R XIV , and R XV are either all H or they are bonded together so that the respective nucleoside subunit has a structure of the aforementioned Formula II-a-tc (in a building block B-1, B-2 or B-x of Formula II-1-a) or Formula II-b-tc (in a building block B-1, B-2 or B-x of Formula II-1-b).
- PG is the protecting group PG-1 (for building block B-1) or PG-2 (for building block B-2) or PG-x (for a building block B-x) and is a di(p-methoxyphenyl)phenylmethyl (DMT) protecting group;
- q is an integer in the range of 0 ⁇ 50, 0 ⁇ 35, 0 ⁇ 30, 0 ⁇ 25, 0 ⁇ 20, 0 ⁇ 15, 0 ⁇ 10, or 0 ⁇ 5, or q is 0;
- Y 2 is selected independently for each repetitive unit q from the group consisting of O and S;
- Z 2 is selected independently for each repetitive unit q from the group consisting of O-R z-2 and S-R z-2 , and Z 2 preferably is for each repetitive unit q O-R z-2 ;
- R z-2 is at each occurrence a 2-cyanoethyl group;
- Z 3 is
- R XIII is independently at each occurrence H or R XIII and R XI of the same nucleoside subunit (i.e.
- R XII , R XIV , and R XV are either all H or they are bonded together so that the respective nucleoside subunit has a structure of the aforementioned Formula II-a-tc (in a block B-1, B-2 or B-x of Formula II-1-a) or Formula II-b-tc (in a B-1, B-2 or B-x of Formula II-1-b).
- PG is the protecting group PG-1 (for building block B-1) or PG-2 (for building block B-2) or PG-x (for a building block B-x) and is a di(p-methoxyphenyl)phenylmethyl (DMT) protecting group;
- q is an integer in the range of 0 ⁇ 50, 0 ⁇ 35, 0 ⁇ 30, 0 ⁇ 25, 0 ⁇ 20, 0 ⁇ 15, 0 ⁇ 10, or 0 ⁇ 5, or q is 0;
- B N is a nucleobase, which may be the same or different at each occurrence;
- Y 2 is selected independently for each repetitive unit q from the group consisting of O and S;
- Z 2 is selected independently for each repetitive unit q from the group consisting of O-R z-2 and S-R z-2 , and Z 2 preferably is for each repetitive unit q O-R z-2 ;
- R is the protecting group PG-1 (for building block B-1) or PG-2 (for building block B
- the building block B-x may be the same or different (i.e. have the same or a different chemical structure) for each coupling cycle comprising steps (b-x) to (h-x) (as far as present), unless indicated differently in the context of specific embodiments.
- a building block B-1, B-2 or B-x for use in the method of the invention may, for example, be obtained commercially, in particular, if said phosphorus moiety is a phosphoramidite moiety or a H-phosphonate monoester moiety.
- a building block B-1, B-2 or B-x for use in the method of the invention may be obtained by means of chemical synthesis. The person skilled in the art is aware of methods of synthesizing such compounds, wherein the synthesis route will obviously depend on the chemical structure of said phosphorus moiety.
- Building blocks B-1, B-2 or B-x e.g. of any one of Formulae II, II-a, and II-b, in which said phosphorus moiety, e.g.
- said phosphorus moiety PM is a phosphoramidite moiety, in particular building blocks B-1, B-2 or B-x of any one of Formulae II-1, II-1-a, and II-1-b, may, for example, be synthesized as disclosed in, in K.V. Gothelf et al., Nature Communications 2021, 12, 2760, and in X. Wei et al., Tetrahedron 2013, 69, 3615 ⁇ 3637.
- said phosphorus moiety PM is a H-phosphonate monoester moiety, may, for example, be synthesized as disclosed in J. Stawinski and R. Strömberg (2005), Di- and Oligonucleotide Synthesis Using H-Phosphonate Chemistry, in Methods in Molecular Biology, vol. 288: Oligonucleotide Synthesis: Methods and Applications, edited by P. Herdewijn, Humana Press Inc., Totowa NJ, https://doi.org/10.1385/1-59259-823-4:081.
- Building blocks B-1, B-2 or B-x e.g. of any one of Formulae II, II-a, and II-b, in which said phosphorus moiety, e.g.
- step (e-1) is a P (V) moiety suitable for chiral phosphorothioate synthesis may, for example, be synthesized as disclosed in P.S. Baran et al., Science 2018, 361, 1234 ⁇ 1238 (also see Supplementary Materials for this reference).
- the term “reacting” in step (e-1) may be understood in the broadest sense as any operation during which the component (C-0) # and the building block B-1 are present in the same reaction vessel or reactor and engage in the bond forming reaction of step (e-1).
- step (e-2) may be understood in the broadest sense as any operation during which the first cycle oligonucleotide (O-1) # and the building block B-2 are present in the same reaction vessel or reactor and engage in the bond forming reaction of step (e-2).
- step (e-x) may be understood in the broadest sense as any operation during which the (x ⁇ 1)-th cycle oligonucleotide (O-(x ⁇ 1)) # and the building block B-x are present in the same reaction vessel or reactor and engage in the bond forming reaction of step (e-x).
- the component (C-0) # , the first cycle oligonucleotide (O-1) # or the (x ⁇ 1)-th cycle oligonucleotide (O-(x ⁇ 1)) # may already be contained in a reaction vessel or reactor, to which the building block B-1, B-2 or B-x is then added.
- the building block B-1, B-2 or B-x or a solution thereof may already be contained in a reaction vessel or reactor, to which the component (C-0) # , the first cycle oligonucleotide (O-1) # or the (x ⁇ 1)-th cycle oligonucleotide (O-(x ⁇ 1)) # is then added.
- steps (e-1), (e-2), and (e-x) a covalent (chemical) bond is formed between said free backbone hydroxyl group of the component (C-0) # , the first cycle oligonucleotide (O-1) # or the (x ⁇ 1)-th cycle oligonucleotide (O-(x ⁇ 1)) # and the phosphorus atom of said phosphorus moiety of the building block B-1, B-2 or B-x.
- the bond forming reaction of steps (e-1), (e-2), and (e-x) is herein also referred to as coupling or coupling reaction or condensation or condensation reaction, and steps (e-1), (e-2), and (e-x) are also referred to as coupling steps or condensation steps.
- the product obtained from the bond forming reaction of step (e-1) is the first cycle oligonucleotide O-1, which comprises the nucleoside sequence of the component (C-0) # and of the building block B-1, wherein these two are now interconnected by an internucleosidic linkage group derived from the phosphorus moiety of the building block B-1.
- the product obtained from the bond forming reaction of step (e-2) is the second cycle oligonucleotide O-2, which comprises the nucleoside sequence of the first cycle oligonucleotide (O-1) # and of the building block B-2, wherein these two are now interconnected by an internucleosidic linkage group derived from the phosphorus moiety of the building block B-2.
- the product obtained from the bond forming reaction of step (e-x) is the x-th cycle oligonucleotide O-x, which comprises the nucleoside sequence of the (x ⁇ 1)-th cycle oligonucleotide (O-(x ⁇ 1)) # and of the building block B-x, wherein these two are now interconnected by an internucleosidic linkage group derived from the phosphorus moiety of the building block B-x.
- the “conditions suitable” to form a covalent bond between said free backbone hydroxyl group of the component (C-0) # , the first cycle oligonucleotide (O-1) # or the (x ⁇ 1)-th cycle oligonucleotide (O-(x ⁇ 1)) # and the phosphorus atom of said phosphorus moiety of the building block B-1, B-2 or B-x form part of the common knowledge of those skilled in the art. These conditions may, for example, depend on the chemical structure of said phosphorus moiety which is to engage in the bond forming reaction.
- a bond forming reaction of step (e-1), (e-2) or (e-x), in which the phosphorus moiety engaging in said reaction is a phosphoramidite moiety, as e.g. present in building blocks B-1, B-2 or B-x of any one of Formulae II-1, II-1-a, and II-1-b, is herein also referred to as phosphoramidite coupling (reaction).
- phosphoramidite couplings may preferably be performed in the presence of an activator. Any activator used in oligonucleotide synthesis by the so-called phosphoramidite method may be used in the method of the invention.
- the activator may, for example, be selected from the group consisting of: - a tetrazole type activator such as 1H-tetrazole, 5-ethylthio-1H-tetrazole (ETT), 5-benzylthio-1H-tetrazole (BTT), 5-methylthio-1H-tetrazole (MTT), 1-methyl-5-mercaptotetrazole, 1-phenyl-5-mercaptotetrazole, and 5-(4-nitrophenyl)-1H-tetrazole, - an imidazole type activator such as 4,5-dicyanoimidazole (DCI) and 2-bromo- 4,5-dicyanoimidacole (2-Br-DCI), - a 1-hydroxybenzotriazole type activator such as 1-hydroxybenzotriazole, 1-hydroxy-6-trifluorobenzotriazole, and 1-hydroxy-6-trifluoro-4- nitrobenzotriazole, - a pyr
- salts obtained from reacting saccharin with an organic base such as pyridine, collidine, lutidine, picoline, N-methylimidazole, and triethylamine.
- organic base such as pyridine, collidine, lutidine, picoline, N-methylimidazole, and triethylamine.
- BTT 5-Benzylthio-1H-tetrazole
- ETT 5-ethylthio-1H-tetrazole
- DCI 4,5-dicyanoimidacole
- N-methylimidazole (NMI) or pyridine may be added alongside the activator, which may help to adjust the acidity of the solution.
- 1.0 ⁇ 100.0 mol, 1.0 ⁇ 50.0 mol, 1.0 ⁇ 40.0 mol, 1.0 ⁇ 30.0 mol, 1.0 ⁇ 20.0 mol, 1.0 ⁇ 15.0 mol, 1.0 ⁇ 10.0 mol, or 2.5 ⁇ 10.0 mol of the activator may be used per 1 mol of the building block B-1, B-2 or B-x engaging in the respective coupling reaction with its phosphoramidite moiety.
- NMI may be used alongside a protic acid, for example a carboxylic acid such as trifluoroacetic acid, without an additional activator.
- Acetonitrile may also be added.
- a mixture of acetonitrile, NMI, and TFA e.g.
- steps (e-1), (e-2), and (e-x) are carried out using a tetrazole-type activator, preferably ETT, most preferably in combination with a base, in particular pyridine. It will be understood that this does not imply that a step (e-2) and/or (e-x) must be performed.
- a bond forming reaction of step (e-1), (e-2) or (e-x), in which the phosphorus moiety engaging in said reaction is a H-phosphonate monoester moiety, is herein also referred to as H-phosphonate coupling (reaction).
- Such H-phosphonate couplings may typically be performed using a condensing agent.
- Any condensing agent used in oligonucleotide synthesis by the so-called H-phosphonate method may be used in the method of the invention.
- the condensing agent may, for example, be selected from the group consisting of pivaloyl chloride (PvCl), 1-adamantanecarbonyl chloride (AdCl), 2,2-dimethylbutyryl chloride, isobutyryl chloride, diphenyl chlorophosphate, 2,4,6-triisopropylbenzenesulfonyl chloride, bis(pentafluorophenyl) carbonate, 2-chloro-5,5-dimethyl-1,3,2-dioxaphosphorinane 2-oxide (also referred to as 5,5-dimethyl-2-oxo-2-chloro-1,3,2-dioxaphosphinane, DMOCP), and bis(2-ox
- the building block B-1, B-2 or B-x comprising said H-phosphonate monoester moiety may be pre-activated with (i.e. incubated with) said condensing agent, and then be combined with the component (C-0) # , the first cycle oligonucleotide (O-1) # or the (x ⁇ 1)-th cycle oligonucleotide (O-(x ⁇ 1)) # .
- a nucleophile such as pyridine may be added.
- EP3378869A1 discloses suitable reaction conditions.
- a bond forming reaction of step (e-1), (e-2) or (e-x), in which the phosphorus moiety engaging in said reaction is an arylphosphate diester moiety is herein also referred to as phosphotriester coupling (reaction).
- the arylphosphate diester moieties may typically be activated with an arylsulfonyl chloride activator, such as mesitylene- 2-sulfonyl chloride (MsCl), usually in the presence of an auxiliary nucleophile such as 1-methylimidazole.
- MsCl mesitylene- 2-sulfonyl chloride
- pre-formed or in-situ generated 1-hydroxybenzotriazole-phosphotriesters may, for example, be used as phosphorus moiety instead of an aryl phosphate diester moiety in a phosphotriester coupling, again in the presence of an auxiliary nucleophile such as 1-methylimidazole.
- a bond forming reaction of step (e-1), (e-2) or (e-x), in which the phosphorus moiety engaging in said reaction is a P (V) moiety allowing for chiral phosphorothioate synthesis may, for example, be performed as disclosed in Baran et al.,Science 2018, 361, 1234 ⁇ 1238 (see also the Supplementary Materials of said publication) and ACS Central Science 2021, 7, 1473 ⁇ 1485), or in a similar fashion.
- the solutions in which steps (e-1), (e-2), and (e-x) are carried out are substantially anhydrous, which may mean that they preferably comprise equal to or less than 2000 ppm, 1900 ppm, 1800 ppm, 1700 ppm, 1600 ppm, 1500 ppm, 1400 ppm, 1300 ppm, 1200 ppm, 1100 ppm, 1000 ppm, 900 ppm, 800 ppm, 700 ppm, 600 ppm, 500 ppm, 400 ppm, 300 ppm, 250 ppm, 200 ppm, 150 ppm, or 100 ppm of water.
- the water content may be determined by means of standard Karl Fischer titration at 25 °C, as known to those skilled in the art.
- the set up may, for example, be as follows:
- the titration cell may be filled with HYDRANAL TM -Coulomat Oil for anolyte reagent, and the inner burette may be filled with HYDRANAL TM - Coulomat CG for catholyte reagent.
- sample 100 ⁇ L
- the measurement may be started.
- the water content of solutions is herein reported in parts per million (ppm).
- 1.0 ⁇ 20.0 mol, 1.0 ⁇ 10.0 mol, 1.0 ⁇ 5.0 mol, 2.0 ⁇ 5.0 mol, or 2.0 ⁇ 4.0 mol, or 1.0 ⁇ 2.5 mol, or 1.0 ⁇ 2.0 mol of the building block B-1, B-2 or B-x may be used per mol of the component (C-0) # , the first cycle oligonucleotide (O-1) # or the (x ⁇ 1)-th cycle oligonucleotide (O-(x ⁇ 1)) # .
- Steps (e-1), (e-2), and (e-x) may, for example, be performed at a temperature in the range of 0 ⁇ 90 °C, 10 ⁇ 70 °C, 15 ⁇ 60 °C, 15 ⁇ 50 °C, or 20 ⁇ 50 °C.
- steps (e-1), (e-2), and (e-x) may simply be performed at room temperature. Increased temperatures may result in shorter reaction times.
- the reaction time may also depend on the chemical structure of the reactants and will routinely be selected by a skilled person, for example based on reaction monitoring using, e.g., thin-layer chromatography and/or high performance liquid chromatography (HPLC), optionally coupled to mass spectrometry.
- HPLC high performance liquid chromatography
- the internucleosidic linkage group formed in a bond forming reaction of a step (e-1), (e-2) or (e-x) is a P(III) linkage group as defined herein, e.g. a phosphite triester linkage group (as e.g. obtained from a phosphoramidite coupling), and supposed to be sulfurized (by incubation with a sulfurizing agent) to a P (V) linkage group, e.g. a phosphorothioate linkage group, in the subsequent step (f-1), (f-2) or (f-x), the respective step (e-1), (e-2) or (e-x) may be carried out in the presence of an antioxidant .
- a P(III) linkage group as defined herein, e.g. a phosphite triester linkage group (as e.g. obtained from a phosphoramidite coupling), and supposed to be sulfurized (by incubation with a
- Said antioxidant may suppress unwanted oxidation of a formed P(III) linkage group to a P (V) linkage group, which may not be amenable to sulfurization in the subsequent step.
- suitable antioxidants are, e.g., disclosed in EP3950698A1 and include inter alia triphenylphosphine, methyldiphenylphosphine, triethyl phosphite, ethoxydiphenylphosphine, diethoxyphenylphosphine, 9,10-dihydro-9-oxa-10-phosphaphenanthrene 10-oxide, and isobutylene sulfide.
- the solvents used may be controlled so that they contain no or a low level of oxidizing species.
- a quencher may optionally be added, wherein said quencher may be used to consume (i.e. react with) the phosphorus moiety of unreacted (excess) molecules of the building block B-1, B-2 or B-x employed.
- the quencher will typically be a nucleophilic compound capable of reacting with the phosphorus moiety. Water may, for example, be used as a quencher. Lactamide may, for example, also be used as a quencher.
- Step (f-1) of some methods of the invention is: optionally, incubating the first cycle oligonucleotide O-1 with an oxidizing or sulfurizing agent, thereby converting any P (III) atoms within said first cycle oligonucleotide O-1 to P (V) atoms.
- Step (f-2) of some of the methods of the invention is: optionally, incubating the second cycle oligonucleotide O-2 with an oxidizing or sulfurizing agent, thereby converting any P (III) atoms within said second cycle oligonucleotide O-2 to P (V) atoms.
- Step (f-x) of some of the methods of the invention is: optionally, incubating the x-th cycle oligonucleotide O-x with an oxidizing or sulfurizing agent, thereby converting any P (III) atoms within said x-th cycle oligonucleotide O-x to P (V) atoms.
- P (III) atom and “P (V) atom” have been defined above.
- the “oxidation state” may be “the charge of an atom after its homonuclear bonds have been divided equally and heteronuclear bonds assigned to the bond partners according to Allen electronegativity, except when the electronegative atom is bonded reversibly as a Lewis-acid ligand, in which case it does not obtain that bonds electrons”, as described in the IUPAC Recommendations 2016 (P. Karen et al., Pure and Applied Chemistry 2016, 88(8), 831 ⁇ 839).
- the Allen electronegativities given in said reference may be used and the P ⁇ H bond electron pair is assigned to H.
- the phosphorus atom of the H-phosphonate monoester moiety has the oxidation state III.
- the phosphorus atom of the phosphoramidite moiety in any one of Formulae II-1, II-1-a, and II-1-b has the oxidation state III.
- the phosphorus atoms in the phosphodiester linkage groups of DNA and RNA have the oxidation state V.
