WO2025081094A2 - Methods, kits and systems for determining the er status of cancer and methods for treating cancer based on same - Google Patents
Methods, kits and systems for determining the er status of cancer and methods for treating cancer based on same Download PDFInfo
- Publication number
- WO2025081094A2 WO2025081094A2 PCT/US2024/051117 US2024051117W WO2025081094A2 WO 2025081094 A2 WO2025081094 A2 WO 2025081094A2 US 2024051117 W US2024051117 W US 2024051117W WO 2025081094 A2 WO2025081094 A2 WO 2025081094A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- cancer
- positive
- sample
- subject
- negative
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6876—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes
- C12Q1/6883—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for diseases caused by alterations of genetic material
- C12Q1/6886—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for diseases caused by alterations of genetic material for cancer
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/154—Methylation markers
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/158—Expression markers
Definitions
- Progesterone receptor (PR) expression in normal breast epithelium is regulated by ER (Jensen, Cancer (1980) 46:2759-2761). Presence of ER, PR and human epidermal growth factor receptor-2 (HER2) status in invasive breast carcinoma is now routinely estimated as these markers are considered to be important prognostic factors.
- ER and PR status has been used for many years to determine a patient’s suitability for treatment with endocrine therapy (e.g., tamoxifen).
- Such methods focus only on a small region at a single tumor site at a given time and therefore do not accurately capture tumor heterogeneity or receptor evolution and therefore only partially characterize the relevant patient population.
- diagnostic methods for determining ER status including methods that are independent of IHC testing.
- Improved diagnostic methods would also better support future clinical trials that seek to identify subpopulations of patients that respond to ER-targeted agents. They would also expand our understanding of the underlying biology of ER-positive cancer and help identify new treatments.
- the present disclosure includes, among other things, histone modification measurements in cfDNA that are characteristic of ER-positive and ER-negative cancers, which in various embodiments are useful, e.g., in detecting, monitoring, selecting treatment for, and/or treating an ER-positive and ER-negative cancers.
- histone modification measurements in cfDNA can be used to detect or determine resistance of a cancer (e.g., breast, ovarian, or endometrial cancer) to a therapy or transformation of a cancer from one subtype to another.
- the present disclosure includes exemplary genomic loci that are differentially modified in ER-positive vs. ER-negative cancer, e.g., breast, ovarian, or endometrial cancer.
- histone methylation can be or include H3K4me3.
- histone acetylation can be or include histone acetylation marks selected from H3K9ac, H3K14ac, H3K18ac, H3K23ac, H3K27ac, or a combination thereof.
- histone acetylation can be or include H3K27ac.
- the present disclosure further relates, in various embodiments, to the measurement of transcription factor binding in cell-free DNA (cfDNA) to determine ER status.
- cfDNA cell-free DNA
- the present disclosure includes, among other things, transcription factor binding measurements in cfDNA that are characteristic of ER-positive cancers, which in various embodiments are useful, e.g., in detecting, monitoring, selecting treatment for, and/or treating an ER-positive cancer.
- transcription factor binding measurements in cfDNA can be used to detect or determine resistance of a cancer (e.g., breast, ovarian, or endometrial cancer) to a therapy or transformation of a cancer from one subtype to another.
- a cancer e.g., breast, ovarian, or endometrial cancer
- histone acetylation corresponds and/or is correlated with transcription factor binding.
- DNA methylation corresponds and/or is correlated with transcription factor binding.
- a genomic locus is differentially bound by transcription factors if it is characterized by increased or decreased transcription factor binding as compared to a reference (e.g., a sample from an ER-negative or healthy subject). Increased or decreased
- transcription factor binding can be or include, e.g., increased or decreased transcription factor binding as determined by various transcription factor binding assays known in the art.
- the present disclosure provides a method of determining the ER status of a cancer in a subject, the method comprising: quantifying, at one or more genomic loci in a biological sample, optionally in cell-free DNA (cfDNA) from a liquid biopsy sample, obtained or derived from the subject: (i) one or more histone modifications, (ii) chromatin accessibility, (iii) binding of one or more transcription factors, and/or (iv) DNA methylation.
- cfDNA cell-free DNA
- the one or more histone modifications are quantified using a histone modification assay that measures one or more of H3K9ac, H3K14ac, H3K18ac, H3K23ac, H3K27ac, H3K4me1, H3K4me2, H3K4me3, and pan-acetylation.
- the histone modification assay detects H3K4me3 modifications.
- the histone modification assay detects H3K27ac modifications.
- chromatin accessibility is quantified using a chromatin accessibility assay selected from ATAC-seq (Assay of Transpose Accessible Chromatin sequencing), NOMe-seq (Nucleosome Occupancy and Methylome sequencing), FAIRE-seq (Formaldehyde-Assisted Isolation of Regulatory Elements sequencing), MNase-seq (Micrococcal Nuclease digestion with sequencing), and a DNase hypersensitivity assay.
- ATAC-seq Assay of Transpose Accessible Chromatin sequencing
- NOMe-seq Nucleosome Occupancy and Methylome sequencing
- FAIRE-seq Formmaldehyde-Assisted Isolation of Regulatory Elements sequencing
- MNase-seq Merococcal Nuclease digestion with sequencing
- binding of one or more transcription factors is quantified using a transcription factor binding assay that detects binding of one or more of p300, mediator complex, cohesin complex, RNA pol II, FOXA1, ESR1, PR, MYC, EN1, FOXM1, KLF4, AP-2, RARa, or RUNX1.
- the transcription factor binding assay is selected from ChIP-seq (Chromatin ImmunoPrecipitation sequencing), CUT&RUN (Cleavage Under Targets and Release Using Nuclease) sequencing, and CUT&Tag (Cleavage Under Targets and Tagmentation) sequencing.
- DNA methylation is quantified using Bisulfite sequencing (BS-Seq), Whole Genome Bisulfite Sequencing (WGBS), Methylated DNA ImmunoPrecipitation sequencing (MeDIP-seq), or Methyl-CpG-Binding Domain sequencing (MBD-seq).
- BS-Seq Bisulfite sequencing
- WGBS Whole Genome Bisulfite Sequencing
- MBD-seq Methyl-CpG-Binding Domain sequencing
- the method comprises quantifying two or more of the following, each at one or more genomic loci in cell-free DNA (cfDNA) from a liquid biopsy sample obtained or derived from the subject: (i) one or more histone modifications, (ii) chromatin accessibility, (iii) transcription factor binding, and/or (iv) DNA methylation.
- the method comprises quantifying two or more histone modifications, e.g., quantifying H3K4me3 and H3K27ac modifications.
- the method comprises quantifying one or more histone modifications and DNA methylation, e.g., quantifying H3K4me3 and/or H3K27ac modifications and DNA methylation. In some embodiments, the method comprises quantifying H3K4me3 modifications, H3K27ac modifications and DNA methylation.
- the biological sample is a liquid biopsy sample, e.g., a plasma sample, serum sample, or urine sample.
- quantification of one or more histone modifications, chromatin accessibility, binding of one or more transcription factors, and/or DNA methylation at the one or more genomic loci as compared to a reference indicates that the subject has an ER- positive cancer.
- quantification of one or more histone modifications, chromatin accessibility, binding of one or more transcription factors, and/or DNA methylation at the one or more genomic loci as compared to a reference indicates that the subject has an ER- negative cancer.
- the cancer is breast cancer, ovarian cancer, or endometrial cancer. In some embodiments, the cancer is breast cancer.
- the reference is a predetermined threshold, a measurement from a liquid biopsy sample, and/or a normalized value, optionally wherein the reference is a measurement from a liquid biopsy sample obtained from a cohort of subjects who have previously been determined to have an ER-negative cancer or to be cancer free.
- cfDNA comprising H3K4me3 modifications is enriched using a method that comprises incubating a sample with an agent (e.g., an antibody) that binds H3K4me3 modifications;
- agent e.g., an antibody
- cfDNA comprising H3K27ac modifications is enriched using a method that comprises incubating a sample with an agent (e.g., an antibody) that binds H3K27ac modifications;
- methylated cfDNA is enriched using a method that comprises incubating a sample with an agent (e.g., an antibody or a methyl binding domain) that binds methylated DNA.
- an agent that binds H3K4me3 modifications, an agent that binds H3K27ac modifications, and/or an agent that binds methylated DNA can be attached (e.g., via a covalent or noncovalent bond) to a physical support (e.g., a bead, a magnetic bead, an agarose bead, or a magnetic epoxy bead) prior to incubating with a sample.
- a physical support e.g., a bead, a magnetic bead, an agarose bead, or a magnetic epoxy bead
- sequence reads are mapped to a reference genome, and one or more genomic loci correspond to sequence read peaks, wherein a sequence read peak corresponds to a region of the genome that has a higher number of sequence reads that the local background.
- peaks in high noise regions are ignored when identifying genomic loci with a higher number of sequence reads than the local background.
- peaks in regions likely to be artifactual are removed.
- peaks that are less than 50 bp in length are removed.
- peaks in regions with high levels of one or more epigenetic markers in white blood cells are removed.
- a method comprises determining an ER-positive/ER- negative ratio score for two or more epigenetic biomarkers. In some embodiments, a method comprises determining an ER-positive/ER-negative ratio score for two or more epigenetic biomarkers, wherein the ER-positive/ER-negative ratio scores are combined. In some embodiments, a method comprises determining an ER-positive/ER-negative ratio score each of H3K4me3 modifications, H3K27ac modifications, and methylated DNA, and combining the ratio scores. In some embodiments, two or more ratio scores can be combined using fitted values determined using a logistic regression.
- a method further comprises comparing one or more quantified epigenetic biomarkers to a reference, and wherein an increase or decrease in the one or more epigenetic markers as compared to the reference indicates that a subject has an ER- positive or an ER-negative cancer.
- a sample comprises a detectable amount of ctDNA (e.g., wherein estimated tumor fraction is >3% for the cfDNA, e.g., as determined by iChorCNA).
- the method comprises quantifying one or more histone modifications, chromatin accessibility, binding of one or more transcription factors, and/or DNA methylation at one or more genomic loci in Tables 1-3.
- the method comprises quantifying H3K4me3 modifications for at least 5, 10, 20, 30, 40, or 50 genomic loci in Table 1.
- the method comprises quantifying H3K27ac modifications for at least 5, 10, 20, 30, 40, or 50 genomic loci in Table 2. In some embodiments, the method comprises quantifying DNA methylation for at least 5, 10, 20, 30, 40, or 50 genomic loci in Table 3.
- the area under the receiver operating characteristic (AUROC) for determining if a subject has an ER-positive cancer vs. an ER-negative cancer is greater than 0.5 (e.g., greater than 0.55, greater than 0.6, greater than 0.65, greater than 0.7, greater than 0.75, greater than 0.8, greater than 0.85, greater than 0.9, or greater than 0.95).
- the ER-positive cancer is an ER-positive cancer based on IHC testing and the ER-negative cancer is an ER-negative cancer based on IHC testing.
- the subject has previously been determined to have cancer.
- a sample is obtained from a subject having cancer wherein a biopsy of the cancer is not possible and/or feasible.
- the present disclosure provides a method of treating a subject having a cancer, the method comprising: administering a cancer therapy to the subject based on the ER status of the cancer, wherein the ER status of the cancer has been determined using any one of the aforementioned methods of determining ER status.
- the method further comprises determining the ER status of the cancer using any one of the aforementioned methods of determining ER status.
- the cancer has been determined to be ER-positive and the cancer therapy comprises an ER-targeted agent.
- the cancer therapy is one appropriate for an ER- negative cancer.
- the cancer therapy does not comprise administering an ER-targeted agent.
- the method further comprises determining the ER status of the cancer using any one of the aforementioned methods of determining ER status.
- the cancer therapy comprises an ER-targeted agent.
- the cancer therapy does not comprise administering an ER-targeted agent.
- the present disclosure provides a method of monitoring the ER status of a cancer in a subject, and optionally treating the cancer, the method comprising: determining the ER status of the cancer using any one of the aforementioned methods of determining ER status at first and second time points.
- the subject has been administered an ER-targeted agent after the first time point and before the second time point.
- the method further comprises administering a cancer therapy, optionally an ER-targeted agent, to the subject based on the ER status of the cancer at the second time point, optionally wherein the type, dose and/or frequency of administration of the cancer therapy is adjusted based on the ER status of the cancer at the second time point.
- the present disclosure provides a method of treating a subject having a cancer, the method comprising: administering an ER-targeted agent to the subject if the subject has been determined to have a validated epigenetic profile indicative of an ER-positive cancer based on analysis of a biological sample, optionally of cell-free DNA (cfDNA) from a liquid biopsy sample, obtained or derived from the subject, and, if the subject has not been determined to a validated epigenetic profile indicative of an ER-positive cancer, not administering an ER-targeted agent, wherein the presence of the validated epigenetic profile has been determined using a validated classifier, wherein the validated classifier has been obtained by: (a) determining a genomic profile of one or more histone modifications, chromatin accessibility, binding of one or more transcription factors, and/or DNA methylation in (i) one or more ER-positive cell lines or (ii) biological samples obtained from a first cohort of subjects who have previously been determined to have an ER
- the classifier in step (d) was trained on two or more histone modification levels in the differential loci. In some embodiments, the two or more histone modification levels comprise H3K4me3 and H3K27ac modification levels. [0059] In some embodiments, the classifier in step (d) was trained on one or more histone modification levels and DNA methylation in the differential loci. In some embodiments, the one or more histone modification levels comprise H3K4me3 and/or H3K27ac modification levels. In some embodiments, the classifier in step (d) was trained using ridge regression, elastic- net regression, or lasso regression.
- the kit comprises reagents for isolation of cell-free DNA (cfDNA) from a liquid biopsy sample. In some embodiments, the kit comprises reagents for library preparation for sequencing. In some embodiments, the kit comprises reagents for sequencing. In some embodiments, the kit comprises instructions for determining if a subject has an ER-positive cancer.
- the present disclosure provides a non-transitory computer readable storage medium encoded with a computer program, wherein the program comprises instructions that when executed by one or more processors cause the one or more processors to
- the sequencer is configured to generate a Whole Genome Sequencing (WGS) data set from the sample.
- the system further comprises a sample preparation device.
- the sample preparation device is configured to prepare the sample for sequencing from a biological sample, optionally a liquid biopsy sample.
- the sample preparation device comprises reagents for quantifying one or more histone modifications, chromatin accessibility, binding of one or more transcription factors, and/or DNA methylation at one or more genomic loci in cell-free DNA (cfDNA) from the biological sample, optionally the liquid biopsy sample.
- the one or more genomic loci are selected from Tables 1-3.
- the reagents comprise one or more methyl-binding domains for use in MBD-seq.
- the device comprises reagents for isolation of cell-free DNA (cfDNA) from the biological sample, optionally the liquid biopsy sample.
- the device comprises reagents for library preparation for sequencing.
- the sequencer comprises reagents for sequencing.
- a method is for determining ER status of a cancer in a subject (e.g., patient).
- the method may include receiving (e.g., by a processor of a computing device) one or more genomic profiles of one or more histone modifications, chromatin accessibility, binding of one or more transcription factors, and/or DNA methylation for the subject.
- the method may further include determining whether the subject has an epigenetic profile indicative of an ER- positive cancer by classifying the genomic profile using an ER classifier.
- an ER classifier has been validated using liquid biopsy sample data.
- a non-transitory computer readable storage medium may be encoded with a computer program, where the program may comprise instructions that when executed by one or more processors cause the one or more processors to perform operations to perform a method for determining ER status of a cancer in a subject (e.g., patient).
- a computer system may include a memory and one or more processors coupled to the memory, wherein the one or more processors are configured to perform operations to perform a method for determining ER status of a cancer in a subject (e.g., patient).
- a method of treating a subject having a cancer includes administering an ER-targeted agent to the subject, wherein the subject has been determined to have a validated epigenetic profile indicative of an ER-positive cancer based on analysis of a biological sample, optionally of cell-free DNA (cfDNA) from a liquid biopsy sample, obtained or derived from the subject.
- the presence of the validated epigenetic profile has been determined using a classifier (e.g., a validated classifier) according to a method for determining ER status of a cancer in a subject (e.g., patient).
- genomic loci from Tables 1-3 for different modifications, namely (i) H3K4me3 modifications, (ii) H3K27ac modifications, (iii) DNA methylation (DNAme) or (iv) all of the above (All) and (b) using different subsets of genomic loci in Tables 1-3 for a particular modification, namely (i) all genomic loci with an absolute log2(fold-change) ⁇ 0.5, (ii) all genomic loci with an absolute log2(fold-change) ⁇ 1, (iii) all genomic loci with an absolute log2(fold-change) ⁇ 2, (iv) all genomic loci with an absolute log2(fold-change) ⁇ 3, and (v) all genomic loci with an absolute log2(fold-change) ⁇ 4.
- Fig.2 shows representative, non-limiting graphs that demonstrate the accuracy of ER status (based on AUCROC) determination using the classifiers that were generated in accordance with Example 2.
- Fig.3 (A) shows a heatmap representation of z-scored, ctDNA- and background- normalized counts at differential peaks (DE-seq, FDR ⁇ 0.05, log2(fold change) > 1) across ER +/- patients (status determined by IHC). Each row corresponds to signal observed in an individual patient, and each column represents an enhancer/promoter/MBD locus.
- (B) shows ROC curves for an exemplary ER status classifier generated in accordance with Example 3 and applied to plasma samples obtained from patients previously diagnosed with metastatic breast cancer. ROC curves assessing performance of a regularized logistic regression model to classify
- Fig.4 is a block diagram of an example network environment for use in the methods and systems described herein, according to illustrative embodiments of the present disclosure.
- Fig.5 is a block diagram of an example computing device and an example mobile computing device, for use in illustrative embodiments of the present disclosure.
- DETAILED DESCRIPTION [0079] The present disclosure is based, at least in part, on the demonstration that the ER status of a cancer in a subject can be determined by detecting and quantifying the presence of histone modifications and/or DNA methylation at one or more genomic loci in cell-free DNA (cfDNA) from a liquid biopsy sample, e.g., a plasma sample obtained or derived from the subject.
- cfDNA cell-free DNA
- the present disclosure also encompasses methods where chromatin accessibility and/or binding of one or more transcription factors are detected at the one or more genomic loci instead of (or in addition to) histone modifications and/or DNA methylation.
- the present disclosure is also based, at least in part, on the demonstration that genomic loci that are differentially modified based on different types of histone modifications (e.g., histone methylation marks such as H3K4me3 and histone acetylation marks such as H3K27ac) and/or DNA methylation can be combined into multimodal classifiers to determine ER status.
- histone methylation marks such as H3K4me3
- histone acetylation marks such as H3K27ac
- DNA methylation can be combined into multimodal classifiers to determine ER status.
- Estrogens are steroidal hormones that function as the primary female sex hormone. There are three major forms of estrogen, namely estrone (E1), estradiol (E2) and estriol (E3). Estradiol (E2) is the predominant estrogen in nonpregnant females, while estrone (E1) and estriol (E3) are primarily produced during pregnancy and following the onset of menopause, respectively. All estrogens are produced from androgens through actions of enzymes such as aromatase. Follicle-stimulating hormone and luteinizing hormone stimulate the synthesis of estrogen in the ovaries.
- estrogens are also produced in smaller amounts by other tissues such as the liver, adrenal glands, and mammary gland.
- PR progesterone receptor
- ER ⁇ and ER ⁇ are members of the nuclear receptor superfamily of transcription factors that are characterized by highly conserved DNA- and ligand-binding domains (Wang et al., J Hematol Oncol (2017) 10:168).
- the DNA binding domain which is extremely well conserved between ER ⁇ and ER ⁇ (97% homology), contains two functionally distinct zinc finger motifs that are responsible for specific DNA binding, as well as mediating receptor dimerization (Hewitt and Korach, Endocr Rev (2016) 39(5):664-675).
- the unliganded ER has been shown to be present in a cytosolic complex with hsp90 and associated proteins, with ligand binding allowing dissociation from the hsp90 complex, receptor dimerization, nuclear localization and binding to estrogen response elements (EREs) in promoters of estrogen- regulated genes (Pratt and Toft, Endocr Rev (1997) 18:306-360).
- EEEs estrogen response elements
- Genome-wide chromatin immunoprecipitation studies have confirmed that the majority of ER-binding sites in estrogen responsive genes conform well to this consensus sequence (Welboren et al., EMBO J (2009) 28:1418-1428).
- a subject has one or more biomarkers and/or risk factors for cancer, e.g., ER-positive cancer, e.g., ER-positive breast cancer, etc.
- a human subject is identified as in need of ER status screening based on an initial cancer diagnosis, e.g., a breast cancer, etc. diagnosis.
- a human subject is a subject
- a sample from a subject e.g., a human can be obtained from a liquid biopsy.
- a sample and/or reference is obtained from serum, plasma, or urine.
- the sample is serum.
- a sample comprises circulating tumor DNA (ctDNA).
- a sample is derived from about 1 mL of blood obtained from the subject.
- a sample is derived from about 0.5-5 mL of blood obtained from the subject, e.g., about 0.5 to about 2 mL, about 0.5 to 1.75 mL, about 0.5 to 1.5 mL, about 0.75 to 1.25 mL, about 0.9 to 1.1 mL, about 1 mL, about 2 mL, about 3 mL, about 4 mL, or about 5 mL of blood.
- a sample is a sample of cell-free DNA (cfDNA).
- cfDNA is typically found in human biofluids (e.g., plasma, serum, or urine) in short, double-stranded fragments.
- cfDNA Circulating tumor DNA
- ctDNA Circulating tumor DNA
- ctDNA can be present in human biofluids bound to leukocytes and erythrocytes or not bound to leukocytes and erythrocytes.
- Various tests for detection of tumor-derived ctDNA are based on detection of genetic or epigenetic modifications that are characteristic of cancer (e.g., of a relevant cancer).
- ctDNA comprises less than 30%, less than 20%, or less than 10% of the cfDNA in the liquid biopsy sample obtained from the subject, e.g., less than 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2% or less than 1% of the cfDNA in the sample.
- the percentage of ctDNA in the liquid biopsy sample is assessed using ichorCNA which estimates the percentage of ctDNA in a sample probabilistically (see Adalsteinsson et al., Nat Commun (2017) 8(1):1324 the entire contents of which are incorporated herein by reference).
- a method comprises isolating DNA (e.g., cfDNA) from a liquid biopsy sample (e.g., from 1, 2, 3, 4, or 5 mL of a liquid biopsy sample).
- a liquid biopsy sample e.g., from 1, 2, 3, 4, or 5 mL of a liquid biopsy sample.
- nucleic acids can be isolated using, without limitation, standard DNA purification techniques, by direct gene capture (e.g., by clarification of a sample to remove assay-inhibiting agents and capturing a target nucleic acid, if present, from the clarified sample with a capture agent to produce a capture complex and isolating the capture complex to recover the target nucleic acid).
- direct gene capture e.g., by clarification of a sample to remove assay-inhibiting agents and capturing a target nucleic acid, if present, from the clarified sample with a capture agent to produce a capture complex and isolating the capture complex to recover the target nucleic acid.
- Samples include materials prepared by processes including, without limitation, steps such as concentration, dilution, adjustment of pH, removal of high abundance polypeptides (e.g., albumin, gamma globulin, and transferrin, etc.), addition of preservatives, addition of calibrants, addition of protease inhibitors, addition of denaturants, desalting, concentration and/or extraction of sample nucleic acids, and/or amplification of sample nucleic acids (e.g., by PCR or other nucleic acid amplification techniques).
- steps such as concentration, dilution, adjustment of pH, removal of high abundance polypeptides (e.g., albumin, gamma globulin, and transferrin, etc.), addition of preservatives, addition of calibrants, addition of protease inhibitors, addition of denaturants, desalting, concentration and/or extraction of sample nucleic acids, and/or amplification of sample nucleic acids (e.g., by PCR or other nucleic
- Separation and purification in the present disclosure may include any procedure known in the art, such as capillary electrophoresis (e.g., in capillary or on-chip) or chromatography (e.g., in capillary, column or on a chip).
- Electrophoresis is a method that can be
- Histone methylation is understood to increase or decrease expression of associated coding sequences, depending on which histone residue is methylated. Histone methylation is an essential modification that can cause monomethylation (me1), dimethylation (me2), and trimethylation (me3) of several amino acids, thus directly affecting heterochromatin formation, gene imprinting, X chromosome inactivation, and gene transcriptional regulation.
- genomic locus can refer to, or be determined by or detected as, a comparative difference or change in modification status of one or more genomic loci between a first sample, condition, disease, or state and a second or reference sample, condition, disease, or state.
- a reference is a normalized sample.
- a reference is a measurement obtained from liquid biopsy samples obtained from a cohort of subjects who have previously been determined to have an ER-positive or ER-negative cancer, including, e.g., an ER-positive or ER-negative breast cancer.
- a reference is a non-contemporaneous sample from the same source, e.g., a prior sample from the same source, e.g., from the same subject.
- a reference for the accessibility status of one or more genomic loci can be the accessibility status of the one or more genomic loci (e.g., one or more differentially accessible genomic loci) in a sample (e.g., a sample from a subject), or a plurality of samples, known to represent a particular state (e.g., an ER-positive cancer or ER-negative cancer).
- differential modification or differential accessibility can refer to a differential (e.g., between a sample and a reference) with an absolute log2(fold-change) that is greater than or equal to 0.5, 1.0, 1.5, 2.0, 2.5, 3.0, 3.5, 4.0 or more, or any range in between, inclusive, e.g., as measured according to an assay provided herein.
- the log2(fold-change) values are based on ratios of ER-positive to ER-negative reads, i.e., positive log2(fold-change) values indicate that sequencing reads in a particular genomic locus are associated with an ER-positive status while a negative log2(fold-change) value indicates that sequencing reads in a particular genomic locus are associated with an ER-negative status.
- Enhancers are genomic loci that can be differentially modified or differentially accessible in and/or between conditions, diseases, and other states. Enhancers are cis-acting DNA regulatory regions that are thought to bind trans-acting proteins that contribute to expression patterns of associated genes.
- Chromatin ImmunoPrecipitation sequencing of histone modifications (e.g., acetylation) have identified millions of enhancers in mammalian genomes.
- the number of active enhancers in any given cell type is estimated to be in the tens of thousands.
- Certain transcription factors TFs
- master transcription factors associate with active enhancers with important impacts on gene expression and cell function.
- Certain such transcription factors preferentially associate with enhancers that regulate genes required for establishing cell identity and function, including enhancer domains known as “super-enhancers”.
- master TFs can participate in inter-connected auto-regulatory circuitries or “cliques” that are self-reinforcing, show marked cell selectivity, and function to maintain cell state and/or cell survival.
- Techniques for Detecting and Quantifying Histone Modifications and Transcription Factor Binding [0130] Various techniques of molecular biology are well known in the art and/or disclosed in the present application for detecting and quantifying histone modifications and/or transcription factor binding. In some embodiments, the methods, kits and systems of present disclosure involve the detection and quantification of histone modifications and/or transcription factor binding in samples, e.g., in liquid biopsy samples including cfDNA such as plasma samples including cfDNA. Chromatin ImmunoPrecipitation (ChIP) is one technique of
- ChIP-chip, ChIP-exo, ChIP Re-ChIP, and ChIPmentation are other alternative techniques that could be used. [0131] ChIP can involve various steps including one or more of fixation, sonication, immunoprecipitation, and analysis of the immunoprecipitated DNA. ChIP has become a very widely used tissue-based technique for determining the in vivo location of binding sites of various transcription factors and histones.
- ChIP helps to detect DNA-protein interactions that take place in living cells. More importantly, ChIP can be coupled to many commonly used molecular biology techniques such as PCR and real-time PCR, PCR with single-stranded conformational polymorphism, Southern blot analysis, Western blot analysis, cloning, and microarray. The resulting versatility has increased the potential of this technique. [0132] ChIP of tissue samples usually involves cross-linking of the chromatin-bound proteins by formaldehyde, followed by sonication or nuclease treatment to obtain small DNA fragments. Immunoprecipitation can be then carried out using specific antibodies to the DNA- binding protein of interest.
- the DNA can be then released from the proteins and analyzed using various methods. ChIP has also been used to study RNA-protein interactions. X-ChIP methods utilize fixed chromatin fragmented by sonication, while the N-ChIP methods utilize native chromatin, which can be unfixed and nuclease digested. [0133]
- the first step of the technique can be the cross-linking of DNA and proteins. Formaldehyde is one of the most used cross-linking agents.
- formaldehyde can be the ease of reversibility of the cross-links and its ability to form bonds that span approximately 2 angstroms. This means that formaldehyde can bind molecules in close association with each other.
- Harvested chromatin can be sonicated in one or more sonication cycles. DNA can be typically broken into to 100–500 bp fragments to pinpoint the location of the DNA sequence of interest.
- An alternative to sonication can be nuclease digestion of the chromatin, e.g., in N- ChIP methods. Purification of chromatin can be achieved using a cesium chloride (CsCl) gradient centrifugation.
- Chromatin can be enriched for a particular histone modification using an agent that binds the histone modification (e.g., immunoprecipitating using one or more antibodies that bind a target epitope).
- an antibody used in ChIP can selectively bind a particular transcription factor or one or more particular histone modifications, such as one or more particular histone acetylation modifications or histone methylation modifications.
- an antibody used to bind a target epitope can be a “pan” antibody (e.g., a pan- acetylation antibody, a pan-methylation antibody, an antibody that binds a group of histone modifications associated with increased transcription activation, and/or an antibody that binds a group of histone modifications associated with increased transcription repression).
- the antibody against the protein of interest is allowed to bind to the protein-DNA complex, and the complex can be then precipitated.
- Immunosorbants commonly used to separate the antigen-antibody complex from the lysate include salmon sperm DNA-protein A-Sepharose®, protein G, magnetic beads, and other engineered immunoprecipitation systems known to those of skill in the art.
- Immunoprecipitated DNA can be eluted. Once the DNA of interest is isolated, many detection and quantification methods can be used to study the isolated gene fragments. Commonly utilized methods include PCR, real-time PCR, slot blot hybridization, microarray techniques, and deep or next-generation sequencing. ChIP-seq combines chromatin immunoprecipitation (ChIP) with massively parallel DNA sequencing to identify the binding sites of DNA-associated proteins.
- ChIP chromatin immunoprecipitation
- ChIP-seq can be used to map DNA-binding proteins, e.g., transcription factor binding sites and histone modifications in a genome-wide manner.
- Cell-free Chromatin ImmunoPrecipitation sequencing involves applying ChIP-seq to samples that include cell-free DNA, e.g., liquid biopsy samples including cfDNA such as plasma samples including cfDNA (e.g., see Sadeh et al., Nat Biotechnol (2021) 39: 586–598 and Jang et al., Life Sci Alliance (2023) 6(12):e202302003 the entire contents of each of which are incorporated herein by reference).
- cfChIP-seq uses
- exemplary antibodies that bind H3K4me3 include PA5-27029 (available from Thermo Fisher Scientific in Waltham, MA) and C15410003 (available from Diagenode in Denville, NJ) and exemplary antibodies that bind H3K27ac include ab21623 or ab4729 (both available from Abcam in Cambridge, UK) and C15210016 (available from Diagenode in Denville, NJ).
- the antibodies or antibody fragments can be covalently coupled to beads, e.g., epoxy beads.
- the antibodies or antibody fragments can be non-covalently coupled to beads, e.g., Protein A or Protein G beads such as Dynabeads® Protein A or Dynabeads® Protein G beads.
- a cfDNA library is then typically prepared from the captured cfDNA.
- Library preparation can be done on-bead or after releasing the captured cfDNA by digestion of bound histones, e.g., using proteinase K.
- the cfDNA library is then sequenced to generate reads of captured cfDNA sequences, e.g., by next-generation sequencing (NGS) as is known in the art.
- NGS next-generation sequencing
- the reads are then analyzed, e.g., aligned and counted using standard bioinformatic techniques as is known in the art.
- a cfChIP-seq bioinformatic pipeline can include, e.g., alignment of sequence reads to a reference genome with BWA or Bowtie2.
- Aligned reads can be used to call and quantify peaks as compared to a reference.
- histone modifications at a given genomic loci can be quantified using sequencing data.
- histone modifications can be quantified by counting the number of sequence reads that fall within a genomic loci (e.g., have at least one nucleotide overlapping with a genomic loci).
- non-uniquely mapped and/or redundant sequence reads are discarded prior to quantifying histone modifications.
- sequence reads that fall within high noise regions of the genome are ignored.
- sequence reads are adjusted on the basis of sequencing depth prior to counting.
- Adjusting on the basis of sequencing depth can include, e.g., quantile normalizing sequence reads to a common reference distribution.
- sequence reads are adjusted on the basis of ChIP quality prior to counting.
- sequence reads are normalized relative to aggregate counts across a set of regions (e.g., 1,000, 2,000,
- CUT&Tag involves antibody-based binding of a target protein, e.g., transcription factor or histone modification of interest, where antibody incubation is directly followed by the shearing of the chromatin and library preparation (see Kaya-Okur et al., Nat Comm (2019) 10:1930).
- a target protein e.g., transcription factor or histone modification of interest
- a method described herein comprises attaching (e.g., ligating) DNA adapters to cfDNA.
- DNA adapters can be attached prior to, during, or after enrichment for a histone modification.
- a method comprises amplifying cfDNA after attaching DNA adapters.
- the methods, kits and systems of the present disclosure involve the detection and quantification of chromatin accessibility in samples, e.g., in liquid biopsy samples including cfDNA such as plasma samples including cfDNA.
- ATAC-seq Assay of Transpose Accessible Chromatin sequencing
- NOMe-seq Nucleosome Occupancy and Methylome sequencing
- FAIRE-seq Formmaldehyde-Assisted Isolation of Regulatory Elements sequencing
- MNase-seq Merococcal Nuclease digestion with sequencing
- DNase hypersensitivity assays are exemplary techniques of molecular biology useful in detecting and quantifying chromatin accessibility in samples. Sono-Seq is another alternative method that could be used (see Auerbach et al., Proc Natl Acad USA (2009) 106(35):14926-14931).
- FAIRE-seq is a method in which nucleosome-depleted regions of DNA (NDRs) are isolated from chromatin.
- a typical FAIRE-seq assay can include a first step in which cells are fixed using formaldehyde so that histones are crosslinked to interacting DNA.
- Crosslinked chromatin can then be sheared by sonication that generates protein-free DNA and protein- crosslinked DNA fragments.
- Protein-free DNA can be isolated using a phenol–chloroform extraction: DNA crosslinked with protein stays in organic phase, while protein-free DNA stays in aqueous phase. Highly crosslinked DNA remains in the organic phase and the non-crosslinked DNA is pulled to the aqueous phase. Non-crosslinked DNA from the aqueous phase can then be amplified and sequenced. Reads enriched in the sequencing pool tend to have lower nucleosome and transcription factor binding and are therefore inferred to come from accessible regions.
- NOMe-seq is a method to identify nucleosome-depleted regions of DNA (NDRs) with M.CviPI methyltransferase that methylates cytosine in GpC dinucleotides not protected by
- a typical NOMe-seq protocol can include a step in which samples are treated with M.CviPI and S-adenosylhomocysteine (SAM) to methylate accessible GpC sites.
- SAM S-adenosylhomocysteine
- DNA is treated with bisulfite, which converts unmethylated cytosine to uracil using sodium bisulfite, while methylated cytosine is unaffected.
- a library is generated using adapters and sequenced. Accessible chromatin is expected to have high levels of GpC m but low levels of C m pG. Therefore, NOMe-seq identifies NDRs using the two separate methylation analyses that serve as independent (but opposite) measures, providing matched chromatin designations for each regulatory element.
- ATAC-seq uses hyperactive Tn5 transposase that preferentially cuts accessible chromatin regions and simultaneously inserts adapters to the fragmented region (Buenrostro et al., Nat Methods (2013) 10(12):1213-1218 the entirety of which is incorporated herein by reference).
- a typical ATAC-seq assay can include a first step in which samples are incubated with Tn5 transposase. DNA can then be isolated and purified. DNA fragmented and tagged by Tn5 transposase can be purified and then amplified to generate a library and sequenced for analysis.
- kits and systems of the present disclosure involve the detection and quantification of chromatin accessibility in samples, e.g., in liquid biopsy samples including cfDNA such as plasma samples including cfDNA.
- Bisulfite sequencing (BS-Seq), Whole Genome Bisulfite Sequencing (WGBS), Methylated DNA ImmunoPrecipitation sequencing (MeDIP-seq), or Methyl-CpG-Binding Domain sequencing (MBD-seq) are exemplary techniques of molecular biology useful in detecting and quantifying chromatin accessibility in samples.
- Reduced representation bisulfite sequencing (RRBS) is another alternative method that could be used (see Meissner et al., Nucleic Acids Res (2005) 33(18):5868-5877).
- Illumina Infinium arrays could also be used to detect and quantify DNA methylation.
- DNA methylation typically refers to the methylation of the 5’ position of cytosine (mC) by DNA methyltransferases (DNMT). It is a major epigenetic modification in humans and many other species. In mammals, most DNA methylations occur within the context of CpG dinucleotides. DNA methylation is thought to be a repressive chromatin modification. Aberrant methylation can lead to many diseases including cancers (Robertson, Nat Rev Genet (2005) 6:597–610 and Bergman and Cedar, Nat Struct Mol Biol (2013) 20:274–281).
- BS-Seq Bisulfite sequencing
- WGBS Whole-Genome Bisulfite Sequencing
- genomic DNA is treated with sodium bisulfite and then sequenced, providing single-base resolution of methylated cytosines in the genome.
- unmethylated cytosines are deaminated to uracil which, upon sequencing, are converted to thymidine.
- methylated cytosines resist deamination and are read as cytosines. The location of the methylated cytosines can then be determined by comparing treated and untreated sequences.
- an agent that binds methylated DNA is attached (e.g., via a covalent or noncovalent bond) to a physical support (e.g., a bead, a magnetic bead, an agarose bead, or a magnetic epoxy bead), wherein the attaching can be prior to, during, or after incubation with a sample.
- a physical support e.g., a bead, a magnetic bead, an agarose bead, or a magnetic epoxy bead
- ER-negative cancer are provided in Table 1 which shows the chromosomal coordinates of each genomic locus and its observed log2(fold-change) (ER-positive/ER-negative).
- the genomic loci are sorted based on their chromosomal coordinates which are based on human genome build hg19.
- a person of skill in the art will recognize that the methods disclosed herein do not require that every genomic locus listed in Table 1 be assessed for H3K4me3 modification. Instead, a subset of loci may be assessed for H3K4me3 modification.
- Subsets of the genomic loci of Table 1 can be selected (e.g., for use in determining ER status) based on various performance criteria, e.g., to select genomic loci that demonstrate differential modification with a particular level of statistical significance and/or a particular threshold of differential between relevant states (e.g., a measured log2(fold-change)). Subsets of the genomic loci may also be selected based on an algorithm, e.g., during the process of obtaining a classifier. Those of skill in the art will
- the present disclosure particularly includes, among other things, subsets of the genomic loci of Table 1, which have an absolute log2(fold-change) of 6.0 or higher, 5.5 or higher, 5.0 or higher, 4.5 or higher, 4.0 or higher, 3.5 or higher, 3.0 or higher, 2.5 or higher, 2.0 or higher, 1.9 or higher, 1.8 or higher, 1.7 or higher, 1.6 or higher, 1.5 or higher, 1.4 or higher, 1.3 or higher, 1.2 or higher, 1.1 or higher, 1.0 or higher, 0.9 or higher, 0.8 or higher, 0.7 or higher, 0.6 or higher, or 0.5 or higher.
- the present disclosure also includes subsets of the genomic loci of Table 1, which have an absolute log2(fold-change) of 6.0 or higher, 5.5 to less than 6.0, 5.0 to less than 5.5, 4.5 to less than 5.0, 4.0 to less than 4.5, 3.8 to less than 4.0, 3.6 to less than 3.8, 3.4 to less than 3.6, 3.2 to less than 3.4, 3.0 to less than 3.2, 2.8 to less than 3.0, 2.6 to less than 2.8, 2.4 to less than 2.6, 2.2 to less than 2.4, 2.0 to less than 2.2, 1.8 to less than 2.0, 1.6 to less than 1.8, 1.4 to less than 1.6, 1.2 to less than 1.4, 1.0 to less than 1.2, 0.8 to less than 1.0, or 0.6 to less than 0.8.
- a sample or subject from which the sample is obtained or derived is determined to have a particular ER status (e.g., ER-positive) if at least 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 150, 200, 250, 300, 350, 400, 450, 500, 750, 1000, 1500, 2000, 2500, or 3000 loci identified in Table 1 (or any subset thereof) are differentially H3K4me3 modified as compared to a reference (e.g., a sample from an ER-negative or healthy subject).
- a reference e.g., a sample from an ER-negative or healthy subject.
- a sample or subject from which the sample is derived is determined to have a particular ER status (e.g., ER-positive) if at least one (e.g., at least 2, at least 3, at least 4, at least 5, at least 6, at least 7, at least 8, at least 9, or at least 10) of the top 3, 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 150, 200, 250, 300, 350, 400, 450, 500, 750, 1000, 1500, 2000, 2500, or 3000 loci identified in Table 2 are H3K27ac modified as compared to a reference (e.g., a sample from an ER-negative or healthy subject) (wherein, e.g., the “top” 10 loci refers to the loci with 10 highest absolute log2(fold-change) in Table 2).
- a reference e.g., a sample from an ER-negative or healthy subject
- a sample or subject from which the sample is derived is determined to have a particular ER status (e.g., ER-positive) if at least one of the top 10 loci (e.g., at least 1, at least 2, at least 3, at least 4, at least 5, at least 6, at least 7, at least 8, at least 9, or 10) identified in Table 2 and at least 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95,
- ER-positive e.g., ER-positive
- a sample or subject from which the sample is derived is determined to have a particular ER status (e.g., ER-positive) if at least one of the top 25 loci identified in Table 2 (e.g., at least 1, at least 2, at least 3, at least 4, at least 5, at least 6, at least 7, at least 8, at least 9, or at least 10, at least 15, at least 20, or 25) and at least 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 150, 200, 250, 300, 350, 400, 450, 500, 750, 1000, 1500, 2000, 2500, or 3000 loci identified in Table 2 (or any subset thereof) in total are H3K27ac modified as compared to a reference (e.g., a sample from an ER- negative or healthy subject).
- a reference e.g., a sample from an ER- negative or healthy subject.
- a sample or subject from which the sample is derived is determined to have a particular ER status (e.g., ER-positive) if at least one of the top 50 loci (e.g., at least 1, at least 2, at least 3, at least 4, at least 5, at least 6, at least 7, at least 8, at least 9, or at least 10, at least 15, at least 20, or at least 25, at least 30, at least 35, at least 40, at least 45, or 50) identified in Table 2 and at least 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 150, 200, 250, 300, 350, 400, 450, 500, 750, 1000, 1500, 2000, 2500, or 3000 loci identified in Table 2 (or any subset thereof) in total are H3K27ac modified as compared to a reference (e.g., a sample from an ER-negative or healthy subject).
- a reference e.g., a sample from an ER-negative or healthy subject.
- a sample or subject from which the sample is derived is determined to have a particular ER status (e.g., ER-positive) if at least five of the top 25 loci identified in Table 2 and at least 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 150, 200, 250, 300, 350, 400, 450, 500, 750, 1000, 1500, 2000, 2500, or 3000 loci identified in Table 2 (or any subset thereof) in total are H3K27ac modified as compared to a reference (e.g., a sample from an ER-negative or healthy subject).
- a reference e.g., a sample from an ER-negative or healthy subject.
- a sample or subject from which the sample is derived is determined to have a particular ER status (e.g., ER-positive) if at least five of the top 50 loci identified in Table 2 and at least 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 150, 200, 250, 300, 350, 400, 450, 500, 750, 1000, 1500, 2000, 2500, or 3000 loci identified in Table 2 (or any subset thereof) in total are H3K27ac modified as compared to a reference (e.g., a sample from an ER-negative or healthy subject).
- a reference e.g., a sample from an ER-negative or healthy subject.
- differentially H3K27ac modified refers to an acetylation status characterized by an increase or decrease in a value measuring acetylation (e.g., of read counts and/or normalized read counts for a given genomic locus), and/or a mean, median and/or
- 12366150v1 Attorney Docket No.2014191-0027 mode thereof, and/or a log thereof (e.g., log base 2 (log2)), of at least 1%, 2%, 3%, 4%, 5%, 6%, 7%, 8%, 9%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 75%, 100%, 2-fold, 3-fold, 4- fold, 5-fold, 6-fold, 7-fold, 8-fold, 9-fold, 10-fold, 15-fold, 20-fold, 25-fold, 30-fold, 35-fold, 40- fold, 45-fold, 50-fold, or greater, or any range in between, inclusive, such as 1% to 50%, 50% to 2-fold, 25% to 50-fold, 25% to 30-fold, 25% to 20-fold, 25% to 16-fold, 30% to 16-fold, 50% to 16-fold, 70% to 16-fold, 2-fold to 16-fold, 2.2-fold to 16-fold, 2.6-fold to 16-fold, 3-fold to 16- fold, 3.4
- an increase or decrease in a value measuring acetylation can be, or is expressed as, a log2(fold-change), e.g., a log2(fold-change) of at least 1%, 2%, 3%, 4%, 5%, 6%, 7%, 8%, 9%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 75%, 100%, 2-fold, 3-fold, 4-fold, 5-fold, 6-fold, 7-fold, 8-fold, 9-fold, 10-fold, 15-fold, 20-fold, or greater, or any range in between, inclusive, such as an increase or decrease of 0.1-fold to 10- fold, 0.2-fold to 5-fold, 0.2-fold to 4.0-fold, 0.4-4.0-fold, 0.4-fold to 4.0-fold, 0.6-fold to 4.0- fold, 0.8-fold to 4.0-fold, 1.0-fold to 4.0-fold.1.2-fold to 4.0-fold.1.4-fold to 4.0-fold, 1.6
- Genomic loci demonstrating differential DNA methylation in ER-positive vs. ER- negative cancer are provided in Table 3, which shows the chromosomal coordinates of each genomic locus and its observed log2(fold-change) (ER-positive/ER-negative). The genomic loci are sorted based on their chromosomal coordinates which are based on human genome build hg19.
- Table 3 shows the chromosomal coordinates of each genomic locus and its observed log2(fold-change) (ER-positive/ER-negative). The genomic loci are sorted based on their chromosomal coordinates which are based on human genome build hg19.
- a person of skill in the art will recognize that the methods disclosed herein do not require that every genomic locus listed in Table 3 be assessed for DNA methylation. Instead, a subset of loci may be assessed for DNA methylation. Subsets of the genomic loci of Table 3 can be selected (e.g., for use in determining ER status) based on various performance criteria,
- genomic loci that demonstrate differential modification with a particular level of statistical significance and/or a particular threshold of differential between relevant states (e.g., a measured log2(fold-change)).
- Subsets of the genomic loci may also be selected based on an algorithm, e.g., during the process of obtaining a classifier.
- Those of skill in the art will appreciate that such subsets of loci of Table 3, and loci included in such subsets, are together, individually, and/or in randomly selected subsets, at least as informative (e.g., as statistically significant and/or reliable) for uses disclosed herein, e.g., for determining ER status.
- the present disclosure particularly includes, among other things, subsets of the genomic loci of Table 3, which have an absolute log2(fold-change) of 6.0 or higher, 5.5 or higher, 5.0 or higher, 4.5 or higher, 4.0 or higher, 3.5 or higher, 3.0 or higher, 2.5 or higher, 2.0 or higher, 1.9 or higher, 1.8 or higher, 1.7 or higher, 1.6 or higher, 1.5 or higher, 1.4 or higher, 1.3 or higher, 1.2 or higher, 1.1 or higher, 1.0 or higher, 0.9 or higher, 0.8 or higher, 0.7 or higher, 0.6 or higher, or 0.5 or higher.
- the present disclosure also includes subsets of the genomic loci of Table 3, which have an absolute log2(fold-change) of 6.0 or higher, 5.5 to less than 6.0, 5.0 to less than 5.5, 4.5 to less than 5.0, 4.0 to less than 4.5, 3.8 to less than 4.0, 3.6 to less than 3.8, 3.4 to less than 3.6, 3.2 to less than 3.4, 3.0 to less than 3.2, 2.8 to less than 3.0, 2.6 to less than 2.8, 2.4 to less than 2.6, 2.2 to less than 2.4, 2.0 to less than 2.2, 1.8 to less than 2.0, 1.6 to less than 1.8, 1.4 to less than 1.6, 1.2 to less than 1.4, 1.0 to less than 1.2, 0.8 to less than 1.0, or 0.6 to less than 0.8.
- a sample or subject from which the sample is obtained or derived is determined to have a particular ER status (e.g., ER-positive) if at least 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 150, 200, 250, 300, 350, 400, 450, 500, 750, 1000, 1500, 2000, 2500, or 3000 loci identified in Table 3 (or any subset thereof) are differentially DNA methylated as compared to a reference (e.g., a sample from an ER-negative or healthy subject).
- a reference e.g., a sample from an ER-negative or healthy subject.
- a subject from which the sample is obtained or derived is determined to have a particular ER status (e.g., ER-positive) if at least a number of loci identified in a Table 3 (or any subset thereof) having a lower bound selected from 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 150, 200, 250, or 300 and an upper bound selected from 10, 15, 20, 25, 50, 75, 100, 150, 200, 250, 300, 350, 400, 450, 500, 750, 1000, 1500, 2000, 2500, or 3000 is found to be
- a sample or subject from which the sample is obtained or derived is determined to have a particular ER status (e.g., ER-positive) if at least a percent of loci identified in Table 3 having a lower bound selected from 0.1%, 0.2%, 0.3%, 0.4%, 0.5%, 1%, 2%, 3%, 4%, 5%, or 10%, and an upper bound selected from 1%, 2%, 3%, 4%, 5%, 10%, 20%, 30%, 40%, 50%, 75%, or 100% is found to be differentially DNA methylated as compared to a reference (e.g., a sample from an ER-negative or healthy subject).
- a reference e.g., a sample from an ER-negative or healthy subject.
- a sample or subject from which the sample is derived is determined to have a particular ER status (e.g., ER-positive) if at least one (e.g., at least 2, at least 3, at least 4, at least 5, at least 6, at least 7, at least 8, at least 9, or at least 10) of the top 3, 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 150, 200, 250, 300, 350, 400, 450, 500, 750, 1000, 1500, 2000, 2500, or 3000 loci identified in Table 3 are differentially DNA methylated as compared to a reference (e.g., a sample from an ER-negative or healthy subject) (wherein, e.g., the “top” 10 loci refers to the loci with 10 highest absolute log2(fold- change) in Table 3).
- a reference e.g., a sample from an ER-negative or healthy subject
- a subject from which the sample is obtained or derived is determined to have a particular ER status (e.g., ER-positive) if at least one of the top 10 loci identified in Table 3 is differentially DNA methylated as compared to a reference (e.g., a sample from an ER-negative or healthy subject).
- a subject from which the sample is obtained or derived is determined to have a particular ER status (e.g., ER-positive) if at least one of the top 25 loci identified in Table 3 is differentially DNA methylated as compared to a reference (e.g., a sample from an ER-negative or healthy subject).
- a reference e.g., a sample from an ER-negative or healthy subject.
- a reference e.g., a sample from an ER-negative or healthy subject.
- 12366150v1 Attorney Docket No.2014191-0027 subject from which the sample is obtained or derived, is determined to have a particular ER status (e.g., ER-positive) if at least one of the top 50 loci identified in Table 3 is differentially DNA methylated as compared to a reference (e.g., a sample from an ER-negative or healthy subject).
- a subject from which the sample is obtained or derived is determined to have a particular ER status (e.g., ER-positive) if at least five of the top 10 loci identified in Table 3 are differentially DNA methylated as compared to a reference (e.g., a sample from an ER-negative or healthy subject).
- a subject from which the sample is obtained or derived is determined to have a particular ER status (e.g., ER-positive) if at least five of the top 25 loci identified in Table 3 are differentially DNA methylated as compared to a reference (e.g., a sample from an ER-negative or healthy subject).
- a subject from which the sample is obtained or derived is determined to have a particular ER status (e.g., ER-positive) if at least five of the top 50 loci identified in Table 3 are differentially DNA methylated as compared to a reference (e.g., a sample from an ER-negative or healthy subject).
- a sample or subject from which the sample is derived is determined to have a particular ER status (e.g., ER-positive) if at least one of the top 10 loci (e.g., at least 1, at least 2, at least 3, at least 4, at least 5, at least 6, at least 7, at least 8, at least 9, or 10) identified in Table 3 and at least 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 150, 200, 250, 300, 350, 400, 450, 500, 750, 1000, 1500, 2000, 2500, or 3000 loci identified in Table 3 (or any subset thereof) in total are differentially DNA methylated as compared to a reference (e.g., a sample from an ER-negative or healthy subject).
- a reference e.g., a sample from an ER-negative or healthy subject.
- a sample or subject from which the sample is derived is determined to have a particular ER status (e.g., ER-positive) if at least one of the top 25 loci identified in Table 3 (e.g., at least 1, at least 2, at least 3, at least 4, at least 5, at least 6, at least 7, at least 8, at least 9, or at least 10, at least 15, at least 20, or 25) and at least 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 150, 200, 250, 300, 350, 400, 450, 500, 750, 1000, 1500, 2000, 2500, or 3000 loci identified in Table 3 (or any subset thereof) in total are differentially DNA methylated as compared to a reference (e.g., a sample from an ER-negative or healthy subject).
- a reference e.g., a sample from an ER-negative or healthy subject.
- a sample or subject from which the sample is derived is determined to have a particular ER status (e.g., ER- positive) if at least one of the top 50 loci (e.g., at least 1, at least 2, at least 3, at least 4, at least 5, at least 6, at least 7, at least 8, at least 9, or at least 10, at least 15, at least 20, or at least 25, at
- Responsiveness can be measured quantitatively (e.g., as in the case of tumor size; as in the case of measurement of histone modification, chromatin accessibility, transcription factor binding, or DNA methylation at one or more genomic loci; or as in the calculation of clinical benefit (CBR)), or qualitatively (e.g., by measures such as “pathological complete response” (pCR), “clinical complete remission” (cCR), “clinical partial remission” (cPR), “clinical stable disease” (cSD), “clinical progressive disease” (cPD), or other qualitative criteria).
- CBR clinical benefit
- kits and systems can be used to detect the clinical efficacy of a course of therapy for cancer, e.g., breast, ovarian, or endometrial cancer.
- a course of therapy for cancer e.g., breast, ovarian, or endometrial cancer.
- methods and/or compositions of the present disclosure could be used to determine the presence, absence, or ER status of a cancer in a subject over the course of treatment.
- compositions of the present disclosure could be used in conjunction with, or confirmed by, other means of determining the presence, absence, or ER status of a cancer including, for example measurements of tumor size or character by techniques such as CT, PET, mammogram, ultrasound, palpation, histology, caliper measurement after biopsy or surgical resection, or by various qualitative, quantitative, or semi quantitative scoring systems including without limitation based on IHC or ISH testing, residual cancer burden (Symmans et al., J Clin Oncol (2007) 25:4414-4422, incorporated by reference herein in its entirety) or Miller-Payne score (Ogston et al., Breast (2003) 12:320-327, incorporated by reference herein in its entirety) in a qualitative fashion like “pathological complete response” (pCR), “clinical complete remission” (cCR), “clinical partial remission” (cPR), “clinical stable disease” (cSD), or “clinical progressive disease”
- treatment efficacy can be monitored, e.g., by using a method described herein to determine a decrease or increase in disease state signal, which can be useful, e.g., for determining whether an administered therapy is effective and/or whether a change in therapy should be made.
- a cancer has gone into remission for a subject (e.g., the subject has minimal residual disease).
- methods, kits, and systems described herein can be useful, e.g., for detecting reoccurrence of
- methods, kits and systems for ER status determination provided herein can inform treatment and/or payment (e.g., reimbursement for or reduction of cost of medical care, such as detecting or treatment) decisions and/or actions, e.g., by individuals, healthcare facilities, healthcare practitioners, health insurance providers, governmental bodies, or other parties interested in healthcare cost.
- payment e.g., reimbursement for or reduction of cost of medical care, such as detecting or treatment
- decisions and/or actions e.g., by individuals, healthcare facilities, healthcare practitioners, health insurance providers, governmental bodies, or other parties interested in healthcare cost.
- methods, kits and systems for ER status determination can inform decision making relating to whether health insurance providers reimburse a healthcare cost payer or recipient (or not), e.g., for (1) ER status determination itself (e.g., reimbursement for detecting otherwise unavailable, available only for periodic/regular detecting, or available only for temporally- and/or incidentally- motivated detecting); and/or for (2) treatment, including initiating, maintaining, and/or altering therapy, e.g., based on the determined ER status.
- ER status determination e.g., reimbursement for detecting otherwise unavailable, available only for periodic/regular detecting, or available only for temporally- and/or incidentally- motivated detecting
- treatment including initiating, maintaining, and/or altering therapy, e.g., based on the determined ER status.
- methods, kits and systems for ER status determination provided herein are used as the basis for, to contribute to, or support a determination as to whether a reimbursement or cost reduction will be provided to a healthcare cost payer or recipient.
- a party seeking reimbursement or cost reduction can provide results of ER status determination conducted in accordance with the present disclosure together with a request for such reimbursement or reduction of a healthcare cost.
- a party making a determination as to whether or not to provide a reimbursement or reduction of a healthcare cost will reach a determination based in whole or in part upon receipt and/or review of results of ER status determination conducted in accordance with the present disclosure.
- ER status determination using methods, kits and systems disclosed herein can be used in classifying subjects, samples, and/or tumors (e.g., breast cancer subjects, samples, and/or tumors).
- methods, kits and systems disclosed herein can be used to generate a set of subjects, samples, and/or tumors identified according to the present methods, kits and systems each classified as corresponding to a particular ER status, and optionally using two or more of such classified subjects, samples, and/or tumors to identify biomarkers that distinguish the classes (i.e., distinguish the subjects, samples, and/or tumors according to their class, e.g., according to their ER status).
- one or more samples obtained from a subject are analyzed by a method comprising enriching for cfDNA comprising a particular histone modification, wherein enriching is performed by a method that comprises incubating the sample with a reagent that specifically binds the histone modification being enriched for, and sequencing the enriched cfDNA.
- ChIP-seq for a histone modification (e.g., H3K4me3 and/or H3K27ac).
- Sequence reads e.g., ChIP-seq sequence reads
- BWA Burrows-Wheeler Aligner
- Non-uniquely mapping and redundant reads are optionally discarded.
- MACS v2.1.1.20140616 can be used for sequence (e.g., ChIP-seq) peak calling with a q-value (FDR) threshold of 0.01.
- Sequence (e.g., ChIP-seq) data quality can optionally be evaluated by any of one or more of a variety of measures, including total peak number, FRiP (fraction of reads in peak) score, number of high- confidence peaks (e.g., enriched > ten-fold over background), and percent of peak overlap with “blacklist” DHS peaks derived from the ENCODE project (Amemiya et al., Sci Rep (2019) 9(1):9354). If the sequence (e.g., ChIP-seq) data quality is below a particular threshold, the data may be discarded and the assay repeated.
- measures including total peak number, FRiP (fraction of reads in peak) score, number of high- confidence peaks (e.g., enriched > ten-fold over background), and percent of peak overlap with “blacklist” DHS peaks derived from the ENCODE project (Amemiya et al., Sci Rep (2019) 9(1):9354). If the sequence
- Sequence e.g., ChIP-seq
- selected genomic loci that are differentially modified as provided herein for the relevant histone modification Tables 1-2
- the number of reads overlapping the selected genomic loci for the relevant histone modification can be summed, e.g., in some embodiments all the genomic loci that are differentially modified with an absolute log2(fold-change) ⁇ 4.0 are selected.
- the average number of reads in the local background of each ChIP-seq peak is subtracted to improve signal to noise.
- a sequence read density for one or more histone modifications can be calculated by a method that comprises (1) summing background adjusted sequence counts at at one or more genomic loci and dividing the resulting sum by the total number of kilobases of the one or more genomic loci, or (2) for each genomic loci, determining the ratio of background adjusted fragment counts to the number of kilobases of the genomic loci, and then summing the ratios for each loci.
- a method comprises determining an ER-positive/ ER-negative ratio score, e.g., by a method that comprises (a) calculating an ER-positive sequence read
- an ER- positive sequence read density can be determined by a method that comprises calculating sequence read density using one or more genomic loci with an increased level of one or more epigenetic biomarkers in sample(s) obtained from one or more subjects with an ER -positive cancer as compared to one or more sample(s) obtained from subjects with an ER-negative cancer.
- an ER -negative sequence read density can be determined by a method that comprises calculating sequence read density using one or more genomic loci with an increased level of one or more epigenetic biomarkers in sample(s) obtained from one or more subjects with an ER-negative cancer as compared to one or more sample(s) obtained from subjects with an ER-positive cancer.
- an ER-positive/ER-negative ratio score is determined for H3K4me3 modifications.
- an ER-positive/ER-negative ratio score is determined for H3K27ac modifications.
- an ER-positive/ER- negative ratio score is determined for methylated DNA.
- an ER- positive/ER-negative ratio score is determined for H3K4me3 modifications and H3K27ac modifications, H3K4me3 and methylated DNA, or H3K27ac and methylated DNA. In some embodiments, an ER-positive/ER-negative ratio score is determined for each of H3K4me3 modifications, H3K27ac modifications, and methylated DNA. In some embodiments, two or more ER-positive/ER-negative ratio scores for different epigenetic biomarkers can be combined. In some embodiments, each ratio score can be combined using fitted values that have been determined using a logistic regression. [0218] The data can then be log2-transformed and quantile normalized to match the distribution of the data used to train a classifier.
- Normalized data can be used as input into a classifier that was trained using the same histone modification(s) and selected genomic loci. The classifier can then use inputted data to determine ER status of a subject’s cancer. It will be appreciated that this or similar approaches can be applied to assays of the present disclosure that quantify chromatin accessibility, transcription factor binding and/or DNA methylation.
- multiple epigenetic biomarkers e.g., one or more histone modifications, chromatin accessibility, binding of one or more transcription factors, and/or DNA methylation
- H3K4me3 and H3K27ac histone modifications are quantified in a single sample.
- kits and systems for ER status determination of the present disclosure are at least for in vitro use. Accordingly, all aspects and embodiments of the present disclosure can be performed and/or used at least in vitro.
- methods of the present disclosure can be implemented on and/or in conjunction with a computer program and computer system. In some embodiments, methods of the present disclosure can be implemented on and/or in conjunction with a non-transitory computer readable storage medium encoded with the computer program, wherein the program comprises instructions that when executed by one or more processors cause the one or more processors to perform operations to perform the method.
- a computer system comprises a database for storage of genomic locus modification status and/or accessibility status data. Such stored profiles can be accessed and used to perform comparisons of interest at a later point in time.
- exemplary program structures and computer systems described herein other, alternative program structures and computer systems will be readily apparent to the skilled artisan.
- Solutions can be formulated, e.g., using distilled water, physiological saline, or an isotonic solution containing glucose and other supplements such as D- sorbitol, D-mannose, D-mannitol, or sodium chloride as an aqueous solution for injection, optionally in combination with a suitable solubilizing agent, for example, an alcohol such as ethanol and/or a polyalcohol such as propylene glycol or polyethylene glycol, and/or a nonionic surfactant such as polysorbate 80TM or HCO-50, and the like.
- a suitable solubilizing agent for example, an alcohol such as ethanol and/or a polyalcohol such as propylene glycol or polyethylene glycol, and/or a nonionic surfactant such as polysorbate 80TM or HCO-50, and the like.
- Route of administration can be parenteral, for example, administration by injection.
- Administration by injection can be by intravenous injection, intramuscular injection, intraperitoneal injection, subcutaneous injection.
- Administration can be systemic or local.
- a composition described herein can be therapeutically delivered to a subject by way of local administration.
- local administration or “local delivery,” can refer to delivery that does not rely upon transport of the composition or therapeutic agent to its intended target tissue or site via the vascular system.
- the composition may be delivered by injection or implantation of the composition or therapeutic agent or by injection or implantation of a device containing the composition or therapeutic agent.
- subcutaneous administration can be accomplished by means of a device, such as a syringe, a prefilled syringe, an auto-injector (e.g., disposable or reusable), a pen injector, a patch injector, a wearable injector, an ambulatory syringe infusion pump with subcutaneous infusion sets, or other device for combining with a therapeutic agent for subcutaneous injection.
- a device such as a syringe, a prefilled syringe, an auto-injector (e.g., disposable or reusable), a pen injector, a patch injector, a wearable injector, an ambulatory syringe infusion pump with subcutaneous infusion sets, or other device for combining with a therapeutic agent for subcutaneous injection.
- An injection system of the present disclosure may employ a delivery pen as described in U.S. Pat. No.5,308,341.
- Pen devices most commonly used for self-delivery of insulin to patients with diabetes, are well known in the art. Such devices can include at least one injection needle, are typically pre-filled with one or more therapeutic unit doses of a solution that includes the therapeutic agent and are useful for rapidly delivering solution to a subject with as little pain as possible.
- One medication delivery pen includes a vial holder into which a vial of a therapeutic or other medication may be received.
- the pen may be an entirely mechanical device or it may be combined with electronic circuitry to accurately set and/or indicate the dosage of medication that is injected into the user. See, e.g., U.S. Pat.
- a composition can be formulated for storage at a temperature below 0°C (e.g., -20°C or -80°C).
- the composition can be formulated for storage for up to 2 years (e.g., one month, two months, three months, four months, five months, six months, seven months, eight months, nine months, 10 months, 11 months, 1 year, or 2 years) at 2-8°C (e.g., 4°C).
- compositions including certain therapeutic agents can be administered as a fixed dose, or in a milligram per kilogram (mg/kg) dose.
- an exemplary single dose of certain pharmaceutical compositions described herein can include certain therapeutic agents as described herein in an amount equal to, e.g., 0.001 to 1000 mg/kg, 1-1000 mg/kg, 1-100 mg/kg, 0.5-50 mg/kg, 0.1-100 mg/kg, 0.5-25 mg/kg, 1-20 mg/kg, and 1-10 mg/kg body weight.
- Exemplary dosages of a composition described herein include, without limitation, 0.1 mg/kg, 0.5 mg/kg, 1 mg/kg, 2 mg/kg, 4 mg/kg, 8 mg/kg, or 20 mg/kg. The present disclosure is not limited to such ranges or dosages.
- the present disclosure further includes methods of preparing pharmaceutical compositions of the present disclosure and kits including pharmaceutical compositions of the present disclosure.
- therapeutic agents of the present disclosure can be administered to a subject in a course of treatment that further includes administration of one or more additional therapeutic agents or therapies that are not therapeutic agents (e.g., surgery or radiation).
- Combination therapies of the present disclosure can include simultaneous exposure of a subject to therapeutic agents of two or more therapeutic regimens.
- a therapeutic agent as described herein can be administered together with (e.g., at the same time and/or in the same composition as) an additional agent or therapy.
- a therapeutic agent of the present disclosure can be administered separately from an additional therapeutic agent or therapy (e.g., at a different time and/or in a different composition than the additional therapeutic agent or therapy). Dosing regimens of a therapeutic agent and one or more additional therapeutic agents with which it is administered in combination can be coordinated or independently determined. In various embodiments, an additional therapeutic agent or therapy administered in combination with a therapeutic agent as described herein can be administered at the same time as therapeutic agent, on the same day as therapeutic agent, or in the same week as therapeutic agent. In various embodiments, an additional therapeutic agent or therapy administered in combination with a therapeutic agent as described herein can be administered such that administration of the
- the administration frequency and/or dosage of one or more additional therapeutic agents can be the same as, similar to, or different from the administration frequency of a therapeutic agent.
- the two or more regimens can be administered simultaneously; in some embodiments, such regimens can be administered sequentially (e.g., all “doses” of a first regimen are administered prior to administration of any doses of a second regimen); in some embodiments, such therapeutic agents are administered in overlapping dosing regimens.
- administration of a therapeutic agent can be to a subject having previously received, scheduled to receive, or in the course of a treatment regimen including an additional cancer therapy.
- Administration of a therapeutic agent can, in some instances, improve delivery or efficacy of another therapeutic agent or therapy with which it is administered in combination.
- therapeutic agent combination therapies can demonstrate synergy and/or greater-than-additive effects between a therapeutic agent and one or more additional therapeutic agents with which it is administered in combination.
- a therapeutic agent can be administered in any effective amount as determined independently or as determined by the joint action of therapeutic agent and any of one or more additional therapeutic agents or therapies administered.
- Administration of the therapeutic agent may, in some embodiments, reduce the therapeutically effective dosage, required dosage, or administered dosage of the additional therapeutic agent or therapy relative to a reference regimen for administration of additional therapeutic agent or therapy or therapy absent the therapeutic agent.
- a composition described herein can replace or augment other previously or currently administered therapy.
- administration of one or more additional therapeutic agents or therapies can cease or diminish, e.g., be administered at lower levels.
- Kits of the present disclosure can include, e.g., reagents such as buffers and/or antibodies useful in the detection and quantification of histone modifications.
- a kit of the present disclosure can include at least one antibody that selective binds a histone modification selected from H3K9ac, H3K14ac, H3K18ac, H3K23ac, H3K27ac, H3K4me1, H3K4me2, or H3K4me3, or pan acetylation.
- kits of the present disclosure can include at least one antibody that selective binds H3K4me3 modifications. In certain embodiments, a kit of the present disclosure can include at least one antibody that selective binds H3K27ac modifications.
- a kit of the present disclosure can include instructional materials disclosing or describing the use of the kit in a method of determining ER status and/or treatment disclosed herein.
- a kit of the present disclosure can include one or more therapeutic agents useful in the treatment of cancer, e.g., as disclosed herein, optionally in combination with instruction materials for treatment of cancer, e.g., breast cancer, ovarian cancer, or endometrial cancer based on ER status.
- a kit of the present disclosure comprises reagents for quantifying one or more histone modifications, chromatin accessibility, binding of one or more transcription factors, and/or DNA methylation at one or more genomic loci, wherein the one or more genomic loci are selected from Tabled 1-3.
- the kit comprises reagents for quantifying H3K4me3 for at least 5, 10, 20, 30, 40, or 50 genomic loci in Table 1.
- the kit comprises reagents for quantifying H3K27ac for at least 5, 10, 20, 30, 40, or 50 genomic loci in Table 2.
- the kit comprises one or more antibodies for use in ChIP-seq, optionally wherein the one or more antibodies specifically bind H3K4me3- or H3K27ac-modified histones.
- the kit comprises reagents for quantifying DNA methylation for at least 5, 10, 20, 30, 40, or 50 genomic loci in Table 3.
- the kit comprises one or more methyl-binding domains for use in MBD-seq.
- the kit comprises one or more antibodies that can bind methylated DNA (e.g., for use in MeDIP).
- the kit comprises reagents for isolation of cell-free DNA (cfDNA) from a liquid biopsy sample.
- the kit comprises reagents for isolation of cell-free DNA (cfDNA) from a liquid biopsy sample.
- the kit comprises reagents for
- the kit comprises reagents for sequencing. In some embodiments, the kit comprises instructions for determining if a subject has an ER-positive cancer.
- the present disclosure includes systems for detecting modification and/or accessibility of one or more genomic loci. In some embodiments, the present disclosure provides systems for quantifying one or more histone modifications, chromatin accessibility, binding of one or more transcription factors, and/or DNA methylation at one or more genomic loci. Systems of the present disclosure can include a sequencer configured to generate a sequencing dataset from a sample; and a non-transitory computer readable storage medium and/or a computer system.
- the non-transitory computer readable storage medium is encoded with a computer program, wherein the program comprises instructions that when executed by one or more processors cause the one or more processors to perform operations to perform a method of the present disclosure.
- the computer system comprises a memory and one or more processors coupled to the memory, wherein the one or more processors are configured to perform a method of the present disclosure.
- the sequencer is configured to generate a Whole Genome Sequencing (WGS) dataset from the sample.
- the system also includes a sample preparation device configured to prepare the sample for sequencing from a biological sample, optionally a liquid biopsy sample.
- the sample preparation device may include reagents for quantifying one or more histone modifications, chromatin accessibility, binding of one or more transcription factors, and/or DNA methylation at one or more genomic loci in cell-free DNA (cfDNA) from the biological sample, optionally the liquid biopsy sample.
- reagents for quantifying one or more histone modifications, chromatin accessibility, binding of one or more transcription factors, and/or DNA methylation at one or more genomic loci in cell-free DNA (cfDNA) from the biological sample, optionally the liquid biopsy sample.
- Systems of the present disclosure can include, e.g., reagents such as buffers and/or antibodies useful in the detection and quantification of histone modifications.
- a system of the present disclosure can include at least one antibody that selective binds a histone modification selected from H3K9ac, H3K14ac, H3K18ac, H3K23ac, H3K27ac, H3K4me1, H3K4me2, or H3K4me3, or pan acetylation.
- a system of the present disclosure can include at least one antibody that selective binds a histone modification selected from H3K9ac, H3K14ac, H3K18ac, H3K23ac, H3K27ac, H3K4me1, H3K4me2, or H3K4me3, or pan acetylation.
- a system of the present disclosure can include at least one antibody that selective binds H3K4me3 modifications.
- a system of the present disclosure can include at least one antibody that selective binds H3K27ac modifications.
- a system of the present disclosure can include instructional materials disclosing or describing the use of the system in a method of determining ER status and/or treatment disclosed herein.
- a system of the present disclosure comprises reagents for quantifying one or more histone modifications, chromatin accessibility, binding of one or more transcription factors, and/or DNA methylation at one or more genomic loci, wherein the one or more genomic loci are selected from Tabled 1-3.
- the system comprises reagents for quantifying H3K4me3 for at least 5, 10, 20, 30, 40, or 50 genomic loci in Table 1.
- the system comprises reagents for quantifying H3K27ac for at least 5, 10, 20, 30, 40, or 50 genomic loci in Table 2.
- the system comprises one or more antibodies for use in ChIP- seq, optionally wherein the one or more antibodies specifically bind H3K4me3- or H3K27ac- modified histones.
- the system comprises reagents for quantifying DNA methylation for at least 5, 10, 20, 30, 40, or 50 genomic loci in Table 3.
- the system comprises one or more methyl-binding domains for use in MBD-seq.
- the system comprises reagents for isolation of cell-free DNA (cfDNA) from a liquid biopsy sample.
- the sequencer comprises reagents for library preparation for sequencing.
- the sequencer comprises reagents for sequencing.
- the system comprises instructions for determining if a subject has an ER-positive cancer.
- the cloud computing environment 400 may include one or more resource providers 402a, 402b, 402c (collectively, 402). Each resource provider 402 may include computing resources. In some implementations, computing resources may include any hardware and/or
- the resource manager 406 may be connected to the resource providers 402 and the computing devices 404 over the computer network 408. In some implementations, the resource manager 406 may facilitate the provision of computing resources by one or more resource providers 402 to one or more computing devices 404.
- the resource manager 406 may receive a request for a computing resource from a particular computing device 404.
- the resource manager 406 may identify one or more resource providers 402 capable of providing the computing resource requested by the computing device 404.
- the resource manager 406 may select a resource provider 402 to provide the computing resource.
- the resource manager 406 may facilitate a connection between the resource provider 402 and a particular computing device 404. In some implementations, the resource manager 406 may establish a connection between a particular resource provider 402 and a particular computing device 404.
- Fig.5 shows an example of a computing device 500 and a mobile computing device 550 that can be used in the methods and systems described in this disclosure.
- the computing device 500 is intended to represent various forms of digital computers, such as laptops, desktops, workstations, personal digital assistants, servers, blade servers, mainframes, and other appropriate computers.
- the mobile computing device 550 is intended to represent various forms of mobile devices, such as personal digital assistants, cellular telephones, smart-phones, and other similar computing devices.
- the components shown here, their connections and relationships, and their functions, are meant to be examples only, and are not meant to be limiting.
- the processor 502 can process instructions for execution within the computing device 500, including instructions stored in the memory 504 or on the storage device 506 to display graphical information for a GUI on an external input/output device, such as a display 516 coupled to the high-speed interface 508.
