WO2025078841A2 - Antibodies, conjugates, and uses thereof - Google Patents
Antibodies, conjugates, and uses thereof Download PDFInfo
- Publication number
- WO2025078841A2 WO2025078841A2 PCT/GB2024/052627 GB2024052627W WO2025078841A2 WO 2025078841 A2 WO2025078841 A2 WO 2025078841A2 GB 2024052627 W GB2024052627 W GB 2024052627W WO 2025078841 A2 WO2025078841 A2 WO 2025078841A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- identity
- seq
- antibody
- antigen
- binding fragment
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2863—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against receptors for growth factors, growth regulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6801—Drug-antibody or immunoglobulin conjugates defined by the pharmacologically or therapeutically active agent
- A61K47/6803—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6801—Drug-antibody or immunoglobulin conjugates defined by the pharmacologically or therapeutically active agent
- A61K47/6803—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates
- A61K47/68031—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates the drug being an auristatin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6801—Drug-antibody or immunoglobulin conjugates defined by the pharmacologically or therapeutically active agent
- A61K47/6803—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates
- A61K47/68037—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates the drug being a camptothecin [CPT] or derivatives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6849—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a receptor, a cell surface antigen or a cell surface determinant
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6883—Polymer-drug antibody conjugates, e.g. mitomycin-dextran-Ab; DNA-polylysine-antibody complex or conjugate used for therapy
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6889—Conjugates wherein the antibody being the modifying agent and wherein the linker, binder or spacer confers particular properties to the conjugates, e.g. peptidic enzyme-labile linkers or acid-labile linkers, providing for an acid-labile immuno conjugate wherein the drug may be released from its antibody conjugated part in an acidic, e.g. tumoural or environment
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/505—Medicinal preparations containing antigens or antibodies comprising antibodies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/54—Medicinal preparations containing antigens or antibodies characterised by the route of administration
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/545—Medicinal preparations containing antigens or antibodies characterised by the dose, timing or administration schedule
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/20—Immunoglobulins specific features characterized by taxonomic origin
- C07K2317/24—Immunoglobulins specific features characterized by taxonomic origin containing regions, domains or residues from different species, e.g. chimeric, humanized or veneered
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/55—Fab or Fab'
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/60—Immunoglobulins specific features characterized by non-natural combinations of immunoglobulin fragments
- C07K2317/62—Immunoglobulins specific features characterized by non-natural combinations of immunoglobulin fragments comprising only variable region components
- C07K2317/622—Single chain antibody (scFv)
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/90—Immunoglobulins specific features characterized by (pharmaco)kinetic aspects or by stability of the immunoglobulin
- C07K2317/92—Affinity (KD), association rate (Ka), dissociation rate (Kd) or EC50 value
Definitions
- Cancer is a leading cause of global deaths, and according to the World Health Organisation (https://qco.iarc.fr') accounted for 10 million deaths in 2020.
- the most common cancers include lung ( ⁇ 1.8 million cases/year), colon and rectum ( ⁇ 935 000), liver ( ⁇ 830 000), stomach ( ⁇ 760 000) and breast ( ⁇ 685 000).
- Poor permeation [1] and multi-drug resistance [2] in solid tumours are two important challenges which lead to the failure of current treatment methods.
- the tumour microenvironment (TME) in solid tumours poses physical challenges for the effective and homogenous delivery of cancer treatments.
- Intra- tumoral drug penetration is hindered by the physical barriers brought on by the tumour stroma, disruption and disorganisation of the extracellular matrix increasing tissue stiffness/solid stress, impaired lymphatic drainage which consequently increases hydrostatic pressure and increased vascular permeability 'leaky vasculature' causing irregular intra-tumour blood flow [3]. Poor drug penetration and distribution leads to marginal doses reaching the tumour resulting in acquired resistance and treatment failure [1].
- ADCs Antibody-Drug Conjugates
- MAb monoclonal antibody
- An alternative approach is to improve penetration and diffusion using smaller formats such as antibody fragments (e.g., Fab fragments ( ⁇ 50 kDa), single-chain variable fragments (scFv ⁇ 30 kDa) and single-domain antibodies (sdAb 12-15 kDa)) and peptides [11].
- antibody fragments e.g., Fab fragments ( ⁇ 50 kDa), single-chain variable fragments (scFv ⁇ 30 kDa) and single-domain antibodies (sdAb 12-15 kDa)
- peptides e.g., Fab fragments ( ⁇ 50 kDa), single-chain variable fragments (scFv ⁇ 30 kDa) and single-domain antibodies (sdAb 12-15 kDa)
- peptides e.g., Fab fragments ( ⁇ 50 kDa), single-chain variable fragments (scFv ⁇ 30 kDa) and single-domain antibodies (sdAb 12-15 kDa
- ADCs Due to its targeting/localization effect, ADCs increase the tumour concentration of the targeted payload, therefore lower doses are required to reach the tumour to effectively destroy the targeted cells. More recent thinking suggests that ADCs have a similar maximum tolerated dose as the equivalent payload but improve efficacy though targeted delivery [13]. Moreover, depending on the chemical nature of the drug and its release in the tumour (either intracellular or extracellular), some payloads can subsequently diffuse and kill surrounding non-targeted tumour cells (“bystander killing”) [14]. Consequently, these features, in some way, help ameliorate the drawbacks of the heterogeneous tumour distribution, but does not solve the problem, especially for intractable or poorly vascularised tumours.
- ADCs An estimated 100 or more other ADCs are currently under clinical development (www.clinicaltrials.gov but the failure rate for solid tumours is disproportionately higher compared to haematological cancers (https ://beacon-intelligence.com).
- the availability of target antigens expressed on the surface of tumour cells and the approval of the use of ADCs have seen success in the area of haematological malignancies.
- Solid tumours however display antigens which are tumour associated and have varying levels of overexpression. These antigens are not exclusively found on the surface of these solid tumours but are also expressed on normal tissues at lower levels bringing about challenges with on-target off-tumour toxicity [6].
- trastuzumab-emtansine was able to broadly improve patient outcomes compared to the non-conjugated trastuzumab MAb and this was further improved by trastuzumab-deruxtecan [5,6,18] which is now widely acknowledged to have a transformative clinical impact in HER2-positive breast cancer [18].
- cMET overexpression relative to that of normal tissues has been observed in many types of solid human malignancies including gastric, colorectal, pancreatic, lung, head and neck, ovarian, breast, renal, prostate, bladder, nasopharyngeal, gliomas, osteosarcomas and melanomas (Table 1). Due to the poor clinical prognosis of solid tumours overexpressing cMET, and the toxicities caused by broad-spectrum chemotherapeutic treatments used today as well as insufficient drug dosage reaching all parts of the tumour, the inventors identified an opportunity which is presented for the development of targeted therapeutics against this receptor. The inventors have developed novel anti-cMET antibodies and antigen-binding fragments thereof, along with antibody-drug and fragment-drug conjugates. These antibodies have surprising beneficial properties in terms of cMET binding, and tumour penetration and clearance.
- the invention provides an antibody or antigen-binding fragment thereof that specifically binds to the MET receptor (cMET), wherein the antibody or antigen-binding fragment thereof comprises a heavy chain variable region (VH) comprising the Complementarity Determining Region (CDRs) CDR-H1, CDR-H2, and CDR-H3 of SEQ ID NO. 1, or a variant thereof having at least 80% sequence identity to SEQ ID NO. 1, and comprises a light chain variable region (VL) comprising the CDRs CDR-L1, CDR-L2, and CDR-L3 of SEQ ID NOs. 196 or 2, or a variant thereof having at least 80% sequence identity with SEQ ID NOs. 196 or 2.
- VH heavy chain variable region
- CDRs Complementarity Determining Region
- CDR-H2 Complementarity Determining Region
- VL light chain variable region
- an antibody or antigen-binding fragment thereof that specifically binds to the MET receptor (cMET), wherein: (i) the antibody or antigen-binding fragment thereof comprises a heavy chain variable region (VH) comprising the Complementarity Determining Regions (CDRs) CDR- Hl, CDR-H2, and CDR-H3 of SEQ ID NO. 1, or a variant thereof having at least 90% sequence identity to SEQ ID NO.
- VH heavy chain variable region
- CDRs Complementarity Determining Regions
- VL light chain variable region
- CDRs Complementarity Determining Regions
- CDRs Complementarity Determining Regions
- the antibody or antigen-binding fragment thereof comprises a VL comprising the CDRs of SEQ ID NO. 196.
- the antibodies and antigen-binding fragments disclosed herein have advantageous properties in terms of cMET binding and blocking of cMET signalling.
- the application also discloses the antibodies and antigen-binding fragments thereof conjugated to payloads which demonstrate advantageous properties in tumour penetration, tumour payload delivery, serum half-life and bioavailability, normal tissue exposure and tumour clearance.
- cMET we mean the MET receptor, which is also referred to as hepatocyte growth factor receptor, HGF receptor, HGFR, and MET.
- the mesenchymal-epithelial transition factor gene referred to as MET is a proto-oncogene which encodes a cell surface tyrosine kinase receptor (cMET) for hepatocyte growth factor (HGF)/scattering factor (SF) ligand.
- cMET Upon ligand binding cMET forms a homo-dimer leading to trans-phosphorylation which activates several pathways including phosphoinositide 3-kinase (P13K)/AKT (protein kinase B), mitogen-activated protein kinase Ras/(MAPK), Janus kinase/signal transducers and activators of transcription (JAK/STAT), SRC and WNT/P catenin [19,20].
- P13K phosphoinositide 3-kinase
- AKT protein kinase B
- mitogen-activated protein kinase Ras/(MAPK) mitogen-activated protein kinase Ras/(MAPK)
- JK/STAT Janus kinase/signal transducers and activators of transcription
- SRC WNT/P catenin
- the HGF/cMET pathway in normal physiology is involved in embryogenesis, normal cellular growth, cell motility, cell survival, wound healing, angiogenesis and tissue regeneration.
- the HGF/cMET pathway regulates disease progression by reducing oxidative stress, inflammation, apoptosis and fibrosis.
- cMET is expressed in vascular, lymphatic endothelial cells and hematopoietic cells, skin, GI tract and lungs at low normal levels and can increase upon tissue repair, regeneration or during inflammation.
- cMET is also expressed at low levels in normal liver, kidney, pancreas, prostate, muscle and bone marrow, during embryogenesis and adulthood [19].
- MET overexpression, genomic amplification, translocations, sub-gene deletions, point mutations and alternative splicing can lead to the aberrant expression of the MET protooncogene. This is a major cause of the dysregulation of the cMET autocrine and paracrine signalling pathway [21].
- Over-expression of the HGF ligand can also lead to aberrant cMET signalling. These alterations, leading to cMET activation have been observed in many human malignancies associated with invasive growth, tumour cell motility, angiogenesis, and poor clinical prognosis [20-23].
- cMET expression strongly and significantly correlates with poor prognosis and patient outcomes in multiple clinical examples of cancer and is highly implicated in other cancers (Table 1).
- MET gene amplification frequency in solid tumours ( ⁇ 2%) is described in the AACR GENIE registry (https://www.aacr.org/professionals/research/aacr-proiect-qenie) [23].
- the MET gene is translated as a single chain precursor of 1390 residues and transported to the golgi for glycosylation.
- Furin cleaves the protein to form a disulphide-linked alpha and beta chain heterodimer.
- the extracellular portion of cMET comprises 3 domains, SEMA (semaphorin, a 7-bladed beta propeller structure which includes the HGF-binding site) domain, PSI (plexin-semaphorin-integrin-domain) and 4 IPT (immunoglobulin-plexin- transcription) repeats.
- the intracellular domains comprise a juxtamembrane sequence, the tyrosine kinase catalytic domain and carboxyl-terminal sequences [19].
- cMET overexpression relative to that of normal tissues has been observed in many types of solid human malignancies including gastric, colorectal, pancreatic, lung, head and neck, ovarian, breast, renal, prostate, bladder, nasopharyngeal, gliomas, osteosarcomas and melanomas (Table 1). Due to the poor clinical prognosis of solid tumours overexpressing cMET, and the toxicities caused by broad-spectrum chemotherapeutic treatments used today as well as insufficient drug dosage reaching all parts of the tumour, an opportunity is presented for the development of targeted therapeutics against this receptor.
- cMET pathway inhibitors e.g., crizotinib, cabozantinib, capmatinib, tepotinib, and glesatinib.
- Cabozantinib (Cabometyx®/Cometriq®) and crizotinib (Xalkori®) are FDA/EMA-approved and marketed drugs but many others have failed clinical development primarily due to poor tolerability or low efficacy (often related to poor patient selection resulting in patients whose tumours are not oncogene addicted and less sensitive to cMET inhibition) [22,51].
- SMIs Small molecule inhibitors block ATP-phosphorylation of the cMET tyrosine kinase preventing downstream signalling and broad specificity for ATP-binding sites makes them specific for related kinases such as VEGFR, RET, AXL and others.
- Cabozantinib is approved for thyroid, liver and kidney cancer but has shown disappointing results in other major solid tumours with side effects such as severe bleeding, blood clots, hypertension, gastro-intestinal toxicities.
- Crizotinib is approved for ALK/ROS-mutated non small-cell lung cancer with serious adverse effects including liver toxicity (fatal in 0.1% patients), interstitial lung disease (severe/fatal in 0.5%), pneumonitis, cardiac effects and neutropenia. This indicates a need for better tolerated drugs.
- cMET expression in tumours is also associated with resistance to targeted therapies against other receptors or signalling pathways.
- EGFR epidermal growth factor receptor
- MET-gene amplification occurring as a by-pass mechanism in 5-20% of resistant patients and HGF over-expression occurring in a higher number of patients.
- VEGF Vascular endothelial growth factor pathway resistance is also seen [20]. This has resulted in therapeutic strategies utilizing cMET cross-reactive SMI's and bispecific MAbs (e.g. amivantamab) and ADCs.
- Single agent anti-cMET MAbs e.g., onartuzumab, emibetuzumab, SAIT-301, and ABT700
- single agent anti-HGF MAbs e.g. ficlatuzumab, rilotumumab
- Total cMET protein levels does not correlate well with pathway activation leading to lack of efficacy in the selected patient population [54]
- onartuzumab (MetMAb) is a humanized antagonistic anti-cMET MAb [22].
- Clinical trials in Phase III for lung cancer in combination with Erlotinib was terminated due to lack of efficacy [55].
- Rilotumumab which binds to HGF and inhibits HGF/cMET signalling, was terminated in Phase III due to increased deaths compared to chemotherapy alone [55].
- ADCs and MAbs
- a number of ADCs are in development where the MAb component, due to its bivalent structure must be designed not to cause cMET cross-linking and induced signalling agonism [51]. This can be achieved by engineering the hinge region or using biparatopic MAbs which enable cMET clustering but not signalling.
- high affinity or high avidity MAbs can inhibit tumour penetration due to the binding site barrier effect [9].
- Cross-linking receptors can induce or speed up their internalization but too rapid internalization can also inhibit tumour mass penetration [56].
- W02018/098035 describes an anti-cMET ADC comprising MMAE payload which demonstrated tumour killing efficacy in a range of human tumour xenograft models at 1- 3mg/kg given intraperitoneally one to four doses.
- Abbvie are currently developing a cMET ADC, called telisotuzumab vedotin, ABBV399, comprising the MMAE payload, (EP3636273B1) which demonstrated tumour cure efficacy in human tumour xenograft models at six doses of 3mg/kg (cumulative dose of 18mg/kg) in tumours which express high to moderate levels of cMET target.
- Telisotuzumab vedotin is in advanced stage (Phase II) clinical trials for NSCLC and has demonstrated good tolerability at 2.7mg/kg [58] and RECIST-qualified tumour regression responses in patients expressing high (3+) to moderate (2+) cMET levels with high-cMET expressing patients responding more favourably (LUMINOSITY Phase II Trial, EGFR-Wild type, cMET- high, ORR-52.2%, EGFR-Wild type, cMET-intermediate, ORR-24.1% [59].
- cMET ADCs are in development at earlier stages such as TR1801 (PBD payload) which is in Phase I [35], SHRA1403 (auristatin payload, [60]), cIRCR201-dPBD (PBD payload, [61]) and BYON3521 (duocarmycin payload, [62]).
- Bispecific cMET ADCs include AZD9592 [63] specifically to address EGFR co-expression and resistance to EGFR inhibitors in lung cancer.
- ultra-potent alkylating, DNA damaging payloads and ADCs against other targets using highly potent payloads such as PBDs have been discontinued due to poor tolerability and an almost 50% failure rate [15].
- ADCs are complex molecules to discover and develop. There are many parameters that need to be optimized, sometimes empirically, but increasingly, it is well established that there are some key design features [5,6].
- the physical and chemical stability (e.g. aggregation, pl, hydrophobicity) of an ADC can potentially impact its efficacy, toxicity and immunogenicity and many other potentially dose-limiting toxicities involved Fc-receptor binding and prolonged exposure to normal tissues [15,64] and general methods for designing and making ADCs are described [5,6,65].
- Idiopathic pulmonary fibrosis is a progressive lung fibrosing disease which is poorly understood. It is characterized by progressive lung scarring leading to respiratory failure and death, with a median survival from diagnosis of ⁇ 3 years [66]. Many therapies have been tested for this harmful condition, yet none have demonstrated effectiveness in modifying respiratory-specific or all-cause mortality in IPF [67].
- the aberrant proliferative events in IPF resemble that of malignant transformation in cancer [66] which could encourage the use of cancer drugs for IPF.
- the key histological feature of IPF is shown in the "fibroblast foci", aggregates of actively proliferating fibroblasts and myofibroblasts- cells which can also harbour MET in its activated form. Therefore, RTK inhibitors such as those against cMET could be efficacious in IPF [66].
- Pirfenidone is a pyridone derivative which exhibits antiinflammatory and anti-fibrotic effects (via downregulation of transforming growth factor- p (TGF-p), a central signalling pathway in fibrosis).
- TGF-p transforming growth factor- p
- Nintedanib is a broad-spectrum- tyrosine kinase inhibitor, originally used as anti-vascular cancer chemotherapy.
- Nintedanib 's anti-fibrotic activity via inhibition of key growth factor signalling pathways involved in pulmonary fibrosis (e.g., platelet-derived growth factor (PDGF), vascular endothelial growth factor (VEGF), and fibroblast growth factor (FGF).
- pulmonary fibrosis e.g., platelet-derived growth factor (PDGF), vascular endothelial growth factor (VEGF), and fibroblast growth factor (FGF).
- PDGF platelet-derived growth factor
- VEGF vascular endothelial growth factor
- FGF fibroblast growth factor
- Nintedanib also competitively inhibits both non-receptor tyrosine kinases (nRTKs) including Lek, Lyn, and Src. Therefore, nintedanib action resembles the broad spectrum tyrosine kinase inhibitors used in anti-cMET cancer therapy. Pirfenidone and nintedanib have demonstrated efficacy in clinical trials, slowing the decline
- TAS-115 a highly specific, SMI of VEGF receptor 2 (VEGFR2) and cMET originally discovered as an antitumor therapeutic [68] has shown therapeutic promise with good tolerability. HGF signals through cMET to upregulate expression of proangiogenic VEGF. Blocking both pathways by inhibiting RTK phosphorylation with TAS- 115 was shown to effectively decrease tumour vascularization and size with minimal toxicity and off-target effects. Applied in the IPF setting, TAS-115 could modulate inflammation associated with fibrosis as demonstrated in preclinical models, better than nintedanib [69].
- cMET-induced invasive growth is emerging as a target for IPF, due to the proliferative nature of this disease in the lung.
- Signalling inhibitors aim to reduce the proliferation of damaging fibroblasts.
- cMET receptor could be a candidate for the targeted delivery of chemical modulator-drugs to dampen down inflammation or proliferation. This would increase the local concentration of such therapeutic drugs and reduce the off-target systemic adverse effects of broadly-acting SMI drugs.
- the antibody or antigen-binding fragment thereof is capable of blocking cMET signalling.
- the antibody or antigen-binding fragment thereof binds cMET with high affinity, or with higher affinity than an alternative anti-cMET antibody.
- Affinity can be expressed by measuring the KD (dissociation constant, or equilibrium dissociation constant, expressed in M) or KA (association constant, or equilibrium association constant, expressed in M -1 ) of a particular antibody or antigen-binding fragment when binding cMET.
- the KD and KA may be calculated by measuring the association rate constant (ka, expressed in M -1 s _1 ) and dissociation rate constant (kd, expressed in s -1 ).
- KA is calculated by ka/kd.
- KD is calculated by kd/ka.
- the antibody or antigen binding fragment thereof has a higher KA and/or a lower KD than an alternative anti-cMET antibody or antigen binding fragment.
- the antibody or antigen binding fragment has a lower KD (i.e. a higher binding affinity) than an alternative anti- cMET antibody or antigen binding fragment.
- the antibody or antigen binding fragment thereof has a higher ka and/or a lower kd than an alternative anti-cMET antibody or antigen binding fragment.
- the antibody or antigen-binding fragment thereof binds cMET with an affinity KD of less than lOnM. In some embodiments, the antibody or antigen-binding fragment thereof binds cMET with an affinity KD of less than 5nM. In some embodiments, the antibody or antigen-binding fragment thereof binds cMET with an affinity KD of less than 4nM. In some embodiments, the antibody or antigen-binding fragment thereof binds cMET with an affinity KD of less than 3nM. In some embodiments, the antibody or antigenbinding fragment thereof binds cMET with an affinity KD of less than 2nM.
- the antibody or antigen-binding fragment thereof of the first aspect comprises a heavy chain variable region (VH) comprising the Complementarity Determining Region (CDRs) CDR-H1, CDR-H2, and CDR-H3 of SEQ ID NO. 21, or a variant thereof having at least 90% sequence identity to SEQ ID NO. 21, for example a variant having at least 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity.
- the VH has at least 90% identity with SEQ ID NO. 21 (i.e. up to 10% variance).
- the VH has at least 91% identity with SEQ ID NO. 21 (i.e. up to 9% variance).
- the antibody or antigen-binding fragment thereof of the first aspect comprises a light chain variable region (VL) comprising the CDRs CDR-L1, CDR-L2, and CDR-L3 of SEQ ID NO. 22, or a variant thereof having at least 90% sequence identity with SEQ ID NO. 22, for example a variant having at least 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity.
- VL light chain variable region
- the VH has at least 90% identity with SEQ ID NO. 22 (i.e. up to 10% variance).
- the VH has at least 91% identity with SEQ ID NO. 22 (i.e. up to 9% variance).
- the antibody or antigen-binding fragment thereof of the first aspect comprises any combination of 6 CDR sequences set out in Table A and/or Table B.
- the antibody or antigen-binding fragment thereof of the first aspect comprises the 6 CDR sequences of any clone listed in Table A and/or Table B.
- the antibody or antigen-binding fragment thereof of the first aspect comprises any pair of VH and VL sequences set out in Table A and/or Table B.
- the antibody or antigen-binding fragment thereof of the first aspect comprises the VH and VL sequences of any clone listed in Table A and/or Table B.
- the VH comprises the CDRs of SEQ ID NO. 1 and the VL comprises the CDRs of SEQ ID NO. 2.
- the VH comprises the CDRs of SEQ ID NO. 1 and the VL comprises the CDRs of SEQ ID NO. 196.
- the VH comprises the CDRs of SEQ ID NO. 3 and the VL comprises the CDRs of SEQ ID NO. 4.
- the VH comprises the CDRs of SEQ ID NO. 3 and the VL comprises the CDRs of SEQ ID NO. 6.
- the VH comprises the CDRs of SEQ ID NO. 3 and the VL comprises the CDRs of SEQ ID NO. 8.
- the VH comprises the CDRs of SEQ ID NO. 5 and the VL comprises the CDRs of SEQ ID NO. 6. In some embodiments of the first aspect of the invention, the VH comprises the CDRs of SEQ ID NO. 7 and the VL comprises the CDRs of SEQ ID NO. 8.
- the VH comprises the CDRs of SEQ ID NO. 9 and the VL comprises the CDRs of SEQ ID NO. 10.
- the VH comprises the CDRs of SEQ ID NO. 11 and the VL comprises the CDRs of SEQ ID NO. 12.
- the VH comprises the CDRs of SEQ ID NO. 13 and the VL comprises the CDRs of SEQ ID NO. 12.
- the VH comprises the CDRs of SEQ ID NO. 14 and the VL comprises the CDRs of SEQ ID NO. 15.
- the VH comprises the CDRs of SEQ ID NO. 16 and the VL comprises the CDRs of SEQ ID NO. 17.
- the VH comprises the CDRs of SEQ ID NO. 18 and the VL comprises the CDRs of SEQ ID NO. 17.
- the VH comprises the CDRs of SEQ ID NO. 19 and the VL comprises the CDRs of SEQ ID NO. 20.
- the VH comprises the CDRs of SEQ ID NO. 21 and the VL comprises the CDRs of SEQ ID NO. 22.
- the VH comprises SEQ ID NO. 3 and the VL comprises SEQ ID NO. 8.
- the VH comprises SEQ ID NO. 5 and the VL comprises SEQ ID NO. 6.
- the VH comprises SEQ ID NO. 7 and the VL comprises SEQ ID NO. 8.
- the VH comprises SEQ ID NO.
- the VH comprises SEQ ID NO.
- VL comprises SEQ ID NO. 12.
- the VH comprises SEQ ID NO.
- VL comprises SEQ ID NO. 12.
- the VH comprises SEQ ID NO.
- the VH comprises SEQ ID NO.
- VL comprises SEQ ID NO. 17.
- the VH comprises SEQ ID NO.
- the VH comprises SEQ ID NO.
- VL comprises SEQ ID NO. 20.
- the VH comprises SEQ ID NO.
- VL comprises SEQ ID NO. 22.
- the antibody or antigen-binding fragment of any of the preceding claims wherein the VH comprises the following CDRs: a) CDR-H1: [SEQ ID NO. 23] or an amino acid sequence having at least 80% identity therewith, for example at least 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably with at least 80% identity; and/or b) CDR-H2: [SEQ ID NO.
- CDR-H3 [SEQ ID NO. 32] or an amino acid sequence having at least 65% identity therewith, for example at least 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably with at least 67% identity.
- the antibody or antigen-binding fragment of any of the preceding claims wherein the VH comprises the following CDRs: a) CDR-H1: [SEQ ID NO. 27] or an amino acid sequence having at least 60% identity therewith, for example at least 60%, 65%, 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably with at least 60% identity; and/or b) CDR-H2: [SEQ ID NO.
- CDR-H3 [SEQ ID NO. 35] or an amino acid sequence having at least 60% identity therewith, for example at least 65%, 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably with at least 69% identity.
- the VH comprises the CDRs of SEQ ID NOs 23, 28, and 32. In some preferred embodiments, the VH comprises the CDRs of SEQ ID NOs 27, 29, and 35.
- the VH comprises the CDRs of any of the following combinations:
- the antibody or antigen-binding fragment comprises a VH having the amino acid sequence of SEQ ID NO. 1, or a variant having at least 80% sequence identity therewith, for example at least 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity.
- the variant has around 93% identity with SEQ ID NO. 1.
- the antibody or antigen-binding fragment comprises a VH having the amino acid sequence of SEQ ID NO. 1.
- the antibody or antigen-binding fragment comprises a VH having the amino acid sequence of SEQ ID NO.
- the variant has around 91% identity with SEQ ID NO. 21.
- the antibody or antigen-binding fragment comprises a VH having the amino acid sequence of SEQ ID NO. 21.
- the antibody or antigen-binding fragment comprises a VH having the amino acid sequence of SEQ ID NOs 3, 5, 7, 9, 11, 13, 14, 16, 18, 19, or 21.
- the VL comprises the following CDRs: a) CDR-L1 : [SEQ ID NO. 37] or an amino acid sequence having at least 80% identity therewith, for example at least 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably having at least 85% identity; and/or b) CDR-L2: [SEQ ID NOs.
- CDR-L3 [SEQ ID NOs. 198 or 46] or an amino acid sequence having at least 55% identity therewith, for example at least 60%, 65%, 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably having at least 71% identity; and/or c) CDR-L3: [SEQ ID NOs. 198 or 46] or an amino acid sequence having at least 55% identity therewith, for example at least 60%, 65%, 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably having at least 55% identity.
- the VL comprises the following CDRs: a) CDR-L1 : [SEQ ID NO. 37] or an amino acid sequence having at least 80% identity therewith, for example at least 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably having at least 85% identity; and/or b) CDR-L2: [SEQ ID NO.
- CDR-L3 [SEQ ID NO. 198] or an amino acid sequence having at least 55% identity therewith, for example at least 60%, 65%, 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably having at least 71% identity; and/or c) CDR-L3: [SEQ ID NO. 198] or an amino acid sequence having at least 55% identity therewith, for example at least 60%, 65%, 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably having at least 55% identity.
- the VL comprises the following CDRs: a) CDR-L1 : [SEQ ID NO. 39] or an amino acid sequence having at least 80% identity therewith, for example at least 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably having at least 92% identity; and/or b) CDR-L2: [SEQ ID NO.
- CDR-L3 [SEQ ID NO. 47] or an amino acid sequence having at least 55% identity therewith, for example at least 60%, 65%, 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably having at least 57% identity; and/or c) CDR-L3: [SEQ ID NO. 47] or an amino acid sequence having at least 55% identity therewith, for example at least 60%, 65%, 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably having at least 55% identity.
- the VL comprises the CDRs of SEQ ID NOs 37, 41, and 46. In some embodiments, the VL comprises the CDRs of SEQ ID NOs 39, 44, and 47.
- the VL comprises the CDRs of any of the following combinations:
- the antibody or antigen-binding fragment comprises a VL having the amino acid sequence of SEQ ID NO. 2, or a variant having at least 90% sequence identity therewith, for example at least 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity.
- the variant has around 91% identity with SEQ ID NO. 2.
- the antibody or antigen-binding fragment comprises a VL having the amino acid sequence of SEQ ID NO. 2.
- the antibody or antigen-binding fragment comprises a VL having the amino acid sequence of SEQ ID NO.
- the variant has around 91% identity with SEQ ID NO. 196.
- the antibody or antigen-binding fragment comprises a VL having the amino acid sequence of SEQ ID NO. 196.
- the antibody or antigen-binding fragment comprises a VL having the amino acid sequence of SEQ ID NO. 22, or a variant having at least 90% sequence identity therewith, for example at least 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity.
- the variant has around 91% identity with SEQ ID NO. 22.
- the antibody or antigen-binding fragment comprises a VL having the amino acid sequence of SEQ ID NO. 22.
- the antibody or antigen-binding fragment comprises a VL having the amino acid sequence of SEQ ID NOs 196, 2, 4, 6, 8, 10, 12, 15, 17, 20, or 22.
- the antibody or antigen-binding fragment comprising the following CDRs: a) CDR-H1: [SEQ ID NO. 23] or an amino acid sequence having at least 80% identity therewith, for example at least 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably with at least 80% identity; and/or b) CDR-H2: [SEQ ID NO.
- CDR-H3 [SEQ ID NO. 32] or an amino acid sequence having at least 60% identity therewith, for example at least 65%, 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably with at least 67% identity; and/or d) CDR-L1 : [SEQ ID NO.
- CDR-L2 [SEQ ID NO. 41] or an amino acid sequence having at least 70% identity therewith, for example at least 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably having at least 71% identity; and/or f) CDR-L3: [SEQ ID NO.
- amino acid sequence having at least 55% identity therewith for example at least 60%, 65%, 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably having at least 55% identity.
- the antibody or antigen-binding fragment comprising the following CDRs: a) CDR-H1: [SEQ ID NO. 23] or an amino acid sequence having at least 80% identity therewith, for example at least 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably with at least 80% identity; and/or b) CDR-H2: [SEQ ID NO.
- CDR-H3 [SEQ ID NO. 32] or an amino acid sequence having at least 60% identity therewith, for example at least 65%, 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably with at least 67% identity; and/or d) CDR-L1 : [SEQ ID NO.
- CDR-L2 [SEQ ID NO. 197] or an amino acid sequence having at least 70% identity therewith, for example at least 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably having at least 71% identity; and/or f) CDR-L3: [SEQ ID NO.
- amino acid sequence having at least 55% identity therewith for example at least 60%, 65%, 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably having at least 55% identity.
- the antibody or antigen-binding fragment comprising the following CDRs: a) CDR-H1: [SEQ ID NO. 27] or an amino acid sequence having at least 60% identity therewith, for example at least 60%, 65%, 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably with at least 60% identity; and/or b) CDR-H2: [SEQ ID NO.
- CDR-H3 [SEQ ID NO. 35] or an amino acid sequence having at least 60% identity therewith, for example at least 65%, 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably with at least 69% identity; and/or d) CDR-L1 : [SEQ ID NO.
- CDR-L2 [SEQ ID NO. 44] or an amino acid sequence having at least 55% identity therewith, for example at least 60%, 65%, 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably having at least 57% identity; and/or f) CDR-L3: [SEQ ID NO.
- amino acid sequence having at least 55% identity therewith for example at least 60%, 65%, 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably having at least 55% identity.
- the antibody or antigen-binding fragment thereof preferably comprises the following combination of CDRs: SEQ ID NOs. 23, 28, 32, 37, 41, and 46.
- the antibody or antigen-binding fragment thereof preferably comprises the following combination of CDRs: SEQ ID NOs. 23, 28, 32, 37, 197, and 198.
- the antibody or antigen-binding fragment thereof preferably comprises the following combination of CDRs: SEQ ID NOs. 27, 29, 35, 39, 44, and 47.
- the antibody or antigen-binding fragment thereof comprises one of the following combinations of CDRs:
- CDR-L2 may have SEQ ID NO. 41 replaced by SEQ ID NO. 197 and/or CDR-L3 may have SEQ ID NO. 46 replaced by CDR SEQ ID NO. 198.
- the antibody or antigen-binding fragment thereof comprises a heavy chain variable region having the amino acid sequence of SEQ ID NO: 1 and a light chain variable region having the amino acid sequence of SEQ ID NO: 2, or an amino acid sequence having at least 80% sequence identity therewith, for example at least 85%, 90%, or 95% sequence identity.
- the antibody or antigen-binding fragment thereof comprises a heavy chain variable region having the amino acid sequence of SEQ ID NO: 1 and a light chain variable region having the amino acid sequence of SEQ ID NO: 2, or an amino acid sequence having at least 90% sequence identity therewith, preferably at least 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% sequence identity therewith.
- the antibody or antigen-binding fragment thereof comprises a heavy chain variable region having the amino acid sequence of SEQ ID NO: 1 and a light chain variable region having the amino acid sequence of SEQ ID NO: 196, or an amino acid sequence having at least 80% sequence identity therewith, for example at least 85%, 90%, or 95% sequence identity.
- the antibody or antigen-binding fragment thereof comprises a heavy chain variable region having the amino acid sequence of SEQ ID NO: 1 and a light chain variable region having the amino acid sequence of SEQ ID NO: 196, or an amino acid sequence having at least 90% sequence identity therewith, preferably at least 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% sequence identity therewith.
- the antibody or antigen-binding fragment thereof comprises a heavy chain variable region having the amino acid sequence of SEQ ID NO: 21 and a light chain variable region having the amino acid sequence of SEQ ID NO: 22, or an amino acid sequence having at least 80% sequence identity therewith, for example at least 85%, 90%, or 95% sequence identity.
- the antibody or antigen-binding fragment thereof comprises a heavy chain variable region having the amino acid sequence of SEQ ID NO: 21 and a light chain variable region having the amino acid sequence of SEQ ID NO: 22, or an amino acid sequence having at least 90% sequence identity therewith, preferably at least 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% sequence identity therewith.
- the antibody or antigen-binding fragment thereof is an antigenbinding fragment.
- the antigen-binding fragment is selected from: scFv, Fv, Fab, F(ab)2, Fab-SH, dsFv, sdAb, di-scFvs bi-scFv, Fcabs, diabodies, scFv-Fc/minibody, triabody, tetrabody, tandAb, half antibody (Unibody) and domain antibodies.
- the antigen-binding fragment is an scFv.
- antigen-binding fragment we mean a functional fragment of an antibody that is capable of binding to cMET.
- the antibody fragment excludes the Fc region of a whole antibody. In particular, it is preferred if the antibody fragment does not include the CH2 and CH3 regions of a whole antibody.
- antibody fragments rather than whole antibodies
- the smaller size of the fragments may lead to improved pharmacological properties, such as better penetration of solid tissue.
- antigen-binding fragments such as Fab, Fv, ScFv and dAb antibody fragments can be expressed in and secreted from E. coli, thus allowing the economic production of large amounts of the said fragments.
- Antibody fragments are functional portions of whole immunoglobulins that possess advantageous properties over complete antibodies such as faster penetration into dense or solid tumours, reduced cross-reactivity with normal tissues and more rapid clearance from the circulation, thus reducing normal tissue exposure overall. It is well known in the art that antibody fragments demonstrate faster pharmacokinetics, dispersing into tissues and eliminating more rapidly (ADME-adsorption, distribution, metabolism and excretion properties). They are also easier to produce in more cost-effective systems such as microbial expression systems [70].
- Antibody fragments can be produced by chemical or enzymatic cleavage, but, more preferably, are produced using recombinant DNA technology. The latter allows for indefinite protein expression in prokaryotic or eukaryotic cell lines and genetic modification leading to fragments with enhanced or additional properties.
- Antibody fragments normally possess at least one variable (V-) domain because V-domains contain the complementarity-determining regions (CDRs) or loops for antigen binding [71]. More recently, CDR-like loops have been inserted into non-variable domains (e.g. constantheavy-3, CH3 domains) enabling these domains to bind to useful or predetermined targets [72],
- antibody fragments to be used effectively as carrier vehicles for cytotoxic drugs they must possess biophysical properties that allow high drug loading via chemical conjugation (or strong and specific non-covalent interactions) without detrimentally affecting protein stability, antibody-antigen binding, and drug-favourable properties such as solubility, aggregation and immunogenicity.
- biophysical properties that allow high drug loading via chemical conjugation (or strong and specific non-covalent interactions) without detrimentally affecting protein stability, antibody-antigen binding, and drug-favourable properties such as solubility, aggregation and immunogenicity.
- additional benefits must be engineered into antibody fragments, or preferably be selected from naturally-occurring antibodies to make them practically useful [74,75].
- One example of such a feature is the incorporation of additional or more optimally distributed surface lysine residues onto antibody fragments, thus increasing its capacity for drug conjugation using amine-directed chemistry.
- amino acids could be used, such as optimally distributed cysteines, tyrosines, glutamates, aspartates, arginines, asparagines, histidines and serines, but lysines are more preferable due to the well- established and successful chemical approaches for conjugation and relative inertness to conjugation without specific activating groups (chapter 10 in [76]).
- Non-natural amino acids such as p-Acetylphenylalanine and formyl-glycine can also be used [77].
- the identification of positions for antibody fragment modification can be by direct analysis of the 3-dimensional structure of the antibody fragment (or parental whole antibody), if available, or by homology modelling using a number of software resources such as Phyre
- the criteria for selecting positions include: (1) the use of amino acids already favoured or conserved at that position (identified from databases such as IMGT or Kabat
- fragments other than scFvs that also comprise a VH and VL region would be expected to conjugate and behave in a similar way to an scFv as has been particularly exemplified in the specification.
- fragments discussed herein are all structurally very similar to the exemplified scFv fragments, with only subtle differences in their formats, as discussed below:
- Fvs including ds-Fvs, are structurally very similar to scFvs, and have almost identical surfaces where the conjugation occurs, differing only in the linker which is not involved in the conjugation reaction.
- Fab fragments comprise the VH and VL regions together with the first constant region, and they therefore are very structurally similar to scFvs.
- Fab (and Fab-SH) fragments are structurally similar to scFvs, such that the claimed conjugates produced using Fab or Fab-SH fragments would reasonably be expected to function in a similar manner to the exemplified scFvs.
- Bi-specific scFvs (bs-scFv) and di/bi-scFvs have the scFv format discussed above, but consist of two scFvs tethered by a linker. It is widely acknowledged in the art that the structure of one of the scFv units in a bivalent or trivalent scFv multimer would be the same as the parental scFv.
- Diabodies are bi-valent scFvs where the VH-VL domains are arranged in a head-to-tail format.
- the antibodies and antigen binding fragments of the invention may alternatively comprise variants of the above-defined sequences.
- the antibodies and antigen binding fragments thereof may have at least 60% sequence identity with any of the sequences disclosed herein. For example, they may have at least 60%, at least 65%, at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, at least 95%, or at least 99% sequence identity.
- the antibodies and antigen binding fragments thereof may alternatively be a variant of a specific sequence disclosed herein, wherein said variant comprises mutations at one or more positions relative to the parent sequence.
- mutations we include insertions, deletions and substitutions.
- a variant may be a substitution, deletion or addition variant.
- a variant polypeptide may comprise 1, 2, 3, 4, 5, 6, 7, 8, 9, up to 10, up to 20, up to 30, up to 40, up to 50, up to 75 or more amino acid mutations, in a preferred embodiment.
- the mutations may be either conservative or non-conservative. For example, conservative substitution refers to the substitution of an amino acid within the same general class (e.g.
- “Deletion” variants may comprise the deletion of individual amino acids, deletion of small groups of amino acids such as 2, 3, 4 or 5 amino acids, or deletion of larger amino acid regions, such as the deletion of specific amino acid domains or other features.
- “Substitution” variants preferably involve the replacement of one or more amino acids with the same number of amino acids and making conservative amino acid substitutions.
- an amino acid may be substituted with an alternative amino acid having similar properties, for example, another basic amino acid, another acidic amino acid, another neutral amino acid, another charged amino acid, another hydrophilic amino acid, another hydrophobic amino acid, another polar amino acid, another aromatic amino acid or another aliphatic amino acid.
- Amino acids herein may be referred to by full name, three letter code or single letter code.
- Preferred "variants" include those in which instead of the naturally occurring amino acid the amino acid which appears in the sequence is a structural analog thereof.
- Amino acids used in the sequences may also be derivatised or modified, e.g. labelled, providing the function of the antibody is not significantly adversely affected.
- Derivatives and variants as described above may be prepared during synthesis of the antibody or by post-production modification or by peptide synthesis or by native chemical ligation of peptides, or when the antibody is in recombinant form using the known techniques of site-directed mutagenesis, random mutagenesis, or enzymatic cleavage and/or ligation of nucleic acids.
- Suitable variants may be at least 70% homologous to a sequence disclosed herein, preferably at least 80% or 90% and more preferably at least 95%, 97% or 99% homologous thereto.
- variants have an amino acid sequence which has more than 60%, or more than 70%, e.g. 75 or 80%, preferably more than 85%, e.g. more than 90 or 95% amino acid identity to a sequence as shown in the sequences disclosed herein (e.g. the VH or VL region sequences, or CDR sequences therein). This level of amino acid identity may be seen across the full length of the relevant SEQ ID NO sequence or over a part of the sequence, such as across 20, 30, 50, 75, 100, 150, 200 or more amino acids, depending on the size of the full-length polypeptide.
- variants of the above CDR sequences may comprise one, two three, four, five, six, seven, eight or more amino acid mutations relative to the reference sequence (such as a deletion, substitution and/or insertion of an amino acid).
- the percent sequence identity between two polypeptides may be determined using suitable computer programs, for example the GAP program of the University of Wisconsin Genetic Computing Group and it will be appreciated that percent identity is calculated in relation to polypeptides whose sequences have been aligned optimally. Methods for determining sequence identity are known to those skilled in the art.
- the alignment may alternatively be carried out using the Clustal W program (as described in [80], which is incorporated herein by reference). The parameters used may be as follows:
- Fast pairwise alignment parameters K-tuple(word) size; 1, window size; 5, gap penalty; 3, number of top diagonals; 5. Scoring method: x percent.
- the BESTFIT program may be used to determine local sequence alignments.
- modified versions of antibodies and antigen-binding fragments thereof e.g. modified by the covalent attachment of polyethylene glycol or other suitable polymers (see below).
- antibodies may be generated via any one of several methods which employ induction of in vivo production of antibody molecules, screening of immunoglobulin libraries [75,81] or generation of monoclonal antibody molecules by cell lines in culture. These include, but are not limited to, the hybridoma technique, the human B-cell hybridoma technique, and the Epstein-Barr virus (EBV)-hybridoma technique [82-85].
- EBV Epstein-Barr virus
- Suitable monoclonal antibodies to selected antigens may be prepared by known techniques, for example those disclosed in [86].
- antibody fragments can be obtained using methods well known in the art (see, for example [87].
- antibody fragments according to the present invention can be prepared by proteolytic hydrolysis of the antibody or by expression in E. coli or mammalian cells (e.g. Chinese hamster ovary cell culture or other protein expression systems) of DNA encoding the fragment.
- antibody fragments can be obtained by pepsin or papain digestion of whole antibodies by conventional methods.
- human or humanised antibodies are preferably used.
- Humanised forms of non-human (e.g. murine) antibodies are genetically engineered chimeric antibodies or antibody fragments having preferably minimal-portions derived from non-human antibodies.
- Humanised antibodies include antibodies in which complementary determining regions of a human antibody (recipient antibody) are replaced by residues from a complementary determining region of a non-human species (donor antibody) such as mouse, rat of rabbit having the desired functionality.
- donor antibody such as mouse, rat of rabbit having the desired functionality.
- Fv framework residues of the human antibody are replaced by corresponding non-human residues.
- Humanised antibodies may also comprise residues which are found neither in the recipient antibody nor in the imported complementarity determining region or framework sequences.
- the humanised antibody will comprise substantially all of at least one, and typically two, variable domains, in which all or substantially all of the complementarity determining regions correspond to those of a non-human antibody and all, or substantially all, of the framework regions correspond to those of a relevant human consensus sequence.
- Humanised antibodies optimally also include at least a portion of an antibody constant region, such as an Fc region, typically derived from a human antibody (see, for example, [88-90].
- humanised antibody has one or more amino acid residues introduced into it from a source which is non-human. These non-human amino acid residues, often referred to as imported residues, are typically taken from an imported variable domain. Humanisation can be essentially performed as described (see, for example, [88,89,91]; US 4,816,567) by substituting human complementarity determining regions with corresponding rodent complementarity determining regions. Accordingly, such humanised antibodies are chimaeric antibodies, wherein substantially less than an intact human variable domain has been substituted by the corresponding sequence from a non-human species. In practice, humanised antibodies may be typically human antibodies in which some complementarity determining region residues and possibly some framework residues are substituted by residues from analogous sites in rodent antibodies.
- Human antibodies can also be identified using various techniques known in the art, including phage display libraries (see, for example, [92-95]).
- suitable antibodies may be tested for binding activity, for example by ELISA.
- the antibody or antigen-binding fragment thereof disclosed herein comprises a heavy chain constant region, or part thereof.
- the heavy chain constant region is of an immunoglobulin subtype selected from the group consisting of IgGl, IgG2, IgG3 and IgG4.
- the heavy chain constant region is of an immunoglobulin subtype IgGl.
- the antibody or antigen-binding fragment thereof disclosed herein comprises a light chain constant region, or part thereof.
- the light chain constant region is of a kappa or lambda light chain. In some embodiments, the constant region is a human or humanised constant region.
- the antibodies, antigen-binding fragments and conjugates disclosed herein have an IC50 of lOOnM or lower.
- the half maximal inhibitory concentration is a measure of the effectiveness of a substance in inhibiting a specific biological or biochemical function. This measure indicates how much of a particular drug or other substance (inhibitor) is needed to inhibit a given biological process (or component thereof) by half. Determination of the IC50 for a given compound is a routine matter, and typically is determined by constructing a dose-response curve and examining the effect of different concentrations of antagonist on reversing agonist activity. The IC50 value is calculated by determining the concentration needed to inhibit half of the maximum biological response of the agonist.
- the antibodies, antigen-binding fragments and conjugates disclosed herein have a serum half-life of at least 2 hours, optionally the serum half-life of at least 2 hours is measured in rodents or in humans. In some embodiments, the serum half-life is preferably 4 hours, alternatively 8, 16, 32, 64 or 128 hours. In some preferred embodiments, the serum half-life is measured in humans.
- the conjugates disclosed herein have a longer serum-half life than the unconjugated antibodies or antigen-binding fragments.
- serum half-life is also referred to as bioavailability.
- Serum half-life is the calculated duration of time for a serum level of a compound to be reduced to half its initial value.
- Bioavailability can be defined as the area-under-curve (AUC) in a plasma concentration vs time pharmacokinetic plot and is related to serum halflife. Determination of the serum half-life or bioavailability for a given compound is a routine matter, and typically is determined by measuring the amounts of drug in the serum over time following compound administration to an organism. Serum half-life or bioavailability is important clinically, as it will determine the dosage regime required in order to consistently achieve a serum level of drug within a clinically effective range.
- the payloads, when coupled to the antibody or antigen-binding fragment are separated by a distance of at least two amino acids (3.5 to 7.5 angstroms). In some embodiments, the payloads, when coupled to the antibody or antigen-binding fragment, are separated by a distance of two amino acids (3.5 to 7.5 angstroms), three amino acids (9 to 12 angstroms), four amino acids (10 to 15 angstroms), five amino acids (15 to 20 angstroms) or six amino acids (20 to 25 angstroms).
- the antibody or antigen-binding fragment thereof disclosed herein further comprises a payload.
- payload we mean a drug molecule that is conjugated to the antibody or antigen-binding fragment thereof.
- the drug molecule may be a cytotoxic or cytostatic agent.
- the drug molecule is in some preferred embodiments a small molecule payload.
- the payload is conjugated to the antibody or antigen-binding fragment via a linker.
- the antibody/antigen- binding fragment conjugated to the payload is referred to as an antibody-drug conjugate (ADC).
- ADC antibody-drug conjugate
- an anti-CD33 ADC (gemtuzumab ozogamicin) was approved to treat acute myeloid leukaemia in 2000 but was withdrawn in 2010 due to liver toxicity, patients' deaths and poor efficacy compared to existing therapies [5].
- trastuzumab emtansine already approved for HER2-expressing breast cancer in 2013, was not approved for HER2-expressing gastric cancer in 2015 due to lack of efficacy [18].
- traztuzumab deruxtecan demonstrated high efficacy in HER2-expressing breast cancer but progression-free survival and other key outcomes are much poorer for HER2-expressing gastric cancer [96] which is known to be more heterogenous and difficult to treat [97,98].
- a review carried out by the FDA concluded that the major dose-limiting toxicities of ADCs were due to payload exposure to normal tissues [99] and many examples of so-called ultra-potent (low pM IC50) ADCs have failed due to a narrow therapeutic window compared to ADCs built with less potent payloads [15].
- scFvs single-chain Fvs
- Fabs Fabs
- DAR drug:antibody ratio
- Antikor Biopharma Ltd has previously described an approach using antibody fragments such as scFvs with a DAR of at least 5 coupled in such a way that leads to antibody fragment drug conjugates (FDCs) with high affinity, high potency, manageable tolerability, good pharmacokinetics and good biophysical properties that lead to FDC products that viable commercial products [WO2016/046574].
- FDCs antibody fragment drug conjugates
- ADCs based on antibody fragments herein known as antibody Fragment Drug Conjugates (FDCs)
- FDCs antibody Fragment Drug Conjugates
- cMET FDCs enable delivery of payloads to cMET-expressing cell surface receptor, and subsequent internalization of the FDC to deliver the payload intracellularly, whilst exhibiting no agonist activity.
- cMET FDCs internalise as well as bivalent ADCs resulting in effective payload delivery to tumours.
- cMET FDCs have a faster plasma halflife and lower bioavailability than comparable ADCs but surprisingly demonstrate a higher quantity of payload delivered to tumours in the early time points and furthermore demonstrate lower liver payload uptake as would be expected for a format carrying a high quantity of payload.
- cMET FDCs are able to elicit complete tumour regression after four doses of Img/kg (cumulative dose of 4mg/kg) whereas an ADC against the same target with the same payload was ineffective at a cumulative dose of 6mg/kg [57].
- cMET FDCs demonstrate high biophysical and thermal stability which is unexpectedly higher than other phage-display selected human scFvs conjugated to similar linker-payloads.
- cMET FDCs can be made with a variety of linker payloads which suggests that cMET FDCs can have utility in many types of tumours irrespective of their sensitivity to particular payload classes and diseases beyond cancer such as other proliferative conditions like Idiopathic Pulmonary Fibrosis where a payload also needs to be delivered to cMET-overexpressing tissues.
- cMET FDCs do not possess an Fc- domain which removes any cross-reaction with Fc-receptors.
- the Fc-gamma receptor binding is an important factor in many dose limiting ADC toxicities such as thrombocytopenia [64], and can also be a factor in damaging Fc-gamma receptor bearing immune cells (e.g. macrophages) which are needed for mounting an effective immune response to cancer or infections.
- the cMET FDC binds cMET with an affinity KD of less than 50nM, 40nM, 30nM, 20nM, lOnM, 5nM or less than InM.
- the cMET FDC binds cMET with an affinity KD of less than lOnM.
- the payload is coupled to the antibody or antigen-binding fragment thereof with a coupling ratio (payload :antibody or antigen-binding fragment) of at least 2: 1, at least 3: 1, at least 4: 1, at least 5: 1, at least 6: 1, at least 7: 1, at least 8: 1, at least 9: 1 or at least 10: 1.
- the coupling ratio (payload :antibody or antigen-binding fragment) is at least 6: 1.
- the payload comprises, but is not limited to, at least one of:
- a toxin cytotoxic and/or cytostatic agent
- a protein-degradation agent (PROTAC)
- the cytotoxic and/or cytostatic agents may be any agents known to inhibit the growth and/or replication of and/or kill cells and in particular cancer/tumour cells. Numerous agents having cytotoxic and/or cytostatic properties are known in the literature. Nonlimiting examples of classes of cytotoxic and/or cytostatic agents include antimitotic agents, topoisomerase I inhibitors, topoisomerase II inhibitors, DNA alkylating agents, DNA cross-linking agents, DNA intercalating agents, DNA groove binding agents, DNA strand-cleavage agents including enediynes, kinase inhibitors, RNA/DNA anti-metabolites, RNA polymerase (transcription) inhibitors, protein synthesis inhibitors, cell-cycle modulators, apoptosis regulators, mitochondria inhibitors, nuclear export inhibitors and N- myristoyl transferase inhibitors.
- the toxin may be selected from the group consisting of:
- An auristatin such as MMAE (monomethyl auristatin E) and MMAF (monomethyl auristatin F);
- a maytansinoid such as maytansine, DM1, DM4 and DM21;
- a camptothecin (exatecan, DXd, Belotecan, SN-38, 7-aminomethyl-(10,ll- methylenedioxy)-camptothecin, 7-aminoethyl-(10,ll-methylenedioxy)- camptothecin, 7-Aminomethyl-(10-Methyl, ll-Fluoro)-camptothecin, 7- Aminoethyl-(10-Methyl, ll-Fluoro)-camptothecin;
- a kinase inhibitor such as nintedanib
- a transcription inhibitor triptolide, o-amanatin
- Immunoregulatory agent such as glucocorticoids (Dexamethasone, Budesonide);
- Immunostimulatory agents such as toll-like receptors TLR.7/8 (imidazoquinoline, T785) and STING agonists such as XMT-1621, IMSA172;
- a DNA alkylating agent such as Duocarmycin, Duocarmycin-SA, seco-DUBA;
- An anthracycline derivative such as PNU-159682, doxorubicin;
- NAMPT nicotinamide phosphoribosylytransferase
- KSP pyrrole-based kinesin spindle protein
- a PROTAC such as BET/BRD4 degraders (MZ1 (BRD4/VHL)), GNE-987 (BRD4/VHL);
- HSP90 inhibitor protein synthesis inhibitor like geldanamycin, (a splicing inhibitor) such as Thailanstatin A;
- a Bcl-xL inhibitor such as clezutoclax.
- the payload is exatecan.
- Microtubules are long filamentous protein polymers consisting of heterodimers of a- and P-tubulin which are involved in a number of cellular processes critical to cellular function, including organelle and vesicle transport, cell migration and mitosis. These polymers are assembled and disassembled in a highly controlled process within cells.
- the main role of microtubules is to separate and segregate chromosomes during cell division.
- tubulin inhibitors interfere with cell division by preventing the two pairs of chromosomes from separating into two daughter cells [101].
- the toxin may be an auristatin which refers to a family of antimitotic agents derived from the natural product dolastatin-10 which is a highly potent linear pentapeptide isolated from Dolabella Auricularia, a sea hare [102].
- Dolastatin-10 and its analogues are extremely potent cytotoxic antineoplastic agents, exhibiting picomolar GIso values in most cancer cell proliferation assays [103].
- the potent antiproliferative activity is caused by the ability to strongly bind to tubulin and inhibit microtubule assembly and tubulin-dependent GTP hydrolysis, resulting in accumulation of cells in the G2/M phase of the cell proliferation cycle resulting in cell cycle arrest and apoptosis.
- dolastatin-10 and the auristatins bind to a region which overlaps with that of the vinca site but extends significantly further and near the exchangeable GTP binding site termed the peptide site.
- Dolastatin-10 and the auristatins bind at the o,p-tubulin interphase, inducing a curved conformation that is incompatible with the straight structure of microtubules.
- dolastatin-10 and related synthetic auristatin analogues also have strong anti-vascular effects.
- dolastatin-10 and other derivatives failed to demonstrate meaningful clinical activities as single-agents due to severe dose-limiting adverse effects.
- targeted delivery to the tumour can reduce systemic toxicity and result in clinical benefit.
- auristatins examples include monomethyl auristatin E (MMAE), monomethyl auristatin F (MMAF), auristatin F, auristatin F hydroxylpropylamide (AF-HPA), monomethyl auristatin D (MMAD), monomethyl auristatin F methylester (MMAF-OMe), PF-06380101.
- MMAE monomethyl auristatin E
- MMAF monomethyl auristatin F
- AF-HPA auristatin F hydroxylpropylamide
- MMAD monomethyl auristatin D
- MMAF-OMe monomethyl auristatin F methylester
- Dolastatin-15 is a related seven-subunit depsipeptide also obtained from Dolabella Auricularia see [US4879278]. Many synthetic analogues of this natural product have been prepared, among which cemadotin (LU-103793) and tasidotin (ILX651) have entered clinical trials [104]. For exemplary drugs and analogues from dolastatin-10 see, US5410024, US5599902, US5635483 and US6323315.
- the toxin may be from the hemiasterlin family.
- Hemiasterlins are a family of cytotoxic tripeptides that were initially isolated from the South African marine sponge Hemiasterella minor [105].
- Hemiasterlin and its synthetic analogues like Taltobulin (HTI-286) show nanomolar cytotoxic potency and probably bind to the 'peptide binding site' shared with dolastatins and cryptophycins and inhibit tubulin assembly triggering mitotic arrest and subsequent apoptosis.
- hemiasterlins have the advantage of a simpler structure allowing the preparation of many synthetic analogues like taltobutin which is less sensitive to P-glycoprotein drug transporters.
- exemplary hemiasterlin derivatives as ADC payloads see WO2016123582 which describes 3-aminophenylhemiasterlin and its derivatives.
- the toxin may be a maytansinoid.
- the term maytansinoid refers to benzoansamacrolide, a class of highly potent antimitotics originally isolated from the African shrub Maytenus ovatus.
- Maytansinol which has the same macrocyclic system as in maytansine but with a C3-hydroxyl instead of the C3-acetyl-N-methyl-alanine side chain.
- Maytansinoids are derivatives of maytansinol with modified C3-side chains that are typically C3-esters [106,107].
- Maytansine binds tubulin at the vinca-binding site, similar to vinca alkaloids, thereby depolymerizing tubulin and inducing mitotic arrest in the G2/M phase [108]. Similar to the auristatins, maytansine in its original form yielded a narrow therapeutic window due to associated neurological and gastrointestinal toxicities. Consequently, synthetic maytansine derivatives were synthesized that possessed 100- 1000-fold increases in potency while also including modifications to enable targeted delivery [109].
- Exemplary maytansinoids or maytansinoid analogues including maytansinol are described [110] and US5416064.
- C3-esters of simple carboxylic acids are described in US4317821.
- C3-esters with derivatives of N-methyl-L-alanine are described in US4260608.
- Exemplary maytansinoids such as DM1, DM3, DM4 and DM21 or maytansinoid analogues including maytansinol are described in US6716821, US7276497 and US20180296694.
- the toxin may be a cryptophycin.
- Cryptopycins are a family of 16- membered cyclic depsipeptides, first isolated from terrestrial blue-green algae. They are potent tubulin-binding antimitotic agents, inhibiting mitotic spindle function of cells in the G2M phase of the cell cycle resulting in apoptotic cell death [111].
- Cryptophycins have demonstrated excellent activity against a broad spectrum of solid tumours with potency in the picomolar range [112] and unlike other antitubulin agents they have a major advantage in that they are insensitive to the ABC transporters P-gP and MRP, which are implicated in multidrug resistance [113].
- cryptophycins have failed as stand-alone drugs due to toxicity, their very high potency (picomolar potency on a panel of tumor cells) and lack of PgP susceptibility make them potential candidates as payloads in targeted delivery [114].
- Exemplary cryptophycin compounds are described in US6680311 and US20180078656.
- the toxin may be a tubulysin.
- Tubulysins are a family of naturally occurring cytotoxic tetrapeptides isolated from mycobacterium cultures [115]. Tubulysins are potent inhibitors microtubule polymerisation, causing rapid disintegration of the cytoskeleton of dividing cells in the G2/M phase leading to apoptosis. Binding is to the peptide binding site like the dolostatins making tubulysins extremely potent cytotoxic agents at sub-nanomolar concentrations against a variety of cancer types especially multidrug resistant lines [116]. This activity has led to significant interest in the tubulysins as targeted anticancer agents. Exemplary tubulysin compounds are described in, US20210188906, WO2021262910, US20170326247, WO2017134547, US20160340386, and WO2015157594.
- the toxin may be a taxane.
- Paclitaxel (Taxol), Docetaxel and Cabazitaxel, Larotaxel, TPI 287 and MAC-321 represent the taxane family of drugs which have demonstrated remarkable efficacy against solid tumours such as ovarian and breast cancer.
- Paclitaxel was first isolated from the Pacific Yew tree Taxus brevifolia and its antitumour activity was first reported in 1971. Paclitaxel works by promoting tubulin polymerisation by stabilising microtubules from depolymerisation, thus causing cell cycle arrest in the G2M stage of the cell cycle, leading to apoptotic cell death. Exemplary paclitaxel and analogues are described in [117].
- the toxin may be a vinca alkaloid and their synthetic analogues.
- Vincristine and vinblastine are complex molecule produced by the leaves of the rosy periwinkle plant. They block the polymerisation of tubulin into microtubules and inhibit mitotic progression resulting in apoptotic cell death.
- the lead compounds vinblastine and vincristine have been employed in clinical practice for more than thirty years and remain widely used to this day. Several hundred derivatives have been synthesised and evaluated for their pharmacological activities [118].
- Vinorelbine (Navelbine) is a semi-synthetic vinca-alkaloid with broad spectrum of activity in breast and non-small cell lung cancer with fewer side effects than other vinca alkaloids.
- kits for treating a wide range of diseases and conditions include 4-deacetyl- vinblastine-3-carboxyhydrazide which was evaluated as a conjugate [119] and N-(3- hydroxypropyl)vindesine alanine which was conjugated to a polyacetal polymer.
- Exemplary vinca alkaloids include vincristine, vinblastine, vindesine and navelbine and those disclosed in US20020103136 and US20100305149.
- eribulin appears to act through some non-mitotic pathways which may contribute to its overall antitumour activity.
- An eribulin derivative modified with a cathepsin-cleavable dipeptide valine-citrulline is described in [121].
- Exemplary eribulin compounds are described in WO2017151979 and WO2023061466.
- the toxin may be an epothilone and their synthetic analogues.
- the epothilones are a group of closely related antitubulin cytotoxic macrocylic lactones with a mechanism of action similar to the taxanes in which they bind to the same 00- tubulin heterodimer, promoting tubulin polymerisation by stabilising microtubules from depolymerisation, thus causing cell cycle arrest in the G2M stage of the cell cycle, leading to apoptotic cell death [122].
- the epothilones were originally identified as metabolites produced by the soil-dwelling myxobacterium Sorangium cellularosum and by 2008 epothilones A-F were identified and characterised.
- Epothilones A and B have strong antiproliferative activity against different types of cancer cells and have been found to also exert their effects on taxol resistant cancers.
- Exemplary epothilone compounds include epithilone A, B, C, D, E, and F and derivatives thereof. Suitable epithilone compounds and derivatives are Ixabepilone (Ixempra, BMS-247550), Utidelone (depoxythilone), Sagopilone (ZK-EPO) and Patupilone (EPO906). Additional exemplary derivatives are described in US6989450, WO9719086, WO9907692, WO9927890 and W02017066606.
- Topoisomerase I inhibitors are nuclear enzymes that regulate the three- dimensional geometry (topology) of DNA and enable supercoiled DNA to relax. Regulation of DNA supercoiling is essential to DNA transcription and replication, when the DNA helix must unwind to permit proper function of the enzymatic machinery involved these processes. In order to relieve the topological stress caused by supercoiling, the enzymes topoisomerase I and topoisomerase II (Topo I and II) produce single or double-strand breaks respectively, followed by re-sealing, thus reducing the tension in the DNA strand without leaving damaging nicks.
- Topoisomerase I works by breaking only one strand of DNA, followed by attachment of the free phosphate residue of the broken strand to a tyrosine residue of the enzyme. The complex then rotates, relieving the supercoiled tension of the DNA, and the two ends are then re-sealed.
- Topoisomerase II works in a related manner but cleaves both strands of DNA simultaneously, passing a complete duplex strand through the cut, followed by re-sealing of both ends.
- Topoisomerase inhibitors affect the activity of Topo I and II, with most acting to block the re-sealing process, leading to cell death by apoptosis [123]. Topoisomerases are considered essential proteins for all human cell division to take place properly.
- camptothecin and its analogues are due the inhibition of the religation step that normally reseals the parent strand of DNA causing an irreversible double-strand DNA break, arresting the process of cell division, resulting in cell death [124].
- camptothecins that may be used in accordance with the present invention include topotecan, exatecan, DX-8951f, DXd, irinotecan, SN28, belotecan, 9- aminocamptothecin, rubitecan, silatecan, lurtotecan, diflometotecan, namitecan, gimatecan, cositecan, rubitecan, FL118 (7-methyl-10,ll-methylenedioxy-20(RS)- camptothecin and its 7-alkylamino derivatives (7-methyl, ethyl, propyl, butyl), 7-n-butyl- 10-aminocamptothecin, 7-n-butyl-9-amino-10,ll-methylenedioxy camptothecin, 4- Amino-9-ethyl-5-fluoro-9-hydroxy-l,2,3,9,12,15-hexahydro-camptothecin, 7- methylamino
- Exemplary camptothecin compounds also include those described in [125]. Exemplary camptothecin compounds are further described in WO2022232834, W02022093800, WO2022015656, WO2022155347, WO2022253035, WO2022246576, WO2022058395, WO2020219287 and WO2019236954.
- Camptothecin has a novel pentacyclic ring structure of and to distinguish modifications and derivatives each ring given a label A-E.
- Exemplary ring modified camptothecins are described in [126].
- camptothecin the presence of a 6-membered o-hydrocy lactone E- ring is regarded as an important feature as it interacts with human Topo I through Arg364 and Asp533 residues and confers antiproliferative activity.
- this lactone E ring is unstable in plasma at physiological pH resulting short half-life and low therapeutic efficacy.
- E-ring modified camptothecin derivatives such as an expanded 7-membered 0-hydroxy lactone like diflomotecan have been developed and also derivatives where the size of the E-ring has been reduced from 6-membered to 5-membered.
- One such active derivative is S39625 [127]. Derivatives where the E ring has been opened are also being explored.
- Non-camptothecin topoisomerase I inhibitors Several non-camptothecin derivatives have recently been developed as topo I inhibitors including indenoisoquinolines including exemplary compounds NSC 725776 and NSC 724998 [128], dibenzonaphthyridione (benzophenanthridine) [129]. Exemplary compounds include GENZ-644282 [130], ARC- 111 (topovale) and other derivatives. Evodiamine is a quinazoline-carbolin alkaloid isolated from the Chinese herb Evodia rutaecarpa and is a potent topo I inhibitor. Many synthetic derivatives of evodiamine have been found to manifest potent antitumour activity [131].
- Nemorubicin a doxorubicin analogue bearing a 2(S)-methoxy-4-morpholinyl chain is a potent antitumour agent that induces DNA strand breaks primarily through topoisomerase-
- nemorubicin also exerts its antitumour action through a novel mechanism different to that of other anthracyclines, involving the inhibition of DNA nucleotide excision repair (NER) [132].
- NER DNA nucleotide excision repair
- Exemplary nemorubicin analogues are described in US20140227299 and W02008092796.
- Topoisomerase II inhibitors Exemplary approved topoisomerase II inhibitors that can be used in accordance with the present invention are etoposide, teniposide, doxorubicin, idarubicin, epirubicin, and mitoxantrone.
- Topoisomerase II is a multi-subunit enzyme which uses ATP to pass an intact helix through a transient double-stranded break in DNA to modulate DNA topology [133]. After strand passage, the DNA backbone is religated and DNA structure restored. It has been suggested that cancer cells rely on the topo II enzyme more than healthy cells since they divide more rapidly.
- Etoposide is a semi-synthetic glucoside analogue of epi-podophyllotoxin, a toxin lignan (from the rhizome of the wild mandrake, Podophyllum peltatum). Etoposide and derivatives act as nonintercalating topo II poisons, preventing topoisomerase II from religating cleaved DNA [135].
- Anthracyclines also intercalate into DNA and form reactive metabolites that interact with many intracellular molecules and biologic effects of the anthracyclines are not based solely on topoisomerase
- the toxin may be a topoisomerase II inhibitor.
- the toxin may be doxorubicin.
- Doxorubicin refers to members of the family of anthracyclines derived from a naturally occurring group of antitumour antibiotics and include daunorubicin, epirubicin and idarubicin.
- the mechanism of action of doxorubicin is common to all anthracyclines and involves DNA intercalation.
- the planar anthraquinone aromatic ring nucleus inserts between the base pairs of the DNA double helix, perpendicular to the double-strand axis. The complex is then stabilised by various hydrogen bonds, hydrophobic interactions and Van der Waals forces.
- doxorubicin By forming intercalated adducts, doxorubicin inhibits both DNA and RNA polymerases, leading to arrest of DNA replication and RIMA transcription. Doxorubucin also binds to the DNA- topoisomerase complex in proximity to the scission site of DNA, thus preventing its repair and triggering apoptosis by inhibiting cellular division at the G1 and G2 phases [137]. Doxorubicin and the other anthracyclines can undergo one- and two-electron reduction, since they are members of the quinone family, producing reactive compounds that damage macromolecules and lipid membranes. Exemplary anthracyclines are described in [138].
- Mitoxantrone is a simplified anthracenedione that binds to topoisomerase II resulting in cleavable complexes that induce DNA strand breaks. Mitoxantrone lacks the ability to form the quinone-type free radicals thought to account for anthracycline cardiotoxicity and is the only agent of its class approved for clinical use. In some embodiments, the toxin is mitoxantrone.
- PNU-159682 is liver metabolite of nemorubicin and is about three orders of magnitude more potent than its parent molecule on cultured human tumour cells. It is a potent topo II inhibitor amongst other poorly understood modes of action and is effective regardless of the cell cycle unlike doxorubicin. PNU-159682 is not an efflux pump substrate and is able to bypass resistance mechanisms observed with known tubulin inhibitors like MMAE and DM1 [139]. In some embodiments, the toxin is PNU-159682.
- PNU-159548 an alkycycline daunorubicin derivative
- Ethonafide is an anthracene-containing derivative of amonafide which inhibits topoisomerase II and may have less toxicity than other anthracene-containing agents.
- Exemplary PNU-159682 derivatives are described in [141].
- the toxin is PNU-159548 or ethonafide.
- the toxin may be a duocarmycin.
- Duocarmycins are a family of DNA minor groove-binding compounds with extraordinarily cytotoxicity originally identified in Streptomyces [142]. Their unique mechanism of action involves DNA minor-groove binding and alkylation of adenine forming DNA-adducts which disrupt the DNA helix ultimately leading to cell death.
- a key advantage of duocarmycins and its analogues is the ability to exert their effects on any phase of the cell cycle acting on both dividing and nondividing cells.
- duocarmycins contain a DNA-binding moiety that selectively interacts with DNA strands and a DNA-alkylating unit that possess a spirocyclic cyclopropapyrroloindole (CPI) moiety and many synthetic duocarmycin analogues maintain these structural features.
- the alkylating CPI unit can also be derivatised in its ring-opened chloromethyl aromatic (seco) form, forming prodrugs that contain a phenolic hydroxyl group producing a cyclopropane-containing cytotoxin, via a process known as spirocyclisation, to prevent spontaneous cyclisation, the phenolic hydroxyl group must be modified usually via a pro-drug strategy.
- Duocarmycins are DNA mono-alkylators, by connecting two duocarmycin- type units per molecule, DNA-cross-linking via double alkylation is achieved and these derivatives display significantly greater potency than duocarmycins.
- the CXI dimers [dimers containing, for example cyclopropapyrroloindole (CPI), cyclopropabenzindole (CBI) or cyclopropathienoindole (CTI) moieties] are the main class of potent bis-alkylating payloads. Exemplary CXI dimers are described [145].
- the toxin may be a Pyrrolobenzodiazepine (PBDs).
- PBDs Pyrrolobenzodiazepine
- the PBD monomers are remarkable in possessing a 3-dimensional shape that allows them to fit perfectly within the minor groove of DNA, once located in a position of low energy in the groove (i.e., a preferred DNA sequence), largely dictated by substituents, the electrophilic imine then alkylates the amine group of an adjacent guanine base, thus producing a robust covalent adduct capable of blocking biological processes such as transcription factor binding and RNA polymerase progression.
- the PBD monomers have both antibacterial properties and selective cytotoxicity toward tumour cells.
- the second subfamily, the PBD dimers are not naturally occurring but are synthetic derivatives, the first examples were designed to span greater lengths of DNA than the PBD monomers, to have enhanced sequence-selectivity, and to form DNA cross-links that might be more difficult for tumour cells to repair. It is now known that PBD dimers can form both inter-strand and intra-strand cross-links, as well as monoadducts under certain conditions, although the interstrand cross-linked adduct is still thought to be the most toxic in cells. The perfect fit of a PBD dimer in the DNA minor groove results in negligible distortion of the DNA helix, thus potentially avoiding DNA- repair mechanisms and drug resistance.
- Duocarmycin-PBD dimers are heterodimeric compounds fomed by linking together two different alkylating subunits derived from duocarmycin and pyrrolobenzodiazepine (PBD) classes of DNA-minor groovealkylating agents.
- Exemplary duocarmycin-PBD dimers are described in [148].
- the toxin may be a mitomycin.
- Mitomycins are a family of aziridine-containing natural products isolated from Streptomyces caespitosus, the best- known members being mitomycin A, B and C.
- Mitomycin C has significant antitumour properties and is active against a variety of tumours including breast, stomach, oesophagus, bladder and NSCLC.
- the main mechanism of action of mitomycin is through in situ bioreduction resulting in a bis-electrophile that can alkylate a range of cellular nucleophiles with DNA alkylation as the main mechanism of action.
- the reductive activation mechanism confers selectivity towards particular tumour types, especially hypoxic solid tumours. Exemplary mitomycins are described in [149,150].
- the toxin may be a nitrogen mustard.
- Nitrogen mustards are some of the earliest examples of DNA alkylating agents, these act either as mono-alkylators or as intra- and interstrand cross-linkers.
- Exemplary nitrogen mustards are chlorambucil (LeukeranTM) and bendamustine (LevactTM).
- Site-directed nitrogen mustards include melphalan (AlkeranTM) which is an L-phenylalanine derivative designed for selective uptake by tumour cells in which protein synthesis may be occurring.
- the oligopyrrole antibiotics netropsin and distamycin A (and related synthetic heteropyrrole/imidazole 'lexitropsin' analogues) are well-documented minor groove binders.
- Cyclophosphamide is one example of a metabolic prodrug that requires enzymic activation by phosphoramide hydrolysis by phosophoramidases, releasing the active agent which alkylates guanine bases resulting in mono-adducts and both intra-and inter-strand crosslinks.
- Other mechanisms of alkylation of DNA are through aziridines and epoxides.
- the active species involved in DNA alkylation by nitrogen mustards is an aziridinium cation and several aziridine derivatives have been tested as antitumour agents like Carboquone, Diaziquone (AZQ) and BZQ.
- nitrosoureas are a class of alkylating agents that decompose to produce alkylating compounds under physiologic conditions, the predominant mechanism being base catalysed decomposition to a chloroethyl diazonium moiety which reacts with DNA to form a unique inter-strand DNA cross-link.
- BCNU Carmustine
- ACNU Nimustine
- Streptozotocin a group of alkylating agents that decompose to produce alkylating compounds under physiologic conditions, the predominant mechanism being base catalysed decomposition to a chloroethyl diazonium moiety which reacts with DNA to form a unique inter-strand DNA cross-link.
- BCNU Carmustine
- ACNU Nimustine
- Streptozotocin Streptozotocin.
- the platinum antitumor agents are complexes of platinum with ligands that can be displaced by nucleophilic (electron-rich) nitrogens in nucleic acids. All platinum agents act as pro-drugs, in that they require the removal of their labile chloride or carboxylate ligands, through their displacement by water, before they can bind DNA. When binding to DNA, platinum complexes predominantly do so through the formation of a coordination bond at the N7 site of guanosine residues. Simultaneous binding at a second, adjacent nucleotide, typically another guanosine base, results in inter-strand cross-linking, significant bending and unwinding of the DNA which prevents transcription and replication.
- platinum drugs are cisplatin, carboplatin (Paraplatin), oxaliplatin (Eloxatin), nedaplatin, loboplatin, picoplatin, heptaplatin, teraplatin, iproplatin, satraplatin and ormaplatin
- the toxin may be an enediyne.
- Enediynes are highly cytotoxic DNA damaging natural products produced through microbial secondary metabolism.
- the enediyne natural products share a common structural motif containing two acetylenic groups conjugated to a double bond or incipient double bond within a 9- or 10-membered carbocycle [152].
- Exemplary 9-membered enediynes include neocarzinostatin, kedarcidin, maduropeptin, C-1027 and N1999A2. These molecules, except for N1999A2, generally are found in complex with a cognate apoprotein that binds the enediyne chromophore to generate a stable chromoprotein.
- Exemplary 10-membered enediynes can be divided into two subfamilies: the calicheamicin-like enediynes including the calicheamicins, the esperamicins, namenamicin and shishijimicins, and the anthraquinone-fused enediynes including dynemicin, uncialamycin, the tiancimycins and the yangpumicins [153].
- the conjugated carbocycle drives the shared mode of action for the enediynes.
- Electronic rearrangement of the enediyne carbocycle (cycloaromatization) generates a benzenoid diradical that drives enediyne-induced cytotoxicity.
- Enediyne natural products with a 9- membered carbocycle can proceed through either the Myers-Saito or Bergman rearrangement pathways depending on their architecture, whereas all known 10- membered enediynes proceed through the Bergman rearrangement.
- These DNA lesions are responsible for the cytotoxicity of enediyne natural products [154].
- Exemplary enediyne antibiotics are described in [155,156].
- Intercalation involves the insertion between the base pairs of DNA, perpendicular to the axis of the helix of a flat, fused aromatic molecule. This unwinding results in local structural changes in the helix such as lengthening or twisting of the base pairs and lead to the interference of recognition and function of DNA-associated proteins such as polymerases, transcription factors and DNA repair systems.
- Exemplary monofunctional intercalating agent is ellipticine and its analogues. Ellipticine is an alkaloid isolated from the leaves of Ochrosia elliptica and has potent anticancer properties.
- Ellipticines are multimodal anticancer agents because they exert their biological activity via several modes of action with intercalation and topoisomerase II inhibition the best established.
- Exemplary ellipticine derivatives are celiptium, datelleptium, the olivacine derivative S-16020 and intoplicine.
- Bifunctional intercalators contain two intercalating units, normally cationic, separated by a spacer chain that must be long enough to allow double intercalation to take place.
- Exemplary bis- intercalators are ditercalinium and elinafide. Further examples of both mono-and bis- intercalators are described in [157].
- Protein kinases are enzymes that catalyse phosphorylation and are divided into three categories: serine, threonine or tyrosine kinases.
- Tyrosine kinases function by catalysing the transfer of a phosphoryl group from a nucleoside triphosphate donor to the hydroxyl group of tyrosine residues on protein substrates and then triggering the activation of downstream signalling cascades.
- a variety of kinase families are involved in cell cycle progression, cell proliferation, motility and angiogenesis. Abnormal activation of tyrosine kinases due to mutations, translocations, or amplifications is implicated in tumorigenesis, progression, invasion, and metastasis of malignancies [158].
- Fibrosis denotes an excessive deposition of collagen and other extracellular matrix (ECM) components in tissue. Deposition of collagen is part of physiological wound healing but when this process becomes abnormal, connective tissue replaces normal parenchyma, leading to tissue destruction and impairment of organ function. This pathologic process can affect several organs, causing diverse chronic diseases such as idiopathic pulmonary fibrosis (IPF) [160].
- IPF idiopathic pulmonary fibrosis
- An exemplary compound for the treatment of IPF is nintedanib.
- Nintedanib is a small molecule, competitive, triple angiokinase inhibitor that targets multiple receptor tyrosine kinases (RTKs) and non-receptor tyrosine kinases (nRTKs). Many of these RTKs are implicated in lung fibrosis and tumour angiogenesis, so nintedanib is therefore used in the treatment of proliferative diseases such as idiopathic pulmonary fibrosis, non-small cell lung cancer, and systemic sclerosis-associated interstitial lung disease.
- RTKs receptor tyrosine kinases
- nRTKs non-receptor tyrosine kinases
- Nintedanib also inhibits kinase signalling pathways in various cells within tumour tissues, including endothelial cells, pericytes, smooth muscle cells, and cells contributing to angiogenesis, culminating in an inhibition of cell proliferation and apoptosis of affected tumour cells [161].
- exemplary derivatives of nintedanib include the ov[36 targeting- nintedanib conjugates [162].
- the payload is a tyrosine kinase inhibitor selected from the group consisting of nintedanib, afatinib, axitinib, bosutinib, crizotinib, dasatinib [163] erlotinib, fostamatinib, gefitinib, ibrutinib, imanitib, lapatinib, mubritinib, nilotinib, pazopanib, pegaptanib, sorafenib, sunitinib, Lenvatinib, linifanib, tivozanibvandetanib, vemurafenib, CEP-701 (lesaurtinib), INCB018424 (ruxolitinib), ARRY-142886 (selemetinib), ARRY- 438162 (binimetinib).
- a tyrosine kinase inhibitor
- RNA polymerase II RNA polymerase II
- transcription factors that form complexes with RNApol II to initiate transcription and co-regulators that mediate chromatin structure and accessibility.
- the toxin maybe an amatoxin.
- Amatoxins including alpha- amanitin, beta-amanitin and amanitin
- Amatoxins are natural and highly potent RNApol II inhibitors derived from the Amanita phalloides mushroom. They are cyclic peptides composed of 8 amino acids and inhibit specifically the DNA-dependent RIMA polymerase II of mammalian cells, affecting transcription and protein biosynthesis, stopping growth and proliferation [164].
- amatoxins include alpha-amanitins, beta-amanitins, gamma amanitins, eta amanitins, amanulin, amanullic acid, amanisamide, amanon and proamanulin. Amatoxins are described in [165].
- RNApol II is essential to RNApol II attachment to the DNA at the initiation step.
- Triptolide a natural compound derived from the Chinese medicinal herb called "thunder god vine"
- Minnelide is a water-soluble pro-drug of triptolide currently being evaluated as an anticancer drug.
- Exemplary triptolide derivatives are described in [166].
- Histone deacetylases are epigenetic regulators that regulate the histone tail, chromatin conformation, protein-DNA interaction and are therefore involved in various cellular processes including transcription and protein regulation. They have been found to be overexpressed or overactivated in cancer cells and are thought to be involved in increased proliferation, migration and invasion.
- Exemplary HDAC inhibitors that may be used in accordance with the present invention include Vorinostat, Romidepsin, Panobinostat, and Belinostat.
- the toxin maybe a thailanstatin, or an analogue or derivative thereof.
- Thailanstatin A and its analogues are RNA splicing inhibitors. RNA splicing controls metabolism, angiogenesis, cancer cell proliferation and metastasis mainly by excision of introns and exons that are responsible for the complex cellular mechanism of transforming RNA to mRNA. They can also directly control the initiation, elongation, and termination of transcription and are potent antiproliferative drugs capable of targeting cells that are actively dividing and quiescent.
- Thailanstatin is a natural product originally isolated from Brucella Thailandensis msmb43 and activated by binding to the SF3b subunit of the spliceosome U2 snRNA subcomplex.
- the thailanstatin family has a strong binding and inhibitory effect on spliceosomes by inhibiting the eukaryotic mRNA splicing pathway, resulting in low nanomolar IC50 values for a variety of cancer cell lines.
- Exemplary Thailanstatin A derivatives are described in [167].
- the toxin maybe a kinesin spindle protein inhibitor.
- Kinesin spindle protein (KSP, synonyms: Eg5, KIF11) is an ATP-dependent motor protein involved in the separation of centrosomes in the G2/M phase of the cell cycle. High expression in certain solid tumours (breast, pancreatic and bladder cancers) is associated with poor prognosis. The blockade of this essential event in mitosis with small molecule KSP inhibitors (KSPis), results in high antitumor potency.
- Eg5 expression is specific to proliferating cells and is not expressed in the cells of the nervous system so avoiding the neurological side effects of classic microtubule targeting agents.
- Exemplary small molecule KSP inhibitors are Ispinesib and Filanesib. Further examples are described in [168].
- the toxin maybe a NAM PT inhibitor.
- Niacinamide phosphate ribose transferase (NAMPT), which converts nicotinamide into nicotinamide mononucleotide, is a rate-limiting enzyme that controls the concentration of NAD + within cells.
- NAMPT Niacinamide phosphate ribose transferase
- NAD + levels drop below the level required for metabolism, leading to an energy crisis and eventually, cell death.
- the cell death following depletion of NAD proceeds via a nonapoptotic and proinflammatory mechanism termed oncosis.
- NAMPT inhibitors that may be used in accordance with the present invention include FK-866, CHS-828, GMX-1777, GMX-17778, GPP 78 hydrochloride, ST 118804, STF31. Further examples are described in [169].
- the toxin maybe a Bcl-xL inhibitor.
- Bcl-xL is an anti-apoptotic protein that plays an important role in tumour formation, metastasis, and drug resistance. Apoptosis is a highly regulated cellular process. Aberration in apoptosis is a common characteristic of various disorders. Therefore, proteins involved in apoptosis are prime targets in multiple therapies. Compared to other antiapoptotic proteins, the expression of Bcl-xL is common in solid tumours and apoptosis of cancer cells can theoretically be triggered by drugs blocking the BH3 binding domain on Bcl-xL. However, Bcl-xL is critical for platelet survival, and pan-inhibitors of Bcl-xL may produce platelet toxicity [170].
- Exemplary BcL-xL and Bcl-2 inhibitors are navitoclax, venetoclax, GX-070 (obatoclax), gossypol acetic acid (AT-101, sabutoclax), WEHI-539), ABT-737 and those described in [171]; exemplary compounds are described in W02017/214301 and WO2017/214282.
- the toxin maybe an immune stimulating agent such as toll-like receptor agonists (e.g. TLR 7/8 agonists) or STING agonists.
- TLR 7/8 agonists toll-like receptor agonists
- STING agonists e.g. TLR 7/8 agonists
- Tumour immunity involves T cells, macrophages, dendritic cells, etc., which transform cold tumours (immunologically inactive) into hot tumours (immunologically active) through immunomodulation, and ultimately enhance the effect of immunotherapy.
- Novel cancer immunotherapies such as checkpoint inhibitors, have proven to be a vital approach for the treatment of an increasing number of cancers [172].
- TLR toll-like receptor
- Natural agonist, cGAMP and other synthetic cyclic dinucleotide (CDN) derivatives have been the main starting point for the development of STING agonists
- non-CDN small molecule STING agonists have also been identified, such as dimeric amidobenzimidazole (diABZI) like XMT-1621, these function by mimicking the native ligand of STING, 2'3'- cGAMP.
- diABZI dimeric amidobenzimidazole
- Exemplary compounds are MZ1 (BRD4/VHL), GNE-9787 (BRD4/VHL), BRD4/VHL, BRD4/CRBN, Era/VHL, Era/XIP, BRM/VHL, TGFbR2/CRBN, other examples are described in [177].
- the toxin maybe an anti-inflammatory agent.
- glucocorticoid receptor modulators In the treatment of immunological disease (inflammation associated with various diseases), glucocorticoid receptor modulators (GRMs) are often the first line of treatment.
- GRM can cause musculoskeletal (osteoporosis and decreased bone density), endocrine, and gastrointestinal side effects and other toxicities, limiting their beneficial therapeutic effects (especially in long-term use).
- Multiple medicinal chemistry programs have targeted small molecule selective glucocorticoid receptor modulators which would minimize GR transactivation (believed to be responsible for the unwanted side effects) while maintaining GR transrepression (believed to be responsible for the desired antiinflammatory effects).
- the toxin may be an antimetabolite.
- Antimetabolites are anticancer agents that are analogues of naturally occurring essential metabolites. They work by either blocking biochemical pathways essential for cell growth and division i.e., as enzyme inhibitors) or by incorporating themselves into nucleic acids (i.e., DNA or RNA) and act as 'false substrates' for relevant polymerases, thus blocking relevant down-stream processes such as replication and transcription.
- the enzymes inhibited by antimetabolites are also present in normal cells, some selectivity towards cancer cells is possible due to their faster cell division rates.
- antimetabolites interfere with nucleic acid synthesis and the production of DNA or RNA by two main mechanisms: By competing for sites of enzymes that participate in essential biosynthetic processes or by incorporating into nucleic acids, inhibiting their normal function and triggering apoptosis.
- One class of antimetabolites are the antifolates, these bind tightly to dihydrofolate reductase (DHFR) thereby inhibiting folate metabolism.
- DHFR dihydrofolate reductase
- antifolates examples include methotrexate, pemetrexed, pralatrexate, raltitrexed, nolatrexed, lometrexol, ZD9331, PT523.
- Purine antimetabolites inhibit several enzymes at various points in the purine synthesis pathway and examples include 6-mercaptopurine, azathioprine, fludarabine phosphate, cladribine, clofarabine, nelarabine.
- pyrimidine anti metabolites Similar to the purine antimetabolites are the pyrimidine anti metabolites, these inhibit the synthesis of DNA in the S-phase and block the movement of cells through the Gl/S part of the cell cycle.
- exemplary drugs include 5- fluoroucil, tegafur, capecitabine, doxifluridine, cytarabine, gemcitabine, and decitabine, azacytidine [180-182].
- the toxin maybe a N-myristolytransferase (NMT) inhibitor.
- NMT N-myristolytransferase
- Myristoylation is a lipid modification to a specific group of proteins and involves the N- terminal modification of proteins with myristic acid, a 14-carbon fatty acid, that allows them to interact with other proteins or membranes. Myristoylation plays vital roles in protein-protein interactions, signal transduction, immune regulation and tumour development. It is catalysed by the enzyme /V-myristoyltransferase (NMT) which introduces irreversible changes to human proteins.
- NMT inhibitors have been shown to inhibit the viability and growth of various cancers and represent a novel class of ADC payloads that exploit cancer cell dependency on myristloylated proteins [183]. Exemplary small-molecule NMT inhibitors are described in WO 2022/058745.
- the antigen-binding fragment is a single chain Fv and the payload is MMAE, and wherein the payload is coupled to the scFv with a coupling ratio (payload :scFv) of 6: 1.
- the antigen-binding fragment is a single chain Fv and the payload is a mono-methyl auristatin E (MMAE) comprising a glucuronide and branched PEG linker, wherein the MMAE is conjugated onto surface lysine residues of the scFv.
- MMAE mono-methyl auristatin E
- Specific examples of compounds according to the invention include, but are not limited to, where:
- the antigen-binding fragment is an scFv and the payload is MMAE;
- the antigen-binding fragment is an scFv and the payload is MMAF;
- the antigen-binding fragment is an scFv and the payload is SN38;
- the antigen-binding fragment is an scFv and the payload is exatecan;
- the antigen-binding fragment is an scFv and the payload is Dxd;
- the antigen-binding fragment is an scFv and the payload is belotecan;
- the antigen-binding fragment is an scFv and the payload is triplotide
- the antigen-binding fragment is an scFv and the payload is nintedanib;
- the antigen-binding fragment is an scFv and the payload is GENZ-644282.
- the term 'linker' refers to any chemical moiety capable of linking a payload as described to an antibody or antigen binding fragment thereof.
- the linkers may be monovalent such that they covalently link a single payload to a single site on the antibody fragment, or the linker maybe polyvalent such that they link more than one payload to a single site on the antibody fragment.
- the linkers connect the payload to the antibody fragment by forming a covalent linkage to the payload at one location and a covalent linkage to the antibody fragment at another.
- the covalent linkages are formed by reaction between functional groups on the linker and functional groups on the payload and antibody fragments.
- the term 'linker' includes (i) unconjugated forms of the linker that include a functional group capable of covalently linking the linker to the payload and a functional group capable of covalently linking the linker to the antibody fragment (ii) partially conjugated forms of the linker that includes a functional group capable of covalently linking the linker to the antibody fragment that is covalently linked to a payload, or vice versa, and (iii) fully conjugated forms of the linker that is covalently linked to both payload and antibody fragment.
- the linker may be chemically or biologically stable or resistant to cleavage (a 'non- cleavable linker') or the linker may be susceptible to cleavage or include linkages that are designed to release the payload upon internalisation within the cell (a 'cleavable linker').
- These cleavable linkers are designed to cleave and/or self-immolate or otherwise breakdown specifically or non-specifically inside cells.
- the term 'self-immolate' refers to an at least bifunctional molecule that can be included in a linker that degrades spontaneously after an initial reaction has taken place and thereby releasing the payload.
- Polyvalent linkers that may be used to link many payloads to a single antibody molecule include Mersanas FleximerTM technology which incorporated payloads into a solubilising polyacetal backbone via a sequence of ester bonds, other approaches use dendritic type linkers.
- the linker is cleavable in vivo.
- Cleavable linkers may include chemically or enzymatically unstable or degradable linkages.
- Cleavable linkers generally rely on processes inside cells to liberate the payload, such as reduction in the cytoplasm, acid induced cleavage in the lysosomes or endosomes, or cleavage by specific proteases or other proteolytic enzymes within the cell [184].
- a linker comprises a chemically labile group such as hydrazone and/or disulphide groups. These types of linkers exploit differential properties between the plasma and some cytoplasmic compartments.
- the intracellular conditions to facilitate drug release for hydrazone containing linkers are the acidic environment of endosomes and lysosomes, while disulphide containing linkers are reduced in the cytosol, which contain high concentrations of reductive agents like glutathione.
- Acid cleavable linkers aim to exploit the acidity of the endosomes (pH 5.5-6.2) and lysosomes (pH 4.5-5.0) whilst maintaining stability in circulation at pH 7.4. Acid-labile groups such as hydrazone remain intact during systemic circulation in the blood's neutral pH and undergo hydrolysis, releasing the payload once internalised into the endosome and lysosome compartments of the cell. Other acid sensitive functional groups used in linkers include a carbonates and esters.
- Disulphide containing linkers are designed to release the payload upon internalisation inside cells, they thermodynamically stable at physiological pH but are susceptible to nucleophilic attack from thiols.
- the dominant thiol species is the reduced form of human serum albumin (HSA), however its reactivity towards large molecules is hampered because the free thiol-containing residue (Cys34) is in a crevice and has limited solvent exposure.
- the cytosol contains high levels of glutathione, a thiol- containing small molecule tripeptide. This difference between the reductive potential of the blood plasma and the cytosol enables selective intracellular release of the payload.
- tumours can have hypoxic regions, resulting in enhanced or elevated levels of reductive enzymes and higher glutathione concentrations.
- the in vivo stability of disulphide linkers may be further enhanced by chemical modification of the linker, e.g., increasing steric protection around the disulphide which makes the linker less susceptible to reduction.
- Enzyme cleavable linkers utilise the high or elevated levels of unique hydrolytic enzymes like cathepsins that reside in the lysosomes of cells and offer an opportunity for the selective cleavage of linkers and the intracellular release of the payload [185].
- Such linkers are typically peptide-based or include peptidic regions that act as substrates for these enzymes.
- Cathepsin B is a ubiquitous cysteine protease whose properties do not differ very much from species to species. It is never found extracellularly, except in pathological conditions such as metastatic tumours. Therefore, conjugates produced with cathepsin B- cleavable linkers are likely to be stable in circulation. Release of a drug from an antibody occurs specifically due to the action of lysosomal proteases, e.g, cathepsin and plasmin.
- Exemplary cleavable peptides that can be used in accordance with the present invention include, but are not limited to dipeptides, tripeptides, tetrapeptides and pentapeptides. Dipeptides are often preferred over longer peptides due to the hydrophobicity of longer peptides.
- Exemplary dipeptides that may be used in some embodiments of the invention include but are not limited to alanine-alanine (ala-ala), valine-alanine (val-ala), valinecitrulline (val-cit), alanine-phenylalanine (ala-phe), phenylalanine-lysine (phe-lys), phenlyalanine-homolysine (phe-homolys) and N-metyl-valine-citrulline(Me-val-cit).
- Non-natural amino acids include, by way of non-limiting examples, homoserine, homoarginine, citrulline, phenylglycine, taurine, iodotyrosine, selenocysteine, norleucine (Nle), norvaline (Nva), beta-alanine, L- or D-naphthalanine, ornithine (Orn), and the like.
- Peptides can be designed and optimized for enzymatic cleavage by a particular enzyme, for example, a tumour-associated protease, cathepsin B, C and D, or a plasmin protease.
- Amino acids also include the D-forms of natural and non-natural amino acids.
- D- designates an amino acid having the “D” (dextrorotary) configuration, as opposed to the configuration in the naturally occurring (“L-”) amino acids.
- L- naturally occurring amino acids.
- a typical self-immolative spacer is the bifunctional paraaminobenzyl alcohol group, which is linked to the peptide through the amino group, forming an amide bond, while amine containing payloads may be attached through carbamate functionalities to the benzylic hydroxyl group of the linker (PABC).
- PABC linker
- the resulting prodrugs are activated upon protease-mediated cleavage (for example with cathepsin B), payload release is thermodynamically driven and takes place after amide bond hydrolysis via either an electron cascade for PABC type linkers (1,4-, 1,6-, or 1,8- elimination) or an intramolecular nucleophilic cyclisation.
- 1,6-elimination from a typical PABC spacer result in the release of the unmodified payload, carbon dioxide, an azaquinone methide and remnants of the linker group [187].
- the self-immolative spacer maintains enzymic activity, independent from the payload and variants of the self- immolative spacer where the PABC has been replaced by a heterocyclic spacer like aminothiazole have also been described in US7754681.
- the linker in accordance with the present invention is a glycosidase- cleavable linker.
- Another class of proteolytic enzymes that are only found in lysosomes are the p-glucuronidases.
- p-glucuronidases are hydrolytic enzymes in the glycosidase class that catalyse the breakdown of p-glucuronic acid residues in polysaccharides. Their exclusivity and abundance in the lysosomal compartment of cells, overexpression in some tumour types and little enzyme activity outside of cells make them ideal candidates for use in enzyme cleavable linkers [188].
- p-Glucuronic acid-based linkers can also be linked to a self-immolative spacer (similar to the PABC used with peptidic linkers) but using a para-hydroxybenzyl alcohol group. However, in this case attachment to the antibody is achieved through substituents on the space molecule (an amine or carboxylic acid group) ortho- to the enzyme-cleavable moiety.
- hydrophilicity of 0-glucuronic acidbased linkers and the overall antibody-drug conjugate can be further increase by the incorporation of linear or branched polyethylene glycols, crown or aza-crown ethers, cyclodextrins, polysarcosines or chito-oligosaccharides like ChetosensarTM through branching points on the linker. This can greatly reduce plasma clearance, thereby increasing exposure and in vivo efficacy [192-195].
- 0-glucuronide linker By modifying the 0-glucuronide linker to contain a dimethylethylene diamine (DMED) self- immolative spacer phenolic or alcohol containing drugs could be enzymatically released. Drug release involves enzymatic deglucuronidation, 1,6-elimination, decarboxylation, and cyclization of the DMED carbamate to the cyclic urea to liberate the free phenol or alcohol functionalised drug [196]. Similar to p-glucuronidase cleavable linkers, p-galactosidase cleavable linkers have also been described. This lysosomal enzyme is also over expressed in certain tumour types and is analogous to p-glucuronidase in its hydrolytic activity but instead hydrolyses p-galactoside [197].
- DMED dimethylethylene diamine
- lysosomal enzymes that have been exploited as enzymatic triggers to release payloads in cancer cells include lysosomal acid pyrophosphatase and acid phosphatase, these enzymes hydrolyse pyrophosphates and terminal mono-phosphates respectively to their parent alcohols.
- a pyrophosphate containing linker has been described for use with alcohol-linked glucocorticoid payloads [198].
- Sulfatases are another group of lysosomal enzymes which have high activity in the lysosome and low activity in human and rodent plasma.
- a number of different sulfatases reside in the lysosome, catalysing the hydrolysis of alkylsulfate esters to alcohols but they also display arylsulfatase activity.
- Arylsulfate linker motifs have been designed that after hydrolysis, a alkoxybenzyl carbamate is revealed which is primed for spontaneous 1,6- elimination of an amine containing payload [199].
- the linker for use in the present invention is a non-cleavable linker.
- non-cleavable linkers do not have a triggerable moiety which ultimately separates the active drug from the antibody through a specific cleavage mechanism but instead release an active metabolite upon complete digestion of the antibody component after internalisation and lysosomal processing
- the linker With a non-cleavable linker, the linker, along with the amino acid residue of the antibody to which the linker is attached, remains connected to the payload, and this whole construct becomes the active metabolite. Since the linker stays attached to the drug after cellular processing, it must be linked at a position which does not interfere significantly with binding of the drug to its target (activity) [200].
- the payload is indirectly conjugated to the antibody or antigenbinding fragment thereof at an amino acid, which may be a lysine or cysteine reside.
- the indirect coupling is via a thiol or maleimide.
- conjugation onto lysines can be direct (Table 2), using drugs or drug-linkers that possess and N- hydroxysuccinimide esters (and their more soluble 3-sulfonated analogues), a tetrafluorophenyl ester (and its sulphonated analogue) pentafluorophenyl ester, an isothiocyanate, a p- lactam reactive group or a mixed anhydride.
- Other lesser known/used direct lysine conjugations involve sulfonyl halides, acylfluorides, iminoboronates, diazonium salts, aldehydes and sulphonylacrylates [202].
- Indirect methods for lysine conjugation include derivatising the amino group with a bifunctional linker (such as those available from Pierce Chemicals (Thermo) and Quanta Bioscience) to generate a secondary reactive group, such as 2-iminothiolane to generate a reactive thiol for conjugating to drugs or drug-linkers with thiol or maleimide reactive groups.
- a bifunctional linker such as those available from Pierce Chemicals (Thermo) and Quanta Bioscience
- 2-iminothiolane to generate a reactive thiol for conjugating to drugs or drug-linkers with thiol or maleimide reactive groups.
- lysine residues via a two-step methodology such as via a phospha-Mannich reaction whereby the lysine amine first undergoes imine formation with an aldehyde reagent, followed by attack of the imine with a nucleophilic triethylphosphite reagent to generate a stable link. Further functionalisation can then be carried out using a hydroxylamine terminated payload [204].
- Reactive lysines can also be modified with methylsulfone phenyloxadiazole (MS-PODA) modified payloads.
- MS-PODA methylsulfone phenyloxadiazole
- MTG microbial transglutaminase
- Another enzyme that can be used for bioconjugation is microbial transglutaminase (MTG), which catalyses the formation of amide bonds between the y-carboxyamide group of glutamine and the e-amine group of lysine. This occurs by initial thioesterification, by attack of the active site cysteine on the y- carboxyamide and loss of ammonia; and finally amide formation by reaction with the primary amine of the lysine.
- MTG is a cheap, readily available enzyme that functions under a diverse range of different temperatures, salt concentrations, and pH. Using this approach that one can utilise MTG into targeting lysine residues as acyl acceptors on a antibodies and proteins.
- LACE conjugation enzymes'
- SUMO small ubiquitin- like modifier
- Another approach one can use involves transforming the E-amino sidechain of lysine into a biorthogonal moiety either directly by transforming the e-amino group into an azide or by a two-step approach involving a bis-functional cross-linker carrying a lysine reactive group and a biorthogonal reactive moiety.
- These bifunctional linkers will be well known to the skilled person.
- Cysteine residues also offer a particularly attractive target for protein bioconjugation due to the exceptionally high nucleophilicity of the deprotonated thiolate side chain at physiological pH and the ability via mutagenesis to insert cysteine residues.
- the go-to method for cysteine modification usually remains the use of maleimides.
- Cysteine modification occurs most commonly by 1,4-conjugate addition to N-substituted maleimides.
- Maleimides are particularly attractive reagents due to their synthetic accessibility and rapid reaction rates with cysteine under mild conditions Maleimide reagents allow extraordinary fast labelling with acceptable cysteine selectivity.
- a variety of maleimide derivatives, including dyes, affinity probes and crosslinkers, are commercially available, allowing easy access without the need of synthetic expertise.
- maleimides suffer from stability issues, thus rendering conjugation products unstable under certain conditions.
- maleimides are the most common cysteine-selective reagents, a variety of novel linkers have been used to functionalise cysteines, avoiding the instability issues of traditional maleimides, for example, iodoacetamides bromomaleimides, carbonylacrylic reagents and N-alkyl vinylpyridine salts [206,207].
- Recent conjugation methods include the use of methylsulphonylphenyloxadiazole reactive linkers to form thioethers [208] and disulphide bridging technologies.
- GCE genetic code expansion
- bioconjugation strategies involve tyrosine selective labelling via the use of a tyrosine-click reaction [210].
- Site-selective histidine modification which poses a challenge due to competition from other more nucleophilic residues such as lysine or cysteine has been developed involving a "chemical linchpin".
- This linchpin is a bifunctional reagent containing both aldehyde and epoxide reactive groups.
- All available lysine residues were transiently protected via reaction with the aldehyde moiety.
- proximal histidine residues react with the pendant epoxide to afford irreversible modification.
- the payload is conjugated to the antibody or antigen-binding fragment thereof by direct conjugation of payloads bearing an N- hydroxy-succinimide ester to multiple lysine residues. In some other embodiments, the payload is conjugated to the antibody or antigen-binding fragment thereof by indirect conjugation to lysine residues wherein the cross-linker SMCC is used to modify surface lysine residues, generating a reducible thiol for conjugating to drugs or drug-linkers bearing a thiol or maleimide group.
- the antibody or antigen-binding fragment thereof is conjugated to multiple types of payloads.
- the same antibody or antigenbinding fragment may be conjugated to two types of payloads, three types of payloads, four types of payloads, five types of payloads, or six types of payloads.
- Combination therapy is important for maximizing patient outcomes by combining different mechanisms of drug action to obtain a larger therapeutic effect without significantly increasing adverse effects and thus widening the therapeutic window.
- Combining the action of trastuzumab antibody with chemotherapy lapatinib is well known in the art and accepted clinical practice for breast cancer (https ://www.esmo.orq/quidelines/quidelines- bv-topic/breast-cancer).
- Immuno-oncology drugs notably checkpoint inhibitors (CPI) such as anti-CTLA4 (ipilimumab), anti-PDl (nivolumab, pembrolizumab) and anti-PDLl (atezolizumab) have transformed the landscape of oncology therapy for diseases such as melanoma, lung and renal cancer by reversing tumour-mediated immune suppression [172].
- CPI drugs combined with standard-of- care cytotoxic chemotherapy can lead to increased toxicity, therefore CPI drugs combined with targeted chemotherapy (i.e., antibody drug conjugates) have gained much interest [213].
- ADCs are known to kill tumours by a variety of mechanisms in addition to the concept of delivering a damaging payload to a targeted cell.
- One key mechanism the 'immunogenic cell death' where the payload-mediated cytotoxicity causes tumour cells to release immunogenic signals that elicit a separate adaptive and innate immune response.
- Immunoglobulin-based ADCs often retain inherent immune-activating functions due to the presence of an IgGl Fc-domain. This can elicit toxicities and damage the specific cells that are required for immune function. For example, Fc-interaction with megakaryocytes Fc-receptors is one reason why ADCs cause dose-limiting thrombocytopenia [15,64,213]. Therefore, many developers have chosen to use immunologically-silent Fc domain (e.g. IgG4) to reduce inadvertent immune cell damage. Antibody fragments such as single-chain Fvs lack an Fc-domain and are not expected to cause Fc/Fc-gamma-receptor mediated toxicities. There is therefore a rationale for combining cMET FDCs in this invention with IO/CPI drugs which may elicit a superior combination effect compared to ADCs due to the lack of an Fc-domain.
- immunologically-silent Fc domain e.g. IgG4
- the antibodies, antigen-binding fragments and ADCs disclosed herein may be combined in a formulation with one or more of the following: a chemotherapeutic agent, an immune-oncology drug, a monoclonal antibody, or a checkpoint inhibitor.
- Another aspect of the invention provides a pharmaceutical composition
- a pharmaceutical composition comprising the antibody or antigen-binding fragment or ADC disclosed herein and a pharmaceutically- acceptable carrier, excipient or diluent.
- Another aspect of the invention provides an antibody or antigen-binding fragment or ADC or pharmaceutical composition as defined herein for use in medicine.
- Another aspect of the invention provides an antibody or antigen-binding fragment or pharmaceutical composition or ADC as defined herein for use in the diagnosis, treatment and/or prevention of a disease.
- the disease is selected from cancer, benign tumours, infectious diseases including bacterial, viral, fungal, trypanosome, nematode and prion infections, cardiovascular disease, autoimmune disease.
- Another aspect of the invention provides an antibody or antigen-binding fragment or ADC or pharmaceutical composition as defined herein for use in the manufacture of a medicament for the treatment and/or prevention of a disease selected from cancer, benign tumours, infectious diseases including bacterial, viral, fungal, trypanosome, nematode and prion infections, cardiovascular disease, and autoimmune disease.
- a disease selected from cancer, benign tumours, infectious diseases including bacterial, viral, fungal, trypanosome, nematode and prion infections, cardiovascular disease, and autoimmune disease.
- the disease is cancer. In some preferred embodiments, the disease is idiopathic pulmonary fibrosis (IPF).
- IPF idiopathic pulmonary fibrosis
- the disease is cancer of the colon, lung, breast, head/neck, prostate, skin, stomach/gastrointestinal, oesophageal, bladder, glioma, renal, ovarian, thyroid and bone.
- the disease is a gastric cancer or gastric tumour.
- Another aspect of the invention provides a process of making a compound (i.e. an ADC or FDC) as defined herein comprising the steps of:
- the conjugation takes place in the presence of at least one polar aprotic solvent and an aqueous buffer. In some embodiments, the conjugation is via a linker between the payload and the antibody or antigen-binding fragment.
- the linker can be any linker as described herein.
- the process comprises the step of:
- Nguyen TD Bordeau BM, Balthasar JP. Cancers (Basel). 2023;15:713.
- Figure 5 Inhibition of HGF binding to human cMET by scFvs 129D5B and 131D5S by ELISA. Clone 131D5S, but not 129D5B, inhibits HGF ligand binding.
- Amino acid sequence of cMET scFv 77F3 (a) VH and (b) VL showing positions of lysines for randomisation (amino acids 13, 23, 74 and 239). Underneath each lysine position are listed the amino acid alternatives that were built into the 77F3 mutant library.
- Biophysical properties of 129D5BTY (A) SEC trace showing predominantly monomeric scFv; (B) DLS trace showing a uniform particle size with no aggregation; (C) melting curve showing that 129D5BTY scFv is highly thermostable with a T m of 74°C.
- Figure 12 ELISA of parent 129D5BTY scFv and a range of mutations at position VH98 some of which reduce or abolish binding to human cMET and all which abolish binding to mouse cMET demonstrating the importance of Arginine-98.
- Figure 23 (A) Characterization of 129D5B-Belotecan-[3GA-PEG2 FDC by SEC-LCMS showing DAR distribution and an average DAR of 5.9 and (B) average DAR of 13; (C) SEC-HPLC analyses demonstrating >98% monomeric FDC of the DAR 13 FDC; (D) Characterization of 129D5B- Triptolide-PEGe FDC by SEC-LCMS showing DAR distribution and an average DAR of 5.0.
- Representative histograms of antibodies binding on the cell surface of high cMET receptor-expressing cell lines Hs746T, SNU5, KATO III and AsPCl are shown. Binding on moderate to low cMET- expressing cell lines is also illustrated.
- Histogram represents untreated cell lines;
- Histogram shows the fluorescence intensity of the secondary antibody when used as a control or the fluorescence intensity of the cMET antibody binding on the cell surface when conjugated to a fluorophore.
- Quantification for cMET receptors by QIFIT Quantification for cMET receptors by QIFIT.
- A Fluorescence calibration using provided beads and fluorescently-labelled 5D5 IgG;
- B Calibration of high to low window;
- C Calibration curve determined for fluorescent intensity vs antibody binding capacity (receptor count);
- D to I shows the flow cytometry of the various cell lines using the 5D5 IgG compared to the calibrated beads. The mean fluorescence for each cell line was compared to the calibration curve to determine the receptor count as shown in Table 9.
- Figure 40 Inhibition of cMET receptor phosphorylation of 4 cMET binding antibodies (129D5BTY scFv, 131D5S scFv, ABT-700 IgG and 5D5 IgG and a non-cMET binding IgG4. All 4 cMET binders inhibit with IC50 values of 1.4nM, 4.3nM, 1.4nM and 1.5nM respectively.
- A Aspartate transaminase
- B Alaine transaminase
- C Urea
- D Creatinine
- E Bilirubin levels were determined 2 days after each dose and plotted vs time. The three black arrows indicate the dosing event (once/3 weeks).
- WBCB White Blood Cells-B
- WBCP White Blood Cell-P
- Platelets counts were determined 2 days after each dose and plotted vs time. The three black arrows indicate the dosing event (once/3 weeks).
- Cynomolgus monkey haematological analyses (A) Lymphocytes counts after four 3- weekly doses of (Dose-1), 0.25mg/kg, (Dose-2) 0.375mg/kg, (Dose-3) 0.5mg/kg and (Dose-4) Img/kg 129D5BTY-MMAE-pGA-BrPEGi2-PEG 4 FDC (DAR6) over 66 days; (B) Total white blood cells made up of neutrophils and other lymphocytes were also counted. Black arrows indicate each of the 4 doses.
- Figure 64 Haematological measurements from cynomolgus taken pre-dose (1), 48h after dose (2) and 10 days (3)
- Figure 65 Clinical Chemistry measurements for key liver markers from cynomolgus taken pre-dose (1), 48h after dose (2) and 10 days (3)
- Figure 66 Clinical Chemistry measurements for key kidney markers from cynomolgus taken pre-dose (1), 48h after dose (2) and 10 days (3)
- a non-immune scFv phage display library 7xl0 9 in size, was generated in-house.
- This library consisted of an antibody scaffold of human VH5 and VL1 genes, with CDR regions diversified based on sequence diversity observed in natural human antibody sequences. Phage display selections with this scFv library were performed using recombinant purified cMET protein and following published methods [216]. Briefly, phage particles displaying the Antikor VH5-VL1 library were incubated with cMET antigens either immobilised directly on maxisorp plates, or captured via protein G immobilised onto maxisorp plates.
- undesirable scFvs recognising the Fc region of the fusion protein were removed by the addition of human IgGl to the selection mixture.
- unbound phage particles were removed by multiple wash steps, and then specifically bound phage eluted by addition of chaotropic agents such as triethlyamine and/or hydrochloric acid.
- Eluted phage were then recovered by re-infection into E. coli TGI cells and plated onto large format 2xTY agar bioassay dishes supplemented with antibiotic.
- Phage display selections for the above strategies were carried out following published methods [216], using solution phase selections with biotinylated human cMET to control antigen concentration and increase selection stringency to favour higher affinity binders. Increased stringency was further applied by lengthening the wash steps to enrich for scFv with slower off rates, and by introducing heat stress to favour scFvs with higher (Tm) melting temperatures.
- Binding affinity kinetic constants for scFv preparations of 3A2 and 26B5 and their affinity optimised variants were measured using a BIAcore T200 instrument. Approximately 100- 150R.U of cMET Fc was captured on a protein G chip by injecting recombinant human, cynomolgus and rat cMET at 2.5
- ScFv were prepared in dilution series (typically 1 :3 dilution series starting with 25-100nM scFv at the highest concentration), and then injected over the antigen coated surfaces and also a blank surface, starting with the lowest concentration of scFv and then working progressively up to the highest concentration. ScFv binding kinetics were then determined from the (blank subtracted) sensorgram traces using 1: 1 binding models and BIAevaluation software. Using this approach, affinities for 3A2 and 26B5 and several affinity optimised variants were determined ( Figure 2a, 2b, 2c and Table 4).
- 129D5B and 131D5S were analysed by scFv ELISA against cMET and a range of other unrelated antigens.
- Each test antigen was immobilised on maxisorb plates (Nunc 443404) by adding 50
- a solution of 3% skimmed milk powder (MarvelTM) in PBS was added to the wells and the plate was incubated for at least Ih at room temperature.
- Purified 129D5B and 131D5S scFv were prepared in 3% Marvel/PBS at lOpig/mL and incubated for at least Ih at room temperature prior to transfer to the blocked ELISA plate where a further incubation of at least Ih took place. Unbound scFv was then washed away using repetitive washes with PBS/Tween followed by PBS.
- 129D5B and 131D5S scFv were shown to bind to unique epitopes of human cMET by Biacore.
- Human cMET Fc 2.5
- Human cMET Fc was captured onto a protein G chip by injecting for 30s at 30
- 129D5B scFv, at Ipig/mL in HBS buffer was then injected over the cMET coupled surface and a blank control surface for 180s, followed immediately by a second injection of 129D5B scFv, at Ipig/mL in HBS buffer, for 180 s.
- 131D5S scFv was injected, at Ipig/mL in HBS buffer, for 180 s followed by buffer only for 600 s. Both scFvs were observed to bind simultaneously to human cMET, indicating that they were binding to different epitopes of the protein (Figure 4).
- Example 4 Inhibition of Hepatocyte growth factor binding to human cMET by 129D5B and 131D5S
- Hepatocyte growth factor regulates cell growth, cell motility, and morphogenesis by activating the cMET receptor and both are implicated in oncogenesis [19,22].
- HGF Hepatocyte growth factor
- a ligand: receptor binding assay was developed.
- HGF Seo Biological 10463-HNAS
- 129D5B and 131D5S scFvs were expressed overnight in E.coli strain TGI using Terrific autoinduction medium. Supernatant and periplasmic fractions were harvested by centrifugation and loaded onto Strep-Tactin® FPLC cartridges via Cytiva Akta chromatography systems. Bound scFv were eluted with free biotin and then loaded onto SuperdexTM S75 gel filtration column to separate monomeric scFv protein from larger oligomers and impurities ( Figure 6a and Figure 6b).
- Particle size and distribution of molecules in solution can be obtained by dynamic light scattering (DLS).
- LDS dynamic light scattering
- Molecules scatter light due to Brownian motion.
- the intensity fluctuations of the scattered light are used to calculate an autocorrelation function which allows determination of the hydrodynamic radius of molecules and dispersity of the solution as well as detection of any aggregates.
- Clones 129D5B and 131D5S scFvs were assessed by DLS (using a Nanotemper Prometheus Panta) and shown to be highly monodispersed and homogeneous samples with no indication of larger aggregates.
- Mean particle size was 2.8- 3nm consistent with small monomeric proteins (Figure 6c).
- Nano-differential scanning fluorimetry measures changes in intrinsic fluorescence while a thermal ramp is applied, leading to unfolding and/or aggregation of the protein.
- the melting temperature (Tm) of the protein is defined as the inflection point where half of the protein is still in its folded state.
- Tm melting temperature
- 77F3 was firstly made by bacterial fermentation and purified by affinity chromatography (>95% pure).
- a cytotoxic payload (MMAE-pGA-BrPEGs-PEGz- NHS Ester) was made in-house and conjugated to the scFv to make 77F3-MMAE-PGA- BrPEGs-PEGz-NHS FDC conjugate (DAR 6) using methods described in Example 39.
- 77F3 FDC was digested into peptide fragments and analysed by mass spectrometry to produce a peptide map and to identify which lysine residues were attached to payload.
- This analysis identified lysine residues in 77F3 that were rarely coupled to payload, namely at positions 13, 23, 74 and 239. These positions were then targeted for mutagenesis in a new scFv library that aimed to change the lysines to alternative amino acids (Figure 8).
- the 77F3 lysine variant library (total diversity 1.2xl0 4 ) was generated by Twist Bioscience Inc and then subcloned into a phagemid vector for phage display selections following standard methods as described in Example 1 [216,217].
- specific 77F3 variant scFv were identified by scFv ELISA using crude bacterial periplasmic extracts and by BIAcore following the procedure described in Example 2. From these selections, three candidate scFvs were isolated, 139B2, 139D2 and 139D7, that were of similar affinity and stability to 77F3 and contained well tolerated amino acid variations from lysine at positions 13, 23, 74 and 239 ( Figure 9 and Table 5).
- 129D5B Two potential deamidation sites were identified in 129D5B: one in VL-CDR2 (NS, Kabat VL51 and VL52); and a second in VL-CDR3 (NS, Kabat VL93 and VL94). These sites were de-risked by converting the motifs to NY in VL-CDR2 and NT in VL-CDR3 using standard mutagenesis techniques [221,222].
- the new molecule, named 129D5BTY was unaffected by these changes and retained the same affinity and specificity for cMET as 129D5B ( Figure 10), and also the biophysical properties, i.e., thermal and colloidal stability, were unchanged ( Figures 6, 7 and 11).
- the Fmoc-protected derivative 8 (35 mg, 0.0187 mmol) was dissolved in dry DMF (2 ml) and cooled to 0°C in a water/ice bath. A solution of DBU (8.4 pl, 0.0560 mmol) in dry DMF (1 mL) was added dropwise over 1 h. After the DBU addition was complete, the mixture was allowed to stir for one further hour under nitrogen. The reaction was then quenched with AcOH (5.3 pl, 0.0934 mmol), and the mixture concentrated under reduced pressure.
- MMAE-pGA-alanine derivative 7 88.0 mg, 0.0707 mmol was dissolved in DMF (1 ml).
- Fmoc-Lys-PEGi2 97.0 mg, 0.0934 mmol was then added to the substrate solution, followed by DIPEA (32 pL, 0.184 mmol).
- the Fmoc-protected derivative 13 (34 mg, 0.0166 mmol) was dissolved in dry DMF (2 ml) and cooled to 0°C in a water/ice bath. To this, a solution of DBU (7.4 pl, 0.0497 mmol) dissolved in dry DMF (0.5 ml) was added dropwise over 1 h, whilst stirring and under nitrogen. After the end of the DBU addition, the mixture was allowed to stir for a further 1.5 h after which the ice bath was removed, and the reaction quenched with AcOH (4.7 pl, 0.0166 mmol).
- the deprotected exatecan derivative 20 (16 mg, 0.0190 mmol) was dissolved in dry DMF (500 pl) and added dropwise over 1 h (using a syringe pump) to a solution of bis-PEGz NHS ester (76 mg, 0.190 mmol) dissolved in dry DMF (500 pl) and DIPEA (10 pl, 0.0571 mmol). After addition was complete, the reaction mixture was stirred for a further 1 h at RT, the solvent was then removed and the crude residue purified by RP automated chromatography [Biotage (water + 0.1% TFA/ACN + 0.1% TFA, 10-90%)], to give the desired product 21 as a yellow solid 9.16 mg (48 %).
- LCMS M+ l) 1126.90; (M+Na) 1149.86
- the exatecan derivative 20 (13 mg, 0.0155 mmol) was dissolved in dry DMF (1 ml) and added dropwise over 1 h (using a syringe pump) to a solution of bis-PEG? NHS ester (96 mg, 0.1546 mmol) dissolved in dry DMF (1 ml) and DIPEA (8.1 pl, 0.0461 mmol). After addition was complete, the reaction mixture was stirred for a further 1 h at RT, the solvent was then removed and the crude residue purified by RP automated chromatography [Biotage (water + 0.1% TFA/ACN + 0.1% TFA, 10-90%)], to give the desired DXd-GGFG- PEG? derivative 22 as a yellow solid 8.15 mg (39 %).
- the exatecan derivative 20 (98.50 mg, 0.117 mmol) and the activated lysine branching point 12 (121 mg, 0.141 mmol) were dissolved in dry DMF (10 ml) under nitrogen. DIPEA (49 pl, 0.281) was added and the mixture was stirred at RT under nitrogen for 22 hours after which the reaction mixture was concentrated under high vacuum and the resulting residue purified by automated RP chromatography [Biotage (water + 0.1% TFA/ACN + 0.1% TFA, 0-100%)] to give the branched-Fmoc lysine DXd derivative 23 as a light-yellow solid 71 mg (38%). LCMS m/z (M+l) 1586.50. The Fmoc-deprotected derivative 24 21 mg (13%) was also isolated. LCMS m/z 1365.20 (M+l).
- the Fmoc-derivative 23 (38.70 mg, 24.40 pmol) was dissolved in dry DCM (4 ml) piperidine (53 pL, 0.537 mmol) was added and the mixture stirred at RT under nitrogen for 2 hours. The reaction was was concentrated under high vacuum and the residue purified by RP automated chromatography [Biotage, (water + 0.1% TFA/ACN + 0.1% TFA, 0-100%) to give Fmoc-deprotected derivative 24 as a yellow solid 33 mg (quantitative yield). LCMS m/z (M+l) 1365.20
- Bis-PEGz-NHS 38 mg, 0.095 mmol was dissolved in dry DMF (500 pl), DIPEA (5 pl, 0.029 mmol) was added. Separately, DXd-PEGs amine 24 (13 mg, 0.095 mmol) was dissolved in dry DMF (500 pl). This was then slowly added (1 h) dropwise using a syringe pump to the stirred solution of the bis-PEGz-NHS and DIPEA.
- the reaction mixture was concentrated in vacuo and purified by RP automated chromatography [Biotage, (water + 0.1% TFA/ACN + 0.1% TFA, 0-100%)] to give the globally deprotected exatecan-pGA amine derivative 27 as a pale-yellow solid 18 mg (71 %).
- LCMS (RT 1.923 min.) m/z (M+l) 848.50.
- the partially deprotected (Fmoc-alanine) exatecan-pGA was also isolated 7 mg (24 %).
- Boc-ValAla-PABA 33 50 mg, 127 pmol was dissolved in anhydrous THF (5 mL). Triphenylphosphine (40 mg, 152 pmol) and NBS (27 mg, 152 pmol) were then added and the reaction mixture was allowed to stir at RT under nitrogen for 7 hours. The solvent was then removed at reduced pressure, and the resulting crude purified on NP automated chromatography [Biotage, (DCM/MeOH (0- 50%)]. Combined fractions were concentrated to give the brominated derivative 34 as a yellow solid 49 mg (84 %). LCMS m/z 469 (M+Na).
- the deprotected nintedanib-Val-Ala-amine 36 (2.7 mg, 3.3 pmol) was dissolved in dry DMF (1 ml) and added dropwise (syringe pump) over 70 min to a stirred solution of bis- PEG2 -NHS ester (13.1 mg, 32.8 pmol) and DIPEA (1.7 pl, 9.8 pmol) in dry DMF (0.5 ml) at RT under nitrogen. After the addition was completed, the reaction mixture was allowed to stir for a further 35 minutes at RT after which the DMF was removed under high vacuum.
- the galactose tetraacetate amine 41 (226 mg, 4.8 pmol), Fmoc-0-alanine (165 mg, 5.3 pmol), and EEDQ (238 mg, 963 pmol) were placed in a round bottom flask and dissolved in dry DCM (12 ml). The reaction mixture was stirred for 21 h under an inert atmosphere after which it was quenched by the addition of sodium bicarbonate saturated solution (15 mL) and the phases separated. The aqueous layer was further back extracted with EtOAc (3 x 20 mL), the organics were then combined, dried over MgSC and concentrated in vacuo.
- reaction was diluted with 1 N HCI to pH-7 and purified by RP preparative chromatography (25% MeCN/water with 0.1% HCOOH) to give the Fmoc- deprotected dexamethasone ethylamino-dihydrogen pyrophosphate 49 as a white solid 200 mg (24%).
- the reaction mixture was concentrated in vacuo and the crude purified by NP automated chromatography [Biotage, DCM/MeOH (0-40%)]. The combined fractions were concentrated to the glucuronide-imidazoquinoline derivative 51 as an off-white solid 31 mg (62 %).
- the Fmoc-protected imidazoquinoline-pGA 52 (8.8 mg, 0.0088 mmol) was dissolved in dry DMF (1 ml)and DIPEA (31 pL, 0.301 mmol) was added. The reaction mixture was stirred for 50 minutes under nitrogen and concentrated in vacuo. The crude was purified by RP automated chromatography [Biotage (water + 0.1% TFA/ACN + 0.1% TFA, 10- 100%)] and appropriate fractions combined and lyophilised to give the desired fully- deprotected imidazoquinoline-pGA amine 53 as a white solid (quantitative yield). LCMS m/z 772.50 (M + l); 386.81 (M/2); 377.81 (M - payload).
- triptolide succinate 55 (3 mg, 6.5 pmol) was dissolved in dry DMF (0.2 ml) and dry DCM (1.6 ml), DCC (1.6 mg, 7.8pmol) and n- hydroxysuccinimide (0.83 mg, 7.2 pmol) were sequentially added and the mixture was allowed to stir at RT under an inert atmosphere for 24 hours. The mixture was concentrated under a vacuum and the residue purified by RP automated chromatography [Biotage (water + 0.1% TFA/ACN + 0.1% TFA, 10-90%)] to give NHS-activated ester triptolide derivative 56 2.57 mg (71%).
- triptolide fragment 558.39 (M+l), 558.40 (M+Na), 1115.81 (dimer), 361.22 (triptolide fragment).
- the triptolide succinic acid NHS ester 56 (1.2 mg, 2.2 pmol) was dissolved in dry DMF (1 ml) and DIPEA (1.1 pL, 6.5 pmol) was added followed by the addition of the amino-PEGe- COOH. The mixture was allowed to stir at RT under inert atmosphere for 2 hours after which the solvent was removed under a vacuum to give triptolide-succinate-PEGe-acid 57 which was used directly in the next step without purification.
- LCMS m/z 796.61 (M+l), 818.64 (M+ Na).
- Triptolide-succinate-PEGe acid 57 (1.71 mg, 2.2 pmol) was stirred with TSTU (1.3 mg, 4.3 pmol) and DIPEA (1.9 pl, 10.7 pmol) in dry DMF (1 ml) for two hours under nitrogen. The solvent was removed at high vacuum and the residue was purified by RP automated chromatography [Biotage (water + 0.1% TFA/ACN + 0.1% TFA, 0-90%)] to give the triptolide-succinate-PEGe NHS ester 58 as a white solid after lyophilisation 0.5 mg.
- LCMS m/z 893.70 (M + l), 915.70 (M + Na).
- MMAE 40 mg, 55.71
- commercially available maleimidocaproyl-Val-Cit-PAB-PNP 60 49.32 mg, 66.85 pl
- HOBt 7.68 mg, 56.83 pl
- DIPEA 10.67 pl, 61.28 pmol
- MMAE derivative 65 (760 mg, 0.64 mmol) in MeOH (3 ml) was added a solution of LiOH.HzO (86 mg, 2.03 mmol) in H2O (3 ml) dropwise at 0 °C, and the mixture stirred at rt for 2h.
- the mixture was acidified with IN HCI to pH-2, filtered with H2O and concentrated to afford the MMAE-mandelic acid derivative 66 as a white solid 700 mg (93%).
Landscapes
- Health & Medical Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Immunology (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Organic Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Cell Biology (AREA)
- Biotechnology (AREA)
- Biochemistry (AREA)
- Biophysics (AREA)
- Genetics & Genomics (AREA)
- Molecular Biology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Peptides Or Proteins (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
Abstract
The present invention provides novel anti-cMET antibodies and antigen-binding fragments thereof. The invention also provides antibody-drug conjugates (ADCs) and fragment-drug conjugates (FDCs) derived from said anti-cMET antibodies and antigen-binding fragments thereof. The invention also provides pharmaceutical compositions, methods of making, and methods of using the disclosed compounds for use in the treatment of cancer and other diseases.
Description
Antibodies, conjugates, and uses thereof
Field of the invention
The present invention relates to antibodies, antigen-binding fragments thereof and fragment-drug conjugates (FDCs) which bind to the human MET-receptor (cMET), methods for discovering and making anti-cMET FDCs and their use in the treatment of cancer and other diseases.
Background of the invention
Cancer is a leading cause of global deaths, and according to the World Health Organisation (https://qco.iarc.fr') accounted for 10 million deaths in 2020. The most common cancers include lung (~1.8 million cases/year), colon and rectum (~935 000), liver (~830 000), stomach (~760 000) and breast (~685 000). Poor permeation [1] and multi-drug resistance [2] in solid tumours are two important challenges which lead to the failure of current treatment methods. The tumour microenvironment (TME) in solid tumours poses physical challenges for the effective and homogenous delivery of cancer treatments. Intra- tumoral drug penetration is hindered by the physical barriers brought on by the tumour stroma, disruption and disorganisation of the extracellular matrix increasing tissue stiffness/solid stress, impaired lymphatic drainage which consequently increases hydrostatic pressure and increased vascular permeability 'leaky vasculature' causing irregular intra-tumour blood flow [3]. Poor drug penetration and distribution leads to marginal doses reaching the tumour resulting in acquired resistance and treatment failure [1].
One approach to increase cancer therapy effectiveness is to use combination of drugs with different or complementary mechanisms of action and non-overlapping clinical toxicities [4]. Antibody-Drug Conjugates (ADCs) are a well-established form of targeted therapy, making an impact in cancer primarily, but also in infectious diseases, inflammatory diseases and other indications [5,6]. Here, a specific monoclonal antibody (MAb) enables the selective accumulation of an effector payload in the required target tissue (e.g., tumour) whilst normal, non-targeted organs eliminate and clear the ADC or its components. ADCs, designed around the standard whole Immunoglobulin (Ig) structure, primarily IgG, have a long circulatory half-life due to the presence of the Fc-domain (which interacts with the neonatal Fc-receptor, FcRn). However, the large size also presents many limitations such as slow and ineffective tumour and organ penetration and long-term exposure of payloads to non-targeted tissues [6,7].
cMET overexpression relative to that of normal tissues has been observed in many types of solid human malignancies including gastric, colorectal, pancreatic, lung, head and neck, ovarian, breast, renal, prostate, bladder, nasopharyngeal, gliomas, osteosarcomas and melanomas (Table 1). Due to the poor clinical prognosis of solid tumours overexpressing cMET, and the toxicities caused by broad-spectrum chemotherapeutic treatments used today as well as insufficient drug dosage reaching all parts of the tumour, an opportunity is presented for the development of targeted therapeutics against this receptor.
Furthermore, MAb or ADC distribution following extravasation from the vasculature is further impeded by cellular internalization and subsequent endocytic clearance at the tumour periphery (an effect known as the "binding-site barrier"), leading to poor penetration and regions of marginal antibody concentrations [8-10]. It follows that higher binding-affinity, multi-valent avidity and higher antigen expression, especially at the tumour edge, can retard MAb tumour percolation and impair homogeneous distribution. Although this barrier can be overcome by increasing the dose of therapeutic agent administered to patients, this can lead to significant side effects, particularly with cytotoxic agents such as ADCs.
An alternative approach is to improve penetration and diffusion using smaller formats such as antibody fragments (e.g., Fab fragments (~50 kDa), single-chain variable fragments (scFv ~30 kDa) and single-domain antibodies (sdAb 12-15 kDa)) and peptides [11]. Although these formats possess higher extravasation and intra-tumoural diffusion rates, the overall percentage tumour uptake is low because the elimination rates for smaller formats is higher compared to full-size antibody molecules. Immunoglobulins possess a half-life of at least 20 days for most therapeutic MAbs [9,12]. Antibody fragments lacking an Fc region have half-lives of minutes-hours for formats whose size falls below the glomerular filtration cut-off (approx. 30-50 kDa) [7,11]. The rapid elimination rate after systemic delivery prevents most antibody fragments from saturating the tumour and achieving a uniform distribution.
Due to its targeting/localization effect, ADCs increase the tumour concentration of the targeted payload, therefore lower doses are required to reach the tumour to effectively destroy the targeted cells. More recent thinking suggests that ADCs have a similar maximum tolerated dose as the equivalent payload but improve efficacy though targeted delivery [13]. Moreover, depending on the chemical nature of the drug and its release in the tumour (either intracellular or extracellular), some payloads can subsequently diffuse and kill surrounding non-targeted tumour cells ("bystander killing") [14]. Consequently, these features, in some way, help ameliorate the drawbacks of the heterogeneous tumour
distribution, but does not solve the problem, especially for intractable or poorly vascularised tumours.
Despite >90% of cancers being solid tumours, only 6/12 of the approved ADCs are for solid tumours (trastuzumab-emtansine, famtrastuzumab-deruxtecan, directed against HER.2, enfortumab-vedotin against Nectin-4, Sacituzumab govitecan against TR.OP-2, tisotumab vedotin against tissue factor and mirvetuximab sorvtansine against folate receptor alpha) which highlights the difficulty in applying ADCs to these diseases [5]. An estimated 100 or more other ADCs are currently under clinical development (www.clinicaltrials.gov but the failure rate for solid tumours is disproportionately higher compared to haematological cancers (https ://beacon-intelligence.com). The availability of target antigens expressed on the surface of tumour cells and the approval of the use of ADCs have seen success in the area of haematological malignancies. Solid tumours however display antigens which are tumour associated and have varying levels of overexpression. These antigens are not exclusively found on the surface of these solid tumours but are also expressed on normal tissues at lower levels bringing about challenges with on-target off-tumour toxicity [6]. High doses, poor biophysical properties (e.g., aggregating) and the presence of an Fc-region can lead to off-target toxicities [15]. Biochemical improvements to ADCs have driven the success in this area such as improved linker chemistry preventing premature release, increased payload loading achieving higher drug:antibody ratios (DAR) for more tolerable payloads, and the discovery of new warheads with different mechanisms of action and membrane-permeable payloads [5,6,16,17],
The addition of a cytotoxic payload to pre-existing targeted therapy illustrates the value of tumour-specific cytotoxin delivery. Trastuzumab-emtansine was able to broadly improve patient outcomes compared to the non-conjugated trastuzumab MAb and this was further improved by trastuzumab-deruxtecan [5,6,18] which is now widely acknowledged to have a transformative clinical impact in HER2-positive breast cancer [18]. cMET overexpression relative to that of normal tissues has been observed in many types of solid human malignancies including gastric, colorectal, pancreatic, lung, head and neck, ovarian, breast, renal, prostate, bladder, nasopharyngeal, gliomas, osteosarcomas and melanomas (Table 1). Due to the poor clinical prognosis of solid tumours overexpressing cMET, and the toxicities caused by broad-spectrum chemotherapeutic treatments used today as well as insufficient drug dosage reaching all parts of the tumour, the inventors identified an opportunity which is presented for the development of targeted therapeutics against this receptor.
The inventors have developed novel anti-cMET antibodies and antigen-binding fragments thereof, along with antibody-drug and fragment-drug conjugates. These antibodies have surprising beneficial properties in terms of cMET binding, and tumour penetration and clearance.
Summary of the invention
In a first aspect the invention provides an antibody or antigen-binding fragment thereof that specifically binds to the MET receptor (cMET), wherein the antibody or antigen-binding fragment thereof comprises a heavy chain variable region (VH) comprising the Complementarity Determining Region (CDRs) CDR-H1, CDR-H2, and CDR-H3 of SEQ ID NO. 1, or a variant thereof having at least 80% sequence identity to SEQ ID NO. 1, and comprises a light chain variable region (VL) comprising the CDRs CDR-L1, CDR-L2, and CDR-L3 of SEQ ID NOs. 196 or 2, or a variant thereof having at least 80% sequence identity with SEQ ID NOs. 196 or 2.
In a particular embodiment of the first aspect of the invention there is provided an antibody or antigen-binding fragment thereof that specifically binds to the MET receptor (cMET), wherein: (i) the antibody or antigen-binding fragment thereof comprises a heavy chain variable region (VH) comprising the Complementarity Determining Regions (CDRs) CDR- Hl, CDR-H2, and CDR-H3 of SEQ ID NO. 1, or a variant thereof having at least 90% sequence identity to SEQ ID NO. 1, and comprises a light chain variable region (VL) comprising the CDRs CDR-L1, CDR-L2, and CDR-L3 of SEQ ID NOs.196 or 2, or a variant thereof having at least 90% sequence identity with SEQ ID NOs.196 or 2; or (ii) the antibody or antigen-binding fragment thereof comprises a heavy chain variable region (VH) comprising the Complementarity Determining Regions (CDRs) CDR-H1, CDR-H2, and CDR-H3 of SEQ ID NO. 21, or a variant thereof having at least 90% sequence identity to SEQ ID NO. 21, and comprises a light chain variable region (VL) comprising the CDRs CDR-L1, CDR-L2, and CDR-L3 of SEQ ID NO. 22, or a variant thereof having at least 90% sequence identity with SEQ ID NO. 22.
In some embodiments of the first aspect of the invention, the antibody or antigen-binding fragment thereof comprises a VL comprising the CDRs of SEQ ID NO. 196.
In some embodiments, the antibodies and antigen-binding fragments disclosed herein have advantageous properties in terms of cMET binding and blocking of cMET signalling. The application also discloses the antibodies and antigen-binding fragments thereof conjugated to payloads which demonstrate advantageous properties in tumour
penetration, tumour payload delivery, serum half-life and bioavailability, normal tissue exposure and tumour clearance.
By "cMET" we mean the MET receptor, which is also referred to as hepatocyte growth factor receptor, HGF receptor, HGFR, and MET. The mesenchymal-epithelial transition factor gene referred to as MET is a proto-oncogene which encodes a cell surface tyrosine kinase receptor (cMET) for hepatocyte growth factor (HGF)/scattering factor (SF) ligand. Upon ligand binding cMET forms a homo-dimer leading to trans-phosphorylation which activates several pathways including phosphoinositide 3-kinase (P13K)/AKT (protein kinase B), mitogen-activated protein kinase Ras/(MAPK), Janus kinase/signal transducers and activators of transcription (JAK/STAT), SRC and WNT/P catenin [19,20].
The HGF/cMET pathway in normal physiology is involved in embryogenesis, normal cellular growth, cell motility, cell survival, wound healing, angiogenesis and tissue regeneration. The HGF/cMET pathway regulates disease progression by reducing oxidative stress, inflammation, apoptosis and fibrosis. cMET is expressed in vascular, lymphatic endothelial cells and hematopoietic cells, skin, GI tract and lungs at low normal levels and can increase upon tissue repair, regeneration or during inflammation. cMET is also expressed at low levels in normal liver, kidney, pancreas, prostate, muscle and bone marrow, during embryogenesis and adulthood [19].
MET overexpression, genomic amplification, translocations, sub-gene deletions, point mutations and alternative splicing can lead to the aberrant expression of the MET protooncogene. This is a major cause of the dysregulation of the cMET autocrine and paracrine signalling pathway [21]. Over-expression of the HGF ligand can also lead to aberrant cMET signalling. These alterations, leading to cMET activation have been observed in many human malignancies associated with invasive growth, tumour cell motility, angiogenesis, and poor clinical prognosis [20-23]. cMET expression strongly and significantly correlates with poor prognosis and patient outcomes in multiple clinical examples of cancer and is highly implicated in other cancers (Table 1). In some cases, genetic defects in MET lead to activation which renders tumour cells 'addicted' (i.e. highly dependent on this pathway for growth and survival) to MET-oncogene signalling [22]. More often than not cMET protein over-expression is not the outcome of MET gene mutations which occur in ~6% of cMET-over-expressing cancers [21]. MET gene amplification frequency in solid tumours (<2%) is described in the AACR GENIE registry (https://www.aacr.org/professionals/research/aacr-proiect-qenie) [23].
The MET gene is translated as a single chain precursor of 1390 residues and transported to the golgi for glycosylation. Furin cleaves the protein to form a disulphide-linked alpha
and beta chain heterodimer. The extracellular portion of cMET comprises 3 domains, SEMA (semaphorin, a 7-bladed beta propeller structure which includes the HGF-binding site) domain, PSI (plexin-semaphorin-integrin-domain) and 4 IPT (immunoglobulin-plexin- transcription) repeats. The intracellular domains comprise a juxtamembrane sequence, the tyrosine kinase catalytic domain and carboxyl-terminal sequences [19]. cMET overexpression relative to that of normal tissues has been observed in many types of solid human malignancies including gastric, colorectal, pancreatic, lung, head and neck, ovarian, breast, renal, prostate, bladder, nasopharyngeal, gliomas, osteosarcomas and melanomas (Table 1). Due to the poor clinical prognosis of solid tumours overexpressing cMET, and the toxicities caused by broad-spectrum chemotherapeutic treatments used today as well as insufficient drug dosage reaching all parts of the tumour, an opportunity is presented for the development of targeted therapeutics against this receptor.

