WO2025072330A1 - Kit inhibitor compounds and methods of use thereof - Google Patents
Kit inhibitor compounds and methods of use thereof Download PDFInfo
- Publication number
- WO2025072330A1 WO2025072330A1 PCT/US2024/048409 US2024048409W WO2025072330A1 WO 2025072330 A1 WO2025072330 A1 WO 2025072330A1 US 2024048409 W US2024048409 W US 2024048409W WO 2025072330 A1 WO2025072330 A1 WO 2025072330A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- cycloalkyl
- pyridine
- disease
- alkyl
- cancer
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
Definitions
- mast cells Upon activation, degranulation occurs, releasing these proinflammatory and immunomodulatory mediators into the surrounding tissues, generally in response to a perceived pathogen (e.g., parasitic, bacterial and viral infections, allergens, toxins, etc.).
- pathogen e.g., parasitic, bacterial and viral infections, allergens, toxins, etc.
- the activation of mast cells serves to induce an immune response to protect the body from pathogens, and to aid in wound healing, and tissue repair.
- misfunctioning mast cells underlie the etiology of many allergic and chronic inflammatory diseases and are implicated in a broad spectrum of conditions.
- SCF Stem Cell Factor
- the present disclosure relates to compounds that are inhibitors of the receptor tyrosine kinase KIT.
- the compounds are represented by Formula III and associated sub- formulas described herein: .
- this disclosure is directed to methods of inhibiting KIT in a subject comprising administering to the subject an effective amount of a compound of Formula III or an associated sub-formula described herein.
- this disclosure is directed to methods of reducing the activity and/or quantity of systemic mast cells in a subject comprising administering to the subject an effective amount of a compound of Formula III or an associated sub-formula described herein.
- this disclosure provides methods for treating a disease, disorder, or condition mediated at least in part by KIT in a subject, comprising administering to the subject a therapeutically effective amount of a compound of Formula III or an associated sub-formula described herein.
- KIT Diseases, disorders, and conditions mediated by KIT include e.g., an allergic disease, disorder, or condition; an inflammatory disease, disorder, or condition; a neuroinflammatory disease, disorder, or condition; a neurological disease, disorder, or condition; an immune related disease, disorder, or condition; an autoimmune related disease, disorder, or condition; a dermatological disease, disorder, or condition; a respiratory disease, disorder, or condition; a metabolic disease, disorder, or condition; a cardiovascular disease, disorder, or condition; a fibrotic disease, disorder, or condition; or cancer.
- Certain aspects of the present disclosure further comprise the administration of one or more additional therapeutic agents as set forth herein below.
- the near or approximating unrecited number can be a number which, in the context in which it is presented, provides the substantial equivalent of the specifically recited number.
- “about” means either within plus or minus 10% of the provided value, or rounded to the nearest significant figure, in all cases inclusive of the provided value. Where ranges are provided, they are inclusive of the boundary values.
- alkyl by itself or as part of another substituent, means, unless otherwise stated, a saturated hydrocarbon radical, having, in some embodiments, one to eight (e.g., C1-C8- alkyl), or one to six (e.g., C 1 -C 6 -alkyl), or one to four carbon atoms (e.g., C 1 -C 4 -alkyl), or one to three carbon atoms (e.g., C 1 -C 3 -alkyl), respectively.
- alkyl encompasses straight and branched-chain hydrocarbon groups.
- alkyl groups include, but are not limited to, methyl (Me, -CH 3 ), ethyl (Et, -CH 2 CH 3 ), n-propyl, isopropyl, n-butyl, t-butyl, isobutyl, sec-butyl, isopentyl, tert-pentyl, n-pentyl, isohexyl, n-hexyl, n-heptyl, 4-isopropylheptane, n-octyl, and the like.
- the alkyl groups are C1-C4 alkyl groups (e.g., methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl, or t-butyl).
- alkylene refers to a straight or branched, saturated, hydrocarbon radical having, in some embodiments, one to six carbon atoms (e.g., C1-C6-alkylene), or one to four carbon atoms (e.g., C1-C4-alkylene), or one to three carbon atoms (e.g., C1-C3-alkylene) and linking at least two other groups, i.e., a divalent hydrocarbon radical.
- two moieties are linked to the alkylene they can be linked to the same carbon atom (i.e., geminal), or different carbon atoms of the alkylene group.
- a straight chain alkylene can be the bivalent radical of -(CH2)n- , where n is 1, 2, 3, 4, 5 or 6 (i.e., a C 1 -C 6 -alkylene).
- Representative alkylene groups include, but are not limited to, methylene, ethylene, propylene, isopropylene, butylene, isobutylene, secbutylene, pentylene, hexylene and the like.
- the alkylene groups are C1- 3 alkylene groups (e.g., methylene, ethylene, or propylene).
- cycloalkyl refers to a monocyclic, bicyclic or polycyclic hydrocarbon ring system having, in some embodiments, 3 to 14 carbon atoms (e.g., C3-C14-cycloalkyl), or 3 to 10 carbon atoms (e.g., C3-C10-cycloalkyl), or 3 to 8 carbon atoms (e.g., C3-C8-cycloalkyl), or 3 to 6 carbon atoms (e.g., C 3 -C 6 -cycloalkyl) or 5 to 6 carbon atoms (e.g., C 5 -C 6 -cycloalkyl).
- 3 to 14 carbon atoms e.g., C3-C14-cycloalkyl
- 10 carbon atoms e.g., C3-C10-cycloalkyl
- 3 to 8 carbon atoms e.g., C3-C8-cycloalkyl
- 3 to 6 carbon atoms e
- Cycloalkyl groups can be saturated or characterized by one or more points of unsaturation (i.e., carbon-carbon double and/or triple bonds), provided that the points of unsaturation do not result in an aromatic system.
- monocyclic cycloalkyl groups include, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cyclohexenyl, cyclohexynyl, cycloheptyl, cycloheptenyl, cycloheptadienyl, cyclooctyl, cyclooctenyl, cyclooctadienyl and the like.
- the rings of bicyclic and polycyclic cycloalkyl groups can be fused, bridged, or spirocyclic.
- Non-limiting examples of bicyclic, spirocyclic and polycyclic hydrocarbon groups include bicyclo[1.1.1]pentane, bicyclo[2.1.1]hexane, bicyclo[2.2.1]heptane, bicyclo[2.2.2]octane, adamantyl, spiro[5.5]undecane, spiro[2.2]pentane, spiro[2.2]pentadiene, spiro[2.5]octane, spiro[2.2]pentadiene, and the like.
- the cycloalkyl groups of the present disclosure are monocyclic C 3 -C 6 -cycloalkyl moieties (e.g., cyclopropyl, cyclobutyl, cyclopentyl, or cyclohexyl).
- a wavy line, " ", that intersects a single, double or triple bond in any chemical structure depicted herein, represents that the point of attachment of the single, double, or triple bond to the remainder of the molecule is through either one of the atoms that make up the single, double or triple bond.
- a bond extending from a substituent to the center of a ring is meant to indicate attachment of that substituent to the ring at any of the available ring vertices, i.e., such that attachment of the substituent to the ring results in a chemically stable arrangement.
- halogen or “halo” are used interchangeably and refer to, unless otherwise stated, a fluorine, chlorine, bromine, or iodine atom.
- haloalkyl refers to alkyl groups, as defined herein, that are substituted with one or more halogen(s) (e.g., 1-3 halogen(s)).
- C 1 -C 6 haloalkyl is meant to include trifluoromethyl, difluoromethyl, 2,2,2-trifluoroethyl, 4-chlorobutyl, 3-bromopropyl, and the like.
- hydroxyalkyl refers to an alkyl group, as defined herein, that is substituted with one or more hydroxyl groups (e.g., 1-3 hydroxyl groups).
- Exemplary hydroxyalkyl groups include methanol, ethanol, 1,2-propanediol, 1,2-hexanediol, glycerol, and the like.
- cyanoalkyl refers to an alkyl group, as defined herein, that is substituted with one cyano group.
- Exemplary cyanoalkyl groups include cyanomethyl, cyanoethyl, cyanopropyl, and the like.
- alkoxy refers to an alkyl group, as defined herein, that is attached to the remainder of the molecule via an oxygen atom (e.g., -O-C1-C12 alkyl, -O-C1-C8 alkyl, -O-C 1 -C 6 alkyl, or -O-C 1 -C 3 alkyl).
- oxygen atom e.g., -O-C1-C12 alkyl, -O-C1-C8 alkyl, -O-C 1 -C 6 alkyl, or -O-C 1 -C 3 alkyl.
- alkoxy groups include methoxy (OMe, -OCH 3 ), ethoxy (OEt, -OCH 2 CH 3 ), n-propoxy, iso-propoxy, n-butoxy, sec- butoxy, n-pentoxy, n-hexoxy, and the like.
- heteroaryl refers to monocyclic or fused bicyclic aromatic groups (or rings) having, in some embodiments, from 5 to 14 (i.e., 5- to 14-membered heteroaryl), or from 5 to 10 (i.e., 5- to 10-membered heteroaryl), or from 5 to 6 (i.e., 5- to 6-membered heteroaryl) members (i.e., ring vertices), and containing from one to five, one to four, one to three, one to two or one ring heteroatom independently selected from nitrogen (N), oxygen (O), and sulfur (S).
- N nitrogen
- O oxygen
- S sulfur
- a heteroaryl group can be attached to the remainder of the molecule through a carbon atom or a heteroatom of the heteroaryl group, when chemically permissible.
- heteroaryl groups include pyridyl, pyridazinyl, pyrazinyl, pyrimidinyl, triazinyl, purinyl, thienopyridinyl, thienopyrimidinyl, pyrazolopyrimidinyl, imidazopyridines, isothiazolyl, pyrazolyl, indazolyl, pteridinyl, imidazolyl, triazolyl, tetrazolyl, oxazolyl, isoxazolyl, thiadiazolyl, pyrrolyl, thiazolyl, furyl, thienyl and the like.
- the heteroaryl groups of the present disclosure are monocyclic 5- to 6-membered heteroaryl moieties having 1-4 ring nitrogen atoms (e.g., pyridinyl, pyrimidinyl, pyrazinyl, pyridazinyl, triazolyl, imidazolyl, pyrazolyl, tetrazolyl, and the like).
- the heteroaryl groups of the present disclosure are 5-membered heteroaryl moieties having 1-4 ring heteroatoms independently selected from N, O, and S (e.g., imidazolyl, pyrazolyl, thiazolyl, oxazolyl, oxadiazole, thiadiazolyl, triazolyl, tetrazolyl, and the like).
- heterocycloalkyl refers to a non-aromatic monocyclic, bicyclic or polycyclic cycloalkyl ring having, in some embodiments, 3 to 14 members (e.g., 3- to 14-membered heterocycle), or 3 to 10 members (e.g., 3- to 10-membered heterocycle), or 4 to 8 members (e.g., 4- to 8-membered heterocycle), or 4 to 6 members (e.g., 4- to 6-membered heterocycle), or 5 to 6 members (e.g., 5- to 6-membered heterocycle), and having from one to five, one to four, one to three, one to two or one ring heteroatom independently selected from nitrogen (N), oxygen (O), and sulfur (S).
- N nitrogen
- O oxygen
- S sulfur
- Heterocycloalkyl groups are saturated or characterized by one or more points of unsaturation (e.g., one or more carbon-carbon double bonds, carbon-carbon triple bonds, carbon- nitrogen double bonds, and/or nitrogen-nitrogen double bonds), provided that the points of unsaturation do not result in an aromatic system.
- the rings of bicyclic and polycyclic heterocycloalkyl groups can be fused, bridged, or spirocyclic.
- heterocycloalkyl groups include aziridine, oxirane, thiirane, azetidine, oxetane, pyrrolidine, imidazolidine, pyrazolidine, dioxolane, phthalimide, piperidine, 1,4-dioxane, morpholine, thiomorpholine, thiomorpholine-S-oxide, thiomorpholine-S,S-oxide, piperazine, 3,4,5,6- tetrahydropyridazine, pyran, decahydroisoquinoline, 3-pyrroline, thiopyran, tetrahydrofuran, tetrahydrothiophene, quinuclidine, 2,6-diazaspiro[3.3]heptane, 2-azaspiro[3.3]heptane, 1- oxaspiro[3.3]heptane, 6-azaspiro[3.4]octane,
- a heterocycloalkyl group can be attached to the remainder of the molecule through a ring carbon atom, or a ring heteroatom, when chemically permissible.
- the heterocycloalkyl groups of the present disclosure are monocyclic 4- to 8-membered heterocycloalkyl moieties having one or two heteroatoms independently selected from N, O, and S, (e.g., azetidine, oxetane, pyrrolidine, tetrahydrofuran, tetrahydropyran, piperidine, piperazine, morpholine, and the like).
- salts of the compounds according to this disclosure are prepared with suitably nontoxic acids or bases, depending on the particular substituents found on the compounds described herein.
- base addition salts can be obtained by contacting the neutral form of such compounds with a sufficient amount of the desired base, either neat or in a suitable inert solvent.
- salts derived from pharmaceutically-acceptable inorganic bases include aluminum, ammonium, calcium, copper, ferric, ferrous, lithium, magnesium, manganic, manganous, potassium, sodium, zinc and the like.
- Salts derived from pharmaceutically-acceptable organic bases include salts of primary, secondary and tertiary amines, including substituted amines, cyclic amines, naturally-occurring amines and the like, such as arginine, betaine, caffeine, choline, N,N’-dibenzylethylenediamine, diethylamine, 2-diethylaminoethanol, 2-dimethylaminoethanol, ethanolamine, ethylenediamine, N- ethylmorpholine, N-ethylpiperidine, glucamine, glucosamine, histidine, hydrabamine, isopropylamine, lysine, methylglucamine, morpholine, piperazine, piperidine, polyamine resins, procaine, purines, theobromine, triethylamine, trimethylamine, tripropylamine, tromethamine and the like.
- acid addition salts can be obtained by contacting the neutral form of such compounds with a sufficient amount of the desired acid, either neat or in a suitable inert solvent.
- suitable inert solvent examples include those derived from inorganic acids like hydrochloric, hydrobromic, nitric, carbonic, monohydrogencarbonic, phosphoric, monohydrogenphosphoric, dihydrogenphosphoric, sulfuric, monohydrogensulfuric, hydriodic, or phosphorous acids and the like, as well as the salts derived from suitably nontoxic organic acids like acetic, propionic, isobutyric, malonic, benzoic, succinic, suberic, fumaric, mandelic, phthalic, benzenesulfonic, p- tolylsulfonic, citric, tartaric, methanesulfonic, and the like.
- salts of amino acids such as arginate and the like, and salts of organic acids like glucuronic or galacturonic acids and the like (see, for example, Berge, S.M., et al, “Pharmaceutical Salts”, Journal of Pharmaceutical Science, 1977, 66, 1-19).
- Certain specific compounds of the present disclosure contain both basic and acidic functionalities that allow the compounds to be converted into either base or acid addition salts.
- the neutral forms of the compounds may be regenerated by contacting the salt with a base or acid and isolating the parent compound in the conventional manner.
- the parent form of the compound differs from the various salt forms in certain physical properties, such as solubility in polar solvents, but otherwise the salts are equivalent to the parent form of the compound for the purposes of the present disclosure.
- This disclosure also contemplates isomers of the compounds described herein (e.g., stereoisomers and/or atropisomers).
- certain compounds of the present disclosure possess asymmetric carbon atoms (chiral centers), or hindered rotation about a single bond; the racemates, diastereomers, and enantiomers, and atropisomers (e.g., Ra, Sa, P and M isomers) of which are all intended to be encompassed within the scope of the present disclosure.
- Stereoisomeric forms may be defined, in terms of absolute stereochemistry, as (R) or (S), depicted uses dashes and/or wedges, and/or in terms of the direction the stereoisomer rotates plane- polarized light (e.g., dextrorotary ((+) or (d)), or levorotary ((-) or (l))).
- stereochemical depiction e.g., using dashes, , and/or wedges,
- a stereochemical assignment e.g., using (R) and (S) notation, or (d) and (l) notation
- isomer(s) e.g., enantiomers and diastereomers, when present.
- “Substantially free of” other isomer(s) indicates at least an 70/30 ratio of the indicated isomer to the other isomer(s), more preferably 80/20, 90/10, or 95/5 or more.
- the indicated isomer will be present in an amount of at least 99%.
- a chemical bond to an asymmetric carbon that is depicted as a solid line ( ) indicates that all possible stereoisomers (e.g., enantiomers, diastereomers, racemic mixtures, etc.) at that carbon atom are included.
- the compound may be present as a racemic mixture, scalemic mixture, or a mixture of diastereomers.
- the compounds of the present disclosure may also contain unnatural proportions of atomic isotopes at one or more of the atoms that constitute such compounds. Unnatural proportions of an isotope may be defined as ranging from the amount found in nature to an amount consisting of 100% of the atom in question.
- the compounds may incorporate radioactive isotopes, such as for example tritium ( 3 H), iodine-125 ( 125 I) or carbon-14 ( 14 C), or non-radioactive isotopes, such as deuterium ( 2 H) or carbon-13 ( 13 C).
- radioactive isotopes such as for example tritium ( 3 H), iodine-125 ( 125 I) or carbon-14 ( 14 C), or non-radioactive isotopes, such as deuterium ( 2 H) or carbon-13 ( 13 C).
- isotopic variants of the compounds of the disclosure may find additional utility, including but not limited to, as diagnostic and/or imaging reagents, or as cytotoxic/radiotoxic therapeutic agents.
- isotopic variants of the compounds of the disclosure can have altered pharmacokinetic and pharmacodynamic characteristics which can contribute to enhanced safety, tolerability or efficacy during treatment.
- the compounds according to this disclosure are characterized by one or more deuterium atoms.
- patient or “subject” are used interchangeably to refer to a human or a non- human animal (e.g., a mammal).
- non- human animal e.g., a mammal.
- treat refers to a course of action that eliminates, reduces, suppresses, mitigates, ameliorates, or prevents the worsening of, either temporarily or permanently, a disease, disorder or condition to which the term applies, or at least one of the symptoms associated therewith.
- Treatment includes alleviation of symptoms, diminishment of extent of disease, inhibiting (e.g., arresting the development or further development of the disease, disorder or condition or clinical symptoms association therewith) an active disease, delaying or slowing of disease progression, improving the quality of life, and/or prolonging survival of a subject as compared to expected survival if not receiving treatment or as compared to a published standard of care therapy for a particular disease.
- the term “in need of treatment” as used herein refers to a judgment made by a physician or similar professional that a subject requires or will benefit from treatment. This judgment is made based on a variety of factors that are in the realm of the physician’s expertise, which may include a positive diagnosis of a disease, disorder or condition.
- the terms “prevent”, “preventing”, “prevention”, “prophylaxis” and the like refer to a course of action initiated in a manner (e.g., prior to the onset of a disease, disorder, condition or symptom thereof) so as to prevent, suppress, inhibit or reduce, either temporarily or permanently, a subject’s risk of developing a disease, disorder, condition or the like (as determined by, for example, the absence of clinical symptoms) or delaying the onset thereof, generally in the context of a subject predisposed to having a particular disease, disorder or condition. In certain instances, the terms also refer to slowing the progression of the disease, disorder or condition or inhibiting progression thereof to a harmful or otherwise undesired state.
- Prevention also refers to a course of action initiated in a subject after the subject has been treated for a disease, disorder, condition or a symptom associated therewith in order to prevent relapse of that disease, disorder, condition or symptom.
- the preventative course of action is taken based on anticipation of a condition or event.
- prevention refers to the prevention, suppression, inhibition or reduction of an allergic, immune, or autoimmune response in a subject suffering from an allergic, inflammatory, neuroinflammatory, neurological, immune, autoimmune, dermatological, respiratory, metabolic, cardiovascular or fibrotic disease, disorder, or condition.
- response may refer to a symptom initiated by an irritant or trigger (e.g., an antigen originating from within the body, or from the external environment) in a subject.
- a reaction e.g., hives, rash, welts, itchy skin, stinging skin, skin fissures, skin lesions, skin blisters, swelling (e.g., in joints, glands, or tissues), vertigo, fatigue, dizziness, fainting, lightheadedness, muscle weakness, headache, dry skin, dry eyes, hair loss, numbness or tingling in extremities, joint pain and/or stiffness, sneezing, runny nose, stuffy nose, chest tightness and/or pain, shortness of breath, wheezing, itchy eyes, watery eyes, blurred vision, sensitivity to light, stomach cramping, abdominal pain, bloating, diarrhea, constipation, indigestion,
- a reaction e.g., hives, rash, welts, itchy skin,
- Subjects susceptible to a trigger are generally those suffering from an allergic, inflammatory, neuroinflammatory, neurological, immune, autoimmune, dermatological, respiratory, metabolic, cardiovascular or fibrotic disease, disorder, or condition, such as those described elsewhere herein.
- the term “in need of prevention” as used herein refers to a judgment made by a physician or other caregiver that a subject requires or will benefit from preventative care. This judgment is made based on a variety of factors that are in the realm of a physician’s or caregiver’s expertise.
- “Substantially pure” indicates that a component (e.g., a compound according to this disclosure) makes up greater than about 50% of the total content of the composition, and typically greater than about 60% of the total content.
- substantially pure refers to compositions in which at least 75%, at least 85%, at least 90% or more of the total composition is the component of interest. In some cases, the component of interest will make up greater than about 90%, or greater than about 95% of the total content of the composition.
- inhibitor of KIT and “KIT inhibitor” may be used interchangeably, and refer to the ability of a molecule to decrease the activation of KIT either directly or indirectly, thereby decreasing activation and/or quantity of systemic mast cells.
- Compounds that are selective for KIT may be particularly useful in the treatment of certain disorders or may offer a reduced likelihood of undesired side effects.
- compounds of the present disclosure are selective over one or more other receptor tyrosine kinases.
- specific examples include, but are not limited to, PDGFR ⁇ , PDGFR ⁇ , CSF1R, and FLT3.
- Selectivity may be determined, for example, by comparing the inhibition of a compound as described herein against KIT against the inhibition of a compound as described herein against another kinase.
- the selective inhibition of a compound of Formula III or an associated sub-formula is at least 1000 times greater, 500 times greater, 100 times greater, 50 times greater, or 20 times greater than inhibition of one or more kinases selected from PDGFR ⁇ , PDGFR ⁇ , CSF1R, and FLT3.
- Compounds provided herein may have advantageous pharmacokinetic profiles including, for example, hepatocyte stability, clearance, inhibition against CYP, and inhibition against hERG.
- Compounds of the Disclosure [0037] The present disclosure relates to compounds that inhibit the activity of KIT.
- this disclosure is directed to a compound having a structure according to Formula III: (Formula III) or a pharmaceutically acceptable salt thereof, wherein: A is: (i) selected from the group consisting of: , , membered heteroaryl is substituted with 0-2 R 1d ; or (iii) -NHC(O)-R 1g ; each R 1 is independently selected from the group consisting of halo, -CN, -OH, C 1 -C 6 - alkyl, C1-C6-haloalkyl, C1-C6-hydroxyalkyl, C1-C6-cyanoalkyl, C1-C6 alkoxy, C3-C6-cycloalkyl, - O-C3-C6-cycloalkyl, 4- to 8-membered heterocycloalkyl, -NR 1a R 1b , phenyl, 5- to 6-membered 12 heteroaryl, -X 1 -NR
- A is selected from the group consisting of: , , , [0040] In some embodiments, . In some embodiments, A is some embodiments, A is . In some embodiments, A is . In some embodiments, A is . In some embodiments, A . In some embodiments, , . In some embodiments, A is . In some embodiments, A is . In some embodiments, A is . In some embodiments, A is . In some embodiments, A is . In some embodiments, A is . In some embodiments, A is . In some embodiments, A is . In some embodiments, A is . In some embodiments, A is . In some embodiments, A is . In some embodiments, A is . In some embodiments, A is . In some embodiments, A is . In some embodiments, A is . In some embodiments, A is . In some embodiments, A is . In some embodiments, A is . In some embodiments, A is . In some embodiments, A
- A is . In some embodiments, A is . some embodiments, A is . some embodiments, . In some embodiments, A is . In some embodiments, A is . In some embodiments, A is . In some embodiments, A is . In some embodiments, A is . In some embodiments, A is . In some embodiments, A is . In some embodiments, A is . In some embodiments, A is . In some embodiments, A is . , . , . In some embodiments, A is . In some embodiments, . In some embodiments, some embodiments, A is . In some embodiments, . In some embodiments, some embodiments, some embodiments, some embodiments, A is . In some embodiments, . In some embodiments, some embodiments, some embodiments, A is . , . In some embodiments, A is some embodiments, A is . In some embodiments, . In some embodiments, some embodiments, A is some embodiments, A is . In some embodiment
- A is . In some embodiments, A is . In some embodiments, A is . In some embodiments, A is . In some embodiments, A is . In some embodiments, A , . In some embodiments, A is . , . In some embodiments, . In some embodiments, . In some embodiments, . In some embodiments, A is . In some embodiments, . In some embodiments, A is some embodiments, A is . In some embodiments, . In some embodiments, . In some embodiments, A is .
- A is a 6-membered heteroaryl having 1-3 ring nitrogen atoms, wherein the 6-membered heteroaryl is substituted with 0-2 R 1d .
- A is a 6- membered heteroaryl having 1-3 ring nitrogen atoms, wherein the 6-membered heteroaryl is substituted with 1-2 R 1d .
- A is pyridazinyl, pyrimidinyl, pyrazinyl, or pyridinyl, wherein A is substituted with 0-2 R 1d .
- A is pyridazinyl, pyrimidinyl, pyrazinyl, or pyridinyl, wherein A is substituted with 1-2 R 1d . In some embodiments, .
- each R 1d when present, is independently halo, C 1 -C 6 alkoxy, C 1 - C3 haloalkoxy, cyclopropyl, -O-cyclopropyl, or -NR 1e R 1f ; and R 1e and R 1f , when present, are independently H, C1-C6 haloalkyl, -(methylene)-(cyclopropyl), or tetrahydrofuranyl; or R 1e and R 1f together with the nitrogen atom to which they are attached form an azetidinyl group.
- A is -NHC(O)-R 1g .
- R 1g when present, is cyclopropyl, -(methylene)-(cyclopropyl), pyridinyl, or pyrazinyl; wherein R 1g is optionally substituted with 0-2 R 1h .
- R 1g when present, is , , , .
- this disclosure is directed to a compound having a structure according to Formula III: (Formula III) or a pharmaceutically acceptable salt thereof, wherein: ring A is a 5-membered heteroaryl having 1-4 ring heteroatoms independently selected from N, O, and S, wherein the heteroaryl is substituted with 0-2 R 1 ; each R 1 is independently selected from the group consisting of halo, -CN, -OH, C1-C6- alkyl, C 1 -C 6 -haloalkyl, C 1 -C 6 -hydroxyalkyl, C 1 -C 6 -cyanoalkyl, C 1 -C 6 alkoxy, C 3 -C 6 -cycloalkyl, - O-C 3 -C 6 -cycloalkyl, 4- to 8-membered heterocycloalkyl, -NR 1a R 1b , phenyl, 5- to 6-membered heteroaryl, -X 1 -NR 1
- ring A is selected from the group consisting of oxadiazolyl, thiazolyl, thiadiazolyl, triazolyl, imidazolyl, pyrazolyl, and tetrazolyl, each of which is substituted with 0-2 R 1 .
- ring A is selected from the group consisting of thiazolyl, thiadiazolyl, triazolyl, imidazolyl, pyrazolyl, and tetrazolyl, each of which is substituted with 0-2 R 1 .
- ring A is imidazolyl, pyrazolyl, triazolyl, or tetrazolyl, each of which is substituted with 0-2 R 1 .
- ring A is selected from the group consisting of , , , , , , , , , , each of which is substituted with 0-2 R 1 .
- ring A , , with 0-2 R 1 ring A is selected from the group consisting of, , , , , , , , , , , , each of which is substituted with 0-2 R 1 .
- ring A is each of which is substituted with 0-2 R 1 . In some embodiments, ring A is substituted with 0-2 R 1 . In some embodiments, ring . [0049] In some embodiments, ring A is a 5-membered heteroaryl having 1-4 ring nitrogen atoms, wherein the heteroaryl is substituted with two adjacent R 1 groups that combine with the two ring atoms to which they are attached to form a 5- or 6-membered heterocycloalkyl group having 1-2 ring heteroatoms independently selected from N and O.
- ring A is pyrazolyl or triazolyl substituted with two adjacent R 1 groups that combine with the two ring atoms to which they are attached to form a 5- or 6-membered heterocycloalkyl group having 1-2 ring heteroatoms independently selected from N and O.
- ring r .
- R 6 is H, or C1-C3-alkyl. In some embodiments, R 6 is H or methyl. In some embodiments, R 6 is methyl. In some embodiments, R 6 is H.
- R 5 is H, C1-C3-alkyl, or -NR 5a R 5b ; wherein R 5a and R 5b are independently H or C1-C3 alkyl.
- R 5 is H, methyl, or -NH2.
- R 5 is H.
- R 5 is methyl.
- R 5 is -NR 5a R 5b , wherein at least one of R 5a and R 5b is H.
- R 5 is -NH2.
- R 5 and R 6 are H.
- m is 0, 1, or 2; and each R 4 , when present is independently halo, C1-C6 alkyl, C1-C6-alkoxy, C3-C6 cycloalkyl, 4- to 8-membered heterocycloalkyl having 1-3 ring heteroatoms independently selected from N, O, and S, or phenyl, wherein said phenyl is optionally substituted with 1-3 halo.
- m is 0.
- m is 1; and R 4 is halo, C 1 -C 6 alkyl, C 1 -C 6 -alkoxy, C 3 -C 6 cycloalkyl, 4- to 8-membered heterocycloalkyl having 1-3 ring heteroatoms independently selected from N, O, and S, or phenyl, wherein said phenyl is optionally substituted with 1-3 halo.
- m is 2; and each R 4 is independently halo, C 1 -C 6 alkyl, C 1 -C 6 -alkoxy, C 3 -C 6 cycloalkyl, 4- to 8-membered heterocycloalkyl having 1-3 ring heteroatoms independently selected from N, O, and S, or phenyl, wherein said phenyl is optionally substituted with 1-3 halo.
- m is 0, 1, or 2; and at least one R 4 , when present is halo.
- R 4 is -F, -Cl, -Br, or -I.
- R 4 is -F, or -Cl.
- R 4 is -F. In some embodiments, R 4 is -Cl. In some embodiments, m is 0, 1, or 2, and at least one R 4 , when present, is C1-C6 alkyl. In some embodiments, R 4 is C1- C 3 alkyl. In some embodiments, R 4 is methyl, ethyl, n-propyl, or isopropyl. In some embodiments, R 4 is methyl, or ethyl. In some embodiments, R 4 is methyl. In some embodiments, R 4 is ethyl. In some embodiments, m is 0, 1, or 2; and at least one R 4 , when present, is C1-C6-alkoxy.
- R 4 is C1-C3-alkoxy. In some embodiments, R 4 is methoxy, ethoxy, n-propoxy, or isopropoxy. In some embodiments, R 4 is methoxy or ethoxy. In some embodiments R 4 is methoxy. In some embodiments, R 4 is ethoxy. In some embodiments, m is 0, 1, or 2; and at least one R 4 , when present, is C3-C6 cycloalkyl. In some embodiments, R 4 is cyclopropyl, cyclobutyl, cyclopentyl, or cyclohexyl. In some embodiments, R 4 is cyclopropyl.
- m is 0, 1, or 2; and at least one R 4 , when present, is 4- to 8-membered heterocycloalkyl having 1-3 ring heteroatoms independently selected from N, O, and S.
- R 4 is a 5- to 6- membered heterocycloalkyl having 1-3 ring heteroatoms independently selected from N, O, and S.
- R 4 is a 6-membered heterocycloalkyl having 1-3 ring heteroatoms independently selected from N, and O.
- R 4 is piperidine, piperazine, or morpholine. In some embodiments, R 4 is morpholine.
- m is 0, 1, or 2; and at least one R 4 , when present, is phenyl optionally substituted with 1-3 halo. In some embodiments, R 4 is phenyl substituted with one halo. In some embodiments, R 4 is phenyl substituted with -F, - Cl, -Br, or -I. In some embodiments, R 4 is phenyl substituted with -F. In some embodiments, m is 0, 1, or 2; and each R 4 , when present, is independently -F, -Cl, -CH 3 , -CH 2 CH 3 , -OCH 3 , - OCH 2 CH 3 , cyclopropyl, .
- m is 1; and R 4 , is independently -F, -Cl, -CH 3 , -CH 2 CH 3 , -OCH 3 , -OCH 2 CH 3 , cyclopropyl, r .
- m is 2; and each R 4 is independently -F, -Cl, -CH 3 , -CH 2 CH 3 , - OCH 3 , -OCH 2 CH 3 , cyclopropyl, .
- m is 0, 1, or 2; and each R 4 , when present is independently halo, C 1 -C 6 alkyl, C 1 -C 6 -alkoxy, or C 3 -C 6 cycloalkyl. In some embodiments, m is 0 or 1; and R 4 , when present is halo, C 1 -C 6 alkyl, C 1 -C 6 -alkoxy, or C 3 -C 6 cycloalkyl. In some embodiments, m is 0. In some embodiments, m is 1, and R 4 is halo, C1-C6 alkyl, C1-C6-alkoxy, or C3-C6 cycloalkyl.
- R 4 is halo, C1-C3-alkyl, C1-C3-alkoxy, or C3-C5-cycloalkyl. In some embodiments, R 4 is -F, -Cl, -CH 3 , -CH 2 CH 3 , -OCH 3 , -OCH 2 CH 3 , or cyclopropyl. [0055] In some embodiments, m is 0, 1, or 2; and each R 4 is independently halo, or C1-C6-alkoxy. In some embodiments, at least one R 4 is halo. In some embodiments, at least one R 4 is Cl, Br, or F. In some embodiments, at least one R 4 is Cl.
- At least one R 4 is C 1 -C 6 - alkoxy. In some embodiments, at least one R 4 is C 1 -C 3 -alkoxy. In some embodiments, at least one R 4 is -OCH3. In some embodiments, m is 0, 1, or 2; and each R 4 is independently Cl, or - OCH 3 . [0056] In some embodiments, R 3 is halo, C 1 -C 6 alkyl, or C 1 -C 6 hydroxyalkyl. In some embodiments, R 3 is halo, C1-C3 alkyl, or C1-C3 hydroxyalkyl. In some embodiments, R 3 is halo.
- R 3 is -F, -Cl, or -Br. In some embodiments, R 3 is -F, or -Cl. In some embodiments, R 3 is -F. In some embodiments, R 3 is -Cl. In some embodiments, R 3 is C1-C3 alkyl. In some embodiments, R 3 is -CH3. In some embodiments, R 3 is C1-C3 hydroxyalkyl. In some embodiments, R 3 is -CH 2 -OH, -CH 2 CH 2 -OH, or -CH 2 CH 2 CH 2 -OH. In some embodiments, R 3 is -CH2-OH.
- R 3 is -F, -Cl, -CH3, or -CH2-OH. [0057] In some embodiments, R 3 is halo or C1-C6-alkyl. In some embodiments, R 3 is halo, or C1- C 3 -alkyl. In some embodiments, R 3 is halo. In some embodiments, R 3 is -F, -Cl, or -Br. In some embodiments, R 3 is -Cl. In some embodiments, R 3 is C 1 -C 3 alkyl. In some embodiments, R 3 is - CH3. In some embodiments, R 3 is -Cl, or -CH3.
- n is 0 or 1, and R 2 , when present is halo, -CN, -C 1 -C 3 -alkyl, -C 1 - C 3 -alkoxy, or C 3 -C 6 -cycloalkyl. In some embodiments, n is 0. In some embodiments, n is 1. In some embodiments, R 2 , when present, is halo. In some embodiments, R 2 , when present, is -F, - Cl, -Br, or -I. In some embodiments, R 2 , when present, is -F, or -Cl. In some embodiments, R 2 , when present, is -F.
- R 2 when present, is -Cl. In some embodiments, R 2 , when present, is -C1-C3-alkyl. In some embodiments, R 2 , when present, is methyl, ethyl, n-propyl, or isopropyl. In some embodiments, R 2 , when present, is methyl. In some embodiments, R 2 , when present, is -C 1 -C 3 -alkoxy. In some embodiments, R 2 , when present, is methoxy, ethoxy, n- propoxyl, or isopropoxy. In some embodiments, R 2 , when present, is methoxy.
- R 2 when present, is C3-C6-cycloalkyl. In some embodiments, R 2 , when present, is cyclopropyl, cyclobutyl, cyclopentyl, or cyclohexyl. In some embodiments, R 2 , when present, is cyclopropyl. In some embodiments, n is 0 or 1, and R 2 , when present, is -F, -Cl, -CN, -CH 3 , - OCH3, or cyclopropyl. [0059] In some embodiments, n is 1, and R 2 is halo. In some embodiments, n is 1 and R 2 is -F or -Cl.
- each R 1 is independently halo, -CN, -OH, C1-C6-alkyl, C1-C6- haloalkyl, C1-C6-hydroxyalkyl, C1-C6 alkoxy, C3-C6-cycloalkyl, -O-C3-C6-cycloalkyl, 4- to 6- membered heterocycloalkyl, -NR 1a R 1b , phenyl, 6-membered heteroaryl, -X 1 -C(O)NR 1a R 1b , -X 1 -O- (C1-C3-alkyl), -X 1 -C3-C6-cycloalkyl, and -X 1 -phenyl; wherein each 6-membered heteroaryl has 1- 2 ring heteroatoms independently selected from N and O; each 4- to 6-membered heterocycloalkyl has 1-3 ring heteroatoms independently selected from N, O, and S;
- each R 1 is independently selected from the group consisting of C1-C6-alkyl, C1-C6-haloalkyl, C1-C6 alkoxy, C3-C6-cycloalkyl, 4- to 8-membered heterocycloalkyl having 1-2 ring heteroatoms independently selected from N and O, -NR 1a R 1b , and -X 1 -C3-C6- cycloalkyl; wherein X 1 is C 1 -C 3 -alkylene; and R 1a and R 1b , when present, are independently H, or C 1 -C 3 alkyl.
- R 1 is selected from the group consisting of C1-C6-alkyl, C1-C6- haloalkyl, C 3 -C 6 -cycloalkyl, and -X 1 -C 3 -C 6 -cycloalkyl, wherein X 1 is C 1 -C 3 -alkylene.
- R 1 is C 1 -C 6 -alkyl.
- R 1 is C 1 -C 4 -alkyl.
- R 1 is methyl, ethyl, n-propyl, iso-propyl, iso-butyl, or tert-butyl.
- R 1 is C1-C6-haloalkyl. In some embodiments, R 1 is C1-C3- haloalkyl. In some embodiments, R 1 is difluoromethyl, trifluoromethyl, trifluoroethyl, difluoroethyl, trifluoropropyl, or difluoropropyl. In some embodiments, R 1 is difluoromethyl, trifluoromethyl, trifluoroethyl, or trifluoropropyl. In some embodiments, R 1 is -CF2H, CF3, - CH 2 CF 2 H, -CH 2 CF 3 , or -CH 2 CH 2 CF 3 .
- R 1 is C 1 -C 6 alkoxy. In some embodiments, R 1 is C 1 -C 3 alkoxy. In some embodiments, R 1 is methoxy. [0066] In some embodiments, R 1 is C 3 -C 6 -cycloalkyl optionally substituted with 1-3 R 1c . In some embodiments, R 1 is C 3 -C 6 -cycloalkyl optionally substituted with 1-2 R 1c . In some embodiments, R 1 is C3-C5-cycloalkyl optionally substituted with 1-3 R 1c . In some embodiments, R 1 is C3-C5-cycloalkyl optionally substituted with 1-2 R 1c .
- R 1 is cyclopropyl, cyclobutyl, cyclopentyl, or cyclohexyl, each of which is optionally substituted with 1-3 R 1c .
- R 1 is cyclopropyl, cyclobutyl, cyclopentyl, or cyclohexyl, each of which is optionally substituted with 1-2 R 1c .
- R 1 is cyclopropyl, cyclobutyl, or cyclopentyl, each of which is optionally substituted with 1-3 R 1c .
- R 1 is cyclopropyl, cyclobutyl, or cyclopentyl, each of which is optionally substituted with 1-2 R 1c .
- each R 1c when present, is independently halo or -OH.
- each R 1c when present, is independently -F, or -OH.
- R 1 is , . [0067]
- R 1 is C 3 -C 6 -cycloalkyl.
- R 1 is C 3 -C 5 - cycloalkyl.
- R 1 is cyclopropyl, cyclobutyl, cyclopentyl, or cyclohexyl.
- R 1 is cyclopropyl, cyclobutyl, or cyclopentyl.
- R 1 is -O-C 3 -C 6 -cycloalkyl.
- R 1 is -O- cyclopropyl, -O-cyclobutyl, -O-cyclopentyl, or -O-cyclohexyl.
- R 1 is -O- cyclopropyl.
- R 1 is 4- to 8-membered heterocycloalkyl having 1-2 ring heteroatoms independently selected from N and O.
- R 1 is 5- to 6-membered heterocycloalkyl having 1-2 ring heteroatoms independently selected from N and O.
- R 1 is pyrrolidinyl, tetrahydrofuranyl, tetrahydropyranyl, piperidinyl, or morpholinyl.
- R 1 is tetrahydrofuranyl, tetrahydropyranyl, or morpholinyl.
- R 1 is , , . In some embodiments, R 1 is .
- R 1 is -NR 1a R 1b , wherein R 1a and R 1b are independently H, C 1 -C 3 alkyl, or C 3 -C 6 cycloalkyl.
- R 1 is -NR 1a R 1b , wherein R 1a and R 1b are independently H, or C1-C3 alkyl.
- at least one of R 1a and R 1b is C1-C3 alkyl.
- both R 1a and R 1b are C1-C3 alkyl.
- at least one of R 1a and R 1b is C 3 -C 6 cycloalkyl.
- anti-inflammatory agents include non-steroidal anti-inflammatory drugs (NSAIDs) (e.g., ibuprofen, aspirin, naproxen, and celecoxib, etodolac, meloxicam, nabumetone, diclofenac, diflunisal, fenoprofen, and flurbiprofen); corticosteroids (e.g., cortisone, prednisone, prednisolone, methylprednisolone, dexamethasone, deflazacort, betamethasone, hydrocortisone, etc.); disease-modifying antirheumatic drugs (DMARDs) (e.g., methotrexate, sulfasalazine, hydroxychloroquine, and leflunomide); anti-tumor necrosis factor (anti-TNF) agents (e.g., infliximab, adalimumab, and
- the additional therapeutic agent comprises an antipsychotic.
- antipsychotics include haloperidol, loxapine, thioridazine, molindone, thiothixene, fluphenazine, mesoridazine, trifluoperazine, perphenazine, chlorpromazine, aripiprazole, clozapine, ziprasidone, risperidone, quetiapine, and olanzapine.
- the additional therapeutic agent comprises one or more anti- anxiety agents.
- anti-anxiety agents include alprazolam, chlordiazepoxide, clonazepam, diazepam, lorazepam, and buspirone.
- the additional therapeutic agent comprises one or more anticonvulsants (e.g., valproic acid, phenytoin, clonazepam, and carbamazepine).
- the additional therapeutic agent comprises one or more respiratory agents.
- the respiratory agent is a bronchodilator (e.g., adrenergic bronchodilator, anticholinergic bronchodilator, methylxanthines, and combinations thereof), an inhaled corticosteroid (e.g., beclomethasone, fluticasone, ciclesonide, mometasone, and budesonide), a beta andrenergic agonist (e.g., albuterol, metaproterenol, pirbuterol, terbutaline, isoetharine and levalbuterol), or leukotriene modifier (e.g., montelukast, zafirlukast, and zileuton).
- a bronchodilator e.g., adrenergic bronchodilator, anticholinergic bronchodilator, methylxanthines, and combinations thereof
- an inhaled corticosteroid e.g., beclomet
- the additional therapeutic agent comprises one or more nasal decongestants.
- exemplary decongestants include oxymetazoline, phenylephrine, and pseudoephedrine.
- the additional therapeutic agent comprises a cough suppressant.
- exemplary cough suppressants include dextromethorphan, guaifenesin, and codeine.
- the compounds according to this disclosure are combined with a proton pump inhibitor (PPI).
- PPIs include lansoprazole, omeprazole, pantoprazole, rebaprazole, and esomeprazole.
- the additional therapeutic agent comprises an agent that modulates cognitive function, e.g., cholinesterase inhibitors (e.g., donepezil, rivastigmine, galantamine), N-methyl-D-aspartate (NMDA) receptor antagonists (e.g., memantine), and agents targeting aggregated soluble and insoluble forms of amyloid beta (e.g., aducanumab).
- cholinesterase inhibitors e.g., donepezil, rivastigmine, galantamine
- NMDA N-methyl-D-aspartate
- amyloid beta e.g., aducanumab
- the additional therapeutic agent comprises an agent that targets thyroid function, such as, for example, anti-thyroid agents (e.g., radioiodine, propylthiouracil (PTU), and methimazole), or thyroid hormone replacement therapy (e.g., levothyrozine, or cytomel).
- anti-thyroid agents e.g., radioiodine, propylthiouracil (PTU), and methimazole
- thyroid hormone replacement therapy e.g., levothyrozine, or cytomel.
- the additional therapeutic agent comprises one or more agents useful in the treatment of diabetes, such as, e.g., insulin; synthetic glucagon; hyperglycemic agents (e.g., metformin, sulfonylureas, glinides, thiazolidinediones, dipeptidyl peptidase-4 (DPP- 4) inhibitors; anti-hyperglycemic agents (e.g., sodium glucose cotransporter-2 (SGLT2) inhibitors including, e.g., canagliflozin, dapagliflozin, and empagliflozin); and GLP-1 receptor agonists (e.g., semaglutide, exenatide, dulaglutide, liraglutide, or lixisenatide).
- agents useful in the treatment of diabetes such as, e.g., insulin; synthetic glucagon; hyperglycemic agents (e.g., metformin, sulfonylureas, glin
- the additional therapeutic agent comprises a diuretic.
- diuretics include spironolactone, bumetanide, torsemide, hydrochlorothiazide, furosemide, and metolazone, and aldosterone antagonists (e.g., spironolactone and eplerenone).
- the additional therapeutic agent comprises one or more of an antidiarrheal (e.g., eluxadoline, or alosetron), a laxative (lubiprostone, or a guanylate cyclase- C (GC-C agonist (e.g., linaclotide).
- an antidiarrheal e.g., eluxadoline, or alosetron
- laxative lubiprostone
- GC-C agonist e.g., linaclotide
