WO2025050069A1 - Programmable gene insertion using engineered integration enzymes - Google Patents
Programmable gene insertion using engineered integration enzymes Download PDFInfo
- Publication number
- WO2025050069A1 WO2025050069A1 PCT/US2024/044901 US2024044901W WO2025050069A1 WO 2025050069 A1 WO2025050069 A1 WO 2025050069A1 US 2024044901 W US2024044901 W US 2024044901W WO 2025050069 A1 WO2025050069 A1 WO 2025050069A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- integration
- engineered
- enzyme
- integration enzyme
- atgrna
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/14—Hydrolases (3)
- C12N9/16—Hydrolases (3) acting on ester bonds (3.1)
- C12N9/22—Ribonucleases [RNase]; Deoxyribonucleases [DNase]
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/87—Introduction of foreign genetic material using processes not otherwise provided for, e.g. co-transformation
- C12N15/90—Stable introduction of foreign DNA into chromosome
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/87—Introduction of foreign genetic material using processes not otherwise provided for, e.g. co-transformation
- C12N15/90—Stable introduction of foreign DNA into chromosome
- C12N15/902—Stable introduction of foreign DNA into chromosome using homologous recombination
- C12N15/907—Stable introduction of foreign DNA into chromosome using homologous recombination in mammalian cells
Definitions
- the present disclosure describes integration enzymes engineered such that upon being introduced into a cell, the integration enzyme has increased stability (e.g., half-life) compared to a control integration enzyme not engineered to have increased stability.
- the increase in stability extends the capacity of the integration enzyme to mediate integration.
- the engineered integration enzyme includes an integration enzyme or fragment thereof, an at least first stabilization domain, and a nuclear localization signal.
- the present disclosure provides integration enzymes engineered to have increased stability for use in site-specific genetic engineering using Programmable Addition via Site-Specific Targeting Elements (PASTE) (see lonnidi et al. doi: 10.1101/2021.11.01.466786; the entirety of lonnidi et al. is incorporated by reference), transposon-mediated gene editing, or other suitable gene editing or gene incorporation technology.
- PASTE Programmable Addition via Site-Specific Targeting Elements
- this disclosure features an engineered integration enzyme, comprising: (a) an integration enzyme or fragment thereof; (b) an at least first stabilization domain; and (c) a nuclear localization signal (NLS).
- an integration enzyme or fragment thereof comprising: (a) an integration enzyme or fragment thereof; (b) an at least first stabilization domain; and (c) a nuclear localization signal (NLS).
- NLS nuclear localization signal
- the integration enzyme is a large serine integrase.
- the method further comprises a second atgRNA.
- the first atgRNA and the second atgRNA are an at least first pair of atgRNAs, wherein the at least first pair of atgRNAs have domains that are capable of guiding the gene editor polypeptide to the target DNA sequence;
- the first atgRNA further includes a first RT template that comprises at least a portion of an at least first integration recognition site;
- the second atgRNA further includes a second RT template that comprises at least a portion of the first integration recognition site, and the first atgRNA and the second atgRNAs collectively encode the entirety of the first integration recognition site, whereby the least first integration recognition site is integrated into the genome of the cell at the target sequence.
- the method enhances integration efficiency of the donor polynucleotide template at the site-specifically integrated integration recognition site at the double-stranded target DNA sequence as compared to the integration efficiency of the donor polynucleotide template at the site-specifically integrated recognition site when using a method that does not comprise the engineered integration enzyme.
- FIG. 1 shows a non-limiting illustration of a gene editor construct packaged within a lipid nanoparticle (LNP).
- LNP lipid nanoparticle
- FIG. 2 illustrates the donor polynucleotide (“donor template”) (synonymously, “cargo” or “payload” or “template polynucleotide”) packaged within a vector.
- donor template synthetic polynucleotide
- FIG. 3 illustrates integrase-mediated self-circularization of the donor template (template polynucleotide) within viral genome.
- the circularized donor template is capable of being genomically incorporated into an orthogonal integrase target recognition site (i.e., “beacon”).
- FIG. 4 shows non-limiting illustrations of a gene editor construct packaged within a lipid nanoparticle and an atgRNA, ngRNA, and donor template (i.e., template polynucleotide encoding a gene of interest) packaged within a vector.
- GOI gene of interest.
- PGI programmable gene insertion.
- U6 U6 promoter.
- atgRNA attachment site-containing guide RNA (atgRNA).
- FIG. 5 shows non-limiting illustrations of a gene editor construct (e.g., mRNA encoding PE2-BxBl) and a nicking guide RNA (ngRNA) packaged within a lipid nanoparticle (LNP) and an atgRNA and donor template (i.e., template polynucleotide encoding a gene of interest) packaged within a vector.
- a gene editor construct e.g., mRNA encoding PE2-BxBl
- ngRNA nicking guide RNA
- LNP lipid nanoparticle
- an atgRNA and donor template i.e., template polynucleotide encoding a gene of interest
- FIG. 6B shows non-limiting examples of sequences that enable self-circularization (e.g., LoxP AttP GT (SEQ ID NO: 568 and SEQ ID NO: 569); FRT AttP GT (SEQ ID NO: 570 and SEQ ID NO: 571); and AttB CC AttP GT (SEQ ID NO: 572 and SEQ ID NO: 573)).
- GT indicates an AttP site with a GT dinucleotide.
- AttB CC indicates an AttB site with a CC dinucleotide.
- LoxP a LoxP recombinase recognition site.
- FRT a FRT recombinase recognition site.
- FIG. 7 shows a non-limiting illustration of recombinase/integrase-mediated intramolecular circularization products.
- FIGs. 8A-8B show non-limiting illustrations of a ddPCR assay and intramolecular circularization ddPCR detection probes.
- FIG. 8A shows a non-limiting illustration of the ddPCR strategy.
- FIG. 8B shows non-limiting examples of the universal probe (SEQ ID NO: 574 and SEQ ID NO: 575) and an AttR probe (SEQ ID NO: 576 and SEQ ID NO: 577) that can be used in the assay shown in FIG. 8A.
- FIG. 9 shows a non-limiting illustration of a pDNA genome and AAV transfection and screening protocol.
- FIG. 10 shows data for circularization of AAV pDNA and packaged AAV genomic DNA with Bxb 1.
- FIG. 11 shows data for Cre-, FLPe-, and Bxb 1 -mediated circularization of AAV pDNA confirmed by ddPCR.
- FIG. 12 shows Cre-, FLPe-, and Bxb 1 -mediated circularization of packaged AAV confirmed by ddPCR
- FIG. 13 shows percent circularization between the Bxb 1 -mediated attR scar ddPCR probe (“attR probe” described in FIG. 8B) and the universal ddPCR probe (“universal probe” described in FIG. 8B)
- FIGs. 14A-14E shows analysis of AttP variants.
- FIG. 14A shows a non-limiting schematic of AttP mutations tested for improving integration efficiency (SEQ ID NOS: 394 and 540-542, respectively, in order of appearance).
- FIG. 14B shows integration efficiencies of wildtype and mutant AttP sites across a panel of AttB lengths.
- FIG. 14C shows a non-limiting schematic of multiplexed integration of different cargo sets at specific genomic loci. Three fluorescent cargos (GFP, mCherry, and YFP) are inserted orthogonally at three different loci (ACTB, LMNB1, NOLC1) for in-frame gene tagging.
- FIG. 14A shows a non-limiting schematic of AttP mutations tested for improving integration efficiency (SEQ ID NOS: 394 and 540-542, respectively, in order of appearance).
- FIG. 14B shows integration efficiencies of wildtype and mutant AttP sites across a panel of AttB lengths.
- FIG. 14C shows
- FIG. 14D shows orthogonality of top 4 AttB/AttP dinucleotide pairs evaluated for GFP integration with PASTE at the ACTB locus.
- FIG. 15 illustrates a schematic of single atgRNA and dual atgRNA approaches for beacon placement (“integration recognition site”).
- FIG. 21A shows ddPCR data for percent in vivo beacon placement in the Nolcl locus of neonatal mice at 6 weeks post-delivery of a single dose of a mixture of two LNPs.
- First LNP contained mRNA encoding a prime editing system and a first synthetic atgRNA (atgRNAl) at a 1 : 1 ratio.
- Second LNP contained mRNA encoding a prime editing system and a second synthetic atgRNA (atgRNA2) at a 1 : 1 ratio.
- Each of the first and second atgRNAs targeted the mouse Nolcl locus, encoded a portion of an integration recognition site (“beacon”), and together included a 6bp overlap.
- the first and second LNPs were combined 1 : 1 as mixture and administered at either 1 mg/kg or 3 mg/kg.
- LNP #F2 LNP formulation #F2.
- Prime editing or CRISPR system Examples of prime editors can be found in the following: W02020/191153, W02020/191171, WO2020/191233,
- an integrase or recombinase is directly linked or fused, for example by a peptide linker, which may be cleavable or non-cleavable, to the prime editor fusion protein (i.e., fused Cas9 nickase-reverse transcriptase) or Gene Writer protein.
- a peptide linker which may be cleavable or non-cleavable, to the prime editor fusion protein (i.e., fused Cas9 nickase-reverse transcriptase) or Gene Writer protein.
- Suitable linkers for example between the Cas9, RT, and integrase, may be selected from Table 3:
- SpCas9 Streptococcus pyogenes Cas9
- REC recognition
- NUC nuclease
- the RuvC domain is assembled from the three split RuvC motifs (RuvC I— III) and interfaces with the PI domain to form a positively charged surface that interacts with the 30 tail of the sgRNA.
- the HNH domain lies between the RuvC II— III motifs and forms only a few contacts with the rest of the protein. Structural aspects of SpCas9 are described by Nishimasu et al., Crystal Structure of Cas9 in Complex with Guide RNA and Target DNA, Cell 156, 935-949, February 27, 2014.
- PAM-interacting domain The NUC lobe contains the PAM-interacting (PI) domain that is positioned to recognize the PAM sequence on the noncomplementary DNA strand.
- the PI domain of SpCas9 is required for the recognition of 5’-NGG-3’ PAM, and deletion of the PI domain (A1099-1368) abolished the cleavage activity, indicating that the PI domain is critical for SpCas9 function and a major determinant for the PAM specificity.
- RuvC domain- The NUC lobe contains the PAM-interacting (PI) domain that is positioned to recognize the PAM sequence on the noncomplementary DNA strand.
- the PI domain of SpCas9 is required for the recognition of 5’-NGG-3’ PAM, and deletion of the PI domain (A1099-1368) abolished the cleavage activity, indicating that the PI domain is critical for SpCas9 function and a major determinant for the PAM specificity.
- SpCas9 HNH nucleases have three catalytic residues, Asp839, His840, and Asn863 and cleave the complementary strand of the target DNA through a single-metal mechanism.
- sgRNA:DNA recognition' The sgRNA guide region is primarily recognized by the REC lobe.
- the backbone phosphate groups of the guide region interact with the RECI domain (Argl65, Glyl66, Arg403, Asn407, Lys510, Tyr515, and Arg661) and the bridge helix (Arg63, Arg66, Arg70, Arg71, Arg74, and Arg78).
- the 20-hydroxyl groups of Gl, Cl 5, U16, and G19 hydrogen bond with Vai 1009, Tyr450, Arg447/Ile448, and Thr404, respectively.
- nucleobases of G21 and U50 in the G21 :U50 wobble pair stack with the terminal C20:G10 pair in the guide:target heteroduplex and Tyr72 on the bridge helix, respectively, with the U50 04 atom hydrogen bonded with Arg75.
- A51 adopts the syn conformation and is oriented in the direction opposite to U50.
- the nucleobase of A51 is sandwiched between Phel 105 and U63, with its Nl, N6, and N7 atoms hydrogen bonded with G62, Glyl l03, and Phel 105, respectively.
- Stem-loop recognition' Stem loop 1 is primarily recognized by the REC lobe, together with the PI domain.
- the backbone phosphate groups of stem loop 1 (nucleotides 52, 53, and 59- 61) interact with the RECI domain (Leu455, Ser460, Arg467, Thr472, and Ile473), the PI domain (Lysl 123 and Lysl 124), and the bridge helix (Arg70 and Arg74), with the 20-hydroxyl group of G58 hydrogen bonded with Leu455.
- A52 interacts with Phel 105 through a face-to-edge p-p stacking interaction, and the flipped U59 nucleobase hydrogen bonds with Asn77.
- the single-stranded linker and stem loops 2 and 3 are primarily recognized by the NUC lobe.
- the backbone phosphate groups of the linker (nucleotides 63-65 and 67) interact with the RuvC domain (Glu57, Lys742, and Lysl097), the PI domain (Thrl l02), and the bridge helix (Arg69), with the 20-hydroxyl groups of U64 and A65 hydrogen bonded with Glu57 and His721, respectively.
- the C67 nucleobase forms two hydrogen bonds with Vail 100.
- Stem loop 2 is recognized by Cas9 via the interactions between the NUC lobe and the non-Watson-Crick A68:G81 pair, which is formed by direct (between the A68 N6 and G81 06 atoms) and water-mediated (between the A68 Nl and G81 Nl atoms) hydrogen-bonding interactions.
- the A68 and G81 nucleobases contact Serl351 and Tyrl356, respectively, whereas the A68:G81 pair interacts with Thrl358 via a water-mediated hydrogen bond.
- the 20-hydroxyl group of A68 hydrogen bonds with His 1349, whereas the G81 nucleobase hydrogen bonds with Lys33.
- Stem loop 3 interacts with the NUC lobe more extensively, as compared to stem loop 2.
- the backbone phosphate group of G92 interacts with the RuvC domain (Arg40 and Lys44), whereas the G89 and U90 nucleobases hydrogen bond with Glnl272 and Glul225/Alal227, respectively.
- the A88 and C91 nucleobases are recognized by Asn46 via multiple hydrogenbonding interactions.
- Cas9 proteins smaller than SpCas9 allow more efficient packaging of nucleic acids encoding CRISPR systems, e.g., Cas9 and sgRNA into one rAAV (“all-in-one-AAV”) particle.
- efficient packaging of CRISPR systems can be achieved in other viral vector systems (i.e., lentiviral, integration deficient lentiviral, hd-AAV, etc.) and non-viral vector systems (i.e., lipid nanoparticle).
- Small Cas9 proteins can be advantageous for multidomain-Cas-nuclease-based systems for prime editing.
- Cas9 proteins include Staphylococcus aureus (SauCas9, 1053 amino acid residues) and Campylobacter jejuni (CjCas9, 984 amino residues).
- SerCas9 Staphylococcus aureus
- CjCas9 Campylobacter jejuni
- Staphylococcus lugdunensis (Siu) Cas9 as having genome-editing activity and provided homology mapping to SpCas9 and SauCas9 to facilitate generation of nickases and inactive (“dead”) enzymes (Schmidt et al., 2021, Improved CRISPR genome editing using small highly active and specific engineered RNA-guided nucleases. Nat Commun 12, 4219. doi.org/10.1038/s41467-021-24454-5) and engineered nucleases with higher cleavage activity by fragmenting and shuffling Cas9 DNAs.
- the small Cas9s and nickases are useful in the instant invention.
- the Cas9 proteins used herein may also include other “Cas9 variants” having at least about 70% identical, at least about 80% identical, at least about 90% identical, at least about 95% identical, at least about 96% identical, at least about 97% identical, at least about 98% identical, at least about 99% identical, at least about 99.5% identical, or at least about 99.9% identical to any reference Cas9 protein, including any wild type Cas9, or mutant Cas9 (e.g., a dead Cas9 or Cas9 nickase), or fragment Cas9, or circular permutant Cas9, or other variant of Cas9 disclosed herein or known in the art.
- Cas9 variants having at least about 70% identical, at least about 80% identical, at least about 90% identical, at least about 95% identical, at least about 96% identical, at least about 97% identical, at least about 98% identical, at least about 99% identical, at least about 99.5% identical, or at least about 99.9% identical to any reference Cas9 protein, including any wild
- the crRNA-target strand heteroduplex is terminated by a stacking interaction with a conserved aromatic residue (Tyr410). This prevents base pairing between the crRNA and the target strand beyond nucleotides U20 and dA(-20), respectively. Beyond this point, the target DNA strand nucleotides re-engage the non-target DNA strand, forming a PAM-distal DNA duplex comprising nucleotides dC(-21)-dA(-27) and dG21*-dT27*, respectively. The duplex is confined between the REC2 and Nuc domains at the end of the central channel formed by the REC and NUC lobes.
- the RuvC active site contains three catalytic residues (D917, E1006, and D1255). Structural observations suggest that both the target and non-target DNA strands are cleaved by the same catalytic mechanism in a single active site in Cpfl/Casl2a enzymes.
- Another type V CRISPR is AsCpfl from Acidaminococcus sp BV3L6 (Yamano et al., Crystal structure of Cpfl in complex with guide RNA and target DNA, Cell 165, 949-962, May 5, 2016)
- the nuclease comprises a Casl2f effector.
- Small CRISPR- associated effector proteins belonging to the type V-F subtype have been identified through the mining of sequence databases and members classified into Casl2fl (Casl4a and type V-U3), Casl2f2 (Casl4b) and Casl2f3 (Casl4c, type V-U2 and U4).
- Casl2fl Casl2fl
- Casl4b Casl2f2
- Casl4c type V-U2 and U4
- protospacer adjacent sequence or “protospacer adjacent motif’ or “PAM” refers to an approximately 2-6 base pair DNA sequence (or a 2-, 3-, 4-, 5-, 6-, 7-, 8-, 9-, 10-, 11-, 12-long nucleotide sequence) that is an important targeting component of a Cas9 nuclease.
- PAM sequence is on either strand, and is downstream in the 5' to 3' direction of Cas9 cut site.
- the PAM specificity can be modified by introducing one or more mutations, including (a) DI 135 V, R1335Q, and T1337R “the VQR variant”, which alters the PAM specificity to NGAN or NGNG, (b) D1135E, R1335Q, and T1337R “the EQR variant”, which alters the PAM specificity to NGAG, and (c) DI 135V, G1218R, R1335E, and T1337R “the VRER variant”, which alters the PAM specificity to NGCG.
- the DI 135E variant of canonical SpCas9 still recognizes NGG, but it is more selective compared to the wild type SpCas9 protein.
- Cas9 enzymes from different bacterial species can have varying PAM specificities and some embodiments are therefore chosen based on the desired PAM recognition.
- Cas9 from Staphylococcus aureus (SaCas9) recognizes NGRRT or NGRRN.
- Cas9 from Neisseria meningitis (NmCas) recognizes NNNNGATT.
- Cas9 from Streptococcus thermophilis (StCas9) recognizes NNAGAAW.
- Cas9 from Treponema denticola (TdCas) recognizes NAAAAC. These examples are not meant to be limiting.
- Oh, Y. et al. describe linking reverse transcriptase to a Francisella novicida Cas9 [FnCas9(H969A)] nickase module.
- FeCas9(H969A) Francisella novicida Cas9
- nickase module By increasing the distance to the PAM, the FnCas9(H969A) nickase module expands the region of a reverse transcription template (RTT) following the primer binding site.
- Prime editor fusion protein describes a protein that is used in prime editing.
- Prime editing uses CRISPR enzyme that nicks or cuts only single strand of double stranded DNA, i.e., a nickase; and a nickase can occur either naturally or by mutation or modification of a nuclease that makes double stranded cuts.
- Such an enzyme can be a catalytically-impaired Cas9 endonuclease (a nickase).
- a nickase can be a Casl2a/b, MAD7, or variant thereof.
- the nickase is fused to an engineered reverse transcriptase (RT).
- the nickase is programmed (directed) with a primeediting guide RNA (pegRNA).
- pegRNA primeediting guide RNA
- the pegRNA both specifies the target site and encodes the desired edit.
- the nickase is a catalytically-impaired Cas9 endonuclease, a Cas9 nickase, that is fused to the reverse transcriptase.
- the Cas9 nickase part of the protein is guided to the DNA target site by the pegRNA, whereby a nick or single stranded cut occurs.
- the reverse transcriptase domain then uses the pegRNA to template reverse transcription of the desired edit, directly polymerizing DNA onto the nicked target DNA strand.
- the edited DNA strand replaces the original DNA strand, creating a heteroduplex containing one edited strand and one unedited strand.
- the prime editor guides resolution of the heteroduplex to favor copying the edit onto the unedited strand, completing the process (typically achieved with a nickase gRNA).
- PEI refers to a PE complex comprising a fusion protein comprising Cas9(H840A) and a wild type MMLV RT having the following N-terminus to C-terminus structure: [NLS]-[Cas9(H840A)]- [linker] -[MMLV_RT(wt)] + a desired atgRNA (or PEgRNA).
- the prime editors disclosed herein is comprised of PEI.
- PE2 refers to a PE complex comprising a fusion protein comprising Cas9(H840A) and a variant MMLV RT having the following N-terminus to C-terminus structure:
- PE3b refers to PE3 but wherein the second-strand nicking guide RNA is designed for temporal control such that the second strand nick is not introduced until after the installation of the desired edit. This is achieved by designing a gRNA with a spacer sequence with mismatches to the unedited original allele that matches only the edited strand. Using this strategy, mismatches between the protospacer and the unedited allele should disfavor nicking by the sgRNA until after the editing event on the PAM strand takes place.
- a prime editing complex consists of a type II CRISPR PE protein containing an RNA-guided DNA-nicking domain fused to a reverse transcriptase (RT) domain and complexed with a pegRNA.
- the pegRNA comprises (5’ to 3’) a spacer that is complementary to the target sequence of a genomic DNA, a nickase (e.g. Cas9) binding site, a reverse transcriptase template including editing positions, and primer binding site (PBS).
- the PE-pegRNA complex binds the target DNA and the CRISPR protein nicks the PAM- containing strand.
- the resulting 3' end of the nicked target hybridizes to the primer-binding site (PBS) of the pegRNA, then primes reverse transcription of new DNA containing the desired edit using the RT template of the pegRNA.
- PBS primer-binding site
- the overall structure of the pegRNA is like that of a typical type II sgRNA with a reverse transcriptase template/primer binding site appended to the 3’ end. The structure leaves the PBS at the 3’ end of the pegRNA free to bind to the nicked strand complementary to the target which forms the primer for reverse transcription.
- Guide RNAs of CRISPRs differ in overall structure. For example, while the spacer of a type II gRNA is located at the 5’ end, the spacer of a type V gRNA is located towards the 3 ’ end, with the CRISPR protein (e.g. Casl2a) binding region located toward the 5’ end. Accordingly, the regions of a type V pegRNA are rearranged compared to a type II pegRNA.
- the overall structure of the pegRNA is like that of a typical type II sgRNA with a reverse transcriptase template/primer binding site appended to the 3’ end.
- the pegRNA comprises (5’ to 3’) a CRISPR protein-binding region, a spacer which is complementary to the target sequence of a genomic DNA, a reverse transcriptase template including editing positions, and primer binding site (PBS).
- a first integration recognition site e.g., any of the integration recognition sites described herein
- a second integration recognition site e.g., any of the integration recognition sites described herein
- a non-limiting example of a cognate pair include an attB site and an attP site, whereby a serine integrase mediates recombination between the attB site and the attP site.
- an atgRNA comprises a reverse transcriptase template that encodes, partially or in its entirety, an integration recognition site (also referred to as an integration target recognition site) or a recombinase recognition site (also referred to as a recombinase target recognition site).
- the integration target recognition site which is to be placed at a desired location in the genome or intracellular nucleic acid, is referred to as a “beacon,” a “beacon” site or an “attachment site” or a “landing pad” or “landing site.”
- An integration target recognition site or recombinase target recognition site incorporated into the pegRNA is referred to as an attachment site containing guide RNA (atgRNA).
- the primer binding site allows the 3’ end of the nicked DNA strand to hybridize to the atgRNA, while the RT template serves as a template for the synthesis of edited genetic information.
- the atgRNA is capable for instance, without limitation, of (i) identifying the target nucleotide sequence to be edited and (ii) encoding new genetic information that replaces (or in some cases adds) the targeted sequence.
- the atgRNA is capable of (i) identifying the target nucleotide sequence to be edited and (ii) encoding an integration site that replaces (or inserts/ deletes within) the targeted sequences.
- the co-delivery system described herein includes a polynucleotide sequence encoding an attachment site-containing guide RNA (atgRNA) packaged in an LNP.
- the co-delivery system described herein includes a vector comprising a polynucleotide sequence encoding an atgRNA.
- the atgRNA comprises a domain that is capable of guiding the prime editor fusion protein to a target sequence, thereby identifying the target nucleotide sequence to be edited; and a reverse transcriptase (RT) template that comprises a first integration recognition site.
- the atgRNA comprises a domain that is capable of guiding the prime editor fusion protein (or prime editor system) to a target sequence, thereby identifying the target nucleotide sequence to be edited; and a reverse transcriptase (RT) template that comprises at least a portion first integration recognition site.
- a domain that is capable of guiding the prime editor fusion protein (or prime editor system) to a target sequence, thereby identifying the target nucleotide sequence to be edited
- RT reverse transcriptase
- the co-delivery system described herein includes a polynucleotide sequence encoding a first attachment site-containing guide RNA (atgRNA) and a polynucleotide nucleotide sequence encoding a second attachment site-containing guide RNA (atgRNA) packaged into the same LNP.
- the co-delivery system described herein includes a polynucleotide sequence encoding a first attachment site-containing guide RNA (atgRNA) packaged into a first LNP and a polynucleotide nucleotide sequence encoding a second attachment site-containing guide RNA (atgRNA) packaged into a second LNP.
- the co-delivery system described herein includes a vector comprising a polynucleotide sequence encoding a first attachment site-containing guide RNA (atgRNA), a polynucleotide sequence encoding a second atgRNA, or both.
- atgRNA attachment site-containing guide RNA
- the co-delivery system described herein includes a polynucleotide sequence encoding a first attachment site-containing guide RNA (atgRNA) packaged into a first LNP and a vector comprising a polynucleotide sequence encoding a second atgRNA.
- atgRNA attachment site-containing guide RNA
- the co-delivery system contains a first atgRNA and a second atgRNA
- the first atgRNA and the second atgRNA are an at least first pair of atgRNAs, where the at least first pair of atgRNAs have domains that are capable of guiding the gene editor protein or prime editor fusion protein to a target sequence
- the first atgRNA further includes a first RT template that comprises at least a portion of the first integration recognition site
- the second atgRNA further includes a second RT template that comprises at least a portion of the first integration recognition site
- the first atgRNA and the second atgRNAs collectively encode the entirety of the first integration recognition site.
- the first atgRNA’ s reverse transcriptase template encodes for a first single-stranded DNA sequence (i.e., a first DNA flap) that contains a complementary region to a second single-stranded DNA sequence (i.e., a second DNA flap) encoded by a second atgRNA comprising a second reverse transcriptase template.
- the complementary region between the first and second single-stranded DNA sequences is comprised of more than 5 consecutive bases of an integrase target recognition site.
- the complementary region between the first and second single-stranded DNA sequences is comprised of more than 10 consecutive bases of an integrase target recognition site.
- the complementary region between the first and second single-stranded DNA sequences is comprised of more than 20 consecutive bases of an integrase target recognition site. In certain embodiments, the complementary region between the first and second single-stranded DNA sequences is comprised of more than 30 consecutive bases of an integrase target recognition site.
- Use of two guide RNAs that are (or encode DNA that is) partially complementarity to each other and comprised of consecutive bases of an integrase target recognition site are referred to as dual, paired, annealing, complementary, or twin attachment site-containing guide RNAs (atgRNAs).
- RNAs that are (or encode DNA that is) full complementarity to each other and comprised of consecutive bases of an integrase target recognition site are referred to as dual, paired, annealing, complementary, or twin attachment sitecontaining guide RNAs (atgRNAs).
- the first atgRNA upon introducing the nucleic acid construct into a cell, incorporates the first integration recognition site into the cell’s genome at the target sequence.
- Table 9 includes atgRNAs, sgRNAs and nicking guides that can be used herein. Spacers are labeled in capital font (SPACER), RT regions in bold capital (RT REGION), AttB sites in bold lower case (attB site), and PBS in capital italics (PBS). Unless otherwise denoted, the AttB is for Bxbl.
- the co-delivery system described herein contains an integrase and/or a recombinase.
- the co-delivery system includes an integrase and/or a recombinase packaged in a LNP.
- the co-delivery system includes a polynucleotide encoding an integrase and/or a recombinase.
- the co-delivery system includes an integrase or a recombinase packaged in a vector (e.g., a viral vector).
- the co-delivery system includes at least a first integrase (e.g., a first integrase and a second integrase) and/or at least a first recombinase (e.g., a first recombinase and a second recombinase).
- a first integrase e.g., a first integrase and a second integrase
- a first recombinase e.g., a first recombinase and a second recombinase
- the integration enzyme (e.g., the integrase or recombinase) is selected from the group consisting of Dre, Vika, Bxbl, ⁇ pC31, RDF, cpBTl, Rl, R2, R3, R4, R5, TP901-1, Al 18, cpFCl, cpCl, MR11, TGI, cp370.1, W , BL3, SPBc, K38, Peaches, Veracruz, Rebeuca, Theia, Benedict, KSSJEB, PattyP, Doom, Scowl, Lockley, Switzer, Bob3, Troube, Abrogate, Anglerfish, Sarfire, SkiPole, Concept!, Museum, Severus, Airmid, Benedict, Hinder, ICleared, Sheen, Mundrea, BxZ2, cpRV, retrotransposases encoded by a Tcl/mariner family member including but not limited to retrotransposases encoded by
- LSRs serine recombinases
- embodiments can include any serine recombinase such as BceINT, SSCINT, SACINT, and INT10 (see lonnidi et al., 2021; Drag-and-drop genome insertion without DNA cleavage with CRISPR directed integrases.
- the integration site can be selected from an attB site, an attP site, an attL site, an attR site, a lox71 site a Vox site, or a FRT site.
- the Cre-lox system is referred to either as a control for programmable gene insertion or as a tool for a recombinase-mediated event separate and distinct from insertion of the donor polynucleotide template (or exogenous nucleic acid) into the integrated recognition site.
- integrases, transposases and the like can depend on nuclear localization.
- prokaryotic enzymes are adapted to modulate nuclear localization.
- eukaryotic or vertebrate enzymes are adapted to modulate nuclear localization.
- the invention provides fusion or hybrid proteins. Such modulation can comprise addition or removal of one or more nuclear localization signal (NLS) and/or addition or removal of one or more nuclear export signal (NES).
- NLS nuclear localization signal
- NES nuclear export signal
- nuclear export signal (NES) of transposases affects the transposition activity of mariner-like elements Ppmarl and Ppmar2 of moso bamboo. Mob DNA. 2019 Aug 19; 10:35. doi: 10.1186/sl3100-019-0179-y).
- the methods and constructs are used to modulate nuclear localization of system components of the invention.
- the integrase used herein is selected from below (Table 10).
- FIGs. 14A-14E shows analysis of effect of variant AttP sites on integration efficiency.
- the integration enzyme is selected from one of the about 27,000 Serine integrases described in International Patent Publication No. WO 2023/070031 A2, which is hereby incorporated by reference in its entirety.
- This disclosure features integration enzymes (also referred to integrases) engineered such that upon being introduced into a cell, the integration enzyme has increased stability (e.g., half- life) compared to a control integration enzyme not engineered to have increased stability.
- An increase in stability includes an increase in half-life and a reduction in protein degradation rates, thereby extending the capacity of the integration enzyme to mediate integration.
- ubiquitin proteosome system proteolysis by ubiquitin proteosome system (UPS) and autophagy.
- proteins can be ubiquitinated on lysine residues, thereby leading to degradation.
- Proteins can also have specific domains (degrons) that can be ubiquitinated, which also leads to protein degradation.
- Degrons also referred to as degradation signals
- Degrons are recognized and polyubiquitinated, targeting the protein for degradation.
- Degrons can be present at both the N-terminus and/or C-terminus of a protein.
- Polyubiquitinated protein is recognized by receptor subunits of the 26S proteosome (RpnlO (yeast)/p54 (drosophila), Rpnl3 and Rpnl).
- the engineered integration enzymes is selected from the engineered integration enzymes described in FIGs. 23, 25, 28, 30B, 32A, 33, 35, and 40-42. In some embodiments, the engineered integration enzymes is selected from the engineered integration enzymes described in Table 26. In some embodiments, the engineered integration enzyme comprises an amino acid sequence that is at least 80% (e.g., 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 99%, or 100%) identical to an amino acid sequence listed in Table 26.
- the engineered integration enzyme is encoded by a nucleic acid sequence that is at least 80% (e.g., 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 99%, or 100%) identical to a nucleic acid sequence listed in Table 26.
- This disclosure features integration enzymes engineered to include a domain (e.g., a stabilization domain) that increases stability of the integration enzyme when compared to an integration enzyme not engineered to include the domain (e.g., the stabilization domain).
- a domain e.g., a stabilization domain
- the integration enzyme or fragment thereof and the stabilization domain are fused, thereby by creating a fusion protein.
- an engineered integration enzyme comprises an integration enzyme or fragment thereof; an at least first stabilization domain; and a nuclear localization signal (NLS).
- the integration enzyme or fragment thereof, the stabilization domain, and the NLS are fused, thereby by creating a fusion protein.
- the engineered integration enzyme comprises an integration enzyme or fragment thereof comprising at least two (e.g., at least three, at least four, at least five, at least six, at least seven, at least eight, at least nine, or at least ten) amino acid modification such that the combination of the at least two modifications increase the stability of the integration enzyme as compared to the integration enzyme not comprising the at least first amino acid modification.
- the amino acid modification can be in a degron motif in the integration enzyme.
- the amino acid modification can be a substitution, deletion, or insertion that increases the stability of the integration enzyme as compared to an integration enzyme not comprising the at least first amino acid modification.
- the engineered integration enzyme is an integration enzyme described in Table 10 and comprises an amino acid modification that increases the stability of the integration enzyme as compared to an integration enzyme selected from Table 10 and not engineered to include the amino acid modification.
- the amino acid modification can be a substitution, deletion, or insertion in an integration enzyme having an amino acid sequence of SEQ ID NO: 388 that increases the stability of the integration enzyme as compared to an integration enzyme having a sequence of SEQ ID NO: 388 that does not comprise the at least first amino acid modification.
- the first amino acid modification is an amino acid substitution of L275V in SEQ ID NO: 388.
- the engineered integration enzyme comprises an amino acid sequence that is at least 80% (e.g., 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 99%, or 100%) identical to an amino acid sequence of SEQ ID NO: 378 and an amino acid modification in the sequence of SEQ ID NO: 378, where the amino acid modification increases the stability of the integration enzyme as compared to an integration enzyme having an amino acid sequence selected of SEQ ID NO: 378.
- the engineered integration enzyme comprises an amino acid sequence that is at least 80% (e.g., 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 99%, or 100%) identical to an amino acid sequence of SEQ ID NO: 382 and an amino acid modification in the sequence of SEQ ID NO: 382, where the amino acid modification increases the stability of the integration enzyme as compared to an integration enzyme having an amino acid sequence selected of SEQ ID NO: 382.
- the engineered integration enzyme comprises an amino acid sequence that is at least 80% (e.g., 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 99%, or 100%) identical to an amino acid sequence of SEQ ID NO: 383 and an amino acid modification in the sequence of SEQ ID NO: 383, where the amino acid modification increases the stability of the integration enzyme as compared to an integration enzyme having an amino acid sequence selected of SEQ ID NO: 383.
- the engineered integration enzyme comprises an amino acid sequence that is at least 80% (e.g., 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 99%, or 100%) identical to an amino acid sequence of SEQ ID NO: 384 and an amino acid modification in the sequence of SEQ ID NO: 384, where the amino acid modification increases the stability of the integration enzyme as compared to an integration enzyme having an amino acid sequence selected of SEQ ID NO: 384.
- the engineered integration enzyme comprises an amino acid sequence that is at least 80% (e.g., 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 99%, or 100%) identical to an amino acid sequence of SEQ ID NO: 385 and an amino acid modification in the sequence of SEQ ID NO: 385, where the amino acid modification increases the stability of the integration enzyme as compared to an integration enzyme having an amino acid sequence selected of SEQ ID NO: 385.
- the engineered integration enzyme comprises an amino acid sequence that is at least 80% (e.g., 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 99%, or 100%) identical to an amino acid sequence of SEQ ID NO: 386 and an amino acid modification in the sequence of SEQ ID NO: 386, where the amino acid modification increases the stability of the integration enzyme as compared to an integration enzyme having an amino acid sequence selected of SEQ ID NO: 386.
- the engineered integration enzyme comprises an amino acid sequence that is at least 80% (e.g., 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 99%, or 100%) identical to an amino acid sequence of SEQ ID NO: 387 and an amino acid modification in the sequence of SEQ ID NO: 387, where the amino acid modification increases the stability of the integration enzyme as compared to an integration enzyme having an amino acid sequence selected of SEQ ID NO: 387.
- the engineered integration enzyme comprises an amino acid sequence that is at least 80% (e.g., 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 99%, or 100%) identical to an amino acid sequence of SEQ ID NO: 388 and an amino acid modification in the sequence of SEQ ID NO: 388, where the amino acid modification increases the stability of the integration enzyme as compared to an integration enzyme having an amino acid sequence selected of SEQ ID NO: 388.
- the engineered integration enzyme comprises an amino acid sequence that is at least 80% (e.g., 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 99%, or 100%) identical to an amino acid sequence of SEQ ID NO: 389 and an amino acid modification in the sequence of SEQ ID NO: 389, where the amino acid modification increases the stability of the integration enzyme as compared to an integration enzyme having an amino acid sequence selected of SEQ ID NO: 389.
- the engineered integration enzyme comprises an amino acid sequence that is at least 80% (e.g., 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 99%, or 100%) identical to an amino acid sequence of SEQ ID NO: 390 and an amino acid modification in the sequence of SEQ ID NO: 390, where the amino acid modification increases the stability of the integration enzyme as compared to an integration enzyme having an amino acid sequence selected of SEQ ID NO: 390.
- the engineered integration enzyme comprises an amino acid sequence that is at least 80% (e.g., 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 99%, or 100%) identical to an amino acid sequence of SEQ ID NO: 391 and an amino acid modification in the sequence of SEQ ID NO: 391, where the amino acid modification increases the stability of the integration enzyme as compared to an integration enzyme having an amino acid sequence selected of SEQ ID NO: 391.
- the engineered integration enzyme comprises an amino acid sequence that is at least 80% (e.g., 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 99%, or 100%) identical to an amino acid sequence of SEQ ID NO: 392 and an amino acid modification in the sequence of SEQ ID NO: 392, where the amino acid modification increases the stability of the integration enzyme as compared to an integration enzyme having an amino acid sequence selected of SEQ ID NO: 392.
- the engineered integration enzyme comprises an amino acid sequence that is at least 80% (e.g., 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 99%, or 100%) identical to an amino acid sequence of SEQ ID NO: 393 and an amino acid modification in the sequence of SEQ ID NO: 393, where the amino acid modification increases the stability of the integration enzyme as compared to an integration enzyme having an amino acid sequence selected of SEQ ID NO: 393.
- This disclosure also features engineered integration enzymes comprising a stabilization domain and an amino acid modification that increases the engineered integration enzymes stability.
- the engineered integration enzyme comprises an integration enzyme as described in International Patent Publication No. WO 2023/070031A2, which is hereby incorporated by reference in its entirety, where the integration enzyme includes a stabilization domain and an amino acid modification, where the stabilization domain, the amino acid modification, or both, increase the stability of the integration enzyme as compared to an integration enzyme not engineered to have the amino acid modification and/or the stabilization domain.
- the engineered integration enzyme is an integration enzyme described in Table 10 and comprises a stabilization domain and an amino acid modification in the same integration enzyme selected from Table 10, wherein the stabilization domain, the amino acid modification, or both, increase the stability of the integration enzyme as compared to an integration enzyme not engineered to have the amino acid modification and/or stabilization domain.
- the engineered integration enzyme is a BxB 1 integration enzyme comprising a stabilization domain and an amino acid modification in the BxB 1 integration enzyme, wherein the stabilization domain, the amino acid modification, or both, increase the stability integration enzyme as compared to an integration enzyme not engineered to have the amino acid modification and/or stabilization domain.
- the engineered integration enzyme comprises an amino acid sequence that is at least 80% (e.g., 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 99%, or 100%) identical to an amino acid sequence selected from SEQ ID NOs: 378-393, a stabilization domain, and an amino acid modification in the amino acid sequence, where the stabilization domain, the amino acid modification, or both, increase the stability of the engineered integration enzyme as compared to an integration enzyme not engineered to have the at amino acid modification and/or the stabilization domain.
- the engineered integration enzyme comprises an amino acid sequence that is at least 80% (e.g., 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 99%, or 100%) identical to an amino acid sequence of SEQ ID NO: 388, a stabilization domain and an amino acid modification in SEQ ID NO: 388, wherein the stabilization domain, the amino acid modification, or both, increase the stability of integration enzyme as compared to an integration enzyme not engineered to have the amino acid modification and/or stabilization domain.
- This disclosure features a polynucleotide comprising a nucleic acid sequence encoding any of the engineered integration enzymes described herein (see Section 4.9.1, 4.9.2, and 4.9.3) or any of the engineered integration enzyme linked to the gene editor polypeptide or fusion proteins described herein (see Section 4.9.5).
- the nucleic acid sequence encoding the engineered integration enzyme is codon optimized. In some embodiments, the codon optimization is performed using an algorithm.
- the nucleic acid sequence of the engineered integration enzyme is optimized based on second structure such that the structure confers increased stability of the polynucleotide.
- the optimization based on second structure relies on an algorithm to determine the optimal mRNA sequence.
- a non-limiting example of an algorithm capable of being used to optimize the mRNA encoding any of the any of the engineered integration enzymes described herein is as described in Zhang et al. (Algorithm for Optimized mRNA Design Improves Stability and Immunogenicity, Nature 2023, doi.org/10.1038/s41586-023-06127-z), which is hereby incorporated by reference in its entirety. Zhang et al. refer to an algorithm called LinearDesign that optimizes both structural stability (via secondary structure) and codon usage.
- the nucleic acid sequence of the engineered integration enzyme is optimized based on the LinearDesign algorithm.
- This disclosure also features vector comprising the nucleic acid sequence encoding any of the engineered integration enzymes described herein (see Section 4.9.1, 4.9.2, and 4.9.3) or any of the engineered integration enzyme linked to the gene editor polypeptide or fusion proteins described herein (see Section 4.9.5).
