WO2025076240A1 - Solid forms of modulators of cystic fibrosis transmembrane conductance regulator - Google Patents
Solid forms of modulators of cystic fibrosis transmembrane conductance regulator Download PDFInfo
- Publication number
- WO2025076240A1 WO2025076240A1 PCT/US2024/049821 US2024049821W WO2025076240A1 WO 2025076240 A1 WO2025076240 A1 WO 2025076240A1 US 2024049821 W US2024049821 W US 2024049821W WO 2025076240 A1 WO2025076240 A1 WO 2025076240A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- theta
- ppm
- degrees
- solvate
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D519/00—Heterocyclic compounds containing more than one system of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring system not provided for in groups C07D453/00 or C07D455/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
Definitions
- crystalline and amorphous solid forms of a Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) modulator crystalline and amorphous solid forms of a Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) modulator, pharmaceutical compositions thereof, methods of treating cystic fibrosis with any of the foregoing, and processes for making crystalline and amorphous forms.
- CFTR Cystic Fibrosis Transmembrane Conductance Regulator
- Cystic fibrosis is a recessive genetic disease that affects approximately 83,000 children and adults worldwide. Despite progress in the treatment of CF, there is no cure.
- the most prevalent disease-causing mutation is a deletion of phenylalanine at position 508 of the CFTR amino acid sequence and is commonly referred to as the F508del mutation. This mutation occurs in approximately 70% of the cases of cystic fibrosis and is associated with severe disease.
- CFTR is a cAMP/ATP-mediated anion channel that is expressed in a variety of cell types, including absorptive and secretory epithelia cells, where it regulates anion flux across the membrane, as well as the activity of other ion channels and proteins.
- epithelial cells normal functioning of CFTR is critical for the maintenance of electrolyte transport throughout the body, including respiratory and digestive tissue.
- CFTR is composed of approximately 1480 amino acids that encode a protein which is made up of a tandem repeat of transmembrane domains, each containing six transmembrane helices and a nucleotide binding domain. The two transmembrane domains are linked by a large, polar, regulatory (R)-domain with multiple phosphorylation sites that regulate channel activity and cellular trafficking.
- Chloride transport takes place by the coordinated activity of ENaC and CFTR present on the apical membrane and the Na + -K + -ATPase pump and Cl- channels expressed on the basolateral surface of the cell. Secondary active transport of chloride from the luminal side leads to the accumulation of intracellular chloride, which can then passively leave the cell via Cl’ channels, resulting in a vectorial transport. Arrangement of Na + /2C17K + cotransporter, Na + -K + -ATPase pump and the basolateral membrane K + channels on the basolateral surface and CFTR on the luminal side coordinate the secretion of chloride via CFTR on the luminal side. Because water is probably never actively transported itself, its flow across epithelia depends on tiny transepithelial osmotic gradients generated by the bulk flow of sodium and chloride.
- Solid forms of pharmaceutical compounds are of interest to the industry due to ease of manufacture, storage, and administration. A number of distinct solid forms may be contemplated for a particular compound, depending upon its chemical and physical properties.
- Crystalline forms are of interest in the pharmaceutical industry, where the control of the crystalline form(s) of the active ingredient may be desirable or even required.
- Reproducible processes for producing a compound with a particular crystalline form in high purity may be desirable for compounds intended to be used in pharmaceuticals, as different crystalline forms may possess different properties.
- different crystalline forms may possess different chemical, physical, and/or pharmaceutical properties.
- one or more crystalline forms disclosed herein may exhibit a higher level of purity, chemical stability, and/or physical stability.
- Certain crystalline forms e.g., crystalline free form, crystalline salt, crystalline salt solvate, and crystalline salt hydrate forms of Compound I (collectively referred to as “crystalline forms”)
- crystalline forms may exhibit lower hygroscopicity.
- the crystalline forms of this disclosure may provide advantages during drug substance manufacturing, storage, and handling.
- pharmaceutically acceptable crystalline forms of Compound I may be particularly useful for the production of drugs for the treatment of CFTR-mediated diseases.
- Amorphous forms of therapeutic compounds may also be of interest in the pharmaceutical industry, where crystalline forms are not especially bioavailable. Some amorphous forms may improve bioavailability and thus allow for administration of reduced dosages. For some compounds, amorphous forms provide the most biologically accessible form of the therapeutic.
- one aspect of the disclosure provides solid forms (crystalline and amorphous) of a CFTR-modulating compound (R)-7-(bicyclo[l. l.l]pentan-l-ylmethyl)-6- ((6-(tert-butyl)furo[2,3-b]pyrazin-2-yl)methyl)-16-(2,6-dimethylphenyl)-9-oxa-3-thia-2,6- diaza-l(2,4)-pyrimidina-4(l,3)-benzenacyclononaphan-5-one 3,3-dioxide (Compound I) and pharmaceutically acceptable salts thereof.
- Compound I can be depicted as having the following structure:
- the crystalline form of Compound I is Compound I Neat Form A. In some embodiments, the crystalline form of Compound I is Compound I Neat Form B. In some embodiments, the crystalline form of Compound I is Compound I Neat Form C. In some embodiments, the crystalline form of Compound I is Compound I Neat Form D. In some embodiments, the crystalline form of compound I is Compound I Neat Form E. In some embodiments, the crystalline form of compound I is Compound I Compressed form A. In some embodiments, the crystalline form of compound I is Compound I Compressed form E. In some embodiments, the crystalline form of Compound I is Compound I EtOH Solvate Form B.
- the crystalline form of Compound I is Compound I MeOH solvate. In some embodiments, the crystalline form of Compound I is Compound I NPA solvate. In some embodiments, the crystalline form of Compound I is Compound I MeOAc Solvate Form A. In some embodiments, the crystalline form of Compound I is Compound I MeOAc Solvate Form B. In some embodiments, the crystalline form of Compound I is Compound I MeOAc Solvate Form C. In some embodiments, the crystalline form of Compound I is Compound I Hydrate Form A. In some embodiments, the crystalline form of Compound I is Compound I Hydrate Form B. In some embodiments, the crystalline form of Compound I is Compound I Hydrate Form C.
- the crystalline form of Compound I is Compound I EtOH Solvate Hydrate. In some embodiments, the crystalline form of Compound I is Compound I MeOH Solvate Hydrate. In some embodiments, the crystalline form of Compound I is Compound I IPA Solvate Hydrate. In some embodiments, the crystalline form of Compound I is Compound I MeOAc Solvate Hydrate.
- the solid form of Compound I is in amorphous form.
- Compound I is formulated as a solid (e.g., spray-dried) dispersion.
- compositions comprising Compound I in any of the pharmaceutically acceptable crystalline forms disclosed herein, which compositions may further include at least one additional active pharmaceutical ingredient and/or at least one carrier.
- methods of treating the CFTR-mediated disease cystic fibrosis comprising administering Compound I in any of the pharmaceutically acceptable solid forms disclosed herein, optionally as part of a pharmaceutical composition comprising at least one additional component (such as a carrier or additional active agent), to a subject in need thereof.
- a further aspect of the disclosure provides processes of making the solid forms of Compound I disclosed herein.
- compositions comprising combinations of a solid form Compound I as described herein or amorphous Compound I formulated as a solid (e.g., spray-dried) dispersion with (A)-l-(2,2-difluorobenzo[d][l,3]dioxol-5-yl)-N-(l-(2,3- dihydroxypropyl)-6-fluoro-2-(l -hydroxy -2-methylpropan-2-yl)-lH-indol-5- yl)cyclopropanecarboxamide (Compound II) and/or pharmaceutically acceptable salts thereof and/or with A-(5-hydroxy-2,4-di-/er/-butyl-phenyl)-4-oxo-lH-quinoline-3-carboxamide
- the solid form of Compound I as disclosed herein or amorphous Compound I formulated as a solid (e.g., spray-dried) dispersion is administered with Compound II and/or Compound III or Compound Ill-d, either in a single pharmaceutical composition or in multiple compositions to treat cystic fibrosis.
- a solid form of Compound I as described herein or amorphous Compound I formulated as a solid (e.g., spray-dried) dispersion is used in combination with (6R,12R)-17-amino-12-methyl-6,15- bis(trifluoromethyl)-13,19-dioxa-3,4,18-triazatricyclo[12.3.1.12,5]nonadeca-l(18),2,4,14,16- pentaen-6-ol (Compound IV), a deuterated derivative, or pharmaceutically acceptable salt thereof in a the treatment of cystic fibrosis.
- a solid form of Compound I as described herein or amorphous Compound I formulated as a solid (e.g., spray-dried) dispersion is used in combination with (67?)-17-amino-12,12-dimethyl-6,15- bis(trifluoromethyl)-19-oxa-3,4,13,18-tetrazatricyclo [12.3.1.12,5]nonadeca-l(18),2,4,14,16- pentaen-6-ol (Compound V), a deuterated derivative, or pharmaceutically acceptable salt thereof to treat cystic fibrosis.
- a further aspect of the disclosure provides processes of making the solid forms of Compound I disclosed herein.
- FIG. 1 provides an XRPD pattern of Compound I Neat Form A.
- FIG. 2 provides a TGA curve for crystalline Compound I Neat Form A.
- FIG. 3 provides a DSC analysis of Compound I Neat Form A.
- FIG. 4 provides a 13 C ssNMR spectrum of Compound I Neat Form A.
- FIG. 5 provides an XRPD pattern of Compound I Neat Form B.
- FIG. 6 provides a 13 C ssNMR spectrum of Compound I Neat Form B.
- FIG. 7 provides an XRPD pattern of Compound I Neat Form C.
- FIG. 8 provides a TGA curve for Compound I Neat Form C.
- FIG. 9 provides a DSC analysis of Compound I Neat Form C.
- FIG. 10 provides a 13 C ssNMR spectrum of Compound I Neat Form C.
- FIG. 11 provides a 13 C ssNMR spectrum of Compound I Neat Form D.
- FIG. 12 provides an XRPD pattern of Compound I Neat Form E.
- FIG. 13 provides a TGA curve for Compound I Neat Form E.
- FIG. 14 provides a DSC analysis of Compound I Neat Form E.
- FIG. 15 provides a 13 C ssNMR spectrum of Compound I Neat Form E.
- FIG. 16 provides an XRPD pattern of Compound I Compressed Form E.
- FIG. 17 provides a 13 C ssNMR spectrum of Compound I Compressed Form E.
- FIG. 18 provides an XRPD pattern of Compound I Compressed Form A.
- FIG. 19 provides a 13 C ssNMR spectrum of Compound I Compressed Form A.
- FIG. 20 provides an XRPD pattern of Compound I EtOH Solvate Form B.
- FIG. 21 provides a TGA curve for Compound I EtOH Solvate Form B.
- FIG. 22 provides a DSC analysis of Compound I EtOH Solvate Form B.
- FIG. 23 provides a 13 C ssNMR spectrum of Compound I EtOH Solvate Form B.
- FIG. 24 provides an XRPD pattern of Compound I MeOH Solvate.
- FIG. 25 provides a 13 C ssNMR spectrum of Compound I MeOH Solvate.
- FIG. 26 provides an XRPD pattern of Compound I NPA solvate.
- FIG. 27 provides a 13 C ssNMR spectrum of Compound I NPA solvate.
- FIG. 28 provides an XRPD pattern of Compound I MeOAc Solvate Form A.
- FIG. 29 provides a TGA curve for Compound I MeOAc Solvate Form A.
- FIG. 30 provides a DSC analysis of Compound I MeOAc Solvate Form A.
- FIG. 31 provides a 13 C ssNMR spectrum of Compound I MeOAc Solvate Form A.
- FIG. 32 provides an XRPD pattern of Compound I MeOAc Solvate Form B.
- FIG. 33 provides a TGA curve for Compound I MeOAc Solvate Form B.
- FIG. 34 provides a DSC analysis of Compound I MeOAc Solvate Form B.
- FIG. 35 provides a 13 C ssNMR spectrum of Compound I MeOAc Solvate Form B.
- FIG. 36 provides an XRPD pattern of Compound I MeOAc Solvate Form C.
- FIG. 37 provides a 13 C ssNMR spectrum of Compound I MeOAc Solvate Form C.
- FIG. 38 provides an XRPD pattern of Compound I Hydrate Form A.
- FIG. 39 provides a 13 C ssNMR spectrum of Compound I Hydrate Form A.
- FIG. 41 provides a 13 C ssNMR spectrum of Compound I Hydrate Form B.
- FIG. 42 provides an XRPD pattern of Compound I Hydrate Form C.
- FIG. 43 provides a 13 C ssNMR spectrum of Compound I Hydrate Form C.
- FIG. 44 provides an XRPD pattern of Compound I EtOH Solvate Hydrate.
- FIG. 45 provides a 13 C ssNMR spectrum of Compound I EtOH Solvate Hydrate.
- FIG. 46 provides an XRPD pattern of Compound I MeOH Solvate Hydrate.
- FIG. 47 provides a 13 C ssNMR spectrum of Compound I MeOH Solvate Hydrate.
- FIG. 48 provides an XRPD pattern of Compound I IPA Solvate Hydrate.
- FIG. 49 provides a 13 C CPMAS spectrum of Compound I IPA Solvate Hydrate.
- FIG. 50 provides an XRPD pattern of Compound I MeOAc Solvate Hydrate.
- FIG. 51 provides a 13 C ssNMR spectrum of Compound I MeOAc Solvate Hydrate.
- FIG. 52 provides an XRPD pattern of amorphous Compound I.
- FIG. 53 provides a 13 C ssNMR spectrum of amorphous Compound I.
- FIG. 54 provides an XRPD pattern of Compound I SDD 1A.
- FIG. 55 provides a 13 C ssNMR spectrum of Compound I SDD 1A.
- FIG. 56 provides a DSC analysis of Compound I SDD 1 A.
- FIG. 57 provides an XRPD pattern of Compound I SDD IB.
- FIG. 58 provides a DSC analysis of Compound I SDD IB.
- FIG. 59 provides an XRPD pattern of Compound I SDD 1C.
- FIG. 60 provides a DSC analysis of Compound I SDD 1C.
- FIG. 61 provides an XRPD pattern of Compound I SDD ID.
- FIG. 62 provides a DSC analysis of Compound I SDD ID.
- FIG. 63 provides an XRPD pattern of Compound I SDD IE.
- FIG. 64 provides a DSC analysis of Compound I SDD IE.
- Compound I refers to the CFTR corrector, (7?)-7-(bicyclo[l.l.l]pentan-l-ylmethyl)-6-((6-(tert-butyl)furo[2,3-b]pyrazin-2-yl)methyl)- 16-(2,6-dimethylphenyl)-9-oxa-3-thia-2,6-diaza-l(2,4)-pyrimidina-4(l,3)- benzenacyclononaphan-5-one 3,3-dioxide, which can be depicted as having the following structure:
- Compound II refers to (R)-l-(2,2- difluorobenzo[d][l, 3]di oxol-5-yl)-,V-( l-(2, 3-dihydroxypropyl)-6-fluoro-2-(l -hydroxy -2- methylpropan-2-yl)-lH-indol-5-yl)cyclopropanecarboxamide, which can be depicted as having the following structure:
- Compound II may be in the form of a pharmaceutically acceptable salt.
- Compound II and methods of making and using Compound II are disclosed in WO 2010/053471, WO 2011/119984, and WO 2015/160787, each incorporated herein by reference.
- “Compound III” as used throughout this disclosure refers to N-[2,4-bis(l, 1- dimethylethyl)-5-hydroxyphenyl]-l,4-dihydro-4-oxoquinoline-3-carboxamide (also known as /V-(5-hydroxy-2,4-di-tert-butyl-phenyl)-4-oxo-l H-quinoline-3-carboxamide) which can be depicted as having the following structure:
- Compound III may also be in the form of a pharmaceutically acceptable salt.
- Compound III and methods of making and using Compound III are disclosed in WO 2006/002421, WO 2007/079139, and WO 2010/019239, each incorporated herein by reference.
- a deuterated derivative of Compound III (Compound Ill-d) is employed in the compositions and methods disclosed herein.
- a chemical name for Compound Ill-d is A-(2-(ferCbutyl)-5-hydroxy-4-(2-(methyl-d 3 )propan-2-yl-l,l,l,3,3,3- d6)phenyl)-4-oxo-l,4-dihydroquinoline-3 -carboxamide, which can be depicted as having the following structure:
- Compound Ill-d may be in the form of a pharmaceutically acceptable salt.
- Compound Ill-d and methods of making and using Compound Ill-d are disclosed in WO 2012/158885 and WO 2014/078842, incorporated herein by reference.
- Compound IV refers to (6R,12R)-17-amino- 12-methyl-6, 15-bis(trifluoromethyl)-13, 19-dioxa-3,4, 18-tri azatri cyclo[12.3.1.12,5]nonadeca- l(18),2,4,14,16-pentaen-6-ol.
- Compound IV may also be in the form of a deuterated derivative or pharmaceutically acceptable salt. Methods of making and using Compound IV, deuterated derivatives and pharmaceutically acceptable salts thereof, are described in WO 2022/032068, incorporated herein by reference.
- Compound V refers to (6J?)-17-amino-12,12- dimethyl-6,15-bis(trifluoromethyl)-19-oxa-3,4,13,18-tetrazatricyclo [12.3.1.12,5]nonadeca- l(18),2,4,14,16-pentaen-6-ol.
- Compound V may also be in the form of a deuterated derivative or pharmaceutically acceptable salt. Methods of making and using Compound V, deuterated derivatives and pharmaceutically acceptable salts thereof, are described in WO 2022/109573, incorporated herein by reference.
- CFTR cystic fibrosis transmembrane conductance regulator
- CFTR modulator and “CFTR modulating compound” interchangeably refer to a compound that increases the activity of CFTR.
- the increase in activity resulting from a CFTR modulator includes but is not limited to compounds that correct, potentiate, stabilize, and/or amplify CFTR.
- CFTR corrector refers to a compound that facilitates the processing and trafficking of CFTR to increase the amount of CFTR at the cell surface.
- Compounds I and II disclosed herein are CFTR correctors.
- CFTR potentiator refers to a compound that increases the channel activity of CFTR protein located at the cell surface, resulting in enhanced ion transport.
- Compounds III, Ill-d, IV, and V disclosed herein are CFTR potentiators. It will be appreciated that when a description of a combination of Compound I and other specified CFTR modulating agents is provided herein, reference to “Compound III or Ill-d” in connection with the combination means that either Compound III or Compound Ill-d, but not both, is included in the combination.
- active pharmaceutical ingredient refers to a biologically active compound.
- a “wedge” ( *) or “hash” (•••"') bond to a stereogenic atom indicates a chiral center of known absolute stereochemistry (i.e. one stereoisomer).
- a “wavy” bond ( ) to a stereogenic atom indicates a chiral center of unknown absolute stereochemistry (i.e. one stereoisomer).
- a “wavy” bond ( ) to a double-bonded carbon indicates a mixture of E/Z isomers.
- a ' (“straight”) bond to a stereogenic atom indicates where there is a mixture (e.g., a racemate or enrichment).
- two (“straight”) bonds to a double-bonded carbon indicates that the double bond possesses the E/Z stereochemistry as A drawn.
- a ' i.e., a “wavy” line perpendicular to a “straight” bond to group “A” indicates that group “A” is a substituent whose point of attachment is at the end of the bond that terminates at the “wavy” line.
- the term “pharmaceutically acceptable solid form” refers to a solid form of Compound I of this disclosure that includes the crystalline form (e g., crystalline free form, crystalline salt, crystalline salt solvate, and crystalline salt hydrate) of Compound I that is nontoxic and suitable for use in pharmaceutical compositions.
- the term “pharmaceutically acceptable solid form” as used herein also refers to the amorphous form of Compound I and solid dispersions comprising that amorphous form.
- patient and “subject” are used interchangeably and refer to an animal including humans.
- an effective dose and “effective amount” are used interchangeably herein and refer to that amount of a compound that produces the desired effect for which it is administered (e.g., improvement in CF or a symptom of CF, or lessening the severity of CF or a symptom of CF).
- the exact amount of an effective dose will depend on the purpose of the treatment and will be ascertainable by one skilled in the art using known techniques (see, e.g., Lloyd (1999) The Art, Science and Technology of Pharmaceutical Compounding).
- treatment generally mean the improvement of CF or one or more of its symptoms or lessening the severity of CF or one or more of its symptoms in a subject.
- Treatment includes, but is not limited to, the following: increased growth of the subject, increased weight gain, reduction of mucus in the lungs, improved pancreatic and/or liver function, reduction of chest infections, and/or reductions in coughing or shortness of breath. Improvements in or lessening the severity of any of these symptoms can be readily assessed according to standard methods and techniques known in the art.
- the term “in combination with,” when referring to two or more compounds, agents, or additional active pharmaceutical ingredients, means the administration of two or more compounds, agents, or active pharmaceutical ingredients to the patient prior to, concurrently with, or subsequent to each other.
- the terms “about” and “approximately,” when used in connection with doses, amounts, or weight percents of ingredients of a composition or a dosage form, include the value of a specified dose, amount, or weight percent or a range of the dose, amount, or weight percent that is recognized by one of ordinary skill in the art to provide a pharmacological effect equivalent to that obtained from the specified dose, amount, or weight percent.
- the terms “about” and “approximately” may refer to an acceptable error for a particular value as determined by one of skill in the art, which depends in part on how the values is measured or determined. In some embodiments, the terms “about” and “approximately” mean within 15%, 10%, 5%, 4%, 3%, 2%, 1%, or 0.5% of a given value or range. In some embodiments, the terms “about” and “approximately” mean within 15% of a given value or range. In some embodiments, the terms “about” and “approximately” mean within 10% of a given value or range.
- amorphous refers to a solid material having no long- range order in the position of its molecules.
- Amorphous solids are generally glasses or supercooled liquids in which the molecules are arranged in a random manner so that there is no well-defined arrangement, e.g., molecular packing, and no long-range order.
- Amorphous solids are generally rather isotropic, i.e., exhibit similar properties in all directions and do not have definite melting points. Instead, they typically exhibit a glass transition temperature which marks a transition from glassy amorphous state to supercooled liquid amorphous state upon heating.
- a solid material may comprise an amorphous compound, and the material may, for example, be characterized by a lack of sharp characteristic crystalline peak(s) in its XRPD spectrum (i.e., the material is not crystalline, but is amorphous, as determined by XRPD). Instead, one or several broad peaks (e.g., halos) may appear in the XRPD pattern of the material. Broad peaks are characteristic of an amorphous solid. See US 2004/0006237 for a comparison of XRPDs of an amorphous material and crystalline material. Other techniques, such as solid-state NMR, may also be used to characterize crystalline or amorphous forms.
- crystal form As used herein, the terms “crystal form,” “crystalline form,” and “form” interchangeably refer to a crystal structure (or polymorph) having a particular molecular packing arrangement in the crystal lattice. Crystalline forms can be identified and distinguished from each other by the presence or absence of a specific salt, solvate, or hydrate, as well as by one or more characterization techniques including, for example, X-ray powder diffraction (XRPD), single crystal X-ray diffraction, and 13 C solid-state nuclear magnetic resonance ( 13 C ssNMR).
- XRPD X-ray powder diffraction
- 13 C ssNMR 13 C solid-state nuclear magnetic resonance
- crystalline form A of Compound I and “crystalline potassium salt form A of Compound I” refer to unique crystalline forms that can be identified and distinguished from each other by the presence or absence of potassium alone as well as by other characterization techniques.
- the novel crystalline forms are characterized by an X-ray powder diffractogram having one or more signals at specified two-theta values (°20).
- free form refers to a non-ionized version of the compound in the solid state.
- free forms include free bases and free acids.
- compressed form refers to a crystalline form that has been mechanically compressed to become a different crystalline form.
- solvate refers to a crystal form comprising one or more molecules of a compound of the present disclosure and, incorporated into the crystal lattice, one or more molecules of a solvent or solvents in stoichiometric or nonstoichiometric amounts.
- the solvent is water
- the solvate is referred to as a “hydrate.”
- Other, nonlimiting examples of solvate forms include those derived from ethanol (“EtOH solvate”), methanol (“MeOH solvate”), //-propanol ( “NPA solvate”), methyl acetate (“MeOAc solvate), and isopropanol (“IP A solvate”).
- a solid material may comprise a mixture of crystalline solids and amorphous solids.
- a solid material comprising an amorphous compound may also, for example, contain up to 30% of a crystalline solid.
- a solid material prepared to comprise an amorphous compound may also, for example, contain up to 25%, 20%, 15%, 10%, 5%, or 2% of a crystalline solid.
- the characterizing data such as XRPD and ssNMR, may contain indicators of both crystalline and amorphous solids.
- a crystalline form of this disclosure may contain up to 30% amorphous compound.
- a crystalline preparation of Compound I may contain up to 25%, 20%, 15%, 10%, 5%, or 2% of an amorphous solid.
- substantially amorphous refers to a solid material having little or no long-range order in the position of its molecules.
- substantially amorphous materials have less than 15% crystallinity (e.g., less than 10% crystallinity, less than 5% crystallinity, or less than 2% crystallinity).
- substantially amorphous includes the descriptor, “amorphous,” which refers to materials having no (0%) crystallinity.
- substantially crystalline refers to a solid material having little or no amorphous molecules.
- substantially crystalline materials have less than 15% amorphous molecules (e.g., less than 10% amorphous molecules, less than 5% amorphous molecules, or less than 2% amorphous molecules).
- substantially crystalline includes the descriptor “crystalline,” which refers to materials that are 100% crystalline form.
- a crystalline form is “substantially pure” when it accounts for an amount by weight equal to or greater than 90% of the sum of all solid form(s) of a given compound in a sample as determined by a method in accordance with the art, such as quantitative XRPD.
- the solid form is “substantially pure” when it accounts for an amount by weight equal to or greater than 95% of the sum of all solid form(s) in a sample.
- the solid form is “substantially pure” when it accounts for an amount by weight equal to or greater than 99% of the sum of all solid form(s) in a sample.
- XRPD refers to the analytical characterization method of X-ray powder diffraction. XRPD patterns disclosed herein were recorded at ambient conditions in transmission or reflection geometry using a diffractometer.
- ambient conditions means room temperature, open air condition and uncontrolled humidity condition.
- room temperature and “ambient temperature” mean 15 °C to 30 °C.
- X-ray powder diffractogram As used herein, the terms “X-ray powder diffractogram,” “X-ray powder diffraction pattern,” and “XRPD pattern,” interchangeably refer to an experimentally obtained pattern plotting signal positions (on the abscissa) versus signal intensities (on the ordinate).
- a “signal” or “peak” as used herein refers to a point in the XRPD pattern where the intensity as measured in counts is at a local maximum.
- An XRPD peak is identified by its angular value as measured in degrees two-theta (° 20), depicted on the abscissa of an X-ray powder diffractogram, which may be expressed, for example, as “a signal at . . . degrees two- theta,” “a signal at [a] two-theta value(s)of ...” and/or “a signal at at least .. . two-theta value(s) selected from . .
- the repeatability of the measured angular values is in the range of ⁇ 0.2° 20, i.e., the angular value can be at the recited angular value + 0.2 degrees two-theta, the angular value - 0.2 degrees two-theta, or any value between those two end points (angular value + 0.2 degrees two-theta and angular value - 0.2 degrees two-theta).
- one or more signals (or peaks) in an XRPD pattern may overlap and may, for example, not be apparent to the naked eye. Indeed, one of ordinary skill in the art would recognize that some art-recognized methods are capable of and suitable for determining whether a signal exists in a pattern, such as Rietveld refinement.
- signal intensities and “peak intensities” interchangeably refer to relative signal intensities within a given X-ray powder diffractogram. Factors that can affect the relative signal or peak intensities include sample thickness and preferred orientation (e.g., the crystalline particles are not distributed randomly).
- an X-ray powder diffractogram is “substantially similar to that in [a particular] Figure” when at least 90%, such as at least 95%, at least 98%, or at least 99% of the signals in the two diffractograms overlap.
- determining “substantial similarity” one of ordinary skill in the art will understand that there may be variation in the intensities and/or signal positions in XRPD diffractograms even for the same crystalline form
- the signal maximum values in XRPD diffractograms in degrees two-theta generally mean that value is identified as ⁇ 0.2 degrees two-theta of the reported value, an art-recognized variance.
- X-ray powder diffractogram having a signal at ... two-theta values refers to an XRPD pattern that contains X-ray reflection positions as measured and observed in X-ray powder diffraction experiments (degrees two-theta).
- TGA thermogravimetric analysis
- TGA/DSC thermogravimetric analysis and differential scanning calorimetry
- DSC differential scanning calorimetry
- ssNMR refers to the analytical method of solid-state nuclear magnetic resonance (NMR).
- CPMAS cross-polarization magic-angle spinning NMR.
- solvent refers to any liquid in which the product is at least partially soluble (solubility of product >1 g/1).
- glass transition temperature or “T g ” refers to the temperature above which a hard and brittle “glassy” amorphous solid becomes viscous or rubbery.
- melting temperature As used herein, the term “melting temperature”, “melting point”, or “T m ” refers to the temperature at which the solid and liquid state of a material are at equilibrium.
- the term "dispersion” refers to a disperse system in which one substance, the dispersed phase, is distributed, in discrete units, throughout a second substance (the continuous phase or vehicle).
- the size of the dispersed phase can vary considerably (e.g., colloidal particles of nanometer dimension, to multiple microns in size).
- the dispersed phases can be solids, liquids, or gases. In the case of a solid dispersion, the dispersed and continuous phases are both solids.
- a solid dispersion can include, inter alia, a crystalline drug in an amorphous polymer; an amorphous drug in an amorphous polymer; an amorphous drug dispersed in an amorphous drug; or, alternatively, an amorphous drug dispersed in one or more excipients.
- a solid dispersion includes the polymer constituting the dispersed phase, and the drug constitute the continuous phase.
- a solid dispersion includes the drug constituting the dispersed phase, and the polymer constituting the continuous phase.
- Compound I in any one of the pharmaceutically acceptable crystalline forms disclosed herein, acts as a CFTR modulator, i.e., it modulates CFTR activity in the body. Individuals suffering from a mutation in the gene encoding CFTR may benefit from receiving a CFTR modulator.
- a CFTR mutation may affect the CFTR quantity, i.e., the number of CFTR channels at the cell surface, or it may impact CFTR function, i.e., the functional ability of each channel to open and transport ions.
- Mutations affecting CFTR quantity include mutations that cause defective synthesis (Class I defect), mutations that cause defective processing and trafficking (Class II defect), mutations that cause reduced synthesis of CFTR (Class V defect), and mutations that reduce the surface stability of CFTR (Class VI defect). Mutations that affect CFTR function include mutations that cause defective gating (Class III defect) and mutations that cause defective conductance (Class IV defect). Some CFTR mutations exhibit characteristics of multiple classes. Certain mutations in the CFTR gene result in cystic fibrosis.
- the invention provides methods of treating, lessening the severity of, or symptomatically treating cystic fibrosis in a patient comprising administering to the patient an effective amount of Compound I in any one of the pharmaceutically acceptable solid forms disclosed herein, alone or in combination with another active ingredient, such as another CFTR modulating agent.
- the patient has an F508del/minimal function (MF) genotype, F508del/F508del genotype (homozygous for the F508del mutation), F508del/gating genotype, or F508del/residual function (RF) genotype.
- MF F508del/minimal function
- F508del/F508del genotype homozygous for the F508del mutation
- F508del/gating genotype F508del/gating genotype
- F508del/residual function (RF) genotype F508del/residual function
- the patient is
- the invention provides methods of treating, lessening the severity of, or symptomatically treating cystic fibrosis in a patient comprising administering to the patient an effective amount of Compound I as crystalline Compound I Neat Form A.
- the method of treating, lessening the severity of, or symptomatically treating cystic fibrosis in a patient comprises administering to the patient an effective amount of Compound I as crystalline Compound I Neat Form B.
- the method of treating, lessening the severity of, or symptomatically treating cystic fibrosis in a patient comprises administering to the patient an effective amount of Compound I as crystalline Compound I Neat Form C
- the method of treating, lessening the severity of, or symptomatically treating cystic fibrosis in a patient comprises administering to the patient an effective amount of Compound I as crystalline Compound I Neat Form D.
- the method of treating, lessening the severity of, or symptomatically treating cystic fibrosis in a patient comprises administering to the patient an effective amount of Compound I as crystalline Compound I Neat Form E.
- the method of treating, lessening the severity of, or symptomatically treating cystic fibrosis in a patient comprises administering to the patient an effective amount of Compound I as crystalline Compound I Compressed Form A.
- the method of treating, lessening the severity of, or symptomatically treating cystic fibrosis in a patient comprises administering to the patient an effective amount of Compound I as crystalline Compound I Compressed Form E.
- the method of treating, lessening the severity of, or symptomatically treating cystic fibrosis in a patient comprises administering to the patient an effective amount of Compound I as crystalline Compound I EtOH Solvate Form B.
- the method of treating, lessening the severity of, or symptomatically treating cystic fibrosis in a patient comprises administering to the patient an effective amount of Compound I as crystalline Compound I MeOH solvate.
- the method of treating, lessening the severity of, or symptomatically treating cystic fibrosis in a patient comprises administering to the patient an effective amount of Compound I as crystalline Compound I NPA solvate.
- the method of treating, lessening the severity of, or symptomatically treating cystic fibrosis in a patient comprises administering to the patient an effective amount of Compound I as crystalline Compound I MeOAc Solvate Form A.
- the method of treating, lessening the severity of, or symptomatically treating cystic fibrosis in a patient comprises administering to the patient an effective amount of Compound I as crystalline Compound I MeOAc Solvate Form B.
- the method of treating, lessening the severity of, or symptomatically treating cystic fibrosis in a patient comprises administering to the patient an effective amount of Compound I as crystalline Compound I MeOAc Solvate Form C.
- the method of treating, lessening the severity of, or symptomatically treating cystic fibrosis in a patient comprises administering to the patient an effective amount of Compound I as crystalline Compound I Hydrate Form A.
- the method of treating, lessening the severity of, or symptomatically treating cystic fibrosis in a patient comprises administering to the patient an effective amount of Compound I as crystalline Compound I Hydrate Form B.
- the method of treating, lessening the severity of, or symptomatically treating cystic fibrosis in a patient comprises administering to the patient an effective amount of Compound I as crystalline Compound I Hydrate Form C.
- the method of treating, lessening the severity of, or symptomatically treating cystic fibrosis in a patient comprises administering to the patient an effective amount of Compound I as crystalline Compound I EtOH Solvate Hydrate.
- the method of treating, lessening the severity of, or symptomatically treating cystic fibrosis in a patient comprises administering to the patient an effective amount of Compound I as crystalline Compound I MeOH Solvate Hydrate.
- the method of treating, lessening the severity of, or symptomatically treating cystic fibrosis in a patient comprises administering to the patient an effective amount of Compound I as crystalline Compound I IPA Solvate Hydrate.
- the method of treating, lessening the severity of, or symptomatically treating cystic fibrosis in a patient comprises administering to the patient an effective amount of Compound I as amorphous Compound I.
- the method of treating, lessening the severity of, or symptomatically treating cystic fibrosis in a patient comprises administering to the patient an effective amount of Compound I as Compound I formulated as a solid (e.g., spray-dried) dispersion.
- One aspect disclosed herein provides methods of treating cystic fibrosis and other CFTR-mediated diseases with Compound I in any one of the pharmaceutically acceptable solid forms disclosed herein or amorphous Compound I formulated as a solid (e.g., spray- dried) dispersion, in combination with other pharmaceutically active agents, including CFTR modulating agents.
- Compound I in any one of the pharmaceutically acceptable solid forms disclosed herein or amorphous Compound I formulated as a solid (e.g., spray-dried) dispersion can be administered in combination with at least one additional active pharmaceutical ingredient, such as, e.g., a CFTR modulating agent.
- the at least one additional active pharmaceutical ingredient is a CFTR corrector. In some embodiments, the at least one additional active pharmaceutical ingredient is a CFTR potentiator. In some embodiments, the methods of treating cystic fibrosis and other CFTR-mediated diseases with Compound I in any one of the pharmaceutically acceptable solid forms disclosed herein, include combination with at least two additional active pharmaceutical ingredients, one of which is a CFTR corrector and one of which is a CFTR potentiator. [00160] In some embodiments, at least one additional active pharmaceutical agent is selected from mucolytic agents, bronchodilators, antibiotics, anti-infective agents, and antiinflammatory agents
- the at least one additional active pharmaceutical agent is selected from (a) Compound IV or Compound V, deuterated derivatives of Compound IV or Compound V, and pharmaceutically acceptable salts of Compound IV, Compound V, and their deuterated derivatives; and optionally (b) Compound II and pharmaceutically acceptable salts thereof.
- the combination therapies provided herein comprise Compound I in any one of the pharmaceutically acceptable crystalline forms disclosed herein or as amorphous Compound I formulated as a solid (e.g., spray-dried) dispersion, and an additional active pharmaceutical agent selected from Compound IV or V, deuterated derivatives, and pharmaceutically acceptable salts thereof.
- the combination optionally includes Compound II.
- the combination therapies provided herein comprise at least one compound selected from Compound I in any one of the pharmaceutically acceptable solid forms disclosed herein or amorphous Compound I formulated as a solid (e.g., spray-dried) dispersion and at least one compound selected from Compound IV or Compound V, and pharmaceutically acceptable salts thereof and at least one compound selected from Compound II and pharmaceutically acceptable salts thereof.
- the at least one additional active pharmaceutical ingredient is selected from (a) Compound II and pharmaceutically acceptable salts thereof; and (b) Compound III or Compound Ill-d and pharmaceutically acceptable salts of Compound III or Compound Ill-d.
- the combination therapies provided herein comprise Compound I in any one of the pharmaceutically acceptable crystalline forms disclosed herein or amorphous Compound I formulated as a solid (e.g., spray-dried) dispersion, and at least one compound selected from Compound II, (Compound III or Ill-d), and pharmaceutically acceptable salts thereof.
- the combination therapies provided herein comprise at least one compound selected from Compound I in any one of the pharmaceutically acceptable solid forms disclosed herein or amorphous Compound I formulated as a solid (e.g., spray-dried) dispersion and at least one compound selected from Compound III or ni-d and/or pharmaceutically acceptable salts thereof.
- At least one compound selected from Compound I in any one of the pharmaceutically acceptable solid forms disclosed herein or amorphous Compound I formulated as a solid (e.g., spray-dried) dispersion is administered in combination with at least one compound selected from Compound II and pharmaceutically acceptable salts thereof.
- At least one compound selected from Compound I in any one of the pharmaceutically acceptable solid forms disclosed herein or amorphous Compound I formulated as a solid (e.g., spray-dried) dispersion is administered in combination with at least one compound selected from Compound III and pharmaceutically acceptable salts thereof.
- at least one compound selected from Compound I in any one of the pharmaceutically acceptable solid forms disclosed herein or amorphous Compound I formulated as a solid (e.g., spray-dried) dispersion is administered in combination with at least one compound selected from Compound Ill-d and pharmaceutically acceptable salts thereof.
- At least one compound selected from Compound I in any one of the pharmaceutically acceptable solid forms disclosed herein or amorphous Compound I formulated as a solid (e g., spray-dried) dispersion is administered in combination with Compounds II or a pharmaceutically acceptable salt thereof and at least one compound selected from Compound III and pharmaceutically acceptable salts thereof.
- at least one compound selected from Compound I in any one of the pharmaceutically acceptable solid forms disclosed herein or amorphous Compound I formulated as a solid (e.g., spray-dried) dispersion is administered in combination with at least one compound selected from Compound II and pharmaceutically acceptable salts thereof and at least one compound selected from Compound Ill-d and pharmaceutically acceptable salts thereof.
- At least one compound selected from Compound I in any one of the pharmaceutically acceptable solid forms disclosed herein or amorphous Compound I formulated as a solid (e.g., spray-dried) dispersion is administered in a first pharmaceutical composition; at least one compound selected from Compound II and pharmaceutically acceptable salts thereof is administered in a second pharmaceutical composition; and at least one compound selected from Compound III, Compound Ill-d, Compound IV, Compound V and pharmaceutically acceptable salts of Compounds III, Ill-d, IV, and V is administered in a third pharmaceutical composition.
- At least one compound selected from Compound I in any one of the pharmaceutically acceptable solid forms disclosed herein or amorphous Compound I formulated as a solid (e.g., spray-dried) dispersion is administered in a first pharmaceutical composition; at least one compound selected from Compound IV or V and pharmaceutically acceptable salts thereof is administered in a second pharmaceutical composition.
- At least one compound selected from Compound I in any one of the pharmaceutically acceptable solid forms disclosed herein or amorphous Compound I formulated as a solid dispersion is administered in a first pharmaceutical composition; at least one compound selected from Compound III or Ill-d and pharmaceutically acceptable salts thereof is administered in a second pharmaceutical composition.
- At least one compound selected from Compound I in any one of the pharmaceutically acceptable solid forms disclosed herein or amorphous Compound I formulated as a solid (e.g., spray-dried) dispersion is administered in a first pharmaceutical composition; and at least one compound selected from Compound IV or V, and pharmaceutically acceptable salts thereof and optionally at least one compound selected from Compound II and pharmaceutically acceptable salts thereof are administered in a second pharmaceutical composition.
- At least one compound selected from Compound I in any one of the pharmaceutically acceptable solid forms disclosed herein or amorphous Compound I formulated as a solid (e.g., spray-dried) dispersion is administered in a first pharmaceutical composition; and (a) at least one compound selected from Compound IV, V, and pharmaceutically acceptable salts thereof and (b) at least one compound selected from Compound II and pharmaceutically acceptable salts thereof are administered in a second pharmaceutical composition.
- At least one compound selected from Compound I in any one of the pharmaceutically acceptable solid forms disclosed herein or amorphous Compound I formulated as a solid (e.g., spray-dried) dispersion is administered in a first pharmaceutical composition; and at least one compound selected from Compound II and pharmaceutically acceptable salts thereof and at least one compound selected from Compound III or Ill-d, and pharmaceutically acceptable salts thereof are administered in a second pharmaceutical composition.
- the second pharmaceutical composition comprises a half of a daily dose of said at least one compound selected from Compound III, Ill-d, and pharmaceutically acceptable salts thereof, and the other half of said at least one compound selected from Compound III, Ill-d, and pharmaceutically acceptable salts thereof is administered in a third pharmaceutical composition.
- Any suitable pharmaceutical formulations can be used for Compound I (in any one of the pharmaceutically acceptable solid forms disclosed herein or amorphous Compound I formulated as a solid (e.g., spray-dried) dispersion), Compound II, Compound III, Compound Ill-d, and pharmaceutically acceptable salts thereof.
- Some exemplary pharmaceutical compositions for Compound II and its pharmaceutically acceptable salts can be found in WO 2011/119984 and WO 2014/014841, incorporated herein by reference.
- compositions for Compound III and its pharmaceutically acceptable salts can be found in WO 2007/134279, WO 2010/019239, WO 2011/019413, WO 2012/027731, and WO 2013/130669, and some exemplary pharmaceutical compositions for Compound Ill-d and its pharmaceutically acceptable salts can be found in US 8,865,902, US 9,181,192, US 9,512,079, WO 2017/053455, and WO 2018/080591, all of which are incorporated herein by reference.
- Exemplary formulations for Compounds IV and V, deuterated derivatives and pharmaceutically acceptable salts thereof can be found in WO 2022/032068 and WO 2022/109573, respectively, both incorporated herein by reference.
- the solid form of Compound I used in the combination therapies of the invention is Compound I Neat Form A.
- the solid form of Compound I used in the combination therapies of the invention is Compound I Neat Form B.
- the solid form of Compound I used in the combination therapies of the invention is Compound I Neat Form C.
- the solid form of Compound I used in the combination therapies of the invention is Compound I Neat Form D.
- the solid form of Compound I used in the combination therapies of the invention is Compound I Neat Form E.
- the solid form of Compound I used in the combination therapies of the invention is Compound I Compressed Form A.
- the solid form of Compound I used in the combination therapies of the invention is Compound I Compressed Form E.
- the solid form of Compound I used in the combination therapies of the invention is Compound I EtOH Solvate Form B.
- the solid form of Compound I used in the combination therapies of the invention is Compound I MeOH Solvate.
- the solid form of Compound I used in the combination therapies of the invention is Compound I NPA Solvate.
- the solid form of Compound I used in the combination therapies of the invention is Compound I MeOAc Solvate Form A.
- the solid form of Compound I used in the combination therapies of the invention is Compound I MeOAc Solvate Form B. In some embodiments, the solid form of Compound I used in the combination therapies of the invention is Compound I MeOAc Solvate Form C. In some embodiments, the solid form of Compound I used in the combination therapies of the invention is Compound I Hydrate Form A. In some embodiments, the solid form of Compound I used in the combination therapies of the invention is Compound I Hydrate Form B. In some embodiments, the solid form of Compound I used in the combination therapies of the invention is Compound I Hydrate Form C. In some embodiments, the solid form of Compound I used in the combination therapies of the invention is Compound I EtOH Solvate Hydrate.
- the solid form of Compound I used in the combination therapies of the invention is Compound I MeOH Solvate Hydrate. In some embodiments, the solid form of Compound I used in the combination therapies of the invention is Compound I IPA Solvate Hydrate. In some embodiments, the solid form of Compound I used in the combination therapies of the invention is Compound I MeOAc Solvate Hydrate. In some embodiments, the solid form of Compound I used in the combination therapies of the invention is amorphous Compound I.
- the form of Compound I used in the combination therapies of the invention is Compound I in a solid (e.g., spray-dried) dispersion.
- compositions comprising Compound I as any one of the pharmaceutically acceptable solid forms disclosed herein or amorphous Compound I formulated as a solid (e.g., spray-dried) dispersion.
- the invention provides pharmaceutical compositions comprising Compound I as any one of the pharmaceutically acceptable solid forms disclosed herein in combination or amorphous Compound I formulated as a solid (e.g., spray-dried) dispersion, with at least one additional active pharmaceutical agent.
- the at least one additional active pharmaceutical ingredient is a CFTR modulator.
- the at least one additional active pharmaceutical agent is a CFTR corrector.
- the at least one additional active pharmaceutical agent is a CFTR potentiator.
- the pharmaceutical composition comprises Compound I as any one of the pharmaceutically acceptable crystalline forms disclosed herein or amorphous Compound I formulated as a solid (e.g., spray-dried) dispersion and at least two additional active pharmaceutical agents, one of which is a CFTR corrector and one of which is a CFTR potentiator.
- At least one additional active pharmaceutical agent is selected from mucolytic agents, bronchodilators, antibiotics, anti-infective agents, and antiinflammatory agents.
- the invention provides a pharmaceutical composition
- a pharmaceutical composition comprising at least one compound selected from Compound I as any one of the pharmaceutically acceptable crystalline forms disclosed herein or amorphous Compound I formulated as a solid (e.g., spray-dried) dispersion, and at least one pharmaceutically acceptable carrier.
- the invention provides a pharmaceutical composition
- a pharmaceutical composition comprising (a) Compound I, as any one of the pharmaceutically acceptable solid forms disclosed herein or amorphous Compound I formulated as a solid (e.g., spray-dried) dispersion, (b) at least one compound selected from Compound IV, Compound V, deuterated derivatives thereof and pharmaceutically acceptable salts thereof, and (c) at least one pharmaceutically acceptable carrier.
- the disclosure provides a pharmaceutical composition
- a pharmaceutical composition comprising (a) Compound I as any one of the pharmaceutically acceptable solid forms disclosed herein or amorphous Compound I formulated as a solid (e.g., spray-dried) dispersion, (b) at least one compound selected from Compound HI, Ill-d, and pharmaceutically acceptable salts thereof, and (c) at least one pharmaceutically acceptable carrier.
- the disclosure provides a pharmaceutical composition
- a pharmaceutical composition comprising (a) Compound I as any one of the pharmaceutically acceptable solid forms disclosed herein or amorphous Compound I formulated as a solid (e.g., spray-dried) dispersion, (b) at least one compound selected from Compound n and pharmaceutically acceptable salts thereof, (c) at least one compound selected from Compound III, Ill-d, VI, and V and pharmaceutically acceptable salts thereof, and (d) at least one pharmaceutically acceptable carrier.
- the disclosure provides a pharmaceutical composition
- a pharmaceutical composition comprising (a) Compound I as any one of the pharmaceutically acceptable solid forms disclosed herein or amorphous Compound I formulated as a solid (e.g., spray-dried) dispersion, (b) at least one compound selected from Compound n and pharmaceutically acceptable salts thereof, (c) at least one compound selected from Compound IV, and pharmaceutically acceptable salts of Compound IV, and (d) at least one pharmaceutically acceptable carrier.
- the disclosure provides a pharmaceutical composition
- a pharmaceutical composition comprising (a) Compound I as any one of the pharmaceutically acceptable solid forms disclosed herein or amorphous Compound I formulated as a solid (e.g., spray-dried) dispersion, (b) at least one compound selected from Compound II and pharmaceutically acceptable salts thereof, (c) at least one compound selected from Compound V, and pharmaceutically acceptable salts of Compound V, and (d) at least one pharmaceutically acceptable carrier.
- any pharmaceutical composition disclosed herein may comprise at least one pharmaceutically acceptable carrier.
- the at least one pharmaceutically acceptable carrier is selected from pharmaceutically acceptable vehicles and pharmaceutically acceptable adjuvants.
- the at least one pharmaceutically acceptable is selected from pharmaceutically acceptable fillers, disintegrants, surfactants, binders, lubricants.
- the solid form of Compound I used in the pharmaceutical compositions of the invention is Compound I Neat Form A.
- the crystalline form of Compound I used in the pharmaceutical compositions of the invention is Compound I Neat Form B.
- the solid form of Compound I used in the pharmaceutical compositions of the invention is Compound I Neat Form C.
- the solid form of Compound I used in the pharmaceutical compositions of the invention is Compound I Neat Form D.
- the solid form of Compound I used in the pharmaceutical compositions of the invention is Compound I Neat Form E.
- the solid form of Compound I used in the pharmaceutical compositions of the invention is Compound I Compressed Form A.
- the solid form of Compound I used in the pharmaceutical compositions of the invention is Compound I Compressed Form E.
- the solid form of Compound I used in the pharmaceutical compositions of the invention is Compound I EtOH Solvate Form B.
- the solid form of Compound I used in the pharmaceutical compositions of the invention is Compound I MeOH Solvate.
- the solid form of Compound I used in the pharmaceutical compositions of the invention is Compound I NPA Solvate.
- the solid form of Compound I used in the pharmaceutical compositions of the invention is Compound I MeOAc Solvate Form A.
- the solid form of Compound I used in the pharmaceutical compositions of the invention is Compound I MeOAc Solvate Form B.
- the solid form of Compound I used in the pharmaceutical compositions of the invention is Compound I MeOAc Solvate Form C.
- the solid form of Compound I used in the pharmaceutical compositions of the invention is Compound I Hydrate Form A.
- the solid form of Compound I used in the pharmaceutical compositions of the invention is Compound I Hydrate Form B.
- the solid form of Compound I used in the pharmaceutical compositions of the invention is Compound I Hydrate Form C.
- the solid form of Compound I used in the pharmaceutical compositions of the invention is Compound I EtOH Solvate Hydrate.
- the solid form of Compound I used in the pharmaceutical compositions of the invention is Compound I MeOH Solvate Hydrate.
- the solid form of Compound I used in the pharmaceutical compositions of the invention is Compound I IPA Solvate Hydrate. In some embodiments, the solid form of Compound I used in the pharmaceutical compositions of the invention is Compound I MeOAc Solvate Hydrate. In some embodiments, the solid form of Compound I used in the pharmaceutical compositions of the invention is amorphous Compound I.
- the form of Compound I used in the pharmaceutical compositions of the invention is Compound I in a solid (e.g., spray-dried) dispersion.
- compositions described herein are useful for treating cystic fibrosis and other CFTR-mediated diseases.
- pharmaceutical compositions disclosed herein may optionally further comprise at least one pharmaceutically acceptable carrier.
- the at least one pharmaceutically acceptable carrier may be selected from adjuvants and vehicles.
- the at least one pharmaceutically acceptable carrier includes any and all solvents, diluents, other liquid vehicles, dispersion aids, suspension aids, surface active agents, isotonic agents, thickening agents, emulsifying agents, preservatives, solid binders, and lubricants, as suited to the particular dosage form desired. Remington: The Science and Practice of Pharmacy, 21st edition, 2005, ed. D.B.
- Non-limiting examples of suitable pharmaceutically acceptable carriers include, but are not limited to, ion exchangers, alumina, aluminum stearate, lecithin, serum proteins (such as human serum albumin), buffer substances (such as phosphates, glycine, sorbic acid, and potassium sorbate), partial glyceride mixtures of saturated vegetable fatty acids, water, salts, and electrolytes (such as protamine sulfate, disodium hydrogen phosphate, potassium hydrogen phosphate, sodium chloride, and zinc salts), colloidal silica, magnesium trisilicate, polyvinyl pyrrolidone, polyacrylates, waxes, polyethylene-polyoxypropylene-block polymers, wool fat, sugars (such as lactose, glucose and sucrose), starches (such as corn starch and potato starch), cellulose and its derivatives (such as sodium carboxymethyl cellulose, ethyl cellulose and cellulose acetate), powdered tragacanth, malt, ge
- Compound I as substantially crystalline Compound I Neat Form A (i.e., wherein less than 15% of Compound I is in amorphous form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous form).
- X-ray powder diffractogram having (a) signals at 9.2 ⁇ 0.2 degrees two-theta, 14.1 ⁇ 0.2 degrees two-theta, and 15.9 ⁇ 0.2 degrees two-theta and (b) two or more two-theta values selected from 16.6 ⁇ 0.2 degrees two-theta, 13.0 ⁇ 0.2 degrees two-theta, 18.4 ⁇ 0.2 degrees two-theta, 10 3 ⁇ 0.2 degrees two-theta, and 22.6 ⁇ 0.2 degrees two- theta.
- the Compound I Neat Form A of any one of Embodiments 1 to 27, characterized by a monoclinic crystal system, aF2i2i2i space group, and unit cell dimensions, measured at 100 K on a Rigaku diffractometer equipped with Cu I ⁇ « radiation (X 1.54178 A) and an HP AD detector, of:
- Compound I as substantially crystalline Neat Form B (i.e., wherein less than 15% of Compound I is in amorphous form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous form).
- the Compound I Neat Form B of any one of Embodiments 30 to 32 characterized by an X-ray powder diffractogram having (a) a signal at 26 3 degrees two-theta and (b) signals at one or more two-theta values selected from: 11.1 degrees two-theta, 19.6 degrees two-theta, and 7.2 degrees two-theta.
- the Compound I Neat Form B of any one of Embodiments 30 to 32 characterized by an X-ray powder diffractogram having (a) a signal at 26.3 ⁇ 0.2 degrees two-theta and (b) signals at two or more two-theta values selected from: 11.1 ⁇ 0.2 degrees two- theta, 19.6 ⁇ 0.2 degrees two-theta, and 7.2 ⁇ 0.2 degrees two-theta.
- the Compound I Neat Form B of any one of Embodiments 30 to 32 characterized by an X-ray powder diffractogram having signals at two-theta values 26.3 ⁇ 0.2 degrees two-theta, 11.1 ⁇ 0.2 degrees two-theta, 19.6 ⁇ 0.2 degrees two-theta, and 7.2 ⁇ 0.2 degrees two-theta.
- the Compound I Neat Form B of any one of Embodiments 30 to 32 characterized by an X-ray powder diffractogram having (a) signals at two-theta values 26.3 ⁇ 0.2 degrees two-theta, 11.
- the Compound I Neat Form B of any one of Embodiments 30 to 32 characterized by an X-ray powder diffractogram having signals at two-theta values 26.3 ⁇ 0.2 degrees two-theta, 11 1 ⁇ 0.2 degrees two-theta, 19.6 ⁇ 0.2 degrees two-theta, 7.2 ⁇ 0.2 degrees two-theta, 18.5 ⁇ 0.2 degrees two-theta, 20.8 ⁇ 0.2 degrees two-theta, 14.3 ⁇ 0.2 degrees two-theta, and 17.1 ⁇ 0.2 degrees two-theta.
- the Compound I Neat Form B of any one of Embodiments 30 to 39 characterized by an X-ray powder diffractogram substantially similar to FIG. 5.
- the Compound I Neat Form B of any one of Embodiments 30 to 40 characterized by a 13 C ssNMR spectrum with a peak at 142.6 ⁇ 0.2 ppm.
- the Compound I Neat Form B of any one of Embodiments 30 to 40 characterized by a 13 C ssNMR spectrum with (a) a peak at 142.6 ⁇ 0.2 ppm and (b) one or more peaks selected from 27.1 ⁇ 0.2 ppm, 18.4 ⁇ 0.2 ppm, 134.9 ⁇ 0.2 ppm, 132.4 ⁇ 0.2 ppm,
- the Compound I Neat Form B of any one of Embodiments 30 to 40 characterized by a 13 C ssNMR spectrum with (a) a peak at 142.6 ⁇ 0.2 ppm and (b) three or more peaks selected from 27.1 ⁇ 0.2 ppm, 18.4 ⁇ 0.2 ppm, 134.9 ⁇ 0.2 ppm, 132.4 ⁇ 0.2 ppm, 128.2 ⁇ 0.2 ppm, 126.6 ⁇ 0.2 ppm, and 133.4 ⁇ 0.2 ppm.
- the Compound I Neat Form B of any one of Embodiments 30 to 40 characterized by a 13 C ssNMR spectrum with (a) a peak at 142.6 ⁇ 0.2 ppm and (b) four or more peaks selected from 27.1 ⁇ 0.2 ppm, 18.4 ⁇ 0.2 ppm, 134.9 ⁇ 0.2 ppm, 132.4 ⁇ 0.2 ppm,
- the Compound I Neat Form B of any one of Embodiments 30 to 40 characterized by a 13 C ssNMR spectrum with (a) a peak at 142.6 ⁇ 0.2 ppm and (b) five or more peaks selected from 27.1 ⁇ 0.2 ppm, 18.4 ⁇ 0.2 ppm, 134.9 ⁇ 0.2 ppm, 132.4 ⁇ 0.2 ppm, 128.2 ⁇ 0.2 ppm, 126.6 ⁇ 0.2 ppm, and 133.4 ⁇ 0.2 ppm.
- the Compound I Neat Form B of any one of Embodiments 30 to 40 characterized by a 13 C ssNMR spectrum with (a) a peak at 142.6 ⁇ 0.2 ppm and (b) six or more peaks selected from 27.1 ⁇ 0.2 ppm, 18.4 ⁇ 0.2 ppm, 134.9 ⁇ 0.2 ppm, 132.4 ⁇ 0.2 ppm, 128.2 ⁇ 0.2 ppm, 126.6 ⁇ 0.2 ppm, and 133.4 ⁇ 0.2 ppm.
- the Compound I Neat Form B of any one of Embodiments 30 to 40 characterized by a 13 C ssNMR spectrum with peaks at 142.6 ⁇ 0.2 ppm, 27.1 ⁇ 0.2 ppm, 18.4 ⁇ 0.2 ppm, 134.9 ⁇ 0.2 ppm, 132.4 ⁇ 0.2 ppm, 128.2 ⁇ 0.2 ppm, 126.6 ⁇ 0.2 ppm, and 133.4 ⁇ 0.2 ppm.
- the Compound I Neat Form B of any one of Embodiments 30 to 48 characterized by a 13 C ssNMR spectrum substantially similar to FIG. 6.
- Compound I as substantially crystalline Neat Form C (i.e., wherein less than 15% of Compound I is in amorphous form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous Form).
- the Compound I of Embodiment 51 wherein Compound I is 100% crystalline Compound I Neat Form C.
- the Compound I Neat Form C of any one of Embodiments 51 to 53 characterized by an X-ray powder diffractogram having a signal at 5.4 ⁇ 0.2 degrees two-theta.
- the Compound I Neat Form C of any one of Embodiments 51 to 53 characterized by an X-ray powder diffractogram having signals at (a) 5.4 ⁇ 0.2 degrees two-theta and (b) one or more of 10.2 ⁇ 0.2 degrees two-theta, 15.2 ⁇ 0.2 degrees two-theta, 12.6 ⁇ 0.2 degrees two-theta, 16.7 ⁇ 0.2 degrees two-theta, 11.7 ⁇ 0.2 degrees two-theta, 23.0 ⁇ 0.2 degrees two-theta, and 7.6 ⁇ 0.2 degrees two-theta.
- the Compound I Neat Form C of any one of Embodiments 51 to 53 characterized by an X-ray powder diffractogram having signals at (a) 5.4 ⁇ 0.2 degrees two-theta and (b) two or more of 10.2 ⁇ 0.2 degrees two-theta, 15.2 ⁇ 0.2 degrees two-theta, 12 6 ⁇ 0.2 degrees two-theta, 16.7 ⁇ 0.2 degrees two-theta, 11.7 ⁇ 0.2 degrees two-theta, 23.0 ⁇ 0.2 degrees two-theta, and 7.6 ⁇ 0.2 degrees two-theta.
- the Compound I Neat Form C of any one of Embodiments 51 to 53 characterized by an X-ray powder diffractogram having signals at (a) 5.4 ⁇ 0.2 degrees two-theta and (b) three or more of 10.2 ⁇ 0.2 degrees two-theta, 15.2 ⁇ 0.2 degrees two-theta, 12.6 ⁇ 0.2 degrees two-theta, 16.7 ⁇ 0.2 degrees two-theta, 11.7 ⁇ 0.2 degrees two-theta, 23.0 ⁇ 0.2 degrees two-theta, and 7.6 ⁇ 0.2 degrees two-theta.
- the Compound I Neat Form C of any one of Embodiments 51 to 53 characterized by an X-ray powder diffractogram having signals at (a) 5.4 ⁇ 0.2 degrees two-theta and (b) four or more of 10.2 ⁇ 0.2 degrees two-theta, 15.2 ⁇ 0.2 degrees two-theta, 12.6 ⁇ 0.2 degrees two-theta, 16.7 ⁇ 0.2 degrees two-theta, 11.7 ⁇ 0.2 degrees two-theta, 23.0 ⁇ 0.2 degrees two-theta, and 7.6 ⁇ 0.2 degrees two-theta.
- the Compound I Neat Form C of any one of Embodiments 51 to 53 characterized by an X-ray powder diffractogram having signals at (a) 5.4 ⁇ 0.2 degrees two-theta and (b) five or more of 10.2 ⁇ 0.2 degrees two-theta, 15.2 ⁇ 0.2 degrees two-theta, 12.6 ⁇ 0.2 degrees two-theta, 16.7 ⁇ 0.2 degrees two-theta, 11.7 ⁇ 0.2 degrees two-theta, 23.0 ⁇ 0.2 degrees two-theta, and 7.6 ⁇ 0.2 degrees two-theta.
- the Compound I Neat Form C of any one of Embodiments 51 to 53 characterized by an X-ray powder diffractogram having signals at (a) 5.4 ⁇ 0.2 degrees two-theta and (b) six or more of 10.2 ⁇ 0.2 degrees two-theta, 15.2 ⁇ 0.2 degrees two-theta, 12.6 ⁇ 0.2 degrees two-theta, 16.7 ⁇ 0.2 degrees two-theta, 11.7 ⁇ 0.2 degrees two-theta, 23.0 ⁇ 0.2 degrees two-theta, and 7.6 ⁇ 0.2 degrees two-theta.
- the Compound I Neat Form C of any one of Embodiments 51 to 53 characterized by an X-ray powder diffractogram having signals at 5.4 ⁇ 0.2 degrees two-theta, 10.2 ⁇ 0.2 degrees two-theta, 15 2 ⁇ 0.2 degrees two-theta, 12.6 ⁇ 0.2 degrees two-theta, 16.7 ⁇ 0.2 degrees two-theta, 11.7 ⁇ 0.2 degrees two-theta, 23.0 ⁇ 0.2 degrees two-theta, and 7.6 ⁇ 0.2 degrees two-theta.
- the Compound I Neat Form C of any one of Embodiments 51 to 61 characterized by an X-ray powder diffractogram substantially similar to FIG. 7.
- the Compound I Neat Form C of any one of Embodiments 51 to 62 characterized by a TGA showing negligible weight loss from ambient temperature up to about 40 °C and about 0.16% weight loss from 40 °C to 240 °C.
- the Compound I Neat Form C of any one of Embodiments 51 to 63 characterized by TGA data substantially similar to FIG. 8.
- the Compound I Neat Form C of any one of Embodiments 51 to 64 characterized by a DSC analysis showing an endothermic peak at about 237 °C.
- the Compound I Neat Form C of any one of Embodiments 51 to 65 characterized by a DSC analysis substantially similar to FIG. 9.
- the Compound I Neat Form C of any one of Embodiments 51 to 66 characterized by a 13 C ssNMR spectrum with peaks at 43.8 ⁇ 0.2 ppm and 132.3 ⁇ 0.2 ppm.
- the Compound I Neat Form C of any one of Embodiments 51 to 67 characterized by a 13 C ssNMR spectrum with (a) peaks at 43.8 ⁇ 0.2 ppm and 132.3 ⁇ 0.2 ppm and (b) one or more peaks selected from 28.4 ⁇ 0.2 ppm, 29.4 ⁇ 0.2 ppm, 129.1 ⁇ 0.2 ppm, 135.1 ⁇ 0.2 ppm, 139.6 ⁇ 0.2 ppm, and 28.2 ⁇ 0.2 ppm.
- the Compound I Neat Form C of any one of Embodiments 51 to 68 characterized by a 13 C ssNMR spectrum with (a) peaks at 43.8 ⁇ 0.2 ppm and 132.3 ⁇ 0.2 ppm and (b) two or more peaks selected from 28.4 ⁇ 0.2 ppm, 29.4 ⁇ 0.2 ppm, 129.1 ⁇ 0.2 ppm,
- the Compound I Neat Form D of any one of Embodiments 76 to 78 characterized by a 13 C ssNMR spectrum with (a) a peak at 50.8 ⁇ 0.2 ppm and (b) one or more peaks selected from 28.3 ⁇ 0.2 ppm, 127.2 ⁇ 0.2 ppm, 129.8 ⁇ 0.2 ppm, 170.6 ⁇ 0.2 ppm.
- the Compound I Neat Form D of any one of Embodiments 76 to 78 characterized by a 13 C ssNMR spectrum with (a) a peak at 50.8 ⁇ 0.2 ppm and (b) three or more peaks selected from 28.3 ⁇ 0.2 ppm, 127.2 ⁇ 0.2 ppm, 129.8 ⁇ 0.2 ppm, 170.6 ⁇ 0.2 ppm.
- the Compound I Neat Form D of any one of Embodiments 76 to 78 characterized by a 13 C ssNMR spectrum with peaks at 50.8 ⁇ 0.2 ppm, 28.3 ⁇ 0.2 ppm, 127.2 ⁇ 0.2 ppm, 129.8 ⁇ 0.2 ppm, 170.6 ⁇ 0.2 ppm.
- the Compound I Neat Form D of any one of Embodiments 76 to 78 characterized by a 13 C ssNMR spectrum with (a) peaks at 50.8 ⁇ 0.2 ppm, 28.3 ⁇ 0.2 ppm, 127.2 ⁇ 0.2 ppm, 129.8 ⁇ 0.2 ppm, and 170.6 ⁇ 0.2 ppm and (b) one or more peaks selected from 135.3 ⁇ 0.2 ppm, 136.8 ⁇ 0.2 ppm, and 129.2 ⁇ 0.2 ppm.
- the Compound I Neat Form D of any one of Embodiments 76 to 78 characterized by a 13 C ssNMR spectrum with (a) peaks at 50.8 ⁇ 0.2 ppm, 28.3 ⁇ 0.2 ppm, 127.2 ⁇ 0.2 ppm, 129.8 ⁇ 0 2 ppm, and 170.6 ⁇ 02 ppm and (b) two or more peaks selected from 135.3 ⁇ 0.2 ppm, 136.8 ⁇ 0.2 ppm, and 129.2 ⁇ 0.2 ppm.
- the Compound I Neat Form D of any one of Embodiments 76 to 78 characterized by a 13 C ssNMR spectrum with peaks at 50.8 ⁇ 0.2 ppm, 28.3 ⁇ 0.2 ppm, 127.2 ⁇ 0.2 ppm, 129.8 ⁇ 0.2 ppm, 170.6 ⁇ 0.2 ppm, 135.3 ⁇ 0.2 ppm, 136.8 ⁇ 0.2 ppm, and 129.2 ⁇ 0.2 ppm
- the Compound I Neat Form D of any one of Embodiments 76 to 86 characterized by a 13 C ssNMR spectrum substantially similar to FIG. 11.
- Compound I as substantially crystalline Neat Form E i.e., wherein less than 15% of Compound I is in amorphous form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous Form).
- the Compound I of Embodiment 90 wherein Compound I is 100% crystalline Compound I Neat Form E.
- Substantially pure Compound I Neat Form E The Compound I Neat Form E of any one of Embodiments 90 to 92, characterized by an X-ray powder diffractogram having (a) a signal at 13.4 ⁇ 0.2 degrees two-theta and (b) a signal at 14.7 ⁇ 0.2 degrees two-theta or 19.6 ⁇ 0.2 degrees two-theta.
- the Compound I Neat Form E of any one of Embodiments 90 to 92 characterized by an X-ray powder diffractogram having signals at 13.4 ⁇ 0.2 degrees two-theta, 14.7 ⁇ 0.2 degrees two-theta, and 19.6 ⁇ 0.2 degrees two-theta
- the Compound I Neat Form E of any one of Embodiments 90 to 92 characterized by an X-ray powder diffractogram having (a) signals at 13.4 ⁇ 0.2 degrees two-theta,
- the Compound I Neat Form E of any one of Embodiments 90 to 92 characterized by an X-ray powder diffractogram having (a) signals at 13.4 ⁇ 0.2 degrees two-theta,
- the Compound I Neat Form E of any one of Embodiments 90 to 92 characterized by an X-ray powder diffractogram having signals at (a) signals at 13.4 ⁇ 0.2 degrees two-theta, 14.7 ⁇ 0.2 degrees two-theta, and 19.6 ⁇ 0.2 degrees two-theta and (b) three or more two-theta values selected from 10.3 degrees two-theta, 8.5 degrees two- theta, 16.0 degrees two-theta, 16.2 degrees two-theta, and 21.4 degrees two-theta.
- the Compound I Neat Form E of any one of Embodiments 90 to 92 characterized by an X-ray powder diffractogram having (a) signals at 13.4 ⁇ 0.2 degrees two-theta,
- the Compound I Neat Form E of any one of Embodiments 90 to 99 characterized by an X-ray powder diffractogram substantially similar to FIG. 12.
- the Compound I Neat Form E of any one of Embodiments 90 to 100 characterized by a TGA showing negligible weight loss from 30 °C to 120 °C.
- the Compound I Neat Form E of any one of Embodiments 90 to 101 characterized by TGA data substantially similar to FIG. 13.
- the Compound I Neat Form E of any one of Embodiments 90 to 102 characterized by a DSC analysis showing an endothermic peak at about 212 °C.
- the Compound I Neat Form E of any one of Embodiments 90 to 103 characterized by a DSC analysis substantially similar to FIG. 14.
- the Compound I Neat Form E of any one of Embodiments 90 to 104 characterized by a 13 C ssNMR spectrum with peaks at 20.5 ⁇ 0.2 ppm and 43.6 ⁇ 0.2 ppm.
- the Compound I Neat Form E of any one of Embodiments 90 to 104 characterized by a 13 C ssNMR spectrum with (a) peaks at 20.5 ⁇ 0.2 ppm and 43.6 ⁇ 0.2 ppm and (b) one or more peaks selected from 28.3 ⁇ 0.2 ppm, 43.6 ⁇ 0.2 ppm, 20.5 ⁇ 0.2 ppm, 135.0 ⁇ 0.2 ppm, 128.2 ⁇ 0.2 ppm, and 139.6 ⁇ 0.2 ppm.
- the Compound I Neat Form E of any one of Embodiments 90 to 104 characterized by a 13 C ssNMR spectrum with (a) peaks at 20.5 ⁇ 0.2 ppm and 43.6 ⁇ 0.2 ppm and (b) two or more peaks selected from 28.3 ⁇ 0.2 ppm, 43.6 ⁇ 0.2 ppm, 20.5 ⁇ 0.2 ppm, 135.0 ⁇ 0.2 ppm, 128.2 ⁇ 0.2 ppm, and 139.6 ⁇ 0.2 ppm.
- the Compound I Neat Form E of any one of Embodiments 90 to 104 characterized by a 13 C ssNMR spectrum with (a) peaks at 20.5 ⁇ 0.2 ppm and 43.6 ⁇ 0.2 ppm and (b) three or more peaks selected from 28.3 ⁇ 0.2 ppm, 43.6 ⁇ 0.2 ppm, 20.5 ⁇ 0.2 ppm, 135.0 ⁇ 0.2 ppm, 128.2 ⁇ 0.2 ppm, and 139.6 ⁇ 0.2 ppm.
- the Compound I Neat Form E of any one of Embodiments 90 to 104 characterized by a 13 C ssNMR spectrum with (a) peaks at 20.5 ⁇ 0.2 ppm and 43.6 ⁇ 0.2 ppm and (b) four or more peaks selected from 28.3 ⁇ 0.2 ppm, 43.6 ⁇ 0.2 ppm, 20.5 ⁇ 0.2 ppm, 135.0 ⁇ 0.2 ppm, 128.2 ⁇ 0.2 ppm, and 139.6 ⁇ 0.2 ppm.
- the Compound I Neat Form E of any one of Embodiments 90 to 104 characterized by a 13 C ssNMR spectrum with (a) peaks at 20.5 ⁇ 0.2 ppm and 43.6 ⁇ 0.2 ppm and (b) five or more peaks selected from 28.3 ⁇ 0.2 ppm, 43.6 ⁇ 0.2 ppm, 20.5 ⁇ 0.2 ppm, 135.0 ⁇ 0.2 ppm, 128.2 ⁇ 0 2 ppm, and 139.6 ⁇ 02 ppm.
- the Compound I Neat Form E of any one of Embodiments 90 to 104 characterized by a 13 C ssNMR spectrum with peaks at 20.5 ⁇ 0.2 ppm, 43.6 ⁇ 0.2 ppm, 28.3 ⁇ 0.2 ppm, 43.6 ⁇ 0.2 ppm, 20.5 ⁇ 0.2 ppm, 135.0 ⁇ 0.2 ppm, 128.2 ⁇ 0.2 ppm, and 139.6 ⁇ 0.2 ppm.
- the Compound I Neat Form E of any one of Embodiments 90 to 111 characterized by a 13 C ssNMR spectrum substantially similar to FIG. 14.
- the Compound I Neat Form E of any one of Embodiments 90 to 113 prepared by a process comprising (i) stirring of Compound I Neat Form A in MeOAc, and (ii) isolation of the solid via centrifugation, and (iii) drying of the solid at about 50 °C.
- Compound I as substantially crystalline compressed Form E (i.e., wherein less than 15% of Compound I is in amorphous form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous Form).
- the Compound I of Embodiment 115 wherein Compound I is 100% crystalline Compound I compressed Form E.
- the Compound I compressed Form E of any one of Embodiments 115 to 117 characterized by an X-ray powder diffractogram having (a) signals at two-theta values
- the Compound I compressed Form E of any one of Embodiments 115 to 117 characterized by an X-ray powder diffractogram having signals at two-theta values 7.5 ⁇ 0.2 degrees two-theta, 20.8 ⁇ 0.2 degrees two-theta, 16.1 ⁇ 0.2 degrees two- theta, 8.6 ⁇ 0.2 degrees two-theta, 18.4 ⁇ 0.2 degrees two-theta, 20.1 ⁇ 0.2 degrees two-theta, 14.2 ⁇ 0.2 degrees two-theta, and 23.1 ⁇ 0.2 degrees two-theta.
- the Compound I compressed Form E of any one of Embodiments 115 to 123 characterized by an X-ray powder diffractogram substantially similar to FIG. 16.
- the Compound I compressed Form E of any one of Embodiments 115 to 124 characterized by a 13 C ssNMR spectrum with peaks at 41.8 ⁇ 0.2 ppm and 135.7 ⁇ 0.2 ppm.
- the Compound I compressed Form E of any one of Embodiments 115 to 124 characterized by a 13 C ssNMR spectrum with (a) peaks at 41.8 ⁇ 0.2 ppm and 135.7 ⁇ 0.2 ppm and (b) one or more peaks selected from 28.2 ⁇ 0.2 ppm, 22.0 ⁇ 0.2 ppm,
- the Compound I compressed Form E of any one of Embodiments 115 to 124 characterized by a 13 C ssNMR spectrum with peaks at 41.8 ⁇ 0.2 ppm, 135.7 ⁇ 0.2 ppm, 28.2 ⁇ 0.2 ppm, 22.0 ⁇ 0.2 ppm, 129.8 ⁇ 0.2 ppm, 136.2 ⁇ 0.2 ppm, 141.1 ⁇ 0.2 ppm, and 127.3 ⁇ 0.2 ppm.
- the Compound I compressed Form E of any one of Embodiments 115 to 131 characterized by a 13 C ssNMR spectrum substantially similar to FIG. 17.
- Compound I as substantially crystalline compressed Form A (i.e., wherein less than 15% of Compound I is in amorphous form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous Form).
- the Compound I of Embodiment 134 wherein Compound I is 100% crystalline Compound I Compressed Form A. Substantially pure Compound I Compressed Form A.
- the Compound I Compressed Form A of any one of Embodiments 134 to 136 characterized by an X-ray powder diffractogram having signals at (a) 8.9 ⁇ 0.2 degrees two-theta, 15.2 ⁇ 0.2 degrees two-theta, 15.8 ⁇ 0.2 degrees two-theta and (b) one or more selected from 18.2 ⁇ 0.2 degrees two-theta, 20.9 ⁇ 0.2 degrees two-theta, and 16.7 ⁇ 0.2 degrees two-theta.
- the Compound I Compressed Form A of any one of Embodiments 134 to 136 characterized by an X-ray powder diffractogram having signals at (a) 8.9 ⁇ 0.2 degrees two-theta, 15.2 ⁇ 0.2 degrees two-theta, 15.8 ⁇ 0.2 degrees two-theta and (b) two or more selected from 18.2 ⁇ 0.2 degrees two-theta, 20.9 ⁇ 0.2 degrees two-theta, and 16.7 ⁇ 0.2 degrees two-theta.
- the Compound I Compressed Form A of any one of Embodiments 134 to 136 characterized by an X-ray powder diffractogram having signals at 8.9 ⁇ 0.2 degrees two-theta, 15.2 ⁇ 0.2 degrees two-theta, 15.8 ⁇ 0.2 degrees two-theta, 18.2 ⁇ 0.2 degrees two-theta, 20.9 ⁇ 0.2 degrees two-theta, and 16.7 ⁇ 0.2 degrees two-theta.
- the Compound I Compressed Form A of any one of Embodiments 134 to 136 characterized by an X-ray powder diffractogram having signals at (a) 8.9 ⁇ 0.2 degrees two-theta, 15.2 ⁇ 0.2 degrees two-theta, 15.8 ⁇ 0.2 degrees two-theta, 18.2 ⁇ 0.2 degrees two-theta, 20.9 ⁇ 0.2 degrees two-theta, and 16.7 ⁇ 0.2 degrees two-theta and (b) 21.5 ⁇ 0.2 degrees two-theta or 23.0 ⁇ 0.2 degrees two-theta.
- the Compound I Compressed Form A of any one of Embodiments 134 to 136 characterized by an X-ray powder diffractogram having signals at 8.9 ⁇ 0.2 degrees two-theta, 15.2 ⁇ 0.2 degrees two-theta, 15.8 ⁇ 0.2 degrees two-theta, 18.2 ⁇ 0.2 degrees two-theta, 20.9 ⁇ 0.2 degrees two-theta, and 16.7 ⁇ 0.2 degrees two-theta, 21.5 ⁇ 0.2 degrees two-theta, and 23.0 ⁇ 0.2 degrees two-theta.
- the Compound I Compressed Form A of any one of Embodiments 134 to 141 characterized by an X-ray powder diffractogram substantially similar to FIG. 18.
- Compressed Form A of any one of Embodiments 134 to 142 characterized by a 13 C ssNMR spectrum with (a) a peak at 28.9 ⁇ 0.2 ppm and (b) one or more peaks selected from 41.9 ⁇ 0.2 ppm, 21.6 ⁇ 0.2 ppm, 132.9 ⁇ 0.2 ppm, and 127.5 ⁇ 0.2 ppm.
- the Compound I Compressed Form A of any one of Embodiments 134 to 142, characterized by a 13 C ssNMR spectrum with (a) a peak at 28.9 ⁇ 0.2 ppm and (b) two or more peaks selected from 41.9 ⁇ 0.2 ppm, 21.6 ⁇ 0.2 ppm, 132.9 ⁇ 0.2 ppm, and 127.5 ⁇ 0.2 ppm.
- the Compound I Compressed Form A of any one of Embodiments 134 to 142, characterized by a 13 C ssNMR spectrum with (a) a peak at 28.9 ⁇ 0.2 ppm and (b) three or more peaks selected from 41.9 ⁇ 0.2 ppm, 21.6 ⁇ 0.2 ppm, 132 9 ⁇ 0.2 ppm, and 127.5 ⁇ 0.2 ppm.
- the Compound I Compressed Form A of any one of Embodiments 134 to 142, characterized by a 13 C ssNMR spectrum with peaks at 28.9 ⁇ 0.2 ppm, 41.9 ⁇ 0.2 ppm, 21.6 ⁇ 0.2 ppm, 132.9 ⁇ 0.2 ppm, and 127.5 ⁇ 0.2 ppm.
- the Compound I Compressed Form A of any one of Embodiments 134 to 142, characterized by a 13 C ssNMR spectrum with peaks at (a) 28.9 ⁇ 0.2 ppm, 41.9 ⁇ 0.2 ppm, 21.6 ⁇ 0.2 ppm, 132.9 ⁇ 0.2 ppm, and 127.5 ⁇ 0.2 ppm and (b) one or more selected from 130.7 ⁇ 0.2 ppm, 133.7 ⁇ 0.2 ppm, and 22.5 ⁇ 0.2 ppm.
- the Compound I Compressed Form A of any one of Embodiments 134 to 142, characterized by a 13 C ssNMR spectrum with peaks at (a) 28.9 ⁇ 0.2 ppm, 41.9 ⁇ 0.2 ppm, 21.6 ⁇ 0.2 ppm, 132.9 ⁇ 0.2 ppm, and 127.5 ⁇ 0.2 ppm and (b) two or more selected from 130.7 ⁇ 0.2 ppm, 133.7 ⁇ 0.2 ppm, and 22.5 ⁇ 0.2 ppm.
- the Compound I Compressed Form A of any one of Embodiments 134 to 142 characterized by a 13 C ssNMR spectrum with peaks at 28.9 ⁇ 0.2 ppm, 41.9 ⁇ 0.2 ppm, 21.6 ⁇ 0.2 ppm, 132.9 ⁇ 0.2 ppm, 127.5 ⁇ 0.2 ppm, 130.7 ⁇ 0.2 ppm, 133.7 ⁇ 0.2 ppm, and 22.5 ⁇ 0.2 ppm.
- the Compound I Compressed Form A of any one of Embodiments 134 to 149 characterized by a 13 C ssNMR spectrum substantially similar to FIG. 18.
- the Compound I Compressed Form A of any one of Embodiments 134 to 150 prepared by a process comprising mechanical compression of Compound I Neat Form A.
- Compound I as substantially crystalline EtOH Solvate Form B (i.e., wherein less than 15% of Compound I is in amorphous Form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous form).
- the Compound I of Embodiment 152 wherein Compound I is 100% crystalline Compound I EtOH Solvate Form B. Substantially pure Compound I EtOH Solvate Form B.
- the Compound I EtOH Solvate Form B of any one of Embodiments 152 to 154 characterized by an X-ray powder diffractogram having a signal at one or more two- theta values selected from 6.4 ⁇ 0.2 degrees two-theta, 10.8 ⁇ 0.2 degrees two-theta,
- the Compound I EtOH Solvate Form B of any one of Embodiments 152 to 164 characterized by TGA data substantially similar to FIG. 21
- the Compound I EtOH Solvate Form B of any one of Embodiments 152 to 165 characterized by a DSC analysis showing an endothermic peak at about 109 °C and at about 197 °C.
- the Compound I EtOH Solvate Form B of any one of Embodiments 152 to 166 characterized by a DSC analysis substantially similar to FIG. 22.
- the Compound I EtOH Solvate Form B of any one of Embodiments 152 to 167 characterized by a 13 C ssNMR spectrum with one or more peaks selected from: 28.1 ⁇ 0.2 ppm, 18.7 ⁇ 0.2 ppm, 44.1 ⁇ 0.2 ppm, 128.9 ⁇ 0.2 ppm, 21.6 ⁇ 0.2 ppm, 142.8 ⁇ 0.2 ppm, 132.0 ⁇ 0.2 ppm and 130.4.
- the Compound I EtOH Solvate Form B of any one of Embodiments 152 to 168 characterized by a 13 C ssNMR spectrum with two or more peaks selected from: 28.1 ⁇ 0.2 ppm, 18.7 ⁇ 0.2 ppm, 44.1 ⁇ 0.2 ppm, 128.9 ⁇ 0.2 ppm, 21.6 ⁇ 0.2 ppm, 142.8 ⁇ 0.2 ppm, 132.0 ⁇ 0.2 ppm and 130.4.
- the Compound I EtOH Solvate Form B of any one of Embodiments 152 to 169 characterized by a 13 C ssNMR spectrum with three or more peaks selected from: 28.1 ⁇ 0.2 ppm, 18.7 ⁇ 0.2 ppm, 44.1 ⁇ 0.2 ppm, 128.9 ⁇ 0.2 ppm, 21.6 ⁇ 0.2 ppm, 142.8 ⁇ 0.2 ppm, 132.0 ⁇ 0.2 ppm and 130.4.
- the Compound I EtOH Solvate Form B of any one of Embodiments 152 to 170 characterized by a 13 C ssNMR spectrum with four or more peaks selected from: 28.1 ⁇ 0.2 ppm, 18.7 ⁇ 0.2 ppm, 44.1 ⁇ 0.2 ppm, 128.9 ⁇ 0.2 ppm, 21.6 ⁇ 0.2 ppm, 142.8 ⁇ 0.2 ppm, 132.0 ⁇ 0.2 ppm and 130.4.
- the Compound I EtOH Solvate Form B of any one of Embodiments 152 to 172 characterized by a 13 C ssNMR spectrum with six or more peaks selected from: 28.1 ⁇ 0.2 ppm, 18.7 ⁇ 0.2 ppm, 44.1 ⁇ 0.2 ppm, 128.9 ⁇ 0.2 ppm, 21.6 ⁇ 0.2 ppm, 142.8 ⁇ 0.2 ppm, 132 0 ⁇ 0.2 ppm and 130.4.
- the Compound I EtOH Solvate Form B of any one of Embodiments 152 to 174 characterized by a 13 C ssNMR spectrum with peaks at: 28.1 ⁇ 0.2 ppm, 18.7 ⁇ 0.2 ppm, 44.1 ⁇ 0.2 ppm, 128.9 ⁇ 0.2 ppm, 21.6 ⁇ 0.2 ppm, 142.8 ⁇ 0.2 ppm, 132.0 ⁇ 0.2 ppm and 130.4.
- the Compound I EtOH Solvate Form B of any one of Embodiments 152 to 177 prepared by a process comprising (i) stirring of Compound I in ethanol, and (ii) isolation of the solid via centrifugation or vacuum filtration.
- Compound I as substantially crystalline MeOH Solvate (i.e., wherein less than 15% of Compound I is in amorphous Form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous form).
- the Compound I of Embodiment 179 wherein Compound I is 100% crystalline Compound I MeOH Solvate.
- Substantially pure Compound I MeOH Solvate The Compound I MeOH Solvate of any one of Embodiments 179 to 181, characterized by an X-ray powder diffractogram having a signal at one or more two- theta values selected from 26.1 ⁇ 0.2 degrees two-theta, 12.2 ⁇ 0.2 degrees two-theta,
- the Compound I MeOH Solvate of any one of Embodiments 179 to 184 characterized by an X-ray powder diffractogram having signals at 26.1 ⁇ 0.2 degrees two-theta, 12.2 ⁇ 0.2 degrees two-theta, 22.8 ⁇ 0.2 degrees two-theta, and 21.1 ⁇ 0.2 degrees two-theta.
- the Compound I MeOH Solvate of any one of Embodiments 179 to 185 characterized by an X-ray powder diffractogram substantially similar to FIG. 24.
- the Compound I MeOH Solvate of any one of Embodiments 179 to 196 prepared by a process comprising (i) dissolving Compound I in MeOH and increasing temperature to about 62 °C, (ii) cooling the solution to about 10 °C, and (iii) isolation of the solid via centrifugation.
- Compound I as substantially crystalline NPA Solvate (i.e., wherein less than 15% of Compound I is in amorphous Form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous form).
- the Compound I NPA Solvate of any one of Embodiments 198 to 200 characterized by an X-ray powder diffractogram having a signal at one or more two-theta values selected from 10.6 ⁇ 0.2 degrees two-theta, 6.3 ⁇ 0.2 degrees two-theta, 19.1 ⁇ 0.2 degrees two-theta, 19.6 ⁇ 0.2 degrees two-theta, 17 3 ⁇ 0.2 degrees two-theta, 17.2 ⁇ 0.2 degrees two-theta, 10.0 ⁇ 0.2 degrees two-theta, and 14.0 ⁇ 0.2 degrees two-theta.
- the Compound I NPA Solvate of any one of Embodiments 198 to 201 characterized by an X-ray powder diffractogram having signals at two or more two-theta values selected from 10.6 ⁇ 0.2 degrees two-theta, 6.3 ⁇ 0.2 degrees two-theta, 19.1 ⁇ 0.2 degrees two-theta, 19.6 ⁇ 0.2 degrees two-theta, 17 3 ⁇ 0.2 degrees two-theta, 17.2 ⁇ 0.2 degrees two-theta, 10.0 ⁇ 0.2 degrees two-theta, and 14.0 ⁇ 0.2 degrees two-theta.
- the Compound I NPA Solvate of any one of Embodiments 198 to 202 characterized by an X-ray powder diffractogram having signals at three or more two-theta values selected from 10.6 ⁇ 0.2 degrees two-theta, 6.3 ⁇ 0.2 degrees two-theta, 19.1 ⁇ 0.2 degrees two-theta, 19.6 ⁇ 0.2 degrees two-theta, 17.3 ⁇ 0.2 degrees two-theta, 17.2 ⁇ 0.2 degrees two-theta, 10.0 ⁇ 0.2 degrees two-theta, and 14.0 ⁇ 0.2 degrees two-theta.
- the Compound I NPA Solvate of any one of Embodiments 198 to 203 characterized by an X-ray powder diffractogram having signals at four or more two-theta values selected from 10.6 ⁇ 0.2 degrees two-theta, 6.3 ⁇ 0.2 degrees two-theta, 19.1 ⁇ 0.2 degrees two-theta, 19.6 ⁇ 0.2 degrees two-theta, 17.3 ⁇ 0.2 degrees two-theta, 17.2 ⁇ 0.2 degrees two-theta, 10.0 ⁇ 0.2 degrees two-theta, and 14.0 ⁇ 0.2 degrees two-theta.
- the Compound I NPA Solvate of any one of Embodiments 198 to 204 characterized by an X-ray powder diffractogram having signals at five or more two-theta values selected from 10.6 ⁇ 0.2 degrees two-theta, 6.3 ⁇ 0.2 degrees two-theta, 19.1 ⁇ 0.2 degrees two-theta, 19.6 ⁇ 0.2 degrees two-theta, 17.3 ⁇ 0.2 degrees two-theta, 17.2 ⁇ 0.2 degrees two-theta, 10.0 ⁇ 0.2 degrees two-theta, and 14.0 ⁇ 0.2 degrees two-theta.
- the Compound I NPA Solvate of any one of Embodiments 198 to 205 characterized by an X-ray powder diffractogram having signals at six or more two-theta values selected from 10.6 ⁇ 0.2 degrees two-theta, 6.3 ⁇ 0.2 degrees two-theta, 19.1 ⁇ 0.2 degrees two-theta, 19.6 ⁇ 0.2 degrees two-theta, 17.3 ⁇ 0.2 degrees two-theta, 17.2 ⁇ 0.2 degrees two-theta, 10 0 ⁇ 0.2 degrees two-theta, and 14 0 ⁇ 0.2 degrees two-theta.
- the Compound I NPA Solvate of any one of Embodiments 198 to 206 characterized by an X-ray powder diffractogram having signals at seven or more two-theta values selected from 10.6 ⁇ 0.2 degrees two-theta, 6.3 ⁇ 0.2 degrees two-theta, 19.1 ⁇ 0.2 degrees two-theta, 19.6 ⁇ 0.2 degrees two-theta, 17.3 ⁇ 0.2 degrees two-theta, 17.2 ⁇ 0.2 degrees two-theta, 10 0 ⁇ 0.2 degrees two-theta, and 14 0 ⁇ 0.2 degrees two-theta.
- the Compound I NPA Solvate of any one of Embodiments 198 to 207 characterized by an X-ray powder diffractogram having signals at 10.6 ⁇ 0.2 degrees two-theta, 6.3 ⁇ 0.2 degrees two-theta, 19.1 ⁇ 0.2 degrees two-theta, 19.6 ⁇ 0.2 degrees two-theta, 17.3 ⁇ 0.2 degrees two-theta, 17.2 ⁇ 0.2 degrees two-theta, 10.0 ⁇ 0.2 degrees two- theta, and 14.0 ⁇ 0.2 degrees two-theta.
- the Compound I NPA Solvate of any one of Embodiments 198 to 208 characterized by an X-ray powder diffractogram substantially similar to FIG. 26.
- the Compound I NPA Solvate of any one of Embodiments 198 to 216 characterized by a 13 C ssNMR spectrum with peaks at: 28.0 ⁇ 0.2 ppm, 30.0 ⁇ 0.2 ppm, 128.7 ⁇ 0.2 ppm, 44.0 ⁇ 0.2 ppm, 130.0 ⁇ 0.2 ppm, 131.0 ⁇ 0.2 ppm, 12.7 ⁇ 0.2 ppm, and 10.8 ⁇ 0.2 ppm.
- the Compound I NPA Solvate of any one of Embodiments 198 to 217 characterized by a 13 C ssNMR spectrum substantially similar to FIG. 27.
- Compound I as substantially crystalline MeOAc Solvate Form A (i.e., wherein less than 15% of Compound I is in amorphous form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous form).
- the Compound I MeOAc Solvate Form A of any one of Embodiments 220 to 222 characterized by an X-ray powder diffractogram having (a) a signal at 8.5 ⁇ 0.2 degrees two-theta and (b) a signal at one or more two-theta values selected from 19.9 ⁇ 0.2 degrees two-theta, 21.3 ⁇ 0.2 degrees two-theta, and 10.1 ⁇ 0.2 degrees two- theta.
- the Compound I MeOAc Solvate Form A of any one of Embodiments 220 to 222 characterized by an X-ray powder diffractogram having (a) a signal at 8.5 ⁇ 0.2 degrees two-theta and (b) a signal at two or more two-theta values selected from 19.9 ⁇ 0.2 degrees two-theta, 21.3 ⁇ 0.2 degrees two-theta, and 10.1 ⁇ 0.2 degrees two- theta.
- the Compound I MeOAc Solvate Form A of any one of Embodiments 220 to 222 characterized by an X-ray powder diffractogram having signals at 8.5 ⁇ 0.2 degrees two-theta, 19.9 ⁇ 0.2 degrees two-theta, 21.3 ⁇ 0.2 degrees two-theta, and 10.1 ⁇ 0.2 degrees two-theta.
- the Compound I MeOAc Solvate Form A of any one of Embodiments 220 to 222 characterized by an X-ray powder diffractogram having signals at (a) signals at 8.5 ⁇ 0.2 degrees two-theta, 19.9 ⁇ 0.2 degrees two-theta, 21.3 ⁇ 0.2 degrees two-theta, and
- the Compound I MeOAc Solvate Form A of any one of Embodiments 220 to 222 characterized by an X-ray powder diffractogram having signals at 8.5 ⁇ 0.2 degrees two-theta, 19.9 ⁇ 0.2 degrees two-theta, 21.3 ⁇ 0.2 degrees two-theta, and 10.1 ⁇ 0.2 degrees two-theta, 15.9 ⁇ 0.2 degrees two-theta, and 13.2 ⁇ 0.2 degrees two-theta.
- the Compound I MeOAc Solvate Form A of any one of Embodiments 220 to 222 characterized by an X-ray powder diffractogram having signals at 8.5 ⁇ 0.2 degrees two-theta, 19 9 ⁇ 0.2 degrees two-theta, 21.3 ⁇ 0.2 degrees two-theta, and 10.1 ⁇ 0.2 degrees two-theta, 15.9 ⁇ 0.2 degrees two-theta, 13.2 ⁇ 0.2 degrees two-theta, 16.0 ⁇ 0.2 degrees two-theta, and 19.4 ⁇ 0.2 degrees two-theta.
- the Compound I MeOAc Solvate Form A of any one of Embodiments 220 to 229 characterized by a TGA showing 1.68% weight loss from 30 °C to 120 °C.
- the Compound I MeOAc Solvate Form A of any one of Embodiments 220 to 230 characterized by TGA data substantially similar to FIG. 29.
- the Compound I MeOAc Solvate Form A of any one of Embodiments 220 to 232 characterized by a DSC analysis substantially similar to FIG. 30.
- the Compound I MeOAc Solvate Form A of any one of Embodiments 220 to 233 characterized by a 13 C ssNMR spectrum with (a) a peak at 129.3 ⁇ 0.2 ppm and (b) one or more peaks selected from: 43.3 ⁇ 0.2 ppm, 29.8 ⁇ 0.2 ppm, 33.2 ⁇ 0.2 ppm, 171.5 ⁇ 0.2 ppm, and 45.1 ⁇ 0.2 ppm.
- the Compound I MeOAc Solvate Form A of any one of Embodiments 220 to 233 characterized by a 13 C ssNMR spectrum with (a) a peak at 129.3 ⁇ 0.2 ppm and (b) two or more peaks selected from: 43.3 ⁇ 0.2 ppm, 29.8 ⁇ 0.2 ppm, 33.2 ⁇ 0.2 ppm, 171.5 ⁇ 0.2 ppm, and 45.1 ⁇ 0.2 ppm.
- the Compound I MeOAc Solvate Form A of any one of Embodiments 220 to 233 characterized by a 13 C ssNMR spectrum with (a) a peak at 129.3 ⁇ 0.2 ppm and (b) three or more peaks selected from: 43.3 ⁇ 0.2 ppm, 29.8 ⁇ 0.2 ppm, 33.2 ⁇ 0.2 ppm, 171.5 ⁇ 0.2 ppm, and 45.1 ⁇ 0.2 ppm.
- the Compound I MeOAc Solvate Form A of any one of Embodiments 220 to 233 characterized by a 13 C ssNMR spectrum with (a) a peak at 129.3 ⁇ 0.2 ppm and (b) four or more peaks selected from: 43.3 ⁇ 0.2 ppm, 29.8 ⁇ 02 ppm, 33.2 ⁇ 0.2 ppm, 171.5 ⁇ 0.2 ppm, and 45.1 ⁇ 0.2 ppm.
- the Compound I MeOAc Solvate Form A of any one of Embodiments 220 to 233 characterized by a 13 C ssNMR spectrum with (a) peaks at 129.3 ⁇ 0.2 ppm, 43.3 ⁇ 0.2 ppm, 29.8 ⁇ 0.2 ppm, 33.2 ⁇ 0.2 ppm, 171.5 ⁇ 0.2 ppm, 45.1 ⁇ 0.2 ppm and (b) a peak at 27.7 ⁇ 0.2 ppm and/or 129.7 ⁇ 0.2 ppm.
- the Compound I MeOAc Solvate Form A of any one of Embodiments 220 to 233 characterized by a 13 C ssNMR spectrum with peaks at 129.3 ⁇ 0.2 ppm, 43.3 ⁇ 0.2 ppm, 29.8 ⁇ 0.2 ppm, 33.2 ⁇ 0.2 ppm, 171.5 ⁇ 0.2 ppm, 45.1 ⁇ 0.2 ppm, 51.3 ⁇ 0.2 ppm, 27.7 ⁇ 0.2 ppm, and 129.7 ⁇ 0.2 ppm.
- the Compound I MeOAc Solvate Form A of any one of Embodiments 220 to 240 characterized by a 13 C ssNMR spectrum substantially similar to FIG. 31.
- Compound I as substantially crystalline MeOAc Solvate Form B (i.e., wherein less than 15% of Compound I is in amorphous form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous form).
- the Compound I MeOAc Solvate Form B of any one of Embodiments 243 to 247 characterized by an X-ray powder diffractogram having (a) a signal at 4.6 ⁇ 0.2 degrees two-theta and/or a signal at 7.7 ⁇ 0.2 degrees two-theta, and (b) a signal at one or more two-theta values selected from 8.6 ⁇ 0.2 degrees two-theta, 8.2 ⁇ 0.2 degrees two-theta, 5.9 ⁇ 0.2 degrees two-theta, 15.7 ⁇ 0.2 degrees two-theta, 12.0 ⁇ 0.2 degrees two-theta, and 12.5 ⁇ 0.2 degrees two-theta.
- the Compound I MeOAc Solvate Form B of any one of Embodiments 243 to 247 characterized by an X-ray powder diffractogram having (a) a signal at 4.6 ⁇ 0.2 degrees two-theta and/or a signal at 7.7 ⁇ 0.2 degrees two-theta, and (b) signals at two or more two-theta values selected from 8.6 ⁇ 0.2 degrees two-theta, 8.2 ⁇ 0.2 degrees two-theta, 5.9 ⁇ 0.2 degrees two-theta, 15.7 ⁇ 0.2 degrees two-theta, 12.0 ⁇ 0.2 degrees two-theta, and 12.5 ⁇ 0.2 degrees two-theta.
- the Compound I MeOAc Solvate Form B of any one of Embodiments 243 to 247 characterized by an X-ray powder diffractogram having (a) a signal at 4.6 ⁇ 0.2 degrees two-theta and/or a signal at 7.7 ⁇ 0.2 degrees two-theta, and (b) signals at three or more two-theta values selected from 8.6 ⁇ 0.2 degrees two-theta, 8.2 ⁇ 0.2 degrees two-theta, 5.9 ⁇ 0.2 degrees two-theta, 15.7 ⁇ 0.2 degrees two-theta, 12.0 ⁇ 0.2 degrees two-theta, and 12.5 ⁇ 0.2 degrees two-theta.
- the Compound I MeOAc Solvate Form B of any one of Embodiments 243 to 247 characterized by an X-ray powder diffractogram having (a) a signal at 4.6 ⁇ 0.2 degrees two-theta and/or a signal at 7.7 ⁇ 0.2 degrees two-theta, and (b) signals at four or more two-theta values selected from 8.6 ⁇ 0.2 degrees two-theta, 8.2 ⁇ 0.2 degrees two-theta, 5 9 ⁇ 0.2 degrees two-theta, 15.7 ⁇ 0.2 degrees two-theta, 12.0 ⁇ 0.2 degrees two-theta, and 12.5 ⁇ 0.2 degrees two-theta.
- the Compound I MeOAc Solvate Form B of any one of Embodiments 243 to 247 characterized by an X-ray powder diffractogram having (a) a signal at 4.6 ⁇ 0.2 degrees two-theta and/or a signal at 7.7 ⁇ 0.2 degrees two-theta, and (b) signals at five or more two-theta values selected from 8.6 ⁇ 0.2 degrees two-theta, 8.2 ⁇ 0.2 degrees two-theta, 5.9 ⁇ 0.2 degrees two-theta, 15.7 ⁇ 0.2 degrees two-theta, 12.0 ⁇ 0.2 degrees two-theta, and 12.5 ⁇ 0.2 degrees two-theta.
- the Compound I MeOAc Solvate Form B of any one of Embodiments 243 to 247 characterized by an X-ray powder diffractogram having signals at 4.6 ⁇ 0.2 degrees two-theta, 7.7 ⁇ 0.2 degrees two-theta, 8.6 ⁇ 0.2 degrees two-theta, 8.2 ⁇ 0.2 degrees two-theta, 5.9 ⁇ 0.2 degrees two-theta, 15.7 ⁇ 0.2 degrees two-theta, 12.0 ⁇ 0.2 degrees two-theta, and 12.5 ⁇ 0.2 degrees two-theta.
- the Compound I MeOAc Solvate Form B of any one of Embodiments 243 to 256 characterized by a DSC analysis showing an endothermic peak at about 51 °C and about 187 °C
- the Compound I MeOAc Solvate Form B of any one of Embodiments 243 to 257 characterized by a DSC analysis substantially similar to FIG. 34.
- Compound I as substantially crystalline MeOAc Solvate Form C (i.e., wherein less than 15% of Compound I is in amorphous form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous form).
- Substantially pure Compound I MeOAc Solvate Form C The Compound I MeOAc Solvate Form C of any one of Embodiments 269 to 271, characterized by an X-ray powder diffractogram having a signal at 6.4 ⁇ 0.2 degrees two-theta.
- the Compound I Hydrate Form A of any one of Embodiments 292 to 295 characterized by an X-ray powder diffractogram having (a) a signal at 21.4 ⁇ 0.2 degrees two-theta, and (b) a signal at one or more two-theta values selected from: 14.0 ⁇ 0.2 degrees two-theta, 19.5 ⁇ 0.2 degrees two-theta, 7.0 ⁇ 0.2 degrees two- theta, 18.6 ⁇ 0.2 degrees two-theta, 11.0 ⁇ 0.2 degrees two-theta, 18.2 ⁇ 0.2 degrees two-theta, and 20.5 ⁇ 0.2 degrees two-theta.
- the Compound I Hydrate Form A of any one of Embodiments 292 to 295 characterized by an X-ray powder diffractogram having (a) a signal at 21.4 ⁇ 0.2 degrees two-theta, and (b) signals at two or more two-theta values selected from: 14.0 ⁇ 0.2 degrees two-theta, 19.5 ⁇ 0.2 degrees two-theta, 7.0 ⁇ 0.2 degrees two-theta, 18.6 ⁇ 0.2 degrees two-theta, 11.0 ⁇ 0.2 degrees two-theta, 18.2 ⁇ 0.2 degrees two- theta, and 20.5 ⁇ 0.2 degrees two-theta.
- the Compound I Hydrate Form A of any one of Embodiments 292 to 295 characterized by an X-ray powder diffractogram having (a) a signal at 21.4 ⁇ 0.2 degrees two-theta, and (b) signals at three or more two-theta values selected from: 14.0 ⁇ 0.2 degrees two-theta, 19.5 ⁇ 0.2 degrees two-theta, 7.0 ⁇ 0.2 degrees two- theta, 18.6 ⁇ 0.2 degrees two-theta, 11.0 ⁇ 0.2 degrees two-theta, 18.2 ⁇ 0.2 degrees two-theta, and 20.5 ⁇ 0.2 degrees two-theta.
- the Compound I Hydrate Form A of any one of Embodiments 292 to 295 characterized by an X-ray powder diffractogram having (a) a signal at 21.4 ⁇ 0.2 degrees two-theta, and (b) signals at four or more two-theta values selected from: 14.0 ⁇ 0.2 degrees two-theta, 19.5 ⁇ 0.2 degrees two-theta, 7.0 ⁇ 0.2 degrees two-theta, 18.6 ⁇ 0.2 degrees two-theta, 11.0 ⁇ 0.2 degrees two-theta, 18.2 ⁇ 0.2 degrees two- theta, and 20.5 ⁇ 0.2 degrees two-theta.
- the Compound I Hydrate Form A of any one of Embodiments 292 to 295 characterized by an X-ray powder diffractogram having (a) a signal at 21.4 ⁇ 0.2 degrees two-theta, and (b) signals at five or more two-theta values selected from: 14.0 ⁇ 0.2 degrees two-theta, 19.5 ⁇ 0.2 degrees two-theta, 7.0 ⁇ 0.2 degrees two-theta, 18.6 ⁇ 0.2 degrees two-theta, 11.0 ⁇ 0.2 degrees two-theta, 18.2 ⁇ 0.2 degrees two- theta, and 20.5 ⁇ 0.2 degrees two-theta.
- the Compound I Hydrate Form A of any one of Embodiments 292 to 295 characterized by an X-ray powder diffractogram having (a) a signal at 21.4 ⁇ 0.2 degrees two-theta, and (b) signals at six or more two-theta values selected from: 14.0 ⁇ 0.2 degrees two-theta, 19.5 ⁇ 0.2 degrees two-theta, 7.0 ⁇ 0.2 degrees two-theta, 18.6 ⁇ 0.2 degrees two-theta, 11 0 ⁇ 0.2 degrees two-theta, 18.2 ⁇ 0.2 degrees two- theta, and 20.5 ⁇ 0.2 degrees two-theta.
- the Compound I Hydrate Form A of any one of Embodiments 292 to 295 characterized by an X-ray powder diffractogram having signals at 21.4 ⁇ 0.2 degrees two-theta, 14.0 ⁇ 0.2 degrees two-theta, 19.5 ⁇ 0.2 degrees two-theta, 7.0 ⁇ 0.2 degrees two-theta, 18.6 ⁇ 0.2 degrees two-theta, 11.0 ⁇ 0.2 degrees two-theta, 18.2 ⁇ 0.2 degrees two-theta, and 20.5 ⁇ 0.2 degrees two-theta.
- the Compound I Hydrate Form A of any one of Embodiments 292 to 307 characterized by a 13 C ssNMR spectrum with (a) peaks at 26.8 ⁇ 0.2 ppm, 18.3 ⁇ 0.2 ppm, and 126.5 ⁇ 0.2 ppm and (b) two or more peaks selected from 131.5 ⁇ 0.2 ppm, 135.0 ⁇ 0.2 ppm, 20.0 ⁇ 0.2 ppm, 142.9 ⁇ 0.2 ppm, and 128.3 ⁇ 0.2 ppm.
- the Compound I Hydrate Form A of any one of Embodiments 292 to 309 characterized by a 13 C ssNMR spectrum with (a) peaks at 26.8 ⁇ 0.2 ppm, 18.3 ⁇ 0.2 ppm, and 126.5 ⁇ 0.2 ppm and (b) four or more peaks selected from 131.5 ⁇ 0.2 ppm, 135.0 ⁇ 0.2 ppm, 20.0 ⁇ 0.2 ppm, 142.9 ⁇ 0.2 ppm, and 128.3 ⁇ 0.2 ppm.
- the Compound I Hydrate Form A of any one of Embodiments 292 to 310 characterized by a 13 C ssNMR spectrum with peaks at 26.8 ⁇ 0.2 ppm, 18.3 ⁇ 0.2 ppm, 126.5 ⁇ 0.2 ppm, 131.5 ⁇ 0.2 ppm, 135.0 ⁇ 0.2 ppm, 20.0 ⁇ 0.2 ppm, 142.9 ⁇ 0.2 ppm, and 128.3 ⁇ 0.2 ppm.
- the Compound I Hydrate Form A of any one of Embodiments 292 to 312, characterized by a monoclinic crystal system, aP2i2i2i space group, and unit cell dimensions, measured at 100 K on a Bruker diffractometer equipped with Cu K a radiation ( 1.54178 A) and an CP AD detector, of:
- Compound I as substantially crystalline Hydrate Form B (i.e., wherein less than 15% of Compound I is in amorphous form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous form).
- the Compound I of Embodiment 315 wherein Compound I is 100% crystalline Compound I Hydrate Form B.
- Substantially pure Compound I Hydrate Form B The Compound I Hydrate Form B of any one of Embodiments 315 to 317, characterized by an X-ray powder diffractogram having a signal at 24.5 ⁇ 0.2 degrees two-theta.
- the Compound I Hydrate Form B of any one of Embodiments 315 to 317 characterized by an X-ray powder diffractogram having (a) a signal at 24.5 ⁇ 0.2 degrees two-theta, and (b) a signal at one or more two-theta values selected from: 7.0 ⁇ 0.2 degrees two-theta, 19.3 ⁇ 0.2 degrees two-theta, 19.4 ⁇ 0.2 degrees two-theta, 15.1 ⁇ 0.2 degrees two-theta, 13.9 ⁇ 0.2 degrees two-theta, 10.9 ⁇ 0.2 degrees two- theta, and 19.7 ⁇ 0.2 degrees two-theta.
- the Compound I Hydrate Form B of any one of Embodiments 315 to 317 characterized by an X-ray powder diffractogram having (a) a signal at 24.5 ⁇ 0.2 degrees two-theta, and (b) signals at three or more two-theta values selected from: 7.0 ⁇ 0.2 degrees two-theta, 19.3 ⁇ 0.2 degrees two-theta, 19.4 ⁇ 0.2 degrees two-theta,
- the Compound I Hydrate Form B of any one of Embodiments 315 to 317 characterized by an X-ray powder diffractogram having (a) a signal at 24.5 ⁇ 0.2 degrees two-theta, and (b) signals at four or more two-theta values selected from: 7.0 ⁇ 0.2 degrees two-theta, 19 3 ⁇ 0.2 degrees two-theta, 19.4 ⁇ 0.2 degrees two-theta,
- the Compound I Hydrate Form B of any one of Embodiments 315 to 317 characterized by an X-ray powder diffractogram having (a) a signal at 24.5 ⁇ 0.2 degrees two-theta, and (b) signals at five or more two-theta values selected from : 7.0 ⁇ 0.2 degrees two-theta, 19.3 ⁇ 0.2 degrees two-theta, 19.4 ⁇ 0.2 degrees two-theta,
- the Compound I Hydrate Form B of any one of Embodiments 315 to 317 characterized by an X-ray powder diffractogram having (a) a signal at 24.5 ⁇ 0.2 degrees two-theta, and (b) signals at six or more two-theta values selected from: 7.0 ⁇ 0.2 degrees two-theta, 19.3 ⁇ 0.2 degrees two-theta, 19.4 ⁇ 0.2 degrees two-theta, 15.1 ⁇ 0.2 degrees two-theta, 13.9 ⁇ 0.2 degrees two-theta, 10.9 ⁇ 0.2 degrees two-theta, and 19.7 ⁇ 0.2 degrees two-theta.
- the Compound I Hydrate Form B of any one of Embodiments 315 to 326 characterized by a 13 C ssNMR spectrum with a peak at 19.3 ⁇ 0.2 ppm.
- the Compound I Hydrate Form B of any one of Embodiments 315 to 327 characterized by a 13 C ssNMR spectrum with (a) a peak at 19.3 ⁇ 0.2 ppm and (b) one or more peaks selected from: 29.4 ⁇ 0.2 ppm, 128.6 ⁇ 0.2 ppm, 130.3 ⁇ 0.2 ppm, 42.3 ⁇ 0.2 ppm, 128.9 ⁇ 0.2 ppm, 134.6 ⁇ 0.2 ppm, and 136.4 ⁇ 0.2 ppm.
- the Compound I Hydrate Form B of any one of Embodiments 315 to 328 characterized by a 13 C ssNMR spectrum with (a) a peak at 19.3 ⁇ 0.2 ppm and (b) two or more peaks selected from: 29.4 ⁇ 0.2 ppm, 128.6 ⁇ 0.2 ppm, 130.3 ⁇ 0.2 ppm, 42.3 ⁇ 0.2 ppm, 128.9 ⁇ 0.2 ppm, 134.6 ⁇ 0.2 ppm, and 136.4 ⁇ 0.2 ppm.
- the Compound I Hydrate Form B of any one of Embodiments 315 to 333 characterized by a 13 C ssNMR spectrum with peaks at 19.3 ⁇ 0.2 ppm, 29.4 ⁇ 0.2 ppm, 128.6 ⁇ 0.2 ppm, 130.3 ⁇ 0.2 ppm, 42.3 ⁇ 0.2 ppm, 128.9 ⁇ 0.2 ppm, 134.6 ⁇ 0.2 ppm, and 136.4 ⁇ 0.2 ppm.
- the Compound I Hydrate Form B of any one of Embodiments 315 to 334 characterized by a 13 C ssNMR spectrum substantially similar to FIG. 41.
- the Compound I Hydrate Form B of any one of Embodiments 315 to 336 prepared by a process comprising (i) stirring Compound I in a MeOH/water mixture at about 80 °C, (ii) isolation of the solid via filtration, and (iii) exposure of the solid to about 11% humidity.
- Compound I as substantially crystalline Hydrate Form C (i.e., wherein less than 15% of Compound I is in amorphous form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous form).
- the Compound I of Embodiment 338 wherein Compound I is 100% crystalline Compound I Hydrate Form C.
- Substantially pure Compound I Hydrate Form C The Compound I Hydrate Form C of any one of Embodiments 338 to 340, characterized by an X-ray powder diffractogram having a signal at 16.1 ⁇ 0.2 degrees two-theta.
- the Compound I Hydrate Form C of any one of Embodiments 338 to 340 characterized by an X-ray powder diffractogram having (a) a signal at 16.1 ⁇ 0.2 degrees two-theta and (b) a signal at one or more two-theta values selected from: 7.0 ⁇ 0.2 degrees two-theta, 21.9 ⁇ 0.2 degrees two-theta, 11.2 ⁇ 0.2 degrees two-theta,
- the Compound I Hydrate Form C of any one of Embodiments 338 to 340 characterized by an X-ray powder diffractogram having (a) a signal at 16.1 ⁇ 0.2 degrees two-theta and (b) a signal at two or more two-theta values selected from: 7.0 ⁇ 0.2 degrees two-theta, 21.9 ⁇ 0.2 degrees two-theta, 11.2 ⁇ 0.2 degrees two-theta,
- the Compound I Hydrate Form C of any one of Embodiments 338 to 340 characterized by an X-ray powder diffractogram having (a) a signal at 16.1 ⁇ 0.2 degrees two-theta and (b) signals at three or more two-theta values selected from: 7.0 ⁇ 0.2 degrees two-theta, 21.9 ⁇ 0.2 degrees two-theta, 11.2 ⁇ 0.2 degrees two-theta,
- the Compound I Hydrate Form C of any one of Embodiments 338 to 340 characterized by an X-ray powder diffractogram having (a) a signal at 16.1 ⁇ 0.2 degrees two-theta and (b) signals at four or more two-theta values selected from: 7.0 ⁇ 0.2 degrees two-theta, 21.9 ⁇ 0.2 degrees two-theta, 11.2 ⁇ 0.2 degrees two-theta,
- the Compound I Hydrate Form C of any one of Embodiments 338 to 340 characterized by an X-ray powder diffractogram having (a) a signal at 16.1 ⁇ 0.2 degrees two-theta and (b) signals at five or more two-theta values selected from: 7.0 ⁇ 0.2 degrees two-theta, 21.9 ⁇ 0.2 degrees two-theta, 11.2 ⁇ 0.2 degrees two-theta, 15.1 ⁇ 0.2 degrees two-theta, 14.9 ⁇ 0.2 degrees two-theta, 15.4 ⁇ 0.2 degrees two-theta, and 18.2 ⁇ 0.2 degrees two-theta.
- the Compound I Hydrate Form C of any one of Embodiments 338 to 340 characterized by an X-ray powder diffractogram having (a) a signal at 16.1 ⁇ 0.2 degrees two-theta and (b) signals at six or more two-theta values selected from: 7.0 ⁇ 0.2 degrees two-theta, 21 9 ⁇ 0.2 degrees two-theta, 11.2 ⁇ 0.2 degrees two-theta, 15.1 ⁇ 0.2 degrees two-theta, 14.9 ⁇ 0.2 degrees two-theta, 15.4 ⁇ 0.2 degrees two-theta, and 18.2 ⁇ 0.2 degrees two-theta.
- the Compound I Hydrate Form C of any one of Embodiments 338 to 340 characterized by an X-ray powder diffractogram having signals at 16.1 ⁇ 0.2 degrees two-theta, 7.0 ⁇ 0.2 degrees two-theta, 21.9 ⁇ 0.2 degrees two-theta, 11.2 ⁇ 0.2 degrees two-theta, 15.1 ⁇ 0.2 degrees two-theta, 14.9 ⁇ 0.2 degrees two-theta, 15.4 ⁇ 0.2 degrees two-theta, and 18.2 ⁇ 0.2 degrees two-theta.
- the Compound I Hydrate Form C of any one of Embodiments 338 to 348 characterized by an X-ray powder diffractogram substantially similar to FIG. 42.
- Compound I as substantially crystalline EtOH Solvate Hydrate (i.e., wherein less than 15% of Compound I is in amorphous form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous form).
- the Compound I EtOH Solvate Hydrate of any one of Embodiments 361 to 365 characterized by an X-ray powder diffractogram having signals at three or more two- theta values selected from 6.7 ⁇ 0.2 degrees two-theta, 11.5 ⁇ 0.2 degrees two-theta, 8.6 ⁇ 0.2 degrees two-theta, 14.7 ⁇ 0.2 degrees two-theta, 20.0 ⁇ 0.2 degrees two- theta, 11.3 ⁇ 0.2 degrees two-theta, 18.7 ⁇ 0.2 degrees two-theta, and 19.1 ⁇ 0.2 degrees two-theta.
- the Compound I EtOH Solvate Hydrate of any one of Embodiments 361 to 366 characterized by an X-ray powder diffractogram having signals at four or more two- theta values selected from 6.7 ⁇ 0.2 degrees two-theta, 11.5 ⁇ 0.2 degrees two-theta,
- the Compound I EtOH Solvate Hydrate of any one of Embodiments 361 to 370 characterized by an X-ray powder diffractogram having signals at 6.7 ⁇ 0.2 degrees two-theta, 11.5 ⁇ 0.2 degrees two-theta, 8.6 ⁇ 0.2 degrees two-theta, 14.7 ⁇ 0.2 degrees two-theta, 20.0 ⁇ 0.2 degrees two-theta, 11.3 ⁇ 0.2 degrees two-theta, 18.7 ⁇ 0.2 degrees two-theta, and 19.1 ⁇ 0.2 degrees two-theta.
- the Compound I EtOH Solvate Hydrate of any one of Embodiments 361 to 373 characterized by a 13 C ssNMR spectrum with two or more peaks selected from: 29.8 ⁇ 0.2 ppm, 127.0 ⁇ 0.2 ppm, 17.9 ⁇ 0.2 ppm, 42.9 ⁇ 0.2 ppm, 18.0 ⁇ 0.2 ppm, 20.9 ⁇ 0.2 ppm, 135.7 ⁇ 0.2 ppm, and 129.1 ⁇ 0.2 ppm.
- the Compound I EtOH Solvate Hydrate of any one of Embodiments 361 to 375 characterized by a 13 C ssNMR spectrum with four or more peaks selected from: 29.8 ⁇ 0.2 ppm, 127.0 ⁇ 0.2 ppm, 17.9 ⁇ 0.2 ppm, 42.9 ⁇ 0.2 ppm, 18.0 ⁇ 0.2 ppm, 20.9 ⁇ 0.2 ppm, 135.7 ⁇ 0.2 ppm, and 129.1 ⁇ 0.2 ppm.
- the Compound I EtOH Solvate Hydrate of any one of Embodiments 361 to 376 characterized by a 13 C ssNMR spectrum with five or more peaks selected from: 29.8 ⁇ 0.2 ppm, 127.0 ⁇ 0.2 ppm, 17.9 ⁇ 0.2 ppm, 42.9 ⁇ 0.2 ppm, 18.0 ⁇ 0.2 ppm, 20.9 ⁇ 0.2 ppm, 135.7 ⁇ 0.2 ppm, and 129.1 ⁇ 0.2 ppm.
- the Compound I EtOH Solvate Hydrate of any one of Embodiments 361 to 377 characterized by a 13 C ssNMR spectrum with six or more peaks selected from: 29.8 ⁇ 0.2 ppm, 127.0 ⁇ 0.2 ppm, 17.9 ⁇ 0.2 ppm, 42.9 ⁇ 0.2 ppm, 18.0 ⁇ 0.2 ppm, 20.9 ⁇ 0.2 ppm, 135.7 ⁇ 0.2 ppm, and 129.1 ⁇ 0.2 ppm.
- the Compound I EtOH Solvate Hydrate of any one of Embodiments 361 to 378 characterized by a 13 C ssNMR spectrum with seven or more peaks selected from: 29.8 ⁇ 0.2 ppm, 127.0 ⁇ 0.2 ppm, 17.9 ⁇ 0.2 ppm, 42.9 ⁇ 0.2 ppm, 18.0 ⁇ 0.2 ppm, 20.9 ⁇ 0.2 ppm, 135.7 ⁇ 0.2 ppm, and 129.1 ⁇ 0.2 ppm.
- the Compound I EtOH Solvate Hydrate of any one of Embodiments 361 to 379 characterized by a 13 C ssNMR spectrum with peaks at: 29.8 ⁇ 0.2 ppm, 127.0 ⁇ 0.2 ppm, 17.9 ⁇ 0.2 ppm, 42.9 ⁇ 0.2 ppm, 18.0 ⁇ 0.2 ppm, 20.9 ⁇ 0.2 ppm, 135.7 ⁇ 0.2 ppm, and 129.1 ⁇ 0.2 ppm.
- the Compound I EtOH Solvate Hydrate of any one of Embodiments 361 to 380 characterized by a 13 C ssNMR spectrum substantially similar to FIG. 45.
- the Compound I EtOH Solvate Hydrate of any one of Embodiments 361 to 381, characterized by a monoclinic crystal system, aP2i2i2i space group, and unit cell dimensions, measured at 100 K on a Bruker diffractometer equipped with Cu K « radiation ( 1.54178 A) and an CP AD detector, of:
- Compound I as substantially crystalline MeOH Solvate Hydrate (i.e., wherein less than 15% of Compound I is in amorphous form, wherein less than 10% of Compound I is in amorphous Form, wherein less than 5% of Compound I is in amorphous Form).
- the Compound I of Embodiment 384 wherein Compound I is 100% crystalline Compound I MeOH hydrate.
- the Compound I MeOH Solvate Hydrate of any one of Embodiments 384 to 386 characterized by an X-ray powder diffractogram having a signal at one or more two- theta values selected from 8.6 ⁇ 0.2 degrees two-theta, 6.7 ⁇ 0.2 degrees two-theta,
- the Compound I MeOH Solvate Hydrate of any one of Embodiments 384 to 395 characterized by a 13 C ssNMR spectrum with one or more peaks selected from: 29.3 ⁇ 0.2 ppm, 126.9 ⁇ 0.2 ppm, 43.1 ⁇ 0.2 ppm, 20.9 ⁇ 0.2 ppm, 17.2 ⁇ 0.2 ppm, 127.8 ⁇ 0.2 ppm, 29.6 ⁇ 0.2 ppm, and 135.7 ⁇ 0.2 ppm.
- the Compound I MeOH Solvate Hydrate of any one of Embodiments 384 to 396 characterized by a 13 C ssNMR spectrum with two or more peaks selected from: 29.3 ⁇ 0.2 ppm, 126.9 ⁇ 0.2 ppm, 43.1 ⁇ 0.2 ppm, 20.9 ⁇ 0.2 ppm, 17.2 ⁇ 0.2 ppm, 127.8 ⁇ 0.2 ppm, 29.6 ⁇ 0.2 ppm, and 135.7 ⁇ 0.2 ppm.
- the Compound I MeOH Solvate Hydrate of any one of Embodiments 384 to 397 characterized by a 13 C ssNMR spectrum with three or more peaks selected from: 29.3 ⁇ 0.2 ppm, 126.9 ⁇ 0.2 ppm, 43.1 ⁇ 0.2 ppm, 20.9 ⁇ 0.2 ppm, 17.2 ⁇ 0.2 ppm, 127.8 ⁇ 0.2 ppm, 29.6 ⁇ 0.2 ppm, and 135.7 ⁇ 0.2 ppm.
- the Compound I MeOH Solvate Hydrate of any one of Embodiments 384 to 400 characterized by a 13 C ssNMR spectrum with six or more peaks selected from: 29.3 ⁇ 0.2 ppm, 126.9 ⁇ 0.2 ppm, 43.1 ⁇ 0.2 ppm, 20.9 ⁇ 0.2 ppm, 17.2 ⁇ 0.2 ppm, 127.8 ⁇ 0.2 ppm, 29.6 ⁇ 0.2 ppm, and 135.7 ⁇ 0.2 ppm.
- the Compound I MeOH Solvate Hydrate of any one of Embodiments 384 to 402 characterized by a 13 C ssNMR spectrum with peaks at: 29.3 ⁇ 0.2 ppm, 126.9 ⁇ 0.2 ppm, 43.1 ⁇ 0.2 ppm, 20.9 ⁇ 0.2 ppm, 17.2 ⁇ 0.2 ppm, 127.8 ⁇ 0.2 ppm, 29.6 ⁇ 0.2 ppm, and 135.7 ⁇ 0.2 ppm.
- Compound I as substantially crystalline IPA Solvate Hydrate (i.e., wherein less than 15% of Compound I is in amorphous form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous form).
- the Compound I IPA Solvate Hydrate of any one of Embodiments 406 to 409 characterized by an X-ray powder diffractogram having signals at two or more two- theta values selected to 6.7 ⁇ 0.2 degrees two-theta, 8.6 ⁇ 0.2 degrees two-theta, 11.5 ⁇ 0.2 degrees two-theta, 19.9 ⁇ 0.2 degrees two-theta, 14.8 ⁇ 0.2 degrees two-theta, 18.7 ⁇ 0.2 degrees two-theta, 17.7 ⁇ 0.2 degrees two-theta, and 17.3 ⁇ 0.2 degrees two-theta.
- the Compound I IPA Solvate Hydrate of any one of Embodiments 406 to 410 characterized by an X-ray powder diffractogram having signals at three or more two- theta values selected from 6.7 ⁇ 0.2 degrees two-theta, 8.6 ⁇ 0.2 degrees two-theta,
- the Compound I IPA Solvate Hydrate of any one of Embodiments 406 to 415 characterized by an X-ray powder diffractogram having signals at 6.7 ⁇ 0.2 degrees two-theta, 8.6 ⁇ 0.2 degrees two-theta, 11.5 ⁇ 0.2 degrees two-theta, 19.9 ⁇ 0.2 degrees two-theta, 14.8 ⁇ 0.2 degrees two-theta, 18.7 ⁇ 0.2 degrees two-theta, 17.7 ⁇ 0.2 degrees two-theta, and 17.3 ⁇ 0.2 degrees two-theta.
- the Compound I IPA Solvate Hydrate of any one of Embodiments 406 to 416 characterized by an X-ray powder diffractogram substantially similar to FIG. 48.
- the Compound I IPA Solvate Hydrate of any one of Embodiments 406 to 420 characterized by a 13 C ssNMR spectrum with four or more peaks selected from: 30.0 ⁇ 0.2 ppm, 127.4 ⁇ 0.2 ppm, 24.9 ⁇ 0.2 ppm, 18.6 ⁇ 0.2 ppm, 24.2 ⁇ 0.2 ppm, 20.3 ⁇ 0.2 ppm, 136.8 ⁇ 0.2 ppm, and 42.7 ⁇ 0.2 ppm.
- the Compound I IPA Solvate Hydrate of any one of Embodiments 406 to 424 characterized by a 13 C ssNMR spectrum with peaks at: 30.0 ⁇ 0.2 ppm, 127.4 ⁇ 0.2 ppm, 24.9 ⁇ 0.2 ppm, 18.6 ⁇ 0.2 ppm, 24.2 ⁇ 0.2 ppm, 20.3 ⁇ 0.2 ppm, 136.8 ⁇ 0.2 ppm, and 42.7 ⁇ 0.2 ppm.
- the Compound I IPA Solvate Hydrate of any one of Embodiments 406 to 425 characterized by a 13 C ssNMR spectrum substantially similar to FIG. 49.
- Compound I as substantially crystalline MeOAc Solvate Hydrate (i.e., wherein less than 15% of Compound I is in amorphous form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous form).
- the Compound I MeOAc Solvate Hydrate of any one of Embodiments 429 to 438 characterized by an X-ray powder diffractogram having signals at 6.5 ⁇ 0.2 degrees two-theta, 10.7 ⁇ 0.2 degrees two-theta, 14.3 ⁇ 0.2 degrees two-theta, 17.5 ⁇ 0.2 degrees two-theta, 7.1 ⁇ 0.2 degrees two-theta, 19.9 ⁇ 0.2 degrees two-theta, 19.5 ⁇ 0.2 degrees two-theta, and 14.2 ⁇ 0.2 degrees two-theta.
- the Compound I MeOAc Solvate Hydrate of any one of Embodiments 429 to 439 characterized by an X-ray powder diffractogram substantially similar to FIG.
- the Compound I MeOAc Solvate Hydrate of any one of Embodiments 429 to 444 characterized by a 13 C ssNMR spectrum with five or more peaks selected from: 28.1 ⁇ 0.2 ppm, 43.6 ⁇ 0.2 ppm, 130.5 ⁇ 0.2 ppm, 21.4 ⁇ 0.2 ppm, 20.7 ⁇ 0.2 ppm, 19.0 ⁇ 0.2 ppm, 135.9 ⁇ 0.2 ppm, and 143.4 ⁇ 0.2 ppm.
- the Compound I MeOAc Solvate Hydrate of any one of Embodiments 429 to 445 characterized by a 13 C ssNMR spectrum with six or more peaks selected from: 28.1 ⁇ 0.2 ppm, 43.6 ⁇ 0.2 ppm, 130.5 ⁇ 0.2 ppm, 21.4 ⁇ 0.2 ppm, 20.7 ⁇ 0.2 ppm, 19.0 ⁇ 0.2 ppm, 135.9 ⁇ 0.2 ppm, and 143.4 ⁇ 0.2 ppm.
- the Compound I MeOAc Solvate Hydrate of any one of Embodiments 429 to 446 characterized by a 13 C ssNMR spectrum with seven or more peaks selected from: 28.1 ⁇ 0.2 ppm, 43.6 ⁇ 0.2 ppm, 130.5 ⁇ 0.2 ppm, 21.4 ⁇ 0.2 ppm, 20.7 ⁇ 0.2 ppm, 19.0 ⁇ 0.2 ppm, 135.9 ⁇ 0.2 ppm, and 143.4 ⁇ 0.2 ppm.
- the Compound I MeOAc Solvate Hydrate of any one of Embodiments 429 to 447 characterized by a 13 C ssNMR spectrum with peaks at: 28.1 ⁇ 0.2 ppm, 43.6 ⁇ 0.2 ppm, 130.5 ⁇ 0.2 ppm, 21.4 ⁇ 0.2 ppm, 20.7 ⁇ 0.2 ppm, 19.0 ⁇ 0.2 ppm, 135.9 ⁇ 0.2 ppm, and 143.4 ⁇ 0.2 ppm.
- the Compound I MeOAc Solvate Hydrate of any one of Embodiments 429 to 448 characterized by a 13 C ssNMR spectrum substantially similar to FIG. 51.
- Compound I as substantially amorphous Compound I (i.e., wherein less than 15% of Compound I is in crystalline form, wherein less than 10% of Compound I is in crystalline form, wherein less than 5% of Compound I is in crystalline form)
- Amorphous Compound I of Embodiment 451 or Embodiment 452 characterized by an X-ray powder diffractogram substantially similar to FIG. 52.
- Amorphous Compound I of any one of Embodiments 451 to 453 characterized by a 13 C ssNMR spectrum substantially similar to FIG. 53.
- a pharmaceutical composition comprising Compound I according to any one of Embodiments 1 to 465 and a pharmaceutically acceptable carrier.
- the pharmaceutical composition according to Embodiment 466 further comprising one or more additional therapeutic agents.
- the pharmaceutical composition according to Embodiment 467 wherein the pharmaceutical composition comprises one or more additional CFTR modulating compounds.
- the pharmaceutical composition according to Embodiment 467 or Embodiment 468, wherein the pharmaceutical composition comprises one or more compounds selected from Compound II, Compound III, Compound Ill-d, Compound IV, and Compound V, and pharmaceutically acceptable salts and deuterated derivatives of any of the foregoing.
- Embodiment 472 wherein the medicament is formulated for administration in combination with one or more compounds selected from Compound II, Compound III, Compound Ill-d, Compound IV, and pharmaceutically acceptable salts and deuterated derivatives of any of the foregoing.
- a method of treating cystic fibrosis comprising administering the Compound I according to any one of Embodiments 1 to 463, or the pharmaceutical composition according to any one of Embodiments 466 to 469, to a subject in need thereof.
- the method of treating according to Embodiment 474, wherein the method further comprises administration of one or more compounds selected from Compound II, Compound III, Compound Ill-d, Compound IV, and pharmaceutically acceptable salts and deuterated derivatives of any of the foregoing.
- a process for preparing Compound I Neat Form A according to any one of Embodiments 1 to 28 comprising (i) dissolving amorphous Compound I in ethanol, (ii) increasing the temperature to about 73 °C, (iii) decreasing the temperature to about 20 °C, (iv) isolation of the solid via filtration, and (v) drying of the solid at 50 °C.
- a process for preparing Compound I Neat Form B according to any one of Embodiments 30 to 49 comprising exposing Compound I Hydrate Form A to about 3% humidity.
- a process for preparing Compound I Neat Form C according to any one of Embodiments 51 to 74 comprising (i) stirring of Compound I in hexadecane at about 80 °C, and (ii) centrifugation and drying of the resulting solid.
- a process for preparing Compound I Neat Form D according to any one of Embodiments 76 to 87 comprising the drying of Compound I hydrate C at about 80 °C.
- a process for preparing Compound I Neat Form D according to any one of Embodiments 76 to 87 comprising (i) stirring of Compound I Neat Form A in IP A, and (ii) isolation of the solid via filtration, and (iii) drying of the solid at about 125 °C.
- a process for preparing Compound I Neat Form E according to any one of Embodiments 90 to 112 comprising (i) stirring of Compound I Neat Form A in MeOAc, and (ii) isolation of the solid via centrifugation, and (iii) drying of the solid at about 50 °C.
- a process for preparing Compound I EtOH Solvate Form B according to any one of Embodiments 152 to 177 comprising (i) stirring of Compound I in ethanol, and (ii) isolation of the solid via centrifugation.
- a process for preparing Compound I MeOH Solvate according to any one of Embodiments 179 to 196 comprising (i) dissolving Compound I in MeOH and increasing temperature to about 62 °C, (ii) cooling the solution to about 10 °C, and (iii) isolation of the solid via centrifugation.
- a process for preparing Compound I NPA Solvate according to any one of Embodiments 198 to 218 comprising (i) stirring of Compound I in NPA, and (ii) isolation of the solid via centrifugation.
- a process for preparing Compound I MeOAc Solvate Form A according to any one of Embodiments 220 to 241 comprising (i) stirring Compound I Neat Form A in MeOAc, and (ii) isolation of the solid via filtration.
- a process for preparing Compound I MeOAc Solvate form B according to any one of Embodiments 243 to 258 comprising (i) stirring of Compound I Neat Form A in MeOAc at about 5 °C, (ii) isolation of the solid via centrifugation, and (iii) drying of the solid at room temperature.
- a process for preparing Compound I MeOAc Solvate form C according to any one of Embodiments 269 to 281 comprising (i) stirring of Compound I Neat Form A in MeOAc at about 5 °C, and (ii) isolation of the solid via filtration.
- a process for preparing Compound I Hydrate Form A according to any one of Embodiments 292 to 313 comprising (i) stirring Compound I EtOH solvate form B in water, and (ii) isolation via filtration and air drying of the solid.
- a process for preparing Compound I Hydrate form B according to any one of Embodiments 315 to 336 comprising (i) stirring Compound I in a MeOH/water mixture at about 80 °C, (ii) isolation of the solid via filtration, and (iii) exposure of the solid to about 11% humidity.
- a process for preparing Compound I Hydrate Form C according to any one of Embodiments 338 to 359 comprising (i) stirring Compound I in a MeOH/water mixture at about 80 °C, (ii) isolation of the solid via filtration, and (iii) exposure of the solid to about 75% humidity.
- a process for preparing Compound I EtOH Solvate Hydrate according to any one of Embodiments 361 to 382 comprising (i) stirring Compound I in an EtOH/water mixture at about 60 °C, and (ii) isolation of the solid via centrifugation.
- a process for preparing Compound I MeOH Solvate Hydrate according to any one of Embodiments 384 to 404 comprising (i) the stirring of Compound I in a MeOH/water mixture at about 80 °C, and (ii) isolation of the solid via centrifugation.
- a process for preparing Compound I IPA Solvate Hydrate according to any one of Embodiments 406 to 427 comprising (i) stirring Compound I in an IPA/water mixture, and (ii) isolation of the solid via centrifugation.
- a process for preparing Compound I MeOAc Solvate Hydrate according to any one of Embodiments 429 to 449 comprising (i) stirring amorphous Compound I in a MeOAc/water mixture, (ii) addition of Compound I MeOAc form A, Compound I MeOAc form B, Compound I MeOAc form C, Compound I Neat Form E, Compound I Hydrate form A, and Compound I Hydrate Form B, (iii) further stirring, and (iv) isolation of the solid via centrifugation.
- a process for preparing amorphous Compound I according to any one of Embodiments 451 to 454 comprising (i) purification of Compound I via column chromatography, (ii) dissolving the solid in DCM, (iii) removal of the solvent, and (iv) drying the solid under high vacuum.
- a process for preparing amorphous Compound I formulated as a spray-dried dispersion according to any one of Embodiments 456 to 462 comprising (i) preparation of a solution of Compound I and polymer in a DCM/MeOH mixture, and (ii) spray drying the resulting solution using a spray dryer.
- Compounds II, III, Ill-d, and IV can be prepared by any suitable method in the art, for example, PCT Publication Nos. WO 2011/133751, WO 2011/133951, WO 2015/160787, and U.S. Patent No. 8,865,902.
- Solution-phase proton and carbon NMR spectra were acquired on either a Bruker Biospin DRX 400 MHz FTNMR spectrometer operating at a 1 H and 13 C resonant frequency of 400 and 100 MHz respectively, or on a 300 MHz NMR spectrometer.
- One dimensional proton and carbon spectra were acquired using a broadband observe (BBFO) probe with 20 Hz sample rotation at 0.1834 and 0.9083 Hz/Pt digital resolution respectively. All proton and carbon spectra were acquired with temperature control at 30 °C using standard, previously published pulse sequences and routine processing parameters.
- BBFO broadband observe
- Solution-phase NMR spectra were also recorded on a Bruker Avance III HD NMR instrument at 400 MHz for 'H using a 30 degree pulse angle, a spectral width of 8000 Hz and 128k points of acquisition. FID were zero-filled to 256k points and a line broadening of 0.3 Hz was applied before Fourier transform.
- LC method A Analytical reverse phase UPLC using an Acquity UPLC BEH Cis column (50 x 2.1 mm, 1.7 pm particle) made by Waters (pn: 186002350), and a dual gradient run from 1-99% mobile phase B over 3.0 minutes.
- Mobile phase A H2O (0.05 % CF3CO2H).
- Mobile phase B CH3CN (0.035 % CF3CO2H).
- LC method C Reversed phase UPLC using an Acquity UPLC BEH Cis column (50 x 2.1 mm, 1.7 pm particle) made by Waters (pn: 186002350), and a dual gradient run from 30-99% mobile phase B over 2.9 minutes.
- Mobile phase A H2O (0.05 % CF3CO2H).
- Mobile phase B CH3CN (0.035 % CF3CO2H).
- LC method I UPLC Luna Cis(2) 50 x 3 mm 3 pm. run: 2.5 min.
- Mobile phase Initial 95% H2O 0.1% FA / 5%MeCN 0.1% FA, linear grad to 95% MeCN 0.1% FA over 1.3 min, hold 1.2 min 95% CH3CN 0.1% FA,.T: 45C, Flow: 1.5 mL/min.
- LC method J UPLC SunFire Cis 75 x 4.6 mm 3.5 pm, run: 6 min. Mobile phase conditions Initial 95% H2O + 0.1% FA/5% CH3CN + 0.1% FA, linear gradient to 95% CFLCN for 4 min, hold for 2 min at 95% CH3CN. T:45 °C, Flow: 1.5 mL/min.
- LC method L Luna Cis 3.0 x 50 mm 3.0 pM, Temp: 45 °C, Flow: 2.0 mL/min, Run Time: 3 minutes.
- Mobile Phase Initial 95% H2O (0.1% Formic Acid) and 5% CH3CN (0.1% FA) linear gradient to 95% CH3CN (0.1% FA) for 2.0 min then hold at 95% CH3CN (0.1% FA) for 1.0 min.
- X-ray powder diffraction (XRPD) spectra were recorded at room temperature (25 ⁇ 2 °C) in transmission mode using a PANalytical Empyrean system equipped with a sealed tube source and a PIXcel ID Medipix-3 detector (Malvern PANalytical Inc, Westborough, Massachusetts).
- the X-Ray generator operated at a voltage of 45 kV and a current of 40 mA with copper radiation (1.54060 A).
- the powder sample was placed on a 96 well sample holder with Mylar fdm and loaded into the instrument. The sample was scanned over a range of about 3 to about 40 °20 with a step size of 0.0131303 °20 and 49 s per step.
- Bruker-Biospin 400 MHz wide-bore spectrometer equipped with Bruker-Biospin 4 mm HFX probe was used. Samples were packed into 4 mm ZrCh rotors and spun under Magic Angle Spinning (MAS) conditions with spinning speed typically set to 12.5 kHz. The proton relaxation time was measured using a 'H MAS Ti saturation recovery relaxation experiment in order to set up proper recycle delay of the 13 C cross-polarization (CP) MAS experiments. The CP contact time of carbon CPMAS experiments was set to 2 ms. A CP proton pulse with linear ramp (from 50% to 100%) was employed. The carbon Hartmann- Hahn match was optimized on an external reference sample (glycine). All carbon spectra were recorded with proton decoupling using the TPPM15 decoupling sequence with a field strength of approximately 100 kHz.
- MAS Magic Angle Spinning
- Thermal gravimetric analysis was performed using a TA5500 Discovery TGA instrument. A sample of approximately 1-10 mg was scanned from room temperature to 300 °C at a heating rate of 10 °C/min while purging with nitrogen.
- CDMT 2-chloro-4,6-dimethoxy-l,3,5-triazine
- DIEA A/f-diisopropylethylamine
- HPLC high-performance liquid chromatography
- HPMCAS hydroxypropylmethylcellulose acetate succinate
- the organic phase was washed with water (2.1 L), 2.1 L of brine, dried over magnesium sulfate, filtered over Celite and concentrated in vacuo affording a light orange oil which had a silt in the slurry.
- the mixture was diluted with -500 mL of heptane and filtered using an M filter. The precipitate was washed with 250 mL of heptane.
- Step 2 tert-ButylN-tert-butoxycarbonyl-N-[4-chloro-6-(2,6- dimethylphenyl)pyrinudin-2-yl]carbamate.
- Step 2 Methyl (2R)-2-(tert-butoxycarbonylanuno)-3-(3-iodo-l- bicyclo[l.1.1 ]pentanyl)propanoate
- reaction mixture was concentrated under reduced pressure, and the resulting residue was purified by reverse phase chromatography on 50 g Cis RediSep Rf gold column using a 5-100% gradient of acetonitrile in pure water to provide methyl (2/ )-2-(Z /7-butoxycarbonyla ino)-3-(3-iodo- l - bicyclo[l .l .l]pentanyl)propanoate (1.64 g, 45%) clear oil that crystallized on standing.
- Triethylborane (in hexanes) (0.5 mL of 1 M, 0.5000 mmol) was added dropwise to a solution of methyl (27?)-2-(tert-butoxycarbonylamino)-3-(3-iodo-l- bicyclo[l.l.l]pentanyl)propanoate (1.64 g, 4.1287 mmol), 2,6-lutidine (1.288 g, 1.4 mL, 12.020 mmol), and tris(trimethylsilyl)silane (3.0628 g, 3.8 mL, 12.317 mmol) in anhydrous THF (9 mL), and left stirring at room temperature over week-end.
- reaction mixture was concentrated under reduced pressure and the resulting residue was purified on silica gel with a 40 g column using a 0-50% ethyl acetate in heptanes gradient to provide methyl (2/?)-3-( l - bicyclo[ 1.1.1] pentanyl)-2-(terLbutoxycarbonylamino)propanoate (766 mg, 65%) as a pale yellow powder.
- iBH4 (350 mg, 16.067 mmol) was added to a solution of methyl (2 ?)-3-(l- bicyclo[ l .l . l]pentanyl)-2-(/c/7-butoxycarbonylamino)propanoate (1.46 g, 5.1497 mmol) in THF (15 mL) maintained at 0 °C with an ice water bath. The reaction mixture was maintained at this temperature for 30 min and then left to warm up and stirred at room temperature for 2 h. The reaction was then cooled down to 0 °C and quenched by the addition of an aqueous saturated solution of ammonium chloride (15 mL).
- Retention time 1.73 minutes.
- the rest of the crude material was dissolved back in DCM (50 mL), and washed with 0.2 M aqueous HC1 (50 mL) until hydrogen gas stopped evolving.
- the aqueous phase was separated, and washed with DCM (50 mL).
- the organic phases were combined, dried with magnesium sulfate, filtered, and concentrated under reduced pressure to provide /c/ -butyl A-[(l/ )- l-( l-bicyclo[ l .1 , l]pentanyl methyl)-2-hydroxy-ethyl]carbamate (1.15 g, 88%) as a white solid.
- Step 1 3-[[4-[(2R)-2-Atnino-3-(l-bicyclo[l.1.1 ]pentanyl)propoxy]-6-(2, 6- dimethylphenyl)pyrinudin-2-yl]sulfamoyl]benzoic acid
- Step 2 3-[[4-[(2R)-3-(l-Bicyclo[l.l.l]pentanyl)-2-[(6-tert-butylfuro[2,3-b]pyrazin-
- Step 3 (11R)-11-(1 -Bicyclofl.1.1 ]pentanylmethyl)-l 2-[( 6-tert-butylfuro[2, 3- b]pyrazin-2-yl)methyl]-6-(2,6-dimethylphenyl)-2,2-dioxo-9-oxa-216-thia-3,5,12,19-
- amorphous Compound I 3.5 g was loaded into a 100 mL reactor (Mettler Toledo Easy Max 102), followed by 50 mL of EtOH. The temperature was increased as fast as possible to 73 °C and held at 73 °C for 1.5 h.
- X-ray powder diffraction (XRPD) results are shown in FIG. 1 and the table below.
- thermogram (FIG. 2) showed 0.04% weight loss from 30 °C to 200 °C.
- thermogram (FIG. 3) showed an endothermic peak around 198 °C with a heat of fusion of 17 J/g. 5.
- a single crystal of Compound I Neat Form A was obtained via desolvation of Compound I EtOH solvate.
- the structure was solved and refined using SHELX programs (Sheldrick, G.M., Acta Cryst., (2008) A64, 112-122) and results are summarized in the table below.
- Compound I Neat Form B was prepared by exposing Compound I Hydrate Form A to 3% humidity for 3 h using an Anton Paar CHC+ chamber (humidity XRD).
- Compound I Neat Form B was prepared by placing Compound I Hydrate Form A in an ssNMR rotor into an oven at 125 °C for 3 h.
- X-ray powder diffraction (XRPD) spectrum of Compound I Neat Form B is shown in FIG. 5 and in the table below.
- thermogram (FIG. 8) showed 0.05% weight loss from 30 °C to 40 °C and
- thermogram (FIG. 9) showed an endothermic peak around 237 °C with a heat of fusion of 42 J/g.
- thermogram (FIG. 14) showed an endothermic peak around 212 °C with a heat of fusion of 30 J/g.
- a single crystal of Compound I Neat Form E was grown by dissolving Compound I Neat Form A in methyl acetate, and crystallizing with addition of heptane. Once isolated, this material was vacuum dried at 50 °C for 2 days.
- X-ray powder diffraction (XRPD) results are shown in FIG. 16 and the table below.
- X-ray powder diffraction (XRPD) results are shown in FIG. 18 and the table below.
- thermogram (FIG. 21) showed 5.29% weight loss from 30 °C to 90 °C.
- X-ray powder diffraction (XRPD) results are shown in FIG. 24 and the table below.
- X-ray powder diffraction (XRPD) results are shown in FIG. 26 and the table below.
- thermogram (FIG. 30) showed an endothermic peak around 100 °C with a heat of fusion of 91 J/g, followed by a second endothermic peak around 216 °C with a heat of fusion of 33 J/g.
- X-ray powder diffraction (XRPD) results are shown in FIG. 32 and the table below.
- thermogram (FIG. 33) showed 0.39% weight loss from 30 °C to 60 °C, and 0.20% weight loss from 60 °C to 190 °C.
- FIG. 35 and in the table below.
- X-ray powder diffraction (XRPD) results are shown in FIG. 38 and the table below.
- X-ray powder diffraction (XRPD) results are shown in FIG. 40 and the table below.
- X-ray powder diffraction (XRPD) results are shown in FIG. 42 and the table below.
- X-ray powder diffraction (XRPD) results are shown in FIG. 44 and the table below.
- FIG. 45 and in the table below.
- X-ray powder diffraction (XRPD) results are shown in FIG. 46 and the table below.
- X-ray powder diffraction (XRPD) results are shown in FIG. 50 and the table below.
- X-ray powder diffraction (XRPD) spectrum for Compound I Spray Dried Dispersion (SDD) 1A was recorded at room temperature in transmission mode using a PANalytical Empyrean system equipped with a sealed tube source and a PIXcellD detector (Malvern PANalytical Inc, Almelo, The Netherlands).
- the X-Ray generator operated at a voltage of 45 kV and a current of 40 mA with copper radiation (1.540598 A).
- the powder sample was placed on an indented area within a zero background holder and flattened with a glass slide then loaded into the instrument. The sample was scanned over the range of about 4° to about 40° 20 with a step size of 0.0131° and 14s per step.
- SDD 1A 50% Amorphous Compound I and 50% Polymer Solvent: DCM/MeOH (50/50 %w/w) Polymer: HPMCAS-H
- X-ray powder diffraction (XRPD) data of Compound I SDD IB were recorded at room temperature in transmission mode using a PANalytical Empyrean system and are shown in FIG. 57.
- X-ray powder diffraction (XRPD) data of Compound I SDD IE was collected at room temperature in transmission mode using a PANalytical Empyrean system, and is shown in FIG. 63 .
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pulmonology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Engineering & Computer Science (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Crystalline forms of Compound I, pharmaceutically acceptable salts, solvates, hydrates, and cocrystals thereof are disclosed. Pharmaceutical compositions comprising the same, methods of treating cystic fibrosis using the same, and methods for making the same are also disclosed.
Description
SOLID FORMS OF MODULATORS OF CYSTIC FIBROSIS TRANSMEMBRANE CONDUCTANCE REGULATOR
[0001] This application claims the benefit of priority of U.S. Provisional Application No. 63/587,829, filed October 4, 2023, the contents of which are incorporated herein by reference in their entirety.
[0002] Disclosed herein are crystalline and amorphous solid forms of a Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) modulator, pharmaceutical compositions thereof, methods of treating cystic fibrosis with any of the foregoing, and processes for making crystalline and amorphous forms.
[0003] Cystic fibrosis (CF) is a recessive genetic disease that affects approximately 83,000 children and adults worldwide. Despite progress in the treatment of CF, there is no cure.
[0004] In patients with CF, mutations in CFTR endogenously expressed in respiratory epithelia lead to reduced apical anion secretion causing an imbalance in ion and fluid transport. The resulting decrease in anion transport contributes to enhanced mucus accumulation in the lung and accompanying microbial infections that ultimately cause death in CF patients. In addition to respiratory disease, CF patients typically suffer from gastrointestinal problems and pancreatic insufficiency that, if left untreated, result in death. In addition, the majority of males with cystic fibrosis are infertile, and fertility is reduced among females with cystic fibrosis.
[0005] Sequence analysis of the CFTR gene has revealed a variety of disease-causing mutations (Cutting, G. R. et al. (1990) Nature 346:366-369; Dean, M. et al. (1990) Cell 61 :863:870; and Kerem, B-S. et al. (1989) Science 245:1073-1080; Kerem, B-S et al. (1990) Proc. Natl. Acad. Sci. USA 87:8447-8451). To date, greater than 2000 mutations in the CF gene have been identified; currently, the CFTR2 database contains information on only 322 of these identified mutations, with sufficient evidence to define 281 mutations as disease causing. The most prevalent disease-causing mutation is a deletion of phenylalanine at position 508 of the CFTR amino acid sequence and is commonly referred to as the F508del mutation. This mutation occurs in approximately 70% of the cases of cystic fibrosis and is associated with severe disease.
[0006] The deletion of residue 508 in CFTR prevents the nascent protein from folding correctly. This results in the inability of the mutant protein to exit the endoplasmic reticulum (ER) and traffic to the plasma membrane. As a result, the number of CFTR channels for
anion transport present in the membrane is far less than observed in cells expressing wildtype CFTR, i.e., CFTR having no mutations. In addition to impaired trafficking, the mutation results in defective channel gating. Together, the reduced number of channels in the membrane and the defective gating lead to reduced anion and fluid transport across epithelia. (Quinton, P. M. (1990), FASEB J. 4: 2709-2727). The channels that are defective because of the F508del mutation are still functional, albeit less functional than wild-type CFTR channels. (Dalemans et al. (1991), Nature Lond. 354: 526-528; Pasyk and Foskett (1995), J. Cell. Biochem. 270: 12347-50). In addition to F508del, other disease-causing mutations in CFTR that result in defective trafficking, synthesis, and/or channel gating could be up- or down-regulated to alter anion secretion and modify disease progression and/or severity.
[0007] CFTR is a cAMP/ATP-mediated anion channel that is expressed in a variety of cell types, including absorptive and secretory epithelia cells, where it regulates anion flux across the membrane, as well as the activity of other ion channels and proteins. In epithelial cells, normal functioning of CFTR is critical for the maintenance of electrolyte transport throughout the body, including respiratory and digestive tissue. CFTR is composed of approximately 1480 amino acids that encode a protein which is made up of a tandem repeat of transmembrane domains, each containing six transmembrane helices and a nucleotide binding domain. The two transmembrane domains are linked by a large, polar, regulatory (R)-domain with multiple phosphorylation sites that regulate channel activity and cellular trafficking.
[0008] Chloride transport takes place by the coordinated activity of ENaC and CFTR present on the apical membrane and the Na+-K+-ATPase pump and Cl- channels expressed on the basolateral surface of the cell. Secondary active transport of chloride from the luminal side leads to the accumulation of intracellular chloride, which can then passively leave the cell via Cl’ channels, resulting in a vectorial transport. Arrangement of Na+/2C17K+ cotransporter, Na+-K+-ATPase pump and the basolateral membrane K+ channels on the basolateral surface and CFTR on the luminal side coordinate the secretion of chloride via CFTR on the luminal side. Because water is probably never actively transported itself, its flow across epithelia depends on tiny transepithelial osmotic gradients generated by the bulk flow of sodium and chloride.
[0009] A number of CFTR-modulating compounds have recently been identified.
However, compounds that can treat or reduce the severity of the cystic fibrosis and other
CFTR-mediated diseases, and particularly the more severe forms of these diseases, are still needed.
[0010] Solid forms of pharmaceutical compounds are of interest to the industry due to ease of manufacture, storage, and administration. A number of distinct solid forms may be contemplated for a particular compound, depending upon its chemical and physical properties.
[0011] Crystalline forms are of interest in the pharmaceutical industry, where the control of the crystalline form(s) of the active ingredient may be desirable or even required.
Reproducible processes for producing a compound with a particular crystalline form in high purity may be desirable for compounds intended to be used in pharmaceuticals, as different crystalline forms may possess different properties. For example, different crystalline forms may possess different chemical, physical, and/or pharmaceutical properties. In some embodiments, one or more crystalline forms disclosed herein may exhibit a higher level of purity, chemical stability, and/or physical stability. Certain crystalline forms (e.g., crystalline free form, crystalline salt, crystalline salt solvate, and crystalline salt hydrate forms of Compound I (collectively referred to as “crystalline forms”)) may exhibit lower hygroscopicity. Thus, the crystalline forms of this disclosure may provide advantages during drug substance manufacturing, storage, and handling. Thus, pharmaceutically acceptable crystalline forms of Compound I may be particularly useful for the production of drugs for the treatment of CFTR-mediated diseases.
[0012] Amorphous forms of therapeutic compounds may also be of interest in the pharmaceutical industry, where crystalline forms are not especially bioavailable. Some amorphous forms may improve bioavailability and thus allow for administration of reduced dosages. For some compounds, amorphous forms provide the most biologically accessible form of the therapeutic.
[0013] Thus, one aspect of the disclosure provides solid forms (crystalline and amorphous) of a CFTR-modulating compound (R)-7-(bicyclo[l. l.l]pentan-l-ylmethyl)-6- ((6-(tert-butyl)furo[2,3-b]pyrazin-2-yl)methyl)-16-(2,6-dimethylphenyl)-9-oxa-3-thia-2,6- diaza-l(2,4)-pyrimidina-4(l,3)-benzenacyclononaphan-5-one 3,3-dioxide (Compound I) and pharmaceutically acceptable salts thereof. Compound I can be depicted as having the following structure:

[0014] In some embodiments, the crystalline form of Compound I is Compound I Neat Form A. In some embodiments, the crystalline form of Compound I is Compound I Neat Form B. In some embodiments, the crystalline form of Compound I is Compound I Neat Form C. In some embodiments, the crystalline form of Compound I is Compound I Neat Form D. In some embodiments, the crystalline form of compound I is Compound I Neat Form E. In some embodiments, the crystalline form of compound I is Compound I Compressed form A. In some embodiments, the crystalline form of compound I is Compound I Compressed form E. In some embodiments, the crystalline form of Compound I is Compound I EtOH Solvate Form B. In some embodiments, the crystalline form of Compound I is Compound I MeOH solvate. In some embodiments, the crystalline form of Compound I is Compound I NPA solvate. In some embodiments, the crystalline form of Compound I is Compound I MeOAc Solvate Form A. In some embodiments, the crystalline form of Compound I is Compound I MeOAc Solvate Form B. In some embodiments, the crystalline form of Compound I is Compound I MeOAc Solvate Form C. In some embodiments, the crystalline form of Compound I is Compound I Hydrate Form A. In some embodiments, the crystalline form of Compound I is Compound I Hydrate Form B. In some embodiments, the crystalline form of Compound I is Compound I Hydrate Form C. In some embodiments, the crystalline form of Compound I is Compound I EtOH Solvate Hydrate. In some embodiments, the crystalline form of Compound I is Compound I MeOH Solvate Hydrate. In some embodiments, the crystalline form of Compound I is Compound I IPA Solvate Hydrate. In some embodiments, the crystalline form of Compound I is Compound I MeOAc Solvate Hydrate.
[0015] In some embodiments, the solid form of Compound I is in amorphous form.
[0016] In some embodiments, Compound I is formulated as a solid (e.g., spray-dried) dispersion.
[0017] Other aspects of the disclosure provide pharmaceutical compositions comprising Compound I in any of the pharmaceutically acceptable crystalline forms disclosed herein,
which compositions may further include at least one additional active pharmaceutical ingredient and/or at least one carrier. Yet other aspects of the disclosure are methods of treating the CFTR-mediated disease cystic fibrosis comprising administering Compound I in any of the pharmaceutically acceptable solid forms disclosed herein, optionally as part of a pharmaceutical composition comprising at least one additional component (such as a carrier or additional active agent), to a subject in need thereof. A further aspect of the disclosure provides processes of making the solid forms of Compound I disclosed herein.
[0018] Also disclosed are pharmaceutical compositions comprising combinations of a solid form Compound I as described herein or amorphous Compound I formulated as a solid (e.g., spray-dried) dispersion with (A)-l-(2,2-difluorobenzo[d][l,3]dioxol-5-yl)-N-(l-(2,3- dihydroxypropyl)-6-fluoro-2-(l -hydroxy -2-methylpropan-2-yl)-lH-indol-5- yl)cyclopropanecarboxamide (Compound II) and/or pharmaceutically acceptable salts thereof
 and/or with A-(5-hydroxy-2,4-di-/er/-butyl-phenyl)-4-oxo-lH-quinoline-3-carboxamide
and/or with A-(5-hydroxy-2,4-di-/er/-butyl-phenyl)-4-oxo-lH-quinoline-3-carboxamide
(Compound III)
 or A-(2-(tert-butyl)-5-hydroxy-4-(2-(methyl-d3)propan-2-yl-l,l,l,3,3,3-d6)phenyl)-4-oxo- l,4-dihydroquinoline-3 -carboxamide (Compound Ill-d)
or A-(2-(tert-butyl)-5-hydroxy-4-(2-(methyl-d3)propan-2-yl-l,l,l,3,3,3-d6)phenyl)-4-oxo- l,4-dihydroquinoline-3 -carboxamide (Compound Ill-d)