- the additional therapeutic agent comprises a cholinergic modulator, such as a cholinergic agonist (e.g., chantix, pilocarpine, or bethanechol), or an anticholinergic agent (e.g., atropine, belladonna alkaloids, benztropine mesylate, clidinium, cyclopentolate, darifenacin, dicylomine, fesoterodine, flavoxate, glycopyrrolate, homatropine hydrobromide, hyoscyamine, ipratropium, orphenadrine, oxybutynin, propantheline, scopolamine, methscopolamine, solifenacin, tiotropium, tolterodine, trihexphenidyl, and trospium).
- a cholinergic modulator such as a cholinergic agonist (e.g., chantix, pilocarpine, or bethanechol), or
- the additional therapeutic agent comprises a vasodilator.
- vasodilators include, but are not limited to nitrates (e.g., nitroprusside, nitroglycerine, isosorbide, and amyl nitrate), hydralazine, treprostinil, minoxidil, angiotensin-converting enzyme (ACE) inhibitors (e.g., benazepril, captopril, enalapril, fosinopril, lisinopril, moexipril, perindopril, quinapril, ramipril, and trandolapril), and angiotensin receptor blockers (ARBs) (e.g., azilsartan, candesartan, eprosartan, irbesartan, losartan, olmesartan, telmisartan, and valsartan).
- ACE angiotensin-converting
- the additional therapeutic agent comprises a thrombolytic agent (e.g., streptokinase, alteplase, reteplase, Tenecteplase, urokinase, prourokinase, and anistreplase).
- a thrombolytic agent e.g., streptokinase, alteplase, reteplase, Tenecteplase, urokinase, prourokinase, and anistreplase.
- the additional therapeutic agent comprises an anticoagulant. Exemplary anticoagulants include rivaroxaban, dabigatran, apixaban, eboxaban, and warfarin.
- the additional therapeutic agent comprises an agent useful in the treatment of fibrosis. Certain such agents include pirfenidone and nintedanib.
- the additional therapeutic agent comprises a targeted agent useful in the treatment of pulmonary arterial hypertension.
- Targeted agents useful in the treatment of pulmonary arterial hypertension include phosphodiesterase-5 (PDE5) inhibitors (e.g., sildenafil, tadalafil and vardenafil); guanylate cyclase stimulators (GCS) (e.g., adempas, riociguat, vericiguat and verquvo); endothelin receptor antagonists (e.g., bosentan, ambrisentan, and macitentan), and prostacyclin and analogues thereof.
- PDE5 phosphodiesterase-5
- GCS guanylate cyclase stimulators
- endothelin receptor antagonists e.g., bosentan, ambrisentan, and macitentan
- Exemplary CFTR modulators include ivacaftor, elexacaftor, lumacaftor, and tezacaftor.
- the additional therapeutic agent comprises an antibiotic.
- Exemplary antibiotics include, but are not limited to phenoxymethylpenicillin, dicloxacillin, amoxicillin, ampicillin, nafcillin, oxacillin, penicillin, cefaclor, cefazolin, cefadroxil, cephalexin, cefuroxime, cefixime, ceroxitin, ceftriaxone, doxycycline, minocycline, sarecycline, erythromycin, clarithromycin, azithromycin, fidaxomicin, roxithromycin, ciprofloxacin, ofloxacin, levofloxacin, moxifloxacin, sulfamethoxazole with trimethoprim, sulfasalazine
- the additional therapeutic agent comprises one or more agents selected from the groups consisting of anti-inflammatory agents, analgesic agents, agents that target one or more cytokines, immunosuppressants, agents that targets one or more mast-cell derived immunomodulators, BTK inhibitors, IgE inhibitors, anti-depressants, anti-psychotics, anti-anxiety agents, anticonvulsants, respiratory agents, nasal decongestants, cough suppressants, proton pump inhibitors (PPIs), agents that modulate cognitive function, agents that target thyroid function, agents useful in the treatment of diabetes, diuretics, antidiarrheals, laxatives, GC-C agonists, cholinergic modulators, antiarrhythmics, vasodilators, cholesterol modifiers, thrombolytic agents, anticoagulants, agents useful in the treatment of fibrosis, agents useful in the treatment of arterial hypertension, mucolytic agents, pancreatic enzymes, CFTR modulators, and/or antibiotics.
- BTK inhibitors IgE inhibitors
- the additional therapeutic agent comprises one or more agents selected from the group consisting of an anti-inflammatory agent, an analgesic agent, an immunosuppressant, and/or an agent that targets one or more cytokines (e.g., IL-12, IL-17, and/or IL-23).
- the additional therapeutic agent comprises one or more agents selected from the group consisting of a respiratory agent, an anti-inflammatory agent, an agent that targets one or more cytokines (e.g., IL-4 and/or IL-13), a mast-cell stabilizer, and/or an agent that targets a mast-cell derived immunomodulator (e.g., leukotrienes).
- the additional therapeutic agent comprises one or more agents selected from the group consisting of an anti-depressant, an anti-psychotic, an anti-anxiety agent, an anticonvulsant, an agent that modulates cognitive function, an anti-CD20 antibody (e.g., ocrelizumab), an anti-inflammatory, and/or an immunosuppressant.
- the additional therapeutic agent comprises an anti-inflammatory and/or an immunosuppressant.
- the additional therapeutic agent comprises an anti-inflammatory agent, an immunosuppressant, and/or an agent that targets a mast-cell derived immunomodulator (e.g., antihistamine and/or leukotriene modulators).
- the additional therapeutic agent comprises an antihistamine, a BTK inhibitor, and/or an IgE inhibitor.
- the additional therapeutic agent comprises an antihistamine, an anti-inflammatory agent (e.g., a corticosteroid), and IgE inhibitor, and/or an immunosuppressant (e.g., cyclosporine).
- the additional therapeutic agent comprises an anti-inflammatory agent, an immunosuppressive agent, an agent that targets one or more cytokines, or a combination thereof.
- the additional therapeutic agent comprises a cholesterol modifier, a diuretic, an antiarrhythmic, a vasodilator, an anti-inflammatory, an analgesic agent, or any combination thereof.
- the compounds described herein are combined with one or more additional therapeutic agents that are considered to be the standard of care (SOC) for one or more of the inflammatory, immune, and/or autoimmune-related indications described herein.
- SOC standard of care
- Exemplary SOC therapies for the indications described herein are summarized in Table 1 and Table 2 below. Table 1 66 Table 2 68 Cancer Therapies [0186]
- the present disclosure contemplates the use of the KIT inhibitors described herein in combination with one or more additional therapies useful in the treatment of cancer.
- one or more of the additional therapies is an additional treatment modality.
- Exemplary treatment modalities include but are not limited to surgical resection of a tumor, bone marrow transplant, radiation therapy, and photodynamic therapy.
- one or more of the additional therapies is a therapeutic agent.
- Exemplary therapeutic agents include chemotherapeutic agents, radiopharmaceuticals, hormone therapies, epigenetic modulators, ATP-adenosine axis-targeting agents (e.g., CD73 inhibitors, CD39 inhibitors, A 2A R inhibitors, and/or A 2B R inhibitors), signal transduction inhibitors (e.g., inhibitors of one or more of TYRO3, MERTK, EGFR, FGFR, VEGFR, HER-2, HER-3, BRAF, RET, MET, ABL, ALK, FLT-3, JAK, STAT, NF-kB), RAS signaling inhibitors (e.g., inhibitors of one or more of KRAS, HRAS, RAF, MEK, ERK, PTEN, SOS (e.g., SOS1), mTORC1, SHP2 (PTPN11), and AKT), PI3K inhibitors, arginase inhibitors, HIF inhibitors (e.g., inhibitors of HIF- 2 ⁇ ), A
- one or more of the additional therapeutic agents is a chemotherapeutic agent.
- chemotherapeutic agents include, but are not limited to, alkylating agents such as thiotepa and cyclosphosphamide; alkyl sulfonates such as busulfan, improsulfan and piposulfan; aziridines such as benzodopa, carboquone, meturedopa, and uredopa; ethylenimines and methylamelamines including altretamine, triethylenemelamine, triethylenephosphoramide, triethylenethiophosphoramide and trimethylolomelamime; nitrogen mustards such as chlorambucil, chlornaphazine, cholophosphamide, estramustine, ifosfamide, mechlorethamine, mechlorethamine oxide hydrochloride, melphalan, novembichin, phenesterine, prednimustine, tro
- combination therapy comprises a chemotherapy regimen that includes one or more chemotherapeutic agents.
- combination therapy comprises a chemotherapeutic regimen comprising FOLFOX (folinic acid, fluorouracil, and oxaliplatin), FOLFIRI (e.g., folinic acid, fluorouracil, and irinotecan), a taxoid (e.g., docetaxel, paclitaxel, nab- paclitaxel, etc.), and gemcitabine.
- FOLFOX folinic acid, fluorouracil, and oxaliplatin
- FOLFIRI e.g., folinic acid, fluorouracil, and irinotecan
- a taxoid e.g., docetaxel, paclitaxel, nab- paclitaxel, etc.
- gemcitabine e.g., gemcitabine.
- one or more of the additional therapeutic agents is an immune checkpoint inhibitor.
- immune checkpoint inhibitor refers to an antagonist of an inhibitory or co-inhibitory immune checkpoint.
- immunoreactive checkpoint inhibitor refers to an antagonist of an inhibitory or co-inhibitory immune checkpoint.
- checkpoint inhibitor checkpoint inhibitor
- CPI CPI-associated receptor -ligand binding and/or altering receptor signaling.
- immune checkpoints ligands and receptors
- PD-1 programmed cell death protein 1
- PD-L1 PD1 ligand
- BTLA B and T lymphocyte attenuator
- CTLA-4 cytotoxic T-lymphocyte associated antigen 4
- TIM-3 T cell immunoglobulin and mucin domain containing protein 3
- LAG-3 lymphocyte activation gene 3
- TIGIT T cell immunoreceptor with Ig and ITIM domains
- CD276 B7-H3
- PD-L2 Galectin 9, CEACAM-1, CD69, Galectin-1, CD113, GPR56, VISTA, 2B4, CD48, GARP, PD1H, LAIR1, TIM-1, and TIM-4, and Killer Inhibitory Receptors, which can be divided into two classes based on their structural features: i) killer cell immunoglobulin- like receptors (KIRs), and
- an immune checkpoint inhibitor is a CTLA-4 antagonist.
- the CTLA-4 antagonist can be an antagonistic CTLA-4 antibody.
- Suitable antagonistic CTLA-4 antibodies include, for example, monospecific antibodies such as ipilimumab or tremelimumab, as well as bispecific antibodies such as MEDI5752 and KN046.
- an immune checkpoint inhibitor is a PD-1 antagonist.
- the PD-1 antagonist can be an antagonistic PD-1 antibody, small molecule or peptide.
- Suitable antagonistic PD-1 antibodies include, for example, monospecific antibodies such as balstilimab, budigalimab, camrelizumab, cosibelimab, dostarlimab, cemiplimab, ezabenlimab (BI-754091), MEDI-0680 (AMP-514; WO2012/145493), nivolumab, pembrolizumab, pidilizumab (CT-011), pimivalimab, retifanlimab, sasanlimab, spartalizumab, sintilmab, tislelizumab, toripalimab, and zimberelimab; as well as bi-specific antibodies such as LY3434172.
- monospecific antibodies such as balstilimab, budigalimab, camrelizumab, cosibelimab, dostarlimab, cemiplimab, ezabenlimab (
- the PD-1 antagonist can be a recombinant protein composed of the extracellular domain of PD-L2 (B7-DC) fused to the Fc portion of IgGl (AMP-224).
- an immune checkpoint inhibitor is zimberelimab.
- an immune checkpoint inhibitor is a PD-L1 antagonist.
- the PD-L1 antagonist can be an antagonistic PD-L1 antibody.
- Suitable antagonistic PD-Ll antibodies include, for example, monospecific antibodies such as avelumab, atezolizumab, durvalumab, BMS-936559, and envafolimab as well as bi-specific antibodies such as LY3434172 and KN046.
- an immune checkpoint inhibitor is a TIGIT antagonist.
- the TIGIT antagonist can be an antagonistic TIGIT antibody.
- Suitable antagonistic anti-TIGIT antibodies include monospecific antibodies such as AGEN1327, AB308 (WO2021247591), BMS 986207, COM902, domvanalimab, EOS-448, etigilimab, IBI-929, JS006, M6223, ociperlimab, SEA-TGT, tiragolumab, vibostolimab; as well as bi-specific antibodies such as AGEN1777 and AZD2936.
- an immune checkpoint inhibitor is an antagonistic anti-TIGIT antibody disclosed in WO2017152088 or WO2021247591.
- an immune checkpoint inhibitor is domvanalimab or AB308.
- an immune checkpoint inhibitor is a LAG-3 antagonist.
- the LAG-3 antagonist can be an antagonistic LAG-3 antibody.
- Suitable antagonistic LAG-3 antibodies include, for example, BMS-986016 (WO10/19570, WO14/08218), or IMP-731 or IMP-321 (WO08/132601, WO09/44273).
- an immune checkpoint inhibitor is a B7-H3 antagonist.
- the B7-H3 antagonist is an antagonistic B7-H3 antibody.
- Suitable antagonist B7-H3 antibodies include, for example, MGA271 (WO11/109400), omburtumab, enoblituzumab, DS-7300a, ABBV-155, and SHR-A1811.
- one or more of the additional therapeutic agents activates a stimulatory or co-stimulatory immune checkpoint.
- stimulatory or co-stimulatory immune checkpoints include B7-1, B7-2, CD28, 4-1BB (CD137), 4-1BBL, ICOS, ICOS-L, OX40, OX40L, GITR, GITRL, CD70, CD27, CD40, DR3 and CD2.
- an agent that activates a stimulatory or co-stimulatory immune checkpoint is a CD137 (4-1BB) agonist.
- the CD137 agonist can be an agonistic CD137 antibody.
- Suitable CD137 antibodies include, for example, urelumab and PF- 05082566 (WO12/32433).
- an agent that activates a stimulatory or co- stimulatory immune checkpoint is a GITR agonist.
- the GITR agonist can be an agonistic GITR antibody.
- Suitable GITR antibodies include, for example, BMS-986153, BMS-986156, TRX-518 (WO06/105021, WO09/009116) and MK-4166 (WO11/028683).
- an agent that activates a stimulatory or co-stimulatory immune checkpoint is an OX40 agonist.
- the OX40 agonist can be an agonistic OX40 antibody.
- Suitable OX40 antibodies include, for example, MEDI-6383, MEDI-6469, MEDI-0562, PF- 04518600, GSK3174998, BMS-986178, and MOXR0916.
- an agent that activates a stimulatory or co-stimulatory immune checkpoint is a CD40 agonist.
- the CD40 agonist can be an agonistic CD40 antibody.
- an agent that activates a stimulatory or co-stimulatory immune checkpoint is a CD27 agonist.
- the CD27 agonist can be an agonistic CD27 antibody.
- Suitable CD27 antibodies include, for example, varlilumab.
- one or more of the additional therapeutic agents is an ATP- adenosine axis-targeting agent.
- an ATP-adenosine axis-targeting agent is an inhibitor of an ectonucleotidase involved in the conversion of ATP to adenosine or an antagonist of adenosine receptor, e.g., ectonucleoside triphosphate diphosphohydrolase 1 (ENTPD1, also known as CD39 or Cluster of Differentiation 39) and the ecto-5'-nucleotidase (NT5E or 5NT, also known as CD73 or Cluster of Differentiation 73).
- ENTPD1 ectonucleoside triphosphate diphosphohydrolase 1
- N5E or 5NT also known as CD73 or Cluster of Differentiation 73
- Exemplary small molecule CD73 inhibitors include CB-708, ORIC-533, LY3475070 and AB680.
- Exemplary anti-CD39 and anti-CD73 antibodies include ES002023, TTX-030, IPH-5201, SRF-617, CPI-006, oleclumab (MEDI9447), NZV930, IPH5301, GS-1423, uliledlimab (TJD5, TJ004309), BMS-986179, and AB598.
- the present disclosure contemplates combination of the compounds described herein with a CD73 inhibitor such as those described in WO 2017/120508, WO 2018/067424, WO 2018/094148, and WO 2020/046813.
- the CD73 inhibitor is quemliclustat (AB680).
- Adenosine can bind to and activate four different G-protein coupled receptors: A 1 R, A2AR, A2BR, and A3R.
- A2R antagonists include etrumadenant, inupadenant, taminadenant, caffeine citrate, NUV-1182, TT-702, DZD-2269, INCB-106385, EVOEXS-21546, AZD-4635, imaradenant, RVU-330, ciforadenant, PBF-509, PBF-999, PBF-1129, and CS-3005.
- the present disclosure contemplates the combination of the compounds described herein with an A2AR antagonist, an A2BR antagonist, or an antagonist of A2AR and A2BR.
- the present disclosure contemplates the combination of the compounds described herein with the adenosine receptor antagonists described in WO 2018/136700, WO 2018/204661, WO 2018/213377, or WO 2020/023846.
- the adenosine receptor antagonist is etrumadenant.
- one or more of the additional therapeutic agents is an inhibitor of a hypoxia-inducible factor (HIF) transcription factor, particularly HIF-2 ⁇ .
- HIF-2 ⁇ inhibitors include belzutifan, ARO-HIF2, PT-2385, and those described in WO 2021113436, WO 2021188769, and WO 2023077046.
- the HIF-2 ⁇ inhibitor is AB521.
- one or more of the additional therapeutic agents is an inhibitor of anexelekto (AXL).
- AXL anexelekto
- the AXL signaling pathway is associated with tumor growth and metastasis, and is believed to mediate resistance to a variety of cancer therapies.
- AXL inhibitors under development that also inhibit other kinases in the TAM family (i.e., TYRO3, MERTK), as well as other receptor tyrosine kinases including MET, FLT3, RON and AURORA, among others.
- Exemplary multikinase inhibitors include sitravatinib, rebastinib, glesatinib, gilteritinib, merestinib, cabozantinib, foretinib, BMS777607, LY2801653, S49076, and RXDX- 106.
- AXL specific inhibitors have also been developed, e.g., small molecule inhibitors including DS-1205, SGI-7079, SLC-391, TP-0903 (i.e., dubermatinib), BGB324 (i.e., bemcentinib), DP3975, and AB801; anti-AXL antibodies such as ADCT-601; and antibody drug conjugates (ADCs) such as BA3011.
- Another strategy to inhibit AXL signaling involves targeting AXL’s ligand, GAS6.
- batiraxcept (AVB-500) is under development as is a Fc fusion protein that binds the GAS6 ligand thereby inhibiting AXL signaling.
- the additional therapeutic agent is an AXL inhibitor described in WO 2022246177, WO 2022246179, or PCT/US2023/069124. In some embodiments, the AXL inhibitor is AB801. [0202] In some embodiments, the additional therapeutic agent comprises chemotherapy, radiation therapy, or both. [0203] In one or more embodiments, the additional therapeutic agent comprises domvanalimab, etrumadenant, quemliclustat, zimberelimab, AB308, AB521, AB598, or AB801, or any combinations thereof.
- the additional therapeutic agent comprises one or more of an immune checkpoint inhibitor, an A2R antagonist, a CD73 inhibitor, a HIF-2 ⁇ inhibitor, a chemotherapeutic agent, radiation therapy, or any combinations thereof.
- the immune checkpoint inhibitor comprises one or more inhibitors that block the activity of at least one of PD-1, PD-L1, BTLA, LAG-3, a B7 family member, TIM-3, TIGIT or CTLA-4,
- the immune checkpoint inhibitor comprises an inhibitor of PD-1 or PD-L1;
- the immune checkpoint inhibitor is selected from the group consisting of avelumab, atezolizumab, durvalumab, dostarlimab, cemiplimab, nivolumab, pembrolizumab, sintilmab, toripalimab, and zimberelimab;
- the immune checkpoint inhibitor is zimberelima
- NCN National Comprehensive Cancer Network
- NCCN Melanoma Cutaneous v2.2021, NCCN Melanoma: Uveal v2.2021, NCCN Prostate Cancer v1.2022, NCCN Squamous Cell Skin Cancer v1.2022, NCCN Hodgkin Lymphoma v2.2022, NCCN Acute Lymphoblastic Leukemia v4.2021, NCCN Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma v2.2022, NCCN Chronic Myeloid Leukemia v3.2022, NCCN Hairy Cell Leukemia v1.2022, NCCN Pediatric Acute Lymphoblastic Leukemia v1.2022, NCNN Small Bowel Adenocarcinoma v1.2022, NCCN Thyroid Carcinoma v3.2021, NCCN Non-Small Cell Lung Cancer v3.2022, NCCN Small Cell Lung Cancer v2.2022, NCCN Breast Cancer v2.2022, NCCN Colon Cancer v3.2021, NCCN Hepatobiliary Cancer v5.2
- Equation 1 illustrates a retrosynthetic disconnection of the compounds of the invention into fragments a, b and c, which are useful for the construction of the compounds according to this disclosure.
- Equation 2 demonstrates one method of forming the bond between fragments a and b.
- a readily available carboxylic acid derived from fragment a is converted to an activated intermediate, such as an acid chloride or an activated ester, using a suitable amide coupling reagent(s).
- carboxylic acids can be accomplished using a wide range of conditions and reagents, such as oxalyl chloride or thionyl chloride.
- carboxylic acids are readily converted to activated esters through the use of amide coupling reagents, such as HATU, CDI, EDC, HOBt, T3P or various other reagents (see, e.g., “Synthesis of amides” in https://www.organic- chemistry.org/synthesis/C1N/amides.shtm).
- the activated ester or acid chloride derived from fragment a can be coupled with a wide variety of amines, including but not limited to anilines (fragment b).
- the transformation may be assisted or accelerated by heating and/or addition of a base.
- Equations 3–6 demonstrate useful methods of synthesizing an appropriately substituted 1,2,4-triazole and connecting fragments b and c.
- an appropriately substituted aryl hydrazide readily condenses with an appropriately substituted carboximidamide or carboximidic acid ester to afford the corresponding 1,2,4-triazole, wherein “OR” typically consists of—but is not limited to—a methoxy or ethoxy group.
- Equation 5 demonstrates another method of forming the bond between fragments b and c via a Pd- catalyzed coupling reaction, such as a Suzuki reaction.
- -B(OR)2 represents a boronic acid or ester and X2 may be chosen from an appropriate halogen atom, such as Cl, Br, or I.
- the coupling is mediated by a transition metal catalyst, such as palladium with an appropriate ligand, and may be facilitated by the use of an organic or inorganic base and heating.
- a transition metal catalyst such as palladium with an appropriate ligand
- the functionalization of the coupling partners may be reversed, as exemplified in Eq. 6, and a wide variety of conditions are known in the art to effect these transformations.
- There are other possible combinations and synthetic sequences that will also give rise to the targeted products. Formation of the bond between fragments a and b may occur before or after connection of the b and c fragments, and each of these fragments may be further modified before or after connection of fragments a, b and c.
- ATP adenosine triphosphate
- BSA bovine serum albumin
- AcOH acetic acid
- DCM and CH 2 Cl 2 dichloromethane
- DIPEA and EtNiPr 2 N,N-diisopropylethylamine
- DMF N,N-dimethylformamide
- DMSO dimethyl sulfoxide
- DMSO-d6 perdeuterated dimethyl sulfoxide
- NMP N-methyl-2-pyrrolidone
- H 2 O lithium hydroxide monohydrate
- Cs2CO3 cesium carbonate
- POCl3 phosphoryl chloride
- MTBE tert-butyl methyl ether
- TBS-Cl tert-butyldimethylsilyl chloride
- Me3SiCF3 (trifluoromethyl)trimethylsilane
- TFA trifluoroacetic acid
- tBuBrettPhos 2-(di-tert- butylphosphino)-2′,4′,6′-triisopropyl-3,6-dimethoxy-1,1′-biphenyl
- PCy3 Pd G2 chloro[(tricyclohexylphosphine)-2-(2′-aminobiphenyl)]palladium(II)
- XPhos 2- dicyclohexylphosphino-2',4',6'-triisopropylbiphenyl
- XPhos Pd G3
- Flash chromatography was conducted on silica gel using an automated system (CombiFlash® RF+ manufactured by Teledyne ISCO), with detection wavelengths of 254 and 280 nm, and optionally equipped with an evaporative light scattering detector.
- Reverse phase preparative HPLC was conducted on an Agilent® 1260 or 1290 Infinity series HPLC.
- Step a A 40 mL vial was charged with pyrazolo[1,5-a]pyridine-3-carboxylic acid (486 mg, 3.0 mmol, 1.0 equiv.), HATU (1.71 g, 4.5 mmol, 1.5 equiv.), EtNiPr2 (1.53 mL, 9.0 mmol, 3.0 equiv.) and DMF (8.6 mL, 0.35 M).
- Step b A mixture of 2-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)aniline HCl (320 mg, 1.14 mmol, 1.0 equiv.), EtNiPr2 (0.36 ml, 2.0 equiv.) and triazolo[4,5-b]pyridin-3- yl pyrazolo[1,5-a]pyridine-3-carboxylate (280 mg, 1.04 mmol, 1.1 equiv.) in NMP (2 mL, 0.5 M) was stirred at 120 °C for approximately 3 h.
- step b Upon complete conversion, as judged by LCMS analysis, the reaction mixture was cooled to rt and diluted with water. The resulting precipitated solid was collected by vacuum filtration, rinsed with water, and dried in vacuo to afford N-[2- methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]pyrazolo[1,5-a]pyridine-3- carboxamide, which was used directly in the next step without further purification.
- the product of step b could also be accessed via an alternative, two-step protocol involving an initial amidation with 5-bromo-2-methylaniline followed by Pd-catalyzed borylation.
- Step c The crude intermediate obtained via step b (20 mg in 0.1 mL NMP, 0.053 mmol, 1.0 eq.) was combined with triazole bromide (20 mg, 0.106 mmol, 2.0 equiv.), XPhos Pd G3 (20 mg, 0.024 mmol, 45 mol%), 1 M aq. Na 2 CO 3 solution (0.4 mL, 0.4 mmol, 7.5 equiv.), and dioxane (0.4 mL, 0.07 M). The reaction mixture was briefly degassed by evacuation/back-filling with N2 3x. The reaction mixture was stirred at 100 °C for approximately 2 h, at which time LCMS analysis indicated complete consumption of starting material.
- Example 2 N-[5-(5-Ethyl-4H-1,2,4-triazol-3-yl)-2-methylphenyl]pyrazolo[1,5-a]pyridine-3- carboxamide [0216] The title compound was prepared from 3-bromo-5-ethyl-4H-1,2,4-triazole in a similar fashion to Ex.1.
- Example 4 N-[5-(5-tert-butyl-4H-1,2,4-triazol-3-yl)-2-methylphenyl]pyrazolo[1,5- a]pyridine-3-carboxamide [0218] The title compound was prepared from 3-bromo-5-tert-butyl-4H-1,2,4-triazole in a similar fashion to Ex.1.
- Example 6 N-[4-Fluoro-2-methyl-5-[5-(trifluoromethyl)-4H-1,2,4-triazol-3- yl]phenyl]pyrazolo[1,5-a]pyridine-3-carboxamide [0220] The title compound was prepared from 3-bromo-5-(trifluoromethyl)-4H-1,2,4-triazole and 5-bromo-4-fluoro-2-methylaniline in a similar fashion to Ex. 1.
- Example 7 N-[5-(5-Cyclobutyl-4H-1,2,4-triazol-3-yl)-2-methylphenyl]pyrazolo[1,5- a]pyridine-3-carboxamide [0221] The title compound was prepared from 3-bromo-5-cyclobutyl-4H-1,2,4-triazole in a similar fashion to Ex.1.
- Step b A 40 mL vial was charged with the 5-[5-(cyclopropylmethyl)-4H-1,2,4-triazol- 3-yl]-4-fluoro-2-methylaniline obtained in step a (36 mg, 0.146 mmol, 1.0 equiv.), triazolo[4,5- b]pyridin-3-yl pyrazolo[1,5-a]pyridine-3-carboxylate (41 mg, 0.146 mmol, 1.0 equiv.), and NMP (0.150 mL, 1 M). The resulting mixture was stirred at 120 °C for 2 h.
- Example 17 6-Fluoro-N-[4-fluoro-5-(5-methoxy-4H-1,2,4-triazol-3-yl)-2- methylphenyl]pyrazolo[1,5-a]pyridine-3-carboxamide
- the title compound was prepared from 6-fluoropyrazolo[1,5-a]pyridine-3-carboxylic acid, 3-bromo-5-methoxy-4H-1,2,4-triazole and 5-bromo-4-fluoro-2-methylaniline in a similar fashion to Ex.1.
- Example 18 N-[5-[5-(Dimethylamino)-4H-1,2,4-triazol-3-yl]-4-fluoro-2- methylphenyl]pyrazolo[1,5-a]pyridine-3-carboxamide [0235] The title compound was prepared from 5-bromo-N,N-dimethyl-4H-1,2,4-triazol-3-amine and 5-bromo-4-fluoro-2-methylaniline in a similar fashion to Ex. 1.
- Example 19 N-[5-(5-Cyclopropyl-4H-1,2,4-triazol-3-yl)-4-fluoro-2-methylphenyl]-6- fluoropyrazolo[1,5-a]pyridine-3-carboxamide
- the title compound was prepared from triazolo[4,5-b]pyridin-3-yl 6-fluoropyrazolo[1,5- a]pyridine-3-carboxylate in a similar fashion to Ex. 12, which was prepared from 6- fluoropyrazolo[1,5-a]pyridine-3-carboxylic acid in a similar fashion to Ex.1, step a.
- Example 23 N-[5-(5-Cyclopropyl-4H-1,2,4-triazol-3-yl)-4-fluoro-2-methylphenyl]-5- methoxypyrazolo[1,5-a]pyridine-3-carboxamide
- the title compound was prepared from triazolo[4,5-b]pyridin-3-yl 5- methoxypyrazolo[1,5-a]pyridine-3-carboxylate in a similar fashion to Ex.12, which was prepared from 5-methoxypyrazolo[1,5-a]pyridine-3-carboxylic acid in a similar fashion to Ex.1, step a.
- Step a To a solution of ethyl O-(2-mesitylenesulfonyl)acethydroxamate (2.00 g, 7.0 mmol) in dioxane (8 mL) was added 70% perchloric acid (1.2 mL) dropwise at 0 °C. The reaction was stirred at rt for 2 h, then the mixture was poured into ice/water (100 mL). The solution was stirred for 10 min until white solid precipitated.
- Step b To a mixture of O-(mesitylenesulfonyl)hydroxylamine (1.08 g, 5.0 mmol) and 3-ethylpyridine (536 mg, 5.0 mmol) was added DCM (20 mL), then the solution was stirred at rt for 36 h.
- Step c To a solution of 2-mesitylenesulfonate-1-amino-3-ethyl-pyridinium (5.0 mmol) in DMF (10 mL) at 0 °C, was added potassium carbonate (1.04 g, 7.5 mmol) and methyl propiolate (631 mg, 7.5 mmol). The reaction was stirred at rt for 18 h.
- Step d To a solution of methyl 6-ethylpyrazolo[1,5-a]pyridine-3-carboxylate (79 mg, 0.383 mmol) in 1:1:1 THF/MeOH/water (1.2 mL) at 0 °C, was added LiOH . H 2 O (81 mg, 1.92 mmol). The reaction was stirred at 50 °C for 2 h. Upon cooling to rt, the mixture was concentrated to remove THF and MeOH. The resultant mixture was adjusted to pH ⁇ 2 by addition of 2 M HCl (aq) . The mixture was diluted with EtOAc (10 mL) and washed with water (5 mL).
- Step e The desired product was prepared in a similar manner to Example 1, step a.
- Step f The desired product was prepared in a similar manner to Example 12, step c.
- Example 25 6-Cyclopropyl-N-[5-(5-cyclopropyl-4H-1,2,4-triazol-3-yl)-4-fluoro-2- methylphenyl]pyrazolo[1,5-a]pyridine-3-carboxamide [0247] The title compound was prepared from 3-cyclopropylpyridine in a similar fashion to Ex. 24.
- Step a To a mixture of 6-bromopyrazolo[1,5-a]pyridine (985 mg, 5.0 mmol), ethanol (0.58 mL, 10.0 mmol), palladium acetate (57 mg, 0.25 mmol), tBuBrettPhos (243 mg, 0.5 mmol) and cesium carbonate (3.26 g, 10.0 mmol) was added degassed toluene (10 mL).
- Step b To a solution of 6-ethoxypyrazolo[l,5-a]pyridine (747 mg, 4.6 mmol) in DMF (9.2 mL) at 0 °C, was added POCl 3 (1.29 mL, 13.8 mmol). The reaction was stirred at rt for 2 h.
- Step c To a solution of 6-ethoxypyrazolo[1,5-a]pyridine-3-carboxaldehyde (700 mg, 3.68 mmol) in water (20 mL), was added NaH2PO4 (1.33 g, 11.04 mmol). After 5 min, 1:1 THF/tBuOH (40 mL), 2-methyl-2-butene (2.34 mL, 22.08 mmol) and sodium chlorite (1.34 g, 14.72 mmol) were added sequentially. The reaction was stirred at rt for 24 h. The mixture was diluted with EtOAc (100 mL) and washed with water (50 mL) and brine (20 mL).
- Step d The desired product was prepared in a similar manner to Example 1, step a.
- Step e The desired product was prepared in a similar manner to Example 12, step c.
- Example 28 N-[4-Fluoro-2-methyl-5-(5-methyl-4H-1,2,4-triazol-3-yl)phenyl]-6- methoxypyrazolo[1,5-a]pyridine-3-carboxamide
- the title compound was prepared in a similar fashion to Ex. 27 from triazolo[4,5- b]pyridin-3-yl 6-methoxypyrazolo[1,5-a]pyridine-3-carboxylate, which was prepared from 6- methoxypyrazolo[1,5-a]pyridine-3-carboxylic acid in a similar fashion to Ex.1, step a.
- Example 29 6-Fluoro-N-[4-fluoro-2-methyl-5-[5-(oxolan-2-yl)-4H-1,2,4-triazol-3- yl]phenyl]pyrazolo[1,5-a]pyridine-3-carboxamide
- the title compound was prepared in a similar fashion to Ex. 12 from tetrahydro-2- furancarboximidamide and triazolo[4,5-b]pyridin-3-yl 6-fluoropyrazolo[1,5-a]pyridine-3- carboxylate, which was prepared from 6-fluoropyrazolo[1,5-a]pyridine-3-carboxylic acid in a similar fashion to Ex.1, step a.
- Example 32 N-[4-Fluoro-2-methyl-5-(5-propyl-4H-1,2,4-triazol-3-yl)phenyl]pyrazolo[1,5- a]pyridine-3-carboxamide [0258] The title compound was prepared from butyramidine hydrochloride in a similar fashion to Ex.12.
- Example 34 N-[5-[5-(Difluoromethyl)-4H-1,2,4-triazol-3-yl]-4-fluoro-2-methylphenyl]-6- methoxypyrazolo[1,5-a]pyridine-3-carboxamide [0260]
- the title compound was prepared from triazolo[4,5-b]pyridin-3-yl 6- methoxypyrazolo[1,5-a]pyridine-3-carboxylate (which was prepared from 6- methoxypyrazolo[1,5-a]pyridine-3-carboxylic acid in a similar fashion to Ex.1 step a) and 5-[5- (difluoromethyl)-4H-1,2,4-triazol-3-yl]-4-fluoro-2-methylaniline in a similar fashion to Ex.33.
- Example 35 N-[5-[5-(Difluoromethyl)-4H-1,2,4-triazol-3-yl]-4-fluoro-2-methylphenyl]-6- fluoropyrazolo[1,5-a]pyridine-3-carboxamide [0261]
- the title compound was prepared from triazolo[4,5-b]pyridin-3-yl 6-fluoropyrazolo[1,5- a]pyridine-3-carboxylate (which was prepared from 6-fluoropyrazolo[1,5-a]pyridine-3-carboxylic acid in a similar fashion to Ex.
- Example 40 6-Chloro-N-[5-(5-ethyl-4H-1,2,4-triazol-3-yl)-4-fluoro-2- methylphenyl]pyrazolo[1,5-a]pyridine-3-carboxamide
- the title compound was prepared from triazolo[4,5-b]pyridin-3-yl 6-chloropyrazolo[1,5- a]pyridine-3-carboxylate (which was prepared from 6-chloropyrazolo[1,5-a]pyridine-3- carboxylic acid in a similar fashion to Ex. 1, step a) and propionimidamide hydrochloride in a similar fashion to Ex.12.
- Example 41 N-[5-(5-Ethyl-4H-1,2,4-triazol-3-yl)-4-fluoro-2-methylphenyl]-6- fluoropyrazolo[1,5-a]pyridine-3-carboxamide [0267]
- the title compound was prepared from triazolo[4,5-b]pyridin-3-yl 6-fluoropyrazolo[1,5- a]pyridine-3-carboxylate (which was prepared from 6-fluoropyrazolo[1,5-a]pyridine-3-carboxylic acid in a similar fashion to Ex.1, step a) and propionimidamide hydrochloride in a similar fashion to Ex.12.
- Example 42 N-[5-(5-Ethyl-4H-1,2,4-triazol-3-yl)-4-fluoro-2-methylphenyl]-6- methoxypyrazolo[1,5-a]pyridine-3-carboxamide
- the title compound was prepared from triazolo[4,5-b]pyridin-3-yl 6- methoxypyrazolo[1,5-a]pyridine-3-carboxylate (which was prepared from - methoxypyrazolo[1,5-a]pyridine-3-carboxylic acid in a similar fashion to Ex. 1, step a) and propionimidamide hydrochloride in a similar fashion to Ex.12.
- Example 43 6-Fluoro-N-[4-fluoro-2-methyl-5-(5-propan-2-yl-4H-1,2,4-triazol-3- yl)phenyl]pyrazolo[1,5-a]pyridine-3-carboxamide
- the title compound was prepared in a similar fashion to Ex.12 using isobutyrimidamide hydrochloride and triazolo[4,5-b]pyridin-3-yl 6-fluoropyrazolo[1,5-a]pyridine-3-carboxylate, which was prepared from 6-fluoropyrazolo[1,5-a]pyridine-3-carboxylic acid in a similar fashion to Ex.1, step a.
- Example 45 6-Chloro-N-[4-fluoro-2-methyl-5-(5-propan-2-yl-4H-1,2,4-triazol-3- yl)phenyl]pyrazolo[1,5-a]pyridine-3-carboxamide
- the title compound was prepared in a similar fashion to Ex.12 using isobutyrimidamide hydrochloride and triazolo[4,5-b]pyridin-3-yl 6-chloropyrazolo[1,5-a]pyridine-3-carboxylate, which was prepared from 6-chloropyrazolo[1,5-a]pyridine-3-carboxylic acid in a similar fashion to Ex.1, step a.
- Step a A round-bottom flask was charged with methyl 4-chloro-2-fluoro-5- nitrobenzoate (1.00 g, 4.28 mmol) and Fe (500 mg, 8.95 mmol, 2.1 equiv.) in AcOH (5 mL). The resulting mixture was then stirred at reflux under a nitrogen atmosphere for 2 h. Upon complete conversion, as judged by LCMS analysis, the reaction mixture was cooled to rt, diluted with EtOAc, filtered, and concentrated in vacuo. The residue was diluted with EtOAc and sat. aq. NaHCO3. The organic phase was separated, dried, and concentrated in vacuo.
- Steps b-d The title compound was prepared from methyl 5-amino-4-chloro-2- fluorobenzoate obtained in step a in a similar fashion to Ex.12.
- Step a A round-bottom flask was charged with methyl 5-amino-4-bromo-2- chlorobenzoate (1.0 g, 3.78 mmol, 1.0 equiv.), trimethylboroxine (0.58 mL, 4.15 mmol, 1.1 equiv.) and PdCl 2 (dppf) (277 mg, 0.378 mmol, 10 mol% equiv.) in 2 M Na 2 CO 3 (6 mL) and dioxane (18 mL).
- Steps b-d The title compound was prepared from methyl 5-amino-2-chloro-4- methylbenzoate obtained in step a in a similar fashion to Ex.12.
- Example 52 N-[5-(5-Cyclopropyl-4H-1,2,4-triazol-3-yl)-4-fluoro-2-methylphenyl]-4- fluoropyrazolo[1,5-a]pyridine-3-carboxamide [0281] The title compound was prepared from 4-fluoropyrazolo[1,5-a]pyridine-3-carboxylic acid in a similar fashion to Ex.12.
- Example 53 N-[2-Chloro-5-(5-cyclopropyl-4H-1,2,4-triazol-3-yl)-4- methylphenyl]pyrazolo[1,5-a]pyridine-3-carboxamide 109 [0282] The title compound was prepared from methyl 5-amino-2-bromo-4-chlorobenzoate in a similar fashion to Ex.50.
- Example 54 N-[2-Chloro-5-(5-cyclopropyl-4H-1,2,4-triazol-3-yl)-4-methylphenyl]-6- fluoropyrazolo[1,5-a]pyridine-3-carboxamide [0283] The title compound was prepared from methyl 5-amino-2-bromo-4-chlorobenzoate and 6-fluoropyrazolo[1,5-a]pyridine-3-carboxylic acid in a similar fashion to Ex. 50.
- Step a Methyl 6-bromopyrazolo[1,5-a]pyridine-3-carboxylate (510 mg, 2 mmol, 1.0 equiv.), RuPhos Pd G4 (340 mg, 0.4 mmol, 20 mol%) and Cs2CO3 (1.95 g, 6 mmol, 3.0 equiv.) were placed in a pressure vial. The vial was placed under N 2 , then dioxane (10 mL) and morpholine (348 mg, 4 mmol, 2.0 equiv.) were added. The reaction mixture was degassed again and stirred at 100 °C for 14 h.
- Step b A solution of the product from step a (450 mg, 1.72 mmol, 1.0 equiv.) in 2:1:1 THF:MeOH:H2O (20 mL) was treated with LiOH ⁇ H2O (206 mg, 8.62 mmol, 5.0 equiv). The thick mixture was allowed to stir at rt for 14 h. The resulting solution was diluted with water (6 mL) and treated with 1N aq. HCl (5 mL) dropwise over a few min. The mixture was allowed to stir for several additional min. to break up any clumps.
- Step a A mixture of methyl 6-bromopyrazolo[1,5-a]pyridine-3-carboxylate compound (510 mg, 2.0 mmol), B 2 pin 2 (762 mg, 1.5 mmol), Pd(dppf)Cl 2 (73 mg, 0.1 mmol), and KOAc (392 mg, 4 mmol) was placed under N 2 . Degassed dioxane (20 mL) was added and the reaction mixture was stirred at 100 °C for 4 h. The mixture was cooled to rt, concentrated, diluted with CH2Cl2 (50 mL), filtered through Celite® to remove solids, and again concentrated to afford the desired intermediate, which was used directly in part 2 of step a without further purification.
- Step b Ester hydrolysis was carried out similar to Ex.58, step b.
- Steps c-d The title compound was prepared in a similar fashion to Ex.24, steps e-f.
- Example 62 N-[5-[5-(2,2-Difluorocyclopropyl)-4H-1,2,4-triazol-3-yl]-4-fluoro-2- methylphenyl]-6-fluoropyrazolo[1,5-a]pyridine-3-carboxamide [0295] The title compound was prepared from 2,2-difluorocyclopropane-1-carboximidamide and triazolo[4,5-b]pyridin-3-yl 6-fluoropyrazolo[1,5-a]pyridine-3-carboxylate in a similar fashion to Ex.12.
- Example 63 N-[5-[5-(3,3-Difluorocyclobutyl)-4H-1,2,4-triazol-3-yl]-4-fluoro-2- methylphenyl]-6-fluoropyrazolo[1,5-a]pyridine-3-carboxamide [0296] The title compound was prepared from 3,3-difluorocyclobutane-1-carboximidamide and triazolo[4,5-b]pyridin-3-yl 6-fluoropyrazolo[1,5-a]pyridine-3-carboxylate in a similar fashion to Ex.12.
- Example 64 N-[5-[5-(3,3-Difluorocyclopentyl)-4H-1,2,4-triazol-3-yl]-4-fluoro-2- methylphenyl]-6-fluoropyrazolo[1,5-a]pyridine-3-carboxamide [0297] The title compound was prepared from 3,3-difluorocyclopentane-1-carboximidamide and triazolo[4,5-b]pyridin-3-yl 6-fluoropyrazolo[1,5-a]pyridine-3-carboxylate in a similar fashion to Ex.12.
- Example 66 4-Fluoro-N-[4-fluoro-2-methyl-5-(5-propan-2-yl-4H-1,2,4-triazol-3- yl)phenyl]pyrazolo[1,5-a]pyridine-3-carboxamide [0299] The title compound was prepared from 2-methylpropionamidine and triazolo[4,5- b]pyridin-3-yl 4-fluoropyrazolo[1,5-a]pyridine-3-carboxylate in a similar fashion to Ex. 12.
- Example 68 N-[5-(5-Chloro-4H-1,2,4-triazol-3-yl)-4-fluoro-2-methylphenyl]pyrazolo[1,5- a]pyridine-3-carboxamide [0301] The title compound was prepared from N-[4-fluoro-2-methyl-5-(4,4,5,5-tetramethyl- 1,3,2-dioxaborolan-2-yl)phenyl]pyrazolo[1,5-a]pyridine-3-carboxamide and 3-bromo-5-chloro- 4H-1,2,4-triazole in a similar fashion to Ex. 1 and Ex. 67.
- Example 69 N-[5-(5-Bromo-4H-1,2,4-triazol-3-yl)-4-fluoro-2-methylphenyl]pyrazolo[1,5- a]pyridine-3-carboxamide [0302] The title compound was prepared from N-[4-fluoro-2-methyl-5-(4,4,5,5-tetramethyl- 1,3,2-dioxaborolan-2-yl)phenyl]pyrazolo[1,5-a]pyridine-3-carboxamide and 3,5-dibromo-4- (oxan-2-yl)-1,2,4-triazole in a similar fashion to Ex.1 and Ex.67 and followed by removal of the THP protecting group.
- Example 70 N-[4-Fluoro-5-[5-(methoxymethyl)-4H-1,2,4-triazol-3-yl]-2- methylphenyl]pyrazolo[1,5-a]pyridine-3-carboxamide [0303] The title compound was prepared from 2-methoxyethanimidamide in a similar fashion to Ex.12.
- Example 71 N-[5-[5-[2-(Dimethylamino)-2-oxoethyl]-4H-1,2,4-triazol-3-yl]-4-fluoro-2- methylphenyl]pyrazolo[1,5-a]pyridine-3-carboxamide [0304]
- the title compound was prepared from 2-cyano-N,N-dimethylacetamide in a similar fashion to Ex.15.
- Example 73 N-[4-Fluoro-5-[5-(4-fluorophenyl)-4H-1,2,4-triazol-3-yl]-2- methylphenyl]pyrazolo[1,5-a]pyridine-3-carboxamide [0306] The title compound was prepared from 4-fluorobenzenecarboximidamide in a similar fashion to Ex.12.
- Example 74 N-[4-Fluoro-2-methyl-5-(5-pyridin-3-yl-4H-1,2,4-triazol-3- yl)phenyl]pyrazolo[1,5-a]pyridine-3-carboxamide [0307] The title compound was prepared from pyridine-3-carboximidamide in a similar fashion to Ex.12.
- Example 77 N-[5-[5-(Dimethylamino)-4H-1,2,4-triazol-3-yl]-4-fluoro-2-methylphenyl]-5- fluoropyrazolo[1,5-a]pyridine-3-carboxamide [0310] The title compound was prepared in a similar fashion to Ex.
- Example 78 N-[5-[5-(Cyclopropylamino)-4H-1,2,4-triazol-3-yl]-4-fluoro-2- methylphenyl]pyrazolo[1,5-a]pyridine-3-carboxamide [0311] The title compound was prepared from 1-cyclopropylguanidine in a similar fashion to Ex.12.
- Example 79 N-[4-Fluoro-2-methyl-5-(5-pyrrolidin-1-yl-4H-1,2,4-triazol-3- yl)phenyl]pyrazolo[1,5-a]pyridine-3-carboxamide [0312] The title compound was prepared from N-[4-fluoro-2-methyl-5-(4,4,5,5-tetramethyl- 1,3,2-dioxaborolan-2-yl)phenyl]pyrazolo[1,5-a]pyridine-3-carboxamide and 3-bromo-5- pyrrolidin-1-yl-4H-1,2,4-triazole in a similar fashion to Ex. 1 and Ex. 67.
- Example 80 N-[4-Fluoro-2-methyl-5-(5-piperazin-1-yl-4H-1,2,4-triazol-3- yl)phenyl]pyrazolo[1,5-a]pyridine-3-carboxamide [0313] The title compound was prepared from N-[4-fluoro-2-methyl-5-(4,4,5,5-tetramethyl- 1,3,2-dioxaborolan-2-yl)phenyl]pyrazolo[1,5-a]pyridine-3-carboxamide and 1-(5-bromo-4H- 1,2,4-triazol-3-yl)piperazine in a similar fashion to Ex. 1 and Ex. 67.
- Example 81 N-[5-[5-(Cyclopropylmethyl)-4H-1,2,4-triazol-3-yl]-4-fluoro-2-methylphenyl]- 6-methoxypyrazolo[1,5-a]pyridine-3-carboxamide [0314] The title compound was prepared from triazolo[4,5-b]pyridin-3-yl 6- methoxypyrazolo[1,5-a]pyridine-3-carboxylate in a similar fashion to Ex.15.
- Example 83 N-[4-Fluoro-2-methyl-5-(5-methyl-4H-1,2,4-triazol-3-yl)phenyl]-6- methylpyrazolo[1,5-a]pyridine-3-carboxamide [0316] The title compound was prepared from ethanimidamide and triazolo[4,5-b]pyridin-3-yl 6-methylpyrazolo[1,5-a]pyridine-3-carboxylate in a similar fashion to Ex.12.
- Example 84 6-Ethyl-N-[4-fluoro-5-(5-methoxy-4H-1,2,4-triazol-3-yl)-2- methylphenyl]pyrazolo[1,5-a]pyridine-3-carboxamide
- the title compound was prepared in a similar fashion to Ex.1 from 3-bromo-5-methoxy- 4H-1,2,4-triazole and 6-ethyl-N-[4-fluoro-2-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)phenyl]pyrazolo[1,5-a]pyridine-3-carboxamide, which was accessed from 6-ethylpyrazolo[1,5- a]pyridine-3-carboxylic acid and 5-bromo-4-fluoro-2-methylaniline in a similar fashion to step b of Ex.1.
- Example 85 5-Fluoro-N-[4-fluoro-5-(5-methoxy-4H-1,2,4-triazol-3-yl)-2- methylphenyl]pyrazolo[1,5-a]pyridine-3-carboxamide
- the title compound was prepared from 5-fluoro-N-[4-fluoro-2-methyl-5-(4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]pyrazolo[1,5-a]pyridine-3-carboxamide and 3- bromo-5-methoxy-4H-1,2,4-triazole in a similar fashion to Ex.1 and Ex.77.
- Example 86 N-[4-Fluoro-5-(5-methoxy-4H-1,2,4-triazol-3-yl)-2-methylphenyl]-6- methoxypyrazolo[1,5-a]pyridine-3-carboxamide
- the title compound was prepared in a similar fashion to Ex.1 from 3-bromo-5-methoxy- 4H-1,2,4-triazole and N-[4-fluoro-2-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)phenyl]-6-methoxypyrazolo[1,5-a]pyridine-3-carboxamide, which was accessed from 6- methoxypyrazolo[1,5-a]pyridine-3-carboxylic acid and 5-bromo-4-fluoro-2-methylaniline in a similar fashion to step b, Ex.1.
- Example 87 6-Fluoro-N-[4-fluoro-2-methyl-5-[5-(oxolan-3-yl)-4H-1,2,4-triazol-3- yl]phenyl]pyrazolo[1,5-a]pyridine-3-carboxamide
- the title compound was prepared from oxolane-3-carboximidamide and triazolo[4,5- b]pyridin-3-yl 6-fluoropyrazolo[1,5-a]pyridine-3-carboxylate in a similar fashion to Ex. 12.
- Example 88 N-[5-(5-Cyclopropyl-2-methyl-1,2,4-triazol-3-yl)-4-fluoro-2- methylphenyl]pyrazolo[1,5-a]pyridine-3-carboxamide [0321] The title compound was prepared from 5-bromo-3-cyclopropyl-1-methyl-1,2,4-triazole and N-[4-fluoro-2-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]pyrazolo[1,5- a]pyridine-3-carboxamide in a similar fashion to Ex.1 and Ex.67.
