WO2025059113A1 - Treatments for enhancing immune response to clostridioides difficile infections - Google Patents
Treatments for enhancing immune response to clostridioides difficile infections Download PDFInfo
- Publication number
- WO2025059113A1 WO2025059113A1 PCT/US2024/046099 US2024046099W WO2025059113A1 WO 2025059113 A1 WO2025059113 A1 WO 2025059113A1 US 2024046099 W US2024046099 W US 2024046099W WO 2025059113 A1 WO2025059113 A1 WO 2025059113A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- tcdb2
- tcdb
- cxcr4
- difficile
- cells
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/4709—Non-condensed quinolines and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7048—Compounds having saccharide radicals and heterocyclic rings having oxygen as a ring hetero atom, e.g. leucoglucosan, hesperidin, erythromycin, nystatin, digitoxin or digoxin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/02—Bacterial antigens
- A61K39/08—Clostridium, e.g. Clostridium tetani
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/51—Medicinal preparations containing antigens or antibodies comprising whole cells, viruses or DNA/RNA
- A61K2039/52—Bacterial cells; Fungal cells; Protozoal cells
- A61K2039/521—Bacterial cells; Fungal cells; Protozoal cells inactivated (killed)
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/545—Medicinal preparations containing antigens or antibodies characterised by the dose, timing or administration schedule
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/555—Medicinal preparations containing antigens or antibodies characterised by a specific combination antigen/adjuvant
- A61K2039/55505—Inorganic adjuvants
Definitions
- Clostridioides difficile is the largest cause of nosocomial infection globally, surpassing that caused by methicillin-resistant Staphylococcus aureus.
- C. difficile infection (CDI) survivors commonly suffer from long-lasting complications due to high rates of disease recurrence, with the chances of relapse increasing after each infection. Prolonged dysbiosis caused by antibiotic therapy and pathogen re-exposure likely contribute to CDI recurrence.
- CDI C. difficile infection
- TcdA Toxin A
- TcdB Toxin B
- TcdB -induced glucosylation of small- GTPases in epithelial cells results in cell rounding and ultimately cell death by apoptosis, diminishing the structural integrity of the colonic lining, allowing tissue infection and dissemination of C. difficile and its toxins.
- C. difficile spores are transmitted via the fecal-oral route and remain viable on surfaces under diverse environmental conditions. Germination of ingested spores is facilitated by dysbiosis of the host microbiome, often induced by broad-spectrum antibiotic therapy.
- CDI establishes in the colon and symptoms range from mild diarrhea to severe pseudomembranous colitis, sepsis, and death. CDI can be successfully cleared in animal models and patients by the innate immune response.
- MyD88-mediated activated neutrophils are necessary for clearance as are Type 1, 2, and 3 innate-like lymphocytes (ILC1, ILC2, ILC3) which have been reported to mitigate disease severity. Additionally, IL-25-regulated eosinophils contribute to protection.
- TcdB-specific IgG constitutes the clearest correlate of protection against recurrent CDI.
- analysis of the TcdB-specific memory B cell compartment in individuals who have recovered from CDI shows an apparent deficiency in Ig class switch and poor TcdB-neutralizing capability of the limited IgG that is produced.
- FIG. 1 shows a scheme of a treatment protocol for investigating delayed IgG class switch and inhibited IgG recall response following TcdB2 treatment in vivo.
- FIG. 2 shows results over time for primary serum B2 -specific IgM, IgGl, IgG2b, and IgG2c endpoint titers as determined by ELISA for the mice of FIG. 1. Significant differences by one way ANOVA in titers and neutralization are as follows: *, P ⁇ 0.05 ****, P ⁇ 0.0001.
- FIG. 3 shows results pre- and post-bleed for serum B2A-specific IgM, IgGl, IgG2b, and IgG2c endpoint titers as determined by ELISA for the mice of FIG. 1. Significant differences depicted were determined by matched pairs t-test.
- Serum B2A-specific IgM, IgGl, IgG2b and IgG2c endpoint titers were determined by ELISA.
- FIG. 6 shows results for CHO cell viability determined using the CCK-8 assay as a measure of TcdB2 neutralization. CHO cells were incubated with TcdB2, sera, or sera and TcdB2 for 24 hours. Significant differences by one way ANOVA in titers and neutralization are as follows: *, P ⁇ 0.05 ****, P ⁇ 0.0001.
- FIG. 7 shows CD40 activation-associated restoration of IgG recall responses in TcdB2-treated mice.
- a booster vaccine was administered on day 60 and consisted of 20 pg of B2A in PBS. Blood samples were collected before (day 60) and after (day 74) the booster. Data shows IgM B2A- specific endpoint titers ⁇ SD (upper panels) and IgGl B2A-specific endpoint titers ⁇ SD (lower panels). Matched pairs t-tests were used to measure significance.
- FIG. 8 shows CD40 activation-associated restoration of IgG recall responses in TcdB2-treated mice.
- a booster vaccine was administered on day 60 and consisted of 20 pg of B2A in PBS. Blood samples were collected before (day 60) and after (day 74) the booster. Data shows IgG2b B2A- specific endpoint titers ⁇ SD (upper panels) and IgG2c B2A-specific endpoint titers ⁇ SD (lower panels). Matched pairs t-tests were used to measure significance.
- FIG. 9 shows results from re-analysis of the data from FIGS. 7-8. Data were reanalyzed by calculating fold change in endpoint IgM, IgGl, IgG2b, and IgG2c titers following booster vaccine administration and comparing the three experimental groups. One way ANOVA with Tukey’s multiple comparison post-test was used to measure significance (*, P ⁇ 0.05).
- FIG. 10 shows representative images of ELISPOT wells with spots attributable to B2A-specific IgGl . Graphs depict number of B2A-specific spots per million cells. Each symbol represents an individual mouse. Significance was determined by one-way ANOVA, *, P ⁇ 0.05.
- FIG. 11 shows representative images of ELISPOT wells with spots attributable to B2A-specific IgG2b. Graphs depict number of B2A-specific spots per million cells. Each symbol represents an individual mouse. Significance was determined by one-way ANOVA, ** P ⁇ 0.01.
- FIG. 12 shows lack of killing but direct intoxication of lymphocytes by TcdB2.
- Graphs represent absolute CD4 + T cell and B cell count from spleens 14 days post- treatment and show results from two pooled experiments (cell count per half spleen is depicted).
- FIG. 13 shows flow cytometry results of B cell splenocytes after being cultured in vitro with vehicle or 0.01, 0.1 or 1 pM TcdB2 or B2A for 6 hours. A subset of splenocytes were incubated at 45 °C for 1 hour as a positive control. Graphs depicts AnnV / 7AAD dye uptake by cells. Statistical significance was determined by one-way ANOVA, **, P ⁇ 0.01. Results are representative of two pooled experiments. Lysates were prepared from splenocytes and analyzed using the capillary automated electrophoresis and blotting system (as shown in FIG. 15).
- FIG. 14 shows flow cytometry results of CD4 + T cell splenocytes after being cultured in vitro with vehicle or 0.01, 0.1 or 1 pM TcdB2 or B2A for 6 hours.
- a subset of splenocytes were incubated at 45 °C for 1 hour as a positive control.
- Graphs depicts AnnV / 7AAD dye uptake by cells. Statistical significance was determined by one-way ANOVA, ****, P ⁇ 0.0001 . Results are representative of two pooled experiments. Lysates were prepared from splenocytes and analyzed using the capillary automated electrophoresis and blotting system (as shown in FIG. 15).
- FIG. 15 shows blot detection of non-glucosylated Rael and a human recombinant Rael for comparison to FIGS. 13-14.
- FIG. 16 shows results of splenocytes which were untreated or treated in vitro for 2 or 4 hr with TcdB2 at a 10 pM final concentration before preparing cell lysates and performing immunoblots for non-glucosylated Rael and the GAPDH total protein loading control (left panel).
- B cells (right) were isolated by magnetic separation before treatment with TcdB2 and a D270N-treated control was added. Flow cytometry histogram indicates degree of B cell enrichment in the samples. Left and right panels are representative of two similar experiments.
- FIGS. 17 shows results of TcdB2 blockade of immunization-induced germinal center formation.
- FIG. 20 representative immunofluorescent sections from mice described in FIG. 17. B220 + total B cells (purple) and Ki67 + proliferating GC B cells (green) are shown. Arrows indicate germinal centers.
- FIG. 21 graph represents relative fluorescence signal associated with GCs.
- Relative signal [(mean fluorescent intensity of GC) - (mean fluorescent intensity of background) x area of GC].
- Statistical significance was determined by one-way ANOVA, **, P ⁇ 0.01
- FIG. 22 shows differentially expressed genes following TcdB2 exposure include CXCR4.
- RNA was purified from axillary and inguinal lymph nodes (aLNs and iLNs). Gene expression was quantified using the Nanostring nCounter SPRINT profiler platform.
- A Upper left panel shows differentially expressed genes (DEGs) comparing TcdB2 to PBS.
- B Upper right panel shows differentially expressed genes (DEGs) comparing TcdB2 to D270N.
- Lower left panel shows differentially expressed genes (DEGs) comparing D270N to PBS.
- FIG. 23A shows differentially expressed genes following TcdB2 exposure include CXCR4.
- Gene expression was quantified using the Nanostring nCounter SPRINT profiler platform as described for FIG. 22. Values for cxcr4, cxcr5, ccr7, and their ligands are depicted.
- RNA was purified from axillary lymph nodes (aLNs) and inguinal lymph nodes (iLNs). Gene expression was quantified using the Nanostring Counter SPRINT profiler platform.
- FIG. 23C shows results of an analysis of all chemokine receptor and ligand genes in the gene panel in TcdB2 vs. PBS treatments. No significant changes in expression of the genes were revealed.
- FIG. 25 shows CXCR4 and CXCR5 expression in CD4 + T cells following TcdB2 and D270N exposure as measured by flow cytometry.
- Flow plots show CXCR4 and CXCR5 expression, while graphs depict the percentage of each cell type expressing CXCR4.
- FIG. 27 shows increased splenocyte migration towards the CXCR4 chemoattractant CXCL12 following TcdB2 treatment.
- FIG. 28 shows increased B cell migration towards the CXCR4 chemoattractant CXCL12 following TcdB2 treatment.
- Representative flow cytometry plots of isolated B cells are shown and graphs depict quantification of migratory B cells and CD4 + T cells. Results are pooled from two independent experiments. Statistical significance was determined by one-way ANOVA, ****, P ⁇ 0.0001.
- FIG. 29A shows increased B cell migration towards the CXCR4 chemoattractant CXCL12 following TcdB2 treatment.
- Representative flow cytometry plots of isolated CD4 + T cells are shown and graphs depict quantification of migratory B cells and CD4 + T cells. Results are pooled from two independent experiments.
- FIG. 29B shows increased B cell migration toward the CXCR4 chemoattractant CXCL12 following TcdB2 treatment.
- Isolated B cells from vehicle-, D270N-, and TcdB2- treated mice were stimulated in vitro with ligands for CXCR4, CXCR5, and CCR7 (CXCL12, CCL19/CCL21, and CXCL13, respectively).
- Statistical significance was determined by one-way ANOVA, ***, P ⁇ 0.001 , ****, P ⁇ 0.0001 .
- FIG. 29C shows increased B cell migration toward the CXCR4 chemoattractant CXCL12 following TcdB2 treatment.
- Graph shows mean ⁇ SD, and data are representative of 2 similar experiments.
- Statistical significance was determined by one-way ANOVA: ***p ⁇ 0.001, ****p ⁇ 0.0001.
- FIG. 30 is a scheme showing the experimental protocol for testing CXCR4 expression on B cells and migration of lymphocytes towards the CXCR4 chemoattractant CXCL12 following C. difficile infection in mice.
- FIG. 31 shows weights (mean ⁇ SD) of the mice of FIG. 30 starting two days before gavage.
- FIG. 32 shows C. difficile CFUs (mean ⁇ SD) in fecal samples collected 2 days post-gavage (left); representative images (right) of cecum and colon from control mouse and infected mouse.
- FIG. 34 shows graphs depicting percent of CXCR4 + B cells from mLN (left) and iLN (right), determined by flow cytometry. Statistical significance was determined by two- tailed T-test.
- FIG. 35 shows graphs depicting percent of CXCR4 + B cells from spleen (left) and aLN (right), determined by flow cytometry. Statistical significance was determined by two- tailed T-test.
- FIG. 36 shows graphs depicting quantification of migratory lymphocytes from mLNs averaged from 4 fields of view from each trans well membrane. Statistical significance was determined by one-way ANOVA , ****, P ⁇ 0.0001.
- FIG. 37 shows that TcdB2 does not impact average affinity of B2A-specific IgG.
- ELIS were performed as described in Materials and Methods except that plates were coated with B2A at final concentrations of 0.1 and 10 pg/ml. Sera were applied to coated and blocked plates at a 1 : 1000 dilution and detected with IgGl-, IgG2b-, and IgG2c-specific detection Abs as described (left, center, right, respectively).
- Graphs depict the absorbance ratios for sera applied to wells coated with 0.1 and 10 pg / ml B2A.
- FIGS. 38-40 show that TcdB2 does not impact total IgG abundance or bone marrow plasma cell numbers. Bone marrow ELISPOTS were performed to detect all Ig specificities simultaneously with assays to detect antigen specific cells.
- FIG. 38 shows that TcdB2 does not impact total IgG abundance or bone marrow plasma cell numbers. Bone marrow ELISPOTS were performed to detect all Ig specificities simultaneously with assays to detect antigen specific cells. Images show representative triplicate wells and graph depicts total IgGl secretion for each mouse analyzed.
- FIG. 39 shows, as in FIG. 38, that TcdB2 does not impact total IgG abundance or bone marrow plasma cell numbers. Bone marrow ELISPOTS were performed to detect all Ig specificities simultaneously with assays to detect antigen specific cells. Images show representative triplicate wells and graph depicts total IgG2b secretion for each mouse analyzed. [0052] FIG. 40 shows background in the assays of FIGS. 38 and 39 using anti-Ig coating, and B2A coating in conjunction with anti-IgGl or IgG2b detection Abs.
- FIG. 41 shows that TcdB2 does not affect splenic B cell and CD4 + T cell numbers.
- Representative flow cytometry plots depict gating strategies for B cell subtypes and CD4 + T cells.
- FIG. 44 shows that TcdB2 does not affect lymph node B cell and CD4 + T cell numbers. Representative flow cytometry plots depict gating strategies for B cell subtypes and CD4 + T cells.
- FIG. 46 shows gating strategies for detection of Annexin V + and 7-AAD + cells and effect of TcdB2 at early and late time points.
- Representative flow cytometry plots depict gating strategies for apoptotic/necrotic B and CD4 + T cells in FIGS. 47-48.
- FIG. 47 shows results for splenocytes cultured in vitro with vehicle or 0.01, 0.1 or 1 pM TcdB2 or B2A for 30 min or 12 hr, then examined by flow cytometry (the 6 hr time point is shown in FIGS. 13-14).
- a subset of splenocytes were incubated at 45°C for 1 hr to induce apoptosis as a positive control.
- Graphs depict AnnV / 7AAD dye uptake by B cells, identifying apoptotic cells.
- FIG. 48 shows results for splenocytes cultured in vitro with vehicle or 0.01, 0.1 or 1 pM TcdB2 or B2A for 30 min or 12 hr, then examined by flow cytometry (the 6 hr time point is shown in FIGS. 13-14).
- a subset of splenocytes were incubated at 45°C for 1 hr to induce apoptosis as a positive control.
- Graphs depict Annexin V / 7AAD dye uptake by CD4 + T cells, identifying apoptotic cells.
- FIG. 49 shows CXCR5 expression by B and CD4 + T cells following TcdB2 toxin treatment Graphs represent percent of CXCR5 on B cells and CD4 + T cells from iLNs post TcdB2, D270N, or PBS treatment.
- FIG. 50 shows graphs representing the percent of CXCR5 on B cells from mLN (left), iLN (right) in uninfected controls or post infection by C.difficile.
- FIG. 51 graphs represent the percent of CXCR5 on B cells from spleen (left), and aLN (right) in uninfected controls or post infection by C. difficile.
- FIG. 52 shows that the CXCR4 antagonist AMD3100 restores normal CXCR4- mediated cell migration.
- Female B6 mice were given 1 ng TcdB2 or PBS vehicle control (i.p.) and then given either PBS vehicle or AMD3100 (1 or 10 mg/g of body weight) by the s.c. route. After 48 h, splenocytes were isolated, and migration toward CXCL12 was measured as described for FIGS. 26-27.
- FIG. 53 shows that the AMD3100 rescues TcdB2-suppressed GC formation
- mice were treated with AMD3100 or PBS vehicle control (i.p.).
- Graphs depict the mean ⁇ SD area and number of GCs in iLNs collected 21 days post treatment. Statistical significance was determined by two-tailed t test. Images show representative H&E sections from lymph nodes. Yellow arrows indicate GCs. Thin dark lines were due to a crease in the section. The scale bar depicts 500 mm.
- Mice were treated with AMD3100 (1 ug/g; i.p.) at hour -2, 24, and 48 post-gavage.
- Relative weight loss is normalized to 100% at day 0 post-gavage.
- Asterisks represent significant change in weight compared to uninfected control. Not depicted is significant change in weight of CDI vs AMD3100+CD1 (*) on days 4 and 5 post gavage.
- FIG. 55 depicts the same data as shown in FIG. 54 but contains SD of weight loss.
- FIG. 56 depicts probability of survival of mice post C. difficile infection in the experiments of FIGS. 54-55. Significance determined by two-way ANOVA: *, P ⁇ 0.05; ***, P ⁇ 0.001.
- AEC 3-amino-9-ethyl-carbazole
- aLN axillary Lymph Node
- APC Allophycocyanin
- BSA Bovine serum albumen
- B2A B2A Tcdb2 mutant antigen
- CD40 Cluster of Differentiation 40
- CD4 + Cluster of Differentiation 4 positive
- CDI Clostridioides difficile infection
- CXCR4 chemokine (C-X-C motif) receptor type 4
- CXCR5 chemokine (C-X-C motif) receptor type 5
- FBS fetal bovine serum
- GAPDH Glyceraldehyde-3 -phosphate dehydrogenase (human)
- H&E Hematoxylin and eosin
- HTS High-throughput Satellite
- Ig Immunoglobulin
- IgG Immunoglobulin G
- IgG 1 Immunoglobulin G 1 ,
- IgG2b Immunoglobulin G2b
- IgG2c Immunoglobulin G2c
- IgM Immunoglobulin M
- IL-25 Interleukin-25
- ILC1 Type 1 innate-like lymphocyte
- ILC2 Type 2 innate-like lymphocyte
- ILC3 Type 3 innate-like lymphocyte
- iLN inguinal Lymph Node
- mAb monoclonal antibody
- MFI mean fluorescent intensity
- mLN mesenteric Lymph Node
- PBS Phosphate-buffered saline solution
- PE Phycoerythrin
- qPCR quantitative Polymerase Chain Reaction
- SDS sodium dodecyl sulfate
- TCA Taurocholic acid
- TCCFA Taurocholate Cycloserine Cefoxitin Fructose Agar
- TcdA C. difficile Toxin A
- TcdB C. difficile Toxin B
- TcdB2 C. difficile Toxin B2. DETAILED DESCRIPTION
- the TcdB may be selected from the group TcdB2, TcdBl, TcdB3, TcdB4, TcdB5, TcdB6, TcdB7, TcdB8, TcdB9, TcdB 10, TcdB 11, and TcdB 12.
- a TcdB2 variant from a highly virulent C. difficile strain delays IgG class switch following vaccination, blocks IgG recall to a vaccine booster, and prevents germinal center formation.
- the mechanism includes TcdB2-dependent increases in B cell expression of CXCR4 and responsiveness to its ligand CXCL12, accounting for altered cell migration and a failure of GC-dependent Bmem.
- TcdB2 exerts a deleterious impact on the mechanisms essential for the establishment of host B cell memory.
- TcdB2 delayed IgG class switch, blocked IgG recall responses, and GC formation in secondary lymphoid organs.
- DEGs differentially expressed genes
- the present disclosure is directed to treatments for a CDI by the administration of a CXCR4 antagonist, wherein TcdB- induced increases in CXCR4-mediated B cell migration above the normal levels is inhibited.
- C. difficile Toxin B refers to a large class of variants having a molecular weight of about 270 kDa.
- the TcdB class includes at least 12 subtypes (including subtypes TcdBl, TcdB2, TcdB3, TcdB4, TcdB5, TcdB6, TcdB7, TcdB8, TcdB9, TcdBlO, TcdBl 1, and TcdBl 2) of varying virulence, each of which includes one or more subvariants.
- CXCR4 antagonists which may be used in embodiments of the present disclosure include, but are not limited to, Plerixafor (AMD3100), Mavorixafor (AMD070), AMD1170, AMD3465, Balixafortide, BPRCX714, BPRCX807, CSV18742, CTCE-9908, CXCR4 Antagonist III, FC122, FC131, HF51116, HZ515H7, ITlt, LFC131, LY2510924, LY2624587, MiRNA-146, MiRNA-193-5p, Motixafortide (BKT-140, 4F-benzoyl-TN 14003, BL-8040), MSX-122, Naringin, PF-06747143, Saikosaponin A, TN14003, Ulocuplumab (MDX-1338, BMS-936564), WZ811, X4-136, and 3OD8, and other CXCR4 antagonists not listed herein.
- the subject may be treated by co-administering a CXCR4 antagonist and an inhibitor of a TcdB.
- the inhibitor of TcdB toxin may be an anti- TcdB monoclonal antibody.
- the anti-TcdB monoclonal antibody may be selected from, but is not limited to, Bezlotoxumab (Zinplava), CANmAbB4, CANmAbBl, CDB1, ABA, A13I, E74F, and PA41.
- the co-administered CXCR4 antagonist may be selected from Plerixafor (AMD3100), Mavorixafor (AMD070), AMD1170, AMD3465, Balixafortide, BPRCX714, BPRCX807, CSV18742, CTCE-9908, CXCR4 Antagonist 111, FC122, FC131, HF51116, HZ515H7, ITlt, LFC131, LY2510924, LY2624587, MiRNA-146, MiRNA-193-5p, Motixafortide (BKT-140, 4F-benzoyl-TN14003, BL-8040), MSX-122, Naringin, PF-
- Table 1A Examples of CXCR4 antagonist+anti-TcdB monoclonal antibody combination pairs 1
- the table represents 124 unique CXCR4 antagonist+anti-TcdB monoclonal antibody pairs, including 31 pairs consisting of a CXCR4 antagonist and Bezlotoxumab (e.g., Mavorixafor and Bezlotoxumab), 31 pairs consisting of a CXCR4 antagonist and CANmAbB4 (e.g., Plerixafor and CANmAbB4), 31 pairs consisting of a CXCR4 antagonist and CANmAbBl (e.g., Balixafortide and CANmAbBl), and 31 pairs consisting of a CXCR4 antagonist and
- 31 pairs consisting of a CXCR4 antagonist and Bezlotoxumab e.g., Mavorixafor and Bezlotoxumab
- CANmAbB4 e.g., Plerixafor and CANmAbB4
- 31 pairs consisting of a CXCR4 antagonist and CANmAbBl e.g.
- CDB1 (e.g., CTCE-9908 and CDB1).
- Table IB Examples of CXCR4 antagonist+anti-TcdB monoclonal antibody combination pairs. 1
- the table represents 124 unique CXCR4 antagonist+anti-TcdB monoclonal antibody pairs, including 31 pairs consisting of a CXCR4 antagonist and ABA, 31 pairs consisting of aCXCR4 antagonist and A13I, 31 pairs consisting of a CXCR4 antagonist and E74F, and 31 pairs consisting of a CXCR4 antagonist and PA41.
- the subject may be treated by co-administering a CXCR4 antagonist and an anti-C. difficile antibiotic.
- the anti-C For example, the anti-C.
- the difficile antibiotic may be selected from, but is not limited to, vancomycin, metronidazole, fidaxomicin, surotomycin, and CB- 183315.
- the co-administered CXCR4 antagonist may be selected from Plerixafor (AMD3100), Mavorixafor (AMD070), AMD1170, AMD3465, Basxafortide, BPRCX714,
- the table represents 155 unique CXCR4 antagonist+anti-C. difficile antibiotic pairs, including 31 pairs consisting of a CXCR4 antagonist and vancomycin, 31 pairs consisting of a CXCR4 antagonist and metronidazole, 31 pairs consisting of a CXCR4 antagonist and fidaxomicin, 31 pairs consisting of a CXCR4 antagonist and surotomycin, and 31 pairs consisting of a CXCR4 antagonist and CB-183315.
- the subject may be treated by co-administering a CXCR4 antagonist and an agonistic anti-CD40 monoclonal antibody to provide an immune system boost against the C. difficile.
- agonistic anti-CD40 monoclonal antibodies which may be used include but are not limited to Selicrelumab (CP-870893, R07009789), Dacetuzmumab (SGN-40), ChiLob 7/4, 2141-V11, APX005M (sotigalimab), JNJ-64457107 (ADC-1013), ABBV-428, CDX-1140H, and SEA-CD40.
- the co-adminstered CXCR4 antagonist may be selected from Plerixafor (AMD3100), Mavorixafor (AMD070), AMD 1170, AMD3465, Balixafortide, BPRCX714, BPRCX807, CSV18742, CTCE-9908, CXCR4 Antagonist III, FC122, FC131, HF51116, HZ515H7, ITlt, LFC131, LY2510924, LY2624587, MiRNA-146, MiRNA-193-5p, Motixafortide (BKT-140, 4F-benzoyl-TN 14003, BL-8040), MSX-122, Naringin, PF-06747143, Saikosaponin A, TN14003, Ulocuplumab (MDX-1338,
- the table represents 155 unique CXCR4 antagonist+agonistic anti-CD40 monoclonal antibody pairs, including 31 pairs consisting of a CXCR4 antagonist and Selicrelumab, 31 pairs consisting of a CXCR4 antagonist and Dacetuzmumab, 31 pairs consisting of a CXCR4 antagonist and Sotigalimab, 31 pairs consisting of a CXCR4 antagonist and JNJ-64457107, and 31 pairs consisting of a CXCR4 antagonist and ABBV-428.
- the table represents 124 unique CXCR4 antagonist+agonistic anti-CD40 monoclonal antibody pairs, including 31 pairs consisting of a CXCR4 antagonist and CDX-1 140H, 31 pairs consisting of a CXCR4 antagonist and SEA-CD40, 31 pairs consisting of a CXCR4 antagonist and ChiLob 7/4, and 31 pairs consisting of a CXCR4 antagonist and 2141-V11.
- compositions and methods of the present disclosure are not limited in application to the details of specific embodiments and examples as set forth in the following description.
- the description provided herein is intended for purposes of illustration only and is not intended to be construed in a limiting sense. As such, the language used herein is intended to be given the broadest possible scope and meaning, and the embodiments and examples are meant to be exemplary, not exhaustive. Also, it is to be understood that the phraseology and terminology employed herein is for the purpose of description and should not be regarded as limiting unless otherwise indicated as so.
- compositions and methods of the present disclosure have been described in terms of particular embodiments, it will be apparent to those of skill in the art that variations may be applied to the compositions and methods and in the steps or in the sequence of steps of the methods described herein without departing from the concept, spirit, and scope of the inventive concepts.
- At least one may extend up to 100 or 1000 or more, depending on the term to which it is attached; in addition, the quantities of 100/1000 are not to be considered limiting, as higher limits may also produce satisfactory results.
- the use of the term “at least one of X, Y, and Z” will be understood to include X alone, Y alone, and Z alone, as well as any combination of X, Y, and Z.
- Reference to a series of ranges includes ranges which combine the values of the boundaries of different ranges within the series.
- a series of ranges for example, of 1-10, 10-20, 20-30, 30-40, 40-50, 50-60, 60-75, 75-100, 100-150, 150- 200, 200-250, 250-300, 300-400, 400-500, 500-750, 750-1,000, includes ranges of 1-20, 10- 50, 50-100, 100-500, and 500-1,000, for example.
- Reference to an integer with more (greater) or less than includes any number greater or less than the reference number, respectively.
- reference to less than 100 includes 99, 98, 97, etc.
- a range of 1 to 50 for example also refers to any range bounded by two different integers including 1 , 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, ,21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, and 50, including for example 18 to 25, 20 to 24, or 20-22.
- the words “comprising” (and any form of comprising, such as “comprise” and “comprises”), “having” (and any form of having, such as “have” and “has”), “including” (and any form of including, such as “includes” and “include”) or “containing” (and any form of containing, such as “contains” and “contain”) are inclusive or open-ended and do not exclude additional, unrecited elements or method steps.
- the terms “about” or “approximately” are used to indicate that a value includes the inherent variation of error for the composition, the method used to administer the composition, or the variation that exists among the study subjects.
- the qualifiers “about” or “approximately” are intended to include not only the exact value, amount, degree, orientation, or other qualified characteristic or value, but are intended to include some slight variations due to measuring error, manufacturing tolerances, stress exerted on various parts or components, observer error, wear and tear, and combinations thereof, for example.
- the term “about” or “approximately,” where used herein when referring to a measurable value such as an amount, a temporal duration, and the like, is meant to encompass, for example, variations of ⁇ 25%, or ⁇ 20%, or ⁇ 15%, ⁇ 10%, or ⁇ 5%, or ⁇ 1%, or ⁇ 0.1% from the specified value, as such variations are appropriate to perform the disclosed methods and as understood by persons having ordinary skill in the art.
- the term “substantially” means that the subsequently described event or circumstance completely occurs or that the subsequently described event or circumstance occurs to a great extent or degree. For example, the term “substantially” means that the subsequently described event or circumstance occurs at least 90% of the time, or at least 95% of the time, or at least 98% of the time.
- any reference to "one embodiment” or “an embodiment” means that a particular element, feature, structure, or characteristic described in connection with the embodiment is included in at least one embodiment and may be included in other embodiments.
- the appearances of the phrase “in one embodiment” in various places in the specification are not necessarily all referring to the same embodiment and are not necessarily limited to a single or particular embodiment.
- CXCR4 antagonist refers to a substance or agent that directly or indirectly inhibits CXCR4 activity, for example by reducing or inhibiting the binding of CXCL12 to CXCR4.
- the term CXCR4 antagonist may also refer to a substance or agent that directly or indirectly inhibits CXCR4 expression.
- CXCR5 antagonist refers to a substance or agent that directly or indirectly inhibits CXCR5 activity, for example by reducing or inhibiting the binding of a CXCR5 ligand (CXCL13) to CXCR5.
- CXCR5 antagonist may also refer to a substance or agent that directly or indirectly inhibits CXCR5 expression.
- CCR7 antagonist refers to a substance or agent that directly or indirectly inhibits CCR7 activity, for example by reducing or inhibiting the binding of a CCR7 ligands (CCL19 and CCL21) to CCR7.
- CCR7 antagonist may also refer to a substance or agent that directly or indirectly inhibits CCR7 expression.
- the term “activity” refers to the ability of a substance or agent to modify the molecular, biochemical, or physiological system of a cell, organ, or organism, without reference to how the substance or agent has its physiological effects.
- biologically active refers to a substance that has activity in a biological system (e.g., in a cell (e.g., isolated, in culture, in a tissue, in an organism), in a cell culture, in a tissue, in an organism, etc.).
- a substance that, when administered to an organism, has a biological effect on that organism is considered to be biologically active.
- a biologically active substance is required (e.g., is necessary and sufficient) for the activity to be present; in such circumstances, that portion or fragment is considered to be a "biologically active" portion or fragment.
- pharmaceutically acceptable refers to compounds and compositions which are suitable for administration to humans and/or animals without undue adverse side effects such as toxicity, irritation and/or allergic response commensurate with a reasonable benefit/risk ratio.
- the compounds of the present disclosure may be combined with one or more pharmaceutically-acceptable excipients, including carriers, vehicles, and diluents which may improve solubility, deliverability, dispersion, stability, and/or conformational integrity of the compounds or conjugates thereof.
- pure or “substantially pure” means an object species is the predominant species present (i.e., on a molar basis it is more abundant than any other object species in the composition thereof), and particularly a substantially purified fraction is a composition wherein the object species comprises at least about 50 percent (on a molar basis) of all macromolecular species present.
- a substantially pure composition will comprise more than about 80% of all macromolecular species present in the composition, more particularly more than about 85%, more than about 90%, more than about 95%, or more than about 99%.
- pure or “substantially pure” also refers to preparations where the object species is at least 60% (w/w) pure, or at least 70% (w/w) pure, or at least 75% (w/w) pure, or at least 80% (w/w) pure, or at least 85% (w/w) pure, or at least 90% (w/w) pure, or at least 92% (w/w) pure, or at least 95% (w/w) pure, or at least 96% (w/w) pure, or at least 97% (w/w) pure, or at least 98% (w/w) pure, or at least 99% (w/w) pure, or 100% (w/w) pure.
- the pronoun “we” is intended to refer to all persons involved in a particular aspect of the investigation disclosed herein and as such may include non- inventor laboratory assistants and non-inventor collaborators working under the supervision of the inventors.
- Non-limiting examples of animals within the scope and meaning of this term include dogs, cats, rats, mice, guinea pigs, chinchillas, horses, goats, cattle, sheep, zoo animals, Old and New World monkeys, non-human primates, and humans.
- Treatment refers to therapeutic treatments.
- prevention refers to prophylactic or preventative treatment measures or reducing the onset of a condition or disease.
- treating refers to administering the composition to a subject for therapeutic purposes and/or for prevention.
- modes of administration include oral, topical, retrobulbar, subconjunctival, transdermal, parenteral, subcutaneous, intranasal, intramuscular, intraperitoneal, intravitreal, and intravenous routes, including both local and systemic applications.
- topical is used herein to define a mode of administration through an epithelial surface, such as but not limited to, the skin, eye, or internal epithelial surfaces.
- the compositions of the present disclosure may be designed to provide delayed, controlled, extended, and/or sustained release using formulation techniques which are well known in the art.
- compositions containing a peptide as described herein refer to a composition containing a peptide as described herein that may be administered to a subject by any method known in the art or otherwise contemplated herein, wherein administration of the composition brings about a therapeutic effect as described elsewhere herein.
- effective amount refers to an amount of a peptide or peptide compound which is sufficient to exhibit a detectable therapeutic, amelioration, or treatment effect in a subject without excessive adverse side effects (such as substantial toxicity, irritation and allergic response) commensurate with a reasonable benefit/risk ratio when used in the manner of the present disclosure.
- the effective amount for a subject will depend upon the subject’s type, size and health, the nature and severity of the condition to be treated, the method of administration, the duration of treatment, the nature of concurrent therapy (if any), the specific formulations employed, and the like. Thus, it is not possible to specify an exact effective amount in advance. However, the effective amount for a given situation can be determined by one of ordinary skill in the art using routine experimentation based on the information provided herein.
- Ameliorate means a detectable or measurable improvement in a subject’s condition or a symptom thereof.
- a detectable or measurable improvement includes a subjective or objective decrease, reduction, inhibition, suppression, limit or control in the occurrence, frequency, severity, progression, or duration of the condition, or an improvement in a symptom or an underlying cause or a consequence of the condition, or a reversal of the condition.
- a successful treatment outcome can lead to a “therapeutic effect,” or “benefit” of ameliorating, decreasing, reducing, inhibiting, suppressing, limiting, controlling, or preventing the occurrence, frequency, severity, progression, or duration of a condition, or consequences of the condition in a subject.
- a decrease or reduction in worsening, such as stabilizing the condition is also a successful treatment outcome.
- a therapeutic benefit therefore need not be complete ablation or reversal of the condition, or any one, most or all adverse symptoms, complications, consequences or underlying causes associated with the condition.
- a satisfactory endpoint may be achieved when there is an incremental improvement such as a partial decrease, reduction, inhibition, suppression, limit, control or prevention in the occurrence, frequency, severity, progression, or duration, or inhibition or reversal of the condition (e.g., stabilizing), over a short or long duration of time (e.g., seconds, minutes, hours).
- small molecule means a low molecular weight organic compound that may serve as an enzyme substrate or regulator of biological processes.
- a "small molecule” is a molecule that is less than about 5 kilodaltons (kD) in size.
- provided nanoparticles further include one or more small molecules.
- the small molecule is less than about 4 kD, 3 kD, about 2 kD, or about 1 kD.
- the small molecule is less than about 800 daltons (D), about 600 D, about 500 D, about 400 D, about 300 D, about 200 D, or about 100 D.
- a small molecule is less than about 2000 g/mol, less than about 1500 g/mol, less than about 1000 g/mol, less than about 800 g/mol, or less than about 500 g/mol.
- one or more small molecules are encapsulated within the nanoparticle.
- small molecules are non-poly meric.
- small molecules are not proteins, polypeptides, oligopeptides, peptides, polynucleotides, oligonucleotides, polysaccharides, glycoproteins, proteoglycans, etc.
- a small molecule is a therapeutic.
- a small molecule is an adjuvant.
- a small molecule is a drug.
- provided agents and/or compositions comprising such agents may be provided in particles.
- Particles as used in this context means nanoparticles or microparticles (or in some instances larger particles) which can consist in whole or in part of provided agent(s) and/or other therapeutic agent(s) as described herein.
- Such particles may contain the agent(s) and/or compositions in a core surrounded by a coating, including, but not limited to, an enteric coating.
- the agent(s) and/or compositions also may be dispersed throughout the particles.
- the agent(s) and/or compositions also maybe adsorbed into the particles.
- the particles maybe of any order release kinetics, including zero-order release, first-order release, second-order release, delayed release, sustained release, immediate release, and any combination thereof, etc.
- the particle may include, in addition to the agent(s) and/or compositions, any of those materials routinely used in the art of pharmacy and medicine, including, but not limited to, erodible, nonerodible, biodegradable, or nonbiodegradable material or combinations thereof.
- the particles maybe microcapsules which comprise one or more provided agents in a solution or in a semi-solid state.
- the particles may be of virtually any shape.
- both non-biodegradable and biodegradable polymeric materials can be used in the manufacture of particles for delivering provided agent(s) and/or compositions.
- Such polymers maybe natural or synthetic polymers.
- a polymer is selected based on the period of time over which release is desired.
- Bioadhesive polymers of particular interest include bioerodible hydrogels which may comprise, for example, polyhyaluronic acids, casein, gelatin, glutin, poly anhydrides, polyacrylic acid, alginate, chitosan, poly(methylmethacrylates), poly(ethylmethacrylates), poly(butylmethacrylate), poly(isobutylmethacrylate), poly(hexylmethacrylate), poly(isodecylmethacrylate), poly(laurylmethacrylate), poly (phenylmethacry late), poly (methylacrylate), poly(isopropylacrylate), poly(isobutylacrylate), and poly(octadecylacrylate).
- provided agents and/or compositions comprising such agents maybe contained in controlled release systems.
- controlled release in this context is intended to refer to any drugcontaining formulation in which the manner and profile of drug release from the formulation are controlled. This refers to immediate as well as non-immediate release formulations, with non-immediate release formulations including but not limited to sustained release and delayed release formulations.
- sustained release also referred to as “extended release” is used in this context in its conventional sense to refer to a drug formulation that provides for gradual release of a drug over an extended period of time, and that in certain particular (but non-limiting) embodiments, although not necessarily, results in substantially constant blood levels of a drug over an extended time period.
- delayed release is used in this context its conventional sense to refer to a drug formulation in which there is a time delay between administration of the formulation and the release of the drug there from.
- “Delayed release” may or may not involve gradual release of drug over an extended period of time, and thus may or may not be “sustained release.”
- use of a long-term sustained release implant maybe particularly suitable for treatment of chronic conditions with one or more provided agents.
- “Long-term” release as used in this context, means that an implant is constructed and arranged to deliver therapeutic levels of the active ingredient for at least 7 days, and in certain non-limiting embodiments, 30-60 days. Long-term sustained release implants are well-known to those of ordinary skill in the art and include some of the release systems described elsewhere herein.
- mutant or “variant” is intended to refer to a protein, peptide, nucleic acid or organism which has at least one amino acid or nucleotide which is different from the wild type version of the protein, peptide, nucleic acid, or organism and includes, but is not limited to, point substitutions, multiple contiguous or non-contiguous substitutions, chimeras, or fusion proteins, and the nucleic acids which encode them.
- homologous or “% identity” as used herein means a nucleic acid (or fragment thereof) or a protein (or a fragment thereof) having a degree of homology to the corresponding natural reference nucleic acid or protein that may be in excess of 70%, or in excess of 80%, or in excess of 85%, or in excess of 90%, or in excess of 91%, or in excess of 92%, or in excess of 93%, or in excess of 94%, or in excess of 95%, or in excess of 96%, or in excess of 97%, or in excess of 98%, or in excess of 99%.
- the percentage of homology or identity as described herein is typically calculated as the percentage of amino acid residues found in the smaller of the two sequences which align with identical amino acid residues in the sequence being compared, when four gaps in a length of 100 amino acids may be introduced to assist in that alignment (as set forth by Dayhoff, in Atlas of Protein Sequence and Structure, Vol. 5, p. 124, National Biochemical Research Foundation, Washington, D.C. (1972)).
- the percentage homology as described above is calculated as the percentage of the components found in the smaller of the two sequences that may also be found in the larger of the two sequences (with the introduction of gaps), with a component being defined as a sequence of four, contiguous amino acids.
- sequence identity or homology can be determined by comparing the sequences when aligned so as to maximize overlap and identity while minimizing sequence gaps.
- sequence identity may be determined using any of a number of mathematical algorithms.
- a non-limiting example of a mathematical algorithm used for comparison of two sequences is the algorithm of Karlin & Altschul (Proc. Natl. Acad. Sci. USA (1990) 87:2264-2268), modified as in Karlin & Altschul (Proc. Natl. Acad. Sci. USA (1993) 90:5873-5877).
- % identity represents the number of amino acids or nucleotides which are identical at corresponding positions in two sequences of a protein having the same activity or encoding similar proteins. For example, two amino acid sequences each having 100 residues will have 95% identity when 95 of the amino acids at corresponding positions are the same.
- Another example of a mathematical algorithm used for comparison of sequences is the algorithm of Myers & Miller (CABIOS (1988) 4:1 1- 17). Such an algorithm is incorporated into the ALIGN program (version 2.0) which is part of the GCG sequence alignment software package. When utilizing the ALIGN program for comparing amino acid sequences, a PAM 120 weight residue table, a gap length penalty of 12, and a gap penalty of 4 can be used. Yet another useful algorithm for identifying regions of local sequence similarity and alignment is the FAST A algorithm as described in Pearson & Lipman (Proc. Natl. Acad. Sci. USA (1988) 85:2444-2448).