 cMET overexpression can be defined by an immunohistochemistry (IHC) H-score (range 0-300) of 200-300 (High, 3+), 100-200 (Medium, 2+), 50-100 (Low, 1+) or 0-50 (Negative, 0) [49]. Briefly, IHC staining protocol for cMET overexpression has been developed using the Ventana cMet CONFIRM (SP44) kit. Tissue samples are stained with the Ventana antibody and then scored by determining the percentages of target tissue cells staining at various intensity levels of low to high [50].
cMET overexpression can be defined by an immunohistochemistry (IHC) H-score (range 0-300) of 200-300 (High, 3+), 100-200 (Medium, 2+), 50-100 (Low, 1+) or 0-50 (Negative, 0) [49]. Briefly, IHC staining protocol for cMET overexpression has been developed using the Ventana cMet CONFIRM (SP44) kit. Tissue samples are stained with the Ventana antibody and then scored by determining the percentages of target tissue cells staining at various intensity levels of low to high [50].
Other methods to determine cMET over-expression levels can also include RNA In Situ Hybridization Assays, silver In Situ Hybridization Assays (SISH) or Quantitative real-time polymerase chain reaction (qPCR). cMET expression determination by protein-based methods such as IHC is cost-effective and well established in clinical settings but does not discriminate between MET-oncogene addicted and non-addicted tumour cells [21,22].
The MET properties described above position this oncogene target as an important prognostic factor and a valuable drug target for numerous applications in clinical oncology [51-55].
There has been a major effort to develop small molecule cMET pathway inhibitors (e.g., crizotinib, cabozantinib, capmatinib, tepotinib, and glesatinib). Cabozantinib (Cabometyx®/Cometriq®) and crizotinib (Xalkori®) are FDA/EMA-approved and marketed drugs but many others have failed clinical development primarily due to poor tolerability or low efficacy (often related to poor patient selection resulting in patients whose tumours are not oncogene addicted and less sensitive to cMET inhibition) [22,51]. Small molecule inhibitors (SMIs) block ATP-phosphorylation of the cMET tyrosine kinase preventing downstream signalling and broad specificity for ATP-binding sites makes them specific for related kinases such as VEGFR, RET, AXL and others.
Cabozantinib is approved for thyroid, liver and kidney cancer but has shown disappointing results in other major solid tumours with side effects such as severe bleeding, blood clots,
hypertension, gastro-intestinal toxicities. Crizotinib is approved for ALK/ROS-mutated non small-cell lung cancer with serious adverse effects including liver toxicity (fatal in 0.1% patients), interstitial lung disease (severe/fatal in 0.5%), pneumonitis, cardiac effects and neutropenia. This indicates a need for better tolerated drugs. cMET expression in tumours is also associated with resistance to targeted therapies against other receptors or signalling pathways. For example, in NSCLC, patients become resistant to epidermal growth factor receptor (EGFR) inhibitors [27] with MET-gene amplification occurring as a by-pass mechanism in 5-20% of resistant patients and HGF over-expression occurring in a higher number of patients. Vascular endothelial growth factor (VEGF) pathway resistance is also seen [20]. This has resulted in therapeutic strategies utilizing cMET cross-reactive SMI's and bispecific MAbs (e.g. amivantamab) and ADCs.
Single agent anti-cMET MAbs (e.g., onartuzumab, emibetuzumab, SAIT-301, and ABT700), or single agent anti-HGF MAbs (e.g. ficlatuzumab, rilotumumab) which aim to inhibit cMET signalling have met with limited clinical success [20,22] for many of the reasons mentioned above. Total cMET protein levels (determined by IHC) does not correlate well with pathway activation leading to lack of efficacy in the selected patient population [54]
For example, onartuzumab (MetMAb) is a humanized antagonistic anti-cMET MAb [22]. Clinical trials in Phase III for lung cancer in combination with Erlotinib was terminated due to lack of efficacy [55]. Rilotumumab, which binds to HGF and inhibits HGF/cMET signalling, was terminated in Phase III due to increased deaths compared to chemotherapy alone [55].
Delivering a cytotoxic payload directly to tumour cells via a MAb against the cMET receptor ensures cell death via irreversible cellular damage independent of cMET-mediated proliferative cell signalling and thus overcomes many of the limitations of inhibitory MAbs or SMIs. A number of ADCs (and MAbs) are in development where the MAb component, due to its bivalent structure must be designed not to cause cMET cross-linking and induced signalling agonism [51]. This can be achieved by engineering the hinge region or using biparatopic MAbs which enable cMET clustering but not signalling. However, high affinity or high avidity MAbs can inhibit tumour penetration due to the binding site barrier effect [9]. Cross-linking receptors can induce or speed up their internalization but too rapid internalization can also inhibit tumour mass penetration [56].
W02018/098035 describes an anti-cMET ADC comprising MMAE payload which demonstrated tumour killing efficacy in a range of human tumour xenograft models at 1- 3mg/kg given intraperitoneally one to four doses. However, the development of this ADC was discontinued. Abbvie are currently developing a cMET ADC, called telisotuzumab
vedotin, ABBV399, comprising the MMAE payload, (EP3636273B1) which demonstrated tumour cure efficacy in human tumour xenograft models at six doses of 3mg/kg (cumulative dose of 18mg/kg) in tumours which express high to moderate levels of cMET target. No efficacy was seen at six doses of Img/kg (cumulative dose of 6mg/kg). ABBV399 was ineffective in cancer models with low-cMET expression, with a threshold of >100,000 receptors/ cel I being described. Greater than 50% maximal cell killing was not observed in call lines with less than 50,000 receptors/cell [51,57]. Telisotuzumab vedotin is in advanced stage (Phase II) clinical trials for NSCLC and has demonstrated good tolerability at 2.7mg/kg [58] and RECIST-qualified tumour regression responses in patients expressing high (3+) to moderate (2+) cMET levels with high-cMET expressing patients responding more favourably (LUMINOSITY Phase II Trial, EGFR-Wild type, cMET- high, ORR-52.2%, EGFR-Wild type, cMET-intermediate, ORR-24.1% [59]. This suggests stratification of patients based on cMET protein expression may lead to better patient outcomes when using an ADC and a high-DAR FDC approach with the potential to deliver more cytotoxic payload per receptor could be additionally beneficial for patients expressing lower levels of cMET. The most common any-grade adverse events were peripheral sensory neuropathy, nausea, and hypoalbuminemia (20-25%).
Other cMET ADCs are in development at earlier stages such as TR1801 (PBD payload) which is in Phase I [35], SHRA1403 (auristatin payload, [60]), cIRCR201-dPBD (PBD payload, [61]) and BYON3521 (duocarmycin payload, [62]). Bispecific cMET ADCs include AZD9592 [63] specifically to address EGFR co-expression and resistance to EGFR inhibitors in lung cancer. cMET ADCs with ultra-potent (IC50 in the pM range) DNA damaging payloads pose a toxicity concern as Abbvie discontinued development (EP3636273B1) as did Immunogen (HucMET27-DGN549). Generally, ultra-potent alkylating, DNA damaging payloads and ADCs against other targets using highly potent payloads such as PBDs have been discontinued due to poor tolerability and an almost 50% failure rate [15].
ADCs are complex molecules to discover and develop. There are many parameters that need to be optimized, sometimes empirically, but increasingly, it is well established that there are some key design features [5,6]. The physical and chemical stability (e.g. aggregation, pl, hydrophobicity) of an ADC can potentially impact its efficacy, toxicity and immunogenicity and many other potentially dose-limiting toxicities involved Fc-receptor binding and prolonged exposure to normal tissues [15,64] and general methods for designing and making ADCs are described [5,6,65].
Idiopathic pulmonary fibrosis (IPF) is a progressive lung fibrosing disease which is poorly understood. It is characterized by progressive lung scarring leading to respiratory failure
and death, with a median survival from diagnosis of ~3 years [66]. Many therapies have been tested for this harmful condition, yet none have demonstrated effectiveness in modifying respiratory-specific or all-cause mortality in IPF [67]. The aberrant proliferative events in IPF resemble that of malignant transformation in cancer [66] which could encourage the use of cancer drugs for IPF. The key histological feature of IPF is shown in the "fibroblast foci", aggregates of actively proliferating fibroblasts and myofibroblasts- cells which can also harbour MET in its activated form. Therefore, RTK inhibitors such as those against cMET could be efficacious in IPF [66].
There are currently two FDA-approved drugs for the treatment of IPF: pirfenidone (Esbriet®) and nintedanib (Ofev®). Pirfenidone is a pyridone derivative which exhibits antiinflammatory and anti-fibrotic effects (via downregulation of transforming growth factor- p (TGF-p), a central signalling pathway in fibrosis). Nintedanib is a broad-spectrum- tyrosine kinase inhibitor, originally used as anti-vascular cancer chemotherapy. Nintedanib's anti-fibrotic activity via inhibition of key growth factor signalling pathways involved in pulmonary fibrosis (e.g., platelet-derived growth factor (PDGF), vascular endothelial growth factor (VEGF), and fibroblast growth factor (FGF). Nintedanib also competitively inhibits both non-receptor tyrosine kinases (nRTKs) including Lek, Lyn, and Src. Therefore, nintedanib action resembles the broad spectrum tyrosine kinase inhibitors used in anti-cMET cancer therapy. Pirfenidone and nintedanib have demonstrated efficacy in clinical trials, slowing the decline in forced vital capacity typical of IPF progression. Despite these two agents, therapeutic options for patients living with this condition remain very limited. More recently, TAS-115, a highly specific, SMI of VEGF receptor 2 (VEGFR2) and cMET originally discovered as an antitumor therapeutic [68] has shown therapeutic promise with good tolerability. HGF signals through cMET to upregulate expression of proangiogenic VEGF. Blocking both pathways by inhibiting RTK phosphorylation with TAS- 115 was shown to effectively decrease tumour vascularization and size with minimal toxicity and off-target effects. Applied in the IPF setting, TAS-115 could modulate inflammation associated with fibrosis as demonstrated in preclinical models, better than nintedanib [69].
Therefore, as in cancer, cMET-induced invasive growth is emerging as a target for IPF, due to the proliferative nature of this disease in the lung. Signalling inhibitors aim to reduce the proliferation of damaging fibroblasts. This suggests that cMET receptor could be a candidate for the targeted delivery of chemical modulator-drugs to dampen down inflammation or proliferation. This would increase the local concentration of such therapeutic drugs and reduce the off-target systemic adverse effects of broadly-acting SMI drugs.
In some embodiments, the antibody or antigen-binding fragment thereof is capable of blocking cMET signalling.
In some embodiments, the antibody or antigen-binding fragment thereof binds cMET with high affinity, or with higher affinity than an alternative anti-cMET antibody. Affinity can be expressed by measuring the KD (dissociation constant, or equilibrium dissociation constant, expressed in M) or KA (association constant, or equilibrium association constant, expressed in M-1) of a particular antibody or antigen-binding fragment when binding cMET. The KD and KA may be calculated by measuring the association rate constant (ka, expressed in M-1s_1) and dissociation rate constant (kd, expressed in s-1). KA is calculated by ka/kd. KD is calculated by kd/ka.
Therefore, by "higher affinity" we mean that the antibody or antigen binding fragment thereof has a higher KA and/or a lower KD than an alternative anti-cMET antibody or antigen binding fragment. In some preferred embodiments, the antibody or antigen binding fragment has a lower KD (i.e. a higher binding affinity) than an alternative anti- cMET antibody or antigen binding fragment. In some embodiments, the antibody or antigen binding fragment thereof has a higher ka and/or a lower kd than an alternative anti-cMET antibody or antigen binding fragment.
In some embodiments, the antibody or antigen-binding fragment thereof binds cMET with an affinity KD of less than lOnM. In some embodiments, the antibody or antigen-binding fragment thereof binds cMET with an affinity KD of less than 5nM. In some embodiments, the antibody or antigen-binding fragment thereof binds cMET with an affinity KD of less than 4nM. In some embodiments, the antibody or antigen-binding fragment thereof binds cMET with an affinity KD of less than 3nM. In some embodiments, the antibody or antigenbinding fragment thereof binds cMET with an affinity KD of less than 2nM.
In some embodiments, the antibody or antigen-binding fragment thereof binds cMET with an affinity KD of less than InM.
In some embodiments, the antibody or antigen-binding fragment thereof of the first aspect comprises a heavy chain variable region (VH) comprising the Complementarity Determining Region (CDRs) CDR-H1, CDR-H2, and CDR-H3 of SEQ ID NO. 1, or a variant thereof having at least 80% sequence identity to SEQ ID NO. 1, for example a variant having at least 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity. In some embodiments, the VH has at least 90% identity with SEQ ID NO. 1 (i.e. up to 10% variance). In some preferred embodiments, the VH has at least 93% identity with SEQ ID NO. 1 (i.e. up to 7% variance).
The CDR sequences described herein are defined according to the Kabat system.
In some embodiments, the antibody or antigen-binding fragment thereof of the first aspect comprises a light chain variable region (VL) comprising the CDRs CDR-L1, CDR-L2, and CDR-L3 of SEQ ID NOs. 196 or 2, or a variant thereof having at least 80% sequence identity with SEQ ID NOs. 196 or 2, for example a variant having at least 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity. In some embodiments, the VL has at least 90% identity with SEQ ID NOs. 196 or 2 (i.e. up to 10% variance). In some preferred embodiments, the VL has at least 91% identity with SEQ ID NOs. 196 or 2 (i.e. up to 9% variance).
In some embodiments, the antibody or antigen-binding fragment thereof of the first aspect comprises a heavy chain variable region (VH) comprising the Complementarity Determining Region (CDRs) CDR-H1, CDR-H2, and CDR-H3 of SEQ ID NO. 21, or a variant thereof having at least 90% sequence identity to SEQ ID NO. 21, for example a variant having at least 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity. In some embodiments, the VH has at least 90% identity with SEQ ID NO. 21 (i.e. up to 10% variance). In some preferred embodiments, the VH has at least 91% identity with SEQ ID NO. 21 (i.e. up to 9% variance).
In some embodiments, the antibody or antigen-binding fragment thereof of the first aspect comprises a light chain variable region (VL) comprising the CDRs CDR-L1, CDR-L2, and CDR-L3 of SEQ ID NO. 22, or a variant thereof having at least 90% sequence identity with SEQ ID NO. 22, for example a variant having at least 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity. In some embodiments, the VH has at least 90% identity with SEQ ID NO. 22 (i.e. up to 10% variance). In some preferred embodiments, the VH has at least 91% identity with SEQ ID NO. 22 (i.e. up to 9% variance).
In some embodiments, the antibody or antigen-binding fragment thereof of the first aspect comprises any combination of 6 CDR sequences set out in Table A and/or Table B. For example, the antibody or antigen-binding fragment thereof of the first aspect comprises the 6 CDR sequences of any clone listed in Table A and/or Table B.
In some embodiments, the antibody or antigen-binding fragment thereof of the first aspect comprises any pair of VH and VL sequences set out in Table A and/or Table B. For example, the antibody or antigen-binding fragment thereof of the first aspect comprises the VH and VL sequences of any clone listed in Table A and/or Table B.
Table A