- This disclosure also features engineered integration enzymes (e.g., any of the engineered integration enzymes described herein (see, e.g., Section 4.9.1, 4.9.2, and 4.9.3) linked to a gene editor polypeptide (e.g., any of the gene editor polypeptides described herein).
- the linked engineered integration enzyme-gene editor polypeptide can be used to incorporate an integration recognition site into the genome of a cell and for integrating a donor polynucleotide template into the genome.
- the linker is a peptide fused in-frame between the engineered integration enzyme and the gene editor polypeptide.
- the gene editor polypeptide comprises a DNA binding domain and a reverse transcriptase.
- This disclosure also features a fusion protein comprising (a) a DNA binding domain, optionally comprising a nickase activity; (b) a reverse transcriptase; and (c) any of the engineered integration enzymes described herein (see, e.g., Sections 4.9.1, 4.9.2, and 4.9.3), wherein at least any two of elements (a), (b), or (c) are linked via at least a first C-terminal linker comprising.
- the fusion protein comprises from N-terminus to C-terminus: (a), (b), and (c); (b), (a), and (c); (c), (a), and (b); (c), (b), and (a); (b), (c), and (a); and (b), (a), and (c).
- the engineered integration enzyme is linked to a gene editor is described in FIG. 46.
- Non-limiting examples of metrics that can be used as indication of protein stability include the following.
- Protein Half-Life This is the time required for half of the amount of protein in a cell to be degraded. This can be used to measure protein turnover.
- One non-limiting example of measuring protein half-life includes using pulse-chase experiments where the protein is labeled with a radioactive or other traceable isotope, then followed over time to see how rapidly it disappears.
- Tm Melting Temperature
- thermodynamic Stability This is the change in Gibbs free energy between the folded and unfolded state of the protein. It measures how much energy is required to unfold the protein, with a larger value indicating a more stable protein.
- thermodynamic stability include equilibrium denaturation experiments, where the protein is exposed to varying concentrations of a denaturant and the degree of unfolding is monitored.
- Protein Degradation Rate This method measures how quickly a protein is degraded in a cell. It can be quantified using a variety of methods, for example, without limitation: western blot analysis (with or without the use of protease inhibitors) to assess protein levels over time.
- This disclosure features a system for site-specifically integrating a donor polynucleotide template into a mammalian cell genome at a target DNA sequence, comprising: an attachment site containing gRNA (atgRNA) comprising at least a portion of an at least first integration recognition site; a gene editor polypeptide comprising a DNA binding nickase domain linked to a reverse transcriptase domain capable of incorporating the integration recognition site into the target DNA sequence, any of the engineered integration enzymes described herein (e.g., an engineered integration enzyme comprising an integration enzyme and a stabilization domain (see Section 4.9.1) or an engineered integration enzyme comprising an integration enzyme comprising an amino acid modification that increases stability (see Section 4.9.2)); a donor polynucleotide template linked to a sequence that is an integration cognate of the integration recognition site present in the atgRNA, whereby the gene editor polypeptide site-specifically integrates the integration recognition site into the target DNA sequence, whereby the engineered integration enzyme integrates the
- the first atgRNA comprises: (i) a domain that is capable of guiding the gene editor polypeptide to the target DNA sequence; and (ii) a reverse transcriptase (RT) template that comprises at least a portion of an at least first integration recognition site, whereby the at least portion of the at least first integration recognition site is integrated into the genome of the cell at the target sequence.
- RT reverse transcriptase
- the system also includes a second atgRNA.
- the first atgRNA and the second atgRNA are an at least first pair of atgRNAs, wherein the at least first pair of atgRNAs have domains that are capable of guiding the gene editor polypeptide to the target DNA sequence;
- the first atgRNA further includes a first RT template that comprises at least a portion of an at least first integration recognition site;
- the second atgRNA further includes a second RT template that comprises at least a portion of the first integration recognition site, and the first atgRNA and the second atgRNAs collectively encode the entirety of the first integration recognition site, whereby the least first integration recognition site is integrated into the genome of the cell at the target sequence.
- the engineered integration enzyme upon introducing the system into the cell, enhances integration efficiency of the donor polynucleotide template at the site-specifically integrated integration recognition site as compared to the integration efficiency of a system using a non-engineered integration enzyme to integrate donor polynucleotide template at the site-specifically integrated integration recognition site.
- This disclosure also features a method for site-specifically integrating a donor polynucleotide template into a mammalian cell genome at a target DNA sequence, comprising: incorporating an integration recognition site into the genome by delivering into the cell: an attachment site containing guide RNA (atgRNA) comprising at least a portion of an at least first integration recognition site; and a gene editor polypeptide or polynucleotide encoding a gene editor polypeptide, wherein the gene editor polypeptide comprises a DNA binding nickase domain linked to a reverse transcriptase domain and is capable of incorporating the integration recognition site into the target DNA sequence; and optionally, a nicking gRNA; and integrating the donor polynucleotide template into the genome by delivering into the cell: any of the engineered integration enzymes described herein (e.g., an engineered integration enzyme comprising an integration enzyme and a stabilization domain (see Section 4.9.1) or an engineered integration enzyme comprising an integration enzyme comprising an integration enzyme compris
- the first atgRNA comprises: (i) a domain that is capable of guiding the gene editor polypeptide to the target DNAsequence; and (ii) a reverse transcriptase (RT) template that comprises at least a portion of an at least first integration recognition site, whereby the at least portion of the at least first integration recognition site is integrated into the genome of the cell at the target sequence.
- RT reverse transcriptase
- the method also includes a second atgRNA.
- the first atgRNA and the second atgRNA are an at least first pair of atgRNAs, wherein the at least first pair of atgRNAs have domains that are capable of guiding the gene editor polypeptide to the target DNA sequence;
- the first atgRNA further includes a first RT template that comprises at least a portion of an at least first integration recognition site;
- the second atgRNA further includes a second RT template that comprises at least a portion of the first integration recognition site, and the first atgRNA and the second atgRNAs collectively encode the entirety of the first integration recognition site, whereby the least first integration recognition site is integrated into the genome of the cell at the target sequence.
- the method enhances integration efficiency of the donor polynucleotide template at the site-specifically integrated integration recognition site at the double-stranded target DNA sequence as compared to the integration efficiency of the donor polynucleotide template at the site-specifically integrated recognition site when using a method that does not comprise the engineered integration enzyme.
- This disclosure features methods of delivering (e.g., co-delivery or dual delivery) a system capable of site-specifically integrating a template polynucleotide into the genome of a cell, where the methods include delivering to a (i) gene editor construct and a (ii) template polynucleotide, and (iii) at least a first attachment site-containing guide (atgRNA).
- a system capable of site-specifically integrating a template polynucleotide into the genome of a cell
- the methods include delivering to a (i) gene editor construct and a (ii) template polynucleotide, and (iii) at least a first attachment site-containing guide (atgRNA).
- This disclosure also features a method for delivering a system capable of site-specifically integrating a template polynucleotide into the genome of a cell, where the method includes: delivering a lipid nanoparticle (LNP) comprising a gene editor polynucleotide (e.g., a gene editor polynucleotide construct) and a vector comprising a template polynucleotide and at least a first attachment site-containing guide RNA (atgRNA).
- the first atgRNA comprises (i) a domain that is capable of guiding the prime editor system to a target sequence; and (ii) a reverse transcriptase (RT) template that comprises at least a portion of a first integration recognition site.
- the RT template comprises the entirety of the first integration recognition site. In some embodiments, where the vector comprises a polynucleotide encoding a first atgRNA, the vector also includes a sequence encoding a nicking guide RNA (ngRNA).
- ngRNA nicking guide RNA
- This disclosure also features a method for delivering a system capable of site-specifically integrating a template polynucleotide into the genome of a cell, where the method includes: delivering a lipid nanoparticle (LNP) comprising a gene editor polynucleotide (e.g., a gene editor polynucleotide construct) and a vector comprising a template polynucleotide and a first attachment site-containing guide RNA (atgRNA) and a second attachment site-containing guide RNA (atgRNA).
- LNP lipid nanoparticle
- the first atgRNA and the second atgRNA are an at least first pair of atgRNAs, wherein the at least first pair of atgRNAs have domains that are capable of guiding the prime editor system to a target sequence, the first atgRNA further includes a first RT template that comprises at least a portion of the at least first integration recognition site; and the second atgRNA further includes a second RT template that comprises at least a portion of the first integration recognition site, and the first atgRNA and the second atgRNAs collectively encode the entirety of the first integration recognition site.
- the first atgRNA and second atgRNA include at least a 6bp overlap (e.g., 6bp of complementarity).
- This disclosure also features a method for delivering a system capable of site-specifically integrating a template polynucleotide into the genome of a cell, where the method includes: delivering into a cell a lipid nanoparticle (LNP) comprising: (i) a gene editor polynucleotide (e.g., a gene editor polynucleotide construct), and (ii) a first attachment site-containing guide RNA (atgRNA); and a vector comprising: (i) a template polynucleotide, and (ii) a second atgRNA.
- LNP lipid nanoparticle
- the first atgRNA and the second atgRNA are an at least first pair of atgRNAs, wherein the at least first pair of atgRNAs have domains that are capable of guiding the prime editor system to a target sequence, the first atgRNA further includes a first RT template that comprises at least a portion of the a first integration recognition site; and the second atgRNA further includes a second RT template that comprises at least a portion of the first integration recognition site, and the first atgRNA and the second atgRNAs collectively encode the entirety of the first integration recognition site.
- the first atgRNA and second atgRNA include at least a 6bp overlap (e.g., 6bp of complementarity).
- This disclosure also features a method for delivering a system capable of site-specifically integrating a template polynucleotide into the genome of a cell, where the method includes delivering: a lipid nanoparticle (LNP) comprising: (i) a gene editor polynucleotide (e.g., a gene editor polynucleotide construct), (ii) a first attachment site-containing guide RNA (atgRNA), and (iii) a second atgRNA; and a vector comprising (i) a template polynucleotide.
- LNP lipid nanoparticle
- the first atgRNA and the second atgRNA are an at least first pair of atgRNAs, wherein the at least first pair of atgRNAs have domains that are capable of guiding the prime editor system to a target sequence, the first atgRNA further includes a first RT template that comprises at least a portion of the at least first integration recognition site; and the second atgRNA further includes a second RT template that comprises at least a portion of the at least first integration recognition site, and the first atgRNA and the second atgRNAs collectively encode the entirety of the first integration recognition site.
- the first atgRNA and second atgRNA include at least a 6bp overlap (e.g., 6bp of complementarity).
- This disclosure also features a method for delivering a system capable of site-specifically integrating a template polynucleotide into the genome of a cell, where the method includes delivering: a lipid nanoparticle (LNP) comprising: (i) a gene editor polynucleotide (e.g., a gene editor polynucleotide construct) and (ii) a first attachment site-containing guide RNA (atgRNA); and a vector comprising: (i) a template polynucleotide, and (ii) a nicking atgRNA.
- LNP lipid nanoparticle
- a gene editor polynucleotide e.g., a gene editor polynucleotide construct
- atgRNA first attachment site-containing guide RNA
- a vector comprising: (i) a template polynucleotide, and (ii) a nicking atgRNA.
- the first atgRNA comprises (i) a domain that is capable of guiding the prime editor system to a target sequence; and (ii) a reverse transcriptase (RT) template that comprises at least a portion of a first integration recognition site.
- the RT template comprises the entirety of the first integration recognition site.
- the LNP and the first vector are delivered at least 1 day, at least 2 days, at least 3 days, at least 4 days, at least 5 days, at least 6 days, at least 7 days, at least 2 weeks, at least 3 weeks, at least 4 weeks, at least 5 weeks, at least 6 weeks, at least 7 weeks, or at least 8 weeks apart.
- the LNP and the second vector are delivered a different times on the same day, at least 1 day, at least 2 days, at least 3 days, at least 4 days, at least 5 days, at least 6 days, at least 7 days, at least 2 weeks, at least 3 weeks, at least 4 weeks, at least 5 weeks, at least 6 weeks, at least 7 weeks, or 8 weeks apart.
- the LNP and the first vector are delivered about 6 weeks apart.
- This disclosure also features a method for delivering a system capable of site-specifically integrating a template polynucleotide into the genome of a cell, where the method includes delivering the system in vivo.
- the system is delivered to a fetus or a neonate to site-specifically integrate in vivo a template polynucleotide into the genome of a cell.
- Delivering the system to a fetus or a neonate provides advantages over delivering the system later in life (e.g., after the neonate phase ends), including: (i) fewer number of cells that need to be treated (e.g., in the adult, there are trillions of cells, but in a fetus, there are significantly fewer cells); (ii) developmental benefits: the early stage of development of a fetus or a neonate means that if a genetic disease is treated successfully, the individual could potentially develop normally, with significant reduction or even complete removal of any of the disease manifestations; (iii) preventing disease progression: in certain genetic conditions the physiological damage is irreversible damage and in some instances is exacerbated as the disease progresses, therefore, intervening at the fetal (or neonate) stage, it is possible to prevent or reduce the progression of the disease and potentially prevent any irreversible damage from occurring; (iv) higher cell turnover and cell division rate: in a developing fetus, cells are dividing rapidly as the
- the method includes delivering an LNP and a first vector, the LNP and the first vector are delivered to a cell in vivo.
- the in vivo cells are present in a fetus or a neonate.
- the LNP is delivered between age 0 (day of birth) and age 7 days and the vector is delivered between age 5 weeks and age 7 weeks.
- the LNP is delivered at about at 2 days and the vector is delivered at about age 6 weeks.
- neonus refers to an unborn offspring.
- nonate refers to a newborn infant, which includes the first 90 days of life. When referring to mice, neonate can refer to animals up to 10 days of age.
- This disclosure also features a system capable of site-specifically integrating a template polynucleotide into the genome of a cell, the system comprising: a lipid nanoparticle (LNP) comprising a gene editor polynucleotide (e.g., a gene editor polynucleotide construct) and a vector comprising a template polynucleotide and at least a first attachment site-containing guide RNA (atgRNA).
- the first atgRNA comprises (i) a domain that is capable of guiding the prime editor system to a target sequence; and (ii) a reverse transcriptase (RT) template that comprises at least a portion of a first integration recognition site.
- the RT template comprises the entirety of the first integration recognition site. In some embodiments, where the vector comprises a polynucleotide encoding a first atgRNA, the vector also includes a sequence encoding a nicking guide RNA (ngRNA).
- ngRNA nicking guide RNA
- This disclosure also features a system capable of site-specifically integrating a template polynucleotide into the genome of a cell, the system comprising: a lipid nanoparticle (LNP) comprising a gene editor polynucleotide (e.g., a gene editor polynucleotide construct) and a vector comprising a template polynucleotide and a first attachment site-containing guide RNA (atgRNA) and a second attachment site-containing guide RNA (atgRNA).
- LNP lipid nanoparticle
- the first atgRNA and the second atgRNA are an at least first pair of atgRNAs, wherein the at least first pair of atgRNAs have domains that are capable of guiding the prime editor system to a target sequence, the first atgRNA further includes a first RT template that comprises at least a portion of the a first integration recognition site; and the second atgRNA further includes a second RT template that comprises at least a portion of the first integration recognition site, and the first atgRNA and the second atgRNAs collectively encode the entirety of the first integration recognition site.
- the first atgRNA and second atgRNA include at least a 6bp overlap.
- This disclosure also features a system capable of site-specifically integrating a template polynucleotide into the genome of a cell, the system comprising: a lipid nanoparticle (LNP) comprising: (i) a gene editor polynucleotide (e.g., a gene editor polynucleotide construct), and (ii) a first attachment site-containing guide RNA (atgRNA); and a vector comprising: (i) a template polynucleotide, and (ii) a second atgRNA.
- LNP lipid nanoparticle
- a gene editor polynucleotide e.g., a gene editor polynucleotide construct
- atgRNA first attachment site-containing guide RNA
- a vector comprising: (i) a template polynucleotide, and (ii) a second atgRNA.
- the first atgRNA and the second atgRNA are an at least first pair of atgRNAs, wherein the at least first pair of atgRNAs have domains that are capable of guiding the prime editor system to a target sequence, the first atgRNA further includes a first RT template that comprises at least a portion of the a first integration recognition site; and the second atgRNA further includes a second RT template that comprises at least a portion of the first integration recognition site, and the first atgRNA and the second atgRNAs collectively encode the entirety of the first integration recognition site.
- the first atgRNA and second atgRNA include at least a 6bp overlap.
- This disclosure also features a system capable of site-specifically integrating a template polynucleotide into the genome of a cell, the system comprising: co-delivering: a lipid nanoparticle (LNP) comprising: (i) a gene editor polynucleotide (e.g., a gene editor polynucleotide construct), (ii) a first attachment site-containing guide RNA (atgRNA), and (iii) a second atgRNA; and a vector comprising (i) a template polynucleotide.
- LNP lipid nanoparticle
- the first atgRNA and the second atgRNA are an at least first pair of atgRNAs, wherein the at least first pair of atgRNAs have domains that are capable of guiding the prime editor system to a target sequence, the first atgRNA further includes a first RT template that comprises at least a portion of the a first integration recognition site; and the second atgRNA further includes a second RT template that comprises at least a portion of the first integration recognition site, and the first atgRNA and the second atgRNAs collectively encode the entirety of the first integration recognition site.
- the first atgRNA and second atgRNA include at least a 6bp overlap.
- This disclosure also features a system capable of site-specifically integrating a template polynucleotide into the genome of a cell, the system comprising: a lipid nanoparticle (LNP) comprising: (i) a gene editor polynucleotide (e.g., a gene editor polynucleotide construct) and (ii) a first attachment site-containing guide RNA (atgRNA); and a vector comprising: (i) a template polynucleotide, and (ii) a nicking atgRNA.
- LNP lipid nanoparticle
- a gene editor polynucleotide e.g., a gene editor polynucleotide construct
- atgRNA first attachment site-containing guide RNA
- a vector comprising: (i) a template polynucleotide, and (ii) a nicking atgRNA.
- the first atgRNA comprises (i) a domain that is capable of guiding the prime editor system to a target sequence; and (ii) a reverse transcriptase (RT) template that comprises at least a portion of a first integration recognition site.
- the RT template comprises the entirety of the first integration recognition site.
- the LNP comprising a gene editor polynucleotide construct is capable delivering to a cell cytoplasm the gene editor polynucleotide construct. In some embodiments, the LNP comprising a gene editor polynucleotide construct is capable delivering to a cell nucleus the gene editor polynucleotide construct. In some embodiments, the LNP comprises a gene editor protein and associated guide nucleic acids. In some embodiments, the LNP comprises a gene editor protein and associated guide nucleic acids that are capable of localizing to cell nucleus.
- a gene editor polynucleotide construct is delivered to a cell by a fusosome. In some embodiments, a gene editor polynucleotide construct is delivered to a cell cytoplasm by a fusosome. In some embodiments, the fusosome comprises a gene editor protein and associated guide nucleic acids.
- a gene editor polynucleotide construct is delivered to a cell by an exosome.
- a gene editor polynucleotide construct is delivered to a cell cytoplasm by an exosome.
- the exosome comprises a gene editor protein and associated guide nucleic acids.
- the prime editor or Gene Writer protein fusion is incorporated (i.e., packaged) into LNP as protein.
- associated atgRNA and optional ngRNAs may be co-packaged with gene editor proteins in LNP.
- the gene editor polynucleotide construct comprises (a) a polynucleotide sequence encoding a prime editor fusion protein or a Gene WriterTM protein, (b) a polynucleotide sequence encoding an attachment site-containing guide RNA (atgRNA), (c) optionally, a polynucleotide sequence encoding a nickase guide RNA (ngRNA), (d) a polynucleotide sequence encoding an integrase, (e) and optionally, a polynucleotide sequence encoding a recombinase.
- atgRNA attachment site-containing guide RNA
- ngRNA nickase guide RNA
- a polynucleotide sequence encoding an integrase e
- a polynucleotide sequence encoding a recombinase optionally, a polynucleotide sequence encoding a recombina
- the prime editor or Gene Writer protein fusion is expressed as a split construct.
- the split construct in reconstituted in a cell.
- the split construct can be fused or ligated via intein protein splicing.
- the split construct can be reconstituted via protein-protein inter-molecular bonding and/or interactions.
- the split construct can be reconstituted via chemical, biological, or environmental induced oligomerization.
- the split construct can be adapted into one or more nucleic acid constructs described herein.
- the systems described include a gene editor polynucleotide that is delivered to a cell using the methods described herein.
- the gene editor polynucleotide is delivered as a polynucleotide (e.g., an mRNA).
- the gene editor polynucleotide is delivered as a protein.
- the gene editor polynucleotide or protein is packaged, and thereby vectorized, within a lipid nanoparticle (LNP).
- LNP lipid nanoparticle
- the gene editor polynucleotide or protein is packaged in a LNP and is codelivered with a template polynucleotide (i.e., nucleic acid “cargo” or nucleic acid “payload”) packaged into a separate vector (e.g., a viral vector (e.g., an AAV or adenovirus)) or a second lipid nanoparticle (LNP).
- a template polynucleotide i.e., nucleic acid “cargo” or nucleic acid “payload” packaged into a separate vector (e.g., a viral vector (e.g., an AAV or adenovirus)) or a second lipid nanoparticle (LNP).
- a separate vector e.g., a viral vector (e.g., an AAV or adenovirus)
- LNP second lipid nanoparticle
- the gene editor polynucleotide is delivered to the cells as a polynucleotide.
- the gene editor polynucleotide is delivered to the cells as an mRNA encoding the gene editor polynucleotide (e.g., the gene editor protein or the prime editor system).
- the mRNA comprises one or more modified uridines.
- the mRNA comprises a sequence where each of the uridines is a modified uridine.
- the mRNA is uridine depleted.
- the mRNA encoding the nickase comprises one or more modified uridines.
- the mRNA encoding the reverse transcriptase comprises one or more modified uridines. In some embodiments, the mRNA encoding the nickase comprises one or more modified uridines, and the mRNA encoding the reverse transcriptase comprises one or more modified uridines. In some embodiments, where the integrase is encoded in an mRNA, the mRNA comprises modified uridines. In some embodiments, a modified uridine is a Nl-Methylpseudouridine-5’ -Triphosphate. In some embodiments, a modified uridine is a pseudouridine. In some embodiments, the mRNA comprises a 5’ cap. In some embodiments, the 5’ cap comprises a molecular formula of C32H43N15O24P4 (free acid).
- the gene editor polynucleotide (e.g., a gene editor polynucleotide construct) comprises a polynucleotide sequence encoding a primer editor system (e.g., any of the prime editor systems described herein).
- the prime editor system comprises a nucleotide sequence encoding a nickase (e.g., any of the Cas proteins or variants thereof (e.g., nickases) and nickases described herein, see Tables 4-8) and a nucleotide sequence encoding a reverse transcriptase (e.g., any of the reverse transcriptases described herein).
- the nucleotide sequence encoding the nickase and the nucleotide sequence encoding the reverse transcriptase are positioned in the construct such that when expressed the nickase is linked to the reverse transcriptase.
- the nickase is linked to the reverse transcriptase by in-frame fusion.
- the nickase is linked to the reverse transcriptase by a linker.
- the linker is a peptide fused in-frame between the nickase and reverse transcriptase.
- the gene editor polynucleotide (e.g., a gene editor polynucleotide construct) further comprises a polynucleotide sequence encoding at least a first integrase (e.g., any of the integrases described herein, e.g., as described in Table 10 and also in Yamall et al., Nat. BiotechnoL, 2022, doi.org/10.1038/s41587-022-01527-4 and Durrant et al., Nat. BiotechnoL, 2022, doi.org/10.1038/s41587-022-01494-w, each of which are herein incorporated by reference in their entireties).
- the linked nickase-reverse transcriptase are further linked to the first integrase.
- the gene editor polynucleotide construct further comprises a polynucleotide sequence encoding at least a first recombinase (e.g., any of the recombinases described herein).
- nucleotide sequence encoding the nickase and the nucleotide sequence encoding the reverse transcriptase are positioned in a construct such that when expressed the nickase is not linked to the reverse transcriptase.
- a sequence encoding a self-cleaving peptide e.g., a P2A
- a sequence encoding a self-cleaving peptide is positioned between the nucleotide sequence encoding the nickase and the nucleotide sequence encoding the reverse transcriptase such that when expressed the nickase is not linked to the reverse transcriptase (see, e.g., FIG. 34).
- nucleotide sequence encoding the nickase and the nucleotide sequence encoding the reverse transcriptase are on separate polynucleotides.
- the nickase and/or the reverse transcriptase can be engineered such that when expressed they are linked to a binding domain, where the binding domain is part of a binding pair that enables recruitment of the reverse transcriptase to the genomic location of the nickase (e.g., nCas9).
- a binding pair can be any pair of molecules wherein upon forming the binding pair the binding domains enable recruitment of the reverse transcriptase to the genomic location of the nickase. Once in sufficient proximity with each other the binding domains can dimerize. This dimerization (or other type of interaction) brings the nickase and RT into proximity with each other, thereby enhancing the reverse transcription of the RT template in the atgRNA.
- Non-limiting examples of binding pairs that can aid recruitment of the RT to the genomic location of the nickase include without limitation: dimerizing leucine zippers, an antigen and a corresponding antigen binding domains (e.g., antibody), RNA aptamers and corresponding RNA aptamer binding proteins, affinity tags and corresponding affinity tag binding proteins.
- dimerizing leucine zippers that can serve as a binding pair includes two binding domains: a Leucine Zip LZ1 domain having a sequence with at least 80%, 85%, 90%, 95%, or 99% sequence identity to a sequence of SEQ ID NO: 568 and a Leucine Zip LZ2 domain having a sequence with at least 80%, 85%, 90%, 95%, or 99% sequence identity to a sequence of SEQ ID NO: 569.
- the Leucine Zip LZ1 domain is positioned either N- terminally or C-terminally to the nickase and the Leucine Zip LZ2 is positioned either N-terminally or C-terminally to the RT.
- the Leucine Zip LZ1 domain is positioned either N-terminally or C-terminally to the RT and the Leucine Zip LZ2 domain is positioned either N- terminally or C-terminally to the Cas9.
- the system and methods described herein include a construct that encodes a polypeptide having a sequence with at least 80%, 85%, 90%, 95%, or 99% sequence identity to a sequence of SEQ ID NO: 576 (a Leucine Zip LZ1 domain positioned C-terminally to the nickase), and a construct that encodes a polypeptide having a sequence with at least 80%, 85%, 90%, 95%, or 99% sequence identity to a sequence of SEQ ID NO: 577 (a Leucine Zip LZ2 domain positioned N-terminally to the RT).
- the system and methods described herein include a construct that encodes a polypeptide having a sequence with at least 80%, 85%, 90%, 95%, or 99% sequence identity to a sequence of SEQ ID NO: 578 (a Leucine Zip LZ1 domain positioned N-terminally to the nickase), and a construct that encodes a polypeptide having a sequence with at least 80%, 85%, 90%, 95%, or 99% sequence identity to a sequence of SEQ ID NO: 577 (a Leucine Zip LZ2 domain positioned N-terminally to the RT).
- dimerizing leucine zippers that can serve as a binding pair includes two binding domains: a EE1234L peptide having a sequence with at least 80%, 85%, 90%, 95%, or 99% sequence identity to a sequence of SEQ ID NO: 570 and a RR1234L peptide having a sequence with at least 80%, 85%, 90%, 95%, or 99% sequence identity to a sequence of SEQ ID NO: 571.
- the EE1234L peptide is positioned either N-terminally or C-terminally to the nickase and the RR1234L peptide is positioned either N-terminally or C-terminally to the RT.
- the EE1234L peptide is positioned either N-terminally or C-terminally to the RT and the RR1234L peptide is positioned either N-terminally or C-terminally to the Cas9.
- the system and methods described herein include a construct that encodes a polypeptide having a sequence with at least 80%, 85%, 90%, 95%, or 99% sequence identity to a sequence of SEQ ID NO: 579 (a EE1234L peptide positioned N-terminally to the nickase), and a construct that encodes a polypeptide having a sequence with at least 80%, 85%, 90%, 95%, or 99% sequence identity to a sequence of SEQ ID NO: 580 (a RR1234L peptide positioned N-terminally to the RT).
- an affinity tag is a SpyTag and the corresponding affinity tag binding protein is SpyCatcher, where upon binding of the SpyTag with the SpyCatcher the fusion protein is a covalently stabilized multi-protein complex.
- the affinity tag e.g., SpyTag
- the affinity tag binding protein e.g.,. SpyCatcher
- the affinity tag binding protein e.g., SpyCatcher
- the affinity tag binding protein is positioned either N-terminally or C-terminally to the nickase.
- an antigen and corresponding antigen binding domain includes an antigen such as a peptide present within a tag or a tag itself and an antigen binding domain that binds specifically to the antigen.
- an antigen is a GCN4 peptide within a SunTag and the antigen binding domain is an anti-GCN4 scFv.
- the SunTag (GCN4 peptide) is positioned either N-terminally or C-terminally to the nickase and the anti-GCN4 antigen binding domain is positioned either N-terminally or C-terminally to the RT.
- the SunTag (GCN4 peptide) is positioned either N-terminally or C-terminally to the RT and the anti-GCN4 antigen binding domain is positioned either N-terminally or C-terminally to the nickase.
- the system and methods described herein include a construct that encodes a polypeptide having a sequence with at least 80%, 85%, 90%, 95%, or 99% sequence identity to a sequence of SEQ ID NO: 574 (SunTag (GCN4 peptide) positioned C-terminally to the nickase), and a construct that encodes a polypeptide having a sequence with at least 80%, 85%, 90%, 95%, or 99% sequence identity to a sequence of SEQ ID NO: 575 (anti-GCN4 antigen binding domain positioned N-terminally to the RT).
- a construct includes a nucleotide sequence encoding a self-cleaving peptide (e.g., a P2A) positioned between the nucleotide sequence encoding the nickase and the nucleotide sequence encoding the RT.
- a nucleotide sequence encoding a self-cleaving peptide e.g., a P2A
- the nucleotide sequence encoding the RT is such that when expressed the nickase is not linked to the reverse transcriptase.
- the construct includes the nucleotide sequences positioned 5 ’-3’: nucleotide sequence encoding the nickase, nucleotide sequence encoding the self-cleaving peptide (e.g., a P2A), and nucleotide sequence encoding the RT. In one embodiment, the construct includes the nucleotide sequences positioned 5 ’-3’: nucleotide sequence encoding the RT, nucleotide sequence encoding the selfcleaving peptide (e.g., a P2A), and nucleotide sequence encoding the nickase.
- the construct that includes a nucleotide sequence encoding a selfcleaving peptide (e.g., a P2A) positioned between the nucleotide sequence encoding the nickase and the nucleotide sequence encoding the RT has an amino acid sequence with at least 80%, 85%, 90%, 95%, or 99% sequence identity to a sequence of SEQ ID NO: 581 or 582 (see FIG. 34).
- This disclosure also features systems and methods that include supplementing with reverse transcriptase.
- the system and methods include supplementation with a polynucleotide encoding a supplemental reverse transcriptase (i.e., a separate polynucleotide from the gene editor polynucleotide, which includes a polynucleotide encoding a reverse transcriptase).
- Systems and methods that include supplementation with reverse transcriptase increase integration efficiency of the at least first integration recognition site into the genome of the cell when compared to systems and methods that do not include supplementation with reverse transcriptase.
- These systems and methods refer to the supplemental or supplemental reverse transcriptase molecule as “supplementing” or “supplementation” with reverse transcriptase.
- the supplemental reverse transcriptase is the same as the reverse transcriptase that is included in the gene editor polynucleotide. In some embodiments, the supplemental reverse transcriptase is different from the reverse transcriptase that is included in the gene editor polynucleotide.
- the polynucleotide encoding the supplemental reverse transcriptase is an mRNA.
- the supplemental reverse transcriptase is delivered concurrently with the one or more LNP, one or more atgRNA, and the template polynucleotide. In some embodiments, the supplemental reverse transcriptase is delivered to the cell prior to delivering the one or more LNP, one or more atgRNA, and the template polynucleotide to the cell. In some embodiments, the supplemental reverse transcriptase is delivered to the cell after delivering the one or more LNP, one or more atgRNA, and the template polynucleotide to the cell.
- the method includes a ratio of 1 : 1, 1:2, 1 :3, 1 :4, 1 :5, 1 :6, 1 :7, 1 :8, 1 :9, or 1 : 10 of gene editor polynucleotide to polynucleotide encoding the supplemental reverse transcriptase.
- Also provided herein are methods of increasing integration efficiency of the at least first integration recognition site into the genome of the cell where the method comprises delivering to the cell one or more LNPs, one or more vectors, one or more template polynucleotides, where the gene editor polynucleotide comprises a ratio of a ratio of 1:1, 1:2, 1:3, 1:4, 1:5, 1:6, 1:7, 1:8, 1:9, 1:10 of the nucleotide sequence encoding the nickase to the nucleotide sequence encoding the reverse transcriptase, and where the increase in integration efficiency is in comparison to methods that do not include supplementation with a polynucleotide encoding RT.
- the system and methods described herein include supplementation with a polynucleotide encoding a reverse transcriptase, where the polynucleotide is in addition to the gene editor polynucleotide that also includes a polynucleotide encoding a reverse transcriptase.
- a method for delivering a system capable of site-specifically integrating a template polynucleotide into the genome of a cell includes delivering one or more LNPs and one or more vectors, and also includes delivering a separate polynucleotide encoding a supplemental reverse transcriptase (RT).
- RT supplemental reverse transcriptase
- a method for delivering a system capable of site-specifically integrating a template polynucleotide into the genome of a cell comprises delivering the gene editor polynucleotide (e.g., a polynucleotide encoding a nickase and a reverse transcriptase either linked or not linked), one or more atgRNA, a template polynucleotide, and a polynucleotide encoding a supplemental reverse transcriptase.
- the gene editor polynucleotide e.g., a polynucleotide encoding a nickase and a reverse transcriptase either linked or not linked
- the method includes delivering into the cell a ratio of gene editor polynucleotide to polynucleotide encoding the supplemental RT of 1:1, 1:2, 1:3, 1:4, 1:5, 1:6, 1:7, 1:8, 1:9, 1:10, 1:11, 1:12, 1:13, 1:14, 1:15, 1:16, 1:17, 1:18, 1:19, 1:20, 1:21, 1:22, 1:23, 1:24, 1:25, 1:26, 1:27, 1:28, 1:29, 1:30, 1:32, 1:34, 1:36, 1:38, or 1:40 or more.
- the method includes delivering into the cell a gene editor polynucleotide and a polynucleotide encoding the supplemental RT, where the polynucleotide encoding the supplemental RT is delivered at 1.5 times (1.5X), 2X, 3X, 4X, 5X, 6X, 7X, 8X, 9X, 10X, 20X, 30X, 40X, 50X, 60X, 70X, 80X, 90X, or 100X or more the amount of the gene editor polynucleotide.
- the method includes delivering into the cell a ratio of polynucleotide encoding the nickase to polynucleotide encoding the RT of 1:1, 1:2, 1:3, 1:4, 1:5, 1:6, 1:7, 1:8, 1:9, 1:10, 1:11, 1:12, 1:13, 1:14, 1:15, 1:16, 1:17, 1:18, 1:19, 1:20, 1:21, 1:22, 1:23, 1:24, 1:25, 1:26, 1:27, 1:28, 1:29, 1:30, 1:32, 1:34, 1:36, 1:38, or 1:40 or more.
- the method includes delivering into the cell a polynucleotide encoding the nickase to polynucleotide encoding the RT, where the polynucleotide encoding the RT is delivered at 1.5 times (1.5X), 2X, 3X, 4X, 5X, 6X, 7X, 8X, 9X, 10X, 20X, 30X, 40X, 50X, 60X, 70X, 80X, 90X, or 100X or more the amount of the nickase polynucleotide.
- the systems and methods that include delivering of the gene editor polynucleotide supplemented with a polynucleotide encoding a supplemental RT increases integration efficiency of the at least first integration recognition site into the genome of the cell when compared to systems and methods that include delivering to the cell a gene editor polynucleotide not supplemented with a polynucleotide encoding supplemental reverse transcriptase.
- the system and methods described herein include supplementation with a polynucleotide encoding a reverse transcriptase, where the system includes a gene editor polynucleotide split over two polynucleotides: a first polynucleotide encoding the nickase and a second polynucleotide encoding the reverse transcriptase.
- the first and second polynucleotides can be delivered to the cell at a ratio of 1 : 1 and supplementation is achieved by adding a polynucleotide encoding a supplemental reverse transcriptase.
- the supplementation comprises adding the polynucleotide encoding the supplemental reverse transcriptase at a ratio of 1 : 1, 1 :2, 1 :3, 1 :4, 1 :5, 1 :6, 1 :7, 1 :8, 1 :9, 1 : 10, 1 : 11, 1 : 12, 1 : 13, 1 : 14, 1 : 15, 1 : 16, 1 : 17, 1 : 18, 1 : 19, 1 :20, 1 :21, 1 :22, 1 :23, 1 :24, 1 :25, 1 :26, 1 :27, 1 :28, 1 :29, 1 :30, 1 :32, 1 :34, 1 :36, 1 :38, or 1 :40 (gene editor polynucleotide (e.g., the first and second polynucleotides delivered to the cell at a ratio of 1 : 1) to the polynucleotide encoding the supplemental reverse transcriptase).
- a method of delivering to the cell the one or more atgRNA and the gene editor polynucleotide (where the nickase and the RT are encoded on different polynucleotides and delivered at a ratio 1 : 1 (nickase to RT)) and supplementing with a polynucleotide encoding a supplemental RT increases integration efficiency of the at least first integration recognition site into the genome of the cell when compared to methods that do not include supplementing with a polynucleotide encoding a supplemental RT.
- the system and methods described herein include supplementation with a polynucleotide encoding a reverse transcriptase, where the system includes a gene editor polynucleotide split over two polynucleotides: a first polynucleotide encoding the nickase and a second polynucleotide encoding the reverse transcriptase, and supplementation is achieved by increasing the amount of the second polynucleotide encoding the reverse transcriptase.
- supplementation of a supplemental reverse transcriptase refers to delivering a greater than 1 : 1 ratio of nickase to reverse transcriptase (e.g., a 1 :2 or greater ratio).
- the gene editor polynucleotide delivered to the cell in a ratio 1 : 1, 1 :2, 1 :3, 1 :4, 1 :5, 1 :6, 1 :7, 1 :8, 1 :9, 1 : 10, 1 : 11, 1 : 12, 1 : 13, 1 : 14, 1 : 15, 1 : 16, 1 : 17, 1 : 18, 1 : 19, 1 :20, 1 :21, 1 :22, 1 :23, 1 :24, 1 :25, 1 :26, 1 :27, 1 :28, 1 :29, 1 :30, 1 :32, 1 :34, 1 :36, 1 :38, or 1 :40 of polynucleotide sequence encoding the nickase to the polynucleotide sequence encoding the reverse transcriptase.
- a method of delivering to the cell the one or more atgRNA and the gene editor polynucleotide where the nickase and the RT are encoded on different polynucleotides and delivered at a ratio greater than 1 : 1 (nickase to RT) increases integration efficiency of the at least first integration recognition site into the genome of the cell when compared to methods comprising delivering to the cell a gene editor polynucleotide at a ratio of 1 : 1 (nickase to RT).
- the system and methods described herein include supplementing with reverse transcriptase, where in supplementing comprises using controllable expression (e.g., inducible promoters, strong promoters) of a polynucleotide encoding a reverse transcriptase.
- controllable expression e.g., inducible promoters, strong promoters
- the polynucleotide encoding the nickase and the polynucleotide encoding the RT are on a dual promoter vector, where each polynucleotide is controlled by a different promoter.
- the polynucleotide encoding the RT is operably linked to a stronger promoter than the polynucleotide encoding the nickase, whereby the RT is expressed at higher levels than the nickase.
- the polynucleotide encoding the RT is operably linked to an inducible promoter, whereby inducing expression of the RT results in higher levels of the RT compared to the nickase.
- the polynucleotide encoding the nCas9 is operably linked to an inducible promoter, whereby inducing expressing of the nCas9 results in lower levels of the nCas9 than the RT.
- the systems and methods described herein include a vector that is capable of co-delivering a template polynucleotide, one or more attachment site-containing gRNA, one or more integrases, one or more recombinases, a gene editor polynucleotide, one or more integration recognition sites, one or more recombinase recognition sites, or a combination thereof.
- Non-limiting examples of vectors that can be used in the methods or systems described herein include the vectors described in FIGs. 3-6.
- the vector includes a polynucleotide sequence encoding an attachment site-containing guide RNA (atgRNA).
- the polynucleotide sequence encoding the attachment site-containing guide RNA (atgRNA) is operably linked to a regulatory element (e.g., a U6 promoter) that is capable of driving expression of the atgRNA.
- the atgRNA comprises (i) a domain that is capable of guiding the prime editor system to a target sequence; and (ii) a reverse transcriptase (RT) template that comprises at least a portion of a first integration recognition site.
- the RT template comprises the entirety of the first integration recognition site.
- the vector or the LNP includes a polynucleotide sequence encoding a nicking gRNA.
- the vector includes a polynucleotide sequence encoding a first attachment site-containing guide RNA (atgRNA) and a polynucleotide sequence encoding a second attachment site-containing guide RNA (atgRNA).
- atgRNA first attachment site-containing guide RNA
- atgRNA second attachment site-containing guide RNA
- the first atgRNA and the second atgRNA are an at least first pair of atgRNAs, wherein the at least first pair of atgRNAs have domains that are capable of guiding the prime editor system to a target sequence, the first atgRNA further includes a first RT template that comprises at least a portion of the a first integration recognition site; the second atgRNA further includes a second RT template that comprises at least a portion of the first integration recognition site, and the first atgRNA and the second atgRNAs collectively encode the entirety of the first integration recognition site.
- the first atgRNA and second atgRNA include at least a 6bp overlap.
- the vector includes a template polynucleotide and a sequence that is an integration cognate of an integration recognition site site-specifically incorporated into the genome of a cell.
- the vector includes a template polynucleotide and a second integration recognition site that is a cognate pair with the first integration recognition site site- specifically incorporated into the genome of the cell.
- the sequence that is an integration cognate e.g., a second integration recognition site
- the vector comprising a template polynucleotide is a recombinant adenovirus, a helper dependent adenovirus, an AAV, a lentivirus, an HSV, an annelovirus, a retrovirus, a DoggyboneTM DNA (dbDNA), a minicircle, a plasmid, a miniDNA, an exosome, a fusosome, or an nanoplasmid.