- WU-BLAST Wired University BLAST
- WU-BLAST version 2.0 software WU-BLAST version 2.0 executable programs for several UNIX platforms.
- This program is based on WU-BLAST version 1.4, which in turn is based on the public domain NCBI-BLAST version 1.4 (Altschul & Gish, Methods in Enzymology (1996) 266:460-480; Altschul et al., J Molec Biol. (1990) 215:403-410; Gish & States, Nature Genetics (1993) 3:266-272; Karlin & Altschul, Proc. Natl. Acad. Sci. USA (1993) 90:5873-5877; all of which are incorporated by reference herein).
- the default amino acid comparison matrix is BLOSUM62, but other amino acid comparison matrices such as PAM can be utilized.
- Specific amino acids may be referred to herein by the following designations: alanine: ala or A; arginine: arg or R; asparagine: asn or N; aspartic acid: asp or D; cysteine: cys or C; glutamic acid: glu or E; glutamine: gin or Q; glycine: gly or G; histidine: his or H; isoleucine: ile or I; leucine: leu or L; lysine: lys or K; methionine: met or M; phenylalanine: phe or F; proline: pro or P; serine: ser or S; threonine: thr or T; tryptophan: trp or W; tyrosine: tyr or Y ; and valine: val or V.
- oligonucleotide include any nucleotide sequence which encodes a variant, chimeric, or mutant peptide including polynucleotides in the form of RNA, such as mRNA, or in the form of DNA, including, for instance, cDNA and genomic DNA obtained by cloning or produced by chemical synthetic techniques or by a combination thereof.
- the DNA may be double- stranded or singlestranded. Single-stranded DNA may be the coding strand, also known as the sense strand, or it may be the non-coding strand, also referred to as the anti-sense strand.
- the polynucleotide sequence encoding a mutant peptide or encoding a therapeutically-effective fragment of a mutant peptide can be substantially the same as the coding sequence of the endogenous coding sequence as long as it encodes a biologically active mutant peptide. Further, the mutant peptide, or therapeutically-effective fragment of a mutant peptide may be expressed using polynucleotide sequence(s) which differ in codon usage due to the degeneracies of the genetic code or allelic variations.
- the peptides of the present disclosure include peptide and nucleic acid variants which comprise additional conservative substitutions.
- the variant peptides include, but are not limited to, variants that are not exactly the same as the sequences disclosed herein, but which have, in addition to the substitutions explicitly described for various sequences listed herein, conservative substitutions of amino acid residues which do substantially not impair the agonistic or antagonistic activity or properties of the variants described herein.
- conservative amino acid substitutions include, but are not limited to, ala to gly, ser, or thr; arg to gin, his, or lys; asn to asp, gin, his, lys, ser, or thr; asp to asn or glu; cys to ser; gin to arg, asn, glu, his, lys, or met; glu to asp, gin, or lys; gly to pro or ala; his to arg, asn, gin, or tyr; ile to leu, met, or val; leu to ile, met, phe, or val; lys to arg, asn, gin, or glu; met to gin, ile, leu, or val; phe to leu, met, trp, or tyr; ser to ala, asn, met, or thr; thr to ala, asn, ser, or
- the present constructs or antigen-binding portions thereof can be formulated into compositions for delivery to a mammalian subject.
- the composition can be administered alone and/or mixed with a pharmaceutically acceptable vehicle or excipient.
- Suitable vehicles are, for example (but not by way of limitation), water, saline, dextrose, glycerol, ethanol, or the like, and combinations thereof.
- the vehicle can contain minor amounts of auxiliary substances such as (but not limited to) wetting or emulsifying agents, pH buffering agents, or adjuvants.
- the compositions of the present disclosure can also include ancillary substances, such as (but not limited to) pharmacological agents, cytokines, or other biological response modifiers.
- compositions can be formulated into compositions in either neutral or salt forms.
- Pharmaceutically acceptable salts include (but are not limited to) the acid addition salts (formed with the free amino groups of the active polypeptides) and which are formed with inorganic acids such as, for example, hydrochloric or phosphoric acids, or organic acids such as acetic, oxalic, tartaric, mandelic, and the like. Salts formed from free carboxyl groups can also be derived from inorganic bases such as, for example, sodium, potassium, ammonium, calcium, or ferric hydroxides, and such organic bases as isopropylamine, trimethylamine, 2-ethylamino ethanol, histidine, and procaine.
- compositions can be administered in a single dose treatment or in multiple dose treatments on a schedule and over a time period appropriate to the age, weight, and condition of the subject, the particular composition used, and the route of administration.
- a single dose of the composition according to the disclosure is administered.
- multiple doses are administered.
- the frequency of administration can vary depending on any of a variety of factors, e.g., severity of the symptoms, degree of immunoprotection desired, or whether the composition is used for prophylactic or curative purposes.
- the composition is administered once per month, twice per month, three times per month, every other week, once per week, twice per week, three times per week, four times per week, five times per week, six times per week, every other day, daily, twice a day, or three times a day.
- the duration of treatment i.e., the period of time over which the composition is administered
- the composition can be administered over a period of time ranging from about one day to about one week, from about two weeks to about four weeks, from about one month to about two months, from about two months to about four months, from about four months to about six months, from about six months to about eight months, from about eight months to about 1 year, from about 1 year to about 2 years, or from about 2 years to about 4 years, or more.
- the dosage of an administered active agent for humans will vary depending upon factors such as (but not limited to) the patient's age, weight, height, sex, general medical condition, and previous medical history.
- the recipient is provided with a dosage of the active agent that is in the range of from about 1 mg to about 1000 mg as a single infusion or single or multiple injections, although a lower or higher dosage also may be administered.
- the dosage may be in the range of from about 25 mg to about 100 mg of the active agent per square meter (m 2 ) of body surface area for a typical adult, although a lower or higher dosage also may be administered.
- Non-limiting examples of dosages of the active agent that may be administered to a human subject further include 1 to 500 mg, 1 to 70 mg, or 1 to 20 mg, although higher or lower doses may be used. Dosages may be repeated as needed, for example (but not by way of limitation), once per week for 4-10 weeks, once per week for 8 weeks, or once per week for 4 weeks. It may also be given less frequently, such as (but not limited to) every other week for several months, or more frequently, such as twice weekly or by continuous infusion.
- the present disclosure is directed to a dosing regimen involving administration of the multispecific construct such as disclosed elsewhere herein.
- the dosing regimen may comprise multiple dosing cycles (e.g., wherein the first dosing cycle is a step-up, fractionated dosing cycle).
- the doses may range from about 0.02 mg to about 2.0 mg (e.g., from about 0.02 to about 1.8 mg, from about 0.02 to about 1.6 mg, from about 0.02 to about 1.4 mg, from about 0.02 to about 1.2 mg, from about 0.05 to about 1.8 mg, from about 0.1 to about 1.8 mg, from about 0.4 to about 1.8 mg, from about 0.6 to about 1.8 mg, from about 0.8 to about 1.8 mg, from about 0.5 to about 1.5 mg, from about 0.8 to about 1.2 mg; e.g., about 1 mg), from about 0.05 mg to about 4.0 mg (e.g., from about 0.05 to about 3.5 mg, from about 0.05 to about 3.0 mg, from about 0.05 to about 2.5 mg, from about 0.05 to about 2.2 mg, from about 0.1 to about 3.5 mg, from about 0.5 to about 3.5 mg, from about 1.0 to about 3.5 mg, from about 1 .5 to about 3.5 mg, from about 1 .8 to about 3.5 mg, from about 1 .0
- the dose may range from 50 mg to 200 mg (e.g., from 50 mg to 175 mg, from 50 mg to 150 mg, from 50 mg to 125 mg, from 50 mg to 100 mg, from 50 mg to 75 mg, from 50 mg to 70 mg, from 52 mg to 100 mg, from 52 mg to 75 mg, from 50 mg to 180 mg, from 55 mg to 150 mg, from 55 mg to 100 mg, from 55 mg to 70 mg, from 55 mg to 65 mg, from 58 mg to 62 mg; e.g., about 60 mg).
- the dose may be about 60 mg.
- the dose is about 1 mg. In some embodiments, the dose is about 2 mg.
- the dose is from 20 mg to 200 mg (e.g., from 20 mg to 175 mg, from 20 mg to 150 mg, from 20 mg to 100 mg, from 20 mg to 75 mg, from 30 mg to 175 mg, from 40 mg to 175 mg, from 45 mg to 175 mg, from 50 mg to 175 mg, from 30 mg to 150 mg, from 40 mg to 100 mg, from 45 mg to 75 mg, from 50 mg to 70 mg, from 55 mg to 65 mg, from 58 mg to 62 mg; about 20 mg, about 30 mg, about 45 mg, or e.g., about 60 mg).
- 20 mg to 200 mg e.g., from 20 mg to 175 mg, from 20 mg to 150 mg, from 20 mg to 100 mg, from 20 mg to 75 mg, from 30 mg to 175 mg, from 40 mg to 175 mg, from 45 mg to 175 mg, from 50 mg to 70 mg, from 55 mg to 65 mg, from 58 mg to 62 mg; about 20 mg, about 30 mg, about 45 mg, or e.g., about 60 mg
- the dose is from about 12 mg to about 48 mg (e.g., from about 12 mg to about 42 mg, from about 12 mg to about 36 mg, from about 12 mg to about 30 mg, from about 18 mg to about 48 mg, from about 18 mg to about 42 mg, from about 24 mg to about 42 mg, from about 27 mg to about 42 mg, from about 24 mg to about 36 mg, from about 27 mg to about 33 mg, from about 28 mg to about 32 mg; e.g., about 24 mg, about 27 mg, about 30 mg, about 33 mg, or about 36 mg).
- the dosing regimen comprises administration of a loading dose, such as from 20 mg to 200 mg (e.g., from 20 mg to 175 mg, from 20 mg to 150 mg, from 20 mg to 100 mg, from 20 mg to 75 mg, from 30 mg to 175 mg, from 40 mg to 175 mg, from 45 mg to 175 mg, from 50 mg to 175 mg, from 30 mg to 150 mg, from 40 mg to 100 mg, from 45 mg to 75 mg, from 50 mg to 70 mg, from 55 mg to 65 mg, from 58 mg to 62 mg; e.g., about 60 mg).
- a loading dose such as from 20 mg to 200 mg (e.g., from 20 mg to 175 mg, from 20 mg to 150 mg, from 20 mg to 100 mg, from 20 mg to 75 mg, from 30 mg to 175 mg, from 40 mg to 175 mg, from 45 mg to 175 mg, from 50 mg to 70 mg, from 55 mg to 65 mg, from 58 mg to 62 mg; e.g., about 60 mg).
- the dose is from about 12 mg to about 48 mg (e.g., from about 12 mg to about 42 mg, from about 12 mg to about 36 mg, from about 12 mg to about 30 mg, from about 18 mg to about 48 mg, from about 18 mg to about 42 mg, from about 24 mg to about 42 mg, from about 27 mg to about 42 mg, from about 24 mg to about 36 mg, from about 27 mg to about 33 mg, from about 28 mg to about 32 mg; e.g., about 24 mg, about 27 mg, about 30 mg, about 33 mg, or about 36 mg).
- the active agent is provided in a concentration of about 1 nM, about 5 nM, about 10 nM, about 25 nM, about 50 nM, about 75 nM, about 100 nM, about 150 nM, about 200 nM, about 250 nM, about 300 nM, about 350 nM, about 400 nM, about 500 nM, about 550 nM, about 600 nM, about 700 nM, about 800 nM, about 900 nM, about 1 pM, about 2 pM, about 3 pM, about 4 pM, about 5 pM, about 6 pM, about 7 pM, about 8 pM, about 9 pM, about 10 pM, about 15 pM, about 20 pM, about 25 pM, about 30 pM, about 35 pM, about 40 pM, about 45 pM, about 50 pM, about 60 pM, about 70
- the present compositions When administered orally, the present compositions may be protected from digestion. This can be accomplished either by complexing the construct or antigen-binding portion thereof with a composition to render it resistant to acidic and enzymatic hydrolysis or by packaging the construct or antigen-binding portion thereof in an appropriately resistant carrier such as (but not limited to) a liposome, e.g., such as shown in U.S. Patent No. 5,391 ,377.
- penetrants appropriate to the barrier to be permeated can be used in the formulation.
- Such penetrants are generally known in the art, and include, e.g., for transmucosal administration, bile salts and fusidic acid derivatives.
- detergents can be used to facilitate permeation.
- Transmucosal administration can be through nasal sprays or using suppositories.
- the agents are formulated into ointments, creams, salves, powders, and gels.
- Transdermal delivery systems can also include (for example but not by way of limitation) patches.
- the present compositions can also be administered in sustained delivery or sustained release mechanisms.
- biodegradeable microspheres or capsules or other biodegradeable polymer configurations capable of sustained delivery of a peptide can be included herein.
- the present compositions can be delivered using any system known in the art, including (but not limited to) dry powder aerosols, liquids delivery systems, air jet nebulizers, propellant systems, and the like.
- the pharmaceutical formulation can be administered in the form of an aerosol or mist.
- the formulation can be supplied in finely divided form along with a surfactant and propellant.
- the device for delivering the formulation to respiratory tissue is an inhaler in which the formulation vaporizes.
- Other liquid delivery systems include (for example but not by way of limitation) air jet nebulizers.
- the active agents may be incorporated in lipid monolayers or bilayers, such as (but not limited to) liposomes, such as shown in U.S. Patent Nos. 6,110,490; 6,096,716; 5,283,185; and 5,279,833.
- non-limiting embodiments of the disclosure include formulations in which the active agents have been attached to the surface of the monolayer or bilayer of the liposomes. Liposomes and liposomal formulations can be prepared according to standard methods and are also well known in the art, such as (but not limited to) those disclosed in U.S. Patent Nos. 4,235,871; 4,501,728; and 4,837,028.
- compositions are prepared with carriers that will protect the construct or fragment thereof against rapid elimination from the body, such as (but not limited to) a controlled release formulation, including implants and microencapsulated delivery systems.
- a controlled release formulation including implants and microencapsulated delivery systems.
- Biodegradable, biocompatible polymers can be used, such as (but not limited to) ethylene vinyl acetate, poly anhydrides, poly glycolic acid, collagen, polyorthoesters, and polylactic acid. Methods for preparation of such formulations will be apparent to those skilled in the art.
- the constructs and fragments thereof in general may be formulated to obtain compositions that include one or more pharmaceutically suitable excipients, surfactants, polyols, buffers, salts, amino acids, or additional ingredients, or some combination of these.
- Non-limiting examples of routes of administration of the active agents described herein include parenteral injection, e.g., by subcutaneous, intramuscular, or transdermal delivery.
- Other forms of parenteral administration include (but are not limited to) intravenous, intraarterial, intralymphatic, intrathecal, intraocular, intracerebral, or intracavitary injection.
- the compositions will be formulated in a unit dosage injectable form such as (but not limited to) a solution, suspension, or emulsion, in association with a pharmaceutically acceptable excipient. Such excipients are inherently nontoxic and nontherapeutic.
- Non-limiting examples of such excipients include saline, Ringer's solution, dextrose solution, and Hanks' solution.
- Nonaqueous excipients such as (but not limited to) fixed oils and ethyl oleate may also be used.
- An alternative non-limiting excipient is 5% dextrose in saline.
- the excipient may contain minor amounts of additives such as (but not limited to) substances that enhance isotonicity and chemical stability, including buffers and preservatives.
- constructs can be delivered or administered alone or as pharmaceutical compositions by any means known in the art, such as (but not limited to) systemically, regionally, or locally; by intra-arterial, intrathecal (IT), intravenous (IV), parenteral, intra-pleural cavity, topical, oral, or local administration, as subcutaneous, intra-tracheal (e.g., by aerosol) or transmucosal (e.g., buccal, bladder, vaginal, uterine, rectal, nasal mucosa).
- Administration can be (for example but not by way of limitation) parenteral, intravenous, oral, subcutaneous, intra-arterial, intracranial, intrathecal, intraperitoneal, topical, intranasal, or intramuscular. Administration can also be localized directly into a tumor. Administration into the systemic circulation by intravenous or subcutaneous administration is typical. Intravenous administration can be, for example (but not by way of limitation), by infusion over a period such as (but not limited to) 30-90 min or by a single bolus injection.
- compositions comprising the constructs can be used (for example but not by way of limitation) for subcutaneous, intramuscular, or transdermal administration.
- Compositions can be presented in unit dosage form, e.g., in ampoules or in multi-dose containers, with an added preservative.
- Compositions can also take such forms as suspensions, solutions, or emulsions in oily or aqueous vehicles, and can contain formulatory agents such as suspending, stabilizing, and/or dispersing agents.
- co-administration refers to administration of the CXCR4 antagonist or other antagonist with one or more additional therapeutic agents within the same dosage form, or in separate dosage forms, simultaneously or at essentially the same time, or at different times such that the CXCR4 antagonist or other antagonist is administered before the one or more additional agents is administered or after the one or more additional agents is administered.
- “Essentially at the same time” as used herein generally means within 30 minutes, within 20 minutes, within five minutes, within two minutes, or within in one minute.
- combination therapy refers to a treatment protocol in which two or more therapeutic agents are coadministered, as that term is defined above.
- compositions may be administered in solution.
- the formulation thereof may be in a solution having a suitable pharmaceutically acceptable buffer, such as (but not limited to) phosphate, Tris (hydroxymethyl) aminomethane-HCl, or citrate, and the like. Buffer concentrations should be in the range of 1 to 100 mM.
- the formulated solution may also contain a salt, such as (but not limited to) sodium chloride or potassium chloride in a concentration of 50 to 150 mM.
- a stabilizing agent such as (but not limited to) mannitol, trehalose, sorbitol, glycerol, albumin, a globulin, a detergent, a gelatin, a protamine, or a salt of protamine may also be included.
- RNA interference refers to the silencing or decreasing of gene expression by siRNAs. It is the process of sequence-specific, post- transcriptional gene silencing in animals and plants, initiated by siRNA that is homologous in its duplex region to the sequence of the silenced gene.
- the gene may be endogenous or exogenous to the organism, present integrated into a chromosome or present in a transfection vector that is not integrated into the genome. The expression of the gene is either completely or partially inhibited. RNAi inhibits the gene by compromising the function of a target RNA, completely or partially.
- RISC RNA-induced silencing complex
- RISC RNA-induced silencing complex
- RISC RNA-induced silencing complex
- short RNAs e.g., approximately 22 nucleotides
- the 22-nucleotide RNA sequences are homologous to the target gene that is being suppressed.
- the 22-nucleotide sequences appear to serve as guide sequences to instruct a multicomponent nuclease, RISC, to destroy the specific mRNAs.
- RNA fragments of 21 to 23 nucleotides from the double-stranded RNA These stably associate with an RNA endonuclease, and probably serve as a discriminator to select mRNAs. Once selected, mRNAs are cleaved at sites 21 to 23 nucleotides apart.
- siRNA refers to a short interfering RNA.
- siRNAs comprise a duplex, or double- stranded region, of about 18-25 nucleotides long; often siRNAs contain from about two to four unpaired nucleotides at the 3' end of each strand.
- At least one strand of the duplex or double-stranded region of a siRNA is substantially homologous to or substantially complementary to a target RNA molecule.
- the strand complementary to a target RNA molecule is the "antisense strand"; the strand homologous to the target RNA molecule is the "sense strand", and is also complementary to the siRNA antisense strand.
- siRNAs may also contain additional sequences; non-limiting examples of such sequences include linking sequences, or loops, as well as stem and other folded structures. siRNAs appear to function as key intermediaries in triggering RNA interference in invertebrates and in vertebrates, and in triggering sequence- specific RNA degradation during posttranscriptional gene silencing in plants.
- the active agents may have strand lengths comprising, for example, approximately 12 to 50, or 18 to 40, or 20 to 30 nucleotides, including a targeting sequence (i.e., a seed sequence) that is complementary to a target sequence of a nucleic acid which comprises a portion of an AR coregulator, such as an AR coregulator as listed elsewhere herein, a pre-mRNA transcribed from an AR coregulator, and/or (2) a mature mRNA processed from said pre-mRNA.
- a targeting sequence i.e., a seed sequence
- an oligonucleotide when an oligonucleotide binds to the target sequence of a preprocessed mRNA, it effectively inhibits splicing at the normal splice acceptor site and thus produces a splice variant mRNA, leading to truncated or otherwise aberrant versions of the encoded protein upon translation, or when the oligonucleotide binds to the target region of a mature mRNA, it effectively inhibits proper translation of the mRNA into an encoded protein.
- nucleic acid encompasses the terms “oligonucleotide” and “polynucleotide,” each as a subgenus of the term “nucleic acid.”
- oligonucleotide generally refers to a molecule of between about 3 and about 100 nucleobases in length.
- polynucleotide generally refers to at least one molecule of greater than about 100 nucleobases in length.
- a nucleic acid may encompass a double-stranded molecule that comprises a complementary strand or "complement" of a particular sequence comprising a molecule.
- a singlestranded nucleic acid may be denoted by the prefix "ss,” and a double-stranded nucleic acid by the prefix "ds.
- polynucleotide sequence or “nucleic acid,” as used herein, include any polynucleotide sequence which encodes a peptide or fusion protein (or polypeptide) including polynucleotides in the form of RNA, such as mRNA, or in the form of DNA, including, for instance, cDNA and genomic DNA obtained by cloning or produced by chemical synthetic techniques or by a combination thereof.
- the RNA or DNA may be double-stranded or single-stranded. Single-stranded DNA may be the coding strand, also known as the sense strand, or it may be the non-coding strand, also referred to as the anti-sense strand.
- RNA uracil
- T thymine
- nucleoside is a base-sugar combination.
- the base portion of the nucleoside is normally a heterocyclic base.
- the two most common classes of such heterocyclic bases are the purines and the pyrimidines.
- Nucleotides are nucleosides that further include a phosphate group covalently linked to the sugar portion of the nucleoside.
- the phosphate group can be linked to either the 2', 3' or 5' hydroxyl moiety of the sugar.
- the phosphate groups covalently link adjacent nucleosides to one another to form a linear polymeric compound.
- this linear polymeric structure can be further joined to form a circular structure, however, open linear structures are generally preferred.
- the phosphate groups are commonly referred to as forming the intemucleoside backbone of the oligonucleotide.
- the normal linkage or backbone of RNA and DNA is a 3' to 5' phosphodiester linkage.
- oligonucleotide refers to an oligomer or polymer of RNA or DNA or mimetics thereof. This term includes oligonucleotides composed of naturally -occurring nucleobases, sugars and covalent internucleoside (backbone) linkages as well as oligonucleotides having non-naturally-occurring nucleobases, sugars and synthetic heterocycles and covalent internucleoside (backbone) linkages which function similarly.
- modified or substituted non-natural oligonucleotides as compared to native (natural) forms may have desirable properties such as, for example, enhanced cellular uptake, enhanced affinity for nucleic acid target and increased stability in the presence of nucleases.
- oligonucleotide is also intended to include linked nucleobase sequences containing modified backbones comprising non-natural internucleoside linkages.
- oligonucleotides having modified backbones include those that retain a phosphorus atom in the backbone and those that do not have a phosphorus atom in the backbone.
- nucleoside is intended to refer to a nucleobase linked to a ribose or deoxyribose sugar (a natural nucleoside), and to a nucleobase linked to a non-ribose or non-deoxyribose heterocycle, e.g., a morpholine structure (a non-natural, or modified, nucleoside or other structures described elsewhere herein).
- a series of such modified, non-natural, nucleosides linked together via an internucleoside backbone can also be considered to be an oligonucleotide (a non-natural, or modified, oligonucleotide).
- sucrose where used herein in the context of a nucleoside, is intended to include “non-sugar” heterocyclic compounds, such as morpholines, as the portion of the internucleoside backbone which is linked to the nucleobase.
- Oligonucleotides useful in the compounds and methods disclosed herein also include those comprising entirely or partially of naturally occurring nucleobases.
- Naturally occurring nucleobases as defined herein include adenine, guanine, thymine, cytosine, and uracil.
- 5-methylcytosine (5-me-C) is technically a naturally occurring nucleobase, for the purposes of the present disclosure it will be included in the list of non-natural (a.k.a., modified) nucleobases.
- oligonucleotides of the present disclosure may further include those comprised entirely or partially of modified nucleobases and their corresponding nucleosides.
- modified nucleobases include, but are not limited to, 5-uracil (pseudouridine), dihydrouracil, inosine, ribothymine, 5-me-C, 7-methylguanine, hypoxanthine, xanthine, 5- hydroxymethyl cytosine, 2- aminoadenine, 2-methyladenine, 6-methyladenine, 2- propyladenine, N6-adenine, N6-isopentenyladenine, 2-methylthio-N6-isopentenyladenine, 2- methylguanine, 6-methylguanine, 2-propylguanine, 1-methylguanine, 7-methylguanine, 2,2- dimethylguanine, 2-thiouracil, 2-thiothymine, 2-thiocytosine, 5-fluorouracil, 5 -brom
- the present disclosure also encompasses oligonucleotides which comprise targeting sequences (base sequences) that are complementary to particular nucleic acid target sequences taught herein.
- a nucleic acid is a "complement” or is “complementary” to another nucleic acid when it is capable of base-pairing with the other nucleic acid according to the standard Watson-Crick, Hoogsteen or reverse Hoogsteen binding complementarity rules.
- Polynucleotides (nucleic acids) are described as “complementary” to one another when hybridization occurs in an antiparallel configuration between two single-stranded polynucleotides.
- complementary refers to the capacity for precise pairing between two nucleotides. For example, if a nucleotide at a certain position of an oligonucleotide is capable of hydrogen bonding with a nucleotide at the same position of a DNA or RNA molecule, then the oligonucleotide and the DNA or RNA are considered to be complementary to each other at that position. The oligonucleotide and the DNA or RNA are complementary to each other when a sufficient number of corresponding positions in each molecule are occupied by nucleotides which can hydrogen bond with each other.
- specifically hybridizable and “complementary” are terms which are used to indicate a sufficient degree of complementarity or precise pairing such that stable and specific binding occurs between the oligonucleotide and the DNA or RNA target, and as such, as is understood in the art, the targeting sequence of an antisense oligonucleotide of the present disclosure need not be 100% complementary to that of its target sequence to be specifically hybridizable.
- An oligonucleotide is specifically hybridizable when binding of the oligonucleotide to the target sequence of the DNA or RNA molecule interferes with the normal function of the target DNA or RNA to cause a loss of utility, and there is a sufficient degree of complementarity to avoid non-specific binding of the oligonucleotide to non-target sequences under conditions in which specific binding is desired, i.e., under physiological conditions in the case of in vivo assays or therapeutic treatment, and in the case of in vitro assays, under conditions in which the assays are performed.
- An oligonucleotide and a target sequence are thus complementary to each other when a sufficient number of nucleobases of the oligonucleotide can hydrogen bond with the corresponding nucleobases of the target sequence, such that a desired effect will occur (e.g., antisense inhibition of a target nucleic acid).
- an oligonucleotide in which 18 of 20 nucleobases of the oligonucleotide are complementary to a target sequence, and would therefore specifically hybridize would represent 90 percent complementarity.
- the remaining noncomplementary nucleobases may be clustered or interspersed with complementary nucleobases and need not be contiguous to each other or to complementary nucleobases.
- an oligonucleotide which is 18 nucleobases in length having three noncomplementary nucleobases which are flanked by two regions of complete complementarity with the target nucleic acid, or are distributed in non-contiguous positions would have 83% overall complementarity with the target sequence.
- the seed sequence of the antisense oligonucleotide provided herein is fully complementary (i.e. 100% complementary) to a target sequence of a nucleic acid.
- "fully complementary” means each nucleobase of the referenced portion of an oligonucleotide (e.g., the seed sequence) is capable of precise base pairing with the corresponding nucleobases of a target nucleic acid.
- target sequence refers to a contiguous series of nucleobases in a specific nucleotide sequence (target region), for example of an mRNA.
- target sequence refers to a sequence that is a subsequence (portion or segment) of the target region, or to the entire sequence of the target region.
- a target sequence may include the 5’ terminal nucleobase of a nucleic acid sequence plus adjacent internal nucleobases of the sequence, or the 3’ terminal nucleobase plus adjacent internal nucleobases of the sequence, or only internal nucleobases within the sequence, or the target sequence may be 100% identical to the target region.
- a nucleic acid compound of the present disclosure comprises an oligonucleotide having a nucleobase sequence that, when written in the 5' to 3' direction, comprises the reverse complement of a target sequence of a nucleic acid target region to which it is targeted.
- Complementary and “antisense” can be used interchangeably.
- Complementary also refers to polynucleotide sequences that are substantially complementary (antisense) over their entire length and have very few base mismatches. For example, sequences of fifteen bases in length may be termed complementary when they have complementary nucleotides at thirteen or fourteen positions. Naturally, sequences which are completely complementary will be sequences which are entirely complementary throughout their entire length and have no base mismatches.
- oligonucleotides of the present disclosure are synthesized using one or more modified nucleotides.
- modified and “modification” when used in the context of the constituents of a nucleotide monomer, i.e., sugar, nucleobase and internucleoside linkage (backbone), refer to non-natural changes to the chemical structure of these naturally occurring constituents or the substitutions of these constituents with non-naturally occurring ones, i.e., mimetics.
- the "unmodified” or “naturally occurring” sugar ribose (of RNA) can be modified by replacing the hydrogen at the 2'-position of ribose with a methyl group.
- the naturally occurring internucleoside linkage of nucleic acids is a 3' to 5' phosphodiester linkage that can be modified, in one embodiment, by replacing one of the non-bridging oxygen atoms of the phosphate linker with a sulfur atom to create a phosphorothioate linkage.
- Modified oligonucleotides are structurally distinguishable, but functionally interchangeable with naturally occurring or synthetic unmodified oligonucleotides and usually have enhanced properties such as increased resistance to degradation by exonucleases and endonucleases, or increased binding affinity.
- modifications to the oligonucleotides of the present disclosure encompass substitutions or changes in internucleoside linkages, sugar moieties, or nucleobases.
- nonnatural or “unnatural” refers to an oligonucleotide which comprises at least one modification in an internucleoside linkage, a sugar, and/or a nucleobase thereof, wherein such modified internucleoside linkage, modified sugar, and/or modified nucleobase is not found naturally in DNA or RNA (unless specifically defined otherwise herein).
- Non-naturally occurring internucleoside linkages of the oligonucleotides of the present disclosure include those that contain a phosphorus atom and also those that do not contain a phosphorus atom.
- Numerous phosphorus-containing modified oligonucleotide backbones are known in the art and may be used in the oligonucleotides of the present disclosure.
- Examples of phosphorus-containing internucleoside linkages of non-natural (modified) oligonucleotide backbones which may occur in the presently disclosed oligonucleotides include, but are not limited to, phosphorothioate, phosphorodithioate, phosphoramidite, phosphorodiamidate, morpholino, phosphotriester, aminoalkylphosphotriester, phosphonate, chiral phosphorothioates, methyl and other alkyl phosphonates including 3’-alkylene phosphonate, 5'-alkylene phosphonate and chiral phosphonate, phosphinate, phosphoramidates including 3'-amino phosphoramidate and aminoalkylphosphoramidate, thionophosphoramidate, thionoalkylphosphonate, thionoalkylphosphotriester, selenophosphates and boranophosphates having normal 3 '-5' linkages, 2'-5
- the internucleoside linkages are without phosphorus atoms and may instead comprise short chain alkyl or cycloalkyl internucleoside linkages, mixed heteroatom and alkyl or cycloalkyl intemucleoside linkages, or one or more short chain heteroatomic or heterocyclic intemucleoside linkages.
- the non-naturally occurring internucleoside linkages are uncharged and in others, the linkages are achiral.
- the non-naturally occurring internucleoside linkages are uncharged and achiral, such as peptide nucleic acids (PNAs).
- antisense oligonucleotides of the present disclosure may be defined by a complementary correspondence to a sequence or SEQ ID NO disclosed herein, or segment thereof, and may comprise, independently, one or more modifications to a sugar moiety, an intemucleoside linkage, or a nucleobase.
- oligonucleotide backbones include siloxane backbones; sulfide, sulfoxide and sulfone backbones; formacetyl and thioformacetyl backbones; methylene formacetyl and thioformacetyl backbones; riboacetyl backbones; alkene containing backbones; sulfamate backbones; methyleneimino and methylenehydrazino backbones; sulfonate and sulfonamide backbones; amide backbones; and others having mixed N, 0, S and CH2 component parts. Examples of U.S.
- patents that teach the preparation of such non-phosphorus containing oligonucleotides include, but are not limited to, U.S. Pat. Nos. 5,034,506; 5,166,315; 5,185,444; 5,214,134; 5,216,141 ; 5,235,033; 5,264,562; 5,264,564; 5,405,938; 5,434,257; 5,466,677;
- both the sugar moiety and the internucleoside linkage, i.e., the backbone, of the nucleotide units are replaced with nonnatural groups.
- One such oligomeric compound is referred to as a peptide nucleic acid (PNA).
- PNA peptide nucleic acid
- the sugar-backbone of an oligonucleotide is replaced with an amide containing backbone, in particular an aminoethylglycine backbone.
- the oligonucleotides described herein stabilized against nucleolytic degradation such as by the incorporation of a modification, e.g., a nucleotide modification.
- the oligonucleotides can include a non-natural nucleoside linkage such as a phosphorothioate linkage as the first, second, and/or third internucleotide linkage at the 5' or 3' end of the oligonucleotide sequence.
- the oligonucleotides can include a 2'- modified nucleotide, e.g., a 2'-deoxy, 2'-deoxy-2'-fluoro, 2'-O-methyl, 2'-O- methoxyethyl (2'- O-MOE), 2’-O-aminopropyl (2'-O-AP), 2’-O-dimethylaminoethyl (2'- O-DMAOE), 2’-O- dimethylaminopropyl (2’-O-DMAP), 2'-O- dimethylaminoethyloxyethyl (2'-O-DMAEOE), or 2'-O— N-methylacetamido (2'-0-NMA) nucleotide.
- the oligonucleotides include at least one 2'-O-methyl-modified nucleotide, and in some embodiments, all of the nucleotides include a 2'-O-
- the oligonucleotide can be further modified so as to be conjugated to an organic moiety such as a biogenic molecule that is selected to improve stability, distribution and/or cellular uptake of the oligonucleotide, e.g., cholesterol, forming the nucleic acid compound of the present disclosure.
- an organic moiety can be attached, e.g., to the 3’ or 5' end of the oligonucleotide, and/or at the 2' position of the sugar moiety of a nucleotide of the oligonucleotide, such as the 2' ribose position.
- the nucleic acid compound can further be in isolated form or can be part of a pharmaceutical composition, such as a pharmaceutical composition formulated for parental administration.
- the pharmaceutical compositions can contain one or more nucleic acid compounds, and in some embodiments will contain two or more inhibitory nucleic acid compounds, each one directed to a different target gene.
- the oligonucleotides can be delivered in any of a variety of forms, including in liposomes as described above, and via expression vectors.
- the oligonucleotide can be endogenously expressed from transcription units inserted into DNA or RNA vectors.
- the recombinant vectors can be DNA plasmids or viral vectors for example.
- Viral vectors suitable for producing the presently disclosed oligonucleotides capable of reducing expression or activity of an AR coregulator can be constructed based on, but not limited to, adeno-associated virus, retrovirus, lentivirus, adenovirus, or alphavirus.
- the recombinant vectors which contain a nucleic acid for expressing the oligonucleotides disclosed herein can be delivered as described above and can persist in target cells.
- viral vectors can be used that provide for transient expression of the oligonucleotides.
- Such vectors can be repeatedly administered as necessary.
- the oligonucleotides may interact with the target RNA and inhibit mRNA activity for example.
- the delivery vehicles (vectors) for the oligonucleotides optionally comprise an expression construct which includes an enhancer sequence, a promoter sequence, and other sequences necessary for expression of the products of the oligonucleotide sequence desired to be produced.
- the promoter is cell- specific.
- cell-specific means that the particular promoter selected for the recombinant vector can direct expression of the selected transgene only in a particular cell type.
- the promoter is specific for expression in prostate cells.
- viruses can be used in connection with the methods described herein, including papovaviruses, e.g., SV40, adenovirus, vaccinia virus, adeno-associated virus, herpesviruses including HSV and EBV, and retroviruses of avian, murine, and human origin.
- lentiviral vectors can be used in connection with the methods described herein.
- the lentiviral vector can be a doxycycline-inducible lentiviral vector engineered to express one or more shRNAs or siRNAs.
- Specific vectors which may be used include, but are not limited to, adeno- associated virus vectors (e.g., as disclosed in U.S. Pat. Nos. 5,139,941, 5,436,146, and 5,622,856), an attenuated or gutless adenoviral vectors, (e.g., as disclosed in U.S. Pat. No. 5,935,935), lentiviral vectors (such as are disclosed in U.S. Pat. Nos. 5,665,577; 5,994,136; and 6,013,516), plasmids or synthetic (non-viral) vectors (such as disclosed in U.S. Pat. Nos.
- the vectors may be either monocistronic, bicistronic, or multicistronic.
- a recombinant vector (e.g., lend-, parvo-, AAV) sequence can be packaged as a “particle” for subsequent infection (transduction) of a cell, ex vivo, in vitro or in vivo.
- a recombinant vector sequence is encapsulated or packaged into an AAV particle, the particle can also be referred to as a “rAAV.”
- Such particles include proteins that encapsulate or package the vector genome. Particular examples include viral envelope proteins, and in the case of AAV, capsid proteins.
- the oligonucleotides of the present disclosure may be used as a form of gene therapy.
- gene therapy means genetic modification of cells by the introduction of exogenous DNA or RNA into these cells, such as via an expression vector containing the oligonucleotide, for the purpose of expressing or replicating one or more peptides, polypeptides, proteins, oligonucleotides, or polynucleotides in vivo for the treatment or prevention of disease or deficiencies in humans or animals. Examples of gene therapy are disclosed for example in U.S. Pat. No. 5,399,346. Any suitable route of administration of the oligonucleotide-containing vector may be employed. For example, parenteral (subcutaneous, subretinal, suprachoroidal, intramuscular, intravenous, transdermal) and like forms of administration may be employed. Dosage formulations include injections, implants, or other known and effective gene therapy delivery methods.
- Delivery of the oligonucleotide-expressing vectors can be systemic, such as by intravenous or intra-muscular administration, direct administration to a tumor site, such as a prostate tumor, by administration to target cells ex-planted from a subject followed by reintroduction into the subject, or by any other means that would allow for introduction into the desired target cell.
- the therapeutic and/or pharmaceutical compositions in non-limiting embodiments, contain viral particles per dose in a range of, for example, from about 10 4 to about 10 11 particles, from about 10 s to about 10 10 particles, or from about 10 6 to about 10 9 particles.
- vector genomes are provided in in a range of, for example, from about 10 4 to about 10 14 vector genomes, from about 10 5 to about 10 13 vector genomes, from about 10 6 to about 10 13 vector genomes, from about 10 7 to about 10 13 vector genomes, from about 10 s to about 10 13 vector genomes, or from about 10 9 to about 10 13 vector genomes.
- doses/quantities of AAV vector are useful in the methods set forth herein.
- biogenic molecules may be conjugated to the oligonucleotides to improve their ability to resist degradation, target certain cells, or to cross barriers like cell membranes or the blood brain barrier.
- biogenic molecules that can be conjugated to the oligonucelotides include lipids such as, but not limited to, stearic acid, palmitic acid, docosanoic acid, docosahexanoic acid, docosahexaenoic acid, cholesterol, tocopherol, and other C12-C22 saturated or unsaturated fatty acids; peptides such as but not limited to, cell-penetrating peptides (CPPs) such as penetratin, HIV-1 Tat peptides, pVEC-Cadherin 615-634, polyarginines (6-12), and transportan, linear and cyclic RGD-containing peptides, and SPACE peptide; receptor-specific ligands; aptamers (synthetic oligoribonucleotides);
- the biogenic molecule may be conjugated to the oligonucleotide by any suitable means, such as via linker or a cleavable bond such as but not limited to disulfide, thioether, pH sensitive (e.g., hydrazone or carboxymethylmaleic anhydride), or ethylene glycol.
- suitable means such as via linker or a cleavable bond such as but not limited to disulfide, thioether, pH sensitive (e.g., hydrazone or carboxymethylmaleic anhydride), or ethylene glycol.
- the oligonucleotides or nucleic acid compounds of the present disclosure may be delivered in the form of nanoparticles and microparticles which encapsulate the nucleic acid compounds within liposomes of cationic lipids or within PEG, for example.
- These delivery systems can enhance intracellular delivery either by protecting the nucleic acid compound from nuclease degradation and/or by promoting absorptive endocytosis.
- dioleylphosphatidylethanolamine to liposome delivery systems results in the destabilization of endosomal membranes and promotion of release of the oligonucleotide after endocytosis.
- the nucleic acid compounds can be administered to cells by a variety of other methods known to those of skill in the art, including, but not limited to, ionophoresis, or by incorporation into other vehicles, such as hydrogels, cyclodextrins, biodegradable nanocapsules, and bioadhesive microspheres, or by proteinaceous vectors.
- the nucleic acid compounds can be delivered via the nanoparticle system shown in U.S. Patent Application Publication 2019/0255088.
- the liposomes may comprise amphipathic agents such as lipids which exist in aggregated form as micelles, insoluble monolayers, liquid crystals, or lamellar layers in aqueous solution.
- Suitable lipids for liposomal formulation include, without limitation, monoglycerides, diglycerides, sulfatides, lysolecithin, phospholipids, saponin, bile acids, and the like. Preparation of such liposomal formulations is within the level of skill in the art, as disclosed, for example, in U.S. Patent Nos. 4,235,871; 4,501,728; 4,837,028; and 4,737,323.