Table B

In some embodiments of the first aspect of the invention, the VH comprises the CDRs of SEQ ID NO. 1 and the VL comprises the CDRs of SEQ ID NO. 2.
In some embodiments of the first aspect of the invention, the VH comprises the CDRs of SEQ ID NO. 1 and the VL comprises the CDRs of SEQ ID NO. 196.
In some embodiments of the first aspect of the invention, the VH comprises the CDRs of SEQ ID NO. 3 and the VL comprises the CDRs of SEQ ID NO. 4.
In some embodiments of the first aspect of the invention, the VH comprises the CDRs of SEQ ID NO. 3 and the VL comprises the CDRs of SEQ ID NO. 6.
In some embodiments of the first aspect of the invention, the VH comprises the CDRs of SEQ ID NO. 3 and the VL comprises the CDRs of SEQ ID NO. 8.
In some embodiments of the first aspect of the invention, the VH comprises the CDRs of SEQ ID NO. 5 and the VL comprises the CDRs of SEQ ID NO. 6.
In some embodiments of the first aspect of the invention, the VH comprises the CDRs of SEQ ID NO. 7 and the VL comprises the CDRs of SEQ ID NO. 8.
In some embodiments of the first aspect of the invention, the VH comprises the CDRs of SEQ ID NO. 9 and the VL comprises the CDRs of SEQ ID NO. 10.
In some embodiments of the first aspect of the invention, the VH comprises the CDRs of SEQ ID NO. 11 and the VL comprises the CDRs of SEQ ID NO. 12.
In some embodiments of the first aspect of the invention, the VH comprises the CDRs of SEQ ID NO. 13 and the VL comprises the CDRs of SEQ ID NO. 12.
In some embodiments of the first aspect of the invention, the VH comprises the CDRs of SEQ ID NO. 14 and the VL comprises the CDRs of SEQ ID NO. 15.
In some embodiments of the first aspect of the invention, the VH comprises the CDRs of SEQ ID NO. 16 and the VL comprises the CDRs of SEQ ID NO. 17.
In some embodiments of the first aspect of the invention, the VH comprises the CDRs of SEQ ID NO. 18 and the VL comprises the CDRs of SEQ ID NO. 17.
In some embodiments of the first aspect of the invention, the VH comprises the CDRs of SEQ ID NO. 19 and the VL comprises the CDRs of SEQ ID NO. 20.
In some embodiments of the first aspect of the invention, the VH comprises the CDRs of SEQ ID NO. 21 and the VL comprises the CDRs of SEQ ID NO. 22.
In some embodiments of the first aspect of the invention, the VH comprises SEQ ID NO. 3 and the VL comprises SEQ ID NO. 8.
In some embodiments of the first aspect of the invention, the VH comprises SEQ ID NO. 5 and the VL comprises SEQ ID NO. 6.
In some embodiments of the first aspect of the invention, the VH comprises SEQ ID NO. 7 and the VL comprises SEQ ID NO. 8.
In some embodiments of the first aspect of the invention, the VH comprises SEQ ID NO.
9 and the VL comprises SEQ ID NO. 10.
In some embodiments of the first aspect of the invention, the VH comprises SEQ ID NO.
11 and the VL comprises SEQ ID NO. 12.
In some embodiments of the first aspect of the invention, the VH comprises SEQ ID NO.
13 and the VL comprises SEQ ID NO. 12.
In some embodiments of the first aspect of the invention, the VH comprises SEQ ID NO.
14 and the VL comprises SEQ ID NO. 15.
In some embodiments of the first aspect of the invention, the VH comprises SEQ ID NO.
16 and the VL comprises SEQ ID NO. 17.
In some embodiments of the first aspect of the invention, the VH comprises SEQ ID NO.
18 and the VL comprises SEQ ID NO. 17.
In some embodiments of the first aspect of the invention, the VH comprises SEQ ID NO.
19 and the VL comprises SEQ ID NO. 20.
In some embodiments of the first aspect of the invention, the VH comprises SEQ ID NO.
21 and the VL comprises SEQ ID NO. 22.
In some embodiments, the antibody or antigen-binding fragment of any of the preceding claims, wherein the VH comprises the following CDRs: a) CDR-H1: [SEQ ID NO. 23] or an amino acid sequence having at least 80% identity therewith, for example at least 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably with at least 80% identity; and/or b) CDR-H2: [SEQ ID NO. 28] or an amino acid sequence having at least 80% identity therewith, for example at least 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably with at least 88% identity; and/or c) CDR-H3: [SEQ ID NO. 32] or an amino acid sequence having at least 65% identity therewith, for example at least 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably with at least 67% identity.
In some embodiments, the antibody or antigen-binding fragment of any of the preceding claims, wherein the VH comprises the following CDRs:
a) CDR-H1: [SEQ ID NO. 27] or an amino acid sequence having at least 60% identity therewith, for example at least 60%, 65%, 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably with at least 60% identity; and/or b) CDR-H2: [SEQ ID NO. 29] or an amino acid sequence having at least 80% identity therewith, for example at least 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably with at least 82% identity; and/or c) CDR-H3: [SEQ ID NO. 35] or an amino acid sequence having at least 60% identity therewith, for example at least 65%, 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably with at least 69% identity.
In some preferred embodiments, the VH comprises the CDRs of SEQ ID NOs 23, 28, and 32. In some preferred embodiments, the VH comprises the CDRs of SEQ ID NOs 27, 29, and 35.
In some other embodiments, the VH comprises the CDRs of any of the following combinations:
(i) SEQ ID NOs 23, 115, and 30 (clones 3A2, 20F6, and 53D11);
(ii) SEQ ID NOs 23, 115, and 31 (clones 49G5, 139B2, 139D2, 139D7 and 77F3);
(iii) SEQ ID NOs 23, 29, and 33 (clone 26B5);
(iv) SEQ ID NOs 23, 29, and 33 (clone 83B11D);
(v) SEQ ID NOs 23, 29, and 34 (clone 94C2);
(vi) SEQ ID NOs 23, 29, and 35 (clone 115D1D);
(vii) SEQ ID NOs 27, 29, and 35 (clone 131D5s);
(viii) SEQ ID NOs 23, 28, and 32 (clone 129D5B/129D5BTY).
In some embodiments, the antibody or antigen-binding fragment comprises a VH having the amino acid sequence of SEQ ID NO. 1, or a variant having at least 80% sequence identity therewith, for example at least 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity. In some preferred embodiments, the variant has around 93% identity with SEQ ID NO. 1. In some preferred embodiments, the antibody or antigen-binding fragment comprises a VH having the amino acid sequence of SEQ ID NO. 1.
In some embodiments, the antibody or antigen-binding fragment comprises a VH having the amino acid sequence of SEQ ID NO. 21, or a variant having at least 90% sequence identity therewith, for example at least 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity. In some preferred embodiments, the variant has around 91% identity with SEQ ID NO. 21. In some preferred embodiments, the antibody or antigen-binding fragment comprises a VH having the amino acid sequence of SEQ ID NO. 21.
In some preferred embodiments, the antibody or antigen-binding fragment comprises a VH having the amino acid sequence of SEQ ID NOs 3, 5, 7, 9, 11, 13, 14, 16, 18, 19, or 21.
In some embodiments, the VL comprises the following CDRs: a) CDR-L1 : [SEQ ID NO. 37] or an amino acid sequence having at least 80% identity therewith, for example at least 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably having at least 85% identity; and/or b) CDR-L2: [SEQ ID NOs. 197 or 41] or an amino acid sequence having at least 70% identity therewith, for example at least 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably having at least 71% identity; and/or c) CDR-L3: [SEQ ID NOs. 198 or 46] or an amino acid sequence having at least 55% identity therewith, for example at least 60%, 65%, 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably having at least 55% identity.
In some embodiments, the VL comprises the following CDRs: a) CDR-L1 : [SEQ ID NO. 37] or an amino acid sequence having at least 80% identity therewith, for example at least 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably having at least 85% identity; and/or b) CDR-L2: [SEQ ID NO. 197] or an amino acid sequence having at least 70% identity therewith, for example at least 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably having at least 71% identity; and/or c) CDR-L3: [SEQ ID NO. 198] or an amino acid sequence having at least 55% identity therewith, for example at least 60%, 65%, 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably having at least 55% identity.
In some embodiments, the VL comprises the following CDRs: a) CDR-L1 : [SEQ ID NO. 39] or an amino acid sequence having at least 80% identity therewith, for example at least 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably having at least 92% identity; and/or b) CDR-L2: [SEQ ID NO. 44] or an amino acid sequence having at least 55% identity therewith, for example at least 60%, 65%, 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably having at least 57% identity; and/or c) CDR-L3: [SEQ ID NO. 47] or an amino acid sequence having at least 55% identity therewith, for example at least 60%, 65%, 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably having at least 55% identity.
In some embodiments, the VL comprises the CDRs of SEQ ID NOs 37, 41, and 46. In some embodiments, the VL comprises the CDRs of SEQ ID NOs 39, 44, and 47.
In some other embodiments, the VL comprises the CDRs of any of the following combinations:
(i) SEQ ID NOs 36, 40, and 45 (clone 3A2);
(ii) SEQ ID NOs 37, 41, and 45 (clones 20F6 and 49G5);
(iii) SEQ ID NOs 37, 41, and 46 (clones 129D5B, 53D11, 139B2, 139D2, 139D7 and
77F3);
(iv) SEQ ID NOs 37, 197, and 198 (clone 129D5BTY);
(v) SEQ ID NOs 38, 42, and 47 (clone 26B5);
(vi) SEQ ID NOs 38, 43, and 48 (clones 83B11D and 94C2);
(vii) SEQ ID NOs 39, 43, and 47 (clone 115D1D); and
(viii) SEQ ID NOs 39, 44, and 47 (clone 131D5s).
In some embodiments, the antibody or antigen-binding fragment comprises a VL having the amino acid sequence of SEQ ID NO. 2, or a variant having at least 90% sequence identity therewith, for example at least 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity. In some preferred embodiments, the variant has around 91% identity with SEQ ID NO. 2. In some preferred embodiments, the antibody or antigen-binding fragment comprises a VL having the amino acid sequence of SEQ ID NO. 2.
In some embodiments, the antibody or antigen-binding fragment comprises a VL having the amino acid sequence of SEQ ID NO. 196, or a variant having at least 90% sequence identity therewith, for example at least 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity. In some preferred embodiments, the variant has around 91% identity with SEQ ID NO. 196. In some preferred embodiments, the antibody or antigen-binding fragment comprises a VL having the amino acid sequence of SEQ ID NO. 196.
In some embodiments, the antibody or antigen-binding fragment comprises a VL having the amino acid sequence of SEQ ID NO. 22, or a variant having at least 90% sequence identity therewith, for example at least 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity. In some preferred embodiments, the variant has around 91% identity with SEQ ID NO. 22. In some preferred embodiments, the antibody or antigen-binding fragment comprises a VL having the amino acid sequence of SEQ ID NO. 22.
In some preferred embodiments, the antibody or antigen-binding fragment comprises a VL having the amino acid sequence of SEQ ID NOs 196, 2, 4, 6, 8, 10, 12, 15, 17, 20, or 22.
In some embodiments, the antibody or antigen-binding fragment comprising the following CDRs: a) CDR-H1: [SEQ ID NO. 23] or an amino acid sequence having at least 80% identity therewith, for example at least 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably with at least 80% identity; and/or b) CDR-H2: [SEQ ID NO. 28] or an amino acid sequence having at least 80% identity therewith, for example at least 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably with at least 88% identity; and/or c) CDR-H3: [SEQ ID NO. 32] or an amino acid sequence having at least 60% identity therewith, for example at least 65%, 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably with at least 67% identity; and/or d) CDR-L1 : [SEQ ID NO. 37] or an amino acid sequence having at least 80% identity therewith, for example at least 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably having at least 85% identity; and/or e) CDR-L2: [SEQ ID NO. 41] or an amino acid sequence having at least 70% identity therewith, for example at least 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably having at least 71% identity; and/or
f) CDR-L3: [SEQ ID NO. 46] or an amino acid sequence having at least 55% identity therewith, for example at least 60%, 65%, 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably having at least 55% identity.
In some embodiments, the antibody or antigen-binding fragment comprising the following CDRs: a) CDR-H1: [SEQ ID NO. 23] or an amino acid sequence having at least 80% identity therewith, for example at least 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably with at least 80% identity; and/or b) CDR-H2: [SEQ ID NO. 28] or an amino acid sequence having at least 80% identity therewith, for example at least 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably with at least 88% identity; and/or c) CDR-H3: [SEQ ID NO. 32] or an amino acid sequence having at least 60% identity therewith, for example at least 65%, 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably with at least 67% identity; and/or d) CDR-L1 : [SEQ ID NO. 37] or an amino acid sequence having at least 80% identity therewith, for example at least 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably having at least 85% identity; and/or e) CDR-L2: [SEQ ID NO. 197] or an amino acid sequence having at least 70% identity therewith, for example at least 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably having at least 71% identity; and/or f) CDR-L3: [SEQ ID NO. 198] or an amino acid sequence having at least 55% identity therewith, for example at least 60%, 65%, 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably having at least 55% identity.
In some embodiments, the antibody or antigen-binding fragment comprising the following CDRs: a) CDR-H1: [SEQ ID NO. 27] or an amino acid sequence having at least 60% identity therewith, for example at least 60%, 65%, 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably with at least 60% identity; and/or b) CDR-H2: [SEQ ID NO. 29] or an amino acid sequence having at least 80% identity therewith, for example at least 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably with at least 82% identity; and/or
c) CDR-H3: [SEQ ID NO. 35] or an amino acid sequence having at least 60% identity therewith, for example at least 65%, 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably with at least 69% identity; and/or d) CDR-L1 : [SEQ ID NO. 39] or an amino acid sequence having at least 80% identity therewith, for example at least 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably having at least 92% identity; and/or e) CDR-L2: [SEQ ID NO. 44] or an amino acid sequence having at least 55% identity therewith, for example at least 60%, 65%, 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably having at least 57% identity; and/or f) CDR-L3: [SEQ ID NO. 47] or an amino acid sequence having at least 55% identity therewith, for example at least 60%, 65%, 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably having at least 55% identity.
In some embodiments, the antibody or antigen-binding fragment thereof preferably comprises the following combination of CDRs: SEQ ID NOs. 23, 28, 32, 37, 41, and 46.
In some embodiments, the antibody or antigen-binding fragment thereof preferably comprises the following combination of CDRs: SEQ ID NOs. 23, 28, 32, 37, 197, and 198.
In some embodiments, the antibody or antigen-binding fragment thereof preferably comprises the following combination of CDRs: SEQ ID NOs. 27, 29, 35, 39, 44, and 47.
In some other embodiments, the antibody or antigen-binding fragment thereof comprises one of the following combinations of CDRs:
(i) SEQ ID NOs. 23, 115, 30, 36, 40, and 45 (clone 3A2);
(ii) SEQ ID NOs. 23, 115, 30, 37, 41, and 45 (clone 20F6);
(iii) SEQ ID NOs. 23, 115, 31, 37, 41, and 45 (clone 49G5);
(iv) SEQ ID NOs. 23, 115, 30, 37, 41, and 46 (clone 53D11);
(v) SEQ ID NOs. 23, 115, 31, 37, 41, and 46 (clone 77F3);
(vi) SEQ ID NOs. 23, 29, 33, 38, 42, and 47 (clone 26B5);
(vii) SEQ ID NOs. 23, 29, 33, 38, 43, and 48 (clone 83B11D);
(viii) SEQ ID NOs. 23, 29, 34, 38, 43, and 48 (clone 94C2);
(ix) SEQ ID NOs. 23, 29, 35, 39, 43, and 47 (clone 115D1D);
(x) SEQ ID NOs. 27, 29, 35, 39, 44, and 47 (clone 131D5s);
(xi) SEQ ID NOs. 23, 28, 32, 37, 41, and 46 (clone 129D5B); and
(xii) SEQ ID NOs. 23, 28, 32, 37, 197, and 198 (clone 129D5BTY).
In any listed embodiment or any listed clone, CDR-L2 may have SEQ ID NO. 41 replaced by SEQ ID NO. 197 and/or CDR-L3 may have SEQ ID NO. 46 replaced by CDR SEQ ID NO. 198.
In some embodiments, the antibody or antigen-binding fragment thereof comprises a heavy chain variable region having the amino acid sequence of SEQ ID NO: 1 and a light chain variable region having the amino acid sequence of SEQ ID NO: 2, or an amino acid sequence having at least 80% sequence identity therewith, for example at least 85%, 90%, or 95% sequence identity. In some preferred embodiments, the antibody or antigen-binding fragment thereof comprises a heavy chain variable region having the amino acid sequence of SEQ ID NO: 1 and a light chain variable region having the amino acid sequence of SEQ ID NO: 2, or an amino acid sequence having at least 90% sequence identity therewith, preferably at least 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% sequence identity therewith.
In some embodiments, the antibody or antigen-binding fragment thereof comprises a heavy chain variable region having the amino acid sequence of SEQ ID NO: 1 and a light chain variable region having the amino acid sequence of SEQ ID NO: 196, or an amino acid sequence having at least 80% sequence identity therewith, for example at least 85%, 90%, or 95% sequence identity. In some preferred embodiments, the antibody or antigen-binding fragment thereof comprises a heavy chain variable region having the amino acid sequence of SEQ ID NO: 1 and a light chain variable region having the amino acid sequence of SEQ ID NO: 196, or an amino acid sequence having at least 90% sequence identity therewith, preferably at least 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% sequence identity therewith.
In some embodiments, the antibody or antigen-binding fragment thereof comprises a heavy chain variable region having the amino acid sequence of SEQ ID NO: 21 and a light chain variable region having the amino acid sequence of SEQ ID NO: 22, or an amino acid sequence having at least 80% sequence identity therewith, for example at least 85%, 90%, or 95% sequence identity. In some preferred embodiments, the antibody or antigen-binding fragment thereof comprises a heavy chain variable region having the amino acid sequence of SEQ ID NO: 21 and a light chain variable region having the amino acid sequence of SEQ ID NO: 22, or an amino acid sequence having at least 90% sequence
identity therewith, preferably at least 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% sequence identity therewith.
In some embodiments, the antibody or antigen-binding fragment thereof is an antigenbinding fragment.
In some embodiments, the antigen-binding fragment is selected from: scFv, Fv, Fab, F(ab)2, Fab-SH, dsFv, sdAb, di-scFvs bi-scFv, Fcabs, diabodies, scFv-Fc/minibody, triabody, tetrabody, tandAb, half antibody (Unibody) and domain antibodies.
In some particularly preferred embodiments, the antigen-binding fragment is an scFv.
By "antibody" we include substantially intact antibody molecules, as well as chimeric antibodies, humanised antibodies, human antibodies (wherein at least one amino acid is mutated relative to the naturally occurring human antibodies), single chain antibodies, biparatopic antibodies, antibody heavy chains, antibody light chains, homodimers and heterodimers of antibody heavy and/or light chains, and antigen binding fragments and derivatives of the same.
By "antigen-binding fragment" we mean a functional fragment of an antibody that is capable of binding to cMET.
It is preferred if the antibody fragment excludes the Fc region of a whole antibody. In particular, it is preferred if the antibody fragment does not include the CH2 and CH3 regions of a whole antibody.
The advantages of using antibody fragments, rather than whole antibodies, are several- fold. The smaller size of the fragments may lead to improved pharmacological properties, such as better penetration of solid tissue. Moreover, antigen-binding fragments such as Fab, Fv, ScFv and dAb antibody fragments can be expressed in and secreted from E. coli, thus allowing the economic production of large amounts of the said fragments.
Antibody fragments are functional portions of whole immunoglobulins that possess advantageous properties over complete antibodies such as faster penetration into dense or solid tumours, reduced cross-reactivity with normal tissues and more rapid clearance from the circulation, thus reducing normal tissue exposure overall. It is well known in the art that antibody fragments demonstrate faster pharmacokinetics, dispersing into tissues and eliminating more rapidly (ADME-adsorption, distribution, metabolism and excretion
properties). They are also easier to produce in more cost-effective systems such as microbial expression systems [70].
Antibody fragments can be produced by chemical or enzymatic cleavage, but, more preferably, are produced using recombinant DNA technology. The latter allows for indefinite protein expression in prokaryotic or eukaryotic cell lines and genetic modification leading to fragments with enhanced or additional properties. Antibody fragments normally possess at least one variable (V-) domain because V-domains contain the complementarity-determining regions (CDRs) or loops for antigen binding [71]. More recently, CDR-like loops have been inserted into non-variable domains (e.g. constantheavy-3, CH3 domains) enabling these domains to bind to useful or predetermined targets [72],
For antibody fragments to be used effectively as carrier vehicles for cytotoxic drugs, they must possess biophysical properties that allow high drug loading via chemical conjugation (or strong and specific non-covalent interactions) without detrimentally affecting protein stability, antibody-antigen binding, and drug-favourable properties such as solubility, aggregation and immunogenicity. Very rarely are these features inherent to antibody fragments [73] so these additional benefits must be engineered into antibody fragments, or preferably be selected from naturally-occurring antibodies to make them practically useful [74,75]. One example of such a feature is the incorporation of additional or more optimally distributed surface lysine residues onto antibody fragments, thus increasing its capacity for drug conjugation using amine-directed chemistry. Other amino acids could be used, such as optimally distributed cysteines, tyrosines, glutamates, aspartates, arginines, asparagines, histidines and serines, but lysines are more preferable due to the well- established and successful chemical approaches for conjugation and relative inertness to conjugation without specific activating groups (chapter 10 in [76]). Non-natural amino acids such as p-Acetylphenylalanine and formyl-glycine can also be used [77]. The identification of positions for antibody fragment modification can be by direct analysis of the 3-dimensional structure of the antibody fragment (or parental whole antibody), if available, or by homology modelling using a number of software resources such as Phyre
[78]. The criteria for selecting positions include: (1) the use of amino acids already favoured or conserved at that position (identified from databases such as IMGT or Kabat
[79]) or through practical demonstration by making and testing antibody fragment mutants; (2) Distribution of residues away from positions that would interfere with antigen binding; and, (3) Separation of conjugating residues so that they do not sterically hinder (or predicted to hinder) each other during chemical reactions or drug release reactions or form highly hydrophobic patches leading to aggregation.
All of the antigen-binding fragments described herein are built from the same building blocks, i.e. the variable domains of the heavy and light chains, and therefore would be expected to function in a similar manner. Variable domains of antibodies (i.e. the VH and VL domains that form an scFv), are compact, discrete domains that are highly conserved.
Therefore, fragments other than scFvs that also comprise a VH and VL region would be expected to conjugate and behave in a similar way to an scFv as has been particularly exemplified in the specification. Furthermore, the fragments discussed herein are all structurally very similar to the exemplified scFv fragments, with only subtle differences in their formats, as discussed below:
Fvs, including ds-Fvs, are structurally very similar to scFvs, and have almost identical surfaces where the conjugation occurs, differing only in the linker which is not involved in the conjugation reaction.
Fab fragments comprise the VH and VL regions together with the first constant region, and they therefore are very structurally similar to scFvs.
Therefore, Fab (and Fab-SH) fragments are structurally similar to scFvs, such that the claimed conjugates produced using Fab or Fab-SH fragments would reasonably be expected to function in a similar manner to the exemplified scFvs.
Bi-specific scFvs (bs-scFv) and di/bi-scFvs have the scFv format discussed above, but consist of two scFvs tethered by a linker. It is widely acknowledged in the art that the structure of one of the scFv units in a bivalent or trivalent scFv multimer would be the same as the parental scFv.
As these fragments have double the identical surface area to scFvs, the lysine content and distribution would be expected to be identical to the scFv, and it would therefore be expected that a bs-scFv or di/bi-scFv to work in the same way.
Diabodies are bi-valent scFvs where the VH-VL domains are arranged in a head-to-tail format.
As discussed herein, it will be appreciated that the antibodies and antigen binding fragments of the invention may alternatively comprise variants of the above-defined sequences. The antibodies and antigen binding fragments thereof may have at least 60%
sequence identity with any of the sequences disclosed herein. For example, they may have at least 60%, at least 65%, at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, at least 95%, or at least 99% sequence identity.
The antibodies and antigen binding fragments thereof may alternatively be a variant of a specific sequence disclosed herein, wherein said variant comprises mutations at one or more positions relative to the parent sequence. By "mutation" we include insertions, deletions and substitutions. Accordingly a variant may be a substitution, deletion or addition variant. A variant polypeptide may comprise 1, 2, 3, 4, 5, 6, 7, 8, 9, up to 10, up to 20, up to 30, up to 40, up to 50, up to 75 or more amino acid mutations, in a preferred embodiment. The mutations may be either conservative or non-conservative. For example, conservative substitution refers to the substitution of an amino acid within the same general class (e.g. an acidic amino acid, a basic amino acid, a non-polar amino acid, a polar amino acid or an aromatic amino acid) by another amino acid within the same class. Thus, the meaning of a conservative amino acid substitution and non-conservative amino acid substitution is well known in the art.
"Deletion" variants may comprise the deletion of individual amino acids, deletion of small groups of amino acids such as 2, 3, 4 or 5 amino acids, or deletion of larger amino acid regions, such as the deletion of specific amino acid domains or other features. "Substitution" variants preferably involve the replacement of one or more amino acids with the same number of amino acids and making conservative amino acid substitutions. For example, an amino acid may be substituted with an alternative amino acid having similar properties, for example, another basic amino acid, another acidic amino acid, another neutral amino acid, another charged amino acid, another hydrophilic amino acid, another hydrophobic amino acid, another polar amino acid, another aromatic amino acid or another aliphatic amino acid. Some properties of the 20 main amino acids which can be used to select suitable substituents are as follows:


Amino acids herein may be referred to by full name, three letter code or single letter code. Preferred "variants" include those in which instead of the naturally occurring amino acid the amino acid which appears in the sequence is a structural analog thereof. Amino acids used in the sequences may also be derivatised or modified, e.g. labelled, providing the function of the antibody is not significantly adversely affected.
Derivatives and variants as described above may be prepared during synthesis of the antibody or by post-production modification or by peptide synthesis or by native chemical ligation of peptides, or when the antibody is in recombinant form using the known techniques of site-directed mutagenesis, random mutagenesis, or enzymatic cleavage and/or ligation of nucleic acids.
Suitable variants may be at least 70% homologous to a sequence disclosed herein, preferably at least 80% or 90% and more preferably at least 95%, 97% or 99% homologous thereto.
Preferably variants have an amino acid sequence which has more than 60%, or more than 70%, e.g. 75 or 80%, preferably more than 85%, e.g. more than 90 or 95% amino acid identity to a sequence as shown in the sequences disclosed herein (e.g. the VH or VL region sequences, or CDR sequences therein). This level of amino acid identity may be seen across the full length of the relevant SEQ ID NO sequence or over a part of the sequence, such as across 20, 30, 50, 75, 100, 150, 200 or more amino acids, depending on the size of the full-length polypeptide.
For example, variants of the above CDR sequences may comprise one, two three, four, five, six, seven, eight or more amino acid mutations relative to the reference sequence (such as a deletion, substitution and/or insertion of an amino acid).
The percent sequence identity between two polypeptides may be determined using suitable computer programs, for example the GAP program of the University of Wisconsin Genetic Computing Group and it will be appreciated that percent identity is calculated in relation to polypeptides whose sequences have been aligned optimally. Methods for determining sequence identity are known to those skilled in the art.
The alignment may alternatively be carried out using the Clustal W program (as described in [80], which is incorporated herein by reference). The parameters used may be as follows:
Fast pairwise alignment parameters: K-tuple(word) size; 1, window size; 5, gap penalty; 3, number of top diagonals; 5. Scoring method: x percent.
Multiple alignment parameters: gap open penalty; 10, gap extension penalty; 0.05. Scoring matrix: BLOSUM.
Alternatively, the BESTFIT program may be used to determine local sequence alignments. Also included within the scope of the invention are modified versions of antibodies and antigen-binding fragments thereof, e.g. modified by the covalent attachment of polyethylene glycol or other suitable polymers (see below).
Methods of generating antibodies and antibody fragments are well known in the art. For example, antibodies may be generated via any one of several methods which employ induction of in vivo production of antibody molecules, screening of immunoglobulin libraries [75,81] or generation of monoclonal antibody molecules by cell lines in culture. These include, but are not limited to, the hybridoma technique, the human B-cell hybridoma technique, and the Epstein-Barr virus (EBV)-hybridoma technique [82-85].
Suitable monoclonal antibodies to selected antigens may be prepared by known techniques, for example those disclosed in [86].
Likewise, antibody fragments can be obtained using methods well known in the art (see, for example [87]. For example, antibody fragments according to the present invention can be prepared by proteolytic hydrolysis of the antibody or by expression in E. coli or mammalian cells (e.g. Chinese hamster ovary cell culture or other protein expression systems) of DNA encoding the fragment. Alternatively, antibody fragments can be obtained by pepsin or papain digestion of whole antibodies by conventional methods.
It will be appreciated by persons skilled in the art that for human therapy or diagnostics, human or humanised antibodies are preferably used. Humanised forms of non-human (e.g. murine) antibodies are genetically engineered chimeric antibodies or antibody fragments having preferably minimal-portions derived from non-human antibodies. Humanised antibodies include antibodies in which complementary determining regions of a human antibody (recipient antibody) are replaced by residues from a complementary determining region of a non-human species (donor antibody) such as mouse, rat of rabbit having the desired functionality. In some instances, Fv framework residues of the human
antibody are replaced by corresponding non-human residues. Humanised antibodies may also comprise residues which are found neither in the recipient antibody nor in the imported complementarity determining region or framework sequences. In general, the humanised antibody will comprise substantially all of at least one, and typically two, variable domains, in which all or substantially all of the complementarity determining regions correspond to those of a non-human antibody and all, or substantially all, of the framework regions correspond to those of a relevant human consensus sequence. Humanised antibodies optimally also include at least a portion of an antibody constant region, such as an Fc region, typically derived from a human antibody (see, for example, [88-90].
Methods for humanising non-human antibodies are well known in the art. Generally, the humanised antibody has one or more amino acid residues introduced into it from a source which is non-human. These non-human amino acid residues, often referred to as imported residues, are typically taken from an imported variable domain. Humanisation can be essentially performed as described (see, for example, [88,89,91]; US 4,816,567) by substituting human complementarity determining regions with corresponding rodent complementarity determining regions. Accordingly, such humanised antibodies are chimaeric antibodies, wherein substantially less than an intact human variable domain has been substituted by the corresponding sequence from a non-human species. In practice, humanised antibodies may be typically human antibodies in which some complementarity determining region residues and possibly some framework residues are substituted by residues from analogous sites in rodent antibodies.
Human antibodies can also be identified using various techniques known in the art, including phage display libraries (see, for example, [92-95]).
Once suitable antibodies are obtained, they may be tested for binding activity, for example by ELISA.
In some embodiments, the antibody or antigen-binding fragment thereof disclosed herein comprises a heavy chain constant region, or part thereof.
In some embodiments, the heavy chain constant region is of an immunoglobulin subtype selected from the group consisting of IgGl, IgG2, IgG3 and IgG4.
In some embodiments, the heavy chain constant region is of an immunoglobulin subtype IgGl.
In some embodiments, the antibody or antigen-binding fragment thereof disclosed herein comprises a light chain constant region, or part thereof.
In some embodiments, the light chain constant region is of a kappa or lambda light chain. In some embodiments, the constant region is a human or humanised constant region.
In some embodiments, the antibodies, antigen-binding fragments and conjugates disclosed herein have an IC50 of lOOnM or lower.
The half maximal inhibitory concentration (IC50) is a measure of the effectiveness of a substance in inhibiting a specific biological or biochemical function. This measure indicates how much of a particular drug or other substance (inhibitor) is needed to inhibit a given biological process (or component thereof) by half. Determination of the IC50 for a given compound is a routine matter, and typically is determined by constructing a dose-response curve and examining the effect of different concentrations of antagonist on reversing agonist activity. The IC50 value is calculated by determining the concentration needed to inhibit half of the maximum biological response of the agonist.
In some embodiments, the antibodies, antigen-binding fragments and conjugates disclosed herein have a serum half-life of at least 2 hours, optionally the serum half-life of at least 2 hours is measured in rodents or in humans. In some embodiments, the serum half-life is preferably 4 hours, alternatively 8, 16, 32, 64 or 128 hours. In some preferred embodiments, the serum half-life is measured in humans.
In some embodiments, the conjugates disclosed herein have a longer serum-half life than the unconjugated antibodies or antigen-binding fragments. In some embodiments, serum half-life is also referred to as bioavailability.
Serum half-life is the calculated duration of time for a serum level of a compound to be reduced to half its initial value. Bioavailability can be defined as the area-under-curve (AUC) in a plasma concentration vs time pharmacokinetic plot and is related to serum halflife. Determination of the serum half-life or bioavailability for a given compound is a routine matter, and typically is determined by measuring the amounts of drug in the serum over time following compound administration to an organism. Serum half-life or bioavailability is important clinically, as it will determine the dosage regime required in order to consistently achieve a serum level of drug within a clinically effective range.
In some embodiments, the payloads, when coupled to the antibody or antigen-binding fragment, are separated by a distance of at least two amino acids (3.5 to 7.5 angstroms). In some embodiments, the payloads, when coupled to the antibody or antigen-binding fragment, are separated by a distance of two amino acids (3.5 to 7.5 angstroms), three amino acids (9 to 12 angstroms), four amino acids (10 to 15 angstroms), five amino acids (15 to 20 angstroms) or six amino acids (20 to 25 angstroms).
In some embodiments, the antibody or antigen-binding fragment thereof disclosed herein further comprises a payload. By "payload" we mean a drug molecule that is conjugated to the antibody or antigen-binding fragment thereof. The drug molecule may be a cytotoxic or cytostatic agent. The drug molecule is in some preferred embodiments a small molecule payload. In some embodiments, the payload is conjugated to the antibody or antigen-binding fragment via a linker. In some embodiments, the antibody/antigen- binding fragment conjugated to the payload is referred to as an antibody-drug conjugate (ADC).
Despite the many advances made in the ADC field over recent years, it is clear that significant problems associated with the biophysical or biological properties of ADCs that result in ineffective or poorly tolerated compounds are often encountered. For example, an anti-CD33 ADC (gemtuzumab ozogamicin) was approved to treat acute myeloid leukaemia in 2000 but was withdrawn in 2010 due to liver toxicity, patients' deaths and poor efficacy compared to existing therapies [5]. In another example, trastuzumab emtansine, already approved for HER2-expressing breast cancer in 2013, was not approved for HER2-expressing gastric cancer in 2015 due to lack of efficacy [18]. In another example, traztuzumab deruxtecan demonstrated high efficacy in HER2-expressing breast cancer but progression-free survival and other key outcomes are much poorer for HER2-expressing gastric cancer [96] which is known to be more heterogenous and difficult to treat [97,98]. A review carried out by the FDA concluded that the major dose-limiting toxicities of ADCs were due to payload exposure to normal tissues [99] and many examples of so-called ultra-potent (low pM IC50) ADCs have failed due to a narrow therapeutic window compared to ADCs built with less potent payloads [15].
To date, all of the approved and advanced clinical stage ADCs are based upon full-length immunoglobulins coupled to a payload. The immunoglobulin format has size and avidity properties which limits its penetration into solid tumours [9,56] and ineffective penetration is a noted as a significant factor in cancer therapy [100]. Highly penetrating peptides and small molecule ligands have been developed as alternative delivery vehicles for drugconjugates but quite often these have to be administered more frequently and due to kidney clearance, can induce toxicities related to renal excretion [9]. Recombinant
antibody fragments such as single-chain Fvs (scFvs) and Fabs have also been considered as smaller delivery vehicles for drug-conjugates [7,11] but their design and low drug:antibody ratio (DAR) has not made them viable drug candidates. Antikor Biopharma Ltd has previously described an approach using antibody fragments such as scFvs with a DAR of at least 5 coupled in such a way that leads to antibody fragment drug conjugates (FDCs) with high affinity, high potency, manageable tolerability, good pharmacokinetics and good biophysical properties that lead to FDC products that viable commercial products [WO2016/046574].
Accordingly, another aspect of the present invention provides ADCs based on antibody fragments, herein known as antibody Fragment Drug Conjugates (FDCs), against cMET using the anti-cMET antibody sequences described herein which avoids the complication of hinge manipulation or bi-valency or multi-valency leading to possible cMET agonism. cMET FDCs enable delivery of payloads to cMET-expressing cell surface receptor, and subsequent internalization of the FDC to deliver the payload intracellularly, whilst exhibiting no agonist activity. Surprisingly, cMET FDCs internalise as well as bivalent ADCs resulting in effective payload delivery to tumours. cMET FDCs have a faster plasma halflife and lower bioavailability than comparable ADCs but surprisingly demonstrate a higher quantity of payload delivered to tumours in the early time points and furthermore demonstrate lower liver payload uptake as would be expected for a format carrying a high quantity of payload. Notably and surprisingly, cMET FDCs are able to elicit complete tumour regression after four doses of Img/kg (cumulative dose of 4mg/kg) whereas an ADC against the same target with the same payload was ineffective at a cumulative dose of 6mg/kg [57].
Described herein, cMET FDCs demonstrate high biophysical and thermal stability which is unexpectedly higher than other phage-display selected human scFvs conjugated to similar linker-payloads. cMET FDCs can be made with a variety of linker payloads which suggests that cMET FDCs can have utility in many types of tumours irrespective of their sensitivity to particular payload classes and diseases beyond cancer such as other proliferative conditions like Idiopathic Pulmonary Fibrosis where a payload also needs to be delivered to cMET-overexpressing tissues. As a further advantage, cMET FDCs do not possess an Fc- domain which removes any cross-reaction with Fc-receptors. Fc-gamma receptor binding is an important factor in many dose limiting ADC toxicities such as thrombocytopenia [64], and can also be a factor in damaging Fc-gamma receptor bearing immune cells (e.g. macrophages) which are needed for mounting an effective immune response to cancer or infections.
In some embodiments, the cMET FDC binds cMET with an affinity KD of less than 50nM, 40nM, 30nM, 20nM, lOnM, 5nM or less than InM.
In certain embodiments, the cMET FDC binds cMET with an affinity KD of less than lOnM.
In some embodiments, the payload is coupled to the antibody or antigen-binding fragment thereof with a coupling ratio (payload :antibody or antigen-binding fragment) of at least 2: 1, at least 3: 1, at least 4: 1, at least 5: 1, at least 6: 1, at least 7: 1, at least 8: 1, at least 9: 1 or at least 10: 1.
In some preferred embodiments, the coupling ratio (payload :antibody or antigen-binding fragment) is at least 6: 1.
In some embodiments, the payload comprises, but is not limited to, at least one of:
• A toxin (cytotoxic and/or cytostatic agent);
• An immunoregulatory/immunostimulatory agent;
• An anti-inflammatory agent;
• A protein-degradation agent (PROTAC);
• A radionucleotide/PET agent;
• A fluorescent dye.
The cytotoxic and/or cytostatic agents may be any agents known to inhibit the growth and/or replication of and/or kill cells and in particular cancer/tumour cells. Numerous agents having cytotoxic and/or cytostatic properties are known in the literature. Nonlimiting examples of classes of cytotoxic and/or cytostatic agents include antimitotic agents, topoisomerase I inhibitors, topoisomerase II inhibitors, DNA alkylating agents, DNA cross-linking agents, DNA intercalating agents, DNA groove binding agents, DNA strand-cleavage agents including enediynes, kinase inhibitors, RNA/DNA anti-metabolites, RNA polymerase (transcription) inhibitors, protein synthesis inhibitors, cell-cycle modulators, apoptosis regulators, mitochondria inhibitors, nuclear export inhibitors and N- myristoyl transferase inhibitors.
The toxin may be selected from the group consisting of:
• An auristatin such as MMAE (monomethyl auristatin E) and MMAF (monomethyl auristatin F);
• Hemiasterlins;
• A maytansinoid such as maytansine, DM1, DM4 and DM21;
• A camptothecin (exatecan, DXd, Belotecan, SN-38, 7-aminomethyl-(10,ll- methylenedioxy)-camptothecin, 7-aminoethyl-(10,ll-methylenedioxy)-
camptothecin, 7-Aminomethyl-(10-Methyl, ll-Fluoro)-camptothecin, 7- Aminoethyl-(10-Methyl, ll-Fluoro)-camptothecin;
• A kinase inhibitor such as nintedanib;
• A transcription inhibitor (triptolide, o-amanatin);
• Immunoregulatory agent such as glucocorticoids (Dexamethasone, Budesonide);
• Immunostimulatory agents such as toll-like receptors TLR.7/8 (imidazoquinoline, T785) and STING agonists such as XMT-1621, IMSA172;
• A DNA alkylating agent such as Duocarmycin, Duocarmycin-SA, seco-DUBA;
• A tubulysin;
• An anthracycline derivative such as PNU-159682, doxorubicin;
• A cryptophycin;
• An enediyne (calicheamicin, uncialamycin);
• A nicotinamide phosphoribosylytransferase (NAMPT) inhibitor such as FK-866 (daporinad) and analogues;
• A pyrrole-based kinesin spindle protein (KSP) inhibitor such as Filasenib;
• A PROTAC such as BET/BRD4 degraders (MZ1 (BRD4/VHL)), GNE-987 (BRD4/VHL);
• A protein synthesis inhibitor (HSP90 inhibitor) like geldanamycin, (a splicing inhibitor) such as Thailanstatin A;
• A Bcl-xL inhibitor such as clezutoclax.
• An inhibitor of N-myristoyl transferase
In some preferred embodiments, the payload is exatecan.
Antimitotic Agents
Microtubules are long filamentous protein polymers consisting of heterodimers of a- and P-tubulin which are involved in a number of cellular processes critical to cellular function, including organelle and vesicle transport, cell migration and mitosis. These polymers are assembled and disassembled in a highly controlled process within cells. The main role of microtubules is to separate and segregate chromosomes during cell division. Thus, tubulin inhibitors interfere with cell division by preventing the two pairs of chromosomes from separating into two daughter cells [101].
In certain embodiments the toxin may be an auristatin which refers to a family of antimitotic agents derived from the natural product dolastatin-10 which is a highly potent linear pentapeptide isolated from Dolabella Auricularia, a sea hare [102]. Dolastatin-10 and its analogues are extremely potent cytotoxic antineoplastic agents, exhibiting picomolar GIso values in most cancer cell proliferation assays [103]. The potent antiproliferative activity is caused by the ability to strongly bind to tubulin and inhibit
microtubule assembly and tubulin-dependent GTP hydrolysis, resulting in accumulation of cells in the G2/M phase of the cell proliferation cycle resulting in cell cycle arrest and apoptosis. Detailed binding studies have shown that dolastatin-10 and the auristatins bind to a region which overlaps with that of the vinca site but extends significantly further and near the exchangeable GTP binding site termed the peptide site. Dolastatin-10 and the auristatins bind at the o,p-tubulin interphase, inducing a curved conformation that is incompatible with the straight structure of microtubules. In addition to effects on tubulin dynamics, it was soon recognised that dolastatin-10 and related synthetic auristatin analogues also have strong anti-vascular effects. However, dolastatin-10 and other derivatives failed to demonstrate meaningful clinical activities as single-agents due to severe dose-limiting adverse effects. However, targeted delivery to the tumour can reduce systemic toxicity and result in clinical benefit.
Examples of auristatins include monomethyl auristatin E (MMAE), monomethyl auristatin F (MMAF), auristatin F, auristatin F hydroxylpropylamide (AF-HPA), monomethyl auristatin D (MMAD), monomethyl auristatin F methylester (MMAF-OMe), PF-06380101. Suitable auristatins are described in W02002088172, W02005081711, US6323315, US5663149, US5530097 and US5504191.
Dolastatin-15 is a related seven-subunit depsipeptide also obtained from Dolabella Auricularia see [US4879278]. Many synthetic analogues of this natural product have been prepared, among which cemadotin (LU-103793) and tasidotin (ILX651) have entered clinical trials [104]. For exemplary drugs and analogues from dolastatin-10 see, US5410024, US5599902, US5635483 and US6323315.
In certain embodiments, the toxin may be from the hemiasterlin family. Hemiasterlins are a family of cytotoxic tripeptides that were initially isolated from the South African marine sponge Hemiasterella minor [105]. Hemiasterlin and its synthetic analogues like Taltobulin (HTI-286) show nanomolar cytotoxic potency and probably bind to the 'peptide binding site' shared with dolastatins and cryptophycins and inhibit tubulin assembly triggering mitotic arrest and subsequent apoptosis. In comparison with other marine peptides, hemiasterlins have the advantage of a simpler structure allowing the preparation of many synthetic analogues like taltobutin which is less sensitive to P-glycoprotein drug transporters. For exemplary hemiasterlin derivatives as ADC payloads see WO2016123582 which describes 3-aminophenylhemiasterlin and its derivatives.
In certain embodiments, the toxin may be a maytansinoid. The term maytansinoid refers to benzoansamacrolide, a class of highly potent antimitotics originally isolated from the
African shrub Maytenus ovatus. Maytansinol, which has the same macrocyclic system as in maytansine but with a C3-hydroxyl instead of the C3-acetyl-N-methyl-alanine side chain. Maytansinoids are derivatives of maytansinol with modified C3-side chains that are typically C3-esters [106,107]. Maytansine binds tubulin at the vinca-binding site, similar to vinca alkaloids, thereby depolymerizing tubulin and inducing mitotic arrest in the G2/M phase [108]. Similar to the auristatins, maytansine in its original form yielded a narrow therapeutic window due to associated neurological and gastrointestinal toxicities. Consequently, synthetic maytansine derivatives were synthesized that possessed 100- 1000-fold increases in potency while also including modifications to enable targeted delivery [109].
Exemplary maytansinoids or maytansinoid analogues including maytansinol are described [110] and US5416064. C3-esters of simple carboxylic acids are described in US4317821. C3-esters with derivatives of N-methyl-L-alanine are described in US4260608. Exemplary maytansinoids such as DM1, DM3, DM4 and DM21 or maytansinoid analogues including maytansinol are described in US6716821, US7276497 and US20180296694.
In certain embodiments the toxin may be a cryptophycin. Cryptopycins are a family of 16- membered cyclic depsipeptides, first isolated from terrestrial blue-green algae. They are potent tubulin-binding antimitotic agents, inhibiting mitotic spindle function of cells in the G2M phase of the cell cycle resulting in apoptotic cell death [111]. Cryptophycins have demonstrated excellent activity against a broad spectrum of solid tumours with potency in the picomolar range [112] and unlike other antitubulin agents they have a major advantage in that they are insensitive to the ABC transporters P-gP and MRP, which are implicated in multidrug resistance [113].
Although cryptophycins have failed as stand-alone drugs due to toxicity, their very high potency (picomolar potency on a panel of tumor cells) and lack of PgP susceptibility make them potential candidates as payloads in targeted delivery [114]. Exemplary cryptophycin compounds are described in US6680311 and US20180078656.
In certain embodiments the toxin may be a tubulysin. Tubulysins are a family of naturally occurring cytotoxic tetrapeptides isolated from mycobacterium cultures [115]. Tubulysins are potent inhibitors microtubule polymerisation, causing rapid disintegration of the cytoskeleton of dividing cells in the G2/M phase leading to apoptosis. Binding is to the peptide binding site like the dolostatins making tubulysins extremely potent cytotoxic agents at sub-nanomolar concentrations against a variety of cancer types especially multidrug resistant lines [116]. This activity has led to significant interest in the tubulysins
as targeted anticancer agents. Exemplary tubulysin compounds are described in, US20210188906, WO2021262910, US20170326247, WO2017134547, US20160340386, and WO2015157594.
In certain embodiments the toxin may be a taxane. Paclitaxel (Taxol), Docetaxel and Cabazitaxel, Larotaxel, TPI 287 and MAC-321 represent the taxane family of drugs which have demonstrated remarkable efficacy against solid tumours such as ovarian and breast cancer. Paclitaxel was first isolated from the Pacific Yew tree Taxus brevifolia and its antitumour activity was first reported in 1971. Paclitaxel works by promoting tubulin polymerisation by stabilising microtubules from depolymerisation, thus causing cell cycle arrest in the G2M stage of the cell cycle, leading to apoptotic cell death. Exemplary paclitaxel and analogues are described in [117].
In certain embodiments the toxin may be a vinca alkaloid and their synthetic analogues. Vincristine and vinblastine are complex molecule produced by the leaves of the rosy periwinkle plant. They block the polymerisation of tubulin into microtubules and inhibit mitotic progression resulting in apoptotic cell death. The lead compounds vinblastine and vincristine have been employed in clinical practice for more than thirty years and remain widely used to this day. Several hundred derivatives have been synthesised and evaluated for their pharmacological activities [118]. Vinorelbine (Navelbine) is a semi-synthetic vinca-alkaloid with broad spectrum of activity in breast and non-small cell lung cancer with fewer side effects than other vinca alkaloids. Other examples include 4-deacetyl- vinblastine-3-carboxyhydrazide which was evaluated as a conjugate [119] and N-(3- hydroxypropyl)vindesine alanine which was conjugated to a polyacetal polymer. Exemplary vinca alkaloids include vincristine, vinblastine, vindesine and navelbine and those disclosed in US20020103136 and US20100305149.
In certain embodiments the toxin may be eribulin. Eribulin is a synthetic analogue of the macrocycle halichondrin B, a complex polyether macrolide first isolated from the Asian marine sponge Halichondria okadai [120]. Eribulin binds specifically to the [3-tubulin subunit on the (+)-end of a microtubule, inhibiting elongation but having no effect on microtubule depolymerisation. Eribulin (as the mesylate salt) was approved by the FDA for the treatment of metastatic breast cancer and is being investigated for use in other tumour types including bladder, lung and. In addition to antimitotic effects, eribulin appears to act through some non-mitotic pathways which may contribute to its overall antitumour activity. An eribulin derivative modified with a cathepsin-cleavable dipeptide valine-citrulline is described in [121]. Exemplary eribulin compounds are described in WO2017151979 and WO2023061466.
In certain embodiments, the toxin may be an epothilone and their synthetic analogues. The epothilones are a group of closely related antitubulin cytotoxic macrocylic lactones with a mechanism of action similar to the taxanes in which they bind to the same 00- tubulin heterodimer, promoting tubulin polymerisation by stabilising microtubules from depolymerisation, thus causing cell cycle arrest in the G2M stage of the cell cycle, leading to apoptotic cell death [122]. The epothilones were originally identified as metabolites produced by the soil-dwelling myxobacterium Sorangium celulosum and by 2008 epothilones A-F were identified and characterised. Epothilones A and B have strong antiproliferative activity against different types of cancer cells and have been found to also exert their effects on taxol resistant cancers. Exemplary epothilone compounds include epithilone A, B, C, D, E, and F and derivatives thereof. Suitable epithilone compounds and derivatives are Ixabepilone (Ixempra, BMS-247550), Utidelone (depoxythilone), Sagopilone (ZK-EPO) and Patupilone (EPO906). Additional exemplary derivatives are described in US6989450, WO9719086, WO9907692, WO9927890 and W02017066606.
DNA targeting agents
Topoisomerase I inhibitors: Topoisomerases are nuclear enzymes that regulate the three- dimensional geometry (topology) of DNA and enable supercoiled DNA to relax. Regulation of DNA supercoiling is essential to DNA transcription and replication, when the DNA helix must unwind to permit proper function of the enzymatic machinery involved these processes. In order to relieve the topological stress caused by supercoiling, the enzymes topoisomerase I and topoisomerase II (Topo I and II) produce single or double-strand breaks respectively, followed by re-sealing, thus reducing the tension in the DNA strand without leaving damaging nicks. Topoisomerase I works by breaking only one strand of DNA, followed by attachment of the free phosphate residue of the broken strand to a tyrosine residue of the enzyme. The complex then rotates, relieving the supercoiled tension of the DNA, and the two ends are then re-sealed. Topoisomerase II works in a related manner but cleaves both strands of DNA simultaneously, passing a complete duplex strand through the cut, followed by re-sealing of both ends. Topoisomerase inhibitors affect the activity of Topo I and II, with most acting to block the re-sealing process, leading to cell death by apoptosis [123]. Topoisomerases are considered essential proteins for all human cell division to take place properly. They are overexpressed in cancer cells, where the higher activity of topoisomerase enzymes accelerates the rate of cell division making the inhibition of these enzymes important anticancer drug targets. In certain embodiments, the toxin may be a topoisomerase I or II inhibitor.
In certain embodiments the toxin may be a camptothecin. The term 'camptothecin' as used herein is intended to mean a camptothecin or camptothecin derivative that functions as a topoisomerase I inhibitor. Camptothecin (CPT) was the first specific topo I inhibitor to be discovered. It is a quinoline plant alkaloid isolated from the Chinese plant Camptotheca acuminata and has a novel pentacyclic structure. The cytotoxic effect of camptothecin and its analogues is due the inhibition of the religation step that normally reseals the parent strand of DNA causing an irreversible double-strand DNA break, arresting the process of cell division, resulting in cell death [124].
Exemplary camptothecins that may be used in accordance with the present invention include topotecan, exatecan, DX-8951f, DXd, irinotecan, SN28, belotecan, 9- aminocamptothecin, rubitecan, silatecan, lurtotecan, diflometotecan, namitecan, gimatecan, cositecan, rubitecan, FL118 (7-methyl-10,ll-methylenedioxy-20(RS)- camptothecin and its 7-alkylamino derivatives (7-methyl, ethyl, propyl, butyl), 7-n-butyl- 10-aminocamptothecin, 7-n-butyl-9-amino-10,ll-methylenedioxy camptothecin, 4- Amino-9-ethyl-5-fluoro-9-hydroxy-l,2,3,9,12,15-hexahydro-camptothecin, 7- methylamino-(10-methyl,ll-fluoro)-camptothecin, 7-trihydroxmethylaminomethyl- (10,ll-methylenedioxy)-camptothecin. Exemplary camptothecin compounds also include those described in [125]. Exemplary camptothecin compounds are further described in WO2022232834, W02022093800, WO2022015656, WO2022155347, WO2022253035, WO2022246576, WO2022058395, WO2020219287 and WO2019236954.
Camptothecin has a novel pentacyclic ring structure of and to distinguish modifications and derivatives each ring given a label A-E. Exemplary ring modified camptothecins are described in [126]. In camptothecin the presence of a 6-membered o-hydrocy lactone E- ring is regarded as an important feature as it interacts with human Topo I through Arg364 and Asp533 residues and confers antiproliferative activity. However, this lactone E ring is unstable in plasma at physiological pH resulting short half-life and low therapeutic efficacy. E-ring modified camptothecin derivatives such as an expanded 7-membered 0-hydroxy lactone like diflomotecan have been developed and also derivatives where the size of the E-ring has been reduced from 6-membered to 5-membered. One such active derivative is S39625 [127]. Derivatives where the E ring has been opened are also being explored.
Non-camptothecin topoisomerase I inhibitors: Several non-camptothecin derivatives have recently been developed as topo I inhibitors including indenoisoquinolines including exemplary compounds NSC 725776 and NSC 724998 [128], dibenzonaphthyridione (benzophenanthridine) [129]. Exemplary compounds include GENZ-644282 [130], ARC- 111 (topovale) and other derivatives. Evodiamine is a quinazoline-carbolin alkaloid
isolated from the Chinese herb Evodia rutaecarpa and is a potent topo I inhibitor. Many synthetic derivatives of evodiamine have been found to manifest potent antitumour activity [131].
Nemorubicin, a doxorubicin analogue bearing a 2(S)-methoxy-4-morpholinyl chain is a potent antitumour agent that induces DNA strand breaks primarily through topoisomerase-
I cleavage and is active in cells resistant to topoisomerase II inhibitors. But nemorubicin also exerts its antitumour action through a novel mechanism different to that of other anthracyclines, involving the inhibition of DNA nucleotide excision repair (NER) [132]. Exemplary nemorubicin analogues are described in US20140227299 and W02008092796.
Topoisomerase II inhibitors: Exemplary approved topoisomerase II inhibitors that can be used in accordance with the present invention are etoposide, teniposide, doxorubicin, idarubicin, epirubicin, and mitoxantrone. Topoisomerase II is a multi-subunit enzyme which uses ATP to pass an intact helix through a transient double-stranded break in DNA to modulate DNA topology [133]. After strand passage, the DNA backbone is religated and DNA structure restored. It has been suggested that cancer cells rely on the topo II enzyme more than healthy cells since they divide more rapidly. Therefore, normally reversible DNA strand breaks are converted into lethal breaks by processes such as transcription and replication causing errors in DNA synthesis and promoting apoptosis [134]. Etoposide is a semi-synthetic glucoside analogue of epi-podophyllotoxin, a toxin lignan (from the rhizome of the wild mandrake, Podophyllum peltatum). Etoposide and derivatives act as nonintercalating topo II poisons, preventing topoisomerase II from religating cleaved DNA [135].
The largest group of topo II inhibitors are the anthracyclines. Anthracyclines also intercalate into DNA and form reactive metabolites that interact with many intracellular molecules and biologic effects of the anthracyclines are not based solely on topoisomerase
II activity [136]. In certain embodiments, the toxin may be a topoisomerase II inhibitor.
In certain embodiments the toxin may be doxorubicin. Doxorubicin refers to members of the family of anthracyclines derived from a naturally occurring group of antitumour antibiotics and include daunorubicin, epirubicin and idarubicin. The mechanism of action of doxorubicin is common to all anthracyclines and involves DNA intercalation. The planar anthraquinone aromatic ring nucleus inserts between the base pairs of the DNA double helix, perpendicular to the double-strand axis. The complex is then stabilised by various hydrogen bonds, hydrophobic interactions and Van der Waals forces. By forming intercalated adducts, doxorubicin inhibits both DNA and RNA polymerases, leading to
arrest of DNA replication and RIMA transcription. Doxorubucin also binds to the DNA- topoisomerase complex in proximity to the scission site of DNA, thus preventing its repair and triggering apoptosis by inhibiting cellular division at the G1 and G2 phases [137]. Doxorubicin and the other anthracyclines can undergo one- and two-electron reduction, since they are members of the quinone family, producing reactive compounds that damage macromolecules and lipid membranes. Exemplary anthracyclines are described in [138].
Mitoxantrone is a simplified anthracenedione that binds to topoisomerase II resulting in cleavable complexes that induce DNA strand breaks. Mitoxantrone lacks the ability to form the quinone-type free radicals thought to account for anthracycline cardiotoxicity and is the only agent of its class approved for clinical use. In some embodiments, the toxin is mitoxantrone.
PNU-159682 is liver metabolite of nemorubicin and is about three orders of magnitude more potent than its parent molecule on cultured human tumour cells. It is a potent topo II inhibitor amongst other poorly understood modes of action and is effective regardless of the cell cycle unlike doxorubicin. PNU-159682 is not an efflux pump substrate and is able to bypass resistance mechanisms observed with known tubulin inhibitors like MMAE and DM1 [139]. In some embodiments, the toxin is PNU-159682.
Several analogues of these anthracyclines are in various stages of clinical development. PNU-159548, an alkycycline daunorubicin derivative, has demonstrated antineoplastic activity in animal models with reduced cardiotoxicity compared to doxorubicin [140]. Ethonafide is an anthracene-containing derivative of amonafide which inhibits topoisomerase II and may have less toxicity than other anthracene-containing agents. Exemplary PNU-159682 derivatives are described in [141]. In some embodiments, the toxin is PNU-159548 or ethonafide.
In certain embodiments, the toxin may be a duocarmycin. Duocarmycins are a family of DNA minor groove-binding compounds with exquisite cytotoxicity originally identified in Streptomyces [142]. Their unique mechanism of action involves DNA minor-groove binding and alkylation of adenine forming DNA-adducts which disrupt the DNA helix ultimately leading to cell death. A key advantage of duocarmycins and its analogues is the ability to exert their effects on any phase of the cell cycle acting on both dividing and nondividing cells. Structurally, duocarmycins contain a DNA-binding moiety that selectively interacts with DNA strands and a DNA-alkylating unit that possess a spirocyclic cyclopropapyrroloindole (CPI) moiety and many synthetic duocarmycin analogues maintain these structural features. The alkylating CPI unit can also be derivatised in its
ring-opened chloromethyl aromatic (seco) form, forming prodrugs that contain a phenolic hydroxyl group producing a cyclopropane-containing cytotoxin, via a process known as spirocyclisation, to prevent spontaneous cyclisation, the phenolic hydroxyl group must be modified usually via a pro-drug strategy.
Exemplary duocarmycins and analogues include CC-1065, duocarmycin SA, duocarmycin TM, duocarmycin A, adozelesin, bizelesin, carzelesin, seco-adezelesin, DC0-NH2, DC1, DCISMe, DC41SMe, (+)-CBI-CDPIl, (+)-CBI-CDPI2, DC41, DC4SMe, DClOSMe, DU-257, DU-86, KW-2189, seco-duocarmycin-SA, duocarmycin MB, duocarmycin GA, duocarmycin B2 [143,144]. Duocarmycins are DNA mono-alkylators, by connecting two duocarmycin- type units per molecule, DNA-cross-linking via double alkylation is achieved and these derivatives display significantly greater potency than duocarmycins. The CXI dimers [dimers containing, for example cyclopropapyrroloindole (CPI), cyclopropabenzindole (CBI) or cyclopropathienoindole (CTI) moieties] are the main class of potent bis-alkylating payloads. Exemplary CXI dimers are described [145].
In certain embodiments, the toxin may be a Pyrrolobenzodiazepine (PBDs). PBDs were discovered in the 1960s and are an important class of sequence-selective DNA-interactive agents that bind covalently to guanine bases within the minor groove of DNA. There are now two recognized subfamilies of pyrrolobenzodiazepines, the first is the PBD monomer subfamily that represents the agents originally discovered in cultures of Streptomyces species (e.g., anthramycin and tomaymycin). The PBD monomers are remarkable in possessing a 3-dimensional shape that allows them to fit perfectly within the minor groove of DNA, once located in a position of low energy in the groove (i.e., a preferred DNA sequence), largely dictated by substituents, the electrophilic imine then alkylates the amine group of an adjacent guanine base, thus producing a robust covalent adduct capable of blocking biological processes such as transcription factor binding and RNA polymerase progression. The PBD monomers have both antibacterial properties and selective cytotoxicity toward tumour cells. The second subfamily, the PBD dimers, are not naturally occurring but are synthetic derivatives, the first examples were designed to span greater lengths of DNA than the PBD monomers, to have enhanced sequence-selectivity, and to form DNA cross-links that might be more difficult for tumour cells to repair. It is now known that PBD dimers can form both inter-strand and intra-strand cross-links, as well as monoadducts under certain conditions, although the interstrand cross-linked adduct is still thought to be the most toxic in cells. The perfect fit of a PBD dimer in the DNA minor groove results in negligible distortion of the DNA helix, thus potentially avoiding DNA- repair mechanisms and drug resistance.
Exemplary pyrrolobenzodiazepine (PBDs), which expressly include dimers and analogues, include but are not limited to those described in [146,147]. Duocarmycin-PBD dimers are heterodimeric compounds fomed by linking together two different alkylating subunits derived from duocarmycin and pyrrolobenzodiazepine (PBD) classes of DNA-minor groovealkylating agents. Exemplary duocarmycin-PBD dimers are described in [148].
In certain embodiments, the toxin may be a mitomycin. Mitomycins are a family of aziridine-containing natural products isolated from Streptomyces caespitosus, the best- known members being mitomycin A, B and C. Mitomycin C has significant antitumour properties and is active against a variety of tumours including breast, stomach, oesophagus, bladder and NSCLC. The main mechanism of action of mitomycin is through in situ bioreduction resulting in a bis-electrophile that can alkylate a range of cellular nucleophiles with DNA alkylation as the main mechanism of action. The reductive activation mechanism confers selectivity towards particular tumour types, especially hypoxic solid tumours. Exemplary mitomycins are described in [149,150].
In some embodiments, the toxin may be a nitrogen mustard. Nitrogen mustards are some of the earliest examples of DNA alkylating agents, these act either as mono-alkylators or as intra- and interstrand cross-linkers. Exemplary nitrogen mustards are chlorambucil (Leukeran™) and bendamustine (Levact™). Site-directed nitrogen mustards include melphalan (Alkeran™) which is an L-phenylalanine derivative designed for selective uptake by tumour cells in which protein synthesis may be occurring. The oligopyrrole antibiotics netropsin and distamycin A (and related synthetic heteropyrrole/imidazole 'lexitropsin' analogues) are well-documented minor groove binders. Attaching nitrogen mustards to their analogues results in increased cytotoxicity, highly specific alkylation at adenines and broad spectrum solid tumour activity. This work culminated in the development of tallimustine. Bis-benzimidazoles are also well-characterised reversible minor groove binding ligands, with extensive studies on the lead compound Hoechst 33258 whose analogues with aniline mustards attached by a variable-length polymethylene chain showed chain length-dependent patterns of DNA alkylation and are potent. Exemplary derivatives are described in [151].
Further approaches utilise selective bioactivation of prodrug forms where there are biochemical differences between tumour and normal tissues. Cyclophosphamide (Endoxana™) is one example of a metabolic prodrug that requires enzymic activation by phosphoramide hydrolysis by phosophoramidases, releasing the active agent which alkylates guanine bases resulting in mono-adducts and both intra-and inter-strand crosslinks. Other mechanisms of alkylation of DNA are through aziridines and epoxides. The
active species involved in DNA alkylation by nitrogen mustards is an aziridinium cation and several aziridine derivatives have been tested as antitumour agents like Carboquone, Diaziquone (AZQ) and BZQ. The high reactivity of epoxides towards nucleophilic groups in biomolecules has led to the development of numerous DNA alkylating drugs like Treosulfan which alkylates DNA at guanine bases and has been used in the treatment of ovarian cancer.
The nitrosoureas are a class of alkylating agents that decompose to produce alkylating compounds under physiologic conditions, the predominant mechanism being base catalysed decomposition to a chloroethyl diazonium moiety which reacts with DNA to form a unique inter-strand DNA cross-link. Currently the most clinically relevant nitrosoureas are Carmustine (BCNU), Lomustine, Nimustine (ACNU) and Streptozotocin.
The platinum antitumor agents are complexes of platinum with ligands that can be displaced by nucleophilic (electron-rich) nitrogens in nucleic acids. All platinum agents act as pro-drugs, in that they require the removal of their labile chloride or carboxylate ligands, through their displacement by water, before they can bind DNA. When binding to DNA, platinum complexes predominantly do so through the formation of a coordination bond at the N7 site of guanosine residues. Simultaneous binding at a second, adjacent nucleotide, typically another guanosine base, results in inter-strand cross-linking, significant bending and unwinding of the DNA which prevents transcription and replication. The cascade effect of this binding is the induction of apoptotic cell death. Exemplary platinum drugs are cisplatin, carboplatin (Paraplatin), oxaliplatin (Eloxatin), nedaplatin, loboplatin, picoplatin, heptaplatin, teraplatin, iproplatin, satraplatin and ormaplatin
In certain embodiments, the toxin may be an enediyne. Enediynes are highly cytotoxic DNA damaging natural products produced through microbial secondary metabolism. The enediyne natural products share a common structural motif containing two acetylenic groups conjugated to a double bond or incipient double bond within a 9- or 10-membered carbocycle [152]. Exemplary 9-membered enediynes include neocarzinostatin, kedarcidin, maduropeptin, C-1027 and N1999A2. These molecules, except for N1999A2, generally are found in complex with a cognate apoprotein that binds the enediyne chromophore to generate a stable chromoprotein. Exemplary 10-membered enediynes can be divided into two subfamilies: the calicheamicin-like enediynes including the calicheamicins, the esperamicins, namenamicin and shishijimicins, and the anthraquinone-fused enediynes including dynemicin, uncialamycin, the tiancimycins and the yangpumicins [153].
The conjugated carbocycle drives the shared mode of action for the enediynes. Electronic rearrangement of the enediyne carbocycle (cycloaromatization) generates a benzenoid
diradical that drives enediyne-induced cytotoxicity. Enediyne natural products with a 9- membered carbocycle can proceed through either the Myers-Saito or Bergman rearrangement pathways depending on their architecture, whereas all known 10- membered enediynes proceed through the Bergman rearrangement. These DNA lesions are responsible for the cytotoxicity of enediyne natural products [154]. Exemplary enediyne antibiotics are described in [155,156].
Although there are many anticancer drugs that interact with DNA by initially intercalating, there are also classes of drugs whose main mode of action is intercalation. Intercalation involves the insertion between the base pairs of DNA, perpendicular to the axis of the helix of a flat, fused aromatic molecule. This unwinding results in local structural changes in the helix such as lengthening or twisting of the base pairs and lead to the interference of recognition and function of DNA-associated proteins such as polymerases, transcription factors and DNA repair systems. Exemplary monofunctional intercalating agent is ellipticine and its analogues. Ellipticine is an alkaloid isolated from the leaves of Ochrosia elliptica and has potent anticancer properties. Ellipticines are multimodal anticancer agents because they exert their biological activity via several modes of action with intercalation and topoisomerase II inhibition the best established. Exemplary ellipticine derivatives are celiptium, datelleptium, the olivacine derivative S-16020 and intoplicine.
In order to increase the binding constant of intercalating compounds, bifunctional of polyfunctional compounds have been designed. Bifunctional intercalators (bis- intercalators) contain two intercalating units, normally cationic, separated by a spacer chain that must be long enough to allow double intercalation to take place. Exemplary bis- intercalators are ditercalinium and elinafide. Further examples of both mono-and bis- intercalators are described in [157].
Tyrosine Kinase Inhibitors
Protein kinases are enzymes that catalyse phosphorylation and are divided into three categories: serine, threonine or tyrosine kinases. Tyrosine kinases function by catalysing the transfer of a phosphoryl group from a nucleoside triphosphate donor to the hydroxyl group of tyrosine residues on protein substrates and then triggering the activation of downstream signalling cascades. In cancer a variety of kinase families are involved in cell cycle progression, cell proliferation, motility and angiogenesis. Abnormal activation of tyrosine kinases due to mutations, translocations, or amplifications is implicated in tumorigenesis, progression, invasion, and metastasis of malignancies [158]. As such, tyrosine kinases have emerged as major targets for drug discovery [159].
Fibrosis denotes an excessive deposition of collagen and other extracellular matrix (ECM) components in tissue. Deposition of collagen is part of physiological wound healing but when this process becomes abnormal, connective tissue replaces normal parenchyma, leading to tissue destruction and impairment of organ function. This pathologic process can affect several organs, causing diverse chronic diseases such as idiopathic pulmonary fibrosis (IPF) [160]. An exemplary compound for the treatment of IPF is nintedanib. Nintedanib is a small molecule, competitive, triple angiokinase inhibitor that targets multiple receptor tyrosine kinases (RTKs) and non-receptor tyrosine kinases (nRTKs). Many of these RTKs are implicated in lung fibrosis and tumour angiogenesis, so nintedanib is therefore used in the treatment of proliferative diseases such as idiopathic pulmonary fibrosis, non-small cell lung cancer, and systemic sclerosis-associated interstitial lung disease. The specific RTKs that nintedanib inhibits are platelet-derived growth factor (PDGFR) a and 0, fibroblast growth factor receptor (FGFR) 1-3, vascular endothelial growth factor receptor (VEGFR) 1-3, and Fns-Like tyrosine kinase-3 (FLT3). Nintedanib binds to the ATP-binding pocket of these receptors and inhibits their activity, thereby blocking signalling cascades that result in the proliferation and migration of lung fibroblasts. Nintedanib also inhibits kinase signalling pathways in various cells within tumour tissues, including endothelial cells, pericytes, smooth muscle cells, and cells contributing to angiogenesis, culminating in an inhibition of cell proliferation and apoptosis of affected tumour cells [161]. Exemplary derivatives of nintedanib include the ov[36 targeting- nintedanib conjugates [162].
In some embodiments, the payload is a tyrosine kinase inhibitor selected from the group consisting of nintedanib, afatinib, axitinib, bosutinib, crizotinib, dasatinib [163] erlotinib, fostamatinib, gefitinib, ibrutinib, imanitib, lapatinib, mubritinib, nilotinib, pazopanib, pegaptanib, sorafenib, sunitinib, Lenvatinib, linifanib, tivozanibvandetanib, vemurafenib, CEP-701 (lesaurtinib), INCB018424 (ruxolitinib), ARRY-142886 (selemetinib), ARRY- 438162 (binimetinib).
Transcription inhibitors
Transcription has a fundamental role in cell development, activity and proliferation. Transcription is regulated by RNA polymerase II (RNApol II) that directly binds to DNA and involves transcription factors that form complexes with RNApol II to initiate transcription and co-regulators that mediate chromatin structure and accessibility.
In certain embodiments, the toxin maybe an amatoxin. Amatoxins (including alpha- amanitin, beta-amanitin and amanitin) are natural and highly potent RNApol II inhibitors derived from the Amanita phalloides mushroom. They are cyclic peptides composed of 8
amino acids and inhibit specifically the DNA-dependent RIMA polymerase II of mammalian cells, affecting transcription and protein biosynthesis, stopping growth and proliferation [164]. Exemplary amatoxins include alpha-amanitins, beta-amanitins, gamma amanitins, eta amanitins, amanulin, amanullic acid, amanisamide, amanon and proamanulin. Amatoxins are described in [165].
Another strategy to stop DNA transcription is to inhibit transcription factors. Transition factors are essential to RNApol II attachment to the DNA at the initiation step. Triptolide, a natural compound derived from the Chinese medicinal herb called "thunder god vine", is highly cytotoxic against a variety of solid tumour cell-lines and one of its main modes of action is as an inhibitor of both RNA Pol I- and RNA Pol II dependent transcriptions. Minnelide is a water-soluble pro-drug of triptolide currently being evaluated as an anticancer drug. Exemplary triptolide derivatives are described in [166].
Histone deacetylases (HDACs) are epigenetic regulators that regulate the histone tail, chromatin conformation, protein-DNA interaction and are therefore involved in various cellular processes including transcription and protein regulation. They have been found to be overexpressed or overactivated in cancer cells and are thought to be involved in increased proliferation, migration and invasion. Exemplary HDAC inhibitors that may be used in accordance with the present invention include Vorinostat, Romidepsin, Panobinostat, and Belinostat.
In certain embodiments, the toxin maybe a thailanstatin, or an analogue or derivative thereof. Thailanstatin A and its analogues are RNA splicing inhibitors. RNA splicing controls metabolism, angiogenesis, cancer cell proliferation and metastasis mainly by excision of introns and exons that are responsible for the complex cellular mechanism of transforming RNA to mRNA. They can also directly control the initiation, elongation, and termination of transcription and are potent antiproliferative drugs capable of targeting cells that are actively dividing and quiescent. Thailanstatin is a natural product originally isolated from Brucella Thailandensis msmb43 and activated by binding to the SF3b subunit of the spliceosome U2 snRNA subcomplex. The thailanstatin family has a strong binding and inhibitory effect on spliceosomes by inhibiting the eukaryotic mRNA splicing pathway, resulting in low nanomolar IC50 values for a variety of cancer cell lines. Exemplary Thailanstatin A derivatives are described in [167].
In certain embodiments, the toxin maybe a kinesin spindle protein inhibitor. Kinesin spindle protein (KSP, synonyms: Eg5, KIF11) is an ATP-dependent motor protein involved in the separation of centrosomes in the G2/M phase of the cell cycle. High expression in
certain solid tumours (breast, pancreatic and bladder cancers) is associated with poor prognosis. The blockade of this essential event in mitosis with small molecule KSP inhibitors (KSPis), results in high antitumor potency. Also, Eg5 expression is specific to proliferating cells and is not expressed in the cells of the nervous system so avoiding the neurological side effects of classic microtubule targeting agents. Exemplary small molecule KSP inhibitors are Ispinesib and Filanesib. Further examples are described in [168].
In certain embodiments, the toxin maybe a NAM PT inhibitor. Niacinamide phosphate ribose transferase (NAMPT), which converts nicotinamide into nicotinamide mononucleotide, is a rate-limiting enzyme that controls the concentration of NAD+ within cells. When NAMPT is suppressed, NAD+ levels drop below the level required for metabolism, leading to an energy crisis and eventually, cell death. In contrast to many other cytotoxic molecules, the cell death following depletion of NAD proceeds via a nonapoptotic and proinflammatory mechanism termed oncosis. Exemplary NAMPT inhibitors that may be used in accordance with the present invention include FK-866, CHS-828, GMX-1777, GMX-17778, GPP 78 hydrochloride, ST 118804, STF31. Further examples are described in [169].
In certain embodiments, the toxin maybe a Bcl-xL inhibitor. Bcl-xL is an anti-apoptotic protein that plays an important role in tumour formation, metastasis, and drug resistance. Apoptosis is a highly regulated cellular process. Aberration in apoptosis is a common characteristic of various disorders. Therefore, proteins involved in apoptosis are prime targets in multiple therapies. Compared to other antiapoptotic proteins, the expression of Bcl-xL is common in solid tumours and apoptosis of cancer cells can theoretically be triggered by drugs blocking the BH3 binding domain on Bcl-xL. However, Bcl-xL is critical for platelet survival, and pan-inhibitors of Bcl-xL may produce platelet toxicity [170].
Exemplary BcL-xL and Bcl-2 inhibitors are navitoclax, venetoclax, GX-070 (obatoclax), gossypol acetic acid (AT-101, sabutoclax), WEHI-539), ABT-737 and those described in [171]; exemplary compounds are described in W02017/214301 and WO2017/214282.
Further exemplary compounds that may be used in accordance with the present invention are G3139 (Oblimersen), Bcl-2-targeting antisense oligonucleotide IPI-194 and IPI-565, HGS1029, GDC-0145, GDC-0152, LCL-161, LBW-242, Bcl-2 family members, death domain proteins, TNF family members, Toll family members and/or NF-kappa-B proteins.
In certain embodiments, the toxin maybe an immune stimulating agent such as toll-like receptor agonists (e.g. TLR 7/8 agonists) or STING agonists. Tumour immunity involves T cells, macrophages, dendritic cells, etc., which transform cold tumours (immunologically inactive) into hot tumours (immunologically active) through immunomodulation, and
ultimately enhance the effect of immunotherapy. Novel cancer immunotherapies, such as checkpoint inhibitors, have proven to be a vital approach for the treatment of an increasing number of cancers [172]. While these therapies successfully modulate the immune system to eradicate metastases, their success does not translate well to "cold" or "non-T cell- inflamed" tumours that lack sufficient T cell infiltrates or exhibit immune suppression prior to treatment. One approach to overcome this resistance is to activate the patients innate immune system/ response via known signalling pathways, including toll-like receptor (TLR) 7/8, nucleotide-binding oligomerization domain, and cGAS-STING and promote tumourspecific T cell recruitment [173].
STING and TLR agonists constitute the two main categories of immune stimulants, however, the systemic administration of potent STING and TLR agonists is associated with severe toxicity caused by cytokine release syndrome. Their conjugation to proteins or antibodies could be one approach in exploiting their strong antitumour potential together with better tolerability. Several chemical scaffolds have been identified as starting points to generate potent TLR7/8 agonists, including purines (loxoribine), imidazoquinolines (imiquimod and resiquimod), deazapurines (compounds described in WO 2015/16827 benzazepines (motolimod) and various others [174]. These compounds all bind in the 2'3- cGMP site of the TLR7/8 proteins (which share a high degree of sequence homology and function). Exemplary compounds are CL264, UC-1V150, D18, and T785. Further exemplary compounds are described in WO 2018/191746 and WO 2020/056192.
The stimulator of interferon genes (STING) is a key regulator in the natural immune signalling pathway and has the function of initiating the body's natural immune defence response and promoting the formation of adaptive immunity by T cells. The STING- mediated type I interferon signalling pathway is a major discovery in the field of natural immunity, providing a new target for tumour immunotherapy. Activating the STING pathway can induce the expression and secretion of type I interferons and a variety of other cytokines, activate the natural immune response, promote the anti-tumour immune response. Natural agonist, cGAMP and other synthetic cyclic dinucleotide (CDN) derivatives have been the main starting point for the development of STING agonists, non-CDN small molecule STING agonists have also been identified, such as dimeric amidobenzimidazole (diABZI) like XMT-1621, these function by mimicking the native ligand of STING, 2'3'- cGAMP. Exemplary compounds are described in [175,176].
In certain embodiments, the toxin maybe a PROTAC. Proteolysis Targeting Chimeric Molecules (PROTACs) are bifunctional molecules that bring together the E3 ligase with the target protein thus allowing its ubiquitination and degradation by the proteasome. Instead
of directly inhibiting its target protein, PROTACs trigger its degradation with several potential clinical advantages such as prolonged effect, catalytic activity and therefore very potent cytotoxicity. PROTACs can benefit from being targeted inside the cell to overcome their limited cell permeability. Exemplary compounds are MZ1 (BRD4/VHL), GNE-9787 (BRD4/VHL), BRD4/VHL, BRD4/CRBN, Era/VHL, Era/XIP, BRM/VHL, TGFbR2/CRBN, other examples are described in [177].
In certain embodiments, the toxin maybe an anti-inflammatory agent. In the treatment of immunological disease (inflammation associated with various diseases), glucocorticoid receptor modulators (GRMs) are often the first line of treatment. However, GRM can cause musculoskeletal (osteoporosis and decreased bone density), endocrine, and gastrointestinal side effects and other toxicities, limiting their beneficial therapeutic effects (especially in long-term use). Multiple medicinal chemistry programs have targeted small molecule selective glucocorticoid receptor modulators which would minimize GR transactivation (believed to be responsible for the unwanted side effects) while maintaining GR transrepression (believed to be responsible for the desired antiinflammatory effects). Exemplary glucocorticoid receptor modulators are dexamethasone, prednisolone, and budesonide. Further examples are described in [178,179], WO 2017/062271, WO2017/210471, WO2019/136487, WO2022/150637.
In certain embodiments the toxin may be an antimetabolite. Antimetabolites are anticancer agents that are analogues of naturally occurring essential metabolites. They work by either blocking biochemical pathways essential for cell growth and division i.e., as enzyme inhibitors) or by incorporating themselves into nucleic acids (i.e., DNA or RNA) and act as 'false substrates' for relevant polymerases, thus blocking relevant down-stream processes such as replication and transcription. Although the enzymes inhibited by antimetabolites are also present in normal cells, some selectivity towards cancer cells is possible due to their faster cell division rates.
Most antimetabolites interfere with nucleic acid synthesis and the production of DNA or RNA by two main mechanisms: By competing for sites of enzymes that participate in essential biosynthetic processes or by incorporating into nucleic acids, inhibiting their normal function and triggering apoptosis. One class of antimetabolites are the antifolates, these bind tightly to dihydrofolate reductase (DHFR) thereby inhibiting folate metabolism. Through this inhibition, thymidylate synthesis is blocked, and thus, purine biosynthesis which can then no longer be incorporated into DNA; examples of antifolates include methotrexate, pemetrexed, pralatrexate, raltitrexed, nolatrexed, lometrexol, ZD9331, PT523. Purine antimetabolites inhibit several enzymes at various points in the purine
synthesis pathway and examples include 6-mercaptopurine, azathioprine, fludarabine phosphate, cladribine, clofarabine, nelarabine. Similar to the purine antimetabolites are the pyrimidine anti metabolites, these inhibit the synthesis of DNA in the S-phase and block the movement of cells through the Gl/S part of the cell cycle. Exemplary drugs include 5- fluoroucil, tegafur, capecitabine, doxifluridine, cytarabine, gemcitabine, and decitabine, azacytidine [180-182].
In certain embodiments the toxin maybe a N-myristolytransferase (NMT) inhibitor. Myristoylation, is a lipid modification to a specific group of proteins and involves the N- terminal modification of proteins with myristic acid, a 14-carbon fatty acid, that allows them to interact with other proteins or membranes. Myristoylation plays vital roles in protein-protein interactions, signal transduction, immune regulation and tumour development. It is catalysed by the enzyme /V-myristoyltransferase (NMT) which introduces irreversible changes to human proteins. NMT inhibitors have been shown to inhibit the viability and growth of various cancers and represent a novel class of ADC payloads that exploit cancer cell dependency on myristloylated proteins [183]. Exemplary small-molecule NMT inhibitors are described in WO 2022/058745.
In some embodiments, the payload is a microtubule inhibitor (MTI), a DNA-damaging agent, a topoisomerase inhibitor, a steroid, a tyrosine kinase inhibitor, or an immunomodulatory drug. In some embodiments, the payload is an MTI, a DNA-damaging agent, or a topoisomerase inhibitor.
In some embodiments, the MTI is an auristatin or a derivative thereof. In some embodiments, the auristatin is mono-methyl auristatin E (MMAE) or mono-methyl auristatin F (MMAF). In some embodiments, the DNA-damaging agent is a nicking agent, an alkylator, or an intercalating agent. In some embodiments, the topoisomerase inhibitor is a camptothecin or a derivative thereof. In some embodiments, the steroid is desxamethasone. In some embodiments, the tyrosine kinase inhibitor is nintedanib. In some embodiments, the immuno-modulatory drug is a Toll-Like Receptor 7/8 agonist drug.
In some embodiments, the antigen-binding fragment is a single chain Fv and the payload is MMAE, and wherein the payload is coupled to the scFv with a coupling ratio (payload :scFv) of 6: 1.
In some embodiments, the antigen-binding fragment is a single chain Fv and the payload is a mono-methyl auristatin E (MMAE) comprising a glucuronide and branched PEG linker, wherein the MMAE is conjugated onto surface lysine residues of the scFv.
Specific examples of compounds according to the invention include, but are not limited to, where:
(i) the antigen-binding fragment is an scFv and the payload is MMAE;
(ii) the antigen-binding fragment is an scFv and the payload is MMAF;
(iii) the antigen-binding fragment is an scFv and the payload is SN38;
(iv) the antigen-binding fragment is an scFv and the payload is exatecan;
(v) the antigen-binding fragment is an scFv and the payload is Dxd;
(vi) the antigen-binding fragment is an scFv and the payload is belotecan;
(vii) the antigen-binding fragment is an scFv and the payload is triplotide;
(viii) the antigen-binding fragment is an scFv and the payload is nintedanib;
(ix) the antigen-binding fragment is an scFv and the payload is dexamethasone;
(x) the antigen-binding fragment is an scFv and the payload is of -benzylamino imidazoquinoline;
(xi) the antigen-binding fragment is an scFv and the payload is GENZ-644282.
In some embodiments, the payload is conjugated to the antibody or antigen-binding fragment thereof via a linker.
In the present invention, the term 'linker' refers to any chemical moiety capable of linking a payload as described to an antibody or antigen binding fragment thereof. The linkers may be monovalent such that they covalently link a single payload to a single site on the antibody fragment, or the linker maybe polyvalent such that they link more than one payload to a single site on the antibody fragment. The linkers connect the payload to the antibody fragment by forming a covalent linkage to the payload at one location and a covalent linkage to the antibody fragment at another. The covalent linkages are formed by reaction between functional groups on the linker and functional groups on the payload and antibody fragments.
The term 'linker' includes (i) unconjugated forms of the linker that include a functional group capable of covalently linking the linker to the payload and a functional group capable of covalently linking the linker to the antibody fragment (ii) partially conjugated forms of the linker that includes a functional group capable of covalently linking the linker to the antibody fragment that is covalently linked to a payload, or vice versa, and (iii) fully conjugated forms of the linker that is covalently linked to both payload and antibody fragment.
The linker may be chemically or biologically stable or resistant to cleavage (a 'non- cleavable linker') or the linker may be susceptible to cleavage or include linkages that are designed to release the payload upon internalisation within the cell (a 'cleavable linker'). These cleavable linkers are designed to cleave and/or self-immolate or otherwise breakdown specifically or non-specifically inside cells. The term 'self-immolate' refers to an at least bifunctional molecule that can be included in a linker that degrades spontaneously after an initial reaction has taken place and thereby releasing the payload. Polyvalent linkers that may be used to link many payloads to a single antibody molecule include Mersanas Fleximer™ technology which incorporated payloads into a solubilising polyacetal backbone via a sequence of ester bonds, other approaches use dendritic type linkers.
Cleavable linkers
In certain embodiments, the linker is cleavable in vivo. Cleavable linkers may include chemically or enzymatically unstable or degradable linkages. Cleavable linkers generally rely on processes inside cells to liberate the payload, such as reduction in the cytoplasm, acid induced cleavage in the lysosomes or endosomes, or cleavage by specific proteases or other proteolytic enzymes within the cell [184]. In certain embodiments, a linker comprises a chemically labile group such as hydrazone and/or disulphide groups. These types of linkers exploit differential properties between the plasma and some cytoplasmic compartments. The intracellular conditions to facilitate drug release for hydrazone containing linkers are the acidic environment of endosomes and lysosomes, while disulphide containing linkers are reduced in the cytosol, which contain high concentrations of reductive agents like glutathione.
Acid cleavable linkers aim to exploit the acidity of the endosomes (pH 5.5-6.2) and lysosomes (pH 4.5-5.0) whilst maintaining stability in circulation at pH 7.4. Acid-labile groups such as hydrazone remain intact during systemic circulation in the blood's neutral pH and undergo hydrolysis, releasing the payload once internalised into the endosome and lysosome compartments of the cell. Other acid sensitive functional groups used in linkers include a carbonates and esters.
Disulphide containing linkers are designed to release the payload upon internalisation inside cells, they thermodynamically stable at physiological pH but are susceptible to nucleophilic attack from thiols. In blood plasma, the dominant thiol species is the reduced form of human serum albumin (HSA), however its reactivity towards large molecules is hampered because the free thiol-containing residue (Cys34) is in a crevice and has limited solvent exposure. In contrast, the cytosol contains high levels of glutathione, a thiol-
containing small molecule tripeptide. This difference between the reductive potential of the blood plasma and the cytosol enables selective intracellular release of the payload. Also, tumours can have hypoxic regions, resulting in enhanced or elevated levels of reductive enzymes and higher glutathione concentrations. The in vivo stability of disulphide linkers may be further enhanced by chemical modification of the linker, e.g., increasing steric protection around the disulphide which makes the linker less susceptible to reduction.
Enzyme cleavable linkers utilise the high or elevated levels of unique hydrolytic enzymes like cathepsins that reside in the lysosomes of cells and offer an opportunity for the selective cleavage of linkers and the intracellular release of the payload [185]. Such linkers are typically peptide-based or include peptidic regions that act as substrates for these enzymes. Cathepsin B is a ubiquitous cysteine protease whose properties do not differ very much from species to species. It is never found extracellularly, except in pathological conditions such as metastatic tumours. Therefore, conjugates produced with cathepsin B- cleavable linkers are likely to be stable in circulation. Release of a drug from an antibody occurs specifically due to the action of lysosomal proteases, e.g, cathepsin and plasmin.
Exemplary cleavable peptides that can be used in accordance with the present invention include, but are not limited to dipeptides, tripeptides, tetrapeptides and pentapeptides. Dipeptides are often preferred over longer peptides due to the hydrophobicity of longer peptides. Exemplary dipeptides that may be used in some embodiments of the invention include but are not limited to alanine-alanine (ala-ala), valine-alanine (val-ala), valinecitrulline (val-cit), alanine-phenylalanine (ala-phe), phenylalanine-lysine (phe-lys), phenlyalanine-homolysine (phe-homolys) and N-metyl-valine-citrulline(Me-val-cit). Exemplary tripeptides include but are not limited to glycine-valine-citrulline (gly-val-cit), glycine-glycine-glycine (gly-gly-gly), valine-lysine-glycine (val-lys-gly) and alanine- alanine-alanine (ala-ala-ala). Exemplary tetrapeptides include but are not limited to glycine-glycine-phenylalanine-glycine (gly-gly-phe-gly) and glycine-phenylalanine- leucine-glycine (gly-phe-leu-gly).
A peptide may comprise naturally occurring and/or non-natural amino acid residues. The term "naturally occurring amino acid" refer to Ala, Asp, Cys, Glu, Phe, Gly, His, He, Lys, Leu, Met, Asn, Pro, Gin, Arg, Ser, Thr, Vai, Trp, and Tyr. "Non-natural amino acids" (i.e., amino acids that do not occur naturally) include, by way of non-limiting examples, homoserine, homoarginine, citrulline, phenylglycine, taurine, iodotyrosine, selenocysteine, norleucine (Nle), norvaline (Nva), beta-alanine, L- or D-naphthalanine, ornithine (Orn), and the like. Peptides can be designed and optimized for enzymatic cleavage by a
particular enzyme, for example, a tumour-associated protease, cathepsin B, C and D, or a plasmin protease.
Amino acids also include the D-forms of natural and non-natural amino acids. "D-" designates an amino acid having the "D" (dextrorotary) configuration, as opposed to the configuration in the naturally occurring ("L-") amino acids. A variety of di-, tri-and tetrapeptides cleavable linkers have been described that link various payloads to antibodies [185,186], WO2019/195665.
The linker described herein may be an enzymatically cleavable linker. Enzymatically cleavable linkers may include a self-immolative spacer to spatially separate the drug from the site of enzymatic cleavage and is a di- or tri-functional chemical moiety that is capable of covalently linking together two or three chemical moieties. The direct attachment of a payload to a peptide linker can impair enzymatic release or result in proteolytic release of an amino acid adduct of the payload, thereby impairing its activity. The use of a self- immolative spacer allows for the elimination of the fully active, chemically unmodified drug upon amide bond hydrolysis. A typical self-immolative spacer is the bifunctional paraaminobenzyl alcohol group, which is linked to the peptide through the amino group, forming an amide bond, while amine containing payloads may be attached through carbamate functionalities to the benzylic hydroxyl group of the linker (PABC). The resulting prodrugs are activated upon protease-mediated cleavage (for example with cathepsin B), payload release is thermodynamically driven and takes place after amide bond hydrolysis via either an electron cascade for PABC type linkers (1,4-, 1,6-, or 1,8- elimination) or an intramolecular nucleophilic cyclisation. 1,6-elimination from a typical PABC spacer result in the release of the unmodified payload, carbon dioxide, an azaquinone methide and remnants of the linker group [187]. The self-immolative spacer maintains enzymic activity, independent from the payload and variants of the self- immolative spacer where the PABC has been replaced by a heterocyclic spacer like aminothiazole have also been described in US7754681.
Glycosidase-cleavable linkers
In some embodiments, the linker in accordance with the present invention is a glycosidase- cleavable linker. Another class of proteolytic enzymes that are only found in lysosomes are the p-glucuronidases. p-glucuronidases are hydrolytic enzymes in the glycosidase class that catalyse the breakdown of p-glucuronic acid residues in polysaccharides. Their exclusivity and abundance in the lysosomal compartment of cells, overexpression in some tumour types and little enzyme activity outside of cells make them ideal candidates for use in enzyme cleavable linkers [188]. The incorporation of a 0-glucuronic acid moiety
into a linker, results in facile release of the drug through cleavage of the 0-glucuronide- glycosidic bond by the lysosomal enzyme p-glucuronidase. Also, p-Glucuronic acid-based linkers can also be linked to a self-immolative spacer (similar to the PABC used with peptidic linkers) but using a para-hydroxybenzyl alcohol group. However, in this case attachment to the antibody is achieved through substituents on the space molecule (an amine or carboxylic acid group) ortho- to the enzyme-cleavable moiety.
A variety of cleavable 0-glucuronic acid-based linkers useful for linking drugs such as auristatins, camptothecins, doxorubicin analogues, CBI minor-groove binders and cryptophycins to antibodies have been described [189-191]. Due to the hydrophilic nature of p-glucuronides, 0-glucuronic acid-based linkers may be used to minimise or overcome payload induced aggregation of antibody conjugates, especially when trying to achieve high drug loadings with very hydrophobic drugs. The hydrophilicity of 0-glucuronic acidbased linkers and the overall antibody-drug conjugate can be further increase by the incorporation of linear or branched polyethylene glycols, crown or aza-crown ethers, cyclodextrins, polysarcosines or chito-oligosaccharides like Chetosensar™ through branching points on the linker. This can greatly reduce plasma clearance, thereby increasing exposure and in vivo efficacy [192-195].
By modifying the 0-glucuronide linker to contain a dimethylethylene diamine (DMED) self- immolative spacer phenolic or alcohol containing drugs could be enzymatically released. Drug release involves enzymatic deglucuronidation, 1,6-elimination, decarboxylation, and cyclization of the DMED carbamate to the cyclic urea to liberate the free phenol or alcohol functionalised drug [196]. Similar to p-glucuronidase cleavable linkers, p-galactosidase cleavable linkers have also been described. This lysosomal enzyme is also over expressed in certain tumour types and is analogous to p-glucuronidase in its hydrolytic activity but instead hydrolyses p-galactoside [197].
Other lysosomal enzymes that have been exploited as enzymatic triggers to release payloads in cancer cells include lysosomal acid pyrophosphatase and acid phosphatase, these enzymes hydrolyse pyrophosphates and terminal mono-phosphates respectively to their parent alcohols. A pyrophosphate containing linker has been described for use with alcohol-linked glucocorticoid payloads [198].
Sulfatases are another group of lysosomal enzymes which have high activity in the lysosome and low activity in human and rodent plasma. A number of different sulfatases reside in the lysosome, catalysing the hydrolysis of alkylsulfate esters to alcohols but they also display arylsulfatase activity. Arylsulfate linker motifs have been designed that after
hydrolysis, a alkoxybenzyl carbamate is revealed which is primed for spontaneous 1,6- elimination of an amine containing payload [199].
Non-cleavable Linkers
In some embodiments, the linker for use in the present invention is a non-cleavable linker. Unlike cleavable linkers, non-cleavable linkers do not have a triggerable moiety which ultimately separates the active drug from the antibody through a specific cleavage mechanism but instead release an active metabolite upon complete digestion of the antibody component after internalisation and lysosomal processing
With a non-cleavable linker, the linker, along with the amino acid residue of the antibody to which the linker is attached, remains connected to the payload, and this whole construct becomes the active metabolite. Since the linker stays attached to the drug after cellular processing, it must be linked at a position which does not interfere significantly with binding of the drug to its target (activity) [200].
Bioconjugation
In some embodiments, the payload is directly conjugated to the antibody or antigenbinding fragment thereof at an amino acid. In some embodiments, the direct coupling is via a lysine or cysteine residue on the surface of the antibody or antigen-binding fragment thereof. In some preferred embodiments, the direct coupling is via a lysine residue on the surface of the antibody or antigen-binding fragment thereof.
In some embodiments, the payload is indirectly conjugated to the antibody or antigenbinding fragment thereof at an amino acid, which may be a lysine or cysteine reside. In some embodiments, the indirect coupling is via a thiol or maleimide.
There are many ways to conjugate cytotoxic drugs to antibodies and antibody fragments [76,201]. This is summarised in Table 2. Lysine residues are favourable for conjugation because they can be present multiply on the surface of antibodies without causing detrimental effects such as unwanted cross-linking and due to the nucleophilicity of the e- amino side chain (see also WO2016/046574 for example). For example, conjugation onto lysines can be direct (Table 2), using drugs or drug-linkers that possess and N- hydroxysuccinimide esters (and their more soluble 3-sulfonated analogues), a tetrafluorophenyl ester (and its sulphonated analogue) pentafluorophenyl ester, an isothiocyanate, a p- lactam reactive group or a mixed anhydride. Other lesser known/used direct lysine conjugations involve sulfonyl halides, acylfluorides, iminoboronates, diazonium salts, aldehydes and sulphonylacrylates [202]. Indirect methods for lysine conjugation include
derivatising the amino group with a bifunctional linker (such as those available from Pierce Chemicals (Thermo) and Quanta Bioscience) to generate a secondary reactive group, such as 2-iminothiolane to generate a reactive thiol for conjugating to drugs or drug-linkers with thiol or maleimide reactive groups. Some lysine residues may be particularly prone to conjugation owing to enhanced nucleophilicity due to the microenvironment around that residue [203]. This allows for more site-selective modifications of lysine residues via a two-step methodology such as via a phospha-Mannich reaction whereby the lysine amine first undergoes imine formation with an aldehyde reagent, followed by attack of the imine with a nucleophilic triethylphosphite reagent to generate a stable link. Further functionalisation can then be carried out using a hydroxylamine terminated payload [204]. Reactive lysines can also be modified with methylsulfone phenyloxadiazole (MS-PODA) modified payloads. Further conjugation methods are known such as native chemical ligation [205], site specific conjugation including using enzymes [206]. Enzymes can provide exquisite specificity for bioconjugation, and one example of a lysine-targeting enzyme is sortase A from Staphylococcus aureus, which catalyses the covalent ligation between two amino acids via an isopeptide bond. Sortase A recognises the primary acid sequence LPXTG - it first cleaves the amide bond between threonine and glycine, undergoing a thioesterification with its active site cysteine and the carbonyl group of threonine. This thioester can be substituted by an amino group to form a stable amide bond. It is possible to use this activity to achieve site-specific modification of lysine residues in a wide range of proteins. Another enzyme that can be used for bioconjugation is microbial transglutaminase (MTG), which catalyses the formation of amide bonds between the y-carboxyamide group of glutamine and the e-amine group of lysine. This occurs by initial thioesterification, by attack of the active site cysteine on the y- carboxyamide and loss of ammonia; and finally amide formation by reaction with the primary amine of the lysine. MTG is a cheap, readily available enzyme that functions under a diverse range of different temperatures, salt concentrations, and pH. Using this approach that one can utilise MTG into targeting lysine residues as acyl acceptors on a antibodies and proteins. Another reported chemoenzymatic method for lysine acylation is called 'lysine acylation using conjugation enzymes' (LACE). This approach uses a small ubiquitin- like modifier (SUMO)-conjugating Ubc9 enzyme, which can identify an IKXE sequence on proteins, alongside a functionalised thioester as an acyl donor. This strategy enables the site-selective modification of the lysine residue within the IKXE tag. Another approach one can use, involves transforming the E-amino sidechain of lysine into a biorthogonal moiety either directly by transforming the e-amino group into an azide or by a two-step approach involving a bis-functional cross-linker carrying a lysine reactive group and a biorthogonal reactive moiety. These bifunctional linkers will be well known to the skilled person.
Cysteine residues also offer a particularly attractive target for protein bioconjugation due to the exceptionally high nucleophilicity of the deprotonated thiolate side chain at physiological pH and the ability via mutagenesis to insert cysteine residues. The go-to method for cysteine modification usually remains the use of maleimides. Cysteine modification occurs most commonly by 1,4-conjugate addition to N-substituted maleimides. Maleimides are particularly attractive reagents due to their synthetic accessibility and rapid reaction rates with cysteine under mild conditions Maleimide reagents allow extraordinary fast labelling with acceptable cysteine selectivity. Furthermore, a variety of maleimide derivatives, including dyes, affinity probes and crosslinkers, are commercially available, allowing easy access without the need of synthetic expertise. However, maleimides suffer from stability issues, thus rendering conjugation products unstable under certain conditions. Although maleimides are the most common cysteine-selective reagents, a variety of novel linkers have been used to functionalise cysteines, avoiding the instability issues of traditional maleimides, for example, iodoacetamides bromomaleimides, carbonylacrylic reagents and N-alkyl vinylpyridine salts [206,207].
Recent conjugation methods include the use of methylsulphonylphenyloxadiazole reactive linkers to form thioethers [208] and disulphide bridging technologies.
In recent years, genetic code expansion (GCE) techniques have enabled the incorporation of non-canonical amino acids (ncAAs) into antibody sequences, resulting in an efficient approach to the site-specific modification of antibodies through bioorthogonal bioconjugation. The most common bioorthogonal approach for bioconjugation involves 'click' chemistry. For various purposes, there are several types of reactions under 'click' chemistry including strain-promoted azido-alkyne cycloaddition (SPAAC) Cu-catalyzed azido-alkyne cycloaddition (CuAAC), Staudinger ligation, cycloaddition reactions between a tetrazine and a trans-alkene or a strained alkyne (leDDA), reactions between an isocyanopropyl group and tetrazine and Diels-Alder reactions between a cyclopentadienone and a strained alkyne [209]. Other biorthogonal chemistries involve oxime-ligation onto para-acetyl-phenylalanine (ncAA).
Other bioconjugation strategies involve tyrosine selective labelling via the use of a tyrosine-click reaction [210]. Site-selective histidine modification which poses a challenge due to competition from other more nucleophilic residues such as lysine or cysteine has been developed involving a "chemical linchpin". This linchpin is a bifunctional reagent containing both aldehyde and epoxide reactive groups. First, all available lysine residues were transiently protected via reaction with the aldehyde moiety. Next, proximal histidine
residues react with the pendant epoxide to afford irreversible modification. Finally, reformation of the aldehyde enables modification of this installed bioorthogonal handle via oxime formation. Selective modification of arginine residues via condensation of its guanidine side chain with a 4-azidophenyl glyoxal (APG) reagent enables the direct introduction of a bioorthogonal azido group onto the antibody, which can subsequently be modified via SPAAC reaction to conjugate various payloads [211].
In some embodiments, the payload is conjugated to the antibody or antigen-binding fragment thereof by direct conjugation of payloads bearing an N- hydroxy-succinimide ester to multiple lysine residues. In some other embodiments, the payload is conjugated to the antibody or antigen-binding fragment thereof by indirect conjugation to lysine residues wherein the cross-linker SMCC is used to modify surface lysine residues, generating a reducible thiol for conjugating to drugs or drug-linkers bearing a thiol or maleimide group. Mixtures of drugs with the same reactive group can be used in the chemical conjugation reaction to generate conjugates with more than one cytotoxic therapeutic drug type or a combination of therapeutic drug and diagnostic agent such as a fluorescent dye [212]. Such conjugates could potentially be useful for overcoming drug resistance or allowing combined imaging and treatment (theranostic).
Therefore, in some embodiments, the antibody or antigen-binding fragment thereof is conjugated to multiple types of payloads. For example, the same antibody or antigenbinding fragment may be conjugated to two types of payloads, three types of payloads, four types of payloads, five types of payloads, or six types of payloads.



