WO2025057088A1 - Rna compositions for delivery of incretin agents - Google Patents
Rna compositions for delivery of incretin agents Download PDFInfo
- Publication number
- WO2025057088A1 WO2025057088A1 PCT/IB2024/058845 IB2024058845W WO2025057088A1 WO 2025057088 A1 WO2025057088 A1 WO 2025057088A1 IB 2024058845 W IB2024058845 W IB 2024058845W WO 2025057088 A1 WO2025057088 A1 WO 2025057088A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- incretin
- composition
- agent
- seq
- domain
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/575—Hormones
- C07K14/605—Glucagons
Definitions
- Obesity is the most prevalent chronic disease worldwide, affecting approximately 650 million adults today. Obesity is considered a starting point for and critical contributor to pre-diabetes, type 2 diabetes (T2D, with its complications), non-alcoholic fatty liver disease (NAFLD), non-alcoholic steatohepatitis (NASH), cardiovascular and renal diseases, and premature mortality. It is estimated that the number of obese (BMI>30kg/m 2 ) people will exceed one billion by 2030, about 10% of which will suffer from severe class III obesity (BMI>40kg/m 2 ). Half of all men living with obesity live in just nine countries: USA, China, India, Brazil, Mexico, Russia, Egypt, Germany, and Turkey. In addition, childhood obesity is sharply on the rise worldwide. T2D, NAFLD, NASH, cardiovascular and renal diseases are also prevalent independent of obesity. There exists a need to develop further therapies for treatment and/or prevention of obesity and other related diseases.
- the present disclosure provides, among other things, polyribonucleotide precursors of said incretins as molecular entities, their production, formulation, and administration to treat obesity and its sequelae, including T2D, early T1D, cardiovascular diseases, renal diseases, NASH and NAFLD.
- the present disclosure also provides methods where these agents are used to treat diseases including T2D, early T1D, cardiovascular diseases, renal diseases, NASH and NAFLD independent of obesity.
- the present disclosure also provides methods where these agents are used to treat sequelae of NASH, including liver fibrosis and cirrhosis.
- the present disclosure also recognizes that such a treatment approach, i.e., delivery of polyribonucleotide precursors of incretins presents additional benefits over current treatments, including but not limited to, broader accessibility for obese people who will otherwise have no access to current products due to limited supply, high price, lack of health insurance, formulations that require lower injection volume compared to marketed peptide-based products, a lower rate of treatment discontinuation by patients due to factors such as gastrointestinal side-effects, and improved properties such as an improved pharmacokinetic profile.
- the improved pharmacokinetic profile has the advantage of lower administration frequency by virtue of a longer-acting therapeutic.
- the present disclosure provides a composition comprising a polyribonucleotide encoding an incretin agent.
- the incretin agent is a GLP 1 receptor agonist.
- the incretin agent is a GIP receptor agonist.
- the incretin agent is a GLP1/GIP dual receptor agonist.
- the incretin agent is a GLP1/GCG dual receptor agonist.
- the incretin agent is a GLP1/GIP/GCG triple receptor agonist.
- an incretin agent comprises an incretin peptide having an amino acid sequence according to any one of SEQ ID NOs: 5-7, 63-64, 69-70, and 74-75. In some embodiments, an incretin agent comprises an incretin peptide having an amino acid sequence according to any one of SEQ ID NOs: 8-9, 62, and 72. In some embodiments, an incretin agent comprises an incretin peptide having an amino acid sequence according SEQ ID NO: 11. In some embodiments, an incretin agent comprises an incretin peptide having an amino acid sequence according to any one of SEQ ID NOs: 12-14. In some embodiments, an incretin agent comprises an incretin peptide having an amino acid sequence according to SEQ ID NO: 15.
- an incretin peptide is fused to a signal peptide, optionally via the N-terminus of the incretin peptide, optionally via a linker.
- the signal peptide has an amino acid sequence according to any one of SEQ ID NOs: 16-39 and 65-67.
- the signal peptide has an amino acid sequence according to any one of SEQ ID NOs: 16-21 and 65-67.
- the signal peptide has an amino acid sequence according to SEQ ID NO: 17.
- the signal peptide has an amino acid sequence according to SEQ ID NO: 65.
- the signal peptide has an amino acid sequence according to SEQ ID NO: 66.
- the incretin agent comprises an amino acid sequence according to any one of SEQ ID NOs: 41-45, 52-61, and 108-152.
- the incretin agent comprises an incretin peptide fused to one or more additional incretin peptides, optionally via one or more linkers.
- the one or more linkers comprise an amino acid sequence according of any one of SEQ ID NOs: 1-5, 68, or 156.
- the incretin agent comprises an incretin peptide fused to two or more incretin peptides.
- the incretin agent comprises at least one GLP1 receptor agonist and at least one GIP receptor agonist.
- the incretin agent comprises at least two GLP1 receptor agonists.
- the incretin agent comprises at least two GIP receptor agonists.
- the incretin agent comprises one or more furin cleavage sites.
- the one or more furin cleavage sites are located between adjacent incretin peptides.
- the one or more furin cleavage sites comprise an amino acid sequence according to SEQ ID NO: 153.
- the incretin agent comprises one or more units that each comprise, from N-terminus to C- terminus: GLP1 receptor agonist-linker-furin cleavage site-GIP receptor agonist, e.g., wherein the incretin agent comprises one unit (e.g., SEQ ID NOs: 76, 77, 78, 79, 80, 81), two units (e.g., SEQ ID NOs: 82); or four units (e.g., SEQ ID NO: 83).
- the incretin agent comprises an amino acid sequence according to any one of SEQ ID NOs: 76-83, 94-97, 102-107.
- the incretin agent comprises a half-life extending moiety.
- the half-life extending moiety comprises albumin (e.g., human serum albumin).
- the human serum albumin comprises an amino acid sequence having at least 90%, 95%, or 99% identity to SEQ ID NO: 159.
- the human serum albumin comprises an amino acid sequence according to SEQ ID NO: 159.
- the incretin agent comprises albumin (e.g., human serum albumin) fused to one or more units that each comprise from, N-terminus to C- terminus: (i) GLP1 receptor agonist-linker (e.g., SEQ ID NO: 98); (ii) GIP receptor agonistlinker (e.g., SEQ ID NO: 100); (iii) GLP1 receptor agonist-linker-furin cleavage site (e.g., SEQ ID NO: 102); or (iv) GLP1 receptor agonist-linker-furin cleavage site-GIP receptor agonist, e.g., wherein the incretin agent comprises one unit (e.g., SEQ ID NO: 104), two units (e.g., SEQ ID NO: 106) or four units (e.g., SEQ ID NO: 107).
- the incretin agent comprises an amino acid sequence according to any one of SEQ ID NOs: 98, 100,
- the half-life extending moiety comprises an albumin binding domain (ABD).
- the ABD is derived from protein G of Streptococcus strain GI48 and/or from protein PAB of Finegoldia magnet, such as ABD035 and SA21.
- the half-life extending moiety comprises an ABD that binds to domain II of human serum albumin and does not overlap or interfere with binding to the FcRn-binding site on albumin.
- the half-life extending moiety comprises ABDCon.
- the half-life extending moiety comprises an albumin binding domain (ABD) derived from the bacterial protein Sso7d from the hyperthermophilic archaeon Sulfolobus solfataricus, such as Ml 1.12 and Ml 8.2.5.
- the half-life extending moiety comprises a DARPin that binds albumin.
- the ABD comprises an immunoglobulin domain or fragment thereof that binds albumin.
- the ABD comprises a fully human domain antibody (dAb) that binds albumin, such as AlbudAb.
- the ABD comprises a Fab that binds albumin, such as dsFv CA645.
- the ABD comprises a heavy chain only (VHH) antibody, such as a nanobody, that binds albumin.
- the VHH antibody comprises a VHH domain having the complementarity determining region (CDR) sequences HCDR1, HCDR2, and/or HCDR3 according to SEQ ID NO: 191 (GFTLDYYA), SEQ ID NO: 192 (IASSGGST), and/or SEQ ID NO: 193 (AAAVLECRTVVRGYDY), respectively.
- the VHH antibody comprises an amino acid sequence having at least 90%, 95%, or 99% identity to SEQ ID NO: 154.
- the VHH antibody comprises an amino acid sequence according to SEQ ID NO: 154.
- the incretin agent comprises a VHH antibody that binds albumin fused to a unit that comprises, from N-terminus to C- terminus: (i) GLPl-linker (e.g., SEQ ID NO: 99); (ii) GIP receptor agonist-linker (e.g., SEQ ID NO: 101); or (iii) GLP1 receptor agonist-linker-furin-GIP receptor agonist-linker (e.g., SEQ ID NO: 103 or 105).
- the incretin agent comprises an amino acid sequence according to any one of SEQ ID NOs: 99, 101, 103, 105.
- the half-life extending moiety does not comprise a Fc domain, such as from a human IgG, optionally from a human IgGl, IgG2, IgG3, or IgG4.
- the half-life extending moiety comprises a Fc domain, such as from a human IgG, optionally from a human IgGl, IgG2, IgG3, or IgG4.
- the human IgG is a human IgG4.
- the incretin agent comprises an IgG4 Fc domain fused to a unit comprising, from N-terminus to C-terminus: (i) GLP1 receptor agonist-linker (e.g., SEQ ID NOs: 10, 89, 90, 91); (ii) GIP receptor agonist-linker (e.g., SEQ ID NO: 92, 93); or (iii) GLP1 receptor agonist-linker-furin-GIP receptor agonist-linker (e.g., SEQ ID NO: 94, 95, 96, 97).
- the IgG4 Fc domain comprises an amino acid sequence that is at least 90%, 95%, or 99% identical to SEQ ID NO: 155.
- the IgG4 Fc domain comprises an amino acid sequence according to SEQ ID NO: 155.
- the incretin agent comprises an amino acid sequence according to any one of SEQ ID NOs: 10, and 89-97.
- the Fc domain comprises one or more mutations in one or both Fc constant domains to increase half-life of the incretin agent and/or induce dimerization.
- the one or more mutations comprises one or more mutations in a CH3 domain.
- the one or more mutations to induce dimerization comprises: (i) Y349C, T366S, L368A, and/or Y407V, according to EU numbering; or (ii) S354C and/or T366W, according to EU numbering.
- the one or more mutations comprises Y349C, T366S, L368A, and Y407V (“FcKIH-b”), according to EU numbering, or S354C and T366W (“FcKIH-a”), according to EU numbering.
- the incretin agent comprises a first polypeptide chain and a second polypeptide chain, wherein the first polypeptide chain comprises an incretin peptide fused to a first Fc domain, wherein the first Fc domain comprises the mutations Y349C, T366S, L368A, and Y407V (“FcKIH-b”), according to EU numbering and wherein the second polypeptide chain comprises an incretin peptide fused to a second Fc domain, wherein the second Fc domain comprises the mutations S354C and T366W (“FcKIH-a”), according to EU numbering.
- the one or more mutations to increase half-life of the incretin agent comprises M428L and N434S (“LS”), according to EU numbering.
- the incretin agent comprises an Fc domain with FcKIH-a mutations on a first polypeptide chain and an Fc domain with FcKIH-b mutations on a second polypeptide chain, where the Fc domain on each polypeptide chain is independently fused with one or more units that comprise, from N-terminus to C-terminus: (i) GLP1 receptor agonist-linker (e.g., SEQ ID NO: 84, 85, 86, 87); or (ii) GIP receptor agonist-linker (e.g., SEQ ID NO: 88).
- the incretin agent comprises an amino acid sequence according to any one of SEQ ID NOs: 84-88.
- the Fc domain comprises one or more mutations that ablate effector activity of the Fc domain (e.g., binding to Fey receptors or Clq).
- the one or more mutations that ablate effector activity of the Fc domain comprise the following mutations: L234S, L235T, and G236R (“STR”) according to EU numbering.
- the one or more mutations that ablate effector activity of the Fc domain comprise the following mutations: L234A and L235A (“LALA”) according to EU numbering.
- the one or more mutations that ablate effector activity of the Fc domain comprise the following mutations: L234A/L235A/P329G (“LALAPG”) according to EU numbering.
- the half-life extending moiety comprises a VNAR that binds albumin. In some embodiments, the half-life extending moiety comprises an XTEN sequence.
- the polyribonucleotide has a ribonucleic acid sequence that is at least 90% identical to any one of SEQ ID NOs: 177-185 and 224-256. In some embodiments, the polyribonucleotide has a ribonucleic acid sequence according to any one of SEQ ID Nos: 177-185 and 224-256.
- the polyribonucleotide comprises at least one noncoding sequence element that enhances RNA stability and/or translation efficiency.
- the at least one non-coding sequence element comprises a 5’ cap structure, a 5’ UTR, a 3 ’ UTR, and/or a polyA tail.
- the polyribonucleotide comprises, in a 5 ’ to 3 ’ direction: a. a 5’ UTR; b. a signal peptide-coding sequence; c. an incretin peptide-coding sequence; d. a 3’ UTR; and e. a polyAtail.
- the polyribonucleotide comprises, in a 5 ’ to 3 ’ direction: (1) a. a 5’ UTR; b. a signal peptide-coding sequence; c. an incretin peptide-coding sequence; d. a linker-coding sequence; e. a half-life extending moiety-coding sequence; f. a 3’ UTR; and g. a polyAtail; or (2) a. a 5’ UTR; b. a signal peptide-coding sequence; c. a halflife extending moiety-coding sequence; d. a linker-coding sequence; e. an incretin peptide- coding sequence; f. a 3’ UTR; and g. a polyAtail.
- the incretin peptide is encoded by a coding sequence which is codon-optimized and/or the G/C content of which is increased compared to wild type coding sequence, wherein the codon-optimization and/or the increase in the G/C content does not change the sequence of the encoded amino acid sequence.
- the polyribonucleotide comprises at least one modified ribonucleotide.
- the polyribonucleotide comprises a modified nucleoside in place of uridine.
- the polyribonucleotide comprises a modified nucleoside in place of each uridine.
- the modified nucleoside is selected from pseudouridine (y), N1 -methyl -pseudouridine (m h
- the modified nucleoside is N1 -methyl -pseudouridine (ml ⁇
- the polyribonucleotide comprises a 5’ cap structure. In some embodiments, the polyribonucleotide comprises a 5 ’ UTR. In some embodiments, the polyribonucleotide comprises a 3’ UTR. In some embodiments, the polyribonucleotide comprises a polyAtail. In some embodiments, the polyAtail comprises at least 100 nucleotides. In some embodiments, the polyribonucleotide is mRNA.
- the polyribonucleotide is formulated as a liquid, formulated as a solid, or a combination thereof. In some embodiments, the polyribonucleotide is formulated for injection. In some embodiments, the polyribonucleotide is formulated for intraperitoneal or intravenous administration. [0026] In some embodiments, the polyribonucleotide is formulated or is to be formulated as lipid particles. In some embodiments, the polyribonucleotide is formulated or is to be formulated as lipid nanoparticles. In some embodiments, the polyribonucleotide is encapsulated within the lipid nanoparticles. In some embodiments, the lipid nanoparticles are pancreas-targeting and/or gut-targeting lipid nanoparticles. In some embodiments, the lipid nanoparticles are cationic lipid nanoparticles.
- lipids that form the lipid nanoparticles comprise a. a polymer-conjugated lipid; b. a cationic lipid; and c. a neutral lipid.
- the polymer-conjugated lipid is a PEG-conjugated lipid.
- the cationic lipid is an ionizable lipid-like material (lipidoid).
- the cationic lipid has one of the following structures:
- the neutral lipid comprises a helper lipid such as 1,2- distearoyl-sn-glycero-3-phosphocholine (DPSC) and/or cholesterol.
- helper lipid such as 1,2- distearoyl-sn-glycero-3-phosphocholine (DPSC) and/or cholesterol.
- the cationic lipid is selected from cationic lipid X-2, X- 3, or X-4 and the neutral lipid comprises a helper lipid such as DOTAP, DOPE, or PS, and cholesterol.
- the polymer-conjugated lipid is C14-PEG2000.
- the lipid nanoparticles comprise: i) about 30 mol% to about 50 mol% of a cationic lipid; ii) about 1 mol% to 5 mol% of a PEG-conjugated lipid; iii) about 30 mol% to about 50 mol% of a helper lipid; and iv) about 20 mol% to about 40 mol% of cholesterol.
- the lipid nanoparticles comprise about 35 mol% of a cationic lipid; about 40 mol% of a helper lipid, about 22.5 mol% of cholesterol, and about 2.5 mol% of a PEG-conjugated lipid.
- the lipid nanoparticles comprise about 35 mol% of cationic lipid X-2, X-3, or X-4, about 40 mol% of DOTAP, DOPE, or PS, about 22.5 mol% of cholesterol, and about 2.5 mol% of C14-PEG2000.
- the lipid nanoparticles comprise about 35 mol% of cationic lipid X-2; about 40 mol% of DOTAP; about 22.5 mol% of cholesterol, and about 2.5 mol% of C14-PEG2000. In some embodiments, the lipid nanoparticles comprise about 35 mol% of cationic lipid X-3; about 40 mol% of DOTAP; about 22.5 mol% of cholesterol; and about 2.5 mol% of C14-PEG2000. In some embodiments, the lipid nanoparticles comprise about 35 mol% of cationic lipid X-4; about 40 mol% of DOTAP; about 22.5 mol% of cholesterol; and about 2.5 mol% of C14-PEG2000.
- the lipid nanoparticles comprise about 35 mol% of cationic lipid X-2; about 40 mol% of DOPE; about 22.5 mol% of cholesterol; and about 2.5 mol% of C14-PEG2000. In some embodiments, the lipid nanoparticles comprise about 35 mol% of cationic lipid X-3; about 40 mol% of DOPE; about 22.5 mol% of cholesterol; and about 2.5 mol% of C14-PEG2000. In some embodiments, the lipid nanoparticles comprise about 35 mol% of cationic lipid X-4; about 40 mol% of DOPE; about 22.5 mol% of cholesterol; and about 2.5 mol% of C14-PEG2000.
- the lipid nanoparticles comprise about 35 mol% of cationic lipid X-2; about 40 mol% of PS; about 22.5 mol% of cholesterol; and about 2.5 mol% of C14- PEG2000. In some embodiments, the lipid nanoparticles comprise about 35 mol% of cationic lipid X-3; about 40 mol% of PS; about 22.5 mol% of cholesterol; and about 2.5 mol% of C14-PEG2000. In some embodiments, the lipid nanoparticles comprise about 35 mol% of cationic lipid X-4; about 40 mol% of PS; about 22.5 mol% of cholesterol; and about 2.5 mol% of C14-PEG2000.
- the lipid nanoparticles are formulated for intraperitoneal (i.p.) delivery. In some embodiments, the lipid nanoparticles have an average size of about 50-150 nm.
- the composition comprises one or more pharmaceutically acceptable carriers, diluents and/or excipients. In some embodiments, the composition further comprise a cryoprotectant. In some embodiments, the cryoprotectant is sucrose. In some embodiments, the composition comprises an aqueous buffered solution. In some embodiments, the aqueous buffered solution includes sodium ions.
- the present disclosure provides, a method of treating a disease-state in a subject in need thereof, comprising administering to said subject a therapeutically effective amount of a composition comprising one or more polyribonucleotides described here.
- the method further comprises administering one or more DPP -4 inhibitors.
- the one or more DPP-4 inhibitors and the composition are administered concurrently.
- the one or more DPP -4 inhibitors and the composition are administered sequentially.
- the one or more DPP-4 inhibitors are administered prior to the composition.
- one or more DPP-4 inhibitors are administered after the composition.
- one or more DPP-4 inhibitors comprises sitagliptin, vildagliptin, saxagliptin, linagliptin, gemigliptin, anagliptin, teneligliptin, alogliptin, trelagliptin, omarigliptin, evogliptin, gosogliptin, dutogliptin, neogliptin, retagliptin, denagliptin, cofroglipin, fotagliptin, prusogliptin, berberine, or any combination thereof.
- one or more DPP- 4 inhibitors are administered orally.
- disease-state is obesity or an obesity-related disorder.
- obesity-related disorder is pre-diabetes, type 2 diabetes (T2D), early type 1 diabetes (T1D), non-alcoholic fatty liver disease (NAFLD), non-alcoholic steatohepatitis (NASH), a cardiovascular (CV) disease, a renal disease, or elevated risk of premature mortality.
- cardiovascular (CV) disease comprises a major cardiovascular event (MACE), including CV death, non-fatal myocardial infarction, non-fatal stroke, and/or heart failure with preserved ejection fraction (HFpEF).
- MACE major cardiovascular event
- HFpEF heart failure with preserved ejection fraction
- the disease-state is a cardiovascular (CV) disease.
- the cardiovascular disease comprises a major cardiovascular event (MACE), including CV death, non-fatal myocardial infarction, non-fatal stroke, and/or heart failure with preserved ejection fraction (HfpEF).
- MACE major cardiovascular event
- the method improves a subject’s blood pressure and/or blood lipids in said subject.
- the disease-state is a renal disease.
- the disease-state is non-alcoholic fatty liver disease (NAFLD).
- NAFLD non-alcoholic fatty liver disease
- NASH non-alcoholic steatohepatitis
- optionally its sequelae, liver fibrosis and cirrhosis optionally its sequelae, liver fibrosis and cirrhosis.
- administering the composition to the subject comprises administering one or more doses of the composition to the subject.
- the one or more doses of the composition are administered to the subject daily, every other day or once a week.
- the one or more doses of the composition are administered to the subject less frequently than once a week.
- the one or more doses of the composition are administered to the subject once every 2, 3 or 4 weeks.
- the composition is administered via injection.
- the composition is administered subcutaneously, intravenously, intramuscularly, or intraperitoneally.
- the composition is administered intraperitoneally.
- the composition is administered non-invasively (e.g., orally or nasally). In some embodiments, administration of the composition results in expression of the incretin agent in the subject. In some embodiments, the composition is administered in a volume that is less than 0.5 mb
- the present disclosure provides, use of the composition of any comprising one or more polyribonucleotides described herein for the treatment of a disease-state in a subject in need thereof.
- the present disclosure provides, a method of producing an incretin agent comprising administering to cells a composition comprising a polyribonucleotide described herein so that the cells express and secrete the incretin agent.
- an incretin agent that comprises an incretin peptide fused to a signal peptide.
- the incretin peptide is fused to the signal peptide via the N-terminus of the incretin peptide, optionally via a linker.
- the signal peptide has an amino acid sequence according to any one of SEQ ID NOs: 16-39 and 65-67.
- the signal peptide has an amino acid sequence according to any one of SEQ ID NOs: 16-21 and 65-67.
- the signal peptide has an amino acid sequence according to SEQ ID NO: 17.
- the signal peptide has an amino acid sequence according to SEQ ID NO: 65. In some embodiments, the signal peptide has an amino acid sequence according to SEQ ID NO: 66. In some embodiments, the incretin agent comprises an incretin peptide fused to a signal peptide that comprises an amino acid sequence according to any one of SEQ ID NOs: 41-45, 52-61, and 108-152.
- the incretin agent comprises an incretin peptide fused to one or more additional incretin peptides, optionally via one or more linkers.
- the one or more linkers comprise an amino acid sequence according of any one of SEQ ID NOs: 1-5, 68, or 156.
- the incretin agent comprises an incretin peptide fused to two or more incretin peptides.
- the incretin agent comprises at least one GLP 1 receptor agonist and at least one GIP receptor agonist. In some embodiments, the incretin agent comprises at least two GLP1 receptor agonists. In some embodiments, the incretin agent comprises at least two GIP receptor agonists. In some embodiments, the incretin agent comprises one or more furin cleavage sites. In some embodiments, the one or more furin cleavage sites are located between adjacent incretin peptides. In some embodiments, the one or more furin cleavage sites comprise an amino acid sequence according to SEQ ID NO: 153.
- the incretin agent comprises one or more units that each comprise, from N-terminus to C-terminus: GLP1 receptor agonist-linker-furin cleavage site-GIP receptor agonist, e.g., wherein the incretin agent comprises one unit (e.g., SEQ ID NOs: 76, 77, 78, 79, 80, 81), two units (e.g., SEQ ID NOs: 82); or four units (e.g., SEQ ID NO: 83).
- the incretin agent comprises an amino acid sequence according to any one of SEQ ID NOs: 76-83, 94-97, 102-107.
- the incretin agent comprises a half-life extending moiety.
- the half-life extending moiety comprises albumin (e.g., human serum albumin).
- the human serum albumin comprises an amino acid sequence having at least 90%, 95%, or 99% identity to SEQ ID NO: 159.
- the human serum albumin comprises an amino acid sequence according to SEQ ID NO: 159.
- the incretin agent comprises albumin (e.g., human serum albumin) fused to one or more units that each comprise from, N-terminus to C- terminus: (i) GLP1 receptor agonist-linker (e.g., SEQ ID NO: 98); (ii) GIP receptor agonistlinker (e.g., SEQ ID NO: 100); (iii) GLP1 receptor agonist-linker-furin cleavage site (e.g., SEQ ID NO: 102); or (iv) GLP1 receptor agonist-linker-furin cleavage site-GIP receptor agonist, e.g., wherein the incretin agent comprises one unit (e.g., SEQ ID NO: 104), two units (e.g., SEQ ID NO: 106) or four units (e.g., SEQ ID NO: 107)In some embodiments, the incretin agent comprises an amino acid sequence according to any one of SEQ ID NOs: 98,
- the half-life extending moiety comprises an albumin binding domain (ABD).
- the ABD is derived from protein G of Streptococcus strain GI48 and/or from protein PAB of Finegoldia magnet, such as ABD035 and SA21.
- the half-life extending moiety comprises an ABD that binds to domain II of human serum albumin and does not overlap or interfere with binding to the FcRn-binding site on albumin.
- the half-life extending moiety comprises ABDCon.
- the half-life extending moiety comprises an ABD derived from the bacterial protein Sso7d from the hyperthermophilic archaeon Sulfolobus solfataricus, such as Mil.12 and M18.2.5.
- the half-life extending moiety comprises a DARPin that binds albumin.
- the ABD comprises an immunoglobulin domain or fragment thereof that binds albumin.
- the ABD comprises a fully human domain antibody (dAb) that binds albumin, such as AlbudAb.
- the ABD comprises a Fab that binds albumin, such as dsFv CA645.
- the ABD comprises a heavy chain only (VHH) antibody, such as a nanobody, that binds albumin.
- VHH antibody comprises a VHH domain having the complementarity determining region (CDR) sequences HCDR1, HCDR2, and/or HCDR3 according to SEQ ID NO: 191 (GFTLDYYA), SEQ ID NO: 192 (IASSGGST), and/or SEQ ID NO: 193 (AAAVLECRTVVRGYDY), respectively.
- the VHH antibody comprises an amino acid sequence having at least 90%, 95%, or 99% identity to SEQ ID NO: 154.
- the VHH antibody comprises an amino acid sequence according to SEQ ID NO: 154.
- the incretin agent comprises a VHH antibody that binds albumin fused to a unit that comprises, from N-terminus to C-terminus: (i) GLPl-linker (e.g., SEQ ID NO: 99); (ii) GIP receptor agonist-linker (e.g., SEQ ID NO: 101); or (iii) GLP1 receptor agonist-linker-furin-GIP receptor agonist-linker (e.g., SEQ ID NO: 103 or 105).
- the incretin agent comprises an amino acid sequence according to any one of SEQ ID NOs: 99, 101, 103, 105.
- the half-life extending moiety does not comprise a Fc domain, such as from a human IgG, optionally from a human IgGl, IgG2, IgG3, or IgG4.
- the half-life extending moiety comprises a Fc domain, such as from a human IgG, optionally from a human IgGl, IgG2, IgG3, or IgG4.
- the human IgG is a human IgG4.
- the incretin agent comprises an IgG4 Fc domain fused to a unit comprising, from N-terminus to C-terminus: i) GLP1 receptor agonistlinker (e.g., SEQ ID NOs: 10, 89, 90, 91); (ii) GIP receptor agonist-linker (e.g., SEQ ID NO: 92, 93); or (iii) GLP1 receptor agonist-linker-furin-GIP receptor agonist-linker (e.g., SEQ ID NO: 94, 95, 96, 97).
- the IgG4 Fc domain comprises an amino acid sequence that is at least 90%, 95%, or 99% identical to SEQ ID NO: 155.
- the IgG4 Fc domain comprises an amino acid sequence according to SEQ ID NO: 155.
- the incretin agent comprises an amino acid sequence according to any one of SEQ ID NOs: 10, 89-97.
- the Fc domain comprises one or more mutations in one or both Fc constant domains to increase half-life of the incretin agent and/or induce dimerization.
- the one or more mutations comprises one or more mutations in a CH3 domain.
- the one or more mutations to induce dimerization comprises: (i) Y349C, T366S, L368A, and/or Y407V, according to EU numbering; or (ii) S354C and/or T366W, according to EU numbering.
- the one or more mutations comprises Y349C, T366S, L368A, and Y407V (“FcKIH-b”), according to EU numbering, or S354C and T366W (“FcKIH-a”), according to EU numbering.
- the incretin agent comprises a first polypeptide chain and a second polypeptide chain, wherein the first polypeptide chain comprises an incretin peptide fused to a first Fc domain, wherein the first Fc domain comprises the mutations Y349C, T366S, L368A, and Y407V (“FcKIH-b”), according to EU numbering and wherein the second polypeptide chain comprises an incretin peptide fused to a second Fc domain, wherein the second Fc domain comprises the mutations S354C and T366W (“FcKIH-a”), according to EU numbering.
- the one or more mutations to increase half-life of the incretin agent comprises M428L and N434S (“LS”), according to EU numbering.
- the incretin agent comprises an Fc domain with FcKIH-a mutations on a first polypeptide chain and an Fc domain with FcKIH-b mutations on a second polypeptide chain, where the Fc domain on each polypeptide chain is independently fused with one or more units that comprise, from N-terminus to C-terminus: (i) GLP1 receptor agonist-linker (e.g., SEQ ID NO: 84, 85, 86, 87); or (ii) GIP receptor agonist-linker (e.g., SEQ ID NO: 88).
- the incretin agent comprises an amino acid sequence according to any one of SEQ ID NOs: 84-88.
- the Fc domain comprises one or more mutations that ablate effector activity of the Fc domain (e.g., binding to Fey receptors or Clq).
- the one or more mutations that ablate effector activity of the Fc domain comprise the following mutations: L234S, L235T, and G236R (“STR”) according to EU numbering.
- the one or more mutations that ablate effector activity of the Fc domain comprise the following mutations: L234A and L235A (“LALA”) according to EU numbering.
- the one or more mutations that ablate effector activity of the Fc domain comprise the following mutations: L234A/L235A/P329G (“LALAPG”) according to EU numbering.
- the half-life extending moiety comprises a VNAR that binds albumin.
- the half-life extending moiety comprises an XTEN sequence.
- the present disclosure provides an incretin agent comprising: a husec signal peptide; an incretin peptide comprising a GLP1 incretin peptide, or fragment or variant thereof; wherein the GLP1 incretin peptide comprises an amino acid sequence having an A8G substitution mutation compared to a wildtype GLP1 amino acid sequence.
- the present disclosure provides a polyribonucleotide encoding the incretin agent comprising a husec signal peptide; an incretin peptide comprising a GLP1 incretin peptide, or fragment or variant thereof; wherein the GLP1 incretin peptide comprises an amino acid sequence having an A8G substitution mutation compared to a wildtype GLP1 amino acid sequence.
- an incretin agent comprising: a husec signal peptide; an incretin peptide comprising a GIP incretin peptide, or fragment or variant thereof; wherein the GIP incretin peptide comprises an amino acid sequence having an A2G substitution mutation compared to a wildtype GIP amino acid sequence.
- the present disclosure provides a polyribonucleotide encoding the incretin agent an incretin agent comprising: a husec signal peptide; an incretin peptide comprising a GIP incretin peptide, or fragment or variant thereof; wherein the GIP incretin peptide comprises an amino acid sequence having an A2G substitution mutation compared to a wildtype GIP amino acid sequence.
- Figure 1 shows an exemplary therapeutic strategy utilizing polyribonucleotides as described herein for delivery and in vivo expression of incretin agents.
- Figure 3 shows an exemplary design of an incretin agent described herein. Specifically, Figure 3 shows a schematic of a polyribonucleotide encoding a signal peptide (“SP”) and a single incretin peptide (which configuration is referred to herein as “I: lx”) (top) and a schematic of the translated incretin protein (bottom).
- SP signal peptide
- I: lx single incretin peptide
- Figure 5 shows an exemplary design of an incretin agent described herein. Specifically, Figure 5 shows a schematic of a polyribonucleotide encoding a signal peptide (“SP”) and four incretin peptides, each separated by a linker (“LI”) furin cleavage site (“F”) (which configuration is referred to herein as “I:4x”) (top) and a schematic of the translated protein (bottom).
- SP signal peptide
- F furin cleavage site
- Figures 6A-B show exemplary incretin agents including an incretin agent that has a signal peptide (“SP”), a GLP1 incretin peptide and a (GGGGS)2 linker ( Figure 6A), and an incretin agent that has a signal peptide (“SP”), a GLP1 incretin peptide, a (GGGGS)2 linker, a furin cleavage site, and a GIP incretin peptide ( Figure 6B).
- Signal peptide cleavage sites are indicated in Figure 6A and Figure 6B.
- a furin cleavage site is indicated in Figure 6B so that upon expression, the GIP incretin peptide is cleaved from the GLP1 incretin peptide.
- Figure 7 shows an exemplary design of an incretin agent described herein.
- Figure 7 shows a schematic of a polyribonucleotide encoding a signal peptide (“SP”) and an incretin agent (e.g., a I: lx, I:2x, or I:4x incretin agent as described herein) fused to a half-life extension (“HLE”) domain, e.g., human serum albumin (“HSA”) or an albumin binding domain (‘ ‘ABD”) (top) via a linker (“L2”) and a schematic of the translated protein (bottom).
- HLE half-life extension
- HSA human serum albumin
- ABD albumin binding domain
- L2 linker
- Figures 8A-B show exemplary incretin agents that may be encoded by polyribonucleotides described herein, that include more than one incretin peptide and a half life extension (HLE) domain.
- Figure 8A shows an incretin agent that has a signal peptide (“SP”), a first GLP1 incretin peptide, a linker, a second GLP1 incretin peptide, a second linker (GGGGS)3, and a half life extension (HLE) domain that is human serum albumin (HSA).
- SP signal peptide
- GGGGS second linker
- HLE half life extension domain that is human serum albumin
- Figure 8B shows an incretin agent that has a signal peptide (“SP”), a first GLP1 incretin peptide, a linker, a second GLP1 incretin peptide, a second linker (GGGGS)3, and a half life extension (HLE) domain that is a VHH domain that binds to HSA.
- SP signal peptide
- GGGGS linker
- HLE half life extension
- Figure 9 shows an exemplary incretin agent that may be encoded by one or more polyribonucleotides described herein, that includes more than one incretin peptide and a half life extension (HLE) domain.
- the incretin agent in Figure 9 has a signal peptide (“SP”), a first GLP1 incretin peptide, a linker (GGGGS)2, a first GIP incretin peptide, a second linker (GGGGS)2, a second GLP1 incretin peptide, a third linker (GGGGS)2, a second GIP incretin peptide, a fourth linker (GGGGS)?
- SP signal peptide
- HLE domain human serum albumin (HSA).
- HSA human serum albumin
- Furin and SP cleavage sites within the incretin agent are indicated with arrows.
- Such a design produces four separate incretin peptides, where the second GIP incretin peptide remains fused to the HLE domain.
- Figure 10 shows an exemplary design of a polyribonucleotide encoding an incretin agent that includes an incretin peptide fused to an Fc domain, where the incretin peptide could be one (I: lx), two (I:2x) or four (I:4x) incretin peptides (top).
- the two polypeptide chains associate and result in dimeric (e.g., homodimeric) structure (bottom).
- Each polypeptide chain also includes a signal peptide (SP) and a linker (L2).
- SP signal peptide
- L2 linker
- the Fc domains include mutations to abolish effector function (e.g., STR, LALA, LALAPG, etc. mutations) and/or to extend half-life (e.g., YTE, LS, etc. mutations).
- mutations to abolish effector function e.g., STR, LALA, LALAPG, etc. mutations
- half-life e.g., YTE, LS, etc. mutations
- Figure 11 shows an exemplary incretin agent that may be encoded by one or more polyribonucleotides described herein, that includes more than one incretin peptides on more than one polypeptide chains.
- each polypeptide chain of the incretin agent in Figure 11 has a signal peptide (“SP”), a GLP1 incretin peptide, a linker (GGGGS)3, and an Fc domain.
- SP signal peptide
- GLP1 incretin peptide GLP1 incretin peptide
- GGGGS linker
- Fc domain Fc domain
- One or both of the Fc domains contains “LS” mutations (M428L/N434S according to the EU numbering scheme) to extend half-life of the incretin agent.
- SP cleavage sites within the incretin agent are indicated with arrows.
- FIG 12 shows an exemplary incretin agent that may be encoded by one or more polyribonucleotides described herein, that includes more than one incretin peptide on more than one polypeptide chain.
- each polypeptide chain of the incretin agent has a signal peptide (SP), a GLP1 incretin peptide, a linker, a GIP peptide, a second linker (GGGGS)3, and an Fc domain.
- SP signal peptide
- GLP1 incretin peptide a linker
- GIP peptide GIP peptide
- Fc domain Fc domain
- Figure 13 shows an exemplary design of a two polyribonucleotides, each encoding a polypeptide chain that includes an incretin peptide fused to an Fc domain (top).
- each polypeptide chain incretin-Fc fusion
- SP signal peptide
- I: lx, I:2x, or I:4x incretin peptides fused to an Fc domain via a link (L2)
- L2 link
- each Fc domain has a modification that induces heterodimerization (e.g., a knob-in-hole mutation).
- Figure 14 shows an exemplary incretin agent that may be encoded by one or more polyribonucleotides described herein, that includes more than one incretin peptides on more than one polypeptide chains.
- each polypeptide chain of the incretin agent in Figure 14 has a signal peptide (SP), a GLP1 or GIP incretin peptide, a linker (GGGGS)3, and an Fc domain.
- SP signal peptide
- GLP1 or GIP incretin peptide a linker (GGGGS)3
- Fc domain Fc domain
- Fc domains contain “LS” mutations (M428L/N434S), “STR” mutations (L234S, L235T, and G236R mutations according to the EU numbering scheme) to silence Fc effector function, and “knob-in-hole” mutations to induce heterodimerization.
- the two polypeptide chains When the two polypeptide chains are expressed, they associate to form a heterodimeric incretin agent that contains two polypeptide chains with different incretin peptides. SP cleavage sites within the incretin agent are indicated with arrows.
- Figure 18 shows concentrations (pg/ml) of exemplary GLP1 incretin agents in supernatant of HEK29tl7 cells transfected with polyribonucleotides encoding for incretin agents containing a viral signal peptide (“viral SP”) or a husec signal peptide (“husec”) and codon-optimized using different strategies (“optl” vs. “optp”).
- viral SP viral signal peptide
- husec husec signal peptide
- Figure 19 shows concentrations (ng/ml) of exemplary GIP incretin agents in supernatant of HEK29tl7 cells transfected with polyribonucleotides encoding for incretin agents containing a viral signal peptide (“viral SP”) or a husec signal peptide (“husec”) and codon-optimized using different strategies (“optl” vs. “optp”).
- the specific incretin agents include: viral SP - GIP (1-42), husec - GIP (l-42)-A2G (optl), and GIP (l-42)-A2G (optp).
- Figure 20 shows a schematic of where the theoretical cleavage sites of the various signal peptides he within an incretin agent amino acid sequence.
- Figure 20 also indicates that the A8G mutation facilitates correct N-terminal processing of GLP1 incretin agents with husec signal peptides
- Figure 21 shows a schematic of where the theoretical cleavage sites of the various signal peptides he within an incretin agent amino acid sequence.
- Figure 21 also indicates that the A2G mutation facilitates correct N-terminal processing of GIP incretin agents with husec signal peptides.
- Figures 22A-B show a schematic of the GLP1R (A) and GIPR (B) overexpressing HEK293 reporter cells lines to be utilized in an assay to determine bioactivity of exemplary GLP1 and GIP incretin agents in Example 7.
- Figure 23 shows results from a bioactivity assay of exemplary GLP1 incretin agents. Specifically, results are expressed as fold induction over control samples.
- Figure 24 shows results from a bioactivity assay of exemplary GIP incretin agents. Specifically, results are expressed as fold induction over control samples.
- Figure 25 shows in vitro activity (GIP expression) for certain exemplary incretin agents tested.
- Figure 26 shows GIP bioactivity for certain exemplary GIP-containing incretin agents tested.
- Figure 27 shows in vitro activity (GLP1 expression) for certain exemplary incretin agents tested.
- Figure 28 shows GLP1 bioactivity for certain exemplary GLP1 -containing incretin agents tested.
- Figure 29 shows a comparison of GIP expression (A) and GIP bioactivity (B) in exemplary candidates with different signal peptides (husec vs. gDl).
- Figure 30 shows a comparison of GLP1 expression (A) and GLP1 bioactivity (B) in exemplary candidates with different signal peptides (husec vs. gDl).
- Figure 31 shows a comparison of GIP expression (A) and GIP bioactivity (B) with and without various half-life extension (HLE) moieties.
- Figure 32 shows a comparison of GLP1 expression (A) and GLP1 bioactivity (32B) with and without various half-life extension (HLE) moieties.
- Figure 33 shows a comparison of GIP expression (A) and GIP bioactivity (B) in exemplary incretin agents that contain both GIP and GLP1, where the order of the GIP and GLP1 peptides encoded by a single polyribonucleotide is varied.
- Figure 34 shows a comparison of GLP1 expression (A) and GLP1 bioactivity (B) in exemplary incretin agents that contain both GIP and GLP1, where the order of the GIP and GLP1 peptides encoded by a single polyribonucleotide is varied.
- agent may refer to a physical entity.
- an agent may be characterized by a particular feature and/or effect.
- therapeutic agent refers to a physical entity has a therapeutic effect and/or elicits a desired biological and/or pharmacological effect.
- an agent may be a compound, molecule, or entity of any chemical class including, for example, a small molecule, polypeptide, nucleic acid, saccharide, lipid, metal, or a combination or complex thereof.
- Aliphatic refers to a straight-chain (i.e., unbranched) or branched, substituted or unsubstituted hydrocarbon chain that is completely saturated or that contains one or more units of unsaturation, or a monocyclic hydrocarbon or bicyclic hydrocarbon that is completely saturated or that contains one or more units of unsaturation, but which is not aromatic (also referred to herein as “cycloaliphatic”), that has a single point or more than one points of attachment to the rest of the molecule.
- aliphatic groups contain 1-12 aliphatic carbon atoms. In some embodiments, aliphatic groups contain 1-6 aliphatic carbon atoms (e.g., Ci-e).
- aliphatic groups contain 1-5 aliphatic carbon atoms (e.g., C1-5). In other embodiments, aliphatic groups contain 1-4 aliphatic carbon atoms (e.g., C1-4). In still other embodiments, aliphatic groups contain 1-3 aliphatic carbon atoms (e.g., C1-3), and in yet other embodiments, aliphatic groups contain 1-2 aliphatic carbon atoms (e.g., C1-2). Suitable aliphatic groups include, but are not limited to, linear or branched, substituted or unsubstituted alkyl, alkenyl, or alkynyl groups and hybrids thereof. A preferred aliphatic group is C1-6 alkyl.
- Alkyl refers to a saturated, optionally substituted straight or branched chain hydrocarbon group having (unless otherwise specified) 1-12, 1-10, 1-8, 1-6, 1-4, 1-3, or 1-2 carbon atoms (e.g., C1-12, C1-10, C1-8, C1-6, C1-4, C1-3, or C1-2).
- exemplary alkyl groups include methyl, ethyl, propyl, butyl, pentyl, hexyl, and heptyl.
- Alkylene refers to a bivalent alkyl group. In some embodiments, “alkylene” is a bivalent straight or branched alkyl group. In some embodiments, an “alkylene chain” is a polymethylene group, i.e., -(CH2)n-, wherein n is a positive integer, e.g., from 1 to 6, from 1 to 4, from 1 to 3, from 1 to 2, or from 2 to 3.
- An optionally substituted alkylene chain is a polymethylene group in which one or more methylene hydrogen atoms is optionally replaced with a substituent. Suitable substituents include those described below for a substituted aliphatic group and also include those described in the specification herein.
- two substituents of the alkylene group may be taken together to form a ring system.
- two substituents can be taken together to form a 3 - to 7-membered ring.
- the substituents can be on the same or different atoms.
- the suffix “-ene” or “-enyl” when appended to certain groups herein are intended to refer to a bifunctional moiety of said group.
- “-ene” or “- enyl”, when appended to “cyclopropyl” becomes “cyclopr is intended to refer to a bifunctional cyclopropyl group, e.
- Alkenyl refers to an optionally substituted straight or branched chain or cyclic hydrocarbon group having at least one double bond and having (unless otherwise specified) 2-12, 2-10, 2-8, 2-6, 2-4, or 2- 3 carbon atoms (e.g., C2-12, C2-10, C2-8, C2-6, C2-4, or C2-3).
- Exemplary alkenyl groups include ethenyl, propenyl, butenyl, pentenyl, hexenyl, and heptenyl.
- cycloalkenyl refers to an optionally substituted non-aromatic monocyclic or multicyclic ring system containing at least one carbon-carbon double bond and having about 3 to about 10 carbon atoms.
- Exemplary monocyclic cycloalkenyl rings include cyclopentenyl, cyclohexenyl, and cycloheptenyl.
- Alkynyl refers to an optionally substituted straight or branched chain hydrocarbon group having at least one triple bond and having (unless otherwise specified) 2-12, 2-10, 2-8, 2-6, 2-4, or 2-3 carbon atoms (e.g., C2-12, C2-10, C2-8, C2-6, C2-4, or C2-3).
- exemplary alkynyl groups include ethynyl, propynyl, butynyl, pentynyl, hexynyl, and heptynyl.
- amino acid refers to a compound and/or substance that can be, is, or has been incorporated into a polypeptide chain, e.g., through formation of one or more peptide bonds.
- an amino acid has the general structure H2N-C(H)(R)-COOH.
- an amino acid is a naturally-occurring amino acid.
- an amino acid is a non-natural amino acid; in some embodiments, an amino acid is a D-amino acid; in some embodiments, an amino acid is an L-amino acid.
- Standard amino acid refers to any of the twenty standard L-amino acids commonly found in naturally occurring peptides.
- Nonstandard amino acid refers to any amino acid, other than the standard amino acids, regardless of whether it is prepared synthetically or obtained from a natural source.
- an amino acid, including a carboxy- and/or amino-terminal amino acid in a polypeptide can contain a structural modification as compared with the general structure above.
- an amino acid may be modified by methylation, amidation, acetylation, pegylation, glycosylation, phosphorylation, and/or substitution (e.g., of the amino group, the carboxylic acid group, one or more protons, and/or the hydroxyl group) as compared with the general structure.
- such modification may, for example, alter the circulating half-life of a polypeptide containing the modified amino acid as compared with one containing an otherwise identical unmodified amino acid.
- such modification does not significantly alter a relevant activity of a polypeptide containing the modified amino acid, as compared with one containing an otherwise identical unmodified amino acid.
- the term “amino acid” may be used to refer to a free amino acid; in some embodiments it may be used to refer to an amino acid residue of a polypeptide.
- Aryl refers to monocyclic and bicyclic ring systems having a total of six to fourteen ring members (e.g., C6-C14), wherein at least one ring in the system is aromatic and wherein each ring in the system contains three to seven ring members. In some embodiments, an “aryl” group contains between six and twelve total ring members (e.g., C6- CI2). The term “aryl” may be used interchangeably with the term “aryl ring”. In certain embodiments, “aryl” refers to an aromatic ring system which includes, but not limited to, phenyl, biphenyl, naphthyl, anthracyl and the like, which may bear one or more substituents.
- aryl groups are hydrocarbons.
- an “aryl” ring system is an aromatic ring (e.g., phenyl) that is fused to a non-aromatic ring (e.g., cycloalkyl). Examples of aryl rings include that are fused include
- Two events or entities are “associated” with one another, as that term is used herein, if the presence, level, degree, type and/or form of one is correlated with that of the other.
- a particular entity e.g., polypeptide, genetic signature, metabolite, microbe, etc.
- a particular entity e.g., polypeptide, genetic signature, metabolite, microbe, etc.
- two or more entities are physically “associated” with one another if they interact, directly or indirectly, so that they are and/or remain in physical proximity with one another.
- two or more entities that are physically associated with one another are covalently linked to one another; in some embodiments, two or more entities that are physically associated with one another are not covalently linked to one another but are non-covalently associated, for example by means of hydrogen bonds, van der Waals interaction, hydrophobic interactions, magnetism, and combinations thereof.
- Co-administration refers to use of a composition (e.g., a pharmaceutical composition) described herein and one or more additional therapeutic agents.
- one or more additional therapeutic agents comprises at least one polyribonucleotide encoding another therapeutic agent (e.g., an incretin agent).
- the combined use of a composition (e.g., a pharmaceutical composition) described herein and an additional therapeutic agent may be performed concurrently or separately (e.g., sequentially in any order).
- a composition e.g., a pharmaceutical composition described herein and an additional therapeutic agent may be combined in one pharmaceutically-acceptable excipient, or they may be placed in separate excipient and delivered to a target cell or administered to a subject at different times.
- a composition e.g., a pharmaceutical composition
- an additional therapeutic agent may be delivered or administered sufficiently close in time that there is at least some temporal overlap in biological effect(s) generated by each on a target cell or a subject being treated.
- Combination therapy refers to those situations in which a subject is simultaneously exposed to two or more therapeutic regimens (e.g., two or more therapeutic agents (e.g., two or more incretin agents)).
- the two or more regimens may be administered simultaneously; in some embodiments, such regimens may be administered sequentially (e.g., all “doses” of a first regimen are administered prior to administration of any doses of a second regimen); in some embodiments, such agents are administered in overlapping dosing regimens.
- administration of combination therapy may involve administration of one or more agent(s) or modality(ies) to a subject receiving the other agent(s) or modality(ies) in the combination.
- combination therapy does not require that individual agents be administered together in a single composition (or even necessarily at the same time), although in some embodiments, two or more agents, or active moieties thereof, may be administered together in a combination composition.
- a combination therapy comprises polyribonucleotides encoding two or more incretin agents.
- Comparable refers to two or more agents, entities, situations, sets of conditions, etc., that may not be identical to one another but that are sufficiently similar to permit comparison there between so that one skilled in the art will appreciate that conclusions may reasonably be drawn based on differences or similarities observed.
- comparable sets of conditions, circumstances, individuals, or populations are characterized by a plurality of substantially identical features and one or a small number of varied features.
- the term “corresponding to” refers to a relationship between two or more entities.
- the term “corresponding to” may be used to designate the position/identity of a structural element in a compound or composition relative to another compound or composition (e.g., to an appropriate reference compound or composition).
- a monomeric residue in a polymer e.g., an amino acid residue in a polypeptide or a nucleic acid residue in a polynucleotide
- a residue in an appropriate reference polymer may be identified as “corresponding to” a residue in an appropriate reference polymer.
- residues in a polypeptide are often designated using a canonical numbering system based on a reference related polypeptide, so that an amino acid “corresponding to” a residue at position 190, for example, need not actually be the 190 th amino acid in a particular amino acid chain but rather corresponds to the residue found at 190 in the reference polypeptide; those of ordinary skill in the art readily appreciate how to identify “corresponding” amino acids.
- sequence alignment strategies including software programs such as, for example, BLAST, CS-BLAST, CUSASW++, DIAMOND, FASTA, GGSEARCH/GLSEARCH, Genoogle, HMMER, HHpred/HHsearch, IDF, Infernal, KLAST, USEARCH, parasail, PSI-BLAST, PSI-Search, ScalaBLAST, Sequilab, SAM, SSEARCH, SWAPHI, SWAPHI-LS, SWIMM, or SWIPE that can be utilized, for example, to identify “corresponding” residues in polypeptides and/or nucleic acids in accordance with the present disclosure.
- software programs such as, for example, BLAST, CS-BLAST, CUSASW++, DIAMOND, FASTA, GGSEARCH/GLSEARCH, Genoogle, HMMER, HHpred/HHsearch, IDF, Infernal, KLAST, USEARCH, parasail, PSI-BLAST, PSI-Search, Scala
- corresponding to may be used to describe an event or entity that shares a relevant similarity with another event or entity (e.g., an appropriate reference event or entity).
- a gene or protein in one organism may be described as “corresponding to” a gene or protein from another organism in order to indicate, in some embodiments, that it plays an analogous role or performs an analogous function and/or that it shows a particular degree of sequence identity or homology, or shares a particular characteristic sequence element.
- Cycloaliphatic refers to a monocyclic C3-8 hydrocarbon or a bicyclic Ce-io hydrocarbon that is completely saturated or that contains one or more units of unsaturation, but which is not aromatic, that has a single point or more than one points of attachment to the rest of the molecule.
- Cycloalkyl refers to an optionally substituted saturated ring monocyclic or polycyclic system of about 3 to about 10 ring carbon atoms.
- Exemplary monocyclic cycloalkyl rings include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, and cycloheptyl.
- amino acid sequence “derived from” a designated amino acid sequence (peptide or polypeptide) “derived from” a designated amino acid sequence (peptide or polypeptide), refers to a structural analogue of a designated amino acid sequence.
- an amino acid sequence which is derived from a particular amino acid sequence has an amino acid sequence that is identical, essentially identical or homologous to that particular sequence or a fragment thereof.
- Amino acid sequences derived from a particular amino acid sequence may be variants of that particular sequence or a fragment thereof.
- incretin agents utilized according to the present disclosure may include amino acid sequences derived from two or more incretin agents, e.g., two or more naturally produced incretins.
- Detecting is used broadly herein to include appropriate means of determining the presence or absence of an entity of interest or any form of measurement of an entity of interest in a sample. Thus, “detecting” may include determining, measuring, assessing, or assaying the presence or absence, level, amount, and/or location of an entity of interest. Quantitative and qualitative determinations, measurements or assessments are included, including semi-quantitative. Such determinations, measurements or assessments may be relative, for example when an entity of interest is being detected relative to a control reference, or absolute. As such, the term “quantifying” when used in the context of quantifying an entity of interest can refer to absolute or to relative quantification.
- Absolute quantification may be accomplished by correlating a detected level of an entity of interest to known control standards (e.g., through generation of a standard curve).
- relative quantification can be accomplished by comparison of detected levels or amounts between two or more different entities of interest to provide a relative quantification of each of the two or more different entities of interest, i.e., relative to each other.
- Dosing regimen may be used to refer to a set of unit doses (typically more than one) that are administered individually to a subject, typically separated by periods of time.
- a given therapeutic agent has a recommended dosing regimen, which may involve one or more doses.
- Encode refers to sequence information of a first molecule that guides production of a second molecule having a defined sequence of nucleotides (e.g., a polyribonucleotide) or a defined sequence of amino acids.
- a DNA molecule can encode an RNA molecule (e.g., by a transcription process that includes a DNA-dependent RNA polymerase enzyme).
- An RNA molecule can encode a polypeptide (e.g., by a translation process).
- a gene, a cDNA, or an RNA molecule encodes a polypeptide if transcription and translation of RNA corresponding to that gene produces the polypeptide in a cell or other biological system.
- a coding region of a polyribonucleotide encoding a target antigen refers to a coding strand, the nucleotide sequence of which is identical to the polyribonucleotide sequence of such a target antigen.
- a coding region of a polyribonucleotide encoding a target antigen refers to a non-coding strand of such a target antigen, which may be used as a template for transcription of a gene or cDNA.
- Engineered refers to the aspect of having been manipulated by the hand of man.
- a polynucleotide is considered to be “engineered” when two or more sequences that are not linked together in that order in nature are manipulated by the hand of man to be directly linked to one another in the engineered polynucleotide and/or when a particular residue in a polynucleotide is non-naturally occurring and/or is caused through action of the hand of man to be linked with an entity or moiety with which it is not linked in nature.
- a gene product can be a transcript, e.g., a polyribonucleotide as provided herein.
- a gene product can be a polypeptide.
- expression of a nucleic acid sequence involves one or more of the following: (1) production of an RNA template from a DNA sequence (e.g., by transcription); (2) processing of an RNA transcript (e.g., by splicing, editing, etc.); (3) translation of an RNA into a polypeptide or protein; and/or (4) post-translational modification of a polypeptide or protein.
- Heteroaliphatic denotes an optionally substituted hydrocarbon moiety having, in addition to carbon atoms, from one to five heteroatoms, that may be straight-chain (i.e., unbranched), branched, or cyclic (“heterocyclic”) and may be completely saturated or may contain one or more units of unsaturation, but which is not aromatic.
- heteroatom refers to nitrogen, oxygen, or sulfur, and includes any oxidized form of nitrogen or sulfur, and any quatemized form of a basic nitrogen.
- nitrogen also includes a substituted nitrogen.
- heteroaliphatic groups contain 1-10 carbon atoms wherein 1-3 carbon atoms are optionally and independently replaced with heteroatoms selected from oxygen, nitrogen, and sulfur. In some embodiments, heteroaliphatic groups contain 1-4 carbon atoms, wherein 1-2 carbon atoms are optionally and independently replaced with heteroatoms selected from oxygen, nitrogen, and sulfur. In yet other embodiments, heteroaliphatic groups contain 1-3 carbon atoms, wherein 1 carbon atom is optionally and independently replaced with a heteroatom selected from oxygen, nitrogen, and sulfur. Suitable heteroaliphatic groups include, but are not limited to, linear or branched, heteroalkyl, heteroalkenyl, and heteroalkynyl groups.
- a 1- to 10 atom heteroaliphatic group includes the following exemplary groups: -O-CH3, -CH2-O-CH3, -O- CH2-CH2-O-CH2-CH2-O-CH3, and the like.
- Heteroaryl The terms “heteroaryl” and “heteroar-”, used alone or as part of a larger moiety, e.g., “heteroaralkyl”, or “heteroaralkoxy”, refer to monocyclic or bicyclic ring groups having 5 to 10 ring atoms (e.g., 5- to 6-membered monocyclic heteroaryl or 9- to 10- membered bicyclic heteroaryl); having 6, 10, or 14 n-electrons shared in a cyclic array; and having, in addition to carbon atoms, from one to five heteroatoms.
- Heteroaryl groups include, without limitation, thienyl, furanyl, pyrrolyl, imidazolyl, pyrazolyl, triazolyl, tetrazolyl, oxazolyl, isoxazolyl, oxadiazolyl, thiazolyl, isothiazolyl, thiadiazolyl, pyridyl, pyridazinyl, pyrimidinyl, pyrazinyl, indolizinyl, purinyl, naphthyridinyl, pteridinyl, imidazo[l,2- a]pyrimidinyl, imidazo[l,2-a]pyridyl, imidazo[4,5-b]pyridyl, imidazo[4,5-c]pyridyl, pyrrolopyridyl, pyrrolopyrazinyl, thienopyrimidinyl, triazolopyridyl, and benzois
- heteroaryl and “heteroar-”, as used herein, also include groups in which a heteroaromatic ring is fused to one or more aryl, cycloaliphatic, or heterocyclyl rings, where the radical or point of attachment is on the heteroaromatic ring (i.e., a bicyclic heteroaryl ring having 1 to 3 heteroatoms).
- Nonlimiting examples include indolyl, isoindolyl, benzothienyl, benzofuranyl, dibenzofuranyl, indazolyl, benzimidazolyl, benzotriazolyl, benzothiazolyl, benzothiadiazolyl, benzoxazolyl, quinolyl, isoquinolyl, cinnolinyl, phthalazinyl, quinazolinyl, quinoxalinyl, 4H- quinolizinyl.
- heteroaryl may be used interchangeably with the terms “heteroaryl ring”, “heteroaryl group”, or “heteroaromatic”, any of which terms include rings that are optionally substituted.
- Heteroatom refers to nitrogen, oxygen, or sulfur, and includes any oxidized form of nitrogen or sulfur, and any quatemized form of a basic nitrogen.
- Heterocycle As used herein, the terms “heterocycle”, “heterocyclyl”, “heterocyclic radical”, and “heterocyclic ring” are used interchangeably and refer to a stable 3- to 8-membered monocyclic, a 6- to 10-membered bicyclic, or a 10- to 16-membered polycyclic heterocyclic moiety that is either saturated or partially unsaturated, and having, in addition to carbon atoms, one or more, such as one to four, heteroatoms, as defined above.
- nitrogen includes a substituted nitrogen.
- the nitrogen may be N (as in 3,4- dihydro-2H-pyrrolyl), NH (as in pyrrolidinyl), or NR + (as in N-substituted pyrrolidinyl).
- a heterocyclic ring can be attached to its pendant group at any heteroatom or carbon atom that results in a stable structure and any of the ring atoms can be optionally substituted.
- saturated or partially unsaturated heterocyclic radicals include, without limitation, azetidinyl, oxetanyl, tetrahydrofuranyl, tetrahydrothienyl, pyrrolidinyl, piperidinyl, decahydroquinolinyl, oxazolidinyl, piperazinyl, dioxanyl, dioxolanyl, diazepinyl, oxazepinyl, thiazepinyl, morpholinyl, and thiamorpholinyl.
- a heterocyclyl group may be mono-, bi-, tri-, or polycyclic, preferably mono-, bi-, or tricyclic, more preferably mono- or bicyclic.
- a bicyclic heterocyclic ring also includes groups in which the heterocyclic ring is fused to one or more aryl rings.
- Exemplary bicyclic heterocyclic groups include indolinyl, isoindolinyl, benzodioxolyl, 1,3-dihydroisobenzofuranyl, 2,3-dihydrobenzofuranyl, and tetrahydroquinolinyl.
- a bicyclic heterocyclic ring can also be a spirocyclic ring system (e.g., 7- to 11 -membered spirocyclic fused heterocyclic ring having, in addition to carbon atoms, one or more heteroatoms as defined above (e.g., one, two, three or four heteroatoms)).
- a bicyclic heterocyclic ring can also be a bridged ring system (e.g., 7- to 11-membered bridged heterocyclic ring having one, two, or three bridging atoms.
- homolog refers to the overall relatedness between polynucleotide molecules (e.g., DNA molecules and/or RNA molecules) and/or between polypeptide molecules.
- polynucleotide molecules e.g., DNA molecules and/or RNA molecules
- polypeptide molecules are considered to be “homologous” to one another if their sequences are at least 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, or 99% identical.
- polynucleotide molecules e.g., DNA molecules and/or RNA molecules
- polypeptide molecules are considered to be “homologous” to one another if their sequences are at least 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, or 99% similar (e.g., containing residues with related chemical properties at corresponding positions).
- certain amino acids are typically classified as similar to one another as “hydrophobic” or “hydrophilic” amino acids, and/or as having “polar” or “non-polar” side chains.
- Identity refers to the overall relatedness between polynucleotide molecules (e.g., DNA molecules and/or RNA molecules) and/or between polypeptide molecules.
- polynucleotide molecules e.g., DNA molecules and/or RNA molecules
- polypeptide molecules are considered to be “substantially identical” to one another if their sequences are at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, or 99% identical.
- Calculation of the percent identity of two nucleic acid or polypeptide sequences can be performed by aligning the two sequences for optimal comparison purposes (e.g., gaps can be introduced in one or both of a first and a second sequence for optimal alignment and non-identical sequences can be disregarded for comparison purposes).
- the length of a sequence aligned for comparison purposes is at least 30%, at least 40%, at least 50%, at least 60%, at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, at least 95%, at least 96%, at least 97%, at least 98%, at least 99%, or substantially 100% of the length of a reference sequence.
- the nucleotides at corresponding positions are then compared.
- the percent identity between the two sequences is a function of the number of identical positions shared by the sequences, taking into account the number of gaps, and the length of each gap, which needs to be introduced for optimal alignment of the two sequences.
- the comparison of sequences and determination of percent identity between two sequences can be accomplished using a mathematical algorithm. For example, the percent identity between two nucleotide sequences can be determined using the algorithm of Meyers and Miller, 1989, which has been incorporated into the ALIGN program (version 2.0).
- nucleic acid sequence comparisons made with the ALIGN program use a PAM120 weight residue table, a gap length penalty of 12 and a gap penalty of 4.
- the percent identity between two nucleotide sequences can, alternatively, be determined using the GAP program in the GCG software package using an NWSgapdna.CMP matrix.
- Increased, Induced, or Reduced indicate values that are relative to a comparable reference measurement.
- an assessed value achieved with a provided composition e.g., a pharmaceutical composition
- an assessed value achieved in a subject may be “increased” relative to that obtained in the same subject under different conditions (e.g., prior to or after an event; or presence or absence of an event such as administration of a composition (e.g., a pharmaceutical composition) as described herein, or in a different, comparable subject (e.g., in a comparable subject that differs from the subject of interest in prior exposure to a condition, e.g., absence of administration of a composition (e.g., a pharmaceutical composition) as described herein.).
- comparative terms refer to statistically relevant differences (e.g., that are of a prevalence and/or magnitude sufficient to achieve statistical relevance).
- the term “reduced” or equivalent terms refers to a reduction in the level of an assessed value by at least 5%, at least 10%, at least 20%, at least 50%, at least 75% or higher, as compared to a comparable reference. In some embodiments, the term “reduced” or equivalent terms refers to a complete or essentially complete inhibition, i.e., a reduction to zero or essentially to zero.
- the term “increased” or “induced” refers to an increase in the level of an assessed value by at least 10%, at least 20%, at least 30%, at least 40%, at least 50%, at least 80%, at least 100%, at least 200%, at least 500%, or higher, as compared to a comparable reference.
- in order refers to the order of features from 5' to 3' along the polynucleotide or polyribonucleotide.
- in order refers to the order of features moving from the N-terminal-most of the features to the C-terminal-most of the features along the polypeptide. “In order” does not mean that no additional features can be present among the listed features.
- Ionizable refers to a compound or group or atom that is charged at a certain pH.
- an ionizable amino lipid such a lipid or a function group or atom thereof bears a positive charge at a certain pH.
- an ionizable amino lipid is positively charged at an acidic pH.
- an ionizable amino lipid is predominately neutral at physiological pH values, e.g., in some embodiments about 7.0-7.4, but becomes positively charged at lower pH values.
- an ionizable amino lipid may have a pKa within a range of about 5 to about 7.
- Isolated means altered or removed from the natural state.
- a nucleic acid or a peptide naturally present in a living animal is not “isolated,” but the same nucleic acid or peptide partially or completely separated from the coexisting materials of its natural state is “isolated.”
- An isolated nucleic acid or protein can exist in substantially purified form, or can exist in a non-native environment such as, for example, a host cell.
- Lipid As used herein, the terms “lipid” and “lipid-like material” are broadly defined as molecules which comprise one or more hydrophobic moieties or groups and optionally also one or more hydrophilic moieties or groups. Molecules comprising hydrophobic moieties and hydrophilic moieties are also typically denoted as amphiphiles.
- RNA lipid nanoparticle refers to a nanoparticle comprising at least one lipid and RNA molecule(s), e.g., one or more polyribonucleotides as provided herein.
- an RNA lipid nanoparticle comprises at least one cationic amino lipid.
- an RNA lipid nanoparticle comprises at least one cationic amino lipid, at least one helper lipid, and at least one polymer- conjugated lipid (e.g., PEG-conjugated lipid).
- RNA lipid nanoparticles as described herein can have an average size (e.g., Z-average) of about 100 nm to 1000 nm, or about 200 nm to 900 nm, or about 200 nm to 800 nm, or about 250 nm to about 700 nm.
- Z-average average size
- RNA lipid nanoparticles can have a particle size (e.g., Z-average) of about 30 nm to about 200 nm, or about 30 nm to about 150 nm, about 40 nm to about 150 nm, about 50 nm to about 150 nm, about 60 nm to about 130 nm, about 70 nm to about 110 nm, about 70 nm to about 100 nm, about 80 nm to about 100 nm, about 90 nm to about 100 nm, about 70 to about 90 nm, about 80 nm to about
- a particle size e.g., Z-average
- RNA lipid nanoparticles may be prepared by mixing lipids with RNA molecules described herein.
- Neutralization refers to an event in which binding agents such as antibodies bind to a biological active site of a virus such as a receptor binding protein, thereby inhibiting the parasitic infection of cells. In some embodiments, the term “neutralization” refers to an event in which binding agents eliminate or significantly reduce ability of infecting cells.
- nucleic acid refers to a polymer of at least 10 nucleotides or more.
- a nucleic acid is or comprises DNA.
- a nucleic acid is or comprises RNA.
- a nucleic acid is or comprises peptide nucleic acid (PNA).
- PNA peptide nucleic acid
- a nucleic acid is or comprises a single stranded nucleic acid.
- a nucleic acid is or comprises a double-stranded nucleic acid.
- a nucleic acid comprises both single and double-stranded portions.
- a nucleic acid comprises a backbone that comprises one or more phosphodiester linkages. In some embodiments, a nucleic acid comprises a backbone that comprises both phosphodiester and non-phosphodiester linkages. For example, in some embodiments, a nucleic acid may comprise a backbone that comprises one or more phosphorothioate or 5'-N-phosphoramidite linkages and/or one or more peptide bonds, e.g., as in a “peptide nucleic acid”.
- a nucleic acid comprises one or more, or all, natural residues (e.g., adenine, cytosine, deoxyadenosine, deoxycytidine, deoxyguanosine, deoxythymidine, guanine, thymine, uracil). In some embodiments, a nucleic acid comprises on or more, or all, non-natural residues.
- natural residues e.g., adenine, cytosine, deoxyadenosine, deoxycytidine, deoxyguanosine, deoxythymidine, guanine, thymine, uracil.
- a non-natural residue comprises a nucleoside analog (e.g., 2-aminoadenosine, 2-thiothymidine, inosine, pyrrolo-pyrimidine, 3 -methyl adenosine, 5 -methylcytidine, C-5 propynyl-cytidine, C-5 propynyl-uridine, 2-aminoadenosine, C5 -bromouridine, C5-fluorouridine, C5 -iodouridine, C5-propynyl -uridine, C5 -propynyl-cytidine, C5 -methylcytidine, 2-aminoadenosine, 7- deazaadenosine, 7-deazaguanosine, 8-oxoadenosine, 8-oxoguanosine, 6-O-methylguanine, 2- thiocytidine, methylated bases, intercalated bases, and combinations
- a non-natural residue comprises one or more modified sugars (e.g., 2'- fluororibose, ribose, 2'-deoxyribose, arabinose, and hexose) as compared to those in natural residues.
- a nucleic acid has a nucleotide sequence that encodes a functional gene product such as an RNA or polypeptide.
- a nucleic acid has a nucleotide sequence that comprises one or more introns.
- a nucleic acid may be prepared by isolation from a natural source, enzymatic synthesis (e.g., by polymerization based on a complementary template, e.g., in vivo or in vitro), reproduction in a recombinant cell or system, or chemical synthesis.
- a nucleic acid is at least 3, 4, 5, 6, 7, 8, 9, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 1 10, 120, 130, 140, 150, 160, 170, 180, 190, 20, 225, 250, 275, 300, 325, 350, 375, 400, 425, 450, 475, 500, 600, 700, 800, 900, 1000, 1500, 2000, 2500, 3000, 3500, 4000, 4500, 5000, 5500, 6000, 6500, 7000, 7500, 8000, 8500, 9000, 9500, 10,000, 10,500, 11,000, 11,500, 12,000, 12,500, 13,000, 13,500, 14,000, 14,500, 15,000, 15,500, 16,000, 16,500, 17,000, 17,500, 18,000, 18,500, 19,000, 19,500, or 20,000 or more residues or nucleotides long.
- compositions comprising: a desired reaction or a desired effect alone or together with further doses.
- a desired reaction in some embodiments relates to inhibition of the course of the disease (e.g., obesity). In some embodiments, such inhibition may comprise slowing down the progress of a disease (e.g., obesity) and/or interrupting or reversing the progress of the disease (e.g., obesity).
- a desired reaction in a treatment of a disease may be or comprise delay or prevention of the onset of a disease (e.g., obesity) or a condition (e.g., a condition associated with obesity).
- an effective amount of a composition (e.g., a pharmaceutical composition) described herein will depend, for example, on disease (e.g., obesity) or a condition (e.g., a condition associated with obesity) to be treated, the severity of such a disease (e.g., obesity) or a condition (e.g., a condition associated with obesity), individual parameters of the patient, including, e.g., age, physiological condition, size and weight, the duration of treatment, the type of an accompanying therapy (if present), the specific route of administration and similar factors. Accordingly, doses of a composition (e.g., a pharmaceutical composition) described herein may depend on various of such parameters. In the case that a reaction in a patient is insufficient with an initial dose, higher doses (or effectively higher doses achieved by a different, more localized route of administration) may be used.
- Polypeptide refers to a polymeric chain of amino acids.
- a polypeptide has an amino acid sequence that occurs in nature.
- a polypeptide has an amino acid sequence that does not occur in nature.
- a polypeptide has an amino acid sequence that is engineered in that it is designed and/or produced through action of the hand of man.
- a polypeptide may comprise or consist of natural amino acids, non-natural amino acids, or both.
- a polypeptide may comprise or consist of only natural amino acids or only non-natural amino acids.
- a polypeptide may comprise D-amino acids, L-amino acids, or both. In some embodiments, a polypeptide may comprise only D-amino acids. In some embodiments, a polypeptide may comprise only L-amino acids. In some embodiments, a polypeptide may include one or more pendant groups or other modifications, e.g., modifying or attached to one or more amino acid side chains, at the polypeptide’s N-terminus, at the polypeptide’s C-terminus, or any combination thereof. In some embodiments, such pendant groups or modifications comprise acetylation, amidation, lipidation, methylation, pegylation, etc., including combinations thereof.
- a polypeptide may be cyclic, and/or may comprise a cyclic portion. In some embodiments, a polypeptide is not cyclic and/or does not comprise any cyclic portion. In some embodiments, a polypeptide is linear. In some embodiments, a polypeptide may be or comprise a stapled polypeptide. In some embodiments, the term “polypeptide” may be appended to a name of a reference polypeptide, activity, or structure; in such instances it is used herein to refer to polypeptides that share the relevant activity or structure and thus can be considered to be members of the same class or family of polypeptides.
- exemplary polypeptides within the class whose amino acid sequences and/or functions are known; in some embodiments, such exemplary polypeptides are reference polypeptides for the polypeptide class or family.
- a member of a polypeptide class or family shows significant sequence homology or identity with, shares a common sequence motif (e.g., a characteristic sequence element) with, and/or shares a common activity (in some embodiments at a comparable level or within a designated range) with a reference polypeptide of the class; in some embodiments with all polypeptides within the class).
- a member polypeptide shows an overall degree of sequence homology or identity with a reference polypeptide that is at least about 30-40%, and is often greater than about 50%, 60%, 70%, 80%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% or more and/or includes at least one region (e.g., a conserved region that may in some embodiments be or comprise a characteristic sequence element) that shows very high sequence identity, often greater than 90% or even 95%, 96%, 97%, 98%, or 99%.
- a conserved region that may in some embodiments be or comprise a characteristic sequence element
- Such a conserved region usually encompasses at least 3-4 and often up to 35 or more amino acids; in some embodiments, a conserved region encompasses at least one stretch of at least 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35 or more contiguous amino acids.
- a relevant polypeptide may comprise or consist of a fragment of a parent polypeptide.
- Prevent As used herein, the term “prevent” or “prevention” when used in connection with the occurrence of a disease, disorder, and/or condition, refers to reducing the risk of developing the disease, disorder and/or condition and/or to delaying onset of one or more characteristics or symptoms of the disease, disorder or condition. Prevention may be considered complete when onset of a disease, disorder or condition has been delayed for a predefined period of time.
- reference describes a standard or control relative to which a comparison is performed.
- an agent, animal, individual, population, sample, sequence or value of interest is compared with a reference or control agent, animal, individual, population, sample, sequence or value.
- a reference or control is tested and/or determined substantially simultaneously with the testing or determination of interest.
- a reference or control is a historical reference or control, optionally embodied in a tangible medium.
- a reference or control is determined or characterized under comparable conditions or circumstances to those under assessment.
- RNA Ribonucleic acid
- RNA Polyribonucleotide
- an RNA is single stranded.
- an RNA is double stranded.
- an RNA comprises both single and double stranded portions.
- an RNA can comprise a backbone structure as described in the definition of “Nucleic acid/ Polynucleotide” above.
- An RNA can be a regulatory RNA (e.g., siRNA, microRNA, etc.), or a messenger RNA (mRNA).
- an RNA is a mRNA. In some embodiments, where an RNA is a mRNA, an RNA typically comprises at its 3' end a poly(A) region. In some embodiments, where an RNA is a mRNA, an RNA typically comprises at its 5' end an art-recognized cap structure, e.g., for recognizing and attachment of a mRNA to a ribosome to initiate translation. In some embodiments, a RNA is a synthetic RNA. Synthetic RNAs include RNAs that are synthesized in vitro (e.g., by enzymatic synthesis methods and/or by chemical synthesis methods).
- Ribonucleotide encompasses unmodified ribonucleotides and modified ribonucleotides.
- unmodified ribonucleotides include the purine bases adenine (A) and guanine (G), and the pyrimidine bases cytosine (C) and uracil (U).
- Modified ribonucleotides may include one or more modifications including, but not limited to, for example, (a) end modifications, e.g., 5' end modifications (e.g., phosphorylation, dephosphorylation, conjugation, inverted linkages, etc.), 3' end modifications (e.g., conjugation, inverted linkages, etc.), (b) base modifications, e.g. , replacement with modified bases, stabilizing bases, destabilizing bases, or bases that base pair with an expanded repertoire of partners, or conjugated bases, (c) sugar modifications (e.g., at the 2' position or 4' position) or replacement of the sugar, and (d) intemucleoside linkage modifications, including modification or replacement of the phosphodiester linkages.
- end modifications e.g., 5' end modifications (e.g., phosphorylation, dephosphorylation, conjugation, inverted linkages, etc.), 3' end modifications (e.g., conjugation, inverted linkages, etc.)
- base modifications
- risk of a disease, disorder, and/or condition refers to a likelihood that a particular individual will develop the disease, disorder, and/or condition. In some embodiments, risk is expressed as a percentage. In some embodiments, risk is expressed as a risk relative to a risk associated with a reference sample or group of reference samples. In some embodiments, a reference sample or group of reference samples have a known risk of a disease, disorder, condition and/or event. In some embodiments, a reference sample or group of reference samples are from individuals comparable to a particular individual.
- risk may reflect one or more genetic attributes, e.g., which may predispose an individual toward development (or not) of a particular disease, disorder and/or condition.
- risk may reflect one or more epigenetic events or attributes and/or one or more lifestyle or environmental events or attributes.
- an agent when used herein in reference to an agent having an activity, is understood by those skilled in the art to mean that the agent discriminates between potential target entities, states, or cells.
- an agent is said to bind “specifically” to its target if it binds preferentially with that target in the presence of one or more competing alternative targets.
- specific interaction is dependent upon the presence of a particular structural feature of the target entity (e.g., an epitope, a cleft, a binding site). It is to be understood that specificity need not be absolute.
- specificity may be evaluated relative to that of a target-binding moiety for one or more other potential target entities (e.g., competitors).
- specificity is evaluated relative to that of a reference specific binding moiety.
- specificity is evaluated relative to that of a reference non-specific binding moiety.
- Substituted or optionally substituted As described herein, compounds of the invention may contain “optionally substituted” moieties.
- the term “substituted,” whether preceded by the term “optionally” or not, means that one or more hydrogens of the designated moiety are replaced with a suitable substituent. “Substituted” applies to one or more hydrogens that are either explicit or implicit from the structure (e.g., refers to at least ).
- an “optionally substituted” group may have a suitable substituent at each substitutable position of the group, and when more than one position in any given structure may be substituted with more than one substituent selected from a specified group, the substituent may be either the same or different at every position.
- Combinations of substituents envisioned by this invention are preferably those that result in the formation of stable or chemically feasible compounds.
- stable refers to compounds that are not substantially altered when subjected to conditions to allow for their production, detection, and, in certain embodiments, their recovery, purification, and use for one or more of the purposes provided herein.
- Groups described as being “substituted” preferably have between 1 and 4 substituents, more preferably 1 or 2 substituents.
- Groups described as being “optionally substituted” may be unsubstituted or be “substituted” as described above.
- Suitable monovalent substituents on R° are independently halogen, -(CH2K2R 1 , -(haloR 1 ), -(CH 2 )o 2OH, -(CH 2 )o 2OR 1 , -(CH 2 )o 2 CH(OR')2, -OChaloR 1 ), -CN, -N 3 , -(CH 2 )o- 2 C(0)R 1 , -(CH 2 )o 2 C(O)OH, -(CH 2 )O 2C(O)OR‘, -(CH 2 )O 2SR 1 , -(CH 2 )o 2SH, -(CH 2 )o 2NH2, -(CH 2 )o 2NHR 1 , -(CH 2 )O-2NR 1 2, -NO2, -SiR* 3 , - OSiR'y -C(
- Suitable divalent substituents that are bound to vicinal substitutable carbons of an “optionally substituted” group include: -O(CR*2)2 3O-, wherein each independent occurrence of R* is selected from hydrogen, C1-6 aliphatic which may be substituted as defined below, or an unsubstituted 5-6-membered saturated, partially unsaturated, or aryl ring having 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur.
- Suitable substituents on the aliphatic group of R* include halogen, - R 1 , -(haloR 1 ), -OH, -OR 1 , -O(haloR'), -CN, -C(O)OH, -C(O)OR.
- each R 1 is unsubstituted or where preceded by “halo” is substituted only with one or more halogens, and is independently C1-4 aliphatic, -CH2PI1, -0(CH2)o iPh, or a 3- to 6-membered saturated, partially unsaturated, or aryl ring having 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur.
- Suitable substituents on a substitutable nitrogen of an “optionally substituted” group include -R ⁇ -NR ⁇ , -C(O)R ⁇ , -C(O)OR ⁇ , -C(0)C(0M -
- each R 1 ' is independently hydrogen, C1-6 aliphatic which may be substituted as defined below, unsubstituted -OPh, or an unsubstituted 3- to 6-membered saturated, partially unsaturated, or aryl ring having 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur, or, notwithstanding the definition above, two independent occurrences of R 1 ', taken together with their intervening atom(s) form an unsubstituted 3- to 12-membered saturated, partially unsaturated, or aryl mono- or bicyclic ring having 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur.
- Suitable substituents on the aliphatic group of R’ are independently halogen, - R 1 , -(haloR 1 ), -OH, -OR 1 , -O(haloR'), -CN, -C(O)OH, -C(O)OR‘, -NH 2 , -NHR 1 , -NR* 2 , or -NO2, wherein each R 1 is unsubstituted or where preceded by “halo” is substituted only with one or more halogens, and is independently C1-4 aliphatic, -CH2PI1, -0(CH2)o iPh, or a 3- to 6-membered saturated, partially unsaturated, or aryl ring having 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur.
- Subject refers to an organism to be administered with a composition described herein, e.g., for experimental, diagnostic, prophylactic, and/or therapeutic purposes. Typical subjects include animals (e.g., mammals such as mice, rats, rabbits, non-human primates, domestic pets, etc.) and humans. In some embodiments, a subject is a human subject. In some embodiments, a subject is suffering from a disease, disorder, or condition (e.g., obesity, a condition associated with obesity, etc.). In some embodiments, a subject is susceptible to a disease, disorder, or condition (e.g., obesity, a condition associated with obesity, etc.).
- a disease, disorder, or condition e.g., obesity, a condition associated with obesity, etc.
- a subject displays one or more symptoms or characteristics of a disease, disorder, or condition (e.g., obesity, a condition associated with obesity, etc.). In some embodiments, a subject displays one or more non-specific symptoms of a disease, disorder, or condition (e.g., obesity, a condition associated with obesity, etc.). In some embodiments, a subject does not display any symptom or characteristic of a disease, disorder, or condition (e.g., obesity, a condition associated with obesity, etc.). In some embodiments, a subject is someone with one or more features characteristic of susceptibility to or risk of a disease, disorder, or condition (e.g., obesity, a condition associated with obesity, etc.). In some embodiments, a subject is a patient. In some embodiments, a subject is an individual to whom diagnosis and/or therapy is and/or has been administered.
- Susceptible to An individual who is “susceptible to” a disease, disorder, and/or condition (e.g., obesity, a condition associated with obesity, etc.) is one who has a higher risk of developing the disease, disorder, and/or condition (e.g., obesity, a condition associated with obesity, etc.) than does a member of the general public.
- an individual who is susceptible to a disease, disorder and/or condition e.g., obesity, a condition associated with obesity, etc.
- an individual who is susceptible to a disease, disorder, and/or condition e.g., obesity, a condition associated with obesity, etc.
- will develop the disease, disorder, and/or condition e.g., obesity, a condition associated with obesity, etc.
- an individual who is susceptible to a disease, disorder, and/or condition e.g., obesity, a condition associated with obesity, etc.
- will not develop the disease, disorder, and/or condition e.g., obesity, a condition associated with obesity, etc.
- a therapeutic agent or therapy is any substance that can be used to alleviate, ameliorate, relieve, inhibit, prevent, delay onset of, reduce severity of, and/or reduce incidence of one or more symptoms or features of a disease, disorder, and/or condition (e.g., obesity, a condition associated with obesity, etc.).
- a therapeutic agent or therapy is a medical intervention (e.g., surgery, radiation, phototherapy) that can be performed to alleviate, relieve, inhibit, present, delay onset of, reduce severity of, and/or reduce incidence of one or more symptoms or features of a disease, disorder, and/or condition.
- a medical intervention e.g., surgery, radiation, phototherapy
- Treat refers to any method used to partially or completely alleviate, ameliorate, relieve, inhibit, prevent, delay onset of, reduce severity of, and/or reduce incidence of one or more symptoms or features of a disease, disorder, and/or condition (e.g., obesity, a condition associated with obesity, etc.). Treatment may be administered to a subject who does not exhibit signs of a disease, disorder, and/or condition (e.g., obesity, a condition associated with obesity, etc.).
- a disease, disorder, and/or condition e.g., obesity, a condition associated with obesity, etc.
- treatment may be administered to a subject who exhibits only early signs of the disease, disorder, and/or condition (e.g., obesity, a condition associated with obesity, etc.), for example for the purpose of decreasing the risk of developing pathology associated with the disease, disorder, and/or condition.
- treatment may be administered to a subject at a later-stage of disease, disorder, and/or condition (e.g., obesity, a condition associated with obesity, etc.).
- structures depicted herein are meant to include all stereoisomeric (e.g., enantiomeric or diastereomeric) forms of the structure, as well as all geometric or conformational isomeric forms of the structure.
- the R and S configurations of each stereocenter are contemplated as part of the disclosure. Therefore, single stereochemical isomers, as well as enantiomeric, diastereomeric, and geometric (or conformational) mixtures of provided compounds are within the scope of the disclosure.
- provided compounds show one or more stereoisomers of a compound, and unless otherwise indicated, represents each stereoisomer alone and/or as a mixture.
- all tautomeric forms of provided compounds are within the scope of the disclosure.
- structures depicted herein are meant to include compounds that differ only in the presence of one or more isotopically enriched atoms.
- compounds having the present structures including replacement of hydrogen by deuterium or tritium, or replacement of a carbon by 13C- or 14C-enriched carbon are within the scope of this disclosure.
- Incretins are peptide hormones that are released in the gastrointestinal (GI) tract in response to glucose consumption, which stimulates insulin secretion by the pancreas and decreases glucagon production, lowering blood sugar levels. Incretins exert their effects by binding to their respective receptors on pancreatic beta cells, leading to insulin release.
- Glucagon-like peptide-1 (GLP1) and glucose-dependent insulinotropic polypeptide (GIP) are two incretins that have been identified for their role in postprandial insulin secretion.
- GIP is largely responsible for insulin release in response to glucose intake.
- GLP1 stimulates satiety, slows gastric emptying, lowers glucagon secretion, and decreases food intake, resulting in weight loss. Agonizing GIP and GLP1 receptors has been shown to produce an additive effect when insulin is secreted. See Chim, USPharm. 2022; 47(10): 18-22, which is incorporated herein by reference in its entirety.
- incretins and incretin mimetics have the potential to treat various diseases including obesity, pre-diabetes, type 2 diabetes (T2D, with its complications), early type 1 diabetes (e.g., within 3 months after diagnosis of T1D), non-alcoholic fatty liver disease (NAFLD), non-alcoholic steatohepatitis (NASH), cardiovascular (CV) disease (e.g., characterized by a major cardiovascular event (MACE), including CV death, non-fatal myocardial infarction, non-fatal stroke, or heart failure with preserved ejection fraction (HFpEF)), renal disease, and elevated risk of premature mortality.
- MACE major cardiovascular event
- HFpEF heart failure with preserved ejection fraction
- Obesity is the most prevalent chronic disease worldwide, affecting approximately 650 million adults. Obesity is considered a starting point and critical contributor to pre-diabetes, type 2 diabetes (T2D, with its complications), non-alcoholic fatty liver disease (NAFLD), non-alcoholic steatohepatitis (NASH), cardiovascular and renal disease, and premature mortality. Obesity imposes a considerable economic burden with additional direct medical costs, productivity costs (absenteeism, presenteeism, disability support, premature mortality), transportation costs (including increased CO2 footprint), human capital accumulation costs (school absenteeism, highest educational degree achieved).
- productivity costs abenteeism, presenteeism, disability support, premature mortality
- transportation costs including increased CO2 footprint
- human capital accumulation costs school absenteeism, highest educational degree achieved.
- BMI>30kg/m 2 It is estimated that the number of obese (BMI>30kg/m 2 ) people will exceed one billion by 2030, about 10% of which will suffer from severe class III obesity (BMI>40kg/m 2 ). Half of all men living with obesity live in just nine countries: USA, China, India, Brazil, Mexico, Russia, Egypt, Germany and Turkey. Childhood obesity is also sharply on the rise worldwide.
- Obesity was declared a disease by the American Association of Clinical Endocrinologists (AACE) only in 2011, and is managed based on severity, starting with lifestyle/behavioral intervention, and increased physical activity, then pharmacotherapy, and finally bariatric surgery.
- AACE American Association of Clinical Endocrinologists
- GLP1 Glucagon-like peptide- 1 receptor agonists like Trulicity® (dulaglutide), Byetta® (exenatide), Ozempic®/Rybelsus® (semaglutide injectable/oral), Victoza® (liraglutide) and Suliqua® (lixisenatide, only in combination with insulin glargine), which are approved for lowering blood sugar in people living with T2D without the need to continuously check blood sugar levels.
- GLP1 Glucagon-like peptide- 1 receptor agonists like Trulicity® (dulaglutide), Byetta® (exenatide), Ozempic®/Rybelsus® (semaglutide injectable/oral), Victoza® (liraglutide) and Suliqua® (lixisenatide, only in combination with insulin glargine), which are approved for lowering blood sugar in people living with T2D without the need to continuously check blood sugar levels.
- An added benefit was the loss of body weight (
- GLP1 receptor agonists with GIP receptor agonists and/or glucagon (GCG) receptor agonists (dual/triple agonists), aiming at even better control of blood sugar and greater weight loss.
- Blood sugar control and weight loss was demonstrated with the GLP1/GCG receptor dual agonist SAR425899; however, the program was discontinued due to unacceptable gastrointestinal side effects in 2019.
- GLP1/GIP receptor dual agonist tirzepatide now in the market as Mounjaro®, was approved as an injectable medicine for adults with T2D used along with diet and exercise to improve blood sugar.
- GIP GIP’s function is to regulate energy balance through cell-surface receptor signaling in the brain and adipose tissue.
- the SURPASS-2 study demonstrated non-inferiority and superiority of tirzepatide against semaglutide in lowering blood sugar. Weight loss however was only a secondary endpoint.
- Table 1 Exemplary incretin mimetics for Treatment of Obesitv/T2D
- the present disclosure recognizes, among other things, the current problems in the market of incretin mimetics for the treatment of obesity, pre-diabetes, T2D, early T1D, NAFLD, NASH, cardiovascular disease, renal disease, and elevated risk of premature mortality, including but not limited to, limited supply, high price, lack of health insurance covering such treatments, frequent injections (e.g., once weekly), high injection volumes, and gastrointestinal side effects.
- the present disclosure provides, among other things, a more efficacious and cost effective way to use incretins and incretin mimetics for treatment of obesity, prediabetes, T2D, early T1D, NAFLD, NASH, cardiovascular disease, renal disease, and elevated risk of premature mortality through the delivery of incretins and incretin mimetics (collectively encompassed by the term “incretin agents”) encoded by one or more polyribonucleotides.
- polyribonucleotides encoding incretin agents provide for a therapeutic treatment of obesity, and/or pre-diabetes, T2D, early T1D, NAFLD, NASH, cardiovascular disease, renal disease, and elevated risk of premature mortality (e.g., diseases related to obesity), which have improved properties compared to known incretin mimetic therapies, including the need for fewer injections (no more than once-weekly injection), lower injection volumes (e.g., no more than 0.5 ml), and fewer or less severe side effects.
- polyribonucleotides for delivery of incretin agents provide for expression of incretin agents in a cell at therapeutically relevant levels, comparable to doses of current peptide-based therapies.
- RNA technologies as a modality to express incretin agents directly in a subject as a novel class of therapeutics that agonize GLP1, GIP, and/or GCG receptors to effectively treat a disease state such as obesity, pre-diabetes, T2D, early T1D, NAFLD, NASH, cardiovascular disease, renal disease, and/or elevated risk of premature mortality.
- a polyribonucleotide as described herein encodes an incretin agent found in nature, or a fragment or variant thereof.
- a polyribonucleotide as described herein encodes an incretin agent, or a fragment or variant thereof, that has been modified from its natural form.
- incretin agents refers to an agent that comprises an incretin or incretin mimetic (where incretins and incretin mimetics are collectively designated “incretin peptides” herein).
- exemplary incretins include GLP1, GIP, and GCG.
- Exemplary incretin mimetics are shown in, e.g., Table 1.
- an incretin agent is a biologically active portion or fragment of an incretin or incretin mimetic.
- an incretin agent comprises an incretin peptide that is part of a fusion.
- an incretin agent is an incretin peptide that is fused to another peptide moiety (e.g., a half-life extending (HLE) domain).
- HLE half-life extending
- an incretin agent comprises a GLP 1 receptor agonist such as GLP 1.
- an incretin agent comprises a GIP receptor agonist such as GIP.
- an incretin agent comprises a dual GIP and GLP1 receptor agonist.
- an incretin agent comprises a triple GIP, GLP1 and GCG receptor agonist.
- an incretin agent comprises a wild type (i.e., unmutated) incretin peptide sequence, or fragment thereof.
- an incretin agent comprises any one of the incretin peptides as represented in SEQ ID NOs: 5-15 and 62-64.
- an incretin agent comprises an incretin peptide, or fragment thereof with at least one mutated amino acid residue in comparison to a wild type reference sequence.
- a mutated amino acid residue comprises a substitution of a natural amino acid residue with another natural amino acid residue.
- an incretin agent comprises any one of the incretin peptides as represented in SEQ ID NOs: 5-10.
- a mutated amino acid residue may confer dual or triple agonizing properties to an incretin agent.
- one or more amino acid substitutions are introduced into a GLP1, GIP or GCG peptide sequence (e.g., as shown in SEQ ID NOs: 12-15 and 62-64) in order to confer binding properties to two or more of the GLP1, GIP and/or GCG receptors.
- an incretin agent has an amino acid sequence that is at least 85%, at least 90%, at least 95%, or 100% identical to any one of the incretin peptides detailed in Table 2.
- an incretin agent comprises any one of the incretin peptides detailed in Table 2 below, or combinations or variants thereof. Table 2: Exemplary Incretin Peptides (with mutations in bold)
- an incretin agent described herein includes a single incretin peptide (which configuration is referred to herein as “I: lx”) (see e.g., Figure 3).
- an incretin peptide is fused to another peptide (e.g., a half-life extending (HLE) domain) via a linker (see e.g., Figures 4-14).
- a linker contains at least one Gly (G) amino acid residue.
- Suitable linkers can be readily selected and can be of various lengths, such as from 1 amino acid (e.g., Gly) to 25 amino acids, from 2 amino acids to 15 amino acids, from 3 amino acids to 12 amino acids, including 4 amino acids to 10 amino acids, 5 amino acids to 9 amino acids, 6 amino acids to 8 amino acids, or 7 amino acids to 8 amino acids (e.g., at least 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24 or 25 amino acids).
- 1 amino acid e.g., Gly
- amino acids to 15 amino acids from 2 amino acids to 15 amino acids
- from 3 amino acids to 12 amino acids including 4 amino acids to 10 amino acids, 5 amino acids to 9 amino acids, 6 amino acids to 8 amino acids, or 7 amino acids to 8 amino acids (e.g., at least 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24 or 25 amino acids).
- Exemplary linkers include glycine polymers (G)n, glycine-serine polymers (including, for example, (GS) n , (GGGGS: SEQ ID NO: l) n and (GGGS: SEQ ID NO: 2) n , where n is an integer of at least one), glycine-alanine polymers, alanine-serine polymers, and other flexible linkers known in the art.
- Glycine and glycineserine polymers are relatively unstructured, and therefore may be able to serve as a neutral tether between components. Glycine accesses significantly more phi-psi space than even alanine and is much less restricted than residues with longer side chains (see Scheraga, Rev.
- a linker comprises an amino acid sequence of SEQ ID NO: 3 (GGGGSGGGGSGGGGSGGGGS or “(G4S)4” linker) or SEQ ID NO: 4 (GGGGSGGGGSGGGGSGGGGSGGGGS or “(G4S)5” linker).
- a linker comprises an amino acid sequence of SEQ ID NO: 68 (GGGGSGGGGS or “(G4S)2” linker), SEQ ID NO: 156 (GGGSGGGS or “(G3S)2” linker), SEQ ID NO: 157 (GGGGSGGGGSGGGGS or “(G4S)3” linker), or GGGGSGGGS (SEQ ID NO: 186).
- an incretin agent includes an incretin peptide connected to another peptide (e.g., a HLE domain described herein) using a (G4S)3 linker.
- an incretin agent described herein includes an incretin peptide connected to another incretin peptide using a (G4S)2 linker.
- an incretin peptide is fused to another peptide (e.g., another incretin peptide and/or a half-life extending (HLE) domain) via a protease cleavage site, e.g., a furin cleavage site (e.g., a peptide that includes the motif R-X-K/R-R SEQ ID NO: 158, e.g., SEQ ID NO: 160 RRKR or SEQ ID NO: 153 NVRRKR) and optionally any of the aforementioned linkers.
- a protease cleavage site e.g., a furin cleavage site
- R-X-K/R-R SEQ ID NO: 158 e.g., SEQ ID NO: 160 RRKR or SEQ ID NO: 153 NVRRKR
- a protease cleavage site e.g., a furin cleavage site comprises any one of SEQ ID NO: 160 RRKR, SEQ ID NO: 153 NVRRKR, SEQ ID NO: 189 RKKR, SEQ ID NO: 190 RMQR, or SEQ ID NO: 191 VFRR.
- the term “furin cleavage site” and “furin recognition site” are used interchangeably herein and refer to sequences that facilitate furin cleavage.
- a furin cleavage site is operably linked to (e.g., on C- terminal side of) a linker (e.g., a glycine linker, e.g., a (G4S)2 linker).
- a linker e.g., a glycine linker, e.g., a (G4S)2 linker.
- an incretin agent includes multiple (e.g., 2, 3, 4 or more) incretin peptides each separated by furin cleavage sites and optionally linkers (e.g., (G4S)2 linkers that are on N-terminal side of the furin cleavage site).
- incretin cleavage site within incretin agents described herein, particularly where an incretin peptide is fused to another peptide (e.g., another incretin peptide and/or a half-life extending (HLE) domain) facilitates the proper cleavage of the incretin peptide from the other peptide and allows the incretin peptide to be fully processed and functional after it is expressed.
- another peptide e.g., another incretin peptide and/or a half-life extending (HLE) domain
- the site of cleavage and type of cleavage site or recognition site in the context of polyribonucleotides encoding incretin agents is important to ensure that the N-terminal end of the incretin peptide is processed correctly.
- Certain furin cleavage and recognition sites, and placement of those sites with respect to an incretin peptide within an incretin agent, may lead to alternative processing or cleavage sites, ultimately changing the final amino acid sequence of the mature incretin peptide.
- any change in amino acid residue could impact bioactivity of the peptide.
- a furin recognition/cleavage site is chosen and positioned within an incretin agent in order to facilitate proper cleavage of the N-terminal end of the incretin peptide, or in other words, create a “scarless” N-terminal end of the incretin peptide, in order to maintain the incretin peptide’s bioactivity.
- Figure 20 and Figure 21 show a schematic of where the theoretical cleavage sites of certain exemplary signal peptides lie.
- Figure 20 indicates that A8G mutation facilitates correct N-terminal processing of GLP1 incretin peptides with husec signal peptides.
- Figure 21 indicates that the A2G mutation facilitates correct N-terminal processing of GIP incretin peptides with husec signal peptides.
- Such a concept and utilization of a furin cleavage site at the N-terminus of an incretin peptide linked to another peptide could also be applied to other gut peptides (e.g., glucagon) and/or other peptides of comparable size/properties as GLP1 and GIP described herein. This is particularly important in the context of delivering incretin agents (or other similar peptides) as one or more polyribonucleotides encoding the incretin agents. Such delivery requires the proper translation of the protein within a cell, in addition to the post- translational processing, including proper cleavage of one incretin peptide from another.
- incretin agents comprising one or more incretin peptides fused to another peptide described herein have been designed and generated to include protease cleavage sites placed within the incretin agent such that the cleavage of the incretin peptide(s) from the other peptide(s) is accurate and does not affect the amino acid sequence of the mature peptide (i.e., creates a scarless N-terminus).
- a “scarless” N-terminus includes a peptide that has been cleaved from another peptide via a cleavage site, and where cleavage occurs in a way such that there are no remaining amino acids that are not part of the mature peptide and all of the amino acids of the mature peptide remain at the N-terminus of the peptide.
- a scarless N-terminus of an incretin peptide (and other similar peptides, e.g., other gut peptides, e.g., glucagon), allows for the proper functionality of the peptide after it is processed into a mature peptide.
- a furin cleavage site is placed immediately 5’ of a second incretin peptide in an incretin agent to ensure that cleavage results in a scarless N- terminus on the second incretin peptide.
- Such cleavage may be important to maintain function of the mature incretin peptide.
- a furin cleavage site is chosen for being compatible with the N-terminal sequence of the incretin peptide (e.g., a wildtype or variant incretin peptide, e.g., GIP with an A2G mutation or a GLP1 with a H1Y mutation and/or a A8G mutation). Mutations as described herein may be introduced into an incretin peptide in order to promote effective cleavage and to maintain a scarless N-terminal of the incretin peptide.
- the incretin peptide e.g., a wildtype or variant incretin peptide, e.g., GIP with an A2G mutation or a GLP1 with a H1Y mutation and/or a A8G mutation.
- a furin cleavage site is e.g., NVRRKR (SEQ ID NO: 153), which is derived from the human MT-MMP 1 protein.
- NVRRKR SEQ ID NO: 153
- Such a furin cleavage site is derived from a human protein and it is compatible with the N-terminal sequence of GIP and GLP1 incretin peptides, including wildtype and variant GIP and GLP1 incretin peptides.
- a mutation is introduced into an incretin peptide described herein to facilitate signal peptide cleavage and to generate a mature incretin peptide with a scarless N-terminus.
- such a mutation includes an A2G mutation in a GIP incretin peptide (e.g., a GIP (1-42) incretin peptide).
- such a mutation includes an A8G mutation in a GLP1 (7-37) incretin peptide.
- such mutations may also increase the half-life of the incretin agents, e.g., by preventing proteolysis of the amino acid in the second position of the incretin peptide (leaving a mature incretin peptide that has been improperly cleaved at its N-terminus and is truncated by 1 or 2 amino acids).
- mutations are selected such that they increase the probability of correct cleavage (i.e., cleavage at the N-terminus such that the mature incretin peptide is not truncated).
- compatibility of signal peptide and cleavage site sequence used in incretin agents described herein depends on the particular adjacent incretin peptide amino acid sequence, particularly the amino acid residues at the N-terminus.
- an incretin agent comprises a single incretin peptide. In some embodiments, an incretin agent comprises or more than one incretin peptides. In some embodiments, two or more incretin peptides that are included in an incretin agent described herein are the same or derived from the same incretin peptide (e.g., are GLP1 receptor agonists). In some embodiments, two or more incretin peptides that are included in an incretin agent described herein are different or derived from different incretin peptides.
- an incretin agent comprises a combination of incretin peptides, e.g., fused on a single polypeptide chain.
- an incretin agent comprises a GLP1 receptor agonist (e.g., GLP1 peptide, or fragment or variant thereof) and a GIP receptor agonist (e.g., a GIP peptide, or fragment or variant thereof).
- an incretin agent comprises one or more incretin peptides selected from SEQ ID NOs: 5-15 and 62-64 and one or more incretin peptides selected from SEQ ID NOs: 5-15 and 62-64.
- an incretin agent comprises a GLP1 receptor agonist (e.g., GLP1 peptide, or fragment or variant thereof), a GIP receptor agonist (e.g., a GIP peptide, or fragment or variant thereof), and a GCG receptor agonist (e.g., a GCG peptide, or fragment of variant thereof).
- an incretin agent comprises more than one GLP1 receptor agonist (e.g., GLP1 peptide, or fragment or variant thereof) that may be the same or different.
- an incretin agent comprises more than one GIP receptor agonist (e.g., GIP peptide, or fragment or variant thereof) that may be the same or different.
- an incretin agent comprises more than one copy of the same incretin peptide and/or a combination of different incretin peptides.
- an incretin peptide is fused to another incretin peptide via a linker.
- a linker contains at least one Gly (G) amino acid residue.
- G Gly
- Suitable linkers can be readily selected and can be of various lengths, such as from 1 amino acid (e.g., Gly) to 25 amino acids, from 2 amino acids to 15 amino acids, from 3 amino acids to 12 amino acids, including 4 amino acids to 10 amino acids, 5 amino acids to 9 amino acids, 6 amino acids to 8 amino acids, or 7 amino acids to 8 amino acids (e.g., at least 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24 or 25 amino acids).
- Exemplary linkers include glycine polymers (G)n, glycine-serine polymers (including, for example, (GS) n , (GGGGS: SEQ ID NO: l) n and (GGGS: SEQ ID NO: 2) n , where n is an integer of at least one), glycine-alanine polymers, alanine -serine polymers, and other flexible linkers known in the art.
- Glycine and glycine-serine polymers are relatively unstructured, and therefore may be able to serve as a neutral tether between components. Glycine accesses significantly more phi-psi space than even alanine and is much less restricted than residues with longer side chains (see Scheraga, Rev.
- a linker comprises an amino acid sequence of SEQ ID NO: 3 (GGGGSGGGGSGGGGSGGGGS or “(G4S)4” linker) or SEQ ID NO: 4 (GGGGSGGGGSGGGGSGGGGSGGGGS or “(G4S)5” linker).
- a linker comprises an amino acid sequence of SEQ ID NO: 68 (GGGGSGGGGS or “(G4S)2” linker), SEQ ID NO: 156 (GGGSGGGS or “(G3S)2” linker), or SEQ ID NO: 157 (GGGGSGGGGSGGGGS or “(G4S)3” linker).
- an incretin peptide is fused to another incretin peptide via a protease cleavage site, e.g., a furin cleavage site (e.g., a peptide that includes the motif R-X-K/R-R SEQ ID NO: 158, e g., RRKR SEQ ID NO: 160 or SEQ ID NO: 153 NVRRKR) and optionally any of the aforementioned linkers.
- a protease cleavage site e.g., a furin cleavage site (e.g., a peptide that includes the motif R-X-K/R-R SEQ ID NO: 158, e g., RRKR SEQ ID NO: 160 or SEQ ID NO: 153 NVRRKR) and optionally any of the aforementioned linkers.
- a protease cleavage site e.g., a furin cleavage site comprises any one of SEQ ID NO: 160 RRKR, SEQ ID NO: 153 NVRRKR, SEQ ID NO: 189 RKKR, SEQ ID NO: 190 RMQR, or SEQ ID NO: 191 VFRR.
- a furin cleavage site is operably linked to (e.g., 3’ of) a linker (e.g., a glycine linker, e.g., a (G4S)2 linker).
- an incretin agent includes multiple (e.g., 2, 3, 4 or more) incretin peptides each separated by furin cleavage sites and optionally linkers (e.g., (G4S)2 linkers that are 5’ of the furin cleavage site).
- linkers e.g., (G4S)2 linkers that are 5’ of the furin cleavage site.
- sequence and placement of cleavage sites (e.g., furin cleavage sites) within an incretin agent is important in order to facilitate proper cleavage of the peptides and generate a scarless N-terminus for the incretin peptide.
- a polyribonucleotide encodes an incretin agent that comprises a signal peptide and a single incretin peptide (which configuration is referred to herein as “I: lx”) (see e.g., Figure 3).
- a polyribonucleotide encodes an incretin agent that comprises a signal peptide and two incretin agents, separated by a linker and a cleavage site, e.g., a furin cleavage site (which configuration is referred to herein as “I:2x”) (see e.g., Figure 4).
- a polyribonucleotide encodes an incretin agent that comprises a signal peptide and four incretin agents, each separated by a linker and a cleavage site, e.g., a furin cleavage site (which configuration is referred to herein as “I:4x”) (see e.g., Figure 5).
- I:4x furin cleavage site
- an incretin peptide can be any incretin peptide described herein (e.g., a GLP1 or GIP incretin peptide, e.g., any one of the incretin peptides shown in Table 2).
- a polyribonucleotide described herein encodes a GLP 1 incretin peptide (e.g., any one of the GLP1 incretin peptides described in Table 2) upstream or 5’ of a GIP incretin peptide (e.g., any one of the GIP incretin peptides described in Table 2).
- a polyribonucleotide described herein encodes a GIP incretin peptide (e.g., any one of the GIP incretin peptides described in Table 2) upstream or 5’ of a GLP1 incretin peptide (e.g., any one of the GLP1 incretin peptides described in Table 2).
- order of incretin peptides is determined by how the incretin peptides are expected to be cleaved, such that the incretin peptides maintain their amino acid sequence and a scarless N-terminus.
- a polyribonucleotide described herein encodes an incretin agent that has an amino acid sequence that is at least 85%, at least 90%, at least 95%, or 100% identical to any one of the incretin agents detailed in Table 3. In some embodiments, a polyribonucleotide described herein encodes an incretin agent that has an amino acid sequence according to any one of the incretin agents detailed in Table 3.
- Table 3 Exemplary Incretin Agents including more than one incretin peptide (with mutations in bold, linkers underlined. Furin cleavage site italicized) where x2 and x4 examples include a linker and Furin cleavage site in between each repeat unit
- the present disclosure provides one or more polyribonucleotides encoding an incretin agent that comprises a combination of incretin peptides.
- one or more polyribonucleotides may encode an incretin agent.
- a first polyribonucleotide may encode a first incretin peptide of an incretin agent and a second polyribonucleotide may encode a second incretin peptide of an incretin agent.
- a polyribonucleotide described herein encodes an incretin agent comprising one or more incretin peptides fused to a half-life extending (HLE) domain (see e.g., incretin agents shown in Figures 7-14).
- HLE domain may be included in an incretin agent described herein to increase the half-life of one of the incretin peptides, or of each incretin peptide.
- HSA Human Serum Albumin
- an incretin agent comprises one or more incretin peptides fused to an HLE domain that comprises albumin, e.g., human serum albumin (HSA).
- HSA human serum albumin
- a half-life extending moiety comprises albumin, e.g., human serum albumin.
- a human serum albumin (HSA) sequence is at least 90%, 95%, or 99% identical to an amino acid sequence according to SEQ ID NO: 159 (DAHKSEVAHRFKDLGEENFKALVLIAFAQYLQQCPFEDHVKLVNEVTEFAKTCVAD ESAENCDKSLHTLFGDKLCTVATLRETYGEMADCCAKQEPERNECFLQHKDDNPNL PRLVRPEVDVMCTAFHDNEETFLKKYLYEIARRHPYFYAPELLFFAKRYKAAFTECCQ AADKAACLLPKLDELRDEGKASSAKQRLKCASLQKFGERAFKAWAVARLSQRFPKA EFAEVSKLVTDLTKVHTECCHGDLLECADDRADLAKYICENQDSISSKLKECCEKPLL EKSHCIAEVENDEMPADLPSLAADFVESKDVCKNYAEAKDVFLGMFLYEYARRHPD YSVVLLLRLAKTYETTLEKCCAAADPHECYAKVFDE
- a HSA sequence comprises or consists of an amino acid sequence according to SEQ ID NO: 159, or a fragment or variant thereof. In some embodiments, a HSA sequence comprises or consists of an amino acid sequence that is a variant of wildtype HSA (i.e., SEQ ID NO: 159) comprising one or more amino acid mutations. In some embodiments, the one or more mutations comprises a mutation at position 573 in SEQ ID NO: 159.
- the K residue at position 573 of SEQ ID NO: 159 is substituted with any one of the following amino acid residues: A, C, D, F, G, H, I, L, M, N, P, Q, R, S, V, W, and Y (SEQ ID NO: 187). In some embodiments, the K residue at position 573 of SEQ ID NO: 159 is substituted with a P residue (SEQ ID NO: 188).
- an HSA variant comprises any one of the HSA variants disclosed in U.S. Patent No. 8,748,380, which is hereby incorporated by reference in its entirety.
- a polyribonucleotide described herein encodes an incretin agent as shown in Figure 7, including a I: lx, I:2x or I:4x configuration (i.e., 1, 2, or 4 incretin peptides), fused to an HLE domain, e.g., HSA or an HSA variant as described herein.
- an incretin peptide may be a GLP1 or GIP incretin peptide described herein, or a variant thereof.
- an incretin agent comprises more than one incretin peptides
- the incretin peptide adjacent to the HLE domain will remain fused to the HLE domain after post-translational processing, and the incretin peptide(s) that are not adjacent to the HLE domain will be cleaved from the adjacent incretin peptide and the HLE domain.
- Such designs may be used when administration of multiple incretin peptides with various half-lives is desirable.
- Such designs may also be desirable where one of the incretin peptides is meant to traverse the blood brain barrier (i.e., where an HLE domain is not desirable) and one of the incretin peptides is meant to remain in circulation for a longer period of time (i.e., where an HLE remains attached).
- an incretin agent comprises more than one incretin peptide separated by a linker and a protease cleavage site (e.g., a furin cleavage site), and one of the incretin peptides is a GLP1 peptide described herein adjacent to an HLE domain, e.g., HSA or an HSA variant, and one of the incretin peptides is a GIP peptide that is not adjacent to the HLE (e.g., at the N-terminus of the polypeptide chain).
- the GIP peptide when the incretin agent is expressed from a polyribonucleotide described herein, the GIP peptide will be cleaved from the GLP1 peptide connected to the HLE domain, such that the GLP1 incretin peptide will have a longer half-life than the GIP incretin peptide.
- an incretin agent comprises more than one incretin peptide separated by a linker and a protease cleavage site (e.g., a furin cleavage site), and one of the incretin peptides is a GIP incretin peptide described herein adjacent to an HLE domain, e.g., HSA or an HSA variant, and one of the incretin peptides is a GLP1 peptide that is not adjacent to the HLE (e.g., at the N-terminus of the polypeptide chain).
- the GLP1 incretin peptide when expressed from a polyribonucleotide described herein, the GLP1 incretin peptide will be cleaved from the GIP peptide connected to the HLE domain, such that the GIP incretin peptide will have a longer half-life than the GLP1 incretin peptide (see e.g., Figure 9).
- a polyribonucleotide described herein encodes an incretin agent as shown in Figure 8A, where the incretin agent has a signal peptide (“SP”), a GLP1 incretin peptide, a linker, a second GLP1 incretin peptide, a second linker (GGGS)3, and half-life extension (HLE) domain that is human serum albumin (HSA) or an HSA variant.
- the incretin agent may include a protease cleavage site (e.g., a furin cleavage site) between the GLP1 incretin peptides.
- the first (N-terminal) GLP1 incretin peptide will be cleaved from the second incretin GLP1 incretin peptide that is adjacent to the HLE domain.
- the resulting GLP1 incretin peptides will have two different half lives (i.e., the GLP1 incretin peptide that remains attached to the HLE domain will have a longer half-life than the GLP 1 incretin peptide that was cleaved).
- a polyribonucleotide described herein encodes an incretin agent as shown in Figure 9, where the incretin agent includes a signal peptide (SP), a first GLP1 incretin peptide, a linker (GGGS)2, a first GIP incretin peptide, a second linker (GGGS)2, a second GLP 1 incretin peptide, a third linker (GGGS)2, a second GIP incretin agent, a fourth linker (GGGSfi and half-life extension (HLE) domain that is human serum albumin (HSA) or an HSA variant.
- HSA human serum albumin
- Furin and SP cleavage sites within the incretin agent are indicated with arrows.
- the incretin agent when expressed, the first GLP1 incretin peptide, the first GIP incretin peptide, and the second GLP1 incretin peptide will be cleaved, and the second GIP incretin peptide will remain fused to the HLE domain.
- the resulting second GIP-HLE fusion will have a longer half-life than the other incretin peptides.
- a polyribonucleotide described herein encodes an incretin agent comprising an amino acid sequence that is at least 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or at least 99% identical to any one of the incretin agent sequences in Table 4.
- an incretin agent comprises any one of the incretin agents detailed in Table 4 below, or combinations or variants thereof.
- Table 4 Exemplary Incretin Agents including hAlbumin (with mutations in bold, linkers underlined. Furin cleavage site italicized) where x2 and x4 examples include a linker and Furin cleavage site in between each repeat unit
- an incretin peptide is fused to a half-life extending moiety that binds albumin.
- albumin binding moieties i.e., albumin binding protein domains
- an albumin binding protein domain comprises an albumin-binding domain (ABD) derived from protein G of Streptococcus strain GI48 and/or from protein PAB of Finegoldia magna.
- an ABD binds to domain II of human serum albumin and does not overlap or interfere with binding to the FcRn-binding site on albumin.
- an ABD comprises ABDCon, a three-helix bundle albumin-binding domain, as described in Jacobs et al., Protein Eng., Des. Sei., 2015, 28(10), 385-393, which is incorporated herein by reference in its entirety.
- an ABD is derived from the bacterial protein Sso7d from the hyperthermophilic archaeon Sulfolobus solfataricus, e.g., Mil.12 and M18.2.5 (as described in Gao et al., Nat. Struct. Biol., 1998, 5(9), 782-786 and Traxlmayr et al., J. Biol.
- an ABD comprises a DARPin, as described in Pluckthun, Annu. Rev. Pharmacol. Toxicol., 2015, 55, 489-511, which is incorporated herein by reference in its entirety.
- an ABD comprises an immunoglobulin domain or fragment thereof.
- an ABD comprises a fully human domain antibody (dAb).
- dAb fully human domain antibody
- an ABD comprises an AlbudAb, as described in Holt et al., Protein Eng., Des. Sei., 2008, 21(5), 283-288, which is incorporated herein by reference in its entirety.
- an ABD comprises a Fab, e.g., dsFv CA645, as described in Adams et al., mAbs, 2016, 8(7), 1336-1346, which is incorporated herein by reference in its entirety.
- an ABD comprises a heavy chain only (VHH) antibody, i.e., nanobody, as described in Steeland et al., Drug Discovery Today, 2016, 21(7), 1076-1113, which is incorporated herein by reference in its entirety.
- VHH heavy chain only
- an ABD comprises a VHH domain comprising one or more of the complementarity determining region (CDR) sequences HCDR1, HCDR2, and/or HCDR3 according to SEQ ID NO: 191 (GFTLDYYA), SEQ ID NO: 192 (IASSGGST), and/or SEQ ID NO: 193 (AAAVLECRTWRGYDY), respectively.
- CDR complementarity determining region
- an ABD comprises a VHH domain comprising the CDR sequences HCDR1, HCDR2, and HCDR3 according to SEQ ID NO: 191 (GFTLDYYA), SEQ ID NO: 192 (IASSGGST), and SEQ ID NO: 193 (AAAVLECRTWRGYDY), respectively.
- an ABD comprises a VHH domain that is at least 90%, 95%, or 99% identical to an amino acid sequence according to SEQ ID NO: 154
- a VHH domain comprises an amino acid sequence according to SEQ ID NO: 154.
- an ABD comprises a VNAR, as described in Muller et al., mAbs, 2012, 4(6), 673-685, which is incorporated herein by reference in its entirety.
- a polyribonucleotide described herein encodes an incretin agent as shown in Figure 8B, which shows an incretin agent that has a signal peptide (SP), a first GLP1 incretin peptide, a linker, a second GLP1 incretin peptide, a second linker (GGGS)3, and half-life extension (HLE) domain that is a VHH domain that binds to HSA.
- SP signal peptide
- GGGS second linker
- HLE half-life extension
- an incretin agent when the incretin agent is expressed, the first GLP1 incretin peptide is cleaved from the second GLP1 incretin peptide, and the second GLP1 incretin peptide remains fused to the HLE domain (anti -HSA VHH domain). As such, the second GLP1 incretin peptide will have a longer half-life than the first GLP1 incretin peptide.
- an incretin agent is the incretin agent shown in Figure 8B, where either or both of the GLP1 incretin peptides may instead be a different incretin agent (e.g., a GIP incretin peptide described herein).
- a single incretin peptide is fused to a VHH domain that binds to HSA.
- albumin-binding domains are known in the art, see e.g., Zorzi et al., MedChemComm, 2019, 10.7, 1068-1081, which is incorporated herein by reference in its entirety.
- a polyribonucleotide described herein encodes an incretin agent comprising an amino acid sequence that is at least 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or at least 99% identical to any one of the incretin agent sequences in Table 5.
- an incretin agent comprises any one of the incretin agents detailed in Table 5 below, or combinations or variants thereof.
- Table 5 Exemplary Incretin Agents including an aHSA-VHH domain (with mutations in bold, linkers underlined)
- a half-life extending moiety is an XTEN sequence as described in U.S. Patent No. 8,673,860 and Podust et al., Journal of Controlled Release, 2016, 240, 52-66, which are incorporated herein by reference in their entirety.
- an XTEN sequence comprises about 100 to about 3000 amino acid residues, preferably 400 to about 3000 residues, wherein at least about 80%, or at least about 90%, or about 91%, or about 92%, or about 93%, or about 94%, or about 95%, or about 96%, or about 97%, or about 98%, or about 99% to about 100% of the sequence consists of multiple units of two or more non-overlapping sequence motifs selected from the amino acid sequences of Table 6 or Table 7.
- the XTEN comprises nonoverlapping sequence motifs in which about 80%, or at least about 90%, or about 91%, or about 92%, or about 93%, or about 94%, or about 95%, or about 96%, or about 97%, or about 98%, or about 99% to about 100% of the sequence consists of two or more non-overlapping sequences selected from a single motif family of Table 6 or Table 7, resulting in a “family” sequence in which the overall sequence remains substantially non-repetitive.
- an XTEN sequence can comprise multiple units of non-overlapping sequence motifs of the AD motif family, or the AE motif family, or the AF motif family, or the AG motif family, or the AM motif family, or the AQ motif family, or the BC family, or the BD family of sequences of Table 6.
- the XTEN comprises motif sequences from two or more of the motif families of Table 6.
- the XTEN comprises motif sequences from one or more of the motif families of Table 7.
- a half-life extending moiety is or comprises an Fc domain, e.g., of a human IgG (e.g., a human IgGl, IgG2, IgG3, or IgG4). In some embodiments, a half-life extending moiety does not comprise an Fc domain, e.g., of a human IgG (e.g., a human IgGl, IgG2, IgG3, or IgG4). In some embodiments, a half-life extending moiety comprises an Fc domain of a human IgG4 or a variant thereof (e.g., as included in Dulaglutide). In some embodiments, an Fc domain of an IgG4 sequence is at least 90, 95, 96, 97, 97, or 99% identical to SEQ ID NO: 155
- an Fc domain of an IgG4 sequence is or comprises an amino acid sequence according to SEQ ID NO: 155.
- Homodimeric Incretin Agents are or comprises an amino acid sequence according to SEQ ID NO: 155.
- an incretin agent comprises more than one incretin peptide
- an incretin agent comprises two or more incretin peptides on a single polypeptide chain.
- an incretin agent comprises one or more incretin peptides on separate polypeptide chains.
- the separate polypeptide chains multimerize (e.g., dimerize).
- the separate polypeptide chains comprise two polypeptide chains that each comprise an immunoglobulin constant domain, and the two polypeptide chains dimerize via two constant domains that combine to make an Fc domain.
- an Fc domain comprises an IgG4 Fc domain (e.g., as included in Dulaglutide). In some embodiments, an Fc domain comprises an IgGl Fc domain.
- An exemplary design of an incretin agent comprising multiple polypeptide chains that include an Fc domain, so that the polypeptide chains dimerize, is shown in e.g., Figure 10. The design in Figure 10 could include incretin peptides in a I: lx, I:2x, or I:4x configuration (or other numbers of incretin peptides could be utilized) and each could be a GLP1 or GIP incretin peptide (or any of the variants described herein).
- the two Fc domains are the same.
- the incretins on each polypeptide chain can be the same or different (or in the case of multiple incretins on a single chain, may contain different combinations of incretin peptides).
- An exemplary incretin agent that includes two polypeptide chains that form a homodimer through dimerization of the Fc domains on each polypeptide chain is shown in Figure 11.
- the incretin peptide contains two polypeptide chains, each includes a signal peptide (SP), a GLP1 incretin peptide, a linker (GGGGS)3, an Fc domain.
- each polypeptide chain may comprise two or more incretin peptides (see e.g., Figure 12).
- the two or more incretin peptides could be the same incretin peptide. In some embodiments, the two or more incretin peptides could be different incretin peptides (see e.g., Figure 12). Where two or more incretin peptides are included on each polypeptide chain, cleavage sites may be introduced to cleave the incretin peptides, leaving one incretin peptide attached to the Fc domain. It will be understood by one skilled in the art that the incretin peptide that remains attached to the Fc domain will have a longer halflife and different activity that the other cleaved incretin peptides.
- Fc domains included in incretin agents described herein not only allow the two polypeptide chains to dimerize, but also may increase half-life of the incretin peptides. Other mutations may also be introduced into the Fc domains to increase half-life of the incretin agent.
- an Fc domain within an incretin agent comprises one or more mutations to increase the half-life of the incretin agent.
- an Fc domain may include an LS mutation within a CH3 region (for enhanced FcRn binding) (see Zalevsky, et al., Nature biotechnology, 2010, 28.2: 157-159, which is incorporated herein by reference).
- Such mutations are noted as M428L and N434S according to EU numbering (i.e., M88L and N94S within the CH3 domain), and referred to herein as “LS”.
- Exemplary incretin agents with such a mutation are shown in e.g., Figure 11, Figure 12, and Figure 14.
- an Fc domain of an IgG4 (LS) sequence is identical to SEQ ID NO: 299 (AESKYGPPCPPCPAPEAAGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSQEDPEVQF NWYVDGVEVHNAKTKPREEQFNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKGLPS SIEKTISKAKGQPREPQVYTLPPSQEEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPE NNYKTTPPVLDSDGSFFLYSRLTVDKSRWQEGNVFSCSVLHEALHSHYTQKSLSLSL G).
- an Fc domain in an incretin agent includes one or more mutated amino acid residues that increase half-life.
- an Fc domain comprises one of the following mutated amino acid residues: M252Y, S254T, and T256E (“YTE”), according to the EU numbering scheme to increase half-life.
- YTE mutated amino acid residues
- an Fc domain comprises a combination of the following mutated amino acid residues: M252Y, S254T, and T256E, according to the EU numbering scheme to increase half-life.
- an Fc domain comprises one of the following mutated amino acid residues: T250Q and M428L (“QL”), according to the EU numbering scheme to increase half-life.
- an Fc domain comprises one of the following mutated amino acid residues: H433K and N434F (“KF”), according to the EU numbering scheme to increase half-life.
- a second Fc domain comprises one of the following mutated amino acid residues: T307A, E380A and N434A (“AAA”), according to the EU numbering scheme to increase half-life.
- an Fc domain comprises the following mutated amino acid residues: V308P, according to the EU numbering scheme to increase half-life. In some embodiments, an Fc domain comprises one of the following mutated amino acid residues: M252Y, V308P, and N434Y (“YPY”), according to the EU numbering scheme to increase half-life. In some embodiments, an Fc domain comprises one of the following mutated amino acid residues: H285D, T307Q, and A378V (“DQV”), according to the EU numbering scheme to increase half-life.
- an Fc domain comprises one of the following mutated amino acid residues: L309D, Q311H, N434S (“DHS”), according to the EU numbering scheme to increase halflife.
- DHS mutated amino acid residues
- Exemplary Fc mutations are described in e.g., Liu et al., Antibodies 9.4: 64 (2020), which is hereby incorporated by reference in its entirety.
- a polyribonucleotide described herein encodes an incretin agent comprising an amino acid sequence that is at least 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or at least 99% identical to any one of the incretin agent sequences in Table 8.
- an incretin agent comprises any one of the incretin agents detailed in Table 8 below, or combinations or variants thereof.
- Table 8 Exemplary Incretin Agents including an IgG4 Fc domain (with mutations in bold, linkers underlined. Furin cleavage site italicized) where x4 example includes a linker and Furin cleavage site in between each repeat unit
- an Fc domain includes one more mutations in order to ablate effector function of the Fc domain included in an incretin agent. By ablating the Fc effector function of the incretin agent, delivery of the incretin agent will be less likely to cause an unwanted immune response, due to triggering cytotoxic and other effector activity by immune cells.
- an Fc domain of a molecule includes one or more mutations that silence effector function.
- one or more mutations in an Fc domain include an “STR” modification, or a combination of mutations comprising L234S, L235T, and G236R, according to the EU numbering scheme.
- an Fc domain will show little or no detectable binding to Fey receptors or to Clq, and do not promote an inflammatory cytokine response (see e.g., Wilkinson et al., (2021) PLoS One 16.12: e0260954, which is herein incorporated by reference in its entirety).
- Exemplary incretin agent comprising an STR modification is shown in e.g., Figure 14.
- a modification that reduces or silences effector function of an Fc domain included in an incretin agent described herein comprises one or more of the following mutations: L234A, L235A, P329G, P329A, N297A, orN297D, according to the EU numbering scheme.
- a modification that silences effector function of an Fc domain comprises the following mutated amino acid residues: L234A and L235A (“LALA”), according to the EU numbering scheme.
- mutations used to ablate effector function of an Fc domain include the following: L234A/L235A/P329G (“LALAPG”), according to EU numbering.
- a modification that silences effector function of an Fc domain includes the following Fc mutations: L234A/L235A/P329G and N297D, according to EU numbering. In some embodiments, a modification that silences effector function of an Fc domain includes the following Fc mutations: L234A, L235A, and N297A, according to EU numbering. In some embodiments, a modification that silences effector function of an Fc domain includes the following Fc mutations: L234A, L235A, and N297D, according to EU numbering.
- a modification that silences effector function of an Fc domain includes the following Fc mutations: L234A, L235A, P329A, and N297A, according to EU numbering. In some embodiments, a modification that silences effector function of an Fc domain includes the following Fc mutations: L234A, L235A, P329A, and N297D, according to EU numbering.
- a modification that reduces or silences effector function of an Fc domain included in an incretin agent described herein comprises the following mutations: L234F/L235E/P331S (“FES”), according to the EU numbering scheme.
- a modification that reduces or silences effector function of an Fc domain included in an incretin agent described herein comprises the following mutations: L234F/L235Q/K322Q (“FQQ”), according to the EU numbering scheme.
- a modification that reduces or silences effector function of an Fc domain included in an incretin agent described herein comprises the following mutations: A330S/P331S, according to the EU numbering scheme.
- one or more polyribonucleotides described herein encode an incretin agent that comprises more than one incretin peptides on separate polypeptide chains.
- separate polypeptide chains multimerize (e.g., dimerize).
- the separate polypeptide chains comprise two polypeptide chains that each comprise an immunoglobulin constant domain, and the two polypeptide chains dimerize via two constant domains that combine to make an Fc domain.
- an Fc domain within an incretin agent comprises one or more mutations to induce dimerization.
- an incretin agent comprises a first polypeptide chain that comprises an incretin peptide fused to the constant domain of an immunoglobulin, where the constant domain comprises one more mutations that induce dimerization with a second polypeptide chain that comprises an incretin peptide fused to the constant domain of an immunoglobulin.
- the constant domain of the first and second polypeptides both contain one or more mutations to induce dimerization.
- the incretin peptide or incretin peptides in the first and second polypeptides are different.
- KH knock-into-holes technology
- a two constant domains by introducing mutations into the CH3 domains to modify the contact interface.
- bulky amino acids are replaced by amino acids with short side chains to create a “hole” and amino acids with large side chains are introduced into the other CH3 domain, to create a “knob”.
- Co-expression of these two constant domains induces dimerization.
- an Fc domain described herein utilizes KIH technology as described in, e.g., WO1998/050431, which is incorporated herein by reference in its entirety.
- an Fc domain of an incretin agent may comprise certain mutations that utilize KIH technology that include, but are not limited to, a CH3 modification.
- an Fc domain of an incretin agent comprises a CH3 domain comprising one or more of the following mutations: Y349C, T366S, L368A, and Y407V (according to EU numbering).
- an Fc domain of an incretin agent comprises a CH3 domain, wherein the CH3 domain comprises each of the following mutations: Y349C, T366S, L368A, and Y407V (according to EU numbering).
- Such a combination of mutations is referred to herein as “FcKIH-b”.
- an Fc domain of an incretin agent comprises a CH3 domain comprising one or more mutations selected from: S354C and T366W (according to EU numbering).
- an Fc domain of an incretin agent comprises a CH3 domain comprising each of the following mutations: S354C and T366W (according to EU numbering).
- Such a combination of CH3 mutations is referred to herein as “FcKIH-a”.
- an incretin agent comprises an Fc domain that includes an FcKIH-a sequence and an FcKIH-b sequence.
- an Fc domain within an incretin agent comprises one or more “KiH” mutations and an LS mutation.
- an incretin agent encoded by one or more polyribonucleotides as described herein comprises one or more incretin peptides fused to an Fc domain, where the CH3 domain of the Fc domain comprises one or more of the following mutations: Y349C, T366S, L368A, and Y407V (according to EU numbering).
- an incretin agent encoded by one or more polyribonucleotides as described herein comprises one or more incretin peptides fused to an Fc domain, where the Fc domain comprises a CH3 domain comprising one or more mutations selected from: S354C and T366W (according to EU numbering).
- an incretin agent comprises a heterodimer, e.g., as shown in Figure 13 or Figure 14.
- Figure 13 shows an exemplary design of two polypeptide chains that include an incretin peptide fused to an Fc domain.
- each polypeptide chain incretin-Fc fusion
- SP signal peptide
- I: lx, I:2x, or I:4x incretin peptides fused to an Fc domain
- each incretin agent includes an Fc mutation that induces heterodimerization (e.g., a knob-in-hole mutation).
- the Fc domains could also include modifications to ablate effector function and/or to increase half life as described herein.
- the 14 has a signal peptide (SP), a GLP1 or GIP incretin peptide, a linker (GGGS)i, and an Fc domain.
- Fc domains contain “LS” mutations (M428L/N434S) to extend half-life of the incretin agent, “STR” mutations to silence Fc effector function, and “knob-in- hole” mutations to promote heterodimerization.
- any mutations described herein may be included instead of or in addition to the LS and/or STR mutations.
- an incretin agent comprises two polypeptide chains that associate and form a heterodimer incretin agent wherein one of the polypeptide chains comprises the sequence of SEQ ID NO: 300 (DKTHTCPPCPAPESTRGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNW YVDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIE KTISKAKGQPREPQVYTLPPCREEMTKNQVSLWCLVKGFYPSDIAVEWESNGQPENN YKTTPPVLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVLHEALHSHYTQKSLSLSPGK) (“FcKIH-a (LS and STR)”) and the other polypeptide chain comprises the sequence of SEQ ID NO:
- a polyribonucleotide described herein encodes an incretin agent comprising an amino acid sequence that is at least 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or at least 99% identical to any one of the incretin agent sequences in Table 9.
- an incretin agent comprises any one of the incretin agents detailed in Table 9 below, or combinations or variants thereof.
- Table 9 Exemplary Incretin Agents including an FcKIH-a or FcKIH-b domain, that form heterodimers (with mutations in bold, linkers underlined)
- any one of the exemplary incretin agents that includes an FcKIH-a domain can be combined with the exemplary incretin agent that includes an FcKIH-b domain, e.g., in some embodiments an incretin agent of SEQ ID NO: 84, 85, 86, 87, 173 or 174 can be combined with an incretin agent of SEQ ID NO: 88.
- a signal peptide is fused, either directly or through a linker, to an encoded incretin peptide described herein.
- an open reading frame of a polyribonucleotide described herein encodes an incretin agent with a signal peptide, e.g., that is functional in mammalian cells.
- signal peptides are sequences, which are typically characterized by a length of about 15 to 30 amino acids. In many embodiments, signal peptides are positioned at the N-terminus of an incretin agent, without being limited thereto.
- signal peptides preferably allow the transport of an incretin agent encoded by polyribonucleotides of the present disclosure with which they are associated into a defined cellular compartment, preferably the cell surface, the endoplasmic reticulum (ER) or the endosomal-lysosomal compartment.
- a defined cellular compartment preferably the cell surface, the endoplasmic reticulum (ER) or the endosomal-lysosomal compartment.
- a ribonucleic acid sequence encoding a signal peptide allows an incretin agent encoded by the polyribonucleotide to be secreted upon translation by cells, e.g., present in a subject, thus yielding a plasma concentration of a biologically active incretin agent.
- a ribonucleic acid sequence encoding a signal peptide included in a polyribonucleotide consists of or comprises a nucleotide sequence that encodes a human signal peptide.
- a ribonucleic acid sequence encoding a secretion signal included in a polyribonucleotide consists of or comprises a nucleotide sequence that encodes a non-human secretion signal.
- a signal peptide may be or comprises a viral signal peptide.
- such a signal peptide may be or comprises the amino acid sequence of MRVLVLLACLAAASNA (SP1-2; SEQ ID NO: 17).
- a signal peptide may be or comprises an amino acid sequence of MRVMAPRTLILLLSGALALTETWA (husec signal peptide delta GS; SEQ ID NO: 65).
- a signal peptide sequence is selected from those included in the Table 10 below, or a fragment or variant thereof:
- a signal peptide may be important for prediction of the cleavage site between a signal peptide and an incretin peptide.
- a signal peptide is, in some embodiments, selected and included in incretin agents to effect proper cleavage of the incretin into mature form.
- the site of cleavage of the signal peptide and type or sequence of the signal peptide in the context of polyribonucleotides encoding incretin agents described herein may be important to ensure that the N-terminal end of the incretin peptide is processed correctly.
- Signal peptides may contain particular sequences or structure that leads to alternative processing or cleavage sites, ultimately changing the final amino acid sequence of the mature incretin peptide. In such relatively small peptides such as GLP1 or GIP (or variants thereof, and other peptides of similar size/properties), a change in amino acid residue could impact bioactivity of the peptide.
- a signal peptide is chosen for inclusion in incretin agents described herein in order to facilitate proper cleavage of the N-terminal end of the incretin peptide, or in other words, create a “scarless” N-terminal end of the incretin peptide, in order to maintain the incretin peptide’s bioactivity.
- Figure 20 and Figure 21 show a schematic of where the theoretical cleavage sites of certain exemplary signal peptides lie with the incretin agent.
- Figure 20 indicates that A8G mutation facilitates correct N-terminal processing of GLP1 incretin agents with a husec signal peptide.
- Figure 21 indicates that the A2G mutation facilitates correct N-terminal processing of GIP incretin agents with a husec signal peptide.
- Such a concept and utilization of a particular signal peptides to facilitate cleavage to generate a mature peptide with a scarless N-terminus could also be applied to other gut peptides (e.g., glucagon) and/or other peptides of comparable size/properties as GLP1 and GIP described herein. This may be important in the context of delivering incretin agents (or other similar peptides) as one or more polyribonucleotides encoding the incretin agents. Such delivery requires the proper translation of the protein within a cell, in addition to the post-translational processing, including proper cleavage of a peptide post-translation.
- Incretin agents comprising one or more incretin peptides fused to another peptide described herein may be designed and generated so that signal peptide cleavage is accurate and does not affect the amino acid sequence of the mature peptide (i.e., creates a “scarless” N terminal).
- a scarless N-terminal of incretin peptides (and other similar peptides, e.g., other gut peptides, e.g., glucagon), allows for the proper functionality of the peptide after it is processed into a mature peptide.
- a polyribonucleotide comprises, in a 5’ to 3’ direction a signal peptide -coding sequence and one or more incretin peptide -coding sequences, in some embodiments, a signal peptide-coding sequence and one or more incretin peptide-codmg sequences encode any one of the sequences as shown in Table 11 below, or a sequence that is at least 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or at least 99% identical to any one of the sequence shown in Table 11 below.
- Table 11 Exemplary Incretin Agents where x2 and x4 examples include a linker and Furin cleavage site in between each repeat unit
- a polyribonucleotide comprises, in a 5’ to 3' direction a signal peptide-coding sequence; an incretin peptide-coding sequence; a linker-coding sequence; and a half-life extending moiety-coding sequence.
- a polyribonucleotide comprises, in a 5’ to 3“ direction a signal peptide-coding sequence; a halflife extending moiety-coding sequence: a linker-coding sequence; and an incretin peptide- coding sequence.
- a polyribonucleotide comprises, in a 5’ to 3 ?
- a signal peplide-coding sequence and one or more incretin peptide-coding sequences each independently separated by a linker-coding sequence and a protease cleavage site-coding sequence, e.g., a farm cleavage site-coding sequence.
- one or more of the incretin peptide-coding sequences is preceded or followed by a linker-coding sequence and a half-lite extending moiety-coding sequence.
- a polyribonucleotide comprises a ribonucleic acid sequence that is at least 80%, 85%, 90%, 91 %, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or at least 99% identical to any of the sequences shown below in Table 12.
- Table 12 Exemplar ⁇ ' Polyribonucleotides encoding Incretin Agents where x2 and x4 examples include a linker and Furin cleavage site in between each repeat unit
- Polyribonucleotides described herein encode an incretin agent as described herein. Additionally, polyribonucleotides described herein, in some embodiments, include encode other elements such as a signal peptide. In some embodiments, polyribonucleotides described herein can comprise a nucleotide sequence that encodes a 5’UTR and/or a 3’ UTR. In some embodiments, polynucleotides described herein can comprise a nucleotide sequence that encodes a polyAtail. In some embodiments, polyribonucleotides described herein may comprise a 5’ cap, which may be incorporated during transcription, or joined to a polyribonucleotide post-transcription.
- a structural feature of mRNAs is a cap structure at the 5 ’-end.
- Natural eukaryotic mRNA comprises a 7-methylguanosine cap linked to the mRNA via a 5 ' to 5 '- triphosphate bridge resulting in capO structure (m7GpppN).
- capO structure m7GpppN
- further modifications can occur at the 2 ’-hydroxy-group (2 ’-OH) (e.g., the 2 ’-hydroxyl group may be methylated to form 2’-0-Me) of the first and subsequent nucleotides producing “capl” and “cap2” five-prime ends, respectively.
- RNA capping is well researched and is described, e.g., in Decroly et al., (2012) Nature Reviews 10: 51-65; and in Ramanathan et al., (2016) Nucleic Acids Res; 44(16): 7511-7526, the entire contents of each of which is hereby incorporated by reference.
- a 5’ cap structure which may be suitable in the context of the present invention is a capO (methylation of the first nucleobase, e.g.
- 5 ’-cap refers to a structure found on the 5 ’-end of an RNA, e.g., mRNA, and generally includes a guanosine nucleotide connected to an RNA, e.g., mRNA, via a 5’- to 5 ’-triphosphate linkage (also referred to as Gppp or G(5’)ppp(5’)).
- a guanosine nucleoside included in a 5 ’ cap may be modified, for example, by methylation at one or more positions (e.g., at the 7-position) on a base (guanine), and/or by methylation at one or more positions of a ribose.
- a guanosine nucleoside included in a 5’ cap comprises a 3’0 methylation at a ribose (3’0meG).
- a guanosine nucleoside included in a 5’ cap comprises methylation at the 7-position of guanine (m7G).
- a guanosine nucleoside included in a 5’ cap comprises methylation at the 7-position of guanine and a 3’ O methylation at a ribose (m7(3’OmeG)).
- m7(3’OmeG) methylation at the 7-position of guanine and a 3’ O methylation at a ribose
- providing an RNA with a 5 ’-cap disclosed herein may be achieved by in vitro transcription, in which a 5 ’-cap is co-transcriptionally expressed into an RNA strand, or may be attached to an RNA post-transcriptionally using capping enzymes.
- co-transcriptional capping with a cap disclosed herein improves the capping efficiency of an RNA compared to co-transcriptional capping with an appropriate reference comparator.
- improving capping efficiency can increase translation efficiency and/or translation rate of an RNA, and/or increase expression of an encoded polypeptide.
- alterations to polynucleotides generates a non- hydrolyzable cap structure which can, for example, prevent decapping and increase RNA half-life.
- a utilized 5’ cap is a capO, a capl, or cap2 structure. See, e.g., Fig. 1 of Ramanathan et al., and Fig. 1 of Decroly et al., each of which is incorporated herein by reference in its entirety.
- an RNA described herein comprises a capl structure. In some embodiments, an RNA described herein comprises a cap2 structure.
- an RNA described herein comprises a capO structure.
- a capO structure comprises a guanosine nucleoside methylated at the 7-position of guanine ((m 7 )G).
- such a capO structure is connected to an RNA via a 5’- to 5 ’-triphosphate linkage and is also referred to herein as (m 7 )Gppp.
- a capO structure comprises a guanosine nucleoside methylated at the 2’- position of the ribose of guanosine In some embodiments, a capO structure comprises a guanosine nucleoside methylated at the 3 ’-position of the ribose of guanosine. In some embodiments, a guanosine nucleoside included in a 5’ cap comprises methylation at the 7- position of guanine and at the 2 ’-position of the ribose ((m2 7 ’ 2 ’°)G).
- a guanosine nucleoside included in a 5’ cap comprises methylation at the 7-position of guanine and at the 2 ’-position of the ribose ((m2 7 ’ 3 ’°)G).
- a cap 1 structure comprises a guanosine nucleoside methylated at the 7-position of guanine ((m 7 )G) and optionally methylated at the 2’ or 3’ position of the ribose, and a 2’0 methylated first nucleotide in an RNA ((m 2 ’°)Ni).
- a capl structure comprises a guanosine nucleoside methylated at the 7-position of guanine ((m 7 )G) and the 3’ position of the ribose, and a 2’0 methylated first nucleotide in an RNA ((m 2 ’°)Ni).
- a capl structure is connected to an RNA via a 5’- to 5 ’-triphosphate linkage and is also referred to herein as, e.g., ((m 7 )Gppp( 2 ’°)Ni) or (m2 7 ’ 3 ' °)Gppp( 2 ’°)Ni), wherein Ni is as defined and described herein.
- a capl structure comprises a second nucleotide, N2, which is at position 2 and is chosen from A, G, C, or U, e.g., (m 7 )Gppp( 2 '°)NipN2 or (m2 7 ’ 3 "°)Gppp( 2 "°)NipN2, wherein each of Ni and N2 is as defined and described herein.
- a cap2 structure comprises a guanosine nucleoside methylated at the 7-position of guanine ((m 7 )G) and optionally methylated at the 2’ or 3’ position of the ribose, and a 2’0 methylated first and second nucleotides in an RNA ((m 2 ' °)Nip(m 2 ’°)N2).
- a cap2 structure comprises a guanosine nucleoside methylated at the 7-position of guanine ((m 7 )G) and the 3 ’ position of the ribose, and a 2’0 methylated first and second nucleotide in an RNA.
- a cap2 structure is connected to an RNA via a 5’- to 5 ’-triphosphate linkage and is also referred to herein as, e.g., ((m 7 )Gppp( 2 '°)Nip( 2 '°)N2) or (m2 7 ’ 3 '°)Gppp( 2 '°)Nip( 2 ’°)N2), wherein each ofNi and N2 is as defined and described herein.
- the 5’ cap is a dinucleotide cap structure. In some embodiments, the 5’ cap is a dinucleotide cap structure comprising Ni, wherein Ni is as defined and described herein. In some embodiments, the 5’ cap is a dinucleotide cap G*Ni, wherein Ni is as defined above and herein, and G* comprises a structure of Formula (I): or a salt thereof, wherein each R 2 and R 3 is -OH or -OCH3; and
- X is O or S.
- R 2 is -OH. In some embodiments, R 2 is -OCH3. In some embodiments, R 3 is -OH. In some embodiments, R 3 is -OCH3. In some embodiments, R 2 is -OH and R 3 is -OH. In some embodiments, R 2 is -OH and R 3 is -CH3. In some embodiments, R 2 is -CH3 and R 3 is -OH. In some embodiments, R 2 is -CH3 and R 3 is -CH3.
- X is O. In some embodiments, X is S.
- the 5’ cap is a dinucleotide capO structure (e.g., (m 7 )GpppNi, (m2 7 ’ 2 '°)GpppNi, (m2 7 ’ 3 '°)GpppNi, (m 7 )GppSpNi, (m2 7 ’ 2 '°)GppSpNi, or (m2 7 ’ 3 '°)GppSpNi), wherein Ni is as defined and described herein.
- the 5’ cap is a dinucleotide capO structure (e.g., (m 7 )GpppNi, (m2 7 ’ 2 '°)GpppNi, (m2 7 ’ 3 '°)GpppNi, (m 7 )GppSpNi, (m2 7 ’ 2 '°)GppSpNi, or (m2 7 ’ 3 '°)GppSpNi), wherein Ni is G.
- a dinucleotide capO structure e.g., (m 7 )GpppNi, (m2 7 ’ 2 '°)GpppNi, (m2 7 ’ 3 '°)GpppNi
- Ni is G.
- the 5’ cap is a dinucleotide capO structure (e.g., (m 7 )GpppNi, (m2 7 ’ 2 '°)GpppNi, (m2 7 ’ 3 '°)GpppNi, (m 7 )GppSpNi, (m2 7 ’ 2 '°)GppSpNi, or (m2 7 ’ 3 '°)GppSpNi), wherein Ni is A, U, or C.
- a dinucleotide capO structure e.g., (m 7 )GpppNi, (m2 7 ’ 2 '°)GpppNi, (m2 7 ’ 3 '°)GpppNi
- Ni is A, U, or C.
- the 5’ cap is a dinucleotide capl structure (e.g., (m 7 )Gppp(m 2 ’-°)Ni, (m2 7 ’ 2 ’-°)Gppp(m 2 ’-°)Ni, (m2 7 ’ 3 ’-°)Gppp(m 2 ’-°)Ni, (m 7 )GppSp(m 2 ’-°)Ni, (m2 7 ’ 2 '°)GppSp(m 2 "°)Ni, or (m2 7 ’ 3 '°)GppSp(m 2 ’’°)Ni), wherein Ni is as defined and described herein.
- Ni is as defined and described herein.
- the 5’ cap is selected from the group consisting of (m 7 )GpppG (“EcapO”), (m 7 )Gppp(m 2 ’°)G (“Ecapl”), (m2 73 '°)GpppG (“ARCA”), and (m2 72 '°)GppSpG (“beta-S-ARCA”).
- the 5’ cap is (m 7 )GpppG (“EcapO”), having a structure: or a salt thereof.
- the 5’ cap is (m 7 )Gppp(m 2 ’°)G (“Ecapl”), having a structure: or a salt thereof.
- the 5’ cap is (m2 7 ’ 3 '°)GpppG (“ARCA”), having a structure: or a salt thereof.
- the 5’ cap is (m2 72 '°)GppSpG (“beta-S-ARCA”), having a structure: or a salt thereof.
- the 5’ cap is a trinucleotide cap structure. In some embodiments, the 5’ cap is a trinucleotide cap structure comprising NipN2, wherein Ni and N2 are as defined and described herein. In some embodiments, the 5’ cap is a dinucleotide cap G*NipN2, wherein Ni and N2 are as defined above and herein, and G* comprises a structure of Formula (I): or a salt thereof, wherein R 2 , R 3 , and X are as defined and described herein.
- the 5’ cap is a trinucleotide capO structure (e.g. (m 7 )GpppNipN2, (m2 7 ’ 2 '°)GpppNipN2, or (m2 7 ’ 3 '°)GpppNipN2), wherein Ni and N2 are as defined and described herein).
- a trinucleotide capO structure e.g. (m 7 )GpppNipN2, (m2 7 ’ 2 '°)GpppNipN2, or (m2 7 ’ 3 '°)GpppNipN2
- the 5’ cap is a trinucleotide capl structure (e.g., (m 7 )Gppp(m 2 "°)NipN2, (m2 7 ’ 2 '°)Gppp(m 2 ’’°)NipN2, (m2 7 ’ 3 '°)Gppp(m 2 ' °)NipN2), wherein Ni and N2 are as defined and described herein.
- a trinucleotide capl structure e.g., (m 7 )Gppp(m 2 "°)NipN2, (m2 7 ’ 2 '°)Gppp(m 2 ’’°)NipN2, (m2 7 ’ 3 '°)Gppp(m 2 ' °)NipN2
- the 5’ cap is a trinucleotide cap2 structure (e.g., (m 7 )Gppp(m 2 '°)Nip(m 2 ’°)N2, (m2 7 ’ 2 ’ °)Gppp(m 2 '°)Nip(m 2 '°)N2, (m2 7 ’ 3 '°)Gppp(m 2 '°)Nip(m 2 ' O )N2), wherein NI and N2 are as defined and described herein.
- NI and N2 are as defined and described herein.
- the 5’ cap is selected from the group consisting of (m2 7 ’ 3 '°)Gppp(m 2 "°)ApG (“CleanCap AG”, “CC413”), (m2 7 ’ 3 '°)Gppp(m 2 ' °)GpG (“CleanCap GG”), (m 7 )Gppp(m 2 '°)ApG, (m 7 )Gppp(m 2 '°)GpG, (m2 73 '°)Gppp(m2 6 ’ 2 ' °)ApG, and (m 7 )Gppp(m 2 ’-°)ApU.
- the 5’ cap is (m2 7 ’ 3 '°)Gppp(m 2 '°)ApG (“CleanCap
- the 5’ cap is (m2 73 '°)Gppp(m 2 '°)GpG (“CleanCap GG”), having a structure:
- the 5’ cap is (m 7 )Gppp(m 2 '°)ApG, having a structure: or a salt thereof.
- the 5’ cap is (m 7 )Gppp(m 2 '°)GpG, having a structure:
- the 5 ’ cap is (m2 7 ’ 3 ’ 0 )Gppp(m2 6 ’ 2 ’’°)ApG, having a structure: or a salt thereof.
- the 5’ cap is (m 7 )Gppp(m 2 ’’°)ApU, having a structure: or a salt thereof.
- the 5’ cap is a tetranucleotide cap structure.
- the 5’ cap is a tetranucleotide cap structure comprising NipN2pNs, wherein Ni, N2, and Ns are as defined and described herein.
- the 5’ cap is a tetranucleotide cap G*NipN2pN ⁇ wherein Ni, N2, and N3 are as defined above and herein, and G* comprises a structure of Formula (I):
- the 5’ cap is a tetranucleotide capO structure (e.g. (m 7 )GpppNipN2pN3, (m2 7 ’ 2 '°)GpppNipN2pN3, or (m2 7 ’ 3 '°)GpppNiN2pN3), wherein Ni, N2, and N3 are as defined and described herein).
- a tetranucleotide capO structure e.g. (m 7 )GpppNipN2pN3, (m2 7 ’ 2 '°)GpppNipN2pN3, or (m2 7 ’ 3 '°)GpppNiN2pN3
- the 5’ cap is a tetranucleotide Capl structure (e.g., (m 7 )Gppp(m 2 '°)NipN2pN3, (m2 7 ’ 2 '°)Gppp(m 2 ' °)NipN2pN3, (m2 7 ’ 3 '°)Gppp(m 2 '°)NipN2N3), wherein Ni, N2, and N3 are as defined and described herein.
- tetranucleotide Capl structure e.g., (m 7 )Gppp(m 2 '°)NipN2pN3, (m2 7 ’ 2 '°)Gppp(m 2 ' °)NipN2pN3, (m2 7 ’ 3 '°)Gppp(m 2 '°)NipN2N3
- the 5’ cap is a tetranucleotide Cap2 structure (e.g., (m 7 )Gppp(m 2 ’-°)Nip(m 2 ’-°)N2pN3, (m2 7 ’ 2 ’- 0 )Gppp(m 2 ’- 0 )Nip(m 2 ’- 0 )N2pN3, (m2 7 ’ 3 ’-°)Gppp(m 2 ’- °)Nip(m 2 ’-°)N 2 pN3), wherein Ni, N2, and N3 are as defined and described herein.
- tetranucleotide Cap2 structure e.g., (m 7 )Gppp(m 2 ’-°)Nip(m 2 ’-°)N2pN3, (m2 7 ’ 2 ’- 0 )Gppp(m 2 ’- 0 )Nip(m 2 ’- 0 )N
- the 5’ cap is selected from the group consisting of (m2 7 ’ 3 '°)Gppp(m 2 '°)Ap(m 2 °)GpG, (m2 7 ’ 3 '°)Gppp(m 2 '°)Gp(m 2 '°)GpC, (m 7 )Gppp(m 2 ’-°)Ap(m 2 ’-°)UpA, and (m 7 )Gppp(m 2 ’-°)Ap(m 2 ’-°)GpG.
- the 5’ cap is (m2 73 '°)Gppp(m 2 '°)Ap(m 2 '°)GpG, having a structure: or a salt thereof.
- the 5’ cap is (m2 73 '°)Gppp(m 2 '°)Gp(m 2 '°)GpC, having a structure:
- the 5 ’ cap is (m 7 )Gppp(m 2 '°)Ap(m 2 '°)UpA, having a structure: or a salt thereof.
- the 5 ’ cap is (m 7 )Gppp(m 2 '°)Ap(m 2 '°)GpG, having a structure: or a salt thereof.
- a 5 ’ UTR utilized in accordance with the present disclosure comprises a cap proximal sequence, e.g., as disclosed herein.
- a cap proximal sequence comprises a sequence adjacent to a 5’ cap.
- a cap proximal sequence comprises nucleotides in positions +1, +2, +3, +4, and/or +5 of an RNA polynucleotide.
- a cap structure comprises one or more polynucleotides of a cap proximal sequence.
- a cap structure comprises an m 7 Guanosine cap and nucleotide +1 (Ni) of an RNA polynucleotide.
- a cap structure comprises an m 7 Guanosine cap and nucleotide +2 (N2) of an RNA polynucleotide.
- a cap structure comprises an m 7 Guanosine cap and nucleotides +1 and +2 (Ni and N2) of an RNA polynucleotide.
- a cap structure comprises an m 7 Guanosine cap and nucleotides +1, +2, and +3 (Ni, N2, and N3) of an RNA polynucleotide.
- one or more residues of a cap proximal sequence may be included in an RNAby virtue of having been included in a cap entity (e.g., a capl or cap2 structure, etc.); alternatively, in some embodiments, at least some of the residues in a cap proximal sequence may be enzymatically added (e.g., by a polymerase such as a T7 polymerase).
- +1 i.e., Ni
- +2 i.e. N2
- +3, +4, and +5 are added by polymerase (e.g., T7 polymerase).
- the 5’ cap is a dinucleotide cap structure, wherein the cap proximal sequence comprises Ni of the 5’ cap, where Ni is any nucleotide, e.g., A, C, G or U.
- the 5’ cap is a trinucleotide cap structure (e.g., the trinucleotide cap structures described above and herein), wherein the cap proximal sequence comprises Ni and N2 of the 5’ cap, wherein Ni and N2 are independently any nucleotide, e.g., A, C, G or U.
- the 5’ cap is a tetranucleotide cap structure (e.g., the trinucleotide cap structures described above and herein), wherein the cap proximal sequence comprises Ni, N2, and N3 of the 5’ cap, wherein Ni, N2, and N3 are any nucleotide, e.g., A, C, G or U.
- the cap proximal sequence comprises Ni, N2, and N3 of the 5’ cap, wherein Ni, N2, and N3 are any nucleotide, e.g., A, C, G or U.
- a cap proximal sequence comprises Ni of a the 5’ cap, and N2, N3, N4 and Ns, wherein Ni to Ns correspond to positions +1, +2, +3, +4, and/or +5 of an RNA polynucleotide.
- a cap proximal sequence comprises Ni and N2 of a the 5’ cap, and N3, N4 and Ns, wherein Ni to Ns correspond to positions +1, +2, +3, +4, and/or +5 of an RNA polynucleotide.
- a cap proximal sequence comprises Ni, N2, and N3 of a the 5’ cap, and N4 and Ns, wherein Ni to Ns correspond to positions +1, +2, +3, +4, and/or +5 of an RNA polynucleotide.
- Ni is A. In some embodiments, Ni is C. In some embodiments, Ni is G. In some embodiments, Ni is U. In some embodiments, N2 is A. In some embodiments, N2 is C. In some embodiments, N2 is G. In some embodiments, N2 is U. In some embodiments, N3 is A. In some embodiments, N3 is C. In some embodiments, N3 is G. In some embodiments, N3 is U. In some embodiments, N4 is A. In some embodiments, N4 is C. In some embodiments, N4 is G. In some embodiments, N4 is U. In some embodiments, Ns is A. In some embodiments, Ns is C. In some embodiments, Ns is G. In some embodiments, N4 is U. In some embodiments, Ns is A. In some embodiments, Ns is C. In some embodiments, Ns is G.
- Ns is U. It will be understood that, each of the embodiments described above and herein (e.g., for Ni through Ns) may be taken singly or in combination and/or may be combined with other embodiments of variables described above and herein (e.g., 5’ caps).
- 5 ’ UTR may comprise a plurality of distinct sequence elements; in some embodiments, such plurality may be or comprise multiple copies of one or more particular sequence elements (e.g., as may be from a particular source or otherwise known as a functional or characteristic sequence element).
- a 5’ UTR comprises multiple different sequence elements.
- untranslated region or “UTR” is commonly used in the art to reference a region in a DNA molecule which is transcribed but is not translated into an amino acid sequence, or to the corresponding region in an RNA polynucleotide, such as an mRNA molecule.
- An untranslated region (UTR) can be present 5’ (upstream) of an open reading frame (5’ UTR) and/or 3’ (downstream) of an open reading frame (3’ UTR).
- the terms “five prime untranslated region” or “5 ’ UTR” refer to a sequence of a polyribonucleotide between the 5’ end of the polyribonucleotide (e.g., a transcription start site) and a start codon of a coding region of the polyribonucleotide.
- Exemplary 5 ’ UTRs include a human alpha globin (hAg) 5 ’ UTR or a fragment thereof, a TEV 5 ’ UTR or a fragment thereof, a HSP70 5 ’ UTR or a fragment thereof, or a c-Jun 5’ UTR or a fragment thereof.
- an RNA disclosed herein comprises a hAg 5 ’ UTR or a fragment thereof.
- an RNA disclosed herein comprises a 5 ’ UTR having at least 80%, at least 85%, at least 90%, at least 95%, at least 96%, at least 97%, at least 98%, or at least 99% identity to a 5 ’ UTR with the sequence according to AACUAGUAUUCUUCUGGUCCCCACAGACUCAGAGAGAACCCGCCACC (SEQ ID NO: 49).
- an RNA disclosed herein comprises a 5’ UTR provided in SEQ ID NO: 49.
- a polynucleotide e.g., DNA, RNA
- a polyadenylate or poly(A) sequence e.g., as described herein.
- a poly(A) sequence is situated downstream of a 3’ UTR, e.g., adjacent to a 3’ UTR.
- RNAs disclosed herein can have a poly(A) sequence attached to the free 3 ’-end of the RNA by a template -independent RNA polymerase after transcription or a poly(A) sequence encoded by DNA and transcribed by a template-dependent RNA polymerase.
- a poly(A) sequence of about 120 A nucleotides has a beneficial influence on the levels of RNA in transfected eukaryotic cells, as well as on the levels of protein that is translated from an open reading frame that is present upstream (5’) of the poly(A) sequence (Holtkamp et al., 2006, Blood, vol. 108, pp. 4009-4017, which is incorporated herein by reference).
- a poly(A) sequence in accordance with the present disclosure is not limited to a particular length; in some embodiments, a poly(A) sequence is any length.
- a poly(A) sequence comprises, essentially consists of, or consists of at least 20, at least 30, at least 40, at least 80, or at least 100 and up to 500, up to 400, up to 300, up to 200, or up to 150 A nucleotides, and, in particular, about 120 A nucleotides.
- nucleotides in the poly(A) sequence typically at least 75%, at least 80%, at least 85%, at least 90%, at least 95%, at least 96%, at least 97%, at least 98%, or at least 99% by number of nucleotides in the poly(A) sequence are A nucleotides, but permits that remaining nucleotides are nucleotides other than A nucleotides, such as U nucleotides (uridylate), G nucleotides (guanylate), or C nucleotides (cytidylate).
- consists of means that all nucleotides in the poly(A) sequence, i.e., 100% by number of nucleotides in the poly(A) sequence, are A nucleotides.
- a nucleotide or “A” refers to adenylate.
- a poly(A) sequence is attached during RNA transcription, e.g., during preparation of in vitro transcribed RNA, based on a DNA template comprising repeated dT nucleotides (deoxythymidylate) in the strand complementary to the coding strand.
- the DNA sequence encoding a poly(A) sequence (coding strand) is referred to as poly(A) cassette.
- the poly(A) cassette present in the coding strand of DNA essentially consists of dA nucleotides, but is interrupted by a random sequence of the four nucleotides (dA, dC, dG, and dT). Such random sequence may be 5 to 50, 10 to 30, or 10 to 20 nucleotides in length.
- a cassette is disclosed in WO2016/005324, hereby incorporated by reference. Any poly(A) cassette disclosed in WO2016/005324 may be used in accordance with the present disclosure.
- a poly(A) cassette that essentially consists of dA nucleotides, but is interrupted by a random sequence having an equal distribution of the four nucleotides (dA, dC, dG, dT) and having a length of e.g., 5 to 50 nucleotides shows, on DNA level, constant propagation of plasmid DNA in E. coli and is still associated, on RNA level, with the beneficial properties with respect to supporting RNA stability and translational efficiency is encompassed.
- the poly(A) sequence contained in an RNA polynucleotide described herein essentially consists of A nucleotides, but is interrupted by a random sequence of the four nucleotides (A, C, G, U).
- Such random sequence may be 5 to 50, 10 to 30, or 10 to 20 nucleotides in length.
- no nucleotides other than A nucleotides flank a poly(A) sequence at its 3 ’-end, i.e., the poly(A) sequence is not masked or followed at its 3 ’-end by a nucleotide other than A.
- the poly(A) sequence may comprise at least 20, at least 30, at least 40, at least 80, or at least 100 and up to 500, up to 400, up to 300, up to 200, or up to 150 nucleotides. In some embodiments, the poly(A) sequence may essentially consist of at least 20, at least 30, at least 40, at least 80, or at least 100 and up to 500, up to 400, up to 300, up to 200, or up to 150 nucleotides. In some embodiments, the poly(A) sequence may consist of at least 20, at least 30, at least 40, at least 80, or at least 100 and up to 500, up to 400, up to 300, up to 200, or up to 150 nucleotides. In some embodiments, the poly(A) sequence comprises at least 100 nucleotides. In some embodiments, the poly(A) sequence comprises about 150 nucleotides. In some embodiments, the poly(A) sequence comprises about 120 nucleotides.
- a polyA tail comprises a specific number of
- Adenosines such as about 50 or more, about 60 or more, about 70 or more, about 80 or more, about 90 or more, about 100 or more, about 120, or about 150 or about 200.
- a polyA tail of a string construct may comprise 200 A residues or less.
- a polyA tail of a string construct may comprise about 200 A residues.
- a polyA tail of a string construct may comprise 180 A residues or less.
- a polyA tail of a string construct may comprise about 180 A residues.
- a polyA tail may comprise 150 residues or less.
- RNA comprises a poly(A) sequence comprising the nucleotide sequence of AAAAAAAAAAAAAAAAAAAAAAAAAAGCAUAUGACUAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA
- a polyA tail comprises a plurality of A residues interrupted by a linker.
- a linker comprises the nucleotide sequence GCAUAUGACU (SEQ ID NO: 3’ UTR
- an RNA utilized in accordance with the present disclosure comprises a 3’ UTR.
- the terms “three prime untranslated region,” “3 ’ untranslated region,” or “3 ’ UTR” refer to a sequence of an mRNA molecule that begins following a stop codon of a coding region of an open reading frame sequence.
- the 3 ’ UTR begins immediately after a stop codon of a coding region of an open reading frame sequence, e.g., in its natural context.
- the 3’ UTR does not begin immediately after stop codon of the coding region of an open reading frame sequence, e.g., in its natural context.
- the term “3’ UTR” does preferably not include the poly(A) sequence.
- the 3’ UTR is upstream of the poly(A) sequence (if present), e.g., directly adjacent to the poly(A) sequence.
- an RNA construct comprises an F element.
- a F element sequence is a 3’ UTR of amino-terminal enhancer of split (AES).
- an RNA disclosed herein comprises a 3 ’ UTR having at least 80%, at least 85%, at least 90%, at least 95%, at least 96%, at least 97%, at least 98%, or at least 99% identity to a 3 ’ UTR with the sequence according to CUGGUACUGCAUGCACGCAAUGCUAGCUGCCCCUUUCCCGUCCUGGGUACCCC GAGUCUCCCCCGACCUCGGGUCCCAGGUAUGCUCCCACCUCCACCUGCCCCACU CACCACCUCUGCUAGUUCCAGACACCUCCCAAGCACGCAGCAAUGCAGCUCAA AACGCUUAGCCUAGCCACACCCCCACGGGAAACAGCAGUGAUUAACCUUUAGC AAUAAACGAAAGUUUAACUAAGCUAUACUAACCCCAGGGUUGGUCAAUUUCG UGCCAGCCACACC (SEQ ID NO: 51).
- an RNA disclosed herein comprises a 3’ UTR provided in SEQ ID NO: 51
- a 3’UTR is an FI element as described in W02017/060314, which is incorporated herein by reference in its entirety.
- RNA compositions e.g., pharmaceutical compositions
- uRNA non-modified uridine containing mRNA
- modRNA nucleoside-modified mRNA
- saRNA self-amplifying mRNA
- RNA is capped, contains open reading frames (ORFs) flanked by untranslated regions (UTR), and have a polyA-tail at the 3’ end.
- ORFs open reading frames flanked by untranslated regions
- An ORF of an uRNA and modRNA vectors encode an incretin agent.
- An saRNA has multiple ORFs.
- the RNA described herein may have modified nucleosides.
- the RNA comprises a modified nucleoside in place of at least one (e.g., every) uridine.
- uracil describes one of the nucleobases that can occur in the nucleic acid of RNA.
- the structure of uracil is:
- uridine describes one of the nucleosides that can occur in RNA.
- the structure of uridine is:
- UTP (uridine 5 ’-triphosphate) has the following structure:
- Pseudo-UTP (pseudouridine 5 ’-triphosphate) has the following structure:
- ‘Pseudouridine” is one example of a modified nucleoside that is an isomer of uridine, where the uracil is attached to the pentose ring via a carbon-carbon bond instead of a nitrogen-carbon glycosidic bond.
- nucleoside is N1 -methyl -pseudouridine (m I T). which has the structure:
- N1 -methyl -pseudo-UTP has the following structure:
- RNA comprises a modified nucleoside in place of at least one uridine. In some embodiments, RNA comprises a modified nucleoside in place of each uridine.
- the modified nucleoside is independently selected from pseudouridine ( ⁇ ), N1-methyl-pseudouridine (m1 ⁇ ), and 5-methyl-uridine (m5U).
- the modified nucleoside comprises pseudouridine ( ⁇ ). In some embodiments, the modified nucleoside comprises N1-methyl-pseudouridine (m1 ⁇ ). In some embodiments, the modified nucleoside comprises 5-methyl-uridine (m5U). In some embodiments, RNA may comprise more than one type of modified nucleoside, and the modified nucleosides are independently selected from pseudouridine ( ⁇ ), N1-methyl-pseudouridine (m1 ⁇ ), and 5- methyl-uridine (m5U). In some embodiments, the modified nucleosides comprise pseudouridine ( ⁇ ) and N1-methyl-pseudouridine (m1 ⁇ ).
- the modified nucleosides comprise pseudouridine ( ⁇ ) and 5-methyl-uridine (m5U). In some embodiments, the modified nucleosides comprise N1-methyl-pseudouridine (m1 ⁇ ) and 5-methyl-uridine (m5U). In some embodiments, the modified nucleosides comprise pseudouridine ( ⁇ ), N1- methyl-pseudouridine (m1 ⁇ ), and 5-methyl-uridine (m5U).
- the modified nucleoside replacing one or more, e.g., all, uridine in the RNA may be any one or more of 3-methyl-uridine (m3U), 5-methoxy- uridine (mo5U), 5-aza-uridine, 6-aza-uridine, 2-thio-5-aza-uridine, 2-thio-uridine (s2U), 4- thio-uridine (s4U), 4-thio-pseudouridine, 2-thio-pseudouridine, 5 -hydroxy-uridine (ho5U), 5- aminoallyl-uridine, 5-halo-uridine (e.g., 5-iodo-uridine or 5-bromo-uridine), uridine 5- oxyacetic acid (cmo5U), uridine 5-oxyacetic acid methyl ester (mcmo5U), 5 -carboxymethyl - uridine (cm5U), 1-carboxymethyl-pseudouridine, 5-car
- 2-thio-2'-O- methyl-uridine (s2Um), 5-methoxycarbonylmethyl-2'-O-methyl-uridine (mcm5Um), 5- carbamoyhnethyl-2'-O-methyl-uridine (ncm5Um), 5-carboxymethylaminomethyl-2'-O- methyl-uridine (cmnm5Um), 3,2'-O-dimethyl-uridine (m3Um), 5-(isopentenylaminomethyl)- 2'-O-methyl-uridine (inm5Um), 1 -thio-uridine, deoxythymidine, 2'-F-ara-uridine, 2'-F- uridine, 2'-OH-ara-uridine, 5-(2-carbomethoxyvinyl) uridine, 5-[3-(l-E- propenylamino)uridine, or any other modified uridine known in the art.
- the RNA comprises other modified nucleosides or comprises further modified nucleosides, e.g., modified cytidine.
- modified cytidine in the RNA 5 -methylcytidine is substituted partially or completely, preferably completely, for cytidine.
- the RNA comprises 5 -methylcytidine and one or more selected from pseudouridine ( ⁇ ), N1-methyl-pseudouridine (m1 ⁇ ), and 5- methyl-uridine (m5U).
- the RNA comprises 5-methylcytidine and N1- methyl-pseudouridine (m1 ⁇ ).
- the RNA comprises 5-methylcytidine in place of each cytidine and N1-methyl-pseudouridine (m1 ⁇ ) in place of each uridine.
- the RNA is “replicon RNA” or simply a “replicon,” in particular “self-replicating RNA” or “self-amplifying RNA.”
- the replicon or self-replicating RNA is derived from or comprises elements derived from a single-stranded (ss) RNA virus, in particular a positive- stranded ssRNA virus, such as an alphavirus. Alphaviruses are typical representatives of positive-stranded RNA viruses.
- Alphaviruses replicate in the cytoplasm of infected cells (for review of the alphaviral life cycle see Jose et al., Future Microbiol., 2009, vol.4, pp.837– 856, which is incorporated herein by reference in its entirety).
- the total genome length of many alphaviruses typically ranges between 11,000 and 12,000 nucleotides, and the genomic RNA typically has a 5’ cap, and a 3’ poly(A) tail.
- the genome of alphaviruses encodes non- structural proteins (involved in transcription, modification and replication of viral RNA and in protein modification) and structural proteins (forming the virus particle). There are typically two open reading frames (ORFs) in the genome.
- the four non-structural proteins are typically encoded together by a first ORF beginning near the 5′ terminus of the genome, while alphavirus structural proteins are encoded together by a second ORF which is found downstream of the first ORF and extends near the 3’ terminus of the genome.
- first ORF is larger than the second ORF, the ratio being roughly 2:1.
- RNA RNA molecule that resembles eukaryotic messenger RNA
- mRNA messenger RNA
- the (+) stranded genomic RNA directly acts like a messenger RNA for the translation of the open reading frame encoding the non-structural polyprotein (nsP1234).
- Alphavirus-derived vectors have been proposed for delivery of foreign genetic information into target cells or target organisms.
- a first ORF encodes an alphavirus-derived RNA-dependent RNA polymerase (replicase), which upon translation mediates self-amplification of the RNA.
- a second ORF encoding alphaviral structural proteins is replaced by an open reading frame encoding a protein of interest, e.g., an incretin agent.
- Alphavirus-based trans-replication systems rely on alphavirus nucleotide sequence elements on two separate nucleic acid molecules: one nucleic acid molecule encodes a viral replicase, and the other nucleic acid molecule is capable of being replicated by said replicase in trans (hence the designation trans-replication system).
- Trans-replication requires the presence of both these nucleic acid molecules in a given host cell.
- the nucleic acid molecule capable of being replicated by the replicase in trans must comprise certain alphaviral sequence elements to allow recognition and RNA synthesis by the alphaviral replicase.
- non-modified uridine platform may include, for example, one or more of intrinsic adjuvant effect, as well as good tolerability and safety.
- modified uridine (e.g., pseudouridine) platform may include reduced adjuvant effect, blunted immune innate immune sensor activating capacity and thus good tolerability and safety.
- self-amplifying platform may include, for example, long duration of protein expression, good tolerability and safety, higher likelihood for efficacy with very low vaccine dose.
- RNA constructs optimized for example, for improved manufacturability, encapsulation, expression level (and/or timing), etc. Certain components are discussed below, and certain preferred embodiments are exemplified herein.
- coding regions are codon-optimized for optimal expression in a subject to be treated using the RNA molecules described herein.
- codon-optimization may be performed such that codons for which frequently occurring tRNAs are available are inserted in place of “rare codons.”
- codon-optimization may include increasing guanosine/cytosine (G/C) content of a coding region of RNA described herein as compared to the G/C content of the corresponding coding sequence of a wild type RNA, wherein the amino acid sequence encoded by the RNA is preferably not modified compared to the amino acid sequence.
- G/C guanosine/cytosine
- a coding sequence (also referred to as a “coding region”) is codon optimized for expression in the subject to whom a composition (e.g., a pharmaceutical composition) is to be administered (e.g., a human).
- a composition e.g., a pharmaceutical composition
- sequences in such a polynucleotide may differ from wild type sequences encoding the relevant incretin agent, even when the amino acid sequence of the incretin agent is wild type.
- strategies for codon optimization for expression in a relevant subject e.g., a human
- a relevant subject e.g., a human
- codon bias differs in codon usage between organisms
- mRNA messenger RNA
- tRNA transfer RNA
- the predominance of selected tRNAs in a cell may generally be a reflection of the codons used most frequently in peptide synthesis. Accordingly, genes may be tailored for optimal gene expression in a given organism based on codon optimization.
- Codon usage tables are available, for example, at the “Codon Usage Database” available at www.kazusa.oijp/codon/ and these tables may be adapted in a number of ways.
- Computer algorithms for codon optimizing a particular sequence for expression in a particular subject or its cells are also available, such as Gene Forge (Aptagen; Jacobus, PA), are also available.
- a polynucleotide (e.g., a polyribonucleotide) of the present disclosure is codon optimized, wherein the codons in the polynucleotide (e.g., the polyribonucleotide) are adapted to human codon usage (herein referred to as “human codon optimized polynucleotide”). Codons encoding the same amino acid occur at different frequencies in a subject, e.g., a human.
- the coding sequence of a polynucleotide of the present disclosure is modified such that the frequency of the codons encoding the same amino acid corresponds to the naturally occurring frequency of that codon according to the human codon usage, e.g., as shown in Table 13.
- the wild type coding sequence is preferably adapted in a way that the codon “GCC” is used with a frequency of 0.40, the codon “GCT” is used with a frequency of 0.28, the codon “GCA” is used with a frequency of 0.22 and the codon “GCG” is used with 30 a frequency of 0. 10 etc. (see Table 13).
- such a procedure (as exemplified for Ala) is applied for each amino acid encoded by the coding sequence of a polynucleotide to obtain sequences adapted to human codon usage.
- Table 13 Human codon usage table with frequencies indicated for each amino acid.
- a coding sequence may be optimized using a multiparametric optimization strategy.
- optimization parameters may include parameters that influence protein expression, which can be, for example, impacted on a transcription level, an mRNA level, and/or a translational level.
- exemplary optimization parameters include, but are not limited to transcription-level parameters (including, e.g., GC content, consensus splice sites, cryptic splice sites, SD sequences, TATA boxes, termination signals, artificial recombination sites, and combinations thereof); mRNA-level parameters (including, e.g., RNA instability motifs, ribosomal entry sites, repetitive sequences, and combinations thereof); translation-level parameters (including, e.g., codon usage, premature poly(A) sites, ribosomal entry sites, secondary structures, and combinations thereof); or combinations thereof.
- a coding sequence may be optimized by a GeneOptimizer algorithm as described in Fath et al.
- a coding sequence may be optimized by Eurofms’ adaption and optimization algorithm “GENEius” as described in Eurofms’ Application Notes: Eurofms’ adaption and optimization software “GENEius” in comparison to other optimization algorithms, the entire content of which is incorporated by reference for the purposes described herein.
- a coding sequence utilized in accordance with the present disclosure has G/C content which is increased compared to a wild type coding sequence for a relevant incretin agent.
- guanosine/cytidine (G/C) content of a coding region is modified relative to a wild type coding sequence for a relevant incretin agent, but the amino acid sequence encoded by the polyribonucleotide is not modified.
- GC enrichment may improve translation of a payload sequence.
- sequences having an increased G (guanosine )/C (cytidine) content are more stable than sequences having an increased A (adenosine )/U (uridine) content.
- the most favorable codons for the stability can be determined (so-called alternative codon usage).
- alternative codon usage the amino acid to be encoded by a polyribonucleotide, there are various possibilities for modification of the ribonucleic acid sequence, compared to its wild type sequence.
- codons which contain A and/or U nucleosides can be modified by substituting these codons by other codons, which code for the same amino acids but contain no A and/or U or contain a lower content of A and/or U nucleosides.
- G/C content of a coding region of a polyribonucleotide described herein is increased by at least 1%, at least 2%, at least 3%, at least 4%, at least 5%, at least 6%, or even more compared to the G/C content of the coding region prior to codon optimization, e.g., of the wild type RNA.
- G/C content of a coding region of a polyribonucleotide described herein is decreased by at least 1%, at least 2%, at least 3%, at least 4%, at least 5%, at least 6%, or even more compared to the G/C content of the coding region prior to codon optimization, e.g., of the wild type RNA.
- a polyribonucleotide comprises a ribonucleic acid sequence that is at least 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or at least 99% identical to any of the sequences shown below in Table 14.
- the polyribonucleotides include exemplary' polyribonucleotide features described herein in addition to a sequence encoding die incretin agent and signal peptide, including a Cap proximal sequence (AAUA), a 5 " UTR sequence
- one or more polyribonucleotides can be formulated with lipid nanoparticles for delivery (e.g., administration).
- lipid nanoparticles can be designed to protect polyribonucleotides from extracellular RNases and/or engineered for systemic delivery of the RNA to target cells (e.g., liver, gut or pancreas cells). In some embodiments, such lipid nanoparticles may be particularly useful to deliver polyribonucleotides when polyribonucleotides are intraperitoneally, intravenously or intramuscularly administered to a subject. Particles for Delivery of at Least One Polyribonucleotide [0336] Polyribonucleotides provided herein can be delivered by particles. In the context of the present disclosure, the term “particle” relates to a structured entity formed by molecules or molecule complexes.
- nucleic acid particles comprise more than one type of nucleic acid molecules (e.g., polyribonucleotides), where the molecular parameters of the nucleic acid molecules may be similar or different from each other, like with respect to molar mass or fundamental structural elements such as molecular architecture, capping, coding regions or other features.
- provided nucleic acid particles e.g., ribonucleic acid particles
- nanoparticle refers to a particle having an average diameter suitable for parenteral administration.
- lipid nanoparticles can have an average size (e.g., mean diameter) of about 30 nm to about 150 nm, about 40 nm to about 150 nm, about 50 nm to about 150 nm, about 60 nm to about 130 nm, about 70 nm to about 110 nm, about 70 nm to about 100 nm, about 70 to about 90 nm, or about 70 nm to about 80 nm.
- lipid nanoparticles in accordance with the present disclosure can have an average size (e.g., mean diameter) of about 50 nm to about 100 nm.
- lipid nanoparticles may have an average size (e.g., mean diameter) of about 50 nm to about 150 nm. In some embodiments, lipid nanoparticles may have an average size (e.g., mean diameter) of about 60 nm to about 120 nm.
- Nucleic acid particles e.g., ribonucleic acid particles described herein may exhibit a polydispersity index less than about 0.5, less than about 0.4, less than about 0.3, or about 0.2 or less.
- the nucleic acid particles e.g., ribonucleic acid particles
- nucleic acid particle e.g., a ribonucleic acid particle
- N/P ratio greater than or equal to 5.
- nucleic acid particle e.g., a ribonucleic acid particle
- nucleic acid particle e.g., a ribonucleic acid particle
- an N/P ratio for a nucleic acid particle (e.g., a ribonucleic acid particle) described herein is from about 10 to about 50. In some embodiments, an N/P ratio for a nucleic acid particle (e.g., a ribonucleic acid particle) described herein is from about 10 to about 70. In some embodiments, an N/P ratio for a nucleic acid particle (e.g., a ribonucleic acid particle) described herein is from about 10 to about 120.
- Nucleic acid particles e.g., ribonucleic acid particles
- Nucleic acid particles can be prepared using a wide range of methods that may involve obtaining a colloid from at least one cationic or cationically ionizable lipid or lipid-like material and/or at least one cationic polymer and mixing the colloid with nucleic acid to obtain nucleic acid particles.
- the term “colloid” as used herein relates to a type of homogeneous mixture in which dispersed particles do not settle out.
- the insoluble particles in the mixture can be microscopic, with particle sizes between 1 and 1000 nanometers.
- the mixture may be termed a colloid or a colloidal suspension.
- the “polydispersity index” is preferably calculated based on dynamic light scattering measurements by the so-called cumulant analysis as mentioned in the definition of the “average diameter.” Under certain prerequisites, it can be taken as a measure of the size distribution of an ensemble of ribonucleic acid nanoparticles (e.g., ribonucleic acid nanoparticles).
- ribonucleic acid nanoparticles e.g., ribonucleic acid nanoparticles.
- nucleic acid particles have been described previously to be suitable for delivery of nucleic acid in particulate form (e.g., Kaczmarek et al., 2017, Genome Medicine 9, 60, which is incorporated herein by reference).
- the nucleic acid particles may comprise nucleic acid (e.g., a polyribonucleotide) which is complexed in different forms by non- covalent interactions to the particle.
- the particles described herein are not viral particles, in particular, they are not infectious viral particles, i.e., they are not able to virally infect cells.
- Some embodiments described herein relate to compositions, methods and uses involving more than one, e.g., 2, 3, 4, 5, 6 or even more nucleic acid species (e.g., polyribonucleotide species).
- nucleic acid particle e.g., ribonucleic acid particle, e.g., ribonucleic acid nanoparticle
- each nucleic acid species e.g., polyribonucleotide species
- each individual nucleic acid particle e.g., ribonucleic acid particle, e.g., ribonucleic acid nanoparticle
- each individual nucleic acid particle e.g., ribonucleic acid particle, e.g., ribonucleic acid nanoparticle
- each individual nucleic acid particle e.g., ribonucleic acid particle, e.g., ribonucleic acid nanoparticle
- formulation will comprise one nucleic acid species (e.g., polyribonucleotide species).
- the individual nucleic acid particle (e.g., ribonucleic acid particle, e.g., ribonucleic acid nanoparticle) formulations may be present as separate entities, e.g., in separate containers.
- Such formulations are obtainable by providing each nucleic acid species (e.g., polyribonucleotide species) separately (typically each in the form of a nucleic acid-containing solution) together with a particle-forming agent, thereby allowing the formation of particles.
- Respective particles will contain exclusively the specific nucleic acid species (e.g., polyribonucleotide species) that is being provided when the particles are formed (individual particulate formulations).
- a composition such as a pharmaceutical composition comprises more than one individual nucleic acid particle (e.g., ribonucleic acid particle, e.g., ribonucleic acid nanoparticle) formulation.
- Respective pharmaceutical compositions are referred to as “mixed particulate formulations.”
- Mixed particulate formulations according to the invention are obtainable by forming, separately, individual nucleic acid particle (e.g., ribonucleic acid particle, e.g., ribonucleic acid nanoparticle) formulations, as described above, followed by a step of mixing of the individual nucleic acid particle (e.g., ribonucleic acid particle, e.g., ribonucleic acid nanoparticle) formulations.
- a formulation comprising a mixed population of nucleic acid-containing particles is obtainable.
- Individual nucleic acid particle e.g., ribonucleic acid particle, e.g., ribonucleic acid nanoparticle
- populations may be together in one container, comprising a mixed population of individual nucleic acid particle (e.g., ribonucleic acid particle, e.g., ribonucleic acid nanoparticle) formulations.
- nucleic acid species e.g., polyribonucleotide species
- a “combined particulate formulation” Such formulations are obtainable by providing a combined formulation (typically combined solution) of different nucleic acid species (e.g., polyribonucleotide species) species together with a particle-forming agent, thereby allowing the formation of particles.
- a “combined particulate formulation” will typically comprise particles that comprise more than one nucleic acid species (e.g., polyribonucleotide species) species.
- nucleic acid species e.g., polyribonucleotide species
- nucleic acids e.g., polyribonucleotides
- nucleic acid particles e.g., ribonucleic acid particles, e.g., lipid nanoparticles
- nucleic acid particles are lipid nanoparticles.
- lipid nanoparticles are liver-targeting lipid nanoparticles.
- lipid nanoparticles are cationic lipid nanoparticles comprising one or more cationic lipids (e.g., ones described herein).
- cationic lipid nanoparticles may comprise at least one cationic lipid, at least one polymer- conjugated lipid, and at least one helper lipid (e.g., at least one neutral lipid).
- Cationic Polymeric Materials [0355] Cationic polymers have been recognized as useful for developing particle delivery vehicles, as reported in PCT App. Pub. No. WO2021/001417, the entirety of which is incorporated herein by reference.
- polymer refers to a composition comprising one or more molecules that comprise repeating units of one or more monomers.
- polymer refers to a composition of polymer molecules.
- a polymer composition comprises polymer molecules having molecules of different lengths (e.g., comprising varying amounts of monomers).
- Polymer compositions described herein are characterized by one or more of a normalized molecular weight (Mn), a weight average molecular weight (Mw), and/or a polydispersity index (PDI).
- Mn normalized molecular weight
- Mw weight average molecular weight
- PDI polydispersity index
- such repeat units can all be identical (a “homopolymer”); alternatively, in some cases, there can be more than one type of repeat unit present within the polymeric material (a “heteropolymer” or a “copolymer”).
- a polymer is biologically derived, e.g., a biopolymer such as a protein.
- additional moieties can also be present in the polymeric material, for example targeting moieties such as those described herein.
- a polymer utilized in accordance with the present disclosure may be a copolymer. Repeat units forming the copolymer can be arranged in any fashion.
- repeat units can be arranged in a random order; alternatively or additionally, in some embodiments, repeat units may be arranged in an alternating order, or as a “block” copolymer, e.g., comprising one or more regions each comprising a first repeat unit (e.g., a first block), and one or more regions each comprising a second repeat unit (e.g., a second block), etc.
- Block copolymers can have two (a diblock copolymer), three (a triblock copolymer), or more numbers of distinct blocks.
- a polymeric material for use in accordance with the present disclosure is biocompatible.
- a biocompatible material is biodegradable, e.g., is able to degrade, chemically and/or biologically, within a physiological environment, such as within the body.
- a polymeric material may be or comprise protamine or polyalkyleneimine.
- protamine is often used to refer to any of various strongly basic proteins of relatively low molecular weight that are rich in arginine and are found associated especially with DNA in place of somatic histones in the sperm cells of various animals (as fish).
- protamine is often used to refer to proteins found in fish sperm that are strongly basic, are soluble in water, are not coagulated by heat, and yield chiefly arginine upon hydrolysis. In purified form, they are used in a long-acting formulation of insulin and to neutralize the anticoagulant effects of heparin.
- protamine as used herein is refers to a protamine amino acid sequence obtained or derived from natural or biological sources, including fragments thereof and/or multimeric forms of said amino acid sequence or fragment thereof, as well as (synthesized) polypeptides which are artificial and specifically designed for specific purposes and cannot be isolated from native or biological sources.
- a polyalkyleneimine comprises polyethylenimine (PEI) and/or polypropylenimine.
- PEI polyethylenimine
- a preferred polyalkyleneimine is polyethyleneimine (PEI).
- the average molecular weight of PEI is preferably 0.75 ⁇ 102 to 107 Da, preferably 1000 to 105 Da, more preferably 10000 to 40000 Da, more preferably 15000 to 30000 Da, even more preferably 20000 to 25000 Da.
- Cationic materials e.g., polymeric materials, including polycationic polymers
- contemplated for use herein include those which are able to electrostatically bind nucleic acid.
- cationic polymeric materials contemplated for use herein include any cationic polymeric materials with which nucleic acid can be associated, e.g., by forming complexes with the nucleic acid or forming vesicles in which the nucleic acid is enclosed or encapsulated.
- particles described herein may comprise polymers other than cationic polymers, e.g., non-cationic polymeric materials and/or anionic polymeric materials. Collectively, anionic and neutral polymeric materials are referred to herein as non- cationic polymeric materials.
- Lipid Compositions Lipids and Lipid-Like Materials Lipids and lipid-like materials have also been recognized as useful for developing particle delivery vehicles.
- lipid and “lipid-like material” are broadly defined herein as molecules which comprise one or more hydrophobic moieties or groups and optionally also one or more hydrophilic moieties or groups. Molecules comprising hydrophobic moieties and hydrophilic moieties are also frequently denoted as amphiphiles. Lipids are usually poorly soluble in water. In an aqueous environment, the amphiphilic nature allows the molecules to self-assemble into organized structures and different phases. One of those phases consists of lipid bilayers, as they are present in vesicles, multilamellar/unilamellar liposomes, or membranes in an aqueous environment.
- the term includes compounds that are able to form amphiphilic layers as they are present in vesicles, multilamellar/unilamellar liposomes, or membranes in an aqueous environment and includes surfactants, or synthesized compounds with both hydrophilic and hydrophobic moieties.
- the term refers to molecules which comprise hydrophilic and hydrophobic moieties with different structural organization, which may or may not be similar to that of lipids.
- Specific examples of amphiphilic compounds that may be included in an amphiphilic layer include, but are not limited to, phospholipids, aminolipids and sphingolipids.
- lipids may be divided into eight categories: fatty acids, glycerolipids, glycerophospholipids, sphingolipids, saccharolipids, polyketides (derived from condensation of ketoacyl subunits), sterols and prenol lipids (derived from condensation of isoprene subunits).
- lipid is sometimes used as a synonym for fats, fats are a subgroup of lipids called triglycerides.
- Lipids also encompass molecules such as fatty acids and their derivatives (including tri-, di-, monoglycerides, and phospholipids), as well as sterol-containing metabolites such as cholesterol.
- Fatty acids are a diverse group of molecules made of a hydrocarbon chain that terminates with a carboxylic acid group; this arrangement confers the molecule with a polar, hydrophilic end, and a nonpolar, hydrophobic end that is insoluble in water.
- the carbon chain typically between four and 24 carbons long, may be saturated or unsaturated, and may be attached to functional groups containing oxygen, halogens, nitrogen, and sulfur. If a fatty acid contains a double bond, there is the possibility of either a cis or trans geometric isomerism, which significantly affects the molecule’s configuration. Cis-double bonds cause the fatty acid chain to bend, an effect that is compounded with more double bonds in the chain.
- Glycerolipids are composed of mono-, di-, and tri-substituted glycerols, the best-known being the fatty acid triesters of glycerol, called triglycerides.
- triglycerides The word “triacylglycerol” is sometimes used synonymously with “triglyceride”. In these compounds, the three hydroxyl groups of glycerol are each esterified, typically by different fatty acids.
- glycosylglycerols are characterized by the presence of one or more sugar residues attached to glycerol via a glycosidic linkage.
- Glycerophospholipids are amphipathic molecules (containing both hydrophobic and hydrophilic regions) that contain a glycerol core linked to two fatty acid- derived “tails” by ester linkages and to one “head” group by a phosphate ester linkage.
- glycerophospholipids usually referred to as phospholipids (though sphingomyelins are also classified as phospholipids) are phosphatidylcholine (also known as PC, GPCho or lecithin), phosphatidylethanolamine (PE or GPEtn) and phosphatidylserine (PS or GPSer).
- Sphingolipids are members of a complex family of compounds that share a common structural feature, a sphingoid base backbone. The major sphingoid base in mammals is commonly referred to as sphingosine.
- Ceramides are a major subclass of sphingoid base derivatives with an amide-linked fatty acid.
- the fatty acids are typically saturated or mono-unsaturated with chain lengths from 16 to 26 carbon atoms.
- the major phosphosphingolipids of mammals are sphingomyelins (ceramide phosphocholines), whereas insects contain mainly ceramide phosphoethanolamines and fungi have phytoceramide phosphoinositols and mannose-containing headgroups.
- the glycosphingolipids are a diverse family of molecules composed of one or more sugar residues linked via a glycosidic bond to the sphingoid base.
- glycosphingolipids such as cerebrosides and gangliosides.
- Sterols such as cholesterol and its derivatives, or tocopherol and its derivatives, are important components of membrane lipids, along with the glycerophospholipids and sphingomyelins.
- Saccharolipids are compounds in which fatty acids are linked directly to a sugar backbone, forming structures that are compatible with membrane bilayers. In the saccharolipids, a monosaccharide substitutes for the glycerol backbone present in glycerolipids and glycerophospholipids.
- Polyketides are synthesized by polymerization of acetyl and propionyl subunits by classic enzymes as well as iterative and multimodular enzymes that share mechanistic features with the fatty acid synthases. They comprise a large number of secondary metabolites and natural products from animal, plant, bacterial, fungal and marine sources, and have great structural diversity. Many polyketides are cyclic molecules whose backbones are often further modified by glycosylation, methylation, hydroxylation, oxidation, or other processes. [0376] Lipids and lipid-like materials may be cationic, anionic or neutral. Neutral lipids or lipid-like materials exist in an uncharged or neutral zwitterionic form at a selected pH.
- suitable lipids or lipid-like materials for use in the present disclosure include those described in WO2020/128031 and US2020/0163878, the entire contents of each of which are incorporated herein by reference for the purposes described herein.
- Cationic or Cationically Ionizable Lipids or Lipid-like Materials [0378] In some embodiments cationic or cationically ionizable lipids or lipid-like materials contemplated for use herein include any cationic or cationically ionizable lipids or lipid-like materials which are able to electrostatically bind nucleic acid.
- cationic or cationically ionizable lipids or lipid-like materials contemplated for use herein can be associated with nucleic acid, e.g., by forming complexes with the nucleic acid or forming vesicles in which the nucleic acid is enclosed or encapsulated.
- Cationic lipids or lipid-like materials are characterized in that they have a net positive charge (e.g., at a relevant pH). Cationic lipids or lipid-like materials bind negatively charged nucleic acid by electrostatic interaction.
- a cationic or cationically ionizable lipid or lipid-like material comprises a head group which includes at least one nitrogen atom (N) which is positive charged or capable of being protonated.
- cationic lipids include, but are not limited to 1,2-dioleoyl-3- trimethylammonium propane (DOTAP); N,N-dimethyl-2,3-dioleyloxypropylamine (DODMA), 1,2-di-O-octadecenyl-3-trimethylammonium propane (DOTMA), 3-(N—(N′,N′- dimethylaminoethane)-carbamoyl)cholesterol (DC-Chol), dimethyldioctadecylammonium (DDAB); 1,2-dioleoyl-3-dimethylammonium-propane (DODAP); 1,2-diacyloxy-3- dimethylammonium propanes; 1,2-dioleoyl-3- trimethylam
- Suitable cationic lipids for use in the present disclosure include those described in WO2020/128031 and US2020/0163878, the entire contents of each of which are incorporated herein by reference for the purposes described herein.
- Further suitable cationic lipids for use in the present disclosure include those described in WO2010/053572 (including C12-200 described at paragraph [00225]) and WO2012/170930, both of which are incorporated herein by reference for the purposes described herein.
- Additional suitable cationic lipids for use in the present disclosure include HGT4003, HGT5000, HGTS001, HGT5001, HGT5002 (see US2015/0140070, which is incorporated herein by reference in its entirety).
- formulations that are useful for pharmaceutical compositions can comprise at least one cationic lipid.
- Representative cationic lipids include, but are not limited to, 1 ,2-dilinoleyoxy-3-(dimethylamino)acetoxypropane (DLin-DAC), 1 ,2- dilinoleyoxy-3morpholinopropane (DLin-MA), 1,2-dilinoleoyl-3-dimethylaminopropane (DLinDAP), 1 ,2-dilinoleylthio-3-dimethylaminopropane (DLin-S-DMA), 1 -linoleoyl-2- linoleyloxy-3dimethylaminopropane (DLin-2-DMAP), 1 ,2-dilinoleyloxy-3- trimethylaminopropane chloride salt (DLin-TMA.CI),
- a cationic lipid may comprise from about 10 mol % to about 100 mol %, about 20 mol % to about 100 mol %, about 30 mol % to about 100 mol %, about 40 mol % to about 100 mol %, or about 50 mol % to about 100 mol % of total lipid present in a lipid composition utilized in accordance with the present disclosure.
- formulations utilized in accordance with the present disclosure may comprise lipids or lipid-like materials other than cationic or cationically ionizable lipids or lipid-like materials, i.e., non-cationic lipids or lipid-like materials (including non-cationically ionizable lipids or lipid-like materials).
- non-cationic lipids or lipid-like materials including non-cationically ionizable lipids or lipid-like materials.
- anionic and neutral lipids or lipid-like materials are referred to herein as non-cationic lipids or lipid-like materials.
- optimizing a formulation of nucleic acid particles by addition of other hydrophobic moieties, such as cholesterol and lipids, in addition to an ionizable/cationic lipid or lipid-like material may, for example, enhance particle stability and efficacy of nucleic acid delivery.
- a lipid or lipid-like material may be incorporated which may or may not affect the overall charge of particles.
- such lipid or lipid-like material is a non-cationic lipid or lipid-like material.
- a non-cationic lipid may comprise, e.g., one or more anionic lipids and/or neutral lipids.
- a “anionic lipid” is negatively charged (e.g., at a selected pH).
- a “helper lipid” exists either in an uncharged, neutral zwitterionic form (e.g., at a selected pH), or, in some embodiments, as having a cationic or positive charge at physiological pH.
- a formulation comprises one of the following helper lipid components: (1) a phospholipid, (2) cholesterol or a derivative thereof; or (3) a mixture of a phospholipid and cholesterol or a derivative thereof.
- cholesterol derivatives include, but are not limited to, cholestanol, cholestanone, cholestenone, coprostanol, cholesteryl-2’-hydroxyethyl ether, cholesteryl-4’-hydroxybutyl ether, tocopherol and derivatives thereof, and mixtures thereof.
- Specific exemplary phospholipids that can be used include, but are not limited to, phosphatidylcholines, phosphatidylethanolamines, phosphatidylglycerols, phosphatidic acids, phosphatidylserines or sphingomyelin.
- Such phospholipids include in particular diacylphosphatidylcholines, such as distearoylphosphatidylcholine (DSPC), dioleoylphosphatidylcholine (DOPC), dimyristoylphosphatidylcholine (DMPC), dipentadecanoylphosphatidylcholine, dilauroylphosphatidylcholine, dipalmitoylphosphatidylcholine (DPPC), diarachidoylphosphatidylcholine (DAPC), dibehenoylphosphatidylcholine (DBPC), ditricosanoylphosphatidylcholine (DTPC), dilignoceroylphatidylcholine (DLPC), palmitoyloleoyl-phosphatidylcholine (POPC), 1,2-di- O-octadecenyl-sn-glycero-3-phosphocholine (18:0 Diether PC), 1-oleoyl-2
- a formulation utilized in accordance with the present disclosure includes DSPC or DSPC and cholesterol.
- a formulation utilized in accordance with the present disclosure comprises a helper lipid that is ionizable or cationic.
- formulations utilized in accordance with the present disclosure include both a cationic lipid and an additional (non-cationic) lipid.
- formulations herein include a polymer conjugated lipid such as a pegylated lipid. “Pegylated lipids” or “PEG-conjugated lipids” comprise both a lipid portion and a polyethylene glycol portion.
- RNA lipoplex particle contains lipid, in particular cationic lipid, and RNA. Electrostatic interactions between positively charged liposomes and negatively charged RNA results in complexation and spontaneous formation of RNA lipoplex particles. Positively charged liposomes may be generally synthesized using a cationic lipid, such as DOTMA, and additional lipids, such as DOPE. In one embodiment, a RNA lipoplex particle is a nanoparticle. [0399] In certain embodiments, RNA lipoplex particles include both a cationic lipid and an additional lipid. In an exemplary embodiment, the cationic lipid is DOTMA and the additional lipid is DOPE.
- the molar ratio of the at least one cationic lipid to the at least one additional lipid is from about 10:0 to about 1:9, about 4:1 to about 1:2, or about 3:1 to about 1:1. In specific embodiments, the molar ratio may be about 3:1, about 2.75:1, about 2.5:1, about 2.25:1, about 2:1, about 1.75:1, about 1.5:1, about 1.25:1, or about 1:1. In an exemplary embodiment, the molar ratio of the at least one cationic lipid to the at least one additional lipid is about 2:1.
- RNA lipoplex particles have an average diameter that in one embodiment ranges from about 200 nm to about 1000 nm, from about 200 nm to about 800 nm, from about 250 to about 700 nm, from about 400 to about 600 nm, from about 300 nm to about 500 nm, or from about 350 nm to about 400 nm.
- RNA lipoplex particles have an average diameter that ranges from about 250 nm to about 700 nm. In another embodiment, the RNA lipoplex particles have an average diameter that ranges from about 300 nm to about 500 nm. In an exemplary embodiment, the RNA lipoplex particles have an average diameter of about 400 nm.
- RNA lipoplex particles and compositions comprising RNA lipoplex particles described herein are useful for delivery of RNA to a target tissue after parenteral administration, in particular after intravenous administration.
- the RNA lipoplex particles may be prepared using liposomes that may be obtained by injecting a solution of the lipids in ethanol into water or a suitable aqueous phase.
- the aqueous phase has an acidic pH. In one embodiment, the aqueous phase comprises acetic acid, e.g., in an amount of about 5 mM.
- Liposomes may be used for preparing RNA lipoplex particles by mixing the liposomes with RNA. In one embodiment, the liposomes and RNA lipoplex particles comprise at least one cationic lipid and at least one additional lipid. In one embodiment, the at least one cationic lipid comprises 1,2-di-O-octadecenyl-3-trimethylammonium propane (DOTMA) and/or 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP).
- DOTMA 1,2-di-O-octadecenyl-3-trimethylammonium propane
- DOTAP 1,2-dioleoyl-3-trimethylammonium-propane
- the at least one additional lipid comprises 1,2-di-(9Z-octadecenoyl)-sn-glycero- 3-phosphoethanolamine (DOPE), cholesterol (Chol) and/or 1,2-dioleoyl-sn-glycero-3- phosphocholine (DOPC).
- the at least one cationic lipid comprises 1,2-di- O-octadecenyl-3-trimethylammonium propane (DOTMA) and the at least one additional lipid comprises 1,2-di-(9Z-octadecenoyl)-sn-glycero-3-phosphoethanolamine (DOPE).
- DOPE 1,2-di-(9Z-octadecenoyl)-sn-glycero-3-phosphoethanolamine
- the liposomes and RNA lipoplex particles comprise 1,2-di-O-octadecenyl-3- trimethylammonium propane (DOTMA) and 1,2-di-(9Z-octadecenoyl)-sn-glycero-3- phosphoethanolamine (DOPE).
- DOTMA 1,2-di-O-octadecenyl-3- trimethylammonium propane
- DOPE 1,2-di-(9Z-octadecenoyl)-sn-glycero-3- phosphoethanolamine
- Spleen targeting RNA lipoplex particles are described in WO2013/143683, incorporated herein by reference. It has been found that RNA lipoplex particles having a net negative charge may be used to preferentially target spleen tissue or spleen cells such as antigen-presenting cells, in particular dendritic cells.
- RNA lipoplex particles of the disclosure may be used for expressing RNA in the spleen.
- no or essentially no RNA accumulation and/or RNA expression in the lung and/or liver occurs.
- RNA accumulation and/or RNA expression in antigen presenting cells such as professional antigen presenting cells in the spleen occurs.
- RNA lipoplex particles of the disclosure may be used for expressing RNA in such antigen presenting cells.
- the antigen presenting cells are dendritic cells and/or macrophages.
- LNPs Lipid Nanoparticles
- nucleic acid such as RNA described herein is administered in the form of lipid nanoparticles (LNPs).
- LNPs may comprise any lipid capable of forming a particle to which the one or more nucleic acid molecules are attached, or in which the one or more nucleic acid molecules are encapsulated.
- an LNP comprises one or more cationic lipids, and one or more stabilizing lipids. Stabilizing lipids include neutral lipids, helper lipids, and pegylated lipids.
- an LNP comprises a cationic lipid, a helper lipid, a sterol, a polymer conjugated lipid; and an RNA, encapsulated within or associated with the lipid nanoparticle.
- a helper lipid is a phosphotidylcholine such as 1,2- distearoyl-sn-glycero-3-phosphocholine (DSPC), 1,2-Dipalmitoyl-sn-glycero-3- phosphocholine (DPPC), 1,2-Dimyristoyl-sn-glycero-3-phosphocholine (DMPC), 1- palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), l ,2-dioleoyl-sn-glycero-3- phosphocholine (DOPC), phophatidylethanolamines such as 1,2-dioleoyl-sn-glycero-3- phosphocholine (DOPC), pho
- a helper lipid is a 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP), 1- ⁇ -phosphatidylserine (PS), or DOPE.
- DOTAP 1,2-dioleoyl-3-trimethylammonium-propane
- PS 1- ⁇ -phosphatidylserine
- DOPE 1,2-dioleoyl-3-trimethylammonium-propane
- a helper lipid is selected from the group consisting of DSPC, DPPC, DMPC, DOPC, POPC, DOPE, DOPE, DOTAP, PS, and SM.
- the helper lipid is DSPC.
- the helper lipid is DOTAP. In some embodiments, the helper lipid is DOPE. In some embodiments, the helper lipid is PS. [0408] In some embodiments, a sterol is cholesterol. [0409] In some embodiments, a polymer conjugated lipid is a pegylated lipid (a PEG lipid).
- a pegylated lipid is 1,2- dimyristoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (C14-PEG2000).
- a pegylated lipid has the following structure: or a pharmaceutically acc thereof, wherein: R 12 and R 13 are each independently a straight or branched, saturated or unsaturated alkyl chain containing from 10 to 30 carbon atoms, wherein the alkyl chain is optionally interrupted by one or more ester bonds; and w has a mean value ranging from 30 to 60.
- R 12 and R 13 are each independently straight, saturated alkyl chains containing from 12 to 16 carbon atoms.
- w has a mean value ranging from 40 to 55. In some embodiments, the average w is about 45. In some embodiments, R 12 and R 13 are each independently a straight, saturated alkyl chain containing about 14 carbon atoms, and w has a mean value of about 45. [0410] In some embodiments, a pegylated lipid is DMG-PEG 2000, e.g., having the following structure: .
- a pegylated lipid is or comprises 2-[(Polyethylene glycol)-2000]-N,N-ditetradecylacetamide with a chemical structure as shown below: or a pharmaceutically acceptable salt thereof, where n’ is an integer from 45 to 50
- the lipid has one of the following structures (IIIA) or (IIIB): wherein: A is a 3 to 8-membered cycloalkyl or cycloalkylene ring; R 6 is, at each occurrence, independently H, OH or C 1 -C 24 alkyl; n is an integer ranging from 1 to 15. [0414] In some of the foregoing embodiments of Formula (III), the lipid has structure (IIIA), and in other embodiments, the lipid has structure (IIIB).
- the lipid has one of the following structures (IIIC) or (IIID): wherein y and z are each independently integers ranging from 1 to 12.
- the lipid has one of the following structures (IIIE) or (IIIF): .
- the lipid has one of the following structures (IIIG), (IIIH), (IIII), or (IIIJ): ; .
- n is an integer ranging from 2 to 12, for example from 2 to 8 or from 2 to 4.
- n is 3, 4, 5 or 6.
- n is 3.
- n is 4.
- n is 5.
- n is 6.
- y and z are each independently an integer ranging from 2 to 10.
- y and z are each independently an integer ranging from 4 to 9 or from 4 to 6.
- R 6 is H.
- R 6 is C1-C24 alkyl.
- R 6 is OH.
- G 3 is unsubstituted.
- G3 is substituted.
- G 3 is linear C1-C24 alkylene or linear C 1 -C 24 alkenylene.
- R 1 or R 2 is C6-C24 alkenyl.
- R 1 and R 2 each, independently have the following structure: , wherein: R 7a and R 7b are, at each occurrence, independently H or C1-C12 alkyl; and a is an integer from 2 to 12, wherein R 7a , R 7b and a are each selected such that R 1 and R 2 each independently comprise from 6 to 20 carbon atoms.
- a is an integer ranging from 5 to 9 or from 8 to 12
- at least one occurrence of R 7a is H.
- R 7a is H at each occurrence.
- at least one occurrence of R 7b is C1-C8 alkyl.
- C 1 -C 8 alkyl is methyl, ethyl, n-propyl, iso-propyl, n- butyl, iso-butyl, tert-butyl, n-hexyl or n-octyl.
- R 1 or R 2 has one of the following structures: ;
- R 4 is methyl or ethyl.
- the cationic lipid of Formula (III) has one of the structures set forth in in Table 15 below. Table 15: Exemplary Cationic Lipid Structures of Formula (III) No. Structure No. Structure No. Structure No. Structure No. Structure No.
- a cationic lipid has one of the structures set forth in Table 16 below.
- Table 16 Exemplary Cationic Lipid Structures of Formula A-F No. Structure O N
- an LNP comprises a cationic lipid that is an ionizable lipid-like material (lipidoid).
- a cationic lipid has one of the following structures as described in Melamed et al., Science Advances, 2023, 9, eade1444 (the entire contents of which are incorporated herein by reference):
- lipid nanoparticles can have an average size (e.g., mean diameter) of about 30 nm to about 150 nm, about 40 nm to about 150 nm, about 50 nm to about 150 nm, about 60 nm to about 130 nm, about 70 nm to about 110 nm, about 70 nm to about 100 nm, about 70 to about 90 nm, or about 70 nm to about 80 nm.
- lipid nanoparticles in accordance with the present disclosure can have an average size (e.g., mean diameter) of about 50 nm to about 100 nm.
- lipid nanoparticles may have an average size (e.g., mean diameter) of about 50 nm to about 150 nm. In some embodiments, lipid nanoparticles may have an average size (e.g., mean diameter) of about 60 nm to about 120 nm.
- lipid nanoparticles in accordance with the present disclosure can have an average size (e.g., mean diameter) of about 30 nm, 35 nm, 40 nm, 45 nm, 50 nm, 55 nm, 60 nm, 65 nm, 70 nm, 75 nm, 80 nm, 85 nm, 90 nm, 95 nm, 100 nm, 105 nm, 110 nm, 115 nm, 120 nm, 125 nm, 130 nm, 135 nm, 140 nm, 145 nm, or 150 nm.
- average size e.g., mean diameter
- average diameter refers to the mean hydrodynamic diameter of particles as measured by dynamic laser light scattering (DLS) with data analysis using the so-called cumulant algorithm, which provides as results the so-called Z-average with the dimension of a length, and the polydispersity index (PI), which is dimensionless (Koppel, Chem. Phys.57, 1972, pp 4814-4820, ISO 13321, which is incorporated herein by reference).
- PI polydispersity index
- lipid nanoparticles described herein may exhibit a polydispersity index less than about 0.5, less than about 0.4, less than about 0.3, or about 0.2 or less.
- lipid nanoparticles can exhibit a polydispersity index in a range of about 0.1 to about 0.3 or about 0.2 to about 0.3.
- the “polydispersity index” is preferably calculated based on dynamic light scattering measurements by the so-called cumulant analysis as mentioned in the definition of the “average diameter.” Under certain prerequisites, it can be taken as a measure of the size distribution of an ensemble of ribonucleic acid nanoparticles (e.g., ribonucleic acid nanoparticles).
- Lipid nanoparticles described herein can be characterized by an “N/P ratio,” which is the molar ratio of cationic (nitrogen) groups (the “N” in N/P) in the cationic polymer to the anionic (phosphate) groups (the “P” in N/P) in RNA.
- N/P ratio is the molar ratio of cationic (nitrogen) groups (the “N” in N/P) in the cationic polymer to the anionic (phosphate) groups (the “P” in N/P) in RNA.
- N/P ratio is the molar ratio of cationic (nitrogen) groups (the “N” in N/P) in the cationic polymer to the anionic (phosphate) groups (the “P” in N/P) in RNA.
- N + cationic form
- Use of a single number in an N/P ratio e.g., an N/P ratio of about 5 is intended to refer to that number over 1, e.g., an N/P
- a lipid nanoparticle described herein has an N/P ratio greater than or equal to 5. In some embodiments, a lipid nanoparticle described herein has an N/P ratio that is about 5, 6, 7, 8, 9, or 10. In some embodiments, an N/P ratio for a lipid nanoparticle described herein is from about 10 to about 50. In some embodiments, an N/P ratio for a lipid nanoparticle described herein is from about 10 to about 70. In some embodiments, an N/P ratio for a lipid nanoparticle described herein is from about 10 to about 120.
- a formulation described herein comprising lipid nanoparticles is formulated for intramuscular (i.m.) or intravenous (i.v.) delivery, and a lipid nanoparticle comprises i) about 30 to about 50 mol% of the cationic lipid; ii) about 1 to about 5 mol% of a PEG-conjugated lipid; iii) about 5 to about 15 mol% of a helper lipid; and iv) about 30 to about 50 mol% of a steroid.
- a formulation described herein comprising lipid nanoparticles is formulated for intraperitoneal (i.p.) delivery, and a lipid nanoparticle comprises: i) about 30 mol% to about 50 mol% of a cationic lipid; ii) about 1 mol% to 5 mol% of a PEG-conjugated lipid; iii) about 30 mol% to about 50 mol% of a helper lipid; and iv) about 20 mol% to about 40 mol% of cholesterol.
- a formulation for i.p. delivery comprises lipid nanoparticles, wherein a lipid nanoparticle comprises about 35 mol% of a cationic lipid; about 40 mol% of a helper lipid, about 22.5 mol% of cholesterol, and about 2.5 mol% of a PEG-conjugated lipid.
- a lipid nanoparticle comprises about 35 mol% of cationic lipid X-2, X-3, or X-4, about 40 mol% of DOTAP, DOPE, or PS, about 22.5 mol% of cholesterol, and about 2.5 mol% of C14-PEG2000. In some embodiments, a lipid nanoparticle comprises about 35 mol% of cationic lipid X-2; about 40 mol% of DOTAP; about 22.5 mol% of cholesterol, and about 2.5 mol% of C14-PEG2000.
- a lipid nanoparticle comprises about 35 mol% of cationic lipid X-3; about 40 mol% of DOTAP; about 22.5 mol% of cholesterol; and about 2.5 mol% of C14-PEG2000. In some embodiments, a lipid nanoparticle comprises about 35 mol% of cationic lipid X-4; about 40 mol% of DOTAP; about 22.5 mol% of cholesterol; and about 2.5 mol% of C14- PEG2000. In some embodiments, a lipid nanoparticle comprises about 35 mol% of cationic lipid X-2; about 40 mol% of DOPE; about 22.5 mol% of cholesterol; and about 2.5 mol% of C14-PEG2000.
- a lipid nanoparticle comprises about 35 mol% of cationic lipid X-3; about 40 mol% of DOPE; about 22.5 mol% of cholesterol; and about 2.5 mol% of C14-PEG2000. In some embodiments, a lipid nanoparticle comprises about 35 mol% of cationic lipid X-4; about 40 mol% of DOPE; about 22.5 mol% of cholesterol; and about 2.5 mol% of C14-PEG2000. In some embodiments, a lipid nanoparticle comprises about 35 mol% of cationic lipid X-2; about 40 mol% of PS; about 22.5 mol% of cholesterol; and about 2.5 mol% of C14-PEG2000.
- a lipid nanoparticle comprises about 35 mol% of cationic lipid X-3; about 40 mol% of PS; about 22.5 mol% of cholesterol; and about 2.5 mol% of C14-PEG2000. In some embodiments, a lipid nanoparticle comprises about 35 mol% of cationic lipid X-4; about 40 mol% of PS; about 22.5 mol% of cholesterol; and about 2.5 mol% of C14-PEG2000.
- these formulations are expected to lead to enhanced delivery of the lipid nanoparticles to pancreatic beta cells and in vivo expression of encoded incretin agents in pancreatic beta cells, particularly when administered via intraperitoneal (i.p.) delivery.
- Lipids and lipid nanoparticles comprising nucleic acids and their method of preparation are known in the art, including, e.g., as described in U.S. Patent Nos.8,569,256, 5,965,542 and U.S.
- an excipient is approved for use in humans and for veterinary use. In some embodiments, an excipient is approved by the United States Food and Drug Administration. In some embodiments, an excipient is pharmaceutical grade. In some embodiments, an excipient meets the standards of the United States Pharmacopoeia (USP), the European Pharmacopoeia (EP), the British Pharmacopoeia, and/or the International Pharmacopoeia.
- USP United States Pharmacopoeia
- EP European Pharmacopoeia
- British Pharmacopoeia the British Pharmacopoeia
- International Pharmacopoeia International Pharmacopoeia
- compositions include, but are not limited to, inert diluents, dispersing and/or granulating agents, surface active agents and/or emulsifiers, disintegrating agents, binding agents, preservatives, buffering agents, lubricating agents, and/or oils. Such excipients may optionally be included in pharmaceutical formulations. Excipients such as cocoa butter and suppository waxes, coloring agents, coating agents, sweetening, flavoring, and/or perfuming agents can be present in the composition, according to the judgment of the formulator.
- the composition can be formulated as a solution, microemulsion, lipid nanoparticles, or other ordered structure suitable to high drug concentration.
- the carrier can be a solvent or dispersion medium containing, for example, water, ethanol, polyol (for example, glycerol, propylene glycol, and liquid polyethylene glycol, and the like), and suitable mixtures thereof. Proper fluidity can be maintained, for example, by the use of surfactants. In many cases, it will be preferable to include isotonic agents, for example, sugars, polyalcohols such as mannitol, sorbitol, or sodium chloride in the composition.
- Relative amounts of polyribonucleotides encapsulated in lipid nanoparticles, a pharmaceutically acceptable excipient, and/or any additional ingredients in a pharmaceutical composition can vary, depending upon the subject to be treated, target cells, diseases or disorders, and may also further depend upon the route by which the composition is to be administered.
- pharmaceutical compositions described herein are formulated into pharmaceutically acceptable dosage forms by conventional methods known to those of skill in the art.
- a pharmaceutical composition described herein may further comprises one or more active agents other than at least one polyribonucleotide encoding an incretin agent.
- an other active agent may be or comprise another known treatment for obesity or a condition or disease related to obesity.
- an exemplary treatment may be one included in Table 1 herein.
- the present disclosure provides the recognition that incretins may be useful in combination with polyribonucleotides and/or compositions provided herein, for example, for treating or preventing obesity and diseases or disorders associated with obesity.
- Exemplary incretins that can be used with compositions described herein include, but are not limited to, those provided in Table 1, fragments thereof, or combinations thereof.
- a pharmaceutical composition provided herein is a preservative-free, sterile RNA-lipid nanoparticle dispersion in an aqueous buffer for intravenous or intramuscular administration.
- compositions are principally directed to pharmaceutical compositions that are suitable for administration to humans, it will be understood by the skilled artisan that such compositions are generally suitable for administration to animals of all sorts. Modification of pharmaceutical compositions suitable for administration to humans in order to render the compositions suitable for administration to various animals is well understood, and the ordinarily skilled veterinary pharmacologist can design and/or perform such modification with merely ordinary, if any, experimentation.
- Patient Populations [0467] Technologies provided herein can be useful for treatment and/or prevention of obesity, or a disease or disorder associated with obesity. As described herein, technologies include polyribonucleotides encoding incretin agents, immunoglobulin chains thereof, or fragments thereof.
- a pharmaceutical composition for treatment and/or prevention of obesity and diseases or disorders associated with obesity (e.g., Type 2 Diabetes (T2D), early T1D (e.g., within 3 months after diagnosis of T1D), non- alcoholic fatty liver disease (NAFLD), non-alcoholic steatohepatitis (NASH), cardiovascular disease, or renal disease).
- a pharmaceutical composition comprises a polyribonucleotides as described herein.
- a subject is one suffering from and/or susceptible to obesity or a disease or disorder associate with obesity.
- a subject may be defined by one or more criterion such as age group, gender, genetic background, preexisting clinical conditions, and/or prior exposure to therapy.
- a subject may be determined to be classified as needing a pharmaceutical composition described herein in accordance with the screening tools for obesity and diseases and disorders associated with obesity.
- a subject may be determined to be classified as needing a pharmaceutical composition described herein according to the results obtained in enzyme immunoassays (EIA), western blot, and/or PCR test, and/or body weight, and/or waist circumference, and/or body mass index.
- EIA enzyme immunoassays
- PCR test enzyme immunoassays
- body weight and/or waist circumference, and/or body mass index.
- a subject is a model organism.
- a subject is a human. In some embodiments, a subject is between 18-65 years of age. In some embodiments, a subject is an age in a range of from about 0 months to about 6 months old, from about 6 to about 12 months old, from about 6 to about 18 months old, from about 18 to about 36 months old, from about 1 to about 5 years old, from about 5 to about 10 years old, from about 10 to about 15 years old, from about 15 to about 20 years old, from about 20 to about 25 years old, from about 25 to about 30 years old, from about 30 to about 35 years old, from about 35 to about 40 years old, from about 40 to about 45 years old, from about 45 to about 50 years old, from about 50 to about 55 years old, from about 55 to about 60 years old, from about 60 to about 65 years old, from about 65 to about 70 years old, from about 70 to about 75 years old, from about 75 to about 80 years old, from about 80 to about 85 years old, from about 85 to about 90 years old, from about 90 to about 95 years old,
- a subject is a human infant. In some embodiments, a subject is a human toddler. In some embodiments, a subject is a human child. In some embodiments, a subject is a human adult. In some embodiments, a subject is an elderly human. [0472] In some embodiments, a subject is not currently considered obese. In some embodiments, a subject has or it as risk of developing obesity.
- a subject is suffering from and/or susceptible to obesity, pre-diabetes, type 2 diabetes (T2D, with its complications), early T1D, non-alcoholic fatty liver disease (NAFLD), non-alcoholic steatohepatitis (NASH), cardiovascular (CV) disease (e.g., characterized by, e.g., a major cardiovascular event (MACE), including CV death, non-fatal myocardial infarction, non-fatal stroke, heart failure with preserved ejection fraction (HFpEF)), renal disease, or elevated risk of premature mortality.
- MACE major cardiovascular event
- CV death non-fatal myocardial infarction
- non-fatal stroke non-fatal stroke
- HFpEF heart failure with preserved ejection fraction
- renal disease or elevated risk of premature mortality.
- a subject is suffering from and/or susceptible to additional co-morbidities related or unrelated to obesity, including any of the following: pre- diabetes, T2D, early T1D, non-alcoholic fatty liver disease (NAFLD), non-alcoholic steatohepatitis (NASH), cardiovascular (CV) disease (e.g., characterized by, e.g., a major cardiovascular event (MACE), including CV death, non-fatal myocardial infarction, non-fatal stroke, heart failure with preserved ejection fraction (HFpEF)), renal disease, and elevated risk of premature mortality.
- MACE major cardiovascular event
- CV death non-fatal myocardial infarction
- non-fatal stroke non-fatal stroke
- HFpEF heart failure with preserved ejection fraction
- renal disease and elevated risk of premature mortality.
- a subject has not previously received treatment for obesity or a disease related to obesity.
- a subject suffering from and/or susceptible to obesity or a disease related to obesity may have received or is currently receiving other therapies for obesity.
- a subject is currently receiving or has received one or more treatments listed in Table 1.
- a subject suffering from and/or susceptible to obesity or a disease related to obesity may have received or is currently receiving a lifestyle intervention, e.g., reduced calorie intake and/or increased physical activity.
- a subject has been receiving one or more treatments listed in Table 1 for greater than 1 week, greater than 2 weeks, greater than 3 weeks, greater than 4 weeks, greater than 5 weeks, greater than 6 weeks, greater than 7 weeks, greater than 8 weeks, greater than 12 weeks, greater than 4 months, greater than 5 months, greater than 6 months, greater than 7 months, greater than 8 months, greater than 9 months, greater than 10 months, or greater than 1 year.
- a subject is responsive to another treatment at the time a polyribonucleotide, composition or pharmaceutical composition described herein is administered.
- a subject is not responsive to another treatment at the time a polyribonucleotide, composition or pharmaceutical composition described herein is administered.
- a subject has previously received one or more treatments for obesity or a disease related to obesity (e.g., one or more treatments listed in Table 1).
- a subject received prior treatment for obesity greater than 1 week, greater than 2 weeks, greater than 3 weeks, greater than 4 weeks, greater than 5 weeks, greater than 6 weeks, greater than 7 weeks, greater than 8 weeks, greater than 12 weeks, greater than 4 months, greater than 5 months, greater than 6 months, greater than 7 months, greater than 8 months, greater than 9 months, greater than 10 months, or greater than 1 year.
- a subject was responsive to the prior treatment. In some embodiments, a subject was not responsive to the prior treatment.
- a subject is characterized by any one of the following characteristics: a BMI of 30 or higher, a BMI of 40 or higher, waist circumference of greater than 35 inches (89 cm) (in women) or greater than 40 inches (102 cm) (in men), high blood pressure, high glucose levels and/or high cholesterol in a blood sample, high HbA1c level, underactive thyroid, liver problems and/or diabetes.
- a subject has received no other treatments for obesity or a disease related to obesity in the last month. In some embodiments, a subject has received no other treatments for obesity or a disease related to obesity in the last year. In some embodiments, a subject has received no treatments for obesity or a disease related to obesity in the last 2 years.
- a pharmaceutical composition described herein can be taken up by cells for production of an encoded incretin agent at therapeutically relevant serum concentrations. Accordingly, the present disclosure provides methods of using pharmaceutical compositions described herein.
- a method provided herein comprises administering a pharmaceutical composition described herein to a [0482]
- administering typically refers to the administration of a composition to a subject to achieve delivery of an agent (e.g., at least one polyribonucleotide encoding an incretin agent described herein) that is, or is included in, a composition to a target site or a site to be treated.
- Administration may be, for example, bronchial (e.g., by bronchial instillation), buccal, dermal (which may be or comprise, for example, one or more of topical to the dermis, intradermal, interdermal, transdermal, etc.), enteral, intra-arterial, intradermal, intragastric, intramedullary, intramuscular, intranasal, intraperitoneal, intrathecal, intravenous, intraventricular, within a specific organ (e.g., intrahepatic), mucosal, nasal, oral, rectal, subcutaneous, sublingual, topical, tracheal (e.g., by intratracheal instillation), vaginal, vitreal, etc.
- bronchial e.g., by bronchial instillation
- buccal which may be or comprise, for example, one or more of topical to the dermis, intradermal, interdermal, transdermal, etc.
- enteral intra-arterial, intraderma
- administration may be intramuscular, intraperitoneal, intravenous, or subcutaneous.
- administration of a pharmaceutical composition results in delivery of one or more polyribonucleotides as described herein (e.g., encoding an incretin agent) to a subject.
- administering a pharmaceutical composition to a subject results in expression in the subject of an incretin agent encoded by an administered polyribonucleotide.
- administering a pharmaceutical composition to a subject results in expression in the subject of an incretin agent encoded by an administered polyribonucleotide.
- administration may involve only a single dose.
- administration may involve administration of a fixed number of doses.
- administration may involve dosing that is intermittent (e.g., a plurality of doses separated in time) and/or periodic (e.g., individual doses separated by a common period of time) dosing.
- administration may involve continuous dosing (e.g., perfusion) for at least a selected period of time.
- a dosing regimen comprises a plurality of doses each of which is separated in time from other doses.
- a dosing regimen comprises a plurality of doses and at least two different time periods ti i di id l d I b di t ll d ithi d i i f the same unit dose amount.
- different doses within a dosing regimen are of different amounts.
- a dosing regimen comprises a first dose in a first dose amount, followed by one or more additional doses in a second dose amount different from the first dose amount.
- a dosing regimen comprises a first dose in a first dose amount, followed by one or more additional doses in a second dose amount same as the first dose amount.
- a dosing regimen is correlated with a desired or beneficial outcome when administered across a relevant population (i.e., is a therapeutic dosing regimen).
- therapies can be administered in dosing cycles.
- pharmaceutical compositions described herein are administered in one or more dosing cycles.
- one dosing cycle is at least 3 or more days (including, e.g., at least 4, at least 5, at least 6, at least 7, at least 8, at least 9, at least 10, at least 11, at least 12, at least 13, at least 14, at least 15, at least 16, at least 17, at least 18, at least 19, at least 20, at least 21, at least 22, at least 23, at least 24, at least 25, at least 26, at least 27, at least 28, at least 29, or at least 30 days).
- one dosing cycle is at least 21 days.
- one dosing cycle may involve multiple doses, e.g., according to a pattern such as, for example, a dose may be administered daily within a dosing cycle, or a dose may be administered every 2 days, every 3 days, every 4 days, every 5 days, every 6 days, every 7 days, every 2 weeks, monthly, every 2 months within a cycle.
- multiple dosing cycles may be administered.
- At least 2 dosing cycles can be administered.
- the number of dosing cycles to be administered may vary with types of treatment (e.g., monotherapy vs. combination therapy).
- at least 3-8 dosing cycles may be administered.
- an RNA polymerase typically traverses at least a portion of a single-stranded DNA template in the 3' ⁇ 5' direction to produce a single- stranded complementary RNA in the 5' ⁇ 3' direction.
- a polyribonucleotide comprises a polyA tail
- a polyA tail may be encoded in a DNA template, e.g., by using an appropriately tailed PCR primer, or it can be added to a polyribonucleotide after in vitro transcription, e.g., by enzymatic treatment (e.g., using a poly(A) polymerase such as an E. coli Poly(A) polymerase).
- a poly(A) tail comprises a plurality of A residues interrupted by a linker.
- capping may be performed after in vitro transcription in the presence of a capping system (e.g., an enzyme- based capping system such as, e.g., capping enzymes of vaccinia virus).
- a capping system e.g., an enzyme- based capping system such as, e.g., capping enzymes of vaccinia virus.
- a cap may be introduced during in vitro transcription, along with a plurality of ribonucleotide triphosphates such that a cap is incorporated into a polyribonucleotide during transcription (also known as co-transcriptional capping).
- a GTP fed- batch procedure with multiple additions in the course of the reaction may be used to maintain a low concentration of GTP in order to effectively cap the RNA.
- polyribonucleotides can be purified (e.g., in some embodiments after in vitro transcription reaction), for example, to remove components utilized or formed in the course of the production, like, e.g., proteins, DNA fragments, and/or or nucleotides.
- Various nucleic acid purifications that are known in the art can be used in accordance with the present disclosure. Certain purification steps may be or include, for example, one or more of precipitation, column chromatography (including, e.g., but not limited to anionic, cationic, hydrophobic interaction chromatography (HIC)), solid substrate- based purification (e.g., magnetic bead-based purification).
- polyribonucleotides (e.g., ones described herein) and/or compositions thereof may be stored stable at a sub-zero temperature (e.g., -20°C or below) for at least 1 month or longer including, at least 2 months, at least 3 months, at least 4 months, at least 5 months, at least 6 months, at least 7 months, at least 8 months, at least 9 months, at least 10 months, at least 11 months, or at least 12 months or longer.
- polyribonucleotides (e.g., ones described herein) and/or compositions thereof may be stored stable at room temperature (e.g., at about 25°C) for at least 1 month or longer.
- one or more assessments may be utilized during manufacture, or other preparation or use of polyribonucleotides (e.g., as a release test).
- one or more quality control parameters may be assessed to determine whether polyribonucleotides described herein meet or exceed acceptance criteria (e.g., for subsequent formulation and/or release for distribution).
- quality control parameters may include, but are not limited to RNA integrity, RNA concentration, residual DNA template and/or residual dsRNA. Certain methods for assessing RNA quality are known in the art; for example, one of skill in the art will recognize that in some embodiments, one or more analytical tests can be used for RNA quality assessment.
- a batch of polyribonucleotides may be assessed for one or more features as described herein to determine next action step(s). For example, a batch of polyribonucleotides can be designated for one or more further steps of manufacturing and/or formulation and/or distribution if RNA quality assessment indicates that such a batch of polyribonucleotides meet or exceed the relevant acceptance criteria. Otherwise, an alternative action can be taken (e.g., discarding the batch) if such a batch of polyribonucleotides does not meet or exceed the acceptance criteria.
- a batch of polyribonucleotides that satisfy assessment results can be utilized for one or more further steps of manufacturing and/or formulation and/or distribution.
- DNA Constructs [0525] Among other things, the present disclosure provides DNA constructs, for example that may encode one or more incretin agents as described herein, or components thereof. In some embodiments, DNA constructs provided by and/or utilized in accordance with the present disclosure are comprised in a vector.
- Non-limiting examples of a vector include plasmid vectors, cosmid vectors, phage vectors such as lambda phage, viral vectors such as retroviral, adenoviral or baculoviral vectors, or artificial chromosome vectors such as bacterial artificial chromosomes (BAC), yeast artificial chromosomes (YAC), or P1 artificial chromosomes (PAC).
- a vector is an expression vector.
- a vector is a cloning vector.
- a vector is a nucleic acid construct that can receive or otherwise become linked to a nucleic acid element of interest (e.g., a construct that is or encodes a payload, or that imparts a particular functionality, etc.).
- Expression vectors which may be plasmid or viral or other vectors, typically include an expressible sequence of interest (e.g., a coding sequence) that is functionally linked with one or more control elements (e.g., promoters, enhancers, transcription terminators, etc.). Typically, such control elements are selected for expression in a system of interest.
- a system is ex vivo (e.g., an in vitro transcription system); in some embodiments, a system is in vivo (e.g., a bacterial, yeast, plant, insect, fish, vertebrate, mammalian cell or tissue, etc.).
- Cloning vectors are generally used to modify, engineer, and/or duplicate (e.g., by replication in vivo, for example in a simple system such as bacteria or yeast, or in vitro, such as by amplification such as polymerase chain reaction or other amplification process).
- a cloning vector may lack expression signals.
- a vector may include replication elements such as primer binding site(s) and/or origin(s) of replication. In many embodiments, a vector may include insertion or modification sites such as restriction endonuclease recognition sites and/or guide RNA binding sites, etc.
- a vector is a viral vector (e.g., an AAV vector). In some embodiments, a vector is a non-viral vector. In some embodiments, a vector is a plasmid. [0531] Those skilled in the art are aware of a variety of technologies useful for the production of recombinant polynucleotides (e.g., DNA or RNA) as described herein.
- polynucleotide(s) of the present disclosure are included in a DNA construct (e.g., a vector) amenable to transcription and/or translation.
- an expression vector comprises a polynucleotide that encodes proteins and/or polypeptides of the present disclosure operatively linked to a sequence or sequences that control expression (e.g., promoters, start signals, stop signals, polyadenylation signals, activators, repressors, etc.).
- a sequence or sequences that control expression are selected to achieve a desired level of expression.
- more than one sequence that controls expression are utilized.
- more than one sequence that controls expression are utilized to achieve a desired level of expression of a plurality of polynucleotides that encode a plurality proteins and/or polypeptides.
- a plurality of recombinant proteins and/or polypeptides are expressed from the same vector (e.g., a bi-cistronic vector, a tri-cistronic vector, multi-cistronic).
- a plurality of polypeptides are expressed, each of which is expressed from a separate vector.
- an expression vector comprising a polynucleotide of the present disclosure is used to produce a RNA and/or protein and/or polypeptide in a host cell.
- a host cell may be in vitro (e.g., a cell line) – for example a cell or cell line (e.g., Human Embryonic Kidney (HEK cells), Chinese Hamster Ovary cells, etc.) suitable for producing polynucleotides of the present disclosure and proteins and/or polypeptides encoded by said polynucleotides.
- HEK cells Human Embryonic Kidney
- Chinese Hamster Ovary cells etc.
- an expression vector is an RNA expression vector.
- an RNA expression vector comprises a polynucleotide template used to produce a RNA in cell-free enzymatic mix.
- an RNA expression vector comprising a polynucleotide template is enzymatically linearized prior to in vitro transcription.
- a polynucleotide template is generated through PCR as a linear polynucleotide template.
- a linearized polynucleotide is mixed with enzymes suitable for RNA synthesis, RNA capping and/or purification.
- the resulting RNA is suitable for producing proteins encoded by the RNA.
- a vector may be introduced into host cells using transfection.
- transfection is completed, for example, using calcium phosphate transfection, lipofection, or polyethylenimine-mediated transfection.
- a vector may be introduced into a host cell using transduction.
- transformed host cells are cultured following introduction of a vector into a host cell to allow for expression of said recombinant polynucleotides.
- Transformed host cells are cultured in growth conditions (e.g., temperature, carbon-dioxide levels, growth medium) in accordance with the requirements of a host cell selected.
- growth conditions e.g., temperature, carbon-dioxide levels, growth medium
- EXEMPLIFICATION Example 1 Generating polyribonucleotides encoding exemplary incretin agents
- the present Example describes methods for generating polyribonucleotide sequences encoding an incretin agent.
- the present Example further describes the design of polyribonucleotides that can achieve transient in vivo incretin agent production following i.p./i.v./i.m./s.c. delivery.
- Methods of the present example include: (1) Cloning of DNA fragments encoding incretin agents (e.g., GLP1, GIP or variants thereof) into a DNA plasmid appropriate to use for RNA expression.
- An appropriate DNA plasmid may encode RNA features including, for example, a 5’ untranslated region (5’ UTR), a Kozak sequence, a 3’ untranslated region (3’ UTR), and/or a polyA tail sequence.
- the DNA plasmid also typically includes restriction sites that enable cloning of DNA fragments encoding incretin agents downstream of regions encoding the 5’ UTR and Kozak sequence and upstream of the regions encoding the 3’ UTR and polyA tail sequence. Examples of appropriate DNA plasmids can also be found in WO2021/214204, which is hereby incorporated by reference in its entirety.
- Verification of selected clones by control digestion and optionally sequencing. Linearization of DNA plasmids encoding incretin agents.
- DNA sequences are generated based on the amino acid sequences of GLP1 (7-37), GIP (1-42) and truncation or mutation variants thereof fused to exemplary signal peptides (SP) including sequence (SP1-2, MRVLVLLACLAAASNA) and the SP shown in SEQ ID NOs: 65 and 66, as shown in Table 11 above.
- SP signal peptides
- the amino acid sequences are translated to DNA nucleotide sequences. Restriction sites, e.g., for Eam1104I (GAAGAG), BamHI (GGATCC), PstI (CTGCAG), SbfI (CCTGCAGG), XhoI (CTCGAG), SpeI (ACTAGT), BspEI (TCCGGA), SacI (GAGCTC), Ear1 (CTCTTCN ⁇ NNN) and NheI (GCTAGC) are optionally eliminated after optimization if any of these enzymes (or others) are used either for linearization or for cloning of the plasmid.
- Restriction sites e.g., for Eam1104I (GAAGAG), BamHI (GGATCC), PstI (CTGCAG), SbfI (CCTGCAGG), XhoI (CTCGAG), SpeI (ACTAGT), BspEI (TCCGGA), SacI (GAGCTC), Ear1 (CTCTTCN ⁇ NNN) and
- GeneOptimizer® removes sequence repeats, introns, cryptic splice sites, internal ribosome entry sites and RNA destabilizing sequence elements (e.g., UpA-dinucleotides), adds RNA stabilizing sequence elements (e.g., CpG-dinucleotides) and avoids stable RNA secondary structures as well as unwanted sequences such as restriction sites.
- the output sequence is then used for ordering of DNA fragment strings.
- RNA destabilizing sequence elements e.g., UpA-dinucleotides
- CpG-dinucleotides CpG-dinucleotides
- Plasmid DNA Preparation Plasmid DNA is prepared by selecting clones for inoculation in culture media. Selected clones are optionally verified by control digestion and optionally sequencing. Cultures are grown and following cell harvest, purification is done, e.g., using the QIAGEN Plasmid Plus Maxi Kit according to the manufacturer’s instructions. DNA concentration may be determined by UV spectroscopy. DNA is stored in certified RNase- and DNase-free reaction tubes.
- RNA optionally capped RNA, is then produced, e.g., following the process as disclosed, e.g., in Kreiter et al., Cancer Immunol. Immunother.2007, 56, 1577–87 and WO2021/214204, each of which is incorporated herein by reference in its entirety.
- Methyl pseudo-uridine may be used in the in vitro transcription reaction and incorporated into the produced RNA.
- Cellulose purification of the resulting RNA is performed to isolate single- stranded RNA, followed by concentration measurement by UV spectroscopy. RNA integrity is determined by microfluidic-based electrophoresis. Further biochemical characterization of the resulting RNA is optionally performed.
- Transfection and Expression [0545] RNA encoding incretin agents is transfected into HEK cells, e.g., via electroporation and resulting incretin agent levels are quantified.
- HEK cells e.g., HEK293T cells are washed with cooled medium. Electroporation is performed in pre-cooled cuvettes.
- Cells and RNA in each sample are at typical concentrations for RNA electroporation. Cells are incubated on ice after electroporation. [0546] Cells are then transferred into expression medium, e.g., Expi293 Medium and counted. Cells are seeded at typical concentrations for expression and incubated, e.g., for 48 hours at 37°C. Supernatants are then harvested by centrifugation of the cells, followed by careful aspiration so as to not disturb the cell pellet, and then stored at 4°C. [0547] Expression of incretin agents is quantified, e.g., via ELISA or Western blot analysis of the cell culture supernatants.
- Example 2 Generating reporter cell lines to monitor incretin activity
- the present Example describes methods for generating reporter cell lines to monitor incretin activity.
- Methods of the present example include: (1) Cloning of DNA fragments encoding incretin receptors (e.g., GLP1R and/or GIPR) into a DNA plasmid (e.g., pT2). (2) Stably transfecting HEK293 cells with the DNA plasmid encoding incretin receptors and mRNA transposase. (3) Sorting cells by FACS for high, medium and low incretin receptor expression, e.g., GLP1R and/or GIPR expression, optionally with a bulk sort followed by a single cell sort.
- incretin receptors e.g., GLP1R and/or GIPR
- incretin receptors e.g., GLP1R and/or GIPR.
- Generating a master cell bank e.g., a DNA plasmid, e.g., pT2.
- GLP1R_mutR and GLP1R_mutL encode slightly different variants of GLP1R (GLP1R_mutR encodes a GLP1R with a L260F mutation relative to the GLP1R encoded by GLP1R_mutL). This can for example be done by in vivo assembly.
- Plasmid DNA is prepared by selecting clones for inoculation in culture media. Selected clones are optionally verified by control digestion and optionally sequencing. Cultures are grown and following cell harvest, purification is done, e.g., using the QIAGEN Plasmid Plus Maxi Kit according to the manufacturer’s instructions.
- DNA concentration may be determined by UV spectroscopy. DNA is stored in certified RNase- and DNase-free reaction tubes.
- Transfection Plasmid DNA encoding incretin receptor(s) and RNA encoding a transposase, e.g., SB100X transposase are transfected into HEK cells, e.g., via electroporation. HEK cells, e.g., HEK293 cells are washed with cooled medium. Electroporation is performed in pre- cooled cuvettes. Cells, plasmid DNA and RNA in each sample are at typical concentrations for electroporation. Cells are incubated on ice after electroporation.
- Cell Sorting [0553] Cells are then sorted by FACS, e.g., using a BD FACSMelodyTM Cell Sorter for high, medium and low incretin receptor expression, e.g., GLP1R and/or GIPR expression, optionally with a bulk sort followed by a single cell sort. Stable expression of incretin receptors is confirmed for selected clones and master cell banks are generated.
- Resulting cell lines may include one or more of the following: HEK293_GLP1R hi , HEK293_GLP1R med , HEK293_GLP1R low , HEK293_GIPR hi , HEK293_GIPR med , HEK293_GIPR low , HEK293_GLP1R hi /GIPR hi , HEK293_GLP1R med /GIPR med , and HEK293_GLP1R low /GIPR low .
- Example 3 Assessing functionality of polyribonucleotide encoded incretins
- Methods of the present example include: (1) Transfecting HEK293_GLP1R/GIPR reporter cells of Example 2 with GLP1 or GIP RNA of Example 1. Reporter cells may be incubated for, e.g., 24 hours after transfection before supernatant is collected. (2) Quantifying downstream signal induction by measuring cAMP release, e.g., in the supernatant, e.g., using a cAMP-GloTM assay or an ELISA.
- Methods of the present example also include: (1) Transfecting wild type HEK293 cells with GLP1 or GIP RNA of Example 1. (2) Collecting supernatant containing GLP1 and GIP. (3) Incubating HEK293_GLP1R/GIPR reporter cells of Example 2 with supernatant. Reporter cells may be incubated for, e.g., 24 hours before supernatant is collected. (4) Quantifying downstream signal induction by measuring cAMP release, e.g., in the supernatant, e.g., using a cAMP-GloTM assay or an ELISA.
- Example 4 In vitro functionality of Polyribonucleotides Encoding Incretin Agents [0557] The present Example demonstrates that polyribonucleotides encoding incretin agents, as described herein, can induce the production of incretin agents. [0558] Methods: In this Example, 6x10 4 HEK293t17 cells were seeded per well in three different 48 well plates and grown over night at 37°C, in 5% CO2 incubator.
- Cells were transfected with 0.6 ⁇ g of polyribonucleotide candidates (polyribonucleotides encoding GLP1 (7-37), GLP1 (7-37)-(K34R), GIP (1-30) and GIP (1-42)) using Lipofectamine Messenger MAX kit (ThermoFisher Scientific, Cat.LMRNA003). The cells were further incubated. After a period of 3 hours, 6 hours, 24 hours, 48 hours, and 72 hours post transfection. For the time points of 3 hours and 6 hours post transfection, supernatant was collected and frozen at - 80°C. At the timepoint of 24 hours post transfection, supernatant was collected and fresh medium was replaced in the well plate.
- polyribonucleotide candidates polyribonucleotides encoding GLP1 (7-37), GLP1 (7-37)-(K34R), GIP (1-30) and GIP (1-42)
- Lipofectamine Messenger MAX kit ThermoFisher Scientific, Cat.
- the well plate was further incubated until 48 hours and 72 hours post transfection.
- the supernatant collected at the 24 hour, 48 hour and 72 hour post transfection time points was stored at -80°C until further analysis.
- the concentration of GIP and GLP1 in the supernatant was then quantified using ELISA. (Human GIP (Total) ELISA Kit and GLP1 (7-36) Active ELISA kit, Merck Millipore). Statistical analysis was performed by One-Way ANOVA followed by post-hoc Tukeys test.
- Results from the ELISA show that the polyribonucleotides encoding incretin agents GLP1 (7-37), GLP1 (7-37)-(K34R), and GIP (1-42) were translated into protein, reaching a maximum concentration after 24 hours (see Figs.10, 11, and 12, respectively). Surprisingly, the results show that GLP1 (7-37)-(K34R) was translated more efficiently that GLP1 (7-37), indicated by a significant increase in the GLP1 (7-37)-(K34R) concentration within 6 hours.
- Example 5 Generating polyribonucleotides encoding exemplary incretin agents
- the present Example describes generation of polyribonucleotides encoding various incretin agents.
- the present Example further describes the design of polyribonucleotides that can achieve transient in vivo incretin agent production following i.p./i.v./i.m./s.c. delivery.
- Methods of the present example include: (1) Cloning of DNA fragments encoding incretin agents (e.g., GLP1, GIP or variants thereof) into a DNA plasmid appropriate to use for RNA expression.
- An appropriate DNA plasmid may encode RNA features including, for example, a 5’ untranslated region (5’ UTR), a Kozak sequence, a 3’ untranslated region (3’ UTR), and/or a polyA tail sequence.
- the DNA plasmid also typically includes restriction sites that enable cloning of DNA fragments encoding incretin agents downstream of regions encoding the 5’ UTR and Kozak sequence and upstream of the regions encoding the 3’ UTR and polyA tail sequence. Examples of appropriate DNA plasmids can also be found in WO2021/214204, which is hereby incorporated by reference in its entirety.
- Verification of selected clones by control digestion and optionally sequencing. Linearization of DNA plasmids encoding incretin agents.
- incretin agents were designed to observe whether a linker fused to an incretin peptide affects expression and functionality of the incretin agent.
- Exemplary incretin agents generated in this Example are shown in Table 18 below.
- RNA In Vitro Transcription [0570] RNA, optionally capped RNA, was then produced, e.g., following the process as disclosed, e.g., in Kreiter et al., Cancer Immunol. Immunother.2007, 56, 1577–87 and WO2021/214204, each of which is incorporated herein by reference in its entirety. Methyl pseudo-uridine was used in the in vitro transcription reaction and incorporated into the produced RNA. Cellulose purification of the resulting RNA was performed to isolate single- stranded RNA, followed by concentration measurement by UV spectroscopy. RNA integrity was determined by microfluidic-based electrophoresis. Further biochemical characterization of the resulting RNA was optionally performed.
- RNA encoding incretin agents were transfected into HEK cells, e.g., via electroporation and resulting incretin agent levels were quantified.
- HEK cells e.g., HEK293T cells were washed with cooled medium. Electroporation was performed in pre-cooled cuvettes. Cells and RNA in each sample were at typical concentrations for RNA electroporation. Cells were incubated on ice after electroporation.
- Cells were then transferred into expression medium, e.g., Expi293 Medium and counted. Cells were seeded at typical concentrations for expression and incubated, e.g., for 48 hours at 37°C.
- Example 6 In vitro functionality of Additional Polyribonucleotides Encoding Incretin Agents
- the present Example examines in vitro functionality of polyribonucleotides encoding incretin agents generated and described in Example 5, and compares level of peptide secretion. The present Example demonstrates that each of the polyribonucleotides generated in Example 5 can induce the production of incretin agents at varying levels.
- Figure 18 shows concentrations (pg/ml) of the exemplary GLP1 incretin agents in supernatant of HEK29t17 cells transfected with polyribonucleotides encoding for the exemplary GLP1 incretin agents containing a viral signal peptide (“viral SP”) or a husec signal peptide (“husec”).
- viral SP viral signal peptide
- husec a husec signal peptide
- Figure 19 shows concentrations (ng/ml) of the exemplary GIP incretin agents in supernatant of HEK29t17 cells transfected with polyribonucleotides encoding for incretin agents containing a viral signal peptide or a husec signal peptide.
- Example 7 Bioactivity of Incretin Agents translated from Polyribonucleotides [0580] The present Example confirms the bioactivity of incretin agents described herein delivered as polyribonucleotides to cells and subsequently translated. Various incretin agents encoded by polyribonucleotides as described in Example 5 were tested.
- Results from the bioactivity assay of GIP incretin agents is shown in Figure 24.
- Results show that the GIP incretin agents with a husec signal peptide and A2G mutation showed better bioactivity than the GIP incretin agents with a viral signal peptide. Additionally, the results show that codon optimization strategy did not impact bioactivity of the GIP incretin agents. Specific bioactivity of mRNA-encoded GIP incretin agents (using either a husec or viral signal peptide) was lower than the controls (GIP and tirzepatide).
- signal peptide in the context of polyribonucleotides encoding incretin agents described herein may affect how the N-terminal end of the incretin peptide is cleaved. Certain signal peptides may lead to alternative processing or cleavage sites, ultimately changing the final amino acid sequence of the mature incretin peptide. In such a relatively small peptide, a change in amino acid sequence could greatly impact bioactivity.
- Figure 21 indicates that the A2G mutation facilitates correct N-terminal processing in GIP incretin agents containing husec signal peptides.
- the results in this example show that signal peptide selection affects translation and bioactivity, potentially due to the way the signal peptide is cleaved from the incretin peptide.
- Example 8 Generating polyribonucleotides encoding exemplary incretin agents [0589]
- the present Example describes generation of polyribonucleotides encoding various incretin agents.
- the present Example further describes the design of polyribonucleotides that can achieve transient in vivo incretin agent production following i.p./i.v./i.m./s.c. delivery.
- Exemplary incretin agents generated in the present Example utilize various strategies described herein for improving activity and half-life of the incretin agent.
- Methods of the present example include: (1) Cloning of DNA fragments encoding incretin agents (e.g., GLP1, GIP or variants thereof) into a DNA plasmid appropriate to use for RNA expression.
- An appropriate DNA plasmid may encode RNA features including, for example, a 5’ untranslated region (5’ UTR), a Kozak sequence, a 3’ untranslated region (3’ UTR), and/or a polyA tail sequence.
- the DNA plasmid also typically includes restriction sites that enable cloning of DNA fragments encoding incretin agents downstream of regions encoding the 5’ UTR and Kozak sequence and upstream of the regions encoding the 3’ UTR and polyA tail sequence. Examples of appropriate DNA plasmids can also be found in WO2021/214204, which is hereby incorporated by reference in its entirety.
- Verification of selected clones by control digestion and optionally sequencing (3) Linearization of DNA plasmids encoding incretin agents.
- Synthesis of polyribonucleotides encoding incretin agents (5) Biochemical characterization of polyribonucleotides encoding incretin agents.
- Table 21 Exemplary polyribonucleotides encoding Incretin Agents where x2 and x4 examples include a linker and Furin cleavage site in between each repeat unit Construct Nucleic acid Description sequence - payload Construct Nucleic acid ID Description sequence - payload Construct Nucleic acid ID Description sequence - payload Plasmid DNA Preparation [0596] Plasmid DNA as prepared by selecting clones for inoculation in culture media. Selected clones were optionally verified by control digestion and optionally sequencing. Cultures were grown and following cell harvest, purification as done, e.g., using the QIAGEN Plasmid Plus Maxi Kit according to the manufacturer’s instructions. DNA concentration was determined by UV spectroscopy.
- RNA was then produced, e.g., following the process as disclosed, e.g., in Kreiter et al., Cancer Immunol.
- RNA encoding incretin agents were transfected into HEK cells, e.g., via electroporation and resulting incretin agent levels were quantified.
- HEK cells e.g., HEK293T cells were washed with cooled medium. Electroporation was performed in pre-cooled cuvettes. Cells and RNA in each sample were at typical concentrations for RNA electroporation. Cells were incubated on ice after electroporation. [0600] Cells were then transferred into expression medium, e.g., Expi293 Medium and counted. Cells were seeded at typical concentrations for expression and incubated, e.g., for 48 hours at 37°C. Supernatants were then harvested by centrifugation of the cells, followed by careful aspiration so as to not disturb the cell pellet, and then stored at 4°C.
- expression medium e.g., Expi293 Medium
- Example 9 In vitro functionality and Bioactivity of Additional Polyribonucleotides Encoding Incretin Agents
- the present Example examines in vitro functionality and bioactivity of polyribonucleotides encoding incretin agents generated as described in Example 8. The present Example compares level of peptide secretion and demonstrates that each of the polyribonucleotides generated in Example 8 can induce the production of incretin agents at varying levels. Additionally, the present Example also confirms the bioactivity of incretin agents described herein delivered as polyribonucleotides to cells and subsequently translated.
- Bioactivity of various incretin agents encoded by polyribonucleotides as described in Example 8 were tested.
- the present Example also examines various design strategies, to improve properties such as half-life, N-terminal cleavage, stability, translation efficiency and bioactivity of the expressed incretin agents.
- Supernatant was collected after a period of 3 hours, 6 hours, 24 hours, 48 hours, and 72 hours post transfection. For the time points of 3 hours and 6 hours post transfection, supernatant was collected and frozen at -80°C. At the timepoint of 24 hours post transfection, supernatant was collected and fresh medium was replaced in the well plate. The well plate was further incubated until 48 hours and 72 hours post transfection. The supernatant collected at the 24 hour, 48 hour and 72 hour post transfection time points was stored at -80°C until further analysis.
- GLP1R-CRE and GIPR-CRE Luciferase Reporter HEK293 cells were each seeded in a white clear-bottomed 96-well plate at a density of ⁇ 38,000 cells per well in 100 ⁇ l of their specific medium. Cells were incubated at 37°C in a CO2 incubator for 2 days.
- FIGIP (1-42) with A2G mutation was used as control for the GIP assay. The assay was repeated in triplicate. Results: [0610]
- Figure 25 shows GIP expression for all of the incretin agents tested. Constructs including two or more incretin peptides (i.e., 4081 and 4082) and those fused to Dula_IgG4 (i.e., 4092 and 4093) showed the highest expression, including at the later timepoint of 72 hours.
- Figure 26 shows GIP bioactivity for all of the GIP-containing incretin agents tested (in each of the three replicates).
- Figure 29 shows a comparison of GIP expression (A) and GIP bioactivity (B) in candidates with different signal peptides (husec vs. gD1).
- Incretin agents with a gD1 signaling peptide showed increased GIP expression (about 2X) and increased GIP bioactivity (about 5X) compared to the same incretin agent with a husec signaling peptide.
- Figure 30 shows a comparison of GLP1 expression (A) and GLP1 bioactivity (B) in candidates with different signal peptides (husec vs. gD1).
- Incretin agents with a gD1 signaling peptide showed increased GLP1 expression compared to incretin agents including a husec signaling peptide (see Figure 30A). However, the incretin agents that utilized a gD1 signaling peptide did not necessarily show better GLP1 bioactivity. Incretin agents utilizing a husec signaling peptide (4071, 3815, and 4073) showed lower GLP1 expression, and as such, less GLP1 peptide was utilized in the bioactivity assay, but even with less GLP1 peptide present, 3815 showed comparable bioactivity to gD1-containing incretin agents 4074, 4075 and 4076 (which samples contained higher levels of GLP1 peptide in the bioactivity assay).
- Figure 31 shows a comparison of GIP expression (A) and GIP bioactivity (B) of GIP with and without various half-life extension (HLE) moieties.
- the results show that the Dula_IgG4-containing incretin agents (4086 and 4087) were well expressed (A). Without wishing to be bound by any theory, the high expression may be due to the overall size of the polyribonucleotide. However, the results show that the higher expression did not necessarily lead to an improvement in GIP bioactivity, which was comparatively lower (B).
- Albumin- containing incretin agent (4096) showed consistent levels of GIP expression and GIP bioactivity compared to the other incretin agents. Additionally, the incretin agent containing a VHH that binds to albumin (a-HSA VHH) (4097) had comparatively lower levels of GIP expression (A), but even with less protein assayed, showed higher GIP bioactivity (B). The incretin agent containing an Fc-fusion with KIH mutation (FcKIH-b) (4089) also showed consistent GIP expression and GIP bioactivity. [0617] Figure 32 shows a comparison of GLP1 expression (A) and GLP1 bioactivity (B) with and without various half-life extension (HLE) moieties.
- HLE half-life extension
- Figure 33 and Figure 34 shows a comparison of expression (Figure 33A and Figure 34A) and bioactivity (Figure 33B and Figure 34B) of GIP and GLP1, respectively in exemplary incretin agents that contain both GIP and GLP1, where the order of the GIP and GLP1 peptides encoded by a single polyribonucleotide was varied.
- incretin agent 4093 contained from N-term to C-term: a GLP1 peptide (with H7Y, A8G, R36G mutations), a furin cleavage site, a GIP peptide (with the A2G mutation), fused to Dula_IgG4 (with LS mutations) while incretin agent 4094 contained from N-term to C-term: a GIP peptide (with the A2G mutation), a furin cleavage site, a GLP1 peptide (with H7Y, A8G, R36G mutations), fused to Dula_IgG4 (with LS mutations).
- FIG 33 shows that GIP in both 4093 and 4094 was expressed (A), however, 4094 (where GIP was at the N-term), had slightly better GIP bioactivity, taking into account the amount of GIP peptide assayed (B).
- GLP1 was more highly expressed in the 4094 construct (where GLP1 was after GIP and adjacent to the Furin cleavage site) than in the 4093 construct (A), however, GLP1 showed higher bioactivity in the 4093 construct, where GL1 was adjacent to the husec signal peptide at the N-terminus (B).
- GIP appears to show better bioactivity with Furin cleavage
- GLP1 appears to show better bioactivity when processed with the husec signaling peptide.
- incretin peptides in order for incretin peptides to be functional and interact with their cognate receptors, GIPR and GLP1R, they are preferably properly cleaved (i.e., with signal peptide cleavage or Furin cleavage) and have a scarless N-terminus (i.e., the cleavage/processing at the N-terminus must not change the peptide structure).
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Biophysics (AREA)
- Medicinal Chemistry (AREA)
- Zoology (AREA)
- Biochemistry (AREA)
- Toxicology (AREA)
- General Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Gastroenterology & Hepatology (AREA)
- Molecular Biology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Endocrinology (AREA)
- Peptides Or Proteins (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
The present disclosure provides compositions (e.g., pharmaceutical compositions) for delivery of incretin agents and related technologies (e.g., components thereof and/or methods relating thereto). Among other things, the present disclosure provides treatment methods using polyribonucleotides encoding incretin agents for various diseases.
Description
RNA COMPOSITIONS FOR DELIVERY OF INCRETIN AGENTS
RELATED APPLICATIONS
[0001] This application claims priority to and the benefit of U.S. Provisional Patent Application No. 63/662,890, filed on June 21, 2024, and PCT Application No.
PCT/IB2023/059007, filed on September 11, 2023, the entire contents of which are hereby incorporated by reference in their entirety.
BACKGROUND
[0002] Obesity is the most prevalent chronic disease worldwide, affecting approximately 650 million adults today. Obesity is considered a starting point for and critical contributor to pre-diabetes, type 2 diabetes (T2D, with its complications), non-alcoholic fatty liver disease (NAFLD), non-alcoholic steatohepatitis (NASH), cardiovascular and renal diseases, and premature mortality. It is estimated that the number of obese (BMI>30kg/m2) people will exceed one billion by 2030, about 10% of which will suffer from severe class III obesity (BMI>40kg/m2). Half of all men living with obesity live in just nine countries: USA, China, India, Brazil, Mexico, Russia, Egypt, Germany, and Turkey. In addition, childhood obesity is sharply on the rise worldwide. T2D, NAFLD, NASH, cardiovascular and renal diseases are also prevalent independent of obesity. There exists a need to develop further therapies for treatment and/or prevention of obesity and other related diseases.
SUMMARY
[0003] Treatment of obesity and T2D with incretins and incretin mimetics, in particular GLP1 and GIP receptor agonists or combinations thereof has shown tremendous benefits for people living with the diseases. Due to production and cost limitations, access to this new class of active agents is limited, depriving millions of people of the necessary treatment. The present disclosure recognizes these shortcomings and provides a new therapeutic modality to deliver incretin mimetics by using polyribonucleotide precursors of said incretins, obviating the need of producing the incretins themselves. The present disclosure provides, among other things, polyribonucleotide precursors of said incretins as molecular entities, their production, formulation, and administration to treat obesity and its sequelae, including T2D, early T1D, cardiovascular diseases, renal diseases, NASH and NAFLD. The present disclosure also provides methods where these agents are used to treat
diseases including T2D, early T1D, cardiovascular diseases, renal diseases, NASH and NAFLD independent of obesity. The present disclosure also provides methods where these agents are used to treat sequelae of NASH, including liver fibrosis and cirrhosis.
[0004] The present disclosure also recognizes that such a treatment approach, i.e., delivery of polyribonucleotide precursors of incretins presents additional benefits over current treatments, including but not limited to, broader accessibility for obese people who will otherwise have no access to current products due to limited supply, high price, lack of health insurance, formulations that require lower injection volume compared to marketed peptide-based products, a lower rate of treatment discontinuation by patients due to factors such as gastrointestinal side-effects, and improved properties such as an improved pharmacokinetic profile. In some embodiments, the improved pharmacokinetic profile has the advantage of lower administration frequency by virtue of a longer-acting therapeutic.
[0005] In one aspect, the present disclosure provides a composition comprising a polyribonucleotide encoding an incretin agent. In some embodiments, the incretin agent is a GLP 1 receptor agonist. In some embodiments, the incretin agent is a GIP receptor agonist. In some embodiments, the incretin agent is a GLP1/GIP dual receptor agonist. In some embodiments, the incretin agent is a GLP1/GCG dual receptor agonist. In some embodiments, the incretin agent is a GLP1/GIP/GCG triple receptor agonist.
[0006] In some embodiments, an incretin agent comprises an incretin peptide having an amino acid sequence according to any one of SEQ ID NOs: 5-7, 63-64, 69-70, and 74-75. In some embodiments, an incretin agent comprises an incretin peptide having an amino acid sequence according to any one of SEQ ID NOs: 8-9, 62, and 72. In some embodiments, an incretin agent comprises an incretin peptide having an amino acid sequence according SEQ ID NO: 11. In some embodiments, an incretin agent comprises an incretin peptide having an amino acid sequence according to any one of SEQ ID NOs: 12-14. In some embodiments, an incretin agent comprises an incretin peptide having an amino acid sequence according to SEQ ID NO: 15.
[0007] In some embodiments, an incretin peptide is fused to a signal peptide, optionally via the N-terminus of the incretin peptide, optionally via a linker. In some embodiments, the signal peptide has an amino acid sequence according to any one of SEQ ID NOs: 16-39 and 65-67. In some embodiments, the signal peptide has an amino acid sequence
according to any one of SEQ ID NOs: 16-21 and 65-67. In some embodiments, the signal peptide has an amino acid sequence according to SEQ ID NO: 17. In some embodiments, the signal peptide has an amino acid sequence according to SEQ ID NO: 65. In some embodiments, the signal peptide has an amino acid sequence according to SEQ ID NO: 66. In some embodiments, the incretin agent comprises an amino acid sequence according to any one of SEQ ID NOs: 41-45, 52-61, and 108-152.
[0008] In some embodiments, the incretin agent comprises an incretin peptide fused to one or more additional incretin peptides, optionally via one or more linkers. In some embodiments, the one or more linkers comprise an amino acid sequence according of any one of SEQ ID NOs: 1-5, 68, or 156. In some embodiments, the incretin agent comprises an incretin peptide fused to two or more incretin peptides. In some embodiments, the incretin agent comprises at least one GLP1 receptor agonist and at least one GIP receptor agonist. In some embodiments, the incretin agent comprises at least two GLP1 receptor agonists. In some embodiments, the incretin agent comprises at least two GIP receptor agonists.
[0009] In some embodiments, the incretin agent comprises one or more furin cleavage sites. In some embodiments, the one or more furin cleavage sites are located between adjacent incretin peptides. In some embodiments, the one or more furin cleavage sites comprise an amino acid sequence according to SEQ ID NO: 153. In some embodiments, the incretin agent comprises one or more units that each comprise, from N-terminus to C- terminus: GLP1 receptor agonist-linker-furin cleavage site-GIP receptor agonist, e.g., wherein the incretin agent comprises one unit (e.g., SEQ ID NOs: 76, 77, 78, 79, 80, 81), two units (e.g., SEQ ID NOs: 82); or four units (e.g., SEQ ID NO: 83). In some embodiments, the incretin agent comprises an amino acid sequence according to any one of SEQ ID NOs: 76-83, 94-97, 102-107.
[0010] In some embodiments, the incretin agent comprises a half-life extending moiety. In some embodiments, the half-life extending moiety comprises albumin (e.g., human serum albumin). In some embodiments, the human serum albumin comprises an amino acid sequence having at least 90%, 95%, or 99% identity to SEQ ID NO: 159. In some embodiments, the human serum albumin comprises an amino acid sequence according to SEQ ID NO: 159. In some embodiments, the incretin agent comprises albumin (e.g., human serum albumin) fused to one or more units that each comprise from, N-terminus to C-
terminus: (i) GLP1 receptor agonist-linker (e.g., SEQ ID NO: 98); (ii) GIP receptor agonistlinker (e.g., SEQ ID NO: 100); (iii) GLP1 receptor agonist-linker-furin cleavage site (e.g., SEQ ID NO: 102); or (iv) GLP1 receptor agonist-linker-furin cleavage site-GIP receptor agonist, e.g., wherein the incretin agent comprises one unit (e.g., SEQ ID NO: 104), two units (e.g., SEQ ID NO: 106) or four units (e.g., SEQ ID NO: 107). In some embodiments, the incretin agent comprises an amino acid sequence according to any one of SEQ ID NOs: 98, 100, 102, 104, 106, 107, or any combination thereof.
[0011] In some embodiments, the half-life extending moiety comprises an albumin binding domain (ABD). In some embodiments, the ABD is derived from protein G of Streptococcus strain GI48 and/or from protein PAB of Finegoldia magnet, such as ABD035 and SA21. In some embodiments, the half-life extending moiety comprises an ABD that binds to domain II of human serum albumin and does not overlap or interfere with binding to the FcRn-binding site on albumin. In some embodiments, the half-life extending moiety comprises ABDCon. In some embodiments, the half-life extending moiety comprises an albumin binding domain (ABD) derived from the bacterial protein Sso7d from the hyperthermophilic archaeon Sulfolobus solfataricus, such as Ml 1.12 and Ml 8.2.5. In some embodiments, the half-life extending moiety comprises a DARPin that binds albumin.
[0012] In some embodiments, the ABD comprises an immunoglobulin domain or fragment thereof that binds albumin. In some embodiments, the ABD comprises a fully human domain antibody (dAb) that binds albumin, such as AlbudAb. In some embodiments, the ABD comprises a Fab that binds albumin, such as dsFv CA645. In some embodiments, the ABD comprises a heavy chain only (VHH) antibody, such as a nanobody, that binds albumin. In some embodiments, the VHH antibody comprises a VHH domain having the complementarity determining region (CDR) sequences HCDR1, HCDR2, and/or HCDR3 according to SEQ ID NO: 191 (GFTLDYYA), SEQ ID NO: 192 (IASSGGST), and/or SEQ ID NO: 193 (AAAVLECRTVVRGYDY), respectively. In some embodiments, the VHH antibody comprises an amino acid sequence having at least 90%, 95%, or 99% identity to SEQ ID NO: 154. In some embodiments, the VHH antibody comprises an amino acid sequence according to SEQ ID NO: 154. In some embodiments, the incretin agent comprises a VHH antibody that binds albumin fused to a unit that comprises, from N-terminus to C- terminus: (i) GLPl-linker (e.g., SEQ ID NO: 99); (ii) GIP receptor agonist-linker (e.g., SEQ ID NO: 101); or (iii) GLP1 receptor agonist-linker-furin-GIP receptor agonist-linker (e.g.,
SEQ ID NO: 103 or 105). In some embodiments, the incretin agent comprises an amino acid sequence according to any one of SEQ ID NOs: 99, 101, 103, 105.
[0013] In some embodiments, the half-life extending moiety does not comprise a Fc domain, such as from a human IgG, optionally from a human IgGl, IgG2, IgG3, or IgG4. In some embodiments, the half-life extending moiety comprises a Fc domain, such as from a human IgG, optionally from a human IgGl, IgG2, IgG3, or IgG4. In some embodiments, the human IgG is a human IgG4. In some embodiments, the incretin agent comprises an IgG4 Fc domain fused to a unit comprising, from N-terminus to C-terminus: (i) GLP1 receptor agonist-linker (e.g., SEQ ID NOs: 10, 89, 90, 91); (ii) GIP receptor agonist-linker (e.g., SEQ ID NO: 92, 93); or (iii) GLP1 receptor agonist-linker-furin-GIP receptor agonist-linker (e.g., SEQ ID NO: 94, 95, 96, 97). In some embodiments, the IgG4 Fc domain comprises an amino acid sequence that is at least 90%, 95%, or 99% identical to SEQ ID NO: 155. In some embodiments, the IgG4 Fc domain comprises an amino acid sequence according to SEQ ID NO: 155. In some embodiments, the incretin agent comprises an amino acid sequence according to any one of SEQ ID NOs: 10, and 89-97.
[0014] In some embodiments, the Fc domain comprises one or more mutations in one or both Fc constant domains to increase half-life of the incretin agent and/or induce dimerization. In some embodiments, the one or more mutations comprises one or more mutations in a CH3 domain. In some embodiments, the one or more mutations to induce dimerization comprises: (i) Y349C, T366S, L368A, and/or Y407V, according to EU numbering; or (ii) S354C and/or T366W, according to EU numbering. In some embodiments, the one or more mutations comprises Y349C, T366S, L368A, and Y407V (“FcKIH-b”), according to EU numbering, or S354C and T366W (“FcKIH-a”), according to EU numbering. In some embodiments, the incretin agent comprises a first polypeptide chain and a second polypeptide chain, wherein the first polypeptide chain comprises an incretin peptide fused to a first Fc domain, wherein the first Fc domain comprises the mutations Y349C, T366S, L368A, and Y407V (“FcKIH-b”), according to EU numbering and wherein the second polypeptide chain comprises an incretin peptide fused to a second Fc domain, wherein the second Fc domain comprises the mutations S354C and T366W (“FcKIH-a”), according to EU numbering.
[0015] In some embodiments, the one or more mutations to increase half-life of the incretin agent comprises M428L and N434S (“LS”), according to EU numbering. In some embodiments, the incretin agent comprises an Fc domain with FcKIH-a mutations on a first polypeptide chain and an Fc domain with FcKIH-b mutations on a second polypeptide chain, where the Fc domain on each polypeptide chain is independently fused with one or more units that comprise, from N-terminus to C-terminus: (i) GLP1 receptor agonist-linker (e.g., SEQ ID NO: 84, 85, 86, 87); or (ii) GIP receptor agonist-linker (e.g., SEQ ID NO: 88). In some embodiments, the incretin agent comprises an amino acid sequence according to any one of SEQ ID NOs: 84-88.
[0016] In some embodiments, the Fc domain comprises one or more mutations that ablate effector activity of the Fc domain (e.g., binding to Fey receptors or Clq). In some embodiments, the one or more mutations that ablate effector activity of the Fc domain (e.g., binding to Fey receptors or Clq) comprise the following mutations: L234S, L235T, and G236R (“STR”) according to EU numbering. In some embodiments, the one or more mutations that ablate effector activity of the Fc domain (e.g., binding to Fey receptors or Clq) comprise the following mutations: L234A and L235A (“LALA”) according to EU numbering. In some embodiments, the one or more mutations that ablate effector activity of the Fc domain (e.g., binding to Fey receptors or Clq) comprise the following mutations: L234A/L235A/P329G (“LALAPG”) according to EU numbering.
[0017] In some embodiments, the half-life extending moiety comprises a VNAR that binds albumin. In some embodiments, the half-life extending moiety comprises an XTEN sequence.
[0018] In some embodiments, the polyribonucleotide has a ribonucleic acid sequence that is at least 90% identical to any one of SEQ ID NOs: 177-185 and 224-256. In some embodiments, the polyribonucleotide has a ribonucleic acid sequence according to any one of SEQ ID Nos: 177-185 and 224-256.
[0019] In some embodiments, the polyribonucleotide comprises at least one noncoding sequence element that enhances RNA stability and/or translation efficiency. In some embodiments, the at least one non-coding sequence element comprises a 5’ cap structure, a 5’ UTR, a 3 ’ UTR, and/or a polyA tail.
[0020] In some embodiments, the polyribonucleotide comprises, in a 5 ’ to 3 ’ direction: a. a 5’ UTR; b. a signal peptide-coding sequence; c. an incretin peptide-coding sequence; d. a 3’ UTR; and e. a polyAtail.
[0021] In some embodiments, the polyribonucleotide comprises, in a 5 ’ to 3 ’ direction: (1) a. a 5’ UTR; b. a signal peptide-coding sequence; c. an incretin peptide-coding sequence; d. a linker-coding sequence; e. a half-life extending moiety-coding sequence; f. a 3’ UTR; and g. a polyAtail; or (2) a. a 5’ UTR; b. a signal peptide-coding sequence; c. a halflife extending moiety-coding sequence; d. a linker-coding sequence; e. an incretin peptide- coding sequence; f. a 3’ UTR; and g. a polyAtail.
[0022] In some embodiments, the incretin peptide is encoded by a coding sequence which is codon-optimized and/or the G/C content of which is increased compared to wild type coding sequence, wherein the codon-optimization and/or the increase in the G/C content does not change the sequence of the encoded amino acid sequence.
[0023] In some embodiments, the polyribonucleotide comprises at least one modified ribonucleotide. In some embodiments, the polyribonucleotide comprises a modified nucleoside in place of uridine. In some embodiments, the polyribonucleotide comprises a modified nucleoside in place of each uridine. In some embodiments, the modified nucleoside is selected from pseudouridine (y), N1 -methyl -pseudouridine (m h|/). and 5 -methyl -uridine (m5U). In some embodiments, the modified nucleoside is N1 -methyl -pseudouridine (ml\|/).
[0024] In some embodiments, the polyribonucleotide comprises a 5’ cap structure. In some embodiments, the polyribonucleotide comprises a 5 ’ UTR. In some embodiments, the polyribonucleotide comprises a 3’ UTR. In some embodiments, the polyribonucleotide comprises a polyAtail. In some embodiments, the polyAtail comprises at least 100 nucleotides. In some embodiments, the polyribonucleotide is mRNA.
[0025] In some embodiments, the polyribonucleotide is formulated as a liquid, formulated as a solid, or a combination thereof. In some embodiments, the polyribonucleotide is formulated for injection. In some embodiments, the polyribonucleotide is formulated for intraperitoneal or intravenous administration.
[0026] In some embodiments, the polyribonucleotide is formulated or is to be formulated as lipid particles. In some embodiments, the polyribonucleotide is formulated or is to be formulated as lipid nanoparticles. In some embodiments, the polyribonucleotide is encapsulated within the lipid nanoparticles. In some embodiments, the lipid nanoparticles are pancreas-targeting and/or gut-targeting lipid nanoparticles. In some embodiments, the lipid nanoparticles are cationic lipid nanoparticles.
[0027] In some embodiments, lipids that form the lipid nanoparticles comprise a. a polymer-conjugated lipid; b. a cationic lipid; and c. a neutral lipid. In some embodiments, the polymer-conjugated lipid is a PEG-conjugated lipid. In some embodiments, the cationic lipid is an ionizable lipid-like material (lipidoid).
[0028] In some embodiments, the cationic lipid has one of the following structures:
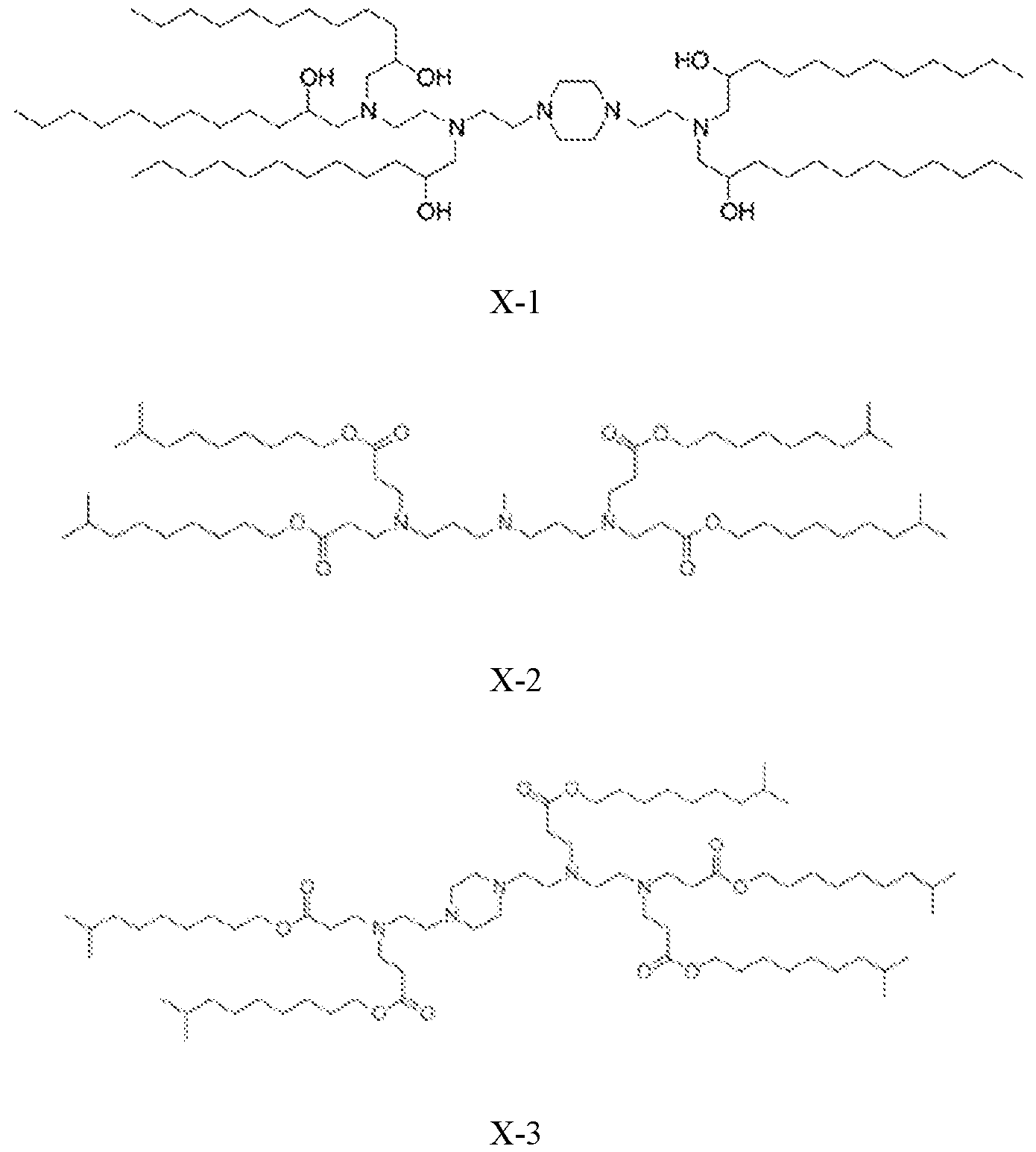
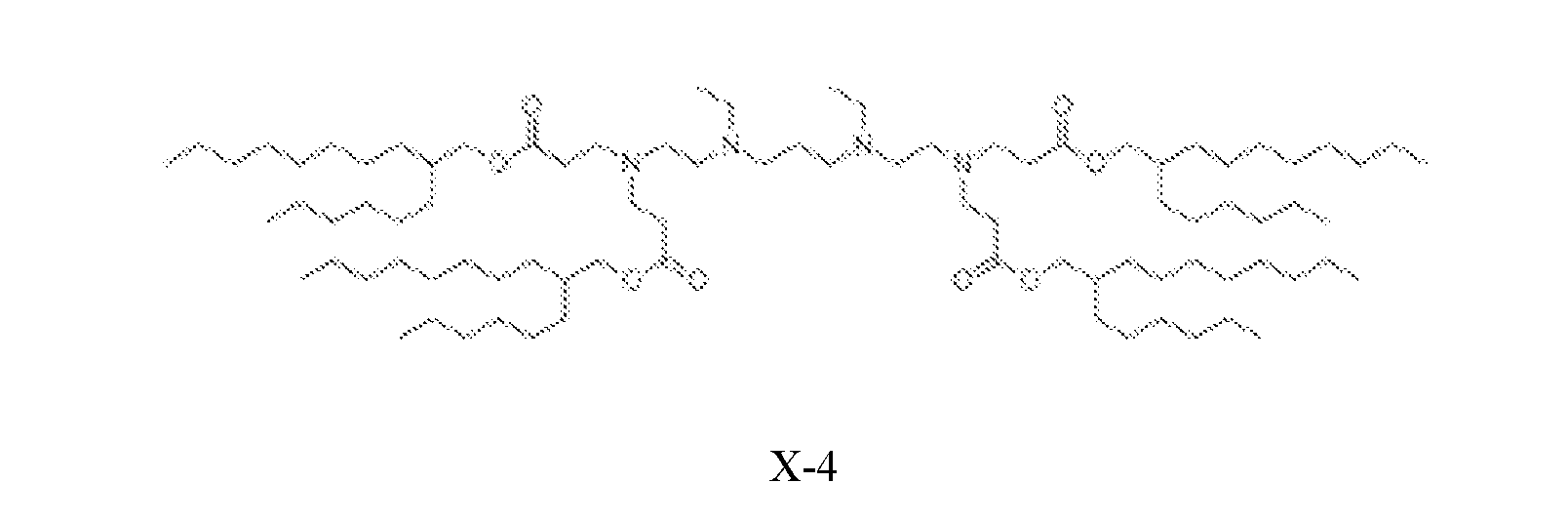
[0029] In some embodiments, the neutral lipid comprises a helper lipid such as 1,2- distearoyl-sn-glycero-3-phosphocholine (DPSC) and/or cholesterol.
[0030] In some embodiments, the cationic lipid is selected from cationic lipid X-2, X- 3, or X-4 and the neutral lipid comprises a helper lipid such as DOTAP, DOPE, or PS, and cholesterol.
[0031] In some embodiments, the polymer-conjugated lipid is C14-PEG2000.
[0032] In some embodiments, the lipid nanoparticles comprise: i) about 30 mol% to about 50 mol% of a cationic lipid; ii) about 1 mol% to 5 mol% of a PEG-conjugated lipid; iii) about 30 mol% to about 50 mol% of a helper lipid; and iv) about 20 mol% to about 40 mol% of cholesterol.
[0033] In some embodiments, the lipid nanoparticles comprise about 35 mol% of a cationic lipid; about 40 mol% of a helper lipid, about 22.5 mol% of cholesterol, and about 2.5 mol% of a PEG-conjugated lipid.
[0034] In some embodiments, the lipid nanoparticles comprise about 35 mol% of cationic lipid X-2, X-3, or X-4, about 40 mol% of DOTAP, DOPE, or PS, about 22.5 mol% of cholesterol, and about 2.5 mol% of C14-PEG2000.
[0035] In some embodiments, the lipid nanoparticles comprise about 35 mol% of cationic lipid X-2; about 40 mol% of DOTAP; about 22.5 mol% of cholesterol, and about 2.5 mol% of C14-PEG2000. In some embodiments, the lipid nanoparticles comprise about 35 mol% of cationic lipid X-3; about 40 mol% of DOTAP; about 22.5 mol% of cholesterol; and about 2.5 mol% of C14-PEG2000. In some embodiments, the lipid nanoparticles comprise about 35 mol% of cationic lipid X-4; about 40 mol% of DOTAP; about 22.5 mol% of cholesterol; and about 2.5 mol% of C14-PEG2000. In some embodiments, the lipid
nanoparticles comprise about 35 mol% of cationic lipid X-2; about 40 mol% of DOPE; about 22.5 mol% of cholesterol; and about 2.5 mol% of C14-PEG2000. In some embodiments, the lipid nanoparticles comprise about 35 mol% of cationic lipid X-3; about 40 mol% of DOPE; about 22.5 mol% of cholesterol; and about 2.5 mol% of C14-PEG2000. In some embodiments, the lipid nanoparticles comprise about 35 mol% of cationic lipid X-4; about 40 mol% of DOPE; about 22.5 mol% of cholesterol; and about 2.5 mol% of C14-PEG2000. In some embodiments, the lipid nanoparticles comprise about 35 mol% of cationic lipid X-2; about 40 mol% of PS; about 22.5 mol% of cholesterol; and about 2.5 mol% of C14- PEG2000. In some embodiments, the lipid nanoparticles comprise about 35 mol% of cationic lipid X-3; about 40 mol% of PS; about 22.5 mol% of cholesterol; and about 2.5 mol% of C14-PEG2000. In some embodiments, the lipid nanoparticles comprise about 35 mol% of cationic lipid X-4; about 40 mol% of PS; about 22.5 mol% of cholesterol; and about 2.5 mol% of C14-PEG2000.
[0036] In some embodiments, the lipid nanoparticles are formulated for intraperitoneal (i.p.) delivery. In some embodiments, the lipid nanoparticles have an average size of about 50-150 nm. In some embodiments, the composition comprises one or more pharmaceutically acceptable carriers, diluents and/or excipients. In some embodiments, the composition further comprise a cryoprotectant. In some embodiments, the cryoprotectant is sucrose. In some embodiments, the composition comprises an aqueous buffered solution. In some embodiments, the aqueous buffered solution includes sodium ions.
[0037] In another aspect, the present disclosure provides, a method of treating a disease-state in a subject in need thereof, comprising administering to said subject a therapeutically effective amount of a composition comprising one or more polyribonucleotides described here.
[0038] In some embodiments, the method further comprises administering one or more DPP -4 inhibitors. In some embodiments, the one or more DPP-4 inhibitors and the composition are administered concurrently. In some embodiments, the one or more DPP -4 inhibitors and the composition are administered sequentially. In some embodiments, the one or more DPP-4 inhibitors are administered prior to the composition. In some embodiments, one or more DPP-4 inhibitors are administered after the composition. In some embodiments, one or more DPP-4 inhibitors comprises sitagliptin, vildagliptin, saxagliptin, linagliptin,
gemigliptin, anagliptin, teneligliptin, alogliptin, trelagliptin, omarigliptin, evogliptin, gosogliptin, dutogliptin, neogliptin, retagliptin, denagliptin, cofroglipin, fotagliptin, prusogliptin, berberine, or any combination thereof. In some embodiments, one or more DPP- 4 inhibitors are administered orally.
[0039] In some embodiments, disease-state is obesity or an obesity-related disorder. In some embodiments, obesity-related disorder is pre-diabetes, type 2 diabetes (T2D), early type 1 diabetes (T1D), non-alcoholic fatty liver disease (NAFLD), non-alcoholic steatohepatitis (NASH), a cardiovascular (CV) disease, a renal disease, or elevated risk of premature mortality. In some embodiments, cardiovascular (CV) disease comprises a major cardiovascular event (MACE), including CV death, non-fatal myocardial infarction, non-fatal stroke, and/or heart failure with preserved ejection fraction (HFpEF).
[0040] In some embodiments, method improves weight management in said subject. In some embodiments, the method reduces weight gain or induces weight loss in said subject. In some embodiments, the disease-state is diabetes. In some embodiments, the method improves glycemic control in said subject. In some embodiments, the method lowers HbAlc in said subject. In some embodiments, the diabetes is pre-diabetes, type 2 diabetes (T2D), or early type 1 diabetes (T1D).
[0041] In some embodiments, the disease-state is a cardiovascular (CV) disease. In some embodiments, the cardiovascular disease comprises a major cardiovascular event (MACE), including CV death, non-fatal myocardial infarction, non-fatal stroke, and/or heart failure with preserved ejection fraction (HfpEF). In some embodiments, the method improves a subject’s blood pressure and/or blood lipids in said subject.
[0042] In some embodiments, the disease-state is a renal disease. In some embodiments, the disease-state is non-alcoholic fatty liver disease (NAFLD). In some embodiments, the disease-state is non-alcoholic steatohepatitis (NASH) and optionally its sequelae, liver fibrosis and cirrhosis.
[0043] In some embodiments, administering the composition to the subject comprises administering one or more doses of the composition to the subject. In some embodiments, the one or more doses of the composition are administered to the subject daily, every other day or once a week. In some embodiments, the one or more doses of the composition are
administered to the subject less frequently than once a week. In some embodiments, the one or more doses of the composition are administered to the subject once every 2, 3 or 4 weeks. In some embodiments, the composition is administered via injection. In some embodiments, the composition is administered subcutaneously, intravenously, intramuscularly, or intraperitoneally. In some embodiments, the composition is administered intraperitoneally. In some embodiments, the composition is administered non-invasively (e.g., orally or nasally). In some embodiments, administration of the composition results in expression of the incretin agent in the subject. In some embodiments, the composition is administered in a volume that is less than 0.5 mb
[0044] In another aspect, the present disclosure provides, use of the composition of any comprising one or more polyribonucleotides described herein for the treatment of a disease-state in a subject in need thereof.
[0045] In another aspect, the present disclosure provides, a method of producing an incretin agent comprising administering to cells a composition comprising a polyribonucleotide described herein so that the cells express and secrete the incretin agent.
[0046] In another aspect, the present disclosure provides, an incretin agent that comprises an incretin peptide fused to a signal peptide. In some embodiments, the incretin peptide is fused to the signal peptide via the N-terminus of the incretin peptide, optionally via a linker. In some embodiments, the signal peptide has an amino acid sequence according to any one of SEQ ID NOs: 16-39 and 65-67. In some embodiments, the signal peptide has an amino acid sequence according to any one of SEQ ID NOs: 16-21 and 65-67. In some embodiments, the signal peptide has an amino acid sequence according to SEQ ID NO: 17. In some embodiments, the signal peptide has an amino acid sequence according to SEQ ID NO: 65. In some embodiments, the signal peptide has an amino acid sequence according to SEQ ID NO: 66. In some embodiments, the incretin agent comprises an incretin peptide fused to a signal peptide that comprises an amino acid sequence according to any one of SEQ ID NOs: 41-45, 52-61, and 108-152.
[0047] In some embodiments, the incretin agent comprises an incretin peptide fused to one or more additional incretin peptides, optionally via one or more linkers. In some embodiments, the one or more linkers comprise an amino acid sequence according of any one
of SEQ ID NOs: 1-5, 68, or 156. In some embodiments, the incretin agent comprises an incretin peptide fused to two or more incretin peptides.
[0048] In some embodiments, the incretin agent comprises at least one GLP 1 receptor agonist and at least one GIP receptor agonist. In some embodiments, the incretin agent comprises at least two GLP1 receptor agonists. In some embodiments, the incretin agent comprises at least two GIP receptor agonists. In some embodiments, the incretin agent comprises one or more furin cleavage sites. In some embodiments, the one or more furin cleavage sites are located between adjacent incretin peptides. In some embodiments, the one or more furin cleavage sites comprise an amino acid sequence according to SEQ ID NO: 153. In some embodiments, the incretin agent comprises one or more units that each comprise, from N-terminus to C-terminus: GLP1 receptor agonist-linker-furin cleavage site-GIP receptor agonist, e.g., wherein the incretin agent comprises one unit (e.g., SEQ ID NOs: 76, 77, 78, 79, 80, 81), two units (e.g., SEQ ID NOs: 82); or four units (e.g., SEQ ID NO: 83). In some embodiments, the incretin agent comprises an amino acid sequence according to any one of SEQ ID NOs: 76-83, 94-97, 102-107.
[0049] In some embodiments, the incretin agent comprises a half-life extending moiety. In some embodiments, the half-life extending moiety comprises albumin (e.g., human serum albumin). In some embodiments, the human serum albumin comprises an amino acid sequence having at least 90%, 95%, or 99% identity to SEQ ID NO: 159. In some embodiments, the human serum albumin comprises an amino acid sequence according to SEQ ID NO: 159.
[0050] In some embodiments, the incretin agent comprises albumin (e.g., human serum albumin) fused to one or more units that each comprise from, N-terminus to C- terminus: (i) GLP1 receptor agonist-linker (e.g., SEQ ID NO: 98); (ii) GIP receptor agonistlinker (e.g., SEQ ID NO: 100); (iii) GLP1 receptor agonist-linker-furin cleavage site (e.g., SEQ ID NO: 102); or (iv) GLP1 receptor agonist-linker-furin cleavage site-GIP receptor agonist, e.g., wherein the incretin agent comprises one unit (e.g., SEQ ID NO: 104), two units (e.g., SEQ ID NO: 106) or four units (e.g., SEQ ID NO: 107)In some embodiments, the incretin agent comprises an amino acid sequence according to any one of SEQ ID NOs: 98, 100, 102, 104, 106, 107, or any combination thereof.
[0051] In some embodiments, the half-life extending moiety comprises an albumin binding domain (ABD). In some embodiments, the ABD is derived from protein G of Streptococcus strain GI48 and/or from protein PAB of Finegoldia magnet, such as ABD035 and SA21. In some embodiments, the half-life extending moiety comprises an ABD that binds to domain II of human serum albumin and does not overlap or interfere with binding to the FcRn-binding site on albumin. In some embodiments, the half-life extending moiety comprises ABDCon. In some embodiments, the half-life extending moiety comprises an ABD derived from the bacterial protein Sso7d from the hyperthermophilic archaeon Sulfolobus solfataricus, such as Mil.12 and M18.2.5. In some embodiments, the half-life extending moiety comprises a DARPin that binds albumin. In some embodiments, the ABD comprises an immunoglobulin domain or fragment thereof that binds albumin. In some embodiments, the ABD comprises a fully human domain antibody (dAb) that binds albumin, such as AlbudAb. In some embodiments, the ABD comprises a Fab that binds albumin, such as dsFv CA645.
[0052] In some embodiments, the ABD comprises a heavy chain only (VHH) antibody, such as a nanobody, that binds albumin. In some embodiments, the VHH antibody comprises a VHH domain having the complementarity determining region (CDR) sequences HCDR1, HCDR2, and/or HCDR3 according to SEQ ID NO: 191 (GFTLDYYA), SEQ ID NO: 192 (IASSGGST), and/or SEQ ID NO: 193 (AAAVLECRTVVRGYDY), respectively. In some embodiments, the VHH antibody comprises an amino acid sequence having at least 90%, 95%, or 99% identity to SEQ ID NO: 154. In some embodiments, the VHH antibody comprises an amino acid sequence according to SEQ ID NO: 154. In some embodiments, the incretin agent comprises a VHH antibody that binds albumin fused to a unit that comprises, from N-terminus to C-terminus: (i) GLPl-linker (e.g., SEQ ID NO: 99); (ii) GIP receptor agonist-linker (e.g., SEQ ID NO: 101); or (iii) GLP1 receptor agonist-linker-furin-GIP receptor agonist-linker (e.g., SEQ ID NO: 103 or 105). In some embodiments, the incretin agent comprises an amino acid sequence according to any one of SEQ ID NOs: 99, 101, 103, 105.
[0053] In some embodiments, the half-life extending moiety does not comprise a Fc domain, such as from a human IgG, optionally from a human IgGl, IgG2, IgG3, or IgG4. In some embodiments, the half-life extending moiety comprises a Fc domain, such as from a human IgG, optionally from a human IgGl, IgG2, IgG3, or IgG4. In some embodiments, the
human IgG is a human IgG4. In some embodiments, the incretin agent comprises an IgG4 Fc domain fused to a unit comprising, from N-terminus to C-terminus: i) GLP1 receptor agonistlinker (e.g., SEQ ID NOs: 10, 89, 90, 91); (ii) GIP receptor agonist-linker (e.g., SEQ ID NO: 92, 93); or (iii) GLP1 receptor agonist-linker-furin-GIP receptor agonist-linker (e.g., SEQ ID NO: 94, 95, 96, 97). In some embodiments, the IgG4 Fc domain comprises an amino acid sequence that is at least 90%, 95%, or 99% identical to SEQ ID NO: 155. In some embodiments, the IgG4 Fc domain comprises an amino acid sequence according to SEQ ID NO: 155. In some embodiments, the incretin agent comprises an amino acid sequence according to any one of SEQ ID NOs: 10, 89-97.
[0054] In some embodiments, the Fc domain comprises one or more mutations in one or both Fc constant domains to increase half-life of the incretin agent and/or induce dimerization. In some embodiments, the one or more mutations comprises one or more mutations in a CH3 domain. In some embodiments, the one or more mutations to induce dimerization comprises: (i) Y349C, T366S, L368A, and/or Y407V, according to EU numbering; or (ii) S354C and/or T366W, according to EU numbering.
[0055] In some embodiments, the one or more mutations comprises Y349C, T366S, L368A, and Y407V (“FcKIH-b”), according to EU numbering, or S354C and T366W (“FcKIH-a”), according to EU numbering. In some embodiments, the incretin agent comprises a first polypeptide chain and a second polypeptide chain, wherein the first polypeptide chain comprises an incretin peptide fused to a first Fc domain, wherein the first Fc domain comprises the mutations Y349C, T366S, L368A, and Y407V (“FcKIH-b”), according to EU numbering and wherein the second polypeptide chain comprises an incretin peptide fused to a second Fc domain, wherein the second Fc domain comprises the mutations S354C and T366W (“FcKIH-a”), according to EU numbering.
[0056] In some embodiments, the one or more mutations to increase half-life of the incretin agent comprises M428L and N434S (“LS”), according to EU numbering. In some embodiments, the incretin agent comprises an Fc domain with FcKIH-a mutations on a first polypeptide chain and an Fc domain with FcKIH-b mutations on a second polypeptide chain, where the Fc domain on each polypeptide chain is independently fused with one or more units that comprise, from N-terminus to C-terminus: (i) GLP1 receptor agonist-linker (e.g., SEQ ID NO: 84, 85, 86, 87); or (ii) GIP receptor agonist-linker (e.g., SEQ ID NO: 88). In
some embodiments, the incretin agent comprises an amino acid sequence according to any one of SEQ ID NOs: 84-88.
[0057] In some embodiments, the Fc domain comprises one or more mutations that ablate effector activity of the Fc domain (e.g., binding to Fey receptors or Clq). In some embodiments, the one or more mutations that ablate effector activity of the Fc domain (e.g., binding to Fey receptors or Clq) comprise the following mutations: L234S, L235T, and G236R (“STR”) according to EU numbering. In some embodiments, the one or more mutations that ablate effector activity of the Fc domain (e.g., binding to Fey receptors or Clq) comprise the following mutations: L234A and L235A (“LALA”) according to EU numbering. In some embodiments, the one or more mutations that ablate effector activity of the Fc domain (e.g., binding to Fey receptors or Clq) comprise the following mutations: L234A/L235A/P329G (“LALAPG”) according to EU numbering.
[0058] In some embodiments, the half-life extending moiety comprises a VNAR that binds albumin.
[0059] In some embodiments, the half-life extending moiety comprises an XTEN sequence.
[0060] In another aspect, the present disclosure provides an incretin agent comprising: a husec signal peptide; an incretin peptide comprising a GLP1 incretin peptide, or fragment or variant thereof; wherein the GLP1 incretin peptide comprises an amino acid sequence having an A8G substitution mutation compared to a wildtype GLP1 amino acid sequence.
[0061] In another aspect, the present disclosure provides a polyribonucleotide encoding the incretin agent comprising a husec signal peptide; an incretin peptide comprising a GLP1 incretin peptide, or fragment or variant thereof; wherein the GLP1 incretin peptide comprises an amino acid sequence having an A8G substitution mutation compared to a wildtype GLP1 amino acid sequence.
[0062] In another aspect, the present disclosure provides, an incretin agent comprising: a husec signal peptide; an incretin peptide comprising a GIP incretin peptide, or fragment or variant thereof; wherein the GIP incretin peptide comprises an amino acid
sequence having an A2G substitution mutation compared to a wildtype GIP amino acid sequence.
[0063] In another aspect, the present disclosure provides a polyribonucleotide encoding the incretin agent an incretin agent comprising: a husec signal peptide; an incretin peptide comprising a GIP incretin peptide, or fragment or variant thereof; wherein the GIP incretin peptide comprises an amino acid sequence having an A2G substitution mutation compared to a wildtype GIP amino acid sequence.
BRIEF DESCRIPTION OF THE DRAWING
[0064] Figure 1 shows an exemplary therapeutic strategy utilizing polyribonucleotides as described herein for delivery and in vivo expression of incretin agents.
[0065] Figure 2 shows a schematic of an exemplary polyribonucleotide that encodes an incretin agent.
[0066] Figure 3 shows an exemplary design of an incretin agent described herein. Specifically, Figure 3 shows a schematic of a polyribonucleotide encoding a signal peptide (“SP”) and a single incretin peptide (which configuration is referred to herein as “I: lx”) (top) and a schematic of the translated incretin protein (bottom).
[0067] Figure 4 shows an exemplary design of an incretin agent described herein. Specifically, Figure 4 shows a schematic of a polyribonucleotide encoding a signal peptide (“SP”) and two incretin peptides separated by a linker (“LI”) and furin cleavage site (“F”) (which configuration is referred to herein as “I:2x”) (top) and a schematic of the translated protein (bottom).
[0068] Figure 5 shows an exemplary design of an incretin agent described herein. Specifically, Figure 5 shows a schematic of a polyribonucleotide encoding a signal peptide (“SP”) and four incretin peptides, each separated by a linker (“LI”) furin cleavage site (“F”) (which configuration is referred to herein as “I:4x”) (top) and a schematic of the translated protein (bottom).
[0069] Figures 6A-B show exemplary incretin agents including an incretin agent that has a signal peptide (“SP”), a GLP1 incretin peptide and a (GGGGS)2 linker (Figure 6A), and an incretin agent that has a signal peptide (“SP”), a GLP1 incretin peptide, a (GGGGS)2
linker, a furin cleavage site, and a GIP incretin peptide (Figure 6B). Signal peptide cleavage sites are indicated in Figure 6A and Figure 6B. Additionally, a furin cleavage site is indicated in Figure 6B so that upon expression, the GIP incretin peptide is cleaved from the GLP1 incretin peptide.
[0070] Figure 7 shows an exemplary design of an incretin agent described herein. Specifically, Figure 7 shows a schematic of a polyribonucleotide encoding a signal peptide (“SP”) and an incretin agent (e.g., a I: lx, I:2x, or I:4x incretin agent as described herein) fused to a half-life extension (“HLE”) domain, e.g., human serum albumin (“HSA”) or an albumin binding domain (‘ ‘ABD”) (top) via a linker (“L2”) and a schematic of the translated protein (bottom).
[0071] Figures 8A-B show exemplary incretin agents that may be encoded by polyribonucleotides described herein, that include more than one incretin peptide and a half life extension (HLE) domain. Figure 8A shows an incretin agent that has a signal peptide (“SP”), a first GLP1 incretin peptide, a linker, a second GLP1 incretin peptide, a second linker (GGGGS)3, and a half life extension (HLE) domain that is human serum albumin (HSA). Figure 8B shows an incretin agent that has a signal peptide (“SP”), a first GLP1 incretin peptide, a linker, a second GLP1 incretin peptide, a second linker (GGGGS)3, and a half life extension (HLE) domain that is a VHH domain that binds to HSA. Furin and SP cleavage sites within the incretin agent are indicated with arrows, so that upon expression, the signal peptide is cleaved and the first GLP1 incretin peptide is cleaved from the second GLP1 incretin peptide, and the second GLP1 incretin peptide remains fused to the HLE domain (HSA or anti -HSA VHH). Such a design produces two incretin peptides with varying halflives and activity.
[0072] Figure 9 shows an exemplary incretin agent that may be encoded by one or more polyribonucleotides described herein, that includes more than one incretin peptide and a half life extension (HLE) domain. Specifically, the incretin agent in Figure 9 has a signal peptide (“SP”), a first GLP1 incretin peptide, a linker (GGGGS)2, a first GIP incretin peptide, a second linker (GGGGS)2, a second GLP1 incretin peptide, a third linker (GGGGS)2, a second GIP incretin peptide, a fourth linker (GGGGS)? and half-life extension (HLE) domain that is human serum albumin (HSA). Furin and SP cleavage sites within the incretin agent are
indicated with arrows. Such a design produces four separate incretin peptides, where the second GIP incretin peptide remains fused to the HLE domain.
[0073] Figure 10 shows an exemplary design of a polyribonucleotide encoding an incretin agent that includes an incretin peptide fused to an Fc domain, where the incretin peptide could be one (I: lx), two (I:2x) or four (I:4x) incretin peptides (top). When two of the polyribonucleotides encoding the two separate chains are expressed, the two polypeptide chains associate and result in dimeric (e.g., homodimeric) structure (bottom). Each polypeptide chain also includes a signal peptide (SP) and a linker (L2). In some embodiments, the Fc domains include mutations to abolish effector function (e.g., STR, LALA, LALAPG, etc. mutations) and/or to extend half-life (e.g., YTE, LS, etc. mutations).
[0074] Figure 11 shows an exemplary incretin agent that may be encoded by one or more polyribonucleotides described herein, that includes more than one incretin peptides on more than one polypeptide chains. Specifically, each polypeptide chain of the incretin agent in Figure 11 has a signal peptide (“SP”), a GLP1 incretin peptide, a linker (GGGGS)3, and an Fc domain. One or both of the Fc domains contains “LS” mutations (M428L/N434S according to the EU numbering scheme) to extend half-life of the incretin agent. When the two polypeptide chains are expressed, they associate to form a homodimeric structure as shown in Figure 11. SP cleavage sites within the incretin agent are indicated with arrows.
[0075] Figure 12 shows an exemplary incretin agent that may be encoded by one or more polyribonucleotides described herein, that includes more than one incretin peptide on more than one polypeptide chain. Specifically, each polypeptide chain of the incretin agent has a signal peptide (SP), a GLP1 incretin peptide, a linker, a GIP peptide, a second linker (GGGGS)3, and an Fc domain. One or both Fc domains contain an LS mutation. When the two polypeptide chains are expressed, they associate to form a homodimeric structure as shown in Figure 12. Furin and SP cleavage sites within the incretin agent are indicated with arrows.
[0076] Figure 13 shows an exemplary design of a two polyribonucleotides, each encoding a polypeptide chain that includes an incretin peptide fused to an Fc domain (top). In each polypeptide chain (incretin-Fc fusion), there is a signal peptide (SP) and one, two or four incretin peptides (I: lx, I:2x, or I:4x) fused to an Fc domain via a link (L2), and each Fc domain has a modification that induces heterodimerization (e.g., a knob-in-hole mutation).
When the two polypeptide chains are expressed, they associate with each other to form a heterodimeric incretin agent.
[0077] Figure 14 shows an exemplary incretin agent that may be encoded by one or more polyribonucleotides described herein, that includes more than one incretin peptides on more than one polypeptide chains. Specifically, each polypeptide chain of the incretin agent in Figure 14 has a signal peptide (SP), a GLP1 or GIP incretin peptide, a linker (GGGGS)3, and an Fc domain. One or both Fc domains contain “LS” mutations (M428L/N434S), “STR” mutations (L234S, L235T, and G236R mutations according to the EU numbering scheme) to silence Fc effector function, and “knob-in-hole” mutations to induce heterodimerization.
When the two polypeptide chains are expressed, they associate to form a heterodimeric incretin agent that contains two polypeptide chains with different incretin peptides. SP cleavage sites within the incretin agent are indicated with arrows.
[0078] Figure 15 shows exemplary concentrations (pg/ml) of GLP1 (7-37) in supernatant of HEK293tl7 cells transfected with polyribonucleotides encoding for GLP1 (7- 37) at 3, 6, 24, 48, and 72 hours after transfection (GLP1 n=4 +/- SD; ns = not significant; * p<0.05, ** p<0.01).
[0079] Figure 16 shows exemplary concentrations (pg/ml) of GLP1 (7-37) with K34R mutation in supernatant of HEK293tl7 cells transfected with polyribonucleotides encoding for GLP1 (7-37)-(K34R) at 3, 6, 24, 48, and 72 hours after transfection (GLP1 n=4 +/- SD; ns = not significant; * p<0.05, ** p<0.01).
[0080] Figure 17 shows exemplary concentrations (pg/ml) of GIP (1-42) in supernatant of HEK293tl7 cells transfected with polyribonucleotides encoding for GIP (1-42) at 3, 6, 24, 48, and 72 hours after transfection (GIP n=6 +/- SD; ns = not significant; * p<0.05, ** p<0.01).
[0081] Figure 18 shows concentrations (pg/ml) of exemplary GLP1 incretin agents in supernatant of HEK29tl7 cells transfected with polyribonucleotides encoding for incretin agents containing a viral signal peptide (“viral SP”) or a husec signal peptide (“husec”) and codon-optimized using different strategies (“optl” vs. “optp”). The specific incretin agents include: viral SP - GLP1 (7-37), viral SP - GLP1 (7-37)-(K34R), husec - GLP-1 (7-37)-A8G
(optl), husec - GLP-1 (7-37)-A8G-linker (optl), husec - GLP-1 (7-37)-A8G (optp), and husec - GLP-1 (7-37)-A8G-linker (optp) incretin agents.
[0082] Figure 19 shows concentrations (ng/ml) of exemplary GIP incretin agents in supernatant of HEK29tl7 cells transfected with polyribonucleotides encoding for incretin agents containing a viral signal peptide (“viral SP”) or a husec signal peptide (“husec”) and codon-optimized using different strategies (“optl” vs. “optp”). The specific incretin agents include: viral SP - GIP (1-42), husec - GIP (l-42)-A2G (optl), and GIP (l-42)-A2G (optp).
[0083] Figure 20 shows a schematic of where the theoretical cleavage sites of the various signal peptides he within an incretin agent amino acid sequence. Figure 20 also indicates that the A8G mutation facilitates correct N-terminal processing of GLP1 incretin agents with husec signal peptides
[0084] Figure 21 shows a schematic of where the theoretical cleavage sites of the various signal peptides he within an incretin agent amino acid sequence. Figure 21 also indicates that the A2G mutation facilitates correct N-terminal processing of GIP incretin agents with husec signal peptides.
[0085] Figures 22A-B show a schematic of the GLP1R (A) and GIPR (B) overexpressing HEK293 reporter cells lines to be utilized in an assay to determine bioactivity of exemplary GLP1 and GIP incretin agents in Example 7.
[0086] Figure 23 shows results from a bioactivity assay of exemplary GLP1 incretin agents. Specifically, results are expressed as fold induction over control samples.
[0087] Figure 24 shows results from a bioactivity assay of exemplary GIP incretin agents. Specifically, results are expressed as fold induction over control samples.
[0088] Figure 25 shows in vitro activity (GIP expression) for certain exemplary incretin agents tested.
[0089] Figure 26 shows GIP bioactivity for certain exemplary GIP-containing incretin agents tested.
[0090] Figure 27 shows in vitro activity (GLP1 expression) for certain exemplary incretin agents tested.
[0091] Figure 28 shows GLP1 bioactivity for certain exemplary GLP1 -containing incretin agents tested.
[0092] Figure 29 shows a comparison of GIP expression (A) and GIP bioactivity (B) in exemplary candidates with different signal peptides (husec vs. gDl).
[0093] Figure 30 shows a comparison of GLP1 expression (A) and GLP1 bioactivity (B) in exemplary candidates with different signal peptides (husec vs. gDl).
[0094] Figure 31 shows a comparison of GIP expression (A) and GIP bioactivity (B) with and without various half-life extension (HLE) moieties.
[0095] Figure 32 shows a comparison of GLP1 expression (A) and GLP1 bioactivity (32B) with and without various half-life extension (HLE) moieties.
[0096] Figure 33 shows a comparison of GIP expression (A) and GIP bioactivity (B) in exemplary incretin agents that contain both GIP and GLP1, where the order of the GIP and GLP1 peptides encoded by a single polyribonucleotide is varied.
[0097] Figure 34 shows a comparison of GLP1 expression (A) and GLP1 bioactivity (B) in exemplary incretin agents that contain both GIP and GLP1, where the order of the GIP and GLP1 peptides encoded by a single polyribonucleotide is varied.
DEFINITIONS
[0098] About: The term “about”, when used herein in reference to a value, refers to a value that is similar, in context to the referenced value. In general, those skilled in the art, familiar with the context, will appreciate the relevant degree of variance encompassed by “about” in that context. For example, in some embodiments, the term “about” may encompass a range ofvalues that within 25%, 20%, 19%, 18%, 17%, 16%, 15%, 14%, 13%, 12%, 11%, 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1%, or less of the referred value.
[0099] Agent: As used herein, the term “agent,” may refer to a physical entity. In some embodiments, an agent may be characterized by a particular feature and/or effect. For example, as used herein, the term “therapeutic agent” refers to a physical entity has a therapeutic effect and/or elicits a desired biological and/or pharmacological effect. In some embodiments, an agent may be a compound, molecule, or entity of any chemical class
including, for example, a small molecule, polypeptide, nucleic acid, saccharide, lipid, metal, or a combination or complex thereof.
[0100] Aliphatic: The term “aliphatic” refers to a straight-chain (i.e., unbranched) or branched, substituted or unsubstituted hydrocarbon chain that is completely saturated or that contains one or more units of unsaturation, or a monocyclic hydrocarbon or bicyclic hydrocarbon that is completely saturated or that contains one or more units of unsaturation, but which is not aromatic (also referred to herein as “cycloaliphatic”), that has a single point or more than one points of attachment to the rest of the molecule. Unless otherwise specified, aliphatic groups contain 1-12 aliphatic carbon atoms. In some embodiments, aliphatic groups contain 1-6 aliphatic carbon atoms (e.g., Ci-e). In some embodiments, aliphatic groups contain 1-5 aliphatic carbon atoms (e.g., C1-5). In other embodiments, aliphatic groups contain 1-4 aliphatic carbon atoms (e.g., C1-4). In still other embodiments, aliphatic groups contain 1-3 aliphatic carbon atoms (e.g., C1-3), and in yet other embodiments, aliphatic groups contain 1-2 aliphatic carbon atoms (e.g., C1-2). Suitable aliphatic groups include, but are not limited to, linear or branched, substituted or unsubstituted alkyl, alkenyl, or alkynyl groups and hybrids thereof. A preferred aliphatic group is C1-6 alkyl.
[0101] Alkyl: The term “alkyl,” used alone or as part of a larger moiety, refers to a saturated, optionally substituted straight or branched chain hydrocarbon group having (unless otherwise specified) 1-12, 1-10, 1-8, 1-6, 1-4, 1-3, or 1-2 carbon atoms (e.g., C1-12, C1-10, C1-8, C1-6, C1-4, C1-3, or C1-2). Exemplary alkyl groups include methyl, ethyl, propyl, butyl, pentyl, hexyl, and heptyl.
[0102] Alkylene: The term “alkylene” is refers to a bivalent alkyl group. In some embodiments, “alkylene” is a bivalent straight or branched alkyl group. In some embodiments, an “alkylene chain” is a polymethylene group, i.e., -(CH2)n-, wherein n is a positive integer, e.g., from 1 to 6, from 1 to 4, from 1 to 3, from 1 to 2, or from 2 to 3. An optionally substituted alkylene chain is a polymethylene group in which one or more methylene hydrogen atoms is optionally replaced with a substituent. Suitable substituents include those described below for a substituted aliphatic group and also include those described in the specification herein. It will be appreciated that two substituents of the alkylene group may be taken together to form a ring system. In certain embodiments, two substituents can be taken together to form a 3 - to 7-membered ring. The substituents can be
on the same or different atoms. The suffix “-ene” or “-enyl” when appended to certain groups herein are intended to refer to a bifunctional moiety of said group. For example, “-ene” or “- enyl”, when appended to “cyclopropyl” becomes “cyclopr is intended to refer to a bifunctional cyclopropyl group, e.
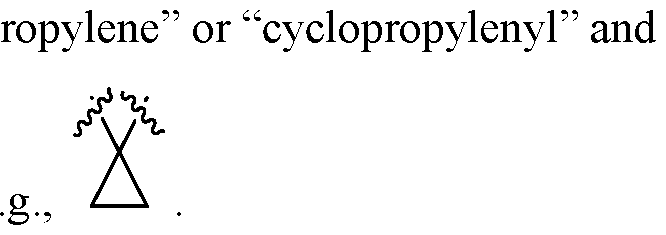
[0103] Alkenyl: The term “alkenyl”, used alone or as part of a larger moiety, refers to an optionally substituted straight or branched chain or cyclic hydrocarbon group having at least one double bond and having (unless otherwise specified) 2-12, 2-10, 2-8, 2-6, 2-4, or 2- 3 carbon atoms (e.g., C2-12, C2-10, C2-8, C2-6, C2-4, or C2-3). Exemplary alkenyl groups include ethenyl, propenyl, butenyl, pentenyl, hexenyl, and heptenyl. The term “cycloalkenyl” refers to an optionally substituted non-aromatic monocyclic or multicyclic ring system containing at least one carbon-carbon double bond and having about 3 to about 10 carbon atoms.
Exemplary monocyclic cycloalkenyl rings include cyclopentenyl, cyclohexenyl, and cycloheptenyl.
[0104] Alkynyl: The term “alkynyl”, used alone or as part of a larger moiety, refers to an optionally substituted straight or branched chain hydrocarbon group having at least one triple bond and having (unless otherwise specified) 2-12, 2-10, 2-8, 2-6, 2-4, or 2-3 carbon atoms (e.g., C2-12, C2-10, C2-8, C2-6, C2-4, or C2-3). Exemplary alkynyl groups include ethynyl, propynyl, butynyl, pentynyl, hexynyl, and heptynyl.
[0105] Amino acid: In its broadest sense, as used herein, the term “amino acid” refers to a compound and/or substance that can be, is, or has been incorporated into a polypeptide chain, e.g., through formation of one or more peptide bonds. In some embodiments, an amino acid has the general structure H2N-C(H)(R)-COOH. In some embodiments, an amino acid is a naturally-occurring amino acid. In some embodiments, an amino acid is a non-natural amino acid; in some embodiments, an amino acid is a D-amino acid; in some embodiments, an amino acid is an L-amino acid. “Standard amino acid” refers to any of the twenty standard L-amino acids commonly found in naturally occurring peptides. “Nonstandard amino acid” refers to any amino acid, other than the standard amino acids, regardless of whether it is prepared synthetically or obtained from a natural source. In some embodiments, an amino acid, including a carboxy- and/or amino-terminal amino acid in a polypeptide, can contain a structural modification as compared with the general structure above. For example, in some
embodiments, an amino acid may be modified by methylation, amidation, acetylation, pegylation, glycosylation, phosphorylation, and/or substitution (e.g., of the amino group, the carboxylic acid group, one or more protons, and/or the hydroxyl group) as compared with the general structure. In some embodiments, such modification may, for example, alter the circulating half-life of a polypeptide containing the modified amino acid as compared with one containing an otherwise identical unmodified amino acid. In some embodiments, such modification does not significantly alter a relevant activity of a polypeptide containing the modified amino acid, as compared with one containing an otherwise identical unmodified amino acid. As will be clear from context, in some embodiments, the term “amino acid” may be used to refer to a free amino acid; in some embodiments it may be used to refer to an amino acid residue of a polypeptide.
[0106] Aryl: The term “aryl” refers to monocyclic and bicyclic ring systems having a total of six to fourteen ring members (e.g., C6-C14), wherein at least one ring in the system is aromatic and wherein each ring in the system contains three to seven ring members. In some embodiments, an “aryl” group contains between six and twelve total ring members (e.g., C6- CI2). The term “aryl” may be used interchangeably with the term “aryl ring”. In certain embodiments, “aryl” refers to an aromatic ring system which includes, but not limited to, phenyl, biphenyl, naphthyl, anthracyl and the like, which may bear one or more substituents. Unless otherwise specified, “aryl” groups are hydrocarbons. In some embodiments, an “aryl” ring system is an aromatic ring (e.g., phenyl) that is fused to a non-aromatic ring (e.g., cycloalkyl). Examples of aryl rings include that are fused include