- the nanoparticles which contain the nucleic acid compounds of the present disclosure may comprise a pharmaceutically acceptable carrier such as, but not limited to, poly (ethylene-co- vinyl acetate), PVA, partially hydrolyzed poly(ethylene-co-vinyl acetate), poly(ethylene-co-vinyl acetate-co-vinyl alcohol), a crosslinked poly(ethylene-co- vinyl acetate), a cross-linked partially hydrolyzed poly(ethylene-co- vinyl acetate), a cross-linked poly(ethylene-co- vinyl acetate-co-vinyl alcohol), poly-D,L-lactic acid, poly-L-lactic acid, poly glycolic acid, PGA, copolymers of lactic acid and glycolic acid, polycaprolactone, poly valerolactone, poly (anhydrides), copolymers of polycaprolactone with polyethylene glycol, copolymers of polylactic acid with polyethylene glycol, polyethylene glycol; fibrin, GelfoamTM (which is
- Copolymers can comprise from about 1 % to about 99% by weight of a first monomer unit such as ethylene oxide and from 99% to about 1% by weight of a second monomer unit such as propylene oxide.
- Blends of a first polymer such as gelatin and a second polymer such as poly-L-lactic acid or poly glycolic acid can comprise from about 1% to about 99% by weight of the first polymer and from about 99% to about 1 % of the second polymer.
- the oligonucleotides or nucleic acid compounds can be delivered directly by systemic administration such as using oral formulations or stereotactic injection into prostate or prostate tumor, typically in saline with chemical modifications to enable uptake, or other methods described elsewhere herein.
- systemic administration such as using oral formulations or stereotactic injection into prostate or prostate tumor, typically in saline with chemical modifications to enable uptake, or other methods described elsewhere herein.
- the oligonucleotide of the nucleic acid compound has a phosphorothioate backbone
- the oligonucleotide binds to serum proteins, slowing excretion by the kidney.
- the aromatic nucleobases also interact with other hydrophobic molecules in serum and on cell surfaces.
- siRNA delivery systems involve complexing the RNA with cationic and neutral lipids, although encouraging results have also been obtained using peptide transduction domains and cationic polymers.
- Including PEGylated lipids in the formulation prolongs the circulating half-life of the particles.
- one type of optimization of single-stranded DNA or RNA oligonucleotides is the use of chemical modifications to increase the nuclease resistance such as the introduction of phosphorothioate ("PS") linkages in place of the phosphodiester bond.
- PS linkages also improved binding to serum proteins in vivo, increasing half-life and permitting greater delivery of active compound to tissues.
- Chemical modifications to subunits of the nucleotides can also improve potency and selectivity by increasing binding affinity of oligonucleotides for their complementary sequences.
- nucleoside sugars examples include 2'-0-methyl (2'-0-Me), 2'-fluoro (2'-F), and 2'-O-methoxyethyl (2'-M0E) RNA, and others as discussed elsewhere herein.
- LNA locked nucleic acid
- BNA bridged nucleic acid
- Therapeutic administration of the active agents described herein include any method by which a nucleic acid (e.g., DNA or RNA), as known to one of ordinary skill in the art.
- a nucleic acid e.g., DNA or RNA
- delivery may be via, for example, oral administration and/or injection into the prostate gland or tumor or both.
- the active agents can be delivered to an organelle, a cell, a tissue, a tumor or an organism via one or more injections (i.e., a needle injection), such as, for example, orally, subcutaneously, intradermally, intramuscularly, intravenously, or intraperitoneally.
- injections i.e., a needle injection
- a described inhibitory nucleic acid or other active agent can be incorporated into pharmaceutical compositions suitable for administration.
- pharmaceutical compositions can comprise one or more the active agents and a pharmaceutically acceptable carrier.
- active agent may be provided in a sustained release composition.
- immediate or sustained release compositions depends on the nature of the condition being treated. If the condition consists of an acute or over-acute disorder, treatment with an immediate release form can be conducted over a prolonged release composition. Alternatively, for certain preventative or long-term treatments, a sustained release composition may be appropriate.
- the active agent can be administered in a single dose or in multiple doses. Where the administration of the active agent is by infusion, the infusion can be a single sustained dose or can be delivered by multiple infusions. Injection of the active agent can be directly into the tissue at or near the site of aberrant or unwanted target gene expression. Multiple injections of the active agent can be made into the tissue, for example, into the prostate gland, into the prostate tumor, or near the tumor.
- active agents of the disclosure can be administered prophylactically in order to prevent or slow the conversion of a non-aggressive prostate cancer to an aggressive form.
- the active agent can be employed in combination therapies, meaning that the present compositions can be administered concurrently with, prior to, or subsequent to, one or more other desired therapeutic agents or medical procedures.
- the combination of therapies (therapeutic agents or procedures) to employ in a combination regimen will take into account compatibility of the desired therapeutic agents and/or procedures and the desired therapeutic effect to be achieved.
- thetherapies employed can achieve a desired effect for the same disorder (for example, a compound described herein can be administered concurrently with another therapeutic agent used to treat the same disorder), or they can achieve different effects (e.g., control of any adverse effects).
- the advantages of the presently disclosed technology include increased cellular uptake without the requirement of transfection reagents, improved stability and circulation, reduction of adverse stimulation of the immune response, reduced off- target toxicity, specific silencing of a cancer sternness and EMT promoting genes, and improved efficacy against solid tumor cancers. These advantages represent a substantial improvement of nanoparticle and liposome-based RNA silencing based delivery platforms.
- This technology can be used in the cancer related context depending on the contribution of DCLKl-related signaling on tumor related outcomes including metastasis, inhibition, or prevention.
- the route of administration can be, but is not limited to, intravenous, intraperitoneal, or intertumoral, or by any other effective means such as oral, subcutaneous, or transdermal administration. Additionally, given the critical role of tumor sternness in promoting the highly proliferative features of tumorigenesis, these agents can be used in combination with traditional and or other therapeutic modalities.
- SAMiRNA is a self-assembling, micelle-forming double-conjugated RNAi with the oligonucleotide in the middle flanked by a hydrophobic and hydrophilic end that is stable in the circulation due to reduced metabolic clearance.
- the hydrophobic interactions drive the molecular assembly into the micellar structure, allowing for targeting moleties lo be added to the hydrophilic end.
- the linker chemistry used in the present disclosure allows for effective endosomal escape and the SAMiRNA hides the unmodified double- stranded RNA from triggering innate immune stimulation, thus reducing the side effects associated with excessive stimulation of the immune system.
- the siRNA design platform comprises a unique siRNA synthesis algorithm that allows for creation of highly specific siRNAs that can knockdown specific RNA moieties of virtually any gene product.
- Large scale synthesis and high-quality detection protocols, RT- qPCR enables quantitative detection of RNA silencing efficacy.
- HRP horseradish peroxidase
- APC- Cy7-conjugated anti-CD19 (6D5) and Alexa 488-conjugated anti-Ki67 (1 lf6) mAbs were purchased from Biolegend (San Diego, CA).
- PE-conjugated anti-CD19 (1D3) and VioletFluor 450-conjugated anti-CD4 (GK1.5) was purchased from Tonbo (Cytek, San Diego, CA).
- AlexaFluor 750-conjugated anti-CD93 (223427) was purchased from R&D Systems (Minneapolis, MN).
- PE-conjugated anti-Annexin and 7AAD were from Stemcell Technologies (Cambridge, MA).
- FcR-blocking mAh 2.4G2 was from BioXCell (Lebanon, NH).
- APC- conjugated anti-CXCR4 (2B11) was purchased from Invitrogen, (Carlsbad, CA).
- TcdB2, B2A, and D270N were expressed in a Bacillus megaterium expression system (MoBiTec, Gottingen, Germany) and purified by nickel affinity chromatography (GE Life Sciences, Boston, MA) as described in Bland, S.J., et al., Deletion of a 19-Amino-Acid Region in Clostridioides difficile TcdB2 Results in Spontaneous Autoprocessing and Reduced Cell Binding and Provides a Nontoxic Immunogen for Vaccination. Infect Immun, 2019, 87(8).
- mice were anesthetized with a vaporized 4% isoflurane / 96% medical air mixture and injection by the intraperitoneal (i.p.) route with 1 ng TcdB2 in sterile PBS.
- Control mice were injected with PBS or 1 ng of an enzymatically inactivated TcdB mutant known as D270N [38].
- mice were immunized subcutaneously (s.c.) with 10 pg B2A adsorbed to 100 pl of a 2% w/v Alhydrogel alum adjuvant suspension (Invivogen, San Diego, CA) and adjusted to a final volume of 200 pl with sterile PBS. The injection volume was divided equally over both flanks.
- mice were immunized with B2A I alum on day 0 and then boosted on day 67.
- Retro-orbital blood was collected on days 14, 28, 42, 67 (pre-boost), and 81 (14 days postboost). Blood samples were incubated for 2 hr at room temperature, then centrifuged at 13,000 ref for 15 minutes. Sera were collected, aliquoted, and stored at -20°C. Where indicated TcdB2-treated and B2A-immunized mice were injected s.c.
- Bacteria shed were quantified on day 2 post-gavage. Fecal pellets were homogenized with IX PBS, serially diluted, plated on TCCFA and cultured under anaerobic conditions at 37°C. CFUs were counted within 48 hours.
- Wells were incubated with 0.2 pg I ml HRP-conjugated IgGl, IgG2b, IgG2c, or IgM for 1 hr. Wells were washed with PBS- T and developed for 5 min at room temperature by addition of 90 pl of ABTS substrate to each well (SeraCare, Milford, MA). To stop the reaction, 110 pl of 10% w/v SDS solution was added to each well. OD of the samples at an absorbance of 405 nm was measured using the Spectrostar NanoTM spectrophotometer (BMG Labtech, Cary, NC) within 30 min.
- Spectrostar NanoTM spectrophotometer BMG Labtech, Cary, NC
- the hamster epithelial cell line CHO-K1 (American Type Culture Collection, Manassas, VA) was cultured in F12-K medium (Gibco, Life Technologies Corporation, Grand Island, NY) supplemented with 10% FBS, 100 units / ml penicillin, and 100 pg / ml streptomycin (Coming, Manassas, VA). Cells were cultured at 37°C in 5% CO2 and passed every 48 hr using tryptic digestion. Ninety six-well plates (Falcon®, Corning, Durham, NC) were seeded with CHO-K1 cells at a density of 3 x 10 4 cells / well and incubated overnight at 37°C in 5% CO2.
- Sera were diluted to 1 :500 then incubated for 1 hr at 37°C with cell culture media containing TcdB2 at a final concentration of 250 nM.
- the CHO cell culture media was replaced with the sera-TcdB2-media solution (or media lacking TcdB2 or sera).
- Cells were incubated for 24 hr at 37°C before addition of 100 pl media containing 10 pl CCK-8 (Sigma- Aldrich®, Millipore Sigma, Saint Louis, MO). Cells were incubated for a further 2-3 hr at 37°C until the absorbance at 450 nm associated with untreated control cells reached 3.0. Background absorbance was near zero and subtracted from all samples. The absorbance associated with the TcdB-treated control cells was then subtracted. The percent viability was calculated as absorbance (experimental sample) / absorbance (untreated controls) x 100.
- Murine spleens, lymph nodes (axillary, inguinal, and mesenteric) and bone marrow cells were isolated from B6 mice. Cells from spleen and lymph nodes were isolated by mechanical disruption. Bone marrow cells were obtained by trimming the ends of the long bones and flushing with media using a 27-gauge syringe needle. Erythrocytes were removed by hypotonic lysis with ACT.
- Multiscreen HTS ELISPOT wells (Millipore, Bedford, MA) were prepared for antigen coating by incubating with 35 % v/v ethanol for 30 s and washed twice with PBS. The plates were coated overnight with anti-mouse Ig or B2A (10 pg I ml final concentration) at 4”C. Plates were washed 3 times with PBS and blocked with RPMI 1640 containing 10% FBS (Atlanta Biologicals, Flowery Branch, GA) for 2 hr at room temperature.
- FBS Antlanta Biologicals, Flowery Branch, GA
- Isolated bone marrow cells (3xl0 6 cells per well) were added then subjected to a 1 :3 serial dilution such that the wells contained 2.00xl0 6 , 6.67x10 s , 2.22x10 s , or 7.41xl0 4 cells.
- the plates were then incubated in 5% CO2 at 37°C, for 4.5 hr.
- the cells were lysed and removed by 4 washes with PBS-T. Plates were incubated overnight at 4°C with 5% v/v FBS in PBS containing HRP-goat anti-mouse IgGl and IgG2b Ab at a final concentration of 1.0 pg / mL (Southern Biotech, Birmingham, AL).
- the plates were then washed with PBS-T and colorimetric detection performed.
- the developing solution was prepared by dissolving one tablet of AEC (Sigma Chemical Co., St. Louis, MO) in 2.5 mL dimethylformamide (Sigma Chemical Co., St. Louis, MO) and mixing with 47.5 mL of a 0.0075 N acetic acid, 0.0175 M sodium acetate buffer. The solution was passed through a 0.2 pm syringe filter before adding hydrogen peroxide to a final concentration of 0.0005% v/v. One hundred microliters of developing solution was added to each well and incubated at room temperature for 10 min. The reaction was stopped by addition of deionized water.
- Splenocytes and lymph node cells were incubated with anti-FcR-blocking antibody at a final concentration of 2.4G2, 20 pg / ml for 5 min. Cells were then stained with fluorochrome-conjugated mAb cocktails to detect CD4 + T cells (anti-CD4) and B cell (antiCD 19 or anti-B220) populations. After incubating at 4°C (unless noted otherwise) for 1 hr, cells were washed with PBS 3 times by centrifugation at 250 ref for 5 minutes. Cells were then fixed with 1 % w/v paraformaldehyde in PB S . Data were collected using a Stratedigm S 1200Ex flow cytometer (Stratedigm, San Jose, CA) and analyzed with FlowJo software (Version 2.0.1, Tree Star, Ashland, OR).
- splenocytes in RPMI 1640 were added to each well of a 48-well tissue culture plate.
- Vehicle (media), TcdB2, or D270N was added to a final concentration of 0.1, 1, or 10 pM.
- Cells were incubated at 37°C for 30 min, 6, 24, or 48 hr before removal of TcdB2 or D270N by washing.
- Cells were re-suspended in RPMI 1640 medium containing 1% v/v FBS and incubated with an anti-FcR-blocking antibody (2.4G2) at a final concentration of 20 pg / ml for 5 min.
- an anti-FcR-blocking antibody 2.5G2
- Annexin V Binding Buffer (Stemcell Technologies, Cambridge, MA) for 15 min at room temperature. The cells were washed with 1 ml of Annexin V Binding Buffer (250 ref for 5 min), then re-suspended in 200 pl of Annexin V Binding Buffer. The cells were analyzed immediately by flow cytometry.
- Splenocytes or splenic B cells isolated with the STEMCELL Pan B cell Isolation Kit were incubated for 4 hr at 37°C in RPMI 1640 with 5% v/v FBS in the absence or presence of TcdB2 or D270N at a final concentration of 10 pM.
- Cells were washed twice in PBS and re-suspended at IxlO 8 cells / ml in Mammalian Protein Extraction Reagent (M-PER) (Thermo Scientific, Rockford, IL) containing IX protease inhibitor cocktail set 1 (EMD Millipore, Billerica, MA).
- M-PER Mammalian Protein Extraction Reagent
- the Jess Simple Western machine (Protein Simple, San Jose CA) is an automated capillary-based protein separation and immunoblotting system.
- the manufacturer’s standard protocol for the 12-230-kDa Jess separation module was followed.
- Cell lysates were prepared in 0.1X Sample buffer and Fluorescent 5X master mix containing dithiothreitol (400 mM final concentration) to achieve a final loading of 1.5 - 5 pg of protein per capillary. Samples were denatured at 95 °C for 5 min before being loaded into each well.
- the 12-230-kDa ladder and cellular lysates were separated by electrophoresis in their respective capillaries and then fixed.
- Capillary bound proteins were washed, blocked and incubated with 1 : 100 dilution of anti-Racl (Clone 102/Racl, Becton Dickinson, San Diego, CA) and a 1 :500 dilution of anti-GAPDH (Clone 6C5, Abeam, Cambridge, MA) antibodies, then washed again and incubated with a 1 :100 dilution of HRP-conjugated anti-mouse IgG2b (Rael) or 1:200 IgGl (GAPDH) (Southern Biotech, Birmingham, AL).
- the HRP-labeled antibodies were detected using peroxide/luminol-S reagent (Protein Simple, San Jose, CA). Imaging of the chemiluminescence from the capillary bound proteins was performed using Compass for Simple Western software (Version 6.1.0, Protein Simple, San Jose, CA). An internal system control was included in each run.
- mice were injected s.c. with 1 ng TcdB2 in 200 pl sterile PBS, divided equally between the left and right flanks. Controls were given 200 pl sterile PBS. Inguinal lymph nodes were isolated and fixed in 70% v/v ethanol. Paraffin sections (5 pm thick) were mounted on slides by Excalibur Pathology Inc. (Norman, OK). A portion of the slides were stained with H&E and imaged. The remaining slides were used for IHC staining. For IHC staining, slides were deparaffinized by incubating for 3 min in xylene, 1 : 1 xylene:ethanol, then 95-50% ethanol concentrations.
- Slides were then submerged in sodium citrate buffer (10 nM NA Citrate, 0.05% Tween 20, pH 6.0) and microwaved on a low power setting for 1 hr. Slides were washed 3 times with PBS for 5 min and incubated with 0.03% Triton X-100 in PBS for 15 min. After washing with PBS, slides were blocked for 30 min in a humidity chamber by incubation with 10% v/v normal goat serum in 1% w/v BSA. Tissue sections were stained with fluorochrome - conjugated mAbs to detect B cells and proliferating cells to identify GCs. Slides were washed with PBS and mounted in prolong gold medium, dried for 24 hr, then visualized using the Leica M205-MFC THUNDER microscope (Leica, Deerfield, IL).
- mice were injected s.c. with 10 ng TcdB2 in 200 pl PBS (controls were given 200 pl PBS or 10 ng D270N in 200 pl PBS). Each injection was divided equally between the left and right flanks. Axillary and inguinal nodes were isolated after 7 days. Lymph nodes were placed immediately in DNA / RNA Shield (Zymo Research, Irvine, CA). RNA was purified from the lymph nodes using Zymo Quick-RNA Miniprep Plus Kit (Zymo Research, Irvine, CA). Purified RNA was stored at -80°C until needed.
- RNA analysis was performed by the OUHSC core facility using a NanoString nCounter SPRINT profilerTM (NanoString, Seattle, WA) in conjunction with the nCounter Myeloid Innate Immunity PanelTM. Data were analyzed with nSolver 4.0 Advanced Analysis SoftwareTM (Version 2.0.134, NanoString, Seattle, WA). Analysis was performed by normalizing the raw transcript counts utilizing negative synthetic sequences to account for background noise and positive synthetic sequences to account for technical variations.
- B cells and CD4 + T cells were isolated from splenocytes using magnetic bead separation following the manufacturer’s protocol for the EasySepTM Mouse Pan-B Cell and EasySepTM Mouse Naive CD4 + T Cell Isolation Kits (STEMCELL Technologies, Cambridge, MA). Five hundred microliters of serum free media (RPMI 1640) or media containing CXCR12 (Sino Biological, Wayne, PA) at a final concentration of 100 ng / ml was added to the lower chamber of a 48-well transwell plate. Thirty thousand cells in a 300 pl volume, consisting of either total splenocytes, isolated B cells, or isolated CD4 + T cells in serum free media were added to the upper chamber of the transwell plates.
- Transwell inserts were then placed in plate wells containing 700 pl of 70% ethanol for 10 min and allowed to air dry for 10 min to fix migratory cells. Once dry, transwell inserts were placed in plate wells containing 600 pl of a 0.02% w/v crystal violet solution for 10 min. The inserts were gently dipped in distilled water to remove excess crystal violet and left to dry overnight. The membrane inserts were imaged using a light microscope at 20X magnification selecting four different randomized fields of view per well. Migratory cells in each field were counted.
- C. difficile spore preparation
- C. difficile R20291 spores were prepared and isolated. Briefly, a single colony grown on BHI+TCA was confluently streaked onto a pre-reduced 70:30 agar plate and incubated at 37°C for 5 days in an anaerobic chamber. Spores were harvested from the agar surface after 5 days by adding 1.5 ml sterile water and scraping the cell/spore material into a 1.5 ml microcentrifuge tube. Cell / spore material was stored at 4°C for 1 week and then separated in a 50% w/v sucrose gradient (centrifuged at 250 ref for 20 min).
- spore pellet was washed 5 times with sterile distilled water and placed in a glass vial at 4°C.
- C. difficile spores were enumerated by plating on TCCFA.
- a pre-calculated concentration of spore inoculum was heated at 65 °C for 20 min, then allowed to cool for 5 min at room temperature.
- mice were housed in sterile cages with sterile food. The animals were provided with Cefoperazone sodium salt (Millipore Sigma, St. Louis, MO) in distilled drinking water (0.5 g / 1) for ten days, followed by two days of distilled water (Thermofisher, Waltham, MA). Mice were infected by oral gavage with 1 X 10 7 heat-treated C. difficile R20291 spores or distilled water to control for the gavage procedure. C. difficile -associated pathology was assessed by monitoring daily weights, and other clinical signs such as lethargy, hunched posture, and diarrhea. Animals were euthanized if the weight loss reached 20.0% or the mice were moribund.
- Bacteria shed were quantified on day 2 post-gavage by homogenizing fecal pellets with IX PBS, serially diluted, plated on TCCFA and cultured under anaerobic conditions at 37°C. CFUs were counted within 48 hr.
- TcdB2 delays IgG class switch during primary responses and inhibits IgG recall responses: [0199] B6 mice were treated with TcdB2 before immunization to evaluate its impact on humoral immunity to the C. difficile vaccine antigen B2A (FIG. 1). TcdB2 had no demonstrable effect on B2A-specific IgM titers at 14-, 28-, and 42-days following immunization, but delayed IgGl and IgG2b production (FIG. 2). Primary IgG2c titers were minimal, with the effects of TcdB2 unable to be determined (FIG. 2). The initial treatment with TcdB2 exerted a profound impact on B2A-specific IgG recall responses (FIG.
- mice were treated with TcdB2 or an equivalent amount of TcdB2 with a D270N point mutation rendering it glucosyl transferase null (subsequently referred to as D270N) (FIG. 4). It was observed that only bioactive TcdB2 was able to inhibit primary IgG responses (FIG. 4).
- D270N-treated mice had higher IgGl titers than the TcdB2-treated mice (FIG. 5).
- CD40 activation restores and enhances IgG recall responses in TcdB2-treated mice:
- TcdB2 Given the inhibitory action of TcdB2 on vaccination-induced primary and recall IgG titers, the effects of a higher dose of B2A antigen for immunization and supplemental B cell help were tested. This was achieved by administration of an agonistic anti-CD40 mAb clone FGK4.5. that mimics the effects of CD40L ligation of CD40 in vivo. When using twice the vaccine dose as in the previous experiment, TcdB2 resulted in minimal effects on IgM titers as expected (FIG. 7, upper panel). Primary and recall IgGl titers were not significantly influenced by TcdB2 but CD40 activation resulted in a 31% increase in endpoint titer (FIG.
- TcdB2 exerted strong effects on IgG2b reducing recall IgG2b titers by 84% (FIG. 8, upper panel).
- Administration of the anti-CD40 mAb restored and enhanced recall IgG2b titers such that they were 330% higher than that observed in control mice (FIG. 8, upper panel).
- a more pronounced effect was observed when measuring IgG2c responses in that TcdB2 completely abrogated the response, which was rescued by CD40 activation (FIG. 8, lower panel).
- the data were also expressed as fold change between primary and recall titers and each experimental group was compared (FIG. 9).
- TcdB2 had no significant effect on the magnitude of the IgM or IgGl recall response, likely due to the higher dose of B2A than in FIGS. 1-6. In contrast, TcdB2 inhibited the IgG2b response and eliminated IgG2c recall responses. Activation of CD40, rescued the IgG2b and IgG2c recall titers and enhanced them to levels above that observed in controls.
- bone marrow cells were harvested and analyzed by ELISPOT assay to enumerate memory B cell-derived long-lived plasma cells.
- the numbers of B2A-specific IgGl- and IgG2b-secreting plasma cells were consistent with the serum titers (FIGS. 10-11, respectively).
- Total plasma cells (of all specificities) did not differ significantly between experimental groups (FIGS. 38-39).
- the assay background was zero spots (FIG. 40) and TcdB2 did not alter total numbers of cells recovered from the bone marrow (FIG. 40).
- TcdB2-exposed B cells were intrinsically capable of IgG class switch provided alternative T helper (Th) cell type signals were provided and that TcdB2 had a long-term impact on the establishment of a long-lived plasma cell compartment following immunization.
- Th T helper
- TcdB2 is known to induce apoptosis in host epithelial cells through the glucosylation of small GTPases. We determined if TcdB2 altered the number of recoverable cells from lymphoid organs, indicating possible death and clearance in vivo. B6 mice were treated with vehicle, inactive TcdB2 known as B2A (a TcdB2 mutant unable to bind to the primary host receptor) or active TcdB2 before analysis of lymph nodes and spleens by flow cytometry 48 hr and 14 days later (FIGS. 12, 41-45). There were no TcdB2-induced changes in numbers of B cells or CD4 + T cells in secondary lymphoid organs observed.
- TcdB2 induced apoptosis or necrosis of B cells and CD4 + T cells directly
- splenocytes isolated from naive B6 mice were cultured in vitro with or without TcdB2 for up to 12 hours (FIGS. 13-14, and 46-48). The cells were then stained with an Annexin V / 7AAD cocktail and analyzed via flow cytometry to identify apoptotic and necrotic cells.
- TcdB2 did not induce necrosis or apoptosis in B cells or CD4 + T cells above the background level in the cultures. These data demonstrates that TcdB2 did not induce apoptosis of lymphocytes in vitro and suggests a lack of apoptosis in lymphocytes in vivo.
- TcdB2 blocks immunization-induced germinal center formation:
- TcdB2 inhibited IgG recall responses but did not induce B or CD4 + T cell death, or render B cells unresponsive to class switch signals
- the effects TcdB2 has on immunization-induced changed in lymphoid architecture were determined.
- B6 mice were treated with TcdB2 or the inactive D270N mutant, then immunized with Alhydrogel-adsorbed B2A. After 21 days, time for sufficient GC formation, the draining lymph nodes, iLNs, were collected and sections mounted on slides for analysis. From H&E-stained sections (FIG. 17), it was demonstrated that TcdB2 but not D270N resulted in the formation of significantly smaller GC structures (FIG.
- TcdB2 exposure increases CXCR4 gene expression and cell surface expression by lymphocytes:
- CXCR4 gene expression was significantly upregulated when comparing TcdB2 to PBS and TcdB2 to D270N, but not when comparing D270N to PBS, showing that TcdB2 must be enzymatically active to alter CXCR4 expression (FIG. 23A).
- Analysis of CXCR5 and CCR7 expression and their corresponding ligands did not reveal any significant alteration following TcdB2 or D270N treatment (FIG. 23A).
- FIG. 23B shows DEGs following TcdB2 exposure include CXCR4.
- Graphs show the increase in expression relative to vehicle-treated control mice and are normalized to gapdh expression using the DDCT method.
- Graphs show the increase in expression relative to vehicle-treated control mice and are normalized to gapdh expression using the DDCT method.
- FIGS. 23C-23E depict changes in expression of all chemokine receptors and their corresponding ligands in the NanostringTM assay.
- FIG. 23C shows results of an analysis of all chemokine receptor and ligand genes in the gene panel in TcdB2 vs. PBS treatments. No significant changes in expression of the genes were revealed.
- FIG. 23D shows results of an analysis of all chemokine receptor and ligand genes in the gene panel in TcdB2 vs. D270N treatments. No significant changes in expression of the genes were revealed.
- TcdB2 leads to increased migration of B cells to the CXCR4 ligand CXCL12:
- transwell migration assays were performed to measure responsiveness to the CXCR4 ligand CXCL12.
- B6 mice were treated with vehicle, D270N, or TcdB2 before isolation of total splenocytes, B cells, or CD4 + T cells (FIG. 26).
- Background levels of migration increased in splenocytes from TcdB2-treated mice, but the effect was not statistically significant (FIG. 27).
- Cells from vehicle control mice showed a statistically significant response to the CXCL12 ligand, as did cells from D270N- and TcdB2-treated mice (FIG. 27).
- the highest level of migration was observed in splenocytes from TcdB2-treated mice, which was significantly different from all other experimental conditions (FIG. 27).
- TcdB2 had no effect on those responses, demonstrating selective effects on CXCR4-mediated migration (FIG. 29B).
- In vitro treatment of B cells with TcdB2 also revealed a glucosyltransferase-dependent and direct effect on CXCR4-mediated migration (FIG. 29C).
- mice were given TcdB2-secreting C. difficile R20291 spores via oral gavage after antibiotic-induced dysbiosis (FIG. 30). Successful infection was confirmed by measuring weight loss (FIG. 31), C. difficile CFUs in fecal samples (FIG. 32, left), and measurement of cecum and colon length (FIG. 32, right). After 2 days, mice were euthanized and spleen and aLN, iLN, and mLN lymph nodes collected for CXCR4 cell surface expression and lymphocyte migration analyses (FIGS. 33-36).
- a CXCR4-dependent cell migration assay was performed (FIG. 36) using lymphocytes from the draining mLNs.
- Cells from mice infected with C. difficile showed significantly higher migration towards CXCL12 than those mLN cells from the uninfected control mice (FIG. 36).
- the effect was dependent on addition of CXCL12 to the cell culture media.
- CXCR4 antagonists reduce weight loss and circumvent the increase in cellular migration towards CXCL12 in CDI:
- a C. difficile mouse model was used to test the ability of AMD3100 to suppress the increased cellular migration towards CXCL12 post intoxication with TcdB2.
- C57BL/6 (B6) mice were treated with TcdB2 alone or with AMD3100 (at standard dose or low dose). After 24 hours, mice treated with AMD3100 received a second dose.
- Inguinal and axillary lymph nodes (FIG. 52) and spleens (FIG. 53) were collected from the mice 48 hours post intoxication and cell migration towards CXCL12 was measured via transwell migration assay. To identify the effects AMD3100 has during a CDI, B6 mice were infected with TcdB2- secreting C.
- mice were treated with AMD3100 (low dose) at hour 0, 24, and 48 post-gavage. Mouse weight was measured daily (FIGS. 54-55). Mice were euthanized if weight loss reached 20% (FIG. 56).
- TcdB2 the secreted toxin TcdB2
- TcdB2 the main driver of disease pathology in CDI, exerts a profound and deleterious effect on the humoral immune response.
- TcdB2 is shown herein to delay IgG class switch, block GC formation, and curtail IgG recall responses.
- Serum TcdB2-neutralizing IgG titers remain the best-known correlate of protection against recurrent CDI but prior to the present work it has remained unclear why infection fails to stimulate an immune response that adequately prevents CDI recurrence.
- Several factors likely contribute to recurrence, including continued antibiotic therapy- maintained dysbiosis, continued C. difficile exposure in the environment, and possible germination of spores that are resident in host epithelial cells.
- the present work indicates that insufficiently protective B cell memory following infection likely contributes to the overall risk of recurrence.
- TcdB2 The inhibition of IgG class switch and recall responses by TcdB2 was consistent with disrupted GC reactions. The observation that immunization-induced GC formation was blocked by TcdB2 but not the D270N mutant was particularly striking. The effects of TcdB2 were apparent both in GC numbers and in their size, such that fewer, smaller GCs were observed. Given that GCs have inherent complexity, such as dark and light zones, the latter being the location of affinity maturation, it is reasonable to suggest that TcdB2 could differentially impact light and dark zone formation.
- a targeted transcriptomic approach identified the chemokine receptor CXCR4 as being upregulated in draining lymph node cells following TcdB2 administration.
- CXCR4 protein expression was confirmed by flow cytometry to increase following TcdB2 treatment, while CXCR5 expression did not change.
- the conformation of the 7 transmembrane receptor CXCR4 and thus its ligand-binding and signaling properties are regulated by Rael.
- Rael is involved in several important processes, including cellular migration. Rael is responsible for regulating conformational changes in CXCR4 that alter receptor activation and migration towards the chemoattractant CXCL12.
- TcdB2 targets Rael
- TcdB2 may enhance CXCR4 activation and/or induce an upregulation in CXCR4 gene expression, which may also contribute to increased chemotaxis.
- the present results firmly implicate altered CXCR4 B cell migration as a mechanism underpinning the TcdB 2- subverted humoral immune response.
- TcdB2 suppresses host humoral immune response, particularly the inhibition of antibody recall response through the stunting of GC formation. As demonstrated by results shown above, this blockage in GC formation is in part due to TcdB2 upregulating the chemokine receptor CXCR4. Therefore, inhibiting CXCR4 can provide a treatment for decreasing the toxic effects of TcdB2 and minimizing the potential for CDI reinfection.
- AMD3100 (Plerixafor) is an FDA approved drug which functions as an CXCR4 antagonist causing inhibition of binding with its chemokine CXCL12. Therefore, using AMD3100, or other CXCR4 antagonists, such as but not limited to those listed elsewhere herein, will reduce CDI recurrence after initial infection and will reduce the toxic effect during C. difficile infection.
- a C. difficile mouse model was used to test the ability of AMD3100 to suppress the increased cellular migration towards CXCL12 post intoxication with TcdB2.
- C57BL/6 (B6) mice were treated with TcdB2 alone or with AMD3100 (at standard dose or low dose). After 24 hours, mice treated with AMD3100 received a second dose. Inguinal and axillary lymph nodes and spleens were collected from the mice 48 hours post intoxication and cell migration towards CXCL12 was measured.
- B6 mice were infected with TcdB2-secreting C. difficile R20291 spores via oral gavage after antibiotic-induced dysbiosis. A portion of mice were treated with AMD3100 (low dose) at hour 0, 24, and 48 post-gavage. Mouse weight was measured daily.
- Results showed that inhibition of CXCR4 activity with AMD3100 reduces weight loss and circumvents the increase in cellular migration towards CXCL12 induced by TcdB2, and reduces morbidity in initial CDI.
- anti-CXCR4 treatments i.e., including but not limited to the CXCR4 antagonists described elsewhere herein
- CXCR4 antagonists which may be used in the anti-CDI treatments of the present disclosure include, but are not limited to, Plerixafor (AMD3100), Mavorixafor (AMD070), AMD1170, AMD3465, Balixafortide, BPRCX714, BPRCX807, CS VI 8742, CTCE-9908, CXCR4 Antagonist III, FC 122, FC131, HF51116, HZ515H7, ITlt, LFC131, LY2510924, LY2624587, MiRNA-146, MiRNA-193-5p, Motixafortide (BKT-140, 4F-benzoyl-TN 14003, BL-8040), MSX-122, Naringin, PF-06747143, Saikosaponin A, TN14003, Ulocuplumab (MDX-1338, BMS-936564), WZ811, X4-136, and 3OD8.
- Plerixafor AMD3
- the CXCR4 antagonists can be administered in a combination therapy with an agonistic anti-CD40 mAb to provide an immune system boost against the C. difficile.
- agonistic anti-CD40 mAbs which may be used include but are not limited to Selicrelumab (CP-870893, R07009789), Dacetuzmumab (SGN-40), ChiLob 7/4, 2141-V11, APX005M (sotigalimab), JNJ-64457107 (ADC-1013), ABBV-428, CDX-1140H, and SEA-CD40.
- agonistic anti-CD40 mAbs which may be used in a combination treatment with a CXCR4 antagonist is not intended to be exclusive of agonistic anti-CD40 mAbs which may be in use in current clinical trials or which may be developed in the future.
- an anti-C. difficile antibiotic may be co-administered to the subject with the CXCR4 antagonist.
- antibiotics that may be used to treat a CDI include, but are not limited to, vancomycin, metronidazole, fidaxomicin, surotomycin, and CB- 183315. This list of antibiotics which may be used in a combination treatment with a CXCR4 antagonist is not intended to be exclusive of anti-CDI antibiotics which may be in use in current clinical trials or which may be developed in the future.
- the subject may be treated by co-administering a CXCR4 antagonist and an inhibitor of TcdB, including TcdB2.
- the inhibitor of TcdB toxin may be an anti-TcdB monoclonal antibody.
- the monoclonal antibody may be at least one of Bezlotoxumab, CANmAbB4, CANmAbBl, CDB1, ABA, Al 31, E74F, and PA41.
- the present disclosure is directed to a method of treating a C. difficile infection (CDI) in a subject in need of such treatment comprising administering to the subject a CXCR4 antagonist, which may be selected from the group which includes, but is not limited to, Plerixafor, Mavorixafor, AMD1170, AMD3465, Balixafortide, BPRCX714, BPRCX807, CSV18742, CTCE-9908, CXCR4 Antagonist III, FC122, FC131, HF51116, HZ515H7, ITlt, LFC131, LY2510924, LY2624587, MiRNA-146, MiRNA-193-5p, Motixafortide, MSX-122, Naringin, PF-06747143, Saikosaponin A, TN14003, Ulocuplumab, WZ811, X4-136, and 30D8.
- a CXCR4 antagonist which may be selected from the group which includes, but is not limited to,
- the CDI may becaused by a C. difficile strain that secretes a C. difficile Toxin B (TcdB).
- the TcdB may be selected from the group TcdB2, TcdBl, TcdB3, TcdB4, TcdB5, TcdB6, TcdB7, TcdB8, TcdB9, TcdB 10, TcdBl l, and TcdB12.
- the subject’s B cell memory (Bmem) against a TcdB may be enhanced by the administration of the CXCR4 antagonist.
- the CDI may be a recurrent CDI.
- an anti-C. difficile antibiotic may be co-administered to the subject with the CXCR4 antagonist.
- an agonistic anti-CD40 monoclonal antibody may be co-administered to the subject with the CXCR4 antagonist.
- the agonistic anti-CD40 monoclonal antibody may be selected from the group which includes, but is not limited to, Selicrelumab, Dacetuzmumab, Sotigalimab, JNJ-64457107, ABBV-428, CDX-1140H, SEA- CD40, ChiLob 7/4, and 2141-Vl l.
- an inhibitor of TcdB may be co-administered to the subject with the CXCR4 antagonist.
- the inhibitor of TcdB toxin may be an anti-TcdB mAb.
- the anti-TcdB mAb may be at least one of bezlotoxumab, CANmAbB4, CANmAbBl, CDB 1 , ABA, Al 31, E74F, and PA41.
- the present disclosure is directed to a substance for use in treating a CDI in a subject, wherein the substance is a CXCR4 antagonist.
- the CXCR4 antagonist may be one or more of Plerixafor, Mavorixafor, AMDI 170, AMD3465, Basxafortide, BPRCX714, BPRCX807, CSV18742, CTCE-9908, CXCR4 Antagonist III, FC 122, FC131 , HF51 116, HZ515H7, ITlt, LFC131 , LY2510924, LY2624587, MiRNA-146, MiRNA-193-5p, Motixafortide, MSX-122, Naringin, PF- 06747143, Saikosaponin A, TN14003, Ulocuplumab, WZ811, X4-136, and 3OD8.
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Epidemiology (AREA)
- Microbiology (AREA)
- Mycology (AREA)
- Immunology (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Organic Chemistry (AREA)
- Molecular Biology (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
Abstract
A method and substance for treating a Clostridioides difficile infection (CDI) in a subject in need of such treatment, comprising administering to the subject a C-X-C chemokine receptor type 4 (CXCR4) antagonist. The CDI may be caused by a C. difficile strain that secretes C. difficile Toxin B (TcdB). The TcdB may be, for example, TcdB2, TcdB1, TcdB3, TcdB4, TcdB5, TcdB6, TcdB7, TcdB8, TcdB9, TcdB10, TcdB11, or TcdB12. The CXCR4 antagonist may be coadministered with an anti-C. difficile antibiotic, an agonistic anti-CD40 monoclonal antibody, and/or an inhibitor of a TcdB such as an anti-TcdB mAb.
Description
TREATMENTS FOR ENHANCING IMMUNE RESPONSE TO CLOSTRIDIOIDES DIFFICILE INFECTIONS
CROSS REFERENCE TO RELATED APPLICATIONS/ INCORPORATION BY REFERENCE STATEMENT
[0001] This application claims benefit of U.S. Provisional Application Ser. No. 63/582,138 filed September 12, 2023, which is hereby expressly incorporated herein by reference in its entirety.
STATEMENT REGARDING FEDERALLY SPONSORED RESEARCH OR DEVELOPMENT
[0002] This invention was made with Government support under Contract Numbers AH74994, AI134719, and AH 19048 awarded by the National Institutes of Health. The Government has certain rights in the invention.
BACKGROUND
[0003] The spore-forming bacterium Clostridioides difficile is the largest cause of nosocomial infection globally, surpassing that caused by methicillin-resistant Staphylococcus aureus. C. difficile infection (CDI) survivors commonly suffer from long-lasting complications due to high rates of disease recurrence, with the chances of relapse increasing after each infection. Prolonged dysbiosis caused by antibiotic therapy and pathogen re-exposure likely contribute to CDI recurrence. However, recent, and compelling evidence indicates that poor B cell memory responses following infection leave murine and human hosts vulnerable to CDI recurrence. Despite these key observations, mechanistic insights as to how C. difficile prevents protective B cell memory following infection are lacking.
[0004] C. difficile secretes large single subunit toxins known as Toxin A (TcdA) and Toxin B (TcdB), both of which are significant virulence factors. Analysis of clinical C. difficile strains and studies in animal models establish TcdB as the main driver of disease pathology. TcdB enters host cells, primarily epithelial cells, by receptor mediated endocytosis. Cytosol- localized TcdB glucosylates small-GTPase family members including Rho, Rac, and Cdc42, leading to altered signaling and cytoskeletal function. TcdB -induced glucosylation of small- GTPases in epithelial cells results in cell rounding and ultimately cell death by apoptosis,
diminishing the structural integrity of the colonic lining, allowing tissue infection and dissemination of C. difficile and its toxins. Critically, disease severity has increased in recent years concomitant with the emergence of C. difficile strains, such as R20291 (ribotype 027), expressing a more virulent version of TcdB known as TcdB2.