Combination therapies
Combination therapy is important for maximizing patient outcomes by combining different mechanisms of drug action to obtain a larger therapeutic effect without significantly increasing adverse effects and thus widening the therapeutic window. Combining the action of trastuzumab antibody with chemotherapy lapatinib is well known in the art and accepted clinical practice for breast cancer (https ://www.esmo.orq/quidelines/quidelines- bv-topic/breast-cancer). Immuno-oncology drugs, notably checkpoint inhibitors (CPI) such as anti-CTLA4 (ipilimumab), anti-PDl (nivolumab, pembrolizumab) and anti-PDLl (atezolizumab) have transformed the landscape of oncology therapy for diseases such as melanoma, lung and renal cancer by reversing tumour-mediated immune suppression [172]. However, not all patients benefit from durable responses and immune-oncology drugs are being increasingly used in combination. CPI drugs combined with standard-of- care cytotoxic chemotherapy can lead to increased toxicity, therefore CPI drugs combined with targeted chemotherapy (i.e., antibody drug conjugates) have gained much interest [213]. ADCs are known to kill tumours by a variety of mechanisms in addition to the concept of delivering a damaging payload to a targeted cell. One key mechanism the 'immunogenic cell death' where the payload-mediated cytotoxicity causes tumour cells to release immunogenic signals that elicit a separate adaptive and innate immune response.
Damage-associate molecular patterns (DAMPs) released by tumour cells dying due to ADC killing include signals such as heat-shock proteins, calreticulin, ATP and high mobility group-1 (HMGB1) proteins which stimulated immune-mediated clearance. Less than 10% of chemotherapy drugs are ICD-inducing, but payloads such as auristatins and anthracyclins stimulate immune cells in vitro and in vivo and can enhance the effect of CPIs. Tubulin destabilizing payloads such as maytanines can directly activate and stimulate the maturation of dendritic cells, thus enhancing the immune system. Taken together, there is a rationale for killing tumours with an ADC which enhances immunity which is
further enhanced by 10 drugs. This has been demonstrated in preclinical animal models [214]. Immunoglobulin-based ADCs often retain inherent immune-activating functions due to the presence of an IgGl Fc-domain. This can elicit toxicities and damage the specific cells that are required for immune function. For example, Fc-interaction with megakaryocytes Fc-receptors is one reason why ADCs cause dose-limiting thrombocytopenia [15,64,213]. Therefore, many developers have chosen to use immunologically-silent Fc domain (e.g. IgG4) to reduce inadvertent immune cell damage. Antibody fragments such as single-chain Fvs lack an Fc-domain and are not expected to cause Fc/Fc-gamma-receptor mediated toxicities. There is therefore a rationale for combining cMET FDCs in this invention with IO/CPI drugs which may elicit a superior combination effect compared to ADCs due to the lack of an Fc-domain.
Clinical trials in breast, urothelial, lung and ovarian cancer are ongoing with promising results in terms of early responses and safety profile. Cancers which do not respond well to 10 drugs, the so-called immunologically cold cancers (low tumour mutational burden such as ovarian or prostate cancer [215]), could benefit more significantly from an ADC- mediated stimulation of the immune system.
Therefore, in some embodiments, the antibodies, antigen-binding fragments and ADCs disclosed herein may be combined in a formulation with one or more of the following: a chemotherapeutic agent, an immune-oncology drug, a monoclonal antibody, or a checkpoint inhibitor.
Combination with other therapeutic modalities such as CART-cells, gene therapy, other forms of chemotherapy, other forms of monoclonal antibody therapy are also possible with the antibodies, antigen-binding fragments and ADCs disclosed herein.
Another aspect of the invention provides a pharmaceutical composition comprising the antibody or antigen-binding fragment or ADC disclosed herein and a pharmaceutically- acceptable carrier, excipient or diluent.
Another aspect of the invention provides an antibody or antigen-binding fragment or ADC or pharmaceutical composition as defined herein for use in medicine.
Another aspect of the invention provides an antibody or antigen-binding fragment or pharmaceutical composition or ADC as defined herein for use in the diagnosis, treatment and/or prevention of a disease. In some embodiments, the disease is selected from cancer, benign tumours, infectious diseases including bacterial, viral, fungal,
trypanosome, nematode and prion infections, cardiovascular disease, autoimmune disease.
Another aspect of the invention provides an antibody or antigen-binding fragment or ADC or pharmaceutical composition as defined herein for use in the manufacture of a medicament for the treatment and/or prevention of a disease selected from cancer, benign tumours, infectious diseases including bacterial, viral, fungal, trypanosome, nematode and prion infections, cardiovascular disease, and autoimmune disease.
In some preferred embodiments, the disease is cancer. In some preferred embodiments, the disease is idiopathic pulmonary fibrosis (IPF).
In some embodiments, the disease is cancer of the colon, lung, breast, head/neck, prostate, skin, stomach/gastrointestinal, oesophageal, bladder, glioma, renal, ovarian, thyroid and bone. In some preferred embodiments, the disease is a gastric cancer or gastric tumour.
Another aspect of the invention provides a process of making a compound (i.e. an ADC or FDC) as defined herein comprising the steps of:
(i) providing a payload as defined herein;
(ii) providing the antibody or antigen-binding fragment as defined herein;
(iii) conjugating the payload and the antibody or antigen binding fragment thereof.
In some embodiments, the conjugation takes place in the presence of at least one polar aprotic solvent and an aqueous buffer. In some embodiments, the conjugation is via a linker between the payload and the antibody or antigen-binding fragment. The linker can be any linker as described herein.
In some embodiments, the process comprises the step of:
(iv) combining the compound with a pharmaceutically-acceptable carrier to form a pharmaceutical composition.
References
1. Minchinton Al, Tannock IF. Nat Rev Cancer. 2006 6:583-92.
2. Lei ZN, Tian Q, Teng QX, Wurpel JND, Zeng L, Pan Y, Chen ZS. MedComm 2023;4:e265.
3. Brown JM and Giaccia AJ. Cancer Research. 1998; 58: 1408-1416.
4. Cruz E and Kayser V. Biologies: Target and Therapy. 2019; 13: 33-51
5. Fu Z, Li S, Han S, Shi C, Zhang Y. Signal Transduct Target Ther. 2022;7:93.
6. Teicher BA, Morris J. Curr Cancer Drug Targets. 2022;22(6):463-529..
7. Deonarain MP. Drug Discov. Today Technol. 2018, 30:47-53.
8. Baker JHE, Kyle AH, Reinsberg SA, et al. Clin Exp Metastasis. 2018;35:691-705
9. Thurber GM, Schmidt MM, Wittrup KD. Adv Drug Deliv Rev. 2008;60: 1421-1434.
10. Fujimori K, Covell DG, Fletcher JE, Weinstein. J Nucl Med. 1990;31 : 1191-1198
11. Deonarain MP, Yahioglu G. Expert Opin Drug Discov. 2021,16:613-624.
12. Chiu ML, Goulet DR, Teplyakov A, Gilliland GL. Antibody Structure and Function: The Basis for Engineering Therapeutics. Antibodies (Basel). 2019,8:55
13. Colombo R and Rich JR Cancer Cell 2022;40: 1255-1263.
14. Li F, Emmerton KK, Jonas M, et al. Cancer Res. 2016;76(9):2710-2719.
15. Nguyen TD, Bordeau BM, Balthasar JP. Cancers (Basel). 2023;15:713.
16. Tumey L.N. in, Innovations for Next-Generation Antibody-Drug Conjugates, Cancer Drug Discovery and Development, Springer International Publishing, 2018. ppl87 (M. Damelin, Ed)
17. Conilh L, Sadilkova L et al. J. Haemaol Oncol 2023;16:3
18. Von Arx C, De Placido P et al Cancer Treat Rev. 2023;113: 102500.
19. Petrini I. Annals Trans Med 2015, 3;82.
20. Rivas S et al. Int J Mol Sci. 2022, 23;13898
21. Raghav K et al. Cancer Treat Rev, 2018, 66;95-103.
22. Comoglio PM et al. Nat Rev cancer. 2018, 18; 341
23. Peng Z, Zhu Y, Wang Q, et al., PLoS One. 2014; 9(1).
24. Lu J and Teng L. Int. J. of Molecular Medicine. 2013; 32: 1247-1254.
25. Wu ZW, Sha Y, Chen Q et al., World Journal of Clinical Cases. 2021; 9: 3350-3355.
26. Salgia R. Mol. Can. Ther. 2017; 16(4).
27. Wu YL, Soo RA et al. Cancer Treat Rev. 2017 Dec;61:70-81.
28. Mesteri I, Schoppmann SF, et al., European Journal of Cancer. 2014; 50(7): 1354- 1360.
29. Chan E, Alkhasawneh A, et al., Journal of Gastrointestinal Oncology. 2016; 7(6): 838-847.
30. Wang H, Tan M, et al., Int. J. Molecular Sciences. 2015; 16(2): 3391-3404.
31. Yamamoto S, Tsuda Hitoshi, Miyai K. Modern Pathology. 2011; 24: 1146-1155.
32. Kim JH, Kim JB and Kim HS. Oncotarget. 2017; 8(43): 75478-75487.
33. Macher-Goeppinger S, Keith M, Endris V et al., Oncotarget. 2017; 8: 1046-1057.
34. Zanetti A, Stoppacciaro A, et al., Journal of Pathology. 1998; 186: 287-291.
35. Gymnopoulos M, Betancourt O et al. J. Mol Oncol. 2020;14:54-68.
36. Vsiansky V, Gumulec J et al. Sci Rep. 2018;8: 10370.
37. Szparecki G, Ilczuk T, et al., Folia Biologica (Praha). 2017; 63: 146-154.
38. Mukae Y, Miyata Y, Nakamura Y et al., Oncology Letters. 2020; 20(1): 135-144.
39. Jia L, Yang X, Tian W et al., Medical Science Monitor. 2018; 24: 8239-8249.
40. Melo Gagliato D, Fontes Jardim D, et al., Clinical Breast Cancer. 2014; 14(6): 468- 474.
41. Ohara M, Saito K et al. Cancers (Basel). 2021;13: 1104
42. Chen W, Wu S et al. Onco Targets Ther. 2021;14:4791-4804
43. Rath P, Lal B et al. Transl Oncol. 2013;6: 104-11
44. Luan T and Yu Y. Int J Clin Exp Med. 2014;7:5583-7.
45. Lee SJ, Lee J et al. Clin Colorectal Cancer. 2018;17: 165-169
46. Gayyed MF, Abd El-Maqsoud NM et al. J Gastrointest Oncol. 2015;6:618-27.
47. Yao HP, Tong XM, Hudson R, Wang MH. J Exp Clin Cancer Res. 2020;39: 198
48. Knudsen BS and Edlund M. Adv Cancer Res. 2004;91 :31-67
49. Immunohistochemistry in tumor diagnostics Tuffaha M, Guski H & Kristiansen G Eds), Springer Publishing 2018
50. Wang M et al. Annals Diag. Pathol 2018, 35;69-78.
51. Yao H-P et al. Drug Discovery Today, 2021, 26, 106-121.
52. Puccini A et al. Drug Safety, 2019; 42;211-233.
53. De Silva DM, Roy A, et al. Biochem Soc Trans. 2017;45:855-870.
54. Hughes VS & Siemann DW. Trends in Cancer, 2018, 4, 94-97
55. Bradley CA, Salto-Tellez M et al. Nat Rev Clin Oncol. 2017;14:562-576.
56. Vasalou C, Helmlinger G, Gomes B. PLoS One. 2015;10:e0118977
57. Wang J, Anderson MG et al. Clin Cancer Res. 2017;23:992-1000
58. Strickler JH, Weekes CD et al. J Clin Oncol. 2018;36:3298-3306.
59. Camidge DR, Barlesi F et al. J Clin Oncol. 2023 Feb 10;41(5): 1105-1115.
60. Yang C, Zhao X et al. Xenobiotica. 2019;49: 1097-1105.
61. Min B, Jin J et al. ACS Omega. 2020;5:25798-25809.
62. Groothuis PG, Jacobs DCH et al. Mol. Cancer Ther. 2023;22:765-777.
63. Comer F et al (2023). AZD9592: An EGFR-cMET bispecific antibody-drug conjugate (ADC) targeting key oncogenic drivers in non-small-cell lung cancer (NSCLC) and beyond. Cancer Research 83 (suppl) abtract 5736
64. Mahalingaiah PK, Ciurlionis R et al. Pharmacol Ther. 2019;200: 110-125.
65. Dumontet C, Reichert JM et al. Nat Rev Drug Discov. 2023;22:641-661.
66. Stella GM, Gentile A et al. J Transl Med. 2016;14:256.
67. Canestaro WJ, Forrester SH et al. Mol Cancer Ther. 2013;12:2685-96
68. Fujita H, Miyadera K et al., Mol. Cancer Ther. 2013: 12:2685-96
69. Koyama K, Goto H et al. Am J Respir Cell Mol Biol. 2019 Apr;60(4):478-487.
70. De Marco A. Microb Cell Fact. 2009, 8:26
71. Carter PJ. Nat Rev Immunol. 2006, 6: 343-57
72. Wozniak-Knopp, G et al. Protein Eng. Des. Sei. 2010, 23, 289-297
73. Worn A & Pluckthun A. J Mol Biol. 2001, 305:989-1010
74. Schaefer JV & Pluckthun A. Protein Eng Des Sei. 2012, 25:485-506
75. Winter G et al., 1991, Nature 349:293-299
76. Ducry, L (Ed) (2013), Antibody-Drug Conjugates, Methods in Molecular Biology volume
1045, Humana Press
77. Behrens CR and Liu B. MAbs. 2014, 6:46-53.
78. Kelley LA & Sternberg MJ. Nat Protoc. 2009; 4: 363-71
79. Chames, P (Ed.), Antibody Engineering: Methods and Protocols, Second Edition, Methods in Molecular Biology, vol. 907, Chapter 1
80. Thompson JD et al., Nuc. Acid Res. 1994, 22:4673-4680
81. Orlandi R et al., Proc. Natl. Acad. Sci. U.S.A. 1989, 86:3833-3837;
82. Kohler D et al., 1975. Nature 256:4950497;
83. Kozbor D et al., 1985. J. Immunol. Methods 81:31-42;
84. Cote RJ et al., Proc. Natl. Acad. Sci. USA 1983, 80:2026-2030;
85. Cole SPC et al., Mol. Cell. Biol. 1984, 62: 109-120.
86. Hurrell, J.G.R in Monoclonal Hybridoma Antibodies: Techniques and Applications", (CRC Press, 1982).
87. Harlow & Lane, 1988, Antibodies: A Laboratory Manual (Cold Spring Harbor Laboratory, New York).
88. Jones PT et al., 1986. Nature 321:522-525;
89. Riechmann L et al., 1988, Nature 332:323-329
90. Presta LG Curr. Op. Struct. Biol. 1992, 2:593-596).
91. Verhoeyen M et al., Science 1998, 239: 1534-1536
92. Hoogenboom & Winter, 1991, J. Mol. Biol. 227:381
93. Marks JD et al., J. Mol. Biol. 1991, 222:581
94. Cole et al., 1985, In: Monoclonal antibodies and Cancer Therapy, Alan R. Liss, pp. 77
95. Boerner P et al., J. Immunol. 1991, 147:86-95.
96. Thuss-Patience PC, Shah MA et al., Lancet Oncol. 2017;18:640-653.
97. Lordick F, Allum W et al., Cancer Treat Rev. 2014;40:692-700.
98. Yver A, Agatsuma T, Soria JC. Ann Oncol. 2020;31:430-434.
99. Saber H, Leighton JK. Regul Toxicol Pharmacol. 2015;71:444-52.
100. Bartelink IH, Jones EF et al., Clin Pharmacol Ther. 2019 ;106: 148-163.
101. Alberts, B et. al. in Molecular Biology of the Cell, Garland Science, Ney York, 4th edition, 2002, E. B. Garon
102. Pettit GR, Kamano Y et al., J. Am. Chem. Soc. 1987, 109(22), 6883-6885
103. E. Flahive, J. Srirangam, Anticancer Agents from Natural Products 2nd ed. CRC Press, 2011, 263-290
104. Kerbrat P, Dieras V et al., Eur. J. Cancer 2003, 39, 317-320
105. Chen H, Arnst KE, Miller DD, Li W Molecules, 2017, 22, 1281
106. Yu, T-W, H. G. Floss, G. M. Gragg and D. J. Newman. Anticancer Agents from Natural Products 2nd ed. CRC Press, 2011, 407-427
107. Zafar S, Armaghan M et al. Biomed. Pharmacother. 2023;165, 115039
108. Remillard, S, Rebhun LI et al. Science (New York, NY. 1975;189(4207): 1002-5
109. Oroudjev E, Lopus M et al. Mol. Can. Ther. 2010, 9(10), 2700-2713
110. Kupchan SM, Sneden AT et al. J. Med. Chem. 1978, 21(1), 31-37]
111. Moobery SL, Busquets L, Tien G Int. J. Cancer 1997, 73, 440-448
112. Trimurtulu G, Ohtani I, Patterson GML, Moore RE, Corbett TH, Valeriote FA, Demchik L J. Am. Chem. Soc., 1994, 116, 4729-4737
113. Smith CD, Zhang X et al., Cancer Res. 1994, 54, 3779-3784
114. Verma VA, Pillow TH et al., Bioorg. Med. Chem. Lett. 2015, 25(4), 864-868
115. Sasie F, Steinmetz H et al., J. Antibiot. 2000, 53, 879-885
116. Murray BC, Peterson MT, Fecik RA Nat. Prod. Reps. 2015, 32, 654-662
117. Ojima I, Geng X et al., J. Med. Chem. 2002, 45, 5620-5623
118. Fahy J Curr. Pharm. Design 2001, 7(13), 1181-1197
119. Laguzza BC, Nichols CL et al., J. Med. Chem. 1989, 32, 548-555
120. Hirata, S, Y, Uemura D Pure Appl. Chem. 1986, 58, 701-710
121. Cheng X, Li J et al., Mol. Cancer Ther. 2018, 17(12) 2665-2675
122. Goodin S, Kane MP, Rubin EH J. Clin. Oncol. 2004, 15(22), 15-25
123. Pommier Y ACS Chem. Biol. 2013, 8, 82-95
124. Iyer L and Ratain MJ Cancer Chemother. Pharmacol., 1998, 42, S31-S43
125. Wani MC, Nicholas AW et al., J. Med. Chem. 1987, 30, 1774-1779
126. Talukdar A, Kundu B et al., Eur. J. Med. Chem. 2022, 236, 114304
127. Tagakagi K, Dexheimer TS et al., Mol. Can. Ther. 2007, 6, 3229-3238
128. Kummar S, Chen A et al. Cancer Chemother. Pharmacol., 2016, 78, 73-81
129. Kurtzberg LS, Roth S et al. Clin. Can. Res. 2011, 17, 2777-2787
130. Nishida M, terayabashi T et al. Nat. Comm. Biology, 2022, 5, 982-995
131. Liang Z, Lei F et al. J. Med. Cem. 2022, 228, 113960
132. M. Broggini Top. Curr. Chem. 208, 283, 191
133. Watt PM and Hickson ID. Biochem. J. 1994, 303, 681-695
134. Broggini M, Anthracycline Chemistry II, Topics in Current Chemistry 2007, vol. 283, 191-206
135. Fortune JM and Osheroff N Proc. Nucleic Acid Res. Mol. Biol. 2000, 64, 221-253
136. Swift LP, Rephali A et al., Cancer Res. 2006, 66, 4863-4871
137. Tacar, O et. al. J. Pharm. Pharmacol. 2013, 61(8), 933-938
138. Swedan HK, Kassab AE et al., Bioorganic Chemistry 2023, 136, 106548
139. Yu SF, Zheng B et al., Clin. Cancer Res. 2015, 21, 3298-306
140. Geroni C, Ripamonti M et al. Cancer Res. 2001, 61, 1983-1990
141. Yu SF, Zheng B et al., Clin. Cancer Res. 2015, 21(14), 3298-306
142. Hanka LJ, Dietz A et al., J. Antibiot. 1978, 31, 1211-1217
143. Yao HP, Zhao H, Hudson R et al., Drug Disc. Today 2021, 26(8), 1857-1874
144. Beekman, A.M. et al. Cytotoxic Payloads for Antibody-Drug Conjugates, Drug Discovery Series no. 71, Ed. D. E. Thurston and P. M. Jackson, 2019, RSC, 187-208
145. Procopiou, G. et al. Cytotoxic Payloads for Antibody-Drug Conjugates, Drug Discovery Series no. 71, Ed. D. E. Thurston and P. M. Jackson, 2019, RSC, 209-240.
146. Mantaj J, Jackson PJM, Rahman KM, Thurston DE. Angew. Chem. Int. Ed. Engl. 2017, 56(2), 462-488
147. Hartley JA Expert Opin. Biol. Ther. 2021, 21(7), 931-943
148. Pillow, T.H and Tercel, M. Cytotoxic Payloads for Antibody-Drug Conjugates, Drug Discovery Series no. 71, Ed. D. E. Thurston and P. M. Jackson, 2019, RSC, 241-258
149. Pyysz, I, et. al. Cytotoxic Payloads for Antibody-Drug Conjugates, Drug Discovery Series no. 71, Ed. D. E. Thurston and P. M. Jackson, 2019, RSC, 137-165
150. Yang Q, Deng Z et al., J. Am. Chem. Soc. 2020, 142, 2532-2540
151. Denny WA Expt. Opin. Ther. Patents 2000, 10(4), 459-474
152. Nicolaou KC Smith AL, Yue EW PNAS 1993, 90(13), 5881-5888
153. Adhikari A, Shen B, Rader C Antibody Therapeutics, 2021, 4(1), 1-15
154. Walker S, Landowitz R, Ding WD PNAS 1992, 89(10), 4608-4612
155. Dushin, R.G. Cytotoxic Payloads for Antibody-Drug Conjugates, Drug Discovery Series no. 71, Ed. D. E. Thurston and P. M. Jackson, 2019, RSC, 259-278
156. Nicolaou KC, Rigol S et al., PNAS 2021, 118(25) e2107042118
157. Venugopal S, Sharma Vet al., Chem. Biol. Drug Des. 2022, 100, 580-598.
158. Robinson DR, Wu YM, Lin SF Oncogene. 2000, 19(49), 5548-57
159. Drake JM, Lee JK, Witte ON Mol. Cell Biol. 2014, 34(10), 1722-32
160. King T, Pardo A Lancet 2011, 378, 1949-1961
161. Wind S, Schmid U et al., Clin. Pharmacokinetics. 2019, 58(9), 1131-1147
162. Bugatti K, Andreucci E et al., ACS Omega 2022, 7, 17658-17669
163. Wang RE, Liu T et al., J. Am. Chem. Soc. 2015, 137, 3229-32
164. Lindell TJ, Weinberg F et al., Science 1970, 170, 447-9
165. Pahl, A. et al. Cytotoxic Payloads for Antibody-Drug Conjugates, Drug Discovery Series no. 71, Ed. D. E. Thurston and P. M. Jackson, 2019, RSC, 398-426
166. Noel P. Trends Pharmacol. Sci. 2019, 40(5), 327-341
167. Subramanyam, C. Cytotoxic Payloads for Antibody-Drug Conjugates, Drug Discovery Series no. 71, Ed. D. E. Thurston and P. M. Jackson, 2019, RSC, 364-379
168. Garcia-Saez I and Skoufias DA Biochem. Pharmacol. 2021, 184, 114364
169. Wei Y, Xiang H, Zhang W Front. Oncol. 2022, 13, 970553
170. Kale J, Osterlund EJ, Andrews DW. Cell Death Differ. 2018, 25, 65-80.
171. Tanaka Y, Aikawa K et al., J. Med. Chem. 2013, 56, 9625-9645
172. Robert C. Nat Commun. 2020;ll(l):3801.
173. Demaria O, Cornen S et al., Nature 2019, 574, 45-56
174. McGowan DC Curr. Top. Med. Chem. 2019, 2228-2238
175. Motedayen L, Pease JE et al., J. Med. Chem. 2023, 66(15), 10715-10733
176. Duwall JR, Thomas JD, Bukhalid RA, et al J. Med. Chem. 2023, 66(15), 10715-10733
177. Dragovich PS Chem. Soc. Rev., 2022,51, 3886-3897
178. Hobson A. The Medicinal Chemistry of Glucocorticoid Receptor Modulators, Springer Briefs in Molecular Science, 2023, Springer, 59-97
179. Hobson AD, McPherson MJ et al. J. Med. Chem. 2022, 65, 4500-4533, Hobson AD, McPherson MJ et al. J. Med. Chem. 2022, 65, 15893-15934]
180. Kovalev IS, Zyryanov GV et al. molecules, 2022, 27(19), 6229
181. Hruba L, Das V, Hajduch M, Dzubak P. Biochem. Pharmacol. 2023, 115741
182. Elgemeie GH. Curr. Pharm. Des. 2003, 9(31), 2627-2642
183. Wang H, Xu X, Wang J, Qiao Y Ann. Med. 2023, 55(1), 1422-1430
184. Bargh JD, Isidro-Lobet A et al. Chem. Soc. Rev. 2019, 48, 4361-4374
185. Johannes, S. et al., Chemical Linkers in Antibody-Drug Conjugates (ADCs), Drug
Discovery Series no. 81, Ed. F.L van Delft and J. M. Lambert 2022, RSC, 173-212
186. Lyski RD, Bou LB et al., Mol. Can. Then 2021, 20(2), 329-339
187. Edupuganti VVSR, Tyndall JDA et al., Rec. Pat Anticancer Drug Disc. 2021, 16, 479- 497
188. deGraaf M, Boven E et al. Curr. Pharm. Design 2002, 8(15), 1391-1403
189. Jeffrey SC, Nguyen MT et al., Bioorg. Med. Chem. Lett. 2007, 17(8), 2278-2280
190. Anselmi M, Borbely A et al. Chem. Eur. J. 2021, 27(3), 1015-1022
191. Shin SH, Park YH et al. Adv. Sci. 2021, 8(23), 2102414
192. Lyon RP, Bovee TD et al. Nat. Bio. 2015, 33(7), 733-735
193. Burke PJ, Hamilton JZ et al. Mol. Can. Ther. 2017, 16(1), 116-123
194. Viricel W, Fournet G et al., Chem. Sci. 2019, 14(10), 4048-4053
195. Evans, N. et al. Front. Pharmacol. 2022, 13. 764540
196. Jeffrey SC, Brabender JD et al., ACS Med. Chem. Lett. 2010, 1(6), 277-280
197. Tancy-Opalinski I, Legigan T et al. Eur. J. Med. Chem. 2014, 74, 302-313
198. Kern JC, Cancilla M et al. J. Am. Chem. Soc. 2016, 138, 1430-1445
199. Bargh JD, Walsh SJ et al. Chem. Sci. 2020, 11(19), 2375-2380].
200. Dugal-Tessier, J. et al. Chemical Linkers in Antibody-Drug Conjugates (ADCs), Drug Discovery Series no. 81, Ed. F.L van Delft and J. M. Lambert 2022, RSC, 136-172.
201. Hermanson, G. (2013) Bioconjugate Techniques, Chapters 2-6, Academic Press
202. Haque M, Forte N, Baker JR. Chem. Comm. 2021, 57, 10689-10702
203. Doppalapudi VR, Huang J et al. Proc. Natl. Acad. Sci. U S A 2010, 107(52), 22611-6
204. Chilamari M, Kalra N, Shukla S, Rai V. Chem. Comm. 2018, 54, 7302-7305
205. Hackenberger CP and Schwarzer D. Angew Chem Int Ed Engl. 2008;47(52) 10030- 74.
206. Chalker JM, Bernardes GJL et al. Chem. Asian J. 2009, 4(5), 630-640
207. Ochtrop P and Hackenberger CP. Curr. Opin. Biol. Chem. 2020, 58, 28-36.
208. Patterson JT, Asano S et al. Bioconj. Chem, 2014, 25, 1402-1407]
209. Kondengadan SM, Bansal S et al. Acta Pharmaceutica Sinica B 2023, 13(5), 1990- 2016
210. Hitoshi B, Nagano M et al. Bioconjugate Chemistry 2013, 24, 520-532
211. Walsh SJ, Bargh JD et al. Chem. Soc. Rev. 2021, 50, 1305-1353].
212. Fernandez-Fernandez A, Manchanda R et al., Appl. Biochem Biotechnol. 2011, 165, 1628-51
213. Nicolo E, Giugliano F et al. Cancer Treat Rev. 2022;106: 102395
214. Muller P, Kreuzaler M et al. Sci Transl Med. 2015;7(315):315ral88.
215. Castle JC, Uduman M, Front Immunol. 2019;10: 1856.
216. Antibody Engineering, Edited by Benny Lo, chapter 8, pl61-176, 2004
217. Phage display, Edited by Tim Clackson and Henry B. Lowman, chapter 13, p243-288, 2004
218. Antibody Engineering, Edited by Benny Lo, chapter 19, p327-343, 200
219. Main et al., J Pharmacol Exp Then, 319(3), pl395-404, 2006
220. Cadwell RC & Joyce GF, PCR Methods, Appl 2, p28- 33, 1992
221. Carter, P., Biochem J, 237, pl-7, 1986
222. Sarker, G & Sommer, SS., Biotechniques, 8(4), p404-407, 1990
223. Jeffrey SC, Andreyka JB et al. Bioconj. Chem, 2006,17(3), 831-840
224. Li Y, Gong L et al. Chemm. Comm. 2022, 58, 2568-2571.
225. Hamilton JZ, Pires TA et al. ChemMedChem, 2021, 16(7), 1077-1081
226. Zhang K, Ma Y et al. Molecular Therapy: Oncolytics, 2020, 18, 304-316
227. Quintana JM, Arboleda D et al. Bioconj. Chem. 2022, 33(8), 1474-1484
228. Li W, Veale KH, Qiu Q et al. ACS Med. Chem. Lett. 2019, 10(10, 1386-1392
229. Meyer Y, Richard JA et al. Org. Biomol. Chem. 2010,8, 1777-1780
230. Zhu Q, Yu X et al. Bioorg. Med. Chem. 2018, 26(16), 4706-4715
231. Chan KF, Wong ILK et al. J. Med. Chem. 2012, 55(5), 1999-2014
232. Agren JKM, Billing JF et al. Synthesis 2006, 3141-3145
233. Liao-Chan S, Daine-Matsuoka B et al. PLoS One. 2015;10(4):e0124708.
234. Wang J et al, BMC Cancer 2016; 16: 105
235. Park H et al (2016) Lab Anim Res 32:79-86, Reference values of clinical pathology parameters in cynomolgus monkeys Macaca fascicularis) used in preclinical studies.
236. Nair AB, Jacob S. (2016). A simple practice guide for dose conversion between animals and human. J Basic Clin Pharm. 7:27-31
237. Deng R et al. (2011) Projecting human pharmacokinetics of therapeutic antibodies from nonclinical data: what have we learned? Mabs. 3:61-6
238. Mocquot P et al. (2022). The pharmacology of blinatumomab: state of the art on pharmacodynamics, pharmacokinetics, adverse drug reactions and evaluation in clinical trials. J Clin Pharm Ther. 47: 1337-1351.
Figure Legends
Figure 1
Binding of scFv 3A2 (left) and 26B5 (right) to cMET and unrelated antigens in phage ELISA
Figure 2a
Binding affinities for 3A2 and affinity optimised variants for human cMET as measured by BIAcore. KDs are shown.
Figure 2b
Binding affinities for 26B5 and affinity optimised variants for human cMET as measured by BIAcore. KDs are shown.
Figure 2c
Binding affinities for (A) 129D5B and (B) 131D5s for human, cynomolgus and rat cMET as measured by BIAcore. KDs are shown.
Figure 3
ELISA of (A) 129D5B and (B) 131D5S, demonstrating specificity of both scFvs for cMET against unrelated antigens.
Figure 4
Epitope binning of 129D5B and 131D5S by BIAcore. (A) 129D5B binding to human cMET blocks further binding by (B) 129D5B as they target the same epitope. However, (C) 131D5S is not blocked by 129D5B indicating it is binding to a different epitope of human cMET
Figure 5
Inhibition of HGF binding to human cMET by scFvs 129D5B and 131D5S by ELISA. Clone 131D5S, but not 129D5B, inhibits HGF ligand binding.
Figure 6
(A) SEC traces of 129D5B (A) and 131D5S (B) showing predominantly monomeric scFv;
(C) Duplicate DLS traces for 129D5B (left) and 131D5S (right) showing a uniform and monomeric particle size (2.8-3nm) for both scFvs with no larger particles or aggregation.
Figure 7a
129D5B scFv is highly thermostable. 129D5B scFv melting curve determined by fluorescence change at 330 nm (top, Tm = 76.9°C), and onset of thermal aggregation determined by DLS (bottom, T onset = 62°C).
Figure 7b
131D5S scFv is highly thermostable. 131D5S melting curve determined by fluorescence change at 330 nm (top, Tm = 68.7°C), and onset of thermal aggregation determined by DLS (bottom, T onset = 62°C).
Figure 8
Amino acid sequence of cMET scFv 77F3 (a) VH and (b) VL showing positions of lysines for randomisation (amino acids 13, 23, 74 and 239). Underneath each lysine position are listed the amino acid alternatives that were built into the 77F3 mutant library.
Figure 9
Binding affinities and kinetics of 77F3 and lysine variants 139B2, 139D2 and 139D7 for human cMET as measured by BIAcore. KDs are shown.
Figure 10
(A) Binding affinities of 129D5B and 129D5BTY for human cMET, and (B) Binding specificity of scFv 129D5B and 129D5BTY to cMET and unrelated antigens, demonstrating that 129D5BTY and 129D5B are equivalent cMET scFv clones. KDs are shown.
Figure 11
Biophysical properties of 129D5BTY: (A) SEC trace showing predominantly monomeric scFv; (B) DLS trace showing a uniform particle size with no aggregation; (C) melting curve showing that 129D5BTY scFv is highly thermostable with a Tm of 74°C.
Figure 12
ELISA of parent 129D5BTY scFv and a range of mutations at position VH98 some of which reduce or abolish binding to human cMET and all which abolish binding to mouse cMET demonstrating the importance of Arginine-98.
Figure 13
BIACore binding analyses of selected 129D5BTY mutants at position VH98 on (A) human and (B) mouse cMET demonstrating the importance of Arginine-98.
Figure 14
Characterization of 129D5BTY-MMAE-pGA-brPEG8-PEG2 FDC. (A) AKTA-Superdex-75 purification traces showing a monomeric FDC which was pooled and analysed; (B) SEC- HPLC analyses demonstrating >98% monomeric FDC; (C) SEC-LCMS showing DAR distribution and an average DAR of 5.9; (D) DLS analyses showing uniform and monomeric particle size of this FDC (~3.4nM) with little signs of larger species and aggregates; (E) NanoDSF between room temperature and 85°C showing the onset of unfolding and Tm of unfolding (63.1°C); (F) BIAcore binding analyses measuring high affinity of 2.16nM and associated kinetic constants.
Figure 15
Characterization of 131D5S-MMAE-pGA-brPEG8-PEG2 FDC. (A) SEC-HPLC analyses demonstrating >88% monomeric FDC; (B) SEC-LCMS showing DAR distribution and an average DAR of 5.4; (C) DLS analyses showing uniform and monomeric particle size of this FDC (~3.8-4nM) with little signs of larger species and aggregates; (D) BIAcore binding analyses measuring high affinity of 112pM and associated kinetic constants.
Figure 16
Characterization of 77F3-MMAE-pGA-brPEG4-PEG2 FDC. (A) SEC-HPLC analyses demonstrating >98% monomeric FDC; (B) SEC-LCMS showing DAR distribution and an average DAR of 8.4.
Figure 17
Characterization of 129D5BTY-MMAE-pGA-brPEGi2-PEG4 FDC. (A) AKTA-Superdex-75 purification traces showing a monomeric FDC which was pooled and analysed; (B) SEC- HPLC analyses demonstrating >98% monomeric FDC; (C) SEC-LCMS showing DAR distribution and an average DAR of 6.4; (D) DLS analyses showing uniform and monomeric particle size of this FDC (~3.4nM) compared with the scFv (3nm) with little signs of larger species and aggregates; (E) NanoDSF between room temperature and 85°C showing the
onset of unfolding and Tm of unfolding (64.9°C); (F) BIAcore binding analyses measuring high affinity of 1.87nM and associated kinetic constants.
Figure 18
Characterization of 129D5B-MMAE-|3GA-brPEGi2-PEG7 FDC. (A) SEC-LCMS showing DAR distribution and an average DAR of 7.5; (B) NanoDSF between room temperature and 85°C showing the onset of unfolding and Tm of unfolding (63.2°C); (C) BIAcore binding analyses measuring high affinity of 4.9nM and associated kinetic constants.
Figure 19
Characterization of 129D5B-MMAE-pGA-brPEG8-PEG2 FDC (A) SEC-HPLC analyses demonstrating >98% monomeric FDC; (B) HIC chromatography showing the resolution of the various DAR species; (C) SEC-LCMS showing DAR distribution and an average DAR of 5.8; (D) NanoDSF between room temperature and 85°C showing the onset of unfolding and Tm of unfolding (64.2°C); (E) BIAcore binding analyses measuring high affinity of 1.7nM and associated kinetic constants.
Figure 20
Characterization of 129D5BTY- MMAE-pGA-brLys(BisPEG8)-PEG2 FDC (A) SEC-HPLC analyses demonstrating >98% monomeric FDC; (B) SEC-LCMS showing DAR distribution and an average DAR of 5.9; (C) DLS analyses showing uniform and monomeric particle size of this FDC (~3.3nM) with little signs of larger species and aggregates; (D) NanoDSF between room temperature and 85°C showing the onset of unfolding and Tm of unfolding (64°C); (E) BIAcore binding analyses measuring high affinity of 1.4nM and associated kinetic constants.
Figure 21
(A) Characterization of 129D5B-DXd-GGFG-PEG7 FDC by SEC-LCMS showing DAR distribution and an average DAR of 7.6; (B) Characterization of 129D5B-Exatecan-[3GA- PEG? FDC by LC-MS showing DAR distribution and an average DAR of 7.9 and a BIACore binding affinity of 254pM (C).
Figure 22
(A) Characterization of 131D5S-DXd-GGFG-brPEG8-PEG2 FDC by SEC-LCMS showing DAR distribution and an average DAR of 9.3; (B) Characterization of 129D5B-N-MeGlucamine- GGFG-PEG2 FDC by LC-MS showing DAR distribution and an average DAR of 4.3
Figure 23
(A) Characterization of 129D5B-Belotecan-[3GA-PEG2 FDC by SEC-LCMS showing DAR distribution and an average DAR of 5.9 and (B) average DAR of 13; (C) SEC-HPLC analyses demonstrating >98% monomeric FDC of the DAR 13 FDC; (D) Characterization of 129D5B- Triptolide-PEGe FDC by SEC-LCMS showing DAR distribution and an average DAR of 5.0.
Figure 24
(A) Characterization of -Benzylamino Imidazoquinoline-PEG? FDC by SEC-LCMS showing DAR distribution and an average DAR of 1.35; (B) Characterization of 129D5B-Nintedanib o-Galactose-PEG? FDC by SEC-LCMS showing DAR distribution and an average DAR of 1.9.
Figure 25
(A) Characterization of 129D5B-GENZ-644282-Val-Ala-brPEGi2-PEG4 FDC by SEC-LCMS showing DAR distribution and an average DAR of 2.4; (B) Characterization of 129D5B- Dexamethasone-dipyrophosphate-PEG2 FDC by SEC-LCMS showing DAR distribution and an average DAR of 3.74; (C) BIAcore binding analyses of 129D5B-Dexamethasone- dipyrophosphate-PEG2 FDC measuring high affinity of 298pM and associated kinetic constants.
Figure 26
(A) Characterization of 129D5B Fab by BIAcore binding analyses measuring high affinity of 12.4pM and associated kinetic constants; (B) Characterization of 129D5B Fab-MMAE- PGA-brPEGs-PEG2 FDC by BIAcore binding analyses measuring high affinity of 41pM and associated kinetic constants; (C) Characterization of 129D5B Fab-mc-Val-Cit-MMAE FDC by BIAcore binding analyses measuring high affinity of 35pM and associated kinetic constants.
Figure 7
(A) Characterization of 129D5B Fab-MMAE-pGA-brPEG8-PEG2 FDC by SEC-LCMS showing DAR distribution and an average DAR of 5.9; (A) Characterization of 129D5B Fab-mc-Val- Cit-MMAE FDC by SEC-LCMS showing DAR distribution and an average DAR of 2.0;
(C) Characterization of 129D5B Fab-MMAE-pGA-brPEG8-PEG2 FDC by SEC-HPLC analyses demonstrating >98% monomeric FDC.
Figure 28
(A) Characterization of 107-A01-MMAF-PEG2 FDC by RP-LCMS showing DAR distribution and an average DAR of 6. The FDC species were separated by LC and individual peaks analysed by mass spectrometry with 2 examples shown (B 8<. C). The 107-A01-MMAF-PEG2 FDC demonstrated >10% aggregation as analysed by SEC-HPLC (D). The 20F6-MMAF-
PEG2 conjugated under similar conditions and analysed by LC-MS showed an average DAR of 6.95 and < 1% aggregation by SEC-HPLC (F).
Figure 29
Representative flow cytometry plots showing cMET scFv clones 129D5B/129D5BTY and 131D5S when compared to known benchmark IgGs ABT-700 and 5D5. Representative histograms of antibodies binding on the cell surface of high cMET receptor-expressing cell lines Hs746T, SNU5, KATO III and AsPCl are shown. Binding on moderate to low cMET- expressing cell lines is also illustrated. (1) Histogram represents untreated cell lines; (2) Histogram shows the fluorescence intensity of the secondary antibody when used as a control or the fluorescence intensity of the cMET antibody binding on the cell surface when conjugated to a fluorophore. (3) Represents cMET antibody binding on the cell surface. Percentage binding by fluorescence shift is shown in Table 8.
Figure 30
Quantification for cMET receptors by QIFIT. (A) Fluorescence calibration using provided beads and fluorescently-labelled 5D5 IgG; (B) Calibration of high to low window; (C) Calibration curve determined for fluorescent intensity vs antibody binding capacity (receptor count); (D to I) shows the flow cytometry of the various cell lines using the 5D5 IgG compared to the calibrated beads. The mean fluorescence for each cell line was compared to the calibration curve to determine the receptor count as shown in Table 9.
Figure 31
Cell killing potency of cMET scFvs on Hs746T and T47D cells. (A) 129D5B scFv; (B) 77F3 scFv; (C) 131D5S scFv, demonstrating no killing.
Figure 32
Cell killing potency of 129D5B-MMAE-pGA-brPEG8-PEG2 FDC (DAR 5.9) on (A) Hs746T , SNU5 & KATOIII cells. (B) NUGC4, SNU16 & NCI-N87; (C) SKOV3, NCI-H1975 & AsPC-1 cells; (D) T47D & JIMT-1 cells. ICso values are shown in Table 10.
Figure 33
(A) Cell killing potency of 129D5BTY-MMAE-0GA-brPEG8-PEG2 FDC (DAR 6.1) on Hs746T, T47D & NCI-N87 cells. (B) Cell killing potency of 129D5BTY-MMAE-0GA-brPEG8-PEG2 FDC (DAR 7.4) on Hs746T &T47D cells; (C) Cell killing potency of 129D5B-MMAE-0GA-brPEGi2- PEG7 FDC (DAR 7.5) on Hs746T & T47D cells; (D) Cell killing potency of 129D5BTY-MMAE- PGA-brPEGi2-PEG4 FDC (DAR 6.4) on Hs746T & T47D cells. ICso values are shown in Table 10.
Figure 34
Cell killing potency of 131D5S-MMAE-pGA-brPEG8-PEG2 FDC (DAR 5.4) on (A) Hs746T, SNU5 & KATOIII cells. (B) NUGC4, SNU16 & NCI-N87 cells; (C) T47D & JIMT-1 cells; (D) SKOV3, NCI-H1975 & AsPC-1 cells. IC50 values are shown in Table 10.
Figure 35
Cell killing potency of (A) 129D5BTY-MMAE-pGA-br(bisPEG8)-PEG2 FDC (DAR 5.3) on Hs746T & T47D cells; (B) 129D5BTY-MMAE-pGA-br(bisPEG8)-PEG2 FDC (DAR 3.8) on Hs746T & T47D cells; (C) 129D5BTY-MMAF-0GA-PEG2 FDC (DAR 7.8) on Hs746T & T47D cells. IC50 values are shown in Table 10.
Figure 36
Cell killing potency of (A) 77F3-MMAE-0GA-brPEG8-PEG2 FDC (DAR 5.6) on Hs746T, & T47D cells; (B) 77F3-MMAE-0GA-brPEG4-PEG2 FDC (DAR 8.1) on Hs746T, & T47D cells; (C) (A) 82H8-MMAE-pGA-brPEG4-PEG2 FDC (DAR 6.5) on Hs746T, &T47D cells. IC50 values are shown in Table 10.
Figure 37
Cell killing potency of (A) 5D5 IgG on Hs746T, & T47D cells; (B) 5D5-MMAE-0GA-brPEG8- PEG2ADC (DAR 4) on Hs746T, &T47D cells; (C) ABT-700 IgG on Hs746T, NCI-N87 & T47D cells; (D) telisotuzumab vedotin (ABBV-399) on Hs746T, SNU5, NCI-N87 & T47D cells. IC50 values are shown in Table 10.
Figure 38
Representative plots showing cell killing potency from 9/50 cell lines of varying cMET expression level as part of the Omniscreen panel (Crown Biosciences Inc). 129D5B-MMAE- PGA-brPEG8-PEG2 FDC (DAR 5.6) was compared to Cis-Platin. IC50 values of all 50 cell lines are shown in Table 10. The FDC is highly potent on tumour cell lines of high, medium and low cMET expression level.
Figure 39
Internalization kinetics over 5h of (A) 129D5B scFv; (B) 129D5B-MMAE-[3GA-brPEG8-PEG2 FDC (DAR 5.6); (C) 5D5 IgG and (D) ABT-700 IgG. Internalization half-lives are shown.
Figure 40
Inhibition of cMET receptor phosphorylation of 4 cMET binding antibodies (129D5BTY scFv, 131D5S scFv, ABT-700 IgG and 5D5 IgG and a non-cMET binding IgG4. All 4 cMET binders inhibit with IC50 values of 1.4nM, 4.3nM, 1.4nM and 1.5nM respectively.
Figure 41
Pharmacokinetics of various 129D5BTY FDCs after an IV single dose in rats. Different linkers were evaluated and plasma concentrations vs time were plotted for each FDC compared with the scFv. Separate immuno-detection was employed to follow the clearance of the scFv component and the linker payload. The AUC (Area-under-curve) was determined using GraphPad Prism and listed in Table 12. Similar AUCs (Strep-Tag vs MMAE detection) indicated conjugate stability.
Figure 42
Metabolic markers detected in the plasma of rats after 3 cycles of Img/kg of 129D5B- MMAE-pGA-brPEG8-PEG2, Img/kg of 129D5B-MMAE-pGA-brPEGi2-PEG4, 4mg/kg of telisotuzumab vedotin or saline doses. (A) Aspartate transaminase; (B) Alaine transaminase; (C) Urea; (D) Creatinine and (E) Bilirubin levels were determined 2 days after each dose and plotted vs time. The three black arrows indicate the dosing event (once/3 weeks).
Figure 43
Metabolic markers detected in the plasma of rats after 3 cycles of Img/kg of 129D5B- MMAE-pGA-brPEG8-PEG2, Img/kg of 129D5B-MMAE-pGA-brPEGi2-PEG4, 4mg/kg of telisotuzumab vedotin or saline doses. (A) White Blood Cells-B (WBCB); (B) White Blood Cell-P (WBCP) and (C) Platelets counts were determined 2 days after each dose and plotted vs time. The three black arrows indicate the dosing event (once/3 weeks).
Figure 44
H&E-stained Kidney and liver sections, after the completion of the 3-cycle rat dosing study where animals were treated with 3 cycles of (A+D) Saline; (B+E) Img/kg of 129D5B- MMAE-pGA-brPEG8-PEG2 and (C+F) Img/kg of 129D5B-MMAE-|3GA-brPEGi2-PEG4. No abnormal pathological features were observed in the FDC treated animals compared to the saline group.
Figure 45
(A) In vivo tumour regression efficacy and (B) body weight study of 129D5BTY FDCs compared to a 5D5 IgG-ADC in a SNU5 gastric cancer xenograft (Table 18, Table 19, study Ref S-l). Animals/group (n=6).
Figure 46
(A) In vivo tumour regression efficacy and (B) body weight study of 131D5S FDCs compared to a 5D5 IgG-ADC in a SNU5 gastric cancer xenograft (Table 18, Table 19, study Ref S-2). Animals/group (n=6).
Figure 47
(A) In vivo tumour regression efficacy and (B) body weight study of 129D5BTY FDCs compared to a 5D5 IgG-ADC in a Hs746T gastric cancer xenograft (Table 18, Table 19, study Ref H-l). Animals/group (n=6).
Figure 48
(A) In vivo tumour regression efficacy and (B) body weight study of 131D5S FDCs compared to a 5D5 IgG-ADC in a Hs746T gastric cancer xenograft (Table 18, Table 19, study Ref H-2). Animals/group (n=6).
Figure 49
(A) In vivo tumour regression efficacy and (B) body weight study of 129D5BTY FDCs compared to a telisotuzumab vedotin (TV) ADC in a cMET low NCI-N87 gastric cancer xenograft (Table 18, Table 19, study Ref N-l). Animals/group (n=4).
Figure 50
Survival plot of 129D5BTY FDCs compared to a telisotuzumab vedotin (TV) ADC in a cMET low NCI-N87 gastric cancer xenograft (Table 18, Table 19, study Ref N-l). Animals/group (n=4).
Figure 51
In vivo tumour regression efficacy study of 129D5BTY and 131D5S FDCs in Hs746T gastric cancer xenograft of large tumours (~500mm3), (Table 18, Table 19, study Ref H-3). The v2 FDC was an alternative (129D5B derivative). Animals/group (n=6).
Figure 52
(A) In vivo tumour regression efficacy and (B) body weight study of 129D5BTY FDCs compared to a telisotuzumab vedotin (ABBV-399) in a SNU5 gastric cancer xenograft followed out to 100 days (Table 18, Table 19, study Ref S-3). Animals/group (n=8).
Figure 53
(A) In vivo tumour regression efficacy and (B) body weight study of 129D5BTY FDCs compared to a telisotuzumab vedotin (ABBV-399) in a Hs746T gastric cancer xenograft followed out to 100 days (Table 18, Table 19, study Ref H-4). Animals/group (n=8).
Figure 54
(A) In vivo tumour regression efficacy and (B) body weight study of 129D5BTY FDCs compared to a telisotuzumab vedotin (ABBV-399) and a non-binding (COVID spike protein specific) FDC in a cMET-moderate NUGC-4 gastric cancer xenograft followed out to 65 days (Table 18, Table 19, study Ref NG-1). Animals/group (n=8).
Figure 55
(A) In vivo tumour regression efficacy and (B) body weight study of 129D5BTY FDCs compared to a telisotuzumab vedotin (ABBV-399) in a cMET-low NCI-N87 gastric cancer xenograft followed out to 65 days (Table 18, Table 19, study Ref S-2). Animals/group (n=8).
Figure 56
(A) 129D5B-MMAE-pGA-brPEGi2-PEG4 FDC DAR stability over 7 days after in vitro incubation with cynomolgus, human, mouse and rat plasma. The DAR remained constant and similar to the starting DAR of 5.6. The full data set for human plasma is shown in Figure 56b as a representative example.
(B) 129D5B-MMAE-pGA-brPEGi2-PEG4 FDC DAR stability over 7 days after in vitro incubation with human plasma. The DAR distribution and average DAR was determined by SEC-LCMS and DAR 5 and DAR 6 is labelled for clarity. The average DAR and distribution remained constant and similar to the starting average DAR of 5.6. Two technical replicates of duplicate runs are shown for each time point.
Figure 57
Quantification of released MMAE payload uptake (over 3 days) into (A) tumours and (B) normal tissues after a single dose of Img/kg 129D5B-MMAE-pGA-brPEGs-PEG2 FDC DAR (6) compared to a single dose of 4mg/kg telisotuzumab vedotin ADC. Peak payload uptake for the FDC was around 24h with peak uptake for the ADC delivered payload beyond 3 days. Normal tissue uptake/accumulation of the payload was very low marginally higher
amounts for the FDC delivered payload in liver and kidney during the first 24h. The quantification is listed in Table 20.
Figure 58
Representative plots showing CD64, CD32a and CD16a binding to 129D5BTY scFvs, FDCs and various reference IgGs. The full set of data with binding affinities and kinetic constants is listed in Table 21.
Figure 59
Cynomolgus monkey haematological analyses. (A) Lymphocytes counts after four 3- weekly doses of (Dose-1), 0.25mg/kg, (Dose-2) 0.375mg/kg, (Dose-3) 0.5mg/kg and (Dose-4) Img/kg 129D5BTY-MMAE-pGA-BrPEGi2-PEG4 FDC (DAR6) over 66 days; (B) Total white blood cells made up of neutrophils and other lymphocytes were also counted. Black arrows indicate each of the 4 doses.
Figure 60
Cynomolgus monkey clinical chemistry analyses (A) Metabolic markers ALT, AST and GLDH determined after four 3-weekly doses of (Dose-1), 0.25mg/kg, (Dose-2) 0.375mg/kg (Dose-3) 0.5mg/kg and (Dose-4) Img/kg 129D5BTY-MMAE-pGA-BrPEGi2-PEG4 FDC (DAR6) over 66 days; (B) Body weights of the cynomolgus monkeys during the four-cycle dosing study.
Figure 61
Clone 129D5B family sequence comparison showing: (A) VH alignment; (B) VL alignment (C) summary of diversity.
Figure 62
Clone 131D5s family sequence comparison showing: (A) VH alignment; (B) VL alignment (C) summary of diversity.
Figure 63
Clones 129D5B and 131D5s sequence comparison.
Figure 64. Haematological measurements from cynomolgus taken pre-dose (1), 48h after dose (2) and 10 days (3)
(a) Average white blood cell count over the entire study (xlO9/L), showing the expected normal range ± 1 standard deviation
(b) Average neutrophil count over the entire study (xlO9/L), showing the expected normal range ± 1 standard deviation
(c) Average lymphocyte count over the entire study (xlO9/L), showing the expected normal range ± 1 standard deviation
(d) Average monocyte, eosinophil, basophil and large unstained cell counts over the entire study (xlO9/mL)
(e) Average platelet count over the entire study (xlO9/L), showing the expected normal range ± 1 standard deviation
(f) Average reticulocyte count over the entire study (xlO9/L), showing the expected normal range ± 1 standard deviation
Figure 65. Clinical Chemistry measurements for key liver markers from cynomolgus taken pre-dose (1), 48h after dose (2) and 10 days (3)
(a) Average alanine aminotransaminase, aspartate transaminase and glutamate dehydrogenase levels over the entire study (U/L)
(b) Average alanine aminotransaminase levels over the entire study (U/L) showing the expected normal range ± 0.5 standard deviation
(c) Average aspartate aminotransaminase levels over the entire study (U/L) showing the expected normal range ± 1 standard deviation
(d) Average alkaline phosphatase levels over the entire study (U/L) showing the expected normal range ± 1 standard deviation
(e) Average bilirubin levels over the entire study ( (mmol/L)
Figure 66. Clinical Chemistry measurements for key kidney markers from cynomolgus taken pre-dose (1), 48h after dose (2) and 10 days (3)
(a) Average urine creatinine levels over the entire study (mmol/L) showing the expected normal range ± 1 standard deviation
(b) Average urine total protein levels over the entire study (mg/L)
(c) Average urine total proteimcreatinine ratio over the entire study (mg/mmol)
(d) Average urine glucose levels over the entire study (mmol/L) showing the expected normal range ± 1 standard deviation
(e) Average urine glucose:creatinine ratio over the entire study (mmol/mmol)
Figure 67
(a) Average body weights for the entire non-human primate study, before and during the 5 escalating doses
(b) Percentage change in average body weights for the entire non-human primate study, before and during the 5 escalating doses
Figure 68
(a) Pharmacokinetic clearance profile for cMET FDC at all 5 escalating doses detected by ELISA anti-MMAE detection
(b) Pharmacokinetic clearance profile for cMET FDC at all 5 escalating doses detected by ELISA anti-Strep Tag detection
(c) Pharmacokinetic clearance profile for cMET FDC at Img/kg dose detected by ELISA anti-MMAE and Strep Tag detection
(d) Bioavailability (AUC) vs Dose plot to illustrate linearity of exposure over the 5 doses with TMDD being observed at the 0.25-0.5mg/kg dose (lower than expected AUC)
Figure 69
Detection of functional, binding cMET FDC by ELISA in urine in the 3 individual animals (M101, M102, M103) after 5 escalating FDC doses, compared to reference quantities.
Figure 70
BIACore binding profiles for the 129D5BTY-MMAE-[3GA-brPEG12-PEG4 FDC used in the non-human primate study showing comparable affinities for (a) human and (b) cynomolgus cMET.
Figure 71
BIACore binding analyses of 129D5BTY scFv compared to (a) ABT-700 IgG and (b) 5D5 IgG. All samples were injected at 400nM.
Figure 72
Purification and DAR characterization of 129D5B-Exatecan FDCs
(a) HPLC-SEC profile of FDC showing the pure Exatecan-MA-PEG12-Val-Ala-PEG4 FDC eluting at 25 minutes (fraction A9)
(b) RP-MS analysis of fraction A9 containing the Exatecan-MA-PEG12-Val-Ala-PEG4 FDC of an average DAR of 5.03 (DAR range 3-7).
(c) BIACore SPR binding profile and derived kinetic and affinity constants for fraction A9 containing the Exatecan-MA-PEG12-Val-Ala-PEG4 FDC
(d) HPLC-SEC profile of FDC showing the pure Exatecan-[3GA-BrPEG12-PEG4 FDC eluting at 23.81 minutes (fraction All)
(e) RP-MS analysis of fraction All containing the Exatecan-[3GA-BrPEG12-PEG4 FDC of an average DAR of 5.03 (DAR range 4-11).
(f) BIACore SPR binding profile and derived kinetic and affinity constants for fraction All containing the Exatecan-[3GA-BrPEG12-PEG4 FDC
(g) HPLC-SEC profile of FDC showing the pure DXd-GGFG-BrPEG8-PEG2 FDC eluting at 25.46 minutes (fraction D4)
(h) RP-MS analysis of fraction D4 containing the DXd-GGFG-BrPEG8-PEG2 FDC of an average DAR of 3.72 (DAR range 2-5).
(i) BIACore SPR binding profile and derived kinetic and affinity constants for DXd-GGFG- BrPEG8-PEG2 FDC
Figure 73
Characterization of the 129D5B scFv-2-(Methylsulphonyl)ethylamino-MMAF-[3GA-PEG2 FDC.
(a) Superdex-75 purification of the FDC which elutes as a pure monomer at 168.14 min. Free payload and small molecule contaminants elute after 250 min; (b) SEC-MS analyses showing the DAR distribution of the various FDC species; (c) BIACore SPR binding analysis on human cMET showing kinetic and binding constants; (d) Cell killing potency analysis on Hs746T (cMET-positive) and T47D cells (cMET negative).
Figure 74
Characterization of the 129D5B scFv-Belotecan-[3GA-PEG2 FDC.
(a) BIACore SPR binding analysis on human cMET showing kinetic and binding constants;
(b) Cell killing potency analysis on Hs746T (cMET-positive) and T47D cells (cMET negative). DAR analysis is shown in Figure 23.
Figure 75
DAR characterization of 129D5B-Nl-Benzylamino Imidazoquinoline-PEG7 FDC. RP-MS analysis showing an average DAR of 1.81 (DAR range 0-5).
Figure 76.
Evaluation of cMET FDC 129D5BTY-MMAE-0GA-BrPEG12-PEG4 (DAR6) in a cMET-low gastric cancer PDX model, (a) SP44-immunohistochemical staining of Crown Biosciences gastric cancer PDX (GA6885) showing high percentage of cMET-negative (IHC 0) and cMET-low (IHC 1+) tumour cells resulting in a low overall cMET IHC score (103.22). (b) Early readout after 2 doses of Img/kg and 2.5mg/kg FDC compared to 3mg/kg ABBV-399 and saline/untreated controls, showing tumour growth inhibition of the test agents and superior efficacy of the higher FDC dose.
Figure 77.
Evaluation of cMET FDC 129D5BTY-MMAE-0GA-BrPEG12-PEG4 (DAR6) in a cMET-low pancreatic cancer PDX model, (a) SP44-immunohistochemical staining of Crown
Biosciences gastric cancer PDX (PA3142) showing high percentage of cMET-negative (IHC 0) and cMET-low (IHC 1+) tumour cells resulting in a low overall cMET IHC score (114.27). (b) Early readout after 2 doses of Img/kg and 2.5mg/kg FDC compared to 3mg/kg ABBV- 399 and saline/untreated controls, showing tumour growth inhibition and superior efficacy of the higher FDC dose.
Examples
Example 1: Discovery of cMET binding scFvs
A non-immune scFv phage display library, 7xl09 in size, was generated in-house. This library consisted of an antibody scaffold of human VH5 and VL1 genes, with CDR regions diversified based on sequence diversity observed in natural human antibody sequences. Phage display selections with this scFv library were performed using recombinant purified cMET protein and following published methods [216]. Briefly, phage particles displaying the Antikor VH5-VL1 library were incubated with cMET antigens either immobilised directly on maxisorp plates, or captured via protein G immobilised onto maxisorp plates. For cMET antigens carrying a human Fc domain, undesirable scFvs recognising the Fc region of the fusion protein were removed by the addition of human IgGl to the selection mixture. After allowing phage binding to occur at room temperature for 2h, unbound phage particles were removed by multiple wash steps, and then specifically bound phage eluted by addition of chaotropic agents such as triethlyamine and/or hydrochloric acid. Eluted phage were then recovered by re-infection into E. coli TGI cells and plated onto large format 2xTY agar bioassay dishes supplemented with antibiotic.
Following the cMET selections, specific scFv were identified by phage ELISA following published methods [216], or by scFv ELISA using crude bacterial periplasmic extracts [217]. ELISAs were performed against cMET proteins and unrelated antigens to identify specific scFvs. From these selections, two candidate scFvs were isolated, 3A2 and 26B5, that were unique in VH CDR.3 sequence and specific for human cMET (Figure 1 and Table 3). Both 3A2 and 26B5 also showed binding to cynomolgus monkey cMET, and 3A2 also showed binding to rat cMET. Both clones have diverse VH CDR.3 sequences and both possess 11 framework lysines in total.
 Example 2: Affinity optimisation of cMET binding scFvs
Example 2: Affinity optimisation of cMET binding scFvs
For both 3A2 and 26B5, a number of strategies were employed to increase scFv binding affinity for cMET, using techniques well described in the art. In summary:
Light chain shuffling [218],
Targeted randomisation of VH and VL CDR.3 regions using randomised oligonucleotides [219],
Error prone mutagenesis of scFv sequences [220]
Phage display selections for the above strategies were carried out following published methods [216], using solution phase selections with biotinylated human cMET to control antigen concentration and increase selection stringency to favour higher affinity binders. Increased stringency was further applied by lengthening the wash steps to enrich for scFv with slower off rates, and by introducing heat stress to favour scFvs with higher (Tm) melting temperatures.
Binding affinity kinetic constants for scFv preparations of 3A2 and 26B5 and their affinity optimised variants were measured using a BIAcore T200 instrument. Approximately 100- 150R.U of cMET Fc was captured on a protein G chip by injecting recombinant human, cynomolgus and rat cMET at 2.5|j.g/mL in HBS buffer for 30s at 30|j.L/min flow rate. Binding kinetics of anti-cMET scFv were then determined by single-cycle kinetics. ScFv were prepared in dilution series (typically 1 :3 dilution series starting with 25-100nM scFv at the highest concentration), and then injected over the antigen coated surfaces and also a blank surface, starting with the lowest concentration of scFv and then working progressively up to the highest concentration. ScFv binding kinetics were then determined from the (blank subtracted) sensorgram traces using 1: 1 binding models and BIAevaluation software. Using this approach, affinities for 3A2 and 26B5 and several affinity optimised variants were determined (Figure 2a, 2b, 2c and Table 4).
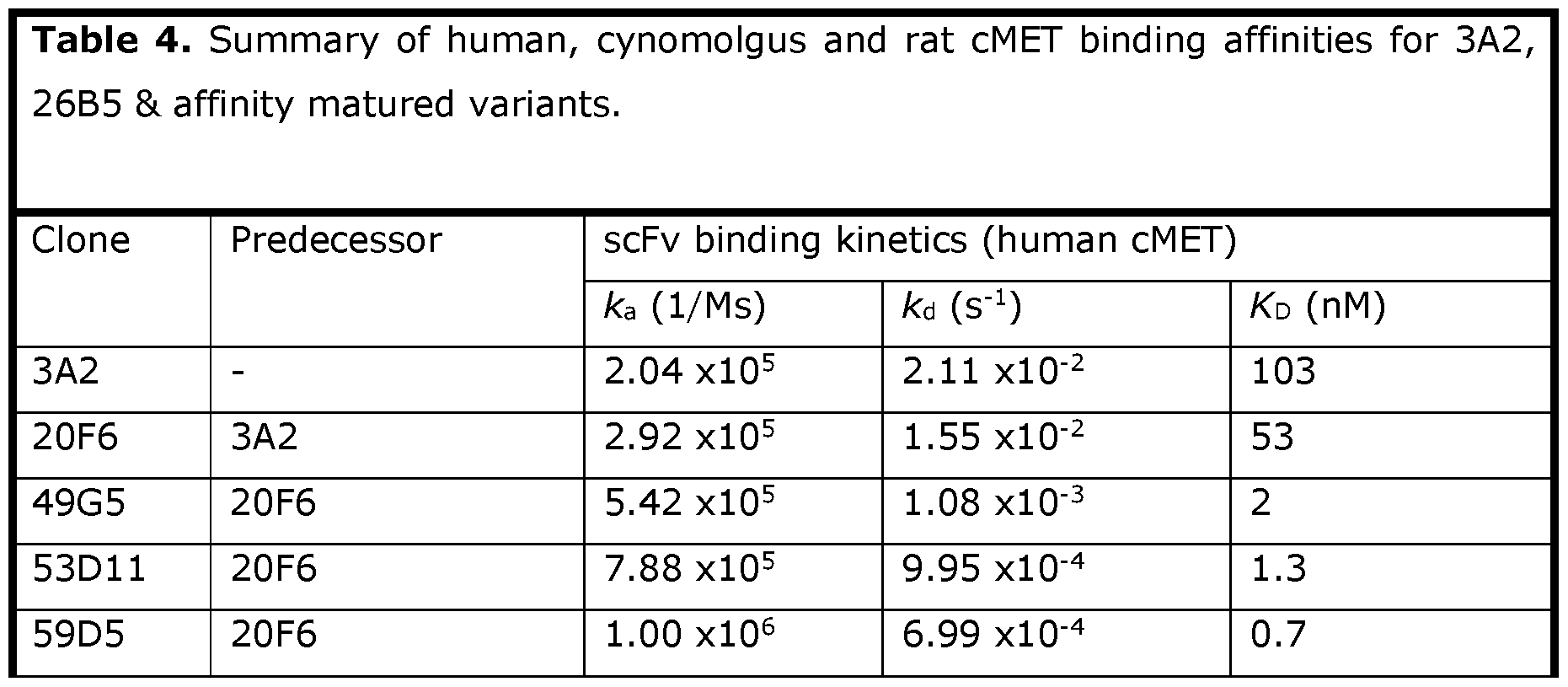