- the vector is capable of localizing to the nucleus.
- the template polynucleotide is delivered to the cytoplasm and localizes to the nucleus. In certain embodiments, the template polynucleotide is delivered to the cytoplasm by LNP. In certain embodiments, the donor template polynucleotide construct comprises a recognition sequence that is recognized by a DNA binding protein (DNA binding domain) or a transcription factor binding domain. In certain embodiments, the donor template polynucleotide construct is delivered to the nucleus by an integrase or recombinase.
- the template polynucleotide is delivered to the mitochondria.
- the donor template polynucleotide construct comprises a mitochondria targeting sequence.
- the vector comprising a template polynucleotide is AAV.
- the AAV contains a 5’ inverted terminal repeat (ITR).
- the AAV contains a 3’ inverted terminal repeat (ITR).
- the AAV contains a 5’ and a 3’ ITR.
- the 5’ and 3’ ITR are not derived from the same serotype of virus.
- the ITRs are derived from adenovirus, AAV2, and/or AAV5.
- the vector comprising a template polynucleotide is single stranded AAV (ssAAV).
- the vector comprising a donor template polynucleotide construct is self-complementary AAV (scAAV).
- the template polynucleotide is capable of being integrated into a genomic locus that contains an integrase target recognition site or a recombinase target recognition site.
- the template polynucleotide comprises at least one of the following: a gene, a gene fragment, an expression cassette, a logic gate system, or any combination thereof. In some embodiments, the template polynucleotide comprises at least one intron or exon.
- the template polynucleotide further comprises at least one integrase target recognition site or a recombinase target integrase site.
- at least one integrase target recognition site or a recombinase target integrase site is placed within the donor template vector inverted terminal repeat.
- the delivery system (e.g., co-delivery system) includes a vector having a sub-sequence that is capable of self-circularizing to form a self-circular nucleic acid.
- the vector comprises a physical portion or region of the vector that is capable of self-circularizing to form a circular construct.
- sub -sequence refers to a portion of the vector that is capable of self-circularizing, where the sub-sequence is flanked by integration recognition sites or recombinase recognition sites positioned to enable selfcircularization.
- self-circular nucleic acid refers to a double-stranded, circular nucleic acid construct produced as a result of recombination of a cognate pair of integrase or recombinase recognition sites present on the vector. Recombination occurs when the vector is contacted with an integrase or a recombinase under conditions that allow for recombination of the cognate pair of integrase or recombinase recognition sites.
- the sub-sequence of the vector includes a first recombinase recognition site and a second recombinase recognition site, wherein the first and second recombinase recognition sites are capable of being recombined by a recombinase.
- the sub-sequence of the vector includes a first recombinase recognition site, a second recombinase recognition site, and a second integration recognition site (e.g., the second integration recognition site is a cognate pair of the first integration recognition site), where the first and second recombinase recognition sites flank the integration recognition site.
- the first recombinase recognition site, the second recombinase recognition, and a recombinase enable the self-circularizing and formation of the circular construct.
- the sub-sequence of the vector includes a third integration recognition site and a fourth integration recognition site, wherein the third and fourth integration recognition sites are a cognate pair.
- the subsequence of the vector includes the second integration recognition site, the third integration recognition site, the fourth integration recognition site, where the third and fourth integration recognition sites flank the second integration recognition site (where the second integration recognition site is a cognate pair of the first integration recognition site).
- the third integration recognition site, the fourth integration recognition site, and an integrase enable self -circularization and formation of the circular construct.
- the third integration recognition site and/or the fourth integration recognition sites cannot recombine with the first integration recognition site and/or the second integration recognition site due, in part, to having different central dinucleotides than the first and second integration recognition sites.
- each integration recognition site or each pair of integration recognition is capable of being recognized by a different integrase. In some embodiments where the subsequence includes three or more integration recognition sites, each integration recognition site or each pair of integration recognition comprises a different central dinucleotide.
- self-circularizing is mediated at the integration recognition sites or recombinase recognition sites. In some embodiments, the self-circularizing is mediated by an integrase or a recombinase.
- the self-circular nucleic acid comprising the second integration recognition site upon introducing the vector into a cell and after selfcircularizing to form the self-circular nucleic acid, the self-circular nucleic acid comprising the second integration recognition site is capable of being integrated into the cell’s genome at the target sequence that contains the first integration recognition site.
- the self-circular nucleic acid comprises one or more additional integration recognition sites that enable integration of an additional nucleic acid cargo.
- the additional nucleic acid cargo includes a sequence that is a cognate pair with one or more of the additional integration recognition sites in the selfcircular nucleic acid.
- integration of the self-circular nucleic acid into the genome of a cell results in integration of the one or more additional integration recognition sites into the genome along with the nucleic acid cargo.
- the integrated one or more additional integration recognition sites serve as an integration recognition site (beacon) for placing the additional nucleic acid cargo.
- the additional nucleic acid cargo is integrated into the cell’s genome.
- the self-circularized nucleic acid comprises a DNA cargo
- the DNA cargo is a gene or gene fragment.
- the DNA cargo is an expression cassette.
- the DNA cargo is a logic gate or logic gate system.
- the logic gate or logic gate system may be DNA based, RNA based, protein based, or a mix of DNA, RNA, and protein.
- the nucleic acid cargo is a genetic, protein, or peptide tag and/or barcode.
- the system or methods described herein include a second vector.
- the gene editor polynucleotide encodes a prime editor system comprising a nickase (e.g., any of the Cas proteins or variants thereof (e.g., nickases) and nickases described herein, see Tables 4-8) and a reverse transcriptase (e.g., any of the reverse transcriptase described herein)
- the second vector comprises a polynucleotide sequence encoding an integrase (e.g., any of the integrases described herein, e.g., as described in Table 10 and also in Yamall et al., Nat.
- the second vector comprises a polynucleotide sequence encoding at least a first recombinase.
- the second vector comprises a polynucleotide sequence encoding at least a first recombinase. In some embodiments, where the gene editor polynucleotide encodes a prime editor system comprising a nickase, a reverse transcriptase, and an integrase, the second vector comprises a polynucleotide sequence encoding at least a second integrase.
- the second vector includes a template polynucleotide and a sequence that is an integration cognate of an integration recognition site site-specifically incorporated into the genome of a cell.
- the second vector includes a template polynucleotide and a second integration recognition site that is a cognate pair with the first integration recognition site site-specifically incorporated into the genome of the cell.
- the sequence that is an integration cognate e.g., a second integration recognition site
- the second vector is a vector selected from: adenovirus, AAV, lentivirus, HSV, annelovirus, retrovirus, DoggyboneTM DNA (dbDNA), minicircle, plasmid, miniDNA, exosome, fusosome, or nanoplasmid.
- the polynucleotide sequence encoding the prime editor system is encoded on at least two different vectors.
- a first vector comprises a polynucleotide sequence encoding a nickase and a second vector comprises a polynucleotide sequence encoding a reverse transcriptase. In such cases, the first vector and second are delivered concurrently.
- the polynucleotide sequence(s) encoding the prime editor system is encoded on at least two (non-contiguous) polynucleotide sequences.
- a first polynucleotide sequence encodes a nickase and a second polynucleotide sequence encodes a reverse transcriptase.
- the first vector and second are delivered concurrently (e.g., in a first LNP).
- LNPs Split Lipid Nanoparticles
- the method includes co-delivering to a cell a first gene editor polynucleotide construct and a first attachment site-containing guide RNA (atgRNA) are packaged, and thereby vectorized, within the first LNP, and a second gene editor polynucleotide construct and a second attachment site containing guide RNR (atgRNA) are packaged, and thereby vectorized, within the second LNP, where the first atgRNA and the second atgRNA are an at least first pair of atgRNA.
- the at least first pair of atgRNAs comprise domains that are capable of guiding the prime editor system to a target sequence.
- the first atgRNA further includes a first RT template that comprises at least a portion of a first integration recognition site.
- the second atgRNA further includes a second RT template that comprises at least a portion of the first integration recognition site.
- the first atgRNA and the second atgRNAs collectively encode the entirety of the first integration recognition site.
- the first atgRNA and second atgRNA include at least a 6bp overlap.
- the method includes delivering a first LNP (e.g., a first LNP comprising a first gene editor polynucleotide construct and a first atgRNA) and a second LNP (e.g., a second LNP comprising a second gene editor polynucleotide construct and a second atgRNA), the first LNP and the second LNP are mixed prior to delivering to a cell.
- a first LNP e.g., a first LNP comprising a first gene editor polynucleotide construct and a first atgRNA
- a second LNP e.g., a second LNP comprising a second gene editor polynucleotide construct and a second atgRNA
- the first LNP and the second LNP are mixed at a ratio of first LNP to second LNP of l:10, 1 :9, 1 :8, 1 :7, 1 :6, 1 :5, 1 :4, 1 :3, 1 :2, 1 : 1, 1 :0.75, 0.75: 1, 2: 1, 3: 1, 4: 1, 5: 1, 6: 1, 7: 1, 8: 1, 9: 1, or 10: 1.
- the first LNP and the second LNP are mixed at a ratio of 1 : 1.
- a first LNP comprising a first gene editor polynucleotide construct and a first attachment site-containing guide RNA (atgRNAl) comprises a ratio of ratio of gene editor polynucleotide construct (e.g., mRNA) to atgRNAl of 1 : 10, 1 :9, 1 :8, 1 :7, 1 :6, 1 :5, 1 :4, 1 :3, 1 :2, 1 : 1, 1 :0.75, 0.75: 1, 2: 1, 3: 1, 4: 1, 5: 1, 6: 1, 7: 1, 8: 1, 9: 1, or 10: 1.
- the first LNP comprises a ratio of mRNA to atgRNAl of 2: 1.
- a second LNP comprising a second gene editor polynucleotide construct and a second attachment site-containing guide RNA (atgRNA2) comprises a ratio of gene editor polynucleotide construct (e.g., mRNA) to atgRNA2 of 1 : 10, 1 :9, 1 :8, 1 :7, 1 :6, 1 :5, 1 :4, 1 :3, 1 :2, 1 : 1, 1 :0.75, 0.75: 1, 2: 1, 3: 1, 4:1, 5: 1, 6: 1, 7: 1, 8: 1, 9: 1, or 10: 1.
- the second LNP comprises a ratio of mRNA to atgRNA2 of 2: 1.
- the method includes delivering a first LNP (e.g., a first LNP comprising a first gene editor polynucleotide construct and a first atgRNA) and a second LNP (e.g., a second LNP comprising a second gene editor polynucleotide construct and a second atgRNA)
- the first LNP and the second LNP are mixed such that the ratio of gene editor polynucleotide construct (e.g., mRNA) to first atgRNA (atgRNAl) to second atgRNA (atgRNA2) is 1 :0.25:0.25, l :0.5:0.5, 1 :0.75:0.75, or 1 : 1 : 1.
- the method of co-delivering to a cell a mixture of LNPs includes co-delivering three or more LNPs, four or more LNPs, five or more LNPs, six or more LNPs, seven or more LNPs, eight or more LNPs, nine or more LNPs, or ten or more LNPs.
- a system capable of site-specifically integrating at least a first integration recognition site into the genome of a cell, the system comprising: a first gene editor polynucleotide construct and a first attachment site-containing guide RNA (atgRNA) are packaged, and thereby vectorized, within the first LNP, and a second gene editor polynucleotide construct and a second attachment site containing guide RNR (atgRNA) are packaged, and thereby vectorized, within the second LNP, where the first atgRNA and the second atgRNA are an at least first pair of atgRNA.
- the at least first pair of atgRNAs comprise domains that are capable of guiding the prime editor system to a target sequence.
- the first atgRNA further includes a first RT template that comprises at least a portion of a first integration recognition site.
- the second atgRNA further includes a second RT template that comprises at least a portion of the first integration recognition site.
- the first atgRNA and the second atgRNAs collectively encode the entirety of the first integration recognition site.
- the first atgRNA and second atgRNA include at least a 6bp overlap.
- the system comprises a first LNP (e.g., any of the first LNPs described herein) and a second LNP (e.g., any of the second LNPs described herein) at a ratio of first LNP to second LNP of 1 : 10, 1 :9, 1 :8, 1 :7, 1 :6, 1 :5, 1 :4, 1 :3, 1 :2, 1 : 1, 1 :0.75, 0.75: 1, 2: 1, 3: 1, 4: 1, 5: 1, 6: 1, 7: 1, 8: 1, 9:1, or 10: 1.
- the system comprise the first LNP and the second LNP at a ratio of 1 : 1.
- the system comprises a first LNP having a ratio of a first gene editor polynucleotide construct to a first attachment site-containing guide RNA (atgRNAl) of 1 : 10, 1 :9, 1 :8, 1 :7, 1 :6, 1 :5, 1:4, 1 :3, 1 :2, 1 : 1, 1 :0.75, 0.75: 1, 2: 1, 3: 1, 4: 1, 5: 1, 6: 1, 7: 1, 8: 1, 9: 1, or 10: 1.
- the system includes a first LNP having a ratio of mRNA (i.e., mRNA encoding the gene editor protein) to atgRNAl of 2: 1.
- the system comprise a second LNP having a ratio of a second gene editor polynucleotide construct to a second attachment site-containing guide RNA (atgRNA2) of 1 : 10, 1 :9, 1 :8, 1 :7, 1:6, 1 :5, 1 :4, 1 :3, 1 :2, 1 : 1, 1 :0.75, 0.75: 1, 2: 1, 3: 1, 4: 1, 5: 1, 6: 1, 7: 1, 8: 1, 9: 1, or 10: 1.
- the system includes a second LNP having a ratio of mRNA (i.e., mRNA encoding the gene editor protein) to atgRNA2 of 2: 1.
- the system comprises a ratio of gene editor polynucleotide construct (e.g., mRNA encoding the gene editor protein) to first atgRNA (atgRNAl) to second atgRNA (atgRNA2) of 1 :0.25:0.25, l :0.5:0.5, 1 :0.75:0.75, or 1 : 1 : 1.
- gene editor polynucleotide construct e.g., mRNA encoding the gene editor protein
- the system comprises a mixture of LNPs comprising three or more LNPs, four or more LNPs, five or more LNPs, six or more LNPs, seven or more LNPs, eight or more LNPs, nine or more LNPs, or ten or more LNPs.
- a vector comprising a template polynucleotide and a sequence that is an integration cognate (i.e., cognate to an integration recognition site site-specifically incorporated into the genome of a cell) can be delivered to the cell concurrently with the split LNPs or after delivery of the split LNPs.
- a vector that includes a template polynucleotide and a second integration recognition site that is a cognate pair with the first integration recognition site is delivered to the cell.
- the sequence that is an integration cognate e.g., a second integration recognition site
- a second integration recognition site enables integration of the template polynucleotide or portion thereof when contacted with an integrase and the site- specifically incorporated first integration recognition site.
- the invention involves vectors, e.g. for delivering or introducing in a cell, but also for propagating these components (e.g. in prokaryotic cells).
- a "vector” is a tool that allows or facilitates the transfer of an entity from one environment to another. It is a replicon, such as a plasmid, phage, or cosmid, into which another DNA segment may be inserted so as to bring about the replication of the inserted segment.
- a vector is capable of replication when associated with the proper control elements.
- the term “vector” refers to a nucleic acid molecule capable of transporting another nucleic acid to which it has been linked.
- Vectors include, but are not limited to, nucleic acid molecules that are single-stranded, doublestranded, or partially double-stranded; nucleic acid molecules that comprise one or more free ends, no free ends (e.g. circular); nucleic acid molecules that comprise DNA, RNA, or both; and other varieties of polynucleotides known in the art.
- a ’’plasmid refers to a circular double stranded DNA loop into which additional DNA segments can be inserted, such as by standard molecular cloning techniques.
- viral vector wherein virally- derived DNA or RNA sequences are present in the vector for packaging into a virus (e.g.
- Viral vectors also include polynucleotides carried by a virus for transfection into a host cell.
- Certain vectors are capable of autonomous replication in a host cell into which they are introduced (e.g. bacterial vectors having a bacterial origin of replication and episomal mammalian vectors).
- Other vectors e.g., non-episomal mammalian vectors are integrated into the genome of a host cell upon introduction into the host cell, and thereby are replicated along with the host genome.
- vectors are capable of directing the expression of genes to which they are operatively-linked. Such vectors are referred to herein as "expression vectors.” Vectors for and that result in expression in a eukaryotic cell can be referred to herein as “eukaryotic expression vectors.” Common expression vectors of utility in recombinant DNA techniques are often in the form of plasmids.
- Recombinant expression vectors can comprise a nucleic acid of the invention in a form suitable for expression of the nucleic acid in a host cell, which means that the recombinant expression vectors include one or more regulatory elements, which may be selected on the basis of the host cells to be used for expression, that is operatively-linked to the nucleic acid sequence to be expressed.
- "operably linked" is intended to mean that the nucleotide sequence of interest is linked to the regulatory element(s) in a manner that allows for expression of the nucleotide sequence (e.g. in an in vitro transcription/translation system or in a host cell when the vector is introduced into the host cell).
- Vector delivery e.g., plasmid, viral delivery:
- the CRISPR enzyme for instance a Type V protein such as C2cl or C2c3, and/or any of the present RNAs, for instance a guide RNA
- Effector proteins and one or more guide RNAs can be packaged into one or more vectors, e.g., plasmid or viral vectors.
- the vector e.g., plasmid or viral vector is delivered to the tissue of interest by, for example, an intramuscular injection, while other times the delivery is via intravenous, transdermal, intranasal, oral, mucosal, or other delivery methods. Such delivery may be either via a single dose, or multiple doses.
- the actual dosage to be delivered herein may vary greatly depending upon a variety of factors, such as the vector choice, the target cell, organism, or tissue, the general condition of the subject to be treated, the degree of transformation/modification sought, the administration route, the administration mode, the type of transformation/modification sought, etc.
- Such a dosage may further contain, for example, a carrier (water, saline, ethanol, glycerol, lactose, sucrose, calcium phosphate, gelatin, dextran, agar, pectin, peanut oil, sesame oil, etc.), a diluent, a pharmaceutically-acceptable carrier (e.g., phosphate-buffered saline), a pharmaceutically-acceptable excipient, and/or other compounds known in the art.
- a carrier water, saline, ethanol, glycerol, lactose, sucrose, calcium phosphate, gelatin, dextran, agar, pectin, peanut oil, sesame oil, etc.
- a pharmaceutically-acceptable carrier e.g., phosphate-buffered saline
- a pharmaceutically-acceptable excipient e.g., phosphate-buffered saline
- the dosage may further contain one or more pharmaceutically acceptable salts such as, for example, a mineral acid salt such as a hydrochloride, a hydrobromide, a phosphate, a sulfate, etc.; and the salts of organic acids such as acetates, propionates, malonates, benzoates, etc.
- auxiliary substances such as wetting or emulsifying agents, pH buffering substances, gels or gelling materials, flavorings, colorants, microspheres, polymers, suspension agents, etc. may also be present herein.
- Suitable exemplary ingredients include microcrystalline cellulose, carboxymethylcellulose sodium, polysorbate 80, phenylethyl alcohol, chlorobutanol, potassium sorbate, sorbic acid, sulfur dioxide, propyl gallate, the parabens, ethyl vanillin, glycerin, phenol, parachlorophenol, gelatin, albumin and a combination thereof.
- the delivery is via an adenovirus, which may be at a single booster dose containing at least 1 x 10 5 particles (also referred to as particle units, pu) of adenoviral vector.
- the dose preferably is at least about 1 x 10 6 particles (for example, about 1 x 10 6 - 1 x 10 11 particles), more preferably at least about 1 x 10 7 particles, more preferably at least about 1 x 10 8 particles (e.g., about 1 x 10 8 -l x 10 11 particles or about 1 x 10 9 -l x 10 12 particles), and most preferably at least about 1 x IO 10 particles (e.g., about 1 x 10 9 - 1 x IO 10 particles or about 1 x 10 9 -l x 10 12 particles), or even at least about 1 x IO 10 particles (e.g., about 1 x 10 10 -l x 10 12 particles) of the adenoviral vector.
- the dose comprises no more than about 1 x 10 14 particles, preferably no more than about 1 x 10 13 particles, even more preferably no more than about 1 x 10 12 particles, even more preferably no more than about 1 x 10 11 particles, and most preferably no more than about 1 x IO 10 particles (e.g., no more than about 1 x 10 9 particles).
- the dose may contain a single dose of adenoviral vector with, for example, about 1 x 10 6 particle units (pu), about 2 x 10 6 pu, about 4 x 10 6 pu, about 1 x 10 7 pu, about 2 x 10 7 pu, about 4 x 10 7 pu, about 1 x 10 8 pu, about 2 x 10 8 pu, about 4 x 10 8 pu, about 1 x 10 9 pu, about 2 x 10 9 pu, about 4 x 10 9 pu, about 1 x IO 10 pu, about 2 x IO 10 pu, about 4 x IO 10 pu, about 1 x 10 11 pu, about 2 x 10 11 pu, about 4 x 10 11 pu, about 1 x 10 12 pu, about 2 x 10 12 pu, or about 4 x 10 12 pu of adenoviral vector.
- adenoviral vector with, for example, about 1 x 10 6 particle units (pu), about 2 x 10 6 pu, about 4 x 10 6 pu, about 1 x 10 7 pu, about 2 x
- the adenoviral vectors in U.S. Pat. No. 8,454,972 B2 to Nabel, et. al., granted on Jun. 4, 2013; incorporated by reference herein, and the dosages at col 29, lines 36-58 thereof.
- the adenovirus is delivered via multiple doses.
- the delivery is via an AAV.
- a therapeutically effective dosage for in vivo delivery of the AAV to a human is believed to be in the range of from about 20 to about 50 ml of saline solution containing from about 1 x 10 10 to about 1 x 10 50 functional AAV/ml solution. The dosage may be adjusted to balance the therapeutic benefit against any side effects.
- the AAV dose is generally in the range of concentrations of from about 1 x 10 5 to 1 x 10 50 genomes AAV (sometimes referred to herein as “vector genomes” or “vg”), from about 1 x 10 8 to 1 x IO 20 genomes AAV, from about 1 x 10 10 to about 1 x 10 16 genomes, or about 1 x 10 11 to about 1 x 10 16 genomes AAV.
- a human dosage may be about 1 x 10 13 genomes AAV.
- concentrations may be delivered in from about 0.001 ml to about 100 ml, about 0.05 to about 50 ml, or about 10 to about 25 ml of a carrier solution.
- the promoter used to drive nucleic acid-targeting effector protein coding nucleic acid molecule expression can include: AAV ITR can serve as a promoter: this is advantageous for eliminating the need for an additional promoter element (which can take up space in the vector). The additional space freed up can be used to drive the expression of additional elements (gRNA, etc.). Also, ITR activity is relatively weaker, so can be used to reduce potential toxicity due to over expression of nucleic acid-targeting effector protein. For ubiquitous expression, can use promoters: CMV, CAG, CBh, PGK, SV40, Ferritin heavy or light chains, etc.
- promoters For brain or other CNS expression, can use promoters: SynapsinI for all neurons, CaMKIIalpha for excitatory neurons, GAD67 or GAD65 or VGAT for GABAergic neurons, etc.
- For liver expression can use Albumin promoter.
- For lung expression can use SP-B.
- For endothelial cells can use ICAM.
- For hematopoietic cells can use IFNbeta or CD45.
- Osteoblasts can use OG-2.
- the promoter used to drive guide RNA can include: Pol III promoters such as U6 or Hl Use of Pol II promoter and intronic cassettes to express guide RNA Adeno Associated Virus (AAV).
- Pol III promoters such as U6 or Hl Use of Pol II promoter and intronic cassettes to express guide RNA Adeno Associated Virus (AAV).
- AAV Addeno Associated Virus
- Nucleic acid-targeting effector protein and one or more guide RNA can be delivered using adeno associated virus (AAV), lentivirus, adenovirus or other plasmid or viral vector types, in particular, using formulations and doses from, for example, U.S. Pat. No. 8,454,972 (formulations, doses for adenovirus), U.S. Pat. No. 8,404,658 (formulations, doses for AAV) and U.S. Pat. No. 5,846,946 (formulations, doses for DNA plasmids) and from clinical trials and publications regarding the clinical trials involving lentivirus, AAV and adenovirus.
- AAV adeno associated virus
- the route of administration, formulation and dose can be as in U.S. Pat. No. 8,454,972 and as in clinical trials involving AAV.
- the route of administration, formulation and dose can be as in U.S. Pat. No. 8,404,658 and as in clinical trials involving adenovirus.
- the route of administration, formulation and dose can be as in U.S. Pat. No. 5,846,946 and as in clinical studies involving plasmids.
- Doses may be based on or extrapolated to an average 70 kg individual (e.g., a male adult human), and can be adjusted for patients, subjects, mammals of different weight and species.
- Frequency of administration is within the ambit of the medical or veterinary practitioner (e.g., physician, veterinarian), depending on usual factors including the age, sex, general health, other conditions of the patient or subject and the particular condition or symptoms being addressed.
- the viral vectors can be injected into the tissue of interest.
- the expression of nucleic acid-targeting effector can be driven by a cell-type specific promoter.
- liver-specific expression might use the Albumin promoter and neuron-specific expression (e.g., for targeting CNS disorders) might use the Synapsin I promoter.
- AAV is advantageous over other viral vectors for a couple of reasons: Low toxicity (this may be due to the purification method not requiring ultra centrifugation of cell particles that can activate the immune response) and Low probability of causing insertional mutagenesis because it doesn't integrate into the host genome.
- AAV has a packaging limit of 4.5 or 4.75 Kb.
- nucleic acid-targeting effector protein such as a Type V protein such as C2cl or C2c3
- a promoter and transcription terminator have to be all fit into the same viral vector. Therefore embodiments of the invention include utilizing homologs of nucleic acid-targeting effector protein (such as a Type V protein such as C2cl or C2c3) that are shorter.
- the AAV can be AAV1, AAV2, AAV5 or any combination thereof.
- AAV8 is useful for delivery to the liver. The herein promoters and vectors are preferred individually.
- Packaging cells are typically used to form virus particles that are capable of infecting a host cell. Such cells include 293 cells, which package adenovirus, and psi2 cells or PA317 cells, which package retrovirus.
- Viral vectors used in gene therapy are usually generated by producing a cell line that packages a nucleic acid vector into a viral particle. The vectors typically contain the minimal viral sequences required for packaging and subsequent integration into a host, other viral sequences being replaced by an expression cassette for the polynucleotide(s) to be expressed. The missing viral functions are typically supplied in trans by the packaging cell line. For example, AAV vectors used in gene therapy typically only possess ITR sequences from the AAV genome which are required for packaging and integration into the host genome.
- Viral DNA is packaged in a cell line, which contains a helper plasmid encoding the other AAV genes, namely rep and cap, but lacking ITR sequences.
- the cell line may also be infected with adenovirus as a helper.
- the helper virus promotes replication of the AAV vector and expression of AAV genes from the helper plasmid.
- the helper plasmid is not packaged in significant amounts due to a lack of ITR sequences. Contamination with adenovirus can be reduced by, e.g., heat treatment to which adenovirus is more sensitive than AAV. Additional methods for the delivery of nucleic acids to cells are known to those skilled in the art. See, for example, US20030087817, incorporated herein by reference.
- Millington-Ward et al. (Molecular Therapy, vol. 19 no. 4, 642-649 April 2011) describes adeno-associated virus (AAV) vectors to deliver an RNA interference (RNAi)-based rhodopsin suppressor and a codon-modified rhodopsin replacement gene resistant to suppression due to nucleotide alterations at degenerate positions over the RNAi target site.
- RNAi RNA interference
- An injection of either 6.0 x 10 8 vp or 1.8 x IO 10 vp AAV were subretinally injected into the eyes by Millington-Ward et al.
- the AAV vectors of Millington-Ward et al. may be applied to the system of the present invention, contemplating a dose of about 2 x 10 11 to about 6 x 10 11 vp administered to a human.
- Dalkara et al. also relates to in vivo directed evolution to fashion an AAV vector that delivers wild-type versions of defective genes throughout the retina after noninjurious injection into the eyes' vitreous humor.
- Dalkara describes a 7 mer peptide display library and an AAV library constructed by DNA shuffling of cap genes from AAV1, 2, 4, 5, 6, 8, and 9.
- the rcAAV libraries and rAAV vectors expressing GFP under a CAG or Rho promoter were packaged and deoxyribonuclease-resistant genomic titers were obtained through quantitative PCR.
- the libraries were pooled, and two rounds of evolution were performed, each consisting of initial library diversification followed by three in vivo selection steps.
- P30 rho-GFP mice were intravitreally injected with 2 ml of iodixanol -purified, phosphate- buffered saline (PBS)-dialyzed library with a genomic titer of about 1. times.10. sup.12 vg/ml.
- PBS phosphate- buffered saline
- the AAV vectors of Dalkara et al. may be applied to the nucleic acid-targeting system of the present invention, contemplating a dose of about 1 x 10 15 to about 1 x 10 16 vg/ml administered to a human.
- Lentiviral vectors are retroviral vectors that are able to transduce or infect non-dividing cells and typically produce high viral titers. Selection of a retroviral gene transfer system would therefore depend on the target tissue. Retroviral vectors are comprised of cis-acting long terminal repeats with packaging capacity for up to 6-10 kb of foreign sequence. The minimum cis-acting LTRs are sufficient for replication and packaging of the vectors, which are then used to integrate the therapeutic gene into the target cell to provide permanent transgene expression.
- Widely used retroviral vectors include those based upon murine leukemia virus (MuLV), gibbon ape leukemia virus (GaLV), Simian Immuno deficiency virus (SW), human immuno deficiency virus (HIV), and combinations thereof (see, e.g., Buchscher et al., J. Virol. 66:2731-2739 (1992); Johann et al., J. Virol. 66: 1635-1640 (1992); Sommnerfelt et al., Virol. 176:58-59 (1990); Wilson et al., J. Virol. 63:2374-2378 (1989); Miller et al., J. Virol.
- MiLV murine leukemia virus
- GaLV gibbon ape leukemia virus
- SW Simian Immuno deficiency virus
- HAV human immuno deficiency virus
- adenoviral based systems may be used.
- Adenoviral based vectors are capable of very high transduction efficiency in many cell types and do not require cell division. With such vectors, high titer and levels of expression have been obtained. This vector can be produced in large quantities in a relatively simple system.
- Adeno-associated virus vectors may also be used to transduce cells with target nucleic acids, e.g., in the in vitro production of nucleic acids and peptides, and for in vivo and ex vivo gene therapy procedures (see, e.g., West et al., Virology 160:38-47 (1987); U.S. Pat. No. 4,797,368; WO 93/24641; Kotin, Human Gene Therapy 5:793-801 (1994); Muzyczka, J. Clin. Invest. 94: 1351 (1994). Construction of recombinant AAV vectors are described in a number of publications, including U.S. Pat. No.
- Packaging cells are typically used to form virus particles that are capable of infecting a host cell. Such cells include 293 cells, which package adenovirus, and yr2 cells or PA317 cells, which package retrovirus.
- Viral vectors used in gene therapy are usually generated by producing a cell line that packages a nucleic acid vector into a viral particle. The vectors typically contain the minimal viral sequences required for packaging and subsequent integration into a host, other viral sequences being replaced by an expression cassette for the polynucleotide(s) to be expressed. The missing viral functions are typically supplied in trans by the packaging cell line. For example, AAV vectors used in gene therapy typically only possess ITR sequences from the AAV genome which are required for packaging and integration into the host genome.
- Viral DNA is packaged in a cell line, which contains a helper plasmid encoding the other AAV genes, namely rep and cap, but lacking ITR sequences.
- the cell line may also be infected with adenovirus as a helper.
- the helper virus promotes replication of the AAV vector and expression of AAV genes from the helper plasmid.
- the helper plasmid is not packaged in significant amounts due to a lack of ITR sequences. Contamination with adenovirus can be reduced by, e.g., heat treatment to which adenovirus is more sensitive than AAV. Additional methods for the delivery of nucleic acids to cells are known to those skilled in the art. See, for example, US20030087817, incorporated herein by reference.
- a host cell is transiently or non-transiently transfected with one or more vectors described herein.
- a cell is transfected as it naturally occurs in a subject.
- a cell that is transfected is taken from a subject.
- Cells taken from a subject include, but are not limited to, hepatocytes or cells isolated from muscle, the CNS, eye or lung.
- Immunological cells are also contemplated, such as but not limited to T cells, HSCs, B-cells and NK cells.
- mRNA delivery methods and compositions that may be utilized in the present disclosure including, for example, PCT/US2014/028330, US8822663B2, NZ700688A, ES2740248T3, EP2755693A4, EP2755986A4, WO2014152940A1, EP3450553B1, BRI 12016030852A2, and EP3362461A1.
- Expression of CRISPR systems in particular is described by W02020014577.
- Each of these publications are incorporated herein by reference in their entireties. Additional disclosure hereby incorporated by reference can be found in Kowalski et al., “Delivering the Messenger: Advances in Technologies for Therapeutic mRNA Delivery,” Mol Therap., 2019; 27(4): 710-728.
- the cell is derived from cells taken from a subject, such as a cell line.
- a cell line A wide variety of cell lines for tissue culture are known in the art. Examples of cell lines include, but are not limited to, C8161, CCRF-CEM, MOLT, mIMCD-3, NHDF, HeLa-S3, Huhl, Huh4, Huh7, HUVEC, HASMC, HEKn, HEKa, MiaPaCell, Panel, PC-3, TF1, CTLL-2, CIR, Rat6, CV1, RPTE, A10, T24, J82, A375, ARH-77, Calul, SW480, SW620, SKOV3, SK-UT, CaCo2, P388D1, SEM-K2, WEHL231, HB56, TIB55, Jurkat, J45.01, LRMB, Bcl-1, BC-3, IC21, DLD2, Raw264.7, NRK, NRK-52E, MRC5, MEF, Hep
- a cell transfected with one or more vectors described herein is used to establish a new cell line comprising one or more vector-derived sequences.
- one or more vectors described herein are used to produce a nonhuman transgenic animal or transgenic plant.
- the transgenic animal is a mammal, such as a mouse, rat, or rabbit.
- the organism or subject is a plant.
- the organism or subject or plant is algae. Methods for producing transgenic plants and animals are known in the art, and generally begin with a method of cell transfection, such as described herein.
- the invention provides for methods of modifying a target polynucleotide in a prokaryotic or eukaryotic cell, which may be in vivo, ex vivo or in vitro.
- the method comprises sampling a cell or population of cells from a human or non-human animal or plant (including micro-algae) and modifying the cell or cells. Culturing may occur at any stage ex vivo. The cell or cells may even be re-introduced into the non-human animal or plant (including micro-algae).
- pathogens are often host-specific.
- Fusariumn oxysporum f. sp. lycopersici causes tomato wilt but attacks only tomato
- Plants have existing and induced defenses to resist most pathogens. Mutations and recombination events across plant generations lead to genetic variability that gives rise to susceptibility, especially as pathogens reproduce with more frequency than plants.
- there can be non-host resistance e.g., the host and pathogen are incompatible.
- Horizontal Resistance e.g., partial resistance against all races of a pathogen, typically controlled by many genes
- Vertical Resistance e.g., complete resistance to some races of a pathogen but not to other races, typically controlled by a few genes.
- Plant and pathogens evolve together, and the genetic changes in one balance changes in other. Accordingly, using Natural Variability, breeders combine most useful genes for Yield. Quality, Uniformity, Hardiness, Resistance.
- the sources of resistance genes include native or foreign Varieties, Heirloom Varieties, Wild Plant Relatives, and Induced Mutations, e.g., treating plant material with mutagenic agents.
- plant breeders are provided with a new tool to induce mutations. Accordingly, one skilled in the art can analyze the genome of sources of resistance genes, and in Varieties having desired characteristics or traits employ the present invention to induce the rise of resistance genes, with more precision than previous mutagenic agents and hence accelerate and improve plant breeding programs.
- target polynucleotides include a sequence associated with a signaling biochemical pathway, e.g., a signaling biochemical pathway-associated gene or polynucleotide.
- target polynucleotides include a disease associated gene or polynucleotide.
- a “disease-associated” gene or polynucleotide refers to any gene or polynucleotide which is yielding transcription or translation products at an abnormal level or in an abnormal form in cells derived from a disease-affected tissues compared with tissues or cells of a non disease control.
- a disease-associated gene also refers to a gene possessing mutation(s) or genetic variation that is directly responsible or is in linkage disequilibrium with a gene(s) that is responsible for the etiology of a disease.
- the transcribed or translated products may be known or unknown, and may be at a normal or abnormal level.
- the delivery system is packaged in one or more LNPs and administered intravenously.
- the co-delivery system is packaged in one or more LNPs and administered intrathecally.
- the co-delivery system is packaged in one or more LNPs and administered by intracerebral ventricular injection.
- the co-delivery system is packaged in one or more LNPs and administered by intraci sternal magna administration.
- the co-delivery system is packaged in one or more LNPs and administered by intravitreal injection.
- lipidmucleic acid complexes including targeted liposomes such as immunolipid complexes
- crystal Science 270:404-410 (1995); Blaese et al., Cancer Gene Ther. 2:291-297 (1995); Behr et al., Bioconjugate Chem. 5:382-389 (1994); Remy et al., Bioconjugate Chem. 5:647-654 (1994); Gao et al., Gene Therapy 2:710-722 (1995); Ahmad et al., Cancer Res. 52:4817-4820 (1992); U.S. Pat. Nos.
- the LNP formulations are selected from LP01 (Cas No. 1799316-64-5), ALC-0315 (Cas No. 2036272-55-4), and cKK-E12 (Cas No. 1432494-65-9).
- the LNP formulation is LP01.
- the LNP formulation is ALC-0315.
- the LNP formulation is cKK-E12.
- LNP doses range from about 0.1 mg/kg to about 100 mg/kg (or any of the values or subranges therein). In some embodiments, LNP doses is about 0.1 mg/kg, about 0.2 mg/kg, about 0.3 mg/kg, about 0.4 mg/kg, about 0.5 mg/kg, about 0.6 mg/kg, about 0.7 mg/kg, about 0.8 mg/kg, about 0.9 mg/kg, about 1.0 mg/kg, 1.5 mg/kg, about 2 mg/kg, about 2.5 mg/kg, about 3 mg/kg, about 3.5 mg/kg, about 4 mg/kg, about 4.5 mg/kg, about 5 mg/kg, about 6 mg/kg, about7 mg/kg, about 7 mg/kg, about 8 mg/kg, about 9 mg/kg, about 10 mg/kg, about 15 mg/kg, about 20 mg/kg, about 25 mg/kg, about 30 mg/kg, about 35 mg/kg, about 40 mg/kg, about 45 mg/kg, or about 50 mg/kg or more
- LNP doses of about 0.01 to about 1 mg per kg of body weight administered intravenously are contemplated.
- Medications to reduce the risk of infusion-related reactions are contemplated, such as dexamethasone, acetaminophen, diphenhydramine or cetirizine, and ranitidine are contemplated.
- Multiple doses of about 0.3 mg per kilogram every 4 weeks for five doses are also contemplated.
- the charge of the LNP must be taken into consideration. As cationic lipids combined with negatively charged lipids to induce nonbilayer structures that facilitate intracellular delivery. Because charged LNPs are rapidly cleared from circulation following intravenous injection, ionizable cationic lipids with pKa values below 7 were developed (see, e.g., Rosin et al, Molecular Therapy, vol. 19, no. 12, pages 1286-2200, December 2011). Negatively charged polymers such as RNA may be loaded into LNPs at low pH values (e.g., pH 4) where the ionizable lipids display a positive charge. However, at physiological pH values, the LNPs exhibit a low surface charge compatible with longer circulation times.
- pH 4 e.g., pH 4
- ionizable cationic lipids Four species of ionizable cationic lipids have been focused upon, namely l,2-dilineoyl-3-dimethylammonium-propane (DLinDAP), 1,2- dilinoleyloxy-3-N,N-dimethylaminopropane (DLinDMA), l,2-dilinoleyloxy-keto-N,N-dimethyl- 3 -aminopropane (DLinKDMA), and l,2-dilinoleyl-4-(2-dimethylaminoethyl)-[l,3]-dioxolane (DLinKC2-DMA).
- DLinDAP 1,2- dilinoleyloxy-3-N,N-dimethylaminopropane
- DLinKDMA 1,2- dilinoleyloxy-keto-N,N-dimethyl- 3 -aminopropane
- LNP siRNA systems containing these lipids exhibit remarkably different gene silencing properties in hepatocytes in vivo, with potencies varying according to the series DLinKC2-DMA>DLinKDMA>DLinDMA»DLinDAP employing a Factor VII gene silencing model (see, e.g., Rosin et al, Molecular Therapy, vol. 19, no. 12, pages 1286-2200, December 2011).
- a dosage of 1 pg/ml of LNP in or associated with the LNP may be contemplated, especially for a formulation containing DLinKC2-DMA.
- the LNP composition comprises one or more one or more ionizable lipids.
- ionizable lipid has its ordinary meaning in the art and may refer to a lipid comprising one or more charged moieties.
- an ionizable lipid may be positively charged or negatively charged.
- the one or more ionizable lipids are selected from the group consisting of 3-(didodecylamino)- N1 ,N1 ,4-tridodecyl- 1 -piperazineethanamine (KL 10), N1 -[2-(didodecylamino)ethyl]-N 1 ,N4,N4- tridodecyl-l,4-piperazinediethanami- ne (KL22), 14,25-ditridecyl-15,18,21,24-tetraaza- octatriacontane (KL25), l,2-dilinoleyloxy-N,N-dimethylaminopropane (DLin-DMA), 2,2- dilinoleyl-4-dimethylaminomethyl-[l,3]-dioxolane (DLin-K-DMA), heptatriaconta-6,9,28,31-
- the lipid nanoparticle may include one or more (e.g., 1, 2, 3, 4, 5, 6, 7, or 8) cationic and/or ionizable lipids.