[0005] C. difficile spores are transmitted via the fecal-oral route and remain viable on surfaces under diverse environmental conditions. Germination of ingested spores is facilitated by dysbiosis of the host microbiome, often induced by broad-spectrum antibiotic therapy. CDI establishes in the colon and symptoms range from mild diarrhea to severe pseudomembranous colitis, sepsis, and death. CDI can be successfully cleared in animal models and patients by the innate immune response. MyD88-mediated activated neutrophils are necessary for clearance as are Type 1, 2, and 3 innate-like lymphocytes (ILC1, ILC2, ILC3) which have been reported to mitigate disease severity. Additionally, IL-25-regulated eosinophils contribute to protection. While innate mechanisms can resolve disease, there is a failure to establish adequate adaptive immune memory following infection to prevent recurrence. TcdB-specific IgG constitutes the clearest correlate of protection against recurrent CDI. Furthermore, analysis of the TcdB- specific memory B cell compartment in individuals who have recovered from CDI shows an apparent deficiency in Ig class switch and poor TcdB-neutralizing capability of the limited IgG that is produced. These observations are recapitulated in mouse models of recurrent CDI wherein infection poorly stimulates TcdB-specific IgG responses, B cell memory, or expansion of T follicular helper cells. It is to improving the innate immune response against recurrent CDI that the present disclosure is directed.
BRIEF DESCRIPTION OF THE DRAWINGS
[0006] FIG. 1 shows a scheme of a treatment protocol for investigating delayed IgG class switch and inhibited IgG recall response following TcdB2 treatment in vivo. Female B6 mice (n=10 per group) were given PBS vehicle or 1 ng TcdB2 (i.p.) then immunized with 10 pg of Alhydrogel-adsorbed B2A (s.c.) after 5 hours. Mice were bled on days 14, 28, 42, and 67 (primary). On day 67 mice were boosted with 10 pg of Alhydrogel-adsorbed B2A (s.c.) and then bled on day 81 (recall).
[0007] FIG. 2 shows results over time for primary serum B2 -specific IgM, IgGl, IgG2b, and IgG2c endpoint titers as determined by ELISA for the mice of FIG. 1. Significant differences by one way ANOVA in titers and neutralization are as follows: *, P < 0.05 ****, P < 0.0001.
[0008] FIG. 3 shows results pre- and post-bleed for serum B2A-specific IgM, IgGl, IgG2b, and IgG2c endpoint titers as determined by ELISA for the mice of FIG. 1. Significant differences depicted were determined by matched pairs t-test. Significant differences for all groups were determined by one way ANOVA and are as follows: *, P < 0.05 ****, P < 0.0001. [0009] FIG. 4 shows over-time results for female B6 mice (n=5 per group) given 1 ng D270N (left-hand bars) or 1 ng TcdB2 (right-hand bars) in PBS (i.p.) then immunized with 10 pg of Alhydrogel-absorbed B2A (s.c.) after 5 hours. Serum B2A-specific IgM, IgGl, IgG2b and IgG2c endpoint titers were determined by ELISA. Significant differences by one way ANOVA in titers are as follows: *, P < 0.05 ****, P < 0.0001.
[0010] FIG. 5 shows results pre-bleed and post-bleed for female B6 mice (n=5 per group) given 1 ng D270N or 1 ng TcdB2 in PBS (i.p.) then immunized with 10 pg of Alhydrogel- absorbed B2A (s.c.) after 5 hours. Serum B2A-specific IgM, IgGl, IgG2b and IgG2c endpoint titers were determined by ELISA.
[0011] FIG. 6 shows results for CHO cell viability determined using the CCK-8 assay as a measure of TcdB2 neutralization. CHO cells were incubated with TcdB2, sera, or sera and TcdB2 for 24 hours. Significant differences by one way ANOVA in titers and neutralization are as follows: *, P < 0.05 ****, P < 0.0001.
[0012] FIG. 7 shows CD40 activation-associated restoration of IgG recall responses in TcdB2-treated mice. Female B6 mice were given PBS vehicle (Left panels, n=6) or 1 ng TcdB (i.p.) (center and right panels, n=10 in total) then immunized after 5 hr with 20 pg of AlhydrogeLadsorbed B2A (s.c.). TcdB2-treated mice were injected s.c. with 100 pg isotype control mAb (center panels, n=5) or 100 pg anti-CD40 mAb (right panels, n=5) on days 1 and 8. A booster vaccine was administered on day 60 and consisted of 20 pg of B2A in PBS. Blood samples were collected before (day 60) and after (day 74) the booster. Data shows IgM B2A- specific endpoint titers ±SD (upper panels) and IgGl B2A-specific endpoint titers ±SD (lower panels). Matched pairs t-tests were used to measure significance.
[0013] FIG. 8 shows CD40 activation-associated restoration of IgG recall responses in TcdB2-treated mice. Female B6 mice were given PBS vehicle (Left panels, n=6) or 1 ng TcdB (i.p.) (center and right panels, n=10 in total) then immunized after 5 hr with 20 pg of AlhydrogeLadsorbed B2A (s.c.). TcdB2-treated mice were injected s.c. with 100 pg isotype control mAb (center panels, n=5) or 100 pg anti-CD40 mAb (right panels, n=5) on days 1 and 8. A booster vaccine was administered on day 60 and consisted of 20 pg of B2A in PBS. Blood samples were collected before (day 60) and after (day 74) the booster. Data shows IgG2b B2A-
specific endpoint titers ±SD (upper panels) and IgG2c B2A-specific endpoint titers ±SD (lower panels). Matched pairs t-tests were used to measure significance.
[0014] FIG. 9 shows results from re-analysis of the data from FIGS. 7-8. Data were reanalyzed by calculating fold change in endpoint IgM, IgGl, IgG2b, and IgG2c titers following booster vaccine administration and comparing the three experimental groups. One way ANOVA with Tukey’s multiple comparison post-test was used to measure significance (*, P <0.05).
[0015] FIG. 10 shows representative images of ELISPOT wells with spots attributable to B2A-specific IgGl . Graphs depict number of B2A-specific spots per million cells. Each symbol represents an individual mouse. Significance was determined by one-way ANOVA, *, P <0.05. [0016] FIG. 11 shows representative images of ELISPOT wells with spots attributable to B2A-specific IgG2b. Graphs depict number of B2A-specific spots per million cells. Each symbol represents an individual mouse. Significance was determined by one-way ANOVA, ** P <0.01.
[0017] FIG. 12 shows lack of killing but direct intoxication of lymphocytes by TcdB2. Graphs represent absolute CD4+ T cell and B cell count from spleens 14 days post- treatment and show results from two pooled experiments (cell count per half spleen is depicted).
[0018] FIG. 13 shows flow cytometry results of B cell splenocytes after being cultured in vitro with vehicle or 0.01, 0.1 or 1 pM TcdB2 or B2A for 6 hours. A subset of splenocytes were incubated at 45 °C for 1 hour as a positive control. Graphs depicts AnnV / 7AAD dye uptake by cells. Statistical significance was determined by one-way ANOVA, **, P < 0.01. Results are representative of two pooled experiments. Lysates were prepared from splenocytes and analyzed using the capillary automated electrophoresis and blotting system (as shown in FIG. 15).
[0019] FIG. 14 shows flow cytometry results of CD4+ T cell splenocytes after being cultured in vitro with vehicle or 0.01, 0.1 or 1 pM TcdB2 or B2A for 6 hours. A subset of splenocytes were incubated at 45 °C for 1 hour as a positive control. Graphs depicts AnnV / 7AAD dye uptake by cells. Statistical significance was determined by one-way ANOVA, ****, P < 0.0001 . Results are representative of two pooled experiments. Lysates were prepared from splenocytes and analyzed using the capillary automated electrophoresis and blotting system (as shown in FIG. 15).
[0020] FIG. 15 shows blot detection of non-glucosylated Rael and a human recombinant Rael for comparison to FIGS. 13-14.
[0021] FIG. 16 shows results of splenocytes which were untreated or treated in vitro for 2 or 4 hr with TcdB2 at a 10 pM final concentration before preparing cell lysates and performing immunoblots for non-glucosylated Rael and the GAPDH total protein loading control (left panel). B cells (right) were isolated by magnetic separation before treatment with TcdB2 and a D270N-treated control was added. Flow cytometry histogram indicates degree of B cell enrichment in the samples. Left and right panels are representative of two similar experiments.
[0022] FIGS. 17 shows results of TcdB2 blockade of immunization-induced germinal center formation. Representative H&E sections of iLNs from mice treated with: PBS vehicle control (n=4); immunized with 10 pg of Alhydrogel-adsorbed B2A (n=4); treated with 1 ng TcdB2 then immunized (n=4); or with 1 ng D270N then immunized (n=2). Arrows indicate GCs.
[0023] FIG. 18 shows TcdB2 blockade of immunization-induced Germinal Centers. Mean ± SD GC count. Mice were treated as follows: immunized s.c. with PBS vehicle control (n = 7), immunized s.c. with 10 mg of B2A/Alum (n = 7), injected i.p. with 1 ng TcdB2 and then immunized s.c. (n = 7), or injected with 1 ng D270N and then immunized s.c. (n = 6). Data are from three pooled experiments. Statistical significance was determined by one-way ANOVA with Dunnett’s multiple-comparisons post-test. For post-tests: *p < 0.05.
[0024] FIG. 19 shows TcdB2 blockade of immunization-induced Germinal Centers. Mean ± SD GC area per mouse. Mice were treated as follows: immunized s.c. with PBS vehicle control (n = 7), immunized s.c. with 10 mg of B2A/Ahim (n = 7), injected i.p. with 1 ng TcdB2 and then immunized s.c. (n = 7), or injected with 1 ng D270N and then immunized s.c. (n = 6). Data are from three pooled experiments. Statistical significance was determined by one-way ANOVA with Dunnett’s multiple-comparisons post-test. For post-tests: ***p < 0.001, ****p < 0.0001.
[0025] FIG. 20: representative immunofluorescent sections from mice described in FIG. 17. B220+ total B cells (purple) and Ki67+ proliferating GC B cells (green) are shown. Arrows indicate germinal centers.
[0026] FIG. 21: graph represents relative fluorescence signal associated with GCs. Relative signal = [(mean fluorescent intensity of GC) - (mean fluorescent intensity of background) x area of GC]. Statistical significance was determined by one-way ANOVA, **, P < 0.01
[0027] FIG. 22 shows differentially expressed genes following TcdB2 exposure include CXCR4. Female mice (n = 4 per group) were given 200 mL PBS vehicle control, 10 ng D270N,
or 10 ng TcdB2 in PBS by the s.c. route. Seven days post treatment, RNA was purified from axillary and inguinal lymph nodes (aLNs and iLNs). Gene expression was quantified using the Nanostring nCounter SPRINT profiler platform. (A) Upper left panel shows differentially expressed genes (DEGs) comparing TcdB2 to PBS. (B) Upper right panel shows differentially expressed genes (DEGs) comparing TcdB2 to D270N. (C) Lower left panel shows differentially expressed genes (DEGs) comparing D270N to PBS.
[0028] FIG. 23A shows differentially expressed genes following TcdB2 exposure include CXCR4. Gene expression was quantified using the Nanostring nCounter SPRINT profiler platform as described for FIG. 22. Values for cxcr4, cxcr5, ccr7, and their ligands are depicted. Female mice (n = 4 per group) were given 200 mL PBS vehicle control, 10 ng D270N, or 10 ng TcdB2 in PBS by the s.c. route. Seven days post treatment, RNA was purified from axillary lymph nodes (aLNs) and inguinal lymph nodes (iLNs). Gene expression was quantified using the Nanostring Counter SPRINT profiler platform. Summary of the log2 fold change, raw p values, and adjusted p values (Benjamin-Yekutieli method) for each two-way comparison in the experiment. A full list of chemokines and their receptors is shown in FIGS. 23C-23E.
[0029] FIG. 23B shows differentially expressed genes following TcdB2 exposure include CXCR4. Relative expression of cxcr4, cxcr5, and ccr7 in isolated B cells as determined by qPCR. Graphs show the increase in expression relative to vehicle-treated control mice and are normalized to gapdh expression using the DDCT method. Data show mean ± SD for 5 mice per group. Female mice (n = 4 per group) were given 200 mL PBS vehicle control, 10 ng D270N, or 10 ng TcdB2 in PBS by the s.c. route. Seven days post treatment, RNA was purified from aLNs and iLNs. Relative expression of cxcr4, cxcr5, and ccr7 in isolated B cells as determined by qPCR. Graphs show the increase in expression relative to vehicle-treated control mice and are normalized to gapdh expression using the AACT method. Data show mean ± SD for 5 mice per group.
[0030] FIG. 23C shows results of an analysis of all chemokine receptor and ligand genes in the gene panel in TcdB2 vs. PBS treatments. No significant changes in expression of the genes were revealed.
[0031] FIG. 23D shows results of an analysis of all chemokine receptor and ligand genes in the gene panel in TcdB2 vs. D270N treatments. No significant changes in expression of the genes were revealed.
[0032] FIG. 23E shows results of an analysis of all chemokine receptor and ligand genes in the gene panel in D270N vs. PBS treatments. No significant changes in expression of the genes were revealed.
[0033] FIG. 24 shows CXCR4 and CXCR5 expression in B cells following TcdB2 and D270N exposure as measured by flow cytometry. Flow plots show CXCR4 and CXCR5 expression, while graphs depict the percentage of each cell type expressing CXCR4. Female mice (n = 4 per group) were given 200 mL PBS vehicle control, 10 ng D270N, or 10 ng TcdB2 in PBS by the s.c. route.
[0034] FIG. 25 shows CXCR4 and CXCR5 expression in CD4+ T cells following TcdB2 and D270N exposure as measured by flow cytometry. Flow plots show CXCR4 and CXCR5 expression, while graphs depict the percentage of each cell type expressing CXCR4. Female mice (n = 4 per group) were given 200 mL PBS vehicle control, 10 ng D270N, or 10 ng TcdB2 in PBS by the s.c. route. Seven days post treatment, RNA was purified from aLNs and iLNs.
[0035] FIG. 26 shows increased B cell migration towards the CXCR4 chemoattractant CXCL12 following TcdB2 treatment. Female B6 mice (n=4 per group) were given 1 ng TcdB2, 1 ng D270N, or PBS vehicle control by the s.c. route. After 48 hours, splenocytes, B cells, or CD4+ T cells were isolated (B and CD4+ T cells by magnetic separation) and seeded into the top of a transwell. The bottom of the transwells contained serum free media with or without CXCR12. Cells were incubated for 6 hours, then migratory cells were stained with crystal violet and counted.
[0036] FIG. 27 shows increased splenocyte migration towards the CXCR4 chemoattractant CXCL12 following TcdB2 treatment. Female B6 mice (n=4 per group) were given 1 ng TcdB2, 1 ng D270N, or PBS vehicle control by the s.c. route. After 48 hours, splenocytes were seeded into the top of a transwell. The graph depicts quantification of migratory splenocytes averaged from 4 fields of view from each transwell membrane. Results are pooled from two independent experiments. Statistical significance was determined by oneway ANOVA, *, P < 0.05, **, P < 0.01.
[0037] FIG. 28 shows increased B cell migration towards the CXCR4 chemoattractant CXCL12 following TcdB2 treatment. Representative flow cytometry plots of isolated B cells are shown and graphs depict quantification of migratory B cells and CD4+ T cells. Results are pooled from two independent experiments. Statistical significance was determined by one-way ANOVA, ****, P < 0.0001.
[0038] FIG. 29A shows increased B cell migration towards the CXCR4 chemoattractant CXCL12 following TcdB2 treatment. Representative flow cytometry plots of isolated CD4+ T
cells are shown and graphs depict quantification of migratory B cells and CD4+ T cells. Results are pooled from two independent experiments.
[0039] FIG. 29B shows increased B cell migration toward the CXCR4 chemoattractant CXCL12 following TcdB2 treatment. Isolated B cells from vehicle-, D270N-, and TcdB2- treated mice were stimulated in vitro with ligands for CXCR4, CXCR5, and CCR7 (CXCL12, CCL19/CCL21, and CXCL13, respectively). Data are pooled from two independent experiments (mean ± SD, n = 8 per group). Statistical significance was determined by one-way ANOVA, ***, P < 0.001 , ****, P < 0.0001 .
[0040] FIG. 29C shows increased B cell migration toward the CXCR4 chemoattractant CXCL12 following TcdB2 treatment. Isolated splenic B cells were cultured with vehicle, D270N, or TcdB2 for 6 h (n = 3). Graph shows mean ± SD, and data are representative of 2 similar experiments. Statistical significance was determined by one-way ANOVA: ***p < 0.001, ****p < 0.0001.
[0041] FIG. 30 is a scheme showing the experimental protocol for testing CXCR4 expression on B cells and migration of lymphocytes towards the CXCR4 chemoattractant CXCL12 following C. difficile infection in mice. Female B6 mice (n=5 infected, n=4 control) were given 0.5 g / L cefoperazone in distilled drinking water for 10 days, then regular distilled drinking water for two days. Mice were then given 1 X 107 heat treated C. difficile R20291 spores or distilled water via oral gavage and lymphatic organs, colons and cecum, and fecal samples collected 2 days post-gavage.
[0042] FIG. 31 shows weights (mean ± SD) of the mice of FIG. 30 starting two days before gavage.
[0043] FIG. 32 shows C. difficile CFUs (mean ± SD) in fecal samples collected 2 days post-gavage (left); representative images (right) of cecum and colon from control mouse and infected mouse.
[0044] FIG. 33 shows representative flow cytometry plots of CXCR4 versus CXCR5 gating strategy (left plot = control, right plot = infected).
[0045] FIG. 34 shows graphs depicting percent of CXCR4+ B cells from mLN (left) and iLN (right), determined by flow cytometry. Statistical significance was determined by two- tailed T-test.
[0046] FIG. 35 shows graphs depicting percent of CXCR4+ B cells from spleen (left) and aLN (right), determined by flow cytometry. Statistical significance was determined by two- tailed T-test.
[0047] FIG. 36 shows graphs depicting quantification of migratory lymphocytes from mLNs averaged from 4 fields of view from each trans well membrane. Statistical significance was determined by one-way ANOVA , ****, P < 0.0001.
[0048] FIG. 37 shows that TcdB2 does not impact average affinity of B2A-specific IgG. ELIS As were performed as described in Materials and Methods except that plates were coated with B2A at final concentrations of 0.1 and 10 pg/ml. Sera were applied to coated and blocked plates at a 1 : 1000 dilution and detected with IgGl-, IgG2b-, and IgG2c-specific detection Abs as described (left, center, right, respectively). Graphs depict the absorbance ratios for sera applied to wells coated with 0.1 and 10 pg / ml B2A.
[0049] FIGS. 38-40 show that TcdB2 does not impact total IgG abundance or bone marrow plasma cell numbers. Bone marrow ELISPOTS were performed to detect all Ig specificities simultaneously with assays to detect antigen specific cells.
[0050] FIG. 38 shows that TcdB2 does not impact total IgG abundance or bone marrow plasma cell numbers. Bone marrow ELISPOTS were performed to detect all Ig specificities simultaneously with assays to detect antigen specific cells. Images show representative triplicate wells and graph depicts total IgGl secretion for each mouse analyzed.
[0051] FIG. 39 shows, as in FIG. 38, that TcdB2 does not impact total IgG abundance or bone marrow plasma cell numbers. Bone marrow ELISPOTS were performed to detect all Ig specificities simultaneously with assays to detect antigen specific cells. Images show representative triplicate wells and graph depicts total IgG2b secretion for each mouse analyzed. [0052] FIG. 40 shows background in the assays of FIGS. 38 and 39 using anti-Ig coating, and B2A coating in conjunction with anti-IgGl or IgG2b detection Abs.
[0053] FIG. 41 shows that TcdB2 does not affect splenic B cell and CD4+ T cell numbers. Representative flow cytometry plots depict gating strategies for B cell subtypes and CD4+ T cells.
[0054] FIG. 42 shows graphs representing absolute CD4+ T cell and B cell (including subtypes) count from spleen of TcdB2 (1 ng i.p.) pre-treated mice (n=4) or PBS (i.p.) control mice (n=3) 48 hours post-treatment, each symbol represents an individual mouse.
[0055] FIG. 43 shows graphs representing absolute CD4+ T cell and B cell (including subtypes) count from spleen of TcdB2 (1 ng s.c.) pre-treated mice (n=5) or PBS (s.c.) control mice (n=5) 48 hours post-treatment. Total cell count from one half of a spleen per mouse. Graphs represent data from two pooled independent experiments, each symbol represents an individual mouse.
[0056] FIG. 44 shows that TcdB2 does not affect lymph node B cell and CD4+ T cell numbers. Representative flow cytometry plots depict gating strategies for B cell subtypes and CD4+ T cells.
[0057] FIG. 45 shows graphs representing absolute B cell subtypes count from spleens (total cell count from one half of a spleen per mouse), mLNs, and aLNs of TcdB2 (1 ng i.p.) pre-treated mice (n=8), B2A (1 ng i.p.) (n=6), or PBS (i.p.) control mice (n=6) 14 days posttreatment. Data are representative of two independent experiments. Each symbol represents an individual mouse.
[0058] FIG. 46 shows gating strategies for detection of Annexin V+ and 7-AAD+ cells and effect of TcdB2 at early and late time points. Representative flow cytometry plots depict gating strategies for apoptotic/necrotic B and CD4+ T cells in FIGS. 47-48.
[0059] FIG. 47 shows results for splenocytes cultured in vitro with vehicle or 0.01, 0.1 or 1 pM TcdB2 or B2A for 30 min or 12 hr, then examined by flow cytometry (the 6 hr time point is shown in FIGS. 13-14). A subset of splenocytes were incubated at 45°C for 1 hr to induce apoptosis as a positive control. Graphs depict AnnV / 7AAD dye uptake by B cells, identifying apoptotic cells.
[0060] FIG. 48 shows results for splenocytes cultured in vitro with vehicle or 0.01, 0.1 or 1 pM TcdB2 or B2A for 30 min or 12 hr, then examined by flow cytometry (the 6 hr time point is shown in FIGS. 13-14). A subset of splenocytes were incubated at 45°C for 1 hr to induce apoptosis as a positive control. Graphs depict Annexin V / 7AAD dye uptake by CD4+ T cells, identifying apoptotic cells.
[0061] FIG. 49 shows CXCR5 expression by B and CD4+ T cells following TcdB2 toxin treatment Graphs represent percent of CXCR5 on B cells and CD4+ T cells from iLNs post TcdB2, D270N, or PBS treatment.
[0062] FIG. 50 shows graphs representing the percent of CXCR5 on B cells from mLN (left), iLN (right) in uninfected controls or post infection by C.difficile.
[0063] FIG. 51 : graphs represent the percent of CXCR5 on B cells from spleen (left), and aLN (right) in uninfected controls or post infection by C. difficile.
[0064] FIG. 52 shows that the CXCR4 antagonist AMD3100 restores normal CXCR4- mediated cell migration. Female B6 mice were given 1 ng TcdB2 or PBS vehicle control (i.p.) and then given either PBS vehicle or AMD3100 (1 or 10 mg/g of body weight) by the s.c. route. After 48 h, splenocytes were isolated, and migration toward CXCL12 was measured as described for FIGS. 26-27. The graph depicts mean ± SD numbers of migratory splenocytes averaged from 4 fields of view from each Transwell membrane. Data are from 2 pooled
experiments (n = 7 mice per group). Statistical significance was determined by one-way ANOVA with Dunnett’s multiple-comparison post-test; ****p < 0.0001.
[0065] FIG. 53 shows that the AMD3100 rescues TcdB2-suppressed GC formation Mice (n = 7 per group from 2 pooled experiments) were given 1 ng TcdB2 or PBS vehicle control (i.p.) and then immunized s.c. with 10 mg of B2D/Alum after 5 h. At 0, 24, and 48 h post PBS or TcdB2 treatment, mice were treated with AMD3100 or PBS vehicle control (i.p.). Graphs depict the mean ± SD area and number of GCs in iLNs collected 21 days post treatment. Statistical significance was determined by two-tailed t test. Images show representative H&E sections from lymph nodes. Yellow arrows indicate GCs. Thin dark lines were due to a crease in the section. The scale bar depicts 500 mm.
[0066] FIG. 54 shows results (relative weight loss) from mice (n=5 uninfected control; n=10 in infected groups) infected with C. difficile spores (2 x 106) via oral gavage following 10 days of cefeperazone antibiotic treatment in drinking water. Mice were treated with AMD3100 (1 ug/g; i.p.) at hour -2, 24, and 48 post-gavage. Relative weight loss is normalized to 100% at day 0 post-gavage. Asterisks represent significant change in weight compared to uninfected control. Not depicted is significant change in weight of CDI vs AMD3100+CD1 (*) on days 4 and 5 post gavage.
[0067] FIG. 55 depicts the same data as shown in FIG. 54 but contains SD of weight loss.
[0068] FIG. 56 depicts probability of survival of mice post C. difficile infection in the experiments of FIGS. 54-55. Significance determined by two-way ANOVA: *, P < 0.05; ***, P < 0.001.
Abbreviations:
Ab: antibody,
ABTS: 2,2’azinobis(3-ethylbenzthiazolinesulfonic acid),
ACT : Tris-buffered ammonium-chloride,
AEC: 3-amino-9-ethyl-carbazole, aLN: axillary Lymph Node,
AMD3 100: Plerixafor HC1,
AnnV / 7AAD: Annexin V/7 Aminoactinomycin D,
ANOVA: Analysis of Variance,
APC: Allophycocyanin,
BHI: Brain Heart Infusion,
Bmem: B cell memory,
BSA: Bovine serum albumen,
B2A: B2A Tcdb2 mutant antigen,
CCK-8: Cell counting kit 8,
CCR7 : C-C chemokine receptor type 7,
CD40: Cluster of Differentiation 40,
CD4+: Cluster of Differentiation 4 positive,
C. difficile-. Clostridioides difficile,
CDI: Clostridioides difficile infection,
CFU : Colony forming unit,
CHO: Chinese hamster ovary,
CXCL12: CXCR4 ligand,
CXCR4: chemokine (C-X-C motif) receptor type 4,
CXCR5: chemokine (C-X-C motif) receptor type 5,
DDCT: delta-delta CT algorithm,
DEG: differentially expressed gene,
D270N: Tcdb2 D270N mutant antigen,
ELISA: Enzyme-linked Immunosorbent Assay,
ELISPOT: Enzyme- linked Immunospot,
FBS: fetal bovine serum,
FITC: Fluorescein isothiocyanate,
GAPDH: Glyceraldehyde-3 -phosphate dehydrogenase (human),
GC: Germinal center,
H&E: Hematoxylin and eosin,
HRP: Horseradish peroxidase,
HTS: High-throughput Satellite,
Ig: Immunoglobulin,
IgG: Immunoglobulin G,
IgG 1 : Immunoglobulin G 1 ,
IgG2b: Immunoglobulin G2b,
IgG2c: Immunoglobulin G2c,
IgM: Immunoglobulin M,
IHC: immunohistochemical,
IL-25: Interleukin-25,
ILC1: Type 1 innate-like lymphocyte,
ILC2: Type 2 innate-like lymphocyte,
ILC3: Type 3 innate-like lymphocyte, iLN: inguinal Lymph Node, mAb: monoclonal antibody,
MFI: mean fluorescent intensity, mLN: mesenteric Lymph Node,
OD: optical density,
OUHSC: University of Oklahoma Health Sciences Center,
PBS: Phosphate-buffered saline solution,
PE: Phycoerythrin, qPCR: quantitative Polymerase Chain Reaction,
Rac 1 : Rac family small GTPase 1 ,
SD: Standard deviation,
SDS: sodium dodecyl sulfate,
TCA: Taurocholic acid,
TCCFA: Taurocholate Cycloserine Cefoxitin Fructose Agar,
TcdA: C. difficile Toxin A,
TcdB: C. difficile Toxin B,
TcdB2: C. difficile Toxin B2.
DETAILED DESCRIPTION
[0069] Recurrent CDI results in significant morbidity and mortality. We previously established that CDI in mice did not protect against reinfection and was associated with poor B cell memory (Bmem) to the secreted toxin (C. difficile Toxin B (TcdB)). Those observations recapitulated our observations in human TcdB-specific Bmem. A mechanism by which C. difficile suppresses the immune response and leaves the host subject susceptible to recurrent infection, including by subversion of Bmem responses, has been discovered. Results described in the present disclosure therefore provide mechanistic insights into the pathogenesis associated with TcdB. In the present disclosure, the CDI may becaused by a C. difficile strain that secretes a C. difficile Toxin B (TcdB). The TcdB may be selected from the group TcdB2, TcdBl, TcdB3, TcdB4, TcdB5, TcdB6, TcdB7, TcdB8, TcdB9, TcdB 10, TcdB 11, and TcdB 12. In a non-limiting example, a TcdB2 variant from a highly virulent C. difficile strain delays IgG class switch following vaccination, blocks IgG recall to a vaccine booster, and prevents germinal center formation. The mechanism includes TcdB2-dependent increases in B cell expression of CXCR4 and responsiveness to its ligand CXCL12, accounting for altered cell migration and a failure of GC-dependent Bmem. These results, which have been reproduced in a CDI model, demonstrate a TcdB -dependent mechanism that can be targeted to increase Bmem and limit disease recurrence.
[0070] In experiments described below, we tested the hypothesis that TcdB2 exerts a deleterious impact on the mechanisms essential for the establishment of host B cell memory. Using a mouse model in which immunization with an inactivated TcdB2 vaccine antigen was preceded by active TcdB2 treatment, it was observed that TcdB2 delayed IgG class switch, blocked IgG recall responses, and GC formation in secondary lymphoid organs. Analysis of differentially expressed genes (DEGs) revealed an increased expression of CXCR4. An increase in migration of B cells in response to the CXCL12 ligand was also observed, explaining the lack of GC formation and the suppression of B cell memory. Effects of TcdB2 were recapitulated in a CDI challenge model. The present work demonstrates that part of the pathogenic mechanism deployed by TcdB2 includes disruption of B cell migration and formation of GCs, thus subverting the production of B cell memory.
[0071] Therefore, in at least certain non-limiting embodiments, the present disclosure is directed to treatments for a CDI by the administration of a CXCR4 antagonist, wherein TcdB- induced increases in CXCR4-mediated B cell migration above the normal levels is inhibited.
[0072] C. difficile Toxin B (TcdB) refers to a large class of variants having a molecular weight of about 270 kDa. As noted, the TcdB class includes at least 12 subtypes (including
subtypes TcdBl, TcdB2, TcdB3, TcdB4, TcdB5, TcdB6, TcdB7, TcdB8, TcdB9, TcdBlO, TcdBl 1, and TcdBl 2) of varying virulence, each of which includes one or more subvariants.
[0073] CXCR4 antagonists which may be used in embodiments of the present disclosure include, but are not limited to, Plerixafor (AMD3100), Mavorixafor (AMD070), AMD1170, AMD3465, Balixafortide, BPRCX714, BPRCX807, CSV18742, CTCE-9908, CXCR4 Antagonist III, FC122, FC131, HF51116, HZ515H7, ITlt, LFC131, LY2510924, LY2624587, MiRNA-146, MiRNA-193-5p, Motixafortide (BKT-140, 4F-benzoyl-TN 14003, BL-8040), MSX-122, Naringin, PF-06747143, Saikosaponin A, TN14003, Ulocuplumab (MDX-1338, BMS-936564), WZ811, X4-136, and 3OD8, and other CXCR4 antagonists not listed herein.
[0074] In certain embodiments, the subject may be treated by co-administering a CXCR4 antagonist and an inhibitor of a TcdB. For example, the inhibitor of TcdB toxin may be an anti- TcdB monoclonal antibody. The anti-TcdB monoclonal antibody may be selected from, but is not limited to, Bezlotoxumab (Zinplava), CANmAbB4, CANmAbBl, CDB1, ABA, A13I, E74F, and PA41. The co-administered CXCR4 antagonist may be selected from Plerixafor (AMD3100), Mavorixafor (AMD070), AMD1170, AMD3465, Balixafortide, BPRCX714, BPRCX807, CSV18742, CTCE-9908, CXCR4 Antagonist 111, FC122, FC131, HF51116, HZ515H7, ITlt, LFC131, LY2510924, LY2624587, MiRNA-146, MiRNA-193-5p, Motixafortide (BKT-140, 4F-benzoyl-TN14003, BL-8040), MSX-122, Naringin, PF-
06747143, Saikosaponin A, TN14003, Ulocuplumab (MDX-1338, BMS-936564), WZ811, X4-136, and 3OD8, and other CXCR4 antagonists not listed herein. Particular pairwise examples of combinations of CXCR4 antagonists and anti-TcdB monoclonal antibodies are shown in Tables 1A and IB.
Table 1A: Examples of CXCR4 antagonist+anti-TcdB monoclonal antibody combination pairs 1


'The table represents 124 unique CXCR4 antagonist+anti-TcdB monoclonal antibody pairs, including 31 pairs consisting of a CXCR4 antagonist and Bezlotoxumab (e.g., Mavorixafor and Bezlotoxumab), 31 pairs consisting of a CXCR4 antagonist and CANmAbB4 (e.g., Plerixafor and CANmAbB4), 31 pairs consisting of a CXCR4 antagonist and CANmAbBl (e.g., Balixafortide and CANmAbBl), and 31 pairs consisting of a CXCR4 antagonist and
CDB1 (e.g., CTCE-9908 and CDB1).
Table IB: Examples of CXCR4 antagonist+anti-TcdB monoclonal antibody combination pairs.1


'The table represents 124 unique CXCR4 antagonist+anti-TcdB monoclonal antibody pairs, including 31 pairs consisting of a CXCR4 antagonist and ABA, 31 pairs consisting of aCXCR4 antagonist and A13I, 31 pairs consisting of a CXCR4 antagonist and E74F, and 31 pairs consisting of a CXCR4 antagonist and PA41.
[0075] See the following for further source information: H. Qiu, et al. “Novel Clostridium difficile Anti-Toxin (TcdA and TcdB) Humanized Monoclonal Antibodies Demonstrate In Vitro Neutralization across a Broad Spectrum of Clinical Strains and In Vivo Potency in a Hamster Spore Challenge Model” PLoS One. 2016; 11(6): e0157970. J. Steele “Antibody Against TcdB, but Not TcdA, Prevents Development of Gastrointestinal and Systemic Clostridium difficile Disease” The Journal of Infectious Diseases, Volume 207, Issue 2, 15 January 2013, Pages 323-330. H. Kroh, et al. “A neutralizing antibody that blocks delivery of the enzymatic cargo of Clostridium difficile toxin TcdB into host cells” J. Biol. Chem, Vol. 293(3), Jan. 2018, 941-952. Z. Yang et al., “A novel multivalent, single-domain antibody targeting TcdA and TcdB prevents fulminant Clostridium difficile infection in mice” J. Infect. Dis, 210 (2014), pp. 964-972.
[0076] In certain embodiments, the subject may be treated by co-administering a CXCR4 antagonist and an anti-C. difficile antibiotic. For example, the anti-C. difficile antibiotic may be selected from, but is not limited to, vancomycin, metronidazole, fidaxomicin, surotomycin, and CB- 183315. The co-administered CXCR4 antagonist may be selected from Plerixafor (AMD3100), Mavorixafor (AMD070), AMD1170, AMD3465, Balixafortide, BPRCX714,
BPRCX807, CSV18742, CTCE-9908, CXCR4 Antagonist III, FC122, FC131, HF51116, HZ515H7, ITlt, LFC131, LY2510924, LY2624587, MiRNA-146, MiRNA-193-5p, Motixafortide (BKT-140, 4F-benzoyl -TN 14003, BL-8040), MSX-122, Naringin, PF-
06747143, Saikosaponin A, TN14003, Ulocuplumab (MDX-1338, BMS-936564), WZ811, X4-136, and 3OD8, and other CXCR4 antagonists not listed herein. Particular pairwise examples of combinations of CXCR4 antagonists and anti-C. difficile antibiotics are shown in Table 2.
Table 2: Examples of CXCR4 antagonist+anti-C. difficile antibiotic combination pairs1


'The table represents 155 unique CXCR4 antagonist+anti-C. difficile antibiotic pairs, including 31 pairs consisting of a CXCR4 antagonist and vancomycin, 31 pairs consisting of a CXCR4 antagonist and metronidazole, 31 pairs consisting of a CXCR4 antagonist and fidaxomicin, 31 pairs consisting of a CXCR4 antagonist and surotomycin, and 31 pairs consisting of a CXCR4 antagonist and CB-183315.
[0077] In certain embodiments, the subject may be treated by co-administering a CXCR4 antagonist and an agonistic anti-CD40 monoclonal antibody to provide an immune system boost against the C. difficile. Examples of agonistic anti-CD40 monoclonal antibodies which may be used include but are not limited to Selicrelumab (CP-870893, R07009789), Dacetuzmumab (SGN-40), ChiLob 7/4, 2141-V11, APX005M (sotigalimab), JNJ-64457107 (ADC-1013), ABBV-428, CDX-1140H, and SEA-CD40. The co-adminstered CXCR4
antagonist may be selected from Plerixafor (AMD3100), Mavorixafor (AMD070), AMD 1170, AMD3465, Balixafortide, BPRCX714, BPRCX807, CSV18742, CTCE-9908, CXCR4 Antagonist III, FC122, FC131, HF51116, HZ515H7, ITlt, LFC131, LY2510924, LY2624587, MiRNA-146, MiRNA-193-5p, Motixafortide (BKT-140, 4F-benzoyl-TN 14003, BL-8040), MSX-122, Naringin, PF-06747143, Saikosaponin A, TN14003, Ulocuplumab (MDX-1338,
BMS-936564), WZ811, X4-136, and 30D8, and other CXCR4 antagonists not listed herein. Particular pairwise examples of combinations of CXCR4 antagonists and agonistic anti-CD40 monoclonal antibody are shown in Tables 3A-3B. Table 3A. Examples of CXCR4 antagonist+agonistic anti-CD40 monoclonal antibody combination pairs.1
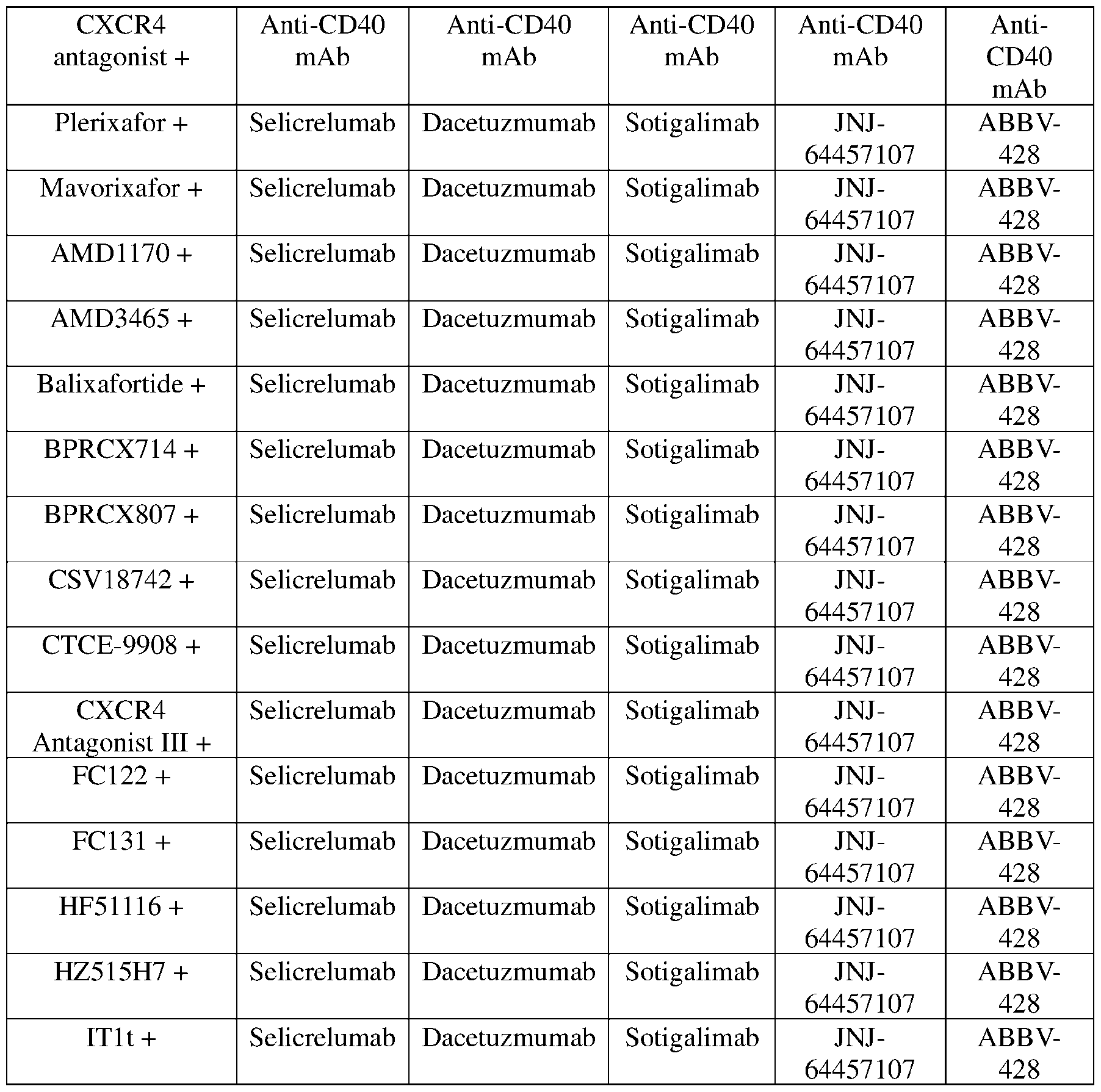
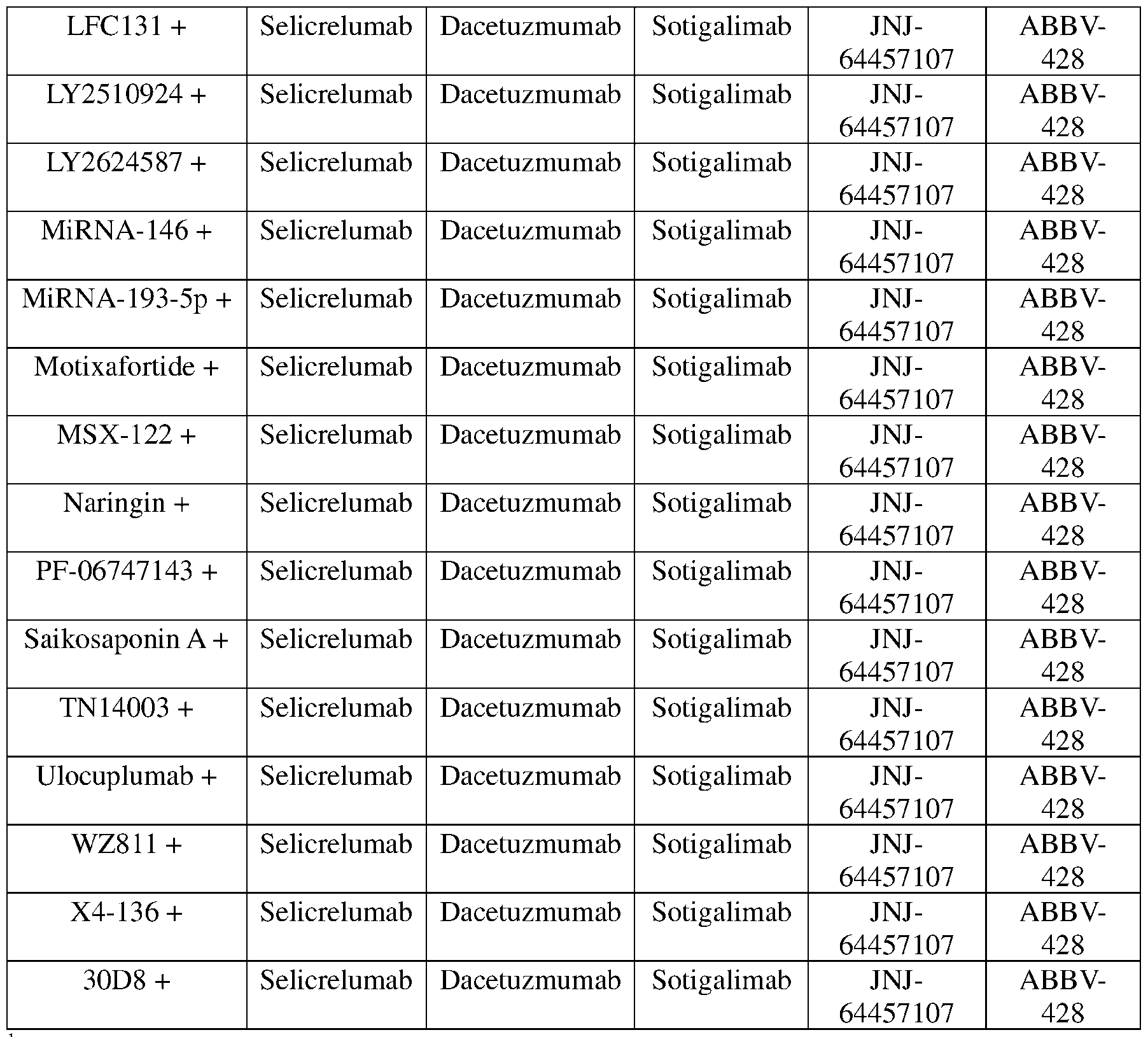
'The table represents 155 unique CXCR4 antagonist+agonistic anti-CD40 monoclonal antibody pairs, including 31 pairs consisting of a CXCR4 antagonist and Selicrelumab, 31 pairs consisting of a CXCR4 antagonist and Dacetuzmumab, 31 pairs consisting of a CXCR4 antagonist and Sotigalimab, 31 pairs consisting of a CXCR4 antagonist and JNJ-64457107, and 31 pairs consisting of a CXCR4 antagonist and ABBV-428.