- the term “optionally” in step (f-1), (f-2), and (f-x) denotes that the respective step may or may not be carried out in a given coupling cycle, unless indicated differently in the context of specific embodiments.
- oligonucleotides comprising one or more P (III) linkage groups are typically less stable than related oligonucleotides comprising only P (V) linkage groups (as e.g. present in DNA and RNA).
- the target oligonucleotide O T comprises only P (V) linkage groups. If the first cycle oligonucleotide O-1 obtained in step (e-1) does not comprise any P (III) atoms, in particular not any P (III) linkage groups, it may not be necessary to carry out step (f-1). If the second cycle oligonucleotide O-2 obtained in step (e-2) does not comprise any P (III) atoms, in particular not any P (III) linkage groups, it may not be necessary to carry out step (f-2).
- step (e-x) of a coupling cycle does not comprise any P (III) atoms, in particular not any P (III) linkage groups, it may not be necessary to carry out step (f-x) in said coupling cycle.
- the oxidation state of the phosphorus atom within an internucleosidic linkage group formed in the bond forming reaction of a step (e-1), (e-2) or (e-x) will typically depend on the chemical structure of said phosphorus moiety of said building block B-1, B-2 or B-x, which engaged in the respective bond forming reaction.
- the oxidation state of the phosphorus atom within such a phosphorus moiety will be preserved in the course of the bond forming reaction of step (e-1), (e-2) or (e-x).
- step (e-1), (e-2) or (e-x) the phosphorus atom of the resulting phosphotriester internucleosidic linkage group will typically be present as P (V) atom, i.e. the phosphotriester linkage group is a P (V) linkage group.
- P (V) chemistry utilized by Baran et al.
- the bond forming reaction of a step (e-1), (e-2) or (e-x) may typically afford a phophite triester product (comprising a phosphite triester internucleosidic linkage group, i.e. a P (III) linkage group), if said phosphorus moiety covalently bonded via its phosphorus atom to an oxygen atom of the backbone of the building block B-1, B-2 or B-x, e.g.
- the phosphorus moiety PM of any one of Formulae II, II-a, and II-b is a phosphoramidite moiety such as, e.g., present in any one of Formulae II-1, II-1-a, and II-1-b, while a H-phosphonate diester product (comprising a H-phosphonate diester internucleosidic linkage group, i.e. a P (III) linkage group) may typically be obtained, if said phosphorus moiety covalently bonded via its phosphorus atom to an oxygen atom of the backbone of the building block B-1, B-2 or B-x, e.g.
- the phosphorus moiety PM of any one of Formulae II, II-a, and II-b is a H-phosphonate monoester moiety.
- H-phosphonate diester linkage groups may be more stable, e.g. under the conditions of steps (b-1), (b-2) or (b-x), than phosphite triester linkage groups, so that a step (f-1) or (f-2) or (f-x) may not need to be performed in each coupling cycle, in which said phosphorus moiety covalently bonded via its phosphorus atom to an oxygen atom of the backbone of the building block B-1, B-2 or B-x, e.g.
- the phosphorus moiety PM of any one of Formulae II, II-a, and II-b is a H-phosphonate monoester moiety.
- step (f-1) or (f-2) or (f-x) may be carried out, so that any P (III) atoms are converted to P (V) atoms.
- - the phosphorus moiety of the building blocks B-1, B-2, and each building block B-x e.g.
- the phosphorus moiety PM of any one of Formulae II, II, II-a, and II-b is independently selected from the group consisting of a phosphoramidite moiety and a H-phosphonate monoester moiety; - in each coupling cycle, in which said phosphorus moiety of the building block B-1, B-2 or B-x is a phosphoramidite moiety, step (f-1) or (f-2) or (f-x) is carried out; and - at least in the final coupling cycle, step (f-1) or (f-2) or (f-x) is carried out.
- - the phosphorus moiety of the building blocks B-1, B-2, and each building block B-x e.g.
- the phosphorus moiety PM of any one of Formulae II, II-a, and II-b is a phosphoramidite moiety; and - in each coupling cycle, step (f-1) or (f-2) or (f-x) is carried out.
- - the phosphorus moiety of the building blocks B-1, B-2, and each building block B-x e.g. the phosphorus moiety PM of any one of Formulae II, II-a, and II-b, is a H-phosphonate monoester moiety; and - at least in the final coupling cycle, step (f-1) or (f-2) or (f-x) is carried out.
- each building block B-1, B-2, and B-x is independently a compound of Formula II-1, preferably of Formula II-1-a, in particular of Formula II-1-b; and - in each coupling cycle, step (f-1) or (f-2) or (f-x) is carried out.
- the “oxidizing agent” or the “sulfurizing agent” to be used in any one of steps (f-1), (f-2), and (f-x) are not particularly limited in terms of their chemical structure as long as the respective agent is capable of converting any P (III) atoms within said first cycle oligonucleotide O-1, said second cycle oligonucleotide O-2, and said x-th cycle oligonucleotide O-x to P (V) atoms.
- An “oxidizing agent” and a “sulfurizing agent” may differ in the means of how P (III) atoms are converted to P (V) atoms.
- an “oxidizing agent” may introduce one or more covalent bonds between the P (III) atom to be oxidized and an oxygen atom.
- a “sulfurizing agent” may introduce one or more covalent bonds between the P (III) atom to be sulfurized and a sulfur atom.
- the term “oxidizing agent” preferably refers to any agent capable of converting a phosphite triester linkage group to a phosphate triester linkage group and a H-phosphonate diester linkage group to a phosphate diester (i.e. a phosphodiester) linkage group.
- sulfurizing agent preferably refers to any agent capable of converting a phosphite triester linkage group to a thiophosphate triester linkage group and a H-phosphonate diester linkage group to a thiophosphate diester linkage group (i.e. a phosphorothioate) linkage group.
- Iodine may be a preferred oxidizing agent.
- an aqueous solution of iodine may be used, preferably in combination with a co-solvent such as an ether solvent, e.g.
- an ether solvent S E (vide infra), a nitrile solvent such as acetonitrile, a (hetero)aromatic solvent such as pyridine, or a mixture thereof.
- a solution of iodine (e.g. 1 mol/L) in a mixed solvent of 4-methyltetrahydropyran (MTHP), water, and, optionally, acetonitrile may be used.
- peroxides such as tert-butyl hydroperoxide, cumene hydroperoxides, bis-trimethylsilyl peroxide, 2-butanone peroxide, and hydrogen peroxide, or peroxy acids such as m-chloroperbenzoic acid (mCPBA) may, for example, be used as oxidizing agents.
- mCPBA m-chloroperbenzoic acid
- a solution of tert-butyl hydroperoxide in a non-polar solvent may be used.
- a solution (e.g. 5 ⁇ 6 mol/L) of tert-butyl hydroperoxide in nonane may be used.
- an aqueous solution of hydrogen peroxide may be used.
- an aqueous solution of hydrogen peroxide and, optionally, potassium iodide may be mixed with an aqueous phosphoric acid buffer (e.g. pH 6.8) to obtain an oxidizing agent.
- an aqueous phosphoric acid buffer e.g. pH 6.8
- an aqueous phosphoric acid buffer e.g. pH 6.8
- (1S)-(+)-(10- camphorsulfonyl)-oxaziridine (CSO) may, for example, be used as oxidizing agent, e.g. as a 0.5 M (i.e.0.5 mol/L) solution in, e.g., acetonitrile.
- the oxidizing agent may, for example, be applied in form of a 0.005 ⁇ 10.0 M (i.e.
- oxidizing agent preferably a 0.01 ⁇ 5.0 M solution in a suitable solvent.
- neat oxidizing agent may, for example, be added (i.e. the oxidizing agent need not be dissolved in a suitable solvent).
- Xanthane hydride (5-amino-3H-1,2,4-dithiazole-3-thione), 3H-1,2-benzodithiol-3- one, and 3-(N,N-dimethylamino-methylidene)amino)-3H-1,2,4-dithiazole-5-thione (CAS RN: 1192027-04-5, DDTT) may be preferred sulfurizing agents.
- 1,4-dithiothreitol DTT
- phenylacetyl disulfide PADS
- 3H-1,2-benzodithiol-3-one 1,1-dioxide Beaucage Reagent
- neat sulfurizing agent i.e. not in solution
- neat DDTT may be added.
- neat xanthane hydride may be added.
- a solution of the sulfurizing agent in a suitable solvent may be added.
- the sulfurizing agent may, for example, be applied in form of a 0.005 ⁇ 5.0 M solution, preferably a 0.01 ⁇ 1.0 M solution in a suitable solvent, for example, selected from the group consisting of pyridine, acetonitrile, water, tetrahydrofuran, and mixtures thereof.
- a suitable solvent for example, selected from the group consisting of pyridine, acetonitrile, water, tetrahydrofuran, and mixtures thereof.
- a solution of xanthane hydride in pyridine may be used, optionally in combination with a co-solvent such as acetonitrile.
- a solution of xanthane hydride (e.g. 0.2 M) in pyridine may be used.
- a solution of xanthane hydride (e.g.0.1 M) in a mixture of pyridine and acetonitrile (e.g.1:1, v/v) may be used.
- Steps (f-1), (f-2), and (f-x) may, for example, be performed at a temperature in the range of 0 ⁇ 90 °C, 10 ⁇ 70 °C, 15 ⁇ 60 °C, or 15 ⁇ 50 °C.
- steps (f-1), (f-2), and (f-x) may simply be performed at room temperature. Increased temperatures may result in shorter reaction times.
- reaction time may also depend on the chemical structure of the reactants and will routinely be selected by a skilled person, for example, based on reaction monitoring using, e.g., thin-layer chromatography and/or high performance liquid chromatography (HPLC), optionally coupled to mass spectrometry.
- the term “incubating” in steps (f-1), (f-2), and (f-x) may be understood in the broadest sense to refer to any process of contacting the first cycle oligonucleotide O-1, the second cycle oligonucleotide O-2 or the x-th cycle oligonucleotide O-x, respectively, and the respective oxidizing or sulfurizing agent, preferably inside the same reaction vessel or reactor.
- the first cycle oligonucleotide O-1, the second cycle oligonucleotide O-2 or the x-th cycle oligonucleotide O-x may already be contained in a reaction vessel or reactor, to which the respective oxidizing or sulfurizing agent is then added.
- the first cycle oligonucleotide O-1, the second cycle oligonucleotide O-2 or the x-th cycle oligonucleotide O-x may be added to the reaction vessel or reactor containing the respective oxidizing or sulfurizing agent.
- excess oxidizing or sulfurizing agent may typically be quenched (i.e. reduced), e.g., by treatment with a reducing agent.
- a reducing agent such as triethylphosphite or a solution thereof may be added directly into the reaction mixture of the respective step (f-1) or (f-2) or (f-x).
- a reducing agent may also be comprised in one or more of the one or more aqueous solutions used for the one or more aqueous extractions.
- Step (g-1) of some methods of the invention is: optionally, subjecting a solution comprising the first cycle oligonucleotide O-1 to one or more aqueous extractions, wherein the organic phase comprises the first cycle oligonucleotide O-1.
- Step (g-2) of some of the methods of the invention is: optionally, subjecting a solution comprising the second cycle oligonucleotide O-2 to one or more aqueous extractions, wherein the organic phase comprises the second cycle oligonucleotide O-2.
- Step (g-x) of some of the methods of the invention is: optionally, subjecting a solution comprising the x-th cycle oligonucleotide O-x to one or more aqueous extractions, wherein the organic phase comprises the x-th cycle oligonucleotide O-x.
- the term “optionally” in steps (g-1), (g-2), and (g-x) denotes that the respective step may or may not be carried out in a given coupling cycle, unless indicated differently in the context of specific embodiments.
- Steps (g-1), (g-2), and (g-x) may, for example, be carried out to (at least partly) remove one or more side products and/or by-products and/or excess reagents and/or water-miscible solvents.
- step (g-1) is carried out (i.e. is not optional).
- step (g-2) is carried out (i.e. is not optional).
- step (g-x) is carried out (i.e. is not optional).
- said solution comprising the first cycle oligonucleotide O-1 in step (g-1) may refer to the reaction mixture obtained from carrying out step (e-1), if step (f-1) is not performed, or may refer to the reaction mixture obtained from carrying out step (f-1) (wherein one or more quenchers or reducing agents may, for example, have been added beforehand as explained above).
- said solution comprising the second cycle oligonucleotide O-2 in step (g-2) may refer to the reaction mixture obtained from carrying out step (e-2), if step (f-2) is not performed, or may refer to the reaction mixture obtained from carrying out step (f-2) (wherein one or more quenchers or reducing agents may, for example, have been added beforehand as explained above).
- said solution comprising the x-th cycle oligonucleotide O-x in step (g-x) may refer to the reaction mixture obtained from carrying out step (e-x), if step (f-x) is not performed, or may refer to the reaction mixture obtained from carrying out step (f-x) (wherein one or more quenchers or reducing agents may, for example, have been added beforehand as explained above).
- said solutions comprising the first cycle oligonucleotide O-1, the second cycle oligonucleotide O-2 or the x-th cycle oligonucleotide O-x may be obtained from the respective reaction mixture by addition of one or more non-polar solvents, for example, in order to facilitate the phase separation.
- non-polar solvents may also be added during or in between the one or more aqueous extractions of any one of steps (g-1), (g-2), and (g-x).
- non-polar solvents comprise those listed above in the context of steps (c-1), (c-2), and (c-x).
- 4-Methyltetrahydropyran (MTHP) may be a preferred non-polar ether solvent which may be added prior to or during or in between the aqueous extraction(s) of any one of steps (g-1), (g-2), and (g-x).
- one or more amide solvents S A may be added prior to or during or in between the aqueous extraction(s) of any one of steps (g-1), (g-2), and (g-x).
- aqueous extraction in steps (g-1), (g-2), and (g-x) of the method of the invention may be understood in the broadest sense as any liquid-liquid extraction operation during which the respective solution comprising the first cycle oligonucleotide O-1, the second cycle oligonucleotide O-2 or the x-th cycle oligonucleotide O-x is extracted with water or an aqueous solution.
- aqueous solution has been defined above.
- the aqueous solution may be the same or different (i.e. comprise the same or different components in the same or different ratios). It will also be understood that the aqueous solutions may be the same or different for different steps (g-x) of different coupling cycles. In case of more than one organic phases (either from a single aqueous extraction or from back-extraction of one or more aqueous phase), these more than one organic phases may be combined to obtain “the organic phase” mentioned in steps (g-1), (g-2), and (g-x). In general, the means of performing aqueous extractions are well-known to the skilled person.
- the organic phase comprises the first cycle oligonucleotide O-1 in step (g-1) may be understood in the broadest sense to mean that some of the molecules of said first cycle oligonucleotide O-1 are dissolved in the organic phase.
- the expression “the organic phase comprises the second cycle oligonucleotide O-2” in step (g-2) may be understood in the broadest sense to mean that some of the molecules of said second cycle oligonucleotide O-2 are dissolved in the organic phase.
- the organic phase comprises the x-th cycle oligonucleotide O-x” in step (g-x) may be understood in the broadest sense to mean that some of the molecules of said x-th cycle oligonucleotide O-x are dissolved in the organic phase. It is preferred that most molecules (e.g. more than 50 %, 60 %, 70 %, 80 %, or 90 % of the molecules) of the said first cycle oligonucleotide O-1 or said second cycle oligonucleotide O-2 or said x-th cycle oligonucleotide O-x are comprised in the organic phase (and thus not in the aqueous phase).
- Step (h-1) of some methods of the invention is: if step (g-1) has been carried out, optionally reducing the water content of the organic phase comprising the first cycle oligonucleotide O-1.
- Step (h-2) of some of the methods of the invention is: if step (g-2) has been carried out, optionally reducing the water content of the organic phase comprising the second cycle oligonucleotide O-2.
- Step (h-x) of some of the methods of the invention is: if step (g-x) has been carried out, optionally reducing the water content of the organic phase comprising the x-th cycle oligonucleotide O-x.
- step (h-1) means that in the first coupling cycle, step (h-1) may only be carried out, if step (g-1) has been carried out.
- step (h-2) has been carried out in step (h-2) means that in the second coupling cycle, step (h-2) may only be carried out, if step (g-2) has been carried out.
- step (g-x) has been carried out in step (h-x) means that in a given coupling cycle comprising steps (b-x) to (h-x) (as far as present), step (h-x) may only be carried out, if step (g-x) has been carried out (in the same coupling cycle).
- steps (h-1), (h-2), and (h-x) denotes that, even if the respective step (g-1), (g-2) or (g-x) has been carried out, the respective step (h-1), (h-2) or (h-x) may or may not be carried, unless indicated differently in the context of specific embodiments.
- step (h-1) is carried out (i.e. is not optional), if step (g-1) has been carried out.
- step (h- 2) is carried out (i.e. is not optional), if step (g-2) has been carried out.
- step (h-x) in at least one, or each, coupling cycle comprising steps (b-x) to (h-x) (as far as present), step (h-x) is carried out (i.e. is not optional), if step (g-x) has been carried out in the same coupling cycle.
- the term “the organic phase” in steps (h-1), (h-2), and (h-x) refers to “the organic phase” of the respective step (g-1) or (g-2) or (g-x) of the same coupling cycle.
- the term “reducing the water content” has been defined above.
- step (h-1) is: if step (g-1) has been carried out, optionally reducing the water content of the organic phase comprising the first cycle oligonucleotide O-1, wherein the water content is adjusted to be equal to or less than 2000 ppm, 1900 ppm, 1800 ppm, 1700 ppm, 1600 ppm, 1500 ppm, 1400 ppm, 1300 ppm, 1200 ppm, 1100 ppm, 1000 ppm, 900 ppm, 800 ppm, 700 ppm, 600 ppm, 500 ppm, 400 ppm, 300 ppm, 250 ppm, 200 ppm, 150 ppm, or 100 ppm.
- step (h-2) is: if step (g-2) has been carried out, optionally reducing the water content of the organic phase comprising the second cycle oligonucleotide O-2, wherein the water content is adjusted to be equal to or less than 2000 ppm, 1900 ppm, 1800 ppm, 1700 ppm, 1600 ppm, 1500 ppm, 1400 ppm, 1300 ppm, 1200 ppm, 1100 ppm, 1000 ppm, 900 ppm, 800 ppm, 700 ppm, 600 ppm, 500 ppm, 400 ppm, 300 ppm, 250 ppm, 200 ppm, 150 ppm, or 100 ppm.