- multiple processors and/or multiple buses may be used, as appropriate, along with multiple memories and types of memory.
- multiple computing devices may be connected, with each device providing portions of the necessary operations (e.g., as a server bank, a group of blade servers, or a multi-processor system).
- multiple computing devices may be connected, with each device providing portions of the necessary operations (e.g., as a server bank, a group of blade servers, or a multi-processor system).
- the memory 504 is a non-volatile memory unit or units.
- the memory 504 may also be another form of computer-readable medium, such as a magnetic or optical disk.
- the storage device 506 is capable of providing mass storage for the computing device 500.
- the storage device 506 may be or contain a computer- readable medium, such as a hard disk device, an optical disk device, a flash memory or other similar solid state memory device, or an array of devices, including devices in a storage area network or other configurations. Instructions can be stored in an information carrier.
- the high-speed interface 508 manages bandwidth-intensive operations for the computing device 500, while the low-speed interface 512 manages lower bandwidth-intensive operations. Such allocation of functions is an example only.
- the high- speed interface 508 is coupled to the memory 504, the display 516 (e.g., through a graphics processor or accelerator), and to the high-speed expansion ports 510, which may accept various expansion cards (not shown).
- the low-speed interface 512 is coupled to the storage device 506 and the low-speed expansion port 514.
- the low-speed expansion port 514 which may include various communication ports (e.g., USB, Bluetooth®, Ethernet, wireless Ethernet) may be coupled to one or more input/output devices, such as a keyboard, a pointing device, a scanner, or a networking device such as a switch or router, e.g., through a network adapter.
- the computing device 500 may be implemented in a number of different forms, as shown in the figure. For example, it may be implemented as a standard server 520, or multiple times in a group of such servers. In addition, it may be implemented in a personal computer such as a laptop computer 522. It may also be implemented as part of a rack server system 524. Alternatively, components from the computing device 500 may be combined with other components in a mobile device (not shown), such as a mobile computing device 550. Each of such devices may contain one or more of the computing device 500 and the mobile computing device 550, and an entire system may be made up of multiple computing devices communicating with each other.
- the mobile computing device 550 includes a processor 552, a memory 564, an input/output device such as a display 554, a communication interface 566, and a transceiver 568, among other components.
- the mobile computing device 550 may also be provided with a storage device, such as a micro-drive or other device, to provide additional storage.
- a storage device such as a micro-drive or other device, to provide additional storage.
- Each of the processor 552, the memory 564, the display 554, the communication interface 566, and the transceiver 568 are interconnected using various buses, and several of the components may be mounted on a common motherboard or in other manners as appropriate.
- the processor 552 can execute instructions within the mobile computing device 550, including instructions stored in the memory 564.
- the processor 552 may be implemented as a chipset of chips that include separate and multiple analog and digital processors.
- the processor 552 may provide, for example, for coordination of the other components of the mobile computing device 550, such as control of user interfaces, applications run by the mobile computing device 550, and wireless communication by the mobile computing device 550.
- the processor 552 may communicate with a user through a control interface 558 and a display interface 556 coupled to the display 554.
- the display 554 may be, for example, a TFT (Thin-Film-Transistor Liquid Crystal Display) display or an OLED (Organic Light Emitting Diode) display, or other appropriate display technology.
- the display interface 556 may comprise appropriate circuitry for driving the display 554 to present graphical and other information to a user.
- the control interface 558 may receive commands from a user and convert them for submission to the processor 552.
- an external interface 562 may provide communication with the processor 552, so as to enable near area communication of the mobile computing device 550 with other devices.
- the external interface 562 may provide, for example, for wired communication in some implementations, or for wireless communication in other implementations, and multiple interfaces may also be used.
- the expansion memory 574 may be provided as a security module for the mobile computing device 550, and may be programmed with instructions that permit secure use of the mobile computing device 550.
- secure applications may be provided via the SIMM cards, along with additional information, such as placing identifying information on the SIMM card in a non- hackable manner.
- the memory may include, for example, flash memory and/or NVRAM memory (non-volatile random access memory), as discussed below.
- instructions are stored in an information carrier and, when executed by one or more processing devices (for example, processor 552), perform one or more methods, such as those described above.
- the instructions can also be stored by one or more storage devices, such as one or more computer- or machine-readable mediums (for example, the memory 564, the expansion memory 574, or memory on the processor 552).
- the instructions can be received in a propagated signal, for example, over the transceiver 568 or the external interface 562.
- the mobile computing device 550 may communicate wirelessly through the communication interface 566, which may include digital signal processing circuitry where necessary.
- the communication interface 566 may provide for communications under various modes or protocols, such as GSM voice calls (Global System for Mobile communications), SMS (Short Message Service), EMS (Enhanced Messaging Service), or MMS messaging (Multimedia Messaging Service), CDMA (code division multiple access), TDMA (time division multiple access), PDC (Personal Digital Cellular), WCDMA (Wideband Code Division Multiple Access), CDMA2000, or GPRS (General Packet Radio Service), among others.
- GSM voice calls Global System for Mobile communications
- SMS Short Message Service
- EMS Enhanced Messaging Service
- MMS messaging Multimedia Messaging Service
- CDMA code division multiple access
- TDMA time division multiple access
- PDC Personal Digital Cellular
- WCDMA Wideband Code Division Multiple Access
- CDMA2000 Code Division Multiple Access
- GPRS General Packet Radio Service
- a GPS (Global Positioning System) receiver module 570 may provide additional navigation- and location-related wireless data to the mobile computing device 550, which may be used as appropriate by applications running on the mobile computing device 550.
- the mobile computing device 550 may also communicate audibly using an audio codec 560, which may receive spoken information from a user and convert it to usable digital information.
- the audio codec 560 may likewise generate audible sound for a user, such as through a speaker, e.g., in a handset of the mobile computing device 550.
- Such sound may include sound from voice telephone calls, may include recorded sound (e.g., voice messages, music files, etc.) and may also include sound generated by applications operating on the mobile computing device 550.
- the mobile computing device 550 may be implemented in a number of different forms, as shown in the figure. For example, it may be implemented as a cellular telephone 580. It
- 12366150v1 Attorney Docket No.2014191-0027 may also be implemented as part of a smart-phone 582, personal digital assistant, or other similar mobile device.
- Various implementations of the systems and techniques described here can be realized in digital electronic circuitry, integrated circuitry, specially designed ASICs (application specific integrated circuits), computer hardware, firmware, software, and/or combinations thereof. These various implementations can include implementation in one or more computer programs that are executable and/or interpretable on a programmable system including at least one programmable processor, which may be special or general purpose, coupled to receive data and instructions from, and to transmit data and instructions to, a storage system, at least one input device, and at least one output device.
- machine-readable medium and computer- readable medium refer to any computer program product, apparatus and/or device (e.g., magnetic discs, optical disks, memory, Programmable Logic Devices (PLDs)) used to provide machine instructions and/or data to a programmable processor, including a machine-readable medium that receives machine instructions as a machine-readable signal.
- machine-readable signal refers to any signal used to provide machine instructions and/or data to a programmable processor.
- Systems and techniques described here can be implemented in a computing system that includes a back end component (e.g., as a data server), or that includes a middleware component (e.g., an application server), or that includes a front end component (e.g., a client computer having a graphical user interface or a Web browser through which a user can interact
- a back end component e.g., as a data server
- a middleware component e.g., an application server
- a front end component e.g., a client computer having a graphical user interface or a Web browser through which a user can interact
- a machine learning module refers to a computer implemented process (e.g., a software function) that implements one or more specific machine learning techniques, e.g., artificial neural networks (ANNs), e.g., convolutional neural networks (CNNs), random forest, decision trees, support vector machines, and the like, in order to determine, for a given input, one or more output values.
- ANNs artificial neural networks
- CNNs convolutional neural networks
- RNNs convolutional neural networks
- a machine learning module is trained, e.g., to accomplish a specific task such as identifying certain response strings, values of determined parameters are fixed and the (e.g., unchanging, static) machine learning module is used to process new data (e.g., different from the training data) and accomplish its trained task without further updates to its parameters (e.g., the machine learning module does not receive feedback and/or updates).
- available input data includes training data and validation data, e.g., where
- training data is used during the training process to optimize a model, whereas validation data is used to check the accuracy of the model while operating on previously unseen data.
- training data is divided into batches (e.g., portions) that is sequentially used (e.g., in random order) as sets of inputs to train a model.
- a model is trained multiple times (e.g., epochs) on the entire set of training data.
- machine learning modules may receive feedback, e.g., based on user review of accuracy, and such feedback may be used as additional training data, to dynamically update the machine learning module.
- two or more machine learning modules may be combined and implemented as a single module and/or a single software application.
- two or more machine learning modules may also be implemented separately, e.g., as separate software applications.
- a machine learning module may be software and/or hardware.
- a machine learning module may be implemented entirely as software, or certain functions of a ANN module may be carried out via specialized hardware (e.g., via an application specific integrated circuit (ASIC) and/or field programmable gate arrays (FPGAs)).
- ASIC application specific integrated circuit
- FPGAs field programmable gate arrays
- machine learning modules implementing machine learning techniques may be composed of individual nodes (e.g. units, neurons).
- a node may receive a set of inputs that may include at least a portion of a given input data for the machine learning module and/or at least one output of another node.
- a node may have at least one parameter to apply and/or a set of instructions to perform (e.g., mathematical functions to execute) over the set of inputs.
- node instructions may include a step to provide various relative importance to the set of inputs using various parameters, such as weights.
- Non-limiting examples of the activation function include Rectified Linear Activation (ReLu), logistic (e.g., sigmoid), hyperbolic tangent (tanh), and softmax.
- a node may have a capability of remembering previous states
- the machine learning module comprises a deep learning architecture composed of nodes organized into layers.
- a layer is a set of nodes that receives data input (e.g., weighted or non-weighted input), transforms it (e.g., by carrying out instructions, e.g., applying a set of functions e.g., linear and/or non-linear functions), and passes transformed values as output (e.g., to the next layer).
- a machine learning module may be composed of at least one layer (e.g., ordered).
- layers e.g., ordered
- types of layers include convolutional layers (e.g., layers with a kernel, a matrix of parameters that is slid across an input to be multiplied with multiple input values to reduce them to a single output value); fully connected (FC) layers (e.g.
- the performance of a machine learning module may be characterized by its ability to produce an output data with specific accuracy.
- a training process is performed to find optimal parameters, such as weights, for each node in each layer of the machine learning module.
- the training process of a machine learning module may involve using output data to calculate an objective function (e.g., cost function, loss function, error function) that needs to be optimized (e.g., minimized, maximized).
- an objective function e.g., cost function, loss function, error function
- a machine learning objective function may be a combination of a loss function and regularization parameter. The loss function is related to how well the output is able to predict the input.
- objective function optimization of a machine learning module may involve finding at least one (e.g., all) of the present global optima (e.g., as opposed to local optima).
- algorithm for objective function optimization follows principles of mathematical optimization for a multi-variable function and relies on achieving specific accuracy of the process.
- Methods disclosed herein may utilize one or more machine-learned models as a classifier.
- a machine-learned model may be or include an artificial neural network.
- a machine- learned model may employ, for example, an attention-based model (e.g., a transformer model, such as, for example, a vision transformer), a transformer model (e.g., a vision transformer), a regression-based model (e.g., a logistic regression model), a regularization-based model (e.g., an elastic net model or a ridge regression model), an instance-based model (e.g., a support vector machine or a k-nearest neighbor model), a Bayesian-based model (e.g., a naive-based model or a Gaussian naive-based model), a clustering-based model (e.g., an expectation maximization model), an ensemble-based model (e.g., an adaptive boosting model, a random forest model, a bootstrap-aggregation model, or a gradient boosting machine model), or a neural-network-based model (e.g., a convolutional neural network, a recurrent neural network,
- a machine-learned model used as a classifier is or is derived from a decision tree methodology, a neural boosted methodology, a bootstrap forest methodology, a boosted tree methodology, a k nearest neighbors methodology, a generalized regression forward selection methodology, a generalized regression pruned forward selection methodology, a fit stepwise methodology, a generalized regression lasso methodology, a generalized regression elastic net methodology, a generalized regression ridge methodology, a nominal logistic methodology, a support vector machines methodology, a discriminant methodology, a na ⁇ ve Bayes methodology, or a combination thereof.
- a machine-learned model is or is derived from a decision tree methodology, a neural boosted methodology, a bootstrap forest methodology, a boosted tree methodology, a k nearest neighbors methodology, a generalized regression forward selection methodology, a generalized regression pruned forward selection methodology, a fit stepwise methodology, a generalized regression lasso methodology, a generalized regression elastic net methodology, a generalized regression ridge methodology, a nominal
- a machine-learned model is or is derived from a decision tree methodology, a neural boosted methodology, a bootstrap forest methodology, a boosted tree methodology, a support vector machines methodology, or a combination thereof.
- the term “about” can encompass a range of values that within 25%, 20%, 19%, 18%, 17%, 16%, 15%, 14%, 13%, 12%, 11%, 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1%, or within a fraction of a percent, of the referenced value.
- “Accessibility Status” or “Chromatin Accessibility Status” As used herein, “accessibility status” or “chromatin accessibility status” of a genomic locus refers to the frequency with which DNA sequences corresponding to the genomic locus are identified in an assay for detection of accessible chromatin.
- Antibody refers to a polypeptide that includes one or more canonical immunoglobulin sequence elements sufficient to confer specific binding to a particular antigen (e.g., a heavy chain variable domain, a light chain variable domain, and/or one or more CDRs).
- a particular antigen e.g., a heavy chain variable domain, a light chain variable domain, and/or one or more CDRs.
- the term antibody includes, without limitation, human antibodies, non-human antibodies, synthetic and/or engineered antibodies, fragments thereof, and agents including the same.
- Antibodies can be naturally occurring immunoglobulins (e.g., generated by an organism reacting to an antigen). Synthetic, non-naturally occurring, or engineered antibodies can be produced by recombinant engineering, chemical synthesis, or other artificial systems or methodologies known to those of skill in the art.
- antibody can include (unless otherwise stated or clear from context) any art-known constructs or formats utilizing antibody structural and/or functional features including without limitation intrabodies, domain antibodies, antibody mimetics, Zybodies®, Fab fragments, Fab’ fragments, F(ab’)2 fragments, Fd’ fragments, Fd fragments, isolated CDRs or sets thereof, single chain antibodies, single-chain Fvs (scFvs), disulfide-linked Fvs (sdFv), polypeptide-Fc fusions, single domain antibodies (e.g., shark single domain antibodies such as IgNAR or fragments thereof), cameloid antibodies, camelized antibodies, masked antibodies (e.g., Probodies®), affybodies, anti-idiotypic (anti-Id) antibodies (including, e.g., anti-anti-Id antibodies), Small Modular ImmunoPharmaceuticals (SMIPs), single chain or Tandem diabodies (TandAb®), VHHs
- SMIPs single
- an antibody includes one or more structural elements recognized by those skilled in the art as a complementarity determining region (CDR) or variable domain.
- CDR complementarity determining region
- an antibody can be a covalently modified (“conjugated”) antibody (e.g., an antibody that includes a polypeptide including one or more canonical immunoglobulin sequence elements sufficient to confer specific binding to a particular antigen, where the polypeptide is covalently linked with one or more of a therapeutic agent, a detectable moiety, another polypeptide, a glycan, or a polyethylene glycol molecule).
- conjugated antibody e.g., an antibody that includes a polypeptide including one or more canonical immunoglobulin sequence elements sufficient to confer specific binding to a particular antigen, where the polypeptide is covalently linked with one or more of a therapeutic agent, a detectable moiety, another polypeptide, a glycan, or a polyethylene glycol molecule.
- antibody sequence elements are humanized, primatized, chimeric, etc., as is known in the art.
- An antibody including a heavy chain constant domain can be, without limitation, an antibody of any known class, including but not limited to, IgA, secretory IgA, IgG, IgE and IgM, based on heavy chain constant domain amino acid sequence (e.g., alpha ( ⁇ ), delta ( ⁇ ), epsilon ( ⁇ ), gamma ( ⁇ ) and mu ( ⁇ )).
- IgG subclasses are also well known to those in the art and include but are not limited to human IgG1, IgG2, IgG3 and IgG4.
- “Isotype” refers to the Ab class or subclass (e.g., IgM or IgG1) that is encoded by the heavy chain constant region genes.
- a “light chain” can be of a distinct type, e.g., kappa ( ⁇ ) or lambda ( ⁇ ), based on the amino acid sequence of the light chain constant domain.
- an antibody has constant region sequences that are characteristic of mouse, rabbit, primate, or human immunoglobulins. Naturally produced immunoglobulins are glycosylated, typically on the CH2 domain. As is known in the art, affinity and/or other binding attributes of Fc regions for Fc receptors can be modulated through glycosylation or other modification.
- an antibody may lack a covalent modification (e.g., attachment of a glycan) that it would have if produced naturally.
- a pan antibody is a pan-acetylation antibody (e.g., an antibody that can bind a histone, e.g., H3 that comprises at least one acetylated lysine, wherein the at least one acetylated lysine can be at any one of a plurality of amino acid positions, e.g., a pan-acetylation antibody can bind an H3 protein comprising an acetylated lysine at any position).
- a pan antibody can bind one or more histone modifications that are associated with transcription activation.
- a pan antibody can bind one or more histone modifications that are associated with transcription silencing.
- an “antibody fragment” refers to a portion of an antibody or antibody agent as described herein, and typically refers to a portion that includes an antigen-binding portion or variable region thereof.
- An antibody fragment can be produced by any means. For example, in some embodiments, an antibody fragment can be enzymatically or chemically produced by fragmentation of an intact antibody or antibody agent. Alternatively, in some embodiments, an antibody fragment can be recombinantly produced, i.e., by expression of an engineered nucleic acid sequence. In some embodiments, an antibody fragment can be wholly or partially synthetically produced.
- two or more entities that are physically associated with one another are covalently linked to one another; in some embodiments, two or more entities that are physically associated with one another are not covalently linked to one another but are non- covalently associated, for example by means of hydrogen bonds, van der Waals interaction, hydrophobic interactions, magnetism, or a combination thereof.
- Between or “From” As used herein, the term “between” refers to content that falls between indicated upper and lower, or first and second, boundaries, inclusive of the boundaries. Similarly, the term “from”, when used in the context of a range of values, indicates that the range includes content that falls between indicated upper and lower, or first and second, boundaries, inclusive of the boundaries.
- biological sample typically refers to a sample obtained or derived from a biological source (e.g., a tissue or organism or cell) of interest, as described herein.
- a biological source is or includes an organism, such as a human subject.
- a biological sample is or includes a biological tissue or fluid.
- a biological sample can be or include cells, tissue, or bodily fluid.
- a biological sample is or includes DNA obtained from a single subject or from a plurality of subjects.
- a biological sample can be a “primary sample” obtained directly from a biological source or can be a “processed sample”, i.e., a sample that was derived from a primary sample, e.g., via dilution, purification, mixing with one or more reagents, or any other processing step(s) as described herein.
- diagnosing includes the act, process, and/or outcome of determining whether, and/or the qualitative of quantitative probability that, a subject has or will develop the condition, disease, or related state.
- diagnosing can include a determination relating to
- Differentially accessible describes a genomic locus for which chromatin accessibility status differs between a first condition or sample and a second condition or sample (e.g., a standard or reference).
- a differentially accessible genomic locus can include a greater or smaller measured accessibility under a selected condition of interest, such as ER-positive state, as compared to a reference state, such as ER-negative state.
- chromatin accessibility and/or transcription factor binding can be used as a measure of epigenetic modifications at a given locus.
- the term “epigenetic marker” refers to an indicator of epigenetic state, and includes, e.g., epigenetic modifications (e.g., histone modifications and DNA methylation, transcription factor biding, chromatin accessibility.
- the term “epigenetic biomarker” refers to an epigenetic marker that can be used in the detection of a disease or condition.
- identity refers to the overall relatedness between polymeric molecules, e.g., between nucleic acid molecules (e.g., DNA molecules) and/or between polypeptide molecules. Methods for the calculation of a percent identity as between two provided sequences are known in the art.
- % sequence identity refers to a relationship between two or more sequences, as determined by comparing the sequences. In the art, “identity” also means the degree of sequence relatedness between protein and nucleic acid sequences as determined by the match between strings of such sequences.
- the percent identity between the two sequences is a function of the number of identical positions shared by the sequences, optionally accounting for the number of gaps, and the length of each gap, which may need to be introduced for optimal alignment of the two sequences.
- the comparison of sequences and determination of percent identity between two sequences can be accomplished using a computational algorithm, such as BLAST (basic local alignment search tool). Sequence alignments and percent identity calculations may be performed using the Megalign program of the LASERGENE bioinformatics computing suite (DNASTAR, Inc., Madison, Wisconsin). Multiple alignment of the sequences
- GCG Genetics Computer Group
- BLASTP BLASTN
- BLASTX Altschul et al., J Mol Biol (1990) 215:403-410
- DNASTAR DNASTAR, Inc., Madison, Wisconsin
- FASTA program incorporating the Smith-Waterman algorithm (Pearson, Comput Methods Genome Res [Proc Int Symp] (1994), Meeting Date 1992, 111-120. Eds. Suhai, Sandor. Plenum, New York, NY (the contents of each of which is separately incorporated herein by reference in its entirety).
- a regulatory sequence can control or impact one or more aspects of gene expression (e.g., cell- type-specific expression, inducible expression, etc.).
- subject refers to an organism, typically a mammal (e.g., a human).
- a subject is suffering from a disease, disorder or condition (e.g., ER-positive cancer, e.g., ER-positive breast cancer, etc.).
- a subject is susceptible to a disease, disorder, or condition.
- a subject displays one or more symptoms or characteristics of a disease, disorder or condition.
- a subject is not suffering from a disease, disorder or condition.
- a subject does not display any symptom or characteristic of a disease, disorder, or condition.
- a subject has one or more features characteristic of susceptibility to or risk of a disease, disorder, or condition.
- a subject is a subject that has been tested for a disease, disorder, or condition, and/or to whom therapy has been administered.
- a human subject can be interchangeably referred to as a “patient” or “individual”.
- therapeutic agent refers to any agent that elicits a desired pharmacological effect when administered to a subject.
- an agent is considered to be a therapeutic agent if it demonstrates a statistically significant effect across an appropriate population.
- the appropriate population can be a population of model organisms or a human population.
- an appropriate population can be defined by various criteria, such as a certain age group, gender, genetic background, preexisting clinical conditions, etc.
- a therapeutic agent is a substance that can be used for treatment of a disease, disorder, or condition (e.g., ER-positive cancer, e.g., ER-positive breast cancer, etc.).
- a therapeutic agent is an agent that has been or is required to be approved by a government agency before it can be marketed for administration to humans.
- a therapeutic agent is an agent for which a medical prescription is required for administration to humans.
- Therapeutically effective amount refers to an amount that produces the desired effect for which it is administered.
- the term refers to an amount that is sufficient, when administered to a population suffering from or susceptible to a disease, disorder, and/or condition (e.g., ER- positive cancer, e.g., ER-positive breast cancer, etc.) in accordance with a therapeutic dosing regimen, to treat the disease, disorder, and/or condition.
- a therapeutically effective amount is one that reduces the incidence and/or severity of, and/or delays onset of, one or more symptoms of the disease, disorder, and/or condition.
- a therapeutically effective amount does not in fact require successful treatment be achieved in a particular individual.
- a therapeutically effective amount may be that amount that provides a particular desired pharmacological response in a significant number of subjects when administered to patients in need of such treatment.
- reference to a therapeutically effective amount may be a reference to an amount as measured in one or more specific tissues (e.g., a tissue affected by the disease, disorder or condition) or fluids (e.g., blood, saliva, serum, sweat, tears, urine, etc.).
- tissue e.g., a tissue affected by the disease, disorder or condition
- fluids e.g., blood, saliva, serum, sweat, tears, urine, etc.
- a therapeutically effective amount of a particular agent or therapy may be formulated and/or administered in a single dose.
- a therapeutically effective amount of a particular agent or therapy may be formulated and/or administered in a plurality of doses, for example, as part of a dosing regimen.
- treatment refers to administration of a therapy that partially or completely alleviates, ameliorates, relieves, inhibits, delays onset of, reduces severity of, and/or reduces incidence of one or more symptoms, features, and/or causes of a particular disease, disorder, or condition, or is administered for the purpose of achieving any such result.
- a “prophylactic treatment” includes a treatment administered to a subject who does not display signs or symptoms of a condition to be treated or displays only early signs or symptoms of the condition to be treated such that treatment is administered for the purpose of diminishing, preventing, or decreasing the risk of developing the condition. Thus, a prophylactic treatment functions as a preventative treatment against a condition.
- a “therapeutic treatment” includes a treatment administered to a subject who displays symptoms or signs of a condition and is administered to the subject for the purpose of reducing the severity or progression of the condition.
- the histone modification assay is selected from ChIP-seq (Chromatin ImmunoPrecipitation sequencing), CUT&RUN (Cleavage Under Targets and Release Using Nuclease) sequencing, and CUT&Tag (Cleavage Under Targets and Tagmentation) sequencing. 6.
- chromatin accessibility is quantified using a chromatin accessibility assay selected from ATAC-seq (Assay of Transpose Accessible Chromatin sequencing), NOMe-seq (Nucleosome Occupancy and Methylome sequencing), FAIRE-seq (Formaldehyde-Assisted Isolation of Regulatory Elements sequencing), MNase-seq (Micrococcal Nuclease digestion with sequencing), and a DNase hypersensitivity assay. 7.
- ATAC-seq Assay of Transpose Accessible Chromatin sequencing
- NOMe-seq Nucleosome Occupancy and Methylome sequencing
- FAIRE-seq Formmaldehyde-Assisted Isolation of Regulatory Elements sequencing
- MNase-seq Merococcal Nuclease digestion with sequencing
- DNase hypersensitivity assay selected from ATAC-seq (Assay of Transpose Accessible Chromatin sequencing)
- NOMe-seq Nucle
- any one of embodiments 1-6 wherein the binding of one or more transcription factors is quantified using a transcription factor binding assay that detects binding of one or more of p300, mediator complex, cohesin complex, RNA pol II, FOXA1, ESR1, PR, MYC, EN1, FOXM1, KLF4, AP-2, RARa, or RUNX1.
- the transcription factor binding assay is selected from ChIP-seq (Chromatin ImmunoPrecipitation sequencing), CUT&RUN (Cleavage Under Targets and Release Using Nuclease) sequencing, and CUT&Tag (Cleavage Under Targets and Tagmentation) sequencing.
- the method of embodiment 14, comprising quantifying H3K4me3 modifications, H3K27ac modifications and DNA methylation.
- the liquid biopsy sample is a plasma sample, serum sample, or urine sample.
- the method comprises isolating DNA (e.g., cfDNA) from 1, 2, 3, 4, or 5 mL of the liquid biopsy sample (e.g., plasma sample). 18.
- the cfDNA comprising H3K4me3 modifications is enriched using a method that comprises incubating the sample with an agent (e.g., an antibody) that binds H3K4me3 modifications;
- the cfDNA comprising H3K27ac modifications is enriched using a method that comprises incubating the sample with an agent (e.g., an antibody) that binds H3K27ac modifications; and/or
- methylated cfDNA is enriched using a method that comprises incubating the sample with an agent (e.g., an antibody or a methyl binding domain) that binds methylated DNA.
- the method comprises incubating with two or more of the agent that binds H3K4 modifications, the agent that binds H3K27ac modifications, and the agent that binds methylated DNA, the sample is incubated with the two or more agents (a) in sequence, or (b) in parallel (e.g., wherein the sample is divided into fractions and each fraction is incubated with a different agent). 22. The method of any one of embodiments 18-21, wherein the sequencing is performed using a next generation sequencing method. 23.
- the method comprises attaching (e.g., ligating) adapters to cfDNA obtained from the subject (e.g., attaching after cfDNA has been enriched for cfDNA comprising one or more H3K4me3 modifications, cfDNA comprising one or more H3K27ac modifications, and/or methylated cfDNA).
- attaching e.g., ligating
- the method comprises amplifying the plurality of converted DNA fragments after attaching adapters to the plurality of DNA fragments.
- mapping sequence reads to a reference genome e.g., mapping sequence reads to a reference genome.
- 12366150v1 Attorney Docket No.2014191-0027 wherein a sequence read peak corresponds to a region of the genome that has a higher number of sequence reads that the local background.
- 28. The method of embodiment 27, wherein peaks in high noise regions are removed and/or where peaks in regions that having increased levels of one or more epigenetic markers in white blood cells are removed.
- 29. The method of embodiments 27 or 28, wherein peaks in regions likely to be artifactual are removed.
- 30 The method of any one of embodiments 27-29, wherein peaks that are less than 50 bp in length are removed. 31.
- the method of any one of embodiments 18-30, wherein quantifying H3K4me3 modifications, H3K27ac modifications, and/or DNA methylation comprises summing the number of sequence reads having at least one nucleotide overlap the one or more genomic loci.
- sequence reads are adjusting on the basis of sequencing depth (e.g., quantile normalizing sequence reads to a common reference distribution) and/or ChIP quality prior to summing.
- sequence counts are normalized to aggregate counts in a given sample across a set of regions (e.g., 10,000 regions) previously determined to have DNAse hypersensitivity in most cell types. 34.
- sequence read density is calculated by: (a) summing background adjusted sequence counts at each of the one or more genomic loci and dividing by the sum of the kilobases of the one or more genomic loci; or (b) for each genomic loci, dividing background adjusted fragment count by the number of kilobases of the genomic loci, and then summing for each loci. 38.
- the one or more genomic loci include one or more genomic loci with an increased level of the one or more epigenetic biomarkers in (a) sample(s) obtained from a subject with an ER-positive cancer as compared to a sample obtained from a subject with an ER-negative cancer, and/or (b) sample(s) obtained from a subject with an ER-negative cancer as compared to a sample obtained from a subject with an ER-positive cancer. 39.
- the method of embodiment 38 comprising calculating an ER-positive/ER-negative ratio score, by a method comprising: (a) calculating an ER-positive sequence read density by a method comprising summing background adjusted sequence counts at each of the one or more genomic loci with an increased level of one or more epigenetic biomarkers in sample(s) obtained from subjects with an ER-positive cancer as compared to samples obtained from subjects with ER-negative cancer; (b) calculating an ER-negative sequence read density by a method comprising summing background adjusted sequence counts at each of the one or more genomic loci with an increased level of the one or more epigenetic biomarkers in sample(s) obtained from subjects with an ER-negative cancer as compared to samples obtained from subjects with an ER-positive cancer; and (c) dividing the ER-positive sequence read density by the ER-negative sequence read density.
- a system for determining the ER status of a cancer in a subject comprising a sequencer configured to generate a sequencing dataset from a sample; and a non-transitory computer readable storage medium of embodiment 79 and/or a computer system of embodiment 80.
- WGS Whole Genome Sequencing
- the system of embodiment 81 or 82 further comprising a sample preparation device configured to prepare the sample for sequencing from a biological sample, optionally a liquid biopsy sample.
- the device comprises reagents for quantifying: (a) H3K4me3, e.g., for at least 5, 10, 20, 30, 40, or 50 genomic loci in Table 1; (b) H3K27ac, e.g., for at least 5, 10, 20, 30, 40, or 50 genomic loci in Table 2; (c) DNA methylation, e.g., for at least 5, 10, 20, 30, 40, or 50 genomic loci in Table 3; or (d) any combination of (a)-(c).
- a method of determining the ER status of a cancer in a subject comprising: receiving (e.g., by a processor of a computing device) one or more genomic profiles of one or more histone modifications, chromatin accessibility, binding of one or more transcription factors, and/or DNA methylation for the subject; and determining whether the subject has an epigenetic profile indicative of an ER-positive cancer by classifying (e.g., by the processor) the genomic profile using the ER classifier.
- receiving e.g., by a processor of a computing device
- one or more genomic profiles of one or more histone modifications, chromatin accessibility, binding of one or more transcription factors, and/or DNA methylation for the subject e.g., by a processor of a computing device
- the method comprising: receiving (e.g., by a processor of a computing device) one or more genomic profiles of one or more histone modifications, chromatin accessibility, binding of one or more transcription factors, and/or DNA methylation for the subject; and determining whether
- the one or more genomic profiles used to train the ER classifier comprise one or more genomic profiles generated by in silico diluting sequence data from ER-positive or ER-negative cell lines with sequence data obtained from healthy donor plasma samples so as to achieve a simulated ctDNA percentage ranging from 0.5% to 50%.
- the method of embodiment 95 wherein the differential loci were identified by comparing genomic profiles of one or more histone modifications and/or DNA methylation in (i) one or more ER-positive cell lines and (ii) one or more ER-negative cell lines.
- 97 The method of embodiment 95 or 96, wherein the ER classifier has been trained on the levels of one or more histone modifications, chromatin accessibility, binding of one or more transcription factors, and/or DNA methylation in the differential loci for the genomic profiles that were generated by in silico diluting sequence data from one or more ER-positive cell lines and sequence data obtained from liquid biopsy samples of healthy subjects.
- ER classifier has been validated by selecting a threshold such that the validated classifier predicts ER-positive cancers with an area under the receiver operating characteristic (AUROC) greater than 0.5 (e.g., greater than 0.55, greater than 0.6, greater than 0.65, greater than 0.7, greater than 0.75, greater than 0.8, greater than 0.85, greater than 0.9, or greater than 0.95).
- AUROC receiver operating characteristic
- a computer system comprising a memory and one or more processors coupled to the memory, wherein the one or more processors are configured to perform operations to perform the method of any one of embodiments 92-110.
- 113. A method of treating a subject having a cancer, the method comprising: administering an ER-targeted agent to the subject, wherein the subject has been determined to have a validated epigenetic profile indicative of an ER-positive cancer based on analysis of a biological sample, optionally of cell-free DNA (cfDNA) from a liquid biopsy sample, obtained or derived from the subject, wherein the presence of the validated epigenetic profile has been determined using a classifier (e.g., a validated classifier) according to a method of any one of embodiments 92-110.
- a classifier e.g., a validated classifier
- ER-positive cell lines ZR751, ZR7530, BT483, T47D, BT474, CAMA1, MCF7, HCC1428, and HCC1500.
- ER-negative cell lines were used: BT549, DU4475, HS578T, BT20, UACC893, HCC38, HCC70, HCC202, HCC1143, HCC1187, HCC1419, HCC1599, HCC1806, HCC1954, HCC2157, HCC2218, and SKBR3.
- Plasma samples were prepared from whole blood collected in EDTA blood collection tubes or Streck cell-free DNA BCT with 4-6 hours of collection and plasma was stored at -80 ⁇ C until use.
- Whole blood was obtained from breast cancer patients under a protocol approved by an IRB. Breast cancer patients had previously been determined to be ER-positive or ER-negative. Informed content was obtained in each case and samples were de-identified.
- Chromatin immunoprecipitation ChIP
- Chromatin immunoprecipitation (ChIP) for histone marks (H3K4me3 and H3K27ac) in cell lines was performed using methods similar to those previously described in Schones et al., Cell (2008) 132(5):887-898, which is incorporated by reference herein in its entirety. Briefly, the cells were lysed and the chromatin was MNase digested to generate approximately 80% mononucleosomes. Nucleosomes were then incubated with antibodies that bind H3K4me3 modifications or H3K27ac modifications that were previously conjugated to magnetic epoxy beads (Invitrogen) with constant mild shaking overnight. The beads were then washed and rinsed.
- ChIP-seq and DNA methylation data analysis [0329] ChIP-sequencing reads and MeDIP-sequencing reads were aligned to the human genome build hg19 using the Burrows-Wheeler Aligner (BWA) version 0.7.15. Non-uniquely mapping and redundant reads were discarded. MACS v2.2.7.1 was used for peak calling with a q- value (FDR) threshold of 0.01. Data quality was evaluated by a variety of measures, including
- Example 2 ER status classifiers based on complex modeling of signals across different subsets of individual genomic loci that are differentiated based on ER-positive and ER- negative status [0330]
- genomic loci likely to differentiate ER- positive and ER-negative samples based on H3K4me3 modification, H3K27ac modification or DNA methylation were first identified.
- union peak maps were created by merging peak coordinates for all of the cell lines, removing regions likely to be artifactual (the ENCODE “blacklist” regions, see Amemiya et al., Sci Rep (2019) 9(1):9354) and discarding all peaks less than 50 bp in length.
- Genomic loci that had differential analyte signal between ER-positive and ER-negative cell lines were determined using DESeq2 (Love et al., Genome Biol (2014) 15(12):550), with an FDR cutoff of 5%. These differential loci are shown in Table 1 (H3K4me3), Table 2 (H3K27ac) and Table 3 (DNA methylation).
- genomic loci from Tables 1-3 for different modifications, namely (i) H3K4me3 modifications, (ii) H3K27ac modifications, (iii) DNA methylation (DNAme) or (iv) all of the above (All) and (b) using different subsets of genomic loci in Tables 1-3 for a particular modification, namely (i) all genomic loci with an absolute log2(fold-change) ⁇ 0.5, (ii) all genomic loci with an absolute log2(fold-change) ⁇ 1, (iii) all genomic loci with an absolute log2(fold-change) ⁇ 2, (iv) all genomic loci with an absolute log2(fold-change) ⁇ 3, and (v) all genomic loci with an absolute log2(fold-change) ⁇ 4.
- Fig.2 shows a representative, non-limiting graphs that demonstrates the accuracy of ER status (based on AUCROC) determination using the classifiers that were generated in accordance with this Example.
- Example 3 ER Status Determination in Plasma Samples [0334] The present example provides data demonstrating that technologies provided in the present disclosure can be used to determine ER status in a subject having cancer, using samples comprising cfDNA. In the present example, plasma samples from patients diagnosed with metastatic breast cancer were characterized. [0335] Plasma samples were obtained from 91 subjects.
- Table 4 shows genes that were determined to be associated with ER status using the disclosed classifiers. As indicated below, genes previously associated with ER status (left column); genes previously shown to have a biological link, but not directly associated with ER
- an ER classifier described herein measures an epigenomic marker (e.g., promoter, enhancer, or DNA methylation marker) associated with one or more of the genes listed in Table 4.
- an epigenomic marker e.g., promoter, enhancer, or DNA methylation marker
- Table 1 Exemplary genomic loci that are differentially H3K4me3 modified in ER-positive vs. ER-negative cancer.
- Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1 chr1:793725-794444 -1.41 676 chr13:99710786-99710933 1.60 2.00 3.08 2.44 1.72 1.59 2.20 2.87 3.23 1.80 1.91 2.59 1.86 1.95 1.91 1.73 1.54 1.61 1.02 1.75 1.86 1.66 1.85 2.12 1.76 1.72 2.40 2.12 3.27 2.01 0.62 1.65 0.57 0.55 1.54 2.30 1.55 2.62 2.35 2.02 1.96 2.14 0.51 2.39 0.62 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 46 chr1:11761594-11762165 -2.11 721 chr14:55543845-55
- compositions or methods are described as having, including, or comprising specific elements, it is to be understood that compositions or methods that consist essentially of, consist of, or do not comprise the recited elements are likewise hereby disclosed. All references cited herein are hereby incorporated by reference.
Landscapes
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Engineering & Computer Science (AREA)
- Immunology (AREA)
- Pathology (AREA)
- Analytical Chemistry (AREA)
- Zoology (AREA)
- Genetics & Genomics (AREA)
- Wood Science & Technology (AREA)
- Physics & Mathematics (AREA)
- Biotechnology (AREA)
- Microbiology (AREA)
- Molecular Biology (AREA)
- Hospice & Palliative Care (AREA)
- Biophysics (AREA)
- Oncology (AREA)
- Biochemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
Abstract
The present disclosure includes, among other things, methods, kits, and systems for determining ER status of cancer, e.g., a breast cancer. In various embodiments, the present disclosure relates to the use of one or more histone modifications, chromatin accessibility, binding of one or more transcription factors, and/or DNA methylation that that are characteristic of ER status of cancer. In some embodiments, differential modifications and/or differential accessibility are detected and quantified at one or more genomic loci of a biological sample, e.g., in cell-free DNA (cfDNA) from a liquid biopsy sample obtained or derived from a subject with cancer. In various embodiments a determined ER status is useful, e.g., in selecting treatment for and/or treating a cancer, e.g., a breast cancer.
Description
Attorney Docket No.2014191-0027 METHODS, KITS AND SYSTEMS FOR DETERMINING THE ER STATUS OF CANCER AND METHODS FOR TREATING CANCER BASED ON SAME Cross-Reference to Related Applications [0001] This application claims the benefit of U.S. Provisional Application No. 63/590,158, filed on 13 October 2023; and U.S. Provisional Application No.63/650,860, filed on 22 May 2024; the entire contents of each application is incorporated herein in its entirety by this reference BACKGROUND [0002] It has long been recognized that some human breast cancers are hormone dependent. Estrogen regulates the differentiation and proliferation of breast epithelial cells and interacts with the estrogen receptor (ER) in the nucleus. Prolonged exposure of estrogen is an important risk factor for cancer. Progesterone receptor (PR) expression in normal breast epithelium is regulated by ER (Jensen, Cancer (1980) 46:2759-2761). Presence of ER, PR and human epidermal growth factor receptor-2 (HER2) status in invasive breast carcinoma is now routinely estimated as these markers are considered to be important prognostic factors. ER and PR status has been used for many years to determine a patient’s suitability for treatment with endocrine therapy (e.g., tamoxifen). [0003] To determine if a cancer is ER-positive, medical practitioners currently order testing that is conducted on a tissue sample using immunohistochemistry (IHC). Samples are reviewed by a pathologist and typically reported as (a) the word positive or negative, (b) a percentage that tells you how many cells out of 100 stained positive for hormone receptors, i.e., a number between 0% (none have receptors) and 100% (all have receptors), and/or (c) an Allred score between 0 and 8. The Allred scoring system looks at what percentage of cells test positive for hormone receptors, along with how well the receptors show up after staining, called intensity (Allred et al., Breast Cancer Res (2004) 6:240-245). This information is then combined to score the sample on a scale from 0 to 8 where, the higher the score, the more receptors were found and the easier they were to see in the sample. [0004] ER-positive cancers can be treated with ER-targeted agents that lower estrogen levels or block estrogen receptors. Conversely, treatment with ER-targeted agents is not helpful 12366150v1
Attorney Docket No.2014191-0027 for ER-negative cancers. These cancers may instead be treated with one or more of surgery and/or radiation, HER2-targeted therapy (if HER2-positive), chemotherapy and immunotherapy. [0005] Current methods are invasive and rely on a single tissue biopsy to characterize the ER status in metastatic breast cancer. Such methods focus only on a small region at a single tumor site at a given time and therefore do not accurately capture tumor heterogeneity or receptor evolution and therefore only partially characterize the relevant patient population. There remains a need in the art for more comprehensive and precise diagnostic methods for determining ER status including methods that are independent of IHC testing. Improved diagnostic methods would also better support future clinical trials that seek to identify subpopulations of patients that respond to ER-targeted agents. They would also expand our understanding of the underlying biology of ER-positive cancer and help identify new treatments. SUMMARY [0006] The present disclosure is based, at least in part, on the demonstration that the ER status of a cancer in a subject can be determined by detecting and quantifying the presence of histone modifications and/or DNA methylation at one or more genomic loci in cell-free DNA (cfDNA) from a liquid biopsy sample, e.g., a plasma sample obtained or derived from the subject. The present disclosure also encompasses methods where chromatin accessibility and/or binding of one or more transcription factors are detected at the one or more genomic loci instead of (or in addition to) histone modifications and/or DNA methylation. The present disclosure is also based, at least in part, on the demonstration that genomic loci that are differentially modified based on different types of histone modifications (e.g., histone methylation marks such as H3K4me3 and histone acetylation marks such as H3K27ac) and/or DNA methylation can be combined into multimodal classifiers to determine ER status. These new monomodal and multimodal classifiers provide minimally invasive ways of determining ER status that are more accurate, objective, and comprehensive than the current tissue-based approaches. No liquid biopsy platform to date has been able to provide actionable resolution on a transcriptionally regulated phenotype relevant for therapy such as ER status. [0007] The present disclosure includes, among other things, technologies for the determination of ER status and for the detection, monitoring, and/or treatment of cancer (including, e.g., breast, ovarian, or endometrial cancer) based on ER status. In various
12366150v1
Attorney Docket No.2014191-0027 embodiments, the present disclosure relates to the measurement of histone modifications in a sample obtained or derived from a subject to detect and/or treat cancer (including, e.g., breast, ovarian, or endometrial cancer) based on ER status. The present disclosure includes, among other things, histone modification measurements in cell-free DNA (cfDNA) that are characteristic of cancer, and which in various embodiments are useful, e.g., for detecting, monitoring, selecting treatment for, and/or treating cancer (including, e.g., breast, ovarian, or endometrial cancer) based on ER status. The present disclosure includes, among other things, histone modification measurements in cfDNA that are characteristic of ER-positive and ER-negative cancers, which in various embodiments are useful, e.g., in detecting, monitoring, selecting treatment for, and/or treating an ER-positive and ER-negative cancers. In some embodiments, histone modification measurements in cfDNA can be used to detect or determine resistance of a cancer (e.g., breast, ovarian, or endometrial cancer) to a therapy or transformation of a cancer from one subtype to another. In various embodiments, the present disclosure includes exemplary genomic loci that are differentially modified in ER-positive vs. ER-negative cancer, e.g., breast, ovarian, or endometrial cancer. In various embodiments, genomic loci differentially modified in cfDNA are or include one or more enhancers. In various embodiments, genomic loci differentially modified in cfDNA are or include one or more promoters. [0008] In various embodiments, a genomic locus is differentially modified if it is characterized by increased or decreased histone modification as compared to a reference (e.g., a sample from an ER-negative or healthy subject). Increased or decreased histone modification can be or include, e.g., increased or decreased histone methylation (hypermethylation or hypomethylation, respectively) of one or more particular methylation marks, or a combination thereof; increased or decreased pan-methylation; increased or decreased histone acetylation (hyperacetylation or hypoacetylation, respectively) of one or more particular acetylation marks, or a combination thereof; and/or increased or decreased pan-acetylation (e.g., pan-H3 acetylation). In various embodiments, histone methylation can be or include histone methylation marks selected from H3K4me1, H3K4me2, H3K4me3, or a combination thereof. In various embodiments, histone methylation can be or include H3K4me3. In various embodiments, histone acetylation can be or include histone acetylation marks selected from H3K9ac, H3K14ac, H3K18ac, H3K23ac, H3K27ac, or a combination thereof. In various embodiments, histone acetylation can be or include H3K27ac.
12366150v1
Attorney Docket No.2014191-0027 [0009] In various embodiments, the present disclosure relates to the measurement of DNA methylation in a sample obtained or derived from a subject to detect and/or treat cancer (including, e.g., breast, ovarian, or endometrial cancer) based on ER status. The present disclosure includes, among other things, DNA methylation measurements in cell-free DNA (cfDNA) that are characteristic of cancer, and which in various embodiments are useful, e.g., for detecting, monitoring, selecting treatment for, and/or treating cancer (including, e.g., breast, ovarian, or endometrial cancer) based on ER status. In some embodiments, DNA methylation measurements in cfDNA can be used to detect or determine resistance of a cancer (e.g., breast, ovarian, or endometrial cancer) to a therapy or transformation of a cancer from one subtype to another. In various embodiments, the present disclosure includes exemplary genomic loci that are differentially DNA methylated in ER-positive vs. ER-negative cancer, e.g., breast, ovarian, or endometrial cancer. In various embodiments, a genomic locus is differentially modified if it is characterized by increased or decreased DNA methylation as compared to a reference (e.g., a sample from an ER-negative or healthy subject). In various embodiments, genomic loci differentially modified in cfDNA are or include one or more enhancers. In various embodiments, genomic loci differentially modified in cfDNA are or include one or more promoters. [0010] The present disclosure further relates, in various embodiments, to the measurement of chromatin accessibility in cell-free DNA (cfDNA) to determine ER status. The present disclosure includes, among other things, chromatin accessibility measurements in cfDNA that are characteristic of ER-positive cancers, which in various embodiments are useful, e.g., in detecting, monitoring, selecting treatment for, and/or treating an ER-positive cancer. In some embodiments, chromatin accessibility measurements in cfDNA can be used to detect or determine resistance of a cancer (e.g., breast, ovarian, or endometrial cancer) to a therapy or transformation of a cancer from one subtype to another. In various embodiments, the present disclosure includes genomic loci that are differentially accessible in ER-positive vs. ER-negative cancers. In various embodiments, genomic loci differentially accessible in cfDNA are or include one or more enhancers. In various embodiments, genomic loci differentially accessible in cfDNA are or include one or more promoters. [0011] In various embodiments, without wishing to be bound by any particular scientific theory, histone methylation (e.g., H3K4me3) corresponds and/or is correlated with chromatin accessibility. In various embodiments, without wishing to be bound by any particular scientific
12366150v1
Attorney Docket No.2014191-0027 theory, histone acetylation (e.g., H3K27ac) corresponds and/or is correlated with chromatin accessibility. In various embodiments, without wishing to be bound by any particular scientific theory, DNA methylation corresponds and/or is correlated with chromatin accessibility. [0012] In various embodiments, a genomic locus is differentially accessible if it is characterized by increased or decreased chromatin accessibility as compared to a reference (e.g., a sample from an ER-negative or healthy subject). Increased or decreased histone modification can be or include, e.g., increased or decreased accessibility as determined by various chromatin accessibility assays known in the art. [0013] The present disclosure further relates, in various embodiments, to the measurement of transcription factor binding in cell-free DNA (cfDNA) to determine ER status. The present disclosure includes, among other things, transcription factor binding measurements in cfDNA that are characteristic of ER-positive cancers, which in various embodiments are useful, e.g., in detecting, monitoring, selecting treatment for, and/or treating an ER-positive cancer. In some embodiments, transcription factor binding measurements in cfDNA can be used to detect or determine resistance of a cancer (e.g., breast, ovarian, or endometrial cancer) to a therapy or transformation of a cancer from one subtype to another. In various embodiments, the present disclosure includes genomic loci that are differentially bound by transcription factors in ER-positive vs. ER-negative cancers. In various embodiments, genomic loci that are differentially bound by transcription factors in cfDNA are or include one or more enhancers. In various embodiments, genomic loci that are differentially bound by transcription factors in cfDNA are or include one or more promoters. [0014] In various embodiments, without wishing to be bound by any particular scientific theory, histone methylation (e.g., H3K4me3) corresponds and/or is correlated with transcription factor binding. In various embodiments, without wishing to be bound by any particular scientific theory, histone acetylation (e.g., H3K27ac) corresponds and/or is correlated with transcription factor binding. In various embodiments, without wishing to be bound by any particular scientific theory, DNA methylation corresponds and/or is correlated with transcription factor binding. [0015] In various embodiments, a genomic locus is differentially bound by transcription factors if it is characterized by increased or decreased transcription factor binding as compared to a reference (e.g., a sample from an ER-negative or healthy subject). Increased or decreased
12366150v1
Attorney Docket No.2014191-0027 transcription factor binding can be or include, e.g., increased or decreased transcription factor binding as determined by various transcription factor binding assays known in the art. [0016] In one aspect, the present disclosure provides a method of determining the ER status of a cancer in a subject, the method comprising: quantifying, at one or more genomic loci in a biological sample, optionally in cell-free DNA (cfDNA) from a liquid biopsy sample, obtained or derived from the subject: (i) one or more histone modifications, (ii) chromatin accessibility, (iii) binding of one or more transcription factors, and/or (iv) DNA methylation. [0017] In some embodiments, the one or more histone modifications are quantified using a histone modification assay that measures one or more of H3K9ac, H3K14ac, H3K18ac, H3K23ac, H3K27ac, H3K4me1, H3K4me2, H3K4me3, and pan-acetylation. In some embodiments, the histone modification assay detects H3K4me3 modifications. In some embodiments, the histone modification assay detects H3K27ac modifications. In some embodiments, the histone modification assay is selected from ChIP-seq (Chromatin ImmunoPrecipitation sequencing), CUT&RUN (Cleavage Under Targets and Release Using Nuclease) sequencing, and CUT&Tag (Cleavage Under Targets and Tagmentation) sequencing. [0018] In some embodiments, chromatin accessibility is quantified using a chromatin accessibility assay selected from ATAC-seq (Assay of Transpose Accessible Chromatin sequencing), NOMe-seq (Nucleosome Occupancy and Methylome sequencing), FAIRE-seq (Formaldehyde-Assisted Isolation of Regulatory Elements sequencing), MNase-seq (Micrococcal Nuclease digestion with sequencing), and a DNase hypersensitivity assay. [0019] In some embodiments, binding of one or more transcription factors is quantified using a transcription factor binding assay that detects binding of one or more of p300, mediator complex, cohesin complex, RNA pol II, FOXA1, ESR1, PR, MYC, EN1, FOXM1, KLF4, AP-2, RARa, or RUNX1. In some embodiments, the transcription factor binding assay is selected from ChIP-seq (Chromatin ImmunoPrecipitation sequencing), CUT&RUN (Cleavage Under Targets and Release Using Nuclease) sequencing, and CUT&Tag (Cleavage Under Targets and Tagmentation) sequencing. [0020] In some embodiments, DNA methylation is quantified using Bisulfite sequencing (BS-Seq), Whole Genome Bisulfite Sequencing (WGBS), Methylated DNA ImmunoPrecipitation sequencing (MeDIP-seq), or Methyl-CpG-Binding Domain sequencing (MBD-seq).
12366150v1
Attorney Docket No.2014191-0027 [0021] In some embodiments, the method comprises quantifying two or more of the following, each at one or more genomic loci in cell-free DNA (cfDNA) from a liquid biopsy sample obtained or derived from the subject: (i) one or more histone modifications, (ii) chromatin accessibility, (iii) transcription factor binding, and/or (iv) DNA methylation. In some embodiments, the method comprises quantifying two or more histone modifications, e.g., quantifying H3K4me3 and H3K27ac modifications. In some embodiments, the method comprises quantifying one or more histone modifications and DNA methylation, e.g., quantifying H3K4me3 and/or H3K27ac modifications and DNA methylation. In some embodiments, the method comprises quantifying H3K4me3 modifications, H3K27ac modifications and DNA methylation. [0022] In some embodiments, the biological sample is a liquid biopsy sample, e.g., a plasma sample, serum sample, or urine sample. [0023] In some embodiments, quantification of one or more histone modifications, chromatin accessibility, binding of one or more transcription factors, and/or DNA methylation at the one or more genomic loci as compared to a reference indicates that the subject has an ER- positive cancer. [0024] In some embodiments, quantification of one or more histone modifications, chromatin accessibility, binding of one or more transcription factors, and/or DNA methylation at the one or more genomic loci as compared to a reference indicates that the subject has an ER- negative cancer. [0025] In some embodiments, the cancer is breast cancer, ovarian cancer, or endometrial cancer. In some embodiments, the cancer is breast cancer. [0026] In some embodiments, the reference is a predetermined threshold, a measurement from a liquid biopsy sample, and/or a normalized value, optionally wherein the reference is a measurement from a liquid biopsy sample obtained from a cohort of subjects who have previously been determined to have an ER-negative cancer or to be cancer free. [0027] In some embodiments, a sample is a liquid biopsy sample comprising cfDNA, and a method comprises: (a) quantifying H3K4me3 modifications at one or more genomic loci using an assay that comprises enriching for cfDNA comprising one or more H3K4me3 modifications and sequencing the cfDNA enriched for H3K4me3 modifications (e.g., using a cfChIP-seq assay);
12366150v1
Attorney Docket No.2014191-0027 (b) quantifying H3K27ac modifications at one or more genomic loci using an assay that comprises enriching for cfDNA comprising one or more H3K27ac modifications and sequencing the cfDNA enriched for H3K27ac modifications (e.g., using a cfChIP-seq assay); and/or; (c) quantifying methylated DNA using an assay that comprises enriching for methylated cfDNA and sequencing the enriched cfDNA to determine a count of sequences with one or more methylated nucleotides (e.g., using a MBD-seq assay). [0028] In some embodiments, [0029] (a) cfDNA comprising H3K4me3 modifications is enriched using a method that comprises incubating a sample with an agent (e.g., an antibody) that binds H3K4me3 modifications; [0030] (b) cfDNA comprising H3K27ac modifications is enriched using a method that comprises incubating a sample with an agent (e.g., an antibody) that binds H3K27ac modifications; and/or [0031] (c) methylated cfDNA is enriched using a method that comprises incubating a sample with an agent (e.g., an antibody or a methyl binding domain) that binds methylated DNA. [0032] In some embodiments, an agent that binds H3K4me3 modifications, an agent that binds H3K27ac modifications, and/or an agent that binds methylated DNA can be attached (e.g., via a covalent or noncovalent bond) to a physical support (e.g., a bead, a magnetic bead, an agarose bead, or a magnetic epoxy bead) prior to incubating with a sample. [0033] In embodiments where a method comprises incubating a sample with two or more of (a) an agent that binds H3K4 modifications, (b) an agent that binds H3K27ac modifications, and (c) an agent that binds methylated DNA, the sample is incubated with the two or more agents (1) in sequence, or (2) in parallel (e.g., wherein the sample is divided into fractions and each fraction is incubated with a different agent). [0034] In some embodiments, sequencing is performed using a next generation sequencing method. [0035] In some embodiments, a method comprises attaching (e.g., ligating) adapters to cfDNA obtained from the subject (e.g., attaching after cfDNA has been enriched for cfDNA
12366150v1
Attorney Docket No.2014191-0027 comprising one or more H3K4me3 modifications, cfDNA comprising one or more H3K27ac modifications, and/or methylated cfDNA). [0036] In some embodiments, a plurality of converted DNA fragments are amplified after attaching adapters to the plurality of DNA fragments. [0037] In some embodiments, sequence reads are mapped to a reference genome. In some embodiments, non-uniquely mapped and redundant sequence reads are discarded prior to quantifying one or more epigenetic biomarkers. In some embodiments, sequence reads are mapped to a reference genome, and one or more genomic loci correspond to sequence read peaks, wherein a sequence read peak corresponds to a region of the genome that has a higher number of sequence reads that the local background. In some embodiments, peaks in high noise regions are ignored when identifying genomic loci with a higher number of sequence reads than the local background. In some embodiments, peaks in regions likely to be artifactual are removed. In some embodiments, peaks that are less than 50 bp in length are removed. In some embodiments, peaks in regions with high levels of one or more epigenetic markers in white blood cells are removed. [0038] In some embodiments, quantifying H3K4me3 modifications, H3K27ac modifications, and/or DNA methylation comprises summing the number of sequence reads having at least one nucleotide overlap one or more genomic loci. In some embodiments, sequence reads are adjusted on the basis of sequencing depth (e.g., quantile normalizing sequence reads to a common reference distribution) and/or ChIP quality prior to summing. In some embodiments, sequence counts are normalized to aggregate counts in a given sample across a set of regions (e.g., 10,000 regions) previously determined to have DNAse hypersensitivity in most cell types. In some embodiments, an estimate of local background signal is subtracted from the sequence reads at each genomic loci prior to summing. [0039] In some embodiments, a method comprises comparing a measure of one or more epigenetic biomarkers to a reference. In some embodiments, a reference is a predetermined threshold, a measurement from a liquid biopsy sample, a measurement from liquid biopsy samples obtained from a cohort of subjects, and/or a normalized value. In some embodiments, a predetermined threshold or a normalized value were previously shown to distinguish an ER- positive and an ER-negative cancer (e.g., distinguish with an AUROC of greater than 0.5). In
12366150v1
Attorney Docket No.2014191-0027 some embodiments, a reference is a measurement from a liquid biopsy sample obtained from a cohort of subjects who have previously been determined to have an ER-positive or ER-negative cancer. In some embodiments, a cohort of subjects has previously been determined to have cancer (e.g., breast cancer). [0040] In some embodiments, a method comprises calculating a sequence read density at one or more genomic loci. In some embodiments, sequence read density can be calculated by a process comprising: (a) summing background adjusted sequence counts at each of one or more genomic loci and dividing by the sum of the kilobases of the one or more genomic loci; or (b) for each genomic loci, dividing background adjusted fragment count by the number of kilobases of the genomic loci, and then summing for each loci. [0041] In some embodiments, one or more genomic loci include one or more genomic loci with an increased level of the one or more epigenetic biomarkers in (a) sample(s) obtained from a subject with an ER-positive cancer as compared to a sample obtained from a subject with an ER-negative cancer, and/or (b) sample(s) obtained from a subject with an ER-negative cancer as compared to a sample obtained from a subject with an ER-positive cancer. [0042] In some embodiments, a method comprises calculating an ER-positive/ER- negative ratio score. In some embodiments, an ER-positive/ER-negative ratio score can be calculated by a method comprising: (a) calculating an ER-positive sequence read density by a method comprising summing background adjusted sequence counts at each of one or more genomic loci with an increased level of one or more epigenetic biomarkers in sample(s) obtained from subjects with an ER-positive cancer as compared to samples obtained from subjects with ER-negative cancer; (b) calculating an ER-negative sequence read density by a method comprising summing background adjusted sequence counts at each of one or more genomic loci with an increased level of one or more epigenetic biomarkers in sample(s) obtained from subjects with an ER-negative cancer as compared to samples obtained from subjects with an ER-positive cancer; and
12366150v1
Attorney Docket No.2014191-0027 (c) dividing the ER-positive sequence read density by the ER-negative sequence read density. [0043] In some embodiments, a method comprises determining an ER-positive/ER- negative ratio score for one or more epigenetic biomarkers. In some embodiments, a method comprises: (a) determining an ER-positive/ER-negative ratio score for H3K4me3 modifications; (b) determining an ER-positive/ER-negative ratio score for H3K27ac modifications; and/or (c) determining an ER-positive/ER-negative ratio score for methylated DNA. [0044] In some embodiments, a method comprises determining an ER-positive/ER- negative ratio score for two or more epigenetic biomarkers. In some embodiments, a method comprises determining an ER-positive/ER-negative ratio score for two or more epigenetic biomarkers, wherein the ER-positive/ER-negative ratio scores are combined. In some embodiments, a method comprises determining an ER-positive/ER-negative ratio score each of H3K4me3 modifications, H3K27ac modifications, and methylated DNA, and combining the ratio scores. In some embodiments, two or more ratio scores can be combined using fitted values determined using a logistic regression. [0045] In some embodiments, quantification of one or more histone modifications, chromatin accessibility, or binding of one or more transcription factors, or any combination thereof at the one or more genomic loci as compared to a reference indicates that the subject has an ER-positive cancer. In some embodiments, quantification of one or more histone modifications, chromatin accessibility, binding of one or more transcription factors, and/or DNA methylation at the one or more genomic loci as compared to a reference indicates that the subject has an ER-negative cancer. [0046] In some embodiments, a method further comprises comparing one or more quantified epigenetic biomarkers to a reference, and wherein an increase or decrease in the one or more epigenetic markers as compared to the reference indicates that a subject has an ER- positive or an ER-negative cancer.
12366150v1
Attorney Docket No.2014191-0027 [0047] In some embodiments, a sample comprises a detectable amount of ctDNA (e.g., wherein estimated tumor fraction is >3% for the cfDNA, e.g., as determined by iChorCNA). [0048] In some embodiments, the method comprises quantifying one or more histone modifications, chromatin accessibility, binding of one or more transcription factors, and/or DNA methylation at one or more genomic loci in Tables 1-3. In some embodiments, the method comprises quantifying H3K4me3 modifications for at least 5, 10, 20, 30, 40, or 50 genomic loci in Table 1. In some embodiments, the method comprises quantifying H3K27ac modifications for at least 5, 10, 20, 30, 40, or 50 genomic loci in Table 2. In some embodiments, the method comprises quantifying DNA methylation for at least 5, 10, 20, 30, 40, or 50 genomic loci in Table 3. [0049] In some embodiments, the area under the receiver operating characteristic (AUROC) for determining if a subject has an ER-positive cancer vs. an ER-negative cancer is greater than 0.5 (e.g., greater than 0.55, greater than 0.6, greater than 0.65, greater than 0.7, greater than 0.75, greater than 0.8, greater than 0.85, greater than 0.9, or greater than 0.95). [0050] In some embodiments, the ER-positive cancer is an ER-positive cancer based on IHC testing and the ER-negative cancer is an ER-negative cancer based on IHC testing. In some embodiments, the subject has previously been determined to have cancer. In some embodiments, a sample is obtained from a subject having cancer wherein a biopsy of the cancer is not possible and/or feasible. [0051] In another aspect, the present disclosure provides a method of treating a subject having a cancer, the method comprising: administering a cancer therapy to the subject based on the ER status of the cancer, wherein the ER status of the cancer has been determined using any one of the aforementioned methods of determining ER status. In some embodiments, the method further comprises determining the ER status of the cancer using any one of the aforementioned methods of determining ER status. In some embodiments, the cancer has been determined to be ER-positive and the cancer therapy comprises an ER-targeted agent. In some embodiments, if a cancer has been determined to be ER-negative, the cancer therapy is one appropriate for an ER- negative cancer. In some embodiments, if the cancer has been determined to be ER-negative, the cancer therapy does not comprise administering an ER-targeted agent. [0052] In another aspect, the present disclosure provides a method of treating a subject having a cancer, the method comprising: administering a cancer therapy to the subject based on
12366150v1
Attorney Docket No.2014191-0027 the ER status of the cancer, wherein the ER status of the cancer has been determined using any one of the aforementioned methods of determining ER status. In some embodiments, the method further comprises determining the ER status of the cancer using any one of the aforementioned methods of determining ER status. In some embodiments, if the cancer has been determined to be ER-positive, the cancer therapy comprises an ER-targeted agent. In some embodiments, if the cancer has been determined to be ER-negative, the cancer therapy does not comprise administering an ER-targeted agent. [0053] In another aspect, the present disclosure provides a method of monitoring the ER status of a cancer in a subject, and optionally treating the cancer, the method comprising: determining the ER status of the cancer using any one of the aforementioned methods of determining ER status at first and second time points. In some embodiments, the subject has been administered an ER-targeted agent after the first time point and before the second time point. In some embodiments, the method further comprises administering a cancer therapy, optionally an ER-targeted agent, to the subject based on the ER status of the cancer at the second time point, optionally wherein the type, dose and/or frequency of administration of the cancer therapy is adjusted based on the ER status of the cancer at the second time point. [0054] In another aspect, the present disclosure provides a method of treating a subject having a cancer, the method comprising: administering an ER-targeted agent to the subject if the subject has been determined to have a validated epigenetic profile indicative of an ER-positive cancer based on analysis of a biological sample, optionally of cell-free DNA (cfDNA) from a liquid biopsy sample, obtained or derived from the subject, and, if the subject has not been determined to a validated epigenetic profile indicative of an ER-positive cancer, not administering an ER-targeted agent, wherein the presence of the validated epigenetic profile has been determined using a validated classifier, wherein the validated classifier has been obtained by: (a) determining a genomic profile of one or more histone modifications, chromatin accessibility, binding of one or more transcription factors, and/or DNA methylation in (i) one or more ER-positive cell lines or (ii) biological samples obtained from a first cohort of subjects who have previously been determined to have an ER-positive cancer; (b) determining a genomic profile of one or more histone modifications, chromatin accessibility, binding of one or more transcription factors, and/or DNA methylation in (i) one or more ER-negative cell lines or (ii) biological samples obtained from a second cohort of healthy subjects or subjects who have
12366150v1
Attorney Docket No.2014191-0027 previously been determined to have an ER-negative cancer; (c) comparing the genomic profile determined in step (a) and the genomic profile determined in step (b), to identify genomic loci that have statistically different histone modification, chromatin accessibility, binding of transcription factor, and/or DNA methylation levels (“differential loci”); (d) training a classifier on histone modification, chromatin accessibility, binding of transcription factor, and/or DNA methylation levels in the differential loci to distinguish between (i) samples from one or more ER-positive cell lines or biological samples obtained from the first cohort, and (ii) samples from one or more ER-negative cell lines or biological samples obtained from the second cohort, to identify samples having a profile of histone modification, chromatin accessibility, binding of transcription factor, and/or DNA methylation levels (“epigenetic profile”) that indicates that the samples are likely obtained from an ER-positive cell line or from the first cohort; and (e) obtaining the validated classifier by validating the classifier from step (d) on a third cohort comprising an independent and group of subjects with ER-positive and ER-negative cancers and selecting a threshold such that the validated classifier predicts ER-positive cancers with an area under the receiver operating characteristic (AUROC) greater than 0.5 (e.g., greater than 0.55, greater than 0.6, greater than 0.65, greater than 0.7, greater than 0.75, greater than 0.8, greater than 0.85, greater than 0.9, or greater than 0.95), wherein subjects falling within the group of predicted ER-positive cancers display the validated epigenetic profile and subjects that do not fall within the group of ER-positive cancers lack the validated epigenetic profile. [0055] In some embodiments, the differential loci in step (c) were identified by comparing the genomic profile of one or more histone modifications and/or DNA methylation in (i) one or more ER-positive cell lines and (ii) one or more ER-negative cell lines. [0056] In some embodiments, the classifier in step (d) was trained on histone modification, chromatin accessibility, binding of transcription factor, and/or DNA methylation levels in the differential loci that were obtained by in silico mixing sequence data from one or more ER-positive cell lines and sequence data obtained from liquid biopsy samples of healthy subjects. [0057] In some embodiments, the validated classifier in step (e) was validated using liquid biopsy samples from the third cohort.
12366150v1
Attorney Docket No.2014191-0027 [0058] In some embodiments, the classifier in step (d) was trained on two or more histone modification levels in the differential loci. In some embodiments, the two or more histone modification levels comprise H3K4me3 and H3K27ac modification levels. [0059] In some embodiments, the classifier in step (d) was trained on one or more histone modification levels and DNA methylation in the differential loci. In some embodiments, the one or more histone modification levels comprise H3K4me3 and/or H3K27ac modification levels. In some embodiments, the classifier in step (d) was trained using ridge regression, elastic- net regression, or lasso regression. In some embodiments, the one or more histone modification levels comprise H3K4me3 and H3K27ac modification levels. In some embodiments, the biological sample is a liquid biopsy sample, e.g., a plasma sample, serum sample, or urine sample. [0060] In another aspect, the present disclosure provides a kit comprising reagents for quantifying one or more histone modifications, chromatin accessibility, binding of one or more transcription factors, and/or DNA methylation at one or more genomic loci, wherein the one or more genomic loci are selected from Tables 1-3. In some embodiments, the kit comprises reagents for quantifying H3K4me3 for at least 5, 10, 20, 30, 40, or 50 genomic loci in Table 1. In some embodiments, the kit comprises reagents for quantifying H3K27ac for at least 5, 10, 20, 30, 40, or 50 genomic loci in Table 2. In some embodiments, the kit comprises reagents for quantifying DNA methylation for at least 5, 10, 20, 30, 40, or 50 genomic loci in Table 3. [0061] In some embodiments, the kit comprises one or more antibodies for use in ChIP- seq, optionally wherein the one or more antibodies specifically bind H3K4me3- or H3K27ac- modified histones. In some embodiments, the kit comprises one or more methyl-binding domains for use in MBD-seq. [0062] In some embodiments, the kit comprises reagents for isolation of cell-free DNA (cfDNA) from a liquid biopsy sample. In some embodiments, the kit comprises reagents for library preparation for sequencing. In some embodiments, the kit comprises reagents for sequencing. In some embodiments, the kit comprises instructions for determining if a subject has an ER-positive cancer. [0063] In another aspect, the present disclosure provides a non-transitory computer readable storage medium encoded with a computer program, wherein the program comprises instructions that when executed by one or more processors cause the one or more processors to
12366150v1
Attorney Docket No.2014191-0027 perform operations to perform the method of any one of the aforementioned methods of determining ER status. [0064] In another aspect, the present disclosure provides a computer system comprising a memory and one or more processors coupled to the memory, wherein the one or more processors are configured to perform operations to perform the method of any one of the aforementioned methods of determining ER. [0065] In another aspect, the present disclosure provides a system for determining the ER status of a cancer in a subject, the system comprising a sequencer configured to generate a sequencing dataset from a sample; and a non-transitory computer readable storage medium of and/or a computer system of the present disclosure. In some embodiments, the sequencer is configured to generate a Whole Genome Sequencing (WGS) data set from the sample. In some embodiments, the system further comprises a sample preparation device. In some embodiments, the sample preparation device is configured to prepare the sample for sequencing from a biological sample, optionally a liquid biopsy sample. In some embodiments, the sample preparation device comprises reagents for quantifying one or more histone modifications, chromatin accessibility, binding of one or more transcription factors, and/or DNA methylation at one or more genomic loci in cell-free DNA (cfDNA) from the biological sample, optionally the liquid biopsy sample. In some embodiments, the one or more genomic loci are selected from Tables 1-3. In some embodiments, the device comprises reagents for quantifying H3K4me3, e.g., for at least 5, 10, 20, 30, 40, or 50 genomic loci in Table 1. In some embodiments, the device comprises reagents for quantifying H3K27ac, e.g., for at least 5, 10, 20, 30, 40, or 50 genomic loci in Table 2. In some embodiments, the device comprises reagents for quantifying DNA methylation, e.g., for at least 5, 10, 20, 30, 40, or 50 genomic loci in Table 3. In some embodiments, the reagents comprise one or more antibodies for use in ChIP-seq, optionally wherein the one or more antibodies specifically bind H3K4me3- or H3K27ac-modified histones. In some embodiments, the reagents comprise one or more methyl-binding domains for use in MBD-seq. In some embodiments, the device comprises reagents for isolation of cell-free DNA (cfDNA) from the biological sample, optionally the liquid biopsy sample. In some embodiments, the device comprises reagents for library preparation for sequencing. In some embodiments, the sequencer comprises reagents for sequencing.
12366150v1
Attorney Docket No.2014191-0027 [0066] In some embodiments a method is for determining ER status of a cancer in a subject (e.g., patient). The method may include receiving (e.g., by a processor of a computing device) one or more genomic profiles of one or more histone modifications, chromatin accessibility, binding of one or more transcription factors, and/or DNA methylation for the subject. The method may further include determining whether the subject has an epigenetic profile indicative of an ER- positive cancer by classifying the genomic profile using an ER classifier. [0067] In some embodiments, an ER classifier has been trained using one or more genomic profiles of one or more histone modifications, chromatin accessibility, binding of one or more transcription factors, and/or DNA methylation for (i) one or more ER-positive cell lines and one or more ER-negative cell lines and/or (ii) one or more biological samples obtained from one or more cohorts of subjects who have previously been determined to have an ER-positive cancer and one or more biological samples obtained from one or more cohorts of subjects who have previously been determined to have an ER-negative cancer. In some embodiments, genomic profiles correspond to samples that have been diluted in silico by mixing different proportions of sequencing fragments from healthy donor plasma samples and cell lines to achieve a simulated ctDNA percentage ranging from 0.5% to 50%. [0068] In some embodiments, one or more genomic profiles are for differential genomic loci found to have statistically significant different levels of one or more histone modifications, chromatin accessibility, binding of one or more transcription factors, and/or DNA methylation levels between one or more ER-positive cell lines and one or more ER-negative cell lines and/or between one or more biological samples obtained from one or more cohorts of subjects who have previously been determined to have an ER-positive cancer and one or more biological samples obtained from one or more cohorts of subjects who have previously been determined to have an ER-negative cancer. In some embodiments, differential loci were identified by comparing the genomic profile of one or more histone modifications and/or DNA methylation in (i) one or more ER-positive cell lines and (ii) one or more ER-negative cell lines. In some embodiments, an ER classifier has been trained on histone modification, chromatin accessibility, binding of transcription factor, and/or DNA methylation levels in the differential loci that were obtained by in silico mixing sequence data from one or more ER-positive cell lines and sequence data obtained from liquid biopsy samples of healthy subjects.