Table 3B. Examples of CXCR4 antagonist+agonistic anti-CD40 monoclonal antibody combination pairs.1



'The table represents 124 unique CXCR4 antagonist+agonistic anti-CD40 monoclonal antibody pairs, including 31 pairs consisting of a CXCR4 antagonist and CDX-1 140H, 31 pairs consisting of a CXCR4 antagonist and SEA-CD40, 31 pairs consisting of a CXCR4 antagonist and ChiLob 7/4, and 31 pairs consisting of a CXCR4 antagonist and 2141-V11.
[0078] Before further describing various embodiments of the compositions and methods of the present disclosure in more detail by way of exemplary description, examples, and results, it is to be understood that the compositions and methods of the present disclosure are not limited in application to the details of specific embodiments and examples as set forth in the following description. The description provided herein is intended for purposes of illustration only and is not intended to be construed in a limiting sense. As such, the language used herein is intended to be given the broadest possible scope and meaning, and the embodiments and examples are meant to be exemplary, not exhaustive. Also, it is to be understood that the phraseology and terminology employed herein is for the purpose of description and should not be regarded as limiting unless otherwise indicated as so. Moreover, in the following detailed description, numerous specific details are set forth in order to provide a more thorough understanding of the present disclosure. However, it will be apparent to a person having ordinary skill in the art that the present disclosure may be practiced without these specific details. In other instances, features which are well known to persons of ordinary skill in the art have not been described in detail to avoid unnecessary complication of the description. It is intended that all alternatives, substitutions, modifications, and equivalents apparent to those having ordinary skill in the art are included within the scope of the present disclosure. Thus, while the compositions and methods of the present disclosure have been described in terms of particular embodiments, it will be apparent to those of skill in the art that variations may be applied to the compositions and methods and in the steps or in the sequence of steps of the methods described herein without departing from the concept, spirit, and scope of the inventive concepts.
[0079] All patents, published patent applications, and non-patent publications mentioned in the specification or referenced in any portion of this application are herein expressly incorporated by reference in their entirety to the same extent as if each individual patent or publication was specifically and individually indicated to be incorporated by reference.
[0080] Unless otherwise defined herein, scientific and technical terms used in connection with the present disclosure shall have the meanings that are commonly understood by those
having ordinary skill in the art. Further, unless otherwise required by context, singular terms shall include pluralities and plural terms shall include the singular.
[0081] As utilized in accordance with the methods and compositions of the present disclosure, the following terms, unless otherwise indicated, shall be understood to have the following meanings:
[0082] The use of the word “a” or “an” when used in conjunction with the term “comprising” in the claims and/or the specification may mean “one,” but it is also consistent with the meaning of “one or more,” “at least one,” and “one or more than one.” The use of the term “or” in the claims is used to mean “and/or” unless explicitly indicated to refer to alternatives only or when the alternatives are mutually exclusive, although the disclosure supports a definition that refers to only alternatives and “and/or.” The use of the term “at least one” will be understood to include one as well as any quantity more than one, including but not limited to, 2, 3, 4, 5, 6, 7, 8, 9, 10, 15, 20, 30, 40, 50, 100, or any integer inclusive therein. The term “at least one” may extend up to 100 or 1000 or more, depending on the term to which it is attached; in addition, the quantities of 100/1000 are not to be considered limiting, as higher limits may also produce satisfactory results. In addition, the use of the term “at least one of X, Y, and Z” will be understood to include X alone, Y alone, and Z alone, as well as any combination of X, Y, and Z.
[0083] As used herein, all numerical values or ranges include fractions of the values and integers within such ranges and fractions of the integers within such ranges unless the context clearly indicates otherwise. Thus, to illustrate, reference to a numerical range, such as 1-10 includes 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, as well as 1.1, 1.2, 1.3, 1.4, 1.5, etc., and so forth. Reference to a range of 1-50 therefore includes 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, etc., up to and including 50, as well as 1.1, 1.2, 1.3, 1.4, 1.5, etc., 2.1, 2.2, 2.3, 2.4, 2.5, etc., and so forth. Reference to a series of ranges includes ranges which combine the values of the boundaries of different ranges within the series. Thus, to illustrate reference to a series of ranges, for example, of 1-10, 10-20, 20-30, 30-40, 40-50, 50-60, 60-75, 75-100, 100-150, 150- 200, 200-250, 250-300, 300-400, 400-500, 500-750, 750-1,000, includes ranges of 1-20, 10- 50, 50-100, 100-500, and 500-1,000, for example. Reference to an integer with more (greater) or less than includes any number greater or less than the reference number, respectively. Thus, for example, reference to less than 100 includes 99, 98, 97, etc. all the way down to the number one (1); and less than 10 includes 9, 8, 7, etc. all the way down to the number one (1). A range of 1 to 50 for example, also refers to any range bounded by two different integers including 1 , 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, ,21, 22, 23, 24, 25, 26, 27, 28, 29, 30,
31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, and 50, including for example 18 to 25, 20 to 24, or 20-22.
[0084] As used in this specification and claims, the words “comprising” (and any form of comprising, such as “comprise” and “comprises”), “having” (and any form of having, such as “have” and “has”), “including” (and any form of including, such as “includes” and “include”) or “containing” (and any form of containing, such as “contains” and “contain”) are inclusive or open-ended and do not exclude additional, unrecited elements or method steps.
[0085] The term “or combinations thereof’ as used herein refers to all permutations and combinations of the listed items preceding the term. For example, “A, B, C, or combinations thereof’ is intended to include at least one of: A, B, C, AB, AC, BC, or ABC, and if order is important in a particular context, also BA, CA, CB, CBA, BCA, ACB, BAC, or CAB. Continuing with this example, expressly included are combinations that contain repeats of one or more item or term, such as BB, AAA, AAB, BBC, AAABCCCC, CBBAAA, CABABB, and so forth. The skilled artisan will understand that typically there is no limit on the number of items or terms in any combination, unless otherwise apparent from the context.
[0086] Throughout this application, the terms “about” or “approximately” are used to indicate that a value includes the inherent variation of error for the composition, the method used to administer the composition, or the variation that exists among the study subjects. As used herein the qualifiers “about” or “approximately” are intended to include not only the exact value, amount, degree, orientation, or other qualified characteristic or value, but are intended to include some slight variations due to measuring error, manufacturing tolerances, stress exerted on various parts or components, observer error, wear and tear, and combinations thereof, for example. The term “about” or “approximately,” where used herein when referring to a measurable value such as an amount, a temporal duration, and the like, is meant to encompass, for example, variations of ± 25%, or ± 20%, or ± 15%, ± 10%, or ± 5%, or ± 1%, or ± 0.1% from the specified value, as such variations are appropriate to perform the disclosed methods and as understood by persons having ordinary skill in the art. As used herein, the term “substantially” means that the subsequently described event or circumstance completely occurs or that the subsequently described event or circumstance occurs to a great extent or degree. For example, the term “substantially” means that the subsequently described event or circumstance occurs at least 90% of the time, or at least 95% of the time, or at least 98% of the time.
[0087] As used herein any reference to "one embodiment" or "an embodiment" means that a particular element, feature, structure, or characteristic described in connection with the embodiment is included in at least one embodiment and may be included in other embodiments.
The appearances of the phrase "in one embodiment" in various places in the specification are not necessarily all referring to the same embodiment and are not necessarily limited to a single or particular embodiment.
[0088] Where used herein, the term CXCR4 antagonist refers to a substance or agent that directly or indirectly inhibits CXCR4 activity, for example by reducing or inhibiting the binding of CXCL12 to CXCR4. The term CXCR4 antagonist may also refer to a substance or agent that directly or indirectly inhibits CXCR4 expression. Similarly, where used herein, the term CXCR5 antagonist refers to a substance or agent that directly or indirectly inhibits CXCR5 activity, for example by reducing or inhibiting the binding of a CXCR5 ligand (CXCL13) to CXCR5. The term CXCR5 antagonist may also refer to a substance or agent that directly or indirectly inhibits CXCR5 expression. Similarly, where used herein, the term CCR7 antagonist refers to a substance or agent that directly or indirectly inhibits CCR7 activity, for example by reducing or inhibiting the binding of a CCR7 ligands (CCL19 and CCL21) to CCR7. The term CCR7 antagonist may also refer to a substance or agent that directly or indirectly inhibits CCR7 expression.
[0089] The term “activity” refers to the ability of a substance or agent to modify the molecular, biochemical, or physiological system of a cell, organ, or organism, without reference to how the substance or agent has its physiological effects.
[0090] As used herein, the phrase "biologically active" refers to a substance that has activity in a biological system (e.g., in a cell (e.g., isolated, in culture, in a tissue, in an organism), in a cell culture, in a tissue, in an organism, etc.). For instance, a substance that, when administered to an organism, has a biological effect on that organism, is considered to be biologically active. It will be appreciated by those skilled in the art that often only a portion or fragment of a biologically active substance is required (e.g., is necessary and sufficient) for the activity to be present; in such circumstances, that portion or fragment is considered to be a "biologically active" portion or fragment.
[0091] The term “pharmaceutically acceptable” refers to compounds and compositions which are suitable for administration to humans and/or animals without undue adverse side effects such as toxicity, irritation and/or allergic response commensurate with a reasonable benefit/risk ratio. The compounds of the present disclosure may be combined with one or more pharmaceutically-acceptable excipients, including carriers, vehicles, and diluents which may improve solubility, deliverability, dispersion, stability, and/or conformational integrity of the compounds or conjugates thereof.
[0092] As used herein, “pure,” or “substantially pure” means an object species is the predominant species present (i.e., on a molar basis it is more abundant than any other object species in the composition thereof), and particularly a substantially purified fraction is a composition wherein the object species comprises at least about 50 percent (on a molar basis) of all macromolecular species present. Generally, a substantially pure composition will comprise more than about 80% of all macromolecular species present in the composition, more particularly more than about 85%, more than about 90%, more than about 95%, or more than about 99%. The term “pure” or “substantially pure” also refers to preparations where the object species is at least 60% (w/w) pure, or at least 70% (w/w) pure, or at least 75% (w/w) pure, or at least 80% (w/w) pure, or at least 85% (w/w) pure, or at least 90% (w/w) pure, or at least 92% (w/w) pure, or at least 95% (w/w) pure, or at least 96% (w/w) pure, or at least 97% (w/w) pure, or at least 98% (w/w) pure, or at least 99% (w/w) pure, or 100% (w/w) pure.
[0093] Where used herein, the pronoun “we” is intended to refer to all persons involved in a particular aspect of the investigation disclosed herein and as such may include non- inventor laboratory assistants and non-inventor collaborators working under the supervision of the inventors.
[0094] Non-limiting examples of animals within the scope and meaning of this term include dogs, cats, rats, mice, guinea pigs, chinchillas, horses, goats, cattle, sheep, zoo animals, Old and New World monkeys, non-human primates, and humans.
[0095] “Treatment” refers to therapeutic treatments. “Prevention” refers to prophylactic or preventative treatment measures or reducing the onset of a condition or disease. The term “treating” refers to administering the composition to a subject for therapeutic purposes and/or for prevention. Non-limiting examples of modes of administration include oral, topical, retrobulbar, subconjunctival, transdermal, parenteral, subcutaneous, intranasal, intramuscular, intraperitoneal, intravitreal, and intravenous routes, including both local and systemic applications. The term “topical” is used herein to define a mode of administration through an epithelial surface, such as but not limited to, the skin, eye, or internal epithelial surfaces. In addition, the compositions of the present disclosure may be designed to provide delayed, controlled, extended, and/or sustained release using formulation techniques which are well known in the art.
[0096] The terms “therapeutic composition” and “pharmaceutical composition” refer to a composition containing a peptide as described herein that may be administered to a subject by any method known in the art or otherwise contemplated herein, wherein administration of the composition brings about a therapeutic effect as described elsewhere herein.
[0097] The term “effective amount” refers to an amount of a peptide or peptide compound which is sufficient to exhibit a detectable therapeutic, amelioration, or treatment effect in a subject without excessive adverse side effects (such as substantial toxicity, irritation and allergic response) commensurate with a reasonable benefit/risk ratio when used in the manner of the present disclosure. The effective amount for a subject will depend upon the subject’s type, size and health, the nature and severity of the condition to be treated, the method of administration, the duration of treatment, the nature of concurrent therapy (if any), the specific formulations employed, and the like. Thus, it is not possible to specify an exact effective amount in advance. However, the effective amount for a given situation can be determined by one of ordinary skill in the art using routine experimentation based on the information provided herein.
[0098] The term “ameliorate” means a detectable or measurable improvement in a subject’s condition or a symptom thereof. A detectable or measurable improvement includes a subjective or objective decrease, reduction, inhibition, suppression, limit or control in the occurrence, frequency, severity, progression, or duration of the condition, or an improvement in a symptom or an underlying cause or a consequence of the condition, or a reversal of the condition. A successful treatment outcome can lead to a “therapeutic effect,” or “benefit” of ameliorating, decreasing, reducing, inhibiting, suppressing, limiting, controlling, or preventing the occurrence, frequency, severity, progression, or duration of a condition, or consequences of the condition in a subject.
[0099] A decrease or reduction in worsening, such as stabilizing the condition, is also a successful treatment outcome. A therapeutic benefit therefore need not be complete ablation or reversal of the condition, or any one, most or all adverse symptoms, complications, consequences or underlying causes associated with the condition. Thus, a satisfactory endpoint may be achieved when there is an incremental improvement such as a partial decrease, reduction, inhibition, suppression, limit, control or prevention in the occurrence, frequency, severity, progression, or duration, or inhibition or reversal of the condition (e.g., stabilizing), over a short or long duration of time (e.g., seconds, minutes, hours).
[0100] As used herein, the term "small molecule" means a low molecular weight organic compound that may serve as an enzyme substrate or regulator of biological processes. In general, a "small molecule" is a molecule that is less than about 5 kilodaltons (kD) in size. In some embodiments, provided nanoparticles further include one or more small molecules. In some embodiments, the small molecule is less than about 4 kD, 3 kD, about 2 kD, or about 1 kD. In some embodiments, the small molecule is less than about
800 daltons (D), about 600 D, about 500 D, about 400 D, about 300 D, about 200 D, or about 100 D. In some embodiments, a small molecule is less than about 2000 g/mol, less than about 1500 g/mol, less than about 1000 g/mol, less than about 800 g/mol, or less than about 500 g/mol. In some embodiments, one or more small molecules are encapsulated within the nanoparticle. In some embodiments, small molecules are non-poly meric. In some embodiments, in accordance with the present disclosure, small molecules are not proteins, polypeptides, oligopeptides, peptides, polynucleotides, oligonucleotides, polysaccharides, glycoproteins, proteoglycans, etc. In some embodiments, a small molecule is a therapeutic. In some embodiments, a small molecule is an adjuvant. In some embodiments, a small molecule is a drug.
[0101] In some embodiments, provided agents and/or compositions comprising such agents may be provided in particles. Particles as used in this context means nanoparticles or microparticles (or in some instances larger particles) which can consist in whole or in part of provided agent(s) and/or other therapeutic agent(s) as described herein. Such particles may contain the agent(s) and/or compositions in a core surrounded by a coating, including, but not limited to, an enteric coating. The agent(s) and/or compositions also may be dispersed throughout the particles. The agent(s) and/or compositions also maybe adsorbed into the particles. The particles maybe of any order release kinetics, including zero-order release, first-order release, second-order release, delayed release, sustained release, immediate release, and any combination thereof, etc. The particle may include, in addition to the agent(s) and/or compositions, any of those materials routinely used in the art of pharmacy and medicine, including, but not limited to, erodible, nonerodible, biodegradable, or nonbiodegradable material or combinations thereof. The particles maybe microcapsules which comprise one or more provided agents in a solution or in a semi-solid state. The particles may be of virtually any shape.
[0102] According to various embodiments, both non-biodegradable and biodegradable polymeric materials can be used in the manufacture of particles for delivering provided agent(s) and/or compositions. Such polymers maybe natural or synthetic polymers. In many embodiments, a polymer is selected based on the period of time over which release is desired. Bioadhesive polymers of particular interest include bioerodible hydrogels which may comprise, for example, polyhyaluronic acids, casein, gelatin, glutin, poly anhydrides, polyacrylic acid, alginate, chitosan, poly(methylmethacrylates), poly(ethylmethacrylates), poly(butylmethacrylate), poly(isobutylmethacrylate), poly(hexylmethacrylate), poly(isodecylmethacrylate),
poly(laurylmethacrylate), poly (phenylmethacry late), poly (methylacrylate), poly(isopropylacrylate), poly(isobutylacrylate), and poly(octadecylacrylate). In some embodiments, provided agents and/or compositions comprising such agents maybe contained in controlled release systems.
[0103] The term "controlled release" in this context is intended to refer to any drugcontaining formulation in which the manner and profile of drug release from the formulation are controlled. This refers to immediate as well as non-immediate release formulations, with non-immediate release formulations including but not limited to sustained release and delayed release formulations.
[0104] The term "sustained release" (also referred to as "extended release") is used in this context in its conventional sense to refer to a drug formulation that provides for gradual release of a drug over an extended period of time, and that in certain particular (but non-limiting) embodiments, although not necessarily, results in substantially constant blood levels of a drug over an extended time period. The term "delayed release" is used in this context its conventional sense to refer to a drug formulation in which there is a time delay between administration of the formulation and the release of the drug there from. "Delayed release" may or may not involve gradual release of drug over an extended period of time, and thus may or may not be "sustained release." In some embodiments, use of a long-term sustained release implant maybe particularly suitable for treatment of chronic conditions with one or more provided agents. "Long-term” release, as used in this context, means that an implant is constructed and arranged to deliver therapeutic levels of the active ingredient for at least 7 days, and in certain non-limiting embodiments, 30-60 days. Long-term sustained release implants are well-known to those of ordinary skill in the art and include some of the release systems described elsewhere herein.
[0105] The term “mutant” or “variant” is intended to refer to a protein, peptide, nucleic acid or organism which has at least one amino acid or nucleotide which is different from the wild type version of the protein, peptide, nucleic acid, or organism and includes, but is not limited to, point substitutions, multiple contiguous or non-contiguous substitutions, chimeras, or fusion proteins, and the nucleic acids which encode them.
[0106] The term "homologous" or “% identity” as used herein means a nucleic acid (or fragment thereof) or a protein (or a fragment thereof) having a degree of homology to the corresponding natural reference nucleic acid or protein that may be in excess of 70%, or in excess of 80%, or in excess of 85%, or in excess of 90%, or in excess of 91%, or in excess of 92%, or in excess of 93%, or in excess of 94%, or in excess of 95%, or in excess of 96%, or in
excess of 97%, or in excess of 98%, or in excess of 99%. For example, in regard to peptides or polypeptides, the percentage of homology or identity as described herein is typically calculated as the percentage of amino acid residues found in the smaller of the two sequences which align with identical amino acid residues in the sequence being compared, when four gaps in a length of 100 amino acids may be introduced to assist in that alignment (as set forth by Dayhoff, in Atlas of Protein Sequence and Structure, Vol. 5, p. 124, National Biochemical Research Foundation, Washington, D.C. (1972)). In one embodiment, the percentage homology as described above is calculated as the percentage of the components found in the smaller of the two sequences that may also be found in the larger of the two sequences (with the introduction of gaps), with a component being defined as a sequence of four, contiguous amino acids. Also included as substantially homologous is any protein product which may be isolated by virtue of cross-reactivity with antibodies to the native protein product. Sequence identity or homology can be determined by comparing the sequences when aligned so as to maximize overlap and identity while minimizing sequence gaps. In particular, sequence identity may be determined using any of a number of mathematical algorithms. A non-limiting example of a mathematical algorithm used for comparison of two sequences is the algorithm of Karlin & Altschul (Proc. Natl. Acad. Sci. USA (1990) 87:2264-2268), modified as in Karlin & Altschul (Proc. Natl. Acad. Sci. USA (1993) 90:5873-5877).
[0107] In one embodiment “% identity” represents the number of amino acids or nucleotides which are identical at corresponding positions in two sequences of a protein having the same activity or encoding similar proteins. For example, two amino acid sequences each having 100 residues will have 95% identity when 95 of the amino acids at corresponding positions are the same.
[0108] Another example of a mathematical algorithm used for comparison of sequences is the algorithm of Myers & Miller (CABIOS (1988) 4:1 1- 17). Such an algorithm is incorporated into the ALIGN program (version 2.0) which is part of the GCG sequence alignment software package. When utilizing the ALIGN program for comparing amino acid sequences, a PAM 120 weight residue table, a gap length penalty of 12, and a gap penalty of 4 can be used. Yet another useful algorithm for identifying regions of local sequence similarity and alignment is the FAST A algorithm as described in Pearson & Lipman (Proc. Natl. Acad. Sci. USA (1988) 85:2444-2448).
[0109] Another algorithm is the WU-BLAST (Washington University BLAST) version 2.0 software (WU-BLAST version 2.0 executable programs for several UNIX platforms). This program is based on WU-BLAST version 1.4, which in turn is based on the public domain
NCBI-BLAST version 1.4 (Altschul & Gish, Methods in Enzymology (1996) 266:460-480; Altschul et al., J Molec Biol. (1990) 215:403-410; Gish & States, Nature Genetics (1993) 3:266-272; Karlin & Altschul, Proc. Natl. Acad. Sci. USA (1993) 90:5873-5877; all of which are incorporated by reference herein).
[0110] In addition to those otherwise mentioned herein, mention is made also of the programs BLAST, gapped BLAST, BLASTN, BLASTP, and PSLBLAST, provided by the National Center for Biotechnology Information. These programs are widely used in the art for this purpose and can align homologous regions of two amino acid sequences. In all search programs in the suite, the gapped alignment routines are integral to the database search itself. Gapping can be turned off if desired. The default penalty (Q) for a gap of length one is Q=9 for proteins and BLASTP, and Q=10 for BLASTN, but may be changed to any integer. The default per-residue penalty for extending a gap (R) is R=2 for proteins and BLASTP, and R=10 for BLASTN, but may be changed to any integer. Any combination of values for Q and R can be used in order to align sequences so as to maximize overlap and identity while minimizing sequence gaps. The default amino acid comparison matrix is BLOSUM62, but other amino acid comparison matrices such as PAM can be utilized.
[0111] Specific amino acids may be referred to herein by the following designations: alanine: ala or A; arginine: arg or R; asparagine: asn or N; aspartic acid: asp or D; cysteine: cys or C; glutamic acid: glu or E; glutamine: gin or Q; glycine: gly or G; histidine: his or H; isoleucine: ile or I; leucine: leu or L; lysine: lys or K; methionine: met or M; phenylalanine: phe or F; proline: pro or P; serine: ser or S; threonine: thr or T; tryptophan: trp or W; tyrosine: tyr or Y ; and valine: val or V.
[0112] The terms “oligonucleotide,” "polynucleotide," or “nucleic acid,” as used herein, include any nucleotide sequence which encodes a variant, chimeric, or mutant peptide including polynucleotides in the form of RNA, such as mRNA, or in the form of DNA, including, for instance, cDNA and genomic DNA obtained by cloning or produced by chemical synthetic techniques or by a combination thereof. The DNA may be double- stranded or singlestranded. Single-stranded DNA may be the coding strand, also known as the sense strand, or it may be the non-coding strand, also referred to as the anti-sense strand. The polynucleotide sequence encoding a mutant peptide or encoding a therapeutically-effective fragment of a mutant peptide can be substantially the same as the coding sequence of the endogenous coding sequence as long as it encodes a biologically active mutant peptide. Further, the mutant peptide, or therapeutically-effective fragment of a mutant peptide may be expressed using
polynucleotide sequence(s) which differ in codon usage due to the degeneracies of the genetic code or allelic variations.
[0113] As noted above, the peptides of the present disclosure, and the nucleic acids which encode them, include peptide and nucleic acid variants which comprise additional conservative substitutions. For example, the variant peptides include, but are not limited to, variants that are not exactly the same as the sequences disclosed herein, but which have, in addition to the substitutions explicitly described for various sequences listed herein, conservative substitutions of amino acid residues which do substantially not impair the agonistic or antagonistic activity or properties of the variants described herein. Examples of such conservative amino acid substitutions include, but are not limited to, ala to gly, ser, or thr; arg to gin, his, or lys; asn to asp, gin, his, lys, ser, or thr; asp to asn or glu; cys to ser; gin to arg, asn, glu, his, lys, or met; glu to asp, gin, or lys; gly to pro or ala; his to arg, asn, gin, or tyr; ile to leu, met, or val; leu to ile, met, phe, or val; lys to arg, asn, gin, or glu; met to gin, ile, leu, or val; phe to leu, met, trp, or tyr; ser to ala, asn, met, or thr; thr to ala, asn, ser, or met; trp to phe or tyr; tyr to his, phe or trp; and val to ile, leu, or met.
[0114] The present constructs or antigen-binding portions thereof can be formulated into compositions for delivery to a mammalian subject. The composition can be administered alone and/or mixed with a pharmaceutically acceptable vehicle or excipient. Suitable vehicles are, for example (but not by way of limitation), water, saline, dextrose, glycerol, ethanol, or the like, and combinations thereof. In addition, the vehicle can contain minor amounts of auxiliary substances such as (but not limited to) wetting or emulsifying agents, pH buffering agents, or adjuvants. The compositions of the present disclosure can also include ancillary substances, such as (but not limited to) pharmacological agents, cytokines, or other biological response modifiers.
[0115] Furthermore, the compositions can be formulated into compositions in either neutral or salt forms. Pharmaceutically acceptable salts include (but are not limited to) the acid addition salts (formed with the free amino groups of the active polypeptides) and which are formed with inorganic acids such as, for example, hydrochloric or phosphoric acids, or organic acids such as acetic, oxalic, tartaric, mandelic, and the like. Salts formed from free carboxyl groups can also be derived from inorganic bases such as, for example, sodium, potassium, ammonium, calcium, or ferric hydroxides, and such organic bases as isopropylamine, trimethylamine, 2-ethylamino ethanol, histidine, and procaine.
[0116] Compositions can be administered in a single dose treatment or in multiple dose treatments on a schedule and over a time period appropriate to the age, weight, and condition of
the subject, the particular composition used, and the route of administration. In one non- limiting embodiment, a single dose of the composition according to the disclosure is administered. In other non-limiting embodiments, multiple doses are administered. The frequency of administration can vary depending on any of a variety of factors, e.g., severity of the symptoms, degree of immunoprotection desired, or whether the composition is used for prophylactic or curative purposes. For example, in certain non-limiting embodiments, the composition is administered once per month, twice per month, three times per month, every other week, once per week, twice per week, three times per week, four times per week, five times per week, six times per week, every other day, daily, twice a day, or three times a day. The duration of treatment (i.e., the period of time over which the composition is administered) can vary, depending on any of a variety of factors, e.g., subject response. For example, the composition can be administered over a period of time ranging from about one day to about one week, from about two weeks to about four weeks, from about one month to about two months, from about two months to about four months, from about four months to about six months, from about six months to about eight months, from about eight months to about 1 year, from about 1 year to about 2 years, or from about 2 years to about 4 years, or more.
[0117] The dosage of an administered active agent for humans will vary depending upon factors such as (but not limited to) the patient's age, weight, height, sex, general medical condition, and previous medical history. In certain non-limiting embodiments, the recipient is provided with a dosage of the active agent that is in the range of from about 1 mg to about 1000 mg as a single infusion or single or multiple injections, although a lower or higher dosage also may be administered. In certain non-limiting embodiments, the dosage may be in the range of from about 25 mg to about 100 mg of the active agent per square meter (m2) of body surface area for a typical adult, although a lower or higher dosage also may be administered. Non-limiting examples of dosages of the active agent that may be administered to a human subject further include 1 to 500 mg, 1 to 70 mg, or 1 to 20 mg, although higher or lower doses may be used. Dosages may be repeated as needed, for example (but not by way of limitation), once per week for 4-10 weeks, once per week for 8 weeks, or once per week for 4 weeks. It may also be given less frequently, such as (but not limited to) every other week for several months, or more frequently, such as twice weekly or by continuous infusion.
[0118] In at least one embodiment, the present disclosure is directed to a dosing regimen involving administration of the multispecific construct such as disclosed elsewhere herein. The dosing regimen may comprise multiple dosing cycles (e.g., wherein the first dosing cycle is a step-up, fractionated dosing cycle). The doses may range from about 0.02 mg to about 2.0 mg
(e.g., from about 0.02 to about 1.8 mg, from about 0.02 to about 1.6 mg, from about 0.02 to about 1.4 mg, from about 0.02 to about 1.2 mg, from about 0.05 to about 1.8 mg, from about 0.1 to about 1.8 mg, from about 0.4 to about 1.8 mg, from about 0.6 to about 1.8 mg, from about 0.8 to about 1.8 mg, from about 0.5 to about 1.5 mg, from about 0.8 to about 1.2 mg; e.g., about 1 mg), from about 0.05 mg to about 4.0 mg (e.g., from about 0.05 to about 3.5 mg, from about 0.05 to about 3.0 mg, from about 0.05 to about 2.5 mg, from about 0.05 to about 2.2 mg, from about 0.1 to about 3.5 mg, from about 0.5 to about 3.5 mg, from about 1.0 to about 3.5 mg, from about 1 .5 to about 3.5 mg, from about 1 .8 to about 3.5 mg, from about 1 .0 to about 3.0 mg, from about 1.5 to about 2.5 mg; e.g., about 2 mg).
[0119] In some embodiments, the dose may range from 50 mg to 200 mg (e.g., from 50 mg to 175 mg, from 50 mg to 150 mg, from 50 mg to 125 mg, from 50 mg to 100 mg, from 50 mg to 75 mg, from 50 mg to 70 mg, from 52 mg to 100 mg, from 52 mg to 75 mg, from 50 mg to 180 mg, from 55 mg to 150 mg, from 55 mg to 100 mg, from 55 mg to 70 mg, from 55 mg to 65 mg, from 58 mg to 62 mg; e.g., about 60 mg). In some embodiments, the dose may be about 60 mg. In some embodiments, the dose is about 1 mg. In some embodiments, the dose is about 2 mg.
[0120] In some embodiments, the dose is from 20 mg to 200 mg (e.g., from 20 mg to 175 mg, from 20 mg to 150 mg, from 20 mg to 100 mg, from 20 mg to 75 mg, from 30 mg to 175 mg, from 40 mg to 175 mg, from 45 mg to 175 mg, from 50 mg to 175 mg, from 30 mg to 150 mg, from 40 mg to 100 mg, from 45 mg to 75 mg, from 50 mg to 70 mg, from 55 mg to 65 mg, from 58 mg to 62 mg; about 20 mg, about 30 mg, about 45 mg, or e.g., about 60 mg). In some embodiments, the dose is from about 12 mg to about 48 mg (e.g., from about 12 mg to about 42 mg, from about 12 mg to about 36 mg, from about 12 mg to about 30 mg, from about 18 mg to about 48 mg, from about 18 mg to about 42 mg, from about 24 mg to about 42 mg, from about 27 mg to about 42 mg, from about 24 mg to about 36 mg, from about 27 mg to about 33 mg, from about 28 mg to about 32 mg; e.g., about 24 mg, about 27 mg, about 30 mg, about 33 mg, or about 36 mg).
[0121] In some embodiments, the dosing regimen comprises administration of a loading dose, such as from 20 mg to 200 mg (e.g., from 20 mg to 175 mg, from 20 mg to 150 mg, from 20 mg to 100 mg, from 20 mg to 75 mg, from 30 mg to 175 mg, from 40 mg to 175 mg, from 45 mg to 175 mg, from 50 mg to 175 mg, from 30 mg to 150 mg, from 40 mg to 100 mg, from 45 mg to 75 mg, from 50 mg to 70 mg, from 55 mg to 65 mg, from 58 mg to 62 mg; e.g., about 60 mg). In some embodiments, the dose is from about 12 mg to about 48 mg (e.g., from about 12 mg to about 42 mg, from about 12 mg to about 36 mg, from about 12 mg to about 30 mg,
from about 18 mg to about 48 mg, from about 18 mg to about 42 mg, from about 24 mg to about 42 mg, from about 27 mg to about 42 mg, from about 24 mg to about 36 mg, from about 27 mg to about 33 mg, from about 28 mg to about 32 mg; e.g., about 24 mg, about 27 mg, about 30 mg, about 33 mg, or about 36 mg).
[0122] In some non-limiting embodiments, the active agent is provided in a concentration of about 1 nM, about 5 nM, about 10 nM, about 25 nM, about 50 nM, about 75 nM, about 100 nM, about 150 nM, about 200 nM, about 250 nM, about 300 nM, about 350 nM, about 400 nM, about 500 nM, about 550 nM, about 600 nM, about 700 nM, about 800 nM, about 900 nM, about 1 pM, about 2 pM, about 3 pM, about 4 pM, about 5 pM, about 6 pM, about 7 pM, about 8 pM, about 9 pM, about 10 pM, about 15 pM, about 20 pM, about 25 pM, about 30 pM, about 35 pM, about 40 pM, about 45 pM, about 50 pM, about 60 pM, about 70 pM, about 75 pM, about 80 pM, about 90 pM, about 100 pM, about 125 pM, about 150 pM, about 175 pM, about 200 pM, about 250 pM, about 300 pM, about 350 pM, about 400 pM, about 500 pM, about 600 pM, about 700 pM, about 750 pM, about 800 pM, about 900 pM, about 1 mM, about 2 mM, about 3 mM, about 4 mM, about 5 mM, about 6 mM, about 7 mM, about 8 mM, about 9 mM, about 10 mM, about 11 mM, about 12 mM, about 13 mM, about 14 mM, about 15 mM, about 20 mM, about 25 mM, about 30 mM, about 35 mM, about 40 mM, about 45 mM, about 50 mM, about 55 mM, about 60 mM, about 65 mM, about 70 mM, about 75 mM, about 80 mM, about 85 mM, about 90 mM, about 95 mM, about 100 mM, about 100 mM, about 110 mM, about 120 mM, about 130 mM, about 140 mM, about 150 mM, about 160 mM, about 170 mM, about 180 mM, about 190 mM, about 200 mM, about 250 mM, about 300 mM, about 400 mM, about 500mM, about 600 mM, about 700 mM, about 800 mM, about 900 mM, about 1000 mM, about 1 M, about 1.1 M, about 1.2 M, about 1.3 M, about 1.4 M, about 1.5 M, about 1.6 M, about 1.7 M, about 1.8 M, about 1.9 M, about 2 M, about 3 M, about 4 M, about 5 M, about 6 M, about 7 M, about 8 M, about 9 M, about 10 M, about 15 M, about 20 M, about 25 M, about 30 M, about 35 M, about 40 M, about 45 M, about 50 M, about 75 M, about 100 M, or any range in between any two of the aforementioned concentrations, including said two concentrations as endpoints of the range, or any number in between any two of the aforementioned concentrations.
[0123] When administered orally, the present compositions may be protected from digestion. This can be accomplished either by complexing the construct or antigen-binding portion thereof with a composition to render it resistant to acidic and enzymatic hydrolysis or
by packaging the construct or antigen-binding portion thereof in an appropriately resistant carrier such as (but not limited to) a liposome, e.g., such as shown in U.S. Patent No. 5,391 ,377. [0124] For transmucosal or transdermal administration, penetrants appropriate to the barrier to be permeated can be used in the formulation. Such penetrants are generally known in the art, and include, e.g., for transmucosal administration, bile salts and fusidic acid derivatives. In addition, detergents can be used to facilitate permeation. Transmucosal administration can be through nasal sprays or using suppositories. For topical transdermal administration, the agents are formulated into ointments, creams, salves, powders, and gels. Transdermal delivery systems can also include (for example but not by way of limitation) patches. The present compositions can also be administered in sustained delivery or sustained release mechanisms. For example, biodegradeable microspheres or capsules or other biodegradeable polymer configurations capable of sustained delivery of a peptide can be included herein.
[0125] For inhalation, the present compositions can be delivered using any system known in the art, including (but not limited to) dry powder aerosols, liquids delivery systems, air jet nebulizers, propellant systems, and the like. For example (but not by way of limitation), the pharmaceutical formulation can be administered in the form of an aerosol or mist. For aerosol administration, the formulation can be supplied in finely divided form along with a surfactant and propellant. In another aspect, the device for delivering the formulation to respiratory tissue is an inhaler in which the formulation vaporizes. Other liquid delivery systems include (for example but not by way of limitation) air jet nebulizers.
[0126] In one aspect, the active agents may be incorporated in lipid monolayers or bilayers, such as (but not limited to) liposomes, such as shown in U.S. Patent Nos. 6,110,490; 6,096,716; 5,283,185; and 5,279,833. In other aspects, non-limiting embodiments of the disclosure include formulations in which the active agents have been attached to the surface of the monolayer or bilayer of the liposomes. Liposomes and liposomal formulations can be prepared according to standard methods and are also well known in the art, such as (but not limited to) those disclosed in U.S. Patent Nos. 4,235,871; 4,501,728; and 4,837,028.
[0127] In one aspect, the compositions are prepared with carriers that will protect the construct or fragment thereof against rapid elimination from the body, such as (but not limited to) a controlled release formulation, including implants and microencapsulated delivery systems. Biodegradable, biocompatible polymers can be used, such as (but not limited to) ethylene vinyl acetate, poly anhydrides, poly glycolic acid, collagen, polyorthoesters, and polylactic acid. Methods for preparation of such formulations will be apparent to those skilled in the art.
[0128] The constructs and fragments thereof in general may be formulated to obtain compositions that include one or more pharmaceutically suitable excipients, surfactants, polyols, buffers, salts, amino acids, or additional ingredients, or some combination of these. This can be accomplished by known methods to prepare pharmaceutically useful dosages, whereby the active compound is combined in a mixture with one or more pharmaceutically suitable excipients. Sterile phosphate-buffered saline is one non-limiting example of a pharmaceutically suitable excipient.
[0129] Non-limiting examples of routes of administration of the active agents described herein include parenteral injection, e.g., by subcutaneous, intramuscular, or transdermal delivery. Other forms of parenteral administration include (but are not limited to) intravenous, intraarterial, intralymphatic, intrathecal, intraocular, intracerebral, or intracavitary injection. In parenteral administration, the compositions will be formulated in a unit dosage injectable form such as (but not limited to) a solution, suspension, or emulsion, in association with a pharmaceutically acceptable excipient. Such excipients are inherently nontoxic and nontherapeutic. Non-limiting examples of such excipients include saline, Ringer's solution, dextrose solution, and Hanks' solution. Nonaqueous excipients such as (but not limited to) fixed oils and ethyl oleate may also be used. An alternative non-limiting excipient is 5% dextrose in saline. The excipient may contain minor amounts of additives such as (but not limited to) substances that enhance isotonicity and chemical stability, including buffers and preservatives. The constructs can be delivered or administered alone or as pharmaceutical compositions by any means known in the art, such as (but not limited to) systemically, regionally, or locally; by intra-arterial, intrathecal (IT), intravenous (IV), parenteral, intra-pleural cavity, topical, oral, or local administration, as subcutaneous, intra-tracheal (e.g., by aerosol) or transmucosal (e.g., buccal, bladder, vaginal, uterine, rectal, nasal mucosa).
[0130] Administration can be (for example but not by way of limitation) parenteral, intravenous, oral, subcutaneous, intra-arterial, intracranial, intrathecal, intraperitoneal, topical, intranasal, or intramuscular. Administration can also be localized directly into a tumor. Administration into the systemic circulation by intravenous or subcutaneous administration is typical. Intravenous administration can be, for example (but not by way of limitation), by infusion over a period such as (but not limited to) 30-90 min or by a single bolus injection.