- step (h-x) is: if step (g-x) has been carried out, optionally reducing the water content of the organic phase comprising the x-th cycle oligonucleotide O-x, wherein the water content is adjusted to be equal to or less than 2000 ppm, 1900 ppm, 1800 ppm, 1700 ppm, 1600 ppm, 1500 ppm, 1400 ppm, 1300 ppm, 1200 ppm, 1100 ppm, 1000 ppm, 900 ppm, 800 ppm, 700 ppm, 600 ppm, 500 ppm, 400 ppm, 300 ppm, 250 ppm, 200 ppm, 150 ppm, or 100 ppm.
- - step (h-1) is carried out (i.e. is not optional), if the organic phase comprising the first cycle oligonucleotide O-1 of step (g-1) comprises more than 2000 ppm, 1900 ppm, 1800 ppm, 1700 ppm, 1600 ppm, 1500 ppm, 1400 ppm, 1300 ppm, 1200 ppm, 1100 ppm, 1000 ppm, 900 ppm, 800 ppm, 700 ppm, 600 ppm, 500 ppm, 400 ppm, 300 ppm, 250 ppm, 200 ppm, 150 ppm, or 100 ppm of water; - step (h-2) is carried out (i.e.
- the organic phase comprising the second cycle oligonucleotide O-2 of step (g-2) comprises more than 2000 ppm, 1900 ppm, 1800 ppm, 1700 ppm, 1600 ppm, 1500 ppm, 1400 ppm, 1300 ppm, 1200 ppm, 1100 ppm, 1000 ppm, 900 ppm, 800 ppm, 700 ppm, 600 ppm, 500 ppm, 400 ppm, 300 ppm, 250 ppm, 200 ppm, 150 ppm, or 100 ppm of water; and - step (h-x) is carried out (i.e.
- the organic phase comprising the x-th cycle oligonucleotide O-x of step (g-x) of the same coupling cycle comprises more than 2000 ppm, 1900 ppm, 1800 ppm, 1700 ppm, 1600 ppm, 1500 ppm, 1400 ppm, 1300 ppm, 1200 ppm, 1100 ppm, 1000 ppm, 900 ppm, 800 ppm, 700 ppm, 600 ppm, 500 ppm, 400 ppm, 300 ppm, 250 ppm, 200 ppm, 150 ppm, or 100 ppm of water.
- reducing the water content is achieved by means of azeotropic distillation and/or by contacting said organic phase comprising the first cycle oligonucleotide O-1, the second cycle oligonucleotide O-2 or the x-th cycle oligonucleotide O-x with a drying agent.
- reducing the water content is achieved by means of azeotropic distillation.
- reducing the water content is achieved by means of contacting said organic phase comprising the first cycle oligonucleotide O-1, the second cycle oligonucleotide O-2 or the x-th cycle oligonucleotide O-x with a drying agent.
- any one of steps (h-1), (h-2), and (h-x) one or more solvents and/or further components may be added to said organic phase comprising the first cycle oligonucleotide O-1, the second cycle oligonucleotide O-2 or the x-th cycle oligonucleotide O-x prior to, during, and/or after reduction of the water content.
- reducing the water content is achieved by means of azeotropic distillation.
- the first coupling cycle further comprises a step (i-1) of reacting free hydroxyl groups with a blocking agent, wherein step (i-1) is carried out after step (e-1) or after step (f-1); and/or - the second coupling cycle further comprises a step (i-2) of reacting free hydroxyl groups with a blocking agent, wherein step (i-2) is carried out after step (e-2) or after step (f-2); and/or - at least one coupling cycle comprising steps (b-x) to (h-x) (as far as present) further comprises a step (i-x) of reacting free hydroxyl groups with a blocking agent, wherein step (i-x) is carried out after step (e-x) or after step (f-x).
- the free backbone hydroxyl group present in the component (C-0) # , the first cycle oligonucleotide (O-1) # , and the (x ⁇ 1)-th cycle oligonucleotide (O-(x ⁇ 1)) # is supposed to engage in the bond forming reaction of step (e-1), (e-2) or (e-x) (of the same coupling cycle), which consumes said free backbone hydroxyl group in the sense of incorporating it into a newly-formed internucleosidic linkage group.
- a typically very small, e.g.
- ⁇ 1 %, ⁇ 0.5 % or ⁇ 0.1 %) fraction of the free backbone hydroxyl groups may not engage in the bond forming reaction at the first occasion, i.e. in the same coupling cycle, in which the respective free hydroxyl group has been generated by cleaving a protecting group.
- Such free hydroxyl groups are herein also referred to as unreacted free hydroxyl groups.
- These groups would be available to participate in the bond forming reactions of the coupling steps of following coupling cycles. This may not be desirable, since it would afford an oligonucleotide product lacking one nucleoside subunit. Such oligonucleotide products may be difficult to remove from the target oligonucleotide O T later on.
- blocking agent and “capping agent” are used interchangeably to denote any chemical reagent capable of acylating, preferably acetylating, a free hydroxyl group.
- Capping agents for oligonucleotide synthesis form part of the common knowledge of those skilled in the art. Any blocking (i.e. capping) agents known from oligonucleotide synthesis may be used in the method of the invention.
- Preferred examples of such blocking agents comprise anhydrides of carboxylic acids, in particular acetic anhydride. It is known to those skilled in the art that free carboxylic acids may also be used in combination with activating agents such as carbodiimides (e.g.
- dicyclohexylcarbdiimide or diisopropylcarbodiimide and additives such as, e.g., 4-dimethylaminopyridine (DMAP).
- DMAP 4-dimethylaminopyridine
- acetylation may be achieved by treating the growing oligonucleotide chains with neat acetic anhydride or a solution thereof.
- An organic base such as pyridine, lutidine (e.g.2,6-lutidine), collidine, N-methylimidazole, or a mixture thereof may be added.
- acetic anhydride e.g.4 moles of acetic anhydride per 1 mol of employed component (C-0) #
- a base such as pyridine (e.g.5 moles of base per 1 mol of employed component (C-0) # ).
- the blocking agents may be added directly into the reaction vessel or reactor, in which step (e-1), (e-2), (e-x), (f-1), (f-2) or (f-x) was carried out.
- step (i-1), (i-2), and (i-x) may be understood in the broadest sense as any operation during which the growing oligonucleotide chains and the blocking agent are present in the same reaction vessel or reactor and engage in the blocking reaction.
- the method further comprises - a step (k-1) of incubating the first cycle oligonucleotide O-1 with a deprotection mixture M-(k-1), thereby cleaving the protecting group PG-1 from the first cycle oligonucleotide O-1, so as to obtain a first cycle oligonucleotide (O-1) # having a free backbone hydroxyl group; and/or - a step (m-1) of incubating the first cycle oligonucleotide O-1 or (O-1) # with a base, thereby cleaving the pseudo solid-phase protecting group PG-s and, optionally, one or more further protecting groups from the first cycle oligonucleotide O-1 or (O-1) # ; and/or - a step (p-1) of modifying the first cycle oligonucleotide O-1 or (O-1) # ; wherein, if more than one of steps (k-1), (k-1), (k-1), (k-1
- any one of steps (k-1), (m-1), and (p-1) may only be carried out after all steps of the first coupling cycle (optionally excluding steps indicated as optional) have been performed. It will also be understood that, if any one of steps (k-1), (m-1), and (p-1) is carried out, the second coupling cycle comprising steps (b-2) to (h-2) and thus any further coupling cycles comprising steps (b-x) to (h-x) (as far as present) may not be carried out.
- the method further comprises - a step (k-2) of incubating the second cycle oligonucleotide O-2 with a deprotection mixture M-(k-2), thereby cleaving the protecting group PG-2 from the second cycle oligonucleotide O-2, so as to obtain a second cycle oligonucleotide (O-2) # having a free backbone hydroxyl group; and/or - a step (m-2) of incubating the second cycle oligonucleotide O-2 or (O-2) # with a base, thereby cleaving the pseudo solid-phase protecting group PG-s and, optionally, one or more further protecting groups from the second cycle oligonucleotide O-2 or (O-2) # ; and/or - a step (p-2) of modifying the second cycle oligonucleotide O-2 or (O-2) # ; wherein, if more than one of steps (k-2), (
- steps (k-2), (m-2), and (p-2) may only be carried out after all steps of the second coupling cycle (optionally excluding steps indicated as optional) have been performed. It will also be understood that, if any one of steps (k-2), (m-2), and (p-2) is carried out, further coupling cycles comprising steps (b-x) to (h-x) (as far as present) may not be carried out.
- the method further comprises - a step (k-n) of incubating the n-th cycle oligonucleotide O-n with a deprotection mixture M-(k-n), thereby cleaving the protecting group PG-n from the n-th cycle oligonucleotide O-n, so as to obtain a n-th cycle oligonucleotide (O-n) # having a free backbone hydroxyl group; and/or - a step (m-n) of incubating the n-th cycle oligonucleotide O-n or (O-n) # with a base, thereby cleaving the pseudo solid-phase protecting group PG-s and, optionally, one or more further protecting groups from the n-th cycle oligonucleotide O-n or (O-n) # ; and/or - a step (p-n) of modifying the n-th cycle oligon
- any one of steps (k-n), (m-n), and (p-n) may only be carried out after all steps of the (n ⁇ 2)-th iteration of the coupling cycle comprising steps (b- x) to (h-x) (optionally excluding steps indicated as optional) have been performed.
- the term "deprotection mixture M-(k-1)" refers to any mixture which may be used to effect cleavage of the protecting group PG-1 from the first cycle oligonucleotide O-1. Any definitions and embodiments pertaining to the deprotection mixture M-(b-2) may also apply to the deprotection mixture M-(k-1).
- deprotection mixture M-(k-n) refers to any mixture which may be used to effect cleavage of the protecting group PG-n from the n-th cycle oligonucleotide O-n.
- any definitions and embodiments pertaining to the deprotection mixture M-(b-x) may also apply to the deprotection mixture M-(k-n).
- the term “base” in steps (m-1), (m-2), and (m-n) may be understood in the broadest sense to refer to any base (a proton acceptor in the sense of the Br ⁇ nsted-Lowry theory), unless indicated differently in the context of specific embodiments.
- Non- limiting examples of such a base comprise ammonia, a (C1 ⁇ C6-alkyl)NH2 monoalkylamine, in particular methylamine, ethylamine or tert-butylamine, a (C 1 ⁇ C 6 -alkyl) 2 NH dialkylamine, in particular dimethylamine or diethylamine, a source of hydroxide ions, and mixtures thereof.
- a base will typically not be employed as such in a step (m-1), (m-2) or (m-n), but rather in form of a solution of the base in a suitable solvent, in particular water, i.e. as an aqueous solution of the respective base.
- Such an aqueous solution may further comprise one or more water-miscible organic solvents such as, e.g., acetonitrile, tetrahydrofuran, methanol, ethanol, and the like.
- a source of hydroxide ions may refer to a salt which comprises hydroxide ions, wherein sodium hydroxide, potassium hydroxide, and ammonium hydroxide may be preferred examples.
- an aqueous solution of ammonia may also be referred to as an ammonium hydroxide solution, since it comprises ammonium ions and hydroxide ions.
- a solution comprising a source of hydroxide anions may comprise said source of hydroxide ions in solvated form.
- said base of any one of steps (m-1), (m-2), and (m-n) is an aqueous solution of one or more compounds selected from the group consisting of ammonia, a (C 1 ⁇ C 6 -alkyl)NH 2 monoalkylamine, a (C 1 ⁇ C 6 -alkyl) 2 NH dialkylamine, sodium hydroxide, potassium hydroxide, and mixtures thereof.
- said base of any one of steps (m-1), (m-2), and (m-n) is an aqueous solution of one or more compounds selected from the group consisting of ammonia, methylamine, ethylamine, tert-butylamine, sodium hydroxide, potassium hydroxide, and mixtures thereof.
- said base of any one of steps (m-1), (m-2), and (m-n) may be an aqueous solution of ammonia, e.g.25 ⁇ 30 wt-% aqueous ammonia.
- said base of any one of steps (m-1), (m-2), and (m-n) may be an aqueous solution of tert-butylamine, e.g.
- said base of any one of steps (m-1), (m-2), and (m-n) may be an aqueous solution of methylamine, e.g. a 1:1 (v:v) mixture of water and methylamine.
- said base of any one of steps (m-1), (m-2), and (m-n) may be a solution of sodium hydroxide (e.g.1 mol/L) in a mixture of water and tetrahydrofuran (e.g. 1:3, v:v).
- hydrazine e.g. used in form of its hydrate
- nucleophilic base e.g. used as nucleophilic base.
- the method comprises exactly one step selected from the group consisting of step (m-1), step (m-2), and step (m-n), and wherein - step (m-1) is carried out by incubating the first cycle oligonucleotide O-1 or (O-1) # with an aqueous solution of an organic amine, preferably an aliphatic amine, more preferably a primary or secondary aliphatic amine, in particular a primary aliphatic amine such as tert-butylamine, and an aliphatic alcohol, preferably an aliphatic alcohol comprising 1 ⁇ 6 carbon atoms, in particular methanol; - step (m-2) is carried out by incubating the second cycle oligonucleotide O-2 or (O-2) # with an aqueous solution of an organic amine, preferably an aliphatic amine, more preferably a primary or secondary aliphatic amine, in particular a primary aliphatic amine such as ter
- the respective oligonucleotide i.e., the first cycle oligonucleotide O-1 or (O-1) # in a step (m-1) and the second cycle oligonucleotide O-2 or (O-2) # in a step (m-2) and the n-th cycle oligonucleotide O-n or (O-n) # in a step (m-n) preferably comprises at least one internucleosidic linkage group selected from the group consisting of a thiophosphate triester group and a thiophosphate diester group, preferably a linkage group of Formula B, where X 1 is S and X 2 is either OH or O-CH2-CH2-CN.
- the method comprises exactly one step selected from the group consisting of step (m-1), step (m-2), and step (m-n), and wherein - step (m-1) is carried out by incubating the first cycle oligonucleotide O-1 or (O-1) # with an aqueous solution of a (C 1 ⁇ C 6 -alkyl)NH 2 monoalkylamine, in particular tert-butylamine, and an aliphatic alcohol comprising 1 ⁇ 6 carbon atoms, in particular methanol; - step (m-2) is carried out by incubating the second cycle oligonucleotide O-2 or (O-2) # with an aqueous solution of a (C1 ⁇ C6-alkyl)NH2 monoalkylamine, in particular tert-butylamine, and an aliphatic alcohol comprising 1 ⁇ 6 carbon atoms, in particular methanol; and - step (m-n) is carried out by incubating the
- the respective oligonucleotide i.e., the first cycle oligonucleotide O-1 or (O-1) # in a step (m-1) and the second cycle oligonucleotide O-2 or (O-2) # in a step (m-2) and the n-th cycle oligonucleotide O-n or (O-n) # in a step (m-n) preferably comprises at least one internucleosidic linkage group selected from the group consisting of a thiophosphate triester group and a thiophosphate diester group, preferably a linkage group of Formula B, where X 1 is S and X 2 is either OH or O-CH 2 -CH 2 -CN.
- the method comprises exactly one step selected from the group consisting of step (m-1), step (m-2), and step (m-n), and wherein - step (m-1) is carried out by incubating the first cycle oligonucleotide O-1 or (O-1) # with an aqueous solution of a (C 1 ⁇ C 6 -alkyl)NH 2 monoalkylamine, in particular tert-butylamine, and an aliphatic alcohol comprising 1 ⁇ 6 carbon atoms, in particular methanol, wherein the volume-ratio of monoalkylamine to aliphatic alcohol to water is 1:1:2 (v:v:v); - step (m-2) is carried out by incubating the second cycle oligonucleotide O-2 or (O-2) # with an aqueous solution of a (C1 ⁇ C6-alkyl)NH2 monoalkylamine, in particular tert-butylamine, and an aliphatic alcohol compris
- the respective oligonucleotide i.e., the first cycle oligonucleotide O-1 or (O-1) # in a step (m-1) and the second cycle oligonucleotide O-2 or (O-2) # in a step (m-2) and the n-th cycle oligonucleotide O-n or (O-n) # in a step (m-n) preferably comprises at least one internucleosidic linkage group selected from the group consisting of a thiophosphate triester group and a thiophosphate diester group, preferably a linkage group of Formula B, where X 1 is S and X 2 is either OH or O-CH2-CH2-CN.
- aqueous solution of a (C 1 ⁇ C 6 -alkyl)NH 2 monoalkylamine, in particular tert-butylamine, and an aliphatic alcohol comprising 1 ⁇ 6 carbon atoms, in particular methanol is particularly preferred, if the pseudo solid-phase protecting group PG-s is a protecting group of Formula P-1-a below, in particular, with the integer a being 1.
- Steps (m-1), (m-2), and (m-n) may, for example, be performed at a temperature in the range of 5 ⁇ 95 °C, 10 ⁇ 95 °C, 15 ⁇ 95 °C, 20 ⁇ 95 °C, 25 ⁇ 95 °C, 30 ⁇ 95 °C, 35 ⁇ 95 °C, 50 ⁇ 90 °C, 55 ⁇ 90 °C, 60 ⁇ 85 °C, or 60 ⁇ 80 °C.
- steps (m-1), (m-2), and (m-n) may preferably be performed in a sealed reaction vessel or reactor, e.g. in an autoclave, when operating at elevated temperatures.
- steps (m-1), (m-2), and (m-n) “one or more further protecting groups” (i.e.
- any common protecting groups removable under alkaline conditions will typically be removed during the incubation with said base in steps (m-1), (m-2), and (m-n).
- Such protecting groups removable under alkaline conditions e.g. under the conditions of steps (m-1), (m-2), and (m-n)
- protecting groups at the internucleosidic linkage groups e.g.
- any protecting groups R Z-1 and R Z-2 may typically be removed under the conditions of steps (m-1), (m-2), and (m-n).
- the term “modifying” may be understood in the broadest sense to embrace “chemically modifying” and/or “biotechnologically modifying” the respective oligonucleotides.