12366150v1
Attorney Docket No.2014191-0027 [0069] In some embodiments, an ER classifier has been trained on two or more histone modification levels in the differential loci. In some embodiments, an ER classifier has been trained on one or more histone modification levels and DNA methylation levels in the differential loci. In some embodiments, a genomic profile of a subject used to classify comprises two or more histone modification levels. In some embodiments, two or more histone modification levels comprise H3K4me3 and H3K27ac modification levels. In some embodiments, a genomic profile of a subject used to classify comprises one or more histone modification levels and DNA methylation levels. In some embodiments, one or more histone modification levels comprise H3K4me3 and/or H3K27ac modification levels. In some embodiments, one or more histone modification levels comprise H3K4me3 and H3K27ac modification levels. [0070] In some embodiments, an ER classifier has been trained with data derived from plasma. In some embodiments, an ER classifier has been trained with data derived from liquid biopsy samples. [0071] In some embodiments, an ER classifier is a validated classifier. In some embodiments, an ER classifier has been validated by selecting a threshold such that the validated classifier predicts ER-positive cancers with an area under the receiver operating characteristic (AUROC) greater than 0.5 (e.g., greater than 0.55, greater than 0.6, greater than 0.65, greater than 0.7, greater than 0.75, greater than 0.8, greater than 0.85, greater than 0.9, or greater than 0.95). In some embodiments, an ER classifier has been validated on a cohort of an independent group of subjects with ER-positive and ER-negative cancers, wherein subjects falling within a group of predicted ER-positive cancers display a validated epigenetic profile and subjects that do not fall within a group of ER-positive cancers lack the validated epigenetic profile. In some embodiments, an ER classifier has been validated using liquid biopsy sample data. [0072] A non-transitory computer readable storage medium may be encoded with a computer program, where the program may comprise instructions that when executed by one or more processors cause the one or more processors to perform operations to perform a method for determining ER status of a cancer in a subject (e.g., patient). A computer system may include a memory and one or more processors coupled to the memory, wherein the one or more processors are configured to perform operations to perform a method for determining ER status of a cancer in a subject (e.g., patient).
12366150v1
Attorney Docket No.2014191-0027 [0073] In some embodiments, a method of treating a subject having a cancer includes administering an ER-targeted agent to the subject, wherein the subject has been determined to have a validated epigenetic profile indicative of an ER-positive cancer based on analysis of a biological sample, optionally of cell-free DNA (cfDNA) from a liquid biopsy sample, obtained or derived from the subject. In some embodiments, the presence of the validated epigenetic profile has been determined using a classifier (e.g., a validated classifier) according to a method for determining ER status of a cancer in a subject (e.g., patient). BRIEF DESCRIPTION OF THE DRAWING [0074] Fig.1 shows ROC curves for exemplary ER status classifiers that were generated in accordance with Example 2. As shown, different classifiers were generated using (i) ridge regression (all genomic loci from the relevant subset of genomic loci) (alpha = 0); (ii) elastic-net regression (many genomic loci from the relevant subset of genomic loci) (alpha = 0.25), or (iii) lasso regression (few genomic loci from the relevant subset of genomic loci) (alpha = 1). As shown, different classifiers were generated with (a) genomic loci from Tables 1-3 for different modifications, namely (i) H3K4me3 modifications, (ii) H3K27ac modifications, (iii) DNA methylation (DNAme) or (iv) all of the above (All) and (b) using different subsets of genomic loci in Tables 1-3 for a particular modification, namely (i) all genomic loci with an absolute log2(fold-change) ≥ 0.5, (ii) all genomic loci with an absolute log2(fold-change) ≥ 1, (iii) all genomic loci with an absolute log2(fold-change) ≥ 2, (iv) all genomic loci with an absolute log2(fold-change) ≥ 3, and (v) all genomic loci with an absolute log2(fold-change) ≥ 4. [0075] Fig.2 shows representative, non-limiting graphs that demonstrate the accuracy of ER status (based on AUCROC) determination using the classifiers that were generated in accordance with Example 2. [0076] Fig.3 (A) shows a heatmap representation of z-scored, ctDNA- and background- normalized counts at differential peaks (DE-seq, FDR < 0.05, log2(fold change) > 1) across ER +/- patients (status determined by IHC). Each row corresponds to signal observed in an individual patient, and each column represents an enhancer/promoter/MBD locus. (B) shows ROC curves for an exemplary ER status classifier generated in accordance with Example 3 and applied to plasma samples obtained from patients previously diagnosed with metastatic breast cancer. ROC curves assessing performance of a regularized logistic regression model to classify
12366150v1
Attorney Docket No.2014191-0027 ER IHC status (positive vs negative) in all samples collected (left curve), all samples with detectable ctDNA (as determined by iChor) (center curve), and samples with detectable ctDNA obtained from one particular source (right curve) are shown. Without wishing to be bound by theory, it is believed that the improved performance observed in samples obtained from the one particular source is due to higher quality of the samples and the pathology of the cancer. (C) shows the summed weights of the negative (left) and positive (right) coefficients in the ER model, stratified by analyte. Genes of known biological importance that were found proximal to enhancers/promoters are highlighted. [0077] Fig.4 is a block diagram of an example network environment for use in the methods and systems described herein, according to illustrative embodiments of the present disclosure. [0078] Fig.5 is a block diagram of an example computing device and an example mobile computing device, for use in illustrative embodiments of the present disclosure. DETAILED DESCRIPTION [0079] The present disclosure is based, at least in part, on the demonstration that the ER status of a cancer in a subject can be determined by detecting and quantifying the presence of histone modifications and/or DNA methylation at one or more genomic loci in cell-free DNA (cfDNA) from a liquid biopsy sample, e.g., a plasma sample obtained or derived from the subject. The present disclosure also encompasses methods where chromatin accessibility and/or binding of one or more transcription factors are detected at the one or more genomic loci instead of (or in addition to) histone modifications and/or DNA methylation. The present disclosure is also based, at least in part, on the demonstration that genomic loci that are differentially modified based on different types of histone modifications (e.g., histone methylation marks such as H3K4me3 and histone acetylation marks such as H3K27ac) and/or DNA methylation can be combined into multimodal classifiers to determine ER status. These new monomodal and multimodal classifiers provide minimally invasive ways of determining ER status that are more accurate, objective, and comprehensive than the current tissue-based approaches. No liquid biopsy platform to date has been able to provide actionable resolution on a transcriptionally regulated phenotype relevant for therapy such as ER status.
12366150v1
Attorney Docket No.2014191-0027 ER status and cancer [0080] Estrogens are steroidal hormones that function as the primary female sex hormone. There are three major forms of estrogen, namely estrone (E1), estradiol (E2) and estriol (E3). Estradiol (E2) is the predominant estrogen in nonpregnant females, while estrone (E1) and estriol (E3) are primarily produced during pregnancy and following the onset of menopause, respectively. All estrogens are produced from androgens through actions of enzymes such as aromatase. Follicle-stimulating hormone and luteinizing hormone stimulate the synthesis of estrogen in the ovaries. However, some estrogens are also produced in smaller amounts by other tissues such as the liver, adrenal glands, and mammary gland. Studies have shown that estrogen is associated with mammary tumorigenesis, ovarian and endometrial carcinogenesis (Folkerd and Dowsett, J Clin Oncol (2010) 28:4038-4044). Also, mounting evidence suggests that estrogen and its target gene encoding progesterone receptor (PR) play critical roles in regulating breast cancer progression (Knutson et al., J Hematol Oncol (2017) 10:89). [0081] The biological effects of estrogen are mostly mediated by its binding and activation of ERα and ERβ, which are members of the nuclear receptor superfamily of transcription factors that are characterized by highly conserved DNA- and ligand-binding domains (Wang et al., J Hematol Oncol (2017) 10:168). The DNA binding domain, which is extremely well conserved between ERα and ERβ (97% homology), contains two functionally distinct zinc finger motifs that are responsible for specific DNA binding, as well as mediating receptor dimerization (Hewitt and Korach, Endocr Rev (2018) 39(5):664-675). The unliganded ER has been shown to be present in a cytosolic complex with hsp90 and associated proteins, with ligand binding allowing dissociation from the hsp90 complex, receptor dimerization, nuclear localization and binding to estrogen response elements (EREs) in promoters of estrogen- regulated genes (Pratt and Toft, Endocr Rev (1997) 18:306-360). Genome-wide chromatin immunoprecipitation studies have confirmed that the majority of ER-binding sites in estrogen responsive genes conform well to this consensus sequence (Welboren et al., EMBO J (2009) 28:1418-1428). While ERα and ERβ can bind to most ERE identically, the differences in ERα and ERβ may lead to tethering differential transcription factors and then modulating different target genes. Thus, the activation of ERα or ERβ can produce both unique and overlapping effects.
12366150v1
Attorney Docket No.2014191-0027 [0082] ERα has also been shown to modulate gene transcription through heterodimerizing with other transcription factors such as activating protein 1 (AP1) and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB). There is a large profile of estrogen-responsive genes, including pS2, cathepsin D, c-fos, c-jun, c-myc, TGF-α, retinoic acid receptor α1, efp, progesterone receptor (PR), insulin-like growth factor 1 (IGF1) (Ikeda et al., Acta Pharmacol Sin (2015) 36:24-31). Many of these ER-regulated genes, including IGF1, cyclin D1, c-myc, and efp, are important for cell proliferation and survival. C-myc is a bona-fide oncogene that is amplified or overexpressed in a variety of human tumors. Efp is an ubiquitin ligase that promotes proteasomal degradation of 14-3-3 sigma thereby stimulating cellular proliferation. While PR is an estrogen-responsive gene, it may antagonize ERα action to inhibit tumor growth, particularly through interacting with RNA polymerase III and inhibiting tRNA transcription. [0083] A pool of ERα are located in the plasma membrane and cytoplasm (Adlanmerini et al., Proc Natl Acad Sci USA (2014) 111:E283-290), where it binds to diverse membrane or cytoplasmic signaling molecules such as the p85 regulatory subunit of class I phosphoinositide 3-kinase, mitogen-activated protein kinase (MAPK) and Src (Omarjee et al., Oncogene (2017) 36:2503-2514). Activation of these signal transduction pathways by estrogen initiates cell survival and proliferation signals. Additionally, these signaling molecules are able to phosphorylate the ERα and its co-regulators to augment nuclear ERα signaling (Arnal et al., Physiol Rev (2017) 97:1045-1087). The genomic and non-genomic actions of ERα play a crucial role in breast epithelial cell proliferation and survival, as well as mammary tumorigenesis [28]. The purpose of this review is to decipher the complex mechanisms underlying the aberrant expression of ERα and ERβ in human cancer. [0084] Based on the ER status, breast tumors can be classified as ER-positive and ER- negative. About 75% of breast cancer cases are ERα positive at diagnosis (Allred et al., Breast Cancer Res (2004) 6:240-245). To determine if a cancer is ER-positive, medical practitioners currently order testing that is conducted on a tissue sample using immunohistochemistry (IHC). Samples are reviewed by a pathologist and typically reported as (a) the word positive or negative, (b) a percentage that tells you how many cells out of 100 stained positive for hormone receptors, i.e., a number between 0% (none have receptors) and 100% (all have receptors), and/or (c) an Allred score between 0 and 8. The Allred scoring system looks at what percentage
12366150v1
Attorney Docket No.2014191-0027 of cells test positive for hormone receptors, along with how well the receptors show up after staining, called intensity (Allred et al., Breast Cancer Res (2004) 6:240-245). This information is then combined to score the sample on a scale from 0 to 8 where, the higher the score, the more receptors were found and the easier they were to see in the sample. The terms “ER-positive” and “ER-negative” as used herein can correspond to any of these traditional approaches for determining ER status. [0085] ER-positive cancers can be treated with ER-targeted agents that lower estrogen levels or block estrogen receptors. ER-positive cancers tend to grow more slowly than those that are ER-negative. Women with hormone receptor-positive breast cancers tend to have a better outlook in the short-term, but these cancers can sometimes come back many years after treatment. [0086] Treatment with ER-targeted agents is not helpful for ER-negative cancers. These cancers may instead be treated with one or more of surgery and/or radiation, HER2-targeted therapy (if HER2-positive), chemotherapy and immunotherapy. These cancers tend to grow faster than ER-positive cancers. If they come back after treatment, it is often in the first few years. ER-negative breast cancers are more common in women who have not yet gone through menopause. ER-targeted agents [0087] The introduction of ER-targeted agents has dramatically influenced the outcome of patients with ER-positive breast cancers. ER-targeted agents block or degrade estrogen receptors or lower estrogen levels. Many ER-targeted agents have already been approved and others are in development or being tested in clinical trials for ER-positive breast cancer and other ER-positive cancers. Agents that block or degrade estrogen receptors (ER) [0088] These agents stop estrogen from fueling breast cancer cells to grow. These agents work by preventing estrogen from activating estrogen receptors. They do this by blocking estrogen from binding to estrogen receptors or by degrading estrogen receptors. The former are called Selective Estrogen Receptor Modulators (SERMs) while the latter are called Selective Estrogen Receptor Degraders (SERDs).
12366150v1
Attorney Docket No.2014191-0027 Selective Estrogen Receptor Modulators (SERMs) [0089] SERMs bind estrogen receptors and block them from binding to estrogen. These agents are pills, taken orally. Tamoxifen [0090] Tamoxifen can be used to treat women with breast cancer who have or have not gone through menopause. This agent can be used in several ways. In women at high risk of breast cancer, tamoxifen can be used to help lower the risk of developing breast cancer. [0091] For women who have been treated with breast-conserving surgery for ductal carcinoma in situ (DCIS) that is ER-positive, taking tamoxifen for 5 years lowers the chance of the DCIS coming back in the same breast. It also lowers the chance of getting an invasive breast cancer or another DCIS in both breasts. [0092] For women with ER-positive invasive breast cancer treated with surgery, tamoxifen can help lower the chances of the cancer coming back and improve the chances of living longer. It can also lower the risk of a new cancer developing in the other breast. Tamoxifen can be started either after (adjuvant) or before (neoadjuvant) surgery. When given after surgery, it is usually taken for 5 to 10 years. This drug is used mainly for women with early-stage breast cancer who have not yet gone through menopause. If the subject has gone through menopause, aromatase inhibitors (see below) are often used instead. [0093] For women with ER-positive breast cancer that has spread to other parts of the body, tamoxifen can often help slow or stop the growth of the cancer and might even shrink some tumors. Toremifene [0094] Toremifene is a SERM that works in a similar way to tamoxifen, but it is used less often and is only approved to treat post-menopausal women with metastatic breast cancer. It is not likely to work if tamoxifen has already been used and has stopped working. Selective estrogen receptor degraders (SERDs)
12366150v1
Attorney Docket No.2014191-0027 [0095] Like SERMs, these agents bind estrogen receptors but do so in a manner that causes them to be degraded. SERDs are used most often in post-menopausal women. When given to pre-menopausal women, they need to be combined with a luteinizing-hormone releasing hormone (LHRH) agonist to turn off the ovaries. Fulvestrant [0096] This ER-targeted agent can be used (i) alone to treat advanced breast cancer that has not been treated with other hormone therapy, (ii) alone to treat advanced breast cancer after other hormone drugs (like tamoxifen and often an aromatase inhibitor) have stopped working, or (iii) in combination with a CDK 4/6 inhibitor or PI3K inhibitor to treat metastatic breast cancer as initial hormone therapy or after other hormone treatments have been tried. It is given as two injections into the buttocks (bottom). For the first month, the two shots are given two weeks apart. After that, they are given once a month. Elacestrant [0097] This ER-targeted can be used to treat advanced, ER-positive, HER2-negative breast cancer when the cancer cells have an ESR1 gene mutation, and the cancer has grown after at least one other type of hormone therapy. Elacestrant is taken daily as pills, orally. Drugs that lower estrogen levels [0098] Because estrogen stimulates ER-positive cancers to grow, lowering the estrogen level can help slow the cancer’s growth or help prevent it from coming back. Aromatase inhibitors (AIs) [0099] Aromatase inhibitors (AIs) are drugs that stop most estrogen production in the body. Before menopause, most estrogen is made by the ovaries. But in women whose ovaries are not working, either because they have gone through menopause or because of certain treatments, estrogen is still made in body fat by an enzyme called aromatase. AIs work by preventing aromatase from making estrogen. [0100] These drugs are useful for women who have gone through menopause, although they can also be used in pre-menopausal women when they are combined with ovarian
12366150v1
Attorney Docket No.2014191-0027 suppression. These AIs are pills taken orally every day to treat breast cancer and include letrozole, anastrozole, and exemestane. Other ER-targeted agents and other cancers [0101] While the sections above focus on FDA approved ER-targeted agents, many other ER-targeted agents are being developed and/or assessed in clinical trials. It is to be understood that these other ER-targeted agents can also be used in treatment methods of the present disclosure. In addition, while the sections above focus on the treatment of ER-positive breast cancer, many of these ER-targeted agents can also be used to treat other ER-positive cancers, e.g., ovarian or endometrial ER-positive cancers. Subjects and Samples [0102] A sample analyzed using methods, kits and systems provided herein can be any biological sample including any processed sample that includes circulating tumor DNA (ctDNA) derived from a biological sample. In various embodiments, a sample analyzed using methods, kits and systems provided herein can be a sample obtained from a mammalian subject. In various embodiments, a sample analyzed using methods, kits and systems provided herein can be a sample obtained from a human subject. [0103] In various instances, a human subject is a subject diagnosed or seeking diagnosis as having, diagnosed as, or seeking diagnosis as at risk of having, and/or diagnosed as or seeking diagnosis as at immediate risk of having, an ER-positive cancer, e.g., ER-positive breast cancer, etc. In various instances, a human subject is a subject identified as needing ER status screening. In certain instances, a human subject is a subject identified as needing ER status screening by a medical practitioner. [0104] The subject may not have undergone previous treatments for cancer, such as the treatments recited in this disclosure. In other embodiments, the subject has undergone previous treatments for cancer, such as the treatments recited in this disclosure. [0105] In various embodiments a subject has one or more biomarkers and/or risk factors for cancer, e.g., ER-positive cancer, e.g., ER-positive breast cancer, etc. In certain embodiments, a human subject is identified as in need of ER status screening based on an initial cancer diagnosis, e.g., a breast cancer, etc. diagnosis. In various instances, a human subject is a subject
12366150v1
Attorney Docket No.2014191-0027 not yet diagnosed as having, not at risk of having, not at immediate risk of having, not diagnosed as having, and/or not seeking diagnosis for a cancer. Genetic factors may also contribute to ER- positive cancer risk, as evidenced by individuals with a family history of ER-positive cancer. [0106] In various embodiments, a sample from a subject, e.g., a human can be obtained from a liquid biopsy. In certain embodiments, a sample and/or reference is obtained from serum, plasma, or urine. In certain embodiments, the sample is serum. In certain embodiments, a sample comprises circulating tumor DNA (ctDNA). In certain embodiments, a sample is derived from about 1 mL of blood obtained from the subject. In certain embodiments, a sample is derived from about 0.5-5 mL of blood obtained from the subject, e.g., about 0.5 to about 2 mL, about 0.5 to 1.75 mL, about 0.5 to 1.5 mL, about 0.75 to 1.25 mL, about 0.9 to 1.1 mL, about 1 mL, about 2 mL, about 3 mL, about 4 mL, or about 5 mL of blood. [0107] In various embodiments, a sample is a sample of cell-free DNA (cfDNA). cfDNA is typically found in human biofluids (e.g., plasma, serum, or urine) in short, double-stranded fragments. The concentration of cfDNA is typically low, but can significantly increase under particular conditions, including without limitation pregnancy, autoimmune disorders, myocardial infarction, and cancer. Circulating tumor DNA (ctDNA) is the component of cell-free DNA specifically derived from cancer cells. ctDNA can be present in human biofluids bound to leukocytes and erythrocytes or not bound to leukocytes and erythrocytes. Various tests for detection of tumor-derived ctDNA are based on detection of genetic or epigenetic modifications that are characteristic of cancer (e.g., of a relevant cancer). Genetic or epigenetic factors characteristic of cancer can include, without limitation, oncogenic or cancer-associated mutations in tumor-suppressor genes, activated oncogenes, chromosomal disorders, histone modifications (e.g., histone methylation and/or histone acetylation), chromatin accessibility, binding of one or more transcription factors and/or DNA methylation. [0108] In various embodiments, ctDNA comprises less than 30%, less than 20%, or less than 10% of the cfDNA in the liquid biopsy sample obtained from the subject, e.g., less than 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2% or less than 1% of the cfDNA in the sample. In some embodiments, the percentage of ctDNA in the liquid biopsy sample is assessed using ichorCNA which estimates the percentage of ctDNA in a sample probabilistically (see Adalsteinsson et al., Nat Commun (2017) 8(1):1324 the entire contents of which are incorporated herein by reference).
12366150v1
Attorney Docket No.2014191-0027 [0109] cfDNA and ctDNA can provide a real-time or nearly real time metric of status of a source tissue. cfDNA and ctDNA demonstrate a half-life in blood of about 2 hours, such that a sample taken at a given time provides a relatively timely reflection of the status of a source tissue. [0110] In some embodiments, a method comprises isolating DNA (e.g., cfDNA) from a liquid biopsy sample (e.g., from 1, 2, 3, 4, or 5 mL of a liquid biopsy sample). Various methods of isolating nucleic acids from a sample (e.g., of isolating cfDNA from blood or plasma) are known in the art. Nucleic acids can be isolated using, without limitation, standard DNA purification techniques, by direct gene capture (e.g., by clarification of a sample to remove assay-inhibiting agents and capturing a target nucleic acid, if present, from the clarified sample with a capture agent to produce a capture complex and isolating the capture complex to recover the target nucleic acid). [0111] Reagents and protocols for obtaining and analyzing cfDNA and ctDNA, such as circulating in blood or other tissue, are commercially available as described in the Examples and well-known in the art (see, for example, Anker et al., Cancer and Metastasis Rev (1999) 18:65- 73; Wua et al., Clin Chim Acta (2002) 321:77-87; Fiegl et al., Cancer Res (2005) 15:1141-1145; Pathak et al., Clin Chem (2006) 52:1833-1842; Schwarzenbach et al., Clin Cancer Res (2009) 15:1032-1038; Schwarzenbach et al., Nat Rev Cancer (2011) 11:426-437) the contents of each of which is separately incorporated herein by reference in their entirety). [0112] In various embodiments, samples can be collected from individuals repeatedly over a period of time (e.g., once daily, weekly, monthly, annually, biannually, etc.). In various embodiments, such samples can be used to verify results from earlier detections and/or to identify an alteration in biological pattern because of, for example, disease progression, resistance to therapy, treatment, remission, and the like. For example, subject samples can be taken and monitored every month, every two months, or combinations of one, two, or three- month intervals according to the present disclosure. In various embodiments, samples can be collected for monitoring over time beginning at or at certain clinically determined stages, such as at resistance to a therapy, before radiographic progression, after radiographic progression, and/or at tissue biopsy. In addition, the ER status obtained at different points in time can be conveniently compared with each other, as well as with those of normal controls during the
12366150v1
Attorney Docket No.2014191-0027 monitoring period, thereby providing the subject’s own values, as an internal, or personal, control for long-term monitoring. [0113] Samples include materials prepared by processes including, without limitation, steps such as concentration, dilution, adjustment of pH, removal of high abundance polypeptides (e.g., albumin, gamma globulin, and transferrin, etc.), addition of preservatives, addition of calibrants, addition of protease inhibitors, addition of denaturants, desalting, concentration and/or extraction of sample nucleic acids, and/or amplification of sample nucleic acids (e.g., by PCR or other nucleic acid amplification techniques). Samples also include materials prepared by techniques that isolate, e.g., nucleosomes or transcription factors and/or nucleic acids associated with nucleosomes or transcription factors. [0114] Removal from a sample of proteins that are not desirable for a relevant purpose or context (e.g., high abundance, uninformative, or undetectable proteins) can be achieved using high affinity reagents, high molecular weight filters, ultracentrifugation and/or electrodialysis. High affinity reagents include antibodies or other reagents (e.g., aptamers) that selectively bind to high abundance proteins. Sample preparation can also include ion exchange chromatography, metal ion affinity chromatography, gel filtration, hydrophobic chromatography, chromatofocusing, adsorption chromatography, isoelectric focusing and related techniques. Molecular weight filters include membranes that separate molecules based on size and molecular weight. Such filters may further employ reverse osmosis, nanofiltration, ultrafiltration and microfiltration. Ultracentrifugation is the centrifugation of a sample at about 15,000-60,000 rpm while monitoring with an optical system the sedimentation (or lack thereof) of particles. Electrodialysis is a procedure which uses an electromembrane or semipermeable membrane in a process in which ions are transported through semi-permeable membranes from one solution to another under the influence of a potential gradient. Since the membranes used in electrodialysis may have the ability to selectively transport ions having positive or negative charge, reject ions of the opposite charge, or to allow species to migrate through a semipermeable membrane based on size and charge, it renders electrodialysis useful for concentration, removal, or separation of electrolytes. [0115] Separation and purification in the present disclosure may include any procedure known in the art, such as capillary electrophoresis (e.g., in capillary or on-chip) or chromatography (e.g., in capillary, column or on a chip). Electrophoresis is a method that can be
12366150v1
Attorney Docket No.2014191-0027 used to separate ionic molecules under the influence of an electric field. Electrophoresis can be conducted in a gel, capillary, or in a microchannel on a chip. Examples of gels used for electrophoresis include starch, acrylamide, polyethylene oxides, agarose, or combinations thereof. A gel can be modified by its cross-linking, addition of detergents, or denaturants, immobilization of enzymes or antibodies (affinity electrophoresis) or substrates (zymography) and incorporation of a pH gradient. Examples of capillaries used for electrophoresis include capillaries that interface with an electrospray. [0116] Capillary electrophoresis (CE) is preferred for separating complex hydrophilic molecules and highly charged solutes. CE technology can also be implemented on microfluidic chips. Depending on the types of capillary and buffers used, CE can be further segmented into separation techniques such as capillary zone electrophoresis (CZE), capillary isoelectric focusing (CIEF), capillary isotachophoresis (CITP) and capillary electrochromatography (CEC). An embodiment to couple CE techniques to electrospray ionization involves the use of volatile solutions, for example, aqueous mixtures containing a volatile acid and/or base and an organic such as an alcohol or acetonitrile. [0117] Capillary isotachophoresis (CITP) is a technique in which the analytes move through the capillary at a constant speed but are nevertheless separated by their respective mobilities. Capillary zone electrophoresis (CZE), also known as free-solution CE (FSCE), is based on differences in the electrophoretic mobility of the analytes, determined by the charge on the analytes, and the frictional resistance the analytes encounter during migration, which is often directly proportional to the size of the analytes. Capillary isoelectric focusing (CIEF) allows weakly-ionizable amphoteric molecules, to be separated by electrophoresis in a pH gradient. CEC is a hybrid technique between traditional high performance liquid chromatography (HPLC) and CE. [0118] Separation and purification techniques used in the present disclosure can include any chromatography procedures known in the art. Chromatography can be based on the differential adsorption and elution of certain analytes or partitioning of analytes between mobile and stationary phases. Different examples of chromatography include, but not limited to, liquid chromatography (LC), gas chromatography (GC), high performance liquid chromatography (HPLC), etc.
12366150v1
Attorney Docket No.2014191-0027 [0119] In some embodiments, whole blood is collected from a subject, and a plasma layer is separated by centrifugation. cfDNA may be then extracted from the plasma using methods known in the art. Histone Modifications, Chromatin Accessibility and Transcription Factor Binding [0120] Histone methylation is understood to increase or decrease expression of associated coding sequences, depending on which histone residue is methylated. Histone methylation is an essential modification that can cause monomethylation (me1), dimethylation (me2), and trimethylation (me3) of several amino acids, thus directly affecting heterochromatin formation, gene imprinting, X chromosome inactivation, and gene transcriptional regulation. Histone methyltransferases promote monomethylation, dimethylation, or trimethylation of histones while histone demethylases promote demethylation of histones. In general, lysine (Lys or K), arginine (Arg or R), and rarely histidine (His or H) are the most common histone methyl acceptors. Histone methylation only occurs at specific lysine and arginine sites of histone H3 and H4. In histone H3, lysine 4, 9, 26, 27, 36, 56, and 79 and arginine 2, 8, and 17 can be methylated. By comparison, histone H4 has fewer methylation sites, in which only lysine 5, 12, and 20 and arginine 3 can be methylated. Histone methylation is often associated with transcriptional activation or inhibition of downstream genes. The methylation of histone H3K4, R8, R17, K26, K36, K79, H4R3, and K12 can activate gene transcription. However, the methylation of histone H3K9, K27, K56, H4K5, and K20 can inhibit gene transcription. Thus, for example, H3K4 methylation generally activates gene expression, while H3K27 methylation generally represses gene expression. [0121] Histone acetylation occurs predominantly at lysine residues and is generally understood to increase expression of associated coding sequences. Without wishing to be bound by any theory, acetylation of lysine residues is thought to neutralize lysine’s positive charge and thereby cause histones to drift away from DNA, which has a negative charge. The released structure facilitates access to transcriptional machinery such as transcription factors and RNA polymerase II. Histone acetylation and deacetylation are generally catalyzed by histone acetyltransferases (HATs) and HDACs, respectively. Acetyl-CoA can be a source and co-factor of acetylation. In regulatory regions, HATs can acetylate histones and recruit HAT-containing complexes to activate the transcriptional process. For instance, H3K9ac and H3K27ac levels can
12366150v1
Attorney Docket No.2014191-0027 be associated with promoter and enhancer activities. Furthermore, H3K27ac enhances not only the kinetics of transcriptional activation, but also accelerates the transition of RNA polymerase II from the initiation state to the elongation state. [0122] Differential modification of a genomic locus (e.g., differential histone methylation and/or differential histone acetylation) can refer to, or be determined by or detected as, a comparative difference or change in modification status of one or more genomic loci between a first sample, condition, disease, or state and a second or reference sample, condition, disease, or state. Those of skill in the art will appreciate that a reference is typically produced by measurement using a methodology identical, similar, or comparable to that by which a compared non-reference measurement was taken. [0123] Chromatin accessibility can refer to the degree to which nuclear macromolecules are able to physically contact DNA and is determined in part by the occupancy and modification status of nucleosomes. Modified histones can regulate chromatin accessibility through a variety of mechanisms, such as altering transcription factor (TF) binding through steric hindrance and modulating nucleosome affinity for active chromatin remodelers. The topological organization of nucleosomes across the genome is non-uniform: while histones can be densely arranged within facultative and constitutive heterochromatin, histones can be depleted at regulatory loci, including within enhancers, insulators and transcribed gene bodies. Active regulatory elements of the genome are generally accessible. [0124] Differential accessibility of a genomic locus can refer to, or be determined by or detected as, a comparative difference or change in modification status of one or more genomic loci between a first sample, condition, disease, or state and a second or reference sample, condition, disease, or state. Those of skill in the art will appreciate that a reference is typically produced by measurement using a methodology identical, similar, or comparable to that by which a compared non-reference measurement was taken. [0125] A reference can be a value or set of values that are predetermined or derived from a sample or set of samples. A reference can be a sample or set of samples. A reference value can be a predetermined threshold value, a value that varies in accordance with circumstances (e.g., according to patient subpopulation, age, weight, or other variables), or a ratio. Reference ratios can be ratios relating to the modification and/or accessibility of multiple loci within individual samples and/or references, or across or between samples and/or references. In various
12366150v1
Attorney Docket No.2014191-0027 embodiments, a reference can have or represent a normal, non-diseased state. In some embodiments, such as for staging of disease or for evaluating the efficacy of treatment, a reference can have or represent a diseased state, e.g., a cancer, stage of cancer, or subtype of cancer, e.g., ER-positive cancer or ER-negative cancer. In some embodiments, a reference can represent an ER+ cancer with an Allred score of 3, 4, 5, 6, 7 or 8 based on IHC testing or an ER+ cancer with an Allred score of at least 3, at least 4, at least 5, at least 6, at least 7 or 8 based on IHC testing. In some embodiments, a reference can represent a represent an ER- cancer with an Allred score of 0, 1, or 2 based on IHC testing. In some embodiments, a reference can correspond to a subject having breast cancer and/or a breast cancer subtype, e.g., ER-positive or ER-negative breast cancer. [0126] In some embodiments, a reference is a predetermined threshold. In some embodiments, the predetermined threshold has previously been shown to be capable of distinguishing ER-positive and ER-negative cancers (e.g., distinguish with an AUROC of greater than 0.5). In some embodiments, a reference is a measurement from a liquid biopsy sample. In some embodiments, a reference is a measurement from liquid biopsy samples obtained from a cohort of subjects. In some embodiments, a reference is a normalized sample. In some embodiments, a reference is a measurement obtained from liquid biopsy samples obtained from a cohort of subjects who have previously been determined to have an ER-positive or ER-negative cancer, including, e.g., an ER-positive or ER-negative breast cancer. [0127] In certain instances, a reference is a non-contemporaneous sample from the same source, e.g., a prior sample from the same source, e.g., from the same subject. In certain instances, a reference for the modification status of one or more genomic loci (e.g., one or more differentially modified genomic loci) can be the modification status of the one or more genomic loci (e.g., one or more differentially modified genomic loci) in a sample (e.g., a sample from a subject), or a plurality of samples, known to represent a particular state (e.g., an ER-positive cancer or ER-negative cancer). In certain instances, a reference for the accessibility status of one or more genomic loci (e.g., one or more differentially accessible genomic loci) can be the accessibility status of the one or more genomic loci (e.g., one or more differentially accessible genomic loci) in a sample (e.g., a sample from a subject), or a plurality of samples, known to represent a particular state (e.g., an ER-positive cancer or ER-negative cancer).
12366150v1
Attorney Docket No.2014191-0027 [0128] In some illustrative but non-limiting embodiments of the present disclosure differential modification or differential accessibility can refer to a differential (e.g., between a sample and a reference) with an absolute log2(fold-change) that is greater than or equal to 0.5, 1.0, 1.5, 2.0, 2.5, 3.0, 3.5, 4.0 or more, or any range in between, inclusive, e.g., as measured according to an assay provided herein. In Tables 1-3, the log2(fold-change) values are based on ratios of ER-positive to ER-negative reads, i.e., positive log2(fold-change) values indicate that sequencing reads in a particular genomic locus are associated with an ER-positive status while a negative log2(fold-change) value indicates that sequencing reads in a particular genomic locus are associated with an ER-negative status. [0129] Enhancers are genomic loci that can be differentially modified or differentially accessible in and/or between conditions, diseases, and other states. Enhancers are cis-acting DNA regulatory regions that are thought to bind trans-acting proteins that contribute to expression patterns of associated genes. Chromatin ImmunoPrecipitation sequencing (ChIP-seq) of histone modifications (e.g., acetylation) have identified millions of enhancers in mammalian genomes. The number of active enhancers in any given cell type is estimated to be in the tens of thousands. Certain transcription factors (TFs), sometimes referred to as “master” transcription factors, associate with active enhancers with important impacts on gene expression and cell function. Certain such transcription factors preferentially associate with enhancers that regulate genes required for establishing cell identity and function, including enhancer domains known as “super-enhancers”. Moreover, master TFs can participate in inter-connected auto-regulatory circuitries or “cliques” that are self-reinforcing, show marked cell selectivity, and function to maintain cell state and/or cell survival. Techniques for Detecting and Quantifying Histone Modifications and Transcription Factor Binding [0130] Various techniques of molecular biology are well known in the art and/or disclosed in the present application for detecting and quantifying histone modifications and/or transcription factor binding. In some embodiments, the methods, kits and systems of present disclosure involve the detection and quantification of histone modifications and/or transcription factor binding in samples, e.g., in liquid biopsy samples including cfDNA such as plasma samples including cfDNA. Chromatin ImmunoPrecipitation (ChIP) is one technique of
12366150v1
Attorney Docket No.2014191-0027 molecular biology useful in detecting and quantifying histone modifications and transcription factor binding in samples. CUT&RUN or CUT&Tag are other more recent techniques that can also be used to detect and quantify histone modifications and transcription factor binding sites. ChIP-chip, ChIP-exo, ChIP Re-ChIP, and ChIPmentation are other alternative techniques that could be used. [0131] ChIP can involve various steps including one or more of fixation, sonication, immunoprecipitation, and analysis of the immunoprecipitated DNA. ChIP has become a very widely used tissue-based technique for determining the in vivo location of binding sites of various transcription factors and histones. Because the proteins are captured at the sites of their binding with DNA, ChIP helps to detect DNA-protein interactions that take place in living cells. More importantly, ChIP can be coupled to many commonly used molecular biology techniques such as PCR and real-time PCR, PCR with single-stranded conformational polymorphism, Southern blot analysis, Western blot analysis, cloning, and microarray. The resulting versatility has increased the potential of this technique. [0132] ChIP of tissue samples usually involves cross-linking of the chromatin-bound proteins by formaldehyde, followed by sonication or nuclease treatment to obtain small DNA fragments. Immunoprecipitation can be then carried out using specific antibodies to the DNA- binding protein of interest. The DNA can be then released from the proteins and analyzed using various methods. ChIP has also been used to study RNA-protein interactions. X-ChIP methods utilize fixed chromatin fragmented by sonication, while the N-ChIP methods utilize native chromatin, which can be unfixed and nuclease digested. [0133] The first step of the technique can be the cross-linking of DNA and proteins. Formaldehyde is one of the most used cross-linking agents. One advantage of using formaldehyde can be the ease of reversibility of the cross-links and its ability to form bonds that span approximately 2 angstroms. This means that formaldehyde can bind molecules in close association with each other. Generally, formaldehyde can be added to the medium in the cell culture flask or plate. It enters the cells through the cell membrane and cross-links the proteins to the chromatin. Formaldehyde fixation of tumor tissues has also been done. Other cross-linking agents that have been used include chemicals such as methylene blue and acridine orange, cisplatin, dimethylarsinic acid, potassium chromate, and ultraviolet (UV) light and lasers.
12366150v1
Attorney Docket No.2014191-0027 [0134] Harvested chromatin can be sonicated in one or more sonication cycles. DNA can be typically broken into to 100–500 bp fragments to pinpoint the location of the DNA sequence of interest. An alternative to sonication can be nuclease digestion of the chromatin, e.g., in N- ChIP methods. Purification of chromatin can be achieved using a cesium chloride (CsCl) gradient centrifugation. [0135] Chromatin can be enriched for a particular histone modification using an agent that binds the histone modification (e.g., immunoprecipitating using one or more antibodies that bind a target epitope). [0136] For example, an antibody used in ChIP can selectively bind a particular transcription factor or one or more particular histone modifications, such as one or more particular histone acetylation modifications or histone methylation modifications. In some embodiments, an antibody used to bind a target epitope can be a “pan” antibody (e.g., a pan- acetylation antibody, a pan-methylation antibody, an antibody that binds a group of histone modifications associated with increased transcription activation, and/or an antibody that binds a group of histone modifications associated with increased transcription repression). The antibody against the protein of interest is allowed to bind to the protein-DNA complex, and the complex can be then precipitated. Immunosorbants commonly used to separate the antigen-antibody complex from the lysate include salmon sperm DNA-protein A-Sepharose®, protein G, magnetic beads, and other engineered immunoprecipitation systems known to those of skill in the art. [0137] Immunoprecipitated DNA can be eluted. Once the DNA of interest is isolated, many detection and quantification methods can be used to study the isolated gene fragments. Commonly utilized methods include PCR, real-time PCR, slot blot hybridization, microarray techniques, and deep or next-generation sequencing. ChIP-seq combines chromatin immunoprecipitation (ChIP) with massively parallel DNA sequencing to identify the binding sites of DNA-associated proteins. ChIP-seq can be used to map DNA-binding proteins, e.g., transcription factor binding sites and histone modifications in a genome-wide manner. [0138] Cell-free Chromatin ImmunoPrecipitation sequencing (cfChIP-seq) involves applying ChIP-seq to samples that include cell-free DNA, e.g., liquid biopsy samples including cfDNA such as plasma samples including cfDNA (e.g., see Sadeh et al., Nat Biotechnol (2021) 39: 586–598 and Jang et al., Life Sci Alliance (2023) 6(12):e202302003 the entire contents of each of which are incorporated herein by reference). In some embodiments, cfChIP-seq uses
12366150v1
Attorney Docket No.2014191-0027 antibodies or antibody fragments that bind specific histone modifications (e.g., H3K4me3 and/or H3K27ac) and/or transcription factors that are coupled (covalently or non-covalently) to beads, e.g., magnetic beads such as Dynabeads® magnetic beads and incubated with a volume, e.g., about 1 mL of thawed plasma obtained from a subject. Without limitation, exemplary antibodies that bind H3K4me3 include PA5-27029 (available from Thermo Fisher Scientific in Waltham, MA) and C15410003 (available from Diagenode in Denville, NJ) and exemplary antibodies that bind H3K27ac include ab21623 or ab4729 (both available from Abcam in Cambridge, UK) and C15210016 (available from Diagenode in Denville, NJ). [0139] In some embodiments, the antibodies or antibody fragments can be covalently coupled to beads, e.g., epoxy beads. In some embodiments, the antibodies or antibody fragments can be non-covalently coupled to beads, e.g., Protein A or Protein G beads such as Dynabeads® Protein A or Dynabeads® Protein G beads. After washing, a cfDNA library is then typically prepared from the captured cfDNA. Library preparation can be done on-bead or after releasing the captured cfDNA by digestion of bound histones, e.g., using proteinase K. The cfDNA library is then sequenced to generate reads of captured cfDNA sequences, e.g., by next-generation sequencing (NGS) as is known in the art. The reads are then analyzed, e.g., aligned and counted using standard bioinformatic techniques as is known in the art. A cfChIP-seq bioinformatic pipeline can include, e.g., alignment of sequence reads to a reference genome with BWA or Bowtie2. Aligned reads can be used to call and quantify peaks as compared to a reference. In some embodiments, histone modifications at a given genomic loci can be quantified using sequencing data. E.g., in some embodiments, histone modifications can be quantified by counting the number of sequence reads that fall within a genomic loci (e.g., have at least one nucleotide overlapping with a genomic loci). In some embodiments, non-uniquely mapped and/or redundant sequence reads are discarded prior to quantifying histone modifications. In some embodiments, when quantifying histone modifications, sequence reads that fall within high noise regions of the genome are ignored. [0140] In some embodiments, sequence reads are adjusted on the basis of sequencing depth prior to counting. Adjusting on the basis of sequencing depth can include, e.g., quantile normalizing sequence reads to a common reference distribution. In some embodiments, sequence reads are adjusted on the basis of ChIP quality prior to counting. In some embodiments, sequence reads are normalized relative to aggregate counts across a set of regions (e.g., 1,000, 2,000,
12366150v1
Attorney Docket No.2014191-0027 3,000, 4,000, 5,000, 6,000, 7,000, 8,000, 9,000, 10,000 or more regions) previously determined to have DNAse hypersensitivity in most cell types. In some embodiments, an estimate of local background signal is subtracted from the count of sequence reads at each genomic loci. [0141] CUT&Tag involves antibody-based binding of a target protein, e.g., transcription factor or histone modification of interest, where antibody incubation is directly followed by the shearing of the chromatin and library preparation (see Kaya-Okur et al., Nat Comm (2019) 10:1930). CUT&Tag assays take advantage of a Tn5 transposase that is fused with Protein A to direct the enzyme to the antibody bound to its target on chromatin. Tn5 transposase is pre-loaded with sequencing adapters (generating the assembled pA-Tn5 adapter transposome) to carry out antibody-targeted tagmentation. In a typical CUT&Tag assay samples are incubated with an antibody immobilized on Concanavalin A-coated magnetic beads to facilitate subsequent washing steps. Cells can be incubated with a primary antibody specific for the target protein of interest followed by incubation with a secondary antibody. Samples can then be incubated with assembled transposomes, which consist of Protein A fused to the Tn5 transposase enzyme that is conjugated to NGS adapters. After incubation, unbound transposome can be washed away using stringent conditions. Tn5 is a Mg2+-dependent enzyme so Mg2+ can be added to activate the reaction, which results in the chromatin being cut close to the protein binding site and simultaneous addition of the NGS adapter DNA sequences. Chromatin cleavage and library preparation can be achieved in one single step. [0142] CUT&RUN is an epigenomic profiling strategy in which antibody-targeted controlled cleavage by micrococcal nuclease releases specific protein-DNA complexes into the supernatant for paired-end DNA sequencing (see Skene and Henikoff, Elife (2017) 6:1-35, Skene et al., Nat Protoc (2018) 13:1006-1019). As only targeted fragments enter into solution, and the vast majority of DNA is left behind, CUT&RUN has low background levels. In an example CUT&RUN assay, a sample is incubated with an antibody or antibody fragment that binds the target protein, e.g., transcription factor or histone modification of interest. The sample is then incubated with Protein-A-MNase after which CaCl2 can be added to initiate the calcium dependent nuclease activity of MNase to cleave the DNA around the target protein. The protein- A-MNase reaction can be quenched by adding chelating agents (EDTA and EGTA). Cleaved DNA fragments are then liberated, extracted, and used to construct a sequencing library.
12366150v1
Attorney Docket No.2014191-0027 [0143] A person of ordinary skill in the art is aware of suitable DNA sequencing technologies for use in methods described herein that comprise a sequencing step. Suitable DNA sequencing technologies include, e.g., next generation sequencing (NGS) approaches. Additional steps that are required to prepare DNA for sequencing via an appropriate sequencing approach can be incorporated into methods described herein. For example, in some embodiments, a method described herein comprises attaching (e.g., ligating) DNA adapters to cfDNA. In some embodiments, DNA adapters can be attached prior to, during, or after enrichment for a histone modification. In some embodiments, a method comprises amplifying cfDNA after attaching DNA adapters. Techniques for Detecting and Quantifying Chromatin Accessibility [0144] Various techniques of molecular biology are well known in the art and/or disclosed in the present application for detecting and quantifying chromatin accessibility. In some embodiments, the methods, kits and systems of the present disclosure involve the detection and quantification of chromatin accessibility in samples, e.g., in liquid biopsy samples including cfDNA such as plasma samples including cfDNA. ATAC-seq (Assay of Transpose Accessible Chromatin sequencing), NOMe-seq (Nucleosome Occupancy and Methylome sequencing), FAIRE-seq (Formaldehyde-Assisted Isolation of Regulatory Elements sequencing), MNase-seq (Micrococcal Nuclease digestion with sequencing), and DNase hypersensitivity assays are exemplary techniques of molecular biology useful in detecting and quantifying chromatin accessibility in samples. Sono-Seq is another alternative method that could be used (see Auerbach et al., Proc Natl Acad USA (2009) 106(35):14926-14931). [0145] DNase hypersensitivity assays can use the non-specific DNA endonuclease Deoxyribonuclease I (DNase I), which selectively digests accessible DNA regions. DNase I hypersensitivity sites (DHS) identified by DNase-seq include open chromatin regulatory regions. A typical DNase hypersensitivity assay can include a first step in which nuclei are isolated from cells using lysis buffer, and nuclei are digested using DNase I. DNA fragment sizes are measured to identify optimal digestion using gel electrophoresis. Biotinylated linkers can be ligated to the ends of digested DNA after polishing to make blunt ends, and the DNA can then be isolated. DNA with biotinylated linker can be digested by restriction endonuclease MmeI and captured by streptavidin coated Dynabeads® to generate short tags to which a second sequencing adaptor can
12366150v1
Attorney Docket No.2014191-0027 be ligated. A second linker can be ligated and amplified to generate a library for sequencing. A DNase-seq bioinformatic pipeline can include, e.g., alignment of sequence reads to a reference genome with BWA or Bowtie2. Aligned reads can be used to call and quantify peaks as compared to a reference. [0146] MNase-seq determines chromatin accessibility with micrococcal nuclease (MNase) that preferentially digests nucleosome-free, protein-unbound DNA. A typical MNase- seq assay can include a first step in which nuclei are isolated from either native or crosslinked chromatin and digested using MNase with titration. In vivo formaldehyde crosslinking step that is designed to capture the interaction between proteins and DNA. This crosslinking allows bound proteins to shield their associated DNA from digestion by MNase. Following crosslinking, samples are digested with MNase, which can be specifically activated by addition of Ca2+ to the buffer. Digestion can be halted by chelating the reaction, at which point the samples are RNase treated, crosslinks are reversed, and proteins are digested away from the chromatin. DNA can then be isolated via a phenol-chloroform extraction. Uncut DNA is purified and mononucleosome bands are isolated and excised through gel electrophoresis. Isolated DNA can be amplified by adding adapters to generate a library, and sequenced. MNase-seq primarily sequences regions of DNA bound by histones or other proteins. Therefore, it indirectly determines which regions of DNA are accessible by directly determining which regions are bound to nucleosomes or proteins. [0147] FAIRE-seq is a method in which nucleosome-depleted regions of DNA (NDRs) are isolated from chromatin. A typical FAIRE-seq assay can include a first step in which cells are fixed using formaldehyde so that histones are crosslinked to interacting DNA. Crosslinked chromatin can then be sheared by sonication that generates protein-free DNA and protein- crosslinked DNA fragments. Protein-free DNA can be isolated using a phenol–chloroform extraction: DNA crosslinked with protein stays in organic phase, while protein-free DNA stays in aqueous phase. Highly crosslinked DNA remains in the organic phase and the non-crosslinked DNA is pulled to the aqueous phase. Non-crosslinked DNA from the aqueous phase can then be amplified and sequenced. Reads enriched in the sequencing pool tend to have lower nucleosome and transcription factor binding and are therefore inferred to come from accessible regions. [0148] NOMe-seq is a method to identify nucleosome-depleted regions of DNA (NDRs) with M.CviPI methyltransferase that methylates cytosine in GpC dinucleotides not protected by
12366150v1
Attorney Docket No.2014191-0027 nucleosomes or other proteins. Unlike CmpG, GpCm in the human genome does not occur naturally in most cell types. GpCm levels at open chromatin regions can be compared to background signals and used to detect and quantify NDRs. A typical NOMe-seq protocol can include a step in which samples are treated with M.CviPI and S-adenosylhomocysteine (SAM) to methylate accessible GpC sites. M.CviPI treated DNA can be sheared using a sonicator, so that DNA fragments can be sequenced. DNA is treated with bisulfite, which converts unmethylated cytosine to uracil using sodium bisulfite, while methylated cytosine is unaffected. A library is generated using adapters and sequenced. Accessible chromatin is expected to have high levels of GpCm but low levels of CmpG. Therefore, NOMe-seq identifies NDRs using the two separate methylation analyses that serve as independent (but opposite) measures, providing matched chromatin designations for each regulatory element. [0149] ATAC-seq uses hyperactive Tn5 transposase that preferentially cuts accessible chromatin regions and simultaneously inserts adapters to the fragmented region (Buenrostro et al., Nat Methods (2013) 10(12):1213-1218 the entirety of which is incorporated herein by reference). A typical ATAC-seq assay can include a first step in which samples are incubated with Tn5 transposase. DNA can then be isolated and purified. DNA fragmented and tagged by Tn5 transposase can be purified and then amplified to generate a library and sequenced for analysis. Techniques for Detecting and Quantifying DNA Methylation [0150] Various techniques of molecular biology are well known in the art and/or disclosed in the present application for detecting and quantifying DNA methylation. In some embodiments, the methods, kits and systems of the present disclosure involve the detection and quantification of chromatin accessibility in samples, e.g., in liquid biopsy samples including cfDNA such as plasma samples including cfDNA. Bisulfite sequencing (BS-Seq), Whole Genome Bisulfite Sequencing (WGBS), Methylated DNA ImmunoPrecipitation sequencing (MeDIP-seq), or Methyl-CpG-Binding Domain sequencing (MBD-seq) are exemplary techniques of molecular biology useful in detecting and quantifying chromatin accessibility in samples. Reduced representation bisulfite sequencing (RRBS) is another alternative method that could be used (see Meissner et al., Nucleic Acids Res (2005) 33(18):5868-5877). Illumina Infinium arrays could also be used to detect and quantify DNA methylation.
12366150v1
Attorney Docket No.2014191-0027 [0151] DNA methylation typically refers to the methylation of the 5’ position of cytosine (mC) by DNA methyltransferases (DNMT). It is a major epigenetic modification in humans and many other species. In mammals, most DNA methylations occur within the context of CpG dinucleotides. DNA methylation is thought to be a repressive chromatin modification. Aberrant methylation can lead to many diseases including cancers (Robertson, Nat Rev Genet (2005) 6:597–610 and Bergman and Cedar, Nat Struct Mol Biol (2013) 20:274–281). [0152] Bisulfite sequencing (BS-Seq) or Whole-Genome Bisulfite Sequencing (WGBS) is a well-established protocol to detect methylated cytosines in genomic DNA. In this method, genomic DNA is treated with sodium bisulfite and then sequenced, providing single-base resolution of methylated cytosines in the genome. Upon bisulfite treatment, unmethylated cytosines are deaminated to uracil which, upon sequencing, are converted to thymidine. Simultaneously, methylated cytosines resist deamination and are read as cytosines. The location of the methylated cytosines can then be determined by comparing treated and untreated sequences. [0153] In some embodiments, methylated DNA can be sequenced using a method that comprises enriching for cfDNA that comprises methylated DNA. Enrichment can be accomplished e.g., using an agent that selectively binds methylated DNA (e.g., an antibody as in MeDIP-seq or a methyl-CpG-Binding Domain (MBD), as in MBD-seq). In some embodiments, an agent that binds methylated DNA is attached (e.g., via a covalent or noncovalent bond) to a physical support (e.g., a bead, a magnetic bead, an agarose bead, or a magnetic epoxy bead), wherein the attaching can be prior to, during, or after incubation with a sample. [0154] MeDIP-seq was first reported by Weber et al., Nat Genet (2005) 37:853–862. In a typical MeDIP-seq protocol, antibody or antibody-fragment that binds 5-methylcytidine (5mC) is used to enrich methylated DNA fragments, then these fragments are sequenced and analyzed. If using 5mC-specific antibodies or antibody fragments, methylated DNA is isolated from genomic DNA via immunoprecipitation. Anti-5mC antibodies are incubated with fragmented genomic DNA and precipitated, followed by DNA purification and sequencing. [0155] Methyl-CpG-Binding Domain sequencing (MBD-seq) is similar to MeDIP-seq except that it uses methyl binding domain (MBD) proteins instead of antibodies or antibody fragments to bind methylated DNA. In a typical MBD-seq protocol, genomic DNA is first sonicated and incubated with tagged MBD proteins that can bind methylated cytosines. The protein-DNA
12366150v1
Attorney Docket No.2014191-0027 complex is then precipitated with antibody-conjugated beads that are specific to the MBD protein tag, followed by DNA purification and sequencing. [0156] In some embodiments, DNA methylation at a given genomic loci can be quantified by sequencing methylated DNA. For example, in some embodiments, DNA methylation at a genomic loci can be quantified by counting the number of sequence reads that overlap with the genomic loci (e.g., comprise at least one nucleotide that overlaps with the genomic loci). [0157] A person of skill in the art is aware of suitable DNA sequencing technologies for use in methods described herein that comprise a sequencing step. Suitable DNA sequencing technologies include, e.g., next generation sequencing (NGS) approaches. Additional steps that are required to prepare DNA for sequencing via an appropriate sequencing approach can be incorporated into methods described herein. For example, in some embodiments, a method described herein comprises attaching (e.g., ligating) DNA adapters to cfDNA. In some embodiments, DNA adapters can be attached prior to, during, or after enrichment for a histone modification. Classifiers [0158] In some embodiments, the present disclosure provides methods for obtaining a classifier, e.g., a validated classifier that can be used to determine ER status. In some embodiments, a subject is determined to have a validated epigenetic profile indicative of an ER- positive cancer based on analysis of a biological sample, optionally of cell-free DNA (cfDNA) from a liquid biopsy sample, obtained or derived from the subject, wherein the presence of the validated epigenetic profile has been determined using a validated classifier. [0159] For illustration purposes and without limitation, in an exemplary embodiment of the present disclosure, the validated classifier may be obtained by: [0160] (a) determining a genomic profile of one or more histone modifications, chromatin accessibility, binding of one or more transcription factors, and/or DNA methylation in (i) one or more ER-positive cell lines or (ii) biological samples obtained from a first cohort of subjects who have previously been determined to have an ER-positive cancer; [0161] (b) determining a genomic profile of one or more histone modifications, chromatin accessibility, binding of one or more transcription factors, and/or DNA methylation in
12366150v1
Attorney Docket No.2014191-0027 (i) one or more ER-negative cell lines or (ii) biological samples obtained from a second cohort of healthy subjects or subjects who have previously been determined to have an ER-negative cancer; [0162] (c) comparing the genomic profile determined in step (a) and the genomic profile determined in step (b), to identify genomic loci that have statistically different histone modification, chromatin accessibility, binding of transcription factor, and/or DNA methylation levels (“differential loci”); [0163] (d) training a classifier on histone modification, chromatin accessibility, binding of transcription factor, and/or DNA methylation levels in the differential loci to distinguish between (i) samples from one or more ER-positive cell lines or biological samples obtained from the first cohort, and (ii) samples from one or more ER-negative cell lines or biological samples obtained from the second cohort, to identify samples having a profile of histone modification, chromatin accessibility, binding of transcription factor, and/or DNA methylation levels (“epigenetic profile”) that indicates that the samples are likely obtained from an ER-positive cell line or from the first cohort; and [0164] (e) obtaining the validated classifier by validating the classifier from step (d) on a third cohort comprising an independent and group of subjects with ER-positive and ER-negative cancers and selecting a threshold such that the validated classifier predicts ER-positive cancers, with an area under the receiver operating characteristic (AUROC) greater than 0.5 (e.g., greater than 0.55, greater than 0.6, greater than 0.65, greater than 0.7, greater than 0.75, greater than 0.8, greater than 0.85, greater than 0.9, or greater than 0.95), wherein subjects falling within the group of predicted ER-positive cancers display the validated epigenetic profile and subjects that do not fall within the group of ER-positive cancers lack the validated epigenetic profile. [0165] A person of ordinary skill will appreciate that other methods can be used to obtain a classifier, e.g., a validated classifier that can be used to determine ER status and that the present disclosure is not limited to classifiers obtained in accordance with this method. Exemplary Genomic Loci [0166] The present disclosure includes the identification of exemplary genomic loci that are differentially modified and/or differentially accessible in ER-positive vs. ER-negative cancer. See Tables 1-3 which show the chromosomal coordinates of each genomic locus and its observed
12366150v1
Attorney Docket No.2014191-0027 log2(fold-change) (ER-positive/ER-negative) which shows whether modification of a locus was correlated with ER-positive status (log2(FC) > 0) or correlated with ER-negative status (log2(FC) < 0). The genomic loci are sorted based on their chromosomal coordinates which are based on human genome build hg19. [0167] The present disclosure is not limited to methods that use the exact same chromosomal coordinates that are recited in Tables 1-3. The present disclosure encompasses methods that use any of the genomic loci in Table 1-3 and also subregions thereof, i.e., references herein to methods that involve detecting and/or quantifying one or more histone modifications, chromatin accessibility, binding of one or more transcription factors, and/or DNA methylation at one or more genomic loci of Table 1-3 encompasses methods that detect these marks anywhere within these genomic loci including within any subregions. For example, where Table 2 references chr1:901178-903765 as a genomic locus for detecting and/or quantifying H3K27ac modification, this encompasses methods that detect and/or quantify H3K27ac modification at any position or sub-region of chr1:901178-903765, e.g., methods that detect and/or quantify H3K27ac modification within chr1:902178-903565, etc. In some embodiments, a subregion may span at least 100, 200, 300, 400, 500, 600, 700, 800, 900, 1000, 1500, 2000, 2500 or at least 3000 contiguous base pairs that are located between the lower and upper coordinates of a genomic locus recited in Tables 1-3. In some embodiments, a subregion may span less than 100, 200, 300, 400, 500, 600, 700, 800, 900, 1000, 1500, 2000, 2500 or at least 3000 contiguous base pairs that are located between the lower and upper coordinates of a genomic locus recited in Tables 1-3. In some embodiments, a subregion may have the same central coordinate as a genomic locus recited in Tables 1-3. In some embodiments, a subregion may have a different central coordinate as a genomic locus recited in Tables 1-3. It is also to be understood that the lower/upper coordinates of the genomic loci in Tables 1-3 are approximate and that the present disclosure encompasses methods where any one or more of the genomic loci are expanded by increasing the size of the genomic locus by 5%, 10%, 15%, 20%, 25%, 30%, 35%, 40% or up to 50% in one or both directions. [0168] In some embodiments a classifier is generated using a set of differentially modified and/or differentially accessible genomic loci that are correlated with ER-positive status and a set of differentially modified and/or differentially accessible loci that are correlated with ER-negative status. Sequence reads that fall into each selected genomic locus are analyzed and
12366150v1
Attorney Docket No.2014191-0027 counted, e.g., as described herein including the Examples. In some embodiments, counts from genomic loci that are correlated with ER-positive status are aggregated and counts from genomic loci that are correlated with ER-negative status are aggregated. In some embodiments, a ratio of the aggregated ER-positive and ER-negative counts is used to determine ER status. Other ways of using the genomic loci and related sequencing data to generate and apply a classifier to determine ER status are described herein and known in the art, e.g., without limitation, methods that use a learning statistical classifier system or a combination of learning statistical classifier systems. [0169] In some embodiments, exemplary genomic loci from Table 1, 2 or 3 are used in a monomodal classifier, e.g., a classifier that uses a single histone modification (e.g., H3K4me3 or H3K27ac) or DNA methylation at one or more genomic loci for purposes of determining ER status. In some embodiments, exemplary genomic loci from Table 1, 2 and/or 3 are used in combination in a multimodal classifier, e.g., a classifier that uses more than one histone modification (e.g., H3K4me3 and H3K27ac) or one or more histone modifications (e.g., H3K4me3 and/or H3K27ac) and DNA methylation at one or more genomic loci for purposes of determining ER status. Differential H3K4me3 modification [0170] Genomic loci demonstrating differential H3K4 methylation (in particular H3K4 trimethylation, H3K4me3) in ER-positive vs. ER-negative cancer are provided in Table 1 which shows the chromosomal coordinates of each genomic locus and its observed log2(fold-change) (ER-positive/ER-negative). The genomic loci are sorted based on their chromosomal coordinates which are based on human genome build hg19. [0171] A person of skill in the art will recognize that the methods disclosed herein do not require that every genomic locus listed in Table 1 be assessed for H3K4me3 modification. Instead, a subset of loci may be assessed for H3K4me3 modification. Subsets of the genomic loci of Table 1 can be selected (e.g., for use in determining ER status) based on various performance criteria, e.g., to select genomic loci that demonstrate differential modification with a particular level of statistical significance and/or a particular threshold of differential between relevant states (e.g., a measured log2(fold-change)). Subsets of the genomic loci may also be selected based on an algorithm, e.g., during the process of obtaining a classifier. Those of skill in the art will
12366150v1
Attorney Docket No.2014191-0027 appreciate that such subsets of loci of Table 1, and loci included in such subsets, are together, individually, and/or in randomly selected subsets, at least as informative (e.g., as statistically significant and/or reliable) for uses disclosed herein, e.g., for determining ER status. See also the Examples of the present disclosure for experiments showing that informative classifiers can be generated using many different combinations of the loci. The present disclosure particularly includes, among other things, subsets of the genomic loci of Table 1, which have an absolute log2(fold-change) of 6.0 or higher, 5.5 or higher, 5.0 or higher, 4.5 or higher, 4.0 or higher, 3.5 or higher, 3.0 or higher, 2.5 or higher, 2.0 or higher, 1.9 or higher, 1.8 or higher, 1.7 or higher, 1.6 or higher, 1.5 or higher, 1.4 or higher, 1.3 or higher, 1.2 or higher, 1.1 or higher, 1.0 or higher, 0.9 or higher, 0.8 or higher, 0.7 or higher, 0.6 or higher, or 0.5 or higher. The present disclosure also includes subsets of the genomic loci of Table 1, which have an absolute log2(fold-change) of 6.0 or higher, 5.5 to less than 6.0, 5.0 to less than 5.5, 4.5 to less than 5.0, 4.0 to less than 4.5, 3.8 to less than 4.0, 3.6 to less than 3.8, 3.4 to less than 3.6, 3.2 to less than 3.4, 3.0 to less than 3.2, 2.8 to less than 3.0, 2.6 to less than 2.8, 2.4 to less than 2.6, 2.2 to less than 2.4, 2.0 to less than 2.2, 1.8 to less than 2.0, 1.6 to less than 1.8, 1.4 to less than 1.6, 1.2 to less than 1.4, 1.0 to less than 1.2, 0.8 to less than 1.0, or 0.6 to less than 0.8. [0172] In various embodiments, a sample or subject from which the sample is obtained or derived, is determined to have a particular ER status (e.g., ER-positive) if at least 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 150, 200, 250, 300, 350, 400, 450, 500, 750, 1000, 1500, 2000, 2500, or 3000 loci identified in Table 1 (or any subset thereof) are differentially H3K4me3 modified as compared to a reference (e.g., a sample from an ER-negative or healthy subject). In certain embodiments, a subject from which the sample is obtained or derived, is determined to have a particular ER status (e.g., ER-positive) if at least a number of loci identified in a Table 1 (or any subset thereof) having a lower bound selected from 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 150, 200, 250, or 300 and an upper bound selected from 10, 15, 20, 25, 50, 75, 100, 150, 200, 250, 300, 350, 400, 450, 500, 750, 1000, 1500, 2000, 2500, or 3000 is found to be differentially H3K4me3 modified as compared to a reference (e.g., a sample from an ER- negative or healthy subject). In certain particular embodiments, a sample or subject from which the sample is obtained or derived, is determined to have a particular ER status (e.g., ER-positive) if at least 1, 2, 3, 4, 5, 10, 20, 30, 40, or 50 loci identified in Table 1 (e.g., about 1 to about 1,000,
12366150v1
Attorney Docket No.2014191-0027 about 5 to about 3,000, about 10 to about 1000, about 25 to about 200, about 5, about 10, about 20, or about 50 loci) are differentially H3K4me3 modified as compared to a reference (e.g., a sample from an ER-negative or healthy subject). In various embodiments a sample or subject from which the sample is obtained or derived, is determined to have a particular ER status (e.g., ER-positive) if at least 0.1%, 0.2%, 0.3%, 0.4%, 0.5%, 1%, 2%, 3%, 4%, 5%, 10%, 20%, 30%, 40%, 50%, 75%, or 100% of loci identified in Table 1 are differentially H3K4me3 modified as compared to a reference (e.g., a sample from an ER-negative or healthy subject). In certain embodiments, a sample or subject from which the sample is obtained or derived, is determined to have a particular ER status (e.g., ER-positive) if at least a percent of loci identified in Table 1 having a lower bound selected from 0.1%, 0.2%, 0.3%, 0.4%, 0.5%, 1%, 2%, 3%, 4%, 5%, or 10%, and an upper bound selected from 1%, 2%, 3%, 4%, 5%, 10%, 20%, 30%, 40%, 50%, 75%, or 100% is found to be differentially H3K4me3 modified as compared to a reference (e.g., a sample from an ER-negative or healthy subject). [0173] In various embodiments, a sample or subject from which the sample is derived, is determined to have a particular ER status (e.g., ER-positive) if at least one (e.g., at least 2, at least 3, at least 4, at least 5, at least 6, at least 7, at least 8, at least 9, or at least 10) of the top 3, 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 150, 200, 250, 300, 350, 400, 450, 500, 750, 1000, 1500, 2000, 2500, or 3000 loci identified in Table 1 are differentially H3K4me3 modified as compared to a reference (e.g., a sample from an ER-negative or healthy subject) (wherein, e.g., the “top” 10 loci refers to the loci with 10 highest absolute log2(fold- change) in Table 1). In some embodiments, a subject from which the sample is obtained or derived, is determined to have a particular ER status (e.g., ER-positive) if at least one of the top 10 loci identified in Table 1 is differentially H3K4me3 modified as compared to a reference (e.g., a sample from an ER-negative or healthy subject). In some embodiments, a subject from which the sample is obtained or derived, is determined to have a particular ER status (e.g., ER-positive) if at least one of the top 25 loci identified in Table 1 is differentially H3K4me3 modified as compared to a reference (e.g., a sample from an ER-negative or healthy subject). In some embodiments, a subject from which the sample is obtained or derived, is determined to have a particular ER status (e.g., ER-positive) if at least one of the top 50 loci identified in Table 1 is differentially H3K4me3 modified as compared to a reference (e.g., a sample from an ER- negative or healthy subject). In some embodiments, a subject from which the sample is obtained
12366150v1
Attorney Docket No.2014191-0027 or derived, is determined to have a particular ER status (e.g., ER-positive) if at least five of the top 10 loci identified in Table 1 are differentially H3K4me3 modified as compared to a reference (e.g., a sample from an ER-negative or healthy subject). In some embodiments, a subject from which the sample is obtained or derived, is determined to have a particular ER status (e.g., ER- positive) if at least five of the top 25 loci identified in Table 1 are differentially H3K4me3 modified as compared to a reference (e.g., a sample from an ER-negative or healthy subject). In some embodiments, a subject from which the sample is obtained or derived, is determined to have a particular ER status (e.g., ER-positive) if at least five of the top 50 loci identified in Table 1 are differentially H3K4me3 modified as compared to a reference (e.g., a sample from an ER- negative or healthy subject). [0174] In various embodiments, a sample or subject from which the sample is derived, is determined to have a particular ER status (e.g., ER-positive) if at least one of the top 10 loci (e.g., at least 1, at least 2, at least 3, at least 4, at least 5, at least 6, at least 7, at least 8, at least 9, or 10) identified in Table 1 and at least 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 150, 200, 250, 300, 350, 400, 450, 500, 750, 1000, 1500, 2000, 2500, or 3000 loci identified in Table 1 (or any subset thereof) in total are differentially H3K4me3 modified as compared to a reference (e.g., a sample from an ER-negative or healthy subject). In various embodiments, a sample or subject from which the sample is derived, is determined to have a particular ER status (e.g., ER-positive) if at least one of the top 25 loci identified in Table 1 (e.g., at least 1, at least 2, at least 3, at least 4, at least 5, at least 6, at least 7, at least 8, at least 9, or at least 10, at least 15, at least 20, or 25) and at least 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 150, 200, 250, 300, 350, 400, 450, 500, 750, 1000, 1500, 2000, 2500, or 3000 loci identified in Table 1 (or any subset thereof) in total are differentially H3K4me3 modified as compared to a reference (e.g., a sample from an ER-negative or healthy subject). In various embodiments, a sample or subject from which the sample is derived, is determined to have a particular ER status (e.g., ER- positive) if at least one of the top 50 loci (e.g., at least 1, at least 2, at least 3, at least 4, at least 5, at least 6, at least 7, at least 8, at least 9, or at least 10, at least 15, at least 20, or at least 25, at least 30, at least 35, at least 40, at least 45, or 50) identified in Table 1 and at least 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 150, 200, 250, 300, 350, 400, 450, 500, 750, 1000, 1500, 2000, 2500, or 3000 loci identified in Table 1 (or any subset thereof) in total are differentially H3K4me3 modified as compared to a reference (e.g., a sample from an ER-
12366150v1
Attorney Docket No.2014191-0027 negative or healthy subject). In various embodiments, a sample or subject from which the sample is derived, is determined to have a particular ER status (e.g., ER-positive) if at least five of the top 25 loci identified in Table 1 and at least 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 150, 200, 250, 300, 350, 400, 450, 500, 750, 1000, 1500, 2000, 2500, or 3000 loci identified in Table 1 (or any subset thereof) in total are differentially H3K4me3 modified as compared to a reference (e.g., a sample from an ER-negative or healthy subject). In various embodiments, a sample or subject from which the sample is derived, is determined to have a particular ER status (e.g., ER-positive) if at least five of the top 50 loci identified in Table 1 and at least 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 150, 200, 250, 300, 350, 400, 450, 500, 750, 1000, 1500, 2000, 2500, or 3000 loci identified in Table 1 (or any subset thereof) in total are differentially H3K4me3 modified as compared to a reference (e.g., a sample from an ER-negative or healthy subject). [0175] In various embodiments, differentially H3K4me3 modified refers to a methylation status characterized by an increase or decrease in a value measuring methylation (e.g., of read counts and/or normalized read counts for a given genomic locus), and/or a mean, median and/or mode thereof, and/or a log thereof (e.g., log base 2 (log2)), of at least 1%, 2%, 3%, 4%, 5%, 6%, 7%, 8%, 9%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 75%, 100%, 2-fold, 3-fold, 4- fold, 5-fold, 6-fold, 7-fold, 8-fold, 9-fold, 10-fold, 15-fold, 20-fold, 25-fold, 30-fold, 35-fold, 40- fold, 45-fold, 50-fold, or greater, or any range in between, inclusive, such as 1% to 50%, 50% to 2-fold, 25% to 50-fold, 25% to 30-fold, 25% to 20-fold, 25% to 16-fold, 30% to 16-fold, 50% to 16-fold, 70% to 16-fold, 2-fold to 16-fold, 2.2-fold to 16-fold, 2.6-fold to 16-fold, 3-fold to 16- fold, 3.4-fold to 16-fold, 4-fold to 16-fold, 4.5-fold to 16-fold, 5.2-fold to 16-fold, 6-fold to 16- fold, 7-fold to 16-fold, or 8-fold to 16-fold, as compared to a reference, optionally where the statistical significance of the increase or decrease is at least 5e-2, 1e-2, 5e-3, 1e-3, 5e-4, 1e-4, 5e- 5, 1e-5, 5e-6, or 1e-6. In various embodiments, an increase or decrease in a value measuring methylation can be, or is expressed as, a log2(fold-change), e.g., a log2(fold-change) of at least 1%, 2%, 3%, 4%, 5%, 6%, 7%, 8%, 9%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 75%, 100%, 2-fold, 3-fold, 4-fold, 5-fold, 6-fold, 7-fold, 8-fold, 9-fold, 10-fold, 15-fold, 20-fold, or greater, or any range in between, inclusive, such as an increase or decrease of 0.1-fold to 10- fold, 0.2-fold to 5-fold, 0.2-fold to 4.0-fold, 0.4-4.0-fold, 0.4-fold to 4.0-fold, 0.6-fold to 4.0- fold, 0.8-fold to 4.0-fold, 1.0-fold to 4.0-fold.1.2-fold to 4.0-fold.1.4-fold to 4.0-fold, 1.6-fold to
12366150v1
Attorney Docket No.2014191-0027 4.0-fold, 1.8-fold to 4.0-fold, 2.0-fold to 4.0-fold, 2.2-fold to 4.0-fold, 2.4-fold to 4.0-fold, 2.6- fold to 4.0-fold, 2.8-fold to 4.0-fold, or 3.0-fold to 4.0-fold, optionally where the statistical significance of the increase or decrease is at least 5e-2, 1e-2, 5e-3, 1e-3, 5e-4, 1e-4, 5e-5, 1e-5, 5e-6, or 1e-6. Differential H3K27ac modification [0176] Genomic loci demonstrating differential H3K27ac modification in ER-positive vs. ER-negative cancer are provided in Table 2, which shows the chromosomal coordinates of each genomic locus and its observed log2(fold-change) (ER-positive/ER-negative). The genomic loci are sorted based on their chromosomal coordinates which are based on human genome build hg19. [0177] A person of skill in the art will recognize that the methods disclosed herein do not require that every genomic locus listed in Table 2 be assessed for H3K27ac modification. Instead, a subset of loci may be assessed for H3K27ac modification. Subsets of the genomic loci of Table 2 can be selected (e.g., for use in determining ER status) based on various performance criteria, e.g., to select genomic loci that demonstrate differential modification with a particular level of statistical significance and/or a particular threshold of differential between relevant states (e.g., a measured log2(fold-change)). Subsets of the genomic loci may also be selected based on an algorithm, e.g., during the process of obtaining a classifier. Those of skill in the art will appreciate that such subsets of loci of Table 2, and loci included in such subsets, are together, individually, and/or in randomly selected subsets, at least as informative (e.g., as statistically significant and/or reliable) for uses disclosed herein, e.g., for determining ER status. See also the Examples of the present disclosure for experiments showing that informative classifiers can be generated using many different combinations of the loci. The present disclosure particularly includes, among other things, subsets of the genomic loci of Table 2, which have an absolute log2(fold-change) of 6.0 or higher, 5.5 or higher, 5.0 or higher, 4.5 or higher, 4.0 or higher, 3.5 or higher, 3.0 or higher, 2.5 or higher, 2.0 or higher, 1.9 or higher, 1.8 or higher, 1.7 or higher, 1.6 or higher, 1.5 or higher, 1.4 or higher, 1.3 or higher, 1.2 or higher, 1.1 or higher, 1.0 or higher, 0.9 or higher, 0.8 or higher, 0.7 or higher, 0.6 or higher, or 0.5 or higher. The present disclosure also includes subsets of the genomic loci of Table 2, which have an absolute log2(fold-change) of 6.0 or higher, 5.5 to less than 6.0, 5.0 to less than 5.5, 4.5 to less than 5.0, 4.0 to less than 4.5, 3.8 to
12366150v1
Attorney Docket No.2014191-0027 less than 4.0, 3.6 to less than 3.8, 3.4 to less than 3.6, 3.2 to less than 3.4, 3.0 to less than 3.2, 2.8 to less than 3.0, 2.6 to less than 2.8, 2.4 to less than 2.6, 2.2 to less than 2.4, 2.0 to less than 2.2, 1.8 to less than 2.0, 1.6 to less than 1.8, 1.4 to less than 1.6, 1.2 to less than 1.4, 1.0 to less than 1.2, 0.8 to less than 1.0, or 0.6 to less than 0.8. [0178] In various embodiments, a sample or subject from which the sample is obtained or derived, is determined to have a particular ER status (e.g., ER-positive) if at least 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 150, 200, 250, 300, 350, 400, 450, 500, 750, 1000, 1500, 2000, 2500, or 3000 loci identified in Table 2 (or any subset thereof) are H3K27ac modified as compared to a reference (e.g., a sample from an ER- negative or healthy subject). In certain embodiments, a subject from which the sample is obtained or derived, is determined to have a particular ER status (e.g., ER-positive) if at least a number of loci identified in a Table 2 (or any subset thereof) having a lower bound selected from 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 150, 200, 250, or 300 and an upper bound selected from 10, 15, 20, 25, 50, 75, 100, 150, 200, 250, 300, 350, 400, 450, 500, 750, 1000, 1500, 2000, 2500, or 3000 is found to be H3K27ac modified as compared to a reference (e.g., a sample from an ER-negative or healthy subject). In certain particular embodiments, a sample or subject from which the sample is obtained or derived, is determined to have a particular ER status (e.g., ER-positive) if at least 1, 2, 3, 4, 5, 10, 20, 30, 40, or 50 loci identified in Table 2 (e.g., about 1 to about 1,000, about 5 to about 3,000, about 10 to about 1000, about 25 to about 200, about 5, about 10, about 20, or about 50 loci) are H3K27ac modified as compared to a reference (e.g., a sample from an ER-negative or healthy subject). In various embodiments a sample or subject from which the sample is obtained or derived, is determined to have a particular ER status (e.g., ER-positive) if at least 0.1%, 0.2%, 0.3%, 0.4%, 0.5%, 1%, 2%, 3%, 4%, 5%, 10%, 20%, 30%, 40%, 50%, 75%, or 100% of loci identified in Table 2 are H3K27ac modified as compared to a reference (e.g., a sample from an ER-negative or healthy subject). In certain embodiments, a sample or subject from which the sample is obtained or derived, is determined to have a particular ER status (e.g., ER-positive) if at least a percent of loci identified in Table 2 having a lower bound selected from 0.1%, 0.2%, 0.3%, 0.4%, 0.5%, 1%, 2%, 3%, 4%, 5%, or 10%, and an upper bound selected from 1%, 2%, 3%, 4%, 5%, 10%, 20%, 30%, 40%, 50%, 75%, or 100% is found to be H3K27ac modified as compared to a reference (e.g., a sample from an ER-negative or healthy subject).