- cationic and/or ionizable lipids include, but are not limited to, 3-(didodecylamino)-Nl,Nl,4-tridodecyl-l-piperazineethanamine (KL10), Nl-[2- (didodecylamino)ethyl]-Nl,N4,N4-tridodecyl-l,4-piperazinediethanami- ne (KL22), 14,25- ditridecyl- 15,18,21 ,24-tetraaza-octatriacontane (KL25), 1 ,2-dilinoleyloxy-N,N- dimethylaminopropane (DLin-DMA), 2,2-dilinoleyl-4-dimethylamin
- DOSPA (sperminecarboxamido)ethyl)-N,N-dimethyl- -ammonium trifluoracetate
- DOGS dioctadecylamidoglycyl carboxyspermine
- DODAP 1, 2-di oleoyl-3 -dimethylammonium propane
- DODMA N,N-dimethyl-2,3-dioleyloxy
- DMRIE N-(l,2- dimyristyloxyprop-3-yl)-N,N-dimethyl-N-hydroxyethyl ammonium bromide
- lipids e.g., LIPOFECTIN.RTM. (including DOTMA and DOPE, available from GIBCO/BRL), and LIPOFECTAMINE.RTM. (including DOSPA and DOPE, available from GIBCO/BRL).
- LIPOFECTIN.RTM including DOTMA and DOPE, available from GIBCO/BRL
- LIPOFECTAMINE.RTM. including DOSPA and DOPE, available from GIBCO/BRL
- KL10, KL22, and KL25 are described, for example, in U.S. Pat. No. 8,691,750.
- the LNP composition comprises one or more amino lipids.
- amino lipid and “cationic lipid” are used interchangeably herein to include those lipids and salts thereof having one, two, three, or more fatty acid or fatty alkyl chains and a pH-titratable amino head group (e.g., an alkylamino or dialkylamino head group).
- a pH-titratable amino head group e.g., an alkylamino or dialkylamino head group.
- the cationic lipid is typically protonated (i.e., positively charged) at a pH below the pKa of the cationic lipid and is substantially neutral at a pH above the pKa.
- the cationic lipids can also be termed titratable cationic lipids.
- the one or more cationic lipids include: a protonatable tertiary amine (e.g., pH-titratable) head group; alkyl chains, wherein each alkyl chain independently has 0 to 3 (e.g., 0, 1, 2, or 3) double bonds; and ether, ester, or ketal linkages between the head group and alkyl chains.
- cationic lipids include, but are not limited to, DSDMA, DODMA, DOTMA, DLinDMA, DLenDMA, . gamma.
- -DLenDMA DLin-K-DMA
- DLin-K-C2- DMA also known as DLin-C2K-DMA, XTC2, and C2K
- DLin-K-C3-DMA also known as DLin-K-C4-DMA
- DLen-C2K-DMA y-DLen-C2-DMA
- C12-200 cKK-E12, cKK-A12, cKK-012
- DLin-MC2- DMA also known as MC2
- DLin-MC3-DMA also known as MC3
- Anionic lipids suitable for use in lipid nanoparticles include, but are not limited to, phosphatidylglycerol, cardiolipin, diacylphosphatidylserine, diacylphosphatidic acid, N- dodecanoyl phosphatidylethanoloamine, N-succinyl phosphatidylethanolamine, N-glutaryl phosphatidylethanolamine, lysylphosphatidylglycerol, and other anionic modifying groups joined to neutral lipids.
- Neutral lipids suitable for use in lipid nanoparticles include, but are not limited to, diacylphosphatidylcholine, diacylphosphatidylethanolamine, ceramide, sphingomyelin, dihydrosphingomyelin, cephalin, sterols (e.g., cholesterol) and cerebrosides.
- the lipid nanoparticle comprises cholesterol.
- Lipids having a variety of acyl chain groups of varying chain length and degree of saturation are available or may be isolated or synthesized by well-known techniques. Additionally, lipids having mixtures of saturated and unsaturated fatty acid chains and cyclic regions can be used.
- the neutral lipids used in the disclosure are DOPE, DSPC, DPPC, POPC, or any related phosphatidylcholine.
- the neutral lipid may be composed of sphingomyelin, dihydrosphingomyeline, or phospholipids with other head groups, such as serine and inositol.
- amphipathic lipids are included in nanoparticles.
- Exemplary amphipathic lipids suitable for use in nanoparticles include, but are not limited to, sphingolipids, phospholipids, fatty acids, and amino lipids.
- the lipid composition of the pharmaceutical composition may comprise one or more phospholipids, for example, one or more saturated or (poly)unsaturated phospholipids or a combination thereof.
- phospholipids comprise a phospholipid moiety and one or more fatty acid moieties.
- a phospholipid moiety can be selected, for example, from the non-limiting group consisting of phosphatidyl choline, phosphatidyl ethanolamine, phosphatidyl glycerol, phosphatidyl serine, phosphatidic acid, 2-lysophosphatidyl choline, and a sphingomyelin.
- a fatty acid moiety can be selected, for example, from the non-limiting group consisting of lauric acid, myristic acid, myristoleic acid, palmitic acid, palmitoleic acid, stearic acid, oleic acid, linoleic acid, alpha-linolenic acid, erucic acid, phytanoic acid, arachidic acid, arachidonic acid, eicosapentaenoic acid, behenic acid, docosapentaenoic acid, and docosahexaenoic acid.
- Particular amphipathic lipids can facilitate fusion to a membrane.
- a cationic phospholipid can interact with one or more negatively charged phospholipids of a membrane (e.g., a cellular or intracellular membrane). Fusion of a phospholipid to a membrane can allow one or more elements (e.g., a therapeutic agent) of a lipid-containing composition (e.g., LNPs) to pass through the membrane permitting, e.g., delivery of the one or more elements to a target tissue.
- a cationic phospholipid can interact with one or more negatively charged phospholipids of a membrane (e.g., a cellular or intracellular membrane). Fusion of a phospholipid to a membrane can allow one or more elements (e.g., a therapeutic agent) of a lipid-containing composition (e.g., LNPs) to pass through the membrane permitting, e.g., delivery of the one or more elements to a target tissue.
- elements e.g., a
- Non-natural amphipathic lipid species including natural species with modifications and substitutions including branching, oxidation, cyclization, and alkynes are also contemplated.
- a phospholipid can be functionalized with or cross-linked to one or more alkynes (e.g., an alkenyl group in which one or more double bonds is replaced with a triple bond).
- alkynes e.g., an alkenyl group in which one or more double bonds is replaced with a triple bond.
- an alkyne group can undergo a copper-catalyzed cycloaddition upon exposure to an azide.
- Phospholipids include, but are not limited to, glycerophospholipids such as phosphatidylcholines, phosphatidylethanolamines, phosphatidylserines, phosphatidylinositols, phosphatidy glycerols, and phosphatidic acids. Phospholipids also include phosphosphingolipid, such as sphingomyelin.
- the LNP composition comprises one or more phospholipids.
- the phospholipid is selected from the group consisting of 1,2-dilinoleoyl-sn- glycero-3 -phosphocholine (DLPC), 1,2-dimyristoyl-sn-glycero-phosphocholine (DMPC), 1,2- dioleoyl-sn-glycero-3 -phosphocholine (DOPC), 1 ,2-dipalmitoyl-sn-glycero-3 -phosphocholine (DPPC), l,2-distearoyl-sn-glycero-3-phosphocholine (DSPC), 1,2-diundecanoyl-sn-glycero- phosphocholine (DUPC), l-palmitoyl-2-oleoyl-sn-glycero-3 -phosphocholine (POPC), 1,2-di-O- octadecenyl-sn-g
- DLPC 1,2-dilino
- phosphorus-lacking compounds such as sphingolipids, glycosphingolipid families, diacylglycerols, and P-acyloxyacids, may also be used. Additionally, such amphipathic lipids can be readily mixed with other lipids, such as triglycerides and sterols.
- the LNP composition comprises one or more helper lipids.
- helper lipid refers to lipids that enhance transfection (e.g., transfection of an LNP comprising an mRNA that encodes a site-directed endonuclease, such as a SpCas9 polypeptide).
- site-directed endonuclease such as a SpCas9 polypeptide
- the mechanism by which the helper lipid enhances transfection includes enhancing particle stability.
- the helper lipid enhances membrane fusogenicity.
- helper lipid of the LNP compositions disclosure herein can be any helper lipid known in the art.
- helper lipids suitable for the compositions and methods include steroids, sterols, and alkyl resorcinols.
- helper lipids suitable for use in the present disclosure include, but are not limited to, saturated phosphatidylcholine (PC) such as distearoyl-PC (DSPC) and dipalymitoyl-PC (DPPC), di oleoylphosphatidylethanolamine (DOPE), 1,2-dilinoleoyl-sn- glycero-3 -phosphocholine (DLPC), cholesterol, 5-heptadecylresorcinol, and cholesterol hemisuccinate.
- PC saturated phosphatidylcholine
- DSPC distearoyl-PC
- DPPC dipalymitoyl-PC
- DOPE di oleoylphosphatidylethanolamine
- DLPC 1,2-dilinoleoyl-sn- glycero-3 -phosphocholine
- cholesterol 5-heptadecylresorcinol
- cholesterol hemisuccinate hemisuccinate.
- the LNP composition comprises one or more structural lipids.
- structural lipid refers to sterols and also to lipids containing sterol moi eties. Without being bound to any particular theory, it is believed that the incorporation of structural lipids into the LNPs mitigates aggregation of other lipids in the particle.
- Structural lipids can be selected from the group including but not limited to, cholesterol, fecosterol, sitosterol, ergosterol, campesterol, stigmasterol, brassicasterol, tomatidine, tomatine, ursolic acid, alpha-tocopherol, hopanoids, phytosterols, steroids, and mixtures thereof.
- the structural lipid is a sterol.
- sterols are a subgroup of steroids consisting of steroid alcohols.
- the structural lipid is a steroid.
- the structural lipid is cholesterol.
- the structural lipid is an analog of cholesterol.
- the lipid component of a lipid nanoparticle composition may include one or more molecules comprising polyethylene glycol, such as PEG or PEG-modified lipids.
- the LNP composition disclosed herein comprise one or more polyethylene glycol (PEG) lipid.
- PEG-lipid refers to polyethylene glycol (PEG)-modified lipids. Such lipids are also referred to as PEGylated lipids.
- PEG-lipids include PEG- modified phosphatidylethanolamine and phosphatidic acid, PEG-ceramide conjugates (e.g., PEG- CerC14 or PEG-CerC20), PEG-modified dialkylamines and PEG-modified 1,2-diacyloxypropan- 3-amines
- a PEG lipid can be PEG-c-DOMG, PEG-DMG, PEG-DLPE, PEG-DMPE, PEG-DPPC, or a PEG-DSPE lipid.
- the PEG-lipid includes, but not limited to 1,2-dimyristoyl-sn-glycerol methoxypolyethylene glycol (PEG-DMG), 1,2-distearoyl-sn- glycero-3-phosphoethanolamine-N-[amino(polyethylene glycol)] (PEG-DSPE), PEG-disteryl glycerol (PEG-DSG), PEG-dipalmetoleyl, PEG-dioleyl, PEG-distearyl, PEG-diacylglycamide (PEG-DAG), PEG-dipalmitoyl phosphatidylethanolamine (PEG-DPPE), or PEG- 1,2- dimyristyloxlpropyl-3-amine (PEG-c-DMA).
- PEG-DMG 1,2-dimyristoyl-sn-glycerol methoxypolyethylene glycol
- PEG-DSPE 1,2-distearoyl-sn-
- the PEG-lipid is selected from the group consisting of a PEG-modified phosphatidylethanolamine, a PEG-modified phosphatidic acid, a PEG-modified ceramide, a PEG-modified dialkylamine, a PEG-modified diacylglycerol, a PEG-modified dialkylglycerol, and mixtures thereof.
- the lipid moiety of the PEG-lipids includes those having lengths of from about C. sub.14 to about C. sub.22, preferably from about C. sub.14 to about C. sub.16.
- a PEG moiety for example a mPEG-NH.sub.2, has a size of about 1000, 2000, 5000, 10,000, 15,000 or 20,000 daltons.
- the PEG-lipid is PEG2k-DMG.
- the one or more PEG lipids of the LNP composition comprises PEG-DMPE.
- the one or more PEG lipids of the LNP composition comprises PEG-DMG.
- the ratio between the lipid components and the nucleic acid molecules of the LNP composition is sufficient for (i) formation of LNPs with desired characteristics, e.g., size, charge, and (ii) delivery of a sufficient dose of nucleic acid at a dose of the lipid component(s) that is tolerable for in vivo administration as readily ascertained by one of skill in the art.
- a nanoparticle e.g., a lipid nanoparticle
- a targeting moiety that is specific to a cell type and/or tissue type.
- a nanoparticle may be targeted to a particular cell, tissue, and/or organ using a targeting moiety.
- a nanoparticle comprises a targeting moiety.
- targeting moieties include ligands, cell surface receptors, glycoproteins, vitamins (e.g., riboflavin) and antibodies (e.g., full-length antibodies, antibody fragments (e.g., Fv fragments, single chain Fv (scFv) fragments, Fab' fragments, or F(ab')2 fragments), single domain antibodies, cam elid antibodies and fragments thereof, human antibodies and fragments thereof, monoclonal antibodies, and multispecific antibodies (e.g., bispecific antibodies)).
- the targeting moiety may be a polypeptide.
- the targeting moiety may include the entire polypeptide (e.g., peptide or protein) or fragments thereof.
- a targeting moiety is typically positioned on the outer surface of the nanoparticle in such a manner that the targeting moiety is available for interaction with the target, for example, a cell surface receptor.
- a variety of different targeting moieties and methods are known and available in the art, including those described, e.g., in Sapra et al., Prog. Lipid Res. 42(5):439-62, 2003 and Abra et al., J. Liposome Res. 12: 1-3, 2002.
- a lipid nanoparticle may include a surface coating of hydrophilic polymer chains, such as polyethylene glycol (PEG) chains (see, e.g., Allen et al., Biochimica et Biophysica Acta 1237: 99-108, 1995; DeFrees et al., Journal of the American Chemistry Society 118: 6101-6104, 1996; Blume et al., Biochimica et Biophysica Acta 1149: ISO- 184, 1993; Klibanov et al., Journal of Liposome Research 2: 321-334, 1992; U.S. Pat. No.
- PEG polyethylene glycol
- a targeting moiety for targeting the lipid nanoparticle is linked to the polar head group of lipids forming the nanoparticle.
- the targeting moiety is attached to the distal ends of the PEG chains forming the hydrophilic polymer coating (see, e.g., Klibanov et al., Journal of Liposome Research 2: 321-334, 1992; Kirpotin et al., FEBS Letters 388: 115-118, 1996).
- Standard methods for coupling the targeting moiety or moieties may be used.
- phosphatidylethanolamine which can be activated for attachment of targeting moieties
- derivatized lipophilic compounds such as lipid-derivatized bleomycin
- Antibody- targeted liposomes can be constructed using, for instance, liposomes that incorporate protein A (see, e.g., Renneisen et al., J. Bio. Chem., 265: 16337-16342, 1990 and Leonetti et al., Proc. Natl. Acad. Sci. (USA), 87:2448-2451, 1990).
- Other examples of antibody conjugation are disclosed in U.S. Pat. No.
- targeting moieties can also include other polypeptides that are specific to cellular components, including antigens associated with neoplasms or tumors.
- Polypeptides used as targeting moieties can be attached to the liposomes via covalent bonds (see, for example Heath, Covalent Attachment of Proteins to Liposomes, 149 Methods in Enzymology 111-119 (Academic Press, Inc. 1987)).
- Other targeting methods include the biotin-avidin system.
- a lipid nanoparticle includes a targeting moiety that targets the lipid nanoparticle to a cell including, but not limited to, hepatocytes, colon cells, epithelial cells, hematopoietic cells, epithelial cells, endothelial cells, lung cells, bone cells, stem cells, mesenchymal cells, neural cells, cardiac cells, adipocytes, vascular smooth muscle cells, cardiomyocytes, skeletal muscle cells, beta cells, pituitary cells, synovial lining cells, ovarian cells, testicular cells, fibroblasts, B cells, T cells, reticulocytes, leukocytes, granulocytes, and tumor cells (including primary tumor cells and metastatic tumor cells).
- the targeting moiety targets the lipid nanoparticle to a hepatocyte.
- the lipid nanoparticles described herein may be lipidoid-based.
- the synthesis of lipidoids has been extensively described and formulations containing these compounds are particularly suited for delivery of polynucleotides (see Mahon et al., Bioconjug Chem. 2010 21 : 1448-1454; Schroeder et al., J Intern Med. 2010 267:9-21; Akinc et al., Nat. Biotechnol. 2008 26:561-569; Love et al., Proc Natl Acad Sci USA. 2010 107: 1864-1869; Siegwart et al., Proc Natl Acad Sci USA. 2011 108: 12996-3001).
- lipidoid formulations for intramuscular or subcutaneous routes may vary significantly depending on the target cell type and the ability of formulations to diffuse through the extracellular matrix into the blood stream. While a particle size of less than 150 nm may be desired for effective hepatocyte delivery due to the size of the endothelial fenestrae (see e.g., Akinc et al., Mol Ther. 2009 17:872-879), use of lipidoid oligonucleotides to deliver the formulation to other cells types including, but not limited to, endothelial cells, myeloid cells, and muscle cells may not be similarly size-limited.
- lipidoid formulations may have a similar component molar ratio.
- Different ratios of lipidoids and other components including, but not limited to, a neutral lipid (e.g., diacylphosphatidylcholine), cholesterol, a PEGylated lipid (e.g., PEG-DMPE), and a fatty acid (e.g., an omega-3 fatty acid) may be used to optimize the formulation of the mRNA or system for delivery to different cell types including, but not limited to, hepatocytes, myeloid cells, muscle cells, etc.
- a neutral lipid e.g., diacylphosphatidylcholine
- cholesterol e.g., a PEGylated lipid
- PEG-DMPE PEGylated lipid
- a fatty acid e.g., an omega-3 fatty acid
- Exemplary lipidoids include, but are not limited to, DLin-DMA, DLin-K-DMA, DLin-KC2-DMA, 98N12-5, C12-200 (including variants and derivatives), DLin-MC3-DMA and analogs thereof.
- lipidoid formulations for the localized delivery of nucleic acids to cells may also not require all of the formulation components which may be required for systemic delivery, and as such may comprise the lipidoid and the mRNA or system.
- a system described herein may be formulated by mixing the mRNA or system, or individual components of the system, with the lipidoid at a set ratio prior to addition to cells.
- In vivo formulations may require the addition of extra ingredients to facilitate circulation throughout the body.
- a system or individual components of a system is added and allowed to integrate with the complex. The encapsulation efficiency is determined using a standard dye exclusion assays.
- In vivo delivery of systems may be affected by many parameters, including, but not limited to, the formulation composition, nature of particle PEGylation, degree of loading, oligonucleotide to lipid ratio, and biophysical parameters such as particle size (Akinc et al., Mol Ther. 2009 17:872-879; herein incorporated by reference in its entirety).
- particle size Akinc et al., Mol Ther. 2009 17:872-879; herein incorporated by reference in its entirety.
- small changes in the anchor chain length of poly(ethylene glycol) (PEG) lipids may result in significant effects on in vivo efficacy.
- Formulations with the different lipidoids including, but not limited to penta[3-(l- laurylaminopropionyl)]-triethylenetetramine hydrochloride (TETA-5LAP; aka 98N12-5, see Murugaiah et al., Analytical Biochemistry, 401 :61 (2010)), C12-200 (including derivatives and variants), MD1, DLin-DMA, DLin-K-DMA, DLin-KC2-DMA and DLin-MC3-DMA can be tested for in vivo activity.
- the lipidoid referred to herein as "98N12-5" is disclosed by Akinc et al., Mol Ther. 2009 17:872-879).
- the lipidoid referred to herein as "C12-200” is disclosed by Love et al., Proc Natl Acad Sci USA. 2010 107: 1864-1869 and Liu and Huang, Molecular Therapy. 2010 669-670.
- the LNPs of the present disclosure in which a nucleic acid is entrapped within the lipid portion of the particle and is protected from degradation, can be formed by any method known in the art including, but not limited to, a continuous mixing method, a direct dilution process, and an in-line dilution process. Additional techniques and methods suitable for the preparation of the LNPs described herein include coacervation, microemulsions, supercritical fluid technologies, phase-inversion temperature (PIT) techniques.
- PIT phase-inversion temperature
- the LNPs used herein are produced via a continuous mixing method, e.g., a process that includes providing an aqueous solution a nucleic acid described herein in a first reservoir, providing an organic lipid solution in a second reservoir (wherein the lipids present in the organic lipid solution are solubilized in an organic solvent, e.g., a lower alkanol such as ethanol), and mixing the aqueous solution with the organic lipid solution such that the organic lipid solution mixes with the aqueous solution so as to substantially instantaneously produce a lipid vesicle (e.g., liposome) encapsulating the nucleic acid molecule within the lipid vesicle.
- a continuous mixing method e.g., a process that includes providing an aqueous solution a nucleic acid described herein in a first reservoir, providing an organic lipid solution in a second reservoir (wherein the lipids present in the organic lipid solution are solubilized in an organic solvent
- the LNPs used herein are produced via a direct dilution process that includes forming a lipid vesicle (e.g., liposome) solution and immediately and directly introducing the lipid vesicle solution into a collection vessel containing a controlled amount of dilution buffer.
- the collection vessel includes one or more elements configured to stir the contents of the collection vessel to facilitate dilution.
- the amount of dilution buffer present in the collection vessel is substantially equal to the volume of lipid vesicle solution introduced thereto.
- the LNPs are produced via an in-line dilution process in which a third reservoir containing dilution buffer is fluidly coupled to a second mixing region.
- the lipid vesicle (e.g., liposome) solution formed in a first mixing region is immediately and directly mixed with dilution buffer in the second mixing region.
- This disclosure is not limited to systems and methods described herein. Any delivery method that is capable of delivering the systems described herein can be used as long as it is capable of site-specifically integrating a template polynucleotide into the genome of a cell.
- compositions, systems and methods for correcting or replacing genes or gene fragments (including introns or exons) or inserting genes in new locations comprises recombination or integration into a safe harbor site (SHS).
- SHS safe harbor site
- a frequently used human SHS is the AAVS1 site on chromosome 19q, initially identified as a site for recurrent adeno-associated virus insertion.
- Another locus comprises the human homolog of the murine Rosa26 locus.
- Yet another SHS comprises the human Hl 1 locus on chromosome 22.
- a complete gene may be prohibitively large and replacement of an entire gene impractical.
- a method of the disclosure comprises recombining corrective gene fragments into a defective locus.
- the methods and compositions can be used to target, without limitation, stem cells for example induced pluripotent stem cells (iPSCs), HSCs, HSPCs, mesenchymal stem cells, or neuronal stem cells and cells at various stages of differentiation.
- stem cells for example induced pluripotent stem cells (iPSCs), HSCs, HSPCs, mesenchymal stem cells, or neuronal stem cells and cells at various stages of differentiation.
- methods and compositions of the disclosure are adapted to target organoids, including patient derived organoids.
- methods and compositions of the disclosure are adapted to treat muscle cells, not limited to cardiomyocytes for Duchene Muscular Dystrophy (DMD).
- the dystrophin gene is the largest gene in the human genome, spanning ⁇ 2.3 Mb of DNA. DMD is composed of 79 exons resulting in a 14-kb full-length mRNA. Common mutations include mutations that disrupt the reading frame of generate a premature stop codon.
- An aspect of DMD that lends it to gene editing as a therapeutic approach is the modular structure of the dystrophin protein. Redundancy in the central rod domain permits the deletion of internal segments of the gene that may harbor loss-of-function mutations, thereby restoring the open reading frame (ORFs).
- the methods and systems described herein are used to treat DMD by site- specifically integrating in the genome a polynucleotide template that repairs or replaces all or a portion of the defective DMD gene.
- MN Membranous Nephropathy
- C3GN C3 glomerulonephritis
- HCU Homocystinuria
- CFTR cystic fibrosis transmembrane conductance regulator
- the most common cystic fibrosis (CF) mutation F508del removes a single amino acid.
- recombining human CFTR into an SHS of a cell that expresses CFTR F508del is a corrective treatment path.
- the methods and systems described herein are used to CF by site-specifically integrating in the genome a polynucleotide template that corrects the mutation causing CF. Proposed validation is detection of persistent CFTR mRNA and protein expression in transduced cells.
- Sickle cell disease is caused by mutation of a specific amino acid — valine to glutamic acid at amino acid position 6.
- SCD is corrected by recombination of the HBB gene into a safe harbor site (SHS) and by demonstrating correction in a proportion of target cells that is high enough to produce a substantial benefit.
- the methods and systems described herein are used to sickle cell disease by site-specifically integrating in the genome a polynucleotide template that corrects the mutation causing the disease.
- validation is detection of persistent HBB mRNA and protein expression in transduced cells.
- DMD Duchenne Muscular Dystrophy.
- the dystrophin gene is the largest gene in the human genome, spanning ⁇ 2.3 Mb of DNA. DMD is composed of 79 exons resulting in a 14- kb full-length mRNA. Common mutations include mutations that disrupt the reading frame of generate a premature stop codon.
- An aspect of DMD that lends it to gene editing as a therapeutic approach is the modular structure of the dystrophin protein. Redundancy in the central rod domain permits the deletion of internal segments of the gene that may harbor loss-of-function mutations, thereby restoring the open reading frame (ORFs).
- recombination will be into safe harbor sites (SHS).
- SHS safe harbor sites
- a frequently used human SHS is the A4FS7 site on chromosome 19q, initially identified as a site for recurrent adeno-associated virus insertion.
- the site is the human homolog of the e murine Rosa26 locus (pubmed. ncbi.nlm.nih.gov/18037879).
- the site is the human Hl 1 locus on chromosome 22.
- Proposed target cells for recombination include stem cells for example induced pluripotent stem cells (iPSCs) and cells at various stages of differentiation. In some cases, a complete gene may be prohibitively large and replacement of an entire gene impractical. In such instances, rescuing mutants by recombining in corrected gene fragments with the methods and systems described herein is a corrective option.
- iPSCs induced pluripotent stem cells
- correcting mutations in exon 44 (or 51) by recombining in a corrective coding sequence downstream of exon 43 (or 50), using the methods and systems described herein is a corrective option.
- Proposed validation is detection of persistent DMD mRNA and protein expression in transduced cells.
- F8 Factor VIII.
- F8 A large proportion of severe hemophilia A patients harbor one of two types of chromosomal inversions in the F VIII gene.
- the recombinase technology and methods described herein are well suited to correcting such inversions (and other mutations) by recombining of the FVIII gene into a SHS.
- correcting factor VIII deficiency by recombining the FVIII gene into an SHS is a corrective path.
- the methods and systems described herein are used to correct factor VIII deficiency by site-specifically integrating in the genome a polynucleotide template that corrects the mutation causing the FIX deficiency. Proposed validation is detection of persistent FVIII mRNA and protein expression in transduced cells.
- Factor 9 Hemophilia B, also called factor IX (FIX) deficiency is a genetic disorder caused by missing or defective factor IX, a clotting protein.
- the methods and systems described herein are used to correct factor IX deficiency by site-specifically integrating in the genome a polynucleotide template that corrects the mutation causing the FIX deficiency.
- Proposed validation is detection of persistent FiX mRNA and protein expression in transduced cells.
- Ornithine transcarbamylase deficiency is a rare genetic condition that causes ammonia to build up in the blood.
- the condition - more commonly called OTC deficiency - is more common in boys than girls and tends to be more severe when symptoms emerge shortly after birth.
- the methods and systems described herein are used to correct OTC deficiency by site-specifically integrating in the genome a polynucleotide template that corrects the mutation causing the OTC deficiency or integrates a polynucleotide encoding a functional ornithine transcarbamylase enzyme.
- Proposed validation is detection of persistent OTC mRNA and protein expression in transduced cells.
- Phenylketonuria also called PKU, is a rare inherited disorder that causes an amino acid called phenylalanine to build up in the body. PKU is caused by a change in the phenylalanine hydroxylase (PAH) gene. This gene helps create the enzyme needed to break down phenylalanine.
- PKU phenylalanine hydroxylase
- the methods and systems described herein are used to correct PKU by site-specifically integrating in the genome a polynucleotide template that corrects the mutation causing the PKU deficiency or integrates a polynucleotide encoding a functional phenylalanine hydroxylase (PAH) gene.
- Proposed validation is detection of persistent PAH mRNA and protein expression in transduced cells.
- Homocystinuria is elevation of the amino acid, homocysteine (protein building block coming from our diet) in the urine or blood.
- Common causes of HCU include: problems with the enzyme cystathionine beta synthase (CBS), which converts homocysteine to the amino acid cystathionine (which then becomes cysteine) and needs the vitamin B6 (pyridoxine); and problems with converting homocysteine to the amino acid methionine.
- CBS cystathionine beta synthase
- pyridoxine pyridoxine
- the methods and systems described herein are used to correct HCU by site-specifically integrating in the genome a polynucleotide template that corrects the mutation causing the HCU or integrates a polynucleotide encoding a functional copy of a gene (e.g., CBS) able to reduce or prevent buildup of homocysteine in the urine.
- Proposed validation is detection of persistent CBS mRNA and protein expression in transduced cells.
- IgA Nephropathy (Berger’s disease). IgA nephropathy, also known as Berger's disease, is a kidney/autoimmune disease that occurs when an antibody called immunoglobulin A (IgA) builds up in the kidneys.
- the methods and systems described herein are used to treat Berger’s disease by administering to a patient an iPSC-derived Natural Killer cell that includes a polynucleotide site-specifically integrated in the genome of the cell using the methods described herein.
- the iPSC-NK cell Upon administering the iPSC-NK cell to the patient, the iPSC-NK cell is capable of removing native cells (e.g., B cells) that are responsible, at least in part, for the symptoms of Berger’s disease.
- ANCA vasculitis is an autoimmune disease affecting small blood vessels in the body. It is caused by autoantibodies called ANCAs, or Anti-Neutrophilic Cytoplasmic Autoantibodies. ANCAs target and attack a certain kind of white blood cells called neutrophils.
- the methods and systems described herein are used to treat ANCA vasculitis by administering to a patient an iPSC-derived Natural Killer cell that includes a polynucleotide site-specifically integrated in the genome of the cell using the methods described herein.
- the iPSC-NK cell Upon administering the iPSC-NK cell to the patient, the iPSC-NK cell is capable of removing native cells (e.g., B cells) that are responsible, at least in part, for the symptoms of ANCA vasculitis.
- Lupus is an autoimmune — a disorder in which the body’s immune system attacks the body’s own cells and organs.
- the methods and systems described herein are used to treat SLE/LN by administering to a patient an iPSC-derived Natural Killer cell that includes a polynucleotide site-specifically integrated in the genome of the cell using the methods described herein.
- the iPSC-NK cell Upon administering the iPSC-NK cell to the patient, the iPSC-NK cell is capable of removing native cells (e.g., B cells) that are responsible, at least in part, for the symptoms of SLE/LN.
- MN Membranous Nephropathy
- the methods and systems described herein are used to treat MN by administering to a patient an iPSC-derived Natural Killer cell that includes a polynucleotide site-specifically integrated in the genome of the cell using the methods described herein.
- the iPSC-NK cell Upon administering the iPSC-NK cell to the patient, the iPSC-NK cell is capable of removing native cells (e.g., B cells) that are responsible, at least in part, for the symptoms of MN.
- C3 glomerulonephritis C3GN.
- C3 glomerulopathy is a group of related conditions that cause the kidneys to malfunction.
- the major features of C3 glomerulopathy include high levels of protein in the urine (proteinuria), blood in the urine (hematuria), reduced amounts of urine, low levels of protein in the blood, and swelling in many areas of the body.
- Affected individuals may have particularly low levels of a protein called complement component 3 (or C3) in the blood.
- the methods and systems described herein are used to treat C3 glomerulopathy by administering to a patient an iPSC-derived Natural Killer cell that includes a polynucleotide site-specifically integrated in the genome of the cell using the methods described herein.
- the iPSC-NK cell Upon administering the iPSC-NK cell to the patient, the iPSC-NK cell is capable of removing native cells (e.g., B cells) that are responsible, at least in part, for the symptoms of C3 glomerulopathy.
- methods of treatment comprises administering an effective amount of the pharmaceutical composition comprising the nucleic acid construct or vectorized nucleic acid construct described above to a patient in need thereof.
- the system e.g., any of the systems described herein
- the systems are delivered to a patient, thereby delivering to a cell in vivo.
- DNA or RNA viral vectors can be administered directly to patients (in vivo) or they can be used to treat cells in vitro, and the modified cells may optionally be administered to patients (ex vivo).
- Conventional viral based systems to be used herein could include retroviral, lentivirus, adenoviral, adeno-associated and herpes simplex virus vectors for gene transfer. Integration in the host genome is possible with the retrovirus, lentivirus, and adeno-associated virus gene transfer methods, often resulting in long term expression of the inserted transgene. Additionally, high transduction efficiencies have been observed in many different cell types and target tissues.
- the co-delivery system described herein e.g., a gene editor construct packaged in a LNP and a donor template packaged in a vector
- the co-delivery system described herein is administered intravenously.
- the co-delivery system described herein e.g., a gene editor construct packaged in a LNP and a donor template packaged in a vector
- the co-delivery system described herein e.g., a gene editor construct packaged in a LNP and a donor template packaged in a vector
- the co-delivery system described herein e.g., a gene editor construct packaged in a LNP and a donor template packaged in a vector
- the co-delivery system described herein e.g., a gene editor construct packaged in a LNP and a donor template packaged in a vector
- Methods of non-viral delivery of the donor DNA template described herein include lipofection, nucleofection, microinjection, biolistics, virosomes, liposomes, immunoliposomes, polycation or lipidmucleic acid conjugates, naked DNA, artificial virions, and agent-enhanced uptake of DNA.
- Lipofection is described in e.g., U.S. Pat. Nos. 5,049,386, 4,946,787; and 4,897,355) and lipofection reagents are sold commercially (e.g., TransfectamTM andLipofectinTM).
- Cationic and neutral lipids that are suitable for efficient receptor-recognition lipofection of polynucleotides include those of Feigner, WO 91/17424; WO 91/16024. Delivery can be to cells (e.g. in vitro or ex vivo administration) or target tissues (e.g. in vivo administration).
- mRNA delivery methods and compositions that may be utilized in the present disclosure including, for example, PCT/US2014/028330, US8822663B2, NZ700688A, ES2740248T3, EP2755693A4, EP2755986A4, WO2014152940A1, EP3450553B1, BRI 12016030852A2, and EP3362461A1.
- Expression of CRISPR systems in particular is described by W02020014577.
- Each of these publications are incorporated herein by reference in their entireties. Additional disclosure hereby incorporated by reference can be found in Kowalski et al., “Delivering the Messenger: Advances in Technologies for Therapeutic mRNA Delivery,” Mol Therap., 2019; 27(4): 710-728. 6.
- Example 1 Delivery of gene editor polynucleotide sequence packaged in LNP and donor template packaged in AAV
- a gene editor polynucleotide construct is packaged into a LNP (FIG. 1), wherein the gene editor polynucleotide sequence comprises a polynucleotide sequence encoding a prime editor protein linked to an integrase via peptide linker a polynucleotide sequence encoding an attachment site-containing guide RNA (atgRNA); a polynucleotide sequence encoding a nickase guide RNA (ngRNA).
- atgRNA attachment site-containing guide RNA
- ngRNA nickase guide RNA
- a donor template polynucleotide construct is packaged in an AAV vector (FIG. 2).
- Co-administration of the gene editor construct packaged LNP and the donor template packaged AAV co-delivers the gene editor construct to a cell cytoplasm and the donor template to a cell nucleus.
- the direct activity of the associated integrase to the specific genomic site is guided.
- Gene editor construct expression, with template co-delivery, results in integration of template “cargo” at a precisely defined target location.
- Example 2 Delivery of gene editor polynucleotide sequence packaged in LNP and donor template capable of self-circularization packaged in AAV
- a gene editor polynucleotide construct is packaged into a LNP (FIG. 1), wherein the gene editor polynucleotide sequence comprises a polynucleotide sequence encoding a prime editor protein linked to an integrase via peptide linker a polynucleotide sequence encoding an attachment site-containing guide RNA (atgRNA); a polynucleotide sequence encoding a nickase guide RNA (ngRNA).
- atgRNA attachment site-containing guide RNA
- ngRNA nickase guide RNA
- a donor template polynucleotide construct is packaged in an AAV vector (FIG. 2).
- Co-administration of the gene editor construct packaged LNP and the donor template packaged AAV co-delivers the gene editor construct to a cell cytoplasm and the donor template to a cell nucleus.
- Integrase-mediated self-circularization of donor template occurs at integration target recognition sites within the AAV genome (FIG. 3).
- an orthogonal integrase landing site i.e., distinct att site from att sites used for selfcircularization
- Gene editor construct expression, with template co-delivery and integrase-mediated circularization of template results in integration of template “cargo” at a precisely defined target location.
- Example 3 Delivery of gene editor polynucleotide sequence packaged in LNP and atgRNA, ngRNA, and donor template co-packaged in AAV
- a gene editor polynucleotide construct is packaged into a LNP (FIG. 4), wherein the gene editor polynucleotide sequence comprises a polynucleotide sequence encoding a prime editor protein linked to an integrase via peptide linker.
- a polynucleotide sequence encoding an attachment site-containing guide RNA (atgRNA), a polynucleotide sequence encoding a nicking guide RNA (ngRNA), and donor template are packaged in an AAV vector (FIG. 4).
- Co-administration of the gene editor construct packaged LNP and the atgRNA, ngRNA, donor template packaged AAV co-delivers the gene editor construct to a cell.
- Integrase- mediated self-circul arization of donor template occurs at integration target recognition sites within the AAV genome (FIG. 3).
- an orthogonal integrase landing site i.e., distinct att site from att sites used for self-circularization
- Gene editor construct expression, with atgRNA, ngRNA, and template co-delivery and integrase-mediated circularization of template results in integration of template “cargo” at a precisely defined target location.
- Example 4 Delivery of gene editor polynucleotide sequence and ngRNA packaged in LNP and atgRNA and donor template co-packaged in AAV
- a gene editor polynucleotide construct and a nicking guide RNA (ngRNA) are packaged into a LNP (FIG. 5), wherein the gene editor polynucleotide sequence comprises a polynucleotide sequence encoding a prime editor protein linked to an integrase via peptide linker.
- a polynucleotide sequence encoding an attachment site-containing guide RNA (atgRNA) and donor template are packaged in an AAV vector (FIG. 5).
- Example 5 Intramolecular circularization of plasmid and packaged AAV genomes
- FIG. 7 A universal ddPCR probe capable of binding any linear or circularized AAV genome was designed, wherein the universal ddPCR probe is designed to only give signal upon cognate recombinase/integrase mediated circularization (FIGs. 8A-8B).
- Circularization products are amplified by use of a circle junction PCR primer set that is designed to amplify only circular products due to primer direction constraints.
- an attR scar quencher-fluorophore probe was designed.
- a template reference primer set was designed and generated to quantify total template DNA (linear or circular confirmation) (FIGs. 8A-8B).
- FIG. 10 demonstrates circularization of AAV pDNA and packaged AAV genomic DNA for both IX Bxbl and 2X Bxbl conditions (confirmed by use of attR ddPCR primer set). Further, replicates that lacked either Bxbl or AAV pDNA substrate demonstrated insignificant circularization. All three of the Cre-, FLPe-, and Bxbl -targeted AAV pDNA substrates demonstrated circularization upon cognate recombinase/integrase introduction, as confirmed by using the universal ddPCR probe (FIG. 11). Moreover, Cre-, FLPe-, and Bxbl-mediated circularization of packaged AAV DJ genomes substrates were demonstrated and confirmed using the universal ddPCR probe (FIG. 12).
- the Bxbl-mediated attR scar probe provided similar percent circularization quantification compared to the universal probe.
- Example 6 In vitro beacon placement in primary mouse hepatocytes and primary human hepatocytes using mRNA and AAV for co-delivery
- This example assessed the efficiency of in vitro beacon placement in primary human hepatocytes using mRNA delivering of a polynucleotide encoding a gene editor polynucleotide construct and AAV to deliver the first and second atgRNA. See FIG. 15 for a non-limiting example of a dual atgRNA-mediated insertion of an integration recognition site.
- the mRNA and AAV were delivered into the primary mouse hepatocytes (PMH) using (i) concurrent delivery (“co-dose”), (ii) AAV delivery followed by a “1- day delay” before delivery of the mRNA, or (iii) AAV delivery followed by a “2-day delay” before delivery of the mRNA.
- Beacon placement was then assessed using next-generation sequencing of DNA isolated from cells subjected to the delivery conditions mentioned above.
- the mRNA encoding the gene editor polynucleotide construct was delivered in various amounts per well: 2000 ng, 1000 ng, 500 ng, 250 ng, 125 ng, 62.5 ng, and 31.25 ng.
- AAV encoding the first and second atgRNA see Table 12).
- the primary mouse hepatocyte data is shown in FIG. 16 and the human primary hepatocyte data is shown in FIG. 17.
- Example 7 In vivo beacon placement with mRNA + AAV guide
- mice were administered AAV containing the first atgRNA (SEQ ID NO: 543; Table 12) and the second atgRNA (SEQ ID NO: 544) targeting the Nolcl locus at 3E11 to 1E12 vector genomes (vg) per animal two 2 weeks prior to administration of the mRNA containing the gene editing polynucleotide construct (see FIG. 18).
- mRNA was delivered using various LNP formulations (e.g., LNP #F1 , LNP #F2, and LNP #F3) at concentrations ranging from 5 mg/kg to 0.5 mg/kg via intravenous injection (see FIG. 18).
- LNP formulations e.g., LNP #F1 , LNP #F2, and LNP #F3
- concentrations ranging from 5 mg/kg to 0.5 mg/kg via intravenous injection (see FIG. 18).
- liver tissue was harvested, genomic DNA was isolated, and beacon efficiency was assessed by NGS. As shown in FIG. 18, three conditions resulted in vivo beacon placement efficiency greater than 10%.
- this data provided proof-of-concept for successful in vivo beacon placement using AAV to deliver the first and second atgRNA and LNPs to deliver the mRNA encoding the gene editor polynucleotide construct.
- Example 8 In vivo integration in mice using AAV to deliver the template polynucleotide and Adenovirus to deliver BxBl
- In vivo integration efficiency in AttP mice was assessed using adenovirus to deliver an integrase (e.g., Bxbl) and an AAV to deliver the template polynucleotide.
- adenovirus to deliver an integrase (e.g., Bxbl) and an AAV to deliver the template polynucleotide.