Example 3: Binding characterization of cMET scFvs 129D5B and 131D5S
(a) Specificity for cMET by ELISA
To demonstrate their specificity for cMET, 129D5B and 131D5S were analysed by scFv ELISA against cMET and a range of other unrelated antigens. Each test antigen was immobilised on maxisorb plates (Nunc 443404) by adding 50|nL volumes at 2|j.g/mL in PBS and incubating at 4°C overnight. Following coating, the antigen solution was aspirated and the plates were washed using PBS (prepared from PBS tablets, Oxoid cat no. BR0014G) supplemented with 0.05% Tween 20 (sigma P1379), followed by washes with PBS without added Tween. To block non-specific protein interactions, a solution of 3% skimmed milk powder (Marvel™) in PBS was added to the wells and the plate was incubated for at least Ih at room temperature. Purified 129D5B and 131D5S scFv were prepared in 3% Marvel/PBS at lOpig/mL and incubated for at least Ih at room temperature prior to transfer to the blocked ELISA plate where a further incubation of at least Ih took place. Unbound scFv was then washed away using repetitive washes with PBS/Tween followed by PBS. A solution of HRP-conjugated streptactin (IBA 2-1502-001), prepared at 1: 1000 dilution in PBS/3% Marvel was then added to each well and a further incubation at room temperature for at least one hour took place. Unbound streptactin-HRP was removed by repeated washing using PBS/Tween and PBS. The ELISA was then developed using TMB substrate (Sigma cat. no. T0440) and the reaction was stopped after 10 min by the addition of 0.5M
H2SO4 solution (Sigma cat. no. 320501). Absorbances were determined by reading at 450nm. Both 129D5B and 131D5S were shown to bind specifically to human cMET and not to any of the other antigens tested (Figure 3). For 129D5B, cross-reactivity with rat and mouse cMET species variants was also observed.
(b) Epitope binning of lead candidates 129D5B and 131D5S
129D5B and 131D5S scFv were shown to bind to unique epitopes of human cMET by Biacore. Human cMET Fc (2.5|j.g/mL in HBS buffer) was captured onto a protein G chip by injecting for 30s at 30|j.L/min flow rate. 129D5B scFv, at Ipig/mL in HBS buffer, was then injected over the cMET coupled surface and a blank control surface for 180s, followed immediately by a second injection of 129D5B scFv, at Ipig/mL in HBS buffer, for 180 s. Finally, 131D5S scFv was injected, at Ipig/mL in HBS buffer, for 180 s followed by buffer only for 600 s. Both scFvs were observed to bind simultaneously to human cMET, indicating that they were binding to different epitopes of the protein (Figure 4).
Example 4: Inhibition of Hepatocyte growth factor binding to human cMET by 129D5B and 131D5S
Hepatocyte growth factor (HGF) regulates cell growth, cell motility, and morphogenesis by activating the cMET receptor and both are implicated in oncogenesis [19,22]. To evaluate if 129D5B and 131D5S were capable of blocking this interaction, a ligand: receptor binding assay was developed. In this assay, HGF (Sino Biological 10463-HNAS) was immobilised onto a Nunc maxisorp plates at 0.5|j.g/mL in PBS buffer overnight at 4°C. Following coating, the HGF solution was aspirated and the plates were washed using PBS (prepared from PBS tablets, Oxoid cat no. BR0014G) supplemented with 0.05% Tween 20 (Sigma P1379), followed by washes with PBS without added Tween. To block non-specific protein interactions, a solution of 3% skimmed milk powder (Marvel™) in PBS was added to the wells and the plate was incubated for at least Ih at room temperature. Dilution series of 129D5B and 131D5S scFv were prepared, starting from lOpig/mL at the highest concentration and diluting 1:4 in 3% Marvel/PBS containing human cMET-Fc HIS (Sino Biological 10692-H03H) at lug/ml. Each 129D5B and 131D5S dilution series was incubated for at least Ih at room temperature prior to transfer to the blocked ELISA plate where a further incubation of at least one hour took place. Unbound scFv were then washed away using repetitive washes with PBS/Tween followed by PBS. A solution of HRP- conjugated anti-HIS antibody (Abeam abll87), prepared at 1: 10000 dilution in PBS/3% Marvel was then added to each well and a further incubation at room temperature for at least Ih took place. Unbound anti-HIS-HRP was removed by repeated washing using PBS/Tween and PBS. The ELISA was then developed using TMB substrate (Sigma cat. no.
T0440) and the reaction was stopped after 10 min by the addition of 0.5M H2SO4 solution (Sigma cat. no. 320501). Absorbances were determined by reading at 450nm. In this assay, it was shown that 131D5S completely inhibited HGF binding to human cMET whereas 129D5B had little or no effect (Figure 5).
Example 5: Biophysical characterisation of 129D5B and 131D5S
(a) SEC purification of monomeric scFv
129D5B and 131D5S scFvs were expressed overnight in E.coli strain TGI using Terrific autoinduction medium. Supernatant and periplasmic fractions were harvested by centrifugation and loaded onto Strep-Tactin® FPLC cartridges via Cytiva Akta chromatography systems. Bound scFv were eluted with free biotin and then loaded onto Superdex™ S75 gel filtration column to separate monomeric scFv protein from larger oligomers and impurities (Figure 6a and Figure 6b).
(b) Assessment of aggregation by Dynamic Light Scattering (DLS)
Particle size and distribution of molecules in solution can be obtained by dynamic light scattering (DLS). Molecules scatter light due to Brownian motion. The intensity fluctuations of the scattered light are used to calculate an autocorrelation function which allows determination of the hydrodynamic radius of molecules and dispersity of the solution as well as detection of any aggregates. Clones 129D5B and 131D5S scFvs were assessed by DLS (using a Nanotemper Prometheus Panta) and shown to be highly monodispersed and homogeneous samples with no indication of larger aggregates. Mean particle size was 2.8- 3nm consistent with small monomeric proteins (Figure 6c).
(c) Assessment ofscFv thermal stability
Nano-differential scanning fluorimetry (NanoDSF) measures changes in intrinsic fluorescence while a thermal ramp is applied, leading to unfolding and/or aggregation of the protein. The melting temperature (Tm) of the protein is defined as the inflection point where half of the protein is still in its folded state. To assess the thermal stability of 129D5B and 131D5S, NanoDSF studies were performed, and both proteins were shown to be highly thermostable, with T onset (initiation of unfolding) in excess of 60°C and Tm values 68- 77°C (Figure 7a, 7b).
Example 6: Generation of cMET scFv with reduced lysine content
To facilitate production of cMET FDCs with a more homogenous DAR, the overall lysine content in one cMET scFv (clone 77F3) was reduced from 11 to 7-8 residues.
To determine which lysines residues could be removed, whilst retaining full stability and binding affinity properties, 77F3 was firstly made by bacterial fermentation and purified by affinity chromatography (>95% pure). A cytotoxic payload (MMAE-pGA-BrPEGs-PEGz- NHS Ester) was made in-house and conjugated to the scFv to make 77F3-MMAE-PGA- BrPEGs-PEGz-NHS FDC conjugate (DAR 6) using methods described in Example 39. Using the enzymes trypsin and chymotrypsin, 77F3 FDC was digested into peptide fragments and analysed by mass spectrometry to produce a peptide map and to identify which lysine residues were attached to payload. This analysis identified lysine residues in 77F3 that were rarely coupled to payload, namely at positions 13, 23, 74 and 239. These positions were then targeted for mutagenesis in a new scFv library that aimed to change the lysines to alternative amino acids (Figure 8).
The 77F3 lysine variant library (total diversity 1.2xl04) was generated by Twist Bioscience Inc and then subcloned into a phagemid vector for phage display selections following standard methods as described in Example 1 [216,217]. Following selections on cMET, specific 77F3 variant scFv were identified by scFv ELISA using crude bacterial periplasmic extracts and by BIAcore following the procedure described in Example 2. From these selections, three candidate scFvs were isolated, 139B2, 139D2 and 139D7, that were of similar affinity and stability to 77F3 and contained well tolerated amino acid variations from lysine at positions 13, 23, 74 and 239 (Figure 9 and Table 5).

Example 7: Removal of potential deamidation sites in cMET scFv 129D5B
During drug development, it has become standard practice to analyse biologies for developability characteristics. Properties such as expression level, solubility, thermal and colloidal stability have all become common measurements during drug development. In addition, protein sequences are scanned for potential amino acid sequence liabilities. These are common motifs in proteins that could be subjected to chemical or enzymatic modification and potentially affect a drugs structure, stability or function. One example of
this is deamidation, where the amide group in the side chain of asparagine is removed or converted to aspartic acid or isoaspartic acid. The risk of this is dependent on multiple factors such as pH, temperature, buffer formulation, and in particular the amino acid residue neighbouring the asparagine residue, with a C-terminal glycine or serine presenting the highest risk. Biologies that carry these motifs are typically subjected to further mutagenesis to remove the high-risk sites without affecting the drugs biological and biophysical properties.
Two potential deamidation sites were identified in 129D5B: one in VL-CDR2 (NS, Kabat VL51 and VL52); and a second in VL-CDR3 (NS, Kabat VL93 and VL94). These sites were de-risked by converting the motifs to NY in VL-CDR2 and NT in VL-CDR3 using standard mutagenesis techniques [221,222]. The new molecule, named 129D5BTY, was unaffected by these changes and retained the same affinity and specificity for cMET as 129D5B (Figure 10), and also the biophysical properties, i.e., thermal and colloidal stability, were unchanged (Figures 6, 7 and 11).
Example 8: Mutagenesis of 129D5BTY to abolish mouse cMET binding
An arginine residue in 129D5BTY VH-CDR3 (Kabat VH98) was targeted for mutation to investigate the effects on scFv binding to human and mouse cMET. This single amino acid position was converted to other possible residues using standard mutagenesis techniques [221,222], and the mutants assessed for binding to human and mouse cMET by ELISA and BIAcore (Figure 12,13). Binding to human cMET was severely impacted when Arg98 was substituted by other amino acids, with some residues (proline, lysine and valine) virtually abolishing binding. The amino acid change with least impact at this position was Arg98 to Gln98, but binding to human cMET was still reduced by several hundred-fold (Figure 13). Binding to mouse cMET was abolished when Arg98 was changed to any other amino acid (Figure 13).
Abbreviations:


Experimental:
LCMS were run on an Agilent LCMS6100C SQ system using two main methods:
Method 1 : Run time: 6 min, Water (+ 0.1 % formic acid)/ MeCN (+ 0.1 % formic acid), MeCN = 5% - 95%, Flow rate: 0.500 mL/min, Injection: 5uL, Column oven T = 40C, MWD: 210, 250, 284 nm, Fragmentor voltage: 280 V, Polarity: positive
Method 2: Run time: 5.5 min., Water (+ 0.1 % formic acid)/ MeCN (+ 0.1 % formic acid), MeCN = 5% - 95%, Flow rate: 0.500 mL/min, Injection: 5uL, Column oven T = 40C, MWD: 210, 250, 284 nm, Fragmentor voltage: 140 V, Polarity: positive
Example 9: Synthesis of MMAE-pGA-BrPEG8-PEG2-NHS Ester (10)


To a solution of the 1-bromo-glucuronide methyl ester (40 g, 100.7 mmol) and 4-hydroxy- 3-nitrobenzaldehyde (28.6 g, 171.2 mmol) in dry MeCN (700.0 mL) was added silver oxide (102.7 g, 443.1 mmol) at room temperature. After stirring at RT overnight under N2, the mixture was filtered through celite. The bright yellow solution was then concentrated in vacuo and the residue was redissolved in EtOAc (300 mL). The mixture was washed with sodium bicarbonate (250 mL) saturated solution and the aqueous layer was further extracted with EtOAc (3 x 50 mL). The combined organic layers were dried over Na2SO4 and concentrated under reduced pressure to give the desired product 1 as a yellow solid 41.4 g (85.0%) and used in the next step directly without further purification. TLC: f = 0.48 (silica gel, PE/EtOAc = 1 : 1). LCMS m/z calculated for (M+NH4)+ 501.1.
To a solution of the benzaldehyde-sugar 1 (41.4 g, 85.6 mmol) in IPA/CHCh (1:5, 12000 mL) at 0°C was added NaBH4 (9.72 g, 256.9 mmol). After stirring at 0° for 1 h, the reaction solution was quenched with water (150 mL). The organic layer was separated, and the aqueous layer was back extracted with DCM (80 mL x 2). The organic phases were combined, dried over NazSC , filtered, and then concentrated under vacuo to give the desired product 2 as an off-white solid 38.7 g (93.0%) which was confirmed by LCMS and HNMR and used to next step directly without further purification. TLC: Rf = 0.2 (silica gel, PE/EtOAc 1/1,). LCMS m/z (M + NH4)+ 503.1.
To a solution of 2 (20 g, 41.2 mmol) in MeOH/water (6/5 v/v, 400 mL) were added NH4CI (2.42 g, 45.3 mmol) and iron powder (11.51 g, 206 mmol). After stirring at 80 °C for 3h under a N2 atmosphere, the mixture was cooled to RT and filtered through celite. The filtrate was quenched with sodium bicarbonate saturated solution and then extracted with EtOAc (3 x 100 mL). The combined organic extracts were dried over Na2SO4, filtered and
concentrated in vacuo. The residue was purified by silica gel column chromatography (PE/EA = 1/1, v/v) to give the desired product 3 as an off-white solid 7.7 g (41.0%). TLC: Rf = 0.23 (silica gel, PE/EtOAc 2/3). LCMS m/z (M+l) 456.1
As described in Jeffrey SC et al. [223]. To solution of the amine 3 (7.7 g, 16.9 mmol), Fmoc-0-alanine (5.79 g, 18.6 mmol) in dry DCM (100 ml) EEDQ (8.36 g, 33.8 mmol) was added and the reaction mixture stirred for 24 h under a N2 atmosphere. The mixture was quenched with sodium bicarbonate saturated solution and the phases separated. The aqueous layer was further back extracted with EtOAc (3 x 50 mL). The organics extracts were combined, dried over MgSC and concentrated in vacuo and purified by silica gel column chromatography (DCM:MeOH, 80: 1) to give the desired product 4 as an off-white solid 10.97 g (86.7%). TLC: Rf = 0.38 (silica gel, DCM/MeOH 20/1, v/v). LCMS m/z (M+ l) 749.9
As described in Jeffrey SC et al. [223]. A mixture of the benzyl alcohol 4 (6.5 g, 8.69 mmol) and pyridine (3.43 g, 43.43 mmol) in dry dichloromethane (80 mL) was cooled to 0°C. To this, a solution of bis-p-nitrophenyl carbonate (6.99 g, 34.76 mmol) in 15 ml of dry DCM was then added drop-wise. After addition was complete, the mixture was stirred at 0°C for 2 h under nitrogen after which it was extracted with dichloromethane (30 ml x 3). The combined organic extracts dried over Na2S04, filtered, and concentrated to dryness. The crude product residue was then purified by silica gel column chromatography (DCM:MeOH, 100: 1) to give the desired PNP-derivative 5 as an off-white solid 7.4 g (93.2%). TLC: Rf = 0.50 (silica gel, DCM/MeOH 20/1, v/v). LCMS m/z = 914.50 (M+l); 936.40 (M+Na).
As described in Jeffrey SC et al. [223]. To a stirred solution of 5 (5.4 g, 5.91 mmol), and monomethylauristatin E (MMAE, 4.27 g, 5.91 mmol) in dry THF (40 ml) containing pyridine (27.65 mL) DIPEA (903.69 mg, 7.00 mmol) was added followed by HOBt (820.8 mg, 6.09 mmol) which resulted in a faint colour change of the solution from colourless to a faint yellow. The reaction mixture was stirred at RT under nitrogen for 24 h after which the pH was adjusted to pH 6 by the addition of citric acid. The resulting mixture was then extracted with EtOAc (40 ml x 2), the combined organic phases were combined, dried over Na2SO4, filtered and concentrated in vacuo. The crude residue was purified by silical gel column chromatography (DCM:MeOH, 30: 1) to give the desired MMAE-pGlucuronide 6 as a white solid 6.9 g (78.2%). TLC: Rf = 0.20 (silica gel, DCM/MeOH 20/1, v/v). LCMS m/z 1493.49 (M + l); 1515.50 (M + Na)
As described in Jeffrey SC et al. [223]. To a cooled solution of 6 (6.70 g, 4.490 mmol) in MeOH (50 ml)/THF (50 ml)/H20 (50 mL) at 0 °C, a solution of UOH.H2O (942 mg, 22.450 mmol) and DBU (2.71 g, 17.960 mmol) in water (100 ml) was added dropwise over 1 h.
After the end of the addition, the reaction mixture was removed from the ice/water bath and allowed to stir for one further hour at RT. Glacial acetic acid (3 mL, 50 mmol) was then added, the reaction mixture concentrated in vacuo and purified by automated chromatography [Biotage (water + 0.1% TFA/MeCN, 0-100%)]. The appropriate pure fractions were combined and lyophilized to give the desired globally deprotected derivative MMAE-PGA 7 as a white solid 4.21 g (75% yield). LCMS m/z 1131.19 (M+l); 1153.08 (M + Na); 718.79 (MMAE) 686.71, 506.50 (MMAE fragments)
As described in RP Lyon et al. [192]. To a cooled solution of the MMAE-pA 7 (88.0 mg, 0.0707 mmol) and DIPEA (24.6 pL, 0.1410 mmol) in dry DMF (1 ml) at 0 °C was added a solution of the activated Fmoc-Lys-PEGs -NHS ester 12 (76.0 mg, 0.0849 mmol) in dry DMF (1.4 ml) via syringe pump over 1 hour. After the addition was complete, the reaction mixture was warmed back to RT and stirred for further 4 hours. The solvent was then removed under high vacuum and the residue purified by RP automated chromatography [Biotage (water + 0.1%TFA/MeCN + 0.1%TFA, 0-100%)] to give the desired product Lysine-PEGs derivative 8 as a white solid after lyophilization 111 mg (84 % yield). LCMS m/z = 1877.70 (M + l); 1898.68 (M + Na); 939.20 (M/2); 719.10 (MMAE); 687.06, 506.77 (MMAE fragments).
The Fmoc-protected derivative 8 (35 mg, 0.0187 mmol) was dissolved in dry DMF (2 ml) and cooled to 0°C in a water/ice bath. A solution of DBU (8.4 pl, 0.0560 mmol) in dry DMF (1 mL) was added dropwise over 1 h. After the DBU addition was complete, the mixture was allowed to stir for one further hour under nitrogen. The reaction was then quenched with AcOH (5.3 pl, 0.0934 mmol), and the mixture concentrated under reduced pressure. The residue was purified by RP automated chromatography [Biotage, (water + 0.1% TFA/MeCN + 0.1% TFA 10-90%) to afford the desired amine 9 as the TFA salt 26.30 mg (98%), as a white powder after lyophilization. LCMS m/z [M+l] 1652.9, (M+l /2] 827.2.
To a solution of Bis-PEGz-NHS (53 mg, 0.134 mmol) and DIPEA (7 pl, 0.043 mmol) in DMF (500 pL) under nitrogen was added a solution of MMAE-pGA-BrPEGs TFA salt 9 (22.2 mg, 0.0134 mmol) in DMF (840 pl) via syringe pump over 2 h. Upon end of the addition, the solvent was removed on high vacuum and the crude residue purified by RP automated chromatograph [Biotage, (water + 0.1% TFA/MeCN + 0.1% TFA 10-90%) to afford MMAE- PGA-BrPEGs-PEGz-NHS ester 10 35.27 mg (86%) as a white solid after lyophilization. LCMS m/z (M + l) 1940.00, [1/2M + 1]+ 970.1.
Example 10: Synthesis of Fmoc-PEGs lysine-NHS ester (12)

As described in RP Lyon et al. [192]. To a solution of Fmoc-Lys-OH (300 mg, 0.814 mmol) in dry DCM (13.5 ml) was added a solution of dPEGs-NHS ester (300 mg, 0.589 mmol) in dry DMF (4 ml) followed by DIPEA (513 pL, 2.94 mmol) - the mixture is a suspension. The reaction mixture was stirred at RT under nitrogen for 16 h before being quenched with IM solution NH4CI (10 mL). The mixture was separated and extracted with DCM (100 ml x 3). The combined organic phases were concentrated to dryness and the residue purified by RP automated chromatography [Biotage, water + 0.1% TFA/MeCN + 0.1% TFA, 0-100%) to give 11 as a sticky residue 217 mg (48% yield). TLC: Rf = 0.50 (silica gel, DCM/MeOH 10/1). LCMS m/z [M + l] 763.5.
To a solution of the Fmoc-dPEG8 acid 11 (200 mg, 0.262 mmol) and TSTU (158 mg, 0.524 mmol) in dry DMF (3.3 ml) was added DIPEA (229 pl, 1.31 mmol) and the mixture stirred at RT under nitrogen for 4 h. The mixture was concentrated to dryness and the residue purified by RP automated chromatography [Biotage, water + 0.1% TFA/MeCN + 0.1% TFA, 0-100%) to give the desired NHS-ester derivative 12 as a sticky-white solid 182 mg (81%). TLC: Rf = 0.70 (silica gel, CH2Cl2/MeOH 10/1). LCMS m/z [M+l] 860.5.
Example 11: Synthesis of MMAE-pGA-BrREGi2-PEG4-NHS Ester (15)

As described in RP Lyon et al. [192]. MMAE-pGA-alanine derivative 7 (88.0 mg, 0.0707 mmol) was dissolved in DMF (1 ml). Fmoc-Lys-PEGi2 (97.0 mg, 0.0934 mmol) dissolved in DMF (1.4 ml) was then added to the substrate solution, followed by DIPEA (32 pL, 0.184 mmol). The resulting mixture was stirred for 4 h at RT under nitrogen, the solvent was removed under high vacuum and the residue purified by RP automated chromatography [Biotage, (water + 0.1% TFA/ACN + 0.1% TFA, 0-100%)] to give the Fmoc-protected
derivative 13 as a white solid 120 mg (74%) after lyophilisation. LCMS m/z (M+l) 2053.30; (1/2 M + l)1027.13; 718.82 (MMAE), 686.72, 506.51 (MMAE fragments).
The Fmoc-protected derivative 13 (34 mg, 0.0166 mmol) was dissolved in dry DMF (2 ml) and cooled to 0°C in a water/ice bath. To this, a solution of DBU (7.4 pl, 0.0497 mmol) dissolved in dry DMF (0.5 ml) was added dropwise over 1 h, whilst stirring and under nitrogen. After the end of the DBU addition, the mixture was allowed to stir for a further 1.5 h after which the ice bath was removed, and the reaction quenched with AcOH (4.7 pl, 0.0166 mmol). The mixture was then concentrated under reduced pressure and purified by RP automated chromatography [Biotage, (water + 0.1% TFA/MeCN + 0.1% TFA, 10- 90%) to give the amine 14 as a white powder 27.0 mg (90%) after lyophilisation. LCMS m/z (M + l) 1831.10; (1/2 M + l) 915.60; 718.82 (MMAE); 686.76, 506.60 (MMAE fragmentation).
To a solution of Bis-PEG4-NHS (131 mg, 0.268 mmol) and DIPEA (14 pl, 0.080 mmol) in dry DMF (1 mL), a solution of the MMAE-[3GA-BrPEGi2 14 (49 mg, 0.026 mmol) in dry DMF (1 mL) was added dropwise using a syringe pump over 1 h. After addition was complete, the solvent was removed by high vacuum and the crude purified by RP automated chromatography [Biotage, (water + 0.1% TFA/MeCN + 0.1% TFA, 10-90%) to give the desired compound 15 31.73 mg (54%) as a white solid after lyophilisation. LCMS m/z (M+l) 2204.50; (1/2 M+l) 1102.58; 718.87 (MMAE), 686.79, 506.61 (MMAE fragmentation).
Example 12: Synthesis of Fmoc-PEGi2 lysine NHS ester (17)

As described RP Lyon et al. [192]. To a solution of Fmoc-Lys-OH (53.7 mg, 0.146 mmol) in dry DCM (3.7 mL) was added a solution of dPEGi2-NHS ester (100 mg, 0.146 mmol) in dry DMF (0.37 mL) followed by DIPEA (127 pL, 0.729 mmol) - the mixture is a suspension. The reaction mixture was stirred at RT under nitrogen for 5 h, quenched with IM solution HCI (5 mL) and stirred for 5 min. The reaction mixture was then extracted with DCM (20 ml), this was dried over MgSC , filtered, and evaporated. The residue was purified by RP automated chromatography [Biotage, (water + 0.1% TFA/MeCN + 0.1% TFA, 0-100%) to
give the pegylated lysine acid 16 as a sticky oil 140 mg (quantitative yield). LCMS m/z 939.87 (M + l); 961,85 (M + Na).
The pegylated lysine 16 acid (137 mg, 0.1459 mmol) and TSTU (88 mg, 0.2918 mmol) were dissolved in dry acetonitrile (5 mL), DIPEA (127 pL, 1.31 mmol) was added and the mixture stirred for 4 h at RT under nitrogen. The reaction mixture was concentrated under high vacuum and purified by RP automated chromatography [Biotage, (water + 0.1% TFA/ACN + 0.1% TFA, 0-100%) to give the desired activated ester derivative 17 as an off-white oil 138 mg (91%). LCMS m/z 1036.70 (M+l); 1058.72 (M+Na).
Example 13: Synthesis of DXd-GGFG-PEGz-NHS Ester (21)

As described in EP3101032, to a stirred solution of Fmoc-GGFG-Glycolic acid 18 (204 mg, 0.316 mmol) and exatecan mesylate (140 mg, 0.263 mmol) in dry DMF (20 mL), triethylamine (36.7 pL, 0.263 mmol) was added followed by DMTMM (156 mg, 0.474 mmol). The resulting solution was was stirred at RT under an inert atmosphere and monitored by LC-MS. Upon complete conversion to 19 (approx. Ih 30 min) piperidine (260 pL, 2.63 mmol) was added, and the reaction mixture was stirred at RT under inert atmosphere for a further 1 hour until the consumption of 19 as determined by LC-MS. The reaction mixture was concentrated in vacuo and the crude purified by RP automated chromatography [Biotage (water + 0.1% TFA/ACN + 0.1% TFA, 10-100%)]. The fractions containing the desired product 20 were combined and freeze dried to leave give the deprotected derivative 20 as a yellow solid (214 mg, 97% yield for two steps). LCMS m/z 1063.81 (M + l); 1085.78 (M + Na)
The deprotected exatecan derivative 20 (16 mg, 0.0190 mmol) was dissolved in dry DMF (500 pl) and added dropwise over 1 h (using a syringe pump) to a solution of bis-PEGz
NHS ester (76 mg, 0.190 mmol) dissolved in dry DMF (500 pl) and DIPEA (10 pl, 0.0571 mmol). After addition was complete, the reaction mixture was stirred for a further 1 h at RT, the solvent was then removed and the crude residue purified by RP automated chromatography [Biotage (water + 0.1% TFA/ACN + 0.1% TFA, 10-90%)], to give the desired product 21 as a yellow solid 9.16 mg (48 %). LCMS (M+ l) 1126.90; (M+Na) 1149.86
Example 14: Synthesis of DXd-GGFG-PEG7-NHS Ester (22)

The exatecan derivative 20 (13 mg, 0.0155 mmol) was dissolved in dry DMF (1 ml) and added dropwise over 1 h (using a syringe pump) to a solution of bis-PEG? NHS ester (96 mg, 0.1546 mmol) dissolved in dry DMF (1 ml) and DIPEA (8.1 pl, 0.0461 mmol). After addition was complete, the reaction mixture was stirred for a further 1 h at RT, the solvent was then removed and the crude residue purified by RP automated chromatography [Biotage (water + 0.1% TFA/ACN + 0.1% TFA, 10-90%)], to give the desired DXd-GGFG- PEG? derivative 22 as a yellow solid 8.15 mg (39 %). LCMS m/z (M+l) 1347.00
Example 15: Synthesis of DXd-GGFG-BrPEG8-PEG2 NHS Ester (25)

The exatecan derivative 20 (98.50 mg, 0.117 mmol) and the activated lysine branching point 12 (121 mg, 0.141 mmol) were dissolved in dry DMF (10 ml) under nitrogen. DIPEA (49 pl, 0.281) was added and the mixture was stirred at RT under nitrogen for 22 hours after which the reaction mixture was concentrated under high vacuum and the resulting residue purified by automated RP chromatography [Biotage (water + 0.1% TFA/ACN + 0.1% TFA, 0-100%)] to give the branched-Fmoc lysine DXd derivative 23 as a light-yellow solid 71 mg (38%). LCMS m/z (M+l) 1586.50. The Fmoc-deprotected derivative 24 21 mg (13%) was also isolated. LCMS m/z 1365.20 (M+l).
The Fmoc-derivative 23 (38.70 mg, 24.40 pmol) was dissolved in dry DCM (4 ml) piperidine (53 pL, 0.537 mmol) was added and the mixture stirred at RT under nitrogen for 2 hours. The reaction was was concentrated under high vacuum and the residue purified by RP automated chromatography [Biotage, (water + 0.1% TFA/ACN + 0.1% TFA, 0-100%) to give Fmoc-deprotected derivative 24 as a yellow solid 33 mg (quantitative yield). LCMS m/z (M+l) 1365.20
Bis-PEGz-NHS (38 mg, 0.095 mmol) was dissolved in dry DMF (500 pl), DIPEA (5 pl, 0.029 mmol) was added. Separately, DXd-PEGs amine 24 (13 mg, 0.095 mmol) was dissolved in dry DMF (500 pl). This was then slowly added (1 h) dropwise using a syringe pump to the stirred solution of the bis-PEGz-NHS and DIPEA. After addition was complete, the
solvent was removed under high vacuum and the crude residue purified by RP automated chromatography [Biotage, (water + 0.1% TFA/ACN + 0.1% TFA, 10-90%)], to give the DXd-GGFG-BrPEG8-PEG2 NHS ester 25 as a yellow solid 3.32 (36%). LCMS (M+l) 1650.09
Example 16: Synthesis of Exatecan-pGA-PEG? NHS Ester (28)

As described in WO2019236954, the PNP-carbonate ester 5 (86 mg, 0.094 mmol), HOBt (13 mg, 0.094 mmol) and exatecan mesylate (50 mg, 0.094 mmol) were stirred in dry THF (2.5 ml) and pyridine (0.5 mL), DIPEA (49 pl, 0.282 mmol) was then added and the reaction mixture stirred at RT under nitrogen for 19 h. The reaction mixture concentrated in vacuo and crude purified by RP automated chromatography [Biotage, (water + 0.1% TFA/ACN + 0.1% TFA, 0-100%)]. Combined fractions were concentrated to give the Fmoc- glucuronide exatecan derivative 26 as a yellow solid 71 mg (62% yield). LCMS m/z (M+l) 1210.93, (M + Na) 1234.85
As described in WO2019236954, a stirred solution of the Fmoc-sugar-exatecan 26 (33 mg, 0.0273 mmol) in 1 : 1 MeOH 8<. THF (4 mL) was cooled with an ice/water bath. To this a solution of LiOH.HzO (6.9 mg, 0.1636 mmol) in 2 mL water was dropwise over 1 h 20 min. The reaction was monitored by LCMS and showed complete consumption of 26. The reaction mixture was concentrated in vacuo and purified by RP automated chromatography [Biotage, (water + 0.1% TFA/ACN + 0.1% TFA, 0-100%)] to give the globally deprotected exatecan-pGA amine derivative 27 as a pale-yellow solid 18 mg (71 %). LCMS (RT = 1.923 min.) m/z (M+l) 848.50. The partially deprotected (Fmoc-alanine) exatecan-pGA was also isolated 7 mg (24 %). LCMS (RT = 2.347 min.) m/z (M+ l)1070.70, (M+Na) 1092.68
Bis-PEGyNHS ester (66 mg, 0.1062 mmol) was dissolved in dry DMF (500 pl) and DIPEA (5.6 pl, 0.0318 mmol) was added. Separately, Exatecan-[3GA amine 27 (9 mg, 0.0106 mmol) was dissolved in DMF (500 pl). This was then slowly added (1.5 h) dropwise using a syringe pump to the stirred solution of the bis-PEG?-NHS and DIPEA. After addition was complete, the solvent was removed under high vacuum and the crude residue purified by RP automated chromatography [Biotage, (water + 0.1% TFA/ACN + 0.1% TFA, 10-90%)], to give Exatecan-pGA-PEG? NHS ester 28 as a pale-yellow sticky solid 1.84 mg (12.8 %). LCMS m/z (M+l) 1354.59, (M + Na) 1376.59.
Example 17: Synthesis of Belotecan-pGA-PEGz NHS Ester (32)

As described in WO2019236954, PNP-carbonate ester 5 (194 mg, 0.213 mmol), HOBt (29 mg, 0.217 mmol) and belotecan hydrochloride (150 mg, 0.213 mmol) were dissolved in dry DMF (5 ml) and dry pyridine (1 ml). DIPEA (44 pl, 0.255 mmol) was then added, and the reaction mixture stirred at RT under nitrogen for 24 h. The reaction mixture was concentrated in vacuo and the crude purified by NP automated chromatography [Biotage (DCM/MeOH, 0-20 %)] to give the belotecan -protected sugar derivative 29 as a yellow solid 258 mg (quantitative yield). LCMS m/z (M + l) 1209.99
A stirred solution of the Fmoc-sugar-belotecan derivative 29 (114 mg, 0.0944 mmol) was dissolved in 1 : 1 MeOH & THF (7 ml) and cooled with an ice/water. To this a solution of LiOH.HzO monohydrate (24 mg, 0.5661 mmol) in water (2 ml) was added dropwise over one hour. The reaction mixture was then allowed to stir whilst reaching RT for 45 minutes, acetic acid (38 pl, 0.6608 mmol) was added, the reaction mixture stirred for a further 2
minutes, and then evaporated to dryness under reduced pressure. The crude 30 was used directly in the next reaction. LCMS m/z (M+l) 1069.00
To a stirred solution of 30 dissolved in dry DMF (6 ml), a solution of DBU (27 pl, 0.1827 mmol) dissolved in dry DMF (6 ml) was added dropwise over 1.5 h under nitrogen and at RT. The reaction was then quenched with acetic acid (35 pl, 0.6090 mmol), allowed to stir for a further 5 minutes, and evaporated to dryness under reduced pressure. The crude was purified RP automated chromatography [Biotage, (water 0.1% TFA/ ACN 0.1% TFA 0-100%)]. The appropriate fractions were combined and lyophilized to give the fully deprotected derivative Belotecan-pGA derivative 31 as a yellow solid 32 mg (40 % overall). LCMS m/z (M+l) 846.50
A solution of Belotecan-pGA derivative 31 (24 mg, 0.0284 mmol) in dry DMF (11 ml) was added dropwise over 3 h via syringe pump into a stirred solution of the bis-PEGz-NHS (57 mg, 0.1419 mmol) and DIPEA (25 pl, 0.1419 mmol). After addition was complete, the reaction mixture was stirred for an additional 45 minutes then evaporated to dryness under reduced pressure. The crude product was purified via RP automated chromatography [Biotage, (water 0.1% TFA/ ACN 0.1% TFA 0-100%)]. The appropriate fractions were combined and lyophilized to give Belotecan-pGA-PEGz NHS ester 32 as a yellow solid 22 mg (69%). LCMS m/z (M + l)1132.60
Example 18: Synthesis of Nintedanib-Val-Ala-PEGz NHS Ester (37)

As described in WO2019232449, commercially available Boc-ValAla-PABA 33 (50 mg, 127 pmol) was dissolved in anhydrous THF (5 mL). Triphenylphosphine (40 mg, 152 pmol) and NBS (27 mg, 152 pmol) were then added and the reaction mixture was allowed to stir at RT under nitrogen for 7 hours. The solvent was then removed at reduced pressure, and the resulting crude purified on NP automated chromatography [Biotage, (DCM/MeOH (0- 50%)]. Combined fractions were concentrated to give the brominated derivative 34 as a yellow solid 49 mg (84 %). LCMS m/z 469 (M+Na).
[As described in WO2019232449, the brominated Val-Ala derivative 34 (25 mg, 54.8 pmol) and nintedanib (29.5 mg, 54.8 pmol) were dissolved in anhydrous DMF (2 ml), DIPEA (14.3 pl, 82.2 umol) was then added and the reaction mixture stirred at RT under nitrogen for 17 hours. The solvent was removed under vacuum and the resulting crude purified on NP automated chromatography [Biotage, (Hex/EOAc (0-100%)], to give the quaternary ammonium Boc-val-ala 35 as an off-white solid 3 mg (6% yield). LCMS m/z 916 (M+l), 540 (nintedanib fragment), 408 (M-Boc/2).
To the Boc-protected Val-Ala-nintedanib 35 (3 mg, 3.3 pmol), 10% TFA solution in dry DCM (3.3 ml) was added dropwise over 30 minutes and the reaction mixture stirred at RT under nitrogen. After addition was complete, the solvent and TFA were removed under high vacuum. LCMS analysis showed the desired deprotected val-ala-nintedanib-amine 36 as a single peak pure enough to be telescoped into next reaction without further purification. LCMS m/z 816 (M + l), 540 (nintedanib fragment), 408 (M/2).
The deprotected nintedanib-Val-Ala-amine 36 (2.7 mg, 3.3 pmol) was dissolved in dry DMF (1 ml) and added dropwise (syringe pump) over 70 min to a stirred solution of bis- PEG2 -NHS ester (13.1 mg, 32.8 pmol) and DIPEA (1.7 pl, 9.8 pmol) in dry DMF (0.5 ml) at RT under nitrogen. After the addition was completed, the reaction mixture was allowed to stir for a further 35 minutes at RT after which the DMF was removed under high vacuum. The crude was purified on RP automated chromatography [Biotage (water + 0.1% TFA/ACN + 0.1% TFA, 0-100%)] to give the desired nintedanib-val-ala-PEGz-NHS ester 37 as a bright yellow solid 0.84 mg (23 % overall). LCMS m/z 1101.40 (M+ l), 540 (nintedanib fragment).
Example 19: Synthesis of Nintedanib a-Galactose-PEG? NHS Ester (46)


As described in Li Y et al. [224]. a-Galactose tetraacetate bromide 38 (500 mg, 1.22 mmol), 4-hydroxy-3-nitrobenzaldehyde (341 mg, 2.04 mmol) and silver oxide (1.10 g, 4.75 mmol) were stirred in anhydrous MeCN (12 mL) under nitrogen for 3 h at RT after which it was filtered through celite. The bright yellow solution was concentrated in vacuo to leave a yellow solid, which was redissolved in EtOAc (10 ml) and washed with sodium bicarbonate saturated solution (15 mL). The aqueous layer was further back extracted with EtOAc (3 x 20 mL). The organic layers were combined, dried over MgS04 and dried under reduced pressure to give the galactose tetraacetate-benzaldehyde derivative 39 as a light-yellow solid 551 mg (91%). This was used in the next reaction without further purification. Monitored by TLC (H/EA 1: 1); Rf = 0.17.
As described in Li Y et al. [224], a solution of the benzaldehyde-galactose tetraacetate 39 (551 mg, 1.11 mmol) was dissolved in in IPA/CHCh (1 :5, 30 ml) and cooled to 0°C, NaBh (125 mg, 3.33 mmol) was then added portion wise, and the reaction mixture stirred under N2 at 0° for 1 hour. A second portion (125 mg, 3.33 mmol) of NaBhU was then added and the mixture stirred for a further 2.5 h after which the reaction mixture was quenched by the careful addition of water (30 mL). The organic layer was separated, and the aqueous layer was further extracted with ethyl acetate (3 x 30 mL). The organic phases were combined, dried over MgSCU, filtered, and concentrated under vacuum to give the benzyl alcohol 40 as alight yellow solid 506 mg (91.5% yield). This was used without further purification in the next reaction. Reaction monitored by TLC (H/EA 1: 1); Rf = 0.069. LCMS m/z: 517.30 (M + H2O), 522.30 (M + Na)
Il l
The galactose tetraacetate-nitrobenzene derivative 40 (506 mg, 1.01 mmol) was dissolved in a 6:5 EtOH/water mixture (13 ml), NHUCI (60 mg, 1.11 mmol) and iron powder (283 mg, 5.10 mmol) were then added, and the mixture stirred at 80°C for 3h. The reaction was followed by TLC (silica gel Hexane/EA 1: 1, Rf = 0.13), filtered through celite and concentrated in vacuo to give the amine derivative 41 as a white solid 226 mg of (48%) which was pure enough to use without further purification. LCMS m/z = 470.30 (M+l), 493.30 (M + Na).
The galactose tetraacetate amine 41 (226 mg, 4.8 pmol), Fmoc-0-alanine (165 mg, 5.3 pmol), and EEDQ (238 mg, 963 pmol) were placed in a round bottom flask and dissolved in dry DCM (12 ml). The reaction mixture was stirred for 21 h under an inert atmosphere after which it was quenched by the addition of sodium bicarbonate saturated solution (15 mL) and the phases separated. The aqueous layer was further back extracted with EtOAc (3 x 20 mL), the organics were then combined, dried over MgSC and concentrated in vacuo. The crude was purified by NP automated chromatography [Biotage, Hexane/EA (10-100%)] to give galactose tetraacetate Fmoc-p-alanine derivative 42 as a white solid 188 mg (51 %). LCMS m/z = 763.50 (M + l), 785.46 (M + Na).
As described in Hamilton JZ et al. [225]. The galactose tetraacetate benzyl alcohol derivative 42 (88 mg, 115.4 pmol) was dissolved in dry THF (2 ml), triphenylphosphine (45 mg, 173 pmol) and NBS (31 mg, 173 pmol) were then added sequentially to the solution, and the mixture allowed to stir at RT under nitrogen for 24 h. The reaction mixture was then concentrated in vacuo and crude purified by NP automated chromatography [Biotage, Hex/EtOAc (30-70%)]. Combined fractions were concentrated to give the benzyl galactose tetraacetate benzyl bromide derivative 43 as a white solid (25 mg (26 %). LCMS m/z = 827.40 (M+ l), 849.33 (M + Na).
To a solution of the galactose tetraacetate benzyl bromide 43 (25 mg, 30 pmol) and nintedanib (16.3 mg, 30 pmol) in dry DMF (2 ml) DIPEA (7.9 pL, 45 pmol) was added and the resulting mixture was allowed to stir under nitrogen at RT for 3h. The reaction mixture was then concentrated in vacuo and the crude purified on RP automated chromatography [Biotage (water + 0.1% TFA/ACN + 0.1% TFA, 0-100%)]. Combined fractions were concentrated to give the nintedanib-galactose tetraacetate 44 as a white solid 21 mg (54 %). m/z = 1286.00 (M+l), 540.42 (nintedanib fragment).
To a stirred solution of the nintedanib-galactose tetraacetate 44 (15 mg, 117 pmol) in 5 mL of a 1 : 1 mixture of MeOH and THF, DBU (8.7 pL, 58 pmol) was added and the mixture stirred at RT for 5 min after which a solution of potassium carbonate (16 mg, 117 pmol) dissolved in 2.5 ml water was added at RT and the reaction mixture stirred for a further 25 more min. after which it was purified by RP automated chromatography [Biotage (water
+ 0.1% TFA/ACN + 0.1% TFA, 0-100%)] to give the globally deprotected derivative nintedanib-galactose 45 as a white solid 10 mg (96 %). LCMS m/z = 894.70 (M+l), 540.40 (nintedanib fragment).
To a stirred solution of bis-PEGyNHS ester (83 mg, 134 pmol) in dry DMF (1 ml) and DIPEA (7 pL, 40 |jmol) under nitrogen, a solution of nintedanib-galactose amine 45 (12 mg, 13.4 |jmol) dissolved in dry DMF (1 ml) was added dropwise over 1 h using a syringe pump. After the addition was complete, the mixture was allowed to stir for a further 30 min. The solvent was then removed under high vacuum and the crude purified by RP automated chromatography [Biotage (water + 0.1% TFA/ACN + 0.1% TFA, 10-100%)] to give nintedanib o-Galactose-PEG? NHS Ester 46 as pale-yellow solid 5.8 mg (31 %) after lyophilisation. LCMS m/z = 1400.20 (M+l), 540.40 (nintedanib fragment).
Example 20: Synthesis of Dexamethasone-dipyrophosphate-PEGz TFP Ester (50)

As described in Kern JC et al. [198], to a solution of dexamethasone (2 g, 5.1 mmol) in THF (2 ml) at -40°C was added diphosphoryl chloride (1.8 mL, 12.7 mmol). The reaction mixture was stirred at -40°C for 2 h, quenched with water and treated with saturated sodium bicarbonate solution unti pH~8. The solution was then made acidic by the drop wise addition of IN HCI solution and extracted with EtOAc (300 mL). The combined organic layers were washed with brine, dried over NazSC , filtered and concentrated to afford dexamethasone dihydrogen phosphate 47 as a yellow solid 2.09 g (86%) LCMS m/z = 473.4 [M + H]+, XH NMR (400 MHz, DMSO-de) 6 7.29 (d, J = 10.2 Hz, 1H), 6.22 (dd, J = 10.2, 1.8 Hz, 1H), 6.01 (s, 1H), 4.97 - 4.87 (m, 1H), 4.55 - 4.46 (m, 1H), 4.15 (dd, J = 10.4, 3.2 Hz, 1H), 3.85 - 3.58 (m, 2H), 2.99 - 2.86 (m, 1H), 2.68 - 2.57 (m, 1H), 2.43
- 2.26 (m, 2H), 2.20 - 2.05 (m, 2H), 1.82 - 1.72 (m, 1H), 1.69 - 1.54 (m, 2H), 1.51 - 1.48 (m, 7H), 1.46 - 1.41 (m, 1H), 1.38 - 1.27 (m, 1H), 1.12 - 1.03 (m, 1H), 0.79 (d, J = 7.2 Hz, 3H).
As described in Kern JC et al. [198], to a solution of (9H-fluoren-9-yl)methyl (2- hydroxyethyl)carbamate (2 g, 7.06 mmol) in THF (2 ml) at -40°C was added diphosphoryl chloride (2.4 mL, 17.65 mmol). The reaction mixture was stirred at -40°C for 2 h. The reaction was quenched with water and treated with saturated sodium bicarbonate solution unti pH~8. The solution was made acidic by the addition of IN HCI solution and extracted with EtOAc (300 mL). The combined organic layers were washed with brine, dried over NazSC , filtered and concentrated to afford (9H-fluoren-9-yl)methyl (2- (phosphonooxy)ethyl)carbamate 48 as a white solid 2.6 g (100%). LCMS m/z = 364.3 [M + H]+, XH NMR (400 MHz, DMSO-cfe) 6 7.89 (d, J = 7.6 Hz, 2H), 7.69 (d, J = 7.4 Hz, 2H), 7.43 - 7.39 (m, 2H), 7.36 - 7.31 (m, 2H), 4.29 (d, J = 6.2 Hz, 2H), 4.21 (t, J = 7.0 Hz, 1H), 3.80 (q, J = 6.4 Hz, 2H), 3.21 (q, J = 6.0 Hz, 2H).
As described in Kern JC et al. [198], to a stirred solution of (9H-fluoren-9-yl)methyl (2- (phosphonooxy)ethyl)carbamate 48 (500 mg, 1.38 mmol) in DMF (5 mL) was added triethylamine (348 mg, 3.44 mmol) and CDI (223 mg, 1.38 mmol). The resulting solution was stirred at room temperature for 30 min. To this mixture were added dexamethasone dihydrogen phosphate 47 (520 mg, 1.1 mmol), the mixture was allowed to stir at room temperature overnight. The reaction was diluted with 1 N HCI to pH-7 and purified by RP preparative chromatography (25% MeCN/water with 0.1% HCOOH) to give the Fmoc- deprotected dexamethasone ethylamino-dihydrogen pyrophosphate 49 as a white solid 200 mg (24%). LCMS m/z = 596.4 [M + H]+, XH NMR (400 MHz, DMSO-de) 6 8.27 (s, 2H), 7.29 (d, J = 10.2 Hz, 1H), 6.21 (dd, J = 10.2, 1.8 Hz, 1H), 6.00 (s, 1H), 4.83 - 4.72 (m, 1H), 4.67 - 4.56 (m, 1H), 4.15 (d, J = 11.6 Hz, 1H), 4.11 - 4.03 (m, 2H), 3.88-3.67 (m, 3H), 3.37 - 3.32 (m, 1H), 3.06 - 2.87 (m, 3H), 2.68 - 2.55 (m, 1H), 2.42 - 2.26 (m, 2H), 2.17 - 1.95 (m, 2H), 1.80 - 1.67 (m, 2H), 1.61 (q, J = 11.6 Hz, 1H), 1.49 (s, 3H), 1.41 - 1.28 (m, 1H), 1.11 - 1.01 (m, 1H), 0.88 (s, 3H), 0.78 (d, J = 7.2 Hz, 3H).
To a solution of dexamethasone ethylamino-dihydrogen pyrophosphate 49 (120 mg, 0.2 mmol) in DMF (2 mL) was added bis-PEG2-(2,3,5,6-tetrafluorophenyl) ester (111 mg, 0.22 mmol) and DIPEA (78 mg, 0.6 mmol). The reaction mixture was stirred at room temperature for 1.5 h and purified by RP prep-HPLC (water + 0.1% TFA: MeCN) to give the dexamethasone dihydrogen pyrophosphate 2,3,5,6-tetrafluorophenyl PEG2 ester as a brown solid 16 mg (8%). LCMS m/z = 932.5 [M + H]+, XH NMR (400 MHz, DMSO-de) 6 8.17 (d, J = 5.6 Hz, 1H), 7.99 - 7.86 (m, 1H), 7.29 (d, J = 10.2 Hz, 1H), 6.21 (dd, J = 10.2, 1.8 Hz, 1H), 6.00 (s, 1H), 4.98 (dd, J = 17.8, 8.8 Hz, 1H), 4.63 (dd, J = 17.8, 7.8 Hz, 1H), 4.14 (d, J = 10.2 Hz, 1H), 3.91 (q, J = 6.2 Hz, 2H), 3.76 (t, J = 6.0 Hz, 2H), 3.62 -
3.57 (m, 9H), 3.27 (q, J = 5.8 Hz, 2H), 3.01 (t, J = 5.8 Hz, 2H), 2.96 - 2.87 (m, 1H), 2.67 - 2.57 (m, 1H), 2.46 - 2.37 (m, 1H), 2.36 - 2.27 (m, 4H), 2.17 - 2.04 (m, 2H), 1.80 - 1.72 (m, 1H), 1.68 - 1.50 (m, 2H), 1.48 (s, 3H), 1.44 - 1.28 (m, 1H), 1.10 - 1.02 (m, 1H), 0.88 (s, 3H), 0.78 (d, J = 7.2 Hz, 3H).
Example 21: Synthesis of N1-Benzylamino Imidazoquinoline-PEG? NHS Ester (54)

To a stirred solution of -benzylamino imidazoquinoline hydrochloride (20.3 mg, 0.047 mmol) in dry DMF (0.5 ml) DIPEA (32.8 pl, 0.188 mmol) was added, and the solution was allowed to stir for 15 minutes at RT under inert atmosphere. In another flask, the PNP- carbonate ester 7 (43 mg, 0.047 mmol) and HOBt (7 mg, 0.052 mmol) were dissolved in dry DMF 1 ml and pyridine 0.3 ml at RT under nitrogen. The latter solution was then added to the imidazoquinoline/DIPEA solution and the reaction mixture was stirred at RT for 2h 40 min. The reaction mixture was concentrated in vacuo and the crude purified by NP automated chromatography [Biotage, DCM/MeOH (0-40%)]. The combined fractions were concentrated to the glucuronide-imidazoquinoline derivative 51 as an off-white solid 31 mg (62 %). LCMS m/z 1134.68 (M+l), 1162.70 (M+Na), 739.60
The glucuronide-imidazoquinoline derivative 51 (10 mg, 0.0088 mmol) was dissolved in a mixture of a MeOH/THF 1: 1 (2 ml), this was cooled with an ice/water bath and a solution of LiOH.HzO (2.2 mg, 0.053 mmol) dissolved in 1 ml water added dropwise over 1 h. The reaction mixture was stirred for a further 45 further min. after which it was quenched with acetic acid (3 pL, 0.0053 mmol), and concentrated in vacuo. The crude deprotected [3GA- Fmoc derivative 52 was used without further purification. LCMS m/z 994.60 (M+l); 497.93 (M/2).
The Fmoc-protected imidazoquinoline-pGA 52 (8.8 mg, 0.0088 mmol) was dissolved in dry DMF (1 ml)and DIPEA (31 pL, 0.301 mmol) was added. The reaction mixture was stirred for 50 minutes under nitrogen and concentrated in vacuo. The crude was purified by RP automated chromatography [Biotage (water + 0.1% TFA/ACN + 0.1% TFA, 10- 100%)] and appropriate fractions combined and lyophilised to give the desired fully- deprotected imidazoquinoline-pGA amine 53 as a white solid (quantitative yield). LCMS m/z 772.50 (M + l); 386.81 (M/2); 377.81 (M - payload).
To a stirred solution of bis-PEG?-NHS (28.1 mg, 0.045 mmol) dissolved in dry DMF (1 ml) and DIPEA (2.4 pL, 0.0136 mmol) a solution of the Imidazoquinoline-pGA-amine 53 (3.5 mg, 0.0045 mmol) dissolved in dry DMF (1 ml) was added dropwise over 1 h using a syringe pump. Once addition was complete the solvent was removed on high vacuum and the crude purified by RP automated chromatography [Biotage (water + 0.1% TFA/ACN + 0.1% TFA, 0-100%)] to give the desired N^Benzylamino Imidazoquinoline-PEG? NHS ester 54 as white powder after lyophilisation 3 mg (52%). LCMS m/z = 1278.20 (M+l); 386.81 (M/2); 1300.18 (M + Na).
Example 22: Synthesis of Triptolide-PEGe NHS Ester (58)

As described in Zhang K et al. [226], succinic anhydride (11 mg, 111 pmol), DMAP (0.7 mg, 5.6 pmol) and triptolide (10 mg, 28 pmol) were dissolved in 0.6 ml of dry pyridine and allowed to stir at RT under inert atmosphere for 26h.The mixture was diluted with ethyl acetate (5 ml) and washed with saturated copper sulphate solution (5 ml), water (5 ml), and brine (5 ml). The organic phase was separated, dried over sodium sulphate and the solvent was removed under reduced pressure. The crude was purified by NP automated chromatography [Biotage (DCM/MeOH 0-20%)] to give the triptolide succinate 55 as a white solid 6 mg (47%). Monitored by TLC (H/EA, 3: 1). Rf = 0.25. LCMS m/z = 461.27 (M+l), 483.30 (M+Na), 361.27 (triptolide fragment).
As described in Zhang K et al. [226], triptolide succinate 55 (3 mg, 6.5 pmol) was dissolved in dry DMF (0.2 ml) and dry DCM (1.6 ml), DCC (1.6 mg, 7.8pmol) and n- hydroxysuccinimide (0.83 mg, 7.2 pmol) were sequentially added and the mixture was allowed to stir at RT under an inert atmosphere for 24 hours. The mixture was concentrated under a vacuum and the residue purified by RP automated chromatography [Biotage (water + 0.1% TFA/ACN + 0.1% TFA, 10-90%)] to give NHS-activated ester triptolide derivative 56 2.57 mg (71%). m/z: 558.39 (M+l), 558.40 (M+Na), 1115.81 (dimer), 361.22 (triptolide fragment).
The triptolide succinic acid NHS ester 56 (1.2 mg, 2.2 pmol) was dissolved in dry DMF (1 ml) and DIPEA (1.1 pL, 6.5 pmol) was added followed by the addition of the amino-PEGe- COOH. The mixture was allowed to stir at RT under inert atmosphere for 2 hours after which the solvent was removed under a vacuum to give triptolide-succinate-PEGe-acid 57 which was used directly in the next step without purification. LCMS m/z = 796.61 (M+l), 818.64 (M+ Na).
Triptolide-succinate-PEGe acid 57 (1.71 mg, 2.2 pmol) was stirred with TSTU (1.3 mg, 4.3 pmol) and DIPEA (1.9 pl, 10.7 pmol) in dry DMF (1 ml) for two hours under nitrogen. The solvent was removed at high vacuum and the residue was purified by RP automated chromatography [Biotage (water + 0.1% TFA/ACN + 0.1% TFA, 0-90%)] to give the triptolide-succinate-PEGe NHS ester 58 as a white solid after lyophilisation 0.5 mg. LCMS m/z = 893.70 (M + l), 915.70 (M + Na).
Example 23: Synthesis of Maleimidocaproly-GGFG-Exatecan (59)

As described in EP3101032, DXd-GGFG 20 (50 mg, 59.46 pmol) was dissolved in dry DMF, maleimidocaproyl-NHS hexanoate (20.16 mg, 65.41 pl) was then added followed by TEA (5.42 mg, 53.52 pl) and the reaction mixture stirred under nitrogen for 2 h. The solvent was removed under high vacuum and the residue purified by RP automated chromatography to give Maleimidocaproly-GGFG-Exatecan 59 as a yellow solid 6.42 mg (10.5%). LCMS m/z 1034.80 (M+l), 1056.78 (M + Na)
Example 24: Synthesis of Maleimidocaproly-Val-Cit-MMAE (61)

MMAE (40 mg, 55.71 |jmol), commercially available maleimidocaproyl-Val-Cit-PAB-PNP 60 (49.32 mg, 66.85 pl), HOBt (7.68 mg, 56.83 pl) and DIPEA (10.67 pl, 61.28 pmol) was stirred in dry DMF with a drop of pyridine under nitrogen for 19 h. The solvent was removed under high vacuum and the residue purified by RP automated chromatography to give the desired MMAE derivative 61 as a white solid after lyophilisation 17.7 mg. m/z = 1317.50 (M + l), 659.40 (M/2), 1341.47 (M + Na).
Example 25: Synthesis of MMAE-Val-Ala-Oligosaccharide-PEG2-TFP Ester (69)

As described in Quintana JM et al. [227], to a solution of methyl 2-hydroxy-2-(4- nitrophenyl)acetate (1.5 g, 7.1 mmol) in MeOH (12 ml) was added 10% Pd/C (300 mg). The reaction mixture was stirred at room temperature for 3 h under hydrogen. The reaction was filtered through Celite washed with with EtOAc and concentrated to afford methyl 2- (4-aminophenyl)-2-hydroxyacetate 62 as a yellow solid 760 mg (59%). LCMS m/z = 182.2 [M + H]+, XH NMR (400 MHz, DMSO-de) 6 7.01 (d, J = 8.4 Hz, 2H), 6.50 (m, 2H), 5.67 (d, J = 5.4 Hz, 1H), 5.07 (s, 2H), 4.90 (d, J = 5.2 Hz, 1H), 3.57 (s, 3H).
To a solution of (tert-butoxycarbonyl)-L-valyl-L-alanine (1.21 g, 4.19 mmol) in DMF (15 ml) was added dropwise 4-Methylmorpholine (637 mg, 6.29 mmol) at 0 °C, then HATU (1.75 g, 4.61 mmol) was added, followed by the addition of a solution of methyl 2-(4-
aminophenyl)-2-hydroxyacetate 62 (760 mg, 4.19 mmol) in DMF (6 ml), the ice bath was removed and the reaction mixture was stirred at room temperature for 5 h. The reaction was quenched with H2O and extracted with EtOAc (30 mix 3). The combined organic layers were washed with brine, dried over NazSC , filtered and concentrated, purified by column chromatography on silica gel (DCM/MeOH 50: 1) to afford methyl 2-(4-((S)-2-((S)-2- ((tert-butoxycarbonyl)amino)-3-methylbutanamido)propanamido)phenyl)-2- hydroxyacetate 63 as a yellow solid 1.4 g (74%). LCMS m/z = 474.3 [M + Na]+ H NMR (400 MHz, DMSO-cfe) 6 9.90 (d, J = 75.0 Hz, 1H), 8.14 (dd, J = 71.8, 7.4 Hz, 1H), 7.56 (dd, J = 16.0, 8.4 Hz, 2H), 7.34 - 7.29 (m, 2H), 6.75 (dd, J = 40.6, 8.6 Hz, 1H), 6.01 - 5.98 (m, 1H), 5.11 - 5.05 (m, 1H), 4.48 - 4.38 (m, 1H), 3.87 - 3.75 (m, 1H), 3.59 (s, 3H), 2.02 - 1.87 (m, 1H), 1.37 (d, J = 5.8 Hz, 9H), 1.29 (dd, J = 7.2, 1.8 Hz, 3H), 0.87 - 0.80 (m, 6H).
To a stirred solution of methyl 2-(4-((S)-2-((S)-2-((tert-butoxycarbonyl)amino)-3- methylbutanamido)propanamido)phenyl)-2-hydroxyacetate 63 (500 mg, 1.11 mmol) in dry DMF (8 ml) was added DIPEA (215 mg, 1.66 mmol) and bis(4-nitrophenyl) carbonate (674 mg, 2.21 mmol) at 0 °C. The resulting solution was stirred at room temperature for 4h, quenched with H2O and extracted with EtOAc (30 mLx 3). The combined organic layers were washed with brine, dried over Na2SO4, filtered and concentrated, purified by column chromatography on silica gel (DCM/MeOH 60: 1) to afford methyl 2-(4-((S)-2-((S)-2- ((tert-butoxycarbonyl)amino)-3-methylbutanamido)propanamido)phenyl)-2-(((4- nitrophenoxy)carbonyl)oxy)acetate 64 as a yellow solid 580 mg (85%). LCMS m/z = 639.4 [M+NaJ H NMR (400 MHz, DMSO-de) 6 10.07 (d, J = 71.4 Hz, 1H), 8.34 (d, J = 9.2 Hz, 2H), 8.13 - 8.07 (m, 1H), 7.72 - 7.64 (m, 2H), 7.58 (d, J = 9.2 Hz, 2H), 7.45 (d, J = 8.2 Hz, 2H), 6.09 (d, J = 2.4 Hz, 1H), 4.48 - 4.38 (m, 1H), 3.88 - 3.76 (m, 1H), 3.71 (s, 3H), 2.01 - 1.88 (m, 1H), 1.37 (d, J = 6.0 Hz, 9H), 1.30 (dd, J = 7.2, 2.4 Hz, 3H), 0.88 - 0.80 (m, 6H).
To a solution of methyl 2-(4-((S)-2-((S)-2-((tert-butoxycarbonyl)amino)-3- methylbutanamido)propanamido)phenyl)-2-(((4-nitrophenoxy)carbonyl)oxy)acetate 64 (181 mg, 0.29 mmol) in dry DMF (2 ml) was added DIPEA (114 mg, 0.88 mmol), HOBt (60 mg, 0.44 mmol) and MMAE (253 mg, 0.35 mmol). The mixture was stirred at rt overnight under nitrogen. The mixture was diluted with water (100 ml) and extracted with EtOAc (50 ml x 3). The combined organic layers were washed with brine (150 ml), dried over Na2SO4, filtered and concentrated. The residue was purified by chromatography on silica gel (DCM/MeOH 100: 1 to 20: 1) to afford the MMAE derivative 65 as a white solid 270 mg (77%). LCMS m/z = 1195.9 [M+H]7H NMR (400 MHz, DMSO-de) 6 10.16 - 9.87 (m, 1H), 8.32 - 8.03 (m, 2H), 7.87 (d, J = 8.4 Hz, 1H), 7.67 - 7.59 (m, 2H), 7.54 - 7.39 (m, 2H), 7.33 - 7.13 (m, 5H), 6.74 (dd, J = 36.4, 8.4 Hz, 1H), 5.87 - 5.77 (m, 1H), 5.42
- 5.31 (m, 1H), 4.79 - 4.19 (m, 5H), 4.06 - 3.90 (m, 2H), 3.85 - 3.75 (m, 1H), 3.63 - 3.58 (m, 3H), 3.26 - 3.09 (m, 8H), 3.08 - 2.84 (m, 5H), 2.45 - 2.20 (m, 2H), 2.17 - 1.67 (m, 7H), 1.59 - 1.42 (m, 2H), 1.37 (d, J = 7.8 Hz, 9H), 1.32 - 1.22 (m, 5H), 1.07 - 0.70 (m, 32H), 0.66 - 0.56 (m, 1H).
To a solution of the MMAE derivative 65 (760 mg, 0.64 mmol) in MeOH (3 ml) was added a solution of LiOH.HzO (86 mg, 2.03 mmol) in H2O (3 ml) dropwise at 0 °C, and the mixture stirred at rt for 2h. The mixture was acidified with IN HCI to pH-2, filtered with H2O and concentrated to afford the MMAE-mandelic acid derivative 66 as a white solid 700 mg (93%). LCMS m/z = 1182.6 [M+HP/H NMR (400 MHz, DMSO-de) 6 10.14 - 9.84 (m, 1H), 8.28 - 8.03 (m, 1H), 8.01 - 7.84 (m, 1H), 7.67 - 7.56 (m, 2H), 7.54 - 7.37 (m, 2H), 7.36
- 7.10 (m, 6H), 6.85 - 6.62 (m, 1H), 5.69 (d, J = 12.6 Hz, 1H), 5.43 - 5.30 (m, 1H), 4.74
- 4.24 (m, 4H), 4.07 - 3.90 (m, 2H), 3.85 - 3.74 (m, 1H), 3.63 - 3.52 (m, 1H), 3.26 - 3.15 (m, 9H), 3.11 (d, J = 6.4 Hz, 1H), 3.01 - 2.92 (m, 3H), 2.30 - 2.07 (m, 3H), 2.00 - 1.69 (m, 6H), 1.55 - 1.44 (m, 2H), 1.37 (d, J = 6.8 Hz, 9H), 1.29 (d, J = 6.8 Hz, 5H), 1.05 - 0.74 (m, 33H).
To a solution of the MMAE-mandelic acid derivative 66 (11 mg, 0.009 mmol) in dry DMF (1 ml) was added HATU (4.3 mg, 0.01 mmol), the mixture was stirred at rt for 0.5h, then 4-Methylmorpholine (2 mg, 0.02 mmol) and the neutral oligosaccharide (10 mg, 0.009 mmol) were added, the mixture was stirred at rt under nitrogen for 2h and directly purified by prep-HPLC to afford the MMAE-Oligosaccharide derivative 67 as a white solid 3 mg (14%).
To a solution of the MMAE-Oligosaccharide derivative 67 (3 mg, 0.001 mmol) in H2O (1 ml) was added TFA (0.3 mg, 0.003 mmol), the mixture was stirred at 50°C for 2h after which it was lyophilized to afford the Boc-deprotected derivative 68 which was used directly for the next step.
To a solution of the Boc-deprotected derivative 68 (2.86 mg, 0.001 mmol) in dry DMF (0.5 ml) was added bis(2,3,5,6-tetrafluorophenyl) 3,3'-(ethane-l,2-diylbis(oxy))dipropionate (0.7 mg, 0.001 mmol) and DIPEA (0.5 mg, 0.003 mmol). The reaction mixture was stirred at room temperature for 1 h under nitrogen after which it was directly purified by prep- HPLC to afford the tetrafluorophenyl ester derivative 69 as a white solid 1 mg (30%).
Example 26: Synthesis of Exatecan-MA-PEGi2-Val-Ala-PEG4-NHS Ester (74)

To a solution of methyl 2-(4-((S)-2-((S)-2-((tert-butoxycarbonyl)amino)-3- methylbutanamido)propanamido)phenyl)-2-(((4-nitrophenoxy)carbonyl)oxy)acetate 64 (500 mg, 0.811 mmol) and Exatecan mesylate (331 mg, 0.624 mmol) in dry DMF (10 ml) was added DIPEA (241 mg, 1.87 mmol). The reaction mixture was stirred at RT overnight under nitrogen. The reaction mixture was diluted with water (100 ml), filtered. The filter cake was purified by trituration with EtzO to afford methyl 2-(4-((S)-2-((S)-2-((tert- butoxycarbonyl)amino)-3-methylbutanamido)propanamido)phenyl)-2-((((lS,9S)-9- ethyl-5-fluoro-9-hydroxy-4-methyl-10,13-dioxo-2,3,9,10,13,15-hexahydro-lH,12H- benzo[de]pyrano[3',4':6,7]indolizino[l,2-b]quinolin-l-yl)carbamoyl)oxy)acetate 70 as a yellow solid 500 mg (67.5%). LCMS m/z = 913.60 [M + H]+. XH NMR (400 MHz, DMSO-d6)
6 10.07 (s, 1H), 8.36 (t, J = 9.8 Hz, 1H), 8.07 (s, 1H), 7.80 - 7.73 (m, 1H), 7.62 (t, J = 11.4 Hz, 2H), 7.41 (t, J = 8.6 Hz, 2H), 7.31 (d, J = 11.8 Hz, 1H), 6.68 (d, J = 8.8 Hz, 1H), 6.51 (d, J = 5.6 Hz, 1H), 5.86 (d, J = 4.4 Hz, 1H), 5.43 (d, J = 6.2 Hz, 2H), 5.32 - 5.24 (m, 2H), 4.41 (s, 1H), 3.82 (s, 1H), 3.68 (d, J = 12.4 Hz, 3H), 3.15 (s, 1H), 2.37 (s, 3H), 2.21 (m, 2H), 1.87 (m, 2H), 1.36 (s, 9H), 1.28 (m, 3H), 0.85 (m, 9H).
To a stirred solution of methyl 2-(4-((S)-2-((S)-2-((tert-butoxycarbonyl)amino)-3- methylbutanamido)propanamido)phenyl)-2-((((lS,9S)-9-ethyl-5-fluoro-9-hydroxy-4- methyl-10,13-dioxo-2,3,9,10,13,15-hexahydro-lH,12H- benzo[de]pyrano[3',4':6,7]indolizino[l,2-b]quinolin-l-yl)carbamoyl)oxy)acetate 70 (100 mg, 0.109 mmol) in a mixture of THF (2 mL) and MeOH (1 mL) was added a solution of LiOH (5.25 mg, 0.219 mmol, 2.0 eq.) in water (1 mL) at 0 °C. The resulting solution was stirred at 0 °C for 30 min. then diluted with 1 N HCI to pH7 and then concentrated in vacuo. The crude was purified by prep-TLC (DCM/MeOH 10: 1) to give the exatecan-madelic acid derivative 71 as a yellow solid 30 mg (30.6%). LCMS m/z = 899.20 [M+H]+. XH NMR (400 MHz, DMSO-d6) 6 9.98 (d, J = 75.2 Hz, 1H), 8.29 - 8.02 (m, 2H), 7.78 (d, J = 10.8 Hz, 1H), 7.59 (m, 2H), 7.44 - 7.28 (m, 3H), 6.75 (m, 1H), 6.52 (d, J = 4.2 Hz, 1H), 5.68 (s, 1H), 5.50 - 5.19 (m, 5H), 4.42 (q, J = 7.2 Hz, 1H), 3.80 (dt, J = 16.4, 7.8 Hz, 1H), 3.10 - 2.97 (m, 4H), 2.39 - 2.35 (m, 3H), 2.27 - 2.08 (m, 2H), 1.91 (m, 4H), 1.36 (m, 10H), 1.33 - 1.27 (m, 4H), 0.85 (m, 9H).
To a solution of the exatecan-mandelic acid derivative 71 (60 mg, 0.067 mmol) in DMF (2 ml) was added HATU (45 mg, 0.134 mmol) and DIPEA (26 mg, 0.202 mmol). The reaction mixture was stirred at room temperature for 15 min. after which the PEGi2-amine was added (75 mg, 0.134 mmol) and the mixture and stirred at RT overnight under nitrogen. The reaction was diluted with water (50 ml) and extracted with EtOAc (50 ml x 2). The combined organic layers were washed with brine, dried over NazSC , filtered and concentrated. The crude was purified by prep-TLC (DCM/MeOH 10: 1) to give the exatecan- PEG derivative 72 as a yellow solid 20 mg (20.8%). LCMS m/z = 1440.55 [M+H]+.
To a solution of the exatecan-PEG derivative 72 (20 mg, 0.0139 mmol) in DCM (2 ml) was added TFA (2 ml). The reaction mixture was stirred at room temperature for 1.5 h. The reaction mixture was concentrated in vacuo to afford the Boc-deprotected derivative 73 as the TFA salt (20 mg, crude). LCMS m/z = 1340.85 [M+H]+.
A solution of 73 (20 mg, 0.139 mmol) in dry DMF (1 ml) was added dropwise to a solution of bis(2,5-dioxopyrrolidin-l-yl) 4,7,10,13-tetraoxahexadecanedioate (40 mg, 0.83 mmol) and DIPEA (5.3 mg, 0.417 mmol) at 10 °C. The reaction mixture was stirred at 10 °C for 0.5 h. after which the mixture was purified by prep-HPLC to give the desired Exatecan-
MA-PEGi2-Val-Ala-PEG4-NHS ester activated 74 as a white solid 4.6 mg (19.3%). LCMS m/z = 1713.90 [M + H]+, 857.75 [1/2M + H]+.
Example 27: Synthesis of GENZ-644282-Val-Ala-BrPEG8-PEG2-NHS Ester (80)