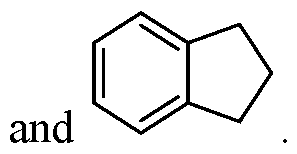
[0107] Associated: Two events or entities are “associated” with one another, as that term is used herein, if the presence, level, degree, type and/or form of one is correlated with that of the other. For example, a particular entity (e.g., polypeptide, genetic signature, metabolite, microbe, etc.) is considered to be associated with a particular disease, disorder, or condition, if its presence, level and/or form correlates with incidence of, susceptibility to, severity of, stage of, etc. the disease, disorder, or condition (e.g., across a relevant population). In some embodiments, two or more entities are physically “associated” with one
another if they interact, directly or indirectly, so that they are and/or remain in physical proximity with one another. In some embodiments, two or more entities that are physically associated with one another are covalently linked to one another; in some embodiments, two or more entities that are physically associated with one another are not covalently linked to one another but are non-covalently associated, for example by means of hydrogen bonds, van der Waals interaction, hydrophobic interactions, magnetism, and combinations thereof.
[0108] Co-administration: As used herein, the term “co-administration” refers to use of a composition (e.g., a pharmaceutical composition) described herein and one or more additional therapeutic agents. In some embodiments, one or more additional therapeutic agents comprises at least one polyribonucleotide encoding another therapeutic agent (e.g., an incretin agent). The combined use of a composition (e.g., a pharmaceutical composition) described herein and an additional therapeutic agent may be performed concurrently or separately (e.g., sequentially in any order). In some embodiments, a composition (e.g., a pharmaceutical composition) described herein and an additional therapeutic agent may be combined in one pharmaceutically-acceptable excipient, or they may be placed in separate excipient and delivered to a target cell or administered to a subject at different times. Each of these situations is contemplated as falling within the meaning of “co-administration” or “combination,” provided that a composition (e.g., a pharmaceutical composition) described herein and an additional therapeutic agent are delivered or administered sufficiently close in time that there is at least some temporal overlap in biological effect(s) generated by each on a target cell or a subject being treated.
[0109] Combination therapy: As used herein, the term “combination therapy” refers to those situations in which a subject is simultaneously exposed to two or more therapeutic regimens (e.g., two or more therapeutic agents (e.g., two or more incretin agents)). In some embodiments, the two or more regimens may be administered simultaneously; in some embodiments, such regimens may be administered sequentially (e.g., all “doses” of a first regimen are administered prior to administration of any doses of a second regimen); in some embodiments, such agents are administered in overlapping dosing regimens. In some embodiments, administration of combination therapy may involve administration of one or more agent(s) or modality(ies) to a subject receiving the other agent(s) or modality(ies) in the combination. For clarity, combination therapy does not require that individual agents be administered together in a single composition (or even necessarily at the same time), although
in some embodiments, two or more agents, or active moieties thereof, may be administered together in a combination composition. In some embodiments, a combination therapy comprises polyribonucleotides encoding two or more incretin agents.
[0110] Comparable: As used herein, the term “comparable” refers to two or more agents, entities, situations, sets of conditions, etc., that may not be identical to one another but that are sufficiently similar to permit comparison there between so that one skilled in the art will appreciate that conclusions may reasonably be drawn based on differences or similarities observed. In some embodiments, comparable sets of conditions, circumstances, individuals, or populations are characterized by a plurality of substantially identical features and one or a small number of varied features. Those of ordinary skill in the art will understand, in context, what degree of identity is required in any given circumstance for two or more such agents, entities, situations, sets of conditions, etc. to be considered comparable. For example, those of ordinary skill in the art will appreciate that sets of circumstances, individuals, or populations are comparable to one another when characterized by a sufficient number and type of substantially identical features to warrant a reasonable conclusion that differences in results obtained or phenomena observed under or with different sets of circumstances, individuals, or populations are caused by or indicative of the variation in those features that are varied.
[0111] Corresponding to: As used herein, the term “corresponding to” refers to a relationship between two or more entities. For example, the term “corresponding to” may be used to designate the position/identity of a structural element in a compound or composition relative to another compound or composition (e.g., to an appropriate reference compound or composition). For example, in some embodiments, a monomeric residue in a polymer (e.g., an amino acid residue in a polypeptide or a nucleic acid residue in a polynucleotide) may be identified as “corresponding to” a residue in an appropriate reference polymer. For example, those of ordinary skill will appreciate that, for purposes of simplicity, residues in a polypeptide are often designated using a canonical numbering system based on a reference related polypeptide, so that an amino acid “corresponding to” a residue at position 190, for example, need not actually be the 190th amino acid in a particular amino acid chain but rather corresponds to the residue found at 190 in the reference polypeptide; those of ordinary skill in the art readily appreciate how to identify “corresponding” amino acids. For example, those skilled in the art will be aware of various sequence alignment strategies, including software programs such as, for example, BLAST, CS-BLAST, CUSASW++, DIAMOND, FASTA,
GGSEARCH/GLSEARCH, Genoogle, HMMER, HHpred/HHsearch, IDF, Infernal, KLAST, USEARCH, parasail, PSI-BLAST, PSI-Search, ScalaBLAST, Sequilab, SAM, SSEARCH, SWAPHI, SWAPHI-LS, SWIMM, or SWIPE that can be utilized, for example, to identify “corresponding” residues in polypeptides and/or nucleic acids in accordance with the present disclosure. Those of skill in the art will also appreciate that, in some instances, the term “corresponding to” may be used to describe an event or entity that shares a relevant similarity with another event or entity (e.g., an appropriate reference event or entity). To give but one example, a gene or protein in one organism may be described as “corresponding to” a gene or protein from another organism in order to indicate, in some embodiments, that it plays an analogous role or performs an analogous function and/or that it shows a particular degree of sequence identity or homology, or shares a particular characteristic sequence element.
[0112] Cycloaliphatic: As used herein, the term “cycloaliphatic” refers to a monocyclic C3-8 hydrocarbon or a bicyclic Ce-io hydrocarbon that is completely saturated or that contains one or more units of unsaturation, but which is not aromatic, that has a single point or more than one points of attachment to the rest of the molecule.
[0113] Cycloalkyl: As used herein, the term “cycloalkyl” refers to an optionally substituted saturated ring monocyclic or polycyclic system of about 3 to about 10 ring carbon atoms. Exemplary monocyclic cycloalkyl rings include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, and cycloheptyl.
[0114] Derived: In the context of an amino acid sequence (peptide or polypeptide) “derived from” a designated amino acid sequence (peptide or polypeptide), refers to a structural analogue of a designated amino acid sequence. In some embodiments, an amino acid sequence which is derived from a particular amino acid sequence has an amino acid sequence that is identical, essentially identical or homologous to that particular sequence or a fragment thereof. Amino acid sequences derived from a particular amino acid sequence may be variants of that particular sequence or a fragment thereof. For example, incretin agents utilized according to the present disclosure may include amino acid sequences derived from two or more incretin agents, e.g., two or more naturally produced incretins.
[0115] Detecting: The term “detecting” is used broadly herein to include appropriate means of determining the presence or absence of an entity of interest or any form of measurement of an entity of interest in a sample. Thus, “detecting” may include determining,
measuring, assessing, or assaying the presence or absence, level, amount, and/or location of an entity of interest. Quantitative and qualitative determinations, measurements or assessments are included, including semi-quantitative. Such determinations, measurements or assessments may be relative, for example when an entity of interest is being detected relative to a control reference, or absolute. As such, the term “quantifying” when used in the context of quantifying an entity of interest can refer to absolute or to relative quantification. Absolute quantification may be accomplished by correlating a detected level of an entity of interest to known control standards (e.g., through generation of a standard curve). Alternatively, relative quantification can be accomplished by comparison of detected levels or amounts between two or more different entities of interest to provide a relative quantification of each of the two or more different entities of interest, i.e., relative to each other.
[0116] Dosing regimen: Those skilled in the art will appreciate that the term “dosing regimen” (or “therapeutic regimen”) may be used to refer to a set of unit doses (typically more than one) that are administered individually to a subject, typically separated by periods of time. In some embodiments, a given therapeutic agent has a recommended dosing regimen, which may involve one or more doses.
[0117] Encode: As used herein, the term “encode” or “encoding” refers to sequence information of a first molecule that guides production of a second molecule having a defined sequence of nucleotides (e.g., a polyribonucleotide) or a defined sequence of amino acids. For example, a DNA molecule can encode an RNA molecule (e.g., by a transcription process that includes a DNA-dependent RNA polymerase enzyme). An RNA molecule can encode a polypeptide (e.g., by a translation process). Thus, a gene, a cDNA, or an RNA molecule encodes a polypeptide if transcription and translation of RNA corresponding to that gene produces the polypeptide in a cell or other biological system. In some embodiments, a coding region of a polyribonucleotide encoding a target antigen refers to a coding strand, the nucleotide sequence of which is identical to the polyribonucleotide sequence of such a target antigen. In some embodiments, a coding region of a polyribonucleotide encoding a target antigen refers to a non-coding strand of such a target antigen, which may be used as a template for transcription of a gene or cDNA.
[0118] Engineered: In general, the term “engineered” refers to the aspect of having been manipulated by the hand of man. For example, a polynucleotide is considered to be
“engineered” when two or more sequences that are not linked together in that order in nature are manipulated by the hand of man to be directly linked to one another in the engineered polynucleotide and/or when a particular residue in a polynucleotide is non-naturally occurring and/or is caused through action of the hand of man to be linked with an entity or moiety with which it is not linked in nature.
[0119] Expression: As used herein, the term “expression” of a nucleic acid sequence refers to the generation of a gene product from the nucleic acid sequence. In some embodiments, a gene product can be a transcript, e.g., a polyribonucleotide as provided herein. In some embodiments, a gene product can be a polypeptide. In some embodiments, expression of a nucleic acid sequence involves one or more of the following: (1) production of an RNA template from a DNA sequence (e.g., by transcription); (2) processing of an RNA transcript (e.g., by splicing, editing, etc.); (3) translation of an RNA into a polypeptide or protein; and/or (4) post-translational modification of a polypeptide or protein.
[0120] Heteroaliphatic: The term “heteroaliphatic” or “heteroaliphatic group,” as used herein, denotes an optionally substituted hydrocarbon moiety having, in addition to carbon atoms, from one to five heteroatoms, that may be straight-chain (i.e., unbranched), branched, or cyclic (“heterocyclic”) and may be completely saturated or may contain one or more units of unsaturation, but which is not aromatic. The term “heteroatom” refers to nitrogen, oxygen, or sulfur, and includes any oxidized form of nitrogen or sulfur, and any quatemized form of a basic nitrogen. The term “nitrogen” also includes a substituted nitrogen. Unless otherwise specified, heteroaliphatic groups contain 1-10 carbon atoms wherein 1-3 carbon atoms are optionally and independently replaced with heteroatoms selected from oxygen, nitrogen, and sulfur. In some embodiments, heteroaliphatic groups contain 1-4 carbon atoms, wherein 1-2 carbon atoms are optionally and independently replaced with heteroatoms selected from oxygen, nitrogen, and sulfur. In yet other embodiments, heteroaliphatic groups contain 1-3 carbon atoms, wherein 1 carbon atom is optionally and independently replaced with a heteroatom selected from oxygen, nitrogen, and sulfur. Suitable heteroaliphatic groups include, but are not limited to, linear or branched, heteroalkyl, heteroalkenyl, and heteroalkynyl groups. For example, a 1- to 10 atom heteroaliphatic group includes the following exemplary groups: -O-CH3, -CH2-O-CH3, -O- CH2-CH2-O-CH2-CH2-O-CH3, and the like.
[0121] Heteroaryl: The terms “heteroaryl” and “heteroar-”, used alone or as part of a larger moiety, e.g., “heteroaralkyl”, or “heteroaralkoxy”, refer to monocyclic or bicyclic ring groups having 5 to 10 ring atoms (e.g., 5- to 6-membered monocyclic heteroaryl or 9- to 10- membered bicyclic heteroaryl); having 6, 10, or 14 n-electrons shared in a cyclic array; and having, in addition to carbon atoms, from one to five heteroatoms. Heteroaryl groups include, without limitation, thienyl, furanyl, pyrrolyl, imidazolyl, pyrazolyl, triazolyl, tetrazolyl, oxazolyl, isoxazolyl, oxadiazolyl, thiazolyl, isothiazolyl, thiadiazolyl, pyridyl, pyridazinyl, pyrimidinyl, pyrazinyl, indolizinyl, purinyl, naphthyridinyl, pteridinyl, imidazo[l,2- a]pyrimidinyl, imidazo[l,2-a]pyridyl, imidazo[4,5-b]pyridyl, imidazo[4,5-c]pyridyl, pyrrolopyridyl, pyrrolopyrazinyl, thienopyrimidinyl, triazolopyridyl, and benzoisoxazolyl. The terms “heteroaryl” and “heteroar-”, as used herein, also include groups in which a heteroaromatic ring is fused to one or more aryl, cycloaliphatic, or heterocyclyl rings, where the radical or point of attachment is on the heteroaromatic ring (i.e., a bicyclic heteroaryl ring having 1 to 3 heteroatoms). Nonlimiting examples include indolyl, isoindolyl, benzothienyl, benzofuranyl, dibenzofuranyl, indazolyl, benzimidazolyl, benzotriazolyl, benzothiazolyl, benzothiadiazolyl, benzoxazolyl, quinolyl, isoquinolyl, cinnolinyl, phthalazinyl, quinazolinyl, quinoxalinyl, 4H- quinolizinyl. carbazolyl, acridinyl, phenazinyl, phenothiazinyl, phenoxazinyl, tetrahydroquinolinyl, tetrahydroisoquinolinyl, pyrido[2,3-b]-l,4-oxazin- 3(4H)-one, 4H-thieno[3,2-b]pyrrole, and benzoisoxazolyl. The term “heteroaryl” may be used interchangeably with the terms “heteroaryl ring”, “heteroaryl group”, or “heteroaromatic”, any of which terms include rings that are optionally substituted.
[0122] Heteroatom: The term “heteroatom” as used herein refers to nitrogen, oxygen, or sulfur, and includes any oxidized form of nitrogen or sulfur, and any quatemized form of a basic nitrogen.
[0123] Heterocycle: As used herein, the terms “heterocycle”, “heterocyclyl”, “heterocyclic radical”, and “heterocyclic ring” are used interchangeably and refer to a stable 3- to 8-membered monocyclic, a 6- to 10-membered bicyclic, or a 10- to 16-membered polycyclic heterocyclic moiety that is either saturated or partially unsaturated, and having, in addition to carbon atoms, one or more, such as one to four, heteroatoms, as defined above. When used in reference to a ring atom of a heterocycle, the term “nitrogen” includes a substituted nitrogen. As an example, in a saturated or partially unsaturated ring having 0-3 heteroatoms selected from oxygen, sulfur or nitrogen, the nitrogen may be N (as in 3,4-
dihydro-2H-pyrrolyl), NH (as in pyrrolidinyl), or NR+ (as in N-substituted pyrrolidinyl). A heterocyclic ring can be attached to its pendant group at any heteroatom or carbon atom that results in a stable structure and any of the ring atoms can be optionally substituted. Examples of such saturated or partially unsaturated heterocyclic radicals include, without limitation, azetidinyl, oxetanyl, tetrahydrofuranyl, tetrahydrothienyl, pyrrolidinyl, piperidinyl, decahydroquinolinyl, oxazolidinyl, piperazinyl, dioxanyl, dioxolanyl, diazepinyl, oxazepinyl, thiazepinyl, morpholinyl, and thiamorpholinyl. A heterocyclyl group may be mono-, bi-, tri-, or polycyclic, preferably mono-, bi-, or tricyclic, more preferably mono- or bicyclic. A bicyclic heterocyclic ring also includes groups in which the heterocyclic ring is fused to one or more aryl rings. Exemplary bicyclic heterocyclic groups include indolinyl, isoindolinyl, benzodioxolyl, 1,3-dihydroisobenzofuranyl, 2,3-dihydrobenzofuranyl, and tetrahydroquinolinyl. A bicyclic heterocyclic ring can also be a spirocyclic ring system (e.g., 7- to 11 -membered spirocyclic fused heterocyclic ring having, in addition to carbon atoms, one or more heteroatoms as defined above (e.g., one, two, three or four heteroatoms)). A bicyclic heterocyclic ring can also be a bridged ring system (e.g., 7- to 11-membered bridged heterocyclic ring having one, two, or three bridging atoms.
[0124] Homology: As used herein, the term “homology” or “homolog” refers to the overall relatedness between polynucleotide molecules (e.g., DNA molecules and/or RNA molecules) and/or between polypeptide molecules. In some embodiments, polynucleotide molecules (e.g., DNA molecules and/or RNA molecules) and/or polypeptide molecules are considered to be “homologous” to one another if their sequences are at least 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, or 99% identical. In some embodiments, polynucleotide molecules (e.g., DNA molecules and/or RNA molecules) and/or polypeptide molecules are considered to be “homologous” to one another if their sequences are at least 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, or 99% similar (e.g., containing residues with related chemical properties at corresponding positions). For example, as is well known by those of ordinary skill in the art, certain amino acids are typically classified as similar to one another as “hydrophobic” or “hydrophilic” amino acids, and/or as having “polar” or “non-polar” side chains. Substitution of one amino acid for another of the same type may often be considered a “homologous” substitution.
[0125] Identity: As used herein, the term “identity” refers to the overall relatedness between polynucleotide molecules (e.g., DNA molecules and/or RNA molecules) and/or between polypeptide molecules. In some embodiments, polynucleotide molecules (e.g., DNA molecules and/or RNA molecules) and/or between polypeptide molecules are considered to be “substantially identical” to one another if their sequences are at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, or 99% identical. Calculation of the percent identity of two nucleic acid or polypeptide sequences, for example, can be performed by aligning the two sequences for optimal comparison purposes (e.g., gaps can be introduced in one or both of a first and a second sequence for optimal alignment and non-identical sequences can be disregarded for comparison purposes). In certain embodiments, the length of a sequence aligned for comparison purposes is at least 30%, at least 40%, at least 50%, at least 60%, at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, at least 95%, at least 96%, at least 97%, at least 98%, at least 99%, or substantially 100% of the length of a reference sequence. The nucleotides at corresponding positions are then compared. When a position in the first sequence is occupied by the same residue (e.g., nucleotide or amino acid) as the corresponding position in the second sequence, then the molecules are identical at that position. The percent identity between the two sequences is a function of the number of identical positions shared by the sequences, taking into account the number of gaps, and the length of each gap, which needs to be introduced for optimal alignment of the two sequences. The comparison of sequences and determination of percent identity between two sequences can be accomplished using a mathematical algorithm. For example, the percent identity between two nucleotide sequences can be determined using the algorithm of Meyers and Miller, 1989, which has been incorporated into the ALIGN program (version 2.0). In some exemplary embodiments, nucleic acid sequence comparisons made with the ALIGN program use a PAM120 weight residue table, a gap length penalty of 12 and a gap penalty of 4. The percent identity between two nucleotide sequences can, alternatively, be determined using the GAP program in the GCG software package using an NWSgapdna.CMP matrix.
[0126] Increased, Induced, or Reduced: As used herein, these terms or grammatically comparable comparative terms, indicate values that are relative to a comparable reference measurement. For example, in some embodiments, an assessed value achieved with a provided composition (e.g., a pharmaceutical composition) may be “increased” relative to that obtained with a comparable reference composition. Alternatively
or additionally, in some embodiments, an assessed value achieved in a subject may be “increased” relative to that obtained in the same subject under different conditions (e.g., prior to or after an event; or presence or absence of an event such as administration of a composition (e.g., a pharmaceutical composition) as described herein, or in a different, comparable subject (e.g., in a comparable subject that differs from the subject of interest in prior exposure to a condition, e.g., absence of administration of a composition (e.g., a pharmaceutical composition) as described herein.). In some embodiments, comparative terms refer to statistically relevant differences (e.g., that are of a prevalence and/or magnitude sufficient to achieve statistical relevance). Those skilled in the art will be aware, or will readily be able to determine, in a given context, a degree and/or prevalence of difference that is required or sufficient to achieve such statistical significance. In some embodiments, the term “reduced” or equivalent terms refers to a reduction in the level of an assessed value by at least 5%, at least 10%, at least 20%, at least 50%, at least 75% or higher, as compared to a comparable reference. In some embodiments, the term “reduced” or equivalent terms refers to a complete or essentially complete inhibition, i.e., a reduction to zero or essentially to zero. In some embodiments, the term “increased” or “induced” refers to an increase in the level of an assessed value by at least 10%, at least 20%, at least 30%, at least 40%, at least 50%, at least 80%, at least 100%, at least 200%, at least 500%, or higher, as compared to a comparable reference.
[0127] In order: As used herein with reference to a polynucleotide or polyribonucleotide, “in order” refers to the order of features from 5' to 3' along the polynucleotide or polyribonucleotide. As used herein with reference to a polypeptide, “in order” refers to the order of features moving from the N-terminal-most of the features to the C-terminal-most of the features along the polypeptide. “In order” does not mean that no additional features can be present among the listed features. For example, if Features A, B, and C of a polynucleotide are described herein as being “in order, Feature A, Feature B, and Feature C,” this description does not exclude, e.g., Feature D being located between Features A and B.
[0128] Ionizable: The term “ionizable” refers to a compound or group or atom that is charged at a certain pH. In the context of an ionizable amino lipid, such a lipid or a function group or atom thereof bears a positive charge at a certain pH. In some embodiments, an ionizable amino lipid is positively charged at an acidic pH. In some embodiments, an
ionizable amino lipid is predominately neutral at physiological pH values, e.g., in some embodiments about 7.0-7.4, but becomes positively charged at lower pH values. In some embodiments, an ionizable amino lipid may have a pKa within a range of about 5 to about 7.
[0129] Isolated: The term “isolated” means altered or removed from the natural state. For example, a nucleic acid or a peptide naturally present in a living animal is not “isolated,” but the same nucleic acid or peptide partially or completely separated from the coexisting materials of its natural state is “isolated.” An isolated nucleic acid or protein can exist in substantially purified form, or can exist in a non-native environment such as, for example, a host cell.
[0130] Lipid: As used herein, the terms “lipid” and “lipid-like material” are broadly defined as molecules which comprise one or more hydrophobic moieties or groups and optionally also one or more hydrophilic moieties or groups. Molecules comprising hydrophobic moieties and hydrophilic moieties are also typically denoted as amphiphiles.
[0131] RNA lipid nanoparticle: As used herein, the term “RNA lipid nanoparticle” refers to a nanoparticle comprising at least one lipid and RNA molecule(s), e.g., one or more polyribonucleotides as provided herein. In some embodiments, an RNA lipid nanoparticle comprises at least one cationic amino lipid. In some embodiments, an RNA lipid nanoparticle comprises at least one cationic amino lipid, at least one helper lipid, and at least one polymer- conjugated lipid (e.g., PEG-conjugated lipid). In various embodiments, RNA lipid nanoparticles as described herein can have an average size (e.g., Z-average) of about 100 nm to 1000 nm, or about 200 nm to 900 nm, or about 200 nm to 800 nm, or about 250 nm to about 700 nm. In some embodiments of the present disclosure, RNA lipid nanoparticles can have a particle size (e.g., Z-average) of about 30 nm to about 200 nm, or about 30 nm to about 150 nm, about 40 nm to about 150 nm, about 50 nm to about 150 nm, about 60 nm to about 130 nm, about 70 nm to about 110 nm, about 70 nm to about 100 nm, about 80 nm to about 100 nm, about 90 nm to about 100 nm, about 70 to about 90 nm, about 80 nm to about
90 nm, or about 70 nm to about 80 nm. In some embodiments, an average size of lipid nanoparticles is determined by measuring the average particle diameter. In some embodiments, RNA lipid nanoparticles may be prepared by mixing lipids with RNA molecules described herein.
[0132] Neutralization : As used herein, the term “neutralization” refers to an event in which binding agents such as antibodies bind to a biological active site of a virus such as a receptor binding protein, thereby inhibiting the parasitic infection of cells. In some embodiments, the term “neutralization” refers to an event in which binding agents eliminate or significantly reduce ability of infecting cells.
[0133] Nucleic acid/ Polynucleotide'. As used herein, the term “nucleic acid” refers to a polymer of at least 10 nucleotides or more. In some embodiments, a nucleic acid is or comprises DNA. In some embodiments, a nucleic acid is or comprises RNA. In some embodiments, a nucleic acid is or comprises peptide nucleic acid (PNA). In some embodiments, a nucleic acid is or comprises a single stranded nucleic acid. In some embodiments, a nucleic acid is or comprises a double-stranded nucleic acid. In some embodiments, a nucleic acid comprises both single and double-stranded portions. In some embodiments, a nucleic acid comprises a backbone that comprises one or more phosphodiester linkages. In some embodiments, a nucleic acid comprises a backbone that comprises both phosphodiester and non-phosphodiester linkages. For example, in some embodiments, a nucleic acid may comprise a backbone that comprises one or more phosphorothioate or 5'-N-phosphoramidite linkages and/or one or more peptide bonds, e.g., as in a “peptide nucleic acid”. In some embodiments, a nucleic acid comprises one or more, or all, natural residues (e.g., adenine, cytosine, deoxyadenosine, deoxycytidine, deoxyguanosine, deoxythymidine, guanine, thymine, uracil). In some embodiments, a nucleic acid comprises on or more, or all, non-natural residues. In some embodiments, a non-natural residue comprises a nucleoside analog (e.g., 2-aminoadenosine, 2-thiothymidine, inosine, pyrrolo-pyrimidine, 3 -methyl adenosine, 5 -methylcytidine, C-5 propynyl-cytidine, C-5 propynyl-uridine, 2-aminoadenosine, C5 -bromouridine, C5-fluorouridine, C5 -iodouridine, C5-propynyl -uridine, C5 -propynyl-cytidine, C5 -methylcytidine, 2-aminoadenosine, 7- deazaadenosine, 7-deazaguanosine, 8-oxoadenosine, 8-oxoguanosine, 6-O-methylguanine, 2- thiocytidine, methylated bases, intercalated bases, and combinations thereof). In some embodiments, a non-natural residue comprises one or more modified sugars (e.g., 2'- fluororibose, ribose, 2'-deoxyribose, arabinose, and hexose) as compared to those in natural residues. In some embodiments, a nucleic acid has a nucleotide sequence that encodes a functional gene product such as an RNA or polypeptide. In some embodiments, a nucleic acid has a nucleotide sequence that comprises one or more introns. In some embodiments, a
nucleic acid may be prepared by isolation from a natural source, enzymatic synthesis (e.g., by polymerization based on a complementary template, e.g., in vivo or in vitro), reproduction in a recombinant cell or system, or chemical synthesis. In some embodiments, a nucleic acid is at least 3, 4, 5, 6, 7, 8, 9, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 1 10, 120, 130, 140, 150, 160, 170, 180, 190, 20, 225, 250, 275, 300, 325, 350, 375, 400, 425, 450, 475, 500, 600, 700, 800, 900, 1000, 1500, 2000, 2500, 3000, 3500, 4000, 4500, 5000, 5500, 6000, 6500, 7000, 7500, 8000, 8500, 9000, 9500, 10,000, 10,500, 11,000, 11,500, 12,000, 12,500, 13,000, 13,500, 14,000, 14,500, 15,000, 15,500, 16,000, 16,500, 17,000, 17,500, 18,000, 18,500, 19,000, 19,500, or 20,000 or more residues or nucleotides long.
[0134] Pharmaceutically effective amount: The term “pharmaceutically effective amount” or “therapeutically effective amount” refers to the amount which achieves a desired reaction or a desired effect alone or together with further doses. In the case of the treatment of a particular disease (e.g., obesity), a desired reaction in some embodiments relates to inhibition of the course of the disease (e.g., obesity). In some embodiments, such inhibition may comprise slowing down the progress of a disease (e.g., obesity) and/or interrupting or reversing the progress of the disease (e.g., obesity). In some embodiments, a desired reaction in a treatment of a disease (e.g., obesity) may be or comprise delay or prevention of the onset of a disease (e.g., obesity) or a condition (e.g., a condition associated with obesity). An effective amount of a composition (e.g., a pharmaceutical composition) described herein will depend, for example, on disease (e.g., obesity) or a condition (e.g., a condition associated with obesity) to be treated, the severity of such a disease (e.g., obesity) or a condition (e.g., a condition associated with obesity), individual parameters of the patient, including, e.g., age, physiological condition, size and weight, the duration of treatment, the type of an accompanying therapy (if present), the specific route of administration and similar factors. Accordingly, doses of a composition (e.g., a pharmaceutical composition) described herein may depend on various of such parameters. In the case that a reaction in a patient is insufficient with an initial dose, higher doses (or effectively higher doses achieved by a different, more localized route of administration) may be used.
[0135] Polypeptide: As used herein, the term “polypeptide” refers to a polymeric chain of amino acids. In some embodiments, a polypeptide has an amino acid sequence that occurs in nature. In some embodiments, a polypeptide has an amino acid sequence that does
not occur in nature. In some embodiments, a polypeptide has an amino acid sequence that is engineered in that it is designed and/or produced through action of the hand of man. In some embodiments, a polypeptide may comprise or consist of natural amino acids, non-natural amino acids, or both. In some embodiments, a polypeptide may comprise or consist of only natural amino acids or only non-natural amino acids. In some embodiments, a polypeptide may comprise D-amino acids, L-amino acids, or both. In some embodiments, a polypeptide may comprise only D-amino acids. In some embodiments, a polypeptide may comprise only L-amino acids. In some embodiments, a polypeptide may include one or more pendant groups or other modifications, e.g., modifying or attached to one or more amino acid side chains, at the polypeptide’s N-terminus, at the polypeptide’s C-terminus, or any combination thereof. In some embodiments, such pendant groups or modifications comprise acetylation, amidation, lipidation, methylation, pegylation, etc., including combinations thereof. In some embodiments, a polypeptide may be cyclic, and/or may comprise a cyclic portion. In some embodiments, a polypeptide is not cyclic and/or does not comprise any cyclic portion. In some embodiments, a polypeptide is linear. In some embodiments, a polypeptide may be or comprise a stapled polypeptide. In some embodiments, the term “polypeptide” may be appended to a name of a reference polypeptide, activity, or structure; in such instances it is used herein to refer to polypeptides that share the relevant activity or structure and thus can be considered to be members of the same class or family of polypeptides. For each such class, the present specification provides and/or those skilled in the art will be aware of exemplary polypeptides within the class whose amino acid sequences and/or functions are known; in some embodiments, such exemplary polypeptides are reference polypeptides for the polypeptide class or family. In some embodiments, a member of a polypeptide class or family shows significant sequence homology or identity with, shares a common sequence motif (e.g., a characteristic sequence element) with, and/or shares a common activity (in some embodiments at a comparable level or within a designated range) with a reference polypeptide of the class; in some embodiments with all polypeptides within the class). For example, in some embodiments, a member polypeptide shows an overall degree of sequence homology or identity with a reference polypeptide that is at least about 30-40%, and is often greater than about 50%, 60%, 70%, 80%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% or more and/or includes at least one region (e.g., a conserved region that may in some embodiments be or comprise a characteristic sequence element) that shows very high sequence identity, often greater than 90% or even 95%, 96%, 97%, 98%, or 99%. Such a
conserved region usually encompasses at least 3-4 and often up to 35 or more amino acids; in some embodiments, a conserved region encompasses at least one stretch of at least 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35 or more contiguous amino acids. In some embodiments, a relevant polypeptide may comprise or consist of a fragment of a parent polypeptide.
[0136] Prevent: As used herein, the term “prevent” or “prevention” when used in connection with the occurrence of a disease, disorder, and/or condition, refers to reducing the risk of developing the disease, disorder and/or condition and/or to delaying onset of one or more characteristics or symptoms of the disease, disorder or condition. Prevention may be considered complete when onset of a disease, disorder or condition has been delayed for a predefined period of time.
[0137] Reference: As used herein, the term “reference” describes a standard or control relative to which a comparison is performed. For example, in some embodiments, an agent, animal, individual, population, sample, sequence or value of interest is compared with a reference or control agent, animal, individual, population, sample, sequence or value. In some embodiments, a reference or control is tested and/or determined substantially simultaneously with the testing or determination of interest. In some embodiments, a reference or control is a historical reference or control, optionally embodied in a tangible medium. Typically, as would be understood by those skilled in the art, a reference or control is determined or characterized under comparable conditions or circumstances to those under assessment. Those skilled in the art will appreciate when sufficient similarities are present to justify reliance on and/or comparison to a particular possible reference or control.
[0138] Ribonucleic acid (RNA) or Polyribonucleotide: As used herein, the term “ribonucleic acid,” “RNA,” or “polyribonucleotide” refers to a polymer of ribonucleotides. In some embodiments, an RNA is single stranded. In some embodiments, an RNA is double stranded. In some embodiments, an RNA comprises both single and double stranded portions. In some embodiments, an RNA can comprise a backbone structure as described in the definition of “Nucleic acid/ Polynucleotide” above. An RNA can be a regulatory RNA (e.g., siRNA, microRNA, etc.), or a messenger RNA (mRNA). In some embodiments, an RNA is a mRNA. In some embodiments, where an RNA is a mRNA, an RNA typically comprises at its 3' end a poly(A) region. In some embodiments, where an RNA is a mRNA, an RNA typically
comprises at its 5' end an art-recognized cap structure, e.g., for recognizing and attachment of a mRNA to a ribosome to initiate translation. In some embodiments, a RNA is a synthetic RNA. Synthetic RNAs include RNAs that are synthesized in vitro (e.g., by enzymatic synthesis methods and/or by chemical synthesis methods).
[0139] Ribonucleotide: As used herein, the term “ribonucleotide” encompasses unmodified ribonucleotides and modified ribonucleotides. For example, unmodified ribonucleotides include the purine bases adenine (A) and guanine (G), and the pyrimidine bases cytosine (C) and uracil (U). Modified ribonucleotides may include one or more modifications including, but not limited to, for example, (a) end modifications, e.g., 5' end modifications (e.g., phosphorylation, dephosphorylation, conjugation, inverted linkages, etc.), 3' end modifications (e.g., conjugation, inverted linkages, etc.), (b) base modifications, e.g. , replacement with modified bases, stabilizing bases, destabilizing bases, or bases that base pair with an expanded repertoire of partners, or conjugated bases, (c) sugar modifications (e.g., at the 2' position or 4' position) or replacement of the sugar, and (d) intemucleoside linkage modifications, including modification or replacement of the phosphodiester linkages. The term “ribonucleotide” also encompasses ribonucleotide triphosphates including modified and non-modified ribonucleotide triphosphates.
[0140] Risk: As will be understood from context, “risk” of a disease, disorder, and/or condition refers to a likelihood that a particular individual will develop the disease, disorder, and/or condition. In some embodiments, risk is expressed as a percentage. In some embodiments, risk is expressed as a risk relative to a risk associated with a reference sample or group of reference samples. In some embodiments, a reference sample or group of reference samples have a known risk of a disease, disorder, condition and/or event. In some embodiments, a reference sample or group of reference samples are from individuals comparable to a particular individual. In some embodiments, risk may reflect one or more genetic attributes, e.g., which may predispose an individual toward development (or not) of a particular disease, disorder and/or condition. In some embodiments, risk may reflect one or more epigenetic events or attributes and/or one or more lifestyle or environmental events or attributes.
[0141] Specific: The term “specific,” when used herein in reference to an agent having an activity, is understood by those skilled in the art to mean that the agent
discriminates between potential target entities, states, or cells. For example, in some embodiments, an agent is said to bind “specifically” to its target if it binds preferentially with that target in the presence of one or more competing alternative targets. In many embodiments, specific interaction is dependent upon the presence of a particular structural feature of the target entity (e.g., an epitope, a cleft, a binding site). It is to be understood that specificity need not be absolute. In some embodiments, specificity may be evaluated relative to that of a target-binding moiety for one or more other potential target entities (e.g., competitors). In some embodiments, specificity is evaluated relative to that of a reference specific binding moiety. In some embodiments, specificity is evaluated relative to that of a reference non-specific binding moiety.
[0142] Substituted or optionally substituted: As described herein, compounds of the invention may contain “optionally substituted” moieties. In general, the term “substituted,” whether preceded by the term “optionally” or not, means that one or more hydrogens of the designated moiety are replaced with a suitable substituent. “Substituted” applies to one or more hydrogens that are either explicit or implicit from the structure (e.g.,
 refers to at least
refers to at least
 ). Unless otherwise indicated, an “optionally substituted” group may have a suitable substituent at each substitutable position of the group, and when more than one position in any given structure may be substituted with more than one substituent selected from a specified group, the substituent may be either the same or different at every position. Combinations of substituents envisioned by this invention are preferably those that result in the formation of stable or chemically feasible compounds. The term “stable,” as used herein, refers to compounds that are not substantially altered when subjected to conditions to allow for their production, detection, and, in certain embodiments, their recovery, purification, and use for one or more of the purposes provided herein. Groups described as being “substituted” preferably have between 1 and 4 substituents, more preferably 1 or 2 substituents. Groups
described as being “optionally substituted” may be unsubstituted or be “substituted” as described above.
). Unless otherwise indicated, an “optionally substituted” group may have a suitable substituent at each substitutable position of the group, and when more than one position in any given structure may be substituted with more than one substituent selected from a specified group, the substituent may be either the same or different at every position. Combinations of substituents envisioned by this invention are preferably those that result in the formation of stable or chemically feasible compounds. The term “stable,” as used herein, refers to compounds that are not substantially altered when subjected to conditions to allow for their production, detection, and, in certain embodiments, their recovery, purification, and use for one or more of the purposes provided herein. Groups described as being “substituted” preferably have between 1 and 4 substituents, more preferably 1 or 2 substituents. Groups
described as being “optionally substituted” may be unsubstituted or be “substituted” as described above.
[0143] Suitable monovalent substituents on a substitutable carbon atom of an “optionally substituted” group are independently halogen; -(CH2)o 4R0; -(CH2)o 4OR0; - 0(CH2)O-4R°, -0-(CH2)O 4C(0)0RO; -(CH2)O 4CH(0RO)2; -(CH2)O 4SRO; -(CH2)O 4Ph, which may be substituted with R°; -(CH2)o 40(CH2)o iPh which may be substituted with R°; - CH=CHPh, which may be substituted with R°; -(CH2)o-40(CH2)o-i-pyridyl which may be substituted with R°; -NO2; -CN; -N3; -(CH2)o-4N(R°)2; -(CH2)o 4N(Ro)C(0)R°; - N(R°)C(S)R°; -(CH2)O 4N(R°)C(0)NRO 2; -N(RO)C(S)NR°2; -(CH2)O 4N(R°)C(O)OR°; - N(R°)N(R°)C(O)R°; -N(R°)N(R°)C(O)NR°2; -N(R°)N(R°)C(O)OR°; -(CH2)o 4C(O)R°; C(S)R°; -(CH2)O 4C(O)OR°; -(CH2)O 4C(O)SR°; -(CH2)O 4C(O)OSiR°3; -(CH2)o 4OC(O)R°; -OC(0)(CH2)o 4SR°; -(CH2)O 4SC(0)RO; -(CH2)O 4C(0)NRO2; -C(S)NRO 2; -C(S)SR°; - SC(S)SR°, -(CH2)O 40C(0)NRO 2; -C(O)N(OR°)R°; -C(O)C(O)R°; -C(O)CH2C(O)R°; - C(NOR°)R°; -(CH2)o 4SSR0; -(CH2)o 4S(O)2R°; -(CH2)o 4S(O)2OR°; -(CH2)o 4OS(O)2R°; - S(O)2NR°2; -(CH2)O 4S(0)RO; -N(RO)S(0)2NR°2; -N(R°)S(O)2R°; -N(OR°)R°; - C(NH)NR°2; -P(O)2R°; -P(O)R°2; -OP(O)R°2; -OP(O)(OR°)2; SiR°3; -(Ci-4 straight or branched alkylene)O-N(R°)2; or-(Ci-4 straight or branched alkylene)C(O)O-N(R°)2, wherein each R° may be substituted as defined below and is independently hydrogen, Ci- 6 aliphatic, -CH2PI1, -0(CH2)o iPh, -CH2-(5- to 6-membered heteroaryl ring), or a 3- to 6- membered saturated, partially unsaturated, or aryl ring having 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur, or, notwithstanding the definition above, two independent occurrences of R°, taken together with their intervening atom(s), form a 3- to 12- membered saturated, partially unsaturated, or aryl mono- or bicyclic ring having 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur, which may be substituted as defined below.
[0144] Suitable monovalent substituents on R° (or the ring formed by taking two independent occurrences of R° together with their intervening atoms), are independently halogen, -(CH2K2R1, -(haloR1), -(CH2)o 2OH, -(CH2)o 2OR1, -(CH2)o 2CH(OR')2, -OChaloR1), -CN, -N3, -(CH2)o-2C(0)R1, -(CH2)o 2C(O)OH, -(CH2)O 2C(O)OR‘, -(CH2)O 2SR1, -(CH2)o 2SH, -(CH2)o 2NH2, -(CH2)o 2NHR1, -(CH2)O-2NR12, -NO2, -SiR*3, - OSiR'y -C(O)SR', -(C1-4 straight or branched alkylene)C(O)OR', or -SSR1 wherein each R1 is unsubstituted or where preceded by “halo” is substituted only with one or more halogens, and
is independently selected from Ci-4 aliphatic, -CH2PI1, -0(CH2)o iPh, or a 3- to 6-membered saturated, partially unsaturated, or aryl ring having 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur. Suitable divalent substituents on a saturated carbon atom of R° include =0 and =S.
[0145] Suitable divalent substituents on a saturated carbon atom of an “optionally substituted” group include the following: =0 (“oxo”), =S, =NNR*2, =NNHC(0)R*, =NNHC(0)0R*, =NNHS(0)2R*, =NR*, =N0R*, -O(C(R*2))2 3O-, or -S(C(R*2))2 -3S-, wherein each independent occurrence of R* is selected from hydrogen, C1-6 aliphatic which may be substituted as defined below, or an unsubstituted 5- to 6-membered saturated, partially unsaturated, or aryl ring having 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur. Suitable divalent substituents that are bound to vicinal substitutable carbons of an “optionally substituted” group include: -O(CR*2)2 3O-, wherein each independent occurrence of R* is selected from hydrogen, C1-6 aliphatic which may be substituted as defined below, or an unsubstituted 5-6-membered saturated, partially unsaturated, or aryl ring having 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur.
[0146] Suitable substituents on the aliphatic group of R* include halogen, - R1, -(haloR1), -OH, -OR1, -O(haloR'), -CN, -C(O)OH, -C(O)OR. -NH2, -NHR1, -NR'2, or - NO2, wherein each R1 is unsubstituted or where preceded by “halo” is substituted only with one or more halogens, and is independently C1-4 aliphatic, -CH2PI1, -0(CH2)o iPh, or a 3- to 6-membered saturated, partially unsaturated, or aryl ring having 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur.
[0147] Suitable substituents on a substitutable nitrogen of an “optionally substituted” group include -R\ -NR^, -C(O)R^, -C(O)OR^, -C(0)C(0M -
C(0)CH2C(0M -S(O)2Rt, -S(O)2NR^2, -C(S)NR^2, -C(NH)NR^2, or -N(R^)S(O)2Rt; wherein each R1' is independently hydrogen, C1-6 aliphatic which may be substituted as defined below, unsubstituted -OPh, or an unsubstituted 3- to 6-membered saturated, partially unsaturated, or aryl ring having 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur, or, notwithstanding the definition above, two independent occurrences of R1', taken together with their intervening atom(s) form an unsubstituted 3- to 12-membered
saturated, partially unsaturated, or aryl mono- or bicyclic ring having 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur.
[0148] Suitable substituents on the aliphatic group of R’ are independently halogen, - R1, -(haloR1), -OH, -OR1, -O(haloR'), -CN, -C(O)OH, -C(O)OR‘, -NH2, -NHR1, -NR*2, or -NO2, wherein each R1 is unsubstituted or where preceded by “halo” is substituted only with one or more halogens, and is independently C1-4 aliphatic, -CH2PI1, -0(CH2)o iPh, or a 3- to 6-membered saturated, partially unsaturated, or aryl ring having 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur.
[0149] Subject: As used herein, the term “subject” refers to an organism to be administered with a composition described herein, e.g., for experimental, diagnostic, prophylactic, and/or therapeutic purposes. Typical subjects include animals (e.g., mammals such as mice, rats, rabbits, non-human primates, domestic pets, etc.) and humans. In some embodiments, a subject is a human subject. In some embodiments, a subject is suffering from a disease, disorder, or condition (e.g., obesity, a condition associated with obesity, etc.). In some embodiments, a subject is susceptible to a disease, disorder, or condition (e.g., obesity, a condition associated with obesity, etc.). In some embodiments, a subject displays one or more symptoms or characteristics of a disease, disorder, or condition (e.g., obesity, a condition associated with obesity, etc.). In some embodiments, a subject displays one or more non-specific symptoms of a disease, disorder, or condition (e.g., obesity, a condition associated with obesity, etc.). In some embodiments, a subject does not display any symptom or characteristic of a disease, disorder, or condition (e.g., obesity, a condition associated with obesity, etc.). In some embodiments, a subject is someone with one or more features characteristic of susceptibility to or risk of a disease, disorder, or condition (e.g., obesity, a condition associated with obesity, etc.). In some embodiments, a subject is a patient. In some embodiments, a subject is an individual to whom diagnosis and/or therapy is and/or has been administered.
[0150] Suffering from: An individual who is “suffering from” a disease, disorder, and/or condition (e.g., obesity, a condition associated with obesity, etc.) has been diagnosed with and/or displays one or more symptoms of a disease, disorder, and/or condition.
[0151] Susceptible to: An individual who is “susceptible to” a disease, disorder, and/or condition (e.g., obesity, a condition associated with obesity, etc.) is one who has a
higher risk of developing the disease, disorder, and/or condition (e.g., obesity, a condition associated with obesity, etc.) than does a member of the general public. In some embodiments, an individual who is susceptible to a disease, disorder and/or condition (e.g., obesity, a condition associated with obesity, etc.) may not have been diagnosed with the disease, disorder, and/or condition (e.g., obesity, a condition associated with obesity, etc.). In some embodiments, an individual who is susceptible to a disease, disorder, and/or condition (e.g., obesity, a condition associated with obesity, etc.) may exhibit symptoms of the disease, disorder, and/or condition (e.g., obesity, a condition associated with obesity, etc.). In some embodiments, an individual who is susceptible to a disease, disorder, and/or condition (e.g., obesity, a condition associated with obesity, etc.) may not exhibit symptoms of the disease, disorder, and/or condition (e.g., obesity, a condition associated with obesity, etc.). In some embodiments, an individual who is susceptible to a disease, disorder, and/or condition (e.g., obesity, a condition associated with obesity, etc.) will develop the disease, disorder, and/or condition (e.g., obesity, a condition associated with obesity, etc.). In some embodiments, an individual who is susceptible to a disease, disorder, and/or condition (e.g., obesity, a condition associated with obesity, etc.) will not develop the disease, disorder, and/or condition (e.g., obesity, a condition associated with obesity, etc.).
[0152] Therapy: The term “therapy” refers to an administration or delivery of an agent or intervention that has a therapeutic effect and/or elicits a desired biological and/or pharmacological effect (e.g., has been demonstrated to be statistically likely to have such effect when administered to a relevant population). In some embodiments, a therapeutic agent or therapy is any substance that can be used to alleviate, ameliorate, relieve, inhibit, prevent, delay onset of, reduce severity of, and/or reduce incidence of one or more symptoms or features of a disease, disorder, and/or condition (e.g., obesity, a condition associated with obesity, etc.). In some embodiments, a therapeutic agent or therapy is a medical intervention (e.g., surgery, radiation, phototherapy) that can be performed to alleviate, relieve, inhibit, present, delay onset of, reduce severity of, and/or reduce incidence of one or more symptoms or features of a disease, disorder, and/or condition.
[0153] Treat: As used herein, the term “treat,” “treatment,” or “treating” refers to any method used to partially or completely alleviate, ameliorate, relieve, inhibit, prevent, delay onset of, reduce severity of, and/or reduce incidence of one or more symptoms or features of a disease, disorder, and/or condition (e.g., obesity, a condition associated with obesity, etc.).
Treatment may be administered to a subject who does not exhibit signs of a disease, disorder, and/or condition (e.g., obesity, a condition associated with obesity, etc.). In some embodiments, treatment may be administered to a subject who exhibits only early signs of the disease, disorder, and/or condition (e.g., obesity, a condition associated with obesity, etc.), for example for the purpose of decreasing the risk of developing pathology associated with the disease, disorder, and/or condition. In some embodiments, treatment may be administered to a subject at a later-stage of disease, disorder, and/or condition (e.g., obesity, a condition associated with obesity, etc.).
[0154] Compounds of this disclosure include those described generally above and are further illustrated by the classes, subclasses, and species disclosed herein. As used herein, the following definitions shall apply unless otherwise indicated. For purposes of this disclosure, the chemical elements are identified in accordance with the Periodic Table of Elements, CAS version, Handbook of Chemistry and Physics, 75th Ed. Additionally, general principles of organic chemistry are described in “Organic Chemistry”, Thomas Sorrell, University Science Books, Sausalito: 1999, and “March’s Advanced Organic Chemistry”, 5th Ed., Ed.: Smith, M.B. and March, J., John Wiley & Sons, New York: 2001, the entire contents of which are hereby incorporated by reference.
[0155] Unless otherwise stated, structures depicted herein are meant to include all stereoisomeric (e.g., enantiomeric or diastereomeric) forms of the structure, as well as all geometric or conformational isomeric forms of the structure. For example, the R and S configurations of each stereocenter are contemplated as part of the disclosure. Therefore, single stereochemical isomers, as well as enantiomeric, diastereomeric, and geometric (or conformational) mixtures of provided compounds are within the scope of the disclosure. For example, in some cases, provided compounds show one or more stereoisomers of a compound, and unless otherwise indicated, represents each stereoisomer alone and/or as a mixture. Unless otherwise stated, all tautomeric forms of provided compounds are within the scope of the disclosure.
[0156] Unless otherwise indicated, structures depicted herein are meant to include compounds that differ only in the presence of one or more isotopically enriched atoms. For example, compounds having the present structures including replacement of hydrogen by
deuterium or tritium, or replacement of a carbon by 13C- or 14C-enriched carbon are within the scope of this disclosure.
DETAILED DESCRIPTION OF CERTAIN EMBODIMENTS
Incretins and Their Use in Treatment of Disease
[0157] Incretins are peptide hormones that are released in the gastrointestinal (GI) tract in response to glucose consumption, which stimulates insulin secretion by the pancreas and decreases glucagon production, lowering blood sugar levels. Incretins exert their effects by binding to their respective receptors on pancreatic beta cells, leading to insulin release. Glucagon-like peptide-1 (GLP1) and glucose-dependent insulinotropic polypeptide (GIP) are two incretins that have been identified for their role in postprandial insulin secretion. GIP is largely responsible for insulin release in response to glucose intake. GLP1 stimulates satiety, slows gastric emptying, lowers glucagon secretion, and decreases food intake, resulting in weight loss. Agonizing GIP and GLP1 receptors has been shown to produce an additive effect when insulin is secreted. See Chim, USPharm. 2022; 47(10): 18-22, which is incorporated herein by reference in its entirety.
[0158] Because of their role in controlling blood glucose, satiety, etc., incretins and incretin mimetics have the potential to treat various diseases including obesity, pre-diabetes, type 2 diabetes (T2D, with its complications), early type 1 diabetes (e.g., within 3 months after diagnosis of T1D), non-alcoholic fatty liver disease (NAFLD), non-alcoholic steatohepatitis (NASH), cardiovascular (CV) disease (e.g., characterized by a major cardiovascular event (MACE), including CV death, non-fatal myocardial infarction, non-fatal stroke, or heart failure with preserved ejection fraction (HFpEF)), renal disease, and elevated risk of premature mortality. Such chronic diseases are prevalent worldwide and often present as co-morbidities.
Obesity
[0159] Obesity is the most prevalent chronic disease worldwide, affecting approximately 650 million adults. Obesity is considered a starting point and critical contributor to pre-diabetes, type 2 diabetes (T2D, with its complications), non-alcoholic fatty liver disease (NAFLD), non-alcoholic steatohepatitis (NASH), cardiovascular and renal disease, and premature mortality. Obesity imposes a considerable economic burden with
additional direct medical costs, productivity costs (absenteeism, presenteeism, disability support, premature mortality), transportation costs (including increased CO2 footprint), human capital accumulation costs (school absenteeism, highest educational degree achieved). It is estimated that the number of obese (BMI>30kg/m2) people will exceed one billion by 2030, about 10% of which will suffer from severe class III obesity (BMI>40kg/m2). Half of all men living with obesity live in just nine countries: USA, China, India, Brazil, Mexico, Russia, Egypt, Germany and Turkey. Childhood obesity is also sharply on the rise worldwide.
[0160] Obesity was declared a disease by the American Association of Clinical Endocrinologists (AACE) only in 2011, and is managed based on severity, starting with lifestyle/behavioral intervention, and increased physical activity, then pharmacotherapy, and finally bariatric surgery.
[0161] Based on short-term studies, weight loss is recommended for overweight or obese patients with T2D. The Look AHEAD study investigating long-term effects of weight loss on cardiovascular disease in more than 5100 people with T2D was halted for futility after almost 10 years in 2012, as it was shown that intensive lifestyle intervention focusing on weight loss did not reduce the rate of cardiovascular events in overweight or obese adults with T2D.
[0162] Overall, the success of pharmacotherapy for obesity has been limited so far, with only modest placebo-corrected weight loss, e.g., 3% for Xenical® (orlistat), 4-5% for Belviq® (lorcaserin) and Contrave® (naltrexone SR/bupropion SR), paired with socially constraining side-effects for Xenical®, and undesirable central nervous effects of the centrally acting agents. Acomplia® (rimonabant) was rejected by FDA due to concerns that using it could increase suicidal thoughts and depression.
[0163] The most effective intervention for obesity remains bariatric surgery, however, only 1% of eligible patients undergo the procedure due to perceived severe complications, including mortality. Bariatric surgery has shown to substantially alter the release of endocrine hormones, which sparked an interest in targeting the pathways of endogenous nutrient- stimulated hormones, essentially mimicking the effect of bariatric surgery with a chemical agent, coined “incretin mimetics”.
Existing treatments using Incretin Mimetics
[0164] Current therapies using incretin mimetics include Glucagon-like peptide- 1 (GLP1) receptor agonists like Trulicity® (dulaglutide), Byetta® (exenatide), Ozempic®/Rybelsus® (semaglutide injectable/oral), Victoza® (liraglutide) and Suliqua® (lixisenatide, only in combination with insulin glargine), which are approved for lowering blood sugar in people living with T2D without the need to continuously check blood sugar levels. An added benefit was the loss of body weight (2-4%) and positive effects on cardiovascular and renal parameters. Recent research combined the activity of GLP1 receptor agonists with GIP receptor agonists and/or glucagon (GCG) receptor agonists (dual/triple agonists), aiming at even better control of blood sugar and greater weight loss. Blood sugar control and weight loss was demonstrated with the GLP1/GCG receptor dual agonist SAR425899; however, the program was discontinued due to unacceptable gastrointestinal side effects in 2019. More recently, the GLP1/GIP receptor dual agonist tirzepatide, now in the market as Mounjaro®, was approved as an injectable medicine for adults with T2D used along with diet and exercise to improve blood sugar. GIP’s function is to regulate energy balance through cell-surface receptor signaling in the brain and adipose tissue. The SURPASS-2 study demonstrated non-inferiority and superiority of tirzepatide against semaglutide in lowering blood sugar. Weight loss however was only a secondary endpoint.
[0165] Phase 3 trials have been performed with weight reduction as a primary endpoint in a population of non-diabetic, overweight/obese patients - see SCALE (liraglutide), STEP-1 (semaglutide) and SURMOUNT-1 (tirzepatide) trials. The SCALE trial demonstrated a 5.4% placebo-corrected weight loss and less progression to pre-diabetes, at a dose of 3 mg liraglutide once daily plus lifestyle intervention. The STEP-1 trial produced 12.4% weight loss in overweight or obese participants on 2.4 mg of semaglutide once weekly plus lifestyle intervention. The SURMOUNT-1 trial demonstrated 17.8% placebo-corrected weight loss on the highest dose of 15 mg tirzepatide once weekly. Cardiometabolic measures improved in all studies. Gastrointestinal side-effects were as expected, especially when starting the treatment, and were manageable. Approved obesity products now include Saxenda® (liraglutide 3 mg daily injection) and Wegovy® (semaglutide once-weekly injection). The FDA granted Fast Track designation for tirzepatide for the treatment of adults with obesity, or overweight with weight-related comorbidities in October 2022 which could lead to approval for this indication in early 2024, based on a rolling submission of further
data from the SURMOUNT study series. Topline results were recently published from the SEUECT trial, testing Wegovy® in patients with established cardiovascular disease and overweight/obesity but without T2D and found that semaglutide at 2.4 mg reduces the risk of major adverse cardiovascular events by 20% in overweight or obese adults. Another study using incretin mimetics is OASIS- 1, a study in non-diabetic overweight/obese patients with oral semaglutide 25 or 50 mg. Oral semaglutide at a maintenance dose of 14 mg is already approved as Rybelsus® for T2D. Phase 2 results for retratutide, LY3437943, a triple receptor agonist (GIP/GLP1/GCG receptors), were recently published showing 22.1% placebo- corrected weight-loss in people living with obesity after 48 weeks of treatment. Semaglutide was also recently shown to show a benefit in early T1D (within 3 months after diagnosis of T ID). In a small study, the need for prandial insulin was eliminated in all participants, and basal insulin in most.
[0166] Exemplary treatments of obesity/T2D using incretin mimetics are shown in Table 1 below.
Table 1: Exemplary incretin mimetics for Treatment of Obesitv/T2D
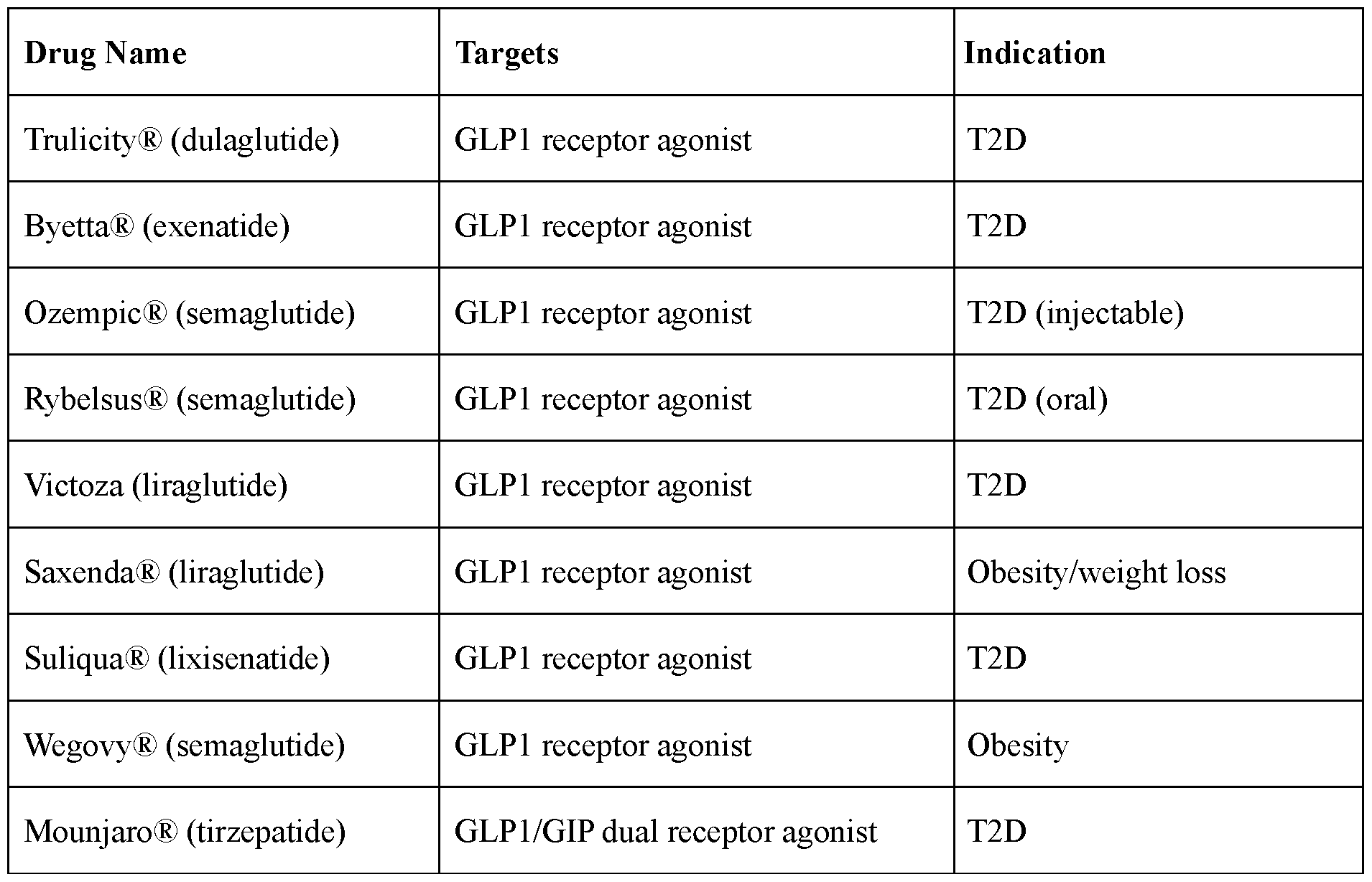 [0167] The present disclosure recognizes, among other things, the current problems in the market of incretin mimetics for the treatment of obesity, pre-diabetes, T2D, early T1D, NAFLD, NASH, cardiovascular disease, renal disease, and elevated risk of premature mortality, including but not limited to, limited supply, high price, lack of health insurance covering such treatments, frequent injections (e.g., once weekly), high injection volumes, and gastrointestinal side effects.
[0167] The present disclosure recognizes, among other things, the current problems in the market of incretin mimetics for the treatment of obesity, pre-diabetes, T2D, early T1D, NAFLD, NASH, cardiovascular disease, renal disease, and elevated risk of premature mortality, including but not limited to, limited supply, high price, lack of health insurance covering such treatments, frequent injections (e.g., once weekly), high injection volumes, and gastrointestinal side effects.
[0168] The present disclosure provides, among other things, a more efficacious and cost effective way to use incretins and incretin mimetics for treatment of obesity, prediabetes, T2D, early T1D, NAFLD, NASH, cardiovascular disease, renal disease, and elevated risk of premature mortality through the delivery of incretins and incretin mimetics (collectively encompassed by the term “incretin agents”) encoded by one or more polyribonucleotides. In some embodiments, polyribonucleotides encoding incretin agents provide for a therapeutic treatment of obesity, and/or pre-diabetes, T2D, early T1D, NAFLD, NASH, cardiovascular disease, renal disease, and elevated risk of premature mortality (e.g., diseases related to obesity), which have improved properties compared to known incretin mimetic therapies, including the need for fewer injections (no more than once-weekly injection), lower injection volumes (e.g., no more than 0.5 ml), and fewer or less severe side effects. Additionally, polyribonucleotides for delivery of incretin agents provide for expression of incretin agents in a cell at therapeutically relevant levels, comparable to doses of current peptide-based therapies.
Polyribonucleotides for Delivery of Incretin Agents
[0169] The present disclosure, among other things, utilizes RNA technologies as a modality to express incretin agents directly in a subject as a novel class of therapeutics that agonize GLP1, GIP, and/or GCG receptors to effectively treat a disease state such as obesity, pre-diabetes, T2D, early T1D, NAFLD, NASH, cardiovascular disease, renal disease, and/or elevated risk of premature mortality. In some embodiments, a polyribonucleotide as described herein encodes an incretin agent found in nature, or a fragment or variant thereof. In some embodiments, a polyribonucleotide as described herein encodes an incretin agent, or a fragment or variant thereof, that has been modified from its natural form.
[0170] As used herein, the term “incretin agents” refers to an agent that comprises an incretin or incretin mimetic (where incretins and incretin mimetics are collectively designated
“incretin peptides” herein). Exemplary incretins include GLP1, GIP, and GCG. Exemplary incretin mimetics are shown in, e.g., Table 1. In some embodiments, an incretin agent is a biologically active portion or fragment of an incretin or incretin mimetic. In some embodiments, an incretin agent comprises an incretin peptide that is part of a fusion. For example, in some embodiments, an incretin agent is an incretin peptide that is fused to another peptide moiety (e.g., a half-life extending (HLE) domain).
[0171] In some embodiments, an incretin agent comprises a GLP 1 receptor agonist such as GLP 1. In some embodiments, an incretin agent comprises a GIP receptor agonist such as GIP. In some embodiments, an incretin agent comprises a dual GIP and GLP1 receptor agonist. In some embodiments, an incretin agent comprises a triple GIP, GLP1 and GCG receptor agonist.
Exemplary Incretin Peptides
[0172] In some embodiments, an incretin agent comprises a wild type (i.e., unmutated) incretin peptide sequence, or fragment thereof. For example, in some embodiments, an incretin agent comprises any one of the incretin peptides as represented in SEQ ID NOs: 5-15 and 62-64.
[0173] In some embodiments, an incretin agent comprises an incretin peptide, or fragment thereof with at least one mutated amino acid residue in comparison to a wild type reference sequence. In some embodiments, a mutated amino acid residue comprises a substitution of a natural amino acid residue with another natural amino acid residue. In some embodiments, an incretin agent comprises any one of the incretin peptides as represented in SEQ ID NOs: 5-10. In some embodiments, a mutated amino acid residue may confer dual or triple agonizing properties to an incretin agent. For example, in some embodiments, one or more amino acid substitutions are introduced into a GLP1, GIP or GCG peptide sequence (e.g., as shown in SEQ ID NOs: 12-15 and 62-64) in order to confer binding properties to two or more of the GLP1, GIP and/or GCG receptors.
[0174] In some embodiments, an incretin agent has an amino acid sequence that is at least 85%, at least 90%, at least 95%, or 100% identical to any one of the incretin peptides detailed in Table 2. In some embodiments, an incretin agent comprises any one of the incretin peptides detailed in Table 2 below, or combinations or variants thereof.
Table 2: Exemplary Incretin Peptides (with mutations in bold)