- “chemically modifying” is defined as subjecting the respective oligonucleotide (which is to be chemically modified) to one or more chemical reactions.
- Non-limiting examples of such chemical reactions are - the removal of one or more protecting groups (including any pseudo solid-phase protecting groups); - conjugation with one or more compounds selected from the group consisting of a nucleoside, an oligonucleotide, a carbohydrate such as a monosaccharide or a polysaccharide, an amino acid, a peptide, a lipid, an active pharmaceutical ingredient, or the like; and - an intramolecular bond forming reaction resulting in cyclization. It will be understood that more than one such chemical reactions may be performed in the course of said chemical modification.
- biotechnologically modifying is defined as subjecting the respective oligonucleotide (which is to be biotechnologically modified) to one or more enzymatic reactions.
- enzymatic reaction refers to any reaction which is enabled by and/or catalyzed by one or more enzymes. A preferred example is enzymatic ligation.
- the method of the invention further comprises a step (z) of isolating the target oligonucleotide O T .
- the means of isolating oligonucleotides upon oligonucleotide synthesis form part of the common knowledge of those skilled in the art.
- such a step of isolating an oligonucleotide comprises one or more purification steps and one or more steps aiming at obtaining the oligonucleotide in solid form.
- chromatographic methods may typically be used, in particular ion exchange (especially anion exchange) chromatography and reversed phase (RP) HPLC, e.g. in form of hydrophobic interaction HPLC.
- RP reversed phase
- a step of isolating an oligonucleotide may comprise ultrafiltration and/or desalting steps.
- the target oligonucleotide to be isolated may be dissolved in a reaction mixture.
- a crude product either by precipitating the target oligonucleotide to be purified from the respective reaction mixture or by evaporation to dryness (e.g. in vacuo). Precipitation may for example be achieved by (partly) removing one or more solvents and/or by addition of one or more antisolvents (in which the target oligonucleotide is not soluble or poorly soluble) and/or by cooling the reaction mixture from which the target oligonucleotide to be isolated.
- the so-obtained crude product may, for example be washed with one or more solvents and/or aqueous solutions.
- the crude product may then be dissolved, so as to obtain a solution, preferably an aqueous solution of the target oligonucleotide to be isolated.
- the reaction mixture containing the target oligonucleotide as such may be used without precipitation or evaporation to dryness.
- a solution, preferably an aqueous solution, containing the target oligonucleotide O T to be isolated may be submitted to ultrafiltration and/or desalting, ion exchange chromatography, and another round of ultrafiltration and/or desalting.
- a crude product containing the target oligonucleotide O T to be isolated may be submitted to reversed phase (RP) HPLC, e.g.
- RP reversed phase
- oligonucleotides in form of hydrophobic interaction HPLC.
- the latter method may preferably be performed, if the oligonucleotides still carry the 5 ⁇ -terminal hydroxyl protecting group, e.g. the DMT group. Said 5 ⁇ -protecting group may even be removed on the RP-HPLC column by passing an acidic solution through the column. If the 5 ⁇ -terminal protecting group has been removed prior to purification, ion exchange chromatography may be preferred. Purification is typically followed by one or more steps aiming at obtaining the oligonucleotide in solid form. Lyophilization or spray drying may, for example, be used. In some cases, it may be desirable to obtain the oligonucleotide in form of a salt with certain counter ions.
- salt exchange may be performed, typically prior to lyophilization or spray drying.
- steps (b-1) to (h-1) no solid-liquid separation is performed and steps (c-1) and (g-1) are carried out in the presence of one or more organic solvents S O , wherein the one or more organic solvents S O are selected so that an organic phase and an aqueous phase are formed during the aqueous extraction.
- steps (c-1) and (g-1) are carried out in the presence of one or more organic solvents S O ” does not imply that step (g-1) must be carried out, i.e., is not optional.
- step (g-1) is carried out, it is carried out in the presence of one or more organic solvents S O .
- the method of the invention comprises performing a second coupling cycle comprising steps (b-2) to (h-2) (as far as present) , during and in between steps (b- 1) to (h-2) (as far as present), no solid-liquid separation is performed and steps (c- 1), (g-1), (c-2), and (g-2) are carried out in the presence of one or more organic solvents S O , wherein the one or more organic solvents S O are selected so that an organic phase and an aqueous phase are formed during the aqueous extraction .
- steps (c-1), (g-1), (c-2), and (g-2) are carried out in the presence of one or more organic solvents S O ” does not imply that steps (g-1) and (g-2) must be carried out, i.e., are not optional. However, if one or both of steps (g-1) and (g-2) are carried out, they are carried out in the presence of one or more organic solvents S O .
- the method of the invention comprises performing (n ⁇ 2) iterations of a coupling cycle comprising the following steps (b-x) to (h-x) (as far as present), wherein n is an integer in the range of 3 to 99, which denotes the total number of coupling cycles performed to obtain to obtain the n-th cycle oligonucleotide O-n, during and in between steps (b-1) to (h-n) (as far as present), no solid-liquid separation is performed, and steps (c-1), (g-1), (c-2), and (g-2), as well as each iteration of steps (c-x) and (g-x) is carried out in the presence of one or more organic solvents S O , wherein the one or more organic solvents S O are selected so that an organic phase and an aqueous phase are formed during the aqueous extraction.
- steps (c-1), (g-1), (c-2), and (g-2), as well as each iteration of steps (c-x) and (g-x) are carried out in the presence of one or more organic solvents S O ” does not imply that steps (g-1), (g-2), and (g-x) must be carried out, i.e., are not optional. However, if one or more of steps (g-1), (g-2) and (g-x) are carried out, they are carried out in the presence of one or more organic solvents S O .
- solid-liquid separation refers to any operation during which a solid is separated from a liquid. Most commonly, solid-liquid separation will be filtration or centrifugation, preferably filtration. It will be understood that such solid-liquid separation may typically not be complete, meaning that some residual liquid may typically be attached to the solid after solid-liquid separation, as known to those skilled in the art. It will be understood that the term “solid-liquid separation” does not embrace evaporation of the liquid.
- a step of, e.g., evaporating a solution to dryness is not considered a “solid-liquid separation”.
- a solid e.g., a drying agent such as molecular sieves as, as, e.g., disclosed in in X. Shi et al., Journal of Organic Chemistry 2022, 87(4), 2087 ⁇ 2110 (cf., e.g., Figure 1 of the cited reference and explanations pertaining thereto)
- a drying agent such as molecular sieves
- the process does not comprise any solid-liquid separation during or in between at least at least 3, 4, 5, 6, 7, 8, 9 consecutive coupling cycles. In some embodiments of the present invention, the process does not comprise any solid-liquid separation of the n-th cycle oligonucleotide O-n or of any oligonucleotidic educts or intermediates involved in the synthesis of the n-th cycle oligonucleotide O-n during or in between at least at least 3, 4, 5, 6, 7, 8, 9 consecutive coupling cycles.
- steps (b-1) to (h-1) means that - none of the steps (b-1), (c-1), (d-1), (e-1), (f-1), (g-1), and (h-1) comprises a solid-liquid separation (each of steps (d-1), (f-1), (g-1), and (h-1) may or may not be carried out as explained above), - if carried out, step (i-1) does not comprise a solid-liquid separation, and - no solid-liquid separation is performed in between any two of these steps.
- steps (b-1) to (h-2) means that - none of the steps (b-1), (c-1), (d-1), (e-1), (f-1), (g-1), (h-1), (b-2), (c-2), (d-2), (e-2), (f-2), (g-2), and (h-2) (as far as present, each of steps (d-1), (f-1), (g-1), (h-1), (d-2), (f-2), (g-2), (h-2) may or may not be carried out as explained above), comprises a solid-liquid separation, - if carried out, step (i-1) does not comprise a solid-liquid separation, - if carried out, step (i-2) does not comprise a solid-liquid separation, and - no solid-liquid separation is performed in between any two of these steps.
- steps (b-1) to (h-n) means that - none of the steps (b-1), (c-1), (d-1), (e-1), (f-1), (g-1), (h-1), (b-2), (c-2), (d-2), (e-2), (f-2), (g-2), and (h-2)(each of steps (d-1), (f-1), (g-1), (h-1), (d-2), (f-2), (g- 2), (h-2) may or may not be carried out as explained above) comprises a solid-liquid separation, - no iteration of step (b-x), no iteration of step (c-x), no iteration of step (d-x), no iteration of step (e-x), no iteration of step (f-x), no iteration of step (g-x), and no iteration of step (h-x) (each of steps (d-x), (f-x) (g-x) and (h-x) may or may not be carried out
- ether solvent may be understood in the broadest sense to refer to any solvent characterized in that its chemical structure comprises at least one oxygen atom covalently bonded to exactly two residues independently selected from the group consisting of an alkyl group, an alkenyl group, an alkynyl group, a heteroalkyl group, a heteroalkenyl group, a heteroalkynyl group, an aryl group, and a heteroaryl group, wherein each of these two residues engages in a covalent chemical bond to said oxygen atom via a carbon atom (C), and wherein these two residues may optionally bond to each other to form a cyclic structure.
- C carbon atom
- an ether solvent S E may be any ether solvent. Examples and further embodiments pertaining to an ether solvent S E will be laid out in a later section of this text.
- steps (b-1) to (h-1) i.e. any one of steps (b-1), (c-1), (d-1), (e-1), (f-1), (g-1), and (h-1) as far as present) are carried out in essentially halogen-free solvents. It will be understood that this does not imply that any optional step must be carried out.
- steps (b-1) to (h-2) i.e. any one of steps (b-1), (c-1), (d-1), (e-1), (f-1), (g-1), (h-1), (b-2), (c-2), (d-2), (e-2), (f-2), (g-2), and (h-2) as far as present
- steps (b-1) to (h-2) i.e. any one of steps (b-1), (c-1), (d-1), (e-1), (f-1), (g-1), (h-1), (b-2), (c-2), (d-2), (e-2), (f-2), (g-2), and (h-2) as far as present
- steps (b-1) to (h-2) i.e. any one of steps (b-1), (c-1), (d-1), (e-1), (f-1), (g-1), (h-1), (b-2), (c-2), (d-2), (e-2), (f-2), (g-2), and (h-2) as far as present
- steps (b-1) to (h-1) (as far as present) are carried out in essentially halogen-free solvents, and further wherein: - if a second coupling cycle comprising steps (b-2) to (h-2) (as far as present) is performed, steps (b-1) to (h-2) (as far as present) are carried out in essentially halogen-free solvents; and - if (n ⁇ 2) iterations of the coupling cycle comprising steps (b-x) to (h-x) (as far as present) are performed, any steps (b-1) to (h-n) (as far as present) are carried out in essentially halogen-free solvents.
- essentially halogen-free solvent refers to a solvent which contains in total equal to or less than 3.0 vol-%, 2.0 vol-%, 1.0 vol-%, 0.1 vol-%, 0.01 vol-%, or 0.001 vol-% of halogenated solvents.
- halogenated solvent refers to any solvent comprising in its chemical structure at least one halogen atom. Examples of halogenated solvents comprise dichloromethane, chloroform, and 1,1-dichloroethane, and 1,2-dichloroethane. To state that a step “is carried out in essentially halogen-free solvents” means that only essentially halogen-free solvents are used in the respective step.
- - said component C-0 comprises exactly one pseudo solid-phase protecting group, which is the pseudo solid-phase protecting group PG-s; and - the building block B-1 does not comprise any pseudo solid-phase protecting groups.
- - said component C-0 comprises exactly one pseudo solid-phase protecting group, which is the pseudo solid-phase protecting group PG-s; and - none of the building block B-1 and B-2 comprise any pseudo solid-phase protecting groups.
- - said component C-0 comprises exactly one pseudo solid-phase protecting group, which is the pseudo solid-phase protecting group PG-s; and - none of the building block B-1, B-2, and B-x comprise any pseudo solid-phase protecting groups.
- the sum of nucleoside subunits comprised in the component C-0 and all building blocks B-1, B-2, and B-x together is in the range of 5 ⁇ 100, 5 ⁇ 90, 5 ⁇ 80, 5 ⁇ 70, 5 ⁇ 60, 5 ⁇ 50, 5 ⁇ 40, 5 ⁇ 30, 5 ⁇ 20, 6 ⁇ 20 or 6 ⁇ 18.
- the amount of nucleoside subunits is determined as 1 + m, where m is the integer m. It will be understood that in each building block B-1, B-2, and B-x of any one of Formulae II, II-a, II-b, II-1, II-1-a, and II-1-b, the amount of nucleoside subunits is determined as 1 + q, where q is the integer q.
- - said component C-0 comprises exactly one pseudo solid-phase protecting group, which is the pseudo solid-phase protecting group PG-s; - none of the building blocks B-1, B-2, and B-x comprises any pseudo solid-phase protecting groups; and - the sum of nucleoside subunits comprised in the component C-0 and all building blocks B-1, B-2, and B-x together is in the range of 5 ⁇ 100, 5 ⁇ 90, 5 ⁇ 80, 5 ⁇ 70, 5 ⁇ 60, 5 ⁇ 50, 5 ⁇ 40, 5 ⁇ 30, 5 ⁇ 20, 6 ⁇ 20 or 6 ⁇ 18.
- - the first cycle oligonucleotide O-1 comprises exactly one pseudo solid-phase protecting group, which is the pseudo solid-phase protecting group PG-s, and - the first cycle oligonucleotide comprises in total 5 ⁇ 100, 5 ⁇ 90, 5 ⁇ 80, 5 ⁇ 70, 5 ⁇ 60, 5 ⁇ 50, 5 ⁇ 40, 5 ⁇ 30, 5 ⁇ 20, 6 ⁇ 20 or 6 ⁇ 18 nucleoside subunits.
- - the second cycle oligonucleotide O-2 comprises exactly one pseudo solid-phase protecting group, which is the pseudo solid-phase protecting group PG-s, and - the second cycle oligonucleotide O-2 comprises in total 5 ⁇ 100, 5 ⁇ 90, 5 ⁇ 80, 5 ⁇ 70, 5 ⁇ 60, 5 ⁇ 50, 5 ⁇ 40, 5 ⁇ 30, 5 ⁇ 20, 6 ⁇ 20 or 6 ⁇ 18 nucleoside subunits.
- - the n-th cycle oligonucleotide O-n comprises exactly one pseudo solid-phase protecting group, which is the pseudo solid-phase protecting group PG-s, and - the n-th cycle oligonucleotide O-n comprises in total 5 ⁇ 100, 5 ⁇ 90, 5 ⁇ 80, 5 ⁇ 70, 5 ⁇ 60, 5 ⁇ 50, 5 ⁇ 40, 5 ⁇ 30, 5 ⁇ 20, 6 ⁇ 20 or 6 ⁇ 18 nucleoside subunits.
- an amide solvent S A is an amide solvent comprising one or more alkyl groups, wherein these one or more alkyl groups together comprise in total 6 ⁇ 48, 8 ⁇ 48, 6 ⁇ 47, 8 ⁇ 47, 6 ⁇ 46, 8 ⁇ 46, 6 ⁇ 45, 8 ⁇ 45, 6 ⁇ 44, 8 ⁇ 44, 6 ⁇ 43, 8 ⁇ 43, 6 ⁇ 42, 8 ⁇ 42, 6 ⁇ 41, 8 ⁇ 41, 6 ⁇ 40, 8 ⁇ 40, 6 ⁇ 39, 8 ⁇ 39, 6 ⁇ 38, 8 ⁇ 38, 6 ⁇ 37, 8 ⁇ 37, 6 ⁇ 36, 8 ⁇ 36, 6 ⁇ 35, 8 ⁇ 35, 6 ⁇ 34, 8 ⁇ 34, 6 ⁇ 33, 8 ⁇ 33, 6 ⁇ 32, 8 ⁇ 32, 6 ⁇ 31, 8 ⁇ 31, 6 ⁇ 30, 8 ⁇ 30, 6 ⁇ 29, 8 ⁇ 29, 6 ⁇ 28, 8 ⁇ 28, 6 ⁇ 27, 8 ⁇ 27, 6 ⁇ 26, 8 ⁇ 26, 6 ⁇ 25, 8 ⁇ 25, 6 ⁇ 24, 8 ⁇ 24, 6–23, 8–23, 6–22, 8–22
- an amide solvent S A is an amide solvent comprising one or more alkyl groups, wherein these one or more alkyl groups together comprise in total 6 ⁇ 24 carbon atoms.
- each amide solvent S A is at each occurrence selected independently from the group consisting of the following Formulae S A -1, S A -2, and S A -3: wherein in Formula S A -1: R A-1 is selected from the group consisting of H and a C1 ⁇ C24-alkyl group, in which exactly one hydrogen residue may optionally be substituted by a C(O)O(C1 ⁇ C5-alkyl) group; and each of R A-2 and R A-3 is independently a C1 ⁇ C24-alkyl group; with the proviso that R A-1 , R A-2 and R A-3 together comprise in total 6 ⁇ 48 carbon atoms; wherein in Formula S A -2: o is an integer of 1 or 2; and R A-4 is a C6–C24-alkyl
- each amide solvent S A is at each occurrence selected independently from the group consisting of the following Formulae S A -1, S A -2, and S A -3: wherein in Formula S A -1: R A-1 is selected from the group consisting of H and a C 1 ⁇ C 22 -alkyl group, in which exactly one hydrogen residue may optionally be substituted by a C(O)O(C 1 ⁇ C 5 -alkyl) group; and each of R A-2 and R A-3 is independently a C 1 ⁇ C 23 -alkyl group; with the proviso that R A-1 , R A-2 and R A-3 together comprise in total 6 ⁇ 24 carbon atoms; wherein in Formula S A -2: o is an integer of 1 or 2; and R A-4 is a C6–C24-alkyl group; and wherein in Formula S A -3: p is an integer of 1 or 2; X A is selected from the group consisting of CH2, O, and NC(O)
- brackets are occasionally used for illustrative purposes, as, e.g., exemplified in the following residue: C(O)O(C 1 C 5 -alkyl). Such brackets are not to be construed to indicate that the group in brackets is only optionally comprised in the respective residue. If an oxygen atom of a chemical residue is depicted in brackets, this indicates that said oxygen atom is covalently bonded only to the carbon atom depicted to the left in the chemical formula of the respective residue.