[0019] Also disclosed are methods of using a solid form of Compound I described herein or amorphous Compound I formulated as a solid (e g., spray-dried) dispersion alone or in
combination with other CFTR modulators to treat cystic fibrosis. In certain embodiments, the solid form of Compound I as disclosed herein or amorphous Compound I formulated as a solid (e.g., spray-dried) dispersion is administered with Compound II and/or Compound III or Compound Ill-d, either in a single pharmaceutical composition or in multiple compositions to treat cystic fibrosis. In some embodiments, a solid form of Compound I as described herein or amorphous Compound I formulated as a solid (e.g., spray-dried) dispersion is used in combination with (6R,12R)-17-amino-12-methyl-6,15- bis(trifluoromethyl)-13,19-dioxa-3,4,18-triazatricyclo[12.3.1.12,5]nonadeca-l(18),2,4,14,16- pentaen-6-ol (Compound IV), a deuterated derivative, or pharmaceutically acceptable salt thereof in a the treatment of cystic fibrosis. In some embodiments, a solid form of Compound I as described herein or amorphous Compound I formulated as a solid (e.g., spray-dried) dispersion is used in combination with (67?)-17-amino-12,12-dimethyl-6,15- bis(trifluoromethyl)-19-oxa-3,4,13,18-tetrazatricyclo [12.3.1.12,5]nonadeca-l(18),2,4,14,16- pentaen-6-ol (Compound V), a deuterated derivative, or pharmaceutically acceptable salt thereof to treat cystic fibrosis.
[0020] A further aspect of the disclosure provides processes of making the solid forms of Compound I disclosed herein.
[0021] Another aspect of the invention provides solid forms of Compound I, or amorphous Compound I formulated as a solid (e.g., spray-dried) dispersion for use in any of the methods described herein.
Brief Description of the Figures
[0022] FIG. 1 provides an XRPD pattern of Compound I Neat Form A.
[0023] FIG. 2 provides a TGA curve for crystalline Compound I Neat Form A.
[0024] FIG. 3 provides a DSC analysis of Compound I Neat Form A.
[0025] FIG. 4 provides a 13C ssNMR spectrum of Compound I Neat Form A.
[0026] FIG. 5 provides an XRPD pattern of Compound I Neat Form B.
[0027] FIG. 6 provides a 13C ssNMR spectrum of Compound I Neat Form B.
[0028] FIG. 7 provides an XRPD pattern of Compound I Neat Form C.
[0029] FIG. 8 provides a TGA curve for Compound I Neat Form C.
[0030] FIG. 9 provides a DSC analysis of Compound I Neat Form C.
[0031] FIG. 10 provides a 13C ssNMR spectrum of Compound I Neat Form C.
[0032] FIG. 11 provides a 13C ssNMR spectrum of Compound I Neat Form D.
[0033] FIG. 12 provides an XRPD pattern of Compound I Neat Form E.
[0034] FIG. 13 provides a TGA curve for Compound I Neat Form E.
[0035] FIG. 14 provides a DSC analysis of Compound I Neat Form E.
[0036] FIG. 15 provides a 13C ssNMR spectrum of Compound I Neat Form E.
[0037] FIG. 16 provides an XRPD pattern of Compound I Compressed Form E.
[0038] FIG. 17 provides a 13C ssNMR spectrum of Compound I Compressed Form E.
[0039] FIG. 18 provides an XRPD pattern of Compound I Compressed Form A.
[0040] FIG. 19 provides a 13C ssNMR spectrum of Compound I Compressed Form A.
[0041] FIG. 20 provides an XRPD pattern of Compound I EtOH Solvate Form B.
[0042] FIG. 21 provides a TGA curve for Compound I EtOH Solvate Form B.
[0043] FIG. 22 provides a DSC analysis of Compound I EtOH Solvate Form B.
[0044] FIG. 23 provides a 13C ssNMR spectrum of Compound I EtOH Solvate Form B.
[0045] FIG. 24 provides an XRPD pattern of Compound I MeOH Solvate.
[0046] FIG. 25 provides a 13C ssNMR spectrum of Compound I MeOH Solvate.
[0047] FIG. 26 provides an XRPD pattern of Compound I NPA solvate.
[0048] FIG. 27 provides a 13C ssNMR spectrum of Compound I NPA solvate.
[0049] FIG. 28 provides an XRPD pattern of Compound I MeOAc Solvate Form A.
[0050] FIG. 29 provides a TGA curve for Compound I MeOAc Solvate Form A.
[0051] FIG. 30 provides a DSC analysis of Compound I MeOAc Solvate Form A.
[0052] FIG. 31 provides a 13C ssNMR spectrum of Compound I MeOAc Solvate Form A.
[0053] FIG. 32 provides an XRPD pattern of Compound I MeOAc Solvate Form B.
[0054] FIG. 33 provides a TGA curve for Compound I MeOAc Solvate Form B.
[0055] FIG. 34 provides a DSC analysis of Compound I MeOAc Solvate Form B.
[0056] FIG. 35 provides a 13C ssNMR spectrum of Compound I MeOAc Solvate Form B.
[0057] FIG. 36 provides an XRPD pattern of Compound I MeOAc Solvate Form C.
[0058] FIG. 37 provides a 13C ssNMR spectrum of Compound I MeOAc Solvate Form C.
[0059] FIG. 38 provides an XRPD pattern of Compound I Hydrate Form A.
[0060] FIG. 39 provides a 13C ssNMR spectrum of Compound I Hydrate Form A.
[0061] FIG. 40 provides an XRPD pattern of Compound I Hydrate Form B.
[0062] FIG. 41 provides a 13C ssNMR spectrum of Compound I Hydrate Form B.
[0063] FIG. 42 provides an XRPD pattern of Compound I Hydrate Form C.
[0064] FIG. 43 provides a 13C ssNMR spectrum of Compound I Hydrate Form C.
[0065] FIG. 44 provides an XRPD pattern of Compound I EtOH Solvate Hydrate.
[0066] FIG. 45 provides a 13C ssNMR spectrum of Compound I EtOH Solvate Hydrate.
[0067] FIG. 46 provides an XRPD pattern of Compound I MeOH Solvate Hydrate.
[0068] FIG. 47 provides a 13C ssNMR spectrum of Compound I MeOH Solvate Hydrate.
[0069] FIG. 48 provides an XRPD pattern of Compound I IPA Solvate Hydrate.
[0070] FIG. 49 provides a 13C CPMAS spectrum of Compound I IPA Solvate Hydrate.
[0071] FIG. 50 provides an XRPD pattern of Compound I MeOAc Solvate Hydrate.
[0072] FIG. 51 provides a 13C ssNMR spectrum of Compound I MeOAc Solvate Hydrate.
[0073] FIG. 52 provides an XRPD pattern of amorphous Compound I.
[0074] FIG. 53 provides a 13C ssNMR spectrum of amorphous Compound I.
[0075] FIG. 54 provides an XRPD pattern of Compound I SDD 1A.
[0076] FIG. 55 provides a 13C ssNMR spectrum of Compound I SDD 1A.
[0077] FIG. 56 provides a DSC analysis of Compound I SDD 1 A.
[0078] FIG. 57 provides an XRPD pattern of Compound I SDD IB.
[0079] FIG. 58 provides a DSC analysis of Compound I SDD IB.
[0080] FIG. 59 provides an XRPD pattern of Compound I SDD 1C.
[0081] FIG. 60 provides a DSC analysis of Compound I SDD 1C.
[0082] FIG. 61 provides an XRPD pattern of Compound I SDD ID.
[0083] FIG. 62 provides a DSC analysis of Compound I SDD ID.
[0084] FIG. 63 provides an XRPD pattern of Compound I SDD IE.
[0085] FIG. 64 provides a DSC analysis of Compound I SDD IE.
Definitions
[0086] “Compound I” as used throughout this disclosure refers to the CFTR corrector, (7?)-7-(bicyclo[l.l.l]pentan-l-ylmethyl)-6-((6-(tert-butyl)furo[2,3-b]pyrazin-2-yl)methyl)- 16-(2,6-dimethylphenyl)-9-oxa-3-thia-2,6-diaza-l(2,4)-pyrimidina-4(l,3)- benzenacyclononaphan-5-one 3,3-dioxide, which can be depicted as having the following structure:
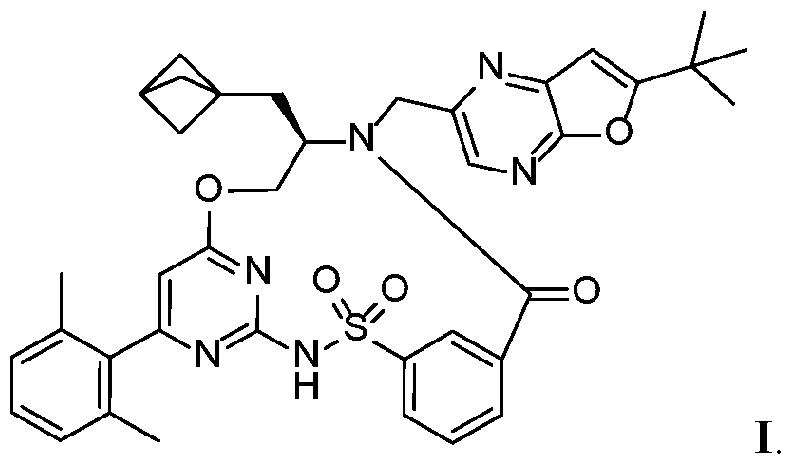
[0087] Compound I and methods for making and using Compound I are disclosed in
PCT/US2023/017627 (incorporated herein by reference).
[0088] “Compound II” as used throughout this disclosure refers to (R)-l-(2,2- difluorobenzo[d][l, 3]di oxol-5-yl)-,V-( l-(2, 3-dihydroxypropyl)-6-fluoro-2-(l -hydroxy -2- methylpropan-2-yl)-lH-indol-5-yl)cyclopropanecarboxamide, which can be depicted as having the following structure:

[0089] Compound II may be in the form of a pharmaceutically acceptable salt. Compound II and methods of making and using Compound II are disclosed in WO 2010/053471, WO 2011/119984, and WO 2015/160787, each incorporated herein by reference.
[0090] “Compound III” as used throughout this disclosure refers to N-[2,4-bis(l, 1- dimethylethyl)-5-hydroxyphenyl]-l,4-dihydro-4-oxoquinoline-3-carboxamide (also known as /V-(5-hydroxy-2,4-di-tert-butyl-phenyl)-4-oxo-l H-quinoline-3-carboxamide) which can be depicted as having the following structure:
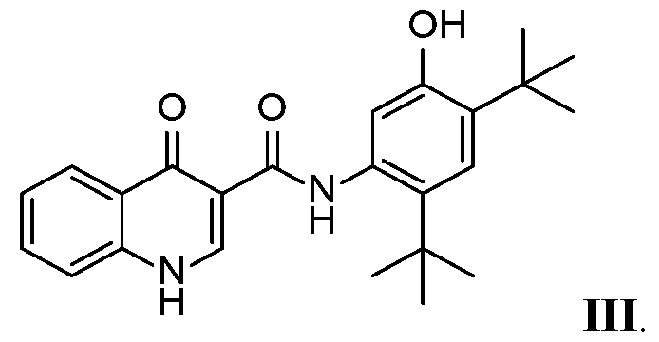
[0091] Compound III may also be in the form of a pharmaceutically acceptable salt. Compound III and methods of making and using Compound III are disclosed in WO 2006/002421, WO 2007/079139, and WO 2010/019239, each incorporated herein by reference.
[0092] In some embodiments, a deuterated derivative of Compound III (Compound Ill-d) is employed in the compositions and methods disclosed herein. A chemical name for Compound Ill-d is A-(2-(ferCbutyl)-5-hydroxy-4-(2-(methyl-d3)propan-2-yl-l,l,l,3,3,3- d6)phenyl)-4-oxo-l,4-dihydroquinoline-3 -carboxamide, which can be depicted as having the following structure:
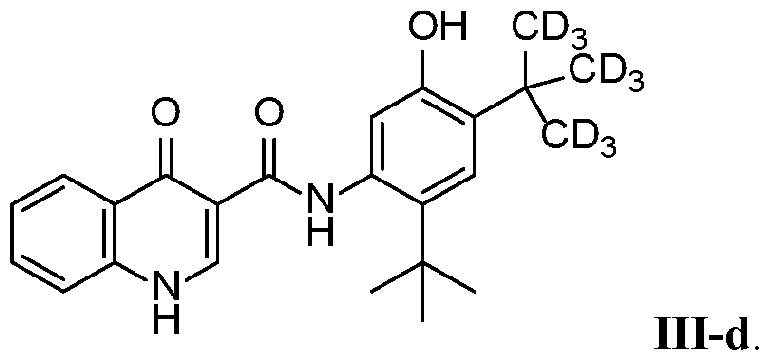
[0093] Compound Ill-d may be in the form of a pharmaceutically acceptable salt. Compound Ill-d and methods of making and using Compound Ill-d are disclosed in WO 2012/158885 and WO 2014/078842, incorporated herein by reference.
[0094] “Compound IV” as used throughout this disclosure refers to (6R,12R)-17-amino- 12-methyl-6, 15-bis(trifluoromethyl)-13, 19-dioxa-3,4, 18-tri azatri cyclo[12.3.1.12,5]nonadeca- l(18),2,4,14,16-pentaen-6-ol. Compound IV may also be in the form of a deuterated derivative or pharmaceutically acceptable salt. Methods of making and using Compound IV, deuterated derivatives and pharmaceutically acceptable salts thereof, are described in WO 2022/032068, incorporated herein by reference.
[0095] “Compound V” as used throughout this disclosure refers to (6J?)-17-amino-12,12- dimethyl-6,15-bis(trifluoromethyl)-19-oxa-3,4,13,18-tetrazatricyclo [12.3.1.12,5]nonadeca- l(18),2,4,14,16-pentaen-6-ol. Compound V may also be in the form of a deuterated derivative or pharmaceutically acceptable salt. Methods of making and using Compound V, deuterated derivatives and pharmaceutically acceptable salts thereof, are described in WO 2022/109573, incorporated herein by reference.
[0096] As used herein, “CFTR” means cystic fibrosis transmembrane conductance regulator.
[0097] As used herein, the terms “CFTR modulator” and “CFTR modulating compound” interchangeably refer to a compound that increases the activity of CFTR. The increase in activity resulting from a CFTR modulator includes but is not limited to compounds that correct, potentiate, stabilize, and/or amplify CFTR.
[0098] As used herein, the term “CFTR corrector” refers to a compound that facilitates the processing and trafficking of CFTR to increase the amount of CFTR at the cell surface. Compounds I and II disclosed herein are CFTR correctors.
[0099] As used herein, the term “CFTR potentiator” refers to a compound that increases the channel activity of CFTR protein located at the cell surface, resulting in enhanced ion transport. Compounds III, Ill-d, IV, and V disclosed herein are CFTR potentiators. It will be appreciated that when a description of a combination of Compound I and other specified CFTR modulating agents is provided herein, reference to “Compound III or Ill-d” in connection with the combination means that either Compound III or Compound Ill-d, but not both, is included in the combination.
[00100] As used herein, the term “active pharmaceutical ingredient” (“API”) or “therapeutic agent” refers to a biologically active compound.
[00101] It will be appreciated that certain compounds of this invention may exist as separate stereoisomers or enantiomers and/or mixtures of those stereoisomers or enantiomers. As used in the chemical structures disclosed herein, a “wedge” ( *) or “hash” (•••"') bond to a stereogenic atom indicates a chiral center of known absolute stereochemistry (i.e. one stereoisomer). As used in the chemical structures disclosed herein, a “wavy” bond ( ) to a stereogenic atom indicates a chiral center of unknown absolute stereochemistry (i.e. one stereoisomer). As used in the chemical structures disclosed herein, a “wavy” bond ( ) to a double-bonded carbon indicates a mixture of E/Z isomers. As used in the chemical structures
disclosed herein, a ' (“straight”) bond to a stereogenic atom indicates where there is a mixture (e.g., a racemate or enrichment). As used herein, two
 (“straight”) bonds to a double-bonded carbon indicates that the double bond possesses the E/Z stereochemistry as A drawn. As used in the chemical structures disclosed herein, a ' (i.e., a “wavy” line perpendicular to a “straight” bond to group “A”) indicates that group “A” is a substituent whose point of attachment is at the end of the bond that terminates at the “wavy” line.
(“straight”) bonds to a double-bonded carbon indicates that the double bond possesses the E/Z stereochemistry as A drawn. As used in the chemical structures disclosed herein, a ' (i.e., a “wavy” line perpendicular to a “straight” bond to group “A”) indicates that group “A” is a substituent whose point of attachment is at the end of the bond that terminates at the “wavy” line.
[00102] As used herein, the term “pharmaceutically acceptable solid form” refers to a solid form of Compound I of this disclosure that includes the crystalline form (e g., crystalline free form, crystalline salt, crystalline salt solvate, and crystalline salt hydrate) of Compound I that is nontoxic and suitable for use in pharmaceutical compositions. The term “pharmaceutically acceptable solid form” as used herein also refers to the amorphous form of Compound I and solid dispersions comprising that amorphous form.
[00103] The terms “patient” and “subject” are used interchangeably and refer to an animal including humans.
[00104] The terms “effective dose” and “effective amount” are used interchangeably herein and refer to that amount of a compound that produces the desired effect for which it is administered (e.g., improvement in CF or a symptom of CF, or lessening the severity of CF or a symptom of CF). The exact amount of an effective dose will depend on the purpose of the treatment and will be ascertainable by one skilled in the art using known techniques (see, e.g., Lloyd (1999) The Art, Science and Technology of Pharmaceutical Compounding).
[00105] As used herein, the terms "treatment," "treating," and the like generally mean the improvement of CF or one or more of its symptoms or lessening the severity of CF or one or more of its symptoms in a subject. “Treatment,” as used herein, includes, but is not limited to, the following: increased growth of the subject, increased weight gain, reduction of mucus in the lungs, improved pancreatic and/or liver function, reduction of chest infections, and/or reductions in coughing or shortness of breath. Improvements in or lessening the severity of any of these symptoms can be readily assessed according to standard methods and techniques known in the art.
[00106] As used herein, the term “in combination with,” when referring to two or more compounds, agents, or additional active pharmaceutical ingredients, means the administration of two or more compounds, agents, or active pharmaceutical ingredients to the patient prior to, concurrently with, or subsequent to each other.
[00107] The terms “about” and “approximately,” when used in connection with doses, amounts, or weight percents of ingredients of a composition or a dosage form, include the value of a specified dose, amount, or weight percent or a range of the dose, amount, or weight percent that is recognized by one of ordinary skill in the art to provide a pharmacological effect equivalent to that obtained from the specified dose, amount, or weight percent. The terms “about” and “approximately” may refer to an acceptable error for a particular value as determined by one of skill in the art, which depends in part on how the values is measured or determined. In some embodiments, the terms “about” and “approximately” mean within 15%, 10%, 5%, 4%, 3%, 2%, 1%, or 0.5% of a given value or range. In some embodiments, the terms “about” and “approximately” mean within 15% of a given value or range. In some embodiments, the terms “about” and “approximately” mean within 10% of a given value or range.
[00108] As used herein, the term “amorphous” refers to a solid material having no long- range order in the position of its molecules. Amorphous solids are generally glasses or supercooled liquids in which the molecules are arranged in a random manner so that there is no well-defined arrangement, e.g., molecular packing, and no long-range order. Amorphous solids are generally rather isotropic, i.e., exhibit similar properties in all directions and do not have definite melting points. Instead, they typically exhibit a glass transition temperature which marks a transition from glassy amorphous state to supercooled liquid amorphous state upon heating. In some embodiments, a solid material may comprise an amorphous compound, and the material may, for example, be characterized by a lack of sharp characteristic crystalline peak(s) in its XRPD spectrum (i.e., the material is not crystalline, but is amorphous, as determined by XRPD). Instead, one or several broad peaks (e.g., halos) may appear in the XRPD pattern of the material. Broad peaks are characteristic of an amorphous solid. See US 2004/0006237 for a comparison of XRPDs of an amorphous material and crystalline material. Other techniques, such as solid-state NMR, may also be used to characterize crystalline or amorphous forms.
[00109] As used herein, the terms “crystal form,” “crystalline form,” and “form” interchangeably refer to a crystal structure (or polymorph) having a particular molecular packing arrangement in the crystal lattice. Crystalline forms can be identified and distinguished from each other by the presence or absence of a specific salt, solvate, or hydrate, as well as by one or more characterization techniques including, for example, X-ray powder diffraction (XRPD), single crystal X-ray diffraction, and 13C solid-state nuclear
magnetic resonance (13C ssNMR). Accordingly, as used herein, the terms “crystalline form A of Compound I” and “crystalline potassium salt form A of Compound I” refer to unique crystalline forms that can be identified and distinguished from each other by the presence or absence of potassium alone as well as by other characterization techniques. In some embodiments, the novel crystalline forms are characterized by an X-ray powder diffractogram having one or more signals at specified two-theta values (°20).
[00110] As used herein, the term “free form” refers to a non-ionized version of the compound in the solid state. Examples of free forms include free bases and free acids.
[00111] As used herein, the term “Neat Form” refers to an unsolvated and unhydrated free form version of a compound in the solid state.
[00112] As used herein, the term “compressed form” refers to a crystalline form that has been mechanically compressed to become a different crystalline form.
[00113] As used herein, the term “solvate” refers to a crystal form comprising one or more molecules of a compound of the present disclosure and, incorporated into the crystal lattice, one or more molecules of a solvent or solvents in stoichiometric or nonstoichiometric amounts. When the solvent is water, the solvate is referred to as a “hydrate.” Other, nonlimiting examples of solvate forms include those derived from ethanol (“EtOH solvate”), methanol (“MeOH solvate”), //-propanol ( “NPA solvate”), methyl acetate (“MeOAc solvate), and isopropanol (“IP A solvate”).
[00114] In some embodiments, a solid material may comprise a mixture of crystalline solids and amorphous solids. A solid material comprising an amorphous compound may also, for example, contain up to 30% of a crystalline solid. In some embodiments, a solid material prepared to comprise an amorphous compound may also, for example, contain up to 25%, 20%, 15%, 10%, 5%, or 2% of a crystalline solid. In embodiments wherein the solid material contains a mixture of crystalline solids and amorphous solids, the characterizing data, such as XRPD and ssNMR, may contain indicators of both crystalline and amorphous solids. In some embodiments, a crystalline form of this disclosure may contain up to 30% amorphous compound. In some embodiments, a crystalline preparation of Compound I may contain up to 25%, 20%, 15%, 10%, 5%, or 2% of an amorphous solid.
[00115] As used herein, the term "substantially amorphous" refers to a solid material having little or no long-range order in the position of its molecules. For example, substantially amorphous materials have less than 15% crystallinity (e.g., less than 10% crystallinity, less
than 5% crystallinity, or less than 2% crystallinity). It is also noted that the term “substantially amorphous” includes the descriptor, “amorphous,” which refers to materials having no (0%) crystallinity.
[00116] As used herein, the term "substantially crystalline" refers to a solid material having little or no amorphous molecules. For example, substantially crystalline materials have less than 15% amorphous molecules (e.g., less than 10% amorphous molecules, less than 5% amorphous molecules, or less than 2% amorphous molecules). It is also noted that the term “substantially crystalline” includes the descriptor “crystalline,” which refers to materials that are 100% crystalline form.
[00117] As used herein, a crystalline form is "substantially pure" when it accounts for an amount by weight equal to or greater than 90% of the sum of all solid form(s) of a given compound in a sample as determined by a method in accordance with the art, such as quantitative XRPD. In some embodiments, the solid form is "substantially pure" when it accounts for an amount by weight equal to or greater than 95% of the sum of all solid form(s) in a sample. In some embodiments, the solid form is "substantially pure" when it accounts for an amount by weight equal to or greater than 99% of the sum of all solid form(s) in a sample.
[00118] As used herein, the term “XRPD” refers to the analytical characterization method of X-ray powder diffraction. XRPD patterns disclosed herein were recorded at ambient conditions in transmission or reflection geometry using a diffractometer.
[00119] As used herein, the term “ambient conditions” means room temperature, open air condition and uncontrolled humidity condition. The terms “room temperature” and “ambient temperature” mean 15 °C to 30 °C.
[00120] As used herein, the terms “X-ray powder diffractogram,” “X-ray powder diffraction pattern,” and “XRPD pattern,” interchangeably refer to an experimentally obtained pattern plotting signal positions (on the abscissa) versus signal intensities (on the ordinate).
[00121] A “signal” or “peak” as used herein refers to a point in the XRPD pattern where the intensity as measured in counts is at a local maximum. An XRPD peak is identified by its angular value as measured in degrees two-theta (° 20), depicted on the abscissa of an X-ray powder diffractogram, which may be expressed, for example, as “a signal at . . . degrees two- theta,” “a signal at [a] two-theta value(s)of ...” and/or “a signal at at least .. . two-theta value(s) selected from . . ..”
[00122] The repeatability of the measured angular values is in the range of ± 0.2° 20, i.e., the angular value can be at the recited angular value + 0.2 degrees two-theta, the angular value - 0.2 degrees two-theta, or any value between those two end points (angular value + 0.2 degrees two-theta and angular value - 0.2 degrees two-theta).
[00123] One of ordinary skill in the art would recognize that one or more signals (or peaks) in an XRPD pattern may overlap and may, for example, not be apparent to the naked eye. Indeed, one of ordinary skill in the art would recognize that some art-recognized methods are capable of and suitable for determining whether a signal exists in a pattern, such as Rietveld refinement.
[00124] The terms “signal intensities” and “peak intensities” interchangeably refer to relative signal intensities within a given X-ray powder diffractogram. Factors that can affect the relative signal or peak intensities include sample thickness and preferred orientation (e.g., the crystalline particles are not distributed randomly).
[00125] As used herein, an X-ray powder diffractogram is “substantially similar to that in [a particular] Figure” when at least 90%, such as at least 95%, at least 98%, or at least 99% of the signals in the two diffractograms overlap. In determining “substantial similarity,” one of ordinary skill in the art will understand that there may be variation in the intensities and/or signal positions in XRPD diffractograms even for the same crystalline form Thus, those of ordinary skill in the art will understand that the signal maximum values in XRPD diffractograms (in degrees two-theta) generally mean that value is identified as ± 0.2 degrees two-theta of the reported value, an art-recognized variance.
[00126] The term “X-ray powder diffractogram having a signal at ... two-theta values” as used herein refers to an XRPD pattern that contains X-ray reflection positions as measured and observed in X-ray powder diffraction experiments (degrees two-theta).
[00127] As used herein, the term “TGA” refers to thermogravimetric analysis and “TGA/DSC” refers to thermogravimetric analysis and differential scanning calorimetry.
[00128] As used herein, the term “DSC” refers to the analytical method of differential scanning calorimetry.
[00129] As used herein, the term “ssNMR” refers to the analytical method of solid-state nuclear magnetic resonance (NMR).
[00130] As used herein, the term “CPMAS” refers to cross-polarization magic-angle spinning NMR.
[00131] As used herein, the term “solvent” refers to any liquid in which the product is at least partially soluble (solubility of product >1 g/1).
[00132] As used herein, the term “glass transition temperature” or “Tg” refers to the temperature above which a hard and brittle “glassy” amorphous solid becomes viscous or rubbery.
[00133] As used herein, the term “melting temperature”, “melting point”, or “Tm” refers to the temperature at which the solid and liquid state of a material are at equilibrium.
[00134] As used herein, the term "dispersion" refers to a disperse system in which one substance, the dispersed phase, is distributed, in discrete units, throughout a second substance (the continuous phase or vehicle). The size of the dispersed phase can vary considerably (e.g., colloidal particles of nanometer dimension, to multiple microns in size). In general, the dispersed phases can be solids, liquids, or gases. In the case of a solid dispersion, the dispersed and continuous phases are both solids. In pharmaceutical applications, a solid dispersion can include, inter alia, a crystalline drug in an amorphous polymer; an amorphous drug in an amorphous polymer; an amorphous drug dispersed in an amorphous drug; or, alternatively, an amorphous drug dispersed in one or more excipients. In some embodiments, a solid dispersion includes the polymer constituting the dispersed phase, and the drug constitute the continuous phase. Or, a solid dispersion includes the drug constituting the dispersed phase, and the polymer constituting the continuous phase.
Methods of Treatment
[00135] Compound I, in any one of the pharmaceutically acceptable crystalline forms disclosed herein, acts as a CFTR modulator, i.e., it modulates CFTR activity in the body. Individuals suffering from a mutation in the gene encoding CFTR may benefit from receiving a CFTR modulator. A CFTR mutation may affect the CFTR quantity, i.e., the number of CFTR channels at the cell surface, or it may impact CFTR function, i.e., the functional ability of each channel to open and transport ions. Mutations affecting CFTR quantity include mutations that cause defective synthesis (Class I defect), mutations that cause defective processing and trafficking (Class II defect), mutations that cause reduced synthesis of CFTR (Class V defect), and mutations that reduce the surface stability of CFTR (Class VI defect). Mutations that affect CFTR function include mutations that cause defective gating (Class III defect) and mutations that cause defective conductance (Class IV defect). Some CFTR mutations exhibit characteristics of multiple classes. Certain mutations in the CFTR gene result in cystic fibrosis.
[00136] Thus, in some embodiments, the invention provides methods of treating, lessening the severity of, or symptomatically treating cystic fibrosis in a patient comprising administering to the patient an effective amount of Compound I in any one of the pharmaceutically acceptable solid forms disclosed herein, alone or in combination with another active ingredient, such as another CFTR modulating agent. In some embodiments, the patient has an F508del/minimal function (MF) genotype, F508del/F508del genotype (homozygous for the F508del mutation), F508del/gating genotype, or F508del/residual function (RF) genotype. In some embodiments the patient is heterozygous and has one F508del mutation In some embodiments the patient is homozygous for the N1303K mutation.
[00137] In some embodiments, the invention provides methods of treating, lessening the severity of, or symptomatically treating cystic fibrosis in a patient comprising administering to the patient an effective amount of Compound I as crystalline Compound I Neat Form A.
[00138] In some embodiments, the method of treating, lessening the severity of, or symptomatically treating cystic fibrosis in a patient comprises administering to the patient an effective amount of Compound I as crystalline Compound I Neat Form B.
[00139] In some embodiments, the method of treating, lessening the severity of, or symptomatically treating cystic fibrosis in a patient comprises administering to the patient an effective amount of Compound I as crystalline Compound I Neat Form C
[00140] In some embodiments, the method of treating, lessening the severity of, or symptomatically treating cystic fibrosis in a patient comprises administering to the patient an effective amount of Compound I as crystalline Compound I Neat Form D.
[00141] In some embodiments, the method of treating, lessening the severity of, or symptomatically treating cystic fibrosis in a patient comprises administering to the patient an effective amount of Compound I as crystalline Compound I Neat Form E.
[00142] In some embodiments, the method of treating, lessening the severity of, or symptomatically treating cystic fibrosis in a patient comprises administering to the patient an effective amount of Compound I as crystalline Compound I Compressed Form A.
[00143] In some embodiments, the method of treating, lessening the severity of, or symptomatically treating cystic fibrosis in a patient comprises administering to the patient an effective amount of Compound I as crystalline Compound I Compressed Form E.
[00144] In some embodiments, the method of treating, lessening the severity of, or symptomatically treating cystic fibrosis in a patient comprises administering to the patient an effective amount of Compound I as crystalline Compound I EtOH Solvate Form B.
[00145] In some embodiments, the method of treating, lessening the severity of, or symptomatically treating cystic fibrosis in a patient comprises administering to the patient an effective amount of Compound I as crystalline Compound I MeOH solvate.
[00146] In some embodiments, the method of treating, lessening the severity of, or symptomatically treating cystic fibrosis in a patient comprises administering to the patient an effective amount of Compound I as crystalline Compound I NPA solvate.
[00147] In some embodiments, the method of treating, lessening the severity of, or symptomatically treating cystic fibrosis in a patient comprises administering to the patient an effective amount of Compound I as crystalline Compound I MeOAc Solvate Form A.
[00148] In some embodiments, the method of treating, lessening the severity of, or symptomatically treating cystic fibrosis in a patient comprises administering to the patient an effective amount of Compound I as crystalline Compound I MeOAc Solvate Form B.
[00149] In some embodiments, the method of treating, lessening the severity of, or symptomatically treating cystic fibrosis in a patient comprises administering to the patient an effective amount of Compound I as crystalline Compound I MeOAc Solvate Form C.
[00150] In some embodiments, the method of treating, lessening the severity of, or symptomatically treating cystic fibrosis in a patient comprises administering to the patient an effective amount of Compound I as crystalline Compound I Hydrate Form A.
[00151] In some embodiments, the method of treating, lessening the severity of, or symptomatically treating cystic fibrosis in a patient comprises administering to the patient an effective amount of Compound I as crystalline Compound I Hydrate Form B.
[00152] In some embodiments, the method of treating, lessening the severity of, or symptomatically treating cystic fibrosis in a patient comprises administering to the patient an effective amount of Compound I as crystalline Compound I Hydrate Form C.
[00153] In some embodiments, the method of treating, lessening the severity of, or symptomatically treating cystic fibrosis in a patient comprises administering to the patient an effective amount of Compound I as crystalline Compound I EtOH Solvate Hydrate.
[00154] In some embodiments, the method of treating, lessening the severity of, or symptomatically treating cystic fibrosis in a patient comprises administering to the patient an effective amount of Compound I as crystalline Compound I MeOH Solvate Hydrate.
[00155] In some embodiments, the method of treating, lessening the severity of, or symptomatically treating cystic fibrosis in a patient comprises administering to the patient an effective amount of Compound I as crystalline Compound I IPA Solvate Hydrate.
[00156] In some embodiments, the method of treating, lessening the severity of, or symptomatically treating cystic fibrosis in a patient comprises administering to the patient an effective amount of Compound I as crystalline Compound I MeOAc Solvate Hydrate.
[00157] In some embodiments, the method of treating, lessening the severity of, or symptomatically treating cystic fibrosis in a patient comprises administering to the patient an effective amount of Compound I as amorphous Compound I.
[00158] In some embodiments, the method of treating, lessening the severity of, or symptomatically treating cystic fibrosis in a patient comprises administering to the patient an effective amount of Compound I as Compound I formulated as a solid (e.g., spray-dried) dispersion.
Combination Therapies
[00159] One aspect disclosed herein provides methods of treating cystic fibrosis and other CFTR-mediated diseases with Compound I in any one of the pharmaceutically acceptable solid forms disclosed herein or amorphous Compound I formulated as a solid (e.g., spray- dried) dispersion, in combination with other pharmaceutically active agents, including CFTR modulating agents. In some embodiments, Compound I in any one of the pharmaceutically acceptable solid forms disclosed herein or amorphous Compound I formulated as a solid (e.g., spray-dried) dispersion, can be administered in combination with at least one additional active pharmaceutical ingredient, such as, e.g., a CFTR modulating agent. In some embodiments, the at least one additional active pharmaceutical ingredient is a CFTR corrector. In some embodiments, the at least one additional active pharmaceutical ingredient is a CFTR potentiator. In some embodiments, the methods of treating cystic fibrosis and other CFTR-mediated diseases with Compound I in any one of the pharmaceutically acceptable solid forms disclosed herein, include combination with at least two additional active pharmaceutical ingredients, one of which is a CFTR corrector and one of which is a CFTR potentiator.
[00160] In some embodiments, at least one additional active pharmaceutical agent is selected from mucolytic agents, bronchodilators, antibiotics, anti-infective agents, and antiinflammatory agents
[00161] In some embodiments, the at least one additional active pharmaceutical agent is selected from (a) Compound IV or Compound V, deuterated derivatives of Compound IV or Compound V, and pharmaceutically acceptable salts of Compound IV, Compound V, and their deuterated derivatives; and optionally (b) Compound II and pharmaceutically acceptable salts thereof. Thus, in some embodiments, the combination therapies provided herein comprise Compound I in any one of the pharmaceutically acceptable crystalline forms disclosed herein or as amorphous Compound I formulated as a solid (e.g., spray-dried) dispersion, and an additional active pharmaceutical agent selected from Compound IV or V, deuterated derivatives, and pharmaceutically acceptable salts thereof. In some embodiments, the combination optionally includes Compound II. In some embodiments, the combination therapies provided herein comprise at least one compound selected from Compound I in any one of the pharmaceutically acceptable solid forms disclosed herein or amorphous Compound I formulated as a solid (e.g., spray-dried) dispersion and at least one compound selected from Compound IV or Compound V, and pharmaceutically acceptable salts thereof and at least one compound selected from Compound II and pharmaceutically acceptable salts thereof.
[00162] In some embodiments, the at least one additional active pharmaceutical ingredient is selected from (a) Compound II and pharmaceutically acceptable salts thereof; and (b) Compound III or Compound Ill-d and pharmaceutically acceptable salts of Compound III or Compound Ill-d. Thus, in some embodiments, the combination therapies provided herein comprise Compound I in any one of the pharmaceutically acceptable crystalline forms disclosed herein or amorphous Compound I formulated as a solid (e.g., spray-dried) dispersion, and at least one compound selected from Compound II, (Compound III or Ill-d), and pharmaceutically acceptable salts thereof. In some embodiments, the combination therapies provided herein comprise at least one compound selected from Compound I in any one of the pharmaceutically acceptable solid forms disclosed herein or amorphous Compound I formulated as a solid (e.g., spray-dried) dispersion and at least one compound selected from Compound III or ni-d and/or pharmaceutically acceptable salts thereof.
[00163] In some embodiments, at least one compound selected from Compound I in any one of the pharmaceutically acceptable solid forms disclosed herein or amorphous Compound I formulated as a solid (e.g., spray-dried) dispersion, is administered in combination with at
least one compound selected from Compound II and pharmaceutically acceptable salts thereof. In some embodiments, at least one compound selected from Compound I in any one of the pharmaceutically acceptable solid forms disclosed herein or amorphous Compound I formulated as a solid (e.g., spray-dried) dispersion is administered in combination with at least one compound selected from Compound III and pharmaceutically acceptable salts thereof In some embodiments, at least one compound selected from Compound I in any one of the pharmaceutically acceptable solid forms disclosed herein or amorphous Compound I formulated as a solid (e.g., spray-dried) dispersion is administered in combination with at least one compound selected from Compound Ill-d and pharmaceutically acceptable salts thereof. In some embodiments, at least one compound selected from Compound I in any one of the pharmaceutically acceptable solid forms disclosed herein or amorphous Compound I formulated as a solid (e g., spray-dried) dispersion, is administered in combination with Compounds II or a pharmaceutically acceptable salt thereof and at least one compound selected from Compound III and pharmaceutically acceptable salts thereof. In some embodiments, at least one compound selected from Compound I in any one of the pharmaceutically acceptable solid forms disclosed herein or amorphous Compound I formulated as a solid (e.g., spray-dried) dispersion, is administered in combination with at least one compound selected from Compound II and pharmaceutically acceptable salts thereof and at least one compound selected from Compound Ill-d and pharmaceutically acceptable salts thereof.
[00164] In some embodiments, at least one compound selected from Compound I in any one of the pharmaceutically acceptable solid forms disclosed herein or amorphous Compound I formulated as a solid (e.g., spray-dried) dispersion is administered in an amount of 2 mg to 1000 mg.
[00165] In some embodiments, at least one compound selected from Compound I in any one of the pharmaceutically acceptable solid forms disclosed herein or amorphous Compound I formulated as a solid (e.g., spray-dried) dispersion is administered in a first pharmaceutical composition; at least one compound selected from Compound II and pharmaceutically acceptable salts thereof is administered in a second pharmaceutical composition; and at least one compound selected from Compound III, Compound Ill-d, Compound IV, Compound V and pharmaceutically acceptable salts of Compounds III, Ill-d, IV, and V is administered in a third pharmaceutical composition.
[00166] In some embodiments, at least one compound selected from Compound I in any one of the pharmaceutically acceptable solid forms disclosed herein or amorphous Compound I formulated as a solid (e.g., spray-dried) dispersion is administered in a first pharmaceutical composition; at least one compound selected from Compound IV or V and pharmaceutically acceptable salts thereof is administered in a second pharmaceutical composition.
[00167] In some embodiments, at least one compound selected from Compound I in any one of the pharmaceutically acceptable solid forms disclosed herein or amorphous Compound I formulated as a solid dispersion is administered in a first pharmaceutical composition; at least one compound selected from Compound III or Ill-d and pharmaceutically acceptable salts thereof is administered in a second pharmaceutical composition.
[00168] In some embodiments, at least one compound selected from Compound I in any one of the pharmaceutically acceptable solid forms disclosed herein or amorphous Compound I formulated as a solid (e.g., spray-dried) dispersion is administered in a first pharmaceutical composition; and at least one compound selected from Compound IV or V, and pharmaceutically acceptable salts thereof and optionally at least one compound selected from Compound II and pharmaceutically acceptable salts thereof are administered in a second pharmaceutical composition. In some embodiments, at least one compound selected from Compound I in any one of the pharmaceutically acceptable solid forms disclosed herein or amorphous Compound I formulated as a solid (e.g., spray-dried) dispersion is administered in a first pharmaceutical composition; and (a) at least one compound selected from Compound IV, V, and pharmaceutically acceptable salts thereof and (b) at least one compound selected from Compound II and pharmaceutically acceptable salts thereof are administered in a second pharmaceutical composition.
[00169] In some embodiments, at least one compound selected from Compound I in any one of the pharmaceutically acceptable solid forms disclosed herein or amorphous Compound I formulated as a solid (e.g., spray-dried) dispersion is administered in a first pharmaceutical composition; and at least one compound selected from Compound II and pharmaceutically acceptable salts thereof and at least one compound selected from Compound III or Ill-d, and pharmaceutically acceptable salts thereof are administered in a second pharmaceutical composition. In some embodiments, the second pharmaceutical composition comprises a half of a daily dose of said at least one compound selected from Compound III, Ill-d, and pharmaceutically acceptable salts thereof, and the other half of said at least one compound
selected from Compound III, Ill-d, and pharmaceutically acceptable salts thereof is administered in a third pharmaceutical composition.
[00170] Any suitable pharmaceutical formulations can be used for Compound I (in any one of the pharmaceutically acceptable solid forms disclosed herein or amorphous Compound I formulated as a solid (e.g., spray-dried) dispersion), Compound II, Compound III, Compound Ill-d, and pharmaceutically acceptable salts thereof. Some exemplary pharmaceutical compositions for Compound II and its pharmaceutically acceptable salts can be found in WO 2011/119984 and WO 2014/014841, incorporated herein by reference. Some exemplary pharmaceutical compositions for Compound III and its pharmaceutically acceptable salts can be found in WO 2007/134279, WO 2010/019239, WO 2011/019413, WO 2012/027731, and WO 2013/130669, and some exemplary pharmaceutical compositions for Compound Ill-d and its pharmaceutically acceptable salts can be found in US 8,865,902, US 9,181,192, US 9,512,079, WO 2017/053455, and WO 2018/080591, all of which are incorporated herein by reference. Exemplary formulations for Compounds IV and V, deuterated derivatives and pharmaceutically acceptable salts thereof can be found in WO 2022/032068 and WO 2022/109573, respectively, both incorporated herein by reference.
[00171] In some embodiments, the solid form of Compound I used in the combination therapies of the invention is Compound I Neat Form A.
[00172] In some embodiments, the solid form of Compound I used in the combination therapies of the invention is Compound I Neat Form B.
[00173] In some embodiments, the solid form of Compound I used in the combination therapies of the invention is Compound I Neat Form C.
[00174] In some embodiments, the solid form of Compound I used in the combination therapies of the invention is Compound I Neat Form D.
[00175] In some embodiments, the solid form of Compound I used in the combination therapies of the invention is Compound I Neat Form E.
[00176] In some embodiments, the solid form of Compound I used in the combination therapies of the invention is Compound I Compressed Form A.
[00177] In some embodiments, the solid form of Compound I used in the combination therapies of the invention is Compound I Compressed Form E.
[00178] In some embodiments, the solid form of Compound I used in the combination therapies of the invention is Compound I EtOH Solvate Form B. In some embodiments, the solid form of Compound I used in the combination therapies of the invention is Compound I MeOH Solvate. In some embodiments, the solid form of Compound I used in the combination therapies of the invention is Compound I NPA Solvate. In some embodiments, the solid form of Compound I used in the combination therapies of the invention is Compound I MeOAc Solvate Form A. In some embodiments, the solid form of Compound I used in the combination therapies of the invention is Compound I MeOAc Solvate Form B. In some embodiments, the solid form of Compound I used in the combination therapies of the invention is Compound I MeOAc Solvate Form C. In some embodiments, the solid form of Compound I used in the combination therapies of the invention is Compound I Hydrate Form A. In some embodiments, the solid form of Compound I used in the combination therapies of the invention is Compound I Hydrate Form B. In some embodiments, the solid form of Compound I used in the combination therapies of the invention is Compound I Hydrate Form C. In some embodiments, the solid form of Compound I used in the combination therapies of the invention is Compound I EtOH Solvate Hydrate. In some embodiments, the solid form of Compound I used in the combination therapies of the invention is Compound I MeOH Solvate Hydrate. In some embodiments, the solid form of Compound I used in the combination therapies of the invention is Compound I IPA Solvate Hydrate. In some embodiments, the solid form of Compound I used in the combination therapies of the invention is Compound I MeOAc Solvate Hydrate. In some embodiments, the solid form of Compound I used in the combination therapies of the invention is amorphous Compound I.
[00179] In some embodiments, the form of Compound I used in the combination therapies of the invention is Compound I in a solid (e.g., spray-dried) dispersion.
Pharmaceutical Compositions
[00180] Another aspect of the invention provides pharmaceutical compositions comprising Compound I as any one of the pharmaceutically acceptable solid forms disclosed herein or amorphous Compound I formulated as a solid (e.g., spray-dried) dispersion. In some embodiments, the invention provides pharmaceutical compositions comprising Compound I as any one of the pharmaceutically acceptable solid forms disclosed herein in combination or amorphous Compound I formulated as a solid (e.g., spray-dried) dispersion, with at least one additional active pharmaceutical agent. In some embodiments, the at least one additional active pharmaceutical ingredient is a CFTR modulator. In some embodiments, the at least
one additional active pharmaceutical agent is a CFTR corrector. In some embodiments, the at least one additional active pharmaceutical agent is a CFTR potentiator. In some embodiments, the pharmaceutical composition comprises Compound I as any one of the pharmaceutically acceptable crystalline forms disclosed herein or amorphous Compound I formulated as a solid (e.g., spray-dried) dispersion and at least two additional active pharmaceutical agents, one of which is a CFTR corrector and one of which is a CFTR potentiator.
[00181] In some embodiments, at least one additional active pharmaceutical agent is selected from mucolytic agents, bronchodilators, antibiotics, anti-infective agents, and antiinflammatory agents.
[00182] In some embodiments, the invention provides a pharmaceutical composition comprising at least one compound selected from Compound I as any one of the pharmaceutically acceptable crystalline forms disclosed herein or amorphous Compound I formulated as a solid (e.g., spray-dried) dispersion, and at least one pharmaceutically acceptable carrier.
[00183] In some embodiments, the invention provides a pharmaceutical composition comprising (a) Compound I, as any one of the pharmaceutically acceptable solid forms disclosed herein or amorphous Compound I formulated as a solid (e.g., spray-dried) dispersion, (b) at least one compound selected from Compound IV, Compound V, deuterated derivatives thereof and pharmaceutically acceptable salts thereof, and (c) at least one pharmaceutically acceptable carrier.
[00184] In some embodiments, the disclosure provides a pharmaceutical composition comprising (a) Compound I as any one of the pharmaceutically acceptable solid forms disclosed herein or amorphous Compound I formulated as a solid (e.g., spray-dried) dispersion, (b) at least one compound selected from Compound HI, Ill-d, and pharmaceutically acceptable salts thereof, and (c) at least one pharmaceutically acceptable carrier.
[00185] In some embodiments, the disclosure provides a pharmaceutical composition comprising (a) Compound I as any one of the pharmaceutically acceptable solid forms disclosed herein or amorphous Compound I formulated as a solid (e.g., spray-dried) dispersion, (b) at least one compound selected from Compound n and pharmaceutically acceptable salts thereof, (c) at least one compound selected from Compound III, Ill-d, VI,
and V and pharmaceutically acceptable salts thereof, and (d) at least one pharmaceutically acceptable carrier.
[00186] In some embodiments, the disclosure provides a pharmaceutical composition comprising (a) Compound I as any one of the pharmaceutically acceptable solid forms disclosed herein or amorphous Compound I formulated as a solid (e.g., spray-dried) dispersion, (b) at least one compound selected from Compound n and pharmaceutically acceptable salts thereof, (c) at least one compound selected from Compound IV, and pharmaceutically acceptable salts of Compound IV, and (d) at least one pharmaceutically acceptable carrier.
[00187] In some embodiments, the disclosure provides a pharmaceutical composition comprising (a) Compound I as any one of the pharmaceutically acceptable solid forms disclosed herein or amorphous Compound I formulated as a solid (e.g., spray-dried) dispersion, (b) at least one compound selected from Compound II and pharmaceutically acceptable salts thereof, (c) at least one compound selected from Compound V, and pharmaceutically acceptable salts of Compound V, and (d) at least one pharmaceutically acceptable carrier.
[00188] Any pharmaceutical composition disclosed herein may comprise at least one pharmaceutically acceptable carrier. In some embodiments, the at least one pharmaceutically acceptable carrier is selected from pharmaceutically acceptable vehicles and pharmaceutically acceptable adjuvants. In some embodiments, the at least one pharmaceutically acceptable is selected from pharmaceutically acceptable fillers, disintegrants, surfactants, binders, lubricants.
[00189] In some embodiments, the solid form of Compound I used in the pharmaceutical compositions of the invention is Compound I Neat Form A.
[00190] In some embodiments, the crystalline form of Compound I used in the pharmaceutical compositions of the invention is Compound I Neat Form B.
[00191] In some embodiments, the solid form of Compound I used in the pharmaceutical compositions of the invention is Compound I Neat Form C.
[00192] In some embodiments, the solid form of Compound I used in the pharmaceutical compositions of the invention is Compound I Neat Form D.
[00193] In some embodiments, the solid form of Compound I used in the pharmaceutical compositions of the invention is Compound I Neat Form E.
[00194] In some embodiments, the solid form of Compound I used in the pharmaceutical compositions of the invention is Compound I Compressed Form A.
[00195] In some embodiments, the solid form of Compound I used in the pharmaceutical compositions of the invention is Compound I Compressed Form E.
[00196] In some embodiments, the solid form of Compound I used in the pharmaceutical compositions of the invention is Compound I EtOH Solvate Form B. In some embodiments, the solid form of Compound I used in the pharmaceutical compositions of the invention is Compound I MeOH Solvate. In some embodiments, the solid form of Compound I used in the pharmaceutical compositions of the invention is Compound I NPA Solvate. In some embodiments, the solid form of Compound I used in the pharmaceutical compositions of the invention is Compound I MeOAc Solvate Form A. In some embodiments, the solid form of Compound I used in the pharmaceutical compositions of the invention is Compound I MeOAc Solvate Form B. In some embodiments, the solid form of Compound I used in the pharmaceutical compositions of the invention is Compound I MeOAc Solvate Form C. In some embodiments, the solid form of Compound I used in the pharmaceutical compositions of the invention is Compound I Hydrate Form A. In some embodiments, the solid form of Compound I used in the pharmaceutical compositions of the invention is Compound I Hydrate Form B. In some embodiments, the solid form of Compound I used in the pharmaceutical compositions of the invention is Compound I Hydrate Form C. In some embodiments, the solid form of Compound I used in the pharmaceutical compositions of the invention is Compound I EtOH Solvate Hydrate. In some embodiments, the solid form of Compound I used in the pharmaceutical compositions of the invention is Compound I MeOH Solvate Hydrate. In some embodiments, the solid form of Compound I used in the pharmaceutical compositions of the invention is Compound I IPA Solvate Hydrate. In some embodiments, the solid form of Compound I used in the pharmaceutical compositions of the invention is Compound I MeOAc Solvate Hydrate. In some embodiments, the solid form of Compound I used in the pharmaceutical compositions of the invention is amorphous Compound I.
[00197] In some embodiments, the form of Compound I used in the pharmaceutical compositions of the invention is Compound I in a solid (e.g., spray-dried) dispersion.
[00198] The pharmaceutical compositions described herein are useful for treating cystic fibrosis and other CFTR-mediated diseases.
[00199] As described above, pharmaceutical compositions disclosed herein may optionally further comprise at least one pharmaceutically acceptable carrier. The at least one pharmaceutically acceptable carrier may be selected from adjuvants and vehicles. The at least one pharmaceutically acceptable carrier, as used herein, includes any and all solvents, diluents, other liquid vehicles, dispersion aids, suspension aids, surface active agents, isotonic agents, thickening agents, emulsifying agents, preservatives, solid binders, and lubricants, as suited to the particular dosage form desired. Remington: The Science and Practice of Pharmacy, 21st edition, 2005, ed. D.B. Troy, Lippincott Williams & Wilkins, Philadelphia, and Encyclopedia of Pharmaceutical Technology, eds. J. Swarbrick and J. C. Boylan, 1988- 1999, Marcel Dekker, New York discloses various carriers used in formulating pharmaceutical compositions and known techniques for the preparation thereof. Except insofar as any conventional carrier is incompatible with the compounds of this disclosure, such as by producing any undesirable biological effect or otherwise interacting in a deleterious manner with any other component(s) of the pharmaceutical composition, its use is contemplated to be within the scope of this disclosure. Non-limiting examples of suitable pharmaceutically acceptable carriers include, but are not limited to, ion exchangers, alumina, aluminum stearate, lecithin, serum proteins (such as human serum albumin), buffer substances (such as phosphates, glycine, sorbic acid, and potassium sorbate), partial glyceride mixtures of saturated vegetable fatty acids, water, salts, and electrolytes (such as protamine sulfate, disodium hydrogen phosphate, potassium hydrogen phosphate, sodium chloride, and zinc salts), colloidal silica, magnesium trisilicate, polyvinyl pyrrolidone, polyacrylates, waxes, polyethylene-polyoxypropylene-block polymers, wool fat, sugars (such as lactose, glucose and sucrose), starches (such as corn starch and potato starch), cellulose and its derivatives (such as sodium carboxymethyl cellulose, ethyl cellulose and cellulose acetate), powdered tragacanth, malt, gelatin, talc, excipients (such as cocoa butter and suppository waxes), oils (such as peanut oil, cottonseed oil, safflower oil, sesame oil, olive oil, corn oil and soybean oil), glycols (such as propylene glycol and polyethylene glycol), esters (such as ethyl oleate and ethyl laurate), agar, buffering agents (such as magnesium hydroxide and aluminum hydroxide), alginic acid, pyrogen-free water, isotonic saline, Ringer's solution, ethyl alcohol, phosphate buffer solutions, non-toxic compatible lubricants (such as sodium lauryl sulfate and magnesium stearate), coloring agents, releasing agents, coating agents, sweetening agents, flavoring agents, perfuming agents, preservatives, and antioxidants.
Non-Limiting List of Exemplary Embodiments
1. Compound I
(Compound I) as substantially crystalline Compound I Neat Form A (i.e., wherein less than 15% of Compound I is in amorphous form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous form).
2. The Compound I of Embodiment 1, wherein Compound I is 100% crystalline Compound I Neat Form A.
3. Substantially pure Compound I Neat Form A.
4. The Compound I Neat Form A of any one of Embodiments 1 to 3, characterized by an X-ray powder diffractogram having signals at (a) 9.2 ± 0.2 degrees two-theta, 14.1 ± 0.2 degrees two-theta, and (b) 15.9 ± 0.2 degrees two-theta and/or 26.2 ± 0.2 degrees two-theta.
5. The Compound I Neat Form A of any one of Embodiments 1 to 3, characterized by an X-ray powder diffractogram having (a) signals at 9.2 ± 0.2 degrees two-theta, 14.1 ± 0.2 degrees two-theta, and 15.9 ± 0.2 degrees two-theta and (b) one or more two-theta values selected from 16.6 ± 0.2 degrees two-theta, 13.0 ± 0.2 degrees two-theta, 18.4 ± 0.2 degrees two-theta, 10 3 ± 0.2 degrees two-theta, and 22.6 ± 0.2 degrees two- theta.
6. The Compound I Neat Form A of any one of Embodiments 1 to 3, characterized by an
X-ray powder diffractogram having (a) signals at 9.2 ± 0.2 degrees two-theta, 14.1 ±
0.2 degrees two-theta, and 15.9 ± 0.2 degrees two-theta and (b) two or more two-theta values selected from 16.6 ± 0.2 degrees two-theta, 13.0 ± 0.2 degrees two-theta, 18.4 ± 0.2 degrees two-theta, 10 3 ± 0.2 degrees two-theta, and 22.6 ± 0.2 degrees two- theta. The Compound I Neat Form A of any one of Embodiments 1 to 3, characterized by an X-ray powder diffractogram having (a) signals at 9.2 ± 0.2 degrees two-theta, 14.1 ± 0.2 degrees two-theta, and 15.9 ± 0.2 degrees two-theta and (b) three or more two- theta values selected from 16 6 ± 0.2 degrees two-theta, 13.0 ± 0.2 degrees two-theta, 18.4 ± 0.2 degrees two-theta, 10.3 ± 0.2 degrees two-theta, and 22.6 ± 0.2 degrees two-theta. The Compound I Neat Form A of any one of Embodiments 1 to 3, characterized by an X-ray powder diffractogram having (a) signals at 9.2 ± 0.2 degrees two-theta, 14.1 ± 0.2 degrees two-theta, and 15.9 ± 0.2 degrees two-theta and (b) four or more two-theta values selected from 16.6 ± 0.2 degrees two-theta, 13.0 ± 0.2 degrees two-theta, 18.4 ± 0.2 degrees two-theta, 10.3 ± 0.2 degrees two-theta, and 22.6 ± 0.2 degrees two- theta. The Compound I Neat Form A of any one of Embodiments 1 to 3, characterized by an X-ray powder diffractogram having signals at 9.2 ± 0.2 degrees two-theta, 14. 1 ± 0.2 degrees two-theta, 15.9 ± 0.2 degrees two-theta, 16.6 ± 0.2 degrees two-theta, 13.0 ± 0.2 degrees two-theta, 18.4 ± 0.2 degrees two-theta, 10.3 ± 0.2 degrees two-theta, and 22.6 ± 0.2 degrees two-theta. The Compound I Neat Form A of any one of Embodiments 1 to 3, characterized by an X-ray powder diffractogram having (a) signals at 9.2 ± 0.2 degrees two-theta, 14.1 ± 0.2 degrees two-theta, and 26.2 ± 0.2 degrees two-theta and (b) one or more two-theta values selected from 16.6 ± 0.2 degrees two-theta, 13.0 ± 0.2 degrees two-theta, 18.4 ± 0.2 degrees two-theta, 10.3 ± 0.2 degrees two-theta, and 22.6 ± 0.2 degrees two- theta. The Compound I Neat Form A of any one of Embodiments 1 to 3, characterized by an X-ray powder diffractogram having (a) signals at 9.2 ± 0.2 degrees two-theta, 14.1 ±
0.2 degrees two-theta, and 26.2 ± 0.2 degrees two-theta and (b) two or more two-theta values selected from 16.6 ± 0.2 degrees two-theta, 13.0 ± 0.2 degrees two-theta, 18.4 ± 0.2 degrees two-theta, 10 3 ± 0.2 degrees two-theta, and 22.6 ± 0.2 degrees two- theta. The Compound I Neat Form A of any one of Embodiments 1 to 3, characterized by an X-ray powder diffractogram having (a) signals at 9.2 ± 0.2 degrees two-theta, 14.1 ± 0.2 degrees two-theta, and 26.2 ± 0.2 degrees two-theta and (b) three or more two- theta values selected from 16 6 ± 0.2 degrees two-theta, 13.0 ± 0.2 degrees two-theta, 18.4 ± 0.2 degrees two-theta, 10.3 ± 0.2 degrees two-theta, and 22.6 ± 0.2 degrees two-theta. The Compound I Neat Form A of any one of Embodiments 1 to 3, characterized by an X-ray powder diffractogram having (a) signals at 9.2 ± 0.2 degrees two-theta, 14.1 ± 0.2 degrees two-theta, and 26.2 ± 0.2 degrees two-theta and (b) four or more two-theta values selected from 16.6 ± 0.2 degrees two-theta, 13.0 ± 0.2 degrees two-theta, 18.4 ± 0.2 degrees two-theta, 10.3 ± 0.2 degrees two-theta, and 22.6 ± 0.2 degrees two- theta. The Compound I Neat Form A of any one of Embodiments 1 to 3, characterized by an X-ray powder diffractogram having signals at 9.2 ± 0.2 degrees two-theta, 14. 1 ± 0.2 degrees two-theta, 26.2 ± 0.2 degrees two-theta, 16.6 ± 0.2 degrees two-theta, 13.0 ± 0.2 degrees two-theta, 18.4 ± 0.2 degrees two-theta, 10.3 ± 0.2 degrees two-theta, and 22.6 ± 0.2 degrees two-theta. The Compound I Neat Form A of any one of Embodiments 1 to 14, characterized by an X-ray powder diffractogram substantially similar to FIG. 1. The Compound I Neat Form A of any one of Embodiments 1 to 15, characterized by a TGA showing negligible weight loss from ambient temperature up to about 200 °C. The Compound I Neat Form A of any one of Embodiments 1 to 16, characterized by TGA data substantially similar to FIG. 2.
The Compound I Neat Form A of any one of Embodiments 1 to 17, characterized by a DSC analysis showing an endothermic peak at about 198 °C. The Compound I Neat Form A of any one of Embodiments 1 to 18, characterized by a DSC analysis substantially similar to FIG. 3. The Compound I Neat Form A of any one of Embodiments 1 to 19, characterized by a 13C ssNMR spectrum with a peak at (a) 126.0 ± 0.2 ppm and (b) one or more peaks selected from 26.6 ± 0.2 ppm, 128.4 ± 0.2 ppm, 20.0 ± 0.2 ppm, 43.7 ± 0.2 ppm,
140.2 ± 0.2 ppm, 168.7 ± 0.2 ppm, and 133.3 ± 0.2 ppm. The Compound I Neat Form A of any one of Embodiments 1 to 19, characterized by a 13C ssNMR spectrum with a peak at (a) 126.0 ± 0.2 ppm and (b) two or more peaks selected from 26.6 ± 0.2 ppm, 128.4 ± 0.2 ppm, 20.0 ± 0.2 ppm, 43.7 ± 0.2 ppm,
140.2 ± 0.2 ppm, 168.7 ± 0.2 ppm, and 133.3 ± 0.2 ppm. The Compound I Neat Form A of any one of Embodiments 1 to 19, characterized by a 13C ssNMR spectrum with a peak at (a) 126.0 ± 0.2 ppm and (b) three or more peaks selected from 26.6 ± 0.2 ppm, 128.4 ± 0.2 ppm, 20.0 ± 0.2 ppm, 43.7 ± 0.2 ppm,
140.2 ± 0.2 ppm, 168.7 ± 0.2 ppm, and 133.3 ± 0.2 ppm. The Compound I Neat Form A of any one of Embodiments 1 to 19, characterized by a 13C ssNMR spectrum with a peak at (a) 126.0 ± 0.2 ppm and (b) four or more peaks selected from 26.6 ± 0.2 ppm, 128.4 ± 0.2 ppm, 20.0 ± 0.2 ppm, 43.7 ± 0.2 ppm,
140.2 ± 0.2 ppm, 168.7 ± 0.2 ppm, and 133.3 ± 0.2 ppm. The Compound I Neat Form A of any one of Embodiments 1 to 19, characterized by a 13C ssNMR spectrum with a peak at (a) 126.0 ± 0.2 ppm and (b) five or more peaks selected from 26.6 ± 0.2 ppm, 128.4 ± 0.2 ppm, 20.0 ± 0.2 ppm, 43.7 ± 0.2 ppm,
140.2 ± 0.2 ppm, 168.7 ± 0.2 ppm, and 133.3 ± 0.2 ppm. The Compound I Neat Form A of any one of Embodiments 1 to 19, characterized by a 13C ssNMR spectrum with a peak at (a) 126.0 ± 0.2 ppm and (b) six or more peaks
selected from 26.6 ± 0.2 ppm, 128.4 ± 0.2 ppm, 20.0 ± 0.2 ppm, 43.7 ± 0.2 ppm, 140.2 ± 0.2 ppm, 168.7 ± 0.2 ppm, and 133.3 ± 0.2 ppm. The Compound I Neat Form A of any one of Embodiments 1 to 19, characterized by a 13C ssNMR spectrum with peaks at 126.0 ± 0.2 ppm, 26.6 ± 0.2 ppm, 128.4 ± 0.2 ppm, 20.0 ± 0.2 ppm, 43.7 ± 0.2 ppm, 140.2 ± 0.2 ppm, 168.7 ± 0.2 ppm, and 133.3 ± 0.2 ppm. The Compound I Neat Form A of any one of Embodiments 1 to 26, characterized by a 13C ssNMR spectrum substantially similar to FIG. 4. The Compound I Neat Form A of any one of Embodiments 1 to 27, characterized by a monoclinic crystal system, aF2i2i2i space group, and unit cell dimensions, measured at 100 K on a Rigaku diffractometer equipped with Cu I<« radiation (X = 1.54178 A) and an HP AD detector, of:
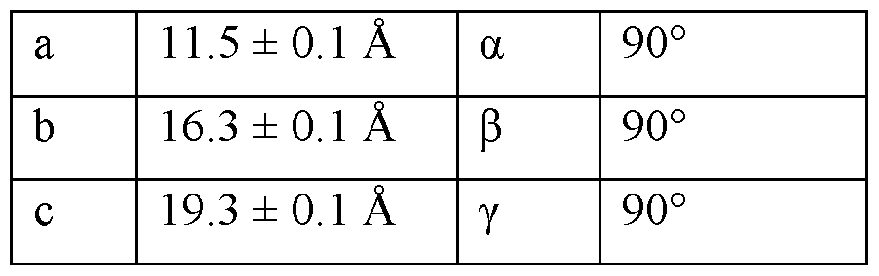 The Compound I Neat Form A according to any one of Embodiments 1 to 28, prepared by a process comprising (i) dissolving amorphous Compound I in ethanol, (ii) increasing the temperature to about 73 °C, (iii) decreasing the temperature to about 20 °C, (iv) isolation of the solid via filtration, and (v) drying of the solid at 50 °C. Compound I as substantially crystalline Neat Form B (i.e., wherein less than 15% of Compound I is in amorphous form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous form). The Compound I of Embodiment 30, wherein Compound I is 100% crystalline Compound I Neat Form B. Substantially pure Compound I Neat Form B.
The Compound I Neat Form B of any one of Embodiments 30 to 32, characterized by an X-ray powder diffractogram having (a) a signal at 26 3 degrees two-theta and (b) signals at one or more two-theta values selected from: 11.1 degrees two-theta, 19.6 degrees two-theta, and 7.2 degrees two-theta. The Compound I Neat Form B of any one of Embodiments 30 to 32, characterized by an X-ray powder diffractogram having (a) a signal at 26.3 ± 0.2 degrees two-theta and (b) signals at two or more two-theta values selected from: 11.1 ± 0.2 degrees two- theta, 19.6 ± 0.2 degrees two-theta, and 7.2 ± 0.2 degrees two-theta. The Compound I Neat Form B of any one of Embodiments 30 to 32, characterized by an X-ray powder diffractogram having signals at two-theta values 26.3 ± 0.2 degrees two-theta, 11.1 ± 0.2 degrees two-theta, 19.6 ± 0.2 degrees two-theta, and 7.2 ± 0.2 degrees two-theta. The Compound I Neat Form B of any one of Embodiments 30 to 32, characterized by an X-ray powder diffractogram having (a) signals at two-theta values 26.3 ± 0.2 degrees two-theta, 11. 1 ± 0.2 degrees two-theta, 19.6 ± 0.2 degrees two-theta, and 7.2 ± 0.2 degrees two-theta and (b) signals at one or more two-theta values selected from: 18.5 ± 0.2 degrees two-theta, 20.8 ± 0.2 degrees two-theta, 14.3 ± 0.2 degrees two- theta, and 17.1 ± 0.2 degrees two-theta. The Compound I Neat Form B of any one of Embodiments 30 to 32, characterized by an X-ray powder diffractogram having (a) signals at two-theta values 26.3 ± 0.2 degrees two-theta, 11. 1 ± 0.2 degrees two-theta, 19.6 ± 0.2 degrees two-theta, and 7.2 ± 0.2 degrees two-theta and (b) signals at two or more two-theta values selected from: 18.5 ± 0.2 degrees two-theta, 20.8 ± 0.2 degrees two-theta, 14.3 ± 0.2 degrees two- theta, and 17.1 ± 0.2 degrees two-theta. The Compound I Neat Form B of any one of Embodiments 30 to 32, characterized by an X-ray powder diffractogram having (a) signals at two-theta values 26.3 ± 0.2 degrees two-theta, 11. 1 ± 0.2 degrees two-theta, 19.6 ± 0.2 degrees two-theta, and 7.2 ± 0.2 degrees two-theta and (b) signals at three or more two-theta values selected
from: 18.5 ± 0.2 degrees two-theta, 20.8 ± 0.2 degrees two-theta, 14.3 ± 0.2 degrees two-theta, and 17.1 ± 0.2 degrees two-theta. The Compound I Neat Form B of any one of Embodiments 30 to 32, characterized by an X-ray powder diffractogram having signals at two-theta values 26.3 ± 0.2 degrees two-theta, 11 1 ± 0.2 degrees two-theta, 19.6 ± 0.2 degrees two-theta, 7.2 ± 0.2 degrees two-theta, 18.5 ± 0.2 degrees two-theta, 20.8 ± 0.2 degrees two-theta, 14.3 ± 0.2 degrees two-theta, and 17.1 ± 0.2 degrees two-theta. The Compound I Neat Form B of any one of Embodiments 30 to 39, characterized by an X-ray powder diffractogram substantially similar to FIG. 5. The Compound I Neat Form B of any one of Embodiments 30 to 40 characterized by a 13C ssNMR spectrum with a peak at 142.6 ± 0.2 ppm. The Compound I Neat Form B of any one of Embodiments 30 to 40, characterized by a 13C ssNMR spectrum with (a) a peak at 142.6 ± 0.2 ppm and (b) one or more peaks selected from 27.1 ± 0.2 ppm, 18.4 ± 0.2 ppm, 134.9 ± 0.2 ppm, 132.4 ± 0.2 ppm,
The Compound I Neat Form A according to any one of Embodiments 1 to 28, prepared by a process comprising (i) dissolving amorphous Compound I in ethanol, (ii) increasing the temperature to about 73 °C, (iii) decreasing the temperature to about 20 °C, (iv) isolation of the solid via filtration, and (v) drying of the solid at 50 °C. Compound I as substantially crystalline Neat Form B (i.e., wherein less than 15% of Compound I is in amorphous form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous form). The Compound I of Embodiment 30, wherein Compound I is 100% crystalline Compound I Neat Form B. Substantially pure Compound I Neat Form B.
The Compound I Neat Form B of any one of Embodiments 30 to 32, characterized by an X-ray powder diffractogram having (a) a signal at 26 3 degrees two-theta and (b) signals at one or more two-theta values selected from: 11.1 degrees two-theta, 19.6 degrees two-theta, and 7.2 degrees two-theta. The Compound I Neat Form B of any one of Embodiments 30 to 32, characterized by an X-ray powder diffractogram having (a) a signal at 26.3 ± 0.2 degrees two-theta and (b) signals at two or more two-theta values selected from: 11.1 ± 0.2 degrees two- theta, 19.6 ± 0.2 degrees two-theta, and 7.2 ± 0.2 degrees two-theta. The Compound I Neat Form B of any one of Embodiments 30 to 32, characterized by an X-ray powder diffractogram having signals at two-theta values 26.3 ± 0.2 degrees two-theta, 11.1 ± 0.2 degrees two-theta, 19.6 ± 0.2 degrees two-theta, and 7.2 ± 0.2 degrees two-theta. The Compound I Neat Form B of any one of Embodiments 30 to 32, characterized by an X-ray powder diffractogram having (a) signals at two-theta values 26.3 ± 0.2 degrees two-theta, 11. 1 ± 0.2 degrees two-theta, 19.6 ± 0.2 degrees two-theta, and 7.2 ± 0.2 degrees two-theta and (b) signals at one or more two-theta values selected from: 18.5 ± 0.2 degrees two-theta, 20.8 ± 0.2 degrees two-theta, 14.3 ± 0.2 degrees two- theta, and 17.1 ± 0.2 degrees two-theta. The Compound I Neat Form B of any one of Embodiments 30 to 32, characterized by an X-ray powder diffractogram having (a) signals at two-theta values 26.3 ± 0.2 degrees two-theta, 11. 1 ± 0.2 degrees two-theta, 19.6 ± 0.2 degrees two-theta, and 7.2 ± 0.2 degrees two-theta and (b) signals at two or more two-theta values selected from: 18.5 ± 0.2 degrees two-theta, 20.8 ± 0.2 degrees two-theta, 14.3 ± 0.2 degrees two- theta, and 17.1 ± 0.2 degrees two-theta. The Compound I Neat Form B of any one of Embodiments 30 to 32, characterized by an X-ray powder diffractogram having (a) signals at two-theta values 26.3 ± 0.2 degrees two-theta, 11. 1 ± 0.2 degrees two-theta, 19.6 ± 0.2 degrees two-theta, and 7.2 ± 0.2 degrees two-theta and (b) signals at three or more two-theta values selected
from: 18.5 ± 0.2 degrees two-theta, 20.8 ± 0.2 degrees two-theta, 14.3 ± 0.2 degrees two-theta, and 17.1 ± 0.2 degrees two-theta. The Compound I Neat Form B of any one of Embodiments 30 to 32, characterized by an X-ray powder diffractogram having signals at two-theta values 26.3 ± 0.2 degrees two-theta, 11 1 ± 0.2 degrees two-theta, 19.6 ± 0.2 degrees two-theta, 7.2 ± 0.2 degrees two-theta, 18.5 ± 0.2 degrees two-theta, 20.8 ± 0.2 degrees two-theta, 14.3 ± 0.2 degrees two-theta, and 17.1 ± 0.2 degrees two-theta. The Compound I Neat Form B of any one of Embodiments 30 to 39, characterized by an X-ray powder diffractogram substantially similar to FIG. 5. The Compound I Neat Form B of any one of Embodiments 30 to 40 characterized by a 13C ssNMR spectrum with a peak at 142.6 ± 0.2 ppm. The Compound I Neat Form B of any one of Embodiments 30 to 40, characterized by a 13C ssNMR spectrum with (a) a peak at 142.6 ± 0.2 ppm and (b) one or more peaks selected from 27.1 ± 0.2 ppm, 18.4 ± 0.2 ppm, 134.9 ± 0.2 ppm, 132.4 ± 0.2 ppm,
128.2 ± 0.2 ppm, 126.6 ± 0.2 ppm, and 133.4 ± 0.2 ppm. The Compound I Neat Form B of any one of Embodiments 30 to 40, characterized by a 13C ssNMR spectrum with (a) a peak at 142.6 ± 0.2 ppm and (b) two or more peaks selected from 27.1 ± 0.2 ppm, 18.4 ± 0.2 ppm, 134.9 ± 0.2 ppm, 132.4 ± 0.2 ppm,
128.2 ± 0.2 ppm, 126.6 ± 0.2 ppm, and 133.4 ± 0.2 ppm. The Compound I Neat Form B of any one of Embodiments 30 to 40, characterized by a 13C ssNMR spectrum with (a) a peak at 142.6 ± 0.2 ppm and (b) three or more peaks selected from 27.1 ± 0.2 ppm, 18.4 ± 0.2 ppm, 134.9 ± 0.2 ppm, 132.4 ± 0.2 ppm, 128.2 ± 0.2 ppm, 126.6 ± 0.2 ppm, and 133.4 ± 0.2 ppm. The Compound I Neat Form B of any one of Embodiments 30 to 40, characterized by a 13C ssNMR spectrum with (a) a peak at 142.6 ± 0.2 ppm and (b) four or more peaks selected from 27.1 ± 0.2 ppm, 18.4 ± 0.2 ppm, 134.9 ± 0.2 ppm, 132.4 ± 0.2 ppm,
128.2 ± 0.2 ppm, 126.6 ± 0.2 ppm, and 133.4 ± 0.2 ppm.
The Compound I Neat Form B of any one of Embodiments 30 to 40, characterized by a 13C ssNMR spectrum with (a) a peak at 142.6 ± 0.2 ppm and (b) five or more peaks selected from 27.1 ± 0.2 ppm, 18.4 ± 0.2 ppm, 134.9 ± 0.2 ppm, 132.4 ± 0.2 ppm, 128.2 ± 0.2 ppm, 126.6 ± 0.2 ppm, and 133.4 ± 0.2 ppm. The Compound I Neat Form B of any one of Embodiments 30 to 40, characterized by a 13C ssNMR spectrum with (a) a peak at 142.6 ± 0.2 ppm and (b) six or more peaks selected from 27.1 ± 0.2 ppm, 18.4 ± 0.2 ppm, 134.9 ± 0.2 ppm, 132.4 ± 0.2 ppm, 128.2 ± 0.2 ppm, 126.6 ± 0.2 ppm, and 133.4 ± 0.2 ppm. The Compound I Neat Form B of any one of Embodiments 30 to 40, characterized by a 13C ssNMR spectrum with peaks at 142.6 ± 0.2 ppm, 27.1 ± 0.2 ppm, 18.4 ± 0.2 ppm, 134.9 ± 0.2 ppm, 132.4 ± 0.2 ppm, 128.2 ± 0.2 ppm, 126.6 ± 0.2 ppm, and 133.4 ± 0.2 ppm. The Compound I Neat Form B of any one of Embodiments 30 to 48, characterized by a 13C ssNMR spectrum substantially similar to FIG. 6. The Compound I Neat Form B of any one of Embodiments 30 to 49, prepared by a process comprising exposing Compound I hydrate Form A to about 3% humidity. Compound I as substantially crystalline Neat Form C (i.e., wherein less than 15% of Compound I is in amorphous form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous Form). The Compound I of Embodiment 51, wherein Compound I is 100% crystalline Compound I Neat Form C. Substantially pure Compound I Neat Form C. The Compound I Neat Form C of any one of Embodiments 51 to 53, characterized by an X-ray powder diffractogram having a signal at 5.4 ± 0.2 degrees two-theta.
The Compound I Neat Form C of any one of Embodiments 51 to 53, characterized by an X-ray powder diffractogram having signals at (a) 5.4 ± 0.2 degrees two-theta and (b) one or more of 10.2 ± 0.2 degrees two-theta, 15.2 ± 0.2 degrees two-theta, 12.6 ± 0.2 degrees two-theta, 16.7 ± 0.2 degrees two-theta, 11.7 ± 0.2 degrees two-theta, 23.0 ± 0.2 degrees two-theta, and 7.6 ± 0.2 degrees two-theta. The Compound I Neat Form C of any one of Embodiments 51 to 53, characterized by an X-ray powder diffractogram having signals at (a) 5.4 ± 0.2 degrees two-theta and (b) two or more of 10.2 ± 0.2 degrees two-theta, 15.2 ± 0.2 degrees two-theta, 12 6 ± 0.2 degrees two-theta, 16.7 ± 0.2 degrees two-theta, 11.7 ± 0.2 degrees two-theta, 23.0 ± 0.2 degrees two-theta, and 7.6 ± 0.2 degrees two-theta. The Compound I Neat Form C of any one of Embodiments 51 to 53, characterized by an X-ray powder diffractogram having signals at (a) 5.4 ± 0.2 degrees two-theta and (b) three or more of 10.2 ± 0.2 degrees two-theta, 15.2 ± 0.2 degrees two-theta, 12.6 ± 0.2 degrees two-theta, 16.7 ± 0.2 degrees two-theta, 11.7 ± 0.2 degrees two-theta, 23.0 ± 0.2 degrees two-theta, and 7.6 ± 0.2 degrees two-theta. The Compound I Neat Form C of any one of Embodiments 51 to 53, characterized by an X-ray powder diffractogram having signals at (a) 5.4 ± 0.2 degrees two-theta and (b) four or more of 10.2 ± 0.2 degrees two-theta, 15.2 ± 0.2 degrees two-theta, 12.6 ± 0.2 degrees two-theta, 16.7 ± 0.2 degrees two-theta, 11.7 ± 0.2 degrees two-theta, 23.0 ± 0.2 degrees two-theta, and 7.6 ± 0.2 degrees two-theta. The Compound I Neat Form C of any one of Embodiments 51 to 53, characterized by an X-ray powder diffractogram having signals at (a) 5.4 ± 0.2 degrees two-theta and (b) five or more of 10.2 ± 0.2 degrees two-theta, 15.2 ± 0.2 degrees two-theta, 12.6 ± 0.2 degrees two-theta, 16.7 ± 0.2 degrees two-theta, 11.7 ± 0.2 degrees two-theta, 23.0 ± 0.2 degrees two-theta, and 7.6 ± 0.2 degrees two-theta. The Compound I Neat Form C of any one of Embodiments 51 to 53, characterized by an X-ray powder diffractogram having signals at (a) 5.4 ± 0.2 degrees two-theta and (b) six or more of 10.2 ± 0.2 degrees two-theta, 15.2 ± 0.2 degrees two-theta, 12.6 ±
0.2 degrees two-theta, 16.7 ± 0.2 degrees two-theta, 11.7 ± 0.2 degrees two-theta, 23.0 ± 0.2 degrees two-theta, and 7.6 ± 0.2 degrees two-theta. The Compound I Neat Form C of any one of Embodiments 51 to 53, characterized by an X-ray powder diffractogram having signals at 5.4 ± 0.2 degrees two-theta, 10.2 ± 0.2 degrees two-theta, 15 2 ± 0.2 degrees two-theta, 12.6 ± 0.2 degrees two-theta, 16.7 ± 0.2 degrees two-theta, 11.7 ± 0.2 degrees two-theta, 23.0 ± 0.2 degrees two-theta, and 7.6 ± 0.2 degrees two-theta. The Compound I Neat Form C of any one of Embodiments 51 to 61, characterized by an X-ray powder diffractogram substantially similar to FIG. 7. The Compound I Neat Form C of any one of Embodiments 51 to 62, characterized by a TGA showing negligible weight loss from ambient temperature up to about 40 °C and about 0.16% weight loss from 40 °C to 240 °C. The Compound I Neat Form C of any one of Embodiments 51 to 63, characterized by TGA data substantially similar to FIG. 8. The Compound I Neat Form C of any one of Embodiments 51 to 64, characterized by a DSC analysis showing an endothermic peak at about 237 °C. The Compound I Neat Form C of any one of Embodiments 51 to 65, characterized by a DSC analysis substantially similar to FIG. 9. The Compound I Neat Form C of any one of Embodiments 51 to 66, characterized by a 13C ssNMR spectrum with peaks at 43.8 ± 0.2 ppm and 132.3 ± 0.2 ppm. The Compound I Neat Form C of any one of Embodiments 51 to 67, characterized by a 13C ssNMR spectrum with (a) peaks at 43.8 ± 0.2 ppm and 132.3 ± 0.2 ppm and (b) one or more peaks selected from 28.4 ± 0.2 ppm, 29.4 ± 0.2 ppm, 129.1 ± 0.2 ppm, 135.1 ± 0.2 ppm, 139.6 ± 0.2 ppm, and 28.2 ± 0.2 ppm.
The Compound I Neat Form C of any one of Embodiments 51 to 68, characterized by a 13C ssNMR spectrum with (a) peaks at 43.8 ± 0.2 ppm and 132.3 ± 0.2 ppm and (b) two or more peaks selected from 28.4 ± 0.2 ppm, 29.4 ± 0.2 ppm, 129.1 ± 0.2 ppm,
135.1 ± 0.2 ppm, 139.6 ± 0.2 ppm, and 28.2 ± 0.2 ppm. The Compound I Neat Form C of any one of Embodiments 51 to 69, characterized by a 13C ssNMR spectrum with (a) peaks at 43.8 ± 0.2 ppm and 132.3 ± 0.2 ppm and (b) three or more peaks selected from 28.4 ± 0.2 ppm, 29.4 ± 0.2 ppm, 129.1 ± 0.2 ppm,
135.1 ± 0.2 ppm, 139.6 ± 0.2 ppm, and 28.2 ± 0.2 ppm. The Compound I Neat Form C of any one of Embodiments 51 to 70, characterized by a 13C ssNMR spectrum with (a) peaks at 43.8 ± 0.2 ppm and 132.3 ± 0.2 ppm and (b) four or more peaks selected from 28.4 ± 0.2 ppm, 29.4 ± 0.2 ppm, 129.1 ± 0.2 ppm,
135.1 ± 0.2 ppm, 139.6 ± 0.2 ppm, and 28.2 ± 0.2 ppm. The Compound I Neat Form C of any one of Embodiments 51 to 71, characterized by a 13C ssNMR spectrum with (a) peaks at 43.8 ± 0.2 ppm and 132.3 ± 0.2 ppm and (b) five or more peaks selected from 28.4 ± 0.2 ppm, 29.4 ± 0.2 ppm, 129.1 ± 0.2 ppm,
135.1 ± 0.2 ppm, 139.6 ± 0.2 ppm, and 28.2 ± 0.2 ppm. The Compound I Neat Form C of any one of Embodiments 51 to 72, characterized by a 13C ssNMR spectrum with peaks at 43.8 ± 0.2 ppm, 132.3 ± 0.2 ppm, 28.4 ± 0.2 ppm, 29.4 ± 0.2 ppm, 129.1 ± 0.2 ppm, 135.1 ± 0.2 ppm, 139.6 ± 0.2 ppm, and 28.2 ± 0.2 ppm. The Compound I Neat Form C of any one of Embodiments 51 to 73, characterized by a 13C ssNMR spectrum substantially similar to FIG. 10. The Compound I Neat Form C of any one of Embodiments 51 to 74, prepared by a process comprising (i) stirring of Compound I in hexadecane at about 80 °C, and (ii) centrifugation and drying of the resulting solid.
Compound I as substantially crystalline Neat Form D (i.e., wherein less than 15% of Compound I is in amorphous form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous Form). The Compound I of Embodiment 76, wherein Compound I is 100% crystalline Compound I Neat Form D. Substantially pure Compound I Neat Form D. The Compound I Neat Form D of any one of Embodiments 76 to 78, characterized by a 13C ssNMR spectrum with a peak at 50.8 ± 0.2 ppm. The Compound I Neat Form D of any one of Embodiments 76 to 78, characterized by a 13C ssNMR spectrum with (a) a peak at 50.8 ± 0.2 ppm and (b) one or more peaks selected from 28.3 ± 0.2 ppm, 127.2 ± 0.2 ppm, 129.8 ± 0.2 ppm, 170.6 ± 0.2 ppm. The Compound I Neat Form D of any one of Embodiments 76 to 78, characterized by a 13C ssNMR spectrum with (a) a peak at 50.8 ± 0.2 ppm and (b) two or more peaks selected from 28.3 ± 0.2 ppm, 127.2 ± 0.2 ppm, 129.8 ± 0.2 ppm, 170.6 ± 0.2 ppm. The Compound I Neat Form D of any one of Embodiments 76 to 78, characterized by a 13C ssNMR spectrum with (a) a peak at 50.8 ± 0.2 ppm and (b) three or more peaks selected from 28.3 ± 0.2 ppm, 127.2 ± 0.2 ppm, 129.8 ± 0.2 ppm, 170.6 ± 0.2 ppm. The Compound I Neat Form D of any one of Embodiments 76 to 78, characterized by a 13C ssNMR spectrum with peaks at 50.8 ± 0.2 ppm, 28.3 ± 0.2 ppm, 127.2 ± 0.2 ppm, 129.8 ± 0.2 ppm, 170.6 ± 0.2 ppm. The Compound I Neat Form D of any one of Embodiments 76 to 78, characterized by a 13C ssNMR spectrum with (a) peaks at 50.8 ± 0.2 ppm, 28.3 ± 0.2 ppm, 127.2 ± 0.2 ppm, 129.8 ± 0.2 ppm, and 170.6 ± 0.2 ppm and (b) one or more peaks selected from 135.3 ± 0.2 ppm, 136.8 ± 0.2 ppm, and 129.2 ± 0.2 ppm.
The Compound I Neat Form D of any one of Embodiments 76 to 78, characterized by a 13C ssNMR spectrum with (a) peaks at 50.8 ± 0.2 ppm, 28.3 ± 0.2 ppm, 127.2 ± 0.2 ppm, 129.8 ± 0 2 ppm, and 170.6 ± 02 ppm and (b) two or more peaks selected from 135.3 ± 0.2 ppm, 136.8 ± 0.2 ppm, and 129.2 ± 0.2 ppm. The Compound I Neat Form D of any one of Embodiments 76 to 78, characterized by a 13C ssNMR spectrum with peaks at 50.8 ± 0.2 ppm, 28.3 ± 0.2 ppm, 127.2 ± 0.2 ppm, 129.8 ± 0.2 ppm, 170.6 ± 0.2 ppm, 135.3 ± 0.2 ppm, 136.8 ± 0.2 ppm, and 129.2 ± 0.2 ppm The Compound I Neat Form D of any one of Embodiments 76 to 86, characterized by a 13C ssNMR spectrum substantially similar to FIG. 11. The Compound I Neat Form D of any one of Embodiments 76 to 87, prepared by a process comprising the drying of Compound I hydrate C at about 80 °C. The Compound I Neat Form D of any one of Embodiments 76 to 87, prepared by a process comprising (i) stirring of Compound I Neat Form A in IP A, and (ii) isolation of the solid via filtration, and (iii) drying of the solid at about 125 °C. Compound I as substantially crystalline Neat Form E (i.e., wherein less than 15% of Compound I is in amorphous form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous Form). The Compound I of Embodiment 90, wherein Compound I is 100% crystalline Compound I Neat Form E. Substantially pure Compound I Neat Form E. The Compound I Neat Form E of any one of Embodiments 90 to 92, characterized by an X-ray powder diffractogram having (a) a signal at 13.4 ± 0.2 degrees two-theta and (b) a signal at 14.7 ± 0.2 degrees two-theta or 19.6 ± 0.2 degrees two-theta.
The Compound I Neat Form E of any one of Embodiments 90 to 92, characterized by an X-ray powder diffractogram having signals at 13.4 ± 0.2 degrees two-theta, 14.7 ± 0.2 degrees two-theta, and 19.6 ± 0.2 degrees two-theta The Compound I Neat Form E of any one of Embodiments 90 to 92, characterized by an X-ray powder diffractogram having (a) signals at 13.4 ± 0.2 degrees two-theta,
14.7 ± 0.2 degrees two-theta, and 19.6 ± 0.2 degrees two-theta and (b) one or more two-theta values selected from 10.3 degrees two-theta, 8.5 degrees two-theta, 16.0 degrees two-theta, 16.2 degrees two-theta, and 21.4 degrees two-theta. The Compound I Neat Form E of any one of Embodiments 90 to 92, characterized by an X-ray powder diffractogram having (a) signals at 13.4 ± 0.2 degrees two-theta,
14.7 ± 0.2 degrees two-theta, and 19.6 ± 0.2 degrees two-theta and (b) two or more two-theta values selected from 10.3 degrees two-theta, 8.5 degrees two-theta, 16.0 degrees two-theta, 16.2 degrees two-theta, and 21.4 degrees two-theta. The Compound I Neat Form E of any one of Embodiments 90 to 92, characterized by an X-ray powder diffractogram having signals at (a) signals at 13.4 ± 0.2 degrees two-theta, 14.7 ± 0.2 degrees two-theta, and 19.6 ± 0.2 degrees two-theta and (b) three or more two-theta values selected from 10.3 degrees two-theta, 8.5 degrees two- theta, 16.0 degrees two-theta, 16.2 degrees two-theta, and 21.4 degrees two-theta. The Compound I Neat Form E of any one of Embodiments 90 to 92, characterized by an X-ray powder diffractogram having (a) signals at 13.4 ± 0.2 degrees two-theta,
14.7 ± 0.2 degrees two-theta, and 19.6 ± 0.2 degrees two-theta and (b) four or more two-theta values selected from 10.3 degrees two-theta, 8.5 degrees two-theta, 16.0 degrees two-theta, 16.2 degrees two-theta, and 21.4 degrees two-theta. The Compound I Neat Form E of any one of Embodiments 90 to 92, characterized by an X-ray powder diffractogram having signals at 13.4 ± 0.2 degrees two-theta, 14.7
± 0.2 degrees two-theta, and 19.6 ± 0.2 degrees two-theta, 10.3 degrees two-theta, 8.5 degrees two-theta, 16.0 degrees two-theta, 16.2 degrees two-theta, and 21.4 degrees two-theta.
The Compound I Neat Form E of any one of Embodiments 90 to 99, characterized by an X-ray powder diffractogram substantially similar to FIG. 12. The Compound I Neat Form E of any one of Embodiments 90 to 100, characterized by a TGA showing negligible weight loss from 30 °C to 120 °C. The Compound I Neat Form E of any one of Embodiments 90 to 101, characterized by TGA data substantially similar to FIG. 13. The Compound I Neat Form E of any one of Embodiments 90 to 102, characterized by a DSC analysis showing an endothermic peak at about 212 °C. The Compound I Neat Form E of any one of Embodiments 90 to 103, characterized by a DSC analysis substantially similar to FIG. 14. The Compound I Neat Form E of any one of Embodiments 90 to 104, characterized by a 13C ssNMR spectrum with peaks at 20.5 ± 0.2 ppm and 43.6 ± 0.2 ppm. The Compound I Neat Form E of any one of Embodiments 90 to 104, characterized by a 13C ssNMR spectrum with (a) peaks at 20.5 ± 0.2 ppm and 43.6 ± 0.2 ppm and (b) one or more peaks selected from 28.3 ± 0.2 ppm, 43.6 ± 0.2 ppm, 20.5 ± 0.2 ppm, 135.0 ± 0.2 ppm, 128.2 ± 0.2 ppm, and 139.6 ± 0.2 ppm. The Compound I Neat Form E of any one of Embodiments 90 to 104, characterized by a 13C ssNMR spectrum with (a) peaks at 20.5 ± 0.2 ppm and 43.6 ± 0.2 ppm and (b) two or more peaks selected from 28.3 ± 0.2 ppm, 43.6 ± 0.2 ppm, 20.5 ± 0.2 ppm, 135.0 ± 0.2 ppm, 128.2 ± 0.2 ppm, and 139.6 ± 0.2 ppm. The Compound I Neat Form E of any one of Embodiments 90 to 104, characterized by a 13C ssNMR spectrum with (a) peaks at 20.5 ± 0.2 ppm and 43.6 ± 0.2 ppm and (b) three or more peaks selected from 28.3 ± 0.2 ppm, 43.6 ± 0.2 ppm, 20.5 ± 0.2 ppm, 135.0 ± 0.2 ppm, 128.2 ± 0.2 ppm, and 139.6 ± 0.2 ppm.
The Compound I Neat Form E of any one of Embodiments 90 to 104, characterized by a 13C ssNMR spectrum with (a) peaks at 20.5 ± 0.2 ppm and 43.6 ± 0.2 ppm and (b) four or more peaks selected from 28.3 ± 0.2 ppm, 43.6 ± 0.2 ppm, 20.5 ± 0.2 ppm, 135.0 ± 0.2 ppm, 128.2 ± 0.2 ppm, and 139.6 ± 0.2 ppm. The Compound I Neat Form E of any one of Embodiments 90 to 104, characterized by a 13C ssNMR spectrum with (a) peaks at 20.5 ± 0.2 ppm and 43.6 ± 0.2 ppm and (b) five or more peaks selected from 28.3 ± 0.2 ppm, 43.6 ± 0.2 ppm, 20.5 ± 0.2 ppm, 135.0 ± 0.2 ppm, 128.2 ± 0 2 ppm, and 139.6 ± 02 ppm. The Compound I Neat Form E of any one of Embodiments 90 to 104, characterized by a 13C ssNMR spectrum with peaks at 20.5 ± 0.2 ppm, 43.6 ± 0.2 ppm, 28.3 ± 0.2 ppm, 43.6 ± 0.2 ppm, 20.5 ± 0.2 ppm, 135.0 ± 0.2 ppm, 128.2 ± 0.2 ppm, and 139.6 ± 0.2 ppm. The Compound I Neat Form E of any one of Embodiments 90 to 111, characterized by a 13C ssNMR spectrum substantially similar to FIG. 14. The Compound I Neat Form E of any one of Embodiments 90 to 112, characterized by an orthorhombic crystal system, a 212121 space group, and unit cell dimensions, measured at 100 K on a Rigaku diffractometer equipped with Cu Kt radiation ( A. = 1.54178 A) and an HP AD detector, of:
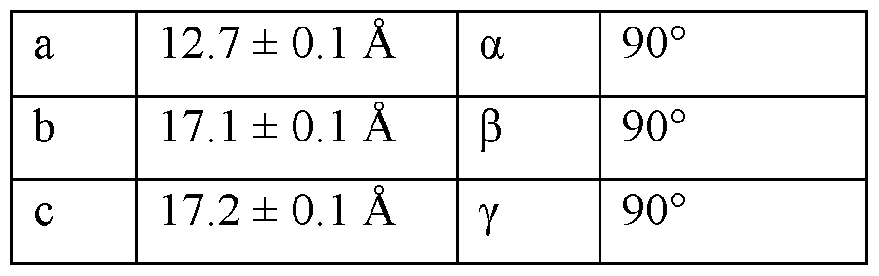 The Compound I Neat Form E of any one of Embodiments 90 to 113, prepared by a process comprising (i) stirring of Compound I Neat Form A in MeOAc, and (ii) isolation of the solid via centrifugation, and (iii) drying of the solid at about 50 °C. Compound I as substantially crystalline compressed Form E (i.e., wherein less than 15% of Compound I is in amorphous form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous Form).
The Compound I of Embodiment 115, wherein Compound I is 100% crystalline Compound I compressed Form E. Substantially pure Compound I compressed Form E. The Compound I compressed Form E of any one of Embodiments 115 to 117, characterized by an X-ray powder diffractogram having (a) signals at two-theta values
The Compound I Neat Form E of any one of Embodiments 90 to 113, prepared by a process comprising (i) stirring of Compound I Neat Form A in MeOAc, and (ii) isolation of the solid via centrifugation, and (iii) drying of the solid at about 50 °C. Compound I as substantially crystalline compressed Form E (i.e., wherein less than 15% of Compound I is in amorphous form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous Form).
The Compound I of Embodiment 115, wherein Compound I is 100% crystalline Compound I compressed Form E. Substantially pure Compound I compressed Form E. The Compound I compressed Form E of any one of Embodiments 115 to 117, characterized by an X-ray powder diffractogram having (a) signals at two-theta values
7.5 ± 0.2 degrees two-theta and 20.8 ± 0.2 degrees two-theta and (b) a signal at 16. 1 ± 0.2 degrees two-theta and/or 8.6 ± 0.2 degrees two-theta. The Compound I compressed Form E of any one of Embodiments 115 to 117, characterized by an X-ray powder diffractogram having signals at two-theta values
7.5 ± 0.2 degrees two-theta, 20.8 ± 0.2 degrees two-theta, 16.1 ± 0.2 degrees two- theta, and 8.6 ± 0.2 degrees two-theta. The Compound I compressed Form E of any one of Embodiments 115 to 117, characterized by an X-ray powder diffractogram having (a) signals at two-theta values
7.5 ± 0.2 degrees two-theta, 20.8 ± 0.2 degrees two-theta, 16.1 ± 0.2 degrees two- theta, and 8.6 ± 0.2 degrees two-theta and (b) signals at one or more two-theta values selected from: 18.4 ± 0.2 degrees two-theta, 20.1 ± 0.2 degrees two-theta, 14.2 ± 0.2 degrees two-theta, and 23.1 ± 0.2 degrees two-theta. The Compound I compressed Form E of any one of Embodiments 115 to 117, characterized by an X-ray powder diffractogram having (a) signals at two-theta values
7.5 ± 0.2 degrees two-theta, 20.8 ± 0.2 degrees two-theta, 16.1 ± 0.2 degrees two- theta, and 8.6 ± 0.2 degrees two-theta and (b) signals at two or more two-theta values selected from: 18.4 ± 0.2 degrees two-theta, 20.1 ± 0.2 degrees two-theta, 14.2 ± 0.2 degrees two-theta, and 23.1 ± 0.2 degrees two-theta. The Compound I compressed Form E of any one of Embodiments 115 to 117, characterized by an X-ray powder diffractogram having (a) signals at two-theta values
7.5 ± 0.2 degrees two-theta, 20.8 ± 0.2 degrees two-theta, 16.1 ± 0.2 degrees two- theta, and 8.6 ± 0.2 degrees two-theta and (b) signals at three or more two-theta
values selected from: 18.4 ± 0.2 degrees two-theta, 20.1 ± 0.2 degrees two-theta, 14.2 ± 0.2 degrees two-theta, and 23.1 ± 0.2 degrees two-theta. The Compound I compressed Form E of any one of Embodiments 115 to 117, characterized by an X-ray powder diffractogram having signals at two-theta values 7.5 ± 0.2 degrees two-theta, 20.8 ± 0.2 degrees two-theta, 16.1 ± 0.2 degrees two- theta, 8.6 ± 0.2 degrees two-theta, 18.4 ± 0.2 degrees two-theta, 20.1 ± 0.2 degrees two-theta, 14.2 ± 0.2 degrees two-theta, and 23.1 ± 0.2 degrees two-theta. The Compound I compressed Form E of any one of Embodiments 115 to 123, characterized by an X-ray powder diffractogram substantially similar to FIG. 16. The Compound I compressed Form E of any one of Embodiments 115 to 124, characterized by a 13C ssNMR spectrum with peaks at 41.8 ± 0.2 ppm and 135.7 ± 0.2 ppm. The Compound I compressed Form E of any one of Embodiments 115 to 124, characterized by a 13C ssNMR spectrum with (a) peaks at 41.8 ± 0.2 ppm and 135.7 ± 0.2 ppm and (b) one or more peaks selected from 28.2 ± 0.2 ppm, 22.0 ± 0.2 ppm,
129.8 ± 0.2 ppm, 136.2 ± 0.2 ppm, and 141.1 ± 0.2 ppm. The Compound I compressed Form E of any one of Embodiments 115 to 124, characterized by a 13C ssNMR spectrum with (a) peaks at 41.8 ± 0.2 ppm and 135.7 ± 0.2 ppm and (b) two or more peaks selected from 28.2 ± 0.2 ppm, 22.0 ± 0.2 ppm,
129.8 ± 0.2 ppm, 136.2 ± 0.2 ppm, and 141.1 ± 0.2 ppm. The Compound I compressed Form E of any one of Embodiments 115 to 124, characterized by a 13C ssNMR spectrum with (a) peaks at 41.8 ± 0.2 ppm and 135.7 ± 0.2 ppm and (b) three or more peaks selected from 28.2 ± 0.2 ppm, 22.0 ± 0.2 ppm,
129.8 ± 0.2 ppm, 136.2 ± 0.2 ppm, and 141.1 ± 0.2 ppm. The Compound I compressed Form E of any one of Embodiments 115 to 124, characterized by a 13C ssNMR spectrum with (a) peaks at 41.8 ± 0.2 ppm and 135.7 ±
0.2 ppm and (b) four or more peaks selected from 28.2 ± 0.2 ppm, 22.0 ± 0.2 ppm, 129.8 ± 0.2 ppm, 136.2 ± 0.2 ppm, and 141.1 ± 0.2 ppm. The Compound I compressed Form E of any one of Embodiments 115 to 124, characterized by a 13C ssNMR spectrum with peaks at 41.8 ± 0.2 ppm, 135.7 ± 0.2 ppm, 28.2 ± 0.2 ppm, 22.0 ± 0.2 ppm, 129.8 ± 0.2 ppm, 136.2 ± 0.2 ppm, and 141.1 ± 0.2 ppm. The Compound I compressed Form E of any one of Embodiments 115 to 124, characterized by a 13C ssNMR spectrum with peaks at 41.8 ± 0.2 ppm, 135.7 ± 0.2 ppm, 28.2 ± 0.2 ppm, 22.0 ± 0.2 ppm, 129.8 ± 0.2 ppm, 136.2 ± 0.2 ppm, 141.1 ± 0.2 ppm, and 127.3 ± 0.2 ppm. The Compound I compressed Form E of any one of Embodiments 115 to 131, characterized by a 13C ssNMR spectrum substantially similar to FIG. 17. The Compound I compressed Form E of any one of Embodiments 115 to 132, prepared by a process comprising mechanical compression of Compound I Neat Form E. Compound I as substantially crystalline compressed Form A (i.e., wherein less than 15% of Compound I is in amorphous form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous Form). The Compound I of Embodiment 134, wherein Compound I is 100% crystalline Compound I Compressed Form A. Substantially pure Compound I Compressed Form A. The Compound I Compressed Form A of any one of Embodiments 134 to 136, characterized by an X-ray powder diffractogram having signals at (a) 8.9 ± 0.2 degrees two-theta, 15.2 ± 0.2 degrees two-theta, 15.8 ± 0.2 degrees two-theta and (b) one or more selected from 18.2 ± 0.2 degrees two-theta, 20.9 ± 0.2 degrees two-theta, and 16.7 ± 0.2 degrees two-theta.
The Compound I Compressed Form A of any one of Embodiments 134 to 136, characterized by an X-ray powder diffractogram having signals at (a) 8.9 ± 0.2 degrees two-theta, 15.2 ± 0.2 degrees two-theta, 15.8 ± 0.2 degrees two-theta and (b) two or more selected from 18.2 ± 0.2 degrees two-theta, 20.9 ± 0.2 degrees two-theta, and 16.7 ± 0.2 degrees two-theta. The Compound I Compressed Form A of any one of Embodiments 134 to 136, characterized by an X-ray powder diffractogram having signals at 8.9 ± 0.2 degrees two-theta, 15.2 ± 0.2 degrees two-theta, 15.8 ± 0.2 degrees two-theta, 18.2 ± 0.2 degrees two-theta, 20.9 ± 0.2 degrees two-theta, and 16.7 ± 0.2 degrees two-theta. The Compound I Compressed Form A of any one of Embodiments 134 to 136, characterized by an X-ray powder diffractogram having signals at (a) 8.9 ± 0.2 degrees two-theta, 15.2 ± 0.2 degrees two-theta, 15.8 ± 0.2 degrees two-theta, 18.2 ± 0.2 degrees two-theta, 20.9 ± 0.2 degrees two-theta, and 16.7 ± 0.2 degrees two-theta and (b) 21.5 ± 0.2 degrees two-theta or 23.0 ± 0.2 degrees two-theta. The Compound I Compressed Form A of any one of Embodiments 134 to 136, characterized by an X-ray powder diffractogram having signals at 8.9 ± 0.2 degrees two-theta, 15.2 ± 0.2 degrees two-theta, 15.8 ± 0.2 degrees two-theta, 18.2 ± 0.2 degrees two-theta, 20.9 ± 0.2 degrees two-theta, and 16.7 ± 0.2 degrees two-theta, 21.5 ± 0.2 degrees two-theta, and 23.0 ± 0.2 degrees two-theta. The Compound I Compressed Form A of any one of Embodiments 134 to 141, characterized by an X-ray powder diffractogram substantially similar to FIG. 18. Compressed Form A of any one of Embodiments 134 to 142, characterized by a 13C ssNMR spectrum with (a) a peak at 28.9 ± 0.2 ppm and (b) one or more peaks selected from 41.9 ± 0.2 ppm, 21.6 ± 0.2 ppm, 132.9 ± 0.2 ppm, and 127.5 ± 0.2 ppm. The Compound I Compressed Form A of any one of Embodiments 134 to 142, characterized by a 13C ssNMR spectrum with (a) a peak at 28.9 ± 0.2 ppm and (b) two
or more peaks selected from 41.9 ± 0.2 ppm, 21.6 ± 0.2 ppm, 132.9 ± 0.2 ppm, and 127.5 ± 0.2 ppm. The Compound I Compressed Form A of any one of Embodiments 134 to 142, characterized by a 13C ssNMR spectrum with (a) a peak at 28.9 ± 0.2 ppm and (b) three or more peaks selected from 41.9 ± 0.2 ppm, 21.6 ± 0.2 ppm, 132 9 ± 0.2 ppm, and 127.5 ± 0.2 ppm. The Compound I Compressed Form A of any one of Embodiments 134 to 142, characterized by a 13C ssNMR spectrum with peaks at 28.9 ± 0.2 ppm, 41.9 ± 0.2 ppm, 21.6 ± 0.2 ppm, 132.9 ± 0.2 ppm, and 127.5 ± 0.2 ppm. The Compound I Compressed Form A of any one of Embodiments 134 to 142, characterized by a 13C ssNMR spectrum with peaks at (a) 28.9 ± 0.2 ppm, 41.9 ± 0.2 ppm, 21.6 ± 0.2 ppm, 132.9 ± 0.2 ppm, and 127.5 ± 0.2 ppm and (b) one or more selected from 130.7 ± 0.2 ppm, 133.7 ± 0.2 ppm, and 22.5 ± 0.2 ppm. The Compound I Compressed Form A of any one of Embodiments 134 to 142, characterized by a 13C ssNMR spectrum with peaks at (a) 28.9 ± 0.2 ppm, 41.9 ± 0.2 ppm, 21.6 ± 0.2 ppm, 132.9 ± 0.2 ppm, and 127.5 ± 0.2 ppm and (b) two or more selected from 130.7 ± 0.2 ppm, 133.7 ± 0.2 ppm, and 22.5 ± 0.2 ppm. The Compound I Compressed Form A of any one of Embodiments 134 to 142, characterized by a 13C ssNMR spectrum with peaks at 28.9 ± 0.2 ppm, 41.9 ± 0.2 ppm, 21.6 ± 0.2 ppm, 132.9 ± 0.2 ppm, 127.5 ± 0.2 ppm, 130.7 ± 0.2 ppm, 133.7 ± 0.2 ppm, and 22.5 ± 0.2 ppm. The Compound I Compressed Form A of any one of Embodiments 134 to 149, characterized by a 13C ssNMR spectrum substantially similar to FIG. 18. The Compound I Compressed Form A of any one of Embodiments 134 to 150, prepared by a process comprising mechanical compression of Compound I Neat Form A.
Compound I as substantially crystalline EtOH Solvate Form B (i.e., wherein less than 15% of Compound I is in amorphous Form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous form). The Compound I of Embodiment 152, wherein Compound I is 100% crystalline Compound I EtOH Solvate Form B. Substantially pure Compound I EtOH Solvate Form B. The Compound I EtOH Solvate Form B of any one of Embodiments 152 to 154, characterized by an X-ray powder diffractogram having a signal at one or more two- theta values selected from 6.4 ± 0.2 degrees two-theta, 10.8 ± 0.2 degrees two-theta,
7.1 ± 0.2 degrees two-theta, 14.2 ± 0.2 degrees two-theta, 19.9 ± 0.2 degrees two- theta, 10.2 ± 0.2 degrees two-theta, 17.5 ± 0.2 degrees two-theta, and 19.5 ± 0.2 degrees two-theta. The Compound I EtOH Solvate Form B of any one of Embodiments 152 to 155, characterized by an X-ray powder diffractogram having signals at two or more two- theta values selected from 6.4 ± 0.2 degrees two-theta, 10.8 ± 0.2 degrees two-theta,
7.1 ± 0.2 degrees two-theta, 14.2 ± 0.2 degrees two-theta, 19.9 ± 0.2 degrees two- theta, 10.2 ± 0.2 degrees two-theta, 17.5 ± 0.2 degrees two-theta, and 19.5 ± 0.2 degrees two-theta. The Compound I EtOH Solvate Form B of any one of Embodiments 152 to 156, characterized by an X-ray powder diffractogram having signals at three or more two- theta values selected from 6.4 ± 0.2 degrees two-theta, 10.8 ± 0.2 degrees two-theta,
7.1 ± 0.2 degrees two-theta, 14.2 ± 0.2 degrees two-theta, 19.9 ± 0.2 degrees two- theta, 10.2 ± 0.2 degrees two-theta, 17.5 ± 0.2 degrees two-theta, and 19.5 ± 0.2 degrees two-theta. The Compound I EtOH Solvate Form B of any one of Embodiments 152 to 157, characterized by an X-ray powder diffractogram having signals at four or more two- theta values selected from 6.4 ± 0.2 degrees two-theta, 10.8 ± 0.2 degrees two-theta,
7.1 ± 0.2 degrees two-theta, 14.2 ± 0.2 degrees two-theta, 19.9 ± 0.2 degrees two-
theta, 10.2 ± 0.2 degrees two-theta, 17.5 ± 0.2 degrees two-theta, and 19.5 ± 0.2 degrees two-theta. The Compound I EtOH Solvate Form B of any one of Embodiments 152 to 158, characterized by an X-ray powder diffractogram having signals at five or more two- theta values selected from 6.4 ± 0.2 degrees two-theta, 10.8 ± 0.2 degrees two-theta,
7. 1 ± 0.2 degrees two-theta, 14.2 ± 0.2 degrees two-theta, 19.9 ± 0.2 degrees two- theta, 10.2 ± 0.2 degrees two-theta, 17.5 ± 0.2 degrees two-theta, and 19.5 ± 0.2 degrees two-theta. The Compound I EtOH Solvate Form B of any one of Embodiments 152 to 159, characterized by an X-ray powder diffractogram having signals at six or more two- theta values selected from 6.4 ± 0.2 degrees two-theta, 10.8 ± 0.2 degrees two-theta,
7. 1 ± 0.2 degrees two-theta, 14.2 ± 0.2 degrees two-theta, 19.9 ± 0.2 degrees two- theta, 10.2 ± 0.2 degrees two-theta, 17.5 ± 0.2 degrees two-theta, and 19.5 ± 0.2 degrees two-theta. The Compound I EtOH Solvate Form B of any one of Embodiments 152 to 160, characterized by an X-ray powder diffractogram having signals at seven or more two- theta values selected from 6.4 ± 0.2 degrees two-theta, 10.8 ± 0.2 degrees two-theta,
7. 1 ± 0.2 degrees two-theta, 14.2 ± 0.2 degrees two-theta, 19.9 ± 0.2 degrees two- theta, 10.2 ± 0.2 degrees two-theta, 17.5 ± 0.2 degrees two-theta, and 19.5 ± 0.2 degrees two-theta. The Compound I EtOH Solvate Form B of any one of Embodiments 152 to 161, characterized by an X-ray powder diffractogram having signals at two-theta values 6.4 ± 0.2 degrees two-theta, 10.8 ± 0.2 degrees two-theta, 7.1 ± 0.2 degrees two-theta, 14.2 ± 0.2 degrees two-theta, 19.9 ± 0.2 degrees two-theta, 10.2 ± 0.2 degrees two- theta, 17.5 ± 0.2 degrees two-theta, and 19.5 ± 0.2 degrees two-theta. The Compound I EtOH Solvate Form B of any one of Embodiments 152 to 162, characterized by an X-ray powder diffractogram substantially similar to FIG. 20.
The Compound I EtOH Solvate Form B of any one of Embodiments 152 to 163, characterized by a TGA showing about 5.29% weight loss from ambient temperature up to about 90 °C. The Compound I EtOH Solvate Form B of any one of Embodiments 152 to 164, characterized by TGA data substantially similar to FIG. 21 The Compound I EtOH Solvate Form B of any one of Embodiments 152 to 165, characterized by a DSC analysis showing an endothermic peak at about 109 °C and at about 197 °C. The Compound I EtOH Solvate Form B of any one of Embodiments 152 to 166, characterized by a DSC analysis substantially similar to FIG. 22. The Compound I EtOH Solvate Form B of any one of Embodiments 152 to 167, characterized by a 13C ssNMR spectrum with one or more peaks selected from: 28.1 ± 0.2 ppm, 18.7 ± 0.2 ppm, 44.1 ± 0.2 ppm, 128.9 ± 0.2 ppm, 21.6 ± 0.2 ppm, 142.8 ± 0.2 ppm, 132.0 ± 0.2 ppm and 130.4. The Compound I EtOH Solvate Form B of any one of Embodiments 152 to 168, characterized by a 13C ssNMR spectrum with two or more peaks selected from: 28.1 ± 0.2 ppm, 18.7 ± 0.2 ppm, 44.1 ± 0.2 ppm, 128.9 ± 0.2 ppm, 21.6 ± 0.2 ppm, 142.8 ± 0.2 ppm, 132.0 ± 0.2 ppm and 130.4. The Compound I EtOH Solvate Form B of any one of Embodiments 152 to 169, characterized by a 13C ssNMR spectrum with three or more peaks selected from: 28.1 ± 0.2 ppm, 18.7 ± 0.2 ppm, 44.1 ± 0.2 ppm, 128.9 ± 0.2 ppm, 21.6 ± 0.2 ppm, 142.8 ± 0.2 ppm, 132.0 ± 0.2 ppm and 130.4. The Compound I EtOH Solvate Form B of any one of Embodiments 152 to 170, characterized by a 13C ssNMR spectrum with four or more peaks selected from: 28.1 ± 0.2 ppm, 18.7 ± 0.2 ppm, 44.1 ± 0.2 ppm, 128.9 ± 0.2 ppm, 21.6 ± 0.2 ppm, 142.8 ± 0.2 ppm, 132.0 ± 0.2 ppm and 130.4.
The Compound I EtOH Solvate Form B of any one of Embodiments 152 to 171, characterized by a 13C ssNMR spectrum with five or more peaks selected from: 28.1 ± 0.2 ppm, 18.7 ± 0.2 ppm, 44.1 ± 0.2 ppm, 128.9 ± 0.2 ppm, 21.6 ± 0.2 ppm, 142.8 ± 0.2 ppm, 132.0 ± 0.2 ppm and 130.4. The Compound I EtOH Solvate Form B of any one of Embodiments 152 to 172, characterized by a 13C ssNMR spectrum with six or more peaks selected from: 28.1 ± 0.2 ppm, 18.7 ± 0.2 ppm, 44.1 ± 0.2 ppm, 128.9 ± 0.2 ppm, 21.6 ± 0.2 ppm, 142.8 ± 0.2 ppm, 132 0 ± 0.2 ppm and 130.4. The Compound I EtOH Solvate Form B of any one of Embodiments 152 to 173, characterized by a 13C ssNMR spectrum with seven or more peaks selected from: 28.1 ± 0.2 ppm, 18.7 ± 0.2 ppm, 44.1 ± 0.2 ppm, 128.9 ± 0.2 ppm, 21.6 ± 0.2 ppm, 142.8 ± 0.2 ppm, 132.0 ± 0.2 ppm and 130.4. The Compound I EtOH Solvate Form B of any one of Embodiments 152 to 174, characterized by a 13C ssNMR spectrum with peaks at: 28.1 ± 0.2 ppm, 18.7 ± 0.2 ppm, 44.1 ± 0.2 ppm, 128.9 ± 0.2 ppm, 21.6 ± 0.2 ppm, 142.8 ± 0.2 ppm, 132.0 ± 0.2 ppm and 130.4. The Compound I EtOH Solvate Form B of any one of Embodiments 152 to 175, characterized by a 13C ssNMR spectrum substantially similar to FIG. 23. The Compound I EtOH Solvate Form B of any one of Embodiments 152 to 176, characterized by a monoclinic crystal system, aP2i2i2i space group, and unit cell dimensions, measured at 100 K on a Bruker diffractometer equipped with Cu K« radiation ( = 1.54178 A) and an CP AD detector, of:
 The Compound I EtOH Solvate Form B of any one of Embodiments 152 to 177, prepared by a process comprising (i) stirring of Compound I in ethanol, and (ii) isolation of the solid via centrifugation or vacuum filtration. Compound I as substantially crystalline MeOH Solvate (i.e., wherein less than 15% of Compound I is in amorphous Form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous form). The Compound I of Embodiment 179, wherein Compound I is 100% crystalline Compound I MeOH Solvate. Substantially pure Compound I MeOH Solvate The Compound I MeOH Solvate of any one of Embodiments 179 to 181, characterized by an X-ray powder diffractogram having a signal at one or more two- theta values selected from 26.1 ± 0.2 degrees two-theta, 12.2 ± 0.2 degrees two-theta,
The Compound I EtOH Solvate Form B of any one of Embodiments 152 to 177, prepared by a process comprising (i) stirring of Compound I in ethanol, and (ii) isolation of the solid via centrifugation or vacuum filtration. Compound I as substantially crystalline MeOH Solvate (i.e., wherein less than 15% of Compound I is in amorphous Form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous form). The Compound I of Embodiment 179, wherein Compound I is 100% crystalline Compound I MeOH Solvate. Substantially pure Compound I MeOH Solvate The Compound I MeOH Solvate of any one of Embodiments 179 to 181, characterized by an X-ray powder diffractogram having a signal at one or more two- theta values selected from 26.1 ± 0.2 degrees two-theta, 12.2 ± 0.2 degrees two-theta,
22.8 ± 0.2 degrees two-theta, and 21.1 ± 0.2 degrees two-theta. The Compound I MeOH Solvate of any one of Embodiments 179 to 182, characterized by an X-ray powder diffractogram having signals at two or more two- theta values selected from 26.1 ± 0.2 degrees two-theta, 12.2 ± 0.2 degrees two-theta,
22.8 ± 0.2 degrees two-theta, and 21.1 ± 0.2 degrees two-theta. The Compound I MeOH Solvate of any one of Embodiments 179 to 183, characterized by an X-ray powder diffractogram having signals at three or more two- theta values selected from 26.1 ± 0.2 degrees two-theta, 12.2 ± 0.2 degrees two-theta,
22.8 ± 0.2 degrees two-theta, and 21.1 ± 0.2 degrees two-theta. The Compound I MeOH Solvate of any one of Embodiments 179 to 184, characterized by an X-ray powder diffractogram having signals at 26.1 ± 0.2 degrees two-theta, 12.2 ± 0.2 degrees two-theta, 22.8 ± 0.2 degrees two-theta, and 21.1 ± 0.2 degrees two-theta.
The Compound I MeOH Solvate of any one of Embodiments 179 to 185, characterized by an X-ray powder diffractogram substantially similar to FIG. 24. The Compound I MeOH Solvate of any one of Embodiments 179 to 186, characterized by a 13C ssNMR spectrum with one or more peaks selected from: 129.1 ± 0.2 ppm, 136.6 ± 0.2 ppm, 136.5 ± 0.2 ppm, 129.6 ± 0.2 ppm, 142.6 ± 0.2 ppm,
130.5 ± 0.2 ppm, 128.2 ± 0.2 ppm, and 130.0 ± 0.2 ppm. The Compound I MeOH Solvate of any one of Embodiments 179 to 187, characterized by a 13C ssNMR spectrum with two or more peaks selected from: 129.1 ± 0.2 ppm, 136.6 ± 0.2 ppm, 136.5 ± 0.2 ppm, 129.6 ± 0.2 ppm, 142.6 ± 0.2 ppm,
130.5 ± 0.2 ppm, 128.2 ± 0.2 ppm, and 130.0 ± 0.2 ppm. The Compound I MeOH Solvate of any one of Embodiments 179 to 188, characterized by a 13C ssNMR spectrum with three or more peaks selected from: 129.1 ± 0.2 ppm, 136.6 ± 0.2 ppm, 136.5 ± 0.2 ppm, 129.6 ± 0.2 ppm, 142.6 ± 0.2 ppm, 130.5 ± 0.2 ppm, 128.2 ± 0.2 ppm, and 130.0 ± 0.2 ppm. The Compound I MeOH Solvate of any one of Embodiments 179 to 189, characterized by a 13C ssNMR spectrum with four or more peaks selected from: 129.1 ± 0.2 ppm, 136.6 ± 0.2 ppm, 136.5 ± 0.2 ppm, 129.6 ± 0.2 ppm, 142.6 ± 0.2 ppm,
130.5 ± 0.2 ppm, 128.2 ± 0.2 ppm, and 130.0 ± 0.2 ppm. The Compound I MeOH Solvate of any one of Embodiments 179 to 190, characterized by a 13C ssNMR spectrum with five or more peaks expressed in ppm ± 0.2 selected from: 129.1 ± 0.2 ppm, 136.6 ± 0.2 ppm, 136.5 ± 0.2 ppm, 129.6 ± 0.2 ppm, 142.6 ± 0.2 ppm, 130.5 ± 0.2 ppm, 128.2 ± 0.2 ppm, 130.0 ± 0.2 ppm. The Compound I MeOH Solvate of any one of Embodiments 179 to 191, characterized by a 13C ssNMR spectrum with six or more peaks selected from: 129.1 ± 0.2 ppm, 136.6 ± 0.2 ppm, 136.5 ± 0.2 ppm, 129.6 ± 0.2 ppm, 142.6 ± 0.2 ppm,
130.5 ± 0.2 ppm, 128.2 ± 0.2 ppm, and 130.0 ± 0.2 ppm.
The Compound I MeOH Solvate of any one of Embodiments 179 to 192, characterized by a 13C ssNMR spectrum with seven or more peaks selected from: 129.1 ± 0.2 ppm, 136.6 ± 0.2 ppm, 136.5 ± 0.2 ppm, 129.6 ± 0.2 ppm, 142.6 ± 0.2 ppm, 130.5 ± 0.2 ppm, 128.2 ± 0.2 ppm, and 130.0 ± 0.2 ppm. The Compound I MeOH Solvate of any one of Embodiments 179 to 193, characterized by a 13C ssNMR spectrum with peaks at: 129.1 ± 0.2 ppm, 136.6 ± 0.2 ppm, 136.5 ± 0.2 ppm, 129.6 ± 0.2 ppm, 142.6 ± 0.2 ppm, 130.5 ± 0.2 ppm, 128.2 ± 0.2 ppm, and 130.0 ± 02 ppm The Compound I MeOH Solvate of any one of Embodiments 179 to 194, characterized by a 13C ssNMR spectrum substantially similar to FIG. 25. The Compound I MeOH Solvate of any one of Embodiments 179 to 195, characterized by a monoclinic crystal system, aP2i2i2i space group, and unit cell dimensions, measured at 100 K on a Bruker diffractometer equipped with Cu K« radiation ( = 1.54178 A) and an CP AD detector, of:
 The Compound I MeOH Solvate of any one of Embodiments 179 to 196, prepared by a process comprising (i) dissolving Compound I in MeOH and increasing temperature to about 62 °C, (ii) cooling the solution to about 10 °C, and (iii) isolation of the solid via centrifugation. Compound I as substantially crystalline NPA Solvate (i.e., wherein less than 15% of Compound I is in amorphous Form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous form). The Compound I of Embodiment 198, wherein Compound I is 100% crystalline Compound I NPA Solvate.
Substantially pure Compound I NPA Solvate. The Compound I NPA Solvate of any one of Embodiments 198 to 200, characterized by an X-ray powder diffractogram having a signal at one or more two-theta values selected from 10.6 ± 0.2 degrees two-theta, 6.3 ± 0.2 degrees two-theta, 19.1 ± 0.2 degrees two-theta, 19.6 ± 0.2 degrees two-theta, 17 3 ± 0.2 degrees two-theta, 17.2 ± 0.2 degrees two-theta, 10.0 ± 0.2 degrees two-theta, and 14.0 ± 0.2 degrees two-theta. The Compound I NPA Solvate of any one of Embodiments 198 to 201, characterized by an X-ray powder diffractogram having signals at two or more two-theta values selected from 10.6 ± 0.2 degrees two-theta, 6.3 ± 0.2 degrees two-theta, 19.1 ± 0.2 degrees two-theta, 19.6 ± 0.2 degrees two-theta, 17 3 ± 0.2 degrees two-theta, 17.2 ± 0.2 degrees two-theta, 10.0 ± 0.2 degrees two-theta, and 14.0 ± 0.2 degrees two-theta. The Compound I NPA Solvate of any one of Embodiments 198 to 202, characterized by an X-ray powder diffractogram having signals at three or more two-theta values selected from 10.6 ± 0.2 degrees two-theta, 6.3 ± 0.2 degrees two-theta, 19.1 ± 0.2 degrees two-theta, 19.6 ± 0.2 degrees two-theta, 17.3 ± 0.2 degrees two-theta, 17.2 ± 0.2 degrees two-theta, 10.0 ± 0.2 degrees two-theta, and 14.0 ± 0.2 degrees two-theta. The Compound I NPA Solvate of any one of Embodiments 198 to 203, characterized by an X-ray powder diffractogram having signals at four or more two-theta values selected from 10.6 ± 0.2 degrees two-theta, 6.3 ± 0.2 degrees two-theta, 19.1 ± 0.2 degrees two-theta, 19.6 ± 0.2 degrees two-theta, 17.3 ± 0.2 degrees two-theta, 17.2 ± 0.2 degrees two-theta, 10.0 ± 0.2 degrees two-theta, and 14.0 ± 0.2 degrees two-theta. The Compound I NPA Solvate of any one of Embodiments 198 to 204, characterized by an X-ray powder diffractogram having signals at five or more two-theta values selected from 10.6 ± 0.2 degrees two-theta, 6.3 ± 0.2 degrees two-theta, 19.1 ± 0.2 degrees two-theta, 19.6 ± 0.2 degrees two-theta, 17.3 ± 0.2 degrees two-theta, 17.2 ± 0.2 degrees two-theta, 10.0 ± 0.2 degrees two-theta, and 14.0 ± 0.2 degrees two-theta. The Compound I NPA Solvate of any one of Embodiments 198 to 205, characterized by an X-ray powder diffractogram having signals at six or more two-theta values
selected from 10.6 ± 0.2 degrees two-theta, 6.3 ± 0.2 degrees two-theta, 19.1 ± 0.2 degrees two-theta, 19.6 ± 0.2 degrees two-theta, 17.3 ± 0.2 degrees two-theta, 17.2 ± 0.2 degrees two-theta, 10 0 ± 0.2 degrees two-theta, and 14 0 ± 0.2 degrees two-theta. The Compound I NPA Solvate of any one of Embodiments 198 to 206, characterized by an X-ray powder diffractogram having signals at seven or more two-theta values selected from 10.6 ± 0.2 degrees two-theta, 6.3 ± 0.2 degrees two-theta, 19.1 ± 0.2 degrees two-theta, 19.6 ± 0.2 degrees two-theta, 17.3 ± 0.2 degrees two-theta, 17.2 ± 0.2 degrees two-theta, 10 0 ± 0.2 degrees two-theta, and 14 0 ± 0.2 degrees two-theta. The Compound I NPA Solvate of any one of Embodiments 198 to 207, characterized by an X-ray powder diffractogram having signals at 10.6 ± 0.2 degrees two-theta, 6.3 ± 0.2 degrees two-theta, 19.1 ± 0.2 degrees two-theta, 19.6 ± 0.2 degrees two-theta, 17.3 ± 0.2 degrees two-theta, 17.2 ± 0.2 degrees two-theta, 10.0 ± 0.2 degrees two- theta, and 14.0 ± 0.2 degrees two-theta. The Compound I NPA Solvate of any one of Embodiments 198 to 208, characterized by an X-ray powder diffractogram substantially similar to FIG. 26. The Compound I NPA Solvate of any one of Embodiments 198 to 209, characterized by a 13C ssNMR spectrum with one or more peaks selected from: 28.0 ± 0.2 ppm, 30.0 ± 0.2 ppm, 128.7 ± 0.2 ppm, 44.0 ± 0.2 ppm, 130.0 ± 0.2 ppm, 131.0 ± 0.2 ppm,
The Compound I MeOH Solvate of any one of Embodiments 179 to 196, prepared by a process comprising (i) dissolving Compound I in MeOH and increasing temperature to about 62 °C, (ii) cooling the solution to about 10 °C, and (iii) isolation of the solid via centrifugation. Compound I as substantially crystalline NPA Solvate (i.e., wherein less than 15% of Compound I is in amorphous Form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous form). The Compound I of Embodiment 198, wherein Compound I is 100% crystalline Compound I NPA Solvate.
Substantially pure Compound I NPA Solvate. The Compound I NPA Solvate of any one of Embodiments 198 to 200, characterized by an X-ray powder diffractogram having a signal at one or more two-theta values selected from 10.6 ± 0.2 degrees two-theta, 6.3 ± 0.2 degrees two-theta, 19.1 ± 0.2 degrees two-theta, 19.6 ± 0.2 degrees two-theta, 17 3 ± 0.2 degrees two-theta, 17.2 ± 0.2 degrees two-theta, 10.0 ± 0.2 degrees two-theta, and 14.0 ± 0.2 degrees two-theta. The Compound I NPA Solvate of any one of Embodiments 198 to 201, characterized by an X-ray powder diffractogram having signals at two or more two-theta values selected from 10.6 ± 0.2 degrees two-theta, 6.3 ± 0.2 degrees two-theta, 19.1 ± 0.2 degrees two-theta, 19.6 ± 0.2 degrees two-theta, 17 3 ± 0.2 degrees two-theta, 17.2 ± 0.2 degrees two-theta, 10.0 ± 0.2 degrees two-theta, and 14.0 ± 0.2 degrees two-theta. The Compound I NPA Solvate of any one of Embodiments 198 to 202, characterized by an X-ray powder diffractogram having signals at three or more two-theta values selected from 10.6 ± 0.2 degrees two-theta, 6.3 ± 0.2 degrees two-theta, 19.1 ± 0.2 degrees two-theta, 19.6 ± 0.2 degrees two-theta, 17.3 ± 0.2 degrees two-theta, 17.2 ± 0.2 degrees two-theta, 10.0 ± 0.2 degrees two-theta, and 14.0 ± 0.2 degrees two-theta. The Compound I NPA Solvate of any one of Embodiments 198 to 203, characterized by an X-ray powder diffractogram having signals at four or more two-theta values selected from 10.6 ± 0.2 degrees two-theta, 6.3 ± 0.2 degrees two-theta, 19.1 ± 0.2 degrees two-theta, 19.6 ± 0.2 degrees two-theta, 17.3 ± 0.2 degrees two-theta, 17.2 ± 0.2 degrees two-theta, 10.0 ± 0.2 degrees two-theta, and 14.0 ± 0.2 degrees two-theta. The Compound I NPA Solvate of any one of Embodiments 198 to 204, characterized by an X-ray powder diffractogram having signals at five or more two-theta values selected from 10.6 ± 0.2 degrees two-theta, 6.3 ± 0.2 degrees two-theta, 19.1 ± 0.2 degrees two-theta, 19.6 ± 0.2 degrees two-theta, 17.3 ± 0.2 degrees two-theta, 17.2 ± 0.2 degrees two-theta, 10.0 ± 0.2 degrees two-theta, and 14.0 ± 0.2 degrees two-theta. The Compound I NPA Solvate of any one of Embodiments 198 to 205, characterized by an X-ray powder diffractogram having signals at six or more two-theta values
selected from 10.6 ± 0.2 degrees two-theta, 6.3 ± 0.2 degrees two-theta, 19.1 ± 0.2 degrees two-theta, 19.6 ± 0.2 degrees two-theta, 17.3 ± 0.2 degrees two-theta, 17.2 ± 0.2 degrees two-theta, 10 0 ± 0.2 degrees two-theta, and 14 0 ± 0.2 degrees two-theta. The Compound I NPA Solvate of any one of Embodiments 198 to 206, characterized by an X-ray powder diffractogram having signals at seven or more two-theta values selected from 10.6 ± 0.2 degrees two-theta, 6.3 ± 0.2 degrees two-theta, 19.1 ± 0.2 degrees two-theta, 19.6 ± 0.2 degrees two-theta, 17.3 ± 0.2 degrees two-theta, 17.2 ± 0.2 degrees two-theta, 10 0 ± 0.2 degrees two-theta, and 14 0 ± 0.2 degrees two-theta. The Compound I NPA Solvate of any one of Embodiments 198 to 207, characterized by an X-ray powder diffractogram having signals at 10.6 ± 0.2 degrees two-theta, 6.3 ± 0.2 degrees two-theta, 19.1 ± 0.2 degrees two-theta, 19.6 ± 0.2 degrees two-theta, 17.3 ± 0.2 degrees two-theta, 17.2 ± 0.2 degrees two-theta, 10.0 ± 0.2 degrees two- theta, and 14.0 ± 0.2 degrees two-theta. The Compound I NPA Solvate of any one of Embodiments 198 to 208, characterized by an X-ray powder diffractogram substantially similar to FIG. 26. The Compound I NPA Solvate of any one of Embodiments 198 to 209, characterized by a 13C ssNMR spectrum with one or more peaks selected from: 28.0 ± 0.2 ppm, 30.0 ± 0.2 ppm, 128.7 ± 0.2 ppm, 44.0 ± 0.2 ppm, 130.0 ± 0.2 ppm, 131.0 ± 0.2 ppm,
12.7 ± 0.2 ppm, and 10.8 ± 0.2 ppm. The Compound I NPA Solvate of any one of Embodiments 198 to 210, characterized by a 13C ssNMR spectrum with two or more peaks selected from: 28.0 ± 0.2 ppm, 30.0 ± 0.2 ppm, 128.7 ± 0.2 ppm, 44.0 ± 0.2 ppm, 130.0 ± 0.2 ppm, 131.0 ± 0.2 ppm,
12.7 ± 0.2 ppm, and 10.8 ± 0.2 ppm. The Compound I NPA Solvate of any one of Embodiments 198 to 211, characterized by a 13C ssNMR spectrum with three or more peaks selected from: 28.0 ± 0.2 ppm, 30.0 ± 0.2 ppm, 128.7 ± 0.2 ppm, 44.0 ± 0.2 ppm, 130.0 ± 0.2 ppm, 131.0 ± 0.2 ppm,
12.7 ± 0.2 ppm, and 10.8 ± 0.2 ppm.
The Compound I NPA Solvate of any one of Embodiments 198 to 212, characterized by a 13C ssNMR spectrum with four or more peaks selected from: 28.0 ± 0.2 ppm, 30.0 ± 0.2 ppm, 128.7 ± 0.2 ppm, 44.0 ± 0.2 ppm, 130.0 ± 0.2 ppm, 131.0 ± 0.2 ppm,
12.7 ± 0.2 ppm, and 10.8 ± 0.2 ppm. The Compound I NPA Solvate of any one of Embodiments 198 to 213, characterized by a 13C ssNMR spectrum with five or more peaks selected from: 28.0 ± 0.2 ppm, 30.0 ± 0.2 ppm, 128.7 ± 0.2 ppm, 44.0 ± 0.2 ppm, 130.0 ± 0.2 ppm, 131.0 ± 0.2 ppm,
12.7 ± 0.2 ppm, and 10 8 ± 0.2 ppm. The Compound I NPA Solvate of any one of Embodiments 198 to 214, characterized by a 13C ssNMR spectrum with six or more peaks selected from: 28.0 ± 0.2 ppm, 30.0 ± 0.2 ppm, 128.7 ± 0.2 ppm, 44.0 ± 0.2 ppm, 130.0 ± 0.2 ppm, 131.0 ± 0.2 ppm, 12.7 ± 0.2 ppm, and 10.8 ± 0.2 ppm. The Compound I NPA Solvate of any one of Embodiments 198 to 215, characterized by a 13C ssNMR spectrum with seven or more peaks selected from: 28.0 ± 0.2 ppm, 30.0 ± 0.2 ppm, 128.7 ± 0.2 ppm, 44.0 ± 0.2 ppm, 130.0 ± 0.2 ppm, 131.0 ± 0.2 ppm,
12.7 ± 0.2 ppm, and 10.8 ± 0.2 ppm. The Compound I NPA Solvate of any one of Embodiments 198 to 216, characterized by a 13C ssNMR spectrum with peaks at: 28.0 ± 0.2 ppm, 30.0 ± 0.2 ppm, 128.7 ± 0.2 ppm, 44.0 ± 0.2 ppm, 130.0 ± 0.2 ppm, 131.0 ± 0.2 ppm, 12.7 ± 0.2 ppm, and 10.8 ± 0.2 ppm. The Compound I NPA Solvate of any one of Embodiments 198 to 217, characterized by a 13C ssNMR spectrum substantially similar to FIG. 27. The Compound I NPA Solvate of any one of Embodiments 198 to 218, prepared by a process comprising (i) stirring of Compound I in NPA, and (ii) isolation of the solid via centrifugation.
Compound I as substantially crystalline MeOAc Solvate Form A (i.e., wherein less than 15% of Compound I is in amorphous form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous form). The Compound I of Embodiment 220, wherein Compound I is 100% crystalline Compound I MeOAc Solvate Form A. Substantially pure Compound I MeOAc Solvate Form A. The Compound I MeOAc Solvate Form A of any one of Embodiments 220 to 222, characterized by an X-ray powder diffractogram having (a) a signal at 8.5 ± 0.2 degrees two-theta and (b) a signal at one or more two-theta values selected from 19.9 ± 0.2 degrees two-theta, 21.3 ± 0.2 degrees two-theta, and 10.1 ± 0.2 degrees two- theta. The Compound I MeOAc Solvate Form A of any one of Embodiments 220 to 222, characterized by an X-ray powder diffractogram having (a) a signal at 8.5 ± 0.2 degrees two-theta and (b) a signal at two or more two-theta values selected from 19.9 ± 0.2 degrees two-theta, 21.3 ± 0.2 degrees two-theta, and 10.1 ± 0.2 degrees two- theta. The Compound I MeOAc Solvate Form A of any one of Embodiments 220 to 222, characterized by an X-ray powder diffractogram having signals at 8.5 ± 0.2 degrees two-theta, 19.9 ± 0.2 degrees two-theta, 21.3 ± 0.2 degrees two-theta, and 10.1 ± 0.2 degrees two-theta. The Compound I MeOAc Solvate Form A of any one of Embodiments 220 to 222, characterized by an X-ray powder diffractogram having signals at (a) signals at 8.5 ± 0.2 degrees two-theta, 19.9 ± 0.2 degrees two-theta, 21.3 ± 0.2 degrees two-theta, and
10.1 ± 0.2 degrees two-theta and (b) a signal at 15.9 ± 0.2 degrees two-theta and/or
13.2 ± 0.2 degrees two-theta. The Compound I MeOAc Solvate Form A of any one of Embodiments 220 to 222, characterized by an X-ray powder diffractogram having signals at 8.5 ± 0.2 degrees
two-theta, 19.9 ± 0.2 degrees two-theta, 21.3 ± 0.2 degrees two-theta, and 10.1 ± 0.2 degrees two-theta, 15.9 ± 0.2 degrees two-theta, and 13.2 ± 0.2 degrees two-theta. The Compound I MeOAc Solvate Form A of any one of Embodiments 220 to 222, characterized by an X-ray powder diffractogram having signals at 8.5 ± 0.2 degrees two-theta, 19 9 ± 0.2 degrees two-theta, 21.3 ± 0.2 degrees two-theta, and 10.1 ± 0.2 degrees two-theta, 15.9 ± 0.2 degrees two-theta, 13.2 ± 0.2 degrees two-theta, 16.0 ± 0.2 degrees two-theta, and 19.4 ± 0.2 degrees two-theta. The Compound I MeOAc Solvate Form A of any one of Embodiments 220 to 228, characterized by an X-ray powder diffractogram substantially similar to FIG. 28. The Compound I MeOAc Solvate Form A of any one of Embodiments 220 to 229, characterized by a TGA showing 1.68% weight loss from 30 °C to 120 °C. The Compound I MeOAc Solvate Form A of any one of Embodiments 220 to 230, characterized by TGA data substantially similar to FIG. 29. The Compound I MeOAc Solvate Form A of any one of Embodiments 220 to 231, characterized by a DSC analysis showing an endothermic peak at about 100 °C and about 216 °C. The Compound I MeOAc Solvate Form A of any one of Embodiments 220 to 232, characterized by a DSC analysis substantially similar to FIG. 30. The Compound I MeOAc Solvate Form A of any one of Embodiments 220 to 233, characterized by a 13C ssNMR spectrum with (a) a peak at 129.3 ± 0.2 ppm and (b) one or more peaks selected from: 43.3 ± 0.2 ppm, 29.8 ± 0.2 ppm, 33.2 ± 0.2 ppm, 171.5 ± 0.2 ppm, and 45.1 ± 0.2 ppm. The Compound I MeOAc Solvate Form A of any one of Embodiments 220 to 233, characterized by a 13C ssNMR spectrum with (a) a peak at 129.3 ± 0.2 ppm and (b) two or more peaks selected from: 43.3 ± 0.2 ppm, 29.8 ± 0.2 ppm, 33.2 ± 0.2 ppm, 171.5 ± 0.2 ppm, and 45.1 ± 0.2 ppm.
The Compound I MeOAc Solvate Form A of any one of Embodiments 220 to 233, characterized by a 13C ssNMR spectrum with (a) a peak at 129.3 ± 0.2 ppm and (b) three or more peaks selected from: 43.3 ± 0.2 ppm, 29.8 ± 0.2 ppm, 33.2 ± 0.2 ppm, 171.5 ± 0.2 ppm, and 45.1 ± 0.2 ppm. The Compound I MeOAc Solvate Form A of any one of Embodiments 220 to 233, characterized by a 13C ssNMR spectrum with (a) a peak at 129.3 ± 0.2 ppm and (b) four or more peaks selected from: 43.3 ± 0.2 ppm, 29.8 ± 02 ppm, 33.2 ± 0.2 ppm, 171.5 ± 0.2 ppm, and 45.1 ± 0.2 ppm. The Compound I MeOAc Solvate Form A of any one of Embodiments 220 to 233, characterized by a 13C ssNMR spectrum with peaks at 129.3 ± 0.2 ppm, 43.3 ± 0.2 ppm, 29.8 ± 0.2 ppm, 33.2 ± 0.2 ppm, 171.5 ± 0.2 ppm, and 45.1 ± 0.2 ppm. The Compound I MeOAc Solvate Form A of any one of Embodiments 220 to 233, characterized by a 13C ssNMR spectrum with (a) peaks at 129.3 ± 0.2 ppm, 43.3 ± 0.2 ppm, 29.8 ± 0.2 ppm, 33.2 ± 0.2 ppm, 171.5 ± 0.2 ppm, 45.1 ± 0.2 ppm and (b) a peak at 27.7 ± 0.2 ppm and/or 129.7 ± 0.2 ppm. The Compound I MeOAc Solvate Form A of any one of Embodiments 220 to 233, characterized by a 13C ssNMR spectrum with peaks at 129.3 ± 0.2 ppm, 43.3 ± 0.2 ppm, 29.8 ± 0.2 ppm, 33.2 ± 0.2 ppm, 171.5 ± 0.2 ppm, 45.1 ± 0.2 ppm, 51.3 ± 0.2 ppm, 27.7 ± 0.2 ppm, and 129.7 ± 0.2 ppm. The Compound I MeOAc Solvate Form A of any one of Embodiments 220 to 240, characterized by a 13C ssNMR spectrum substantially similar to FIG. 31. The Compound I MeOAc Solvate Form A of any one of Embodiments 220 to 241, prepared by a process comprising (i) stirring Compound I Neat Form A in MeOAc, and (ii) isolation of the solid via filtration.
Compound I as substantially crystalline MeOAc Solvate Form B (i.e., wherein less than 15% of Compound I is in amorphous form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous form). The Compound I of Embodiment 243, wherein Compound I is 100% crystalline Compound I MeOAc Form B. Substantially pure Compound I MeOAc Solvate Form B. The Compound I MeOAc Solvate Form B of any one of Embodiments 243 to 245, characterized by an X-ray powder diffractogram having a signal at 4.6 ± 0.2 degrees two-theta and/or a signal at 7.7 ± 0.2 degrees two-theta. The Compound I MeOAc Solvate Form B of any one of Embodiments 243 to 246, characterized by an X-ray powder diffractogram having signals at 4.6 ± 0.2 degrees two-theta and 7.7 ± 0.2 degrees two-theta. The Compound I MeOAc Solvate Form B of any one of Embodiments 243 to 247, characterized by an X-ray powder diffractogram having (a) a signal at 4.6 ± 0.2 degrees two-theta and/or a signal at 7.7 ± 0.2 degrees two-theta, and (b) a signal at one or more two-theta values selected from 8.6 ± 0.2 degrees two-theta, 8.2 ± 0.2 degrees two-theta, 5.9 ± 0.2 degrees two-theta, 15.7 ± 0.2 degrees two-theta, 12.0 ± 0.2 degrees two-theta, and 12.5 ± 0.2 degrees two-theta. The Compound I MeOAc Solvate Form B of any one of Embodiments 243 to 247, characterized by an X-ray powder diffractogram having (a) a signal at 4.6 ± 0.2 degrees two-theta and/or a signal at 7.7 ± 0.2 degrees two-theta, and (b) signals at two or more two-theta values selected from 8.6 ± 0.2 degrees two-theta, 8.2 ± 0.2 degrees two-theta, 5.9 ± 0.2 degrees two-theta, 15.7 ± 0.2 degrees two-theta, 12.0 ± 0.2 degrees two-theta, and 12.5 ± 0.2 degrees two-theta. The Compound I MeOAc Solvate Form B of any one of Embodiments 243 to 247, characterized by an X-ray powder diffractogram having (a) a signal at 4.6 ± 0.2 degrees two-theta and/or a signal at 7.7 ± 0.2 degrees two-theta, and (b) signals at
three or more two-theta values selected from 8.6 ± 0.2 degrees two-theta, 8.2 ± 0.2 degrees two-theta, 5.9 ± 0.2 degrees two-theta, 15.7 ± 0.2 degrees two-theta, 12.0 ± 0.2 degrees two-theta, and 12.5 ± 0.2 degrees two-theta. The Compound I MeOAc Solvate Form B of any one of Embodiments 243 to 247, characterized by an X-ray powder diffractogram having (a) a signal at 4.6 ± 0.2 degrees two-theta and/or a signal at 7.7 ± 0.2 degrees two-theta, and (b) signals at four or more two-theta values selected from 8.6 ± 0.2 degrees two-theta, 8.2 ± 0.2 degrees two-theta, 5 9 ± 0.2 degrees two-theta, 15.7 ± 0.2 degrees two-theta, 12.0 ± 0.2 degrees two-theta, and 12.5 ± 0.2 degrees two-theta. The Compound I MeOAc Solvate Form B of any one of Embodiments 243 to 247, characterized by an X-ray powder diffractogram having (a) a signal at 4.6 ± 0.2 degrees two-theta and/or a signal at 7.7 ± 0.2 degrees two-theta, and (b) signals at five or more two-theta values selected from 8.6 ± 0.2 degrees two-theta, 8.2 ± 0.2 degrees two-theta, 5.9 ± 0.2 degrees two-theta, 15.7 ± 0.2 degrees two-theta, 12.0 ± 0.2 degrees two-theta, and 12.5 ± 0.2 degrees two-theta. The Compound I MeOAc Solvate Form B of any one of Embodiments 243 to 247, characterized by an X-ray powder diffractogram having signals at 4.6 ± 0.2 degrees two-theta, 7.7 ± 0.2 degrees two-theta, 8.6 ± 0.2 degrees two-theta, 8.2 ± 0.2 degrees two-theta, 5.9 ± 0.2 degrees two-theta, 15.7 ± 0.2 degrees two-theta, 12.0 ± 0.2 degrees two-theta, and 12.5 ± 0.2 degrees two-theta. The Compound I MeOAc Solvate Form B of any one of Embodiments 243 to 253, characterized by an X-ray powder diffractogram substantially similar to FIG. 32. The Compound I MeOAc Solvate Form B of any one of Embodiments 243 to 254, characterized by a TGA showing about 1.39% weight loss from 30 °C to 60 °C and about 0.20% weight loss from 60 °C to 190 °C. The Compound I MeOAc Solvate Form B of any one of Embodiments 243 to 255, characterized by TGA data substantially similar to FIG. 33.
The Compound I MeOAc Solvate Form B of any one of Embodiments 243 to 256, characterized by a DSC analysis showing an endothermic peak at about 51 °C and about 187 °C The Compound I MeOAc Solvate Form B of any one of Embodiments 243 to 257, characterized by a DSC analysis substantially similar to FIG. 34. The Compound I MeOAc Solvate Form B of any one of Embodiments 243 to 258, characterized by a 13C ssNMR spectrum with a peak at 50.4 ± 0.2 ppm. The Compound I MeOAc Solvate Form B of any one of Embodiments 243 to 258, characterized by a 13C ssNMR spectrum with (a) a peak at 50.4 ± 0.2 ppm and (b) one or more peaks selected from 51.4 ± 0.2 ppm, 27.8 ± 0.2 ppm, 43.4 ± 0.2 ppm, 130.5 ± 0.2 ppm, 130.0 ± 0.2 ppm, 128.8 ± 0.2 ppm, and 127.6 ± 0.2 ppm. The Compound I MeOAc Solvate Form B of any one of Embodiments 243 to 258, characterized by a 13C ssNMR spectrum with (a) a peak at 50.4 ± 0.2 ppm and (b) two or more peaks selected from 51.4 ± 0.2 ppm, 27.8 ± 0.2 ppm, 43.4 ± 0.2 ppm, 130.5 ± 0.2 ppm, 130.0 ± 0.2 ppm, 128.8 ± 0.2 ppm, and 127.6 ± 0.2 ppm. The Compound I MeOAc Solvate Form B of any one of Embodiments 243 to 258, characterized by a 13C ssNMR spectrum with (a) a peak at 50.4 ± 0.2 ppm and (b) three or more peaks selected from 51.4 ± 0.2 ppm, 27.8 ± 0.2 ppm, 43.4 ± 0.2 ppm,
130.5 ± 0.2 ppm, 130.0 ± 0.2 ppm, 128.8 ± 0.2 ppm, and 127.6 ± 0.2 ppm. The Compound I MeOAc Solvate Form B of any one of Embodiments 243 to 258, characterized by a 13C ssNMR spectrum with (a) a peak at 50.4 ± 0.2 ppm and (b) four or more peaks selected from 51.4 ± 0.2 ppm, 27.8 ± 0.2 ppm, 43.4 ± 0.2 ppm,
130.5 ± 0.2 ppm, 130.0 ± 0.2 ppm, 128.8 ± 0.2 ppm, and 127.6 ± 0.2 ppm. The Compound I MeOAc Solvate Form B of any one of Embodiments 243 to 258, characterized by a 13C ssNMR spectrum with (a) a peak at 50.4 ± 0.2 ppm and (b) five or more peaks selected from 51.4 ± 0.2 ppm, 27.8 ± 0.2 ppm, 43.4 ± 0.2 ppm,
130.5 ± 0.2 ppm, 130.0 ± 0.2 ppm, 128.8 ± 0.2 ppm, and 127.6 ± 0.2 ppm.
The Compound I MeOAc Solvate Form B of any one of Embodiments 243 to 258, characterized by a 13C ssNMR spectrum with (a) a peak at 50.4 ± 0.2 ppm and (b) six or more peaks selected from 51.4 ± 0.2 ppm, 27.8 ± 0.2 ppm, 43.4 ± 0.2 ppm, 130.5 ± 0.2 ppm, 130.0 ± 0.2 ppm, 128.8 ± 0.2 ppm, and 127.6 ± 0.2 ppm. The Compound I MeOAc Solvate Form B of any one of Embodiments 243 to 258, characterized by a 13C ssNMR spectrum with peaks at 50.4 ± 0.2 ppm, 51.4 ± 0.2 ppm, 27.8 ± 0.2 ppm, 43.4 ± 0.2 ppm, 130.5 ± 0.2 ppm, 130.0 ± 0.2 ppm, 128.8 ± 0.2 ppm, and 127.6 ± 0.2 ppm. The Compound I MeOAc Solvate Form B of any one of Embodiments 243 to 258, characterized by a 13C ssNMR spectrum substantially similar to FIG. 35. The Compound I MeOAc Solvate Form B of any one of Embodiments 243 to 267, prepared by a process comprising (i) stirring of Compound I Neat Form A in MeOAc at about 5 °C, (ii) isolation of the solid via centrifugation, and (iii) drying of the solid at room temperature. Compound I as substantially crystalline MeOAc Solvate Form C (i.e., wherein less than 15% of Compound I is in amorphous form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous form). The Compound I of Embodiment 269, wherein Compound I is 100% crystalline Compound I MeOAc Form C. Substantially pure Compound I MeOAc Solvate Form C. The Compound I MeOAc Solvate Form C of any one of Embodiments 269 to 271, characterized by an X-ray powder diffractogram having a signal at 6.4 ± 0.2 degrees two-theta. The Compound I MeOAc Solvate Form C of any one of Embodiments 269 to 271, characterized by an X-ray powder diffractogram having (a) a signal at 6.4 ± 0.2
degrees two-theta and (b) a signal at one or more two-theta values selected from 18.7 ± 0.2 degrees two-theta, 18.9 ± 0.2 degrees two-theta, and 19.8 ± 0.2 degrees two- theta. The Compound I MeOAc Solvate Form C of any one of Embodiments 269 to 271, characterized by an X-ray powder diffractogram having (a) a signal at 6.4 ± 0.2 degrees two-theta and (b) a signal at two or more two-theta values selected from 18.7 ± 0.2 degrees two-theta, 18.9 ± 0.2 degrees two-theta, and 19.8 ± 0.2 degrees two- theta. The Compound I MeOAc Solvate Form C of any one of Embodiments 269 to 271, characterized by an X-ray powder diffractogram having signals (a) a signal at 6.4 ± 0.2 degrees two-theta and (b) a signal at three or more two-theta values selected from
18.7 ± 0.2 degrees two-theta, 18.9 ± 0.2 degrees two-theta, and 19.8 ± 0.2 degrees two-theta. The Compound I MeOAc Solvate Form C of any one of Embodiments 269 to 271, characterized by an X-ray powder diffractogram having signals at 6.4 ± 0.2 degrees two-theta, 18.7 ± 0.2 degrees two-theta, 18.9 ± 0.2 degrees two-theta, and 19.8 ± 0.2 degrees two-theta. The Compound I MeOAc Solvate Form C of any one of Embodiments 269 to 271, characterized by an X-ray powder diffractogram having signals at (a) signals at 6.4 ± 0.2 degrees two-theta, 18.7 ± 0.2 degrees two-theta, 18.9 ± 0.2 degrees two-theta, and
19.8 ± 0.2 degrees two-theta and (b) signals at one or more two-theta values selected from 16.8 ± 0.2 degrees two-theta, 19.0 ± 0.2 degrees two-theta, 12.9 ± 0.2 degrees two-theta, and 14.1 ± 0.2 degrees two-theta. The Compound I MeOAc Solvate Form C of any one of Embodiments 269 to 271, characterized by an X-ray powder diffractogram having (a) signals at 6.4 ± 0.2 degrees two-theta, 18.7 ± 0.2 degrees two-theta, 18.9 ± 0.2 degrees two-theta, and
19.8 ± 0.2 degrees two-theta and (b) signals at two or more two-theta values selected from 16.8 ± 0.2 degrees two-theta, 19.0 ± 0.2 degrees two-theta, 12.9 ± 0.2 degrees two-theta, and 14.1 ± 0.2 degrees two-theta.
The Compound I MeOAc Solvate Form C of any one of Embodiments 269 to 271, characterized by an X-ray powder diffractogram having (a) signals at 6 4 ± 0.2 degrees two-theta, 18.7 ± 0.2 degrees two-theta, 18.9 ± 0.2 degrees two-theta, and 19.8 ± 0.2 degrees two-theta and (b) signals at three or more two-theta values selected from 16.8 ± 0.2 degrees two-theta, 19.0 ± 0.2 degrees two-theta, 14.1 ± 0.2 degrees two-theta, and 19.2 ± 0.2 degrees two-theta. The Compound I MeOAc Solvate Form C of any one of Embodiments 269 to 271, characterized by an X-ray powder diffractogram having signals at 6.4 ± 0.2 degrees two-theta, 18.7 ± 0.2 degrees two-theta, 18.9 ± 0.2 degrees two-theta, 19.8 ± 0.2 degrees two-theta, 16.8 ± 0.2 degrees two-theta, 19.0 ± 0.2 degrees two-theta, 14.1 ± 0.2 degrees two-theta, and 19.2 ± 0.2 degrees two-theta. The Compound I MeOAc Solvate Form C of any one of Embodiments 269 to 279, characterized by an X-ray powder diffractogram substantially similar to FIG. 36. The Compound I MeOAc Solvate Form C of any one of Embodiments 269 to 281, characterized by a 13C ssNMR spectrum with one or more peaks selected from: 28.3 ± 0.2 ppm, 49.6 ± 0.2 ppm, and 150.1 ± 0.2 ppm. The Compound I MeOAc Solvate Form C of any one of Embodiments 269 to 282, characterized by a 13C ssNMR spectrum with two or more peaks selected from 28.3 ± 0.2 ppm, 49.6 ± 0.2 ppm, and 150.1 ± 0.2 ppm. The Compound I MeOAc Solvate Form C of any one of Embodiments 269 to 283, characterized by a 13C ssNMR spectrum with peaks at 28.3 ± 0.2 ppm, 49.6 ± 0.2 ppm, and 150.1 ± 0.2 ppm. The Compound I MeOAc Solvate Form C of any one of Embodiments 269 to 284, characterized by a 13C ssNMR spectrum with (a) peaks at 28.3 ± 0.2 ppm, 49.6 ± 0.2 ppm, and 150.1 ± 0.2 ppm and (b) one or more peaks selected from 28.9 ± 0.2 ppm, 51.4 ± 0.2 ppm, 129.9 ± 0.2 ppm, 51.8 ± 0.2 ppm, and 42.4 ± 0.2 ppm.
The Compound I MeOAc Solvate Form C of any one of Embodiments 269 to 285, characterized by a 13C ssNMR spectrum with (a) peaks at 28.3 ± 0.2 ppm, 49.6 ± 0.2 ppm, and 150.1 ± 0.2 ppm and (b) two or more peaks selected from 28.9 ± 0.2 ppm,
51.4 ± 0.2 ppm, 129.9 ± 0.2 ppm, 51.8 ± 0.2 ppm, and 42.4 ± 0.2 ppm. The Compound I MeOAc Solvate Form C of any one of Embodiments 269 to 286, characterized by a 13C ssNMR spectrum with (a) peaks at 28.3 ± 0.2 ppm, 49.6 ± 0.2 ppm, and 150.1 ± 0.2 ppm and (b) three or more peaks selected from 28.9 ± 0.2 ppm,
51.4 ± 0.2 ppm, 129.9 ± 0.2 ppm, 51.8 ± 0.2 ppm, and 42.4 ± 0.2 ppm. The Compound I MeOAc Solvate Form C of any one of Embodiments 269 to 287, characterized by a 13C ssNMR spectrum with (a) peaks at 28.3 ± 0.2 ppm, 49.6 ± 0.2 ppm, and 150.1 ± 0.2 ppm and (b) four or more peaks selected from 28.9 ± 0.2 ppm,
51.4 ± 0.2 ppm, 129.9 ± 0.2 ppm, 51.8 ± 0.2 ppm, and 42.4 ± 0.2 ppm. The Compound I MeOAc Solvate Form C of any one of Embodiments 269 to 288, characterized by a 13C ssNMR spectrum with peaks at 28.3 ± 0.2 ppm, 49.6 ± 0.2 ppm, 150.1 ± 0.2 ppm, 28.9 ± 0.2 ppm, 51.4 ± 0.2 ppm, 129.9 ± 0.2 ppm, 51.8 ± 0.2 ppm, and 42.4 ± 0.2 ppm. The Compound I MeOAc Solvate Form C of any one of Embodiments 269 to 289, characterized by a 13C ssNMR spectrum substantially similar to FIG. 37. The Compound I MeOAc Solvate Form C of any one of Embodiments 269 to 290, prepared by a process comprising (i) stirring of Compound I Neat Form A in MeOAc at about 5 °C, and (ii) isolation of the solid via filtration. Compound I as substantially crystalline hydrate Form A (i.e., wherein less than 15% of Compound I is in amorphous form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous form). The Compound I of Embodiment 292, wherein Compound I is 100% crystalline Compound I Hydrate Form A.
Substantially pure Compound I Hydrate Form A. The Compound I Hydrate Form A of any one of Embodiments 292 to 294, characterized by an X-ray powder diffractogram having a signal at 21.4 ± 0.2 degrees two-theta. The Compound I Hydrate Form A of any one of Embodiments 292 to 295, characterized by an X-ray powder diffractogram having (a) a signal at 21.4 ± 0.2 degrees two-theta, and (b) a signal at one or more two-theta values selected from: 14.0 ± 0.2 degrees two-theta, 19.5 ± 0.2 degrees two-theta, 7.0 ± 0.2 degrees two- theta, 18.6 ± 0.2 degrees two-theta, 11.0 ± 0.2 degrees two-theta, 18.2 ± 0.2 degrees two-theta, and 20.5 ± 0.2 degrees two-theta. The Compound I Hydrate Form A of any one of Embodiments 292 to 295, characterized by an X-ray powder diffractogram having (a) a signal at 21.4 ± 0.2 degrees two-theta, and (b) signals at two or more two-theta values selected from: 14.0 ± 0.2 degrees two-theta, 19.5 ± 0.2 degrees two-theta, 7.0 ± 0.2 degrees two-theta, 18.6 ± 0.2 degrees two-theta, 11.0 ± 0.2 degrees two-theta, 18.2 ± 0.2 degrees two- theta, and 20.5 ± 0.2 degrees two-theta. The Compound I Hydrate Form A of any one of Embodiments 292 to 295, characterized by an X-ray powder diffractogram having (a) a signal at 21.4 ± 0.2 degrees two-theta, and (b) signals at three or more two-theta values selected from: 14.0 ± 0.2 degrees two-theta, 19.5 ± 0.2 degrees two-theta, 7.0 ± 0.2 degrees two- theta, 18.6 ± 0.2 degrees two-theta, 11.0 ± 0.2 degrees two-theta, 18.2 ± 0.2 degrees two-theta, and 20.5 ± 0.2 degrees two-theta. The Compound I Hydrate Form A of any one of Embodiments 292 to 295, characterized by an X-ray powder diffractogram having (a) a signal at 21.4 ± 0.2 degrees two-theta, and (b) signals at four or more two-theta values selected from: 14.0 ± 0.2 degrees two-theta, 19.5 ± 0.2 degrees two-theta, 7.0 ± 0.2 degrees two-theta, 18.6 ± 0.2 degrees two-theta, 11.0 ± 0.2 degrees two-theta, 18.2 ± 0.2 degrees two- theta, and 20.5 ± 0.2 degrees two-theta.
The Compound I Hydrate Form A of any one of Embodiments 292 to 295, characterized by an X-ray powder diffractogram having (a) a signal at 21.4 ± 0.2 degrees two-theta, and (b) signals at five or more two-theta values selected from: 14.0 ± 0.2 degrees two-theta, 19.5 ± 0.2 degrees two-theta, 7.0 ± 0.2 degrees two-theta, 18.6 ± 0.2 degrees two-theta, 11.0 ± 0.2 degrees two-theta, 18.2 ± 0.2 degrees two- theta, and 20.5 ± 0.2 degrees two-theta. The Compound I Hydrate Form A of any one of Embodiments 292 to 295, characterized by an X-ray powder diffractogram having (a) a signal at 21.4 ± 0.2 degrees two-theta, and (b) signals at six or more two-theta values selected from: 14.0 ± 0.2 degrees two-theta, 19.5 ± 0.2 degrees two-theta, 7.0 ± 0.2 degrees two-theta, 18.6 ± 0.2 degrees two-theta, 11 0 ± 0.2 degrees two-theta, 18.2 ± 0.2 degrees two- theta, and 20.5 ± 0.2 degrees two-theta. The Compound I Hydrate Form A of any one of Embodiments 292 to 295, characterized by an X-ray powder diffractogram having signals at 21.4 ± 0.2 degrees two-theta, 14.0 ± 0.2 degrees two-theta, 19.5 ± 0.2 degrees two-theta, 7.0 ± 0.2 degrees two-theta, 18.6 ± 0.2 degrees two-theta, 11.0 ± 0.2 degrees two-theta, 18.2 ± 0.2 degrees two-theta, and 20.5 ± 0.2 degrees two-theta. The Compound I Hydrate Form A of any one of Embodiments 292 to 302, characterized by an X-ray powder diffractogram substantially similar to FIG. 38 The Compound I Hydrate Form A of any one of Embodiments 292 to 303, characterized by a 13C ssNMR spectrum with one or more peaks selected from: 26.8 ± 0.2 ppm, 18.3 ± 0.2 ppm, and 126.5 ± 0.2 ppm. The Compound I Hydrate Form A of any one of Embodiments 292 to 304, characterized by a 13C ssNMR spectrum with two or more peaks selected from: 26.8 ± 0.2 ppm, 18.3 ± 0.2 ppm, and 126.5 ± 0.2 ppm. The Compound I Hydrate Form A of any one of Embodiments 292 to 305, characterized by a 13C ssNMR spectrum with peaks at 26.8 ± 0.2 ppm, 18.3 ± 0.2 ppm, and 126.5 ± 0.2 ppm.
The Compound I Hydrate Form A of any one of Embodiments 292 to 306, characterized by a 13C ssNMR spectrum with (a) peaks at 26.8 ± 0.2 ppm, 18.3 ± 0.2 ppm, and 126.5 ± 0.2 ppm and (b) one or more peaks selected from 131.5 ± 0.2 ppm, 135.0 ± 0.2 ppm, 20.0 ± 0.2 ppm, 142.9 ± 0.2 ppm, and 128.3 ± 0.2 ppm. The Compound I Hydrate Form A of any one of Embodiments 292 to 307, characterized by a 13C ssNMR spectrum with (a) peaks at 26.8 ± 0.2 ppm, 18.3 ± 0.2 ppm, and 126.5 ± 0.2 ppm and (b) two or more peaks selected from 131.5 ± 0.2 ppm, 135.0 ± 0.2 ppm, 20.0 ± 0.2 ppm, 142.9 ± 0.2 ppm, and 128.3 ± 0.2 ppm. The Compound I Hydrate Form A of any one of Embodiments 292 to 308, characterized by a 13C ssNMR spectrum with (a) peaks at 26.8 ± 0.2 ppm, 18.3 ± 0.2 ppm, and 126.5 ± 0.2 ppm and (b) three or more peaks selected from 131.5 ± 0.2 ppm, 135.0 ± 0.2 ppm, 20.0 ± 0.2 ppm, 142.9 ± 0.2 ppm, and 128.3 ± 0.2 ppm. The Compound I Hydrate Form A of any one of Embodiments 292 to 309, characterized by a 13C ssNMR spectrum with (a) peaks at 26.8 ± 0.2 ppm, 18.3 ± 0.2 ppm, and 126.5 ± 0.2 ppm and (b) four or more peaks selected from 131.5 ± 0.2 ppm, 135.0 ± 0.2 ppm, 20.0 ± 0.2 ppm, 142.9 ± 0.2 ppm, and 128.3 ± 0.2 ppm. The Compound I Hydrate Form A of any one of Embodiments 292 to 310, characterized by a 13C ssNMR spectrum with peaks at 26.8 ± 0.2 ppm, 18.3 ± 0.2 ppm, 126.5 ± 0.2 ppm, 131.5 ± 0.2 ppm, 135.0 ± 0.2 ppm, 20.0 ± 0.2 ppm, 142.9 ± 0.2 ppm, and 128.3 ± 0.2 ppm. The Compound I Hydrate Form A of any one of Embodiments 292 to 311, characterized by a 13C ssNMR spectrum substantially similar to FIG. 39. The Compound I Hydrate Form A of any one of Embodiments 292 to 312, characterized by a monoclinic crystal system, aP2i2i2i space group, and unit cell dimensions, measured at 100 K on a Bruker diffractometer equipped with Cu Ka radiation ( = 1.54178 A) and an CP AD detector, of:
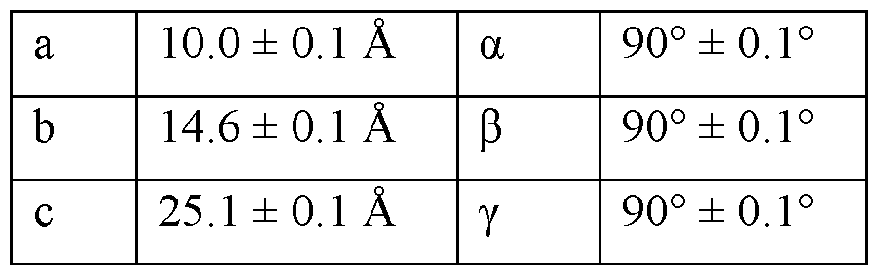 The Compound I Hydrate Form A of any one of Embodiments 292 to 313, prepared by a process comprising (i) stirring Compound I EtOH Solvate Form B in water, and (ii) isolation via filtration and air drying of the solid. Compound I as substantially crystalline Hydrate Form B (i.e., wherein less than 15% of Compound I is in amorphous form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous form). The Compound I of Embodiment 315, wherein Compound I is 100% crystalline Compound I Hydrate Form B. Substantially pure Compound I Hydrate Form B. The Compound I Hydrate Form B of any one of Embodiments 315 to 317, characterized by an X-ray powder diffractogram having a signal at 24.5 ± 0.2 degrees two-theta. The Compound I Hydrate Form B of any one of Embodiments 315 to 317, characterized by an X-ray powder diffractogram having (a) a signal at 24.5 ± 0.2 degrees two-theta, and (b) a signal at one or more two-theta values selected from: 7.0 ± 0.2 degrees two-theta, 19.3 ± 0.2 degrees two-theta, 19.4 ± 0.2 degrees two-theta, 15.1 ± 0.2 degrees two-theta, 13.9 ± 0.2 degrees two-theta, 10.9 ± 0.2 degrees two- theta, and 19.7 ± 0.2 degrees two-theta. The Compound I Hydrate Form B of any one of Embodiments 315 to 317, characterized by an X-ray powder diffractogram having (a) a signal at 24.5 ± 0.2 degrees two-theta, and (b) signals at two or more two-theta values selected from: 7.0 ± 0.2 degrees two-theta, 19.3 ± 0.2 degrees two-theta, 19.4 ± 0.2 degrees two-theta, 15.1 ± 0.2 degrees two-theta, 13.9 ± 0.2 degrees two-theta, 10.9 ± 0.2 degrees two- theta, and 19.7 ± 0.2 degrees two-theta.
The Compound I Hydrate Form B of any one of Embodiments 315 to 317, characterized by an X-ray powder diffractogram having (a) a signal at 24.5 ± 0.2 degrees two-theta, and (b) signals at three or more two-theta values selected from: 7.0 ± 0.2 degrees two-theta, 19.3 ± 0.2 degrees two-theta, 19.4 ± 0.2 degrees two-theta,
The Compound I Hydrate Form A of any one of Embodiments 292 to 313, prepared by a process comprising (i) stirring Compound I EtOH Solvate Form B in water, and (ii) isolation via filtration and air drying of the solid. Compound I as substantially crystalline Hydrate Form B (i.e., wherein less than 15% of Compound I is in amorphous form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous form). The Compound I of Embodiment 315, wherein Compound I is 100% crystalline Compound I Hydrate Form B. Substantially pure Compound I Hydrate Form B. The Compound I Hydrate Form B of any one of Embodiments 315 to 317, characterized by an X-ray powder diffractogram having a signal at 24.5 ± 0.2 degrees two-theta. The Compound I Hydrate Form B of any one of Embodiments 315 to 317, characterized by an X-ray powder diffractogram having (a) a signal at 24.5 ± 0.2 degrees two-theta, and (b) a signal at one or more two-theta values selected from: 7.0 ± 0.2 degrees two-theta, 19.3 ± 0.2 degrees two-theta, 19.4 ± 0.2 degrees two-theta, 15.1 ± 0.2 degrees two-theta, 13.9 ± 0.2 degrees two-theta, 10.9 ± 0.2 degrees two- theta, and 19.7 ± 0.2 degrees two-theta. The Compound I Hydrate Form B of any one of Embodiments 315 to 317, characterized by an X-ray powder diffractogram having (a) a signal at 24.5 ± 0.2 degrees two-theta, and (b) signals at two or more two-theta values selected from: 7.0 ± 0.2 degrees two-theta, 19.3 ± 0.2 degrees two-theta, 19.4 ± 0.2 degrees two-theta, 15.1 ± 0.2 degrees two-theta, 13.9 ± 0.2 degrees two-theta, 10.9 ± 0.2 degrees two- theta, and 19.7 ± 0.2 degrees two-theta.
The Compound I Hydrate Form B of any one of Embodiments 315 to 317, characterized by an X-ray powder diffractogram having (a) a signal at 24.5 ± 0.2 degrees two-theta, and (b) signals at three or more two-theta values selected from: 7.0 ± 0.2 degrees two-theta, 19.3 ± 0.2 degrees two-theta, 19.4 ± 0.2 degrees two-theta,
15.1 ± 0.2 degrees two-theta, 13 9 ± 0.2 degrees two-theta, 10.9 ± 0.2 degrees two- theta, and 19.7 ± 0.2 degrees two-theta. The Compound I Hydrate Form B of any one of Embodiments 315 to 317, characterized by an X-ray powder diffractogram having (a) a signal at 24.5 ± 0.2 degrees two-theta, and (b) signals at four or more two-theta values selected from: 7.0 ± 0.2 degrees two-theta, 19 3 ± 0.2 degrees two-theta, 19.4 ± 0.2 degrees two-theta,
15.1 ± 0.2 degrees two-theta, 13.9 ± 0.2 degrees two-theta, 10.9 ± 0.2 degrees two- theta, and 19.7 ± 0.2 degrees two-theta. The Compound I Hydrate Form B of any one of Embodiments 315 to 317, characterized by an X-ray powder diffractogram having (a) a signal at 24.5 ± 0.2 degrees two-theta, and (b) signals at five or more two-theta values selected from : 7.0 ± 0.2 degrees two-theta, 19.3 ± 0.2 degrees two-theta, 19.4 ± 0.2 degrees two-theta,
15.1 ± 0.2 degrees two-theta, 13.9 ± 0.2 degrees two-theta, 10.9 ± 0.2 degrees two- theta, and 19.7 ± 0.2 degrees two-theta. The Compound I Hydrate Form B of any one of Embodiments 315 to 317, characterized by an X-ray powder diffractogram having (a) a signal at 24.5 ± 0.2 degrees two-theta, and (b) signals at six or more two-theta values selected from: 7.0 ± 0.2 degrees two-theta, 19.3 ± 0.2 degrees two-theta, 19.4 ± 0.2 degrees two-theta, 15.1 ± 0.2 degrees two-theta, 13.9 ± 0.2 degrees two-theta, 10.9 ± 0.2 degrees two-theta, and 19.7 ± 0.2 degrees two-theta. The Compound I Hydrate Form B of any one of Embodiments 315 to 317, characterized by an X-ray powder diffractogram having signals at 24.5 ± 0.2 degrees two-theta, 7.0 ± 0.2 degrees two-theta, 19.3 ± 0.2 degrees two-theta, 19.4 ± 0.2 degrees two-theta, 15.1 ± 0.2 degrees two-theta, 13.9 ± 0.2 degrees two-theta, 10.9 ± 0.2 degrees two-theta, and 19.7 ± 0.2 degrees two-theta.
The Compound I Hydrate Form B of any one of Embodiments 315 to 325, characterized by an X-ray powder diffractogram substantially similar to FIG. 40. The Compound I Hydrate Form B of any one of Embodiments 315 to 326, characterized by a 13C ssNMR spectrum with a peak at 19.3 ± 0.2 ppm. The Compound I Hydrate Form B of any one of Embodiments 315 to 327, characterized by a 13C ssNMR spectrum with (a) a peak at 19.3 ± 0.2 ppm and (b) one or more peaks selected from: 29.4 ± 0.2 ppm, 128.6 ± 0.2 ppm, 130.3 ± 0.2 ppm, 42.3 ± 0.2 ppm, 128.9 ± 0.2 ppm, 134.6 ± 0.2 ppm, and 136.4 ± 0.2 ppm. The Compound I Hydrate Form B of any one of Embodiments 315 to 328, characterized by a 13C ssNMR spectrum with (a) a peak at 19.3 ± 0.2 ppm and (b) two or more peaks selected from: 29.4 ± 0.2 ppm, 128.6 ± 0.2 ppm, 130.3 ± 0.2 ppm, 42.3 ± 0.2 ppm, 128.9 ± 0.2 ppm, 134.6 ± 0.2 ppm, and 136.4 ± 0.2 ppm. The Compound I Hydrate Form B of any one of Embodiments 315 to 329, characterized by a 13C ssNMR spectrum with (a) a peak at 19.3 ± 0.2 ppm and (b) three or more peaks selected from: 29.4 ± 0.2 ppm, 128.6 ± 0.2 ppm, 130.3 ± 0.2 ppm,
42.3 ± 0.2 ppm, 128.9 ± 0.2 ppm, 134.6 ± 0.2 ppm, and 136.4 ± 0.2 ppm. The Compound I Hydrate Form B of any one of Embodiments 315 to 330, characterized by a 13C ssNMR spectrum with (a) a peak at 19.3 ± 0.2 ppm and (b) four or more peaks selected from: 29.4 ± 0.2 ppm, 128.6 ± 0.2 ppm, 130.3 ± 0.2 ppm,
42.3 ± 0.2 ppm, 128.9 ± 0.2 ppm, 134.6 ± 0.2 ppm, and 136.4 ± 0.2 ppm. The Compound I Hydrate Form B of any one of Embodiments 315 to 331, characterized by a 13C ssNMR spectrum with (a) a peak at 19.3 ± 0.2 ppm and (b) five or more peaks selected from: 29.4 ± 0.2 ppm, 128.6 ± 0.2 ppm, 130.3 ± 0.2 ppm,
42.3 ± 0.2 ppm, 128.9 ± 0.2 ppm, 134.6 ± 0.2 ppm, and 136.4 ± 0.2 ppm. The Compound I Hydrate Form B of any one of Embodiments 315 to 332, characterized by a 13C ssNMR spectrum with (a) a peak at 19.3 ± 0.2 ppm and (b) six
or more peaks selected from: 29.4 ± 0.2 ppm, 128.6 ± 0.2 ppm, 130.3 ± 0.2 ppm, 42.3 ± 0.2 ppm, 128.9 ± 0.2 ppm, 134.6 ± 0.2 ppm, and 136.4 ± 0.2 ppm. The Compound I Hydrate Form B of any one of Embodiments 315 to 333, characterized by a 13C ssNMR spectrum with peaks at 19.3 ± 0.2 ppm, 29.4 ± 0.2 ppm, 128.6 ± 0.2 ppm, 130.3 ± 0.2 ppm, 42.3 ± 0.2 ppm, 128.9 ± 0.2 ppm, 134.6 ± 0.2 ppm, and 136.4 ± 0.2 ppm. The Compound I Hydrate Form B of any one of Embodiments 315 to 334, characterized by a 13C ssNMR spectrum substantially similar to FIG. 41. The Compound I Hydrate Form B of any one of Embodiments 315 to 335, characterized by a monoclinic crystal system, aP2i2i2i space group, and unit cell dimensions, measured at 100 K on a Bruker diffractometer equipped with Cu Ka radiation ( = 1.54178 A) and an CP AD detector, of:
 The Compound I Hydrate Form B of any one of Embodiments 315 to 336, prepared by a process comprising (i) stirring Compound I in a MeOH/water mixture at about 80 °C, (ii) isolation of the solid via filtration, and (iii) exposure of the solid to about 11% humidity. Compound I as substantially crystalline Hydrate Form C (i.e., wherein less than 15% of Compound I is in amorphous form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous form). The Compound I of Embodiment 338, wherein Compound I is 100% crystalline Compound I Hydrate Form C. Substantially pure Compound I Hydrate Form C.
The Compound I Hydrate Form C of any one of Embodiments 338 to 340, characterized by an X-ray powder diffractogram having a signal at 16.1 ± 0.2 degrees two-theta. The Compound I Hydrate Form C of any one of Embodiments 338 to 340, characterized by an X-ray powder diffractogram having (a) a signal at 16.1 ± 0.2 degrees two-theta and (b) a signal at one or more two-theta values selected from: 7.0 ± 0.2 degrees two-theta, 21.9 ± 0.2 degrees two-theta, 11.2 ± 0.2 degrees two-theta,
The Compound I Hydrate Form B of any one of Embodiments 315 to 336, prepared by a process comprising (i) stirring Compound I in a MeOH/water mixture at about 80 °C, (ii) isolation of the solid via filtration, and (iii) exposure of the solid to about 11% humidity. Compound I as substantially crystalline Hydrate Form C (i.e., wherein less than 15% of Compound I is in amorphous form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous form). The Compound I of Embodiment 338, wherein Compound I is 100% crystalline Compound I Hydrate Form C. Substantially pure Compound I Hydrate Form C.
The Compound I Hydrate Form C of any one of Embodiments 338 to 340, characterized by an X-ray powder diffractogram having a signal at 16.1 ± 0.2 degrees two-theta. The Compound I Hydrate Form C of any one of Embodiments 338 to 340, characterized by an X-ray powder diffractogram having (a) a signal at 16.1 ± 0.2 degrees two-theta and (b) a signal at one or more two-theta values selected from: 7.0 ± 0.2 degrees two-theta, 21.9 ± 0.2 degrees two-theta, 11.2 ± 0.2 degrees two-theta,
15.1 ± 0.2 degrees two-theta, 14.9 ± 0.2 degrees two-theta, 15.4 ± 0.2 degrees two- theta, and 18.2 ± 0.2 degrees two-theta. The Compound I Hydrate Form C of any one of Embodiments 338 to 340, characterized by an X-ray powder diffractogram having (a) a signal at 16.1 ± 0.2 degrees two-theta and (b) a signal at two or more two-theta values selected from: 7.0 ± 0.2 degrees two-theta, 21.9 ± 0.2 degrees two-theta, 11.2 ± 0.2 degrees two-theta,
15.1 ± 0.2 degrees two-theta, 14.9 ± 0.2 degrees two-theta, 15.4 ± 0.2 degrees two- theta, and 18.2 ± 0.2 degrees two-theta. The Compound I Hydrate Form C of any one of Embodiments 338 to 340, characterized by an X-ray powder diffractogram having (a) a signal at 16.1 ± 0.2 degrees two-theta and (b) signals at three or more two-theta values selected from: 7.0 ± 0.2 degrees two-theta, 21.9 ± 0.2 degrees two-theta, 11.2 ± 0.2 degrees two-theta,
15.1 ± 0.2 degrees two-theta, 14.9 ± 0.2 degrees two-theta, 15.4 ± 0.2 degrees two- theta, and 18.2 ± 0.2 degrees two-theta. The Compound I Hydrate Form C of any one of Embodiments 338 to 340, characterized by an X-ray powder diffractogram having (a) a signal at 16.1 ± 0.2 degrees two-theta and (b) signals at four or more two-theta values selected from: 7.0 ± 0.2 degrees two-theta, 21.9 ± 0.2 degrees two-theta, 11.2 ± 0.2 degrees two-theta,
15.1 ± 0.2 degrees two-theta, 14.9 ± 0.2 degrees two-theta, 15.4 ± 0.2 degrees two- theta, and 18.2 ± 0.2 degrees two-theta. The Compound I Hydrate Form C of any one of Embodiments 338 to 340, characterized by an X-ray powder diffractogram having (a) a signal at 16.1 ± 0.2
degrees two-theta and (b) signals at five or more two-theta values selected from: 7.0 ± 0.2 degrees two-theta, 21.9 ± 0.2 degrees two-theta, 11.2 ± 0.2 degrees two-theta, 15.1 ± 0.2 degrees two-theta, 14.9 ± 0.2 degrees two-theta, 15.4 ± 0.2 degrees two-theta, and 18.2 ± 0.2 degrees two-theta. The Compound I Hydrate Form C of any one of Embodiments 338 to 340, characterized by an X-ray powder diffractogram having (a) a signal at 16.1 ± 0.2 degrees two-theta and (b) signals at six or more two-theta values selected from: 7.0 ± 0.2 degrees two-theta, 21 9 ± 0.2 degrees two-theta, 11.2 ± 0.2 degrees two-theta, 15.1 ± 0.2 degrees two-theta, 14.9 ± 0.2 degrees two-theta, 15.4 ± 0.2 degrees two-theta, and 18.2 ± 0.2 degrees two-theta. The Compound I Hydrate Form C of any one of Embodiments 338 to 340, characterized by an X-ray powder diffractogram having signals at 16.1 ± 0.2 degrees two-theta, 7.0 ± 0.2 degrees two-theta, 21.9 ± 0.2 degrees two-theta, 11.2 ± 0.2 degrees two-theta, 15.1 ± 0.2 degrees two-theta, 14.9 ± 0.2 degrees two-theta, 15.4 ± 0.2 degrees two-theta, and 18.2 ± 0.2 degrees two-theta. The Compound I Hydrate Form C of any one of Embodiments 338 to 348, characterized by an X-ray powder diffractogram substantially similar to FIG. 42. The Compound I Hydrate Form C of any one of Embodiments 338 to 349, characterized by a 13C ssNMR spectrum with a peak at 127.7 ± 0.2 ppm. The Compound I Hydrate Form C of any one of Embodiments 338 to 350, characterized by a 13C ssNMR spectrum with (a) a peak at 127.7 ± 0.2 ppm and (b) one or more peaks selected from 51.6 ± 0.2 ppm, 29.6 ± 0.2 ppm, 42.2 ± 0.2 ppm, 102.4 ± 0.2 ppm, 130.5 ± 0.2 ppm, 19.9 ± 0.2 ppm, 142.7 ± 0.2 ppm. The Compound I Hydrate Form C of any one of Embodiments 338 to 351, characterized by a 13C ssNMR spectrum with (a) a peak at 127.7 ± 0.2 ppm and (b) two or more peaks selected from 51.6 ± 0.2 ppm, 29.6 ± 0.2 ppm, 42.2 ± 0.2 ppm, 102.4 ± 0.2 ppm, 130.5 ± 0.2 ppm, 19.9 ± 0.2 ppm, 142.7 ± 0.2 ppm.
The Compound I Hydrate Form C of any one of Embodiments 338 to 352, characterized by a 13C ssNMR spectrum with (a) a peak at 127.7 ± 0.2 ppm and (b) three or more peaks selected from 51.6 ± 0.2 ppm, 29.6 ± 0.2 ppm, 42.2 ± 0.2 ppm,
102.4 ± 0.2 ppm, 130.5 ± 0.2 ppm, 19.9 ± 0.2 ppm, 142.7 ± 0.2 ppm. The Compound I Hydrate Form C of any one of Embodiments 338 to 353, characterized by a 13C ssNMR spectrum with (a) a peak at 127.7 ± 0.2 ppm and (b) four or more peaks selected from 51.6 ± 0.2 ppm, 29.6 ± 0.2 ppm, 42.2 ± 0.2 ppm,
102.4 ± 0.2 ppm, 130.5 ± 0.2 ppm, 19.9 ± 0.2 ppm, 142.7 ± 0.2 ppm. The Compound I Hydrate Form C of any one of Embodiments 338 to 354, characterized by a 13C ssNMR spectrum with (a) a peak at 127.7 ± 0.2 ppm and (b) five or more peaks selected from 51.6 ± 0.2 ppm, 29.6 ± 0.2 ppm, 42.2 ± 0.2 ppm,
102.4 ± 0.2 ppm, 130.5 ± 0.2 ppm, 19.9 ± 0.2 ppm, 142.7 ± 0.2 ppm. The Compound I Hydrate Form C of any one of Embodiments 338 to 355, characterized by a 13C ssNMR spectrum with (a) a peak at 127.7 ± 0.2 ppm and (b) six or more peaks selected from 51.6 ± 0.2 ppm, 29.6 ± 0.2 ppm, 42.2 ± 0.2 ppm,
102.4 ± 0.2 ppm, 130.5 ± 0.2 ppm, 19.9 ± 0.2 ppm, 142.7 ± 0.2 ppm. The Compound I Hydrate Form C of any one of Embodiments 338 to 356, characterized by a 13C ssNMR spectrum with peaks at 127.7 ± 0.2 ppm, 51.6 ± 0.2 ppm, 29.6 ± 0.2 ppm, 42.2 ± 0.2 ppm, 102.4 ± 0.2 ppm, 130.5 ± 0.2 ppm, 19.9 ± 0.2 ppm, 142.7 ± 0.2 ppm. The Compound I hydrate Form C of any one of Embodiments 338 to 357, characterized by a 13C ssNMR spectrum substantially similar to FIG. 43. The Compound I Hydrate Form C of any one of Embodiments 338 to 358, characterized by a monoclinic crystal system, aP2i2i2i space group, and unit cell dimensions, measured at 100 K on a Bruker diffractometer equipped with Cu K« radiation ( = 1.54178 A) and an CP AD detector, of:

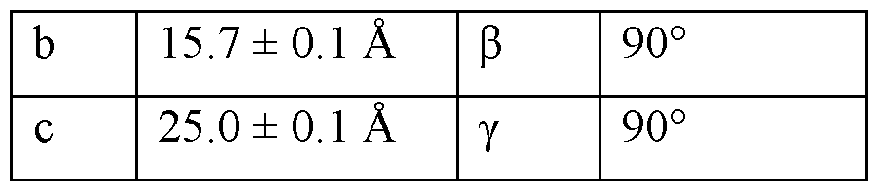 The Compound I Hydrate Form C of any one of Embodiments 338 to 359, prepared by a process comprising (i) stirring Compound I in a MeOH/water mixture at about 80 °C, (ii) isolation of the solid via filtration, and (iii) exposure of the solid to about 75% humidity. Compound I as substantially crystalline EtOH Solvate Hydrate (i.e., wherein less than 15% of Compound I is in amorphous form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous form). The Compound I of Embodiment 361, wherein Compound I is 100% crystalline Compound I EtOH Solvate Hydrate. Substantially pure Compound I EtOH Solvate Hydrate. The Compound I EtOH Solvate Hydrate of any one of Embodiments 361 to 363, characterized by an X-ray powder diffractogram having a signal at one or more two- theta values selected from 6.7 ± 0.2 degrees two-theta, 11.5 ± 0.2 degrees two-theta, 8.6 ± 0.2 degrees two-theta, 14.7 ± 0.2 degrees two-theta, 20.0 ± 0.2 degrees two- theta, 11.3 ± 0.2 degrees two-theta, 18.7 ± 0.2 degrees two-theta, and 19.1 ± 0.2 degrees two-theta. The Compound I EtOH Solvate Hydrate of any one of Embodiments 361 to 364, characterized by an X-ray powder diffractogram having signals at two or more two- theta values selected from 6.7 ± 0.2 degrees two-theta, 11.5 ± 0.2 degrees two-theta, 8.6 ± 0.2 degrees two-theta, 14.7 ± 0.2 degrees two-theta, 20.0 ± 0.2 degrees two- theta, 11.3 ± 0.2 degrees two-theta, 18.7 ± 0.2 degrees two-theta, and 19.1 ± 0.2 degrees two-theta. The Compound I EtOH Solvate Hydrate of any one of Embodiments 361 to 365, characterized by an X-ray powder diffractogram having signals at three or more two- theta values selected from 6.7 ± 0.2 degrees two-theta, 11.5 ± 0.2 degrees two-theta,
8.6 ± 0.2 degrees two-theta, 14.7 ± 0.2 degrees two-theta, 20.0 ± 0.2 degrees two- theta, 11.3 ± 0.2 degrees two-theta, 18.7 ± 0.2 degrees two-theta, and 19.1 ± 0.2 degrees two-theta. The Compound I EtOH Solvate Hydrate of any one of Embodiments 361 to 366, characterized by an X-ray powder diffractogram having signals at four or more two- theta values selected from 6.7 ± 0.2 degrees two-theta, 11.5 ± 0.2 degrees two-theta,
The Compound I Hydrate Form C of any one of Embodiments 338 to 359, prepared by a process comprising (i) stirring Compound I in a MeOH/water mixture at about 80 °C, (ii) isolation of the solid via filtration, and (iii) exposure of the solid to about 75% humidity. Compound I as substantially crystalline EtOH Solvate Hydrate (i.e., wherein less than 15% of Compound I is in amorphous form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous form). The Compound I of Embodiment 361, wherein Compound I is 100% crystalline Compound I EtOH Solvate Hydrate. Substantially pure Compound I EtOH Solvate Hydrate. The Compound I EtOH Solvate Hydrate of any one of Embodiments 361 to 363, characterized by an X-ray powder diffractogram having a signal at one or more two- theta values selected from 6.7 ± 0.2 degrees two-theta, 11.5 ± 0.2 degrees two-theta, 8.6 ± 0.2 degrees two-theta, 14.7 ± 0.2 degrees two-theta, 20.0 ± 0.2 degrees two- theta, 11.3 ± 0.2 degrees two-theta, 18.7 ± 0.2 degrees two-theta, and 19.1 ± 0.2 degrees two-theta. The Compound I EtOH Solvate Hydrate of any one of Embodiments 361 to 364, characterized by an X-ray powder diffractogram having signals at two or more two- theta values selected from 6.7 ± 0.2 degrees two-theta, 11.5 ± 0.2 degrees two-theta, 8.6 ± 0.2 degrees two-theta, 14.7 ± 0.2 degrees two-theta, 20.0 ± 0.2 degrees two- theta, 11.3 ± 0.2 degrees two-theta, 18.7 ± 0.2 degrees two-theta, and 19.1 ± 0.2 degrees two-theta. The Compound I EtOH Solvate Hydrate of any one of Embodiments 361 to 365, characterized by an X-ray powder diffractogram having signals at three or more two- theta values selected from 6.7 ± 0.2 degrees two-theta, 11.5 ± 0.2 degrees two-theta,
8.6 ± 0.2 degrees two-theta, 14.7 ± 0.2 degrees two-theta, 20.0 ± 0.2 degrees two- theta, 11.3 ± 0.2 degrees two-theta, 18.7 ± 0.2 degrees two-theta, and 19.1 ± 0.2 degrees two-theta. The Compound I EtOH Solvate Hydrate of any one of Embodiments 361 to 366, characterized by an X-ray powder diffractogram having signals at four or more two- theta values selected from 6.7 ± 0.2 degrees two-theta, 11.5 ± 0.2 degrees two-theta,
8.6 ± 0.2 degrees two-theta, 14.7 ± 0.2 degrees two-theta, 20.0 ± 0.2 degrees two- theta, 11.3 ± 0.2 degrees two-theta, 18.7 ± 0.2 degrees two-theta, and 19.1 ± 0.2 degrees two-theta. The Compound I EtOH Solvate Hydrate of any one of Embodiments 361 to 367, characterized by an X-ray powder diffractogram having signals at five or more two- theta values selected from 6.7 ± 0.2 degrees two-theta, 11.5 ± 0.2 degrees two-theta,
8.6 ± 0.2 degrees two-theta, 14.7 ± 0.2 degrees two-theta, 20.0 ± 0.2 degrees two- theta, 11.3 ± 0.2 degrees two-theta, 18.7 ± 0.2 degrees two-theta, and 19.1 ± 0.2 degrees two-theta. The Compound I EtOH Solvate Hydrate of any one of Embodiments 361 to 368, characterized by an X-ray powder diffractogram having signals at six or more two- theta values selected from 6.7 ± 0.2 degrees two-theta, 11.5 ± 0.2 degrees two-theta,
8.6 ± 0.2 degrees two-theta, 14.7 ± 0.2 degrees two-theta, 20.0 ± 0.2 degrees two- theta, 11.3 ± 0.2 degrees two-theta, 18.7 ± 0.2 degrees two-theta, and 19.1 ± 0.2 degrees two-theta. The Compound I EtOH Solvate Hydrate of any one of Embodiments 361 to 369, characterized by an X-ray powder diffractogram having signals at seven or more two- theta values selected from 6.7 ± 0.2 degrees two-theta, 11.5 ± 0.2 degrees two-theta,
8.6 ± 0.2 degrees two-theta, 14.7 ± 0.2 degrees two-theta, 20.0 ± 0.2 degrees two- theta, 11.3 ± 0.2 degrees two-theta, 18.7 ± 0.2 degrees two-theta, and 19.1 ± 0.2 degrees two-theta. The Compound I EtOH Solvate Hydrate of any one of Embodiments 361 to 370, characterized by an X-ray powder diffractogram having signals at 6.7 ± 0.2 degrees
two-theta, 11.5 ± 0.2 degrees two-theta, 8.6 ± 0.2 degrees two-theta, 14.7 ± 0.2 degrees two-theta, 20.0 ± 0.2 degrees two-theta, 11.3 ± 0.2 degrees two-theta, 18.7 ± 0.2 degrees two-theta, and 19.1 ± 0.2 degrees two-theta. The Compound I EtOH Solvate Hydrate of any one of Embodiments 361 to 371, characterized by an X-ray powder diffractogram substantially similar to FIG. 44. The Compound I EtOH Solvate Hydrate of any one of Embodiments 361 to 372, characterized by a 13C ssNMR spectrum with one or more peaks selected from: 29.8 ± 0.2 ppm, 127.0 ± 0.2 ppm, 17.9 ± 0.2 ppm, 42.9 ± 0.2 ppm, 18.0 ± 0.2 ppm, 20.9 ± 0.2 ppm, 135.7 ± 0.2 ppm, and 129.1 ± 0.2 ppm. The Compound I EtOH Solvate Hydrate of any one of Embodiments 361 to 373, characterized by a 13C ssNMR spectrum with two or more peaks selected from: 29.8 ± 0.2 ppm, 127.0 ± 0.2 ppm, 17.9 ± 0.2 ppm, 42.9 ± 0.2 ppm, 18.0 ± 0.2 ppm, 20.9 ± 0.2 ppm, 135.7 ± 0.2 ppm, and 129.1 ± 0.2 ppm. The Compound I EtOH Solvate Hydrate of any one of Embodiments 361 to 374, characterized by a 13C ssNMR spectrum with three or more peaks selected from: 29.8 ± 0.2 ppm, 127.0 ± 0.2 ppm, 17.9 ± 0.2 ppm, 42.9 ± 0.2 ppm, 18.0 ± 0.2 ppm, 20.9 ± 0.2 ppm, 135.7 ± 0.2 ppm, and 129.1 ± 0.2 ppm. The Compound I EtOH Solvate Hydrate of any one of Embodiments 361 to 375, characterized by a 13C ssNMR spectrum with four or more peaks selected from: 29.8 ± 0.2 ppm, 127.0 ± 0.2 ppm, 17.9 ± 0.2 ppm, 42.9 ± 0.2 ppm, 18.0 ± 0.2 ppm, 20.9 ± 0.2 ppm, 135.7 ± 0.2 ppm, and 129.1 ± 0.2 ppm. The Compound I EtOH Solvate Hydrate of any one of Embodiments 361 to 376, characterized by a 13C ssNMR spectrum with five or more peaks selected from: 29.8 ± 0.2 ppm, 127.0 ± 0.2 ppm, 17.9 ± 0.2 ppm, 42.9 ± 0.2 ppm, 18.0 ± 0.2 ppm, 20.9 ± 0.2 ppm, 135.7 ± 0.2 ppm, and 129.1 ± 0.2 ppm. The Compound I EtOH Solvate Hydrate of any one of Embodiments 361 to 377, characterized by a 13C ssNMR spectrum with six or more peaks selected from: 29.8 ±
0.2 ppm, 127.0 ± 0.2 ppm, 17.9 ± 0.2 ppm, 42.9 ± 0.2 ppm, 18.0 ± 0.2 ppm, 20.9 ± 0.2 ppm, 135.7 ± 0.2 ppm, and 129.1 ± 0.2 ppm. The Compound I EtOH Solvate Hydrate of any one of Embodiments 361 to 378, characterized by a 13C ssNMR spectrum with seven or more peaks selected from: 29.8 ± 0.2 ppm, 127.0 ± 0.2 ppm, 17.9 ± 0.2 ppm, 42.9 ± 0.2 ppm, 18.0 ± 0.2 ppm, 20.9 ± 0.2 ppm, 135.7 ± 0.2 ppm, and 129.1 ± 0.2 ppm. The Compound I EtOH Solvate Hydrate of any one of Embodiments 361 to 379, characterized by a 13C ssNMR spectrum with peaks at: 29.8 ± 0.2 ppm, 127.0 ± 0.2 ppm, 17.9 ± 0.2 ppm, 42.9 ± 0.2 ppm, 18.0 ± 0.2 ppm, 20.9 ± 0.2 ppm, 135.7 ± 0.2 ppm, and 129.1 ± 0.2 ppm. The Compound I EtOH Solvate Hydrate of any one of Embodiments 361 to 380, characterized by a 13C ssNMR spectrum substantially similar to FIG. 45. The Compound I EtOH Solvate Hydrate of any one of Embodiments 361 to 381, characterized by a monoclinic crystal system, aP2i2i2i space group, and unit cell dimensions, measured at 100 K on a Bruker diffractometer equipped with Cu K« radiation ( = 1.54178 A) and an CP AD detector, of:
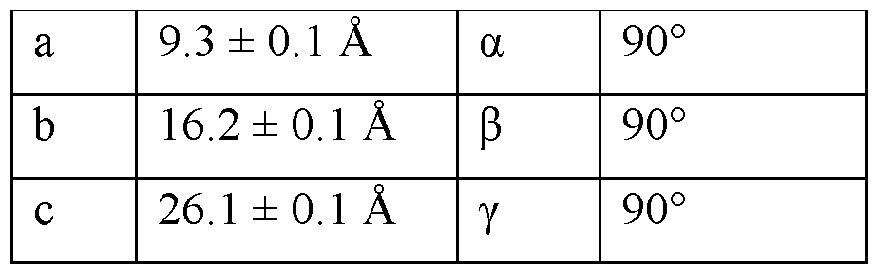 The Compound I EtOH Solvate Hydrate of any one of Embodiments 361 to 382, prepared by a process comprising (i) stirring Compound I in an EtOH/water mixture at about 60 °C, and (ii) isolation of the solid by centrifugation. Compound I as substantially crystalline MeOH Solvate Hydrate (i.e., wherein less than 15% of Compound I is in amorphous form, wherein less than 10% of Compound I is in amorphous Form, wherein less than 5% of Compound I is in amorphous Form).
The Compound I of Embodiment 384, wherein Compound I is 100% crystalline Compound I MeOH hydrate. Substantially pure Compound I MeOH Solvate Hydrate. The Compound I MeOH Solvate Hydrate of any one of Embodiments 384 to 386, characterized by an X-ray powder diffractogram having a signal at one or more two- theta values selected from 8.6 ± 0.2 degrees two-theta, 6.7 ± 0.2 degrees two-theta,
The Compound I EtOH Solvate Hydrate of any one of Embodiments 361 to 382, prepared by a process comprising (i) stirring Compound I in an EtOH/water mixture at about 60 °C, and (ii) isolation of the solid by centrifugation. Compound I as substantially crystalline MeOH Solvate Hydrate (i.e., wherein less than 15% of Compound I is in amorphous form, wherein less than 10% of Compound I is in amorphous Form, wherein less than 5% of Compound I is in amorphous Form).
The Compound I of Embodiment 384, wherein Compound I is 100% crystalline Compound I MeOH hydrate. Substantially pure Compound I MeOH Solvate Hydrate. The Compound I MeOH Solvate Hydrate of any one of Embodiments 384 to 386, characterized by an X-ray powder diffractogram having a signal at one or more two- theta values selected from 8.6 ± 0.2 degrees two-theta, 6.7 ± 0.2 degrees two-theta,
11.5 ± 0.2 degrees two-theta, 19 9 ± 0.2 degrees two-theta, 14.8 ± 0.2 degrees two- theta, 19.2 ± 0.2 degrees two-theta, 18.8 ± 0.2 degrees two-theta, and 19.2 ± 0.2 degrees two-theta. The Compound I MeOH Solvate Hydrate of any one of Embodiments 384 to 387, characterized by an X-ray powder diffractogram having signals at two or more two- theta values selected from 8.6 ± 0.2 degrees two-theta, 6.7 ± 0.2 degrees two-theta,
11.5 ± 0.2 degrees two-theta, 19.9 ± 0.2 degrees two-theta, 14.8 ± 0.2 degrees two- theta, 19.2 ± 0.2 degrees two-theta, 18.8 ± 0.2 degrees two-theta, and 19.2 ± 0.2 degrees two-theta. The Compound I MeOH Solvate Hydrate of any one of Embodiments 384 to 388, characterized by an X-ray powder diffractogram having signals at three or more two- theta values selected from 8.6 ± 0.2 degrees two-theta, 6.7 ± 0.2 degrees two-theta,
11.5 ± 0.2 degrees two-theta, 19.9 ± 0.2 degrees two-theta, 14.8 ± 0.2 degrees two- theta, 19.2 ± 0.2 degrees two-theta, 18.8 ± 0.2 degrees two-theta, and 19.2 ± 0.2 degrees two-theta. The Compound I MeOH Solvate Hydrate of any one of Embodiments 384 to 389, characterized by an X-ray powder diffractogram having signals at four or more two- theta values selected from 8.6 ± 0.2 degrees two-theta, 6.7 ± 0.2 degrees two-theta,
11.5 ± 0.2 degrees two-theta, 19.9 ± 0.2 degrees two-theta, 14.8 ± 0.2 degrees two- theta, 19.2 ± 0.2 degrees two-theta, 18.8 ± 0.2 degrees two-theta, and 19.2 ± 0.2 degrees two-theta.
The Compound I MeOH Solvate Hydrate of any one of Embodiments 384 to 390, characterized by an X-ray powder diffractogram having signals at five or more two- theta values selected from 8.6 ± 0.2 degrees two-theta, 6.7 ± 0.2 degrees two-theta,
11.5 ± 0.2 degrees two-theta, 19.9 ± 0.2 degrees two-theta, 14.8 ± 0.2 degrees two- theta, 19.2 ± 0.2 degrees two-theta, 18.8 ± 0.2 degrees two-theta, and 19.2 ± 0.2 degrees two-theta. The Compound I MeOH Solvate Hydrate of any one of Embodiments 384 to 391, characterized by an X-ray powder diffractogram having signals at six or more two- theta values selected from 8.6 ± 0.2 degrees two-theta, 6.7 ± 0.2 degrees two-theta,
11.5 ± 0.2 degrees two-theta, 19.9 ± 0.2 degrees two-theta, 14.8 ± 0.2 degrees two- theta, 19.2 ± 0.2 degrees two-theta, 18.8 ± 0.2 degrees two-theta, and 19.2 ± 0.2 degrees two-theta. The Compound I MeOH Solvate Hydrate of any one of Embodiments 384 to 392, characterized by an X-ray powder diffractogram having signals at seven or more two- theta values selected from 8.6 ± 0.2 degrees two-theta, 6.7 ± 0.2 degrees two-theta,
11.5 ± 0.2 degrees two-theta, 19.9 ± 0.2 degrees two-theta, 14.8 ± 0.2 degrees two- theta, 19.2 ± 0.2 degrees two-theta, 18.8 ± 0.2 degrees two-theta, and 19.2 ± 0.2 degrees two-theta. The Compound I MeOH Solvate Hydrate of any one of Embodiments 384 to 393, characterized by an X-ray powder diffractogram having signals at 8.6 ± 0.2 degrees two-theta, 6.7 ± 0.2 degrees two-theta, 11.5 ± 0.2 degrees two-theta, 19.9 ± 0.2 degrees two-theta, 14.8 ± 0.2 degrees two-theta, 19.2 ± 0.2 degrees two-theta, 18.8 ± 0.2 degrees two-theta, and 19.2 ± 0.2 degrees two-theta. The Compound I MeOH Solvate Hydrate of any one of Embodiments 384 to 394, characterized by an X-ray powder diffractogram substantially similar to FIG. 46. The Compound I MeOH Solvate Hydrate of any one of Embodiments 384 to 395, characterized by a 13C ssNMR spectrum with one or more peaks selected from: 29.3 ± 0.2 ppm, 126.9 ± 0.2 ppm, 43.1 ± 0.2 ppm, 20.9 ± 0.2 ppm, 17.2 ± 0.2 ppm, 127.8 ± 0.2 ppm, 29.6 ± 0.2 ppm, and 135.7 ± 0.2 ppm.
The Compound I MeOH Solvate Hydrate of any one of Embodiments 384 to 396, characterized by a 13C ssNMR spectrum with two or more peaks selected from: 29.3 ± 0.2 ppm, 126.9 ± 0.2 ppm, 43.1 ± 0.2 ppm, 20.9 ± 0.2 ppm, 17.2 ± 0.2 ppm, 127.8 ± 0.2 ppm, 29.6 ± 0.2 ppm, and 135.7 ± 0.2 ppm. The Compound I MeOH Solvate Hydrate of any one of Embodiments 384 to 397, characterized by a 13C ssNMR spectrum with three or more peaks selected from: 29.3 ± 0.2 ppm, 126.9 ± 0.2 ppm, 43.1 ± 0.2 ppm, 20.9 ± 0.2 ppm, 17.2 ± 0.2 ppm, 127.8 ± 0.2 ppm, 29.6 ± 0.2 ppm, and 135.7 ± 0.2 ppm. The Compound I MeOH Solvate Hydrate of any one of Embodiments 384 to 398 characterized by a 13C ssNMR spectrum with four or more peaks selected from: 29.3 ± 0.2 ppm, 126.9 ± 0.2 ppm, 43.1 ± 0.2 ppm, 20.9 ± 0.2 ppm, 17.2 ± 0.2 ppm, 127.8 ± 0.2 ppm, 29.6 ± 0.2 ppm, and 135.7 ± 0.2 ppm. The Compound I MeOH Solvate Hydrate of any one of Embodiments 384 to 399, characterized by a 13C ssNMR spectrum with five or more peaks selected from: 29.3 ± 0.2 ppm, 126.9 ± 0.2 ppm, 43.1 ± 0.2 ppm, 20.9 ± 0.2 ppm, 17.2 ± 0.2 ppm, 127.8 ± 0.2 ppm, 29.6 ± 0.2 ppm, and 135.7 ± 0.2 ppm. The Compound I MeOH Solvate Hydrate of any one of Embodiments 384 to 400, characterized by a 13C ssNMR spectrum with six or more peaks selected from: 29.3 ± 0.2 ppm, 126.9 ± 0.2 ppm, 43.1 ± 0.2 ppm, 20.9 ± 0.2 ppm, 17.2 ± 0.2 ppm, 127.8 ± 0.2 ppm, 29.6 ± 0.2 ppm, and 135.7 ± 0.2 ppm. The Compound I MeOH Solvate Hydrate of any one of Embodiments 384 to 401, characterized by a 13C ssNMR spectrum with seven or more peaks selected from: 29.3 ± 0.2 ppm, 126.9 ± 0.2 ppm, 43.1 ± 0.2 ppm, 20.9 ± 0.2 ppm, 17.2 ± 0.2 ppm, 127.8 ± 0.2 ppm, 29.6 ± 0.2 ppm, and 135.7 ± 0.2 ppm. The Compound I MeOH Solvate Hydrate of any one of Embodiments 384 to 402, characterized by a 13C ssNMR spectrum with peaks at: 29.3 ± 0.2 ppm, 126.9 ± 0.2
ppm, 43.1 ± 0.2 ppm, 20.9 ± 0.2 ppm, 17.2 ± 0.2 ppm, 127.8 ± 0.2 ppm, 29.6 ± 0.2 ppm, and 135.7 ± 0.2 ppm. The Compound I MeOH Solvate Hydrate of any one of Embodiments 384 to 403, characterized by a 13C ssNMR spectrum substantially similar to FIG. 47. The Compound I MeOH Solvate Hydrate of any one of Embodiments 384 to 404, prepared by a process comprising (i) the stirring of Compound I in a MeOH/water mixture at about 80 °C, and (ii) isolation of the solid via centrifugation. Compound I as substantially crystalline IPA Solvate Hydrate (i.e., wherein less than 15% of Compound I is in amorphous form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous form). The Compound I of Embodiment 406, wherein Compound I is 100% crystalline Compound I IPA Solvate Hydrate. Substantially pure Compound I IPA Solvate Hydrate. The Compound I IPA Solvate Hydrate of any one of Embodiments 406 to 408, characterized by an X-ray powder diffractogram having a signal at one or more two- theta values selected from 6.7 ± 0.2 degrees two-theta, 8.6 ± 0.2 degrees two-theta,
11.5 ± 0.2 degrees two-theta, 19.9 ± 0.2 degrees two-theta, 14.8 ± 0.2 degrees two- theta, 18.7 ± 0.2 degrees two-theta, 17.7 ± 0.2 degrees two-theta, and 17.3 ± 0.2 degrees two-theta. The Compound I IPA Solvate Hydrate of any one of Embodiments 406 to 409, characterized by an X-ray powder diffractogram having signals at two or more two- theta values selected to 6.7 ± 0.2 degrees two-theta, 8.6 ± 0.2 degrees two-theta, 11.5 ± 0.2 degrees two-theta, 19.9 ± 0.2 degrees two-theta, 14.8 ± 0.2 degrees two-theta, 18.7 ± 0.2 degrees two-theta, 17.7 ± 0.2 degrees two-theta, and 17.3 ± 0.2 degrees two-theta.
The Compound I IPA Solvate Hydrate of any one of Embodiments 406 to 410, characterized by an X-ray powder diffractogram having signals at three or more two- theta values selected from 6.7 ± 0.2 degrees two-theta, 8.6 ± 0.2 degrees two-theta,
11.5 ± 0.2 degrees two-theta, 19.9 ± 0.2 degrees two-theta, 14.8 ± 0.2 degrees two- theta, 18.7 ± 0.2 degrees two-theta, 17.7 ± 0.2 degrees two-theta, and 17.3 ± 0.2 degrees two-theta. The Compound I IPA Solvate Hydrate of any one of Embodiments 406 to 411, characterized by an X-ray powder diffractogram having signals at four or more two- theta values selected from 6.7 ± 0.2 degrees two-theta, 8.6 ± 0.2 degrees two-theta,
11.5 ± 0.2 degrees two-theta, 19.9 ± 0.2 degrees two-theta, 14.8 ± 0.2 degrees two- theta, 18.7 ± 0.2 degrees two-theta, 17.7 ± 0.2 degrees two-theta, and 17.3 ± 0.2 degrees two-theta. The Compound I IPA Solvate Hydrate of any one of Embodiments 406 to 412, characterized by an X-ray powder diffractogram having signals at five or more two- theta values selected from 6.7 ± 0.2 degrees two-theta, 8.6 ± 0.2 degrees two-theta,
11.5 ± 0.2 degrees two-theta, 19.9 ± 0.2 degrees two-theta, 14.8 ± 0.2 degrees two- theta, 18.7 ± 0.2 degrees two-theta, 17.7 ± 0.2 degrees two-theta, and 17.3 ± 0.2 degrees two-theta. The Compound I IPA Solvate Hydrate of any one of Embodiments 406 to 413, characterized by an X-ray powder diffractogram having signals at six or more two- theta values selected from 6.7 ± 0.2 degrees two-theta, 8.6 ± 0.2 degrees two-theta,
11.5 ± 0.2 degrees two-theta, 19.9 ± 0.2 degrees two-theta, 14.8 ± 0.2 degrees two- theta, 18.7 ± 0.2 degrees two-theta, 17.7 ± 0.2 degrees two-theta, and 17.3 ± 0.2 degrees two-theta. The Compound I IPA Solvate Hydrate of any one of Embodiments 406 to 414, characterized by an X-ray powder diffractogram having signals at seven or more two- theta values selected from 6.7 ± 0.2 degrees two-theta, 8.6 ± 0.2 degrees two-theta,
11.5 ± 0.2 degrees two-theta, 19.9 ± 0.2 degrees two-theta, 14.8 ± 0.2 degrees two- theta, 18.7 ± 0.2 degrees two-theta, 17.7 ± 0.2 degrees two-theta, and 17.3 ± 0.2 degrees two-theta.
The Compound I IPA Solvate Hydrate of any one of Embodiments 406 to 415, characterized by an X-ray powder diffractogram having signals at 6.7 ± 0.2 degrees two-theta, 8.6 ± 0.2 degrees two-theta, 11.5 ± 0.2 degrees two-theta, 19.9 ± 0.2 degrees two-theta, 14.8 ± 0.2 degrees two-theta, 18.7 ± 0.2 degrees two-theta, 17.7 ± 0.2 degrees two-theta, and 17.3 ± 0.2 degrees two-theta. The Compound I IPA Solvate Hydrate of any one of Embodiments 406 to 416, characterized by an X-ray powder diffractogram substantially similar to FIG. 48. The Compound I IPA Solvate Hydrate of any one of Embodiments 406 to 417, characterized by a 13C ssNMR spectrum with one or more peaks selected from: 30.0 ± 0.2 ppm, 127.4 ± 0.2 ppm, 24.9 ± 0.2 ppm, 18.6 ± 0.2 ppm, 24.2 ± 0.2 ppm, 20.3 ± 0.2 ppm, 136.8 ± 0.2 ppm, and 42.7 ± 0.2 ppm. The Compound I IPA Solvate Hydrate of any one of Embodiments 406 to 418, characterized by a 13C ssNMR spectrum with two or more peaks selected from: 30.0 ± 0.2 ppm, 127.4 ± 0.2 ppm, 24.9 ± 0.2 ppm, 18.6 ± 0.2 ppm, 24.2 ± 0.2 ppm, 20.3 ± 0.2 ppm, 136.8 ± 0.2 ppm, and 42.7 ± 0.2 ppm. The Compound I IPA Solvate Hydrate of any one of Embodiments 406 to 419, characterized by a 13C ssNMR spectrum with three or more peaks selected from: 30.0 ± 0.2 ppm, 127.4 ± 0.2 ppm, 24.9 ± 0.2 ppm, 18.6 ± 0.2 ppm, 24.2 ± 0.2 ppm, 20.3 ± 0.2 ppm, 136.8 ± 0.2 ppm, and 42.7 ± 0.2 ppm. The Compound I IPA Solvate Hydrate of any one of Embodiments 406 to 420, characterized by a 13C ssNMR spectrum with four or more peaks selected from: 30.0 ± 0.2 ppm, 127.4 ± 0.2 ppm, 24.9 ± 0.2 ppm, 18.6 ± 0.2 ppm, 24.2 ± 0.2 ppm, 20.3 ± 0.2 ppm, 136.8 ± 0.2 ppm, and 42.7 ± 0.2 ppm. The Compound I IPA Solvate Hydrate of any one of Embodiments 406 to 421, characterized by a 13C ssNMR spectrum with five or more peaks selected from: 30.0 ± 0.2 ppm, 127.4 ± 0.2 ppm, 24.9 ± 0.2 ppm, 18.6 ± 0.2 ppm, 24.2 ± 0.2 ppm, 20.3 ± 0.2 ppm, 136.8 ± 0.2 ppm, and 42.7 ± 0.2 ppm.
The Compound I IPA Solvate Hydrate of any one of Embodiments 406 to 422, characterized by a 13C ssNMR spectrum with six or more peaks selected from: 30.0 ± 0.2 ppm, 127.4 ± 0.2 ppm, 24.9 ± 0.2 ppm, 18.6 ± 0.2 ppm, 24.2 ± 0.2 ppm, 20.3 ± 0.2 ppm, 136.8 ± 0.2 ppm, and 42.7 ± 0.2 ppm. The Compound I IPA Solvate Hydrate of any one of Embodiments 406 to 423, characterized by a 13C ssNMR spectrum with seven or more peaks selected from: 30.0 ± 0.2 ppm, 127.4 ± 0.2 ppm, 24.9 ± 0.2 ppm, 18.6 ± 0.2 ppm, 24.2 ± 0.2 ppm, 20.3 ± 0.2 ppm, 136.8 ± 0.2 ppm, and 42.7 ± 0.2 ppm. The Compound I IPA Solvate Hydrate of any one of Embodiments 406 to 424, characterized by a 13C ssNMR spectrum with peaks at: 30.0 ± 0.2 ppm, 127.4 ± 0.2 ppm, 24.9 ± 0.2 ppm, 18.6 ± 0.2 ppm, 24.2 ± 0.2 ppm, 20.3 ± 0.2 ppm, 136.8 ± 0.2 ppm, and 42.7 ± 0.2 ppm. The Compound I IPA Solvate Hydrate of any one of Embodiments 406 to 425, characterized by a 13C ssNMR spectrum substantially similar to FIG. 49. The Compound I IPA Solvate Hydrate of any one of Embodiments 406 to 426, characterized by a monoclinic crystal system, aP2i2i2i space group, and unit cell dimensions, measured at 100 K on a Bruker diffractometer equipped with Cu K« radiation ( = 1.54178 A) and an CP AD detector, of:
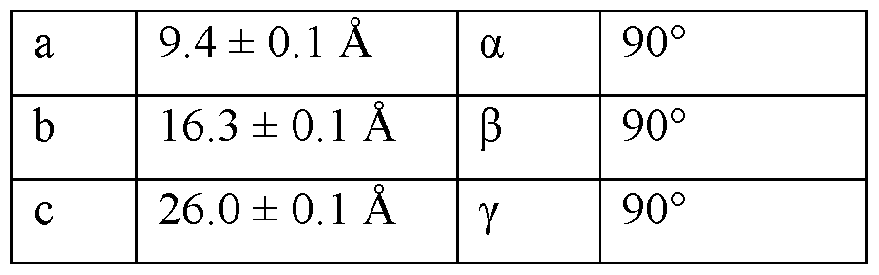 The Compound I IPA Solvate Hydrate of any one of Embodiments 406 to 427, prepared by a process comprising (i) stirring Compound I in an IPA/water mixture, and (ii) isolation of the solid via centrifugation.
Compound I as substantially crystalline MeOAc Solvate Hydrate (i.e., wherein less than 15% of Compound I is in amorphous form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous form). The Compound I of Embodiment 429, wherein Compound I is 100% crystalline Compound I MeOAc hydrate. Substantially pure Compound I MeOAc Solvate Hydrate. The Compound I MeOAc Solvate Hydrate of any one of Embodiments 429 to 431, characterized by an X-ray powder diffractogram having a signal at one or more two- theta values selected from 6.5 ± 0.2 degrees two-theta, 10.7 ± 0.2 degrees two-theta,
The Compound I IPA Solvate Hydrate of any one of Embodiments 406 to 427, prepared by a process comprising (i) stirring Compound I in an IPA/water mixture, and (ii) isolation of the solid via centrifugation.
Compound I as substantially crystalline MeOAc Solvate Hydrate (i.e., wherein less than 15% of Compound I is in amorphous form, wherein less than 10% of Compound I is in amorphous form, wherein less than 5% of Compound I is in amorphous form). The Compound I of Embodiment 429, wherein Compound I is 100% crystalline Compound I MeOAc hydrate. Substantially pure Compound I MeOAc Solvate Hydrate. The Compound I MeOAc Solvate Hydrate of any one of Embodiments 429 to 431, characterized by an X-ray powder diffractogram having a signal at one or more two- theta values selected from 6.5 ± 0.2 degrees two-theta, 10.7 ± 0.2 degrees two-theta,
14.3 ± 0.2 degrees two-theta, 17.5 ± 0.2 degrees two-theta, 7.1 ± 0.2 degrees two- theta, 19.9 ± 0.2 degrees two-theta, 19.5 ± 0.2 degrees two-theta, and 14.2 ± 0.2 degrees two-theta. The Compound I MeOAc Solvate Hydrate of any one of Embodiments 429 to 432, characterized by an X-ray powder diffractogram having signals at two or more two- theta values selected from 6.5 ± 0.2 degrees two-theta, 10.7 ± 0.2 degrees two-theta,
14.3 ± 0.2 degrees two-theta, 17.5 ± 0.2 degrees two-theta, 7.1 ± 0.2 degrees two- theta, 19.9 ± 0.2 degrees two-theta, 19.5 ± 0.2 degrees two-theta, and 14.2 ± 0.2 degrees two-theta. The Compound I MeOAc Solvate Hydrate of any one of Embodiments 429 to 433, characterized by an X-ray powder diffractogram having signals at three or more two- theta values selected from 6.5 ± 0.2 degrees two-theta, 10.7 ± 0.2 degrees two-theta,
14.3 ± 0.2 degrees two-theta, 17.5 ± 0.2 degrees two-theta, 7.1 ± 0.2 degrees two- theta, 19.9 ± 0.2 degrees two-theta, 19.5 ± 0.2 degrees two-theta, and 14.2 ± 0.2 degrees two-theta. The Compound I MeOAc Solvate Hydrate of any one of Embodiments 429 to 434, characterized by an X-ray powder diffractogram having signals at four or more two- theta values selected from 6.5 ± 0.2 degrees two-theta, 10.7 ± 0.2 degrees two-theta,
14.3 ± 0.2 degrees two-theta, 17.5 ± 0.2 degrees two-theta, 7.1 ± 0.2 degrees two-
theta, 19.9 ± 0.2 degrees two-theta, 19.5 ± 0.2 degrees two-theta, and 14.2 ± 0.2 degrees two-theta. The Compound I MeOAc Solvate Hydrate of any one of Embodiments 429 to 435, characterized by an X-ray powder diffractogram having signals at five or more two- theta values selected from 6.5 ± 0.2 degrees two-theta, 10.7 ± 0.2 degrees two-theta,
14.3 ± 0.2 degrees two-theta, 17.5 ± 0.2 degrees two-theta, 7.1 ± 0.2 degrees two- theta, 19.9 ± 0.2 degrees two-theta, 19.5 ± 0.2 degrees two-theta, and 14.2 ± 0.2 degrees two-theta. The Compound I MeOAc Solvate Hydrate of any one of Embodiments 429 to 436, characterized by an X-ray powder diffractogram having signals at six or more two- theta values selected from 6.5 ± 0.2 degrees two-theta, 10.7 ± 0.2 degrees two-theta,
14.3 ± 0.2 degrees two-theta, 17.5 ± 0.2 degrees two-theta, 7.1 ± 0.2 degrees two- theta, 19.9 ± 0.2 degrees two-theta, 19.5 ± 0.2 degrees two-theta, and 14.2 ± 0.2 degrees two-theta. The Compound I MeOAc Solvate Hydrate of any one of Embodiments 429 to 437, characterized by an X-ray powder diffractogram having signals at seven or more two- theta values selected from 6.5 ± 0.2 degrees two-theta, 10.7 ± 0.2 degrees two-theta,
14.3 ± 0.2 degrees two-theta, 17.5 ± 0.2 degrees two-theta, 7.1 ± 0.2 degrees two- theta, 19.9 ± 0.2 degrees two-theta, 19.5 ± 0.2 degrees two-theta, and 14.2 ± 0.2 degrees two-theta. The Compound I MeOAc Solvate Hydrate of any one of Embodiments 429 to 438, characterized by an X-ray powder diffractogram having signals at 6.5 ± 0.2 degrees two-theta, 10.7 ± 0.2 degrees two-theta, 14.3 ± 0.2 degrees two-theta, 17.5 ± 0.2 degrees two-theta, 7.1 ± 0.2 degrees two-theta, 19.9 ± 0.2 degrees two-theta, 19.5 ± 0.2 degrees two-theta, and 14.2 ± 0.2 degrees two-theta. The Compound I MeOAc Solvate Hydrate of any one of Embodiments 429 to 439, characterized by an X-ray powder diffractogram substantially similar to FIG. 50.
The Compound I MeOAc Solvate Hydrate of any one of Embodiments 429 to 440, characterized by a 13C ssNMR spectrum with one or more peaks selected from: 28.1 ± 0.2 ppm, 43.6 ± 0.2 ppm, 130.5 ± 0.2 ppm, 21.4 ± 0.2 ppm, 20.7 ± 0.2 ppm, 19.0 ± 0.2 ppm, 135.9 ± 0.2 ppm, and 143.4 ± 0.2 ppm. The Compound I MeOAc Solvate Hydrate of any one of Embodiments 429 to 441, characterized by a 13C ssNMR spectrum with two or more peaks selected from: 28.1 ± 0.2 ppm, 43.6 ± 0.2 ppm, 130.5 ± 0.2 ppm, 21.4 ± 0.2 ppm, 20.7 ± 0.2 ppm, 19.0 ± 0.2 ppm, 135.9 ± 0.2 ppm, and 143.4 ± 0.2 ppm. The Compound I MeOAc Solvate Hydrate of any one of Embodiments 429 to 442, characterized by a 13C ssNMR spectrum with three or more peaks selected from: 28.1 ± 0.2 ppm, 43.6 ± 0.2 ppm, 130.5 ± 0.2 ppm, 21.4 ± 0.2 ppm, 20.7 ± 0.2 ppm, 19.0 ± 0.2 ppm, 135.9 ± 0.2 ppm, and 143.4 ± 0.2 ppm. The Compound I MeOAc Solvate Hydrate of any one of Embodiments 429 to 443, characterized by a 13C ssNMR spectrum with four or more peaks selected from: 28.1 ± 0.2 ppm, 43.6 ± 0.2 ppm, 130.5 ± 0.2 ppm, 21.4 ± 0.2 ppm, 20.7 ± 0.2 ppm, 19.0 ± 0.2 ppm, 135.9 ± 0.2 ppm, and 143.4 ± 0.2 ppm. The Compound I MeOAc Solvate Hydrate of any one of Embodiments 429 to 444, characterized by a 13C ssNMR spectrum with five or more peaks selected from: 28.1 ± 0.2 ppm, 43.6 ± 0.2 ppm, 130.5 ± 0.2 ppm, 21.4 ± 0.2 ppm, 20.7 ± 0.2 ppm, 19.0 ± 0.2 ppm, 135.9 ± 0.2 ppm, and 143.4 ± 0.2 ppm. The Compound I MeOAc Solvate Hydrate of any one of Embodiments 429 to 445, characterized by a 13C ssNMR spectrum with six or more peaks selected from: 28.1 ± 0.2 ppm, 43.6 ± 0.2 ppm, 130.5 ± 0.2 ppm, 21.4 ± 0.2 ppm, 20.7 ± 0.2 ppm, 19.0 ± 0.2 ppm, 135.9 ± 0.2 ppm, and 143.4 ± 0.2 ppm. The Compound I MeOAc Solvate Hydrate of any one of Embodiments 429 to 446, characterized by a 13C ssNMR spectrum with seven or more peaks selected from: 28.1 ± 0.2 ppm, 43.6 ± 0.2 ppm, 130.5 ± 0.2 ppm, 21.4 ± 0.2 ppm, 20.7 ± 0.2 ppm, 19.0 ± 0.2 ppm, 135.9 ± 0.2 ppm, and 143.4 ± 0.2 ppm.
The Compound I MeOAc Solvate Hydrate of any one of Embodiments 429 to 447, characterized by a 13C ssNMR spectrum with peaks at: 28.1 ± 0.2 ppm, 43.6 ± 0.2 ppm, 130.5 ± 0.2 ppm, 21.4 ± 0.2 ppm, 20.7 ± 0.2 ppm, 19.0 ± 0.2 ppm, 135.9 ± 0.2 ppm, and 143.4 ± 0.2 ppm. The Compound I MeOAc Solvate Hydrate of any one of Embodiments 429 to 448, characterized by a 13C ssNMR spectrum substantially similar to FIG. 51. The Compound I MeOAc Solvate Hydrate of any one of Embodiments 429 to 449, prepared by a process comprising (i) stirring amorphous Compound I in a MeOAc/water mixture, (ii) addition of Compound I MeOAc Form A, Compound I MeOAc Form B, Compound I MeOAc Form C, Compound I Neat Form E, Compound I Hydrate Form A, and Compound I hydrate Form B, (iii) further stirring, and (iv) isolation of the solid via centrifugation. Compound I as substantially amorphous Compound I (i.e., wherein less than 15% of Compound I is in crystalline form, wherein less than 10% of Compound I is in crystalline form, wherein less than 5% of Compound I is in crystalline form) The substantially amorphous Compound I of Embodiment 451, wherein Compound I is 100% amorphous. Amorphous Compound I of Embodiment 451 or Embodiment 452 characterized by an X-ray powder diffractogram substantially similar to FIG. 52. Amorphous Compound I of any one of Embodiments 451 to 453 characterized by a 13C ssNMR spectrum substantially similar to FIG. 53. Amorphous Compound I of any one of Embodiments 451 to 454, prepared by a process comprising (i) purification of Compound I via column chromatography, (ii) dissolving the solid in DCM, (iii) removal of the solvent, and (iv) drying the solid under high vacuum.
Compound I formulated as a solid dispersion. The Compound I of Embodiment 456, wherein the solid dispersion is a spray-dried dispersion. The Compound I formulated as a spray-dried dispersion of Embodiment 456 or Embodiment 457, wherein the spray-dried dispersion comprises a polymer. The Compound I formulated as a spray-dried dispersion of Embodiment 458, wherein the polymer is HPMCAS-H. The Compound I formulated as a spray-dried dispersion of Embodiment 458, wherein the polymer is HPMC-E15. The Compound I formulated as a spray-dried dispersion of Embodiment 459 or Embodiment 460, wherein the w/w ratio of Compound I to polymer is 50/50. The Compound I formulated as a spray-dried dispersion of Embodiment 459 or Embodiment 460, wherein the w/w ratio of Compound I to polymer is 80/20. The Compound I formulated as a spray-dried dispersion of any one of Embodiments 456 to 462, prepared by a process comprising (i) preparation of a solution of Compound I and polymer in a DCM/MeOH mixture, and (ii) spray drying the resulting solution using a spray dryer. The Compound I formulated as a spray-dried dispersion of Embodiment 463, wherein the DCM/MeOH mixture has a w/w ratio of 50/50. The Compound I formulated as a spray-dried dispersion of Embodiment 463, wherein the DCM/MeOH mixture has a w/w ratio of 63/37. A pharmaceutical composition comprising Compound I according to any one of Embodiments 1 to 465 and a pharmaceutically acceptable carrier. The pharmaceutical composition according to Embodiment 466 further comprising one or more additional therapeutic agents.
The pharmaceutical composition according to Embodiment 467, wherein the pharmaceutical composition comprises one or more additional CFTR modulating compounds. The pharmaceutical composition according to Embodiment 467 or Embodiment 468, wherein the pharmaceutical composition comprises one or more compounds selected from Compound II, Compound III, Compound Ill-d, Compound IV, and Compound V, and pharmaceutically acceptable salts and deuterated derivatives of any of the foregoing. The Compound I according to any one of Embodiments 1 to 463, or the pharmaceutical composition according to any one of Embodiments 466 to 469, for use in the treatment of cystic fibrosis. The compound for use of Embodiment 470, wherein the compound is administered in combination with one or more compounds selected from Compound II, Compound III, Compound Ill-d, Compound IV, and Compound V, and pharmaceutically acceptable salts and deuterated derivatives of any of the foregoing. Use of the Compound I according to any one of Embodiments 1 to 463, or the pharmaceutical composition according to any one of Embodiments 466 to 469 in the manufacture of a medicament for the treatment of cystic fibrosis. The use according to Embodiment 472, wherein the medicament is formulated for administration in combination with one or more compounds selected from Compound II, Compound III, Compound Ill-d, Compound IV, and pharmaceutically acceptable salts and deuterated derivatives of any of the foregoing. A method of treating cystic fibrosis comprising administering the Compound I according to any one of Embodiments 1 to 463, or the pharmaceutical composition according to any one of Embodiments 466 to 469, to a subject in need thereof.
The method of treating according to Embodiment 474, wherein the method further comprises administration of one or more compounds selected from Compound II, Compound III, Compound Ill-d, Compound IV, and pharmaceutically acceptable salts and deuterated derivatives of any of the foregoing. A process for preparing Compound I Neat Form A according to any one of Embodiments 1 to 28 comprising (i) dissolving amorphous Compound I in ethanol, (ii) increasing the temperature to about 73 °C, (iii) decreasing the temperature to about 20 °C, (iv) isolation of the solid via filtration, and (v) drying of the solid at 50 °C. A process for preparing Compound I Neat Form B according to any one of Embodiments 30 to 49 comprising exposing Compound I Hydrate Form A to about 3% humidity. A process for preparing Compound I Neat Form C according to any one of Embodiments 51 to 74 comprising (i) stirring of Compound I in hexadecane at about 80 °C, and (ii) centrifugation and drying of the resulting solid. A process for preparing Compound I Neat Form D according to any one of Embodiments 76 to 87 comprising the drying of Compound I hydrate C at about 80 °C. A process for preparing Compound I Neat Form D according to any one of Embodiments 76 to 87 comprising (i) stirring of Compound I Neat Form A in IP A, and (ii) isolation of the solid via filtration, and (iii) drying of the solid at about 125 °C. A process for preparing Compound I Neat Form E according to any one of Embodiments 90 to 112 comprising (i) stirring of Compound I Neat Form A in MeOAc, and (ii) isolation of the solid via centrifugation, and (iii) drying of the solid at about 50 °C.
A process for preparing Compound I EtOH Solvate Form B according to any one of Embodiments 152 to 177 comprising (i) stirring of Compound I in ethanol, and (ii) isolation of the solid via centrifugation. A process for preparing Compound I MeOH Solvate according to any one of Embodiments 179 to 196 comprising (i) dissolving Compound I in MeOH and increasing temperature to about 62 °C, (ii) cooling the solution to about 10 °C, and (iii) isolation of the solid via centrifugation. A process for preparing Compound I NPA Solvate according to any one of Embodiments 198 to 218 comprising (i) stirring of Compound I in NPA, and (ii) isolation of the solid via centrifugation. A process for preparing Compound I MeOAc Solvate Form A according to any one of Embodiments 220 to 241 comprising (i) stirring Compound I Neat Form A in MeOAc, and (ii) isolation of the solid via filtration. A process for preparing Compound I MeOAc Solvate form B according to any one of Embodiments 243 to 258 comprising (i) stirring of Compound I Neat Form A in MeOAc at about 5 °C, (ii) isolation of the solid via centrifugation, and (iii) drying of the solid at room temperature. A process for preparing Compound I MeOAc Solvate form C according to any one of Embodiments 269 to 281 comprising (i) stirring of Compound I Neat Form A in MeOAc at about 5 °C, and (ii) isolation of the solid via filtration. A process for preparing Compound I Hydrate Form A according to any one of Embodiments 292 to 313 comprising (i) stirring Compound I EtOH solvate form B in water, and (ii) isolation via filtration and air drying of the solid. A process for preparing Compound I Hydrate form B according to any one of Embodiments 315 to 336 comprising (i) stirring Compound I in a MeOH/water mixture at about 80 °C, (ii) isolation of the solid via filtration, and (iii) exposure of the solid to about 11% humidity.
A process for preparing Compound I Hydrate Form C according to any one of Embodiments 338 to 359 comprising (i) stirring Compound I in a MeOH/water mixture at about 80 °C, (ii) isolation of the solid via filtration, and (iii) exposure of the solid to about 75% humidity. A process for preparing Compound I EtOH Solvate Hydrate according to any one of Embodiments 361 to 382 comprising (i) stirring Compound I in an EtOH/water mixture at about 60 °C, and (ii) isolation of the solid via centrifugation. A process for preparing Compound I MeOH Solvate Hydrate according to any one of Embodiments 384 to 404 comprising (i) the stirring of Compound I in a MeOH/water mixture at about 80 °C, and (ii) isolation of the solid via centrifugation. A process for preparing Compound I IPA Solvate Hydrate according to any one of Embodiments 406 to 427 comprising (i) stirring Compound I in an IPA/water mixture, and (ii) isolation of the solid via centrifugation. A process for preparing Compound I MeOAc Solvate Hydrate according to any one of Embodiments 429 to 449 comprising (i) stirring amorphous Compound I in a MeOAc/water mixture, (ii) addition of Compound I MeOAc form A, Compound I MeOAc form B, Compound I MeOAc form C, Compound I Neat Form E, Compound I Hydrate form A, and Compound I Hydrate Form B, (iii) further stirring, and (iv) isolation of the solid via centrifugation. A process for preparing amorphous Compound I according to any one of Embodiments 451 to 454 comprising (i) purification of Compound I via column chromatography, (ii) dissolving the solid in DCM, (iii) removal of the solvent, and (iv) drying the solid under high vacuum. A process for preparing amorphous Compound I formulated as a spray-dried dispersion according to any one of Embodiments 456 to 462 comprising (i) preparation of a solution of Compound I and polymer in a DCM/MeOH mixture, and (ii) spray drying the resulting solution using a spray dryer.
Methods of Preparing Compound I and Solid Forms of Compound I
General Experimental Procedures
[00200] Compounds II, III, Ill-d, and IV can be prepared by any suitable method in the art, for example, PCT Publication Nos. WO 2011/133751, WO 2011/133951, WO 2015/160787, and U.S. Patent No. 8,865,902.
[00201] Reagents and starting materials were obtained by commercial sources unless otherwise stated and were used without purification.
[00202] Solution-phase proton and carbon NMR spectra were acquired on either a Bruker Biospin DRX 400 MHz FTNMR spectrometer operating at a 1 H and 13C resonant frequency of 400 and 100 MHz respectively, or on a 300 MHz NMR spectrometer. One dimensional proton and carbon spectra were acquired using a broadband observe (BBFO) probe with 20 Hz sample rotation at 0.1834 and 0.9083 Hz/Pt digital resolution respectively. All proton and carbon spectra were acquired with temperature control at 30 °C using standard, previously published pulse sequences and routine processing parameters.
[00203] Solution-phase NMR spectra were also recorded on a Varian Mercury NMR instrument at 300 MHz for 'H using a 45 degree pulse angle, a spectral width of 4800 Hz and 28860 points of acquisition. FID were zero-filled to 32k points and a line broadening of 0.3 Hz was applied before Fourier transform
[00204] Solution-phase NMR spectra were also recorded on a Bruker Avance III HD NMR instrument at 400 MHz for 'H using a 30 degree pulse angle, a spectral width of 8000 Hz and 128k points of acquisition. FID were zero-filled to 256k points and a line broadening of 0.3 Hz was applied before Fourier transform.
[00205] Solution-phase NMR spectra were also recorded on a Bruker AC 250 MHz instrument equipped with a: 5 mm QNP(H1/C13/F19/P31) probe (type: 250-SB, s#23055/0020) or on a Varian 500 MHz instrument equipped with a ID PFG, 5 mm, 50- 202/500 MHz probe (model/part# 99337300).
General UPLC/HPLC Analytical Methods
[00206] LC method A: Analytical reverse phase UPLC using an Acquity UPLC BEH Cis column (50 x 2.1 mm, 1.7 pm particle) made by Waters (pn: 186002350), and a dual gradient run from 1-99% mobile phase B over 3.0 minutes. Mobile phase A = H2O (0.05 %
CF3CO2H). Mobile phase B = CH3CN (0.035 % CF3CO2H). Flow rate = 1.2 mL/min, injection volume = 1.5 pL, and column temperature = 60 °C.
[00207] LC method C: Reversed phase UPLC using an Acquity UPLC BEH Cis column (50 x 2.1 mm, 1.7 pm particle) made by Waters (pn: 186002350), and a dual gradient run from 30-99% mobile phase B over 2.9 minutes. Mobile phase A = H2O (0.05 % CF3CO2H). Mobile phase B = CH3CN (0.035 % CF3CO2H). Flow rate = 1.2 mL/min, injection volume = 1.5 pL, and column temperature = 60 °C.
[00208] LC method I: UPLC Luna Cis(2) 50 x 3 mm 3 pm. run: 2.5 min. Mobile phase: Initial 95% H2O 0.1% FA / 5%MeCN 0.1% FA, linear grad to 95% MeCN 0.1% FA over 1.3 min, hold 1.2 min 95% CH3CN 0.1% FA,.T: 45C, Flow: 1.5 mL/min.
[00209] LC method J: UPLC SunFire Cis 75 x 4.6 mm 3.5 pm, run: 6 min. Mobile phase conditions Initial 95% H2O + 0.1% FA/5% CH3CN + 0.1% FA, linear gradient to 95% CFLCN for 4 min, hold for 2 min at 95% CH3CN. T:45 °C, Flow: 1.5 mL/min.
[00210] LC method L: Luna Cis 3.0 x 50 mm 3.0 pM, Temp: 45 °C, Flow: 2.0 mL/min, Run Time: 3 minutes. Mobile Phase: Initial 95% H2O (0.1% Formic Acid) and 5% CH3CN (0.1% FA) linear gradient to 95% CH3CN (0.1% FA) for 2.0 min then hold at 95% CH3CN (0.1% FA) for 1.0 min.
[00211] Unless otherwise stated, the following procedures were employed for the analysis of all solid forms.
X-Ray Powder Diffraction
[00212] X-ray powder diffraction (XRPD) spectra were recorded at room temperature (25 ± 2 °C) in transmission mode using a PANalytical Empyrean system equipped with a sealed tube source and a PIXcel ID Medipix-3 detector (Malvern PANalytical Inc, Westborough, Massachusetts). The X-Ray generator operated at a voltage of 45 kV and a current of 40 mA with copper radiation (1.54060 A). The powder sample was placed on a 96 well sample holder with Mylar fdm and loaded into the instrument. The sample was scanned over a range of about 3 to about 40 °20 with a step size of 0.0131303 °20 and 49 s per step.
Solid State NMR
[00213] Bruker-Biospin 400 MHz wide-bore spectrometer equipped with Bruker-Biospin 4 mm HFX probe was used. Samples were packed into 4 mm ZrCh rotors and spun under Magic Angle Spinning (MAS) conditions with spinning speed typically set to 12.5 kHz. The
proton relaxation time was measured using a 'H MAS Ti saturation recovery relaxation experiment in order to set up proper recycle delay of the 13C cross-polarization (CP) MAS experiments. The CP contact time of carbon CPMAS experiments was set to 2 ms. A CP proton pulse with linear ramp (from 50% to 100%) was employed. The carbon Hartmann- Hahn match was optimized on an external reference sample (glycine). All carbon spectra were recorded with proton decoupling using the TPPM15 decoupling sequence with a field strength of approximately 100 kHz.
Thermogravimetric Analysis
[00214] Thermal gravimetric analysis was performed using a TA5500 Discovery TGA instrument. A sample of approximately 1-10 mg was scanned from room temperature to 300 °C at a heating rate of 10 °C/min while purging with nitrogen.
Differential Scanning Calorimetry
[00215] Differential scanning calorimetry was performed using a TA5500 Discovery DSC instrument. A sample of approximately 1-10 mg was scanned from room temperature to 300 °C at a heating rate of 10 °C/min.
Examples
Abbreviations
Boc = Zc/V-butyloxycarbonyl
CDMT = 2-chloro-4,6-dimethoxy-l,3,5-triazine
DCM = dichloromethane
DIEA = A/f-diisopropylethylamine
DMAP = 4-dimethylaminopyridine
DME = dimethoxyethane
DMF = dimethylformamide
DMSO = dimethylsulfoxide
Dppf = 1,1’ -bi s(diphenylphosphino)ferrocene
EtOAc = ethyl acetate
EtOH = ethanol
ESI = electrospray ionization
HPLC = high-performance liquid chromatography
HPMCAS = hydroxypropylmethylcellulose acetate succinate
LC = liquid chromatography
MeOH = methanol
MeTHF = 2-methyltetrahydrofuran
MS = mass spectrometry
THF = tetrahydrofuran
UPLC = ultra-high performance liquid chromatography
Example 1: Synthesis of (U)-16-(2,6-dimethylphenyl)-7-isobutyl-6-(spiro[2.3]hexan-5- yl)-9-oxa-3-thia-2,6-diaza-l(2,4)-pyrimidina-4(l,3)-benzenacyclononaphan-5-one 3,3- dioxide (Compound I)
Part A: Preparation of 3-[[4-chloro-6-(2,6-dimethylphenyl)pyrimidin-2-