Linkers
[0175] In some embodiments, an incretin agent described herein includes a single incretin peptide (which configuration is referred to herein as “I: lx”) (see e.g., Figure 3). In some embodiments, an incretin peptide is fused to another peptide (e.g., a half-life extending (HLE) domain) via a linker (see e.g., Figures 4-14). In some embodiments, a linker contains at least one Gly (G) amino acid residue. Suitable linkers can be readily selected and can be of various lengths, such as from 1 amino acid (e.g., Gly) to 25 amino acids, from 2 amino acids to 15 amino acids, from 3 amino acids to 12 amino acids, including 4 amino acids to 10 amino acids, 5 amino acids to 9 amino acids, 6 amino acids to 8 amino acids, or 7 amino acids to 8 amino acids (e.g., at least 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24 or 25 amino acids). Exemplary linkers include glycine polymers (G)n, glycine-serine polymers (including, for example, (GS)n, (GGGGS: SEQ ID NO: l)n and (GGGS: SEQ ID NO: 2)n, where n is an integer of at least one), glycine-alanine polymers, alanine-serine polymers, and other flexible linkers known in the art. Glycine and glycineserine polymers are relatively unstructured, and therefore may be able to serve as a neutral tether between components. Glycine accesses significantly more phi-psi space than even alanine and is much less restricted than residues with longer side chains (see Scheraga, Rev. Computational Chem. 11: 173-142 (1992)). In some embodiments, a linker comprises an amino acid sequence of SEQ ID NO: 3 (GGGGSGGGGSGGGGSGGGGS or “(G4S)4” linker) or SEQ ID NO: 4 (GGGGSGGGGSGGGGSGGGGSGGGGS or “(G4S)5” linker). In some embodiments, a linker comprises an amino acid sequence of SEQ ID NO: 68 (GGGGSGGGGS or “(G4S)2” linker), SEQ ID NO: 156 (GGGSGGGS or “(G3S)2” linker),
SEQ ID NO: 157 (GGGGSGGGGSGGGGS or “(G4S)3” linker), or GGGGSGGGS (SEQ ID NO: 186).
[0176] In some embodiments, an incretin agent includes an incretin peptide connected to another peptide (e.g., a HLE domain described herein) using a (G4S)3 linker. In some embodiments, an incretin agent described herein includes an incretin peptide connected to another incretin peptide using a (G4S)2 linker.
Cleavage Sites
[0177] In some embodiments, an incretin peptide is fused to another peptide (e.g., another incretin peptide and/or a half-life extending (HLE) domain) via a protease cleavage site, e.g., a furin cleavage site (e.g., a peptide that includes the motif R-X-K/R-R SEQ ID NO: 158, e.g., SEQ ID NO: 160 RRKR or SEQ ID NO: 153 NVRRKR) and optionally any of the aforementioned linkers. In some embodiments, a protease cleavage site, e.g., a furin cleavage site comprises any one of SEQ ID NO: 160 RRKR, SEQ ID NO: 153 NVRRKR, SEQ ID NO: 189 RKKR, SEQ ID NO: 190 RMQR, or SEQ ID NO: 191 VFRR. The term “furin cleavage site” and “furin recognition site” are used interchangeably herein and refer to sequences that facilitate furin cleavage.
[0178] In some embodiments, a furin cleavage site is operably linked to (e.g., on C- terminal side of) a linker (e.g., a glycine linker, e.g., a (G4S)2 linker). In some embodiments, an incretin agent includes multiple (e.g., 2, 3, 4 or more) incretin peptides each separated by furin cleavage sites and optionally linkers (e.g., (G4S)2 linkers that are on N-terminal side of the furin cleavage site).
[0179] Inclusion of a furin cleavage site within incretin agents described herein, particularly where an incretin peptide is fused to another peptide (e.g., another incretin peptide and/or a half-life extending (HLE) domain) facilitates the proper cleavage of the incretin peptide from the other peptide and allows the incretin peptide to be fully processed and functional after it is expressed.
[0180] Without wishing to be bound by any theory, the site of cleavage and type of cleavage site or recognition site in the context of polyribonucleotides encoding incretin agents is important to ensure that the N-terminal end of the incretin peptide is processed correctly. Certain furin cleavage and recognition sites, and placement of those sites with
respect to an incretin peptide within an incretin agent, may lead to alternative processing or cleavage sites, ultimately changing the final amino acid sequence of the mature incretin peptide. In such relatively small peptides such as GLP1 or GIP (or variants thereof, and other peptides of similar size/properties), any change in amino acid residue could impact bioactivity of the peptide. In some embodiments, a furin recognition/cleavage site is chosen and positioned within an incretin agent in order to facilitate proper cleavage of the N-terminal end of the incretin peptide, or in other words, create a “scarless” N-terminal end of the incretin peptide, in order to maintain the incretin peptide’s bioactivity. Figure 20 and Figure 21 show a schematic of where the theoretical cleavage sites of certain exemplary signal peptides lie. Figure 20 indicates that A8G mutation facilitates correct N-terminal processing of GLP1 incretin peptides with husec signal peptides. Figure 21 indicates that the A2G mutation facilitates correct N-terminal processing of GIP incretin peptides with husec signal peptides.
[0181] Such a concept and utilization of a furin cleavage site at the N-terminus of an incretin peptide linked to another peptide could also be applied to other gut peptides (e.g., glucagon) and/or other peptides of comparable size/properties as GLP1 and GIP described herein. This is particularly important in the context of delivering incretin agents (or other similar peptides) as one or more polyribonucleotides encoding the incretin agents. Such delivery requires the proper translation of the protein within a cell, in addition to the post- translational processing, including proper cleavage of one incretin peptide from another. In some embodiments, incretin agents comprising one or more incretin peptides fused to another peptide described herein have been designed and generated to include protease cleavage sites placed within the incretin agent such that the cleavage of the incretin peptide(s) from the other peptide(s) is accurate and does not affect the amino acid sequence of the mature peptide (i.e., creates a scarless N-terminus). A “scarless” N-terminus, referred to herein, includes a peptide that has been cleaved from another peptide via a cleavage site, and where cleavage occurs in a way such that there are no remaining amino acids that are not part of the mature peptide and all of the amino acids of the mature peptide remain at the N-terminus of the peptide. A scarless N-terminus of an incretin peptide (and other similar peptides, e.g., other gut peptides, e.g., glucagon), allows for the proper functionality of the peptide after it is processed into a mature peptide.
[0182] In some embodiments, a furin cleavage site is placed immediately 5’ of a second incretin peptide in an incretin agent to ensure that cleavage results in a scarless N- terminus on the second incretin peptide. Such cleavage may be important to maintain function of the mature incretin peptide.
[0183] In some embodiments, a furin cleavage site is chosen for being compatible with the N-terminal sequence of the incretin peptide (e.g., a wildtype or variant incretin peptide, e.g., GIP with an A2G mutation or a GLP1 with a H1Y mutation and/or a A8G mutation). Mutations as described herein may be introduced into an incretin peptide in order to promote effective cleavage and to maintain a scarless N-terminal of the incretin peptide.
[0184] As disclosed herein, various furin cleavage sequence may be utilized to promote proper cleave. For example, in some embodiments, a furin cleavage site is e.g., NVRRKR (SEQ ID NO: 153), which is derived from the human MT-MMP 1 protein. Such a furin cleavage site is derived from a human protein and it is compatible with the N-terminal sequence of GIP and GLP1 incretin peptides, including wildtype and variant GIP and GLP1 incretin peptides. Other human furin cleavage sequences may be utilized, as different human furin cleavage sequences may show different cleavage efficiencies depending on adjacent amino acid sequences (see Izidoro, et al., Archives of biochemistry and biophysics 2009, 487.2, 105-114, which is herein incorporated by reference in its entirety).
[0185] In some embodiments, a mutation is introduced into an incretin peptide described herein to facilitate signal peptide cleavage and to generate a mature incretin peptide with a scarless N-terminus. In some embodiments, such a mutation includes an A2G mutation in a GIP incretin peptide (e.g., a GIP (1-42) incretin peptide). In some embodiments, such a mutation includes an A8G mutation in a GLP1 (7-37) incretin peptide. In some embodiments, such mutations may also increase the half-life of the incretin agents, e.g., by preventing proteolysis of the amino acid in the second position of the incretin peptide (leaving a mature incretin peptide that has been improperly cleaved at its N-terminus and is truncated by 1 or 2 amino acids). In some embodiments, mutations are selected such that they increase the probability of correct cleavage (i.e., cleavage at the N-terminus such that the mature incretin peptide is not truncated). In some embodiments, compatibility of signal peptide and cleavage site sequence used in incretin agents described herein depends on the particular adjacent incretin peptide amino acid sequence, particularly the amino acid residues at the N-terminus.
Incretin Agents with Multiple Incretin Peptides
[0186] In some embodiments, an incretin agent comprises a single incretin peptide. In some embodiments, an incretin agent comprises or more than one incretin peptides. In some embodiments, two or more incretin peptides that are included in an incretin agent described herein are the same or derived from the same incretin peptide (e.g., are GLP1 receptor agonists). In some embodiments, two or more incretin peptides that are included in an incretin agent described herein are different or derived from different incretin peptides.
[0187] In some embodiments, an incretin agent comprises a combination of incretin peptides, e.g., fused on a single polypeptide chain. For example, in some embodiments, an incretin agent comprises a GLP1 receptor agonist (e.g., GLP1 peptide, or fragment or variant thereof) and a GIP receptor agonist (e.g., a GIP peptide, or fragment or variant thereof). In some embodiments, an incretin agent comprises one or more incretin peptides selected from SEQ ID NOs: 5-15 and 62-64 and one or more incretin peptides selected from SEQ ID NOs: 5-15 and 62-64. In some embodiments, an incretin agent comprises a GLP1 receptor agonist (e.g., GLP1 peptide, or fragment or variant thereof), a GIP receptor agonist (e.g., a GIP peptide, or fragment or variant thereof), and a GCG receptor agonist (e.g., a GCG peptide, or fragment of variant thereof). In some embodiments, an incretin agent comprises more than one GLP1 receptor agonist (e.g., GLP1 peptide, or fragment or variant thereof) that may be the same or different. In some embodiments, an incretin agent comprises more than one GIP receptor agonist (e.g., GIP peptide, or fragment or variant thereof) that may be the same or different. In some embodiments an incretin agent comprises more than one copy of the same incretin peptide and/or a combination of different incretin peptides.
[0188] In some embodiments, an incretin peptide is fused to another incretin peptide via a linker. In some embodiments, a linker contains at least one Gly (G) amino acid residue. Suitable linkers can be readily selected and can be of various lengths, such as from 1 amino acid (e.g., Gly) to 25 amino acids, from 2 amino acids to 15 amino acids, from 3 amino acids to 12 amino acids, including 4 amino acids to 10 amino acids, 5 amino acids to 9 amino acids, 6 amino acids to 8 amino acids, or 7 amino acids to 8 amino acids (e.g., at least 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24 or 25 amino acids). Exemplary linkers include glycine polymers (G)n, glycine-serine polymers (including, for example, (GS)n, (GGGGS: SEQ ID NO: l)n and (GGGS: SEQ ID NO: 2)n, where n is an
integer of at least one), glycine-alanine polymers, alanine -serine polymers, and other flexible linkers known in the art. Glycine and glycine-serine polymers are relatively unstructured, and therefore may be able to serve as a neutral tether between components. Glycine accesses significantly more phi-psi space than even alanine and is much less restricted than residues with longer side chains (see Scheraga, Rev. Computational Chem. 11: 173-142 (1992)). In some embodiments, a linker comprises an amino acid sequence of SEQ ID NO: 3 (GGGGSGGGGSGGGGSGGGGS or “(G4S)4” linker) or SEQ ID NO: 4 (GGGGSGGGGSGGGGSGGGGSGGGGS or “(G4S)5” linker). In some embodiments, a linker comprises an amino acid sequence of SEQ ID NO: 68 (GGGGSGGGGS or “(G4S)2” linker), SEQ ID NO: 156 (GGGSGGGS or “(G3S)2” linker), or SEQ ID NO: 157 (GGGGSGGGGSGGGGS or “(G4S)3” linker).
[0189] In some embodiments, an incretin peptide is fused to another incretin peptide via a protease cleavage site, e.g., a furin cleavage site (e.g., a peptide that includes the motif R-X-K/R-R SEQ ID NO: 158, e g., RRKR SEQ ID NO: 160 or SEQ ID NO: 153 NVRRKR) and optionally any of the aforementioned linkers. In some embodiments, a protease cleavage site, e.g., a furin cleavage site comprises any one of SEQ ID NO: 160 RRKR, SEQ ID NO: 153 NVRRKR, SEQ ID NO: 189 RKKR, SEQ ID NO: 190 RMQR, or SEQ ID NO: 191 VFRR. In some embodiments, a furin cleavage site is operably linked to (e.g., 3’ of) a linker (e.g., a glycine linker, e.g., a (G4S)2 linker). In some embodiments, an incretin agent includes multiple (e.g., 2, 3, 4 or more) incretin peptides each separated by furin cleavage sites and optionally linkers (e.g., (G4S)2 linkers that are 5’ of the furin cleavage site). As described herein, in some embodiments, sequence and placement of cleavage sites (e.g., furin cleavage sites) within an incretin agent is important in order to facilitate proper cleavage of the peptides and generate a scarless N-terminus for the incretin peptide.
[0190] In some embodiments, a polyribonucleotide encodes an incretin agent that comprises a signal peptide and a single incretin peptide (which configuration is referred to herein as “I: lx”) (see e.g., Figure 3). In some embodiments, a polyribonucleotide encodes an incretin agent that comprises a signal peptide and two incretin agents, separated by a linker and a cleavage site, e.g., a furin cleavage site (which configuration is referred to herein as “I:2x”) (see e.g., Figure 4). Such a design allows for cleavage of a first incretin peptide from a second incretin peptide after translation of the peptides from the polyribonucleotide. In some embodiments, a polyribonucleotide encodes an incretin agent that comprises a signal
peptide and four incretin agents, each separated by a linker and a cleavage site, e.g., a furin cleavage site (which configuration is referred to herein as “I:4x”) (see e.g., Figure 5). Such a design allows for cleavage of the four individual incretin peptides after translation of the incretin peptides from the polyribonucleotide. One of skill in the art would understand that while incretin agents comprising 1, 2, and 4 incretin peptides are specifically described herein, incretin agents with a different number of incretin peptides separated by cleavage sites and linkers may be used. In any of the designs shown in Figures 3-5, an incretin peptide can be any incretin peptide described herein (e.g., a GLP1 or GIP incretin peptide, e.g., any one of the incretin peptides shown in Table 2).
[0191] In some embodiments, a polyribonucleotide described herein encodes a GLP 1 incretin peptide (e.g., any one of the GLP1 incretin peptides described in Table 2) upstream or 5’ of a GIP incretin peptide (e.g., any one of the GIP incretin peptides described in Table 2). In some embodiments, a polyribonucleotide described herein encodes a GIP incretin peptide (e.g., any one of the GIP incretin peptides described in Table 2) upstream or 5’ of a GLP1 incretin peptide (e.g., any one of the GLP1 incretin peptides described in Table 2). In some embodiments, order of incretin peptides (N-terminus to C-terminus direction) is determined by how the incretin peptides are expected to be cleaved, such that the incretin peptides maintain their amino acid sequence and a scarless N-terminus.
[0192] In some embodiments, a polyribonucleotide described herein encodes an incretin agent that has an amino acid sequence that is at least 85%, at least 90%, at least 95%, or 100% identical to any one of the incretin agents detailed in Table 3. In some embodiments, a polyribonucleotide described herein encodes an incretin agent that has an amino acid sequence according to any one of the incretin agents detailed in Table 3.
Table 3 : Exemplary Incretin Agents including more than one incretin peptide (with mutations in bold, linkers underlined. Furin cleavage site italicized) where x2 and x4 examples include a linker and Furin cleavage site in between each repeat unit