- a hydrocarbon group such as, e.g., a C1–C5-alkyl group forms part of a residue comprising further atoms
- said hydrocarbon group may be presented in brackets to highlight the fact that it is a separate group within said residue.
- a C(O)O(C1–C5-alkyl) residue is an ester residue, bonded via the carboxyl carbon atom to the respective parent structure, wherein the C 1 –C 5 -alkyl moiety is bonded to the oxygen atom not depicted in brackets.
- a O(C1–C40-alkyl) residue denotes an alkoxy residue in which the alkyl moiety comprises at least one and not more than 40 carbon atoms.
- a C(O)(C1–C40-alkyl) residue denotes an alkanoyl residue bonded to the respective parent structure via a carbonyl carbon atom, wherein the C1–C40-alkyl moiety is bonded directly to the carbonyl carbon atom and not to the oxygen atom depicted in brackets.
- the skilled artisan is capable of identifying and obtaining amide solvents S A fulfilling the aforementioned structural requirements and such amide solvents may also be commercially available.
- Examples of amide solvents S A of Formula S A -1 comprise N,N-dibutylformamide (DBF), N,N-dibutylacetamide, methyl 5-(dimethylamino)-2- methyl-5-oxopentanoate (CAS-RN 1174627-68-9), N,N-dimethyloctanamide, N,N-dimethyldecanamide, N,N-diethyldodecanamide, and N,N-bis(1- methylpropyl)acetamide.
- DPF N,N-dibutylformamide
- N,N-dibutylacetamide methyl 5-(dimethylamino)-2- methyl-5-oxopentanoate
- N,N-dimethyloctanamide N,N-dimethyldecanamide
- N,N-diethyldodecanamide N,N-bis(1- methylpropyl)acetamide.
- Examples of amide solvents S A of Formula S A -2 comprise N-octyl-2-pyrrolidone (NOP), N-octyl-2-piperidone, and 1-cyclohexyl-2-pyrrolidone (CAS RN: 6837-24-7).
- Examples of amide solvents S A of Formula S A -3 comprise N-nonanoylpyrrolidine (CAS-RN 20308-70-7), N-nonanoylpiperidine (CAS-RN 20368-13-2), N-nonanoylmorpholine (CAS-RN 5299-64-9), and 1,4-dipentanoylpiperazine (CAS RN 18903-08-7).
- each amide solvent S A independently of each other is selected from the group consisting of N,N-dibutylformamide (DBF) and N-octyl-2- pyrrolidone (NOP).
- each amide solvent S A is independently of each other selected from the group consisting of, N-octyl-2-pyrrolidone (NOP), N,N-dibutylformamide (DBF), N,N-diethyldodecanamide, and 1-cyclohexyl-2-pyrrolidone.
- each ether solvent S E is at each occurrence selected independently from the group consisting of the following Formulae S E -1 and S E -2: (Formula S E -2), wherein in Formula S E -1: s is an integer of 0 or 1, and each of R E-1 , R E-2 , R E-3 , R E-4 , R E-5 , R E-6 , R E-7 , R E-8 , R E-9 , and R E-10 is independently selected from the group consisting of H and a C1 ⁇ C5-alkyl group, with the proviso that at least one of R E-1 , R E-2 , R E-3 , R E-4 , R E-5 , R E-6 , R E-7 , R E-8 , R E-9 , and R E-10 is a C1 ⁇ C5-alkyl group; and wherein in Formula S E -2: each of R E-11 and R E-12 is independently a C1 ⁇ C6-alkyl group,
- each ether solvent S E is independently of each other a solvent of any one of the aforementioned Formulae S E -1 and S E -2, wherein in Formula S E -1: s is an integer of 0 or 1, and each of R E-1 , R E-2 , R E-3 , R E-4 , R E-5 , R E-6 , R E-7 , R E-8 , R E-9 , and R E-10 is independently selected from the group consisting of H and a C 1 ⁇ C 5 -alkyl group, with the proviso that exactly one, exactly two or exactly three of R E-1 , R E-2 , R E-3 , R E-4 , R E-5 , R E-6 , R E-7 , R E-8 , R E-9 , and R E-10 are a C1 ⁇ C5-alkyl group; and wherein in Formula S E -2: each of R E-11 and R E-12 is independently
- each ether solvent S E is independently of each other a solvent of any one of the aforementioned Formulae S E -1 and S E -2, wherein in Formula S E -1: s is an integer of 0 or 1, and each of R E-1 , R E-2 , R E-3 , R E-4 , R E-5 , R E-6 , R E-7 , R E-8 , R E-9 , and R E-10 is independently selected from the group consisting of H and CH 3 (i.e.
- each of R E-11 and R E-12 is independently a C1 ⁇ C6-alkyl group, with the proviso that R E-11 and R E-12 together comprise in total 6 ⁇ 12 carbon atoms.
- each ether solvent S E is independently of each other a solvent of any one of the aforementioned Formulae S E -1 and S E -2, wherein in Formula S E -1: s is an integer of 0 or 1, and each of R E-1 , R E-2 , R E-3 , R E-4 , R E-5 , R E-6 , R E-7 , R E-8 , R E-9 , and R E-10 is independently selected from the group consisting of H and CH3 (i.e.
- each of R E-11 and R E-12 is independently a C 1 ⁇ C 6 -alkyl group, with the proviso that R E-11 and R E-12 together comprise in total 6 ⁇ 12 carbon atoms.
- each ether solvent S E is independently of each other a solvent of the aforementioned Formula S E -1, wherein in Formula S E -1: s is an integer of 0 or 1, and each of R E-1 , R E-2 , R E-3 , R E-4 , R E-5 , R E-6 , R E-7 , R E-8 , R E-9 , and R E-10 is independently selected from the group consisting of H and CH3 (i.e.
- ether solvents S E of Formula S E-1 comprise 4-methyltetrahydropyran (MTHP), 3-methyltetrahydropyran, 2-methyltetrahydropyran, 2-methyltetrahydrofuran, 3-methyltetrahydrofuran, 2,5-dimethyltetrahydrofuran, 2,2-dimethyltetrahydrofuran, 2,2,5,5-tetramethyltetrahydrofuran, and the like.
- MTHP 4-methyltetrahydropyran
- 3-methyltetrahydropyran 2-methyltetrahydropyran
- 2-methyltetrahydropyran 2-methyltetrahydrofuran
- 3-methyltetrahydrofuran 2,5-dimethyltetrahydrofuran
- 2,2-dimethyltetrahydrofuran 2,2,5,5-tetramethyltetrahydrofuran
- ether solvents S E of Formula S E -2 comprise cyclopentyl methyl ether (CPME), cyclohexyl methyl ether, dibutyl ether, isoamyl ether, methyl tert-butyl ether (MTBE), diisopropyl ether, and the like.
- each ether solvent S E is independently selected from the group consisting of 4-methyltetrahydropyran (MTHP) and cyclopentyl methyl ether (CPME).
- each ether solvent S E is 4-methyltetrahydropyran (MTHP).
- each pseudo solid-phase protecting group e.g. the pseudo solid-phase protecting group PG-s
- a pseudo solid-phase protecting group of Formula P-1 may be bonded to any position of a nucleoside or oligonucleotide, with the proviso that said point of attachment indicated by the asterisk in Formula P-1 (i.e. the atom of the nucleoside or oligonucleotide to which said pseudo solid-phase protecting group is covalently bonded) must be the oxygen atom of a hydroxyl moiety or the nitrogen atom of an amine moiety.
- hydroxyl moieties to which a pseudo solid-phase protecting group of Formula P-1 may be covalently bonded are hydroxyl moieties of the backbone of a nucleoside or oligonucleotide, e.g. any hydroxyl moieties available in the carbohydrate (preferably ribose or 2 ⁇ -deoxyribose) moiety of a nucleoside moiety as well as any hydroxyl moieties present in an internucleosidic linkage group.
- amine moieties to which a pseudo solid-phase protecting group of Formula P-1 may be covalently bonded are amine moieties of nucleobases, in particular the exocyclic amine moieties of adenine, guanine, cytosine, and 5-methylcytosine.
- the component C-0 of any one of Formulae I, I-a, and I-b comprises a pseudo solid-phase protecting group PG-s which is bonded to a hydroxyl moiety.
- the asterisk of Formula P-1 indicates the oxygen atom in the respective Formula I, I-a, and I-b, to which the protecting group PG-s is bonded and the covalent bond between the protecting group PG-s and said oxygen atom in any one of Formulae I, I-a, and I-b is the covalent bond interconnecting the carbonyl carbon atom and the asterisk in Formula P-1 (in other words: only one and not two covalent chemical bonds interconnect the carbonyl carbon atom shown in Formula P-1 and the respective oxygen atom of the respective Formula I, I-a, and I-b).
- the integer a in Formula P-1 is 0. In some embodiments of the invention, the integer a in Formula P-1 is 1. In some embodiments of the invention, the integer b in Formula P-1 is 1. In some embodiments of the invention, the integer a in Formula P-1 is 0. In some embodiments of the invention, the integer b in Formula P-1 is 0. In some embodiments of the invention, the integer a in Formula P-1 is 0 and the integer b in Formula P-1 is 1. In some embodiments of the invention, the integer a in Formula P-1 is 1 and the integer b in Formula P-1 is 1.
- the integer a in Formula P-1 is 1 and the integer b in Formula P-1 is 0. In some embodiments of the invention, the integer a in Formula P-1 is 0 and the integer b in Formula P-1 is 0.
- the linker moiety L P in a pseudo solid-phase protecting group of Formula P-1 may be any chemical residue which interconnects the oxygen atom, if the integer a is 1, or the carbonyl carbon atom, if the integer a is 0, with the phenyl residue to which R P-1 is bonded.
- said linker moiety may be a C 1 ⁇ C 40 -alkylene moiety, in which alkylene moiety one or more carbon atoms and/or one or more hydrogen residues may optionally be substituted for a heteroatom selected from the group consisting of O and N.
- alkylene group or moiety or residue
- alkylene group may be understood in the broadest sense to be similar to an alkyl residue, with the sole difference, that an alkylene residue mandatorily comprises two or more, preferably exactly two, chemical bonds (for the avoidance of doubt: a double bond is not the same as two chemical bonds) to the parent structure comprising said alkylene group.
- An alkylene group may preferably be an alkanediyl residue.
- a methyl group i.e. –CH 3
- the corresponding alkylene group would be a methylene group (i.e. –CH 2 ⁇ ) and differs from the alkyl group in that is bonds to the parent structure via exactly two covalent chemical bonds (indicated by the hyphens; both bonds originate from the methylene carbon atom).
- an alkylene group may be branched, unbranched (i.e. linear) or cyclic.
- each pseudo solid-phase protecting group e.g. the pseudo solid-phase protecting group PG-s
- (Formula P-1-b) wherein in Formula P-1-b: the integers a and i, the asterisk, and R P-1 are defined as for Formula P-1
- L 1 is a C 1 ⁇ C 12 -alkylene group, preferably a C 1 ⁇ C 4 -alkylene group, in particular a C1 ⁇ C2-alkylene group
- X P-2 is selected from the group consisting of ++C(O)
- X P-1 is selected from the group consisting of +C(O)N(R P-B )++, +C(O)O++, and +C(O)S++, where R P-B is selected from the group consisting of H and a C 1 ⁇ C 6 -alkyl group, and where + is the point of attachment to L 1 or O or the carbonyl carbon atom and ++ is the point of attachment to L 2 ;
- L 2 is a C 1 ⁇ C 12 -alkylene group, preferably a C 1 ⁇ C 4 -alkylene group;
- X P-2 is selected from the group consisting of ++C(O)N(R P-C )+, ++C(O)O+, and ++C(O)S+, where R P-C is selected from the group consisting of H and a C1 ⁇ C6-alkyl group, and where + is the point of attachment to L 2 and ++ is the point of attachment to the phenyl moiety
- R P-1 is selected from the group consisting of +C(O)N(R P-B )++, +C(O)O++, and +C(O)S++, where R P-B is selected from the group consisting of H and a C1 ⁇ C6-alkyl group, and where + is the point of attachment to L 1 and ++ is represents the carbon atom to which R P-D is bonded in CHR P-D ; and R P-D is selected from the group consisting of H and a residue of the following Formula P-1-L**: (Formula P-1-L**), wherein in Formula P-1-L**: the hashtag (#) represents the carbon atom to which R P-D is bonded in CHR P-D , k is an integer of 0 to 4; R P-3 is defined as R P-1 (i.e.
- L 1 is a C 1 ⁇ C 12 -alkylene group, preferably a C 1 ⁇ C 4 -alkylene group, in particular a C 2 -alkylene group (i.e.
- X P-1 is selected from the group consisting of +C(O)++, +C(O)N(R P-B )++, +C(O)O++, and +C(O)S++, where R P-B is selected from the group consisting of H and a C 1 ⁇ C 6 -alkyl group, and where + is the point of attachment to L 1 and ++ is the point of attachment to L 2 ;
- L 2 is a C 1 ⁇ C 12 -alkylene group, preferably a C 1 ⁇ C 4 -alkylene group, in particular a C 1 ⁇ C 4 -alkylene group; and
- X P-3 is selected from the group consisting of ++C(O)N(R P-E )+, and ++C(O)O+ where + denotes the point of attachment to L 2 and ++ is the point of attachment to the phenyl moiety substituted with R P-1 , and where R P-E is selected from the group
- each pseudo solid-phase protecting group e.g. the pseudo solid-phase protecting group PG-s
- PG-s is a protecting group of any one of the aforementioned Formulae P-1-a, P-1-b, P-1-c, P-1-d, and P-1-e, wherein in Formula P-1-a: the asterisk and R P-1 are defined as for Formula P-1; a is an integer of 0 or 1; i is an integer of 1 to 3, preferably 2 to 3; and L 1 is a C1 ⁇ C3-alkylene group (i.e.
- Formula P-1-b the asterisk and R P-1 are defined as for Formula P-1; the integer a is 0 (i.e. the oxygen atom is absent and L 1 bonds directly to the carbonyl carbon atom); i is an integer of 1 to 3, preferably 2 to 3; L 1 is a C1 ⁇ C4-alkylene group, in particular a C1 ⁇ C2-alkylene group; and X P-2 is selected from the group consisting of ++C(O)N(R P-C )+ and ++C(O)O+, where R P-C is selected from the group consisting of H and a C1 ⁇ C6-alkyl group, and where + is the point of attachment to L 1 and ++ is the point of attachment to the phenyl moiety substituted with R P-1 ; wherein in Formula P-1-c: the asterisk and R P-1 are defined as for Formula P-1; the integer a is 0 (i.e. the oxygen atom is absent and L 1 bonds directly to the carbonyl carbon
- X P-1 is selected from the group consisting of +C(O)N(R P-B )++ and +C(O)O++, where R P-B is selected from the group consisting of H and a C1 ⁇ C6-alkyl group, and where + is the point of attachment to L 1 or the carbonyl carbon atom and ++ is the point of attachment to L 2 ;
- L 2 is a C1 ⁇ C4-alkylene group;
- X P-2 is selected from the group consisting of ++C(O)N(R P-C )+ and ++C(O)O+, where R P-C is selected from the group consisting of H and a C1 ⁇ C6-alkyl group, and where + is the point of attachment to L 2 and ++ is the point of attachment to the phenyl moiety substituted with R P-1 ; and may optionally together represent the following structure which the dashed line represents the covalent bond to L 1 or the carbonyl carbon atom
- R P-3 of Formula P-1-L** and R P-1 of Formula P-1); and R P-2 is H; and wherein in Formula P-1-e: the integer a is 0; i is an integer of 1 to 3, preferably 2 to 3; L 1 is a C 1 ⁇ C 4 -alkylene group, in particular a C 2 -alkylene group; X P-1 is selected from the group consisting of +C(O)++, +C(O)N(R P-B )++, and +C(O)O++, where R P-B is selected from the group consisting of H and a C1 ⁇ C6-alkyl group, and where + is the point of attachment to L 1 and ++ is the point of attachment to L 2 ; L 2 is a C1 ⁇ C4-alkylene group, in particular a C1 ⁇ C2-alkylene group; and X P-3 is selected from the group consisting of ++C(O)N(R P-E )+, and ++C(
- each pseudo solid-phase protecting group e.g. the pseudo solid-phase protecting group PG-s
- PG-s is a protecting group of any one of the aforementioned Formulae P-1-a, P-1- b, P-1-c, P-1-d, and P-1-e
- Formula P-1-a the asterisk and R P-1 are defined as for Formula P-1
- a is an integer of 0 or 1
- i is an integer of 1 to 3, preferably 2 to 3
- L 1 is C1 ⁇ C3-alkylene group
- the integer a is 0
- i is an integer of 1 to 3, preferably 2 to 3
- L 1 is a C 1 ⁇ C 4 -alkylene group, in particular a C 1 ⁇ C 2 -alkylene group
- X P-2 is selected from the group consisting of ++C(O)N(R P-C
- X P-2 together represent the following structure which the dashed line represents the covalent bond to L 1 and the wavy line represents the covalent bond to the phenyl moiety substituted with R P-1 ; and wherein in Formula P-1-d: the asterisk and R P-1 are defined as for Formula P-1; the integer a is 0; i is an integer of 1 to 3, preferably 2 to 3; L 1 is a C1 ⁇ C4-alkylene group, in particular a C2-alkylene group; X P-1 is selected from the group consisting of +C(O)N(R P-B )++ and +C(O)O++, where R P-B is selected from the group consisting of H and a CH3, and where + is the point of attachment to L 1 and ++ represents the carbon atom to which R P-D is bonded in CHR P-D ; and R P-D is selected from the group consisting of H and a residue of the aforementioned Formula P-1-L**
- R P-3 of Formula P-1-L** and R P-1 of Formula P-1); and R P-2 is H; and wherein in Formula P-1-e: the integer a is 0; i is an integer of 1 to 3, preferably 2 to 3; L 1 is a C1 ⁇ C4-alkylene group, in particular a C2-alkylene group; X P-1 is selected from the group consisting of +C(O)++, +C(O)N(R P-B )++, and +C(O)O++, where R P-B is selected from the group consisting of H and CH3, and where + is the point of attachment to L 1 and ++ is the point of attachment to L 2 ; L 2 is a C1 ⁇ C4-alkylene group, in particular a C1 ⁇ C2-alkylene group; and X P-3 is selected from the group consisting of ++C(O)N(R P-E )+, and ++C(O)O+, where R P-E is selected from the group
- R P-1 in any one of Formulae P-1, P-1-a, P-1-b, P-1-c, P-1-d, and P-1-e as well as R P-3 in any one of Formulae P-1 and P-1-d is at each occurrence independently selected from the group consisting of O(C 1 ⁇ C 40 -alkyl), O(C 2 ⁇ C 40 -alkenyl), a C 1 ⁇ C 40 -alkyl group, a C 2 ⁇ C 40 -alkenyl group, C(O)(C 1 ⁇ C 40 -alkyl), and C(O)(C 2 ⁇ C 40 -alkenyl).