- the adenovirus i.e., adenovirus containing polynucleotide encoding the integrase
- the AAV i.e., AAV containing the template polynucleotide and an attB site
- mice containing dual AttP sites integrated in to the Rosa26 locus (B6.RosaBxb-GT/GA; female, Strain# 036152).
- the Rosa26 locus included a first AttP site comprising a GT dinucleotide and a second AttP site comprising a GA dinucleotide.
- the AAV was a scAAV8 containing a vector having a template polynucleotide and a 38 bp GT AttB site.
- the Adenovirus was an adenovirus-type 5 (Ad5) containing a polynucleotide encoding Bxbl (“Bxbl AdV”) (SEQ ID NO: 563; Table 14). Mice were administered the adenovirus and AAV according to the experimental details in Table 13.
- this data establishes proof-of-concept for in vivo integration using an adenovirus to deliver and drive expression of Bxbl and an AAV to deliver the template polynucleotide to be integrated into a mammalian genome, in this case, the mouse genome.
- the first LNP contained mRNA encoding a prime editing system and a first synthetic atgRNA (atgRNAl).
- the mRNA and atgRNAl were included at 1 : 1 ratio in the first LNP.
- the second LNP contained mRNA encoding a prime editing system and a second synthetic atgRNA (atgRNA2).
- the mRNA and atgRNA2 were included at a 1 : 1 ratio in the second LNP.
- Each of the first and second atgRNAs targeted the mouse Nolcl locus and each encoded a portion of an integration recognition site (a “beacon”).
- the LNP mixture was administered to the neonatal mice (2-5 day old CD-I mice) according to the experimental details in Table 16.
- liver samples either whole liver for groups 1-3 or liver punches from each lobe for groups 4-6 (see Table 13) were collected and genomic DNA was isolated. Beacon placement was detected using ddPCR and NGS.
- Neonates were also assessed at six weeks after administration of the LNP mixture. Beacon placement was detected using ddPCR and NGS. As shown in FIG. 21 A., at six weeks post administration, ddPCR revealed about 4% beacon placement (in Nolcl alleles) for a 3mg/kg dose of the LNP mixture. Confirmation of beacon placement using NGS showed about 15% beacon placement (in Nolcl alleles) for a 3 mg/kg dose of the LNP mixture (see FIG. 2 IB). Assessment of the percent of integrated beacons that included the expected integration recognition site (“perfect beacon”) revealed that about 3.5% of beacons were comprised of perfect beacons (see FIG. 21C).
- this data demonstrated successful in vivo site-specific integration of an integration recognition site.
- this data showed that a split LNP approach can be used for site- specifically integrating an integration recognition site in vivo in a mammalian genome, in this case neonatal mice.
- the first LNP contained mRNA encoding a prime editing system and a first synthetic atgRNA (atgRNAl).
- the mRNA and atgRNAl were included at different ratios (e.g., 1 :0.5, 1: 1, and 1 :2) ratio in the first LNP.
- the second LNP contained mRNA encoding a prime editing system and a second synthetic atgRNA (atgRNA2).
- the mRNA and atgRNA2 were included at different ratios (e.g., 1 :0.5, 1 : 1, and 1 :2) ratio in the second LNP.
- the first and second atgRNAs targeted mouse Factor IX (“mF9”) locus and each encoded a portion of an integration recognition site (“beacon”). Similar to Example 9, atgRNAl and atgRNA2 together included a 6bp overlap and were combined 1 : 1 as mixture prior to administration.
- the first atgRNA and second atgRNA are provide in Table 17, where the atgRNA include one or more 2’O-methyl modifications and one or more phosphorothioate linkages.
- the LNP mixture was administered to female CD-I mice 6-8 weeks old according to the experimental details in Table 18.
- **1:0.5:0.5 mRNA:atgRNAl 1:1; mRNA:atgRNA21:1; LNPs mixed 1:1
- ***1:1:1 mRNA:atgRNAl 1:2; mRNA:atgRNA21:2; LNPs mixed 1:1
- ddPCR revealed about 0.8% beacon placement (in mF9 alleles) following administration of a 1 :0.25:0.25 ratio of mRNA:atgRNAl :atgRNA2.
- beacon placement using NGS showed about 14% beacon placement (in mF9 alleles) following administration of the 1 :0.25:0.25 ratio of mRNA:atgRNAl :atgRNA2 (see FIG. 22B). Similar to Example 9, an NGS-based assay was used to determined what percentages of the integrated beacons included the expected integration recognition site (“perfect beacon”). As shown in FIG. 22C, about 0.02% of the beacons placed in the mF9 locus were “perfect” beacons.
- this data showed successful in vivo site-specific integration of an integration recognition site in adult mice.
- this data showed that the ratio of mRNA to atgRNA is an important consideration in determining efficacy of in vivo site-specific integration of an integration recognition site.
- Engineered integrases described in FIG. 23 were evaluated for ability to mediate programmable gene insertion, measured as Integration % (see FIG. 24B) or “Beacon Occupancy” (see FIG. 24C). Percent beacon placement, which precedes integration, is illustrated in FIG. 24 A.
- the engineered integrases were introduced into primary human hepatocytes (PHH) at a high dose (750ng) and a low dose (250ng) along with an mRNA encoding a gene editor polypeptide (i.e., a Cas9 nickase fused to a reverse transcriptase (SEQ ID NO: 589)); an AAV expressing an atgRNA (the atgRNA comprising a spacer sequence with sequence complementarity to a sequence in the human factor IX locus, a scaffold, an RT template comprising an integration recognition site, and a primer binding site (See Table 19 below); and a donor polynucleotide comprising a cognate integration recognition site.
- the AAV used to deliver the atgRNA also included the donor polynucleotide sequence.
- the AAV sequence is AAVG048 (SEQ ID NO: 592; See Table 20).
- Engineered integrases described in FIG. 25 were evaluated for their ability to mediate programmable gene insertion (as measured by “Occupancy %” (FIG. 26)).
- the engineered integrases were introduced into primary human hepatocytes (PHH) at a high dose (1.3 pmol) and a low dose (0.2 pmol) along with a gene editor polypeptide (i.e., mRNA encoding a gene editor polypeptide (i.e., a Cas9 nickase fused to a reverse transcriptase)); an AAV expressing an atgRNA (the atgRNA comprising a spacer sequence with sequence complementarity to a sequence in the human factor IX locus, a scaffold, an RT template comprising an integration recognition site, and a primer binding site (see Table 19 above for atgRNA sequences); and a donor polynucleotide template comprising a cognate integration recognition site.
- a gene editor polypeptide i.e., mRNA encoding a gene editor polypeptide (i.e., a Cas9 nickase fused to a reverse transcriptase)
- the AAV used to deliver the atgRNA also included the donor polynucleotide template sequence.
- the AAV sequence is AAVG048 (SEQ ID NO: 592; see Table 20 above). Genomic DNA was harvested 3 days after transduction. Data is shown in
- Engineered integrases described in FIG. 23 comprising were evaluated for their ability to mediate programmable gene insertion (as indicated by “Occupancy %” (FIGs. 27A-27D)).
- additional experiments were performed to assess engineered BxBl integrases having a c-terminal tag.
- these engineered integrases were assessed for their ability to mediate programmable gene insertion (e.g., as indicated by “% Beacon Occupancy”).
- the engineered integrases were introduced into a primary human hepatocyte (PHH) at amounts of 1000 ng, 500 ng, and 250 ng, along with a gene editor polypeptide (i.e., mRNA encoding a gene editor polypeptide (i.e., a Cas9 nickase fused to a reverse transcriptase)); an atgRNA comprising a spacer sequence with sequence complementarity to a sequence in the human factor IX locus, a scaffold, an RT template comprising an integration recognition site, and a primer binding site (see Table 19 above for atgRNA sequences); and a donor polynucleotide template comprising a cognate integration recognition site at 40 fmol, 20 fmol, and 10 fmol.
- a gene editor polypeptide i.e., mRNA encoding a gene editor polypeptide (i.e., a Cas9 nickase fused to a reverse transcriptase)
- the donor polynucleotide template was introduced into the cells as an AAV vector (PL 753 (SEQ ID NO: 593)). Genomic DNA was harvested 3 days after transduction. [0578] Data is shown in FIGs. 27A-27D. Overall, this data showed that a c-terminal tag reduced integration efficiency of BxBl.
- the integration enzymes engineered to include stabilization domains were also assessed for their dose dependent impact on PGI. Additional engineered integration enzymes are described in FIG. 28 (see also Table 26).
- the engineered integrases were introduced into a primary human hepatocyte (PHH) at a 250 ng along with a gene editor polypeptide (i.e., mRNA encoding a gene editor polypeptide (i.e., a Cas9 nickase fused to a reverse transcriptase)); an atgRNA comprising a spacer sequence with sequence complementarity to a sequence in the human factor IX locus, a scaffold, an RT template comprising an integration recognition site, and a primer binding site (see Table 19 above in Example 11 for atgRNA sequences); and a donor polynucleotide template comprising a cognate integration recognition site. Genomic DNA was harvested 3 days after transduction.
- a gene editor polypeptide i.e., mRNA encoding a gene editor polypeptide (i.e., a Cas9 nickase fused to a reverse transcriptase)
- an atgRNA comprising a spacer sequence with sequence complementar
- FIGs. 29A-29B Data for programmable gene insertion (PGI) is shown in FIGs. 29A-29B. This data showed that an engineered BxBl comprising a stabilon domain resulted in increased programmable gene insertion (PGI) compared to the other BxB 1 polypeptides, including other engineered BxBl polypeptides.
- a gene editor polypeptide i.e., mRNA encoding a gene editor polypeptide (i.e., a Cas9 nickase fused to a reverse transcriptase)
- an atgRNA comprising a spacer sequence with sequence complementarity to a sequence in the human factor IX locus, a scaffold, an RT template comprising an integration recognition site, and a primer binding site (see Table 19 above for atgRNA sequences)
- a donor polynucleotide template comprising a cognate integration recognition site.
- Engineered integration enzymes were assessed for their ability to mediate PGI in pluripotent stem cells.
- a non-limiting exemplary workflow is as described in FIG. 30A.
- Engineered integration enzymes that were used in these experiments are as described in FIG. 30B (see also Table 26).
- the engineered integrases were introduced into a pluripotent stem cell clone 52 or clone 17 along with a BxBl mRNA ; and a donor polynucleotide template comprising a cognate integration recognition site (PL1113 (SEQ ID NO: 594; see Table 22 below)).
- PGI programmable gene insertion
- FIG. 31A shows ddPCR data for PGI for clone 52 at day 3 and day 6.
- Flow cytometry was used to assess GFP expression which indicates programmable gene insertion (PGI) (see FIG. 31B). This data shows that PL1323 and PL1325 yield % GFP+ cells by flow -50%.
- Engineered integrases were also assessed for their ability to mediate programmable gene insertion in hematopoietic stem cells.
- the engineered integrases were introduced into a hematopoietic stem cell along with a gene editor polypeptide (i.e., mRNA encoding a gene editor polypeptide (i.e., a Cas9 nickase fused to a reverse transcriptase)); an atgRNA comprising a spacer sequence a scaffold, an RT template comprising an integration recognition site, and a primer binding site; and a donor polynucleotide template comprising a cognate integration recognition site.
- a gene editor polypeptide i.e., mRNA encoding a gene editor polypeptide (i.e., a Cas9 nickase fused to a reverse transcriptase)
- an atgRNA comprising a spacer sequence a scaffold, an RT template comprising an integration recognition site, and a primer binding site
- a donor polynucleotide template comprising a cognate integration recognition site.
- the gene editor polypeptide i.e., mRNA encoding a gene editor polypeptide
- the atgRNA were introduced into a cell using a first electroporation and the engineered integrase and the donor polynucleotide template (mini circle) were introduced into the cell using a second electroporation.
- Engineered integrases used in these experiments are as shown in FIG. 32A
- Genomic DNA was harvested 3 days after transduction.
- Data for programmable gene insertion (PGI) is shown in FIG. 32B. This data shows that a BxB 1 integrase engineered to include a stabilon motif improves PGI by 3-5 fold. 6.17.
- BxBl was engineered to include a stabilon domain or an Exin21 domain (see, e.g., engineered integration enzymes shown in FIG. 33 (see also Table 26)).
- the engineered integration enzymes described in FIG. 33 were introduced into a HEK293 cell along with a gene editor polypeptide (i.e., mRNA encoding a gene editor polypeptide (i.e., a Cas9 nickase fused to a reverse transcriptase)); an atgRNA comprising a spacer sequence with sequence complementarity to a sequence in the human factor IX locus, a scaffold, an RT template comprising an integration recognition site, and a primer binding site (see Table 19 above for atgRNA sequences); and a donor polynucleotide template comprising a cognate integration recognition site.
- the data is shown in FIG. 34.
- the sub-set of engineered BxBl integrases shown in FIG. 35 were selected for further study.
- BxBl integrases were engineered to include an amino acid modification with the aim that the modification would improve stability (e.g., half-life).
- FIG. 36A shows degron motifs and lysine residues that are candidate for amino acid modifications that may increase the stability of a BxBl integrase.
- mRNAs encoding BxBl were optimized with the aim of improving stability (i.e., half-life of the mRNA) upon being introduced into a cell.
- FIGs. 37A-37B show first generations attempts at optimizing BxB 1 RNA structure using the linear design algorithm comparing non-optimized mRNA encoding BxB 1 in FIG. 37A with optimized mRNA encoding BxBl in FIG. 37B.
- FIGs. 38A-38B show first generations attempts at optimizing an RNA structure for RNA encoding nCas9-RT using the LinearDesign algorithm comparing non-optimized mRNA encoding nCas9-RT in FIG. 38A with optimized mRNA encoding nCsa9-RT in FIG. 38B.
- nCas9-RTs in FIGs. 38A-38B were tested for their ability to insert integration recognition sites into a genome at the human factor IX locus.
- the nCas9-RTs in FIGs. 38A-38B were introduced into a cell along with an atgRNA comprising a spacer sequence with sequence complementarity to a sequence in the human factor IX locus, a scaffold, an RT template comprising an integration recognition site, and a primer binding site (see Table 19 above in Example 11 for atgRNA sequences).
- Beacon placement data is shown in FIG. 39.
- Fusion proteins comprising an nCas9-RT are fused with engineered integration enzymes (e.g., BxBl) and tested for their ability to mediate PGI.
- engineered integration enzymes e.g., BxBl
- fusion proteins included those as shown in FIG. 40 (see also Table 26). These fusions proteins are introduced into cells along with an atgRNA and a donor polynucleotide template. Genomic DNA is harvested three days after transduction.
- Fusion proteins comprising a nCas9 and RT are fused with engineered integration enzymes (e.g., BxBl) in various combinations and tested for their ability to mediate PGI.
- engineered integration enzymes e.g., BxBl
- fusion proteins included those as shown in FIG. 41 (see also Table 26). These fusions proteins are introduced into cells along with an atgRNA and a donor polynucleotide template. Genomic DNA is harvested three days after transduction.
- a non-limiting exemplary workflow is as described in FIG. 30A.
- the engineered integrases were introduced into a pluripotent stem cell clone 52 or clone 17 along and a donor polynucleotide template comprising a cognate integration recognition site (PL1113 (SEQ ID NO: 591; see Table 22) or PL2134.
- iPSC clones 52 and 17 include beacons site-specifically integrated into their genomes.
- Genomic DNA was harvested 3 days after transduction.
- Digital droplet PCR (ddPCR) data for programmable gene insertion (PGI) with PL1113 donor at day 3 is shown in FIG. 43 for the indicated engineered BxBl polypeptides.
- Flow cytometry data for PGI with PL1113 donor at day 7 is shown in FIGs. 44A and 44B.
- FIG. 44A provides a comparison between percent PGI between ddPCR data and flow cytometry data.
- Flow cytometry was used to assess GFP expression, which indicates programmable gene insertion (PGI).
- FIG. 45 PGI with PL2134 donor was also assessed and compared to PGI with PL1113. This data shows that PGI was higher with PL1113 than PL2134.
- the engineered integration enzymes fused to a gene editor polypeptide are referred to herein as the all-in-one constructs.
- mRNA encoding the engineered integrases from FIG. 46 were introduced into a primary human hepatocytes (line HU8403) along with a pair of atgRNA (AA089: atgRNAl is SEQ ID NO: 611 and atgRNA2 is SEQ ID NO: 612 (see Table 24)). Electroporation was performed as described elsewhere herein (see Example 22). mRNA was electroporated at either 187 fmol or 374 fmol. Controls included mRNA encoding nCas9-RT with and without a stabilization domain and a mRNA encoding RT-IRES-nCas9.
- Genomic DNA was harvested 4 days after electroporation.
- Digital droplet PCR (ddPCR) was used to assess beacon placement for each of the constructs tested (see FIG. 46).
- ddPCR data for beacon placement revealed for the all-in-one mRNAs were slightly lower than the control (PL883) but were comparable.
- the nCas9-RT-Bxbl orientation produced more efficient beacon placement compared to the Bxbl-nCas9-RT orientation (see FIG. 47).
- mRNA encoding PL1931 (SEQ ID NO: 631 (amino acid) and SEQ ID NO: 632 (nucleic acid); see FIG. 46) was introduced into a primary human hepatocyte (line HU8403) along with a pair of atgRNA (AA115 (atgRNAl (SEQ ID NO: 613) and atgRNA2 (SEQ ID NO: 614); see Table below) using MessengerMax lipofection.
- AA115 atgRNAl
- SEQ ID NO: 614 atgRNA2
- MessengerMax lipofection At day 0, 33,000 HU8403 cells were plated per well and MessengerMax, 0.15 pl atgRNA pair, and either 0.15 l or 0.3 pl of various concentrations of Bxbl mRNA (see FIG. 48).
- beacon placement was similar between mRNA PL1931 and mRNA PL883 at equimolar input.
- PGI in PHH was compared following lipofection with an mRNA encoding PL1931 (“all-in-one”) (SEQ ID NO: 631 (amino acid) and SEQ ID NO: 632 (nucleic acid)); see FIG. 46) or two mRNAs one encoding nCas9-RT and a second encoding BxB 1.
- mRNA was introduced into PHH line HU8403 along with a pair of atgRNA (AA115) using MessengerMax lipofection.
- BxBl was introduced at 210 fmol (or -125 ng) (note specific constructs included: PL1323 (cmyc-NLS-no stabilon), PL1325 (SV40-NLS- Stabilon), and PL 1709 (SV40-exin21 -stabilon).
- a 3 lipoplex approach was used: guides 0.15pl per well (20 mM) with 0.15 pl MessengerMax (or 30 pl MessengerMax); BxBl mRNA at 250 ng with 0.15 pl MessengerMax (or 30 pl MessengerMax) per well; and nCas9- RT mRNA PL883 (SEQ ID NO: 617) fixed at 400 ng or 187 fmol with 0.15 pl MesserngerMax (or 30 pl MessengerMax) per well.
- Genomic DNA was harvested 4 days after lipofection.
- Digital droplet PCR (ddPCR) was used to assess PGI. As shown in FIG. 49, PGI was around 1% for each of the conditions tested. Looking closer at each PGI event, FIG. 50 and FIG.
- FIG. 51 show data for total edits (AttB + AttL). Analysis of whether MessengerMax volume impacted beacon placement revealed that increased MessengerMax volume decreased beacon placement but increased integration (FIG. 52). Analysis of the 3 lioplex approach in FIG. 53, revealed a 20-30% hit to beacon placement ratio observed with integration samples (3 lipo) compared to beacon only samples (2 lipo). This data shows that mRNAs encoding a BxBl-stabilon result in increased beacon occupancy compared to mRNA encoding BxBl without a stabilon (see PL1323 (SEQ ID NO: 618 (amino acid) and SEQ ID NO: 619 (nucleic acid)).
- BxB 1 was engineered to include a stabilon peptide motif and/or an Exin21 peptide motif. See FIGs. 54, 57, 59, 61, and 63, see also Table 27.
- the engineered integration enzyme constructs described in FIG. 54, 61, and 63 were introduced into HEK293 mediating integration of a donor polynucleotide (cargo) having an integration site cognate to the integration site in the target genome. Recombination site attL was produced and percent integration was measured by ddPCR. The data is provided in FIG. 55 and FIG. 64. The results showed that various engineered bxbl integrases showed significant improved percent integration as compared to others. Percent integration was measured by ddPCR.
- beacon placement was performed in neonatal mice. Beacon placement was conducted using methods such as those already described in this text. For example, see Example 9. Next, we performed integration of cargo into the genome of the cell using methods previously described herein this document. The results showed that various Bxbl integrases displayed significant beacon conversion compared to others. See FIG. 56. Beacon conversion was measured using ddPCR.
- PGI as measured by beacon conversion mediated by the engineered integration enzymes was assessed in primary human hepatocytes (PHH, line HU8403). Data is shown in FIG. 58 and FIG. 60. See also Table 26 and 27. Results showed that various engineered BxBl constructs resulted in increased integration of the donor polynucleotide compared to the other engineered BxB 1 polypeptides.
- FIG. 62A shows percent integration resulting in recombination site attL.
- FIG. 62B shows percent integration resulting in recombination site attR.
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Genetics & Genomics (AREA)
- Engineering & Computer Science (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Molecular Biology (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biotechnology (AREA)
- General Engineering & Computer Science (AREA)
- Biomedical Technology (AREA)
- Microbiology (AREA)
- Biochemistry (AREA)
- General Health & Medical Sciences (AREA)
- Plant Pathology (AREA)
- Biophysics (AREA)
- Physics & Mathematics (AREA)
- Mycology (AREA)
- Cell Biology (AREA)
- Medicinal Chemistry (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
Abstract
The present disclosure provides compositions comprising engineered integration enzymes and methods of using the same. In certain embodiments, the engineered integration enzyme comprises a stabilization domain that increases the stability of the integration enzyme as compared to integration enzymes not comprising the stabilization domain. In other embodiments, the engineered integration enzyme comprises at least a first amino acid modification that increases the stability of the integration enzyme as compared to an integration enzyme that does not comprise the amino acid modification.
Description
PROGRAMMABLE GENE INSERTION USING ENGINEERED INTEGRATION ENZYMES
1. CROSS-REFERENCE TO RELATED APPLICATIONS
[0001] This application claims the benefit of and priority to U.S. Provisional Application No. 63/580,293, filed September 01, 2023, which is hereby incorporated by reference in its entirety.
2 SEQUENCE LISTING
[0002] The instant application contains a Sequence Listing which has been submitted electronically via Patent Center and is hereby incorporated by reference in its entirety. Said XML copy, created on Month XX, 2024, is named XX, and is XX bytes in size.
2. BACKGROUND
[0003] Programmable, efficient, and multiplexed genome integration of large, diverse DNA cargo independent of DNA repair is a challenge for various genome editing techniques. Many current gene integration approaches require double strand breaks that evoke DNA damage responses and rely on repair pathways that are inactive in terminally differentiated cells. Furthermore, CRISPR-based approaches that bypass double stranded breaks, such as Prime editing, are limited to modification or insertion of short sequences.
[0004] There is a need in the art for techniques that address and overcome these shortcomings and provide varied options that enable the co-delivery of gene editor constructs and associated donor polynucleotides for the insertion and/or deletion of large sequences into cells for therapeutic and circuit-based uses for broad purposes, across eukaryotic as well as prokaryotic systems.
3. SUMMARY
[0005] The present disclosure describes integration enzymes engineered such that upon being introduced into a cell, the integration enzyme has increased stability (e.g., half-life) compared to a control integration enzyme not engineered to have increased stability. The increase in stability extends the capacity of the integration enzyme to mediate integration. In typical embodiments, the engineered integration enzyme includes an integration enzyme or fragment thereof, an at least first stabilization domain, and a nuclear localization signal.
[0006] In one embodiment, the present disclosure provides integration enzymes engineered to have increased stability for use in site-specific genetic engineering using Programmable Addition via Site-Specific Targeting Elements (PASTE) (see lonnidi et al. doi:
10.1101/2021.11.01.466786; the entirety of lonnidi et al. is incorporated by reference), transposon-mediated gene editing, or other suitable gene editing or gene incorporation technology. Non-limiting examples of PASTE are also described in U.S. Pat. 11,572,556 and PCT Publication Nos. WO 2022/087235A and WO 2023/077148A1, each of which are hereby incorporated by reference in their entireties.
[0007] In one aspect, this disclosure features an engineered integration enzyme, comprising: (a) an integration enzyme or fragment thereof; (b) an at least first stabilization domain; and (c) a nuclear localization signal (NLS).
[0008] In some embodiments, the integration enzyme or fragment thereof comprises integration enzyme, recombinase, or transposase activity.
[0009] In some embodiments, the integration enzyme is a large serine integrase.
[0010] In some embodiments, the integration enzyme or fragment thereof is selected from: a BxBl polypeptide, SsuINT, SssINT, SscINT, Ssc2INT, SsdINT, SmcINT, UhmINT, SacINT, RsaINT, Rsa2INT, Bxbl, Tp9INT, BtlINT, BceINT, BcyINT, and SluINT.
[0011] In some embodiments, the integration enzyme or fragment therefor is a BxBl polypeptide or a variant thereof.
[0012] In some embodiments, the integration enzyme or fragment thereof comprises an amino acid sequence that is at least 80% identical to an amino acid sequence selected from SEQ ID NOs: 378-393.
[0013] In some embodiments, the BxBl polypeptide comprises an amino acid sequence having at least 80% sequence identity to a sequence of SEQ ID NO: 388.
[0014] In some embodiments, the BxBl polypeptide has an amino acid sequence of SEQ ID NO: 388.
[0015] In some embodiments, the engineered integration enzyme binds to a cognate pair of integration recognition sites.
[0016] In some embodiments, the cognate pair of integration recognition sites is selected from: an attB and an attP and a modified AttB and a modified AttP.
[0017] In some embodiments, at least one of the integration recognition sites of the cognate pair is integrated into a mammalian cell genome at a target DNA sequence by an attachment site containing guide RNA (atgRNA) and a gene editor polypeptide.
[0018] In some embodiments, the engineered integration enzyme further comprises a second stabilization domain.
[0019] In some embodiments, the first stabilization domain and/or second stabilization domain are positioned in the engineered integration enzyme such that the engineered integration enzyme and the first and/or second stabilization domains are an in-frame fusion.
[0020] In some embodiments, the first stabilization domain and/or second stabilization domain are located N-terminus to the integration enzyme or fragment thereof, C-terminus to the integration enzyme of fragment thereof, or between two consecutive amino acid residues in the amino acid sequence of the integration enzyme or fragment thereof.
[0021] In some embodiments, the first stabilization domain and/or second stabilization domain are located N-terminus to the integration enzyme or fragment thereof.
[0022] In some embodiments, the first stabilization domain and/or second stabilization domain are located C-terminus to the integration enzyme or fragment thereof.
[0023] In some embodiments, the first stabilization domain and/or second stabilization domain are located between two consecutive amino acid residues in the amino acid sequence of the integration enzyme or fragment thereof.
[0024] In some embodiments, the two consecutive amino acid residues in the amino acid sequence of the integration enzyme are located in a N-terminal catalytic domain; a recombinase domain, or one or more zinc ribbon domain.
[0025] In some embodiments, the two consecutive amino acid residues in the amino acid sequence of the integration enzyme correspond amino acid residues between the N-terminal catalytic domain and the recombinase domain or between the recombinase domain and one of the zinc ribbon domains.
[0026] In some embodiments, the stabilization domain is selected from: a stabilon motif and a exin21 motif.
[0027] In some embodiments, the stabilization domain is a stabilon motif having at least 80% sequence identity to a sequence of KDKDKKSDGKDSQKK (SEQ ID NO: 583 or KDKKSDGKDSQKK (SEQ ID NO: 584).
[0028] In some embodiments, the stabilization domain is a exin21 motif having a sequence of QPRFAAA (SEQ ID NO: 585) or a sequence of QPRFAAA (SEQ ID NO: 586) within one, two or three amino acid substitutions.
[0029] In some embodiments, the second stabilization domain is selected from: a stabilon motif and exin21 motif.
[0030] In some embodiments, the engineered integration enzyme further comprises one or more additional stabilization domains.
[0031] In some embodiments, the one or more additional stabilization domains is selected from: a stabilon motif and a exin21 motif.
[0032] In some embodiments, the engineered integration enzyme further comprises at least a first tag.
[0033] In some embodiments, the engineered integration enzyme further comprises a second tag.
[0034] In some embodiments, the first tag and/or the second tag are position in the engineered integration enzyme such that the engineered integration enzyme and the first and/or second tag are an in-frame fusion.
[0035] In some embodiments, the first tag and/or second tag are selected from: an HA tag, a HiBit tag, strep tag, and SUMO tag.
[0036] In some embodiments, the engineered integration enzyme further comprises one or more linkers.
[0037] In some embodiments, the one or more linkers is a glycine serine linker selected from: GS, GSG, or GGGS.
[0038] In some embodiments, the engineered integration enzyme comprises an orientation from N-terminus to C-terminus: N-I-S; I-S-N; S-I-N; N-S-I; N-Xi-I-S; I-Xi-S-N; S-Xi-I-N; N-Xi-S-I; N-I-Xi-S; I-S-Xi-N; S-I-Xi-N; N-S-Xi-I; N-X1-I-X2-S; I-X1-S-X2-N; S-X1-I-X2-N; and N-Xi-S-
X2-I; wherein I is the integration enzyme or fragment thereof, Xi is a first linker, S is the stabilization domain, X2 is a second linker, and N is the NLS.
[0039] In another aspect, this disclosure features an engineered integration enzyme, comprising: (a) an integration enzyme or fragment thereof comprising at least a first amino acid modification that increases the stability of the integration enzyme as compared to an integration enzyme not comprising the at least first amino acid modification; and (b) a nuclear localization signal (NLS).
[0040] In some embodiments, the first amino acid modification is located in a degron motif in the integration enzyme.
[0041] In some embodiments, the first amino acid modification is an amino acid substitution, an insertion, a deletion, or a combination thereof.
[0042] In some embodiments, the first amino acid modification is an amino acid substitution of L275V in SEQ ID NO: 388.
[0043] In some embodiments, the engineered integration enzyme is linked to a gene editor polypeptide.
[0044] In some embodiments, the C-terminus of the engineered integration enzyme is linked to the gene editor polypeptide.
[0045] In some embodiments, the C-terminus of the gene editor polypeptide is linked to the engineered integration enzymes.
[0046] In some embodiments, the engineered integration enzyme is linked to the gene editor polypeptide by in-frame fusion.
[0047] In some embodiments, the engineered integration enzyme is linked to the gene editor polypeptide by a linker.
[0048] In some embodiments, the linker is a peptide fused in-frame between the engineered integration enzyme and the gene editor polypeptide.
[0049] In some embodiments, the one or more linkers is selected from: Table 3.
[0050] In some embodiments, the gene editor polypeptide comprises a DNA binding domain and a reverse transcriptase.
[0051] In another aspect, this disclosure features a polynucleotide comprising a nucleic acid sequence encoding any of the engineered integration enzymes described herein or any of the engineered integration enzymes linked to the gene editor polypeptides described herein.
[0052] In some embodiments, the nucleic acid sequence encoding the engineered integration enzyme is codon optimized.
[0053] In some embodiments, the codon optimization is performed using an algorithm.
[0054] In some embodiments, the nucleic acid sequence of the engineered integration enzyme is optimized based on second structure such that the structure confers increased stability of the polynucleotide.
[0055] In some embodiments, the nucleic acid sequence of the engineered integration enzyme is optimized based on a linear design algorithm.
[0056] In another aspect, this disclosure features a vector comprising any of the nucleic acid sequences described herein.
[0057] In another aspect, this disclosure features a host cell comprising any of the the vectors described herein.
[0058] In another aspect, this disclosure features a fusion protein, comprising: (a) a DNA binding domain, optionally comprising a nickase activity; (b) a reverse transcriptase; and (c) any of the engineered integration enzymes described herein, wherein at least any two of elements (a), (b), or (c) are linked via at least a first C-terminal linker.
[0059] In some embodiments, the C-terminal linker is a linker selected from: Table 3.
[0060] In another aspect, this disclosure features a polynucleotide comprising any of the nucleic acid sequences encoding the all-in-one fusion protein.
[0061] In another aspect, this disclosure features a vector comprising any of the nucleic acid sequences described herein.
[0062] In another aspect, this disclosure features a host cell comprising any of the vectors described herein.
[0063] In another aspect, this disclosure features a system for site-specifically integrating a donor polynucleotide template into a mammalian cell genome at a target DNA sequence, comprising: an attachment site containing gRNA (atgRNA) comprising at least a portion of an at least first integration recognition site; a gene editor polypeptide comprising a DNA binding nickase domain linked to a reverse transcriptase domain capable of incorporating the integration recognition site into the target DNA sequence, any of the engineered integration enzymes described herein; and a donor polynucleotide template linked to a sequence that is an integration cognate of the integration recognition site present in the atgRNA, whereby the gene editor polypeptide site-specifically integrates the integration recognition site into the target DNA sequence, whereby the engineered integration enzyme integrates the donor polynucleotide template into the target DNA sequence.
[0064] In some embodiments, the first atgRNA comprises: (i) a domain that is capable of guiding the gene editor polypeptide to the target DNA sequence; and (ii) a reverse transcriptase (RT) template that comprises at least a portion of an at least first integration recognition site, whereby the at least portion of the at least first integration recognition site is integrated into the genome of the cell at the target sequence.
[0065] In some embodiments, the system further comprises a second atgRNA.
[0066] In some embodiments, the first atgRNA and the second atgRNA are an at least first pair of atgRNAs, wherein the at least first pair of atgRNAs have domains that are capable of guiding the gene editor polypeptide to the target DNA sequence; the first atgRNA further includes a first RT template that comprises at least a portion of an at least first integration recognition site; the second atgRNA further includes a second RT template that comprises at least a portion of the first integration recognition site, and the first atgRNA and the second atgRNAs collectively encode the entirety of the first integration recognition site, whereby the least first integration recognition site is integrated into the genome of the cell at the target sequence.
[0067] In some embodiments, where, upon introducing the system into the cell, the engineered integration enzyme enhances integration efficiency of the donor polynucleotide template at the site-specifically integrated integration recognition site as compared to the integration efficiency of a system using a non-engineered integration enzyme to integrate donor polynucleotide template at the site-specifically integrated integration recognition site.
[0068] In another aspect, this disclosure features a method for site-specifically integrating a donor polynucleotide template into a mammalian cell genome at a target DNA sequence, comprising: incorporating an integration recognition site into the genome by delivering into the cell: an attachment site containing guide RNA (atgRNA) comprising at least a portion of an at least first integration recognition site; and a gene editor polypeptide or polynucleotide encoding a gene editor polypeptide, wherein the gene editor polypeptide comprises a DNA binding nickase domain linked to a reverse transcriptase domain and is capable of incorporating the integration recognition site into the target DNA sequence; and optionally, a nicking gRNA; and integrating the donor polynucleotide template into the genome by delivering into the cell: any of the engineered integration enzymes described herein; and a donor polynucleotide template, wherein the donor polynucleotide template is linked to a sequence that is an integration cognate of the integration recognition site present in the atgRNA, and wherein the donor polynucleotide template is integrated into the genome at the incorporated genomic integration recognition site by the integration enzyme.
[0069] In some embodiments, the atgRNA, the gene editor polypeptide or polynucleotide encoding the gene editor polypeptide, the optional nicking gRNA, the engineered integration enzyme, and the donor polynucleotide template are introduced into the cell concurrently.
[0070] In some embodiments, the first atgRNA comprises: (i) a domain that is capable of guiding the gene editor polypeptide to the target DNAsequence; and (ii) a reverse transcriptase (RT) template that comprises at least a portion of an at least first integration recognition site, whereby the at least portion of the at least first integration recognition site is integrated into the genome of the cell at the target sequence.
[0071] In some embodiments, the method further comprises a second atgRNA.
[0072] In some embodiments, the first atgRNA and the second atgRNA are an at least first pair of atgRNAs, wherein the at least first pair of atgRNAs have domains that are capable of guiding the gene editor polypeptide to the target DNA sequence; the first atgRNA further includes a first RT template that comprises at least a portion of an at least first integration recognition site; the second atgRNA further includes a second RT template that comprises at least a portion of the first integration recognition site, and the first atgRNA and the second atgRNAs collectively encode the entirety of the first integration recognition site, whereby the least first integration recognition site is integrated into the genome of the cell at the target sequence.
[0073] In some embodiments, the method enhances integration efficiency of the donor polynucleotide template at the site-specifically integrated integration recognition stie at the double-stranded target DNA sequence as compared to the integration efficiency of the donor polynucleotide template at the site-specifically integrated recognition site when using a method that does not comprise the engineered integration enzyme.
4. BRIEF DESCRIPTION OF THE DRAWINGS
[0074] These and other features, aspects, and advantages of the present invention will become better understood with regard to the following description, and accompanying drawings, where:
[0075] FIG. 1 shows a non-limiting illustration of a gene editor construct packaged within a lipid nanoparticle (LNP).
[0076] FIG. 2 illustrates the donor polynucleotide (“donor template”) (synonymously, “cargo” or “payload” or “template polynucleotide”) packaged within a vector.
[0077] FIG. 3 illustrates integrase-mediated self-circularization of the donor template (template polynucleotide) within viral genome. The circularized donor template is capable of being genomically incorporated into an orthogonal integrase target recognition site (i.e., “beacon”).
[0078] FIG. 4 shows non-limiting illustrations of a gene editor construct packaged within a lipid nanoparticle and an atgRNA, ngRNA, and donor template (i.e., template polynucleotide encoding a gene of interest) packaged within a vector. GOI = gene of interest. PGI = programmable gene insertion. U6 = U6 promoter. atgRNA = attachment site-containing guide RNA (atgRNA).
[0079] FIG. 5 shows non-limiting illustrations of a gene editor construct (e.g., mRNA encoding PE2-BxBl) and a nicking guide RNA (ngRNA) packaged within a lipid nanoparticle (LNP) and an atgRNA and donor template (i.e., template polynucleotide encoding a gene of interest) packaged within a vector.
[0080] FIGs. 6A-6B show non-limiting illustrations of three self-complementary AAV (scAAV) genomes capable of recombinase/integrase-mediated self-circularization. FIG. 6A shows the structure of the three self-complementary AAV (scAAV) genomes capable of recombinase/integrase-mediated self-circularization. FIG. 6B shows non-limiting examples of sequences that enable self-circularization (e.g., LoxP AttP GT (SEQ ID NO: 568 and SEQ ID NO: 569); FRT AttP GT (SEQ ID NO: 570 and SEQ ID NO: 571); and AttB CC AttP GT (SEQ ID NO: 572 and SEQ ID NO: 573)). GT indicates an AttP site with a GT dinucleotide. AttB CC
indicates an AttB site with a CC dinucleotide. LoxP = a LoxP recombinase recognition site. FRT = a FRT recombinase recognition site.
[0081] FIG. 7 shows a non-limiting illustration of recombinase/integrase-mediated intramolecular circularization products.
[0082] FIGs. 8A-8B show non-limiting illustrations of a ddPCR assay and intramolecular circularization ddPCR detection probes. FIG. 8A shows a non-limiting illustration of the ddPCR strategy. FIG. 8B shows non-limiting examples of the universal probe (SEQ ID NO: 574 and SEQ ID NO: 575) and an AttR probe (SEQ ID NO: 576 and SEQ ID NO: 577) that can be used in the assay shown in FIG. 8A.
[0083] FIG. 9 shows a non-limiting illustration of a pDNA genome and AAV transfection and screening protocol.
[0084] FIG. 10 shows data for circularization of AAV pDNA and packaged AAV genomic DNA with Bxb 1.
[0085] FIG. 11 shows data for Cre-, FLPe-, and Bxb 1 -mediated circularization of AAV pDNA confirmed by ddPCR.
[0086] FIG. 12 shows Cre-, FLPe-, and Bxb 1 -mediated circularization of packaged AAV confirmed by ddPCR
[0087] FIG. 13 shows percent circularization between the Bxb 1 -mediated attR scar ddPCR probe (“attR probe” described in FIG. 8B) and the universal ddPCR probe (“universal probe” described in FIG. 8B)
[0088] FIGs. 14A-14E shows analysis of AttP variants. FIG. 14A shows a non-limiting schematic of AttP mutations tested for improving integration efficiency (SEQ ID NOS: 394 and 540-542, respectively, in order of appearance). FIG. 14B shows integration efficiencies of wildtype and mutant AttP sites across a panel of AttB lengths. FIG. 14C shows a non-limiting schematic of multiplexed integration of different cargo sets at specific genomic loci. Three fluorescent cargos (GFP, mCherry, and YFP) are inserted orthogonally at three different loci (ACTB, LMNB1, NOLC1) for in-frame gene tagging. FIG. 14D shows orthogonality of top 4 AttB/AttP dinucleotide pairs evaluated for GFP integration with PASTE at the ACTB locus. FIG. 14E shows efficiency of multiplexed PASTE insertion of combinations of fluorophores at ACTB, LMNB1, and NOLC1 loci. Data are mean (n= 3) ± s.e.m.
[0089] FIG. 15 illustrates a schematic of single atgRNA and dual atgRNA approaches for beacon placement (“integration recognition site”).
[0090] FIG. 16 shows percent beacon placement in primary mouse hepatocytes (PMH) following delivery of mRNA to deliver a polynucleotide encoding a gene editor polynucleotide construct and an AAV to deliver the first and second atgRNA according to the following conditions: (i) concurrent delivery (“co-dose”), (ii) AAV delivery followed by a “1-day delay” before delivery of the mRNA, or (iii) AAV delivery followed by a “2-day delay” before delivery of the mRNA.
[0091] FIG. 17 shows percent beacon placement in primary human hepatocytes (PHH) following delivering of mRNA to deliver a polynucleotide encoding a gene editor polynucleotide construct and an AAV to deliver the first and second atgRNA. The mRNA and AAV were delivered concurrently.
[0092] FIG. 18 shows percent in vivo beacon placement in the Nolcl locus of mice following delivery of a polynucleotide encoding a gene editor polynucleotide construct using a lipid nanoparticle (LNP) and a first atgRNA and second atgRNA using an AAV. %BP = % beacon placement. LNP were administered at doses of 0.5 mg/kg, 1.5 mg/kg, 3 mg/kg, and 5 mg/kg. AAV was administered at 1E11, 3E11, or 1E12 viral genomes (vg) per animal. LNP #F1 = LNP formulation #1. LNP #F2 = LNP formulation #F2. LNP #F3 = LNP formulation #F3.