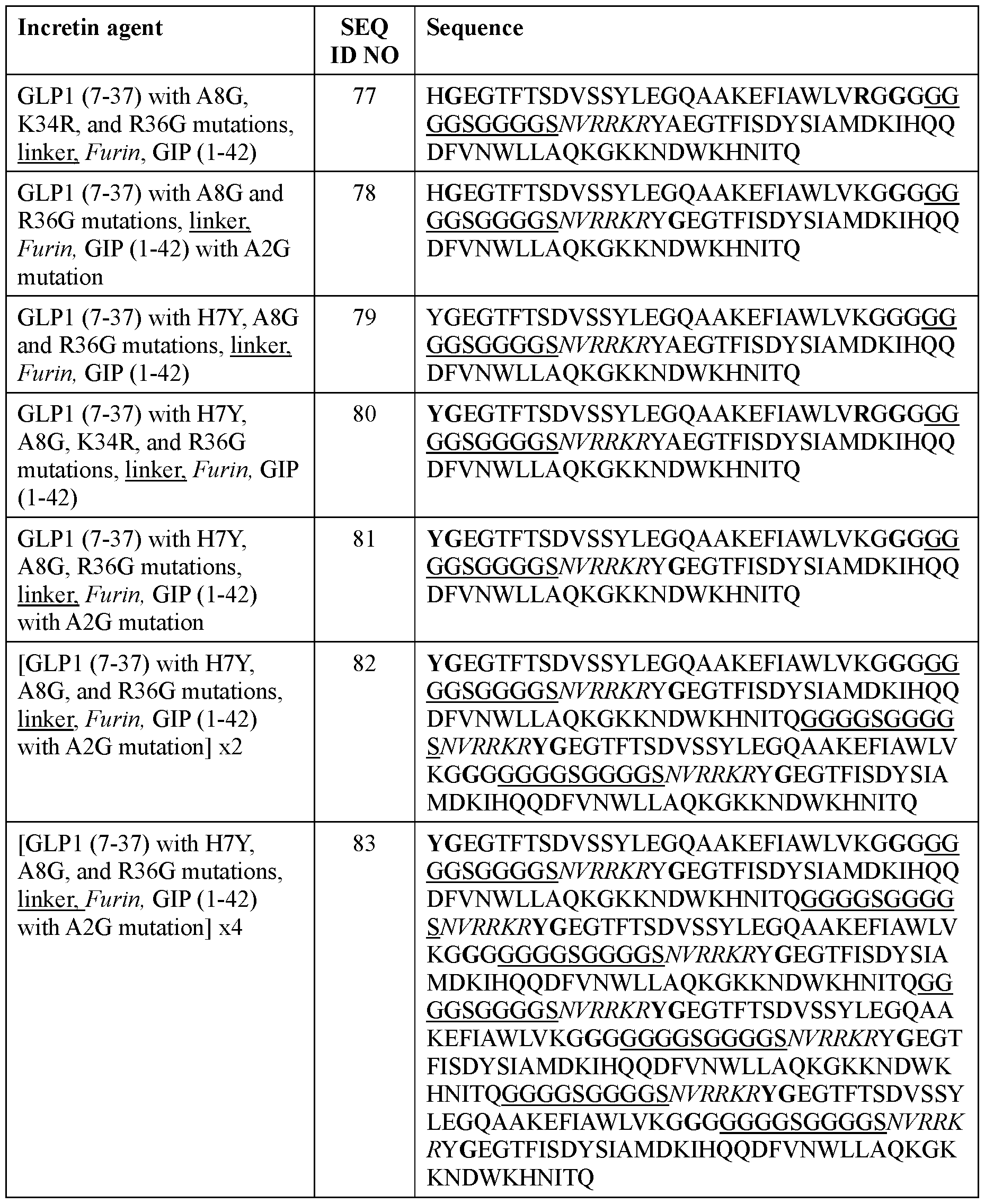
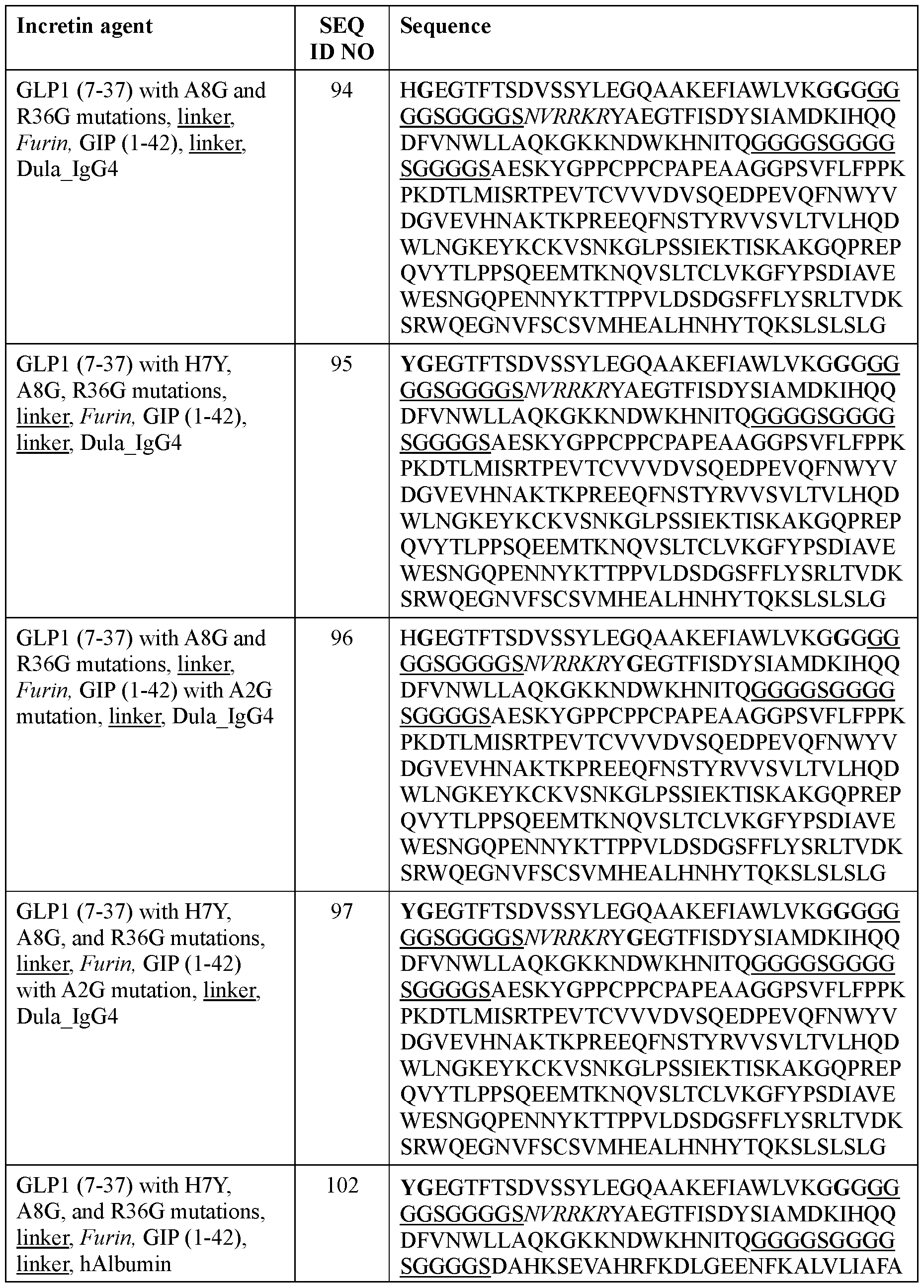
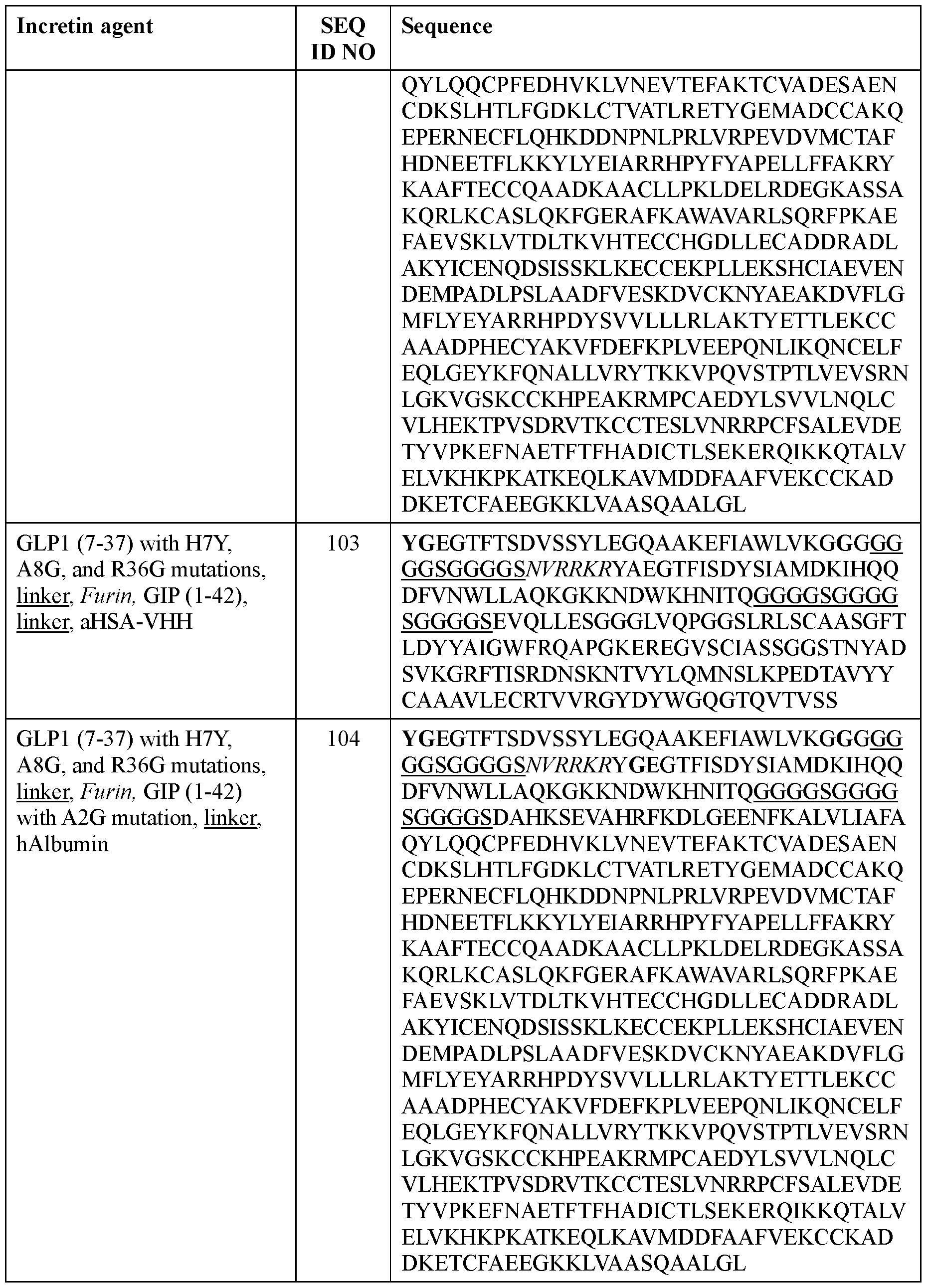
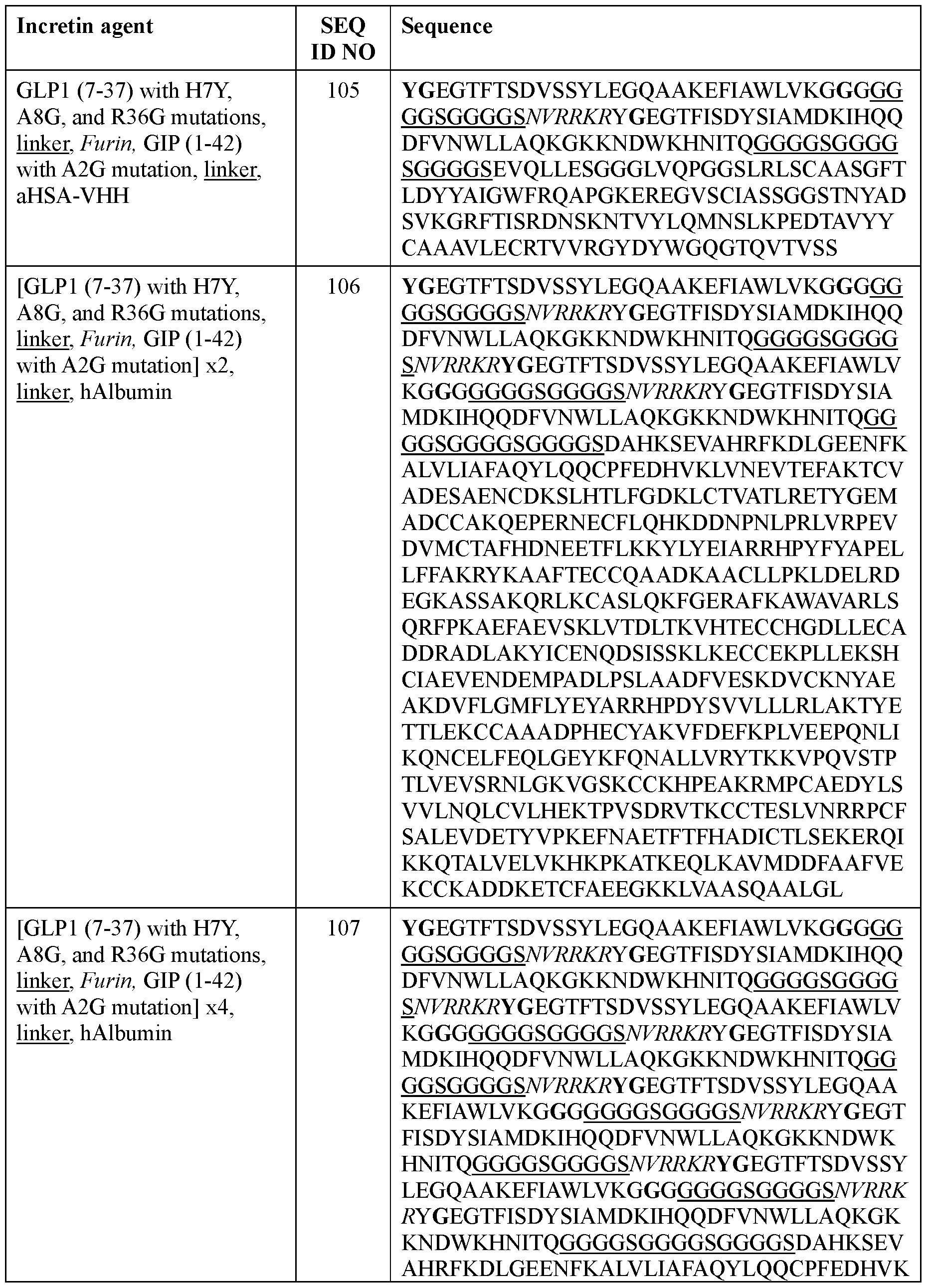
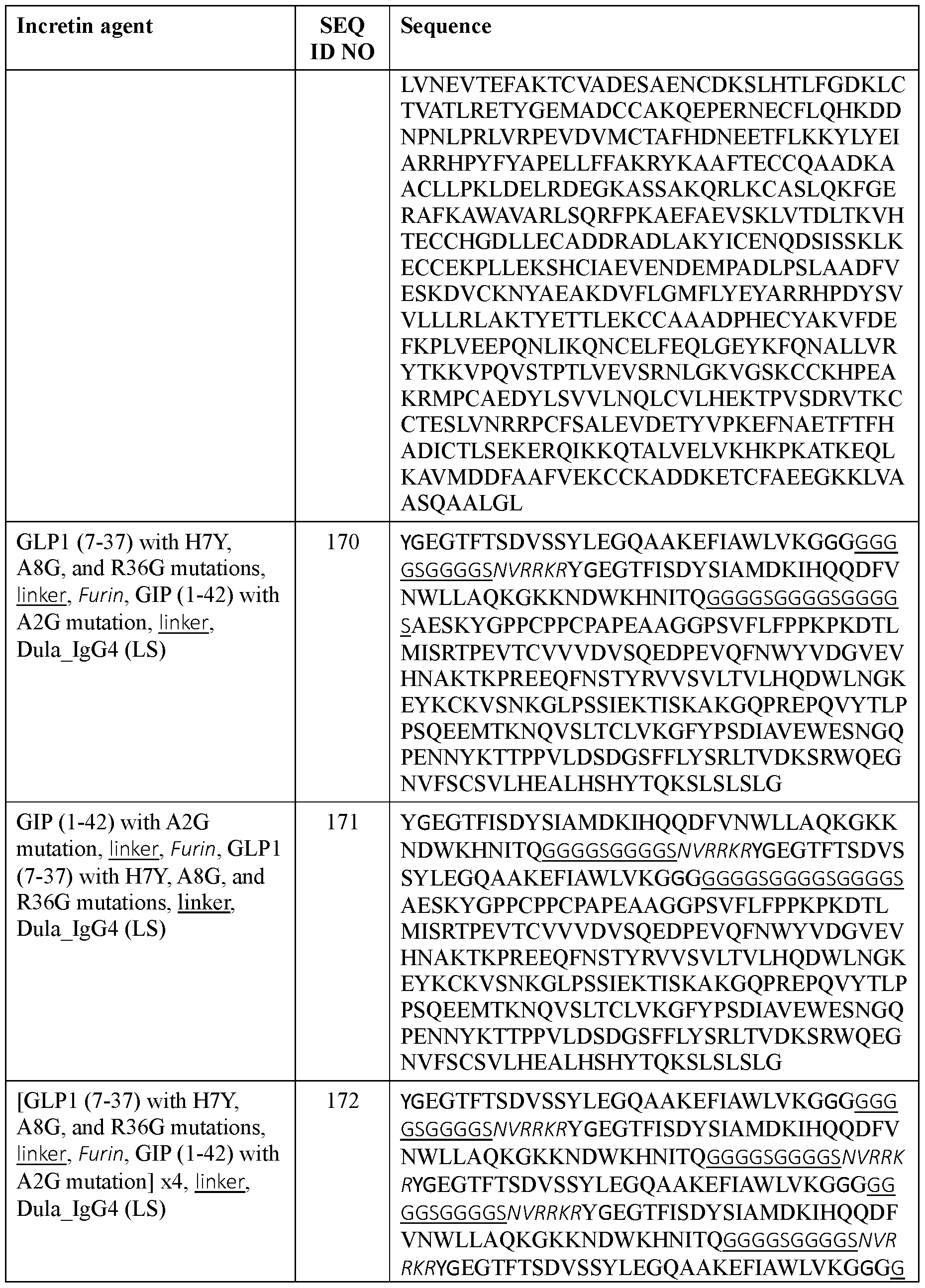