[0131] Formulated compositions comprising the constructs can be used (for example but not by way of limitation) for subcutaneous, intramuscular, or transdermal administration. Compositions can be presented in unit dosage form, e.g., in ampoules or in multi-dose containers, with an added preservative. Compositions can also take such forms as suspensions,
solutions, or emulsions in oily or aqueous vehicles, and can contain formulatory agents such as suspending, stabilizing, and/or dispersing agents.
[0132] The terms “co-administration,” “coadministration,” “co-administer,” and “coadminister,”, as used herein, refer to administration of the CXCR4 antagonist or other antagonist with one or more additional therapeutic agents within the same dosage form, or in separate dosage forms, simultaneously or at essentially the same time, or at different times such that the CXCR4 antagonist or other antagonist is administered before the one or more additional agents is administered or after the one or more additional agents is administered. “Essentially at the same time” as used herein generally means within 30 minutes, within 20 minutes, within five minutes, within two minutes, or within in one minute.
[0133] The term “combination therapy” refers to a treatment protocol in which two or more therapeutic agents are coadministered, as that term is defined above.
[0134] The compositions may be administered in solution. The formulation thereof may be in a solution having a suitable pharmaceutically acceptable buffer, such as (but not limited to) phosphate, Tris (hydroxymethyl) aminomethane-HCl, or citrate, and the like. Buffer concentrations should be in the range of 1 to 100 mM. The formulated solution may also contain a salt, such as (but not limited to) sodium chloride or potassium chloride in a concentration of 50 to 150 mM. An effective amount of a stabilizing agent such as (but not limited to) mannitol, trehalose, sorbitol, glycerol, albumin, a globulin, a detergent, a gelatin, a protamine, or a salt of protamine may also be included.
[0135] As used herein, the term "RNA interference" or "RNAi" refers to the silencing or decreasing of gene expression by siRNAs. It is the process of sequence-specific, post- transcriptional gene silencing in animals and plants, initiated by siRNA that is homologous in its duplex region to the sequence of the silenced gene. The gene may be endogenous or exogenous to the organism, present integrated into a chromosome or present in a transfection vector that is not integrated into the genome. The expression of the gene is either completely or partially inhibited. RNAi inhibits the gene by compromising the function of a target RNA, completely or partially. Both plants and animals mediate RNAi by the RNA-induced silencing complex (RISC); a sequence-specific, multicomponent nuclease that destroys messenger RNAs homologous to the silencing trigger. RISC is known to contain short RNAs (e.g., approximately 22 nucleotides) derived from the double-stranded RNA trigger, although the protein components of this activity are unknown. However, the 22-nucleotide RNA sequences are homologous to the target gene that is being suppressed. Thus, the 22-nucleotide sequences appear to serve as guide sequences to instruct a multicomponent nuclease, RISC, to destroy the
specific mRNAs. Biochemical reactions that recapitulate this phenomenon generate RNA fragments of 21 to 23 nucleotides from the double-stranded RNA. These stably associate with an RNA endonuclease, and probably serve as a discriminator to select mRNAs. Once selected, mRNAs are cleaved at sites 21 to 23 nucleotides apart.
[0136] As used herein, the term "siRNA" refers to a short interfering RNA. In some embodiments, siRNAs comprise a duplex, or double- stranded region, of about 18-25 nucleotides long; often siRNAs contain from about two to four unpaired nucleotides at the 3' end of each strand. At least one strand of the duplex or double-stranded region of a siRNA is substantially homologous to or substantially complementary to a target RNA molecule. The strand complementary to a target RNA molecule is the "antisense strand"; the strand homologous to the target RNA molecule is the "sense strand", and is also complementary to the siRNA antisense strand. siRNAs may also contain additional sequences; non-limiting examples of such sequences include linking sequences, or loops, as well as stem and other folded structures. siRNAs appear to function as key intermediaries in triggering RNA interference in invertebrates and in vertebrates, and in triggering sequence- specific RNA degradation during posttranscriptional gene silencing in plants.
[0137] In at least certain embodiments, the active agents (e.g., oligonucleotides) may have strand lengths comprising, for example, approximately 12 to 50, or 18 to 40, or 20 to 30 nucleotides, including a targeting sequence (i.e., a seed sequence) that is complementary to a target sequence of a nucleic acid which comprises a portion of an AR coregulator, such as an AR coregulator as listed elsewhere herein, a pre-mRNA transcribed from an AR coregulator, and/or (2) a mature mRNA processed from said pre-mRNA. For example, when an oligonucleotide binds to the target sequence of a preprocessed mRNA, it effectively inhibits splicing at the normal splice acceptor site and thus produces a splice variant mRNA, leading to truncated or otherwise aberrant versions of the encoded protein upon translation, or when the oligonucleotide binds to the target region of a mature mRNA, it effectively inhibits proper translation of the mRNA into an encoded protein.
[0138] The term "nucleic acid" is well known in the art. A "nucleic acid" as used herein will generally refer to a molecule (i.e., a strand) of DNA, RNA or a derivative or analog thereof, comprising a nucleobase. A nucleobase includes, for example, a naturally-occurring purine or pyrimidine base found in DNA (e.g., an adenine "A," a guanine "G," a thymine "T" or a cytosine "C") or RNA (e.g., an "A," a "G," a uracil "U" or a "C"). The term nucleobase also includes nonnatural bases as described below. The term "nucleic acid" encompasses the terms "oligonucleotide" and "polynucleotide," each as a subgenus of the term "nucleic acid." The term
"oligonucleotide" generally refers to a molecule of between about 3 and about 100 nucleobases in length. The term "polynucleotide" generally refers to at least one molecule of greater than about 100 nucleobases in length. These definitions generally refer to a single- stranded molecule, but in specific embodiments will also encompass an additional strand that is partially, substantially or fully complementary to the single- stranded molecule. Thus, a nucleic acid may encompass a double-stranded molecule that comprises a complementary strand or "complement" of a particular sequence comprising a molecule. As used herein, a singlestranded nucleic acid may be denoted by the prefix "ss," and a double-stranded nucleic acid by the prefix "ds. The terms "polynucleotide sequence" or “nucleic acid,” as used herein, include any polynucleotide sequence which encodes a peptide or fusion protein (or polypeptide) including polynucleotides in the form of RNA, such as mRNA, or in the form of DNA, including, for instance, cDNA and genomic DNA obtained by cloning or produced by chemical synthetic techniques or by a combination thereof. The RNA or DNA may be double-stranded or single-stranded. Single-stranded DNA may be the coding strand, also known as the sense strand, or it may be the non-coding strand, also referred to as the anti-sense strand.
[0139] Where a strand is designated herein as RNA, and thus comprises uracil (U) nucleobases, the present disclosure is also directed to an equivalent DNA sequence where the U nucleobase is replaced with a thymine (T) nucleobase. For example, where an RNA active agent described herein comprises the sequence GUCUGA, the equivalent DNA active agent comprises the seed sequence GTCTGA.
[0140] As is known in the art, a nucleoside is a base-sugar combination. The base portion of the nucleoside is normally a heterocyclic base. The two most common classes of such heterocyclic bases are the purines and the pyrimidines. Nucleotides are nucleosides that further include a phosphate group covalently linked to the sugar portion of the nucleoside. For those nucleosides that include a pentofuranosyl sugar, the phosphate group can be linked to either the 2', 3' or 5' hydroxyl moiety of the sugar. In forming oligonucleotides, the phosphate groups covalently link adjacent nucleosides to one another to form a linear polymeric compound. In turn the respective ends of this linear polymeric structure can be further joined to form a circular structure, however, open linear structures are generally preferred. Within the oligonucleotide structure, the phosphate groups are commonly referred to as forming the intemucleoside backbone of the oligonucleotide. The normal linkage or backbone of RNA and DNA is a 3' to 5' phosphodiester linkage.
[0141] Therefore, in the context of the present disclosure, the term "oligonucleotide" refers to an oligomer or polymer of RNA or DNA or mimetics thereof. This term includes
oligonucleotides composed of naturally -occurring nucleobases, sugars and covalent internucleoside (backbone) linkages as well as oligonucleotides having non-naturally-occurring nucleobases, sugars and synthetic heterocycles and covalent internucleoside (backbone) linkages which function similarly. Such modified or substituted non-natural oligonucleotides , as compared to native (natural) forms may have desirable properties such as, for example, enhanced cellular uptake, enhanced affinity for nucleic acid target and increased stability in the presence of nucleases.
[0142] Where used herein, the term “oligonucleotide,” is also intended to include linked nucleobase sequences containing modified backbones comprising non-natural internucleoside linkages. As defined in this specification, oligonucleotides having modified backbones include those that retain a phosphorus atom in the backbone and those that do not have a phosphorus atom in the backbone. Further, for the purposes of this specification, the term “nucleoside” is intended to refer to a nucleobase linked to a ribose or deoxyribose sugar (a natural nucleoside), and to a nucleobase linked to a non-ribose or non-deoxyribose heterocycle, e.g., a morpholine structure (a non-natural, or modified, nucleoside or other structures described elsewhere herein). Thus, a series of such modified, non-natural, nucleosides linked together via an internucleoside backbone can also be considered to be an oligonucleotide (a non-natural, or modified, oligonucleotide). Further, the term “sugar” where used herein in the context of a nucleoside, is intended to include “non-sugar” heterocyclic compounds, such as morpholines, as the portion of the internucleoside backbone which is linked to the nucleobase.
[0143] Oligonucleotides useful in the compounds and methods disclosed herein also include those comprising entirely or partially of naturally occurring nucleobases. Naturally occurring nucleobases as defined herein, include adenine, guanine, thymine, cytosine, and uracil. Although 5-methylcytosine (5-me-C) is technically a naturally occurring nucleobase, for the purposes of the present disclosure it will be included in the list of non-natural (a.k.a., modified) nucleobases.
[0144] As noted above, oligonucleotides of the present disclosure may further include those comprised entirely or partially of modified nucleobases and their corresponding nucleosides. These modified nucleobases include, but are not limited to, 5-uracil (pseudouridine), dihydrouracil, inosine, ribothymine, 5-me-C, 7-methylguanine, hypoxanthine, xanthine, 5- hydroxymethyl cytosine, 2- aminoadenine, 2-methyladenine, 6-methyladenine, 2- propyladenine, N6-adenine, N6-isopentenyladenine, 2-methylthio-N6-isopentenyladenine, 2- methylguanine, 6-methylguanine, 2-propylguanine, 1-methylguanine, 7-methylguanine, 2,2- dimethylguanine, 2-thiouracil, 2-thiothymine, 2-thiocytosine, 5-fluorouracil, 5 -bromouracil, 5-
chlorouracil, 5-iodouracil, dihydrouracil, 5-methyl-2-thiouracil, 2-thiouracil, 4-thiouracil, 5- methyluracil, uracil-5 -oxy acetic acid methylester, uracil-5 -oxy acetic acid, 5- carboxymethylaminomethyl-2-thiouracil, hypoxanthine, xanthine, 4-acetylcytosine, 5- (carboxyhydroxylmethyl) uracil, 5 -methoxy carboxyme thy luracil, 5-methoxyuracil, 5-methyl- 2-thiouracil, 3-(3-amino-3-N-2-carboxypropyl) uracil, 5-carboxymethylaminomethyluracil, 5- methylaminomethyluracil, 5-methoxyaminomethyl-2-thiouracil, alkynyl derivatives of pyrimidine bases including 5-propynyl uracil, and 5-propynyl cytosine, 6-azo uracil, 6-azo cytosine, 6-azo thymine, 4-thiouracil, 8-halo-adenines, 8-amino adenine, 8-thiol adenine, 8- thioalkyl adenine, 8-hydroxyl adenine, 5 -trifluoromethyl uracil, 3-methylcytosine, 5- methylcytosine, 5-trifluoromethyl cytosine, 7-methylguanine,7-methyladenine, 2-F-adenine, 2- amino- adenine, 8-azaguanine and 8-azaadenine, 7-deazaguanine, 8-halo-guanines, 8-amino guanine, 8-thiol guanine, 8-thioalkyl guanine, 8-hydroxyl guanine, 7-deazaadenine, 3- deazaguanine, 3-deazaadenine, beta-D-galactosylqueosine, beta-D-mannosylqueosine, 1- methylinosine, 2,6-diaminopurine, queosine, tricyclic pyrimidines, phenoxazine cytidine(lH- pyrimido[5,4-b][l,4]benzoxazin-2(3H)-one), and phenothiazine cytidine (lH-pyrimido[5,4- b 111 ,4 |benzothiazin-2(3H)-one.
[0145] The present disclosure also encompasses oligonucleotides which comprise targeting sequences (base sequences) that are complementary to particular nucleic acid target sequences taught herein. A nucleic acid is a "complement" or is "complementary" to another nucleic acid when it is capable of base-pairing with the other nucleic acid according to the standard Watson-Crick, Hoogsteen or reverse Hoogsteen binding complementarity rules. Polynucleotides (nucleic acids) are described as "complementary" to one another when hybridization occurs in an antiparallel configuration between two single-stranded polynucleotides.
[0146] More particularly, "complementary," as used herein, refers to the capacity for precise pairing between two nucleotides. For example, if a nucleotide at a certain position of an oligonucleotide is capable of hydrogen bonding with a nucleotide at the same position of a DNA or RNA molecule, then the oligonucleotide and the DNA or RNA are considered to be complementary to each other at that position. The oligonucleotide and the DNA or RNA are complementary to each other when a sufficient number of corresponding positions in each molecule are occupied by nucleotides which can hydrogen bond with each other. Thus, "specifically hybridizable" and "complementary" are terms which are used to indicate a sufficient degree of complementarity or precise pairing such that stable and specific binding occurs between the oligonucleotide and the DNA or RNA target, and as such, as is understood in the art, the targeting sequence of an antisense oligonucleotide of the present disclosure need not
be 100% complementary to that of its target sequence to be specifically hybridizable. An oligonucleotide is specifically hybridizable when binding of the oligonucleotide to the target sequence of the DNA or RNA molecule interferes with the normal function of the target DNA or RNA to cause a loss of utility, and there is a sufficient degree of complementarity to avoid non-specific binding of the oligonucleotide to non-target sequences under conditions in which specific binding is desired, i.e., under physiological conditions in the case of in vivo assays or therapeutic treatment, and in the case of in vitro assays, under conditions in which the assays are performed. An oligonucleotide and a target sequence are thus complementary to each other when a sufficient number of nucleobases of the oligonucleotide can hydrogen bond with the corresponding nucleobases of the target sequence, such that a desired effect will occur (e.g., antisense inhibition of a target nucleic acid).
[0147] For example, an oligonucleotide in which 18 of 20 nucleobases of the oligonucleotide are complementary to a target sequence, and would therefore specifically hybridize, would represent 90 percent complementarity. In this example, the remaining noncomplementary nucleobases may be clustered or interspersed with complementary nucleobases and need not be contiguous to each other or to complementary nucleobases. As such, an oligonucleotide which is 18 nucleobases in length having three noncomplementary nucleobases which are flanked by two regions of complete complementarity with the target nucleic acid, or are distributed in non-contiguous positions, would have 83% overall complementarity with the target sequence.
[0148] In other embodiments, the seed sequence of the antisense oligonucleotide provided herein is fully complementary (i.e. 100% complementary) to a target sequence of a nucleic acid. As used herein, "fully complementary" means each nucleobase of the referenced portion of an oligonucleotide (e.g., the seed sequence) is capable of precise base pairing with the corresponding nucleobases of a target nucleic acid.
[0149] The term "target sequence" where used herein refers to a contiguous series of nucleobases in a specific nucleotide sequence (target region), for example of an mRNA. The term “target sequence” refers to a sequence that is a subsequence (portion or segment) of the target region, or to the entire sequence of the target region. A target sequence may include the 5’ terminal nucleobase of a nucleic acid sequence plus adjacent internal nucleobases of the sequence, or the 3’ terminal nucleobase plus adjacent internal nucleobases of the sequence, or only internal nucleobases within the sequence, or the target sequence may be 100% identical to the target region. In certain embodiments, a nucleic acid compound of the present disclosure comprises an oligonucleotide having a nucleobase sequence that, when written in the 5' to 3'
direction, comprises the reverse complement of a target sequence of a nucleic acid target region to which it is targeted.
[0150] The terms "complementary" and "antisense" can be used interchangeably. Complementary also refers to polynucleotide sequences that are substantially complementary (antisense) over their entire length and have very few base mismatches. For example, sequences of fifteen bases in length may be termed complementary when they have complementary nucleotides at thirteen or fourteen positions. Naturally, sequences which are completely complementary will be sequences which are entirely complementary throughout their entire length and have no base mismatches.
[0151] In certain embodiments, oligonucleotides of the present disclosure are synthesized using one or more modified nucleotides. As used herein, the terms "modified" and "modification" when used in the context of the constituents of a nucleotide monomer, i.e., sugar, nucleobase and internucleoside linkage (backbone), refer to non-natural changes to the chemical structure of these naturally occurring constituents or the substitutions of these constituents with non-naturally occurring ones, i.e., mimetics. For example, the "unmodified" or "naturally occurring" sugar ribose (of RNA) can be modified by replacing the hydrogen at the 2'-position of ribose with a methyl group. Similarly, the naturally occurring internucleoside linkage of nucleic acids is a 3' to 5' phosphodiester linkage that can be modified, in one embodiment, by replacing one of the non-bridging oxygen atoms of the phosphate linker with a sulfur atom to create a phosphorothioate linkage. Modified oligonucleotides are structurally distinguishable, but functionally interchangeable with naturally occurring or synthetic unmodified oligonucleotides and usually have enhanced properties such as increased resistance to degradation by exonucleases and endonucleases, or increased binding affinity.
[0152] As noted above, in certain embodiments, modifications to the oligonucleotides of the present disclosure encompass substitutions or changes in internucleoside linkages, sugar moieties, or nucleobases. Where used herein in reference to an oligonucleotide, the term “nonnatural” or "unnatural" refers to an oligonucleotide which comprises at least one modification in an internucleoside linkage, a sugar, and/or a nucleobase thereof, wherein such modified internucleoside linkage, modified sugar, and/or modified nucleobase is not found naturally in DNA or RNA (unless specifically defined otherwise herein).
[0153] Non-naturally occurring internucleoside linkages of the oligonucleotides of the present disclosure include those that contain a phosphorus atom and also those that do not contain a phosphorus atom. Numerous phosphorus-containing modified oligonucleotide backbones are known in the art and may be used in the oligonucleotides of the present
disclosure. Examples of phosphorus-containing internucleoside linkages of non-natural (modified) oligonucleotide backbones which may occur in the presently disclosed oligonucleotides include, but are not limited to, phosphorothioate, phosphorodithioate, phosphoramidite, phosphorodiamidate, morpholino, phosphotriester, aminoalkylphosphotriester, phosphonate, chiral phosphorothioates, methyl and other alkyl phosphonates including 3’-alkylene phosphonate, 5'-alkylene phosphonate and chiral phosphonate, phosphinate, phosphoramidates including 3'-amino phosphoramidate and aminoalkylphosphoramidate, thionophosphoramidate, thionoalkylphosphonate, thionoalkylphosphotriester, selenophosphates and boranophosphates having normal 3 '-5' linkages, 2'-5' linked analogs of these, and those having inverted polarity wherein one or more internucleotide linkages is a 3' to 3’, 5' to 5' or 2' to 2’ linkage, and oligonucleotides having inverted polarity comprise a single 3' to 3' linkage at the 3'-most internucleotide linkage i.e., a single inverted nucleoside residue which may be abasic (the nucleobase is missing or has a hydroxyl group in place thereof) linkages. Examples of U.S. patents that teach the preparation of such phosphorus-containing linkages include, but are not limited to, U.S. Pat. Nos. 3,687,808; 4,469,863; 4,476,301; 5,023,243; 5,177,196; 5,188,897; 5,264,423; 5,276,019; 5,278,302;
5,286,717; 5,321,131; 5,399,676; 5,405,939; 5,453,496; 5,455,233; 5,466,677; 5,476,925;
5,519,126; 5,536,821; 5,541,306; 5,550,111; 5,563,253; 5,571,799; 5,587,361 ; 5,194,599;
5,565,555; 5,527,899; 5,721,218; 5,672,697 and 5,625,050.
[0154] As noted above, in some embodiments, the internucleoside linkages are without phosphorus atoms and may instead comprise short chain alkyl or cycloalkyl internucleoside linkages, mixed heteroatom and alkyl or cycloalkyl intemucleoside linkages, or one or more short chain heteroatomic or heterocyclic intemucleoside linkages. In further embodiments, the non-naturally occurring internucleoside linkages are uncharged and in others, the linkages are achiral. In some embodiments, the non-naturally occurring internucleoside linkages are uncharged and achiral, such as peptide nucleic acids (PNAs).
[0155] It is understood that the sequence set forth in each sequence or SEQ ID NO contained herein is independent of any modification to sugar moieties, intemucleoside linkages, or nucleobases of the sequence, unless otherwise specified. As such, antisense oligonucleotides of the present disclosure may be defined by a complementary correspondence to a sequence or SEQ ID NO disclosed herein, or segment thereof, and may comprise, independently, one or more modifications to a sugar moiety, an intemucleoside linkage, or a nucleobase. Other embodiments of oligonucleotide backbones include siloxane backbones; sulfide, sulfoxide and sulfone backbones; formacetyl and thioformacetyl backbones; methylene formacetyl and
thioformacetyl backbones; riboacetyl backbones; alkene containing backbones; sulfamate backbones; methyleneimino and methylenehydrazino backbones; sulfonate and sulfonamide backbones; amide backbones; and others having mixed N, 0, S and CH2 component parts. Examples of U.S. patents that teach the preparation of such non-phosphorus containing oligonucleotides include, but are not limited to, U.S. Pat. Nos. 5,034,506; 5,166,315; 5,185,444; 5,214,134; 5,216,141 ; 5,235,033; 5,264,562; 5,264,564; 5,405,938; 5,434,257; 5,466,677;
5,470,967; 5,489,677; 5,541,307; 5,561,225; 5,596,086; 5,602,240; 5,610,289; 5,602,240;
5,608,046; 5,610,289; 5,618,704; 5,623,070; 5,663,312; 5,633,360; 5,677,437; 5,792,608;
5,646,269 and 5,677,439.
[0156] In certain oligonucleotide mimetics of the present disclosure, both the sugar moiety and the internucleoside linkage, i.e., the backbone, of the nucleotide units are replaced with nonnatural groups. One such oligomeric compound is referred to as a peptide nucleic acid (PNA). In PNA compounds, the sugar-backbone of an oligonucleotide is replaced with an amide containing backbone, in particular an aminoethylglycine backbone. The nucleobases are retained and are bound directly or indirectly to aza nitrogen atoms of the amide portion of the backbone. Examples of U.S. patents that teach the preparation of PNA compounds include, but are not limited to, U.S. Pat. Nos. 5,539,082; 5,714,331; and 5,719,262.
[0157] The oligonucleotides described herein stabilized against nucleolytic degradation such as by the incorporation of a modification, e.g., a nucleotide modification. The oligonucleotides can include a non-natural nucleoside linkage such as a phosphorothioate linkage as the first, second, and/or third internucleotide linkage at the 5' or 3' end of the oligonucleotide sequence. In certain embodiments, the oligonucleotides can include a 2'- modified nucleotide, e.g., a 2'-deoxy, 2'-deoxy-2'-fluoro, 2'-O-methyl, 2'-O- methoxyethyl (2'- O-MOE), 2’-O-aminopropyl (2'-O-AP), 2’-O-dimethylaminoethyl (2'- O-DMAOE), 2’-O- dimethylaminopropyl (2’-O-DMAP), 2'-O- dimethylaminoethyloxyethyl (2'-O-DMAEOE), or 2'-O— N-methylacetamido (2'-0-NMA) nucleotide. In a particular embodiment, the oligonucleotides include at least one 2'-O-methyl-modified nucleotide, and in some embodiments, all of the nucleotides include a 2'-O-methyl modification.
[0158] As noted elsewhere herein, the oligonucleotide can be further modified so as to be conjugated to an organic moiety such as a biogenic molecule that is selected to improve stability, distribution and/or cellular uptake of the oligonucleotide, e.g., cholesterol, forming the nucleic acid compound of the present disclosure. Such an organic moiety can be attached, e.g., to the 3’ or 5' end of the oligonucleotide, and/or at the 2' position of the sugar moiety of a nucleotide of the oligonucleotide, such as the 2' ribose position.
[0159] The nucleic acid compound can further be in isolated form or can be part of a pharmaceutical composition, such as a pharmaceutical composition formulated for parental administration. The pharmaceutical compositions can contain one or more nucleic acid compounds, and in some embodiments will contain two or more inhibitory nucleic acid compounds, each one directed to a different target gene.
[0160] The oligonucleotides can be delivered in any of a variety of forms, including in liposomes as described above, and via expression vectors. The oligonucleotide can be endogenously expressed from transcription units inserted into DNA or RNA vectors. The recombinant vectors can be DNA plasmids or viral vectors for example. Viral vectors suitable for producing the presently disclosed oligonucleotides capable of reducing expression or activity of an AR coregulator can be constructed based on, but not limited to, adeno-associated virus, retrovirus, lentivirus, adenovirus, or alphavirus. The recombinant vectors which contain a nucleic acid for expressing the oligonucleotides disclosed herein can be delivered as described above and can persist in target cells. Alternatively, viral vectors can be used that provide for transient expression of the oligonucleotides. Such vectors can be repeatedly administered as necessary. Once expressed, in one embodiment, the oligonucleotides may interact with the target RNA and inhibit mRNA activity for example. The delivery vehicles (vectors) for the oligonucleotides optionally comprise an expression construct which includes an enhancer sequence, a promoter sequence, and other sequences necessary for expression of the products of the oligonucleotide sequence desired to be produced. In one embodiment, the promoter is cell- specific. The term "cell-specific" means that the particular promoter selected for the recombinant vector can direct expression of the selected transgene only in a particular cell type. As one example, the promoter is specific for expression in prostate cells. A number of viruses can be used in connection with the methods described herein, including papovaviruses, e.g., SV40, adenovirus, vaccinia virus, adeno-associated virus, herpesviruses including HSV and EBV, and retroviruses of avian, murine, and human origin. In certain embodiments, lentiviral vectors can be used in connection with the methods described herein. In certain embodiments, the lentiviral vector can be a doxycycline-inducible lentiviral vector engineered to express one or more shRNAs or siRNAs.
[0161] Specific vectors which may be used include, but are not limited to, adeno- associated virus vectors (e.g., as disclosed in U.S. Pat. Nos. 5,139,941, 5,436,146, and 5,622,856), an attenuated or gutless adenoviral vectors, (e.g., as disclosed in U.S. Pat. No. 5,935,935), lentiviral vectors (such as are disclosed in U.S. Pat. Nos. 5,665,577; 5,994,136; and 6,013,516), plasmids or synthetic (non-viral) vectors (such as disclosed in U.S. Pat. Nos.
4,394,448 and 5,676,954), and/or nanoparticles (such as disclosed, for example, in U.S. Patents 6,217,912; 7,514,098; and 8,323,618), retroviral vectors (such as are disclosed in U.S. Pat. Nos. 5,672,510; 5,707,865; and 5,817,491), herpes virus vectors (such as are disclosed in U.S. Pat. No. 5,288,641), and sindbis virus vectors and papilloma vims vectors (such as are disclosed in EP 820 773). The vectors may be either monocistronic, bicistronic, or multicistronic. A recombinant vector (e.g., lend-, parvo-, AAV) sequence can be packaged as a “particle” for subsequent infection (transduction) of a cell, ex vivo, in vitro or in vivo. Where a recombinant vector sequence is encapsulated or packaged into an AAV particle, the particle can also be referred to as a “rAAV.” Such particles include proteins that encapsulate or package the vector genome. Particular examples include viral envelope proteins, and in the case of AAV, capsid proteins.
[0162] Thus, the oligonucleotides of the present disclosure may be used as a form of gene therapy. The term “gene therapy” as used herein means genetic modification of cells by the introduction of exogenous DNA or RNA into these cells, such as via an expression vector containing the oligonucleotide, for the purpose of expressing or replicating one or more peptides, polypeptides, proteins, oligonucleotides, or polynucleotides in vivo for the treatment or prevention of disease or deficiencies in humans or animals. Examples of gene therapy are disclosed for example in U.S. Pat. No. 5,399,346. Any suitable route of administration of the oligonucleotide-containing vector may be employed. For example, parenteral (subcutaneous, subretinal, suprachoroidal, intramuscular, intravenous, transdermal) and like forms of administration may be employed. Dosage formulations include injections, implants, or other known and effective gene therapy delivery methods.
[0163] Delivery of the oligonucleotide-expressing vectors can be systemic, such as by intravenous or intra-muscular administration, direct administration to a tumor site, such as a prostate tumor, by administration to target cells ex-planted from a subject followed by reintroduction into the subject, or by any other means that would allow for introduction into the desired target cell. The therapeutic and/or pharmaceutical compositions, in non-limiting embodiments, contain viral particles per dose in a range of, for example, from about 104 to about 1011 particles, from about 10s to about 1010 particles, or from about 106 to about 109 particles. In the context of AAV vectors, vector genomes are provided in in a range of, for example, from about 104 to about 1014 vector genomes, from about 105 to about 1013 vector genomes, from about 106 to about 1013 vector genomes, from about 107 to about 1013 vector genomes, from about 10s to about 1013 vector genomes, or from about 109 to about 1013 vector genomes. Such doses/quantities of AAV vector are useful in the methods set forth herein.
[0164] Because nucleases that cleave the phosphodiester linkages are expressed in almost every cell, unmodified nucleic acid molecules such as the inhibitory oligonucleotides of the present disclosure may be modified to resist degradation, as described above for example. Other biogenic molecules may be conjugated to the oligonucleotides to improve their ability to resist degradation, target certain cells, or to cross barriers like cell membranes or the blood brain barrier. Examples of biogenic molecules that can be conjugated to the oligonucelotides include lipids such as, but not limited to, stearic acid, palmitic acid, docosanoic acid, docosahexanoic acid, docosahexaenoic acid, cholesterol, tocopherol, and other C12-C22 saturated or unsaturated fatty acids; peptides such as but not limited to, cell-penetrating peptides (CPPs) such as penetratin, HIV-1 Tat peptides, pVEC-Cadherin 615-634, polyarginines (6-12), and transportan, linear and cyclic RGD-containing peptides, and SPACE peptide; receptor-specific ligands; aptamers (synthetic oligoribonucleotides); antibodies or antibody fragments; CpG-containing oligonucleotides; polyamines, such as spermine and spermidine; polymers such as dendrimers and polyethylene glycols (e.g., PEG 0.6 kDa -5,000 kDa); and saccharides such as N-acetylgalactosamine (GalNAc) and cyclodextrins. The biogenic molecule may be conjugated to the oligonucleotide by any suitable means, such as via linker or a cleavable bond such as but not limited to disulfide, thioether, pH sensitive (e.g., hydrazone or carboxymethylmaleic anhydride), or ethylene glycol.
[0165] The oligonucleotides or nucleic acid compounds of the present disclosure may be delivered in the form of nanoparticles and microparticles which encapsulate the nucleic acid compounds within liposomes of cationic lipids or within PEG, for example. These delivery systems can enhance intracellular delivery either by protecting the nucleic acid compound from nuclease degradation and/or by promoting absorptive endocytosis. Further, the addition of dioleylphosphatidylethanolamine to liposome delivery systems results in the destabilization of endosomal membranes and promotion of release of the oligonucleotide after endocytosis. The nucleic acid compounds can be administered to cells by a variety of other methods known to those of skill in the art, including, but not limited to, ionophoresis, or by incorporation into other vehicles, such as hydrogels, cyclodextrins, biodegradable nanocapsules, and bioadhesive microspheres, or by proteinaceous vectors. In one example, the nucleic acid compounds can be delivered via the nanoparticle system shown in U.S. Patent Application Publication 2019/0255088. The liposomes may comprise amphipathic agents such as lipids which exist in aggregated form as micelles, insoluble monolayers, liquid crystals, or lamellar layers in aqueous solution. Suitable lipids for liposomal formulation include, without limitation, monoglycerides, diglycerides, sulfatides, lysolecithin, phospholipids, saponin, bile acids, and
the like. Preparation of such liposomal formulations is within the level of skill in the art, as disclosed, for example, in U.S. Patent Nos. 4,235,871; 4,501,728; 4,837,028; and 4,737,323.
[0166] In certain embodiments, the nanoparticles which contain the nucleic acid compounds of the present disclosure may comprise a pharmaceutically acceptable carrier such as, but not limited to, poly (ethylene-co- vinyl acetate), PVA, partially hydrolyzed poly(ethylene-co-vinyl acetate), poly(ethylene-co-vinyl acetate-co-vinyl alcohol), a crosslinked poly(ethylene-co- vinyl acetate), a cross-linked partially hydrolyzed poly(ethylene-co- vinyl acetate), a cross-linked poly(ethylene-co- vinyl acetate-co-vinyl alcohol), poly-D,L-lactic acid, poly-L-lactic acid, poly glycolic acid, PGA, copolymers of lactic acid and glycolic acid, polycaprolactone, poly valerolactone, poly (anhydrides), copolymers of polycaprolactone with polyethylene glycol, copolymers of polylactic acid with polyethylene glycol, polyethylene glycol; fibrin, Gelfoam™ (which is a water-insoluble, off-white, nonelastic, porous, pliable gel foam prepared from purified gelatin and water for injection), and combinations and blends thereof. Copolymers can comprise from about 1 % to about 99% by weight of a first monomer unit such as ethylene oxide and from 99% to about 1% by weight of a second monomer unit such as propylene oxide. Blends of a first polymer such as gelatin and a second polymer such as poly-L-lactic acid or poly glycolic acid can comprise from about 1% to about 99% by weight of the first polymer and from about 99% to about 1 % of the second polymer.
[0167] The oligonucleotides or nucleic acid compounds can be delivered directly by systemic administration such as using oral formulations or stereotactic injection into prostate or prostate tumor, typically in saline with chemical modifications to enable uptake, or other methods described elsewhere herein. In certain embodiments, such as when the oligonucleotide of the nucleic acid compound has a phosphorothioate backbone, the oligonucleotide binds to serum proteins, slowing excretion by the kidney. The aromatic nucleobases also interact with other hydrophobic molecules in serum and on cell surfaces. In certain embodiments, siRNA delivery systems involve complexing the RNA with cationic and neutral lipids, although encouraging results have also been obtained using peptide transduction domains and cationic polymers. Including PEGylated lipids in the formulation prolongs the circulating half-life of the particles.
[0168] As noted, one type of optimization of single-stranded DNA or RNA oligonucleotides is the use of chemical modifications to increase the nuclease resistance such as the introduction of phosphorothioate ("PS") linkages in place of the phosphodiester bond. This modification improves protection from digestion by nucleases. PS linkages also improved binding to serum proteins in vivo, increasing half-life and permitting greater delivery of active
compound to tissues. Chemical modifications to subunits of the nucleotides can also improve potency and selectivity by increasing binding affinity of oligonucleotides for their complementary sequences. Examples of such modifications to the nucleoside sugars include 2'-0-methyl (2'-0-Me), 2'-fluoro (2'-F), and 2'-O-methoxyethyl (2'-M0E) RNA, and others as discussed elsewhere herein. Even more affinity can be gained using oligonucleotides modified with locked nucleic acid (LNA), which contains a methylene bridge between the 2’ and 4' position of the ribose. This bridge "locks" the ribose ring in a conformation that is ideal for binding, leading to high affinity for complementary sequences. Related bridged nucleic acid (BNA) compounds have been developed and share these favorable properties. Their high affinity has permitted the development of far shorter oligonucleotides than previously thought possible which nonetheless retain high potency. The chemistry for introducing 2’-0-Me, 2'- MOE, 2'-F, or LNA into oligonucleotides is compatible with DNA or RNA synthesis, allowing chimeras with DNA or RNA bases to be easily obtained. This compatibility allows the properties of chemically modified oligonucleotides to be fine-tuned for specific applications, which is a major advantage for development that makes LNAs and other BNAs convenient tools for many applications.
[0169] Therapeutic administration of the active agents described herein include any method by which a nucleic acid (e.g., DNA or RNA), as known to one of ordinary skill in the art. For treatment of aggressive prostate cancer, delivery may be via, for example, oral administration and/or injection into the prostate gland or tumor or both.
[0170] In certain embodiments, the active agents can be delivered to an organelle, a cell, a tissue, a tumor or an organism via one or more injections (i.e., a needle injection), such as, for example, orally, subcutaneously, intradermally, intramuscularly, intravenously, or intraperitoneally. A described inhibitory nucleic acid or other active agent can be incorporated into pharmaceutical compositions suitable for administration. For example, pharmaceutical compositions can comprise one or more the active agents and a pharmaceutically acceptable carrier.
[0171] Then active agent may be provided in a sustained release composition. The use of immediate or sustained release compositions depends on the nature of the condition being treated. If the condition consists of an acute or over-acute disorder, treatment with an immediate release form can be conducted over a prolonged release composition. Alternatively, for certain preventative or long-term treatments, a sustained release composition may be appropriate.
[0172] The active agent can be administered in a single dose or in multiple doses. Where the administration of the active agent is by infusion, the infusion can be a single sustained dose
or can be delivered by multiple infusions. Injection of the active agent can be directly into the tissue at or near the site of aberrant or unwanted target gene expression. Multiple injections of the active agent can be made into the tissue, for example, into the prostate gland, into the prostate tumor, or near the tumor.
[0173] In addition to treating pre-existing aggressive or non-aggressive prostate cancers, active agents of the disclosure can be administered prophylactically in order to prevent or slow the conversion of a non-aggressive prostate cancer to an aggressive form. The active agent can be employed in combination therapies, meaning that the present compositions can be administered concurrently with, prior to, or subsequent to, one or more other desired therapeutic agents or medical procedures. The combination of therapies (therapeutic agents or procedures) to employ in a combination regimen will take into account compatibility of the desired therapeutic agents and/or procedures and the desired therapeutic effect to be achieved. It will also be appreciated that thetherapies employed can achieve a desired effect for the same disorder (for example, a compound described herein can be administered concurrently with another therapeutic agent used to treat the same disorder), or they can achieve different effects (e.g., control of any adverse effects).
[0174] In various embodiments, the advantages of the presently disclosed technology include increased cellular uptake without the requirement of transfection reagents, improved stability and circulation, reduction of adverse stimulation of the immune response, reduced off- target toxicity, specific silencing of a cancer sternness and EMT promoting genes, and improved efficacy against solid tumor cancers. These advantages represent a substantial improvement of nanoparticle and liposome-based RNA silencing based delivery platforms. This technology can be used in the cancer related context depending on the contribution of DCLKl-related signaling on tumor related outcomes including metastasis, inhibition, or prevention. The route of administration can be, but is not limited to, intravenous, intraperitoneal, or intertumoral, or by any other effective means such as oral, subcutaneous, or transdermal administration. Additionally, given the critical role of tumor sternness in promoting the highly proliferative features of tumorigenesis, these agents can be used in combination with traditional and or other therapeutic modalities.
[0175] SAMiRNA is a self-assembling, micelle-forming double-conjugated RNAi with the oligonucleotide in the middle flanked by a hydrophobic and hydrophilic end that is stable in the circulation due to reduced metabolic clearance. The hydrophobic interactions drive the molecular assembly into the micellar structure, allowing for targeting moleties lo be added to the hydrophilic end.
[0176] The linker chemistry used in the present disclosure allows for effective endosomal escape and the SAMiRNA hides the unmodified double- stranded RNA from triggering innate immune stimulation, thus reducing the side effects associated with excessive stimulation of the immune system. The siRNA design platform comprises a unique siRNA synthesis algorithm that allows for creation of highly specific siRNAs that can knockdown specific RNA moieties of virtually any gene product. Large scale synthesis and high-quality detection protocols, RT- qPCR enables quantitative detection of RNA silencing efficacy.
EXAMPLES
[0177] Certain embodiments of the present disclosure will now be further discussed in terms of several specific, non-limiting, examples. The examples described below will serve to illustrate the general practice of the present disclosure, it being understood that the particulars shown are merely exemplary for purposes of illustrative discussion of particular embodiments of the present disclosure only and are not intended to be limiting of the claims of the present disclosure.
EXPERIMENTAL
METHODS
Mice:
[0178] Female C57BL/6 (B6) mice at 8 weeks of age were purchased from Charles River Laboratories (Bethesda, MD). All animal procedures were approved by the Institutional Animal Care and Use Committee at the University of Oklahoma Health Sciences Center (OUHSC) (original protocol 20-020- AHI, renewed as 22-082-CHI). This study was performed in accordance with the recommendations of the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health.
Antibodies and Fluorochromes:
[0179] Horseradish peroxidase (HRP)-conjugated anti-mouse IgM, IgGl, IgG2b, and IgG2c were purchased from Southern Biotech (Birmingham, AL). Biotin-conjugated anti- CXCR5 (2G8), FITC-conjugated anti-B220 (RA3-6B2) and FITC-conjugated anti-CD21/23 (7G6) mAbs and PE-conjugated streptavidin were from BD Biosciences (San Jose, CA). APC- Cy7-conjugated anti-CD19 (6D5) and Alexa 488-conjugated anti-Ki67 (1 lf6) mAbs were purchased from Biolegend (San Diego, CA). PE-conjugated anti-CD19 (1D3) and VioletFluor 450-conjugated anti-CD4 (GK1.5) was purchased from Tonbo (Cytek, San Diego, CA).
AlexaFluor 750-conjugated anti-CD93 (223427) was purchased from R&D Systems (Minneapolis, MN). PE-conjugated anti-Annexin and 7AAD were from Stemcell Technologies (Cambridge, MA). FcR-blocking mAh 2.4G2 was from BioXCell (Lebanon, NH). APC- conjugated anti-CXCR4 (2B11) was purchased from Invitrogen, (Carlsbad, CA).