- Step a A vial was charged with N-[4-fluoro-2-methyl-5-(4,4,5,5-tetramethyl-1,3,2- dioxaborolan-2-yl)phenyl]pyrazolo[1,5-a]pyridine-3-carboxamide (494 mg, 1.25 mmol, 1.0 equiv.), 4-benzyl-3-bromo-5-fluoro-1,2,4-triazole (318 mg, 1.25 mmol, 1.0 equiv.), Pd XPhos G3 (105 mg, 0.12 mmol, 10 mol%), 2M Na2CO3 (aq.
- Step b A vial was charged with the product from step a (75 mg, 0.17 mmol, 1.0 equiv.), NaOEt (68 mg, 1.02 mmol, 6.0 equiv.) and EtOH (1 mL). The reaction mixture was stirred at 80 °C for approximately 16 h, at which time LCMS analysis indicated 70% consumption of starting material.
- Step c A reactor jar was charged with the product from step b (65 mg, 0.13 mmol, 1.0 equiv.) and Pd/C (13 mg, 20 wt%) in MeOH (10 mL) containing a few drops of 4N HCl in dioxane. The reaction mixture was shaken under 30 psi of hydrogen gas for 3 h, then filtered over Celite® and concentrated under reduced pressure to afford the crude product. Purification by reverse phase HPLC afforded the title compound.
- Example 90 N-[5-(1-Benzyl-5-fluoro-1,2,4-triazol-3-yl)-4-fluoro-2- methylphenyl]pyrazolo[1,5-a]pyridine-3-carboxamide [0325] The title compound was prepared from N-[4-fluoro-2-methyl-5-(4,4,5,5-tetramethyl- 1,3,2-dioxaborolan-2-yl)phenyl]pyrazolo[1,5-a]pyridine-3-carboxamide and 1-benzyl-3-bromo- 5-fluoro-1,2,4-triazole in a similar fashion to Ex.1 and Ex.67.
- Step a A vial was charged with methyl 2-amino-4-bromo-5-fluorobenzoate (1.5 g, 6 mmol, 1.0 equiv.) and THF (10 mL). To the reaction mixture was added LiBH4 in THF (2M, 6 mL, 12 mmol, 2.0 equiv.).
- reaction mixture was stirred at 50 °C for 2 h, at which time LCMS analysis indicated complete consumption of starting material.
- the reaction mixture was cooled to rt, diluted with water and stirred for 30 min., during which time a few drops of conc. HCl were added.
- the mixture was diluted with EtOAc.
- the organic phase was separated, washed with brine (2x), dried over Na 2 SO 4 and concentrated in vacuo.
- the crude product was purified by column chromatography (0% to 90% EtOAc in hexanes) to provide the desired (2-amino-4-bromo-5- fluorophenyl) methanol.
- Step b A vial was charged with product from step a (1.3 g, 5.9 mmol, 1.0 equiv.), imidazole (1.0 g, 14.7 mmol, 2.5 equiv.) and DMF (15 mL). TBS-Cl (979 mg, 6.5 mmol, 1.1 equiv.) was added to the mixture. The reaction mixture was stirred for 5 h at 23 °C, at which time LCMS analysis indicated complete consumption of starting material. The reaction mixture was diluted with EtOAc, the layers were separated, and the organic layer was washed successively with sat. aq. NH4Cl (2x) and brine (2x).
- Step c A mixture of the product from step b (2.0 g, 5.9 mmol, 1.0 equiv.), and triazolo[4,5-b]pyridin-3-yl pyrazolo[1,5-a]pyridine-3-carboxylate (1.7 g, 5.9 mmol, 1.0 equiv.) in NMP (15.0 mL) was stirred at 120 °C for 1 h. Upon complete conversion, as judged by LCMS analysis, the reaction mixture was cooled to rt and diluted with water. The resulting precipitated solid was collected by vacuum filtration, rinsed with water, and dried in vacuo.
- Step d A vial was charged with the product from step c (1.0 g, 2.0 mmol, 1 equiv.), 4,4,4',4',5,5,5',5'-octamethyl-2,2'-bi(l,3,2-dioxaborolane) (615 mg, 2.4 mmol, 1.2 equiv.), KOAc (600 mg, 6.1 mmol, 3 equiv.) and 1,4-dioxane (15 mL). The reaction mixture was degassed with N 2 for 30 min. before Pd(dppf)C1 2 (74 mg, 0.1 mmol, 5 mmol%) was added.
- reaction mixture was then heated at 100 °C for 12 h, after which time LCMS analysis showed complete conversion of starting material.
- the reaction mixture was cooled to rt and diluted with EtOAc.
- the organic layer was filtered over Celite® and the filtrate was concentrated in vacuo.
- the crude product was purified by column chromatography (0% to 60% EtOAc in hexanes) to provide the desired N-[2- [[tert-butyl(dimethyl)silyl]oxymethyl]-4-fluoro-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)phenyl]-6-fluoropyrazolo[1,5-a]pyridine-3-carboxamide.
- Step e A vial was charged with the product from step d (150 mg, 0.27 mmol, 1.0 equiv.), 3-bromo-5-cyclopropyl-4H-1,2,4-triazole (104 mg, 0.55 mmol, 2.0 equiv.), XPhos Pd G3 (47 mg, 0.055 mmol, 20 mol%), K 3 CO 3 (114 mg, 0.83 mmol, 3.0 equiv.), 1,4-dioxane (2.0 mL) and water (0.2 mL). The reaction mixture was degassed with N 2 and then stirred at 100 °C for 16 h, at which time LCMS analysis indicated complete consumption of starting material.
- Step f A vial was charged with the product from step e (145 mg, 0.276 mmol, 1.0 equiv.) and THF (2 mL). The mixture was cooled to 0 °C and TBAF (1M soln. in THF, 0.83 mL, 0.83 mmol, 3.0 equiv.) was added. The reaction mixture was allowed to warm to rt and stirred for 8 h until complete consumption of starting material was determined by LCMS analysis. The reaction mixture was diluted with water and EtOAc, the layers were separated, and the organic layer was washed with brine (2x). The combined organic layers were collected, dried over Na2SO4 and concentrated in vacuo.
- Step a To a solution of methyl 5-bromo-1H-1,2,4-triazole-3-carboxylate (2.96 g, 14.4 mmol, 1.0 equiv.) and pTsOH ⁇ H2O (547 mg, 2.88 mmol, 20 mol%) in THF (21.8 mL, 0.7 M) under N2 at rt was added dihydropyran (3.94 mL, 43.2 mmol, 3.0 equiv.) portion-wise over 5 min. The resulting mixture was heated at reflux for 6 h, then cooled to rt and concentrated under reduced pressure. The resulting residue was treated with sat. aq.
- Step b A vial was charged with one of the regioisomeric products from step a (211.7 mg, 0.730 mmol, 1.0 equiv.) in THF (2.0 mL, 0.36 M) and placed under N 2 .
- Step c A vial was charged successively with the crude product obtained in step b (51.9 mg, 0.180 mmol, 1.0 equiv.), 6-fluoro-N-[4-fluoro-2-methyl-5-(4,4,5,5-tetramethyl-1,3,2- dioxaborolan-2-yl)phenyl]pyrazolo[1,5-a]pyridine-3-carboxamide (74.4 mg, 0.180 mmol, 1.0 equiv., prepared from 6-fluoropyrazolo[1,5-a]pyridine-3-carboxylic acid and 5-bromo-4-fluoro-2- methylaniline according to Ex.1, steps a-b), Pd XPhos G3 (15.2 mg, 0.018 mmol, 10 mol%), 1M aq.
- Step d The product obtained in step c was taken up in 4N HCl in dioxane (1.5 mL) and the mixture was stirred at rt for 14 h.
- Step b A vial was charged with the product from step a (60 mg, 0.117 mmol, 1.0 equiv.), 4,4,5,5-tetramethyl-2-[1-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)cyclopropyl]-1,3,2- dioxaborolane (51 mg, 0.17 mmol, 1.5 equiv.), Pd XPhos G3 (10 mg, 0.01 mmol, 10 mol%), Cs2CO3 (76 mg, 0.23 mmol, 2.0 equiv.), dioxane (2.0 mL) and water (0.5 mL).
- Step c To a solution of the crude product from step b (0.117 mmol, 1.0 equiv.) in 10% aq. NaOH (1 mL) at 0 °C was added H 2 O 2 (30 wt% in water, 0.16 mL, 28 equiv.) dropwise.
- reaction mixture was allowed to stir for 0.5 h, then the reaction was quenched by addition of sat. aq. Na2S2O3.
- the mixture was diluted with EtOAc and water.
- the organic layer was separated, dried over Na 2 SO 4 and concentrated in vacuo.
- the crude product was purified by column chromatography (0% to 100% EtOAc in hexanes) to provide 6-fluoro-N-[4-fluoro-5-[5-(1- hydroxycyclopropyl)-4-(oxan-2-yl)-1,2,4-triazol-3-yl]-2-methylphenyl]pyrazolo[1,5-a]pyridine- 3-carboxamide, which was treated with 4N HCl in dioxane (2 mL) and stirred overnight to afford the title compound.
- Example 94 N-[5-(5-Cyclopropyl-1,3,4-oxadiazol-2-yl)-4-fluoro-2- methylphenyl]pyrazolo[1,5-a]pyridine-3-carboxamide [0339] A vial was charged with triazolo[4,5-b]pyridin-3-yl pyrazolo[1,5-a]pyridine-3- carboxylate (22.5 mg, 0.080 mmol, 1.0 equiv.), 5-(5-cyclopropyl-1,3,4-oxadiazol-2-yl)-4-fluoro- 2-methylaniline (18.7 mg, 0.080 mmol, 1.0 equiv.), which was obtained as a minor byproduct in the synthesis of Ex.12 (step b), and NMP (0.2 mL, 0.5 M).
- Example 96 N-[5-[1-(2,2-Difluoroethyl)triazol-4-yl]-4-fluoro-2-methylphenyl]pyrazolo[1,5- a]pyridine-3-carboxamide [0341] The title compound was prepared from 4-bromo-1-(2,2-difluoroethyl)triazole and N-[4- fluoro-2-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]pyrazolo[1,5-a]pyridine- 3-carboxamide in a similar fashion to Ex.1 and Ex.67.
- Example 99 N-[5-[1-(2,2-Difluoroethyl)triazol-4-yl]-4-fluoro-2-methylphenyl]-6- fluoropyrazolo[1,5-a]pyridine-3-carboxamide [0344]
- the title compound was prepared from 4-bromo-1-(2,2-difluoroethyl)triazole and 6- fluoro-N-[4-fluoro-2-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)phenyl]pyrazolo[1,5-a]pyridine-3-carboxamide in a similar fashion to Ex. 1 and Ex. 76.
- Example 100 N-[4-Fluoro-5-[1-(4-fluorophenyl)-1,2,4-triazol-3-yl]-2- methylphenyl]pyrazolo[1,5-a]pyridine-3-carboxamide [0345] The title compound was prepared from 3-bromo-1-(4-fluorophenyl)-1H-1,2,4-triazole and N-[4-fluoro-2-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]pyrazolo[1,5- a]pyridine-3-carboxamide in a similar fashion to Ex.1 and Ex.67.
- Example 102 N-[4-Fluoro-5-[1-(4-fluorophenyl)-1,2,4-triazol-3-yl]-2-methylphenyl]-6- methoxypyrazolo[1,5-a]pyridine-3-carboxamide [0347] The title compound was prepared from 3-bromo-1-(4-fluorophenyl)-1H-1,2,4-triazole and N-[4-fluoro-2-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]-6- methoxypyrazolo[1,5-a]pyridine-3-carboxamide in a similar fashion to Ex.1 and Ex.86.
- Example 103 N-[5-(2-Cyclopropyl-1,3-thiazol-4-yl)-4-fluoro-2-methylphenyl]-6- fluoropyrazolo[1,5-a]pyridine-3-carboxamide [0348] The title compound was prepared from 4-bromo-2-cyclopropyl-1,3-thiazole and 6- fluoro-N-[4-fluoro-2-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)phenyl]pyrazolo[1,5-a]pyridine-3-carboxamide in a similar fashion to Ex. 1 and Ex. 76.
- Example 104 N-[5-(5-Cyclopropyl-1,2,4-thiadiazol-3-yl)-4-fluoro-2-methylphenyl]-6- fluoropyrazolo[1,5-a]pyridine-3-carboxamide [0349] The title compound was prepared from 3-bromo-5-cyclopropyl-1,2,4-thiadiazole and 6- fluoro-N-[4-fluoro-2-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)phenyl]pyrazolo[1,5-a]pyridine-3-carboxamide in a similar fashion to Ex.
- Example 105 N-[5-(5-Cyclopropyl-1,3,4-thiadiazol-2-yl)-4-fluoro-2-methylphenyl]-6- fluoropyrazolo[1,5-a]pyridine-3-carboxamide [0350] The title compound was prepared from 2-bromo-5-cyclopropyl-1,3,4-thiadiazole and 6- fluoro-N-[4-fluoro-2-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)phenyl]pyrazolo[1,5-a]pyridine-3-carboxamide in a similar fashion to Ex.
- Example 106 N-[5-(3-Cyclopropyl-1,2,4-thiadiazol-5-yl)-4-fluoro-2-methylphenyl]-6- fluoropyrazolo[1,5-a]pyridine-3-carboxamide [0351] The title compound was prepared from 5-bromo-3-cyclopropyl-1,2,4-thiadiazole and 6- fluoro-N-[4-fluoro-2-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)phenyl]pyrazolo[1,5-a]pyridine-3-carboxamide in a similar fashion to Ex.
- Example 108 N-[5-(4-Cyclopropyl-1H-imidazol-2-yl)-4-fluoro-2- methylphenyl]pyrazolo[1,5-a]pyridine-3-carboxamide [0353] The title compound was prepared from 2-bromo-4-cyclopropyl-1H-imidazole and N-[4- fluoro-2-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]pyrazolo[1,5-a]pyridine- 3-carboxamide in a similar fashion to Ex.1 and Ex.67.
- Example 110 N-[5-(1-Cyclopropylimidazol-4-yl)-4-fluoro-2-methylphenyl]pyrazolo[1,5- a]pyridine-3-carboxamide [0355] The title compound was prepared from 4-bromo-1-cyclopropylimidazole and N-[4- fluoro-2-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]pyrazolo[1,5-a]pyridine- 3-carboxamide in a similar fashion to Ex.1 and Ex.67.
- Example 111 N-[4-Fluoro-2-methyl-5-(3-methyl-1,2,4-oxadiazol-5-yl)phenyl]pyrazolo[1,5- a]pyridine-3-carboxamide [0356] The title compound was prepared from 5-bromo-3-methyl-1,2,4-oxadiazole and N-[4- fluoro-2-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]pyrazolo[1,5-a]pyridine- 3-carboxamide e in a similar fashion to Ex.1 and Ex.67.
- Example 112 N-[5-[1-(2,2-Difluoroethyl)pyrazol-4-yl]-4-fluoro-2-methylphenyl]-6- methylpyrazolo[1,5-a]pyridine-3-carboxamide
- Step a A 20 mL vial was charged with 5-bromo-4-fluoro-2-methylaniline (0.395 g, 1.938 mmol, 1.0 equiv.), 1-(2,2-difluoroethyl)-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)pyrazole (0.5 g, 1.938 mmol, 1.0 equiv.), Pd(dppf)Cl 2 (0.141 g, 0.193 mmol, 10 mol%), and Na2CO3 (2.91 mL, 2M aq. solution, 3.0 equiv.) in of 1,4-dioxane (6 mL).
- Step b A mixture of 5-[1-(2,2-difluoroethyl)pyrazol-4-yl]-4-fluoro-2-methylaniline (0.090 g, 0.306 mmol, 1.0 equiv.) and triazolo[4,5-b]pyridin-3-yl 6-methylpyrazolo[1,5- a]pyridine-3-carboxylate (0.078 g, 0.306 mmol, 1.0 equiv., prepared according to Ex.1, step a) in NMP (2.0 mL) was stirred at 120 °C for 6 h.
- NMP 2.0 mL
- Example 120 6-Chloro-N-[5-[1-(2,2-difluoroethyl)pyrazol-4-yl]-2,4- dimethylphenyl]pyrazolo[1,5-a]pyridine-3-carboxamide [0366] The title compound was prepared from 5-bromo-2,4-dimethylaniline and triazolo[4,5- b]pyridin-3-yl 6-chloropyrazolo[1,5-a]pyridine-3-carboxylate in a similar fashion to Ex.112.
- Example 121 N-[5-[1-(2,2-Difluoroethyl)pyrazol-4-yl]-2,4-dimethylphenyl]-6- fluoropyrazolo[1,5-a]pyridine-3-carboxamide [0367] The title compound was prepared from 5-bromo-2,4-dimethylaniline and triazolo[4,5- b]pyridin-3-yl 6-fluoropyrazolo[1,5-a]pyridine-3-carboxylate in a similar fashion to Ex.112.
- Example 122 N-[5-[1-(2,2-Difluoroethyl)pyrazol-4-yl]-2,4-dimethylphenyl]-6- methoxypyrazolo[1,5-a]pyridine-3-carboxamide [0368] The title compound was prepared from 5-bromo-2,4-dimethylaniline and triazolo[4,5- b]pyridin-3-yl 6-methoxypyrazolo[1,5-a]pyridine-3-carboxylate in a similar fashion to Ex. 112.
- Step a A vial was charged with 5-bromo-4-chloro-2-methylaniline (340 mg, 1.55 mmol, 1.0 equiv.), 1-(2,2-difluoroethyl)-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrazole (400 mg, 1.55 mmol, 1.0 equiv.), Pd(dppf)Cl 2 (117 mg, 0.16 mmol, 10 mol%), Na 2 CO 3 (328 mg, 3.10 mmol, 2.0 equiv.), di
- reaction mixture was degassed with N2, and it was stirred at 95 °C for 2 h, at which time LCMS analysis indicated complete consumption of starting material.
- the reaction mixture was cooled to rt and diluted with EtOAc.
- the organic layer was separated, filtered over Celite®, and concentrated in vacuo.
- the crude product was purified by column chromatography (0% to 60% EtOAc in hexanes) to provide the desired 4-chloro-5-[1-(2,2-difluoroethyl)pyrazol-4-yl]-2-methylaniline.
- Step b A vial was charged with product from step a (100 mg, 0.37 mmol, 1.0 equiv.), cyclopropylboronic acid (47 mg, 0.55 mmol, 1.5 equiv.), PCy3 Pd G2 (22 mg, 0.037 mmol, 10 mol%), K3PO4 (234 mg, 1.10 mmol, 3.0 equiv.), toluene (2.0 mL) and water (0.2 mL). The reaction mixture was degassed with N 2, and it was stirred at 100 °C for 3 h, at which time LCMS analysis indicated partial consumption of starting material. The reaction mixture was cooled to rt and diluted with EtOAc.
- Step c A mixture of 4-cyclopropyl-5-[1-(2,2-difluoroethyl)pyrazol-4-yl]-2- methylaniline (28 mg, 0.10 mmol, 1.0 equiv.), and triazolo[4,5-b]pyridin-3-yl pyrazolo[1,5- a]pyridine-3-carboxylate (28 mg, 0.10 mmol, 1.0 equiv.) in NMP (1.0 mL) was stirred at 120 °C for 1 h. Upon complete conversion, as judged by LCMS analysis, the reaction mixture was cooled to rt and diluted with water.
- Example 125 N-[5-[1-(2,2-Difluoroethyl)-1,2,4-triazol-3-yl]-4-fluoro-2- methylphenyl]pyrazolo[1,5-a]pyridine-3-carboxamide [0373] The title compound was prepared from 1-(2,2-difluoroethyl)-3-(4,4,5,5-tetramethyl- 1,3,2-dioxaborolan-2-yl)-1,2,4-triazole and triazolo[4,5-b]pyridin-3-yl pyrazolo[1,5-a]pyridine- 3-carboxylate in a similar fashion to Ex.112.
- Example 126 N-[5-[1-(2,2-Difluoroethyl)-1,2,4-triazol-3-yl]-2,4-dimethylphenyl]-6- methoxypyrazolo[1,5-a]pyridine-3-carboxamide [0374] The title compound was prepared from 1-(2,2-difluoroethyl)-3-(4,4,5,5-tetramethyl- 1,3,2-dioxaborolan-2-yl)-1,2,4-triazole, 5-bromo-2,4-dimethylaniline, and triazolo[4,5-b]pyridin- 3-yl 6-methoxypyrazolo[1,5-a]pyridine-3-carboxylate in a similar fashion to Ex.
- Example 127 N-[5-[1-(2,2-Difluoroethyl)-1,2,4-triazol-3-yl]-4-fluoro-2-methylphenyl]-6- methylpyrazolo[1,5-a]pyridine-3-carboxamide [0375]
- the title compound was prepared from 1-(2,2-difluoroethyl)-3-(4,4,5,5-tetramethyl- 1,3,2-dioxaborolan-2-yl)-1,2,4-triazole in a similar fashion to Ex. 112.
- Example 128 N-[5-[1-(2,2-Difluoroethyl)-1,2,4-triazol-3-yl]-2,4-dimethylphenyl]-6- methylpyrazolo[1,5-a]pyridine-3-carboxamide [0376]
- the title compound was prepared from 1-(2,2-difluoroethyl)-3-(4,4,5,5-tetramethyl- 1,3,2-dioxaborolan-2-yl)-1,2,4-triazole and 5-bromo-2,4-dimethylaniline in a similar fashion to Ex.112.
- Example 129 N-[5-(1-Cyclopropylpyrazol-4-yl)-4-fluoro-2-methylphenyl]pyrazolo[1,5- a]pyridine-3-carboxamide [0377] The title compound was prepared from 1-cyclopropyl-4-(4,4,5,5-tetramethyl-1,3,2- dioxaborolan-2-yl)pyrazole and triazolo[4,5-b]pyridin-3-yl pyrazolo[1,5-a]pyridine-3- carboxylate.
- Example 132 N-[5-[1-(Cyclopropylmethyl)pyrazol-4-yl]-4-fluoro-2- methylphenyl]pyrazolo[1,5-a]pyridine-3-carboxamide [0380] The title compound was prepared from 1-(cyclopropylmethyl)-4-(4,4,5,5-tetramethyl- 1,3,2-dioxaborolan-2-yl)pyrazole and triazolo[4,5-b]pyridin-3-yl pyrazolo[1,5-a]pyridine-3- carboxylate in a similar fashion to Ex.112.
- Example 133 N-[5-[1-(Cyclopropylmethyl)pyrazol-4-yl]-2,4-difluorophenyl]pyrazolo[1,5- a]pyridine-3-carboxamide [0381] The title compound was prepared from 1-(cyclopropylmethyl)-4-(4,4,5,5-tetramethyl- 1,3,2-dioxaborolan-2-yl)pyrazole, 5-bromo-2,4-difluoroaniline, and triazolo[4,5-b]pyridin-3-yl pyrazolo[1,5-a]pyridine-3-carboxylate in a similar fashion to Ex.
- Example 134 N-[4-Chloro-5-[1-(cyclopropylmethyl)pyrazol-4-yl]-2- methylphenyl]pyrazolo[1,5-a]pyridine-3-carboxamide [0382] The title compound was prepared from 1-(cyclopropylmethyl)-4-(4,4,5,5-tetramethyl- 1,3,2-dioxaborolan-2-yl)-1H-pyrazole, 5-bromo-4-chloro-2-methylaniline, and triazolo[4,5- b]pyridin-3-yl pyrazolo[1,5-a]pyridine-3-carboxylate in a similar fashion to Ex. 112.
- Example 135 N-[2-Chloro-5-[1-(cyclopropylmethyl)pyrazol-4-yl]-4- fluorophenyl]pyrazolo[1,5-a]pyridine-3-carboxamide [0383] The title compound was prepared from 1-(cyclopropylmethyl)-4-(4,4,5,5-tetramethyl- 1,3,2-dioxaborolan-2-yl)-1H-pyrazole, 5-bromo-2-chloro-4-fluoroaniline, and triazolo[4,5- b]pyridin-3-yl pyrazolo[1,5-a]pyridine-3-carboxylate in a similar fashion to Ex.
- Example 136 N-[5-[1-(Cyclopropylmethyl)pyrazol-4-yl]-2,4-dimethylphenyl]pyrazolo[1,5- a]pyridine-3-carboxamide [0384] The title compound was prepared from 1-(cyclopropylmethyl)-4-(4,4,5,5-tetramethyl- 1,3,2-dioxaborolan-2-yl)-1H-pyrazole, 5-bromo-2,4-dimethylaniline, and triazolo[4,5-b]pyridin- 3-yl pyrazolo[1,5-a]pyridine-3-carboxylate in a similar fashion to Ex.
- Example 138 N-[4-Fluoro-2-methyl-5-(1-propan-2-ylpyrazol-4-yl)phenyl]pyrazolo[1,5- a]pyridine-3-carboxamide [0386] The title compound was prepared from 1-propan-2-yl-4-(4,4,5,5-tetramethyl-1,3,2- dioxaborolan-2-yl)pyrazole and triazolo[4,5-b]pyridin-3-yl pyrazolo[1,5-a]pyridine-3- carboxylate in a similar fashion to Ex.112.
- Example 139 N-[4-Fluoro-2-methyl-5-[1-(trifluoromethyl)pyrazol-4- yl]phenyl]pyrazolo[1,5-a]pyridine-3-carboxamide [0387] The title compound was prepared from 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)- 1-(trifluoromethyl)pyrazole and triazolo[4,5-b]pyridin-3-yl pyrazolo[1,5-a]pyridine-3- carboxylate in a similar fashion to Ex.112.
- Example 140 N-[4-Fluoro-2-methyl-5-[1-(oxan-4-yl)pyrazol-4-yl]phenyl]pyrazolo[1,5- a]pyridine-3-carboxamide [0388] The title compound was prepared from 1-(oxan-4-yl)-4-(4,4,5,5-tetramethyl-1,3,2- dioxaborolan-2-yl)pyrazole and triazolo[4,5-b]pyridin-3-yl pyrazolo[1,5-a]pyridine-3- carboxylate in a similar fashion to Ex.112.
- Example 141 N-[4-Fluoro-2-methyl-5-[1-(2,2,2-trifluoroethyl)pyrazol-4- yl]phenyl]pyrazolo[1,5-a]pyridine-3-carboxamide [0389] The title compound was prepared from 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)- 1-(2,2,2-trifluoroethyl)pyrazole and triazolo[4,5-b]pyridin-3-yl pyrazolo[1,5-a]pyridine-3- carboxylate in a similar fashion to Ex.112.
- Example 142 N-[4-Fluoro-2-methyl-5-[1-(3,3,3-trifluoropropyl)pyrazol-4- yl]phenyl]pyrazolo[1,5-a]pyridine-3-carboxamide [0390] The title compound was prepared from 4-bromo-1-(3,3,3-trifluoropropyl)pyrazole and N-[4-fluoro-2-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]pyrazolo[1,5- a]pyridine-3-carboxamide in a similar fashion to Ex.1 and Ex.67.
- Step a A 40 mL vial was charged with 1-ethenyl-4-iodopyrazole (500 mg, 2.27 mmol, 1.0 equiv.), NaI (2.1 g, 13.9 mmol, 6.1 equiv.), and MeCN (15 mL, 0.15 M) under an N2 atmosphere. Me 3 SiCF 3 (2.2 mL, 14.9 mmol, 6.6 equiv.) was added over 45 min to the reaction mixture at 80 °C, and the reaction mixture was stirred at 80 °C for an additional 3 h. Upon complete conversion, as judged by LCMS analysis, the reaction mixture was cooled to rt.
- Step b The crude intermediate obtained in step a was converted to the title compound in a similar fashion to Ex.1.
- Example 144 N-[5-[1-(2,2-Difluorocyclopropyl)pyrazol-4-yl]-4-fluoro-2-methylphenyl]-6- fluoropyrazolo[1,5-a]pyridine-3-carboxamide [0393] The title compound was prepared from 6-fluoro-N-[4-fluoro-2-methyl-5-(4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]pyrazolo[1,5-a]pyridine-3-carboxamide in a similar fashion to Ex. 143.
- Step a N-[4-Fluoro-5-[1-(3-hydroxycyclobutyl)pyrazol-4-yl]-2- methylphenyl]pyrazolo[1,5-a]pyridine-3-carboxamide
- Step a A round-bottom flask was charged with 4-bromo-1H-pyrazole (2.00 g, 13.6 mmol, 1.0 equiv.), 3-bromocyclobutan-1-one (2.60 g, 17.7 mmol, 1.3 equiv.), K 2 CO 3 (2.60 g, 17.7 mmol, 1.30 equiv.), and DMF (39 mL, 0.35 M), and the resulting mixture was stirred at 25 °C for 18 h.
- Step b A 40 mL vial was charged with the crude product obtained in step a (2.7 g) and MeOH (14 mL). NaBH4 (0.573 g, 15.1 mmol) was added to the mixture in small portions at 0 °C, and the resulting mixture was allowed to warm to 25 °C and stir for 4 h.
- Step c The crude intermediate obtained in step b was converted to the title compound in a similar fashion to Ex.1.
- Example 149 N-[4-Chloro-5-[1-(4-fluorophenyl)pyrazol-4-yl]-2-methylphenyl]-6- methoxypyrazolo[1,5-a]pyridine-3-carboxamide [0400]
- the title compound was prepared from [1-(4-fluorophenyl)-1H-pyrazol-4-yl]boronic acid, 5-bromo-4-chloro-2-methylaniline, and triazolo[4,5-b]pyridin-3-yl 6-methoxypyrazolo[1,5- a]pyridine-3-carboxylate in a similar fashion to Ex.112.
- Example 150 N-[4-Fluoro-2-methyl-5-(1-methylpyrazol-3-yl)phenyl]pyrazolo[1,5- a]pyridine-3-carboxamide [0401] The title compound was prepared from 1-methyl-3-(4,4,5,5-tetramethyl-1,3,2- dioxaborolan-2-yl)pyrazole and triazolo[4,5-b]pyridin-3-yl pyrazolo[1,5-a]pyridine-3- carboxylate in a similar fashion to Ex.112.
- Example 151 N-[5-(5-Cyclopropyl-1H-pyrazol-3-yl)-4-fluoro-2-methylphenyl]pyrazolo[1,5- a]pyridine-3-carboxamide [0402] The title compound was prepared from 5-cyclopropyl-3-iodo-1H-pyrazole and N-[4- fluoro-2-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]pyrazolo[1,5-a]pyridine- 3-carboxamide in a similar fashion to Ex.1 and Ex.67.
- Example 153 N-[5-(5-Cyclopropyl-1H-pyrazol-3-yl)-4-fluoro-2-methylphenyl]-6- methoxypyrazolo[1,5-a]pyridine-3-carboxamide [0404] The title compound was prepared from 5-cyclopropyl-3-iodo-1H-pyrazole and N-[4- fluoro-2-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]-6-methoxypyrazolo[1,5- a]pyridine-3-carboxamide in a similar fashion to Ex.1 and Ex.86.
- Step a A mixture of triazolo[4,5-b]pyridin-3-yl pyrazolo[1,5-a]pyridine-3-carboxylate (500 mg, 1.78 mmol, 1.0 equiv.), and 5-amino-2-fluoro-4-methylbenzonitrile (268 mg, 1.78 mmol, 1.0 equiv.) in NMP (6.0 mL) was stirred at 120 °C for 6 h. The reaction mixture was cooled to rt and diluted with water.
- Step b A vial was charged with product from step a (495 mg, 1.68 mmol, 1.0 equiv.), sodium azide (219 mg, 3.36 mmol, 2.0 equiv.), ammonium chloride (180 mg, 3.36 mmol, 2.0 equiv.) and DMF (8 mL). The reaction mixture was stirred at 110 °C for 16 h, at which time LCMS analysis indicated full consumption of starting material.
- Step c To a solution of product from step b (75 mg, 0.22 mmol, 1.0 equiv.) and K 2 CO 3 (60 mg, 0.44 mmol, 2.0 equiv.) in DMF (1 mL) was added bromomethylcyclopropane (59 mg, 0.44 mmol, 2.0 equiv.).
- Example 156 6-Fluoro-N-[4-fluoro-2-methyl-5-(2-methyltetrazol-5-yl)phenyl]pyrazolo[1,5- a]pyridine-3-carboxamide
- the title compound was prepared from triazolo[4,5-b]pyridin-3-yl 6-fluoropyrazolo[1,5- a]pyridine-3-carboxylate and iodomethane in a similar fashion to Ex. 154.
- Example 157 6-Fluoro-N-[4-fluoro-2-methyl-5-(1-methyltetrazol-5-yl)phenyl]pyrazolo[1,5- a]pyridine-3-carboxamide
- the title compound was obtained as a minor byproduct in the synthesis of Ex.157, which was prepared from triazolo[4,5-b]pyridin-3-yl 6-fluoropyrazolo[1,5-a]pyridine-3-carboxylate and iodomethane in a similar fashion to Ex.154.
- Example 158 N-[5-(2-Ethyltetrazol-5-yl)-4-fluoro-2-methylphenyl]pyrazolo[1,5-a]pyridine- 3-carboxamide [0411] The title compound was prepared from bromoethane in a similar fashion to Ex.154.
- Example 161 N-[5-(6-cyclopropylpyridazin-3-yl)-4-fluoro-2-methylphenyl]pyrazolo[1,5- a]pyridine-3-carboxamide [0414] The title compound was prepared from N-[4-fluoro-2-methyl-5-(4,4,5,5-tetramethyl- 1,3,2-dioxaborolan-2-yl)phenyl]pyrazolo[1,5-a]pyridine-3-carboxamide and 3-bromo-6- cyclopropylpyridazine in a similar fashion to Ex. 89 (step a).
- Example 162 N-[5-(2-cyclopropylpyrimidin-5-yl)-4-fluoro-2-methylphenyl]pyrazolo[1,5- a]pyridine-3-carboxamide [0415] The title compound was prepared from N-[4-fluoro-2-methyl-5-(4,4,5,5-tetramethyl- 1,3,2-dioxaborolan-2-yl)phenyl]pyrazolo[1,5-a]pyridine-3-carboxamide and 5-bromo-2- cyclopropylpyrimidine in a similar fashion to Ex. 89 (step a).
- Example 163 N-[5-(5-cyclopropylpyrazin-2-yl)-4-fluoro-2-methylphenyl]pyrazolo[1,5- a]pyridine-3-carboxamide [0416] The title compound was prepared from N-[4-fluoro-2-methyl-5-(4,4,5,5-tetramethyl- 1,3,2-dioxaborolan-2-yl)phenyl]pyrazolo[1,5-a]pyridine-3-carboxamide and 2-bromo-5- cyclopropylpyrazine in a similar fashion to Ex.89 (step a).
- Example 164 N-[5-(2-cyclopropyloxypyrimidin-5-yl)-4-fluoro-2-methylphenyl]-6- fluoropyrazolo[1,5-a]pyridine-3-carboxamide [0417] The title compound was prepared from 6-fluoro-N-[4-fluoro-2-methyl-5-(4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]pyrazolo[1,5-a]pyridine-3-carboxamide and 5- bromo-2-cyclopropyloxypyrimidine in a similar fashion to Ex.92 (step c).
- Example 165 6-fluoro-N-[4-fluoro-2-methyl-5-[2-[[(3S)-oxolan-3-yl]amino]pyrimidin-5- yl]phenyl]pyrazolo[1,5-a]pyridine-3-carboxamide
- Step a A round-bottom flask was charged with 5-bromo-2-chloropyrimidine (1.93 g, 10.0 mmol, 1.0 equiv.), (3S)-oxolan-3-amine hydrochloride (2.47 g, 20.0 mmol, 2.0 equiv.), K 2 CO 3 (5.53 g, 40.0 mmol, 40.0 equiv.), and DMF (20 mL, 0.5 M), and the resulting mixture was stirred at 70 °C for 12 h.
- Step b A vial was charged successively with the crude product obtained in step a (55 mg, 0.225 mmol, 1.5 equiv.), 6-fluoro-N-[4-fluoro-2-methyl-5-(4,4,5,5-tetramethyl-1,3,2- dioxaborolan-2-yl)phenyl]pyrazolo[1,5-a]pyridine-3-carboxamide (62 mg, 0.15 mmol, 1.0 equiv., prepared from 6-fluoropyrazolo[1,5-a]pyridine-3-carboxylic acid and 5-bromo-4-fluoro-2- methylaniline according to Ex.1, steps a-b), Pd(dppf)Cl2 (11 mg, 0.015 mmol, 10 mol%), 2M aq.
- Example 166 N-[5-[2-(2,2-difluoroethylamino)pyrimidin-5-yl]-4-fluoro-2-methylphenyl]-6- fluoropyrazolo[1,5-a]pyridine-3-carboxamide [0420] The title compound was prepared from 6-fluoro-N-[4-fluoro-2-methyl-5-(4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]pyrazolo[1,5-a]pyridine-3-carboxamide and 5- bromo-N-(2,2-difluoroethyl)pyrimidin-2-amine (prepared from 5-bromo-2-chloropyrimidine and 2,2-difluoroethanamine hydrochloride according to Ex.165, step a) in a similar fashion to Ex.165.
- Example 167 6-chloro-N-[5-[1-(2,2-difluoroethyl)triazol-4-yl]-4-fluoro-2- methylphenyl]pyrazolo[1,5-a]pyridine-3-carboxamide [0421] The title compound was prepared from 6-fluoro-N-[4-fluoro-2-methyl-5-(4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]pyrazolo[1,5-a]pyridine-3-carboxamide and 5- bromo-N-(cyclopropylmethyl)pyrimidin-2-amine (prepared from 5-bromo-2-chloropyrimidine and cyclopropylmethanamine according to Ex.
- Example 168 6-fluoro-N-[4-fluoro-2-methyl-5-[6-[[(3S)-oxolan-3-yl]amino]pyridazin-3- yl]phenyl]pyrazolo[1,5-a]pyridine-3-carboxamide
- the title compound was prepared from 6-fluoro-N-[4-fluoro-2-methyl-5-(4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]pyrazolo[1,5-a]pyridine-3-carboxamide and 6- bromo-N-[(3S)-oxolan-3-yl]pyridazin-3-amine (prepared from 3-bromo-6-chloropyridazine and (3S)-oxolan-3-amine hydrochloride according to Ex.165,
- Example 170 N-[5-(6-cyclopropyloxypyridazin-3-yl)-4-fluoro-2-methylphenyl]-6- fluoropyrazolo[1,5-a]pyridine-3-carboxamide [0424] The title compound was prepared from 6-fluoro-N-[4-fluoro-2-methyl-5-(4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]pyrazolo[1,5-a]pyridine-3-carboxamide and 3- bromo-6-cyclopropyloxypyridazine (prepared from 3-bromo-6-fluoropyridazine and cyclopropanol according to Ex.165, step a) in a similar fashion to Ex.165.
- Step a A 40 mL vial was charged with 5-bromo-2-chloro-4-fluoropyridine (250 mg, 0.95 mmol., 1.0 equiv.), cyclopropyl zinc bromide (443 mg, 1.9 mmol., 2 equiv.), Pd(PPh 3 ) 4 (59 mg, 0.05 mmol., 5 mol%) and THF (6.0 mL). The resulting mixture was stirred at 50 °C for 5 h. Upon complete conversion, as judged by LCMS analysis, the reaction mixture was cooled to rt and filtered over Celite® and concentrated in vacuo.
- Step b A vial was charged successively with the product obtained in step a, 5-bromo-2- cyclopropyl-4-fluoropyridine (78 mg, 0.363 mmol., 1.0 equiv.), 6-fluoro-N-[4-fluoro-2-methyl-5- (4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]pyrazolo[1,5-a]pyridine-3-carboxamide (100 mg, 0.242 mmol, 1.0 equiv., prepared from 6-fluoropyrazolo[1,5-a]pyridine-3-carboxylic acid and 5-bromo-4-fluoro-2-methylaniline according to Ex.
- Example 172 N-[2,4-dimethyl-5-(6-propan-2-yloxypyridazin-3-yl)phenyl]-6- fluoropyrazolo[1,5-a]pyridine-3-carboxamide [0427] The title compound was prepared from 6-fluoro-N-[4-fluoro-2-methyl-5-(4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]pyrazolo[1,5-a]pyridine-3-carboxamide and 3- bromo-6-propan-2-yloxypyridazine in a similar fashion to Ex.
- Example 173 N-[5-(5-difluoromethoxy-2-pyridyl)-4-fluoro-2-tolyl]-1,7a-diaza-3- indenecarboxamide [0428] The title compound was prepared from 6-fluoro-N-[4-fluoro-2-methyl-5-(4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]pyrazolo[1,5-a]pyridine-3-carboxamide and 2- bromo-5-(difluoromethoxy)pyridine according to Ex.165 (step a), in a similar fashion to Ex.165.
- Step a A solution of cyclopropanol (280 mg, 4.8 mmol, 1.5 equiv.) in DMF (5.0 mL) was added NatBuO (470 mg, 4.8 mmol, 1.5 equiv.).
- Step b A mixture of product from step a with KOH (10.0 equiv.) in tBuOH (10mL) and water (2mL) was stirred at 105 °C for 48h, then cooled to room temperature. The reaction mixture was acidified with 1N HCl to PH ⁇ 3 and then extracted with EtOAc. The organic layer was separated, dried over Na2SO4 and concentrated in vacuo. The crude product was used directly in the next step without further purification.
- Step c The title compound was prepared from 5-cyclopropyloxypyridine-2-carboxylic acid and 6-N-(5-amino-4-fluoro-2-methylphenyl)-6-fluoropyrazolo[1,5-a]pyridine-3- carboxamide in a similar fashion to Ex. 176.
- Example 175 6-fluoro-N-[4-fluoro-2-methyl-5-[[5-[[(3S)-oxolan-3-yl]amino]pyridine-2- carbonyl]amino]phenyl]pyrazolo[1,5-a]pyridine-3-carboxamide [0432] The title compound was prepared from 5-[[(3S)-oxolan-3-yl]amino]pyridine-2- carboxylic acid and 6-N-(5-amino-4-fluoro-2-methylphenyl)-6-fluoropyrazolo[1,5-a]pyridine-3- carboxamide in a similar fashion to Ex.176.
- Step a A mixture of triazolo[4,5-b]pyridin-3-yl 6-fluoropyrazolo[1,5-a]pyridine-3- carboxylate (298 mg, 1.0 mmol, 1.0 equiv.) and 5-bromo-4-fluoro-2-methylaniline (204 mg, 1.0 mmol, 1.0 equiv.) in NMP (6.0 mL) was stirred at 120 °C for 4 h. The reaction mixture was cooled to rt and diluted with water.
- Step b A vial was charged with product from step a (260 mg, 0.72 mmol, 1.0 equiv.), diphenylmethanimine (155 mg, 0.85 mmol, 1.2 equiv.), Pd 2 (dba) 3 (66 mg, 0.072 mmol, 0.1 equiv.), XantPhos (84 mg, 0.44 mmol, 0.2 equiv.), Cs 2 CO 3 (705 mg, 2.16 mmol, 3.0 equiv.) and dioxane (8 mL). The reaction mixture was stirred at 100 °C for 16 h under N2, at which time LCMS analysis indicated full consumption of starting material.
- Step c To a mixture of product from step b (220 mg, 0.47 mmol, 1.0 equiv.), NaOAc (97 mg, 1.18 mmol, 2.5 equiv.) in MeOH (5 mL) was added NH2OH HCl (66 mg, 0.94 mmol, 2.0 equiv.).
- Step d The desired product was prepared from 5-fluoropyridine-2-carboxylic acid in a similar manner to Example 1, step a.
- Example 177 6-fluoro-N-[4-fluoro-2-methyl-5-[(5-methylpyridine-2- carbonyl)amino]phenyl]pyrazolo[1,5-a]pyridine-3-carboxamide
- the title compound was prepared from 5-methylpyridine-2-carboxylic acid and 6-N-(5- amino-4-fluoro-2-methylphenyl)-6-fluoropyrazolo[1,5-a]pyridine-3-carboxamide in a similar fashion to Ex.176.
- Example 178 6-fluoro-N-[4-fluoro-2-methyl-5-(pyridine-3- carbonylamino)phenyl]pyrazolo[1,5-a]pyridine-3-carboxamide
- the title compound was prepared from pyridine-2-carboxylic acid and 6-N-(5-amino-4- fluoro-2-methylphenyl)-6-fluoropyrazolo[1,5-a]pyridine-3-carboxamide in a similar fashion to Ex.176.
- Example 180 N-[5-[(2,2-difluorocyclopropanecarbonyl)amino]-4-fluoro-2-methylphenyl]- 6-fluoropyrazolo[1,5-a]pyridine-3-carboxamide [0440] The title compound was prepared from 2,2-difluorocyclopropane-1-carboxylic acid and 6-N-(5-amino-4-fluoro-2-methylphenyl)-6-fluoropyrazolo[1,5-a]pyridine-3-carboxamide in a similar fashion to Ex. 176.
- Example 181 N-[5-(cyclopropanecarbonylamino)-4-fluoro-2-methylphenyl]-6- fluoropyrazolo[1,5-a]pyridine-3-carboxamide [0441] The title compound was prepared from cyclopropanecarboxylic acid and 6-N-(5-amino- 4-fluoro-2-methylphenyl)-6-fluoropyrazolo[1,5-a]pyridine-3-carboxamide in a similar fashion to Ex.176.
- Activity was determined as a function of phosphorylated biotinylated TK peptide generated by the transfer of phosphate from ATP as measured by the use of the HTRF KinEASE-TK assay kit (Cisbio, Product #: 62TK0PEJ).
- Levels of phosphorylated biotinylated TK peptide (Part 61TK0BLC of KinEASE assay kit) are quantified by its capture by phosphor-TK-Antibody-Cryptate (Part of KinEASE assay kit) and XL665-labeled Streptavidin (Part 610SAXLG of KinEASE assay kit) followed by measurement of Time-Resolved Fluorescence Resonance Energy Transfer (TR- FRET) signal.
- TR- FRET Time-Resolved Fluorescence Resonance Energy Transfer
- Enzyme solution was prepared for each of the tyrosine kinases in 50 mM HEPES, pH 7.4, 5 mM MgCl2 for c-Kit or 10 mM MgCl2 for FLT3, CSF1R, PDGFR ⁇ and PDGFR ⁇ , 2 mM MnCl2, 0.01% Brij-35 and 0.01% BSA.
- M07e cells (DSMZ, catalog # ACC 104) were serum starved. Cells were centrifuged and cell pellet resuspended in OptiMEM (Gibco, catalog # 31985062) to a density of 1x10 6 - 2.5x10 6 cells per mL. Cells were then incubated overnight at 5% CO2 and 37 °C in an appropriately sized flask. On the day of the experiment, an 11 point, half log titration of test compound was pre-dispensed into 96-well round bottom polypropylene plates (Corning, catalog # 3365).
- Cell pellets were resuspended with either OptiMEM or human serum (Innovative Research, catalog # ISERAB1000ML) to a cell density of 1x10 6 cells per mL. Cells were then added to the appropriate compound plate at 100,000 cells per well in 100 ⁇ L and mixed. Plates were incubated at 5% CO2 and 37 °C for 50 min. Stem Cell Factor, SCF (R&D Systems, #255-SC/CF) was diluted in OptiMEM to 1 ⁇ g/mL and added to all wells as 10 ⁇ L for a final concentration of 90.9 ng/mL. Plates were incubated at 5% CO2 and 37 °C for 10 min.
- SCF Stem Cell Factor
- Plates were centrifuged, supernatant removed by flicking, and cells resuspended in 50 ⁇ L primary antibody, anti-phospho- c-KIT (Tyr703) (Cell Signaling, catalog # 3073), diluted to 1 ⁇ g/mL with permeabilization buffer. Plates were incubated at rt for 1 h. Plates were centrifuged, supernatant removed, and cells resuspended with 200 ⁇ L DPBS + 0.5% BSA.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
The present disclosure is directed to compounds that are inhibitors of KIT having a structure according to Formula (III), and compositions containing those compounds. Methods of using the compounds for the treatment of diseases, disorders, or conditions are also described.
Description
KIT INHIBITOR COMPOUNDS AND METHODS OF USE THEREOF CROSS-REFERENCE TO RELATED APPLICATIONS [0001] This application claims the benefit of priority to U.S. Provisional Patent Application No. 63/540,604, filed on September 26, 2023, the entire content of which is incorporated by reference herein. BACKGROUND [0002] The following discussion is provided to aid the reader in understanding the disclosure and is not admitted to describe or constitute prior art thereto. [0003] Mast cells contain granules containing proinflammatory and immunomodulatory mediators. Upon activation, degranulation occurs, releasing these proinflammatory and immunomodulatory mediators into the surrounding tissues, generally in response to a perceived pathogen (e.g., parasitic, bacterial and viral infections, allergens, toxins, etc.). The activation of mast cells serves to induce an immune response to protect the body from pathogens, and to aid in wound healing, and tissue repair. However, misfunctioning mast cells underlie the etiology of many allergic and chronic inflammatory diseases and are implicated in a broad spectrum of conditions. [0004] The activation of receptor tyrosine kinase KIT on mast cells by its ligand, Stem Cell Factor (SCF), is required for mast cell differentiation, maturation, and survival. For certain diseases, mast cell activation may play a central role in the onset and progression of the disease. Accordingly, inhibition of KIT may lead to mast cell depletion, and provide a promising therapeutic approach for mast cell-driven diseases. Thus, there is a need to develop inhibitors of KIT. SUMMARY [0005] In one aspect, the present disclosure relates to compounds that are inhibitors of the receptor tyrosine kinase KIT. The compounds are represented by Formula III and associated sub- formulas described herein:
 . [0006] In another aspect, this disclosure is directed to methods of inhibiting KIT in a subject comprising administering to the subject an effective amount of a compound of Formula III or an associated sub-formula described herein. [0007] In another aspect, this disclosure is directed to methods of reducing the activity and/or quantity of systemic mast cells in a subject comprising administering to the subject an effective amount of a compound of Formula III or an associated sub-formula described herein. [0008] In yet another aspect, this disclosure provides methods for treating a disease, disorder, or condition mediated at least in part by KIT in a subject, comprising administering to the subject a therapeutically effective amount of a compound of Formula III or an associated sub-formula described herein. Diseases, disorders, and conditions mediated by KIT include e.g., an allergic disease, disorder, or condition; an inflammatory disease, disorder, or condition; a neuroinflammatory disease, disorder, or condition; a neurological disease, disorder, or condition; an immune related disease, disorder, or condition; an autoimmune related disease, disorder, or condition; a dermatological disease, disorder, or condition; a respiratory disease, disorder, or condition; a metabolic disease, disorder, or condition; a cardiovascular disease, disorder, or condition; a fibrotic disease, disorder, or condition; or cancer. [0009] Certain aspects of the present disclosure further comprise the administration of one or more additional therapeutic agents as set forth herein below. DETAILED DESCRIPTION OF THE DISCLOSURE [0010] Before the present disclosure is further described, it is to be understood that the disclosure is not limited to the particular embodiments set forth herein, and it is also to be understood that the terminology used herein is for the purpose of describing particular embodiments only, and is not intended to be limiting.
Definitions [0011] Unless otherwise defined, all terms of art, notations and other scientific terms or terminology used herein are intended to have the meanings commonly understood by those of skill in the art to which this disclosure pertains. [0012] The term “about” as used herein has its original meaning of approximately and is to provide literal support for the exact number that it precedes, as well as a number that is near to or approximately the number that the term precedes. In determining whether a number is near to or approximately a specifically recited number, the near or approximating unrecited number can be a number which, in the context in which it is presented, provides the substantial equivalent of the specifically recited number. For example, if the degree of approximation is not otherwise clear from the context, “about” means either within plus or minus 10% of the provided value, or rounded to the nearest significant figure, in all cases inclusive of the provided value. Where ranges are provided, they are inclusive of the boundary values. [0013] The term "alkyl", by itself or as part of another substituent, means, unless otherwise stated, a saturated hydrocarbon radical, having, in some embodiments, one to eight (e.g., C1-C8- alkyl), or one to six (e.g., C1-C6-alkyl), or one to four carbon atoms (e.g., C1-C4-alkyl), or one to three carbon atoms (e.g., C1-C3-alkyl), respectively. The term “alkyl” encompasses straight and branched-chain hydrocarbon groups. Examples of alkyl groups include, but are not limited to, methyl (Me, -CH3), ethyl (Et, -CH2CH3), n-propyl, isopropyl, n-butyl, t-butyl, isobutyl, sec-butyl, isopentyl, tert-pentyl, n-pentyl, isohexyl, n-hexyl, n-heptyl, 4-isopropylheptane, n-octyl, and the like. In some embodiments, the alkyl groups are C1-C4 alkyl groups (e.g., methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl, or t-butyl). [0014] The term “alkylene” refers to a straight or branched, saturated, hydrocarbon radical having, in some embodiments, one to six carbon atoms (e.g., C1-C6-alkylene), or one to four carbon atoms (e.g., C1-C4-alkylene), or one to three carbon atoms (e.g., C1-C3-alkylene) and linking at least two other groups, i.e., a divalent hydrocarbon radical. When two moieties are linked to the alkylene they can be linked to the same carbon atom (i.e., geminal), or different carbon atoms of the alkylene group. For instance, a straight chain alkylene can be the bivalent radical of -(CH2)n- , where n is 1, 2, 3, 4, 5 or 6 (i.e., a C1-C6-alkylene). Representative alkylene groups include, but are not limited to, methylene, ethylene, propylene, isopropylene, butylene, isobutylene,