[0093] FIG. 19 show percent in vivo integration of a template polynucleotide in AttP mice following delivering of the Bxbl using adenovirus (AdV) and the template polynucleotide using an AAV (“AAV Cargo”). Bxbl Adv was administered to the mice at a dose of either 3E10 or 1E11 vector genomes (vg) per animal. AAV Cargo was administered to the mice at a dose of 1E12.
[0094] FIG. 20A shows ddPCR data for percent in vivo beacon placement in the Nolcl locus of neonatal mice at eight days post-delivery of a single dose of a mixture of two LNPs. First LNP contained mRNA encoding a prime editing system and a first synthetic atgRNA (atgRNAl) at a 1 : 1 ratio. Second LNP contained mRNA encoding a prime editing system and a second synthetic atgRNA (atgRNA2) at a 1 : 1 ratio. Each of the first and second atgRNAs targeted the mouse Nolcl locus, encoded a portion of an integration recognition site (“beacon”), and together included a 6bp overlap. The first and second LNPs were combined 1 : 1 as mixture and administered at either 1 mg/kg or 3 mg/kg. LNP #F2 = LNP formulation #F2.
[0095] FIG. 20B show NGS data for percent in vivo beacon placement in the Nolcl locus of the same neonatal mice and treatment conditions as described in FIG. 20A. NGS data shows beacon placement eight days after administration of the LNP mixture. LNP #F2 = LNP formulation #F2.
[0096] FIG. 20C shows NGS data for percentage of in vivo beacons placed in the Nolcl NGS data is for the same mice with the same treatment conditions as described in FIG. 20A. NGS data shows data for eight days after administration of the LNP mixture. LNP #F2 = LNP formulation #F2.
[0097] FIG. 21A shows ddPCR data for percent in vivo beacon placement in the Nolcl locus of neonatal mice at 6 weeks post-delivery of a single dose of a mixture of two LNPs. First LNP contained mRNA encoding a prime editing system and a first synthetic atgRNA (atgRNAl) at a 1 : 1 ratio. Second LNP contained mRNA encoding a prime editing system and a second synthetic atgRNA (atgRNA2) at a 1 : 1 ratio. Each of the first and second atgRNAs targeted the mouse Nolcl locus, encoded a portion of an integration recognition site (“beacon”), and together included a 6bp overlap. The first and second LNPs were combined 1 : 1 as mixture and administered at either 1 mg/kg or 3 mg/kg. LNP #F2 = LNP formulation #F2.
[0098] FIG. 21B shows NGS data for percent in vivo beacon placement in the Nolcl locus of the same neonatal mice and treatment conditions as described in FIG. 21A. NGS data shows beacon placement 6 weeks after administration of the LNP mixture. LNP #F2 = LNP formulation #F2.
[0099] FIG. 21C shows NGS data for percentage of in vivo beacons placed in the Nolcl locus that included the expected integration recognition site. Data is from the same mice with the same treatment conditions as described in FIG. 22A. NGS data shows data at 6 weeks after administration of the LNP mixture. LNP #F2 = LNP formulation #F2.
[0100] FIG. 22A shows ddPCR data for percent in vivo beacon placement in the Factor IX (“mF9”) locus of 6-8 week old mice at day 8 post-delivery of a single dose of a mixture of two LNPs. First LNP contained mRNA encoding a prime editing system and a first synthetic atgRNA (atgRNAl) at a ratio of 1 :0.5, 1 : 1, or 1 :2. Second LNP contained mRNA encoding a prime editing system and a second synthetic atgRNA (atgRNA2) at a ratio of 1 : 1, 1 :0.5, or 1 :2. Each of the first and second atgRNAs targeted the mouse Factor IX locus, encoded a portion of an integration recognition site (“beacon”), and together included a 6bp overlap. The first and second LNPs were combined 1 : 1 as mixture with the final ratio of mRNA:atgRNAl :atgRNA2 at 1 :0.25:0.25; 1 :0.5:0.5, or 1 : 1 : 1. LNP #F2 = LNP formulation #F2.
[0101] FIG. 22B shows NGS data for percent in vivo beacon placement in the mF9 locus of the same neonatal mice and treatment conditions as described in FIG. 22A. NGS data shows beacon placement 8 days after administration of the LNP mixture. LNP #F2 = LNP formulation #F2.
[0102] FIG. 22C shows NGS data for percent of in vivo beacons placed in the mF9 locus that included the expected integration recognition site. Data is from the same mice with the same treatment conditions as described in FIG. 22A. NGS data shows data at 8 days after administration of the LNP mixture. LNP #F2 = LNP formulation #F2.
[0103] FIG. 23 shows schematics for non-limiting examples of engineered integrases. Abbreviations: 5’ UTR = 5’ untranslated region; XBG = Xenopus beta globin; NLS = nuclear localization signal; BxBl = BxBl integration enzyme; HA = HA Tag; HiBit = tag; 3’ UTR = 3’ untranslated region; 80A’s = poly A tail comprising 80 adenines. L275V indicates a L to V substitution at position 275 of BxBl (SEQ ID NO: 388).
[0104] FIGs. 24A-24C show PGI data for the engineered integration enzymes described in FIG. 23. FIG. 24A shows ddPCR data for beacon placement. FIG. 24B shows ddPCR for integration %. FIG. 24C shows ddPCR data for beacon occupancy %.
[0105] FIG. 25 shows schematics for non-limiting examples of engineered integrases having a codon optimized coding sequence for BxBl and a split poly A. 5’ UTR = 5’ untranslated region; XBG = Xenopus beta globin; NLS = nuclear localization signal; BxBl = BxBl integration enzyme; HA = HA Tag; HiBit = tag; 3’ UTR = 3’ untranslated region; 30-70pA’s = polyA tail comprising 30-70 adenines. 80A’s = poly A tail comprising 80 adenines.
[0106] FIG. 26 shows % Occupancy (an indicator of PGI) in cells treated with the indicated engineered BxBl integration enzymes (e.g., as described in FIG. 25) at high dose (1.3 pmol) or low does (0.2 pmol).
[0107] FIGs. 27A-27D show % Beacon Occupancy (an indicator of PGI) in cells treated with mRNA encoding the indicated engineered BxB 1 integration enzymes: PL760 (FIG. 27A), PL1303 (FIG. 27B), PL 1304 (FIG. 27C), and PL 1305 (FIG. 27D). mRNA was introduced at 1000 ng, 500 ng, 250 ng, and 0 ng (control).
[0108] FIG. 28 shows schematics for non-limiting examples of engineered integrases. 5’ UTR = 5’ untranslated region; XBG = Xenopus beta globin; NLS = nuclear localization signal; BxBl = BxBl integration enzyme; HA = HA Tag; HiBit = tag; 3’ UTR = 3’ untranslated region; 80 A’ s =
poly A tail comprising 80 adenines; Strep = streptavidin tag; Stabilion = stabilion peptide; and Sumo = sumo peptide.
[0109] FIG. 29A shows PGI % in primary human hepatocytes (PHH) transfected with 250 ng of the indicated mRNAs: PL1303; PL1325: PL1326; PL1327; and PL1305 as well as controls Beacon only, nCas9-RT only and untreated cells.
[0110] FIG.29B shows Beacon Occupancy % (an indicator of PGI) in primary human hepatocytes (PHH) transfected with 250 ng of the indicated mRNAs: PL1303; PL1325; PL1326; PL1327; and PL1305 as well as controls Beacon only, nCas9-RT only and untreated cells.
[0111] FIG. 29C shows PGI % in primary human hepatocytes (PHH) transfected with the indicated mRNAs: PL1303; PL1305; and PL1325 as well as controls Beacon only, nCas9-RT only and untreated cells.
[0112] FIG. 29D shows Beacon Occupancy % (an indicator of PGI) in primary human hepatocytes (PHH) transfected with the indicated mRNAs: PL1303; PL1305; and PL1325 as well as controls Beacon only, nCas9-RT only and untreated cells.
[0113] FIG. 30A shows a non-limiting workflow for using the engineered integration enzymes for programmable gene insertion, for example, in a pluripotent stem cell.
[0114] FIG. 30B shows schematics for non-limiting examples of engineered integrases. 5’ UTR = 5’ untranslated region; XBG = Xenopus beta globin; NLS = nuclear localization signal; BxB 1 = BxBl integration enzyme; HA = HA Tag; HiBit = tag; 3’ UTR = 3’ untranslated region; 80 A’ s = poly A tail comprising 80 adenines; Stabilion = stabilion peptide.
[0115] FIG. 30C shows cell viability data in iPSCs (clones 52 and 17) at day 3 after transduction for each of the conditions indicated on the x-axis.
[0116] FIG. 30D shows ddPCR data for percent (%) PGI in iPSC (clones 52 and 17) at day 3 after transduction for each of the conditions indicated on the x-axis.
[0117] FIG. 31 A shows ddPCR data for percent (%) PGI at day 3 and day 6 for iPSC clone 52 for each of the conditions indicated on the x-axis.
[0118] FIG. 31B shows flow cytometry plots of side-scatter versus FITC-A for each of the indicated conditions. FITC-A is an indicator PGI.
[0119] FIG. 32A shows a schematic of non-limiting examples of engineered BxBl integrases assessed for their ability to mediate PGI in hematopoietic stem cells (HSCs).
[0120] FIG. 32B shows ddPCR data for percent (%) PGI in HSCs for each of the conditions indicated on the x-axis (see engineered BxBl integrases described in FIG. 32A).
[0121] FIG. 33 shows a schematic of non-limiting examples of engineered BxBl integrases.
[0122] FIG. 34 shows ddPCR data for % Beacon Occupancy (an indicator of PGI) for each of the conditions indicated on the x-axis.
[0123] FIG. 35 shows a schematic of engineered BxBl integrases selected for further study.
[0124] FIG. 36A shows a schematic of a BxBl integrase with identification of the location of two predicted degron motifs as well as the location of lysine that are candidates for lysine to arginine amino acid substitutions.
[0125] FIG. 36B shows protein expression data in the form of a western blot comparing a BxBl having a lysine at position 10 substituted for an arginine (K10R) (left panel) versus a BxBl having a lysine at position 10 (right panel). Samples were collected at 24 and 48 hours.
[0126] FIGs. 37A-37B show first attempts at optimizing BxBl RNA structure using the LinearDesign algorithm. Non-optimized mRNA encoding BxBl in shown in FIG. 37A with optimized mRNA encoding BxBl shown in FIG. 37B.
[0127] FIGs. 38A-38B show first generations attempts at optimizing an RNA structure for RNA encoding nCas9-RT using the LinearDesign algorithm. Non-optimized mRNA encoding nCas9- RT in shown in FIG. 38A with optimized mRNA encoding nCsa9-RT shown in FIG. 38B.
[0128] FIG. 39 ddPCR data for Beacon placement for the nCas9-RT described in FIG. 38A and FIG. 38B
[0129] FIG. 40 shows non-limiting examples of fusion proteins comprising an nCas9-RT are fused with engineered integration enzymes (e.g., BxBl). 5’ UTR = 5’ untranslated region; XBG = Xenopus beta globin; NLS = nuclear localization signal; BxBl = BxBl integration enzyme; HA = HA Tag; HiBit = tag; 3 ’ UTR = 3 ’ untranslated region; 80A’ s = poly A tail comprising 80 adenines; and Stabilion = stabilion peptide.
[0130] FIG. 41 shows non-limiting examples of fusion proteins. Fusion proteins comprising a nCas9 and RT are fused with engineered integration enzymes (e.g., BxBl). 5’ UTR = 5’ untranslated region; XBG = Xenopus beta globin; NLS = nuclear localization signal; BxBl = BxBl integration enzyme; HA = HA Tag; HiBit = tag; 3’ UTR = 3’ untranslated region; 80 A’ s = poly A tail comprising 80 adenines; and Stabilion = stabilion peptide.
[0131] FIG. 42 shows schematics for non-limiting examples of engineered integrases. 5’ UTR = 5’ untranslated region; XBG = Xenopus beta globin; NLS = nuclear localization signal; BxBl = BxBl integration enzyme; HA = HA Tag; HiBit = tag; 3’ UTR = 3’ untranslated region; 80 A’ s = poly A tail comprising 80 adenines; Stabilion = stabilion peptide
[0132] FIG. 43 shows ddPCR data for percent (%) PGI in iPSC (clones 52) at day 3 after transduction for each of the conditions indicated on the x-axis.
[0133] FIG. 44A shows ddPCR data and flow cytometry data for percent (%) PGI at day 7 for iPSC clone 52 for each of the conditions indicated on the x-axis.
[0134] FIG. 44B shows flow cytometry plots of side-scatter versus FITC-A for each of the indicated conditions. FITC-A is an indicator PGI.
[0135] FIG. 45 shows ddPCR data for percent (%) PGI in iPSC (clones 52) at day 3 after transduction for each of the conditions indicated on the x-axis.
[0136] FIG. 46 shows schematics for non-limiting examples of engineered integrases fused to gene editor polypeptides. 5’ UTR = 5’ untranslated region; XBG = Xenopus beta globin; NLS = nuclear localization signal; BxB 1 = BxB 1 integration enzyme; HA = HA Tag; HiBit = tag; 3 ’ UTR = 3’ untranslated region; 80A’s = poly A tail comprising 80 adenines; Stabilion = stabilion peptide.
[0137] FIG. 47 shows ddPCR data for percent beacon placement in primary human hepatocyte (PHH line HU8412) at day 4 after transduction for each of the conditions indicated on the x-axis. Two different concentrations for each construct were used: 187 firnol and 374 fmol.
[0138] FIG. 48 shows ddPCR data for percent beacon placement in primary human hepatocyte (PHH line HU8412) at day 4 after transduction for each of the conditions indicated on the x-axis. PL883 was used as a control with ddPCR data presented in middle panel. Right panel includes a table converting mass (ng) to fmol for the BxB 1 mRNA used in these transfections.
[0139] FIG. 49 shows ddPCR data for integration data in primary human hepatocyte (PHH line HU8412) at day 4 after transduction for each of the conditions indicated on the x-axis.
[0140] FIG. 50 shows ddPCR data for total edit (AttB + AttL) in primary human hepatocyte (PHH line HU8412) at day 4 after transduction for each of the conditions indicated on the x-axis.
[0141] FIG. 51 shows ddPCR data for total edit (AttB + AttL) in primary human hepatocyte (PHH line HU8412) at day 4 after transduction for each of the conditions indicated on the x-axis.
[0142] FIG. 52 shows ddPCR data for percent beacon placement in primary human hepatocyte (PHH line HU8412) at day 4 after transduction for each of the conditions indicated on the x-axis.
[0143] FIG. 53 shows ddPCR data for integration data, total edit (AttB + AttL), and beacon placement in primary human hepatocyte (PHH line HU8412) at day 4 after transduction for each of the conditions indicated on the x-axis. Two different concentrations for each construct were used: 187 frnol and 210 fmol.
[0144] FIG. 54 shows schematics for non-limiting examples of engineered Bxbl integrase constructs. 5XBG = 5’ Xenopus beta globin; UTR = untranslated region; N = SV40 promoter; SI may be HA Tag; S2 may be HA tag, Exin 21 peptide, or a stabilon peptide; S3 may be a stabilon peptide or Exin 21 peptide; BxBl = BxBl integration enzyme; S4 may be a stabilon peptide; S5 may be a stabilon peptide; 3XBG = 3’ Xenopus beta globin; UTR = untranslated region; 80 poly A = poly A tail comprising 80 adenines. See Table 26.
[0145] FIG. 55 shows percent integration in HEK293 cells producing recombination site attL as mediated by the engineered integration enzyme constructs described in FIG. 54 as compared to previously presented engineered integrase constructs measured by ddPCR.
[0146] FIG. 56 shows percent beacon conversion measured by ddPCR for various engineered integration enzymes in mice with the beacon placed as a neonate.
[0147] FIG. 57 shows schematics for non-limiting examples of engineered integrase constructs. 5XBG = 5’ Xenopus beta globin; UTR = untranslated region; N may be SV40 promoter; S2 may be HA tag; S3 may be a stabilon peptide; BxBl = BxBl integration enzyme; S4 may be a stabilon peptide; 3XBG = 3’ Xenopus beta globin; UTR = untranslated region; 80 polyA = poly A tail comprising 80 adenines. See Table 26.
[0148] FIG. 58 shows percent beacon conversion in primary human hepatocytes (PHH, line HU8403) as mediated by a selection of engineered mRNA integration enzymes.
[0149] FIG. 59 shows schematics for non-limiting examples of engineered integrase constructs. 5XBG = 5’ Xenopus beta globin; UTR = untranslated region; N = SV40 promoter; SI may be Exin21 peptide; S2 may be Exin 21 peptide or a stabilon peptide; S3 may be a stabilon peptide or Exin 21 peptide; BxBl = BxBl integration enzyme; S4 may be a stabilon peptide; S5 may be a stabilon peptide; 3XBG = 3’ Xenopus beta globin; UTR = untranslated region; 80 polyA = poly A tail comprising 80 adenines. See Table 26.
[0150] FIG. 60 shows percent beacon conversion in primary human hepatocytes (PHH, line HU8403) as mediated by a selection of engineered mRNA integration enzymes.
[0151] FIG. 61 shows schematics for non-limiting examples of engineered integrase constructs. 5XBG= 5’ Xenopus beta globin; UTR = untranslated region; N = SV40 promoter; SI = TT1, TT2, 2XG4S, or T3; S2 = Exin21 peptide; S3 = a stabilon peptide; BxBl = BxBl integration enzyme; S4 = a stabilon peptide; 3XBG = 3’ Xenopus beta globin; UTR = untranslated region; 80 polyA = poly A tail comprising 80 adenines. See Table 26.
[0152] FIGs. 62A-62B shows percent integration mediated by engineered integration enzyme constructs. FIG. 62A shows percent integration producing recombination site attL as measured by ddPCR. FIG. 62B shows percent integration producing recombination site attR as measured by ddPCR.
[0153] FIG. 63 shows schematics for non-limiting examples of engineered integrase constructs. 5XBG = 5’ Xenopus beta globin; UTR = untranslated region; N = SV40 promoter; SI = 2XTT3, TT4, TT5, TT6, TT7, or TT8; S2 = Exin21 peptide; S3 = a stabilon peptide; BxBl = BxBl integration enzyme; S4 = a stabilon peptide; 3XBG = 3’ Xenopus beta globin; UTR = untranslated region; 80 polyA = poly A tail comprising 80 adenines. See Table 26.
[0154] FIG. 64 shows percent integration producing recombination product site attL in HEK293T cells for select engineered integration enzyme constructs as measured by ddPCR.
5. DETAILED DESCRIPTION
[0155] Described herein are integration enzymes engineered such that upon being introduced into a cell, the integration enzyme has increased stability (e.g., half-life) compared to a control integration enzyme not engineered to have increased stability. The increase in stability extends
the capacity of the integration enzyme to mediate integration. For example, this disclosure features an integration enzyme engineered to include a stabilization domain on the N-terminus, which resulted in increased integration of a donor polynucleotide template into an integration recognition site (i.e., beacon) placed into a genome of a cell as compared to an integration enzyme not engineered to include the stabilization domain. In typical embodiments, the engineered integration enzyme includes an integration enzyme or fragment thereof, an at least first stabilization domain, and a nuclear localization signal. At least in some case, location (e.g., N-terminus or C-terminus) of the stabilization domain in the integration enzyme impacted the integration enzyme’s ability to mediate integration.
5.1. Terminology
[0156] Unless defined otherwise, all technical and scientific terms used herein have the meaning commonly understood by a person skilled in the art to which this invention belongs. As used herein, the following terms have the meanings ascribed to them below.
[0157] “Gene editor” as used herein, is a protein that that can be used to perform gene editing, gene modification, gene insertion, gene deletion, or gene inversion. As used herein, the terms “gene editor polynucleotide” refers to polynucleotide sequence encoding the gene editor protein. Such an enzyme or enzyme fusion may contain DNA or RNA targetable nuclease protein (i.e., Cas protein, ADAR, or ADAT), wherein target specificity is mediated by a complexed nucleic acid (i.e., guide RNA). Such an enzyme or enzyme fusion may be a DNA/RNA targetable protein, wherein target specificity is mediated by internal, conjugated, fused, or linked amino acids, such as within TALENs, ZFNs, or meganucleases. The skilled person in the art would appreciate that the gene editor can demonstrate targeted nuclease activity, targeted binding with no nuclease activity, or targeted nickase activity (or cleavase activity). A gene editor comprising a targetable protein may be fused, linked, complexed, operate in cis or trans to one or more proteins or protein fragment motifs. Gene editors may be fused or linked to one or more integrase, recombinase, polymerase, telomerase, reverse transcriptase, or invertase. A gene editor can be a prime editor fusion protein or a gene writer fusion protein.
[0158] “Prime editor fusion protein” as used herein, describes a protein that is used in prime editing. “Prime editor system” as used herein describes the components used in prime editing. Prime editing uses CRISPR enzyme that nicks or cuts only single strand of double stranded DNA, i.e., a nickase; the nickase can occur either naturally or by mutation or modification of a nuclease that makes double stranded cuts. The nickase is programmed (directed) with a prime-editing guide
RNA (pegRNA). The skilled person in the art would appreciate that the pegRNA both specifies the target site and encodes the desired edit. Described herein are attachment site containing guide RNA (atgRNA) that both specifies the target and encodes for the desired integrase target recognition site. The nickase may be programmed (directed) with an atgRNA. Advantageously the nickase is a catalytically impaired Cas9 endonuclease, a Cas9 nickase, that is fused to the reverse transcriptase. During genetic editing, the Cas9 nickase part of the protein is guided to the DNA target site by the atgRNA (or pegRNA), whereby a nick or single stranded cut occurs. The reverse transcriptase domain then uses the atgRNA (or pegRNA) to template reverse transcription of the desired edit, directly polymerizing DNA onto the nicked target DNA strand. The edited DNA strand replaces the original DNA strand, creating a heteroduplex containing one edited strand and one unedited strand. Afterward, optionally, the prime editor (PE) guides resolution of the heteroduplex to favor copying the edit onto the unedited strand, completing the process (typically achieved with a nickase gRNA). Other enzymes that can be used to nick or cut only a single strand of double stranded DNA includes a cleavase (e.g., cleavase I enzyme).
[0159] In some embodiments, an additional agent or agents may be added that improve the efficiency and outcome purity of the prime edit. In some embodiments, the agent may be chemical or biological and disrupt DNA mismatch repair (MMR) processes at or near the edit site (i.e., PE4 and PE5 and PEmax architecture by Chen et al. Cell, 184, 1-18, October 28, 2021; Chen et al. is incorporated herein by reference). In typical embodiments, the agent is a MMR-inhibiting protein. In certain embodiments, the MMR-inhibiting protein is dominant negative MMR protein. In certain embodiments, the dominant negative MMR protein is MLHldn. In particular embodiments, the MMR-inhibiting agent is incorporated into the co-delivery method described herein. In some embodiments, the MMR-inhibiting agent is linked or fused to the prime editor protein fusion, which may or may not have a linked or fused integrase. In some embodiments, the MMR-inhibiting agent is linked or fused to the Gene Writer™ protein, which may or may not have a linked or fused integrase.
[0160] The prime editor or gene editor system can be used to achieve DNA deletion and replacement. In some embodiments, the DNA deletion replacement is induced using a pair of atgRNAs or pegRNA that target opposite DNA strands, programming not only the sites that are nicked but also the outcome of the repair (i.e., PrimeDel by Choi et al. Nat. Biotechnology, October 14, 2021; Choi et al. is incorporated herein by reference and TwinPE by Anzalone et al. BioRxiv, November 2, 2021; Anzalone et al. is incorporated herein by reference). In some embodiments described herein, the DNA deletion is induced using a single atgRNA. In some
embodiments, the DNA deletion and replacement is induced using a wild type Cas9 prime editor (PE-Cas9) system (i.e., PED AR by Jiang et al. Nat. Biotechnology, October 14, 2021; Jiang et al. is incorporated herein by reference in its entirety). In some embodiments, the DNA replacement is an integrase target recognition site or recombinase target recognition site. In certain embodiments, the constructs and methods described herein may be utilized to incorporate the pair of pegRNAs (or atgRNAs) used in PrimeDel, TwinPE (WO2021226558 incorporated by reference herein in its entirety), or PED AR, the prime editor fusion protein or Gene Writer protein, optionally a nickase guide RNA (ngRNA), an integrase, a nucleic acid cargo, and optionally a recombinase into a LNP delivery system or vector delivery system (e.g., AAV or Adenovirus). The integrase may be directly linked, for example by a peptide linker, to the prime editor fusion or gene writer protein.
[0161] In some embodiments, the prime editors can refer to a retrovirus or lentivirus reverse transcriptase such as a Moloney Murine Leukemia Virus (M-MLV) reverse transcriptase (RT) fused to a CRISPR enzyme nickase such as a Cas9 H840A nickase, a Cas9nickase. In some embodiments, the prime editors can refer to a retrovirus or lentivirus reverse transcriptase such as a Moloney Murine Leukemia Virus (M-MLV) reverse transcriptase (RT) fused to a cleavase. In some embodiments the RT can be fused at, near or to the C-terminus of a Cas9nickase, e.g., Cas9 H840A. Fusing the RT to the C-terminus region, e.g., to the C-terminus, of the Cas9 nickase may result in higher editing efficiency. Such a complex is called PEI. In some embodiments, the CRISPR enzyme nickase, e.g., Cas9(H840A), i.e., a Cas9nickase, can be linked to a non-M-MLV reverse transcriptase such as an AMV-RT or XRT (Cas9(H840A)-AMV-RT or XRT). In some embodiments, instead of the CRISPR enzyme nickase being a Cas9 (H840A), i.e., instead of being a Cas9 nickase, the CRISPR enzyme nickase instead can be a CRISPR enzyme that naturally is a nickase or cuts a single strand of double stranded DNA; for instance, the CRISPR enzyme nickase can be Casl2a/b. Alternatively, the CRISPR enzyme nickase can be another mutation of Cas9, such as Cas9(D10A). A CRISPR enzyme, such as a CRISPR enzyme nickase, such as Cas9 (wild type), Cas9(H840A), Cas9(D10A) or Cas 12a/b nickase can be fused in some embodiments to a pentamutant of M-MLV RT (D200N/ L603W/ T330P/ T306K/ W313F), whereby there can be up to about 45-fold higher efficiency, and this is called PE2. In some embodiments, the M-MLV RT comprise one or more of the mutations Y8H, P51L, S56A, S67R, E69K, V129P, L139P, T197A, H204R, V223H, T246E, N249D, E286R, Q2911, E302K, E302R, F309N, M320L, P330E, L435G, L435R, N454K, D524A, D524G, D524N, E562Q, D583N, H594Q, E607K, D653N, and L671P. Specific M-MLV RT mutations are shown in Table 1.

[0162] In some embodiments, the reverse transcriptase can also be a wild-type or modified transcription xenopolymerase (RTX), avian myeloblastosis virus reverse transcriptase (AMV RT), Feline Immunodeficiency Virus reverse transcriptase (FIV-RT), FeLV-RT (Feline leukemia virus reverse transcriptase), HIV-RT (Human Immunodeficiency Virus reverse transcriptase). In some embodiments, the reverse transcriptase can be a fusion of MMuLV to the Sto7d DNA binding domain (see lonnidi et al.; https://doi.0rg/lO.l 101/2021.11.01.466786). The fusion of MMuLV to the Sto7d DNA binding domain sequence is given in Table 2.

[0163] PE3, PE3b, PE4, PE5, and/or PEmax, which a skilled person can incorporate into the co-delivery system described herein, involves nicking the non-edited strand, potentially causing the cell to remake that strand using the edited strand as the template to induce HR. The nicking of the non-edited strand can involve the use of a nicking guide RNA (ngRNA).
[0164] The skilled person can readily incorporate into the co-delivery system described herein described herein a prime editing or CRISPR system. Examples of prime editors can be found in the following: W02020/191153, W02020/191171, WO2020/191233,
WO2020/191234, WO2020/191239, W02020/191241, WO2020/191242, WO2020/191243,
WO2020/191245, WO2020/191246, WO2020/191248, WO2020/191249, each of which is incorporated by reference herein in its entirety. In addition, mention is made, and can be used herein, of CRISPR Patent Applications and Patents of the Zhang laboratory and/or Broad Institute, Inc. and Massachusetts Institute of Technology and/or Broad Institute, Inc., Massachusetts Institute of Technology and President and Fellows of Harvard College and/or Editas Medicine, Inc. Broad Institute, Inc., The University of Iowa Research Foundation and
Massachusetts Institute of Technology, including those claiming priority to US Application 61/736,527, filed December 12, 2012, including US Patents 11,104,937, 11,091,798,
11,060,115, 11,041,173, 11,021,740, 11,008,588, 11,001,829, 10,968,257, 10,954,514,
10,946,108, 10,930,367, 10,876,100, 10,851,357, 10,781,444, 10,711,285, 10,689,691,
10,648,020, 10,640,788, 10,577,630, 10,550,372, 10,494,621, 10,377,998, 10,266,887,
10,266,886, 10,190,137, 9,840,713, 9,822,372, 9,790,490, 8,999,641, 8,993,233, 8,945,839,
8,932,814, 8,906,616, 8,895,308, 8,889,418, 8,889,356, 8,871,445, 8,865,406, 8,795,965,
8,771,945, and 8,697,359; CRISPRPatent Applications and Patents of the Doudna laboratory and/or of Regents of the University of California, the University of Vienna and Emmanuelle
Charpentier, including those claiming priority to US application 61/652,086, filed May 25, 2012, and/or 61/716,256, filed October 19, 2012, and/or 61/757,640, filed January 28, 2013, and/or 61/765,576, filed February 15, 2013 and/or 13/842,859, including US Patents
11,028,412, 11,008,590, 11,008,589, 11,001,863, 10,988,782, 10,988,780, 10,982,231,
10,982,230, 10,900,054, 10,793,878, 10,774,344, 10,752,920, 10,676,759, 10,669,560,
10,640,791, 10,626,419, 10,612,045, 10,597,680, 10,577,631, 10,570,419, 10,563,227,
10,550,407, 10,533,190, 10,526,619, 10,519,467, 10,513,712, 10,487,341, 10,443,076,
10,428,352, 10,421,980, 10,415,061, 10,407,697, 10,400,253, 10,385,360, 10,358,659,
10,358,658, 10,351,878, 10,337,029, 10,308,961, 10,301,651, 10,266,850, 10,227,611,
10,113,167, and 10,000,772; CRISPR Patent Applications and Patents of Vilnius University and/or the Siksnys laboratory, including those claiming priority to US application 62/046384 and/or 61/625,420 and/or 61/613,373 and/or PCT/IB2015/056756, including US Patent
10,385,336; CRISPRPatent Applications and Patents of the President and Fellows of Harvard
College, including those of George Church’s laboratory and/or claiming priority to US application 61/738,355, filed December 17, 2012, including 11,111,521, 11,085,072,
11,064,684, 10,959,413, 10,925,263, 10,851,369, 10,787,684, 10,767,194, 10,717,990,
10,683,490, 10,640,789, 10,563,225, 10,435,708, 10,435,679, 10,375,938, 10,329,587,
10,273,501, 10,100,291, 9,970,024, 9,914,939, 9,777,262, 9,587,252, 9,267,135, 9,260,723, 9,074,199, 9,023,649; CRISPR Patent Applications and Patents of the President and Fellows
of Harvard College, including those of David Liu’s laboratory, including 11,111,472,
11,104,967, 11,078,469, 11,071,790, 11,053,481, 11,046,948, 10,954,548, 10,947,530,
10,912,833, 10,858,639, 10,745,677, 10,704,062, 10,682,410, 10,612,011, 10,597,679,
10,508,298, 10,465,176, 10,323,236, 10,227,581, 10,167,457, 10,113,163, 10,077,453,
9,999,671, 9,840,699, 9,737,604, 9,526,784, 9,388,430, 9,359,599, 9,340,800, 9,340,799, 9,322,037, 9,322,006, 9,228,207, 9,163,284, and 9,068,179; and CRISPR Patent Applications and Patents of Toolgen Incorporated and/or the Kim laboratory and/or claiming priority to US application 61/717,324, filed October 23, 2012 and/or 61/803,599, filed March 20, 2013 and/or 61/837,481, filed June 20, 2013 and/or 62/033,852, filed August 6, 2014 and/or PCT/KR2013/009488 and/or PCT/KR2015/008269, including US Patent 10,851,380, and 10,519,454; and CRISPR Patent Applications and Patents of Sigma and/or Millipore and/or the Chen laboratory and/or claiming priority to US application 61/734,256, filed December 6, 2012 and/or 61/758,624, filed January 30, 2013 and/or 61/761,046, filed February 5, 2013 and/or 61/794,422, filed March 15, 2013, including US Patent 10,731,181, each of which is hereby incorporated herein by reference, and from the disclosures of the foregoing, the skilled person can readily make and use a prime editing or CRISPR system, and can especially appreciate impaired endonucleases, such as a mutated Cas9 that only nicks a single strand of DNA and is hence a nickase, or a CRISPR enzyme that only makes a single-stranded cut that can be employed in a PASTE system of the invention. Further, from the disclosures of the foregoing, the skilled person can incorporate the selected CRISPR enzyme, as part of the prime editor fusion or gene editor fusion, into the co-delivery method described herein.
[0165] Prior to RT -mediated edit incorporation, the prime editor protein (or system) (1) site-specifically targets a genomic locus and (2) performs a catalytic cut or nick. These steps are typically performed by a CRISPR-Cas. However, in some embodiments the Cas protein may be substituted by other nucleic acid programmable DNA binding proteins (napDNAbp) such as zinc finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), or meganucleases. In addition, to the extent the “targeting rules” of other napDNAbp are known or are newly determined, it becomes possible to use new napDNAbp, beyond Cas9, to site specifically target and modify genomic sites of interest.
[0166] Similar to a prime editor protein, a Gene Writer can introduce novel DNA elements, such as an integration target site, into a DNA locus. A Gene Writer protein comprises: (A) a polypeptide or a nucleic acid encoding a polypeptide, wherein the polypeptide comprises (i) a reverse transcriptase domain, and either (x) an endonuclease domain that contains DNA binding
functionality or (y) an endonuclease domain and separate DNA binding domain; and (B) a template RNA comprising (i) a sequence that binds the polypeptide and (ii) a heterologous insert sequence. Examples of such Gene Writer™ proteins and related systems can be found in US20200109398, which is incorporated by reference herein in its entirety.
[0167] In some embodiments, the prime editor or Gene Writer protein fusion or prime editor protein linked or fused to an integrase is expressed as a split construct. In typical embodiments, the split construct in reconstituted in a cell. In some embodiments, the split construct can be fused or ligated via intein protein splicing. In some embodiments, the split construct can be reconstituted via protein-protein inter-molecular bonding and/or interactions. In some embodiments, the split construct can be reconstituted via chemical, biological, or environmental induced oligomerization. In certain embodiments, the split construct can be adapted into one or more delivery vectors described herein.
[0168] In some embodiments, an integrase or recombinase is directly linked or fused, for example by a peptide linker, which may be cleavable or non-cleavable, to the prime editor fusion protein (i.e., fused Cas9 nickase-reverse transcriptase) or Gene Writer protein. Suitable linkers, for example between the Cas9, RT, and integrase, may be selected from Table 3:


[0169] In some embodiments, the prime editor or Gene Writer protein fusion or prime editor protein linked or fused to an integrase is expressed as a split construct. In typical embodiments, the split construct in reconstituted in a cell. In some embodiments, the split construct can be fused or ligated via intein protein splicing. In some embodiments, the split construct can be reconstituted via protein-protein inter-molecular bonding and/or interactions. In some embodiments, the split construct can be reconstituted via chemical, biological, or environmental induced oligomerization.
In certain embodiments, the split construct can be adapted into one or more nucleic acid constructs described herein.
5.2. Type II CRISPR proteins
[0170] The skilled person can incorporate a selected CRISPR enzyme, described below, as part of the prime editor fusion, into the co-delivery method described herein. Streptococcus pyogenes Cas9 (SpCas9), the most common enzyme used in genome-editing applications, is a large nuclease of 1368 amino acid residues. Advantages of SpCas9 include its short, 5'-NGG-3' PAM and very high average editing efficiency. SpCas9 consists of two lobes: a recognition (REC) lobe and a nuclease (NUC) lobe. The REC lobe can be divided into three regions, a long a helix referred to as the bridge helix (residues 60-93), the RECI (residues 94-179 and 308-713) domain, and the REC2 (residues 180-307) domain. The NUC lobe consists of the RuvC (residues 1-59, 718-769, and 909-1098), HNH (residues 775-908), and PAM-interacting (PI) (residues 1099- 1368) domains. The negatively charged sgRNA:target DNA heteroduplex is accommodated in a positively charged groove at the interface between the REC and NUC lobes. In the NUC lobe, the RuvC domain is assembled from the three split RuvC motifs (RuvC I— III) and interfaces with the PI domain to form a positively charged surface that interacts with the 30 tail of the sgRNA. The HNH domain lies between the RuvC II— III motifs and forms only a few contacts with the rest of the protein. Structural aspects of SpCas9 are described by Nishimasu et al., Crystal Structure of Cas9 in Complex with Guide RNA and Target DNA, Cell 156, 935-949, February 27, 2014.
[0171] REC lobe: The REC lobe includes the RECI and REC2 domains. The REC2 domain does not contact the bound guide:target heteroduplex, indicating that truncation of REC lobe may be tolerated by SpCas9. Further, SpCas9 mutant lacking the REC2 domain (D175-307) retained -50% of the wild-type Cas9 activity, indicating that the REC2 domain is not critical for DNA cleavage. In striking contrast, the deletion of either the repeat-interacting region (D97-150) or the anti -repeat-interacting region (D312-409) of the RECI domain abolished the DNA cleavage activity, indicating that the recognition of the repeat:anti-repeat duplex by the RECI domain is critical for the Cas9 function.
[0172] PAM-interacting domain: The NUC lobe contains the PAM-interacting (PI) domain that is positioned to recognize the PAM sequence on the noncomplementary DNA strand. The PI domain of SpCas9 is required for the recognition of 5’-NGG-3’ PAM, and deletion of the PI domain (A1099-1368) abolished the cleavage activity, indicating that the PI domain is critical for SpCas9 function and a major determinant for the PAM specificity.
[0173] RuvC domain-. The RuvC nucleases of SpCas9 have an RNase H fold and four catalytic residues, AsplO (Ala), Glu762, His983, and Asp986, that are critical for the two-metal cleavage of the noncomplementary strand of the target DNA. In addition to the conserved RNase H fold, the Cas9 RuvC domain has other structural elements involved in interactions with the guide:target heteroduplex (an end-capping loop between a42 and a43) and the PI domain/stem loop 3 (P hairpin formed by P3 and P4).
[0174] HNH domain'. SpCas9 HNH nucleases have three catalytic residues, Asp839, His840, and Asn863 and cleave the complementary strand of the target DNA through a single-metal mechanism.
[0175] sgRNA:DNA recognition'. The sgRNA guide region is primarily recognized by the REC lobe. The backbone phosphate groups of the guide region (nucleotides 2, 4-6, and 13-20) interact with the RECI domain (Argl65, Glyl66, Arg403, Asn407, Lys510, Tyr515, and Arg661) and the bridge helix (Arg63, Arg66, Arg70, Arg71, Arg74, and Arg78). The 20-hydroxyl groups of Gl, Cl 5, U16, and G19 hydrogen bond with Vai 1009, Tyr450, Arg447/Ile448, and Thr404, respectively.
[0176] A mutational analysis demonstrated that the R66A, R70A, and R74A mutations on the bridge helix markedly reduced the DNA cleavage activities, highlighting the functional significance of the recognition of the sgRNA “seed” region by the bridge helix. Although Arg78 and Argl65 also interact with the “seed” region, the R78A and R165A mutants showed only moderately decreased activities. These results are consistent with the fact that Arg66, Arg70, and Arg74 form multiple salt bridges with the sgRNA backbone, whereas Arg78 and Argl65 form a single salt bridge with the sgRNA backbone. Moreover, the alanine mutations of the repeat: antirepeat duplex-interacting residues (Arg75 and Lysl63) and the stemloop- 1 -interacting residue (Arg69) resulted in decreased DNA cleavage activity, confirming the functional importance of the recognition of the repeat:anti-repeat duplex and stem loop 1 by Cas9.
[0177] RNA-guided DNA targeting'. SpCas9 recognizes the guide:target heteroduplex in a sequence-independent manner. The backbone phosphate groups of the target DNA (nucleotides 1, 9-11, 13, and 20) interact with the REC 1 (Asn497, Trp659, Arg661, and Gln695), RuvC (Gln926), and PI (Glul 108) domains. The C2’ atoms of the target DNA (nucleotides 5, 7, 8, 11, 19, and 20) form van der Waals interactions with the RECI domain (Leul69, Tyr450, Met495, Met694, and His698) and the RuvC domain (Ala728). The terminal base pair of the guide:target heteroduplex (Gl :C20’) is recognized by the RuvC domain via end-capping interactions; the sgRNA Gl and
target DNA C20’ nucleobases interact with the Tyrl013 and Vai 1015 side chains, respectively, whereas the 20-hydroxyl and phosphate groups of sgRNA G1 interact with Vai 1009 and Gln926, respectively.
[0178] Repeat:Anti-Repeat duplex recognition'. The nucleobases of U23/A49 and A42/G43 hydrogen bond with the side chain of Argl l22 and the main-chain carbonyl group of Phe351, respectively. The nucleobase of the flipped U44 is sandwiched between Tyr325 and His328, with its N3 atom hydrogen bonded with Tyr325, whereas the nucleobase of the unpaired G43 stacks with Tyr359 and hydrogen bonds with Asp364.
[0179] The nucleobases of G21 and U50 in the G21 :U50 wobble pair stack with the terminal C20:G10 pair in the guide:target heteroduplex and Tyr72 on the bridge helix, respectively, with the U50 04 atom hydrogen bonded with Arg75. Notably, A51 adopts the syn conformation and is oriented in the direction opposite to U50. The nucleobase of A51 is sandwiched between Phel 105 and U63, with its Nl, N6, and N7 atoms hydrogen bonded with G62, Glyl l03, and Phel 105, respectively.