[0193] In some embodiments, the present disclosure provides one or more polyribonucleotides encoding an incretin agent that comprises a combination of incretin peptides. In such embodiments, one or more polyribonucleotides may encode an incretin agent. In some embodiments, a first polyribonucleotide may encode a first incretin peptide of an incretin agent and a second polyribonucleotide may encode a second incretin peptide of an incretin agent.
Half-life extension (HLE) domains
[0194] In some embodiments, a polyribonucleotide described herein encodes an incretin agent comprising one or more incretin peptides fused to a half-life extending (HLE) domain (see e.g., incretin agents shown in Figures 7-14). In some embodiments, where an incretin agent comprises more than one incretin peptide, an HLE domain may be included in an incretin agent described herein to increase the half-life of one of the incretin peptides, or of each incretin peptide.
Human Serum Albumin (HSA)
[0195] In some embodiments, an incretin agent comprises one or more incretin peptides fused to an HLE domain that comprises albumin, e.g., human serum albumin (HSA). In some embodiments a half-life extending moiety comprises albumin, e.g., human serum albumin. In some embodiments, a human serum albumin (HSA) sequence is at least 90%, 95%, or 99% identical to an amino acid sequence according to SEQ ID NO: 159 (DAHKSEVAHRFKDLGEENFKALVLIAFAQYLQQCPFEDHVKLVNEVTEFAKTCVAD
ESAENCDKSLHTLFGDKLCTVATLRETYGEMADCCAKQEPERNECFLQHKDDNPNL PRLVRPEVDVMCTAFHDNEETFLKKYLYEIARRHPYFYAPELLFFAKRYKAAFTECCQ AADKAACLLPKLDELRDEGKASSAKQRLKCASLQKFGERAFKAWAVARLSQRFPKA EFAEVSKLVTDLTKVHTECCHGDLLECADDRADLAKYICENQDSISSKLKECCEKPLL EKSHCIAEVENDEMPADLPSLAADFVESKDVCKNYAEAKDVFLGMFLYEYARRHPD YSVVLLLRLAKTYETTLEKCCAAADPHECYAKVFDEFKPLVEEPQNLIKQNCELFEQ LGEYKFQNALLVRYTKKVPQVSTPTLVEVSRNLGKVGSKCCKHPEAKRMPCAEDYL SWLNQLCVLHEKTPVSDRVTKCCTESLVNRRPCFSALEVDETYVPKEFNAETFTFHA DICTLSEKERQIKKQTALVELVKHKPKATKEQLKAVMDDFAAFVEKCCKADDKETCF AEEGKKLVAASQAALGL), or a fragment or variant thereof. In some embodiments, a HSA sequence comprises or consists of an amino acid sequence according to SEQ ID NO: 159, or a fragment or variant thereof. In some embodiments, a HSA sequence comprises or consists of an amino acid sequence that is a variant of wildtype HSA (i.e., SEQ ID NO: 159) comprising one or more amino acid mutations. In some embodiments, the one or more mutations comprises a mutation at position 573 in SEQ ID NO: 159. In some embodiments, the K residue at position 573 of SEQ ID NO: 159 is substituted with any one of the following amino acid residues: A, C, D, F, G, H, I, L, M, N, P, Q, R, S, V, W, and Y (SEQ ID NO: 187). In some embodiments, the K residue at position 573 of SEQ ID NO: 159 is substituted with a P residue (SEQ ID NO: 188). In some embodiments, an HSA variant comprises any one of the HSA variants disclosed in U.S. Patent No. 8,748,380, which is hereby incorporated by reference in its entirety.
[0196] In some embodiments, a polyribonucleotide described herein encodes an incretin agent as shown in Figure 7, including a I: lx, I:2x or I:4x configuration (i.e., 1, 2, or 4 incretin peptides), fused to an HLE domain, e.g., HSA or an HSA variant as described herein. In some embodiments, an incretin peptide may be a GLP1 or GIP incretin peptide described herein, or a variant thereof. In some embodiments, where an incretin agent comprises more than one incretin peptides, the incretin peptide adjacent to the HLE domain will remain fused to the HLE domain after post-translational processing, and the incretin peptide(s) that are not adjacent to the HLE domain will be cleaved from the adjacent incretin peptide and the HLE domain. Such designs may be used when administration of multiple incretin peptides with various half-lives is desirable. Such designs may also be desirable where one of the incretin peptides is meant to traverse the blood brain barrier (i.e., where an
HLE domain is not desirable) and one of the incretin peptides is meant to remain in circulation for a longer period of time (i.e., where an HLE remains attached).
[0197] In some embodiments, an incretin agent comprises more than one incretin peptide separated by a linker and a protease cleavage site (e.g., a furin cleavage site), and one of the incretin peptides is a GLP1 peptide described herein adjacent to an HLE domain, e.g., HSA or an HSA variant, and one of the incretin peptides is a GIP peptide that is not adjacent to the HLE (e.g., at the N-terminus of the polypeptide chain). In such an embodiment, when the incretin agent is expressed from a polyribonucleotide described herein, the GIP peptide will be cleaved from the GLP1 peptide connected to the HLE domain, such that the GLP1 incretin peptide will have a longer half-life than the GIP incretin peptide. In some embodiments, an incretin agent comprises more than one incretin peptide separated by a linker and a protease cleavage site (e.g., a furin cleavage site), and one of the incretin peptides is a GIP incretin peptide described herein adjacent to an HLE domain, e.g., HSA or an HSA variant, and one of the incretin peptides is a GLP1 peptide that is not adjacent to the HLE (e.g., at the N-terminus of the polypeptide chain). In such an embodiment, when the incretin agent is expressed from a polyribonucleotide described herein, the GLP1 incretin peptide will be cleaved from the GIP peptide connected to the HLE domain, such that the GIP incretin peptide will have a longer half-life than the GLP1 incretin peptide (see e.g., Figure 9).
[0198] In some embodiments, a polyribonucleotide described herein encodes an incretin agent as shown in Figure 8A, where the incretin agent has a signal peptide (“SP”), a GLP1 incretin peptide, a linker, a second GLP1 incretin peptide, a second linker (GGGS)3, and half-life extension (HLE) domain that is human serum albumin (HSA) or an HSA variant. Additionally, the incretin agent may include a protease cleavage site (e.g., a furin cleavage site) between the GLP1 incretin peptides. In such an embodiment, once the incretin agent is expressed, the first (N-terminal) GLP1 incretin peptide will be cleaved from the second incretin GLP1 incretin peptide that is adjacent to the HLE domain. The resulting GLP1 incretin peptides will have two different half lives (i.e., the GLP1 incretin peptide that remains attached to the HLE domain will have a longer half-life than the GLP 1 incretin peptide that was cleaved).
[0199] In some embodiments, a polyribonucleotide described herein encodes an incretin agent as shown in Figure 9, where the incretin agent includes a signal peptide (SP), a first GLP1 incretin peptide, a linker (GGGS)2, a first GIP incretin peptide, a second linker (GGGS)2, a second GLP 1 incretin peptide, a third linker (GGGS)2, a second GIP incretin agent, a fourth linker (GGGSfi and half-life extension (HLE) domain that is human serum albumin (HSA) or an HSA variant. Furin and SP cleavage sites within the incretin agent are indicated with arrows. In such an embodiment, when the incretin agent is expressed, the first GLP1 incretin peptide, the first GIP incretin peptide, and the second GLP1 incretin peptide will be cleaved, and the second GIP incretin peptide will remain fused to the HLE domain. The resulting second GIP-HLE fusion will have a longer half-life than the other incretin peptides.
[0200] In some embodiments, a polyribonucleotide described herein encodes an incretin agent comprising an amino acid sequence that is at least 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or at least 99% identical to any one of the incretin agent sequences in Table 4. In some embodiments, an incretin agent comprises any one of the incretin agents detailed in Table 4 below, or combinations or variants thereof.
Table 4: Exemplary Incretin Agents including hAlbumin (with mutations in bold, linkers underlined. Furin cleavage site italicized) where x2 and x4 examples include a linker and Furin cleavage site in between each repeat unit

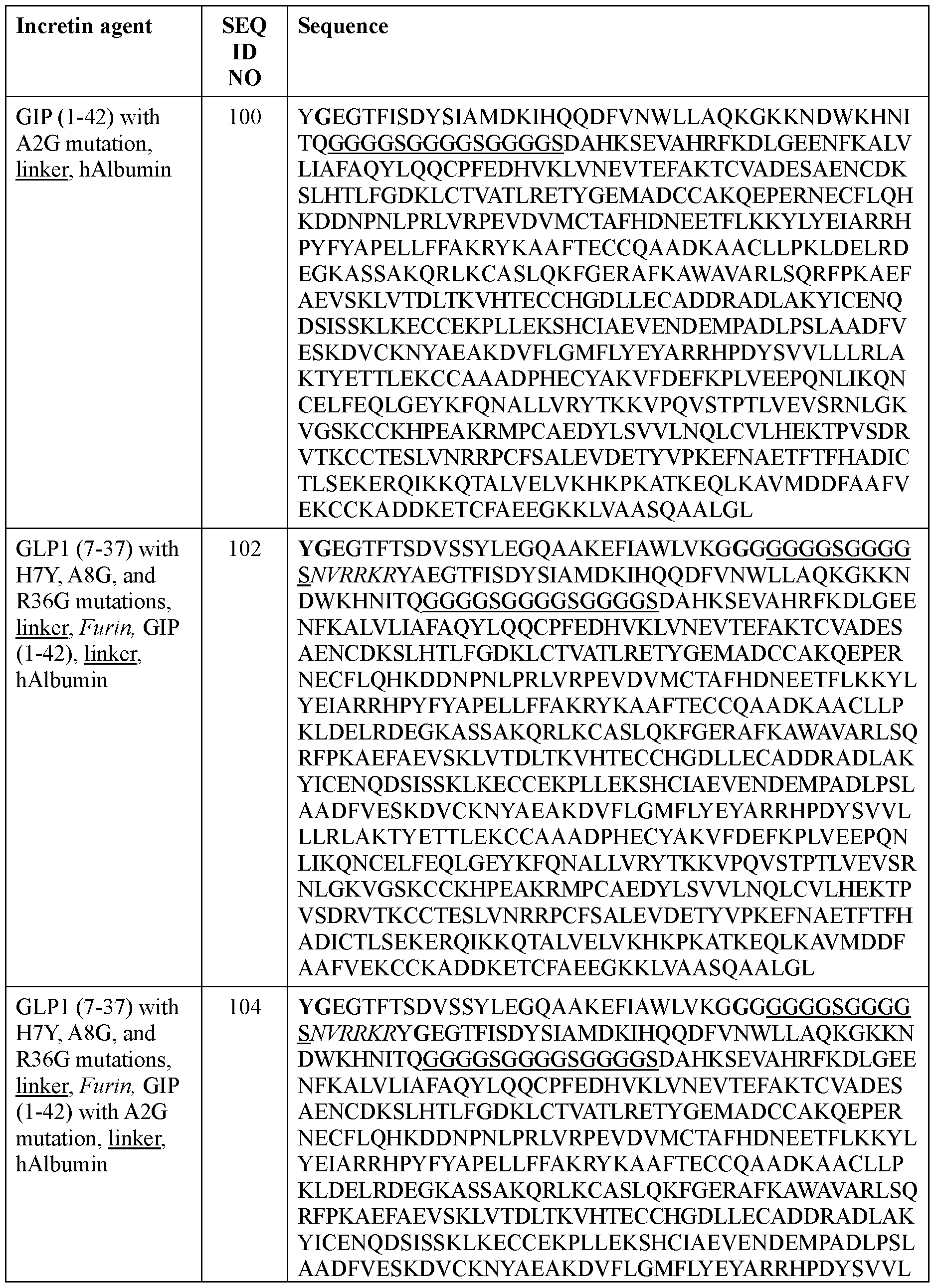


Albumin Binding domains
[0201] In some embodiments, an incretin peptide is fused to a half-life extending moiety that binds albumin. Various albumin binding moieties (i.e., albumin binding protein domains) may be utilized as a half-life extending moiety for incretin agents described herein (see, e.g., Zorzi et al., MedChemComm, 2019, 10.7, 1068-1081, which is incorporated herein by reference in its entirety). In some embodiments, an albumin binding protein domain comprises an albumin-binding domain (ABD) derived from protein G of Streptococcus strain GI48 and/or from protein PAB of Finegoldia magna. such as ABD035 and SA21 (as described in Levy, et al., PLoS One, 2014, 9(2), e87704, which is incorporated herein by reference in its entirety) and ABD094 (NCT02690142) (as described in Frejd and Kim, Exp. Mol. Med., 2017, 49(3), e306, which is incorporated herein by reference in its entirety). In some embodiments, an ABD binds to domain II of human serum albumin and does not overlap or interfere with binding to the FcRn-binding site on albumin.
[0202] In some embodiments, an ABD comprises ABDCon, a three-helix bundle albumin-binding domain, as described in Jacobs et al., Protein Eng., Des. Sei., 2015, 28(10), 385-393, which is incorporated herein by reference in its entirety. In some embodiments, an ABD is derived from the bacterial protein Sso7d from the hyperthermophilic archaeon Sulfolobus solfataricus, e.g., Mil.12 and M18.2.5 (as described in Gao et al., Nat. Struct. Biol., 1998, 5(9), 782-786 and Traxlmayr et al., J. Biol. Chem., 2016, 291(43), 22496-22508, which are incorporated herein by reference in their entirety). In some embodiments, an ABD comprises a DARPin, as described in Pluckthun, Annu. Rev. Pharmacol. Toxicol., 2015, 55, 489-511, which is incorporated herein by reference in its entirety.
[0203] In some embodiments, an ABD comprises an immunoglobulin domain or fragment thereof. In some embodiments, an ABD comprises a fully human domain antibody
(dAb). For example, in some embodiments, an ABD comprises an AlbudAb, as described in Holt et al., Protein Eng., Des. Sei., 2008, 21(5), 283-288, which is incorporated herein by reference in its entirety. In some embodiments, an ABD comprises a Fab, e.g., dsFv CA645, as described in Adams et al., mAbs, 2016, 8(7), 1336-1346, which is incorporated herein by reference in its entirety.
[0204] In some embodiments, an ABD comprises a heavy chain only (VHH) antibody, i.e., nanobody, as described in Steeland et al., Drug Discovery Today, 2016, 21(7), 1076-1113, which is incorporated herein by reference in its entirety. In some embodiments, an ABD comprises a VHH domain comprising one or more of the complementarity determining region (CDR) sequences HCDR1, HCDR2, and/or HCDR3 according to SEQ ID NO: 191 (GFTLDYYA), SEQ ID NO: 192 (IASSGGST), and/or SEQ ID NO: 193 (AAAVLECRTWRGYDY), respectively. In some embodiments, an ABD comprises a VHH domain comprising the CDR sequences HCDR1, HCDR2, and HCDR3 according to SEQ ID NO: 191 (GFTLDYYA), SEQ ID NO: 192 (IASSGGST), and SEQ ID NO: 193 (AAAVLECRTWRGYDY), respectively. In some embodiments, an ABD comprises a VHH domain that is at least 90%, 95%, or 99% identical to an amino acid sequence according to SEQ ID NO: 154
EVQLLESGGGLVQPGGSLRLSCAASGFTLDYYAIGWFRQAPGKEREGVSCIASSGGST NYADSVKGRFTISRDNSKNTVYLQMNSLKPEDTAVYYCAAAVLECRTVVRGYDYW GQGTQVTVSS or “aHSA-VHH”). In some embodiments, a VHH domain comprises an amino acid sequence according to SEQ ID NO: 154. In some embodiments, an ABD comprises a VNAR, as described in Muller et al., mAbs, 2012, 4(6), 673-685, which is incorporated herein by reference in its entirety.
[0205] In some embodiments, a polyribonucleotide described herein encodes an incretin agent as shown in Figure 8B, which shows an incretin agent that has a signal peptide (SP), a first GLP1 incretin peptide, a linker, a second GLP1 incretin peptide, a second linker (GGGS)3, and half-life extension (HLE) domain that is a VHH domain that binds to HSA. Furin and SP cleavage sites within the incretin agent are indicated with arrows. In such an embodiment, when the incretin agent is expressed, the first GLP1 incretin peptide is cleaved from the second GLP1 incretin peptide, and the second GLP1 incretin peptide remains fused to the HLE domain (anti -HSA VHH domain). As such, the second GLP1 incretin peptide will have a longer half-life than the first GLP1 incretin peptide. In some embodiments, an incretin
agent is the incretin agent shown in Figure 8B, where either or both of the GLP1 incretin peptides may instead be a different incretin agent (e.g., a GIP incretin peptide described herein). In some embodiments, a single incretin peptide is fused to a VHH domain that binds to HSA.
[0206] Other albumin-binding domains are known in the art, see e.g., Zorzi et al., MedChemComm, 2019, 10.7, 1068-1081, which is incorporated herein by reference in its entirety.
[0207] In some embodiments, a polyribonucleotide described herein encodes an incretin agent comprising an amino acid sequence that is at least 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or at least 99% identical to any one of the incretin agent sequences in Table 5. In some embodiments, an incretin agent comprises any one of the incretin agents detailed in Table 5 below, or combinations or variants thereof.
Table 5 : Exemplary Incretin Agents including an aHSA-VHH domain (with mutations in bold, linkers underlined)


XTENs
[0208] In some embodiments, a half-life extending moiety is an XTEN sequence as described in U.S. Patent No. 8,673,860 and Podust et al., Journal of Controlled Release, 2016, 240, 52-66, which are incorporated herein by reference in their entirety.
[0209] In some embodiments, an XTEN sequence comprises about 100 to about 3000 amino acid residues, preferably 400 to about 3000 residues, wherein at least about 80%, or at least about 90%, or about 91%, or about 92%, or about 93%, or about 94%, or about 95%, or about 96%, or about 97%, or about 98%, or about 99% to about 100% of the sequence consists of multiple units of two or more non-overlapping sequence motifs selected from the amino acid sequences of Table 6 or Table 7. In some cases, the XTEN comprises nonoverlapping sequence motifs in which about 80%, or at least about 90%, or about 91%, or about 92%, or about 93%, or about 94%, or about 95%, or about 96%, or about 97%, or about 98%, or about 99% to about 100% of the sequence consists of two or more non-overlapping sequences selected from a single motif family of Table 6 or Table 7, resulting in a “family” sequence in which the overall sequence remains substantially non-repetitive. Accordingly, in these embodiments, an XTEN sequence can comprise multiple units of non-overlapping sequence motifs of the AD motif family, or the AE motif family, or the AF motif family, or the AG motif family, or the AM motif family, or the AQ motif family, or the BC family, or the BD family of sequences of Table 6. In other cases, the XTEN comprises motif sequences from two or more of the motif families of Table 6. In other cases, the XTEN comprises motif sequences from one or more of the motif families of Table 7.
Table 6: XTEN Sequence Motifs of 12 Amino Acids and Motif Families


Table 7: XTEN Sequence Motifs of 12 Amino Acids and Motif Families

Fc Domains and Incretin agents with multiple polypeptide chains
[0210] In some embodiments, a half-life extending moiety is or comprises an Fc domain, e.g., of a human IgG (e.g., a human IgGl, IgG2, IgG3, or IgG4). In some embodiments, a half-life extending moiety does not comprise an Fc domain, e.g., of a human IgG (e.g., a human IgGl, IgG2, IgG3, or IgG4). In some embodiments, a half-life extending moiety comprises an Fc domain of a human IgG4 or a variant thereof (e.g., as included in Dulaglutide). In some embodiments, an Fc domain of an IgG4 sequence is at least 90, 95, 96, 97, 97, or 99% identical to SEQ ID NO: 155
(AESKYGPPCPPCPAPEAAGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSQEDPEVQF NWYVDGVEVHNAKTKPREEQFNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKGLPS SIEKTISKAKGQPREPQVYTLPPSQEEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPE NNYKTTPPVLDSDGSFFLYSRLTVDKSRWQEGNVFSCSVMHEALHNHYTQKSLSLSL G). In some embodiments, an Fc domain of an IgG4 sequence is or comprises an amino acid sequence according to SEQ ID NO: 155.
Homodimeric Incretin Agents
[0211] In some embodiments where an incretin agent comprises more than one incretin peptide, an incretin agent comprises two or more incretin peptides on a single polypeptide chain. In some embodiments, where an incretin agent comprises more than one incretin peptide, an incretin agent comprises one or more incretin peptides on separate polypeptide chains.
[0212] In some embodiments, the separate polypeptide chains multimerize (e.g., dimerize). In some embodiments where an incretin agent comprises one or more incretin peptides on separate polypeptide chains, the separate polypeptide chains comprise two polypeptide chains that each comprise an immunoglobulin constant domain, and the two polypeptide chains dimerize via two constant domains that combine to make an Fc domain.
[0213] In some embodiments, an Fc domain comprises an IgG4 Fc domain (e.g., as included in Dulaglutide). In some embodiments, an Fc domain comprises an IgGl Fc domain. An exemplary design of an incretin agent comprising multiple polypeptide chains that include an Fc domain, so that the polypeptide chains dimerize, is shown in e.g., Figure 10. The design in Figure 10 could include incretin peptides in a I: lx, I:2x, or I:4x configuration (or other numbers of incretin peptides could be utilized) and each could be a GLP1 or GIP incretin peptide (or any of the variants described herein). In Figure 10, the two Fc domains are the same. The incretins on each polypeptide chain can be the same or different (or in the case of multiple incretins on a single chain, may contain different combinations of incretin peptides). An exemplary incretin agent that includes two polypeptide chains that form a homodimer through dimerization of the Fc domains on each polypeptide chain is shown in Figure 11. In Figure 11, the incretin peptide contains two polypeptide chains, each includes a signal peptide (SP), a GLP1 incretin peptide, a linker (GGGGS)3, an Fc domain. In some embodiments, each polypeptide chain may comprise two or more incretin peptides (see e.g., Figure 12). In some embodiments, the two or more incretin peptides could be the same incretin peptide. In some embodiments, the two or more incretin peptides could be different incretin peptides (see e.g., Figure 12). Where two or more incretin peptides are included on each polypeptide chain, cleavage sites may be introduced to cleave the incretin peptides, leaving one incretin peptide attached to the Fc domain. It will be understood by one skilled in
the art that the incretin peptide that remains attached to the Fc domain will have a longer halflife and different activity that the other cleaved incretin peptides.
[0214] Fc domains included in incretin agents described herein not only allow the two polypeptide chains to dimerize, but also may increase half-life of the incretin peptides. Other mutations may also be introduced into the Fc domains to increase half-life of the incretin agent.
Fc mutations for HLE
[0215] In some embodiments, an Fc domain within an incretin agent comprises one or more mutations to increase the half-life of the incretin agent. For example, in some embodiments, an Fc domain may include an LS mutation within a CH3 region (for enhanced FcRn binding) (see Zalevsky, et al., Nature biotechnology, 2010, 28.2: 157-159, which is incorporated herein by reference). Such mutations are noted as M428L and N434S according to EU numbering (i.e., M88L and N94S within the CH3 domain), and referred to herein as “LS”. Exemplary incretin agents with such a mutation are shown in e.g., Figure 11, Figure 12, and Figure 14.
[0216] In some embodiments, an Fc domain of an IgG4 (LS) sequence is identical to SEQ ID NO: 299 (AESKYGPPCPPCPAPEAAGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSQEDPEVQF NWYVDGVEVHNAKTKPREEQFNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKGLPS SIEKTISKAKGQPREPQVYTLPPSQEEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPE NNYKTTPPVLDSDGSFFLYSRLTVDKSRWQEGNVFSCSVLHEALHSHYTQKSLSLSL G).
[0217] In some embodiments, an Fc domain in an incretin agent includes one or more mutated amino acid residues that increase half-life. In some embodiments, an Fc domain comprises one of the following mutated amino acid residues: M252Y, S254T, and T256E (“YTE”), according to the EU numbering scheme to increase half-life. In some embodiments, an Fc domain comprises a combination of the following mutated amino acid residues: M252Y, S254T, and T256E, according to the EU numbering scheme to increase half-life.
[0218] In some embodiments, an Fc domain comprises one of the following mutated amino acid residues: T250Q and M428L (“QL”), according to the EU numbering scheme to
increase half-life. In some embodiments, an Fc domain comprises one of the following mutated amino acid residues: H433K and N434F (“KF”), according to the EU numbering scheme to increase half-life. In some embodiments, a second Fc domain comprises one of the following mutated amino acid residues: T307A, E380A and N434A (“AAA”), according to the EU numbering scheme to increase half-life. In some embodiments, an Fc domain comprises the following mutated amino acid residues: V308P, according to the EU numbering scheme to increase half-life. In some embodiments, an Fc domain comprises one of the following mutated amino acid residues: M252Y, V308P, and N434Y (“YPY”), according to the EU numbering scheme to increase half-life. In some embodiments, an Fc domain comprises one of the following mutated amino acid residues: H285D, T307Q, and A378V (“DQV”), according to the EU numbering scheme to increase half-life. In some embodiments, an Fc domain comprises one of the following mutated amino acid residues: L309D, Q311H, N434S (“DHS”), according to the EU numbering scheme to increase halflife. Exemplary Fc mutations are described in e.g., Liu et al., Antibodies 9.4: 64 (2020), which is hereby incorporated by reference in its entirety.
[0219] In some embodiments, a polyribonucleotide described herein encodes an incretin agent comprising an amino acid sequence that is at least 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or at least 99% identical to any one of the incretin agent sequences in Table 8. In some embodiments, an incretin agent comprises any one of the incretin agents detailed in Table 8 below, or combinations or variants thereof.
Table 8: Exemplary Incretin Agents including an IgG4 Fc domain (with mutations in bold, linkers underlined. Furin cleavage site italicized) where x4 example includes a linker and Furin cleavage site in between each repeat unit

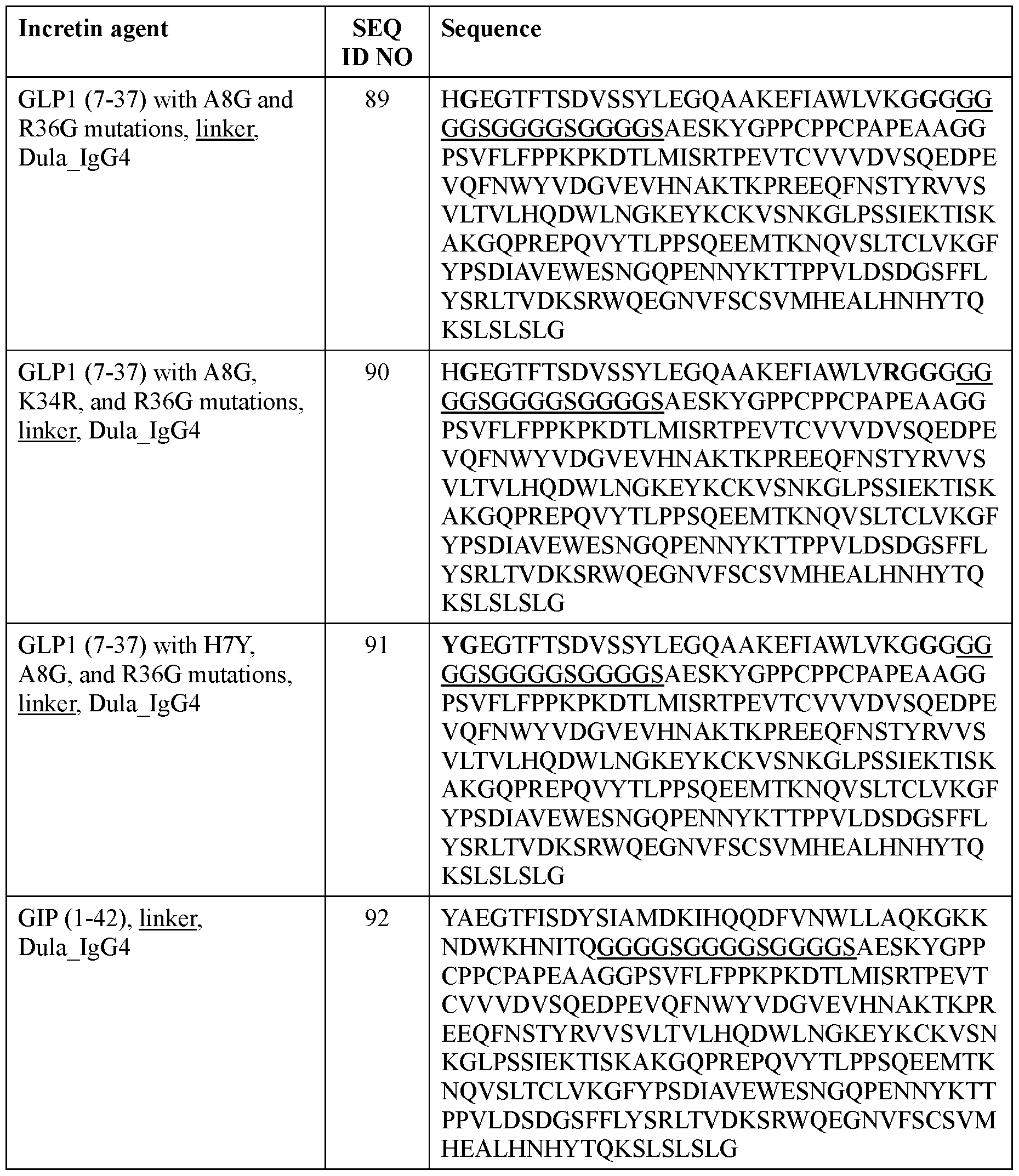
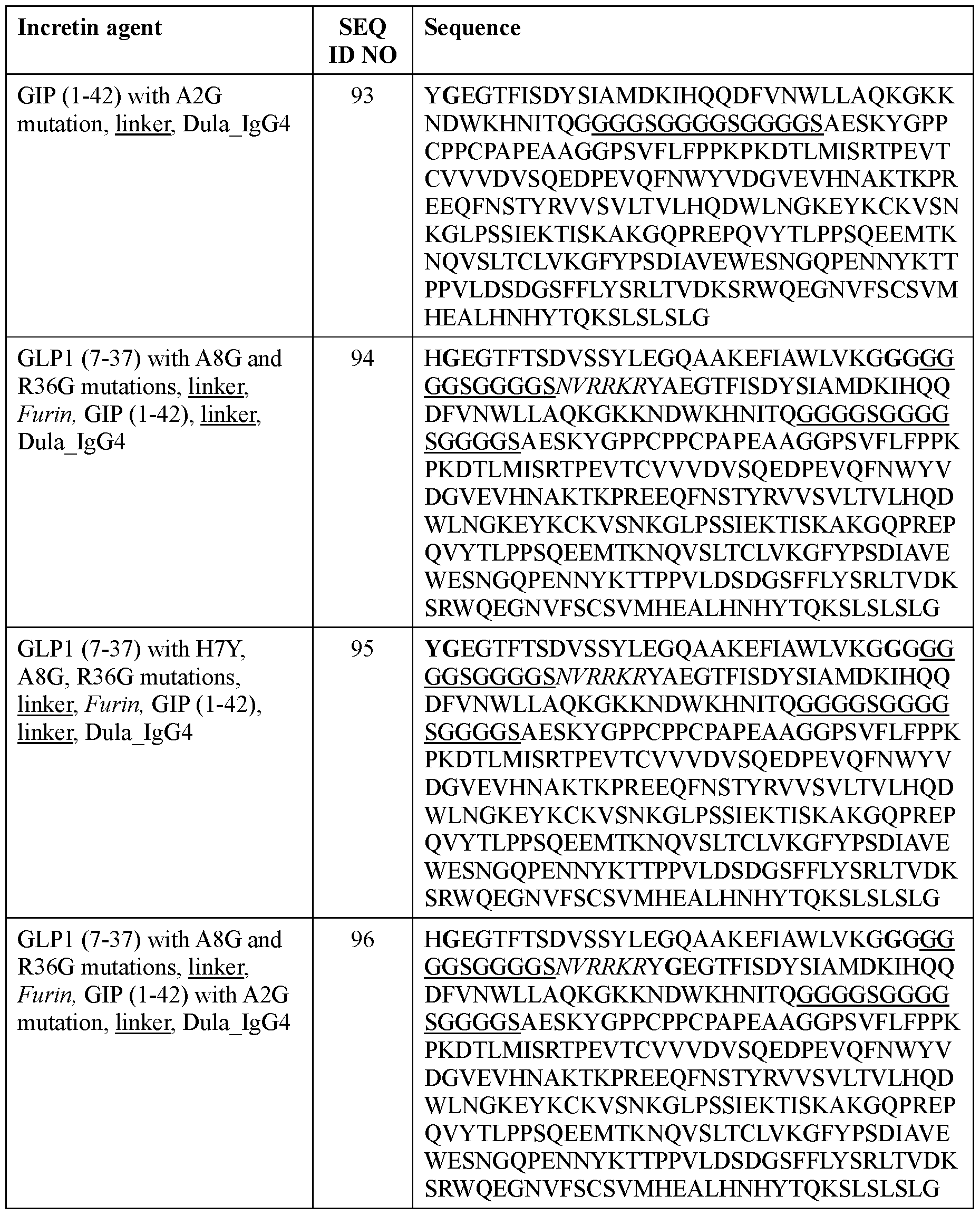
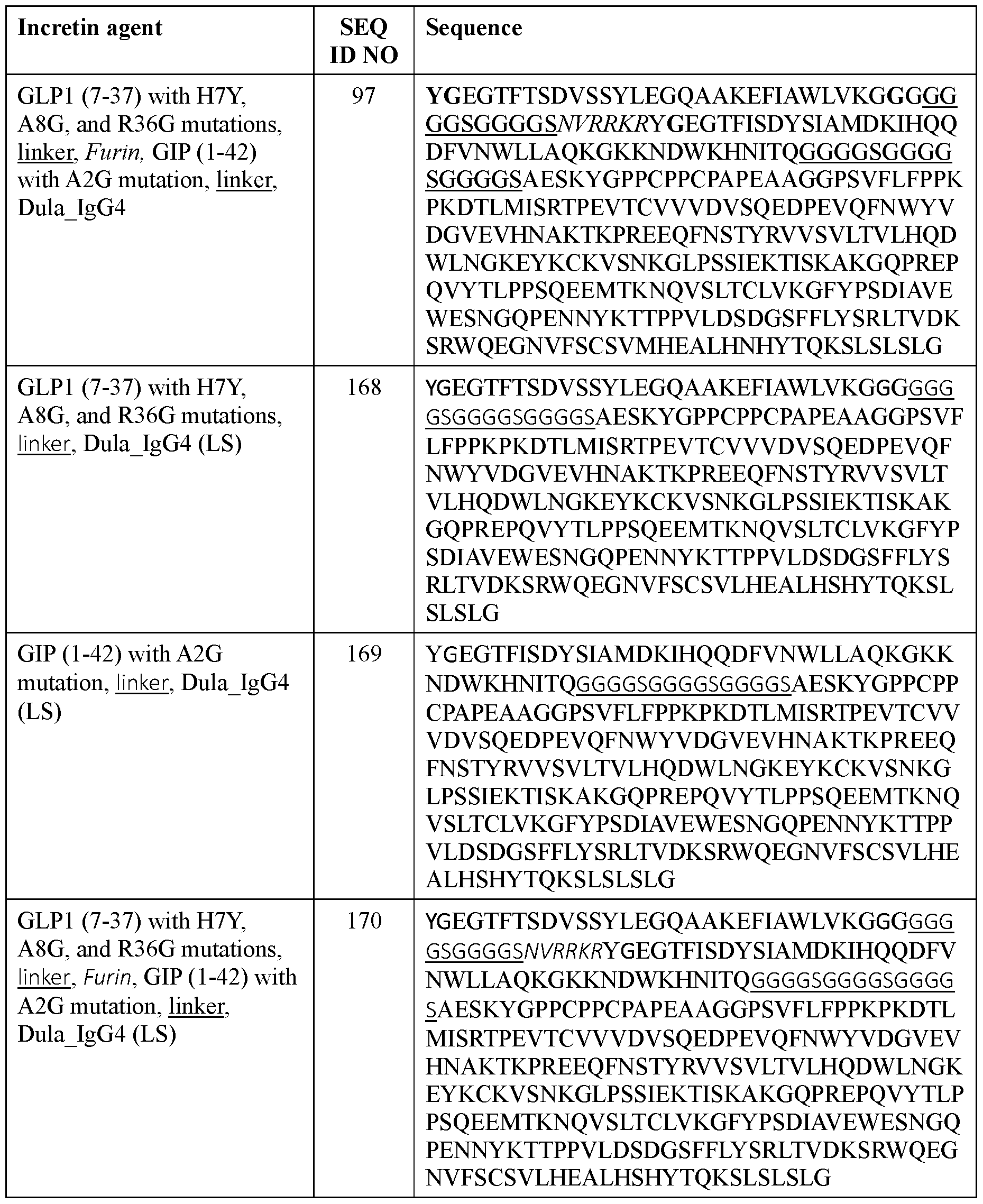

Mutations to ablate Fc effector function
[0220] In some embodiments, an Fc domain includes one more mutations in order to ablate effector function of the Fc domain included in an incretin agent. By ablating the Fc effector function of the incretin agent, delivery of the incretin agent will be less likely to cause an unwanted immune response, due to triggering cytotoxic and other effector activity by immune cells. In some embodiments, an Fc domain of a molecule includes one or more mutations that silence effector function.
[0221] In some embodiments, one or more mutations in an Fc domain include an “STR” modification, or a combination of mutations comprising L234S, L235T, and G236R,
according to the EU numbering scheme. When such mutations are introduced into an Fc domain, an Fc domain will show little or no detectable binding to Fey receptors or to Clq, and do not promote an inflammatory cytokine response (see e.g., Wilkinson et al., (2021) PLoS One 16.12: e0260954, which is herein incorporated by reference in its entirety). Exemplary incretin agent comprising an STR modification is shown in e.g., Figure 14.
[0222] In some embodiments, a modification that reduces or silences effector function of an Fc domain included in an incretin agent described herein comprises one or more of the following mutations: L234A, L235A, P329G, P329A, N297A, orN297D, according to the EU numbering scheme. In some embodiments, a modification that silences effector function of an Fc domain comprises the following mutated amino acid residues: L234A and L235A (“LALA”), according to the EU numbering scheme. In some embodiments, mutations used to ablate effector function of an Fc domain include the following: L234A/L235A/P329G (“LALAPG”), according to EU numbering. In some embodiments, a modification that silences effector function of an Fc domain comprises the following mutated amino acid residues: L234A, L235A, and P329A (“LALAPA”), according to the EU numbering scheme. In some embodiments, a modification that silences effector function of an Fc domain further comprises N297A, or N297D, according to EU numbering. In some embodiments, a modification that silences effector function of an Fc domain includes the following Fc mutations: L234A/L235A/P329G and N297A, according to EU numbering. In some embodiments, a modification that silences effector function of an Fc domain includes the following Fc mutations: L234A/L235A/P329G and N297D, according to EU numbering. In some embodiments, a modification that silences effector function of an Fc domain includes the following Fc mutations: L234A, L235A, and N297A, according to EU numbering. In some embodiments, a modification that silences effector function of an Fc domain includes the following Fc mutations: L234A, L235A, and N297D, according to EU numbering. In some embodiments, a modification that silences effector function of an Fc domain includes the following Fc mutations: L234A, L235A, P329A, and N297A, according to EU numbering. In some embodiments, a modification that silences effector function of an Fc domain includes the following Fc mutations: L234A, L235A, P329A, and N297D, according to EU numbering.
[0223] In some embodiments, a modification that reduces or silences effector function of an Fc domain included in an incretin agent described herein comprises the following
mutations: L234F/L235E/P331S (“FES”), according to the EU numbering scheme. In some embodiments, a modification that reduces or silences effector function of an Fc domain included in an incretin agent described herein comprises the following mutations: L234F/L235Q/K322Q (“FQQ”), according to the EU numbering scheme. In some embodiments, a modification that reduces or silences effector function of an Fc domain included in an incretin agent described herein comprises the following mutations: A330S/P331S, according to the EU numbering scheme.
[0224] One of ordinary skill in the art will appreciate that other modifications known in the art could be used in order to ablate effector function.
Heterodimeric Incretin Agents
[0225] In some embodiments, one or more polyribonucleotides described herein encode an incretin agent that comprises more than one incretin peptides on separate polypeptide chains. In some embodiments, separate polypeptide chains multimerize (e.g., dimerize). In some embodiments where an incretin agent comprises one or more incretin peptides on separate polypeptide chains, the separate polypeptide chains comprise two polypeptide chains that each comprise an immunoglobulin constant domain, and the two polypeptide chains dimerize via two constant domains that combine to make an Fc domain.
[0226] In some embodiments, an Fc domain within an incretin agent comprises one or more mutations to induce dimerization. For example, in some embodiments, an incretin agent comprises a first polypeptide chain that comprises an incretin peptide fused to the constant domain of an immunoglobulin, where the constant domain comprises one more mutations that induce dimerization with a second polypeptide chain that comprises an incretin peptide fused to the constant domain of an immunoglobulin. In some embodiments, the constant domain of the first and second polypeptides both contain one or more mutations to induce dimerization. In some embodiments, the incretin peptide or incretin peptides in the first and second polypeptides are different.
[0227] One approach to inducing dimerization is known as “knob-into-holes technology” (KIH), which aims to force the pairing of two different constant domains by introducing mutations into the CH3 domains to modify the contact interface. On one CH3 domain, bulky amino acids are replaced by amino acids with short side chains to create a
“hole” and amino acids with large side chains are introduced into the other CH3 domain, to create a “knob”. Co-expression of these two constant domains induces dimerization. In some embodiments, an Fc domain described herein utilizes KIH technology as described in, e.g., WO1998/050431, which is incorporated herein by reference in its entirety. As described herein, an Fc domain of an incretin agent may comprise certain mutations that utilize KIH technology that include, but are not limited to, a CH3 modification. In some embodiments, an Fc domain of an incretin agent comprises a CH3 domain comprising one or more of the following mutations: Y349C, T366S, L368A, and Y407V (according to EU numbering). In some embodiments, an Fc domain of an incretin agent comprises a CH3 domain, wherein the CH3 domain comprises each of the following mutations: Y349C, T366S, L368A, and Y407V (according to EU numbering). Such a combination of mutations is referred to herein as “FcKIH-b”. In some embodiments, an Fc domain of an incretin agent comprises a CH3 domain comprising one or more mutations selected from: S354C and T366W (according to EU numbering). In some embodiments, an Fc domain of an incretin agent comprises a CH3 domain comprising each of the following mutations: S354C and T366W (according to EU numbering). Such a combination of CH3 mutations is referred to herein as “FcKIH-a”. In some embodiments, an incretin agent comprises an Fc domain that includes an FcKIH-a sequence and an FcKIH-b sequence.
[0228] In some embodiments, an Fc domain within an incretin agent comprises one or more “KiH” mutations and an LS mutation.
[0229] Accordingly, in some embodiments, an incretin agent encoded by one or more polyribonucleotides as described herein comprises one or more incretin peptides fused to an Fc domain, where the CH3 domain of the Fc domain comprises one or more of the following mutations: Y349C, T366S, L368A, and Y407V (according to EU numbering). In some embodiments, an incretin agent encoded by one or more polyribonucleotides as described herein comprises one or more incretin peptides fused to an Fc domain, where the Fc domain comprises a CH3 domain comprising one or more mutations selected from: S354C and T366W (according to EU numbering).
[0230] In some embodiments, an incretin agent comprises a heterodimer, e.g., as shown in Figure 13 or Figure 14. Figure 13 shows an exemplary design of two polypeptide chains that include an incretin peptide fused to an Fc domain. In each polypeptide chain
(incretin-Fc fusion), there is a signal peptide (SP) and one, two or four incretin peptides (I: lx, I:2x, or I:4x) fused to an Fc domain and each incretin agent includes an Fc mutation that induces heterodimerization (e.g., a knob-in-hole mutation). The Fc domains could also include modifications to ablate effector function and/or to increase half life as described herein. When one or more polyribonucleotides encoding the two polypeptide chains (top) in Figure 13 are expressed, the two polypeptide chains associate and form a heterodimer incretin agent (bottom). An exemplary incretin agent according to the design shown in Figure
13 is shown in Figure 14. Specifically, each polypeptide chain of the incretin agent in Figure
14 has a signal peptide (SP), a GLP1 or GIP incretin peptide, a linker (GGGS)i, and an Fc domain. One or both of the Fc domains contain “LS” mutations (M428L/N434S) to extend half-life of the incretin agent, “STR” mutations to silence Fc effector function, and “knob-in- hole” mutations to promote heterodimerization. In some embodiments, any mutations described herein may be included instead of or in addition to the LS and/or STR mutations. When the two peptide chains are expressed, they associate to form a heterodimeric structure that contains two peptide chains with different incretin peptides. SP cleavage sites within the incretin agent are indicated with arrows.
[0231] In some embodiments, an incretin agent comprises two polypeptide chains that associate and form a heterodimer incretin agent wherein one of the polypeptide chains comprises the sequence of SEQ ID NO: 300 (DKTHTCPPCPAPESTRGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNW YVDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIE KTISKAKGQPREPQVYTLPPCREEMTKNQVSLWCLVKGFYPSDIAVEWESNGQPENN YKTTPPVLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVLHEALHSHYTQKSLSLSPGK) (“FcKIH-a (LS and STR)”) and the other polypeptide chain comprises the sequence of SEQ ID NO:
301 (DKTHTCPPCPAPESTRGPS VFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFN WYVDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKALPAP IEKTISKAKGQPREPQVCTLPPSREEMTKNQVSLSCAVKGFYPSDIAVEWESNGQPEN NYKTTPPVLDSDGSFFLVSKLTVDKSRWQQGNVFSCSVLHEALHSHYTQKSLSLSPG K) (“FcKIH-b (LS and STR)”).
[0232] In some embodiments, a polyribonucleotide described herein encodes an incretin agent comprising an amino acid sequence that is at least 85%, 86%, 87%, 88%, 89%,
90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or at least 99% identical to any one of the incretin agent sequences in Table 9. In some embodiments, an incretin agent comprises any one of the incretin agents detailed in Table 9 below, or combinations or variants thereof.
Table 9: Exemplary Incretin Agents including an FcKIH-a or FcKIH-b domain, that form heterodimers (with mutations in bold, linkers underlined)
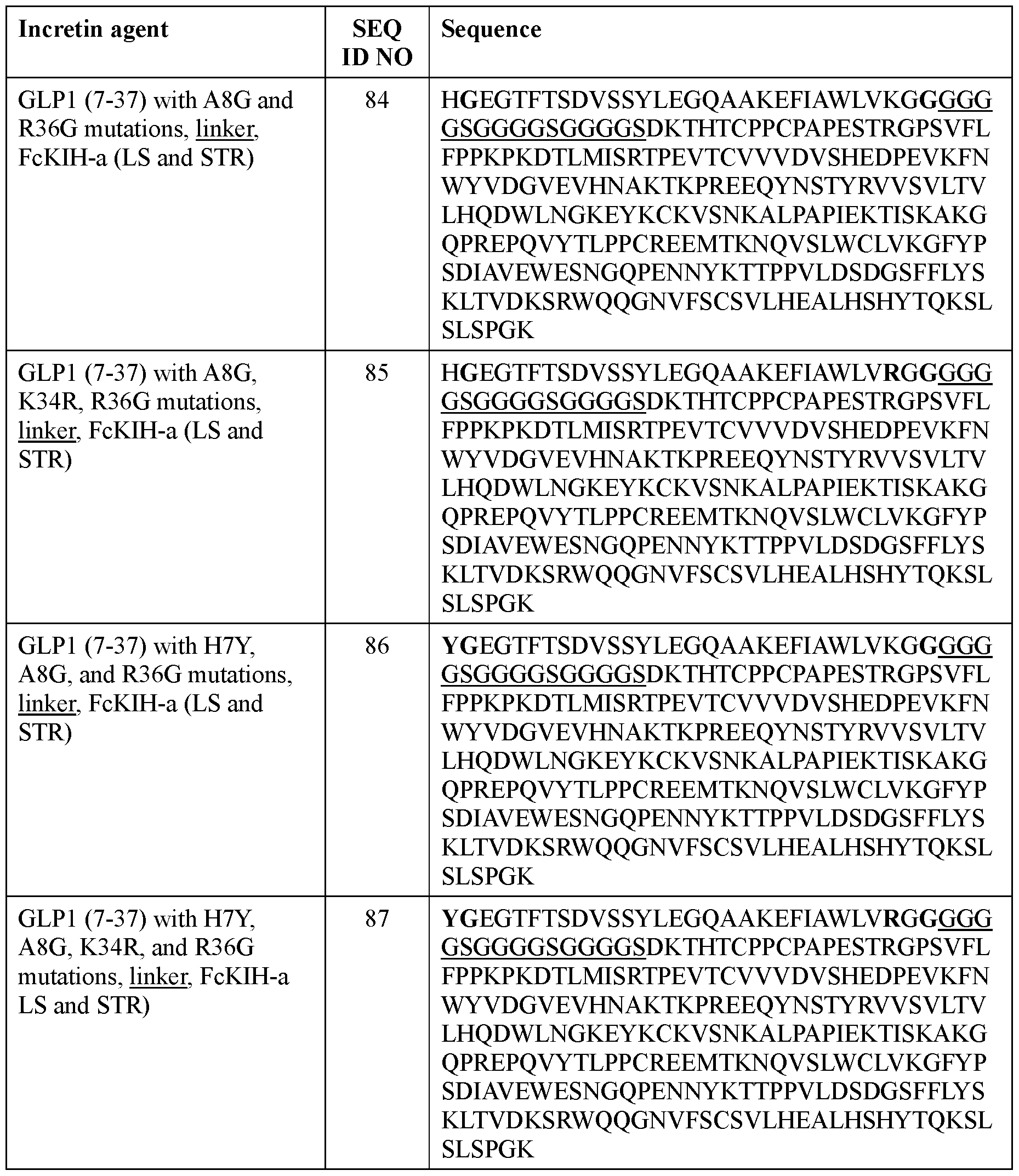
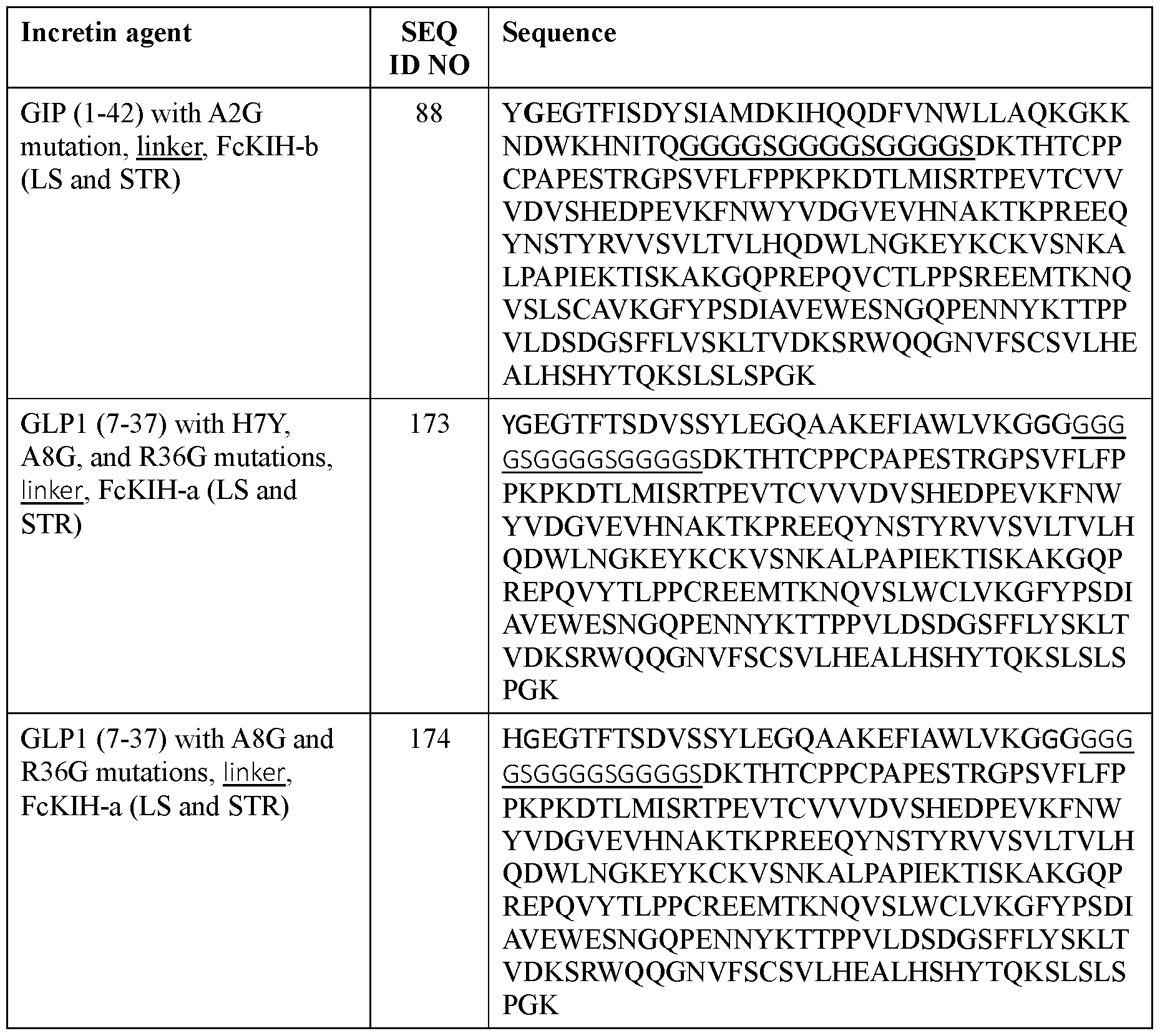
[0233] In some embodiments any one of the exemplary incretin agents that includes an FcKIH-a domain can be combined with the exemplary incretin agent that includes an FcKIH-b domain, e.g., in some embodiments an incretin agent of SEQ ID NO: 84, 85, 86, 87, 173 or 174 can be combined with an incretin agent of SEQ ID NO: 88.
Signal peptides
[0234] According to certain embodiments, a signal peptide is fused, either directly or through a linker, to an encoded incretin peptide described herein. In some embodiments, an open reading frame of a polyribonucleotide described herein encodes an incretin agent with a signal peptide, e.g., that is functional in mammalian cells.
[0235] In some embodiments, signal peptides are sequences, which are typically characterized by a length of about 15 to 30 amino acids. In many embodiments, signal peptides are positioned at the N-terminus of an incretin agent, without being limited thereto. In some embodiments, signal peptides preferably allow the transport of an incretin agent encoded by polyribonucleotides of the present disclosure with which they are associated into a defined cellular compartment, preferably the cell surface, the endoplasmic reticulum (ER) or the endosomal-lysosomal compartment.
[0236] In some embodiments, a ribonucleic acid sequence encoding a signal peptide allows an incretin agent encoded by the polyribonucleotide to be secreted upon translation by cells, e.g., present in a subject, thus yielding a plasma concentration of a biologically active incretin agent.
[0237] In some embodiments, a ribonucleic acid sequence encoding a signal peptide included in a polyribonucleotide consists of or comprises a nucleotide sequence that encodes a human signal peptide. In some embodiments, a ribonucleic acid sequence encoding a secretion signal included in a polyribonucleotide consists of or comprises a nucleotide sequence that encodes a non-human secretion signal. In some embodiments, a signal peptide may be or comprises a viral signal peptide. In some embodiments, such a signal peptide may be or comprises the amino acid sequence of MRVLVLLACLAAASNA (SP1-2; SEQ ID NO: 17). In some embodiments, a signal peptide may be or comprises an amino acid sequence of MRVMAPRTLILLLSGALALTETWA (husec signal peptide delta GS; SEQ ID NO: 65).
[0238] In some embodiments, a signal peptide sequence is selected from those included in the Table 10 below, or a fragment or variant thereof:
Table 10: Exemplary Signal Peptides

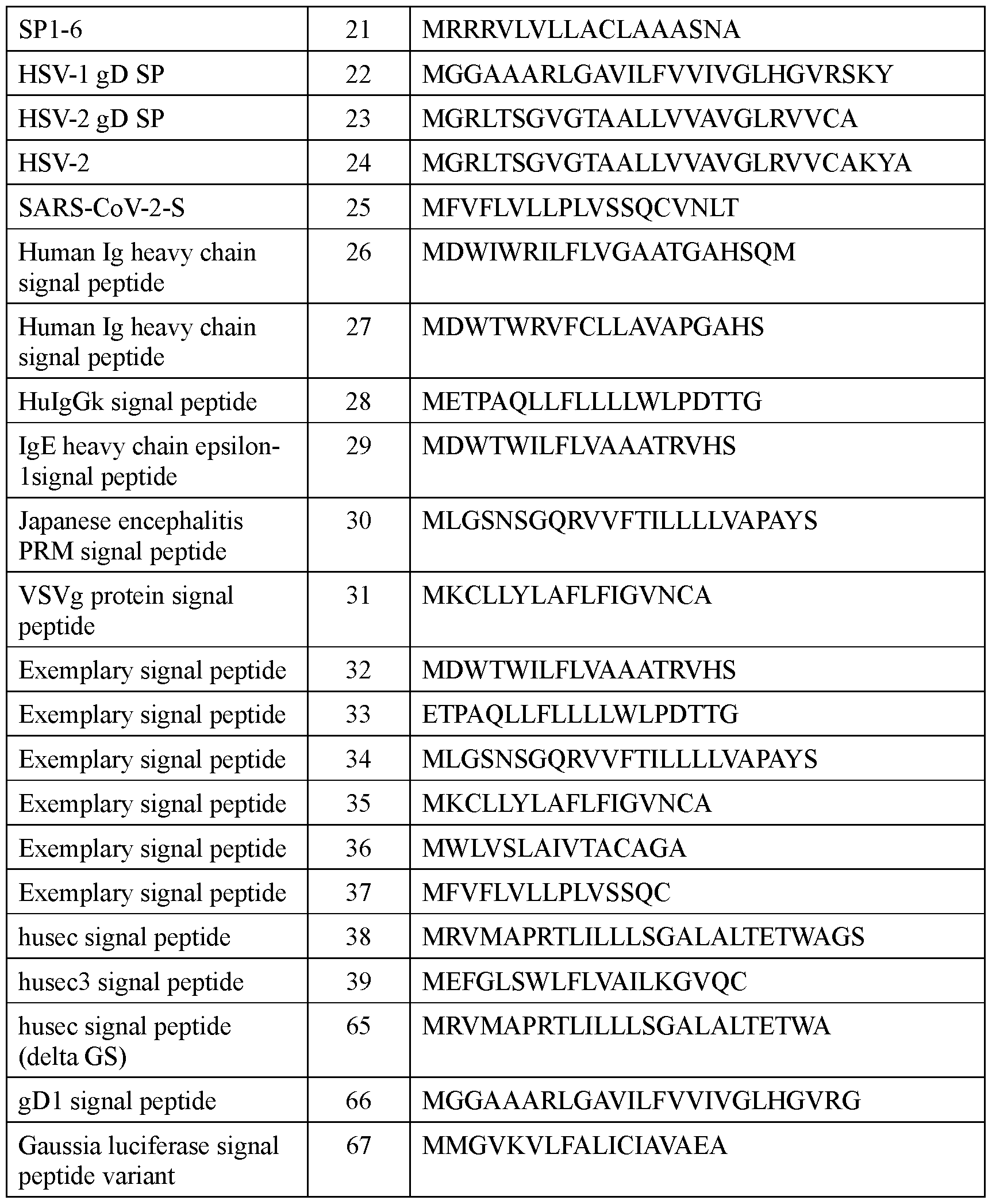
[0239] The present disclosure recognizes, among other things, that selection of a signal peptide may be important for prediction of the cleavage site between a signal peptide and an incretin peptide. In order for polyribonucleotides to deliver and express incretin peptides, where the expressed incretin peptides maintain the proper function and bioactivity, a signal peptide is, in some embodiments, selected and included in incretin agents to effect proper cleavage of the incretin into mature form.
[0240] Without wishing to be bound by any theory, the site of cleavage of the signal peptide and type or sequence of the signal peptide in the context of polyribonucleotides encoding incretin agents described herein may be important to ensure that the N-terminal end of the incretin peptide is processed correctly. Signal peptides may contain particular sequences or structure that leads to alternative processing or cleavage sites, ultimately changing the final amino acid sequence of the mature incretin peptide. In such relatively small peptides such as GLP1 or GIP (or variants thereof, and other peptides of similar size/properties), a change in amino acid residue could impact bioactivity of the peptide. In some embodiments, a signal peptide is chosen for inclusion in incretin agents described herein in order to facilitate proper cleavage of the N-terminal end of the incretin peptide, or in other words, create a “scarless” N-terminal end of the incretin peptide, in order to maintain the incretin peptide’s bioactivity. Figure 20 and Figure 21 show a schematic of where the theoretical cleavage sites of certain exemplary signal peptides lie with the incretin agent. Figure 20 indicates that A8G mutation facilitates correct N-terminal processing of GLP1 incretin agents with a husec signal peptide. Figure 21 indicates that the A2G mutation facilitates correct N-terminal processing of GIP incretin agents with a husec signal peptide.
[0241] Such a concept and utilization of a particular signal peptides to facilitate cleavage to generate a mature peptide with a scarless N-terminus could also be applied to other gut peptides (e.g., glucagon) and/or other peptides of comparable size/properties as GLP1 and GIP described herein. This may be important in the context of delivering incretin agents (or other similar peptides) as one or more polyribonucleotides encoding the incretin agents. Such delivery requires the proper translation of the protein within a cell, in addition to the post-translational processing, including proper cleavage of a peptide post-translation. Incretin agents comprising one or more incretin peptides fused to another peptide described herein may be designed and generated so that signal peptide cleavage is accurate and does not affect the amino acid sequence of the mature peptide (i.e., creates a “scarless” N terminal). A scarless N-terminal of incretin peptides (and other similar peptides, e.g., other gut peptides, e.g., glucagon), allows for the proper functionality of the peptide after it is processed into a mature peptide.
[0242] In some embodiments, a polyribonucleotide comprises, in a 5’ to 3’ direction a signal peptide -coding sequence and one or more incretin peptide -coding sequences, in some embodiments, a signal peptide-coding sequence and one or more incretin peptide-codmg
sequences encode any one of the sequences as shown in Table 11 below, or a sequence that is at least 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or at least 99% identical to any one of the sequence shown in Table 11 below.
Table 11 : Exemplary Incretin Agents where x2 and x4 examples include a linker and Furin cleavage site in between each repeat unit

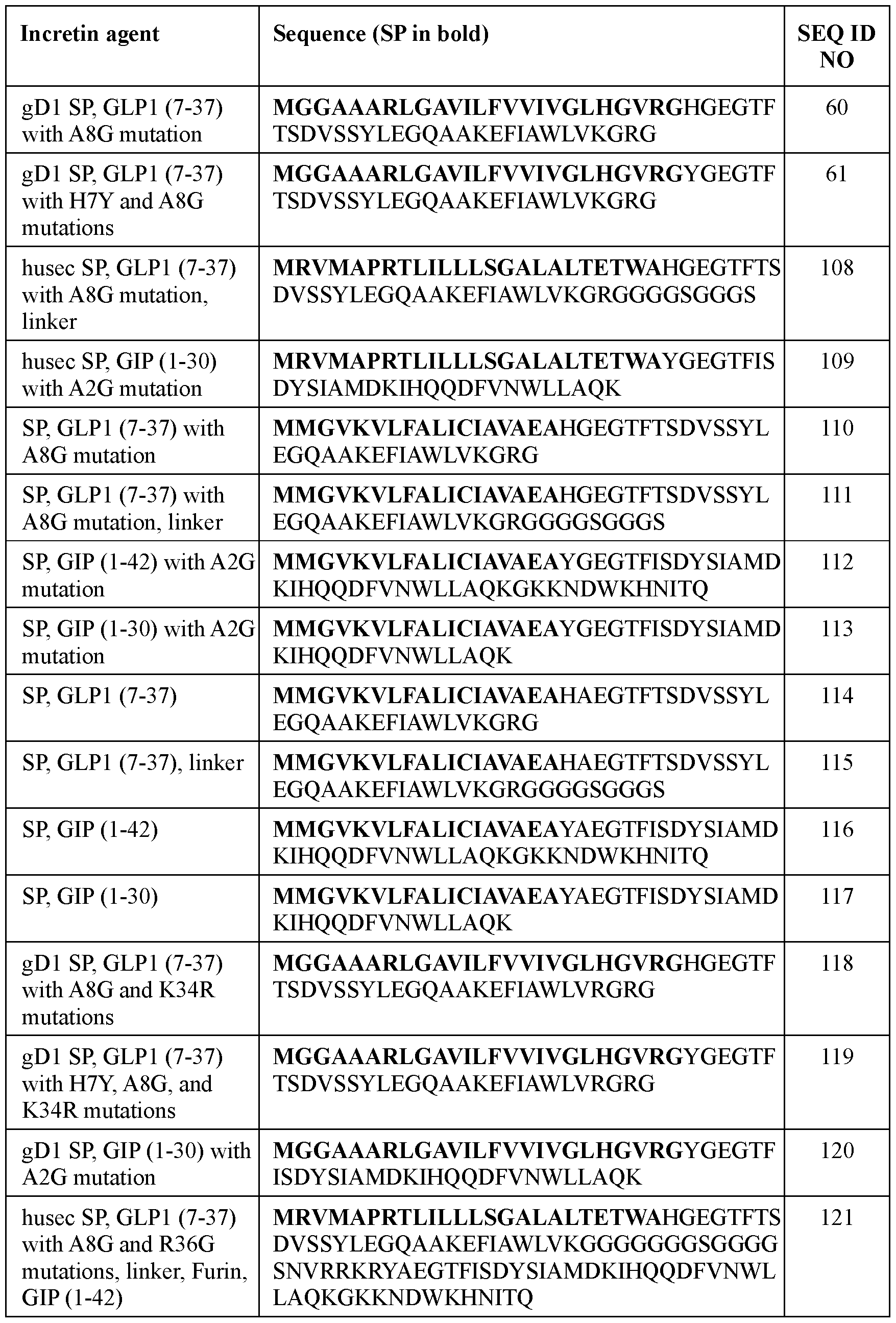

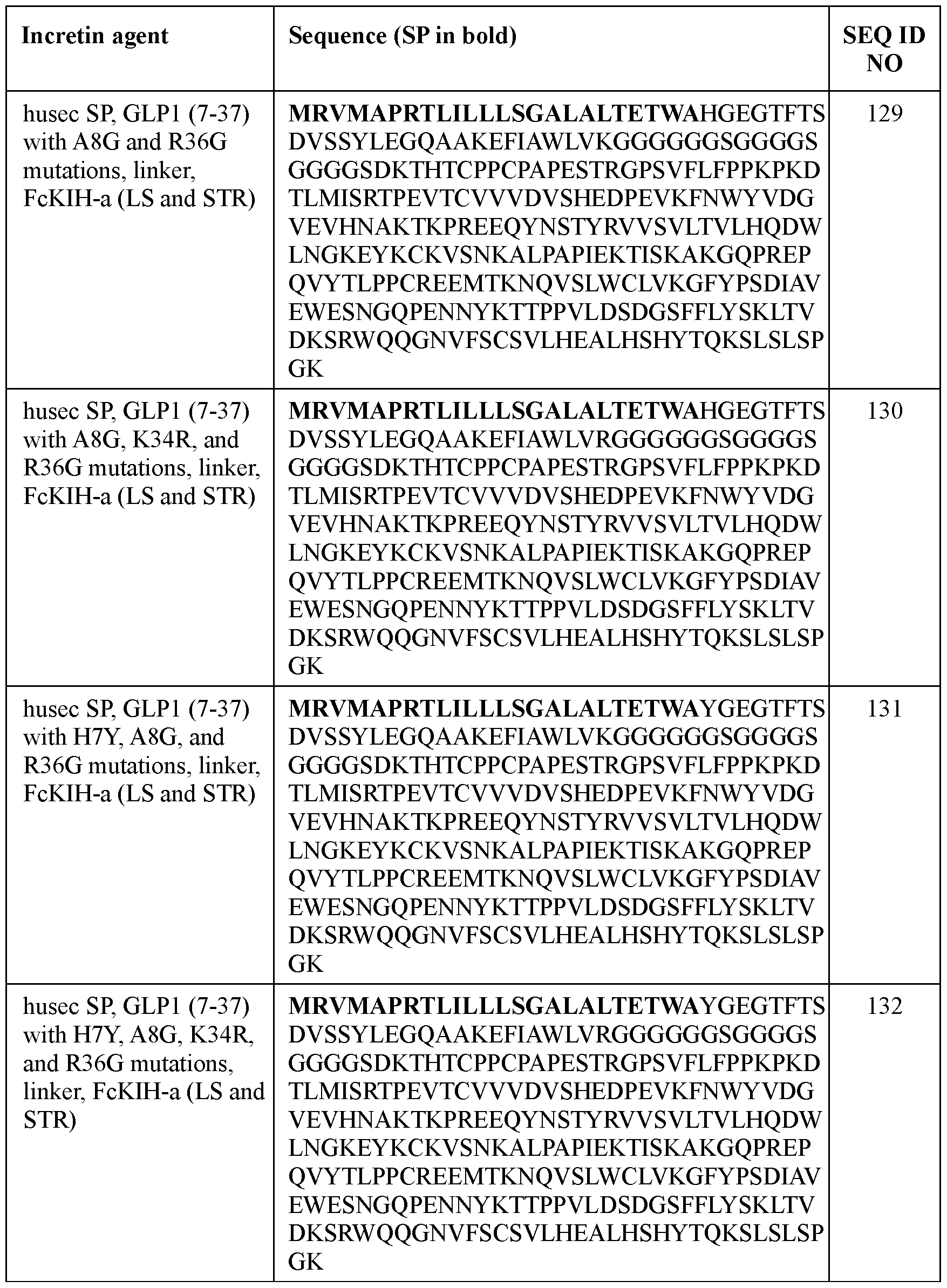
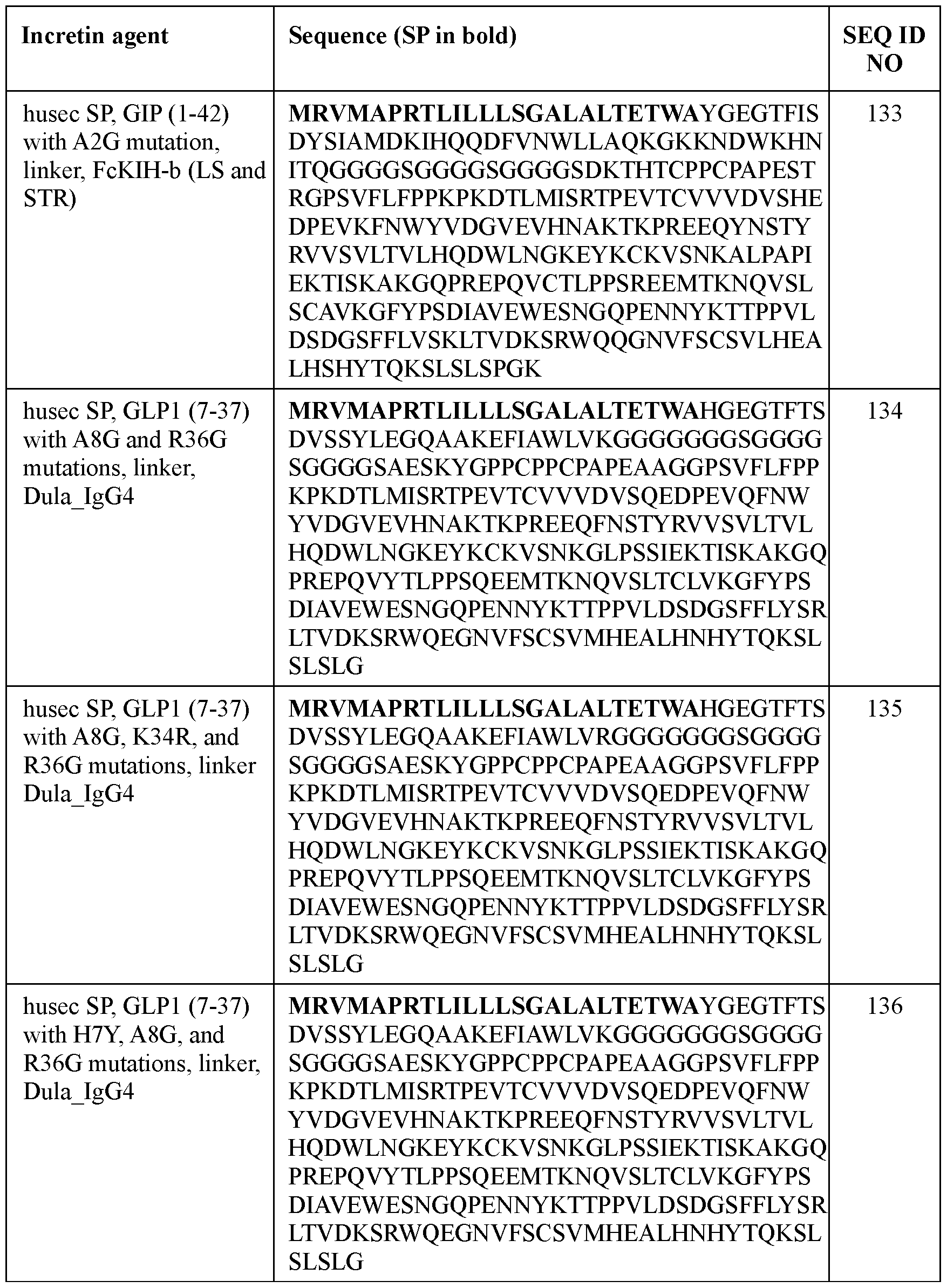
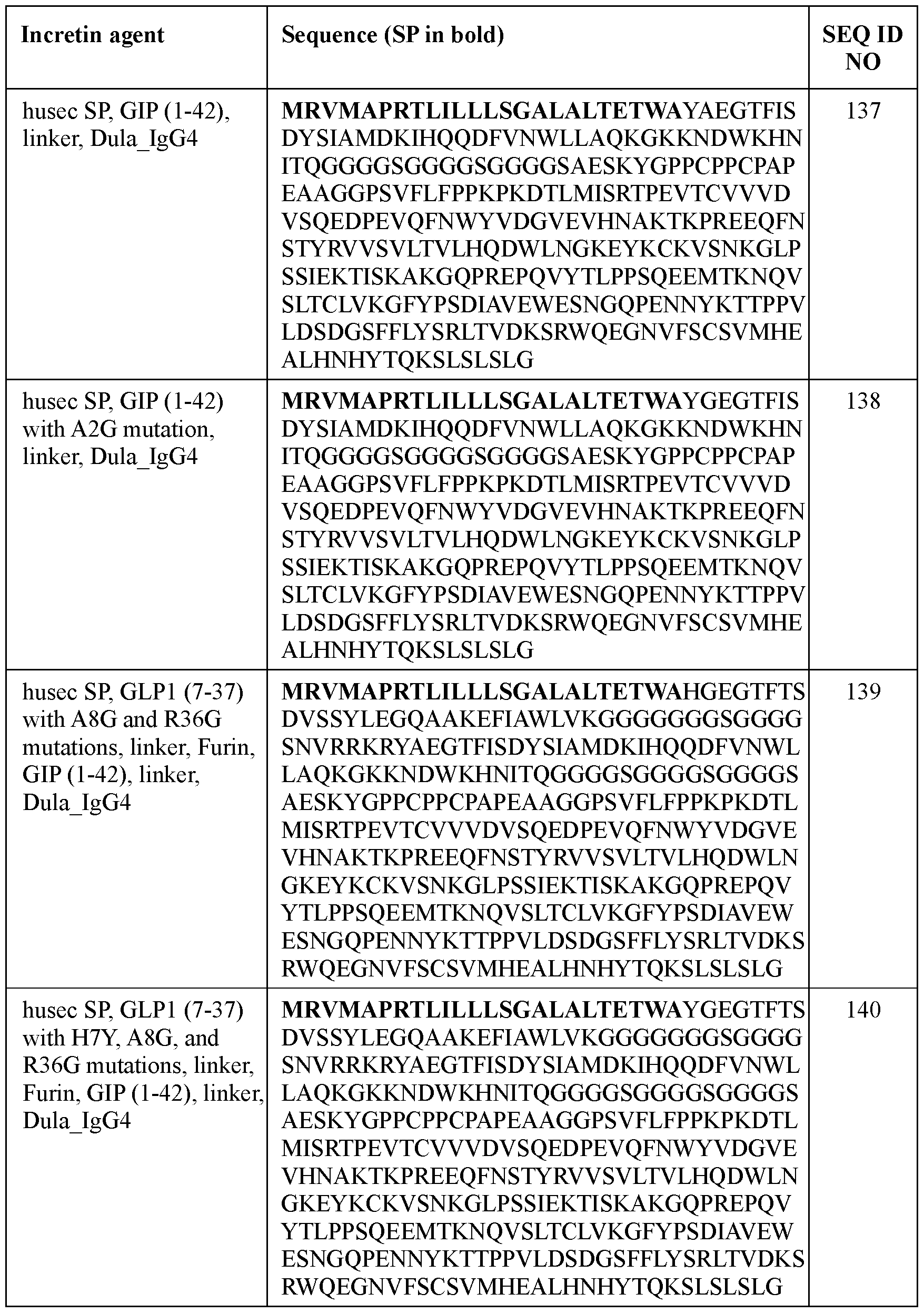

I
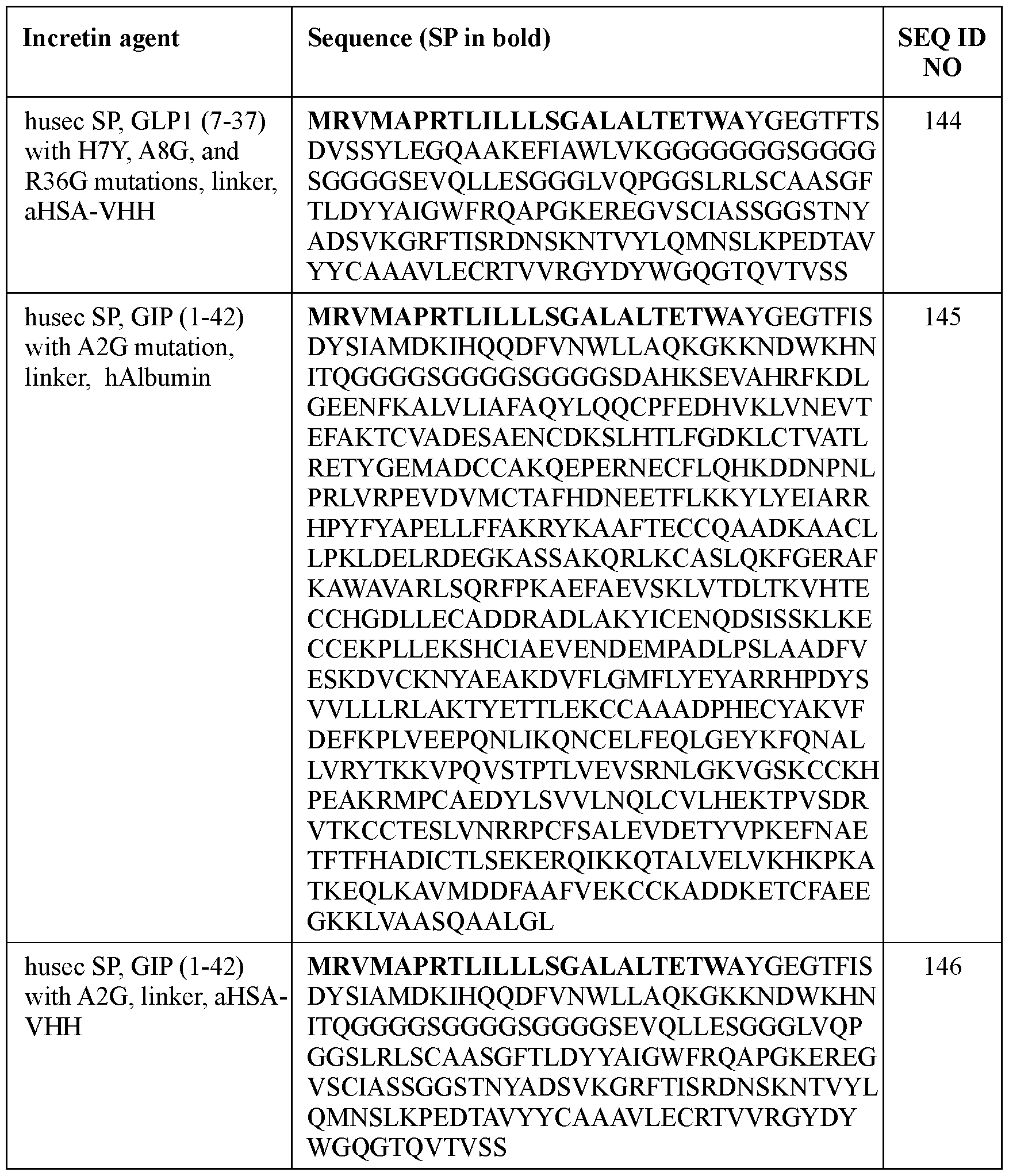
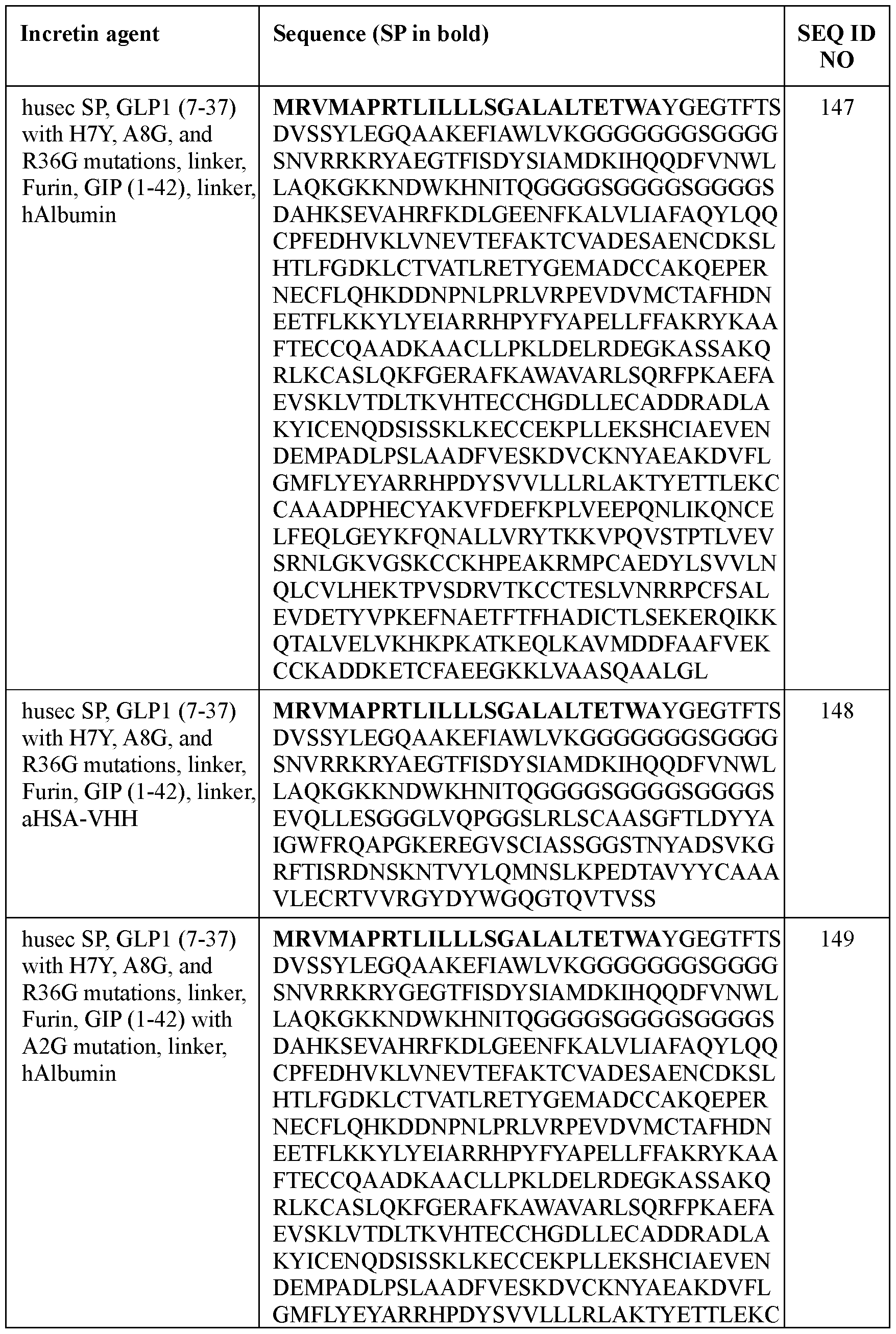

I
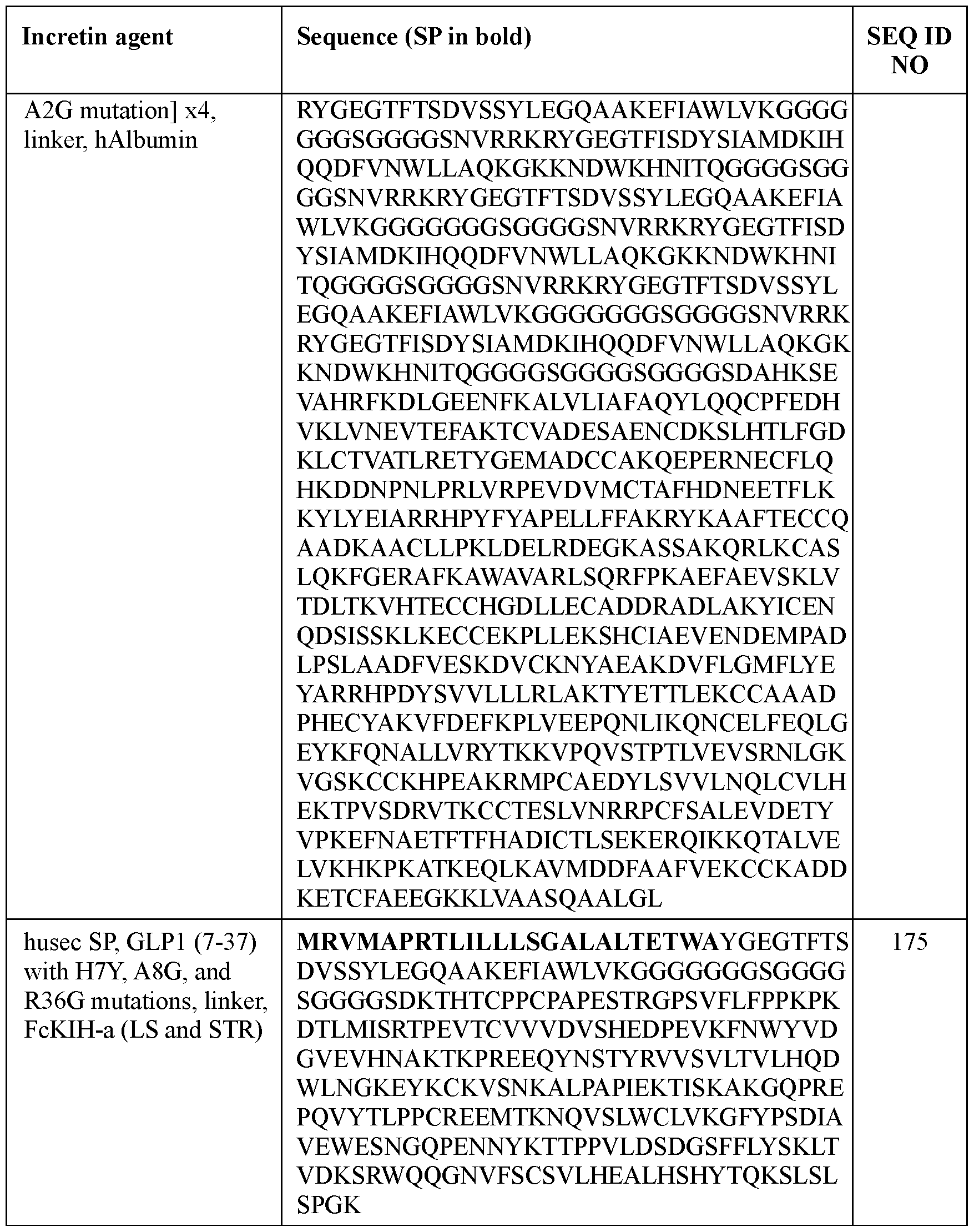 Incretin agent Sequence (SP in bold) SEQ ID
Incretin agent Sequence (SP in bold) SEQ ID
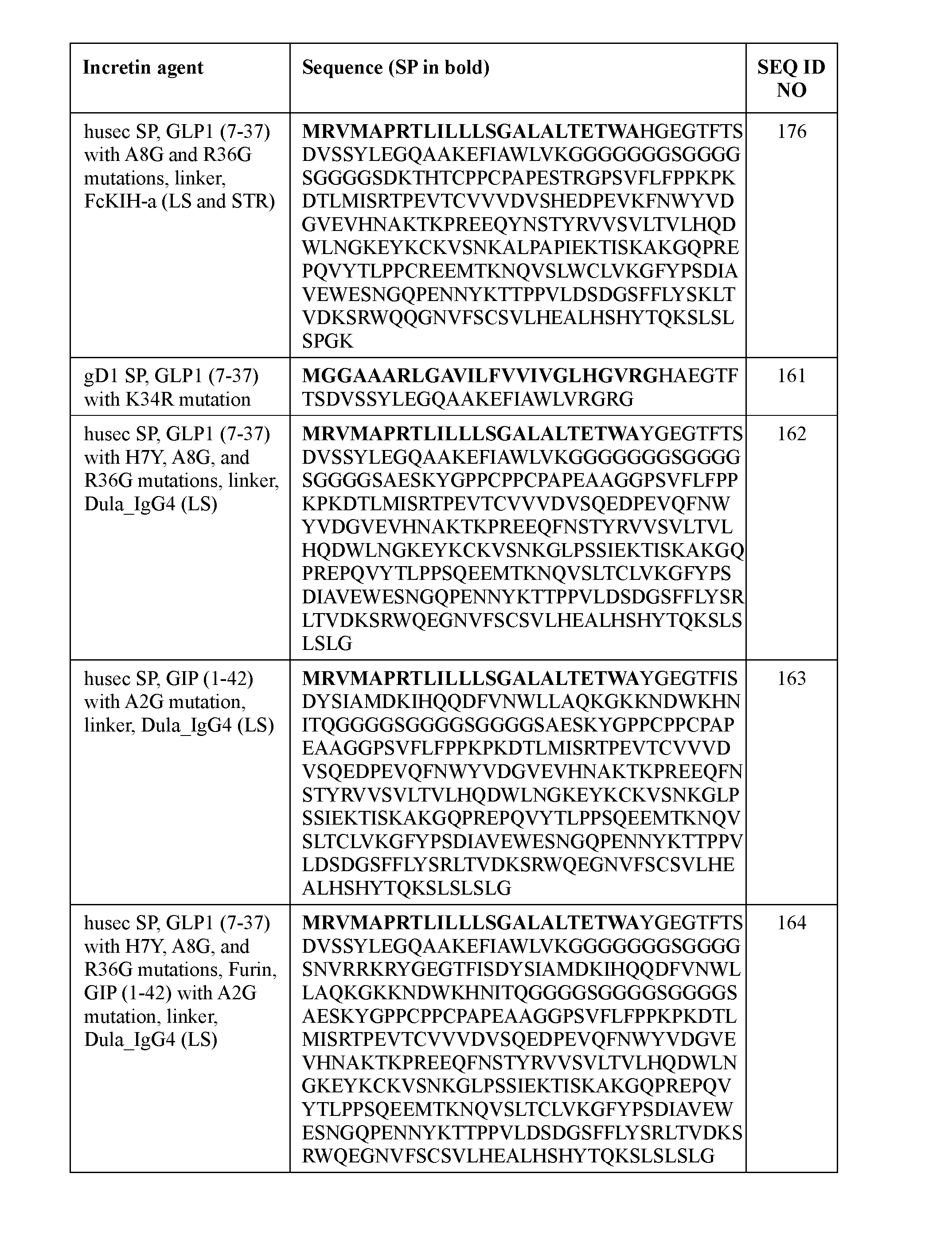

[0243] In some embodiments, a polyribonucleotide comprises, in a 5’ to 3' direction a signal peptide-coding sequence; an incretin peptide-coding sequence; a linker-coding sequence; and a half-life extending moiety-coding sequence. In some embodiments, a polyribonucleotide comprises, in a 5’ to 3“ direction a signal peptide-coding sequence; a halflife extending moiety-coding sequence: a linker-coding sequence; and an incretin peptide- coding sequence.
[0244] In some embodiments, a polyribonucleotide comprises, in a 5’ to 3? direction a signal peplide-coding sequence and one or more incretin peptide-coding sequences, each independently separated by a linker-coding sequence and a protease cleavage site-coding sequence, e.g., a farm cleavage site-coding sequence. In some such embodiments, one or more of the incretin peptide-coding sequences is preceded or followed by a linker-coding sequence and a half-lite extending moiety-coding sequence.
Exemplary polyribonucleotides encoding Incretin Agents
[0245] In some embodiments, a polyribonucleotide comprises a ribonucleic acid sequence that is at least 80%, 85%, 90%, 91 %, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or at least 99% identical to any of the sequences shown below in Table 12.
Table 12: Exemplar}' Polyribonucleotides encoding Incretin Agents where x2 and x4 examples include a linker and Furin cleavage site in between each repeat unit
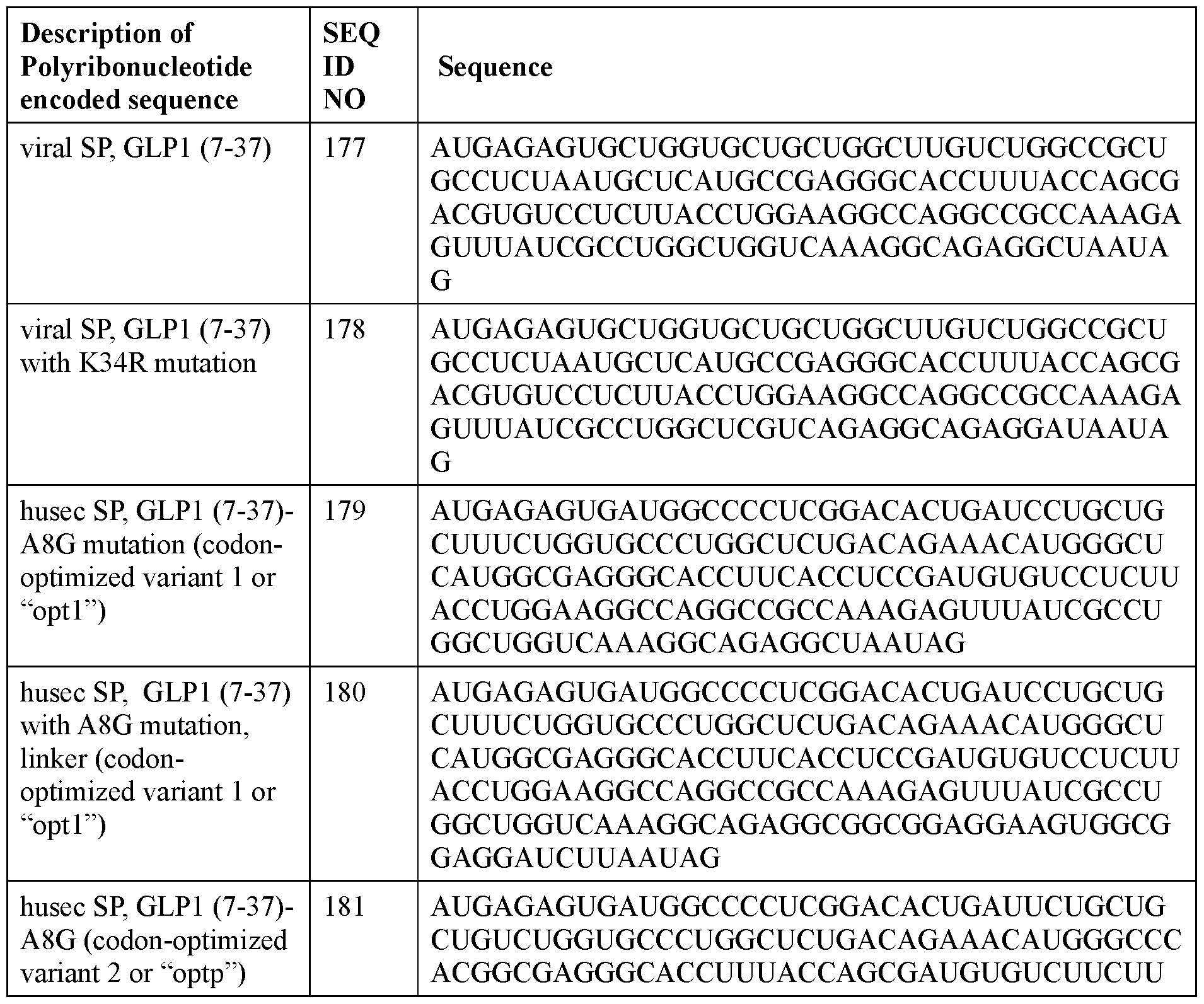
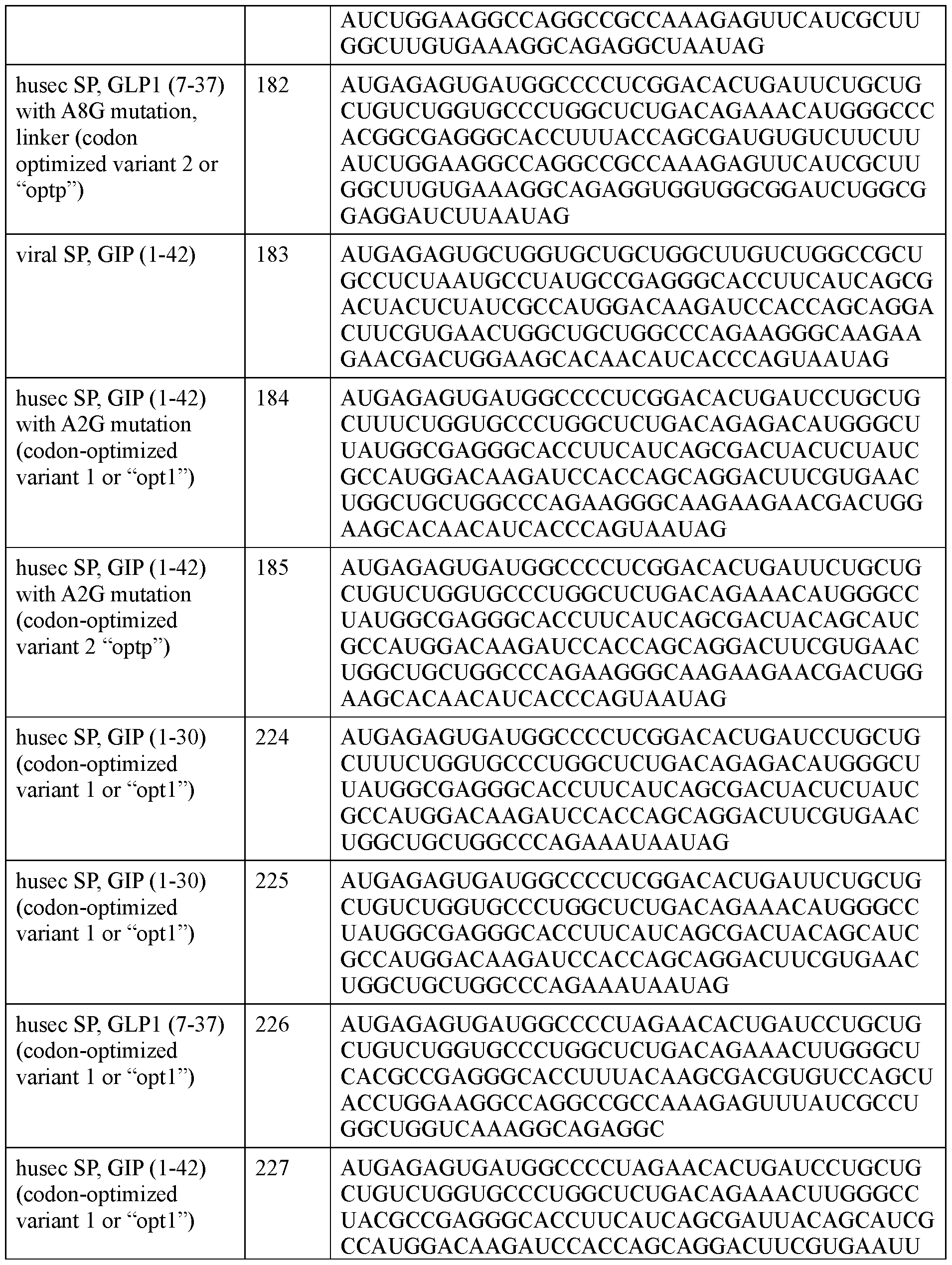
I
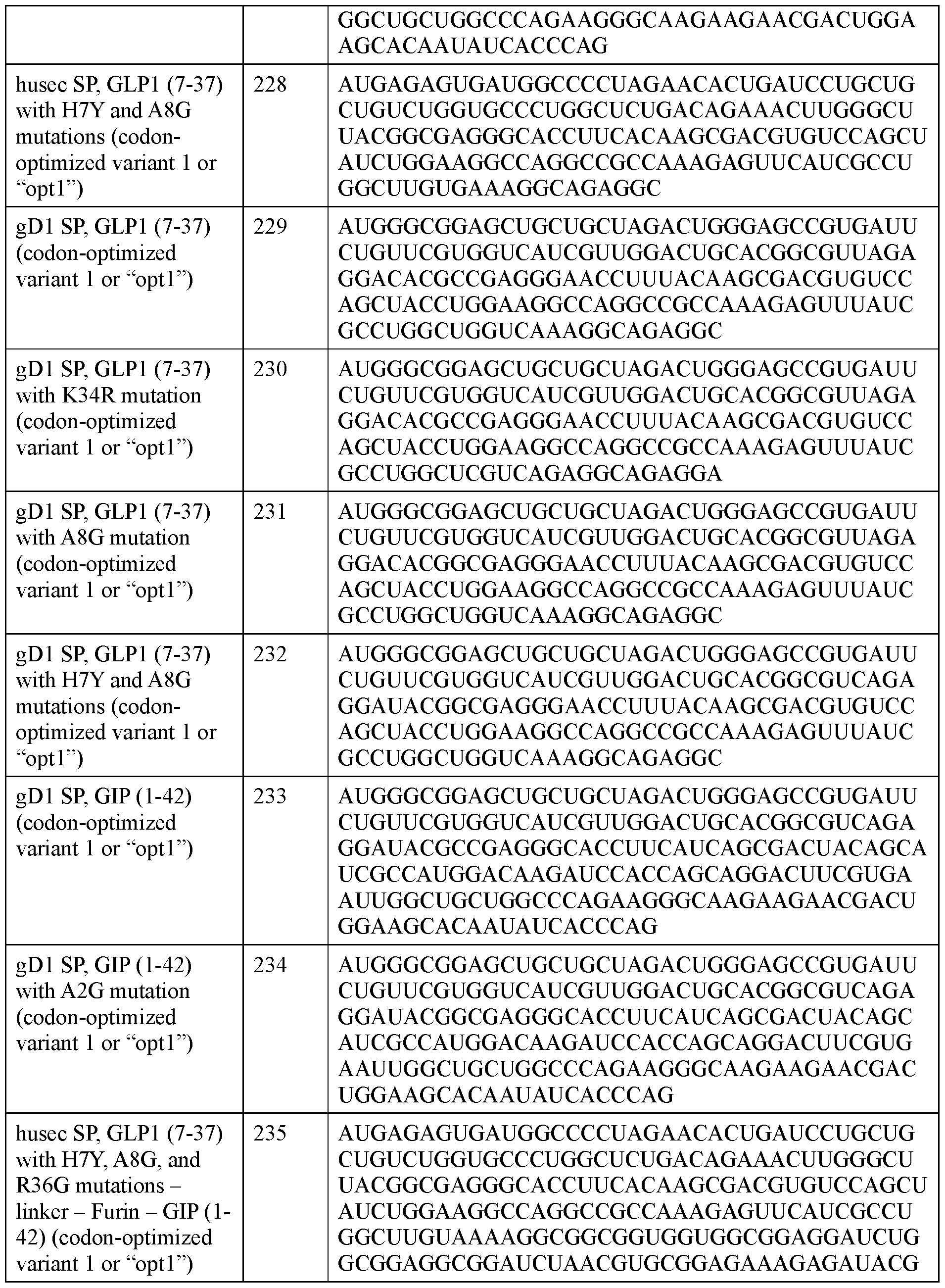
I
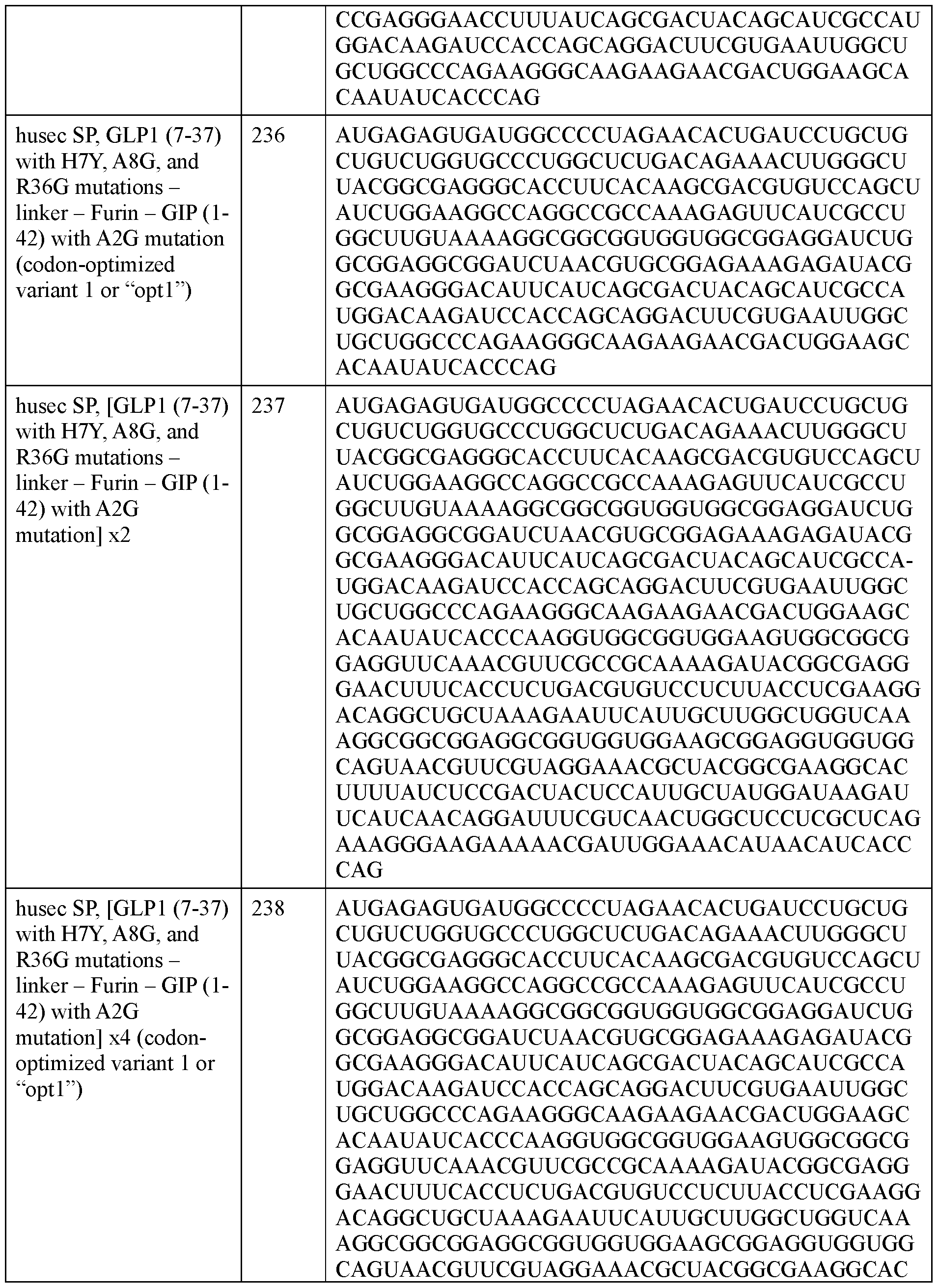
]