Purification of TcdB2, B2A, and D270N:
[0180] TcdB2, B2A, and D270N were expressed in a Bacillus megaterium expression system (MoBiTec, Gottingen, Germany) and purified by nickel affinity chromatography (GE Life Sciences, Boston, MA) as described in Bland, S.J., et al., Deletion of a 19-Amino-Acid Region in Clostridioides difficile TcdB2 Results in Spontaneous Autoprocessing and Reduced Cell Binding and Provides a Nontoxic Immunogen for Vaccination. Infect Immun, 2019, 87(8). Purity and integrity were confirmed by SDS-PAGE and each batch of TcdB2 was tested for toxicity using a CHO cell killing assay (Marion Ehrich, R.L.V.T., Jeffery M. Libby, Tracy D. Wilkins, Production of Clostridium difficile Antitoxin. Infection and Immunity, 1980. 28(3): p. 1041-1043).
Immunization and bleed schedule:
[0181] Mice were anesthetized with a vaporized 4% isoflurane / 96% medical air mixture and injection by the intraperitoneal (i.p.) route with 1 ng TcdB2 in sterile PBS. Control mice were injected with PBS or 1 ng of an enzymatically inactivated TcdB mutant known as D270N [38]. After 5 hr, mice were immunized subcutaneously (s.c.) with 10 pg B2A adsorbed to 100 pl of a 2% w/v Alhydrogel alum adjuvant suspension (Invivogen, San Diego, CA) and adjusted to a final volume of 200 pl with sterile PBS. The injection volume was divided equally over both flanks. The mice were immunized with B2A I alum on day 0 and then boosted on day 67. Retro-orbital blood was collected on days 14, 28, 42, 67 (pre-boost), and 81 (14 days postboost). Blood samples were incubated for 2 hr at room temperature, then centrifuged at 13,000 ref for 15 minutes. Sera were collected, aliquoted, and stored at -20°C. Where indicated TcdB2-treated and B2A-immunized mice were injected s.c. with 100 pg anti-CD40 (rat IgG2a, clone FGK4.5) or isotype control mAbs (rat IgG2a, clone 2A3) formulated for in vivo use (InVivoPlus™, BioXCell, Lebanon, NH). The mAbs were injected on days 1 and 8.
Fecal bacteria enumeration:
[0182] Bacteria shed were quantified on day 2 post-gavage. Fecal pellets were homogenized with IX PBS, serially diluted, plated on TCCFA and cultured under anaerobic conditions at 37°C. CFUs were counted within 48 hours.
ELISA:
[0183] Nunc MaxiSorp ELISA 96-well plates (Thermo Scientific, Waltham, MA) were coated overnight at 4°C with phosphate coating buffer (0.1 M Na2HPO4, pH 9.0) containing B2A at a final concentration of 10 pg / ml. Wells were washed 4 times with PBS-T (lx PBS, 0.05% Tween) and blocked with 1% BSA in PBS-T for 2 hr at room temperature. This was followed by washing 4 times with PBS-T. Wells were incubated overnight with mouse sera (serially diluted 2-fold) and PBS-T, then washed with PBS-T. Wells were incubated with 0.2 pg I ml HRP-conjugated IgGl, IgG2b, IgG2c, or IgM for 1 hr. Wells were washed with PBS- T and developed for 5 min at room temperature by addition of 90 pl of ABTS substrate to each well (SeraCare, Milford, MA). To stop the reaction, 110 pl of 10% w/v SDS solution was added to each well. OD of the samples at an absorbance of 405 nm was measured using the Spectrostar Nano™ spectrophotometer (BMG Labtech, Cary, NC) within 30 min.
Cell culture and in vitro neutralization:
[0184] The hamster epithelial cell line CHO-K1 (American Type Culture Collection, Manassas, VA) was cultured in F12-K medium (Gibco, Life Technologies Corporation, Grand Island, NY) supplemented with 10% FBS, 100 units / ml penicillin, and 100 pg / ml streptomycin (Coming, Manassas, VA). Cells were cultured at 37°C in 5% CO2 and passed every 48 hr using tryptic digestion. Ninety six-well plates (Falcon®, Corning, Durham, NC) were seeded with CHO-K1 cells at a density of 3 x 104 cells / well and incubated overnight at 37°C in 5% CO2. Sera were diluted to 1 :500 then incubated for 1 hr at 37°C with cell culture media containing TcdB2 at a final concentration of 250 nM. The CHO cell culture media was replaced with the sera-TcdB2-media solution (or media lacking TcdB2 or sera). Cells were incubated for 24 hr at 37°C before addition of 100 pl media containing 10 pl CCK-8 (Sigma- Aldrich®, Millipore Sigma, Saint Louis, MO). Cells were incubated for a further 2-3 hr at 37°C until the absorbance at 450 nm associated with untreated control cells reached 3.0. Background absorbance was near zero and subtracted from all samples. The absorbance associated with the TcdB-treated control cells was then subtracted. The percent viability was calculated as absorbance (experimental sample) / absorbance (untreated controls) x 100.
Cell preparation:
[0185] Murine spleens, lymph nodes (axillary, inguinal, and mesenteric) and bone marrow cells were isolated from B6 mice. Cells from spleen and lymph nodes were isolated by mechanical disruption. Bone marrow cells were obtained by trimming the ends of the long bones and flushing with media using a 27-gauge syringe needle. Erythrocytes were removed by hypotonic lysis with ACT.
ELISPOTS:
[0186] Multiscreen HTS ELISPOT wells (Millipore, Bedford, MA) were prepared for antigen coating by incubating with 35 % v/v ethanol for 30 s and washed twice with PBS. The plates were coated overnight with anti-mouse Ig or B2A (10 pg I ml final concentration) at 4”C. Plates were washed 3 times with PBS and blocked with RPMI 1640 containing 10% FBS (Atlanta Biologicals, Flowery Branch, GA) for 2 hr at room temperature. Isolated bone marrow cells (3xl06 cells per well) were added then subjected to a 1 :3 serial dilution such that the wells contained 2.00xl06, 6.67x10 s, 2.22x10s, or 7.41xl04 cells. The plates were then incubated in 5% CO2 at 37°C, for 4.5 hr. The cells were lysed and removed by 4 washes with PBS-T. Plates were incubated overnight at 4°C with 5% v/v FBS in PBS containing HRP-goat anti-mouse IgGl and IgG2b Ab at a final concentration of 1.0 pg / mL (Southern Biotech, Birmingham, AL). The plates were then washed with PBS-T and colorimetric detection performed. The developing solution was prepared by dissolving one tablet of AEC (Sigma Chemical Co., St. Louis, MO) in 2.5 mL dimethylformamide (Sigma Chemical Co., St. Louis, MO) and mixing with 47.5 mL of a 0.0075 N acetic acid, 0.0175 M sodium acetate buffer. The solution was passed through a 0.2 pm syringe filter before adding hydrogen peroxide to a final concentration of 0.0005% v/v. One hundred microliters of developing solution was added to each well and incubated at room temperature for 10 min. The reaction was stopped by addition of deionized water. The plates were then washed 20 times and allowed to air dry at room temperature under light-protected conditions for 24 hr. Antibody secreting cells (spots) were enumerated using an ImmunoSpot® analyzer (Cellular Technology Limited, Shaker Heights, OH).
Flow cytometry:
[0187] Splenocytes and lymph node cells were incubated with anti-FcR-blocking antibody at a final concentration of 2.4G2, 20 pg / ml for 5 min. Cells were then stained with fluorochrome-conjugated mAb cocktails to detect CD4+ T cells (anti-CD4) and B cell (antiCD 19 or anti-B220) populations. After incubating at 4°C (unless noted otherwise) for 1 hr,
cells were washed with PBS 3 times by centrifugation at 250 ref for 5 minutes. Cells were then fixed with 1 % w/v paraformaldehyde in PB S . Data were collected using a Stratedigm S 1200Ex flow cytometer (Stratedigm, San Jose, CA) and analyzed with FlowJo software (Version 2.0.1, Tree Star, Ashland, OR).
Cell viability assay:
[0188] Three million splenocytes in RPMI 1640 were added to each well of a 48-well tissue culture plate. Vehicle (media), TcdB2, or D270N was added to a final concentration of 0.1, 1, or 10 pM. Cells were incubated at 37°C for 30 min, 6, 24, or 48 hr before removal of TcdB2 or D270N by washing. Cells were re-suspended in RPMI 1640 medium containing 1% v/v FBS and incubated with an anti-FcR-blocking antibody (2.4G2) at a final concentration of 20 pg / ml for 5 min. Cell were stained with anti-B220 and anti-CD4 fluorochrome-conjugated antibodies for 30 min then with anti-Annexin/7AAD in Annexin V Binding Buffer (Stemcell Technologies, Cambridge, MA) for 15 min at room temperature. The cells were washed with 1 ml of Annexin V Binding Buffer (250 ref for 5 min), then re-suspended in 200 pl of Annexin V Binding Buffer. The cells were analyzed immediately by flow cytometry.
Capillary Electrophoresis and Western Blot:
[0189] Splenocytes or splenic B cells isolated with the STEMCELL Pan B cell Isolation Kit (Stemcell Technologies, Cambridge, MA) were incubated for 4 hr at 37°C in RPMI 1640 with 5% v/v FBS in the absence or presence of TcdB2 or D270N at a final concentration of 10 pM. Cells were washed twice in PBS and re-suspended at IxlO8 cells / ml in Mammalian Protein Extraction Reagent (M-PER) (Thermo Scientific, Rockford, IL) containing IX protease inhibitor cocktail set 1 (EMD Millipore, Billerica, MA). Cells were shaken for 10 min on ice and then centrifuged at 14,000 rc/'for 15 min. Supernatants were collected and aliquots stored at -80°C. The protein concentration in the lysates was calculated using a Bradford Protein Assay (Bio-Rad, Mountain View, CA).
[0190] The Jess Simple Western machine (Protein Simple, San Jose CA) is an automated capillary-based protein separation and immunoblotting system. The manufacturer’s standard protocol for the 12-230-kDa Jess separation module was followed. Cell lysates were prepared in 0.1X Sample buffer and Fluorescent 5X master mix containing dithiothreitol (400 mM final concentration) to achieve a final loading of 1.5 - 5 pg of protein per capillary. Samples were denatured at 95 °C for 5 min before being loaded into each well. The 12-230-kDa ladder and cellular lysates were separated by electrophoresis in their respective capillaries and then fixed.
Capillary bound proteins were washed, blocked and incubated with 1 : 100 dilution of anti-Racl (Clone 102/Racl, Becton Dickinson, San Diego, CA) and a 1 :500 dilution of anti-GAPDH (Clone 6C5, Abeam, Cambridge, MA) antibodies, then washed again and incubated with a 1 :100 dilution of HRP-conjugated anti-mouse IgG2b (Rael) or 1:200 IgGl (GAPDH) (Southern Biotech, Birmingham, AL). The HRP-labeled antibodies were detected using peroxide/luminol-S reagent (Protein Simple, San Jose, CA). Imaging of the chemiluminescence from the capillary bound proteins was performed using Compass for Simple Western software (Version 6.1.0, Protein Simple, San Jose, CA). An internal system control was included in each run.
Histology and IHC staining:
[0191] Mice were injected s.c. with 1 ng TcdB2 in 200 pl sterile PBS, divided equally between the left and right flanks. Controls were given 200 pl sterile PBS. Inguinal lymph nodes were isolated and fixed in 70% v/v ethanol. Paraffin sections (5 pm thick) were mounted on slides by Excalibur Pathology Inc. (Norman, OK). A portion of the slides were stained with H&E and imaged. The remaining slides were used for IHC staining. For IHC staining, slides were deparaffinized by incubating for 3 min in xylene, 1 : 1 xylene:ethanol, then 95-50% ethanol concentrations. Slides were then submerged in sodium citrate buffer (10 nM NA Citrate, 0.05% Tween 20, pH 6.0) and microwaved on a low power setting for 1 hr. Slides were washed 3 times with PBS for 5 min and incubated with 0.03% Triton X-100 in PBS for 15 min. After washing with PBS, slides were blocked for 30 min in a humidity chamber by incubation with 10% v/v normal goat serum in 1% w/v BSA. Tissue sections were stained with fluorochrome - conjugated mAbs to detect B cells and proliferating cells to identify GCs. Slides were washed with PBS and mounted in prolong gold medium, dried for 24 hr, then visualized using the Leica M205-MFC THUNDER microscope (Leica, Deerfield, IL).
Germinal Center Image Quantification:
[0192] Bright field images of H&E-stained lymph node sections at a 10X magnification were analyzed using ImageJ (NIH, distributed by Fiji, Bethesda, MD). The average diameter of each GC was calculated (length + width / 2). Area was calculated from the average diameter (d) [area = 7i*(d/2)2]. Area (pm2) and MFI)of GC within lymph node sections stained for IHC were analyzed using the LAS X Life Science Microscope Software (Version 5.1.0, Leica, Deerfield, IL). The relative signal of the GC was calculated using the following equation: [(MFI of GC - MFI of background) x Area of GC].
Multi-targeting mRNA profiling:
[0193] Mice were injected s.c. with 10 ng TcdB2 in 200 pl PBS (controls were given 200 pl PBS or 10 ng D270N in 200 pl PBS). Each injection was divided equally between the left and right flanks. Axillary and inguinal nodes were isolated after 7 days. Lymph nodes were placed immediately in DNA / RNA Shield (Zymo Research, Irvine, CA). RNA was purified from the lymph nodes using Zymo Quick-RNA Miniprep Plus Kit (Zymo Research, Irvine, CA). Purified RNA was stored at -80°C until needed. RNA analysis was performed by the OUHSC core facility using a NanoString nCounter SPRINT profiler™ (NanoString, Seattle, WA) in conjunction with the nCounter Myeloid Innate Immunity Panel™. Data were analyzed with nSolver 4.0 Advanced Analysis Software™ (Version 2.0.134, NanoString, Seattle, WA). Analysis was performed by normalizing the raw transcript counts utilizing negative synthetic sequences to account for background noise and positive synthetic sequences to account for technical variations.
Transwell Migration Assays:
[0194] B cells and CD4+ T cells were isolated from splenocytes using magnetic bead separation following the manufacturer’s protocol for the EasySep™ Mouse Pan-B Cell and EasySep™ Mouse Naive CD4+ T Cell Isolation Kits (STEMCELL Technologies, Cambridge, MA). Five hundred microliters of serum free media (RPMI 1640) or media containing CXCR12 (Sino Biological, Wayne, PA) at a final concentration of 100 ng / ml was added to the lower chamber of a 48-well transwell plate. Thirty thousand cells in a 300 pl volume, consisting of either total splenocytes, isolated B cells, or isolated CD4+ T cells in serum free media were added to the upper chamber of the transwell plates. Cells were then incubated for 6 hr at 37°C in 5% CO2. The media was carefully aspirated from the transwell inserts and the interior gently swabbed with a sterile cotton swab to remove any non-migratory cells. Transwell inserts were then placed in plate wells containing 700 pl of 70% ethanol for 10 min and allowed to air dry for 10 min to fix migratory cells. Once dry, transwell inserts were placed in plate wells containing 600 pl of a 0.02% w/v crystal violet solution for 10 min. The inserts were gently dipped in distilled water to remove excess crystal violet and left to dry overnight. The membrane inserts were imaged using a light microscope at 20X magnification selecting four different randomized fields of view per well. Migratory cells in each field were counted.
C. difficile spore preparation:
[0195] C. difficile R20291 spores were prepared and isolated. Briefly, a single colony grown on BHI+TCA was confluently streaked onto a pre-reduced 70:30 agar plate and incubated at 37°C for 5 days in an anaerobic chamber. Spores were harvested from the agar surface after 5 days by adding 1.5 ml sterile water and scraping the cell/spore material into a 1.5 ml microcentrifuge tube. Cell / spore material was stored at 4°C for 1 week and then separated in a 50% w/v sucrose gradient (centrifuged at 250 ref for 20 min). Finally, the spore pellet was washed 5 times with sterile distilled water and placed in a glass vial at 4°C. Before infection of mice, C. difficile spores were enumerated by plating on TCCFA. A pre-calculated concentration of spore inoculum was heated at 65 °C for 20 min, then allowed to cool for 5 min at room temperature.
C. difficile infection:
[0196] Mice were housed in sterile cages with sterile food. The animals were provided with Cefoperazone sodium salt (Millipore Sigma, St. Louis, MO) in distilled drinking water (0.5 g / 1) for ten days, followed by two days of distilled water (Thermofisher, Waltham, MA). Mice were infected by oral gavage with 1 X 107 heat-treated C. difficile R20291 spores or distilled water to control for the gavage procedure. C. difficile -associated pathology was assessed by monitoring daily weights, and other clinical signs such as lethargy, hunched posture, and diarrhea. Animals were euthanized if the weight loss reached 20.0% or the mice were moribund. Bacteria shed were quantified on day 2 post-gavage by homogenizing fecal pellets with IX PBS, serially diluted, plated on TCCFA and cultured under anaerobic conditions at 37°C. CFUs were counted within 48 hr.
Statistics:
[0197] Data were analyzed using GraphPad Prism (Version 9.1.1, La Jolla, CA). A two tailed T-test or a Mann-Whitney test, and One-way ANOVA with Tukey’s multiple comparison test was used for statistical analysis between two and multiple experimental groups respectively. Where indicated, paired t-tests were used. A Two-way Repeated Measure ANOVA with Tukey’s multiple comparisons test was used to determine statistical significance in weight loss measured at multiple time points.
Ethics:
[0198] All animal procedures were approved by the Institutional Animal Care and Use Committee at the OUHSC (original protocol 20-020- AHI, renewed as 22-082-CHI). This study was performed in accordance with the recommendations of the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health.
RESULTS
TcdB2 delays IgG class switch during primary responses and inhibits IgG recall responses: [0199] B6 mice were treated with TcdB2 before immunization to evaluate its impact on humoral immunity to the C. difficile vaccine antigen B2A (FIG. 1). TcdB2 had no demonstrable effect on B2A-specific IgM titers at 14-, 28-, and 42-days following immunization, but delayed IgGl and IgG2b production (FIG. 2). Primary IgG2c titers were minimal, with the effects of TcdB2 unable to be determined (FIG. 2). The initial treatment with TcdB2 exerted a profound impact on B2A-specific IgG recall responses (FIG. 3). By comparing sera collected on day 67 (pre-boost) and day 81 (post-boost), it was observed that TcdB2 blocked or inhibited booster vaccine-induced increases in B2A-specific IgGl, IgG2b, and IgG2c titers (FIG. 3).
[0200] To determine if enzymatic activity of TcdB2 was required for the impact on primary and recall Ab responses, mice were treated with TcdB2 or an equivalent amount of TcdB2 with a D270N point mutation rendering it glucosyl transferase null (subsequently referred to as D270N) (FIG. 4). It was observed that only bioactive TcdB2 was able to inhibit primary IgG responses (FIG. 4). When comparing sera collected on day 67 (pre-boost) and day 81 (post-boost) from mice treated with either TcdB2 or D270N, then the D270N-treated mice had higher IgGl titers than the TcdB2-treated mice (FIG. 5).
[0201] To measure the impact of TcdB2 on Ab function, sera from vehicle- or TcdB2- treated B2A-immunized mice were tested for their ability to prevent in vitro intoxication of the CHO cell line with TcdB2 (FIG. 6). TcdB2 neutralization in primary bleed sera collected on days 14, 28, and 42 at high concentration (1 :100) reflected the primary B2A-specific IgG titers (FIG. 6). Recall sera collected from TcdB2-treated mice had a significantly decreased ability to neutralize TcdB2 in vitro compared to the immunized control sera. The reduced neutralization was apparent at a lower concentration of sera (1 :2000) (FIG. 6). In contrast, the affinity of sera from control and TcdB2-treated mice was not significantly different (FIG. 37), indicating that the amounts of B2A-specific IgG in the sera accounted for differences in TcdB2 neutralization rather than differences in affinity. Therefore, a single bolus of TcdB2 delayed
IgG class switching and the establishment of TcdB2-neutralizing antibodies, and blocked IgG recall responses.
CD40 activation restores and enhances IgG recall responses in TcdB2-treated mice:
[0202] Given the inhibitory action of TcdB2 on vaccination-induced primary and recall IgG titers, the effects of a higher dose of B2A antigen for immunization and supplemental B cell help were tested. This was achieved by administration of an agonistic anti-CD40 mAb clone FGK4.5. that mimics the effects of CD40L ligation of CD40 in vivo. When using twice the vaccine dose as in the previous experiment, TcdB2 resulted in minimal effects on IgM titers as expected (FIG. 7, upper panel). Primary and recall IgGl titers were not significantly influenced by TcdB2 but CD40 activation resulted in a 31% increase in endpoint titer (FIG. 7, lower panel). In contrast, TcdB2 exerted strong effects on IgG2b reducing recall IgG2b titers by 84% (FIG. 8, upper panel). Administration of the anti-CD40 mAb restored and enhanced recall IgG2b titers such that they were 330% higher than that observed in control mice (FIG. 8, upper panel). A more pronounced effect was observed when measuring IgG2c responses in that TcdB2 completely abrogated the response, which was rescued by CD40 activation (FIG. 8, lower panel). The data were also expressed as fold change between primary and recall titers and each experimental group was compared (FIG. 9). TcdB2 had no significant effect on the magnitude of the IgM or IgGl recall response, likely due to the higher dose of B2A than in FIGS. 1-6. In contrast, TcdB2 inhibited the IgG2b response and eliminated IgG2c recall responses. Activation of CD40, rescued the IgG2b and IgG2c recall titers and enhanced them to levels above that observed in controls.
[0203] At the final time point, bone marrow cells were harvested and analyzed by ELISPOT assay to enumerate memory B cell-derived long-lived plasma cells. The numbers of B2A-specific IgGl- and IgG2b-secreting plasma cells were consistent with the serum titers (FIGS. 10-11, respectively). Total plasma cells (of all specificities) did not differ significantly between experimental groups (FIGS. 38-39). The assay background was zero spots (FIG. 40) and TcdB2 did not alter total numbers of cells recovered from the bone marrow (FIG. 40). These results demonstrate that TcdB2-exposed B cells were intrinsically capable of IgG class switch provided alternative T helper (Th) cell type signals were provided and that TcdB2 had a long-term impact on the establishment of a long-lived plasma cell compartment following immunization.
Non-lethal intoxication of lymphocytes by TcdB2:
[0204] TcdB2 is known to induce apoptosis in host epithelial cells through the glucosylation of small GTPases. We determined if TcdB2 altered the number of recoverable cells from lymphoid organs, indicating possible death and clearance in vivo. B6 mice were treated with vehicle, inactive TcdB2 known as B2A (a TcdB2 mutant unable to bind to the primary host receptor) or active TcdB2 before analysis of lymph nodes and spleens by flow cytometry 48 hr and 14 days later (FIGS. 12, 41-45). There were no TcdB2-induced changes in numbers of B cells or CD4+ T cells in secondary lymphoid organs observed.
[0205] To determine if TcdB2 induced apoptosis or necrosis of B cells and CD4+ T cells directly, splenocytes, isolated from naive B6 mice were cultured in vitro with or without TcdB2 for up to 12 hours (FIGS. 13-14, and 46-48). The cells were then stained with an Annexin V / 7AAD cocktail and analyzed via flow cytometry to identify apoptotic and necrotic cells. TcdB2 did not induce necrosis or apoptosis in B cells or CD4+ T cells above the background level in the cultures. These data demonstrates that TcdB2 did not induce apoptosis of lymphocytes in vitro and suggests a lack of apoptosis in lymphocytes in vivo.
[0206] The lack of necrotic or apoptotic B and T cells observed following TcdB2 treatment suggested the possibility of non-lethal effects following TcdB2 exposure. Since the mechanism of action of TcdB2 requires glucosylation of small GTPases including Rael, the impact on amounts of non-glucosylated Rael were measured using a capillary -based automated western blot (FIGS. 15-16). As shown, (FIG. 15), using a mAb, murine Rael in primary lymphocytes could be specifically detected. Treatment of lymphocytes in vitro resulted in a loss of non-glucosylated Rael, consistent with direct intoxication of lymphocytes by TcdB2 (FIG. 16, left)). A further analysis in which B cells were isolated before in vitro culture treatment revealed loss of non-glycosylated Rael following TcdB2 exposure but not D270N exposure (FIG. 16, right). These results demonstrate that TcdB2 glucosylates Rael in B cells and appears to cause non-lethal intoxication.
TcdB2 blocks immunization-induced germinal center formation:
[0207] As TcdB2 inhibited IgG recall responses but did not induce B or CD4+ T cell death, or render B cells unresponsive to class switch signals, the effects TcdB2 has on immunization-induced changed in lymphoid architecture were determined. B6 mice were treated with TcdB2 or the inactive D270N mutant, then immunized with Alhydrogel-adsorbed B2A. After 21 days, time for sufficient GC formation, the draining lymph nodes, iLNs, were collected and sections mounted on slides for analysis. From H&E-stained sections (FIG. 17),
it was demonstrated that TcdB2 but not D270N resulted in the formation of significantly smaller GC structures (FIG. 18) and significantly fewer of them (FIG. 19), following immunization. To confirm the results, draining iLN sections were stained with fluorochrome- conjugated anti-B220 and anti-Ki67 to examine GC structures (FIG. 20). The relative signal associated with Ki67 staining, demonstrative of rapidly proliferating GC B cells, was measured (FIG. 21), which confirmed the observations from the H&E-stained sections. These data demonstrate that TcdB2 inhibited GC formation, consistent with inhibited Ig class switch and recall responses.
TcdB2 exposure increases CXCR4 gene expression and cell surface expression by lymphocytes:
[0208] To identify a potential mechanism by which TcdB2 inhibits GC formation, DEGs in draining lymph node cells were profiled following TcdB2 or D270N treatment (FIGS. 22, 23A and 23B). RNA was purified from aLNs and iLNs 7 days after treatment with PBS, D270N in PBS, or TcdB2 in PBS. Changes in gene expression were analyzed using Nanostring™ nCounter SPRINT profiler and nSolver software. DEGs were curated and represented as volcano plots (FIG. 22). CXCR4 gene expression was significantly upregulated when comparing TcdB2 to PBS and TcdB2 to D270N, but not when comparing D270N to PBS, showing that TcdB2 must be enzymatically active to alter CXCR4 expression (FIG. 23A). Analysis of CXCR5 and CCR7 expression and their corresponding ligands did not reveal any significant alteration following TcdB2 or D270N treatment (FIG. 23A).
[0209] FIG. 23B shows DEGs following TcdB2 exposure include CXCR4. Relative expression of cxcr4, cxcr5, and ccr7 in isolated B cells as determined by qPCR. Graphs show the increase in expression relative to vehicle-treated control mice and are normalized to gapdh expression using the DDCT method. Graphs show the increase in expression relative to vehicle-treated control mice and are normalized to gapdh expression using the DDCT method. These data demonstrate that enzymatically active TcdB2 selectively increases CXCR4- encoding mRNA expression in B cells. Given our focus on the lack of GCs following TcdB2 treatment, the remaining studies were focused on CXCR4.
[0210] TcdB2 does not significantly alter expression of chemokines other than CXCR4 or their ligands FIGS. 23C-23E depict changes in expression of all chemokine receptors and their corresponding ligands in the Nanostring™ assay. FIG. 23C shows results of an analysis of all chemokine receptor and ligand genes in the gene panel in TcdB2 vs. PBS treatments. No significant changes in expression of the genes were revealed. FIG. 23D shows
results of an analysis of all chemokine receptor and ligand genes in the gene panel in TcdB2 vs. D270N treatments. No significant changes in expression of the genes were revealed. FIG. 23E shows results of an analysis of all chemokine receptor and ligand genes in the gene panel in D270N vs. PBS treatments. No significant changes in expression of the genes were revealed. [0211] To determine if TcdB2 altered CXCR4 cell surface protein expression, mice were treated with TcdB2 or D270N and draining lymph node cells were examined by flow cytometry for B cell and CD4+ T cell expression of CXCR4 and the chemokine receptor CXCR5. It was observed that TcdB2 significantly increased CXCR4 cell surface expression by B cells, whereas the D270N mutant had no effect on expression, consistent with DEG and qPCR data (FIG. 24). The increase in CXCR4 expression by B cells was not observed in CD4+ T cells (FIG. 25). TcdB2 did not significantly affect CXCR5 cell surface expression by B cells or CD4+ T cells (FIGS. 49-51).
TcdB2 leads to increased migration of B cells to the CXCR4 ligand CXCL12:
[0212] To determine if increased CXCR4 expression altered B cell or CD4+ T cell migration, transwell migration assays were performed to measure responsiveness to the CXCR4 ligand CXCL12. B6 mice were treated with vehicle, D270N, or TcdB2 before isolation of total splenocytes, B cells, or CD4+ T cells (FIG. 26). Background levels of migration increased in splenocytes from TcdB2-treated mice, but the effect was not statistically significant (FIG. 27). Cells from vehicle control mice showed a statistically significant response to the CXCL12 ligand, as did cells from D270N- and TcdB2-treated mice (FIG. 27). However, the highest level of migration was observed in splenocytes from TcdB2-treated mice, which was significantly different from all other experimental conditions (FIG. 27).
[0213] Isolated B cells from TcdB2-treated mice showed a prominent and significant increase in migration as compared to all other experimental conditions (FIG. 28). In contrast, CD4+ T cell migration was minimal and was not affected by TcdB2 treatment (FIG. 29A). These results demonstrate that enzymatically active TcdB2 increases CXCR4 expression by B cells but not CD4+ T cells and increases their migration towards the CXCL12 chemoattractant. These results are consistent with the observed inhibited GC formation and IgG recall responses by TcdB2. The ligands for CCR7 (CCL19 and CCL21) and CXCR5 (CXCL13) stimulated a low level of B cell migration. TcdB2 had no effect on those responses, demonstrating selective effects on CXCR4-mediated migration (FIG. 29B). In vitro treatment of B cells with TcdB2 also revealed a glucosyltransferase-dependent and direct effect on CXCR4-mediated migration (FIG. 29C). These results demonstrate that enzymatically active TcdB2 increases CXCR4
expression by B cells but not CD4+ T cells and increases their migration toward the CXCL12 chemoattractant. These results are consistent with inhibited GC formation and IgG recall responses by TcdB2.
CXCR4 expression and related migration increased in lymphocytes from draining lymph nodes following CDI:
[0214] To determine if effects on CXCR4 expression and subsequent cell migration by TcdB2 is recapitulated in a CDT model, B6 mice were given TcdB2-secreting C. difficile R20291 spores via oral gavage after antibiotic-induced dysbiosis (FIG. 30). Successful infection was confirmed by measuring weight loss (FIG. 31), C. difficile CFUs in fecal samples (FIG. 32, left), and measurement of cecum and colon length (FIG. 32, right). After 2 days, mice were euthanized and spleen and aLN, iLN, and mLN lymph nodes collected for CXCR4 cell surface expression and lymphocyte migration analyses (FIGS. 33-36).
[0215] To determine if CDI altered CXCR4 cell surface protein expression, lymphocytes from draining mLNs, regional lymphoid organs (iLNs and Spleen), and distal lymph nodes (aLNs), were examined by flow cytometry for B cell CXCR4 and CXCR5 expression (FIGS. 33-35, and FIGS. 50-51). Lymphocytes from proximal (FIG. 34, left) and regional (FIG. 34, right and FIG. 35, left) lymphoid organs had an increase in CXCR4 cell surface expression from the CDI group compared to the uninfected control group while lymphocytes from distal lymphoid organs (FIG. 35, right) did not have a change in CXCR4 expression. CXCR5 expression was not altered in CDI versus the control group (FIGS. 50-51).
[0216] A CXCR4-dependent cell migration assay was performed (FIG. 36) using lymphocytes from the draining mLNs. Cells from mice infected with C. difficile showed significantly higher migration towards CXCL12 than those mLN cells from the uninfected control mice (FIG. 36). The effect was dependent on addition of CXCL12 to the cell culture media. These data demonstrate that the upregulation of CXCR4 on B cells and its effect on migration was recapitulated in a CDI mouse model. This implicates C. difficile -secreted TcdB2 in humoral immune suppression during CDI.
CXCR4 antagonists reduce weight loss and circumvent the increase in cellular migration towards CXCL12 in CDI:
[0217] A C. difficile mouse model was used to test the ability of AMD3100 to suppress the increased cellular migration towards CXCL12 post intoxication with TcdB2. C57BL/6 (B6) mice were treated with TcdB2 alone or with AMD3100 (at standard dose or low dose).
After 24 hours, mice treated with AMD3100 received a second dose. Inguinal and axillary lymph nodes (FIG. 52) and spleens (FIG. 53) were collected from the mice 48 hours post intoxication and cell migration towards CXCL12 was measured via transwell migration assay. To identify the effects AMD3100 has during a CDI, B6 mice were infected with TcdB2- secreting C. difficile R20291 spores via oral gavage after antibiotic-induced dysbiosis. A portion of mice were treated with AMD3100 (low dose) at hour 0, 24, and 48 post-gavage. Mouse weight was measured daily (FIGS. 54-55). Mice were euthanized if weight loss reached 20% (FIG. 56).
[0218] Data in FIGS. 54-56 indicate that inhibition of CXCR4 activity with AMD3100 reduces weight loss and circumvents the increase in cellular migration towards CXCL12 induced by TcdB2, and reduces morbidity in initial CDI. These results demonstrate that anti- CXCR4 treatments (i.e., including but not limited to the CXCR4 antagonists described elsewhere herein) can be used to block TcdB2-induced effects on GC formation, leading to proper B memory formation against C. difficile leading to reduced C. difficile reinfections.
DISCUSSION
[0219] The present work demonstrates that the secreted toxin TcdB2, the main driver of disease pathology in CDI, exerts a profound and deleterious effect on the humoral immune response. TcdB2 is shown herein to delay IgG class switch, block GC formation, and curtail IgG recall responses.
[0220] Serum TcdB2-neutralizing IgG titers remain the best-known correlate of protection against recurrent CDI but prior to the present work it has remained unclear why infection fails to stimulate an immune response that adequately prevents CDI recurrence. Several factors likely contribute to recurrence, including continued antibiotic therapy- maintained dysbiosis, continued C. difficile exposure in the environment, and possible germination of spores that are resident in host epithelial cells. However, the present work indicates that insufficiently protective B cell memory following infection likely contributes to the overall risk of recurrence.
[0221] We observed that TcdB2 administration prior to a standard immunization protocol
(an Alum adjuvant-adsorbed protein antigen), resulted in delayed IgG class switch and severely abrogated IgG recall responses following administration of a booster vaccine. As a result, IgGl titers were lower in TcdB2-treated mice, and this was reflected in the reduced ability of that IgG to neutralize TcdB2 in vitro. However, average affinity of the IgG, measured indirectly by ELISA, suggested no significant difference in affinity maturation of the IgG molecules that
were successfully produced. Although our results showed that TcdB2 blocked GC formation, there is precedent for extra-GC affinity maturation. Our data indicate that B cells under conditions of TcdB2 intoxication are still intrinsically able to undergo GC -independent affinity maturation.
[0222] The inhibition of IgG class switch and recall responses by TcdB2 was consistent with disrupted GC reactions. The observation that immunization-induced GC formation was blocked by TcdB2 but not the D270N mutant was particularly striking. The effects of TcdB2 were apparent both in GC numbers and in their size, such that fewer, smaller GCs were observed. Given that GCs have inherent complexity, such as dark and light zones, the latter being the location of affinity maturation, it is reasonable to suggest that TcdB2 could differentially impact light and dark zone formation. Since CXCR4 expression indicates B and T cells that are not resident in GC light zones, we believe that TcdB2 subverts B and CD4+ T cells interactions in the light zone of GCs, resulting in blockade of light zone formation within GCs.
[0223] A targeted transcriptomic approach identified the chemokine receptor CXCR4 as being upregulated in draining lymph node cells following TcdB2 administration. CXCR4 protein expression was confirmed by flow cytometry to increase following TcdB2 treatment, while CXCR5 expression did not change. The conformation of the 7 transmembrane receptor CXCR4 and thus its ligand-binding and signaling properties are regulated by Rael. By facilitating GTP binding activity, Rael is involved in several important processes, including cellular migration. Rael is responsible for regulating conformational changes in CXCR4 that alter receptor activation and migration towards the chemoattractant CXCL12. Considering TcdB2 targets Rael, these data indicate a link between TcdB2 and CXCR4 activation on B cells. TcdB2 may enhance CXCR4 activation and/or induce an upregulation in CXCR4 gene expression, which may also contribute to increased chemotaxis. The present results firmly implicate altered CXCR4 B cell migration as a mechanism underpinning the TcdB 2- subverted humoral immune response.
[0224] TcdB2 suppresses host humoral immune response, particularly the inhibition of antibody recall response through the stunting of GC formation. As demonstrated by results shown above, this blockage in GC formation is in part due to TcdB2 upregulating the chemokine receptor CXCR4. Therefore, inhibiting CXCR4 can provide a treatment for decreasing the toxic effects of TcdB2 and minimizing the potential for CDI reinfection. In a non- limiting example, AMD3100 (Plerixafor) is an FDA approved drug which functions as an CXCR4 antagonist causing inhibition of binding with its chemokine CXCL12. Therefore,
using AMD3100, or other CXCR4 antagonists, such as but not limited to those listed elsewhere herein, will reduce CDI recurrence after initial infection and will reduce the toxic effect during C. difficile infection.
[0225] A C. difficile mouse model was used to test the ability of AMD3100 to suppress the increased cellular migration towards CXCL12 post intoxication with TcdB2. C57BL/6 (B6) mice were treated with TcdB2 alone or with AMD3100 (at standard dose or low dose). After 24 hours, mice treated with AMD3100 received a second dose. Inguinal and axillary lymph nodes and spleens were collected from the mice 48 hours post intoxication and cell migration towards CXCL12 was measured. To identify the effects AMD3100 has during a CDI, B6 mice were infected with TcdB2-secreting C. difficile R20291 spores via oral gavage after antibiotic-induced dysbiosis. A portion of mice were treated with AMD3100 (low dose) at hour 0, 24, and 48 post-gavage. Mouse weight was measured daily.
[0226] Results showed that inhibition of CXCR4 activity with AMD3100 reduces weight loss and circumvents the increase in cellular migration towards CXCL12 induced by TcdB2, and reduces morbidity in initial CDI. These results demonstrate that anti-CXCR4 treatments (i.e., including but not limited to the CXCR4 antagonists described elsewhere herein) can be used to block TcdB2-induced effects on GC formation, leading to proper B memory formation against C. difficile leading to reduced C. difficile reinfections.
[0227] As noted previously, CXCR4 antagonists which may be used in the anti-CDI treatments of the present disclosure include, but are not limited to, Plerixafor (AMD3100), Mavorixafor (AMD070), AMD1170, AMD3465, Balixafortide, BPRCX714, BPRCX807, CS VI 8742, CTCE-9908, CXCR4 Antagonist III, FC 122, FC131, HF51116, HZ515H7, ITlt, LFC131, LY2510924, LY2624587, MiRNA-146, MiRNA-193-5p, Motixafortide (BKT-140, 4F-benzoyl-TN 14003, BL-8040), MSX-122, Naringin, PF-06747143, Saikosaponin A, TN14003, Ulocuplumab (MDX-1338, BMS-936564), WZ811, X4-136, and 3OD8. This list of CXCR4 antagonists is not intended to be exclusive of CXCR4 antagonists which may be in use in current clinical trials or which may be developed in the future.
[0228] In another application, the CXCR4 antagonists can be administered in a combination therapy with an agonistic anti-CD40 mAb to provide an immune system boost against the C. difficile. Results that indicate that the CD40 pathway is intact in toxin-exposed B cells and is responsive to CD40 ligation. Examples of agonistic anti-CD40 mAbs which may be used include but are not limited to Selicrelumab (CP-870893, R07009789), Dacetuzmumab (SGN-40), ChiLob 7/4, 2141-V11, APX005M (sotigalimab), JNJ-64457107 (ADC-1013), ABBV-428, CDX-1140H, and SEA-CD40. This list of agonistic anti-CD40 mAbs which may
be used in a combination treatment with a CXCR4 antagonist is not intended to be exclusive of agonistic anti-CD40 mAbs which may be in use in current clinical trials or which may be developed in the future.
[0229] In another application, an anti-C. difficile antibiotic may be co-administered to the subject with the CXCR4 antagonist. Examples of antibiotics that may be used to treat a CDI include, but are not limited to, vancomycin, metronidazole, fidaxomicin, surotomycin, and CB- 183315. This list of antibiotics which may be used in a combination treatment with a CXCR4 antagonist is not intended to be exclusive of anti-CDI antibiotics which may be in use in current clinical trials or which may be developed in the future.
[0230] In certain embodiments, the subject may be treated by co-administering a CXCR4 antagonist and an inhibitor of TcdB, including TcdB2. For example, the inhibitor of TcdB toxin may be an anti-TcdB monoclonal antibody. The monoclonal antibody may be at least one of Bezlotoxumab, CANmAbB4, CANmAbBl, CDB1, ABA, Al 31, E74F, and PA41. This list of inhibitors of TcdB which may be used in a combination treatment with a CXCR4 antagonist is not intended to be exclusive of inhibitors of TcdB which may be in use in current clinical trials or which may be developed in the future.
[0231] In summary, in at least certain embodiments, the present disclosure is directed to a method of treating a C. difficile infection (CDI) in a subject in need of such treatment comprising administering to the subject a CXCR4 antagonist, which may be selected from the group which includes, but is not limited to, Plerixafor, Mavorixafor, AMD1170, AMD3465, Balixafortide, BPRCX714, BPRCX807, CSV18742, CTCE-9908, CXCR4 Antagonist III, FC122, FC131, HF51116, HZ515H7, ITlt, LFC131, LY2510924, LY2624587, MiRNA-146, MiRNA-193-5p, Motixafortide, MSX-122, Naringin, PF-06747143, Saikosaponin A, TN14003, Ulocuplumab, WZ811, X4-136, and 30D8. The CDI may becaused by a C. difficile strain that secretes a C. difficile Toxin B (TcdB). The TcdB may be selected from the group TcdB2, TcdBl, TcdB3, TcdB4, TcdB5, TcdB6, TcdB7, TcdB8, TcdB9, TcdB 10, TcdBl l, and TcdB12. The subject’s B cell memory (Bmem) against a TcdB may be enhanced by the administration of the CXCR4 antagonist. The CDI may be a recurrent CDI. In the method, an anti-C. difficile antibiotic may be co-administered to the subject with the CXCR4 antagonist. The anti-C. difficile antibiotic may be selected from vancomycin, metronidazole, fidaxomicin, surotomycin, and CB-183315. In the method, an agonistic anti-CD40 monoclonal antibody may be co-administered to the subject with the CXCR4 antagonist. The agonistic anti-CD40 monoclonal antibody may be selected from the group which includes, but is not limited to, Selicrelumab, Dacetuzmumab, Sotigalimab, JNJ-64457107, ABBV-428, CDX-1140H, SEA-
CD40, ChiLob 7/4, and 2141-Vl l. In the method, an inhibitor of TcdB may be co-administered to the subject with the CXCR4 antagonist. The inhibitor of TcdB toxin may be an anti-TcdB mAb. The anti-TcdB mAb may be at least one of bezlotoxumab, CANmAbB4, CANmAbBl, CDB 1 , ABA, Al 31, E74F, and PA41. In certain embodiments, the present disclosure is directed to a substance for use in treating a CDI in a subject, wherein the substance is a CXCR4 antagonist. As noted, the CXCR4 antagonist may be one or more of Plerixafor, Mavorixafor, AMDI 170, AMD3465, Balixafortide, BPRCX714, BPRCX807, CSV18742, CTCE-9908, CXCR4 Antagonist III, FC 122, FC131 , HF51 116, HZ515H7, ITlt, LFC131 , LY2510924, LY2624587, MiRNA-146, MiRNA-193-5p, Motixafortide, MSX-122, Naringin, PF- 06747143, Saikosaponin A, TN14003, Ulocuplumab, WZ811, X4-136, and 3OD8.