12366150v1
Attorney Docket No.2014191-0027 [0179] In various embodiments, a sample or subject from which the sample is derived, is determined to have a particular ER status (e.g., ER-positive) if at least one (e.g., at least 2, at least 3, at least 4, at least 5, at least 6, at least 7, at least 8, at least 9, or at least 10) of the top 3, 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 150, 200, 250, 300, 350, 400, 450, 500, 750, 1000, 1500, 2000, 2500, or 3000 loci identified in Table 2 are H3K27ac modified as compared to a reference (e.g., a sample from an ER-negative or healthy subject) (wherein, e.g., the “top” 10 loci refers to the loci with 10 highest absolute log2(fold-change) in Table 2). In some embodiments, a subject from which the sample is obtained or derived, is determined to have a particular ER status (e.g., ER-positive) if at least one of the top 10 loci identified in Table 2 is H3K27ac modified as compared to a reference (e.g., a sample from an ER-negative or healthy subject). In some embodiments, a subject from which the sample is obtained or derived, is determined to have a particular ER status (e.g., ER-positive) if at least one of the top 25 loci identified in Table 2 is H3K27ac modified as compared to a reference (e.g., a sample from an ER-negative or healthy subject). In some embodiments, a subject from which the sample is obtained or derived, is determined to have a particular ER status (e.g., ER-positive) if at least one of the top 50 loci identified in Table 2 is H3K27ac modified as compared to a reference (e.g., a sample from an ER-negative or healthy subject). In some embodiments, a subject from which the sample is obtained or derived, is determined to have a particular ER status (e.g., ER-positive) if at least five of the top 10 loci identified in Table 2 are H3K27ac modified as compared to a reference (e.g., a sample from an ER-negative or healthy subject). In some embodiments, a subject from which the sample is obtained or derived, is determined to have a particular ER status (e.g., ER-positive) if at least five of the top 25 loci identified in Table 2 are H3K27ac modified as compared to a reference (e.g., a sample from an ER-negative or healthy subject). In some embodiments, a subject from which the sample is obtained or derived, is determined to have a particular ER status (e.g., ER-positive) if at least five of the top 50 loci identified in Table 2 are H3K27ac modified as compared to a reference (e.g., a sample from an ER-negative or healthy subject). [0180] In various embodiments, a sample or subject from which the sample is derived, is determined to have a particular ER status (e.g., ER-positive) if at least one of the top 10 loci (e.g., at least 1, at least 2, at least 3, at least 4, at least 5, at least 6, at least 7, at least 8, at least 9, or 10) identified in Table 2 and at least 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95,
12366150v1
Attorney Docket No.2014191-0027 100, 150, 200, 250, 300, 350, 400, 450, 500, 750, 1000, 1500, 2000, 2500, or 3000 loci identified in Table 2 (or any subset thereof) in total are H3K27ac modified as compared to a reference (e.g., a sample from an ER-negative or healthy subject). In various embodiments, a sample or subject from which the sample is derived, is determined to have a particular ER status (e.g., ER-positive) if at least one of the top 25 loci identified in Table 2 (e.g., at least 1, at least 2, at least 3, at least 4, at least 5, at least 6, at least 7, at least 8, at least 9, or at least 10, at least 15, at least 20, or 25) and at least 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 150, 200, 250, 300, 350, 400, 450, 500, 750, 1000, 1500, 2000, 2500, or 3000 loci identified in Table 2 (or any subset thereof) in total are H3K27ac modified as compared to a reference (e.g., a sample from an ER- negative or healthy subject). In various embodiments, a sample or subject from which the sample is derived, is determined to have a particular ER status (e.g., ER-positive) if at least one of the top 50 loci (e.g., at least 1, at least 2, at least 3, at least 4, at least 5, at least 6, at least 7, at least 8, at least 9, or at least 10, at least 15, at least 20, or at least 25, at least 30, at least 35, at least 40, at least 45, or 50) identified in Table 2 and at least 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 150, 200, 250, 300, 350, 400, 450, 500, 750, 1000, 1500, 2000, 2500, or 3000 loci identified in Table 2 (or any subset thereof) in total are H3K27ac modified as compared to a reference (e.g., a sample from an ER-negative or healthy subject). In various embodiments, a sample or subject from which the sample is derived, is determined to have a particular ER status (e.g., ER-positive) if at least five of the top 25 loci identified in Table 2 and at least 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 150, 200, 250, 300, 350, 400, 450, 500, 750, 1000, 1500, 2000, 2500, or 3000 loci identified in Table 2 (or any subset thereof) in total are H3K27ac modified as compared to a reference (e.g., a sample from an ER-negative or healthy subject). In various embodiments, a sample or subject from which the sample is derived, is determined to have a particular ER status (e.g., ER-positive) if at least five of the top 50 loci identified in Table 2 and at least 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 150, 200, 250, 300, 350, 400, 450, 500, 750, 1000, 1500, 2000, 2500, or 3000 loci identified in Table 2 (or any subset thereof) in total are H3K27ac modified as compared to a reference (e.g., a sample from an ER-negative or healthy subject). [0181] In various embodiments, differentially H3K27ac modified refers to an acetylation status characterized by an increase or decrease in a value measuring acetylation (e.g., of read counts and/or normalized read counts for a given genomic locus), and/or a mean, median and/or
12366150v1
Attorney Docket No.2014191-0027 mode thereof, and/or a log thereof (e.g., log base 2 (log2)), of at least 1%, 2%, 3%, 4%, 5%, 6%, 7%, 8%, 9%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 75%, 100%, 2-fold, 3-fold, 4- fold, 5-fold, 6-fold, 7-fold, 8-fold, 9-fold, 10-fold, 15-fold, 20-fold, 25-fold, 30-fold, 35-fold, 40- fold, 45-fold, 50-fold, or greater, or any range in between, inclusive, such as 1% to 50%, 50% to 2-fold, 25% to 50-fold, 25% to 30-fold, 25% to 20-fold, 25% to 16-fold, 30% to 16-fold, 50% to 16-fold, 70% to 16-fold, 2-fold to 16-fold, 2.2-fold to 16-fold, 2.6-fold to 16-fold, 3-fold to 16- fold, 3.4-fold to 16-fold, 4-fold to 16-fold, 4.5-fold to 16-fold, 5.2-fold to 16-fold, 6-fold to 16- fold, 7-fold to 16-fold, or 8-fold to 16-fold, as compared to a reference, optionally where the statistical significance of the increase or decrease is at least 5e-2, 1e-2, 5e-3, 1e-3, 5e-4, 1e-4, 5e- 5, 1e-5, 5e-6, or 1e-6. In various embodiments, an increase or decrease in a value measuring acetylation can be, or is expressed as, a log2(fold-change), e.g., a log2(fold-change) of at least 1%, 2%, 3%, 4%, 5%, 6%, 7%, 8%, 9%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 75%, 100%, 2-fold, 3-fold, 4-fold, 5-fold, 6-fold, 7-fold, 8-fold, 9-fold, 10-fold, 15-fold, 20-fold, or greater, or any range in between, inclusive, such as an increase or decrease of 0.1-fold to 10- fold, 0.2-fold to 5-fold, 0.2-fold to 4.0-fold, 0.4-4.0-fold, 0.4-fold to 4.0-fold, 0.6-fold to 4.0- fold, 0.8-fold to 4.0-fold, 1.0-fold to 4.0-fold.1.2-fold to 4.0-fold.1.4-fold to 4.0-fold, 1.6-fold to 4.0-fold, 1.8-fold to 4.0-fold, 2.0-fold to 4.0-fold, 2.2-fold to 4.0-fold, 2.4-fold to 4.0-fold, 2.6- fold to 4.0-fold, 2.8-fold to 4.0-fold, or 3.0-fold to 4.0-fold, optionally where the statistical significance of the increase or decrease is at least 5e-2, 1e-2, 5e-3, 1e-3, 5e-4, 1e-4, 5e-5, 1e-5, 5e-6, or 1e-6. Differential DNA methylation [0182] Genomic loci demonstrating differential DNA methylation in ER-positive vs. ER- negative cancer are provided in Table 3, which shows the chromosomal coordinates of each genomic locus and its observed log2(fold-change) (ER-positive/ER-negative). The genomic loci are sorted based on their chromosomal coordinates which are based on human genome build hg19. [0183] A person of skill in the art will recognize that the methods disclosed herein do not require that every genomic locus listed in Table 3 be assessed for DNA methylation. Instead, a subset of loci may be assessed for DNA methylation. Subsets of the genomic loci of Table 3 can be selected (e.g., for use in determining ER status) based on various performance criteria, e.g., to
12366150v1
Attorney Docket No.2014191-0027 select genomic loci that demonstrate differential modification with a particular level of statistical significance and/or a particular threshold of differential between relevant states (e.g., a measured log2(fold-change)). Subsets of the genomic loci may also be selected based on an algorithm, e.g., during the process of obtaining a classifier. Those of skill in the art will appreciate that such subsets of loci of Table 3, and loci included in such subsets, are together, individually, and/or in randomly selected subsets, at least as informative (e.g., as statistically significant and/or reliable) for uses disclosed herein, e.g., for determining ER status. See also the Examples of the present disclosure for experiments showing that informative classifiers can be generated using many different combinations of the loci. The present disclosure particularly includes, among other things, subsets of the genomic loci of Table 3, which have an absolute log2(fold-change) of 6.0 or higher, 5.5 or higher, 5.0 or higher, 4.5 or higher, 4.0 or higher, 3.5 or higher, 3.0 or higher, 2.5 or higher, 2.0 or higher, 1.9 or higher, 1.8 or higher, 1.7 or higher, 1.6 or higher, 1.5 or higher, 1.4 or higher, 1.3 or higher, 1.2 or higher, 1.1 or higher, 1.0 or higher, 0.9 or higher, 0.8 or higher, 0.7 or higher, 0.6 or higher, or 0.5 or higher. The present disclosure also includes subsets of the genomic loci of Table 3, which have an absolute log2(fold-change) of 6.0 or higher, 5.5 to less than 6.0, 5.0 to less than 5.5, 4.5 to less than 5.0, 4.0 to less than 4.5, 3.8 to less than 4.0, 3.6 to less than 3.8, 3.4 to less than 3.6, 3.2 to less than 3.4, 3.0 to less than 3.2, 2.8 to less than 3.0, 2.6 to less than 2.8, 2.4 to less than 2.6, 2.2 to less than 2.4, 2.0 to less than 2.2, 1.8 to less than 2.0, 1.6 to less than 1.8, 1.4 to less than 1.6, 1.2 to less than 1.4, 1.0 to less than 1.2, 0.8 to less than 1.0, or 0.6 to less than 0.8. [0184] In various embodiments, a sample or subject from which the sample is obtained or derived, is determined to have a particular ER status (e.g., ER-positive) if at least 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 150, 200, 250, 300, 350, 400, 450, 500, 750, 1000, 1500, 2000, 2500, or 3000 loci identified in Table 3 (or any subset thereof) are differentially DNA methylated as compared to a reference (e.g., a sample from an ER-negative or healthy subject). In certain embodiments, a subject from which the sample is obtained or derived, is determined to have a particular ER status (e.g., ER-positive) if at least a number of loci identified in a Table 3 (or any subset thereof) having a lower bound selected from 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 150, 200, 250, or 300 and an upper bound selected from 10, 15, 20, 25, 50, 75, 100, 150, 200, 250, 300, 350, 400, 450, 500, 750, 1000, 1500, 2000, 2500, or 3000 is found to be
12366150v1
Attorney Docket No.2014191-0027 differentially DNA methylated as compared to a reference (e.g., a sample from an ER-negative or healthy subject). In certain particular embodiments, a sample or subject from which the sample is obtained or derived, is determined to have a particular ER status (e.g., ER-positive) if at least 1, 2, 3, 4, 5, 10, 20, 30, 40, or 50 loci identified in Table 3 (e.g., about 1 to about 1,000, about 5 to about 3,000, about 10 to about 1000, about 25 to about 200, about 5, about 10, about 20, or about 50 loci) are differentially DNA methylated as compared to a reference (e.g., a sample from an ER-negative or healthy subject). In various embodiments a sample or subject from which the sample is obtained or derived, is determined to have a particular ER status (e.g., ER-positive) if at least 0.1%, 0.2%, 0.3%, 0.4%, 0.5%, 1%, 2%, 3%, 4%, 5%, 10%, 20%, 30%, 40%, 50%, 75%, or 100% of loci identified in Table 3 are differentially DNA methylated as compared to a reference (e.g., a sample from an ER-negative or healthy subject). In certain embodiments, a sample or subject from which the sample is obtained or derived, is determined to have a particular ER status (e.g., ER-positive) if at least a percent of loci identified in Table 3 having a lower bound selected from 0.1%, 0.2%, 0.3%, 0.4%, 0.5%, 1%, 2%, 3%, 4%, 5%, or 10%, and an upper bound selected from 1%, 2%, 3%, 4%, 5%, 10%, 20%, 30%, 40%, 50%, 75%, or 100% is found to be differentially DNA methylated as compared to a reference (e.g., a sample from an ER-negative or healthy subject). [0185] In various embodiments, a sample or subject from which the sample is derived, is determined to have a particular ER status (e.g., ER-positive) if at least one (e.g., at least 2, at least 3, at least 4, at least 5, at least 6, at least 7, at least 8, at least 9, or at least 10) of the top 3, 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 150, 200, 250, 300, 350, 400, 450, 500, 750, 1000, 1500, 2000, 2500, or 3000 loci identified in Table 3 are differentially DNA methylated as compared to a reference (e.g., a sample from an ER-negative or healthy subject) (wherein, e.g., the “top” 10 loci refers to the loci with 10 highest absolute log2(fold- change) in Table 3). In some embodiments, a subject from which the sample is obtained or derived, is determined to have a particular ER status (e.g., ER-positive) if at least one of the top 10 loci identified in Table 3 is differentially DNA methylated as compared to a reference (e.g., a sample from an ER-negative or healthy subject). In some embodiments, a subject from which the sample is obtained or derived, is determined to have a particular ER status (e.g., ER-positive) if at least one of the top 25 loci identified in Table 3 is differentially DNA methylated as compared to a reference (e.g., a sample from an ER-negative or healthy subject). In some embodiments, a
12366150v1
Attorney Docket No.2014191-0027 subject from which the sample is obtained or derived, is determined to have a particular ER status (e.g., ER-positive) if at least one of the top 50 loci identified in Table 3 is differentially DNA methylated as compared to a reference (e.g., a sample from an ER-negative or healthy subject). In some embodiments, a subject from which the sample is obtained or derived, is determined to have a particular ER status (e.g., ER-positive) if at least five of the top 10 loci identified in Table 3 are differentially DNA methylated as compared to a reference (e.g., a sample from an ER-negative or healthy subject). In some embodiments, a subject from which the sample is obtained or derived, is determined to have a particular ER status (e.g., ER-positive) if at least five of the top 25 loci identified in Table 3 are differentially DNA methylated as compared to a reference (e.g., a sample from an ER-negative or healthy subject). In some embodiments, a subject from which the sample is obtained or derived, is determined to have a particular ER status (e.g., ER-positive) if at least five of the top 50 loci identified in Table 3 are differentially DNA methylated as compared to a reference (e.g., a sample from an ER-negative or healthy subject). [0186] In various embodiments, a sample or subject from which the sample is derived, is determined to have a particular ER status (e.g., ER-positive) if at least one of the top 10 loci (e.g., at least 1, at least 2, at least 3, at least 4, at least 5, at least 6, at least 7, at least 8, at least 9, or 10) identified in Table 3 and at least 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 150, 200, 250, 300, 350, 400, 450, 500, 750, 1000, 1500, 2000, 2500, or 3000 loci identified in Table 3 (or any subset thereof) in total are differentially DNA methylated as compared to a reference (e.g., a sample from an ER-negative or healthy subject). In various embodiments, a sample or subject from which the sample is derived, is determined to have a particular ER status (e.g., ER-positive) if at least one of the top 25 loci identified in Table 3 (e.g., at least 1, at least 2, at least 3, at least 4, at least 5, at least 6, at least 7, at least 8, at least 9, or at least 10, at least 15, at least 20, or 25) and at least 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 150, 200, 250, 300, 350, 400, 450, 500, 750, 1000, 1500, 2000, 2500, or 3000 loci identified in Table 3 (or any subset thereof) in total are differentially DNA methylated as compared to a reference (e.g., a sample from an ER-negative or healthy subject). In various embodiments, a sample or subject from which the sample is derived, is determined to have a particular ER status (e.g., ER- positive) if at least one of the top 50 loci (e.g., at least 1, at least 2, at least 3, at least 4, at least 5, at least 6, at least 7, at least 8, at least 9, or at least 10, at least 15, at least 20, or at least 25, at
12366150v1
Attorney Docket No.2014191-0027 least 30, at least 35, at least 40, at least 45, or 50) identified in Table 3 and at least 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 150, 200, 250, 300, 350, 400, 450, 500, 750, 1000, 1500, 2000, 2500, or 3000 loci identified in Table 3 (or any subset thereof) in total are differentially DNA methylated as compared to a reference (e.g., a sample from an ER-negative or healthy subject). In various embodiments, a sample or subject from which the sample is derived, is determined to have a particular ER status (e.g., ER-positive) if at least five of the top 25 loci identified in Table 3 and at least 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 150, 200, 250, 300, 350, 400, 450, 500, 750, 1000, 1500, 2000, 2500, or 3000 loci identified in Table 3 (or any subset thereof) in total are differentially DNA methylated as compared to a reference (e.g., a sample from an ER-negative or healthy subject). In various embodiments, a sample or subject from which the sample is derived, is determined to have a particular ER status (e.g., ER-positive) if at least five of the top 50 loci identified in Table 3 and at least 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 150, 200, 250, 300, 350, 400, 450, 500, 750, 1000, 1500, 2000, 2500, or 3000 loci identified in Table 3 (or any subset thereof) in total are differentially DNA methylated as compared to a reference (e.g., a sample from an ER-negative or healthy subject). [0187] In various embodiments, differentially DNA methylated refers to a methylation status characterized by an increase or decrease in a value measuring methylation (e.g., of read counts and/or normalized read counts for a given genomic locus), and/or a mean, median and/or mode thereof, and/or a log thereof (e.g., log base 2 (log2)), of at least 1%, 2%, 3%, 4%, 5%, 6%, 7%, 8%, 9%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 75%, 100%, 2-fold, 3-fold, 4- fold, 5-fold, 6-fold, 7-fold, 8-fold, 9-fold, 10-fold, 15-fold, 20-fold, 25-fold, 30-fold, 35-fold, 40- fold, 45-fold, 50-fold, or greater, or any range in between, inclusive, such as 1% to 50%, 50% to 2-fold, 25% to 50-fold, 25% to 30-fold, 25% to 20-fold, 25% to 16-fold, 30% to 16-fold, 50% to 16-fold, 70% to 16-fold, 2-fold to 16-fold, 2.2-fold to 16-fold, 2.6-fold to 16-fold, 3-fold to 16- fold, 3.4-fold to 16-fold, 4-fold to 16-fold, 4.5-fold to 16-fold, 5.2-fold to 16-fold, 6-fold to 16- fold, 7-fold to 16-fold, or 8-fold to 16-fold, as compared to a reference, optionally where the statistical significance of the increase or decrease is at least 5e-2, 1e-2, 5e-3, 1e-3, 5e-4, 1e-4, 5e- 5, 1e-5, 5e-6, or 1e-6. In various embodiments, an increase or decrease in a value measuring methylation can be, or is expressed as, a log2(fold-change), e.g., a log2(fold-change) of at least 1%, 2%, 3%, 4%, 5%, 6%, 7%, 8%, 9%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%,
12366150v1
Attorney Docket No.2014191-0027 75%, 100%, 2-fold, 3-fold, 4-fold, 5-fold, 6-fold, 7-fold, 8-fold, 9-fold, 10-fold, 15-fold, 20-fold, or greater, or any range in between, inclusive, such as an increase of 0.1-fold to 10-fold, 0.2-fold to 5-fold, 0.2-fold to 4.0-fold, 0.4-4.0-fold, 0.4-fold to 4.0-fold, 0.6-fold to 4.0-fold, 0.8-fold to 4.0-fold, 1.0-fold to 4.0-fold.1.2-fold to 4.0-fold.1.4-fold to 4.0-fold, 1.6-fold to 4.0-fold, 1.8- fold to 4.0-fold, 2.0-fold to 4.0-fold, 2.2-fold to 4.0-fold, 2.4-fold to 4.0-fold, 2.6-fold to 4.0-fold, 2.8-fold to 4.0-fold, or 3.0-fold to 4.0-fold, optionally where the statistical significance of the increase or decrease is at least 5e-2, 1e-2, 5e-3, 1e-3, 5e-4, 1e-4, 5e-5, 1e-5, 5e-6, or 1e-6. Differential chromatin accessibility or transcription factor binding [0188] Genomic loci provided in Tables 1-3 can also demonstrate differential chromatin accessibility or transcription factor binding in ER-positive cancer vs. ER-negative cancer. [0189] In various embodiments, without wishing to be bound by any particular scientific theory, histone methylation (e.g., H3K4me3) corresponds and/or is correlated with chromatin accessibility. In various embodiments, without wishing to be bound by any particular scientific theory, histone acetylation (e.g., H3K27ac) corresponds and/or is correlated with chromatin accessibility. In various embodiments, without wishing to be bound by any particular scientific theory, DNA methylation corresponds and/or is correlated with chromatin accessibility. [0190] In some embodiments, without wishing to be limited to any particular scientific theory, chromatin accessibility corresponds and/or is correlated with H3K4me3 modifications. As a result, in some embodiments, ER status may be determined by detecting and quantifying chromatin accessibility at one or more genomic loci in Table 1 in accordance with the section above discussing exemplary genomic loci with differential H3K4me3 modifications. [0191] In some embodiments, without wishing to be limited to any particular scientific theory, chromatin accessibility corresponds and/or is correlated with H3K27ac modifications. As a result, in some embodiments, ER status may be determined by detecting and quantifying chromatin accessibility at one or more genomic loci in Table 2 in accordance with the section above discussing exemplary genomic loci with differential H3K27ac modifications. [0192] In some embodiments, without wishing to be limited to any particular scientific theory, chromatin accessibility corresponds and/or is correlated with DNA methylation. As a result, in some embodiments, ER status may be determined by detecting and quantifying
12366150v1
Attorney Docket No.2014191-0027 chromatin accessibility at one or more genomic loci in Table 3 in accordance with the section above discussing exemplary genomic loci with differential DNA methylation. [0193] In various embodiments, without wishing to be bound by any particular scientific theory, histone methylation (e.g., H3K4me3) corresponds and/or is correlated with transcription factor binding. In various embodiments, without wishing to be bound by any particular scientific theory, histone acetylation (e.g., H3K27ac) corresponds and/or is correlated with transcription factor binding. In various embodiments, without wishing to be bound by any particular scientific theory, DNA methylation corresponds and/or is correlated with transcription factor binding. [0194] In some embodiments, without wishing to be limited to any particular scientific theory, binding of RNA pol II corresponds and/or is correlated with H3K4me3 modifications. As a result, in some embodiments, ER status may be determined by detecting and quantifying binding of RNA pol II at one or more genomic loci in Table 1 in accordance with the section above discussing exemplary genomic loci with differential H3K4me3 modifications. [0195] In some embodiments, without wishing to be limited to any particular scientific theory, binding of p300, mediator complex, cohesin complex or RNA pol II corresponds and/or is correlated with H3K27ac modifications. As a result, in some embodiments, ER status may be determined by detecting and quantifying binding of p300, mediator complex, cohesin complex or RNA pol II at one or more genomic loci in Table 2 in accordance with the section above discussing exemplary genomic loci with differential H3K27ac modifications. [0196] In some embodiments, without wishing to be limited to any particular scientific theory, binding of FOXA1, ESR1, PR, MYC, EN1, FOXM1, KLF4, AP-2, RARa, or RUNX1 corresponds and/or is correlated with histone methylation (e.g., H3K4me3), histone acetylation (e.g., H3K27ac) or DNA methylation. As a result, in some embodiments, ER status may be determined by detecting and quantifying binding of FOXA1, ESR1, PR, MYC, EN1, FOXM1, KLF4, AP-2, RARa, RUNX1 at one or more genomic loci in Tables 1-3 in accordance with the sections above discussing exemplary genomic loci with differential histone methylation (e.g., H3K4me3), histone acetylation (e.g., H3K27ac) or DNA methylation. Applications [0197] Methods, kits and systems of the present disclosure include analysis of differentially modified and/or differentially accessible genomic loci to determine the ER status of
12366150v1
Attorney Docket No.2014191-0027 a cancer. Methods, kits and systems of the present disclosure can be used in any of a variety of applications. For example, methods, kits and systems of the present disclosure can be used in detecting and/or treating cancers based on ER status. Methods, kits and systems of the present disclosure can also be used to detect or determine resistance of a cancer, e.g., breast, ovarian, or endometrial cancer to a therapy or transformation from one cancer subtype to another. [0198] In various embodiments, methods, kits and systems of the present disclosure can be applied to an asymptomatic human subject. As used herein, a subject can be referred to as “asymptomatic” if the subject does not report, and/or demonstrate by non-invasively observable indicia (e.g., without one, several, or all of device-based probing, tissue sample analysis, bodily fluid analysis, surgery, or cancer screening), sufficient characteristics of cancer to support a medically reasonable suspicion that the subject is likely suffering from cancer, e.g., breast, ovarian, or endometrial cancer. Detection of early-stage cancer can be achieved using methods, kits and systems of the present disclosure, with attendant medical benefits including potential for early treatment and attendant improvement in therapeutic outcomes. [0199] In various embodiments, methods, kits, and systems of the present disclosure can be applied to a human subject that has increased susceptibility for cancer (including breast cancer). [0200] In various embodiments, methods, kits and systems of the present disclosure can be applied to a symptomatic human subject. As used herein, a subject can be referred to as “symptomatic” if the subject report, and/or demonstrates by non-invasively observable indicia (e.g., without one, several, or all of device-based probing, tissue sample analysis, bodily fluid analysis, surgery, or cancer screening), sufficient characteristics of cancer to support a medically reasonable suspicion that the subject is likely suffering from cancer, e.g., breast, ovarian, or endometrial cancer. For example, in various embodiments a sample from a subject, optionally where the subject has a cancer that is of unknown ER status, can be assayed according to one or more embodiments of the present disclosure to determine if the cancer is ER-positive or ER- negative. In various embodiments a sample from a subject, where the subject has a cancer that is known or suspected of being ER-positive (or ER-negative), can be assayed according to one or more embodiments of the present disclosure to determine if the cancer is in fact ER-positive (or ER-negative).
12366150v1
Attorney Docket No.2014191-0027 [0201] In some embodiments, methods, kits and systems of the present disclosure can be used to determine that a subject has an ER-positive cancer, optionally an ER-positive cancer that correlates with an ER+ Allred score of 3, 4, 5, 6, 7 or 8 based on IHC testing. In some embodiments, methods, kits and systems of the present disclosure can be used to determine that a subject has an ER-positive cancer, optionally an ER-positive cancer that correlates with an ER+ Allred score of at least 3, at least 4, at least 5, at least 6, at least 7 or 8 based on IHC testing. In some embodiments, methods, kits and systems of the present disclosure can be used to determine that a subject has an ER-negative cancer, optionally an ER-negative cancer that correlates with ER- Allred score of 0, 1, or 2 based on IHC testing. [0202] In some embodiments, methods, kits and systems of the present disclosure can be used to validate or confirm a prior determination that a subject has an ER-positive cancer, optionally an ER-positive cancer that correlates with an ER+ Allred score of 3, 4, 5, 6, 7 or 8 based on IHC testing. In some embodiments, methods, kits and systems of the present disclosure can be used to validate or confirm a prior determination that a subject has an ER-positive cancer, optionally an ER-positive cancer that correlates with an ER+ Allred score of at least 3, at least 4, at least 5, at least 6, at least 7 or 8 based on IHC testing. In some embodiments, methods, kits and systems of the present disclosure can be used to validate or confirm a prior determination that a subject has an ER-negative cancer, optionally an ER-negative cancer that correlates with ER- Allred score of 0, 1, or 2 based on IHC testing. [0203] In some embodiments, methods, kits and systems of the present disclosure are used to identify and detect new ER related categories that are independent of IHC or ISH scoring. For example, instead of training the classifier on samples from cohorts that were defined based on ER IHC or ISH testing, classifiers are trained on samples from cohorts that are defined based on whether they respond or do not respond to a particular ER-targeted agent. The resulting classifiers are then used to identify subjects that are more likely to respond to the particular ER- targeted agent independent of any IHC or ISH scoring. It is therefore to be understood that the term “ER status” as used herein is not limited to ER-positive and ER-negative or the traditional ER scoring based on IHC or ISH testing but can encompass any ER related categories including whether a subject will or will not respond to a particular ER-targeted agent. [0204] Those of skill in the art will appreciate that regular, preventative, and/or prophylactic screening to determine ER status improves diagnosis of cancer, including and/or
12366150v1
Attorney Docket No.2014191-0027 particularly early-stage cancer. Thus, the present disclosure provides, among other things, methods, kits and systems particularly useful for the diagnosis and treatment of early-stage cancer. Generally, and particularly in embodiments in which ER-positive cancer detection in accordance with the present disclosure is carried out annually, and/or in which a subject is asymptomatic at time of detecting, methods, kits and systems of the present disclosure are especially likely to detect early-stage ER-positive cancer. In various embodiments, detecting in accordance with methods, kits and systems of the present disclosure reduces cancer mortality, e.g., by early cancer diagnosis. [0205] In various embodiments ER status determination in accordance with the present disclosure is performed once for a given subject or multiple times for a given subject. In various embodiments, ER status determination in accordance with the present disclosure is performed on a regular basis, e.g., every six months, annually, every two years, every three years, every four years, every five years, or every ten years. [0206] In various embodiments, methods, kits and systems disclosed herein provide a determination of ER status. In other instances, methods, kits and systems disclosed herein will be indicative of ER status but not definitive for ER status. In various instances in which methods, kits and systems of the present disclosure are used to determine ER status, the same can be followed by a further confirmatory assay, which further assay can confirm, support, undermine, or reject a determination resulting from a prior determination, e.g., a determination in accordance with the present disclosure. As used herein, a confirmatory assay can be an ER test that is currently recognized by medical practitioners, e.g., ER scoring based on IHC or ISH testing. [0207] In various embodiments, ER status determination according to one or more methods, kits and/or systems disclosed herein is followed by treatment of cancer. In various embodiments, treatment of cancer includes administration of a therapeutic regimen including one or more cancer therapies provided herein, including without limitation one or more of ER targeted therapy, surgery, radiation, endocrine therapy, chemotherapy, and/or immunotherapy. In various embodiments, treatment of cancer includes administration of a therapeutic regimen including one or more treatments provided herein as available, appropriate, and/or preferred for a particular ER status. [0208] In various embodiments, methods, kits and systems can be used to determine whether a particular subject and/or cancer is likely to be and/or is characterized as responsive to
12366150v1
Attorney Docket No.2014191-0027 ER targeted therapy. In some such embodiments, methods, kits and systems can be followed by treatment of the subject with an ER targeted therapy. [0209] In various embodiments, methods, kits and systems can be used to determine whether a particular subject and/or cancer is likely to be and/or is characterized as resistant to, non-responsive to, or not recommended treatment with to ER targeted therapy. In some such embodiments, methods, kits and systems can be followed by treatment with one or more of surgery and/or radiation, a HER2-targeted agent (if HER2-positive), chemotherapy and immunotherapy instead of ER targeted therapy. [0210] Responsiveness can refer to the ability or likelihood of a therapy to cause a reduction in tumor size or inhibit tumor growth or metastasis. Responsiveness can refer to improvement in prognosis (e.g., increased time to cancer recurrence or increased life expectancy, e.g., overall survival, recurrence-free survival, metastasis-free survival, or disease-free survival). Responsiveness can refer to achievement of a treatment benefit, including e.g., improvement in one or more symptoms of cancer, e.g., breast, ovarian, or endometrial cancer. Responsiveness can be measured quantitatively (e.g., as in the case of tumor size; as in the case of measurement of histone modification, chromatin accessibility, transcription factor binding, or DNA methylation at one or more genomic loci; or as in the calculation of clinical benefit (CBR)), or qualitatively (e.g., by measures such as “pathological complete response” (pCR), “clinical complete remission” (cCR), “clinical partial remission” (cPR), “clinical stable disease” (cSD), “clinical progressive disease” (cPD), or other qualitative criteria). Resistance can refer to the inability or unlikelihood of a therapy to achieve a desired therapeutic effect (e.g., a reduction in tumor size, improvement in prognosis, or other treatment benefit such as, e.g., improvement in one or more symptoms of cancer) in a subject and/or cancer. Resistance includes both acquired and natural resistance. In certain embodiments, resistance includes the extent to which one or more desired therapeutic benefits results from administration of a therapy to a subject and/or cancer is less than that expected and/or achieved in a reference (e.g., less than 90%, 80%, 70%, 60%, 50%, 40%, 30%, 20%, or 10% of benefit achieved in a reference). [0211] In various embodiments, methods, kits and systems can be used to detect the clinical efficacy of a course of therapy for cancer, e.g., breast, ovarian, or endometrial cancer. For example, methods and/or compositions of the present disclosure could be used to determine the presence, absence, or ER status of a cancer in a subject over the course of treatment. Methods
12366150v1
Attorney Docket No.2014191-0027 and/or compositions of the present disclosure could be used in conjunction with, or confirmed by, other means of determining the presence, absence, or ER status of a cancer including, for example measurements of tumor size or character by techniques such as CT, PET, mammogram, ultrasound, palpation, histology, caliper measurement after biopsy or surgical resection, or by various qualitative, quantitative, or semi quantitative scoring systems including without limitation based on IHC or ISH testing, residual cancer burden (Symmans et al., J Clin Oncol (2007) 25:4414-4422, incorporated by reference herein in its entirety) or Miller-Payne score (Ogston et al., Breast (2003) 12:320-327, incorporated by reference herein in its entirety) in a qualitative fashion like “pathological complete response” (pCR), “clinical complete remission” (cCR), “clinical partial remission” (cPR), “clinical stable disease” (cSD), or “clinical progressive disease” (cPD). [0212] In some embodiments, methods, kits, and systems described herein can be used to monitor progression of disease in a subject. In some embodiments, monitoring progression entails obtaining and characterizing samples from a subject at at least a first and a second time point. In some embodiments, at the first time point, a subject has already been diagnosed with lung cancer (e.g., an ER-negative cancer or ER-positive cancer). In some embodiments, at a first time point, a subject has been determined to have cancer and therapy is administered before or close to (e.g., the same day as) the first time point or between the first time point and the second time point; in such embodiments, determination of ER status at at least the first and the second time points can be used to monitor treatment efficacy and/or determine when a change in therapy should be made. For example, in some embodiments, a subject has previously been diagnosed with an ER-positive at the first time point, an ER-positive therapy is being or will be administered to the subject, and disease status can be monitored, which can be useful, e.g., for determining whether a change in therapy should be made. In some embodiments, treatment efficacy can be monitored, e.g., by using a method described herein to determine a decrease or increase in disease state signal, which can be useful, e.g., for determining whether an administered therapy is effective and/or whether a change in therapy should be made. In some embodiments, at the first time point, a cancer has gone into remission for a subject (e.g., the subject has minimal residual disease). In embodiments where a cancer has gone into remission, methods, kits, and systems described herein can be useful, e.g., for detecting reoccurrence of
12366150v1
Attorney Docket No.2014191-0027 cancer, and can be faster, less expensive, and/or less invasive than, e.g., approaches that rely on tissue biopsies and/or imaging techniques. [0213] In some embodiments, methods, kits and systems for ER status determination provided herein can inform treatment and/or payment (e.g., reimbursement for or reduction of cost of medical care, such as detecting or treatment) decisions and/or actions, e.g., by individuals, healthcare facilities, healthcare practitioners, health insurance providers, governmental bodies, or other parties interested in healthcare cost. [0214] In some embodiments, methods, kits and systems for ER status determination provided herein can inform decision making relating to whether health insurance providers reimburse a healthcare cost payer or recipient (or not), e.g., for (1) ER status determination itself (e.g., reimbursement for detecting otherwise unavailable, available only for periodic/regular detecting, or available only for temporally- and/or incidentally- motivated detecting); and/or for (2) treatment, including initiating, maintaining, and/or altering therapy, e.g., based on the determined ER status. For example, in some embodiments, methods, kits and systems for ER status determination provided herein are used as the basis for, to contribute to, or support a determination as to whether a reimbursement or cost reduction will be provided to a healthcare cost payer or recipient. In some instances, a party seeking reimbursement or cost reduction can provide results of ER status determination conducted in accordance with the present disclosure together with a request for such reimbursement or reduction of a healthcare cost. In some instances, a party making a determination as to whether or not to provide a reimbursement or reduction of a healthcare cost will reach a determination based in whole or in part upon receipt and/or review of results of ER status determination conducted in accordance with the present disclosure. [0215] In various embodiments, ER status determination using methods, kits and systems disclosed herein can be used in classifying subjects, samples, and/or tumors (e.g., breast cancer subjects, samples, and/or tumors). In various embodiments, methods, kits and systems disclosed herein can be used to generate a set of subjects, samples, and/or tumors identified according to the present methods, kits and systems each classified as corresponding to a particular ER status, and optionally using two or more of such classified subjects, samples, and/or tumors to identify biomarkers that distinguish the classes (i.e., distinguish the subjects, samples, and/or tumors according to their class, e.g., according to their ER status).
12366150v1
Attorney Docket No.2014191-0027 [0216] For illustration purposes and without limitation, in an exemplary assay of the present disclosure, one or more samples obtained from a subject (e.g., a liquid biopsy sample including cfDNA, e.g., a plasma sample including cfDNA) are analyzed by a method comprising enriching for cfDNA comprising a particular histone modification, wherein enriching is performed by a method that comprises incubating the sample with a reagent that specifically binds the histone modification being enriched for, and sequencing the enriched cfDNA. One example of such an assay is ChIP-seq for a histone modification (e.g., H3K4me3 and/or H3K27ac). Sequence reads (e.g., ChIP-seq sequence reads) can be aligned to human genome build hg19, e.g., using the Burrows-Wheeler Aligner (BWA). Non-uniquely mapping and redundant reads are optionally discarded. [0217] To provide one example of peak calling, MACS v2.1.1.20140616 can be used for sequence (e.g., ChIP-seq) peak calling with a q-value (FDR) threshold of 0.01. Sequence (e.g., ChIP-seq) data quality can optionally be evaluated by any of one or more of a variety of measures, including total peak number, FRiP (fraction of reads in peak) score, number of high- confidence peaks (e.g., enriched > ten-fold over background), and percent of peak overlap with “blacklist” DHS peaks derived from the ENCODE project (Amemiya et al., Sci Rep (2019) 9(1):9354). If the sequence (e.g., ChIP-seq) data quality is below a particular threshold, the data may be discarded and the assay repeated. Sequence (e.g., ChIP-seq) peaks that overlap with selected genomic loci that are differentially modified as provided herein for the relevant histone modification (Tables 1-2) can then be used to determine ER status. The number of reads overlapping the selected genomic loci for the relevant histone modification can be summed, e.g., in some embodiments all the genomic loci that are differentially modified with an absolute log2(fold-change) ≥ 4.0 are selected. In some embodiments, the average number of reads in the local background of each ChIP-seq peak is subtracted to improve signal to noise. In some embodiments, a sequence read density for one or more histone modifications can be calculated by a method that comprises (1) summing background adjusted sequence counts at at one or more genomic loci and dividing the resulting sum by the total number of kilobases of the one or more genomic loci, or (2) for each genomic loci, determining the ratio of background adjusted fragment counts to the number of kilobases of the genomic loci, and then summing the ratios for each loci. In some embodiments, a method comprises determining an ER-positive/ ER-negative ratio score, e.g., by a method that comprises (a) calculating an ER-positive sequence read
12366150v1
Attorney Docket No.2014191-0027 density, calculating an ER-negative sequence read density, and dividing the ER-positive sequence read density by the ER-negative sequence read density. In some embodiments, an ER- positive sequence read density can be determined by a method that comprises calculating sequence read density using one or more genomic loci with an increased level of one or more epigenetic biomarkers in sample(s) obtained from one or more subjects with an ER -positive cancer as compared to one or more sample(s) obtained from subjects with an ER-negative cancer. In some embodiments, an ER -negative sequence read density can be determined by a method that comprises calculating sequence read density using one or more genomic loci with an increased level of one or more epigenetic biomarkers in sample(s) obtained from one or more subjects with an ER-negative cancer as compared to one or more sample(s) obtained from subjects with an ER-positive cancer. An ER-positive/ ER-negative ratio score for determined for one or more histone modifications. In some embodiments an ER-positive/ER-negative ratio score is determined for H3K4me3 modifications. In some embodiments, an ER-positive/ER-negative ratio score is determined for H3K27ac modifications. In some embodiments, an ER-positive/ER- negative ratio score is determined for methylated DNA. In some embodiments, an ER- positive/ER-negative ratio score is determined for H3K4me3 modifications and H3K27ac modifications, H3K4me3 and methylated DNA, or H3K27ac and methylated DNA. In some embodiments, an ER-positive/ER-negative ratio score is determined for each of H3K4me3 modifications, H3K27ac modifications, and methylated DNA. In some embodiments, two or more ER-positive/ER-negative ratio scores for different epigenetic biomarkers can be combined. In some embodiments, each ratio score can be combined using fitted values that have been determined using a logistic regression. [0218] The data can then be log2-transformed and quantile normalized to match the distribution of the data used to train a classifier. Normalized data can be used as input into a classifier that was trained using the same histone modification(s) and selected genomic loci. The classifier can then use inputted data to determine ER status of a subject’s cancer. It will be appreciated that this or similar approaches can be applied to assays of the present disclosure that quantify chromatin accessibility, transcription factor binding and/or DNA methylation. [0219] In some embodiments, multiple epigenetic biomarkers (e.g., one or more histone modifications, chromatin accessibility, binding of one or more transcription factors, and/or DNA methylation) can be quantified for a single sample. In such embodiments, two or more assays for
12366150v1
Attorney Docket No.2014191-0027 assessing the epigenetic biomarkers can be performed in sequence (meaning a single sample can be probed for each modification in sequence) or in parallel (meaning that a single sample can be divided into multiple fractions, and then each fraction analyzed to quantifying an epigenetic biomarker). In some embodiments, H3K4me3 and H3K27ac histone modifications; H3K4me3 modifications and DNA methylation; H3K27ac modifications and DNA methylation; or H3K4me3 modifications, H3K27ac histone modifications, and DNA methylation are quantified in a single sample. [0220] For the avoidance of any doubt, those of skill in the art will appreciate from the present disclosure that methods, kits and systems for ER status determination of the present disclosure are at least for in vitro use. Accordingly, all aspects and embodiments of the present disclosure can be performed and/or used at least in vitro. [0221] Those of skill in the art will also appreciate that, in certain embodiments, methods of the present disclosure can be implemented on and/or in conjunction with a computer program and computer system. In some embodiments, methods of the present disclosure can be implemented on and/or in conjunction with a non-transitory computer readable storage medium encoded with the computer program, wherein the program comprises instructions that when executed by one or more processors cause the one or more processors to perform operations to perform the method. A computer system can also store and manipulate data generated by methods of the present disclosure that comprise a plurality of genomic locus modification status and/or accessibility status changes/profiles, which data can be used by a computer system in implementing methods disclosed herein. In certain embodiments, a computer system (i) receives modification status and/or accessibility status data; (ii) stores the data; and (iii) compares the data in any number of ways described herein (e.g., analysis relative to appropriate references), e.g., to determine ER status. In certain embodiments, a computer system (i) compares the genomic locus modification and/or accessibility status to a reference; and (ii) outputs an indication of whether the modification status and/or accessibility status of the genomic locus is significantly different from the reference and/or provides a determination regarding ER status. [0222] Numerous types of computer systems can be used to implement methods of the present disclosure according to knowledge possessed by a skilled artisan in the bioinformatics and/or computer arts. Several software components can be loaded into memory during operation of such a computer system. The software components can comprise both software components
12366150v1
Attorney Docket No.2014191-0027 that are standard in the art and components that are special to the present disclosure (e.g., dCHIP software described in Lin et al., Bioinformatics (2004) 20:1233-1240, incorporated herein by reference in its entirety; radial basis machine learning algorithms (RBM) known in the art). Methods of the present disclosure can also be programmed or modeled in mathematical software packages that allow symbolic entry of equations and high-level specification of processing, including specific algorithms to be used, thereby freeing a user of the need to procedurally program individual equations and algorithms. Such packages include, e.g., Matlab from Mathworks (Natick, MA), Mathematica from Wolfram Research (Champaign, IL), S-Plus from MathSoft (Seattle, WA), R from R Foundation for Statistical Computing (Vienna, Austria), Python from Python Software Foundation (Wilmington, DE), or Perl from Perl Foundation (Holland, MI). In certain embodiments, a computer system comprises a database for storage of genomic locus modification status and/or accessibility status data. Such stored profiles can be accessed and used to perform comparisons of interest at a later point in time. In addition to the exemplary program structures and computer systems described herein, other, alternative program structures and computer systems will be readily apparent to the skilled artisan. [0223] As demonstrated in the Examples, various algorithms can be applied to the comparison, between samples and references, of the modification status and/or accessibility status of genomic loci that are differentially modified in different ER states. In various embodiments, an algorithm can be a single learning statistical classifier system. Other suitable statistical algorithms are well known to those of skill in the art. For example, learning statistical classifier systems include a machine learning algorithmic technique capable of adapting to complex datasets (e.g., a panel of genomic loci of interest) and making decisions based upon such datasets. In some embodiments, a single learning statistical classifier system such as a classification tree (e.g., random forest) is used. In other embodiments, a combination of 2, 3, 4, 5, 6, 7, 8, 9, 10, or more learning statistical classifier systems are used, preferably in tandem. Examples of learning statistical classifier systems include, but are not limited to, those described in the Examples and also those using inductive learning (e.g., decision/classification trees such as random forests, classification and regression trees (C&RT), boosted trees, etc.), Probably Approximately Correct (PAC) learning, connectionist learning (e.g., neural networks (NN), artificial neural networks (ANN), neuro fuzzy networks (NFN), network structures, perceptrons such as multi-layer perceptrons, multi-layer feed-forward networks, applications of neural
12366150v1
Attorney Docket No.2014191-0027 networks, Bayesian learning in belief networks, etc.), reinforcement learning (e.g., passive learning in a known environment such as naive learning, adaptive dynamic learning, and temporal difference learning, passive learning in an unknown environment, active learning in an unknown environment, learning action-value functions, applications of reinforcement learning, etc.), and genetic algorithms and evolutionary programming. Other learning statistical classifier systems include support vector machines (e.g., Kernel methods), multivariate adaptive regression splines (MARS), Levenberg-Marquardt algorithms, Gauss-Newton algorithms, mixtures of Gaussians, gradient descent algorithms, and learning vector quantization (LVQ). In certain embodiments, methods of the present disclosure can include sending classification results to a medical practitioner, e.g., an oncologist. [0224] In various embodiments, the area under the receiver operating characteristic (AUROC) for determining if a subject has a particular ER cancer status (e.g., an ER-positive cancer vs. an ER-negative cancer) is greater than 0.5 (e.g., greater than 0.55, greater than 0.6, greater than 0.65, greater than 0.7, greater than 0.75, greater than 0.8, greater than 0.85, greater than 0.9, or greater than 0.95). Formulation and Administration of Therapeutic Agents [0225] The present disclosure includes methods where a therapeutic agent or regimen is administered to a subject based on the ER status of a cancer (e.g., breast cancer, ovarian cancer, or endometrial cancer). In general, the therapeutic agent or regimen provided herein will be available, appropriate, and/or preferred for the determined ER status. Those of skill in the art will be aware of recommended and/or governmentally approved formulations and/or dosages for various therapeutic agents provided herein. [0226] The present disclosure includes pharmaceutical compositions for delivery of one or more therapeutic agents to a subject. As disclosed herein, a pharmaceutical composition may be in any form known in the art, including formulations for administration according to any route known in the art. A suitable means of administration can be selected based on the age and condition of a subject. [0227] Pharmaceutical composition forms of the present disclosure can include, e.g., liquid, semi-solid and solid dosage forms. Pharmaceutical composition forms of the present disclosure can include, e.g., liquid solutions (e.g., injectable and infusible solutions), dispersions
12366150v1
Attorney Docket No.2014191-0027 or suspensions, tablets, pills, powders, and liposomes. Selection or use of any particular form may depend, in part, on the intended mode of administration and therapeutic application. Accordingly, the compositions can be formulated for administration by a parenteral mode (e.g., intravenous, subcutaneous, intraperitoneal, or intramuscular injection) or a non-parenteral mode. As used herein, parenteral administration refers to modes of administration other than enteral and topical administration, usually by injection or infusion. [0228] In some embodiments, the compositions provided herein are present in unit dosage form, which unit dosage form can be suitable for self-administration. Such a unit dosage form may be provided within a container, e.g., a pill, vial, cartridge, prefilled syringe, or disposable pen. [0229] A pharmaceutical composition of the present disclosure can be in an injectable or infusible form. For example, the present disclosure includes sterile formulations for injection or infusion, which can be formulated in accordance with conventional pharmaceutical practices. Sterile solutions can be prepared by incorporating a composition described herein in the required amount in an appropriate solvent with one or a combination of ingredients enumerated above, as required, followed by filter sterilization. Solutions can be formulated, e.g., using distilled water, physiological saline, or an isotonic solution containing glucose and other supplements such as D- sorbitol, D-mannose, D-mannitol, or sodium chloride as an aqueous solution for injection, optionally in combination with a suitable solubilizing agent, for example, an alcohol such as ethanol and/or a polyalcohol such as propylene glycol or polyethylene glycol, and/or a nonionic surfactant such as polysorbate 80™ or HCO-50, and the like. In the case of sterile powders for the preparation of sterile injectable solutions, methods for preparation include vacuum drying and freeze-drying that yield a powder of a composition described herein plus any additional desired ingredient (see below) from a previously sterile-filtered solution thereof. The proper fluidity of a solution can be maintained, for example, by the use of a coating such as lecithin, by the maintenance of the required particle size in the case of dispersion and by the use of surfactants. Prolonged absorption of injectable compositions can be brought about by including in the composition a reagent that delays absorption, for example, monostearate salts, and gelatin. In particular instances, a pharmaceutical composition can be formulated, for example, as a buffered solution at a suitable concentration and suitable for storage, e.g., at 2-8°C (e.g., 4°C).
12366150v1
Attorney Docket No.2014191-0027 [0230] In various embodiments, a pharmaceutical composition of the present disclosure can be formulated as a solution, microemulsion, dispersion, liposome, or other ordered structure suitable for stable storage at high concentration. Generally, dispersions are prepared by incorporating a composition described herein into a sterile vehicle that contains a basic dispersion medium and the required other ingredients from those enumerated above. [0231] In various instances, a pharmaceutical composition can be formulated to include a pharmaceutically acceptable carrier or excipient. Examples of pharmaceutically acceptable carriers include, without limitation, any and all solvents, dispersion media, coatings, antibacterial and antifungal agents, isotonic and absorption delaying agents, and the like that are physiologically compatible. [0232] In certain embodiments, compositions can be formulated with a carrier that will protect the therapeutic agent against rapid release, such as a controlled release formulation, including implants and microencapsulated delivery systems. Biodegradable, biocompatible polymers can be used, such as ethylene vinyl acetate, polyanhydrides, polyglycolic acid, collagen, polyorthoesters, and polylactic acid. Many methods for the preparation of such formulations are known in the art. See, e.g., J. R. Robinson (1978) “Sustained and Controlled Release Drug Delivery Systems,” Marcel Dekker, Inc., New York. [0233] Route of administration can be parenteral, for example, administration by injection. Administration by injection can be by intravenous injection, intramuscular injection, intraperitoneal injection, subcutaneous injection. Administration can be systemic or local. In certain embodiments, a composition described herein can be therapeutically delivered to a subject by way of local administration. As used herein, “local administration” or “local delivery,” can refer to delivery that does not rely upon transport of the composition or therapeutic agent to its intended target tissue or site via the vascular system. For example, the composition may be delivered by injection or implantation of the composition or therapeutic agent or by injection or implantation of a device containing the composition or therapeutic agent. In certain embodiments, following local administration in the vicinity of a target tissue or site, the composition or therapeutic agent, or one or more components thereof, may diffuse to an intended target tissue or site that is not the site of administration. [0234] A pharmaceutical composition can be administered parenterally in the form of an injectable formulation comprising a sterile solution or suspension in water or another
12366150v1
Attorney Docket No.2014191-0027 pharmaceutically acceptable liquid. For example, a pharmaceutical composition can be formulated by suitably combining the therapeutic molecule with pharmaceutically acceptable vehicles or media, such as sterile water and physiological saline, vegetable oil, emulsifier, suspension agent, surfactant, stabilizer, flavoring excipient, diluent, vehicle, preservative, binder, followed by mixing in a unit dose form required for generally accepted pharmaceutical practices. Examples of oily liquid include sesame oil and soybean oil, and it may be combined with benzyl benzoate or benzyl alcohol as a solubilizing agent. Other items that may be included are a buffer such as a phosphate buffer, or sodium acetate buffer, a soothing agent such as procaine hydrochloride, a stabilizer such as benzyl alcohol or phenol, and an antioxidant. The formulated injection can be packaged in a suitable ampule. [0235] In various embodiments, subcutaneous administration can be accomplished by means of a device, such as a syringe, a prefilled syringe, an auto-injector (e.g., disposable or reusable), a pen injector, a patch injector, a wearable injector, an ambulatory syringe infusion pump with subcutaneous infusion sets, or other device for combining with a therapeutic agent for subcutaneous injection. [0236] An injection system of the present disclosure may employ a delivery pen as described in U.S. Pat. No.5,308,341. Pen devices, most commonly used for self-delivery of insulin to patients with diabetes, are well known in the art. Such devices can include at least one injection needle, are typically pre-filled with one or more therapeutic unit doses of a solution that includes the therapeutic agent and are useful for rapidly delivering solution to a subject with as little pain as possible. One medication delivery pen includes a vial holder into which a vial of a therapeutic or other medication may be received. The pen may be an entirely mechanical device or it may be combined with electronic circuitry to accurately set and/or indicate the dosage of medication that is injected into the user. See, e.g., U.S. Pat. No.6,192,891. In some embodiments, the needle of the pen device is disposable and the kits include one or more disposable replacement needles. Pen devices suitable for delivery of any one of the presently featured compositions are also described in, e.g., U.S. Pat. Nos.6,277,099; 6,200,296; and 6,146,361, the disclosures of each of which are incorporated herein by reference in their entirety. A microneedle-based pen device is described in, e.g., U.S. Pat. No.7,556,615, the disclosure of which is incorporated herein by reference in its entirety. See also the Precision Pen Injector (PPI) device, MOLLYTM, manufactured by Scandinavian Health Ltd.
12366150v1
Attorney Docket No.2014191-0027 [0237] In certain embodiments, administration of a therapeutic agent as described herein is achieved by administering to a subject a nucleic acid encoding a therapeutic agent described herein. Nucleic acids encoding a therapeutic agent described herein can be incorporated into a gene construct to be used as a part of a gene therapy protocol to deliver nucleic acids that can be used to express and produce therapeutic agent within cells. Expression constructs of such components may be administered in any therapeutically effective carrier, e.g., any formulation or composition capable of effectively delivering the component gene to cells in vivo. Approaches include insertion of the subject gene in viral vectors including recombinant retroviruses, adenovirus, adeno-associated virus, lentivirus, and herpes simplex virus-1 (HSV-1), or recombinant bacterial or eukaryotic plasmids. Viral vectors can transfect cells directly; plasmid DNA can be delivered with the help of, for example, cationic liposomes (lipofectin) or derivatized, polylysine conjugates, gramicidin S, artificial viral envelopes or other such intracellular carriers, as well as direct injection of the gene construct or CaPO4 precipitation. Examples of suitable retroviruses include adenovirus-derived vectors, adeno-associated virus (AAV), pLJ, pZIP, pWE, and pEM which are known to those skilled in the art. [0238] In some embodiments, a composition can be formulated for storage at a temperature below 0°C (e.g., -20°C or -80°C). In some embodiments, the composition can be formulated for storage for up to 2 years (e.g., one month, two months, three months, four months, five months, six months, seven months, eight months, nine months, 10 months, 11 months, 1 year, or 2 years) at 2-8°C (e.g., 4°C). Thus, in some embodiments, the compositions described herein are stable in storage for at least 1 year at 2-8°C (e.g., 4°C). [0239] A pharmaceutical composition can include a therapeutically effective amount of a therapeutic agent described herein. Such effective amounts can be readily determined by one of ordinary skill in the art. A therapeutically effective amount can be an amount at which any toxic or detrimental effects of the composition are outweighed by therapeutically beneficial effects. In some embodiments, a dose can also be chosen to reduce or avoid production of antibodies or other host immune responses against a therapeutic agent. Those of skill in the art will appreciate that data obtained from cell culture assays and animal studies can be used in formulating a range of dosage for use in humans. In various embodiments, the amount of active ingredient included in a pharmaceutical composition is such that a suitable dose within the designated range can be administered to subjects. The dose and method of administration can vary depending on weight,
12366150v1
Attorney Docket No.2014191-0027 age, condition, and other characteristics of a patient, and can be suitably selected as needed by those skilled in the art. [0240] Pharmaceutical compositions including certain therapeutic agents, e.g., therapeutic antibodies, can be administered as a fixed dose, or in a milligram per kilogram (mg/kg) dose. While in no way intended to be limiting, an exemplary single dose of certain pharmaceutical compositions described herein can include certain therapeutic agents as described herein in an amount equal to, e.g., 0.001 to 1000 mg/kg, 1-1000 mg/kg, 1-100 mg/kg, 0.5-50 mg/kg, 0.1-100 mg/kg, 0.5-25 mg/kg, 1-20 mg/kg, and 1-10 mg/kg body weight. Exemplary dosages of a composition described herein include, without limitation, 0.1 mg/kg, 0.5 mg/kg, 1 mg/kg, 2 mg/kg, 4 mg/kg, 8 mg/kg, or 20 mg/kg. The present disclosure is not limited to such ranges or dosages. [0241] The present disclosure further includes methods of preparing pharmaceutical compositions of the present disclosure and kits including pharmaceutical compositions of the present disclosure. [0242] In various embodiments, therapeutic agents of the present disclosure can be administered to a subject in a course of treatment that further includes administration of one or more additional therapeutic agents or therapies that are not therapeutic agents (e.g., surgery or radiation). Combination therapies of the present disclosure can include simultaneous exposure of a subject to therapeutic agents of two or more therapeutic regimens. [0243] In certain embodiments, a therapeutic agent as described herein can be administered together with (e.g., at the same time and/or in the same composition as) an additional agent or therapy. In certain embodiments, a therapeutic agent of the present disclosure can be administered separately from an additional therapeutic agent or therapy (e.g., at a different time and/or in a different composition than the additional therapeutic agent or therapy). Dosing regimens of a therapeutic agent and one or more additional therapeutic agents with which it is administered in combination can be coordinated or independently determined. In various embodiments, an additional therapeutic agent or therapy administered in combination with a therapeutic agent as described herein can be administered at the same time as therapeutic agent, on the same day as therapeutic agent, or in the same week as therapeutic agent. In various embodiments, an additional therapeutic agent or therapy administered in combination with a therapeutic agent as described herein can be administered such that administration of the
12366150v1
Attorney Docket No.2014191-0027 therapeutic agent and the additional therapeutic agent or therapy are separated by one or more hours before or after, one or more days before or after, one or more weeks before or after, or one or more months before or after administration of the therapeutic agent. In various embodiments, the administration frequency and/or dosage of one or more additional therapeutic agents can be the same as, similar to, or different from the administration frequency of a therapeutic agent. In some embodiments, the two or more regimens can be administered simultaneously; in some embodiments, such regimens can be administered sequentially (e.g., all “doses” of a first regimen are administered prior to administration of any doses of a second regimen); in some embodiments, such therapeutic agents are administered in overlapping dosing regimens. [0244] In certain embodiments, administration of a therapeutic agent can be to a subject having previously received, scheduled to receive, or in the course of a treatment regimen including an additional cancer therapy. Administration of a therapeutic agent can, in some instances, improve delivery or efficacy of another therapeutic agent or therapy with which it is administered in combination. [0245] It is contemplated that therapeutic agent combination therapies can demonstrate synergy and/or greater-than-additive effects between a therapeutic agent and one or more additional therapeutic agents with which it is administered in combination. A therapeutic agent can be administered in any effective amount as determined independently or as determined by the joint action of therapeutic agent and any of one or more additional therapeutic agents or therapies administered. Administration of the therapeutic agent may, in some embodiments, reduce the therapeutically effective dosage, required dosage, or administered dosage of the additional therapeutic agent or therapy relative to a reference regimen for administration of additional therapeutic agent or therapy or therapy absent the therapeutic agent. In certain embodiment, a composition described herein can replace or augment other previously or currently administered therapy. For example, upon treating with therapeutic agent, administration of one or more additional therapeutic agents or therapies can cease or diminish, e.g., be administered at lower levels. Kits [0246] The present disclosure includes kits for detecting modification and/or accessibility of one or more genomic loci. In some embodiments, the present disclosure provides kits for
12366150v1
Attorney Docket No.2014191-0027 quantifying one or more histone modifications, chromatin accessibility, binding of one or more transcription factors, and/or DNA methylation at one or more genomic loci. Kits of the present disclosure can include, e.g., reagents such as buffers and/or antibodies useful in the detection and quantification of histone modifications. In certain embodiments, a kit of the present disclosure can include at least one antibody that selective binds a histone modification selected from H3K9ac, H3K14ac, H3K18ac, H3K23ac, H3K27ac, H3K4me1, H3K4me2, or H3K4me3, or pan acetylation. In certain embodiments, a kit of the present disclosure can include at least one antibody that selective binds H3K4me3 modifications. In certain embodiments, a kit of the present disclosure can include at least one antibody that selective binds H3K27ac modifications. A kit of the present disclosure can include instructional materials disclosing or describing the use of the kit in a method of determining ER status and/or treatment disclosed herein. In various embodiments, a kit of the present disclosure can include one or more therapeutic agents useful in the treatment of cancer, e.g., as disclosed herein, optionally in combination with instruction materials for treatment of cancer, e.g., breast cancer, ovarian cancer, or endometrial cancer based on ER status. [0247] In some embodiments, a kit of the present disclosure comprises reagents for quantifying one or more histone modifications, chromatin accessibility, binding of one or more transcription factors, and/or DNA methylation at one or more genomic loci, wherein the one or more genomic loci are selected from Tabled 1-3. [0248] In some embodiments, the kit comprises reagents for quantifying H3K4me3 for at least 5, 10, 20, 30, 40, or 50 genomic loci in Table 1. In some embodiments, the kit comprises reagents for quantifying H3K27ac for at least 5, 10, 20, 30, 40, or 50 genomic loci in Table 2. In some embodiments, the kit comprises one or more antibodies for use in ChIP-seq, optionally wherein the one or more antibodies specifically bind H3K4me3- or H3K27ac-modified histones. [0249] In some embodiments, the kit comprises reagents for quantifying DNA methylation for at least 5, 10, 20, 30, 40, or 50 genomic loci in Table 3. In some embodiments, the kit comprises one or more methyl-binding domains for use in MBD-seq. In some embodiments, the kit comprises one or more antibodies that can bind methylated DNA (e.g., for use in MeDIP). [0250] In some embodiments, the kit comprises reagents for isolation of cell-free DNA (cfDNA) from a liquid biopsy sample. In some embodiments, the kit comprises reagents for
12366150v1
Attorney Docket No.2014191-0027 library preparation for sequencing. In some embodiments, the kit comprises reagents for sequencing. In some embodiments, the kit comprises instructions for determining if a subject has an ER-positive cancer. Systems [0251] The present disclosure includes systems for detecting modification and/or accessibility of one or more genomic loci. In some embodiments, the present disclosure provides systems for quantifying one or more histone modifications, chromatin accessibility, binding of one or more transcription factors, and/or DNA methylation at one or more genomic loci. Systems of the present disclosure can include a sequencer configured to generate a sequencing dataset from a sample; and a non-transitory computer readable storage medium and/or a computer system. [0252] In some embodiments, the non-transitory computer readable storage medium is encoded with a computer program, wherein the program comprises instructions that when executed by one or more processors cause the one or more processors to perform operations to perform a method of the present disclosure. [0253] In some embodiments, the computer system comprises a memory and one or more processors coupled to the memory, wherein the one or more processors are configured to perform a method of the present disclosure. [0254] In some embodiments, the sequencer is configured to generate a Whole Genome Sequencing (WGS) dataset from the sample. In some embodiments, the system also includes a sample preparation device configured to prepare the sample for sequencing from a biological sample, optionally a liquid biopsy sample. The sample preparation device may include reagents for quantifying one or more histone modifications, chromatin accessibility, binding of one or more transcription factors, and/or DNA methylation at one or more genomic loci in cell-free DNA (cfDNA) from the biological sample, optionally the liquid biopsy sample. [0255] Systems of the present disclosure can include, e.g., reagents such as buffers and/or antibodies useful in the detection and quantification of histone modifications. In certain embodiments, a system of the present disclosure can include at least one antibody that selective binds a histone modification selected from H3K9ac, H3K14ac, H3K18ac, H3K23ac, H3K27ac, H3K4me1, H3K4me2, or H3K4me3, or pan acetylation. In certain embodiments, a system of the
12366150v1
Attorney Docket No.2014191-0027 present disclosure can include at least one antibody that selective binds H3K4me3 modifications. In certain embodiments, a system of the present disclosure can include at least one antibody that selective binds H3K27ac modifications. A system of the present disclosure can include instructional materials disclosing or describing the use of the system in a method of determining ER status and/or treatment disclosed herein. [0256] In some embodiments, a system of the present disclosure comprises reagents for quantifying one or more histone modifications, chromatin accessibility, binding of one or more transcription factors, and/or DNA methylation at one or more genomic loci, wherein the one or more genomic loci are selected from Tabled 1-3. [0257] In some embodiments, the system comprises reagents for quantifying H3K4me3 for at least 5, 10, 20, 30, 40, or 50 genomic loci in Table 1. In some embodiments, the system comprises reagents for quantifying H3K27ac for at least 5, 10, 20, 30, 40, or 50 genomic loci in Table 2. In some embodiments, the system comprises one or more antibodies for use in ChIP- seq, optionally wherein the one or more antibodies specifically bind H3K4me3- or H3K27ac- modified histones. [0258] In some embodiments, the system comprises reagents for quantifying DNA methylation for at least 5, 10, 20, 30, 40, or 50 genomic loci in Table 3. In some embodiments, the system comprises one or more methyl-binding domains for use in MBD-seq. [0259] In some embodiments, the system comprises reagents for isolation of cell-free DNA (cfDNA) from a liquid biopsy sample. In some embodiments, the sequencer comprises reagents for library preparation for sequencing. In some embodiments, the sequencer comprises reagents for sequencing. In some embodiments, the system comprises instructions for determining if a subject has an ER-positive cancer. [0260] Illustrative embodiments of systems and methods disclosed herein were described above with reference to computations performed locally by a computing device. However, computations performed over a network are also contemplated. Fig. 4 shows an illustrative network environment 400 for use in the methods and systems described herein. In brief overview, referring now to Fig. 4, a block diagram of an illustrative cloud computing environment 400 is shown and described. The cloud computing environment 400 may include one or more resource providers 402a, 402b, 402c (collectively, 402). Each resource provider 402 may include computing resources. In some implementations, computing resources may include any hardware and/or
12366150v1
Attorney Docket No.2014191-0027 software used to process data. For example, computing resources may include hardware and/or software capable of executing algorithms, computer programs, and/or computer applications. In some implementations, illustrative computing resources may include application servers and/or databases with storage and retrieval capabilities. Each resource provider 402 may be connected to any other resource provider 402 in the cloud computing environment 400. In some implementations, the resource providers 402 may be connected over a computer network 408. Each resource provider 402 may be connected to one or more computing device 404a, 404b, 404c (collectively, 404), over the computer network 408. [0261] The cloud computing environment 400 may include a resource manager 406. The resource manager 406 may be connected to the resource providers 402 and the computing devices 404 over the computer network 408. In some implementations, the resource manager 406 may facilitate the provision of computing resources by one or more resource providers 402 to one or more computing devices 404. The resource manager 406 may receive a request for a computing resource from a particular computing device 404. The resource manager 406 may identify one or more resource providers 402 capable of providing the computing resource requested by the computing device 404. The resource manager 406 may select a resource provider 402 to provide the computing resource. The resource manager 406 may facilitate a connection between the resource provider 402 and a particular computing device 404. In some implementations, the resource manager 406 may establish a connection between a particular resource provider 402 and a particular computing device 404. In some implementations, the resource manager 406 may redirect a particular computing device 404 to a particular resource provider 402 with the requested computing resource. [0262] Fig.5 shows an example of a computing device 500 and a mobile computing device 550 that can be used in the methods and systems described in this disclosure. The computing device 500 is intended to represent various forms of digital computers, such as laptops, desktops, workstations, personal digital assistants, servers, blade servers, mainframes, and other appropriate computers. The mobile computing device 550 is intended to represent various forms of mobile devices, such as personal digital assistants, cellular telephones, smart-phones, and other similar computing devices. The components shown here, their connections and relationships, and their functions, are meant to be examples only, and are not meant to be limiting.
12366150v1
Attorney Docket No.2014191-0027 [0263] The computing device 500 includes a processor 502, a memory 504, a storage device 506, a high-speed interface 508 connecting to the memory 504 and multiple high-speed expansion ports 510, and a low-speed interface 512 connecting to a low-speed expansion port 514 and the storage device 506. Each of the processor 502, the memory 504, the storage device 506, the high-speed interface 508, the high-speed expansion ports 510, and the low-speed interface 512, are interconnected using various busses, and may be mounted on a common motherboard or in other manners as appropriate. The processor 502 can process instructions for execution within the computing device 500, including instructions stored in the memory 504 or on the storage device 506 to display graphical information for a GUI on an external input/output device, such as a display 516 coupled to the high-speed interface 508. In other implementations, multiple processors and/or multiple buses may be used, as appropriate, along with multiple memories and types of memory. Also, multiple computing devices may be connected, with each device providing portions of the necessary operations (e.g., as a server bank, a group of blade servers, or a multi-processor system). Also, multiple computing devices may be connected, with each device providing portions of the necessary operations (e.g., as a server bank, a group of blade servers, or a multi-processor system). Thus, as the term is used herein, where a plurality of functions are described as being performed by “a processor”, this encompasses embodiments wherein the plurality of functions are performed by any number of processors (e.g., one or more processors) of any number of computing devices (e.g., one or more computing devices). Furthermore, where a function is described as being performed by “a processor”, this encompasses embodiments wherein the function is performed by any number of processors (e.g., one or more processors) of any number of computing devices (e.g., one or more computing devices) (e.g., in a distributed computing system). [0264] The memory 504 stores information within the computing device 500. In some implementations, the memory 504 is a volatile memory unit or units. In some implementations, the memory 504 is a non-volatile memory unit or units. The memory 504 may also be another form of computer-readable medium, such as a magnetic or optical disk. [0265] The storage device 506 is capable of providing mass storage for the computing device 500. In some implementations, the storage device 506 may be or contain a computer- readable medium, such as a hard disk device, an optical disk device, a flash memory or other similar solid state memory device, or an array of devices, including devices in a storage area network or other configurations. Instructions can be stored in an information carrier. The
12366150v1
Attorney Docket No.2014191-0027 instructions, when executed by one or more processing devices (for example, processor 502), perform one or more methods, such as those described above. The instructions can also be stored by one or more storage devices such as computer- or machine-readable mediums (for example, the memory 504, the storage device 506, or memory on the processor 502). [0266] The high-speed interface 508 manages bandwidth-intensive operations for the computing device 500, while the low-speed interface 512 manages lower bandwidth-intensive operations. Such allocation of functions is an example only. In some implementations, the high- speed interface 508 is coupled to the memory 504, the display 516 (e.g., through a graphics processor or accelerator), and to the high-speed expansion ports 510, which may accept various expansion cards (not shown). In the implementation, the low-speed interface 512 is coupled to the storage device 506 and the low-speed expansion port 514. The low-speed expansion port 514, which may include various communication ports (e.g., USB, Bluetooth®, Ethernet, wireless Ethernet) may be coupled to one or more input/output devices, such as a keyboard, a pointing device, a scanner, or a networking device such as a switch or router, e.g., through a network adapter. [0267] The computing device 500 may be implemented in a number of different forms, as shown in the figure. For example, it may be implemented as a standard server 520, or multiple times in a group of such servers. In addition, it may be implemented in a personal computer such as a laptop computer 522. It may also be implemented as part of a rack server system 524. Alternatively, components from the computing device 500 may be combined with other components in a mobile device (not shown), such as a mobile computing device 550. Each of such devices may contain one or more of the computing device 500 and the mobile computing device 550, and an entire system may be made up of multiple computing devices communicating with each other. [0268] The mobile computing device 550 includes a processor 552, a memory 564, an input/output device such as a display 554, a communication interface 566, and a transceiver 568, among other components. The mobile computing device 550 may also be provided with a storage device, such as a micro-drive or other device, to provide additional storage. Each of the processor 552, the memory 564, the display 554, the communication interface 566, and the transceiver 568, are interconnected using various buses, and several of the components may be mounted on a common motherboard or in other manners as appropriate.