- R P-1 in any one of Formulae P-1, P-1-a, P-1-b, P-1-c, P-1-d, and P-1-e as well as R P-3 in any one of Formulae P-1 and P-1-d is at each occurrence independently selected from the group consisting of O(C 1 ⁇ C 30 -alkyl), O(C 2 ⁇ C 30 - alkenyl), a C 1 ⁇ C 30 -alkyl group, a C 2 ⁇ C 30 -alkenyl group, C(O)(C 1 ⁇ C 30 -alkyl), and C(O)(C2 ⁇ C30-alkenyl).
- R P-1 in any one of Formulae P-1, P-1-a, P-1-b, P-1-c, P-1-d, and P-1-e as well as R P-3 in any one of Formulae P-1 and P-1-d is at each occurrence independently selected from the group consisting of O(C1 ⁇ C30-alkyl), O(C2 ⁇ C30-alkenyl), C(O)(C1 ⁇ C30-alkyl), and C(O)(C2 ⁇ C30-alkenyl).
- R P-1 in any one of Formulae P-1, P-1-a, P-1-b, P-1-c, P-1-d, and P-1-e as well as R P-3 in any one of Formulae P-1 and P-1-d is at each occurrence independently selected from the group consisting of O(C1 ⁇ C30-alkyl) and C(O)(C1 ⁇ C30-alkyl).
- R P-1 in any one of Formulae P-1, P-1-a, P-1-b, P-1-c, P-1-d, and P- 1-e as well as R P-3 in any one of Formulae P-1 and P-1-d is at each occurrence independently O(C1 ⁇ C30-alkyl).
- R P-1 in any one of Formulae P-1, P-1-a, P-1-b, P-1-c, P-1-d, and P-1-e as well as R P-3 in any one of Formulae P-1 and P-1-d is at each occurrence independently O(C 1 ⁇ C 30 - alkyl), wherein said C 1 ⁇ C 30 -alkyl in O(C 1 ⁇ C 30 -alkyl) is a linear (i.e. unbranched and non-cyclic) alkyl residue.
- Pseudo solid-phase protecting groups of the aforementioned Formula P-1-a methods for their preparation, introduction into nucleosides or oligonucleotides as well as cleavage from oligonucleotides are, e.g., disclosed in PCT/EP2022/059528 published as WO 2022/214692 A1, wherein reference is in particular made to Formulae II, II-a, and III to VII of said reference as well as to Examples 1 to 40 of said reference.
- Pseudo solid-phase protecting groups of the aforementioned Formula P-1-b are, e.g., disclosed in EP3825300A1.
- Pseudo solid-phase protecting groups of the aforementioned Formula P-1-c are, e.g., disclosed in S. Kim et al., Chemistry – A European Journal 2013, 19, 8615 ⁇ 8620 (DOI: 10.1002/chem.201300655), wherein reference is also made to the Supporting Information belonging to said reference and comprising the experimental protocols.
- Pseudo solid-phase protecting groups of the aforementioned Formula P-1-d are, e.g., disclosed in US2013267697A1, EP2711370A1, and EP3398955A1.
- Examples of pseudo solid-phase protecting groups of Formula P-1-d comprise protecting groups of the following structures: , wherein in any one of these structures, the asterisk, R P-1 , and R P-3 are defined as for Formula P-1.
- R P-1 and R P-3 may, e.g., be at each occurrence a C22H45-alkyl group, a C21H43-alkyl group, a C20H41-alkyl group, a C19H39-alkyl group or a C18H37-alkyl group.
- each pseudo solid-phase protecting group e.g. the pseudo solid-phase protecting group PG-s, is a protecting group of the aforementioned Formula P-1-a.
- the method of the invention may comprise a first coupling cycle comprising steps (b-1) to (h-1) (as far as present), wherein steps (d-1), (f-1), (g-1), and (h-1) are optional, which is indicated in Figure 1 by embracing these steps in brackets.
- a component C-0 Prior to the first coupling cycle, a component C-0 is provided [step (a-1)].
- the protecting group PG-0 is removed from said component C-0 [step (b-1)] which yields the component (C-0) # .
- step (c-1) This is followed by one or more aqueous extractions [step (c-1)], optionally followed by reducing the water content of the organic phase which comprises the component (C-0) # [step (d-1)].
- the component (C-0) # is reacted with a provided first building block B-1 [step (e-1)] which yields a first cycle oligonucleotide O-1.
- any P (III) atoms within O-1 may be converted to P (V) atoms by incubation with an oxidizing or sulfurizing agent [step (f-1)], optionally followed by one or more aqueous extractions [step (g-1)], optionally followed by reducing the water content of the organic phase which comprises the first cycle oligonucleotide O-1 [step (h-1)].
- the method of the invention may further comprise a second coupling cycle comprising steps (b-2) to (h-2), wherein steps (d-2), (f-2), (g-2), and (h-2) are optional, which is indicated in Figure 1 by embracing these steps in brackets.
- the first cycle oligonucleotide O-1 may serve as the educt of the second coupling cycle.
- the protecting group PG-1 is removed from the first cycle O-1 [step (b-2)] which yields the first cycle oligonucleotide (O-1) # .
- This is followed by one or more aqueous extractions [step (c-2)], optionally followed by reducing the water content of the organic phase which comprises the first cycle oligonucleotide (O-1) # [step (d-1)].
- the first cycle oligonucleotide (O-1) # is reacted with a provided second building block B-2 [step (e-2)] which yields a second cycle oligonucleotide O-2.
- any P (III) atoms within O-2 may be converted to P (V) atoms by incubation with an oxidizing or sulfurizing agent [step (f-2)], optionally followed by one or more aqueous extractions [step (g-2)], optionally followed by reducing the water content of the organic phase which comprises the second cycle oligonucleotide O-2 [step (h-2)].
- the method of the invention may further comprise (n ⁇ 2) iterations of a coupling cycle comprising steps (b-x) to (h-x) (as far as present), which are referred to as “further coupling cycles” in Figure 1.
- Steps (d-x), (f-x), (g-x), and (h-x) are optional in each of these further coupling cycles, which is indicated in Figure 1 by embracing these steps in brackets.
- the oligonucleotide (O-(x ⁇ 1)) # i.e. (O-2) #
- step (n ⁇ 2) coupling cycles comprising steps (b-x) to (h-x) (as far as present) [“further coupling cycles”] may be performed, in each of which the oligonucleotide O-x of the previous coupling cycle is used as educt O-(x ⁇ 1).
- Target oligonucleotides of the syntheses presented herein The following Examples pertain to the synthesis of target oligonucleotides, wherein these target oligonucleotides are summarized in the following Table T-2.
- Table T-2 Target oligonucleotides of the oligonucleotide syntheses presented herein.
- this pseudo solid-phase protecting group is, for example, depicted in compound (2) below.
- C cytidine
- dC 2 ⁇ -doexycytidine
- MeC or meC 5-methylcytidine (the nucleobase is 5-methylcytosine)
- T or dT thymidine (meaning 2 ⁇ -deoxythymidine and not ribothymidine)
- M indicates that the nucleoside subunit denoted to the right of said letter “M” comprises a 2 ⁇ -O-(2-methoxyethyl) residue (2 ⁇ -O-MOE, i.e. the 2 ⁇ -carbon atom of the carbohydrate moiety is substituted with O-CH2-CH2-O-CH3).
- protecting groups at the nucleobases is indicated in brackets to the right of the nucleoside symbol: “(bz)” and “(Bz)” interchangeably denote a benzoyl protecting group at the exocyclic amine moiety of the respective nucleobase; “(ac)” and “(Ac)” interchangeably denote an acetyl protecting group at the exocyclic amine moiety of the nucleobase; “(ib)” denotes an isobutyryl protecting group at the exocyclic amine moiety of the nucleobase. Unless indicated differently, all internucleosidic linkage groups are phosphodiester linkage groups.
- nucleoside subunits are separated by hyphens to enhance readability.
- all oligonucleotides are denoted in 5 ⁇ to 3 ⁇ direction (i.e. the nucleoside subunit denoted to the very left is the 5 ⁇ -terminal nucleoside subunit). Nonetheless, to enhance readability, the 5 ⁇ - and the 3 ⁇ -termnini are herein typically indicated as such.
- 5 ⁇ -MMeUs-MMeC-3 ⁇ herein denotes a dinucleotide, in which the 3 ⁇ -hydroxyl moiety of a 2 ⁇ -O-MOE substituted 5-methyluridine and the 5 ⁇ -hydroxyl moiety of a 2 ⁇ -O-MOE substituted 5-methylcytidine are interconnected by a phosphorothioate linkage group.
- 5 ⁇ -dA(bz)-dT-3 ⁇ denotes a dinucleotide, in which the 3 ⁇ -hydroxyl moiety of a benzyol-protected 2 ⁇ - deoxyadenosine and the 5 ⁇ -hydroxyl moiety of a (2 ⁇ -deoxy)thymidine are interconnected by a phosphodiester linkage group.
- Method B Solid phase: YMC-Pack Pro C18, 3 ⁇ m, 4.6 ⁇ 100 mm; Mobile phase A: AcOH aq.; Mobile phase B: THF; Flow rate: 1.0 mL/min; Gradient (mobile phase B %): 0-10.0 min; 65 to 90 %, 10.0-15.0 min; 90 %, 15.0-15.1 min; 90 to 65 %, 15.01-20.0 min; 65 %; Column temperature: 40 °C; Detection wavelength: 220 nm.
- Method D Solid phase: YMC-Triart C18, 12 nm, 1.9 ⁇ m, 2.1 ⁇ 150 mm; Mobile phase A: 0.1% AcOH aq.; Mobile phase B: THF; Flow rate: 0.3 mL/min; Gradient (mobile phase B %): 0.0-15.0 min; 65% to 75 %, 15.0-15.1 min; 75 to 90 %, 15.1-20.1 min; 90%, 20.0-20.1 min; 90 to 65%, 20.1-30.0 min; 65%; Detected wavelength: 260 nm.
- Method ESolid phase Waters ACQUITY BEH, 1.7 um, 2.1 ⁇ 100 mm; Mobile phase A: 50 mM di-n- butylammonium acetate aq.; Mobile phase B: Acetonitril:H2O (90:10 v/v) ; Flow rate: 0.2 mL/min; Gradient (mobile phase B %): 0-30 min; 25 to 45 %, 30-31 min; 45 to 90 %, 31-33 min; 90 %, 33-33.1 min; 90 to 25 %, 33.1-45 min; 25 %; Column temperature: 60 °C; Detection wavelength: 260 nm HPLC-MS methods Method 1 Solid phase: Waters ACQUITY peptide BEH, 1.7 um, 2.1 ⁇ 100 mm; Mobile phase A: 50 mM di-n- butylammonium acetate aq.; Mobile phase B: Acetonitril:H2O (90:10 v/v) ; Flow rate:
- N-octyl-2-pyrrolidone N-octyl-2-pyrrolidone (NOP) was obtained commercially. Prior to use, it was treated as follows: L-Ascorbic acid (1 wt-%) and 2,6-di-tert-butyl-p-cresol (1 wt-%) were added, followed by stirring at room temperature (i.e. approximately 25 °C) for 30 min.
- the titration cell was filled with HYDRANAL TM -Coulomat Oil for anolyte reagent, and the inner burette was filled with HYDRANAL TM -Coulomat CG for catholyte reagent.
- sample 100 ⁇ L was added to the titration cell and the measurement started.
- the water content of solutions is herein reported in parts per million (ppm).
- ppm parts per million
- the pH value was determined using a pH-electrode at rt.
- the pH electrode was calibrated by pH buffers (pH 4.01, 6.86, 9.18).
- a LAQUA D-72 water quality meter by HORIBA equipped with a 9680S-10D Long ToupH electrode by HORIBA was used for the pH-measurements in the Examples presented herein.
- Room temperature (rt) As used herein, unless indicated differently, the terms room temperature (rt) and ambient temperature are used interchangeably to denote a temperature of approximately 25 °C. Unless indicated differently, all reactions were performed at rt.
- Pseudo solid-phase protecting groups of Formula P-1-a may be synthesized and introduced into the starting nucleoside by methods disclosed in the prior art. In particular, the methods disclosed in WO 2022/214692 A1 and in PCT/EP2023/078683 may be used.
- the resulting reaction mixture was stirred at rt for 100 min (monitored by HPLC, Method A) and subsequently washed twice with saturated aqueous NH4Cl (150 mL and 75 mL) and then with saturated aqueous NaCl (75 mL).2-Methyltetrahydrofuran (100 mL) was added to the organic phase followed by concentration in vacuo to a volume of 100 mL.
- Acetic anhydride (7.5 mL, 75.67 mmol), pyridine (8.0 mL, 99.12 mmol) and 1-methylimidazole (7.9mL, 99.11 mmol) were added. After stirring at rt for 30 min, methanol (12 mL, 44.7 mmol) was added. After stirring 30 min at rt, the reaction mixture was concentrated in vacuo, followed by addition of acetonitrile (250 mL).
- the resulting reaction mixture was stirred at rt for 120 min and subsequently washed twice with saturated aqueous NH4Cl (150mL and 75 mL) and then with saturated aqueous NaCl (75 mL).2- methyltetrahydrofuran (200 mL) was added to the organic phase followed by concentration in vacuo to a volume of 100 mL.
- 3,5-bis(docosyloxy)benzenemethanol (CAS-RN: 955095-32-6, 5.00 g, 6.60 mmol, 1.0 eq.), DBU (1.48 mL, 9.92 mmol, 1.5 eq.) and 1-methylimidazole (789 ⁇ L, 9.90 mmol, 1.5 eq.) were added to the solution (water content: 108.9 ppm) of DMT- MG(ib)-1H-imidazole-1-carboxyl.
- Syntheses of oligonucleotides Example 1: Synthesis A of target oligonucleotide OT-1 (see Table T-2) Providing DMTr-MG(ib)s-MG(ib)-(Kc-C1-carbonate) (4) as an exemplary component C-0 Dichloroacetic acid (DCA, 30 mL) was added at rt to a stirred solution of DMTr- MG(ib)-(Kc-C1-carbonate) (2, 5.29 g, 3.53 mmol) obtained from Synthesis A laid out above, and thiomalic acid (2.99 g, 19.91 mmol, 5.6 eq.) in cyclopentyl methyl ether (CPME, 90 mL), and the mixture was stirred at rt until reaction monitoring by HPLC (Method D) indicated completion of the DMTr-deprotection.
- DCA Dichloroacetic acid
- N-methylmorpholine NMM, 38 mL
- acetone 15 mL
- water 60 mL
- the organic phase was separated and washed twice with water (45 mL ⁇ 2).
- the so-obtained organic phase was concentrated, followed by precipitation by addition of acetonitrile (300 mL).
- the resulting precipitate was filtrated, then washed twice with acetonitrile (120 mL ⁇ 2) to give MG(ib)-(Kc-C1-carbonate) (3, 3.86 g, 81.6 % yield for 2 steps from 3,5 bis(docosyloxy)benzenemethanol).
- DMTr-MG(ib) phosphoramidite (CAS-RN: 251647-55-9, 1.20 g, 1.31 mmol, 2.0 eq.) and MTHP (5 mL) were added to the so-obtained solution comprising MG(ib)-(Kc-C1-carbonate) (3), followed by concentration in vacuo (bath temperature 45 °C, pressure 40 hPa) to a volume of 7.5 mL.5-Ethylthio-1H-tetrazole (859 mg, 6.60 mmol, 10.0 eq.) and pyridine (1.6 mL, 19.82 mmol, 30.0 eq.) were added and the mixture was stirred at rt until reaction monitoring by HPLC (Method C) indicated completion of the coupling.
- Lactamide (CAS-RN: 2043-43-8, 119 mg, 1.34 mmol, 2.0 eq.) was added, followed by stirring for 30 min.
- 3-(N,N-dimethylaminomethylidene)amino)-3H-1,2,4-dithiazole-5-thione (CAS-RN: 1192027-04-5, DDTT, 407 mg, 1.98 mmol, 3.0 eq) was added.
- triethylphosphite (343 ⁇ L, 3.0 eq) was added.
- propan-2-ol (1.0 mL) was added, followed by stirring for 10 min at rt.
- the so-obtained solution comprising DMTr-MG(ib)s-MG(ib)-(Kc-C1-carbonate) (4) was used directly in the subsequent step (without any precipitation, filtration or purification steps in between).
- a mixture of glutathione (CAS-RN: 70-18-8,1.01 g, 3.29 mmol.5.0 eq.), acetonitrile (5 mL) and trifluoroacetic acid (TFA, 5 mL) was added to the so-obtained solution comprising DMTr-MG(ib)s-MG(ib)-(Kc-C1-carbonate) (4) at 0 °C
- the aqueous phase had a pH-value of 5.0.
- the aqueous phase had a pH-value of 5.1.
- MTHP 7.5 mL was added to the so-obtained organic phase comprising MG(ib)s- MG(ib)-(Kc-C1-carbonate) (5), followed by concentration in vacuo to a volume of 7.5 mL (bath temperature 35 ⁇ 45 °C, pressure 40-200 hPa).
- the MTHP (7.5 mL) addition and subsequent concentration was repeated four times (final volume: 7.5 mL; water content: 160.1 ppm).
- DMTr-MMeU phosphoramidite (CAS-RN: 163878- 63-5, 1.08 g, 1.32 mmol, 2.0 eq.) and MTHP (5 mL) were added to the so-obtained solution comprising MG(ib)s-MG(ib)-(Kc-C1-carbonate) (5), followed by concentration in vacuo (bath temperature 45 °C, pressure 40 hPa) to a volume of 7.5 mL.5-Ethylthio-1H-tetrazole (861 mg, 6.61 mmol, 10.0 eq.) and pyridine (1.6 mL, 19.82 mmol, 30.0 eq.) were added and the mixture was stirred at rt until reaction monitoring by HPLC (Method C) indicated completion of the coupling.