secbutylene, pentylene, hexylene and the like. In some embodiments, the alkylene groups are C1- 3 alkylene groups (e.g., methylene, ethylene, or propylene). [0015] The term "cycloalkyl" refers to a monocyclic, bicyclic or polycyclic hydrocarbon ring system having, in some embodiments, 3 to 14 carbon atoms (e.g., C3-C14-cycloalkyl), or 3 to 10 carbon atoms (e.g., C3-C10-cycloalkyl), or 3 to 8 carbon atoms (e.g., C3-C8-cycloalkyl), or 3 to 6 carbon atoms (e.g., C3-C6-cycloalkyl) or 5 to 6 carbon atoms (e.g., C5-C6-cycloalkyl). Cycloalkyl groups can be saturated or characterized by one or more points of unsaturation (i.e., carbon-carbon double and/or triple bonds), provided that the points of unsaturation do not result in an aromatic system. Examples of monocyclic cycloalkyl groups include, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cyclohexenyl, cyclohexynyl, cycloheptyl, cycloheptenyl, cycloheptadienyl, cyclooctyl, cyclooctenyl, cyclooctadienyl and the like. The rings of bicyclic and polycyclic cycloalkyl groups can be fused, bridged, or spirocyclic. Non-limiting examples of bicyclic, spirocyclic and polycyclic hydrocarbon groups include bicyclo[1.1.1]pentane, bicyclo[2.1.1]hexane, bicyclo[2.2.1]heptane, bicyclo[2.2.2]octane, adamantyl, spiro[5.5]undecane, spiro[2.2]pentane, spiro[2.2]pentadiene, spiro[2.5]octane, spiro[2.2]pentadiene, and the like. In some embodiments, the cycloalkyl groups of the present disclosure are monocyclic C3-C6-cycloalkyl moieties (e.g., cyclopropyl, cyclobutyl, cyclopentyl, or cyclohexyl). [0016] As used herein, a wavy line, " ", that intersects a single, double or triple bond in any chemical structure depicted herein, represents that the point of attachment of the single, double, or triple bond to the remainder of the molecule is through either one of the atoms that make up the single, double or triple bond. Additionally, a bond extending from a substituent to the center of a ring (e.g., a phenyl ring or heteroaryl ring) is meant to indicate attachment of that substituent to the ring at any of the available ring vertices, i.e., such that attachment of the substituent to the ring results in a chemically stable arrangement. [0017] The terms "halogen," or “halo” are used interchangeably and refer to, unless otherwise stated, a fluorine, chlorine, bromine, or iodine atom. Additionally, terms such as "haloalkyl," refer to alkyl groups, as defined herein, that are substituted with one or more halogen(s) (e.g., 1-3 halogen(s)). For example, the term "C1-C6 haloalkyl" is meant to include trifluoromethyl, difluoromethyl, 2,2,2-trifluoroethyl, 4-chlorobutyl, 3-bromopropyl, and the like.
[0018] The term “hydroxyalkyl” refers to an alkyl group, as defined herein, that is substituted with one or more hydroxyl groups (e.g., 1-3 hydroxyl groups). Exemplary hydroxyalkyl groups include methanol, ethanol, 1,2-propanediol, 1,2-hexanediol, glycerol, and the like. [0019] The term “cyanoalkyl” refers to an alkyl group, as defined herein, that is substituted with one cyano group. Exemplary cyanoalkyl groups include cyanomethyl, cyanoethyl, cyanopropyl, and the like. [0020] As used herein, the term “alkoxy” refers to an alkyl group, as defined herein, that is attached to the remainder of the molecule via an oxygen atom (e.g., -O-C1-C12 alkyl, -O-C1-C8 alkyl, -O-C1-C6 alkyl, or -O-C1-C3 alkyl). Non-limiting examples of alkoxy groups include methoxy (OMe, -OCH3), ethoxy (OEt, -OCH2CH3), n-propoxy, iso-propoxy, n-butoxy, sec- butoxy, n-pentoxy, n-hexoxy, and the like. [0021] The term "heteroaryl" refers to monocyclic or fused bicyclic aromatic groups (or rings) having, in some embodiments, from 5 to 14 (i.e., 5- to 14-membered heteroaryl), or from 5 to 10 (i.e., 5- to 10-membered heteroaryl), or from 5 to 6 (i.e., 5- to 6-membered heteroaryl) members (i.e., ring vertices), and containing from one to five, one to four, one to three, one to two or one ring heteroatom independently selected from nitrogen (N), oxygen (O), and sulfur (S). A heteroaryl group can be attached to the remainder of the molecule through a carbon atom or a heteroatom of the heteroaryl group, when chemically permissible. Non-limiting examples of heteroaryl groups include pyridyl, pyridazinyl, pyrazinyl, pyrimidinyl, triazinyl, purinyl, thienopyridinyl, thienopyrimidinyl, pyrazolopyrimidinyl, imidazopyridines, isothiazolyl, pyrazolyl, indazolyl, pteridinyl, imidazolyl, triazolyl, tetrazolyl, oxazolyl, isoxazolyl, thiadiazolyl, pyrrolyl, thiazolyl, furyl, thienyl and the like. In some embodiments, the heteroaryl groups of the present disclosure are monocyclic 5- to 6-membered heteroaryl moieties having 1-4 ring nitrogen atoms (e.g., pyridinyl, pyrimidinyl, pyrazinyl, pyridazinyl, triazolyl, imidazolyl, pyrazolyl, tetrazolyl, and the like). In some embodiments, the heteroaryl groups of the present disclosure are 5-membered heteroaryl moieties having 1-4 ring heteroatoms independently selected from N, O, and S (e.g., imidazolyl, pyrazolyl, thiazolyl, oxazolyl, oxadiazole, thiadiazolyl, triazolyl, tetrazolyl, and the like). [0022] The term "heterocycloalkyl" refers to a non-aromatic monocyclic, bicyclic or polycyclic cycloalkyl ring having, in some embodiments, 3 to 14 members (e.g., 3- to 14-membered
heterocycle), or 3 to 10 members (e.g., 3- to 10-membered heterocycle), or 4 to 8 members (e.g., 4- to 8-membered heterocycle), or 4 to 6 members (e.g., 4- to 6-membered heterocycle), or 5 to 6 members (e.g., 5- to 6-membered heterocycle), and having from one to five, one to four, one to three, one to two or one ring heteroatom independently selected from nitrogen (N), oxygen (O), and sulfur (S). Heterocycloalkyl groups are saturated or characterized by one or more points of unsaturation (e.g., one or more carbon-carbon double bonds, carbon-carbon triple bonds, carbon- nitrogen double bonds, and/or nitrogen-nitrogen double bonds), provided that the points of unsaturation do not result in an aromatic system. The rings of bicyclic and polycyclic heterocycloalkyl groups can be fused, bridged, or spirocyclic. Non-limiting examples of heterocycloalkyl groups include aziridine, oxirane, thiirane, azetidine, oxetane, pyrrolidine, imidazolidine, pyrazolidine, dioxolane, phthalimide, piperidine, 1,4-dioxane, morpholine, thiomorpholine, thiomorpholine-S-oxide, thiomorpholine-S,S-oxide, piperazine, 3,4,5,6- tetrahydropyridazine, pyran, decahydroisoquinoline, 3-pyrroline, thiopyran, tetrahydrofuran, tetrahydrothiophene, quinuclidine, 2,6-diazaspiro[3.3]heptane, 2-azaspiro[3.3]heptane, 1- oxaspiro[3.3]heptane, 6-azaspiro[3.4]octane, and the like. A heterocycloalkyl group can be attached to the remainder of the molecule through a ring carbon atom, or a ring heteroatom, when chemically permissible. In some embodiments, the heterocycloalkyl groups of the present disclosure are monocyclic 4- to 8-membered heterocycloalkyl moieties having one or two heteroatoms independently selected from N, O, and S, (e.g., azetidine, oxetane, pyrrolidine, tetrahydrofuran, tetrahydropyran, piperidine, piperazine, morpholine, and the like). [0023] As referred to herein, "pharmaceutically acceptable salt" is meant to include salts of the compounds according to this disclosure that are prepared with suitably nontoxic acids or bases, depending on the particular substituents found on the compounds described herein. When compounds of the present disclosure contain suitably acidic functionalities, base addition salts can be obtained by contacting the neutral form of such compounds with a sufficient amount of the desired base, either neat or in a suitable inert solvent. Examples of salts derived from pharmaceutically-acceptable inorganic bases include aluminum, ammonium, calcium, copper, ferric, ferrous, lithium, magnesium, manganic, manganous, potassium, sodium, zinc and the like. Salts derived from pharmaceutically-acceptable organic bases include salts of primary, secondary and tertiary amines, including substituted amines, cyclic amines, naturally-occurring amines and the like, such as arginine, betaine, caffeine, choline, N,N’-dibenzylethylenediamine, diethylamine,
2-diethylaminoethanol, 2-dimethylaminoethanol, ethanolamine, ethylenediamine, N- ethylmorpholine, N-ethylpiperidine, glucamine, glucosamine, histidine, hydrabamine, isopropylamine, lysine, methylglucamine, morpholine, piperazine, piperidine, polyamine resins, procaine, purines, theobromine, triethylamine, trimethylamine, tripropylamine, tromethamine and the like. When compounds of the present disclosure contain suitably basic functionalities, acid addition salts can be obtained by contacting the neutral form of such compounds with a sufficient amount of the desired acid, either neat or in a suitable inert solvent. Examples of pharmaceutically acceptable acid addition salts include those derived from inorganic acids like hydrochloric, hydrobromic, nitric, carbonic, monohydrogencarbonic, phosphoric, monohydrogenphosphoric, dihydrogenphosphoric, sulfuric, monohydrogensulfuric, hydriodic, or phosphorous acids and the like, as well as the salts derived from suitably nontoxic organic acids like acetic, propionic, isobutyric, malonic, benzoic, succinic, suberic, fumaric, mandelic, phthalic, benzenesulfonic, p- tolylsulfonic, citric, tartaric, methanesulfonic, and the like. Also included are salts of amino acids such as arginate and the like, and salts of organic acids like glucuronic or galacturonic acids and the like (see, for example, Berge, S.M., et al, “Pharmaceutical Salts”, Journal of Pharmaceutical Science, 1977, 66, 1-19). Certain specific compounds of the present disclosure contain both basic and acidic functionalities that allow the compounds to be converted into either base or acid addition salts. [0024] The neutral forms of the compounds may be regenerated by contacting the salt with a base or acid and isolating the parent compound in the conventional manner. The parent form of the compound differs from the various salt forms in certain physical properties, such as solubility in polar solvents, but otherwise the salts are equivalent to the parent form of the compound for the purposes of the present disclosure. [0025] This disclosure also contemplates isomers of the compounds described herein (e.g., stereoisomers and/or atropisomers). For example, certain compounds of the present disclosure possess asymmetric carbon atoms (chiral centers), or hindered rotation about a single bond; the racemates, diastereomers, and enantiomers, and atropisomers (e.g., Ra, Sa, P and M isomers) of which are all intended to be encompassed within the scope of the present disclosure. Stereoisomeric forms may be defined, in terms of absolute stereochemistry, as (R) or (S), depicted uses dashes and/or wedges, and/or in terms of the direction the stereoisomer rotates plane-
polarized light (e.g., dextrorotary ((+) or (d)), or levorotary ((-) or (l))). When a stereochemical depiction (e.g., using dashes, , and/or wedges, ) is shown in a chemical structure, or a stereochemical assignment (e.g., using (R) and (S) notation, or (d) and (l) notation) is made in a chemical name, it is meant to indicate that the depicted/referred to isomer is present and substantially free of one or more other isomer(s) (e.g., enantiomers and diastereomers, when present). “Substantially free of” other isomer(s) indicates at least an 70/30 ratio of the indicated isomer to the other isomer(s), more preferably 80/20, 90/10, or 95/5 or more. In some embodiments, the indicated isomer will be present in an amount of at least 99%. A chemical bond to an asymmetric carbon that is depicted as a solid line ( ) indicates that all possible stereoisomers (e.g., enantiomers, diastereomers, racemic mixtures, etc.) at that carbon atom are included. In such instances, the compound may be present as a racemic mixture, scalemic mixture, or a mixture of diastereomers. [0026] The compounds of the present disclosure may also contain unnatural proportions of atomic isotopes at one or more of the atoms that constitute such compounds. Unnatural proportions of an isotope may be defined as ranging from the amount found in nature to an amount consisting of 100% of the atom in question. For example, the compounds may incorporate radioactive isotopes, such as for example tritium (3H), iodine-125 (125I) or carbon-14 (14C), or non-radioactive isotopes, such as deuterium (2H) or carbon-13 (13C). Such isotopic variations can provide additional utilities to those described elsewhere herein. For instance, isotopic variants of the compounds of the disclosure may find additional utility, including but not limited to, as diagnostic and/or imaging reagents, or as cytotoxic/radiotoxic therapeutic agents. Additionally, isotopic variants of the compounds of the disclosure can have altered pharmacokinetic and pharmacodynamic characteristics which can contribute to enhanced safety, tolerability or efficacy during treatment. In some embodiments, the compounds according to this disclosure are characterized by one or more deuterium atoms. [0027] The terms “patient” or “subject” are used interchangeably to refer to a human or a non- human animal (e.g., a mammal). [0028] The terms “treat”, “treating”, treatment” and the like refer to a course of action that eliminates, reduces, suppresses, mitigates, ameliorates, or prevents the worsening of, either temporarily or permanently, a disease, disorder or condition to which the term applies, or at least
one of the symptoms associated therewith. Treatment includes alleviation of symptoms, diminishment of extent of disease, inhibiting (e.g., arresting the development or further development of the disease, disorder or condition or clinical symptoms association therewith) an active disease, delaying or slowing of disease progression, improving the quality of life, and/or prolonging survival of a subject as compared to expected survival if not receiving treatment or as compared to a published standard of care therapy for a particular disease. [0029] The term “in need of treatment” as used herein refers to a judgment made by a physician or similar professional that a subject requires or will benefit from treatment. This judgment is made based on a variety of factors that are in the realm of the physician’s expertise, which may include a positive diagnosis of a disease, disorder or condition. [0030] The terms “prevent”, “preventing”, “prevention”, “prophylaxis” and the like refer to a course of action initiated in a manner (e.g., prior to the onset of a disease, disorder, condition or symptom thereof) so as to prevent, suppress, inhibit or reduce, either temporarily or permanently, a subject’s risk of developing a disease, disorder, condition or the like (as determined by, for example, the absence of clinical symptoms) or delaying the onset thereof, generally in the context of a subject predisposed to having a particular disease, disorder or condition. In certain instances, the terms also refer to slowing the progression of the disease, disorder or condition or inhibiting progression thereof to a harmful or otherwise undesired state. Prevention also refers to a course of action initiated in a subject after the subject has been treated for a disease, disorder, condition or a symptom associated therewith in order to prevent relapse of that disease, disorder, condition or symptom. In one or more embodiments, the preventative course of action is taken based on anticipation of a condition or event. In one embodiment, prevention refers to the prevention, suppression, inhibition or reduction of an allergic, immune, or autoimmune response in a subject suffering from an allergic, inflammatory, neuroinflammatory, neurological, immune, autoimmune, dermatological, respiratory, metabolic, cardiovascular or fibrotic disease, disorder, or condition. [0031] As used herein, the term “response” may refer to a symptom initiated by an irritant or trigger (e.g., an antigen originating from within the body, or from the external environment) in a subject. For example, “response” may refer to a reaction (e.g., hives, rash, welts, itchy skin, stinging skin, skin fissures, skin lesions, skin blisters, swelling (e.g., in joints, glands, or tissues), vertigo, fatigue, dizziness, fainting, lightheadedness, muscle weakness, headache, dry skin, dry
eyes, hair loss, numbness or tingling in extremities, joint pain and/or stiffness, sneezing, runny nose, stuffy nose, chest tightness and/or pain, shortness of breath, wheezing, itchy eyes, watery eyes, blurred vision, sensitivity to light, stomach cramping, abdominal pain, bloating, diarrhea, constipation, indigestion, heartburn, excessive flatulence, frequent urination, difficulty swallowing, muscle spasm, muscle tremor, unexpected weight gain or loss, increased thirst, increased hunger, irritability, temperature sensitivity, low blood pressure, constriction of airways, weak pulse, rapid pulse, nausea, vomiting, anemia, depression, and the like) caused by an external or internal trigger in a subject susceptible to the trigger. Subjects susceptible to a trigger are generally those suffering from an allergic, inflammatory, neuroinflammatory, neurological, immune, autoimmune, dermatological, respiratory, metabolic, cardiovascular or fibrotic disease, disorder, or condition, such as those described elsewhere herein. [0032] The term “in need of prevention” as used herein refers to a judgment made by a physician or other caregiver that a subject requires or will benefit from preventative care. This judgment is made based on a variety of factors that are in the realm of a physician’s or caregiver’s expertise. [0033] “Substantially pure” indicates that a component (e.g., a compound according to this disclosure) makes up greater than about 50% of the total content of the composition, and typically greater than about 60% of the total content. More typically, “substantially pure” refers to compositions in which at least 75%, at least 85%, at least 90% or more of the total composition is the component of interest. In some cases, the component of interest will make up greater than about 90%, or greater than about 95% of the total content of the composition. [0034] The terms “inhibitor of KIT” and “KIT inhibitor” may be used interchangeably, and refer to the ability of a molecule to decrease the activation of KIT either directly or indirectly, thereby decreasing activation and/or quantity of systemic mast cells. [0035] Compounds that are selective for KIT may be particularly useful in the treatment of certain disorders or may offer a reduced likelihood of undesired side effects. In one embodiment, compounds of the present disclosure are selective over one or more other receptor tyrosine kinases. Specific examples include, but are not limited to, PDGFRα, PDGFRβ, CSF1R, and FLT3. Selectivity may be determined, for example, by comparing the inhibition of a compound as described herein against KIT against the inhibition of a compound as described herein against another kinase. In one embodiment, the selective inhibition of a compound of Formula III or an
associated sub-formula is at least 1000 times greater, 500 times greater, 100 times greater, 50 times greater, or 20 times greater than inhibition of one or more kinases selected from PDGFRα, PDGFRβ, CSF1R, and FLT3. [0036] Compounds provided herein may have advantageous pharmacokinetic profiles including, for example, hepatocyte stability, clearance, inhibition against CYP, and inhibition against hERG. Compounds of the Disclosure [0037] The present disclosure relates to compounds that inhibit the activity of KIT. [0038] In one aspect, this disclosure is directed to a compound having a structure according to Formula III:
. [0006] In another aspect, this disclosure is directed to methods of inhibiting KIT in a subject comprising administering to the subject an effective amount of a compound of Formula III or an associated sub-formula described herein. [0007] In another aspect, this disclosure is directed to methods of reducing the activity and/or quantity of systemic mast cells in a subject comprising administering to the subject an effective amount of a compound of Formula III or an associated sub-formula described herein. [0008] In yet another aspect, this disclosure provides methods for treating a disease, disorder, or condition mediated at least in part by KIT in a subject, comprising administering to the subject a therapeutically effective amount of a compound of Formula III or an associated sub-formula described herein. Diseases, disorders, and conditions mediated by KIT include e.g., an allergic disease, disorder, or condition; an inflammatory disease, disorder, or condition; a neuroinflammatory disease, disorder, or condition; a neurological disease, disorder, or condition; an immune related disease, disorder, or condition; an autoimmune related disease, disorder, or condition; a dermatological disease, disorder, or condition; a respiratory disease, disorder, or condition; a metabolic disease, disorder, or condition; a cardiovascular disease, disorder, or condition; a fibrotic disease, disorder, or condition; or cancer. [0009] Certain aspects of the present disclosure further comprise the administration of one or more additional therapeutic agents as set forth herein below. DETAILED DESCRIPTION OF THE DISCLOSURE [0010] Before the present disclosure is further described, it is to be understood that the disclosure is not limited to the particular embodiments set forth herein, and it is also to be understood that the terminology used herein is for the purpose of describing particular embodiments only, and is not intended to be limiting.
Definitions [0011] Unless otherwise defined, all terms of art, notations and other scientific terms or terminology used herein are intended to have the meanings commonly understood by those of skill in the art to which this disclosure pertains. [0012] The term “about” as used herein has its original meaning of approximately and is to provide literal support for the exact number that it precedes, as well as a number that is near to or approximately the number that the term precedes. In determining whether a number is near to or approximately a specifically recited number, the near or approximating unrecited number can be a number which, in the context in which it is presented, provides the substantial equivalent of the specifically recited number. For example, if the degree of approximation is not otherwise clear from the context, “about” means either within plus or minus 10% of the provided value, or rounded to the nearest significant figure, in all cases inclusive of the provided value. Where ranges are provided, they are inclusive of the boundary values. [0013] The term "alkyl", by itself or as part of another substituent, means, unless otherwise stated, a saturated hydrocarbon radical, having, in some embodiments, one to eight (e.g., C1-C8- alkyl), or one to six (e.g., C1-C6-alkyl), or one to four carbon atoms (e.g., C1-C4-alkyl), or one to three carbon atoms (e.g., C1-C3-alkyl), respectively. The term “alkyl” encompasses straight and branched-chain hydrocarbon groups. Examples of alkyl groups include, but are not limited to, methyl (Me, -CH3), ethyl (Et, -CH2CH3), n-propyl, isopropyl, n-butyl, t-butyl, isobutyl, sec-butyl, isopentyl, tert-pentyl, n-pentyl, isohexyl, n-hexyl, n-heptyl, 4-isopropylheptane, n-octyl, and the like. In some embodiments, the alkyl groups are C1-C4 alkyl groups (e.g., methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl, or t-butyl). [0014] The term “alkylene” refers to a straight or branched, saturated, hydrocarbon radical having, in some embodiments, one to six carbon atoms (e.g., C1-C6-alkylene), or one to four carbon atoms (e.g., C1-C4-alkylene), or one to three carbon atoms (e.g., C1-C3-alkylene) and linking at least two other groups, i.e., a divalent hydrocarbon radical. When two moieties are linked to the alkylene they can be linked to the same carbon atom (i.e., geminal), or different carbon atoms of the alkylene group. For instance, a straight chain alkylene can be the bivalent radical of -(CH2)n- , where n is 1, 2, 3, 4, 5 or 6 (i.e., a C1-C6-alkylene). Representative alkylene groups include, but are not limited to, methylene, ethylene, propylene, isopropylene, butylene, isobutylene,
secbutylene, pentylene, hexylene and the like. In some embodiments, the alkylene groups are C1- 3 alkylene groups (e.g., methylene, ethylene, or propylene). [0015] The term "cycloalkyl" refers to a monocyclic, bicyclic or polycyclic hydrocarbon ring system having, in some embodiments, 3 to 14 carbon atoms (e.g., C3-C14-cycloalkyl), or 3 to 10 carbon atoms (e.g., C3-C10-cycloalkyl), or 3 to 8 carbon atoms (e.g., C3-C8-cycloalkyl), or 3 to 6 carbon atoms (e.g., C3-C6-cycloalkyl) or 5 to 6 carbon atoms (e.g., C5-C6-cycloalkyl). Cycloalkyl groups can be saturated or characterized by one or more points of unsaturation (i.e., carbon-carbon double and/or triple bonds), provided that the points of unsaturation do not result in an aromatic system. Examples of monocyclic cycloalkyl groups include, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cyclohexenyl, cyclohexynyl, cycloheptyl, cycloheptenyl, cycloheptadienyl, cyclooctyl, cyclooctenyl, cyclooctadienyl and the like. The rings of bicyclic and polycyclic cycloalkyl groups can be fused, bridged, or spirocyclic. Non-limiting examples of bicyclic, spirocyclic and polycyclic hydrocarbon groups include bicyclo[1.1.1]pentane, bicyclo[2.1.1]hexane, bicyclo[2.2.1]heptane, bicyclo[2.2.2]octane, adamantyl, spiro[5.5]undecane, spiro[2.2]pentane, spiro[2.2]pentadiene, spiro[2.5]octane, spiro[2.2]pentadiene, and the like. In some embodiments, the cycloalkyl groups of the present disclosure are monocyclic C3-C6-cycloalkyl moieties (e.g., cyclopropyl, cyclobutyl, cyclopentyl, or cyclohexyl). [0016] As used herein, a wavy line, " ", that intersects a single, double or triple bond in any chemical structure depicted herein, represents that the point of attachment of the single, double, or triple bond to the remainder of the molecule is through either one of the atoms that make up the single, double or triple bond. Additionally, a bond extending from a substituent to the center of a ring (e.g., a phenyl ring or heteroaryl ring) is meant to indicate attachment of that substituent to the ring at any of the available ring vertices, i.e., such that attachment of the substituent to the ring results in a chemically stable arrangement. [0017] The terms "halogen," or “halo” are used interchangeably and refer to, unless otherwise stated, a fluorine, chlorine, bromine, or iodine atom. Additionally, terms such as "haloalkyl," refer to alkyl groups, as defined herein, that are substituted with one or more halogen(s) (e.g., 1-3 halogen(s)). For example, the term "C1-C6 haloalkyl" is meant to include trifluoromethyl, difluoromethyl, 2,2,2-trifluoroethyl, 4-chlorobutyl, 3-bromopropyl, and the like.
[0018] The term “hydroxyalkyl” refers to an alkyl group, as defined herein, that is substituted with one or more hydroxyl groups (e.g., 1-3 hydroxyl groups). Exemplary hydroxyalkyl groups include methanol, ethanol, 1,2-propanediol, 1,2-hexanediol, glycerol, and the like. [0019] The term “cyanoalkyl” refers to an alkyl group, as defined herein, that is substituted with one cyano group. Exemplary cyanoalkyl groups include cyanomethyl, cyanoethyl, cyanopropyl, and the like. [0020] As used herein, the term “alkoxy” refers to an alkyl group, as defined herein, that is attached to the remainder of the molecule via an oxygen atom (e.g., -O-C1-C12 alkyl, -O-C1-C8 alkyl, -O-C1-C6 alkyl, or -O-C1-C3 alkyl). Non-limiting examples of alkoxy groups include methoxy (OMe, -OCH3), ethoxy (OEt, -OCH2CH3), n-propoxy, iso-propoxy, n-butoxy, sec- butoxy, n-pentoxy, n-hexoxy, and the like. [0021] The term "heteroaryl" refers to monocyclic or fused bicyclic aromatic groups (or rings) having, in some embodiments, from 5 to 14 (i.e., 5- to 14-membered heteroaryl), or from 5 to 10 (i.e., 5- to 10-membered heteroaryl), or from 5 to 6 (i.e., 5- to 6-membered heteroaryl) members (i.e., ring vertices), and containing from one to five, one to four, one to three, one to two or one ring heteroatom independently selected from nitrogen (N), oxygen (O), and sulfur (S). A heteroaryl group can be attached to the remainder of the molecule through a carbon atom or a heteroatom of the heteroaryl group, when chemically permissible. Non-limiting examples of heteroaryl groups include pyridyl, pyridazinyl, pyrazinyl, pyrimidinyl, triazinyl, purinyl, thienopyridinyl, thienopyrimidinyl, pyrazolopyrimidinyl, imidazopyridines, isothiazolyl, pyrazolyl, indazolyl, pteridinyl, imidazolyl, triazolyl, tetrazolyl, oxazolyl, isoxazolyl, thiadiazolyl, pyrrolyl, thiazolyl, furyl, thienyl and the like. In some embodiments, the heteroaryl groups of the present disclosure are monocyclic 5- to 6-membered heteroaryl moieties having 1-4 ring nitrogen atoms (e.g., pyridinyl, pyrimidinyl, pyrazinyl, pyridazinyl, triazolyl, imidazolyl, pyrazolyl, tetrazolyl, and the like). In some embodiments, the heteroaryl groups of the present disclosure are 5-membered heteroaryl moieties having 1-4 ring heteroatoms independently selected from N, O, and S (e.g., imidazolyl, pyrazolyl, thiazolyl, oxazolyl, oxadiazole, thiadiazolyl, triazolyl, tetrazolyl, and the like). [0022] The term "heterocycloalkyl" refers to a non-aromatic monocyclic, bicyclic or polycyclic cycloalkyl ring having, in some embodiments, 3 to 14 members (e.g., 3- to 14-membered
heterocycle), or 3 to 10 members (e.g., 3- to 10-membered heterocycle), or 4 to 8 members (e.g., 4- to 8-membered heterocycle), or 4 to 6 members (e.g., 4- to 6-membered heterocycle), or 5 to 6 members (e.g., 5- to 6-membered heterocycle), and having from one to five, one to four, one to three, one to two or one ring heteroatom independently selected from nitrogen (N), oxygen (O), and sulfur (S). Heterocycloalkyl groups are saturated or characterized by one or more points of unsaturation (e.g., one or more carbon-carbon double bonds, carbon-carbon triple bonds, carbon- nitrogen double bonds, and/or nitrogen-nitrogen double bonds), provided that the points of unsaturation do not result in an aromatic system. The rings of bicyclic and polycyclic heterocycloalkyl groups can be fused, bridged, or spirocyclic. Non-limiting examples of heterocycloalkyl groups include aziridine, oxirane, thiirane, azetidine, oxetane, pyrrolidine, imidazolidine, pyrazolidine, dioxolane, phthalimide, piperidine, 1,4-dioxane, morpholine, thiomorpholine, thiomorpholine-S-oxide, thiomorpholine-S,S-oxide, piperazine, 3,4,5,6- tetrahydropyridazine, pyran, decahydroisoquinoline, 3-pyrroline, thiopyran, tetrahydrofuran, tetrahydrothiophene, quinuclidine, 2,6-diazaspiro[3.3]heptane, 2-azaspiro[3.3]heptane, 1- oxaspiro[3.3]heptane, 6-azaspiro[3.4]octane, and the like. A heterocycloalkyl group can be attached to the remainder of the molecule through a ring carbon atom, or a ring heteroatom, when chemically permissible. In some embodiments, the heterocycloalkyl groups of the present disclosure are monocyclic 4- to 8-membered heterocycloalkyl moieties having one or two heteroatoms independently selected from N, O, and S, (e.g., azetidine, oxetane, pyrrolidine, tetrahydrofuran, tetrahydropyran, piperidine, piperazine, morpholine, and the like). [0023] As referred to herein, "pharmaceutically acceptable salt" is meant to include salts of the compounds according to this disclosure that are prepared with suitably nontoxic acids or bases, depending on the particular substituents found on the compounds described herein. When compounds of the present disclosure contain suitably acidic functionalities, base addition salts can be obtained by contacting the neutral form of such compounds with a sufficient amount of the desired base, either neat or in a suitable inert solvent. Examples of salts derived from pharmaceutically-acceptable inorganic bases include aluminum, ammonium, calcium, copper, ferric, ferrous, lithium, magnesium, manganic, manganous, potassium, sodium, zinc and the like. Salts derived from pharmaceutically-acceptable organic bases include salts of primary, secondary and tertiary amines, including substituted amines, cyclic amines, naturally-occurring amines and the like, such as arginine, betaine, caffeine, choline, N,N’-dibenzylethylenediamine, diethylamine,
2-diethylaminoethanol, 2-dimethylaminoethanol, ethanolamine, ethylenediamine, N- ethylmorpholine, N-ethylpiperidine, glucamine, glucosamine, histidine, hydrabamine, isopropylamine, lysine, methylglucamine, morpholine, piperazine, piperidine, polyamine resins, procaine, purines, theobromine, triethylamine, trimethylamine, tripropylamine, tromethamine and the like. When compounds of the present disclosure contain suitably basic functionalities, acid addition salts can be obtained by contacting the neutral form of such compounds with a sufficient amount of the desired acid, either neat or in a suitable inert solvent. Examples of pharmaceutically acceptable acid addition salts include those derived from inorganic acids like hydrochloric, hydrobromic, nitric, carbonic, monohydrogencarbonic, phosphoric, monohydrogenphosphoric, dihydrogenphosphoric, sulfuric, monohydrogensulfuric, hydriodic, or phosphorous acids and the like, as well as the salts derived from suitably nontoxic organic acids like acetic, propionic, isobutyric, malonic, benzoic, succinic, suberic, fumaric, mandelic, phthalic, benzenesulfonic, p- tolylsulfonic, citric, tartaric, methanesulfonic, and the like. Also included are salts of amino acids such as arginate and the like, and salts of organic acids like glucuronic or galacturonic acids and the like (see, for example, Berge, S.M., et al, “Pharmaceutical Salts”, Journal of Pharmaceutical Science, 1977, 66, 1-19). Certain specific compounds of the present disclosure contain both basic and acidic functionalities that allow the compounds to be converted into either base or acid addition salts. [0024] The neutral forms of the compounds may be regenerated by contacting the salt with a base or acid and isolating the parent compound in the conventional manner. The parent form of the compound differs from the various salt forms in certain physical properties, such as solubility in polar solvents, but otherwise the salts are equivalent to the parent form of the compound for the purposes of the present disclosure. [0025] This disclosure also contemplates isomers of the compounds described herein (e.g., stereoisomers and/or atropisomers). For example, certain compounds of the present disclosure possess asymmetric carbon atoms (chiral centers), or hindered rotation about a single bond; the racemates, diastereomers, and enantiomers, and atropisomers (e.g., Ra, Sa, P and M isomers) of which are all intended to be encompassed within the scope of the present disclosure. Stereoisomeric forms may be defined, in terms of absolute stereochemistry, as (R) or (S), depicted uses dashes and/or wedges, and/or in terms of the direction the stereoisomer rotates plane-
polarized light (e.g., dextrorotary ((+) or (d)), or levorotary ((-) or (l))). When a stereochemical depiction (e.g., using dashes, , and/or wedges, ) is shown in a chemical structure, or a stereochemical assignment (e.g., using (R) and (S) notation, or (d) and (l) notation) is made in a chemical name, it is meant to indicate that the depicted/referred to isomer is present and substantially free of one or more other isomer(s) (e.g., enantiomers and diastereomers, when present). “Substantially free of” other isomer(s) indicates at least an 70/30 ratio of the indicated isomer to the other isomer(s), more preferably 80/20, 90/10, or 95/5 or more. In some embodiments, the indicated isomer will be present in an amount of at least 99%. A chemical bond to an asymmetric carbon that is depicted as a solid line ( ) indicates that all possible stereoisomers (e.g., enantiomers, diastereomers, racemic mixtures, etc.) at that carbon atom are included. In such instances, the compound may be present as a racemic mixture, scalemic mixture, or a mixture of diastereomers. [0026] The compounds of the present disclosure may also contain unnatural proportions of atomic isotopes at one or more of the atoms that constitute such compounds. Unnatural proportions of an isotope may be defined as ranging from the amount found in nature to an amount consisting of 100% of the atom in question. For example, the compounds may incorporate radioactive isotopes, such as for example tritium (3H), iodine-125 (125I) or carbon-14 (14C), or non-radioactive isotopes, such as deuterium (2H) or carbon-13 (13C). Such isotopic variations can provide additional utilities to those described elsewhere herein. For instance, isotopic variants of the compounds of the disclosure may find additional utility, including but not limited to, as diagnostic and/or imaging reagents, or as cytotoxic/radiotoxic therapeutic agents. Additionally, isotopic variants of the compounds of the disclosure can have altered pharmacokinetic and pharmacodynamic characteristics which can contribute to enhanced safety, tolerability or efficacy during treatment. In some embodiments, the compounds according to this disclosure are characterized by one or more deuterium atoms. [0027] The terms “patient” or “subject” are used interchangeably to refer to a human or a non- human animal (e.g., a mammal). [0028] The terms “treat”, “treating”, treatment” and the like refer to a course of action that eliminates, reduces, suppresses, mitigates, ameliorates, or prevents the worsening of, either temporarily or permanently, a disease, disorder or condition to which the term applies, or at least
one of the symptoms associated therewith. Treatment includes alleviation of symptoms, diminishment of extent of disease, inhibiting (e.g., arresting the development or further development of the disease, disorder or condition or clinical symptoms association therewith) an active disease, delaying or slowing of disease progression, improving the quality of life, and/or prolonging survival of a subject as compared to expected survival if not receiving treatment or as compared to a published standard of care therapy for a particular disease. [0029] The term “in need of treatment” as used herein refers to a judgment made by a physician or similar professional that a subject requires or will benefit from treatment. This judgment is made based on a variety of factors that are in the realm of the physician’s expertise, which may include a positive diagnosis of a disease, disorder or condition. [0030] The terms “prevent”, “preventing”, “prevention”, “prophylaxis” and the like refer to a course of action initiated in a manner (e.g., prior to the onset of a disease, disorder, condition or symptom thereof) so as to prevent, suppress, inhibit or reduce, either temporarily or permanently, a subject’s risk of developing a disease, disorder, condition or the like (as determined by, for example, the absence of clinical symptoms) or delaying the onset thereof, generally in the context of a subject predisposed to having a particular disease, disorder or condition. In certain instances, the terms also refer to slowing the progression of the disease, disorder or condition or inhibiting progression thereof to a harmful or otherwise undesired state. Prevention also refers to a course of action initiated in a subject after the subject has been treated for a disease, disorder, condition or a symptom associated therewith in order to prevent relapse of that disease, disorder, condition or symptom. In one or more embodiments, the preventative course of action is taken based on anticipation of a condition or event. In one embodiment, prevention refers to the prevention, suppression, inhibition or reduction of an allergic, immune, or autoimmune response in a subject suffering from an allergic, inflammatory, neuroinflammatory, neurological, immune, autoimmune, dermatological, respiratory, metabolic, cardiovascular or fibrotic disease, disorder, or condition. [0031] As used herein, the term “response” may refer to a symptom initiated by an irritant or trigger (e.g., an antigen originating from within the body, or from the external environment) in a subject. For example, “response” may refer to a reaction (e.g., hives, rash, welts, itchy skin, stinging skin, skin fissures, skin lesions, skin blisters, swelling (e.g., in joints, glands, or tissues), vertigo, fatigue, dizziness, fainting, lightheadedness, muscle weakness, headache, dry skin, dry
eyes, hair loss, numbness or tingling in extremities, joint pain and/or stiffness, sneezing, runny nose, stuffy nose, chest tightness and/or pain, shortness of breath, wheezing, itchy eyes, watery eyes, blurred vision, sensitivity to light, stomach cramping, abdominal pain, bloating, diarrhea, constipation, indigestion, heartburn, excessive flatulence, frequent urination, difficulty swallowing, muscle spasm, muscle tremor, unexpected weight gain or loss, increased thirst, increased hunger, irritability, temperature sensitivity, low blood pressure, constriction of airways, weak pulse, rapid pulse, nausea, vomiting, anemia, depression, and the like) caused by an external or internal trigger in a subject susceptible to the trigger. Subjects susceptible to a trigger are generally those suffering from an allergic, inflammatory, neuroinflammatory, neurological, immune, autoimmune, dermatological, respiratory, metabolic, cardiovascular or fibrotic disease, disorder, or condition, such as those described elsewhere herein. [0032] The term “in need of prevention” as used herein refers to a judgment made by a physician or other caregiver that a subject requires or will benefit from preventative care. This judgment is made based on a variety of factors that are in the realm of a physician’s or caregiver’s expertise. [0033] “Substantially pure” indicates that a component (e.g., a compound according to this disclosure) makes up greater than about 50% of the total content of the composition, and typically greater than about 60% of the total content. More typically, “substantially pure” refers to compositions in which at least 75%, at least 85%, at least 90% or more of the total composition is the component of interest. In some cases, the component of interest will make up greater than about 90%, or greater than about 95% of the total content of the composition. [0034] The terms “inhibitor of KIT” and “KIT inhibitor” may be used interchangeably, and refer to the ability of a molecule to decrease the activation of KIT either directly or indirectly, thereby decreasing activation and/or quantity of systemic mast cells. [0035] Compounds that are selective for KIT may be particularly useful in the treatment of certain disorders or may offer a reduced likelihood of undesired side effects. In one embodiment, compounds of the present disclosure are selective over one or more other receptor tyrosine kinases. Specific examples include, but are not limited to, PDGFRα, PDGFRβ, CSF1R, and FLT3. Selectivity may be determined, for example, by comparing the inhibition of a compound as described herein against KIT against the inhibition of a compound as described herein against another kinase. In one embodiment, the selective inhibition of a compound of Formula III or an
associated sub-formula is at least 1000 times greater, 500 times greater, 100 times greater, 50 times greater, or 20 times greater than inhibition of one or more kinases selected from PDGFRα, PDGFRβ, CSF1R, and FLT3. [0036] Compounds provided herein may have advantageous pharmacokinetic profiles including, for example, hepatocyte stability, clearance, inhibition against CYP, and inhibition against hERG. Compounds of the Disclosure [0037] The present disclosure relates to compounds that inhibit the activity of KIT. [0038] In one aspect, this disclosure is directed to a compound having a structure according to Formula III:
 (Formula III) or a pharmaceutically acceptable salt thereof, wherein: A is: (i) selected from the group consisting of: , , ,
(Formula III) or a pharmaceutically acceptable salt thereof, wherein: A is: (i) selected from the group consisting of: , , ,