GENZ-644282 (50 mg, 0.123 mmol), Fmoc-ValAla-PNP ester 75 (84 mg, 0.123 mmol) and HOBt (33 mg, 0.246 mmol) were dissolved in a mixture of anhydrous DMF (2 ml) and pyridine (0.4 mL), and DIPEA (21 pl, 0.123 mmol) was added. The mixture was stirred for 3 h at RT, dried under reduced pressure and the crude purified by NP automated chromatography [Biotage, Hex/EtOAc 0-100%) to give 0.12 mg (quantitative yield) of the desired Fmoc-val-ala-GENZ-644282 derivative 76 as a yellow oil. LCMS m/z: 949.79 (M + 1); 950.79 (M + 2); 972.87 (M+Na).
The Fmoc-protected GENZ-644282 derivative 76 (117 mg, 0.123 mmol) was dissolved in 10 ml of a 4% (v/v) solution of diethylamine in anhydrous DMF and the mixture was stirred at RT over 2 h. The reaction mixture was concentrated in vacuo and the crude purified by RP automated chromatography [Biotage (water + 0.1% TFA/ACN + 0.1% TFA, 0-100%)]. Combined fractions were concentrated to give the desired deprotected derivative 77 as an off-white solid 89 mg (quantitative yield). LCMS m/z: 727.60 (M + 1); 749.54 (M+Na).
To a solution of GENZ-644282-val-ala amine 77 (20 mg, 0.028 mmol) dissolved in dry DMF (1 ml). Fmoc-Lys-PEGs (24 mg, 0.028 mmol) dissolved in dry DMF (1.4 ml) was added followed by the addition of DIPEA (20 pL, 0.056 mmol). The resulting mixture was stirred for 4 h at RT after which the solvent was removed under high vacuum and the crude purified by RP automated chromatography [Biotage (water + 0.1% TFA/ACN + 0.1% TFA, 0-100%)] to give the Fmoc-GENZ-644282-val-ala-PEGs derivative 78 21 mg (53 % yield). LCMS m/z: 1472.40 (M + 1); 1494.39 (M+Na).
The Fmoc-GENZ-644282-val-ala-PEGs derivative 78 (36 mg, 0.025 mmol) was dissolved in dry DMF (3 ml) and cooled to 0°C in a water/ice bath. To this stirred solution under nitrogen, a solution of DBU (7.3 pL, 0.049 mmol) dissolved in dry DMF (2 mL) was slowly added dropwise over 1 h. After the end of the DBU addition, the mixture was quenched with AcOH (4.1 pL, 0.025 mmol), and concentrated under reduced pressure. The crude was purified by RP automated chromatography [Biotage (water + 0.1% TFA/ACN + 0.1% TFA, 0-0%)] and fractions combined and freeze dried to give the deprotected amine derivative 79 as a white solid 25 mg (68 % yield). LCMS m/z: 1250.20 (M + 1); 1272.11 (M + Na); 625.71 (M/2).
To a stirred solution of bis-NHS-PEGz (80 mg, 0.20 mmol) in dry DMF (2 ml), DIPEA (10.5 pL, 0.060 mmol) was added. In a separate flask, GENZ-val-ala-PEGs amine 79 (25 mg, 0.020 mmol) was dissolved in dry DMF (1 ml), this was then added dropwise to the stirred solution of the bis-NHS-PEGz solution over 1 h. Once addition was complete, the solvent was removed on high vacuum and the crude purified by RP automated chromatography [Biotage (water + 0.1% TFA/ACN + 0.1% TFA, 10-90%)]. to give the desired GENZ- 644282-Val-Ala-BrPEGs-PEG2-NHS ester derivative 80 as a white solid 12 mg (39 %). LCMS m/z: 1535.40 (M + 1); 1557.40 (M + Na).
Example 28: Synthesis of Exatecan N-MeGlucamine-GGFG-PEGz-NHS Ester (86)

As described in Li W [228], Fmoc-L-Glu-(2-phenylisopropyloxy)-OH 81 (100 mg, 204.69 pmol) and N-methyl-D-glucamine (60 mg, 307.04 pmol) were weighed in a round bottom flask to which was added DMF (5 ml) and DIPEA (35.6pl, 204.69 pmol), followed by DMTMM dissolved in a mixture of DMF (5 ml) and distilled water (1.25 ml). The mixture was allowed to stir at RT for 2h 25 min. The reaction mixture was concentrated in vacuo and the crude purified by RP automated chromatography [Biotage (water + 0.1% TFA/ACN + 0.1% TFA, 10-100%)]. The appropriate fractions were combined and lyophlised to give the N-methyl-D-glucamine glutamic acid derivative 82 as a white solid 118 mg of (87%). LCMS m/z 687.49 (M+Na), 547.40 iPrPhFmocGIu-N-methyl-D-glucamine 82 (108 mg, 162.23 pmol) was dissolved in a TFA in DCM solution (16 ml, 1% v/v) and stirred in in an ice bath. After 10 min, the reaction mixture was concentrated in vacuo and the deprotected derivative 83 used without purification in the next step, assuming full conversion. LCMS m/z 547.40 (M+l), 569.34 (M+Na).
N-methylglucamine-Glu-OH-Fmoc 83 (60 mg, 109.58 pmol), exatecan-GGFG-amine 4 (92 mg, 109.58 pmol), EDC (25 mg, 131.49 pmol), HOBt (14.8 mg, 109.58 pmol), and DIPEA (38.2 pl, 219.15 pmol) were dissolved in 6 ml DMF and allowed to stir for 20h at RT under nitrogen atmosphere. The reaction mixture was concentrated in vacuo and crude purified by RP automated chromatography [Biotage (water + 0.1% TFA/ACN + 0.1% TFA, 0- 100%)]. The combined fractions were lyophilised to give Fmoc-GluOH-N-methyl-D- glucamine-GGFG-Exatecan derivative 84 as a yellow solid. 83 mg (55 %). LCMS m/z 1371.89 (M + l), 1393.85 (M + Na)
Fmoc-GluOH-N-methyl-D-glucamine-GGFG-Exatecan 84 (41.5 mg, 30.28 pmol) was dissolved in dry DMF (1.5 ml) containing DIEA (106 pl, 1.03 mmol). The mixture was stirred at RT under inert atmosphere for 1.5 h. The reaction mixture was concentrated in vacuo and crude purified RP automated chromatography [Biotage (water + 0.1% TFA/ACN + 0.1% TFA, 0-100%)]. The combined fractions were freeze dried to give the deported glutamic acid derivative 85 as a yellow solid 18 mg (52 %). LCMS m/z 1148 (M+l), 1170 (M+Na)
N-methyl-D-glucamine-GluOH-GGFG-exatecan 85 (18 mg, 16.59 pmol) was dissolved in dry DMF (1 ml) and added dropwise over one hour (syringe pump), to a stirred solution of bis-PEGz-NHS ester(62.9 mg, 165.90 pmol) and DIPEA (8.2 pl, 47.07 pmol) at RT under inert atmosphere. After addition was complete, the reaction mixture was concentrated in vacuo and the crude purified RP automated chromatography [Biotage (water + 0.1% TFA/ACN + 0.1% TFA, 0-100%)]. The combined fractions were lyophilised to give the N- methyl-D-glucamine-GluOH-GGFG-exatecan-PEGz-NHS ester derivative 86 as a lightyellow solid 4.23 mg (34%). LCMS m/zl433.70 (M+l), 1455.72 (M + Na)
Example 29: Synthesis of MMAE-pGA-BrPEGi2-PEG2-NHS Ester (87)
 To a stirred solution of bis-NHS-PEGz (53 mg, 0.134 mmol) dissolved in dry DMF (500 pl) and DIPEA (7 pl, 0.0403 mmol), MMAE-[3GA-BrPEGi2-amine 14 (22.2 mg, 0.0134 mmol) dissolved in dry DMF (840 pl) was added dropwise over 1 h using a syringe pump. Afre addition was complete, the solvent was removed on high vacuum and the crude purified by RP automated chromatography [Biotage (water + 0.1% TFA/ACN + 0.1% TFA, 10- 90%)] to give MMAE-[3GA-BrPEGi2-PEG2-NHS Ester 87 as a white solid after lyophilisation 35.27 mg (86%). LCMS m/z = 2116.50 (M+l); 1058.50 (M/2); 718.90 (MMAE); 506.59 (MMAE fragment).
To a stirred solution of bis-NHS-PEGz (53 mg, 0.134 mmol) dissolved in dry DMF (500 pl) and DIPEA (7 pl, 0.0403 mmol), MMAE-[3GA-BrPEGi2-amine 14 (22.2 mg, 0.0134 mmol) dissolved in dry DMF (840 pl) was added dropwise over 1 h using a syringe pump. Afre addition was complete, the solvent was removed on high vacuum and the crude purified by RP automated chromatography [Biotage (water + 0.1% TFA/ACN + 0.1% TFA, 10- 90%)] to give MMAE-[3GA-BrPEGi2-PEG2-NHS Ester 87 as a white solid after lyophilisation 35.27 mg (86%). LCMS m/z = 2116.50 (M+l); 1058.50 (M/2); 718.90 (MMAE); 506.59 (MMAE fragment).
Example 30: Synthesis of MMAE-pGA-BrPEGi2-PEG7-NHS Ester (88)

Bis-PEGyNHS (78 mg, 0.126 mmol) was dissolved in dry DMF (1 ml), DIPEA (6.6 pl, 0.038 mmol) was then added. To this stirred solution, MMAE-[3GA-BrPEGi2-amine 14 (23 mg, 0.0126 mmol) dissolved in dry DMF (1 ml) was then added dropwise via syringe pump over 1 h. After addition was complete, the solvent was removed on a high vacuum and the crude purified by RP automated chromatography [Biotage (water + 0.1% TFA/ACN + 0.1% TFA, 10-90%)] to give MMAE-pGA-BrPEGi2-PEG7-NHS Ester 88 as a white solid after lyophilisation 11.48 mg (39%). m/z = 2335.87 (M+l), 1167.46 (doubly charged ion), 718.40 (MMAE), 506.19 (MMAE fragmentation).
Example 31: Synthesis of SN38-pGA-DMEDA-PEG2-NHS Ester (95)

As described in Meyer Y et al [229], Phosgene solution (370 pl, 15% in toluene) was placed in a round bottom flask and cooled in an ice bath under nitrogen. In a separate flask, the starting N-Boc-diethylamine 89 (95 mg, 0.505 mmol) and triethylamine (77 pl, 0.555 mmol) were dissolved in dry toluene (1 ml). The solution of the amine 90 was then added dropwise to the phosgene solution over 15 minutes, then allowed to stir for 20 hours under nitrogen. The reaction mixture was evacuated and the crude containing the chlorocarbonyl 90 was used directly in the next reaction without further purification.
SN38 (99 mg 0.2525 mmol) was added to the chlorocarbonyl-boc-amine 90, pyridine (1 ml) was then added, and the resulting mixture allowed to stir at RT under inert atmosphere for 21 hours. The reaction was quenched with water (5 ml) and extracted with ethyl acetate (3 x 5 ml). The organic phases were combined, washed with brine, dried over magnesium sulphate, and evaporated to dryness under reduced pressure. The residue was purified on NPB [Biotage (DCM/MeOH, 0-20 %)] to afford the desired SN-38-Boc-DMEDA derivative 91 as a transparent oil 71 mg (46 %).
SN-38-Boc-DMEDA 91 (71 mg, 0.117 mmol), was dissolved a 1: 1 solution of DCM and trifluoroacetic acid (2 ml), the reaction mixture was stirred in an ice/water bath for 2h under nitrogen. The reaction mixture was concentrated in vacuo and the crude deprotected SN-38-DMEDA 92 used in the next reaction without any further purification.
The PNP-carbonate ester 5 (107 mg, 0.117 mmol), HOBt (16 mg, 0.119 mmol) and crude SN-38-DMEDA 92 (59 mg, 0.117 mmol, quantitative yield assumed) stirred in dry THF (3.3 ml), pyridine (0.65 ml) DIPEA (422 pL, 0.129 mmol) were then added and the reaction mixture stirred at RT under nitrogen for 20 h. Reaction mixture was then concentrated in vacuo and crude purified on NP automated chromatography [Biotage (DCM/MeOH, 0-20 %)]. Fractions were combined and concentrated to give the SN-38-DMEDA-glucuronide 93 as a yellow solid 98 mg (65%), found m/z = 1210.93 (M + 1), 1234.85 (M + Na)
The SN-38-DMEDA-sugar 93 (48 mg, 0.0375 mmol) was dissolved in MeOH (4.3 ml) and potassium carbonate (26 mg, 6.1873 mmol) was added to the solution. The reaction mixture was stirred for 2 hours, and then neutralised with HCI 0.1 M solution. The solvent was removed under high vacuum and the crude purified by RP automated chromatography [Biotage (water + 0.1% TFA/ACN + 0.1% TFA, 0-100%)]. to give globally deprotected SN-38-DMEDA-PGA 94 as a yellow solid 15 mg (44 %).
To a stirred solution of bis- PEG2-NHS ester (15 mg, 0.0163 mmol), and DIPEA (14 pl, 0.0816 mmol) in dry DMF (3 ml,) SN-38-DMEDA-PGA 94 dissolved in DMF (6 ml) was added dropwise to the solution over Ih 40 min under nitrogen atmosphere using a syringe pump. The reaction mixture was then stirred for a further 45 min. The solvent was removed using high vacuum and the crude purified by RP automated chromatography [Biotage (water + 0.1% TFA/ACN + 0.1% TFA, 0-100%)] to give SN38-pGA-DMEDA-PEG2- NHS Ester 95 as a pale-yellow solid, 9 mg (29 %).
Example 32: Synthesis of SN38-BA-Val-Ala-PEG2-NHS Ester (101)

As described in Zhu Q et al. [230], Di-^utyl-dicarbonate (47 mg, 0.2153 mmol) and pyridine (401 pL, 4.9693 mmol) were added to a suspension of SN-38 (65 mg, 0.1656 mmol) in dry DCM (3 ml). The mixture was stirred at room temperature overnight under an inert atmosphere. The solution was washed with 1 N HCI (5 ml), saturated NaHCCh solution (5 ml), brine (5 ml) and dried over NazSC . The solvent was removed under vacuum to give the crude Boc-SN-38 96 which was used without further purification. LCMS m/z = 493.30 (M + l)+
As described in Zhu Q et al. [230], Boc-SN-38 96 (89 mg, 0.1807 mmol), 4-tert- Butoxycarbonylaminobutyric acid (73 mg, 0.3600 mmol), EDCI (0.034 g, 0.21 mmol) and DMAP (0.033g, 0.27 mmol) were dissolved in dry DCM (4 ml) and stirred overnight at room temperature under nitrogen. The solution was washed with water (10 ml), brine (10 ml) dried over Na2SO4 and the solvent was removed under vacuum. The residue was purified by RP automated chromatography [Biotage (water + 0.1% TFA/ACN + 0.1% TFA, 0-100%)] to give Boc-SN-38-Fmoc-butylamine derivative 97 as a yellow powder 27.00 mg (22 %).
Boc-SN-38-Fmoc-butylamine derivative 97 (26.6 mg, 0.050 mmol) was placed in an oven- dried flask under nitrogen and dissolved in dry DCM (1.0 ml). This solution was placed in an ice/water bath and 0.5 ml (7.651 mmol) of TFA in 0.5 ml of dry DCM was added
dropwise at 0°C using a syringe pump. After 1 h the cold bath was removed and the reaction mixture stirred for a further 30 minutes. The reaction mixture was then concentrated under reduced pressure, providing a dark yellow oily residue as the doubledeprotected SN-38-butylamine derivative 98 which was used in the next step without any further purification. LCMSm/z = 478.30 (M + l)+
To a solution of Fmoc-Val-Ala-PAB-PNP (25 mg, 0.0367 mmol) and the crude SN-38- butylamine derivative 98 in dry DMF (1.5 ml), DIPEA (32 pL, 0.1836 mmol) was added, the flask was then covered with aluminium foil and the reaction stirred under nitrogen for 1 h at 4 °C. The reaction mixture was then allowed to warm up and stirred for a further 5 hours at RT. The DMF was evaporated, and the crude purified by RP automated chromatography [Biotage (water 0.1% TFA/ ACN 0.1% TFA 0-100%)] to give SN-38-BA- Fmoc-val-ala 99 as pale-yellow powder 11 mg (30%). LCMS m/z = 1019.72 (M+l)
SN-38-BA-Fmoc-val-ala 99 was dissolved in a round bottom flask containing diethylamine (50 pl) in dry DMF (0.25 mL). After 1 h, the reaction mixture was concentrated, dried under high vacuum to give SN-38-val-ala-amine 1OO which was used in the next step without further purification. LCMS m/z = 797.60 (M+ l)+
SN-38-val-ala-amine 1OO (7 mg, 0.0088 mmol) in dry DMF (1 ml) was added via syringe pump over 1 hour into a stirred solution of bis-PEGz-NHS ester (40 mg, 0.1004 mmol) and DIPEA (5.2 uL, 0.0301 mmol) in DMF (0.5 ml) and under nitrogen. The reaction mixture was stirred for one further hour, and then evaporated to dryness under reduced pressure. The crude was purified via RP automated chromatography [Biotage (water 0.1% TFA/ ACN 0.1% TFA 0-100%)], fractions were combined and lyophilized to give SN38-BA-Val-Ala- PEG2-NHS ester 101 as pale-yellow powder. LCMS m/z = 1082.8 (M+l)
Example 33: Synthesis of MMAE-pGA-BrPEG4-PEG2-NHS Ester (104)

As described in RP Lyon et al. [192]. To a stirred solution of MMAE-pGA-alanine derivative 7 (62 mg, 0.0498 mmol) dissolved in dry DMF (0.7 ml) and DIPEA (17.4 pl, 0.0996 mmol), a solution of the activated Fmoc-Lys-PEG4-NHS ester 106 (40.9 mg, 0.0598 mmol) dissolved in DMF (1 ml) was added. The reaction mixture was stirred for 4 h after which the solvent was removed under high vacuum and the oily residue purified by RP automated chromatography [Biotage: H2O (0.1% TFA)/CHsCN (0.1% TFA), 10-90%] to give the Fmoc-lysine-PEG4 derivative 102 as a white solid 80 mg (94 % yield) after lyophilisation. LCMS m/z = 1699.00 (M+l), 850.00 (1/2 M+l).
To a stirred solution of Fmoc-lysine-PEG4 derivative 102 (80 mg, 0.0471 mmol) was dissolved in dry DMF (3 ml) and cooled to 0°C in a water/ice bath, DBU (21.2 pL, 0.141 mmol) dissolved in dry DMF (1.7 mL) was added dropwise over 1 h under nitrogen. After addition was complete, the mixture was allowed to stir for a further 30 min. the ice bath was then removed and the reaction was quenched with AcOH (19 pl, 0.3325 mmol), and concentrated. The residue was purified by RP automated chromatography [Biotage: H2O (0.1% TFA)/CHsCN (0.1% TFA), 30-100%] to give the deprotected lysine-PEG4 derivative 103 64.2 mg (92 %).
To a solution of Bis-PEGz-NHS (203 mg, 0.0508 mmol) and DIPEA (26.5 pl, 0.1520 mmol) in dry DMF (3 ml) a solution of the MMAE-[3GA-BrPEG4 103 (75 mg, 0.0508 mmol) in dry DMF (2.1 ml) was added drop wise over 1 h using a syringe pump. After addition was complete the mixture was allowed to stir for further 30 min. after which the solvent was removed on high vacuum and the crude purified by RP automated chromatography [Biotage (water + 0.1% TFA/ AON + 0.1% TFA, 10-90%)] to give the desired MMAE-pGA- BrPEG4-PEG2-NHS Ester 104 as a white solid after lyophilisation 65.0 mg (73%). LCMS (M + K) 1800.74, ((1/2 M + K) 901.33.
To Fmoc-Lys-OH (300 mg) in dry DCM (16 mL) was added a solution of the PEG4-NHS ester in dry DCM (4 mL) followed by DIPEA (710 pL) - mixture is a suspension, so dry DMF (2 ml) was added and the mixture stirred, becoming clear after 1.5 h. The clear reaction mixture was stirred at RT under nitrogen overnight after which it was quenched with IM NH4CI solution (20 mL). The phases were separated, and the aqueous layer was further extracted with DCM (3 x 10 ml). The organic phases were combined, dried over MgSC , filtered and concentrated to dryness under reduced pressure to give a colourless oil that was purified by NP automated chromatography [Biotage (DCM/MeOH 0-10%)] to give the lysine-PEG4 105 as an oil 449 mg (94%). TLC, DCM/MeOH (25 %). Rf = 0.064. LCMS m/z = 587.00 (M + l).
Fmoc-Lys-PEG4 105 (140 mg, 0.239 mmol) was dissolved in dry DMF (3 ml), TSTU (143.7 mg, 0.477 mmol) and DIPEA (207.3 pL, 1.19 mmol) were added, and the mixture stirred at RT under nitrogen. After 2.5 h, the solvent was removed under high vacuum and the crude purified by NP automated chromatography [Biotage (DCM/MeOH 5-10%)], to give the lysine-PEG4-NHS ester 106 152 mg (93%). TLC (DCM/MeOH, 5%). Rf = 0.18. LCMS m/z = 685.00 (M + l).
Example 34: Synthesis of MMAE-pGA-BrLys(BisPEG8)-PEG2-NHS Ester (112)

As described in Chan KF et al. [231], in a round bottom flask, di(2, 5, 8, 11, 14, 17, 20,23- octaoxapentacosan-25-yl)amine (131 mg, 0.175 mmol) and succinic anhydride (35 mg, 0.349 mmol) were dissolved in 10 ml of pyridine and stirred at RT, under nitrogen for 3 hours. The reaction mixture was then poured into a separating funnel containing 1 M hydrochloric acid solution and the mixture continuously extracted with DCM. The combined organic layers were dried over MgSO4, filtered, and evaporated to give the Bis-PEGs-amine butanoic acid 107 light brown oil (assumed quantitative yield) that was used without further purification. LCMS m/z: 850.70 (M+l); 872.70 (M+Na); 425.90 (1/2 M+ l).
Bis-PEGs-amine butanoic acid 107 (140 mg, 0.174 mmol), DIPEA (162 uL, 0.152 mmol) and TSTU (105 mg, 0.348 mmol) were dissolved in dry DMF (5 ml) and stirred under nitrogen at RT for 5 hours. The solvent was removed on high vacuum and the crude activated ester 108 used without further purification. LCMS m/z: 947.72 (M+l); 964.80 (M + H2O; 474.43 (1/2 M + l).
To Fmoc-Lys-OH (63.8 mg, 0.173 mmol) in dry DCM (3 ml) was added a solution of the bis-(PEG)s activated ester 108 (164 mg, 0.173 mmol) dissolved in dry DCM 0.5 ml) followed by DIPEA (151 pL, 0.866 mmol). The mixture was stirred at RT under nitrogen for 5 hours and quenched with IM NF CI solution (5 ml). Phases were separated, and the
aqueous layer was further extracted with DCM (3 x 10 mL). The organic phases were combined, dried over MgSC , filtered and concentrated to dryness under reduced pressure to give a colourless oil that was purified by RP automated chromatography [Biotage (water + 0.1% TFA/ACN + 0.1% TFA, 10-90%)] to give the Fmoc-Lys-Bis-(PEG)s-acid as an off- white oil 60 mg (29 %). LCMS m/z = 1200.96 (M + l); 601.14 (M/2). The Fmoc-Lys-Bis- (PEG)s-acid (60 mg, 0.050 mmol), DIPEA (43,5 uL, 0.250 mmol) and TSTU (30.1 mg, 0.100 mmol) were dissolved in dry DMF (3 ml) and stirred under nitrogen at RT for 5 hours. The solvent was removed on high vacuum and the crude purified by RP aoutomated chromatography [Biotage (water + 0.1% TFA/ACN + 0.1% TFA, 0-100%)] to give Fmoc- Lys-Bis-(PEG)s-NHS ester 109 as a clear viscous oil 51 mg (80 %). LCMS m/z: 1299.99 (M + l); 649.61 (1/2 M + l).
To a stirred solution of MMAE-[3A 7 (11 mg, 0.0097 mmol) was dissolved in anhydrous DMF (2 ml), Fmoc-Lys-Bis-(PEG)s-NHS ester 109 (12.6 mg, 0.0097 mmol) was added followed by DIPEA (5.1 pl, 0.029 mmol). After 3 h of stirring at RT under nitrogen atmosphere, the solvent was removed under high vacuum and the crude purified in RP automated chromatography [Biotage (water + 0.1% TFA/ACN + 0.1% TFA, 10-100%]) and appropriate fractions combined and lyophilised to give the Fmoc-bis-(PEGs)-MMAE- glucuronide derivative 110 20 mg (89 % yield) as a white solid. LCMS m/z: 2314.50 (M + l); 1157.45 (1/2 M+ l); 718.80 (MMAE).
The Fmoc-bis-(PEGs)-MMAE-glucuronide derivative 110 (20 mg, 0.0087 mmol) was dissolved in anhydrous DMF (2 ml) and cooled in an ice bath. DBU (3.9 pL, 0.026 mmol) dissolved in anhydrous DMF (1 ml) was added dropwise over one hour under inert atmosphere. After the end of the addition, the mixture was allowed to stir for further 20 min, the solvent was removed under high vacuum and the crude purified by RP automated chromatography [Biotage (water + 0.1% TFA/ACN + 0.1% TFA, 10-100%)] to give bis- (PEGs)-MMAE-glucuronide amine derivative 111 214 mg (78 % yield) as a white solid after lyophilisation. LCMS m/z: 2093.30 (M+l); 1047.20 (1/2 M + l).
To a stirred solution of bis-PEGz-NHS ester (3.8 mg, 0.0010 mmol) dissolved in DMF (1 ml), and DIPEA (0.5 pL, 0.003 mmol, bis-(PEGs)-MMAE-glucuronide amine 111 (2 mg, 0.001 mmol) dissolved in DMF (1 m) was added dropwise over 35 min. using a syringe pump. Once addition was complete, the mixture was allowed to stir for further 1 hour after which the solvent was removed on high vacuum and the crude purified by RP automated chromatography [Biotage (water + 0.1% TFA/ACN + 0.1% TFA, 10-90%)] to give MMAE- PGA-BrLys(BisPEGs)-PEG2-NHS ester 112 as a white solid after lyophilisation 120.91 mg (40 %). LCMS m/z = 2376.60 (M + l), 1188.90 (1/2 M + l), 2398.36 (M + Na).
Example 35: Synthesis of MMAE-pGA-BrGlu(BisPEGs)-PEG2-NHS Ester (118)

As described in Agren JKM et al. [235], Di(2,5,8,ll,14,17,20,23-octaoxapentacosan-25- yl)amine (10 mg, 0.013 mmol), FmocGIuOtBu (5.7 mg, 0.013 mmol), EDC (3 mg, 0.026 mmol), HOBt (1.8 mg, 0.013 mmol) and DIPEA (4. pL, 0.027 mmol) were dissolved in 1 ml dry THF. The mixture was stirred under nitrogen at RT for 21 hours. The solvent was removed on high vacuum and the crude was purified by RP automated chromatography [Biotage (water + 0.1% TFA/ACN + 0.1% TFA, 0-100%)] to give PEGylated glutamic acid derivative 113 15 mg (97 %) as a white powder. LCMS m/z 1159.20 (M+ l); 1182.21 (M+Na).
The starting double-protected PEGylated glutamic acid derivative 113 (15 mg, 0.013 mmol) was dissolved in 1 ml neat TFA and the mixture stirred for 30 min. under nitrogen. The excess TFA was removed on high vacuum and the crude Fmoc-bis(PEGs)-glutamic acid 114 was used without further purification. LCMS m/z: 1103.10 (M+l); 1125.12 (M+Na).
Fmoc-bis(PEG8)-glutamic acid 114 (88 mg, 0.080 mmol) and TSTU (48 mg, 0.161 mmol) were dissolved in dry DMF (1 ml); DIPEA (70 pl, 0.401 mmol) was added and the mixture stirred for 5 h at RT after which it was under high vacuum. The residue was purified by RP automated chromatography [Biotage (water + 0.1% TFA/ACN + 0.1% TFA, 0-100%)] to give Fmoc-bis(PEGs)-glutamic acid NHS ester 115 as a clear oil 80 mg (83%). LCMS m/z 1200.90 (M+l); 1122.90 (M+Na).
To a stirred solution of MMAE-pA 7 (41 mg, 0.036 mmol) dissolved in dry DMF (6 ml). NHS-activated Fmoc-Glu-bis(PEGs) (43.50 mg, 0.036 mmol) dissolved in dry DMF (1 ml) was added solution followed by DIPEA (19 pl, 0.109 mmol). The reaction mixture was stirred for 4 h at RT after which the solvent was removed under high vacuum and crude purified by RP automated chromatography [Biotage (water + 0.1% TFA/ACN + 0.1% TFA, 15-100%)] to give the MMAE-Fmoc-bis(PEG8)-0GA 116 5 mg (31%) as a sticky white solid. LCMS m/z 2215.40 (M + l); 2237.30 (M + Na); 718.83 (MMAE); 506.49 (MMAE fragment).
MMAE-Fmoc-bis(PEG8)-0GA 116 (5 mg, 0.0023 mmol) was dissolved in dry DMF (1 ml) and DBU (1.1 pL, 0.0068 mmol) was added in one portion. The reaction mixture was stirred at RT under nitrogen. After 1.5 h, the solvent was removed under high vacuum and the crude purified by RP automated chromatography [Biotage (water + 0.1% TFA/ACN + 0.1% TFA, 0-100%)] to give MMAE- bis(PEG8)-|3GA amine 117 3 mg (67 %) as a white solid were obtained. LCMS m/z 1992.20 (M+ l); 996.84 (1/2 M+l); 718.80 (MMAE); 506.60 (MMAE fragment).
MMAE- bis(PEG8)-0GA amine 117 (8 mg, 0.004 mmol) was dissolved in dry DMF (1 mL) and added dropwise to a solution of bis-PEG4-NHS ester (16.1 mg, 0.040 mmol) and DIPEA (2.1 pL, 0.012 mmol) in ImL DMF over one hour using a syringe pump. After the addition, was complete, the reaction mixture was stirred at RT under inert atmosphere for 1 further hour. The solvent was then removed under high vacuum and the crude purified by RP automated chromatography [Biotage (water + 0.1% TFA/ACN + 0.1% TFA, 0-100%)] to give the desired MMAE-pGA-BrGlu(BisPEGs)-PEG2-NHS Ester 118 as a white solid after lyophilisation 3.58 mg (67%). LCMS m/z 2217.39 (M + l); 1139.67 (1/2 M + l); 718.80 (MMAE); 686.79, 506.56 (MMAE fragment).
Example 36: Synthesis of MMAE-Val-Ala-BrPEGs-PEG2-NHS Ester (123)

MMAE (100 mg, 0.139 mmol), Fmoc-ValAla-PAB-PNP (104 mg, 0.153 mmol) and HOBt (38 mg, 0.279 mmol) were placed in a round bottom flask and dissolved in a mixture of DMF (2 ml) and pyridine (0.4 ml). DIPEA (24 pL, 0.139 mmol) was added and the solution was allowed to stir at RT under nitrogen for 24 h. The mixture was dried under high vacuum and the crude was washed with ethyl acetate (3 x) to afford Fmoc-val-ala-MMAE 119 as a solid pure enough to carry on with the synthesis without any further purification, 127 mg (74%). LCMS m/z 1260.90 (M + l); 718.80 (MMAE)
Fmoc-ValAla-MMAE 119 (90 mg, 0.071 mmol) was dissolved in dry DMF (10 ml) and diethylamine (370 pL, 3.573 mmol) was added. The solution was allowed to stir at RT under nitrogen for 2 h and then dried under high vacuum. The crude was purified by RP automated chromatography [Biotage (water + 0.1% TFA/ACN + 0.1% TFA, 0-100%)], fractions combine and concentrated to afford the HzN-val-ala-MMAE 120 as a white solid 74 mg (quantitative yield). LCMS m/z 1038.10 (M+l), 1060.06 M+Na), 718.80 (MMAE).
H2N-ValAla-MMAE 120 (81 mg, 0.0781 mmol) and Fmoc-Lys-PEGs-NHS 12 (81 mg, 0.0937 mg) were dissolved in DMF (5 mL) and DIPEA (33 pL, 0.1874 mmol) was added. The solution was allowed to stir at RT under nitrogen for 5 h. and then a further amount (0.0698 mmol) of Fmoc-Lys-PEGs-NHS 12 was added to the reaction mixture, and stirring
continued for a further 18 h. The reaction mixture was evaporated under high vacuum and the crude residue purified by RP automated chromatography [Biotage (water + 0.1% TFA/ACN + 0.1% TFA, 0-100%)]. Fractions were combined and evaporated to give the Fmoc-branched lysine MMAE derivative 121 as an off-white solid 50 mg (36%). LCMS m/z 1783.00 (M+l); 1805.96 (M + Na); 718.84 (MMAE).
DBU (23.4 pL, 0.157 mmol) was dissolved in dry DMF (5 ml) and added dropwise over 1 hour, to a stirred solution of Fmoc-branched lysine MMAE 121 (93 mg, 0.052 mmol) dissolved in dry DMF (5 ml) at °C and under nitrogen. Once addition was complete the reaction mixture was quenched with acetic acid (14.7 pL, 0.261 mmol) and concentrated to dryness under high vacuum. The crude was purified by RP automated chromatography [Biotage (water + 0.1% TFA/ACN + 0.1% TFA, 15-100%)]. Fractions were combined and lyophilised to give the deprotected derivative 122 as a white powder (quantitative yield) LCMS m/z 1560.70 (M + l); 1582.70 (M + Na); 718.81 (MMAE).
Bis-PEGz-NHS ester (110 mg, 0.275 mmol) was dissolved in dry DMF (2 ml) and DIPEA (14.4 uL, 0.083 mmol) was added. To this stirred solution under nitrogen, MMAE-val-ala- Lys-PEG8-NH2 122 (43 mg, 0.0276 mmol) dissolved in dry DMF (2 ml) was added dropwise over 1 h with a syringe pump. Once addition was complete the solvent was removed on high vacuum and the crude purified by RP automated chromatography [Biotage (water + 0.1% TFA/ACN + 0.1% TFA, 10-90%)] to give MMAE-Val-Ala-BrPEG8-PEG2-NHS ester 123 as a white solid after lyophilisation 21 mg (41 %). LCMS m/z 1846.90 (M + l); 923.97 (M/2); 718.70 (MMAE); 686.60 (MMAE fragment).
Example 37: Synthesis of MMAE-Val-Ala- BrLys(BisPEG8)-PEG4-NHS Ester (126)

HzN-ValAla-MMAE 120 (33 mg, 0.032 mmol), the NHS-activated bis-PEGs linker 109 (38 mg, 0.032 mmol), and HATU (24.1 mg, 0.063 mmol) were dissolved in dry DMF (3 ml) and DIPEA (16.6 pl, 0.095 mmol) was added. The mixture was stirred under nitrogen at RT for 2.5 hours. The solvent was removed on high vacuum and the crude was purified by RP automated chromatography [Biotage (water + 0.1% TFA/ACN + 0.1% TFA, 0-100%)] to give the Fmoc-val-ala-MMAE-bisPEGs derivative 124 as a white powder 45 mg (64 %). LCMS m/z 2221.40 (M + l); 2243.37 (M + Na); 1111.18 (1/2 M+l); 718.80 (MMAE); 506.52 (MMAE fragment).
DBU (4.5 pl, 0.030 mmol) was dissolved in dry DMF (1 ml) and added dropwise over 1 hour, to a stirred solution of Fmoc-ValAla-MMAE-bisPEGs 124 (22 mg, 0.010 mmol) in dry DMF (1 ml) at °C and under nitrogen. Once addition was complete, the mixture was quenched with acetic acid (1.80 pL, 0.061 mmol) and concentrated to dryness under high vacuum to give the crude deprotected derivative MMAE-val-ala-bisPEGs amine 125. This was used without further purification. LCMS m/z 1999.28 (M + l); 1000.10 (1/2 M + l); 718.80 (MMAE); 506.50 (MMAE fragment).
Bis-PEG4-NHS ester (9.8 mg, 0.020 mmol) was dissolved in dry DMF (0.5 ml) and DIPEA (1.1 pL, 0.006 mmol) was added. To this stirred solution under nitrogen, MMAE-val-ala- Lys-bisPEGs-NH2 125 (4 mg, 0.002 mmol) dissolved in dry DMF (1 ml) was added dropwise over 1 h with a syringe pump. Once addition was complete, the solvent was removed on high vacuum and the crude purified by RP automated chromatography [Biotage (water + 0.1% TFA/ACN + 0.1% TFA, 10-90%)] to give MMAE-Val-Ala- BrLys(BisPEGs)-PEG4-NHS Ester 126 as a white solid after lyophilisation 2.16 mg (45 %). LCMS m/z 2372.60 (M + l); 1186.56 (M/2); 1208.88 (M/2, Na adduct); 718.85 (MMAE); 506.59 (MMAE fragment).
Example 38: Summary of cMET scFv linker-payload conjugates
Table 6 describes disclosed FDCs using a wide range of auristatin and non-auristatin based linker-payloads. Specific examples are described in Examples 39 to 57. Similar FDCs incorporating the 129D5B and 129D5BTY scFvs are described to demonstrate equivalence. The synthesis of the MMAF-PEG2 linker payload is described in WO2016046574.
Example 39: Conjugation of 129D5BTY scFv to MMAE-(3GA-brPEGs-PEG2-NHS Ester (10)
129D5BTY scFv was conjugated to linker-payload 10 to obtain a DAR 5.9 as per conditions described in WO2016046574. Specifically, a stock solution of pure scFv (>95%) was diluted to 4.3mg/ml into a pre-equilibrated solution of 20% DMSO in bicarbonate buffer pH9.2. Linker-Payload (25mM in DMSO) was added starting with 5 equivalents before adding more paylaod every 90-120 min and the reaction mixture incubated at 20°C, 300rpm until the desired DAR was achieved. The reaction progress was monitored by sizeexclusion chromatography mass spectrometry (SEC-MS, Agilent AdvanceBio SEC 120A 1.9mm 2.1x150mm PEEK isocratic run in 75mM Ammonium Acetate, 10% IPA on an Agilent QTOF 6545XT). A total of 19.2 equivalents (5 additions) were required.
The crude reaction mixture was pH neutralized with IM NaH2PO4 (1.5%) and purified on an AKTA Pure through a Superdex 75 26/600 isocratic run eluting with 10% IPA/PBS (Figure 14a). The monomeric fractions were combined, and buffer exchanged into Histidine/NaCI buffer pH7.5. The concentrated sample was filtered through a sterile 0.2|j.m PES filter, flash frozen in liquid nitrogen and stored at -80°C. The sample was analysed by SEC HPLC (Sepax Zenix C 150 7.8 x 300mm), SEC and RP-LCMS, UV/Vis spectroscopy, DLS and nanoDSF before being used for further studies. The sample was pure and monomeric (>98.5%, Figure 14b), with a DAR distribution of 3-9 (~80% within 5-7, Figure 14c) with a cumulative radius of ~3.4nm (Figure 14d). The NanoDSF data (Figure 14e) showed a decrease in thermal stability compared to the unconjugated parent antibody
129D5BTY but maintained a highly thermal stable folded structure with a Tm of 64.1°C and a binding affinity (/ d) of 2.16nM (Figure 14f), Table 6.
Example 40: Conjugation of 131D5S scFv to MMAE-(3GA-brPEGs-PEG2-NHS Ester (10)
131D5S scFv was conjugated to linker-payload 10 to obtain a DAR 5.4 as per conditions described in Example 39. Specifically, a stock solution of pure scFv (>95%) was diluted to Img/ml into a pre-equilibrated solution of 20% DMSO in bicarbonate buffer pH9.2. Linker- Payload (25mM in DMSO) was added in 5 equivalents every 90-120 min and the reaction mixture incubated at 20°C, 300rpm. The reaction progress was monitored by sizeexclusion chromatography mass spectrometry (SEC-MS, Agilent AdvanceBio SEC 120A 1.9mm 2.1x150mm PEEK isocratic run in 75mM Ammonium Acetate on an Agilent QTOF 6545XT. A total of 21 equivalents (5 additions) were required.
The crude reaction mixture was pH neutralized with 0.1M NaH2PO4 (10%) and purified on an AKTA Avant through a Superdex 75 26/600 isocratic run eluting with 10% IPA/PBS. The monomeric fractions (Figure 15a) were combined and buffer exchanged into Histidine/NaCI buffer pH7.5. The concentrated sample was filtered through a sterile 0.2|j.m PES filter, flash frozen in liquid nitrogen and stored at -20°C. The sample was analysed by SEC HPLC (Sepax Zenix C 150 7.8 x 300mm), SEC-LCMS (Figure 15b, UV/Vis spectroscopy, DLS (Figure 15c) and nanoDSF before being used for further studies. The binding affinity was 112pM (Figure 15d; Table 6).
Example 41: Conjugation of 77F3 scFv to MMAE-(3GA-brPEG4-PEG2-NHS Ester (104)
77F3 scFv was conjugated to Linker-Payload 104 to obtain a DAR 8.4 as per conditions described in Example 39. Specifically, a stock solution of pure scFv (>95%) was diluted to Img/ml into a pre-equilibrated solution of 20% DMSO in bicarbonate buffer pH9.2. Linker- Payload (25mM in DMSO) was added in 5 equivalents every 90-120 min and the reaction mixture incubated at 20°C, 300rpm. A total of 60 equivalents (12 additions) were required.
The crude reaction mixture was pH neutralized with IM NaH2PO4 (10%) and purified on an AKTA Avant through a Superdex 75 26/600 isocratic run eluting with 10% IPA/PBS. The monomeric fractions (Figure 16a) were combined and buffer exchanged into PBS. The concentrated sample was filtered through a sterile 0.2|j.m PES filter and stored at 4°C. The sample was analysed by SEC HPLC (Sepax Zenix C 150 7.8 x 300mm), amino acid analyses, SEC-LCMS (Figure 16b), UV/Vis spectroscopy, DLS and SEC-MALS before being used for further studies. The binding affinity was 10.4nM (Table 6).
Example 42: Conjugation of 129D5BTY scFv to MMAE-pGA-brPEGi2-PEG4-NHS Ester (15)
129D5BTY scFv was conjugated to Linker-Payload 15 to obtain a DAR 6.4 as per conditions described in Example 39. Specifically, a stock solution of pure scFv (>95%) was diluted to 4.8mg/ml into a pre-equilibrated solution of 20% DMSO in bicarbonate buffer pH9.2. Linker-Payload (25mM in DMSO) was added starting with 5 equivalents every 90-120 min and the reaction mixture incubated at 20°C, 300rpm. The reaction progress was monitored by size-exclusion chromatography mass spectrometry (SEC-MS, Agilent AdvanceBio SEC 120A 1.9mm 2.1x150mm PEEK isocratic run in 75mM Ammonium Acetate, 10% IPA on an Agilent QTOF 6545XT). A total of 8.3 equivalents (2 additions) were required.
The crude reaction mixture was pH neutralized with IM NaH2PO4 (1.5%) and purified on an AKTA Avant through a Superdex 75 26/600 isocratic run eluting with 10% IPA/PBS (Figure 17a). The monomeric fractions were combined and buffer exchanged into Histidine/NaCI buffer pH7.5. The concentrated sample was filtered through a sterile 0.2|j.m PES filter, flash frozen in liquid nitrogen and stored at -80°C. The sample was analysed by SEC HPLC (Sepax Zenix C 150 7.8 x 300mm) showing 99% monomeric FDC (Figure 17b), SEC-LCMS DAR 6.4 (Figure 17c), UV/Vis spectroscopy, DLS monomeric particle size (Figure 7d), nanoDSF showing Tm ~64.9 °C (Figure 17e) and binding affinity of 1.8nM (Figure 17f) before being used for further studies (Table 6).
Example 43: Conjugation of 129D5B scFv to MMAE-(3GA-brPEGi2-PEG7-NHS Ester (88)
129D5B scFv was conjugated to Linker-Payload 88 to obtain a DAR 7.5 as per conditions described in Example 39. Specifically, a stock solution of pure scFv (>95%) was diluted to Img/ml into a pre-equilibrated solution of 20% DMSO in bicarbonate buffer pH9.2. Linker- Payload (25mM in DMSO) was added in 5 equivalents every 90-120 min and the reaction mixture incubated at 20°C, 300rpm. The reaction progress was monitored by sizeexclusion chromatography mass spectrometry (SEC-MS, Agilent AdvanceBio SEC 120A 1.9mm 2.1x150mm PEEK isocratic run in 75mM Ammonium Acetate, 10% IPA on an Agilent QTOF 6545XT). A total of 25 equivalents (5 additions) were required.
The crude reaction mixture was pH neutralized with IM NaH2PO4 (1.5%) and purified on an AKTA Avant through a Superdex 75 26/600 isocratic run eluting with 10% IPA/PBS. The monomeric fractions were combined and buffer exchanged into Histidine/NaCI buffer pH7.5. The concentrated sample was filtered through a sterile 0.2|j.m PES filter, flash frozen in liquid nitrogen and stored at -20°C. The sample was analysed by SEC-LCMS (Figure 18a), UV/Vis spectroscopy, DLS and nanoDSF (Figure 18b) before being used for further studies. The binding affinity was 4.9nM (Figure 18c; Table 6).
Example 44: Conjugation of 129D5B scFv to MMAE-(3GA-brPEGs-PEG2-NHS Ester (10)
129D5B scFv was conjugated to linker-payload 10 to obtain a DAR 5.8 as per conditions described in Example 39. Specifically, a stock solution of pure scFv (>95%) was diluted to Img/ml into a pre-equilibrated solution of 20% DMSO in bicarbonate buffer pH9.2. Linker- Payload (25mM in DMSO) was added in 5 equivalents every 90-120 min and the reaction mixture incubated at 20°C, 300rpm. The reaction progress was monitored by sizeexclusion chromatography mass spectrometry (SEC-MS, Agilent AdvanceBio SEC 120A 1.9mm 2.1x150mm PEEK isocratic run in 75mM Ammonium Acetate, 10% IPA on an Agilent QTOF 6545XT). A total of 20 equivalents (4 additions) were required.
The crude reaction mixture was pH neutralized with IM NaH2PO4 (1.5%) and purified on an AKTA Avant through a Superdex 75 26/600 isocratic run eluting with 10% IPA/PBS. The monomeric fractions were combined and buffer exchanged into Histidine/NaCI buffer pH7.5. The concentrated sample was filtered through a sterile 0.2|j.m PES filter, flash frozen in liquid nitrogen and stored at -20°C. The sample was analysed by SEC HPLC (Sepax Zenix C 150 7.8 x 300mm) (Figure 19a, HIC-UV (Figure 19b), SEC-LCMS (Figure 19c), UV/Vis spectroscopy, DLS and nanoDSF (Figure 19d) before being used for further studies. The binding affinity was 1.7nM (Figure 19e; Table 6).
Example 45: Conjugation of 129D5BTY scFv to MMAE-(3GA-brLys(BisPEGs)-PEG2- NHS Ester (112)
129D5BTY scFv was conjugated to linker-payload 112 to obtain a DAR 5.9 as per conditions described in Example 39. Specifically, a stock solution of pure scFv (>95%) was diluted to Img/ml into a pre-equilibrated solution of 20% DMSO in bicarbonate buffer pH9.2. Linker-Payload (25mM in DMSO) was added in 5 equivalents every 90-120 min and the reaction mixture incubated at 20°C, 300rpm. The reaction progress was monitored by size-exclusion chromatography mass spectrometry (SEC-MS, Agilent AdvanceBio SEC 120A 1.9mm 2.1x150mm PEEK isocratic run in 75mM Ammonium Acetate, 10% IPA on an Agilent QTOF 6545XT). A total of 20 equivalents (4 additions) were required.
The crude reaction mixture was pH neutralized with IM NaH2PO4 (1%) and purified on an Agilent 1200 BioInert through a Zenix C150 10mmx300mm column using an isocratic run eluting with 10% IPA/PBS. The monomeric fractions were combined and buffer exchanged into Histidine/NaCI buffer pH7.5. The concentrated sample was filtered through a sterile 0.2|j.m PES filter, flash frozen in liquid nitrogen and stored at -20°C. The sample was analysed by SEC HPLC (Sepax Zenix C 150 7.8 x 300mm) (Figure 20a, SEC-LCMS (Figure
20b), UV/Vis spectroscopy, nanoDSF (Figure 20c) and DLS (Figure 20d) before being used for further studies. The binding affinity was 1.4nM (Figure 20e; Table 6).
Example 46: Conjugation of 129D5B to DXd-GGFG-PEG7-NHS Ester (22)
129D5B scFv was conjugated to linker-payload 22 to obtain a DAR 7.6 as per conditions described above. Specifically, a stock solution of pure scFv (>95%) was diluted to Img/ml into a pre-equilibrated solution of 20% DMSO in bicarbonate buffer pH9.2. Linker-Payload (25mM in DMSO) was added in 5 equivalents every 90-120 min and the reaction mixture incubated at 20°C, 300rpm. The reaction progress was monitored by size-exclusion chromatography mass spectrometry (SEC-MS, Agilent AdvanceBio SEC 120A 1.9mm 2.1x150mm PEEK isocratic run in 75mM Ammonium Acetate, 10% IPA on an Agilent QTOF 6545XT). A total of 25 equivalents (5 additions) were required.
The crude reaction mixture was pH neutralized with 0.1M NaH2PO4 (10%) and purified on an Agilent 1200 BioInert through a Zenix C150 10mmx300mm column using an isocratic run eluting with 10% IPA/PBS. The monomeric fractions were combined and buffer exchanged into Histidine/NaCI buffer pH7 and analysed by SEC-LCMS (Figure 21a; Table 6).
Example 47: Conjugation of 129D5B to Exatecan-pGA-PEG? NHS Ester (28)
129D5B scFv was conjugated to linker-payload 28 to obtain a DAR 7.9 as per conditions described above. Specifically, a stock solution of pure scFv (>95%) was diluted to Img/ml into a pre-equilibrated solution of 20% DMSO in bicarbonate buffer pH9.2. Linker-Payload (25mM in DMSO) was added in 5 equivalents every 90-120 min and the reaction mixture incubated at 20°C, 300rpm. The reaction progress was monitored by size-exclusion chromatography mass spectrometry (SEC-MS, Agilent AdvanceBio SEC 120A 1.9mm 2.1x150mm PEEK isocratic run in 75mM Ammonium Acetate, 10% IPA on an Agilent QTOF 6545XT). A total of 25 equivalents (5 additions) were required.
The crude reaction mixture was pH neutralized with IM NaH2PO4 (1.5%) and purified on an Agilent 1200 BioInert through a Zenix C150 10mmx300mm column using an isocratic run eluting with 10% IPA/PBS. The monomeric fractions were combined and buffer exchanged into Histidine/NaCI buffer pH7.5. The concentrated sample was filtered through a sterile 0.2|j.m PES filter, flash frozen in liquid nitrogen and stored at -20°C. The sample was analysed by SEC HPLC (Sepax Zenix C 150 7.8 x 300mm), SEC-LCMS (Figure 21b) and UV/Vis spectroscopy before being used for further studies. The affinity was 254pM (Figure 21c; Table 6).
Example 48: Conjugation of 131D5S to DXd-GGFG-brPEG8-PEG2 NHS Ester (25)
131D5S scFv was conjugated to linker-payload 25 to obtain a DAR 9.3 as per conditions described above. Specifically, a stock solution of pure scFv (>95%) was diluted to Img/ml into a pre-equilibrated solution of 20% DMSO in bicarbonate buffer pH9.2. Linker-Payload (25mM in DMSO) was added starting with 5 equivalents every 90-120 min and the reaction mixture incubated at 20°C, 300rpm. The reaction progress was monitored by sizeexclusion chromatography mass spectrometry (SEC-MS, Agilent AdvanceBio SEC 120A 1.9mm 2.1x150mm PEEK isocratic run in 75mM Ammonium Acetate on an Agilent QTOF 6545XT). A total of 45 equivalents (9 additions) were required (Figure 22a; Table 6).
Example 49: Conjugation of 129D5B to Exatecan N-MeGlucamine-GGFG-PEGz- NHS Ester (86)
129D5B scFv was conjugated to linker-payload 86 to obtain a DAR 4.3 as per conditions described above. Specifically, a stock solution of pure scFv (>95%) was diluted to Img/ml into a pre-equilibrated solution of 20% DMSO in bicarbonate buffer pH9.2. Linker-Payload (25mM in DMSO) was added in 5 equivalents every 90-120 min and the reaction mixture incubated at 20°C, 300rpm. The reaction progress was monitored by size-exclusion chromatography mass spectrometry (SEC-MS, Agilent AdvanceBio SEC 120A 1.9mm 2.1x150mm PEEK isocratic run in 75mM Ammonium Acetate, 10% IPA on an Agilent QTOF 6545XT. A total of 30 equivalents (6 additions) were required.
The crude reaction mixture was pH neutralized with IM NaH2PO4 (10%) and purified on an Agilent 1200 BioInert through a Zenix C150 10mmx300mm column using an isocratic run eluting with 10% IPA/PBS. The monomeric fractions were combined and buffer exchanged into Histidine/NaCI buffer pH7.5. The concentrated sample was filtered through a sterile 0.2|j.m PES filter, flash frozen in liquid nitrogen and stored at -20°C. The sample was analysed by, SEC-LCMS (Figure 22b) and UV/Vis spectroscopy before being used for further studies (Table 6).
Example 50: Conjugation of 129D5B scFv to Belotecan-(3GA-PEG2 NHS Ester (32)
129D5B scFv was conjugated to linker-payload 32 to obtain a DAR 5.9 as per conditions described above. Specifically, a stock solution of pure scFv (>95%) was diluted to Img/ml into a pre-equilibrated solution of 20% DMSO in bicarbonate buffer pH9.2. Linker-Payload (25mM in DMSO) was added in 20 equivalents every 90-120 min and the reaction mixture incubated at 20°C, 300rpm. The reaction progress was monitored by size-exclusion chromatography mass spectrometry (SEC-MS, Agilent AdvanceBio SEC 120A 1.9mm 2.1x150mm PEEK isocratic run in 75mM Ammonium Acetate, 10% IPA on an Agilent QTOF 6545XT). A total of 25 equivalents (5 additions) were required.
The crude reaction mixture was purified/buffer exchanged in a single step using a Zeba column into PBS with 4% trehalose and flash frozen in liquid nitrogen and stored at -20°C. The sample was analysed by SEC-LCMS (Figure 23a), UV/Vis spectroscopy, SEC-HPLC (Figure 23c) before being used for further studies (Table 6). Higher reactive equivalents (40) produced FDCs with a DAR of 13 as shown by SEC-LCMS (Figure 23b) with no aggregation.
The binding affinity was 12.2nM (Figure 74a, Table 6). Cell killing potency on cMET- expressing Hs746T cells was 733.7pM (Figure 74b, Table 10).
Example 51: Conjugation of 129D5B scFv to Triptolide-PEGe NHS Ester (58)
129D5B scFv was conjugated to linker-payload 58 to obtain a DAR 5.0 as per conditions described above. Specifically, a stock solution of pure scFv (>95%) was diluted to Img/ml into a pre-equilibrated solution of 20% DMSO in bicarbonate buffer pH9.2. Linker-Payload (25mM in DMSO) was added in 5 equivalents every 90-120 min and the reaction mixture incubated at 20°C, 300rpm. The reaction progress was monitored by size-exclusion chromatography mass spectrometry (SEC-MS, Agilent AdvanceBio SEC 120A 1.9mm 2.1x150mm PEEK isocratic run in 75mM Ammonium Acetate, 10% IPA on an Agilent QTOF 6545XT). A total of 20 equivalents (5 additions) were required to obtain a DAR 5 by SEC- LCMS (Figure 23d; Table 6).
Example 52: Conjugation of 129D5B scFv to N1-Benzylamino Imidazoquinoline- PEG7 NHS Ester (54)
129D5B scFv was conjugated to linker-payload 54 to obtain a DAR 1.35 as per conditions described above. Specifically, a stock solution of pure scFv (>95%) was diluted to Img/ml into a pre-equilibrated solution of 20% DMSO in bicarbonate buffer pH 9.2. Linker-Payload (25mM in DMSO) was added in 5 equivalents every 90-120 min and the reaction mixture incubated at 20°C, 300rpm. The reaction progress was monitored by size-exclusion chromatography mass spectrometry (SEC-MS, Agilent AdvanceBio SEC 120A 1.9mm 2.1x150mm PEEK isocratic run in 75mM Ammonium Acetate on an Agilent QTOF 6545XT (Figure 24a, Table 6). A total of 15 equivalents (3 additions) were required. The sample was neutralized with IM NaH2PO4 (1.5% v/v) and stored at 4°C.
A modified conjugation reaction based on Example 77 yielded a slightly higher DAR (1.81). A stock solution of pure scFv (>95%) was diluted to 50pM (1.4mg/mL) into a preequilibrated solution of lOOmM sodium phosphate pH 8.0, 10% DMA. Linker-Payload (25mM in DMSO) was added starting with 1 molar equivalent (compared to scFv lysines) before adding more payload every 60 min and the reaction mixture incubated at 22°C, 500rpm until the DAR was achieved (3 molar equivalents required) (Figure 75).
Example 53: Conjugation of 129D5B scFv to Nintedanib a-Galactose-PEG? NHS Ester (46)
129D5B scFv was conjugated to linker-payload 46 to obtain a DAR 1.9 as per conditions described above. Specifically, a stock solution of pure scFv (>95%) was diluted to Img/ml into a pre-equilibrated solution of 20% DMSO in bicarbonate buffer pH9.2. Linker-Payload (25mM in DMSO) was added starting with 10 equivalents every 90-120 min and the reaction mixture incubated at 20°C, 300rpm. The reaction progress was monitored by reverse-phase chromatography mass spectrometry (RP-MS, Agilent PLRP) eluting with a gradient of 0.1%FA/water/MeCN in in 75mM Ammonium Acetate on an Agilent QTOF 6545XT. A total of 40 equivalents (4 additions) were required. The crude reaction mixture was analysed by LCMS (Figure 24b; Table 6) and HIC-UV chromatography.
Example 54: Conjugation of 129D5B scFv to GENZ-644282-Val-Ala-brPEGi2- PEG4-NHS Ester (80)
129D5B scFv was conjugated to linker-payload 80 to obtain a DAR 2.4 as per conditions described above. Specifically, a stock solution of pure scFv (>95%) was diluted to Img/ml into a pre-equilibrated solution of 20% DMSO in bicarbonate buffer pH9.2. Linker-Payload (25mM in DMSO) was added starting with 5 equivalents every 90-120 min and the reaction mixture incubated at 20°C, 300rpm. The reaction progress was monitored by RP-MS, (RPMS C4 Phenomenex column (100x2.1mm) gradient run in 0.1%FA/water/MeCN on an Agilent QTOF 6545XT). A total of 20 equivalents (4 additions) were required. The crude reaction mixtures were pH neutralized with IM NaH2PO4 (10%) before being used for further studies (Figure 25a; Table 6).
Example 55: Conjugation of 129D5B scFv to Dexamethasone-dipyrophosphate- PEG2 TFP Ester (50)
129D5B scFv was conjugated to linker-payload 50 to obtain a DAR 3.74 as per conditions described above. Specifically, a stock solution of pure scFv (>95%) was diluted to Img/ml into a pre-equilibrated solution of 20% DMSO in bicarbonate buffer pH9.2. Linker-Payload (25mM in DMSO) was added in 10 equivalents every 90-120 min and the reaction mixture incubated at 20°C, 300rpm. The reaction progress was monitored by size-exclusion chromatography mass spectrometry (SEC-MS, Agilent AdvanceBio SEC 120A 1.9mm 2.1x150mm PEEK isocratic run in 75mM Ammonium Acetate, 10% IPA on an Agilent QTOF 6545XT). A total of 10 equivalents (1 addition) were required (Figure 25b). The binding affinity was 298nM (Figure 25c; Table 6).
Example 56: Conjugation of 129D5B Fab to MMAE-(3GA-brPEGs-PEG2-NHS Ester (10)
A Fab version of the 129D5B scFv was constructed by BioIntron Inc by fusing the VH gene to the CHI gene from a rabbit IgGl and the VL gene to the rabbit CL gene and expressed and purified from CHO cells. The binding affinity was confirmed by BIAcore as 30pM (Figure 26a). This was conjugated to linker-payload 10 to obtain a DAR 5.9 as per conditions described in Example 39. Specifically, a stock solution of pure scFv (>95%) was diluted to Img/ml into a pre-equilibrated solution of 20% DMSO in bicarbonate buffer pH9.2. Linker- Payload (25mM in DMSO) was added starting with 5 equivalents every 90-120 min and the reaction mixture incubated at 20°C, 300rpm. The reaction progress was monitored by RP chromatography mass spectrometry RP-MS, RPMS C4 phenomenex column (100x2.1mm) gradient run in 0.1%FA/water/MeCN on an Agilent QTOF 6545XT. A total of 15 equivalents (2 additions) were required (Figure 27a, Figure 27c; Table 6). The binding affinity was 41pM (Figure 26b).
Example 57: Conjugation of 129D5B Fab to Maleimidocaproly-Val-Cit-MMAE (61)
129D5B Fab (example 53) was conjugated to linker-payload 61 to obtain a DAR 2. Under nitrogen, in a round bottomed flask, the antibody was diluted into PBS to a final Img/ml before adding the EDTA (50mM) and TCEP (ImM) and degassing for lOmins at RT before incubating at 4°C overnight. Degassed Payload diluted in anhydrous DMSO were added to the antibody solution at RT and incubated with gentle stirring for Ihr. The reaction was monitored by RP-Mass spectrometry (RPMS C4 Phenomenex column (100x2.1mm) gradient run in 0.1%FA/water/MeCN on an Agilent QTOF 6545XT). The reaction was quenched with acetyl cysteine (1.8 equivalents to the payload) at 20°C at RT. The crude reaction mixture was purified by HIC chromatography on an AKTA Pure, on a 1ml Butyl HP column eluting with a gradient of ammonium sulphate in sodium sulphate pH7.4.
The required fractions were combined, and buffer exchanged into Histidine/NaCI buffer pH7.5. The concentrated sample was filtered through a sterile 0.2|j.m PES filter, flash frozen in liquid nitrogen and stored at -20°C. The sample was analysed by RP-LCMS (Figure 22B) SEC HPLC (Sepax Zenix C 150 7.8 x 300mm), Figure 27b; Table 6). The binding affinity was 35pM (Figure 26c).




Example 58: Conjugation to cMET scFv described in WO2013/079973
Phage display is a common method for scFv discovery but even for those skilled in the art, scFvs against the cMET target may not be suitable for bioconjugation unless following the disclosures herein. For example DiCara DM et al (WO2013079973) described anti-cMET scFvs, discovered from human phage libraries which bind with to cMET with high affinity. Two clones, 107-A07 and 107-A01 are noted.
ScFv clone 107-A01 was constructed and produced as described in WO2013079973 and was conjugated to MMAF-PEG2 Linker-Payload (WO2016046574) to obtain a DAR 6 using the same conditions as described in WO2016046574. Specifically, a stock solution of pure scFv (>95%) was diluted to Img/ml into a pre-equilibrated solution of 20% DMSO in bicarbonate buffer pH9.2. Linker-Payload (25mM in DMSO) was added in 6 equivalents every 90-120 min and the reaction mixture incubated at 20°C, 300rpm. A total of 12 equivalents (2 additions) were required.
The crude reaction mixture was pH neutralized with 0.1M NaH2PO4 (10%) and purified on an Agilent 1200, on a Zenix-C 150 lOmmx 300mm, using an isocratic run eluting with 10% IPA/PBS. The monomeric fractions were combined and buffer exchanged into PBS pH7.5. The concentrated sample was filtered through a sterile 0.2|j.m PES filter and stored at 4°C. The sample was analysed by SEC-LCMS (Figure 28a-c), UV/Vis spectroscopy, SDS- PAGE and SEC-HPLC (Figure 28d) before being used for further studies. A parallel conjugation reaction was carried out using the 20F6 cMET scFv described herein (Example 2) and analysed similarly. The sample was analysed by SEC-LCMS (Figure 28e), UV/Vis spectroscopy, SDS-PAGE and SEC-HPLC (Figure 28f) before being used for further studies. The 107-A01-MMAF-PEG2 FDC demonstrated significant levels of aggregation (>10%) and poor biophysical properties compared to the 20F6-MMAF-PEG2 FDC (< 1% aggregation),
even though the latter FDC had a similar or higher DAR. A similar observation was also seen when scFv clone 107-A07 was used. This demonstrated that scFvs disclosed in this invention display unexpectedly superior bioconjugation and biophysical properties when compared to other human anti-cMET scFvs.
Example 59: Flow cytometry of cMET FDCs on low, medium and high cMET expressing cell lines
A list of cell lines analysed is shown in Table 7, with estimated cMET receptor levels from the literature [57]. For cell surface binding, 50-70% confluent cells were harvested from flasks using cell dissociation buffer (Gibco). Cells were washed and resuspended in PBS, 0.5% BSA, 0.8mM EDTA and 0.02% NaNs. Cells were transferred to a 96-well microplate and incubated with primary antibodies (129D5B/129D5BTY scFvs, 131D5S scFv, ABT-700 IgG and 5D5 IgG) or controls for Ih on ice. Cells were washed and incubated for Ih on ice and in the dark with appropriate detection antibodies (labelled with a fluorophore) except for untreated cells. After incubation, cells were washed, re-suspend in flow buffer and analysed on a Guava EasyCyte HT flow cytometer (Cytek Biosciences).

The results showed that cMET scFv clones 129D5B/129D5BTY and 131D5S bound to Hs746T, SNU5 and other cMET-expressing cell lines from different tissue origins, when compared to known benchmark IgGs ABT-700 and 5D5 (Table 8, Figure 29). Significant binding was also seen in cell lines expressing moderate to low cMET levels.

In a more detailed analyses, cMET receptor cell surface density (antigen-binding capacity per cell) was determined by indirect immunofluorescence staining of cell surface antigens using the QIFIKIT (DAKO-K0078) on 6 cell lines. Anti-cMET 5D5 IgGl (5D5.11.6) hybridoma was purchased from the ATCC. All cell lines were acquired from ATCC (UK-LGC) and were grown in the recommended media and conditions. Cells were treated with cMET 5D5 mouse antibody at saturating concentration (5mg/ml). Surface binding was detected by treating cells with QIFIKIT-provided FITC-conjugated antibody at saturating concentration (Figure 30).
For the construction of the calibration curve, calibration beads were treated with QIFIKIT- provided FITC-conjugated antibody - individual markers are used to determine the MFI (Mean Fluorescence Intensity) of each bead population (Fig 30a). The Set-Up beads were used to establish the window of analysis, as it comprises a mixture of blank and high-level
beads (Fig 30b). Indirect immunofluorescence staining data of the QIFIKIT beads was acquired on a Guava EasyCyte HT flow cytometer (Cytek Biosciences) using standard operating procedures. The standard curve (Fig 30c) was used to assign ABC (antibodybinding capacity) or the number of monoclonal antibody binding sites/cell per cell or microbeads to each of the cell lines tested (Table 9).

Example 60: Cell-killing Potency Assays
Cell lines were obtained from commercial depositaries and grown according to the recommended conditions, harvested during the logarithmic growth period and counted. The cell concentration was adjusted to the optimal conditions for each cell line. Cell suspension (in the range of 1000-10,000 cells/well) was added to three 96-well plates and incubated overnight in a humidified incubator at 37° C with 5% CO2. FDC, ADC and unconjugated antibodies at high working concentrations were prepared in media with 10 (x3) and 100 (x2) fold serial dilutions to achieve 6-8 dose levels. Cells are treated with diluted compounds (x5 replicates for each drug concentration). Control plates are treated with either medium or triton-XlOO and incubated for 96h in a humidified incubator at 37°C with 5% CO2. Celltiter-96 Aqueous One Solution (Promega G5381) reagent is added to each well. After 4 hours incubation at 37° C with 5% CO2 optical density at 490nm is recorded. The data is analysed to calculate relative IC50, and a dose-response curve is fitted using a nonlinear regression model with a sigmoidal dose-response using GraphPad Prism. The cell killing potency is summarised in Table 10 with representative plots shown in Figures 31 to 37. FDC potency was in the range of 0.1 to 2nM IC50 with cMET-negative cell lines showing no or low sensitivity to the FDC. Surprisingly, FDCs were potent against cMET low cell lines (e.g. NCI-N87) whereas telisotuzumab vedotin ADC was much less effective (reported threshold for efficacy is ~100,000 receptors/cell [57] suggesting that
high-DAR FDCs can kill tumour cells with as low as ~8000 receptors/ cel I.




Example 61: Cell-killing Potency on 50 cell-line Omniscreen™ panel
Crown Biosciences (UK) run independent cell potency screens across cell lines with known genetic properties. Fifty cell lines were selected on the basis of cMET RIMA expression to include tumours from 9 tissues of origin with high, medium and low cMET expression (as determined by Crown Biosciences). 129D5B-MMAE-pGA-BrPEG8-PEG2 FDC (DAR 6.75) was incubated at a range of concentrations for 72h and cell viability determined using CellTiter-Glo® Luminescent Cell Viability Assay (Promega). The potency was determined as ICso and compared to reference drug cisplatin. A summary of the FDC's potency against these cell lines is shown in Table 11 and representative plots shown in Figure 38.



The cMET FDC was potently cytotoxic to a range of solid tumour cell lines including gastric/stomach, lung, pancreas, triple-negative breast, kidney, head & neck, colon, oesophagus and liver at a range of cMET-expression levels (high, medium and low).
Example 62: cMET FDC internalization
Cell surface internalization experiments were conducted as described by [236]. Briefly, SNU5 cells (2xl05/well) were incubated in the dark on ice for Ih with Alexa Fluor 488 labelled antibodies/conjugates (0.15 to 2 mg/mL) in cold medium containing FBS. After washing, cells were resuspended in warm medium containing FBS and incubated at 37°C, 5% CO2 for the indicated intervals to promote internalisation of surface fluorescence (0, 15, 30, 45, 60, 120, 180, 240 & 300 min). At each time point cells were rapidly chilled in the dark. Once all time points have been collected, cells were incubated on ice/protected from light for 30 minutes with either, quenching ab (lOpig/ml) or unlabelled antibody (lOmg/mL). Cells were washed and resuspended in flow buffer (PBS/0.5% BSA). The mean fluorescence intensity of 10,000 cells was quantified by flow cytometry (Guava Flow Cytometer). Internalised fluorescence was calculated from quenched and non-quenched sample data by correcting for incomplete surface quenching. Internalised fluorescence was plotted, and a curve fit was obtained in Prism by nonlinear regression with the one-phase association equation: Y = Y0 + (Plateau— Y0) > (1— exp(-K_x)) with Y0 = 0 when X (time) is zero and Plateau = 100. The summary statistic half-life (ti/2) is the time at which internalised fluorescence is equal to fifty per cent and enabled comparison of internalisation for the different dye-conjugated cMET Abs. Half-time is in the time units on the X-axis and is computed as the reciprocal of In (2)/K. For each curve fit the 95% confidence interval for half-time is reported. The tl/2 for internalization for the 129D5B scFv, 129D5B FDC, 5D5 IgG and ABT-700 IgG was 85, 81, 81 and 64 minutes respectively. Linker-payload conjugation did not appear to affect the rate of internalization which itself was similar to bivalent IgGs (Figure 39).
Example 63: Inhibition of cMET phosphorylation by scFvs
Non-small cell lung cancer cell line A549 was obtained from the American Type Culture Collection. (ATCC-CRM-CLL-185). Phosphorylation inhibition was determined according to [237]. A549 cells were seeded at 4.0xl04 per well in a 96-well plate in complete growth medium [F12K+10% FCS]. Twenty-four hours later, cells were pretreated with antibodies in duplicate wells for one hour at 37°C, and then stimulated with InM (~100ng/ml) recombinant human HGF (R&D Systems) for 10 min at 37 °C. Cells were wash with PBS and incubated on ice with lOOul/well of Cell Lysis Buffer (Cell Signalling Technology) supplemented with PMSF protease inhibitor. Cell lysates were collected by centrifugation at 13000 rpm for 10 min at 4°C. and correspond to the supernatant phase. Protein content was quantified using a Pierce 660nm protein assay (Pierce).
The phosphorylation status of c-Met was quantified by ELISA. Briefly, a goat anti-cMET antibody (R&D Systems, ref AF276) was used as a capture antibody. After a saturation
step with a TBS-BSA 5% buffer, 7 mg protein lysates were added to each we!! of the coated 96-weil plate and incubated for 90 min at room temperature (RT). The plates were washed with 0.1% TBS-Tween and the anti-phospho-c-Met Mab, directed against the phosphorylated Tyr residues at position 1230, 1234 and 1235 - Invitrogen) was added. After an additional 1-hour incubation and 4 washes, an anti-rabbit-antibody coupled to HRP was added and incubated for 45 minutes at RT. The plates were washed before SuperSignal ELISA Pico Chemiluminescent Substrate (ThermoScientific) was added and the luminescence was measured using a ClarioSTAR Microplate reader. The data shown in Figure 40 shows that 129D5BTY and 131D5S scFvs inhibit phosphorylation (IC50 1.4nM and 4.3nM respectively) as well as reference IgG antibodies 5D5 and ABT-700 (IC50 1.4nM and 1.5nM respectively). An irrelevant IgG4 antibody does not inhibit.
Example 64: Pharmacokinetics of cMET FDCs after single dose
The pharmacokinetic clearance for a range of FDCs (DAR ~6) was measured after a single IV dose of Img/kg in Sprague-Dawley rats, immuno-detecting the scFv component by anti-StrepTag ELISA and the payload component by anti-MMAE ELISA on cMET protein coated ELISA plates (Table 12; Figure 41). The data was plotted using GraphPad (Prism) and the bioavailability (plasma exposure) was determined as the area under the curve (AUC) of plasma concentration vs time (Table 12). The AUC for the unmodified 129D5B scFv was 2.7 mgmL-1hr and conjugates all had greater plasma exposure due to the presence of the linker payload with the increase in exposure exceeding 20x with the longest PEG-chain containing linker. FDCs with a greater plasma exposure are expected to be more efficacious whilst retaining the benefits of superior penetration compared to ADCs. The pharmacokinetic profile and AUC were very similar whether detected by anti- StrepTag immuno-detection or anti-MMAE immunodetection confirming that the FDCs were stable with no significant payload deconjugation occurring.


Example 65: Toxicological measurements in rats of cMET FDCs after single dose
The clinical chemistry toxicological parameters for a range of FDCs (DAR 6.4 ± 0.1) was measured after a single IV dose of Img/kg in Sprague-Dawley rats, compared to carrier/saline buffer treated controls. The peak value for liver markers aspartate transaminase (AST) and alanine transaminase (ALT) was previously determined to occur at 48-72h, post-IV injection therefore this was selected as the sampling timepoint. The data is shown in Table 13. Rapid elimination of high-DAR FDCs are expected to trigger a transient spike in AST and ALT liver markers, which was observed. ALT levels were elevated 42-64% and AST levels increased 175-233% compared to untreated control animals. Immunohistochemical staining of liver and kidneys one week post-IV dosing showed no significant pathological damage compared to untreated control animals.


Example 66: Toxicological measurements in rats of cMET FDCs after multiple doses
The clinical chemistry and haematological toxicological parameters for two FDCs (DAR 6.1- 6.36) was measured after three IV doses of Img/kg in Sprague-Dawley rats in 3-week cycles, compared to carrier/saline buffer treated controls. AST, ALT, bilirubin, urea, creatine, white blood cells, red blood cells and platelets were measure 48h after each dose. Also compared was experimental ADC telisotuzumab vedotin (DAR 3.1) at 4mg/kg which contains the same MMAE payload type as the FDCs at a similar conjugate molar ratio. The data is shown in Tables 14-17 and plotted in Figures 42-43.







The more rapid elimination of the high DAR FDCs led to an increase in AST levels after each dose, but this was a transient rise and approximately 2-3x higher than untreated controls. This rise was as expected considering the 2x higher DAR, 5x lower molecular weight and 4x lower conjugate mass injected. ALT increases were more moderate with no significant increase after the 3rd dose compared to untreated controls or the ADC. No
significant changes were observed with other metabolic markers. Considering the haematological measurements, there was no significant increase in red or white blood cells or platelet count. At the end of the study 7 days after the 3rd dose, formalin fixed liver and kidney tissues were collected, transferred to 70% isopropanol and paraffin-embedded in order to section into slides for immunohistochemical staining. Consecutive sections 4|j.m thick were stained with haematoxylin and eosin and digitally scanned at x20 objective using the Hamamatsu Nanozoomer (Histologix Ltd, UK). The images were assessed by a Board certified veterinary pathologist (PathCelerate, UK). There were no pathological findings in FDC vs saline treated animals. Representative images from 12 sections are shown in Figure 44.
Example 67: In vivo tumour regression and cure efficacy in human tumour xenograft models cMET FDCs were evaluated in human tumour xenograft models. Hs746T gastric and NCI- N87 gastric tumour cells were implanted into BALB/c nude mice, SNU5 gastric tumour cells were implanted into CB17 SCID mice and NUGC4 gastric tumour cells were implanted into NOD-SCID mice. Approximately l-10xl06 cells were implanted in 1 : 1 mixture of media/matrigel and implanted subcutaneously into the animal flank. When the tumours had reached the required size (150mm3 to 500mm3), dosing commenced by intravenous injection into the tail vein. Tumours were measured by calipers and tumour volume calculated as Length x width2 and body weights recorded every 2-3 days. Independent Contract Research Organizations (Crown Bio, UK/China and Pharmidex UK) carried out the studies adhering to local veterinary/ethical guidelines. Test agents are described in Table 18.


The tumour growth inhibition/cure data is summarised in Table 19 and illustrated in Figures 45 to 55 with p-values for comparing groups.



* TGI: 100% TGI is a complete cure from the tumour size at start of treatment, -100% represents a doubling of tumour size
The data shows that the 129D5B(TY) and 131D5S-based cMET FDCs are efficacious in a range of tumour xenograft models with the 129D5BTY FDC being the most potent and 129D5BTY FDC with a MMAE-[3GA-BrPEGi2-PEG4 linker payload being consistently more active in models compared to MMAE-pGA-BrPEGs-PEGz. In many cases FDCs were equally
or significantly more potent, causing similar or greater tumour inhibition when compared on a similar payload or similar conjugate molar concentration. In many cases, the FDCs were equally or better tolerated (by body weight measurements) than similar ADCs with equal or more MMAE payload content. FDCs were also highly active in tumour models as low as 33,000 (NUGC4) and 8,000 receptors (NCIN87) per cell, unlike telisotuzumab vedotin (ABBV-399) [57],
Example 68: Plasma stability of cMET FDCs cMET FDC 129D5BTY- MMAE-pGA-BrPEGi2-PEG4 (DAR6) (140pmol, lp.1) was incubated in duplicate in plasma (K2-EDTA treated from individual snap frozen aliquots thawed on ice) at 37°C in a thermomixer. Plasma from four species was used: Cynomologus monkey, mouse, human and Sprague-Dawley rat. Plasma was centrifuged at 17000g for 10 minutes to remove precipitate prior to use. After incubation for the desired time, pre-equilibrated MagStrep "type3" XT beads (IBA; catalogue number 2-4090-002- Iptl of packed beads in 50 .l of PBS containing ImM EDTA) were added to the plasma-conjugate samples. The T- zero samples were prepared by adding conjugate to a mixture of plasma and beads and then immediately incubated at 6°C with mixing. The samples with beads were incubated on a thermomixer set to 6°C and 1200rpm for 45 minutes. After incubation the beads were recovered on a magnetic rack and were subjected to three rounds of washing with 200ul of PBSE. The washed beads were then eluted with 50p of 50mM biotin in PBSE for 20 minutes on a thermomixer at 6°C with mixing speed 1200 rpm. Eluates were recovered using the magnetic stand and were filtered using spin filters (0.22|j.m) for 2 minutes at 10,000 rpm. Filtered samples were subjected to LCMS as described above. Eluates (10-15 p.1) were separated on 04 column (Phenomenex Biozen 2.1mmx 10cm, 1.7|j.m bead size) connected to an Agilent 6345 XT QTOF. The column was eluted with a gradient of 0.1% aqueous formic acid, 10% IPA and acetonitrile containing 0.1% formic acid. Duplicate technical replicates were run in duplicate on the system. The resulting mass spectra were deconvoluted using Agilent Masshunter software. The deconvoluted spectra were manually integrated and mean DAR calculated from the four runs (duplicate runs of two technical replicates) for each species timepoint. Figure 56a shows the plot of average DAR over 1 week in all 4 species and Figure 56b shows a representative example of the LC-MS DAR analyses for the human plasma stability study. In all four species, the cMET FDC has a similar and stable DAR profile demonstrating that the FDC is stable to degradation and linker-payload release.
Example 69: Uptake of MMAE payload delivered by cMET FDC
Subcutaneous SNU5 tumours were grown to ~500mm3 and single doses of Img/kg cMET FDC (129D5B-MMAE-pGA-BrPEG8-PEG2, DAR 6) or 4mg/kg cMET ADC (telisotuzumab
vedotin, DAR 3.1) was administered IV. Tumours and normal tissues were collected at various time points and snap frozen in liquid nitrogen. Approximately lOOmg of tumour/tissue was weighed and extracted in 20% methanol with homegenizer beads and homogenized. The extract was centrifuged and the supernatant was lyophilised and redissolved in 10% methanol, 0.1% formic acid for LC-MS analyses. MMAE calibrations and an internal standard of MMAF were used. Analyses was carried out in a Waters TQ-X triple quadrupole (column Waters HSS T3 and mobile phase 0.1% Formic acid in a water/methanol gradient) following and quantifying the MMAE mass of 718.5 Da. The data is shown in Table 20 and plotted in Figure 57.