[00216] To a solution of 4,6-dichloropyrimidin-2-amine (300 g, 1.829 mol) in DCM (2.1 L) was added BOC2O (838 g, 3.840 mol) followed by DMAP (5.6 g, 45.84 mmol). The mixture was stirred at ambient temperature for 6 h. Additional DMAP (5.6 g, 45.84 mmol) was added and the reaction was continued to stir at ambient temperature for 24 h. The mixture was
diluted with water (2.1 L) and the organic phase separated. The organic phase was washed with water (2.1 L), 2.1 L of brine, dried over magnesium sulfate, filtered over Celite and concentrated in vacuo affording a light orange oil which had a silt in the slurry. The mixture was diluted with -500 mL of heptane and filtered using an M filter. The precipitate was washed with 250 mL of heptane. The filtrate was concentrated in vacuo affording a thick orange oil which was seeded with solid from a previous experiment and crystallized on standing, affording a light orange hard solid, tert-butyl A-tertebutoxycarbonyl-A-(4,6- tfichloropyrimidin-2-yl)carbamate (645 g, 97%). 'H NMR (400 MHz, DMSO-cL) 8 8.07 (s, 1H), 1.44 (s, 18H). ESI-MS m/z calc. 363.07526, found 364.1 (M+l)+; Retention time: 2.12 minutes (LC method A).
Step 2: tert-ButylN-tert-butoxycarbonyl-N-[4-chloro-6-(2,6- dimethylphenyl)pyrinudin-2-yl]carbamate.
[00217] All solvents were degassed prior to use. To a slurry of tert-butyl N-tert- butoxycarbonyl-A-(4,6-dichloropyrimidin-2-yl)carbamate (88 g, 241.6 mmol), (2,6- dimethylphenyl)boronic acid (approximately 36.24 g, 241.6 mmol), CS2CO3 (approximately 196.8 g, 604.0 mmol) in DME (704 mb), and water (176 mL) were added. Pd(dppf)Ch (approximately 8.839 g, 12.08 mmol) was added and the mixture was vigorously stirred under nitrogen at 80 °C (reflux) for 1 h (no starting material remained). The reaction was cooled to ambient temperature and diluted with water (704 mL). The aqueous phase was separated and extracted with EtOAc (704 mL). The organic phase was washed with 700 mL of brine, dried over magnesium sulfate, filtered and concentrated in vacuo. The crude product was chromatographed on a 1500 g silica gel column eluting with 0-30% EtOAc/hexanes. The product fractions (eluted at 15% EtOAc) were combined and concentrated in vacuo affording the product as a clear oil which crystallized on standing. / /7-butyl A-ter/-butoxycarbonyl-/V-[4-chloro-6-(2,6-dimethylphenyl)pyrimidin-2- yl]carbamate (81.3 g, 78%). 'H NMR (400 MHz, DMSO-efc) 8 7.88 (s, 1H), 7.30 (dd, J = 8.2, 7.0 Hz, 1H), 7.21 - 7.16 (m, 2H), 2.03 (s, 6H), 1.38 (s, 18H). ESI-MS m/z calc. 433.17682, found 434.1 (M+l)+; Retention time: 2.32 minutes (LC method A).