 membered heteroaryl is substituted with 0-2 R1d; or (iii) -NHC(O)-R1g; each R1 is independently selected from the group consisting of halo, -CN, -OH, C1-C6- alkyl, C1-C6-haloalkyl, C1-C6-hydroxyalkyl, C1-C6-cyanoalkyl, C1-C6 alkoxy, C3-C6-cycloalkyl, - O-C3-C6-cycloalkyl, 4- to 8-membered heterocycloalkyl, -NR1aR1b, phenyl, 5- to 6-membered 12
heteroaryl, -X1-NR1aR1b, -X1-C(O)NR1aR1b, -X1-O-(C1-C3-alkyl), -X1-C3-C6-cycloalkyl, -X1- phenyl, and -X2-(4- to 8-membered heterocycloalkyl), wherein each 5- to 6-membered heteroaryl has 1-2 ring heteroatoms independently selected from N and O; each 4- to 8-membered heterocycloalkyl has 1-3 ring heteroatoms independently selected from N, O, and S, and said 4- to 8-membered heterocycloalkyl is optionally substituted with one oxo; and each R1 is optionally substituted with 1-3 R1c; or two adjacent R1 groups are combined with the two ring atoms to which they are attached to form a 5- or 6-membered heterocycloalkyl group having 1-2 ring heteroatoms independently selected from N, and O; each X1 is C1-C3-alkylene; each X2 is C1-C3-alkylene, or –(C1-C3-alkylene)-C(O)-; R1a and R1b, when present, are independently H, C1-C6 alkyl, or C3-C6 cycloalkyl; each R1c, when present, is independently halo, -OH, -C1-C6-alkyl, -C1-C6-alkoxy, or -C(O)- (C1-C6-alkyl); each R1d, when present, is independently halo, C1-C6 alkoxy, C1-C3 haloalkoxy, C3-C6- cycloalkyl, -O-C3-C6-cycloalkyl, or -NR1eR1f; R1e and R1f, when present, are independently H, C1-C6 haloalkyl, -(C1-C3-alkylene)-(C3- C6-cycloalkyl), or 4- to 8-membered heterocycloalkyl having 1-3 ring heteroatoms independently selected from N, O, and S; or R1e and R1f together with the nitrogen atom to which they are attached form a 4- to 6-membered heterocycloalkyl group having 1-2 ring heteroatoms independently selected from N, and O; R1g, when present, is C3-C6-cycloalkyl, -(C1-C3-alkylene)-(C3-C6-cycloalkyl), or 6- membered heteroaryl having 1-3 ring nitrogen atoms; wherein R1g is optionally substituted with 0-2 R1h; each R1h, when present, is independently halo, C1-C6 alkyl, -O-C3-C6-cycloalkyl, or -NH- (4- to 8-membered heterocycloalkyl), wherein the 4- to 8-membered heterocycloalkyl has 1-2 ring heteroatoms independently selected from N, O, and S; n is 0 or 1; m is 0, 1 or 2; R2, when present, is halo, -CN, -C1-C3-alkyl, -C1-C3-alkoxy, or C3-C6-cycloalkyl;
R3 is -H, halo, -C1-C6-alkyl, -C1-C6-haloalkyl, -C1-C6-hydroxyalkyl, -C1-C6-alkoxy, or - C(O)NR3aR3b; R3a and R3b, when present, are independently H, or C1-C3 alkyl; each R4, when present, is independently halo, C1-C6 alkyl, C1-C6-alkoxy, C3-C6 cycloalkyl, 4- to 8-membered heterocycloalkyl having 1-3 ring heteroatoms independently selected from N, O, and S, or phenyl, wherein said phenyl is optionally substituted with 1-3 halo; R5 is H, C1-C3-alkyl, or -NR5aR5b; R5a and R5b, when present, are independently H or C1-C3 alkyl; and R6 is H, or C1-C3-alkyl. [0039] In some embodiments, A is selected from the group consisting of:
membered heteroaryl is substituted with 0-2 R1d; or (iii) -NHC(O)-R1g; each R1 is independently selected from the group consisting of halo, -CN, -OH, C1-C6- alkyl, C1-C6-haloalkyl, C1-C6-hydroxyalkyl, C1-C6-cyanoalkyl, C1-C6 alkoxy, C3-C6-cycloalkyl, - O-C3-C6-cycloalkyl, 4- to 8-membered heterocycloalkyl, -NR1aR1b, phenyl, 5- to 6-membered 12
heteroaryl, -X1-NR1aR1b, -X1-C(O)NR1aR1b, -X1-O-(C1-C3-alkyl), -X1-C3-C6-cycloalkyl, -X1- phenyl, and -X2-(4- to 8-membered heterocycloalkyl), wherein each 5- to 6-membered heteroaryl has 1-2 ring heteroatoms independently selected from N and O; each 4- to 8-membered heterocycloalkyl has 1-3 ring heteroatoms independently selected from N, O, and S, and said 4- to 8-membered heterocycloalkyl is optionally substituted with one oxo; and each R1 is optionally substituted with 1-3 R1c; or two adjacent R1 groups are combined with the two ring atoms to which they are attached to form a 5- or 6-membered heterocycloalkyl group having 1-2 ring heteroatoms independently selected from N, and O; each X1 is C1-C3-alkylene; each X2 is C1-C3-alkylene, or –(C1-C3-alkylene)-C(O)-; R1a and R1b, when present, are independently H, C1-C6 alkyl, or C3-C6 cycloalkyl; each R1c, when present, is independently halo, -OH, -C1-C6-alkyl, -C1-C6-alkoxy, or -C(O)- (C1-C6-alkyl); each R1d, when present, is independently halo, C1-C6 alkoxy, C1-C3 haloalkoxy, C3-C6- cycloalkyl, -O-C3-C6-cycloalkyl, or -NR1eR1f; R1e and R1f, when present, are independently H, C1-C6 haloalkyl, -(C1-C3-alkylene)-(C3- C6-cycloalkyl), or 4- to 8-membered heterocycloalkyl having 1-3 ring heteroatoms independently selected from N, O, and S; or R1e and R1f together with the nitrogen atom to which they are attached form a 4- to 6-membered heterocycloalkyl group having 1-2 ring heteroatoms independently selected from N, and O; R1g, when present, is C3-C6-cycloalkyl, -(C1-C3-alkylene)-(C3-C6-cycloalkyl), or 6- membered heteroaryl having 1-3 ring nitrogen atoms; wherein R1g is optionally substituted with 0-2 R1h; each R1h, when present, is independently halo, C1-C6 alkyl, -O-C3-C6-cycloalkyl, or -NH- (4- to 8-membered heterocycloalkyl), wherein the 4- to 8-membered heterocycloalkyl has 1-2 ring heteroatoms independently selected from N, O, and S; n is 0 or 1; m is 0, 1 or 2; R2, when present, is halo, -CN, -C1-C3-alkyl, -C1-C3-alkoxy, or C3-C6-cycloalkyl;
R3 is -H, halo, -C1-C6-alkyl, -C1-C6-haloalkyl, -C1-C6-hydroxyalkyl, -C1-C6-alkoxy, or - C(O)NR3aR3b; R3a and R3b, when present, are independently H, or C1-C3 alkyl; each R4, when present, is independently halo, C1-C6 alkyl, C1-C6-alkoxy, C3-C6 cycloalkyl, 4- to 8-membered heterocycloalkyl having 1-3 ring heteroatoms independently selected from N, O, and S, or phenyl, wherein said phenyl is optionally substituted with 1-3 halo; R5 is H, C1-C3-alkyl, or -NR5aR5b; R5a and R5b, when present, are independently H or C1-C3 alkyl; and R6 is H, or C1-C3-alkyl. [0039] In some embodiments, A is selected from the group consisting of:
 , , ,
, , ,
 [0040] In some embodiments,
[0040] In some embodiments,
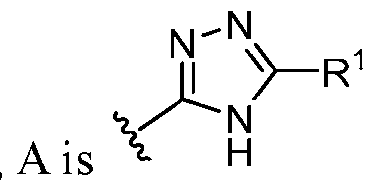 . In some embodiments, A is
. In some embodiments, A is
 some embodiments, A is
some embodiments, A is
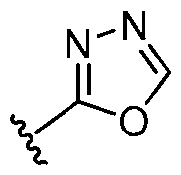 . In some embodiments, A is
. In some embodiments, A is
 . In some embodiments, A is . In some embodiments, A
. In some embodiments, A is . In some embodiments, A
 . In some embodiments,
. In some embodiments,
 , . In some embodiments, A is
, . In some embodiments, A is
 . In some embodiments, A is
. In some embodiments, A is
 . In some embodiments, A is
. In some embodiments, A is
 . In some embodiments, A is
. In some embodiments, A is
 . In some embodiments, A is
. In some embodiments, A is
 . In some embodiments,
A is
. In some embodiments,
A is
 . In some embodiments,
. In some embodiments,
 . In some embodiments, A is
. In some embodiments, A is
 . In some embodiments, A is
. In some embodiments, A is
 . In some embodiments, A is
. In some embodiments, A is
 . In some embodiments, A is
. In some embodiments, A is
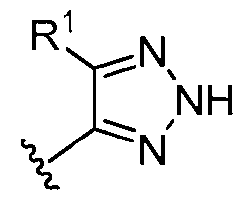 . In some embodiments, A is
. In some embodiments, A is
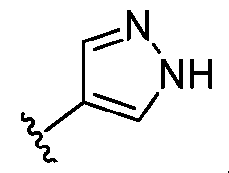 . In some embodiments, . In some embodiments, A is
. In some embodiments, . In some embodiments, A is
 . In some embodiments, A is
. In some embodiments, A is
 . some embodiments,
. some embodiments,
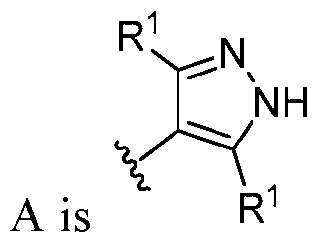 . In some embodiments, A is
. In some embodiments, A is
 . In some embodiments, A is
. In some embodiments, A is
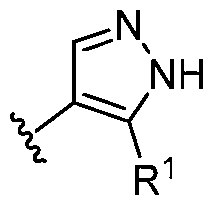 . In some embodiments, A is
. In some embodiments, A is
 . In some embodiments, A is
. In some embodiments, A is
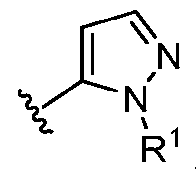 . In some embodiments, A is
. In some embodiments, A is
 . In some embodiments, A is
. In some embodiments, A is
 . , . , . In some embodiments, A is
. , . , . In some embodiments, A is
 . In some embodiments,
. In some embodiments,
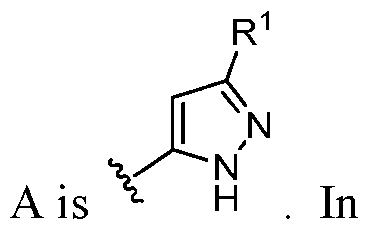 . some embodiments, A is
. some embodiments, A is
 . , . In some embodiments,
. , . In some embodiments,
 . In some embodiments, A is
. In some embodiments, A is
 . In some embodiments,
. In some embodiments,
 . In some embodiments,
. In some embodiments,
 some embodiments,
some embodiments,
 some embodiments, A is
some embodiments, A is
 . , . In some embodiments, A is
. , . In some embodiments, A is
 some embodiments, A is
some embodiments, A is
 . In some embodiments,
. In some embodiments,
 . In some embodiments, A is
. In some embodiments, A is
 . In some embodiments, A is
. In some embodiments, A is
 . In some embodiments, A is
. In some embodiments, A is
 . In some embodiments, A is
. In some embodiments, A is
 . In some embodiments, A
. In some embodiments, A
 , . In some embodiments, A is
, . In some embodiments, A is
 . , . In some embodiments,
. , . In some embodiments,
 . In some embodiments,
. In some embodiments,
 . In some embodiments,
. In some embodiments,
 . In some
embodiments, A is
. In some
embodiments, A is
 . In some embodiments,
. In some embodiments,
 . In some embodiments, A is
. In some embodiments, A is
 some embodiments, A is
some embodiments, A is
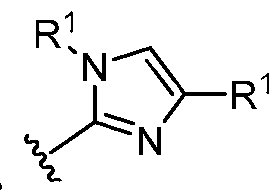 . In some embodiments,
. In some embodiments,
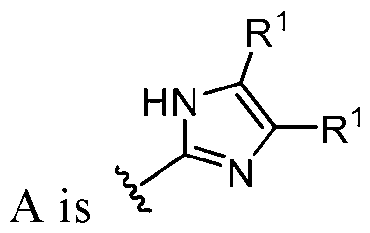 . In some embodiments,
. In some embodiments,
 . In some embodiments,
. In some embodiments,
 . In some embodiments, A is
. In some embodiments, A is
 . [0041] In some embodiments, A is a 6-membered heteroaryl having 1-3 ring nitrogen atoms, wherein the 6-membered heteroaryl is substituted with 0-2 R1d. In some embodiments, A is a 6- membered heteroaryl having 1-3 ring nitrogen atoms, wherein the 6-membered heteroaryl is substituted with 1-2 R1d. In some embodiments, A is pyridazinyl, pyrimidinyl, pyrazinyl, or pyridinyl, wherein A is substituted with 0-2 R1d. In some embodiments, A is pyridazinyl, pyrimidinyl, pyrazinyl, or pyridinyl, wherein A is substituted with 1-2 R1d. In some embodiments,
. [0041] In some embodiments, A is a 6-membered heteroaryl having 1-3 ring nitrogen atoms, wherein the 6-membered heteroaryl is substituted with 0-2 R1d. In some embodiments, A is a 6- membered heteroaryl having 1-3 ring nitrogen atoms, wherein the 6-membered heteroaryl is substituted with 1-2 R1d. In some embodiments, A is pyridazinyl, pyrimidinyl, pyrazinyl, or pyridinyl, wherein A is substituted with 0-2 R1d. In some embodiments, A is pyridazinyl, pyrimidinyl, pyrazinyl, or pyridinyl, wherein A is substituted with 1-2 R1d. In some embodiments,
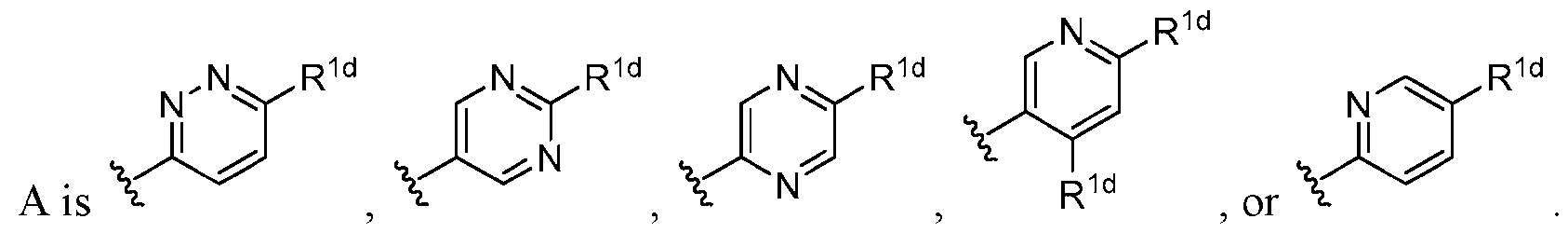 . [0042] In some embodiments, each R1d, when present, is independently halo, C1-C6 alkoxy, C1- C3 haloalkoxy, cyclopropyl, -O-cyclopropyl, or -NR1eR1f; and R1e and R1f, when present, are independently H, C1-C6 haloalkyl, -(methylene)-(cyclopropyl), or tetrahydrofuranyl; or R1e and R1f together with the nitrogen atom to which they are attached form an azetidinyl group.
[ , ,
. [0042] In some embodiments, each R1d, when present, is independently halo, C1-C6 alkoxy, C1- C3 haloalkoxy, cyclopropyl, -O-cyclopropyl, or -NR1eR1f; and R1e and R1f, when present, are independently H, C1-C6 haloalkyl, -(methylene)-(cyclopropyl), or tetrahydrofuranyl; or R1e and R1f together with the nitrogen atom to which they are attached form an azetidinyl group.
[ , ,
 [0044] In some embodiments, A is -NHC(O)-R1g. In some embodiments, R1g, when present, is cyclopropyl, -(methylene)-(cyclopropyl), pyridinyl, or pyrazinyl; wherein R1g is optionally substituted with 0-2 R1h. In some embodiments, R1g, when present, is
[0044] In some embodiments, A is -NHC(O)-R1g. In some embodiments, R1g, when present, is cyclopropyl, -(methylene)-(cyclopropyl), pyridinyl, or pyrazinyl; wherein R1g is optionally substituted with 0-2 R1h. In some embodiments, R1g, when present, is
 , , ,
, , ,
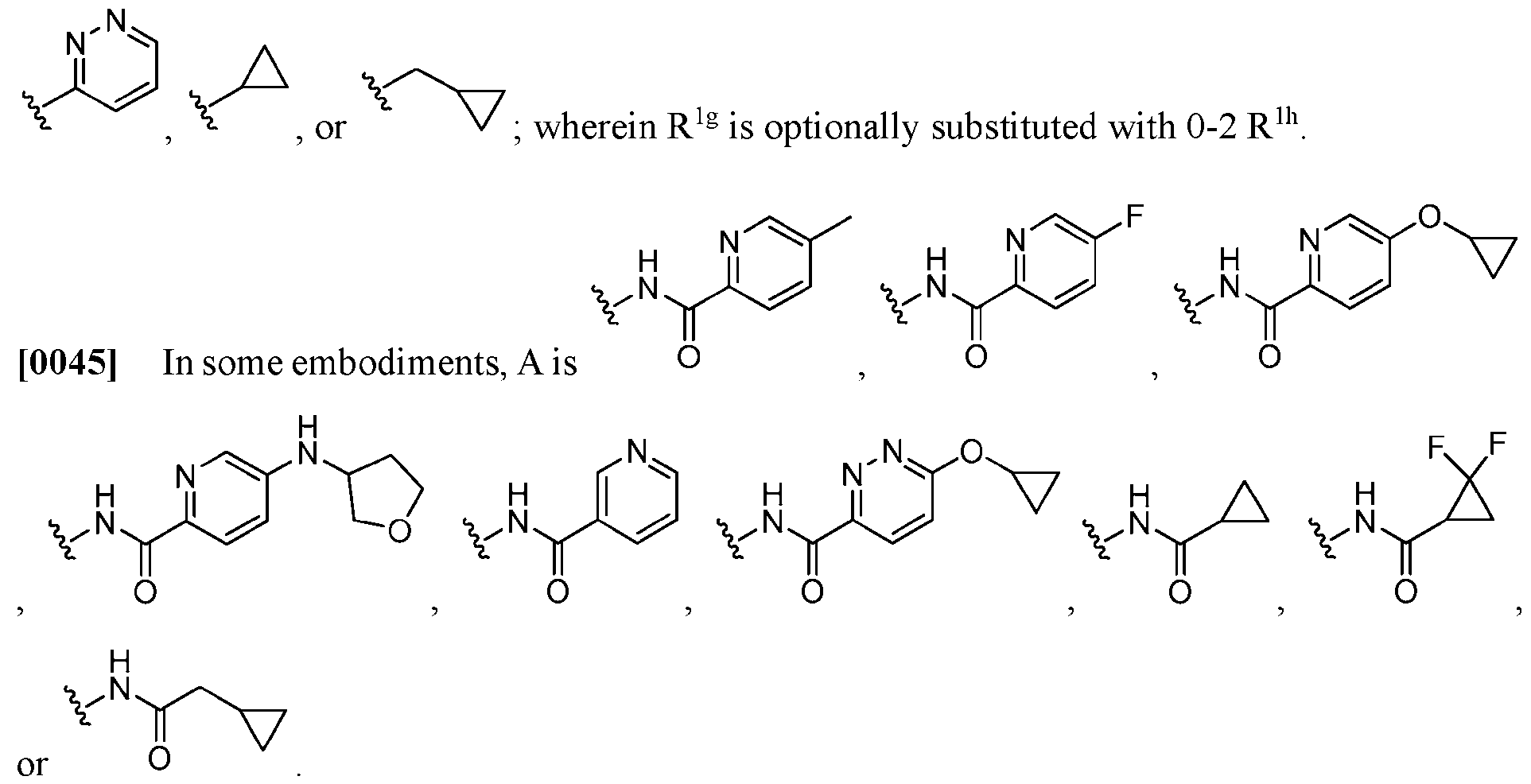 . [0046] In another aspect, this disclosure is directed to a compound having a structure according to Formula III:
. [0046] In another aspect, this disclosure is directed to a compound having a structure according to Formula III:
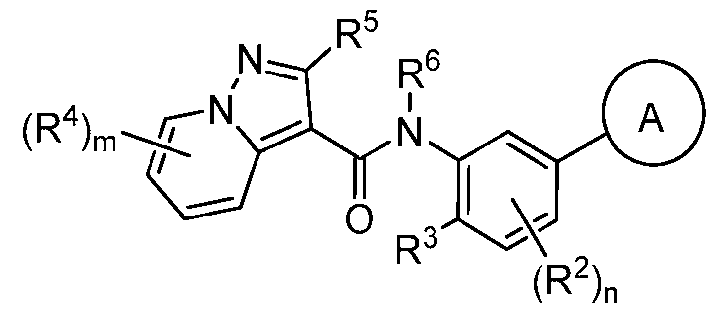 (Formula III) or a pharmaceutically acceptable salt thereof, wherein: ring A is a 5-membered heteroaryl having 1-4 ring heteroatoms independently selected from N, O, and S, wherein the heteroaryl is substituted with 0-2 R1; each R1 is independently selected from the group consisting of halo, -CN, -OH, C1-C6- alkyl, C1-C6-haloalkyl, C1-C6-hydroxyalkyl, C1-C6-cyanoalkyl, C1-C6 alkoxy, C3-C6-cycloalkyl, - O-C3-C6-cycloalkyl, 4- to 8-membered heterocycloalkyl, -NR1aR1b, phenyl, 5- to 6-membered heteroaryl, -X1-NR1aR1b, -X1-C(O)NR1aR1b, -X1-O-(C1-C3-alkyl), -X1-C3-C6-cycloalkyl, -X1- phenyl, and -X2-(4- to 8-membered heterocycloalkyl), wherein each 5- to 6-membered heteroaryl has 1-2 ring heteroatoms independently selected from N and O; each 4- to 8-membered heterocycloalkyl has 1-3 ring heteroatoms independently selected from N, O, and S, and said 4- to 8-membered heterocycloalkyl is optionally substituted with one oxo; and each R1 is optionally substituted with 1-3 R1c; or two adjacent R1 groups are combined with the two ring atoms to which they are attached to form a 5- or 6-membered heterocycloalkyl group having 1-2 ring heteroatoms independently selected from N, and O; each X1 is C1-C3-alkylene; each X2 is C1-C3-alkylene, or –(C1-C3-alkylene)-C(O)-; R1a and R1b, when present, are independently H, C1-C6 alkyl, or C3-C6 cycloalkyl; R1c, when present, is halo, -OH, -C1-C6-alkyl, -C1-C6-alkoxy, or -C(O)-(C1-C6-alkyl); n is 0 or 1; m is 0, 1 or 2; R2, when present, is halo, -CN, -C1-C3-alkyl, -C1-C3-alkoxy, or C3-C6-cycloalkyl; R3 is -H, halo, -C1-C6-alkyl, -C1-C6-haloalkyl, -C1-C6-hydroxyalkyl, -C1-C6-alkoxy, or - C(O)NR3aR3b; R3a and R3b, when present, are independently H, or C1-C3 alkyl;
each R4, when present, is independently halo, C1-C6 alkyl, C1-C6-alkoxy, C3-C6 cycloalkyl, 4- to 8-membered heterocycloalkyl having 1-3 ring heteroatoms independently selected from N, O, and S, or phenyl, wherein said phenyl is optionally substituted with 1-3 halo; R5 is H, C1-C3-alkyl, or -NR5aR5b; R5a and R5b, when present, are independently H or C1-C3 alkyl; and R6 is H, or C1-C3-alkyl. [0047] In some embodiments, ring A is selected from the group consisting of oxadiazolyl, thiazolyl, thiadiazolyl, triazolyl, imidazolyl, pyrazolyl, and tetrazolyl, each of which is substituted with 0-2 R1. In some embodiments, ring A is selected from the group consisting of thiazolyl, thiadiazolyl, triazolyl, imidazolyl, pyrazolyl, and tetrazolyl, each of which is substituted with 0-2 R1. In some embodiments, ring A is imidazolyl, pyrazolyl, triazolyl, or tetrazolyl, each of which is substituted with 0-2 R1. [0048] In some embodiments, ring A is selected from the group consisting of
(Formula III) or a pharmaceutically acceptable salt thereof, wherein: ring A is a 5-membered heteroaryl having 1-4 ring heteroatoms independently selected from N, O, and S, wherein the heteroaryl is substituted with 0-2 R1; each R1 is independently selected from the group consisting of halo, -CN, -OH, C1-C6- alkyl, C1-C6-haloalkyl, C1-C6-hydroxyalkyl, C1-C6-cyanoalkyl, C1-C6 alkoxy, C3-C6-cycloalkyl, - O-C3-C6-cycloalkyl, 4- to 8-membered heterocycloalkyl, -NR1aR1b, phenyl, 5- to 6-membered heteroaryl, -X1-NR1aR1b, -X1-C(O)NR1aR1b, -X1-O-(C1-C3-alkyl), -X1-C3-C6-cycloalkyl, -X1- phenyl, and -X2-(4- to 8-membered heterocycloalkyl), wherein each 5- to 6-membered heteroaryl has 1-2 ring heteroatoms independently selected from N and O; each 4- to 8-membered heterocycloalkyl has 1-3 ring heteroatoms independently selected from N, O, and S, and said 4- to 8-membered heterocycloalkyl is optionally substituted with one oxo; and each R1 is optionally substituted with 1-3 R1c; or two adjacent R1 groups are combined with the two ring atoms to which they are attached to form a 5- or 6-membered heterocycloalkyl group having 1-2 ring heteroatoms independently selected from N, and O; each X1 is C1-C3-alkylene; each X2 is C1-C3-alkylene, or –(C1-C3-alkylene)-C(O)-; R1a and R1b, when present, are independently H, C1-C6 alkyl, or C3-C6 cycloalkyl; R1c, when present, is halo, -OH, -C1-C6-alkyl, -C1-C6-alkoxy, or -C(O)-(C1-C6-alkyl); n is 0 or 1; m is 0, 1 or 2; R2, when present, is halo, -CN, -C1-C3-alkyl, -C1-C3-alkoxy, or C3-C6-cycloalkyl; R3 is -H, halo, -C1-C6-alkyl, -C1-C6-haloalkyl, -C1-C6-hydroxyalkyl, -C1-C6-alkoxy, or - C(O)NR3aR3b; R3a and R3b, when present, are independently H, or C1-C3 alkyl;
each R4, when present, is independently halo, C1-C6 alkyl, C1-C6-alkoxy, C3-C6 cycloalkyl, 4- to 8-membered heterocycloalkyl having 1-3 ring heteroatoms independently selected from N, O, and S, or phenyl, wherein said phenyl is optionally substituted with 1-3 halo; R5 is H, C1-C3-alkyl, or -NR5aR5b; R5a and R5b, when present, are independently H or C1-C3 alkyl; and R6 is H, or C1-C3-alkyl. [0047] In some embodiments, ring A is selected from the group consisting of oxadiazolyl, thiazolyl, thiadiazolyl, triazolyl, imidazolyl, pyrazolyl, and tetrazolyl, each of which is substituted with 0-2 R1. In some embodiments, ring A is selected from the group consisting of thiazolyl, thiadiazolyl, triazolyl, imidazolyl, pyrazolyl, and tetrazolyl, each of which is substituted with 0-2 R1. In some embodiments, ring A is imidazolyl, pyrazolyl, triazolyl, or tetrazolyl, each of which is substituted with 0-2 R1. [0048] In some embodiments, ring A is selected from the group consisting of
 , ,
, ,
 , , , , , , , ,
, , , , , , , ,
 , , each of which is substituted with 0-2 R1. In some embodiments, ring A , ,
, , each of which is substituted with 0-2 R1. In some embodiments, ring A , ,
 with 0-2 R1. In some embodiments, ring A is selected from the group consisting of,
with 0-2 R1. In some embodiments, ring A is selected from the group consisting of,
 ,
,
 , , , , , , , ,
, , , , , , , ,
 , each of which is substituted with 0-2 R1. In some embodiments, ring A is
, each of which is substituted with 0-2 R1. In some embodiments, ring A is
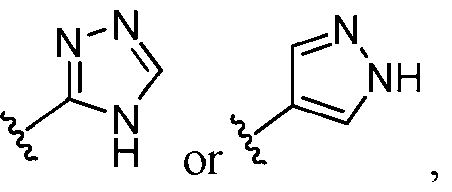 each of which is substituted with 0-2 R1. In some embodiments, ring A is
each of which is substituted with 0-2 R1. In some embodiments, ring A is
 substituted with 0-2 R1. In some embodiments, ring
substituted with 0-2 R1. In some embodiments, ring
 . [0049] In some embodiments, ring A is a 5-membered heteroaryl having 1-4 ring nitrogen atoms, wherein the heteroaryl is substituted with two adjacent R1 groups that combine with the two ring atoms to which they are attached to form a 5- or 6-membered heterocycloalkyl group having 1-2 ring heteroatoms independently selected from N and O. In some embodiments, ring A is pyrazolyl or triazolyl substituted with two adjacent R1 groups that combine with the two ring atoms to which they are attached to form a 5- or 6-membered heterocycloalkyl group having 1-2 ring heteroatoms independently selected from N and O. In some embodiments, ring
. [0049] In some embodiments, ring A is a 5-membered heteroaryl having 1-4 ring nitrogen atoms, wherein the heteroaryl is substituted with two adjacent R1 groups that combine with the two ring atoms to which they are attached to form a 5- or 6-membered heterocycloalkyl group having 1-2 ring heteroatoms independently selected from N and O. In some embodiments, ring A is pyrazolyl or triazolyl substituted with two adjacent R1 groups that combine with the two ring atoms to which they are attached to form a 5- or 6-membered heterocycloalkyl group having 1-2 ring heteroatoms independently selected from N and O. In some embodiments, ring
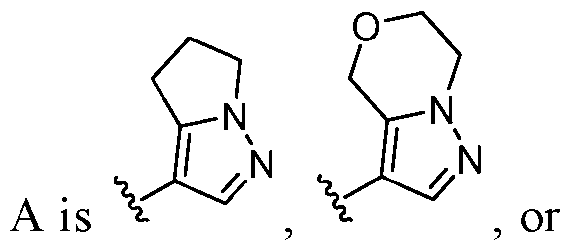 r
r
 . [0050] In some embodiments, R6 is H, or C1-C3-alkyl. In some embodiments, R6 is H or methyl. In some embodiments, R6 is methyl. In some embodiments, R6 is H.
[0051] In some embodiments, R5 is H, C1-C3-alkyl, or -NR5aR5b; wherein R5a and R5b are independently H or C1-C3 alkyl. In some embodiments, R5 is H, methyl, or -NH2. In some embodiments, R5 is H. In some embodiments, R5 is methyl. In some embodiments, R5 is -NR5aR5b, wherein at least one of R5a and R5b is H. In some embodiments, R5 is -NH2. [0052] In some embodiments, R5 and R6 are H. [0053] In some embodiments, m is 0, 1, or 2; and each R4, when present is independently halo, C1-C6 alkyl, C1-C6-alkoxy, C3-C6 cycloalkyl, 4- to 8-membered heterocycloalkyl having 1-3 ring heteroatoms independently selected from N, O, and S, or phenyl, wherein said phenyl is optionally substituted with 1-3 halo. In some embodiments, m is 0. In some embodiments, m is 1; and R4 is halo, C1-C6 alkyl, C1-C6-alkoxy, C3-C6 cycloalkyl, 4- to 8-membered heterocycloalkyl having 1-3 ring heteroatoms independently selected from N, O, and S, or phenyl, wherein said phenyl is optionally substituted with 1-3 halo. In some embodiments, m is 2; and each R4 is independently halo, C1-C6 alkyl, C1-C6-alkoxy, C3-C6 cycloalkyl, 4- to 8-membered heterocycloalkyl having 1-3 ring heteroatoms independently selected from N, O, and S, or phenyl, wherein said phenyl is optionally substituted with 1-3 halo. In some embodiments, m is 0, 1, or 2; and at least one R4, when present is halo. In some embodiments, R4 is -F, -Cl, -Br, or -I. In some embodiments, R4 is -F, or -Cl. In some embodiments, R4 is -F. In some embodiments, R4 is -Cl. In some embodiments, m is 0, 1, or 2, and at least one R4, when present, is C1-C6 alkyl. In some embodiments, R4 is C1- C3 alkyl. In some embodiments, R4 is methyl, ethyl, n-propyl, or isopropyl. In some embodiments, R4 is methyl, or ethyl. In some embodiments, R4 is methyl. In some embodiments, R4 is ethyl. In some embodiments, m is 0, 1, or 2; and at least one R4, when present, is C1-C6-alkoxy. In some embodiments, R4 is C1-C3-alkoxy. In some embodiments, R4 is methoxy, ethoxy, n-propoxy, or isopropoxy. In some embodiments, R4 is methoxy or ethoxy. In some embodiments R4 is methoxy. In some embodiments, R4 is ethoxy. In some embodiments, m is 0, 1, or 2; and at least one R4, when present, is C3-C6 cycloalkyl. In some embodiments, R4 is cyclopropyl, cyclobutyl, cyclopentyl, or cyclohexyl. In some embodiments, R4 is cyclopropyl. In some embodiments, m is 0, 1, or 2; and at least one R4, when present, is 4- to 8-membered heterocycloalkyl having 1-3 ring heteroatoms independently selected from N, O, and S. In some embodiments, R4 is a 5- to 6- membered heterocycloalkyl having 1-3 ring heteroatoms independently selected from N, O, and S. In some embodiments R4 is a 6-membered heterocycloalkyl having 1-3 ring heteroatoms
independently selected from N, and O. In some embodiments, R4 is piperidine, piperazine, or morpholine. In some embodiments, R4 is morpholine. In some embodiments, m is 0, 1, or 2; and at least one R4, when present, is phenyl optionally substituted with 1-3 halo. In some embodiments, R4 is phenyl substituted with one halo. In some embodiments, R4 is phenyl substituted with -F, - Cl, -Br, or -I. In some embodiments, R4 is phenyl substituted with -F. In some embodiments, m is 0, 1, or 2; and each R4, when present, is independently -F, -Cl, -CH3, -CH2CH3, -OCH3, - OCH2CH3, cyclopropyl,
. [0050] In some embodiments, R6 is H, or C1-C3-alkyl. In some embodiments, R6 is H or methyl. In some embodiments, R6 is methyl. In some embodiments, R6 is H.
[0051] In some embodiments, R5 is H, C1-C3-alkyl, or -NR5aR5b; wherein R5a and R5b are independently H or C1-C3 alkyl. In some embodiments, R5 is H, methyl, or -NH2. In some embodiments, R5 is H. In some embodiments, R5 is methyl. In some embodiments, R5 is -NR5aR5b, wherein at least one of R5a and R5b is H. In some embodiments, R5 is -NH2. [0052] In some embodiments, R5 and R6 are H. [0053] In some embodiments, m is 0, 1, or 2; and each R4, when present is independently halo, C1-C6 alkyl, C1-C6-alkoxy, C3-C6 cycloalkyl, 4- to 8-membered heterocycloalkyl having 1-3 ring heteroatoms independently selected from N, O, and S, or phenyl, wherein said phenyl is optionally substituted with 1-3 halo. In some embodiments, m is 0. In some embodiments, m is 1; and R4 is halo, C1-C6 alkyl, C1-C6-alkoxy, C3-C6 cycloalkyl, 4- to 8-membered heterocycloalkyl having 1-3 ring heteroatoms independently selected from N, O, and S, or phenyl, wherein said phenyl is optionally substituted with 1-3 halo. In some embodiments, m is 2; and each R4 is independently halo, C1-C6 alkyl, C1-C6-alkoxy, C3-C6 cycloalkyl, 4- to 8-membered heterocycloalkyl having 1-3 ring heteroatoms independently selected from N, O, and S, or phenyl, wherein said phenyl is optionally substituted with 1-3 halo. In some embodiments, m is 0, 1, or 2; and at least one R4, when present is halo. In some embodiments, R4 is -F, -Cl, -Br, or -I. In some embodiments, R4 is -F, or -Cl. In some embodiments, R4 is -F. In some embodiments, R4 is -Cl. In some embodiments, m is 0, 1, or 2, and at least one R4, when present, is C1-C6 alkyl. In some embodiments, R4 is C1- C3 alkyl. In some embodiments, R4 is methyl, ethyl, n-propyl, or isopropyl. In some embodiments, R4 is methyl, or ethyl. In some embodiments, R4 is methyl. In some embodiments, R4 is ethyl. In some embodiments, m is 0, 1, or 2; and at least one R4, when present, is C1-C6-alkoxy. In some embodiments, R4 is C1-C3-alkoxy. In some embodiments, R4 is methoxy, ethoxy, n-propoxy, or isopropoxy. In some embodiments, R4 is methoxy or ethoxy. In some embodiments R4 is methoxy. In some embodiments, R4 is ethoxy. In some embodiments, m is 0, 1, or 2; and at least one R4, when present, is C3-C6 cycloalkyl. In some embodiments, R4 is cyclopropyl, cyclobutyl, cyclopentyl, or cyclohexyl. In some embodiments, R4 is cyclopropyl. In some embodiments, m is 0, 1, or 2; and at least one R4, when present, is 4- to 8-membered heterocycloalkyl having 1-3 ring heteroatoms independently selected from N, O, and S. In some embodiments, R4 is a 5- to 6- membered heterocycloalkyl having 1-3 ring heteroatoms independently selected from N, O, and S. In some embodiments R4 is a 6-membered heterocycloalkyl having 1-3 ring heteroatoms
independently selected from N, and O. In some embodiments, R4 is piperidine, piperazine, or morpholine. In some embodiments, R4 is morpholine. In some embodiments, m is 0, 1, or 2; and at least one R4, when present, is phenyl optionally substituted with 1-3 halo. In some embodiments, R4 is phenyl substituted with one halo. In some embodiments, R4 is phenyl substituted with -F, - Cl, -Br, or -I. In some embodiments, R4 is phenyl substituted with -F. In some embodiments, m is 0, 1, or 2; and each R4, when present, is independently -F, -Cl, -CH3, -CH2CH3, -OCH3, - OCH2CH3, cyclopropyl,
 . In some embodiments, m is 1; and R4, is independently -F, -Cl, -CH3, -CH2CH3, -OCH3, -OCH2CH3, cyclopropyl,
. In some embodiments, m is 1; and R4, is independently -F, -Cl, -CH3, -CH2CH3, -OCH3, -OCH2CH3, cyclopropyl,
 r
r
 . In some embodiments, m is 2; and each R4 is independently -F, -Cl, -CH3, -CH2CH3, - OCH3, -OCH2CH3, cyclopropyl,
. In some embodiments, m is 2; and each R4 is independently -F, -Cl, -CH3, -CH2CH3, - OCH3, -OCH2CH3, cyclopropyl,
 . [0054] In some embodiments, m is 0, 1, or 2; and each R4, when present is independently halo, C1-C6 alkyl, C1-C6-alkoxy, or C3-C6 cycloalkyl. In some embodiments, m is 0 or 1; and R4, when present is halo, C1-C6 alkyl, C1-C6-alkoxy, or C3-C6 cycloalkyl. In some embodiments, m is 0. In some embodiments, m is 1, and R4 is halo, C1-C6 alkyl, C1-C6-alkoxy, or C3-C6 cycloalkyl. In some embodiments, R4 is halo, C1-C3-alkyl, C1-C3-alkoxy, or C3-C5-cycloalkyl. In some embodiments, R4 is -F, -Cl, -CH3, -CH2CH3, -OCH3, -OCH2CH3, or cyclopropyl. [0055] In some embodiments, m is 0, 1, or 2; and each R4 is independently halo, or C1-C6-alkoxy. In some embodiments, at least one R4 is halo. In some embodiments, at least one R4 is Cl, Br, or F. In some embodiments, at least one R4 is Cl. In some embodiments at least one R4 is C1-C6- alkoxy. In some embodiments, at least one R4 is C1-C3-alkoxy. In some embodiments, at least one R4 is -OCH3. In some embodiments, m is 0, 1, or 2; and each R4 is independently Cl, or - OCH3. [0056] In some embodiments, R3 is halo, C1-C6 alkyl, or C1-C6 hydroxyalkyl. In some embodiments, R3 is halo, C1-C3 alkyl, or C1-C3 hydroxyalkyl. In some embodiments, R3 is halo. In some embodiments, R3 is -F, -Cl, or -Br. In some embodiments, R3 is -F, or -Cl. In some
embodiments, R3 is -F. In some embodiments, R3 is -Cl. In some embodiments, R3 is C1-C3 alkyl. In some embodiments, R3 is -CH3. In some embodiments, R3 is C1-C3 hydroxyalkyl. In some embodiments, R3 is -CH2-OH, -CH2CH2-OH, or -CH2CH2CH2-OH. In some embodiments, R3 is -CH2-OH. In some embodiments R3 is -F, -Cl, -CH3, or -CH2-OH. [0057] In some embodiments, R3 is halo or C1-C6-alkyl. In some embodiments, R3 is halo, or C1- C3-alkyl. In some embodiments, R3 is halo. In some embodiments, R3 is -F, -Cl, or -Br. In some embodiments, R3 is -Cl. In some embodiments, R3 is C1-C3 alkyl. In some embodiments, R3 is - CH3. In some embodiments, R3 is -Cl, or -CH3. [0058] In some embodiments, n is 0 or 1, and R2, when present is halo, -CN, -C1-C3-alkyl, -C1- C3-alkoxy, or C3-C6-cycloalkyl. In some embodiments, n is 0. In some embodiments, n is 1. In some embodiments, R2, when present, is halo. In some embodiments, R2, when present, is -F, - Cl, -Br, or -I. In some embodiments, R2, when present, is -F, or -Cl. In some embodiments, R2, when present, is -F. In some embodiments, R2, when present, is -Cl. In some embodiments, R2, when present, is -C1-C3-alkyl. In some embodiments, R2, when present, is methyl, ethyl, n-propyl, or isopropyl. In some embodiments, R2, when present, is methyl. In some embodiments, R2, when present, is -C1-C3-alkoxy. In some embodiments, R2, when present, is methoxy, ethoxy, n- propoxyl, or isopropoxy. In some embodiments, R2, when present, is methoxy. In some embodiments, R2, when present, is C3-C6-cycloalkyl. In some embodiments, R2, when present, is cyclopropyl, cyclobutyl, cyclopentyl, or cyclohexyl. In some embodiments, R2, when present, is cyclopropyl. In some embodiments, n is 0 or 1, and R2, when present, is -F, -Cl, -CN, -CH3, - OCH3, or cyclopropyl. [0059] In some embodiments, n is 1, and R2 is halo. In some embodiments, n is 1 and R2 is -F or -Cl. [0060] In some embodiments, each R1 is independently halo, -CN, -OH, C1-C6-alkyl, C1-C6- haloalkyl, C1-C6-hydroxyalkyl, C1-C6 alkoxy, C3-C6-cycloalkyl, -O-C3-C6-cycloalkyl, 4- to 6- membered heterocycloalkyl, -NR1aR1b, phenyl, 6-membered heteroaryl, -X1-C(O)NR1aR1b, -X1-O- (C1-C3-alkyl), -X1-C3-C6-cycloalkyl, and -X1-phenyl; wherein each 6-membered heteroaryl has 1- 2 ring heteroatoms independently selected from N and O; each 4- to 6-membered heterocycloalkyl has 1-3 ring heteroatoms independently selected from N, O, and S; and each R1 is unsubstituted or
substituted with 1-3 R1c; each R1a and R1b is independently H, C1-C3 alkyl, or C3-C6 cycloalkyl; each R1c is independently halo or -OH; and each X1 is C1-C3-alkylene. [0061] In some embodiments, each R1 is independently selected from the group consisting of C1-C6-alkyl, C1-C6-haloalkyl, C1-C6 alkoxy, C3-C6-cycloalkyl, 4- to 8-membered heterocycloalkyl having 1-2 ring heteroatoms independently selected from N and O, -NR1aR1b, and -X1-C3-C6- cycloalkyl; wherein X1 is C1-C3-alkylene; and R1a and R1b, when present, are independently H, or C1-C3 alkyl. [0062] In some embodiments, R1 is selected from the group consisting of C1-C6-alkyl, C1-C6- haloalkyl, C3-C6-cycloalkyl, and -X1-C3-C6-cycloalkyl, wherein X1 is C1-C3-alkylene. [0063] In some embodiments, R1 is C1-C6-alkyl. In some embodiments, R1 is C1-C4-alkyl. In some embodiments, R1 is methyl, ethyl, n-propyl, iso-propyl, iso-butyl, or tert-butyl. [0064] In some embodiments, R1 is C1-C6-haloalkyl. In some embodiments, R1 is C1-C3- haloalkyl. In some embodiments, R1 is difluoromethyl, trifluoromethyl, trifluoroethyl, difluoroethyl, trifluoropropyl, or difluoropropyl. In some embodiments, R1 is difluoromethyl, trifluoromethyl, trifluoroethyl, or trifluoropropyl. In some embodiments, R1 is -CF2H, CF3, - CH2CF2H, -CH2CF3, or -CH2CH2CF3. [0065] In some embodiments, R1 is C1-C6 alkoxy. In some embodiments, R1 is C1-C3 alkoxy. In some embodiments, R1 is methoxy. [0066] In some embodiments, R1 is C3-C6-cycloalkyl optionally substituted with 1-3 R1c. In some embodiments, R1 is C3-C6-cycloalkyl optionally substituted with 1-2 R1c. In some embodiments, R1 is C3-C5-cycloalkyl optionally substituted with 1-3 R1c. In some embodiments, R1 is C3-C5-cycloalkyl optionally substituted with 1-2 R1c. In some embodiments, R1 is cyclopropyl, cyclobutyl, cyclopentyl, or cyclohexyl, each of which is optionally substituted with 1-3 R1c. In some embodiments, R1 is cyclopropyl, cyclobutyl, cyclopentyl, or cyclohexyl, each of which is optionally substituted with 1-2 R1c. In some embodiments, R1 is cyclopropyl, cyclobutyl, or cyclopentyl, each of which is optionally substituted with 1-3 R1c. In some embodiments, R1 is cyclopropyl, cyclobutyl, or cyclopentyl, each of which is optionally substituted with 1-2 R1c. In some embodiments, each R1c when present, is independently halo or -OH. In some embodiments,
each R1c, when present, is independently -F, or -OH. In some embodiments, R1 is
. [0054] In some embodiments, m is 0, 1, or 2; and each R4, when present is independently halo, C1-C6 alkyl, C1-C6-alkoxy, or C3-C6 cycloalkyl. In some embodiments, m is 0 or 1; and R4, when present is halo, C1-C6 alkyl, C1-C6-alkoxy, or C3-C6 cycloalkyl. In some embodiments, m is 0. In some embodiments, m is 1, and R4 is halo, C1-C6 alkyl, C1-C6-alkoxy, or C3-C6 cycloalkyl. In some embodiments, R4 is halo, C1-C3-alkyl, C1-C3-alkoxy, or C3-C5-cycloalkyl. In some embodiments, R4 is -F, -Cl, -CH3, -CH2CH3, -OCH3, -OCH2CH3, or cyclopropyl. [0055] In some embodiments, m is 0, 1, or 2; and each R4 is independently halo, or C1-C6-alkoxy. In some embodiments, at least one R4 is halo. In some embodiments, at least one R4 is Cl, Br, or F. In some embodiments, at least one R4 is Cl. In some embodiments at least one R4 is C1-C6- alkoxy. In some embodiments, at least one R4 is C1-C3-alkoxy. In some embodiments, at least one R4 is -OCH3. In some embodiments, m is 0, 1, or 2; and each R4 is independently Cl, or - OCH3. [0056] In some embodiments, R3 is halo, C1-C6 alkyl, or C1-C6 hydroxyalkyl. In some embodiments, R3 is halo, C1-C3 alkyl, or C1-C3 hydroxyalkyl. In some embodiments, R3 is halo. In some embodiments, R3 is -F, -Cl, or -Br. In some embodiments, R3 is -F, or -Cl. In some
embodiments, R3 is -F. In some embodiments, R3 is -Cl. In some embodiments, R3 is C1-C3 alkyl. In some embodiments, R3 is -CH3. In some embodiments, R3 is C1-C3 hydroxyalkyl. In some embodiments, R3 is -CH2-OH, -CH2CH2-OH, or -CH2CH2CH2-OH. In some embodiments, R3 is -CH2-OH. In some embodiments R3 is -F, -Cl, -CH3, or -CH2-OH. [0057] In some embodiments, R3 is halo or C1-C6-alkyl. In some embodiments, R3 is halo, or C1- C3-alkyl. In some embodiments, R3 is halo. In some embodiments, R3 is -F, -Cl, or -Br. In some embodiments, R3 is -Cl. In some embodiments, R3 is C1-C3 alkyl. In some embodiments, R3 is - CH3. In some embodiments, R3 is -Cl, or -CH3. [0058] In some embodiments, n is 0 or 1, and R2, when present is halo, -CN, -C1-C3-alkyl, -C1- C3-alkoxy, or C3-C6-cycloalkyl. In some embodiments, n is 0. In some embodiments, n is 1. In some embodiments, R2, when present, is halo. In some embodiments, R2, when present, is -F, - Cl, -Br, or -I. In some embodiments, R2, when present, is -F, or -Cl. In some embodiments, R2, when present, is -F. In some embodiments, R2, when present, is -Cl. In some embodiments, R2, when present, is -C1-C3-alkyl. In some embodiments, R2, when present, is methyl, ethyl, n-propyl, or isopropyl. In some embodiments, R2, when present, is methyl. In some embodiments, R2, when present, is -C1-C3-alkoxy. In some embodiments, R2, when present, is methoxy, ethoxy, n- propoxyl, or isopropoxy. In some embodiments, R2, when present, is methoxy. In some embodiments, R2, when present, is C3-C6-cycloalkyl. In some embodiments, R2, when present, is cyclopropyl, cyclobutyl, cyclopentyl, or cyclohexyl. In some embodiments, R2, when present, is cyclopropyl. In some embodiments, n is 0 or 1, and R2, when present, is -F, -Cl, -CN, -CH3, - OCH3, or cyclopropyl. [0059] In some embodiments, n is 1, and R2 is halo. In some embodiments, n is 1 and R2 is -F or -Cl. [0060] In some embodiments, each R1 is independently halo, -CN, -OH, C1-C6-alkyl, C1-C6- haloalkyl, C1-C6-hydroxyalkyl, C1-C6 alkoxy, C3-C6-cycloalkyl, -O-C3-C6-cycloalkyl, 4- to 6- membered heterocycloalkyl, -NR1aR1b, phenyl, 6-membered heteroaryl, -X1-C(O)NR1aR1b, -X1-O- (C1-C3-alkyl), -X1-C3-C6-cycloalkyl, and -X1-phenyl; wherein each 6-membered heteroaryl has 1- 2 ring heteroatoms independently selected from N and O; each 4- to 6-membered heterocycloalkyl has 1-3 ring heteroatoms independently selected from N, O, and S; and each R1 is unsubstituted or
substituted with 1-3 R1c; each R1a and R1b is independently H, C1-C3 alkyl, or C3-C6 cycloalkyl; each R1c is independently halo or -OH; and each X1 is C1-C3-alkylene. [0061] In some embodiments, each R1 is independently selected from the group consisting of C1-C6-alkyl, C1-C6-haloalkyl, C1-C6 alkoxy, C3-C6-cycloalkyl, 4- to 8-membered heterocycloalkyl having 1-2 ring heteroatoms independently selected from N and O, -NR1aR1b, and -X1-C3-C6- cycloalkyl; wherein X1 is C1-C3-alkylene; and R1a and R1b, when present, are independently H, or C1-C3 alkyl. [0062] In some embodiments, R1 is selected from the group consisting of C1-C6-alkyl, C1-C6- haloalkyl, C3-C6-cycloalkyl, and -X1-C3-C6-cycloalkyl, wherein X1 is C1-C3-alkylene. [0063] In some embodiments, R1 is C1-C6-alkyl. In some embodiments, R1 is C1-C4-alkyl. In some embodiments, R1 is methyl, ethyl, n-propyl, iso-propyl, iso-butyl, or tert-butyl. [0064] In some embodiments, R1 is C1-C6-haloalkyl. In some embodiments, R1 is C1-C3- haloalkyl. In some embodiments, R1 is difluoromethyl, trifluoromethyl, trifluoroethyl, difluoroethyl, trifluoropropyl, or difluoropropyl. In some embodiments, R1 is difluoromethyl, trifluoromethyl, trifluoroethyl, or trifluoropropyl. In some embodiments, R1 is -CF2H, CF3, - CH2CF2H, -CH2CF3, or -CH2CH2CF3. [0065] In some embodiments, R1 is C1-C6 alkoxy. In some embodiments, R1 is C1-C3 alkoxy. In some embodiments, R1 is methoxy. [0066] In some embodiments, R1 is C3-C6-cycloalkyl optionally substituted with 1-3 R1c. In some embodiments, R1 is C3-C6-cycloalkyl optionally substituted with 1-2 R1c. In some embodiments, R1 is C3-C5-cycloalkyl optionally substituted with 1-3 R1c. In some embodiments, R1 is C3-C5-cycloalkyl optionally substituted with 1-2 R1c. In some embodiments, R1 is cyclopropyl, cyclobutyl, cyclopentyl, or cyclohexyl, each of which is optionally substituted with 1-3 R1c. In some embodiments, R1 is cyclopropyl, cyclobutyl, cyclopentyl, or cyclohexyl, each of which is optionally substituted with 1-2 R1c. In some embodiments, R1 is cyclopropyl, cyclobutyl, or cyclopentyl, each of which is optionally substituted with 1-3 R1c. In some embodiments, R1 is cyclopropyl, cyclobutyl, or cyclopentyl, each of which is optionally substituted with 1-2 R1c. In some embodiments, each R1c when present, is independently halo or -OH. In some embodiments,
each R1c, when present, is independently -F, or -OH. In some embodiments, R1 is
 ,
,
 . [0067] In some embodiments, R1 is C3-C6-cycloalkyl. In some embodiments, R1 is C3-C5- cycloalkyl. In some embodiments, R1 is cyclopropyl, cyclobutyl, cyclopentyl, or cyclohexyl. In some embodiments, R1 is cyclopropyl, cyclobutyl, or cyclopentyl. [0068] In some embodiments, R1 is -O-C3-C6-cycloalkyl. In some embodiments, R1 is -O- cyclopropyl, -O-cyclobutyl, -O-cyclopentyl, or -O-cyclohexyl. In some embodiments, R1 is -O- cyclopropyl. [0069] In some embodiments, R1 is 4- to 8-membered heterocycloalkyl having 1-2 ring heteroatoms independently selected from N and O. In some embodiments, R1 is 5- to 6-membered heterocycloalkyl having 1-2 ring heteroatoms independently selected from N and O. In some embodiments, R1 is pyrrolidinyl, tetrahydrofuranyl, tetrahydropyranyl, piperidinyl, or morpholinyl. In some embodiments, R1 is tetrahydrofuranyl, tetrahydropyranyl, or morpholinyl. In some embodiments, R1 is
. [0067] In some embodiments, R1 is C3-C6-cycloalkyl. In some embodiments, R1 is C3-C5- cycloalkyl. In some embodiments, R1 is cyclopropyl, cyclobutyl, cyclopentyl, or cyclohexyl. In some embodiments, R1 is cyclopropyl, cyclobutyl, or cyclopentyl. [0068] In some embodiments, R1 is -O-C3-C6-cycloalkyl. In some embodiments, R1 is -O- cyclopropyl, -O-cyclobutyl, -O-cyclopentyl, or -O-cyclohexyl. In some embodiments, R1 is -O- cyclopropyl. [0069] In some embodiments, R1 is 4- to 8-membered heterocycloalkyl having 1-2 ring heteroatoms independently selected from N and O. In some embodiments, R1 is 5- to 6-membered heterocycloalkyl having 1-2 ring heteroatoms independently selected from N and O. In some embodiments, R1 is pyrrolidinyl, tetrahydrofuranyl, tetrahydropyranyl, piperidinyl, or morpholinyl. In some embodiments, R1 is tetrahydrofuranyl, tetrahydropyranyl, or morpholinyl. In some embodiments, R1 is
 , , . In some embodiments, R1 is
, , . In some embodiments, R1 is
 . [0070] In some embodiments, R1 is -NR1aR1b, wherein R1a and R1b are independently H, C1-C3 alkyl, or C3-C6 cycloalkyl. In some embodiments, R1 is -NR1aR1b, wherein R1a and R1b are independently H, or C1-C3 alkyl. In some embodiments, at least one of R1a and R1b is C1-C3 alkyl. In some embodiments, both R1a and R1b are C1-C3 alkyl. In some embodiments, at least one of R1a and R1b is C3-C6 cycloalkyl. In some embodiments, at least one of R1a and R1b is cyclopropyl. In some embodiments, R1 is -NH2. In some embodiments, R1 is -N(CH3)2. In some embodiments, R1 i
. [0070] In some embodiments, R1 is -NR1aR1b, wherein R1a and R1b are independently H, C1-C3 alkyl, or C3-C6 cycloalkyl. In some embodiments, R1 is -NR1aR1b, wherein R1a and R1b are independently H, or C1-C3 alkyl. In some embodiments, at least one of R1a and R1b is C1-C3 alkyl. In some embodiments, both R1a and R1b are C1-C3 alkyl. In some embodiments, at least one of R1a and R1b is C3-C6 cycloalkyl. In some embodiments, at least one of R1a and R1b is cyclopropyl. In some embodiments, R1 is -NH2. In some embodiments, R1 is -N(CH3)2. In some embodiments, R1 i
 . [0071] In some embodiments, R1 is phenyl optionally substituted with 1-3 R1c. In some embodiments, R1 is phenyl optionally substituted with 1-2 R1c. In some embodiments, R1 is phenyl
optionally substituted with 1 R1c. In some embodiments R1 is phenyl optionally substituted with 1-3 R1c, wherein each R1c is independently halo. In some embodiments R1 is phenyl optionally substituted with 1-3 R1c, wherein each R1c is independently -F. In some embodiments, R1 is
. [0071] In some embodiments, R1 is phenyl optionally substituted with 1-3 R1c. In some embodiments, R1 is phenyl optionally substituted with 1-2 R1c. In some embodiments, R1 is phenyl
optionally substituted with 1 R1c. In some embodiments R1 is phenyl optionally substituted with 1-3 R1c, wherein each R1c is independently halo. In some embodiments R1 is phenyl optionally substituted with 1-3 R1c, wherein each R1c is independently -F. In some embodiments, R1 is
 . [0072] In some embodiments, R1 is 5- to 6-membered heteroaryl having 1-2 ring heteroatoms independently selected from N, and O. In some embodiments, R1 is a 6-membered heteroaryl having 1-2 ring heteroatoms that are independently N. In some embodiments, R1 is pyridinyl, pyrimidinyl, pyrazinyl, or pyridazinyl. In some embodiments, R1 is pyridyl. In some embodiments,
. [0072] In some embodiments, R1 is 5- to 6-membered heteroaryl having 1-2 ring heteroatoms independently selected from N, and O. In some embodiments, R1 is a 6-membered heteroaryl having 1-2 ring heteroatoms that are independently N. In some embodiments, R1 is pyridinyl, pyrimidinyl, pyrazinyl, or pyridazinyl. In some embodiments, R1 is pyridyl. In some embodiments,
 [0073] In some embodiments, R1 is -X1-phenyl; wherein X1 is -C1-C3-alkylene. In some embodiments, R1 is -X1-phenyl, wherein X1 is methylene, ethylene, n-propylene, or iso-propylene. In some embodiments,
[0073] In some embodiments, R1 is -X1-phenyl; wherein X1 is -C1-C3-alkylene. In some embodiments, R1 is -X1-phenyl, wherein X1 is methylene, ethylene, n-propylene, or iso-propylene. In some embodiments,
 . [0074] In some embodiments, R1 is -X1-C3-C6-cycloalkyl; wherein X1 is C1-C3-alkylene. In some embodiments, R1 is -X1-C3-C6-cycloalkyl; wherein X1 is methylene, ethylene, n-propylene, or iso-propylene. In some embodiments, R1 is -X1-cyclopropyl. In some embodiments, R1 is
. [0074] In some embodiments, R1 is -X1-C3-C6-cycloalkyl; wherein X1 is C1-C3-alkylene. In some embodiments, R1 is -X1-C3-C6-cycloalkyl; wherein X1 is methylene, ethylene, n-propylene, or iso-propylene. In some embodiments, R1 is -X1-cyclopropyl. In some embodiments, R1 is
 . [0075] In some embodiments, each R1 is independently selected from the group consisting of - F, -Cl, -Br, -CN, -OH, , , , , , , , , , , , , , , , , , , , , , , , , , ,
,
. [0075] In some embodiments, each R1 is independently selected from the group consisting of - F, -Cl, -Br, -CN, -OH, , , , , , , , , , , , , , , , , , , , , , , , , , ,
,
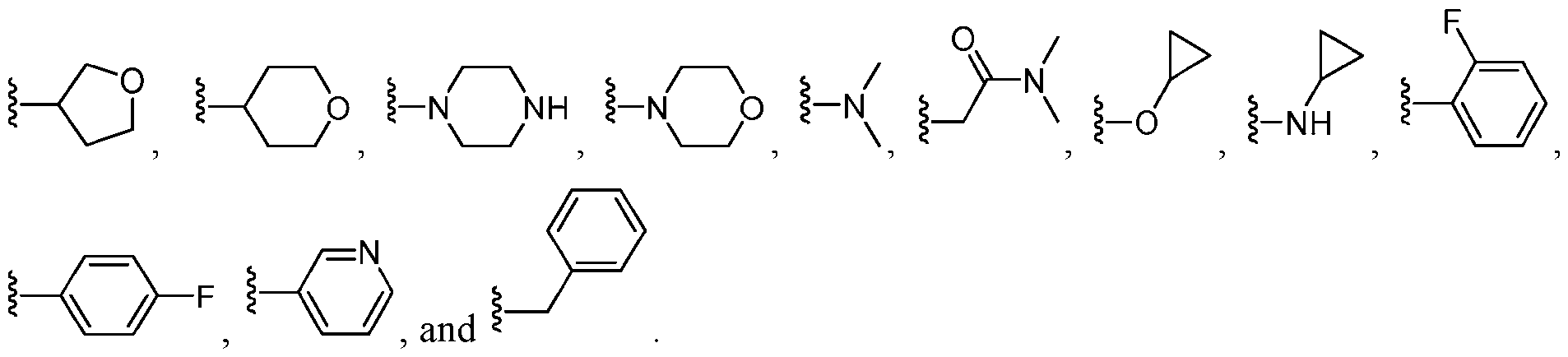 [0076] In some embodiments, the compound, or a pharmaceutically acceptable salt thereof, has a structure according to Formula III:
[0076] In some embodiments, the compound, or a pharmaceutically acceptable salt thereof, has a structure according to Formula III:
 (Formula III) wherein: A is: (i) selected from the group consisting of: , ,
(Formula III) wherein: A is: (i) selected from the group consisting of: , ,
 (ii) a 6-membered heteroaryl having 1 or 2 ring nitrogen atoms, wherein the 6- membered heteroaryl is substituted with 0-2 R1d; or (iii) -NHC(O)-R1g; each R1 is independently selected from the group consisting of halo, CN, -OH, C1-C6-alkyl, C1-C6-haloalkyl, C1-C6-hydroxyalkyl, C1-C6 alkoxy, C3-C6-cycloalkyl, -O-C3-C6-cycloalkyl, 4- to
8-membered heterocycloalkyl, -NR1aR1b, phenyl, 5- to 6-membered heteroaryl, -X1-C(O)NR1aR1b, -X1-O-(C1-C3-alkyl), -X1-C3-C6-cycloalkyl, and -X1-phenyl, wherein each 5- to 6-membered heteroaryl has 1-2 ring heteroatoms independently selected from N and O; each 4- to 8-membered heterocycloalkyl has 1-3 ring heteroatoms independently selected from N, O, and S; and each R1 is optionally substituted with 1-3 R1c; each X1 is C1-C3-alkylene; R1a and R1b, when present, are independently H, C1-C6 alkyl, or C3-C6 cycloalkyl; each R1c, when present, is independently halo, or -OH; each R1d, when present, is independently halo, C1-C6 alkoxy, C3-C6-cycloalkyl, -O-C3-C6- cycloalkyl, or -NR1eR1f; R1e and R1f, when present, are independently H, C1-C6 haloalkyl, -(C1-C3-alkylene)-(C3- C6-cycloalkyl), or 4- to 8-membered heterocycloalkyl having 1-3 ring heteroatoms independently selected from N, O, and S; or R1e and R1f together with the nitrogen atom to which they are attached form a 4- to 6-membered heterocycloalkyl group having 1-2 ring heteroatoms independently selected from N, and O; R1g, when present, is C3-C6-cycloalkyl, -(C1-C3-alkylene)-(C3-C6-cycloalkyl), or 6- membered heteroaryl having 1 or 2 ring nitrogen atoms; wherein R1g is optionally substituted with 0-2 R1h; each R1h, when present, is independently halo, C1-C6 alkyl, -O-C3-C6-cycloalkyl, or -NH- (4- to 8-membered heterocycloalkyl), wherein the 4- to 8-membered heterocycloalkyl has 1-2 ring heteroatoms independently selected from N, O, and S; n is 0 or 1; m is 0, 1 or 2; R2, when present, is halo, -CN, -C1-C3-alkyl, -C1-C3-alkoxy, or C3-C6-cycloalkyl; R3 is halo, C1-C3 alkyl, or C1-C3 hydroxyalkyl; each R4, when present, is independently halo, C1-C6 alkyl, C1-C6-alkoxy, C3-C6 cycloalkyl, 4- to 8-membered heterocycloalkyl having 1-3 ring heteroatoms independently selected from N, O, and S, or phenyl, wherein said phenyl is optionally substituted with 1-3 halo; and R5 and R6 are H. [0077] In some embodiments, the compound, or a pharmaceutically acceptable salt thereof, has a structure according to Formula III:
(ii) a 6-membered heteroaryl having 1 or 2 ring nitrogen atoms, wherein the 6- membered heteroaryl is substituted with 0-2 R1d; or (iii) -NHC(O)-R1g; each R1 is independently selected from the group consisting of halo, CN, -OH, C1-C6-alkyl, C1-C6-haloalkyl, C1-C6-hydroxyalkyl, C1-C6 alkoxy, C3-C6-cycloalkyl, -O-C3-C6-cycloalkyl, 4- to
8-membered heterocycloalkyl, -NR1aR1b, phenyl, 5- to 6-membered heteroaryl, -X1-C(O)NR1aR1b, -X1-O-(C1-C3-alkyl), -X1-C3-C6-cycloalkyl, and -X1-phenyl, wherein each 5- to 6-membered heteroaryl has 1-2 ring heteroatoms independently selected from N and O; each 4- to 8-membered heterocycloalkyl has 1-3 ring heteroatoms independently selected from N, O, and S; and each R1 is optionally substituted with 1-3 R1c; each X1 is C1-C3-alkylene; R1a and R1b, when present, are independently H, C1-C6 alkyl, or C3-C6 cycloalkyl; each R1c, when present, is independently halo, or -OH; each R1d, when present, is independently halo, C1-C6 alkoxy, C3-C6-cycloalkyl, -O-C3-C6- cycloalkyl, or -NR1eR1f; R1e and R1f, when present, are independently H, C1-C6 haloalkyl, -(C1-C3-alkylene)-(C3- C6-cycloalkyl), or 4- to 8-membered heterocycloalkyl having 1-3 ring heteroatoms independently selected from N, O, and S; or R1e and R1f together with the nitrogen atom to which they are attached form a 4- to 6-membered heterocycloalkyl group having 1-2 ring heteroatoms independently selected from N, and O; R1g, when present, is C3-C6-cycloalkyl, -(C1-C3-alkylene)-(C3-C6-cycloalkyl), or 6- membered heteroaryl having 1 or 2 ring nitrogen atoms; wherein R1g is optionally substituted with 0-2 R1h; each R1h, when present, is independently halo, C1-C6 alkyl, -O-C3-C6-cycloalkyl, or -NH- (4- to 8-membered heterocycloalkyl), wherein the 4- to 8-membered heterocycloalkyl has 1-2 ring heteroatoms independently selected from N, O, and S; n is 0 or 1; m is 0, 1 or 2; R2, when present, is halo, -CN, -C1-C3-alkyl, -C1-C3-alkoxy, or C3-C6-cycloalkyl; R3 is halo, C1-C3 alkyl, or C1-C3 hydroxyalkyl; each R4, when present, is independently halo, C1-C6 alkyl, C1-C6-alkoxy, C3-C6 cycloalkyl, 4- to 8-membered heterocycloalkyl having 1-3 ring heteroatoms independently selected from N, O, and S, or phenyl, wherein said phenyl is optionally substituted with 1-3 halo; and R5 and R6 are H. [0077] In some embodiments, the compound, or a pharmaceutically acceptable salt thereof, has a structure according to Formula III:
 (Formula III) wherein: ring A is a 5-membered heteroaryl having 1-4 ring heteroatoms independently selected from N, O, and S, wherein the heteroaryl is substituted with 0-2 R1; each R1 is independently selected from the group consisting of halo, CN, -OH, C1-C6-alkyl, C1-C6-haloalkyl, C1-C6-hydroxyalkyl, C1-C6 alkoxy, C3-C6-cycloalkyl, -O-C3-C6-cycloalkyl, 4- to 8-membered heterocycloalkyl, -NR1aR1b, phenyl, 5- to 6-membered heteroaryl, -X1-C(O)NR1aR1b, -X1-O-(C1-C3-alkyl), -X1-C3-C6-cycloalkyl, and -X1-phenyl, wherein each 5- to 6-membered heteroaryl has 1-2 ring heteroatoms independently selected from N and O; each 4- to 8-membered heterocycloalkyl has 1-3 ring heteroatoms independently selected from N, O, and S; and each R1 is optionally substituted with 1-3 R1c; each X1 is C1-C3-alkylene; R1a and R1b, when present, are independently H, C1-C6 alkyl, or C3-C6 cycloalkyl; R1c, when present, is halo, or -OH; n is 0 or 1; m is 0, 1 or 2; R2, when present, is halo, -CN, -C1-C3-alkyl, -C1-C3-alkoxy, or C3-C6-cycloalkyl; R3 is halo, C1-C3 alkyl, or C1-C3 hydroxyalkyl; each R4, when present, is independently halo, C1-C6 alkyl, C1-C6-alkoxy, C3-C6 cycloalkyl, 4- to 8-membered heterocycloalkyl having 1-3 ring heteroatoms independently selected from N, O, and S, or phenyl, wherein said phenyl is optionally substituted with 1-3 halo; and R5 and R6 are H. [0078] In some embodiments, the compound, or a pharmaceutically acceptable salt thereof, has a structure according to Formula III:
(Formula III) wherein: ring A is a 5-membered heteroaryl having 1-4 ring heteroatoms independently selected from N, O, and S, wherein the heteroaryl is substituted with 0-2 R1; each R1 is independently selected from the group consisting of halo, CN, -OH, C1-C6-alkyl, C1-C6-haloalkyl, C1-C6-hydroxyalkyl, C1-C6 alkoxy, C3-C6-cycloalkyl, -O-C3-C6-cycloalkyl, 4- to 8-membered heterocycloalkyl, -NR1aR1b, phenyl, 5- to 6-membered heteroaryl, -X1-C(O)NR1aR1b, -X1-O-(C1-C3-alkyl), -X1-C3-C6-cycloalkyl, and -X1-phenyl, wherein each 5- to 6-membered heteroaryl has 1-2 ring heteroatoms independently selected from N and O; each 4- to 8-membered heterocycloalkyl has 1-3 ring heteroatoms independently selected from N, O, and S; and each R1 is optionally substituted with 1-3 R1c; each X1 is C1-C3-alkylene; R1a and R1b, when present, are independently H, C1-C6 alkyl, or C3-C6 cycloalkyl; R1c, when present, is halo, or -OH; n is 0 or 1; m is 0, 1 or 2; R2, when present, is halo, -CN, -C1-C3-alkyl, -C1-C3-alkoxy, or C3-C6-cycloalkyl; R3 is halo, C1-C3 alkyl, or C1-C3 hydroxyalkyl; each R4, when present, is independently halo, C1-C6 alkyl, C1-C6-alkoxy, C3-C6 cycloalkyl, 4- to 8-membered heterocycloalkyl having 1-3 ring heteroatoms independently selected from N, O, and S, or phenyl, wherein said phenyl is optionally substituted with 1-3 halo; and R5 and R6 are H. [0078] In some embodiments, the compound, or a pharmaceutically acceptable salt thereof, has a structure according to Formula III:
 (Formula III) wherein: ring A is a 5-membered heteroaryl having 1-4 ring nitrogen atoms, wherein the heteroaryl is substituted with 0-2 R1; each R1 is independently selected from the group consisting of halo, -CN, C1-C6-alkyl, C1- C6-haloalkyl, C1-C6-hydroxyalkyl, C1-C6-cyanoalkyl, C1-C6 alkoxy, C3-C6-cycloalkyl, 4- to 8- membered heterocycloalkyl, -NR1aR1b, phenyl, 5- to 6-membered heteroaryl, -X1-NR1aR1b, -X1- C(O)NR1aR1b, -X1-O-(C1-C3-alkyl), -X1-C3-C6-cycloalkyl, -X1-phenyl, and -X2-(4- to 8-membered heterocycloalkyl), wherein each 5- to 6-membered heteroaryl has 1-2 ring heteroatoms independently selected from N and O; each 4- to 8-membered heterocycloalkyl has 1-3 ring heteroatoms independently selected from N, O, and S, and said heterocycloalkyl is optionally substituted with one oxo; and each R1 is optionally substituted with one R1c; or two adjacent R1 groups are combined with the two ring atoms to which they are attached to form a 5- or 6-membered heterocycloalkyl group having 1-2 ring heteroatoms independently selected from N and O; each X1 is C1-C3-alkylene; each X2 is C1-C3-alkylene, or –(C1-C3-alkylene)-C(O)-; R1a and R1b, when present, are independently H, C1-C6 alkyl, or C3-C6 cycloalkyl; R1c, when present, is halo, -OH, -C1-C6-alkyl, -C1-C6-alkoxy, or -C(O)-(C1-C6-alkyl); n is 0 or 1; m is 0, 1 or 2; R2, when present, is halo, -CN, -C1-C3-alkyl, -C1-C3-alkoxy, or C3-C6-cycloalkyl; R3 is -H, halo, -C1-C6-alkyl, -C1-C6-haloalkyl, C1-C6-alkoxy, or -C(O)NR3aR3b; R3a and R3b, when present, are independently H, or C1-C3 alkyl; each R4, when present, is independently halo, C1-C6 alkyl, C1-C6-alkoxy, or C3-C6 cycloalkyl; R5 is H, C1-C3-alkyl, or -NR5aR5b;
R5a and R5b, when present, are independently H or C1-C3 alkyl; and R6 is H, or C1-C3-alkyl. [0079] In some embodiments, the compound, or a pharmaceutically acceptable salt thereof, has a structure according to Formula IIIa:
(Formula III) wherein: ring A is a 5-membered heteroaryl having 1-4 ring nitrogen atoms, wherein the heteroaryl is substituted with 0-2 R1; each R1 is independently selected from the group consisting of halo, -CN, C1-C6-alkyl, C1- C6-haloalkyl, C1-C6-hydroxyalkyl, C1-C6-cyanoalkyl, C1-C6 alkoxy, C3-C6-cycloalkyl, 4- to 8- membered heterocycloalkyl, -NR1aR1b, phenyl, 5- to 6-membered heteroaryl, -X1-NR1aR1b, -X1- C(O)NR1aR1b, -X1-O-(C1-C3-alkyl), -X1-C3-C6-cycloalkyl, -X1-phenyl, and -X2-(4- to 8-membered heterocycloalkyl), wherein each 5- to 6-membered heteroaryl has 1-2 ring heteroatoms independently selected from N and O; each 4- to 8-membered heterocycloalkyl has 1-3 ring heteroatoms independently selected from N, O, and S, and said heterocycloalkyl is optionally substituted with one oxo; and each R1 is optionally substituted with one R1c; or two adjacent R1 groups are combined with the two ring atoms to which they are attached to form a 5- or 6-membered heterocycloalkyl group having 1-2 ring heteroatoms independently selected from N and O; each X1 is C1-C3-alkylene; each X2 is C1-C3-alkylene, or –(C1-C3-alkylene)-C(O)-; R1a and R1b, when present, are independently H, C1-C6 alkyl, or C3-C6 cycloalkyl; R1c, when present, is halo, -OH, -C1-C6-alkyl, -C1-C6-alkoxy, or -C(O)-(C1-C6-alkyl); n is 0 or 1; m is 0, 1 or 2; R2, when present, is halo, -CN, -C1-C3-alkyl, -C1-C3-alkoxy, or C3-C6-cycloalkyl; R3 is -H, halo, -C1-C6-alkyl, -C1-C6-haloalkyl, C1-C6-alkoxy, or -C(O)NR3aR3b; R3a and R3b, when present, are independently H, or C1-C3 alkyl; each R4, when present, is independently halo, C1-C6 alkyl, C1-C6-alkoxy, or C3-C6 cycloalkyl; R5 is H, C1-C3-alkyl, or -NR5aR5b;
R5a and R5b, when present, are independently H or C1-C3 alkyl; and R6 is H, or C1-C3-alkyl. [0079] In some embodiments, the compound, or a pharmaceutically acceptable salt thereof, has a structure according to Formula IIIa:
 . [0080] In some embodiments, the compound, or a pharmaceutically acceptable salt thereof, has a structure according to Formula II:
. [0080] In some embodiments, the compound, or a pharmaceutically acceptable salt thereof, has a structure according to Formula II:
 (Formula II) wherein: R1 is selected from the group consisting of C1-C6-alkyl, C1-C6-haloalkyl, C1-C6 alkoxy, C3- C6-cycloalkyl, 4- to 8-membered heterocycloalkyl, -NR1aR1b, and -X1-C3-C6-cycloalkyl, wherein said heterocycloalkyl has 1-2 ring heteroatoms independently selected from N and O; X1 is C1-C3-alkylene; R1a and R1b, when present, are independently H, or C1-C3 alkyl; n is 0 or 1; m is 0, 1 or 2; R2, when present, is halo; R3 is halo or C1-C6-alkyl; and each R4, when present, is independently halo, C1-C6 alkyl, C1-C6-alkoxy, or C3-C6 cycloalkyl. [0081] In some embodiments, the compound, or a pharmaceutically acceptable salt thereof, has a structure according to Formula I:
(Formula II) wherein: R1 is selected from the group consisting of C1-C6-alkyl, C1-C6-haloalkyl, C1-C6 alkoxy, C3- C6-cycloalkyl, 4- to 8-membered heterocycloalkyl, -NR1aR1b, and -X1-C3-C6-cycloalkyl, wherein said heterocycloalkyl has 1-2 ring heteroatoms independently selected from N and O; X1 is C1-C3-alkylene; R1a and R1b, when present, are independently H, or C1-C3 alkyl; n is 0 or 1; m is 0, 1 or 2; R2, when present, is halo; R3 is halo or C1-C6-alkyl; and each R4, when present, is independently halo, C1-C6 alkyl, C1-C6-alkoxy, or C3-C6 cycloalkyl. [0081] In some embodiments, the compound, or a pharmaceutically acceptable salt thereof, has a structure according to Formula I:
 (Formula I) wherein: R1 is selected from the group consisting of C1-C6-alkyl, C1-C6-haloalkyl, C3-C6-cycloalkyl, and -X1-C3-C6-cycloalkyl, wherein X1 is C1-C3-alkylene; n is 0 or 1; R2, when present, is halo; R3 is C1-C6-alkyl; and R4 is H, halo, or C1-C6-alkoxy. [0082] In some embodiments, the compound has a structure according to Formula Ia:
(Formula I) wherein: R1 is selected from the group consisting of C1-C6-alkyl, C1-C6-haloalkyl, C3-C6-cycloalkyl, and -X1-C3-C6-cycloalkyl, wherein X1 is C1-C3-alkylene; n is 0 or 1; R2, when present, is halo; R3 is C1-C6-alkyl; and R4 is H, halo, or C1-C6-alkoxy. [0082] In some embodiments, the compound has a structure according to Formula Ia:
 (Formula Ia). [0083] In some embodiments, the compound has a structure according to Formula Ib:
(Formula Ia). [0083] In some embodiments, the compound has a structure according to Formula Ib:
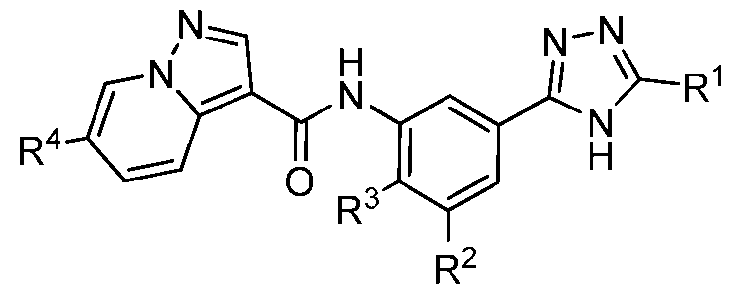 (Formula Ib). [0084] In some embodiments, the compound has a structure according to Formula Ic:
(Formula Ib). [0084] In some embodiments, the compound has a structure according to Formula Ic:
 (Formula Ic). [0085] In some embodiments, the compound, or a pharmaceutically acceptable salt thereof, has a structure according to Formula IV:
(Formula Ic). [0085] In some embodiments, the compound, or a pharmaceutically acceptable salt thereof, has a structure according to Formula IV:
 (Formula IV) wherein: R1 is selected from the group consisting of C1-C6-alkyl; C1-C6-haloalkyl; C3-C6-cycloalkyl; 4- to 8-membered heterocycloalkyl having 1-3 ring heteroatoms independently selected from N, O, and S; phenyl; and -X1-C3-C6-cycloalkyl; wherein X1 is C1-C3-alkylene; and the phenyl and C3- C6-cycloalkyl are optionally substituted with 1-3 R1c; each R1c, when present, is independently halo or -OH; n is 0 or 1; m is 0, 1 or 2; R2, when present, is halo, -CN, -C1-C3-alkyl, -C1-C3-alkoxy, or C3-C6-cycloalkyl; R3 is halo or C1-C6-alkyl; and R4 is H, halo, C1-C6 alkyl, or C1-C6-alkoxy. [0086] In some embodiments, the compound, or a pharmaceutically acceptable salt thereof, has a structure according to Formula IVa:
(Formula IV) wherein: R1 is selected from the group consisting of C1-C6-alkyl; C1-C6-haloalkyl; C3-C6-cycloalkyl; 4- to 8-membered heterocycloalkyl having 1-3 ring heteroatoms independently selected from N, O, and S; phenyl; and -X1-C3-C6-cycloalkyl; wherein X1 is C1-C3-alkylene; and the phenyl and C3- C6-cycloalkyl are optionally substituted with 1-3 R1c; each R1c, when present, is independently halo or -OH; n is 0 or 1; m is 0, 1 or 2; R2, when present, is halo, -CN, -C1-C3-alkyl, -C1-C3-alkoxy, or C3-C6-cycloalkyl; R3 is halo or C1-C6-alkyl; and R4 is H, halo, C1-C6 alkyl, or C1-C6-alkoxy. [0086] In some embodiments, the compound, or a pharmaceutically acceptable salt thereof, has a structure according to Formula IVa:
 . [0087] In some embodiments, the compound is selected from the group consisting of:
. [0087] In some embodiments, the compound is selected from the group consisting of:













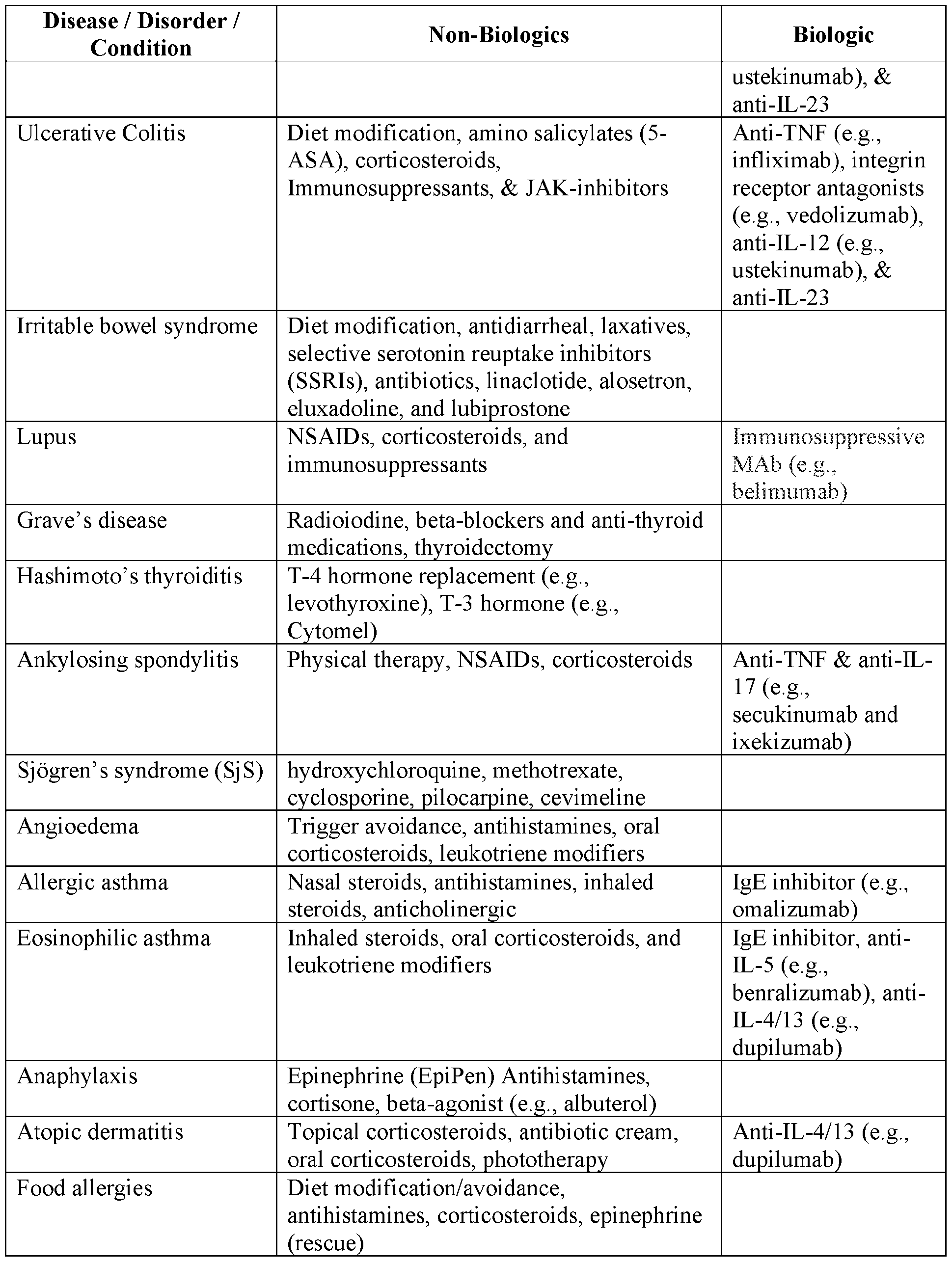


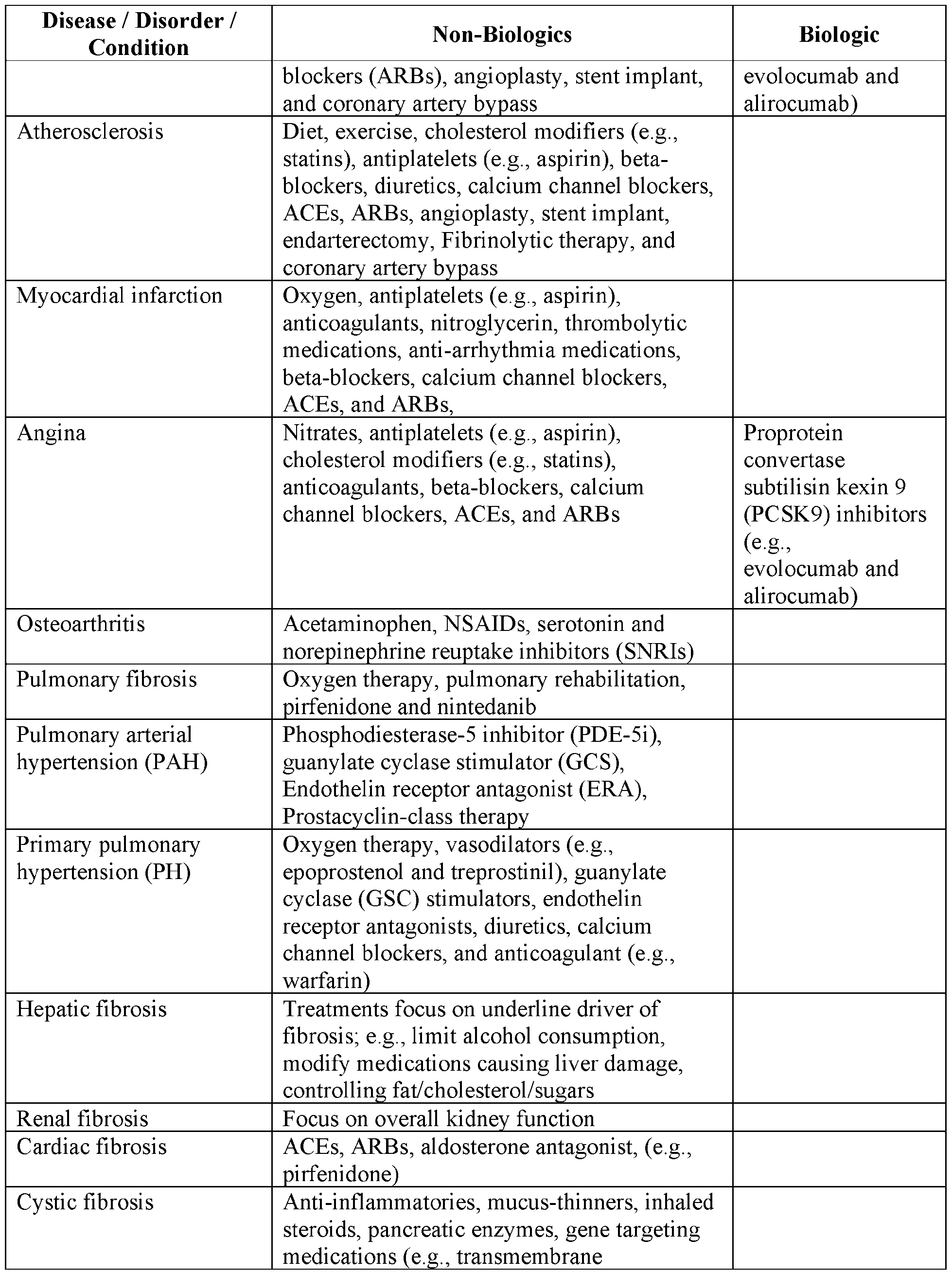




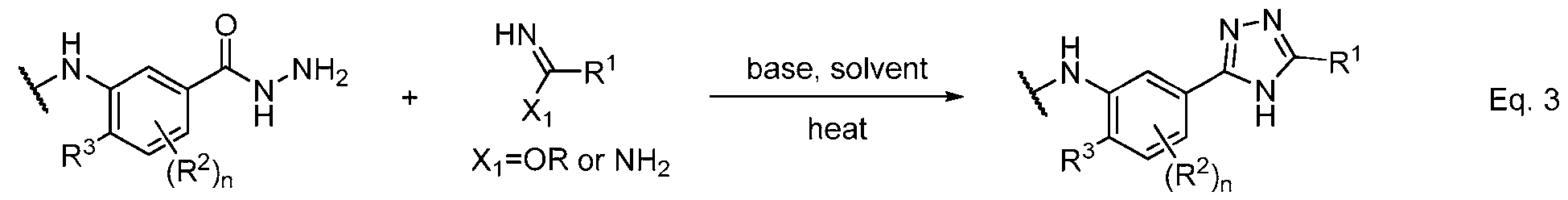

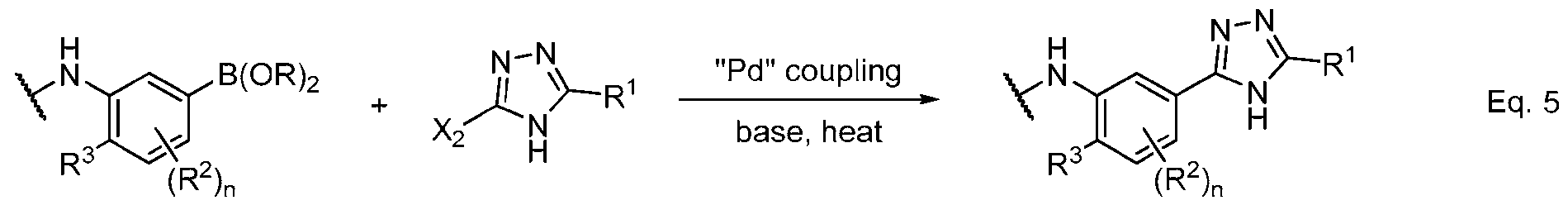
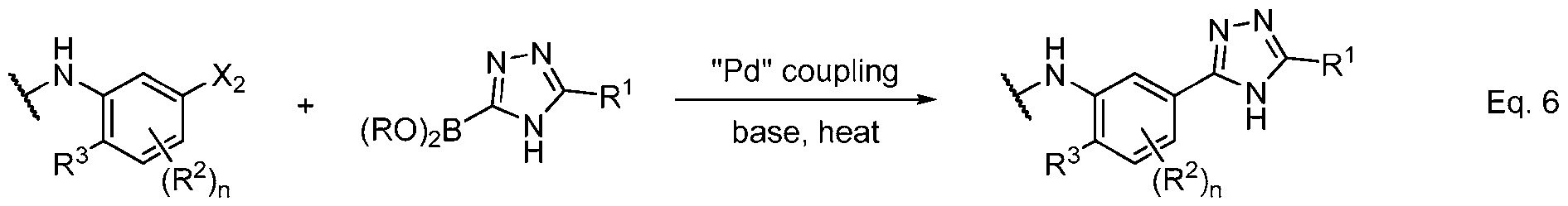
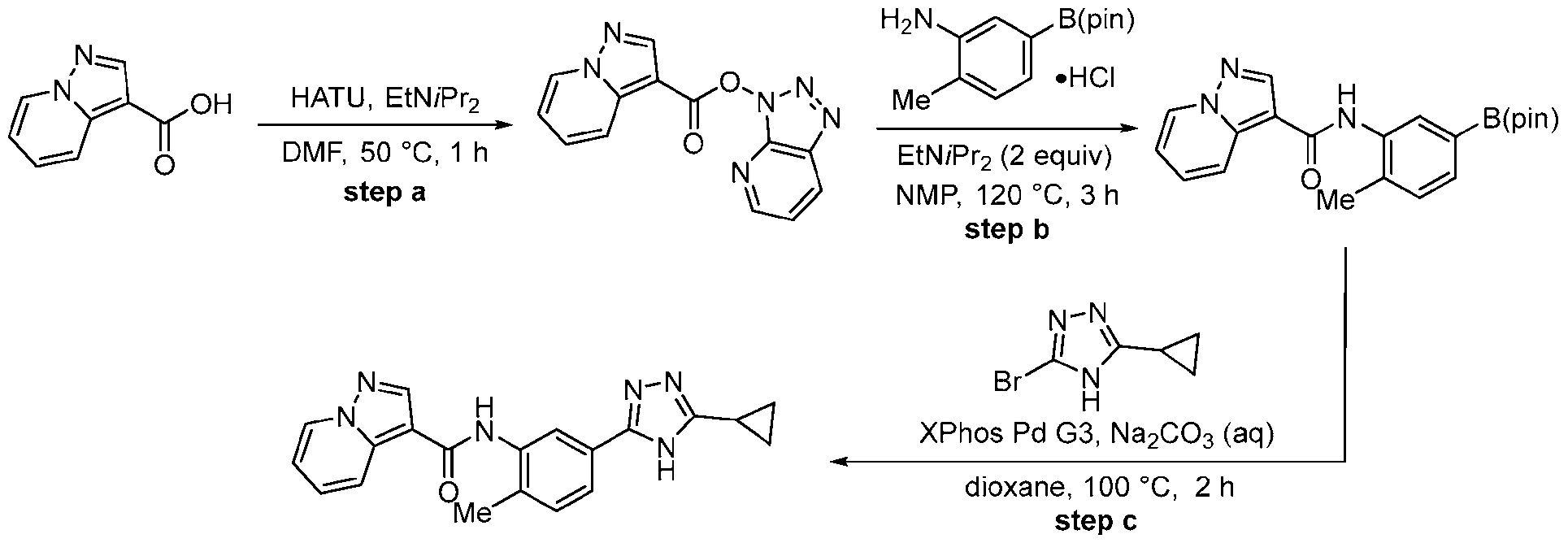








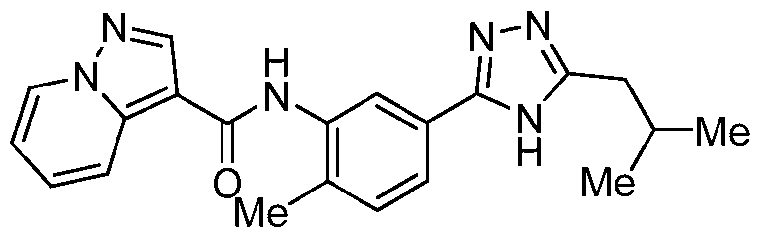






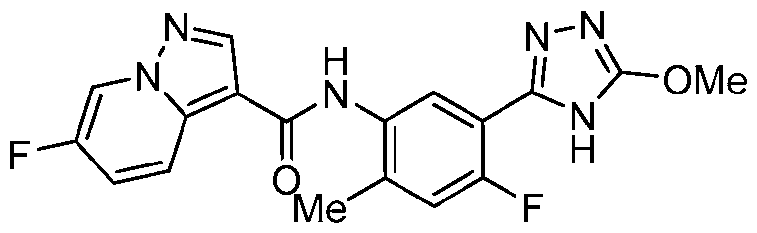

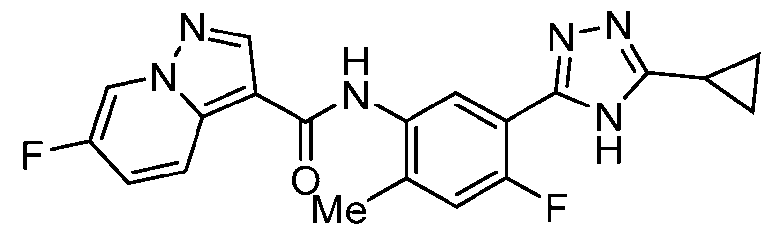
















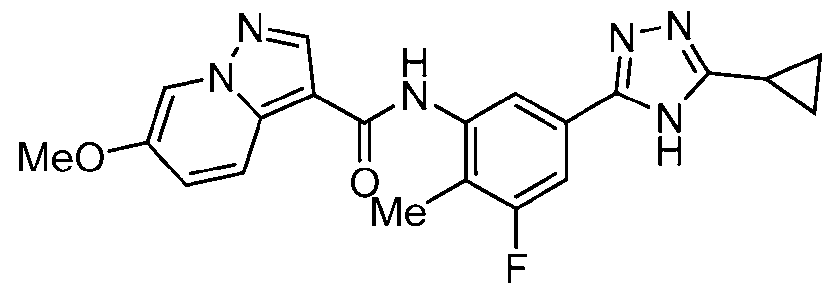















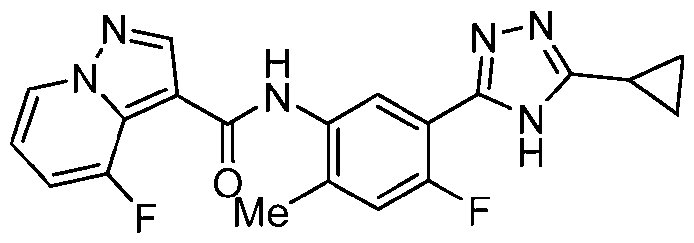


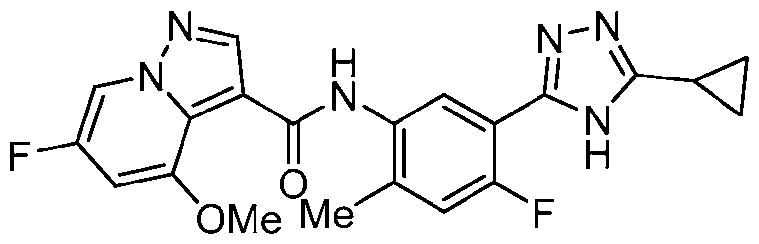
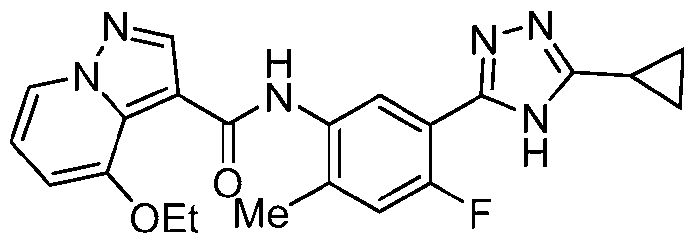
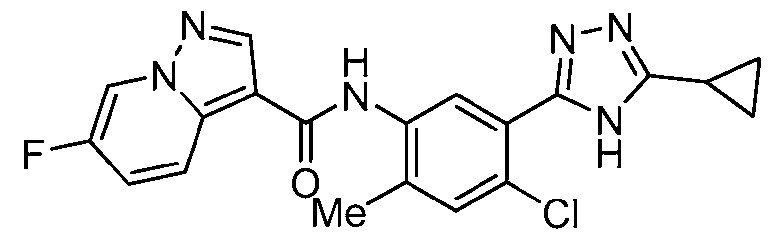



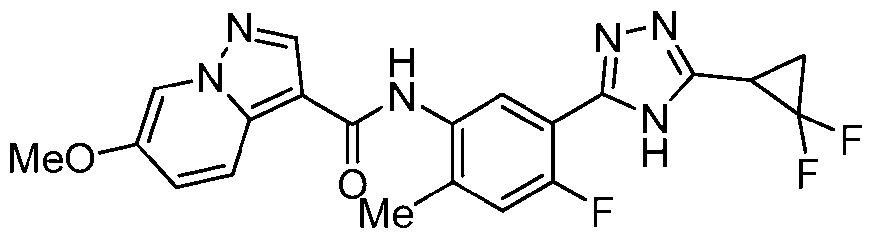
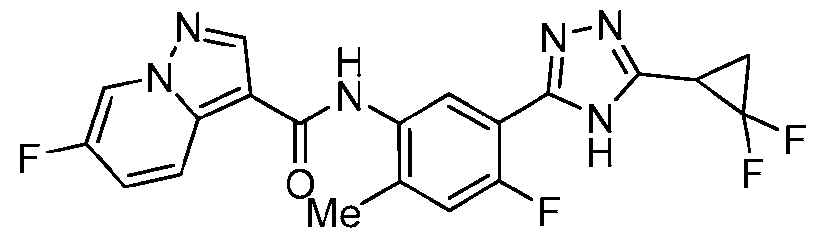
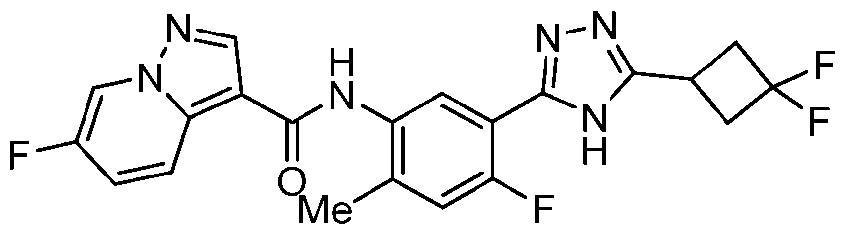


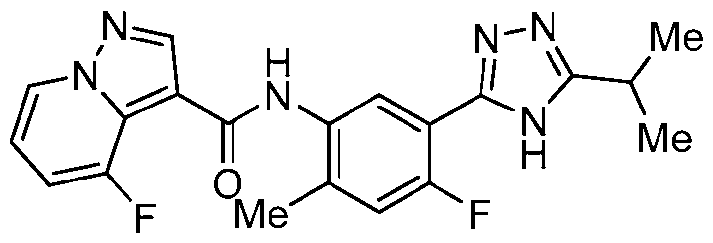
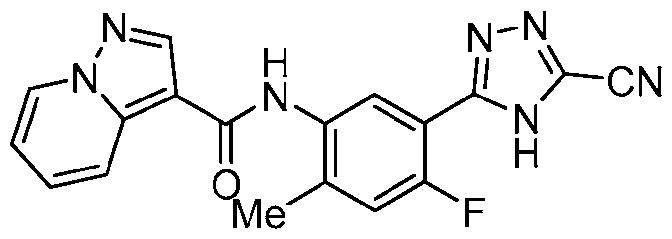




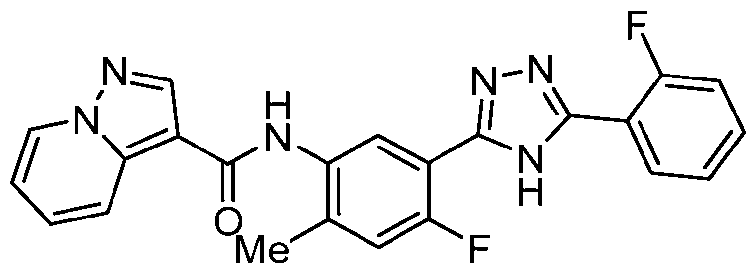







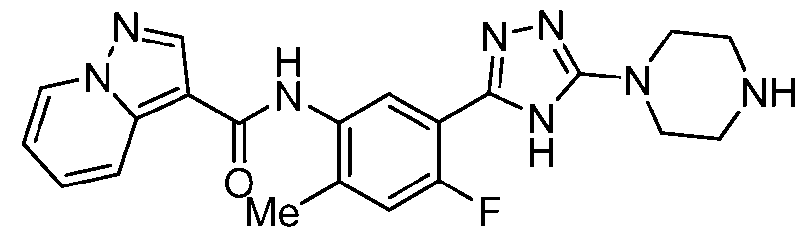



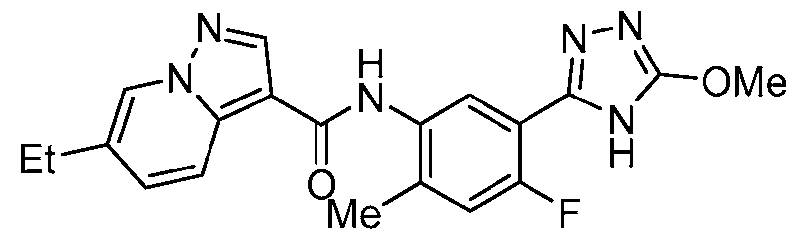

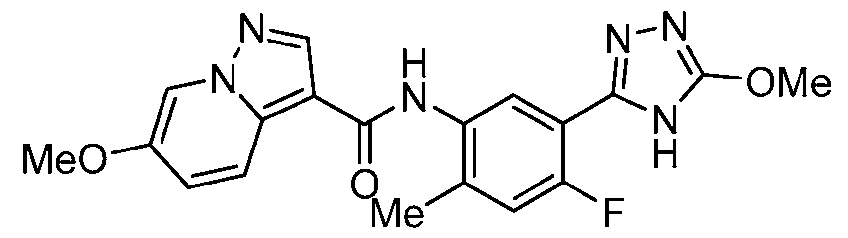


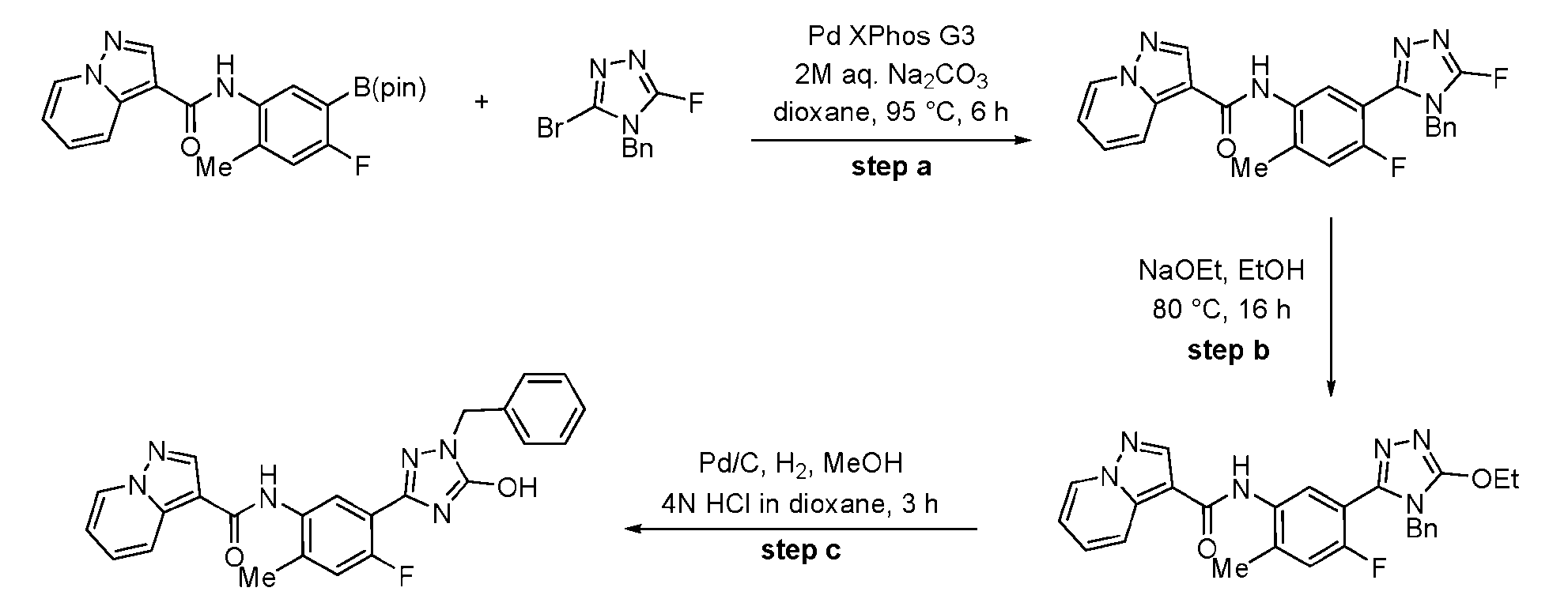

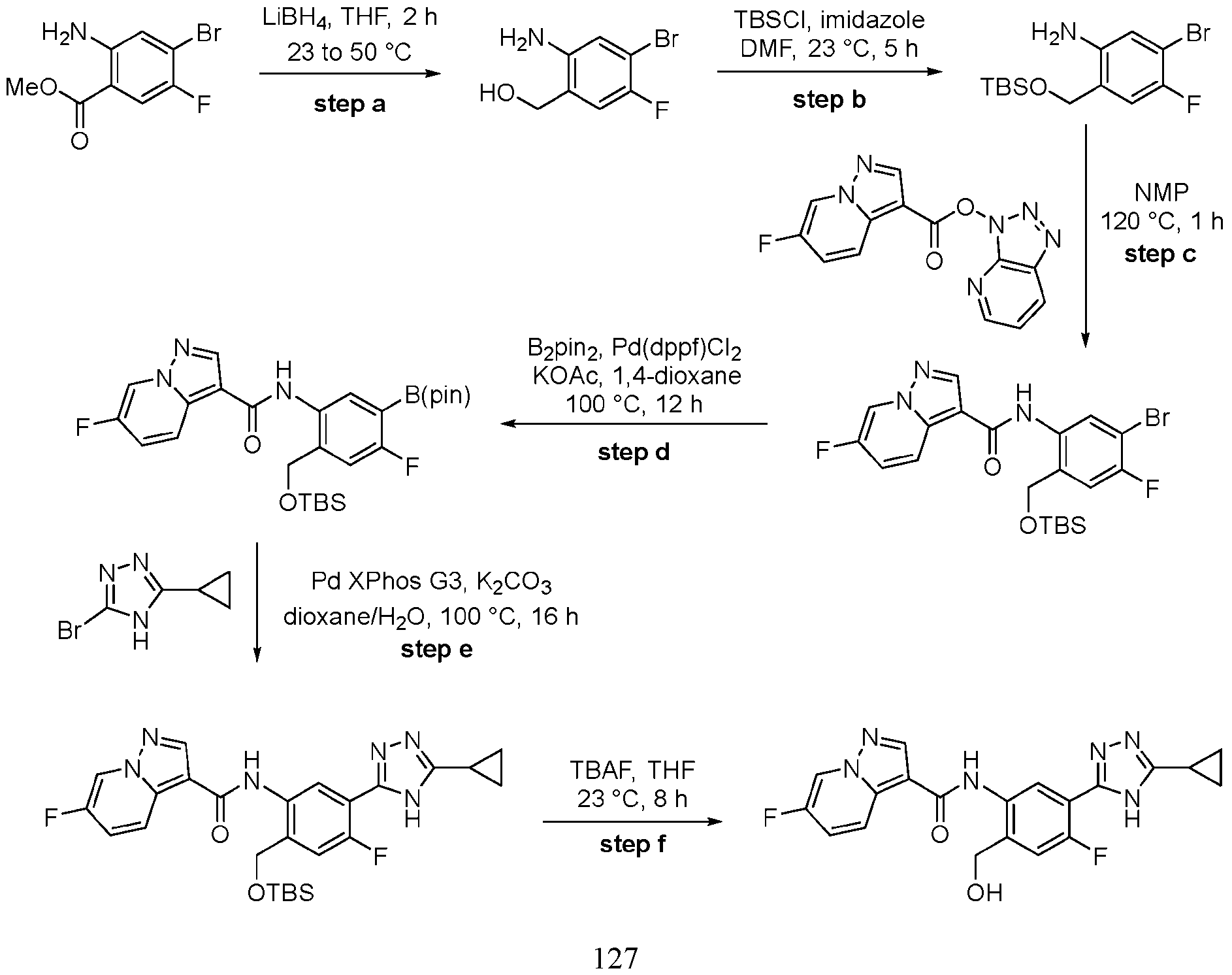

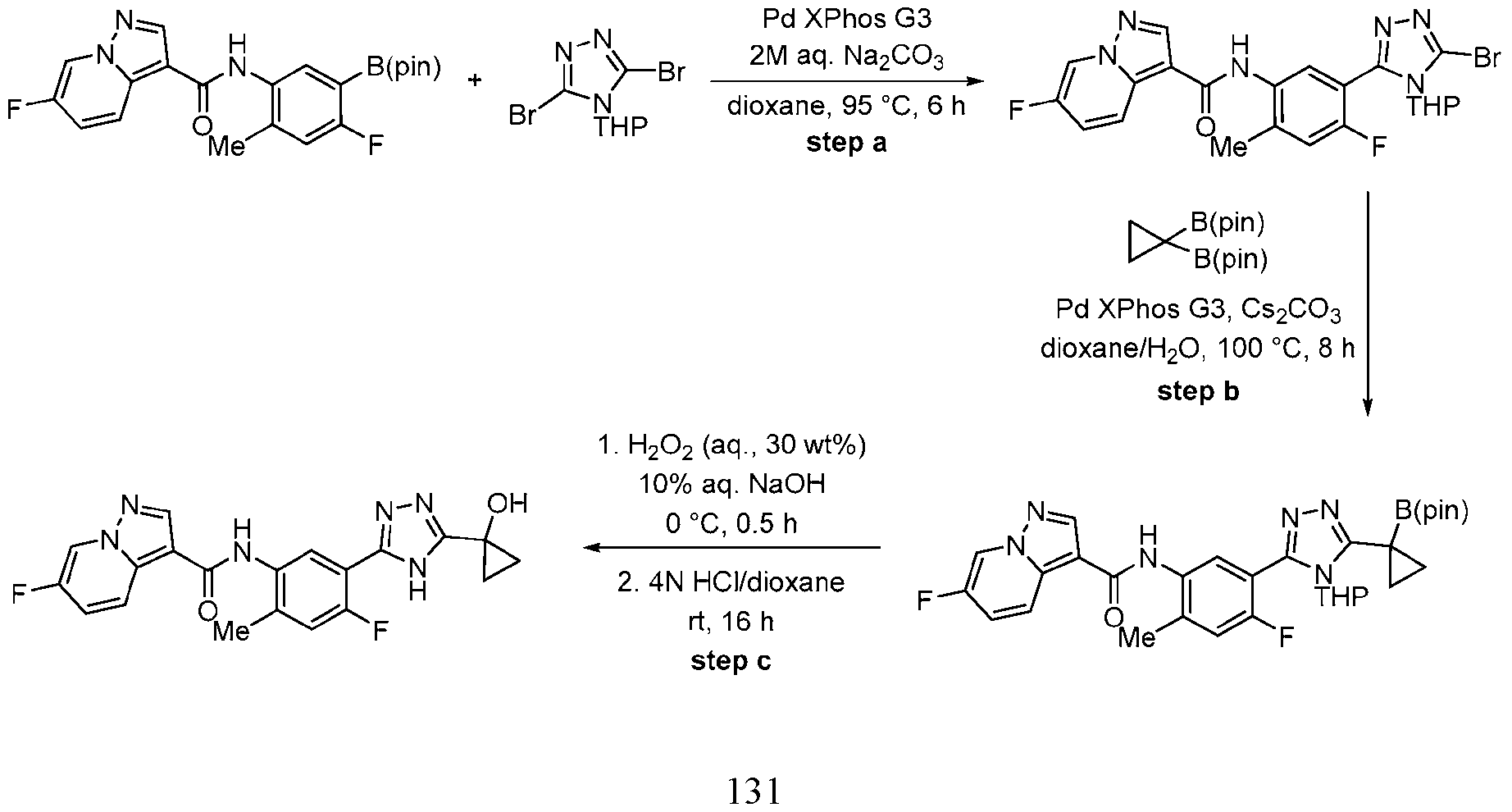














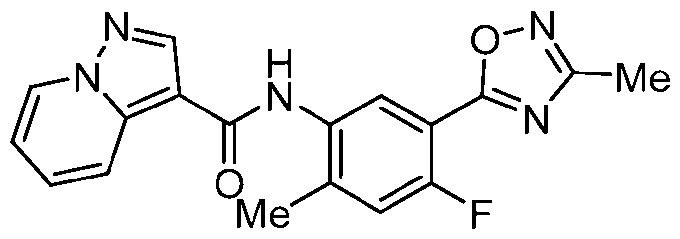

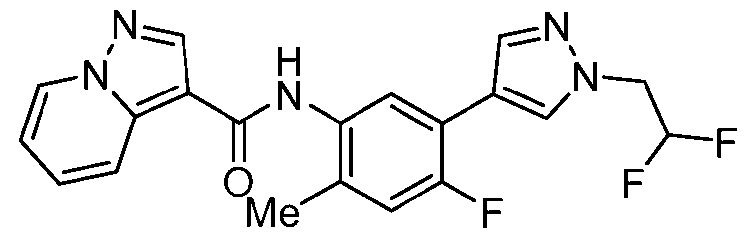





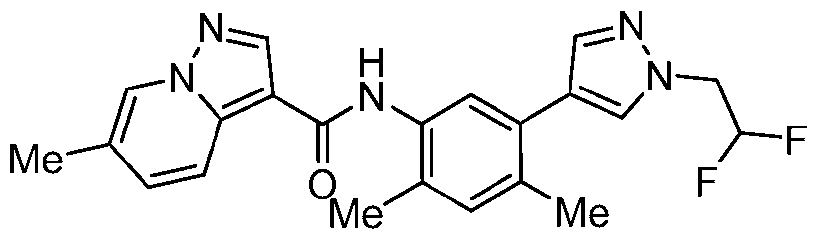

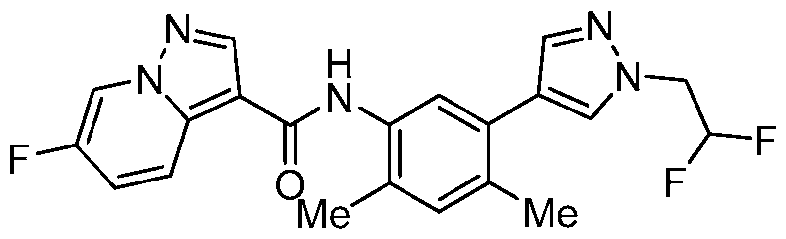
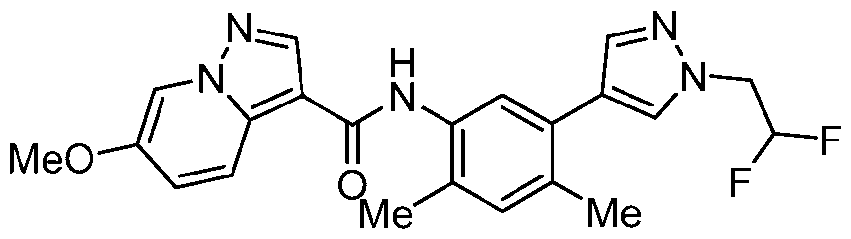




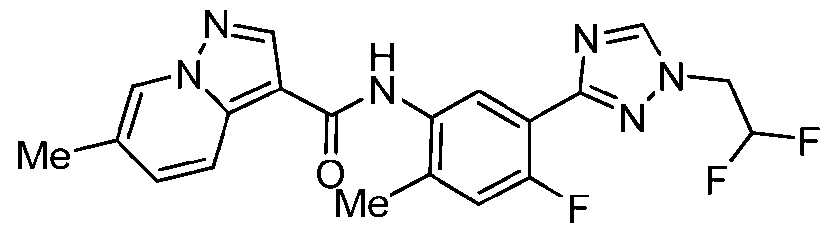
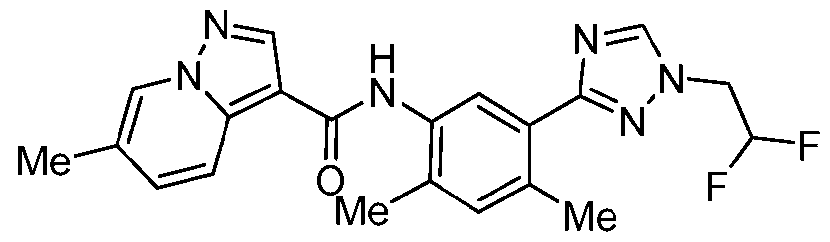