12366150v1
Attorney Docket No.2014191-0027 [0269] The processor 552 can execute instructions within the mobile computing device 550, including instructions stored in the memory 564. The processor 552 may be implemented as a chipset of chips that include separate and multiple analog and digital processors. The processor 552 may provide, for example, for coordination of the other components of the mobile computing device 550, such as control of user interfaces, applications run by the mobile computing device 550, and wireless communication by the mobile computing device 550. [0270] The processor 552 may communicate with a user through a control interface 558 and a display interface 556 coupled to the display 554. The display 554 may be, for example, a TFT (Thin-Film-Transistor Liquid Crystal Display) display or an OLED (Organic Light Emitting Diode) display, or other appropriate display technology. The display interface 556 may comprise appropriate circuitry for driving the display 554 to present graphical and other information to a user. The control interface 558 may receive commands from a user and convert them for submission to the processor 552. In addition, an external interface 562 may provide communication with the processor 552, so as to enable near area communication of the mobile computing device 550 with other devices. The external interface 562 may provide, for example, for wired communication in some implementations, or for wireless communication in other implementations, and multiple interfaces may also be used. [0271] The memory 564 stores information within the mobile computing device 550. The memory 564 can be implemented as one or more of a computer-readable medium or media, a volatile memory unit or units, or a non-volatile memory unit or units. An expansion memory 574 may also be provided and connected to the mobile computing device 550 through an expansion interface 572, which may include, for example, a SIMM (Single In Line Memory Module) card interface. The expansion memory 574 may provide extra storage space for the mobile computing device 550, or may also store applications or other information for the mobile computing device 550. Specifically, the expansion memory 574 may include instructions to carry out or supplement the processes described above, and may include secure information also. Thus, for example, the expansion memory 574 may be provided as a security module for the mobile computing device 550, and may be programmed with instructions that permit secure use of the mobile computing device 550. In addition, secure applications may be provided via the SIMM cards, along with additional information, such as placing identifying information on the SIMM card in a non- hackable manner.
12366150v1
Attorney Docket No.2014191-0027 [0272] The memory may include, for example, flash memory and/or NVRAM memory (non-volatile random access memory), as discussed below. In some implementations, instructions are stored in an information carrier and, when executed by one or more processing devices (for example, processor 552), perform one or more methods, such as those described above. The instructions can also be stored by one or more storage devices, such as one or more computer- or machine-readable mediums (for example, the memory 564, the expansion memory 574, or memory on the processor 552). In some implementations, the instructions can be received in a propagated signal, for example, over the transceiver 568 or the external interface 562. [0273] The mobile computing device 550 may communicate wirelessly through the communication interface 566, which may include digital signal processing circuitry where necessary. The communication interface 566 may provide for communications under various modes or protocols, such as GSM voice calls (Global System for Mobile communications), SMS (Short Message Service), EMS (Enhanced Messaging Service), or MMS messaging (Multimedia Messaging Service), CDMA (code division multiple access), TDMA (time division multiple access), PDC (Personal Digital Cellular), WCDMA (Wideband Code Division Multiple Access), CDMA2000, or GPRS (General Packet Radio Service), among others. Such communication may occur, for example, through the transceiver 568 using a radio-frequency. In addition, short-range communication may occur, such as using a Bluetooth®, Wi-Fi™, or other such transceiver (not shown). In addition, a GPS (Global Positioning System) receiver module 570 may provide additional navigation- and location-related wireless data to the mobile computing device 550, which may be used as appropriate by applications running on the mobile computing device 550. [0274] The mobile computing device 550 may also communicate audibly using an audio codec 560, which may receive spoken information from a user and convert it to usable digital information. The audio codec 560 may likewise generate audible sound for a user, such as through a speaker, e.g., in a handset of the mobile computing device 550. Such sound may include sound from voice telephone calls, may include recorded sound (e.g., voice messages, music files, etc.) and may also include sound generated by applications operating on the mobile computing device 550. [0275] The mobile computing device 550 may be implemented in a number of different forms, as shown in the figure. For example, it may be implemented as a cellular telephone 580. It
12366150v1
Attorney Docket No.2014191-0027 may also be implemented as part of a smart-phone 582, personal digital assistant, or other similar mobile device. [0276] Various implementations of the systems and techniques described here can be realized in digital electronic circuitry, integrated circuitry, specially designed ASICs (application specific integrated circuits), computer hardware, firmware, software, and/or combinations thereof. These various implementations can include implementation in one or more computer programs that are executable and/or interpretable on a programmable system including at least one programmable processor, which may be special or general purpose, coupled to receive data and instructions from, and to transmit data and instructions to, a storage system, at least one input device, and at least one output device. [0277] These computer programs (also known as programs, software, software applications or code) include machine instructions for a programmable processor, and can be implemented in a high-level procedural and/or object-oriented programming language, and/or in assembly/machine language. As used herein, the terms machine-readable medium and computer- readable medium refer to any computer program product, apparatus and/or device (e.g., magnetic discs, optical disks, memory, Programmable Logic Devices (PLDs)) used to provide machine instructions and/or data to a programmable processor, including a machine-readable medium that receives machine instructions as a machine-readable signal. The term machine-readable signal refers to any signal used to provide machine instructions and/or data to a programmable processor. [0278] To provide for interaction with a user, the systems and techniques described here can be implemented on a computer having a display device (e.g., a CRT (cathode ray tube) or LCD (liquid crystal display) monitor) for displaying information to the user and a keyboard and a pointing device (e.g., a mouse or a trackball) by which the user can provide input to the computer. Other kinds of devices can be used to provide for interaction with a user as well; for example, feedback provided to the user can be any form of sensory feedback (e.g., visual feedback, auditory feedback, or tactile feedback); and input from the user can be received in any form, including acoustic, speech, or tactile input. [0279] Systems and techniques described here can be implemented in a computing system that includes a back end component (e.g., as a data server), or that includes a middleware component (e.g., an application server), or that includes a front end component (e.g., a client computer having a graphical user interface or a Web browser through which a user can interact
12366150v1
Attorney Docket No.2014191-0027 with an implementation of the systems and techniques described here), or any combination of such back end, middleware, or front end components. The components of the system can be interconnected by any form or medium of digital data communication (e.g., a communication network). Examples of communication networks include a local area network (LAN), a wide area network (WAN), and the Internet. [0280] The computing system can include clients and servers. A client and server are generally remote from each other and typically interact through a communication network. The relationship of client and server arises by virtue of computer programs running on the respective computers and having a client-server relationship to each other. [0281] Certain embodiments described herein make use of computer algorithms in the form of software instructions executed by a computer processor, for example in a classifier. In certain embodiments, the software instructions include a machine learning (ML) module, for example as a classifier. As used herein, a machine learning module refers to a computer implemented process (e.g., a software function) that implements one or more specific machine learning techniques, e.g., artificial neural networks (ANNs), e.g., convolutional neural networks (CNNs), random forest, decision trees, support vector machines, and the like, in order to determine, for a given input, one or more output values. In certain embodiments, the input comprises image data and/or alphanumeric data which can include 2D and/or 3D datasets, numbers, words, phrases, or lengthier strings, for example. In certain embodiments, the one or more output values comprise image data (e.g.2D and/or 3D datasets) and/or values representing numeric values, words, phrases, or other alphanumeric strings. [0282] In certain embodiments, machine learning modules implementing machine learning techniques are trained, for example, using datasets that include categories of data described herein. Such training may be used to determine various parameters of machine learning algorithms implemented by a machine learning module, such as weights associated with layers in neural networks. In certain embodiments, once a machine learning module is trained, e.g., to accomplish a specific task such as identifying certain response strings, values of determined parameters are fixed and the (e.g., unchanging, static) machine learning module is used to process new data (e.g., different from the training data) and accomplish its trained task without further updates to its parameters (e.g., the machine learning module does not receive feedback and/or updates). In certain embodiments, available input data includes training data and validation data, e.g., where
12366150v1
Attorney Docket No.2014191-0027 the validation data is separate and non-overlapping with the training data. For example, in certain embodiments, training data is used during the training process to optimize a model, whereas validation data is used to check the accuracy of the model while operating on previously unseen data. In certain embodiments, training data is divided into batches (e.g., portions) that is sequentially used (e.g., in random order) as sets of inputs to train a model. In certain embodiments, a model is trained multiple times (e.g., epochs) on the entire set of training data. In certain embodiments, machine learning modules may receive feedback, e.g., based on user review of accuracy, and such feedback may be used as additional training data, to dynamically update the machine learning module. In certain embodiments, two or more machine learning modules may be combined and implemented as a single module and/or a single software application. In certain embodiments, two or more machine learning modules may also be implemented separately, e.g., as separate software applications. A machine learning module may be software and/or hardware. For example, a machine learning module may be implemented entirely as software, or certain functions of a ANN module may be carried out via specialized hardware (e.g., via an application specific integrated circuit (ASIC) and/or field programmable gate arrays (FPGAs)). [0283] In certain embodiments, machine learning modules implementing machine learning techniques may be composed of individual nodes (e.g. units, neurons). A node may receive a set of inputs that may include at least a portion of a given input data for the machine learning module and/or at least one output of another node. A node may have at least one parameter to apply and/or a set of instructions to perform (e.g., mathematical functions to execute) over the set of inputs. In certain embodiments, node instructions may include a step to provide various relative importance to the set of inputs using various parameters, such as weights. The weights may be applied by performing scalar multiplication (e.g., or other mathematical function) between a set of inputs values and the parameters, resulting in a set of weighted inputs. In certain embodiments, a node may have a transfer function to combine the set of weighted inputs into one output value. A transfer function may be implemented by a summation of all the weighted inputs and the addition of an offset (e.g., bias) value. In certain embodiments, a node may have an activation function to introduce non-linearity into the output value. Non-limiting examples of the activation function include Rectified Linear Activation (ReLu), logistic (e.g., sigmoid), hyperbolic tangent (tanh), and softmax. In certain embodiments, a node may have a capability of remembering previous states
12366150v1
Attorney Docket No.2014191-0027 (e.g., recurrent nodes). Previous states may be applied to the input and output values using a set of learning parameters. [0284] In certain embodiments, the machine learning module comprises a deep learning architecture composed of nodes organized into layers. For example, a layer is a set of nodes that receives data input (e.g., weighted or non-weighted input), transforms it (e.g., by carrying out instructions, e.g., applying a set of functions e.g., linear and/or non-linear functions), and passes transformed values as output (e.g., to the next layer). In certain embodiments, the set of nodes in a particular layer may share the same parameters and instructions without interacting with each other. A machine learning module may be composed of at least one layer (e.g., ordered). Examples of types of layers include convolutional layers (e.g., layers with a kernel, a matrix of parameters that is slid across an input to be multiplied with multiple input values to reduce them to a single output value); fully connected (FC) layers (e.g. all nodes are connected to all outputs of the previous layer); recurrent layers, long/short term memory (LSTM) layers, gated recurrent unit (GRU) layers (e.g., nodes with the various abilities to memorize and apply their previous inputs and/or outputs); batch normalization (BN) layers (e.g., layers that normalize a set of outputs from another layer, allowing for more independent learning of individual layers); activation layers (e.g., layers with nodes that only contain an activation function); and/or (un)pooling layers [e.g., layers that reduce (increase) dimensions of an input by summarizing (splitting) input values in defined patches). [0285] In certain embodiments, the performance of a machine learning module may be characterized by its ability to produce an output data with specific accuracy. To achieve specific accuracy, a training process is performed to find optimal parameters, such as weights, for each node in each layer of the machine learning module. In certain embodiments, the training process of a machine learning module may involve using output data to calculate an objective function (e.g., cost function, loss function, error function) that needs to be optimized (e.g., minimized, maximized). For example, a machine learning objective function may be a combination of a loss function and regularization parameter. The loss function is related to how well the output is able to predict the input. The loss function may take various forms, like mean squared error, mean absolute error, binary cross-entropy, categorical cross-entropy, for example. The regularization term may be needed to prevent overfitting and improve generalization of the training process. Examples of regularization techniques include L1 Regularization or Lasso Regression, L2
12366150v1
Attorney Docket No.2014191-0027 Regularization or Ridge Regression, and Dropout (e.g., dropping layer outputs at random during training process). [0286] In certain embodiments, objective function optimization of a machine learning module may involve finding at least one (e.g., all) of the present global optima (e.g., as opposed to local optima). In certain embodiments, the algorithm for objective function optimization follows principles of mathematical optimization for a multi-variable function and relies on achieving specific accuracy of the process. Examples of objective function optimization algorithms include gradient descent, nonlinear conjugate gradient, random search, Levenberg-Marquardt algorithm, limited-memory Broyden-Fietcher-Goldfarb-Shanno algorithm, pattern search, basin hopping method, Krylov method, Adam method, genetic algorithm, particle swarm optimization, surrogate optimization, and simulated annealing. [0287] Methods disclosed herein may utilize one or more machine-learned models as a classifier. A machine-learned model may be or include an artificial neural network. A machine- learned model may employ, for example, an attention-based model (e.g., a transformer model, such as, for example, a vision transformer), a transformer model (e.g., a vision transformer), a regression-based model (e.g., a logistic regression model), a regularization-based model (e.g., an elastic net model or a ridge regression model), an instance-based model (e.g., a support vector machine or a k-nearest neighbor model), a Bayesian-based model (e.g., a naive-based model or a Gaussian naive-based model), a clustering-based model (e.g., an expectation maximization model), an ensemble-based model (e.g., an adaptive boosting model, a random forest model, a bootstrap-aggregation model, or a gradient boosting machine model), or a neural-network-based model (e.g., a convolutional neural network, a recurrent neural network, autoencoder, a back propagation network, or a stochastic gradient descent network). [0288] In some embodiments, a machine-learned model used as a classifier is or is derived from a decision tree methodology, a neural boosted methodology, a bootstrap forest methodology, a boosted tree methodology, a k nearest neighbors methodology, a generalized regression forward selection methodology, a generalized regression pruned forward selection methodology, a fit stepwise methodology, a generalized regression lasso methodology, a generalized regression elastic net methodology, a generalized regression ridge methodology, a nominal logistic methodology, a support vector machines methodology, a discriminant methodology, a naïve Bayes methodology, or a combination thereof. In some embodiments, a machine-learned model is or is
12366150v1
Attorney Docket No.2014191-0027 derived from a decision tree methodology, a neural boosted methodology, a bootstrap forest methodology, a boosted tree methodology, a generalized regression lasso methodology, a generalized regression elastic net methodology, a generalized regression ridge methodology, a nominal logistic methodology, a support vector machines methodology, a discriminant methodology, or a combination thereof. In some embodiments, a machine-learned model is or is derived from a decision tree methodology, a neural boosted methodology, a bootstrap forest methodology, a boosted tree methodology, a support vector machines methodology, or a combination thereof. Definitions [0289] “A” or “An”: The articles “a” and “an” are used herein to refer to one or to more than one (i.e., to at least one) of the grammatical object of the article. By way of example, “an element” refers to one element or more than one element. [0290] About: The term “about”, when used herein in reference to a value, refers to a value that is similar, in context, to the referenced value. In general, those skilled in the art, familiar with the context, will appreciate the relevant degree of variance encompassed by “about” in that context. For example, in some embodiments, the term “about” can encompass a range of values that within 25%, 20%, 19%, 18%, 17%, 16%, 15%, 14%, 13%, 12%, 11%, 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1%, or within a fraction of a percent, of the referenced value. [0291] “Accessibility Status” or “Chromatin Accessibility Status”: As used herein, “accessibility status” or “chromatin accessibility status” of a genomic locus refers to the frequency with which DNA sequences corresponding to the genomic locus are identified in an assay for detection of accessible chromatin. Accessibility status can be determined by various assays known in the art, including without limitation ChIP-seq as one example. Where two samples are separately analyzed by the same assay or comparable assays for detection of accessible DNA sequences, differences in chromatin accessibility status of genomic loci can be detected. Accessibility status can be compared to a standard or reference. A sample that has an accessibility status that differs in accessibility status from a standard or reference can be referred to as differentially modified. Suitable assays for determining chromatin accessibility are known in the art. Exemplary assays include ATAC-seq (Assay of Transpose Accessible Chromatin
12366150v1
Attorney Docket No.2014191-0027 sequencing), NOMe-seq (Nucleosome Occupancy and Methylome sequencing), FAIRE-seq (Formaldehyde-Assisted Isolation of Regulatory Elements sequencing), MNase-seq (Micrococcal Nuclease digestion with sequencing), and/or a DNase hypersensitivity assay. [0292] Administration: As used herein, the term “administration” typically refers to the administration of a disease appropriate (e.g., ER-positive cancer appropriate) treatment. In some embodiments, the disease appropriate treatment may comprise administering a composition to a subject, for example to achieve delivery of an agent that is, is included in, or is otherwise delivered by, the composition. In some embodiments, the disease appropriate treatment may comprise administering an appropriate surgical procedure or radiological procedure, optionally in combination with administration of a composition. [0293] Agent: As used herein, the term “agent” may refer to any chemical or physical entity, including without limitation any of one or more of an atom, e.g., a radioactive atom, molecule, compound, conjugate, polypeptide, polynucleotide, polysaccharide, lipid, cell, or combination or complex thereof. [0294] Antibody: As used herein, the term “antibody” refers to a polypeptide that includes one or more canonical immunoglobulin sequence elements sufficient to confer specific binding to a particular antigen (e.g., a heavy chain variable domain, a light chain variable domain, and/or one or more CDRs). Thus, the term antibody includes, without limitation, human antibodies, non-human antibodies, synthetic and/or engineered antibodies, fragments thereof, and agents including the same. Antibodies can be naturally occurring immunoglobulins (e.g., generated by an organism reacting to an antigen). Synthetic, non-naturally occurring, or engineered antibodies can be produced by recombinant engineering, chemical synthesis, or other artificial systems or methodologies known to those of skill in the art. [0295] As is well known in the art, typical human immunoglobulins are approximately 150 kD tetrameric agents that include two identical heavy (H) chain polypeptides (about 50 kD each) and two identical light (L) chain polypeptides (about 25 kD each) that associate with each other to form a structure commonly referred to as a “Y-shaped” structure. Typically, each heavy chain includes a heavy chain variable domain (VH) and a heavy chain constant domain (CH). The heavy chain constant domain includes three CH domains: CH1, CH2 and CH3. A short region, known as the “switch”, connects the heavy chain variable and constant regions. The “hinge” connects CH2 and CH3 domains to the rest of the immunoglobulin. Each light chain
12366150v1
Attorney Docket No.2014191-0027 includes a light chain variable domain (VL) and a light chain constant domain (CL), separated from one another by another “switch.” Each variable domain contains three hypervariable loops known as “complement determining regions” (CDR1, CDR2, and CDR3) and four somewhat invariant “framework” regions (FR1, FR2, FR3, and FR4). In each VH and VL, the three CDRs and four FRs are arranged from amino-terminus to carboxy-terminus in the following order: FR1, CDR1, FR2, CDR2, FR3, CDR3, and FR4. The variable regions of a heavy and/or a light chain are typically understood to provide a binding moiety that can interact with an antigen. Constant domains can mediate binding of an antibody to various immune system cells (e.g., effector cells and/or cells that mediate cytotoxicity), receptors, and elements of the complement system. Heavy and light chains are linked to one another by a single disulfide bond, and two other disulfide bonds connect the heavy chain hinge regions to one another, so that the dimers are connected to one another and the tetramer is formed. When natural immunoglobulins fold, the FR regions form the beta sheets that provide the structural framework for the domains, and the CDR loop regions from both the heavy and light chains are brought together in three- dimensional space so that they create a single hypervariable antigen binding site located at the tip of the Y structure. [0296] In some embodiments, an antibody is a polyclonal, monoclonal, monospecific, or multispecific antibody (e.g., a bispecific antibody). In some embodiments, an antibody includes at least one light chain monomer or dimer, at least one heavy chain monomer or dimer, at least one heavy chain-light chain dimer, or a tetramer that includes two heavy chain monomers and two light chain monomers. Moreover, the term “antibody” can include (unless otherwise stated or clear from context) any art-known constructs or formats utilizing antibody structural and/or functional features including without limitation intrabodies, domain antibodies, antibody mimetics, Zybodies®, Fab fragments, Fab’ fragments, F(ab’)2 fragments, Fd’ fragments, Fd fragments, isolated CDRs or sets thereof, single chain antibodies, single-chain Fvs (scFvs), disulfide-linked Fvs (sdFv), polypeptide-Fc fusions, single domain antibodies (e.g., shark single domain antibodies such as IgNAR or fragments thereof), cameloid antibodies, camelized antibodies, masked antibodies (e.g., Probodies®), affybodies, anti-idiotypic (anti-Id) antibodies (including, e.g., anti-anti-Id antibodies), Small Modular ImmunoPharmaceuticals (SMIPs), single chain or Tandem diabodies (TandAb®), VHHs, Anticalins®, Nanobodies®, minibodies, BiTE®s, ankyrin repeat proteins or DARPINs®, Avimers®, DARTs, TCR-like antibodies,
12366150v1
Attorney Docket No.2014191-0027 Adnectins®, Affilins®, Trans-bodies®, Affibodies®, TrimerX®, MicroProteins, Fynomers®, Centyrins®, KALBITOR®s, chimeric antigen receptors (CARs), engineered T-cell receptors (TCRs), and antigen-binding fragments of any of the above. [0297] In various embodiments, an antibody includes one or more structural elements recognized by those skilled in the art as a complementarity determining region (CDR) or variable domain. In some embodiments, an antibody can be a covalently modified (“conjugated”) antibody (e.g., an antibody that includes a polypeptide including one or more canonical immunoglobulin sequence elements sufficient to confer specific binding to a particular antigen, where the polypeptide is covalently linked with one or more of a therapeutic agent, a detectable moiety, another polypeptide, a glycan, or a polyethylene glycol molecule). In some embodiments, antibody sequence elements are humanized, primatized, chimeric, etc., as is known in the art. [0298] An antibody including a heavy chain constant domain can be, without limitation, an antibody of any known class, including but not limited to, IgA, secretory IgA, IgG, IgE and IgM, based on heavy chain constant domain amino acid sequence (e.g., alpha (α), delta (δ), epsilon (ε), gamma (γ) and mu (µ)). IgG subclasses are also well known to those in the art and include but are not limited to human IgG1, IgG2, IgG3 and IgG4. “Isotype” refers to the Ab class or subclass (e.g., IgM or IgG1) that is encoded by the heavy chain constant region genes. As used herein, a “light chain” can be of a distinct type, e.g., kappa (κ) or lambda (λ), based on the amino acid sequence of the light chain constant domain. In some embodiments, an antibody has constant region sequences that are characteristic of mouse, rabbit, primate, or human immunoglobulins. Naturally produced immunoglobulins are glycosylated, typically on the CH2 domain. As is known in the art, affinity and/or other binding attributes of Fc regions for Fc receptors can be modulated through glycosylation or other modification. In some embodiments, an antibody may lack a covalent modification (e.g., attachment of a glycan) that it would have if produced naturally. In some embodiments, antibodies produced and/or utilized in accordance with the present disclosure include glycosylated Fc domains, including Fc domains with modified or engineered glycosylation. [0299] In some embodiments, an antibody can be specific for a particular histone modification (e.g., an antibody can bind one histone modification, e.g., H3K27ac with a higher affinity than other histone modifications, under conditions that are commonly used in ChIP-seq
12366150v1
Attorney Docket No.2014191-0027 experiments). In some embodiments, an antibody is specific for an H3K9ac, H3K14ac, H3K18ac, H3K23ac, H3K27ac, H3K4me1, H3K4me2, or H3K4me3 modification. In some embodiments, an antibody is specific for an H3K27ac modification. In some embodiments, an antibody is specific for an H3K4me3 modification. [0300] In some embodiments, an antibody is a “pan” antibody. As used herein, the term pan antibody refers to an antibody that can bind a group of histone modifications having one or more features that are similar. In some embodiments, a pan antibody is a pan-methylation antibody (e.g., an antibody that can bind a histone, e.g., H3 that comprises at least one methylated lysine, wherein the at least one methylated lysine can be at any one of a plurality of amino acid positions, e.g., in some embodiments, a pan-methylation antibody can bind an H3 protein comprising a methylated lysine at any position). In some embodiments, a pan antibody is a pan-acetylation antibody (e.g., an antibody that can bind a histone, e.g., H3 that comprises at least one acetylated lysine, wherein the at least one acetylated lysine can be at any one of a plurality of amino acid positions, e.g., a pan-acetylation antibody can bind an H3 protein comprising an acetylated lysine at any position). In some embodiments, a pan antibody can bind one or more histone modifications that are associated with transcription activation. In some embodiments, a pan antibody can bind one or more histone modifications that are associated with transcription silencing. [0301] Antibody fragment: As used herein, an “antibody fragment” refers to a portion of an antibody or antibody agent as described herein, and typically refers to a portion that includes an antigen-binding portion or variable region thereof. An antibody fragment can be produced by any means. For example, in some embodiments, an antibody fragment can be enzymatically or chemically produced by fragmentation of an intact antibody or antibody agent. Alternatively, in some embodiments, an antibody fragment can be recombinantly produced, i.e., by expression of an engineered nucleic acid sequence. In some embodiments, an antibody fragment can be wholly or partially synthetically produced. In some embodiments, an antibody fragment (particularly an antigen-binding antibody fragment) can have a length of at least about 50, 60, 70, 80, 90, 100, 110, 120, 130, 140, 150, 160, 170, 180, 190 amino acids or more, in some embodiments at least about 200 amino acids. [0302] Associated with: Two events or entities are “associated” with one another, as that term is used herein, if the presence, level and/or form of one is correlated with that of the other.
12366150v1
Attorney Docket No.2014191-0027 For example, a particular entity (e.g., an epigenetic profile comprising one or more histone modifications at a set of genomic loci, etc.) is considered to be associated with a particular disease, disorder, or condition, if its presence, level and/or form correlates with incidence of and/or susceptibility to the disease, disorder, or condition (e.g., across a relevant population). In some embodiments, two or more entities are physically “associated” with one another if they interact, directly or indirectly, so that they are and/or remain in physical proximity with one another. In some embodiments, two or more entities that are physically associated with one another are covalently linked to one another; in some embodiments, two or more entities that are physically associated with one another are not covalently linked to one another but are non- covalently associated, for example by means of hydrogen bonds, van der Waals interaction, hydrophobic interactions, magnetism, or a combination thereof. [0303] “Between” or “From”: As used herein, the term “between” refers to content that falls between indicated upper and lower, or first and second, boundaries, inclusive of the boundaries. Similarly, the term “from”, when used in the context of a range of values, indicates that the range includes content that falls between indicated upper and lower, or first and second, boundaries, inclusive of the boundaries. [0304] Biological Sample: As used herein, the term “biological sample” typically refers to a sample obtained or derived from a biological source (e.g., a tissue or organism or cell) of interest, as described herein. In some embodiments, a biological source is or includes an organism, such as a human subject. In some embodiments, a biological sample is or includes a biological tissue or fluid. In some embodiments, a biological sample can be or include cells, tissue, or bodily fluid. “Bodily fluids” refer to fluids that are excreted or secreted from the body as well as fluids that are normally not (e.g., blood, serum, plasma, Cowper’s fluid or pre- ejaculate fluid, chyle, chyme, stool, interstitial fluid, intracellular fluid, lymph, menses, saliva, sebum, semen, serum, sweat, synovial fluid, tears, urine, vitreous humor, vomit). In some embodiments, a biological sample can be or include blood, blood components, cell-free DNA (cfDNA), circulating-tumor DNA (ctDNA), ascites, biopsy samples, surgical specimens, cell- containing body fluids, sputum, saliva, feces, urine, cerebrospinal fluid, peritoneal fluid, pleural fluid, lymph, gynecological fluids, secretions, excretions, skin swabs, vaginal swabs, oral swabs, nasal swabs, washings or lavages such as a ductal lavages or bronchoalveolar lavages, aspirates, scrapings, or bone marrow. In some embodiments, a biological sample is a liquid biopsy sample
12366150v1
Attorney Docket No.2014191-0027 obtained from a bodily fluid. In some embodiments, a biological sample is or includes DNA obtained from a single subject or from a plurality of subjects. A biological sample can be a “primary sample” obtained directly from a biological source or can be a “processed sample”, i.e., a sample that was derived from a primary sample, e.g., via dilution, purification, mixing with one or more reagents, or any other processing step(s) as described herein. A biological sample can also be referred to as a “sample.” [0305] Blood component: As used herein, the term “blood component” refers to any component of whole blood, including red blood cells, white blood cells, plasma, platelets, endothelial cells, mesothelial cells, epithelial cells, cell-free DNA (cfDNA), and circulating- tumor DNA (ctDNA). Blood components also include the components of plasma, including proteins, metabolites, lipids, nucleic acids, and carbohydrates, and any other cells that can be present in blood, e.g., due to pregnancy, organ transplant, infection, injury, or disease. [0306] Cancer: As used herein, the terms “cancer,” “malignancy,” “tumor,” and “carcinoma,” are used interchangeably to refer to a disease, disorder, or condition in which cells exhibit or exhibited relatively abnormal, uncontrolled, and/or autonomous growth, so that they display or displayed an abnormally elevated proliferation rate and/or aberrant growth phenotype. In some embodiments, a cancer can include one or more tumors. In some embodiments, a cancer can be or include cells that are precancerous (e.g., benign), malignant, pre-metastatic, metastatic, and/or non-metastatic. In some embodiments, a cancer can be or include a solid tumor. In some embodiments, a cancer be associated with ER-positive status, e.g., an ER-positive breast cancer, etc. [0307] Combination therapy: As used herein, the term “combination therapy” refers to administration to a subject of two or more therapeutic agents or therapeutic regimens such that the two or more therapeutic agents or therapeutic regimens together treat a disease, condition, or disorder of the subject. In some embodiments, the two or more therapeutic agents or therapeutic regimens can be administered simultaneously, sequentially, or in overlapping dosing regimens. Those of skill in the art will appreciate that combination therapy includes but does not require that the two therapeutic agents or therapeutic regimens be administered together in a single composition, nor at the same time. [0308] Corresponding to: As used herein, the term “corresponding to” may be used to designate the position/identity of a structural element in a compound or composition through
12366150v1
Attorney Docket No.2014191-0027 comparison with an appropriate reference compound or composition. For example, in some embodiments, a monomeric residue in a polymer (e.g., an amino acid residue in a polypeptide or a nucleic acid residue in a polynucleotide) may be identified as “corresponding to” a residue in an appropriate reference polymer. For example, those of skill in the art appreciate that residues in a provided polypeptide or polynucleotide sequence are often designated (e.g., numbered or labeled) according to the scheme of a related reference sequence (even if, e.g., such designation does not reflect literal numbering of the provided sequence). By way of illustration, if a reference sequence includes a particular amino acid motif at positions 100-110, and a second related sequence includes the same motif at positions 110-120, the motif positions of the second related sequence can be said to “correspond to” positions 100-110 of the reference sequence. Those of skill in the art appreciate that corresponding positions can be readily identified, e.g., by alignment of sequences, and that such alignment is commonly accomplished by any of a variety of known tools, strategies, and/or algorithms, including without limitation software programs such as, for example, BLAST, CS-BLAST, CUDASW++, DIAMOND, FASTA, GGSEARCH/GLSEARCH, Genoogle, HMMER, HHpred/HHsearch, IDF, Infernal, KLAST, USEARCH, parasail, PSI-BLAST, PSI-Search, ScalaBLAST, Sequilab, SAM, SSEARCH, SWAPHI, SWAPHI-LS, SWIMM, or SWIPE. Two sequences can be identified as corresponding if they are identical or if they share substantial identity, e.g., at least 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, e.g., over a length of at least 10, 20, 30, 40, 50, 60, 70, 80, 90, 100, 200, 300, 400, 500 or more residues. In various embodiments, a nucleic acid sequence can correspond to a sequence that is identical or substantially identical (e.g., at least 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical) to the complement of the nucleic acid sequence, e.g., over a length of at least 10, 20, 30, 40, 50, 60, 70, 80, 90, 100, 200, 300, 400, 500 or more nucleic acid residues. [0309] “Diagnosing”, “Detecting”, “Determining” or “Screening for”: As used herein, “diagnosing”, “detecting”, “determining”, “screening for” the presence of a condition or disease (e.g., ER-positive cancer), or a related state (e.g., responsiveness of an ER-positive cancer to one or more ER-targeted therapies) includes the act, process, and/or outcome of determining whether, and/or the qualitative of quantitative probability that, a subject has or will develop the condition, disease, or related state. In some instances, diagnosing can include a determination relating to
12366150v1
Attorney Docket No.2014191-0027 prognosis and/or likely response to one or more general or particular therapeutic agents or regimens. [0310] Differentially accessible: As used herein, the term “differentially accessible” describes a genomic locus for which chromatin accessibility status differs between a first condition or sample and a second condition or sample (e.g., a standard or reference). A differentially accessible genomic locus can include a greater or smaller measured accessibility under a selected condition of interest, such as ER-positive state, as compared to a reference state, such as ER-negative state. [0311] Differentially modified: As used herein, the term “differentially modified” describes a genomic locus for which histone modification status and/or DNA methylation status differs between a first condition or sample and a second condition or sample (e.g., a standard or reference). A differentially modified genomic locus can include a greater or smaller number or frequency of histone modification and/or DNA methylations under a selected condition of interest, such as ER-positive state, as compared to a reference state, such as ER-negative state. [0312] Epigenetic modification: As used herein, the term “epigenetic modifications” refers to heritable alterations to the genome that are not due to changes in DNA sequence. Epigenetic modifications include chemical modifications such as, e.g., DNA methylation and histone modification. In some embodiments, epigenetic modifications can cause a change in chromatin structure, DNA accessibility, and/or transcription factor binding. In some embodiments, epigenetic modifications can be detected or measured directly (e.g., by using an agent that binds an epigenetic modification (e.g., an antibody that binds H3K4me3 or H3K27ac)). In some embodiments, epigenetic modifications can be measured indirectly, e.g., by measuring or detecting one or more attributes, changes in which are indicative of changes in epigenetic modifications. For example, in some embodiments, chromatin accessibility and/or transcription factor binding can be used as a measure of epigenetic modifications at a given locus. As used herein, the term “epigenetic marker” refers to an indicator of epigenetic state, and includes, e.g., epigenetic modifications (e.g., histone modifications and DNA methylation, transcription factor biding, chromatin accessibility. As used herein, the term “epigenetic biomarker” refers to an epigenetic marker that can be used in the detection of a disease or condition.
12366150v1
Attorney Docket No.2014191-0027 [0313] Identity: As used herein, the term “identity” refers to the overall relatedness between polymeric molecules, e.g., between nucleic acid molecules (e.g., DNA molecules) and/or between polypeptide molecules. Methods for the calculation of a percent identity as between two provided sequences are known in the art. The term “% sequence identity” refers to a relationship between two or more sequences, as determined by comparing the sequences. In the art, “identity” also means the degree of sequence relatedness between protein and nucleic acid sequences as determined by the match between strings of such sequences. “Identity” (often referred to as “similarity”) can be readily calculated by known methods, including those described in: Computational Molecular Biology (Lesk, A. M. ed.) Oxford University Press, NY (1988); Biocomputing: Informatics and Genome Projects (Smith, D. W. ed.) Academic Press, NY (1994); Computer Analysis of Sequence Data, Part I (Griffin, A. M. and Griffin, H. G. eds.) Humana Press, NJ (1994); Sequence Analysis in Molecular Biology (Von Heijne, G. ed.) Academic Press (1987); and Sequence Analysis Primer (Gribskov, M. and Devereux, J. eds.) Oxford University Press, NY (1992), each of which are separately incorporated by reference in their entirety. Preferred methods to determine identity are designed to give the best match between the sequences tested. Methods to determine identity and similarity are codified in publicly available computer programs. For example, calculation of the percent identity of two nucleic acid or polypeptide sequences can be performed by aligning the two sequences (or the complement of one or both sequences) for optimal comparison purposes (e.g., gaps can be introduced in one or both of a first and a second sequences for optimal alignment and non- identical sequences can be disregarded for comparison purposes). The nucleotides or amino acids at corresponding positions are then compared. When a position in the first sequence is occupied by the same residue (e.g., nucleotide or amino acid) as the corresponding position in the second sequence, then the molecules are identical at that position. The percent identity between the two sequences is a function of the number of identical positions shared by the sequences, optionally accounting for the number of gaps, and the length of each gap, which may need to be introduced for optimal alignment of the two sequences. The comparison of sequences and determination of percent identity between two sequences can be accomplished using a computational algorithm, such as BLAST (basic local alignment search tool). Sequence alignments and percent identity calculations may be performed using the Megalign program of the LASERGENE bioinformatics computing suite (DNASTAR, Inc., Madison, Wisconsin). Multiple alignment of the sequences
12366150v1
Attorney Docket No.2014191-0027 can also be performed using the Clustal method of alignment (Higgins and Sharp, Comp Appl Biosci (1989) 5(2):151-153), incorporated by reference herein in its entirety, with default parameters (GAP PENALTY=10, GAP LENGTH PENALTY=10). Relevant programs also include the GCG suite of programs (Wisconsin Package Version 9.0, Genetics Computer Group (GCG), Madison, Wisconsin); BLASTP, BLASTN, BLASTX (Altschul et al., J Mol Biol (1990) 215:403-410); DNASTAR (DNASTAR, Inc., Madison, Wisconsin); and the FASTA program incorporating the Smith-Waterman algorithm (Pearson, Comput Methods Genome Res [Proc Int Symp] (1994), Meeting Date 1992, 111-120. Eds. Suhai, Sandor. Plenum, New York, NY (the contents of each of which is separately incorporated herein by reference in its entirety). Within the context of this disclosure, it will be understood that where sequence analysis software is used for analysis, the results of the analysis are based on the “default values” of the program referenced. “Default values” will mean any set of values or parameters, which originally load with the software when first initialized. [0314] “Improve,” “increase,” “inhibit,” or “reduce”: As used herein, the terms “improve”, “increase”, “inhibit”, and “reduce”, and grammatical equivalents thereof, indicate qualitative or quantitative difference from a reference. [0315] Methylation Status: As used herein, “methylation status” of a genomic locus refers to the frequency with which DNA sequences corresponding to the genomic locus are identified in an assay for detection of DNA methylated sequences and/or the density (e.g., the measured density) of DNA methylation corresponding to the genomic locus. Methylation status can be determined by various assays known in the art, including without limitation Bisulfite sequencing (BS-Seq), Whole Genome Bisulfite Sequencing (WGBS), Methylated DNA ImmunoPrecipitation sequencing (MeDIP-seq), or Methyl-CpG-Binding Domain sequencing (MBD-seq). Where two samples are separately analyzed by the same assay or comparable assays for detection of DNA methylated sequences, differences in methylation status of genomic loci can be detected. Methylation status can be compared to a standard or reference. A sample that has a methylation status that differs from a standard or reference can be referred to as differentially modified. [0316] “Modification Status” or “Histone Modification Status”: As used herein, “modification status” or “histone modification status” of a genomic locus refers to the frequency with which DNA sequences corresponding to the genomic locus are identified in an assay for
12366150v1
Attorney Docket No.2014191-0027 detection of DNA sequences associated with histones bearing one or more histone modifications (e.g., one or more particular histone modifications) and/or the density (e.g., the measured density) of histone modifications (e.g., one or more particular histone modifications) corresponding to the genomic locus. Modification status can be determined by various assays known in the art, including without limitation ChIP-seq as one example. Other well-known assays include CUT&RUN (Cleavage Under Targets and Release Using Nuclease) sequencing and CUT&Tag (Cleavage Under Targets and Tagmentation). Where two samples are separately analyzed by the same assay or comparable assays for detection of DNA sequences associated with histones bearing one or more histone modifications (e.g., one or more particular histone modifications), differences in modification status of genomic loci can be detected. Modification status can be compared to a standard or reference. A sample that has a modification status that differs in modification status or histone modification status from a standard or reference can be referred to as differentially modified. [0317] Regulatory sequence: As used herein in the context of expression of a nucleic acid coding sequence, a regulatory sequence is a nucleic acid sequence that controls expression of a coding sequence, e.g., a promoter sequence or an enhancer sequence. In some embodiments, a regulatory sequence can control or impact one or more aspects of gene expression (e.g., cell- type-specific expression, inducible expression, etc.). [0318] Subject: As used herein, the term “subject” refers to an organism, typically a mammal (e.g., a human). In some embodiments, a subject is suffering from a disease, disorder or condition (e.g., ER-positive cancer, e.g., ER-positive breast cancer, etc.). In some embodiments, a subject is susceptible to a disease, disorder, or condition. In some embodiments, a subject displays one or more symptoms or characteristics of a disease, disorder or condition. In some embodiments, a subject is not suffering from a disease, disorder or condition. In some embodiments, a subject does not display any symptom or characteristic of a disease, disorder, or condition. In some embodiments, a subject has one or more features characteristic of susceptibility to or risk of a disease, disorder, or condition. In some embodiments, a subject is a subject that has been tested for a disease, disorder, or condition, and/or to whom therapy has been administered. In some instances, a human subject can be interchangeably referred to as a “patient” or “individual”.
12366150v1
Attorney Docket No.2014191-0027 [0319] Therapeutic agent: As used herein, the term “therapeutic agent” refers to any agent that elicits a desired pharmacological effect when administered to a subject. In some embodiments, an agent is considered to be a therapeutic agent if it demonstrates a statistically significant effect across an appropriate population. In some embodiments, the appropriate population can be a population of model organisms or a human population. In some embodiments, an appropriate population can be defined by various criteria, such as a certain age group, gender, genetic background, preexisting clinical conditions, etc. In some embodiments, a therapeutic agent is a substance that can be used for treatment of a disease, disorder, or condition (e.g., ER-positive cancer, e.g., ER-positive breast cancer, etc.). In some embodiments, a therapeutic agent is an agent that has been or is required to be approved by a government agency before it can be marketed for administration to humans. In some embodiments, a therapeutic agent is an agent for which a medical prescription is required for administration to humans. [0320] Therapeutically effective amount: As used herein, “therapeutically effective amount” refers to an amount that produces the desired effect for which it is administered. In some embodiments, the term refers to an amount that is sufficient, when administered to a population suffering from or susceptible to a disease, disorder, and/or condition (e.g., ER- positive cancer, e.g., ER-positive breast cancer, etc.) in accordance with a therapeutic dosing regimen, to treat the disease, disorder, and/or condition. In some embodiments, a therapeutically effective amount is one that reduces the incidence and/or severity of, and/or delays onset of, one or more symptoms of the disease, disorder, and/or condition. Those of ordinary skill in the art will appreciate that the term “therapeutically effective amount” does not in fact require successful treatment be achieved in a particular individual. Rather, a therapeutically effective amount may be that amount that provides a particular desired pharmacological response in a significant number of subjects when administered to patients in need of such treatment. In some embodiments, reference to a therapeutically effective amount may be a reference to an amount as measured in one or more specific tissues (e.g., a tissue affected by the disease, disorder or condition) or fluids (e.g., blood, saliva, serum, sweat, tears, urine, etc.). Those of ordinary skill in the art will appreciate that, in some embodiments, a therapeutically effective amount of a particular agent or therapy may be formulated and/or administered in a single dose. In some embodiments, a therapeutically effective amount of a particular agent or therapy may be formulated and/or administered in a plurality of doses, for example, as part of a dosing regimen.
12366150v1
Attorney Docket No.2014191-0027 [0321] Treatment: As used herein, the term “treatment” (also “treat” or “treating”) refers to administration of a therapy that partially or completely alleviates, ameliorates, relieves, inhibits, delays onset of, reduces severity of, and/or reduces incidence of one or more symptoms, features, and/or causes of a particular disease, disorder, or condition, or is administered for the purpose of achieving any such result. In some embodiments, such treatment can be of a subject who does not exhibit signs of the relevant disease, disorder, or condition and/or of a subject who exhibits only early signs of the disease, disorder, or condition (e.g., ER-positive cancer, e.g., ER- positive breast cancer, etc.). Alternatively, or additionally, such treatment can be of a subject who exhibits one or more established signs of the relevant disease, disorder and/or condition. In some embodiments, treatment can be of a subject who has been diagnosed as suffering from the relevant disease, disorder, and/or condition. In some embodiments, treatment can be of a subject known to have one or more susceptibility factors that are statistically correlated with increased risk of development of the relevant disease, disorder, or condition. A “prophylactic treatment” includes a treatment administered to a subject who does not display signs or symptoms of a condition to be treated or displays only early signs or symptoms of the condition to be treated such that treatment is administered for the purpose of diminishing, preventing, or decreasing the risk of developing the condition. Thus, a prophylactic treatment functions as a preventative treatment against a condition. A “therapeutic treatment” includes a treatment administered to a subject who displays symptoms or signs of a condition and is administered to the subject for the purpose of reducing the severity or progression of the condition. Certain Exemplary Embodiments 1. A method of determining the ER status of a cancer in a subject, the method comprising: quantifying, at one or more genomic loci in a biological sample, optionally in cell-free DNA (cfDNA) from a liquid biopsy sample, obtained or derived from the subject one or more epigenetic biomarkers, wherein the one or more epigenetic biomarkers comprise: (i) one or more histone modifications, (ii) chromatin accessibility, (iii) binding of one or more transcription factors, and/or (iv) DNA methylation.
12366150v1
Attorney Docket No.2014191-0027 2. The method of embodiment 1, wherein the one or more histone modifications are quantified using a histone modification assay that measures one or more of H3K9ac, H3K14ac, H3K18ac, H3K23ac, H3K27ac, H3K4me1, H3K4me2, H3K4me3, and pan-acetylation. 3. The method of embodiment 2, wherein the histone modification assay detects H3K4me3 modifications. 4. The method of embodiment 2, wherein the histone modification assay detects H3K27ac modifications. 5. The method of any one of embodiments 2-4, wherein the histone modification assay is selected from ChIP-seq (Chromatin ImmunoPrecipitation sequencing), CUT&RUN (Cleavage Under Targets and Release Using Nuclease) sequencing, and CUT&Tag (Cleavage Under Targets and Tagmentation) sequencing. 6. The method of any one of embodiments 1-5, wherein chromatin accessibility is quantified using a chromatin accessibility assay selected from ATAC-seq (Assay of Transpose Accessible Chromatin sequencing), NOMe-seq (Nucleosome Occupancy and Methylome sequencing), FAIRE-seq (Formaldehyde-Assisted Isolation of Regulatory Elements sequencing), MNase-seq (Micrococcal Nuclease digestion with sequencing), and a DNase hypersensitivity assay. 7. The method of any one of embodiments 1-6, wherein the binding of one or more transcription factors is quantified using a transcription factor binding assay that detects binding of one or more of p300, mediator complex, cohesin complex, RNA pol II, FOXA1, ESR1, PR, MYC, EN1, FOXM1, KLF4, AP-2, RARa, or RUNX1. 8. The method of embodiment 7, wherein the transcription factor binding assay is selected from ChIP-seq (Chromatin ImmunoPrecipitation sequencing), CUT&RUN (Cleavage Under Targets and Release Using Nuclease) sequencing, and CUT&Tag (Cleavage Under Targets and Tagmentation) sequencing. 9. The method of any one of embodiments 1-8, wherein DNA methylation is quantified using Bisulfite sequencing (BS-Seq), Whole Genome Bisulfite Sequencing (WGBS), Methylated DNA ImmunoPrecipitation sequencing (MeDIP-seq), or Methyl-CpG-Binding Domain sequencing (MBD-seq).
12366150v1
Attorney Docket No.2014191-0027 10. The method of any one of embodiments 1-9, comprising quantifying two or more of the epigenetic biomarkers, each at one or more genomic loci in cell-free DNA (cfDNA) from a liquid biopsy sample obtained or derived from the subject. 11. The method of embodiment 10, comprising quantifying two or more histone modifications. 12. The method of embodiment 11, comprising quantifying H3K4me3 and H3K27ac modifications. 13. The method of embodiment 10, comprising quantifying one or more histone modifications and DNA methylation. 14. The method of embodiment 13, comprising quantifying H3K4me3 and/or H3K27ac modifications and DNA methylation. 15. The method of embodiment 14, comprising quantifying H3K4me3 modifications, H3K27ac modifications and DNA methylation. 16. The method of any one of embodiments 1-15, wherein the liquid biopsy sample is a plasma sample, serum sample, or urine sample. 17. The method of any one of embodiments 1-16, wherein the method comprises isolating DNA (e.g., cfDNA) from 1, 2, 3, 4, or 5 mL of the liquid biopsy sample (e.g., plasma sample). 18. The method of any one of embodiments 1-17, wherein the sample is a liquid biopsy sample comprising cfDNA, and the method comprises: (a) quantifying H3K4me3 modifications at one or more genomic loci using an assay that comprises enriching for cfDNA comprising one or more H3K4me3 modifications and sequencing the cfDNA enriched for H3K4me3 modifications (e.g., using a cfChIP-seq assay); (b) quantifying H3K27ac modifications at one or more genomic loci using an assay that comprises enriching for cfDNA comprising one or more H3K27ac modifications and sequencing the cfDNA enriched for H3K27ac modifications (e.g., using a cfChIP-seq assay); and/or; (c) quantifying methylated DNA using an assay that comprises enriching for methylated cfDNA and sequencing the enriched cfDNA to determine a count of sequences with one or more methylated nucleotides (e.g., using a MBD-seq assay). 19. The method of embodiment 18, wherein:
12366150v1
Attorney Docket No.2014191-0027 (a) the cfDNA comprising H3K4me3 modifications is enriched using a method that comprises incubating the sample with an agent (e.g., an antibody) that binds H3K4me3 modifications; (b) the cfDNA comprising H3K27ac modifications is enriched using a method that comprises incubating the sample with an agent (e.g., an antibody) that binds H3K27ac modifications; and/or (c) methylated cfDNA is enriched using a method that comprises incubating the sample with an agent (e.g., an antibody or a methyl binding domain) that binds methylated DNA. 20. The method of embodiment 19, wherein the agent that binds H3K4me3 modifications, the agent that binds H3K27ac modifications, and/or the agent that binds methylated DNA are attached (e.g., via a covalent or noncovalent bond) to a physical support (e.g., a bead, a magnetic bead, an agarose bead, or a magnetic epoxy bead) prior to incubating with the sample. 21. The method of embodiment 19 or 20, wherein, if the method comprises incubating with two or more of the agent that binds H3K4 modifications, the agent that binds H3K27ac modifications, and the agent that binds methylated DNA, the sample is incubated with the two or more agents (a) in sequence, or (b) in parallel (e.g., wherein the sample is divided into fractions and each fraction is incubated with a different agent). 22. The method of any one of embodiments 18-21, wherein the sequencing is performed using a next generation sequencing method. 23. The method of any one of embodiments 18-22, wherein the method comprises attaching (e.g., ligating) adapters to cfDNA obtained from the subject (e.g., attaching after cfDNA has been enriched for cfDNA comprising one or more H3K4me3 modifications, cfDNA comprising one or more H3K27ac modifications, and/or methylated cfDNA). 24. The method of embodiment 23, wherein the method comprises amplifying the plurality of converted DNA fragments after attaching adapters to the plurality of DNA fragments. 25. The method of any one of embodiments 18-24, comprising mapping sequence reads to a reference genome. 26. The method of any one of embodiments 18-25, wherein non-uniquely mapped and redundant sequence reads are discarded. 27. The method of any one of embodiments 18-26, wherein the sequence reads are mapped to a reference genome, wherein the one or more genomic loci correspond to sequence read peaks,
12366150v1
Attorney Docket No.2014191-0027 wherein a sequence read peak corresponds to a region of the genome that has a higher number of sequence reads that the local background. 28. The method of embodiment 27, wherein peaks in high noise regions are removed and/or where peaks in regions that having increased levels of one or more epigenetic markers in white blood cells are removed. 29. The method of embodiments 27 or 28, wherein peaks in regions likely to be artifactual are removed. 30. The method of any one of embodiments 27-29, wherein peaks that are less than 50 bp in length are removed. 31. The method of any one of embodiments 18-30, wherein quantifying H3K4me3 modifications, H3K27ac modifications, and/or DNA methylation comprises summing the number of sequence reads having at least one nucleotide overlap the one or more genomic loci. 32. The method of embodiment 31, wherein sequence reads are adjusting on the basis of sequencing depth (e.g., quantile normalizing sequence reads to a common reference distribution) and/or ChIP quality prior to summing. 33. The method of embodiment 31 or 32, wherein sequence counts are normalized to aggregate counts in a given sample across a set of regions (e.g., 10,000 regions) previously determined to have DNAse hypersensitivity in most cell types. 34. The method of any one of embodiments 31-33, wherein an estimate of local background signal is subtracted from the sequence reads at each genomic loci prior to summing. 35. The method of any one of embodiments 1-34, further comprising comparing the measure of the one or more epigenetic biomarkers to a reference. 36. The method of embodiment 35, wherein the reference is a predetermined threshold, a measurement from a liquid biopsy sample, a measurement from liquid biopsy samples obtained from a cohort of subjects, and/or a normalized value, optionally wherein: the predetermined threshold and the normalized value were previously shown to distinguish an ER-positive cancer and an ER-negative cancer (e.g., distinguish with an AUROC of greater than 0.5); the reference is a measurement from a liquid biopsy sample obtained from a cohort of subjects who have previously been determined to have an ER-positive cancer or an ER-negative cancer.
12366150v1
Attorney Docket No.2014191-0027 37. The method of any one of embodiments 18-36, comprising calculating sequence read density at the one or more genomic loci, optionally wherein sequence read density is calculated by: (a) summing background adjusted sequence counts at each of the one or more genomic loci and dividing by the sum of the kilobases of the one or more genomic loci; or (b) for each genomic loci, dividing background adjusted fragment count by the number of kilobases of the genomic loci, and then summing for each loci. 38. The method of embodiment 37, wherein the one or more genomic loci include one or more genomic loci with an increased level of the one or more epigenetic biomarkers in (a) sample(s) obtained from a subject with an ER-positive cancer as compared to a sample obtained from a subject with an ER-negative cancer, and/or (b) sample(s) obtained from a subject with an ER-negative cancer as compared to a sample obtained from a subject with an ER-positive cancer. 39. The method of embodiment 38, comprising calculating an ER-positive/ER-negative ratio score, by a method comprising: (a) calculating an ER-positive sequence read density by a method comprising summing background adjusted sequence counts at each of the one or more genomic loci with an increased level of one or more epigenetic biomarkers in sample(s) obtained from subjects with an ER-positive cancer as compared to samples obtained from subjects with ER-negative cancer; (b) calculating an ER-negative sequence read density by a method comprising summing background adjusted sequence counts at each of the one or more genomic loci with an increased level of the one or more epigenetic biomarkers in sample(s) obtained from subjects with an ER-negative cancer as compared to samples obtained from subjects with an ER-positive cancer; and (c) dividing the ER-positive sequence read density by the ER-negative sequence read density. 40. The method of embodiment 39, comprising: (a) determining an ER-positive/ER-negative ratio score for H3K4me3 modifications; (b) determining an ER-positive/ER-negative ratio score for H3K27ac modifications; and/or (c) determining an ER-positive/ER-negative ratio score for methylated DNA.
12366150v1
Attorney Docket No.2014191-0027 41. The method of embodiment 40, comprising performing each of (a)-(c), and combining each of the ratio scores. 42. The method of embodiment 41, where the ratio scores are combined using fitted values determined using a logistic regression. 43. The method of any one of embodiments 1-42, wherein the method further comprises comparing the one or more quantified genomic markers to a reference, and wherein an increase or decrease in the one or more genomic markers as compared to the reference indicates that the subject has an ER-positive or an ER-negative cancer. 44. The method of any one of embodiments 35-43, wherein the reference is a predetermined threshold, a measurement from a liquid biopsy sample, and/or a normalized value, optionally wherein: the reference is a measurement from liquid biopsy samples obtained from a cohort of subjects who have previously been determined to have an ER-negative cancer or to be cancer free; the predetermined threshold and the normalized value were previously shown to distinguish ER-positive and ER-negative cancers (e.g., to provide an AUROC value of at least 0.5); or the cohort of subjects had previously been determined to have a cancer (e.g., an ER- positive or ER-negative cancer). 45. The method of any one of embodiments 1-44, wherein the method comprises quantifying one or more of the epigenetic biomarkers at one or more genomic loci in Tables 1-3. 46. The method of embodiment 45, wherein the method comprises: (a) quantifying H3K4me3 modifications for at least 5, 10, 20, 30, 40, or 50 genomic loci listed in Table 1; (b) quantifying H3K27ac modifications for at least 5, 10, 20, 30, 40, or 50 genomic loci listed in Table 2; and/or (c) quantifying DNA methylation for at least 5, 10, 20, 30, 40, or 50 genomic loci listed in Table 3. 47. The method of any one of embodiments 1-46, wherein the method provides an area under the receiver operating characteristic (AUROC) for determining if a subject has an ER-positive cancer vs. an ER-negative cancer of greater than 0.5 (e.g., greater than 0.55, greater than 0.6,
12366150v1
Attorney Docket No.2014191-0027 greater than 0.65, greater than 0.7, greater than 0.75, greater than 0.8, greater than 0.85, greater than 0.9, or greater than 0.95). 48. The method of any one of embodiments 1-46, wherein the ER-positive cancer is an ER- positive cancer based on IHC testing and the ER-negative cancer is an ER-negative cancer based on IHC testing. 49. The method of any one of embodiments 1-48, wherein the sample comprises a detectable amount of ctDNA (e.g., wherein estimated tumor fraction is >3% for the cfDNA, e.g., as determined by iChorCNA). 50. The method of any one of embodiments 1-49, wherein the subject has previously been determined to have cancer, the subject has an increased susceptibility to cancer, and/or the method further comprises determining whether the subject has a cancer. 51. The method of any one of embodiments 1-50, wherein the cancer is breast cancer (e.g., metastatic breast cancer), ovarian cancer, or endometrial cancer. 52. The method of embodiment 51, wherein the cancer is breast cancer. 53. A method of treating a subject having a cancer, the method comprising: administering a cancer therapy to the subject based on the ER status of the cancer, wherein the ER status of the cancer has been determined using the method of any one of embodiments 1-52. 54. The method of embodiment 53, further comprising determining the ER status of the cancer using the method of any one of embodiments 1-52. 55. The method of embodiment 54 or 55, wherein, if the cancer has been determined to be ER-positive, the cancer therapy administered is an ER-targeted agent. 56. The method of embodiment 54 or 55, wherein, if the cancer has been determined to be ER-negative, the cancer therapy administered is an ER-targeted agent. 57. The method of embodiment 56, wherein, if the cancer has been determined to be ER- negative, the cancer therapy is one appropriate for an ER-negative cancer (e.g., a therapy that comprises surgery, and/or radiation therapy). 58. A method of monitoring the ER status of a cancer in a subject, and optionally treating the cancer, the method comprising: determining the ER status of the cancer using the method of any one of embodiments 1- 52 at first and second time points.
12366150v1
Attorney Docket No.2014191-0027 59. The method of embodiment 58, wherein the subject has been administered an ER- targeted agent after the first time point and before the second time point. 60. The method of embodiment 58 or 59, further comprising administering a cancer therapy, optionally an ER-targeted agent, to the subject based on the ER status of the cancer at the second time point, optionally wherein the type, dose and/or frequency of administration of the cancer therapy is adjusted based on the ER status of the cancer at the second time point. 61. A method of treating a subject having a cancer, the method comprising: administering an ER-targeted agent to the subject if the subject has been determined to have a validated epigenetic profile indicative of an ER-positive cancer based on analysis of a biological sample, optionally of cell-free DNA (cfDNA) from a liquid biopsy sample, obtained or derived from the subject, and, if the subject has not been determined to have a validated epigenetic profile indicative of an ER-positive cancer, not administering an ER-targeted agent, wherein the presence of the validated epigenetic profile has been determined using a validated classifier, wherein the validated classifier has been obtained by: (a) determining a genomic profile of one or more histone modifications, chromatin accessibility, binding of one or more transcription factors, and/or DNA methylation in (i) one or more ER-positive cell lines or (ii) biological samples obtained from a first cohort of subjects who have previously been determined to have an ER- positive cancer; (b) determining a genomic profile of one or more histone modifications, chromatin accessibility, binding of one or more transcription factors, and/or DNA methylation in (i) one or more ER-negative cell lines or (ii) biological samples obtained from a second cohort of healthy subjects or subjects who have previously been determined to have an ER-negative cancer; (c) comparing the genomic profile determined in step (a) and the genomic profile determined in step (b), to identify genomic loci that have statistically different histone modification, chromatin accessibility, binding of transcription factor, and/or DNA methylation levels (“differential loci”); (d) training a classifier on histone modification, chromatin accessibility, binding of transcription factor, and/or DNA methylation levels in the differential loci to
12366150v1
Attorney Docket No.2014191-0027 distinguish between (i) samples from one or more ER-positive cell lines or biological samples obtained from the first cohort, and (ii) samples from one or more ER-negative cell lines or biological samples obtained from the second cohort, to identify samples having a profile of histone modification, chromatin accessibility, binding of transcription factor, and/or DNA methylation levels (“epigenetic profile”) that indicates that the samples are likely obtained from an ER-positive cell line or from the first cohort; and (e) obtaining the validated classifier by validating the classifier from step (d) on a third cohort comprising an independent and group of subjects with ER-positive and ER-negative cancers and selecting a threshold such that the validated classifier predicts ER-positive cancers, with an area under the receiver operating characteristic (AUROC) greater than 0.5 (e.g., greater than 0.55, greater than 0.6, greater than 0.65, greater than 0.7, greater than 0.75, greater than 0.8, greater than 0.85, greater than 0.9, or greater than 0.95), wherein subjects falling within the group of predicted ER-positive cancers display the validated epigenetic profile and subjects that do not fall within the group of ER- positive cancers lack the validated epigenetic profile. 62. The method of embodiment 61, wherein the differential loci in step (c) were identified by comparing the genomic profile of one or more histone modifications and/or DNA methylation in (i) one or more ER-positive cell lines and (ii) one or more ER-negative cell lines. 63. The method of embodiment 61 or 62, wherein the classifier in step (d) was trained on histone modification, chromatin accessibility, binding of transcription factor, and/or DNA methylation levels in the differential loci that were obtained by in silico mixing sequence data from one or more ER-positive cell lines and sequence data obtained from liquid biopsy samples of healthy subjects. 64. The method of any one of embodiments 61-63, wherein the validated classifier in step (e) was validated using liquid biopsy samples from the third cohort. 65. The method of any one of embodiments 61-64, wherein the classifier in step (d) was trained on two or more histone modification levels in the differential loci. 66. The method of embodiment 65, wherein the two or more histone modification levels comprise H3K4me3 and H3K27ac modification levels. 67. The method of any one of embodiments 61-66, wherein the classifier in step (d) was trained on one or more histone modification levels and DNA methylation in the differential loci.
12366150v1
Attorney Docket No.2014191-0027 68. The method of any one of embodiments 61-66, wherein the classifier in step (d) was trained using ridge regression, elastic-net regression, or lasso regression. 69. The method of embodiment 61 or 62, wherein the one or more histone modification levels comprise H3K4me3 and/or H3K27ac modification levels. 70. The method of any one of embodiments 61-69, wherein the liquid biopsy sample is a plasma sample, serum sample, or urine sample. 71. A kit comprising reagents for quantifying one or more histone modifications, chromatin accessibility, binding of one or more transcription factors, and/or DNA methylation at one or more genomic loci, wherein the one or more genomic loci are selected from Tables 1-3. 72. The kit of embodiment 71, wherein the kit comprises reagents for: (a) quantifying H3K4me3 modifications for at least 5, 10, 20, 30, 40, or 50 genomic loci in Table 1; (b) quantifying H3K27ac modifications for at least 5, 10, 20, 30, 40, or 50 genomic loci in Table 2; (c) quantifying DNA methylation for at least 5, 10, 20, 30, 40, or 50 genomic loci in Table 3; (d) any combination of (a)-(c). 73. The kit of embodiment 71 or 72, wherein the kit comprises one or more antibodies for use in ChIP-seq, optionally wherein the one or more antibodies specifically bind H3K4me3- or H3K27ac-modified histones. 74. The kit of any one of embodiments 71-73, wherein the kit comprises one or more methyl- binding domains for use in MBD-seq or wherein the kit comprises one or more antibodies that bind methylated DNA for use in MeDIP-seq. 75. The kit of any one of embodiments 71-74, wherein the kit comprises reagents for isolation of cell-free DNA (cfDNA) from a liquid biopsy sample. 76. The kit of any one of embodiments 71-75, wherein the kit comprises reagents for library preparation for sequencing. 77. The kit of any one of embodiments 71-76, wherein the kit comprises reagents for sequencing. 78. The kit of any one of embodiments 71-77, wherein the kit comprises instructions for determining if a subject has an ER-positive cancer.
12366150v1
Attorney Docket No.2014191-0027 79. A non-transitory computer readable storage medium encoded with a computer program, wherein the program comprises instructions that when executed by one or more processors cause the one or more processors to perform operations to perform the method of any one of embodiments 1-70. 80. A computer system comprising a memory and one or more processors coupled to the memory, wherein the one or more processors are configured to perform operations to perform the method of any one of embodiments 1-70. 81. A system for determining the ER status of a cancer in a subject, the system comprising a sequencer configured to generate a sequencing dataset from a sample; and a non-transitory computer readable storage medium of embodiment 79 and/or a computer system of embodiment 80. 82. The system of embodiment 81, wherein the sequencer is configured to generate a Whole Genome Sequencing (WGS) dataset from the sample. 83. The system of embodiment 81 or 82 further comprising a sample preparation device configured to prepare the sample for sequencing from a biological sample, optionally a liquid biopsy sample. 84. The system of embodiment 83, wherein the sample preparation device comprises reagents for quantifying one or more histone modifications, chromatin accessibility, binding of one or more transcription factors, and/or DNA methylation at one or more genomic loci in cell-free DNA (cfDNA) from the biological sample, optionally the liquid biopsy sample. 85. The system of embodiment 84, wherein the one or more genomic loci are selected from Tables 1-3. 86. The system of any one of embodiments 83-85, wherein the device comprises reagents for quantifying: (a) H3K4me3, e.g., for at least 5, 10, 20, 30, 40, or 50 genomic loci in Table 1; (b) H3K27ac, e.g., for at least 5, 10, 20, 30, 40, or 50 genomic loci in Table 2; (c) DNA methylation, e.g., for at least 5, 10, 20, 30, 40, or 50 genomic loci in Table 3; or (d) any combination of (a)-(c).
12366150v1
Attorney Docket No.2014191-0027 87. The system of embodiment 85 or 86, wherein the reagents comprise one or more antibodies for use in ChIP-seq, optionally wherein the one or more antibodies specifically bind H3K4me3- or H3K27ac-modified histones. 88. The system of embodiment 86 or 87, wherein the reagents comprise one or more methyl- binding domains for use in MBD-seq. 89. The system of any one of embodiments 83-88, wherein the device comprises reagents for isolation of cell-free DNA (cfDNA) from the biological sample, optionally the liquid biopsy sample. 90. The system of any one of embodiments 81-89, wherein the device comprises reagents for library preparation for sequencing. 91. The system of any one of embodiments 81-90, wherein the sequencer comprises reagents for sequencing. 92. A method of determining the ER status of a cancer in a subject (e.g., patient), the method comprising: receiving (e.g., by a processor of a computing device) one or more genomic profiles of one or more histone modifications, chromatin accessibility, binding of one or more transcription factors, and/or DNA methylation for the subject; and determining whether the subject has an epigenetic profile indicative of an ER-positive cancer by classifying (e.g., by the processor) the genomic profile using the ER classifier. 93. The method of embodiment 92, wherein the ER classifier has been trained using one or more genomic profiles of one or more histone modifications, chromatin accessibility, binding of one or more transcription factors, and/or DNA methylation for (i) one or more ER-positive cell lines and one or more ER-negative cell lines and/or (ii) one or more biological samples obtained from one or more cohorts of subjects who have previously been determined to have an ER-positive cancer and one or more biological samples obtained from one or more cohorts of subjects who have previously been determined to have an ER-negative cancer. 94. The method of embodiment 93, wherein the one or more genomic profiles used to train the ER classifier comprise one or more genomic profiles generated by in silico diluting sequence data from ER-positive or ER-negative cell lines with sequence data obtained from healthy donor plasma samples so as to achieve a simulated ctDNA percentage ranging from 0.5% to 50%.
12366150v1
Attorney Docket No.2014191-0027 95. The method of embodiment 2 or 3, wherein the one or more genomic profiles used to train the ER classifier are for differential loci having statistically significant differences in levels of one or more histone modifications, chromatin accessibility, binding of one or more transcription factors, and/or DNA methylation levels in (a) one or more ER-positive cell lines as compared to one or more ER-negative cell lines and/or (b) one or more biological samples obtained from one or more cohorts of subjects who have previously been determined to have an ER-positive cancer and one or more biological samples obtained from one or more cohorts of subjects who have previously been determined to have an ER-negative cancer. 96. The method of embodiment 95, wherein the differential loci were identified by comparing genomic profiles of one or more histone modifications and/or DNA methylation in (i) one or more ER-positive cell lines and (ii) one or more ER-negative cell lines. 97. The method of embodiment 95 or 96, wherein the ER classifier has been trained on the levels of one or more histone modifications, chromatin accessibility, binding of one or more transcription factors, and/or DNA methylation in the differential loci for the genomic profiles that were generated by in silico diluting sequence data from one or more ER-positive cell lines and sequence data obtained from liquid biopsy samples of healthy subjects. 98. The method of any one of embodiments 95-97, wherein the ER classifier has been trained on two or more histone modification levels in the differential loci. 99. The method of any one of embodiments 95-98, wherein the ER classifier has been trained on one or more histone modification levels and DNA methylation levels in the differential loci. 100. The method of any one of embodiments 92-99, comprising receiving one or more genomic profiles of two or more histone modification levels. 101. The method of embodiment 98 or 100, wherein the two or more histone modifications comprise H3K4me3 and H3K27ac modifications. 102. The method of any one of embodiments 92-101, comprising receiving one or more genomic profiles of one or more histone modifications and DNA methylation. 103. The method of embodiment 99 or 102, wherein the one or more histone modifications comprise H3K4me3 and/or H3K27ac modifications. 104. The method of embodiment 99 or embodiment 102, wherein the one or more histone modifications comprise H3K4me3 and H3K27ac modifications.
12366150v1
Attorney Docket No.2014191-0027 105. The method of any one of embodiments 92-104, wherein the ER classifier has been trained with sequence data derived from plasma samples. 106. The method of any one of embodiments 92-105, wherein the ER classifier has been trained with data derived from liquid biopsy samples. 107. The method of any one of embodiments 92-106, wherein the ER classifier is a validated classifier. 108. The method of embodiment 107, wherein the ER classifier has been validated by selecting a threshold such that the validated classifier predicts ER-positive cancers with an area under the receiver operating characteristic (AUROC) greater than 0.5 (e.g., greater than 0.55, greater than 0.6, greater than 0.65, greater than 0.7, greater than 0.75, greater than 0.8, greater than 0.85, greater than 0.9, or greater than 0.95). 109. The method of embodiment 107 or 108, wherein the ER classifier has been validated on a a group of subjects with ER-positive or ER-negative cancers, wherein subjects falling within a group of predicted ER-positive cancers display a validated epigenetic profile and subjects that do not fall within a group of ER-positive cancers lack the validated epigenetic profile. 110. The method of any one of embodiments 107-109 wherein the ER classifier has been validated using liquid biopsy sample data. 111. A non-transitory computer readable storage medium encoded with a computer program, wherein the program comprises instructions that when executed by one or more processors cause the one or more processors to perform operations to perform the method of any one of embodiments 92-110. 112. A computer system comprising a memory and one or more processors coupled to the memory, wherein the one or more processors are configured to perform operations to perform the method of any one of embodiments 92-110. 113. A method of treating a subject having a cancer, the method comprising: administering an ER-targeted agent to the subject, wherein the subject has been determined to have a validated epigenetic profile indicative of an ER-positive cancer based on analysis of a biological sample, optionally of cell-free DNA (cfDNA) from a liquid biopsy sample, obtained or derived from the subject, wherein the presence of the validated epigenetic profile has been determined using a classifier (e.g., a validated classifier) according to a method of any one of embodiments 92-110.