]

]

]

]

]

]

]

]

]

I

I

I

I

I

I

Exemplary polyribonucleotide Features
[0246] Polyribonucleotides described herein encode an incretin agent as described herein. Additionally, polyribonucleotides described herein, in some embodiments, include encode other elements such as a signal peptide. In some embodiments, polyribonucleotides described herein can comprise a nucleotide sequence that encodes a 5’UTR and/or a 3’ UTR. In some embodiments, polynucleotides described herein can comprise a nucleotide sequence that encodes a polyAtail. In some embodiments, polyribonucleotides described herein may comprise a 5’ cap, which may be incorporated during transcription, or joined to a polyribonucleotide post-transcription.
5’ Cap
[0247] A structural feature of mRNAs is a cap structure at the 5 ’-end. Natural eukaryotic mRNA comprises a 7-methylguanosine cap linked to the mRNA via a 5 ' to 5 '- triphosphate bridge resulting in capO structure (m7GpppN). In most eukaryotic mRNA and some viral mRNA, further modifications can occur at the 2 ’-hydroxy-group (2 ’-OH) (e.g., the 2 ’-hydroxyl group may be methylated to form 2’-0-Me) of the first and subsequent nucleotides producing “capl” and “cap2” five-prime ends, respectively. Diamond et al., (2014) Cytokine & growth Factor Reviews, 25:543-550 reported that capO-mRNA cannot be translated as efficiently as capl -mRNA in which the role of 2’-0-Me in the penultimate position at the mRNA 5’ end is determinant. Lack of the 2’-0-Me has been shown to trigger innate immunity and activate IFN response. Daffis et al., (2010) Nature, 468 :452-456 ; and Ziist et al. (2011) Nature Immunology, 12 : 137-143.
[0248] RNA capping is well researched and is described, e.g., in Decroly et al., (2012) Nature Reviews 10: 51-65; and in Ramanathan et al., (2016) Nucleic Acids Res; 44(16): 7511-7526, the entire contents of each of which is hereby incorporated by reference. For example, in some embodiments, a 5’ cap structure which may be suitable in the context of the present invention is a capO (methylation of the first nucleobase, e.g. m7GpppN), capl (additional methylation of the ribose of the adjacent nucleotide of m7GpppN), cap2 (additional methylation of the ribose of the 2nd nucleotide downstream of the m7GpppN), cap3 (additional methylation of the ribose of the 3rd nucleotide downstream of the m7GpppN), cap4 (additional methylation of the ribose of the 4th nucleotide downstream of the m7GpppN), ARCA (“anti-reverse cap analogue”), modified ARCA (e.g., phosphorothioate modified ARCA), inosine, N1 -methyl -guanosine, 2’-fluoro-guanosine, 7- deaza-guanosine, 8-oxo-guanosine, 2-amino-guanosine, LNA-guanosine, and 2-azido- guanosine.
[0249] The term “5 ’-cap” as used herein refers to a structure found on the 5 ’-end of an RNA, e.g., mRNA, and generally includes a guanosine nucleotide connected to an RNA, e.g., mRNA, via a 5’- to 5 ’-triphosphate linkage (also referred to as Gppp or G(5’)ppp(5’)). In some embodiments, a guanosine nucleoside included in a 5 ’ cap may be modified, for example, by methylation at one or more positions (e.g., at the 7-position) on a base (guanine), and/or by methylation at one or more positions of a ribose. In some embodiments, a guanosine nucleoside included in a 5’ cap comprises a 3’0 methylation at a ribose (3’0meG). In some embodiments, a guanosine nucleoside included in a 5’ cap comprises methylation at the 7-position of guanine (m7G). In some embodiments, a guanosine nucleoside included in a 5’ cap comprises methylation at the 7-position of guanine and a 3’ O methylation at a ribose (m7(3’OmeG)). It will be understood that the notation used in the above paragraph, e.g., “(m27’3 ’°)G” or “m7(3’OmeG)”, applies to other structures described herein.
[0250] In some embodiments, providing an RNA with a 5 ’-cap disclosed herein may be achieved by in vitro transcription, in which a 5 ’-cap is co-transcriptionally expressed into an RNA strand, or may be attached to an RNA post-transcriptionally using capping enzymes. In some embodiments, co-transcriptional capping with a cap disclosed herein improves the capping efficiency of an RNA compared to co-transcriptional capping with an appropriate reference comparator. In some embodiments, improving capping efficiency can increase translation efficiency and/or translation rate of an RNA, and/or increase expression of an
encoded polypeptide. In some embodiments, alterations to polynucleotides generates a non- hydrolyzable cap structure which can, for example, prevent decapping and increase RNA half-life.
[0251] In some embodiments, a utilized 5’ cap is a capO, a capl, or cap2 structure. See, e.g., Fig. 1 of Ramanathan et al., and Fig. 1 of Decroly et al., each of which is incorporated herein by reference in its entirety. In some embodiments, an RNA described herein comprises a capl structure. In some embodiments, an RNA described herein comprises a cap2 structure.
[0252] In some embodiments, an RNA described herein comprises a capO structure. In some embodiments, a capO structure comprises a guanosine nucleoside methylated at the 7-position of guanine ((m7)G). In some embodiments, such a capO structure is connected to an RNA via a 5’- to 5 ’-triphosphate linkage and is also referred to herein as (m7)Gppp. In some embodiments, a capO structure comprises a guanosine nucleoside methylated at the 2’- position of the ribose of guanosine In some embodiments, a capO structure comprises a guanosine nucleoside methylated at the 3 ’-position of the ribose of guanosine. In some embodiments, a guanosine nucleoside included in a 5’ cap comprises methylation at the 7- position of guanine and at the 2 ’-position of the ribose ((m27’2 ’°)G). In some embodiments, a guanosine nucleoside included in a 5’ cap comprises methylation at the 7-position of guanine and at the 2 ’-position of the ribose ((m27’3 ’°)G).
[0253] In some embodiments, a cap 1 structure comprises a guanosine nucleoside methylated at the 7-position of guanine ((m7)G) and optionally methylated at the 2’ or 3’ position of the ribose, and a 2’0 methylated first nucleotide in an RNA ((m2 ’°)Ni). In some embodiments, a capl structure comprises a guanosine nucleoside methylated at the 7-position of guanine ((m7)G) and the 3’ position of the ribose, and a 2’0 methylated first nucleotide in an RNA ((m2 ’°)Ni). In some embodiments, a capl structure is connected to an RNA via a 5’- to 5 ’-triphosphate linkage and is also referred to herein as, e.g., ((m7)Gppp(2 ’°)Ni) or (m27’3 ' °)Gppp(2 ’°)Ni), wherein Ni is as defined and described herein. In some embodiments, a capl structure comprises a second nucleotide, N2, which is at position 2 and is chosen from A, G, C, or U, e.g., (m7)Gppp(2 '°)NipN2 or (m27’3 "°)Gppp(2 "°)NipN2, wherein each of Ni and N2 is as defined and described herein.
[0254] In some embodiments, a cap2 structure comprises a guanosine nucleoside methylated at the 7-position of guanine ((m7)G) and optionally methylated at the 2’ or 3’ position of the ribose, and a 2’0 methylated first and second nucleotides in an RNA ((m2 ' °)Nip(m2 ’°)N2). In some embodiments, a cap2 structure comprises a guanosine nucleoside methylated at the 7-position of guanine ((m7)G) and the 3 ’ position of the ribose, and a 2’0 methylated first and second nucleotide in an RNA. In some embodiments, a cap2 structure is connected to an RNA via a 5’- to 5 ’-triphosphate linkage and is also referred to herein as, e.g., ((m7)Gppp(2 '°)Nip(2 '°)N2) or (m27’3 '°)Gppp(2 '°)Nip(2 ’°)N2), wherein each ofNi and N2 is as defined and described herein.
[0255] In some embodiments, the 5’ cap is a dinucleotide cap structure. In some embodiments, the 5’ cap is a dinucleotide cap structure comprising Ni, wherein Ni is as defined and described herein. In some embodiments, the 5’ cap is a dinucleotide cap G*Ni, wherein Ni is as defined above and herein, and G* comprises a structure of Formula (I):
 or a salt thereof, wherein each R2 and R3 is -OH or -OCH3; and
or a salt thereof, wherein each R2 and R3 is -OH or -OCH3; and
X is O or S.
[0256] In some embodiments, R2 is -OH. In some embodiments, R2 is -OCH3. In some embodiments, R3 is -OH. In some embodiments, R3 is -OCH3. In some embodiments, R2 is -OH and R3 is -OH. In some embodiments, R2 is -OH and R3 is -CH3. In some embodiments, R2 is -CH3 and R3 is -OH. In some embodiments, R2 is -CH3 and R3 is -CH3.
[0257] In some embodiments, X is O. In some embodiments, X is S.
[0258] In some embodiments, the 5’ cap is a dinucleotide capO structure (e.g., (m7)GpppNi, (m27’2 '°)GpppNi, (m27’3 '°)GpppNi, (m7)GppSpNi, (m27’2 '°)GppSpNi, or (m27’3 '°)GppSpNi), wherein Ni is as defined and described herein. In some embodiments, the 5’ cap is a dinucleotide capO structure (e.g., (m7)GpppNi, (m27’2 '°)GpppNi, (m27’3 '°)GpppNi, (m7)GppSpNi, (m27’2 '°)GppSpNi, or (m27’3 '°)GppSpNi), wherein Ni is G. In some embodiments, the 5’ cap is a dinucleotide capO structure (e.g., (m7)GpppNi, (m27’2 '°)GpppNi, (m27’3 '°)GpppNi, (m7)GppSpNi, (m27’2 '°)GppSpNi, or (m27’3 '°)GppSpNi), wherein Ni is A, U, or C. In some embodiments, the 5’ cap is a dinucleotide capl structure (e.g., (m7)Gppp(m2’-°)Ni, (m27’2’-°)Gppp(m2’-°)Ni, (m27’3’-°)Gppp(m2’-°)Ni, (m7)GppSp(m2’-°)Ni, (m27’2 '°)GppSp(m2 "°)Ni, or (m27’3 '°)GppSp(m2 ’’°)Ni), wherein Ni is as defined and described herein. In some embodiments, the 5’ cap is selected from the group consisting of (m7)GpppG (“EcapO”), (m7)Gppp(m2 ’°)G (“Ecapl”), (m273 '°)GpppG (“ARCA”), and (m272 '°)GppSpG (“beta-S-ARCA”). In some embodiments, the 5’ cap is (m7)GpppG (“EcapO”), having a structure:
 or a salt thereof.
or a salt thereof.
[0259] In some embodiments, the 5’ cap is (m7)Gppp(m2 ’°)G (“Ecapl”), having a structure:
 or a salt thereof.
[0260] In some embodiments, the 5’ cap is (m27’3 '°)GpppG (“ARCA”), having a structure:
or a salt thereof.
[0260] In some embodiments, the 5’ cap is (m27’3 '°)GpppG (“ARCA”), having a structure:
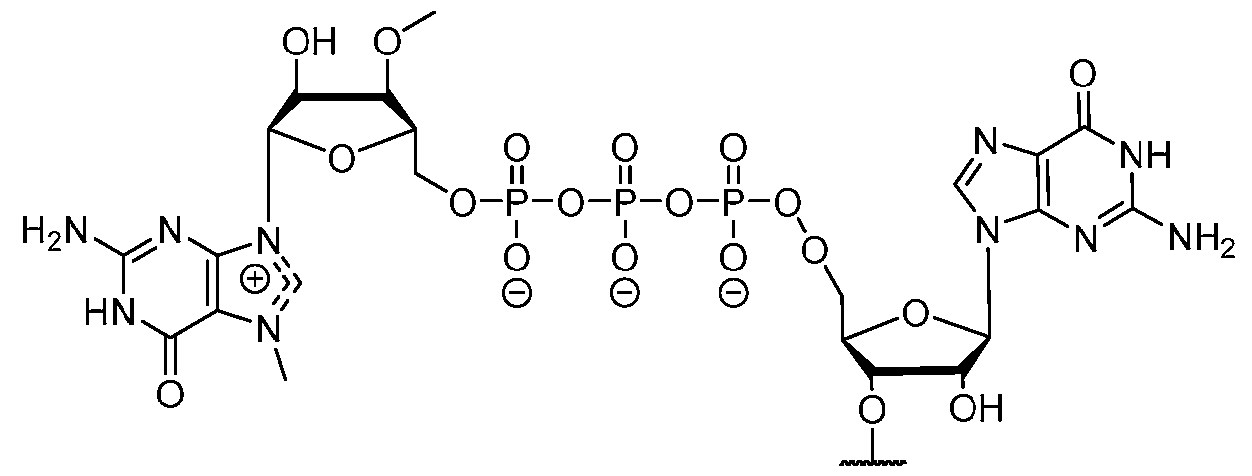 or a salt thereof.
or a salt thereof.
[0261] In some embodiments, the 5’ cap is (m272 '°)GppSpG (“beta-S-ARCA”), having a structure:
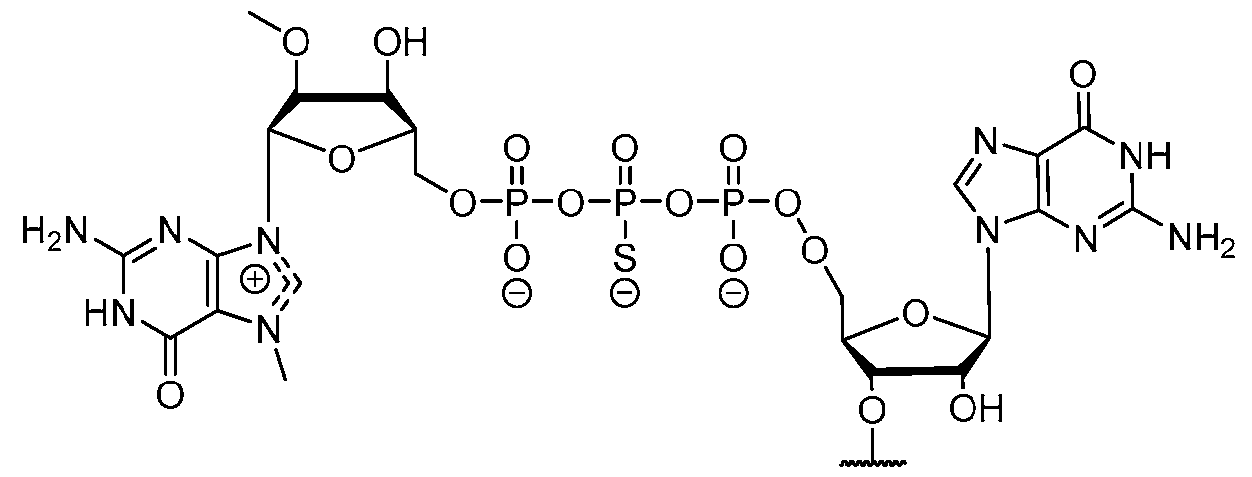 or a salt thereof.
or a salt thereof.
[0262] In some embodiments, the 5’ cap is a trinucleotide cap structure. In some embodiments, the 5’ cap is a trinucleotide cap structure comprising NipN2, wherein Ni and N2 are as defined and described herein. In some embodiments, the 5’ cap is a dinucleotide cap G*NipN2, wherein Ni and N2 are as defined above and herein, and G* comprises a structure of Formula (I):
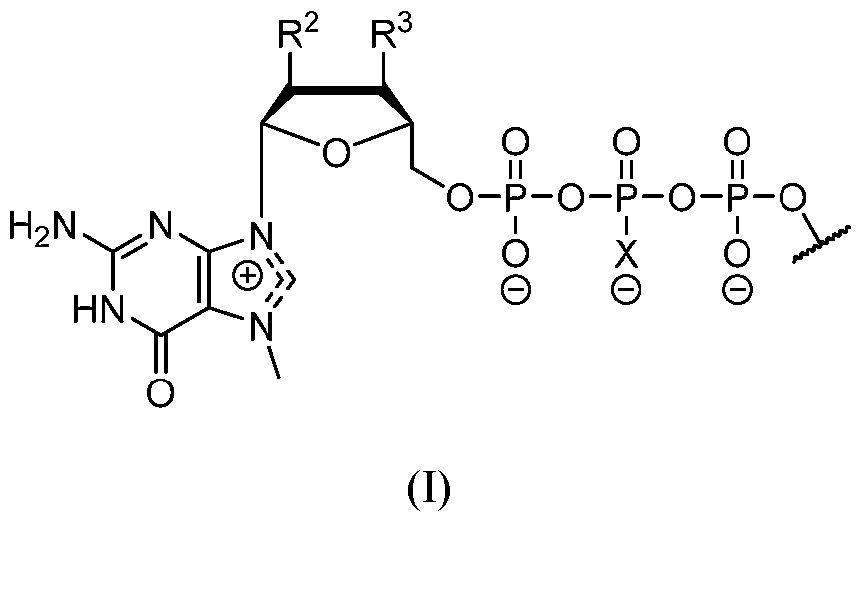 or a salt thereof, wherein R2, R3, and X are as defined and described herein.
or a salt thereof, wherein R2, R3, and X are as defined and described herein.
[0263] In some embodiments, the 5’ cap is a trinucleotide capO structure (e.g. (m7)GpppNipN2, (m27’2 '°)GpppNipN2, or (m27’3 '°)GpppNipN2), wherein Ni and N2 are as defined and described herein). In some embodiments, the 5’ cap is a trinucleotide capl structure (e.g., (m7)Gppp(m2 "°)NipN2, (m27’2 '°)Gppp(m2 ’’°)NipN2, (m27’3 '°)Gppp(m2 ' °)NipN2), wherein Ni and N2 are as defined and described herein. In some embodiments, the 5’ cap is a trinucleotide cap2 structure (e.g., (m7)Gppp(m2 '°)Nip(m2 ’°)N2, (m27’2 ’ °)Gppp(m2 '°)Nip(m2 '°)N2, (m27’3 '°)Gppp(m2 '°)Nip(m2 'O)N2), wherein NI and N2 are as defined and described herein. In some embodiments, the 5’ cap is selected from the group consisting of (m27’3 '°)Gppp(m2 "°)ApG (“CleanCap AG”, “CC413”), (m27’3 '°)Gppp(m2 ' °)GpG (“CleanCap GG”), (m7)Gppp(m2 '°)ApG, (m7)Gppp(m2 '°)GpG, (m273 '°)Gppp(m26’2 ' °)ApG, and (m7)Gppp(m2’-°)ApU.
[0264] In some embodiments, the 5’ cap is (m27’3 '°)Gppp(m2 '°)ApG (“CleanCap
AG”, “CC413”), having a structure:
 or a salt thereof.
or a salt thereof.
[0265] In some embodiments, the 5’ cap is (m273 '°)Gppp(m2 '°)GpG (“CleanCap GG”), having a structure:
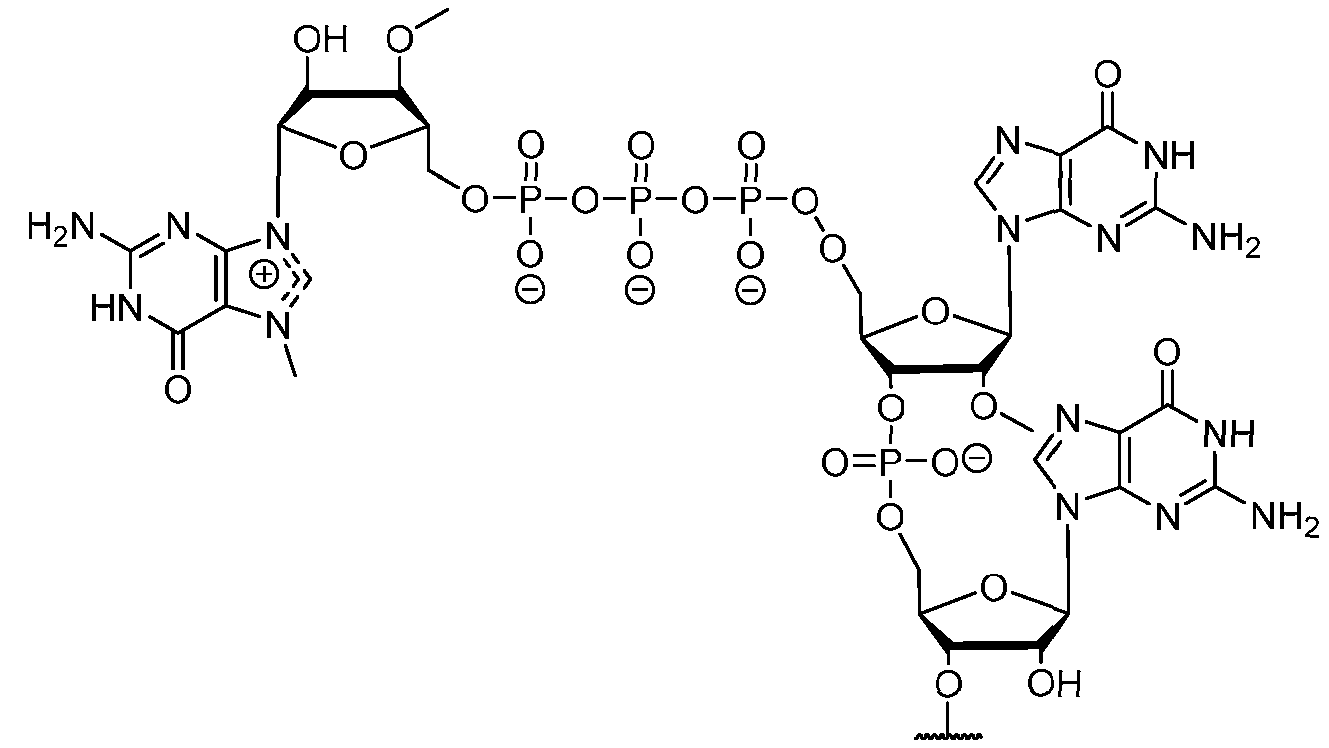
[0266] In some embodiments, the 5’ cap is (m7)Gppp(m2 '°)ApG, having a structure:
 or a salt thereof.
or a salt thereof.
[0267] In some embodiments, the 5’ cap is (m7)Gppp(m2 '°)GpG, having a structure:

[0268] In some embodiments, the 5 ’ cap is (m27’3 ’0)Gppp(m26’2’’°)ApG, having a structure:
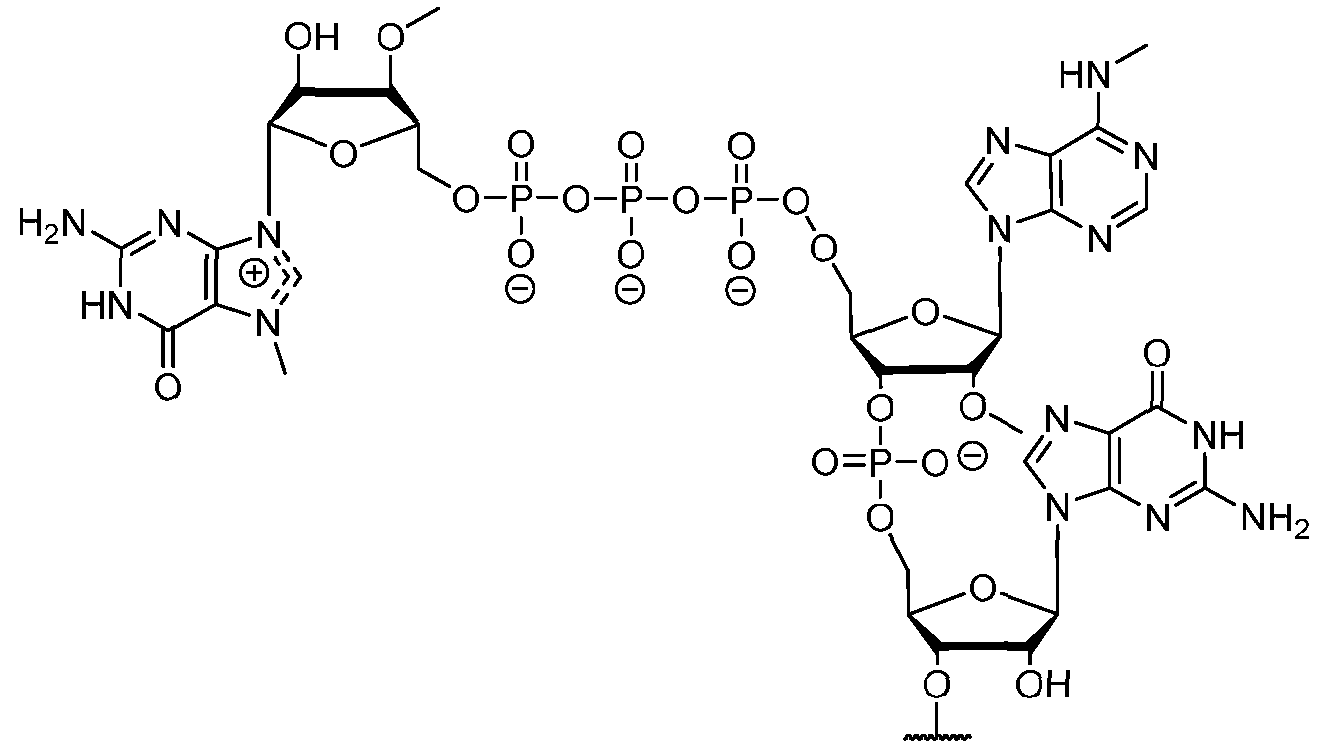 or a salt thereof.
or a salt thereof.
[0269] In some embodiments, the 5’ cap is (m7)Gppp(m2’’°)ApU, having a structure:
 or a salt thereof.
or a salt thereof.
[0270] In some embodiments, the 5’ cap is a tetranucleotide cap structure. In some embodiments, the 5’ cap is a tetranucleotide cap structure comprising NipN2pNs, wherein Ni, N2, and Ns are as defined and described herein. In some embodiments, the 5’ cap is a tetranucleotide cap G*NipN2pN< wherein Ni, N2, and N3 are as defined above and herein, and G* comprises a structure of Formula (I):
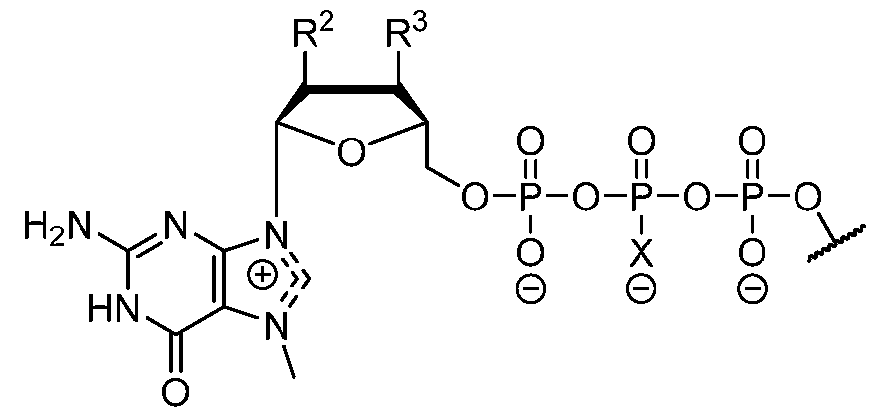
(I) or a salt thereof, wherein R2, R3, and X are as defined and described herein.
[0271] In some embodiments, the 5’ cap is a tetranucleotide capO structure (e.g. (m7)GpppNipN2pN3, (m27’2 '°)GpppNipN2pN3, or (m27’3 '°)GpppNiN2pN3), wherein Ni, N2, and N3 are as defined and described herein). In some embodiments, the 5’ cap is a tetranucleotide Capl structure (e.g., (m7)Gppp(m2 '°)NipN2pN3, (m27’2 '°)Gppp(m2 ' °)NipN2pN3, (m27’3 '°)Gppp(m2 '°)NipN2N3), wherein Ni, N2, and N3 are as defined and described herein. In some embodiments, the 5’ cap is a tetranucleotide Cap2 structure (e.g., (m7)Gppp(m2’-°)Nip(m2’-°)N2pN3, (m27’2’-0)Gppp(m2’-0)Nip(m2’-0)N2pN3, (m27’3’-°)Gppp(m2’-
°)Nip(m2 ’-°)N2pN3), wherein Ni, N2, and N3 are as defined and described herein. In some embodiments, the 5’ cap is selected from the group consisting of (m27’3 '°)Gppp(m2 '°)Ap(m2 °)GpG, (m27’3 '°)Gppp(m2 '°)Gp(m2 '°)GpC, (m7)Gppp(m2’-°)Ap(m2’-°)UpA, and (m7)Gppp(m2’-°)Ap(m2’-°)GpG.
[0272] In some embodiments, the 5’ cap is (m273 '°)Gppp(m2 '°)Ap(m2 '°)GpG, having a structure:
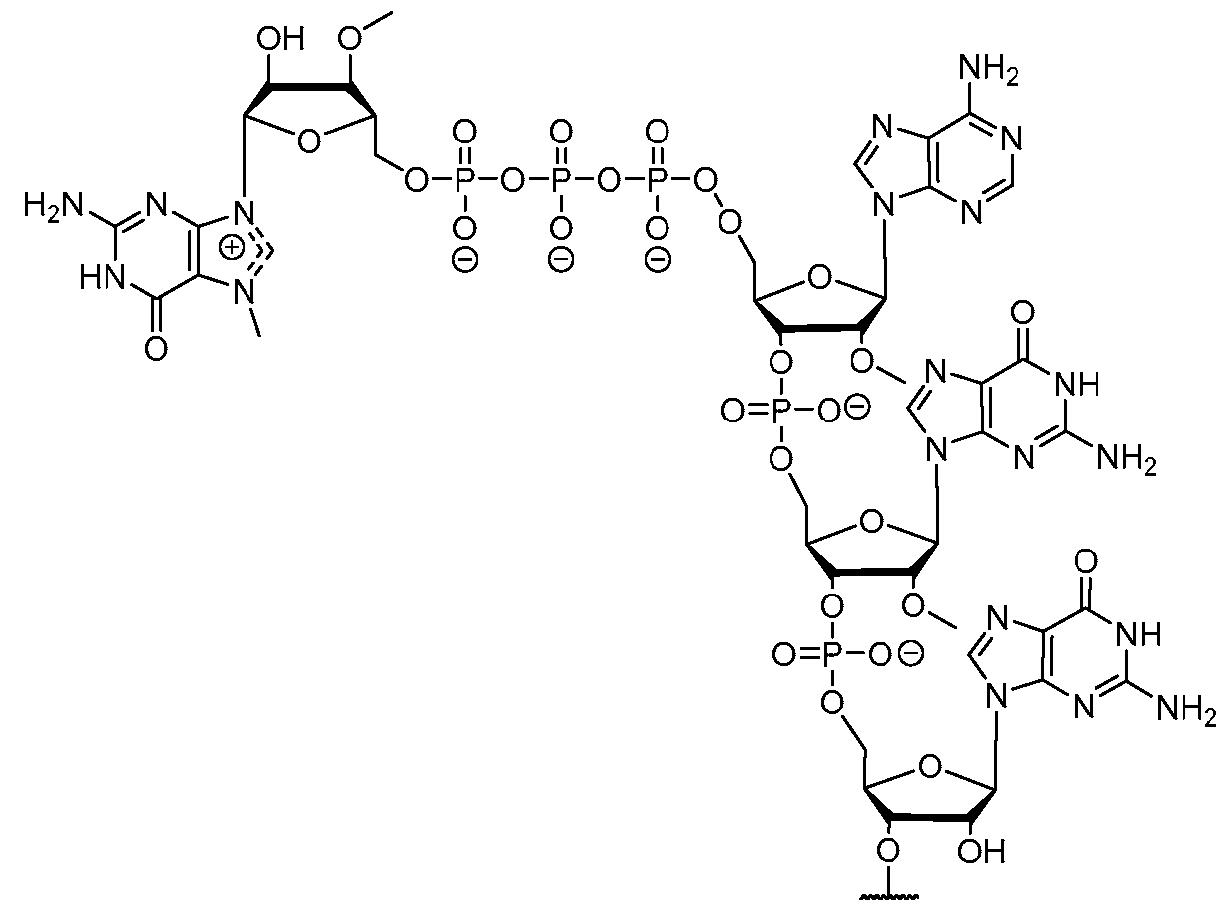 or a salt thereof.
or a salt thereof.
[0273] In some embodiments, the 5’ cap is (m273 '°)Gppp(m2 '°)Gp(m2 '°)GpC, having a structure:

[0274] In some embodiments, the 5 ’ cap is (m7)Gppp(m2 '°)Ap(m2 '°)UpA, having a structure:
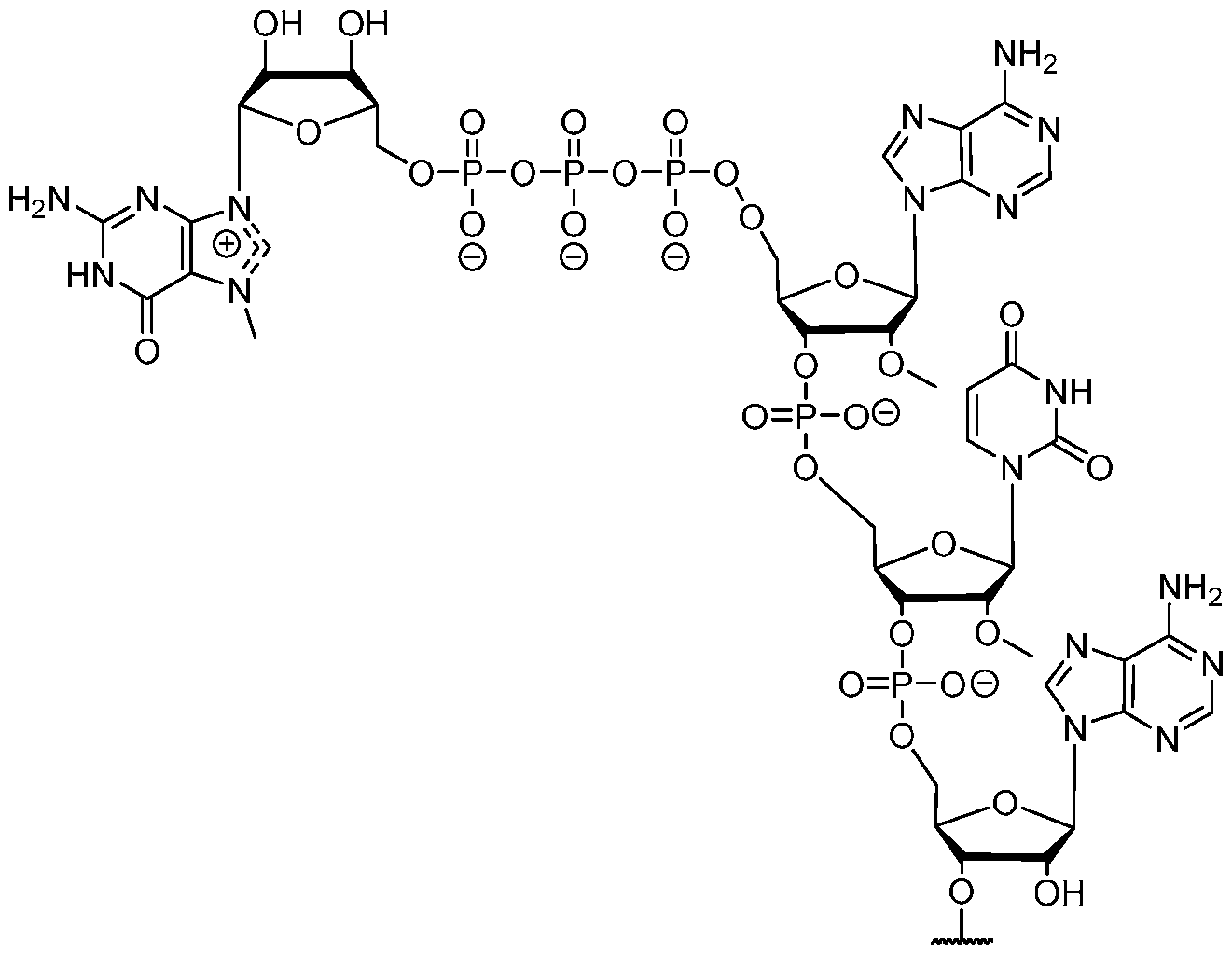 or a salt thereof.
[0275] In some embodiments, the 5 ’ cap is (m7)Gppp(m2 '°)Ap(m2 '°)GpG, having a structure:
or a salt thereof.
[0275] In some embodiments, the 5 ’ cap is (m7)Gppp(m2 '°)Ap(m2 '°)GpG, having a structure:
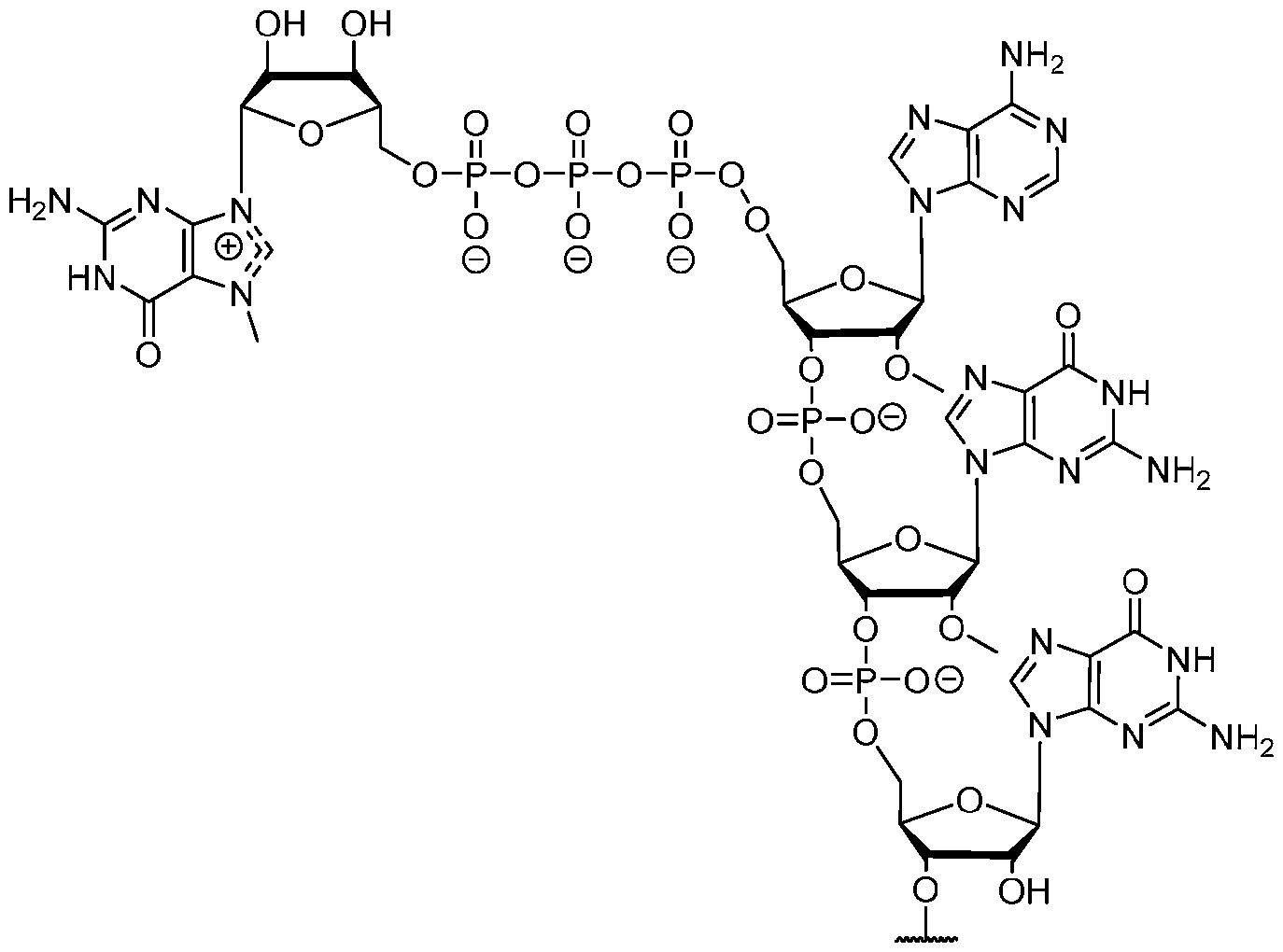 or a salt thereof.
or a salt thereof.
Cap Proximal Sequences
[0276] In some embodiments, a 5 ’ UTR utilized in accordance with the present disclosure comprises a cap proximal sequence, e.g., as disclosed herein. In some embodiments, a cap proximal sequence comprises a sequence adjacent to a 5’ cap. In some embodiments, a cap proximal sequence comprises nucleotides in positions +1, +2, +3, +4, and/or +5 of an RNA polynucleotide.
[0277] In some embodiments, a cap structure comprises one or more polynucleotides of a cap proximal sequence. In some embodiments, a cap structure comprises an m7 Guanosine cap and nucleotide +1 (Ni) of an RNA polynucleotide. In some embodiments, a cap structure comprises an m7 Guanosine cap and nucleotide +2 (N2) of an RNA polynucleotide. In some embodiments, a cap structure comprises an m7 Guanosine cap and nucleotides +1 and +2 (Ni and N2) of an RNA polynucleotide. In some embodiments, a cap structure comprises an m7 Guanosine cap and nucleotides +1, +2, and +3 (Ni, N2, and N3) of an RNA polynucleotide.
[0278] Those skilled in the art, reading the present disclosure, will appreciate that, in some embodiments, one or more residues of a cap proximal sequence (e.g., one or more of residues +1, +2, +3, +4, and/or +5) may be included in an RNAby virtue of having been included in a cap entity (e.g., a capl or cap2 structure, etc.); alternatively, in some embodiments, at least some of the residues in a cap proximal sequence may be enzymatically added (e.g., by a polymerase such as a T7 polymerase). For example, in certain exemplified embodiments where a m27’3 '°Gppp(mi2 '°)ApG cap is utilized, +1 (i.e., Ni) and +2 (i.e. N2) are the (mi2 ’°)A and G residues of the cap, and +3, +4, and +5 are added by polymerase (e.g., T7 polymerase).
[0279] In some embodiments, the 5’ cap is a dinucleotide cap structure, wherein the cap proximal sequence comprises Ni of the 5’ cap, where Ni is any nucleotide, e.g., A, C, G or U. In some embodiments, the 5’ cap is a trinucleotide cap structure (e.g., the trinucleotide cap structures described above and herein), wherein the cap proximal sequence comprises Ni and N2 of the 5’ cap, wherein Ni and N2 are independently any nucleotide, e.g., A, C, G or U. In some embodiments, the 5’ cap is a tetranucleotide cap structure (e.g., the trinucleotide cap structures described above and herein), wherein the cap proximal sequence comprises Ni, N2, and N3 of the 5’ cap, wherein Ni, N2, and N3 are any nucleotide, e.g., A, C, G or U.
[0280] In some embodiments, e.g., where the 5’ cap is a dinucleotide cap structure, a cap proximal sequence comprises Ni of a the 5’ cap, and N2, N3, N4 and Ns, wherein Ni to Ns correspond to positions +1, +2, +3, +4, and/or +5 of an RNA polynucleotide. In some embodiments, e.g., where the 5’ cap is a trinucleotide cap structure, a cap proximal sequence comprises Ni and N2 of a the 5’ cap, and N3, N4 and Ns, wherein Ni to Ns correspond to positions +1, +2, +3, +4, and/or +5 of an RNA polynucleotide. In some embodiments, e.g., where the 5’ cap is a tetranucleotide cap structure, a cap proximal sequence comprises Ni, N2, and N3 of a the 5’ cap, and N4 and Ns, wherein Ni to Ns correspond to positions +1, +2, +3, +4, and/or +5 of an RNA polynucleotide.
[0281] In some embodiments, Ni is A. In some embodiments, Ni is C. In some embodiments, Ni is G. In some embodiments, Ni is U. In some embodiments, N2 is A. In some embodiments, N2 is C. In some embodiments, N2 is G. In some embodiments, N2 is U. In some embodiments, N3 is A. In some embodiments, N3 is C. In some embodiments, N3 is G. In some embodiments, N3 is U. In some embodiments, N4 is A. In some embodiments, N4
is C. In some embodiments, N4 is G. In some embodiments, N4 is U. In some embodiments, Ns is A. In some embodiments, Ns is C. In some embodiments, Ns is G. In some embodiments, Ns is U. It will be understood that, each of the embodiments described above and herein (e.g., for Ni through Ns) may be taken singly or in combination and/or may be combined with other embodiments of variables described above and herein (e.g., 5’ caps).
5 ’ UTR
[0282] In some embodiments, a nucleic acid (e.g., DNA, RNA) utilized in accordance with the present disclosure comprises a 5 ’ UTR. In some embodiments, 5 ’ UTR may comprise a plurality of distinct sequence elements; in some embodiments, such plurality may be or comprise multiple copies of one or more particular sequence elements (e.g., as may be from a particular source or otherwise known as a functional or characteristic sequence element). In some embodiments a 5’ UTR comprises multiple different sequence elements.
[0283] The term “untranslated region” or “UTR” is commonly used in the art to reference a region in a DNA molecule which is transcribed but is not translated into an amino acid sequence, or to the corresponding region in an RNA polynucleotide, such as an mRNA molecule. An untranslated region (UTR) can be present 5’ (upstream) of an open reading frame (5’ UTR) and/or 3’ (downstream) of an open reading frame (3’ UTR). As used herein, the terms “five prime untranslated region” or “5 ’ UTR” refer to a sequence of a polyribonucleotide between the 5’ end of the polyribonucleotide (e.g., a transcription start site) and a start codon of a coding region of the polyribonucleotide. In some embodiments, “5 ’ UTR” refers to a sequence of a polyribonucleotide that begins at the 5 ’ end of the polyribonucleotide (e.g., a transcription start site) and ends one nucleotide (nt) before a start codon (usually AUG) of a coding region of the polyribonucleotide, e.g., in its natural context. In some embodiments, a 5’ UTR comprises a Kozak sequence. A 5’ UTR is downstream of the 5’ cap (if present), e.g., directly adjacent to the 5’ cap. In some embodiments, a 5’ UTR disclosed herein comprises a cap proximal sequence, e.g., as defined and described herein. In some embodiments, a cap proximal sequence comprises a sequence adjacent to a 5’ cap.
[0284] Exemplary 5 ’ UTRs include a human alpha globin (hAg) 5 ’ UTR or a fragment thereof, a TEV 5 ’ UTR or a fragment thereof, a HSP70 5 ’ UTR or a fragment thereof, or a c-Jun 5’ UTR or a fragment thereof.
[0285] In some embodiments, an RNA disclosed herein comprises a hAg 5 ’ UTR or a fragment thereof.
[0286] In some embodiments, an RNA disclosed herein comprises a 5 ’ UTR having at least 80%, at least 85%, at least 90%, at least 95%, at least 96%, at least 97%, at least 98%, or at least 99% identity to a 5 ’ UTR with the sequence according to AACUAGUAUUCUUCUGGUCCCCACAGACUCAGAGAGAACCCGCCACC (SEQ ID NO: 49). In some embodiments, an RNA disclosed herein comprises a 5’ UTR provided in SEQ ID NO: 49.
PolyA Tail
[0287] In some embodiments, a polynucleotide (e.g., DNA, RNA) disclosed herein comprises a polyadenylate or poly(A) sequence, e.g., as described herein. In some embodiments, a poly(A) sequence is situated downstream of a 3’ UTR, e.g., adjacent to a 3’ UTR.
[0288] As used herein, the term “poly(A) sequence” or “polyA tail” refers to an uninterrupted or interrupted sequence of adenylate residues which is typically located at the 3’-end of an RNA polynucleotide. Poly(A) sequences are known to those of skill in the art and may follow the 3’ UTR in the RNAs described herein. An uninterrupted poly(A) sequence is characterized by consecutive adenylate residues. In nature, an uninterrupted poly(A) sequence is typical. In some embodiments, polynucleotides disclosed herein comprise an uninterrupted poly(A) sequence. In some embodiments, polynucleotides disclosed herein comprise interrupted poly(A) sequence. In some embodiments, RNAs disclosed herein can have a poly(A) sequence attached to the free 3 ’-end of the RNA by a template -independent RNA polymerase after transcription or a poly(A) sequence encoded by DNA and transcribed by a template-dependent RNA polymerase.
[0289] It has been demonstrated that a poly(A) sequence of about 120 A nucleotides has a beneficial influence on the levels of RNA in transfected eukaryotic cells, as well as on the levels of protein that is translated from an open reading frame that is present upstream (5’) of the poly(A) sequence (Holtkamp et al., 2006, Blood, vol. 108, pp. 4009-4017, which is incorporated herein by reference).
[0290] In some embodiments, a poly(A) sequence in accordance with the present disclosure is not limited to a particular length; in some embodiments, a poly(A) sequence is any length. In some embodiments, a poly(A) sequence comprises, essentially consists of, or consists of at least 20, at least 30, at least 40, at least 80, or at least 100 and up to 500, up to 400, up to 300, up to 200, or up to 150 A nucleotides, and, in particular, about 120 A nucleotides. In this context, “essentially consists of” means that most nucleotides in the poly(A) sequence, typically at least 75%, at least 80%, at least 85%, at least 90%, at least 95%, at least 96%, at least 97%, at least 98%, or at least 99% by number of nucleotides in the poly(A) sequence are A nucleotides, but permits that remaining nucleotides are nucleotides other than A nucleotides, such as U nucleotides (uridylate), G nucleotides (guanylate), or C nucleotides (cytidylate). In this context, “consists of” means that all nucleotides in the poly(A) sequence, i.e., 100% by number of nucleotides in the poly(A) sequence, are A nucleotides. The term “A nucleotide” or “A” refers to adenylate. [0291] In some embodiments, a poly(A) sequence is attached during RNA transcription, e.g., during preparation of in vitro transcribed RNA, based on a DNA template comprising repeated dT nucleotides (deoxythymidylate) in the strand complementary to the coding strand. The DNA sequence encoding a poly(A) sequence (coding strand) is referred to as poly(A) cassette. [0292] In some embodiments, the poly(A) cassette present in the coding strand of DNA essentially consists of dA nucleotides, but is interrupted by a random sequence of the four nucleotides (dA, dC, dG, and dT). Such random sequence may be 5 to 50, 10 to 30, or 10 to 20 nucleotides in length. Such a cassette is disclosed in WO2016/005324, hereby incorporated by reference. Any poly(A) cassette disclosed in WO2016/005324 may be used in accordance with the present disclosure. A poly(A) cassette that essentially consists of dA nucleotides, but is interrupted by a random sequence having an equal distribution of the four nucleotides (dA, dC, dG, dT) and having a length of e.g., 5 to 50 nucleotides shows, on DNA level, constant propagation of plasmid DNA in E. coli and is still associated, on RNA level, with the beneficial properties with respect to supporting RNA stability and translational efficiency is encompassed. In some embodiments, the poly(A) sequence contained in an RNA polynucleotide described herein essentially consists of A nucleotides, but is interrupted by a random sequence of the four nucleotides (A, C, G, U). Such random sequence may be 5 to 50, 10 to 30, or 10 to 20 nucleotides in length.
[0293] In some embodiments, no nucleotides other than A nucleotides flank a poly(A) sequence at its 3 ’-end, i.e., the poly(A) sequence is not masked or followed at its 3 ’-end by a nucleotide other than A.
[0294] In some embodiments, the poly(A) sequence may comprise at least 20, at least 30, at least 40, at least 80, or at least 100 and up to 500, up to 400, up to 300, up to 200, or up to 150 nucleotides. In some embodiments, the poly(A) sequence may essentially consist of at least 20, at least 30, at least 40, at least 80, or at least 100 and up to 500, up to 400, up to 300, up to 200, or up to 150 nucleotides. In some embodiments, the poly(A) sequence may consist of at least 20, at least 30, at least 40, at least 80, or at least 100 and up to 500, up to 400, up to 300, up to 200, or up to 150 nucleotides. In some embodiments, the poly(A) sequence comprises at least 100 nucleotides. In some embodiments, the poly(A) sequence comprises about 150 nucleotides. In some embodiments, the poly(A) sequence comprises about 120 nucleotides.
[0295] In some embodiments, a polyA tail comprises a specific number of
Adenosines, such as about 50 or more, about 60 or more, about 70 or more, about 80 or more, about 90 or more, about 100 or more, about 120, or about 150 or about 200. In some embodiments a polyA tail of a string construct may comprise 200 A residues or less. In some embodiments, a polyA tail of a string construct may comprise about 200 A residues. In some embodiments, a polyA tail of a string construct may comprise 180 A residues or less. In some embodiments, a polyA tail of a string construct may comprise about 180 A residues. In some embodiments, a polyA tail may comprise 150 residues or less.
[0296] In some embodiments, RNA comprises a poly(A) sequence comprising the nucleotide sequence of AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAGCAUAUGACUAAAAAAAAAAAA AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA AAAAAA (SEQ ID NO: 50), or a nucleotide sequence having at least 99%, 98%, 97%, 96%, 95%, 90%, 85%, or 80% identity to the nucleotide sequence of SEQ ID NO: 50. In some embodiments, a poly(A) tail comprises a nucleotide sequence according to SEQ ID NO: 50.
[0297] In some embodiments, a polyA tail comprises a plurality of A residues interrupted by a linker. In some embodiments, a linker comprises the nucleotide sequence GCAUAUGACU (SEQ ID NO:
3’ UTR
[0298] In some embodiments, an RNA utilized in accordance with the present disclosure comprises a 3’ UTR. As used herein, the terms “three prime untranslated region,” “3 ’ untranslated region,” or “3 ’ UTR” refer to a sequence of an mRNA molecule that begins following a stop codon of a coding region of an open reading frame sequence. In some embodiments, the 3 ’ UTR begins immediately after a stop codon of a coding region of an open reading frame sequence, e.g., in its natural context. In other embodiments, the 3’ UTR does not begin immediately after stop codon of the coding region of an open reading frame sequence, e.g., in its natural context. The term “3’ UTR” does preferably not include the poly(A) sequence. Thus, the 3’ UTR is upstream of the poly(A) sequence (if present), e.g., directly adjacent to the poly(A) sequence.
[0299] In some embodiments, an RNA disclosed herein comprises a 3 ’ UTR comprising an F element and/or an I element. In some embodiments, a 3’ UTR or a proximal sequence thereto comprises a restriction site. In some embodiments, a restriction site is a BamHI site. In some embodiments, a restriction site is a.Xho! site.
[0300] In some embodiments, an RNA construct comprises an F element. In some embodiments, a F element sequence is a 3’ UTR of amino-terminal enhancer of split (AES).
[0301] In some embodiments, an RNA disclosed herein comprises a 3 ’ UTR having at least 80%, at least 85%, at least 90%, at least 95%, at least 96%, at least 97%, at least 98%, or at least 99% identity to a 3 ’ UTR with the sequence according to CUGGUACUGCAUGCACGCAAUGCUAGCUGCCCCUUUCCCGUCCUGGGUACCCC GAGUCUCCCCCGACCUCGGGUCCCAGGUAUGCUCCCACCUCCACCUGCCCCACU CACCACCUCUGCUAGUUCCAGACACCUCCCAAGCACGCAGCAAUGCAGCUCAA AACGCUUAGCCUAGCCACACCCCCACGGGAAACAGCAGUGAUUAACCUUUAGC AAUAAACGAAAGUUUAACUAAGCUAUACUAACCCCAGGGUUGGUCAAUUUCG UGCCAGCCACACC (SEQ ID NO: 51). In some embodiments, an RNA disclosed herein comprises a 3’ UTR provided in SEQ ID NO: 51.
[0302] In some embodiments, a 3’UTR is an FI element as described in W02017/060314, which is incorporated herein by reference in its entirety.
RNA Formats
[0303] At least three distinct formats useful for RNA compositions (e.g., pharmaceutical compositions) have been developed, namely non-modified uridine containing mRNA (uRNA), nucleoside-modified mRNA (modRNA), and self-amplifying mRNA (saRNA). Each of these platforms displays unique features. In general, in all three formats, RNA is capped, contains open reading frames (ORFs) flanked by untranslated regions (UTR), and have a polyA-tail at the 3’ end. An ORF of an uRNA and modRNA vectors encode an incretin agent. An saRNA has multiple ORFs.
[0304] In some embodiments, the RNA described herein may have modified nucleosides. In some embodiments, the RNA comprises a modified nucleoside in place of at least one (e.g., every) uridine.
[0305] The term “uracil,” as used herein, describes one of the nucleobases that can occur in the nucleic acid of RNA. The structure of uracil is:
[0306] The term “uridine,” as used herein, describes one of the nucleosides that can occur in RNA. The structure of uridine is:
[0002]
[0307] UTP (uridine 5 ’-triphosphate) has the following structure:
[0308] Pseudo-UTP (pseudouridine 5 ’-triphosphate) has the following structure:
[0004]
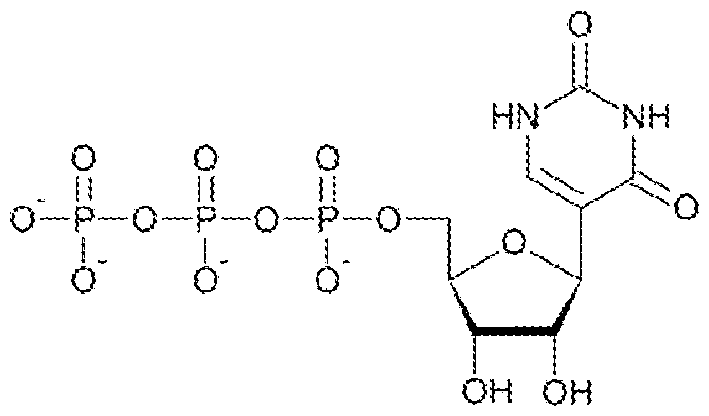
[0309] ‘Pseudouridine” is one example of a modified nucleoside that is an isomer of uridine, where the uracil is attached to the pentose ring via a carbon-carbon bond instead of a nitrogen-carbon glycosidic bond.
[0310] Another exemplary modified nucleoside is N1 -methyl -pseudouridine (m I T). which has the structure:
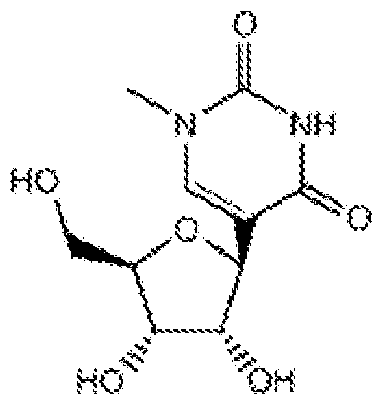
[0005]
[0311] N1 -methyl -pseudo-UTP has the following structure:
[0006]
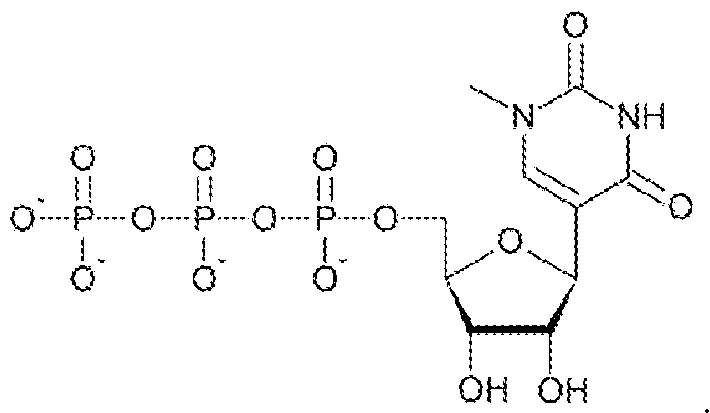 [0312] Another exemplary modified nucleoside is 5-methyl-uridine (m5U), which has the structure: . ts, one or more uridine in the RNA described herein is
[0312] Another exemplary modified nucleoside is 5-methyl-uridine (m5U), which has the structure: . ts, one or more uridine in the RNA described herein is
 replaced by a modified nucleoside. In some embodiments, the modified nucleoside is a modified uridine. [0314] In some embodiments, RNA comprises a modified nucleoside in place of at least one uridine. In some embodiments, RNA comprises a modified nucleoside in place of each uridine. [0315] In some embodiments, the modified nucleoside is independently selected from pseudouridine (ψ), N1-methyl-pseudouridine (m1ψ), and 5-methyl-uridine (m5U). In some embodiments, the modified nucleoside comprises pseudouridine (ψ). In some embodiments, the modified nucleoside comprises N1-methyl-pseudouridine (m1ψ). In some embodiments, the modified nucleoside comprises 5-methyl-uridine (m5U). In some embodiments, RNA may comprise more than one type of modified nucleoside, and the modified nucleosides are independently selected from pseudouridine (ψ), N1-methyl-pseudouridine (m1ψ), and 5- methyl-uridine (m5U). In some embodiments, the modified nucleosides comprise pseudouridine (ψ) and N1-methyl-pseudouridine (m1ψ). In some embodiments, the modified nucleosides comprise pseudouridine (ψ) and 5-methyl-uridine (m5U). In some embodiments, the modified nucleosides comprise N1-methyl-pseudouridine (m1ψ) and 5-methyl-uridine (m5U). In some embodiments, the modified nucleosides comprise pseudouridine (ψ), N1- methyl-pseudouridine (m1ψ), and 5-methyl-uridine (m5U). [0316] In some embodiments, the modified nucleoside replacing one or more, e.g., all, uridine in the RNA may be any one or more of 3-methyl-uridine (m3U), 5-methoxy- uridine (mo5U), 5-aza-uridine, 6-aza-uridine, 2-thio-5-aza-uridine, 2-thio-uridine (s2U), 4-
thio-uridine (s4U), 4-thio-pseudouridine, 2-thio-pseudouridine, 5 -hydroxy-uridine (ho5U), 5- aminoallyl-uridine, 5-halo-uridine (e.g., 5-iodo-uridine or 5-bromo-uridine), uridine 5- oxyacetic acid (cmo5U), uridine 5-oxyacetic acid methyl ester (mcmo5U), 5 -carboxymethyl - uridine (cm5U), 1-carboxymethyl-pseudouridine, 5-carboxyhydroxymethyl-uridine (chm5U), 5-carboxyhydroxymethyl-uridine methyl ester (mchm5U), 5-methoxycarbonyhnethyl-uridine (mcm5U), 5-methoxycarbonylmethyl-2-thio-uridine (mcm5s2U), 5 -aminomethyl -2 -thiouridine (nm5s2U), 5-methylaminomethyl-uridine (mnm5U), 1 -ethyl -pseudouridine, 5- methylaminomethyl-2 -thio-uridine (mnm5s2U), 5-methylaminomethyl-2-seleno-uridine (mnm5se2U), 5 -carbamoylmethyl -uridine (ncm5U), 5-carboxymethylaminomethyl-uridine (cmnm5U), 5-carboxymethylaminomethyl-2-thio-uridine (cmnm5s2U), 5 -propynyl -uridine, 1-propynyl-pseudouridine, 5 -taurinomethyl -uridine (rm5U), 1-taurinomethyl-pseudouridine, 5-taurinomethyl-2-thio-uridine(rm5s2U), l-taurinomethyl-4-thio-pseudouridine), 5-methyl-2- thio-uridine (m5s2U), l-methyl-4-thio-pseudouridine (m I s4v|/)_ 4-thio-l -methylpseudouridine, 3-methyl-pseudouridine (m3 v|/)_ 2-thio-l -methyl -pseudouridine, 1-methyl-l- deaza-pseudouridine, 2-thio-l -methyl- 1-deaza-pseudouridine, dihydrouridine (D), dihydropseudouridine, 5,6-dihydrouridine, 5-methyl-dihydrouridine (m5D), 2-thio- dihydrouridine, 2-thio-dihydropseudouridine, 2-methoxy-uridine, 2-methoxy-4-thio-uridine, 4-methoxy-pseudouridine, 4-methoxy-2-thio-pseudouridine, Nl-methyl-pseudouridine, 3-(3- amino-3-carboxypropyl)uridine (acp3U), l-methyl-3-(3-amino-3- carboxypropyl)pseudouridine (acp3 \|/), 5-(isopentenylaminomethyl)uridine (inm5U), 5- (isopentenylaminomethyl)-2-thio-uridine (inm5s2U), a-thio-uridine, 2'-O-methyl-uridine (Um), 5,2'-O-dimethyl-uridine (m5Um), 2'-O-methyl-pseudouridine (y/m ). 2-thio-2'-O- methyl-uridine (s2Um), 5-methoxycarbonylmethyl-2'-O-methyl-uridine (mcm5Um), 5- carbamoyhnethyl-2'-O-methyl-uridine (ncm5Um), 5-carboxymethylaminomethyl-2'-O- methyl-uridine (cmnm5Um), 3,2'-O-dimethyl-uridine (m3Um), 5-(isopentenylaminomethyl)- 2'-O-methyl-uridine (inm5Um), 1 -thio-uridine, deoxythymidine, 2'-F-ara-uridine, 2'-F- uridine, 2'-OH-ara-uridine, 5-(2-carbomethoxyvinyl) uridine, 5-[3-(l-E- propenylamino)uridine, or any other modified uridine known in the art.
replaced by a modified nucleoside. In some embodiments, the modified nucleoside is a modified uridine. [0314] In some embodiments, RNA comprises a modified nucleoside in place of at least one uridine. In some embodiments, RNA comprises a modified nucleoside in place of each uridine. [0315] In some embodiments, the modified nucleoside is independently selected from pseudouridine (ψ), N1-methyl-pseudouridine (m1ψ), and 5-methyl-uridine (m5U). In some embodiments, the modified nucleoside comprises pseudouridine (ψ). In some embodiments, the modified nucleoside comprises N1-methyl-pseudouridine (m1ψ). In some embodiments, the modified nucleoside comprises 5-methyl-uridine (m5U). In some embodiments, RNA may comprise more than one type of modified nucleoside, and the modified nucleosides are independently selected from pseudouridine (ψ), N1-methyl-pseudouridine (m1ψ), and 5- methyl-uridine (m5U). In some embodiments, the modified nucleosides comprise pseudouridine (ψ) and N1-methyl-pseudouridine (m1ψ). In some embodiments, the modified nucleosides comprise pseudouridine (ψ) and 5-methyl-uridine (m5U). In some embodiments, the modified nucleosides comprise N1-methyl-pseudouridine (m1ψ) and 5-methyl-uridine (m5U). In some embodiments, the modified nucleosides comprise pseudouridine (ψ), N1- methyl-pseudouridine (m1ψ), and 5-methyl-uridine (m5U). [0316] In some embodiments, the modified nucleoside replacing one or more, e.g., all, uridine in the RNA may be any one or more of 3-methyl-uridine (m3U), 5-methoxy- uridine (mo5U), 5-aza-uridine, 6-aza-uridine, 2-thio-5-aza-uridine, 2-thio-uridine (s2U), 4-
thio-uridine (s4U), 4-thio-pseudouridine, 2-thio-pseudouridine, 5 -hydroxy-uridine (ho5U), 5- aminoallyl-uridine, 5-halo-uridine (e.g., 5-iodo-uridine or 5-bromo-uridine), uridine 5- oxyacetic acid (cmo5U), uridine 5-oxyacetic acid methyl ester (mcmo5U), 5 -carboxymethyl - uridine (cm5U), 1-carboxymethyl-pseudouridine, 5-carboxyhydroxymethyl-uridine (chm5U), 5-carboxyhydroxymethyl-uridine methyl ester (mchm5U), 5-methoxycarbonyhnethyl-uridine (mcm5U), 5-methoxycarbonylmethyl-2-thio-uridine (mcm5s2U), 5 -aminomethyl -2 -thiouridine (nm5s2U), 5-methylaminomethyl-uridine (mnm5U), 1 -ethyl -pseudouridine, 5- methylaminomethyl-2 -thio-uridine (mnm5s2U), 5-methylaminomethyl-2-seleno-uridine (mnm5se2U), 5 -carbamoylmethyl -uridine (ncm5U), 5-carboxymethylaminomethyl-uridine (cmnm5U), 5-carboxymethylaminomethyl-2-thio-uridine (cmnm5s2U), 5 -propynyl -uridine, 1-propynyl-pseudouridine, 5 -taurinomethyl -uridine (rm5U), 1-taurinomethyl-pseudouridine, 5-taurinomethyl-2-thio-uridine(rm5s2U), l-taurinomethyl-4-thio-pseudouridine), 5-methyl-2- thio-uridine (m5s2U), l-methyl-4-thio-pseudouridine (m I s4v|/)_ 4-thio-l -methylpseudouridine, 3-methyl-pseudouridine (m3 v|/)_ 2-thio-l -methyl -pseudouridine, 1-methyl-l- deaza-pseudouridine, 2-thio-l -methyl- 1-deaza-pseudouridine, dihydrouridine (D), dihydropseudouridine, 5,6-dihydrouridine, 5-methyl-dihydrouridine (m5D), 2-thio- dihydrouridine, 2-thio-dihydropseudouridine, 2-methoxy-uridine, 2-methoxy-4-thio-uridine, 4-methoxy-pseudouridine, 4-methoxy-2-thio-pseudouridine, Nl-methyl-pseudouridine, 3-(3- amino-3-carboxypropyl)uridine (acp3U), l-methyl-3-(3-amino-3- carboxypropyl)pseudouridine (acp3 \|/), 5-(isopentenylaminomethyl)uridine (inm5U), 5- (isopentenylaminomethyl)-2-thio-uridine (inm5s2U), a-thio-uridine, 2'-O-methyl-uridine (Um), 5,2'-O-dimethyl-uridine (m5Um), 2'-O-methyl-pseudouridine (y/m ). 2-thio-2'-O- methyl-uridine (s2Um), 5-methoxycarbonylmethyl-2'-O-methyl-uridine (mcm5Um), 5- carbamoyhnethyl-2'-O-methyl-uridine (ncm5Um), 5-carboxymethylaminomethyl-2'-O- methyl-uridine (cmnm5Um), 3,2'-O-dimethyl-uridine (m3Um), 5-(isopentenylaminomethyl)- 2'-O-methyl-uridine (inm5Um), 1 -thio-uridine, deoxythymidine, 2'-F-ara-uridine, 2'-F- uridine, 2'-OH-ara-uridine, 5-(2-carbomethoxyvinyl) uridine, 5-[3-(l-E- propenylamino)uridine, or any other modified uridine known in the art.
[0317] In some embodiments, the RNA comprises other modified nucleosides or comprises further modified nucleosides, e.g., modified cytidine. For example, in some embodiments, in the RNA 5 -methylcytidine is substituted partially or completely, preferably completely, for cytidine. In some embodiments, the RNA comprises 5 -methylcytidine and
one or more selected from pseudouridine (ψ), N1-methyl-pseudouridine (m1ψ), and 5- methyl-uridine (m5U). In some embodiments, the RNA comprises 5-methylcytidine and N1- methyl-pseudouridine (m1ψ). In some embodiments, the RNA comprises 5-methylcytidine in place of each cytidine and N1-methyl-pseudouridine (m1ψ) in place of each uridine. [0318] In some embodiments of the present disclosure, the RNA is “replicon RNA” or simply a “replicon,” in particular “self-replicating RNA” or “self-amplifying RNA.” In one particularly preferred embodiment, the replicon or self-replicating RNA is derived from or comprises elements derived from a single-stranded (ss) RNA virus, in particular a positive- stranded ssRNA virus, such as an alphavirus. Alphaviruses are typical representatives of positive-stranded RNA viruses. Alphaviruses replicate in the cytoplasm of infected cells (for review of the alphaviral life cycle see José et al., Future Microbiol., 2009, vol.4, pp.837– 856, which is incorporated herein by reference in its entirety). The total genome length of many alphaviruses typically ranges between 11,000 and 12,000 nucleotides, and the genomic RNA typically has a 5’ cap, and a 3’ poly(A) tail. The genome of alphaviruses encodes non- structural proteins (involved in transcription, modification and replication of viral RNA and in protein modification) and structural proteins (forming the virus particle). There are typically two open reading frames (ORFs) in the genome. The four non-structural proteins (nsP1–nsP4) are typically encoded together by a first ORF beginning near the 5′ terminus of the genome, while alphavirus structural proteins are encoded together by a second ORF which is found downstream of the first ORF and extends near the 3’ terminus of the genome. Typically, the first ORF is larger than the second ORF, the ratio being roughly 2:1. In cells infected by an alphavirus, only the nucleic acid sequence encoding non-structural proteins is translated from the genomic RNA, while the genetic information encoding structural proteins is translatable from a subgenomic transcript, which is an RNA molecule that resembles eukaryotic messenger RNA (mRNA; Gould et al., 2010, Antiviral Res., vol.87 pp.111–124). Following infection, i.e., at early stages of the viral life cycle, the (+) stranded genomic RNA directly acts like a messenger RNA for the translation of the open reading frame encoding the non-structural polyprotein (nsP1234). [0319] Alphavirus-derived vectors have been proposed for delivery of foreign genetic information into target cells or target organisms. In simple approaches, a first ORF encodes an alphavirus-derived RNA-dependent RNA polymerase (replicase), which upon translation mediates self-amplification of the RNA. A second ORF encoding alphaviral structural
proteins is replaced by an open reading frame encoding a protein of interest, e.g., an incretin agent. Alphavirus-based trans-replication systems rely on alphavirus nucleotide sequence elements on two separate nucleic acid molecules: one nucleic acid molecule encodes a viral replicase, and the other nucleic acid molecule is capable of being replicated by said replicase in trans (hence the designation trans-replication system). Trans-replication requires the presence of both these nucleic acid molecules in a given host cell. The nucleic acid molecule capable of being replicated by the replicase in trans must comprise certain alphaviral sequence elements to allow recognition and RNA synthesis by the alphaviral replicase.
[0320] Features of a non-modified uridine platform may include, for example, one or more of intrinsic adjuvant effect, as well as good tolerability and safety. Features of modified uridine (e.g., pseudouridine) platform may include reduced adjuvant effect, blunted immune innate immune sensor activating capacity and thus good tolerability and safety. Features of self-amplifying platform may include, for example, long duration of protein expression, good tolerability and safety, higher likelihood for efficacy with very low vaccine dose.
[0321] The present disclosure provides particular RNA constructs optimized, for example, for improved manufacturability, encapsulation, expression level (and/or timing), etc. Certain components are discussed below, and certain preferred embodiments are exemplified herein.
Codon Optimization and GC Enrichment
[0322] As used herein, the term “codon-optimized” refers to alteration of codons in a coding region of a nucleic acid molecule (e.g., a polyribonucleotide) to reflect the typical codon usage of a host organism (e.g., a subject receiving a polyribonucleotide) without preferably altering the amino acid sequence encoded by the nucleic acid molecule. Within the context of the present disclosure, in some embodiments, coding regions are codon-optimized for optimal expression in a subject to be treated using the RNA molecules described herein. In some embodiments, codon-optimization may be performed such that codons for which frequently occurring tRNAs are available are inserted in place of “rare codons.” In some embodiments, codon-optimization may include increasing guanosine/cytosine (G/C) content of a coding region of RNA described herein as compared to the G/C content of the corresponding coding sequence of a wild type RNA, wherein the amino acid sequence encoded by the RNA is preferably not modified compared to the amino acid sequence.
[0323] In some embodiments, a coding sequence (also referred to as a “coding region”) is codon optimized for expression in the subject to whom a composition (e.g., a pharmaceutical composition) is to be administered (e.g., a human). Thus, in some embodiments, sequences in such a polynucleotide (e.g., a polyribonucleotide) may differ from wild type sequences encoding the relevant incretin agent, even when the amino acid sequence of the incretin agent is wild type.
[0324] In some embodiments, strategies for codon optimization for expression in a relevant subject (e.g., a human), and even, in some cases, for expression in a particular cell or tissue.
[0325] Various species exhibit particular bias for certain codons of a particular amino acid. Without wishing to be bound by any one theory, codon bias (differences in codon usage between organisms) often correlates with the efficiency of translation of messenger RNA (mRNA), which is in turn believed to be dependent on, among other things, the properties of the codons being translated and the availability of particular transfer RNA (tRNA) molecules. The predominance of selected tRNAs in a cell may generally be a reflection of the codons used most frequently in peptide synthesis. Accordingly, genes may be tailored for optimal gene expression in a given organism based on codon optimization. Codon usage tables are available, for example, at the “Codon Usage Database” available at www.kazusa.oijp/codon/ and these tables may be adapted in a number of ways. Computer algorithms for codon optimizing a particular sequence for expression in a particular subject or its cells are also available, such as Gene Forge (Aptagen; Jacobus, PA), are also available.
[0326] In some embodiments, a polynucleotide (e.g., a polyribonucleotide) of the present disclosure is codon optimized, wherein the codons in the polynucleotide (e.g., the polyribonucleotide) are adapted to human codon usage (herein referred to as “human codon optimized polynucleotide”). Codons encoding the same amino acid occur at different frequencies in a subject, e.g., a human. Accordingly, in some embodiments, the coding sequence of a polynucleotide of the present disclosure is modified such that the frequency of the codons encoding the same amino acid corresponds to the naturally occurring frequency of that codon according to the human codon usage, e.g., as shown in Table 13. For example, in the case of the amino acid Ala, the wild type coding sequence is preferably adapted in a way that the codon “GCC” is used with a frequency of 0.40, the codon “GCT” is used with a
frequency of 0.28, the codon “GCA” is used with a frequency of 0.22 and the codon “GCG” is used with 30 a frequency of 0. 10 etc. (see Table 13). Accordingly, in some embodiments, such a procedure (as exemplified for Ala) is applied for each amino acid encoded by the coding sequence of a polynucleotide to obtain sequences adapted to human codon usage.
Table 13: Human codon usage table with frequencies indicated for each amino acid.

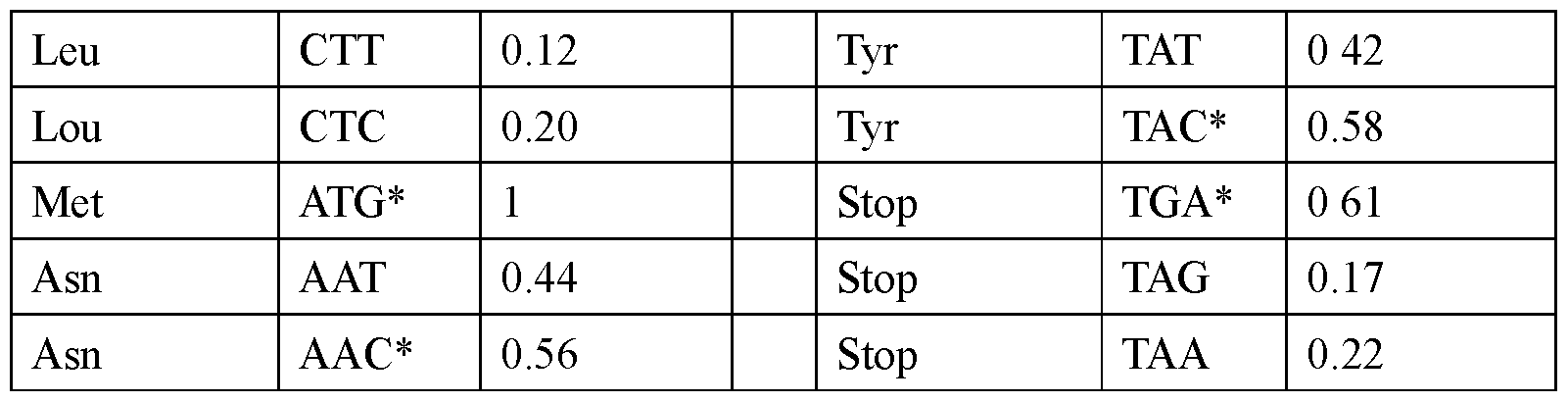
[0327] Certain strategies for codon optimization and/or G/C enrichment for human expression are described in W02002/098443, which is incorporated by reference herein in its entirety. In some embodiments, a coding sequence may be optimized using a multiparametric optimization strategy. In some embodiments, optimization parameters may include parameters that influence protein expression, which can be, for example, impacted on a transcription level, an mRNA level, and/or a translational level. In some embodiments, exemplary optimization parameters include, but are not limited to transcription-level parameters (including, e.g., GC content, consensus splice sites, cryptic splice sites, SD sequences, TATA boxes, termination signals, artificial recombination sites, and combinations thereof); mRNA-level parameters (including, e.g., RNA instability motifs, ribosomal entry sites, repetitive sequences, and combinations thereof); translation-level parameters (including, e.g., codon usage, premature poly(A) sites, ribosomal entry sites, secondary structures, and combinations thereof); or combinations thereof. In some embodiments, a coding sequence may be optimized by a GeneOptimizer algorithm as described in Fath et al. “Multiparameter RNA and Codon Optimization: A Standardized Tool to Assess and Enhance Autologous Mammalian Gene Expression” PloS ONE 6(3): el7596; Rabb et al., “The GeneOptimizer Algorithm: using a sliding window approach to cope with the vast sequence space in multiparameter DNA sequence optimization” Systems and Synthetic Biology (2010) 4:215-225; and Graft et al. “Codon-optimized genes that enable increased heterologous expression in mammalian cells and elicit efficient immune responses in mice after vaccination of naked DNA” Methods Mol Med (2004) 94: 197-210, the entire content of each of which is incorporated herein for the purposes described herein. In some embodiments, a coding sequence may be optimized by Eurofms’ adaption and optimization algorithm “GENEius” as described in Eurofms’ Application Notes: Eurofms’ adaption and optimization software “GENEius” in comparison to other optimization algorithms, the entire content of which is incorporated by reference for the purposes described herein.
[0328] In some embodiments, a coding sequence utilized in accordance with the present disclosure has G/C content which is increased compared to a wild type coding sequence for a relevant incretin agent. In some embodiments, guanosine/cytidine (G/C) content of a coding region is modified relative to a wild type coding sequence for a relevant incretin agent, but the amino acid sequence encoded by the polyribonucleotide is not modified.
[0329] Without wishing to be bound by any particular theory, it is proposed that GC enrichment may improve translation of a payload sequence. Typically, sequences having an increased G (guanosine )/C (cytidine) content are more stable than sequences having an increased A (adenosine )/U (uridine) content. In respect to the fact that several codons code for one and the same amino acid (so-called degeneration of the genetic code), the most favorable codons for the stability can be determined (so-called alternative codon usage). Depending on the amino acid to be encoded by a polyribonucleotide, there are various possibilities for modification of the ribonucleic acid sequence, compared to its wild type sequence. In particular, codons which contain A and/or U nucleosides can be modified by substituting these codons by other codons, which code for the same amino acids but contain no A and/or U or contain a lower content of A and/or U nucleosides.
[0330] In some embodiments, G/C content of a coding region of a polyribonucleotide described herein is increased by at least 1%, at least 2%, at least 3%, at least 4%, at least 5%, at least 6%, or even more compared to the G/C content of the coding region prior to codon optimization, e.g., of the wild type RNA. In some embodiments, G/C content of a coding region of a polyribonucleotide described herein is decreased by at least 1%, at least 2%, at least 3%, at least 4%, at least 5%, at least 6%, or even more compared to the G/C content of the coding region prior to codon optimization, e.g., of the wild type RNA.
[0331] In some embodiments, stability and translation efficiency of an polyribonucleotide may incorporate one or more elements established to contribute to stability and/or translation efficiency of the polyribonucleotide; exemplary such elements are described, for example, in W02007/036366 incorporated herein by reference. In some embodiments, to increase expression of a polyribonucleotide used according to the present disclosure, a polyribonucleotide may be modified within the coding region, i.e., the sequence encoding the expressed peptide or protein, without altering the sequence of the expressed
peptide or protein, for example so as to increase the GC -content to increase mRNA stability and/or to perform a codon optimization and, thus, enhance translation in cells.
Exemplary Polyribonucleotides encoding Incretin Agents
[0332] In some embodiments, a polyribonucleotide comprises a ribonucleic acid sequence that is at least 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or at least 99% identical to any of the sequences shown below in Table 14. The polyribonucleotides include exemplary' polyribonucleotide features described herein in addition to a sequence encoding die incretin agent and signal peptide, including a Cap proximal sequence (AAUA), a 5 " UTR sequence
AACUAGUAUUCUUCUGGUCCCCACAGACUCAGAGAGAACCCGCCACC (SEQ ID NO: 49), a PolyA tail sequence
AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAGCAUAUGACUAAAAAAAAAAAA AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA AAAAAA (SEQ ID NO: 50), and a 3 ’ UTR sequence
CUGGUACUGCAUGCACGCAAUGCUAGCUGCCCCUUUCCCGUCCUGGGUACCCC GAGUCUCCCCCGACCUCGGGUCCCAGGUAUGCUCCCACCUCCACCUGCCCCACU CACCACCUCUGCUAGUUCCAGACACCUCCCAAGCACGCAGCAAUGCAGCUCAA AACGCUUAGCCUAGCCACACCCCCACGGGAAACAGCAGUGAUUAACCUUUAGC AAUAAACGAAAGUUUAACUAAGCUAUACUAACCCCAGGGUUGGUCAAUUUCG UGCCAGCCACACC (SEQ ID NO: 51).
Table 14: Exemplary Polyribonucleotides encoding Incretin Agents where x2 and x4 examples include a linker and Furin cleavage site in between each repeat unit


