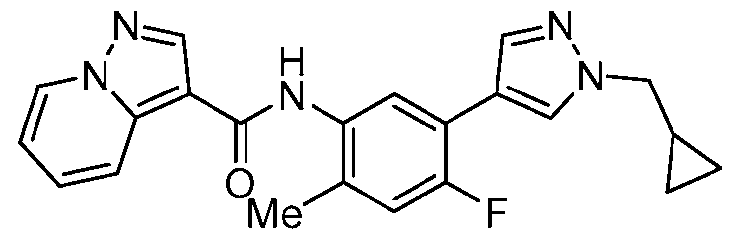

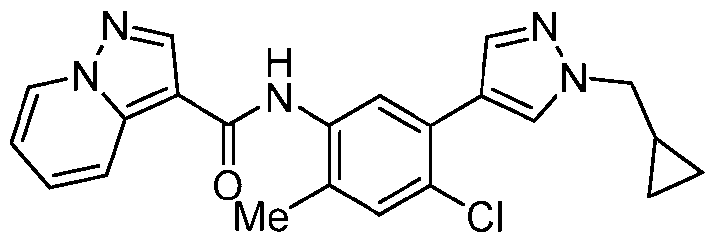




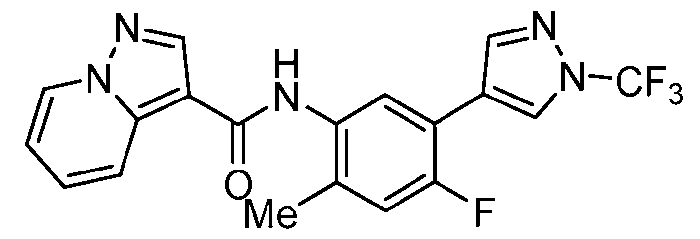



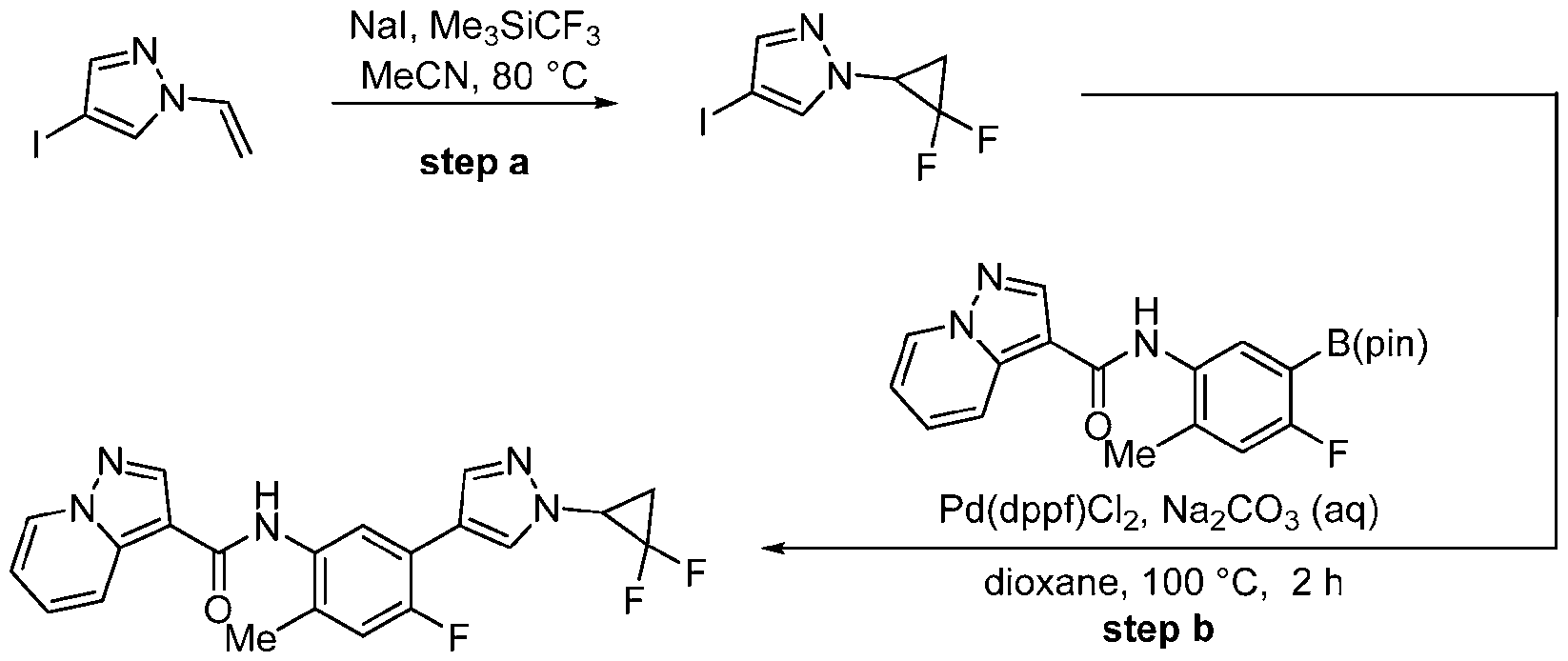








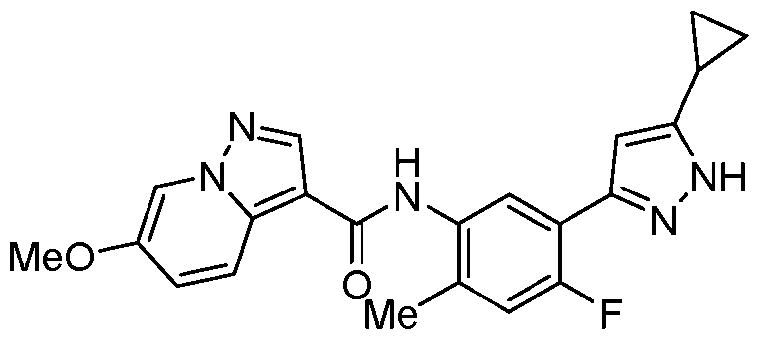

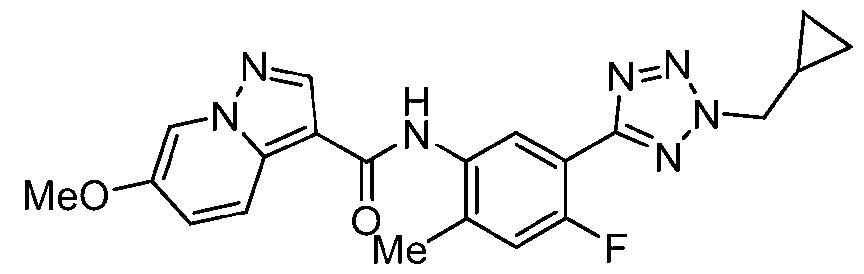









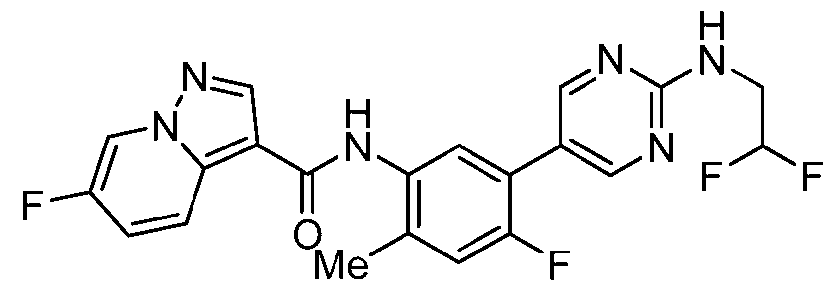
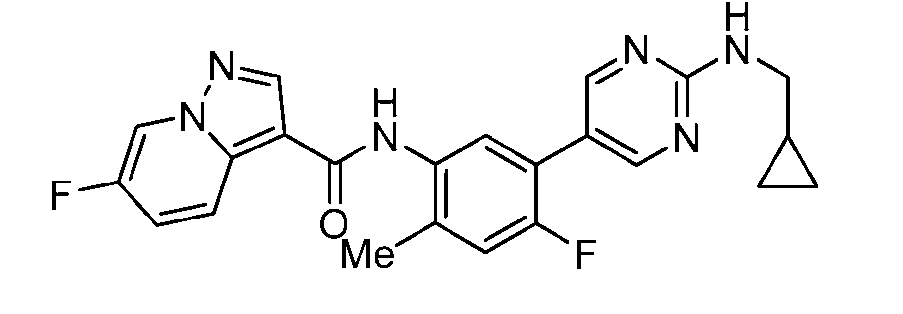







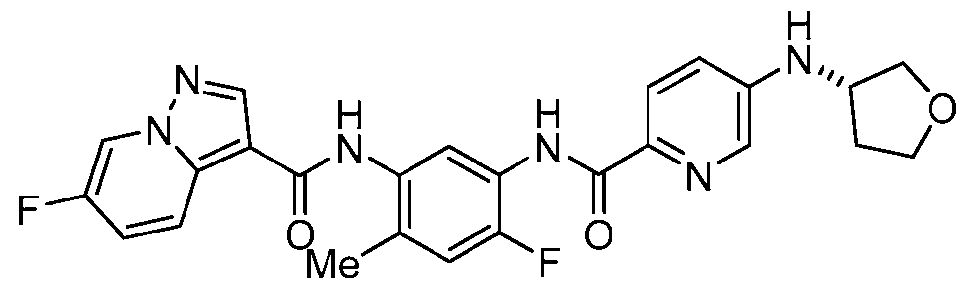
[0433] Step a: A mixture of triazolo[4,5-b]pyridin-3-yl 6-fluoropyrazolo[1,5-a]pyridine-3- carboxylate (298 mg, 1.0 mmol, 1.0 equiv.) and 5-bromo-4-fluoro-2-methylaniline (204 mg, 1.0 mmol, 1.0 equiv.) in NMP (6.0 mL) was stirred at 120 °C for 4 h. The reaction mixture was cooled to rt and diluted with water. The resulting precipitated solid was collected by vacuum filtration, rinsed with water, and dried under vacuum to afford N-(5-bromo-4-fluoro-2-methylphenyl)-6- fluoropyrazolo[1,5-a]pyridine-3-carboxamide, which was used directly in the next step without further purification. [0434] Step b: A vial was charged with product from step a (260 mg, 0.72 mmol, 1.0 equiv.), diphenylmethanimine (155 mg, 0.85 mmol, 1.2 equiv.), Pd2(dba)3 (66 mg, 0.072 mmol, 0.1 equiv.), XantPhos (84 mg, 0.44 mmol, 0.2 equiv.), Cs2CO3 (705 mg, 2.16 mmol, 3.0 equiv.) and dioxane (8 mL). The reaction mixture was stirred at 100 °C for 16 h under N2, at which time LCMS analysis indicated full consumption of starting material. The reaction mixture was cooled to rt and concentrated under reduced pressure. The crude product was purified by column chromatography (10% to 70% EtOAc in MeOH) to provide the desired N-[5-(benzhydrylideneamino)-4-fluoro-2- methylphenyl]-6-fluoropyrazolo[1,5-a]pyridine-3-carboxamide. [0435] Step c: To a mixture of product from step b (220 mg, 0.47 mmol, 1.0 equiv.), NaOAc (97 mg, 1.18 mmol, 2.5 equiv.) in MeOH (5 mL) was added NH2OH HCl (66 mg, 0.94 mmol, 2.0 equiv.). The reaction mixture was stirred at 45 °C for 3 h, then diluted with EtOAc and NaHCO3 aq. The organic layer was separated, dried over Na2SO4 and concentrated in vacuo. The crude product was purified by column chromatography (10% to 70% EtOAc in hexanes) to provide the desired N-(5-amino-4-fluoro-2-methylphenyl)-6-fluoropyrazolo[1,5-a]pyridine-3-carboxamide. [0436] Step d: The desired product was prepared from 5-fluoropyridine-2-carboxylic acid in a similar manner to Example 1, step a.1H NMR (400 MHz, DMSO-d6) δ 10.25 (s, 1H), 9.77 (s, 1H), 9.16 (ddd, J = 4.6, 2.2, 0.7 Hz, 1H), 8.74 (d, J = 2.7 Hz, 1H), 8.26 – 8.17 (m, 2H), 8.04 – 7.92 (m, 2H), 7.61 (ddd, J = 10.2, 8.2, 2.2 Hz, 1H), 7.26 (d, J = 11.5 Hz, 1H), 6.51 (s, 1H), 2.22 (s, 3H). ESI MS [M+H]+ for C21H15F3N5O2, calcd 426.1, found 426.2. Example 177: 6-fluoro-N-[4-fluoro-2-methyl-5-[(5-methylpyridine-2- carbonyl)amino]phenyl]pyrazolo[1,5-a]pyridine-3-carboxamide
 [0437] The title compound was prepared from 5-methylpyridine-2-carboxylic acid and 6-N-(5- amino-4-fluoro-2-methylphenyl)-6-fluoropyrazolo[1,5-a]pyridine-3-carboxamide in a similar fashion to Ex.176. 1H NMR (400 MHz, DMSO-d6) δ 10.28 (s, 1H), 9.77 (s, 1H), 9.16 (dd, J = 4.7, 2.2 Hz, 1H), 8.74 (s, 1H), 8.56 (s, 1H), 8.21 (dd, J = 9.8, 5.9 Hz, 1H), 8.10 (d, J = 7.7 Hz, 1H), 8.03 (d, J = 7.9 Hz, 1H), 7.91 – 7.83 (m, 1H), 7.61 (ddd, J = 10.1, 8.3, 2.2 Hz, 1H), 7.26 (d, J = 11.6 Hz, 1H), 2.39 (s, 3H), 2.21 (s, 3H). ESI MS [M+H]+ for C22H18F2N5O2, calcd 422.1, found 422.2. Example 178: 6-fluoro-N-[4-fluoro-2-methyl-5-(pyridine-3- carbonylamino)phenyl]pyrazolo[1,5-a]pyridine-3-carboxamide
[0437] The title compound was prepared from 5-methylpyridine-2-carboxylic acid and 6-N-(5- amino-4-fluoro-2-methylphenyl)-6-fluoropyrazolo[1,5-a]pyridine-3-carboxamide in a similar fashion to Ex.176. 1H NMR (400 MHz, DMSO-d6) δ 10.28 (s, 1H), 9.77 (s, 1H), 9.16 (dd, J = 4.7, 2.2 Hz, 1H), 8.74 (s, 1H), 8.56 (s, 1H), 8.21 (dd, J = 9.8, 5.9 Hz, 1H), 8.10 (d, J = 7.7 Hz, 1H), 8.03 (d, J = 7.9 Hz, 1H), 7.91 – 7.83 (m, 1H), 7.61 (ddd, J = 10.1, 8.3, 2.2 Hz, 1H), 7.26 (d, J = 11.6 Hz, 1H), 2.39 (s, 3H), 2.21 (s, 3H). ESI MS [M+H]+ for C22H18F2N5O2, calcd 422.1, found 422.2. Example 178: 6-fluoro-N-[4-fluoro-2-methyl-5-(pyridine-3- carbonylamino)phenyl]pyrazolo[1,5-a]pyridine-3-carboxamide
 [0438] The title compound was prepared from pyridine-2-carboxylic acid and 6-N-(5-amino-4- fluoro-2-methylphenyl)-6-fluoropyrazolo[1,5-a]pyridine-3-carboxamide in a similar fashion to Ex.176. 1H NMR (400 MHz, Methanol-d4) δ 9.13 – 9.07 (m, 1H), 8.81 – 8.71 (m, 2H), 8.60 (s, 1H), 8.41 (dt, J = 8.1, 1.9 Hz, 1H), 8.28 (ddd, J = 9.8, 5.6, 0.7 Hz, 1H), 7.81 (d, J = 7.3 Hz, 1H), 7.64 (ddd, J = 8.0, 5.0, 0.9 Hz, 1H), 7.50 (ddt, J = 9.9, 8.0, 2.0 Hz, 1H), 7.18 (d, J = 11.0 Hz, 1H), 2.32 (s, 3H). ESI MS [M+H]+ for C21H16F2N5O2, calcd 408.1, found 408.2. Example 179: N-[5-[(6-cyclopropyloxypyridazine-3-carbonyl)amino]-4-fluoro-2- methylphenyl]-6-fluoropyrazolo[1,5-a]pyridine-3-carboxamide
[0439] The title compound was prepared from 6-cyclopropyloxypyridazine-3-carboxylic acid and 6-N-(5-amino-4-fluoro-2-methylphenyl)-6-fluoropyrazolo[1,5-a]pyridine-3-carboxamide in a similar fashion to Ex.176. 1H NMR (400 MHz, DMSO-d6) δ 10.50 (s, 1H), 9.76 (s, 1H), 9.16 (dd, J = 4.6, 2.2 Hz, 1H), 8.74 (s, 1H), 8.25 – 8.18 (m, 2H), 7.88 (d, J = 7.5 Hz, 1H), 7.61 (ddd, J = 10.1, 8.2, 2.3 Hz, 1H), 7.45 (d, J = 9.2 Hz, 1H), 7.26 (d, J = 11.3 Hz, 1H), 4.47 (m, 1H), 2.23 (s, 3H), 0.92 – 0.74 (m, 4H). ESI MS [M+H]+ for C23H19F2N6O3, calcd 465.1, found 465.2. Example 180: N-[5-[(2,2-difluorocyclopropanecarbonyl)amino]-4-fluoro-2-methylphenyl]- 6-fluoropyrazolo[1,5-a]pyridine-3-carboxamide
[0438] The title compound was prepared from pyridine-2-carboxylic acid and 6-N-(5-amino-4- fluoro-2-methylphenyl)-6-fluoropyrazolo[1,5-a]pyridine-3-carboxamide in a similar fashion to Ex.176. 1H NMR (400 MHz, Methanol-d4) δ 9.13 – 9.07 (m, 1H), 8.81 – 8.71 (m, 2H), 8.60 (s, 1H), 8.41 (dt, J = 8.1, 1.9 Hz, 1H), 8.28 (ddd, J = 9.8, 5.6, 0.7 Hz, 1H), 7.81 (d, J = 7.3 Hz, 1H), 7.64 (ddd, J = 8.0, 5.0, 0.9 Hz, 1H), 7.50 (ddt, J = 9.9, 8.0, 2.0 Hz, 1H), 7.18 (d, J = 11.0 Hz, 1H), 2.32 (s, 3H). ESI MS [M+H]+ for C21H16F2N5O2, calcd 408.1, found 408.2. Example 179: N-[5-[(6-cyclopropyloxypyridazine-3-carbonyl)amino]-4-fluoro-2- methylphenyl]-6-fluoropyrazolo[1,5-a]pyridine-3-carboxamide
[0439] The title compound was prepared from 6-cyclopropyloxypyridazine-3-carboxylic acid and 6-N-(5-amino-4-fluoro-2-methylphenyl)-6-fluoropyrazolo[1,5-a]pyridine-3-carboxamide in a similar fashion to Ex.176. 1H NMR (400 MHz, DMSO-d6) δ 10.50 (s, 1H), 9.76 (s, 1H), 9.16 (dd, J = 4.6, 2.2 Hz, 1H), 8.74 (s, 1H), 8.25 – 8.18 (m, 2H), 7.88 (d, J = 7.5 Hz, 1H), 7.61 (ddd, J = 10.1, 8.2, 2.3 Hz, 1H), 7.45 (d, J = 9.2 Hz, 1H), 7.26 (d, J = 11.3 Hz, 1H), 4.47 (m, 1H), 2.23 (s, 3H), 0.92 – 0.74 (m, 4H). ESI MS [M+H]+ for C23H19F2N6O3, calcd 465.1, found 465.2. Example 180: N-[5-[(2,2-difluorocyclopropanecarbonyl)amino]-4-fluoro-2-methylphenyl]- 6-fluoropyrazolo[1,5-a]pyridine-3-carboxamide
 [0440] The title compound was prepared from 2,2-difluorocyclopropane-1-carboxylic acid and 6-N-(5-amino-4-fluoro-2-methylphenyl)-6-fluoropyrazolo[1,5-a]pyridine-3-carboxamide in a similar fashion to Ex. 176. 1H NMR (400 MHz, DMSO-d6) δ 10.20 (s, 1H), 9.70 (s, 1H), 9.15 (ddd, J = 4.6, 2.3, 0.8 Hz, 1H), 8.71 (s, 1H), 8.19 (ddd, J = 9.8, 5.9, 0.8 Hz, 1H), 7.86 (d, J = 7.7 Hz, 1H), 7.60 (ddd, J = 9.8, 8.2, 2.3 Hz, 1H), 7.19 (d, J = 11.6 Hz, 1H), 3.03 – 2.90 (m, 1H), 2.18 (s, 3H), 2.01 – 1.88 (m, 2H). ESI MS [M+H]+ for C19H15F4N4O2, calcd 407.1, found 407.2. Example 181: N-[5-(cyclopropanecarbonylamino)-4-fluoro-2-methylphenyl]-6- fluoropyrazolo[1,5-a]pyridine-3-carboxamide
[0440] The title compound was prepared from 2,2-difluorocyclopropane-1-carboxylic acid and 6-N-(5-amino-4-fluoro-2-methylphenyl)-6-fluoropyrazolo[1,5-a]pyridine-3-carboxamide in a similar fashion to Ex. 176. 1H NMR (400 MHz, DMSO-d6) δ 10.20 (s, 1H), 9.70 (s, 1H), 9.15 (ddd, J = 4.6, 2.3, 0.8 Hz, 1H), 8.71 (s, 1H), 8.19 (ddd, J = 9.8, 5.9, 0.8 Hz, 1H), 7.86 (d, J = 7.7 Hz, 1H), 7.60 (ddd, J = 9.8, 8.2, 2.3 Hz, 1H), 7.19 (d, J = 11.6 Hz, 1H), 3.03 – 2.90 (m, 1H), 2.18 (s, 3H), 2.01 – 1.88 (m, 2H). ESI MS [M+H]+ for C19H15F4N4O2, calcd 407.1, found 407.2. Example 181: N-[5-(cyclopropanecarbonylamino)-4-fluoro-2-methylphenyl]-6- fluoropyrazolo[1,5-a]pyridine-3-carboxamide
 [0441] The title compound was prepared from cyclopropanecarboxylic acid and 6-N-(5-amino- 4-fluoro-2-methylphenyl)-6-fluoropyrazolo[1,5-a]pyridine-3-carboxamide in a similar fashion to Ex.176. 1H NMR (400 MHz, DMSO-d6) δ 9.92 (s, 1H), 9.67 (s, 1H), 9.15 (ddd, J = 4.7, 2.3, 0.8 Hz, 1H), 8.71 (s, 1H), 8.19 (ddd, J = 9.8, 5.9, 0.8 Hz, 1H), 7.84 (d, J = 7.7 Hz, 1H), 7.59 (ddd, J = 9.8, 8.2, 2.3 Hz, 1H), 7.15 (d, J = 11.6 Hz, 1H), 2.16 (s, 3H), 1.99 – 1.89 (m, 1H), 0.75 (d, J = 6.1 Hz, 4H). ESI MS [M+H]+ for C19H17F2N4O2, calcd 371.1, found 371.2.
Example 182: N-[5-[(2-cyclopropylacetyl)amino]-4-fluoro-2-methylphenyl]-6- fluoropyrazolo[1,5-a]pyridine-3-carboxamide
[0441] The title compound was prepared from cyclopropanecarboxylic acid and 6-N-(5-amino- 4-fluoro-2-methylphenyl)-6-fluoropyrazolo[1,5-a]pyridine-3-carboxamide in a similar fashion to Ex.176. 1H NMR (400 MHz, DMSO-d6) δ 9.92 (s, 1H), 9.67 (s, 1H), 9.15 (ddd, J = 4.7, 2.3, 0.8 Hz, 1H), 8.71 (s, 1H), 8.19 (ddd, J = 9.8, 5.9, 0.8 Hz, 1H), 7.84 (d, J = 7.7 Hz, 1H), 7.59 (ddd, J = 9.8, 8.2, 2.3 Hz, 1H), 7.15 (d, J = 11.6 Hz, 1H), 2.16 (s, 3H), 1.99 – 1.89 (m, 1H), 0.75 (d, J = 6.1 Hz, 4H). ESI MS [M+H]+ for C19H17F2N4O2, calcd 371.1, found 371.2.
Example 182: N-[5-[(2-cyclopropylacetyl)amino]-4-fluoro-2-methylphenyl]-6- fluoropyrazolo[1,5-a]pyridine-3-carboxamide
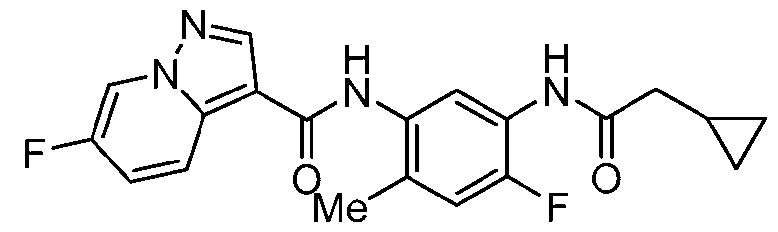 [0442] The title compound was prepared from 2-cyclopropyl acetic acid and 6-N-(5-amino-4- fluoro-2-methylphenyl)-6-fluoropyrazolo[1,5-a]pyridine-3-carboxamide in a similar fashion to Ex.176. 1H NMR (400 MHz, DMSO-d6) δ 9.69 (s, 1H), 9.53 (s, 1H), 9.15 (ddd, J = 4.6, 2.3, 0.8 Hz, 1H), 8.72 (s, 1H), 8.20 (ddd, J = 9.8, 6.0, 0.8 Hz, 1H), 7.83 (d, J = 7.7 Hz, 1H), 7.60 (ddd, J = 9.8, 8.3, 2.3 Hz, 1H), 7.15 (d, J = 11.6 Hz, 1H), 2.23 (d, J = 7.0 Hz, 2H), 2.17 (s, 3H), 1.08 – 0.93 (m, 1H), 0.48 – 0.37 (m, 2H), 0.23 – 0.12 (m, 2H). ESI MS [M+H]+ for C20H19F2N4O2, calcd 385.1, found 385.2. Example 183: N-[5-(cyclopropanecarbonylamino)-4-fluoro-2-methylphenyl]pyrazolo[1,5- a]pyridine-3-carboxamide
[0442] The title compound was prepared from 2-cyclopropyl acetic acid and 6-N-(5-amino-4- fluoro-2-methylphenyl)-6-fluoropyrazolo[1,5-a]pyridine-3-carboxamide in a similar fashion to Ex.176. 1H NMR (400 MHz, DMSO-d6) δ 9.69 (s, 1H), 9.53 (s, 1H), 9.15 (ddd, J = 4.6, 2.3, 0.8 Hz, 1H), 8.72 (s, 1H), 8.20 (ddd, J = 9.8, 6.0, 0.8 Hz, 1H), 7.83 (d, J = 7.7 Hz, 1H), 7.60 (ddd, J = 9.8, 8.3, 2.3 Hz, 1H), 7.15 (d, J = 11.6 Hz, 1H), 2.23 (d, J = 7.0 Hz, 2H), 2.17 (s, 3H), 1.08 – 0.93 (m, 1H), 0.48 – 0.37 (m, 2H), 0.23 – 0.12 (m, 2H). ESI MS [M+H]+ for C20H19F2N4O2, calcd 385.1, found 385.2. Example 183: N-[5-(cyclopropanecarbonylamino)-4-fluoro-2-methylphenyl]pyrazolo[1,5- a]pyridine-3-carboxamide
 [0443] The title compound was prepared from cyclopropanecarboxylic acid and triazolo[4,5- b]pyridin-3-yl pyrazolo[1,5-a]pyridine-3-carboxylate in a similar fashion to Ex. 176. 1H NMR (400 MHz, DMSO-d6) δ 9.92 (s, 1H), 9.61 (s, 1H), 8.79 (dt, J = 7.0, 1.1 Hz, 1H), 8.69 (s, 1H), 8.16 (dt, J = 8.9, 1.3 Hz, 1H), 7.84 (d, J = 7.7 Hz, 1H), 7.47 (ddd, J = 8.9, 6.8, 1.1 Hz, 1H), 7.15 (d, J = 11.8 Hz, 1H), 7.07 (td, J = 6.9, 1.4 Hz, 1H), 2.17 (s, 3H), 1.94 (q, J = 6.2 Hz, 1H), 0.75 (d, J = 6.2 Hz, 4H). ESI MS [M+H]+ for C19H18FN4O2, calcd 353.1, found 353.2. Biological Example Inhibition of c-Kit, PDGFRα, PDGFRβ, CSF1R and FLT3 kinase activity [0444] Compounds were evaluated to determine the potency with which they inhibit the kinase activity of the following panel of tyrosine kinases: c-Kit (SignalChem, Product #: K06-11BG), PDGFRα (SignalChem, Product #: P12-18G), PDGFRβ (SignalChem, Product #: P13-11G),
CSF1R (SignalChem, Product #: C74-11G) and FLT3 (Invitrogen, Product #: PR4666C). Activity was determined as a function of phosphorylated biotinylated TK peptide generated by the transfer of phosphate from ATP as measured by the use of the HTRF KinEASE-TK assay kit (Cisbio, Product #: 62TK0PEJ). Levels of phosphorylated biotinylated TK peptide (Part 61TK0BLC of KinEASE assay kit) are quantified by its capture by phosphor-TK-Antibody-Cryptate (Part of KinEASE assay kit) and XL665-labeled Streptavidin (Part 610SAXLG of KinEASE assay kit) followed by measurement of Time-Resolved Fluorescence Resonance Energy Transfer (TR- FRET) signal. On the day of the assay, compounds were solubilized in DMSO and dispensed into a 384-well white Opti-plate (PerkinElmer, catalog # 6007290) to generate a 22 point 1:2 titration. Enzyme solution was prepared for each of the tyrosine kinases in 50 mM HEPES, pH 7.4, 5 mM MgCl2 for c-Kit or 10 mM MgCl2 for FLT3, CSF1R, PDGFRα and PDGFRβ, 2 mM MnCl2, 0.01% Brij-35 and 0.01% BSA. Working enzyme concentrations were prepared for c-Kit at 2 nM (2x), PDGFRα and PDGFRβ at 10 nM (2x), CSF1R at 2 nM (2x) and FLT3 at 0.4 nM (2x). Five microliters of enzyme dilution of each tyrosine kinase were added to their respective 384-well white Opti-plate pre-dispensed with compound and allowed to incubate for 1 h at rt. Utilizing the same enzyme buffer recipe, 2x substrate mixes were prepared for each enzyme as follows: For c- Kit, 1.6 μM (2x) TK substrate and 16 µM (2x) ATP (Promega, catalog # V915). For PDGFRα, 3.2 μM TK substrate and 1 μM ATP. For PDGFRβ, 3.2 μM TK substrate and 40 μM ATP. For CSF1R, 3.2 μM TK substrate and 20 μM ATP. For FLT3, 3.2 μM TK substrate and 100 μM ATP. Reactions were initiated by addition of 5 µL of the respective 2x substrate mix to each well of the plates containing the respective tyrosine kinase enzymes and were allowed to proceed for 120 minutes at rt, except for FLT3, the reaction time is 45 min. Following the reaction, 10 μL of detection mix consisting of 0.2 μM (2x) of Streptavidin-XL665 and 2 μM (2x) of TK-Antibody-Cryptate prepared in detection buffer (Part 62SDBRDF of KinEASE assay kit) was added and then allowed to incubate for 60 min. The TR-FRET signal was quantified by measuring the ratio of emission at 665 nm to 620 nm after excitation at 320 nm by reading on a PerkinElmer Envision multimode reader. Compound potencies (IC50 values) were determined using a standard 4-parameter non- linear regression fit.
Phospho-KIT (Y703) Cellular Assay in M07e Cells [0445] The day prior to assay, M07e cells (DSMZ, catalog # ACC 104) were serum starved. Cells were centrifuged and cell pellet resuspended in OptiMEM (Gibco, catalog # 31985062) to a density of 1x106 - 2.5x106 cells per mL. Cells were then incubated overnight at 5% CO2 and 37 °C in an appropriately sized flask. On the day of the experiment, an 11 point, half log titration of test compound was pre-dispensed into 96-well round bottom polypropylene plates (Corning, catalog # 3365). The serum starved M07e cells were centrifuged and resuspended to a cell density of 1x106 cells per mL with DPBS (Gibco, catalog # 14190-144). Live/Dead Green Fixable viability dye (Invitrogen, catalog # L34970) was added as 1 µL/ 1 mL of cell suspension. Cells were then incubated in the dark at 4 °C for 30 min. Cells were washed by centrifugation and resuspension with DPBS + 0.5% BSA (Gemini Bio Products, catalog # 700110/100, 30% BSA solution). Cells were resuspended, separated into two equal volumes and again centrifuged. Cell pellets were resuspended with either OptiMEM or human serum (Innovative Research, catalog # ISERAB1000ML) to a cell density of 1x106 cells per mL. Cells were then added to the appropriate compound plate at 100,000 cells per well in 100 μL and mixed. Plates were incubated at 5% CO2 and 37 °C for 50 min. Stem Cell Factor, SCF (R&D Systems, #255-SC/CF) was diluted in OptiMEM to 1 µg/mL and added to all wells as 10 µL for a final concentration of 90.9 ng/mL. Plates were incubated at 5% CO2 and 37 °C for 10 min. Incubation was stopped by addition of 110 µL 4% paraformaldehyde solution in PBS (Life Technologies, catalog # J19943.K2). Plates were incubated at rt for at least 15 min. Plates were then centrifuged and supernatant removed by flicking. Cells were washed once by resuspension in 200 µL DPBS + 0.5% BSA, centrifuged, and supernatant removed by flicking. Cells were resuspended in 100 µL 1X Permeabilization buffer (Invitrogen, catalog # 88882400) and plates incubated at rt for 30 min. Plates were centrifuged, supernatant removed by flicking, and cells resuspended in 50 µL primary antibody, anti-phospho- c-KIT (Tyr703) (Cell Signaling, catalog # 3073), diluted to 1 µg/mL with permeabilization buffer. Plates were incubated at rt for 1 h. Plates were centrifuged, supernatant removed, and cells resuspended with 200 µL DPBS + 0.5% BSA. Plates were again centrifuged, supernatant removed by flicking, and cells resuspended in 100 µL secondary antibody, goat anti-rabbit AlexaFluor647 (AF647) (Invitrogen, catalog # A21244) diluted to 1 µg/mL with DPBS + 0.5% BSA. Plates were incubated at rt for 30 min. followed by centrifugation, supernatant removal, and cell resuspension with 200 µL DPBS + 0.5% BSA. Cells were washed again by centrifugation, supernatant removal,
and cell resuspension with 200 µL DPBS + 0.5% BSA. Samples were then read by flow cytometry. The Forward:Side scatter plot was gated and that population was gated for live cells based on the Live/Dead Green staining. The geometric mean of AF647 of the live cell population was recorded. Percentage maximum activity in each test well was calculated based on DMSO (100% activity) and positive control treated cell wells (0% activity). The potencies (IC50 values) of test compounds were determined using a standard 4-parameter non-linear regression fit. Table 3: Biochemical and cellular potency of specific examples vs. KIT, PDGFRα, PDGFRβ, CSF1R, and FLT3 (IC50: + means > 1 μM, ++ means 100 nM to 1 μM, +++ means < 100 nM). ND=not determined.
[0443] The title compound was prepared from cyclopropanecarboxylic acid and triazolo[4,5- b]pyridin-3-yl pyrazolo[1,5-a]pyridine-3-carboxylate in a similar fashion to Ex. 176. 1H NMR (400 MHz, DMSO-d6) δ 9.92 (s, 1H), 9.61 (s, 1H), 8.79 (dt, J = 7.0, 1.1 Hz, 1H), 8.69 (s, 1H), 8.16 (dt, J = 8.9, 1.3 Hz, 1H), 7.84 (d, J = 7.7 Hz, 1H), 7.47 (ddd, J = 8.9, 6.8, 1.1 Hz, 1H), 7.15 (d, J = 11.8 Hz, 1H), 7.07 (td, J = 6.9, 1.4 Hz, 1H), 2.17 (s, 3H), 1.94 (q, J = 6.2 Hz, 1H), 0.75 (d, J = 6.2 Hz, 4H). ESI MS [M+H]+ for C19H18FN4O2, calcd 353.1, found 353.2. Biological Example Inhibition of c-Kit, PDGFRα, PDGFRβ, CSF1R and FLT3 kinase activity [0444] Compounds were evaluated to determine the potency with which they inhibit the kinase activity of the following panel of tyrosine kinases: c-Kit (SignalChem, Product #: K06-11BG), PDGFRα (SignalChem, Product #: P12-18G), PDGFRβ (SignalChem, Product #: P13-11G),
CSF1R (SignalChem, Product #: C74-11G) and FLT3 (Invitrogen, Product #: PR4666C). Activity was determined as a function of phosphorylated biotinylated TK peptide generated by the transfer of phosphate from ATP as measured by the use of the HTRF KinEASE-TK assay kit (Cisbio, Product #: 62TK0PEJ). Levels of phosphorylated biotinylated TK peptide (Part 61TK0BLC of KinEASE assay kit) are quantified by its capture by phosphor-TK-Antibody-Cryptate (Part of KinEASE assay kit) and XL665-labeled Streptavidin (Part 610SAXLG of KinEASE assay kit) followed by measurement of Time-Resolved Fluorescence Resonance Energy Transfer (TR- FRET) signal. On the day of the assay, compounds were solubilized in DMSO and dispensed into a 384-well white Opti-plate (PerkinElmer, catalog # 6007290) to generate a 22 point 1:2 titration. Enzyme solution was prepared for each of the tyrosine kinases in 50 mM HEPES, pH 7.4, 5 mM MgCl2 for c-Kit or 10 mM MgCl2 for FLT3, CSF1R, PDGFRα and PDGFRβ, 2 mM MnCl2, 0.01% Brij-35 and 0.01% BSA. Working enzyme concentrations were prepared for c-Kit at 2 nM (2x), PDGFRα and PDGFRβ at 10 nM (2x), CSF1R at 2 nM (2x) and FLT3 at 0.4 nM (2x). Five microliters of enzyme dilution of each tyrosine kinase were added to their respective 384-well white Opti-plate pre-dispensed with compound and allowed to incubate for 1 h at rt. Utilizing the same enzyme buffer recipe, 2x substrate mixes were prepared for each enzyme as follows: For c- Kit, 1.6 μM (2x) TK substrate and 16 µM (2x) ATP (Promega, catalog # V915). For PDGFRα, 3.2 μM TK substrate and 1 μM ATP. For PDGFRβ, 3.2 μM TK substrate and 40 μM ATP. For CSF1R, 3.2 μM TK substrate and 20 μM ATP. For FLT3, 3.2 μM TK substrate and 100 μM ATP. Reactions were initiated by addition of 5 µL of the respective 2x substrate mix to each well of the plates containing the respective tyrosine kinase enzymes and were allowed to proceed for 120 minutes at rt, except for FLT3, the reaction time is 45 min. Following the reaction, 10 μL of detection mix consisting of 0.2 μM (2x) of Streptavidin-XL665 and 2 μM (2x) of TK-Antibody-Cryptate prepared in detection buffer (Part 62SDBRDF of KinEASE assay kit) was added and then allowed to incubate for 60 min. The TR-FRET signal was quantified by measuring the ratio of emission at 665 nm to 620 nm after excitation at 320 nm by reading on a PerkinElmer Envision multimode reader. Compound potencies (IC50 values) were determined using a standard 4-parameter non- linear regression fit.
Phospho-KIT (Y703) Cellular Assay in M07e Cells [0445] The day prior to assay, M07e cells (DSMZ, catalog # ACC 104) were serum starved. Cells were centrifuged and cell pellet resuspended in OptiMEM (Gibco, catalog # 31985062) to a density of 1x106 - 2.5x106 cells per mL. Cells were then incubated overnight at 5% CO2 and 37 °C in an appropriately sized flask. On the day of the experiment, an 11 point, half log titration of test compound was pre-dispensed into 96-well round bottom polypropylene plates (Corning, catalog # 3365). The serum starved M07e cells were centrifuged and resuspended to a cell density of 1x106 cells per mL with DPBS (Gibco, catalog # 14190-144). Live/Dead Green Fixable viability dye (Invitrogen, catalog # L34970) was added as 1 µL/ 1 mL of cell suspension. Cells were then incubated in the dark at 4 °C for 30 min. Cells were washed by centrifugation and resuspension with DPBS + 0.5% BSA (Gemini Bio Products, catalog # 700110/100, 30% BSA solution). Cells were resuspended, separated into two equal volumes and again centrifuged. Cell pellets were resuspended with either OptiMEM or human serum (Innovative Research, catalog # ISERAB1000ML) to a cell density of 1x106 cells per mL. Cells were then added to the appropriate compound plate at 100,000 cells per well in 100 μL and mixed. Plates were incubated at 5% CO2 and 37 °C for 50 min. Stem Cell Factor, SCF (R&D Systems, #255-SC/CF) was diluted in OptiMEM to 1 µg/mL and added to all wells as 10 µL for a final concentration of 90.9 ng/mL. Plates were incubated at 5% CO2 and 37 °C for 10 min. Incubation was stopped by addition of 110 µL 4% paraformaldehyde solution in PBS (Life Technologies, catalog # J19943.K2). Plates were incubated at rt for at least 15 min. Plates were then centrifuged and supernatant removed by flicking. Cells were washed once by resuspension in 200 µL DPBS + 0.5% BSA, centrifuged, and supernatant removed by flicking. Cells were resuspended in 100 µL 1X Permeabilization buffer (Invitrogen, catalog # 88882400) and plates incubated at rt for 30 min. Plates were centrifuged, supernatant removed by flicking, and cells resuspended in 50 µL primary antibody, anti-phospho- c-KIT (Tyr703) (Cell Signaling, catalog # 3073), diluted to 1 µg/mL with permeabilization buffer. Plates were incubated at rt for 1 h. Plates were centrifuged, supernatant removed, and cells resuspended with 200 µL DPBS + 0.5% BSA. Plates were again centrifuged, supernatant removed by flicking, and cells resuspended in 100 µL secondary antibody, goat anti-rabbit AlexaFluor647 (AF647) (Invitrogen, catalog # A21244) diluted to 1 µg/mL with DPBS + 0.5% BSA. Plates were incubated at rt for 30 min. followed by centrifugation, supernatant removal, and cell resuspension with 200 µL DPBS + 0.5% BSA. Cells were washed again by centrifugation, supernatant removal,
and cell resuspension with 200 µL DPBS + 0.5% BSA. Samples were then read by flow cytometry. The Forward:Side scatter plot was gated and that population was gated for live cells based on the Live/Dead Green staining. The geometric mean of AF647 of the live cell population was recorded. Percentage maximum activity in each test well was calculated based on DMSO (100% activity) and positive control treated cell wells (0% activity). The potencies (IC50 values) of test compounds were determined using a standard 4-parameter non-linear regression fit. Table 3: Biochemical and cellular potency of specific examples vs. KIT, PDGFRα, PDGFRβ, CSF1R, and FLT3 (IC50: + means > 1 μM, ++ means 100 nM to 1 μM, +++ means < 100 nM). ND=not determined.