12366150v1
Attorney Docket No.2014191-0027 EXAMPLES [0322] The present Examples demonstrate the identification and use of differentially modified and/or differentially accessible genomic loci in ER-positive and ER-negative cell lines and/or from cfDNA in plasma samples obtained from subjects with ER-positive and ER-negative breast cancers. The present Examples show that differentially modified and/or differentially accessible genomic loci of the present disclosure can be used to determine ER status from cfDNA in plasma samples obtained from subjects with ER-positive and ER-negative cancers. Example 1: Materials and Methods [0323] This Example describes the materials and methods that were used to generate sequencing data that was then used in Examples 2 and 3 to create ER status classifiers. Materials Cell lines [0324] The following ER-positive cell lines were used: ZR751, ZR7530, BT483, T47D, BT474, CAMA1, MCF7, HCC1428, and HCC1500. The following ER-negative cell lines were used: BT549, DU4475, HS578T, BT20, UACC893, HCC38, HCC70, HCC202, HCC1143, HCC1187, HCC1419, HCC1599, HCC1806, HCC1954, HCC2157, HCC2218, and SKBR3. Plasma samples [0325] Plasma samples were prepared from whole blood collected in EDTA blood collection tubes or Streck cell-free DNA BCT with 4-6 hours of collection and plasma was stored at -80˚C until use. Whole blood was obtained from breast cancer patients under a protocol approved by an IRB. Breast cancer patients had previously been determined to be ER-positive or ER-negative. Informed content was obtained in each case and samples were de-identified. Methods Chromatin immunoprecipitation (ChIP)
12366150v1
Attorney Docket No.2014191-0027 [0326] Chromatin immunoprecipitation (ChIP) for histone marks (H3K4me3 and H3K27ac) in cell lines was performed using methods similar to those previously described in Schones et al., Cell (2008) 132(5):887-898, which is incorporated by reference herein in its entirety. Briefly, the cells were lysed and the chromatin was MNase digested to generate approximately 80% mononucleosomes. Nucleosomes were then incubated with antibodies that bind H3K4me3 modifications or H3K27ac modifications that were previously conjugated to magnetic epoxy beads (Invitrogen) with constant mild shaking overnight. The beads were then washed and rinsed. Sequencing libraries were generated from purified immunoprecipitated sample DNA and then sequenced. [0327] Chromatin immunoprecipitation (ChIP) for histone marks (H3K4me3 and H3K27ac) in plasma samples was performed using methods similar to those previously described in Sadeh et al., Nat Biotechnol (2021) 39: 586-598 and Jang et al., Life Sci Alliance (2023) 6(12):e202302003. Briefly, about 1 mL frozen plasma was thawed and then prepared for ChIP. The thawed plasma was incubated with antibodies that bind H3K4me3 modifications or H3K27ac modifications that were previously conjugated to magnetic epoxy beads (Invitrogen) with constant mild shaking overnight. The beads were then washed and rinsed. Sequencing libraries were generated from purified immunoprecipitated sample DNA and then sequenced. Methylated DNA enrichment [0328] Enrichment of DNA methylation was performed on DNA extracted from cell lines and human plasma samples using the EpiMark® Methylated DNA Enrichment Kit (E2600S, available from New England Biolabs) following the manufacturer’s protocol. Briefly, cfDNA libraries were prepared and adaptors ligated. Then, the EpiMark® capture reagent was applied to each library sample following the manufacturer’s protocol. Enriched DNA libraries were amplified and sequenced. ChIP-seq and DNA methylation data analysis [0329] ChIP-sequencing reads and MeDIP-sequencing reads were aligned to the human genome build hg19 using the Burrows-Wheeler Aligner (BWA) version 0.7.15. Non-uniquely mapping and redundant reads were discarded. MACS v2.2.7.1 was used for peak calling with a q- value (FDR) threshold of 0.01. Data quality was evaluated by a variety of measures, including
12366150v1
Attorney Docket No.2014191-0027 total peak number, FrIP (fraction of reads in peak) score, number of high-confidence peaks (enriched > ten-fold over background), and percent of peak overlap with “blacklist” DHS peaks derived from the ENCODE project (Amemiya et al., Sci Rep (2019) 9(1):9354). Peaks were assessed for overlap with gene features and CpG islands using annotatr. IGV was used to visualize normalized read counts at specific genomic loci. Overlap of peaks was assessed using BEDTools and the GenomicRanges package in Bioconductor. Peaks were considered overlapping if they shared one or more nucleotide. Example 2: ER status classifiers based on complex modeling of signals across different subsets of individual genomic loci that are differentiated based on ER-positive and ER- negative status [0330] To construct an ER status classifier, genomic loci likely to differentiate ER- positive and ER-negative samples based on H3K4me3 modification, H3K27ac modification or DNA methylation were first identified. To accomplish this, for each analyte, union peak maps were created by merging peak coordinates for all of the cell lines, removing regions likely to be artifactual (the ENCODE “blacklist” regions, see Amemiya et al., Sci Rep (2019) 9(1):9354) and discarding all peaks less than 50 bp in length. The number of sequencing fragments overlapping each peak (by at least 1 bp) were quantified for each analyte, local background signal was subtracted to improve signal-to-noise, the result was log2-transformed, and quantile normalized across the entire dataset (for each analyte separately). Genomic loci that had differential analyte signal between ER-positive and ER-negative cell lines were determined using DESeq2 (Love et al., Genome Biol (2014) 15(12):550), with an FDR cutoff of 5%. These differential loci are shown in Table 1 (H3K4me3), Table 2 (H3K27ac) and Table 3 (DNA methylation). [0331] Next, a simulated dataset was built for each analyte to train an ER status classifier. This dataset was constructed for each analyte by mixing different proportions of sequencing fragments from healthy donor plasma samples and the cell lines to achieve simulated ctDNA percentages ranging from 0.5% to 50%. Sequencing fragments overlapping the union cell line maps for each simulated dataset were then quantified as described above. Quantifications for all genomic loci were then used with a given absolute log2-fold-change cutoff and for a given analyte (or combination of analytes) from the simulated dataset as input to train a regularized logistic regression. More specifically, models were trained on the ER status of the cell lines from
12366150v1
Attorney Docket No.2014191-0027 which the simulated datasets were constructed, using glmnet (Engebretsen and Bohlin, Clin Epigenetics (2019) 11(1):123). Models were trained with different alpha values ranging from 1 (lasso) to 0 (ridge) in order to demonstrate the robustness of the approach. [0332] Fig.1 shows ROC curves for exemplary ER status classifiers that were generated in accordance with this method. As shown, different classifiers were generated using (i) ridge regression (all features from the relevant subset of genomic loci) (alpha = 0); (ii) elastic-net regression (many features from the relevant subset of genomic loci) (alpha = 0.25), or (iii) lasso regression (few features from the relevant subset of genomic loci) (alpha = 1). As shown, different classifiers were generated with (a) genomic loci from Tables 1-3 for different modifications, namely (i) H3K4me3 modifications, (ii) H3K27ac modifications, (iii) DNA methylation (DNAme) or (iv) all of the above (All) and (b) using different subsets of genomic loci in Tables 1-3 for a particular modification, namely (i) all genomic loci with an absolute log2(fold-change) ≥ 0.5, (ii) all genomic loci with an absolute log2(fold-change) ≥ 1, (iii) all genomic loci with an absolute log2(fold-change) ≥ 2, (iv) all genomic loci with an absolute log2(fold-change) ≥ 3, and (v) all genomic loci with an absolute log2(fold-change) ≥ 4. [0333] Fig.2 shows a representative, non-limiting graphs that demonstrates the accuracy of ER status (based on AUCROC) determination using the classifiers that were generated in accordance with this Example. Example 3: ER Status Determination in Plasma Samples [0334] The present example provides data demonstrating that technologies provided in the present disclosure can be used to determine ER status in a subject having cancer, using samples comprising cfDNA. In the present example, plasma samples from patients diagnosed with metastatic breast cancer were characterized. [0335] Plasma samples were obtained from 91 subjects. Of these samples, 43 were obtained from subjects having associated clinical data, 80 provided high quality datasets across all analytes (promoter, enhancer, DNAme), and 49 had ctDNA that could be measured by copy number variation analysis (>3% ctDNA as quantified by ichorCNA (see, e.g., Adalsteinsson et al., Nat Comm., 2017, the contents of which are incorporated by reference herein in their entirety).41 samples were obtained from subjects having HER2- breast cancer and 12 samples were obtained from subjects having HER2+ breast cancer.28 samples were obtained from
12366150v1
Attorney Docket No.2014191-0027 subjects having an ER- breast cancer and 25 were obtained from subjects having an ER+ breast cancer. Samples were collected in accordance with the method provided in Example 1. Blood samples were collected within 6 weeks of tumor biopsy, with ER status determined using immunohistochemistry (IHC). [0336] A multianalyte, liquid biopsy assay performed in accordance with the approaches described herein (e.g., as described in Example 1) was applied to capture genome-wide epigenomic signals, and to map enhancers, promoters, and DNA methylation data from 1 mL of plasma. A logistic regression model was applied to infer ER status from plasma-based epigenomic profiles. Results [0337] The ER status classifier was applied to 19 of 44 samples determined to have detectable ctDNA.11 of the 19 (58%) samples were from subjects having an ER+ cancer as determined by IHC (ER IHC range was 80-95% for these subjects). Overall, 18 of the 19 samples were correctly classified as ER+ or ER- by the ER classifier (AUC=1).3 of the 19 samples were collected from patients who were ER+ at primary diagnosis and ER- at the time of tissue sampling. For each of these samples, the ER classifier correctly predicted ER- status. [0338] AUC plots are shown in Fig.3(A). As shown in the plots, the classifiers provided herein are capable of accurately distinguishing subjects having ER+ vs ER- cancers using plasma samples (even for samples without detectable ctDNA). For samples with detectable ctDNA (e.g., ctDNA > 3%), accuracy increased. As shown in Fig.3(A), accuracy further improved when characterizing samples from a particular source (see right plot, showing an accuracy that was virtually the same as IHC). Fig.3(A) demonstrates that technologies provided herein are capable of accurately determining ER status of different cancers. [0339] As shown in Fig.3(B), classifiers described herein detected analyte features that were previously associated with ER+ and ER- status. This data demonstrates that methods provided herein can detect genes that are biologically relevant, and accurately reflect ER status of a cancer. [0340] Table 4 shows genes that were determined to be associated with ER status using the disclosed classifiers. As indicated below, genes previously associated with ER status (left column); genes previously shown to have a biological link, but not directly associated with ER
12366150v1
Attorney Docket No.2014191-0027 activity (middle column); and genes not previously shown to be associated with ER status (right column) were all found to be associated with ER status. The below table demonstrates that classifiers provided herein can detect biologically relevant features (indicating that they accurately reflect ER status) and can also discover new genes and pathways that may play a role in ER expression. In some embodiments, an ER classifier described herein measures an epigenomic marker (e.g., promoter, enhancer, or DNA methylation marker) associated with one or more of the genes listed in Table 4. [0341] Table 4: Selected Genes Incorporated in Certain ER Status Classifiers Genes Previously Genes Previously Linked Genes Not Previously A i t d ith ER ith ER t t A i t d Li k d ith
 TABLES [0342] The following Tables identify exemplary genomic loci that are differentially modified and/or differentially accessible in ER-positive vs. ER-negative cancer. Table 1 is based on differential H3K4me3 modifications. Table 2 is based on differential H3K27ac modifications. Table 3 is based on differential DNA methylation.
TABLES [0342] The following Tables identify exemplary genomic loci that are differentially modified and/or differentially accessible in ER-positive vs. ER-negative cancer. Table 1 is based on differential H3K4me3 modifications. Table 2 is based on differential H3K27ac modifications. Table 3 is based on differential DNA methylation.
12366150v1
Attorney Docket No.2014191-0027 Table 1: Exemplary genomic loci that are differentially H3K4me3 modified in ER-positive vs. ER-negative cancer. Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1 chr1:793725-794444 -1.41 676 chr13:99710786-99710933 1.60 2.00 3.08 2.44 1.72 1.59 2.20 2.87 3.23 1.80 1.91 2.59 1.86 1.95 1.91 1.73 1.54 1.61 1.02 1.75 1.86 1.66 1.85 2.12 1.76 1.72 2.40 2.12 3.27 2.01 0.62 1.65 0.57 0.55 1.54 2.30 1.55 2.62 2.35 2.02 1.96 2.14 0.51 2.39 0.62
 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 46 chr1:11761594-11762165 -2.11 721 chr14:55543845-55544874 -2.13 1.58 1.62 0.51 2.58 1.86 2.39 2.77 1.89 2.13 1.51 1.38 1.33 1.70 1.53 1.35 1.63 2.07 0.66 1.00 2.14 1.87 1.42 1.08 1.12 1.96 1.73 1.55 1.48 1.50 0.52 0.52 2.21 0.79 2.33 1.73 2.19 1.82 2.01 2.60 2.31 0.54 1.82 2.77 1.70 2.27
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 46 chr1:11761594-11762165 -2.11 721 chr14:55543845-55544874 -2.13 1.58 1.62 0.51 2.58 1.86 2.39 2.77 1.89 2.13 1.51 1.38 1.33 1.70 1.53 1.35 1.63 2.07 0.66 1.00 2.14 1.87 1.42 1.08 1.12 1.96 1.73 1.55 1.48 1.50 0.52 0.52 2.21 0.79 2.33 1.73 2.19 1.82 2.01 2.60 2.31 0.54 1.82 2.77 1.70 2.27
 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 92 chr1:32665518-32667241 -0.91 767 chr14:100624667-100626493 0.85 1.47 1.79 1.04 2.27 2.29 2.83 2.35 1.85 3.40 1.42 0.76 1.75 1.66 1.98 2.50 2.11 1.79 2.20 1.56 2.89 1.66 2.74 1.74 2.91 1.68 3.01 1.99 1.14 1.89 1.85 2.58 1.31 1.35 2.56 1.59 2.59 1.63 1.47 1.58 2.04 2.92 1.24 2.47 2.82 0.91
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 92 chr1:32665518-32667241 -0.91 767 chr14:100624667-100626493 0.85 1.47 1.79 1.04 2.27 2.29 2.83 2.35 1.85 3.40 1.42 0.76 1.75 1.66 1.98 2.50 2.11 1.79 2.20 1.56 2.89 1.66 2.74 1.74 2.91 1.68 3.01 1.99 1.14 1.89 1.85 2.58 1.31 1.35 2.56 1.59 2.59 1.63 1.47 1.58 2.04 2.92 1.24 2.47 2.82 0.91
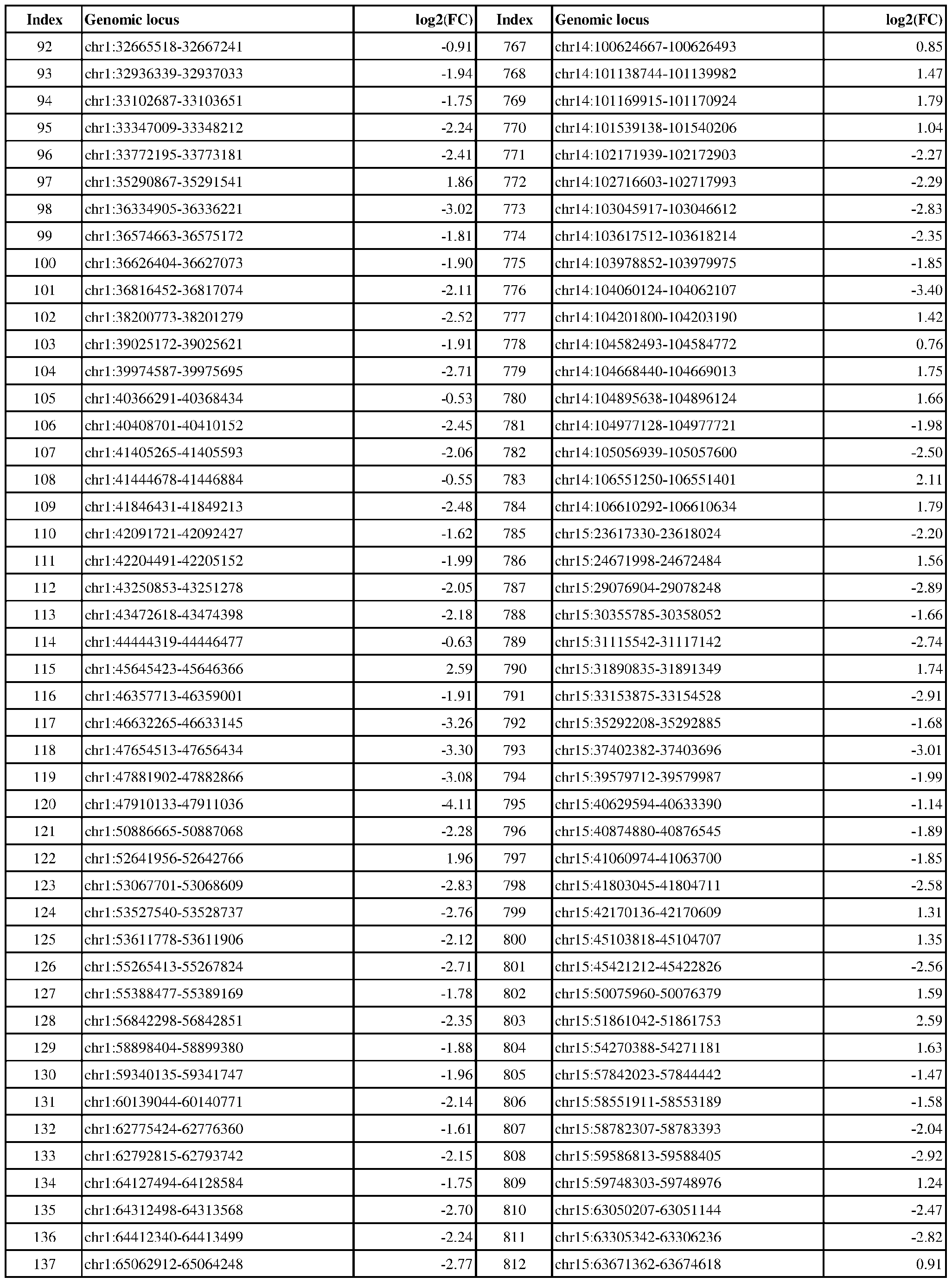 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 138 chr1:65731098-65732136 -3.33 813 chr15:63969997-63970618 2.09 0.67 1.78 2.24 2.25 2.58 2.56 1.82 1.40 2.73 2.19 2.29 2.35 2.03 2.63 3.08 1.72 2.13 2.96 1.47 1.87 2.02 2.53 1.47 2.18 2.48 1.92 1.52 2.18 2.15 1.71 1.88 1.66 2.51 2.29 2.23 1.05 1.90 1.88 1.61 2.70 2.45 2.38 1.92 2.70 1.66
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 138 chr1:65731098-65732136 -3.33 813 chr15:63969997-63970618 2.09 0.67 1.78 2.24 2.25 2.58 2.56 1.82 1.40 2.73 2.19 2.29 2.35 2.03 2.63 3.08 1.72 2.13 2.96 1.47 1.87 2.02 2.53 1.47 2.18 2.48 1.92 1.52 2.18 2.15 1.71 1.88 1.66 2.51 2.29 2.23 1.05 1.90 1.88 1.61 2.70 2.45 2.38 1.92 2.70 1.66
 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 184 chr1:111735041-111736290 -2.18 859 chr16:5699949-5700420 1.70 1.91 0.77 1.67 1.98 1.41 1.86 2.09 2.35 1.89 1.57 2.42 1.75 1.63 3.00 1.74 3.07 2.41 1.70 1.82 1.56 2.23 1.92 2.24 1.96 2.07 2.19 1.96 3.37 1.41 1.86 2.39 1.67 1.97 3.17 1.89 2.63 2.48 2.62 1.63 2.58 1.87 1.68 2.08 2.66 2.23
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 184 chr1:111735041-111736290 -2.18 859 chr16:5699949-5700420 1.70 1.91 0.77 1.67 1.98 1.41 1.86 2.09 2.35 1.89 1.57 2.42 1.75 1.63 3.00 1.74 3.07 2.41 1.70 1.82 1.56 2.23 1.92 2.24 1.96 2.07 2.19 1.96 3.37 1.41 1.86 2.39 1.67 1.97 3.17 1.89 2.63 2.48 2.62 1.63 2.58 1.87 1.68 2.08 2.66 2.23
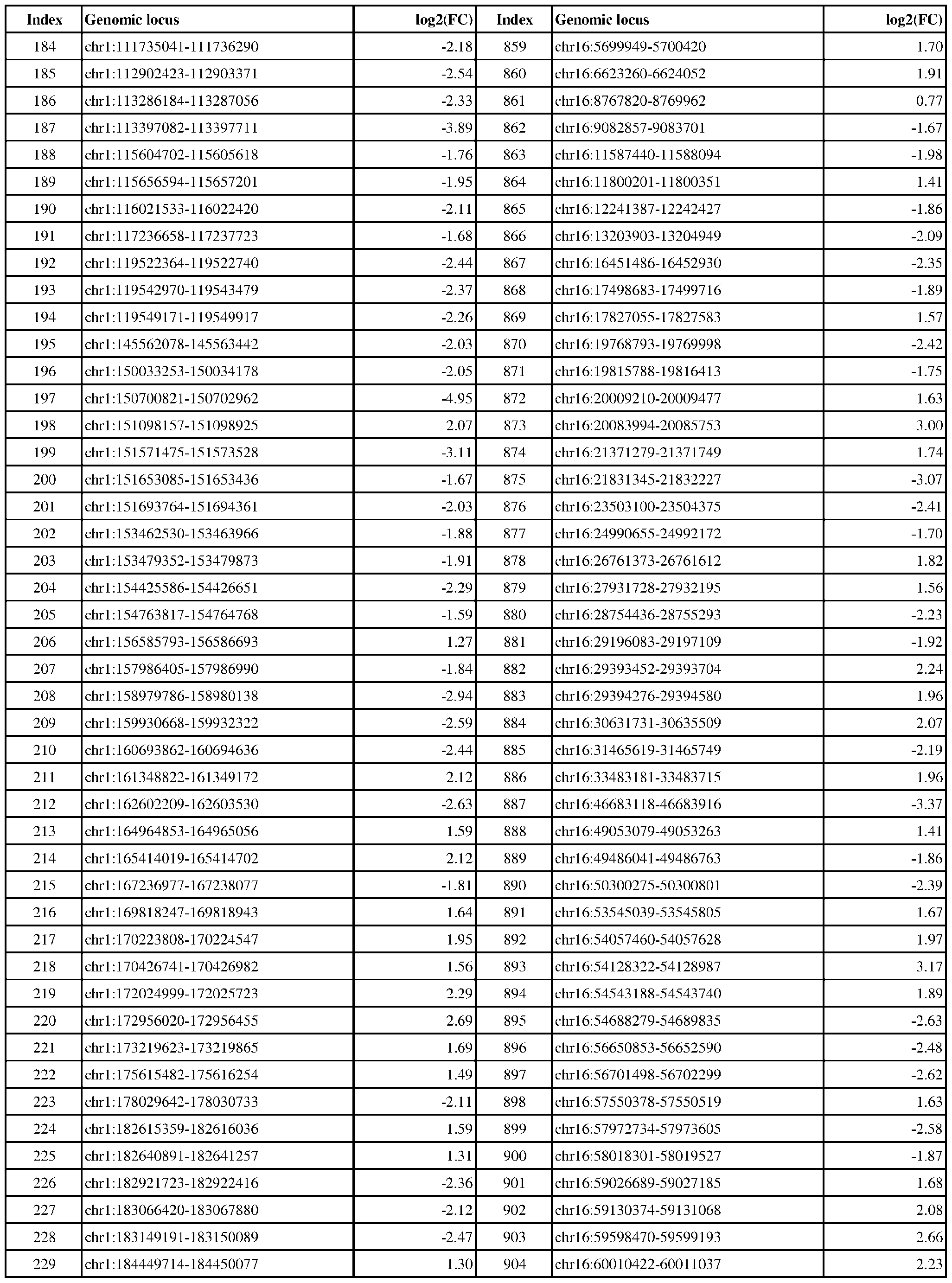 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 230 chr1:185381520-185382485 -2.57 905 chr16:64070755-64070916 1.52 1.42 1.56 2.92 3.01 1.81 1.56 1.75 2.04 2.48 1.63 2.31 3.23 2.10 1.81 2.53 1.89 2.35 2.09 1.99 1.57 2.30 1.93 1.68 2.83 2.20 2.41 3.11 2.22 2.66 1.44 3.79 2.01 2.48 1.77 1.94 2.56 2.28 2.47 2.56 2.23 1.86 2.41 1.58 1.97 0.67
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 230 chr1:185381520-185382485 -2.57 905 chr16:64070755-64070916 1.52 1.42 1.56 2.92 3.01 1.81 1.56 1.75 2.04 2.48 1.63 2.31 3.23 2.10 1.81 2.53 1.89 2.35 2.09 1.99 1.57 2.30 1.93 1.68 2.83 2.20 2.41 3.11 2.22 2.66 1.44 3.79 2.01 2.48 1.77 1.94 2.56 2.28 2.47 2.56 2.23 1.86 2.41 1.58 1.97 0.67
 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 276 chr1:234951364-234954011 -1.57 951 chr17:2262369-2263121 -2.19 2.33 2.20 2.62 2.15 1.71 0.84 0.69 2.08 2.41 2.40 2.36 0.76 2.51 2.70 2.11 3.16 2.21 2.22 3.15 2.22 2.02 2.68 3.71 1.28 1.81 1.72 2.72 2.13 2.93 2.31 1.74 1.65 0.78 2.46 2.02 2.49 1.81 3.20 1.58 2.09 1.86 1.61 1.69 2.31 2.14
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 276 chr1:234951364-234954011 -1.57 951 chr17:2262369-2263121 -2.19 2.33 2.20 2.62 2.15 1.71 0.84 0.69 2.08 2.41 2.40 2.36 0.76 2.51 2.70 2.11 3.16 2.21 2.22 3.15 2.22 2.02 2.68 3.71 1.28 1.81 1.72 2.72 2.13 2.93 2.31 1.74 1.65 0.78 2.46 2.02 2.49 1.81 3.20 1.58 2.09 1.86 1.61 1.69 2.31 2.14
 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 322 chr10:28615271-28616909 -2.14 997 chr17:30821447-30823285 -2.49 2.16 1.73 2.55 1.58 1.75 1.23 1.65 1.86 1.75 1.87 2.29 2.42 3.68 2.41 2.90 2.61 1.97 2.04 2.28 2.44 2.15 1.94 2.17 2.03 1.62 2.18 1.74 1.48 2.25 2.33 0.83 0.82 1.04 1.50 1.69 0.74 2.17 1.73 1.44 2.13 2.03 1.36 1.36 2.66 1.60
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 322 chr10:28615271-28616909 -2.14 997 chr17:30821447-30823285 -2.49 2.16 1.73 2.55 1.58 1.75 1.23 1.65 1.86 1.75 1.87 2.29 2.42 3.68 2.41 2.90 2.61 1.97 2.04 2.28 2.44 2.15 1.94 2.17 2.03 1.62 2.18 1.74 1.48 2.25 2.33 0.83 0.82 1.04 1.50 1.69 0.74 2.17 1.73 1.44 2.13 2.03 1.36 1.36 2.66 1.60
 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 368 chr10:82258115-82259240 -1.54 1043 chr17:56064075-56066314 0.72 0.70 1.89 0.97 1.04 0.81 0.73 1.38 0.82 1.68 2.16 2.60 2.26 2.43 1.75 2.56 2.28 3.18 1.76 1.66 2.19 1.56 2.59 2.26 2.38 1.99 2.14 2.69 2.39 1.33 2.36 2.30 2.79 2.30 2.15 2.12 1.76 2.19 2.43 2.54 2.20 2.80 2.72 2.46 1.75 2.03
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 368 chr10:82258115-82259240 -1.54 1043 chr17:56064075-56066314 0.72 0.70 1.89 0.97 1.04 0.81 0.73 1.38 0.82 1.68 2.16 2.60 2.26 2.43 1.75 2.56 2.28 3.18 1.76 1.66 2.19 1.56 2.59 2.26 2.38 1.99 2.14 2.69 2.39 1.33 2.36 2.30 2.79 2.30 2.15 2.12 1.76 2.19 2.43 2.54 2.20 2.80 2.72 2.46 1.75 2.03
 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 414 chr10:134807613-134808276 -1.78 1089 chr17:78020665-78021305 2.68 2.58 2.51 1.84 1.75 2.50 2.37 3.57 1.80 1.32 1.57 1.80 1.68 1.28 1.95 2.03 2.29 2.49 3.77 2.05 2.08 1.78 2.64 2.18 2.60 2.18 2.07 1.77 1.81 1.70 1.59 2.11 2.69 2.24 2.69 2.77 2.29 1.69 2.20 2.76 2.24 3.06 2.33 4.33 1.98 1.99
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 414 chr10:134807613-134808276 -1.78 1089 chr17:78020665-78021305 2.68 2.58 2.51 1.84 1.75 2.50 2.37 3.57 1.80 1.32 1.57 1.80 1.68 1.28 1.95 2.03 2.29 2.49 3.77 2.05 2.08 1.78 2.64 2.18 2.60 2.18 2.07 1.77 1.81 1.70 1.59 2.11 2.69 2.24 2.69 2.77 2.29 1.69 2.20 2.76 2.24 3.06 2.33 4.33 1.98 1.99
 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 460 chr11:42456087-42456808 2.58 1135 chr18:38519431-38520110 -2.59 1.68 1.89 2.29 2.04 2.34 2.15 1.34 2.47 1.56 3.30 2.36 1.72 2.27 1.55 2.18 3.36 1.59 1.57 1.96 2.76 1.44 1.94 1.48 2.38 2.27 2.03 1.82 1.56 2.46 2.29 3.38 2.48 1.62 2.08 2.38 2.32 3.31 2.46 1.71 2.78 2.82 2.46 2.12 1.60 1.70
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 460 chr11:42456087-42456808 2.58 1135 chr18:38519431-38520110 -2.59 1.68 1.89 2.29 2.04 2.34 2.15 1.34 2.47 1.56 3.30 2.36 1.72 2.27 1.55 2.18 3.36 1.59 1.57 1.96 2.76 1.44 1.94 1.48 2.38 2.27 2.03 1.82 1.56 2.46 2.29 3.38 2.48 1.62 2.08 2.38 2.32 3.31 2.46 1.71 2.78 2.82 2.46 2.12 1.60 1.70
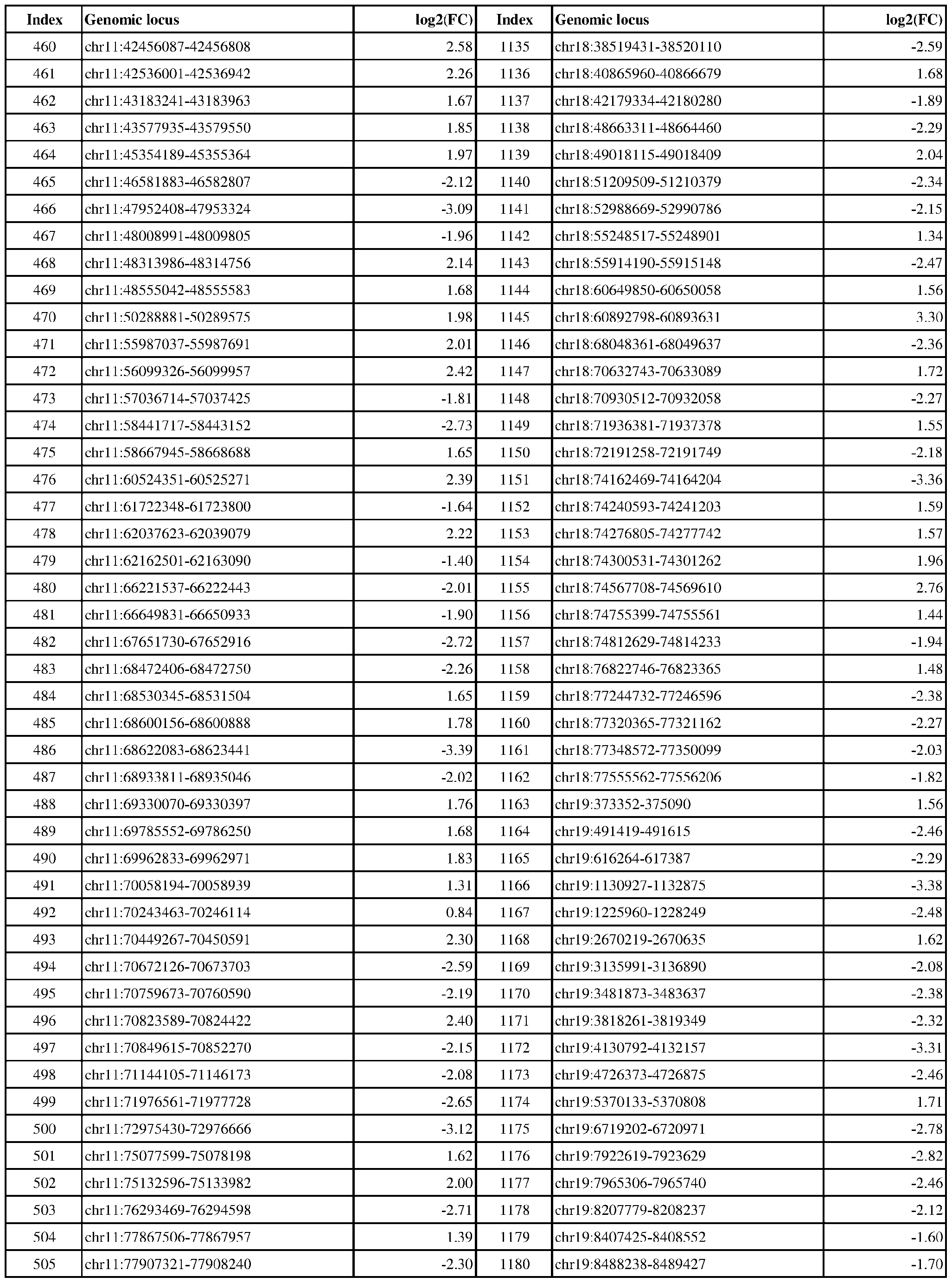 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 506 chr11:82931125-82931914 -2.38 1181 chr19:8656798-8658166 -2.13 2.20 3.60 3.32 2.68 4.04 2.62 2.26 2.11 3.83 2.96 2.21 2.56 1.52 2.05 1.54 2.85 2.03 2.45 1.93 2.44 1.86 2.46 2.20 1.90 3.25 2.20 2.60 2.94 2.26 2.01 1.53 1.92 1.63 2.11 1.28 1.63 1.53 1.62 2.58 3.27 2.22 2.17 2.14 0.66 2.76
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 506 chr11:82931125-82931914 -2.38 1181 chr19:8656798-8658166 -2.13 2.20 3.60 3.32 2.68 4.04 2.62 2.26 2.11 3.83 2.96 2.21 2.56 1.52 2.05 1.54 2.85 2.03 2.45 1.93 2.44 1.86 2.46 2.20 1.90 3.25 2.20 2.60 2.94 2.26 2.01 1.53 1.92 1.63 2.11 1.28 1.63 1.53 1.62 2.58 3.27 2.22 2.17 2.14 0.66 2.76
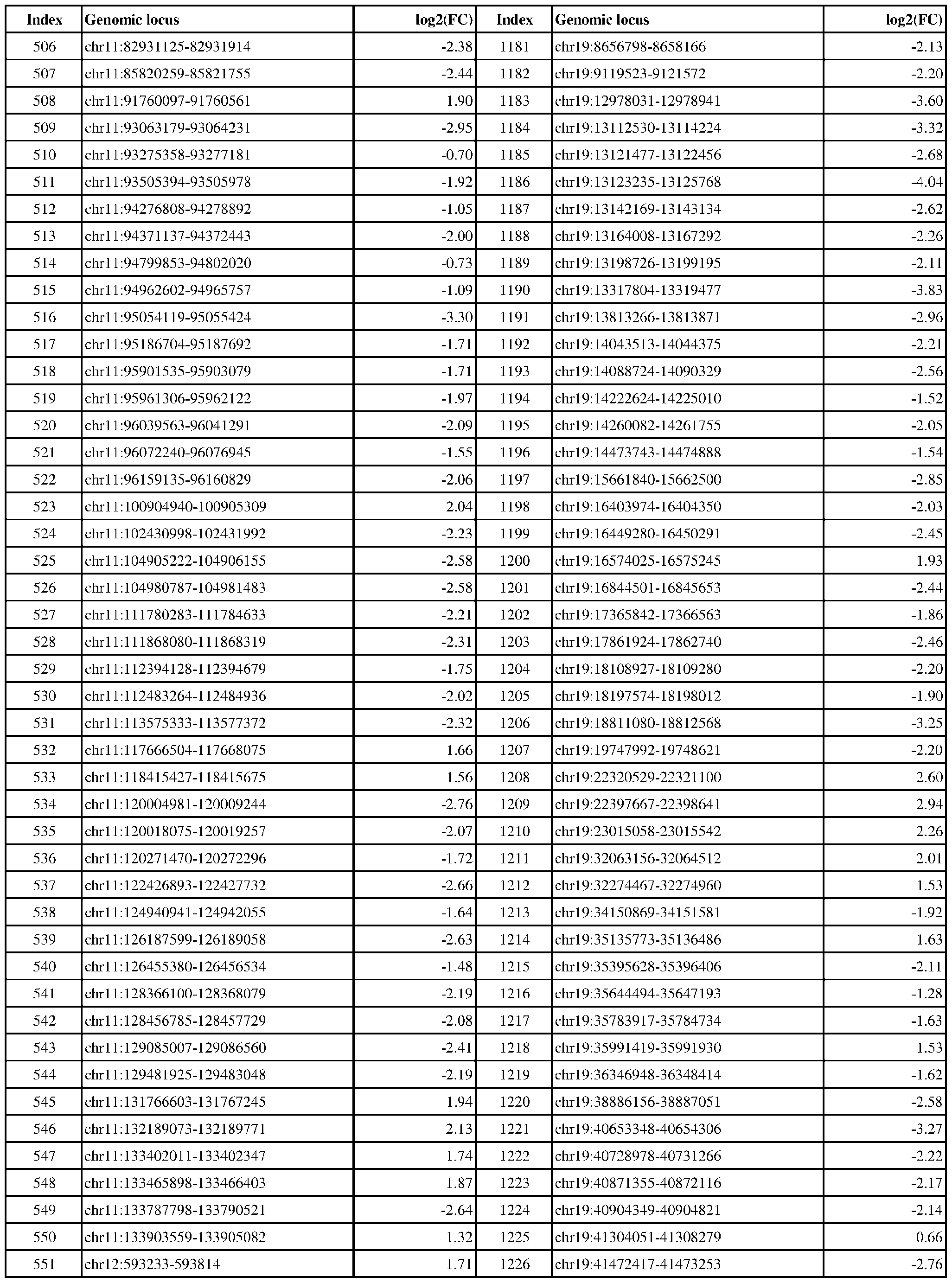 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 552 chr12:651365-652193 1.78 1227 chr19:41726518-41726899 -1.74 2.07 1.67 1.72 1.57 2.40 1.39 2.49 2.48 2.62 2.37 1.95 1.53 2.22 2.42 2.79 4.50 1.54 3.17 2.42 2.06 3.08 1.72 3.33 2.31 2.28 3.40 2.70 2.26 2.31 2.76 2.19 3.12 3.20 2.04 1.70 2.14 1.61 1.91 3.30 1.75 2.06 1.91 1.72 1.83 1.72
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 552 chr12:651365-652193 1.78 1227 chr19:41726518-41726899 -1.74 2.07 1.67 1.72 1.57 2.40 1.39 2.49 2.48 2.62 2.37 1.95 1.53 2.22 2.42 2.79 4.50 1.54 3.17 2.42 2.06 3.08 1.72 3.33 2.31 2.28 3.40 2.70 2.26 2.31 2.76 2.19 3.12 3.20 2.04 1.70 2.14 1.61 1.91 3.30 1.75 2.06 1.91 1.72 1.83 1.72
 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 598 chr12:58870833-58871314 1.85 1273 chr2:19555806-19558754 -2.13 1.84 1.75 2.33 2.05 2.02 2.47 2.77 2.00 1.93 1.68 1.01 2.52 2.16 2.83 1.97 2.85 2.12 2.17 2.07 2.81 3.37 2.40 2.92 2.80 2.64 4.59 1.99 1.73 1.79 2.20 2.49 2.99 2.46 2.95 1.30 2.15 1.59 1.67 2.00 1.57 1.97 1.52 2.88 2.23 2.85
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 598 chr12:58870833-58871314 1.85 1273 chr2:19555806-19558754 -2.13 1.84 1.75 2.33 2.05 2.02 2.47 2.77 2.00 1.93 1.68 1.01 2.52 2.16 2.83 1.97 2.85 2.12 2.17 2.07 2.81 3.37 2.40 2.92 2.80 2.64 4.59 1.99 1.73 1.79 2.20 2.49 2.99 2.46 2.95 1.30 2.15 1.59 1.67 2.00 1.57 1.97 1.52 2.88 2.23 2.85
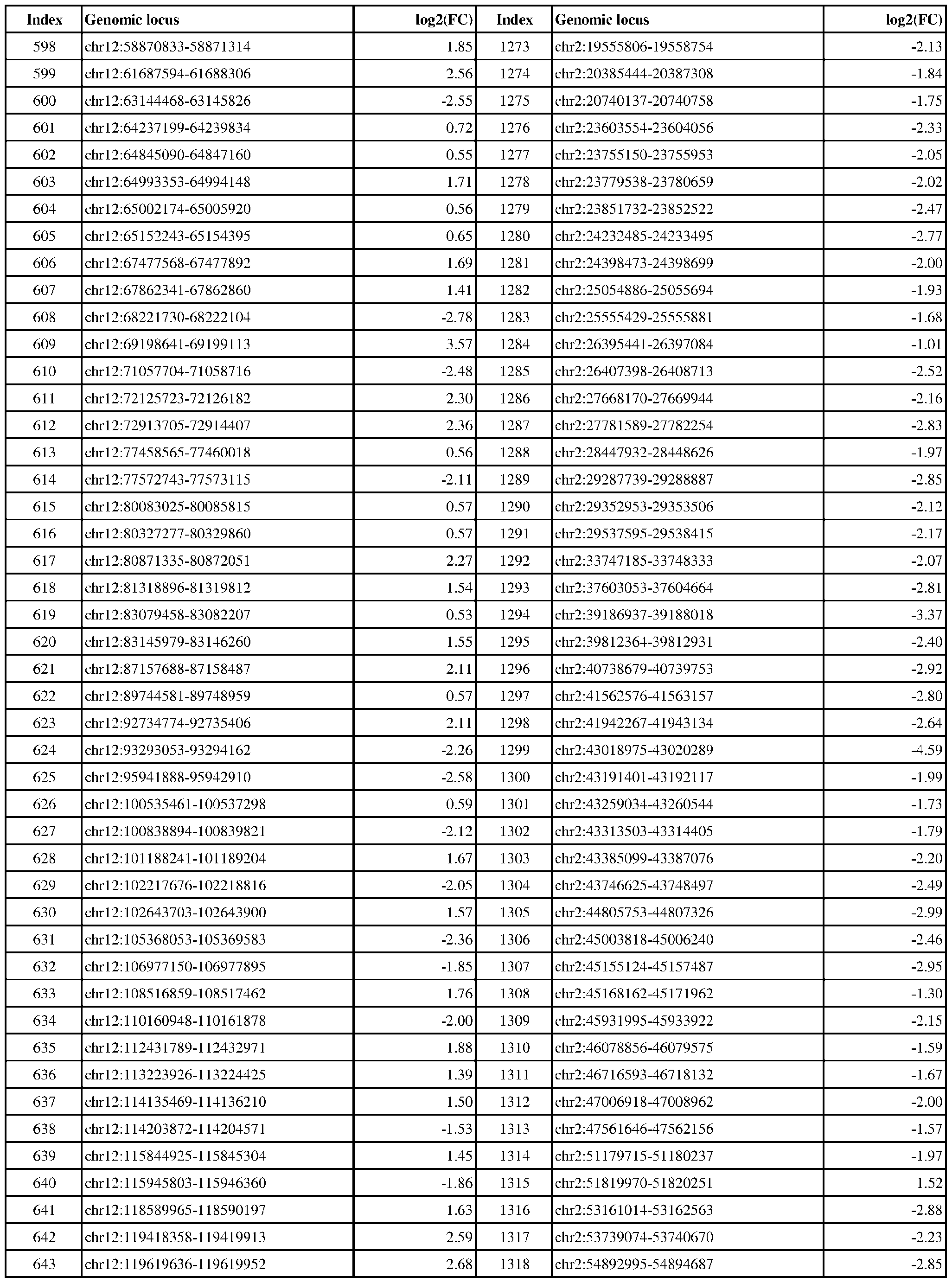 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 644 chr12:120124142-120124712 2.13 1319 chr2:54925268-54926492 -2.92 3.08 1.77 1.69 2.41 1.88 2.21 2.20 2.69 3.05 2.44 2.26 1.75 2.01 2.69 2.14 1.81 1.72 3.00 2.15 2.41 2.44 1.27 2.89 2.12 1.88 2.44 1.91 2.05 2.01 1.99 1.85
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 644 chr12:120124142-120124712 2.13 1319 chr2:54925268-54926492 -2.92 3.08 1.77 1.69 2.41 1.88 2.21 2.20 2.69 3.05 2.44 2.26 1.75 2.01 2.69 2.14 1.81 1.72 3.00 2.15 2.41 2.44 1.27 2.89 2.12 1.88 2.44 1.91 2.05 2.01 1.99 1.85

Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1351 chr2:74641740-74642946 -2.57 2026 chr5:102781317-102781692 1.52 .34 .83 .91 .52 .19 .85 .44 .15 .53 .99 .88 .92 .66 .63 .28 .50 .17 .63 .45 .97 .18 .55 .75 .01 .78 .43 .53 .48 .15 .27 .17 .71 .10 .67 .08 .51 .85 .40 .64 .42 .46 .67 .99 .95
 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1396 chr2:129017981-129019120 -1.82 2071 chr5:171022927-171023975 -1.89 .90 .08 .41 .55 .38 .18 .42 .27 .58 .23 .57 .10 .05 .90 .42 .29 .58 .64 .08 .91 .98 .47 .94 .09 .27 .91 .73 .34 .90 .67 .42 .62 .95 .22 .05 .96 .88 .16 .40 .97 .51 .44 .81 .48 .34
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1396 chr2:129017981-129019120 -1.82 2071 chr5:171022927-171023975 -1.89 .90 .08 .41 .55 .38 .18 .42 .27 .58 .23 .57 .10 .05 .90 .42 .29 .58 .64 .08 .91 .98 .47 .94 .09 .27 .91 .73 .34 .90 .67 .42 .62 .95 .22 .05 .96 .88 .16 .40 .97 .51 .44 .81 .48 .34
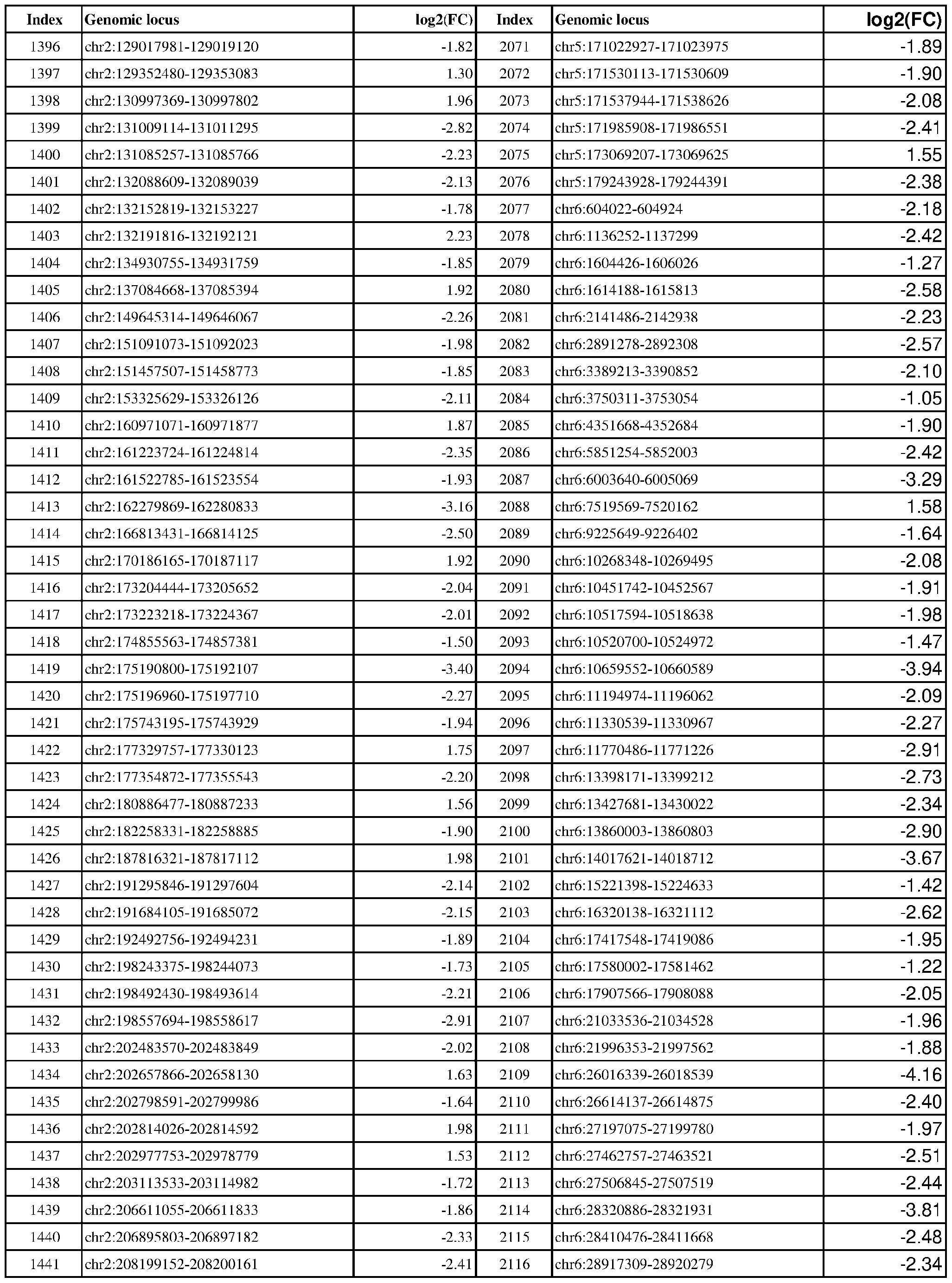 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1442 chr2:208546125-208546854 -3.24 2117 chr6:29647899-29649090 -2.43 .70 .59 .75 .03 .04 .25 .06 .70 .68 .59 .44 .04 .22 .52 .08 .96 .32 .62 .45 .88 .80 .99 .89 .24 .02 .13 .50 .35 .87 .05 .31 .75 .13 .84 .10 .11 .67 .83 .26 .93 .73 .67 .41 .71 .53
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1442 chr2:208546125-208546854 -3.24 2117 chr6:29647899-29649090 -2.43 .70 .59 .75 .03 .04 .25 .06 .70 .68 .59 .44 .04 .22 .52 .08 .96 .32 .62 .45 .88 .80 .99 .89 .24 .02 .13 .50 .35 .87 .05 .31 .75 .13 .84 .10 .11 .67 .83 .26 .93 .73 .67 .41 .71 .53
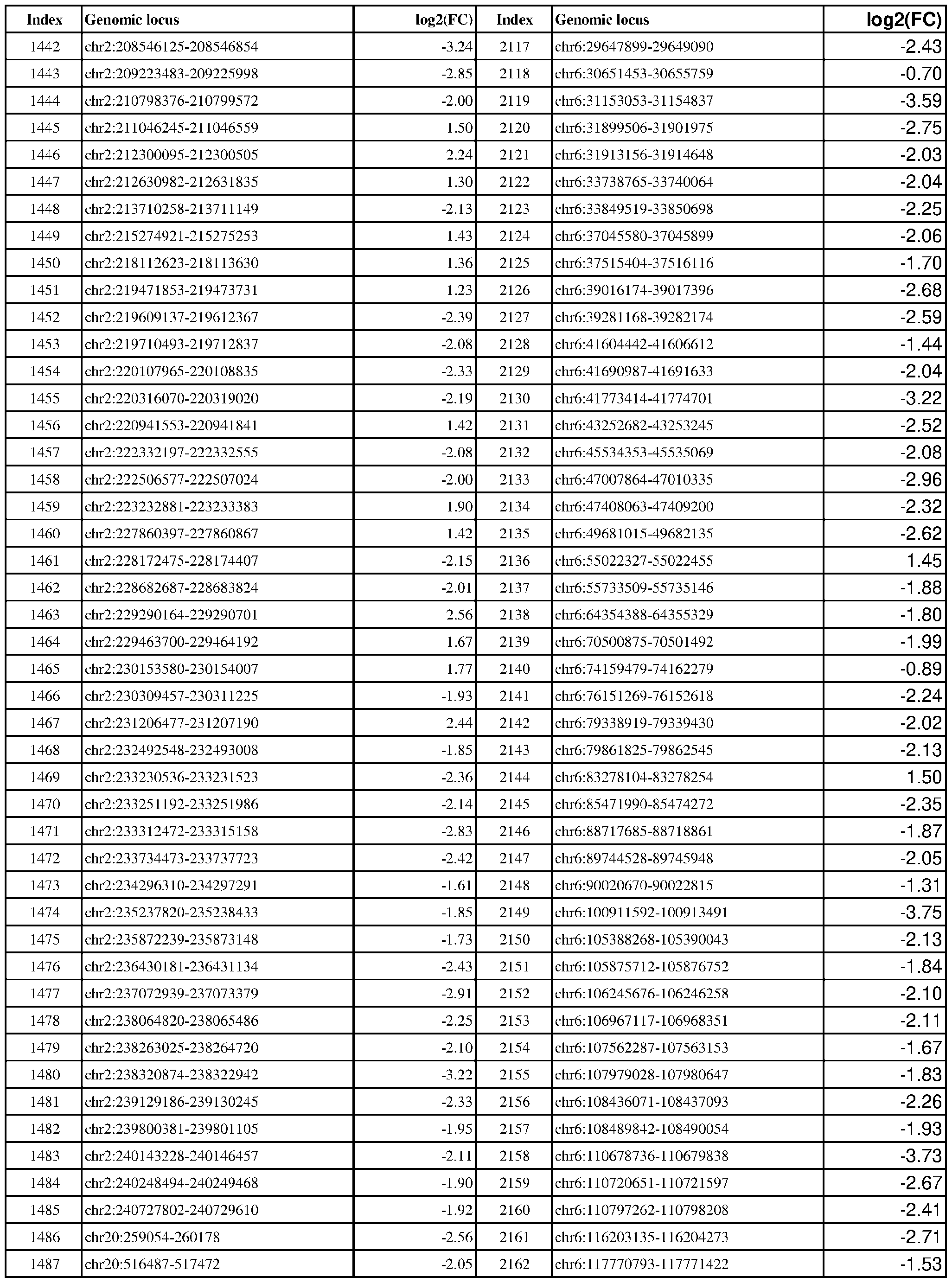 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1488 chr20:589571-591428 -2.15 2163 chr6:126239895-126242707 -2.01 .26 .83 .57 .87 .82 .29 .49 .19 .92 .89 .75 .94 .53 .62 .10 .43 .88 .54 .54 .83 .74 .19 .01 .04 .27 .29 .92 .00 .67 .37 .84 .75 .79 .99 .54 .74 .80 .19 .08 .56 .95 .37 .06 .47 .87
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1488 chr20:589571-591428 -2.15 2163 chr6:126239895-126242707 -2.01 .26 .83 .57 .87 .82 .29 .49 .19 .92 .89 .75 .94 .53 .62 .10 .43 .88 .54 .54 .83 .74 .19 .01 .04 .27 .29 .92 .00 .67 .37 .84 .75 .79 .99 .54 .74 .80 .19 .08 .56 .95 .37 .06 .47 .87
 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1534 chr20:44848451-44849784 -2.19 2209 chr6:166670235-166671484 -2.44 .64 .06 .70 .52 .11 .90 .02 .40 .24 .02 .59 .22 .98 .49 .44 .87 .64 .34 .94 .36 .97 .64 .51 .81 .64 .56 .79 .69 .33 .74 .36 .87 .88 .95 .55 .42 .71 .72 .40 .16 .77 .26 .40 .66 .14
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1534 chr20:44848451-44849784 -2.19 2209 chr6:166670235-166671484 -2.44 .64 .06 .70 .52 .11 .90 .02 .40 .24 .02 .59 .22 .98 .49 .44 .87 .64 .34 .94 .36 .97 .64 .51 .81 .64 .56 .79 .69 .33 .74 .36 .87 .88 .95 .55 .42 .71 .72 .40 .16 .77 .26 .40 .66 .14
 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1580 chr20:53992488-53993003 2.00 2255 chr7:27154735-27155536 -2.36 .17 .26 .29 .63 .95 .73 .88 .86 .55 .78 .91 .71 .58 .71 .52 .88 .96 .94 .89 .19 .10 .06 .38 .71 .84 .31 .15 .57 .05 .88 .11 .51 .04 .66 .13 .00 .55 .91 .88 .28 .74 .31 .51 .39 .88
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1580 chr20:53992488-53993003 2.00 2255 chr7:27154735-27155536 -2.36 .17 .26 .29 .63 .95 .73 .88 .86 .55 .78 .91 .71 .58 .71 .52 .88 .96 .94 .89 .19 .10 .06 .38 .71 .84 .31 .15 .57 .05 .88 .11 .51 .04 .66 .13 .00 .55 .91 .88 .28 .74 .31 .51 .39 .88
 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1626 chr21:35975262-35975736 -1.61 2301 chr7:100063026-100064041 -2.42 .18 .35 .98 .77 .73 .68 .59 .66 .31 .19 .77 .01 .00 .76 .06 .29 .95 .78 .97 .92 .72 .22 .27 .38 .78 .95 .96 .50 .94 .02 .69 .79 .34 .19 .50 .73 .67 .91 .15 .50 .97 .02 .65 .16 .79
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1626 chr21:35975262-35975736 -1.61 2301 chr7:100063026-100064041 -2.42 .18 .35 .98 .77 .73 .68 .59 .66 .31 .19 .77 .01 .00 .76 .06 .29 .95 .78 .97 .92 .72 .22 .27 .38 .78 .95 .96 .50 .94 .02 .69 .79 .34 .19 .50 .73 .67 .91 .15 .50 .97 .02 .65 .16 .79
 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1672 chr22:36044081-36046097 -1.43 2347 chr7:151553324-151554004 2.09 .56 .04 .79 .45 .82 .82 .60 .84 .05 .05 .02 .43 .83 .44 .86 .69 .94 .16 .92 .78 .52 .30 .97 .41 .11 .01 .23 .27 .62 .38 .83 .92 .19 .01 .86 .01 .93 .26 .76 .19 .57 .20 .68 .66 .07
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1672 chr22:36044081-36046097 -1.43 2347 chr7:151553324-151554004 2.09 .56 .04 .79 .45 .82 .82 .60 .84 .05 .05 .02 .43 .83 .44 .86 .69 .94 .16 .92 .78 .52 .30 .97 .41 .11 .01 .23 .27 .62 .38 .83 .92 .19 .01 .86 .01 .93 .26 .76 .19 .57 .20 .68 .66 .07
 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1718 chr22:46828228-46828802 -3.14 2393 chr8:37317841-37318640 2.08 .38 .07 .15 .29 .36 .44 .92 .68 .70 .52 .94 .18 .54 .11 .88 .49 .68 .62 .91 .08 .23 .70 .59 .71 .79 .67 .68 .86 .57 .84 .73 .18 .47 .67 .83 .09 .05 .88 .77 .79 .16 .17 .70 .53 .15
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1718 chr22:46828228-46828802 -3.14 2393 chr8:37317841-37318640 2.08 .38 .07 .15 .29 .36 .44 .92 .68 .70 .52 .94 .18 .54 .11 .88 .49 .68 .62 .91 .08 .23 .70 .59 .71 .79 .67 .68 .86 .57 .84 .73 .18 .47 .67 .83 .09 .05 .88 .77 .79 .16 .17 .70 .53 .15
 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1764 chr3:65582291-65583997 -2.73 2439 chr8:56831617-56833144 -2.19 .19 .23 .48 .97 .21 .87 .55 .22 .32 .37 .94 .27 .12 .63 .21 .18 .34 .79 .55 .06 .50 .14 .06 .56 .10 .94 .53 .92 .08 .56 .65 .19 .36 .58 .39 .32 .49 .07 .29 .43 .69 .37 .83 .39 .94
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1764 chr3:65582291-65583997 -2.73 2439 chr8:56831617-56833144 -2.19 .19 .23 .48 .97 .21 .87 .55 .22 .32 .37 .94 .27 .12 .63 .21 .18 .34 .79 .55 .06 .50 .14 .06 .56 .10 .94 .53 .92 .08 .56 .65 .19 .36 .58 .39 .32 .49 .07 .29 .43 .69 .37 .83 .39 .94
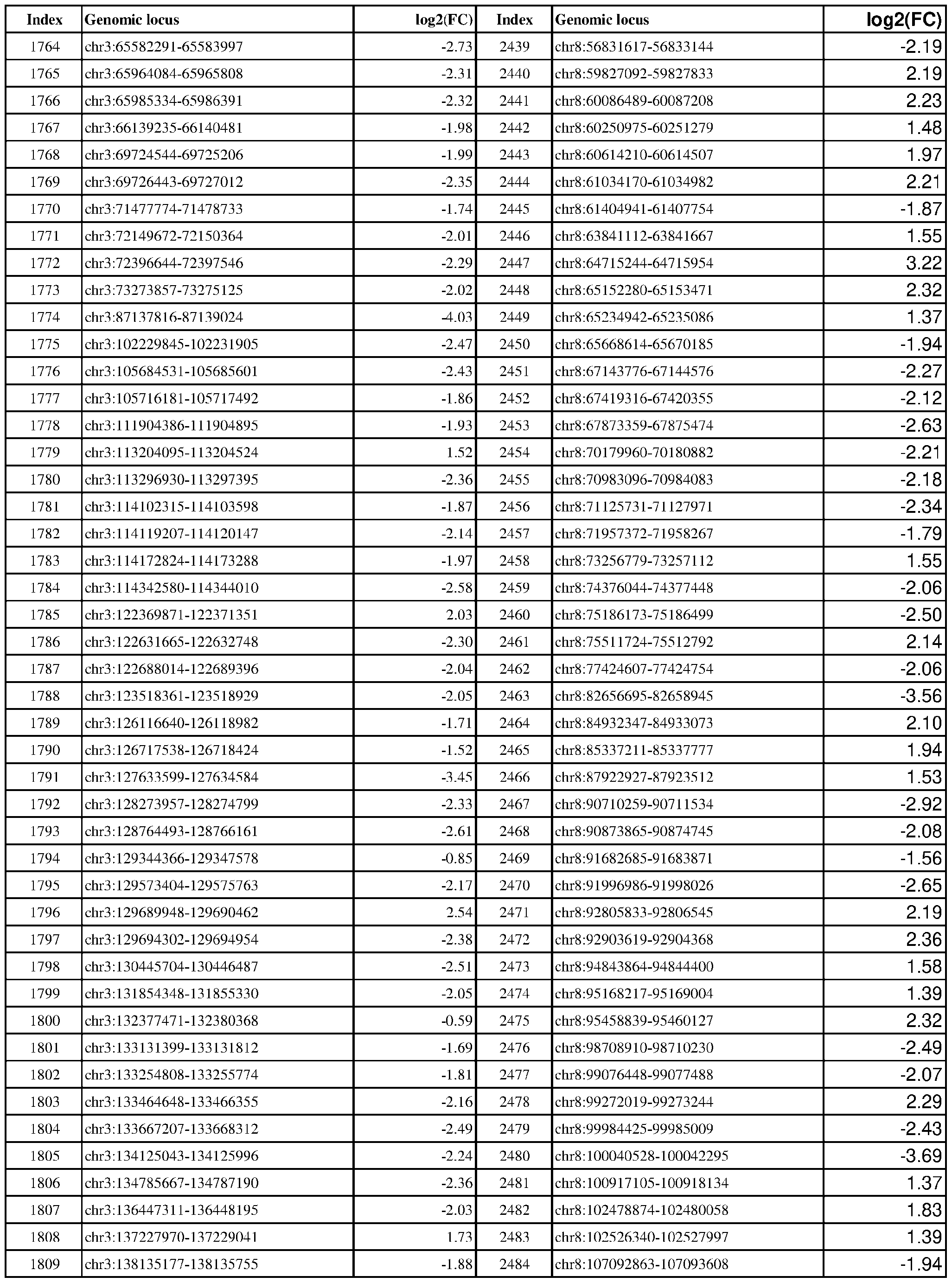 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1810 chr3:138152532-138155082 -1.34 2485 chr8:107511620-107513918 -2.94 .34 .09 .62 .50 .04 .35 .06 .98 .00 .90 .93 .24 .93 .77 .39 .54 .30 .35 .05 .56 .10 .56 .76 .82 .48 .88 .91 .81 .27 .34 .98 .82 .54 .89 .34 .95 .30 .18 .81 .24 .82 .74 .91 .97 .61
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1810 chr3:138152532-138155082 -1.34 2485 chr8:107511620-107513918 -2.94 .34 .09 .62 .50 .04 .35 .06 .98 .00 .90 .93 .24 .93 .77 .39 .54 .30 .35 .05 .56 .10 .56 .76 .82 .48 .88 .91 .81 .27 .34 .98 .82 .54 .89 .34 .95 .30 .18 .81 .24 .82 .74 .91 .97 .61
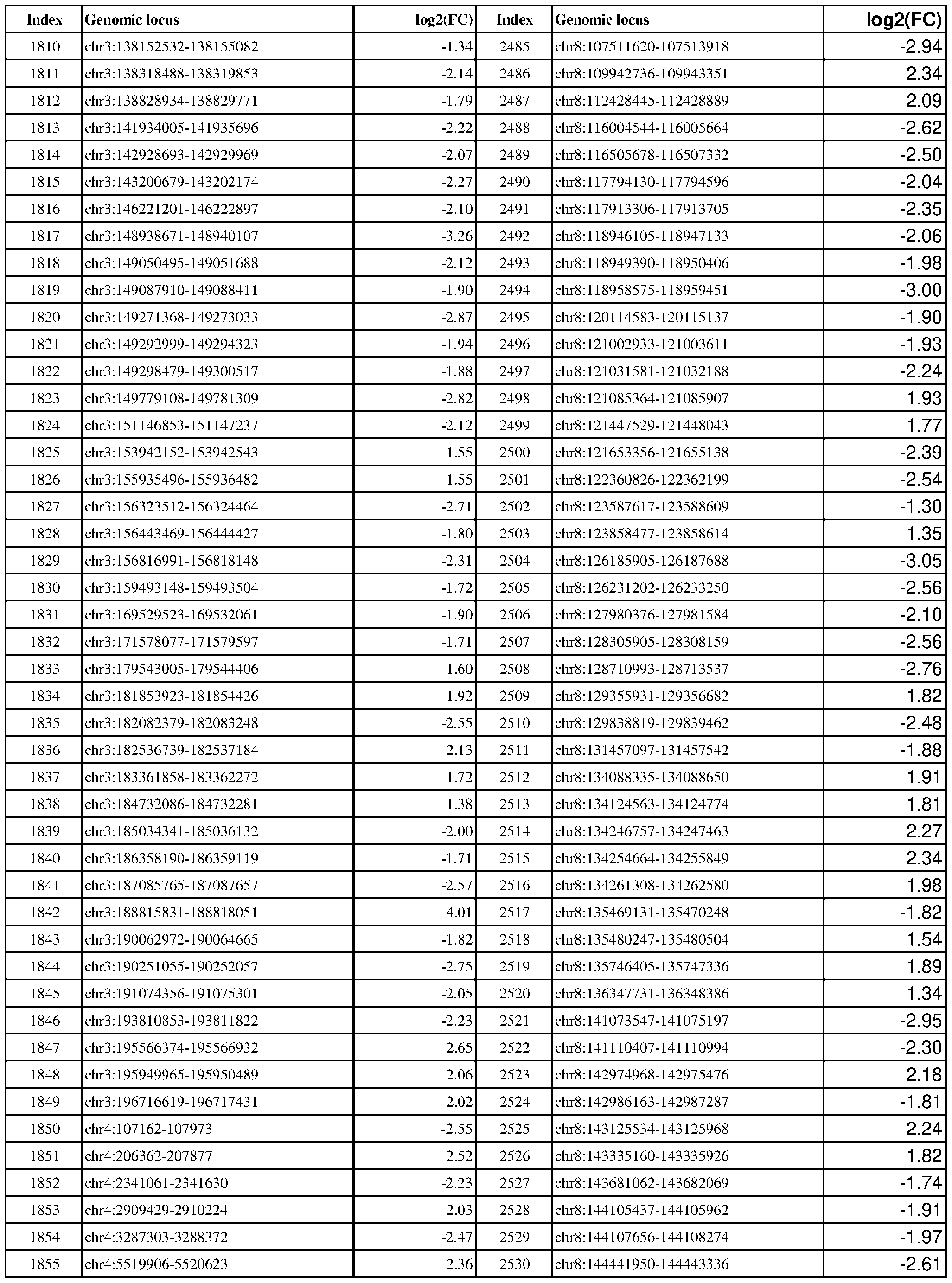 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1856 chr4:5595429-5596184 1.74 2531 chr8:144965238-144966443 -2.35 .73 .25 .10 .91 .33 .15 .00 .24 .40 .86 .83 .02 .36 .25 .78 .97 .89 .60 .35 .84 .39 .19 .89 .33 .26 .53 .40 .48 .13 .14 .30 .34 .70 .07 .38 .73 .72 .95 .86 .02 .82 .69 .89 .95 .44
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1856 chr4:5595429-5596184 1.74 2531 chr8:144965238-144966443 -2.35 .73 .25 .10 .91 .33 .15 .00 .24 .40 .86 .83 .02 .36 .25 .78 .97 .89 .60 .35 .84 .39 .19 .89 .33 .26 .53 .40 .48 .13 .14 .30 .34 .70 .07 .38 .73 .72 .95 .86 .02 .82 .69 .89 .95 .44
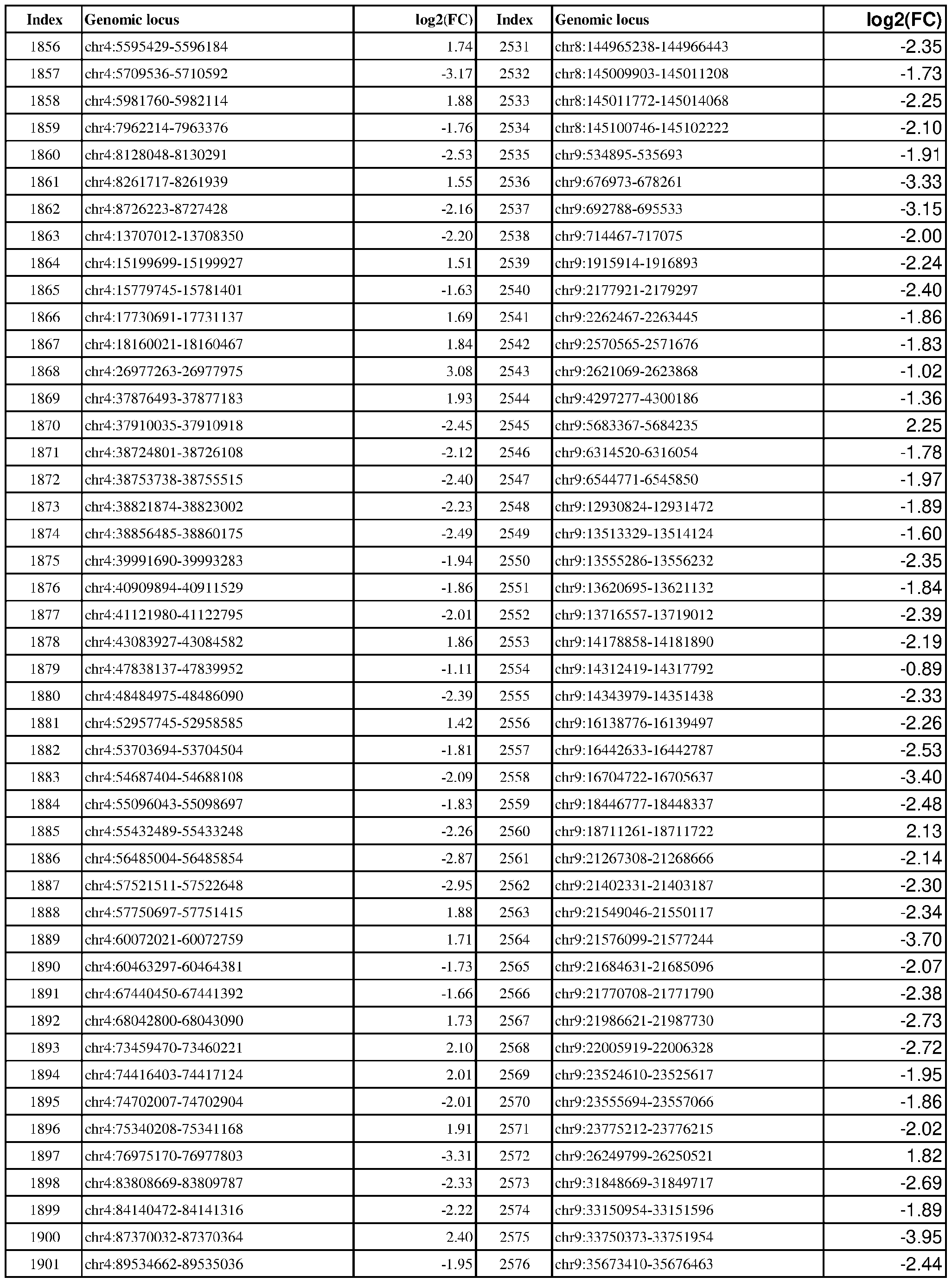 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1902 chr4:93404260-93404471 1.29 2577 chr9:35961997-35963090 -2.23 .00 .86 .48 .10 .34 .44 .72 .91 .28 .28 .19 .55 .68 .98 .05 .78 .85 .40 .16 .30 .89 .03 .18 .28 .94 .10 .61 .73 .85 .05 .29 .50 .04 .07 .50 .65 .97 .28 .17 .46 .41 .94 .43 .86 .96
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1902 chr4:93404260-93404471 1.29 2577 chr9:35961997-35963090 -2.23 .00 .86 .48 .10 .34 .44 .72 .91 .28 .28 .19 .55 .68 .98 .05 .78 .85 .40 .16 .30 .89 .03 .18 .28 .94 .10 .61 .73 .85 .05 .29 .50 .04 .07 .50 .65 .97 .28 .17 .46 .41 .94 .43 .86 .96
 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1948 chr4:186535777-186536188 2.14 2623 chr9:129379042-129379598 2.14 .66 .55 .47 .59 .11 .44 .43 .05 .63 .10 .93 .67 .92 .29 .42 .28 .31 .38 .82 .89 .47 .86 .49 .06 .96 .82 .11 .57 .29 .86 .43 .78 .04 .02 .55 .45 .12 .03 .94 .50 .53 .74 .62 .78 .14
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1948 chr4:186535777-186536188 2.14 2623 chr9:129379042-129379598 2.14 .66 .55 .47 .59 .11 .44 .43 .05 .63 .10 .93 .67 .92 .29 .42 .28 .31 .38 .82 .89 .47 .86 .49 .06 .96 .82 .11 .57 .29 .86 .43 .78 .04 .02 .55 .45 .12 .03 .94 .50 .53 .74 .62 .78 .14
 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1994 chr5:36657653-36658883 -2.09 2669 chrX:36436618-36437039 1.61 .31 .13 .34 .56 .29 .08 .42 .05 .42 .89 .66 .08 .58 .05 .32 .54 .18 .92 .18 .66 .47 .76 .94 .26 .91 .25 .98 .95 .18 .90 .75 .28 .46 .24 .76 .89 .22 .37 .75 .69 .72 .08 .45 .95 .08
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1994 chr5:36657653-36658883 -2.09 2669 chrX:36436618-36437039 1.61 .31 .13 .34 .56 .29 .08 .42 .05 .42 .89 .66 .08 .58 .05 .32 .54 .18 .92 .18 .66 .47 .76 .94 .26 .91 .25 .98 .95 .18 .90 .75 .28 .46 .24 .76 .89 .22 .37 .75 .69 .72 .08 .45 .95 .08
 Attorney Docket No.2014191-0027 Table 2: Exemplary genomic loci that are differentially H3K27ac modified in ER-positive vs. ER-negative cancer. Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1 chr1:901178-903765 -1.14 701 chr14:60097023-60097669 1.94 0.67 1.01 1.99 2.81 2.28 1.26 1.05 0.71 1.61 2.71 0.95 0.65 1.46 1.69 1.98 1.56 1.42 2.20 1.96 0.71 0.59 1.38 1.12 1.18 1.57 2.11 0.86 1.35 1.73 0.85 2.14 1.68 2.08 1.42 1.79 1.72 2.19 3.01 1.11 1.42 1.75 2.48 1.73 1.25
Attorney Docket No.2014191-0027 Table 2: Exemplary genomic loci that are differentially H3K27ac modified in ER-positive vs. ER-negative cancer. Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1 chr1:901178-903765 -1.14 701 chr14:60097023-60097669 1.94 0.67 1.01 1.99 2.81 2.28 1.26 1.05 0.71 1.61 2.71 0.95 0.65 1.46 1.69 1.98 1.56 1.42 2.20 1.96 0.71 0.59 1.38 1.12 1.18 1.57 2.11 0.86 1.35 1.73 0.85 2.14 1.68 2.08 1.42 1.79 1.72 2.19 3.01 1.11 1.42 1.75 2.48 1.73 1.25
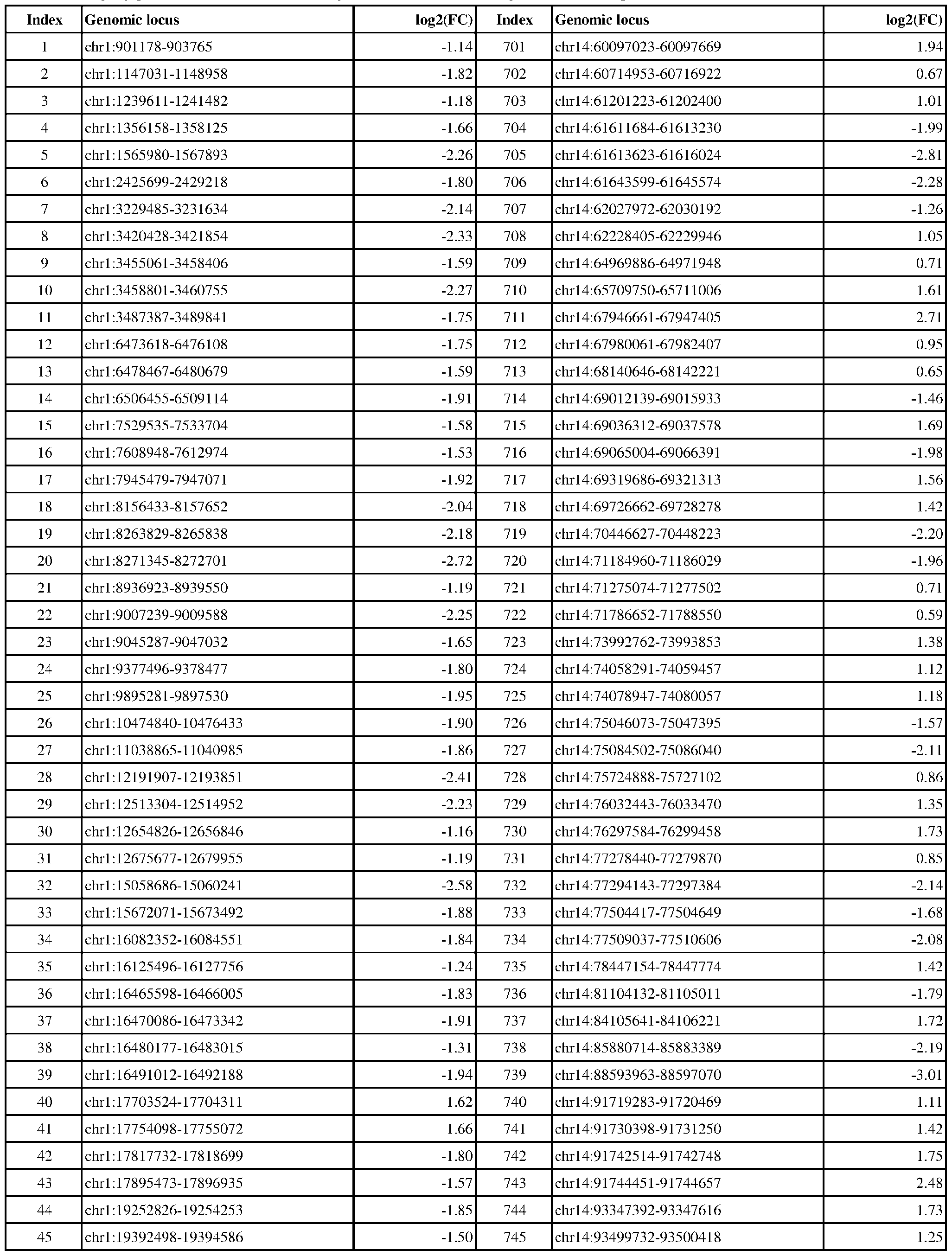 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 46 chr1:19600109-19601084 -2.17 746 chr14:94576232-94578236 -1.97 1.84 1.47 1.85 0.85 1.57 1.54 1.69 1.80 1.36 1.56 2.35 2.09 2.37 1.68 0.77 2.10 0.59 1.99 1.03 2.13 1.05 0.93 2.62 1.90 1.88 3.04 1.88 1.80 1.80 2.03 1.86 1.42 1.42 1.01 0.95 1.30 1.75 1.26 0.64 1.95 0.83 2.35 2.12 2.83 2.23
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 46 chr1:19600109-19601084 -2.17 746 chr14:94576232-94578236 -1.97 1.84 1.47 1.85 0.85 1.57 1.54 1.69 1.80 1.36 1.56 2.35 2.09 2.37 1.68 0.77 2.10 0.59 1.99 1.03 2.13 1.05 0.93 2.62 1.90 1.88 3.04 1.88 1.80 1.80 2.03 1.86 1.42 1.42 1.01 0.95 1.30 1.75 1.26 0.64 1.95 0.83 2.35 2.12 2.83 2.23
 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 92 chr1:48679557-48682538 -2.01 792 chr15:63085966-63087828 -2.22 2.04 0.88 1.60 0.73 0.81 1.67 1.74 1.91 2.46 2.30 1.68 2.13 2.06 2.22 2.19 2.04 2.58 1.71 3.36 0.91 1.22 1.51 1.19 2.30 1.96 1.34 1.78 1.70 2.32 1.99 2.46 2.42 2.02 1.61 2.03 1.97 2.14 2.14 2.53 1.77 2.07 1.50 2.60 1.18 2.05
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 92 chr1:48679557-48682538 -2.01 792 chr15:63085966-63087828 -2.22 2.04 0.88 1.60 0.73 0.81 1.67 1.74 1.91 2.46 2.30 1.68 2.13 2.06 2.22 2.19 2.04 2.58 1.71 3.36 0.91 1.22 1.51 1.19 2.30 1.96 1.34 1.78 1.70 2.32 1.99 2.46 2.42 2.02 1.61 2.03 1.97 2.14 2.14 2.53 1.77 2.07 1.50 2.60 1.18 2.05