[00218] terZ-Butyl A-ter/-butoxycarbonyl-A-[4-chloro-6-(2,6-dimethyl phenyl) pyrimidineyl] carbamate (514.8 g, 915.9 mmol) was dissolved in di chloromethane (4 L). Hydrogen chloride in 1,4-di oxane (1 L, 4 mol) was added and the mixture was stirred overnight at room temperature. The resulting precipitate was collected by vacuum filtration and dried in vacuo to obtain 4-chloro-6-(2,6-dimethylphenyl)pyrimidin-2-amine hydrochloride (213.5 g, 64%) as a white solid (213.5 g, 82%). 'HNMR (250 MHz, DMSO-tL) 5 7.45-6.91 (m, 3H), 6.73 (s, 1H), 2.08 (s, 6H). ESI-MS m/z calc. 233.072, found 234.1 (M+l)+; Retention time: 2.1 minutes (LC Method C).

[00219] 4 -Chloro-6-(2,6-dimethylphenyl)pyrimidin-2-amine (hydrochloride salt) (166 g, 614.5 mmol) and 4-chloro-6-(2,6-dimethylphenyl)pyrimidin-2-amine (hydrochloride salt) (30 g, 111.0 mmol) were suspended in DCM (2.5 L), treated with NaOH (725 mL of 1 M, 725.0 mmol) and stirred at room temperature for 1 hour. The mixture was transferred into a separatory funnel and left standing over night. The DCM phase was separated and the aqueous phase with insoluble material was extracted twice more with DCM (2 x 500 mL). The combined brown DCM phases were stirred over magnesium sulfate and charcoal for 1 hour, filtered, and the yellow solution was concentrated to a volume of - 500 mL. The solution was diluted with heptane (750 mL) and DCM was removed under reduced pressure at 60 °C to give a cream suspension. It was stirred at room temperature for 1 hour, filtered, washed with cold heptane and dried to give 4-chloro-6-(2,6-dimethylphenyl)pyrimidin-2- amine (157 g, 91%) as a cream solid. 'H NMR (400 MHz, DMSO-tL) 5 7.28 - 7.14 (m, 3H), 7.10 (d, J = 7.5 Hz, 2H), 6.63 (s, 1H), 2.06 (s, 6H). ESI-MS m/z calc. 233.07198, found 234.0 (M+l)+; Retention time: 1.45 minutes (LC method A).
[00220] 4 -Chloro-6-(2,6-dimethylphenyl)pyrimidin-2-amine (235 g, 985.5 mmol) was dissolved in MeTHF (2.3 L) and cooled in an ice bath under stirring and nitrogen. To the cold solution methyl 3 -chlorosulfonylbenzoate (347 g, 1.479 mol) was added in one portion (seemed slightly endothermic) and to the cold pale-yellow solution a solution of 2-methyl- butan-2-ol lithium salt (in heptane, 875 mL of 3.1 M, 2.712 mol) was added dropwise over 1.25 h (exothermic, internal temperature from 0 to 10 °C). The ice bath was removed, and the greenish solution was stirred for 4 h at room temperature. To the greenish solution cold HC1 (2 L of 1.5 M, 3.000 mol) was added, the phases separated, and the organic phase was washed once with water (I L) and once with brine (500 mL). The aqueous phases were back extracted once with MeTHF (350 mL), and the organic phases were combined. This yellow MeTHF solution of methyl 3-[[4-chloro-6-(2,6-dimethylphenyl)pyrimidin-2- yl]sulfamoyl]benzoate (ESLMS m/z calc. 431.07065, found 432.0 (M+l)+; Retention time: 1.81 minutes) was treated with NaOH (2.3 L of 2 M, 4.600 mol), and stirred at room temperature for 1 h. The phases were separated, and the NaOH phase was washed twice with MeTHF (2 x 500 mL) and the combined organic phases were extracted once with 2 M NaOH (1 x 250 mL). The combined NaOH phases were combined, stirred in an ice bath, and slowly acidified by addition of aqueous HC1 (416 mL of 36 % w/w, 4.929 mol), while keeping the internal temperature between 10 and 20 °C. At the end of the addition (pH ~5-6) the final pH was adjusted to 2-3 by addition of solid citric acid. The formed yellow tacky suspension was stirred at room temperature overnight to give a cream crisp suspension. The solid was collected by filtration, washed with plenty of water and sucked dry for 3 h. The solid was dried under reduced pressure with a nitrogen leak at 45-50 °C for 120 h. 3-[[4-chloro-6-(2,6- dimethylphenyl)pyrimidin-2-yl]sulfamoyl]benzoic acid (395 g, 96%) was isolated as an off- white solid. 'HNMR (400 MHz, DMSO-cL) 5 13.44 (s, 1H), 12.46 (s, 1H), 8.48 - 8.39 (m, 1H), 8.25 - 8.15 (m, 1H), 8.15 - 8.08 (m, 1H), 7.68 (t, J = 7.8 Hz, 1H), 7.31 (s, 1H), 7.28 - 7.18 (m, 1H), 7.10 (d, J = 7.6 Hz, 2H), 1.84 (s, 6H). ESLMS m/z calc. 417.055, found 418.0 (M+l)+; Retention time: 1.56 minutes. (LC method A).
Part B: Preparation of (2R)-2-Amino-3-(l-bicyclo[l.l.l]pentanyl)propan-l-ol

[00221] MeLi (solution in diethyl ether, 48 mL of 1.6 M, 76.800 mmol) was slowly added to a solution of l,l-dibromo-2,2-bis(chloromethyl)cyclopropane (10 g, 33.691 mmol) in diethyl ether (25 mL) during 1 h, while maintaining the internal reaction temperature between -50 and -60 °C. The reaction mixture was then warmed-up to 0 °C, and stirred at this temperature for 30 min. The reaction flask was attached to a bent tube equipped with vacuum adapter and a receiving flask. The system was placed under vacuum using a water vacuum aspirator and the reaction mixture was distilled into the receiving flask that was cooled with liquid nitrogen while maintaining the distillation flask at around 0 °C with a water-ice bath to provide a distillate solution containing about 3.5% of tricyclo[l .1.1 ,01,3]pentane (53 g, 83%) as a clear solution. JH NMR (400 MHz, CDCh) 8 1.96 (s, 6H) in diethyl ether. This solution was used directly in the next reaction without further purification.
Step 2: Methyl (2R)-2-(tert-butoxycarbonylanuno)-3-(3-iodo-l- bicyclo[l.1.1 ]pentanyl)propanoate
[00222] Methyl (25)-2-(fe/7-butoxycarbonylamino)-3-iodo-propanoate (3 g, 9.1149 mmol) was dissolved in a solution of tricyclofl.1.1.01, 3]pentane (in DCM, 53 g, 28.063 mmol) from
the previous experiment and then tri ethylborane (in hexanes, 1.1 mL of 1 M, 1.1000 mmol) was added. This reaction mixture was stirred 1 h at room temperature. The reaction mixture was concentrated under reduced pressure, and the resulting residue was purified by reverse phase chromatography on 50 g Cis RediSep Rf gold column using a 5-100% gradient of acetonitrile in pure water to provide methyl (2/ )-2-(Z /7-butoxycarbonyla ino)-3-(3-iodo- l - bicyclo[l .l .l]pentanyl)propanoate (1.64 g, 45%) clear oil that crystallized on standing. 'H NMR (400 MHz, CDCh) S 4.98 (d, J = 7.3 Hz, 1H), 4.37 - 4.23 (m, 1H), 3.74 (s, 3H), 2.31 - 2.21 (m, 6H), 2.19 - 2.08 (m, 1H), 1.88 (dd, J = 14.7, 7.6 Hz, 1H), 1.45 (s, 9H). ESI-MS m/z calc. 395.0594, found 296.0 (M-99)+; Retention time: 1.95 minutes, along with a sample of lower purity methyl (2J?)-2-(terLbutoxycarbonylamino)-3-(3-iodo-l-bicyclo[l.l.l]pentanyl) propanoate (1.63 g, 35%) yellow oil that crystallized on standing. 'HNMR (400 MHz, CDCh) 84.98 (d, J = 7.8 Hz, 1H), 4.36 - 4.25 (m, 1H), 3.74 (s, 3H), 2.31 - 2.19 (m, 6H), 2.16 - 2.10 (m, 1H), 1.88 (dd, J = 14.4, 7.6 Hz, 1H), 1.45 (s, 9H). ESI-MS m/z calc. 395.0594, found 296.0 (M-99)+; Retention time: 1.96 minutes; LC method I.
Step 3: Methyl (2R)-3-(l-bicyclo[l.l.l]pentanyl)-2-(tert-

[00223] Triethylborane (in hexanes) (0.5 mL of 1 M, 0.5000 mmol) was added dropwise to a solution of methyl (27?)-2-(tert-butoxycarbonylamino)-3-(3-iodo-l- bicyclo[l.l.l]pentanyl)propanoate (1.64 g, 4.1287 mmol), 2,6-lutidine (1.288 g, 1.4 mL, 12.020 mmol), and tris(trimethylsilyl)silane (3.0628 g, 3.8 mL, 12.317 mmol) in anhydrous THF (9 mL), and left stirring at room temperature over week-end. The reaction mixture was concentrated under reduced pressure and the resulting residue was purified on silica gel with a 40 g column using a 0-50% ethyl acetate in heptanes gradient to provide methyl (2/?)-3-( l - bicyclo[ 1.1.1] pentanyl)-2-(terLbutoxycarbonylamino)propanoate (766 mg, 65%) as a pale yellow powder. JHNMR (400 MHz, CDCh) 64.94 (d, J = 13 Hz, 1H), 4.35 - 4.25 (m, 1H), 3.73 (s, 3H), 2.45 (s, 1H), 2.04 - 1.91 (m, 1H), 1.83 - 1.69 (m, 7H), 1.45 (s, 9H). ESI-MS m/z calc. 269.16272, found 170.2 (M-99)+; Retention time: 1.91 minutes; LC method L.
Step 4: tert-Butyl N-[(lR)-l-(l-bicyclo[l.l.l]pentanylmethyl)-2-hydroxy- ethyl]carbamate

[00224] iBH4 (350 mg, 16.067 mmol) was added to a solution of methyl (2 ?)-3-(l- bicyclo[ l .l . l]pentanyl)-2-(/c/7-butoxycarbonylamino)propanoate (1.46 g, 5.1497 mmol) in THF (15 mL) maintained at 0 °C with an ice water bath. The reaction mixture was maintained at this temperature for 30 min and then left to warm up and stirred at room temperature for 2 h. The reaction was then cooled down to 0 °C and quenched by the addition of an aqueous saturated solution of ammonium chloride (15 mL). The biphasic mixture was stirred for 20 min at 0 °C. Then the layers were separated, and the aqueous layer was extracted with ethyl acetate (4 x 25 mL). The combined organic layers were washed with brine (150 mL), dried over magnesium sulfate, filtered, and concentrated under reduced pressure to afford crude and partially quenched material. A sample of this material terLbutyl /V-[(lA)-l-(l-bicyclo[l.l.l]pentanylmethyl)-2-hydroxy-ethyl]carbamate (100 mg, 7%) (white powder) was kept and set aside. ES MS m/z calc. 241.1678, found 264.2 (M+23)+;
Retention time: 1.73 minutes. The rest of the crude material was dissolved back in DCM (50 mL), and washed with 0.2 M aqueous HC1 (50 mL) until hydrogen gas stopped evolving. The aqueous phase was separated, and washed with DCM (50 mL). The organic phases were combined, dried with magnesium sulfate, filtered, and concentrated under reduced pressure to provide /c/ -butyl A-[(l/ )- l-( l-bicyclo[ l .1 , l]pentanyl methyl)-2-hydroxy-ethyl]carbamate (1.15 g, 88%) as a white solid. 1HNMR (400 MHz, CDCh) 5 4.55 (br. s., 1H), 3.75 - 3.58 (m, 2H), 3.56 - 3.47 (m, 1H), 2.59 - 2.34 (m, 2H), 1.78 - 1.70 (m, 6H), 1.65 (dd, J = 14.7, 4.9 Hz, 1H), 1.53 (dd, J = 14.7, 8.6 Hz, 1H), 1.46 (s, 9H). ESLMS m/z calc. 241.1678, found 264.2 (M+23)+;null (M-)+; Retention time: 1.73 minutes; LC method J.

[00225] /c77-Butyl A'-[( l / )- 1 -(1 -bicyclo[l .1.1 ]pentanylmethyl)-2-hydroxy-ethyl]carbamate (1.15 g, 4.4794 mmol) was added in small portions to a solution of HC1 (in dioxane, 40 mL of 4 M, 160.00 mmol) stirring at ambient temperature then left at this temperature for 1 h. The reaction mixture was concentrated under reduced pressure. The resulting residue was
triturated in anhydrous THF (20 mL), filtered, and washed with more THF (2 x 5 mL). The resulting solid was dried under high vacuum for 1 h to provide (27?)-2-amino-3-(l- bicyclo[l.l.l]pentanyl) propan- l-ol (hydrochloride salt) (730 mg, 87%) as a white powder. 'HNMR (400 MHz, DMSO-tfc) 57.91 (br. s., 3H), 5.31 (t, J = 5.1 Hz, 1H), 3.62 (dt, J = 11.7, 3.7 Hz, 1H), 3.39 (dt, J = 11.7, 6.0 Hz, 1H), 3.07 - 2.96 (m, 1H), 2.45 (s, 1H), 1.78 - 1.61 (m, 8H). ESI-MS m/z calc. 141.11537, found 142.2 (M+l)+; Retention time: 0.57 minutes; LC method I.
Part C: Perparation of (l?)-16-(2,6-dimethylphenyl)-7-isobutyl-6-(spiro[2.3]hexan-5-yl)- 9-oxa-3-thia-2,6-diaza-l(2,4)-pyrimidina-4(l,3)-benzenacyclononaphan-5-one 3,3- dioxide (Compound I)

Step 1 : 3-[[4-[(2R)-2-Atnino-3-(l-bicyclo[l.1.1 ]pentanyl)propoxy]-6-(2, 6- dimethylphenyl)pyrinudin-2-yl]sulfamoyl]benzoic acid

[00226] A flame-dried flask under nitrogen atmosphere was charged with (2J?)-2-amino-3- (l-bicyclo[l.l.l]pentanyl)propan-l-ol (hydrochloride salt) (505 mg, 2.7002 mmol), 3-[[4- chloro-6-(2,6-dimethylphenyl)pyrimidin-2-yl]sulfamoyl]benzoic acid (1.3 g, 3.1110 mmol), DMF (5.8 mL), and MeTHF (58 mL). This suspension was cooled down to 0 °C and sodium Zc/7-butoxide (1.5 g, 15.608 mmol) was added, and left stirring for 5 min at this temperature.
The reaction was then left to warm-up to ambient temperature and stirred for 30 min. The mixture was then cooled down to 0 °C, and diluted with MeTHF (50 mL), quenched with ice cold 1 M HC1 (100 mL) while stirring vigorously. The aqueous phase was separated and extracted with MeTHF (3 x 50 mL). The combined organic phases were dried with sodium sulfate, filtered, and concentrated under reduced pressure. The resulting residue was purified by reverse phase chromatography on 100 g Cis aq. RediSep Rf gold column using a 5-100% gradient of acetonitrile in acidic water (0.1% HC1 content) to provide 3-[[4-[(2A)-2-amino-3- (l-bicyclo[l.l.l]pentanyl)propoxy]-6-(2,6-dimethylphenyl)pyrimidin-2-yl]sulfamoyl]benzoic acid (hydrochloride salt) (1.254 g, 82%) as a white powder. 1 H NMR (400 MHz, DMSO-t/e) 8 13.07 (br. s, 1H), 8.46 (t, J = 1.7 Hz, 1H), 8.25 (br. s, 3H), 8.14 (dd, J = 14.1, 7.7 Hz, 2H), 7.71 (t, J = 7.7 Hz, 1H), 7.31 - 7.21 (m, 1H), 7.18 - 7.07 (m, 2H), 6.30 (br. s, 1H), 4.35 (dd, J = 11.7, 2.2 Hz, 1H), 4.16 (dd, J = 12.0, 6.6 Hz, 1H), 3.49 - 3.45 (m, 1H, overlap with water), 2.46 (s, 1H), 2.07 (s, 1H), 2.01 (br. s, 6H), 1.83 (d, J = 6.8 Hz, 2H), 1.75, 1.70 (ABq. JAB = 9.5 Hz, 6H). ESI-MS m/z calc. 522.19366, found 523.2 (M+l)+; Retention time: 2.48 minutes; LC method J.
Step 2: 3-[[4-[(2R)-3-(l-Bicyclo[l.l.l]pentanyl)-2-[(6-tert-butylfuro[2,3-b]pyrazin-

[00227] A 20 mL vial was charged under nitrogen with 3-[[4-[(2A)-2-amino-3-(l- bicyclo[l.l.l]pentanyl)propoxy]-6-(2,6-dimethylphenyl)pyrimidin-2-yl]sulfamoyl]benzoic acid (hydrochloride salt) (83 mg, 0.1476 mmol), 6-fert-butylfuro[2,3-b]pyrazine-2- carbaldehyde (31.6 mg, 0.1547 mmol), anhydrous DCM (3.0 mL), acetic acid (20 pL, 0.3517 mmol), and DIEA (95 pL, 0.5454 mmol). The mixture was cooled down in an ice bath, sodium tri acetoxyb orohydri de (110 mg, 0.5190 mmol) was added, and the reaction was stirred for 4 h while allowing to warm to ambient temperature. The reaction was quenched with aqueous 1 M HC1 (1 mL), MeOH (0.5 mL) and DMSO (0.5 mL), filtered and purified by reverse phase HPLC (1-99% acetonitrile/5 mM aqueous HC1 over 30 min) to give as a white solid 3-[[4-[(2J?)-3-(l-bicyclo[l.l. l]pentanyl)-2-[(6-terLbutylfuro[2,3-b]pyrazin-2-
yl)methylamino]propoxy]-6-(2,6-dimethylphenyl)pyrimidin-2-yl]sulfamoyl]benzoic acid (hydrochloride salt) (68.3 mg, 62%). ESI-MS m/z calc. 710.28864, found 711.2 (M+l)+; Retention time: 1.61 minutes. LC method A.
Step 3: (11R)-11-(1 -Bicyclofl.1.1 ]pentanylmethyl)-l 2-[( 6-tert-butylfuro[2, 3- b]pyrazin-2-yl)methyl]-6-(2,6-dimethylphenyl)-2,2-dioxo-9-oxa-216-thia-3,5,12,19-

[00228] 3-[[4-[(2A)-3-(l-bicyclo[l.l.l]pentanyl)-2-[(6-tert-butylfuro[2,3-b]pyrazin-2- yl)methylamino]propoxy]-6-(2,6-dimethylphenyl)pyrimidin-2-yl]sulfamoyl]benzoic acid (hydrochloride salt) (67 mg, 0.08966 mmol) was combined under nitrogen with CDMT (43 mg, 0.2449 mmol), and DMF (3.0 mL). The solution was stirred at 0 °C. 4-Methyl- morpholine (65 pL, 0.5912 mmol) was added, and the mixture was stirred in the cooling bath that was allowed to warm to ambient temperature. After 16 h, the reaction was filtered, and purified by reverse phase HPLC (1-99% acetonitrile/5 mM aqueous HC1 over 30 min) to give as a white solid (HA)-l l-(l-bicyclo[l.l.l]pentanylmethyl)-12-[(6-ter/-butylfuro[2,3- b]pyrazin-2-yl)methyl]-6-(2,6-dimethylphenyl)-2,2-dioxo-9-oxa-2X6-thia-3,5,12,19- tetrazatricyclo[12.3.1.14,8]nonadeca-l(18),4(19),5,7,14,16-hexaen-13-one (33.6 mg, 54%). 'HNMR (400 MHz, Chloroform- ) 6 9.06 (s, 1H), 8.72 (t, J = 1.8 Hz, 1H), 8.41 (s, 1H), 8.14 (d, J = 7.9 Hz, 1H), 7.89 (dt, J = 7.7, 1.4 Hz, 1H), 7.67 (t, J = 7.8 Hz, 1H), 7.20 (t, J = 7.6 Hz, 1H), 7.04 (d, J = 7.6 Hz, 2H), 6.61 (s, 1H), 6.20 (s, 1H), 5.40 (dd, J = 11.4, 4.0 Hz, 1H), 5.36 - 5.29 (m, 1H), 4.29 - 4.18 (m, 2H), 4.09 - 4.00 (m, 1H), 2.45 (s, 1H), 2.09 - 1.96 (m, 7H), 1.81 (dd, J = 15.5, 3.3 Hz, 1H), 1.62 - 1.56 (m, 6H), 1.42 (s, 9H). ESI-MS m/z calc. 692.2781, found 693.2 (M+l)+; Retention time: 2.3 minutes. LC method A.
Example 2: Compound I Neat Form A
1. Synthetic Procedure
[00229] 3.5 g of amorphous Compound I was loaded into a 100 mL reactor (Mettler Toledo Easy Max 102), followed by 50 mL of EtOH. The temperature was increased as fast as possible to 73 °C and held at 73 °C for 1.5 h.
[00230] Once the material was dissolved, the temperature was decreased to 20 °C over 5.083 h. The resulting crystals of Compound I EtOH Solvate Form B were fdtered under nitrogen flow. The isolated material was placed in a vacuum oven for 3 d under nitrogen sweep at 50 °C, resulting in the formation of Compound I Neat form A. The crystal phase was confirmed using XRPD analysis.
2. X-Ray Powder Diffraction
[00231] X-ray powder diffraction (XRPD) results are shown in FIG. 1 and the table below.
Table 1. XRPD Signals for Compound I Neat Form A

3. Thermogravimetric Analysis
[00232] The thermogram (FIG. 2) showed 0.04% weight loss from 30 °C to 200 °C.
4. Differential Scanning Calorimetry Analysis
[00233] The thermogram (FIG. 3) showed an endothermic peak around 198 °C with a heat of fusion of 17 J/g.
5. Solid State NMR
[00234] The 13C CPMAS spectrum of Compound I Neat Form A is shown in FIG. 4 and in the table below.
Table 2. 13C CPMAS of Compound I Neat Form A

6. Single Crystal Elucidation
[00235] A single crystal of Compound I Neat Form A was obtained via desolvation of Compound I EtOH solvate. X-ray diffraction data were acquired at 100 K on a Rigaku diffractometer equipped with Cu Ka radiation ( = 1.54178 A) and an HP AD detector. The structure was solved and refined using SHELX programs (Sheldrick, G.M., Acta Cryst., (2008) A64, 112-122) and results are summarized in the table below.
Table 3. Single Crystal Elucidation of Compound I Neat Form A
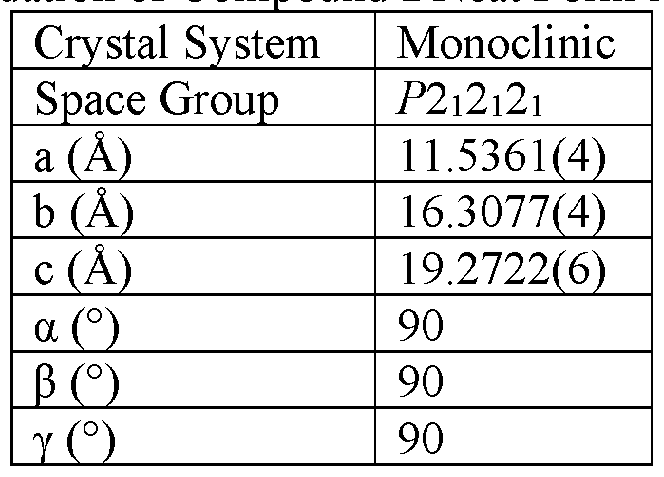

Example 3: Compound I Neat Form B
1. Synthetic Procedure
[00236] Compound I Neat Form B was prepared by exposing Compound I Hydrate Form A to 3% humidity for 3 h using an Anton Paar CHC+ chamber (humidity XRD).
[00237] In another procedure, Compound I Neat Form B was prepared by placing Compound I Hydrate Form A in an ssNMR rotor into an oven at 125 °C for 3 h.
2. X-Ray Powder Diffraction
[00238] X-ray powder diffraction (XRPD) spectrum of Compound I Neat Form B is shown in FIG. 5 and in the table below.
Table 4. XRPD Signals for Compound Neat Form B
 3. Solid State NMR
3. Solid State NMR
[00239] The 13C CPMAS spectrum of Compound I Neat Form B is shown in FIG. 6 and in the table below.
Table 5. 13C CPMAS of Compound I Neat Form B

Example 4: Compound I Neat Form C
1. Synthetic Procedure
[00240] Approximately 60 mg of Compound I was added to a 2 mL HPLC vial, followed by the addition of 250 uL of hexadecane. The vial was left to slurry at 80 °C for 19 d. The slurry was then centrifuged and dried under reduced pressure overnight at 50 °C.
2. X-Ray Powder Diffraction
[00241] X-ray powder diffraction (XRPD) results are shown in FIG. 7 and the table below.
T able 6. XRPD Signals for Compound I Neat Form C
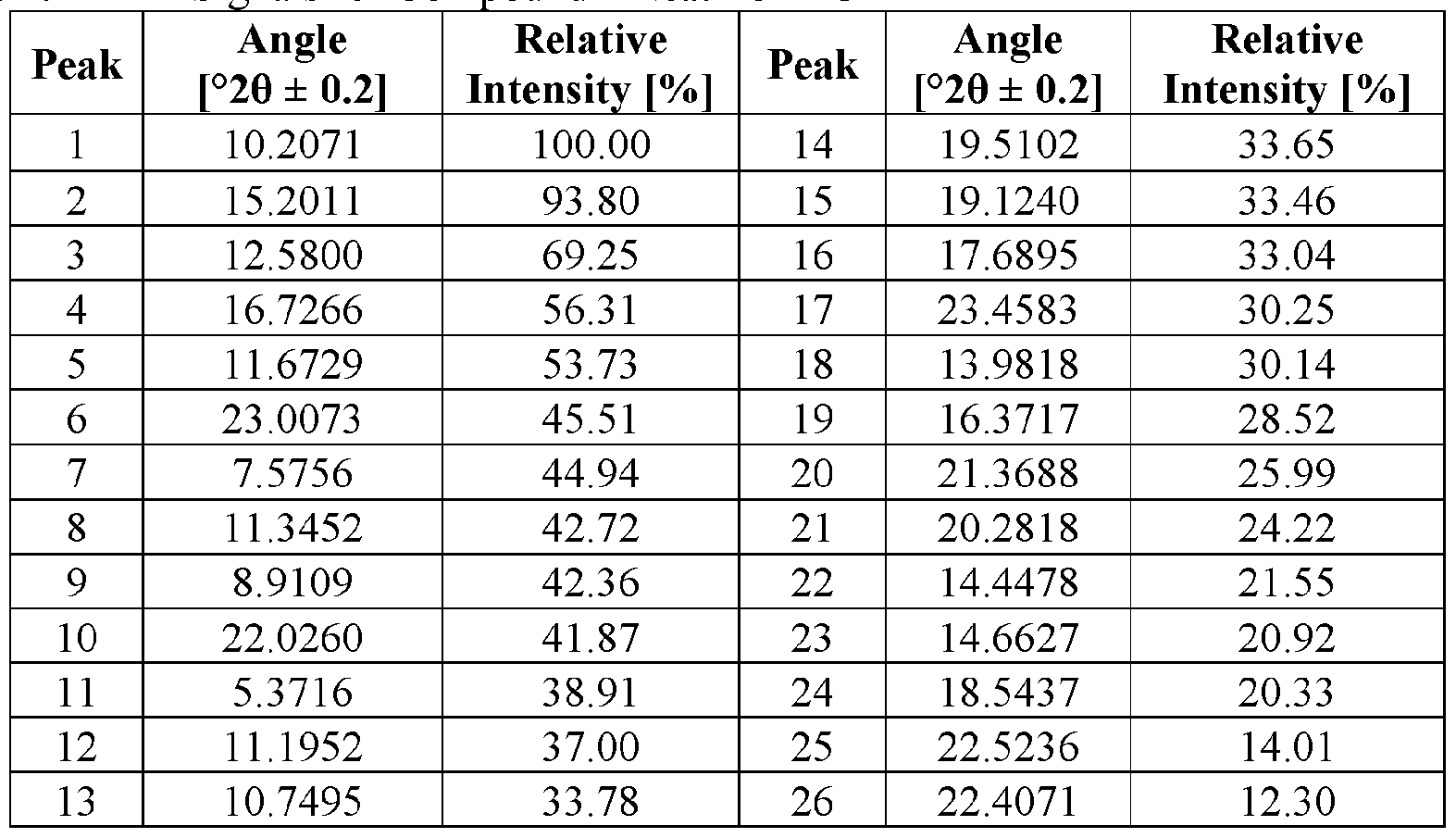
3. Thermogravimetric Analysis
[00242] The thermogram (FIG. 8) showed 0.05% weight loss from 30 °C to 40 °C and
0.16% weight loss from 40 °C to 240 °C.
4. Differential Scanning Calorimetry Analysis
[00243] The thermogram (FIG. 9) showed an endothermic peak around 237 °C with a heat of fusion of 42 J/g.
5. Solid State NMR
[00244] The 13C CPMAS spectrum of Compound I Neat Form C is shown in FIG. 10 and in the table below.
Table 7. 13C CPMAS of Compound I Neat Form C

Example 5: Compound I Neat Form D
1. Synthetic Procedure
[00245] Compound I Neat Form D was prepared by vacuum drying Compound I Hydrate Form C at 80 °C overnight.
[00246] In a separate procedure, Compound I Neat Form D was prepared by slurrying 150 mg of Compound I Neat Form A in 3 mL of IPA overnight. After fdtration, the solids were placed in an oven at 125 °C for 3 h.
3. Solid State NMR
[00247] The 13C CPMAS spectrum of Compound I Neat Form D is shown in FIG. 11 and in the table below.
Table 8, l3C CPMAS of Compound I Neat Form D

Example 6: Compound I Neat Form E
1. Synthetic Procedure
[00248] 100 mg of Compound I Neat Form A was added to a HPLC vial, followed by the addition of 0.5 mh of MeOAc. The mixture was stirred with a magnetic stir bar for 4 d. Then, the slurry was centrifuged, and the resulting solid was dried under vacuum at 50 °C, yielding Compound I Neat Form E.
2. X-Ray Powder Diffraction
[00249] X-ray powder diffraction (XRPD) results are shown in FIG. 12 and the table below.
Table 9. XRPD Signals for Compound I Neat Form E
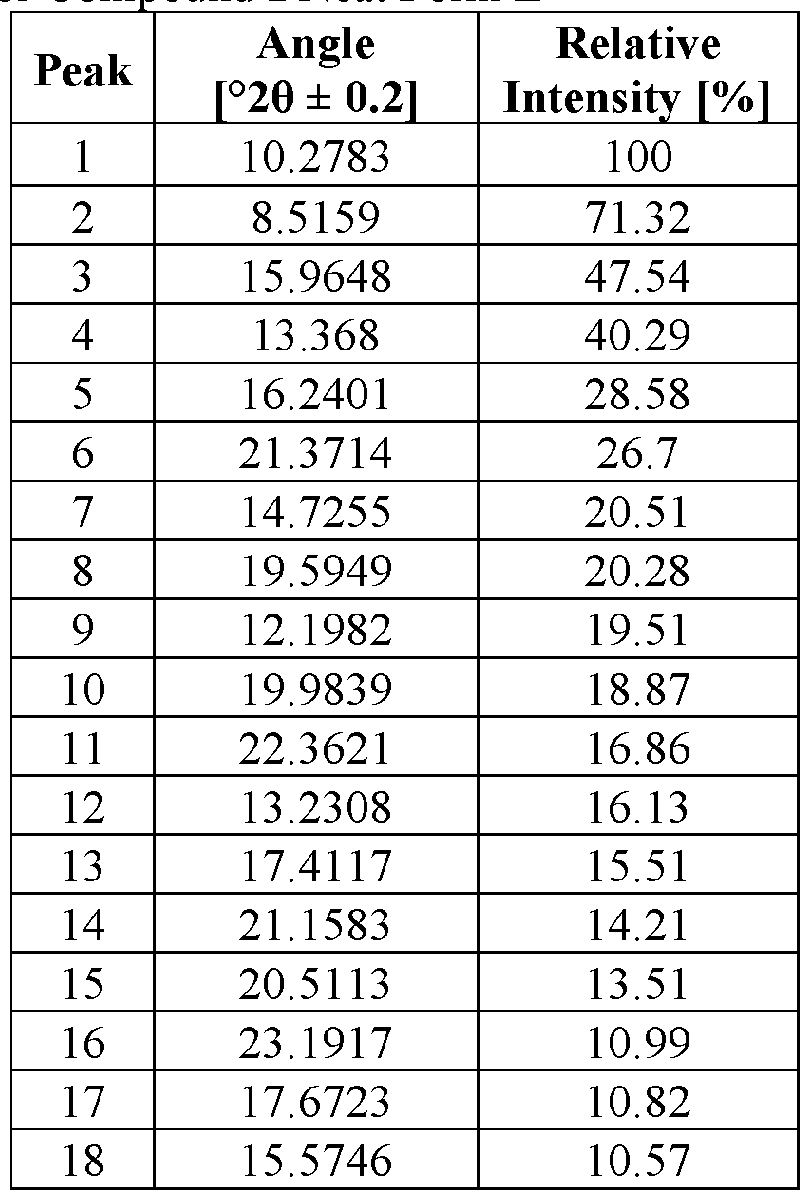
3. Thermogravimetric Analysis
[00250] The thermogram (FIG. 13) showed 0.08% weight loss from 30 °C to 120 °C.
4. Differential Scanning Calorimetry Analysis
[00251] The thermogram (FIG. 14) showed an endothermic peak around 212 °C with a heat of fusion of 30 J/g.
5. Solid State NMR
[00252] The 13C CPMAS spectrum of Compound I Neat Form E is shown in FIG. 15 and in the table below.
Table 10. 13C CPMAS of Compound I Neat Form E


6. Single Crystal Elucidation
[00253] A single crystal of Compound I Neat Form E was grown by dissolving Compound I Neat Form A in methyl acetate, and crystallizing with addition of heptane. Once isolated, this material was vacuum dried at 50 °C for 2 days. X-ray diffraction data were acquired at 100 K on a Rigaku diffractometer equipped with Cu I<« radiation (X=l .54178 A) and a HP AD detector. The structure was solved and refined using SHELX programs (Sheldrick, GM , Acta Cryst., (2008) A64, 112-122) and results are summarized in the table below.
Table 11. Single Crystal Elucidation of Compound I Neat Form E
 Example 7: Compound I Compressed Form E
Example 7: Compound I Compressed Form E
1. Synthetic Procedure
[00254] 100 mg of Compound I Neat Form E was compressed using a Material Transfer System (MTS) and a 5 mm round flat face Natoli #99307. The MTS was set up with tooling. The powder was manually weighed and fdled into a die. The compression happened at 100 mm/min and reached the target force of 21 6 kN and held for the dwell time of 10 s. The compacts were compressed 10 times, where after each compression, the compact was removed and broken up with a spatula. After being broken back up the same material was then put back into the die to compress again until it was completed for a total of 10 compressions to afford Compound I Compressed Form E.
2. X-Ray Powder Diffraction
[00255] X-ray powder diffraction (XRPD) results are shown in FIG. 16 and the table below.

5. Solid State NMR
[00256] The 13C CPMAS spectrum of Compound I Compressed Form E is shown in FIG.
17 and in the table below.
Table 13. 13C CPMAS of Compound I Compressed Form E


Example 8: Compound I Compressed Form A
1. Synthetic Procedure
[00257] 100 mg of Compound I Neat Form A was compressed using the MTS and a 5 mm round flat face Natoli #99307. The MTS was set up with tooling. The powder was manually weighed and filled into a die. The compression happened at 100 mm/min and reached the target force of 21.6 kN and held for the dwell time of 10 s. The compacts were compressed 10 times, where after each compression, the compact was removed and broken up with a spatula. After being broken back up the same material was then put back into the die to compress again until it was completed for a total of 10 compressions to afford Compound I Compressed Form A.
2. X-Ray Powder Diffraction
[00258] X-ray powder diffraction (XRPD) results are shown in FIG. 18 and the table below.
Table 14, XRPD Signals for Compound I Compressed Form A
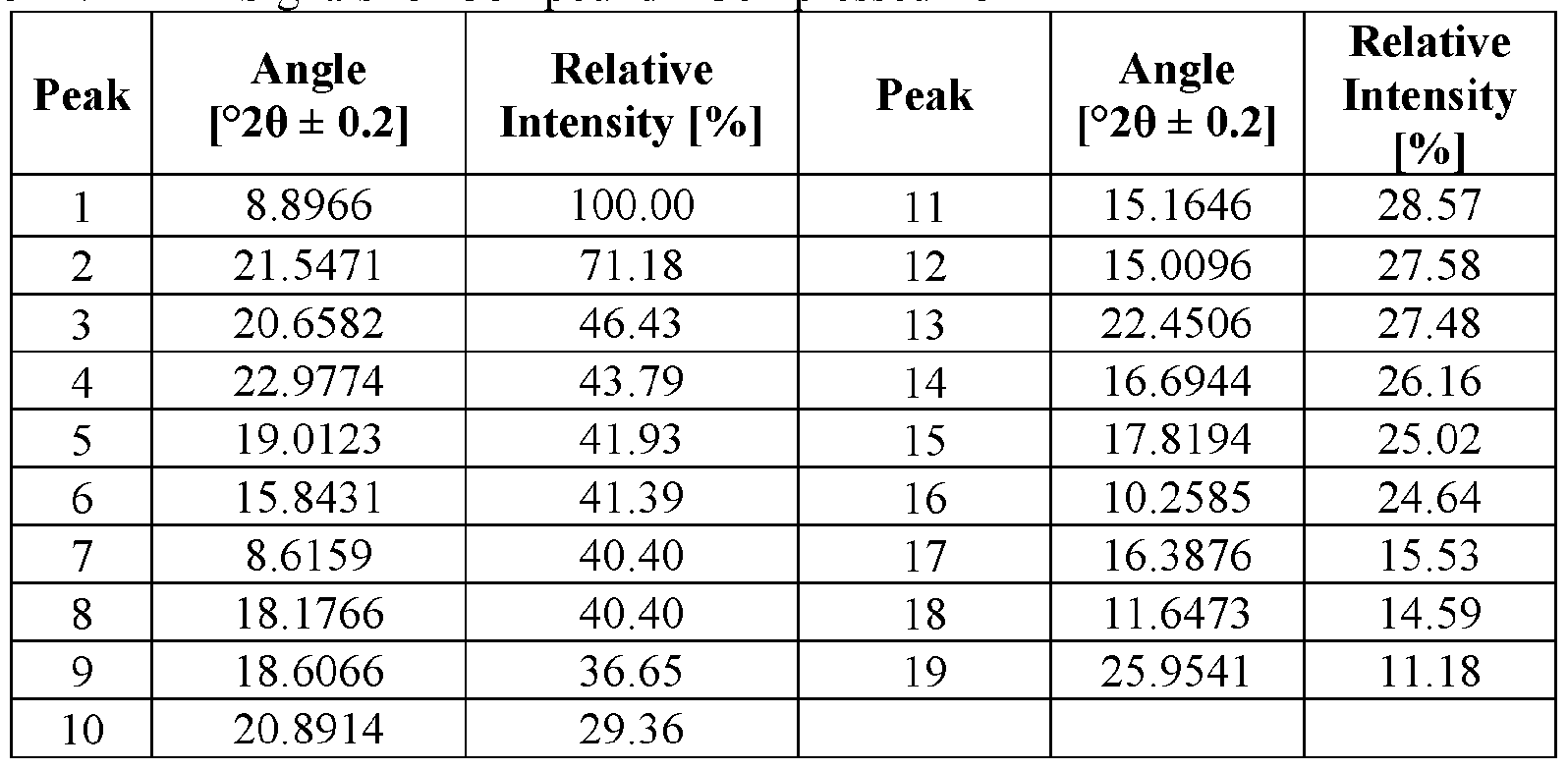 5. Solid State NMR
5. Solid State NMR
[00259] The 13C CPMAS spectrum of Compound I Compressed Form A is shown in FIG.
19 and in the table below.
Table 15. 13C CPMAS of Compound I Compressed form A

Example 9: Compound I EtOH Solvate Form B
1. Synthetic Procedure
[00260] 1000 mg of Compound I Neat Form A was added to a 20 mL scintillation vial, followed by the addition of 10 mL of EtOH. The contents of the vial were stirred with a magnetic stir bar at room temperature for 5 d. The slurry was centrifuged to yield Compound I EtOH Solvate Form B.
2. X-Ray Powder Diffraction
[00261] X-ray powder diffraction (XRPD) results are shown in FIG. 20 and the table below.
Table 16. XRPD Signals for Compound I EtOH Solvate Form B

3. Thermogravimetric Analysis
[00262] The thermogram (FIG. 21) showed 5.29% weight loss from 30 °C to 90 °C.
4. Differential Scanning Calorimetry Analysis
[00263] The thermogram (FIG. 22) showed an endothermic peak around 109 °C with a heat of fusion of 43 J/g, followed by a second endothermic peak around 197 °C with a heat of fusion of 13 J/g.
5. Solid State NMR
[00264] The 13C CPMAS spectrum of Compound I EtOH Solvate Form B is shown in FIG.
23 and in the table below.
Table 17. 13C CPMAS of Compound I EtOH Solvate Form B

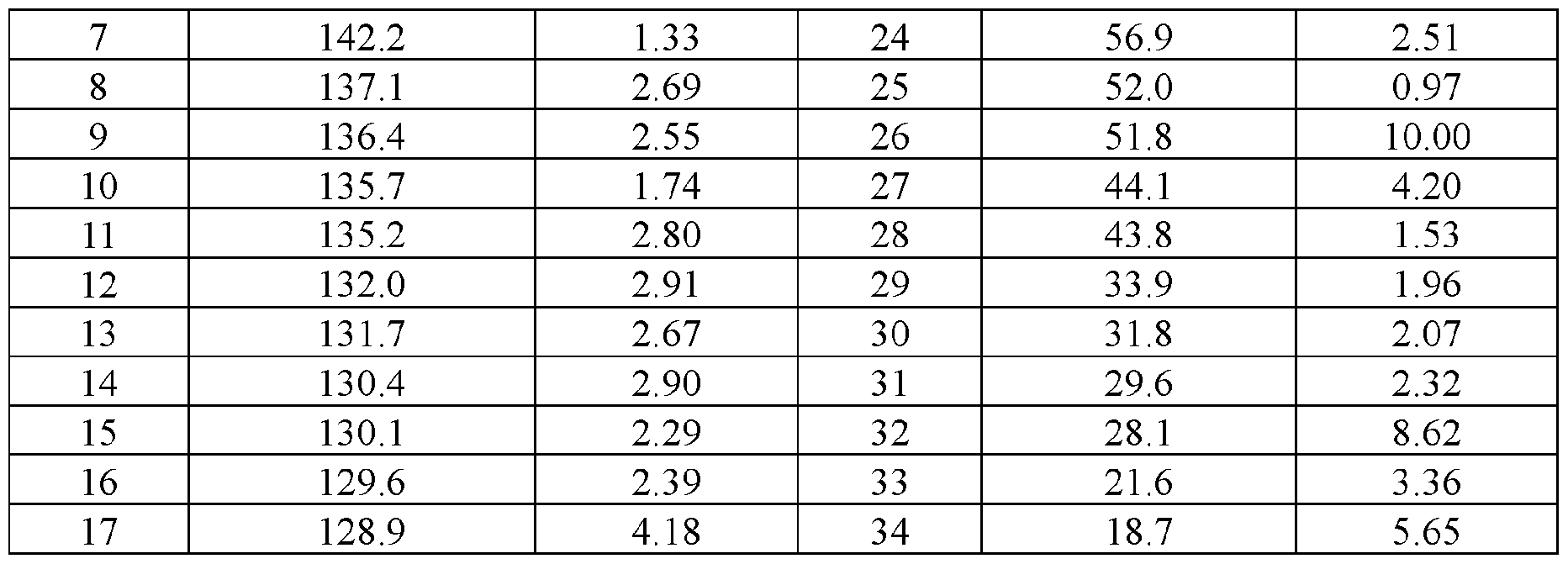
6. Single Crystal Elucidation
[00265] A single crystal of Compound I EtOH Solvate Form B was obtained from EtOH. X-ray diffraction data were acquired at 100 K on a Bruker diffractometer equipped with Cu Kot radiation (X=l .54178 A) and a CP AD detector. The structure was solved and refined using SHELX programs (Sheldrick, G.M., Acta Cryst., (2008) A64, 112-122) and results are summarized in the table below.
Table 18. Single Crystal Elucidation of Compound I EtQEI Solvate Form B

Example 10: Compound I MeOH Solvate
1. Synthetic Procedure
[00266] A reaction vessel was loaded with 600 mg of Compound I Neat Form A. 30 mL of MeOH was added to the reactor and the temperature was increased to 62 °C as fast as possible. The vessel was cooled to 10 °C over 5 h. The slurry was then centrifuged and Compound I MeOH solvate was isolated as a wet cake.
2. X-Ray Powder Diffraction
[00267] X-ray powder diffraction (XRPD) results are shown in FIG. 24 and the table below.
Table 19. XRPD Signals for Compound I MeOH Solvate

3. Solid State NMR
[00268] The 13C CPMAS spectrum of Compound I MeOH solvate is shown in FIG. 25 and in the table below.
Table 20. 13C CPMAS of Compound I MeOH Solvate
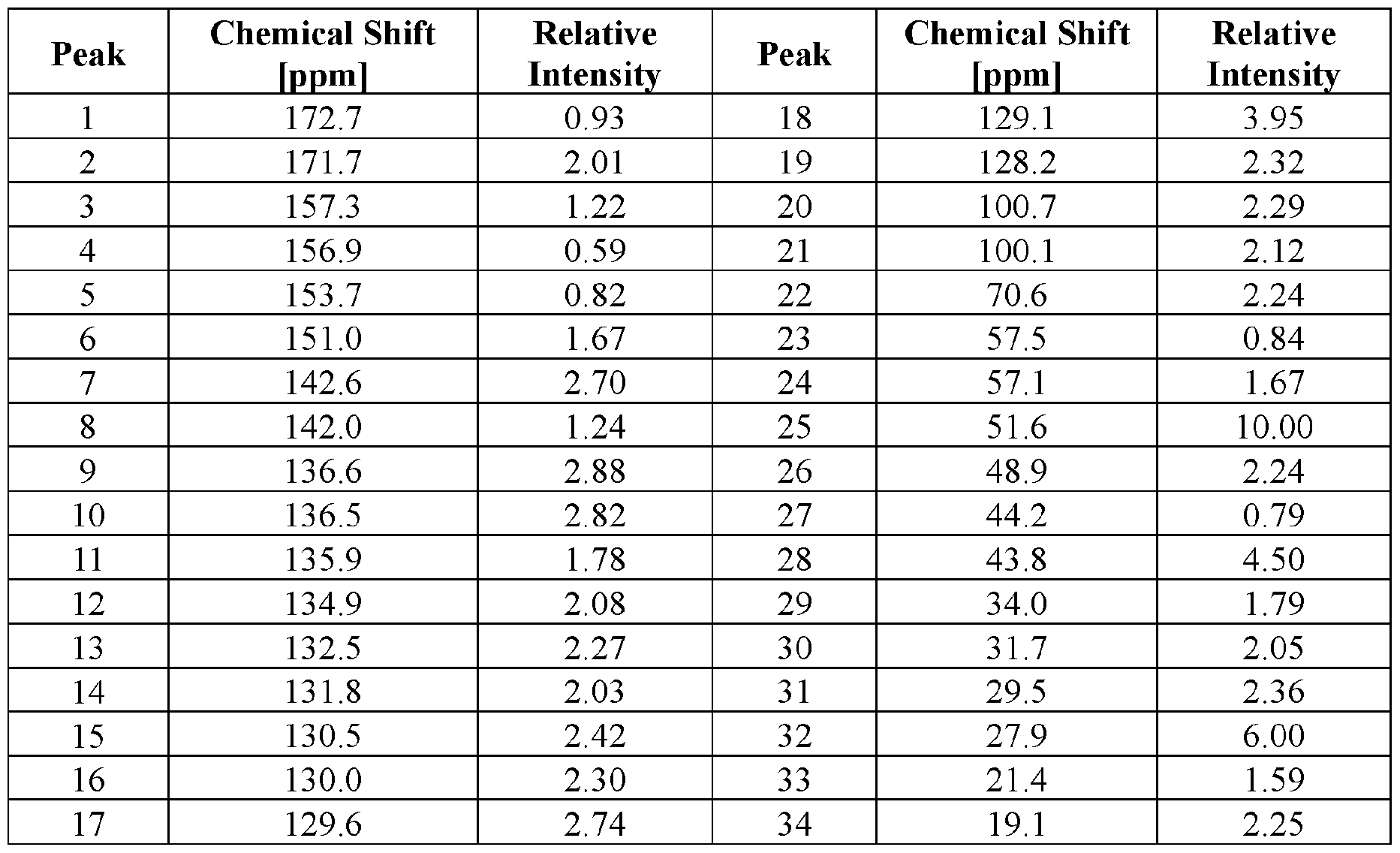
4. Single Crystal Elucidation
[00269] A single crystal of Compound I MeOH solvate was obtained from MeOH. X-ray diffraction data were acquired at 100 K on a Bruker diffractometer equipped with Cu Ka radiation (X=l.54178 A) and a CP AD detector. The structure was solved and refined using SHELX programs (Sheldrick, G.M., Acta Cryst., (2008) A64, 112-122) and results are summarized in the table below.
Table 21. Single Crystal Elucidation of Compound I MeOH Solvate
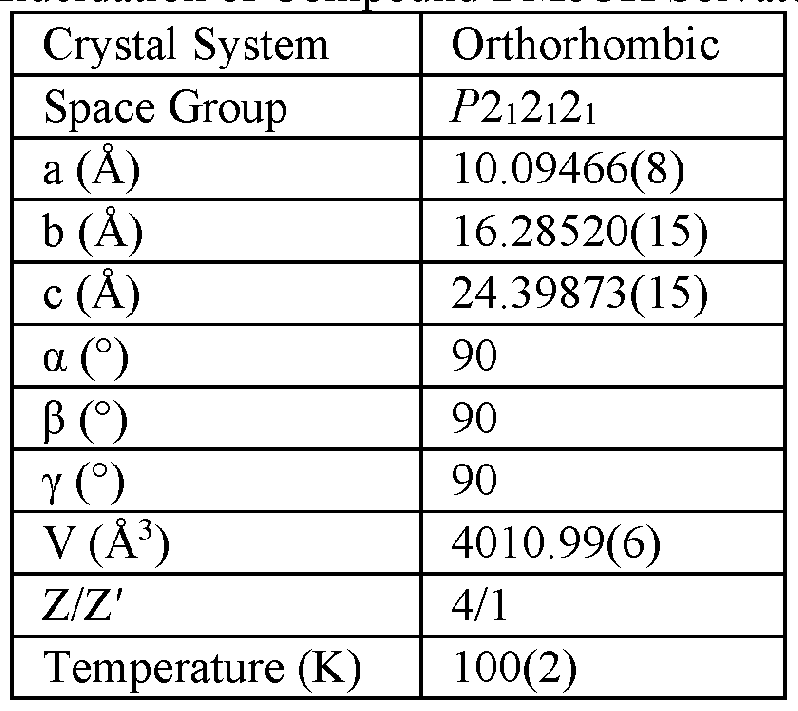 Example 11: Compound I NPA Solvate
Example 11: Compound I NPA Solvate
1. Synthetic Procedure
[00270] Approximately 50 mg of Compound I Neat Form A was added to an HPLC vial, followed by 1 mb of NPA and a magnetic stir bar. The vial was allowed to stir at rt for 2 d. The solids were isolated by centrifugation to yield Compound I NPA solvate.
2. X-Ray Powder Diffraction
[00271] X-ray powder diffraction (XRPD) results are shown in FIG. 26 and the table below.
Table 22. XRPD Signals for Compound I NPA Solvate

3. Solid State NMR
[00272] The 13C CPMAS spectrum of Compound I NPA solvate is shown in FIG. 27 and in the table below.
Table 23. 13C CPMAS of Compound I NPA Solvate

Example 12: Compound I MeOAc Solvate Form A
1. Synthetic Procedure
[00273] 1000 mg of Compound I Neat Form A was weighed in an HPLC vial, followed by the addition of 0.5 mL of MeOAc. After stirring for 3 d at rt, the slurry was filtered, and the resulting Compound I MeOAc Solvate Form A was isolated as a wet cake.
2. X-Ray Powder Diffraction
[00274] X-ray powder diffraction (XRPD) results are shown in FIG. 28 and the table below.
Table 24. XRPD Signals for Compound I MeOAc Solvate Form A

3. Thermogravimetric Analysis
[00275] The thermogram (FIG. 29) showed 1.68% weight loss from 30 °C to 120 °C.
4. Differential Scanning Calorimetry Analysis
[00276] The thermogram (FIG. 30) showed an endothermic peak around 100 °C with a heat of fusion of 91 J/g, followed by a second endothermic peak around 216 °C with a heat of fusion of 33 J/g.
5. Solid State NMR
[00277] The 13C CPMAS spectrum of Compound I MeOAc Solvate Form A is shown in FIG. 31 and in the table below.
Table 25. 13C CPMAS of Compound I MeOAc Solvate Form A

Example 13: Compound I MeOAc Solvate Form B
1. Synthetic Procedure
[00278] 1000 mg of Compound I Neat Form A was weighed in an HPLC vial, followed by the addition of 0.5 mL of MeOAc. After stirring for 5 d at 5 °C, the slurry was centrifuged, and the solid was vacuum dried overnight at rt.
2. X-Ray Powder Diffraction
[00279] X-ray powder diffraction (XRPD) results are shown in FIG. 32 and the table below.
Table 26. XRPD Signals for Compound I MeOAc Solvate Form B
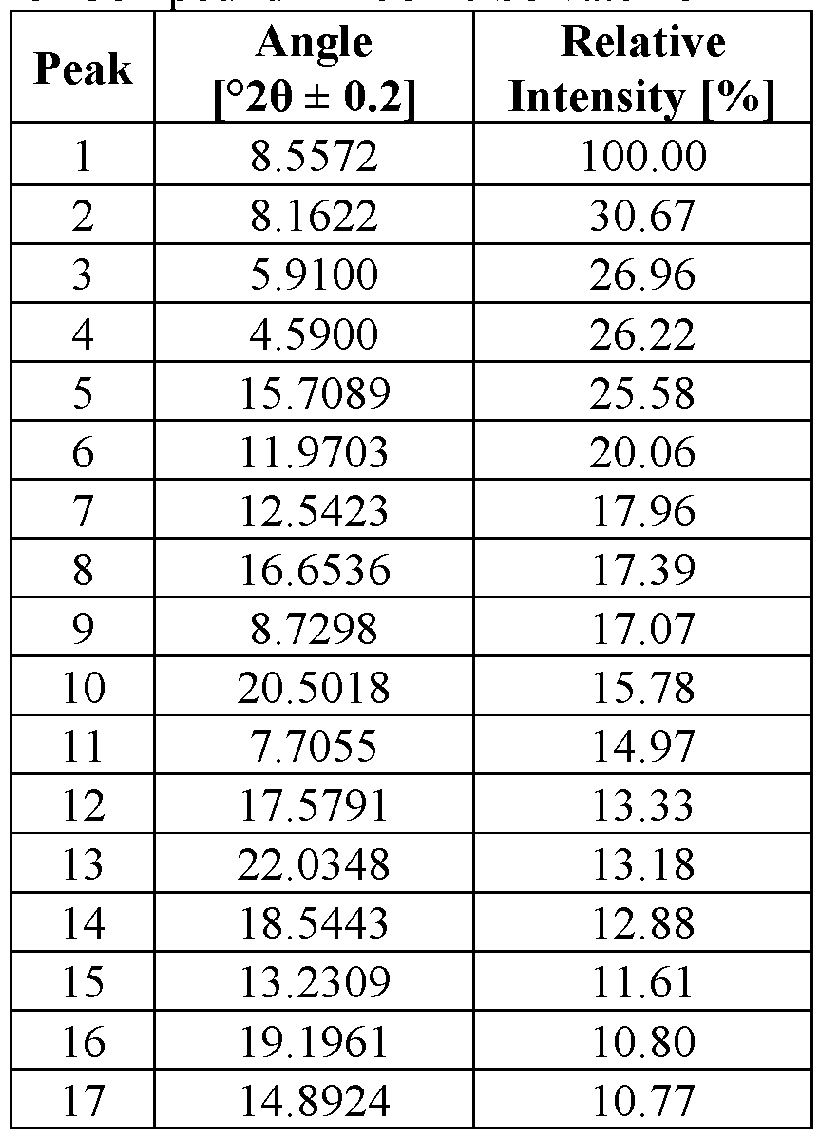
3. Thermogravimetric Analysis
[00280] The thermogram (FIG. 33) showed 0.39% weight loss from 30 °C to 60 °C, and 0.20% weight loss from 60 °C to 190 °C.
4. Differential Scanning Calorimetry Analysis
[00281] The thermogram (FIG. 34) showed an endothermic peak around 51 °C with a heat of fusion of 8 J/g, followed by a second endothermic peak around 187 °C with a heat of fusion of 10.5 J/g.
5. Solid State NMR
[00282] The 13C CPMAS spectrum of Compound I MeOAc Solvate Form B is shown in
FIG. 35 and in the table below.
Table 27. 13C CPMAS of Compound I MeOAc Solvate Form B