 ACGGCGAUCUGCUUGAGUGUGCCGACGACAGAGCUGACCU GGCCAAGUACAUCUGCGAGAAUCAGGACAGCAUCAGCAGC AAGCUGAAAGAGUGCUGCGAGAAGCCUCUGCUGGAAAAGA C G C A C U G C G C U G U A A U U A G A A G
ACGGCGAUCUGCUUGAGUGUGCCGACGACAGAGCUGACCU GGCCAAGUACAUCUGCGAGAAUCAGGACAGCAUCAGCAGC AAGCUGAAAGAGUGCUGCGAGAAGCCUCUGCUGGAAAAGA C G C A C U G C G C U G U A A U U A G A A G
 RNA Delivery Technologies [0333] Provided polyribonucleotides may be delivered for therapeutic applications described herein using any appropriate methods known in the art, including, e.g., delivery as naked RNAs, or delivery mediated by viral and/or non-viral vectors, polymer-based vectors, lipid-based vectors, nanoparticles (e.g., lipid nanoparticles, polymeric nanoparticles, lipid- polymer hybrid nanoparticles, etc.), and/or peptide-based vectors. See, e.g., Wadhwa et al. “Opportunities and Challenges in the Delivery of mRNA-Based Vaccines” Pharmaceutics
(2020) 102 (27 pages), the content of which is incorporated herein by reference, for information on various approaches that may be useful for delivery polyribonucleotides described herein. [0334] In some embodiments, one or more polyribonucleotides can be formulated with lipid nanoparticles for delivery (e.g., administration). [0335] In some embodiments, lipid nanoparticles can be designed to protect polyribonucleotides from extracellular RNases and/or engineered for systemic delivery of the RNA to target cells (e.g., liver, gut or pancreas cells). In some embodiments, such lipid nanoparticles may be particularly useful to deliver polyribonucleotides when polyribonucleotides are intraperitoneally, intravenously or intramuscularly administered to a subject. Particles for Delivery of at Least One Polyribonucleotide [0336] Polyribonucleotides provided herein can be delivered by particles. In the context of the present disclosure, the term “particle” relates to a structured entity formed by molecules or molecule complexes. In some embodiments, the term “particle” relates to a micro- or nano-sized structure, such as a micro- or nano-sized compact structure dispersed in a medium. In some embodiments, a particle is a nucleic acid containing particle such as a particle comprising a polyribonucleotide. [0337] Electrostatic interactions between positively charged molecules such as polymers and lipids and negatively charged nucleic acid (e.g., a polyribonucleotide) are involved in particle formation. This results in complexation and spontaneous formation of nucleic acid particles (e.g., ribonucleic acid particles). In some embodiments, a nucleic acid particle (e.g., ribonucleic acid particle) is a nanoparticle. [0338] A “nucleic acid particle” (e.g., a ribonucleic acid particle) are particles that encompass or contain a nucleic acid, and are used to deliver nucleic acid (e.g., a polyribonucleotide) to a target site of interest (e.g., cell, tissue, organ, and the like). A nucleic acid particle (e.g., a ribonucleic acid particle) may be formed from (i) at least one cationic or cationically ionizable lipid or lipid-like material, (ii) at least one cationic polymer such as protamine, or a mixture of (i) and (ii), and (iii) nucleic acid (e.g., a polyribonucleotide).
Nucleic acid particles (e.g., a ribonucleic acid particle) include lipid nanoparticles (lipid nanoparticle) and lipoplexes (LPX). [0339] In some embodiments, nucleic acid particles (e.g., ribonucleic acid particles) comprise more than one type of nucleic acid molecules (e.g., polyribonucleotides), where the molecular parameters of the nucleic acid molecules may be similar or different from each other, like with respect to molar mass or fundamental structural elements such as molecular architecture, capping, coding regions or other features. [0340] In some embodiments, provided nucleic acid particles (e.g., ribonucleic acid particles) can comprise lipid nanoparticles. As used in the present disclosure, “nanoparticle” refers to a particle having an average diameter suitable for parenteral administration. In various embodiments, lipid nanoparticles can have an average size (e.g., mean diameter) of about 30 nm to about 150 nm, about 40 nm to about 150 nm, about 50 nm to about 150 nm, about 60 nm to about 130 nm, about 70 nm to about 110 nm, about 70 nm to about 100 nm, about 70 to about 90 nm, or about 70 nm to about 80 nm. In some embodiments, lipid nanoparticles in accordance with the present disclosure can have an average size (e.g., mean diameter) of about 50 nm to about 100 nm. In some embodiments, lipid nanoparticles may have an average size (e.g., mean diameter) of about 50 nm to about 150 nm. In some embodiments, lipid nanoparticles may have an average size (e.g., mean diameter) of about 60 nm to about 120 nm. In some embodiments, lipid nanoparticles in accordance with the present disclosure can have an average size (e.g., mean diameter) of about 30 nm, 35 nm, 40 nm, 45 nm, 50 nm, 55 nm, 60 nm, 65 nm, 70 nm, 75 nm, 80 nm, 85 nm, 90 nm, 95 nm, 100 nm, 105 nm, 110 nm, 115 nm, 120 nm, 125 nm, 130 nm, 135 nm, 140 nm, 145 nm, or 150 nm. [0341] Nucleic acid particles (e.g., ribonucleic acid particles) described herein may exhibit a polydispersity index less than about 0.5, less than about 0.4, less than about 0.3, or about 0.2 or less. By way of example, the nucleic acid particles (e.g., ribonucleic acid particles) can exhibit a polydispersity index in a range of about 0.1 to about 0.3 or about 0.2 to about 0.3. [0342] Nucleic acid particles (e.g., ribonucleic acid particles) described herein can be characterized by an “N/P ratio,” which is the molar ratio of cationic (nitrogen) groups (the “N” in N/P) in the cationic polymer to the anionic (phosphate) groups (the “P” in N/P) in
RNA. It is understood that a cationic group is one that is either in cationic form (e.g., N+), or one that is ionizable to become cationic. Use of a single number in an N/P ratio (e.g., an N/P ratio of about 5) is intended to refer to that number over 1, e.g., an N/P ratio of about 5 is intended to mean 5:1. In some embodiments, a nucleic acid particle (e.g., a ribonucleic acid particle) described herein has an N/P ratio greater than or equal to 5. In some embodiments, a nucleic acid particle (e.g., a ribonucleic acid particle) described herein has an N/P ratio that is about 5, 6, 7, 8, 9, or 10. In some embodiments, an N/P ratio for a nucleic acid particle (e.g., a ribonucleic acid particle) described herein is from about 10 to about 50. In some embodiments, an N/P ratio for a nucleic acid particle (e.g., a ribonucleic acid particle) described herein is from about 10 to about 70. In some embodiments, an N/P ratio for a nucleic acid particle (e.g., a ribonucleic acid particle) described herein is from about 10 to about 120. [0343] Nucleic acid particles (e.g., ribonucleic acid particles) described herein can be prepared using a wide range of methods that may involve obtaining a colloid from at least one cationic or cationically ionizable lipid or lipid-like material and/or at least one cationic polymer and mixing the colloid with nucleic acid to obtain nucleic acid particles. [0344] The term “colloid” as used herein relates to a type of homogeneous mixture in which dispersed particles do not settle out. The insoluble particles in the mixture can be microscopic, with particle sizes between 1 and 1000 nanometers. The mixture may be termed a colloid or a colloidal suspension. Sometimes the term “colloid” only refers to the particles in the mixture and not the entire suspension. [0345] The term “average diameter” or “mean diameter” refers to the mean hydrodynamic diameter of particles as measured by dynamic laser light scattering (DLS) with data analysis using the so-called cumulant algorithm, which provides as results the so-called Z-average with the dimension of a length, and the polydispersity index (PI), which is dimensionless (Koppel, D., J. Chem. Phys.57, 1972, pp 4814-4820, ISO 13321, which is incorporated herein by reference). Herein “average diameter,” “mean diameter,” “diameter,” or “size” for particles is used synonymously with this value of the Z-average. [0346] The “polydispersity index” is preferably calculated based on dynamic light scattering measurements by the so-called cumulant analysis as mentioned in the definition of the “average diameter.” Under certain prerequisites, it can be taken as a measure of the size
distribution of an ensemble of ribonucleic acid nanoparticles (e.g., ribonucleic acid nanoparticles). [0347] Different types of nucleic acid particles have been described previously to be suitable for delivery of nucleic acid in particulate form (e.g., Kaczmarek et al., 2017, Genome Medicine 9, 60, which is incorporated herein by reference). For non-viral nucleic acid delivery vehicles, nanoparticle encapsulation of nucleic acid physically protects nucleic acid from degradation and, depending on the specific chemistry, can aid in cellular uptake and endosomal escape. [0348] The present disclosure describes particles comprising nucleic acid (e.g., a polyribonucleotide), at least one cationic or cationically ionizable lipid or lipid-like material, and/or at least one cationic polymer which associate with the nucleic acid (e.g., a polyribonucleotide) to form nucleic acid particles (e.g., ribonucleic acid particles, e.g., ribonucleic acid nanoparticles) and compositions comprising such particles. The nucleic acid particles (e.g., ribonucleic acid particles, e.g., ribonucleic acid nanoparticles) may comprise nucleic acid (e.g., a polyribonucleotide) which is complexed in different forms by non- covalent interactions to the particle. The particles described herein are not viral particles, in particular, they are not infectious viral particles, i.e., they are not able to virally infect cells. [0349] Some embodiments described herein relate to compositions, methods and uses involving more than one, e.g., 2, 3, 4, 5, 6 or even more nucleic acid species (e.g., polyribonucleotide species). [0350] In a nucleic acid particle (e.g., ribonucleic acid particle, e.g., ribonucleic acid nanoparticle) formulation, it is possible that each nucleic acid species (e.g., polyribonucleotide species) is separately formulated as an individual nucleic acid particle (e.g., ribonucleic acid particle, e.g., ribonucleic acid nanoparticle) formulation. In that case, each individual nucleic acid particle (e.g., ribonucleic acid particle, e.g., ribonucleic acid nanoparticle) formulation will comprise one nucleic acid species (e.g., polyribonucleotide species). The individual nucleic acid particle (e.g., ribonucleic acid particle, e.g., ribonucleic acid nanoparticle) formulations may be present as separate entities, e.g., in separate containers. Such formulations are obtainable by providing each nucleic acid species (e.g., polyribonucleotide species) separately (typically each in the form of a nucleic acid-containing solution) together with a particle-forming agent, thereby allowing the formation of particles.
Respective particles will contain exclusively the specific nucleic acid species (e.g., polyribonucleotide species) that is being provided when the particles are formed (individual particulate formulations). [0351] In some embodiments, a composition such as a pharmaceutical composition comprises more than one individual nucleic acid particle (e.g., ribonucleic acid particle, e.g., ribonucleic acid nanoparticle) formulation. Respective pharmaceutical compositions are referred to as “mixed particulate formulations.” Mixed particulate formulations according to the invention are obtainable by forming, separately, individual nucleic acid particle (e.g., ribonucleic acid particle, e.g., ribonucleic acid nanoparticle) formulations, as described above, followed by a step of mixing of the individual nucleic acid particle (e.g., ribonucleic acid particle, e.g., ribonucleic acid nanoparticle) formulations. By the step of mixing, a formulation comprising a mixed population of nucleic acid-containing particles is obtainable. Individual nucleic acid particle (e.g., ribonucleic acid particle, e.g., ribonucleic acid nanoparticle) populations may be together in one container, comprising a mixed population of individual nucleic acid particle (e.g., ribonucleic acid particle, e.g., ribonucleic acid nanoparticle) formulations. [0352] Alternatively, it is possible that different nucleic acid species (e.g., polyribonucleotide species) are formulated together as a “combined particulate formulation.” Such formulations are obtainable by providing a combined formulation (typically combined solution) of different nucleic acid species (e.g., polyribonucleotide species) species together with a particle-forming agent, thereby allowing the formation of particles. As opposed to a “mixed particulate formulation,” a “combined particulate formulation” will typically comprise particles that comprise more than one nucleic acid species (e.g., polyribonucleotide species) species. In a combined particulate composition different nucleic acid species (e.g., polyribonucleotide species) are typically present together in a single particle. [0353] In certain embodiments, nucleic acids (e.g., polyribonucleotides), when present in provided nucleic acid particles (e.g., ribonucleic acid particles, e.g., lipid nanoparticles) are resistant in aqueous solution to degradation with a nuclease. [0354] In some embodiments, nucleic acid particles (e.g., ribonucleic acid particles) are lipid nanoparticles. In some embodiments, lipid nanoparticles are liver-targeting lipid nanoparticles. In some embodiments, lipid nanoparticles are cationic lipid nanoparticles
comprising one or more cationic lipids (e.g., ones described herein). In some embodiments, cationic lipid nanoparticles may comprise at least one cationic lipid, at least one polymer- conjugated lipid, and at least one helper lipid (e.g., at least one neutral lipid). Cationic Polymeric Materials [0355] Cationic polymers have been recognized as useful for developing particle delivery vehicles, as reported in PCT App. Pub. No. WO2021/001417, the entirety of which is incorporated herein by reference. As used herein, the term “polymer” refers to a composition comprising one or more molecules that comprise repeating units of one or more monomers. As used herein, “polymer,” “polymeric material,” and “polymer composition” are used interchangeably, and unless otherwise specified, refer to a composition of polymer molecules. A person of skill in the art will appreciate that a polymer composition comprises polymer molecules having molecules of different lengths (e.g., comprising varying amounts of monomers). Polymer compositions described herein are characterized by one or more of a normalized molecular weight (Mn), a weight average molecular weight (Mw), and/or a polydispersity index (PDI). In some embodiments, such repeat units can all be identical (a “homopolymer”); alternatively, in some cases, there can be more than one type of repeat unit present within the polymeric material (a “heteropolymer” or a “copolymer”). In some cases, a polymer is biologically derived, e.g., a biopolymer such as a protein. In some cases, additional moieties can also be present in the polymeric material, for example targeting moieties such as those described herein. [0356] In some embodiments, a polymer utilized in accordance with the present disclosure may be a copolymer. Repeat units forming the copolymer can be arranged in any fashion. For example, in some embodiments, repeat units can be arranged in a random order; alternatively or additionally, in some embodiments, repeat units may be arranged in an alternating order, or as a “block” copolymer, e.g., comprising one or more regions each comprising a first repeat unit (e.g., a first block), and one or more regions each comprising a second repeat unit (e.g., a second block), etc. Block copolymers can have two (a diblock copolymer), three (a triblock copolymer), or more numbers of distinct blocks. [0357] In certain embodiments, a polymeric material for use in accordance with the present disclosure is biocompatible. In certain embodiments, a biocompatible material is
biodegradable, e.g., is able to degrade, chemically and/or biologically, within a physiological environment, such as within the body. [0358] In certain embodiments, a polymeric material may be or comprise protamine or polyalkyleneimine. [0359] As those skilled in the art are aware term “protamine” is often used to refer to any of various strongly basic proteins of relatively low molecular weight that are rich in arginine and are found associated especially with DNA in place of somatic histones in the sperm cells of various animals (as fish). In particular, the term “protamine” is often used to refer to proteins found in fish sperm that are strongly basic, are soluble in water, are not coagulated by heat, and yield chiefly arginine upon hydrolysis. In purified form, they are used in a long-acting formulation of insulin and to neutralize the anticoagulant effects of heparin. [0360] In some embodiments, the term “protamine” as used herein is refers to a protamine amino acid sequence obtained or derived from natural or biological sources, including fragments thereof and/or multimeric forms of said amino acid sequence or fragment thereof, as well as (synthesized) polypeptides which are artificial and specifically designed for specific purposes and cannot be isolated from native or biological sources. [0361] In some embodiments, a polyalkyleneimine comprises polyethylenimine (PEI) and/or polypropylenimine. In some embodiments, a preferred polyalkyleneimine is polyethyleneimine (PEI). In some embodiments, the average molecular weight of PEI is preferably 0.75∙102 to 107 Da, preferably 1000 to 105 Da, more preferably 10000 to 40000 Da, more preferably 15000 to 30000 Da, even more preferably 20000 to 25000 Da. [0362] Cationic materials (e.g., polymeric materials, including polycationic polymers) contemplated for use herein include those which are able to electrostatically bind nucleic acid. In some embodiments, cationic polymeric materials contemplated for use herein include any cationic polymeric materials with which nucleic acid can be associated, e.g., by forming complexes with the nucleic acid or forming vesicles in which the nucleic acid is enclosed or encapsulated. [0363] In some embodiments, particles described herein may comprise polymers other than cationic polymers, e.g., non-cationic polymeric materials and/or anionic polymeric
materials. Collectively, anionic and neutral polymeric materials are referred to herein as non- cationic polymeric materials. Lipid Compositions Lipids and Lipid-Like Materials [0364] Lipids and lipid-like materials have also been recognized as useful for developing particle delivery vehicles. The terms “lipid” and “lipid-like material” are broadly defined herein as molecules which comprise one or more hydrophobic moieties or groups and optionally also one or more hydrophilic moieties or groups. Molecules comprising hydrophobic moieties and hydrophilic moieties are also frequently denoted as amphiphiles. Lipids are usually poorly soluble in water. In an aqueous environment, the amphiphilic nature allows the molecules to self-assemble into organized structures and different phases. One of those phases consists of lipid bilayers, as they are present in vesicles, multilamellar/unilamellar liposomes, or membranes in an aqueous environment. Hydrophobicity can be conferred by the inclusion of a polar groups that include, but are not limited to, long-chain saturated and unsaturated aliphatic hydrocarbon groups and such groups substituted by one or more aromatic, cycloaliphatic, or heterocyclic group(s). The hydrophilic groups may comprise polar and/or charged groups and include carbohydrates, phosphate, carboxylic, sulfate, amino, sulfhydryl, nitro, hydroxyl, and other like groups. [0365] Often, an amphiphilic compound has a polar head attached to a long hydrophobic tail. In some embodiments, the polar portion is soluble in water, while the non- polar portion is insoluble in water. In addition, the polar portion may have either a formal positive charge, or a formal negative charge. Alternatively, the polar portion may have both a formal positive and a negative charge, and be a zwitterion or inner salt. For purposes of the disclosure, the amphiphilic compound can be, but is not limited to, one or a plurality of natural or non-natural lipids and lipid-like compounds. [0366] A “lipid-like material” is a substance that is structurally and/or functionally related to a lipid but may not be considered a lipid in a strict sense. For example, the term includes compounds that are able to form amphiphilic layers as they are present in vesicles, multilamellar/unilamellar liposomes, or membranes in an aqueous environment and includes surfactants, or synthesized compounds with both hydrophilic and hydrophobic moieties. Generally speaking the term refers to molecules which comprise hydrophilic and
hydrophobic moieties with different structural organization, which may or may not be similar to that of lipids. [0367] Specific examples of amphiphilic compounds that may be included in an amphiphilic layer include, but are not limited to, phospholipids, aminolipids and sphingolipids. [0368] Generally, lipids may be divided into eight categories: fatty acids, glycerolipids, glycerophospholipids, sphingolipids, saccharolipids, polyketides (derived from condensation of ketoacyl subunits), sterols and prenol lipids (derived from condensation of isoprene subunits). Although the term “lipid” is sometimes used as a synonym for fats, fats are a subgroup of lipids called triglycerides. Lipids also encompass molecules such as fatty acids and their derivatives (including tri-, di-, monoglycerides, and phospholipids), as well as sterol-containing metabolites such as cholesterol. [0369] Fatty acids are a diverse group of molecules made of a hydrocarbon chain that terminates with a carboxylic acid group; this arrangement confers the molecule with a polar, hydrophilic end, and a nonpolar, hydrophobic end that is insoluble in water. The carbon chain, typically between four and 24 carbons long, may be saturated or unsaturated, and may be attached to functional groups containing oxygen, halogens, nitrogen, and sulfur. If a fatty acid contains a double bond, there is the possibility of either a cis or trans geometric isomerism, which significantly affects the molecule’s configuration. Cis-double bonds cause the fatty acid chain to bend, an effect that is compounded with more double bonds in the chain. Other major lipid classes in the fatty acid category are the fatty esters and fatty amides. [0370] Glycerolipids are composed of mono-, di-, and tri-substituted glycerols, the best-known being the fatty acid triesters of glycerol, called triglycerides. The word “triacylglycerol” is sometimes used synonymously with “triglyceride”. In these compounds, the three hydroxyl groups of glycerol are each esterified, typically by different fatty acids. Additional subclasses of glycerolipids are represented by glycosylglycerols, which are characterized by the presence of one or more sugar residues attached to glycerol via a glycosidic linkage. [0371] Glycerophospholipids are amphipathic molecules (containing both hydrophobic and hydrophilic regions) that contain a glycerol core linked to two fatty acid-
derived “tails” by ester linkages and to one “head” group by a phosphate ester linkage. Examples of glycerophospholipids, usually referred to as phospholipids (though sphingomyelins are also classified as phospholipids) are phosphatidylcholine (also known as PC, GPCho or lecithin), phosphatidylethanolamine (PE or GPEtn) and phosphatidylserine (PS or GPSer). [0372] Sphingolipids are members of a complex family of compounds that share a common structural feature, a sphingoid base backbone. The major sphingoid base in mammals is commonly referred to as sphingosine. Ceramides (N-acyl-sphingoid bases) are a major subclass of sphingoid base derivatives with an amide-linked fatty acid. The fatty acids are typically saturated or mono-unsaturated with chain lengths from 16 to 26 carbon atoms. The major phosphosphingolipids of mammals are sphingomyelins (ceramide phosphocholines), whereas insects contain mainly ceramide phosphoethanolamines and fungi have phytoceramide phosphoinositols and mannose-containing headgroups. The glycosphingolipids are a diverse family of molecules composed of one or more sugar residues linked via a glycosidic bond to the sphingoid base. Examples of these are the simple and complex glycosphingolipids such as cerebrosides and gangliosides. [0373] Sterols, such as cholesterol and its derivatives, or tocopherol and its derivatives, are important components of membrane lipids, along with the glycerophospholipids and sphingomyelins. [0374] Saccharolipids are compounds in which fatty acids are linked directly to a sugar backbone, forming structures that are compatible with membrane bilayers. In the saccharolipids, a monosaccharide substitutes for the glycerol backbone present in glycerolipids and glycerophospholipids. The most familiar saccharolipids are the acylated glucosamine precursors of the Lipid A component of the lipopolysaccharides in Gram- negative bacteria. Typical lipid A molecules are disaccharides of glucosamine, which are derivatized with as many as seven fatty-acyl chains. The minimal lipopolysaccharide required for growth in E. coli is Kdo2-Lipid A, a hexa-acylated disaccharide of glucosamine that is glycosylated with two 3-deoxy-D-manno-octulosonic acid (Kdo) residues. [0375] Polyketides are synthesized by polymerization of acetyl and propionyl subunits by classic enzymes as well as iterative and multimodular enzymes that share mechanistic features with the fatty acid synthases. They comprise a large number of
secondary metabolites and natural products from animal, plant, bacterial, fungal and marine sources, and have great structural diversity. Many polyketides are cyclic molecules whose backbones are often further modified by glycosylation, methylation, hydroxylation, oxidation, or other processes. [0376] Lipids and lipid-like materials may be cationic, anionic or neutral. Neutral lipids or lipid-like materials exist in an uncharged or neutral zwitterionic form at a selected pH. [0377] In some embodiments, suitable lipids or lipid-like materials for use in the present disclosure include those described in WO2020/128031 and US2020/0163878, the entire contents of each of which are incorporated herein by reference for the purposes described herein. Cationic or Cationically Ionizable Lipids or Lipid-like Materials [0378] In some embodiments cationic or cationically ionizable lipids or lipid-like materials contemplated for use herein include any cationic or cationically ionizable lipids or lipid-like materials which are able to electrostatically bind nucleic acid. In one embodiment, cationic or cationically ionizable lipids or lipid-like materials contemplated for use herein can be associated with nucleic acid, e.g., by forming complexes with the nucleic acid or forming vesicles in which the nucleic acid is enclosed or encapsulated. [0379] Cationic lipids or lipid-like materials are characterized in that they have a net positive charge (e.g., at a relevant pH). Cationic lipids or lipid-like materials bind negatively charged nucleic acid by electrostatic interaction. Generally, cationic lipids possess a lipophilic moiety, such as a sterol, an acyl chain, a diacyl or more acyl chains, and the head group of the lipid typically carries the positive charge. [0380] In certain embodiments, a cationic lipid or lipid-like material has a net positive charge only at certain pH, in particular acidic pH, while it has preferably no net positive charge, preferably has no charge, i.e., it is neutral, at a different, preferably higher pH such as physiological pH. This ionizable behavior is thought to enhance efficacy through helping with endosomal escape and reducing toxicity as compared with particles that remain cationic at physiological pH.
[0381] In some embodiments, a cationic or cationically ionizable lipid or lipid-like material comprises a head group which includes at least one nitrogen atom (N) which is positive charged or capable of being protonated. [0382] Examples of cationic lipids include, but are not limited to 1,2-dioleoyl-3- trimethylammonium propane (DOTAP); N,N-dimethyl-2,3-dioleyloxypropylamine (DODMA), 1,2-di-O-octadecenyl-3-trimethylammonium propane (DOTMA), 3-(N—(N′,N′- dimethylaminoethane)-carbamoyl)cholesterol (DC-Chol), dimethyldioctadecylammonium (DDAB); 1,2-dioleoyl-3-dimethylammonium-propane (DODAP); 1,2-diacyloxy-3- dimethylammonium propanes; 1,2-dialkyloxy-3-dimethylammonium propanes; dioctadecyldimethyl ammonium chloride (DODAC), 1,2-distearyloxy-N,N-dimethyl-3- aminopropane (DSDMA), 2,3-di(tetradecoxy)propyl-(2-hydroxyethyl)-dimethylazanium (DMRIE), 1,2-dimyristoyl-sn-glycero-3-ethylphosphocholine (DMEPC), l,2-dimyristoyl-3- trimethylammonium propane (DMTAP), 1,2-dioleyloxypropyl-3-dimethyl-hydroxyethyl ammonium bromide (DORIE), and 2,3-dioleoyloxy- N-[2(spermine carboxamide)ethyl]-N,N- dimethyl-l-propanamium trifluoroacetate (DOSPA), 1,2-dilinoleyloxy-N,N- dimethylaminopropane (DLinDMA), 1,2-dilinolenyloxy-N,N-dimethylaminopropane (DLenDMA), dioctadecylamidoglycyl spermine (DOGS), 3-dimethylamino-2-(cholest-5-en- 3-beta-oxybutan-4-oxy)-1-(cis,cis-9,12-oc-tadecadienoxy)propane (CLinDMA), 2-[5′- (cholest-5-en-3-beta-oxy)-3′-oxapentoxy)-3-dimethyl-1-(cis,cis-9′,12′- octadecadienoxy)propane (CpLinDMA), N,N-dimethyl-3,4-dioleyloxybenzylamine (DMOBA), 1,2-N,N′-dioleylcarbamyl-3-dimethylaminopropane (DOcarbDAP), 2,3- Dilinoleoyloxy-N,N-dimethylpropylamine (DLinDAP), 1,2-N,N′-Dilinoleylcarbamyl-3- dimethylaminopropane (DLincarbDAP), 1,2-Dilinoleoylcarbamyl-3-dimethylaminopropane (DLinCDAP), 2,2-dilinoleyl-4-dimethylaminomethyl-[1,3]-dioxolane (DLin-K-DMA), 2,2- dilinoleyl-4-dimethylaminoethyl-[1,3]-dioxolane (DLin-K-XTC2-DMA), 2,2-dilinoleyl-4-(2- dimethylaminoethyl)-[1,3]-dioxolane (DLin-KC2-DMA), heptatriaconta-6,9,28,31-tetraen- 19-yl-4-(dimethylamino)butanoate (DLin-MC3-DMA), N-(2-Hydroxyethyl)-N,N-dimethyl- 2,3-bis(tetradecyloxy)-1-propanaminium bromide (DMRIE), (±)-N-(3-aminopropyl)-N,N- dimethyl-2,3-bis(cis-9-tetradecenyloxy)-1-propanaminium bromide (GAP-DMORIE), (±)-N- (3-aminopropyl)-N,N-dimethyl-2,3-bis(dodecyloxy)-1-propanaminium bromide (GAP- DLRIE), (±)-N-(3-aminopropyl)-N,N-dimethyl-2,3-bis(tetradecyloxy)-1-propanaminium bromide (GAP-DMRIE), N-(2-Aminoethyl)-N,N-dimethyl-2,3-bis(tetradecyloxy)-1-
propanaminium bromide (βAE-DMRIE), N-(4-carboxybenzyl)-N,N-dimethyl-2,3- bis(oleoyloxy)propan-1-aminium (DOBAQ), 2-({8-[(3β)-cholest-5-en-3-yloxy]octyl}oxy)- N,N-dimethyl-3-[(9Z,12Z)-octadeca-9,12-dien-1-yloxy]propan-1-amine (Octyl-CLinDMA), 1,2-dimyristoyl-3-dimethylammonium-propane (DMDAP), 1,2-dipalmitoyl-3- dimethylammonium-propane (DPDAP), N1-[2-((1S)-1-[(3-aminopropyl)amino]-4-[di(3- amino-propyl)amino]butylcarboxamido)ethyl]-3,4-di[oleyloxy]-benzamide (MVL5), 1,2- dioleoyl-sn-glycero-3-ethylphosphocholine (DOEPC), 2,3-bis(dodecyloxy)-N-(2- hydroxyethyl)-N,N-dimethylpropan-1-amonium bromide (DLRIE), N-(2-aminoethyl)-N,N- dimethyl-2,3-bis(tetradecyloxy)propan-1-aminium bromide (DMORIE), di((Z)-non-2-en-1- yl) 8,8’-((((2(dimethylamino)ethyl)thio)carbonyl)azanediyl)dioctanoate (ATX), N,N- dimethyl-2,3-bis(dodecyloxy)propan-1-amine (DLDMA), N,N-dimethyl-2,3- bis(tetradecyloxy)propan-1-amine (DMDMA), Di((Z)-non-2-en-1-yl)-9-((4- (dimethylaminobutanoyl)oxy)heptadecanedioate (L319), N-Dodecyl-3-((2- dodecylcarbamoyl-ethyl)-{2-[(2-dodecylcarbamoyl-ethyl)-2-{(2-dodecylcarbamoyl-ethyl)-[2- (2-dodecylcarbamoyl-ethylamino)-ethyl]-amino}-ethylamino)propionamide (lipidoid 98N12- 5), 1-[2-[bis(2-hydroxydodecyl)amino]ethyl-[2-[4-[2-[bis(2 hydroxydodecyl)amino]ethyl]piperazin-1-yl]ethyl]amino]dodecan-2-ol (lipidoid C12-200), LIPOFECTIN® (commercially available cationic liposomes comprising DOTMA and 1 ,2- dioleoyl-sn-3phosphoethanolamine (DOPE), from GIBCO/BRL, Grand Island, N.Y.); LIPOFECTAMINE® (commercially available cationic liposomes comprising N-(1 - (2,3dioleyloxy)propyl)-N-(2-(sperminecarboxamido)ethyl)-N,N-dimethylammonium trifluoroacetate (DOSPA) and (DOPE), from GIBCO/BRL); and TRANSFECTAM® (commercially available cationic lipids comprising dioctadecylamidoglycyl carboxyspermine (DOGS) in ethanol from Promega Corp., Madison, Wis.) or any combination of any of the foregoing. Further suitable cationic lipids for use in the present disclosure include those described in WO2020/128031 and US2020/0163878, the entire contents of each of which are incorporated herein by reference for the purposes described herein. Further suitable cationic lipids for use in the present disclosure include those described in WO2010/053572 (including C12-200 described at paragraph [00225]) and WO2012/170930, both of which are incorporated herein by reference for the purposes described herein. Additional suitable cationic lipids for use in the present disclosure include HGT4003, HGT5000, HGTS001, HGT5001, HGT5002 (see US2015/0140070, which is incorporated herein by reference in its entirety).
[0383] In some embodiments, formulations that are useful for pharmaceutical compositions (e.g., immunogenic compositions, e.g., vaccines) compositions as described herein can comprise at least one cationic lipid. Representative cationic lipids include, but are not limited to, 1 ,2-dilinoleyoxy-3-(dimethylamino)acetoxypropane (DLin-DAC), 1 ,2- dilinoleyoxy-3morpholinopropane (DLin-MA), 1,2-dilinoleoyl-3-dimethylaminopropane (DLinDAP), 1 ,2-dilinoleylthio-3-dimethylaminopropane (DLin-S-DMA), 1 -linoleoyl-2- linoleyloxy-3dimethylaminopropane (DLin-2-DMAP), 1 ,2-dilinoleyloxy-3- trimethylaminopropane chloride salt (DLin-TMA.CI), 1 ,2-dilinoleoyl-3- trimethylaminopropane chloride salt (DLin-TAP.CI), 1 ,2-dilinoleyloxy-3-(N- methylpiperazino)propane (DLin-MPZ), 3-(N,Ndilinoleylamino)-1 ,2-propanediol (DLinAP), 3-(N,N-dioleylamino)-1 ,2-propanediol (DOAP), 1 ,2-dilinoleyloxo-3-(2-N,N- dimethylamino)ethoxypropane (DLin-EG-DMA), and 2,2-dilinoleyl-4- dimethylaminomethyl-[1 ,3]-dioxolane (DLin-K-DMA), 2,2-dilinoleyl-4-(2- dimethylaminoethyl)-[1 ,3]-dioxolane (DLin-KC2-DMA); dilinoleyl-methyl-4- dimethylaminobutyrate (DLin-MC3-DMA); MC3 (US2010/0324120, which is incorporated herein by reference in its entirety). [0384] In some embodiments, amino or cationic lipids useful in accordance with the present disclosure have at least one protonatable or deprotonatable group, such that the lipid is positively charged at a pH at or below physiological pH (e.g., pH 7.4), and neutral at a second pH, preferably at or above physiological pH. It will, of course, be understood that the addition or removal of protons as a function of pH is an equilibrium process, and that the reference to a charged or a neutral lipid refers to the nature of the predominant species and does not require that all of lipids have to be present in the charged or neutral form. Lipids having more than one protonatable or deprotonatable group, or which are zwitterionic, are not excluded and may likewise suitable in the context of the present invention. [0385] In some embodiments, a protonatable lipid has a pKa of the protonatable group in the range of about 4 to about 11, e.g., a pKa of about 5 to about 7. [0386] In some embodiments, a cationic lipid may comprise from about 10 mol % to about 100 mol %, about 20 mol % to about 100 mol %, about 30 mol % to about 100 mol %, about 40 mol % to about 100 mol %, or about 50 mol % to about 100 mol % of total lipid present in a lipid composition utilized in accordance with the present disclosure.
Additional Lipids or Lipid-like materials [0387] In some embodiments, formulations utilized in accordance with the present disclosure may comprise lipids or lipid-like materials other than cationic or cationically ionizable lipids or lipid-like materials, i.e., non-cationic lipids or lipid-like materials (including non-cationically ionizable lipids or lipid-like materials). Collectively, anionic and neutral lipids or lipid-like materials are referred to herein as non-cationic lipids or lipid-like materials. In some embodiments, optimizing a formulation of nucleic acid particles by addition of other hydrophobic moieties, such as cholesterol and lipids, in addition to an ionizable/cationic lipid or lipid-like material may, for example, enhance particle stability and efficacy of nucleic acid delivery. [0388] In some embodiments, a lipid or lipid-like material may be incorporated which may or may not affect the overall charge of particles. In certain embodiments, such lipid or lipid-like material is a non-cationic lipid or lipid-like material. [0389] In some embodiments, a non-cationic lipid may comprise, e.g., one or more anionic lipids and/or neutral lipids. An “anionic lipid” is negatively charged (e.g., at a selected pH). [0390] A “helper lipid” exists either in an uncharged, neutral zwitterionic form (e.g., at a selected pH), or, in some embodiments, as having a cationic or positive charge at physiological pH. In some embodiments, a formulation comprises one of the following helper lipid components: (1) a phospholipid, (2) cholesterol or a derivative thereof; or (3) a mixture of a phospholipid and cholesterol or a derivative thereof. Examples of cholesterol derivatives include, but are not limited to, cholestanol, cholestanone, cholestenone, coprostanol, cholesteryl-2’-hydroxyethyl ether, cholesteryl-4’-hydroxybutyl ether, tocopherol and derivatives thereof, and mixtures thereof. [0391] Specific exemplary phospholipids that can be used include, but are not limited to, phosphatidylcholines, phosphatidylethanolamines, phosphatidylglycerols, phosphatidic acids, phosphatidylserines or sphingomyelin. Such phospholipids include in particular diacylphosphatidylcholines, such as distearoylphosphatidylcholine (DSPC), dioleoylphosphatidylcholine (DOPC), dimyristoylphosphatidylcholine (DMPC), dipentadecanoylphosphatidylcholine, dilauroylphosphatidylcholine,
dipalmitoylphosphatidylcholine (DPPC), diarachidoylphosphatidylcholine (DAPC), dibehenoylphosphatidylcholine (DBPC), ditricosanoylphosphatidylcholine (DTPC), dilignoceroylphatidylcholine (DLPC), palmitoyloleoyl-phosphatidylcholine (POPC), 1,2-di- O-octadecenyl-sn-glycero-3-phosphocholine (18:0 Diether PC), 1-oleoyl-2- cholesterylhemisuccinoyl-sn-glycero-3-phosphocholine (OChemsPC), 1-hexadecyl-sn- glycero-3-phosphocholine (C16 Lyso PC) and phosphatidylethanolamines, in particular diacylphosphatidylethanolamines, such as dioleoylphosphatidylethanolamine (DOPE), distearoyl-phosphatidylethanolamine (DSPE), dipalmitoyl-phosphatidylethanolamine (DPPE), dimyristoyl-phosphatidylethanolamine (DMPE), dilauroyl- phosphatidylethanolamine (DLPE), diphytanoyl-phosphatidylethanolamine (DPyPE), and further phosphatidylethanolamine lipids with different hydrophobic chains. [0392] In certain embodiments, a formulation utilized in accordance with the present disclosure includes DSPC or DSPC and cholesterol. [0393] In some embodiments, a formulation utilized in accordance with the present disclosure comprises a helper lipid that is ionizable or cationic. [0394] In certain embodiments, formulations utilized in accordance with the present disclosure include both a cationic lipid and an additional (non-cationic) lipid. [0395] In some embodiments, formulations herein include a polymer conjugated lipid such as a pegylated lipid. “Pegylated lipids” or “PEG-conjugated lipids” comprise both a lipid portion and a polyethylene glycol portion. Pegylated lipids are known in the art. [0396] Without wishing to be bound by theory, the amount of (total) cationic lipid compared to the amount of other lipid(s) in formulation may affect important characteristics, such as charge, particle size, stability, tissue selectivity, and bioactivity of the nucleic acid. In some embodiments, the molar ratio of the at least one cationic lipid to the at least one additional lipid is from about 10:0 to about 1:9, about 4:1 to about 1:2, or about 3:1 to about 1:1. Lipoplex Particles [0397] In certain embodiments of the present disclosure, the RNA described herein may be present in RNA lipoplex particles.
[0398] An “RNA lipoplex particle” contains lipid, in particular cationic lipid, and RNA. Electrostatic interactions between positively charged liposomes and negatively charged RNA results in complexation and spontaneous formation of RNA lipoplex particles. Positively charged liposomes may be generally synthesized using a cationic lipid, such as DOTMA, and additional lipids, such as DOPE. In one embodiment, a RNA lipoplex particle is a nanoparticle. [0399] In certain embodiments, RNA lipoplex particles include both a cationic lipid and an additional lipid. In an exemplary embodiment, the cationic lipid is DOTMA and the additional lipid is DOPE. [0400] In some embodiments, the molar ratio of the at least one cationic lipid to the at least one additional lipid is from about 10:0 to about 1:9, about 4:1 to about 1:2, or about 3:1 to about 1:1. In specific embodiments, the molar ratio may be about 3:1, about 2.75:1, about 2.5:1, about 2.25:1, about 2:1, about 1.75:1, about 1.5:1, about 1.25:1, or about 1:1. In an exemplary embodiment, the molar ratio of the at least one cationic lipid to the at least one additional lipid is about 2:1. [0401] In some embodiments, RNA lipoplex particles have an average diameter that in one embodiment ranges from about 200 nm to about 1000 nm, from about 200 nm to about 800 nm, from about 250 to about 700 nm, from about 400 to about 600 nm, from about 300 nm to about 500 nm, or from about 350 nm to about 400 nm. In specific embodiments, the RNA lipoplex particles have an average diameter of about 200 nm, about 225 nm, about 250 nm, about 275 nm, about 300 nm, about 325 nm, about 350 nm, about 375 nm, about 400 nm, about 425 nm, about 450 nm, about 475 nm, about 500 nm, about 525 nm, about 550 nm, about 575 nm, about 600 nm, about 625 nm, about 650 nm, about 700 nm, about 725 nm, about 750 nm, about 775 nm, about 800 nm, about 825 nm, about 850 nm, about 875 nm, about 900 nm, about 925 nm, about 950 nm, about 975 nm, or about 1000 nm. In an embodiment, the RNA lipoplex particles have an average diameter that ranges from about 250 nm to about 700 nm. In another embodiment, the RNA lipoplex particles have an average diameter that ranges from about 300 nm to about 500 nm. In an exemplary embodiment, the RNA lipoplex particles have an average diameter of about 400 nm. [0402] RNA lipoplex particles and compositions comprising RNA lipoplex particles described herein are useful for delivery of RNA to a target tissue after parenteral
administration, in particular after intravenous administration. The RNA lipoplex particles may be prepared using liposomes that may be obtained by injecting a solution of the lipids in ethanol into water or a suitable aqueous phase. In one embodiment, the aqueous phase has an acidic pH. In one embodiment, the aqueous phase comprises acetic acid, e.g., in an amount of about 5 mM. Liposomes may be used for preparing RNA lipoplex particles by mixing the liposomes with RNA. In one embodiment, the liposomes and RNA lipoplex particles comprise at least one cationic lipid and at least one additional lipid. In one embodiment, the at least one cationic lipid comprises 1,2-di-O-octadecenyl-3-trimethylammonium propane (DOTMA) and/or 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP). In one embodiment, the at least one additional lipid comprises 1,2-di-(9Z-octadecenoyl)-sn-glycero- 3-phosphoethanolamine (DOPE), cholesterol (Chol) and/or 1,2-dioleoyl-sn-glycero-3- phosphocholine (DOPC). In one embodiment, the at least one cationic lipid comprises 1,2-di- O-octadecenyl-3-trimethylammonium propane (DOTMA) and the at least one additional lipid comprises 1,2-di-(9Z-octadecenoyl)-sn-glycero-3-phosphoethanolamine (DOPE). In one embodiment, the liposomes and RNA lipoplex particles comprise 1,2-di-O-octadecenyl-3- trimethylammonium propane (DOTMA) and 1,2-di-(9Z-octadecenoyl)-sn-glycero-3- phosphoethanolamine (DOPE). [0403] Spleen targeting RNA lipoplex particles are described in WO2013/143683, incorporated herein by reference. It has been found that RNA lipoplex particles having a net negative charge may be used to preferentially target spleen tissue or spleen cells such as antigen-presenting cells, in particular dendritic cells. Accordingly, following administration of the RNA lipoplex particles, RNA accumulation and/or RNA expression in the spleen occurs. Thus, RNA lipoplex particles of the disclosure may be used for expressing RNA in the spleen. In an embodiment, after administration of the RNA lipoplex particles, no or essentially no RNA accumulation and/or RNA expression in the lung and/or liver occurs. In one embodiment, after administration of the RNA lipoplex particles, RNA accumulation and/or RNA expression in antigen presenting cells, such as professional antigen presenting cells in the spleen occurs. Thus, RNA lipoplex particles of the disclosure may be used for expressing RNA in such antigen presenting cells. In one embodiment, the antigen presenting cells are dendritic cells and/or macrophages.
Lipid Nanoparticles (LNPs) [0404] In some embodiments, nucleic acid such as RNA described herein is administered in the form of lipid nanoparticles (LNPs). In some embodiments, LNPs may comprise any lipid capable of forming a particle to which the one or more nucleic acid molecules are attached, or in which the one or more nucleic acid molecules are encapsulated. [0405] In some embodiments, an LNP comprises one or more cationic lipids, and one or more stabilizing lipids. Stabilizing lipids include neutral lipids, helper lipids, and pegylated lipids. [0406] In some embodiments, an LNP comprises a cationic lipid, a helper lipid, a sterol, a polymer conjugated lipid; and an RNA, encapsulated within or associated with the lipid nanoparticle. [0407] In some embodiments, a helper lipid is a phosphotidylcholine such as 1,2- distearoyl-sn-glycero-3-phosphocholine (DSPC), 1,2-Dipalmitoyl-sn-glycero-3- phosphocholine (DPPC), 1,2-Dimyristoyl-sn-glycero-3-phosphocholine (DMPC), 1- palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), l ,2-dioleoyl-sn-glycero-3- phosphocholine (DOPC), phophatidylethanolamines such as 1,2-dioleoyl-sn-glycero-3- phosphoethanolamine (DOPE), sphingomyelins (SM). In some embodiments, a helper lipid is a 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP), 1-α-phosphatidylserine (PS), or DOPE. In some embodiments, a helper lipid is selected from the group consisting of DSPC, DPPC, DMPC, DOPC, POPC, DOPE, DOPG, DPPG, POPE, DPPE, DMPE, DSPE, DOTAP, PS, and SM. In some embodiments, the helper lipid is selected from the group consisting of DSPC, DPPC, DMPC, DOPC, POPC, DOPE, DOTAP, PS, and SM. In some embodiments, the helper lipid is DSPC. In some embodiments, the helper lipid is DOTAP. In some embodiments, the helper lipid is DOPE. In some embodiments, the helper lipid is PS. [0408] In some embodiments, a sterol is cholesterol. [0409] In some embodiments, a polymer conjugated lipid is a pegylated lipid (a PEG lipid). In some embodiments, a PEG lipid is selected from pegylated diacylglycerol (PEG- DAG) such as l-(monomethoxy-polyethyleneglycol)- 2,3-dimyristoylglycerol (PEG-DMG) (e.g., 1,2-dimyristoyl-rac-glycero-3-methoxypolyethylene glycol-2000 (PEG2000-DMG)), a pegylated phosphatidylethanoloamine (PEG-PE) a PEG succinate diacylglycerol (PEG-S-
DAG) such as 4-O-(2',3'-di(tetradecanoyloxy)propyl-1-O-(ω- methoxy(polyethoxy)ethyl)butanedioate (PEG-S-DMG), a pegylated ceramide (PEG-cer), or a PEG dialkoxypropylcarbamate such as ω-m ethoxy (polyethoxy)ethyl-N-(2,3- di(tetradecanoxy)propyl)carbamate, and 2,3-di(tetradecanoxy)propy 1-Ν-(ω methoxy(polyethoxy)ethyl)carbamate. In some embodiments, a pegylated lipid is 1,2- dimyristoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (C14-PEG2000). In some embodiments, a pegylated lipid has the following structure: or a pharmaceutically acc
RNA Delivery Technologies [0333] Provided polyribonucleotides may be delivered for therapeutic applications described herein using any appropriate methods known in the art, including, e.g., delivery as naked RNAs, or delivery mediated by viral and/or non-viral vectors, polymer-based vectors, lipid-based vectors, nanoparticles (e.g., lipid nanoparticles, polymeric nanoparticles, lipid- polymer hybrid nanoparticles, etc.), and/or peptide-based vectors. See, e.g., Wadhwa et al. “Opportunities and Challenges in the Delivery of mRNA-Based Vaccines” Pharmaceutics
(2020) 102 (27 pages), the content of which is incorporated herein by reference, for information on various approaches that may be useful for delivery polyribonucleotides described herein. [0334] In some embodiments, one or more polyribonucleotides can be formulated with lipid nanoparticles for delivery (e.g., administration). [0335] In some embodiments, lipid nanoparticles can be designed to protect polyribonucleotides from extracellular RNases and/or engineered for systemic delivery of the RNA to target cells (e.g., liver, gut or pancreas cells). In some embodiments, such lipid nanoparticles may be particularly useful to deliver polyribonucleotides when polyribonucleotides are intraperitoneally, intravenously or intramuscularly administered to a subject. Particles for Delivery of at Least One Polyribonucleotide [0336] Polyribonucleotides provided herein can be delivered by particles. In the context of the present disclosure, the term “particle” relates to a structured entity formed by molecules or molecule complexes. In some embodiments, the term “particle” relates to a micro- or nano-sized structure, such as a micro- or nano-sized compact structure dispersed in a medium. In some embodiments, a particle is a nucleic acid containing particle such as a particle comprising a polyribonucleotide. [0337] Electrostatic interactions between positively charged molecules such as polymers and lipids and negatively charged nucleic acid (e.g., a polyribonucleotide) are involved in particle formation. This results in complexation and spontaneous formation of nucleic acid particles (e.g., ribonucleic acid particles). In some embodiments, a nucleic acid particle (e.g., ribonucleic acid particle) is a nanoparticle. [0338] A “nucleic acid particle” (e.g., a ribonucleic acid particle) are particles that encompass or contain a nucleic acid, and are used to deliver nucleic acid (e.g., a polyribonucleotide) to a target site of interest (e.g., cell, tissue, organ, and the like). A nucleic acid particle (e.g., a ribonucleic acid particle) may be formed from (i) at least one cationic or cationically ionizable lipid or lipid-like material, (ii) at least one cationic polymer such as protamine, or a mixture of (i) and (ii), and (iii) nucleic acid (e.g., a polyribonucleotide).
Nucleic acid particles (e.g., a ribonucleic acid particle) include lipid nanoparticles (lipid nanoparticle) and lipoplexes (LPX). [0339] In some embodiments, nucleic acid particles (e.g., ribonucleic acid particles) comprise more than one type of nucleic acid molecules (e.g., polyribonucleotides), where the molecular parameters of the nucleic acid molecules may be similar or different from each other, like with respect to molar mass or fundamental structural elements such as molecular architecture, capping, coding regions or other features. [0340] In some embodiments, provided nucleic acid particles (e.g., ribonucleic acid particles) can comprise lipid nanoparticles. As used in the present disclosure, “nanoparticle” refers to a particle having an average diameter suitable for parenteral administration. In various embodiments, lipid nanoparticles can have an average size (e.g., mean diameter) of about 30 nm to about 150 nm, about 40 nm to about 150 nm, about 50 nm to about 150 nm, about 60 nm to about 130 nm, about 70 nm to about 110 nm, about 70 nm to about 100 nm, about 70 to about 90 nm, or about 70 nm to about 80 nm. In some embodiments, lipid nanoparticles in accordance with the present disclosure can have an average size (e.g., mean diameter) of about 50 nm to about 100 nm. In some embodiments, lipid nanoparticles may have an average size (e.g., mean diameter) of about 50 nm to about 150 nm. In some embodiments, lipid nanoparticles may have an average size (e.g., mean diameter) of about 60 nm to about 120 nm. In some embodiments, lipid nanoparticles in accordance with the present disclosure can have an average size (e.g., mean diameter) of about 30 nm, 35 nm, 40 nm, 45 nm, 50 nm, 55 nm, 60 nm, 65 nm, 70 nm, 75 nm, 80 nm, 85 nm, 90 nm, 95 nm, 100 nm, 105 nm, 110 nm, 115 nm, 120 nm, 125 nm, 130 nm, 135 nm, 140 nm, 145 nm, or 150 nm. [0341] Nucleic acid particles (e.g., ribonucleic acid particles) described herein may exhibit a polydispersity index less than about 0.5, less than about 0.4, less than about 0.3, or about 0.2 or less. By way of example, the nucleic acid particles (e.g., ribonucleic acid particles) can exhibit a polydispersity index in a range of about 0.1 to about 0.3 or about 0.2 to about 0.3. [0342] Nucleic acid particles (e.g., ribonucleic acid particles) described herein can be characterized by an “N/P ratio,” which is the molar ratio of cationic (nitrogen) groups (the “N” in N/P) in the cationic polymer to the anionic (phosphate) groups (the “P” in N/P) in
RNA. It is understood that a cationic group is one that is either in cationic form (e.g., N+), or one that is ionizable to become cationic. Use of a single number in an N/P ratio (e.g., an N/P ratio of about 5) is intended to refer to that number over 1, e.g., an N/P ratio of about 5 is intended to mean 5:1. In some embodiments, a nucleic acid particle (e.g., a ribonucleic acid particle) described herein has an N/P ratio greater than or equal to 5. In some embodiments, a nucleic acid particle (e.g., a ribonucleic acid particle) described herein has an N/P ratio that is about 5, 6, 7, 8, 9, or 10. In some embodiments, an N/P ratio for a nucleic acid particle (e.g., a ribonucleic acid particle) described herein is from about 10 to about 50. In some embodiments, an N/P ratio for a nucleic acid particle (e.g., a ribonucleic acid particle) described herein is from about 10 to about 70. In some embodiments, an N/P ratio for a nucleic acid particle (e.g., a ribonucleic acid particle) described herein is from about 10 to about 120. [0343] Nucleic acid particles (e.g., ribonucleic acid particles) described herein can be prepared using a wide range of methods that may involve obtaining a colloid from at least one cationic or cationically ionizable lipid or lipid-like material and/or at least one cationic polymer and mixing the colloid with nucleic acid to obtain nucleic acid particles. [0344] The term “colloid” as used herein relates to a type of homogeneous mixture in which dispersed particles do not settle out. The insoluble particles in the mixture can be microscopic, with particle sizes between 1 and 1000 nanometers. The mixture may be termed a colloid or a colloidal suspension. Sometimes the term “colloid” only refers to the particles in the mixture and not the entire suspension. [0345] The term “average diameter” or “mean diameter” refers to the mean hydrodynamic diameter of particles as measured by dynamic laser light scattering (DLS) with data analysis using the so-called cumulant algorithm, which provides as results the so-called Z-average with the dimension of a length, and the polydispersity index (PI), which is dimensionless (Koppel, D., J. Chem. Phys.57, 1972, pp 4814-4820, ISO 13321, which is incorporated herein by reference). Herein “average diameter,” “mean diameter,” “diameter,” or “size” for particles is used synonymously with this value of the Z-average. [0346] The “polydispersity index” is preferably calculated based on dynamic light scattering measurements by the so-called cumulant analysis as mentioned in the definition of the “average diameter.” Under certain prerequisites, it can be taken as a measure of the size
distribution of an ensemble of ribonucleic acid nanoparticles (e.g., ribonucleic acid nanoparticles). [0347] Different types of nucleic acid particles have been described previously to be suitable for delivery of nucleic acid in particulate form (e.g., Kaczmarek et al., 2017, Genome Medicine 9, 60, which is incorporated herein by reference). For non-viral nucleic acid delivery vehicles, nanoparticle encapsulation of nucleic acid physically protects nucleic acid from degradation and, depending on the specific chemistry, can aid in cellular uptake and endosomal escape. [0348] The present disclosure describes particles comprising nucleic acid (e.g., a polyribonucleotide), at least one cationic or cationically ionizable lipid or lipid-like material, and/or at least one cationic polymer which associate with the nucleic acid (e.g., a polyribonucleotide) to form nucleic acid particles (e.g., ribonucleic acid particles, e.g., ribonucleic acid nanoparticles) and compositions comprising such particles. The nucleic acid particles (e.g., ribonucleic acid particles, e.g., ribonucleic acid nanoparticles) may comprise nucleic acid (e.g., a polyribonucleotide) which is complexed in different forms by non- covalent interactions to the particle. The particles described herein are not viral particles, in particular, they are not infectious viral particles, i.e., they are not able to virally infect cells. [0349] Some embodiments described herein relate to compositions, methods and uses involving more than one, e.g., 2, 3, 4, 5, 6 or even more nucleic acid species (e.g., polyribonucleotide species). [0350] In a nucleic acid particle (e.g., ribonucleic acid particle, e.g., ribonucleic acid nanoparticle) formulation, it is possible that each nucleic acid species (e.g., polyribonucleotide species) is separately formulated as an individual nucleic acid particle (e.g., ribonucleic acid particle, e.g., ribonucleic acid nanoparticle) formulation. In that case, each individual nucleic acid particle (e.g., ribonucleic acid particle, e.g., ribonucleic acid nanoparticle) formulation will comprise one nucleic acid species (e.g., polyribonucleotide species). The individual nucleic acid particle (e.g., ribonucleic acid particle, e.g., ribonucleic acid nanoparticle) formulations may be present as separate entities, e.g., in separate containers. Such formulations are obtainable by providing each nucleic acid species (e.g., polyribonucleotide species) separately (typically each in the form of a nucleic acid-containing solution) together with a particle-forming agent, thereby allowing the formation of particles.
Respective particles will contain exclusively the specific nucleic acid species (e.g., polyribonucleotide species) that is being provided when the particles are formed (individual particulate formulations). [0351] In some embodiments, a composition such as a pharmaceutical composition comprises more than one individual nucleic acid particle (e.g., ribonucleic acid particle, e.g., ribonucleic acid nanoparticle) formulation. Respective pharmaceutical compositions are referred to as “mixed particulate formulations.” Mixed particulate formulations according to the invention are obtainable by forming, separately, individual nucleic acid particle (e.g., ribonucleic acid particle, e.g., ribonucleic acid nanoparticle) formulations, as described above, followed by a step of mixing of the individual nucleic acid particle (e.g., ribonucleic acid particle, e.g., ribonucleic acid nanoparticle) formulations. By the step of mixing, a formulation comprising a mixed population of nucleic acid-containing particles is obtainable. Individual nucleic acid particle (e.g., ribonucleic acid particle, e.g., ribonucleic acid nanoparticle) populations may be together in one container, comprising a mixed population of individual nucleic acid particle (e.g., ribonucleic acid particle, e.g., ribonucleic acid nanoparticle) formulations. [0352] Alternatively, it is possible that different nucleic acid species (e.g., polyribonucleotide species) are formulated together as a “combined particulate formulation.” Such formulations are obtainable by providing a combined formulation (typically combined solution) of different nucleic acid species (e.g., polyribonucleotide species) species together with a particle-forming agent, thereby allowing the formation of particles. As opposed to a “mixed particulate formulation,” a “combined particulate formulation” will typically comprise particles that comprise more than one nucleic acid species (e.g., polyribonucleotide species) species. In a combined particulate composition different nucleic acid species (e.g., polyribonucleotide species) are typically present together in a single particle. [0353] In certain embodiments, nucleic acids (e.g., polyribonucleotides), when present in provided nucleic acid particles (e.g., ribonucleic acid particles, e.g., lipid nanoparticles) are resistant in aqueous solution to degradation with a nuclease. [0354] In some embodiments, nucleic acid particles (e.g., ribonucleic acid particles) are lipid nanoparticles. In some embodiments, lipid nanoparticles are liver-targeting lipid nanoparticles. In some embodiments, lipid nanoparticles are cationic lipid nanoparticles
comprising one or more cationic lipids (e.g., ones described herein). In some embodiments, cationic lipid nanoparticles may comprise at least one cationic lipid, at least one polymer- conjugated lipid, and at least one helper lipid (e.g., at least one neutral lipid). Cationic Polymeric Materials [0355] Cationic polymers have been recognized as useful for developing particle delivery vehicles, as reported in PCT App. Pub. No. WO2021/001417, the entirety of which is incorporated herein by reference. As used herein, the term “polymer” refers to a composition comprising one or more molecules that comprise repeating units of one or more monomers. As used herein, “polymer,” “polymeric material,” and “polymer composition” are used interchangeably, and unless otherwise specified, refer to a composition of polymer molecules. A person of skill in the art will appreciate that a polymer composition comprises polymer molecules having molecules of different lengths (e.g., comprising varying amounts of monomers). Polymer compositions described herein are characterized by one or more of a normalized molecular weight (Mn), a weight average molecular weight (Mw), and/or a polydispersity index (PDI). In some embodiments, such repeat units can all be identical (a “homopolymer”); alternatively, in some cases, there can be more than one type of repeat unit present within the polymeric material (a “heteropolymer” or a “copolymer”). In some cases, a polymer is biologically derived, e.g., a biopolymer such as a protein. In some cases, additional moieties can also be present in the polymeric material, for example targeting moieties such as those described herein. [0356] In some embodiments, a polymer utilized in accordance with the present disclosure may be a copolymer. Repeat units forming the copolymer can be arranged in any fashion. For example, in some embodiments, repeat units can be arranged in a random order; alternatively or additionally, in some embodiments, repeat units may be arranged in an alternating order, or as a “block” copolymer, e.g., comprising one or more regions each comprising a first repeat unit (e.g., a first block), and one or more regions each comprising a second repeat unit (e.g., a second block), etc. Block copolymers can have two (a diblock copolymer), three (a triblock copolymer), or more numbers of distinct blocks. [0357] In certain embodiments, a polymeric material for use in accordance with the present disclosure is biocompatible. In certain embodiments, a biocompatible material is
biodegradable, e.g., is able to degrade, chemically and/or biologically, within a physiological environment, such as within the body. [0358] In certain embodiments, a polymeric material may be or comprise protamine or polyalkyleneimine. [0359] As those skilled in the art are aware term “protamine” is often used to refer to any of various strongly basic proteins of relatively low molecular weight that are rich in arginine and are found associated especially with DNA in place of somatic histones in the sperm cells of various animals (as fish). In particular, the term “protamine” is often used to refer to proteins found in fish sperm that are strongly basic, are soluble in water, are not coagulated by heat, and yield chiefly arginine upon hydrolysis. In purified form, they are used in a long-acting formulation of insulin and to neutralize the anticoagulant effects of heparin. [0360] In some embodiments, the term “protamine” as used herein is refers to a protamine amino acid sequence obtained or derived from natural or biological sources, including fragments thereof and/or multimeric forms of said amino acid sequence or fragment thereof, as well as (synthesized) polypeptides which are artificial and specifically designed for specific purposes and cannot be isolated from native or biological sources. [0361] In some embodiments, a polyalkyleneimine comprises polyethylenimine (PEI) and/or polypropylenimine. In some embodiments, a preferred polyalkyleneimine is polyethyleneimine (PEI). In some embodiments, the average molecular weight of PEI is preferably 0.75∙102 to 107 Da, preferably 1000 to 105 Da, more preferably 10000 to 40000 Da, more preferably 15000 to 30000 Da, even more preferably 20000 to 25000 Da. [0362] Cationic materials (e.g., polymeric materials, including polycationic polymers) contemplated for use herein include those which are able to electrostatically bind nucleic acid. In some embodiments, cationic polymeric materials contemplated for use herein include any cationic polymeric materials with which nucleic acid can be associated, e.g., by forming complexes with the nucleic acid or forming vesicles in which the nucleic acid is enclosed or encapsulated. [0363] In some embodiments, particles described herein may comprise polymers other than cationic polymers, e.g., non-cationic polymeric materials and/or anionic polymeric
materials. Collectively, anionic and neutral polymeric materials are referred to herein as non- cationic polymeric materials. Lipid Compositions Lipids and Lipid-Like Materials [0364] Lipids and lipid-like materials have also been recognized as useful for developing particle delivery vehicles. The terms “lipid” and “lipid-like material” are broadly defined herein as molecules which comprise one or more hydrophobic moieties or groups and optionally also one or more hydrophilic moieties or groups. Molecules comprising hydrophobic moieties and hydrophilic moieties are also frequently denoted as amphiphiles. Lipids are usually poorly soluble in water. In an aqueous environment, the amphiphilic nature allows the molecules to self-assemble into organized structures and different phases. One of those phases consists of lipid bilayers, as they are present in vesicles, multilamellar/unilamellar liposomes, or membranes in an aqueous environment. Hydrophobicity can be conferred by the inclusion of a polar groups that include, but are not limited to, long-chain saturated and unsaturated aliphatic hydrocarbon groups and such groups substituted by one or more aromatic, cycloaliphatic, or heterocyclic group(s). The hydrophilic groups may comprise polar and/or charged groups and include carbohydrates, phosphate, carboxylic, sulfate, amino, sulfhydryl, nitro, hydroxyl, and other like groups. [0365] Often, an amphiphilic compound has a polar head attached to a long hydrophobic tail. In some embodiments, the polar portion is soluble in water, while the non- polar portion is insoluble in water. In addition, the polar portion may have either a formal positive charge, or a formal negative charge. Alternatively, the polar portion may have both a formal positive and a negative charge, and be a zwitterion or inner salt. For purposes of the disclosure, the amphiphilic compound can be, but is not limited to, one or a plurality of natural or non-natural lipids and lipid-like compounds. [0366] A “lipid-like material” is a substance that is structurally and/or functionally related to a lipid but may not be considered a lipid in a strict sense. For example, the term includes compounds that are able to form amphiphilic layers as they are present in vesicles, multilamellar/unilamellar liposomes, or membranes in an aqueous environment and includes surfactants, or synthesized compounds with both hydrophilic and hydrophobic moieties. Generally speaking the term refers to molecules which comprise hydrophilic and
hydrophobic moieties with different structural organization, which may or may not be similar to that of lipids. [0367] Specific examples of amphiphilic compounds that may be included in an amphiphilic layer include, but are not limited to, phospholipids, aminolipids and sphingolipids. [0368] Generally, lipids may be divided into eight categories: fatty acids, glycerolipids, glycerophospholipids, sphingolipids, saccharolipids, polyketides (derived from condensation of ketoacyl subunits), sterols and prenol lipids (derived from condensation of isoprene subunits). Although the term “lipid” is sometimes used as a synonym for fats, fats are a subgroup of lipids called triglycerides. Lipids also encompass molecules such as fatty acids and their derivatives (including tri-, di-, monoglycerides, and phospholipids), as well as sterol-containing metabolites such as cholesterol. [0369] Fatty acids are a diverse group of molecules made of a hydrocarbon chain that terminates with a carboxylic acid group; this arrangement confers the molecule with a polar, hydrophilic end, and a nonpolar, hydrophobic end that is insoluble in water. The carbon chain, typically between four and 24 carbons long, may be saturated or unsaturated, and may be attached to functional groups containing oxygen, halogens, nitrogen, and sulfur. If a fatty acid contains a double bond, there is the possibility of either a cis or trans geometric isomerism, which significantly affects the molecule’s configuration. Cis-double bonds cause the fatty acid chain to bend, an effect that is compounded with more double bonds in the chain. Other major lipid classes in the fatty acid category are the fatty esters and fatty amides. [0370] Glycerolipids are composed of mono-, di-, and tri-substituted glycerols, the best-known being the fatty acid triesters of glycerol, called triglycerides. The word “triacylglycerol” is sometimes used synonymously with “triglyceride”. In these compounds, the three hydroxyl groups of glycerol are each esterified, typically by different fatty acids. Additional subclasses of glycerolipids are represented by glycosylglycerols, which are characterized by the presence of one or more sugar residues attached to glycerol via a glycosidic linkage. [0371] Glycerophospholipids are amphipathic molecules (containing both hydrophobic and hydrophilic regions) that contain a glycerol core linked to two fatty acid-
derived “tails” by ester linkages and to one “head” group by a phosphate ester linkage. Examples of glycerophospholipids, usually referred to as phospholipids (though sphingomyelins are also classified as phospholipids) are phosphatidylcholine (also known as PC, GPCho or lecithin), phosphatidylethanolamine (PE or GPEtn) and phosphatidylserine (PS or GPSer). [0372] Sphingolipids are members of a complex family of compounds that share a common structural feature, a sphingoid base backbone. The major sphingoid base in mammals is commonly referred to as sphingosine. Ceramides (N-acyl-sphingoid bases) are a major subclass of sphingoid base derivatives with an amide-linked fatty acid. The fatty acids are typically saturated or mono-unsaturated with chain lengths from 16 to 26 carbon atoms. The major phosphosphingolipids of mammals are sphingomyelins (ceramide phosphocholines), whereas insects contain mainly ceramide phosphoethanolamines and fungi have phytoceramide phosphoinositols and mannose-containing headgroups. The glycosphingolipids are a diverse family of molecules composed of one or more sugar residues linked via a glycosidic bond to the sphingoid base. Examples of these are the simple and complex glycosphingolipids such as cerebrosides and gangliosides. [0373] Sterols, such as cholesterol and its derivatives, or tocopherol and its derivatives, are important components of membrane lipids, along with the glycerophospholipids and sphingomyelins. [0374] Saccharolipids are compounds in which fatty acids are linked directly to a sugar backbone, forming structures that are compatible with membrane bilayers. In the saccharolipids, a monosaccharide substitutes for the glycerol backbone present in glycerolipids and glycerophospholipids. The most familiar saccharolipids are the acylated glucosamine precursors of the Lipid A component of the lipopolysaccharides in Gram- negative bacteria. Typical lipid A molecules are disaccharides of glucosamine, which are derivatized with as many as seven fatty-acyl chains. The minimal lipopolysaccharide required for growth in E. coli is Kdo2-Lipid A, a hexa-acylated disaccharide of glucosamine that is glycosylated with two 3-deoxy-D-manno-octulosonic acid (Kdo) residues. [0375] Polyketides are synthesized by polymerization of acetyl and propionyl subunits by classic enzymes as well as iterative and multimodular enzymes that share mechanistic features with the fatty acid synthases. They comprise a large number of
secondary metabolites and natural products from animal, plant, bacterial, fungal and marine sources, and have great structural diversity. Many polyketides are cyclic molecules whose backbones are often further modified by glycosylation, methylation, hydroxylation, oxidation, or other processes. [0376] Lipids and lipid-like materials may be cationic, anionic or neutral. Neutral lipids or lipid-like materials exist in an uncharged or neutral zwitterionic form at a selected pH. [0377] In some embodiments, suitable lipids or lipid-like materials for use in the present disclosure include those described in WO2020/128031 and US2020/0163878, the entire contents of each of which are incorporated herein by reference for the purposes described herein. Cationic or Cationically Ionizable Lipids or Lipid-like Materials [0378] In some embodiments cationic or cationically ionizable lipids or lipid-like materials contemplated for use herein include any cationic or cationically ionizable lipids or lipid-like materials which are able to electrostatically bind nucleic acid. In one embodiment, cationic or cationically ionizable lipids or lipid-like materials contemplated for use herein can be associated with nucleic acid, e.g., by forming complexes with the nucleic acid or forming vesicles in which the nucleic acid is enclosed or encapsulated. [0379] Cationic lipids or lipid-like materials are characterized in that they have a net positive charge (e.g., at a relevant pH). Cationic lipids or lipid-like materials bind negatively charged nucleic acid by electrostatic interaction. Generally, cationic lipids possess a lipophilic moiety, such as a sterol, an acyl chain, a diacyl or more acyl chains, and the head group of the lipid typically carries the positive charge. [0380] In certain embodiments, a cationic lipid or lipid-like material has a net positive charge only at certain pH, in particular acidic pH, while it has preferably no net positive charge, preferably has no charge, i.e., it is neutral, at a different, preferably higher pH such as physiological pH. This ionizable behavior is thought to enhance efficacy through helping with endosomal escape and reducing toxicity as compared with particles that remain cationic at physiological pH.
[0381] In some embodiments, a cationic or cationically ionizable lipid or lipid-like material comprises a head group which includes at least one nitrogen atom (N) which is positive charged or capable of being protonated. [0382] Examples of cationic lipids include, but are not limited to 1,2-dioleoyl-3- trimethylammonium propane (DOTAP); N,N-dimethyl-2,3-dioleyloxypropylamine (DODMA), 1,2-di-O-octadecenyl-3-trimethylammonium propane (DOTMA), 3-(N—(N′,N′- dimethylaminoethane)-carbamoyl)cholesterol (DC-Chol), dimethyldioctadecylammonium (DDAB); 1,2-dioleoyl-3-dimethylammonium-propane (DODAP); 1,2-diacyloxy-3- dimethylammonium propanes; 1,2-dialkyloxy-3-dimethylammonium propanes; dioctadecyldimethyl ammonium chloride (DODAC), 1,2-distearyloxy-N,N-dimethyl-3- aminopropane (DSDMA), 2,3-di(tetradecoxy)propyl-(2-hydroxyethyl)-dimethylazanium (DMRIE), 1,2-dimyristoyl-sn-glycero-3-ethylphosphocholine (DMEPC), l,2-dimyristoyl-3- trimethylammonium propane (DMTAP), 1,2-dioleyloxypropyl-3-dimethyl-hydroxyethyl ammonium bromide (DORIE), and 2,3-dioleoyloxy- N-[2(spermine carboxamide)ethyl]-N,N- dimethyl-l-propanamium trifluoroacetate (DOSPA), 1,2-dilinoleyloxy-N,N- dimethylaminopropane (DLinDMA), 1,2-dilinolenyloxy-N,N-dimethylaminopropane (DLenDMA), dioctadecylamidoglycyl spermine (DOGS), 3-dimethylamino-2-(cholest-5-en- 3-beta-oxybutan-4-oxy)-1-(cis,cis-9,12-oc-tadecadienoxy)propane (CLinDMA), 2-[5′- (cholest-5-en-3-beta-oxy)-3′-oxapentoxy)-3-dimethyl-1-(cis,cis-9′,12′- octadecadienoxy)propane (CpLinDMA), N,N-dimethyl-3,4-dioleyloxybenzylamine (DMOBA), 1,2-N,N′-dioleylcarbamyl-3-dimethylaminopropane (DOcarbDAP), 2,3- Dilinoleoyloxy-N,N-dimethylpropylamine (DLinDAP), 1,2-N,N′-Dilinoleylcarbamyl-3- dimethylaminopropane (DLincarbDAP), 1,2-Dilinoleoylcarbamyl-3-dimethylaminopropane (DLinCDAP), 2,2-dilinoleyl-4-dimethylaminomethyl-[1,3]-dioxolane (DLin-K-DMA), 2,2- dilinoleyl-4-dimethylaminoethyl-[1,3]-dioxolane (DLin-K-XTC2-DMA), 2,2-dilinoleyl-4-(2- dimethylaminoethyl)-[1,3]-dioxolane (DLin-KC2-DMA), heptatriaconta-6,9,28,31-tetraen- 19-yl-4-(dimethylamino)butanoate (DLin-MC3-DMA), N-(2-Hydroxyethyl)-N,N-dimethyl- 2,3-bis(tetradecyloxy)-1-propanaminium bromide (DMRIE), (±)-N-(3-aminopropyl)-N,N- dimethyl-2,3-bis(cis-9-tetradecenyloxy)-1-propanaminium bromide (GAP-DMORIE), (±)-N- (3-aminopropyl)-N,N-dimethyl-2,3-bis(dodecyloxy)-1-propanaminium bromide (GAP- DLRIE), (±)-N-(3-aminopropyl)-N,N-dimethyl-2,3-bis(tetradecyloxy)-1-propanaminium bromide (GAP-DMRIE), N-(2-Aminoethyl)-N,N-dimethyl-2,3-bis(tetradecyloxy)-1-
propanaminium bromide (βAE-DMRIE), N-(4-carboxybenzyl)-N,N-dimethyl-2,3- bis(oleoyloxy)propan-1-aminium (DOBAQ), 2-({8-[(3β)-cholest-5-en-3-yloxy]octyl}oxy)- N,N-dimethyl-3-[(9Z,12Z)-octadeca-9,12-dien-1-yloxy]propan-1-amine (Octyl-CLinDMA), 1,2-dimyristoyl-3-dimethylammonium-propane (DMDAP), 1,2-dipalmitoyl-3- dimethylammonium-propane (DPDAP), N1-[2-((1S)-1-[(3-aminopropyl)amino]-4-[di(3- amino-propyl)amino]butylcarboxamido)ethyl]-3,4-di[oleyloxy]-benzamide (MVL5), 1,2- dioleoyl-sn-glycero-3-ethylphosphocholine (DOEPC), 2,3-bis(dodecyloxy)-N-(2- hydroxyethyl)-N,N-dimethylpropan-1-amonium bromide (DLRIE), N-(2-aminoethyl)-N,N- dimethyl-2,3-bis(tetradecyloxy)propan-1-aminium bromide (DMORIE), di((Z)-non-2-en-1- yl) 8,8’-((((2(dimethylamino)ethyl)thio)carbonyl)azanediyl)dioctanoate (ATX), N,N- dimethyl-2,3-bis(dodecyloxy)propan-1-amine (DLDMA), N,N-dimethyl-2,3- bis(tetradecyloxy)propan-1-amine (DMDMA), Di((Z)-non-2-en-1-yl)-9-((4- (dimethylaminobutanoyl)oxy)heptadecanedioate (L319), N-Dodecyl-3-((2- dodecylcarbamoyl-ethyl)-{2-[(2-dodecylcarbamoyl-ethyl)-2-{(2-dodecylcarbamoyl-ethyl)-[2- (2-dodecylcarbamoyl-ethylamino)-ethyl]-amino}-ethylamino)propionamide (lipidoid 98N12- 5), 1-[2-[bis(2-hydroxydodecyl)amino]ethyl-[2-[4-[2-[bis(2 hydroxydodecyl)amino]ethyl]piperazin-1-yl]ethyl]amino]dodecan-2-ol (lipidoid C12-200), LIPOFECTIN® (commercially available cationic liposomes comprising DOTMA and 1 ,2- dioleoyl-sn-3phosphoethanolamine (DOPE), from GIBCO/BRL, Grand Island, N.Y.); LIPOFECTAMINE® (commercially available cationic liposomes comprising N-(1 - (2,3dioleyloxy)propyl)-N-(2-(sperminecarboxamido)ethyl)-N,N-dimethylammonium trifluoroacetate (DOSPA) and (DOPE), from GIBCO/BRL); and TRANSFECTAM® (commercially available cationic lipids comprising dioctadecylamidoglycyl carboxyspermine (DOGS) in ethanol from Promega Corp., Madison, Wis.) or any combination of any of the foregoing. Further suitable cationic lipids for use in the present disclosure include those described in WO2020/128031 and US2020/0163878, the entire contents of each of which are incorporated herein by reference for the purposes described herein. Further suitable cationic lipids for use in the present disclosure include those described in WO2010/053572 (including C12-200 described at paragraph [00225]) and WO2012/170930, both of which are incorporated herein by reference for the purposes described herein. Additional suitable cationic lipids for use in the present disclosure include HGT4003, HGT5000, HGTS001, HGT5001, HGT5002 (see US2015/0140070, which is incorporated herein by reference in its entirety).
[0383] In some embodiments, formulations that are useful for pharmaceutical compositions (e.g., immunogenic compositions, e.g., vaccines) compositions as described herein can comprise at least one cationic lipid. Representative cationic lipids include, but are not limited to, 1 ,2-dilinoleyoxy-3-(dimethylamino)acetoxypropane (DLin-DAC), 1 ,2- dilinoleyoxy-3morpholinopropane (DLin-MA), 1,2-dilinoleoyl-3-dimethylaminopropane (DLinDAP), 1 ,2-dilinoleylthio-3-dimethylaminopropane (DLin-S-DMA), 1 -linoleoyl-2- linoleyloxy-3dimethylaminopropane (DLin-2-DMAP), 1 ,2-dilinoleyloxy-3- trimethylaminopropane chloride salt (DLin-TMA.CI), 1 ,2-dilinoleoyl-3- trimethylaminopropane chloride salt (DLin-TAP.CI), 1 ,2-dilinoleyloxy-3-(N- methylpiperazino)propane (DLin-MPZ), 3-(N,Ndilinoleylamino)-1 ,2-propanediol (DLinAP), 3-(N,N-dioleylamino)-1 ,2-propanediol (DOAP), 1 ,2-dilinoleyloxo-3-(2-N,N- dimethylamino)ethoxypropane (DLin-EG-DMA), and 2,2-dilinoleyl-4- dimethylaminomethyl-[1 ,3]-dioxolane (DLin-K-DMA), 2,2-dilinoleyl-4-(2- dimethylaminoethyl)-[1 ,3]-dioxolane (DLin-KC2-DMA); dilinoleyl-methyl-4- dimethylaminobutyrate (DLin-MC3-DMA); MC3 (US2010/0324120, which is incorporated herein by reference in its entirety). [0384] In some embodiments, amino or cationic lipids useful in accordance with the present disclosure have at least one protonatable or deprotonatable group, such that the lipid is positively charged at a pH at or below physiological pH (e.g., pH 7.4), and neutral at a second pH, preferably at or above physiological pH. It will, of course, be understood that the addition or removal of protons as a function of pH is an equilibrium process, and that the reference to a charged or a neutral lipid refers to the nature of the predominant species and does not require that all of lipids have to be present in the charged or neutral form. Lipids having more than one protonatable or deprotonatable group, or which are zwitterionic, are not excluded and may likewise suitable in the context of the present invention. [0385] In some embodiments, a protonatable lipid has a pKa of the protonatable group in the range of about 4 to about 11, e.g., a pKa of about 5 to about 7. [0386] In some embodiments, a cationic lipid may comprise from about 10 mol % to about 100 mol %, about 20 mol % to about 100 mol %, about 30 mol % to about 100 mol %, about 40 mol % to about 100 mol %, or about 50 mol % to about 100 mol % of total lipid present in a lipid composition utilized in accordance with the present disclosure.
Additional Lipids or Lipid-like materials [0387] In some embodiments, formulations utilized in accordance with the present disclosure may comprise lipids or lipid-like materials other than cationic or cationically ionizable lipids or lipid-like materials, i.e., non-cationic lipids or lipid-like materials (including non-cationically ionizable lipids or lipid-like materials). Collectively, anionic and neutral lipids or lipid-like materials are referred to herein as non-cationic lipids or lipid-like materials. In some embodiments, optimizing a formulation of nucleic acid particles by addition of other hydrophobic moieties, such as cholesterol and lipids, in addition to an ionizable/cationic lipid or lipid-like material may, for example, enhance particle stability and efficacy of nucleic acid delivery. [0388] In some embodiments, a lipid or lipid-like material may be incorporated which may or may not affect the overall charge of particles. In certain embodiments, such lipid or lipid-like material is a non-cationic lipid or lipid-like material. [0389] In some embodiments, a non-cationic lipid may comprise, e.g., one or more anionic lipids and/or neutral lipids. An “anionic lipid” is negatively charged (e.g., at a selected pH). [0390] A “helper lipid” exists either in an uncharged, neutral zwitterionic form (e.g., at a selected pH), or, in some embodiments, as having a cationic or positive charge at physiological pH. In some embodiments, a formulation comprises one of the following helper lipid components: (1) a phospholipid, (2) cholesterol or a derivative thereof; or (3) a mixture of a phospholipid and cholesterol or a derivative thereof. Examples of cholesterol derivatives include, but are not limited to, cholestanol, cholestanone, cholestenone, coprostanol, cholesteryl-2’-hydroxyethyl ether, cholesteryl-4’-hydroxybutyl ether, tocopherol and derivatives thereof, and mixtures thereof. [0391] Specific exemplary phospholipids that can be used include, but are not limited to, phosphatidylcholines, phosphatidylethanolamines, phosphatidylglycerols, phosphatidic acids, phosphatidylserines or sphingomyelin. Such phospholipids include in particular diacylphosphatidylcholines, such as distearoylphosphatidylcholine (DSPC), dioleoylphosphatidylcholine (DOPC), dimyristoylphosphatidylcholine (DMPC), dipentadecanoylphosphatidylcholine, dilauroylphosphatidylcholine,
dipalmitoylphosphatidylcholine (DPPC), diarachidoylphosphatidylcholine (DAPC), dibehenoylphosphatidylcholine (DBPC), ditricosanoylphosphatidylcholine (DTPC), dilignoceroylphatidylcholine (DLPC), palmitoyloleoyl-phosphatidylcholine (POPC), 1,2-di- O-octadecenyl-sn-glycero-3-phosphocholine (18:0 Diether PC), 1-oleoyl-2- cholesterylhemisuccinoyl-sn-glycero-3-phosphocholine (OChemsPC), 1-hexadecyl-sn- glycero-3-phosphocholine (C16 Lyso PC) and phosphatidylethanolamines, in particular diacylphosphatidylethanolamines, such as dioleoylphosphatidylethanolamine (DOPE), distearoyl-phosphatidylethanolamine (DSPE), dipalmitoyl-phosphatidylethanolamine (DPPE), dimyristoyl-phosphatidylethanolamine (DMPE), dilauroyl- phosphatidylethanolamine (DLPE), diphytanoyl-phosphatidylethanolamine (DPyPE), and further phosphatidylethanolamine lipids with different hydrophobic chains. [0392] In certain embodiments, a formulation utilized in accordance with the present disclosure includes DSPC or DSPC and cholesterol. [0393] In some embodiments, a formulation utilized in accordance with the present disclosure comprises a helper lipid that is ionizable or cationic. [0394] In certain embodiments, formulations utilized in accordance with the present disclosure include both a cationic lipid and an additional (non-cationic) lipid. [0395] In some embodiments, formulations herein include a polymer conjugated lipid such as a pegylated lipid. “Pegylated lipids” or “PEG-conjugated lipids” comprise both a lipid portion and a polyethylene glycol portion. Pegylated lipids are known in the art. [0396] Without wishing to be bound by theory, the amount of (total) cationic lipid compared to the amount of other lipid(s) in formulation may affect important characteristics, such as charge, particle size, stability, tissue selectivity, and bioactivity of the nucleic acid. In some embodiments, the molar ratio of the at least one cationic lipid to the at least one additional lipid is from about 10:0 to about 1:9, about 4:1 to about 1:2, or about 3:1 to about 1:1. Lipoplex Particles [0397] In certain embodiments of the present disclosure, the RNA described herein may be present in RNA lipoplex particles.
[0398] An “RNA lipoplex particle” contains lipid, in particular cationic lipid, and RNA. Electrostatic interactions between positively charged liposomes and negatively charged RNA results in complexation and spontaneous formation of RNA lipoplex particles. Positively charged liposomes may be generally synthesized using a cationic lipid, such as DOTMA, and additional lipids, such as DOPE. In one embodiment, a RNA lipoplex particle is a nanoparticle. [0399] In certain embodiments, RNA lipoplex particles include both a cationic lipid and an additional lipid. In an exemplary embodiment, the cationic lipid is DOTMA and the additional lipid is DOPE. [0400] In some embodiments, the molar ratio of the at least one cationic lipid to the at least one additional lipid is from about 10:0 to about 1:9, about 4:1 to about 1:2, or about 3:1 to about 1:1. In specific embodiments, the molar ratio may be about 3:1, about 2.75:1, about 2.5:1, about 2.25:1, about 2:1, about 1.75:1, about 1.5:1, about 1.25:1, or about 1:1. In an exemplary embodiment, the molar ratio of the at least one cationic lipid to the at least one additional lipid is about 2:1. [0401] In some embodiments, RNA lipoplex particles have an average diameter that in one embodiment ranges from about 200 nm to about 1000 nm, from about 200 nm to about 800 nm, from about 250 to about 700 nm, from about 400 to about 600 nm, from about 300 nm to about 500 nm, or from about 350 nm to about 400 nm. In specific embodiments, the RNA lipoplex particles have an average diameter of about 200 nm, about 225 nm, about 250 nm, about 275 nm, about 300 nm, about 325 nm, about 350 nm, about 375 nm, about 400 nm, about 425 nm, about 450 nm, about 475 nm, about 500 nm, about 525 nm, about 550 nm, about 575 nm, about 600 nm, about 625 nm, about 650 nm, about 700 nm, about 725 nm, about 750 nm, about 775 nm, about 800 nm, about 825 nm, about 850 nm, about 875 nm, about 900 nm, about 925 nm, about 950 nm, about 975 nm, or about 1000 nm. In an embodiment, the RNA lipoplex particles have an average diameter that ranges from about 250 nm to about 700 nm. In another embodiment, the RNA lipoplex particles have an average diameter that ranges from about 300 nm to about 500 nm. In an exemplary embodiment, the RNA lipoplex particles have an average diameter of about 400 nm. [0402] RNA lipoplex particles and compositions comprising RNA lipoplex particles described herein are useful for delivery of RNA to a target tissue after parenteral
administration, in particular after intravenous administration. The RNA lipoplex particles may be prepared using liposomes that may be obtained by injecting a solution of the lipids in ethanol into water or a suitable aqueous phase. In one embodiment, the aqueous phase has an acidic pH. In one embodiment, the aqueous phase comprises acetic acid, e.g., in an amount of about 5 mM. Liposomes may be used for preparing RNA lipoplex particles by mixing the liposomes with RNA. In one embodiment, the liposomes and RNA lipoplex particles comprise at least one cationic lipid and at least one additional lipid. In one embodiment, the at least one cationic lipid comprises 1,2-di-O-octadecenyl-3-trimethylammonium propane (DOTMA) and/or 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP). In one embodiment, the at least one additional lipid comprises 1,2-di-(9Z-octadecenoyl)-sn-glycero- 3-phosphoethanolamine (DOPE), cholesterol (Chol) and/or 1,2-dioleoyl-sn-glycero-3- phosphocholine (DOPC). In one embodiment, the at least one cationic lipid comprises 1,2-di- O-octadecenyl-3-trimethylammonium propane (DOTMA) and the at least one additional lipid comprises 1,2-di-(9Z-octadecenoyl)-sn-glycero-3-phosphoethanolamine (DOPE). In one embodiment, the liposomes and RNA lipoplex particles comprise 1,2-di-O-octadecenyl-3- trimethylammonium propane (DOTMA) and 1,2-di-(9Z-octadecenoyl)-sn-glycero-3- phosphoethanolamine (DOPE). [0403] Spleen targeting RNA lipoplex particles are described in WO2013/143683, incorporated herein by reference. It has been found that RNA lipoplex particles having a net negative charge may be used to preferentially target spleen tissue or spleen cells such as antigen-presenting cells, in particular dendritic cells. Accordingly, following administration of the RNA lipoplex particles, RNA accumulation and/or RNA expression in the spleen occurs. Thus, RNA lipoplex particles of the disclosure may be used for expressing RNA in the spleen. In an embodiment, after administration of the RNA lipoplex particles, no or essentially no RNA accumulation and/or RNA expression in the lung and/or liver occurs. In one embodiment, after administration of the RNA lipoplex particles, RNA accumulation and/or RNA expression in antigen presenting cells, such as professional antigen presenting cells in the spleen occurs. Thus, RNA lipoplex particles of the disclosure may be used for expressing RNA in such antigen presenting cells. In one embodiment, the antigen presenting cells are dendritic cells and/or macrophages.
Lipid Nanoparticles (LNPs) [0404] In some embodiments, nucleic acid such as RNA described herein is administered in the form of lipid nanoparticles (LNPs). In some embodiments, LNPs may comprise any lipid capable of forming a particle to which the one or more nucleic acid molecules are attached, or in which the one or more nucleic acid molecules are encapsulated. [0405] In some embodiments, an LNP comprises one or more cationic lipids, and one or more stabilizing lipids. Stabilizing lipids include neutral lipids, helper lipids, and pegylated lipids. [0406] In some embodiments, an LNP comprises a cationic lipid, a helper lipid, a sterol, a polymer conjugated lipid; and an RNA, encapsulated within or associated with the lipid nanoparticle. [0407] In some embodiments, a helper lipid is a phosphotidylcholine such as 1,2- distearoyl-sn-glycero-3-phosphocholine (DSPC), 1,2-Dipalmitoyl-sn-glycero-3- phosphocholine (DPPC), 1,2-Dimyristoyl-sn-glycero-3-phosphocholine (DMPC), 1- palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), l ,2-dioleoyl-sn-glycero-3- phosphocholine (DOPC), phophatidylethanolamines such as 1,2-dioleoyl-sn-glycero-3- phosphoethanolamine (DOPE), sphingomyelins (SM). In some embodiments, a helper lipid is a 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP), 1-α-phosphatidylserine (PS), or DOPE. In some embodiments, a helper lipid is selected from the group consisting of DSPC, DPPC, DMPC, DOPC, POPC, DOPE, DOPG, DPPG, POPE, DPPE, DMPE, DSPE, DOTAP, PS, and SM. In some embodiments, the helper lipid is selected from the group consisting of DSPC, DPPC, DMPC, DOPC, POPC, DOPE, DOTAP, PS, and SM. In some embodiments, the helper lipid is DSPC. In some embodiments, the helper lipid is DOTAP. In some embodiments, the helper lipid is DOPE. In some embodiments, the helper lipid is PS. [0408] In some embodiments, a sterol is cholesterol. [0409] In some embodiments, a polymer conjugated lipid is a pegylated lipid (a PEG lipid). In some embodiments, a PEG lipid is selected from pegylated diacylglycerol (PEG- DAG) such as l-(monomethoxy-polyethyleneglycol)- 2,3-dimyristoylglycerol (PEG-DMG) (e.g., 1,2-dimyristoyl-rac-glycero-3-methoxypolyethylene glycol-2000 (PEG2000-DMG)), a pegylated phosphatidylethanoloamine (PEG-PE) a PEG succinate diacylglycerol (PEG-S-
DAG) such as 4-O-(2',3'-di(tetradecanoyloxy)propyl-1-O-(ω- methoxy(polyethoxy)ethyl)butanedioate (PEG-S-DMG), a pegylated ceramide (PEG-cer), or a PEG dialkoxypropylcarbamate such as ω-m ethoxy (polyethoxy)ethyl-N-(2,3- di(tetradecanoxy)propyl)carbamate, and 2,3-di(tetradecanoxy)propy 1-Ν-(ω methoxy(polyethoxy)ethyl)carbamate. In some embodiments, a pegylated lipid is 1,2- dimyristoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (C14-PEG2000). In some embodiments, a pegylated lipid has the following structure: or a pharmaceutically acc
 thereof, wherein: R12 and R13 are each independently a straight or branched, saturated or unsaturated alkyl chain containing from 10 to 30 carbon atoms, wherein the alkyl chain is optionally interrupted by one or more ester bonds; and w has a mean value ranging from 30 to 60. In some embodiments, R12 and R13 are each independently straight, saturated alkyl chains containing from 12 to 16 carbon atoms. In some embodiments, w has a mean value ranging from 40 to 55. In some embodiments, the average w is about 45. In some embodiments, R12 and R13 are each independently a straight, saturated alkyl chain containing about 14 carbon atoms, and w has a mean value of about 45. [0410] In some embodiments, a pegylated lipid is DMG-PEG 2000, e.g., having the following structure: . [0411]
thereof, wherein: R12 and R13 are each independently a straight or branched, saturated or unsaturated alkyl chain containing from 10 to 30 carbon atoms, wherein the alkyl chain is optionally interrupted by one or more ester bonds; and w has a mean value ranging from 30 to 60. In some embodiments, R12 and R13 are each independently straight, saturated alkyl chains containing from 12 to 16 carbon atoms. In some embodiments, w has a mean value ranging from 40 to 55. In some embodiments, the average w is about 45. In some embodiments, R12 and R13 are each independently a straight, saturated alkyl chain containing about 14 carbon atoms, and w has a mean value of about 45. [0410] In some embodiments, a pegylated lipid is DMG-PEG 2000, e.g., having the following structure: . [0411]
 In some embodiments, a pegylated lipid is or comprises 2-[(Polyethylene glycol)-2000]-N,N-ditetradecylacetamide with a chemical structure as shown below:
In some embodiments, a pegylated lipid is or comprises 2-[(Polyethylene glycol)-2000]-N,N-ditetradecylacetamide with a chemical structure as shown below:
 or a pharmaceutically acceptable salt thereof, where n’ is an integer from 45 to 50 [0412] In some embodiments, a cationic lipid component of LNPs has the structure of Formula (III): or a pharmaceutically acceptable r stereoisomer thereof, wherein: one of L1 or L2 is –O(C=O)-, -(C
or a pharmaceutically acceptable salt thereof, where n’ is an integer from 45 to 50 [0412] In some embodiments, a cationic lipid component of LNPs has the structure of Formula (III): or a pharmaceutically acceptable r stereoisomer thereof, wherein: one of L1 or L2 is –O(C=O)-, -(C
 =O)O-, -C(=O)-, -O-, -S(O)x-, -S-S-, -C(=O)S-, SC(=O)-, - NRaC(=O)-, -C(=O)NRa-, NRaC(=O)NRa-, -OC(=O)NRa- or -NRaC(=O)O-, and the other of L1 or L2 is –O(C=O)-, -(C=O)O-, -C(=O)-, -O-, -S(O)x-, -S-S-, -C(=O)S-, SC(=O)-, - NRaC(=O)-, -C(=O)NRa-, NRaC(=O)NRa-, -OC(=O)NRa- or -NRaC(=O)O- or a direct bond; G1 and G2 are each independently unsubstituted C1-C12 alkylene or C1-C12 alkenylene; G3 is C1-C24 alkylene, C1-C24 alkenylene, C3-C8 cycloalkylene, C3-C8 cycloalkenylene; Ra is H or C1-C12 alkyl; R1 and R2 are each independently C6-C24 alkyl or C6-C24 alkenyl; R3 is H, OR5, CN, -C(=O)OR4, -OC(=O)R4 or –NR5C(=O)R4; R4 is C1-C12 alkyl; R5 is H or C1-C6 alkyl; and x is 0, 1 or 2. [0413] In some of the foregoing embodiments of Formula (III), the lipid has one of the following structures (IIIA) or (IIIB):
=O)O-, -C(=O)-, -O-, -S(O)x-, -S-S-, -C(=O)S-, SC(=O)-, - NRaC(=O)-, -C(=O)NRa-, NRaC(=O)NRa-, -OC(=O)NRa- or -NRaC(=O)O-, and the other of L1 or L2 is –O(C=O)-, -(C=O)O-, -C(=O)-, -O-, -S(O)x-, -S-S-, -C(=O)S-, SC(=O)-, - NRaC(=O)-, -C(=O)NRa-, NRaC(=O)NRa-, -OC(=O)NRa- or -NRaC(=O)O- or a direct bond; G1 and G2 are each independently unsubstituted C1-C12 alkylene or C1-C12 alkenylene; G3 is C1-C24 alkylene, C1-C24 alkenylene, C3-C8 cycloalkylene, C3-C8 cycloalkenylene; Ra is H or C1-C12 alkyl; R1 and R2 are each independently C6-C24 alkyl or C6-C24 alkenyl; R3 is H, OR5, CN, -C(=O)OR4, -OC(=O)R4 or –NR5C(=O)R4; R4 is C1-C12 alkyl; R5 is H or C1-C6 alkyl; and x is 0, 1 or 2. [0413] In some of the foregoing embodiments of Formula (III), the lipid has one of the following structures (IIIA) or (IIIB):
 wherein: A is a 3 to 8-membered cycloalkyl or cycloalkylene ring; R6 is, at each occurrence, independently H, OH or C1-C24 alkyl; n is an integer ranging from 1 to 15.
[0414] In some of the foregoing embodiments of Formula (III), the lipid has structure (IIIA), and in other embodiments, the lipid has structure (IIIB). [0415] In other embodiments of Formula (III), the lipid has one of the following structures (IIIC) or (IIID):
wherein: A is a 3 to 8-membered cycloalkyl or cycloalkylene ring; R6 is, at each occurrence, independently H, OH or C1-C24 alkyl; n is an integer ranging from 1 to 15.
[0414] In some of the foregoing embodiments of Formula (III), the lipid has structure (IIIA), and in other embodiments, the lipid has structure (IIIB). [0415] In other embodiments of Formula (III), the lipid has one of the following structures (IIIC) or (IIID):
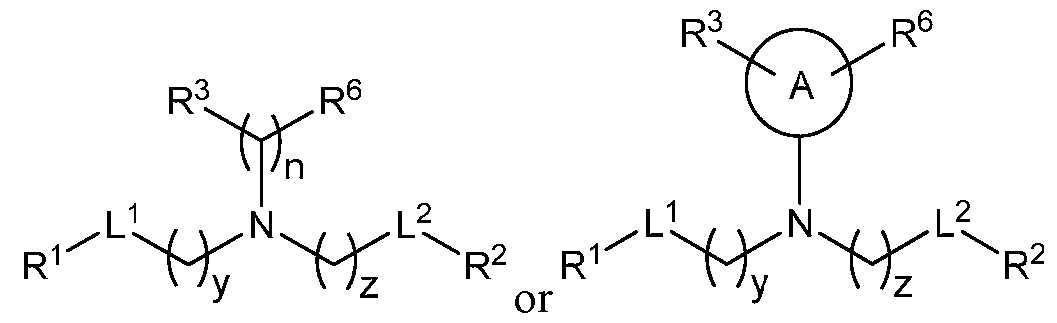 wherein y and z are each independently integers ranging from 1 to 12. [0416] In any of the foregoing embodiments of Formula (III), one of L1 or L2 is -O(C=O)-. For example, in some embodiments each of L1 and L2 are -O(C=O)-. In some different embodiments of any of the foregoing, L1 and L2 are each independently -(C=O)O- or -O(C=O)-. For example, in some embodiments each of L1 and L2 is -(C=O)O-. [0417] In some different embodiments of Formula (III), the lipid has one of the following structures (IIIE) or (IIIF): .
wherein y and z are each independently integers ranging from 1 to 12. [0416] In any of the foregoing embodiments of Formula (III), one of L1 or L2 is -O(C=O)-. For example, in some embodiments each of L1 and L2 are -O(C=O)-. In some different embodiments of any of the foregoing, L1 and L2 are each independently -(C=O)O- or -O(C=O)-. For example, in some embodiments each of L1 and L2 is -(C=O)O-. [0417] In some different embodiments of Formula (III), the lipid has one of the following structures (IIIE) or (IIIF): .
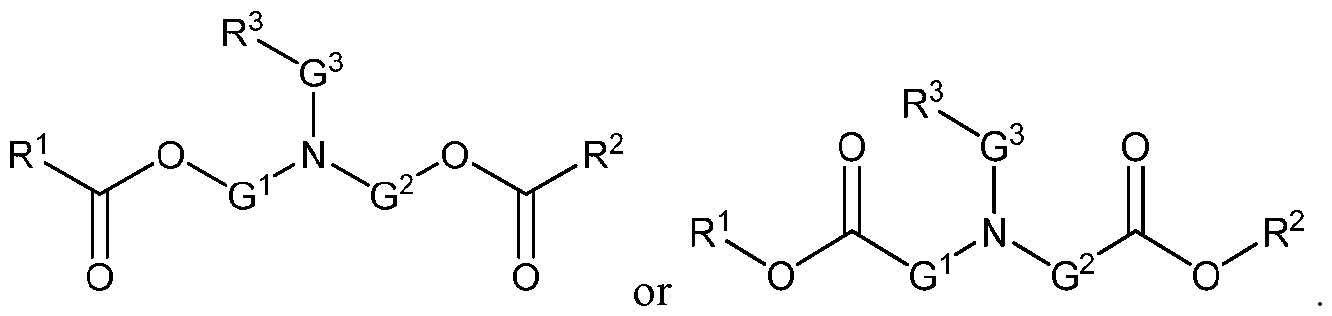 [0418] In some of the foregoing embodiments of Formula (III), the lipid has one of the following structures (IIIG), (IIIH), (IIII), or (IIIJ): ;
[0418] In some of the foregoing embodiments of Formula (III), the lipid has one of the following structures (IIIG), (IIIH), (IIII), or (IIIJ): ;
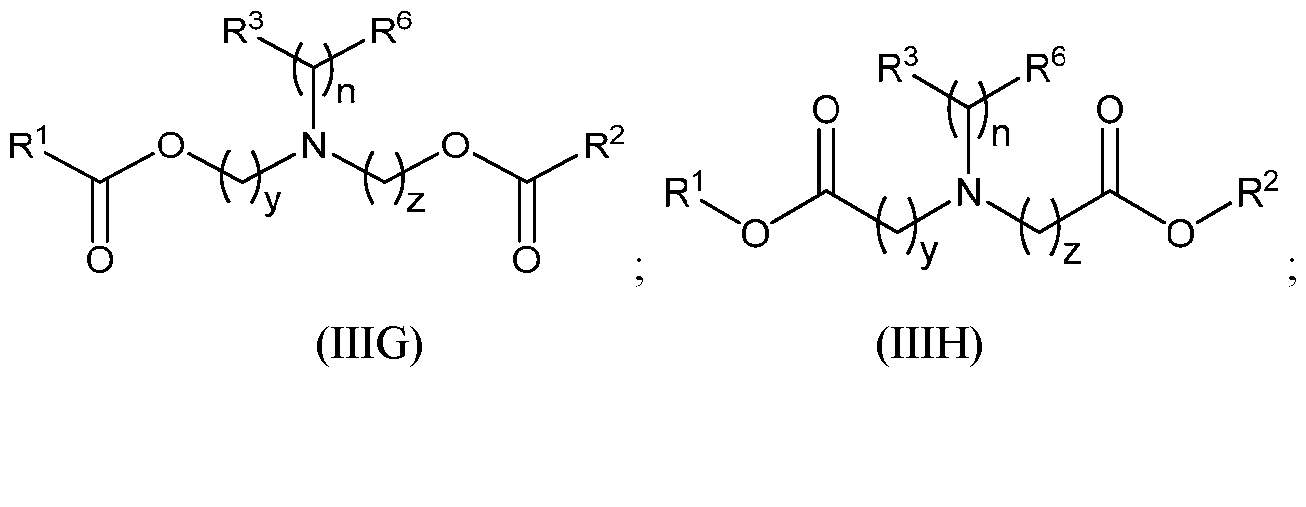 .
.
 [0419] In some of the foregoing embodiments of Formula (III), n is an integer ranging from 2 to 12, for example from 2 to 8 or from 2 to 4. For example, in some embodiments, n is 3, 4, 5 or 6. In some embodiments, n is 3. In some embodiments, n is 4. In some embodiments, n is 5. In some embodiments, n is 6. [0420] In some other of the foregoing embodiments of Formula (III), y and z are each independently an integer ranging from 2 to 10. For example, in some embodiments, y and z are each independently an integer ranging from 4 to 9 or from 4 to 6. [0421] In some of the foregoing embodiments of Formula (III), R6 is H. In other of the foregoing embodiments, R6 is C1-C24 alkyl. In other embodiments, R6 is OH. [0422] In some embodiments of Formula (III), G3 is unsubstituted. In other embodiments, G3 is substituted. In various different embodiments, G3 is linear C1-C24 alkylene or linear C1-C24 alkenylene. [0423] In some other foregoing embodiments of Formula (III), R1 or R2, or both, is C6-C24 alkenyl. For example, in some embodiments, R1 and R2 each, independently have the following structure: , wherein:
[0419] In some of the foregoing embodiments of Formula (III), n is an integer ranging from 2 to 12, for example from 2 to 8 or from 2 to 4. For example, in some embodiments, n is 3, 4, 5 or 6. In some embodiments, n is 3. In some embodiments, n is 4. In some embodiments, n is 5. In some embodiments, n is 6. [0420] In some other of the foregoing embodiments of Formula (III), y and z are each independently an integer ranging from 2 to 10. For example, in some embodiments, y and z are each independently an integer ranging from 4 to 9 or from 4 to 6. [0421] In some of the foregoing embodiments of Formula (III), R6 is H. In other of the foregoing embodiments, R6 is C1-C24 alkyl. In other embodiments, R6 is OH. [0422] In some embodiments of Formula (III), G3 is unsubstituted. In other embodiments, G3 is substituted. In various different embodiments, G3 is linear C1-C24 alkylene or linear C1-C24 alkenylene. [0423] In some other foregoing embodiments of Formula (III), R1 or R2, or both, is C6-C24 alkenyl. For example, in some embodiments, R1 and R2 each, independently have the following structure: , wherein:
 R7a and R7b are, at each occurrence, independently H or C1-C12 alkyl; and a is an integer from 2 to 12, wherein R7a, R7b and a are each selected such that R1 and R2 each independently comprise from 6 to 20 carbon atoms. For example, in some embodiments a is an integer ranging from 5 to 9 or from 8 to 12
[0424] In some of the foregoing embodiments of Formula (III), at least one occurrence of R7a is H. For example, in some embodiments, R7a is H at each occurrence. In other different embodiments of the foregoing, at least one occurrence of R7b is C1-C8 alkyl. For example, in some embodiments, C1-C8 alkyl is methyl, ethyl, n-propyl, iso-propyl, n- butyl, iso-butyl, tert-butyl, n-hexyl or n-octyl. [0425] In different embodiments of Formula (III), R1 or R2, or both, has one of the following structures: ;
R7a and R7b are, at each occurrence, independently H or C1-C12 alkyl; and a is an integer from 2 to 12, wherein R7a, R7b and a are each selected such that R1 and R2 each independently comprise from 6 to 20 carbon atoms. For example, in some embodiments a is an integer ranging from 5 to 9 or from 8 to 12
[0424] In some of the foregoing embodiments of Formula (III), at least one occurrence of R7a is H. For example, in some embodiments, R7a is H at each occurrence. In other different embodiments of the foregoing, at least one occurrence of R7b is C1-C8 alkyl. For example, in some embodiments, C1-C8 alkyl is methyl, ethyl, n-propyl, iso-propyl, n- butyl, iso-butyl, tert-butyl, n-hexyl or n-octyl. [0425] In different embodiments of Formula (III), R1 or R2, or both, has one of the following structures: ;
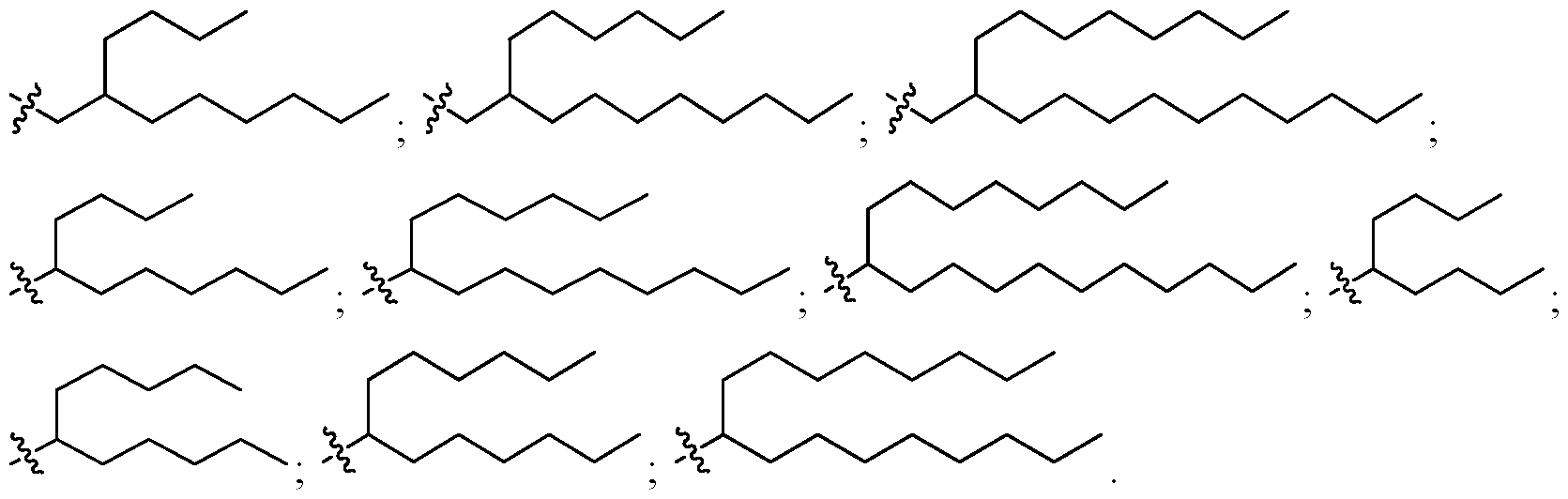 [0426] In some of the foregoing embodiments of Formula (III), R3 is OH, CN, -C(=O)OR4, -OC(=O)R4 or –NHC(=O)R4. In some embodiments, R4 is methyl or ethyl. [0427] In various different embodiments, the cationic lipid of Formula (III) has one of the structures set forth in in Table 15 below. Table 15: Exemplary Cationic Lipid Structures of Formula (III) No. Structure
[0426] In some of the foregoing embodiments of Formula (III), R3 is OH, CN, -C(=O)OR4, -OC(=O)R4 or –NHC(=O)R4. In some embodiments, R4 is methyl or ethyl. [0427] In various different embodiments, the cationic lipid of Formula (III) has one of the structures set forth in in Table 15 below. Table 15: Exemplary Cationic Lipid Structures of Formula (III) No. Structure
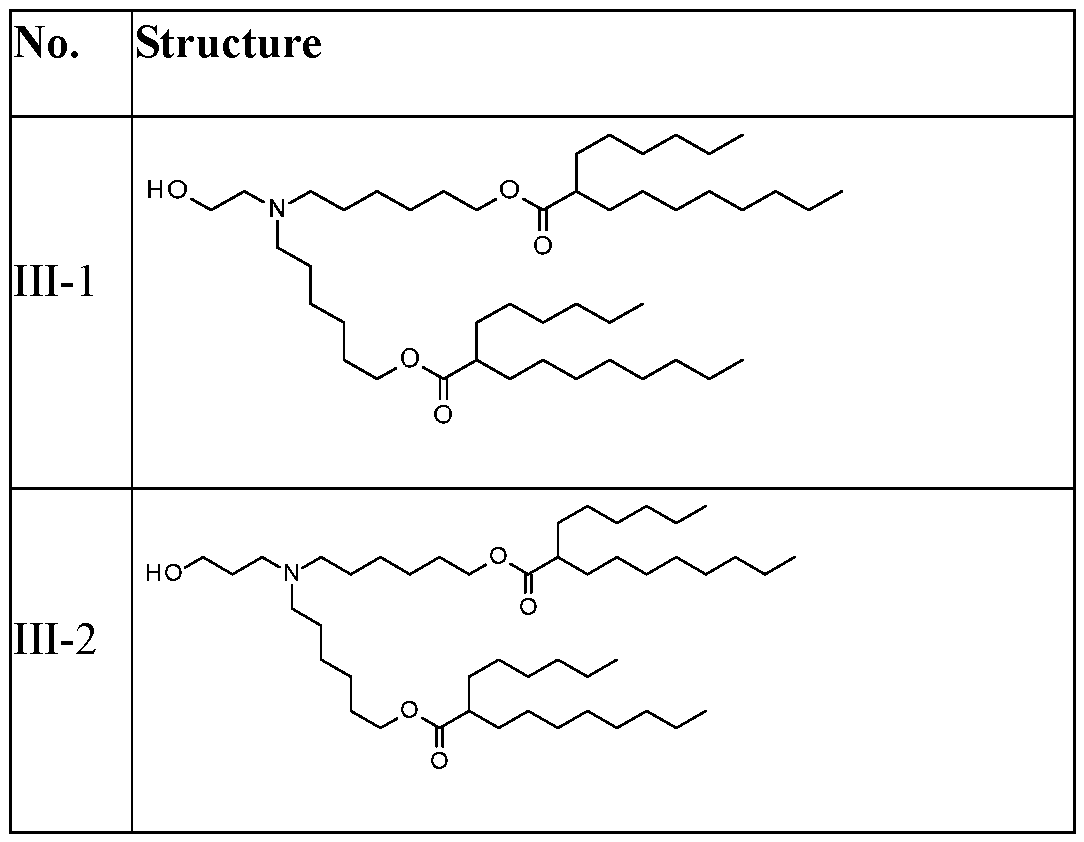 No. Structure
No. Structure
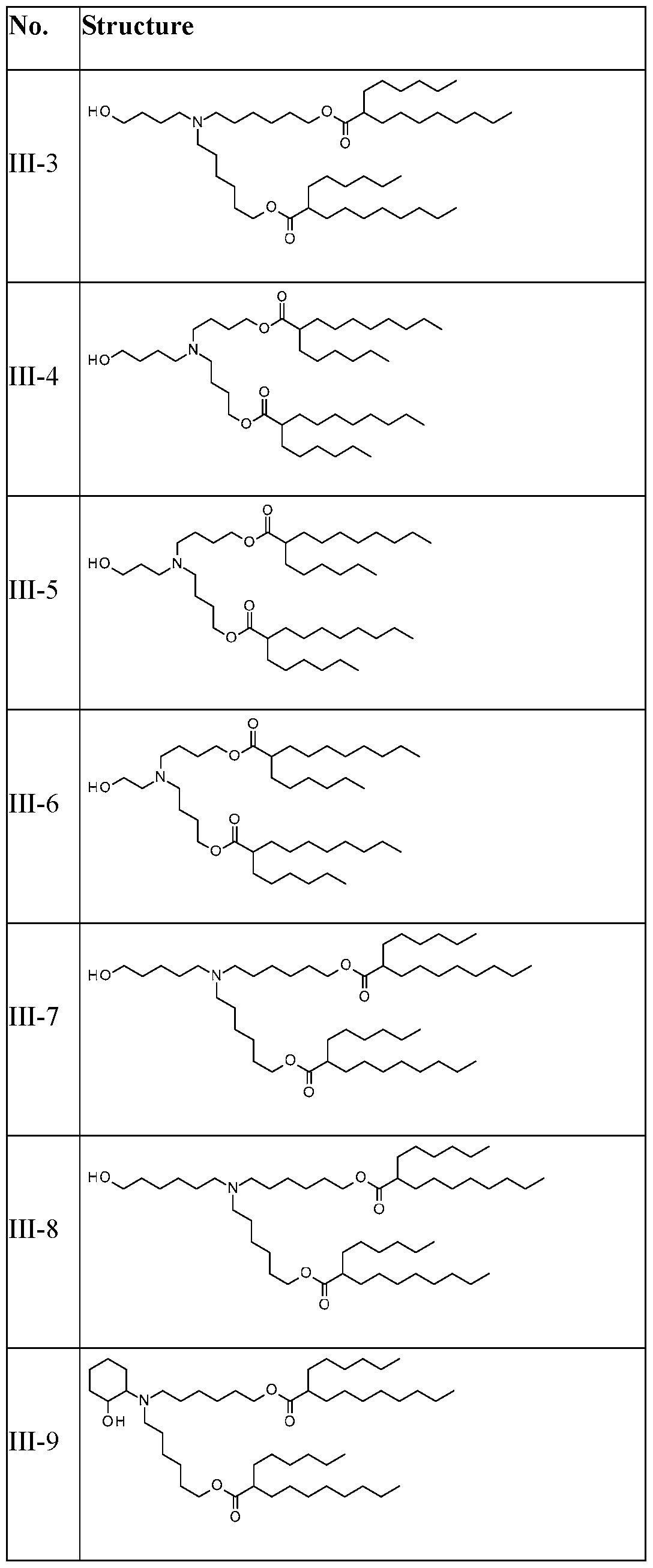 No. Structure
No. Structure
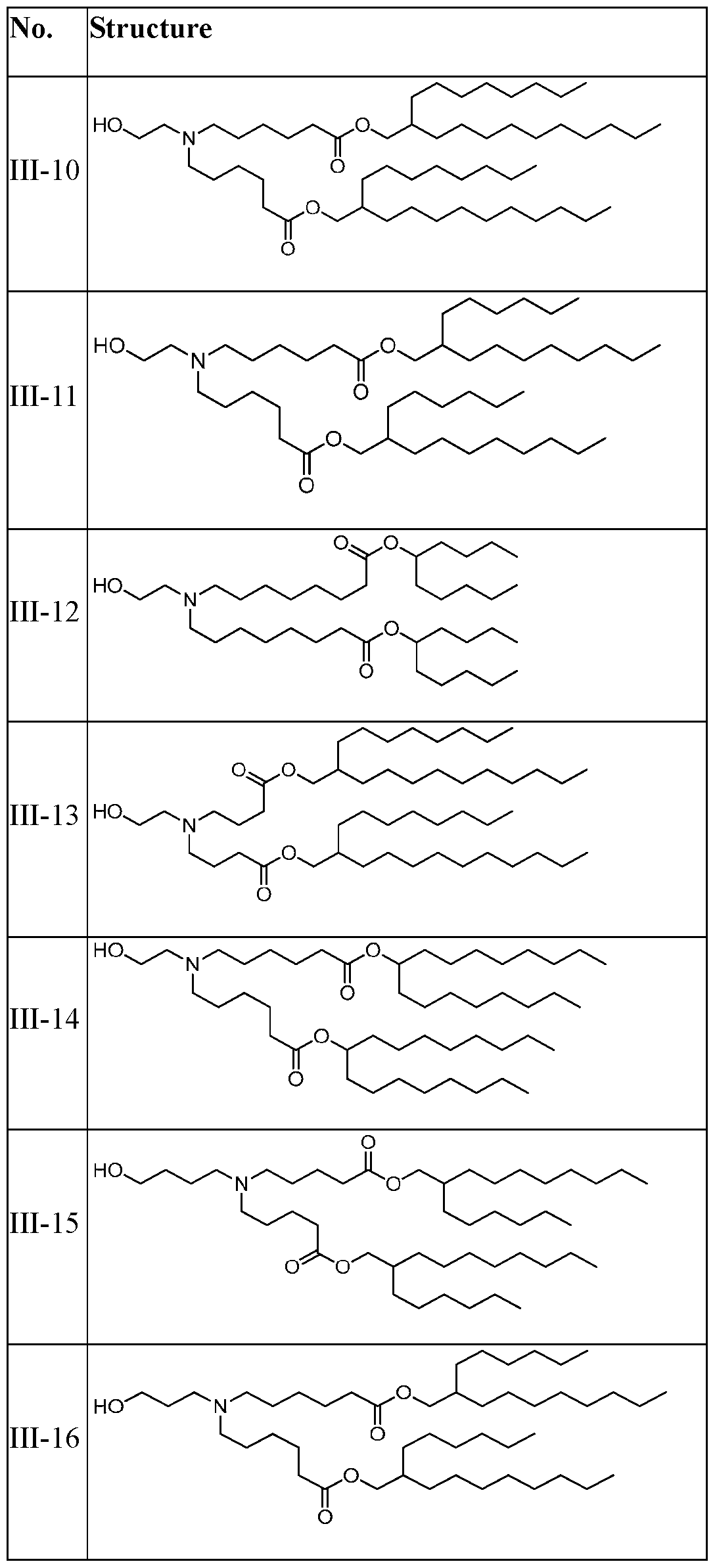 No. Structure
No. Structure
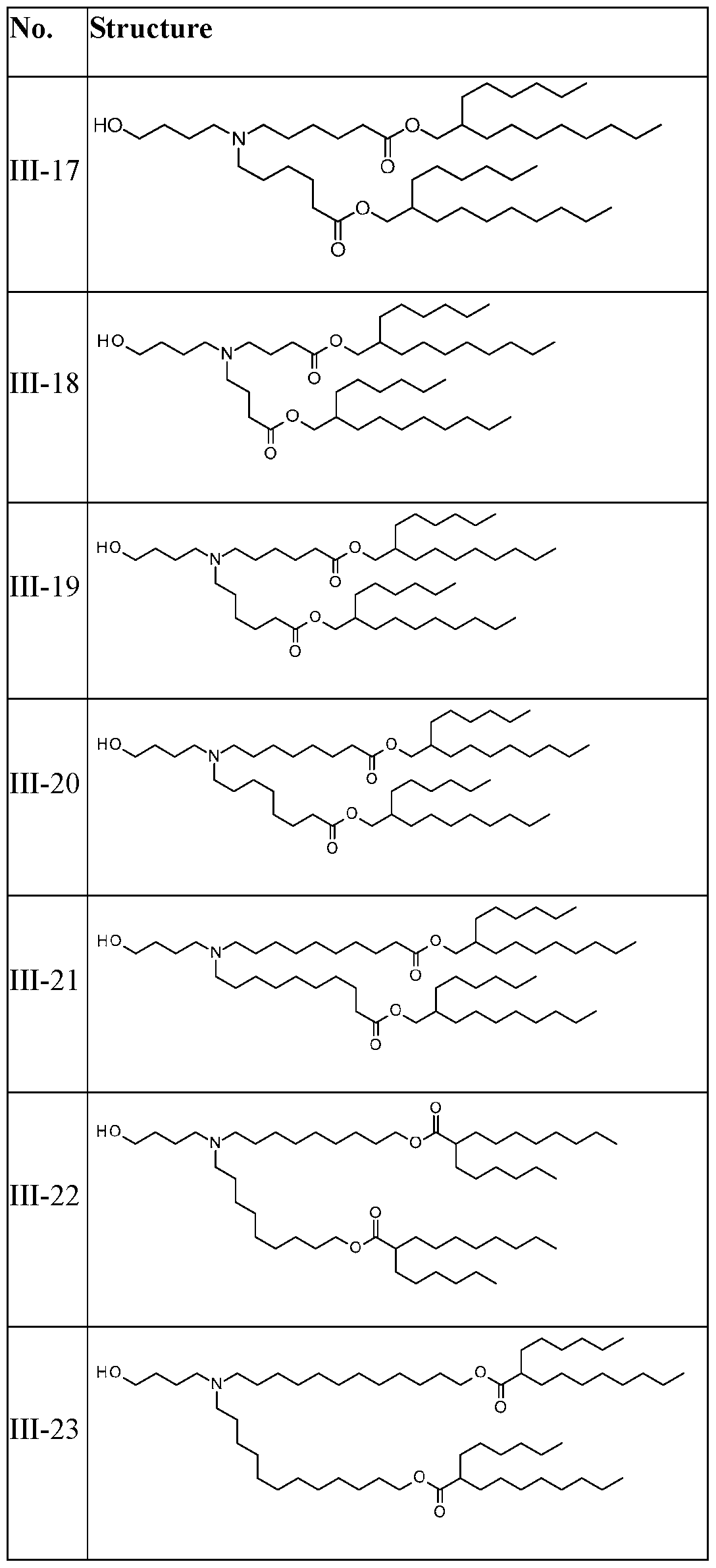 No. Structure
No. Structure
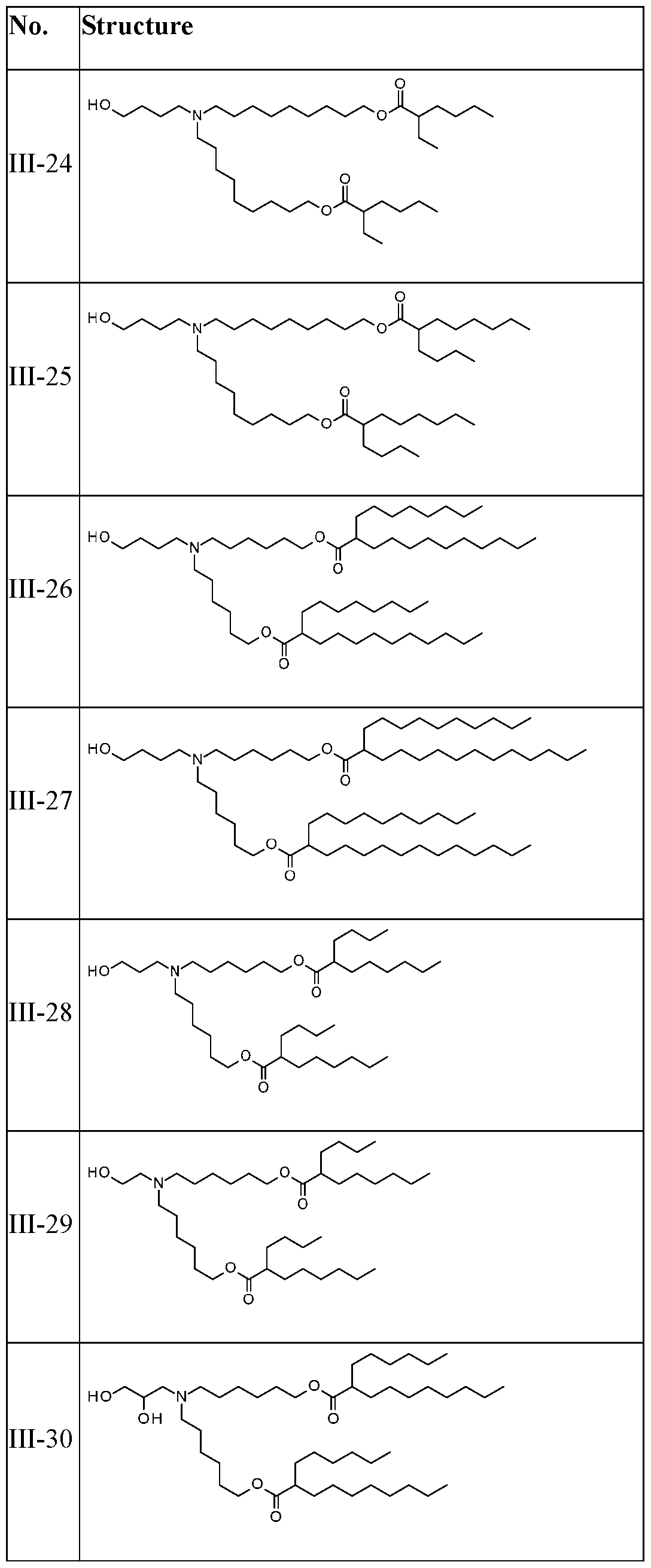 No. Structure [0008]
No. Structure [0008]
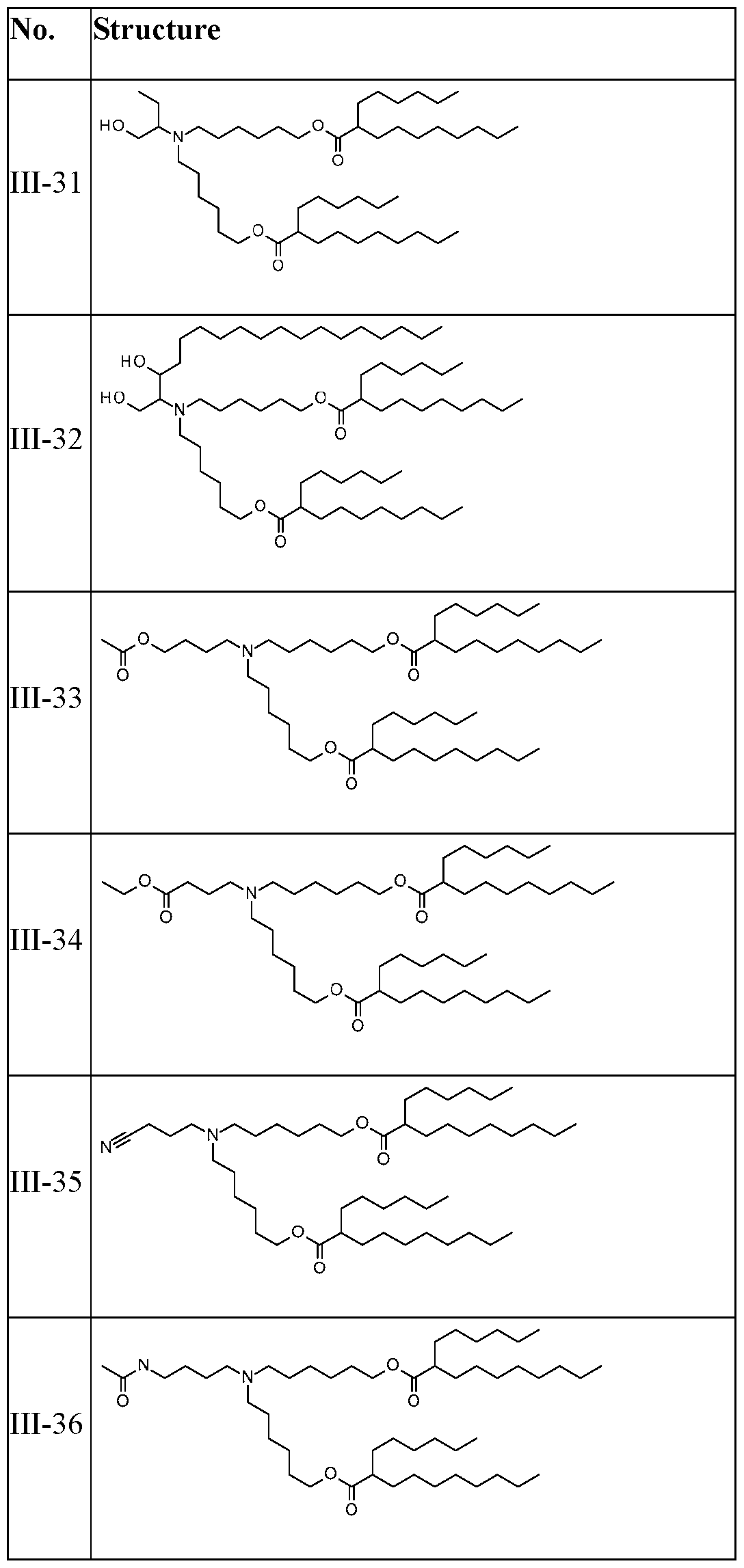 [0428] In various different embodiments, a cationic lipid has one of the structures set forth in Table 16 below.
Table 16: Exemplary Cationic Lipid Structures of Formula A-F No. Structure O N
[0428] In various different embodiments, a cationic lipid has one of the structures set forth in Table 16 below.
Table 16: Exemplary Cationic Lipid Structures of Formula A-F No. Structure O N
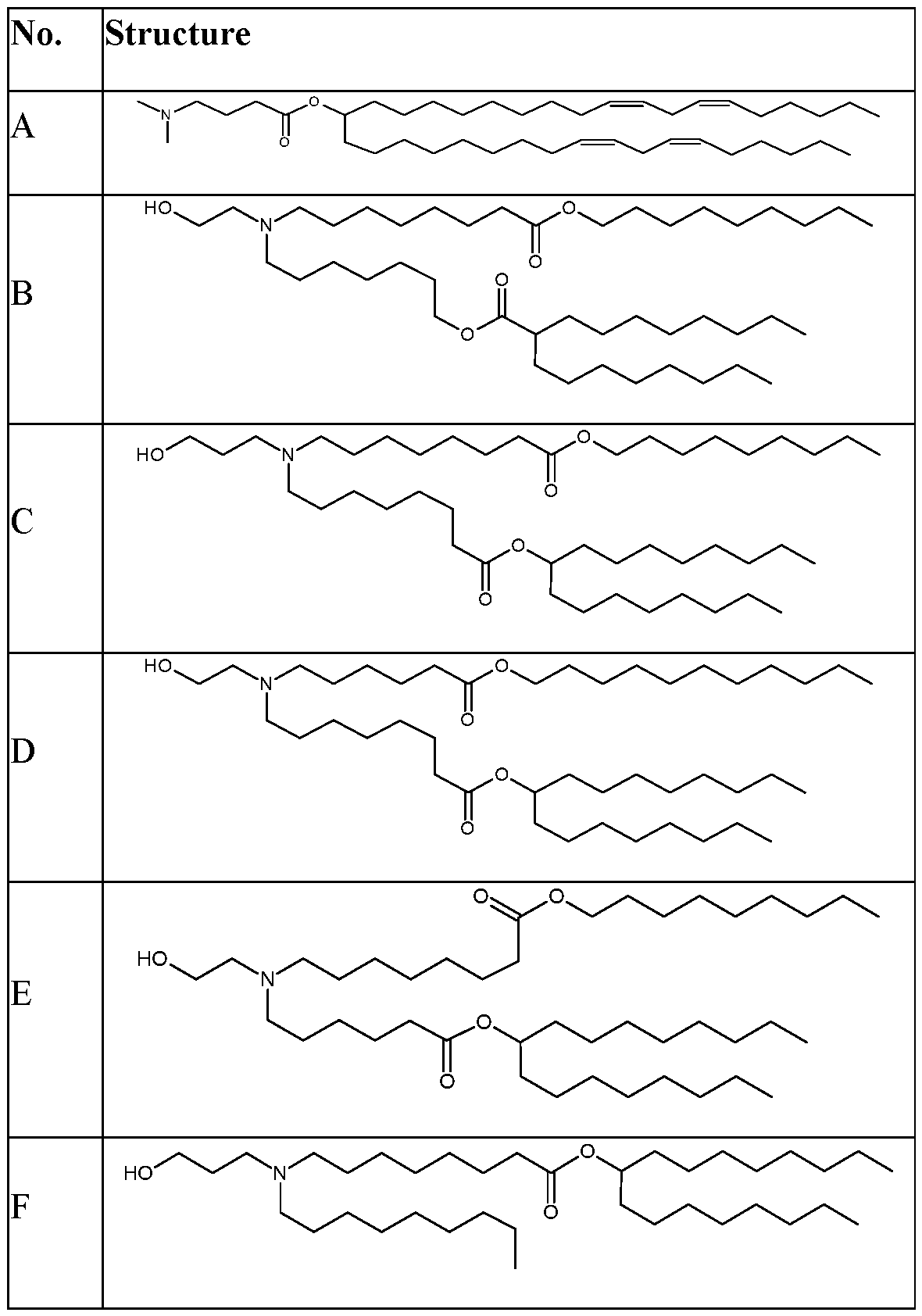 [0429] In some embodiments, an LNP comprises a cationic lipid that is an ionizable lipid-like material (lipidoid). In some embodiments, a cationic lipid has one of the following structures as described in Melamed et al., Science Advances, 2023, 9, eade1444 (the entire contents of which are incorporated herein by reference):
[0429] In some embodiments, an LNP comprises a cationic lipid that is an ionizable lipid-like material (lipidoid). In some embodiments, a cationic lipid has one of the following structures as described in Melamed et al., Science Advances, 2023, 9, eade1444 (the entire contents of which are incorporated herein by reference):