[0232] While the present disclosure has been described in connection with certain embodiments so that aspects thereof may be more fully understood and appreciated, it is not intended that the present disclosure be limited to these particular embodiments. On the contrary, it is intended that all alternatives, modifications and equivalents are included within the scope of the present disclosure. Thus the examples described above, which include particular embodiments, will serve to illustrate the practice of the present disclosure, it being understood that the particulars shown are by way of example and for purposes of illustrative discussion of particular embodiments only and are presented in the cause of providing what is believed to be the most useful and readily understood description of procedures as well as of the principles and conceptual aspects of the presently disclosed methods and compositions. Changes may be made in the formulation of the various compositions described herein, the methods described herein or in the steps or the sequence of steps of the methods described herein without departing from the spirit and scope of the present disclosure.
Claims
1. A method of treating a Clostridioides difficile infection (CDI) in a subject in need of such treatment comprising: administering to the subject a C-X-C chemokine receptor type 4 (CXCR4) antagonist.
2. The method of claim 1, wherein the CDI is caused by a C. difficile strain that secretes a C. difficile Toxin B (TcdB).
3. The method of claim 2, wherein the TcdB is selected from the group TcdB2, TcdBl, TcdB3, TcdB4, TcdB5, TcdB6, TcdB7, TcdB8, TcdB9, TcdBlO, TcdBl 1, and TcdB12.
4. The method of claim 1, wherein the CXCR4 antagonist is selected from the group consisting of Plerixafor, Mavorixafor, AMD1170, AMD3465, Balixafortide, BPRCX714, BPRCX807, CSV18742, CTCE-9908, CXCR4 Antagonist III, FC122, FC131, HF51116, HZ515H7, ITlt, LFC131, LY2510924, LY2624587, MiRNA-146, MiRNA-193-5p, Motixafortide, MSX-122, Naringin, PF-06747143, Saikosaponin A, TN14003, Ulocuplumab, WZ811, X4-136, and 3OD8.
5. The method of claim 1, wherein the subject’s B cell memory against a C. difficile Toxin B (TcdB) is enhanced by the administration of the CXCR4 antagonist.
6. The method of claim 1, wherein the C. difficile is a hypervirulent strain.
7. The method of claim 1, wherein the CDI is a recurrent CDI.
8. The method of claim 1, wherein the CXCR4 antagonist is co-administered to the subject with at least one of an anti-C. difficile antibiotic, an agonistic anti-CD40 monoclonal antibody (mAb), and an inhibitor of a TcdB.
9. The method of claim 8, wherein the anti-C. difficile antibiotic is selected from the group consisting of vancomycin, metronidazole, fidaxomicin, surotomycin, and CB-183315.
10. The method of claim 8, wherein the agonistic anti-CD40 mAh is selected from the group consisting of Selicrelumab, Dacetuzmumab, Sotigalimab, JNJ-64457107, ABBV-428, CDX- 1 I40H, SEA-CD40, ChiLob 7/4, and 2141-Vl l.
11. The method of claim 8, wherein the inhibitor of the TcdB is an anti-TcdB mAh.
12. The method of claim 11, wherein the anti-TcdB mAh is selected from Bezlotoxumab, CANmAbB4, CANmAbB l , CDB1 , ABA, Al 31, E74F, and PA41.
13. A substance for use in treating a Clostridioides difficile infection (CDI) in a subject, wherein the substance is a C-X-C chemokine receptor type 4 (CXCR4) antagonist.
14. The substance of claim 13, wherein the CDI is caused by a C. difficile strain that secretes a C. difficile Toxin B (TcdB).
15. The substance of claim 14, wherein the TcdB is selected from the group TcdB2, TcdBl, TcdB3, TcdB4, TcdB5, TcdB6, TcdB7, TcdB8, TcdB9, TcdBlO, TcdBl 1, and TcdB12.
16. The substance of any one of claims 13-15, wherein the CXCR4 antagonist is selected from the group consisting of Plerixafor, Mavorixafor, AMD1170, AMD3465, Balixafortide, BPRCX714, BPRCX807, CSV18742, CTCE-9908, CXCR4 Antagonist III, FC122, FC131, HF51116, HZ515H7, ITlt, LFC131, LY2510924, LY2624587, MiRNA-146, MiRNA-193-5p, Motixafortide, MSX-122, Naringin, PF-06747143, Saikosaponin A, TN14003, Ulocuplumab, WZ811, X4-136, and 3OD8.
17. The substance of any one of claims 13-16, wherein the CDI is a recurrent CDI.
18. The substance of any one of claims 13-17, wherein the subject’s B cell memory (Bmem) against a TcdB is enhanced by the administration of the CXCR4 antagonist.
19. A substance for use in combination with a C-X-C chemokine receptor type 4 (CXCR4) antagonist for treating a Clostridioides difficile infection (CDI) in a subject, wherein the substance is selected from the group consisting of an anti-C. difficile antibiotic, an agonistic anti-CD40 monoclonal antibody (mAb), and an inhibitor of a C. difficile Toxin B (TcdB).
20. The substance of claim 19, wherein the CDI is caused by a C. difficile strain that secretes a TcdB.
21. The substance of claim 19 or 20, wherein the TcdB is selected from the group TcdB2, TcdBl, TcdB3, TcdB4, TcdB5, TcdB6, TcdB7, TcdB8, TcdB9, TcdBlO, TcdBl l, and TcdB 12.
22. The substance of any one of claims 19-21 , wherein the CXCR4 antagonist is selected from the group consisting of Plerixafor, Mavorixafor, AMD1170, AMD3465, Balixafortide, BPRCX714, BPRCX807, CSV18742, CTCE-9908, CXCR4 Antagonist III, FC122, FC131, HF51116, HZ515H7, ITlt, LFC131, LY2510924, LY2624587, MiRNA-146, MiRNA-193-5p, Motixafortide, MSX-122, Naringin, PF-06747143, Saikosaponin A, TN14003, Ulocuplumab, WZ811, X4-136, and 3OD8.
23. The substance of any one of claims 19-22, wherein the anti-C. difficile antibiotic is selected from the group consisting of vancomycin, metronidazole, fidaxomicin, surotomycin, and CB- 183315, the agonistic anti-CD40 mAh is selected from the group consisting of Selicrelumab, Dacetuzmumab, ChiLob 7/4, 2141-V11, Sotigalimab, JNJ-64457107, ABBV-428, CDX- 1140H, and SEA-CD40, and the inhibitor of the TcdB is an anti-TcdB mAh.
24. The substance of any one of claims 19-23, wherein the anti-TcdB mAb is selected from bezlotoxumab, CANmAbB4, CANmAbBl, CDB1, ABA, A13I, E74F, and PA41.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US202363582138P | 2023-09-12 | 2023-09-12 | |
| US63/582,138 | 2023-09-12 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2025059113A1 true WO2025059113A1 (en) | 2025-03-20 |
Family
ID=92899807
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2024/046099 Pending WO2025059113A1 (en) | 2023-09-12 | 2024-09-11 | Treatments for enhancing immune response to clostridioides difficile infections |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2025059113A1 (en) |
Citations (81)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3687808A (en) | 1969-08-14 | 1972-08-29 | Univ Leland Stanford Junior | Synthetic polynucleotides |
| US4235871A (en) | 1978-02-24 | 1980-11-25 | Papahadjopoulos Demetrios P | Method of encapsulating biologically active materials in lipid vesicles |
| US4394448A (en) | 1978-02-24 | 1983-07-19 | Szoka Jr Francis C | Method of inserting DNA into living cells |
| US4469863A (en) | 1980-11-12 | 1984-09-04 | Ts O Paul O P | Nonionic nucleic acid alkyl and aryl phosphonates and processes for manufacture and use thereof |
| US4476301A (en) | 1982-04-29 | 1984-10-09 | Centre National De La Recherche Scientifique | Oligonucleotides, a process for preparing the same and their application as mediators of the action of interferon |
| US4501728A (en) | 1983-01-06 | 1985-02-26 | Technology Unlimited, Inc. | Masking of liposomes from RES recognition |
| US4737323A (en) | 1986-02-13 | 1988-04-12 | Liposome Technology, Inc. | Liposome extrusion method |
| US4837028A (en) | 1986-12-24 | 1989-06-06 | Liposome Technology, Inc. | Liposomes with enhanced circulation time |
| US5023243A (en) | 1981-10-23 | 1991-06-11 | Molecular Biosystems, Inc. | Oligonucleotide therapeutic agent and method of making same |
| US5034506A (en) | 1985-03-15 | 1991-07-23 | Anti-Gene Development Group | Uncharged morpholino-based polymers having achiral intersubunit linkages |
| US5139941A (en) | 1985-10-31 | 1992-08-18 | University Of Florida Research Foundation, Inc. | AAV transduction vectors |
| US5166315A (en) | 1989-12-20 | 1992-11-24 | Anti-Gene Development Group | Sequence-specific binding polymers for duplex nucleic acids |
| US5177196A (en) | 1990-08-16 | 1993-01-05 | Microprobe Corporation | Oligo (α-arabinofuranosyl nucleotides) and α-arabinofuranosyl precursors thereof |
| US5185444A (en) | 1985-03-15 | 1993-02-09 | Anti-Gene Deveopment Group | Uncharged morpolino-based polymers having phosphorous containing chiral intersubunit linkages |
| US5188897A (en) | 1987-10-22 | 1993-02-23 | Temple University Of The Commonwealth System Of Higher Education | Encapsulated 2',5'-phosphorothioate oligoadenylates |
| US5194599A (en) | 1988-09-23 | 1993-03-16 | Gilead Sciences, Inc. | Hydrogen phosphonodithioate compositions |
| US5214134A (en) | 1990-09-12 | 1993-05-25 | Sterling Winthrop Inc. | Process of linking nucleosides with a siloxane bridge |
| US5216141A (en) | 1988-06-06 | 1993-06-01 | Benner Steven A | Oligonucleotide analogs containing sulfur linkages |
| US5235033A (en) | 1985-03-15 | 1993-08-10 | Anti-Gene Development Group | Alpha-morpholino ribonucleoside derivatives and polymers thereof |
| US5264423A (en) | 1987-03-25 | 1993-11-23 | The United States Of America As Represented By The Department Of Health And Human Services | Inhibitors for replication of retroviruses and for the expression of oncogene products |
| US5264562A (en) | 1989-10-24 | 1993-11-23 | Gilead Sciences, Inc. | Oligonucleotide analogs with novel linkages |
| US5264564A (en) | 1989-10-24 | 1993-11-23 | Gilead Sciences | Oligonucleotide analogs with novel linkages |
| US5276019A (en) | 1987-03-25 | 1994-01-04 | The United States Of America As Represented By The Department Of Health And Human Services | Inhibitors for replication of retroviruses and for the expression of oncogene products |
| US5278302A (en) | 1988-05-26 | 1994-01-11 | University Patents, Inc. | Polynucleotide phosphorodithioates |
| US5279833A (en) | 1990-04-04 | 1994-01-18 | Yale University | Liposomal transfection of nucleic acids into animal cells |
| US5283185A (en) | 1991-08-28 | 1994-02-01 | University Of Tennessee Research Corporation | Method for delivering nucleic acids into cells |
| US5288641A (en) | 1984-06-04 | 1994-02-22 | Arch Development Corporation | Herpes Simplex virus as a vector |
| US5321131A (en) | 1990-03-08 | 1994-06-14 | Hybridon, Inc. | Site-specific functionalization of oligodeoxynucleotides for non-radioactive labelling |
| US5391377A (en) | 1990-10-19 | 1995-02-21 | Cortecs Limited | Biphasic release formations for lipophilic acids |
| US5399676A (en) | 1989-10-23 | 1995-03-21 | Gilead Sciences | Oligonucleotides with inverted polarity |
| US5399346A (en) | 1989-06-14 | 1995-03-21 | The United States Of America As Represented By The Department Of Health And Human Services | Gene therapy |
| US5405938A (en) | 1989-12-20 | 1995-04-11 | Anti-Gene Development Group | Sequence-specific binding polymers for duplex nucleic acids |
| US5405939A (en) | 1987-10-22 | 1995-04-11 | Temple University Of The Commonwealth System Of Higher Education | 2',5'-phosphorothioate oligoadenylates and their covalent conjugates with polylysine |
| US5434257A (en) | 1992-06-01 | 1995-07-18 | Gilead Sciences, Inc. | Binding compentent oligomers containing unsaturated 3',5' and 2',5' linkages |
| US5436146A (en) | 1989-09-07 | 1995-07-25 | The Trustees Of Princeton University | Helper-free stocks of recombinant adeno-associated virus vectors |
| US5455233A (en) | 1989-11-30 | 1995-10-03 | University Of North Carolina | Oligoribonucleoside and oligodeoxyribonucleoside boranophosphates |
| US5466677A (en) | 1993-03-06 | 1995-11-14 | Ciba-Geigy Corporation | Dinucleoside phosphinates and their pharmaceutical compositions |
| US5470967A (en) | 1990-04-10 | 1995-11-28 | The Dupont Merck Pharmaceutical Company | Oligonucleotide analogs with sulfamate linkages |
| US5476925A (en) | 1993-02-01 | 1995-12-19 | Northwestern University | Oligodeoxyribonucleotides including 3'-aminonucleoside-phosphoramidate linkages and terminal 3'-amino groups |
| US5489677A (en) | 1990-07-27 | 1996-02-06 | Isis Pharmaceuticals, Inc. | Oligonucleoside linkages containing adjacent oxygen and nitrogen atoms |
| US5519126A (en) | 1988-03-25 | 1996-05-21 | University Of Virginia Alumni Patents Foundation | Oligonucleotide N-alkylphosphoramidates |
| US5539082A (en) | 1993-04-26 | 1996-07-23 | Nielsen; Peter E. | Peptide nucleic acids |
| US5541307A (en) | 1990-07-27 | 1996-07-30 | Isis Pharmaceuticals, Inc. | Backbone modified oligonucleotide analogs and solid phase synthesis thereof |
| US5550111A (en) | 1984-07-11 | 1996-08-27 | Temple University-Of The Commonwealth System Of Higher Education | Dual action 2',5'-oligoadenylate antiviral derivatives and uses thereof |
| US5561225A (en) | 1990-09-19 | 1996-10-01 | Southern Research Institute | Polynucleotide analogs containing sulfonate and sulfonamide internucleoside linkages |
| US5571799A (en) | 1991-08-12 | 1996-11-05 | Basco, Ltd. | (2'-5') oligoadenylate analogues useful as inhibitors of host-v5.-graft response |
| US5587361A (en) | 1991-10-15 | 1996-12-24 | Isis Pharmaceuticals, Inc. | Oligonucleotides having phosphorothioate linkages of high chiral purity |
| US5596086A (en) | 1990-09-20 | 1997-01-21 | Gilead Sciences, Inc. | Modified internucleoside linkages having one nitrogen and two carbon atoms |
| US5602240A (en) | 1990-07-27 | 1997-02-11 | Ciba Geigy Ag. | Backbone modified oligonucleotide analogs |
| US5608046A (en) | 1990-07-27 | 1997-03-04 | Isis Pharmaceuticals, Inc. | Conjugated 4'-desmethyl nucleoside analog compounds |
| US5610289A (en) | 1990-07-27 | 1997-03-11 | Isis Pharmaceuticals, Inc. | Backbone modified oligonucleotide analogues |
| US5618704A (en) | 1990-07-27 | 1997-04-08 | Isis Pharmacueticals, Inc. | Backbone-modified oligonucleotide analogs and preparation thereof through radical coupling |
| US5622856A (en) | 1995-08-03 | 1997-04-22 | Avigen | High efficiency helper system for AAV vector production |
| US5623070A (en) | 1990-07-27 | 1997-04-22 | Isis Pharmaceuticals, Inc. | Heteroatomic oligonucleoside linkages |
| US5625050A (en) | 1994-03-31 | 1997-04-29 | Amgen Inc. | Modified oligonucleotides and intermediates useful in nucleic acid therapeutics |
| US5633360A (en) | 1992-04-14 | 1997-05-27 | Gilead Sciences, Inc. | Oligonucleotide analogs capable of passive cell membrane permeation |
| US5646269A (en) | 1994-04-28 | 1997-07-08 | Gilead Sciences, Inc. | Method for oligonucleotide analog synthesis |
| US5663312A (en) | 1993-03-31 | 1997-09-02 | Sanofi | Oligonucleotide dimers with amide linkages replacing phosphodiester linkages |
| US5665577A (en) | 1989-02-06 | 1997-09-09 | Dana-Farber Cancer Institute | Vectors containing HIV packaging sequences, packaging defective HIV vectors, and uses thereof |
| US5672697A (en) | 1991-02-08 | 1997-09-30 | Gilead Sciences, Inc. | Nucleoside 5'-methylene phosphonates |
| US5672510A (en) | 1990-01-19 | 1997-09-30 | Genetic Therapy, Inc. | Retroviral vectors |
| US5676954A (en) | 1989-11-03 | 1997-10-14 | Vanderbilt University | Method of in vivo delivery of functioning foreign genes |
| US5677439A (en) | 1990-08-03 | 1997-10-14 | Sanofi | Oligonucleotide analogues containing phosphate diester linkage substitutes, compositions thereof, and precursor dinucleotide analogues |
| US5677437A (en) | 1990-07-27 | 1997-10-14 | Isis Pharmaceuticals, Inc. | Heteroatomic oligonucleoside linkages |
| US5707865A (en) | 1994-12-21 | 1998-01-13 | Kohn; Donald B. | Retroviral vectors for expression in embryonic cells |
| EP0820773A1 (en) | 1995-11-16 | 1998-01-28 | Universidad Autonoma Barcelona | Treatment of diabetes with a glucokinase gene |
| US5714331A (en) | 1991-05-24 | 1998-02-03 | Buchardt, Deceased; Ole | Peptide nucleic acids having enhanced binding affinity, sequence specificity and solubility |
| US5719262A (en) | 1993-11-22 | 1998-02-17 | Buchardt, Deceased; Ole | Peptide nucleic acids having amino acid side chains |
| US5721218A (en) | 1989-10-23 | 1998-02-24 | Gilead Sciences, Inc. | Oligonucleotides with inverted polarity |
| US5792608A (en) | 1991-12-12 | 1998-08-11 | Gilead Sciences, Inc. | Nuclease stable and binding competent oligomers and methods for their use |
| US5817491A (en) | 1990-09-21 | 1998-10-06 | The Regents Of The University Of California | VSV G pseusdotyped retroviral vectors |
| US5935935A (en) | 1993-06-10 | 1999-08-10 | Genetic Therapy, Inc. | Adenoviral vectors for treatment of hemophilia |
| US5994136A (en) | 1997-12-12 | 1999-11-30 | Cell Genesys, Inc. | Method and means for producing high titer, safe, recombinant lentivirus vectors |
| US6013516A (en) | 1995-10-06 | 2000-01-11 | The Salk Institute For Biological Studies | Vector and method of use for nucleic acid delivery to non-dividing cells |
| US6096716A (en) | 1994-12-12 | 2000-08-01 | The Board Of Regents, The University Of Texas System | Liposome-mediated transfection of central nervous system cells |
| US6110490A (en) | 1994-08-05 | 2000-08-29 | The United States Of America As Represented By The Department Of Health And Human Services | Liposomal delivery system for biologically active agents |
| US6217912B1 (en) | 1998-07-13 | 2001-04-17 | Expression Genetics, Inc. | Polyester analogue of poly-L-lysine as a soluble, biodegradable gene delivery carrier |
| US7514098B2 (en) | 2001-05-30 | 2009-04-07 | The Board Of Trustees Of The Leland Stanford Junior University | Use of targeted cross-linked nanoparticles for in vivo gene delivery |
| US8323618B2 (en) | 2007-11-07 | 2012-12-04 | University Of Houston System | Ultrasmall superparamagnetic iron oxide nanoparticles and uses thereof |
| US20190255088A1 (en) | 2018-02-17 | 2019-08-22 | The Board Of Regents Of The University Of Oklahoma | Biocompatible organo-inorganic nanocomposites |
| WO2020079449A1 (en) * | 2018-10-18 | 2020-04-23 | Citrox Biosciences Limited | Bioflavonoid compositions and their use for water purification and food preservation |
-
2024
- 2024-09-11 WO PCT/US2024/046099 patent/WO2025059113A1/en active Pending
Patent Citations (88)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3687808A (en) | 1969-08-14 | 1972-08-29 | Univ Leland Stanford Junior | Synthetic polynucleotides |
| US4235871A (en) | 1978-02-24 | 1980-11-25 | Papahadjopoulos Demetrios P | Method of encapsulating biologically active materials in lipid vesicles |
| US4394448A (en) | 1978-02-24 | 1983-07-19 | Szoka Jr Francis C | Method of inserting DNA into living cells |
| US4469863A (en) | 1980-11-12 | 1984-09-04 | Ts O Paul O P | Nonionic nucleic acid alkyl and aryl phosphonates and processes for manufacture and use thereof |
| US5023243A (en) | 1981-10-23 | 1991-06-11 | Molecular Biosystems, Inc. | Oligonucleotide therapeutic agent and method of making same |
| US4476301A (en) | 1982-04-29 | 1984-10-09 | Centre National De La Recherche Scientifique | Oligonucleotides, a process for preparing the same and their application as mediators of the action of interferon |
| US4501728A (en) | 1983-01-06 | 1985-02-26 | Technology Unlimited, Inc. | Masking of liposomes from RES recognition |
| US5288641A (en) | 1984-06-04 | 1994-02-22 | Arch Development Corporation | Herpes Simplex virus as a vector |
| US5550111A (en) | 1984-07-11 | 1996-08-27 | Temple University-Of The Commonwealth System Of Higher Education | Dual action 2',5'-oligoadenylate antiviral derivatives and uses thereof |
| US5034506A (en) | 1985-03-15 | 1991-07-23 | Anti-Gene Development Group | Uncharged morpholino-based polymers having achiral intersubunit linkages |
| US5235033A (en) | 1985-03-15 | 1993-08-10 | Anti-Gene Development Group | Alpha-morpholino ribonucleoside derivatives and polymers thereof |
| US5185444A (en) | 1985-03-15 | 1993-02-09 | Anti-Gene Deveopment Group | Uncharged morpolino-based polymers having phosphorous containing chiral intersubunit linkages |
| US5139941A (en) | 1985-10-31 | 1992-08-18 | University Of Florida Research Foundation, Inc. | AAV transduction vectors |
| US4737323A (en) | 1986-02-13 | 1988-04-12 | Liposome Technology, Inc. | Liposome extrusion method |
| US4837028A (en) | 1986-12-24 | 1989-06-06 | Liposome Technology, Inc. | Liposomes with enhanced circulation time |
| US5264423A (en) | 1987-03-25 | 1993-11-23 | The United States Of America As Represented By The Department Of Health And Human Services | Inhibitors for replication of retroviruses and for the expression of oncogene products |
| US5286717A (en) | 1987-03-25 | 1994-02-15 | The United States Of America As Represented By The Department Of Health And Human Services | Inhibitors for replication of retroviruses and for the expression of oncogene products |
| US5276019A (en) | 1987-03-25 | 1994-01-04 | The United States Of America As Represented By The Department Of Health And Human Services | Inhibitors for replication of retroviruses and for the expression of oncogene products |
| US5188897A (en) | 1987-10-22 | 1993-02-23 | Temple University Of The Commonwealth System Of Higher Education | Encapsulated 2',5'-phosphorothioate oligoadenylates |
| US5405939A (en) | 1987-10-22 | 1995-04-11 | Temple University Of The Commonwealth System Of Higher Education | 2',5'-phosphorothioate oligoadenylates and their covalent conjugates with polylysine |
| US5519126A (en) | 1988-03-25 | 1996-05-21 | University Of Virginia Alumni Patents Foundation | Oligonucleotide N-alkylphosphoramidates |
| US5278302A (en) | 1988-05-26 | 1994-01-11 | University Patents, Inc. | Polynucleotide phosphorodithioates |
| US5453496A (en) | 1988-05-26 | 1995-09-26 | University Patents, Inc. | Polynucleotide phosphorodithioate |
| US5216141A (en) | 1988-06-06 | 1993-06-01 | Benner Steven A | Oligonucleotide analogs containing sulfur linkages |
| US5194599A (en) | 1988-09-23 | 1993-03-16 | Gilead Sciences, Inc. | Hydrogen phosphonodithioate compositions |
| US5565555A (en) | 1988-09-23 | 1996-10-15 | Gilead Sciences, Inc. | Nucleoside hydrogen phosphonodithioate diesters and activated phosphonodithioate analogues |
| US5665577A (en) | 1989-02-06 | 1997-09-09 | Dana-Farber Cancer Institute | Vectors containing HIV packaging sequences, packaging defective HIV vectors, and uses thereof |
| US5399346A (en) | 1989-06-14 | 1995-03-21 | The United States Of America As Represented By The Department Of Health And Human Services | Gene therapy |
| US5436146A (en) | 1989-09-07 | 1995-07-25 | The Trustees Of Princeton University | Helper-free stocks of recombinant adeno-associated virus vectors |
| US5721218A (en) | 1989-10-23 | 1998-02-24 | Gilead Sciences, Inc. | Oligonucleotides with inverted polarity |
| US5399676A (en) | 1989-10-23 | 1995-03-21 | Gilead Sciences | Oligonucleotides with inverted polarity |
| US5527899A (en) | 1989-10-23 | 1996-06-18 | Gilead Sciences, Inc. | Oligonucleotides with inverted polarity |
| US5264564A (en) | 1989-10-24 | 1993-11-23 | Gilead Sciences | Oligonucleotide analogs with novel linkages |
| US5264562A (en) | 1989-10-24 | 1993-11-23 | Gilead Sciences, Inc. | Oligonucleotide analogs with novel linkages |
| US5676954A (en) | 1989-11-03 | 1997-10-14 | Vanderbilt University | Method of in vivo delivery of functioning foreign genes |
| US5455233A (en) | 1989-11-30 | 1995-10-03 | University Of North Carolina | Oligoribonucleoside and oligodeoxyribonucleoside boranophosphates |
| US5405938A (en) | 1989-12-20 | 1995-04-11 | Anti-Gene Development Group | Sequence-specific binding polymers for duplex nucleic acids |
| US5166315A (en) | 1989-12-20 | 1992-11-24 | Anti-Gene Development Group | Sequence-specific binding polymers for duplex nucleic acids |
| US5672510A (en) | 1990-01-19 | 1997-09-30 | Genetic Therapy, Inc. | Retroviral vectors |
| US5321131A (en) | 1990-03-08 | 1994-06-14 | Hybridon, Inc. | Site-specific functionalization of oligodeoxynucleotides for non-radioactive labelling |
| US5563253A (en) | 1990-03-08 | 1996-10-08 | Worcester Foundation For Biomedical Research | Linear aminoalkylphosphoramidate oligonucleotide derivatives |
| US5541306A (en) | 1990-03-08 | 1996-07-30 | Worcester Foundation For Biomedical Research | Aminoalkylphosphotriester oligonucleotide derivatives |
| US5536821A (en) | 1990-03-08 | 1996-07-16 | Worcester Foundation For Biomedical Research | Aminoalkylphosphorothioamidate oligonucleotide deratives |
| US5279833A (en) | 1990-04-04 | 1994-01-18 | Yale University | Liposomal transfection of nucleic acids into animal cells |
| US5470967A (en) | 1990-04-10 | 1995-11-28 | The Dupont Merck Pharmaceutical Company | Oligonucleotide analogs with sulfamate linkages |
| US5677437A (en) | 1990-07-27 | 1997-10-14 | Isis Pharmaceuticals, Inc. | Heteroatomic oligonucleoside linkages |
| US5541307A (en) | 1990-07-27 | 1996-07-30 | Isis Pharmaceuticals, Inc. | Backbone modified oligonucleotide analogs and solid phase synthesis thereof |
| US5623070A (en) | 1990-07-27 | 1997-04-22 | Isis Pharmaceuticals, Inc. | Heteroatomic oligonucleoside linkages |
| US5618704A (en) | 1990-07-27 | 1997-04-08 | Isis Pharmacueticals, Inc. | Backbone-modified oligonucleotide analogs and preparation thereof through radical coupling |
| US5602240A (en) | 1990-07-27 | 1997-02-11 | Ciba Geigy Ag. | Backbone modified oligonucleotide analogs |
| US5489677A (en) | 1990-07-27 | 1996-02-06 | Isis Pharmaceuticals, Inc. | Oligonucleoside linkages containing adjacent oxygen and nitrogen atoms |
| US5610289A (en) | 1990-07-27 | 1997-03-11 | Isis Pharmaceuticals, Inc. | Backbone modified oligonucleotide analogues |
| US5608046A (en) | 1990-07-27 | 1997-03-04 | Isis Pharmaceuticals, Inc. | Conjugated 4'-desmethyl nucleoside analog compounds |
| US5677439A (en) | 1990-08-03 | 1997-10-14 | Sanofi | Oligonucleotide analogues containing phosphate diester linkage substitutes, compositions thereof, and precursor dinucleotide analogues |
| US5177196A (en) | 1990-08-16 | 1993-01-05 | Microprobe Corporation | Oligo (α-arabinofuranosyl nucleotides) and α-arabinofuranosyl precursors thereof |
| US5214134A (en) | 1990-09-12 | 1993-05-25 | Sterling Winthrop Inc. | Process of linking nucleosides with a siloxane bridge |
| US5561225A (en) | 1990-09-19 | 1996-10-01 | Southern Research Institute | Polynucleotide analogs containing sulfonate and sulfonamide internucleoside linkages |
| US5596086A (en) | 1990-09-20 | 1997-01-21 | Gilead Sciences, Inc. | Modified internucleoside linkages having one nitrogen and two carbon atoms |
| US5817491A (en) | 1990-09-21 | 1998-10-06 | The Regents Of The University Of California | VSV G pseusdotyped retroviral vectors |
| US5391377A (en) | 1990-10-19 | 1995-02-21 | Cortecs Limited | Biphasic release formations for lipophilic acids |
| US5672697A (en) | 1991-02-08 | 1997-09-30 | Gilead Sciences, Inc. | Nucleoside 5'-methylene phosphonates |
| US5714331A (en) | 1991-05-24 | 1998-02-03 | Buchardt, Deceased; Ole | Peptide nucleic acids having enhanced binding affinity, sequence specificity and solubility |
| US5571799A (en) | 1991-08-12 | 1996-11-05 | Basco, Ltd. | (2'-5') oligoadenylate analogues useful as inhibitors of host-v5.-graft response |
| US5283185A (en) | 1991-08-28 | 1994-02-01 | University Of Tennessee Research Corporation | Method for delivering nucleic acids into cells |
| US5587361A (en) | 1991-10-15 | 1996-12-24 | Isis Pharmaceuticals, Inc. | Oligonucleotides having phosphorothioate linkages of high chiral purity |
| US5792608A (en) | 1991-12-12 | 1998-08-11 | Gilead Sciences, Inc. | Nuclease stable and binding competent oligomers and methods for their use |
| US5633360A (en) | 1992-04-14 | 1997-05-27 | Gilead Sciences, Inc. | Oligonucleotide analogs capable of passive cell membrane permeation |
| US5434257A (en) | 1992-06-01 | 1995-07-18 | Gilead Sciences, Inc. | Binding compentent oligomers containing unsaturated 3',5' and 2',5' linkages |
| US5476925A (en) | 1993-02-01 | 1995-12-19 | Northwestern University | Oligodeoxyribonucleotides including 3'-aminonucleoside-phosphoramidate linkages and terminal 3'-amino groups |
| US5466677A (en) | 1993-03-06 | 1995-11-14 | Ciba-Geigy Corporation | Dinucleoside phosphinates and their pharmaceutical compositions |
| US5663312A (en) | 1993-03-31 | 1997-09-02 | Sanofi | Oligonucleotide dimers with amide linkages replacing phosphodiester linkages |
| US5539082A (en) | 1993-04-26 | 1996-07-23 | Nielsen; Peter E. | Peptide nucleic acids |
| US5935935A (en) | 1993-06-10 | 1999-08-10 | Genetic Therapy, Inc. | Adenoviral vectors for treatment of hemophilia |
| US5719262A (en) | 1993-11-22 | 1998-02-17 | Buchardt, Deceased; Ole | Peptide nucleic acids having amino acid side chains |
| US5625050A (en) | 1994-03-31 | 1997-04-29 | Amgen Inc. | Modified oligonucleotides and intermediates useful in nucleic acid therapeutics |
| US5646269A (en) | 1994-04-28 | 1997-07-08 | Gilead Sciences, Inc. | Method for oligonucleotide analog synthesis |
| US6110490A (en) | 1994-08-05 | 2000-08-29 | The United States Of America As Represented By The Department Of Health And Human Services | Liposomal delivery system for biologically active agents |
| US6096716A (en) | 1994-12-12 | 2000-08-01 | The Board Of Regents, The University Of Texas System | Liposome-mediated transfection of central nervous system cells |
| US5707865A (en) | 1994-12-21 | 1998-01-13 | Kohn; Donald B. | Retroviral vectors for expression in embryonic cells |
| US5622856A (en) | 1995-08-03 | 1997-04-22 | Avigen | High efficiency helper system for AAV vector production |
| US6013516A (en) | 1995-10-06 | 2000-01-11 | The Salk Institute For Biological Studies | Vector and method of use for nucleic acid delivery to non-dividing cells |
| EP0820773A1 (en) | 1995-11-16 | 1998-01-28 | Universidad Autonoma Barcelona | Treatment of diabetes with a glucokinase gene |
| US5994136A (en) | 1997-12-12 | 1999-11-30 | Cell Genesys, Inc. | Method and means for producing high titer, safe, recombinant lentivirus vectors |
| US6217912B1 (en) | 1998-07-13 | 2001-04-17 | Expression Genetics, Inc. | Polyester analogue of poly-L-lysine as a soluble, biodegradable gene delivery carrier |
| US7514098B2 (en) | 2001-05-30 | 2009-04-07 | The Board Of Trustees Of The Leland Stanford Junior University | Use of targeted cross-linked nanoparticles for in vivo gene delivery |
| US8323618B2 (en) | 2007-11-07 | 2012-12-04 | University Of Houston System | Ultrasmall superparamagnetic iron oxide nanoparticles and uses thereof |
| US20190255088A1 (en) | 2018-02-17 | 2019-08-22 | The Board Of Regents Of The University Of Oklahoma | Biocompatible organo-inorganic nanocomposites |
| WO2020079449A1 (en) * | 2018-10-18 | 2020-04-23 | Citrox Biosciences Limited | Bioflavonoid compositions and their use for water purification and food preservation |
Non-Patent Citations (17)
| Title |
|---|
| "Publication Only", BONE MARROW TRANSPLANTATION, NATURE PUBLISHING GROUP, GB, vol. 49, no. Suppl 1, 1 March 2014 (2014-03-01), pages S438 - S559, XP037758929, ISSN: 0268-3369, [retrieved on 20140328], DOI: 10.1038/BMT.2014.50 * |
| ALTSCHUL ET AL., J MOLEC BIOL, vol. 215, 1990, pages 403 - 410 |
| ALTSCHULGISH, METHODS IN ENZYMOLOGY, vol. 266, 1996, pages 460 - 480 |
| BLAND, S.J ET AL.: "Deletion of a 19-Amino-Acid Region in Clostridioides difficile TcdB2 Results in Spontaneous Autoprocessing and Reduced Cell Binding and Provides a Nontoxic Immunogen for Vaccination", INFECT IMMUN, vol. 87, no. 8, 2019, XP093008927 |
| DAYHOFF: "National Biochemical Research Foundation", vol. 5, 1972, article "Atlas of Protein Sequence and Structure", pages: 124 |
| GISHSTATES, NATURE GENETICS, vol. 3, 1993, pages 266 - 272 |
| H. KROH ET AL.: "A neutralizing antibody that blocks delivery of the enzymatic cargo of Clostridium difficile toxin TcdB into host cells", J. BIOL. CHEM, vol. 293, no. 3, January 2018 (2018-01-01), pages 941 - 952, XP055584079, DOI: 10.1074/jbc.M117.813428 |
| H. QIU ET AL.: "Novel Clostridium difficile Anti-Toxin (TcdA and TcdB) Humanized Monoclonal Antibodies Demonstrate In Vitro Neutralization across a Broad Spectrum of Clinical Strains and In Vivo Potency in a Hamster Spore Challenge Model", PLOS ONE, vol. 11, no. 6, 2016, pages 0157970 |
| J. STEELE: "Antibody Against TcdB, but Not TcdA, Prevents Development of Gastrointestinal and Systemic Clostridium difficile Disease", THE JOURNAL OF INFECTIOUS DISEASES, vol. 207, 15 January 2013 (2013-01-15), pages 323 - 330, XP055272483, DOI: 10.1093/infdis/jis669 |
| KARLINALTSCHUL, PROC. NATL. ACAD. SCI. USA, vol. 87, 1990, pages 2264 - 2268 |
| KARLINALTSCHUL, PROC. NATL. ACAD. SCI. USA, vol. 90, 1993, pages 5873 - 5877 |
| MARION EHRICH, R.L.V.TJEFFERY M. LIBBYTRACY D. WILKINS: "Production of Clostridium difficile Antitoxin", INFECTION AND IMMUNITY, vol. 28, no. 3, 1980, pages 1041 - 1043 |
| MYERSMILLER, CABIOS, vol. 4, 1988, pages 11 - 17 |
| NORMAN KAYLEE M. ET AL: "Clostridioides difficile toxin B subverts germinal center and antibody recall responses by stimulating a drug-treatable CXCR4-dependent mechanism", CELL REPORTS, vol. 43, no. 5, 1 May 2024 (2024-05-01), US, pages 114245, XP093222817, ISSN: 2211-1247, DOI: 10.1016/j.celrep.2024.114245 * |
| PEARSONLIPMAN, PROC. NATL. ACAD. SCI. USA, vol. 85, 1988, pages 2444 - 2448 |
| VIOLA FÜHNER ET AL: "Development of Neutralizing and Non-neutralizing Antibodies Targeting Known and Novel Epitopes of TcdB of Clostridioides difficile", FRONTIERS IN MICROBIOLOGY, vol. 9, 6 December 2018 (2018-12-06), XP055584106, DOI: 10.3389/fmicb.2018.02908 * |
| Z. YANG ET AL.: "A novel multivalent, single-domain antibody targeting TcdA and TcdB prevents fulminant Clostridium difficile infection in mice", J. INFECT. DIS, vol. 210, 2014, pages 964 - 972, XP055376052, DOI: 10.1093/infdis/jiu196 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6990176B2 (en) | Methods for therapeutic administration of messenger ribonucleic acid drugs | |
| ES2388968T3 (en) | Smad7 inhibitors for the treatment of CNS diseases | |
| EP2594280A1 (en) | Activin neutralisers and uses thereof for treatment of diseases associated with aberrant "host defence response" activation | |
| EP3210611B1 (en) | Methods of treating vascular inflammatory disorders | |
| AU2009302468A1 (en) | Use of inhibitors of toll-like receptors in the prevention and treatment of hypercholesterolemia and hyperlipidemia and diseases related thereto | |
| CN102149390A (en) | Complement antagonists and uses thereof | |
| KR20110039382A (en) | Modulation of toll-like receptor 8 expression by antisense oligonucleotides | |
| US10006029B2 (en) | Methods of treating colorectal cancer | |
| US10478473B2 (en) | Compositions and methods of using anti-mullerian hormone for treatment of infertility | |
| EP3622958B1 (en) | Use of potassium ion channel inhibitor for treatment of depression and pharmaceutical composition | |
| US20130028889A1 (en) | Dosing regimens for treating and preventing ocular disorders using c-raf antisense | |
| WO2025059113A1 (en) | Treatments for enhancing immune response to clostridioides difficile infections | |
| WO2022061112A1 (en) | Combination treatment for cancer | |
| US20100111937A1 (en) | Modulation of Toll-Like Receptor 5 Expression by Antisense Oligonucleotides | |
| JP2012521749A (en) | Kanamycin antisense nucleic acid for the treatment of cancer | |
| US20210180063A1 (en) | Anti-arid3a treatments for inflammatory disorders | |
| JP2013520446A (en) | Top coat protein 2 (EMP2) binding reagent and use thereof in the treatment of eye diseases | |
| US20240148864A1 (en) | Polysaccharide adjuvants for virus vaccines | |
| WO2023240225A1 (en) | Constitutively active polymeric sting mimics for antitumor immunity | |
| US20250327112A1 (en) | Circulating biomarker assay for breast cancer prediction | |
| US20200375965A1 (en) | Targeting Lipid Metabolism and Free Fatty Acid (FFA) Oxidation to Treat Diseases Mediated by Resident Memory T Cells (TRM) | |
| Lazarczyk et al. | Immunostimulatory oligonucleotides in therapy of allergic diseases | |
| US20240424146A1 (en) | Drug screening method and model for monitoring platinum-resistant ovarian cancer tumor growth and metastases | |
| WO2021007329A2 (en) | Treatment of pulmonary fibrosis using inhibitors of neu1 sialidase | |
| WO2017150371A1 (en) | Therapeutic agent for tissue damage |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 24777124 Country of ref document: EP Kind code of ref document: A1 |