[0180] Stem-loop recognition'. Stem loop 1 is primarily recognized by the REC lobe, together with the PI domain. The backbone phosphate groups of stem loop 1 (nucleotides 52, 53, and 59- 61) interact with the RECI domain (Leu455, Ser460, Arg467, Thr472, and Ile473), the PI domain (Lysl 123 and Lysl 124), and the bridge helix (Arg70 and Arg74), with the 20-hydroxyl group of G58 hydrogen bonded with Leu455. A52 interacts with Phel 105 through a face-to-edge p-p stacking interaction, and the flipped U59 nucleobase hydrogen bonds with Asn77.
[0181] The single-stranded linker and stem loops 2 and 3 are primarily recognized by the NUC lobe. The backbone phosphate groups of the linker (nucleotides 63-65 and 67) interact with the RuvC domain (Glu57, Lys742, and Lysl097), the PI domain (Thrl l02), and the bridge helix (Arg69), with the 20-hydroxyl groups of U64 and A65 hydrogen bonded with Glu57 and His721, respectively. The C67 nucleobase forms two hydrogen bonds with Vail 100.
[0182] Stem loop 2 is recognized by Cas9 via the interactions between the NUC lobe and the non-Watson-Crick A68:G81 pair, which is formed by direct (between the A68 N6 and G81 06 atoms) and water-mediated (between the A68 Nl and G81 Nl atoms) hydrogen-bonding interactions. The A68 and G81 nucleobases contact Serl351 and Tyrl356, respectively, whereas the A68:G81 pair interacts with Thrl358 via a water-mediated hydrogen bond. The 20-hydroxyl
group of A68 hydrogen bonds with His 1349, whereas the G81 nucleobase hydrogen bonds with Lys33.
[0183] Stem loop 3 interacts with the NUC lobe more extensively, as compared to stem loop 2. The backbone phosphate group of G92 interacts with the RuvC domain (Arg40 and Lys44), whereas the G89 and U90 nucleobases hydrogen bond with Glnl272 and Glul225/Alal227, respectively. The A88 and C91 nucleobases are recognized by Asn46 via multiple hydrogenbonding interactions.
[0184] Cas9 proteins smaller than SpCas9 allow more efficient packaging of nucleic acids encoding CRISPR systems, e.g., Cas9 and sgRNA into one rAAV (“all-in-one-AAV”) particle. In addition, efficient packaging of CRISPR systems can be achieved in other viral vector systems (i.e., lentiviral, integration deficient lentiviral, hd-AAV, etc.) and non-viral vector systems (i.e., lipid nanoparticle). Small Cas9 proteins can be advantageous for multidomain-Cas-nuclease-based systems for prime editing. Well characterized smaller Cas9 proteins include Staphylococcus aureus (SauCas9, 1053 amino acid residues) and Campylobacter jejuni (CjCas9, 984 amino residues). However, both recognize longer PAMs, 5'-NNGRRT-3' for SauCas9 (R = A or G) and 5'-NNNNRYAC-3' for CjCas9 (Y = C or T), which reduces the number of uniquely addressable target sites in the genome, in comparison to the NGG SpCas9 PAM. Among smaller Cas9s, Schmidt et al. identified Staphylococcus lugdunensis (Siu) Cas9 as having genome-editing activity and provided homology mapping to SpCas9 and SauCas9 to facilitate generation of nickases and inactive (“dead”) enzymes (Schmidt et al., 2021, Improved CRISPR genome editing using small highly active and specific engineered RNA-guided nucleases. Nat Commun 12, 4219. doi.org/10.1038/s41467-021-24454-5) and engineered nucleases with higher cleavage activity by fragmenting and shuffling Cas9 DNAs. The small Cas9s and nickases are useful in the instant invention.
[0185] Besides dead Cas9 and Cas9 nickase variants, the Cas9 proteins used herein may also include other “Cas9 variants” having at least about 70% identical, at least about 80% identical, at least about 90% identical, at least about 95% identical, at least about 96% identical, at least about 97% identical, at least about 98% identical, at least about 99% identical, at least about 99.5% identical, or at least about 99.9% identical to any reference Cas9 protein, including any wild type Cas9, or mutant Cas9 (e.g., a dead Cas9 or Cas9 nickase), or fragment Cas9, or circular permutant Cas9, or other variant of Cas9 disclosed herein or known in the art. In some embodiments, a Cas9 variant may have 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 21, 24,
25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50 or more amino acid changes compared to a reference Cas9. In some embodiments, the Cas9 variant comprises a fragment of a reference Cas9 (e.g., a gRNA binding domain or a DNA-cleavage domain), such that the fragment is at least about 70% identical, at least about 80% identical, at least about 90% identical, at least about 95% identical, at least about 96% identical, at least about 97% identical, at least about 98% identical, at least about 99% identical, at least about 99.5% identical, or at least about 99.9% identical to the corresponding fragment of wild type Cas9. In some embodiments, the fragment is at least 30%, at least 35%, at least 40%, at least 45%, at least 50%, at least 55%, at least 60%, at least 65%, at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, at least 95% identical, at least 96%, at least 97%, at least 98%, at least 99%, or at least 99.5% of the amino acid length of a corresponding wild type Cas9 (e.g., SEQ ID NO: 18).
[0186] In some embodiments, the disclosure also may utilize Cas9 fragments that retain their functionality and that are fragments of any herein disclosed Cas9 protein. In some embodiments, the Cas9 fragment is at least 100 amino acids in length. In some embodiments, the fragment is at least 100, 150, 200, 250, 300, 350, 400, 450, 500, 550, 600, 650, 700, 750, 800, 850, 900, 950, 1000, 1050, 1100, 1150, 1200, 1250, or at least 1300 amino acids in length.
[0187] In various embodiments, the prime editors disclosed herein may comprise one of the Cas9 variants described as follows, or a Cas9 variant thereof having at least about 70% identical, at least about 80% identical, at least about 90% identical, at least about 95% identical, at least about 96% identical, at least about 97% identical, at least about 98% identical, at least about 99% identical, at least about 99.5% identical, or at least about 99.9% identical to any reference Cas9 variants.











































[0188] In some embodiments, prime editors utilized herein comprise CRISPR-Cas system enzymes other than type II enzymes. In certain embodiments, prime editors comprise type V or type VI CRISPR-Cas system enzymes. It will be appreciated that certain CRISPR enzymes exhibit promiscuous ssDNA cleavage activity and appropriate precautions should be considered. In certain embodiments, prime editors comprise a nickase or a dead CRISPR with nuclease function comprised in a different component.
[0189] In various embodiments, the nucleic acid programmable DNA binding proteins utilized herein include, without limitation, Cas9 (e.g., dCas9 and nCas9), Cast 2a (Cpfl), Casl2bl (C2cl), Casl2b2, Casl2c (C2c3), Casl2d (CasY), Casl2e (CasX), C2c4, C2c5, C2c8, C2c9, C2cl0, Cast 3a (C2c2), Cast 3b (C2c6), Cast 3c (C2c7), Cast 3d, and Argonaute. Cas-equivalents further include those described in Makarova et al., “C2c2 is a single-component programmable RNA- guided RNA-targeting CRISPR effector,” Science 2016; 353(6299) and Makarova et al., “Classification and Nomenclature of CRISPR-Cas Systems: Where from Here?,” The CRISPR Journal, Vol. l. No.5, 2018, the contents of which are incorporated herein by reference. One example of a nucleic acid programmable DNA-binding protein that has different PAM specificity than Cas9 is Clustered Regularly Interspaced Short Palindromic Repeats from Prevotella and Francisella 1 (i.e, Casl2a (Cpfl)). Similar to Cas9, Casl2a (Cpfl) is also a Class 2 CRISPR effector, but it is a member of type V subgroup of enzymes, rather than the type II subgroup. It has been shown that Casl2a (Cpfl) mediates robust DNA interference with features distinct from Cas9. Casl2a (Cpfl) is a single RNA-guided endonuclease lacking tracrRNA, and it utilizes a T- rich protospacer-adjacent motif (TTN, TTTN, or YTN). Moreover, Cpfl cleaves DNA via a
staggered DNA double-stranded break. Out of 16 Cpfl -family proteins, two enzymes from Acidaminococcus and Lachnospiraceae are shown to have efficient genome-editing activity in human cells. Cpfl proteins are known in the art and have been described previously, for example Yamano et al., “Crystal structure of Cpfl in complex with guide RNA and target DNA.” Cell (165) 2016, p.949-962; the entire contents of which is hereby incorporated by reference.
5.3. Type V CRISPR proteins
[0190] In some embodiments, prime editors used herein comprise the type V CRISPR family includes Francisella novicida U112 Cpfl (FnCpfl) also known as FnCasl2a. FnCpfl adopts a bilobed architecture with the two lobes connected by the wedge (WED) domain. The N-terminal REC lobe consists of two a-helical domains (RECI and REC2) that have been shown to coordinate the crRNA-target DNA heteroduplex. The C-terminal NUC lobe consists of the C-terminal RuvC and Nuc domains involved in target cleavage, the arginine-rich bridge helix (BH), and the PAM- interacting (PI) domain. The repeat-derived segment of the crRNA forms a pseudoknot stabilized by intra-molecular base-pairing and hydrogen-bonding interactions. The pseudoknot is coordinated by residues from the WED, RuvC, and REC2 domains, as well as by two hydrated magnesium cations. Notably, nucleotides 1-5 of the crRNA are ordered in the central cavity of FnCasl2a and adopt an A-form-like helical conformation. Conformational ordering of the seed sequence is facilitated by multiple interactions between the ribose and phosphate moieties of the crRNA backbone and FnCpfl residues in the WED and RECI domains. These include residues Thrl6, Lys595, His804, and His881 from the WED domain and residues Tyr47, Lys51, Phel82, and Argl86 from the RECI domain. The structure of the FnCasl2a-crRNA complex further reveals that the bases of the seed sequence are solvent exposed and poised for hybridization with target DNA. Structural aspects of FnCpfl are described by Swarts et al., Structural Basis for Guide RNA Processing and Seed-Dependent DNA Targeting by CRISPR-Casl2a, Molecular Cell 66, 221-233, April 20, 2017.
[0191] Pre-crRNA processing'. Essential residues for crRNA processing include His843, Lys852, and Lys869. Structural observations are consistent with an acid-base catalytic mechanism in which Lys869 acts as the general base catalyst to deprotonate the attacking 2’ -hydroxyl group of U(-19), while His843 acts as a general acid to protonate the 5’-oxygen leaving group of A(-18). In turn, the side chain of Lys852 is involved in charge stabilization of the transition state. Collectively, these interactions facilitate the intra-molecular attack of the 20-hydroxyl group of U(-19) on the scissile phosphate and promote the formation of the 2’,3’-cyclic phosphate product.
[0192] R-loop formation'. The crRNA-target DNA strand heteroduplex is enclosed in the central cavity formed by the REC and NUC lobes and interacts extensively with the RECI and REC2 domains. The PAM-containing DNA duplex comprises target strand nucleotides dT0-dT8 and non-target strand nucleotides dA(8)*-dA0* and is contacted by the PI, WED, and RECI domains. The 5’-TTN-3’ PAM is recognized in FnCasl2a by a mechanism combining the shapespecific recognition of a narrowed minor groove, with base-specific recognition of the PAM bases by two invariant residues, Lys671 and Lys613. Directly downstream of the PAM, the duplex of the target DNA is disrupted by the side chain of residue Lys667, which is inserted between the DNA strands and forms a cation-7t stacking interaction with the dAO-dTO* base pair. The phosphate group linking target strand residues dT(-l) and dTO is coordinated by hydrogen-bonding interactions with the side chain of Lys823 and the backbone amide of Gly826. Target strand residue dT(-l) bends away from residue TO, allowing the target strand to interact with the seed sequence of the crRNA. The non-target strand nucleotides dTl*-dT5* interact with the Arg692- Ser702 loop in FnCasl2a through hydrogen-bonding and ionic interactions between backbone phosphate groups and side chains of Arg692, Asn700, Ser702, and Gln704, as well as main-chain amide groups of Lys699, Asn700, and Ser702. Alanine substitution of Q704 or replacement of residues Thr698-Ser702 in FnCasl2a with the sequence Ala-Gly3 (SEQ ID NO: 115) substantially reduced DNA cleavage activity, suggesting that these residues contribute to R-loop formation by stabilizing the displaced conformation of the nontarget DNA strand.
[0193] In the FnCasl2a R-loop complex, the crRNA-target strand heteroduplex is terminated by a stacking interaction with a conserved aromatic residue (Tyr410). This prevents base pairing between the crRNA and the target strand beyond nucleotides U20 and dA(-20), respectively. Beyond this point, the target DNA strand nucleotides re-engage the non-target DNA strand, forming a PAM-distal DNA duplex comprising nucleotides dC(-21)-dA(-27) and dG21*-dT27*, respectively. The duplex is confined between the REC2 and Nuc domains at the end of the central channel formed by the REC and NUC lobes.
[0194] Target DNA cleavage'. FnCpfl can independently accommodate both the target and non-target DNA strands in the catalytic pocket of the RuvC domain. The RuvC active site contains three catalytic residues (D917, E1006, and D1255). Structural observations suggest that both the target and non-target DNA strands are cleaved by the same catalytic mechanism in a single active site in Cpfl/Casl2a enzymes.
[0195] Another type V CRISPR is AsCpfl from Acidaminococcus sp BV3L6 (Yamano et al., Crystal structure of Cpfl in complex with guide RNA and target DNA, Cell 165, 949-962, May 5, 2016)
[0196] In certain embodiments, the nuclease comprises a Casl2f effector. Small CRISPR- associated effector proteins belonging to the type V-F subtype have been identified through the mining of sequence databases and members classified into Casl2fl (Casl4a and type V-U3), Casl2f2 (Casl4b) and Casl2f3 (Casl4c, type V-U2 and U4). (See, e.g., Karvelis et al., PAM recognition by miniature CRISPR-Casl2f nucleases triggers programmable double-stranded DNA target cleavage. Nucleic Acids Research, 21 May 2020, 48(9), 5016-23 doi.org/10.1093/nar/gkaa208). Xu et al. described development of a 529 amino acid Casl2f-based system for mammalian genome engineering through multiple rounds of iterative protein engineering and screening. (Xu, X. et al., Engineered Miniature CRISPR-Cas System for Mammalian Genome Regulation and Editing. Molecular Cell, October 21, 2021, 81(20): 4333-45, doi.org/10.1016/j.molcel.2021.08.008).
[0197] Exemplary CRISPR-Cas proteins and enzymes used in the prime editors herein include the following without limitation.





































Ill








5.4. Protospacer Adjacent Motif
[0198] As used herein, the term “protospacer adjacent sequence” or “protospacer adjacent motif’ or “PAM” refers to an approximately 2-6 base pair DNA sequence (or a 2-, 3-, 4-, 5-, 6-, 7-, 8-, 9-, 10-, 11-, 12-long nucleotide sequence) that is an important targeting component of a Cas9 nuclease. Typically, the PAM sequence is on either strand, and is downstream in the 5' to 3' direction of Cas9 cut site. The canonical PAM sequence (i.e., the PAM sequence that is associated with the Cas9 nuclease of Streptococcus pyogenes or SpCas9) is 5'-NGG-3' wherein “N” is any nucleobase followed by two guanine (“G”) nucleobases. Different PAM sequences can be associated with different Cas9 nucleases or equivalent proteins from different organisms. In addition, any given Cas9 nuclease may be modified to alter the PAM specificity of the nuclease such that the nuclease recognizes alternative PAM sequence.
[0199] For example, with reference to the canonical SpCas9 amino acid sequence, the PAM specificity can be modified by introducing one or more mutations, including (a) DI 135 V, R1335Q, and T1337R “the VQR variant”, which alters the PAM specificity to NGAN or NGNG, (b) D1135E, R1335Q, and T1337R “the EQR variant”, which alters the PAM specificity to NGAG, and (c) DI 135V, G1218R, R1335E, and T1337R “the VRER variant”, which alters the PAM specificity to NGCG. In addition, the DI 135E variant of canonical SpCas9 still recognizes NGG, but it is more selective compared to the wild type SpCas9 protein.
[0200] It will also be appreciated that Cas9 enzymes from different bacterial species (i.e., Cas9 orthologs) can have varying PAM specificities and some embodiments are therefore chosen based on the desired PAM recognition. For example, Cas9 from Staphylococcus aureus (SaCas9) recognizes NGRRT or NGRRN. In addition, Cas9 from Neisseria meningitis (NmCas) recognizes NNNNGATT. In another example, Cas9 from Streptococcus thermophilis (StCas9) recognizes NNAGAAW. In still another example, Cas9 from Treponema denticola (TdCas) recognizes NAAAAC. These examples are not meant to be limiting. It will be further appreciated that non- SpCas9s bind a variety of PAM sequences, which makes them useful to expand the range of sequences that can be targeted according to the invention. Furthermore, non-SpCas9s may have other characteristics that make them more useful than SpCas9. For example, Cas9 from Staphylococcus aureus (SaCas9) is about 1 kilobase smaller than SpCas9, so it can be packaged
into adeno-associated virus (AAV). Further reference may be made to Shah et al., “Protospacer recognition motifs: mixed identities and functional diversity,” RNA Biology, 10(5): 891-899 (which is incorporated herein by reference). Gasiunas used cell-free biochemical screens to identify protospacer adjacent motif (PAM) and guide RNA requirements of 79 Cas9 proteins. (Gasiunas et al., A catalogue of biochemically diverse CRISPR-Cas9 orthologs, Nature Communications 11 :5512 doi.org/10.1038/s41467-020-19344-l) The authors described 7 classes of gRNA and 50 different PAM requirement.
[0201] Oh, Y. et al. describe linking reverse transcriptase to a Francisella novicida Cas9 [FnCas9(H969A)] nickase module. (Oh, Y. et al., Expansion of the prime editing modality with Cas9 from Francisella novicida, bioRxiv 2021.05.25.445577; doi.org/10.1101/2021.05.25.445577). By increasing the distance to the PAM, the FnCas9(H969A) nickase module expands the region of a reverse transcription template (RTT) following the primer binding site.
5.5. Prime Editors
[0202] “Prime editor fusion protein” describes a protein that is used in prime editing. Prime editing uses CRISPR enzyme that nicks or cuts only single strand of double stranded DNA, i.e., a nickase; and a nickase can occur either naturally or by mutation or modification of a nuclease that makes double stranded cuts. Such an enzyme can be a catalytically-impaired Cas9 endonuclease (a nickase). Such an enzyme can be a Casl2a/b, MAD7, or variant thereof. The nickase is fused to an engineered reverse transcriptase (RT). The nickase is programmed (directed) with a primeediting guide RNA (pegRNA). The skilled person in the art would appreciate that the pegRNA both specifies the target site and encodes the desired edit. Advantageously the nickase is a catalytically-impaired Cas9 endonuclease, a Cas9 nickase, that is fused to the reverse transcriptase. During genetic editing, the Cas9 nickase part of the protein is guided to the DNA target site by the pegRNA, whereby a nick or single stranded cut occurs. The reverse transcriptase domain then uses the pegRNA to template reverse transcription of the desired edit, directly polymerizing DNA onto the nicked target DNA strand. The edited DNA strand replaces the original DNA strand, creating a heteroduplex containing one edited strand and one unedited strand. Afterward, optionally, the prime editor (PE) guides resolution of the heteroduplex to favor copying the edit onto the unedited strand, completing the process (typically achieved with a nickase gRNA).
[0203] As used herein, “PEI” refers to a PE complex comprising a fusion protein comprising Cas9(H840A) and a wild type MMLV RT having the following N-terminus to C-terminus
structure: [NLS]-[Cas9(H840A)]- [linker] -[MMLV_RT(wt)] + a desired atgRNA (or PEgRNA).
In various embodiments, the prime editors disclosed herein is comprised of PEI.
[0204] As used herein, “PE2” refers to a PE complex comprising a fusion protein comprising Cas9(H840A) and a variant MMLV RT having the following N-terminus to C-terminus structure:
[NLS]-[Cas9(H840A)]- [linker]-[MMLV_RT(D200N)(T330P)(L603W)(T306K)(W313F)] + a desired atgRNA (or PEgRNA). In various embodiments, the prime editors disclosed herein are comprised of PE2. In various embodiments, the prime editors disclosed herein is comprised of PE2 and co-expression of MMR protein MLHldn, that is PE4.
[0205] As used herein, “PE3” refers to PE2 plus a second-strand nicking guide RNA that complexes with the PE2 and introduces a nick in the non-edited DNA strand. The induction of the second nick increases the chances of the unedited strand, rather than the edited strand, to be repaired. In various embodiments, the prime editors disclosed herein are comprised of PE3. In various embodiments, the prime editors disclosed herein are comprised of PE3 and co-expression of MMR protein MLHldn, that is PE5.
[0206] As used herein, “PE3b” refers to PE3 but wherein the second-strand nicking guide RNA is designed for temporal control such that the second strand nick is not introduced until after the installation of the desired edit. This is achieved by designing a gRNA with a spacer sequence with mismatches to the unedited original allele that matches only the edited strand. Using this strategy, mismatches between the protospacer and the unedited allele should disfavor nicking by the sgRNA until after the editing event on the PAM strand takes place.
5.6. Guides for Prime Editing
[0207] Anzalone et al., 2019 (Nature 576: 149) describes prime editing and a prime editing complex using a type II CRISPR and can be used herein. A prime editing complex consists of a type II CRISPR PE protein containing an RNA-guided DNA-nicking domain fused to a reverse transcriptase (RT) domain and complexed with a pegRNA. The pegRNA comprises (5’ to 3’) a spacer that is complementary to the target sequence of a genomic DNA, a nickase (e.g. Cas9) binding site, a reverse transcriptase template including editing positions, and primer binding site (PBS). The PE-pegRNA complex binds the target DNA and the CRISPR protein nicks the PAM- containing strand. The resulting 3' end of the nicked target hybridizes to the primer-binding site (PBS) of the pegRNA, then primes reverse transcription of new DNA containing the desired edit using the RT template of the pegRNA. The overall structure of the pegRNA is like that of a typical
type II sgRNA with a reverse transcriptase template/primer binding site appended to the 3’ end. The structure leaves the PBS at the 3’ end of the pegRNA free to bind to the nicked strand complementary to the target which forms the primer for reverse transcription.
[0208] Guide RNAs of CRISPRs differ in overall structure. For example, while the spacer of a type II gRNA is located at the 5’ end, the spacer of a type V gRNA is located towards the 3 ’ end, with the CRISPR protein (e.g. Casl2a) binding region located toward the 5’ end. Accordingly, the regions of a type V pegRNA are rearranged compared to a type II pegRNA. The overall structure of the pegRNA is like that of a typical type II sgRNA with a reverse transcriptase template/primer binding site appended to the 3’ end. The pegRNA comprises (5’ to 3’) a CRISPR protein-binding region, a spacer which is complementary to the target sequence of a genomic DNA, a reverse transcriptase template including editing positions, and primer binding site (PBS).
[0209] In typical embodiments, an atgRNA comprises a reverse transcriptase template that encodes, partially or in its entirety, an integration recognition site (also referred to as an integration target recognition site) or a recombinase recognition site (also referred to as a recombinase target recognition site). The integration target recognition site, which is to be placed at a desired location in the genome or intracellular nucleic acid, is referred to as a “beacon” site or an “attachment site” or a “landing pad” or “landing site.” An integration target recognition site or recombinase target recognition site incorporated into the pegRNA is referred to as an attachment site containing guide RNA (atgRNA).
5.7. Attachment Site-Containing Guide RNA (atgRNA)
[0210] As used herein, the term “attachment site-containing guide RNA” (atgRNA) and the like refer to an extended single guide RNA (sgRNA) comprising a primer binding site (PBS), a reverse transcriptase (RT) template sequence, and wherein the RT template encodes for an integration recognition site or a recombinase recognition site that can be recognized by a recombinase, integrase, or transposase. In some embodiments, the RT template comprises a clamp sequence and an integration recognition site. As referred to herein an atgRNA may be referred to as a guide RNA. An integration recognition site or recombinase target recognition site incorporated into the pegRNA is referred to as an attachment site containing guide RNA (atgRNA).
[0211] As used herein, the term “cognate integration recognition site” or “integration cognate” or “cognate pair” refers to a first integration recognition site (e.g., any of the integration recognition sites described herein) and a second integration recognition site (e.g., any of the integration recognition sites described herein) that can be recombined. Recombination between a first
integration recognition site (e.g., any of the integration recognition sites described herein) and a second recognition site (e.g., any of the integration recognition sites described herein) is mediated by functional symmetry between the two integration recognition sites and the central dinucleotide of each of the two integration recognition sites. In some cases, a first integration recognition site (e.g., any of the integration recognition sites described herein) that can be recombined with a second integration recognition site (e.g., any of the integration recognition sites described herein) are referred to as a “cognate pair.” A non-limiting example of a cognate pair include an attB site and an attP site, whereby a serine integrase mediates recombination between the attB site and the attP site.
[0212] In typical embodiments, an atgRNA comprises a reverse transcriptase template that encodes, partially or in its entirety, an integration recognition site (also referred to as an integration target recognition site) or a recombinase recognition site (also referred to as a recombinase target recognition site). The integration target recognition site, which is to be placed at a desired location in the genome or intracellular nucleic acid, is referred to as a “beacon,” a “beacon” site or an “attachment site” or a “landing pad” or “landing site.” An integration target recognition site or recombinase target recognition site incorporated into the pegRNA is referred to as an attachment site containing guide RNA (atgRNA).
[0213] During genome editing, the primer binding site allows the 3’ end of the nicked DNA strand to hybridize to the atgRNA, while the RT template serves as a template for the synthesis of edited genetic information. The atgRNA is capable for instance, without limitation, of (i) identifying the target nucleotide sequence to be edited and (ii) encoding new genetic information that replaces (or in some cases adds) the targeted sequence. In some embodiments, the atgRNA is capable of (i) identifying the target nucleotide sequence to be edited and (ii) encoding an integration site that replaces (or inserts/ deletes within) the targeted sequences.
[0214] In some embodiments, the co-delivery system described herein includes a polynucleotide sequence encoding an attachment site-containing guide RNA (atgRNA) packaged in an LNP. In some embodiments, the co-delivery system described herein includes a vector comprising a polynucleotide sequence encoding an atgRNA. In some embodiments, the atgRNA comprises a domain that is capable of guiding the prime editor fusion protein to a target sequence, thereby identifying the target nucleotide sequence to be edited; and a reverse transcriptase (RT) template that comprises a first integration recognition site. In some embodiments, the atgRNA
comprises a domain that is capable of guiding the prime editor fusion protein (or prime editor system) to a target sequence, thereby identifying the target nucleotide sequence to be edited; and a reverse transcriptase (RT) template that comprises at least a portion first integration recognition site.
[0215] In some embodiments, the co-delivery system described herein includes a polynucleotide sequence encoding a first attachment site-containing guide RNA (atgRNA) and a polynucleotide nucleotide sequence encoding a second attachment site-containing guide RNA (atgRNA) packaged into the same LNP. In some embodiments, the co-delivery system described herein includes a polynucleotide sequence encoding a first attachment site-containing guide RNA (atgRNA) packaged into a first LNP and a polynucleotide nucleotide sequence encoding a second attachment site-containing guide RNA (atgRNA) packaged into a second LNP.
[0216] In some embodiments, the co-delivery system described herein includes a vector comprising a polynucleotide sequence encoding a first attachment site-containing guide RNA (atgRNA), a polynucleotide sequence encoding a second atgRNA, or both.
[0217] In some embodiments, the co-delivery system described herein includes a polynucleotide sequence encoding a first attachment site-containing guide RNA (atgRNA) packaged into a first LNP and a vector comprising a polynucleotide sequence encoding a second atgRNA.
[0218] In some embodiments, where the co-delivery system contains a first atgRNA and a second atgRNA, the first atgRNA and the second atgRNA are an at least first pair of atgRNAs, where the at least first pair of atgRNAs have domains that are capable of guiding the gene editor protein or prime editor fusion protein to a target sequence, the first atgRNA further includes a first RT template that comprises at least a portion of the first integration recognition site; and the second atgRNA further includes a second RT template that comprises at least a portion of the first integration recognition site, and the first atgRNA and the second atgRNAs collectively encode the entirety of the first integration recognition site.
[0219] In some embodiments, the first atgRNA’ s reverse transcriptase template encodes for a first single-stranded DNA sequence (i.e., a first DNA flap) that contains a complementary region to a second single-stranded DNA sequence (i.e., a second DNA flap) encoded by a second atgRNA comprising a second reverse transcriptase template. In certain embodiments, the complementary region between the first and second single-stranded DNA sequences is comprised of more than 5
consecutive bases of an integrase target recognition site. In certain embodiments, the complementary region between the first and second single-stranded DNA sequences is comprised of more than 10 consecutive bases of an integrase target recognition site. In certain embodiments, the complementary region between the first and second single-stranded DNA sequences is comprised of more than 20 consecutive bases of an integrase target recognition site. In certain embodiments, the complementary region between the first and second single-stranded DNA sequences is comprised of more than 30 consecutive bases of an integrase target recognition site. Use of two guide RNAs that are (or encode DNA that is) partially complementarity to each other and comprised of consecutive bases of an integrase target recognition site are referred to as dual, paired, annealing, complementary, or twin attachment site-containing guide RNAs (atgRNAs). In certain embodiments, use of two guide RNAs that are (or encode DNA that is) full complementarity to each other and comprised of consecutive bases of an integrase target recognition site are referred to as dual, paired, annealing, complementary, or twin attachment sitecontaining guide RNAs (atgRNAs).
[0220] In some embodiments, upon introducing the nucleic acid construct into a cell, the first atgRNA incorporates the first integration recognition site into the cell’s genome at the target sequence.
[0221] Table 9 includes atgRNAs, sgRNAs and nicking guides that can be used herein. Spacers are labeled in capital font (SPACER), RT regions in bold capital (RT REGION), AttB sites in bold lower case (attB site), and PBS in capital italics (PBS). Unless otherwise denoted, the AttB is for Bxbl.


















5.8. Integrases/Recombinases and Integration/Recombination Sites
[0222] In typical embodiments, the co-delivery system described herein contains an integrase and/or a recombinase. In some embodiments, the co-delivery system includes an integrase and/or a recombinase packaged in a LNP. In one embodiment, the co-delivery system includes a polynucleotide encoding an integrase and/or a recombinase. In some embodiments, the co-delivery system includes an integrase or a recombinase packaged in a vector (e.g., a viral vector). In some
embodiments, the co-delivery system includes at least a first integrase (e.g., a first integrase and a second integrase) and/or at least a first recombinase (e.g., a first recombinase and a second recombinase).
[0223] In some embodiments, the integration enzyme (e.g., the integrase or recombinase) is selected from the group consisting of Dre, Vika, Bxbl, <pC31, RDF, cpBTl, Rl, R2, R3, R4, R5, TP901-1, Al 18, cpFCl, cpCl, MR11, TGI, cp370.1, W , BL3, SPBc, K38, Peaches, Veracruz, Rebeuca, Theia, Benedict, KSSJEB, PattyP, Doom, Scowl, Lockley, Switzer, Bob3, Troube, Abrogate, Anglerfish, Sarfire, SkiPole, Concept!!, Museum, Severus, Airmid, Benedict, Hinder, ICleared, Sheen, Mundrea, BxZ2, cpRV, retrotransposases encoded by a Tcl/mariner family member including but not limited to retrotransposases encoded by LI, Tol2, Tel, Tc3, Himar 1 (isolated from the horn fly, Haematobia irrilans). Mos (Mosaic element of Drosophila mauritiana), and Minos, and any mutants thereof. As can be used herein, Xu et al describes methods for evaluating integrase activity in E. coli and mammalian cells and confirmed at least R4, cpC31, (pBTl, Bxbl, SPBc, TP901-1 and WP integrases to be active on substrates integrated into the genome of HT1080 cells (Xu et al., 2013, Accuracy and efficiency define Bxbl integrase as the best of fifteen candidate serine recombinases for the integration of DNA into the human genome. BMC Biotechnol. 2013 Oct 20;13:87. doi: 10.1186/1472-6750-13-87). Durrant describes new large serine recombinases (LSRs) divided into three classes distinguished from one another by efficiency and specificity, including landing pad LSRs which outperform wild-type Bxbl in episomal and chromosomal integration efficiency, LSRs that achieve both efficient and sitespecific integration without a landing pad, and multi -targeting LSRs with minimal site-specificity. Additionally, embodiments can include any serine recombinase such as BceINT, SSCINT, SACINT, and INT10 (see lonnidi et al., 2021; Drag-and-drop genome insertion without DNA cleavage with CRISPR directed integrases. bioRxiv 2021.11.01.466786, doi org/10. 1101 /2021 1 1 .01.466786). In some embodiments, the integration site can be selected from an attB site, an attP site, an attL site, an attR site, a lox71 site a Vox site, or a FRT site. In instances in this disclosure that refer to a Cre-lox system, the Cre-lox system is referred to either as a control for programmable gene insertion or as a tool for a recombinase-mediated event separate and distinct from insertion of the donor polynucleotide template (or exogenous nucleic acid) into the integrated recognition site.
[0224] It will be appreciated that desired activity of integrases, transposases and the like can depend on nuclear localization. In certain embodiments, prokaryotic enzymes are adapted to modulate nuclear localization. In certain embodiments, eukaryotic or vertebrate enzymes are
adapted to modulate nuclear localization. In certain embodiments, the invention provides fusion or hybrid proteins. Such modulation can comprise addition or removal of one or more nuclear localization signal (NLS) and/or addition or removal of one or more nuclear export signal (NES). Xu et al compared derivatives of fourteen serine integrases that either possess or lack a nuclear localization signal (NLS) to conclude that certain integrases benefit from addition of an NLS whereas others are transported efficiently without addition, and a major determinant of activity in yeast and vertebrate cells is avoidance of toxicity. (Xu et al., 2016, Comparison and optimization of ten phage encoded serine integrases for genome engineering in Saccharomyces cerevisiae. BMC Biotechnol. 2016 Feb 9; 16: 13. doi: 10.1186/sl2896-016-0241-5). Ramakrishnan et al. systematically studied the effect of different NES mutants developed from mariner-like elements (MLEs) on transposase localization and activity and concluded that nuclear export provides a means of controlling transposition activity and maintaining genome integrity. (Ramakrishnan et al. Nuclear export signal (NES) of transposases affects the transposition activity of mariner-like elements Ppmarl and Ppmar2 of moso bamboo. Mob DNA. 2019 Aug 19; 10:35. doi: 10.1186/sl3100-019-0179-y). The methods and constructs are used to modulate nuclear localization of system components of the invention.
[0225] In typical embodiments, the integrase used herein is selected from below (Table 10).












[0227] In some embodiments, the integration enzyme is selected from one of the about 27,000 Serine integrases described in International Patent Publication No. WO 2023/070031 A2, which is hereby incorporated by reference in its entirety.
5.9. Engineered Integration Enzymes
[0228] This disclosure features integration enzymes (also referred to integrases) engineered such that upon being introduced into a cell, the integration enzyme has increased stability (e.g., half- life) compared to a control integration enzyme not engineered to have increased stability. An increase in stability includes an increase in half-life and a reduction in protein degradation rates, thereby extending the capacity of the integration enzyme to mediate integration.
[0229] Without wishing to be bound by theory, there are two main pathways for protein degradation: proteolysis by ubiquitin proteosome system (UPS) and autophagy. Under the UPS system, proteins can be ubiquitinated on lysine residues, thereby leading to degradation. Proteins can also have specific domains (degrons) that can be ubiquitinated, which also leads to protein degradation. For example, in the UPS, Degrons (also referred to as degradation signals) are recognized and polyubiquitinated, targeting the protein for degradation. Degrons can be present
at both the N-terminus and/or C-terminus of a protein. Polyubiquitinated protein is recognized by receptor subunits of the 26S proteosome (RpnlO (yeast)/p54 (drosophila), Rpnl3 and Rpnl).
[0230] In some embodiments, the engineered integration enzymes is selected from the engineered integration enzymes described in FIGs. 23, 25, 28, 30B, 32A, 33, 35, and 40-42. In some embodiments, the engineered integration enzymes is selected from the engineered integration enzymes described in Table 26. In some embodiments, the engineered integration enzyme comprises an amino acid sequence that is at least 80% (e.g., 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 99%, or 100%) identical to an amino acid sequence listed in Table 26. In some embodiments, the engineered integration enzyme is encoded by a nucleic acid sequence that is at least 80% (e.g., 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 99%, or 100%) identical to a nucleic acid sequence listed in Table 26.
5.9.1. Engineered Integration Enzyme with a Stabilization Domain
[0231] This disclosure features integration enzymes engineered to include a domain (e.g., a stabilization domain) that increases stability of the integration enzyme when compared to an integration enzyme not engineered to include the domain (e.g., the stabilization domain).
[0232] In some embodiments, the integration enzyme or fragment thereof and the stabilization domain are fused, thereby by creating a fusion protein.
[0233] In one embodiment, an engineered integration enzyme comprises an integration enzyme or fragment thereof; an at least first stabilization domain; and a nuclear localization signal (NLS). In some embodiments, the integration enzyme or fragment thereof, the stabilization domain, and the NLS are fused, thereby by creating a fusion protein.
[0234] In some embodiments, the engineered integration enzyme is an integration enzyme described in Table 10 and engineered to include a stabilization domain that increases the stability of the BxB 1 as compared to an Bxb 1 integration enzyme not engineered to include the stabilization domain.
[0235] In some embodiments, the engineered integration enzyme is a BxBl integration enzyme engineered to include a stabilization domain that increases the stability of the BxBl as compared to an Bxbl integration enzyme not engineered to include the stabilization domain.
[0236] In some embodiments, the engineered integration enzyme comprises an integration enzyme as described in International Patent Publication No. WO 2023/070031A2, which is hereby
incorporated by reference in its entirety, where the integration enzyme is engineered to include a stabilization domain that increases the stability of the integration enzyme as compared to an integration enzyme not engineered to include the stabilization domain.
[0237] In some embodiments, the engineered integration enzyme comprises an amino acid sequence that is at least 80% (e.g., 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 99%, or 100%) identical to an amino acid sequence selected from SEQ ID NOs: 378-393 and a stabilization domain, where the stabilization domain increases the stability of the engineered integration enzyme as compared to an integration enzyme not engineered to include the stabilization domain.
[0238] In some embodiments, the engineered integration enzyme comprises an amino acid sequence that is at least 80% (e.g., 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 99%, or 100%) identical to an amino acid sequence of SEQ ID NO: 378 and a stabilization domain that increase the stability of the integration enzyme as compared to an integration enzyme not engineered to include a stabilization domain.
[0239] In some embodiments, the engineered integration enzyme comprises an amino acid sequence that is at least 80% (e.g., 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 99%, or 100%) identical to an amino acid sequence of SEQ ID NO: 379 and a stabilization domain, where the stabilization domain increase the stability of the integration enzyme as compared to an integration enzyme not engineered to include a stabilization domain.
[0240] In some embodiments, the engineered integration enzyme comprises an amino acid sequence that is at least 80% (e.g., 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 99%, or 100%) identical to an amino acid sequence of SEQ ID NO: 380 and a stabilization domain, where the stabilization domain increase the stability of the integration enzyme as compared to an integration enzyme not engineered to include a stabilization domain.
[0241] In some embodiments, the engineered integration enzyme comprises an amino acid sequence that is at least 80% (e.g., 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 99%, or 100%) identical to an amino acid sequence of SEQ ID NO: 381 and a stabilization domain, where the stabilization domain increase the stability of the integration enzyme as compared to an integration enzyme not engineered to include a stabilization domain.
[0242] In some embodiments, the engineered integration enzyme comprises an amino acid sequence that is at least 80% (e.g., 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 99%,
or 100%) identical to an amino acid sequence of SEQ ID NO: 382 and a stabilization domain, where the stabilization domain increase the stability of the integration enzyme as compared to an integration enzyme not engineered to include a stabilization domain.
[0243] In some embodiments, the engineered integration enzyme comprises an amino acid sequence that is at least 80% (e.g., 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 99%, or 100%) identical to an amino acid sequence of SEQ ID NO: 383 and a stabilization domain, where the stabilization domain increase the stability of the integration enzyme as compared to an integration enzyme not engineered to include a stabilization domain.
[0244] In some embodiments, the engineered integration enzyme comprises an amino acid sequence that is at least 80% (e.g., 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 99%, or 100%) identical to an amino acid sequence of SEQ ID NO: 384 and a stabilization domain, where the stabilization domain increase the stability of the integration enzyme as compared to an integration enzyme not engineered to include a stabilization domain.
[0245] In some embodiments, the engineered integration enzyme comprises an amino acid sequence that is at least 80% (e.g., 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 99%, or 100%) identical to an amino acid sequence of SEQ ID NO: 385 and a stabilization domain, where the stabilization domain increase the stability of the integration enzyme as compared to an integration enzyme not engineered to include a stabilization domain.
[0246] In some embodiments, the engineered integration enzyme comprises an amino acid sequence that is at least 80% (e.g., 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 99%, or 100%) identical to an amino acid sequence of SEQ ID NO: 386 and a stabilization domain, where the stabilization domain increase the stability of the integration enzyme as compared to an integration enzyme not engineered to include a stabilization domain.
[0247] In some embodiments, the engineered integration enzyme comprises an amino acid sequence that is at least 80% (e.g., 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 99%, or 100%) identical to an amino acid sequence of SEQ ID NO: 387 and a stabilization domain, where the stabilization domain increase the stability of the integration enzyme as compared to an integration enzyme not engineered to include a stabilization domain.
[0248] In some embodiments, the engineered integration enzyme comprises an amino acid sequence that is at least 80% (e.g., 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 99%, or 100%) identical to an amino acid sequence of SEQ ID NO: 388 and a stabilization domain,
where the stabilization domain increase the stability of the integration enzyme as compared to an integration enzyme not engineered to include a stabilization domain.
[0249] In some embodiments, the engineered integration enzyme comprises an amino acid sequence that is at least 80% (e.g., 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 99%, or 100%) identical to an amino acid sequence of SEQ ID NO: 389 and a stabilization domain, where the stabilization domain increase the stability of the integration enzyme as compared to an integration enzyme not engineered to include a stabilization domain.