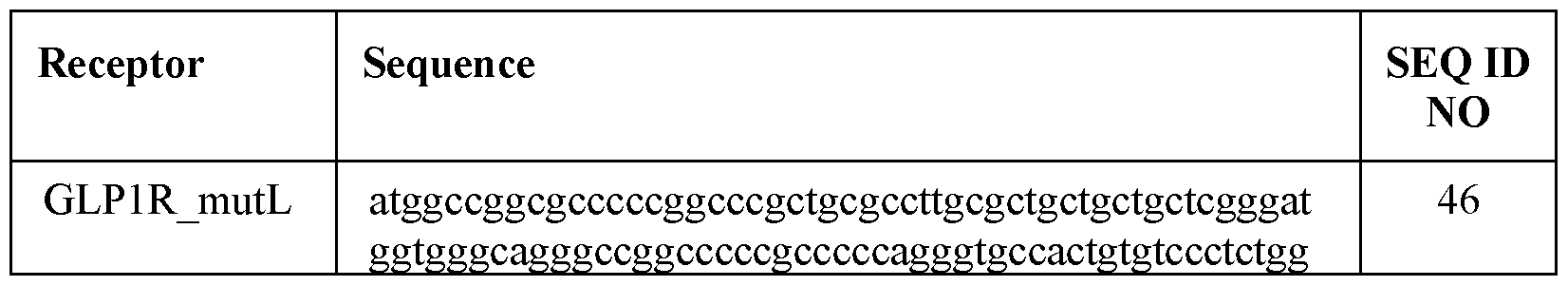




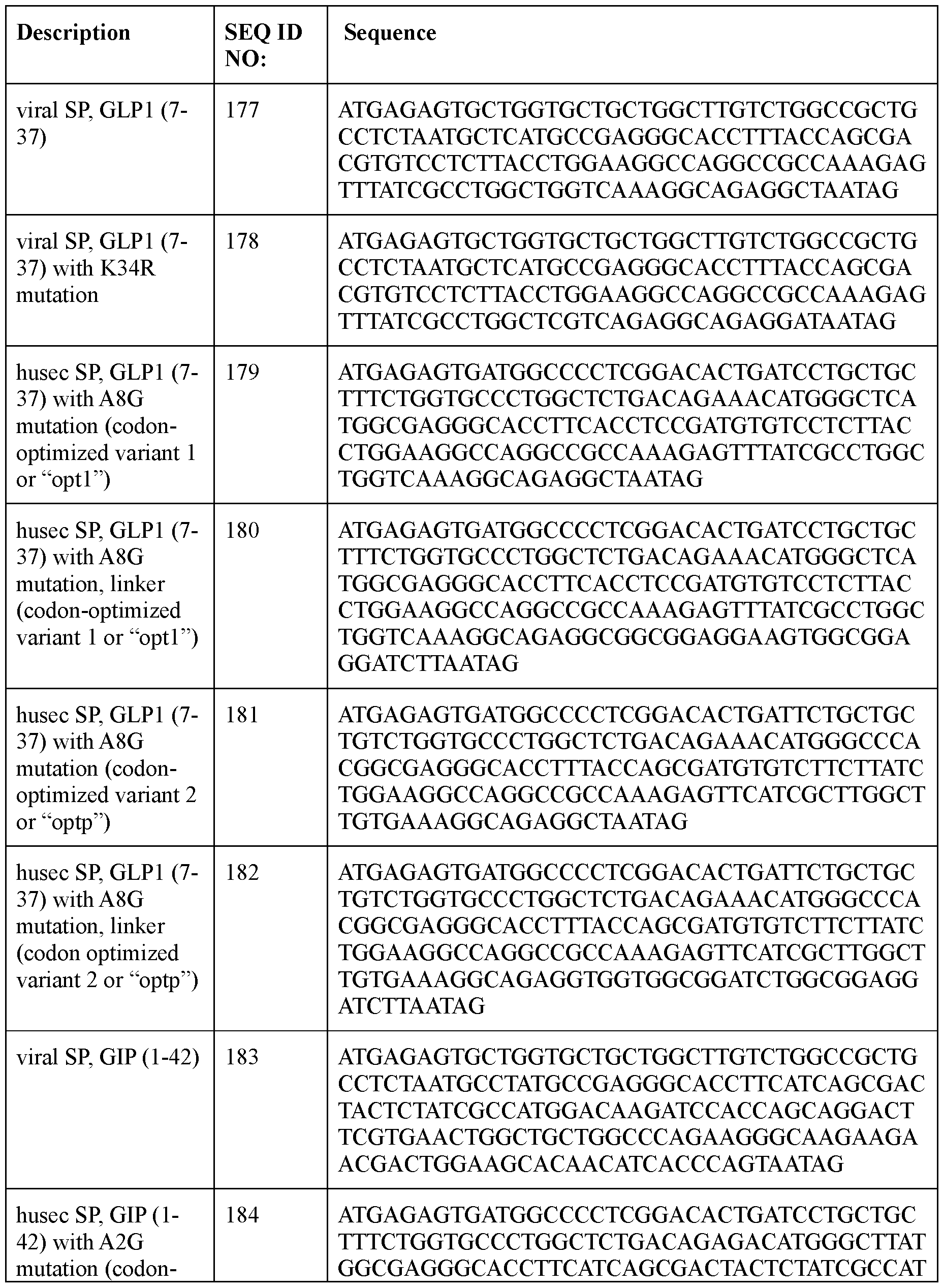


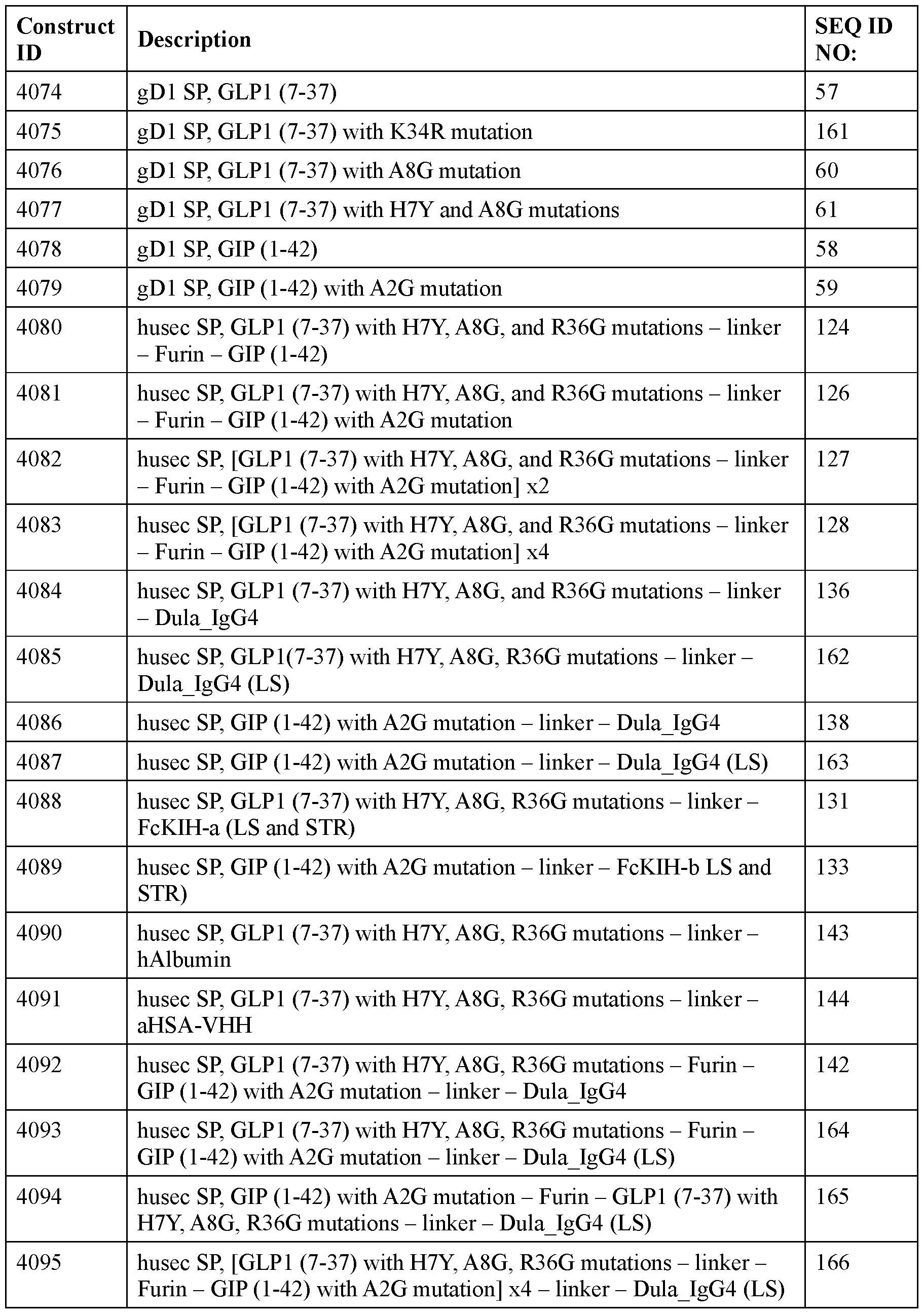
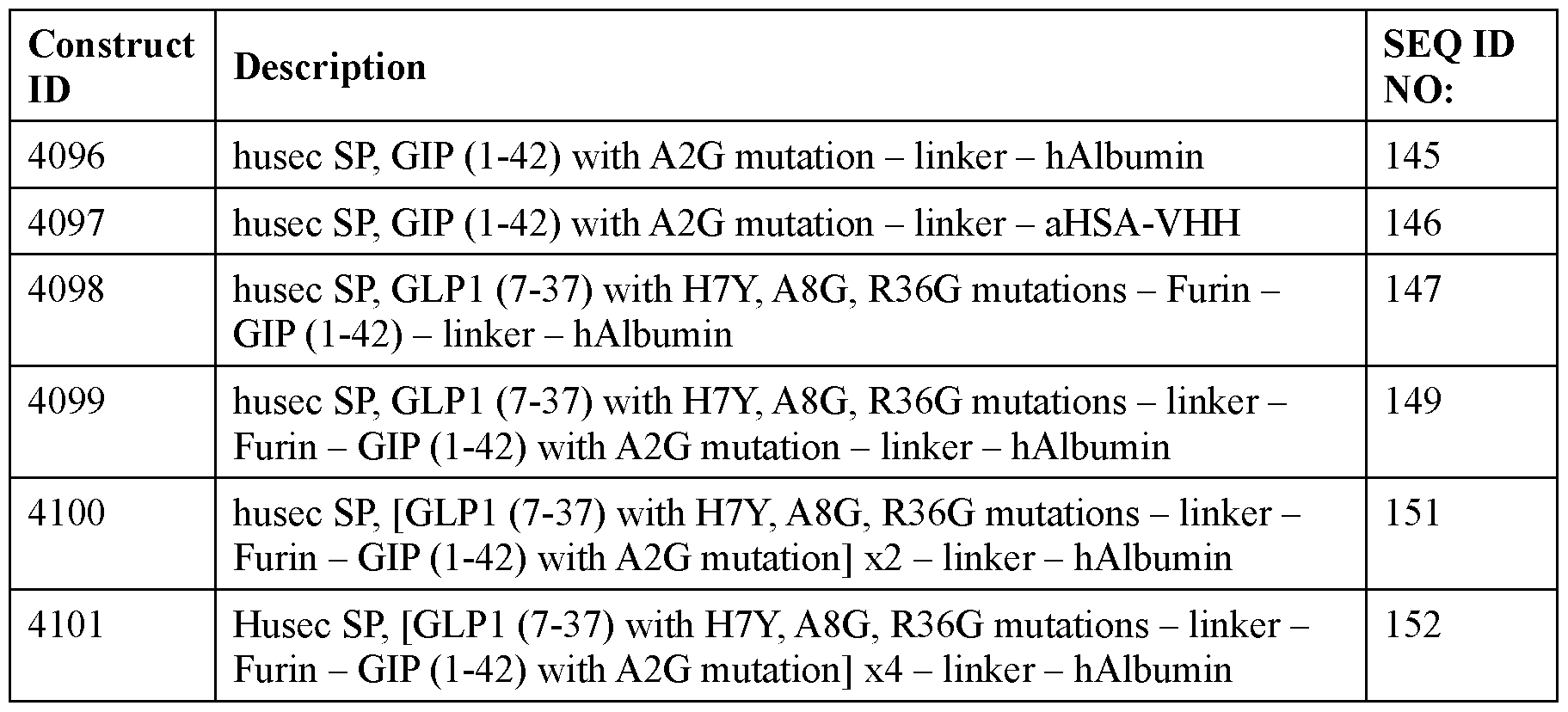
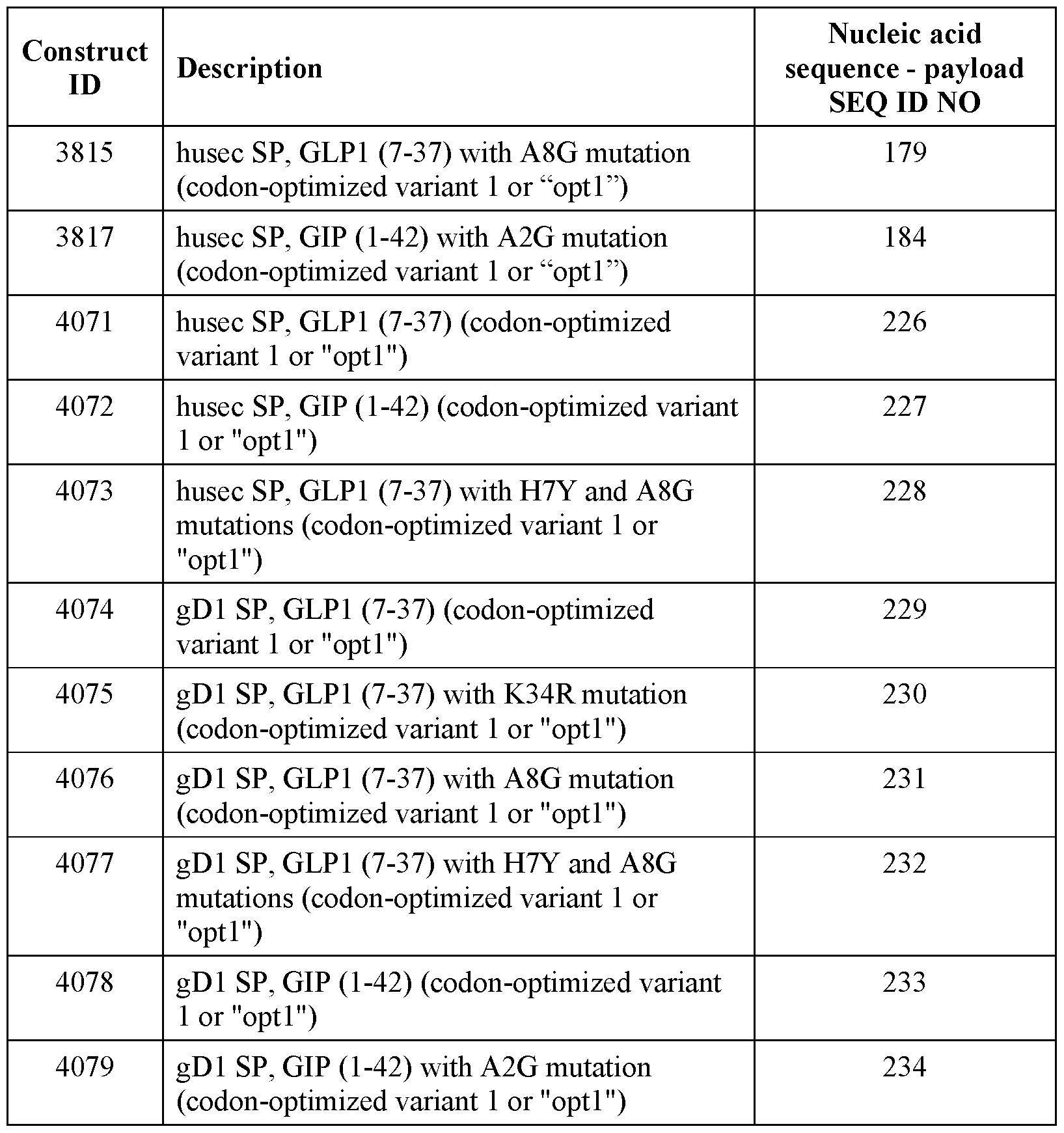

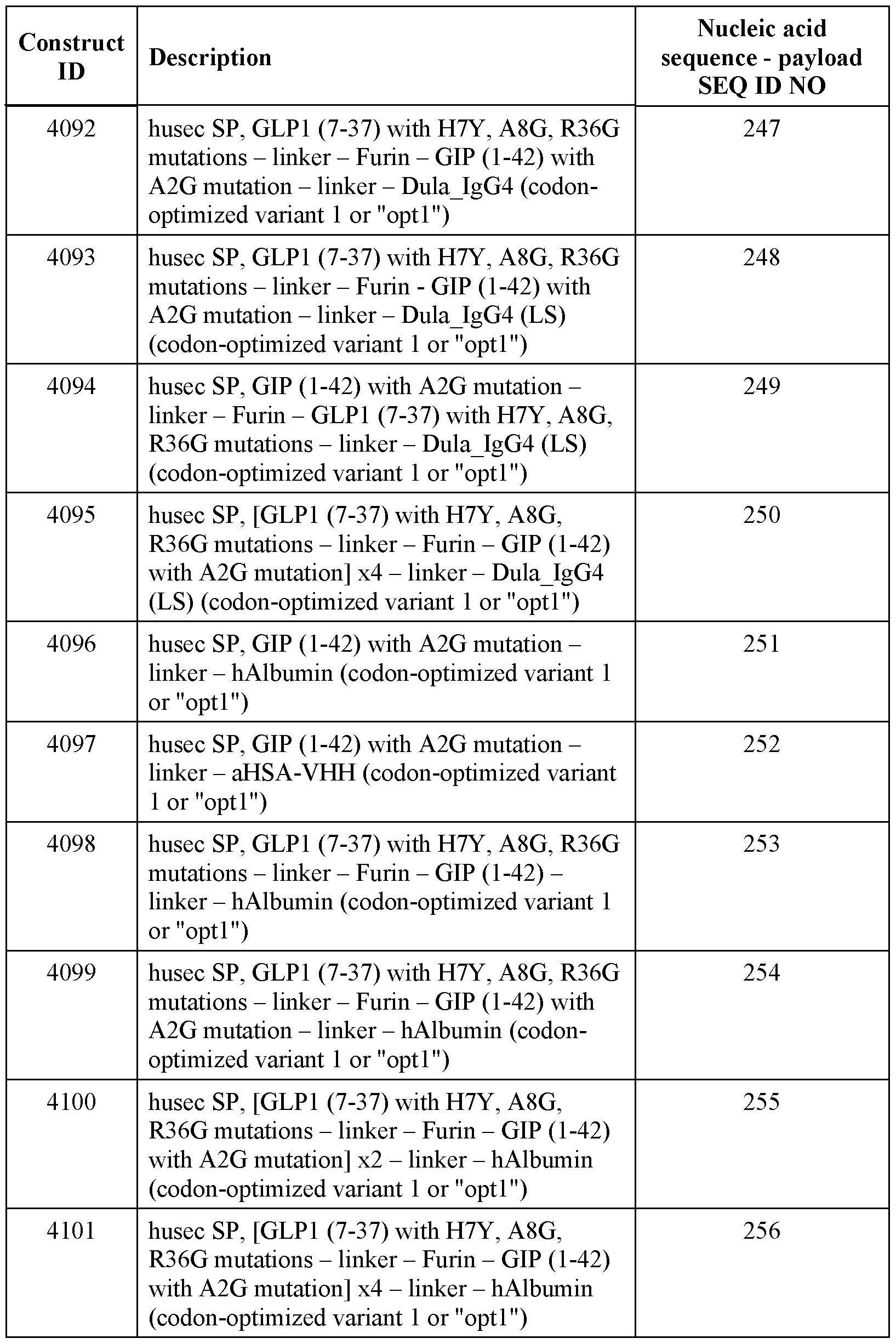
Claims
CLAIMS We claim: 1. A composition comprising a polyribonucleotide encoding an incretin agent.
2. The composition of claim 1, wherein the incretin agent is a GLP1 receptor agonist.
3. The composition of claim 1, wherein the incretin agent is a GIP receptor agonist.
4. The composition of claim 1, wherein the incretin agent is a GLP1/GIP dual receptor agonist.
5. The composition of claim 1, wherein the incretin agent is a GLP1/GCG dual receptor agonist.
6. The composition of claim 1, wherein the incretin agent is a GLP1/GIP/GCG triple receptor agonist.
7. The composition of claim 2, wherein the incretin agent comprises an incretin peptide having an amino acid sequence according to any one of SEQ ID NOs: 5-7, 63-64, 69-70, and 74-75.
8. The composition of claim 3, wherein the incretin agent comprises an incretin peptide having an amino acid sequence according to any one of SEQ ID NOs: 8-9, 62, and 72.
9. The composition of claim 2, wherein the incretin agent comprises an incretin peptide having an amino acid sequence according SEQ ID NO: 11.
10. The composition of claim 4, wherein the incretin agent comprises an incretin peptide having an amino acid sequence according to any one of SEQ ID NOs: 12-14.
11. The composition of claim 6, wherein the incretin agent comprises an incretin peptide having an amino acid sequence according to SEQ ID NO: 15.
12. The composition of any one of claims 7-11, wherein the incretin peptide is fused to a signal peptide, optionally via the N-terminus of the incretin peptide, optionally via a linker.

13. The composition of claim 12, wherein the signal peptide has an amino acid sequence according to any one of SEQ ID NOs: 16-39 and 65-67.
14. The composition of claim 12, wherein the signal peptide has an amino acid sequence according to any one of SEQ ID NOs: 16-21 and 65-67.
15. The composition of claim 12, wherein the signal peptide has an amino acid sequence according to SEQ ID NO: 17.
16. The composition of claim 12, wherein the signal peptide has an amino acid sequence according to SEQ ID NO: 65.
17. The composition of claim 12, wherein the signal peptide has an amino acid sequence according to SEQ ID NO: 66.
18. The composition of claim 1, wherein the incretin agent comprises an amino acid sequence according to any one of SEQ ID NOs: 41-45, 52-61, and 108-152.
19. The composition of any one of claims 1-18, wherein the incretin agent comprises an incretin peptide fused to one or more additional incretin peptides, optionally via one or more linkers.
20. The composition of claim 19, wherein the one or more linkers comprise an amino acid sequence according of any one of SEQ ID NOs: 1-5, 68, or 156.
21. The composition of claim 19 or 20, wherein the incretin agent comprises an incretin peptide fused to two or more incretin peptides.
22. The composition of any one of claims 19-21, wherein the incretin agent comprises at least one GLP1 receptor agonist and at least one GIP receptor agonist.
23. The composition of any one of claims 19-22, wherein the incretin agent comprises at least two GLP1 receptor agonists.
24. The composition of any one of claims 19-23, wherein the incretin agent comprises at least two GIP receptor agonists.

25. The composition of any one of claims 19-24, wherein the incretin agent comprises one or more furin cleavage sites.
26. The composition of claim 25, wherein the one or more furin cleavage sites are located between adjacent incretin peptides.
27. The composition of claim 27 or 28, wherein the one or more furin cleavage sites comprise an amino acid sequence according to SEQ ID NO: 153.
28. The composition of any one of claims 19-27, wherein the incretin agent comprises one or more units that each comprise, from N-terminus to C-terminus: GLP1 receptor agonist-linker-furin cleavage site-GIP receptor agonist, e.g., wherein the incretin agent comprises one unit (e.g., SEQ ID NOs: 76, 77, 78, 79, 80, 81), two units (e.g., SEQ ID NOs: 82); or four units (e.g., SEQ ID NO: 83).
29. The composition of any one of claims 19-30, wherein the incretin agent comprises an amino acid sequence according to any one of SEQ ID NOs: 76-83, 94-97, 102-107.
30. The composition of any one of claims 1-29, wherein the incretin agent comprises a half-life extending moiety.
31. The composition of claim 30, wherein the half-life extending moiety comprises albumin (e.g., human serum albumin).
32. The composition of claim 31, wherein the human serum albumin comprises an amino acid sequence having at least 90%, 95%, or 99% identity to SEQ ID NO: 159.
33. The composition of claim 31 or 32, wherein the human serum albumin comprises an amino acid sequence according to SEQ ID NO: 159.
34. The composition of any one of claims 31-33, wherein the incretin agent comprises albumin (e.g., human serum albumin) fused to one or more units that each comprise from, N-terminus to C-terminus: (i) GLP1 receptor agonist-linker (e.g., SEQ ID NO: 98); (ii) GIP receptor agonist-linker (e.g., SEQ ID NO: 100);
 (iii) GLP1 receptor agonist-linker-furin cleavage site (e.g., SEQ ID NO: 102); or (iv) GLP1 receptor agonist-linker-furin cleavage site-GIP receptor agonist, e.g., wherein the incretin agent comprises one unit (e.g., SEQ ID NO: 104), two units (e.g., SEQ ID NO: 106) or four units (e.g., SEQ ID NO: 107).
(iii) GLP1 receptor agonist-linker-furin cleavage site (e.g., SEQ ID NO: 102); or (iv) GLP1 receptor agonist-linker-furin cleavage site-GIP receptor agonist, e.g., wherein the incretin agent comprises one unit (e.g., SEQ ID NO: 104), two units (e.g., SEQ ID NO: 106) or four units (e.g., SEQ ID NO: 107).
35. The composition of any one of claims 31-34, wherein the incretin agent comprises an amino acid sequence according to any one of SEQ ID NOs: 98, 100, 102, 104, 106, 107, or any combination thereof.
36. The composition of claim 30, wherein the half-life extending moiety comprises an albumin binding domain (ABD).
37. The composition of claim 36, wherein the ABD is derived from protein G of Streptococcus strain GI48 and/or from protein PAB of Finegoldia magna, such as ABD035 and SA21.
38. The composition of claim 36, wherein the half-life extending moiety comprises an ABD that binds to domain II of human serum albumin and does not overlap or interfere with binding to the FcRn-binding site on albumin.
39. The composition of claim 36, wherein the half-life extending moiety comprises ABDCon.
40. The composition of claim 36, wherein the half-life extending moiety comprises an albumin binding domain (ABD) derived from the bacterial protein Sso7d from the hyperthermophilic archaeon Sulfolobus solfataricus, such as M11.12 and M18.2.5.
41. The composition of claim 36, wherein the half-life extending moiety comprises a DARPin that binds albumin.
42. The composition of claim 36, wherein the ABD comprises an immunoglobulin domain or fragment thereof that binds albumin.
43. The composition of claim 36 or 42, wherein the ABD comprises a fully human domain antibody (dAb) that binds albumin, such as AlbudAb.

44. The composition of claim 36 or 42-43, wherein the ABD comprises a Fab that binds albumin, such as dsFv CA645.
45. The composition of claim 36 or 42-44, wherein the ABD comprises a heavy chain only (VHH) antibody, such as a nanobody, that binds albumin.
46. The composition of claim 45, wherein the VHH antibody comprises a VHH domain having the complementarity determining region (CDR) sequences HCDR1, HCDR2, and/or HCDR3 according to SEQ ID NO: 191 (GFTLDYYA), SEQ ID NO: 192 (IASSGGST), and/or SEQ ID NO: 193 (AAAVLECRTVVRGYDY), respectively.
47. The composition of claim 46, wherein the VHH antibody comprises an amino acid sequence having at least 90%, 95%, or 99% identity to SEQ ID NO: 154.
48. The composition of claim 46 or 47, wherein the VHH antibody comprises an amino acid sequence according to SEQ ID NO: 154.
49. The composition of any one of claims 45-47, wherein the incretin agent comprises a VHH antibody that binds albumin fused to a unit that comprises, from N-terminus to C- terminus: (i) GLP1-linker (e.g., SEQ ID NO: 99); (ii) GIP receptor agonist-linker (e.g., SEQ ID NO: 101); or (iii) GLP1 receptor agonist-linker-furin-GIP receptor agonist-linker (e.g., SEQ ID NO: 103 or 105).
50. The composition of any one of claims 45-49, wherein the incretin agent comprises an amino acid sequence according to any one of SEQ ID NOs: 99, 101, 103, 105.
51. The composition of claim 30, wherein the half-life extending moiety does not comprise a Fc domain, such as from a human IgG, optionally from a human IgG1, IgG2, IgG3, or IgG4.
52. The composition of claim 30, wherein the half-life extending moiety comprises a Fc domain, such as from a human IgG, optionally from a human IgG1, IgG2, IgG3, or IgG4.

53. The composition of claim 52, wherein the human IgG is a human IgG4.
54. The composition of claim 52 or 53, wherein the incretin agent comprises an IgG4 Fc domain fused to a unit comprising, from N-terminus to C-terminus: (i) GLP1 receptor agonist-linker (e.g., SEQ ID NOs: 10, 89, 90, 91); (ii) GIP receptor agonist-linker (e.g., SEQ ID NO: 92, 93); or (iii) GLP1 receptor agonist-linker-furin-GIP receptor agonist-linker (e.g., SEQ ID NO: 94, 95, 96, 97).
55. The composition of claim 53 or 54, wherein the IgG4 Fc domain comprises an amino acid sequence that is at least 90%, 95%, or 99% identical to SEQ ID NO: 155.
56. The composition of claim 55, wherein the IgG4 Fc domain comprises an amino acid sequence according to SEQ ID NO: 155.
57. The composition of any one of claims 53-56, wherein the incretin agent comprises an amino acid sequence according to any one of SEQ ID NOs: 10, and 89-97.
58. The composition of any one of claims 52-57, wherein the Fc domain comprises one or more mutations in one or both Fc constant domains to increase half-life of the incretin agent and/or induce dimerization.
59. The composition of claim 58, wherein the one or more mutations comprises one or more mutations in a CH3 domain.
60. The composition of claim 58 or 59, wherein the one or more mutations to induce dimerization comprises: (i) Y349C, T366S, L368A, and/or Y407V, according to EU numbering; or (ii) S354C and/or T366W, according to EU numbering.
61. The composition of any one of claims 58-60, wherein the one or more mutations comprises Y349C, T366S, L368A, and Y407V (“FcKIH-b”), according to EU numbering, or S354C and T366W (“FcKIH-a”), according to EU numbering.

62. The composition of any one of claims 58-61, wherein the incretin agent comprises a first polypeptide chain and a second polypeptide chain, wherein the first polypeptide chain comprises an incretin peptide fused to a first Fc domain, wherein the first Fc domain comprises the mutations Y349C, T366S, L368A, and Y407V (“FcKIH-b”), according to EU numbering and wherein the second polypeptide chain comprises an incretin peptide fused to a second Fc domain, wherein the second Fc domain comprises the mutations S354C and T366W (“FcKIH-a”), according to EU numbering.
63. The composition of any one of claims 58-62, wherein the one or more mutations to increase half-life of the incretin agent comprises M428L and N434S (“LS”), according to EU numbering.
64. The composition of any one of claims 58-63, wherein the incretin agent comprises an Fc domain with FcKIH-a mutations on a first polypeptide chain and an Fc domain with FcKIH-b mutations on a second polypeptide chain, where the Fc domain on each polypeptide chain is independently fused with one or more units that comprise, from N- terminus to C-terminus: (i) GLP1 receptor agonist-linker (e.g., SEQ ID NO: 84, 85, 86, 87); or (ii) GIP receptor agonist-linker (e.g., SEQ ID NO: 88).
65. The composition of any one of claims 58-64, wherein the incretin agent comprises an amino acid sequence according to any one of SEQ ID NOs: 84-88.
66. The composition of any one of claims 58-64, wherein the Fc domain comprises one or more mutations that ablate effector activity of the Fc domain (e.g., binding to Fcγ receptors or C1q).
67. The composition of claim 66, wherein the one or more mutations that ablate effector activity of the Fc domain (e.g., binding to Fcγ receptors or C1q) comprise the following mutations: L234S, L235T, and G236R (“STR”) according to EU numbering.
68. The composition of claim 66, wherein the one or more mutations that ablate effector activity of the Fc domain (e.g., binding to Fcγ receptors or C1q) comprise the following mutations: L234A and L235A (“LALA”) according to EU numbering.

69. The composition of claim 66, wherein the one or more mutations that ablate effector activity of the Fc domain (e.g., binding to Fcγ receptors or C1q) comprise the following mutations: L234A/L235A/P329G (“LALAPG”) according to EU numbering.
70. The composition of claim 30, wherein the half-life extending moiety comprises a VNAR that binds albumin.
71. The composition of claim 30, wherein the half-life extending moiety comprises an XTEN sequence.
72. The composition of any one of claims 1-71, wherein the polyribonucleotide has a ribonucleic acid sequence that is at least 90% identical to any one of SEQ ID NOs: 177-185 and 224-256.
73. The composition of claim 72, wherein the polyribonucleotide has a ribonucleic acid sequence according to any one of SEQ ID Nos: 177-185 and 224-256.
74. The composition of any one of claims 1-73, wherein the polyribonucleotide comprises at least one non-coding sequence element that enhances RNA stability and/or translation efficiency.
75. The composition of claim 74, wherein the at least one non-coding sequence element comprises a 5’ cap structure, a 5’ UTR, a 3’ UTR, and/or a polyA tail.
76. The composition of claim 75, wherein the polyribonucleotide comprises, in a 5’ to 3’ direction: a. a 5’ UTR; b. a signal peptide-coding sequence; c. an incretin peptide-coding sequence; d. a 3’ UTR; and e. a polyA tail.
77. The composition of claim 75, wherein the polyribonucleotide comprises, in a 5’ to 3’ direction: (1)
 a. a 5’ UTR; b. a signal peptide-coding sequence; c. an incretin peptide-coding sequence; d. a linker-coding sequence; e. a half-life extending moiety-coding sequence; f. a 3’ UTR; and g. a polyA tail; or (2) a. a 5’ UTR; b. a signal peptide-coding sequence; c. a half-life extending moiety-coding sequence; d. a linker-coding sequence; e. an incretin peptide-coding sequence; f. a 3’ UTR; and g. a polyA tail.
a. a 5’ UTR; b. a signal peptide-coding sequence; c. an incretin peptide-coding sequence; d. a linker-coding sequence; e. a half-life extending moiety-coding sequence; f. a 3’ UTR; and g. a polyA tail; or (2) a. a 5’ UTR; b. a signal peptide-coding sequence; c. a half-life extending moiety-coding sequence; d. a linker-coding sequence; e. an incretin peptide-coding sequence; f. a 3’ UTR; and g. a polyA tail.
78. The composition of any one of claims 1-77, wherein the incretin peptide is encoded by a coding sequence which is codon-optimized and/or the G/C content of which is increased compared to wild type coding sequence, wherein the codon-optimization and/or the increase in the G/C content does not change the sequence of the encoded amino acid sequence.
79. The composition of any one of claims 1-78, wherein the polyribonucleotide comprises at least one modified ribonucleotide.
80. The composition of claim 79, wherein the polyribonucleotide comprises a modified nucleoside in place of uridine.
81. The composition of claim 80, wherein the polyribonucleotide comprises a modified nucleoside in place of each uridine.
82. The composition of claim 81, wherein the modified nucleoside is selected from pseudouridine (ψ), N1-methyl-pseudouridine (m1ψ), and 5-methyl-uridine (m5U).
83. The composition of any one of claims 80-83, wherein the modified nucleoside is N1- methyl-pseudouridine (m1ψ).
84. The composition of any one of claims 1-83, wherein the polyribonucleotide comprises a 5’ cap structure.
85. The composition of any one of claims 1-84, wherein the polyribonucleotide comprises a 5’ UTR.
86. The composition of any one of claims 1-85, wherein the polyribonucleotide comprises a 3’ UTR.
87. The composition of any one of claims 1-86, wherein the polyribonucleotide comprises a polyA tail.
88. The composition of claim 87, wherein the polyA tail comprises at least 100 nucleotides.
89. The composition of any one of claims 1-88, wherein the polyribonucleotide is mRNA.
90. The composition of any one of claims 1-89, wherein the polyribonucleotide is formulated as a liquid, formulated as a solid, or a combination thereof.
91. The composition of any one of claims 1-90, wherein the polyribonucleotide is formulated for injection.
92. The composition of any one of claims 1-91, wherein the polyribonucleotide is formulated for intraperitoneal or intravenous administration.
93. The composition of any one of claims 1-92, wherein the polyribonucleotide is formulated or is to be formulated as lipid particles.
94. The composition of claim 93, wherein the polyribonucleotide is formulated or is to be formulated as lipid nanoparticles.
95. The composition of claim 94, wherein the polyribonucleotide is encapsulated within the lipid nanoparticles.

96. The composition of claim 94 or 95, wherein the lipid nanoparticles are pancreas- targeting and/or gut-targeting lipid nanoparticles.
97. The composition of any one of claims 94-96, wherein the lipid nanoparticles are cationic lipid nanoparticles.
98. The composition of claim 97, wherein lipids that form the lipid nanoparticles comprise: a. a polymer-conjugated lipid; b. a cationic lipid; and c. a neutral lipid.
99. The composition of claim 98, wherein the polymer-conjugated lipid is a PEG- conjugated lipid.
100. The composition of claim 98 or 99, wherein the cationic lipid is an ionizable lipid- like material (lipidoid).
101. The composition of claim 100, wherein the cationic lipid has one of the following structures:


102. The composition of any one of claims 99-101, wherein the neutral lipid comprises a helper lipid such as 1,2-distearoyl-sn-glycero-3-phosphocholine (DPSC) and/or cholesterol.
103. The composition of any one of claims 99-102, wherein the cationic lipid is selected from cationic lipid X-2, X-3, or X-4 and the neutral lipid comprises a helper lipid such as DOTAP, DOPE, or PS, and cholesterol.
104. The composition of claim 103, wherein the polymer-conjugated lipid is C14- PEG2000.
105. The composition of any one of claims 99-104, wherein the lipid nanoparticles comprise: i) about 30 mol% to about 50 mol% of a cationic lipid; ii) about 1 mol% to 5 mol% of a PEG-conjugated lipid; iii) about 30 mol% to about 50 mol% of a helper lipid; and iv) about 20 mol% to about 40 mol% of cholesterol.
106. The composition of any one of claims 99-104, wherein the lipid nanoparticles comprise about 35 mol% of a cationic lipid; about 40 mol% of a helper lipid, about 22.5 mol% of cholesterol, and about 2.5 mol% of a PEG-conjugated lipid.

107. The composition of claim 106, wherein the lipid nanoparticles comprise about 35 mol% of cationic lipid X-2, X-3, or X-4, about 40 mol% of DOTAP, DOPE, or PS, about 22.5 mol% of cholesterol, and about 2.5 mol% of C14-PEG2000.
108. The composition of claim 107, wherein the lipid nanoparticles comprise about 35 mol% of cationic lipid X-2; about 40 mol% of DOTAP; about 22.5 mol% of cholesterol, and about 2.5 mol% of C14-PEG2000.
109. The composition of claim 107, wherein the lipid nanoparticles comprise about 35 mol% of cationic lipid X-3; about 40 mol% of DOTAP; about 22.5 mol% of cholesterol; and about 2.5 mol% of C14-PEG2000.
110. The composition of claim 107, wherein the lipid nanoparticles comprise about 35 mol% of cationic lipid X-4; about 40 mol% of DOTAP; about 22.5 mol% of cholesterol; and about 2.5 mol% of C14-PEG2000.
111. The composition of claim 107, wherein the lipid nanoparticles comprise about 35 mol% of cationic lipid X-2; about 40 mol% of DOPE; about 22.5 mol% of cholesterol; and about 2.5 mol% of C14-PEG2000.
112. The composition of claim 107, wherein the lipid nanoparticles comprise about 35 mol% of cationic lipid X-3; about 40 mol% of DOPE; about 22.5 mol% of cholesterol; and about 2.5 mol% of C14-PEG2000.
113. The composition of claim 107, wherein the lipid nanoparticles comprise about 35 mol% of cationic lipid X-4; about 40 mol% of DOPE; about 22.5 mol% of cholesterol; and about 2.5 mol% of C14-PEG2000.
114. The composition of claim 107, wherein the lipid nanoparticles comprise about 35 mol% of cationic lipid X-2; about 40 mol% of PS; about 22.5 mol% of cholesterol; and about 2.5 mol% of C14-PEG2000.
115. The composition of claim 107, wherein the lipid nanoparticles comprise about 35 mol% of cationic lipid X-3; about 40 mol% of PS; about 22.5 mol% of cholesterol; and about 2.5 mol% of C14-PEG2000.

116. The composition of claim 107, wherein the lipid nanoparticles comprise about 35 mol% of cationic lipid X-4; about 40 mol% of PS; about 22.5 mol% of cholesterol; and about 2.5 mol% of C14-PEG2000.
117. The composition of any one of claims 94-116, wherein the lipid nanoparticles are formulated for intraperitoneal (i.p.) delivery.
118. The composition of any one of claims 94-117, wherein the lipid nanoparticles have an average size of about 50-150 nm.
119. The composition of any one of claims 1-118, further comprising one or more pharmaceutically acceptable carriers, diluents and/or excipients.
120. The composition of claim 119, further comprising a cryoprotectant.
121. The composition of claim 120, wherein the cryoprotectant is sucrose.
122. The composition of any one of claims 119-121, further comprising an aqueous buffered solution.
123. The composition of claim 122, wherein the aqueous buffered solution includes sodium ions.
124. A method of treating a disease-state in a subject in need thereof, comprising administering to said subject a therapeutically effective amount of a composition according to any one of claims 1-123.
125. The method of claim 124, further comprising administering one or more DPP-4 inhibitors.
126. The method of claim 125, wherein the one or more DPP-4 inhibitors and the composition are administered concurrently.
127. The method of claim 125, wherein the one or more DPP-4 inhibitors and the composition are administered sequentially.
128. The method of claim 127, wherein the one or more DPP-4 inhibitors are administered prior to the composition.

129. The method of claim 127, wherein the one or more DPP-4 inhibitors are administered after the composition.
130. The method of any one of claims 125-129, wherein the one or more DPP-4 inhibitors comprises sitagliptin, vildagliptin, saxagliptin, linagliptin, gemigliptin, anagliptin, teneligliptin, alogliptin, trelagliptin, omarigliptin, evogliptin, gosogliptin, dutogliptin, neogliptin, retagliptin, denagliptin, cofroglipin, fotagliptin, prusogliptin, berberine, or any combination thereof.
131. The method of any one of claims 125-130, wherein the one or more DPP-4 inhibitors are administered orally.
132. The method of claim 124, wherein the disease-state is obesity or an obesity-related disorder.
133. The method of claim 132, wherein the obesity-related disorder is pre-diabetes, type 2 diabetes (T2D), early type 1 diabetes (T1D), non-alcoholic fatty liver disease (NAFLD), non-alcoholic steatohepatitis (NASH), a cardiovascular (CV) disease, a renal disease, or elevated risk of premature mortality.
134. The method of claim 133, wherein the cardiovascular (CV) disease comprises a major cardiovascular event (MACE), including CV death, non-fatal myocardial infarction, non-fatal stroke, and/or heart failure with preserved ejection fraction (HFpEF).
135. The method of claim 132, wherein the method improves weight management in said subject.
136. The method of claim 132, wherein the method reduces weight gain or induces weight loss in said subject.
137. The method of claim 124, wherein the disease-state is diabetes.
138. The method of claim 137, wherein the method improves glycemic control in said subject.
139. The method of claim 137, wherein the method lowers HbA1c in said subject.

140. The method of claim 137, wherein the diabetes is pre-diabetes, type 2 diabetes (T2D), or early type 1 diabetes (T1D).
141. The method of claim 124, wherein the disease-state is a cardiovascular (CV) disease.
142. The method of claim 141, wherein the cardiovascular disease comprises a major cardiovascular event (MACE), including CV death, non-fatal myocardial infarction, non- fatal stroke, and/or heart failure with preserved ejection fraction (HfpEF).
143. The method of claim 141, wherein the method improves a subject’s blood pressure and/or blood lipids in said subject.
144. The method of claim 124, wherein the disease-state is a renal disease.
145. The method of claim 124, wherein the disease-state is non-alcoholic fatty liver disease (NAFLD).
146. The method of claim 124, wherein the disease-state is non-alcoholic steatohepatitis (NASH) and optionally its sequelae, liver fibrosis and cirrhosis.
147. The method of any one of claims 124-146, wherein administering the composition to the subject comprises administering one or more doses of the composition to the subject.
148. The method of claim 147, wherein the one or more doses of the composition are administered to the subject daily, every other day or once a week.
149. The method of claim 147, wherein the one or more doses of the composition are administered to the subject less frequently than once a week.
150. The method of claim 147, wherein the one or more doses of the composition are administered to the subject once every 2, 3 or 4 weeks.
151. The method of any one of claims 124-150, wherein the composition is administered via injection.
152. The method of claim 151, wherein the composition is administered subcutaneously, intravenously, intramuscularly, or intraperitoneally

153. The method of claim 152, wherein the composition is administered intraperitoneally.
154. The method of any one of claims 124-150, wherein the composition is administered non-invasively (e.g., orally or nasally).
155. The method of any one of claims 124-154, wherein administration of the composition results in expression of the incretin agent in the subject.
156. The method of any one of claims 124-155, wherein the composition is administered in a volume that is less than 0.5 mL.
157. Use of the composition of any one of claims 1-123 for the treatment of a disease- state in a subject in need thereof.
158. A method of producing an incretin agent comprising administering to cells the composition of any one of claims 1-123 so that the cells express and secrete the incretin agent.
159. An incretin agent that comprises an incretin peptide fused to a signal peptide.
160. The incretin agent of claim 159, wherein the incretin peptide is fused to the signal peptide via the N-terminus of the incretin peptide, optionally via a linker.
161. The incretin agent of claim 159 or 160, wherein the signal peptide has an amino acid sequence according to any one of SEQ ID NOs: 16-39 and 65-67.
162. The incretin agent of claim 161, wherein the signal peptide has an amino acid sequence according to any one of SEQ ID NOs: 16-21 and 65-67.
163. The incretin agent of claim 161, wherein the signal peptide has an amino acid sequence according to SEQ ID NO: 17.
164. The incretin agent of claim 161, wherein the signal peptide has an amino acid sequence according to SEQ ID NO: 65.
165. The incretin agent of claim 161, wherein the signal peptide has an amino acid sequence according to SEQ ID NO: 66.

166. The incretin agent of any one of claims 159-165, wherein the incretin agent comprises an incretin peptide fused to a signal peptide that comprises an amino acid sequence according to any one of SEQ ID NOs: 41-45, 52-61, and 108-152.
167. The incretin agent of any one of claims 159-166, wherein the incretin agent comprises an incretin peptide fused to one or more additional incretin peptides, optionally via one or more linkers.
168. The incretin agent of claim 167, wherein the one or more linkers comprise an amino acid sequence according of any one of SEQ ID NOs: 1-5, 68, or 156.
169. The incretin agent of claim 167 or 168, wherein the incretin agent comprises an incretin peptide fused to two or more incretin peptides.
170. The incretin agent of any one of claims 167-169, wherein the incretin agent comprises at least one GLP1 receptor agonist and at least one GIP receptor agonist.
171. The incretin agent of any one of claims 167-170, wherein the incretin agent comprises at least two GLP1 receptor agonists.
172. The incretin agent of any one of claims 167-171, where in the incretin agent comprises at least two GIP receptor agonists.
173. The composition of any one of claims 167-172, wherein the incretin agent comprises one or more furin cleavage sites.
174. The incretin agent of claim 173, wherein the one or more furin cleavage sites are located between adjacent incretin peptides.
175. The incretin agent of claim 173 or 174, wherein the one or more furin cleavage sites comprise an amino acid sequence according to SEQ ID NO: 153.
176. The incretin agent of any one of claims 167-175, wherein the incretin agent comprises one or more units that each comprise, from N-terminus to C-terminus: GLP1 receptor agonist-linker-furin cleavage site-GIP receptor agonist, e.g., wherein the incretin agent comprises one unit (e.g., SEQ ID NOs: 76, 77, 78, 79, 80, 81), two units (e.g., SEQ ID NOs: 82); or four units (e.g., SEQ ID NO: 83).

177. The incretin agent of any one of claims 167-176, wherein the incretin agent comprises an amino acid sequence according to any one of SEQ ID NOs: 76-83, 94-97, 102- 107.
178. The incretin agent of any one of claims 159-177, wherein the incretin agent comprises a half-life extending moiety.
179. The incretin agent of claim 178, wherein the half-life extending moiety comprises albumin (e.g., human serum albumin).
180. The incretin agent of claim 180, wherein the human serum albumin comprises an amino acid sequence having at least 90%, 95%, or 99% identity to SEQ ID NO: 159.
181. The incretin agent of claim 179 or 180, wherein the human serum albumin comprises an amino acid sequence according to SEQ ID NO: 159.
182. The incretin agent of any one of claims 172-174, wherein the incretin agent comprises albumin (e.g., human serum albumin) fused to one or more units that each comprise from, N-terminus to C-terminus: (i) GLP1 receptor agonist-linker (e.g., SEQ ID NO: 98); (ii) GIP receptor agonist-linker (e.g., SEQ ID NO: 100); (iii) GLP1 receptor agonist-linker-furin cleavage site (e.g., SEQ ID NO: 102); or (iv) GLP1 receptor agonist-linker-furin cleavage site-GIP receptor agonist, e.g., wherein the incretin agent comprises one unit (e.g., SEQ ID NO: 104), two units (e.g., SEQ ID NO: 106) or four units (e.g., SEQ ID NO: 107).
183. The incretin agent of any one of claims 179-182, wherein the incretin agent comprises an amino acid sequence according to any one of SEQ ID NOs: 98, 100, 102, 104, 106, 107, or any combination thereof.
184. The incretin agent of claim 178, wherein the half-life extending moiety comprises an albumin binding domain (ABD).

185. The incretin agent of claim 184, wherein the ABD is derived from protein G of Streptococcus strain GI48 and/or from protein PAB of Finegoldia magna, such as ABD035 and SA21.
186. The incretin agent of claim 184, wherein the half-life extending moiety comprises an ABD that binds to domain II of human serum albumin and does not overlap or interfere with binding to the FcRn-binding site on albumin.
187. The incretin agent of claim 184, wherein the half-life extending moiety comprises ABDCon.
188. The incretin agent of claim 184, wherein the half-life extending moiety comprises an ABD derived from the bacterial protein Sso7d from the hyperthermophilic archaeon Sulfolobus solfataricus, such as M11.12 and M18.2.5.
189. The incretin agent of claim 178, wherein the half-life extending moiety comprises a DARPin that binds albumin.
190. The incretin agent of claim 184, wherein the ABD comprises an immunoglobulin domain or fragment thereof that binds albumin.
191. The incretin agent of claim 184 or 190, wherein the ABD comprises a fully human domain antibody (dAb) that binds albumin, such as AlbudAb.
192. The incretin agent of any one of claims 184 or 190-191, wherein the ABD comprises a Fab that binds albumin, such as dsFv CA645.
193. The incretin agent of claim 184 or 190-192, wherein the ABD comprises a heavy chain only (VHH) antibody, such as a nanobody, that binds albumin.
194. The incretin agent of claim 193, wherein the VHH antibody comprises a VHH domain having the complementarity determining region (CDR) sequences HCDR1, HCDR2, and/or HCDR3 according to SEQ ID NO: 191 (GFTLDYYA), SEQ ID NO: 192 (IASSGGST), and/or SEQ ID NO: 193 (AAAVLECRTVVRGYDY), respectively.
195. The incretin agent of claim 193 or 194, wherein the VHH antibody comprises an amino acid sequence having at least 90%, 95%, or 99% identity to SEQ ID NO: 154.

196. The incretin agent of claim 195, wherein the VHH antibody comprises an amino acid sequence according to SEQ ID NO: 154.
197. The incretin agent of any one of claims 193-196, wherein the incretin agent comprises a VHH antibody that binds albumin fused to a unit that comprises, from N- terminus to C-terminus: (i) GLP1-linker (e.g., SEQ ID NO: 99); (ii) GIP receptor agonist-linker (e.g., SEQ ID NO: 101); or (iii) GLP1 receptor agonist-linker-furin-GIP receptor agonist-linker (e.g., SEQ ID NO: 103 or 105).
198. The incretin agent of any one of claims 193-197, wherein the incretin agent comprises an amino acid sequence according to any one of SEQ ID NOs: 99, 101, 103, 105.
199. The incretin agent of claim, wherein the half-life extending moiety does not comprise a Fc domain, such as from a human IgG, optionally from a human IgG1, IgG2, IgG3, or IgG4.
200. The incretin agent of claim 178, wherein the half-life extending moiety comprises a Fc domain, such as from a human IgG, optionally from a human IgG1, IgG2, IgG3, or IgG4.
201. The incretin agent of claim 200, wherein the human IgG is a human IgG4.
202. The incretin agent of claim 200 or 201, wherein the incretin agent comprises an IgG4 Fc domain fused to a unit comprising, from N-terminus to C-terminus: (i) GLP1 receptor agonist-linker (e.g., SEQ ID NOs: 10, 89, 90, 91); (ii) GIP receptor agonist-linker (e.g., SEQ ID NO: 92, 93); or (iii) GLP1 receptor agonist-linker-furin-GIP receptor agonist-linker (e.g., SEQ ID NO: 94, 95, 96, 97).

203. The incretin agent of any one of claims 200-202, wherein the IgG4 Fc domain comprises an amino acid sequence that is at least 90%, 95%, or 99% identical to SEQ ID NO: 155.
204. The incretin agent of claim 203, wherein the IgG4 Fc domain comprises an amino acid sequence according to SEQ ID NO: 155.
205. The incretin agent of any one of claims 200-204, wherein the incretin agent comprises an amino acid sequence according to any one of SEQ ID NOs: 10, 89-97.
206. The incretin agent of any one of claims 200-205, wherein the Fc domain comprises one or more mutations in one or both Fc constant domains to increase half-life of the incretin agent and/or induce dimerization.
207. The incretin agent of claim 206, wherein the one or more mutations comprises one or more mutations in a CH3 domain.
208. The incretin agent of claim 206 or 207, wherein the one or more mutations to induce dimerization comprises: (i) Y349C, T366S, L368A, and/or Y407V, according to EU numbering; or (ii) S354C and/or T366W, according to EU numbering.
209. The incretin agent of any one of claims 206-208, wherein the one or more mutations comprises Y349C, T366S, L368A, and Y407V (“FcKIH-b”), according to EU numbering, or S354C and T366W (“FcKIH-a”), according to EU numbering.
210. The incretin agent of any one of claims 206-209, wherein the incretin agent comprises a first polypeptide chain and a second polypeptide chain, wherein the first polypeptide chain comprises an incretin peptide fused to a first Fc domain, wherein the first Fc domain comprises the mutations Y349C, T366S, L368A, and Y407V (“FcKIH-b”), according to EU numbering and wherein the second polypeptide chain comprises an incretin peptide fused to a second Fc domain, wherein the second Fc domain comprises the mutations S354C and T366W (“FcKIH-a”), according to EU numbering.

211. The incretin agent of any one of claims 206-210, wherein the one or more mutations to increase half-life of the incretin agent comprises M428L and N434S (“LS”), according to EU numbering.
212. The incretin agent of any one of claims 206-211, wherein the incretin agent comprises an Fc domain with FcKIH-a mutations on a first polypeptide chain and an Fc domain with FcKIH-b mutations on a second polypeptide chain, where the Fc domain on each polypeptide chain is independently fused with one or more units that comprise, from N- terminus to C-terminus: (i) GLP1 receptor agonist-linker (e.g., SEQ ID NO: 84, 85, 86, 87); or (ii) GIP receptor agonist-linker (e.g., SEQ ID NO: 88).
213. The incretin agent of any one of claims 206-212, wherein the incretin agent comprises an amino acid sequence according to any one of SEQ ID NOs: 84-88.
214 The composition of any one of claims 206-213, wherein the Fc domain comprises one or more mutations that ablate effector activity of the Fc domain (e.g., binding to Fcγ receptors or C1q).
215. The composition of claim 214, wherein the one or more mutations that ablate effector activity of the Fc domain (e.g., binding to Fcγ receptors or C1q) comprise the following mutations: L234S, L235T, and G236R (“STR”) according to EU numbering.
216. The composition of claim 215, wherein the one or more mutations that ablate effector activity of the Fc domain (e.g., binding to Fcγ receptors or C1q) comprise the following mutations: L234A and L235A (“LALA”) according to EU numbering.
217. The composition of claim 216, wherein the one or more mutations that ablate effector activity of the Fc domain (e.g., binding to Fcγ receptors or C1q) comprise the following mutations: L234A/L235A/P329G (“LALAPG”) according to EU numbering.
218. The incretin agent of claim 178, wherein the half-life extending moiety comprises a VNAR that binds albumin.

219. The incretin agent of claim 178, wherein the half-life extending moiety comprises an XTEN sequence.
220. An incretin agent comprising: a husec signal peptide; an incretin peptide comprising a GLP1 incretin peptide, or fragment or variant thereof; wherein the GLP1 incretin peptide comprises an amino acid sequence having an A8G substitution mutation compared to a wildtype GLP1 amino acid sequence.
221. A polyribonucleotide encoding the incretin agent of claim 220.
222. An incretin agent comprising: a husec signal peptide; an incretin peptide comprising a GIP incretin peptide, or fragment or variant thereof; wherein the GIP incretin peptide comprises an amino acid sequence having an A2G substitution mutation compared to a wildtype GIP amino acid sequence.
223. A polyribonucleotide encoding the incretin agent of claim 222.

Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/IB2023/059007 WO2025056938A1 (en) | 2023-09-11 | 2023-09-11 | Rna compositions for delivery of incretin agents |
| IBPCT/IB2023/059007 | 2023-09-11 | ||
| US202463662890P | 2024-06-21 | 2024-06-21 | |
| US63/662,890 | 2024-06-21 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2025057088A1 true WO2025057088A1 (en) | 2025-03-20 |
Family
ID=93037101
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2024/058845 Pending WO2025057088A1 (en) | 2023-09-11 | 2024-09-11 | Rna compositions for delivery of incretin agents |
Country Status (2)
| Country | Link |
|---|---|
| TW (1) | TW202540154A (en) |
| WO (1) | WO2025057088A1 (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN120590288A (en) * | 2025-08-07 | 2025-09-05 | 北京悦康科创医药科技股份有限公司 | Polyamine-based, polyhydroxy-based ionizable cationic lipids, compositions containing same, and uses thereof |
Citations (59)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US907699A (en) | 1906-04-24 | 1908-12-22 | William D Moore | Building-block. |
| WO1998050431A2 (en) | 1997-05-02 | 1998-11-12 | Genentech, Inc. | A method for making multispecific antibodies having heteromultimeric and common components |
| WO1999039741A2 (en) | 1998-02-03 | 1999-08-12 | Inex Pharmaceuticals Corporation | Systemic delivery of serum stable plasmid lipid particles for cancer therapy |
| US5965542A (en) | 1997-03-18 | 1999-10-12 | Inex Pharmaceuticals Corp. | Use of temperature to control the size of cationic liposome/plasmid DNA complexes |
| WO2001007548A1 (en) | 1999-07-26 | 2001-02-01 | The Procter & Gamble Company | Cationic charge boosting systems |
| WO2002098443A2 (en) | 2001-06-05 | 2002-12-12 | Curevac Gmbh | Stabilised mrna with an increased g/c content and optimised codon for use in gene therapy |
| US20040142025A1 (en) | 2002-06-28 | 2004-07-22 | Protiva Biotherapeutics Ltd. | Liposomal apparatus and manufacturing methods |
| US20050017054A1 (en) | 2003-07-23 | 2005-01-27 | Tom Iverson | Flyback transformer wire attach method to printed circuit board |
| US20050064595A1 (en) | 2003-07-16 | 2005-03-24 | Protiva Biotherapeutics, Inc. | Lipid encapsulated interfering RNA |
| US20050118253A1 (en) | 1998-02-03 | 2005-06-02 | Protiva Biotherapeutics, Inc. | Systemic delivery of serum stable plasmid lipid particles for cancer therapy |
| US20050175682A1 (en) | 2003-09-15 | 2005-08-11 | Protiva Biotherapeutics, Inc. | Polyethyleneglycol-modified lipid compounds and uses thereof |
| US20060008910A1 (en) | 2004-06-07 | 2006-01-12 | Protiva Biotherapeuties, Inc. | Lipid encapsulated interfering RNA |
| US20060083780A1 (en) | 2004-06-07 | 2006-04-20 | Protiva Biotherapeutics, Inc. | Cationic lipids and methods of use |
| US20070042031A1 (en) | 2005-07-27 | 2007-02-22 | Protiva Biotherapeutics, Inc. | Systems and methods for manufacturing liposomes |
| WO2007036366A2 (en) | 2005-09-28 | 2007-04-05 | Johannes Gutenberg-Universität Mainz, Vertreten Durch Den Präsidenten | Modification of rna, producing an increased transcript stability and translation efficiency |
| WO2010053572A2 (en) | 2008-11-07 | 2010-05-14 | Massachusetts Institute Of Technology | Aminoalcohol lipidoids and uses thereof |
| US20100130588A1 (en) | 2008-04-15 | 2010-05-27 | Protiva Biotherapeutics, Inc. | Novel lipid formulations for nucleic acid delivery |
| US20100324120A1 (en) | 2009-06-10 | 2010-12-23 | Jianxin Chen | Lipid formulation |
| US20110076335A1 (en) | 2009-07-01 | 2011-03-31 | Protiva Biotherapeutics, Inc. | Novel lipid formulations for delivery of therapeutic agents to solid tumors |
| US20110117125A1 (en) | 2008-01-02 | 2011-05-19 | Tekmira Pharmaceuticals Corporation | Compositions and methods for the delivery of nucleic acids |
| WO2011141705A1 (en) | 2010-05-12 | 2011-11-17 | Protiva Biotherapeutics, Inc. | Novel cationic lipids and methods of use thereof |
| US20110311583A1 (en) | 2008-11-10 | 2011-12-22 | Alnylam Pharmaceuticals, Inc. | Novel lipids and compositions for the delivery of therapeutics |
| US20120027803A1 (en) | 2010-06-03 | 2012-02-02 | Alnylam Pharmaceuticals, Inc. | Biodegradable lipids for the delivery of active agents |
| US20120172411A1 (en) | 2010-09-17 | 2012-07-05 | Protiva Biotherapeutics, Inc. | Novel trialkyl cationic lipids and methods of use thereof |
| US20120295832A1 (en) | 2011-05-17 | 2012-11-22 | Arrowhead Research Corporation | Novel Lipids and Compositions for Intracellular Delivery of Biologically Active Compounds |
| WO2012170930A1 (en) | 2011-06-08 | 2012-12-13 | Shire Human Genetic Therapies, Inc | Lipid nanoparticle compositions and methods for mrna delivery |
| US20130017223A1 (en) | 2009-12-18 | 2013-01-17 | The University Of British Columbia | Methods and compositions for delivery of nucleic acids |
| US20130022649A1 (en) | 2009-12-01 | 2013-01-24 | Protiva Biotherapeutics, Inc. | Snalp formulations containing antioxidants |
| WO2013016058A1 (en) | 2011-07-22 | 2013-01-31 | Merck Sharp & Dohme Corp. | Novel bis-nitrogen containing cationic lipids for oligonucleotide delivery |
| US20130086373A1 (en) | 2011-09-29 | 2013-04-04 | Apple Inc. | Customized content for electronic devices |
| WO2013086322A1 (en) | 2011-12-07 | 2013-06-13 | Alnylam Pharmaceuticals, Inc. | Branched alkyl and cycloalkyl terminated biodegradable lipids for the delivery of active agents |
| WO2013086373A1 (en) | 2011-12-07 | 2013-06-13 | Alnylam Pharmaceuticals, Inc. | Lipids for the delivery of active agents |
| US20130195920A1 (en) | 2011-12-07 | 2013-08-01 | Alnylam Pharmaceuticals, Inc. | Biodegradable lipids for the delivery of active agents |
| US20130245107A1 (en) | 2011-12-16 | 2013-09-19 | modeRNA Therapeutics | Dlin-mc3-dma lipid nanoparticle delivery of modified polynucleotides |
| WO2013143683A1 (en) | 2012-03-26 | 2013-10-03 | Biontech Ag | Rna formulation for immunotherapy |
| US8569256B2 (en) | 2009-07-01 | 2013-10-29 | Protiva Biotherapeutics, Inc. | Cationic lipids and methods for the delivery of therapeutic agents |
| US20130323269A1 (en) | 2010-07-30 | 2013-12-05 | Muthiah Manoharan | Methods and compositions for delivery of active agents |
| US20130338210A1 (en) | 2009-12-07 | 2013-12-19 | Alnylam Pharmaceuticals, Inc. | Compositions for nucleic acid delivery |
| WO2014008334A1 (en) | 2012-07-06 | 2014-01-09 | Alnylam Pharmaceuticals, Inc. | Stable non-aggregating nucleic acid lipid particle formulations |
| US8673860B2 (en) | 2009-02-03 | 2014-03-18 | Amunix Operating Inc. | Extended recombinant polypeptides and compositions comprising same |
| US8748380B2 (en) | 2009-10-30 | 2014-06-10 | Novozymes Biopharma Dk A/S | Albumin variants |
| US20140200257A1 (en) | 2011-01-11 | 2014-07-17 | Alnylam Pharmaceuticals, Inc. | Pegylated lipids and their use for drug delivery |
| US20150140070A1 (en) | 2013-10-22 | 2015-05-21 | Shire Human Genetic Therapies, Inc. | Lipid formulations for delivery of messenger rna |
| US20150203446A1 (en) | 2011-09-27 | 2015-07-23 | Takeda Pharmaceutical Company Limited | Di-aliphatic substituted pegylated lipids |
| WO2015199952A1 (en) | 2014-06-25 | 2015-12-30 | Acuitas Therapeutics Inc. | Novel lipids and lipid nanoparticle formulations for delivery of nucleic acids |
| WO2016005324A1 (en) | 2014-07-11 | 2016-01-14 | Biontech Rna Pharmaceuticals Gmbh | Stabilization of poly(a) sequence encoding dna sequences |
| WO2017004143A1 (en) | 2015-06-29 | 2017-01-05 | Acuitas Therapeutics Inc. | Lipids and lipid nanoparticle formulations for delivery of nucleic acids |
| WO2017060314A2 (en) | 2015-10-07 | 2017-04-13 | Biontech Rna Pharmaceuticals Gmbh | 3' utr sequences for stabilization of rna |
| WO2017075531A1 (en) | 2015-10-28 | 2017-05-04 | Acuitas Therapeutics, Inc. | Novel lipids and lipid nanoparticle formulations for delivery of nucleic acids |
| WO2017182524A1 (en) | 2016-04-22 | 2017-10-26 | Biontech Rna Pharmaceuticals Gmbh | Methods for providing single-stranded rna |
| WO2018081480A1 (en) | 2016-10-26 | 2018-05-03 | Acuitas Therapeutics, Inc. | Lipid nanoparticle formulations |
| WO2019207060A1 (en) * | 2018-04-25 | 2019-10-31 | Ethris Gmbh | Lipid-based formulations for the delivery of rna |
| US20200163878A1 (en) | 2016-10-26 | 2020-05-28 | Curevac Ag | Lipid nanoparticle mrna vaccines |
| WO2020128031A2 (en) | 2018-12-21 | 2020-06-25 | Curevac Ag | Rna for malaria vaccines |
| WO2021001417A1 (en) | 2019-07-02 | 2021-01-07 | Biontech Rna Pharmaceuticals Gmbh | Rna formulations suitable for therapy |
| WO2021214204A1 (en) | 2020-04-22 | 2021-10-28 | BioNTech SE | Rna constructs and uses thereof |
| WO2022125713A1 (en) * | 2020-12-09 | 2022-06-16 | Aravasc Inc. | Polymeric micelle complexes of mrna or zwitterionic agents, and formulations and uses thereof |
| CN114788876A (en) * | 2022-02-24 | 2022-07-26 | 北京医院 | mRNA medicinal preparation for treating diabetes, preparation method and application thereof |
| US20220347111A1 (en) * | 2019-06-21 | 2022-11-03 | Université Catholique de Louvain | Lipid nanocapsules charged with incretin mimetics |
-
2024
- 2024-09-11 TW TW113134412A patent/TW202540154A/en unknown
- 2024-09-11 WO PCT/IB2024/058845 patent/WO2025057088A1/en active Pending
Patent Citations (74)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US907699A (en) | 1906-04-24 | 1908-12-22 | William D Moore | Building-block. |
| US5965542A (en) | 1997-03-18 | 1999-10-12 | Inex Pharmaceuticals Corp. | Use of temperature to control the size of cationic liposome/plasmid DNA complexes |
| WO1998050431A2 (en) | 1997-05-02 | 1998-11-12 | Genentech, Inc. | A method for making multispecific antibodies having heteromultimeric and common components |
| US20050118253A1 (en) | 1998-02-03 | 2005-06-02 | Protiva Biotherapeutics, Inc. | Systemic delivery of serum stable plasmid lipid particles for cancer therapy |
| WO1999039741A2 (en) | 1998-02-03 | 1999-08-12 | Inex Pharmaceuticals Corporation | Systemic delivery of serum stable plasmid lipid particles for cancer therapy |
| WO2001007548A1 (en) | 1999-07-26 | 2001-02-01 | The Procter & Gamble Company | Cationic charge boosting systems |
| WO2002098443A2 (en) | 2001-06-05 | 2002-12-12 | Curevac Gmbh | Stabilised mrna with an increased g/c content and optimised codon for use in gene therapy |
| US20040142025A1 (en) | 2002-06-28 | 2004-07-22 | Protiva Biotherapeutics Ltd. | Liposomal apparatus and manufacturing methods |
| US20110216622A1 (en) | 2002-06-28 | 2011-09-08 | Protiva Biotherapeutics, Inc. | Liposomal apparatus and manufacturing method |
| US20050064595A1 (en) | 2003-07-16 | 2005-03-24 | Protiva Biotherapeutics, Inc. | Lipid encapsulated interfering RNA |
| US20060240093A1 (en) | 2003-07-16 | 2006-10-26 | Protiva Biotherapeutics, Inc. | Lipid encapsulated interfering rna |
| US20120058188A1 (en) | 2003-07-16 | 2012-03-08 | Protiva Biotherapeutics, Inc. | Lipid encapsulated interfering rna |
| US20050017054A1 (en) | 2003-07-23 | 2005-01-27 | Tom Iverson | Flyback transformer wire attach method to printed circuit board |
| US20050175682A1 (en) | 2003-09-15 | 2005-08-11 | Protiva Biotherapeutics, Inc. | Polyethyleneglycol-modified lipid compounds and uses thereof |
| US20110091525A1 (en) | 2003-09-15 | 2011-04-21 | Protiva Biotherapeutics, Inc. | Polyethyleneglycol-modified lipid compounds and uses thereof |
| US20060083780A1 (en) | 2004-06-07 | 2006-04-20 | Protiva Biotherapeutics, Inc. | Cationic lipids and methods of use |
| US20060008910A1 (en) | 2004-06-07 | 2006-01-12 | Protiva Biotherapeuties, Inc. | Lipid encapsulated interfering RNA |
| US20110262527A1 (en) | 2004-06-07 | 2011-10-27 | Protiva Biotherapeutics, Inc. | Cationic lipids and methods of use |
| US20110060032A1 (en) | 2004-06-07 | 2011-03-10 | Protiva Biotherapeutics, Inc. | Lipid encapsulating interfering rna |
| US20070042031A1 (en) | 2005-07-27 | 2007-02-22 | Protiva Biotherapeutics, Inc. | Systems and methods for manufacturing liposomes |
| WO2007036366A2 (en) | 2005-09-28 | 2007-04-05 | Johannes Gutenberg-Universität Mainz, Vertreten Durch Den Präsidenten | Modification of rna, producing an increased transcript stability and translation efficiency |
| US20110117125A1 (en) | 2008-01-02 | 2011-05-19 | Tekmira Pharmaceuticals Corporation | Compositions and methods for the delivery of nucleic acids |
| US20100130588A1 (en) | 2008-04-15 | 2010-05-27 | Protiva Biotherapeutics, Inc. | Novel lipid formulations for nucleic acid delivery |
| US20120183581A1 (en) | 2008-04-15 | 2012-07-19 | Protiva Biotherapeutics, Inc | Novel lipid formulations for nucleic acid delivery |
| WO2010053572A2 (en) | 2008-11-07 | 2010-05-14 | Massachusetts Institute Of Technology | Aminoalcohol lipidoids and uses thereof |
| US20160199485A1 (en) | 2008-11-10 | 2016-07-14 | Tekmira Pharmaceuticals Corporation | Novel lipids and compositions for the delivery of therapeutics |
| US20150265708A1 (en) | 2008-11-10 | 2015-09-24 | Tekmira Pharmaceuticals Corporation | Novel lipids and compositions for the delivery of therapeutics |
| US20110311583A1 (en) | 2008-11-10 | 2011-12-22 | Alnylam Pharmaceuticals, Inc. | Novel lipids and compositions for the delivery of therapeutics |
| US20110311582A1 (en) | 2008-11-10 | 2011-12-22 | Muthiah Manoharan | Novel lipids and compositions for the delivery of therapeutics |
| US8673860B2 (en) | 2009-02-03 | 2014-03-18 | Amunix Operating Inc. | Extended recombinant polypeptides and compositions comprising same |
| US20100324120A1 (en) | 2009-06-10 | 2010-12-23 | Jianxin Chen | Lipid formulation |
| US8569256B2 (en) | 2009-07-01 | 2013-10-29 | Protiva Biotherapeutics, Inc. | Cationic lipids and methods for the delivery of therapeutic agents |
| US20110076335A1 (en) | 2009-07-01 | 2011-03-31 | Protiva Biotherapeutics, Inc. | Novel lipid formulations for delivery of therapeutic agents to solid tumors |
| US8748380B2 (en) | 2009-10-30 | 2014-06-10 | Novozymes Biopharma Dk A/S | Albumin variants |
| US20130022649A1 (en) | 2009-12-01 | 2013-01-24 | Protiva Biotherapeutics, Inc. | Snalp formulations containing antioxidants |
| US20130338210A1 (en) | 2009-12-07 | 2013-12-19 | Alnylam Pharmaceuticals, Inc. | Compositions for nucleic acid delivery |
| US20130017223A1 (en) | 2009-12-18 | 2013-01-17 | The University Of British Columbia | Methods and compositions for delivery of nucleic acids |
| US20130123338A1 (en) | 2010-05-12 | 2013-05-16 | Protiva Biotherapeutics, Inc. | Novel cationic lipids and methods of use thereof |
| WO2011141705A1 (en) | 2010-05-12 | 2011-11-17 | Protiva Biotherapeutics, Inc. | Novel cationic lipids and methods of use thereof |
| US20160009637A1 (en) | 2010-06-03 | 2016-01-14 | Alnylam Pharmaceuticals, Inc. | Biodegradable lipids for the delivery of active agents |
| US20120027803A1 (en) | 2010-06-03 | 2012-02-02 | Alnylam Pharmaceuticals, Inc. | Biodegradable lipids for the delivery of active agents |
| US20130323269A1 (en) | 2010-07-30 | 2013-12-05 | Muthiah Manoharan | Methods and compositions for delivery of active agents |
| US20120172411A1 (en) | 2010-09-17 | 2012-07-05 | Protiva Biotherapeutics, Inc. | Novel trialkyl cationic lipids and methods of use thereof |
| US20140200257A1 (en) | 2011-01-11 | 2014-07-17 | Alnylam Pharmaceuticals, Inc. | Pegylated lipids and their use for drug delivery |
| US20120295832A1 (en) | 2011-05-17 | 2012-11-22 | Arrowhead Research Corporation | Novel Lipids and Compositions for Intracellular Delivery of Biologically Active Compounds |
| WO2012170930A1 (en) | 2011-06-08 | 2012-12-13 | Shire Human Genetic Therapies, Inc | Lipid nanoparticle compositions and methods for mrna delivery |
| WO2013016058A1 (en) | 2011-07-22 | 2013-01-31 | Merck Sharp & Dohme Corp. | Novel bis-nitrogen containing cationic lipids for oligonucleotide delivery |
| US20150203446A1 (en) | 2011-09-27 | 2015-07-23 | Takeda Pharmaceutical Company Limited | Di-aliphatic substituted pegylated lipids |
| US20130086373A1 (en) | 2011-09-29 | 2013-04-04 | Apple Inc. | Customized content for electronic devices |
| WO2013086322A1 (en) | 2011-12-07 | 2013-06-13 | Alnylam Pharmaceuticals, Inc. | Branched alkyl and cycloalkyl terminated biodegradable lipids for the delivery of active agents |
| US20130195920A1 (en) | 2011-12-07 | 2013-08-01 | Alnylam Pharmaceuticals, Inc. | Biodegradable lipids for the delivery of active agents |
| US20140308304A1 (en) | 2011-12-07 | 2014-10-16 | Alnylam Pharmaceuticals, Inc. | Lipids for the delivery of active agents |
| US20150005363A1 (en) | 2011-12-07 | 2015-01-01 | Alnylam Pharmaceuticals, Inc. | Branched Alkyl And Cycloalkyl Terminated Biodegradable Lipids For The Delivery Of Active Agents |
| WO2013086373A1 (en) | 2011-12-07 | 2013-06-13 | Alnylam Pharmaceuticals, Inc. | Lipids for the delivery of active agents |
| US20150273068A1 (en) | 2011-12-07 | 2015-10-01 | Alnylam Pharmaceuticals, Inc. | Biodegradable lipids for the delivery of active agents |
| US20130245107A1 (en) | 2011-12-16 | 2013-09-19 | modeRNA Therapeutics | Dlin-mc3-dma lipid nanoparticle delivery of modified polynucleotides |
| WO2013143683A1 (en) | 2012-03-26 | 2013-10-03 | Biontech Ag | Rna formulation for immunotherapy |
| WO2014008334A1 (en) | 2012-07-06 | 2014-01-09 | Alnylam Pharmaceuticals, Inc. | Stable non-aggregating nucleic acid lipid particle formulations |
| US20150140070A1 (en) | 2013-10-22 | 2015-05-21 | Shire Human Genetic Therapies, Inc. | Lipid formulations for delivery of messenger rna |
| WO2015199952A1 (en) | 2014-06-25 | 2015-12-30 | Acuitas Therapeutics Inc. | Novel lipids and lipid nanoparticle formulations for delivery of nucleic acids |
| WO2016005324A1 (en) | 2014-07-11 | 2016-01-14 | Biontech Rna Pharmaceuticals Gmbh | Stabilization of poly(a) sequence encoding dna sequences |
| WO2017004143A1 (en) | 2015-06-29 | 2017-01-05 | Acuitas Therapeutics Inc. | Lipids and lipid nanoparticle formulations for delivery of nucleic acids |
| WO2017060314A2 (en) | 2015-10-07 | 2017-04-13 | Biontech Rna Pharmaceuticals Gmbh | 3' utr sequences for stabilization of rna |
| WO2017075531A1 (en) | 2015-10-28 | 2017-05-04 | Acuitas Therapeutics, Inc. | Novel lipids and lipid nanoparticle formulations for delivery of nucleic acids |
| WO2017182524A1 (en) | 2016-04-22 | 2017-10-26 | Biontech Rna Pharmaceuticals Gmbh | Methods for providing single-stranded rna |
| WO2018081480A1 (en) | 2016-10-26 | 2018-05-03 | Acuitas Therapeutics, Inc. | Lipid nanoparticle formulations |
| US20200163878A1 (en) | 2016-10-26 | 2020-05-28 | Curevac Ag | Lipid nanoparticle mrna vaccines |
| WO2019207060A1 (en) * | 2018-04-25 | 2019-10-31 | Ethris Gmbh | Lipid-based formulations for the delivery of rna |
| WO2020128031A2 (en) | 2018-12-21 | 2020-06-25 | Curevac Ag | Rna for malaria vaccines |
| US20220347111A1 (en) * | 2019-06-21 | 2022-11-03 | Université Catholique de Louvain | Lipid nanocapsules charged with incretin mimetics |
| WO2021001417A1 (en) | 2019-07-02 | 2021-01-07 | Biontech Rna Pharmaceuticals Gmbh | Rna formulations suitable for therapy |
| WO2021214204A1 (en) | 2020-04-22 | 2021-10-28 | BioNTech SE | Rna constructs and uses thereof |
| WO2022125713A1 (en) * | 2020-12-09 | 2022-06-16 | Aravasc Inc. | Polymeric micelle complexes of mrna or zwitterionic agents, and formulations and uses thereof |
| CN114788876A (en) * | 2022-02-24 | 2022-07-26 | 北京医院 | mRNA medicinal preparation for treating diabetes, preparation method and application thereof |
Non-Patent Citations (41)
| Title |
|---|
| "March's Advanced Organic Chemistry", 2001, JOHN WILEY & SONS |
| "Remington: The Science and Practice of Pharmacy", 2005, LIPPINCOTT WILLIAMS & WILKINS |
| "Remington's The Science and Practice of Pharmacy", 2006, LIPPINCOTT, WILLIAMS & WILKINS |
| ADAMS ET AL., MABS, vol. 8, no. 7, 2016, pages 1336 - 1346 |
| CHIM, US PHARM., vol. 47, no. 10, 2022, pages 18 - 22 |
| DAFFIS ET AL., NATURE, vol. 468, 2010, pages 452 - 456 |
| DECROLY ET AL., NATURE, vol. 10, 2012, pages 51 - 65 |
| DIAMOND ET AL., CYTOKINE & GROWTH FACTOR REVIEWS, vol. 25, 2014, pages 543 - 550 |
| FATH ET AL.: "Multiparameter RNA and Codon Optimization: A Standardized Tool to Assess and Enhance Autologous Mammalian Gene Expression", PLOS ONE, vol. 6, no. 3, pages 17596 |
| FREJDKIM, EXP. MOL. MED., vol. 49, no. 3, 2017, pages 306 |
| GAO ET AL., NAT. STRUCT. BIOL., vol. 5, no. 9, 1998, pages 782 - 786 |
| GARCIA-NAFRIA: "IVA cloning: A single-tube universal cloning system exploiting bacterial In Vivo Assembly", SCIENTIFIC REPORTS, vol. 6, 2016, XP055420185, DOI: 10.1038/srep27459 |
| GOULD ET AL., ANTIVIRAL RES., vol. 87, 2010, pages 111 - 124 |
| GRAFT ET AL.: "Codon-optimized genes that enable increased heterologous expression in mammalian cells and elicit efficient immune responses in mice after vaccination of naked DNA", METHODS MOL MED, vol. 94, 2004, pages 197 - 210, XP001246616 |
| HOLT ET AL., PROTEIN ENG., DES. SEL., vol. 21, no. 5, 2008, pages 283 - 288 |
| HOLTKAMP ET AL., BLOOD, vol. 108, 2006, pages 4009 - 4017 |
| JACOBS ET AL., PROTEIN ENG., DES. SEL., vol. 28, no. 10, 2015, pages 385 - 393 |
| JOSE ET AL., FUTURE MICROBIOL., vol. 4, 2009, pages 837 - 856 |
| KACZMAREK ET AL., GENOME MEDICINE, vol. 9, 2017, pages 60 |
| KOPPEL, CHEM. PHYS., vol. 57, 1972, pages 4814 - 4820 |
| KOPPEL, D., J. CHEM. PHYS., vol. 57, 1972, pages 4814 - 4820 |
| KREITER ET AL., CANCER IMMUNOL. IMMUNOTHER., vol. 56, 2007, pages 1577 - 87 |
| LEVY ET AL., PLOS ONE, vol. 9, no. 2, 2014, pages 87704 |
| LIU ET AL., ANTIBODIES, vol. 9, no. 4, 2020, pages 64 |
| MELAMED ET AL., SCIENCE ADVANCES, vol. 9, 2023, pages 1444 |
| MULLER ET AL., MABS, vol. 4, no. 6, 2012, pages 673 - 685 |
| PLUCKTHUN, ANNU. REV. PHARMACOL. TOXICOL.,, vol. 55, 2015, pages 489 - 511 |
| PODUST ET AL., JOURNAL OF CONTROLLED RELEASE, vol. 240, 2016, pages 52 - 66 |
| RABB ET AL.: "The GeneOptimizer Algorithm: using a sliding window approach to cope with the vast sequence space in multiparameter DNA sequence optimization", SYSTEMS AND SYNTHETIC BIOLOGY, vol. 4, 2010, pages 215 - 225, XP055065511, DOI: 10.1007/s11693-010-9062-3 |
| RAMANATHAN ET AL., NUCLEIC ACIDS RES, vol. 44, no. 16, 2016, pages 7511 - 7526 |
| S. NAGATA ET AL: "Synthesis and biological activity of artificial mRNA prepared with novel phosphorylating reagents", NUCLEIC ACIDS RESEARCH, vol. 38, no. 21, 1 November 2010 (2010-11-01), pages 7845 - 7857, XP055074797, ISSN: 0305-1048, DOI: 10.1093/nar/gkq638 * |
| SCHERAGA, REV. COMPUTATIONAL CHEM., vol. 11, 1992, pages 173 - 142 |
| STEELAND ET AL., DRUG DISCOVERY TODAY, vol. 21, no. 7, 2016, pages 1076 - 1113 |
| THOMAS SORRELL: "Organic Chemistry", 1999, UNIVERSITY SCIENCE BOOKS |
| TRAXLMAYR ET AL., J. BIOL. CHEM., vol. 291, no. 43, 2016, pages 22496 - 22508 |
| VERSPOHL ET AL: "Novel therapeutics for type 2 diabetes: Incretin hormone mimetics (glucagon-like peptide-1 receptor agonists) and dipeptidyl peptidase-4 inhibitors", PHARMACOLOGY & THERAPEUTICS, ELSEVIER, GB, vol. 124, no. 1, 1 October 2009 (2009-10-01), pages 113 - 138, XP026501107, ISSN: 0163-7258, [retrieved on 20090621], DOI: 10.1016/J.PHARMTHERA.2009.06.002 * |
| WADHWA ET AL.: "Opportunities and Challenges in the Delivery of mRNA-Based Vaccines", PHARMACEUTICS, vol. 102, 2020, pages 27 |
| WILKINSON ET AL., PLOS ONE, vol. 16, no. 12, 2021, pages 0260954 |
| ZALEVSKY ET AL., NATURE BIOTECHNOLOGY, vol. 28, no. 2, 2010, pages 157 - 159 |
| ZORZI ET AL., MEDCHEMCOMM, vol. 10, no. 7, 2019, pages 1068 - 1081 |
| ZÜST ET AL., NATURE IMMUNOLOGY, vol. 12, 2011, pages 137 - 143 |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN120590288A (en) * | 2025-08-07 | 2025-09-05 | 北京悦康科创医药科技股份有限公司 | Polyamine-based, polyhydroxy-based ionizable cationic lipids, compositions containing same, and uses thereof |
| CN120590288B (en) * | 2025-08-07 | 2025-11-21 | 北京悦康科创医药科技股份有限公司 | Polyamine-based, polyhydroxy ionizable cationic lipids, compositions comprising same, and uses |
Also Published As
| Publication number | Publication date |
|---|---|
| TW202540154A (en) | 2025-10-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20240041785A1 (en) | Compositions and methods for stabilization of lipid nanoparticle mrna vaccines | |
| CN113939282A (en) | Method for preparing lipid nanoparticles | |
| US12351834B2 (en) | Compositions and methods for the treatment of ornithine transcarbamylase deficiency | |
| US20230219996A1 (en) | Rna vaccines | |
| AU2017271665A1 (en) | Treatment of primary ciliary dyskinesia with synthetic messenger RNA | |
| JP7445657B2 (en) | Compositions and methods for treating ornithine transcarbamylase deficiency | |
| KR20240099279A (en) | RNA molecule | |
| US20210145860A1 (en) | Compositions, methods and uses of messenger rna | |
| WO2025057088A1 (en) | Rna compositions for delivery of incretin agents | |
| WO2025056938A1 (en) | Rna compositions for delivery of incretin agents | |
| AU2023347387A1 (en) | Compositions for delivery of liver stage antigens and related methods | |
| CA3146392A1 (en) | Compositions and methods for the prevention and/or treatment of covid-19 | |
| WO2025213131A1 (en) | Rna compositions for delivery of orthopox antigens and related methods | |
| WO2024228044A1 (en) | Optimized csp variants and related methods | |
| WO2025030154A1 (en) | Pharmaceutical compositions for delivery of herpes simplex virus glycoprotein b antigens and related methods | |
| WO2026015882A1 (en) | Hsv antigen fragments and related methods | |
| IL324297A (en) | Optimal CSP Variants and Related Methods | |
| WO2026003577A1 (en) | Multisubunit hepacivirus vaccines and therapeutics | |
| CA3146411A1 (en) | Compositions and methods for prevention and/or treatment of covid-19 | |
| WO2024130158A1 (en) | Lipid nanoparticles and polynucleotides encoding extended serum half-life interleukin-22 for the treatment of metabolic disease | |
| WO2025054556A1 (en) | Rna compositions for delivery of mpox antigens and related methods | |
| CA3154578A1 (en) | Compositions and methods for the prevention and/or treatment of covid-19 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 24787246 Country of ref document: EP Kind code of ref document: A1 |