Example 14: Compound I MeOAc Solvate Form C
1. Synthetic Procedure
[00283] 1000 mg of Compound I Neat Form A was weighed in an HPLC vial, followed by the addition of 0.5 mL of MeOAc. After stirring for 6 d at 5 °C, the slurry was filtered as a wet cake, yielding Compound I MeOAc Solvate Form C.
2. X-Ray Powder Diffraction
[00284] X-ray powder diffraction (XRPD) results are shown in FIG. 36 and the table below.
Tab e 28, XRPD Signals for Compound I MeOAc Solvate Form C
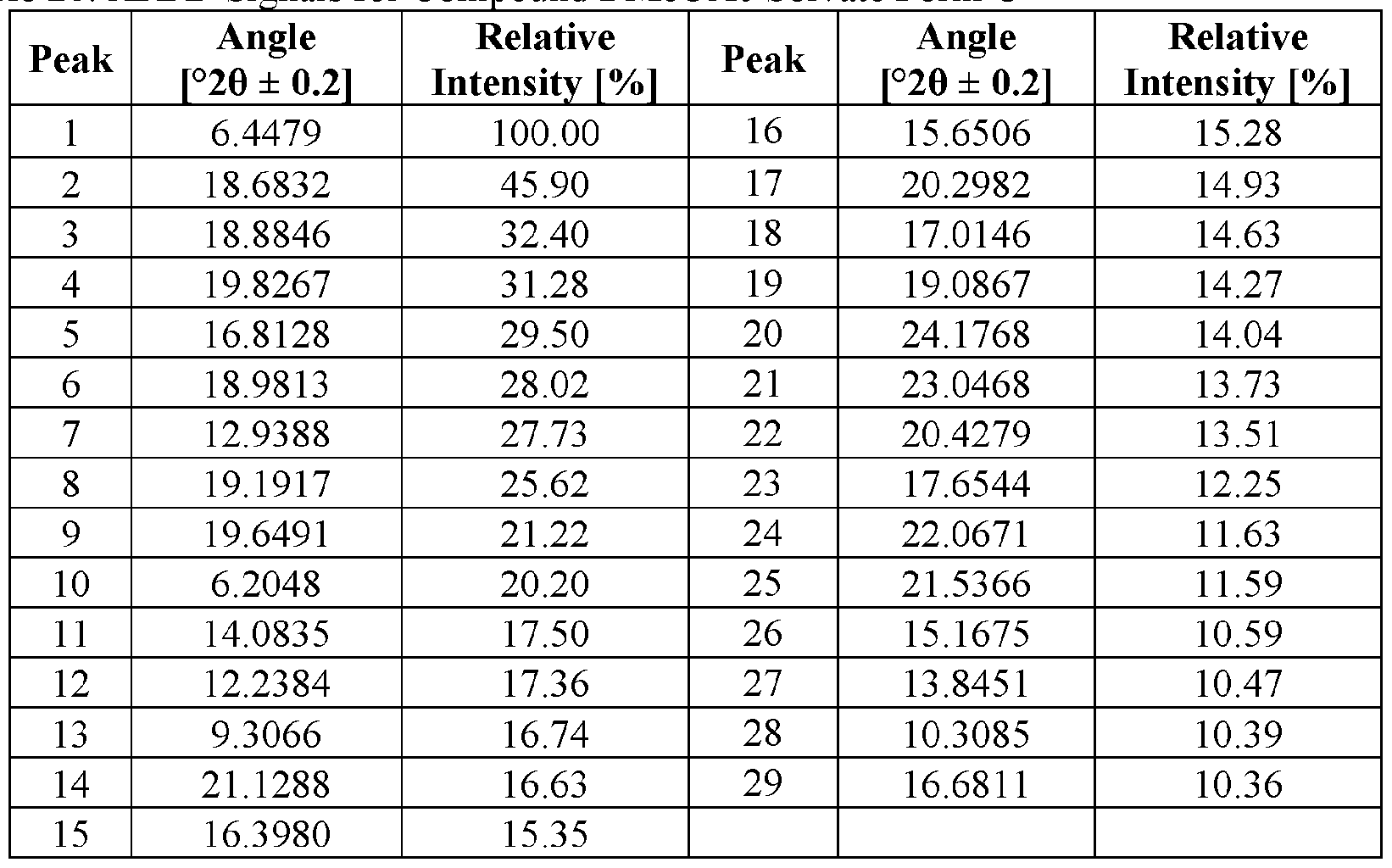
3. Solid State NMR
[00285] The 13C CPMAS spectrum of Compound I MeOAc Solvate Form C is shown in
FIG. 37 and in the table below.
Table 29. 13C CPMAS of Compound I MeOAc Solvate Form C
 Example 15: Compound I Hydrate Form A
Example 15: Compound I Hydrate Form A
1. Synthetic Procedure
[00286] A 400 mL reaction vessel was charged with 16.5 g of Compound I EtOH Solvate Form B and 250 mL of water. The solution was stirred at 25 °C for 4 h. The slurry was then filtered, and the resulting solids were air dried overnight.
2. X-Ray Powder Diffraction
[00287] X-ray powder diffraction (XRPD) results are shown in FIG. 38 and the table below.
Table 30. XRPD Signals for Compound I Hydrate Form A

3. Solid State NMR
[00288] The 13C CPMAS spectrum of Compound I Hydrate Form A is shown in FIG. 39 and in the table below.
Table 31. 13C CPMAS of Compound I Hydrate Form A

4. Single Crystal Elucidation
[00289] A single crystal of Compound I Hydrate Form A was obtained via solvent exchange of Compound I EtOH solvate with water. X-ray diffraction data were acquired at 100 K on a Bruker diffractometer equipped with Cu Ka radiation (k=l.54178 A) and a CP AD detector. The structure was solved and refined using SHELX programs (Sheldrick, G.M., Acta Cryst., (2008) A64, 112-122) and results are summarized the table below.
Table 32. Single Crystal Elucidation of Compound I Hydrate Form A
 Example 16: Compound I Hydrate Form B
Example 16: Compound I Hydrate Form B
1. Synthetic Procedure
[00290] 200 mg of amorphous Compound I was added to an HPLC vial with 1.5 mL of 50:50 MeOH/Water. The vial was placed in a shaker block at 80 °C for 2 d. The slurry was centrifuged and vacuum dried at 80 °C overnight. The sample was then exposed to 11% RH for three days.
2. X-Ray Powder Diffraction
[00291] X-ray powder diffraction (XRPD) results are shown in FIG. 40 and the table below.
T able 33, XRPD Signals for Compound I Hydrate Form B
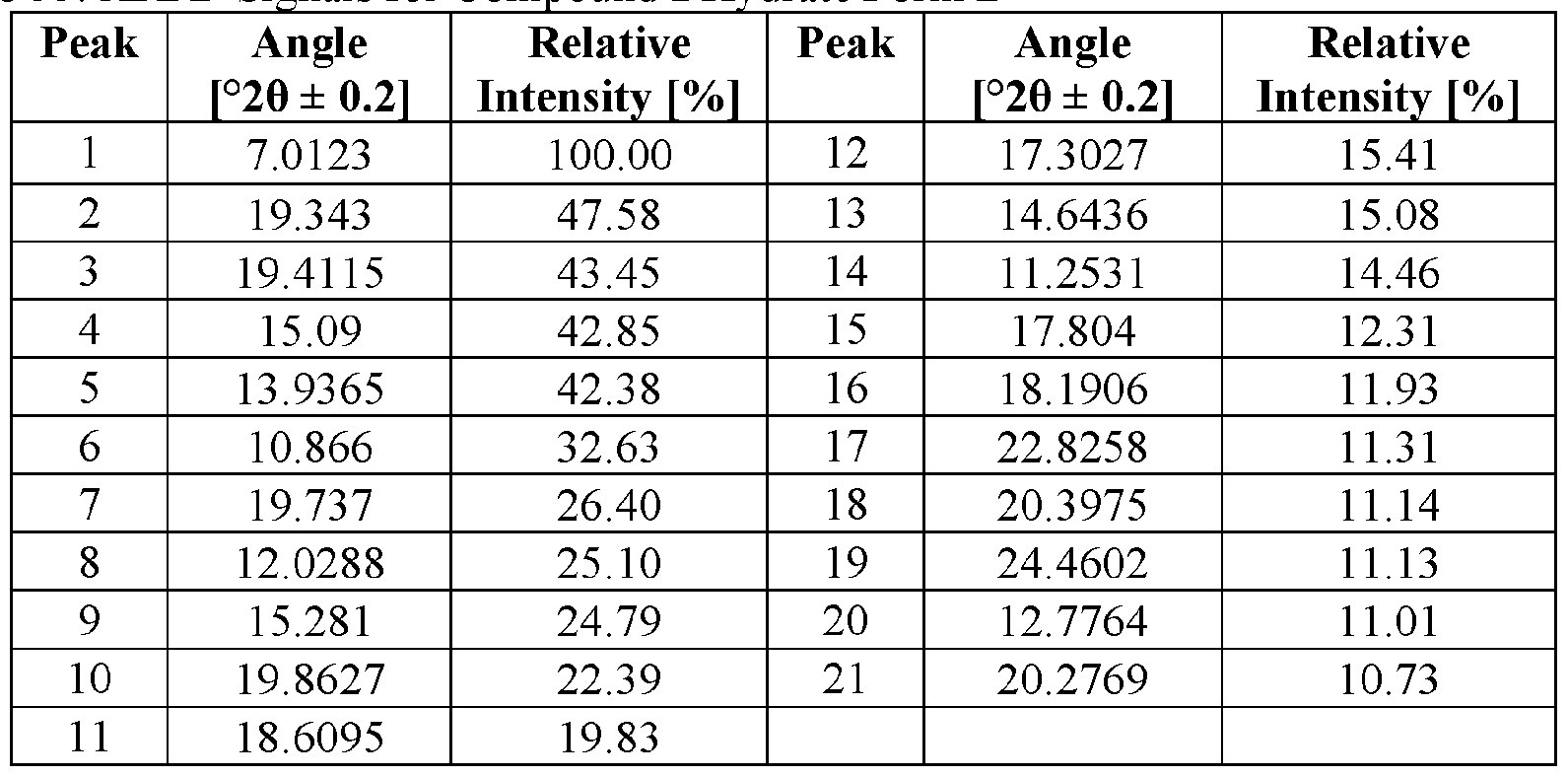
3. Solid State NMR
[00292] The 13C CPMAS spectrum of Compound I Hydrate Form B is shown in FIG. 41 and in the table below.
Table 34. 13C CPMAS of Compound I Hydrate Form B

4. Single Crystal Elucidation
[00293] A single crystal of Compound I Hydrate Form B was obtained via solvent exchange of Compound I EtOH Solvate Hydrate with water. X-ray diffraction data were acquired at 100 K on a Bruker diffractometer equipped with Cu K« radiation ( =l .54178 A) and a CPAD detector. The structure was solved and refined using SHELX programs (Sheldrick, G.M., Acta Cryst., (2008) A64, 112-122) and results are summarized in the table below.
Table 35. Single Crystal Elucidation of Compound I Hydrate Form B
 Example 17: Compound I Hydrate Form C
Example 17: Compound I Hydrate Form C
1. Synthetic Procedure
[00294] 100 mg of amorphous Compound I was dissolved in 1 mL of 50:50 MeOH/Water.
The solution was placed in a shaker block at 80 °C. After 5 d of shaking, the slurry was centrifuged and vacuum dried at 80 °C overnight. The dried sample was then exposed to 75% RH for 20 d.
2. X-Ray Powder Diffraction
[00295] X-ray powder diffraction (XRPD) results are shown in FIG. 42 and the table below.
Table 36, XRPD Signals for Compound I Hydrate Form C
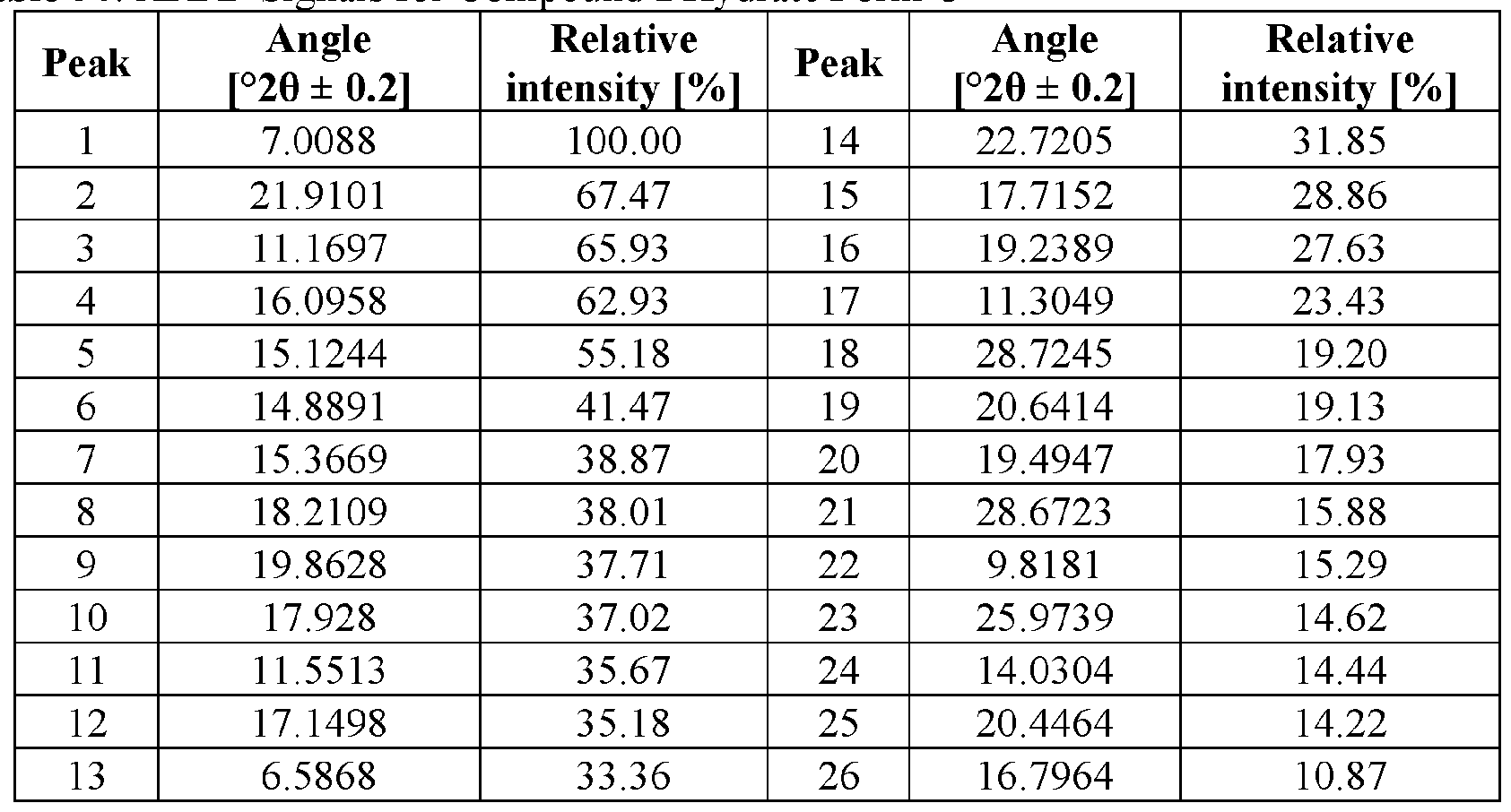
3. Solid State NMR
[00296] The 13C CPMAS spectrum of Compound I Hydrate Form C is shown in FIG. 43 and in the table below.
Table 37. 13C CPMAS of Compound I Hydrate Form C
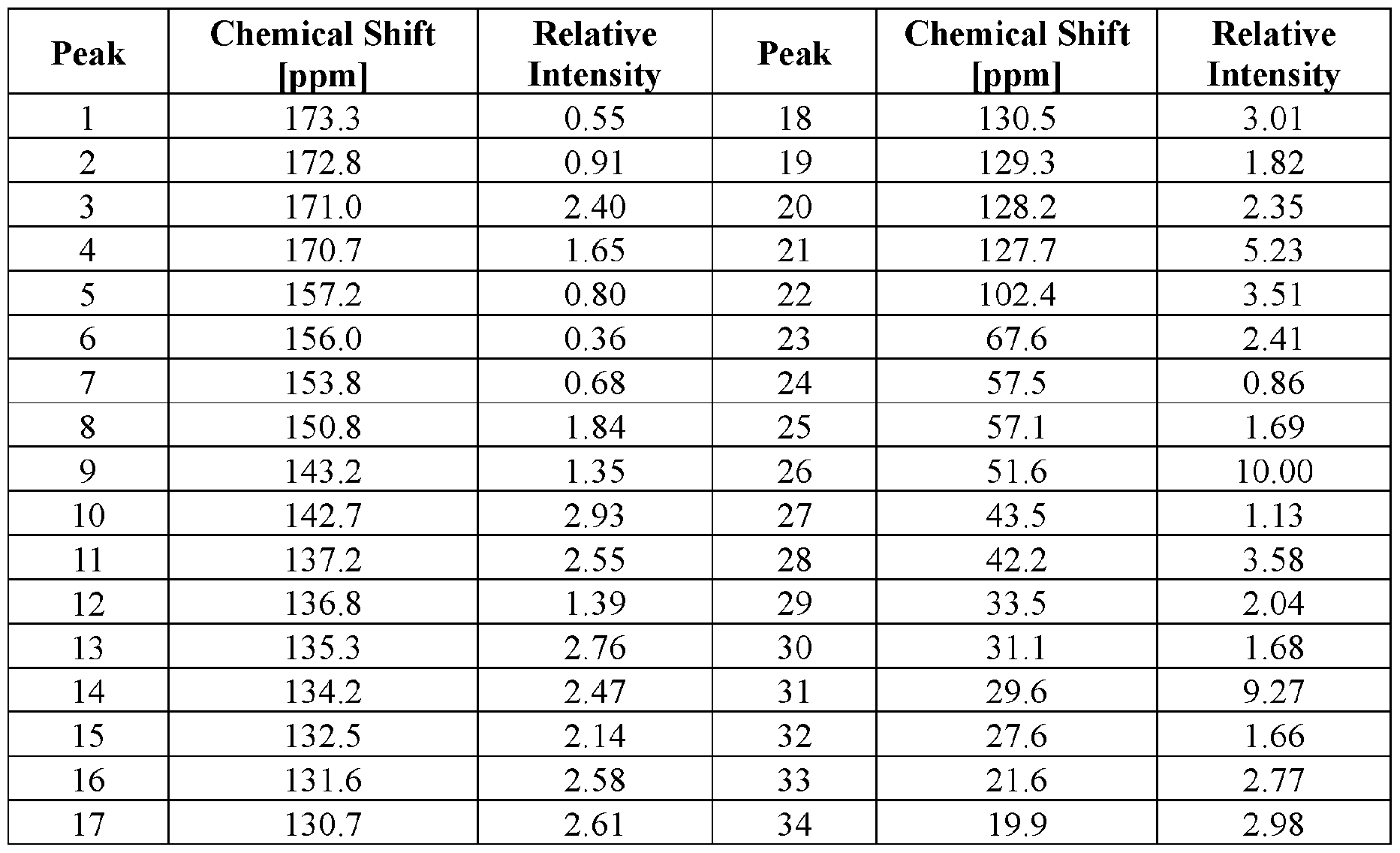
4. Single Crystal Elucidation
[00297] A single crystal of Compound I Hydrate Form C was obtained via solvent exchange of the Compound I EtOH Solvate Hydrate with water. X-ray diffraction data were acquired at 100 K on a Bruker diffractometer equipped with Cu K« radiation ( =l .54178 A) and a CPAD detector. The structure was solved and refined using SHELX programs (Sheldrick, G.M., Acta Cryst., (2008) A64, 112-122) and results are summarized in the table below.
Table 38. Single Crystal Elucidation of Compound I Hydrate Form C
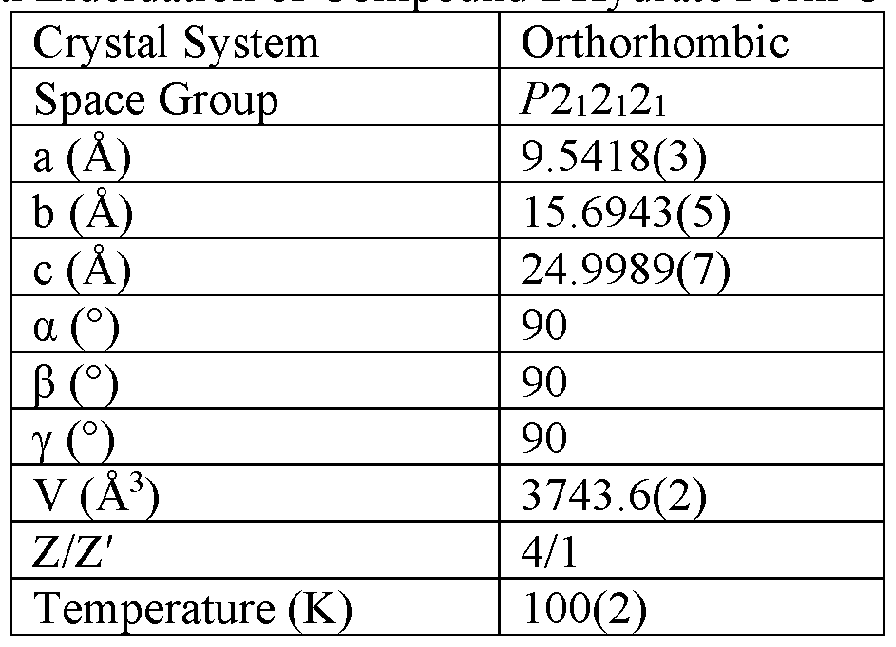
Example 18: Compound I EtOH Solvate Hydrate
1. Synthetic Procedure
[00298] 400 mg of Compound I Neat Form A was added to a scintillation vial. 2 mL of EtOH, 2 mL of water, and a magnetic stir bar were added to the vial. The contents of the vial were stirred at 60 °C overnight. The resulting solid was isolated by centrifugation to yield Compound I EtOH Solvate Hydrate.
2. X-Ray Powder Diffraction
[00299] X-ray powder diffraction (XRPD) results are shown in FIG. 44 and the table below.
Table 39. XRPD Signals for Compound I EtOH Solvate Hydrate
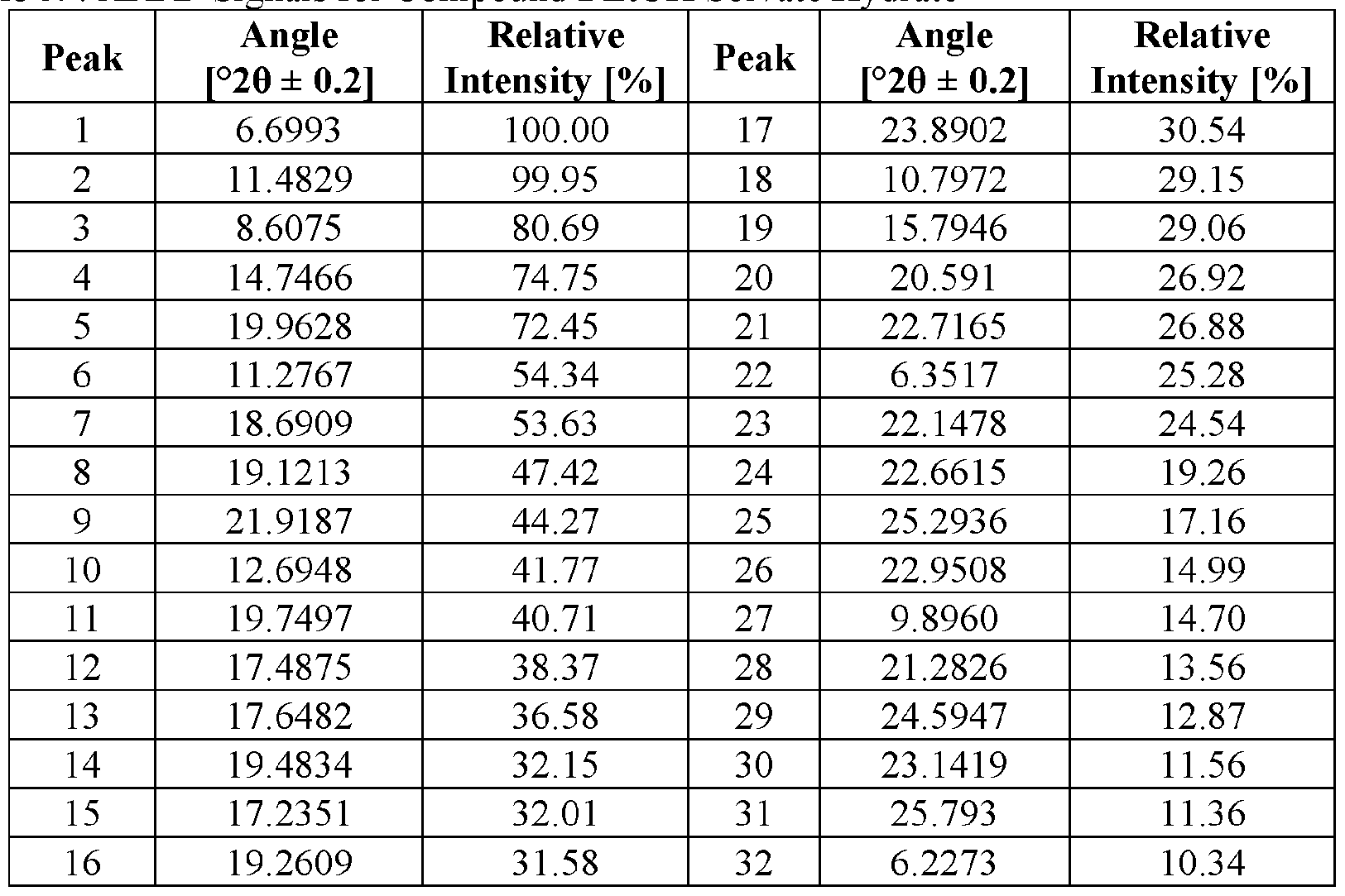
3. Solid State NMR
[00300] The 13C CPMAS spectrum of Compound I EtOH Solvate Hydrate is shown in
FIG. 45 and in the table below.
Table 40. 13C CPMAS of Compound I EtOH Solvate Hydrate
 4. Single Crystal Elucidation
4. Single Crystal Elucidation
[00301] A single crystal of Compound I EtOH Solvate Hydrate was obtained from a slurry of Compound I in a 80/20 mixture of EtOH with water. X-ray diffraction data were acquired at 100 K on a Bruker diffractometer equipped with Cu Ka radiation (X=l.54178 A) and a CP AD detector. The structure was solved and refined using SHELX programs (Sheldrick, GM , Acta Cryst., (2008) A64, 112-122) and results are summarized in the table below.
Table 41. Single Crystal Elucidation of Compound I EtOH Solvate Hydrate

Example 19: Compound I MeOH Solvate Hydrate
1. Synthetic Procedure
[00302] 150 mg of amorphous Compound I was dissolved in 1 m of 50:50 MeOH/water. The solution was slurried at 80 °C in a shaker block overnight. The solid was isolated by centrifugation to yield Compound I MeOH Solvate Hydrate.
2. X-Ray Powder Diffraction
[00303] X-ray powder diffraction (XRPD) results are shown in FIG. 46 and the table below.
T able 42, XRPD Signals for Compound I MeOH Solvate Hydrate

3. Solid State NMR
[00304] The 13C CPMAS spectrum of Compound I MeOH Solvate Hydrate is shown in
FIG. 47 and in the table below.
Table 43. 13C CPMAS of Compound I MeOH Solvate Hydrate
 Example 20: Compound I IPA Solvate Hydrate
Example 20: Compound I IPA Solvate Hydrate
1. Synthetic Procedure
[00305] 100 mg of amorphous Compound I was dissolved in 2 mL of IPA and 1 mL of water. The solution was sonicated for 1 h. The sample was slurried for 3 months. The resulting solid was isolated by centrifugation to yield Compound I IPA Solvate Hydrate.
2. X-Ray Powder Diffraction
[00306] X-ray powder diffraction (XRPD) results are shown in FIG. 48 and the table below.
Table 44, XRPD Signals for Compound I IPA Solvate Hydrate

3. Solid State NMR
[00307] The 13C CPMAS spectrum of Compound I IPA Solvate Hydrate is shown in FIG.
49 and in the table below.
Table 45. 13C CPMAS of Compound I IPA Solvate Hydrate

4. Single Crystal Elucidation
[00308] A single crystal of Compound I IPA Solvate Hydrate was obtained from IPA and water. X-ray diffraction data were acquired at 100 K on a Bruker diffractometer equipped with Cu Ka radiation (X=l.54178 A) and a CPAD detector. The structure was solved and refined using SHELX programs (Sheldrick, G.M., Acta Cryst., (2008) A64, 112-122) and results are summarized in the table below.
Table 46. Single Crystal Elucidation of Compound I IPA Solvate Hydrate
 Example 21: Compound I MeOAc Solvate Hydrate
Example 21: Compound I MeOAc Solvate Hydrate
1. Synthetic Procedure
[00309] 100 mg of amorphous Compound I was added to an HPLC vial, followed by the addition of 0.5 mL of 95:5 MeOAc:water. Then, the vial was placed in a shaker block for 1 d at rt. Then, Compound I MeOAc form A, Compound I MeOAc form B, Compound I MeOAc form C, Compound I Neat Form E, Compound I Hydrate form A, and Compound I Hydrate form B were added to the vial. Then, the slurry was shaken for an additional 4 d at rt. The mixture was then centrifuged, yielding a wet cake of Compound I MeOAc Solvate Hydrate.
2. X-Ray Powder Diffraction
[00310] X-ray powder diffraction (XRPD) results are shown in FIG. 50 and the table below.
Table 47. XRPD Signals for Compound I MeOAc Solvate Hydrate

3. Solid State NMR
[00311] The 13C CPMAS spectrum of Compound I MeOAc Solvate Hydrate is shown in
FIG. 51 and in the table below.
Table 48. 13C CPMAS of Compound I MeOAc Solvate Hydrate
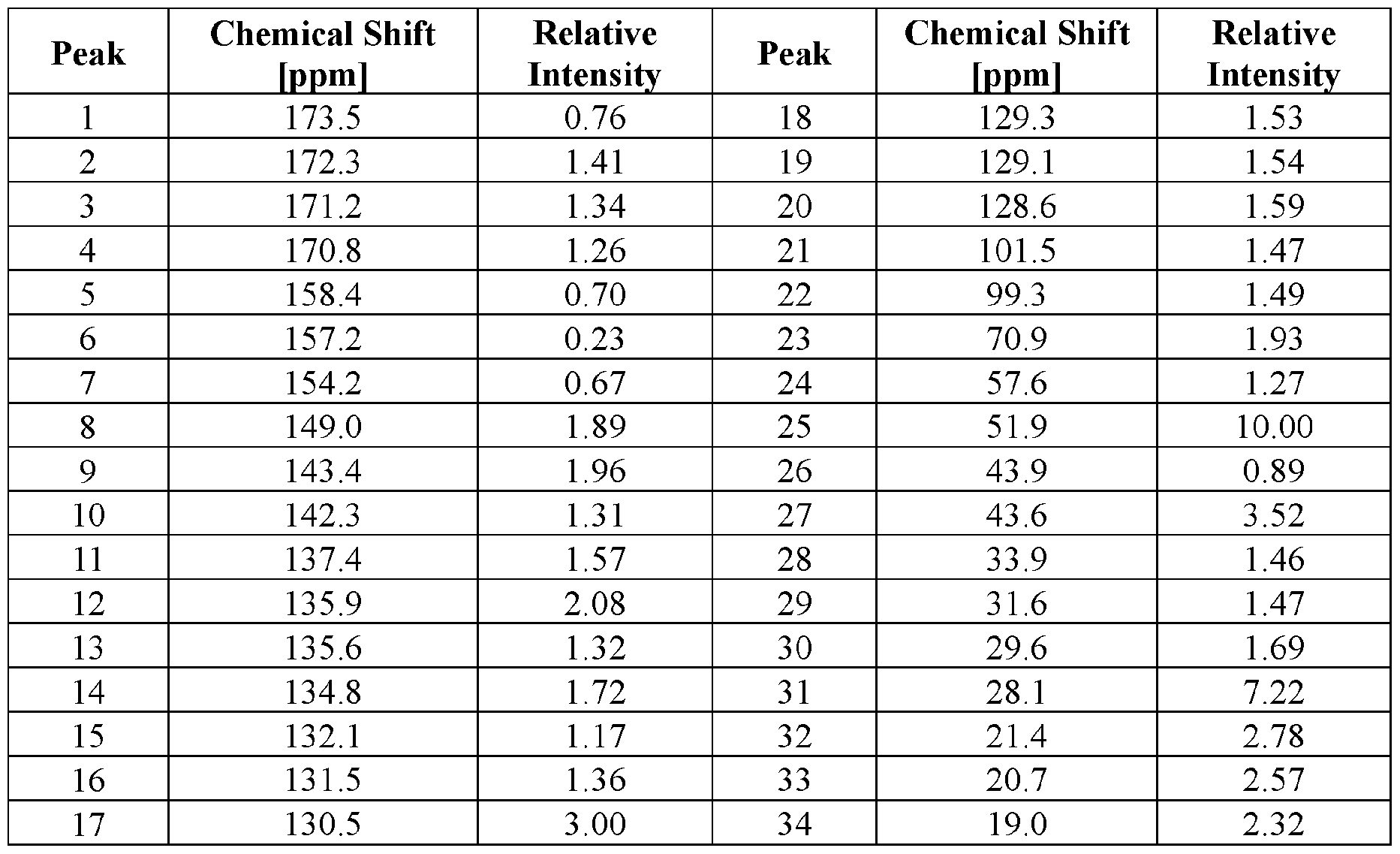
Example 22: Amorphous Compound I
1. Synthetic Procedure
[00312] 147 g of crude Compound I was dissolved in 440 mL DCM with 10 mL toluene and purified via column chromatography (3 kg silica) using a gradient of 15% to 50% EtOAc/hexanes. The combined fractions were concentrated under reduced pressure. The concentrated product was dissolved in DCM and concentrated under reduced pressure four times. After drying under high vacuum, 108.35 g of amorphous Compound I was obtained (98.84% purity).
2. X-Ray Powder Diffraction
[00313] X-ray powder diffraction (XRPD) results are shown in FIG. 52.
3. Solid State NMR
[00314] The 13C CPMAS spectrum of amorphous Compound I is shown in FIG. 53 and in the table below.
Table 49. 13C CPMAS of Amorphous Compound I
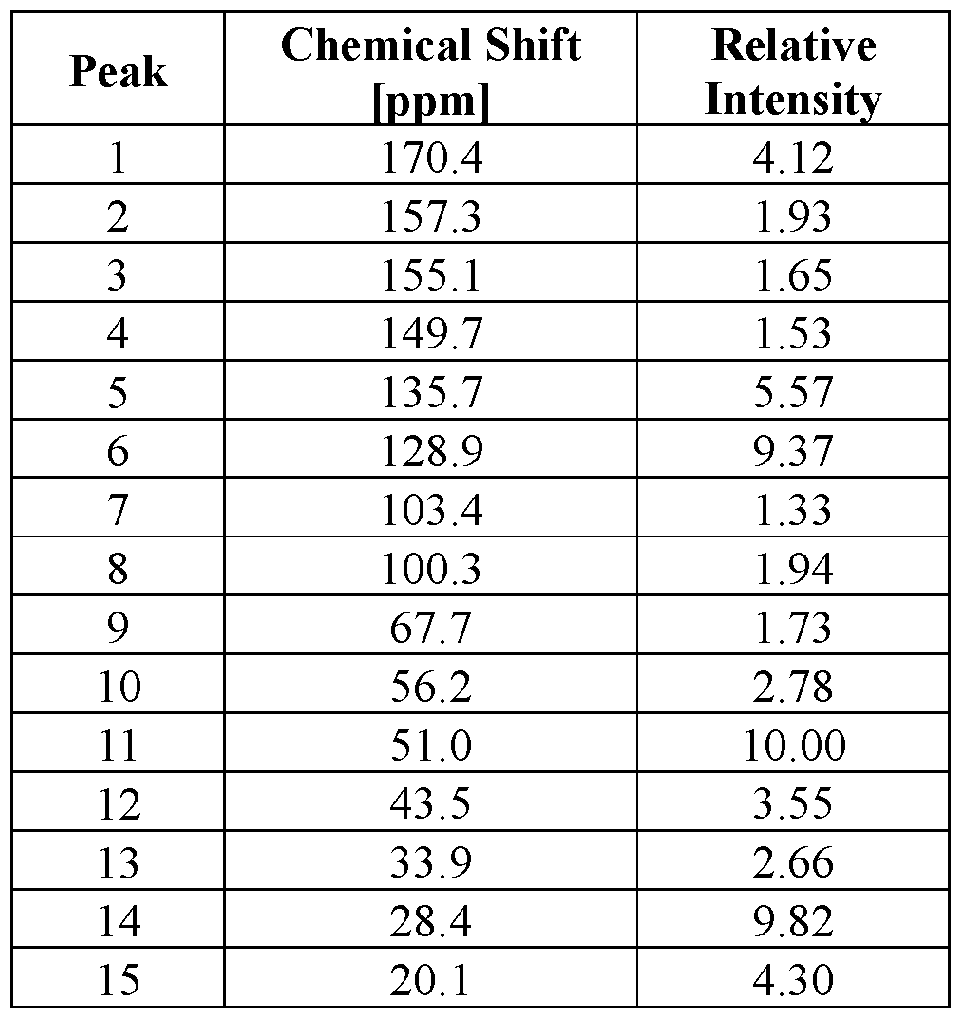
Example 23: Solid Dispersions of Compound I
[00315] Unless otherwise stated, the following procedures were employed for the analysis of all solid dispersions.
X-Ray Powder Diffraction
[00316] X-ray powder diffraction (XRPD) spectrum for Compound I Spray Dried Dispersion (SDD) 1A was recorded at room temperature in transmission mode using a PANalytical Empyrean system equipped with a sealed tube source and a PIXcellD detector (Malvern PANalytical Inc, Almelo, The Netherlands). The X-Ray generator operated at a voltage of 45 kV and a current of 40 mA with copper radiation (1.540598 A). The powder sample was placed on an indented area within a zero background holder and flattened with a glass slide then loaded into the instrument. The sample was scanned over the range of about 4° to about 40° 20 with a step size of 0.0131° and 14s per step.
[00317] XRPD spectra for Compound 1 SDDs 1B-1E were recorded at room temperature in reflection mode using a PANalytical Empyrean system equipped with a sealed tube source and a PIXcellD Medipix3 detector (Malvern PANalytical Inc, Westborough, Massachusetts). The X-Ray generator operated at a voltage of 45 kV and a current of 40 mA with copper radiation (1.540598 A). The powder samples were plated using back-loading sample holders and loaded into the instrument. The samples were scanned over the range of about 3° to about 40° 20 with a step size of 0.0131° and 48 s per step.
Solid State NMR
[00318] Bruker-Biospin 400 MHz wide-bore spectrometer equipped with Bruker-Biospin 4mm HFX probe was used. Samples were packed into 4mm ZrCh rotors and spun under Magic Angle Spinning (MAS) condition with spinning speed typically set to 12.5 kHz. The proton relaxation time was measured using 'H MAS Ti saturation recovery relaxation experiment in order to set up proper recycle delay of the 13C and 31P cross-polarization (CP) MAS experiments. The fluorine relaxation time was measured using 19F MAS Ti saturation recovery relaxation experiment in order to set up proper recycle delay of the 19F MAS experiment. The CP contact time of carbon as well as phosphorus CPMAS experiments was set to 2 ms. A CP proton pulse with linear ramp (from 50% to 100%) was employed. The carbon Hartmann-Hahn match was optimized on external reference sample (glycine), while phosphorus Hartmann-Hahn match was optimized on the actual samples. All carbon, phosphorus and fluorine spectra were recorded with proton decoupling using TPPM15 decoupling sequence with the field strength of approximately 100 kHz.
Differential Scanning Calorimetry
[00319] Modulated differential scanning calorimetry (mDSC) analysis of Compound I SDD 1A was carried out using the Mettler Toledo DSC 3. A sample with a weight of roughly 3 mg was weighed into an aluminum crimp sealed pan with a pinhole. The sample was equilibrated at -20 °C then heated to 250 °C at a rate of 2 °C/min (modulation amplitude ± 0.32 °C, modulation period 60 sec). When the run was completed, the data were analyzed using the STARe thermal analysis software (Mettler Toledo, Columbus, OH). The glass transition was taken from the reversing heat flow.
[00320] MDSC analysis of Compound 1 SDDs 1B-1E was carried out using a Mettler Toledo DSC 3+ instrument. Samples with weights between 5-8 mg were weighed into 40 pL aluminum crimp sealed pans with pinhole lids. The plans were placed on the autosampler tray with an empty pan as a reference. The samples were heated from 25 to 200 °C at a rate of 2 °C/min (modulation amplitude ± 0.32 °C, modulation period 60 sec). When the runs were completed, the data were analyzed using the STARe thermal analysis software (Mettler Toledo, Columbus, OH). The glass transition temperatures were taken from the reversing heat flow.
Residual Solvent Analysis:
[00321] Residual solvents were determined by head-space gas chromatography using an Agilent system (GC 6890N with Headspace unit 7967A, or equivalent). Sample material (50 or 100 mg ± 10%) was dissolved in 1.0 mL of dimethyl acetamide inside of a 10 mb
headspace vial. The vial was then capped. The column used in analysis was DB-624, 30 m x 0.32 mm i.d., 1.8 pm film thickness (manufacturer J & W). Residual solvents were detected by Flame Ionization Detection (FID).
A. SDD 1A: 50% Amorphous Compound I and 50% Polymer Solvent: DCM/MeOH (50/50 %w/w) Polymer: HPMCAS-H
1. Synthetic Procedure
[00322] 15 g of Compound I and 15 g of HPMCAS-H polymer were weighed separately. Compound I was dissolved in 270 g of 1 : 1 DCM:MeOH, followed by the addition of the polymer to the solution. The solution was continuously stirred by magnetic stirring for 30 min, resulting in a light pink clear solution at 50% drug load and 10% solid load.
[00323] The resulting 300 g of solution was then spray-dried using a Buchi B290 spray dryer at 50 mm nozzle gas rotameter, 90% aspiration, 78 °C inlet temperature, 48 °C outlet temperature, 20% pump setting (6 g/min) with 2 pulse setting, -5 °C condenser temperature, and -50 mbar filter pressure. The solution had a run time of 60 min to spray dry. 23.33 g of wet solid was collected and underwent secondary drying at 40 °C for 48 h. 22.77 g of dry 1 : 1 Compound I to polymer spray-dried dispersion was recovered, representing approximately 76% yield.
2. X-Ray Powder Diffraction
[00324] X-ray powder diffraction (XRPD) results are shown in FIG. 54.
3. Solid State NMR
[00325] The 13C CPMAS spectrum of Compound I SDD 1A is shown in FIG. 55 and in the table below.
Table 50. 13C CPMAS of Compound


4. Differential Scanning Calorimetry Analysis
[00326] Modulated differential scanning calorimetry (mDSC) analysis of Compound I SDD 1A was carried out using the Mettler Toledo DSC 3. The thermogram showed a glass transition temperature of 145 °C in the reversing heat flow as shown in FIG. 56.
5. Residual Solvents
[00327] Sample Results (ppm) of residual solvents detected in Compound I SDD 1A samples in the final spray dry dispersion after secondary drying at 40 °C, vacuum:

ND = not detected
B. SDD IB: 50% Compound I and 50% Polymer Solvent: DCM/MeOH (63/37% w/w) Polymer: HPMCAS-H
1. Synthetic Procedure
[00328] 621 g of Compound I Form A and 621 grams of HPMCAS-H were weighed separately. 7042.14 g of DCM was mixed with 4135.86 g of MeOH. Compound I Form A was added to this DCM: MeOH mixture and stirred at sufficient vortex using a magnetic stir plate. The solution was stirred until it became visually clear. HPMCAS-H was added to this solution and continuously stirred, resulting in a visually clear solution.
[00329] The resulting solution was then spray-dried using Anhydro MS-35 spray dryer at the following spray drying parameters. The resulting wet SDD IB was dried in an oven at 40 °C vacuum until residual solvent criteria were met.
MS-35 Parameters

2. X-Ray Powder Diffraction
[00330] X-ray powder diffraction (XRPD) data of Compound I SDD IB were recorded at room temperature in transmission mode using a PANalytical Empyrean system and are shown in FIG. 57.
3. Differential Scanning Calorimetry Analysis
[00331] Modulated differential scanning calorimetry (mDSC) analysis of Compound I SDD IB was carried out using the Mettler Toledo DSC 3+ The thermogram shows a glass transition temperature of 142 °C in the reversing heat flow as shown in FIG. 58.
4. Residual Solvents
[00332] Sample Results (ppm) of residual solvents detected in Compound I SDD IB samples in the final spray dry dispersion after secondary drying at 40 °C, vacuum:
ND = not detected
C. SDD 1C: 80% Compound I and 20% Polymer Solvent: DCM/MeOH (63/37% w/w) Polymer: HPMCAS-H
1. Synthetic Procedure
[00333] 15 g of Compound I Form A and 3.75 g of HPMCAS-H were weighed separately. 106.31 g of DCM was mixed with 62.44 g of MeOH in a glass bottle. Compound I Form A was added to this DCM: MeOH mixture and stirred at a sufficient vortex using a magnetic stirring plate. The solution was stirred until it became visually clear. HPMCAS-H was added to this solution and continuously stirred for ~1 h, resulting in a clear solution.
[00334] The resulting solution was then spray-dried using Buchi B290 spray dryer at 50 mm nozzle gas rotameter, 85% aspiration, 94 °C inlet temperature, 52 °C outlet temperature, 20% pump setting with 2 pulse setting, -10 °C condenser temperature, and -80 mbar filter pressure. 16.07 g of wet solid was collected and underwent secondary drying at 40 °C for 72 h. 15.11 g of dry SDD 1C was recovered after secondary drying.
2. X-Ray Powder Diffraction
[00335] X-ray powder diffraction (XRPD) data of Compound I SDD 1C were recorded at room temperature in transmission mode using a PANalytical Empyrean system, and are shown in FIG. 59.
3. Differential Scanning Calorimetry Analysis
[00336] Modulated differential scanning calorimetry (mDSC) analysis of Compound I SDD 1C was carried out using the Mettler Toledo DSC 3+. The thermogram shows a glass transition temperature of 162 °C in the reversing heat flow as shown in FIG. 60.
4. Residual Solvents
[00337] Sample Results (ppm) of residual solvents detected in Compound I SDD 1C samples after secondary drying for 48 h (T48) in a bottle, and additional 24 h (T72) at 40 °C, vacuum :

ND = not detected
D. SDD ID: 50% Compound I and 50% Polymer Solvent: DCM/MeOH (63/37 w/w) Polymer: HPMC-E15
1. Synthetic Procedure
[00338] 9.0 g of Compound I Form A and 9.0 g of HPMC- El 5 were weighed separately. 102.06 g of DCM was mixed with 59.94 g of MeOH in a glass bottle. Compound I Form A was added to this DCM: MeOH mixture and stirred at a sufficient vortex using magnetic stirring plate until the solution became clear. HPMC -El was added to this solution and continuously stirred for ~1 h, resulting in a clear solution.
[00339] The resulting 180.0 g of the solution was then spray-dried using Buchi B290 spray dryer at 50 mm nozzle gas rotameter, 85% aspiration, 88 °C inlet temperature, 49 °C outlet temperature, 20% pump setting with 2 pulse setting, -10 °C condenser temperature, and -80 mbar filter pressure. 12.0 grams of wet solid was collected and underwent secondary drying at 40 °C for 72 h. 11.8 g of dry SDD ID was recovered, representing approximately an 67.4% dry yield.
2. X-Ray Powder Diffraction
[00340] X-ray powder diffraction (XRPD) data of Compound I SDD ID were collected at room temperature in transmission mode using a PANalytical Empyrean system, and are shown in FIG. 61.
3. Differential Scanning Calorimetry
[00341] Modulated differential scanning calorimetry (mDSC) analysis of Compound I SDD ID was carried out using the Mettler Toledo DSC 3+. The thermogram shows a glass transition temperature of 157 °C in the reversing heat flow as shown in FIG. 62.
4. Residual Solvents
[00342] Sample Results (ppm) of residual solvents detected in Compound I SDD ID samples after secondary drying for 48 h (T48) in a bottle, and additional 24 h in a tray (T72) at 40 °C, vacuum :

ND = not detected
E. SDD IE: 80% Compound I and 20% Polymer Solvent: DCM/MeOH (63/37 w/w) Polymer: HPMC-E15
1. Synthetic Procedure
[00343] 15.0 g of Compound I Form A and 3.75 g of HPMC- E15 were weighed separately. 106.31 g of DCM was mixed with 62.44 g of MeOH in a glass bottle. Compound I Form A was added to this DCM/MeOH mixture and stirred by magnetic stirring method until the solution became clear. HPMC -El 5 was added to this solution and continuously stirred for ~1 h.
[00344] The solution was then spray-dried using Buchi B290 spray dryer at 50 mm nozzle gas rotameter, 85% aspiration, 101 °C inlet temperature, 51 °C outlet temperature, 20% pump setting with 2 pulse setting, -10 °C condenser temperature, and -80 mbar filter pressure. 15.3 g of wet solid was collected and underwent secondary drying at 40 °C for 72 h. 14.4 g of dry SDD IE was recovered after secondary drying.
4. X-Ray Powder Diffraction
[00345] X-ray powder diffraction (XRPD) data of Compound I SDD IE was collected at room temperature in transmission mode using a PANalytical Empyrean system, and is shown in FIG. 63 .
3. Differential Scanning Calorimetry
[00346] Modulated differential scanning calorimetry (mDSC) analysis of Compound I SDD IE was carried out using the Mettler Toledo DSC 3+ The thermogram shows a glass transition temperature of 168 °C in the reversing heat flow as shown in FIG. 64.
4. Residual Solvents
[00347] Sample Results (ppm) of residual solvents detected in Compound I SDD IE samples after secondary drying for 48 h (T48) in a bottle, and additional 24 h in a tray at 40 °C, vacuum (T72):

ND = not detected
Claims
1. A solid form of Compound I selected from Compound I Neat Form A, Compound I Neat Form B, Compound I Neat Form C, Compound I Neat Form D, Compound I Neat Form E, Compound I Compressed Form A, Compound I Compressed Form E, Compound I EtOH Solvate Form B, Compound I MeOH solvate, Compound I NPA solvate, Compound I MeOAc Solvate Form A, MeOAc Solvate Form B, Compound I MeOAc Solvate Form C, Compound I Hydrate Form A, Compound I Hydrate Form B, Compound I Hydrate Form C, Compound I EtOH Solvate Hydrate, Compound I MeOH Solvate Hydrate, Compound I IPA Solvate Hydrate, and Compound I MeOAc Solvate Hydrate.
2. Compound I formulated as a spray-dried dispersion.
3. A pharmaceutical composition comprising Compound I according to claim 1 or claim 2 and a pharmaceutically acceptable carrier.
4. The pharmaceutical composition according to claim 3, further comprising one or more additional therapeutic agents.
5. The pharmaceutical composition according to claim 4, wherein the pharmaceutical composition comprises one or more additional CFTR modulating compounds.
6. The pharmaceutical composition according to claim 4 or claim 5, wherein the pharmaceutical composition comprises one or more compounds selected from Compound II, Compound IV, Compound V, and pharmaceutically acceptable salts and deuterated derivatives of any of the foregoing.
7. The Compound I according to claim 1 or claim 2, or the pharmaceutical composition according to any one of claims 3 to 6, for use in the treatment of cystic fibrosis.
8. The compound for use of claim 7, wherein the compound is administered in combination with one or more compounds selected from Compound II, Compound
IV, and Compound V, and pharmaceutically acceptable salts and deuterated derivatives of any of the foregoing.
9. Use of the Compound I according to claim 1 or claim 2, or the pharmaceutical composition according to any one of claims 3 to 6 in the manufacture of a medicament for the treatment of cystic fibrosis.
10. The use according to claim 9, wherein the medicament is formulated for administration in combination with one or more compounds selected from Compound II, Compound IV, and Compound V, and pharmaceutically acceptable salts and deuterated derivatives of any of the foregoing.
11. A method of treating cystic fibrosis comprising administering the Compound I according to claim 1 or claim 2, or the pharmaceutical composition according to any one of claims 3 to 6, to a subject in need thereof.
12. The method of treating according to claim 11, wherein the method further comprises administration of one or more compounds selected from Compound II, Compound IV, Compound V, and pharmaceutically acceptable salts and deuterated derivatives of any of the foregoing.
13. A process for preparing Compound I Neat Form A according to claim 1 comprising (i) dissolving amorphous Compound I in ethanol, (ii) increasing the temperature to about 73 °C, (iii) decreasing the temperature to about 20 °C, (iv) isolation of the solid via filtration, and (v) drying of the solid at 50 °C.
14. A process for preparing Compound I Neat Form B according to claim 1 comprising exposing Compound I Hydrate Form A to about 3% humidity.
15. A process for preparing Compound I Neat Form C according to claim 1 comprising (i) stirring of Compound I in hexadecane at about 80 °C, and (ii) centrifugation and drying of the resulting solid.
16. A process for preparing Compound I Neat Form D according to claim 1 comprising the drying of Compound I hydrate C at about 80 °C.
17. A process for preparing Compound I Neat Form D according to claim 1 comprising (i) stirring of Compound I Neat Form A in IP A, and (ii) isolation of the solid via fdtration, and (iii) drying of the solid at about 125 °C.
18. A process for preparing Compound I Neat Form E according to claim 1 comprising (i) stirring of Compound I Neat Form A in MeOAc, and (ii) isolation of the solid via centrifugation, and (iii) drying of the solid at about 50 °C.
19. A process for preparing Compound I Compressed Form E according to claim 1 comprising mechanical compression of Compound I Neat Form E.
20. A process for preparing Compound I Compressed Form A according to claim 1 comprising mechanical compression of Compound I Neat Form A.
21. A process for preparing Compound I EtOH Solvate Form B according to claim 1 comprising (i) stirring of Compound I in ethanol, and (ii) isolation of the solid via centrifugation.
22. A process for preparing Compound I MeOH solvate according to claim 1 comprising (i) dissolving Compound I in MeOH and increasing temperature to about 62 °C, (ii) cooling the solution to about 10 °C, and (iii) isolation of the solid via centrifugation.
23. A process for preparing Compound I NPA solvate according to claim 1 comprising (i) stirring of Compound I in NPA, and (ii) isolation of the solid via centrifugation.
24. A process for preparing Compound I MeOAc Solvate Form A according to claim 1 comprising (i) stirring Compound I Neat Form A in MeOAc, and (ii) isolation of the solid via filtration.
25. A process for preparing Compound I MeOAc Solvate Form B according to claim 1 comprising (i) stirring of Compound I Neat Form A in MeOAc at about 5 °C, (ii)
isolation of the solid via centrifugation, and (iii) drying of the solid at room temperature.
26. A process for preparing Compound I MeOAc Solvate Form C according to claim 1 comprising (i) stirring of Compound I Neat Form A in MeOAc at about 5 °C, and (ii) isolation of the solid via filtration.
27. A process for preparing Compound I Hydrate Form A according to claim 1 comprising (i) stirring Compound I EtOH Solvate Form B in water, and (ii) isolation via filtration and air drying of the solid.
28. A process for preparing Compound I Hydrate Form B according to claim 1 comprising (i) stirring Compound I in a MeOH/water mixture at about 80 °C, (ii) isolation of the solid via filtration, and (iii) exposure of the solid to about 11% humidity.
29. A process for preparing Compound I Hydrate Form C according to claim 1 comprising (i) stirring Compound I in a MeOH/water mixture at about 80 °C, (ii) isolation of the solid via filtration, and (iii) exposure of the solid to about 75% humidity.
30. A process for preparing Compound I EtOH Solvate Hydrate according to claim 1 comprising (i) stirring Compound I in an EtOH/water mixture at about 60 °C, and (ii) isolation of the solid.
31. A process for preparing Compound I MeOH Solvate Hydrate according to claim 1 comprising (i) the stirring of Compound I in a MeOH/water mixture at about 80 °C, and (ii) isolation of the solid via centrifugation.
32. A process for preparing Compound I IPA Solvate Hydrate according to claim 1 comprising (i) stirring Compound I in an IPA/water mixture, and (ii) isolation of the solid via centrifugation.
33. A process for preparing Compound I MeOAc Solvate Hydrate according to claim 1 comprising (i) stirring amorphous Compound I in a MeOAc/water mixture, (ii) addition of Compound I MeOAc form A, Compound I MeOAc form B, Compound I MeOAc form C, Compound I Neat Form E, Compound I Hydrate form A, and Compound I Hydrate Form B, (iii) further stirring, and (iv) isolation of the solid via centrifugation.
34. A process for preparing Compound I spray-dried dispersion according to claim 2 comprising (i) preparation of a solution of Compound I and polymer in a DCM/MeOH mixture, and (ii) spray drying the resulting solution using a spray dryer.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US202363587829P | 2023-10-04 | 2023-10-04 | |
| US63/587,829 | 2023-10-04 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2025076240A1 true WO2025076240A1 (en) | 2025-04-10 |
Family
ID=93214866
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2024/049821 Pending WO2025076240A1 (en) | 2023-10-04 | 2024-10-03 | Solid forms of modulators of cystic fibrosis transmembrane conductance regulator |
Country Status (2)
| Country | Link |
|---|---|
| TW (1) | TW202515573A (en) |
| WO (1) | WO2025076240A1 (en) |
Citations (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20040006237A1 (en) | 2001-11-14 | 2004-01-08 | Teva Pharmaceutical Industries Ltd. | Amorphous and crystalline forms of losartan potassium and process for their preparation |
| WO2006002421A2 (en) | 2004-06-24 | 2006-01-05 | Vertex Pharmaceuticals Incorporated | Modulators of atp-binding cassette transporters |
| WO2007079139A2 (en) | 2005-12-28 | 2007-07-12 | Vertex Pharmaceuticals, Inc. | Solid forms of n-[2,4-bis(1,1-dimethylethyl)-5-hydroxyphenyl]-1,4-dihydro-4-oxoquinoline-3-carboxamide |
| WO2007134279A2 (en) | 2006-05-12 | 2007-11-22 | Vertex Pharmaceuticals Incorporated | Compositions of n-[2,4-bis(1,1-dimethylethyl)-5-hydroxyphenyl]-1,4-dihydro-4-oxoquinoline-3-carboxamide |
| WO2010019239A2 (en) | 2008-08-13 | 2010-02-18 | Vertex Pharmaceuticals Incorporated | Pharmaceutical composition and administrations thereof |
| WO2010053471A1 (en) | 2008-11-06 | 2010-05-14 | Vertex Pharmaceuticals Incorporated | Modulators of atp-binding cassette transporters |
| WO2011019413A1 (en) | 2009-08-13 | 2011-02-17 | Vertex Pharmaceuticals Incorporated | Pharmaceutical composition and administrations thereof |
| WO2011119984A1 (en) | 2010-03-25 | 2011-09-29 | Vertex Pharmaceuticals Incorporated | Solid forms of (r)-1(2,2-difluorobenzo[d][1,3]dioxol-5-yl)-n-(1-(2,3-dihyderoxypropyl)-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)-1h-indol-5-yl) cyclopropanecarboxamide |
| WO2011133951A1 (en) | 2010-04-22 | 2011-10-27 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions and administrations thereof |
| WO2011133751A2 (en) | 2010-04-22 | 2011-10-27 | Vertex Pharmaceuticals Incorporated | Process of producing cycloalkylcarboxamido-indole compounds |
| WO2012027731A2 (en) | 2010-08-27 | 2012-03-01 | Vertex Pharmaceuticals Incorporated | Pharmaceutical composition and administrations thereof |
| WO2012158885A1 (en) | 2011-05-18 | 2012-11-22 | Concert Pharmaceuticals Inc. | Deuterated derivatives of ivacaftor |
| WO2013130669A1 (en) | 2012-02-27 | 2013-09-06 | Vertex Pharmaceuticals Incorporated | Pharmaceutical composition and administration thereof |
| WO2014014841A1 (en) | 2012-07-16 | 2014-01-23 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions of (r)-1-(2,2-diflurorbenzo[d][1,3]dioxol-5-yl)-n-(1-(2,3-dihydroxypropyl)-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)-1h-indol-5-yl) cyclopropanecarboxamide and administration thereof |
| WO2014078842A1 (en) | 2012-11-19 | 2014-05-22 | Concert Pharmaceuticals, Inc. | Deuterated cftr potentiators |
| US8865902B2 (en) | 2011-05-18 | 2014-10-21 | Concert Pharmaceuticals, Inc. | Deuterated CFTR potentiators |
| WO2015160787A1 (en) | 2014-04-15 | 2015-10-22 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions for the treatment of cystic fibrosis transmembrane conductance regulator mediated diseases |
| WO2017053455A1 (en) | 2015-09-21 | 2017-03-30 | Concert Pharmaceuticals, Inc. | Administration of deuterated cftr potentiators |
| WO2018080591A1 (en) | 2016-10-27 | 2018-05-03 | Vertex Pharmaceuticals (Europe) Limited | Methods of treatment with deuterated cftr potentiators |
| WO2022032068A1 (en) | 2020-08-07 | 2022-02-10 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| WO2022076622A2 (en) * | 2020-10-07 | 2022-04-14 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| WO2022109573A1 (en) | 2020-11-18 | 2022-05-27 | Vertex Pharmaceuticals Incorporated | Macrocycles containing a 1,3,4-oxadiazole ring for use as modulators of cystic fibrosis transmembrane conductance regulator |
| WO2023196429A1 (en) * | 2022-04-06 | 2023-10-12 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
-
2024
- 2024-10-03 WO PCT/US2024/049821 patent/WO2025076240A1/en active Pending
- 2024-10-04 TW TW113137966A patent/TW202515573A/en unknown
Patent Citations (25)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20040006237A1 (en) | 2001-11-14 | 2004-01-08 | Teva Pharmaceutical Industries Ltd. | Amorphous and crystalline forms of losartan potassium and process for their preparation |
| WO2006002421A2 (en) | 2004-06-24 | 2006-01-05 | Vertex Pharmaceuticals Incorporated | Modulators of atp-binding cassette transporters |
| WO2007079139A2 (en) | 2005-12-28 | 2007-07-12 | Vertex Pharmaceuticals, Inc. | Solid forms of n-[2,4-bis(1,1-dimethylethyl)-5-hydroxyphenyl]-1,4-dihydro-4-oxoquinoline-3-carboxamide |
| WO2007134279A2 (en) | 2006-05-12 | 2007-11-22 | Vertex Pharmaceuticals Incorporated | Compositions of n-[2,4-bis(1,1-dimethylethyl)-5-hydroxyphenyl]-1,4-dihydro-4-oxoquinoline-3-carboxamide |
| WO2010019239A2 (en) | 2008-08-13 | 2010-02-18 | Vertex Pharmaceuticals Incorporated | Pharmaceutical composition and administrations thereof |
| WO2010053471A1 (en) | 2008-11-06 | 2010-05-14 | Vertex Pharmaceuticals Incorporated | Modulators of atp-binding cassette transporters |
| WO2011019413A1 (en) | 2009-08-13 | 2011-02-17 | Vertex Pharmaceuticals Incorporated | Pharmaceutical composition and administrations thereof |
| WO2011119984A1 (en) | 2010-03-25 | 2011-09-29 | Vertex Pharmaceuticals Incorporated | Solid forms of (r)-1(2,2-difluorobenzo[d][1,3]dioxol-5-yl)-n-(1-(2,3-dihyderoxypropyl)-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)-1h-indol-5-yl) cyclopropanecarboxamide |
| WO2011133951A1 (en) | 2010-04-22 | 2011-10-27 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions and administrations thereof |
| WO2011133751A2 (en) | 2010-04-22 | 2011-10-27 | Vertex Pharmaceuticals Incorporated | Process of producing cycloalkylcarboxamido-indole compounds |
| WO2012027731A2 (en) | 2010-08-27 | 2012-03-01 | Vertex Pharmaceuticals Incorporated | Pharmaceutical composition and administrations thereof |
| US9512079B2 (en) | 2011-05-18 | 2016-12-06 | Concert Pharmaceuticals, Inc. | Deuterated CFTR potentiators |
| WO2012158885A1 (en) | 2011-05-18 | 2012-11-22 | Concert Pharmaceuticals Inc. | Deuterated derivatives of ivacaftor |
| US9181192B2 (en) | 2011-05-18 | 2015-11-10 | Concert Pharmaceuticals, Inc. | Deuterated CFTR potentiators |
| US8865902B2 (en) | 2011-05-18 | 2014-10-21 | Concert Pharmaceuticals, Inc. | Deuterated CFTR potentiators |
| WO2013130669A1 (en) | 2012-02-27 | 2013-09-06 | Vertex Pharmaceuticals Incorporated | Pharmaceutical composition and administration thereof |
| WO2014014841A1 (en) | 2012-07-16 | 2014-01-23 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions of (r)-1-(2,2-diflurorbenzo[d][1,3]dioxol-5-yl)-n-(1-(2,3-dihydroxypropyl)-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)-1h-indol-5-yl) cyclopropanecarboxamide and administration thereof |
| WO2014078842A1 (en) | 2012-11-19 | 2014-05-22 | Concert Pharmaceuticals, Inc. | Deuterated cftr potentiators |
| WO2015160787A1 (en) | 2014-04-15 | 2015-10-22 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions for the treatment of cystic fibrosis transmembrane conductance regulator mediated diseases |
| WO2017053455A1 (en) | 2015-09-21 | 2017-03-30 | Concert Pharmaceuticals, Inc. | Administration of deuterated cftr potentiators |
| WO2018080591A1 (en) | 2016-10-27 | 2018-05-03 | Vertex Pharmaceuticals (Europe) Limited | Methods of treatment with deuterated cftr potentiators |
| WO2022032068A1 (en) | 2020-08-07 | 2022-02-10 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| WO2022076622A2 (en) * | 2020-10-07 | 2022-04-14 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| WO2022109573A1 (en) | 2020-11-18 | 2022-05-27 | Vertex Pharmaceuticals Incorporated | Macrocycles containing a 1,3,4-oxadiazole ring for use as modulators of cystic fibrosis transmembrane conductance regulator |
| WO2023196429A1 (en) * | 2022-04-06 | 2023-10-12 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
Non-Patent Citations (9)
| Title |
|---|
| "Encyclopedia of Pharmaceutical Technology", 1988, MARCEL DEKKER |
| "Remington: The Science and Practice of Pharmacy,", 2005, LIPPINCOTT WILLIAMS & WILKINS |
| CUTTING, G. R. ET AL., NATURE, vol. 346, 1990, pages 366 - 369 |
| DALEMANS ET AL., NATURE LOND., vol. 354, 1991, pages 526 - 528 |
| DEAN, M. ET AL., CELL, vol. 61, no. 863, 1990, pages 870 |
| KEREM, B-S ET AL., PROC. NATL. ACAD. SCI. USA, vol. 87, 1990, pages 8447 - 8451 |
| KEREM, B-S. ET AL., SCIENCE, vol. 245, 1989, pages 1073 - 1080 |
| PASYKFOSKETT, J. CELL. BIOCHEM., vol. 270, 1995, pages 12347 - 50 |
| QUINTON, P. M., FASEB J., vol. 4, 1990, pages 2709 - 2727 |
Also Published As
| Publication number | Publication date |
|---|---|
| TW202515573A (en) | 2025-04-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US12319693B2 (en) | Crystalline forms of CFTR modulators | |
| US20230303589A1 (en) | Crystalline forms of cftr modulators | |
| WO2021030555A1 (en) | Modulators of cystic fibrosis transmembrane conductance regulator | |
| WO2020214921A1 (en) | Solid forms of modulators of cftr | |
| EP4225763A1 (en) | Modulators of cystic fibrosis transmembrane conductance regulator | |
| US12186306B2 (en) | Methods of treatment for cystic fibrosis | |
| WO2025076240A1 (en) | Solid forms of modulators of cystic fibrosis transmembrane conductance regulator | |
| US20250340568A1 (en) | Solid forms of a macrocyclic compounds as cftr modulators and their preparation | |
| AU2026200305A1 (en) | Crystalline forms of cftr modulators | |
| CN116829148A (en) | How to treat cystic fibrosis |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 24794595 Country of ref document: EP Kind code of ref document: A1 |