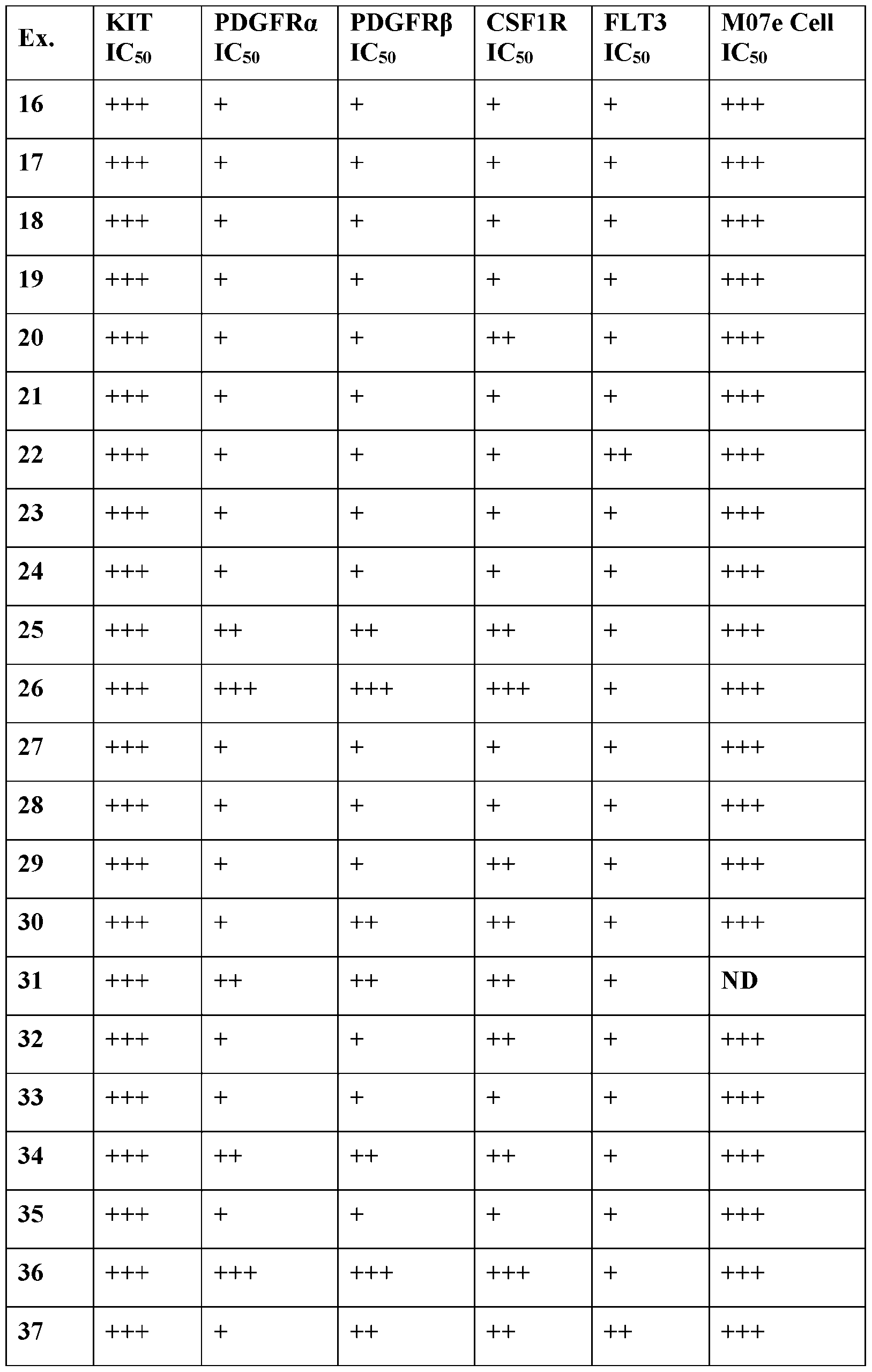


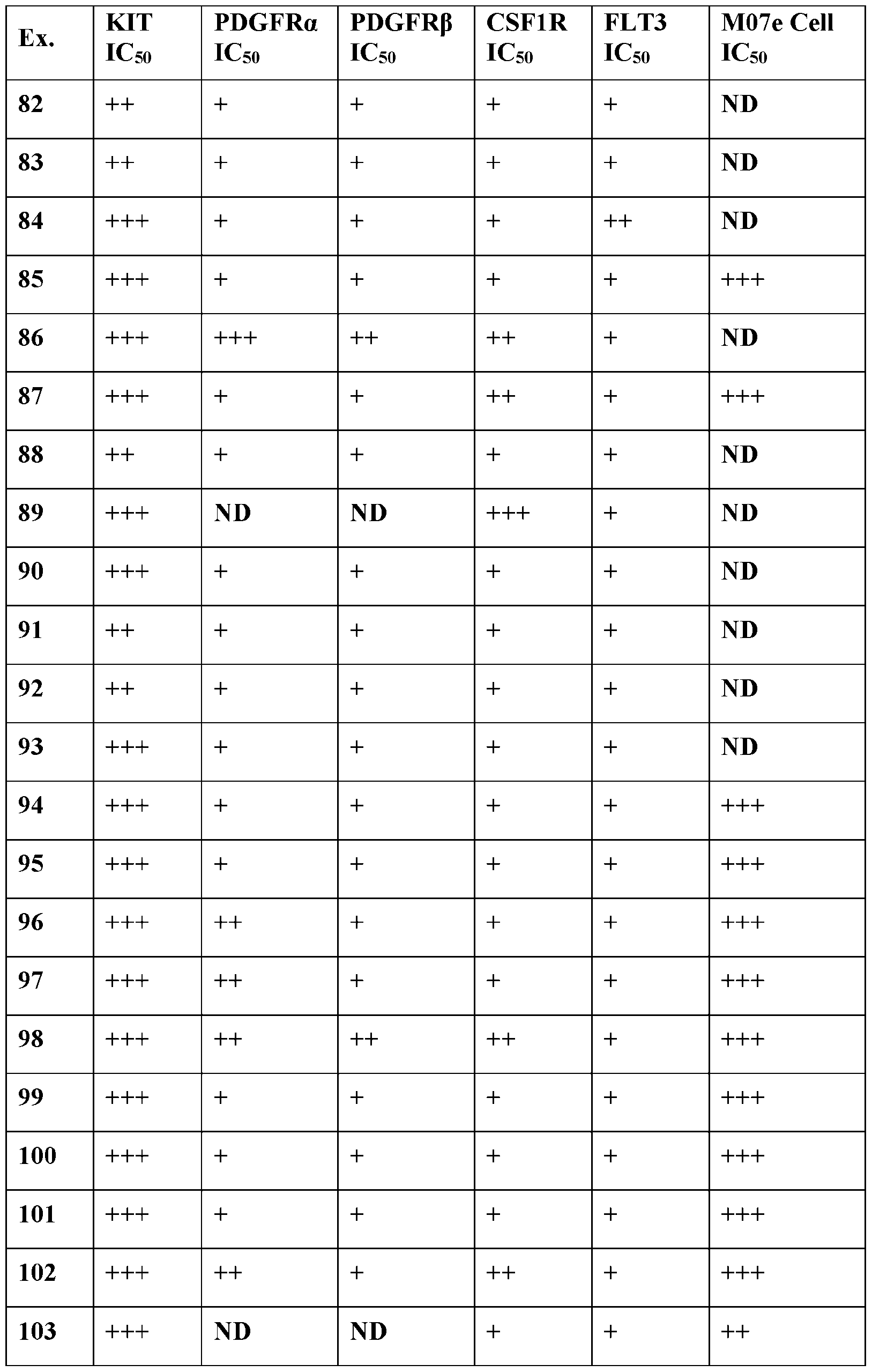 185
185



 [0446] Although the foregoing disclosure has been described in some detail by way of illustration and Example for purposes of clarity of understanding, one of skill in the art will appreciate that certain changes and modifications may be practiced within the scope of the appended claims.
[0446] Although the foregoing disclosure has been described in some detail by way of illustration and Example for purposes of clarity of understanding, one of skill in the art will appreciate that certain changes and modifications may be practiced within the scope of the appended claims.
Claims
CLAIMS What is claimed is: 1. A compound having a structure according to Formula III:
 (Formula III) or a pharmaceutically acceptable salt thereof, wherein: A is: (i) selected from the group consisting of: ,
(Formula III) or a pharmaceutically acceptable salt thereof, wherein: A is: (i) selected from the group consisting of: ,
 , , , , ,
, , , , ,
 r (ii) a 6-membered heteroaryl having 1-3 ring nitrogen atoms, wherein the 6- membered heteroaryl is substituted with 0-2 R1d; or (iii) -NHC(O)-R1g; each R1 is independently selected from the group consisting of halo, -CN, -OH, C1- C6-alkyl, C1-C6-haloalkyl, C1-C6-hydroxyalkyl, C1-C6-cyanoalkyl, C1-C6 alkoxy, C3-C6- cycloalkyl, -O-C3-C6-cycloalkyl, 4- to 8-membered heterocycloalkyl, -NR1aR1b, phenyl, 5- to 6-membered heteroaryl, -X1-NR1aR1b, -X1-C(O)NR1aR1b, -X1-O-(C1-C3-alkyl), -X1-C3- C6-cycloalkyl, -X1-phenyl, and -X2-(4- to 8-membered heterocycloalkyl), wherein each 5- to 6-membered heteroaryl has 1-2 ring heteroatoms independently selected from N and O; each 4- to 8-membered heterocycloalkyl has 1-3 ring heteroatoms independently selected
from N, O, and S, and said 4- to 8-membered heterocycloalkyl is optionally substituted with one oxo; and each R1 is optionally substituted with 1-3 R1c; or two adjacent R1 groups are combined with the two ring atoms to which they are attached to form a 5- or 6-membered heterocycloalkyl group having 1-2 ring heteroatoms independently selected from N, and O; each X1 is C1-C3-alkylene; each X2 is C1-C3-alkylene, or –(C1-C3-alkylene)-C(O)-; R1a and R1b, when present, are independently H, C1-C6 alkyl, or C3-C6 cycloalkyl; each R1c, when present, is independently halo, -OH, -C1-C6-alkyl, -C1-C6-alkoxy, or -C(O)-(C1-C6-alkyl); each R1d, when present, is independently halo, C1-C6 alkoxy, C1-C3 haloalkoxy, C3- C6-cycloalkyl, -O-C3-C6-cycloalkyl, or -NR1eR1f; R1e and R1f, when present, are independently H, C1-C6 haloalkyl, -(C1-C3-alkylene)- (C3-C6-cycloalkyl), or 4- to 8-membered heterocycloalkyl having 1-3 ring heteroatoms independently selected from N, O, and S; or R1e and R1f together with the nitrogen atom to which they are attached form a 4- to 6-membered heterocycloalkyl group having 1-2 ring heteroatoms independently selected from N, and O; R1g, when present, is C3-C6-cycloalkyl, -(C1-C3-alkylene)-(C3-C6-cycloalkyl), or 6- membered heteroaryl having 1-3 ring nitrogen atoms; wherein R1g is optionally substituted with 0-2 R1h; each R1h, when present, is independently halo, C1-C6 alkyl, -O-C3-C6-cycloalkyl, or -NH-(4- to 8-membered heterocycloalkyl), wherein the 4- to 8-membered heterocycloalkyl has 1-2 ring heteroatoms independently selected from N, O, and S; n is 0 or 1; m is 0, 1, or 2; R2, when present, is halo, -CN, -C1-C3-alkyl, -C1-C3-alkoxy, or C3-C6-cycloalkyl; R3 is -H, halo, -C1-C6-alkyl, -C1-C6-haloalkyl, -C1-C6-hydroxyalkyl, -C1-C6-alkoxy, or -C(O)NR3aR3b; R3a and R3b, when present, are independently H, or C1-C3 alkyl; each R4, when present, is independently halo, C1-C6 alkyl, C1-C6-alkoxy, C3-C6 cycloalkyl, 4- to 8-membered heterocycloalkyl having 1-3 ring heteroatoms independently
selected from N, O, and S, or phenyl, wherein said phenyl is optionally substituted with 1- 3 halo; R5 is H, C1-C3-alkyl, or -NR5aR5b; R5a and R5b, when present, are independently H or C1-C3 alkyl; and R6 is H, or C1-C3-alkyl. 2. The compound according to claim 1, or a pharmaceutically acceptable salt thereof, wherein R6 is H. 3. The compound according to claim 1 or claim 2, or a pharmaceutically acceptable salt thereof, wherein R5 is H. 4. The compound according to claim any one of claims 1-3, or a pharmaceutically acceptable salt thereof, wherein: m is 0, 1, or 2; and each R4, when present, is independently -F, -Cl, -CH3, -CH2CH3, -OCH3, -OCH2CH3, cyclopropyl,
r (ii) a 6-membered heteroaryl having 1-3 ring nitrogen atoms, wherein the 6- membered heteroaryl is substituted with 0-2 R1d; or (iii) -NHC(O)-R1g; each R1 is independently selected from the group consisting of halo, -CN, -OH, C1- C6-alkyl, C1-C6-haloalkyl, C1-C6-hydroxyalkyl, C1-C6-cyanoalkyl, C1-C6 alkoxy, C3-C6- cycloalkyl, -O-C3-C6-cycloalkyl, 4- to 8-membered heterocycloalkyl, -NR1aR1b, phenyl, 5- to 6-membered heteroaryl, -X1-NR1aR1b, -X1-C(O)NR1aR1b, -X1-O-(C1-C3-alkyl), -X1-C3- C6-cycloalkyl, -X1-phenyl, and -X2-(4- to 8-membered heterocycloalkyl), wherein each 5- to 6-membered heteroaryl has 1-2 ring heteroatoms independently selected from N and O; each 4- to 8-membered heterocycloalkyl has 1-3 ring heteroatoms independently selected
from N, O, and S, and said 4- to 8-membered heterocycloalkyl is optionally substituted with one oxo; and each R1 is optionally substituted with 1-3 R1c; or two adjacent R1 groups are combined with the two ring atoms to which they are attached to form a 5- or 6-membered heterocycloalkyl group having 1-2 ring heteroatoms independently selected from N, and O; each X1 is C1-C3-alkylene; each X2 is C1-C3-alkylene, or –(C1-C3-alkylene)-C(O)-; R1a and R1b, when present, are independently H, C1-C6 alkyl, or C3-C6 cycloalkyl; each R1c, when present, is independently halo, -OH, -C1-C6-alkyl, -C1-C6-alkoxy, or -C(O)-(C1-C6-alkyl); each R1d, when present, is independently halo, C1-C6 alkoxy, C1-C3 haloalkoxy, C3- C6-cycloalkyl, -O-C3-C6-cycloalkyl, or -NR1eR1f; R1e and R1f, when present, are independently H, C1-C6 haloalkyl, -(C1-C3-alkylene)- (C3-C6-cycloalkyl), or 4- to 8-membered heterocycloalkyl having 1-3 ring heteroatoms independently selected from N, O, and S; or R1e and R1f together with the nitrogen atom to which they are attached form a 4- to 6-membered heterocycloalkyl group having 1-2 ring heteroatoms independently selected from N, and O; R1g, when present, is C3-C6-cycloalkyl, -(C1-C3-alkylene)-(C3-C6-cycloalkyl), or 6- membered heteroaryl having 1-3 ring nitrogen atoms; wherein R1g is optionally substituted with 0-2 R1h; each R1h, when present, is independently halo, C1-C6 alkyl, -O-C3-C6-cycloalkyl, or -NH-(4- to 8-membered heterocycloalkyl), wherein the 4- to 8-membered heterocycloalkyl has 1-2 ring heteroatoms independently selected from N, O, and S; n is 0 or 1; m is 0, 1, or 2; R2, when present, is halo, -CN, -C1-C3-alkyl, -C1-C3-alkoxy, or C3-C6-cycloalkyl; R3 is -H, halo, -C1-C6-alkyl, -C1-C6-haloalkyl, -C1-C6-hydroxyalkyl, -C1-C6-alkoxy, or -C(O)NR3aR3b; R3a and R3b, when present, are independently H, or C1-C3 alkyl; each R4, when present, is independently halo, C1-C6 alkyl, C1-C6-alkoxy, C3-C6 cycloalkyl, 4- to 8-membered heterocycloalkyl having 1-3 ring heteroatoms independently
selected from N, O, and S, or phenyl, wherein said phenyl is optionally substituted with 1- 3 halo; R5 is H, C1-C3-alkyl, or -NR5aR5b; R5a and R5b, when present, are independently H or C1-C3 alkyl; and R6 is H, or C1-C3-alkyl. 2. The compound according to claim 1, or a pharmaceutically acceptable salt thereof, wherein R6 is H. 3. The compound according to claim 1 or claim 2, or a pharmaceutically acceptable salt thereof, wherein R5 is H. 4. The compound according to claim any one of claims 1-3, or a pharmaceutically acceptable salt thereof, wherein: m is 0, 1, or 2; and each R4, when present, is independently -F, -Cl, -CH3, -CH2CH3, -OCH3, -OCH2CH3, cyclopropyl,
 . 5. The compound according to any one of claims 1-4, or a pharmaceutically acceptable salt thereof, wherein R3 is halo, C1-C3 alkyl, or C1-C3 hydroxyalkyl. 6. The compound according to any one of claims 1-5, or a pharmaceutically acceptable salt thereof, wherein R3 is -F, -Cl, -CH3, or -CH2-OH. 7. The compound according to any one of claims 1-6, or a pharmaceutically acceptable salt thereof, wherein n is 0 or 1, and R2, when present, is -F, -Cl, -CN, -CH3, -OCH3, or cyclopropyl. 8. The compound according to claim 7, wherein n is 1. 9. The compound according to any one of claims 1-8, or a pharmaceutically acceptable salt thereof, wherein:
each R1 is independently halo, -CN, -OH, C1-C6-alkyl, C1-C6-haloalkyl, C1-C6- hydroxyalkyl, C1-C6 alkoxy, C3-C6-cycloalkyl, -O-C3-C6-cycloalkyl, 4- to 6-membered heterocycloalkyl, -NR1aR1b, phenyl, 6-membered heteroaryl, -X1-C(O)NR1aR1b, -X1-O- (C1-C3-alkyl), -X1-C3-C6-cycloalkyl, and -X1-phenyl; wherein each 6-membered heteroaryl has 1-2 ring heteroatoms independently selected from N and O; each 4- to 6-membered heterocycloalkyl has 1-3 ring heteroatoms independently selected from N, O, and S; and each R1 is unsubstituted or substituted with 1-3 R1c; each R1a and R1b is independently H, C1-C3 alkyl, or C3-C6 cycloalkyl; each R1c is independently halo or -OH; and and each X1 is C1-C3-alkylene. 10. The compound according to any one of claims 1-9, or a pharmaceutically acceptable salt thereof, wherein R1 is selected from the group consisting of -F, -Cl, -Br, -CN, -OH, , , , , ,
. 5. The compound according to any one of claims 1-4, or a pharmaceutically acceptable salt thereof, wherein R3 is halo, C1-C3 alkyl, or C1-C3 hydroxyalkyl. 6. The compound according to any one of claims 1-5, or a pharmaceutically acceptable salt thereof, wherein R3 is -F, -Cl, -CH3, or -CH2-OH. 7. The compound according to any one of claims 1-6, or a pharmaceutically acceptable salt thereof, wherein n is 0 or 1, and R2, when present, is -F, -Cl, -CN, -CH3, -OCH3, or cyclopropyl. 8. The compound according to claim 7, wherein n is 1. 9. The compound according to any one of claims 1-8, or a pharmaceutically acceptable salt thereof, wherein:
each R1 is independently halo, -CN, -OH, C1-C6-alkyl, C1-C6-haloalkyl, C1-C6- hydroxyalkyl, C1-C6 alkoxy, C3-C6-cycloalkyl, -O-C3-C6-cycloalkyl, 4- to 6-membered heterocycloalkyl, -NR1aR1b, phenyl, 6-membered heteroaryl, -X1-C(O)NR1aR1b, -X1-O- (C1-C3-alkyl), -X1-C3-C6-cycloalkyl, and -X1-phenyl; wherein each 6-membered heteroaryl has 1-2 ring heteroatoms independently selected from N and O; each 4- to 6-membered heterocycloalkyl has 1-3 ring heteroatoms independently selected from N, O, and S; and each R1 is unsubstituted or substituted with 1-3 R1c; each R1a and R1b is independently H, C1-C3 alkyl, or C3-C6 cycloalkyl; each R1c is independently halo or -OH; and and each X1 is C1-C3-alkylene. 10. The compound according to any one of claims 1-9, or a pharmaceutically acceptable salt thereof, wherein R1 is selected from the group consisting of -F, -Cl, -Br, -CN, -OH, , , , , ,
 11. The compound according to claim 1, having a structure according to Formula III:
11. The compound according to claim 1, having a structure according to Formula III:
 or a pharmaceutically acceptable salt thereof, wherein: A is (i) selected from the group consisting of: , ,
or a pharmaceutically acceptable salt thereof, wherein: A is (i) selected from the group consisting of: , ,
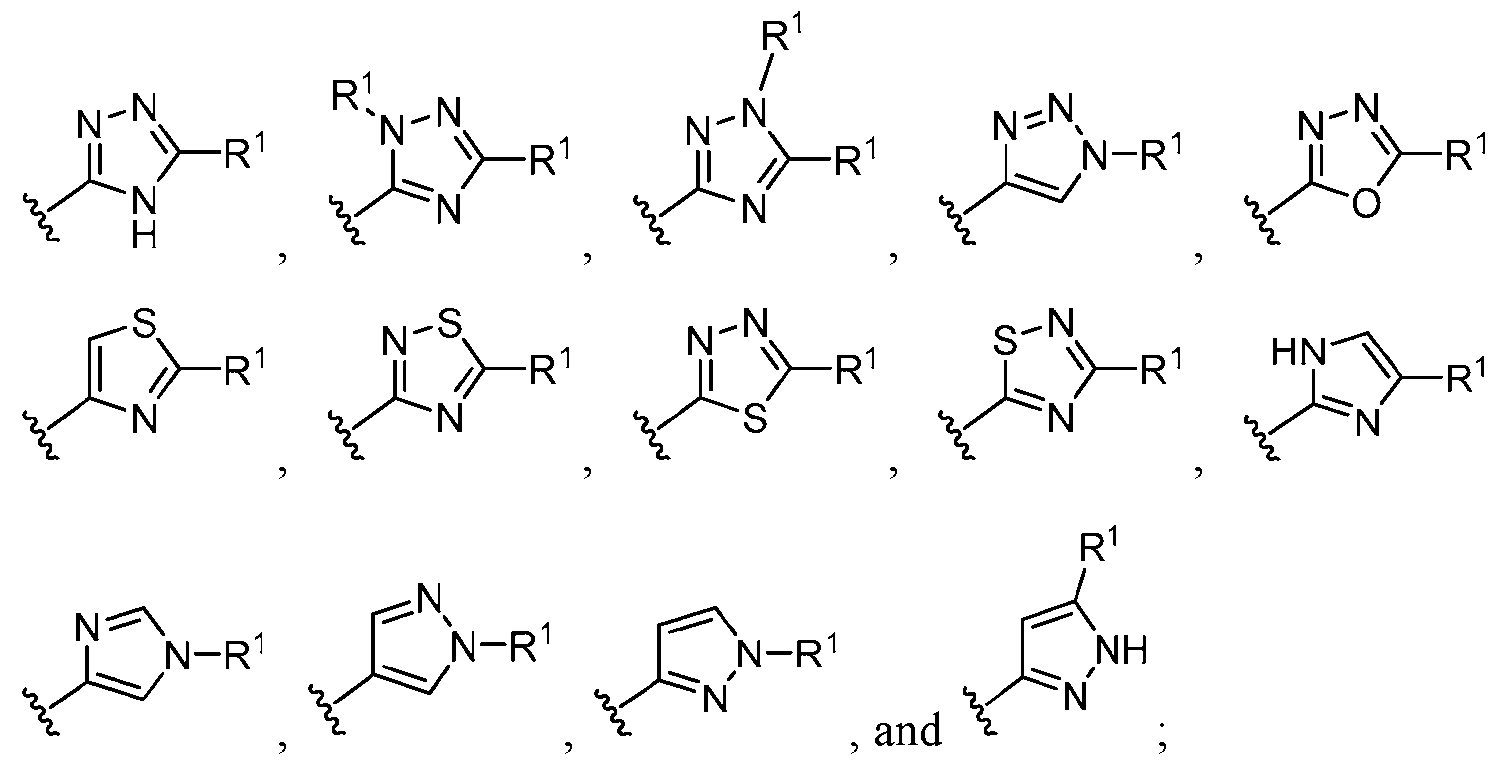 (ii) a 6-membered heteroaryl having 1 or 2 ring nitrogen atoms, wherein the 6- membered heteroaryl is substituted with 0-2 R1d; or (iii) -NHC(O)-R1g; each R1 is independently selected from the group consisting of halo, CN, -OH, C1- C6-alkyl, C1-C6-haloalkyl, C1-C6-hydroxyalkyl, C1-C6 alkoxy, C3-C6-cycloalkyl, -O-C3- C6-cycloalkyl, 4- to 8-membered heterocycloalkyl, -NR1aR1b, phenyl, 5- to 6-membered heteroaryl, -X1-C(O)NR1aR1b, -X1-O-(C1-C3-alkyl), -X1-C3-C6-cycloalkyl, and -X1-phenyl, wherein each 5- to 6-membered heteroaryl has 1-2 ring heteroatoms independently selected from N and O; each 4- to 8-membered heterocycloalkyl has 1-3 ring heteroatoms independently selected from N, O, and S; and each R1 is optionally substituted with 1-3 R1c; each X1 is C1-C3-alkylene; R1a and R1b, when present, are independently H, C1-C6 alkyl, or C3-C6 cycloalkyl; each R1c, when present, is independently halo, or -OH;
each R1d, when present, is independently halo, C1-C6 alkoxy, C3-C6-cycloalkyl, -O-C3-C6-cycloalkyl, or -NR1eR1f; R1e and R1f, when present, are independently H, C1-C6 haloalkyl, -(C1-C3-alkylene)- (C3-C6-cycloalkyl), or 4- to 8-membered heterocycloalkyl having 1-3 ring heteroatoms independently selected from N, O, and S; or R1e and R1f together with the nitrogen atom to which they are attached form a 4- to 6-membered heterocycloalkyl group having 1-2 ring heteroatoms independently selected from N, and O; R1g, when present, is C3-C6-cycloalkyl, -(C1-C3-alkylene)-(C3-C6-cycloalkyl), or 6- membered heteroaryl having 1 or 2 ring nitrogen atoms; wherein R1g is optionally substituted with 0-2 R1h; each R1h, when present, is independently halo, C1-C6 alkyl, -O-C3-C6-cycloalkyl, or -NH-(4- to 8-membered heterocycloalkyl), wherein the 4- to 8-membered heterocycloalkyl has 1-2 ring heteroatoms independently selected from N, O, and S; n is 0 or 1; m is 0, 1, or 2; R2, when present, is halo, -CN, -C1-C3-alkyl, -C1-C3-alkoxy, or C3-C6-cycloalkyl; R3 is halo, C1-C3 alkyl, or C1-C3 hydroxyalkyl; each R4, when present, is independently halo, C1-C6 alkyl, C1-C6-alkoxy, C3-C6 cycloalkyl, 4- to 8-membered heterocycloalkyl having 1-3 ring heteroatoms independently selected from N, O, and S, or phenyl, wherein said phenyl is optionally substituted with 1- 3 halo; and R5 and R6 are H. 12. The compound according to claim 11, having a structure according to Formula IIIa:
(ii) a 6-membered heteroaryl having 1 or 2 ring nitrogen atoms, wherein the 6- membered heteroaryl is substituted with 0-2 R1d; or (iii) -NHC(O)-R1g; each R1 is independently selected from the group consisting of halo, CN, -OH, C1- C6-alkyl, C1-C6-haloalkyl, C1-C6-hydroxyalkyl, C1-C6 alkoxy, C3-C6-cycloalkyl, -O-C3- C6-cycloalkyl, 4- to 8-membered heterocycloalkyl, -NR1aR1b, phenyl, 5- to 6-membered heteroaryl, -X1-C(O)NR1aR1b, -X1-O-(C1-C3-alkyl), -X1-C3-C6-cycloalkyl, and -X1-phenyl, wherein each 5- to 6-membered heteroaryl has 1-2 ring heteroatoms independently selected from N and O; each 4- to 8-membered heterocycloalkyl has 1-3 ring heteroatoms independently selected from N, O, and S; and each R1 is optionally substituted with 1-3 R1c; each X1 is C1-C3-alkylene; R1a and R1b, when present, are independently H, C1-C6 alkyl, or C3-C6 cycloalkyl; each R1c, when present, is independently halo, or -OH;
each R1d, when present, is independently halo, C1-C6 alkoxy, C3-C6-cycloalkyl, -O-C3-C6-cycloalkyl, or -NR1eR1f; R1e and R1f, when present, are independently H, C1-C6 haloalkyl, -(C1-C3-alkylene)- (C3-C6-cycloalkyl), or 4- to 8-membered heterocycloalkyl having 1-3 ring heteroatoms independently selected from N, O, and S; or R1e and R1f together with the nitrogen atom to which they are attached form a 4- to 6-membered heterocycloalkyl group having 1-2 ring heteroatoms independently selected from N, and O; R1g, when present, is C3-C6-cycloalkyl, -(C1-C3-alkylene)-(C3-C6-cycloalkyl), or 6- membered heteroaryl having 1 or 2 ring nitrogen atoms; wherein R1g is optionally substituted with 0-2 R1h; each R1h, when present, is independently halo, C1-C6 alkyl, -O-C3-C6-cycloalkyl, or -NH-(4- to 8-membered heterocycloalkyl), wherein the 4- to 8-membered heterocycloalkyl has 1-2 ring heteroatoms independently selected from N, O, and S; n is 0 or 1; m is 0, 1, or 2; R2, when present, is halo, -CN, -C1-C3-alkyl, -C1-C3-alkoxy, or C3-C6-cycloalkyl; R3 is halo, C1-C3 alkyl, or C1-C3 hydroxyalkyl; each R4, when present, is independently halo, C1-C6 alkyl, C1-C6-alkoxy, C3-C6 cycloalkyl, 4- to 8-membered heterocycloalkyl having 1-3 ring heteroatoms independently selected from N, O, and S, or phenyl, wherein said phenyl is optionally substituted with 1- 3 halo; and R5 and R6 are H. 12. The compound according to claim 11, having a structure according to Formula IIIa:
 ) or a pharmaceutically acceptable salt thereof. 13. A compound selected from the group consisting of:
) or a pharmaceutically acceptable salt thereof. 13. A compound selected from the group consisting of:


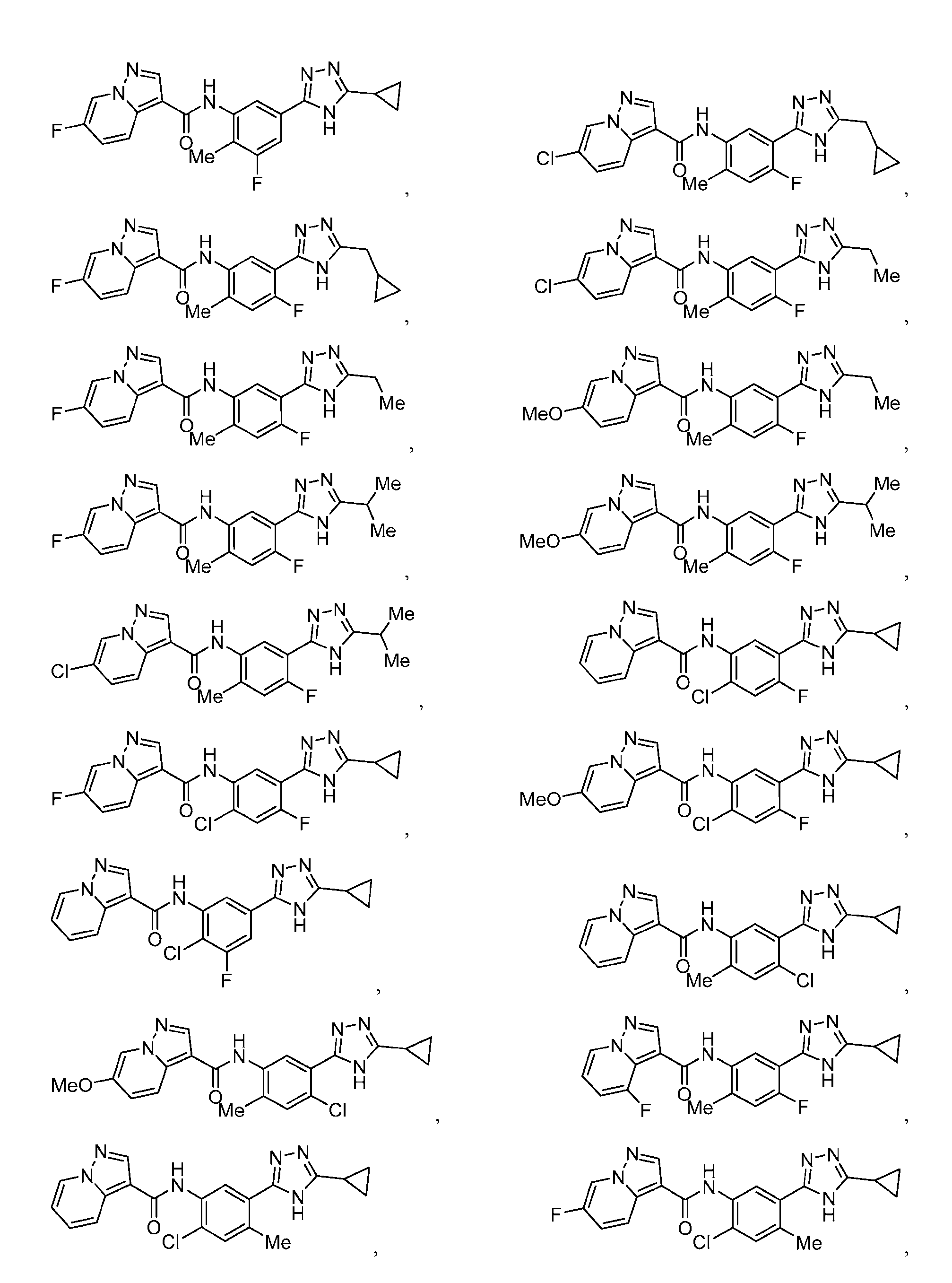


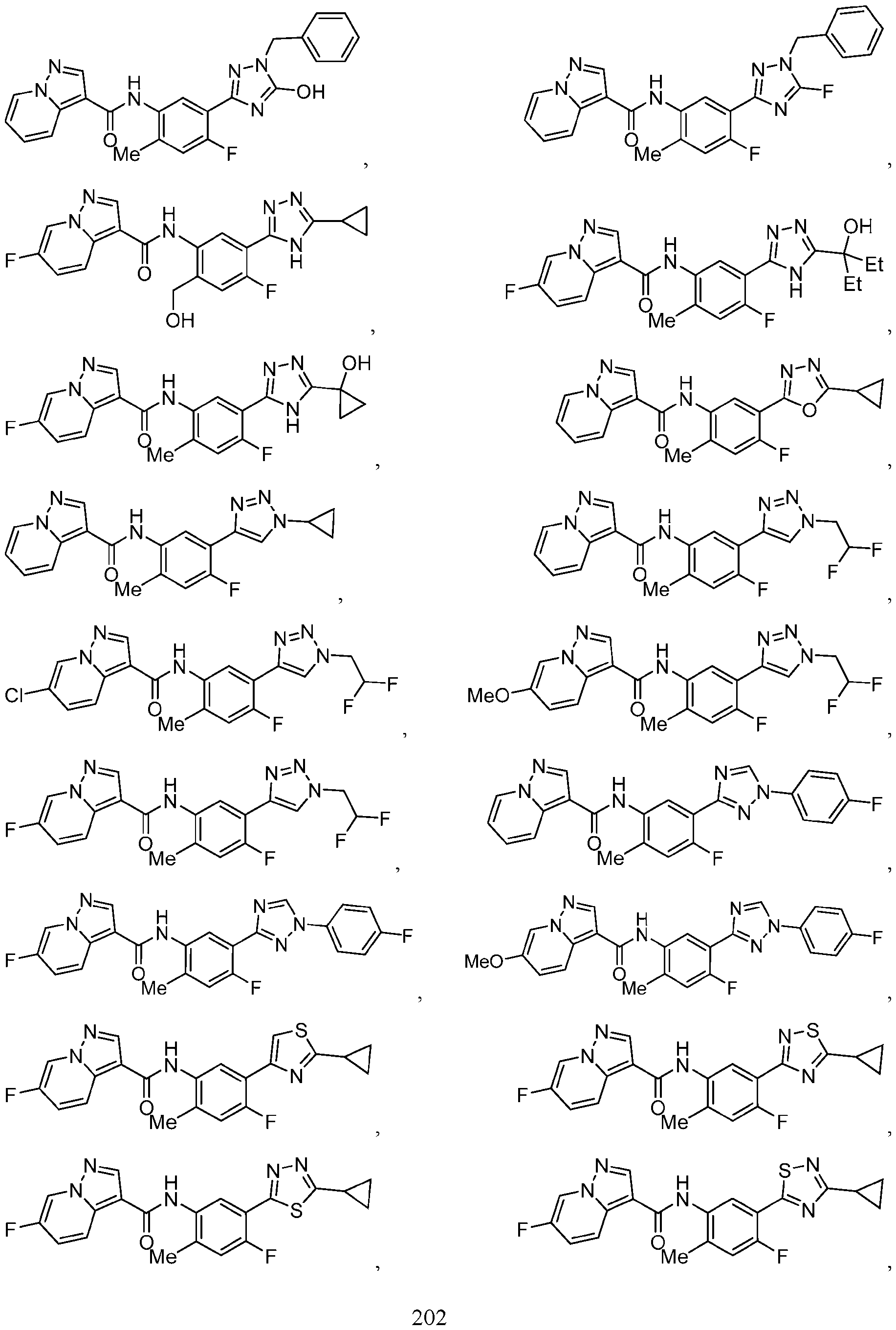
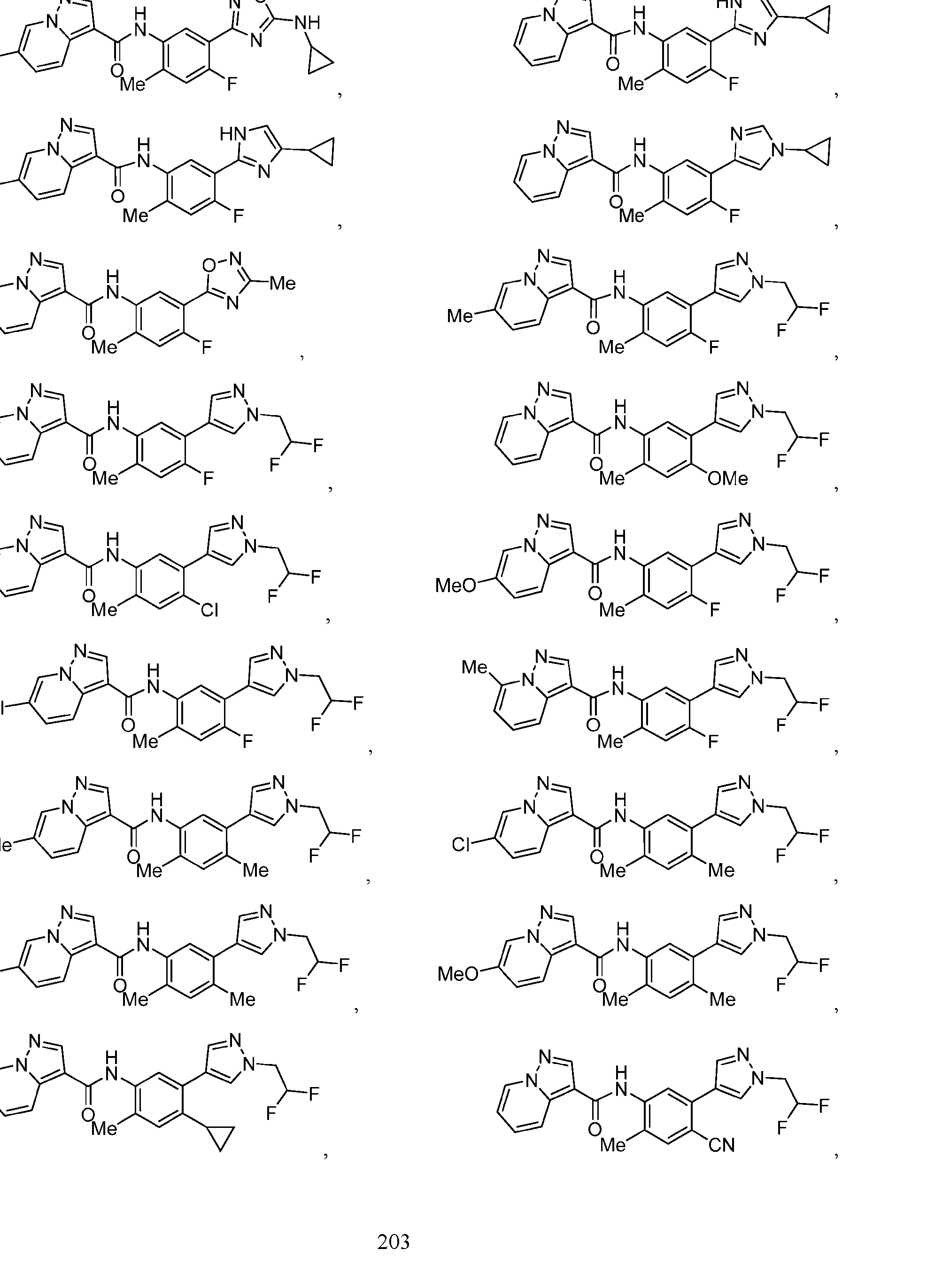




22. The method according to claim 21, wherein the cardiovascular or fibrotic disease, disorder, or condition is coronary heart disease, atherosclerosis, myocardial infarction, angina, osteoarthritis, pulmonary fibrosis, idiopathic pulmonary fibrosis, pulmonary arterial hypertension, primary pulmonary hypertension, hepatic fibrosis, renal fibrosis, cardiac fibrosis, cystic fibrosis, bronchitis, or asthma. 23. The method according to any one of claims 16-22, wherein the disease, disorder or condition is antihistamine-refractory. 24. The method according to any one of claims 16-22, wherein the disease, disorder or condition is refractory to IgE inhibitor therapy. 25. The method according to any one of claims 16-22, wherein the disease, disorder or condition is contraindicated or resistant to antihistamines. 26. The method according to any one of claims 16-25, further comprising administering at least one additional therapeutic agent to the subject. 27. The method according to claim 26, wherein the at least one additional therapeutic agent comprises one or more of an antihistamine, a BTK inhibitor, and/or an IgE inhibitor. 28. The method according to claim 27, wherein the IgE inhibitor is omalizumab, or ligelizumab. 29. The method according to claim 27 or claim 28, wherein the BTK inhibitor is ibrutinib, acalabrutinib, remibrutinib, or zanubrutinib. 30. The method according to any one of claims 26-29, wherein the additional therapeutic agent comprises an anti-inflammatory agent, an immunosuppressive agent, an agent that targets one or more cytokines, or combinations thereof. 31. The method according to claim 30, wherein the anti-inflammatory agent is a corticosteroid. 32. The method according to claim 30 or claim 31, wherein the immunosuppressive agent is cyclosporine.
33. The method according to any one of claims 30-32, wherein the agent that targets one or more cytokines targets IL-4 and/or IL-13. 34. The method according to claim 33, wherein the agent that targets one or more cytokines is dupilumab. 35. The method according to any one of claims 26-34, wherein the additional therapeutic agent comprises a leukotriene modifier. 36. The method according to any one of claims 16-18, 21, or 22, further comprising administering at least one additional therapeutic agent to the subject. 37. The method according to claim 36, wherein the additional therapeutic agent comprises a cholesterol modifier, a diuretic, an antiarrhythmic, a vasodilator, an anti-inflammatory, an analgesic agent, or any combination thereof. 38. The method according to any one of claims 16-18, wherein the disease, disorder, or condition is cancer. 39. The method according to claim 38, wherein the cancer is melanoma, prostate cancer, pancreatic cancer, squamous cell carcinoma, Hodgkin lymphoma, leukemia (e.g., chronic myeloid leukemia, chronic eosinophilic leukemia, chronic myelomonocytic leukemia, myeloid leukemia, chronic myeloid leukemia, acute myeloid leukemia, acute megakaryoblastic leukemia, mast cell leukemia, acute lymphocytic leukemia), gastric cancer (e.g., gastrointenstinal stromal cancer), thyroid cancer, breast cancer, endometrial cancer, cervical cancer, esophageal cancer, lung cancer (e.g., small cell and non-small cell lung cancer), colorectal cancer, prostate cancer, liver cancer, bile duct cancer, gallbladder cancer, neuroendocrine tumors, kidney cancer, head and neck cancer, bone cancer, brain cancer (e.g., glioblastoma, medulloblastoma), mesothelioma, or soft tissue sarcoma. 40. The method according to claim 38 or claim 39, further comprising administering at least one additional therapeutic agent.
41. The method according to claim 40, wherein the at least one additional therapeutic agent comprises an immune checkpoint inhibitor, an A2R antagonist, a CD73 inhibitor, a HIF-2α inhibitor, a chemotherapeutic agent, radiation therapy, or any combinations thereof. 42. The method according to claim 41, wherein the immune checkpoint inhibitor comprises one or more inhibitors that block the activity of at least one of PD-1, PD-L1, BTLA, LAG-3, a B7 family member, TIM-3, TIGIT or CTLA-4. 43. The method according to claim 42, wherein said immune checkpoint inhibitor comprises an inhibitor of PD-1 or PD-L1. 44. The method according to claim 43, wherein said immune checkpoint inhibitor is selected from the group consisting of avelumab, atezolizumab, durvalumab, dostarlimab, cemiplimab, nivolumab, pembrolizumab, sintilmab, toripalimab, and zimberelimab. 45. The method according to claim 44, wherein said immune checkpoint inhibitor is zimberelimab. 46. The method according to any one of claims 42-45, wherein said immune checkpoint inhibitor comprises an inhibitor that blocks the activity of TIGIT. 47. The method according to claim 46, wherein the immune checkpoint inhibitor is domvanalimab or AB308. 48. A method of preventing an allergic, immune, or autoimmune response in a subject in need thereof, said method comprising administering a compound according to any one of claims 1-14, or a pharmaceutically acceptable salt thereof, or the pharmaceutical composition according to claim 15 to the subject. 49. The method according to claim 48, wherein the subject suffers from an allergic, immune, or autoimmune disease, disorder or condition. 50. The method according to claim 49, wherein the allergic, immune, or autoimmune disease, disorder or condition is arthritis, asthma, bullous dermatosis, alopecia areata, chronic rhinosinusitis with nasal polyps (CRSwNP), coeliac disease, systemic sclerosis, netherton
syndrome, idiopathic anaphylaxis, migraine, chronic graft verse host disease, multiple sclerosis, psoriasis, Crohn’s disease, inflammatory bowel disease, irritable bowel syndrome, lupus, Grave’s disease, Hashimoto’s thyroiditis, ankylosing spondylitis, Sjӧgren’s syndrome (SjS), angioedema, allergic asthma, eosinophilic asthma, anaphylaxis, atopic dermatitis, food allergies, allergic conjunctivitis, allergic rhinitis, urticaria (e.g., chronic spontaneous urticaria (CSU), acute urticaria, or physical urticaria including popular urticaria, cold urticaria, cholinergic urticaria, solar urticaria, scleroderma, and dermatographic urticaria), mastocytosis, dermographism, systemic mastocytosis, systemic mastocytosis, mast cell activation syndrome, mast cell gastrointestinal disease, dermatitis herpetiformis, dermatosis, dermatitis, allergic contact dermatitis, eosinophilic gastrointestinal disease, eosinophilic esophagitis, type I diabetes, type II diabetes, or prurigo nodularis. 51. The method according to any one of claims 48-50, wherein the disease, disorder or condition is antihistamine-refractory. 52. The method according to any one of claims 48-50, wherein the disease, disorder or condition is refractory to IgE inhibitor therapy. 53. The method according to any one of claims 48-50, wherein the disease, disorder or condition is contraindicated or resistant to antihistamines. 54. The method according to any one of claims 48-53, wherein the method further comprises administering at least one additional therapeutic agent to the subject. 55. The method according to claim 54, wherein the at least one additional therapeutic agent comprises one or more of an antihistamine, a BTK inhibitor, and/or an IgE inhibitor. 56. The method according to claim 55, wherein the IgE inhibitor is omalizumab, or ligelizumab. 57. The method according to claim 55, wherein the BTK inhibitor is ibrutinib, acalabrutinib, remibrutinib, or zanubrutinib. 58. The method according to any one of claims 54-57, wherein the additional therapeutic agent comprises an anti-inflammatory agent, an immunosuppressive agent, an agent that targets one or more cytokines, or combinations thereof.
59. The method according to claim 58, wherein the anti-inflammatory agent is a corticosteroid. 60. The method according to claim 58 or claim 59, wherein the immunosuppressive agent is cyclosporine. 61. The method according to any one of claims 58-60, wherein the agent that targets one or more cytokines targets IL-4 and/or IL-13. 62. The method according to claim 61, wherein the agent that targets one or more cytokines is dupilumab. 63. The method according to any one of claims 54-62, wherein the additional therapeutic agent comprises a leukotriene modifier.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US202363540604P | 2023-09-26 | 2023-09-26 | |
| US63/540,604 | 2023-09-26 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2025072330A1 true WO2025072330A1 (en) | 2025-04-03 |
Family
ID=93010717
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2024/048409 Pending WO2025072330A1 (en) | 2023-09-26 | 2024-09-25 | Kit inhibitor compounds and methods of use thereof |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2025072330A1 (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US12448379B2 (en) | 2022-11-30 | 2025-10-21 | Blueprint Medicines Corporation | Wild type kit inhibitors |
| WO2025256552A1 (en) * | 2024-06-11 | 2025-12-18 | 上海翰森生物医药科技有限公司 | Pyrazole derivative inhibitor, preparation method therefor and use thereof |
Citations (29)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006105021A2 (en) | 2005-03-25 | 2006-10-05 | Tolerrx, Inc. | Gitr binding molecules and uses therefor |
| WO2008009487A1 (en) * | 2006-03-10 | 2008-01-24 | Novartis Ag | Heterobicyclic carboxamides as inhibitors for kinases |
| WO2008132601A1 (en) | 2007-04-30 | 2008-11-06 | Immutep | Cytotoxic anti-lag-3 monoclonal antibody and its use in the treatment or prevention of organ transplant rejection and autoimmune disease |
| WO2009009116A2 (en) | 2007-07-12 | 2009-01-15 | Tolerx, Inc. | Combination therapies employing gitr binding molecules |
| WO2009044273A2 (en) | 2007-10-05 | 2009-04-09 | Immutep | Use of recombinant lag-3 or the derivatives thereof for eliciting monocyte immune response |
| WO2010019570A2 (en) | 2008-08-11 | 2010-02-18 | Medarex, Inc. | Human antibodies that bind lymphocyte activation gene-3 (lag-3), and uses thereof |
| WO2011028683A1 (en) | 2009-09-03 | 2011-03-10 | Schering Corporation | Anti-gitr antibodies |
| WO2011109400A2 (en) | 2010-03-04 | 2011-09-09 | Macrogenics,Inc. | Antibodies reactive with b7-h3, immunologically active fragments thereof and uses thereof |
| WO2012032433A1 (en) | 2010-09-09 | 2012-03-15 | Pfizer Inc. | 4-1bb binding molecules |
| WO2012145493A1 (en) | 2011-04-20 | 2012-10-26 | Amplimmune, Inc. | Antibodies and other molecules that bind b7-h1 and pd-1 |
| WO2013033167A1 (en) * | 2011-09-01 | 2013-03-07 | Irm Llc | Compounds and compositions as c-kit kinase inhibitors |
| WO2014008218A1 (en) | 2012-07-02 | 2014-01-09 | Bristol-Myers Squibb Company | Optimization of antibodies that bind lymphocyte activation gene-3 (lag-3), and uses thereof |
| WO2017120508A1 (en) | 2016-01-08 | 2017-07-13 | Arcus Biosciences, Inc. | Modulators of 5'-nucleotidase, ecto and the use thereof |
| WO2017152088A1 (en) | 2016-03-04 | 2017-09-08 | JN Biosciences, LLC | Antibodies to tigit |
| WO2018067424A1 (en) | 2016-10-03 | 2018-04-12 | Arcus Biosciences, Inc. | Inhibitors of adenosine 5'-nucleotidase |
| WO2018094148A1 (en) | 2016-11-18 | 2018-05-24 | Arcus Biosciences, Inc. | Inhibitors of cd73-mediated immunosuppression |
| WO2018136700A1 (en) | 2017-01-20 | 2018-07-26 | Arcus Biosciences, Inc. | Azolopyrimidine for the treatment of cancer-related disorders |
| WO2018204661A1 (en) | 2017-05-05 | 2018-11-08 | Arcus Biosciences, Inc. | Quinazoline-pyridine derivatives for the treatment of cancer-related disorders |
| WO2018213377A1 (en) | 2017-05-17 | 2018-11-22 | Arcus Biosciences, Inc. | Quinazoline-pyrazole derivatives for the treatment of cancer-related disorders |
| WO2020023846A1 (en) | 2018-07-27 | 2020-01-30 | Arcus Biosciences, Inc. | Pyridone a2r antagonists |
| WO2020046813A1 (en) | 2018-08-27 | 2020-03-05 | Arcus Biosciences, Inc. | Cd73 inhibitors |
| WO2021113436A1 (en) | 2019-12-04 | 2021-06-10 | Arcus Biosciences, Inc. | Inhibitors of hif-2alpha |
| WO2021188769A1 (en) | 2020-03-19 | 2021-09-23 | Arcus Biosciences, Inc. | Tetralin and tetrahydroquinoline compounds as inhibitors of hif-2alpha |
| WO2021247591A1 (en) | 2020-06-02 | 2021-12-09 | Arcus Biosciences, Inc. | Antibodies to tigit |
| WO2022246177A1 (en) | 2021-05-21 | 2022-11-24 | Arcus Biosciences, Inc. | Axl compounds |
| WO2022246179A1 (en) | 2021-05-21 | 2022-11-24 | Arcus Biosciences, Inc. | Axl inhibitor compounds |
| US20230069124A1 (en) | 2021-08-24 | 2023-03-02 | Red Hat, Inc. | Schema based type-coercion for structured documents |
| WO2023077046A1 (en) | 2021-10-29 | 2023-05-04 | Arcus Biosciences, Inc. | Inhibitors of hif-2alpha and methods of use thereof |
| WO2024118887A1 (en) * | 2022-11-30 | 2024-06-06 | Blueprint Medicines Corporation | N-phenyl-pyrazolo[1,5-a]pyridine-3-carboxamide derivatives as wild type c-kit kinase inhibitors for the treatment of urticaria |
-
2024
- 2024-09-25 WO PCT/US2024/048409 patent/WO2025072330A1/en active Pending
Patent Citations (29)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006105021A2 (en) | 2005-03-25 | 2006-10-05 | Tolerrx, Inc. | Gitr binding molecules and uses therefor |
| WO2008009487A1 (en) * | 2006-03-10 | 2008-01-24 | Novartis Ag | Heterobicyclic carboxamides as inhibitors for kinases |
| WO2008132601A1 (en) | 2007-04-30 | 2008-11-06 | Immutep | Cytotoxic anti-lag-3 monoclonal antibody and its use in the treatment or prevention of organ transplant rejection and autoimmune disease |
| WO2009009116A2 (en) | 2007-07-12 | 2009-01-15 | Tolerx, Inc. | Combination therapies employing gitr binding molecules |
| WO2009044273A2 (en) | 2007-10-05 | 2009-04-09 | Immutep | Use of recombinant lag-3 or the derivatives thereof for eliciting monocyte immune response |
| WO2010019570A2 (en) | 2008-08-11 | 2010-02-18 | Medarex, Inc. | Human antibodies that bind lymphocyte activation gene-3 (lag-3), and uses thereof |
| WO2011028683A1 (en) | 2009-09-03 | 2011-03-10 | Schering Corporation | Anti-gitr antibodies |
| WO2011109400A2 (en) | 2010-03-04 | 2011-09-09 | Macrogenics,Inc. | Antibodies reactive with b7-h3, immunologically active fragments thereof and uses thereof |
| WO2012032433A1 (en) | 2010-09-09 | 2012-03-15 | Pfizer Inc. | 4-1bb binding molecules |
| WO2012145493A1 (en) | 2011-04-20 | 2012-10-26 | Amplimmune, Inc. | Antibodies and other molecules that bind b7-h1 and pd-1 |
| WO2013033167A1 (en) * | 2011-09-01 | 2013-03-07 | Irm Llc | Compounds and compositions as c-kit kinase inhibitors |
| WO2014008218A1 (en) | 2012-07-02 | 2014-01-09 | Bristol-Myers Squibb Company | Optimization of antibodies that bind lymphocyte activation gene-3 (lag-3), and uses thereof |
| WO2017120508A1 (en) | 2016-01-08 | 2017-07-13 | Arcus Biosciences, Inc. | Modulators of 5'-nucleotidase, ecto and the use thereof |
| WO2017152088A1 (en) | 2016-03-04 | 2017-09-08 | JN Biosciences, LLC | Antibodies to tigit |
| WO2018067424A1 (en) | 2016-10-03 | 2018-04-12 | Arcus Biosciences, Inc. | Inhibitors of adenosine 5'-nucleotidase |
| WO2018094148A1 (en) | 2016-11-18 | 2018-05-24 | Arcus Biosciences, Inc. | Inhibitors of cd73-mediated immunosuppression |
| WO2018136700A1 (en) | 2017-01-20 | 2018-07-26 | Arcus Biosciences, Inc. | Azolopyrimidine for the treatment of cancer-related disorders |
| WO2018204661A1 (en) | 2017-05-05 | 2018-11-08 | Arcus Biosciences, Inc. | Quinazoline-pyridine derivatives for the treatment of cancer-related disorders |
| WO2018213377A1 (en) | 2017-05-17 | 2018-11-22 | Arcus Biosciences, Inc. | Quinazoline-pyrazole derivatives for the treatment of cancer-related disorders |
| WO2020023846A1 (en) | 2018-07-27 | 2020-01-30 | Arcus Biosciences, Inc. | Pyridone a2r antagonists |
| WO2020046813A1 (en) | 2018-08-27 | 2020-03-05 | Arcus Biosciences, Inc. | Cd73 inhibitors |
| WO2021113436A1 (en) | 2019-12-04 | 2021-06-10 | Arcus Biosciences, Inc. | Inhibitors of hif-2alpha |
| WO2021188769A1 (en) | 2020-03-19 | 2021-09-23 | Arcus Biosciences, Inc. | Tetralin and tetrahydroquinoline compounds as inhibitors of hif-2alpha |
| WO2021247591A1 (en) | 2020-06-02 | 2021-12-09 | Arcus Biosciences, Inc. | Antibodies to tigit |
| WO2022246177A1 (en) | 2021-05-21 | 2022-11-24 | Arcus Biosciences, Inc. | Axl compounds |
| WO2022246179A1 (en) | 2021-05-21 | 2022-11-24 | Arcus Biosciences, Inc. | Axl inhibitor compounds |
| US20230069124A1 (en) | 2021-08-24 | 2023-03-02 | Red Hat, Inc. | Schema based type-coercion for structured documents |
| WO2023077046A1 (en) | 2021-10-29 | 2023-05-04 | Arcus Biosciences, Inc. | Inhibitors of hif-2alpha and methods of use thereof |
| WO2024118887A1 (en) * | 2022-11-30 | 2024-06-06 | Blueprint Medicines Corporation | N-phenyl-pyrazolo[1,5-a]pyridine-3-carboxamide derivatives as wild type c-kit kinase inhibitors for the treatment of urticaria |
Non-Patent Citations (2)
| Title |
|---|
| BERGE, S.M. ET AL.: "Journal of Pharmaceutical Science", vol. 66, 1977, article "Pharmaceutical Salts", pages: 1 - 19 |
| PARDOLL, NATURE REV. CANCER, vol. 12, April 2012 (2012-04-01), pages 252 - 64 |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US12448379B2 (en) | 2022-11-30 | 2025-10-21 | Blueprint Medicines Corporation | Wild type kit inhibitors |
| WO2025256552A1 (en) * | 2024-06-11 | 2025-12-18 | 上海翰森生物医药科技有限公司 | Pyrazole derivative inhibitor, preparation method therefor and use thereof |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US12410170B2 (en) | 2,3,5-trisubstituted pyrazolo[1,5-A]pyrimidine compounds | |
| US12103907B2 (en) | Tetralin and tetrahydroquinoline compounds as inhibitors of HIF-2alpha | |
| EP4069212A1 (en) | Inhibitors of hif-2alpha | |
| US20230339902A1 (en) | Tricyclic ligands for degradation of ikzf2 or ikzf4 | |
| WO2025072330A1 (en) | Kit inhibitor compounds and methods of use thereof | |
| KR102874347B1 (en) | compound | |
| US20240246967A1 (en) | Axl compounds | |
| AU2018210272A1 (en) | Azolopyrimidine for the treatment of cancer-related disorders | |
| RS56657B1 (en) | Novel amino pyrimidine derivatives | |
| TW202448871A (en) | Azolylpyridine pyridazinone amides as sos1 inhibitors | |
| CA3161339A1 (en) | Cyclic compounds and methods of using same | |
| TW201902895A (en) | 6-5 fused ring as a C5a inhibitor | |
| CA3090150A1 (en) | Ahr modulators | |
| US11396502B2 (en) | Substituted heterocyclic derivatives as PI3K inhibitors | |
| US20260028336A1 (en) | Cbl-b inhibitors and methods of use thereof | |
| US20240300936A1 (en) | Preparation of substituted 1,2-diaminoheterocyclic compound derivatives and their use as pharmaceutical agents | |
| US12384773B2 (en) | Thiazole compounds and methods of use thereof | |
| WO2025054339A1 (en) | Triazolopyridine compounds as inhibitors of kit | |
| RU2800063C2 (en) | Compounds | |
| US20250011318A1 (en) | Cbl-b inhibitors and methods of use thereof | |
| US20250352549A1 (en) | Substituted heterocyclic compound derivatives and their pharmaceutical use | |
| HK40116573A (en) | Compounds as inhibitors of axl | |
| HK40076614A (en) | Inhibitors of hif-2alpha | |
| HK40068316A (en) | 2,3,5-trisubstituted pyrazolo[1,5-a]pyrimidine compounds | |
| HK40027765B (en) | Quinazoline-pyrazole derivatives for the treatment of cancer-related disorders |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 24786333 Country of ref document: EP Kind code of ref document: A1 |