 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 138 chr1:100511730-100513526 -1.96 838 chr15:101666006-101669439 -2.13 1.66 2.32 1.44 1.41 1.47 0.56 1.58 1.47 1.84 1.52 1.95 1.44 1.51 1.77 2.41 1.88 2.27 1.68 2.25 2.52 1.90 2.03 1.91 2.00 1.71 2.74 2.39 2.40 2.25 0.99 1.60 2.36 1.82 2.70 1.79 1.70 1.47 2.42 1.84 2.15 2.02 3.55 2.34 2.52 2.26
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 138 chr1:100511730-100513526 -1.96 838 chr15:101666006-101669439 -2.13 1.66 2.32 1.44 1.41 1.47 0.56 1.58 1.47 1.84 1.52 1.95 1.44 1.51 1.77 2.41 1.88 2.27 1.68 2.25 2.52 1.90 2.03 1.91 2.00 1.71 2.74 2.39 2.40 2.25 0.99 1.60 2.36 1.82 2.70 1.79 1.70 1.47 2.42 1.84 2.15 2.02 3.55 2.34 2.52 2.26
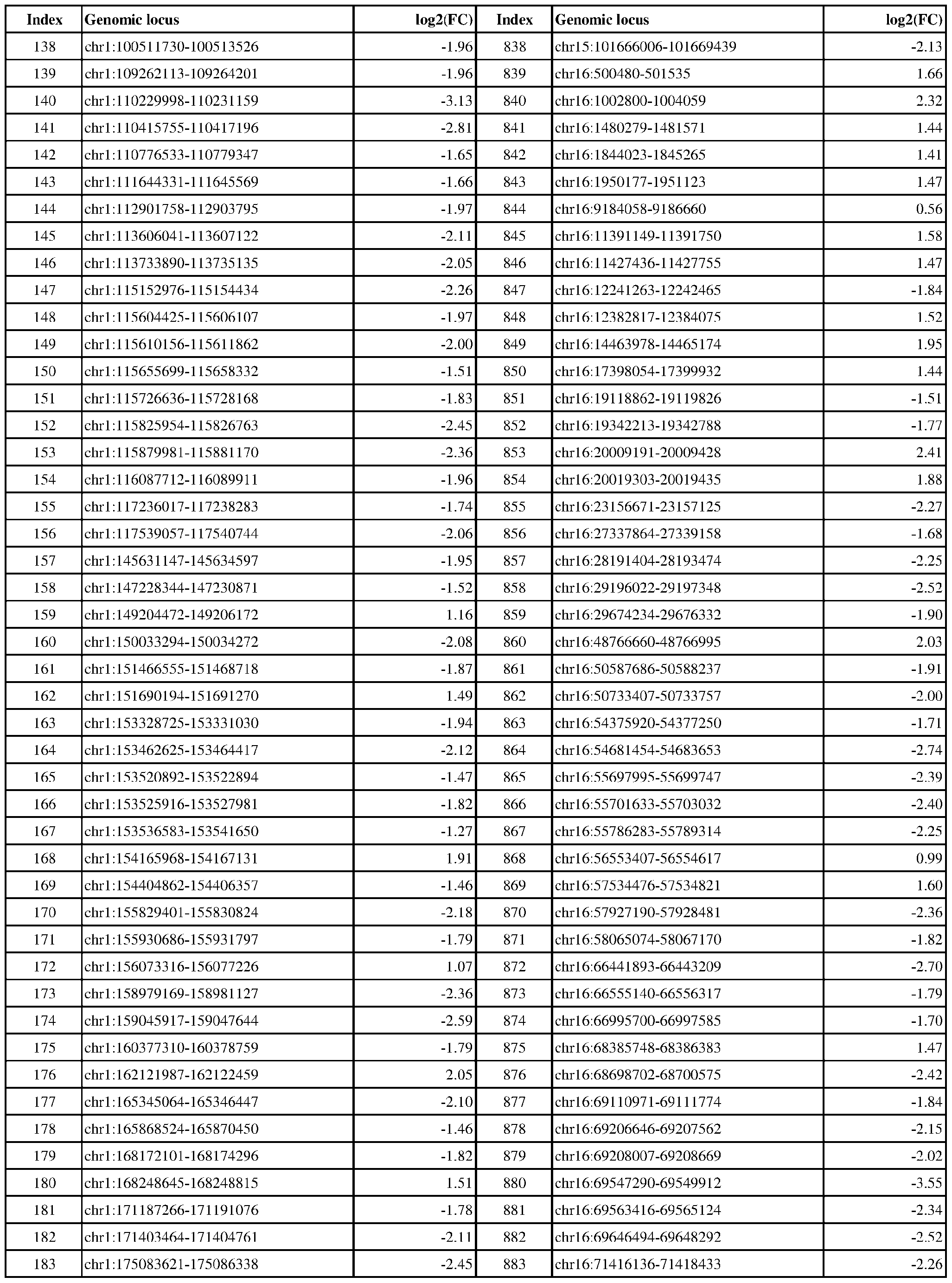 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 184 chr1:176964732-176966659 -2.33 884 chr16:72908342-72910349 -2.12 2.28 2.00 2.25 1.99 1.51 2.99 1.88 2.12 1.95 2.38 2.13 1.71 2.28 2.34 2.18 2.71 1.82 1.89 2.05 1.88 1.28 1.68 2.52 2.21 2.30 2.02 1.96 2.31 2.80 2.34 1.88 1.92 1.97 1.55 0.80 2.12 2.96 2.38 2.80 2.02 1.89 2.85 1.87 1.84 2.14
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 184 chr1:176964732-176966659 -2.33 884 chr16:72908342-72910349 -2.12 2.28 2.00 2.25 1.99 1.51 2.99 1.88 2.12 1.95 2.38 2.13 1.71 2.28 2.34 2.18 2.71 1.82 1.89 2.05 1.88 1.28 1.68 2.52 2.21 2.30 2.02 1.96 2.31 2.80 2.34 1.88 1.92 1.97 1.55 0.80 2.12 2.96 2.38 2.80 2.02 1.89 2.85 1.87 1.84 2.14
 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 230 chr1:222460576-222462480 -2.97 930 chr17:15868689-15870524 -1.99 2.58 2.44 2.19 2.39 1.44 2.86 2.55 1.15 1.01 0.92 1.88 1.46 1.80 0.82 1.94 1.70 2.02 2.40 0.84 1.89 2.38 2.83 2.07 2.15 1.00 0.82 1.44 1.82 1.90 1.34 2.24 1.71 1.31 1.20 1.29 2.01 1.57 1.57 1.59 2.40 2.96 2.63 3.09 2.30 2.71
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 230 chr1:222460576-222462480 -2.97 930 chr17:15868689-15870524 -1.99 2.58 2.44 2.19 2.39 1.44 2.86 2.55 1.15 1.01 0.92 1.88 1.46 1.80 0.82 1.94 1.70 2.02 2.40 0.84 1.89 2.38 2.83 2.07 2.15 1.00 0.82 1.44 1.82 1.90 1.34 2.24 1.71 1.31 1.20 1.29 2.01 1.57 1.57 1.59 2.40 2.96 2.63 3.09 2.30 2.71
 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 276 chr10:4395901-4397300 -2.50 976 chr17:38672943-38675927 -2.00 1.85 3.02 1.90 1.85 1.08 2.19 1.28 2.76 2.06 1.32 0.80 1.34 1.59 1.21 1.23 2.12 2.15 2.02 2.25 2.07 2.14 2.24 2.43 2.08 1.66 1.33 1.22 2.26 1.32 1.11 1.43 1.76 2.05 1.15 1.18 1.08 0.75 1.46 1.48 1.91 1.73 3.34 1.83 2.23 2.39
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 276 chr10:4395901-4397300 -2.50 976 chr17:38672943-38675927 -2.00 1.85 3.02 1.90 1.85 1.08 2.19 1.28 2.76 2.06 1.32 0.80 1.34 1.59 1.21 1.23 2.12 2.15 2.02 2.25 2.07 2.14 2.24 2.43 2.08 1.66 1.33 1.22 2.26 1.32 1.11 1.43 1.76 2.05 1.15 1.18 1.08 0.75 1.46 1.48 1.91 1.73 3.34 1.83 2.23 2.39
 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 322 chr10:32034239-32035289 -1.98 1022 chr17:60885225-60885967 1.97 1.99 0.96 1.54 2.02 2.43 2.35 2.90 3.19 1.21 2.52 2.20 2.39 2.49 2.79 2.74 2.86 1.55 2.26 2.23 3.03 3.03 2.62 2.08 0.84 1.89 1.89 1.62 2.29 1.88 2.19 3.39 1.83 2.72 2.03 2.93 3.26 2.42 2.15 2.93 2.09 1.35 2.22 2.52 0.67 2.13
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 322 chr10:32034239-32035289 -1.98 1022 chr17:60885225-60885967 1.97 1.99 0.96 1.54 2.02 2.43 2.35 2.90 3.19 1.21 2.52 2.20 2.39 2.49 2.79 2.74 2.86 1.55 2.26 2.23 3.03 3.03 2.62 2.08 0.84 1.89 1.89 1.62 2.29 1.88 2.19 3.39 1.83 2.72 2.03 2.93 3.26 2.42 2.15 2.93 2.09 1.35 2.22 2.52 0.67 2.13
 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 368 chr10:95924636-95925614 1.37 1068 chr17:80744054-80748749 -5.21 2.01 1.95 2.49 1.77 1.69 2.49 1.42 2.32 1.61 1.03 1.73 2.27 3.02 1.87 3.26 2.13 1.89 2.68 2.90 1.99 1.71 1.45 1.56 3.32 2.82 2.00 2.52 1.98 2.20 1.56 2.57 1.98 1.85 1.77 1.82 2.32 2.22 2.06 1.85 2.29 1.65 2.31 1.65 2.27 1.92
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 368 chr10:95924636-95925614 1.37 1068 chr17:80744054-80748749 -5.21 2.01 1.95 2.49 1.77 1.69 2.49 1.42 2.32 1.61 1.03 1.73 2.27 3.02 1.87 3.26 2.13 1.89 2.68 2.90 1.99 1.71 1.45 1.56 3.32 2.82 2.00 2.52 1.98 2.20 1.56 2.57 1.98 1.85 1.77 1.82 2.32 2.22 2.06 1.85 2.29 1.65 2.31 1.65 2.27 1.92
 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 414 chr11:12111588-12113377 -1.97 1114 chr18:48663418-48664890 -1.96 2.26 2.02 2.13 1.82 2.08 2.06 2.34 2.92 1.92 2.85 1.48 1.81 2.22 2.31 1.57 3.31 3.64 1.50 2.16 2.61 2.39 2.09 1.66 1.35 1.10 2.23 1.86 1.53 1.91 1.87 3.95 4.28 2.97 3.78 1.92 3.32 2.25 2.30 2.01 2.32 1.90 2.43 2.27 2.03 1.87
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 414 chr11:12111588-12113377 -1.97 1114 chr18:48663418-48664890 -1.96 2.26 2.02 2.13 1.82 2.08 2.06 2.34 2.92 1.92 2.85 1.48 1.81 2.22 2.31 1.57 3.31 3.64 1.50 2.16 2.61 2.39 2.09 1.66 1.35 1.10 2.23 1.86 1.53 1.91 1.87 3.95 4.28 2.97 3.78 1.92 3.32 2.25 2.30 2.01 2.32 1.90 2.43 2.27 2.03 1.87
 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 460 chr11:68108776-68111461 1.06 1160 chr19:17217323-17219524 -1.79 2.13 2.16 2.76 2.57 1.79 2.25 1.33 1.63 2.10 2.64 1.10 2.03 2.23 2.06 1.11 1.43 1.82 1.63 1.87 2.09 1.57 2.38 2.08 2.47 1.77 2.26 1.88 3.20 1.32 1.61 1.87 2.87 1.78 3.23 3.67 1.75 1.86 2.06 3.15 2.39 1.63 3.63 0.91 2.44 1.73
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 460 chr11:68108776-68111461 1.06 1160 chr19:17217323-17219524 -1.79 2.13 2.16 2.76 2.57 1.79 2.25 1.33 1.63 2.10 2.64 1.10 2.03 2.23 2.06 1.11 1.43 1.82 1.63 1.87 2.09 1.57 2.38 2.08 2.47 1.77 2.26 1.88 3.20 1.32 1.61 1.87 2.87 1.78 3.23 3.67 1.75 1.86 2.06 3.15 2.39 1.63 3.63 0.91 2.44 1.73
 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 506 chr11:112483159-112485726 -3.04 1206 chr2:9303845-9307091 -1.49 2.82 1.41 2.12 2.38 1.82 2.15 1.85 1.88 1.77 1.72 0.74 2.02 1.69 1.99 1.46 1.81 2.22 1.58 1.48 2.58 2.42 2.12 1.90 2.47 1.85 2.44 2.25 1.77 1.70 1.42 1.22 1.26 1.02 1.57 2.11 1.94 2.35 3.13 2.98 2.05 2.17 2.13 2.39 2.23 2.43
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 506 chr11:112483159-112485726 -3.04 1206 chr2:9303845-9307091 -1.49 2.82 1.41 2.12 2.38 1.82 2.15 1.85 1.88 1.77 1.72 0.74 2.02 1.69 1.99 1.46 1.81 2.22 1.58 1.48 2.58 2.42 2.12 1.90 2.47 1.85 2.44 2.25 1.77 1.70 1.42 1.22 1.26 1.02 1.57 2.11 1.94 2.35 3.13 2.98 2.05 2.17 2.13 2.39 2.23 2.43
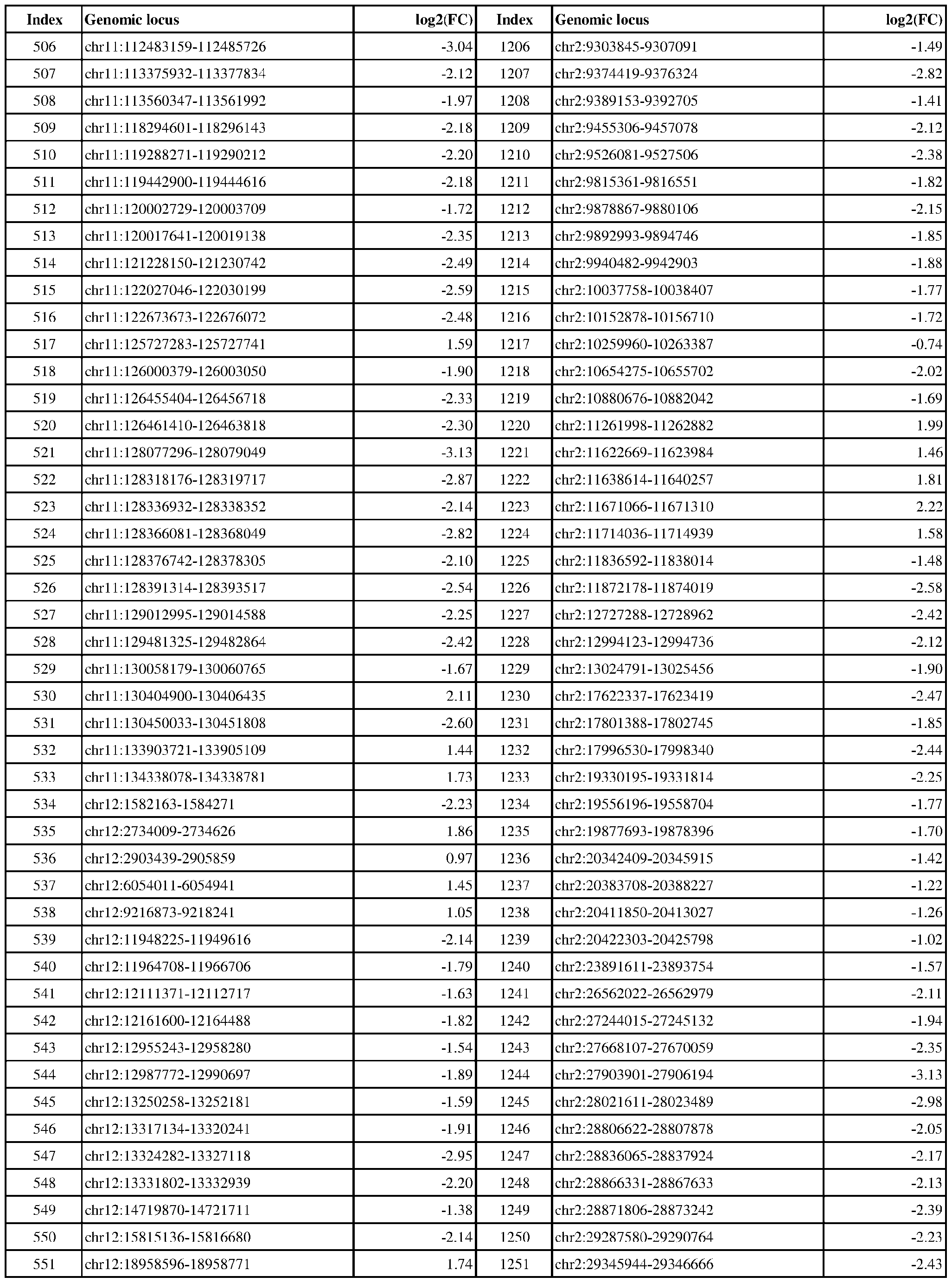 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 552 chr12:19318447-19318781 1.75 1252 chr2:30495754-30498168 -2.04 3.74 1.92 1.83 2.02 1.65 1.93 1.93 1.92 1.39 2.14 1.98 1.51 1.50 2.11 2.46 2.43 1.76 2.14 2.41 1.89 1.87 1.58 2.80 2.58 1.40 2.04 2.14 0.92 2.38 1.89 1.78 2.70 1.51 1.51 1.63 1.42 0.91 2.12 2.27 1.49 2.69 2.52 1.72 1.23 1.96
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 552 chr12:19318447-19318781 1.75 1252 chr2:30495754-30498168 -2.04 3.74 1.92 1.83 2.02 1.65 1.93 1.93 1.92 1.39 2.14 1.98 1.51 1.50 2.11 2.46 2.43 1.76 2.14 2.41 1.89 1.87 1.58 2.80 2.58 1.40 2.04 2.14 0.92 2.38 1.89 1.78 2.70 1.51 1.51 1.63 1.42 0.91 2.12 2.27 1.49 2.69 2.52 1.72 1.23 1.96
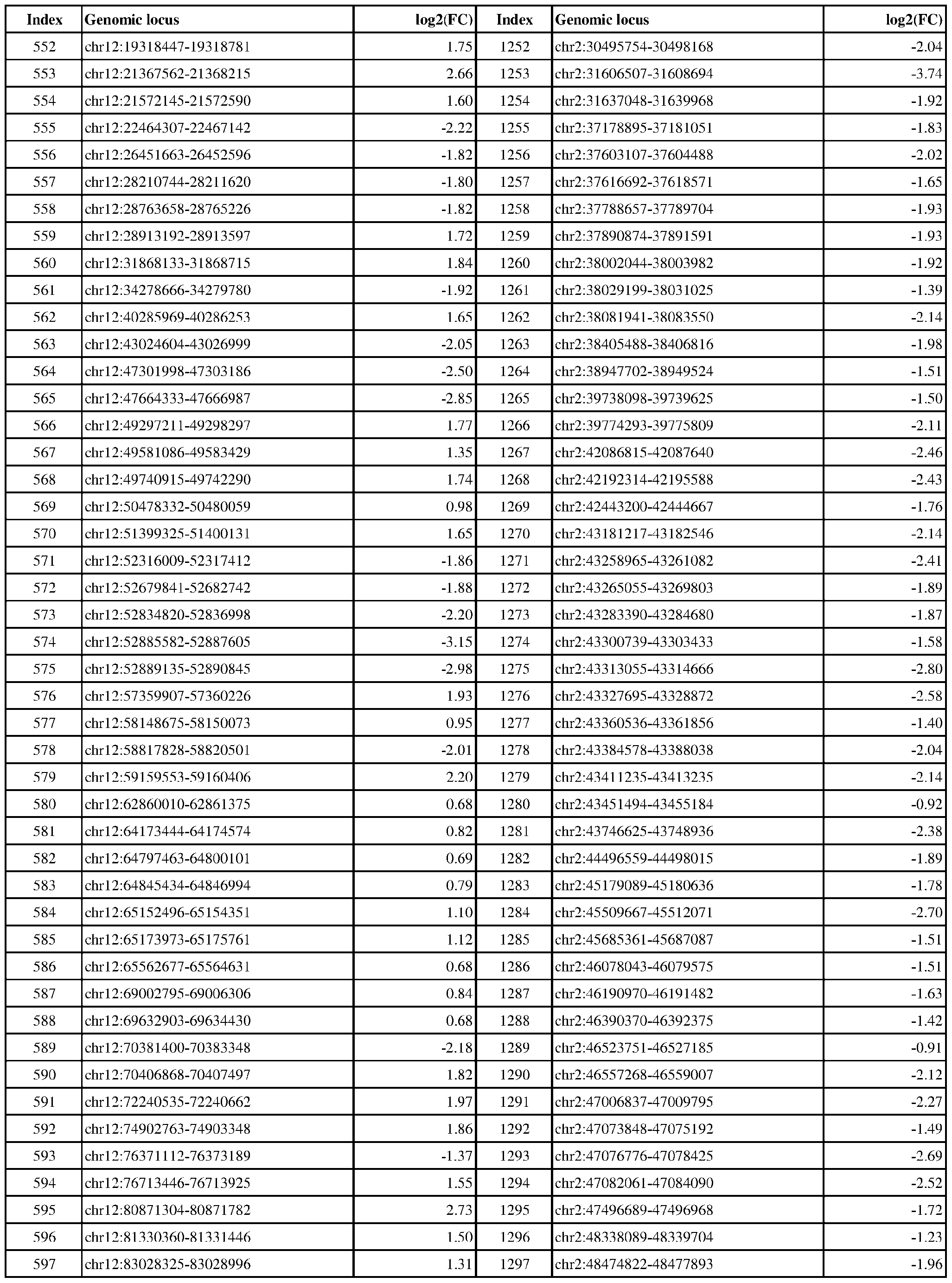 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 598 chr12:83325186-83326255 1.84 1298 chr2:53160084-53162665 -3.18 2.43 2.77 2.02 2.33 3.53 2.53 1.83 2.00 2.71 1.58 1.91 1.69 3.04 2.31 2.06 1.53 3.43 2.09 2.04 1.95 1.52 1.61 1.83 1.87 1.01 2.28 1.47 2.70 1.88 1.63 1.99 2.21 2.21 2.27 2.30 2.05 1.71 2.00 2.07 1.53 2.69 1.79 1.77 2.41 2.59
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 598 chr12:83325186-83326255 1.84 1298 chr2:53160084-53162665 -3.18 2.43 2.77 2.02 2.33 3.53 2.53 1.83 2.00 2.71 1.58 1.91 1.69 3.04 2.31 2.06 1.53 3.43 2.09 2.04 1.95 1.52 1.61 1.83 1.87 1.01 2.28 1.47 2.70 1.88 1.63 1.99 2.21 2.21 2.27 2.30 2.05 1.71 2.00 2.07 1.53 2.69 1.79 1.77 2.41 2.59
 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 644 chr13:45952580-45956649 -1.57 1344 chr2:75787100-75789938 -1.54 2.10 2.12 2.02 2.48 2.41 2.25 2.37 2.83 2.19 1.88 1.91 2.35 1.79 2.07 1.94 1.47 2.03 1.50 1.97 1.71 2.01 2.60 1.53 1.59 1.70 1.56 1.96 1.81 1.82 3.63 2.80 2.38 2.16 2.45 3.02 2.00 2.16 2.72 1.96 1.11 2.41 2.19 1.64 1.50 2.31
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 644 chr13:45952580-45956649 -1.57 1344 chr2:75787100-75789938 -1.54 2.10 2.12 2.02 2.48 2.41 2.25 2.37 2.83 2.19 1.88 1.91 2.35 1.79 2.07 1.94 1.47 2.03 1.50 1.97 1.71 2.01 2.60 1.53 1.59 1.70 1.56 1.96 1.81 1.82 3.63 2.80 2.38 2.16 2.45 3.02 2.00 2.16 2.72 1.96 1.11 2.41 2.19 1.64 1.50 2.31
 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 690 chr14:50777774-50779942 0.70 1390 chr2:128158210-128160046 -2.30 1.45 1.94 3.28 1.67 2.48 2.14 1.96 1.86 2.14 2.54
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 690 chr14:50777774-50779942 0.70 1390 chr2:128158210-128160046 -2.30 1.45 1.94 3.28 1.67 2.48 2.14 1.96 1.86 2.14 2.54

Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1401 chr2:131581394-131582654 -1.88 2101 chr5:140893579-140894455 -1.58 1.88 2.31 1.74 2.46 2.20 1.76 2.38 1.87 2.13 2.26 1.62 1.66 1.68 1.95 1.89 1.83 1.68 1.65 1.91 1.75 2.44 1.74 1.11 1.66 2.42 2.26 2.33 1.74 2.70 1.53 1.73 1.87 2.66 2.11 1.94 2.28 2.06 1.76 1.78 3.06 2.40 2.63 1.70 2.67
 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1446 chr2:192502051-192503535 -1.59 2146 chr6:1702314-1704615 -2.16 1.92 2.69 2.95 2.55 2.33 1.82 2.36 1.84 2.35 1.31 2.16 1.87 1.68 1.53 2.26 2.06 1.93 2.24 2.36 2.08 2.45 0.77 2.26 2.52 2.37 1.74 1.78 1.93 2.34 3.09 2.89 2.52 2.07 2.32 1.51 1.55 1.47 1.73 2.45 2.27 2.56 1.88 1.91 1.68 2.21
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1446 chr2:192502051-192503535 -1.59 2146 chr6:1702314-1704615 -2.16 1.92 2.69 2.95 2.55 2.33 1.82 2.36 1.84 2.35 1.31 2.16 1.87 1.68 1.53 2.26 2.06 1.93 2.24 2.36 2.08 2.45 0.77 2.26 2.52 2.37 1.74 1.78 1.93 2.34 3.09 2.89 2.52 2.07 2.32 1.51 1.55 1.47 1.73 2.45 2.27 2.56 1.88 1.91 1.68 2.21
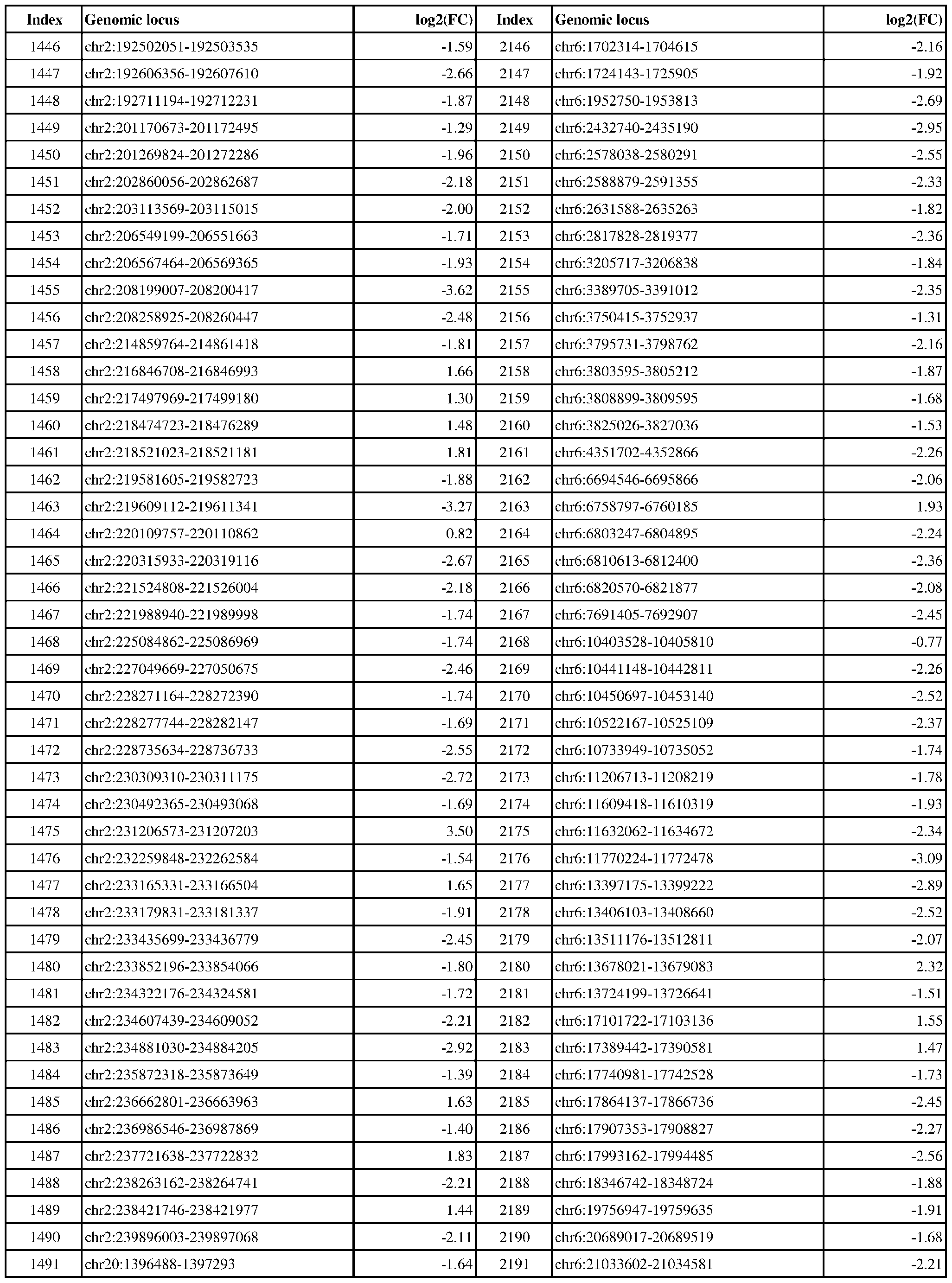 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1492 chr20:1458523-1460014 -3.47 2192 chr6:21219788-21221181 -2.01 2.39 1.84 1.59 2.50 3.05 1.04 1.86 2.26 1.84 1.81 2.95 2.12 2.45 1.74 1.74 1.39 1.92 1.14 2.37 2.22 1.00 1.65 2.10 3.21 2.32 2.01 2.26 1.77 1.63 1.85 2.07 1.25 1.29 2.31 1.96 1.82 1.64 2.92 2.16 2.27 2.46 1.97 2.30 2.47 2.11
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1492 chr20:1458523-1460014 -3.47 2192 chr6:21219788-21221181 -2.01 2.39 1.84 1.59 2.50 3.05 1.04 1.86 2.26 1.84 1.81 2.95 2.12 2.45 1.74 1.74 1.39 1.92 1.14 2.37 2.22 1.00 1.65 2.10 3.21 2.32 2.01 2.26 1.77 1.63 1.85 2.07 1.25 1.29 2.31 1.96 1.82 1.64 2.92 2.16 2.27 2.46 1.97 2.30 2.47 2.11
 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1538 chr20:43229436-43231342 -1.75 2238 chr6:74159715-74162285 -2.12 1.15 2.15 2.31 2.08 2.28 2.93 2.52 1.99 2.46 2.33 2.69 1.74 2.60 2.02 2.19 3.12 3.31 1.78 2.38 1.64 2.32 1.70 2.54 2.71 2.32 1.66 2.68 1.69 2.63 1.56 2.37 2.25 1.67 3.29 3.26 1.72 3.47 1.92 2.74 1.97 2.46 2.37 3.10 1.14 0.96
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1538 chr20:43229436-43231342 -1.75 2238 chr6:74159715-74162285 -2.12 1.15 2.15 2.31 2.08 2.28 2.93 2.52 1.99 2.46 2.33 2.69 1.74 2.60 2.02 2.19 3.12 3.31 1.78 2.38 1.64 2.32 1.70 2.54 2.71 2.32 1.66 2.68 1.69 2.63 1.56 2.37 2.25 1.67 3.29 3.26 1.72 3.47 1.92 2.74 1.97 2.46 2.37 3.10 1.14 0.96
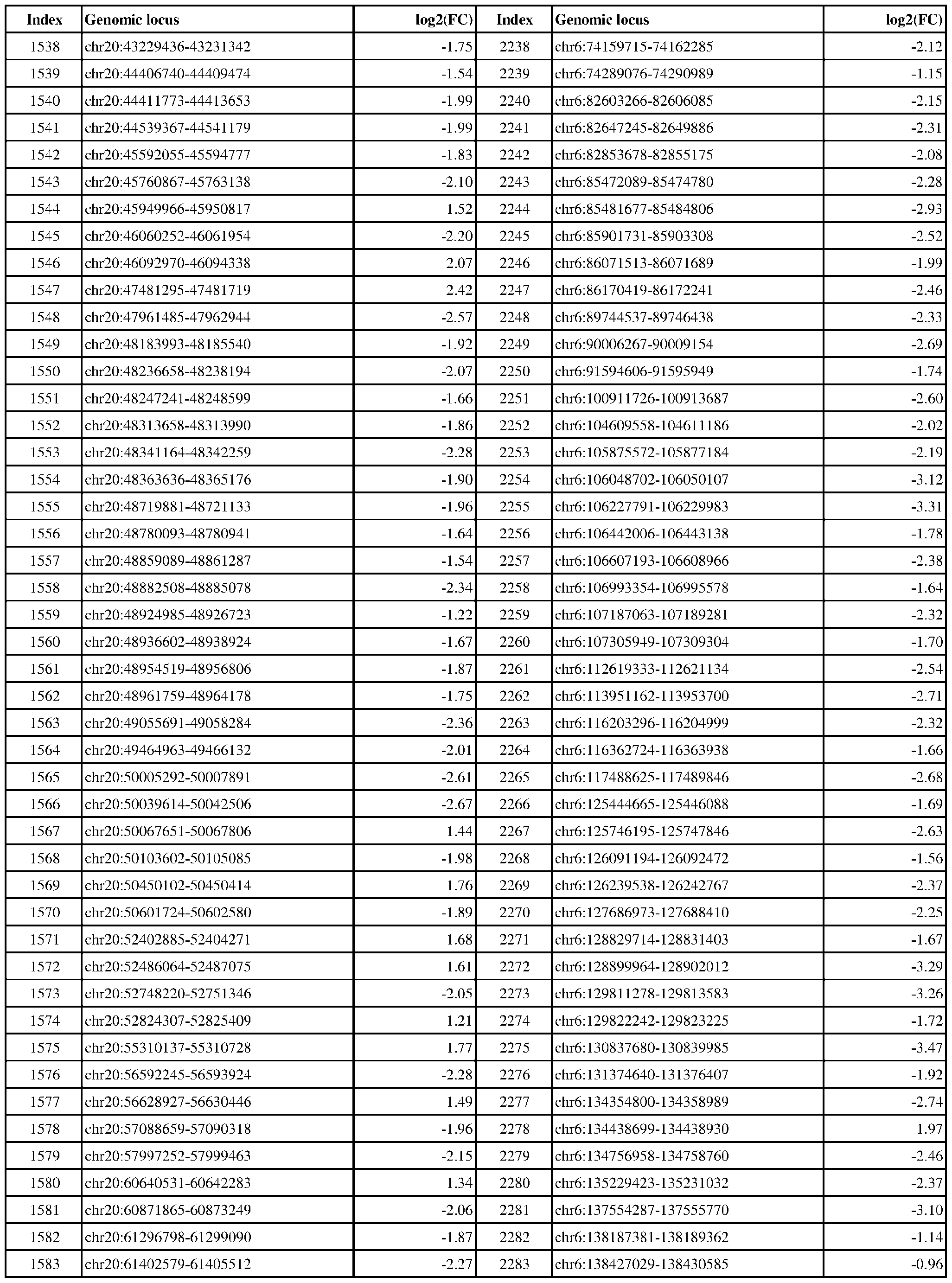 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1584 chr20:61435560-61438400 0.82 2284 chr6:138914088-138915476 -2.62 0.98 2.07 2.69 1.52 1.64 2.59 2.24 2.14 2.10 2.46 2.50 1.82 1.97 1.79 2.32 1.76 2.05 1.87 1.86 2.28 2.00 1.98 1.97 2.34 3.40 3.02 0.71 2.11 1.76 2.18 1.66 2.64 2.09 1.92 2.18 1.61 2.15 1.84 2.41 2.22 1.67 1.86 2.07 1.69 1.69
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1584 chr20:61435560-61438400 0.82 2284 chr6:138914088-138915476 -2.62 0.98 2.07 2.69 1.52 1.64 2.59 2.24 2.14 2.10 2.46 2.50 1.82 1.97 1.79 2.32 1.76 2.05 1.87 1.86 2.28 2.00 1.98 1.97 2.34 3.40 3.02 0.71 2.11 1.76 2.18 1.66 2.64 2.09 1.92 2.18 1.61 2.15 1.84 2.41 2.22 1.67 1.86 2.07 1.69 1.69
 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1630 chr21:43735305-43735925 1.69 2330 chr7:869827-872413 -2.01 1.81 1.95 2.97 1.87 2.29 1.81 1.88 1.95 2.07 2.25 1.93 2.21 1.01 1.14 1.90 2.30 2.08 2.14 1.56 2.60 1.90 2.08 2.21 2.22 3.14 1.58 2.41 1.79 4.27 1.81 1.97 2.05 1.82 2.84 2.62 1.82 1.71 2.19 1.87 2.12 1.88 2.65 1.72 1.93 1.58
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1630 chr21:43735305-43735925 1.69 2330 chr7:869827-872413 -2.01 1.81 1.95 2.97 1.87 2.29 1.81 1.88 1.95 2.07 2.25 1.93 2.21 1.01 1.14 1.90 2.30 2.08 2.14 1.56 2.60 1.90 2.08 2.21 2.22 3.14 1.58 2.41 1.79 4.27 1.81 1.97 2.05 1.82 2.84 2.62 1.82 1.71 2.19 1.87 2.12 1.88 2.65 1.72 1.93 1.58
 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1676 chr22:38021184-38023942 -2.02 2376 chr7:30245860-30247116 -1.84 1.74 1.97 1.85 2.24 1.55 1.77 1.61 1.77 1.76 2.70 2.34 3.19 1.36 1.80 1.72 1.44 1.27 2.74 2.95 2.38 3.32 1.76 1.56 2.06 1.73 2.41 2.13 2.12 2.07 3.00 2.42 2.22 2.08 3.30 2.38 2.21 2.76 2.16 3.84 2.29 1.97 2.11 2.74 1.66 2.11
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1676 chr22:38021184-38023942 -2.02 2376 chr7:30245860-30247116 -1.84 1.74 1.97 1.85 2.24 1.55 1.77 1.61 1.77 1.76 2.70 2.34 3.19 1.36 1.80 1.72 1.44 1.27 2.74 2.95 2.38 3.32 1.76 1.56 2.06 1.73 2.41 2.13 2.12 2.07 3.00 2.42 2.22 2.08 3.30 2.38 2.21 2.76 2.16 3.84 2.29 1.97 2.11 2.74 1.66 2.11
 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1722 chr3:9904116-9908916 -1.71 2422 chr7:73264927-73266810 -1.71 1.92 1.96 2.28 1.34 1.24 1.01 1.98 2.58 2.41 1.99 1.77 1.97 1.55 1.42 1.37 1.85 2.00 1.62 2.64 1.48 2.09 3.05 2.32 1.50 3.44 1.71 1.84 1.69 1.97 1.60 2.14 1.82 1.70 1.52 1.81 1.74 1.79 1.81 2.10 1.81 1.79 1.84 2.57 2.52 2.98
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1722 chr3:9904116-9908916 -1.71 2422 chr7:73264927-73266810 -1.71 1.92 1.96 2.28 1.34 1.24 1.01 1.98 2.58 2.41 1.99 1.77 1.97 1.55 1.42 1.37 1.85 2.00 1.62 2.64 1.48 2.09 3.05 2.32 1.50 3.44 1.71 1.84 1.69 1.97 1.60 2.14 1.82 1.70 1.52 1.81 1.74 1.79 1.81 2.10 1.81 1.79 1.84 2.57 2.52 2.98
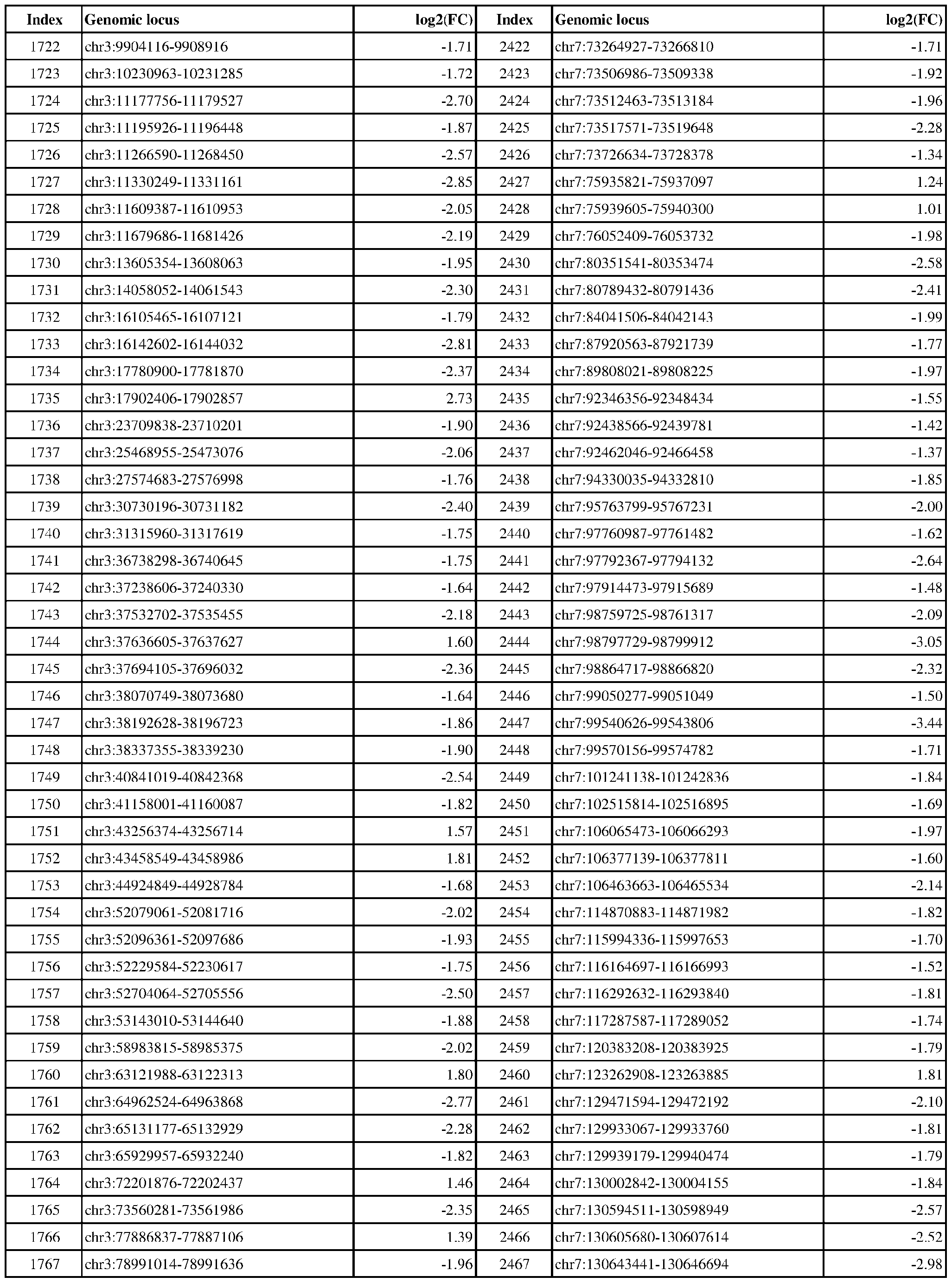 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1768 chr3:87137862-87139128 -2.86 2468 chr7:130848729-130850211 -1.98 1.82 1.64 2.76 2.13 1.58 2.36 2.07 2.29 2.99 2.48 2.19 1.61 1.72 1.56 2.76 3.00 1.79 2.98 1.91 2.28 2.05 2.92 2.45 2.39 1.79 1.86 1.81 2.49 2.08 1.92 2.31 1.94 1.77 2.04 1.72 2.17 2.49 1.96 1.75 3.14 1.60 1.42 2.37 1.90 1.50
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1768 chr3:87137862-87139128 -2.86 2468 chr7:130848729-130850211 -1.98 1.82 1.64 2.76 2.13 1.58 2.36 2.07 2.29 2.99 2.48 2.19 1.61 1.72 1.56 2.76 3.00 1.79 2.98 1.91 2.28 2.05 2.92 2.45 2.39 1.79 1.86 1.81 2.49 2.08 1.92 2.31 1.94 1.77 2.04 1.72 2.17 2.49 1.96 1.75 3.14 1.60 1.42 2.37 1.90 1.50
 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1814 chr3:143200351-143202332 -2.41 2514 chr8:37549026-37549513 2.94 1.32 1.61 1.63 2.86 1.74 1.89 1.84 2.47 1.76 2.04 3.31 1.71 2.19 2.10 2.33 1.57 1.65 2.42 1.46 1.91 1.60 2.96 2.04 1.89 2.38 2.08 1.94 2.80 3.04 2.46 1.55 2.03 2.02 1.15 1.95 2.55 2.86 1.72 1.85 1.97 2.75 1.82 1.98 1.92 2.11
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1814 chr3:143200351-143202332 -2.41 2514 chr8:37549026-37549513 2.94 1.32 1.61 1.63 2.86 1.74 1.89 1.84 2.47 1.76 2.04 3.31 1.71 2.19 2.10 2.33 1.57 1.65 2.42 1.46 1.91 1.60 2.96 2.04 1.89 2.38 2.08 1.94 2.80 3.04 2.46 1.55 2.03 2.02 1.15 1.95 2.55 2.86 1.72 1.85 1.97 2.75 1.82 1.98 1.92 2.11
 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1860 chr3:185540964-185544392 -1.61 2560 chr8:80597254-80598547 -1.84 2.60 2.36 1.88 2.17 2.27 1.79 3.66 1.59 2.00 2.08 2.45 2.80 2.41 2.12 1.67 1.64 1.89 1.91 1.97 1.54 1.49 1.63 1.56 1.25 1.55 2.06 2.34 3.32 1.72 2.08 3.71 2.64 2.04 2.06 1.53 2.37 2.20 2.35 2.39 3.66 1.78 1.59 2.39 3.20 3.00
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1860 chr3:185540964-185544392 -1.61 2560 chr8:80597254-80598547 -1.84 2.60 2.36 1.88 2.17 2.27 1.79 3.66 1.59 2.00 2.08 2.45 2.80 2.41 2.12 1.67 1.64 1.89 1.91 1.97 1.54 1.49 1.63 1.56 1.25 1.55 2.06 2.34 3.32 1.72 2.08 3.71 2.64 2.04 2.06 1.53 2.37 2.20 2.35 2.39 3.66 1.78 1.59 2.39 3.20 3.00
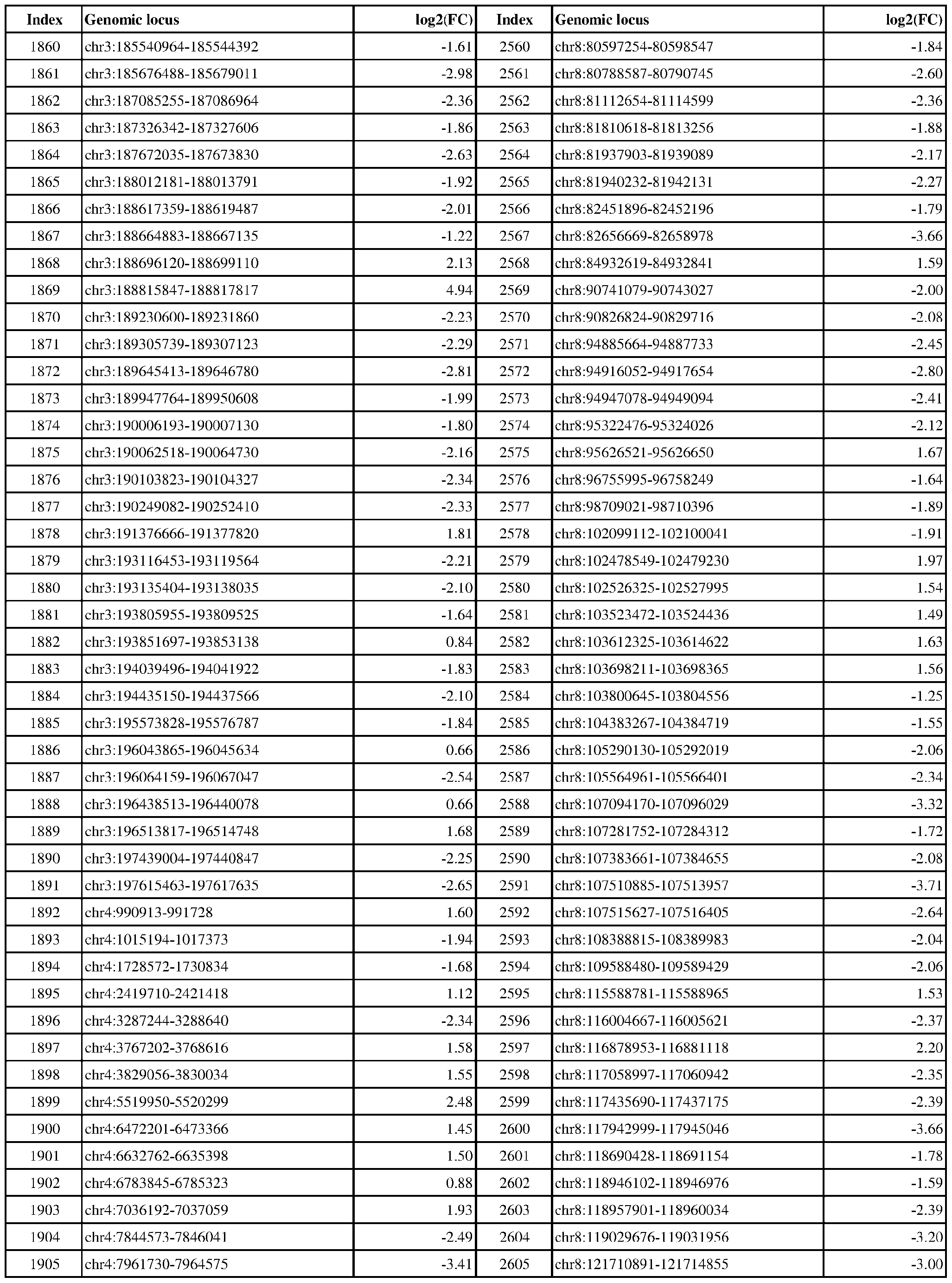 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1906 chr4:7971940-7975169 -1.24 2606 chr8:122557525-122559066 -2.04 2.28 1.93 2.24 2.07 1.87 2.06 2.70 2.01 2.73 1.97 2.09 3.32 3.07 1.67 2.13 1.77 2.24 2.17 2.34 3.13 4.47 2.25 2.11 2.06 2.62 1.51 1.36 1.57 1.96 1.82 1.76 2.22 1.64 1.89 1.85 1.97 1.88 1.50 1.46 2.26 2.19 1.83 1.98 2.23 1.97
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1906 chr4:7971940-7975169 -1.24 2606 chr8:122557525-122559066 -2.04 2.28 1.93 2.24 2.07 1.87 2.06 2.70 2.01 2.73 1.97 2.09 3.32 3.07 1.67 2.13 1.77 2.24 2.17 2.34 3.13 4.47 2.25 2.11 2.06 2.62 1.51 1.36 1.57 1.96 1.82 1.76 2.22 1.64 1.89 1.85 1.97 1.88 1.50 1.46 2.26 2.19 1.83 1.98 2.23 1.97
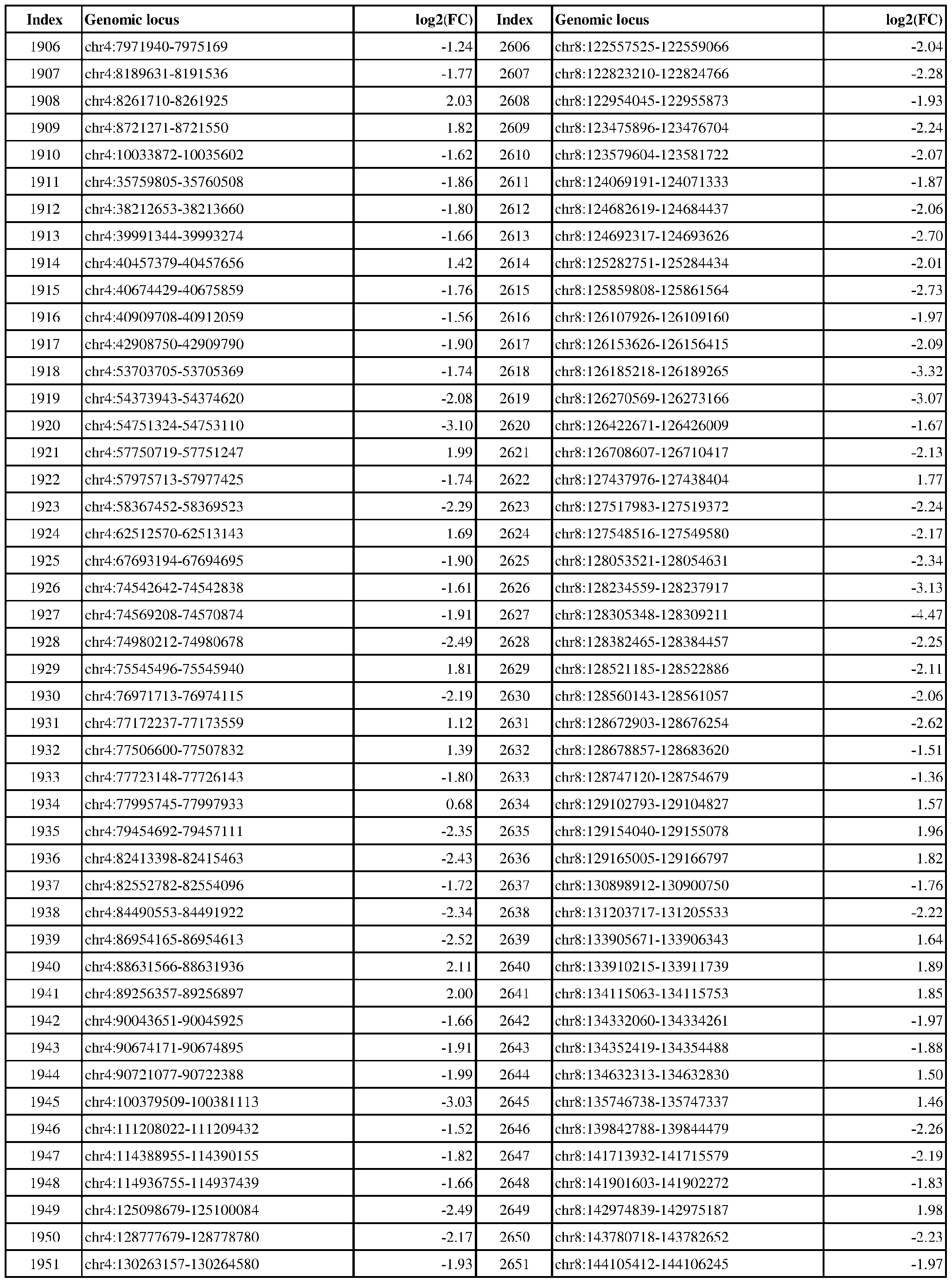 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1952 chr4:140357209-140357729 -2.15 2652 chr8:144107545-144108374 -1.55 1.42 1.74 1.35 1.55 1.27 3.08 2.73 2.41 2.39 2.05 1.18 2.19 2.06 1.91 2.94 2.08 1.65 2.48 2.81 3.23 2.08 1.83 2.32 2.10 2.25 2.51 3.27 2.07 1.74 1.65 2.82 2.31 1.52 1.42 2.84 2.11 1.95 1.76 1.81 1.84 1.27 1.89 1.64 1.59 1.63
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1952 chr4:140357209-140357729 -2.15 2652 chr8:144107545-144108374 -1.55 1.42 1.74 1.35 1.55 1.27 3.08 2.73 2.41 2.39 2.05 1.18 2.19 2.06 1.91 2.94 2.08 1.65 2.48 2.81 3.23 2.08 1.83 2.32 2.10 2.25 2.51 3.27 2.07 1.74 1.65 2.82 2.31 1.52 1.42 2.84 2.11 1.95 1.76 1.81 1.84 1.27 1.89 1.64 1.59 1.63
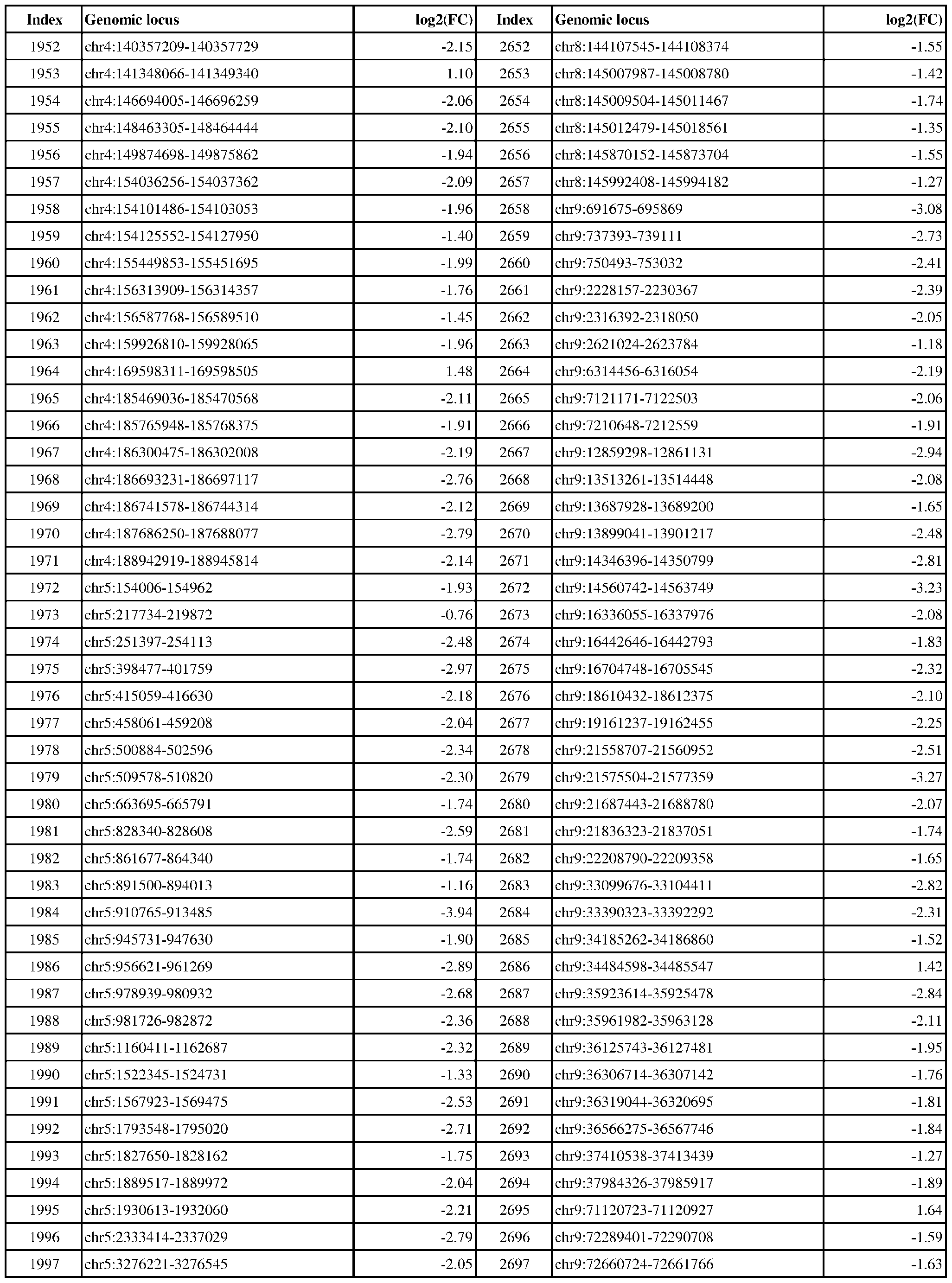 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1998 chr5:3310167-3312625 -3.00 2698 chr9:73168588-73170222 -2.56 1.57 1.66 1.73 1.99 1.77 1.46 0.93 2.35 1.81 1.68 1.46 1.73 2.16 2.48 2.09 2.06 2.24 2.29 1.96 1.71 1.90 2.30 1.71 1.62 1.76 1.88 2.09 1.38 1.70 1.76 2.39 2.87 1.85 2.38 3.26 2.23 0.82 1.81 2.14 1.86 1.97 1.83 1.96 0.76 1.66
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1998 chr5:3310167-3312625 -3.00 2698 chr9:73168588-73170222 -2.56 1.57 1.66 1.73 1.99 1.77 1.46 0.93 2.35 1.81 1.68 1.46 1.73 2.16 2.48 2.09 2.06 2.24 2.29 1.96 1.71 1.90 2.30 1.71 1.62 1.76 1.88 2.09 1.38 1.70 1.76 2.39 2.87 1.85 2.38 3.26 2.23 0.82 1.81 2.14 1.86 1.97 1.83 1.96 0.76 1.66
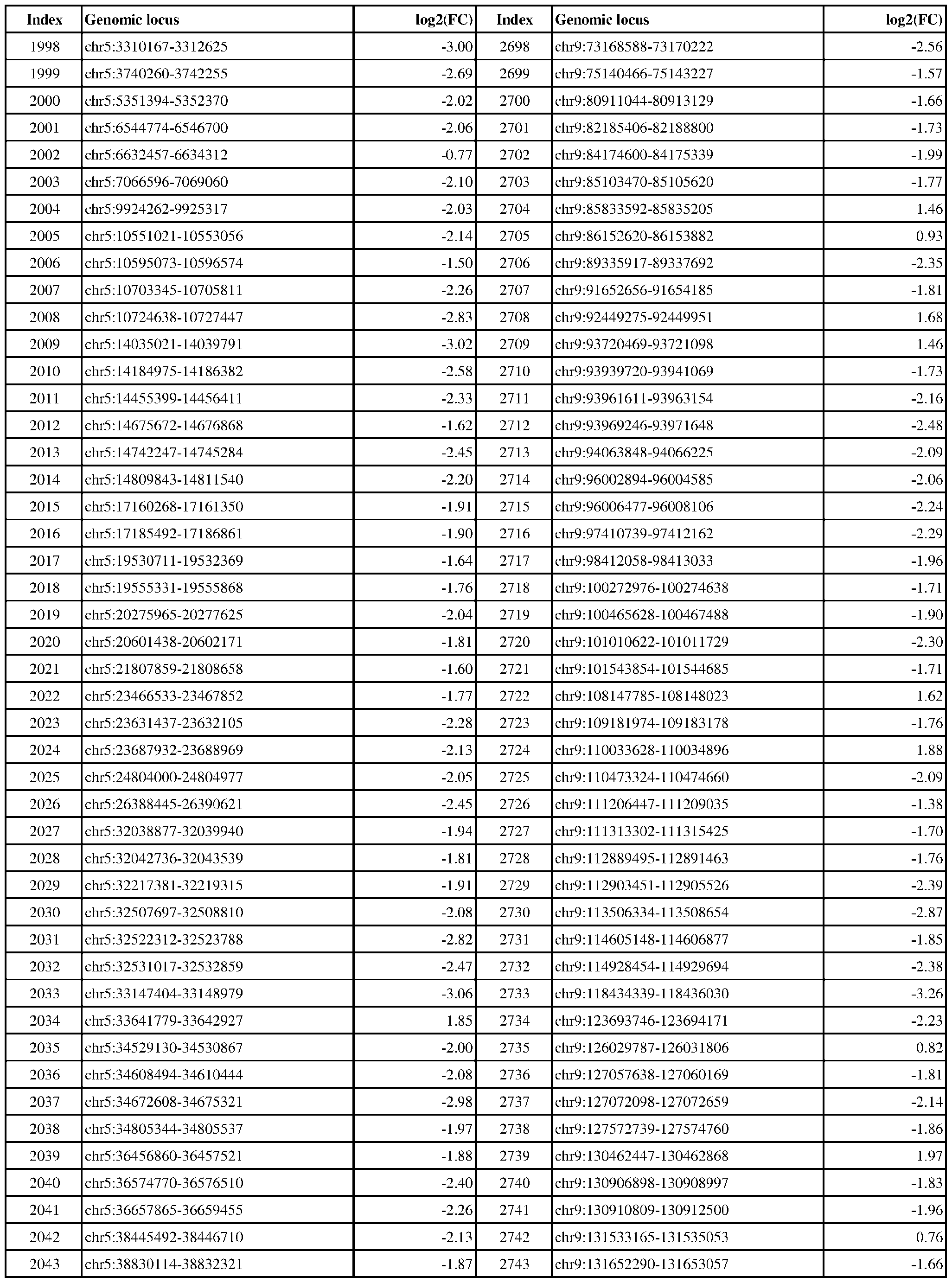 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 2044 chr5:38853746-38855326 -2.21 2744 chr9:131664024-131665677 -2.11 2.48 1.62 1.90 2.31 2.52 2.36 2.77 0.89 2.13 1.58 1.27 1.49 1.20 2.63 1.31 1.02 0.88 1.01 2.24 1.55 2.12 1.51 1.82 1.03 1.75 1.91 0.91 1.18 1.39 2.36 1.68 1.60 1.67 1.13 1.48 1.78 2.12 1.90 1.99 2.03 2.26 1.85 1.93 1.96 2.20
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 2044 chr5:38853746-38855326 -2.21 2744 chr9:131664024-131665677 -2.11 2.48 1.62 1.90 2.31 2.52 2.36 2.77 0.89 2.13 1.58 1.27 1.49 1.20 2.63 1.31 1.02 0.88 1.01 2.24 1.55 2.12 1.51 1.82 1.03 1.75 1.91 0.91 1.18 1.39 2.36 1.68 1.60 1.67 1.13 1.48 1.78 2.12 1.90 1.99 2.03 2.26 1.85 1.93 1.96 2.20
 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 2090 chr5:133388486-133391015 -2.40 2790 chrX:118108583-118110805 -1.30 1.35 1.74 1.96 1.96 1.75 1.79 2.25 2.25 1.90 1.85 1.86 1.94 1.54 2.10 2.14
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 2090 chr5:133388486-133391015 -2.40 2790 chrX:118108583-118110805 -1.30 1.35 1.74 1.96 1.96 1.75 1.79 2.25 2.25 1.90 1.85 1.86 1.94 1.54 2.10 2.14

Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1 chr1:1141525-1142216 -4.76 489 chr14:70512579-70513161 -3.19 1.88 1.45 2.53 1.49 1.26 3.22 3.58 3.68 2.64 4.25 1.83 1.33 2.36 2.17 3.71 3.53 1.13 1.84 1.50 1.88 1.63 2.60 3.71 5.37 3.71 4.52 1.51 2.40 2.80 2.94 2.50 2.20 3.08 5.07 3.14 2.92 5.34 2.03 2.59 3.13 2.47 2.74 3.25 2.75
 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 46 chr1:21644300-21646907 -1.81 534 chr15:33803935-33804459 -3.12 2.44 4.01 3.00 2.75 2.25 2.63 3.47 3.40 5.81 2.49 4.04 4.16 3.15 2.15 1.08 1.72 3.57 2.54 3.40 1.10 1.83 2.98 2.58 3.39 3.81 1.88 3.12 3.46 2.62 2.41 4.34 1.98 1.37 3.09 3.33 4.25 4.96 3.10 3.68 3.90 0.87 2.23 2.23 2.97 5.70
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 46 chr1:21644300-21646907 -1.81 534 chr15:33803935-33804459 -3.12 2.44 4.01 3.00 2.75 2.25 2.63 3.47 3.40 5.81 2.49 4.04 4.16 3.15 2.15 1.08 1.72 3.57 2.54 3.40 1.10 1.83 2.98 2.58 3.39 3.81 1.88 3.12 3.46 2.62 2.41 4.34 1.98 1.37 3.09 3.33 4.25 4.96 3.10 3.68 3.90 0.87 2.23 2.23 2.97 5.70
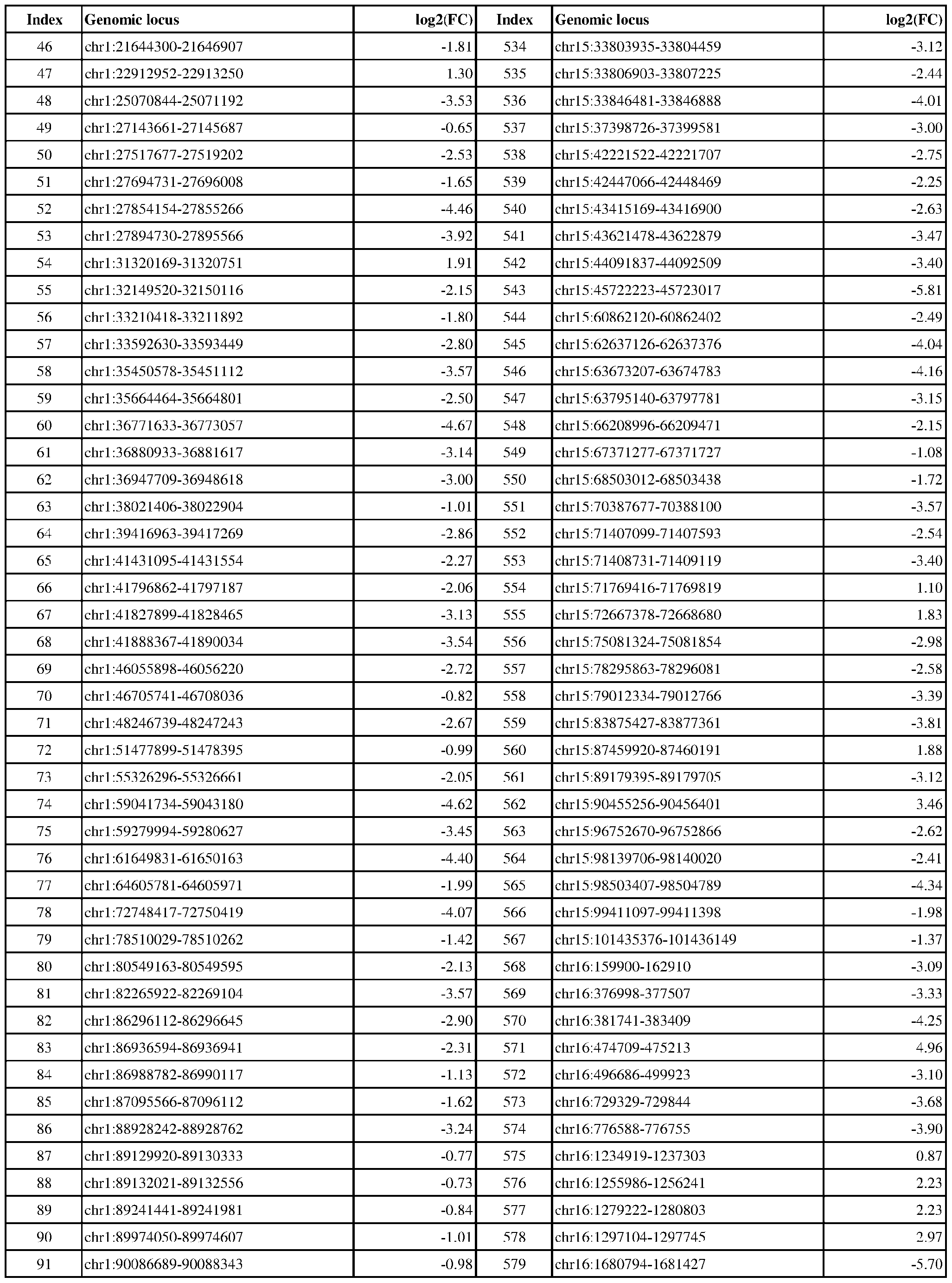 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 92 chr1:90150531-90151046 -1.04 580 chr16:1729719-1733401 -3.62 3.37 3.22 4.01 2.81 2.61 3.45 1.98 2.42 3.02 2.22 1.85 2.01 2.12 2.17 2.32 2.64 1.88 2.81 3.65 1.87 2.15 1.63 2.17 3.38 3.67 2.92 2.27 2.55 2.12 1.51 2.19 2.85 7.54 3.66 2.17 2.29 2.00 2.06 2.53 1.96 2.14 1.48 2.96 2.82 2.85
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 92 chr1:90150531-90151046 -1.04 580 chr16:1729719-1733401 -3.62 3.37 3.22 4.01 2.81 2.61 3.45 1.98 2.42 3.02 2.22 1.85 2.01 2.12 2.17 2.32 2.64 1.88 2.81 3.65 1.87 2.15 1.63 2.17 3.38 3.67 2.92 2.27 2.55 2.12 1.51 2.19 2.85 7.54 3.66 2.17 2.29 2.00 2.06 2.53 1.96 2.14 1.48 2.96 2.82 2.85
 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 138 chr1:208040648-208041092 -2.54 626 chr16:75525286-75525583 -2.00 2.72 2.58 2.20 2.01 3.36 3.63 3.01 3.79 2.97 2.84 3.58 3.72 2.93 3.11 2.63 3.34 3.31 2.86 3.31 4.85 3.23 3.56 4.66 3.64 1.68 2.23 2.77 2.04 2.80 3.55 4.60 2.37 2.37 3.97 2.82 2.79 7.07 3.79 3.01 1.94 2.48 1.68 4.33 1.71 1.19
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 138 chr1:208040648-208041092 -2.54 626 chr16:75525286-75525583 -2.00 2.72 2.58 2.20 2.01 3.36 3.63 3.01 3.79 2.97 2.84 3.58 3.72 2.93 3.11 2.63 3.34 3.31 2.86 3.31 4.85 3.23 3.56 4.66 3.64 1.68 2.23 2.77 2.04 2.80 3.55 4.60 2.37 2.37 3.97 2.82 2.79 7.07 3.79 3.01 1.94 2.48 1.68 4.33 1.71 1.19
 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 184 chr10:15273416-15273718 -3.76 672 chr17:4046537-4046802 2.51 1.41 3.10 4.53 2.20 4.07 3.46 3.09 1.63 1.06 2.86 2.91 5.99 2.55 3.60 2.90 2.21 2.05 2.24 2.47 1.85 1.92 1.88 2.03 1.93 2.10 1.91 1.66 1.69 2.63 2.72 2.19 2.20 3.49 3.11 2.14 2.71 2.72 2.66 3.71 2.94 2.64 3.08 3.76 3.16 2.83
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 184 chr10:15273416-15273718 -3.76 672 chr17:4046537-4046802 2.51 1.41 3.10 4.53 2.20 4.07 3.46 3.09 1.63 1.06 2.86 2.91 5.99 2.55 3.60 2.90 2.21 2.05 2.24 2.47 1.85 1.92 1.88 2.03 1.93 2.10 1.91 1.66 1.69 2.63 2.72 2.19 2.20 3.49 3.11 2.14 2.71 2.72 2.66 3.71 2.94 2.64 3.08 3.76 3.16 2.83
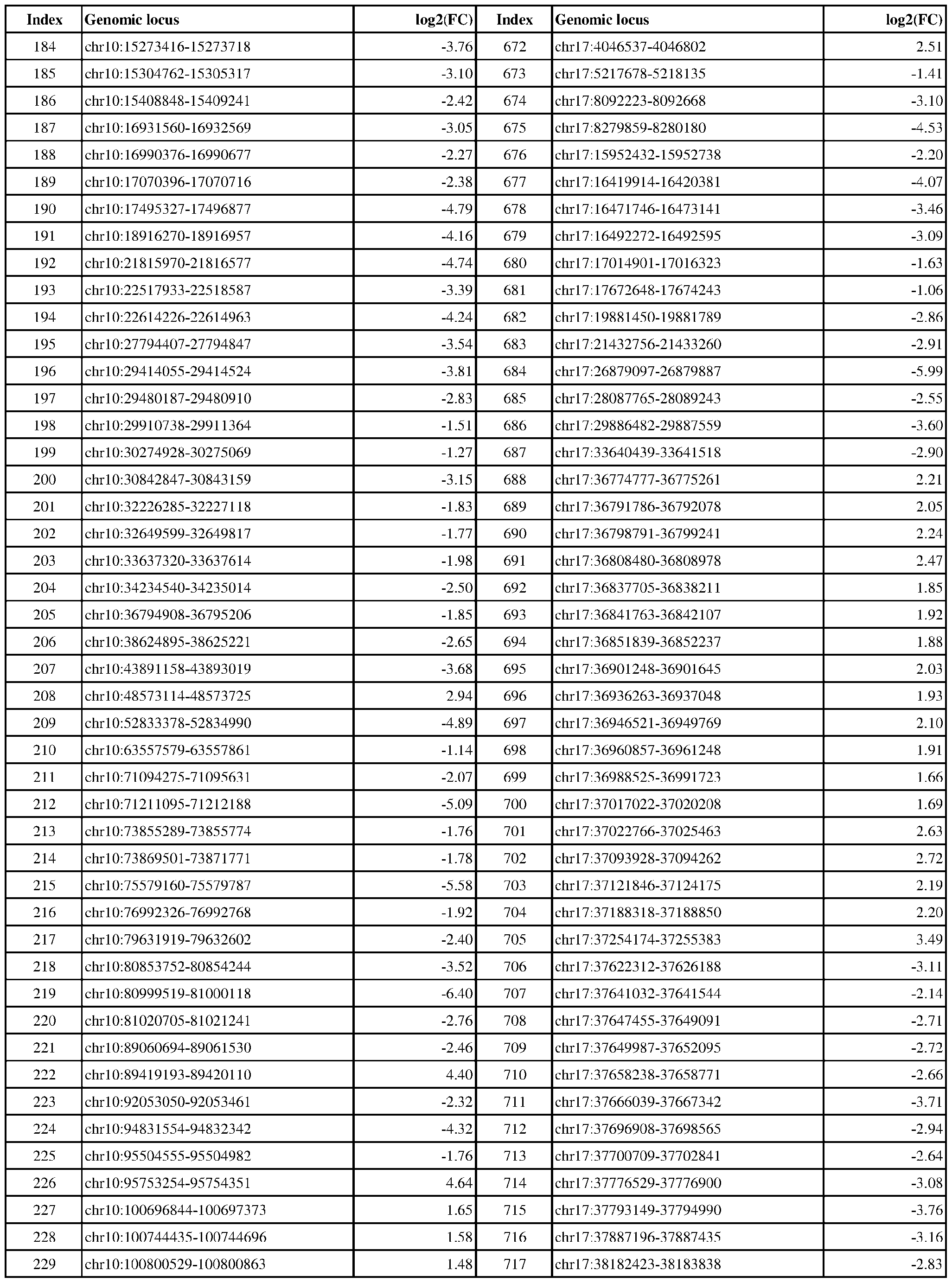 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 230 chr10:100841255-100841754 1.44 718 chr17:38225073-38225494 -3.02 3.85 4.44 2.94 3.73 3.17 3.39 3.82 4.26 3.47 1.68 3.04 2.28 2.84 2.86 3.00 2.47 3.13 3.14 3.17 2.43 2.13 2.05 2.44 3.11 4.59 3.23 6.72 1.55 2.58 2.93 1.46 1.90 1.34 1.38 1.91 1.98 1.98 2.67 1.74 1.89 1.76 1.69 1.58 1.44 2.65
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 230 chr10:100841255-100841754 1.44 718 chr17:38225073-38225494 -3.02 3.85 4.44 2.94 3.73 3.17 3.39 3.82 4.26 3.47 1.68 3.04 2.28 2.84 2.86 3.00 2.47 3.13 3.14 3.17 2.43 2.13 2.05 2.44 3.11 4.59 3.23 6.72 1.55 2.58 2.93 1.46 1.90 1.34 1.38 1.91 1.98 1.98 2.67 1.74 1.89 1.76 1.69 1.58 1.44 2.65
 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 276 chr11:66554827-66555249 -2.06 764 chr17:57884514-57885192 1.56 1.84 1.54 1.75 1.89 1.74 1.30 3.36 1.62 2.70 3.44 4.68 3.47 2.34 2.89 2.57 2.70 2.24 3.13 2.35 1.86 2.84 2.34 2.02 2.21 2.16 2.72 2.68 2.34 2.24 3.25 2.77 2.19 3.76 2.93 3.13 4.01 2.77 2.12 3.62 3.62 2.49 3.00 5.75 2.99 3.54
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 276 chr11:66554827-66555249 -2.06 764 chr17:57884514-57885192 1.56 1.84 1.54 1.75 1.89 1.74 1.30 3.36 1.62 2.70 3.44 4.68 3.47 2.34 2.89 2.57 2.70 2.24 3.13 2.35 1.86 2.84 2.34 2.02 2.21 2.16 2.72 2.68 2.34 2.24 3.25 2.77 2.19 3.76 2.93 3.13 4.01 2.77 2.12 3.62 3.62 2.49 3.00 5.75 2.99 3.54
 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 322 chr11:121694444-121694975 -2.45 810 chr17:75641171-75644120 -4.12 2.66 3.35 3.00 3.39 4.53 3.18 3.99 3.88 3.12 3.57 3.81 4.48 2.07 3.14 4.63 3.01 3.65 5.22 2.77 3.92 2.52 1.89 2.43 2.88 2.32 2.47 3.31 2.94 3.32 2.94 2.21 2.62 2.31 2.70 2.83 2.57 3.28 2.99 2.18 3.45 2.98 2.70 3.65 2.42 2.81
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 322 chr11:121694444-121694975 -2.45 810 chr17:75641171-75644120 -4.12 2.66 3.35 3.00 3.39 4.53 3.18 3.99 3.88 3.12 3.57 3.81 4.48 2.07 3.14 4.63 3.01 3.65 5.22 2.77 3.92 2.52 1.89 2.43 2.88 2.32 2.47 3.31 2.94 3.32 2.94 2.21 2.62 2.31 2.70 2.83 2.57 3.28 2.99 2.18 3.45 2.98 2.70 3.65 2.42 2.81
 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 368 chr11:128470660-128471133 -2.50 856 chr18:6797635-6798494 -2.73 3.41 3.39 3.15 2.77 3.74 2.47 2.51 2.29 3.00 2.07 5.88 4.06 3.18 3.51 3.15 2.84 2.87 4.15 2.23 1.99 2.02 4.74 4.53 3.87 2.83 4.30 3.24 3.74 3.17 3.61 3.21 1.77 4.21 3.51 4.16 3.81 3.80 4.06 2.32 2.93 3.02 4.77 3.17 3.17 2.89
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 368 chr11:128470660-128471133 -2.50 856 chr18:6797635-6798494 -2.73 3.41 3.39 3.15 2.77 3.74 2.47 2.51 2.29 3.00 2.07 5.88 4.06 3.18 3.51 3.15 2.84 2.87 4.15 2.23 1.99 2.02 4.74 4.53 3.87 2.83 4.30 3.24 3.74 3.17 3.61 3.21 1.77 4.21 3.51 4.16 3.81 3.80 4.06 2.32 2.93 3.02 4.77 3.17 3.17 2.89
 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 414 chr12:77272243-77273650 1.93 902 chr19:3390209-3390502 -2.93 5.15 3.77 3.05 2.99 2.92 3.23 4.87 3.45 5.61 5.15 2.71 4.19 2.70 4.31 5.32 3.69 2.66 2.78 2.73 3.57 2.91 4.82 4.09 3.87 2.47 2.91 2.99 1.38 2.52 3.30 2.70 2.65 2.33 4.69 2.29 0.92 2.32 1.31 3.01 3.47 3.31 2.06 2.16 2.53 2.69
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 414 chr12:77272243-77273650 1.93 902 chr19:3390209-3390502 -2.93 5.15 3.77 3.05 2.99 2.92 3.23 4.87 3.45 5.61 5.15 2.71 4.19 2.70 4.31 5.32 3.69 2.66 2.78 2.73 3.57 2.91 4.82 4.09 3.87 2.47 2.91 2.99 1.38 2.52 3.30 2.70 2.65 2.33 4.69 2.29 0.92 2.32 1.31 3.01 3.47 3.31 2.06 2.16 2.53 2.69
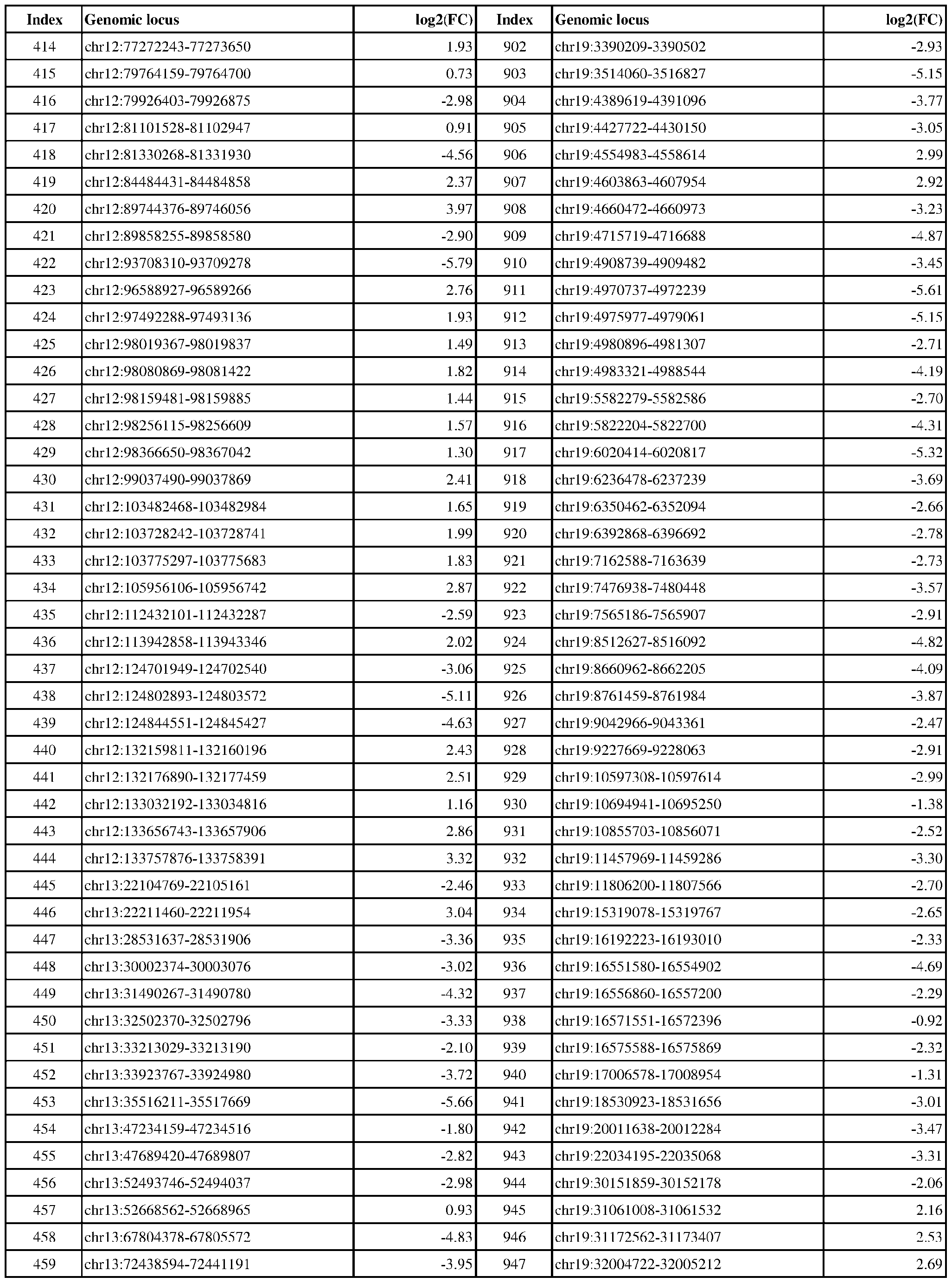 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 460 chr13:76210963-76211380 4.21 948 chr19:34217713-34218896 2.20 1.45 1.81 3.62 4.95 3.27 3.93 2.81 3.43 4.82 2.14 2.05 3.18 5.04 5.94 2.25 2.22 4.02 2.79 3.16 3.32 1.64 2.53 4.85 5.97 3.98 4.85 2.03 1.28
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 460 chr13:76210963-76211380 4.21 948 chr19:34217713-34218896 2.20 1.45 1.81 3.62 4.95 3.27 3.93 2.81 3.43 4.82 2.14 2.05 3.18 5.04 5.94 2.25 2.22 4.02 2.79 3.16 3.32 1.64 2.53 4.85 5.97 3.98 4.85 2.03 1.28
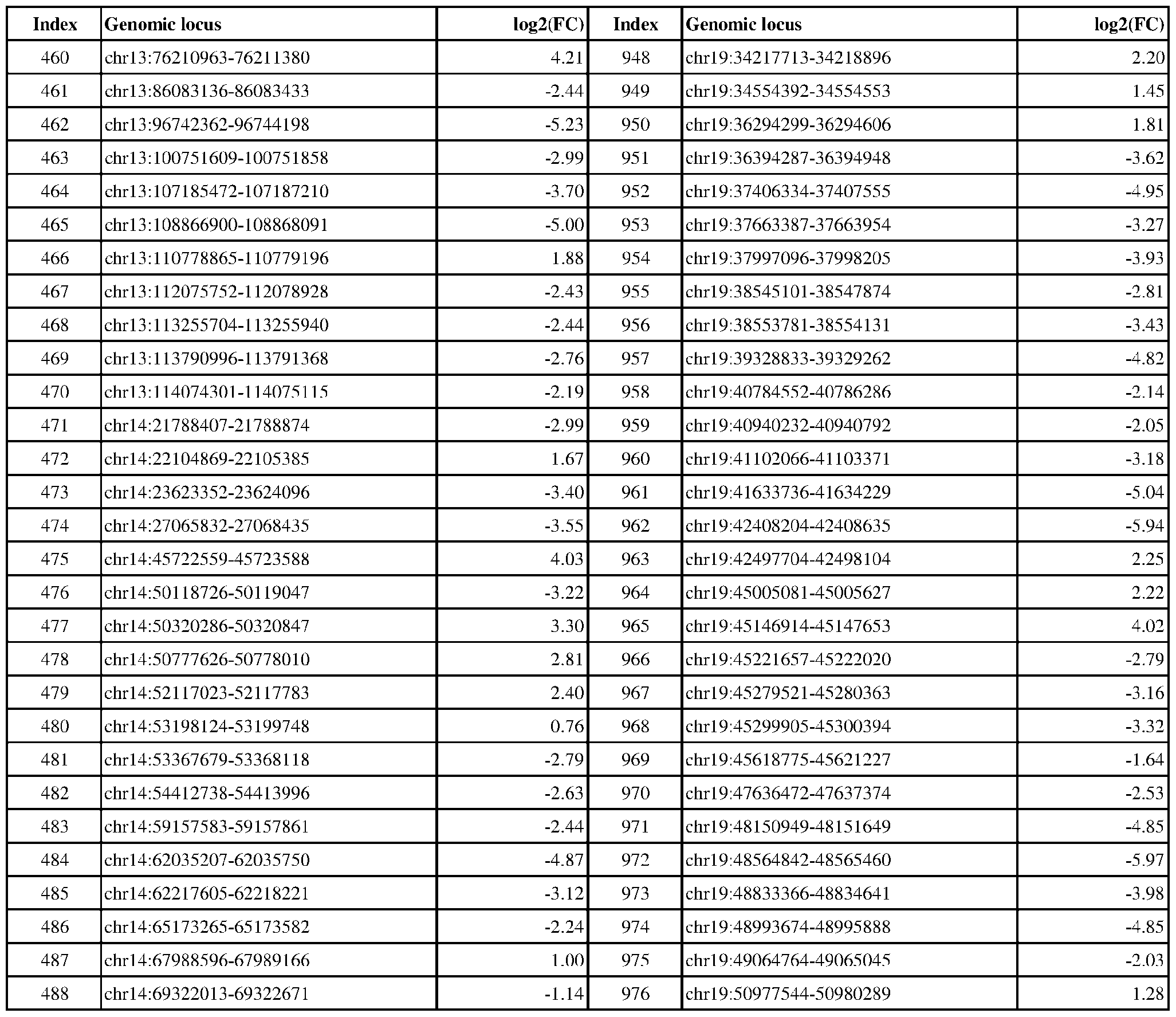
Attorney Docket No.2014191-0027 Table 3: Exemplary genomic loci that are differentially DNA methylated in ER-positive vs. ER-negative cancer. Index Genomic locus log2(FC) Index Genomic locus log2(FC) 977 chr19:51061684-51062328 1.80 1465 chr6:3455585-3456349 -3.89 3.56 3.60 2.26 3.30 3.65 1.59 1.89 1.72 3.31 2.88 2.45 1.93 4.21 2.88 2.61 3.33 3.48 3.61 3.10 2.88 2.06 4.68 4.17 3.74 2.72 3.57 5.42 1.98 6.01 4.26 2.75 2.24 2.49 3.46 4.23 3.78 3.14 1.93 1.94 4.24 1.87 2.96 2.31 2.56
 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1022 chr2:60645074-60645513 -2.04 1510 chr6:114178103-114179089 3.35 3.47 1.43 1.92 3.52 1.22 0.78 0.86 0.67 4.44 0.98 0.85 0.97 0.96 1.00 3.26 2.51 1.78 1.71 3.26 3.43 1.17 2.08 1.84 1.51 0.94 0.93 0.96 2.21 2.74 3.62 3.87 1.53 2.63 2.06 6.02 3.75 1.48 4.14 1.11 1.39 1.81 0.72 3.70 3.43 0.95
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1022 chr2:60645074-60645513 -2.04 1510 chr6:114178103-114179089 3.35 3.47 1.43 1.92 3.52 1.22 0.78 0.86 0.67 4.44 0.98 0.85 0.97 0.96 1.00 3.26 2.51 1.78 1.71 3.26 3.43 1.17 2.08 1.84 1.51 0.94 0.93 0.96 2.21 2.74 3.62 3.87 1.53 2.63 2.06 6.02 3.75 1.48 4.14 1.11 1.39 1.81 0.72 3.70 3.43 0.95
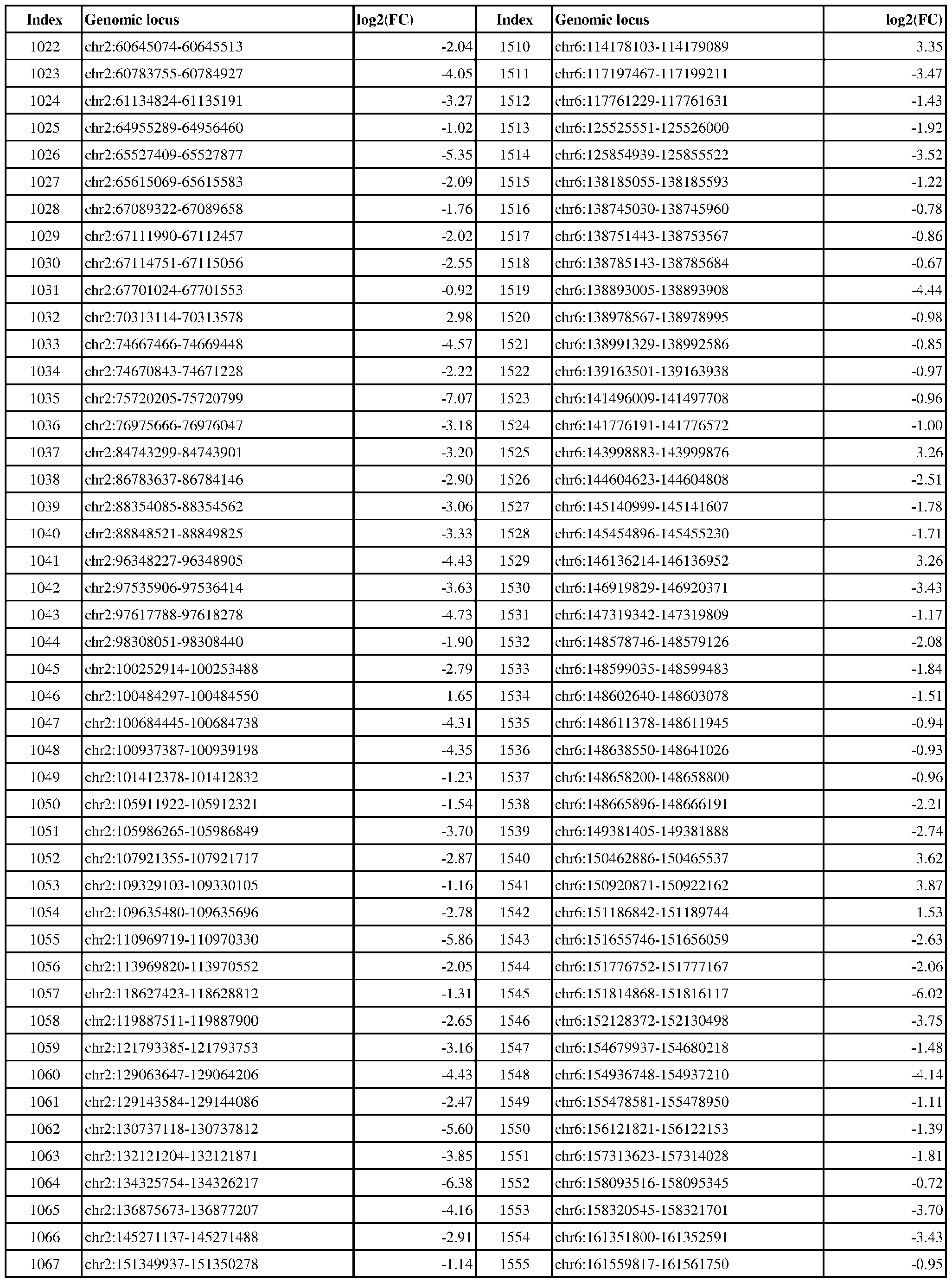 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1068 chr2:152954007-152955861 -4.52 1556 chr6:161572135-161572908 -1.30 2.29 2.58 2.84 2.46 3.39 1.26 2.94 1.34 1.25 3.01 2.13 3.79 4.72 3.46 3.02 3.34 4.68 4.31 2.61 4.66 5.15 3.05 3.06 2.60 4.33 3.29 4.03 3.84 3.54 3.23 3.51 4.39 4.34 3.21 2.63 3.53 4.22 5.38 3.13 4.62 4.25 3.79 3.54 5.02 3.34
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1068 chr2:152954007-152955861 -4.52 1556 chr6:161572135-161572908 -1.30 2.29 2.58 2.84 2.46 3.39 1.26 2.94 1.34 1.25 3.01 2.13 3.79 4.72 3.46 3.02 3.34 4.68 4.31 2.61 4.66 5.15 3.05 3.06 2.60 4.33 3.29 4.03 3.84 3.54 3.23 3.51 4.39 4.34 3.21 2.63 3.53 4.22 5.38 3.13 4.62 4.25 3.79 3.54 5.02 3.34
 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1114 chr20:6759161-6759686 -2.52 1602 chr7:4651658-4652928 -3.74 4.35 2.21 3.85 3.15 3.56 3.22 3.15 3.53 4.35 4.45 3.05 2.22 4.92 3.54 3.61 4.03 3.57 3.08 3.07 3.06 3.38 3.65 3.74 3.18 3.64 2.98 3.13 2.62 4.24 2.61 2.59 3.36 3.20 2.46 3.11 2.84 2.76 3.52 2.87 2.74 1.83 3.39 1.43 2.32 4.05
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1114 chr20:6759161-6759686 -2.52 1602 chr7:4651658-4652928 -3.74 4.35 2.21 3.85 3.15 3.56 3.22 3.15 3.53 4.35 4.45 3.05 2.22 4.92 3.54 3.61 4.03 3.57 3.08 3.07 3.06 3.38 3.65 3.74 3.18 3.64 2.98 3.13 2.62 4.24 2.61 2.59 3.36 3.20 2.46 3.11 2.84 2.76 3.52 2.87 2.74 1.83 3.39 1.43 2.32 4.05
 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1160 chr20:52773523-52774029 2.38 1648 chr7:34717859-34718428 1.85 1.57 1.92 4.03 3.84 3.80 3.37 2.18 3.96 2.18 4.64 2.83 3.00 3.10 3.37 3.13 3.98 2.20 2.45 6.06 1.83 4.38 4.11 2.71 2.58 3.97 3.52 1.63 2.62 3.63 3.19 3.41 1.29 3.00 3.03 2.12 2.36 3.91 2.14 2.84 3.07 4.52 3.40 2.61 2.27 2.72
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1160 chr20:52773523-52774029 2.38 1648 chr7:34717859-34718428 1.85 1.57 1.92 4.03 3.84 3.80 3.37 2.18 3.96 2.18 4.64 2.83 3.00 3.10 3.37 3.13 3.98 2.20 2.45 6.06 1.83 4.38 4.11 2.71 2.58 3.97 3.52 1.63 2.62 3.63 3.19 3.41 1.29 3.00 3.03 2.12 2.36 3.91 2.14 2.84 3.07 4.52 3.40 2.61 2.27 2.72
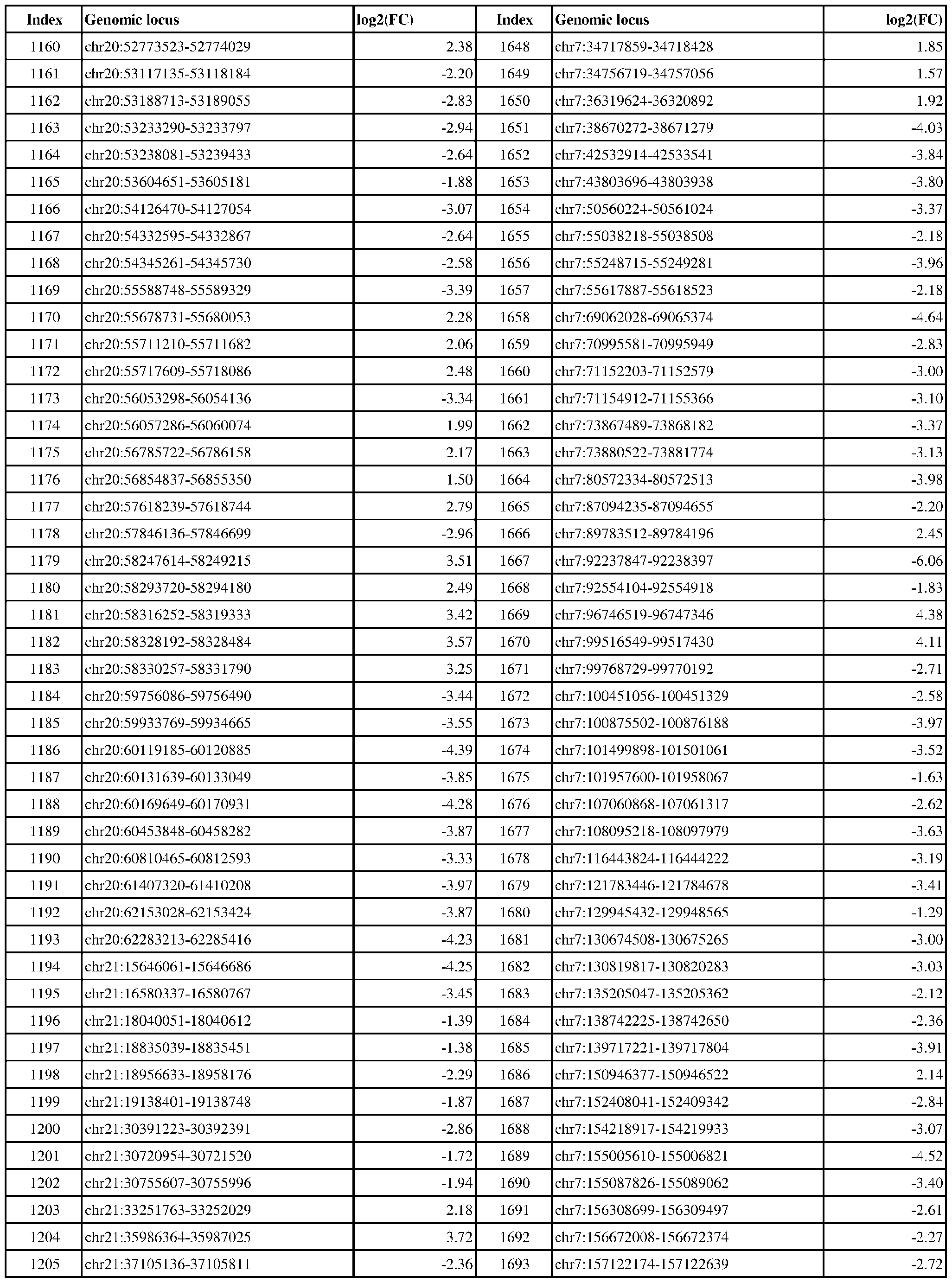 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1206 chr21:37872453-37873502 -2.22 1694 chr7:157128968-157129440 -4.06 2.77 2.58 2.14 3.89 2.24 1.33 2.96 2.57 3.16 2.32 2.39 2.74 2.35 2.60 2.47 2.64 2.22 2.79 2.40 3.39 3.53 2.72 1.94 3.51 1.94 1.93 2.38 2.17 1.99 1.61 2.10 2.25 1.90 2.18 2.29 2.22 2.30 1.89 2.45 2.48 2.46 2.33 2.23 1.89 2.24
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1206 chr21:37872453-37873502 -2.22 1694 chr7:157128968-157129440 -4.06 2.77 2.58 2.14 3.89 2.24 1.33 2.96 2.57 3.16 2.32 2.39 2.74 2.35 2.60 2.47 2.64 2.22 2.79 2.40 3.39 3.53 2.72 1.94 3.51 1.94 1.93 2.38 2.17 1.99 1.61 2.10 2.25 1.90 2.18 2.29 2.22 2.30 1.89 2.45 2.48 2.46 2.33 2.23 1.89 2.24
 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1252 chr22:36748103-36748679 -3.25 1740 chr8:37504905-37506129 2.21 2.24 2.22 2.28 1.98 1.96 2.03 2.32 1.89 1.89 1.75 2.13 1.69 2.31 1.78 1.94 1.79 2.48 2.21 3.71 2.11 2.53 2.32 1.84 1.66 1.86 2.05 1.73 1.96 1.63 1.58 2.29 4.90 2.59 2.50 2.38 2.80 3.62 3.65 3.33 2.06 2.64 7.62 2.88 4.19 3.52
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1252 chr22:36748103-36748679 -3.25 1740 chr8:37504905-37506129 2.21 2.24 2.22 2.28 1.98 1.96 2.03 2.32 1.89 1.89 1.75 2.13 1.69 2.31 1.78 1.94 1.79 2.48 2.21 3.71 2.11 2.53 2.32 1.84 1.66 1.86 2.05 1.73 1.96 1.63 1.58 2.29 4.90 2.59 2.50 2.38 2.80 3.62 3.65 3.33 2.06 2.64 7.62 2.88 4.19 3.52
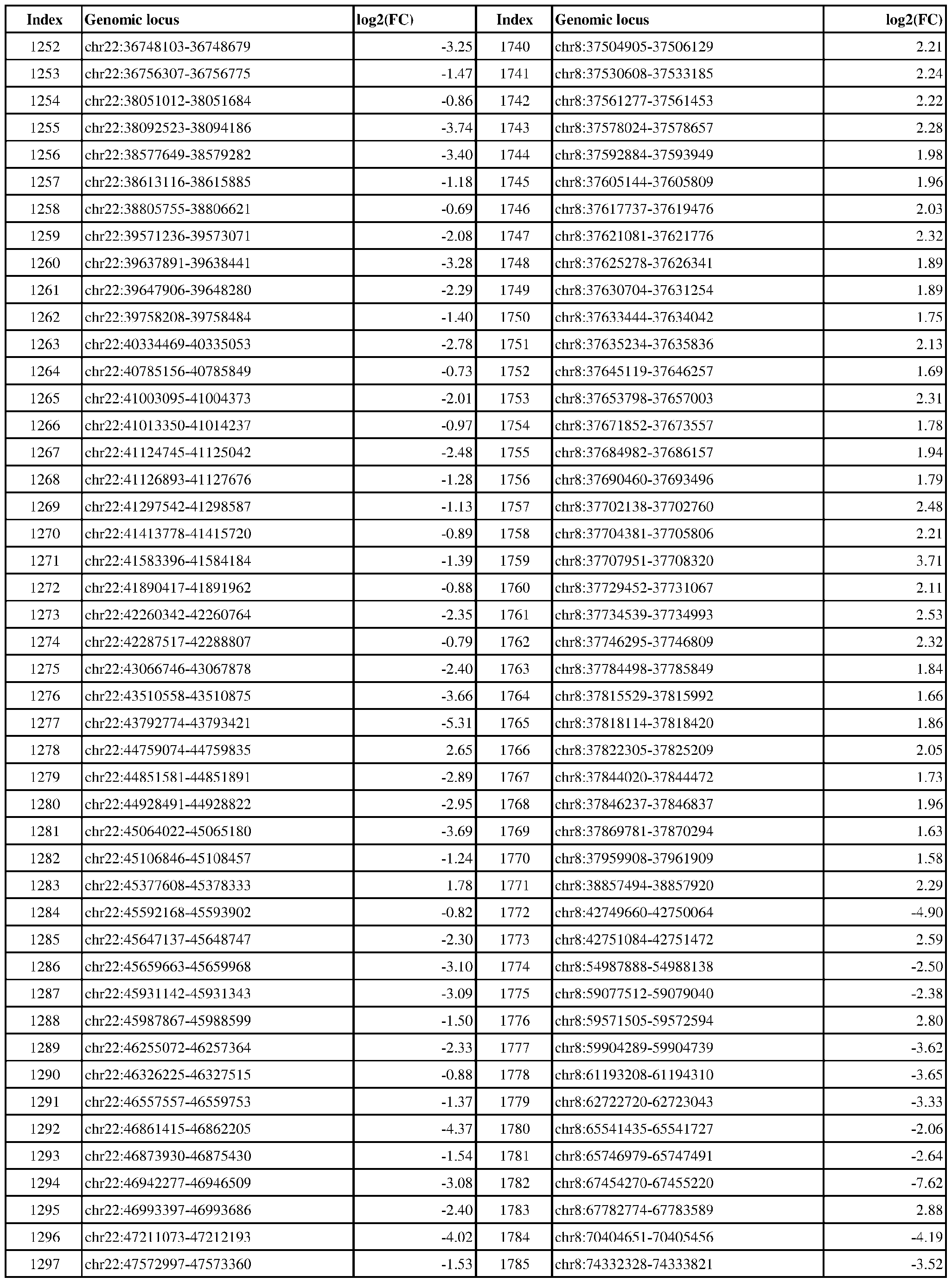 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1298 chr22:47946397-47948802 2.30 1786 chr8:89339016-89341010 -3.92 4.29 4.31 3.48 3.30 2.51 2.57 3.55 2.76 1.90 3.80 3.55 3.29 4.40 3.72 1.56 3.72 2.63 3.69 5.40 2.55 2.20 3.04 3.77 3.29 2.69 3.32 3.04 3.31 4.52 3.53 3.60 3.53 2.55 2.99 3.10 1.89 1.32 2.03 2.75 1.56 2.39 1.56 2.17 2.27 2.18
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1298 chr22:47946397-47948802 2.30 1786 chr8:89339016-89341010 -3.92 4.29 4.31 3.48 3.30 2.51 2.57 3.55 2.76 1.90 3.80 3.55 3.29 4.40 3.72 1.56 3.72 2.63 3.69 5.40 2.55 2.20 3.04 3.77 3.29 2.69 3.32 3.04 3.31 4.52 3.53 3.60 3.53 2.55 2.99 3.10 1.89 1.32 2.03 2.75 1.56 2.39 1.56 2.17 2.27 2.18
 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1344 chr3:170028131-170029230 -1.59 1832 chr9:16340579-16341149 -1.45 1.74 2.14 2.26 2.87 3.13 2.87 2.68 6.50 1.87 1.96 2.90 3.48 4.34 2.57 1.69 1.71 2.51 2.57 2.59 2.64 1.79 2.43 2.12 1.90 2.33 1.87 2.47 2.01 2.64 1.39 2.23 3.10 2.19 3.25 2.94 2.08 3.17 3.34 2.37 5.27 3.52 3.46 1.29 4.62 1.44
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1344 chr3:170028131-170029230 -1.59 1832 chr9:16340579-16341149 -1.45 1.74 2.14 2.26 2.87 3.13 2.87 2.68 6.50 1.87 1.96 2.90 3.48 4.34 2.57 1.69 1.71 2.51 2.57 2.59 2.64 1.79 2.43 2.12 1.90 2.33 1.87 2.47 2.01 2.64 1.39 2.23 3.10 2.19 3.25 2.94 2.08 3.17 3.34 2.37 5.27 3.52 3.46 1.29 4.62 1.44
 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1390 chr4:90031839-90032438 -3.22 1878 chr9:101032569-101032991 1.63 3.20 2.58 2.80 3.18 3.46 1.92 3.27 2.91 3.15 2.28 2.91 2.91 3.34 3.11 3.24 5.26 5.49 4.53 2.31 3.01 2.25 4.96 2.36 3.09 2.78 5.27 4.77 1.22 3.83 1.81 2.21 1.99 3.14 2.81 2.37 2.89 2.62 3.00 4.47 3.70 0.86 1.37 0.93 0.87 3.53
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1390 chr4:90031839-90032438 -3.22 1878 chr9:101032569-101032991 1.63 3.20 2.58 2.80 3.18 3.46 1.92 3.27 2.91 3.15 2.28 2.91 2.91 3.34 3.11 3.24 5.26 5.49 4.53 2.31 3.01 2.25 4.96 2.36 3.09 2.78 5.27 4.77 1.22 3.83 1.81 2.21 1.99 3.14 2.81 2.37 2.89 2.62 3.00 4.47 3.70 0.86 1.37 0.93 0.87 3.53
 Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1436 chr5:72236478-72237635 -1.68 1924 chrX:14047640-14048386 -4.98 1.09 1.26 1.44 1.21 0.88 3.40 4.60 1.25 3.33 3.46 2.91 2.73 2.87 4.25 3.52 3.01 2.93 1.62 1.75 1.52 3.23 2.16 1.77 1.53 2.41 3.01 1.80 3.51
Attorney Docket No.2014191-0027 Index Genomic locus log2(FC) Index Genomic locus log2(FC) 1436 chr5:72236478-72237635 -1.68 1924 chrX:14047640-14048386 -4.98 1.09 1.26 1.44 1.21 0.88 3.40 4.60 1.25 3.33 3.46 2.91 2.73 2.87 4.25 3.52 3.01 2.93 1.62 1.75 1.52 3.23 2.16 1.77 1.53 2.41 3.01 1.80 3.51