- Lactamide (118 mg, 1.33 mmol, 2.0 eq.) was added, followed by stirring for 30 min at rt.
- DDTT (407 mg, 1.98 mmol, 3.0 eq) was added.
- triethylphosphite (343 ⁇ L, 3.0 eq) was added.
- propan- 2-ol (1.0 mL) was added, followed by stirring for 10 min at rt.
- the so-obtained solution comprising DMTr-MMeUs-MG(ib)s-MG(ib)-(Kc-C1-carbonate) (6) was used directly in the subsequent step (without any precipitation, filtration or purification steps in between).
- the aqueous phase had a pH-value of 5.0.
- the aqueous phase had a pH-value of 5.1.
- MTHP 7.5 mL was added to the so-obtained organic phase comprising MMeUs- MG(ib)s-MG(ib)-(Kc-C1-carbonate) (7), followed by concentration in vacuo to a volume of 7.5 mL (bath temperature 35 ⁇ 45 °C, pressure 40-200 hPa).
- the MTHP (7.5 mL) addition and subsequent concentration was repeated four times (final volume: 7.5 mL; water content: 189.4 ppm).
- DMTr-MMeC(bz) phosphoramidite (CAS-RN: 163759-94-2, 1.21 g, 1.31 mmol, 2.0 eq.) and MTHP (5 mL) were added to the so-obtained solution comprising MMeUs-MG(ib)s-MG(ib)-(Kc-C1-carbonate) (7), followed by concentration in vacuo (bath temperature 45 °C, pressure 40 hPa) to a volume of 7.5 mL.5-Ethylthio-1H-tetrazole (860 mg, 6.61 mmol, 10.0 eq.) and pyridine (1.6 mL, 19.82 mmol, 30.0 eq.) were added and the mixture was stirred at rt until reaction monitoring by HPLC (Method C) indicated completion of the coupling.
- Lactamide (116 mg, 1.30 mmol, 2.0 eq.) was added, followed by stirring for 30 min at rt.
- DDTT (410 mg, 2.00 mmol, 3.0 eq) was added.
- triethylphosphite (343 ⁇ L, 3.0 eq) was added.
- propan-2-ol (1.0 mL) was added, followed by stirring for 10 min at rt.
- the aqueous phase had a pH-value of 4.6.
- the aqueous phase had a pH-value of 4.7.
- MTHP 7.5 mL was added to the so-obtained organic phase comprising MMeC(bz)s-MMeUs-MG(ib)s-MG(ib)-(Kc-C1-carbonate) (9), followed by concentration in vacuo to a volume of 7.5 mL (bath temperature 35 ⁇ 45 °C, pressure 40-200 hPa).
- the MTHP (7.5 mL) addition and subsequent concentration was repeated four times (final volume: 7.5 mL; water content: 189.4 ppm).
- DMTr-MG(ib) phosphoramidite (CAS-RN: 251647-55-9, 1.20 g, 1.31 mmol, 2.0 eq.) and MTHP (5 mL) were added to the so-obtained solution comprising MMeC(bz)s-MMeUs- MG(ib)s-MG(ib)-(Kc-C1-carbonate) (9), followed by concentration in vacuo (bath temperature 45 °C, pressure 40 hPa) to a volume of 7.5 mL.5-Ethylthio-1H-tetrazole (858 mg, 6.59 mmol, 10.0 eq.) and pyridine (1.6 mL, 19.82 mmol, 30.0 eq.) were added and the mixture was stirred at rt until reaction monitoring by HPLC (Method C) indicated completion of the coupling.
- Lactamide (118 mg, 1.33 mmol, 2.0 eq.) was added, followed by stirring for 30 min at rt.
- DDTT (407 mg, 1.98 mmol, 3.0 eq) was added.
- triethylphosphite (343 ⁇ L, 3.0 eq) was added.
- propan-2-ol (1.0 mL) was added, followed by stirring for 10 min at rt.
- the so-obtained solution comprising DMTr-MG(ib)s- MMeC(bz)s-MMeUs-MG(ib)s-MG(ib)-(Kc-C1-carbonate) (10) was used directly in the subsequent step (without any precipitation, filtration or purification steps in between).
- the aqueous phase had a pH-value of 4.5.
- the aqueous phase had a pH-value of 4.6.
- MTHP 7.5 mL was added to the so-obtained organic phase comprising MG(ib)s- MMeC(bz)s-MMeUs-MG(ib)s-MG(ib)-(Kc-C1-carbonate) (OT-1), followed by concentration in vacuo to a weight of 11.6 g (bath temperature 35 ⁇ 45 °C, pressure 40-200 hPa), followed by addition of methanol (120 mL).
- the carbocation scavenger molecules which remain in the organic phase, will react with and thereby consume molecules of the phosphoramidite building block in the subsequent coupling reaction.
- the phosphoramidite building block is an expensive starting material.
- a non-extractable carbocation scavenger such as 1-dodecanethiol instead of an extractable carbocation scavenger such as glutathione (present Example)
- glutathione Present Example
- Example 2 (comparative Example): Synthesis B of target oligonucleotide OT-1 (see Table T-2)
- Present Example 2 is a comparative Example, which differs from the Example 1 according to the present invention only with regard to the aqueous extractions, in particular with regard to the pH-values of the aqueous phases of the aqueous extractions.
- DMTr-MG(ib)s-MG(ib)-(Kc-C1-carbonate) (4) as an exemplary component C-0
- the protocol was as described above for Example 1.
- the so-obtained solution comprising DMTr-MG(ib)s-MG(ib)-(Kc-C1-carbonate) (4) was used directly in the subsequent step (without any precipitation, filtration or purification steps in between).
- the aqueous phase had a pH-value of 0.8.
- NOP 1.0 mL
- acetone 10 mL
- the aqueous phase had a pH-value of 6.3.
- the aqueous phase had a pH-value of 8.2.
- the aqueous phase had a pH-value of 8.2.
- the aqueous phase had a pH-value of 0.7.
- NOP 1.0 mL
- acetone 10 mL
- the aqueous phase had a pH-value of 6.8.
- the aqueous phase had a pH-value of 8.0.
- the aqueous phase had a pH-value of 7.5.
- the aqueous phase had a pH-value of 0.7.
- NOP 1.0 mL
- acetone 10 mL
- the aqueous phase had a pH-value of 6.6.
- the aqueous phase had a pH-value of 7.8.
- the aqueous phase had a pH-value of 7.3.
- n-Heptane 7.5 mL
- MTHP 7.5 mL
- acetone 10 mL
- aqueous phase had a pH-value of 0.6.
- NOP 1.0 mL
- the aqueous phase had a pH-value of 6.8.
- the aqueous phase had a pH-value of 8.2.
- the aqueous phase had a pH-value of 8.2.
- MG(ib)s-MMeC(bz)s-MMeUs-MG(ib)s-MG(ib)-(Kc-C1-carbonate) (OT-1, 1.35 g, 62.4 % crude yield from MG(ib)-(Kc-C1-carbonate) (3), 64.8 % purity (HPLC, Method C)).
- Example 1 afforded the target oligonucleotide O T -1 in higher yield (73.6% for Example 1 instead of 62.4% for comparative Example 2) and higher purity (92.8% for Example 1 instead of 64.8% for comparative Example 2) as compared to the comparative Example 2.
- Example 3 Synthesis of target oligonucleotide OT-2 (see Table T-2) Providing DMTr-dA(bz)-dT-(Kc-C1-carbonate) (12) as an exemplary component C-0 Dichloroacetic acid (DCA, 50 mL) was added at rt to a stirred solution of DMTr-dT- (Kc-C1-carbonate) (1, 7.92 g, 5.96 mmol) obtained as laid out above, and thiomalic acid (4.97 g, 33.10 mmol, 5.5 eq.) in cyclopentyl methyl ether (CPME, 150 mL), and the mixture was stirred at rt until reaction monitoring by HPLC (Method D) indicated completion of the DMTr-deprotection.
- DCA Dichloroacetic acid
- N-methylmorpholine NMM, 62 mL
- acetone and water 100 mL were added, followed by extraction.
- the organic phase was separated and washed twice with water (75 mL ⁇ 2).
- the so-obtained organic phase was concentrated to 9.5 g, followed by precipitation by addition of acetonitrile (440 mL).
- the resulting precipitate was filtrated, then washed twice with acetonitrile (175 mL ⁇ 2) to give dT-(Kc-C1-carbonate) (11, 6.26 g, quantitative yield from DMTr-dT- (Kc-C1-carbonate) (1)).
- DMTr-dA(bz) phosphoramidite (CAS-RN: 98796-53-3,1.16 g, 1.35 mmol, 2.0 eq.) and MTHP (5 mL) were added to the so- obtained solution comprising dT-(Kc-C1-carbonate) (11), followed by concentration in vacuo (bath temperature 45 °C, pressure 40 hPa) to a volume of 15 mL.
- the aqueous phase had a pH-value of 5.1.
- the aqueous phase had a pH-value of 5.6.
- MTHP (10 mL) was added to the so-obtained organic phase comprising dA(bz)-dT- (Kc-C1-carbonate) (13), followed by concentration in vacuo to a volume of 15 mL (bath temperature 35 ⁇ 45 °C, pressure 40-200 hPa).
- the MTHP (10 mL) addition and subsequent concentration was repeated four times (final volume: 15 mL; water content: 136.2 ppm).
- DMTr-dC(bz) phosphoramidite (CAS-RN: 102212-98-6, 1.08 g, 1.32 mmol, 2.0 eq.) and MTHP (5 mL) were added to the so-obtained solution comprising dA(bz)-dT-(Kc-C1-carbonate) (13), followed by concentration in vacuo (bath temperature 45 °C, pressure 40 hPa) to a volume of 15 mL.5-Ethylthio-1H- tetrazole (860 mg, 6.61 mmol, 10.0 eq.) and pyridine (1.6 mL, 19.82 mmol, 30.0 eq.) were added and the mixture was stirred at rt until reaction monitoring by HPLC (Method C) indicated completion of the coupling.
- Lactamide 120 mg, 1.35 mmol, 2.0 eq. was added, followed by stirring for 30 min at rt.1 M I 2 in MTHP (1.45 mL, 1.45 mmol, 2.2 eq.), H 2 O (36 ⁇ L), and acetonitrile (1.5 mL) were added. After stirring for 10 min at rt, the reaction mixture was extracted twice: first with 0.5 N Na2S2O3 aq. (10 mL), then with 0.5 N Na2S2O3 aq. (10 mL).
- the so-obtained organic phase comprising DMTr-dC(bz)-dA(bz)-dT-(Kc-C1-carbonate) (14) was used directly in the subsequent step (without any precipitation, filtration or purification steps in between).
- the aqueous phase had a pH-value of 5.1.
- the aqueous phase had a pH-value of 5.6.
- MTHP (10 mL) was added to the so-obtained organic phase comprising dC(bz)- dA(bz)-dT-(Kc-C1-carbonate) (15), followed by concentration in vacuo to a volume of 15 mL (bath temperature 35 ⁇ 45 °C, pressure 40-200 hPa).
- the MTHP (10 mL) addition and subsequent concentration was repeated four times (final volume: 15 mL; water content: 236.2 ppm).
- DMTr-dT phosphoramidite (CAS-RN: 98796-51-1, 0.99 g, 1.32 mmol, 2.0 eq.) and MTHP (5 mL) were added to the so-obtained solution comprising dC(bz)-dA(bz)-dT-(Kc-C1-carbonate) (15), followed by concentration in vacuo (bath temperature 45 °C, pressure 40 hPa) to a volume of 15 mL.5-Ethylthio-1H-tetrazole (857 mg, 6.58 mmol, 10.0 eq.) and pyridine (1.6 mL, 19.82 mmol, 30.0 eq.) were added and the mixture was stirred at rt until reaction monitoring by HPLC (Method C) indicated completion of the coupling.
- Lactamide (118 mg, 1.33 mmol, 2.0 eq.) was added, followed by stirring for 30 min at rt.1 M I2 in MTHP (1.45 mL, 1.45 mmol, 2.2 eq.), H2O (36 ⁇ L), and acetonitrile (1.5 mL) were added. After stirring for 10 min at rt, the reaction mixture was extracted twice: first with 0.5 N Na2S2O3 aq. (10 mL), then with 0.5 N Na2S2O3 aq. (10 mL). The organic phase was concentrated in vacuo to a volume of 15 mL (bath temperature 35 ⁇ 45 °C, pressure 40-200 hPa).
- the so-obtained organic phase comprising DMTr-dT- dC(bz)-dA(bz)-dT-(Kc-C1-carbonate) (16) was used directly in the subsequent step (without any precipitation, filtration or purification steps in between).
- the aqueous phase had a pH-value of 5.2.
- the aqueous phase had a pH-value of 5.7.
- MTHP (10 mL) was added to the so-obtained organic phase comprising dT-dC(bz)- dA(bz)-dT-(Kc-C1-carbonate) (17), followed by concentration in vacuo to a volume of 15 mL (bath temperature 35 ⁇ 45 °C, pressure 40-200 hPa).
- the MTHP (10 mL) addition and subsequent concentration was repeated four times (final volume: 15 mL; water content: 204.0 ppm).
- DMTr-dA(bz) phosphoramidite (CAS-RN: 98796- 53-3, 1.13 g, 1.32 mmol, 2.0 eq.) and MTHP (5 mL) were added to the so-obtained solution comprising dT-dC(bz)-dA(bz)-dT-(Kc-C1-carbonate) (17), followed by concentration in vacuo (bath temperature 45 °C, pressure 40 hPa) to a volume of 15 mL.5-Ethylthio-1H-tetrazole (858 mg, 6.59 mmol, 10.0 eq.) and pyridine (1.6 mL, 19.82 mmol, 30.0 eq.) were added and the mixture was stirred at rt until reaction monitoring by HPLC (Method C) indicated completion of the coupling.
- Lactamide (118 mg, 1.33 mmol, 2.0 eq.) was added, followed by stirring for 30 min at rt.1 M I2 in MTHP (1.45 mL, 1.45 mmol, 2.2 eq.), H2O (36 ⁇ L), and acetonitrile (1.5 mL) were added. After stirring for 10 min at rt, the reaction mixture was extracted twice at 40 °C: first with 0.5 N Na2S2O3 aq. (10 mL), then with 0.5 N Na2S2O3 aq. (10 mL). The organic phase was concentrated in vacuo to a volume of 15 mL (bath temperature 35 ⁇ 45 °C, pressure 40-200 hPa).
- the so-obtained organic phase comprising DMTr- dA(bz)-dT-dC(bz)-dA(bz)-dT-(Kc-C1-carbonate) (18) was used directly in the subsequent step (without any precipitation, filtration or purification steps in between).
- the aqueous phase had a pH-value of 5.0.
- the aqueous phase had a pH-value of 5.5.
- MTHP 7.5 mL was added to the so-obtained organic phase comprising dA(bz)-dT- dC(bz)-dA(bz)-dT-(Kc-C1-carbonate) (19), followed by concentration in vacuo to a volume of 9.0 g (bath temperature 35 ⁇ 45 °C, pressure 40-200 hPa), followed by addition of methanol (120 mL).
- dA(bz)-dT-dC(bz)-dA(bz)-dT-(Kc-C1- carbonate) (19, 895 mg, 48.9 % crude yield based on dT-(Kc-C1-carbonate) (11)) as a white solid.
- Example 4 Synthesis of target oligonucleotide OT-3 (see Table T-2) Providing DMTr-MG(ib)s-MG(ib)-(Kc-C1-carbonate) (4) as an exemplary component C-0 Dichloroacetic acid (DCA, 50 mL) was added at rt to a stirred solution of DMTr- MG(ib)-(Kc-C1-carbonate) (2, 6.98 g, 4.66 mmol) obtained from Synthesis B laid out above, and thiomalic acid (4.96 g, 33.03 mmol, 7.1 eq.) in cyclopentyl methyl ether (CPME, 150 mL), and the mixture was stirred at rt until reaction monitoring by HPLC (Method D) indicated completion of
- N-methylmorpholine NMM, 80 mL
- acetone 25 mL
- water 100 mL
- the organic phase was separated and washed twice with water (75 mL ⁇ 2).
- the so-obtained organic phase was concentrated to 9.72 g, followed by precipitation by addition of acetonitrile (500 mL).
- the resulting precipitate was filtrated, then washed twice with acetonitrile (200 mL ⁇ 2) to give MG(ib)-(Kc-C1- carbonate) (3, 4.65 g, 58.9 % yield for 2 steps from 3,5-bis(docosyloxy)benzenemethanol).
- DMTr-MG(ib) phosphoramidite (CAS-RN: 251647-55-9, 2.41 g, 2.64 mmol, 2.0 eq.) and MTHP (10 mL) were added (water content of the so- obtained solution: 180.3 ppm), followed by concentration in vacuo (bath temperature 45 °C, pressure 40 hPa), then adjustment a volume to 15 mL by addition of MTHP (7.5 mL).
- the liquid contents of the flask were transferred into a separatory funnel, thereby leaving the molecular sieves inside the flask.
- the pH- value of the aqueous phase was 4.6.
- the pH-value of the aqueous phase was 5.1.
- the pH-value of the aqueous phase was 5.5.
- MTHP 7.5 mL
- MG(ib)s-MG(ib)-(Kc-C1-carbonate) MG(ib)s-MG(ib)-(Kc-C1-carbonate) (5)
- concentration in vacuo a volume of 7.5 mL (bath temperature 35 ⁇ 45 °C, pressure 40-200 hPa).
- the MTHP (7.5 mL) addition and subsequent concentration was repeated four times (final volume: 7.5 mL; water content: 247.8 ppm).
- Lactamide (235 mg, 2.64 mmol, 2.0 eq.) was added, followed by stirring for 30 min at rt.
- DDTT (813 mg, 3.96 mmol, 3.0 eq) was added.
- triethylphosphite (686 ⁇ L, 3.96 mmol, 3.0 eq) was added.
- propan-2-ol 2.0 mL was added, followed by stirring for 10 min at rt.