The FDC and ADC were administered at equimolar concentrations but despite the more rapid plasma clearance, surprisingly more MMAE payload was delivered to the tumour when conjugated as an FDC compared to an ADC during the first 24-48h and surprisingly low normal tissue uptake over 72h.
Example 70: FcR binding of cMET FDCs
The BIACore T200 was used for surface plasmon resonance to measure molecular interactions in real-time. CM5 series S sensor chip (Cytiva BR100530) was used to immobilise ligand (human CD16a/FCGR3A protein (176 val - Sino Biological 10389- H08H1), Human CD32a/FCGR2A protein (167 His - Sino Biological 10374-H08C1) and Human CD64/FCGR1A protein (Sino Biological 10256-H0H8) proteins) on the surface through covalent coupling. 1000 RU were immobilized.
To measure kinetic parameters, varying concentrations of the cMET scFvs, antibodies, Fragment Drug Conjugate (FDC) and control antibodies ('analytes') were flowed over the immobilised CD16a, CD32a and CD64 sensor chips. Real-time measurements are made of the association phase, as analyte is flowed, and of the dissociation phase, as buffer is flowed. The on (association), kott (dissociation), and equilibrium binding constants
(affinity, Ka = 1/Kd) were determined using the BIAevaluation software. The chip surface was regenerated using 3M MgCl. The data (representative data shown in Figure 58) shows that the scFvs and corresponding FDC do not cross-react to Fc-Gamma receptors where IgG-based antibodies and conjugates have observable binding summarised in Table 21.

Example 71: Toxicological evaluation in non human primates
A non-GLP pilot study to understand the toxicological profile of cMET FDC 129D5BTY-
MMAE-pGA-BrPEGi2-PEG4 (DAR.6) in cynomolgus non-human primates (Macaca
fascicularis, females, 2.5-3 years old, 2.5-3kg weight) was undertaken. Studies were carried out at CRO Aptuit (Italy). Standard animal housing, lighting and diet (altromin 6024 supplemented with fruit and vegetables) was used. Escalating doses (intravenous bolus) of FDC were administered into 3 animals and a range of metabolic, clinical chemistry and haematological parameters were measured at pre-dose, 2 days post-dose, 7 days post-dose and one day before the 2nd and subsequent doses. Each dose cycle was for 3 weeks. Dose-1 was 0.25mg/kg, dose-2 was 0.375mg/kg, dose-3 was 0.5mg/kg and dose- 4 was Img/kg. Based on an ADC of DAR3, the equivalent MMAE dose on an ADC is 2.5mg/kg, 3.75mg/kg, 5mg/kg and lOmg/kg. The FDC was prepared as in Example 42 with additional checks to ensure an endotoxin level of <1 EU/mg. Haematological measurements are show in Figure 59, clinical chemistry measurements associated with liver metabolism is shown in Figure 60a and body weights shown in Figure 60b. During the study there were no adverse clinical observations, the body weights were normal and the animals did not show signs of toxicity. Lymphocytes were generally in the normal (predose) range except there was a small reduction in neutrophils after the 1st dose which did not worsen with subsequent doses (mild neutropenia) and fully recovered by the 4th dose. There was no significant increase in aspartate or alanine amino-transaminases (normally associated with liver toxicity). Overall, the cMET FDC was well tolerated indicating that higher doses were possible. A dose of Img/kg FDC has the same amount of MMAE payload as telisotuzumab vedotin at lOmg/kg (5-times larger, 2x lower DAR) and lOmg/kg exceeds the range where toxicity is seen in non-human primate models [97]. Therefore this cMET FDC was surprisingly well tolerated.
Example 72: Synthesis of Exatecan-(3GA-BrPEGi2-PEG4 NHS Ester (133)

To a solution of (2S,3R,4S,5S,6S)-2-(2-amino-4-(hydroxymethyl)phenoxy)-6- (methoxycarbonyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate 3 (500 mg, 1.10 mmol) in dry DCM (5 mL) was added 3-((tert-butoxycarbonyl)amino)propanoic acid (249.28 mg, 1.32 mmol) and EEDQ(543.01 mg, 2.20 mmol). The mixture was stirred at room
temperature overnight, diluted with DCM (100 mL) and washed with water (50 mL x 3). The organic phase was dried over Na2SO4, filtered, concentrated and the resulting residue purified by silica gel chromatography (PE:EtOAc 2: 1) to afford (2S,3R,4S,5S,6S)-2-(2-(3- ((tert-butoxycarbonyl)amino)propanamido)-4-(hydroxymethyl)phenoxy)-6-
(methoxycarbonyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate 127 as a yellow solid (383 mg, 55.6%). LCMS m/z =627.10[M + H]+. XH NMR (400 MHz, DMSO-d6) 6 8.67 (s, 1H), 7.82 (s, 1H), 7.02 (d, J = 2.8 Hz, 2H), 6.80 (s, 1H), 5.61 - 5.39 (m, 2H), 5.27 - 4.97 (m, 3H), 4.71 (d, J = 9.6 Hz, 1H), 4.40 (d, J = 5.6 Hz, 2H), 3.64 (d, J = 4.8 Hz, 3H), 3.22 (t, J = 6.4 Hz, 2H), 2.09 - 1.90 (m, 9H), 1.38 (d, J = 5.0 Hz, 9H).
To a solution of (2S,3R,4S,5S,6S)-2-(2-(3-((tert-butoxycarbonyl)amino)propanamido)-4- (hydroxymethyl)phenoxy)-6-(methoxycarbonyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate 127 (200 mg, 0.32 mmol) in DMF (5 mL) was added bis(4-nitrophenyl) carbonate (194.20 mg, 0.62 mmol) and DIPEA(123.76 mg, 0.96 mmol). The resulting mixture was stirred at room temperature for 16 hours, diluted with water (100 mL), and extracted with EtOAc (50 mL x 3). The combined organic phases were dried over Na2SO4, filtered and concentrated. The residue was purified by silica gel chromatography (PE:EtOAc=2: 1) to afford (2S,3R,4S,5S,6S)-2-(2-(3-((tert-butoxycarbonyl)amino)- propanamido)-4-((((4-nitrophenoxy)carbonyl)oxy)methyl)phenoxy)-6- (methoxycarbonyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate 128 (140 mg, 55.3%) as a yellow solid. LCMS m/z =814.40[M + Na]+. XH NMR (400 MHz, DMSO-d6) 6 8.78 (s, 1H), 8.36 - 8.25 (m, 2H), 7.99 (s, 1H), 7.62 - 7.50 (m, 2H), 7.23 (d, J = 8.6 Hz, 1H), 7.12 (d, J = 8.6 Hz, 1H), 6.81 (s, 1H), 5.75 (s, 2H), 5.63 (d, J = 7.8 Hz, 1H), 5.50 (t, J = 9.6 Hz, 1H), 5.22 (d, J = 8.4 Hz, 2H), 5.07 (t, J = 9.8 Hz, 1H), 4.74 (d, J = 10.0 Hz, 1H), 3.64 (s, 3H), 3.22 (d, J = 6.6 Hz, 2H), 3.17 (d, J = 5.2 Hz, 2H), 2.07 - 1.97 (m, 9H), 1.38 (s, 9H).
To a solution of (2S,3R,4S,5S,6S)-2-(2-(3-((tert-butoxycarbonyl)amino)propanamido)-4- ((((4-nitrophenoxy)carbonyl)oxy)methyl)phenoxy)-6-(methoxycarbonyl)tetrahydro-2H- pyran-3,4,5-triyl triacetate 128 (300 mg, 379 pmol), HOBt (4.0 mg, 31.6 pmol) and Exatecan mesylate (167 mg, 316 pmol) in DMF (5 mL) was added DIPEA (122 mg, 948 pmol). The reaction mixture was stirred at room temperature overnight under N2 after which the reaction mixture was diluted with water (50 mL) and extracted with EtOAc (40 mL x 2). The combined organic phases were dried with Na2SO4, concentrated in vacuo. The crude was purified by silica gel chromatography (DCM: MeOH=30: l) to afford (2S,3R,4S,5S,6S)-2-(2-(3-((tert-butoxycarbonyl)amino)propanamido)-4-(((((lS,9S)-9- ethyl-5-fluoro-9-hydroxy-4-methyl-10,13-dioxo-2,3,9,10,13,15-hexahydro-lH,12H- benzo[de]pyrano[3',4':6,7]indolizino[l,2-b]quinolin-l- yl)carbamoyl)oxy)methyl)phenoxy)-6-(methoxycarbonyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate 129 (260 mg, 63.1%) as a yellow solid. LCMS m/z = 1088.15 [M+H]+. XH NMR
(400 MHz, DMSO) 6 8.73 (s, 1H), 8.06 (d, J = 8.8 Hz, 1H), 7.88 (s, 1H), 7.77 (d, J = 10.8 Hz, 1H), 7.31 (s, 1H), 7.18 (d, J = 8.4 Hz, 1H), 7.10 (d, J = 8.6 Hz, 1H), 6.78 (s, 1H), 6.51 (s, 1H), 5.59 (d, J = 7.8 Hz, 1H), 5.53 - 5.41 (m, 3H), 5.27 (s, 3H), 5.17 (dd, J = 9.8, 7.8 Hz, 1H), 5.11 - 5.02 (m, 3H), 4.72 (d, J = 9.8 Hz, 1H), 3.62 (s, 3H), 3.41 - 3.36 (m, 2H), 3.17 (q, J = 6.8 Hz, 4H), 2.46 (d, J = 6.8 Hz, 2H), 2.40 - 2.35 (m, 3H), 2.18 (s, 2H), 2.08 - 1.97 (m, 9H), 1.88 (m, 2H), 1.36 (s, 9H), 0.88 (t, J = 7.4 Hz, 3H).
To a solution of (2S,3R,4S,5S,6S)-2-(2-(3-((tert-butoxycarbonyl)amino)propanamido)-4- (((((lS,9S)-9-ethyl-5-fluoro-9-hydroxy-4-methyl-10,13-dioxo-2,3,9,10,13,15- hexahydro-lH,12H-benzo[de]pyrano[3',4' :6,7]indolizino[l,2-b]quinolin-l- yl)carbamoyl)oxy)methyl)phenoxy)-6-(methoxycarbonyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate 129 (100 mg, 91.6 pmol) in MeCN (2 mL) at 0 °C was added a solution of LiOH (8 mg, 0.335 mmol) in water (2 mL). The reaction mixture was stirred at 0 °C for 1 h after which the reaction mixture was quenched with AcOH (10.5 mg) and concentrated in vacuo to afford (2S,3S,4S,5R,6S)-6-(2-(3-aminopropanamido)-4-((5S,8S,llS,12R)-ll- ((S)-sec-butyl)-12-(2-((S)-2-((lR,2R)-3-(((lS,2R)-l-hydroxy-l-phenylpropan-2- yl)amino)-l-methoxy-2-methyl-3-oxopropyl)pyrrolidin-l-yl)-2-oxoethyl)-5,8-diisopropyl- 4,10-dimethyl-3,6,9-trioxo-2,13-dioxa-4,7,10-triazatetradecyl)phenoxy)-3,4,5- trihydroxytetrahydro-2H-pyran-2-carboxylic acid 130 (100 mg, crude) as a yellow solid, which was used into next step directly. LCMS m/z = 948.10 [M+H]+. XH NMR (400 MHz, DMSO) 6 9.12 (s, 1H), 8.20 - 8.01 (m, 2H), 7.77 (d, J = 10.8 Hz, 1H), 7.31 (s, 1H), 7.11 (t, J = 7.2 Hz, 2H), 6.77 (t, J = 5.8 Hz, 1H), 6.50 (s, 1H), 5.82 - 5.73 (m, 1H), 5.44 (s, 2H), 5.27 (d, J = 13.8 Hz, 4H), 5.06 (s, 2H), 4.77 (d, J = 6.8 Hz, 1H), 3.72 (d, J = 9.0 Hz, 1H), 3.21 - 3.09 (m, 4H), 2.38 (s, 3H), 2.18 (m, 2H), 1.87 (m, 2H), 1.36 (s, 9H), 0.88 (t, J = 7.2 Hz, 3H).
To a solution of (2S,3S,4S,5R,6S)-6-(2-(3-((tert-butoxycarbonyl)amino)propanamido)-4- (((((lS,9S)-9-ethyl-5-fluoro-9-hydroxy-4-methyl-10,13-dioxo-2,3,9,10,13,15- hexahydro-lH,12H-benzo[de]pyrano[3',4' :6,7]indolizino[l,2-b]quinolin-l- yl)carbamoyl)oxy)methyl)phenoxy)-3,4,5-trihydroxytetrahydro-2H-pyran-2-carboxylic acid 130 (100 mg, 105 pmol) in MeCN (2 mL) at 0°C was added HCI/dioxane (4M, 2 mL). The reaction mixture was stirred at 0°C for 1 h then concentrated. The crude residue was purified by prep-HPLC to afford (2S,3S,4S,5R,6S)-6-(2-(3-aminopropanamido)-4- (((((lS,9S)-9-ethyl-5-fluoro-9-hydroxy-4-methyl-10,13-dioxo-2,3,9,10,13,15- hexahydro-lH,12H-benzo[de]pyrano[3',4' :6,7]indolizino[l,2-b]quinolin-l- yl)carbamoyl)oxy)methyl)phenoxy)-3,4,5-trihydroxytetrahydro-2H-pyran-2-carboxylic acid 27 (10 mg, 12.8% two steps) as a white solid. LCMS m/z = 848.10 [M+H]+.
XH NMR (400 MHz, DMSO) 6 9.61 (brs, 1H), 8.16 - 8.02 (m, 2H), 7.74 (d, J = 10.8 Hz, 1H), 7.30 (s, 1H), 7.22 - 7.12 (m, 2H), 6.60 - 6.47 (m, 1H), 5.43 (s, 2H), 5.30 - 5.18
(m, 3H), 5.13 - 4.99 (m, 2H), 4.59 (d, J = 6.8 Hz, 1H), 3.15 - 2.97 (m, 6H), 2.91 - 2.81 (m, 1H), 2.74 - 2.63 (m, 1H), 2.36 (s, 3H), 2.24 - 2.14 (m, 2H), 1.93 - 1.81 (m, 2H), 0.88 (t, J = 7.4 Hz, 3H).
To a solution of (2S,3S,4S,5R,6S)-6-(2-(3-aminopropanamido)-4-(((((lS,9S)-9-ethyl-5- fluoro-9-hydroxy-4-methyl-10,13-dioxo-2,3,9,10,13,15-hexahydro-lH,12H- benzo[de]pyrano[3',4':6,7]indolizino[l,2-b]quinolin-l-yl)carbamoyl)oxy)methyl) phenoxy)-3,4,5-trihydroxytetrahydro-2H-pyran-2-carboxylic acid (300.0 mg, 353 pmol) in DMF (5 mL) was added 2,5-dioxopyrrolidin-l-yl (S)-44-((((9H-fluoren-9- yl)methoxy)carbonyl)amino)-38-oxo-2,5,8,ll,14,17,20,23,26,29,32,35-dodecaoxa-39- azapentatetracontan-45-oate 17 (439 mg, 425 pmol) and DIPEA (136 mg, 1.06 mmol). The mixture was stirred at RT overnight then directly purified RP-column chromatography (40% MeCN in water) and prep-HPLC [SHIMADZU LC 20Column: Agilent 10 Prep-C18, 250*21.2mm, 10 um Mobile Phase A: 0.1% HCOOH in water (v/v)Mobile Phase B: MeCN] to afford (2S,3S,4S,5R,6S)-6-(2-((S)-44-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)- 38,45-dioxo-2,5,8,ll,14,17,20,23,26,29,32,35-dodecaoxa-39,46-diazanonatetracontan- 49-amido)-4-(((((lS,9S)-9-ethyl-5-fluoro-9-hydroxy-4-methyl-10,13-dioxo- 2,3,9,10,13,15-hexahydro-lH,12H-benzo[de]pyrano [3',4':6,7]indolizino[l,2-b]quinolin- l-yl)ca rbamoyl)oxy)methyl)phenoxy)-3,4,5-trihydroxytetrahydro-2H-pyran-2-carboxylic acid 131 (90 mg, 14.4%) as a yellow oil. LCMS m/z = 893.70 [M+H3O+H]+/2. XH NMR (400 MHz, DMSO) 6 9.42 (d, J = 10.4 Hz, 1H), 8.49 (s, 6H), 8.29 - 8.05 (m, 3H), 7.86 (m, 5H), 7.81 - 7.66 (m, 3H), 7.45 - 7.24 (m, 7H), 7.19 - 7.05 (m, 2H), 6.60 (d, J = 42.0 Hz, 1H), 6.28 (s, 1H), 5.44 (d, J = 3.6 Hz, 2H), 5.32 - 5.22 (m, 4H), 5.03 (m, 2H), 4.58 (d, J = 6.8 Hz, 1H), 4.24 - 4.15 (m, 2H), 3.89 (m, 1H), 3.56 (m, 8H), 3.45 - 3.06 (m, 41H), 2.95 - 2.89 (m, 2H), 2.37 (d, J = 5.6 Hz, 4H), 2.27 (m, 2H), 2.21 - 2.15 (m, 2H), 1.86 (m, 2H), 1.59 - 1.44 (m, 2H), 1.27 (m, 6H), 0.86 (d, J = 7.2 Hz, 3H).
To a solution of (2S,3S,4S,5R,6S)-6-(2-((S)-44-((((9H-fluoren-9-yl)methoxy)carbonyl) amino)-38,45-dioxo-2,5,8,ll,14,17,20,23,26,29,32,35-dodecaoxa-39,46- diazanonatetracontan-49-amido)-4-(((((lS,9S)-9-ethyl-5-fluoro-9-hydroxy-4-methyl- 10,13-dioxo-2,3,9,10,13,15-hexahydro-lH,12H-benzo[de]pyrano[3',4' :6,7] indolizino[l,2-b]quinolin-l-yl)carbamoyl)oxy)methyl)phenoxy)-3,4,5- trihydroxytetrahydro-2H-pyran-2-carboxylic acid 131 (70 mg, 39.5 pmol) in dry DMF (3 mL) at 0°C was added drop-wise a solution of morpholine (30 mg) in dry DMF (0.5 mL). The mixture was allowed to warm to room temperature and stirred overnight after which it was purified by Biotage automated chromatography (40% MeCN in water) to afford (2S,3S,4S,5R,6S)-6-(2-((S)-44-amino-38,45-dioxo-2,5,8,ll,14,17,20,23,26,29,32,35- dodecaoxa-39,46-diazanonatetracontan-49-amido)-4-(((((lS,9S)-9-ethyl-5-fluoro-9- hydroxy-4-methyl-10,13-dioxo-2,3,9,10,13,15-hexahydro-lH,12H-benzo[de]pyrano
[3',4':6,7]indolizino[l,2-b]quinolin-l-yl)carbamoyl)oxy)methyl)phenoxy)-3,4,5- trihydroxytetrahydro-2H-pyran-2-carboxylic acid 132 (40 mg, 64.5%) as a white solid. LCMS m/z = 774.05 [M + 2H]+/2. XH NMR (400 MHz, MeOD) 6 8.16 (brs, 1H), 7.63 (s, 1H), 7.26 - 7.19 (m, 1H), 7.17 - 7.08 (m, 1H), 5.59 (d, J = 16.4 Hz, 1H), 5.39 - 5.25 (m, 4H), 5.19 - 5.09 (m, 3H), 4.41 - 4.16 (m, 3H), 3.77 - 3.49 (m, 56H), 3.35 (s, 3H), 3.26 - 3.00 (m, 3H), 2.75 - 2.49 (m, 2H), 2.45 - 2.27 (m, 7H), 2.02 - 1.91 (m, 2H), 1.80 - 1.55 (m, 2H), 1.40 - 1.22 (m, 6H), 1.01 (t, J = 7.4 Hz, 3H).
A solution of bis(2,5-dioxopyrrolidin-l-yl) 4,7,10,13-tetraoxahexadecanedioate (149 mg, 226 pmol) and DIPEA (17 mg, 135 pmol) in DMF (1 mL) at 10°C was added a solution of (2S,3S,4S,5R,6S)-6-(2-((S)-44-amino-38,45-dioxo-2,5,8,ll,14,17,20,23,26,29,32,35- dodecaoxa-39,46-diazanonatetracontan-49-amido)-4-(((((lS,9S)-9-ethyl-5-fluoro-9- hydroxy-4-methyl-10,13-dioxo-2,3,9,10,13,15-hexahydro-lH,12H-benzo[de]pyrano [3',4':6,7]indolizino[l,2-b]quinolin-l-yl)carbamoyl)oxy)methyl) phenoxy)-3,4,5- trihydroxytetrahydro-2H-pyran-2-carboxylic acid 132 (70 mg, 45.2 pmol) in DMF (1 mL) drop-wise. The mixture was stirred at 10°C for 1.0 h and purified directly by prep-HPLC [Instrument: SHIMADZU LC 20Column: Agilent 10 Prep-C18, 250*21.2mm, 10 um Mobile Phase A: 0.1% HCOOH in water (v/v) Mobile Phase B: MeCN]. The appropriate fractions were combined and extracted with DCM (100 mL x 3). The combined organic phase were dried over Na2SO4 and concentrated to afford (2S,3S,4S,5R,6S)-6-(2-((S)-44-(16-((2,5- dioxopyrrolidin-l-yl)oxy)-16-oxo-4,7,10,13-tetraoxahexadecanamido)-38,45-dioxo- 2,5,8,ll,14,17,20,23,26,29,32,35-dodecaoxa-39,46-diazanonatetracontan-49-amido)- 4-(((((lS,9S)-9-ethyl-5-fluoro-9-hydroxy-4-methyl-10,13-dioxo-2,3,9,10,13,15- hexahydro-lH,12H-benzo[de]pyrano[3',4' :6,7] indolizino[l,2-b]quinolin-l- yl)carbamoyl)oxy)methyl)phenoxy)-3,4,5-trihydroxytetrahydro-2H-pyran-2-carboxylic acid 133 (22 mg, 25.3%) as a white solid. LCMS m/z = 1919.65 [M+H]+. XH NMR (400 MHz, DMSO) 6 12.82 (s, 1H), 9.08 (s, 1H), 8.25 - 8.16 (m, 1H), 8.05 (d, J = 8.4 Hz, 1H), 7.93 (d, J = 7.2 Hz, 2H), 7.82 - 7.70 (m, 2H), 7.31 (s, 1H), 7.10 (s, 2H), 6.50 (s, 1H), 5.85 (d, J = 3.8 Hz, 1H), 5.49 - 5.35 (m, 3H), 5.34 - 5.24 (m, 4H), 5.11 - 5.01 (m, 2H), 4.91 - 4.82 (m, 1H), 4.22 - 4.13 (m, 1H), 3.88 (d, J = 9.6 Hz, 1H), 3.71 (t, J = 6.0 Hz, 2H), 3.59 - 3.44 (m, 62H), 3.44 - 3.41 (m, 3H), 3.39 - 3.35 (m, 2H), 3.23 (s, 4H), 3.17 - 3.07 (m, 1H), 3.00 - 2.88 (m, 4H), 2.80 (s, 3H), 2.42 - 2.33 (m, 5H), 2.27 (t, J = 6.6 Hz, 2H), 2.23 - 2.15 (m, 2H), 1.93 - 1.81 (m, 2H), 1.64 - 1.40 (m, 2H), 1.37 - 1.27 (m, 2H), 1.27 - 1.18 (m, 2H), 0.88 (t, J = 7.2 Hz, 3H).
Example 73: Synthesis of 2-(Methylsulphonyl)ethylamino-MMAF-(3GA-PEG2 NHS Ester (138)

MMAF 134 MMAF (100 mg, 0.137 mmol), HATU (103.6 mg, 0.273 mmol), 2- methanesulfonylethan-l-amine (18.5 mg, 0.159 mmol) and triethylamine (45.7 pL, 0.328 mmol) were dissolved in dry DMF (10 mL) and stirred at RT under inert atmosphere for 18 hours. The reaction mixture was concentrated in vacuo and the remaining crude purified on RP chromatography [Biotage (water + 0.1% TFA/ACN + 0.1% TFA, 0-100%)]. The combined fractions were lyophilised to to give the MMAF-methylsulphonyl derivative 135 as an off-white solid (65 mg, 57% yield). RT = 2.534 min. LCMS m/z = 837.70 (M+ l); 838.70 (M+2), 859.60 (M + Na); 731.69 (loss of methanesulfonylethane).
PNP-carbonate ester 5 (30.1 mg, 0.033 mmol), HOBt (4.1 mg, 0.030 mmol) and MMAF sulfonamide 135 (23 mg mg, 0.027 mmol) were dissolved in a mixture of dry DMF (2 mL), pyridine (400 pL) and DIPEA (14.4 pL, 0.824 mmol. The reaction mixture was stirred at rt under N2 for 3 h and then concentrated in vacuo. The was crude was purified on RP chromatography [Biotage (water + 0.1% TFA/ACN + 0.1% TFA, 0-100%)]. The combined fractions were freeze dried to give the MMAF-sugar derivative 136 as an off-white solid (18 mg, 41% yield). RT = 4.259 min. LCMS m/z 1612.59 (M + l); 1634.63 (M + Na); 837.88 (MMAF sulfonamide fragment)
A solution pf the MMAF-(acetate-protected) sugar 136 (18 mg, 0.0112 mmol) dissolved in a 1: 1 mixture of THF/MeOH (1 mL) was cooled with an ice/water bath. To this, a solution of LiOH.HzO (2.81 mg, 0.067 mmol) dissolved in water (1 mL) was added dropwise over 1 h. After the end of the addition, the mixture was allowed to stir while reaching RT for 5h. After LCMS showed incomplete deprotection, a second portion of LiOH.HzO (2.81 mg, 0.067 mmol) dissolved in water (1 mL) was added to the reaction mixture and stirred for a further 3 h at RT, which led to complete deprotection. The reaction was quenched by the addition of Glacial acetic acid (3.8 pL, 0.067 mmol) and the reaction mixture was concentrated in vacuo. The residue was purified by RP automated chromatography [Biotage (water + 0.1% TFA/ACN + 0.1% TFA, 0-100%)] to give the globally deprotected MMAF-glucuronic acid 137 derivative as a white solid 9 mg (64 %). RT = 2.247 min. LCMS m/z = 1250.24 (M+l); 1272.21 (M + Na); 837.86 (MMAF-sulfonamide fragment); 625.66 (MMAF fragment).
To a stirred solution of bis-PEGz-NHS (29.0 mg, 0.0721 mmol) and DIPEA (3.8 pl, 21.63 pmol) in dry DMF (1 mL), a solution of the MMAF-pGA 137 (9.0 mg, 7.21 pmol) dissolved in dry DMF (1 mL) was added dropwise using a syringe pump over 1 h. After addition was complete, the solvent was removed by high vacuum and the crude purified by RP automated chromatography [Biotage, (water + 0.1% TFA/MeCN + 0.1% TFA, 10-90%)] to give the desired NHS-ester derivative 138 7.0 mg (64%) as a white solid after lyophilisation. RT 2.387 min. LCMS m/z = 1535.50 (M + l); 1536.50 (M+2); 1557.50 (M+Na); 837.89 (MMAF-sulfonamide fragment); 625.70 (MMAF fragment).
Example 74: Toxicological evaluation in non-human primates at higher doses
The non-GLP pilot study to understand the toxicological profile of cMET FDC 129D5BTY- MMAE-pGA-BrPEGi2-PEG4 (DAR6) in cynomolgus non-human primates (see Example 71) was continued with 2 further doses. Each additional dose cycle was for 3 weeks. Dose-4 was Img/kg, dose-5 was 2mg/kg. Based on an ADC of DAR3, the equivalent MMAE dose on an ADC is lOmg/kg and 20mg/kg respectively. The FDC was prepared as in Example 42 with additional checks to ensure an endotoxin level of <1 EU/mg. As the same animals were used, the cumulative dose at the end of the study was 4.125mg/kg over 12 weeks.
Haematological measurements are shown in Figure 64, clinical chemistry measurements associated with liver metabolism is shown in Figure 65 and kidney metabolism in Figure 66 and final body weights shown in Figure 67. All tissues were examined by a qualified pathologist (DVM, Dip.RCPath, FRCPath) and any histological observations are described below. All reference (normal) values were derived from Park et al [235].
The following tissues were examined macroscopically: Adrenals, Aorta (thoracic), Brain, Caecum, Colon, Duodenum, Eyes, Femur (femoral head), Gall bladder, Heart, Ileum, Injection sites, Jejunum, Kidneys, Larynx, Liver, Lung (all lobes), Lymph node mandibular, Lymph node mesenteric, Lymph node inguinal, Mammary gland (thoracic), Oesophagus (distal portion), Optic nerves, Ovaries, Pancreas, Parathyroids, Pituitary, Rectum, Salivary glands (mandibular, parotid), Sciatic nerve, Skeletal muscle (hindlimb), Skin, Spinal cord (cervical region, thoracis region, lumbar region), Spleen, Sternum with bone marrow, Stomach, Thymus, Thyroids, Tongue, Tonsils, Trachea, Trachea bifurcation (with main- stem bronchi), Urinary bladder, Uterus and Vagina with no test item related abnormalities detected in any animal.
The following tissues were examined microscopically: Brain, Caecum, Colon, Duodenum, Eyes, Gall bladder, Heart, Ileum, Injection sites, Jejunum, Kidneys, Liver, Lung (all lobes), Lymph node mesenteric, Ovaries, Pancreas, Sciatic nerve, Skeletal muscle (hindlimb), Spleen, Sternum with bone marrow, Stomach and Urinary bladder. No test item abnormalities were observed in the majority of animals other (a) minimal inflammatory cell infiltrate in the bladder mucosa of one animal and (d) minimal to mild observations (oedema, inflammation, basophilia) related to a mild obstructive nephropathy (outflow obstruction) in the renal medulla and collecting tubules in the cortex.
By the 5th and final dose (2mg/kg, cumulative dose of 4.125mg/kg), the cMET FDC was well tolerated by a number of different measures. Haematologically, white blood cells, neutrophils and lymphocytes (Figure 64a-d) remained in the normal range indicating the absence of neutropenia. Platelet counts were also normal indicating the absence of thrombocytopenia (Figure 64e). From clinical chemistry analyses after the final dose, liver enzymes (Figure 65a-d), notably alanine and aspartate aminotransferase (ALT and AST) were in the normal range, indicating no significant liver damage. This correlated with normal levels of bilirubin (Figure 65e). This was surprising given the high quantity of MMAE administered (at least 4x higher than generally tolerated [97]) with the liver being a major organ for MMAE metabolism. Clinical chemistry markers for kidney function were also in the normal range (Figure 66). However, there was evidence of protein in the urine after the highest dose, indicative of abnormal kidney function. This was reversible/recoverable.
During the entire study, there were no adverse clinical observations other than a moderate but reversible loss in body weight after the 5th dose (Figure 67). This was related to the fluid loss caused by the abnormal kidney function and corelates to the mild microscopic findings seen in the kidney histopathology.
The final dose of 2mg/kg was tolerated and the data can be used to make the following conclusions:
• The maximum tolerated dose (MTD) in cynomolgus was not reached and represents at least 4x higher MMAE payload delivery than comparable ADCs [97].
• This is equivalent to 8mg/kg (mouse) [236] and together with a minimum efficacious dose of ~0.25mg/kg (Example 67), this equates to a therapeutic index of at least 32.
• cMET FDC clearance involves the kidneys.
• The human equivalent dose would be in the region of 0.67mg/kg [236].
Example 75: Pharmacokinetic analyses in non-human primates
Plasma samples were collected over 72h for each dosing cycle and the pharmacokinetic clearance was detected by anti-MMAE and anti-StrepTag detection, as described for the rodent studies (Example 66). The clearance profiles are plotted in Figure 68a-c and the kinetic parameters are listed in Table 22. At each escalating dose, the plasma exposure (bioavailability) as determined by the area-under-curve (AUC) calculation increased and this increase was linear from the 3rd dose onwards. This indicates that there was no cMET- target mediated tissue deposition/clearance (TMDD) at the higher doses. The bioavailability as detected by anti-MMAE/payload (intact conjugate) and anti-Strep tag (scFv only) was similar indicating that there was no significant payload deconjugation in the plasma. There was some evidence of clipping of the Strep tag at the lower doses, but the pharmacokinetic profile was very similar when detecting either component as exemplified in Figure 68c. The half-life of the intact conjugate was 7-9h at the higher doses. A plot of AUC vs FDC dose (Figure 68d) illustrates the linearity of the pharmacokinetics at the higher dose and the TMDD at the lower doses.
Urine samples were collected and cMET FDC binding was analysed to see if any intact conjugate was present (Figure 69). Although not quantitative due to the nature of the collections, significant quantities (~1000g) of functional, binding FDC were detected at around 4-48 hours but not 10 days suggesting the FDC clears, at least in part, as an intact molecule via the kidney excretion route.
These data can be used to make the following conclusions:
• The bioavailability and half-life of the cMET FDC is surprisingly long given the small format size (~40kDa) and kidney clearance. Blinatumomab is a scFv-based protein (~55kDa) with a faster half-life of 1-2.5h in non-human primates [238].
• The cMET FDC binds to cynomolgus cMET with a very similar high affinity (3.48nM vs 9.72nM, Figure 70) and one would have expected a small format with rapid clearance pharmacokinetics to be subject to significant TMDD and poor bioavailability in non-human primates. Unexpectedly, this was not observed.
• The half-life of the cMET FDC would translate to ~12h in humans [237] and is surprisingly close to that of IgG-based ADCs such as telisotuzumab vedotin (NHP half-life ~18-24h, human half-life 36-48h) [57,58].

Example 76: Binding of 129D5BTY scFv and 129D5BTY-MMAE-0GA-brPEG12- PEG4 FDC
Binding of the 129D5BTY scFv was compared to the ABT-700 IgG (antibody component of tesilotuzumab vedotin) by BIACore SPR as described in Example 2. Two injections (400nM) of the 129D5BTY scFv were used to saturate the sensor chip surface. A further injection of ABT-700 (400nM) for 5 minutes showed an increased sensorgram response indicating that ABT-700 was able to access cMET epitopes that were non-overlapping and different to that bound by 129D5BTY scFv (Figure 71a). A similar study was carried out to compare binding with the 5D5 IgG (a precursor to Roche's MetMab) where 400nM of 129D5BTY scFv was compared to 400nM of 5D5 IgG. The sensorgrams indicated non-overlapping epitopes (Figure 71b).
BIACore SPR binding analyses was also used to determine the binding affinities and kinetic constants for the 129D5BTY-MMAE-[3GA-brPEG12-PEG4 FDC on human and cynomolgus cMET. The binding association rate, dissociation rate and affinity were similar across the two species indication excellent species cross-reactivity (Figure 70, Table 23).

Example 77: Conjugation of 129D5B scFv to Exatecan-MA-PEGi2-Val-Ala-PEG4- NHS Ester (74)
129D5B scFv was conjugated to linker-payload 74 to obtain a DAR 5.03 as follows. A stock solution of pure scFv (>95%) was diluted to 50pM (1.4mg/mL) into a pre-equilibrated solution of lOOmM sodium phosphate pH 8.0, 10% DMA. Linker-Payload (25mM in DMSO) was added starting with 1 molar equivalent (compared to scFv lysines) before adding more payload every 60 min and the reaction mixture incubated at 22°C, 500rpm until the desired DAR was achieved. The reaction progress was monitored by reverse-phase mass spectrometry (reaction stopped by the addition of Tris-HCI pH 7.5 (final concentration lOmM, 50% of 0.1% formic acid in water was added, the sample was filtered through 0.2mm spin filters and applied to a C4 reverse phase column (Phenomenex bioZen 2.6mm widepore 100x2.1mm) on an Agilent QTOF 6545XT run in 10% IPA/10% acetonitrile in 0.1% fomic acid/water) and. A total of 1.5 molar equivalents (over 11 lysines) was required.
The sample was purified by SEC HPLC (Sepax Zenix C 150 3pm 10 x 300mm). Sample quality was analysed by RP-LCMS, UV/Vis spectroscopy, DLS and nanoDSF before being used for further studies. The sample was pure and monomeric (>98%, Figure 72a), with a DAR distribution of 3-7 (~88% within 4-6) (Figure 72b). The NanoDSF showed a small decrease in thermal stability compared to the unconjugated parent antibody 129D5BTY but maintained a highly thermal stable folded structure with a Tm of 57.05°C and a high binding affinity (kd) of 0.37nM (Figure 72c), Table 6.
Example 78: Conjugation of 129D5B scFv to Exatecan-(3GA-BrPEGi2-PEG4 NHS Ester (133)
129D5B scFv was conjugated, purified and analysed according to the method described in Example 77 to linker-payload 133 to obtain a conjugate. The sample was pure and monomeric (>98%, Figure 72d) with a DAR 7.9 (Figure 72e) with >70% DAR 6-10. A stock solution of pure scFv (>95%) was diluted to 50pM (1.4mg/mL) into a preequilibrated solution of lOOmM sodium phosphate pH 8.0, 10% DMA. Linker-Payload (25mM in DMSO) was added starting with 1 molar equivalent (compared to scFv lysines) before adding more payload every 60 min and the reaction mixture incubated at 22°C,
500rpm until the desired DAR was achieved (2 molar equivalents required). A high binding affinity was retained (/ d) of. 4.5nM (Figure. 72f, Table 6).
Example 79: Conjugation of 129D5B scFv to DXd-GGFG-BrPEGs-PEG2 NHS Ester (25)
129D5B scFv was conjugated, purified and analysed according to the method described in Example 77 to linker-payload 25 to obtain a DAR 3.7 (Figure 72g). The sample was pure and monomeric (>98%, Figure 69h). A stock solution of pure scFv (>95%) was diluted to 50pM (1.4mg/mL) into a pre-equilibrated solution of lOOmM sodium phosphate pH 8.0, 10% DMA. Linker-Payload (25mM in DMSO) was added starting with 1 molar equivalent (compared to scFv lysines) before adding more payload every 60 min and the reaction mixture incubated at 22°C, 500rpm until the desired DAR was achieved (1.6 molar equivalents required). A high binding affinity was retained (/ d) of. 0.34nM (Figure 72i, Table 6).
Example 80: Conjugation of 129D5B scFv 2-(Methylsulphonyl)ethylamino-MMAF- PGA-PEG2 NHS Ester (138)
129D5B scFv was conjugated, purified and analysed according to the method described in Example 39 to linker-payload 138 to obtain a DAR 5.15. Specifically, a stock solution of pure scFv (>95%) was diluted to Img/ml into a pre-equilibrated solution of 20% DMSO in bicarbonate buffer pH9.2. Linker- Payload (25mM in DMSO) was added in 5 molar equivalents every 90-120 min and the reaction mixture incubated at 20°C, 300rpm. The reaction progress was monitored by size- exclusion chromatography mass spectrometry (SEC-MS, Agilent AdvanceBio SEC 120A 1.9mm 2.1x150mm PEEK isocratic run in 75mM Ammonium Acetate, 10% IPA on an Agilent QTOF 6545XT). A total of 30 equivalents (6 additions) were required (~2.7 equivalents over the lysines).
The crude reaction mixture was pH neutralized with IM NaH2PO4 (1.5%) and purified on an AKTA Avant through a Superdex 75 26/600 isocratic run eluting with 10% IPA/PBS. The monomeric fractions were combined and buffer exchanged into Histidine/NaCI buffer pH7.5 (Figure 73a). The concentrated sample was filtered through a sterile 0.2pm PES filter, flash frozen in liquid nitrogen and stored at -20°C. The sample was analysed by SEC (Sepax Zenix C 150 7.8 x 300mm), SEC-LCMS (Figure 73b). The binding affinity was high at 719pM (Figure 73c; Table 6). Cell killing potency on cMET-expressing Hs746T cells was 96.3pM with no significant activity against cMET-negative T47D cells (Figure 73d, Table 10).
Example 81: FDC efficacy in cMET low gastric and pancreatic cancer patient- derived xenograft (PDX) tumour models
A selection of 16 PDX models (supplied by Crown Bioscience, China) for 5 solid tumours (gastric, pancreatic, breast, lung and head & neck) were evaluated by immunohistochemistry (IHC) for cMET protein expression and cMET gene expression. IHC was carried out using the validated SP44 anti-cMET antibody by Crown Bioscience. Two PDX models were chosen based on low gene expression and IHC score (gastric model GA6885, IHC score 103.22 and pancreatic model PA3142, IHC score 114.27, Figure 76a and Figure 77a) that were not expected to demonstrate tumour growth inhibition efficacy when treated with ABBV399 (telisotuzumab vedotin) as disclosed in EP3626273B1 (In Champions NSCLC PDX model CTG-0363, IHC score 105.52, ABBV399 treatment of 6 doses, weekly at 3mg/kg was ineffective and not significantly different to the vehicle- treated group).
The human patient derived tumours were implanted subcutaneously in BALB/c nude mice in 2-3mm3 pieces and groups of 5 animals randomized into 4 groups of tumour sizes 150- 200mm3 and treated with ABBV399 (3mg/kg), cMET FDC 129D5BTY-MMAE-0GA-BrPEGi2- PEG4 (DAR.6) at Img/kg and 2.5mg/kg and a vehicle/saline control which were prepared as described for other animal studies (Examples 67, 69, 71).
After 2 doses (of the planned 6 doses) for all test agents at one dose/week, the higher dose of FDC demonstrated clear tumour growth inhibition in both the gastric and pancreatic cancer PDX models compared to ABBV399 with excellent tolerability as measured by body weight changes (Figure 76b and Figure 77b, Table 24). In the gastric PDX model, all test agents demonstrated tumour growth inhibition, but in the more challenging pancreatic PDX model, only the higher FDC dose demonstrated any observable tumour growth inhibition (Table 24). PDX models, notably gastric and pancreatic are known to better represent the heterogeneity and physiological properties (e.g. drug resistance and dense stroma) of human cancers compared to cell-line models. The observed superior efficacy of the cMET FDC 129D5BTY-MMAE-[3GA-BrPEGi2-PEG4 supports the premise of improved penetration and enhanced payload delivery whilst being well tolerated.
Table 24

Amino acid sequences







The CDR sequences described herein are defined according to the Kabat system.
CDR-H1:



CDR-H2:



CDR-H3:



CDR-L1:


CDR-L2:


CDR-L3:



>3A2 VH (same for 20F6, 59D5, 54E11, 59D4, 59A10, 65C2, 53G1, 65A3, 65G6, 53G9, 58H11, 59E5, 59H2, 59A6, 59A2, 59D10, 58A9, 59H6, 52G1, 58A2, 52D5, 75G7, 7102, 71F11 and 53D11) (SEQ ID NO. 49)
GAGGTTCAGCTGGTTCAGAGCGGTGCCGAAGTTAAAAAACCGGGTGAAAGCCTGAAAATTTCAT GTAAAGGTAGCGGCTATTCTTTCACTACTTATTGGATCGGTTGGGTGCGCCAGATGCCTGGTAAA GGTCTGGAATGGATGGGCATCATCAACCCCGCTGACAGCAACACTCGTTATAACCCGAGCTTCC AGGGTCAAGTTACCATTAGCGCGGATAAAAGCATTAGCACCGCATATCTGCAGTGGTCTAGCCT GAAAGCGAGCGACACCGCCGTGTATTACTGTGCCCGTCATGGTGGTCGTGGTCGTCTGGACGTT TGGGGTCAGGGCACCCTGGTCACCGTCTCGAGT
>49G5 VH (SEQ ID NO. 50)
GAGGTTCAGCTGGTTCAGAGCGGTGCCGAAGTTAAAAAACCGGGTGAAAGCCTGAAAATTTCAT GTAAAGGTAGCGGCTATTCTTTCACTACTTATTGGATCGGTTGGGTGCGCCAGATGCCTGGTAAA GGTCTGGAATGGATGGGCATCATCAACCCCGCTGACAGCAACACTCGTTATAACCCGAGCTTCC AGGGTCAAGTTACCATTAGCGCGGATAAAAGCATTAGCACCGCATATCTGCAGTGGTCTAGCCT GAAAGCGAGCGACACCGCCGTGTATTACTGTGCCCGTCATGGTGGTCGTGGGCGTTTGTGGCC GTGGGGTCAGGGCACCCTGGTCACCGTCTCGAGT
>77F3 VH (SEQ ID NO. 51)
GAGGTTCAGCTGGTTCAGAGCGGTGCCGAAGTTAAAAAACCGGGTGAAAGCCTGAAAATTTCAT GTAAAGGTAGCGGCTATTCTTTCACTACTTATTGGATCGGTTGGGTGCGCCAGATGCCTGGTAAA GGTCTGGAATGGATGGGCATCATCAACCCCGCTGACAGCAACACTCGTTATAACCCGAGCTTCC AGGGTCAAGTTACCATTAGCGCGGATAAAAGCATTAGCACCGCATATCTGCAGTGGTCTAGCCT GAAAGCGAGCGACACCGCCGTGTATTACTGTGCCCGTCATGGCGGTCGTGGGCGTTTGTGGCC GTGGGGTCAGGGCACCCTGGTCACCGTCTCGAGT
>129D5B VH (SEQ ID NO. 52)
GAGGTTCAGCTGGTTCAGAGCGGTGCCGAAGTTAAAAAACCGGGTGAAAGCCTGAAAATTTCAT GTAAAGGTAGCGGCTATTCTTTCACTACTTATTGGATCGGTTGGGTGCGCCAGATGCCTGGTAAA GGTCTGGAATGGATGGGCATCATCAACCCCGCTGACAGCAACACTCGTTATAACCCGGGCTTCC AGGGTCAAGTTACCATTAGCGCGGATAAAAGCATTAGCACCGCATATCTGCAGTGGTCTAGCCT GAAAGCGAGCGACACCGCCGTGTATTACTGTGCCCGTCATAGCGGTCGTGGGCGTTTGTGGCC GTGGGGTCAGGGCACCCTGGTCACCGTCTCGAGT
>139B2 VH (SEQ ID NO. 53)
GAGGTTCAGCTGGTTCAGAGCGGTGCCGAAGTTGCGAAACCGGGTGAAAGCCTGAAAATTTCAT
GTACGGGTAGCGGCTATTCTTTCACTACTTATTGGATCGGTTGGGTGCGCCAGATGCCTGGTAAA
GGTCTGGAATGGATGGGCATCATCAACCCCGCTGACAGCAACACTCGTTATAACCCGAGCTTCC
AGGGTCAAGTTACCATTAGCGCGGATAAAAGCATTAGCACCGCATATCTGCAGTGGTCTAGCCT
GAAAGCGAGCGACACCGCCGTGTATTACTGTGCCCGTCATGGCGGTCGTGGGCGTTTGTGGCC GTGGGGTCAGGGCACCCTGGTCACCGTCTCGAGT
>139D2 VH (SEQ ID NO. 54)
GAGGTTCAGCTGGTTCAGAGCGGTGCCGAAGTTGCGAAACCGGGTGAAAGCCTGAAAATTTCAT
GTGCGGGTAGCGGCTATTCTTTCACTACTTATTGGATCGGTTGGGTGCGCCAGATGCCTGGTAA
AGGTCTGGAATGGATGGGCATCATCAACCCCGCTGACAGCAACACTCGTTATAACCCGAGCTTC
CAGGGTCAAGTTACCATTAGCGCGGATAGTAGCATTAGCACCGCATATCTGCAGTGGTCTAGCC
TGAAAGCGAGCGACACCGCCGTGTATTACTGTGCCCGTCATGGCGGTCGTGGGCGTTTGTGGCC GTGGGGTCAGGGCACCCTGGTCACCGTCTCGAGT
>139D7 VH (SEQ ID NO. 55)
GAGGTTCAGCTGGTTCAGAGCGGTGCCGAAGTTCTGAAACCGGGTGAAAGCCTGAAAATTTCAT
GTACGGGTAGCGGCTATTCTTTCACTACTTATTGGATCGGTTGGGTGCGCCAGATGCCTGGTAAA
GGTCTGGAATGGATGGGCATCATCAACCCCGCTGACAGCAACACTCGTTATAACCCGAGCTTCC
AGGGTCAAGTTACCATTAGCGCGGATAGTAGCATTAGCACCGCATATCTGCAGTGGTCTAGCCT
GAAAGCGAGCGACACCGCCGTGTATTACTGTGCCCGTCATGGCGGTCGTGGGCGTTTGTGGCC GTGGGGTCAGGGCACCCTGGTCACCGTCTCGAGT
>26B5 VH (SEQ ID NO. 56)
GAGGTTCAGCTGGTTCAGAGCGGTGCCGAAGTTAAAAAACCGGGTGAAAGCCTGAAAATTTCAT
GTAAAGGTAGCGGCTATACTTTCGCTACTTATTGGATCGGTTGGGTGCGCCAGATGCCTGGTAA
AGGTCTGGAATGGATGGGCATCATCAACCCCGGTGACAGCGACACTCGTTATAACCCGAGCTTC
CAGGGTCAAGTTACCATTAGCGCGGATAAAAGCATTAGCACCGCATATCTGCAGTGGTCTAGCCT
GAAAGCGAGCGACACCGCCGTGTATTACTGTGCCCGTGCTGACTACGACTACTACCGTGGTTAC
TACATGGACATCTGGGGTCAGGGCACCCTGGTCACCGTCTCGAGT
>83B11D VH (SEQ ID NO. 57)
GAGGTTCAGCTGGTTCAGAGCGGTGCCGAAGTTAAAAAACCGGGTGAAAGCCTGAAAATTTCAT
GTAAAGGTAGCGGCTATACTTTCGATACTTATTGGATCGGTTGGGTGCGCCAGATGCCTGGTAA
AGGTCTGGAATGGATGGGCATCATCAACCCCGGTGACAGCGACACTCGTTATAACCCGAGCTTC
CAGGGTCAAGTTACCATTAGCGCGGATAAAAGCATTAGCACCGCATATCTGCAGTGGTCTAGCCT
GAAAGCGAGCGACACCGCCGTGTATTACTGTGCCCGTGCTGACTACGACTACTACCGTGGTTAC
TACATGGACATCTGGGGTCAGGGCACCCTGGTCACCGTCTCGAGT
>9402 VH (SEQ ID NO. 58)
GAGGTTCAGCTGGTTCAGAGCGGTGCCGAAGTTAAAAAACCGGGTGAAAGCCTGAAAATTTCAT
GTAAAGGTAGCGGCTATACTTTCGATACTTATTGGATCGGTTGGGTGCGCCAGATGCCTGGTAA
AGGTCTGGAATGGATGGGCATCATCAACCCCGGTGACAGCGACACTCGTTATAACCCGAGCTTC
CAGGGTCAAGTTACCATTAGCGCGGATAAAAGCATTAGCACCGCATATCTGCAGTGGTCTAGCCT
GAAAGCGAGCGACACCGCCGTGTATTACTGTGCCCGTGTTGATGATTTTTATTACCGTGGTTACT ACATGGACATCTGGGGTCAGGGCACCCTGGTCACCGTCTCGAGT
>115D1D VH (SEQ ID NO. 59)
GAGGTTCAGCTGGTTCAGAGCGGTGCCGAAGTTAAAAAACCGGGTGAAAGCCTGAAAATTTCAT
GTAAAGGTAGCGGCTATGCTTTCGATACTTATTGGATCGGTTGGGTGCGCCAGATGCCTGGTAA
AGGTCTGGAATGGATGGGCATCATCAACCCCGGTGACAGCGACACTCGTTATAACCCGAGCTTC
CAGGGTCAAGTTACCATTAGCGCGGATAAAAGCATTAGCACCGCATATCTGCAGTGGTCTAGCCT
GAAAGCGAGCGACACCGCCGTGTATTACTGTGCCCGTGTTGATGATTATTATTACCGTGGTTACT ACATGGACATCTGGGGTCAGGGCACCCTGGTCACCGTCTCGAGT
>131D5S VH (SEQ ID NO. 60)
GAGGTTCAGCTGGTTCAGAGCGGTGCCGAAGTTAAAAAACCGGGTGAAAGCCTGAAAATTTCAT
GTAAAGGTAGCGGCTATGCTTTCGATACTTATTGGGTCGGTTGGGTGCGCCAGATGCCAGGTAA
AGGTCTGGAATGGATGGGCATCATCAACCCCGGTGACAGCGACACTCGTTATAACCCGAGCTTC
CAGGGTCAAGTTACCATTAGCGCGGATAAAAGCATTAGCACCGCATATCTGCAGTGGTCTAGCCT
GAAAGCGAGCGACACCGCCGTGTATTACTGTGCCCGTGTTGATGATTATTATTACCGTGGTTACT
ACATGGACATCTGGGGTCAGGGCACCCTGGTCACTGTCTCGAGT
>3A2 VL (SEQ ID NO. 61)
CAGTCTGTGCTGACTCAGCCTCCGAGCGTTAGCGCAGCACCGGGTCAGAAAGTGACCATTAGTT
GTAGCGGTAGCAGCAGCAACATCGGCAACTACTACGTCTCTTGGTATCAGCAGCTGCCTGGCAC
CGCACCGAAACTGCTGATTTATCGTAACTCTAACCGCCCATCTGGTGTGCCGGATCGTTTTAGCG
GTAGTAAAAGCGGCACCAGCGCAAGCCTGGCAATTAGCGGTCTGCAGAGCGAAGATGAAGCAG
ATTATTACTGTGCGGCTTGGGACGATTCTCTTTCTGGTTGGGTTTTTGGTGGTGGGACCAAGCTG ACCGTCCTA
>20F6 VL (same for 49G5, 49A6, 55G9 and 55A3) (SEQ ID NO. 62)
CAGTCTGTGCTGACTCAGCCTCCGAGCGTTAGCGCAGCACCGGGTCAGAAAGTGACCATTAGTT
GTAGCGGTAGCAGCAGCAACATCGGCTCTAACTACGTCTCTTGGTATCAGCAGCTGCCTGGCAC
CGCACCGAAACTGCTGATTTATAGTAACTCTAACCGCCCATCTGGTGTGCCGGATCGTTTTAGCG
GTAGTAAAAGCGGCACCAGCGCAAGCCTGGCAATTAGCGGTCTGCAGAGCGAAGATGAAGCAG
ATTATTACTGTGCGGCTTGGGACGATTCTCTTTCTGGTTGGGTTTTTGGTGGTGGGACCAAGCTG
ACCGTCCTA
>53D11 (same for 77F3 and 126A8B) (SEQ ID NO. 63)
CAGTCTGTGCTGACTCAGCCTCCGAGCGTTAGCGCAGCACCGGGTCAGAAAGTGACCATTAGTT
GTAGCGGTAGCAGCAGCAACATCGGCTCTAACTACGTCTCTTGGTATCAGCAGCTGCCTGGCAC
CGCACCGAAACTGCTGATTTATAGTAACTCTAACCGCCCATCTGGTGTGCCGGATCGTTTTAGCG
GTAGTAAAAGCGGCACCAGCGCAAGCCTGGCAATTAGCGGTCTGCAGAGCGAAGATGAAGCAG
ATTATTACTGTGCGGCTTGGGATAATTCTGTTGCGGGTTGGGTTTTTGGTGGTGGGACCAAGCT GACCGTCCTA
>129D5B VL (SEQ ID NO. 64)
CAGCCTGTGCTGACTCAGCCTCCGAGCGTTAGCGCAGCACCGGGTCAGAAAGTGACCATTAGTT
GCAGCGGTAGCAGCAGCAACATCGGCTCTAACTACGTCTCTTGGTATCAGCAGCTGCCTGGCAC
CGCACCGAAACTGCTGATTTATAGTAACTCTAACCGCCCATCTGGTGTGCCGGATCGTTTTAGCG
GTAGTAAAAGCGGCACCAGCGCAAGCCTGGCAATTAGCGGTCTGCAGAGCGAAGATGAAGCAG
ATTATTACTGTGCGGCTTGGGATAATTCTGTTGCAGGTTGGGTTTTTGGTGGTGGGACCAAGCTG ACCGTCCTA
>129D5BTY VL (SEQ ID NO. 199)
CAGCCTGTGCTGACTCAGCCTCCGAGCGTTAGCGCAGCACCGGGTCAGAAAGTGACCATTAGTT
GCAGCGGTAGCAGCAGCAACATCGGCTCTAACTACGTCTCTTGGTATCAGCAGCTGCCTGGCAC
CGCACCGAAACTGCTGATTTATAGTAACTACAACCGCCCATCTGGTGTGCCGGATCGTTTTAGCG
GTAGTAAAAGCGGCACCAGCGCAAGCCTGGCAATTAGCGGTCTGCAGAGCGAAGATGAAGCAG
ATTATTACTGTGCGGCTTGGGATAATACGGTTGCAGGTTGGGTTTTTGGTGGTGGGACCAAGCT GACCGTCCTA
>139B2 VL (SEQ ID NO. 65)
CAGTCTGTGCTGACTCAGCCTCCGAGCGTTAGCGCAGCACCGGGTCAGAAAGTGACCATTAGTT
GTAGCGGTAGCAGCAGCAACATCGGCTCTAACTACGTCTCTTGGTATCAGCAGCTGCCTGGCAC
CGCACCGAAACTGCTGATTTATAGTAACTCTAACCGCCCATCTGGTGTGCCGGATCGTTTTAGCG
GTAGTAAAAGCGGCACCAGCGCAAGCCTGGCAATTAGCGGTCTGCAGAGCGAAGATGAAGCAG
ATTATTACTGTGCGGCTTGGGATAATTCTGTTGCGGGTTGGGTTTTTGGTGGTGGGACCCAGCT GACCGTCCTA
>139D2 VL (same for 139D7) (SEQ ID NO. 66)
CAGTCTGTGCTGACTCAGCCTCCGAGCGTTAGCGCAGCACCGGGTCAGAAAGTGACCATTAGTT
GTAGCGGTAGCAGCAGCAACATCGGCTCTAACTACGTCTCTTGGTATCAGCAGCTGCCTGGCAC
CGCACCGAAACTGCTGATTTATAGTAACTCTAACCGCCCATCTGGTGTGCCGGATCGTTTTAGCG
GTAGTAAAAGCGGCACCAGCGCAAGCCTGGCAATTAGCGGTCTGCAGAGCGAAGATGAAGCAG
ATTATTACTGTGCGGCTTGGGATAATTCTGTTGCGGGTTGGGTTTTTGGTGGTGGGACCCGGCT GACCGTCCTA
>26B5 VL (SEQ ID NO. 67)
CAGTCTGTGCTGACTCAGCCTCCGAGCGTTAGCGCAGCACCGGGTCAGAAAGTGACCATTAGTT
GTAGCGGTAGCAGCAGCAACATCGGCTCTTACGTTGTCTACTGGTATCAGCAGCTGCCTGGCAC
CGC ACCGAAACTGCTGATTT ATGACAAC ACT AACCGCCCATCTGGTGTGCCGGATCGTTTT AGOG
GTAGTAAAAGCGGCACCAGCGCAAGCCTGGCAATTAGCGGTCTGCAGAGCGAAGATGAAGCAG
ATTATTACTGTGCGACTTGGGACGATTCTCTTAACGGTCCGGTTTTTGGTGGTGGGACCAAGCTG ACCGTCCTA
>83B11D VL (same for 94C2, 94C3, 94E6, 112F2 and 112G4) (SEQ ID NO. 68)
CAGTCTGTGCTGACTCAGCCTCCGAGCGTTAGCGCAGCACCGGGTCAGAAAGTGACCATTAGTT
GTAGCGGTAGCAGCAGCAACATCGGCTCTTACGTTGTCTACTGGTATCAGCAGCTGCCTGGCAC
CGCACCGAAACTGCTGATTTATGACAACAACCAGCGCCCATCTGGTGTGCCGGATCGTTTTAGC
GGTAGTAAAAGCGGCACCAGCGCAAGCCTGGCAATTAGCGGTCTGCAGAGCGAAGATGAAGCA
GATTATTACTGTGCGTCTTGGGACGATTCTCTTAACGGTCCGGTTTTTGGTGGTGGGACCAAGCT GACCGTCCTA
> 115D1D VL (same for 116A5 and 121H3B) (SEQ ID NO. 69)
CAGTCTGTGCTGACTCAGCCTCCGAGCGTTAGCGCAGCACCGGGTCAGAAAGTGACCATTAGTT
GTAGCGGTAGCAGCAGCAACATCGGCCCTTACGTTGTCTACTGGTATCAGCAGCTGCCTGGCAC
CGCACCGAAACTGCTGATTTATGACAACAACCAGCGCCCATCTGGCGTGCCGGATCGTTTTAGC
GGTAGTAAAAGCGGCACCAGCGCAAGCCTGGCAATTAGCGGTCTGCAGAGCGAAGATGAAGCA
GATTATTACTGTGCGACTTGGGACGATTCTCTTAACGGTCCGGTTTTTGGTGGTGGGACCAAGCT GACCGTCCTA
> 131D5S VL (SEQ ID NO. 70)
CAGTCTGTGCTGACGCAGCCTCCGAGCGTTAGCGCAGCACCGGGTCAGAAAGTGACCATTAGTT
GTAGCGGTAGCAGCAGCAACATCGGCCCTTACGTTGTCTACTGGTATCAGCAGCTGCCTGGCAG
CGCACCGAAACTGCTGATTTATGACAACAACCAGCGCCCAACTGGCGTGCCGGATCGTTTTAGC
GGTAGTAAAAGCGGCACCAGCGCAAGCCTGGCAATTAGCGGTCTGCAGAGCGAAGATGAAGCA
GATTATTACTGTGCGACTTGGGACGATTCACTTAACGGTCCGGTTTTTGGTGGTGGGACCAAGCT GACCGTCCTA
>49A6 VH (SEQ ID NO. 154)
GAGGTTCAGCTGGTTCAGAGCGGTGCCGAAGTTAAAAAACCGGGTGAAAGCCTGAAAATTTCAT
GTAAAGGTAGCGGCTATTCTTTCACTACTTATTGGATCGGTTGGGTGCGCCAGATGCCTGGTAAA
GGTCTGGAATGGATGGGCATCATCAACCCCGCTGACAGCAACACTCGTTATAACCCGAGCTTCC
AGGGTCAAGTTACCATTAGCGCGGATAAAAGCATTAGCACCGCAtATCTGCAGTGGTCTAGCCTG
AAAGCGAGCGACACCGCCGTGTATTACTGTGCCCGTCATGGTGGTCGTGGTCGTCTTGCTTATT
GGGGTCAGGGCACCCTGGTCACCGTCTCGAGT
>55G9 VH (SEQ ID NO. 155)
GAGGTTCAGCTGGTTCAGAGCGGTGCCGAAGTTAAAAAACCGGGTGAAAGCCTGAAAATTTCAT
GTAAAGGTAGCGGCTATTCTTTCACTACTTATTGGATCGGTTGGGTGCGCCAGATGCCTGGTAAA
GGTCTGGAATGGATGGGCATCATCAACCCCGCTGACAGCAACACTCGTTATAACCCGAGCTTCC
AGGGTCAAGTTACCATTAGCGCGGATAAAAGCATTAGCACCGCATATCTGCAGTGGTCTAGCCT
GAAAGCGAGCGACACCGCCGTGTATTACTGTGCCCGTCATGGTGGTCGTGGGAGGCTTGAGCG
TTGGGGTCAGGGCACCCTGGTCACCGTCTCGAGT
>55A3 VH (SEQ ID NO. 156)
GAGGTTCAGCTGGTTCAGAGCGGTGCCGAAGTTAAAAAACCGGGTGAAAGCCTGAAAATTTCAT
GTAAAGGTAGCGGCTATTCTTTCACTACTTATTGGATCGGTTGGGTGCGCCAGATGCCTGGTAAA
GGTCTGGAATGGATGGGCATCATCAACCCCGCTGACAGCAACACTCGTTATAACCCGAGCTTCC
AGGGTCAAGTTACCATTAGCGCGGATAAAAGCATTAGCACCGCATATCTGCAGTGGTCTAGCCT
GAAAGCGAGCGACACCGCCGTGTATTACTGTGCCCGTCATGGTGGTCGTGGTCGTCTGCCTTAT
TGGGGTCAGGGCACCCTGGTCACCGTCTCGAGT
>7403 VH (SEQ ID NO. 157)
GAGGTTCAGCTGGTTCAGAGCGGTGCCGAAGTTAAGAAACCGGGTGAAAGCCTGAAAATTTCAT
GTAAAGGTAGCGGCTATTCTTTCACTACTTATTGGATCGGTTGGATGCGCCAGATGCCTGGTAAA
GGTCTGGAATGGATGGGCATCATCAACCCCGCTGACAGCAACACTCGTTATAACCCGAGCTTCC
AGGGTCAAGTTACCATTAGCGCGGATAAAAGCATTAGCACCGCATATCTGCAGTGGTCTAGCCT
GAAAGCGAGCGACACCGCCGTGTATTACTGTGCCCGTCATGGTGGTCGTGGTCGTCTGGACGTT
TGGGGTCAGGGCACCCTGGTCACCGTCTCGAGT
>64B2 VH (SEQ ID NO. 158)
GAGGTTCAGCTGGTTCAGAGCGGTGCCGAAGTTAAAAAACCGGGTGAAAGCCTGAAAATTTCAT
GCGAAGGTAGCGGCTATTCTTTCACTACTTATTGGATCGGTTGGGTGCGCCAGATGCCTGGTAA
AGGTCTGGAATGGATGGGCATCATCAACCCCGCTGACAGCAACACTCGTTATAACCCGAGCTTC
CAGGGTCAAGTTACCATTAGCGCGGATAAAAGCATTAGCACCGCATATCTGCAGTGGTCTAGCCT
GAAAGCGAGCGACACCGCCGTGTATTACTGTGCCCGTCATGGTGGTCGTGGTCGTCTGGACGTT
TGGGGTCAGGGCACCCTGGTCACCGTCTCGAGT
>71D4 VH (SEQ ID NO. 159)
GAGGTTCAGCTGGTTCAGAGCGGTGCCGAAGTTAAAAAACCGGGTGAAAGCCTGAAAATTTCAT
GTAAAGGTAGCGGCTATTCTTTCACTACTTATTGGGTCGGTTGGGTGCGCCAGATGCCTGGTAAA
GGTCTGGAATGGATGGGCATCATCAACCCCGCTGACAGCAACACTCGTTATAACCCGAGCTTCC
AGGGTCAAGTTACCATTAGCGCGGATAAGAGCATTAGCACCGCATATCTGCAGTGGTCTAGCCT
GAAAGCGAGCGACACCGCCGTGTATTACTGTGCCCGTCATGGTGGTCGTGGTCGTCTGGACGTT TGGGGTCAGGGCACCCTGGTCACCGTCTCGAGT
>126A8B VH (SEQ ID NO. 160)
GAGGTTCAGCTGGTTCAGAGCGGTGCCGAAGTTAAAAAACCGGGTGAAAGCCTGAAAATTTCAT
GTAAAGGTAGCGGCTATTCTTTCACTACTTATTGGATCGGTTGGGTGCGCCAGATGCCTGGTAAA
GGTCTGGAATGGATGGGCATCATCAACCCCGCTGACAGCAACACTCGTTATAGCCCGAGCTTCC
AGGGTCAAGTTACCATTAGCGCGGATAAAAGCATTAGCACCGCATATCTGCAGTGGTCTAGCCT
GAAAGCGAGCGACACCGCCGTGTATTACTGTGCCCGTCATAGCGGTCGTGGGCGTTTGTGGCC GTGGGGTCAGGGCACCCTGGTCACCGTCTCGAGT
>9403 VH (same for 116A5) (SEQ ID NO. 161)
GAGGTTCAGCTGGTTCAGAGCGGTGCCGAAGTTAAAAAACCGGGTGAAAGCCTGAAAATTTCAT
GTAAAGGTAGCGGCTATACTTTCGATACTTATTGGATCGGTTGGGTGCGCCAGATGCCTGGTAA
AGGTCTGGAATGGATGGGCATCATCAACCCCGGTGACAGCGACACTCGTTATAACCCGAGCTTC
CAGGGTCAAGTTACCATTAGCGCGGATAAAAGCATTAGCACCGCATATCTGCAGTGGTCCAGCC
TGAAAGCGAGCGACACCGCCGTGTATTACTGTGCCCGTACGGATGATTATTATTACCGTGGTTAC
TACATGGACATCTGGGGTCAGGGCACCCTGGTCACCGTCTCGAGT
>94E6 VH (SEQ ID NO. 162)
GAGGTTCAGCTGGTTCAGAGCGGTGCCGAAGTTAAAAAACCGGGTGAAAGCCTGAAAATTTCAT
GTAAAGGTAGCGGCTATACTTTCGATACTTATTGGATCGGTTGGGTGCGCCAGATGCCTGGTAA
AGGTCTGGAATGGATGGGCATCATCAACCCCGGTGACAGCGACACTCGTTATAACCCGAGCTTC
CAGGGTCAAGTTACCATTAGCGCGGATAAAAGCATTAGCACCGCATATCTGCAGTGGTCTAGCCT
GAAAGCGAGCGACACCGCCGTGTATTACTGTGCCCGTACGAATGATGAGTATTACCGTGGTTAC
TACATGGACATCTGGGGTCAGGGCACCCTGGTCACCGTCTCGAGT
>112F2 VH (same for 113A3) (SEQ ID NO. 163)
GAGGTTCAGCTGGTTCAGAGCGGTGCCGAAGTTAAAAAACCGGGTGAAAGCCTGAAAATTTCAT
GTAAAGGTAGCGGCTATACTTTCGATACTTATGGGATCGGTTGGGTGCGCCAGATGCCTGGTAA
AGGTCTGGAATGGATGGGCATCATCAACCCCGGTGACAGCGACACTCGTTATAACCCGAGCTTC
CAGGGTCAAGTTACCATTAGCGCGGATAAAAGCATTAGCACCGCATATCTGCAGTGGTCTAGCCT
GAAAGCGAGCGACACCGCCGTGTATTACTGTGCCCGTGTTGATGATTTTTATTACCGTGGTTACT
ACATGGACATCTGGGGTCAGGGCACCCTGGTCACCGTCTCGAGT
>112G4 VH (same for 113G8) (SEQ ID NO. 164)
GAGGTTCAGCTGGTTCAGAGCGGTGCCGAAGTTAAAAAACCGGGTGAAAGCCTGAAAATTTCAT
GTAAAGGTAGCGGCTATACTTTCGATACTTATGGGATCGGTTGGGTGCGCCAGATGCCTGGTAA
AGGTCTGGAATGGATGGGCACCATCGACCCCGGTGACAGCGACACTCGTTATAACCCGAGCTTC
CAGGGTCAAGTTACCATTAGCGCGGATAAAAGCATTAGCACCGCATATCTGCAGTGGTCTAGCCT
GAAAGCGAGCGACACCGCCGTGTATTACTGTGCCCGTACGGATGATTATTATTACCGTGGTTACT ACATGGACATCTGGGGTCAGGGCACCCTGGTCACCGTCTCGAGT
>11507 VH (SEQ ID NO. 165)
GAGGTTCAGCTGGTTCAGAGCGGTGCCGAAGTTAAAAAACCGGGTGAAAGCCTGAAAATTTCAT
GTAAAGGTAGCGGCTATACTTTCGATACTTATcAGATCGGTTGGGTGCGCCAGATGCCTGGTAAA
GGTCTGGAATGGATGGGCATCATCAACCCCGGTGACAGCGACACTCGCTATAACCCGAGCTTCC
AGGGTCAAGTTACCATTAGCGCGGATAAAAGCATTAGCACCGCATATCTGCAGTGGTCTAGCCT
GAAAGCGAGCGACACCGCCGTGTATTACTGTGCCCGTGTTGATGATTTTTATTACCGTGGTTACT ACATGGACATCTGGGGTCAGGGCACCCTGGTCACCGTCTCGAGT
>121H3B VH (SEQ ID NO. 166)
GAGGTTCAGCTTGTTCAGAGCGGTGCCGAAGTTAAAAAACCGGGTGAAAGCCTGAAAATTTCAT
GTAAAGGTAGCGGCTATACTTTCGATACTTATTGGATCGGTTGGGTGCGCCAGATGCCTGGTAA
AGGTCTGGAATGGATGGGCATCATCAACCCCGGTGACAGCGACGCTCGTTATAACCCGAGCTTC
CAGGGTCAAGTTACCATTAGCGCGGATAAAAGCATTAGCACCGCATATCTGCAGTGGTCCAGCC
TGAAAGCGAGCGACACCGCCGTGTATTACTGTGCCCGTGCGGATGATTATTATTACCGTGGTTAC TACATGGACATCTGGGGTCAGGGCACCCTGGTCACCGTCTCGAGT
>59D5 VL (SEQ ID NO. 167)
CAGTCTGTGCTGACTCAGCCTCCGAGCGTTAGCGCAGCACCGGGTCAGAAAGTGACCATTAGTT
GTAGCGGTAGCAGCAGCAACATCGGCTCTAACTACGTCTCTTGGTATCAGCAGCTGCCTGGCAC
CGCACCGAAACTGCTGATTTATAGTAACTCTAACCGCCCATCTGGTGTGCCGGATCGTTTTAGCG
GTAGTAAAAGCGGCACCAGCGCAAGCCTGGCAATTAGCGGTCTGCAGAGCGAAGATGAAGCAG
ATTATTACTGTGCGGCTTGGGATAATCCTAGTGGGGGTTGGGTTTTTGGTGGTGGGACCAAGCT GACCGTCCTA
>54E11 VL (SEQ ID NO. 168)
CAGTCTGTGCTGACTCAGCCTCCGAGCGTTAGCGCAGCACCGGGTCAGAAAGTGACCATTAGTT
GTAGCGGTAGCAGCAGCAACATCGGCTCTAACTACGTCTCTTGGTATCAGCAGCTGCCTGGCAC
CGCACCGAAACTGCTGATTTATAGTAACTCTAACCGCCCATCTGGTGTGCCGGATCGTTTTAGCG
GTAGTAAAAGCGGCACCAGCGCAAGCCTGGCAATTAGCGGTCTGCAGAGCGAAGATGAAGCAG
ATTATTACTGTGCGGCTTGGGATCAGATTTCTGGGGGTTGGGTTTTTGGTGGTGGGACCAAGCT
GACCGTCCTA
>59D4 VL (SEQ ID NO. 169)
CAGTCTGTGCTGACTCAGCCTCCGAGCGTTAGCGCAGCACCGGGTCAGAAAGTGACCATTAGTT
GTAGCGGTAGCAGCAGCAACATCGGCTCTAACTACGTCTCTTGGTATCAGCAGCTGCCTGGCAC
CGCACCGAAACTGCTGATTTATAGTAACTCTAACCGCCCATCTGGTGTGCCGGATCGTTTTAGCG
GTAGTAAAAGCGGCACCAGCGCAAGCCTGGCAATTAGCGGTCTGCAGAGCGAAGATGAAGCAG
ATTATTACTGTGCGGCTTGGGATCAGGAGTTTGGGGGTTGGGTTTTTGGTGGTGGGACCAAGCT GACCGTCCTA
>59A10 VL (SEQ ID NO. 170)
CAGTCTGTGCTGACTCAGCCTCCGAGCGTTAGCGCAGCACCGGGTCAGAAAGTGACCATTAGTT
GTAGCGGTAGCAGCAGCAACATCGGCTCTAACTACGTCTCTTGGTATCAGCAGCTGCCTGGCAC
CGCACCGAAACTGCTGATTTATAGTAACTCTAACCGCCCATCTGGTGTGCCGGATCGTTTTAGCG
GTAGTAAAAGCGGCACCAGCGCAAGCCTGGCAATTAGCGGTCTGCAGAGCGAAGATGAAGCAG
ATTATTACTGTGCGGCTTGGGATCAGGGTTATAATGGTTGGGTTTTTGGTGGTGGGACCAAGCT GACCGTCCTA
>65C2 VL (SEQ ID NO. 171)
CAGTCTGTGCTGACTCAGCCTCCGAGCGTTAGCGCAGCACCGGGTCAGAAAGTGACCATTAGTT
GTAGCGGTAGCAGCAGCAACATCGGCTCTAACTACGTCTCTTGGTATCAGCAGCTGCCTGGCAC
CGCACCGAAACTGCTGATTTATAGTAACTCTAACCGCCCATCTGGTGTGCCGGATCGTTTTAGCG
GTAGTAAAAGCGGCACCAGCGCAAGCCTGGCAATTAGCGGTCTGCAGAGCGAAGATGAAGCAG
ATTATTACTGTGCGGCTTGGGATCAGAGTACTGATGGTTGGGTTTTTGGTGGTGGGACCAAGCT GACCGTCCTA
>53G1 VL (SEQ ID NO. 172)
CAGTCTGTGCTGACTCAGCCTCCGAGCGTTAGCGCAGCACCGGGTCAGAAAGTGACCATTAGTT
GTAGCGGTAGCAGCAGCAACATCGGCTCTAACTACGTCTCTTGGTATCAGCAGCTGCCTGGCAC
CGCACCGAAACTGCTGATTTATAGTAACTCTAACCGCCCATCTGGTGTGCCGGATCGTTTTAGCG
GTAGTAAAAGCGGCACCAGCGCAAGCCTGGCAATTAGCGGTCTGCAGAGCGAAGATGAAGCAG
ATTATTACTGTGCGGCTTGGAGTAATTATGCTGCTGGTTGGGTTTTTGGTGGTGGGACCAAGCTG ACCGTCCTA
>65A3 VL (SEQ ID NO. 173)
CAGTCTGTGCTGACTCAGCCTCCGAGCGTTAGCGCAGCACCGGGTCAGAAAGTGACCATTAGTT
GTAGCGGTAGCAGCAGCAACATCGGCTCTAACTACGTCTCTTGGTATCAGCAGCTGCCTGGCAC
CGCACCGAAACTGCTGATTTATAGTAACTCTAACCGCCCATCTGGTGTGCCGGATCGTTTTAGCG
GTAGTAAAAGCGGCACCAGCGCAAGCCTGGCAATTAGCGGTCTGCAGAGCGAAGATGAAGCAG
ATTATTACTGTGCGGCTTGGGAGAATTATGCTGCTGGTTGGGTTTTTGGTGGTGGGACCAAGCT GACCGTCCTA
>65G6 VL (SEQ ID NO. 174)
CAGTCTGTGCTGACTCAGCCTCCGAGCGTTAGCGCAGCACCGGGTCAGAAAGTGACCATTAGTT
GTAGCGGTAGCAGCAGCAACATCGGCTCTAACTACGTCTCTTGGTATCAGCAGCTGCCTGGCAC
CGCACCGAAACTGCTGATTTATAGTAACTCTAACCGCCCATCTGGTGTGCCGGATCGTTTTAGCG
GTAGTAAAAGCGGCACCAGCGCAAGCCTGGCAATTAGCGGTCTGCAGAGCGAAGATGAAGCAG
ATTATTACTGTGCGGCTTGGGAGAATTGGGAGGCGGGTTGGGTTTTTGGTGGTGGGACCAAGCT GACCGTCCTA
>53G9 VL (SEQ ID NO. 175)
CAGTCTGTGCTGACTCAGCCTCCGAGCGTTAGCGCAGCACCGGGTCAGAAAGTGACCATTAGTT
GTAGCGGTAGCAGCAGCAACATCGGCTCTAACTACGTCTCTTGGTATCAGCAGCTGCCTGGCAC
CGCACCGAAACTGCTGATTTATAGTAACTCTAACCGCCCATCTGGTGTGCCGGATCGTTTTAGCG
GTAGTAAAAGCGGCACCAGCGCAAGCCTGGCAATTAGCGGTCTGCAGAGCGAAGATGAAGCAG
ATTATTACTGTGCGGCTTGGGATTTGACGGAGGCTGGTTGGGTTTTTGGTGGTGGGACCAAGCT GACCGTCCTA
>58H11 VL (SEQ ID NO. 176)
CAGTCTGTGCTGACTCAGCCTCCGAGCGTTAGCGCAGCACCGGGTCAGAAAGTGACCATTAGTT
GTAGCGGTAGCAGCAGCAACATCGGCTCTAACTACGTCTCTTGGTATCAGCAGCTGCCTGGCAC
CGCACCGAAACTGCTGATTTATAGTAACTCTAACCGCCCATCTGGTGTGCCGGATCGTTTTAGCG
GTAGTAAAAGCGGCACCAGCGCAAGCCTGGCAATTAGCGGTCTGCAGAGCGAAGATGAAGCAG
ATTATTACTGTGCGGCTTGGGACACGGATCAGGCGGGGTGGGTTTTTGGTGGTGGGACCAAGCT GACCGTCCTA
>59E5 VL (SEQ ID NO. 177)
CAGTCTGTGCTGACTCAGCCTCCGAGCGTTAGCGCAGCACCGGGTCAGAAAGTGACCATTAGTT
GTAGCGGTAGCAGCAGCAACATCGGCTCTAACTACGTCTCTTGGTATCAGCAGCTGCCTGGCAC
CGCACCGAAACTGCTGATTTATAGTAACTCTAACCGCCCATCTGGTGTGCCGGATCGTTTTAGCG
GTAGTAAAAGCGGCACCAGCGCAAGCCTGGCAATTAGCGGTCTGCAGAGCGAAGATGAAGCAG
ATTATTACTGTGCGGCTTGGGATTCTGAGGAGGCGGGTTGGGTTTTTGGTGGTGGGACCAAGCT GACCGTCCTA
>59H2 VL (SEQ ID NO. 178)
CAGTCTGTGCTGACTCAGCCTCCGAGCGTTAGCGCAGCACCGGGTCAGAAAGTGACCATTAGTT
GTAGCGGTAGCAGCAGCAACATCGGCTCTAACTACGTCTCTTGGTATCAGCAGCTGCCTGGCAC
CGCACCGAAACTGCTGATTTATAGTAACTCTAACCGCCCATCTGGTGTGCCGGATCGTTTTAGCG
GTAGTAAAAGCGGCACCAGCGCAAGCCTGGCAATTAGCGGTCTGCAGAGCGAAGATGAAGCAG
ATTATTACTGTGCGGCTTGGGATTCTTCGTATGGTGGTTGGGTTTTTGGTGGTGGGACCAAGCTG ACCGTCCTA
>59A6 VL (SEQ ID NO. 179)
CAGTCTGTGCTGACTCAGCCTCCGAGCGTTAGCGCAGCACCGGGTCAGAAAGTGACCATTAGTT
GTAGCGGTAGCAGCAGCAACATCGGCTCTAACTACGTCTCTTGGTATCAGCAGCTGCCTGGCAC
CGCACCGAAACTGCTGATTTATAGTAACTCTAACCGCCCATCTGGTGTGCCGGATCGTTTTAGCG
GTAGTAAAAGCGGCACCAGCGCAAGCCTGGCAATTAGCGGTCTGCAGAGCGAAGATGAAGCAG
ATTATTACTGTGCGGCTTGGGATTCGGCTAATGCGGGTTGGGTTTTTGGTGGTGGGACCAAGCT GACCGTCCTA
>59A2 VL (SEQ ID NO. 180)
CAGTCTGTGCTGACTCAGCCTCCGAGCGTTAGCGCGGCACCGGGCCAGAAAGTGACCATTAGTT
GTAGCGGTAGCAGCAGCAACATCGGCTCTAACTACGTCTCTTGGTATCAGCAGCTGCCTGGCAC
CGCACCGAAACTGCTGATTTATAGTAACTCTAACCGCCCATCTGGTGTGCCGGATCGTTTTAGCG
GTAGTAAAAGCGGCACCAGCGCAAGCCTGGCAATTAGCGGTCTGCAGAGCGAAGATGAAGCAG
ATTATTACTGTGCGGCTTGGGATAGTAGTGCTGCTGGTTGGGTTTTTGGTGGTGGGACCAAGCT GACCGTCCTA
>59D10 VL (SEQ ID NO. 181)
CAGTCTGTGCTGACTCAGCCTCCGAGCGTTAGCGCAGCACCGGGTCAGAAAGTGACCATTAGTT
GTAGCGGTAGCAGCAGCAACATCGGCTCTAACTACGTCTCTTGGTATCAGCAGCTGCCTGGCAC
CGCACCGAAACTGCTGATTTATAGTAACTCTAACCGCCCATCTGGTGTGCCGGATCGTTTTAGCG
GTAGTAAAAGCGGCACCAGCGCAAGCCTGGCAATTAGCGGTCTGCAGAGCGAAGATGAAGCAG
ATTATTACTGTGCGGCTTGGGATGGTAATGATGATGGTTGGGTTTTTGGTGGTGGGACCAAGCT GACCGTCCTA
>58A9 VL (SEQ ID NO. 182)
CAGTCTGTGCTGACTCAGCCTCCGAGCGTTAGCGCAGCACCGGGTCAGAAAGTGACCATTAGTT
GTAGCGGTAGCAGCAGCAACATCGGCTCTAACTACGTCTCTTGGTATCAGCAGCTGCCTGGCAC
CGCACCGAAACTGCTGATTTATAGTAACTCTAACCGCCCATCTGGTGTGCCGGATCGTTTTAGCG
GTAGTAAAAGCGGCACCAGCGCAAGCCTGGCAATTAGCGGTCTGCAGAGCGAAGATGAAGCAG
ATTATTACTGTGCGGCTTGGGACAATCATCAGGCTAGTTGGGTTTTTGGTGGTGGGACCAAGCT
GACCGTCCTA
>74C3 VL (SEQ ID NO. 183)
CAGTCTGTGCTGACTCAGCCTCCGAGCGTTAGCGCAGCACCGGGTCAGAAAGTGACCATTAGTT
GTAGCGGTAGCAGCAGCAACATCGGCTCTAACTACGTCTCTTGGTATCAGCAGCTGCCTGGCAC
CGCACCGAAGCTGCTGATTTATAGTAACTCTAACCGCCCATCTGGTGTGCCGGATCGTTTTAGCG
GTAGTAAAAGCGGCACCAGCGCAAGCCTGGCAATTAGCGGTCTGCAGAGCGAAGATGAAGCAG
ATTATTACTGTGCGGCTTGGGATAATCCTGAGGCGGGTTGGGTTTTTGGTGGTGGGACCAAGCT GACCGTCCTA
>59H6 VL (SEQ ID NO. 184)
CAGTCTGTGCTGACTCAGCCTCCGAGCGTTAGCGCAGCACCGGGTCAGAAAGTGACCATTAGTT
GTAGCGGTAGCAGCAGCAACATCGGCTCTAACTACGTCTCTTGGTATCAGCAGCTGCCTGGCAC
CGCACCGAAACTGCTGATTTATAGTAACTCTAACCGCCCATCTGGTGTGCCGGATCGTTTTAGCG
GTAGTAAAAGCGGCACCAGCGCAAGCCTGGCAATTAGCGGTCTGCAGAGCGAAGATGAAGCAG
ATTATTACTGTGCGGCTTGGGATCAGGATTGGTCGGGTTGGGTTTTTGGTGGTGGGACCAAGCT GACCGTCCTA
>64B2 VL (SEQ ID NO. 185)
CAGTCTGTGCTGACTCAGCCTCCGAGCGTTAGCGCAGCACCGGGTCAGAAAGTGACCATTAGTT
GTAGCGGTAGCAGCAGCAACATCGGCTCTAACTACGTCTCTTGGTATCAGCAGCTGCCTGGCAC
CGCACCGAAACTGCTGATTTATAGTAACTCTAACCGCCCATCTGGTGTGCCGGATCGTTTTAGCG
GTAGTAAAAGCGGCACCAGCGCAAGCCTGGCAATTAGCGGTCTGCAGAGCGAAGATGAAGCAG
ATTATTACTGTGCGGCTTGGGACCAGCAGTTTGCTGGTTGGGTTTTTGGTGGTGGGACCAAGCT GACCGTCCTA
>52G1 VL (SEQ ID NO. 186)
CAGTCTGTGCTGACTCAGCCTCCGAGCGTTAGCGCAGCACCGGGTCAGAAAGTGACCATTAGTT
GTAGCGGTAGCAGCAGCAACATCGGCTCTAACTACGTCTCTTGGTATCAGCAGCTGCCTGGCAC
CGCACCGAAACTGCTGATTTATAGTAACTCTAACCGCCCATCTGGTGTGCCGGATCGTTTTAGCG
GTAGTAAAAGCGGCACCAGCGCAAGCCTGGCAATTAGCGGTCTGCAGAGCGAAGATGAAGCAG
ATTATTACTGTGCGGCTTGGGACTCGATTGCTCATAATTGGGTTTTTGGTGGTGGGACCAAGCTG ACCGTCCTA
>58A2 VL (SEQ ID NO. 187)
CAGTCTGTGCTGACTCAGCCTCCGAGCGTTAGCGCAGCACCGGGTCAGAAAGTGACCATTAGTT
GTAGCGGTAGCAGCAGCAACATCGGCTCTAACTACGTCTCTTGGTATCAGCAGCTGCCTGGCAC
CGCACCGAAACTGCTGATTTATAGTAACTCTAACCGCCCATCTGGTGTGCCGGATCGTTTTAGCG
GTAGTAAAAGCGGCACCAGCGCAAGCCTGGCAATTAGCGGTCTGCAGAGCGAAGATGAAGCAG
ATTATTACTGTGCGGCTTGGGACTCTAATCCTTCGCATTGGGTTTTTGGTGGTGGGACCAAGCTG ACCGTCCTA
>52D5 VL (SEQ ID NO. 188)
CAGTCTGTGCTGACTCAGCCTCCGAGCGTTAGCGCAGCACCGGGTCAGAAAGTGACCATTAGTT
GTAGCGGTAGCAGCAGCAACATCGGCTCTAACTACGTCTCTTGGTATCAGCAGCTGCCTGGCAC
CGCACCGAAACTGCTGATTTATAGTAACTCTAACCGCCCATCTGGTGTGCCGGATCGTTTTAGCG
GTAGTAAAAGCGGCACCAGCGCAAGCCTGGCAATTAGCGGTCTGCAGAGCGAAGATGAAGCAG
ATTATTACTGTGCGGCTTGGGACATTGCTAGTGCGAATTGGGTTTTTGGTGGTGGGACCAAGCT GACCGTCCTA
>71D4 VL (SEQ ID NO. 189)
CAGTCTGTGCTGACTCAGCCTCCGAGCGTTAGCGCAGCACCGGGTCAGAAAGTGACCATTAGTT
GTAGCGGTAGCAGCAGCAACATCGGCTCTAACTACGTCTCTTGGTATCAGCAGCTGCCTGGCAC
CGCACCGAAACTGCTGATTTATAGTAACTCTAACCGCCCATCTGGTGTGCCGGATCGTTTTAGCG
GTAGTAAAAGCGGCACCAGCGCAAGCCTGGCAATTAGCGGTCTGCAGAGCGAAGATGAAGCAG
ATTATTACTGTGCGGCTTGGGATCAGATTTCGGCGGGTTGGGTTTTTGGTGGTGGGACCAAGCT GACCGTCCTA
>75G7 VL (SEQ ID NO. 190)
CAGTCTGTGCTGACTCAGCCTCCGAGCGTTAGCGCAGCACCGGGTCAGAAAGTGACCATTAGTT
GTAGCGGTAGCAGCAGCAACATCGGCTCTAACTACGTCTCTTGGTATCAGCAGCTGCCTGGCAC
CGCACCGAAACTGCTGATTTATAGTAACTCTAACCGCCCATCTGGTGTGCCGGATCGTTTTAGCG
GTAGTAAAAGCGGCACCAGCGCAAGCCTGGCAATTAGCGGTCTGCAGAGCGAAGATGAAGCAG
ATTATTACTGTGCGGCTTGGGATAATCATTATGCGGGTTGGGTTTTTGGTGGTGGGACCAAGCTG ACCGTCCTA
>71C2 VL (SEQ ID NO. 191)
CAGTCTGTGCTGACTCAGCCTCCGAGCGTTAGCGCAGCACCGGGTCAGAAAGTGACCATTAGTT
GTAGCGGTAGCAGCAGCAACATCGGCTCTAACTACGTCTCTTGGTATCAGCAGCTGCCTGGCAC
CGCACCGAAACTGCTGATTTATAGTAACTCTAACCGCCCATCTGGTGTGCCGGATCGTTTTAGCG
GTAGTAAAAGCGGCACCAGCGCAAGCCTGGCAATTAGCGGTCTGCAGAGCGAAGATGAAGCAG
ATTATTACTGTGCGGCTTGGGATAATACTAGTGCTGGTTGGGTTTTTGGTGGTGGGACCAAGCTG ACCGTCCTA
>71F11 VL (SEQ ID NO. 192)
CAGTCTGTGCTGACTCAGCCTCCGAGCGTTAGCGCAGCACCGGGTCAGAAAGTGACCATTAGTT
GTAGCGGTAGCAGCAGCAACATCGGCTCTAACTACGTCTCTTGGTATCAGCAGCTGCCTGGCAC
CGCACCGAAACTGCTGATTTATAGTAACTCTAACCGCCCATCTGGTGTGCCGGATCGTTTTAGCG
GTAGTAAAAGCGGCACCAGCGCAAGCCTGGCAATTAGCGGTCTGCAGAGCGAAGATGAAGCAG
ATTATTACTGTGCGGCTTGGGATAATTCTCTTGCGGGTTGGGTTTTTGGTGGTGGGACCAAGCTG
ACCGTCCTA
>113A3 VL (SEQ ID NO. 193)
CAGTCTGTGCTGACTCAGCCTCCGAGCGTTAGCGCAGCACCGGGTCAGAAAGTGACCATTAGTT
GTAGCGGTAGCAGCAGCAACATCGGCTCTTACGTTGTCTACTGGTATCAGCAGCTGCCTGGCAC
CGCACCGAAACTGCTGATTTATGACAACAACCAGCGCCCATCTGGTGTGCCGGATCGTTTTAGC
GGTAGTAAAAGCGGCACCAGCGCAAGCCTGGCAATTAGCGGTCTGCAGAGCGAAGATGAAGCA
GATTATTACTGTGCGTCTTGGGACGATGTTGATTCTGCTCCGGTTTTTGGTGGTGGGACCAAGCT GACCGTCCTA
>113G8 VL (SEQ ID NO. 194)
CAGTCTGTGCTGACGCAGCCTCCGAGCGTTAGCGCAGCACCGGGTCAGAAAGTGACCATTAGTT
GTAGCGGTAGCAGCAGCAACATCGGCTCTTACGTTGTCTACTGGTATCAGCAGCTGCCTGGCAC
CGCACCGAAACTGCTGATTTATGACAACAACCAGCGCCCATCTGGTGTGCCGGATCGTTTTAGC
GGTAGTAAAAGCGGCACCAGCGCAAGCCTGGCAATTAGCGGTCTGCAGAGCGAAGATGAAGCA
GATTATTACTGTGCGTCTTGGGACGATCCTAATAATGCGCCGGTTTTTGGTGGTGGGACCAAGCT GACCGTCCTA
>115C7 VL (SEQ ID NO. 195)
CAGTCTGTGCTGACGCAGCCTCCGAGCGTTAGCGCAGCACCGGGTCAGAAAGTGACCATTAGTT
GTAGCGGTAGCAGCAGCAACATCGGCTCTTACGTTGTCTACTGGTATCAGCAGCTGCCTGGCAC
CGCACCGAAACTGCTGATTTATGACAACAACCAGCGCCCATCTGGTGTGCCGGATCGTTTTAGC
GGTAGTAAAAGCGGCACCAGCGCAAGCCTGGCAATTAGCGGTCTGCAGAGCGAAGATGAAGCA
GATTATTACTGTGCGACTTGGGACGATCCTAGTCAGCAGCCTGTTTTTGGTGGTGGGAcCAAGCT GACCGTCCTA
Claims
1. An antibody or antigen-binding fragment thereof that specifically binds to the MET receptor (cMET), wherein : (i) the antibody or antigen-binding fragment thereof comprises a heavy chain variable region (VH) comprising the Complementarity Determining Regions (CDRs) CDR-H1, CDR-H2, and CDR-H3 of SEQ ID NO. 1, or a variant thereof having at least 90% sequence identity to SEQ ID NO. 1, and comprises a light chain variable region (VL) comprising the CDRs CDR-L1, CDR-L2, and CDR-L3 of SEQ ID NO. 196 or SEQ ID NO.
2, or a variant thereof having at least 90% sequence identity with SEQ ID NO. 196 or SEQ ID NO. 2; or (ii) the antibody or antigen-binding fragment thereof comprises a heavy chain variable region (VH) comprising the Complementarity Determining Regions (CDRs) CDR- Hl, CDR-H2, and CDR-H3 of SEQ ID NO. 21, or a variant thereof having at least 90% sequence identity to SEQ ID NO. 21, and comprises a light chain variable region (VL) comprising the CDRs CDR-L1, CDR-L2, and CDR-L3 of SEQ ID NO. 22, or a variant thereof having at least 90% sequence identity with SEQ ID NO. 22.
2. The antibody or antigen-binding fragment of claim 1, wherein the antibody or antigen-binding fragment is an antigen-binding fragment.
3. The antibody or antigen-binding fragment of claim 1, wherein the VH comprises the CDRs of SEQ ID NO. 1 and the VL comprises the CDRs of SEQ ID NOs. 196 or 2.
4. The antibody or antigen-binding fragment of any of the preceding claims, wherein the VH comprises the following CDRs: a) CDR-H1 : [SEQ ID NO. 23] or an amino acid sequence having at least 80% identity therewith, for example at least 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably with at least 80% identity; and/or b) CDR-H2: [SEQ ID NO. 28] or an amino acid sequence having at least 80% identity therewith, for example at least 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably with 88% identity; and/or c) CDR-H3: [SEQ ID NO. 32] or an amino acid sequence having at least 73% identity therewith, for example at least 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably with at least 67% identity.
5. The antibody or antigen-binding fragment of any of the preceding claims, wherein the VH comprises the CDRs of SEQ ID NOs 23, 28, and 32.
6. The antibody or antigen-binding fragment of any of the preceding claims comprising a VH having the amino acid sequence of SEQ ID NO. 1, or a variant having at least 80% sequence identity therewith, for example at least 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably having around 93% identity.
7. The antibody or antigen-binding fragment of any of the preceding claims, wherein the VL comprises the following CDRs: a) CDR-L1 : [SEQ ID NO. 37] or an amino acid sequence having at least 80% identity therewith, for example at least 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably having 85% identity; and/or b) CDR-L2: [SEQ ID NOs. 197 or 41] or an amino acid sequence having at least 80% identity therewith, for example at least 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably having at least 71% identity; and/or c) CDR-L3: [SEQ ID NOs. 198 or 46] or an amino acid sequence having at least 55% identity therewith, for example at least 60%, 65%, 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably having at least 55% identity.
8. The antibody or antigen-binding fragment of any of the preceding claims, wherein the VL comprises the CDRs of SEQ ID NOs 37, 197 or 41, and 198 or 46, preferably wherein the VL comprises the CDRs of SEQ ID NOs 37, 197, and 198 or SEQ ID NOs. 37, 41, and 46.
9. The antibody or antigen-binding fragment of any of the preceding claims comprising a VL having the amino acid sequence of SEQ ID NOs. 196 or 2, or a variant having at least 80% sequence identity therewith, for example at least 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably having around 91% identity.
10. The antibody or antigen-binding fragment of any of the preceding claims comprising the following CDRs: a) CDR-H1: [SEQ ID NO. 23] or an amino acid sequence having at least 80% identity therewith, for example at least 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably with at least 80% identity; and/or
b) CDR-H2: [SEQ ID NO. 28] or an amino acid sequence having at least 80% identity therewith, for example at least 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably with 88% identity; and/or c) CDR-H3: [SEQ ID NO. 32] or an amino acid sequence having at least 73% identity therewith, for example at least 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably with at least 67% identity; and/or d) CDR-L1 : [SEQ ID NO. 37] or an amino acid sequence having at least 80% identity therewith, for example at least 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably having 85% identity; and/or e) CDR-L2: [SEQ ID NO. 197 or 41] or an amino acid sequence having at least 80% identity therewith, for example at least 85%, 90%, 95%, or 99% identity, preferably having at least 71% identity; and/or f) CDR-L3: [SEQ ID NO. 198 or 46] or an amino acid sequence having at least 55% identity therewith, for example at least 60%, 65%, 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably having at least 55% identity.
11. The antibody or antigen-binding fragment of claim 1, wherein the VH comprises the CDRs of SEQ ID NO. 21 and the VL comprises the CDRs of SEQ ID NO. 22.
12. The antibody or antigen-binding fragment of any of the preceding claims, wherein the VH comprises the following CDRs: a) CDR-H1: [SEQ ID NO. 27] or an amino acid sequence having at least 60% identity therewith, for example at least 60%, 65%, 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably with at least 60% identity; and/or b) CDR-H2: [SEQ ID NO. 29] or an amino acid sequence having at least 80% identity therewith, for example at least 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably with at least 82% identity; and/or c) CDR-H3: [SEQ ID NO. 35] or an amino acid sequence having at least 60% identity therewith, for example at least 65%, 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably with at least 69% identity.
13. The antibody or antigen-binding fragment of any of the preceding claims, wherein the VH comprises the CDRs of SEQ ID NOs 27, 29, and 35.
14. The antibody or antigen-binding fragment of any of the preceding claims comprising a VH having the amino acid sequence of SEQ ID NO. 21, or a variant having at least 80% sequence identity therewith, for example at least 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably having around 91% identity.
15. The antibody or antigen-binding fragment of any of the preceding claims, wherein the VL comprises the following CDRs: a) CDR-L1 : [SEQ ID NO. 39] or an amino acid sequence having at least 80% identity therewith, for example at least 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably having at least 92% identity; and/or b) CDR-L2: [SEQ ID NO. 44] or an amino acid sequence having at least 55% identity therewith, for example at least 60%, 65%, 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably having at least 57% identity; and/or c) CDR-L3: [SEQ ID NO. 47] or an amino acid sequence having at least 55% identity therewith, for example at least 60%, 65%, 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably having at least 55% identity.
16. The antibody or antigen-binding fragment of any of the preceding claims, wherein the VL comprises the CDRs of SEQ ID NOs 39, 44, and 47.
17. The antibody or antigen-binding fragment of any of the preceding claims comprising a VL having the amino acid sequence of SEQ ID NO. 22, or a variant having at least 80% sequence identity therewith, for example at least 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably having around 91% identity.
18. The antibody or antigen-binding fragment of any of the preceding claims comprising the following CDRs: a) CDR-H1: [SEQ ID NO. 27] or an amino acid sequence having at least 60% identity therewith, for example at least 60%, 65%, 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably with at least 60% identity; and/or b) CDR-H2: [SEQ ID NO. 29] or an amino acid sequence having at least 80% identity therewith, for example at least 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably with at least 82% identity; and/or
c) CDR-H3: [SEQ ID NO. 35] or an amino acid sequence having at least 60% identity therewith, for example at least 65%, 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably with at least 69% identity; and/or d) CDR-L1 : [SEQ ID NO. 39] or an amino acid sequence having at least 80% identity therewith, for example at least 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably having at least 92% identity; and/or e) CDR-L2: [SEQ ID NO. 44] or an amino acid sequence having at least 55% identity therewith, for example at least 60%, 65%, 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably having at least 57% identity; and/or f) CDR-L3: [SEQ ID NO. 47] or an amino acid sequence having at least 55% identity therewith, for example at least 60%, 65%, 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity, preferably having at least 55% identity.
19. The antibody or antigen-binding fragment thereof according to claim 1 comprising any one of the following combinations of CDRs:
(i) SEQ ID NOs. 23, 28, 32, 37, 197, and 198 (clone 129D5BTY);
(ii) SEQ ID NOs. 23, 28, 32, 37, 41, and 46 (clone 129D5B);
(Hi) SEQ ID NOs. 23, 115, 30, 36, 40, and 45 (clone 3A2);
(iv) SEQ ID NOs. 23, 115, 30, 37, 41, and 45 (clone 20F6);
(v) SEQ ID NOs. 23, 115, 31, 37, 41, and 45 (clone 49G5);
(vi) SEQ ID NOs. 23, 115, 30, 37, 41, and 46 (clone 53D11);
(vii) SEQ ID NOs. 23, 115, 31, 37, 41, and 46 (clone 77F3);
(viii) SEQ ID NOs. 23, 29, 33, 38, 42, and 47 (clone 26B5);
(ix) SEQ ID NOs. 23, 29, 33, 38, 43, and 48 (clone 83B11D);
(x) SEQ ID NOs. 23, 29, 34, 38, 43, and 48 (clone 9402);
(xi) SEQ ID NOs. 23, 29, 35, 39, 43, and 47 (clone 115D1D); and
(xii) SEQ ID NOs. 27, 29, 35, 39, 44, and 47 (clone 131D5s).
20. The antibody or antigen-binding fragment thereof according to any of the preceding claims wherein the antigen-binding fragment is selected from: scFv, Fv, Fab, F(ab)2, Fab- SH, dsFv, sdAb, di-scFvs bi-scFv, Fcabs, diabodies, scFv-Fc/minibody, triabody, tetrabody, tandAb, half antibody (Unibody) and domain antibodies.
21. The antibody or antigen-binding fragment thereof according to any of the preceding claims wherein the antigen-binding fragment binds cMET with an affinity KD of less than InM.
22. The antibody or antigen-binding fragment thereof according to any of the preceding claims further comprising a payload, optionally wherein the payload is a toxin, further optionally wherein the payload is coupled to the antibody or antigen-binding fragment thereof via a linker.
23. The antibody or antigen-binding fragment thereof of claim 22, wherein the payload is coupled to the antibody or antigen-binding fragment thereof with a coupling ratio (payload:antibody or antigen-binding fragment) of at least 2: 1, at least 3: 1, at least 4: 1, at least 5: 1, at least 6: 1, at least 7: 1, at least 8: 1, at least 9: 1, or at least 10: 1.
24. The antibody or antigen-binding fragment thereof of claim 23, wherein the payload is coupled to the antibody or antigen-binding fragment thereof with a coupling ratio (payload:antibody or antigen-binding fragment) of at least 6: 1.
25. The antibody or antigen-binding fragment thereof of claims 22-24, wherein the payload is directly conjugated to the antibody or antigen-binding fragment thereof at an amino acid, optionally wherein the direct coupling is via a lysine or cysteine residue on the surface of the antibody or antigen-binding fragment thereof, preferably wherein the coupling is via a lysine residue.
26. The antibody or antigen-binding fragment thereof of claims 22-24, wherein the payload is indirectly conjugated to the antibody or antigen-binding fragment thereof at an amino acid, optionally wherein the indirect coupling is via a thiol or maleimide.
27. The antibody or antigen-binding fragment thereof of claims 22-26, wherein the payload is a microtubule inhibitor (MTI), a DNA-damaging agent, a topoisomerase inhibitor, a steroid, a tyrosine kinase inhibitor, an immuno-modulatory drug.
28. The antibody or antigen-binding fragment thereof of claim 27 , wherein the MTI is an auristatin or a derivative thereof.
29. The antibody or antigen-binding fragment thereof of claims 27-28, wherein the DNA-damaging agent is a nicking agent, an alkylator, or an intercalating agent.
30. The antibody or antigen-binding fragment thereof of claims 27-29, wherein the topoisomerase inhibitor is a camptothecin or a derivative thereof, optionally wherein the topoisomerase inhibitor is exatecan.
31. The antibody or antigen-binding fragment thereof of claims 27-30, wherein the steroid is dexamethasone.
32. The antibody or antigen-binding fragment thereof of claims 27-31, wherein the tyrosine kinase inhibitor is nintedanib.
33. The antibody or antigen-binding fragment thereof of claims 27-32, wherein the immuno-modulatory drug is a Toll-Like Receptor 7/8 agonist drug.
34. The antibody or antigen-binding fragment thereof of claims 22-28, wherein the antigen-binding fragment is a single chain Fv (scFv) and the payload is MMAE, and wherein the payload is coupled to the scFv with a coupling ratio (payload :scFv) of 6: 1.
35. The antibody or antigen-binding fragment thereof of claims 22-28, wherein the antigen-binding fragment is a single chain Fv (scFv) and the payload is a mono-methyl auristatin E (MMAE) comprising a glucuronide and branched PEG linker, wherein the MMAE is conjugated onto surface lysine residues of the scFv.
36. The antibody or antigen-binding fragment thereof of claims 22-35 wherein the FDC binds cMET with an affinity KD of less than lOnM.
37. A pharmaceutical composition comprising the antibody or antigen-binding fragment of any of Claims 1-36 and a pharmaceutically-acceptable carrier, excipient or diluent.
38. An antibody or antigen-binding fragment or pharmaceutical composition as defined in any of Claims 1 to 37 for use in medicine.
39. An antibody or antigen-binding fragment or pharmaceutical composition as defined in any of Claims 1 to 37 for use in the diagnosis, treatment and/or prevention of a disease wherein the disease is selected from cancer, benign tumours, infectious diseases including bacterial, viral, fungal, trypanosome, nematode and prion infections, cardiovascular disease, autoimmune disease.
40. An antibody or antigen-binding fragment or pharmaceutical composition as defined in any of Claims 1 to 37 for use in the manufacture of a medicament for the treatment and/or prevention of a disease selected from cancer, benign tumours, infectious diseases
including bacterial, viral, fungal, trypanosome, nematode and prion infections, cardiovascular disease, and autoimmune disease.
41. The antibody or antigen-binding fragment for use according to any of Claims 38 to 40 wherein the disease is cancer of the colon, lung, breast, head/neck, prostate, skin, stomach/gastrointestinal, oesophageal, bladder, glioma, renal, ovarian, thyroid and bone and
42. A process of making a compound as defined in any of Claims 1 to 36 comprising the steps of:
(i) providing a small molecule payload;
(ii) providing the antibody or antigen-binding fragment as defined in any of Claims 1 to 21;
(iii) conjugating the payload and the antibody or antigen binding fragment thereof.
43. The process according to Claim 42 further comprising the step of:
(iv) combining the compound with a pharmaceutically-acceptable carrier to form a pharmaceutical composition.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB202315594 | 2023-10-11 | ||
| GB2315594.8 | 2023-10-11 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2025078841A2 true WO2025078841A2 (en) | 2025-04-17 |
| WO2025078841A3 WO2025078841A3 (en) | 2025-05-15 |
Family
ID=93154460
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/GB2024/052627 Pending WO2025078841A2 (en) | 2023-10-11 | 2024-10-11 | Antibodies, conjugates, and uses thereof |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2025078841A2 (en) |
Citations (65)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4260608A (en) | 1978-11-14 | 1981-04-07 | Takeda Chemical Industries, Ltd. | Maytansinoids, pharmaceutical compositions thereof and methods of use thereof |
| US4317821A (en) | 1979-06-08 | 1982-03-02 | Takeda Chemical Industries, Ltd. | Maytansinoids, their use and pharmaceutical compositions thereof |
| US4816567A (en) | 1983-04-08 | 1989-03-28 | Genentech, Inc. | Recombinant immunoglobin preparations |
| US4879278A (en) | 1989-05-16 | 1989-11-07 | Arizona Board Of Regents | Isolation and structural elucidation of the cytostatic linear depsipeptide dolastatin 15 |
| US5410024A (en) | 1993-01-21 | 1995-04-25 | Arizona Board Of Regents Acting On Behalf Of Arizona State University | Human cancer inhibitory pentapeptide amides |
| US5416064A (en) | 1989-10-25 | 1995-05-16 | Immunogen, Inc. | Cytotoxic agents comprising maytansinoids and their therapeutic use |
| US5504191A (en) | 1994-08-01 | 1996-04-02 | Arizona Board Of Regents Acting On Behalf Of Arizona State University | Human cancer inhibitory pentapeptide methyl esters |
| US5530097A (en) | 1994-08-01 | 1996-06-25 | Arizona Board Of Regents Acting On Behalf Of Arizona State University | Human cancer inhibitory peptide amides |
| US5599902A (en) | 1994-11-10 | 1997-02-04 | Arizona Board Of Regents Acting On Behalf Of Arizona State University | Cancer inhibitory peptides |
| WO1997019086A1 (en) | 1995-11-17 | 1997-05-29 | GESELLSCHAFT FüR BIOTECHNOLOGISCHE FORSCHUNG MBH (GBF) | Epothilone derivatives, preparation and use |
| US5635483A (en) | 1992-12-03 | 1997-06-03 | Arizona Board Of Regents Acting On Behalf Of Arizona State University | Tumor inhibiting tetrapeptide bearing modified phenethyl amides |
| US5663149A (en) | 1994-12-13 | 1997-09-02 | Arizona Board Of Regents Acting On Behalf Of Arizona State University | Human cancer inhibitory pentapeptide heterocyclic and halophenyl amides |
| WO1999007692A2 (en) | 1997-08-09 | 1999-02-18 | Schering Aktiengesellschaft | New epothilone derivatives, method for producing same and their pharmaceutical use |
| WO1999027890A2 (en) | 1997-12-04 | 1999-06-10 | Bristol-Myers Squibb Company | A process for the preparation of ring-opened epothilone intermediates which are useful for the preparation of epothilone analogs |
| US6323315B1 (en) | 1999-09-10 | 2001-11-27 | Basf Aktiengesellschaft | Dolastatin peptides |
| US20020103136A1 (en) | 1998-03-05 | 2002-08-01 | Dong-Mei Feng | Conjugates useful in the treatment of prostate cancer |
| WO2002088172A2 (en) | 2001-04-30 | 2002-11-07 | Seattle Genetics, Inc. | Pentapeptide compounds and uses related thereto |
| US6680311B1 (en) | 1996-08-30 | 2004-01-20 | Eli Lilly And Company | Cryptophycin compounds |
| US6716821B2 (en) | 2001-12-21 | 2004-04-06 | Immunogen Inc. | Cytotoxic agents bearing a reactive polyethylene glycol moiety, cytotoxic conjugates comprising polyethylene glycol linking groups, and methods of making and using the same |
| WO2005081711A2 (en) | 2003-11-06 | 2005-09-09 | Seattle Genetics, Inc. | Monomethylvaline compounds capable of conjugation to ligands |
| US6989450B2 (en) | 2000-10-13 | 2006-01-24 | The University Of Mississippi | Synthesis of epothilones and related analogs |
| US7276497B2 (en) | 2003-05-20 | 2007-10-02 | Immunogen Inc. | Cytotoxic agents comprising new maytansinoids |
| WO2008092796A1 (en) | 2007-01-29 | 2008-08-07 | Nerviano Medical Sciences S.R.L. | Antitumor combination comprising a morpholinyl anthracycline and an antibody |
| US7754681B2 (en) | 2004-02-23 | 2010-07-13 | Seattle Genetics, Inc. | Heterocyclic self-immolative linkers and conjugates |
| US20100305149A1 (en) | 2009-05-28 | 2010-12-02 | Mersana Therapeutics, Inc. | Polyal Drug Conjugates Comprising Variable Rate-Releasing Linkers |
| WO2013079973A1 (en) | 2011-12-02 | 2013-06-06 | Di Cara Danielle Marie | Antibodies against hgf - receptor and uses |
| US20140227299A1 (en) | 2008-02-01 | 2014-08-14 | Genentech, Inc. | Nemorubicin metabolite and analog reagents, antibody-drug conjugates and methods |
| WO2015016827A1 (en) | 2013-07-30 | 2015-02-05 | Benemilk Oy | Dietary compositions for ruminants and containers for storing and dispensing same |
| WO2015157594A1 (en) | 2014-04-11 | 2015-10-15 | Medimmune Llc | Tubulysin derivatives |
| WO2016046574A1 (en) | 2014-09-25 | 2016-03-31 | Antikor Biopharma Limited | Biological materials and uses thereof |
| WO2016123582A1 (en) | 2015-01-30 | 2016-08-04 | Sutro Biopharma, Inc. | Hemiasterlin derivatives for conjugation and therapy |
| US20160340386A1 (en) | 2014-01-28 | 2016-11-24 | Tube Pharmaceuticals Gmbh | Cytotoxic tubulysin compounds for conjugation |
| EP3101032A1 (en) | 2014-01-31 | 2016-12-07 | Daiichi Sankyo Company, Limited | Anti-her2 antibody-drug conjugate |
| WO2017062271A2 (en) | 2015-10-06 | 2017-04-13 | Merck Sharp & Dohme Corp. | Antibody drug conjugate for anti-inflammatory applications |
| WO2017066606A1 (en) | 2015-10-16 | 2017-04-20 | William Marsh Rice University | Epothilone analogs, methods of synthesis, methods of treatment, and drug conjugates thereof |
| WO2017134547A1 (en) | 2016-02-01 | 2017-08-10 | Pfizer Inc. | Tubulysin analogs and methods for their preparation |
| WO2017151979A1 (en) | 2016-03-02 | 2017-09-08 | Eisai Inc. | Eribulin-based antibody-drug conjugates and methods of use |
| US20170326247A1 (en) | 2016-05-10 | 2017-11-16 | Bristol-Myers Squibb Company | Antibody-drug conjugates of tubulysin analogs with enhanced stability |
| WO2017210471A1 (en) | 2016-06-02 | 2017-12-07 | Abbvie Inc. | Glucocorticoid receptor agonist and immunoconjugates thereof |
| WO2017214301A1 (en) | 2016-06-08 | 2017-12-14 | Abbvie Inc. | Anti-egfr antibody drug conjugates |
| WO2017214282A1 (en) | 2016-06-08 | 2017-12-14 | Abbvie Inc. | Anti-egfr antibody drug conjugates |
| US20180078656A1 (en) | 2015-03-17 | 2018-03-22 | Exiris S.R.L. | Cryptophycin-based antibody-drug conjugates with novel self-immolative linkers |
| WO2018098035A1 (en) | 2016-11-23 | 2018-05-31 | Eli Lilly And Company | Met antibody drug conjugates |
| US20180296694A1 (en) | 2017-02-28 | 2018-10-18 | Immunogen, Inc. | Maytansinoid derivatives with self-immolative peptide linkers and conjugates thereof |
| WO2018191746A1 (en) | 2017-04-14 | 2018-10-18 | Bolt Biotherapeutics, Inc. | Immunoconjugate synthesis method |
| WO2019136487A2 (en) | 2018-01-08 | 2019-07-11 | Regeneron Pharmaceuticals, Inc. | Steroids and antibody-conjugates thereof |
| WO2019195665A1 (en) | 2018-04-06 | 2019-10-10 | Seattle Genetics, Inc. | Camptothecin peptide conjugates |
| WO2019232449A1 (en) | 2018-06-01 | 2019-12-05 | Eisai R&D Management Co., Ltd. | Splicing modulator antibody-drug conjugates and methods of use |
| WO2019236954A1 (en) | 2018-06-07 | 2019-12-12 | Seattle Genetics, Inc. | Camptothecin conjugates |
| WO2020056192A1 (en) | 2018-09-12 | 2020-03-19 | Silverback Therapeutics, Inc. | Antibody conjugates of toll-like receptor agonists |
| WO2020219287A1 (en) | 2019-04-26 | 2020-10-29 | Immunogen, Inc. | Camptothecin derivatives |
| EP3626273B1 (en) | 2016-05-17 | 2020-12-30 | AbbVie Biotherapeutics Inc. | Anti-cmet antibody drug conjugates and methods for their use |
| US20210188906A1 (en) | 2017-11-29 | 2021-06-24 | William Marsh Rice University | Tubulysin analogues as anticancer agents and payloads for antibody-drug conjugates and methods of treatment therewith |
| EP3636273B1 (en) | 2012-08-21 | 2021-12-08 | California Northstate College Of Pharmacy, LLC | Novel formulations and uses for curcuma extracts |
| WO2021262910A2 (en) | 2020-06-24 | 2021-12-30 | Regeneron Pharmaceuticals, Inc. | Tubulysins and protein-tubulysin conjugates |
| WO2022015656A1 (en) | 2020-07-13 | 2022-01-20 | Regeneron Pharmaceuticals, Inc. | Camptothecin analogs conjugated to a glutamine residue in a protein, and their use |
| WO2022058395A1 (en) | 2020-09-15 | 2022-03-24 | Synaffix B.V. | Antibody-exatecan conjugates |
| WO2022058745A2 (en) | 2020-09-18 | 2022-03-24 | Imperial College Innovations Limited | Novel compounds and their use in therapy |
| WO2022093800A2 (en) | 2020-10-27 | 2022-05-05 | Elucida Oncology, Inc. | Carrier particle-drug conjugates, self-immolative linkers, and uses thereof |
| WO2022150637A1 (en) | 2021-01-07 | 2022-07-14 | Immunext, Inc. | NOVEL STEROID PAYLOADS, STEROID LINKERS, ADCs CONTAINING AND USE THEREOF |
| WO2022155347A1 (en) | 2021-01-15 | 2022-07-21 | R.P. Scherer Technologies, Llc | Camptothecine antibody-drug conjugates and methods of use thereof |
| WO2022232834A1 (en) | 2021-04-29 | 2022-11-03 | Abbvie Inc. | Anti-c-met antibody drug conjugates |
| WO2022246576A1 (en) | 2021-05-27 | 2022-12-01 | Zymeworks Inc. | Camptothecin analogues, conjugates and methods of use |
| WO2022253035A1 (en) | 2021-06-02 | 2022-12-08 | 四川科伦博泰生物医药股份有限公司 | Antibody drug conjugate, and preparation method therefor and use thereof |
| WO2023061466A1 (en) | 2021-10-14 | 2023-04-20 | 江苏恒瑞医药股份有限公司 | Preparation method for eribulin derivative |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US12528870B2 (en) * | 2020-09-01 | 2026-01-20 | Remegen Co., Ltd. | Anti-c-Met antibody-drug conjugate and applications thereof |
-
2024
- 2024-10-11 WO PCT/GB2024/052627 patent/WO2025078841A2/en active Pending
Patent Citations (65)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4260608A (en) | 1978-11-14 | 1981-04-07 | Takeda Chemical Industries, Ltd. | Maytansinoids, pharmaceutical compositions thereof and methods of use thereof |
| US4317821A (en) | 1979-06-08 | 1982-03-02 | Takeda Chemical Industries, Ltd. | Maytansinoids, their use and pharmaceutical compositions thereof |
| US4816567A (en) | 1983-04-08 | 1989-03-28 | Genentech, Inc. | Recombinant immunoglobin preparations |
| US4879278A (en) | 1989-05-16 | 1989-11-07 | Arizona Board Of Regents | Isolation and structural elucidation of the cytostatic linear depsipeptide dolastatin 15 |
| US5416064A (en) | 1989-10-25 | 1995-05-16 | Immunogen, Inc. | Cytotoxic agents comprising maytansinoids and their therapeutic use |
| US5635483A (en) | 1992-12-03 | 1997-06-03 | Arizona Board Of Regents Acting On Behalf Of Arizona State University | Tumor inhibiting tetrapeptide bearing modified phenethyl amides |
| US5410024A (en) | 1993-01-21 | 1995-04-25 | Arizona Board Of Regents Acting On Behalf Of Arizona State University | Human cancer inhibitory pentapeptide amides |
| US5504191A (en) | 1994-08-01 | 1996-04-02 | Arizona Board Of Regents Acting On Behalf Of Arizona State University | Human cancer inhibitory pentapeptide methyl esters |
| US5530097A (en) | 1994-08-01 | 1996-06-25 | Arizona Board Of Regents Acting On Behalf Of Arizona State University | Human cancer inhibitory peptide amides |
| US5599902A (en) | 1994-11-10 | 1997-02-04 | Arizona Board Of Regents Acting On Behalf Of Arizona State University | Cancer inhibitory peptides |
| US5663149A (en) | 1994-12-13 | 1997-09-02 | Arizona Board Of Regents Acting On Behalf Of Arizona State University | Human cancer inhibitory pentapeptide heterocyclic and halophenyl amides |
| WO1997019086A1 (en) | 1995-11-17 | 1997-05-29 | GESELLSCHAFT FüR BIOTECHNOLOGISCHE FORSCHUNG MBH (GBF) | Epothilone derivatives, preparation and use |
| US6680311B1 (en) | 1996-08-30 | 2004-01-20 | Eli Lilly And Company | Cryptophycin compounds |
| WO1999007692A2 (en) | 1997-08-09 | 1999-02-18 | Schering Aktiengesellschaft | New epothilone derivatives, method for producing same and their pharmaceutical use |
| WO1999027890A2 (en) | 1997-12-04 | 1999-06-10 | Bristol-Myers Squibb Company | A process for the preparation of ring-opened epothilone intermediates which are useful for the preparation of epothilone analogs |
| US20020103136A1 (en) | 1998-03-05 | 2002-08-01 | Dong-Mei Feng | Conjugates useful in the treatment of prostate cancer |
| US6323315B1 (en) | 1999-09-10 | 2001-11-27 | Basf Aktiengesellschaft | Dolastatin peptides |
| US6989450B2 (en) | 2000-10-13 | 2006-01-24 | The University Of Mississippi | Synthesis of epothilones and related analogs |
| WO2002088172A2 (en) | 2001-04-30 | 2002-11-07 | Seattle Genetics, Inc. | Pentapeptide compounds and uses related thereto |
| US6716821B2 (en) | 2001-12-21 | 2004-04-06 | Immunogen Inc. | Cytotoxic agents bearing a reactive polyethylene glycol moiety, cytotoxic conjugates comprising polyethylene glycol linking groups, and methods of making and using the same |
| US7276497B2 (en) | 2003-05-20 | 2007-10-02 | Immunogen Inc. | Cytotoxic agents comprising new maytansinoids |
| WO2005081711A2 (en) | 2003-11-06 | 2005-09-09 | Seattle Genetics, Inc. | Monomethylvaline compounds capable of conjugation to ligands |
| US7754681B2 (en) | 2004-02-23 | 2010-07-13 | Seattle Genetics, Inc. | Heterocyclic self-immolative linkers and conjugates |
| WO2008092796A1 (en) | 2007-01-29 | 2008-08-07 | Nerviano Medical Sciences S.R.L. | Antitumor combination comprising a morpholinyl anthracycline and an antibody |
| US20140227299A1 (en) | 2008-02-01 | 2014-08-14 | Genentech, Inc. | Nemorubicin metabolite and analog reagents, antibody-drug conjugates and methods |
| US20100305149A1 (en) | 2009-05-28 | 2010-12-02 | Mersana Therapeutics, Inc. | Polyal Drug Conjugates Comprising Variable Rate-Releasing Linkers |
| WO2013079973A1 (en) | 2011-12-02 | 2013-06-06 | Di Cara Danielle Marie | Antibodies against hgf - receptor and uses |
| EP3636273B1 (en) | 2012-08-21 | 2021-12-08 | California Northstate College Of Pharmacy, LLC | Novel formulations and uses for curcuma extracts |
| WO2015016827A1 (en) | 2013-07-30 | 2015-02-05 | Benemilk Oy | Dietary compositions for ruminants and containers for storing and dispensing same |
| US20160340386A1 (en) | 2014-01-28 | 2016-11-24 | Tube Pharmaceuticals Gmbh | Cytotoxic tubulysin compounds for conjugation |
| EP3101032A1 (en) | 2014-01-31 | 2016-12-07 | Daiichi Sankyo Company, Limited | Anti-her2 antibody-drug conjugate |
| WO2015157594A1 (en) | 2014-04-11 | 2015-10-15 | Medimmune Llc | Tubulysin derivatives |
| WO2016046574A1 (en) | 2014-09-25 | 2016-03-31 | Antikor Biopharma Limited | Biological materials and uses thereof |
| WO2016123582A1 (en) | 2015-01-30 | 2016-08-04 | Sutro Biopharma, Inc. | Hemiasterlin derivatives for conjugation and therapy |
| US20180078656A1 (en) | 2015-03-17 | 2018-03-22 | Exiris S.R.L. | Cryptophycin-based antibody-drug conjugates with novel self-immolative linkers |
| WO2017062271A2 (en) | 2015-10-06 | 2017-04-13 | Merck Sharp & Dohme Corp. | Antibody drug conjugate for anti-inflammatory applications |
| WO2017066606A1 (en) | 2015-10-16 | 2017-04-20 | William Marsh Rice University | Epothilone analogs, methods of synthesis, methods of treatment, and drug conjugates thereof |
| WO2017134547A1 (en) | 2016-02-01 | 2017-08-10 | Pfizer Inc. | Tubulysin analogs and methods for their preparation |
| WO2017151979A1 (en) | 2016-03-02 | 2017-09-08 | Eisai Inc. | Eribulin-based antibody-drug conjugates and methods of use |
| US20170326247A1 (en) | 2016-05-10 | 2017-11-16 | Bristol-Myers Squibb Company | Antibody-drug conjugates of tubulysin analogs with enhanced stability |
| EP3626273B1 (en) | 2016-05-17 | 2020-12-30 | AbbVie Biotherapeutics Inc. | Anti-cmet antibody drug conjugates and methods for their use |
| WO2017210471A1 (en) | 2016-06-02 | 2017-12-07 | Abbvie Inc. | Glucocorticoid receptor agonist and immunoconjugates thereof |
| WO2017214301A1 (en) | 2016-06-08 | 2017-12-14 | Abbvie Inc. | Anti-egfr antibody drug conjugates |
| WO2017214282A1 (en) | 2016-06-08 | 2017-12-14 | Abbvie Inc. | Anti-egfr antibody drug conjugates |
| WO2018098035A1 (en) | 2016-11-23 | 2018-05-31 | Eli Lilly And Company | Met antibody drug conjugates |
| US20180296694A1 (en) | 2017-02-28 | 2018-10-18 | Immunogen, Inc. | Maytansinoid derivatives with self-immolative peptide linkers and conjugates thereof |
| WO2018191746A1 (en) | 2017-04-14 | 2018-10-18 | Bolt Biotherapeutics, Inc. | Immunoconjugate synthesis method |
| US20210188906A1 (en) | 2017-11-29 | 2021-06-24 | William Marsh Rice University | Tubulysin analogues as anticancer agents and payloads for antibody-drug conjugates and methods of treatment therewith |
| WO2019136487A2 (en) | 2018-01-08 | 2019-07-11 | Regeneron Pharmaceuticals, Inc. | Steroids and antibody-conjugates thereof |
| WO2019195665A1 (en) | 2018-04-06 | 2019-10-10 | Seattle Genetics, Inc. | Camptothecin peptide conjugates |
| WO2019232449A1 (en) | 2018-06-01 | 2019-12-05 | Eisai R&D Management Co., Ltd. | Splicing modulator antibody-drug conjugates and methods of use |
| WO2019236954A1 (en) | 2018-06-07 | 2019-12-12 | Seattle Genetics, Inc. | Camptothecin conjugates |
| WO2020056192A1 (en) | 2018-09-12 | 2020-03-19 | Silverback Therapeutics, Inc. | Antibody conjugates of toll-like receptor agonists |
| WO2020219287A1 (en) | 2019-04-26 | 2020-10-29 | Immunogen, Inc. | Camptothecin derivatives |
| WO2021262910A2 (en) | 2020-06-24 | 2021-12-30 | Regeneron Pharmaceuticals, Inc. | Tubulysins and protein-tubulysin conjugates |
| WO2022015656A1 (en) | 2020-07-13 | 2022-01-20 | Regeneron Pharmaceuticals, Inc. | Camptothecin analogs conjugated to a glutamine residue in a protein, and their use |
| WO2022058395A1 (en) | 2020-09-15 | 2022-03-24 | Synaffix B.V. | Antibody-exatecan conjugates |
| WO2022058745A2 (en) | 2020-09-18 | 2022-03-24 | Imperial College Innovations Limited | Novel compounds and their use in therapy |
| WO2022093800A2 (en) | 2020-10-27 | 2022-05-05 | Elucida Oncology, Inc. | Carrier particle-drug conjugates, self-immolative linkers, and uses thereof |
| WO2022150637A1 (en) | 2021-01-07 | 2022-07-14 | Immunext, Inc. | NOVEL STEROID PAYLOADS, STEROID LINKERS, ADCs CONTAINING AND USE THEREOF |
| WO2022155347A1 (en) | 2021-01-15 | 2022-07-21 | R.P. Scherer Technologies, Llc | Camptothecine antibody-drug conjugates and methods of use thereof |
| WO2022232834A1 (en) | 2021-04-29 | 2022-11-03 | Abbvie Inc. | Anti-c-met antibody drug conjugates |
| WO2022246576A1 (en) | 2021-05-27 | 2022-12-01 | Zymeworks Inc. | Camptothecin analogues, conjugates and methods of use |
| WO2022253035A1 (en) | 2021-06-02 | 2022-12-08 | 四川科伦博泰生物医药股份有限公司 | Antibody drug conjugate, and preparation method therefor and use thereof |
| WO2023061466A1 (en) | 2021-10-14 | 2023-04-20 | 江苏恒瑞医药股份有限公司 | Preparation method for eribulin derivative |
Non-Patent Citations (215)
| Title |
|---|
| "Antibody Engineering", 2004, pages: 327 - 343 |
| "Methods in Molecular Biology", vol. 1045, 2013, HUMANA PRESS, article "Antibody Engineering: Methods and Protocols" |
| ADHIKARI ASHEN BRADER C, ANTIBODY THERAPEUTICS, vol. 4, no. 1, 2021, pages 1 - 15 |
| AGREN JKMBILLING JF ET AL., SYNTHESIS, 2006, pages 3141 - 3145 |
| ANSELMI MBORBELY A ET AL., CHEM. EUR. J., vol. 27, no. 3, 2021, pages 1015 - 1022 |
| BAKER JHEKYLE AHREINSBERG SA ET AL., CLIN EXP METASTASIS., vol. 35, 2018, pages 691 - 705 |
| BARGH JDISIDRO-LOBET A ET AL., CHEM. SOC. REV., vol. 48, 2019, pages 4361 - 4374 |
| BARGH JDWALSH SJ ET AL., CHEM. SCI., vol. 11, no. 19, 2020, pages 2375 - 2380 |
| BARTELINK IHJONES EF ET AL., CLIN PHARMACOL THER., vol. 106, 2019, pages 148 - 163 |
| BEHRENS CRLIU B, MABS, vol. 6, 2014, pages 46 - 53 |
| BOERNER P ET AL., J. IMMUNOL., vol. 147, 1991, pages 86 - 95 |
| BRADLEY CASALTO-TELLEZ M ET AL., NAT REV CLIN ONCOL., vol. 14, 2017, pages 562 - 576 |
| BROGGINI M: "Anthracycline Chemistry II", TOPICS IN CURRENT CHEMISTRY, vol. 283, 2007, pages 191 - 206 |
| BROWN JMGIACCIA AJ, CANCER RESEARCH, vol. 58, 1998, pages 1408 - 1416 |
| BUGATTI KANDREUCCI E ET AL., ACS OMEGA, vol. 7, 2022, pages 17658 - 17669 |
| BURKE PJHAMILTON JZ ET AL., MOL. CAN. THER., vol. 16, no. 1, 2017, pages 116 - 123 |
| CADWELL RCJOYCE GF, PCR METHODS, APPL, vol. 2, 1992, pages 28 - 33 |
| CAMIDGE DRBARLESI F ET AL., J CLIN ONCOL., vol. 41, no. 5, 10 February 2023 (2023-02-10), pages 1105 - 1115 |
| CANESTARO WJFORRESTER SH ET AL., MOL CANCER THER., vol. 12, 2013, pages 2685 - 96 |
| CARTER PJ, NAT REV IMMUNOL., vol. 6, 2006, pages 343 - 57 |
| CARTER, P., BIOCHEM J, vol. 237, 1986, pages 1 - 7 |
| CASTLE JCUDUMAN M, FRONT IMMUNOL, vol. 10, 2019, pages 1856 |
| CHALKER JMBERNARDES GJL ET AL., CHEM. ASIAN J., vol. 4, no. 5, 2009, pages 630 - 640 |
| CHAN EALKHASAWNEH A ET AL., JOURNAL OF GASTROINTESTINAL ONCOLOGY., vol. 7, no. 6, 2016, pages 838 - 847 |
| CHAN KFWONG ILK ET AL., J. MED. CHEM., vol. 55, no. 5, 2012, pages 1999 - 2014 |
| CHEN HARNST KEMILLER DDLI W, MOLECULES, vol. 22, 2017, pages 1281 |
| CHEN WWU S ET AL., ONCO TARGETS THER., vol. 14, 2021, pages 4791 - 4804 |
| CHENG XLI J ET AL., MOL. CANCER THER., vol. 17, no. 12, 2018, pages 2665 - 2675 |
| CHILAMARI MKALRA NSHUKLA SRAI V, CHEM. COMM., vol. 54, 2018, pages 7302 - 7305 |
| CHIU MLGOULET DRTEPLYAKOV AGILLILAND GL: "Antibody Structure and Function: The Basis for Engineering Therapeutics", ANTIBODIES, vol. 8, 2019, pages 55 |
| COLE ET AL.: "Monoclonal antibodies and Cancer Therapy", 1985, ALAN R. LISS, pages: 77 |
| COLE SPC ET AL., MOL. CELL. BIOL., vol. 62, 1984, pages 109 - 120 |
| COLOMBO RRICH JR, CANCER CELL, vol. 40, 2022, pages 1255 - 1263 |
| COMER F: "AZD9592: An EGFR-cMET bispecific antibody-drug conjugate (ADC) targeting key oncogenic drivers in non-small-cell lung cancer (NSCLC) and beyond", CANCER RESEARCH, vol. 83, 2023 |
| COMOGLIO PM ET AL., NAT REV CANCER., vol. 18, 2018, pages 341 |
| CONILH LSADILKOVA L ET AL., J. HAEMAOL ONCOL, vol. 16, 2023, pages 3 |
| COTE RJ ET AL., PROC. NATL. ACAD. SCI. USA, vol. 80, 1983, pages 2026 - 2030 |
| CRUZ EKAYSER V, BIOLOGICS: TARGET AND THERAPY, vol. 13, 2019, pages 33 - 51 |
| DE MARCO A, MICROB CELL FACT., vol. 8, 2009, pages 26 |
| DE SILVA DMROY A ET AL., BIOCHEM SOC TRANS., vol. 45, 2017, pages 855 - 870 |
| DEGRAAF MBOVEN E ET AL., CURR. PHARM. DESIGN, vol. 8, no. 15, 2002, pages 1391 - 1403 |
| DEMARIA OCORNEN S ET AL., NATURE, vol. 574, 2019, pages 45 - 56 |
| DENG R ET AL.: "Projecting human pharmacokinetics of therapeutic antibodies from nonclinical data: what have we learned?", MABS., vol. 3, 2011, pages 61 - 6 |
| DENNY WA, EXPT. OPIN. THER. PATENTS, vol. 10, no. 4, 2000, pages 459 - 474 |
| DEONARAIN MP, DRUG DISCOV. TODAY TECHNOL., vol. 30, 2018, pages 47 - 53 |
| DEONARAIN MPYAHIOGLU G, EXPERT OPIN DRUG DISCOV., vol. 16, 2021, pages 613 - 624 |
| DOPPALAPUDI VRHUANG J ET AL., PROC. NATL. ACAD. SCI. U S A, vol. 107, no. 52, 2010, pages 22611 - 6 |
| DRAGOVICH PS, CHEM. SOC. REV., vol. 51, 2022, pages 3886 - 3897 |
| DRAKE JMLEE JKWITTE ON, MOL. CELL BIOL., vol. 34, no. 10, 2014, pages 1722 - 32 |
| DUGAL-TESSIER, J. ET AL.: "Drug Discovery Series no. 81", 2022, RSC, article "Chemical Linkers in Antibody-Drug Conjugates (ADCs", pages: 136 - 172 |
| DUMONTET CREICHERT JM ET AL., NAT REV DRUG DISCOV., vol. 22, 2023, pages 641 - 661 |
| DUWALL JRTHOMAS JDBUKHALID RA ET AL., J. MED. CHEM., vol. 66, no. 15, 2023, pages 10715 - 10733 |
| EDUPUGANTI VVSRTYNDALL JDA ET AL., REC. PAT ANTICANCER DRUG DISC., vol. 16, 2021, pages 479 - 497 |
| ELGEMEIE GH, CURR. PHARM. DES., vol. 9, no. 31, 2003, pages 2627 - 2642 |
| EVANS, N. ET AL., FRONT. PHARMACOL., vol. 13, 2022, pages 764540 |
| FAHY J, CURR. PHARM. DESIGN, vol. 7, no. 13, 2001, pages 1181 - 1197 |
| FERNANDEZ-FERNANDEZ AMANCHANDA R ET AL., APPL. BIOCHEM BIOTECHNOL., vol. 165, 2011, pages 1628 - 51 |
| FORTUNE JMOSHEROFF N, PROC. NUCLEIC ACID RES. MOL. BIOL., vol. 64, 2000, pages 221 - 253 |
| FUJIMORI KCOVELL DGFLETCHER JE, WEINSTEIN. J NUCL MED., vol. 31, 1990, pages 1191 - 1198 |
| FUJITA HMIYADERA K ET AL., MOL. CANCER THER., vol. 12, 2013, pages 2685 - 96 |
| GARCIA-SAEZ ISKOUFIAS DA, BIOCHEM. PHARMACOL., vol. 184, 2021, pages 114364 |
| GAYYED MFABD EL-MAQSOUD NM ET AL., J GASTROINTEST ONCOL., vol. 6, 2015, pages 618 - 27 |
| GERONI CRIPAMONTI M ET AL., CANCER RES., vol. 61, 2001, pages 1983 - 1990 |
| GOODIN SKANE MPRUBIN EH, J. CLIN. ONCOL., vol. 15, no. 22, 2004, pages 15 - 25 |
| GROOTHUIS PGJACOBS DCH ET AL., MOL. CANCER THER., vol. 22, 2023, pages 765 - 777 |
| GYMNOPOULOS MBETANCOURT O ET AL., J. MOL ONCOL., vol. 14, 2020, pages 54 - 68 |
| HACKENBERGER CPSCHWARZER D, ANGEW CHEM INT ED ENGL., vol. 47, no. 52, 2008, pages 10030 - 74 |
| HAMILTON JZPIRES TA ET AL., CHEMMEDCHEM, vol. 16, no. 7, 2021, pages 1077 - 1081 |
| HANKA LJDIETZ A ET AL., J. ANTIBIOT., vol. 31, 1978, pages 1211 - 1217 |
| HAQUE MFORTE NBAKER JR, CHEM. COMM., vol. 57, 2021, pages 10689 - 10702 |
| HARTLEY JA, EXPERT OPIN. BIOL. THER., vol. 21, no. 7, 2021, pages 931 - 943 |
| HIRATA, S, YUEMURA D, PURE APPL. CHEM., vol. 58, 1986, pages 701 - 710 |
| HITOSHI BNAGANO M ET AL., BIOCONJUGATE CHEMISTRY, vol. 24, 2013, pages 520 - 532 |
| HOBSON A: "Springer Briefs in Molecular Science", 2023, SPRINGER, article "The Medicinal Chemistry of Glucocorticoid Receptor Modulators", pages: 59 - 97 |
| HOBSON ADMCPHERSON MJ ET AL., J. MED. CHEM., vol. 65, 2022, pages 15893 - 15934 |
| HOOGENBOOMWINTER, J. MOL. BIOL., vol. 222, 1991, pages 581 |
| HRUBA LDAS VHAJDUCH MDZUBAK P, BIOCHEM. PHARMACOL., 2023, pages 115741 |
| HUGHES VSSIEMANN DW, TRENDS IN CANCER, vol. 4, 2018, pages 94 - 97 |
| HURRELL, J.G.R: "Monoclonal Hybridoma Antibodies: Techniques and Applications", 1982, CRC PRESS |
| IYER LRATAIN MJ, CANCER CHEMOTHER. PHARMACOL., vol. 42, 1998, pages S31 - S43 |
| JEFFREY SCANDREYKA JB ET AL., BIOCONJ. CHEM, vol. 17, no. 3, 2006, pages 831 - 840 |
| JEFFREY SCBRABENDER JD ET AL., ACS MED. CHEM. LETT., vol. 1, no. 6, 2010, pages 277 - 280 |
| JEFFREY SCNGUYEN MT ET AL., BIOORG. MED. CHEM. LETT., vol. 17, no. 8, 2007, pages 2278 - 2280 |
| JIA LYANG XTIAN W ET AL., MEDICAL SCIENCE MONITOR., vol. 24, 2018, pages 8239 - 8249 |
| JONES PT ET AL., NATURE, vol. 321, 1986, pages 522 - 525 |
| KALE JOSTERLUND EJANDREWS DW, CELL DEATH DIFFER., vol. 25, 2018, pages 65 - 80 |
| KELLEY LASTERNBERG MJ, NAT PROTOC., vol. 4, 2009, pages 363 - 71 |
| KERBRAT PDIERAS V ET AL., EUR. J. CANCER, vol. 39, 2003, pages 317 - 320 |
| KERN JCCANCILLA M ET AL., J. AM. CHEM. SOC., vol. 138, 2016, pages 1430 - 1445 |
| KING TPARDO A, LANCET, vol. 378, 2011, pages 1949 - 1961 |
| KNUDSEN BSEDLUND M, ADV CANCER RES., vol. 91, 2004, pages 31 - 67 |
| KOHLER D ET AL., NATURE, vol. 256, 1975, pages 4950497 |
| KONDENGADAN SMBANSAL S ET AL., ACTA PHARMACEUTICA SINICA B, vol. 13, no. 5, 2023, pages 1990 - 2016 |
| KOVALEV ISZYRYANOV GV ET AL., MOLECULES, vol. 27, no. 19, 2022, pages 6229 |
| KOYAMA KGOTO H ET AL., AM J RESPIR CELL MOL BIOL., vol. 60, no. 4, April 2019 (2019-04-01), pages 478 - 487 |
| KOZBOR D ET AL., J. IMMUNOL. METHODS, vol. 81, 1985, pages 31 - 42 |
| KUMMAR SCHEN A ET AL., CANCER CHEMOTHER. PHARMACOL., vol. 78, 2016, pages 73 - 81 |
| KUPCHAN SMSNEDEN AT ET AL., J. MED. CHEM., vol. 21, no. 1, 1978, pages 31 - 37 |
| KURTZBERG LSROTH S ET AL., CLIN. CAN. RES., vol. 17, 2011, pages 2777 - 2787 |
| LAGUZZA BCNICHOLS CL ET AL., J. MED. CHEM., vol. 32, 1989, pages 548 - 555 |
| LEE SJLEE J ET AL., CLIN COLORECTAL CANCER., vol. 17, 2018, pages 165 - 169 |
| LEI ZNTIAN QTENG QXWURPEL JNDZENG LPAN YCHEN ZS, MEDCOMM, vol. 4, 2023, pages e265 |
| LI FEMMERTON KKJONAS M ET AL., CANCER RES., vol. 76, no. 9, 2016, pages 2710 - 2719 |
| LI WVEALE KHQIU Q ET AL., ACS MED. CHEM. LETT., vol. 10, no. 10, 2019, pages 1386 - 1392 |
| LI YGONG L ET AL., CHEMM. COMM., vol. 58, 2022, pages 2568 - 2571 |
| LIANG ZLEI F ET AL., J. MED. CEM., vol. 228, 2022, pages 113960 |
| LIAO-CHAN SDAINE-MATSUOKA B ET AL., PLOS ONE, vol. 10, no. 4, 2015, pages e0124708 |
| LINDELL TJWEINBERG F ET AL., SCIENCE, vol. 170, 1970, pages 447 - 9 |
| LORDICK FALLUM W ET AL., CANCER TREAT REV., vol. 40, 2014, pages 692 - 700 |
| LU JTENG L, INT. J. OF MOLECULAR MEDICINE., vol. 32, 2013, pages 1247 - 1254 |
| LUAN TYU Y, INT J CLIN EXP MED., vol. 7, 2014, pages 5583 - 7 |
| LYON RPBOVEE TD ET AL., NAT. BIO., vol. 33, no. 7, 2015, pages 733 - 735 |
| LYSKI RDBOU LB ET AL., MOL. CAN. THER., vol. 20, no. 2, 2021, pages 329 - 339 |
| M. BROGGINI, TOP. CURR. CHEM., vol. 208, no. 283, pages 191 |
| MACHER-GOEPPINGER SKEITH MENDRIS V ET AL., ONCOTARGET, vol. 8, no. 43, 2017, pages 75478 - 75487 |
| MAHALINGAIAH PKCIURLIONIS R ET AL., PHARMACOL THER., vol. 200, 2019, pages 110 - 125 |
| MAIN ET AL., J PHARMACOL EXP THER., vol. 319, no. 3, 2006, pages 1395 - 404 |
| MANTAJ JJACKSON PJMRAHMAN KMTHURSTON DE, ANGEW. CHEM. INT. ED. ENGL., vol. 56, no. 2, 2017, pages 462 - 488 |
| MCGOWAN DC, CURR. TOP. MED. CHEM., 2019, pages 2228 - 2238 |
| MELO GAGLIATO DFONTES JARDIM D ET AL., CLINICAL BREAST CANCER, vol. 14, no. 6, 2014, pages 468 - 474 |
| MESTERI ISCHOPPMANN SF ET AL., EUROPEAN JOURNAL OF CANCER., vol. 50, no. 7, 2014, pages 1354 - 1360 |
| MEYER YRICHARD JA ET AL., ORG. BIOMOL. CHEM., vol. 8, 2010, pages 1777 - 1780 |
| MILLER PKREUZALER M ET AL., SCI TRANSL MED, vol. 7, no. 315, 2015 |
| MIN BJIN J ET AL., ACS OMEGA., vol. 5, 2020, pages 25798 - 25809 |
| MINCHINTON AITANNOCK IF, NAT REV CANCER., vol. 6, 2006, pages 583 - 92 |
| MOCQUOT P ET AL.: "The pharmacology of blinatumomab: state of the art on pharmacodynamics, pharmacokinetics, adverse drug reactions and evaluation in clinical trials", J CLIN PHARM THER., vol. 47, 2022, pages 1337 - 1351 |
| MOOBERY SLBUSQUETS LTIEN G, INT. J. CANCER, vol. 73, 1997, pages 440 - 448 |
| MUKAE YMIYATA YNAKAMURA Y ET AL., ONCOLOGY LETTERS, vol. 20, no. 1, 2020, pages 135 - 144 |
| MURRAY BCPETERSON MTFECIK RA, NAT. PROD. REPS., vol. 32, 2015, pages 654 - 662 |
| NAIR ABJACOB S.: "A simple practice guide for dose conversion between animals and human", J BASIC CLIN PHARM., vol. 7, 2016, pages 27 - 31 |
| NGUYEN TDBORDEAU BMBALTHASAR JP, CANCERS, vol. 15, 2023, pages 713 |
| NICOLAOU KCRIGOL S ET AL., PNAS, vol. 118, no. 25, 2021, pages e2107042118 |
| NICOLAOU KCSMITH ALYUE EW, PNAS, vol. 90, no. 13, 1993, pages 5881 - 5888 |
| NICOLÒ EGIUGLIANO F ET AL., CANCER TREAT REV., vol. 106, 2022, pages 102395 |
| NISHIDA MTERAYABASHI T ET AL., NAT. COMM. BIOLOGY, vol. 5, 2022, pages 982 - 995 |
| NOEL P, TRENDS PHARMACOL. SCI., vol. 40, no. 5, 2019, pages 327 - 341 |
| OCHTROP PHACKENBERGER CP, CURR. OPIN. BIOL. CHEM., vol. 58, 2020, pages 28 - 36 |
| OHARA MSAITO K ET AL., CANCERS, vol. 13, 2021, pages 1104 |
| OJIMA IGENG X ET AL., J. MED. CHEM., vol. 45, 2002, pages 5620 - 5623 |
| ORLANDI R ET AL., PROC. NATL. ACAD. SCI. U.S.A., vol. 86, 1989, pages 3833 - 3837 |
| OROUDJEV ELOPUS M ET AL., MOL. CAN. THER., vol. 9, no. 10, 2010, pages 2700 - 2713 |
| PARK H ET AL.: "Reference values of clinical pathology parameters in cynomolgus monkeys (Macaca fascicularis) used in preclinical studies", LAB ANIM RES, vol. 32, 2016, pages 79 - 86 |
| PATTERSON JTASANO S ET AL., BIOCONJ. CHEM, vol. 25, 2014, pages 1402 - 1407 |
| PENG ZZHU YWANG Q ET AL., PLOS ONE, vol. 9, no. 1, 2014 |
| PETRINI I, ANNALS TRANS MED, vol. 3, 2015, pages 82 |
| PETTIT GRKAMANO Y ET AL., J. AM. CHEM. SOC., vol. 109, no. 22, 1987, pages 6883 - 6885 |
| PILLOW, T.HTERCEL, M: "Drug Discovery Series no. 71", 2019, RSC, article "Cytotoxic Payloads for Antibody-Drug Conjugates", pages: 364 - 379 |
| POMMIER Y, ACS CHEM. BIOL., vol. 8, 2013, pages 82 - 95 |
| PRESTA LG, CURR. OP. STRUCT. BIOL., vol. 2, 1992, pages 593 - 596 |
| PUCCINI A ET AL., DRUG SAFETY, vol. 42, 2019, pages 211 - 233 |
| QUINTANA JMARBOLEDA D ET AL., BIOCONJ. CHEM., vol. 33, no. 8, 2022, pages 1474 - 1484 |
| RAGHAV K ET AL., CANCER TREAT REV, vol. 66, 2018, pages 95 - 103 |
| RATH PLAL B ET AL., TRANSL ONCOL., vol. 6, 2013, pages 104 - 11 |
| REMILLARD, SREBHUN LI ET AL., SCIENCE, vol. 189, no. 4207, 1975, pages 1002 - 5 |
| RIECHMANN L ET AL., NATURE, vol. 332, 1988, pages 323 - 329 |
| RIVAS S ET AL., INT J MOL SCI., vol. 23, 2022, pages 13898 |
| ROBERT C., NAT COMMUN., vol. 11, no. 1, 2020, pages 3801 |
| ROBINSON DRWU YMLIN SF, ONCOGENE, vol. 19, no. 49, 2000, pages 5548 - 57 |
| SABER HLEIGHTON JK, REGUL TOXICOL PHARMACOL., vol. 71, 2015, pages 444 - 52 |
| SARKER, GSOMMER, SS., BIOTECHNIQUES, vol. 8, no. 4, 1990, pages 404 - 407 |
| SASIE FSTEINMETZ H ET AL., J. ANTIBIOT., vol. 53, 2000, pages 879 - 885 |
| SCHAEFER JVPLUCKTHUN A, PROTEIN ENG DES SEL., vol. 25, 2012, pages 485 - 506 |
| SHIN SHPARK YH ET AL., ADV. SCI., vol. 8, no. 23, 2021, pages 2102414 |
| SMITH CDZHANG X ET AL., CANCER RES., vol. 54, 1994, pages 3779 - 3784 |
| STELLA GMGENTILE A ET AL., J TRANSL MED, vol. 14, 2016, pages 256 |
| STRICKLER JHWEEKES CD ET AL., J CLIN ONCOL., vol. 36, 2018, pages 3298 - 3306 |
| SWEDAN HKKASSAB AE ET AL., BIOORGANIC CHEMISTRY, vol. 136, 2023, pages 106548 |
| SWIFT LPREPHALI A ET AL., CANCER RES., vol. 66, 2006, pages 4863 - 4871 |
| SZPARECKI GILCZUK T ET AL., FOLIA BIOLOGICA, vol. 63, 2017, pages 146 - 154 |
| TACAR, O, J. PHARM. PHARMACOL., vol. 61, no. 8, 2013, pages 933 - 938 |
| TAGAKAGI KDEXHEIMER TS ET AL., MOL. CAN. THER., vol. 6, 2007, pages 3229 - 3238 |
| TALUKDAR AKUNDU B ET AL., EUR. J. MED. CHEM., vol. 236, 2022, pages 114304 |
| TANAKA YAIKAWA K ET AL., J. MED. CHEM., vol. 56, 2013, pages 9625 - 9645 |
| TANCY-OPALINSKI ILEGIGAN T ET AL., EUR. J. MED. CHEM., vol. 74, 2014, pages 302 - 313 |
| TEICHER BAMORRIS J., CURR CANCER DRUG TARGETS, vol. 22, no. 6, 2022, pages 463 - 529 |
| THOMPSON JD ET AL., NUC. ACID RES., vol. 22, 1994, pages 4673 - 4680 |
| THURBER GMSCHMIDT MMWITTRUP KD, ADV DRUG DELIV REV., vol. 60, 2008, pages 1421 - 1434 |
| THUSS-PATIENCE PCSHAH MA ET AL., LANCET ONCOL., vol. 18, 2017, pages 640 - 653 |
| TRIMURTULU GOHTANI IPATTERSON GMLMOORE RECORBETT THVALERIOTE FADEMCHIK L, J. AM. CHEM. SOC., vol. 116, 1994, pages 4729 - 4737 |
| TUMEY L.N.: "Cancer Drug Discovery and Development", 2018, SPRINGER INTERNATIONAL PUBLISHING, article "Innovations for Next-Generation Antibody-Drug Conjugates", pages: 187 |
| VENUGOPAL SSHARMA V ET AL., CHEM. BIOL. DRUG DES., vol. 100, 2022, pages 580 - 598 |
| VERHOEYEN M ET AL., SCIENCE, vol. 239, 1998, pages 1534 - 1536 |
| VERMA VAPILLOW TH ET AL., BIOORG. MED. CHEM. LETT., vol. 25, no. 4, 2015, pages 864 - 868 |
| VIRICEL WFOURNET G ET AL., CHEM. SCI., vol. 14, no. 10, 2019, pages 4048 - 4053 |
| VON ARX CDE PLACIDO P ET AL., CANCER TREAT REV., vol. 113, 2023, pages 102500 |
| VSIANSKY VGUMULEC J ET AL., SCI REP., vol. 8, 2018, pages 10370 |
| WALKER SLANDOWITZ RDING WD, PNAS, vol. 89, no. 10, 1992, pages 4608 - 4612 |
| WALSH SJBARGH JD ET AL., CHEM. SOC. REV., vol. 50, 2021, pages 1305 - 1353 |
| WANG HM ET AL., INT. J. MOLECULAR SCIENCES., vol. 16, no. 2, 2015, pages 3391 - 3404 |
| WANG HXU XWANG JQIAO Y, ANN. MED., vol. 55, no. 1, 2023, pages 1422 - 1430 |
| WANG J ET AL., BMC CANCER, vol. 16, 2016, pages 105 |
| WANG JANDERSON MG ET AL., CLIN CANCER RES., vol. 23, 2017, pages 992 - 1000 |
| WANG M ET AL., ANNALS DIAG. PATHOL, vol. 35, 2018, pages 69 - 78 |
| WANG RELIU T ET AL., J. AM. CHEM. SOC., vol. 137, 2015, pages 3229 - 32 |
| WANI MCNICHOLAS AW ET AL., J. MED. CHEM., vol. 30, 1987, pages 1774 - 1779 |
| WATT PMHICKSON ID, BIOCHEM. J., vol. 303, 1994, pages 681 - 695 |
| WEI YXIANG HZHANG W, FRONT. ONCOL., vol. 13, 2022, pages 970553 |
| WIND SSCHMID U ET AL., CLIN. PHARMACOKINETICS., vol. 58, no. 9, 2019, pages 1131 - 1147 |
| WINTER G ET AL., NATURE, vol. 349, 1991, pages 293 - 299 |
| WORN APLÜCKTHUN A., J MOL BIOL., vol. 305, 2001, pages 989 - 1010 |
| WOZNIAK-KNOPP, G ET AL., PROTEIN ENG. DES. SEL., vol. 23, 2010, pages 289 - 297 |
| WU YLSOO RA ET AL., CANCER TREAT REV., vol. 61, December 2017 (2017-12-01), pages 70 - 81 |
| WU ZWSHA YCHEN Q ET AL., WORLD JOURNAL OF CLINICAL CASES., vol. 9, 2021, pages 3350 - 3355 |
| YAMAMOTO STSUDA HITOSHIMIYAI K, MODERN PATHOLOGY, vol. 24, 2011, pages 1146 - 1155 |
| YANG CZHAO X ET AL., XENOBIOTICA, vol. 49, 2019, pages 1097 - 1105 |
| YANG QDENG Z ET AL., J. AM. CHEM. SOC., vol. 142, 2020, pages 2532 - 2540 |
| YAO H-P ET AL., DRUG DISCOVERY TODAY, vol. 26, 2021, pages 106 - 121 |
| YAO HPTONG XMHUDSON RWANG MH, EXP CLIN CANCER RES., vol. 39, 2020, pages 198 |
| YAO HPZHAO HHUDSON R ET AL., DRUG DISC. TODAY, vol. 26, no. 8, 2021, pages 1857 - 1874 |
| YU SFZHENG B ET AL., CLIN. CANCER RES., vol. 21, no. 14, 2015, pages 3298 - 306 |
| YVER AAGATSUMA TSORIA JC, ANN ONCOL., vol. 31, 2020, pages 430 - 434 |
| ZAFAR SARMAGHAN M ET AL., BIOMED. PHARMACOTHER., vol. 165, 2023, pages 115039 |
| ZANETTI ASTOPPACCIARO A ET AL., JOURNAL OF PATHOLOGY, vol. 186, 1998, pages 287 - 291 |
| ZHANG KMA Y ET AL., MOLECULAR THERAPY: ONCOLYTICS, vol. 18, 2020, pages 304 - 316 |
| ZHU QYU X ET AL., BIOORG. MED. CHEM., vol. 26, no. 16, 2018, pages 4706 - 4715 |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2025078841A3 (en) | 2025-05-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6947630B2 (en) | Biological substances and their use | |
| CN111587124B (en) | ROR1 antibody immunoconjugate | |
| US20230135723A1 (en) | Anti-cd98 antibodies and antibody drug conjugates | |
| KR102434626B1 (en) | Anti-B7-H3 Antibody and Antibody Drug Conjugates | |
| KR20240095316A (en) | BCMA monoclonal antibodies and antibody-drug conjugates | |
| TWI901692B (en) | Anti-bcma antibody-drug conjugates and methods of use | |
| CN110903395A (en) | Antibody, conjugate, preparation method and application thereof | |
| JP2019526529A (en) | Anti-B7-H3 antibody and antibody drug conjugate | |
| WO2019055931A1 (en) | Anti- folate receptor alpha antibody conjugates and their uses | |
| US20200002432A1 (en) | Anti-cd98 antibodies and antibody drug conjugates | |
| EP3469000A1 (en) | Anti-b7-h3 antibodies and antibody drug conjugates | |
| JP2016050204A (en) | Stability-modulating linker used with antibody drug conjugate | |
| JP2019522643A (en) | Anti-CD98 antibodies and antibody drug conjugates | |
| KR20200088402A (en) | Ligand-drug-conjugate as substrate for selective cleavage by cathepsin B exopeptidase activity | |
| JP2024540692A (en) | Conjugates Comprising Phosphorus(V) and Camptothecin Moieties | |
| KR20250117749A (en) | Anti-c-met antibody drug conjugates | |
| WO2025078841A2 (en) | Antibodies, conjugates, and uses thereof | |
| TWI908142B (en) | Nectin-4 antibodies and antibody-drug conjugates | |
| EP4534102A1 (en) | Antibody drug conjugate (adc) targeting nectin 4 and comprising an exatecan payload | |
| EP4534101A1 (en) | Antibody drug conjugate (adc) targeting nectin 4 and comprising an exatecan payload | |
| JP2025542295A (en) | Conjugates Comprising a Phosphorus(V) Moiety and a Drug | |
| Merlino | Characterization of MEN1309/OBT076, a new antibody conjugated to the DM4 maytansinoide toxin | |
| HK40052903A (en) | Anti-b7-h3 antibodies and antibody drug conjugates | |
| HK1237350B (en) | Stability-modulating linkers for use with antibody drug conjugates |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 24791475 Country of ref document: EP Kind code of ref document: A2 |