Attorney Docket No.2014191-0027 OTHER EMBODIMENTS [0344] It will be appreciated that the scope of the present disclosure is to be defined by that which may be understood from the disclosure and claims rather than by the specific embodiments that have been presented by way of example. Elements described with respect to one aspect or embodiment of the present disclosure are also contemplated with respect to other aspects or embodiments of the present disclosure. For example, elements of claims that depend directly or indirectly from a certain independent claim presented herein serve as support for those elements being presented in additional dependent claims of one or more other independent claims. Throughout the description, where compositions or methods are described as having, including, or comprising specific elements, it is to be understood that compositions or methods that consist essentially of, consist of, or do not comprise the recited elements are likewise hereby disclosed. All references cited herein are hereby incorporated by reference.
12366150v1
Claims
Attorney Docket No.2014191-0027 CLAIMS What is claimed is: 1. A method of determining the ER status of a cancer in a subject, the method comprising: quantifying, at one or more genomic loci in cell-free DNA (cfDNA) from a liquid biopsy sample obtained or derived from the subject, one or more epigenetic biomarkers, wherein the one or more epigenetic biomarkers comprise: (i) one or more histone modifications, (ii) chromatin accessibility, (iii) binding of one or more transcription factors, and/or (iv) DNA methylation; and determining the ER status of the cancer in the subject by comparing the level of the one or more epigenetic biomarkers at the one or more genomic loci to a reference; wherein the one or more genomic loci comprise (i) one or more genomic loci with an increased level of the one or more epigenetic biomarkers in subjects with an ER-positive cancer as compared to subjects with an ER-negative cancer, and/or (ii) one or more genomic loci with an increased level of the one or more epigenetic biomarkers in subjects with ER-negative cancer as compared to subjects with an ER positive cancer. 2. The method of claim 1, wherein: (a) the liquid biopsy sample is a plasma sample, serum sample, or urine sample; (b) the method comprises isolating cfDNA from about 1 mL of the liquid biopsy sample (e.g., plasma sample); and/or (c) the sample comprises a detectable amount of ctDNA (e.g., wherein estimated tumor fraction is >3% for the cfDNA, e.g., as determined by iChorCNA). 3. The method of claim 1 or 2, wherein the one or more histone modifications are quantified using a histone modification assay that measures one or more of H3K9ac, H3K14ac, H3K18ac, H3K23ac, H3K27ac, H3K4me1, H3K4me2, H3K4me3, and pan-acetylation. 4. The method of any one of claims 1-3, wherein:
12366150v1
Attorney Docket No.2014191-0027 (a) the one or more histone modifications are quantified using an assay selected from ChIP-seq (Chromatin ImmunoPrecipitation sequencing), CUT&RUN (Cleavage Under Targets and Release Using Nuclease) sequencing, and CUT&Tag (Cleavage Under Targets and Tagmentation) sequencing; (b) the chromatin accessibility is quantified using a chromatin accessibility assay selected from ATAC-seq (Assay of Transpose Accessible Chromatin sequencing), NOMe-seq (Nucleosome Occupancy and Methylome sequencing), FAIRE-seq (Formaldehyde-Assisted Isolation of Regulatory Elements sequencing), MNase-seq (Micrococcal Nuclease digestion with sequencing), and a DNase hypersensitivity assay; (c) the binding of one or more transcription factors is quantified using a transcription factor binding assay that detects binding of one or more of p300, mediator complex, cohesin complex, RNA pol II, FOXA1, ESR1, PR, MYC, EN1, FOXM1, KLF4, AP-2, RARa, or RUNX1, optionally wherein the transcription factor binding assay is selected from ChIP-seq (Chromatin ImmunoPrecipitation sequencing), CUT&RUN (Cleavage Under Targets and Release Using Nuclease) sequencing, and CUT&Tag (Cleavage Under Targets and Tagmentation) sequencing; and/or (d) DNA methylation is quantified using Bisulfite sequencing (BS-Seq), Whole Genome Bisulfite Sequencing (WGBS), Methylated DNA ImmunoPrecipitation sequencing (MeDIP-seq), or Methyl-CpG-Binding Domain sequencing (MBD-seq). 5. The method of any one of claims 1-4, wherein the method comprises: (a) quantifying H3K4me3 modifications at one or more genomic loci using an assay that comprises enriching for cfDNA comprising one or more H3K4me3 modifications (e.g., using a method that comprises incubating with an agent that binds H3K4me3 modifications) and sequencing the cfDNA enriched for H3K4me3 modifications to determine a count of sequences with one or more H3K4me3 modifications; (b) quantifying H3K27ac modifications at one or more genomic loci using an assay that comprises enriching for cfDNA comprising one or more H3K27ac modifications (e.g., using a method that comprises incubating with an agent that that binds H3K27ac modifications) and sequencing the cfDNA enriched for H3K27ac modifications to determine a count of sequences with one or more H3K27ac modifications; and/or
12366150v1
Attorney Docket No.2014191-0027 (c) quantifying methylated DNA at one or more genomic loci using an assay that comprises enriching for methylated cfDNA (e.g., using a method that comprises incubating with an agent that binds methylated DNA) and sequencing the enriched cfDNA to determine a count of sequences with one or more methylated nucleotides; and optionally: (d) if the method comprises use of an agent that binds H3K4me3 modifications, an agent that binds H3K27ac modifications, and/or an agent that binds methylated DNA, the agent(s) are attached (e.g., via a covalent or noncovalent bond) to a physical support (e.g., a bead, a magnetic bead, an agarose bead, or a magnetic epoxy bead) prior to incubating with the sample; and/or (e) if the method comprises incubating with two or more of an agent that binds H3K4 modifications, an agent that binds H3K27ac modifications, and an agent that binds methylated DNA, the sample is incubated with the two or more agents (i) in sequence or (ii) in parallel (e.g., wherein the sample is divided into fractions and each fraction is incubated with a different agent). 6. The method of any one of claims 1-5, comprising mapping sequence reads to a reference genome, optionally wherein non-uniquely mapped and redundant sequence reads are discarded and/or peaks in high noise regions are removed. 7. The method of claim 6, wherein the one or more genomic loci correspond to sequence read peaks, wherein a sequence read peak corresponds to a region of the genome that has a higher number of sequence reads that the local background. 8. The method of any one of claims 5-7, wherein quantifying H3K4me3 modifications, H3K27ac modifications, and/or DNA methylation comprises summing the number of sequence reads having at least one nucleotide overlap with the one or more genomic loci, optionally wherein: sequence reads are adjusting on the basis of sequencing depth (e.g., quantile normalizing sequence reads to a common reference distribution) and/or ChIP quality prior to summing;
12366150v1
Attorney Docket No.2014191-0027 sequence counts are normalized to aggregate counts in a given sample across a set of regions (e.g., 10,000 regions) previously determined to have DNAse hypersensitivity in most cell types; and/or an estimate of local background signal is subtracted from the sequence reads at each genomic loci prior to summing. 9. The method of any one of claims 1-8, wherein the reference is a predetermined threshold, a measurement from a liquid biopsy sample, a measurement from liquid biopsy samples obtained from a cohort of subjects, and/or a normalized value, wherein: the predetermined threshold and the normalized value were previously shown to distinguish ER-positive and ER-negative subjects (e.g., distinguish with an AUROC of greater than 0.5); the reference is a measurement from a liquid biopsy sample obtained from a cohort of subjects who have previously been determined to have an ER-positive or ER-negative cancer. 10. The method of any one of claims 5-9, comprising calculating sequence read density at the one or more genomic loci, optionally wherein sequence read density is calculated by: (a) summing background adjusted sequence counts at each of the one or more genomic loci and dividing by the sum of the kilobases of the one or more genomic loci; or (b) for each genomic loci, dividing background adjusted fragment count by the number of kilobases of the genomic loci, and then summing for each loci. 11. The method of claim 10, comprising calculating an ER-positive/ER-negative ratio score by a method comprising: (a) calculating an ER-positive sequence read density by a method comprising summing background adjusted sequence counts at each of the one or more genomic loci with an increased level of the one or more epigenetic biomarkers in sample(s) obtained from subjects with an ER-positive cancer as compared to samples obtained from subjects with ER-negative cancer; (b) calculating an ER-negative sequence read density by a method comprising summing background adjusted sequence counts at each of the one or more genomic loci with an
12366150v1
Attorney Docket No.2014191-0027 increased level of the one or more epigenetic biomarkers in sample(s) obtained from subjects with an ER-negative cancer as compared to samples obtained from subjects with an ER-positive cancer; and (c) dividing the ER-positive sequence read density by the ER-negative sequence read density. 12. The method of claim 11, comprising: (a) determining an ER-positive/ER-negative ratio score for H3K4me3 modifications; (b) determining an ER-positive/ER-negative ratio score for H3K27ac modifications; and/or (c) determining an ER-positive/ER-negative ratio score for methylated DNA; and if each of (a)-(c) is performed, optionally combining each of the ratio scores (e.g., using fitted values determined using a logistic regression). 13. The method of any one of claims 1-12, wherein the method comprises quantifying the one or more of the epigenetic biomarkers at one or more genomic loci in Tables 1-3; optionally, wherein the method comprises quantifying: (a) H3K4me3 modifications for at least 5, 10, 20, 30, 40, or 50 genomic loci listed in Table 1; (b) H3K27ac modifications for at least 5, 10, 20, 30, 40, or 50 genomic loci listed in Table 2; and/or (c) DNA methylation for at least 5, 10, 20, 30, 40, or 50 genomic loci listed in Table 3. 14. The method of any one of claims 1-13, wherein the method provides an area under the receiver operating characteristic (AUROC) for determining if a subject has an ER-positive cancer vs. an ER-negative cancer of greater than 0.5 (e.g., greater than 0.55, greater than 0.6, greater than 0.65, greater than 0.7, greater than 0.75, greater than 0.8, greater than 0.85, greater than 0.9, or greater than 0.95).
12366150v1
Attorney Docket No.2014191-0027 15. The method of any one of claims 1-14, wherein the subject has previously been determined to have cancer, the subject has an increased susceptibility to cancer, and/or wherein the method further comprises determining whether the subject has cancer. 16. The method of any one of claims 1-15, wherein the ER-positive cancer is an ER-positive cancer based on IHC testing and the ER-negative cancer is an ER-negative cancer based on IHC testing. 17. The method of any one of claims 1-16, wherein the cancer is breast cancer (e.g., metastatic breast cancer), ovarian cancer, or endometrial cancer. 18. A method of treating a subject having a cancer, the method comprising: administering a cancer therapy to the subject based on the ER status of the cancer, wherein the ER status of the cancer has been determined using the method of any one of claims 1-17, and wherein: (a) if the cancer has been determined to be ER-positive, the cancer therapy administered comprises an ER-targeted agent; and (b) if the cancer has been determined to be ER-negative, not administering an ER- targeted agent, optionally wherein the therapy administered is one that is suitable for an ER- negative cancer (e.g., a therapy that (i) does not comprise administering an ER-targeted agent, (ii) comprises surgery, and/or (iii) comprises radiation therapy). 19. A method of monitoring the ER status of a cancer in a subject, and optionally treating the cancer, the method comprising determining the ER status of the cancer using the method of any one of claims 1-17 at first and second time points, optionally wherein the subject has been administered an ER-targeted agent at or prior to the first time point, or after the first time point and before the second time point. 20. The method of claim 18 or 19, further comprising administering a cancer therapy, optionally an ER-targeted agent, to the subject based on the ER status of the cancer at the second
12366150v1
Attorney Docket No.2014191-0027 time point, optionally wherein the type, dose and/or frequency of administration of the cancer therapy is adjusted based on the ER status of the cancer at the second time point. 21. A method of treating a subject having a cancer, the method comprising: administering an ER-targeted agent to the subject if the subject has been determined to have a validated epigenetic profile indicative of an ER-positive cancer based on analysis of a biological sample, optionally of cell-free DNA (cfDNA) from a liquid biopsy sample, obtained or derived from the subject, and not administering the ER-targeted agent if the subject has not been determined to have a validated epigenetic profile indicative of an ER-positive cancer, wherein the presence of the validated epigenetic profile has been determined using a validated classifier, wherein the validated classifier has been obtained by: (a) determining a genomic profile of one or more histone modifications, chromatin accessibility, binding of one or more transcription factors, and/or DNA methylation in (i) one or more ER-positive cell lines or (ii) biological samples obtained from a first cohort of subjects who have previously been determined to have an ER- positive cancer; (b) determining a genomic profile of one or more histone modifications, chromatin accessibility, binding of one or more transcription factors, and/or DNA methylation in (i) one or more ER-negative cell lines or (ii) biological samples obtained from a second cohort of healthy subjects or subjects who have previously been determined to have an ER-negative cancer; (c) comparing the genomic profile determined in step (a) and the genomic profile determined in step (b), to identify genomic loci that have statistically different histone modification, chromatin accessibility, binding of transcription factor, and/or DNA methylation levels (“differential loci”); (d) training a classifier on histone modification, chromatin accessibility, binding of transcription factor, and/or DNA methylation levels in the differential loci to distinguish between (i) samples from one or more ER-positive cell lines or biological samples obtained from the first cohort, and (ii) samples from one or more ER-negative cell lines or biological samples obtained from the second cohort, to identify samples
12366150v1
Attorney Docket No.2014191-0027 having a profile of histone modification, chromatin accessibility, binding of transcription factor, and/or DNA methylation levels (“epigenetic profile”) that indicates that the samples are likely obtained from an ER-positive cell line or from the first cohort; and (e) obtaining the validated classifier by validating the classifier from step (d) on a third cohort comprising an independent and group of subjects with ER-positive and ER-negative cancers and selecting a threshold such that the validated classifier predicts ER-positive cancers, with an area under the receiver operating characteristic (AUROC) greater than 0.5 (e.g., greater than 0.55, greater than 0.6, greater than 0.65, greater than 0.7, greater than 0.75, greater than 0.8, greater than 0.85, greater than 0.9, or greater than 0.95), wherein subjects falling within the group of predicted ER-positive cancers display the validated epigenetic profile and subjects that do not fall within the group of ER- positive cancers lack the validated epigenetic profile. 22. The method of claim 21, wherein: (a) the differential loci in step (c) were identified by comparing the genomic profile of one or more histone modifications and/or DNA methylation in (i) one or more ER-positive cell lines and (ii) one or more ER-negative cell lines; (b) the classifier in step (d) was trained on histone modification, chromatin accessibility, binding of transcription factor, and/or DNA methylation levels in the differential loci that were obtained by in silico mixing sequence data from one or more ER-positive cell lines and sequence data obtained from liquid biopsy samples of healthy subjects; (c) the validated classifier in step (e) was validated using liquid biopsy samples from the third cohort; (d) the classifier in step (d) was trained on one or more (e.g., two or more) histone modification levels and/or DNA methylation levels in the differential loci, optionally wherein the one or more histone modification levels comprise H3K4me3 and H3K27ac modification levels; and/or (e) the classifier in step (d) was trained using ridge regression, elastic-net regression, or lasso regression.
12366150v1
Attorney Docket No.2014191-0027 23. A kit comprising reagents for quantifying one or more histone modifications, chromatin accessibility, binding of one or more transcription factors, and/or DNA methylation at one or more genomic loci, wherein the one or more genomic loci are selected from Tables 1-3; optionally, wherein the kit comprises reagents for quantifying: (a) H3K4me3 modifications for at least 5, 10, 20, 30, 40, or 50 genomic loci in Table 1; (b) H3K27ac modifications for at least 5, 10, 20, 30, 40, or 50 genomic loci in Table 2; (c) DNA methylation for at least 5, 10, 20, 30, 40, or 50 genomic loci in Table 3; or (d) any combination of (a)-(c). 24. The kit of claim 23, wherein the kit comprises: (a) one or more antibodies for use in ChIP-seq, optionally wherein the one or more antibodies specifically bind H3K4me3- or H3K27ac-modified histones; (b) one or more methyl-binding domains for use in MBD-seq or one or more antibodies that bind methylated DNA for use in MeDIP; (c) reagents for isolation of cell-free DNA (cfDNA) from a liquid biopsy sample; (d) reagents for library preparation for sequencing; (e) reagents for sequencing; (f) instructions for determining if a subject has an ER-positive cancer; or (g) any combination of (a)-(f). 25. A non-transitory computer readable storage medium encoded with a computer program, wherein the program comprises instructions that when executed by one or more processors cause the one or more processors to perform operations to perform the method of any one of claims 1- 22. 26. A computer system comprising a memory and one or more processors coupled to the memory, wherein the one or more processors are configured to perform operations to perform the method of any one of claims 1-22.
12366150v1
Attorney Docket No.2014191-0027 27. A system for determining the ER status of a cancer in a subject, the system comprising a sequencer configured to generate a sequencing dataset from a sample; and a non-transitory computer readable storage medium of claim 25 and/or a computer system of claim 26, optionally wherein the sequencer is configured to generate a Whole Genome Sequencing (WGS) dataset from the sample. 28. The system of claim 26 or 27 further comprising: a sample preparation device configured to prepare the sample for sequencing from a biological sample (e.g., a liquid biopsy sample); optionally wherein the sample preparation device comprises reagents for quantifying one or more histone modifications, chromatin accessibility, binding of one or more transcription factors, and/or DNA methylation at one or more genomic loci in cell-free DNA (cfDNA) from the biological sample, optionally the liquid biopsy sample. 29. The system of any one of claims 26-28, wherein the one or more genomic loci are selected from Tables 1-3; optionally, wherein: (a) the device comprises reagents for quantifying H3K4me3, e.g., for at least 5, 10, 20, 30, 40, or 50 genomic loci in Table 1; (b) the device comprises reagents for quantifying H3K27ac, e.g., for at least 5, 10, 20, 30, 40, or 50 genomic loci in Table 2. (c) the device comprises reagents for quantifying DNA methylation, e.g., for at least 5, 10, 20, 30, 40, or 50 genomic loci in Table 3; or (d) a combination of (a)-(c). 30. The system of claim 29, wherein: (a) the reagents comprise one or more antibodies for use in ChIP-seq, optionally wherein the one or more antibodies specifically bind H3K4me3- or H3K27ac-modified histones; (b) the reagents comprise one or more methyl-binding domains for use in MBD-seq; (c) the device comprises reagents for isolation of cell-free DNA (cfDNA) from the biological sample, optionally the liquid biopsy sample;
12366150v1
Attorney Docket No.2014191-0027 (d) the device comprises reagents for library preparation for sequencing; and/or (e) the sequencer comprises reagents for sequencing. 31. A method of determining the ER status of a cancer in a subject (e.g., patient), the method comprising: receiving (e.g., by a processor of a computing device) one or more genomic profiles of one or more histone modifications, chromatin accessibility, binding of one or more transcription factors, and/or DNA methylation for the subject; and determining whether the subject has an epigenetic profile indicative of an ER-positive cancer by classifying (e.g., by the processor) the genomic profile using the ER classifier. 32. The method of claim 31, wherein the ER classifier has been trained using one or more genomic profiles of one or more histone modifications, chromatin accessibility, binding of one or more transcription factors, and/or DNA methylation for (i) one or more ER-positive cell lines and one or more ER-negative cell lines and/or (ii) one or more biological samples obtained from one or more cohorts of subjects who have previously been determined to have an ER-positive cancer and one or more biological samples obtained from one or more cohorts of subjects who have previously been determined to have an ER-negative cancer, optionally wherein the one or more genomic profiles used to train the ER classifier comprise one or more genomic profiles generated by in silico diluting sequence data from ER- positive or ER-negative cell lines with sequence data obtained from healthy donor plasma samples so as to achieve a simulated ctDNA percentage ranging from 0.5% to 50%. 33. The method of claim 32, wherein the one or more genomic profiles used to train the ER classifier are for differential loci having statistically significant differences in levels of one or more histone modifications, chromatin accessibility, binding of one or more transcription factors, and/or DNA methylation levels in (a) one or more ER-positive cell lines as compared to one or more ER- negative cell lines and/or (b) one or more biological samples obtained from one or more cohorts of subjects who have previously been determined to have an ER-positive cancer and one or more biological samples obtained from one or more cohorts of subjects who have previously been determined to have an ER-negative cancer,
12366150v1
Attorney Docket No.2014191-0027 optionally wherein the differential loci were identified by comparing genomic profiles of one or more histone modifications and/or DNA methylation in (i) one or more ER-positive cell lines and (ii) one or more ER-negative cell lines. 34. The method of any one of claims 31-33, wherein the ER classifier has been trained using: (a) genomic profiles of two or more histone modification levels in the differential loci or, (b) genomic profiles of one or more histone modification levels and DNA methylation levels in the differential loci. 35. The method of any one of claims 31-34, comprising receiving: (a) one or more genomic profiles of two or more histone modification levels, optionally wherein the two or more histone modifications comprise H3K4me3 and H3K27ac modifications; (b) one or more genomic profiles of one or more histone modifications and DNA methylation, optionally wherein the one or more histone modifications comprise H3K4me3 and/or H3K27ac modifications. 36. The method of any one of claims 31-35, wherein the ER classifier is a validated classifier, wherein the ER classifier has been validated by selecting a threshold such that the validated classifier predicts ER-positive cancers with an area under the receiver operating characteristic (AUROC) greater than 0.5 (e.g., greater than 0.55, greater than 0.6, greater than 0.65, greater than 0.7, greater than 0.75, greater than 0.8, greater than 0.85, greater than 0.9, or greater than 0.95), and optionally wherein: (a) the ER classifier has been validated on a a group of subjects with ER-positive or ER-negative cancers, wherein subjects falling within a group of predicted ER-positive cancers display a validated epigenetic profile and subjects that do not fall within a group of ER-positive cancers lack the validated epigenetic profile; and/or (b) the ER classifier has been validated using liquid biopsy sample data.
12366150v1
Attorney Docket No.2014191-0027 37. A non-transitory computer readable storage medium encoded with a computer program, wherein the program comprises instructions that when executed by one or more processors cause the one or more processors to perform operations to perform the method of any one of claims 31- 36. 38. A computer system comprising a memory and one or more processors coupled to the memory, wherein the one or more processors are configured to perform operations to perform the method of any one of claims 31-37. 39. A method of treating a subject having a cancer, the method comprising: administering an ER-targeted agent to the subject, wherein the subject has been determined to have a validated epigenetic profile indicative of an ER-positive cancer based on analysis of a biological sample, optionally of cell-free DNA (cfDNA) from a liquid biopsy sample, obtained or derived from the subject, wherein the presence of the validated epigenetic profile has been determined using a classifier (e.g., a validated classifier) according to a method of any one of claims 31-37.
12366150v1
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US202363590158P | 2023-10-13 | 2023-10-13 | |
| US63/590,158 | 2023-10-13 | ||
| US202463650860P | 2024-05-22 | 2024-05-22 | |
| US63/650,860 | 2024-05-22 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2025081094A2 true WO2025081094A2 (en) | 2025-04-17 |
| WO2025081094A3 WO2025081094A3 (en) | 2025-08-07 |
Family
ID=93292057
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2024/051117 Pending WO2025081094A2 (en) | 2023-10-13 | 2024-10-11 | Methods, kits and systems for determining the er status of cancer and methods for treating cancer based on same |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2025081094A2 (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2025245302A1 (en) | 2024-05-22 | 2025-11-27 | Precede Biosciences, Inc. | Methods, kits and systems for determining er activity of cancer and methods for treating cancer based on same |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5308341A (en) | 1993-09-28 | 1994-05-03 | Becton, Dickinson And Company | Method of testing the dose accuracy of a medication delivery device |
| US6146361A (en) | 1996-09-26 | 2000-11-14 | Becton Dickinson And Company | Medication delivery pen having a 31 gauge needle |
| US6192891B1 (en) | 1999-04-26 | 2001-02-27 | Becton Dickinson And Company | Integrated system including medication delivery pen, blood monitoring device, and lancer |
| US6277099B1 (en) | 1999-08-06 | 2001-08-21 | Becton, Dickinson And Company | Medication delivery pen |
| US7556615B2 (en) | 2001-09-12 | 2009-07-07 | Becton, Dickinson And Company | Microneedle-based pen device for drug delivery and method for using same |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2024515565A (en) * | 2021-04-08 | 2024-04-10 | フレッド ハッチンソン キャンサー センター | Cell-free DNA sequencing data analysis methods to investigate nucleosome protection and chromatin accessibility |
-
2024
- 2024-10-11 WO PCT/US2024/051117 patent/WO2025081094A2/en active Pending
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5308341A (en) | 1993-09-28 | 1994-05-03 | Becton, Dickinson And Company | Method of testing the dose accuracy of a medication delivery device |
| US6146361A (en) | 1996-09-26 | 2000-11-14 | Becton Dickinson And Company | Medication delivery pen having a 31 gauge needle |
| US6200296B1 (en) | 1996-09-26 | 2001-03-13 | Becton Dickinson And Company | 5mm injection needle |
| US6192891B1 (en) | 1999-04-26 | 2001-02-27 | Becton Dickinson And Company | Integrated system including medication delivery pen, blood monitoring device, and lancer |
| US6277099B1 (en) | 1999-08-06 | 2001-08-21 | Becton, Dickinson And Company | Medication delivery pen |
| US7556615B2 (en) | 2001-09-12 | 2009-07-07 | Becton, Dickinson And Company | Microneedle-based pen device for drug delivery and method for using same |
Non-Patent Citations (43)
| Title |
|---|
| "Biocomputing: Informatics and Genome Projects", 1994, ACADEMIC PRESS |
| "Computational Molecular Biology", 1988, OXFORD UNIVERSITY PRESS |
| "Sequence Analysis in Molecular Biology", 1987, ACADEMIC PRESS |
| ADALSTEINSSON ET AL., NAT COMM., 2017 |
| ADALSTEINSSON ET AL., NAT COMMUN, vol. 8, no. 1, 2017, pages 1324 |
| ADLANMERINI ET AL., PROC NATL ACAD SCI USA, vol. 111, 2014, pages 283 - 290 |
| ALLRED ET AL., BREAST CANCER RES, vol. 6, 2004, pages 240 - 245 |
| ALTSCHUL ET AL., J MOL BIOL, vol. 215, 1990, pages 403 - 410 |
| AMEMIYA ET AL., SCI REP, vol. 9, no. 1, 2019, pages 9354 |
| ANKER ET AL., CANCER AND METASTASIS REV, vol. 18, 1999, pages 65 - 73 |
| ARNAL ET AL., PHYSIOL REV, vol. 97, 2017, pages 1045 - 1087 |
| AUERBACH ET AL., PROC NATL ACAD USA, vol. 106, no. 35, 2009, pages 14926 - 14931 |
| BERGMANCEDAR, NAT STRUCT MOL BIOL, vol. 20, 2013, pages 274 - 281 |
| BUENROSTRO ET AL., NAT METHODS, vol. 10, no. 12, 2013, pages 1213 - 1218 |
| FIEGL ET AL., CANCER RES, vol. 15, 2005, pages 1141 - 1145 |
| FOLKERDDOWSETT, J CLIN ONCOL, vol. 28, 2010, pages 4038 - 4044 |
| HEWITTKORACH, ENDOCR REV, vol. 39, no. 5, 2018, pages 664 - 675 |
| HIGGINSSHARP, COMP APPL BIOSCI, vol. 5, no. 2, 1989, pages 151 - 153 |
| IKEDA ET AL., ACTA PHARMACOL SIN, vol. 36, 2015, pages 24 - 31 |
| J. R. ROBINSON: "Sustained and Controlled Release Drug Delivery Systems", 1978, MARCEL DEKKER, INC. |
| JANG ET AL., LIFE SCI ALLIANCE, vol. 6, no. 12, 2023, pages e202302003 |
| JENSEN, CANCER, vol. 46, 1980, pages 2759 - 2761 |
| KAYA-OKUR ET AL., NAT COMM, vol. 10, 2019, pages 1930 |
| KNUTSON ET AL., J HEMATOL ONCOL, vol. 10, 2017, pages 168 |
| LIN ET AL., BIOINFORMATICS, vol. 20, 2004, pages 1233 - 1240 |
| LOVE ET AL., GENOME BIOL, vol. 15, no. 12, 2014, pages 550 |
| MEISSNER ET AL., NUCLEIC ACIDS RES, vol. 33, no. 18, 2005, pages 5868 - 5877 |
| OGSTON ET AL., BREAST, vol. 12, 2003, pages 320 - 327 |
| OMARJEE ET AL., ONCOGENE, vol. 36, 2017, pages 2503 - 2514 |
| PATHAK ET AL., CLIN CHEM, vol. 52, 2006, pages 1833 - 1842 |
| PEARSON: "Comput Methods Genome Res [Proc Int Symp] (1994", 1992, OXFORD UNIVERSITY PRESS, pages: 111 - 120 |
| PRATTTOFT, ENDOCR REV, vol. 18, 1997, pages 306 - 360 |
| ROBERTSON, NAT REV GENET, vol. 6, 2005, pages 597 - 610 |
| SADEH ET AL., NAT BIOTECHNOL, vol. 39, 2021, pages 586 - 598 |
| SCHONES ET AL., CELL, vol. 132, no. 5, 2008, pages 887 - 898 |
| SCHWARZENBACH ET AL., CLIN CANCER RES, vol. 15, 2009, pages 1032 - 1038 |
| SCHWARZENBACH ET AL., NAT REV CANCER, vol. 11, 2011, pages 426 - 437 |
| SKENE ET AL., NAT PROTOC, vol. 13, 2018, pages 1006 - 1019 |
| SKENEHENIKOFF, ELIFE, vol. 6, 2017, pages 1 - 35 |
| SYMMANS ET AL., J CLIN ONCOL, vol. 25, 2007, pages 4414 - 4422 |
| WEBER ET AL., NAT GENET, vol. 37, 2005, pages 853 - 862 |
| WELBOREN ET AL., EMBO J, vol. 28, 2009, pages 1418 - 1428 |
| WUA ET AL., CLIN CHIM ACTA, vol. 321, 2002, pages 77 - 87 |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2025245302A1 (en) | 2024-05-22 | 2025-11-27 | Precede Biosciences, Inc. | Methods, kits and systems for determining er activity of cancer and methods for treating cancer based on same |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2025081094A3 (en) | 2025-08-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US10706954B2 (en) | Systems and methods for identifying responders and non-responders to immune checkpoint blockade therapy | |
| US12525318B2 (en) | Method of monitoring anti-TNF therapy in a subject suffering from rheumatoid arthritis based in part on a trained machine learning classifier | |
| JP2023500054A (en) | Classification of the tumor microenvironment | |
| US20220319638A1 (en) | Predicting response to treatments in patients with clear cell renal cell carcinoma | |
| US20240161868A1 (en) | System and method for gene expression and tissue of origin inference from cell-free dna | |
| WO2022197968A1 (en) | Methods of classifying and treating patients | |
| JP2023538963A (en) | Methods and systems for predicting response to anti-TNF therapy | |
| WO2025081094A2 (en) | Methods, kits and systems for determining the er status of cancer and methods for treating cancer based on same | |
| EP4423301A1 (en) | Tumor microenvironment types in breast cancer | |
| JP7772700B2 (en) | Methods for treating glioblastoma | |
| WO2025081100A1 (en) | Methods, kits and systems for determining the her2 status of cancer and methods for treating cancer based on same | |
| WO2025245302A1 (en) | Methods, kits and systems for determining er activity of cancer and methods for treating cancer based on same | |
| WO2025081121A1 (en) | Methods, kits and systems for determining the status of lung cancer and methods for treating lung cancer based on same | |
| WO2025213150A1 (en) | Methods, kits and systems for measuring psa and psma expression and methods for treating cancer based on same | |
| CA3065568C (en) | Systems and methods for identifying responders and non-responders to immune checkpoint blockade therapy | |
| US20260038636A1 (en) | Methods of treating cancer | |
| WO2025111249A2 (en) | Methods, kits and systems for determining sarcomatoid differentiation of renal cell carcinoma and methods for treating based on the same | |
| Honda et al. | A key molecular driver of tumor-infiltrating lymphocytes in invasive breast cancer on machine learning–based meta-mining research | |
| HK40022696B (en) | Systems and methods for identifying responders and non-responders to immune checkpoint blockade therapy | |
| Fröhling et al. | Multi-Layered Molecular Profiling Informs the Diagnosis and Targeted Therapy of Desmoplastic Small Round Cell Tumor |