- Table E-1 Overview of iterations 2 to 8 of the coupling cycle of Examples 4 number of the starting product coupling material cycle 2 DMTr-MMeC(bz)s-MMeUs-MG(ib)s-MG(ib)-(Kc-C1- 6 carbonate) (8) DMTr-MG(ib)s-MMeC(bz)s-MMeUs-MG(ib)s-MG(ib)- 3 8 (Kc-C1-carbonate) (10) DMTr-MMeUs-MG(ib)s-MMeC(bz)s-MMeUs-MG(ib)s- 4 10 MG(ib)-(Kc-C1-carbonate) (20) 5 DMTr-MA(bz)s-MMeUs-MG(ib)s-MMeC(bz)s-MMeUs- 20 MG(ib)s-MG(ib)-(Kc-C1-carbonate) (21) DMTr-MA(
- n-Heptane 15 mL
- MTHP 15 mL
- acetone 20 mL
- the aqueous phase had a pH-value of 4.6.
- NOP 2.0 mL
- was added to the organic phase in the separatory funnel, followed by extraction with a mixture of, 5 wt-% aqueous 2 sodium chloride solution and acetone (1:1 v/v, 90 mL, pH-value 5.2, pH) and phase separation.
- the aqueous phase had a pH-value of 5.1.
- the aqueous phase had a pH-value of 5.5.
- the aqueous phase had a pH-value of 4.6.
- the aqueous phase had a pH-value of 5.1.
- the aqueous phase had a pH- value of 5.5.
- n-Heptane 15 mL
- MTHP 15 mL
- acetone 20 mL
- the aqueous phase had a pH-value of 4.6.
- the aqueous phase had a pH-value of 5.0.
- the aqueous phase had a pH- value of 5.5.
- the aqueous phase had a pH-value of 4.6.
- the aqueous phase had a pH-value of 5.1.
- the aqueous phase had a pH- value of 5.4.
- n-Heptane 15 mL
- MTHP 15 wt-% aqueous sodium chloride solution
- pH-value 4.9
- the aqueous phase had a pH-value of 4.6.
- NOP 2.0 mL
- the aqueous phase had a pH- value of 5.1.
- the aqueous phase had a pH- value of 5.4.
- the aqueous phase had a pH-value of 4.6.
- NOP 2.0 mL
- MTHP 15 mL
- the aqueous phase had a pH- value of 5.1.
- the aqueous phase had a pH- value of 5.3.
- n-Heptane 15 mL
- MTHP 15 mL
- acetone 20 mL
- the aqueous phase had a pH-value of 4.5.
- NOP 2.0 mL
- was added to the organic phase in the separatory funnel, followed by extraction with a mixture of, 5 wt-% aqueous sodium chloride solution and acetone (1:1 v/v, 90 mL, pH-value 5.2, pH) and phase separation.
- the aqueous phase had a pH-value of 5.0.
- the aqueous phase had a pH- value of 5.4.
- Azeotropic distillation In an eggplant flask, MTHP (15 mL) was added to the organic phase obtained from the preceding aqueous extractions, followed by concentration in vacuo (bath temperature 35 ⁇ 45 °C, pressure 40-200 hPa). This MTHP addition and concentration step was typically repeated.
- the water content was adjusted to be less than 400 ppm.
- Coupling reaction including quenching with Lactamide
- Molecular sieves 4A 0.1 g
- Pyridine (30 eq.)
- 5-Ethylthio-1H-tetrazole (10 eq.) were added, and the mixture was stirred at rt until reaction monitoring by HPLC (Method D) indicated completion of the coupling reaction.
- Lactamide (1.0 eq.) was added, and the mixture was stirred for 30 min at rt.
- Sulfurization including quenching with triethyl phosphite and isopropyl alcohol
- DDTT (3.0 eq.
- the so-obtained solution was transferred into a separatory funnel, thereby leaving the molecular sieves inside the flask.
- the aqueous phase had a pH-value of 4.5.
- the aqueous phase had a pH-value of 5.1.
- the aqueous phase had a pH-value of 5.4.
- MTHP 15 mL was added to the so-obtained organic phase comprising MA(bz)s-MMeUs-MA(bz)s- MA(bz)s-MMeUs-MG(ib)s-MMeC(bz)s-MMeUs-MG(ib)s-MG(ib)-(Kc-C1-carbonate) (25), followed by concentration in vacuo to a weight of 11.6 g (bath temperature 35 ⁇ 45 °C, pressure 40-200 hPa), followed by addition of methanol (120 mL), and stirring for 30 min under ice-cooling.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Biochemistry (AREA)
- Molecular Biology (AREA)
- Engineering & Computer Science (AREA)
- Biotechnology (AREA)
- General Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Saccharide Compounds (AREA)
Abstract
Description



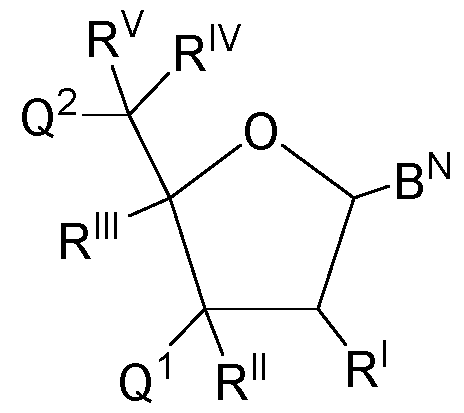
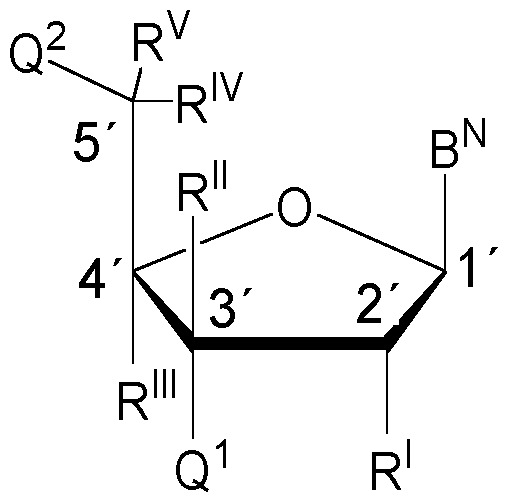




















Claims


Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/EP2023/078683 WO2024083746A1 (en) | 2022-10-17 | 2023-10-16 | Method and composition for oligonucleotide synthesis |
| EPPCT/EP2023/078683 | 2023-10-16 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2025082632A1 true WO2025082632A1 (en) | 2025-04-24 |
Family
ID=90720065
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2024/060228 Pending WO2025082632A1 (en) | 2023-10-16 | 2024-04-16 | Method and composition for oligonucleotide synthesis |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2025082632A1 (en) |
Citations (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009073809A2 (en) | 2007-12-04 | 2009-06-11 | Alnylam Pharmaceuticals, Inc. | Carbohydrate conjugates as delivery agents for oligonucleotides |
| US20130267697A1 (en) | 2012-02-17 | 2013-10-10 | Ajinomoto Co., Inc. | Oligonucleotide with protected base |
| EP2711370A1 (en) | 2011-05-17 | 2014-03-26 | Ajinomoto Co., Inc. | Method for producing oligonucleotide |
| EP2816053A1 (en) | 2012-02-17 | 2014-12-24 | Ajinomoto Co., Inc. | Base-protected oligonucleotide |
| US20150112053A1 (en) | 2012-05-30 | 2015-04-23 | Hokkaido System Science Co., Ltd. | Oligonucleotide synthesis method using highly dispersible liquid-phase support |
| EP2921499A1 (en) | 2012-11-14 | 2015-09-23 | Takeda Pharmaceutical Company Limited | Method for liquid-phase synthesis of nucleic acid |
| WO2016055601A1 (en) | 2014-10-10 | 2016-04-14 | F. Hoffmann-La Roche Ag | Galnac phosphoramidites, nucleic acid conjugates thereof and their use |
| EP3015467A1 (en) | 2013-05-24 | 2016-05-04 | Ajinomoto Co., Inc. | Morpholino oligonucleotide manufacturing method |
| EP3263579A1 (en) | 2015-01-21 | 2018-01-03 | Ajinomoto Co., Inc. | Precipitation accelerator and precipitation method in which same is used |
| EP3378869A1 (en) | 2015-11-17 | 2018-09-26 | Nissan Chemical Corporation | Method for producing oligonucleotide |
| US20180291056A1 (en) | 2015-12-22 | 2018-10-11 | Ajinomoto Co., Inc. | Oligonucleotide manufacturing method |
| EP3398955A1 (en) | 2015-12-16 | 2018-11-07 | Ajinomoto Co., Inc. | Oligonucleotide production method, and nucleoside, nucleotide, or oligonucleotide |
| WO2019075419A1 (en) | 2017-10-13 | 2019-04-18 | Dicerna Pharmaceuticals, Inc. | Methods and compositions for inhibiting expression of ldha |
| EP3733680A1 (en) | 2017-12-27 | 2020-11-04 | KNC Laboratories Co., Ltd | Production of highly fat-soluble phosphoramidite |
| WO2020227618A2 (en) | 2019-05-08 | 2020-11-12 | Biogen Ma Inc. | Convergent liquid phase syntheses of oligonucleotides |
| EP3825300A1 (en) | 2018-07-20 | 2021-05-26 | Fujimoto Chemicals Co. Ltd. | Alkoxyphenyl derivative, nucleoside protector, nucleotide protector, method for producing oligonucleotide, and method for removing substituent |
| EP3925964A1 (en) | 2019-02-15 | 2021-12-22 | Ajinomoto Co., Inc. | Production method for oligonucleotides |
| EP3950698A1 (en) | 2019-03-28 | 2022-02-09 | Ajinomoto Co., Inc. | Method for producing oligonucleotide having phosphorothioate site |
| EP4006045A1 (en) | 2019-08-29 | 2022-06-01 | FUJIFILM Corporation | Nucleic acid compound manufacturing method and nucleic acid compound |
| WO2022214692A1 (en) | 2021-04-09 | 2022-10-13 | Bachem Holding Ag | Pseudo solid phase protecting group and methods for the synthesis of oligonucleotides and oligonucleotide conjugates |
-
2024
- 2024-04-16 WO PCT/EP2024/060228 patent/WO2025082632A1/en active Pending
Patent Citations (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009073809A2 (en) | 2007-12-04 | 2009-06-11 | Alnylam Pharmaceuticals, Inc. | Carbohydrate conjugates as delivery agents for oligonucleotides |
| EP2711370A1 (en) | 2011-05-17 | 2014-03-26 | Ajinomoto Co., Inc. | Method for producing oligonucleotide |
| EP3296306A1 (en) | 2012-02-17 | 2018-03-21 | Ajinomoto Co., Inc. | Base-protected oligonucleotide |
| US20130267697A1 (en) | 2012-02-17 | 2013-10-10 | Ajinomoto Co., Inc. | Oligonucleotide with protected base |
| EP2816053A1 (en) | 2012-02-17 | 2014-12-24 | Ajinomoto Co., Inc. | Base-protected oligonucleotide |
| US20150080565A1 (en) | 2012-02-17 | 2015-03-19 | Ajinomoto Co., Inc. | Oligonucleotide with protected base |
| US20150112053A1 (en) | 2012-05-30 | 2015-04-23 | Hokkaido System Science Co., Ltd. | Oligonucleotide synthesis method using highly dispersible liquid-phase support |
| EP2921499A1 (en) | 2012-11-14 | 2015-09-23 | Takeda Pharmaceutical Company Limited | Method for liquid-phase synthesis of nucleic acid |
| EP3015467A1 (en) | 2013-05-24 | 2016-05-04 | Ajinomoto Co., Inc. | Morpholino oligonucleotide manufacturing method |
| WO2016055601A1 (en) | 2014-10-10 | 2016-04-14 | F. Hoffmann-La Roche Ag | Galnac phosphoramidites, nucleic acid conjugates thereof and their use |
| EP3263579A1 (en) | 2015-01-21 | 2018-01-03 | Ajinomoto Co., Inc. | Precipitation accelerator and precipitation method in which same is used |
| EP3378869A1 (en) | 2015-11-17 | 2018-09-26 | Nissan Chemical Corporation | Method for producing oligonucleotide |
| EP3398955A1 (en) | 2015-12-16 | 2018-11-07 | Ajinomoto Co., Inc. | Oligonucleotide production method, and nucleoside, nucleotide, or oligonucleotide |
| US20180291056A1 (en) | 2015-12-22 | 2018-10-11 | Ajinomoto Co., Inc. | Oligonucleotide manufacturing method |
| WO2019075419A1 (en) | 2017-10-13 | 2019-04-18 | Dicerna Pharmaceuticals, Inc. | Methods and compositions for inhibiting expression of ldha |
| EP3733680A1 (en) | 2017-12-27 | 2020-11-04 | KNC Laboratories Co., Ltd | Production of highly fat-soluble phosphoramidite |
| EP3825300A1 (en) | 2018-07-20 | 2021-05-26 | Fujimoto Chemicals Co. Ltd. | Alkoxyphenyl derivative, nucleoside protector, nucleotide protector, method for producing oligonucleotide, and method for removing substituent |
| EP3925964A1 (en) | 2019-02-15 | 2021-12-22 | Ajinomoto Co., Inc. | Production method for oligonucleotides |
| EP3950698A1 (en) | 2019-03-28 | 2022-02-09 | Ajinomoto Co., Inc. | Method for producing oligonucleotide having phosphorothioate site |
| WO2020227618A2 (en) | 2019-05-08 | 2020-11-12 | Biogen Ma Inc. | Convergent liquid phase syntheses of oligonucleotides |
| EP4006045A1 (en) | 2019-08-29 | 2022-06-01 | FUJIFILM Corporation | Nucleic acid compound manufacturing method and nucleic acid compound |
| WO2022214692A1 (en) | 2021-04-09 | 2022-10-13 | Bachem Holding Ag | Pseudo solid phase protecting group and methods for the synthesis of oligonucleotides and oligonucleotide conjugates |
Non-Patent Citations (15)
| Title |
|---|
| A. FISCHER ET AL., J. CHEM. SOC, 1964, pages 3591 - 3596, Retrieved from the Internet <URL:https://doi.org/10.1039/JR9640003591> |
| A. SCHWENGER ET AL., EUROPEAN JOURNAL OF ORGANIC CHEMISTRY, 2017, pages 5852 - 5864 |
| ACS CENTRAL SCIENCE, vol. 7, 2021, pages 1473 - 1485 |
| H. LONNEBERG, BEILSTEIN JOURNAL OF ORGANIC CHEMISTRY, vol. 13, 2017, pages 1368 - 1387 |
| J. STAWINSKIR. STROMBERG: "Di- and Oligonucleotide Synthesis Using H-Phosphonate Chemistry", METHODS IN MOLECULAR BIOLOGY, vol. 288, 2005 |
| K.V. GOTHELF ET AL., NATURE COMMUNICATIONS, vol. 12, 2021, pages 2760 |
| M.C. DE KONING ET AL., ORGANIC PROCESS RESEARCH & DEVELOPMENT, vol. 10, 2006, pages 1238 - 1245 |
| P. KAREN ET AL., PURE AND APPLIED CHEMISTRY, vol. 88, no. 8, 2016, pages 831 - 839 |
| P.S. BARAN ET AL., SCIENCE, vol. 361, 2018, pages 12341238 - 1238 |
| S. KIM ET AL., A EUROPEAN J, vol. 19, 2013, pages 8615 - 8620 |
| S. KIM ET AL., CHEMISTRY - A EUROPEAN JOURNAL, vol. 19, 2013, pages 8615 - 8620 |
| X. SHI ET AL., J. ORG. CHEM., vol. 87, no. 4, 2022, pages 2087 - 2110 |
| X. SHI ET AL., JOURNAL OF ORGANIC CHEMISTRY, vol. 87, no. 4, 2022, pages 2087 - 2110 |
| X. SHI ET AL., THE JOURNAL OF ORGANIC CHEMISTRY, vol. 87, no. 4, 2022, pages 2087 - 2110 |
| X. WEI ET AL., TETRAHEDRON, vol. 69, 2013, pages 3615 - 3637 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR101281836B1 (en) | Phosphoramidite compound and method for producing oligo-rna | |
| JP6281599B2 (en) | Pseudo solid phase protecting groups and nucleotides | |
| WO2019208571A1 (en) | Amidite compound and method for producing polynucleotide using said compound | |
| AU2011217918A1 (en) | Phosphoramidites for synthetic RNA in the reverse direction | |
| JP2010526835A (en) | Thiocarbon protecting group for RNA synthesis | |
| EP3645544A1 (en) | Multiple coupling&oxidation method | |
| KR101345465B1 (en) | Desorption Method of Nucleic Acid Protector | |
| JP7229539B2 (en) | Production of highly lipophilic phosphoramidites | |
| EP1317466B1 (en) | Synthons for oligonucleotide synthesis | |
| CN114502566B (en) | Method for producing nucleic acid oligomer | |
| EP1995252A1 (en) | Method for removal of nucleic acid-protecting group | |
| GB2366290A (en) | Hexitol Nucleosides | |
| US20260001902A1 (en) | Improved oligonucleotide synthesis | |
| WO2025082632A1 (en) | Method and composition for oligonucleotide synthesis | |
| EP4605403B1 (en) | Method and composition for oligonucleotide synthesis | |
| EP2017282A1 (en) | Method of capping oligonucleic acid | |
| WO2024083746A1 (en) | Method and composition for oligonucleotide synthesis | |
| JP4802712B2 (en) | Solution phase synthesis of ribonucleic acid compounds and oligonucleic acid compounds | |
| CN101522701B (en) | Introduction method of nucleic acid protecting group | |
| WO2021080021A1 (en) | Method for producing oligonucleotide |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 24717724 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2024717724 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2024717724 Country of ref document: EP Effective date: 20251114 |
|
| ENP | Entry into the national phase |
Ref document number: 2024717724 Country of ref document: EP Effective date: 20251114 |
|
| ENP | Entry into the national phase |
Ref document number: 2024717724 Country of ref document: EP Effective date: 20251114 |
|
| ENP | Entry into the national phase |
Ref document number: 2024717724 Country of ref document: EP Effective date: 20251114 |