[0250] In some embodiments, the engineered integration enzyme comprises an amino acid sequence that is at least 80% (e.g., 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 99%, or 100%) identical to an amino acid sequence of SEQ ID NO: 390 and a stabilization domain, where the stabilization domain increase the stability of the integration enzyme as compared to an integration enzyme not engineered to include a stabilization domain.
[0251] In some embodiments, the engineered integration enzyme comprises an amino acid sequence that is at least 80% (e.g., 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 99%, or 100%) identical to an amino acid sequence of SEQ ID NO: 391 and a stabilization domain, where the stabilization domain increase the stability of the integration enzyme as compared to an integration enzyme not engineered to include a stabilization domain.
[0252] In some embodiments, the engineered integration enzyme comprises an amino acid sequence that is at least 80% (e.g., 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 99%, or 100%) identical to an amino acid sequence of SEQ ID NO: 392 and a stabilization domain, where the stabilization domain increase the stability of the integration enzyme as compared to an integration enzyme not engineered to include a stabilization domain.
[0253] In some embodiments, the engineered integration enzyme comprises an amino acid sequence that is at least 80% (e.g., 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 99%, or 100%) identical to an amino acid sequence of SEQ ID NO: 393 and a stabilization domain, where the stabilization domain increase the stability of the integration enzyme as compared to an integration enzyme not engineered to include a stabilization domain.
5.9.1.1 Stabilization Domains
[0254] In one embodiment, an engineered integration enzyme comprises an integration enzyme or fragment thereof; and an at least first stabilization domain. In some embodiments, the engineered integration enzyme comprises an integration enzyme or a fragment thereof, a first stabilization
domain, and a second stabilization domain. In some embodiments, the engineered integration enzyme comprises an integration enzyme or a fragment thereof, a first stabilization domain, a second stabilization domain, and a third stabilization domain.
[0255] In some embodiments, the stabilization domain is selected from a stabilon motif, an exin21 motif, or a combination thereof.
[0256] In some embodiments, the stabilization domain comprises a stabilon domain. The stabilon sequence originated from C-terminus of p54 protein (drosophila). The stabilon amino acid sequence is conserved across species. The stabilon domain can be 13 amino acids (e.g., KDKKSDGKDSQKK (SEQ ID NO: 583) (Drosophila melanogaster)) or 15 amino acids (e.g., KDKDKKSDGKDSQKK (SEQ ID NO: 584)). In some embodiments, the stabilization domain comprises an amino acid sequence that is at least 80% (e.g., 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 99%, or 100%) identical to an amino acid sequence of KDKKSDGKDSQKK (SEQ ID NO: 583). In some embodiments, the stabilization domain comprises an amino acid sequence that is at least 80% (e.g., 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 99%, or 100%) identical to an amino acid sequence of KDKDKKSDGKDSQKK (SEQ ID NO: 584).
[0257] In some embodiments, the stabilization domain comprises a Exin21 domain. In some embodiments, the stabilization domain comprises an amino acid sequence of QPRFAAA (SEQ ID NO: 585). In some embodiments, the stabilization domain comprises an amino acid sequence having one, two, three, four or five or more amino acid substitutions in SEQ ID NO: 585. In some embodiments, the stabilization domain is encoded by a polynucleotide sequence having a nucleic acid sequence that is at least 80% (e.g., 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 99%, or 100%) identical to CAACCGCGGTTCGCGGCCGCT (SEQ ID NO: 586).
[0258] In some embodiments, the engineered integration enzymes comprises an integration enzyme comprising an amino acid sequence that is at least 80% (e.g., 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 99%, or 100%) identical to an amino acid sequence of SEQ ID NO: 388 and a stabilization domain that comprises an amino acid sequence that is at least 80% (e.g., 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 99%, or 100%) identical to an amino acid sequence of KDKDKKSDGKDSQKK (SEQ ID NO: 584) or an amino acid sequence having one, two, three, four or five or more amino acid substitutions in SEQ ID NO: 584.
[0259] In some embodiments, the engineered integration enzymes comprises an integration enzyme comprising an amino acid sequence that is at least 80% (e.g., 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 99%, or 100%) identical to an amino acid sequence of SEQ ID NO: 388 and a stabilization domain comprising an amino acid sequence of QPRFAAA (SEQ ID NO: 585) or an amino acid sequence having one, two, three, four or five or more amino acid substitutions in SEQ ID NO: 585.
[0260] In some embodiments, the stabilization domain is positioned relative to the integration enzyme (e.g., N-terminus, C-terminus, or in an internal position) such that when expressed/present in a cell, the stabilization domain increase stability of the integration enzyme (e.g., increase stability when compared to an integration enzyme not engineered to include the domain). In embodiments where the stabilization domain is placed in an internal position in the integration enzyme, the position in the integration enzyme is selected strategically placed such that it confers increased stability without compromising the integration enzyme’s native function. For example, by positioning the stabilization domain internally, interference with the protein’s natural structure and function can be avoided while still gaining the benefits of the stabilization domain.
[0261] In some embodiments, an engineered integration enzyme comprises an integration enzyme or a fragment thereof and a first stabilization domain, and optionally a second stabilization domain.
[0262] In some embodiments, the first stabilization domain and/or second stabilization domain are positioned in the engineered integration enzyme such that the engineered integration enzyme and the first and/or second stabilization domains are an in-frame fusion.
[0263] In some embodiments, the first stabilization domain and/or second stabilization domain are located N-terminus to the integration enzyme or fragment thereof, C-terminus to the integration enzyme of fragment thereof, or between two consecutive amino acid residues in the amino acid sequence of the integration enzyme or fragment thereof (also referred to as “internal”).
[0264] In some embodiments, the first stabilization domain and/or second stabilization domain are located N-terminus to the integration enzyme or fragment thereof.
[0265] In some embodiments, the first stabilization domain and/or second stabilization domain are located C-terminus to the integration enzyme or fragment thereof.
[0266] In some embodiments, the first stabilization domain and/or second stabilization domain are located between two consecutive amino acid residues in the amino acid sequence of the integration enzyme or fragment thereof.
[0267] In some embodiments, the two consecutive amino acid residues in the amino acid sequence of the integration enzyme are located in a N-terminal catalytic domain; a recombinase domain, or one or more zinc ribbon domain.
[0268] In some embodiments, the two consecutive amino acid residues in the amino acid sequence of the integration enzyme correspond amino acid residues between the N-terminal catalytic domain and the recombinase domain or between the recombinase domain and one of the zinc ribbon domains.
[0269] In some embodiments, the engineered integration enzyme comprises an orientation from N-terminus to C-terminus: N-I-S; I-S-N; S-I-N; N-S-I;N-Xi-I-S; I-Xi-S-N; S-Xi-I-N; N-Xi-S-I; N-I-Xi-S; I-S-Xi-N; S-I-Xi-N; N-S-Xi-I; N-X1-I-X2-S; I-X1-S-X2-N; S-X1-I-X2-N; and N-Xi-S- X2-I; wherein I is the integration enzyme or fragment thereof, Xi is a first linker, S is the stabilization domain, X2 is a second linker, and N is the NLS.
5.9.2. Engineered Integration Enzymes with a Modification that Increase Stabilization
[0270] This disclosure features an integration enzyme where the amino acid sequence of the integration is modified such that the integration is more stable than an integration enzyme not comprising a modification in the amino acid sequence.
[0271] In one embodiment, an engineered integration enzyme comprises: an integration enzyme or fragment thereof comprising at least a first amino acid modification that increases the stability of the integration enzyme as compared to the integration enzyme not comprising the at least first amino acid modification. In some embodiments, the engineered integration enzyme comprises a nuclear localization signal (NLS), where the NLS is positioned in the engineered integration enzyme such that it directs the engineered integration enzyme to the nucleus.
[0272] In some embodiments, the engineered integration enzyme comprises an integration enzyme or fragment thereof comprising at least two (e.g., at least three, at least four, at least five, at least six, at least seven, at least eight, at least nine, or at least ten) amino acid modification such that the combination of the at least two modifications increase the stability of the integration enzyme as compared to the integration enzyme not comprising the at least first amino acid modification.
[0273] In some embodiments, the amino acid modification can be in a degron motif in the integration enzyme. In some embodiments, the amino acid modification can be a substitution, deletion, or insertion that increases the stability of the integration enzyme as compared to an integration enzyme not comprising the at least first amino acid modification.
[0274] In some embodiments, the amino acid modification can be a substitution, deletion, or insertion in a degron motif in the integration enzyme. Computational prediction for BxB 1 showed the existence of two degron domains and several lysine residues (FIG. 36A).
[0275] In some embodiments, the engineered integration enzyme is an integration enzyme described in Table 10 and comprises an amino acid modification that increases the stability of the integration enzyme as compared to an integration enzyme selected from Table 10 and not engineered to include the amino acid modification.
[0276] In some embodiments, the amino acid modification can be a substitution, deletion, or insertion in a BxB 1 integration enzyme that increases the stability of the BxB 1 as compared to an Bxbl integration enzyme that does not comprise the at least first amino acid modification.
[0277] In some embodiments, the amino acid modification can be a substitution, deletion, or insertion in an integration enzyme having an amino acid sequence of SEQ ID NO: 388 that increases the stability of the integration enzyme as compared to an integration enzyme having a sequence of SEQ ID NO: 388 that does not comprise the at least first amino acid modification.
[0278] In one embodiment, the first amino acid modification is an amino acid substitution of L275V in SEQ ID NO: 388.
[0279] In some embodiments, the engineered integration enzyme comprises an integration enzyme as described in International Patent Publication No. WO 2023/070031A2, which is hereby incorporated by reference in its entirety, where the integration enzyme includes an amino acid modification that increases the stability of the integration enzyme as compared to an integration enzyme not engineered to have the amino acid modification.
[0280] In some embodiments, the engineered integration enzyme comprises an amino acid sequence that is at least 80% (e.g., 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 99%, or 100%) identical to an amino acid sequence selected from SEQ ID NOs: 378-393 and an amino acid modification in the amino acid sequence where the modification increases the stability of the
engineered integration enzyme as compared to an integration enzyme not engineered to have the at least first amino acid modification.
[0281] In some embodiments, the engineered integration enzyme comprises an amino acid sequence that is at least 80% (e.g., 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 99%, or 100%) identical to an amino acid sequence of SEQ ID NO: 378 and an amino acid modification in the sequence of SEQ ID NO: 378, where the amino acid modification increases the stability of the integration enzyme as compared to an integration enzyme having an amino acid sequence selected of SEQ ID NO: 378.
[0282] In some embodiments, the engineered integration enzyme comprises an amino acid sequence that is at least 80% (e.g., 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 99%, or 100%) identical to an amino acid sequence of SEQ ID NO: 379 and an amino acid modification in the sequence of SEQ ID NO: 379, where the amino acid modification increases the stability of the integration enzyme as compared to an integration enzyme having an amino acid sequence selected of SEQ ID NO: 379.
[0283] In some embodiments, the engineered integration enzyme comprises an amino acid sequence that is at least 80% (e.g., 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 99%, or 100%) identical to an amino acid sequence of SEQ ID NO: 380 and an amino acid modification in the sequence of SEQ ID NO: 380, where the amino acid modification increases the stability of the integration enzyme as compared to an integration enzyme having an amino acid sequence selected of SEQ ID NO: 381.
[0284] In some embodiments, the engineered integration enzyme comprises an amino acid sequence that is at least 80% (e.g., 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 99%, or 100%) identical to an amino acid sequence of SEQ ID NO: 381 and an amino acid modification in the sequence of SEQ ID NO: 381, where the amino acid modification increases the stability of the integration enzyme as compared to an integration enzyme having an amino acid sequence selected of SEQ ID NO: 381.
[0285] In some embodiments, the engineered integration enzyme comprises an amino acid sequence that is at least 80% (e.g., 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 99%, or 100%) identical to an amino acid sequence of SEQ ID NO: 382 and an amino acid modification in the sequence of SEQ ID NO: 382, where the amino acid modification increases the stability of
the integration enzyme as compared to an integration enzyme having an amino acid sequence selected of SEQ ID NO: 382.
[0286] In some embodiments, the engineered integration enzyme comprises an amino acid sequence that is at least 80% (e.g., 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 99%, or 100%) identical to an amino acid sequence of SEQ ID NO: 383 and an amino acid modification in the sequence of SEQ ID NO: 383, where the amino acid modification increases the stability of the integration enzyme as compared to an integration enzyme having an amino acid sequence selected of SEQ ID NO: 383.
[0287] In some embodiments, the engineered integration enzyme comprises an amino acid sequence that is at least 80% (e.g., 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 99%, or 100%) identical to an amino acid sequence of SEQ ID NO: 384 and an amino acid modification in the sequence of SEQ ID NO: 384, where the amino acid modification increases the stability of the integration enzyme as compared to an integration enzyme having an amino acid sequence selected of SEQ ID NO: 384.
[0288] In some embodiments, the engineered integration enzyme comprises an amino acid sequence that is at least 80% (e.g., 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 99%, or 100%) identical to an amino acid sequence of SEQ ID NO: 385 and an amino acid modification in the sequence of SEQ ID NO: 385, where the amino acid modification increases the stability of the integration enzyme as compared to an integration enzyme having an amino acid sequence selected of SEQ ID NO: 385.
[0289] In some embodiments, the engineered integration enzyme comprises an amino acid sequence that is at least 80% (e.g., 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 99%, or 100%) identical to an amino acid sequence of SEQ ID NO: 386 and an amino acid modification in the sequence of SEQ ID NO: 386, where the amino acid modification increases the stability of the integration enzyme as compared to an integration enzyme having an amino acid sequence selected of SEQ ID NO: 386.
[0290] In some embodiments, the engineered integration enzyme comprises an amino acid sequence that is at least 80% (e.g., 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 99%, or 100%) identical to an amino acid sequence of SEQ ID NO: 387 and an amino acid modification in the sequence of SEQ ID NO: 387, where the amino acid modification increases the stability of
the integration enzyme as compared to an integration enzyme having an amino acid sequence selected of SEQ ID NO: 387.
[0291] In some embodiments, the engineered integration enzyme comprises an amino acid sequence that is at least 80% (e.g., 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 99%, or 100%) identical to an amino acid sequence of SEQ ID NO: 388 and an amino acid modification in the sequence of SEQ ID NO: 388, where the amino acid modification increases the stability of the integration enzyme as compared to an integration enzyme having an amino acid sequence selected of SEQ ID NO: 388.
[0292] In some embodiments, the engineered integration enzyme comprises an amino acid sequence that is at least 80% (e.g., 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 99%, or 100%) identical to an amino acid sequence of SEQ ID NO: 389 and an amino acid modification in the sequence of SEQ ID NO: 389, where the amino acid modification increases the stability of the integration enzyme as compared to an integration enzyme having an amino acid sequence selected of SEQ ID NO: 389.
[0293] In some embodiments, the engineered integration enzyme comprises an amino acid sequence that is at least 80% (e.g., 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 99%, or 100%) identical to an amino acid sequence of SEQ ID NO: 390 and an amino acid modification in the sequence of SEQ ID NO: 390, where the amino acid modification increases the stability of the integration enzyme as compared to an integration enzyme having an amino acid sequence selected of SEQ ID NO: 390.
[0294] In some embodiments, the engineered integration enzyme comprises an amino acid sequence that is at least 80% (e.g., 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 99%, or 100%) identical to an amino acid sequence of SEQ ID NO: 391 and an amino acid modification in the sequence of SEQ ID NO: 391, where the amino acid modification increases the stability of the integration enzyme as compared to an integration enzyme having an amino acid sequence selected of SEQ ID NO: 391.
[0295] In some embodiments, the engineered integration enzyme comprises an amino acid sequence that is at least 80% (e.g., 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 99%, or 100%) identical to an amino acid sequence of SEQ ID NO: 392 and an amino acid modification in the sequence of SEQ ID NO: 392, where the amino acid modification increases the stability of
the integration enzyme as compared to an integration enzyme having an amino acid sequence selected of SEQ ID NO: 392.
[0296] In some embodiments, the engineered integration enzyme comprises an amino acid sequence that is at least 80% (e.g., 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 99%, or 100%) identical to an amino acid sequence of SEQ ID NO: 393 and an amino acid modification in the sequence of SEQ ID NO: 393, where the amino acid modification increases the stability of the integration enzyme as compared to an integration enzyme having an amino acid sequence selected of SEQ ID NO: 393.
5.9.3. Engineered Integration Enzymes with a Combination of a Stabilization Domain and a Modification that Increases Stabilization
[0297] This disclosure also features engineered integration enzymes comprising a stabilization domain and an amino acid modification that increases the engineered integration enzymes stability.
[0298] In some embodiments, the engineered integration enzyme comprises an integration enzyme as described in International Patent Publication No. WO 2023/070031A2, which is hereby incorporated by reference in its entirety, where the integration enzyme includes a stabilization domain and an amino acid modification, where the stabilization domain, the amino acid modification, or both, increase the stability of the integration enzyme as compared to an integration enzyme not engineered to have the amino acid modification and/or the stabilization domain.
[0299] In some embodiments, the engineered integration enzyme is an integration enzyme described in Table 10 and comprises a stabilization domain and an amino acid modification in the same integration enzyme selected from Table 10, wherein the stabilization domain, the amino acid modification, or both, increase the stability of the integration enzyme as compared to an integration enzyme not engineered to have the amino acid modification and/or stabilization domain.
[0300] In some embodiments, the engineered integration enzyme is a BxB 1 integration enzyme comprising a stabilization domain and an amino acid modification in the BxB 1 integration enzyme, wherein the stabilization domain, the amino acid modification, or both, increase the stability integration enzyme as compared to an integration enzyme not engineered to have the amino acid modification and/or stabilization domain.
[0301] In some embodiments, the engineered integration enzyme comprises an amino acid sequence that is at least 80% (e.g., 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 99%, or 100%) identical to an amino acid sequence selected from SEQ ID NOs: 378-393, a stabilization
domain, and an amino acid modification in the amino acid sequence, where the stabilization domain, the amino acid modification, or both, increase the stability of the engineered integration enzyme as compared to an integration enzyme not engineered to have the at amino acid modification and/or the stabilization domain. In some embodiments, the engineered integration enzyme comprises an amino acid sequence that is at least 80% (e.g., 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 99%, or 100%) identical to an amino acid sequence of SEQ ID NO: 388, a stabilization domain and an amino acid modification in SEQ ID NO: 388, wherein the stabilization domain, the amino acid modification, or both, increase the stability of integration enzyme as compared to an integration enzyme not engineered to have the amino acid modification and/or stabilization domain.
5.9.4. Engineered mRNA Encoding an Integration Enzyme
[0302] This disclosure features a polynucleotide comprising a nucleic acid sequence encoding any of the engineered integration enzymes described herein (see Section 4.9.1, 4.9.2, and 4.9.3) or any of the engineered integration enzyme linked to the gene editor polypeptide or fusion proteins described herein (see Section 4.9.5).
[0303] In some embodiments, the nucleic acid sequence encoding the engineered integration enzyme is codon optimized. In some embodiments, the codon optimization is performed using an algorithm.
[0304] In some embodiments, the nucleic acid sequence of the engineered integration enzyme is optimized based on second structure such that the structure confers increased stability of the polynucleotide. In some embodiments, the optimization based on second structure relies on an algorithm to determine the optimal mRNA sequence.
[0305] A non-limiting example of an algorithm capable of being used to optimize the mRNA encoding any of the any of the engineered integration enzymes described herein (see Section 4.9.1, 4.9.2, and 4.9.3) is as described in Zhang et al. (Algorithm for Optimized mRNA Design Improves Stability and Immunogenicity, Nature 2023, doi.org/10.1038/s41586-023-06127-z), which is hereby incorporated by reference in its entirety. Zhang et al. refer to an algorithm called LinearDesign that optimizes both structural stability (via secondary structure) and codon usage.
[0306] In some embodiments, the nucleic acid sequence of the engineered integration enzyme is optimized based on the LinearDesign algorithm.
[0307] This disclosure also features vector comprising the nucleic acid sequence encoding any of the engineered integration enzymes described herein (see Section 4.9.1, 4.9.2, and 4.9.3) or any of the engineered integration enzyme linked to the gene editor polypeptide or fusion proteins described herein (see Section 4.9.5).
[0308] Other non-limiting examples of increasing the stability of an mRNA include: using novel UTR’s or codons that increase the longevity of the mRNA or circular mRNA.
5.9.5. Engineered Integration Enzymes fused to a Gene Editor Polypeptide
[0309] This disclosure also features engineered integration enzymes (e.g., any of the engineered integration enzymes described herein (see, e.g., Section 4.9.1, 4.9.2, and 4.9.3) linked to a gene editor polypeptide (e.g., any of the gene editor polypeptides described herein). In such cases, the linked engineered integration enzyme-gene editor polypeptide can be used to incorporate an integration recognition site into the genome of a cell and for integrating a donor polynucleotide template into the genome.
[0310] In some embodiments, the C-terminus of the engineered integration enzyme is linked to the gene editor polypeptide.
[0311] In some embodiments, the C-terminus of the gene editor polypeptide is linked to the engineered integration enzymes.
[0312] In some embodiments, the engineered integration enzyme is linked to the gene editor polypeptide by in-frame fusion.
[0313] In some embodiments, the engineered integration enzyme is linked to the gene editor polypeptide by a linker.
[0314] In some embodiments, the linker is a peptide fused in-frame between the engineered integration enzyme and the gene editor polypeptide.
[0315] In some embodiments, the one or more linkers is selected from: Table 3.
[0316] In some embodiments, the gene editor polypeptide comprises a DNA binding domain and a reverse transcriptase.
[0317] This disclosure also features a fusion protein comprising (a) a DNA binding domain, optionally comprising a nickase activity; (b) a reverse transcriptase; and (c) any of the engineered
integration enzymes described herein (see, e.g., Sections 4.9.1, 4.9.2, and 4.9.3), wherein at least any two of elements (a), (b), or (c) are linked via at least a first C-terminal linker comprising. In some embodiments, the fusion protein comprises from N-terminus to C-terminus: (a), (b), and (c); (b), (a), and (c); (c), (a), and (b); (c), (b), and (a); (b), (c), and (a); and (b), (a), and (c).
[0318] In some embodiments, the engineered integration enzyme is linked to a gene editor is described in FIG. 46.
5.9.6. Measuring Stability
[0319] Non-limiting examples of metrics that can be used as indication of protein stability include the following.
[0320] Protein Half-Life: This is the time required for half of the amount of protein in a cell to be degraded. This can be used to measure protein turnover. One non-limiting example of measuring protein half-life includes using pulse-chase experiments where the protein is labeled with a radioactive or other traceable isotope, then followed over time to see how rapidly it disappears.
[0321] Melting Temperature (Tm): This is a measure of a protein’s thermal stability. It is the temperature at which half of the protein population is unfolded or denatured. It can be determined through a variety of methods, for example, without limitation, differential scanning calorimetry (DSC), circular dichroism (CD), or fluorescence-based thermal shift assays.
[0322] Thermodynamic Stability (AG): This is the change in Gibbs free energy between the folded and unfolded state of the protein. It measures how much energy is required to unfold the protein, with a larger value indicating a more stable protein. Non-limiting examples of measuring thermodynamic stability include equilibrium denaturation experiments, where the protein is exposed to varying concentrations of a denaturant and the degree of unfolding is monitored.
[0323] Protein Degradation Rate: This method measures how quickly a protein is degraded in a cell. It can be quantified using a variety of methods, for example, without limitation: western blot analysis (with or without the use of protease inhibitors) to assess protein levels over time.
[0324] Resistance to Chemical Denaturation: The concentration of denaturing agent (e,g., urea or guanidinium chloride) needed to unfold a protein can provide a measure of the proteins stability.
5.9.7. Systems Comprising an Engineered Integration Enzyme
[0325] This disclosure features a system for site-specifically integrating a donor polynucleotide template into a mammalian cell genome at a target DNA sequence, comprising: an attachment site containing gRNA (atgRNA) comprising at least a portion of an at least first integration recognition site; a gene editor polypeptide comprising a DNA binding nickase domain linked to a reverse transcriptase domain capable of incorporating the integration recognition site into the target DNA sequence, any of the engineered integration enzymes described herein (e.g., an engineered integration enzyme comprising an integration enzyme and a stabilization domain (see Section 4.9.1) or an engineered integration enzyme comprising an integration enzyme comprising an amino acid modification that increases stability (see Section 4.9.2)); a donor polynucleotide template linked to a sequence that is an integration cognate of the integration recognition site present in the atgRNA, whereby the gene editor polypeptide site-specifically integrates the integration recognition site into the target DNA sequence, whereby the engineered integration enzyme integrates the donor polynucleotide template into the target DNA sequence.
[0326] In some embodiments of the system comprising an engineered integration enzyme, the first atgRNA comprises: (i) a domain that is capable of guiding the gene editor polypeptide to the target DNA sequence; and (ii) a reverse transcriptase (RT) template that comprises at least a portion of an at least first integration recognition site, whereby the at least portion of the at least first integration recognition site is integrated into the genome of the cell at the target sequence.
[0327] In some embodiments of the system comprising an engineered integration enzyme, the system also includes a second atgRNA.
[0328] In some embodiments of the system comprising an engineered integration enzyme, the first atgRNA and the second atgRNA are an at least first pair of atgRNAs, wherein the at least first pair of atgRNAs have domains that are capable of guiding the gene editor polypeptide to the target DNA sequence; the first atgRNA further includes a first RT template that comprises at least a portion of an at least first integration recognition site; the second atgRNA further includes a second RT template that comprises at least a portion of the first integration recognition site, and the first atgRNA and the second atgRNAs collectively encode the entirety of the first integration recognition site, whereby the least first integration recognition site is integrated into the genome of the cell at the target sequence.
[0329] In some embodiments of the system comprising an engineered integration enzyme, upon introducing the system into the cell, the engineered integration enzyme enhances integration
efficiency of the donor polynucleotide template at the site-specifically integrated integration recognition site as compared to the integration efficiency of a system using a non-engineered integration enzyme to integrate donor polynucleotide template at the site-specifically integrated integration recognition site.
5.9.8. Method of Using an Engineered Integration Enzyme
[0330] This disclosure also features a method for site-specifically integrating a donor polynucleotide template into a mammalian cell genome at a target DNA sequence, comprising: incorporating an integration recognition site into the genome by delivering into the cell: an attachment site containing guide RNA (atgRNA) comprising at least a portion of an at least first integration recognition site; and a gene editor polypeptide or polynucleotide encoding a gene editor polypeptide, wherein the gene editor polypeptide comprises a DNA binding nickase domain linked to a reverse transcriptase domain and is capable of incorporating the integration recognition site into the target DNA sequence; and optionally, a nicking gRNA; and integrating the donor polynucleotide template into the genome by delivering into the cell: any of the engineered integration enzymes described herein (e.g., an engineered integration enzyme comprising an integration enzyme and a stabilization domain (see Section 4.9.1) or an engineered integration enzyme comprising an integration enzyme comprising an amino acid modification that increases stability (see Section 4.9.2)); and a donor polynucleotide template, wherein the donor polynucleotide template is linked to a sequence that is an integration cognate of the integration recognition site present in the atgRNA, and wherein the donor polynucleotide template is integrated into the genome at the incorporated genomic integration recognition site by the integration enzyme.
[0331] In some embodiments of the method using the engineered integration enzyme, the first atgRNA comprises: (i) a domain that is capable of guiding the gene editor polypeptide to the target DNAsequence; and (ii) a reverse transcriptase (RT) template that comprises at least a portion of an at least first integration recognition site, whereby the at least portion of the at least first integration recognition site is integrated into the genome of the cell at the target sequence.
[0332] In some embodiments of the method using the engineered integration enzyme, the method also includes a second atgRNA.
[0333] In some embodiments of the method using the engineered integration enzyme, the first atgRNA and the second atgRNA are an at least first pair of atgRNAs, wherein the at least first pair of atgRNAs have domains that are capable of guiding the gene editor polypeptide to the target
DNA sequence; the first atgRNA further includes a first RT template that comprises at least a portion of an at least first integration recognition site; the second atgRNA further includes a second RT template that comprises at least a portion of the first integration recognition site, and the first atgRNA and the second atgRNAs collectively encode the entirety of the first integration recognition site, whereby the least first integration recognition site is integrated into the genome of the cell at the target sequence.
[0334] In some embodiments of the method using the engineered integration enzyme, the method enhances integration efficiency of the donor polynucleotide template at the site-specifically integrated integration recognition stie at the double-stranded target DNA sequence as compared to the integration efficiency of the donor polynucleotide template at the site-specifically integrated recognition site when using a method that does not comprise the engineered integration enzyme.
5.10. Co-delivery of gene editor and donor DNA template
[0335] This disclosure features methods of delivering (e.g., co-delivery or dual delivery) a system capable of site-specifically integrating a template polynucleotide into the genome of a cell, where the methods include delivering to a (i) gene editor construct and a (ii) template polynucleotide, and (iii) at least a first attachment site-containing guide (atgRNA).
[0336] This disclosure also features a method for delivering a system capable of site-specifically integrating a template polynucleotide into the genome of a cell, where the method includes: delivering a lipid nanoparticle (LNP) comprising a gene editor polynucleotide (e.g., a gene editor polynucleotide construct) and a vector comprising a template polynucleotide and at least a first attachment site-containing guide RNA (atgRNA). In some embodiments, the first atgRNA comprises (i) a domain that is capable of guiding the prime editor system to a target sequence; and (ii) a reverse transcriptase (RT) template that comprises at least a portion of a first integration recognition site. In some embodiments, where the vector comprises a polynucleotide encoding a first atgRNA, the RT template comprises the entirety of the first integration recognition site. In some embodiments, where the vector comprises a polynucleotide encoding a first atgRNA, the vector also includes a sequence encoding a nicking guide RNA (ngRNA).
[0337] This disclosure also features a method for delivering a system capable of site-specifically integrating a template polynucleotide into the genome of a cell, where the method includes: delivering a lipid nanoparticle (LNP) comprising a gene editor polynucleotide (e.g., a gene editor polynucleotide construct) and a vector comprising a template polynucleotide and a first attachment site-containing guide RNA (atgRNA) and a second attachment site-containing guide RNA
(atgRNA). In some embodiments, the first atgRNA and the second atgRNA are an at least first pair of atgRNAs, wherein the at least first pair of atgRNAs have domains that are capable of guiding the prime editor system to a target sequence, the first atgRNA further includes a first RT template that comprises at least a portion of the at least first integration recognition site; and the second atgRNA further includes a second RT template that comprises at least a portion of the first integration recognition site, and the first atgRNA and the second atgRNAs collectively encode the entirety of the first integration recognition site. In such embodiments, the first atgRNA and second atgRNA include at least a 6bp overlap (e.g., 6bp of complementarity).
[0338] This disclosure also features a method for delivering a system capable of site-specifically integrating a template polynucleotide into the genome of a cell, where the method includes: delivering into a cell a lipid nanoparticle (LNP) comprising: (i) a gene editor polynucleotide (e.g., a gene editor polynucleotide construct), and (ii) a first attachment site-containing guide RNA (atgRNA); and a vector comprising: (i) a template polynucleotide, and (ii) a second atgRNA. In some embodiments, the first atgRNA and the second atgRNA are an at least first pair of atgRNAs, wherein the at least first pair of atgRNAs have domains that are capable of guiding the prime editor system to a target sequence, the first atgRNA further includes a first RT template that comprises at least a portion of the a first integration recognition site; and the second atgRNA further includes a second RT template that comprises at least a portion of the first integration recognition site, and the first atgRNA and the second atgRNAs collectively encode the entirety of the first integration recognition site. In such embodiments, the first atgRNA and second atgRNA include at least a 6bp overlap (e.g., 6bp of complementarity).
[0339] This disclosure also features a method for delivering a system capable of site-specifically integrating a template polynucleotide into the genome of a cell, where the method includes delivering: a lipid nanoparticle (LNP) comprising: (i) a gene editor polynucleotide (e.g., a gene editor polynucleotide construct), (ii) a first attachment site-containing guide RNA (atgRNA), and (iii) a second atgRNA; and a vector comprising (i) a template polynucleotide. In some embodiments, the first atgRNA and the second atgRNA are an at least first pair of atgRNAs, wherein the at least first pair of atgRNAs have domains that are capable of guiding the prime editor system to a target sequence, the first atgRNA further includes a first RT template that comprises at least a portion of the at least first integration recognition site; and the second atgRNA further includes a second RT template that comprises at least a portion of the at least first integration recognition site, and the first atgRNA and the second atgRNAs collectively encode the entirety of
the first integration recognition site. In such embodiments, the first atgRNA and second atgRNA include at least a 6bp overlap (e.g., 6bp of complementarity).
[0340] This disclosure also features a method for delivering a system capable of site-specifically integrating a template polynucleotide into the genome of a cell, where the method includes delivering: a lipid nanoparticle (LNP) comprising: (i) a gene editor polynucleotide (e.g., a gene editor polynucleotide construct) and (ii) a first attachment site-containing guide RNA (atgRNA); and a vector comprising: (i) a template polynucleotide, and (ii) a nicking atgRNA. In some embodiments, the first atgRNA comprises (i) a domain that is capable of guiding the prime editor system to a target sequence; and (ii) a reverse transcriptase (RT) template that comprises at least a portion of a first integration recognition site. In some embodiments, where the vector comprises a polynucleotide encoding a first atgRNA, the RT template comprises the entirety of the first integration recognition site.
[0341] In some embodiments, where the method includes delivering an LNP and a first vector, the LNP and the first vector are delivered at least 1 day, at least 2 days, at least 3 days, at least 4 days, at least 5 days, at least 6 days, at least 7 days, at least 2 weeks, at least 3 weeks, at least 4 weeks, at least 5 weeks, at least 6 weeks, at least 7 weeks, or at least 8 weeks apart. In some embodiments, where the method includes delivering an LNP and a second vector, the LNP and the second vector are delivered a different times on the same day, at least 1 day, at least 2 days, at least 3 days, at least 4 days, at least 5 days, at least 6 days, at least 7 days, at least 2 weeks, at least 3 weeks, at least 4 weeks, at least 5 weeks, at least 6 weeks, at least 7 weeks, or 8 weeks apart. In some embodiments, the LNP and the first vector are delivered about 6 weeks apart.
[0342] This disclosure also features a method for delivering a system capable of site-specifically integrating a template polynucleotide into the genome of a cell, where the method includes delivering the system in vivo. In a non-limiting example, the system is delivered to a fetus or a neonate to site-specifically integrate in vivo a template polynucleotide into the genome of a cell. Delivering the system to a fetus or a neonate provides advantages over delivering the system later in life (e.g., after the neonate phase ends), including: (i) fewer number of cells that need to be treated (e.g., in the adult, there are trillions of cells, but in a fetus, there are significantly fewer cells); (ii) developmental benefits: the early stage of development of a fetus or a neonate means that if a genetic disease is treated successfully, the individual could potentially develop normally, with significant reduction or even complete removal of any of the disease manifestations; (iii) preventing disease progression: in certain genetic conditions the physiological damage is
irreversible damage and in some instances is exacerbated as the disease progresses, therefore, intervening at the fetal (or neonate) stage, it is possible to prevent or reduce the progression of the disease and potentially prevent any irreversible damage from occurring; (iv) higher cell turnover and cell division rate: in a developing fetus, cells are dividing rapidly as the fetus (or neonate) grows, which means that if programmable gene insertion is achieved in the fetus (or neonate) is introduced, it could be propagated more rapidly throughout the body than in an older child or adult; and (v) immune tolerance: for example, there is evidence to suggest that performing gene therapy (e.g., programmable gene insertion) early in development might result in immune tolerance to the vector, thereby reducing the risk of an immune response against the system.
[0343] In some embodiments, the method includes delivering an LNP and a first vector, the LNP and the first vector are delivered to a cell in vivo. In some embodiments, the in vivo cells are present in a fetus or a neonate. In some embodiments, the LNP is delivered between age 0 (day of birth) and age 7 days and the vector is delivered between age 5 weeks and age 7 weeks. In some embodiments, the LNP is delivered at about at 2 days and the vector is delivered at about age 6 weeks.
[0344] In some embodiments, where the method includes delivering an LNP and a first vector, the LNP and the first vector are delivered to a cell in vivo, the LNP can be delivered to a fetus at a first time point and the vector is delivered to the fetus after the fetus is bom (referred to after birth as a neonate). In some embodiments, the LNP is delivered to a fetus and the vector is delivered to the fetus after birth (i.e., at the neonate stage) at any point between birth and up to age 8 weeks. In some embodiments, the LNP is delivered to the fetus and the vector is delivered at about age 1 week, 2 weeks, 3 weeks, 4 weeks, 5 weeks, 6 weeks, 7 weeks or 8 weeks.
[0345] In some embodiments, where the method includes delivering an LNP and a first vector, the LNP and the first vector are delivered to a cell in vivo, the LNP can be delivered to a fetus at a first time point and the vector is delivered to the fetus (child) after the fetus (child) is bom, for example, when the child is age 90 days or older (e.g., age 6 months, age 9 months, age 1 year, age 2 years, age 3 years, age 4 years, age 5 years, age 6 years, or older).
[0346] As used herein, the term “fetus” refers to an unborn offspring. As used herein, the term “neonate” refers to a newborn infant, which includes the first 90 days of life. When referring to mice, neonate can refer to animals up to 10 days of age.
[0347] This disclosure also features a system capable of site-specifically integrating a template polynucleotide into the genome of a cell, the system comprising: a lipid nanoparticle (LNP) comprising a gene editor polynucleotide (e.g., a gene editor polynucleotide construct) and a vector comprising a template polynucleotide and at least a first attachment site-containing guide RNA (atgRNA). In some embodiments, the first atgRNA comprises (i) a domain that is capable of guiding the prime editor system to a target sequence; and (ii) a reverse transcriptase (RT) template that comprises at least a portion of a first integration recognition site. In some embodiments, where the vector comprises a polynucleotide encoding a first atgRNA, the RT template comprises the entirety of the first integration recognition site. In some embodiments, where the vector comprises a polynucleotide encoding a first atgRNA, the vector also includes a sequence encoding a nicking guide RNA (ngRNA).
[0348] This disclosure also features a system capable of site-specifically integrating a template polynucleotide into the genome of a cell, the system comprising: a lipid nanoparticle (LNP) comprising a gene editor polynucleotide (e.g., a gene editor polynucleotide construct) and a vector comprising a template polynucleotide and a first attachment site-containing guide RNA (atgRNA) and a second attachment site-containing guide RNA (atgRNA). In some embodiments, the first atgRNA and the second atgRNA are an at least first pair of atgRNAs, wherein the at least first pair of atgRNAs have domains that are capable of guiding the prime editor system to a target sequence, the first atgRNA further includes a first RT template that comprises at least a portion of the a first integration recognition site; and the second atgRNA further includes a second RT template that comprises at least a portion of the first integration recognition site, and the first atgRNA and the second atgRNAs collectively encode the entirety of the first integration recognition site. In such embodiments, the first atgRNA and second atgRNA include at least a 6bp overlap.
[0349] This disclosure also features a system capable of site-specifically integrating a template polynucleotide into the genome of a cell, the system comprising: a lipid nanoparticle (LNP) comprising: (i) a gene editor polynucleotide (e.g., a gene editor polynucleotide construct), and (ii) a first attachment site-containing guide RNA (atgRNA); and a vector comprising: (i) a template polynucleotide, and (ii) a second atgRNA. In some embodiments, the first atgRNA and the second atgRNA are an at least first pair of atgRNAs, wherein the at least first pair of atgRNAs have domains that are capable of guiding the prime editor system to a target sequence, the first atgRNA further includes a first RT template that comprises at least a portion of the a first integration recognition site; and the second atgRNA further includes a second RT template that comprises at least a portion of the first integration recognition site, and the first atgRNA and the second
atgRNAs collectively encode the entirety of the first integration recognition site. In such embodiments, the first atgRNA and second atgRNA include at least a 6bp overlap.
[0350] This disclosure also features a system capable of site-specifically integrating a template polynucleotide into the genome of a cell, the system comprising: co-delivering: a lipid nanoparticle (LNP) comprising: (i) a gene editor polynucleotide (e.g., a gene editor polynucleotide construct), (ii) a first attachment site-containing guide RNA (atgRNA), and (iii) a second atgRNA; and a vector comprising (i) a template polynucleotide. In some embodiments, the first atgRNA and the second atgRNA are an at least first pair of atgRNAs, wherein the at least first pair of atgRNAs have domains that are capable of guiding the prime editor system to a target sequence, the first atgRNA further includes a first RT template that comprises at least a portion of the a first integration recognition site; and the second atgRNA further includes a second RT template that comprises at least a portion of the first integration recognition site, and the first atgRNA and the second atgRNAs collectively encode the entirety of the first integration recognition site. In such embodiments, the first atgRNA and second atgRNA include at least a 6bp overlap.
[0351] This disclosure also features a system capable of site-specifically integrating a template polynucleotide into the genome of a cell, the system comprising: a lipid nanoparticle (LNP) comprising: (i) a gene editor polynucleotide (e.g., a gene editor polynucleotide construct) and (ii) a first attachment site-containing guide RNA (atgRNA); and a vector comprising: (i) a template polynucleotide, and (ii) a nicking atgRNA. In some embodiments, the first atgRNA comprises (i) a domain that is capable of guiding the prime editor system to a target sequence; and (ii) a reverse transcriptase (RT) template that comprises at least a portion of a first integration recognition site. In some embodiments, where the vector comprises a polynucleotide encoding a first atgRNA, the RT template comprises the entirety of the first integration recognition site.
[0352] In typical embodiments, the LNP comprising a gene editor polynucleotide construct is capable delivering to a cell cytoplasm the gene editor polynucleotide construct. In some embodiments, the LNP comprising a gene editor polynucleotide construct is capable delivering to a cell nucleus the gene editor polynucleotide construct. In some embodiments, the LNP comprises a gene editor protein and associated guide nucleic acids. In some embodiments, the LNP comprises a gene editor protein and associated guide nucleic acids that are capable of localizing to cell nucleus.
[0353] In some embodiments, a gene editor polynucleotide construct is delivered to a cell by a fusosome. In some embodiments, a gene editor polynucleotide construct is delivered to a cell
cytoplasm by a fusosome. In some embodiments, the fusosome comprises a gene editor protein and associated guide nucleic acids.
[0354] In some embodiments, a gene editor polynucleotide construct is delivered to a cell by an exosome. In some embodiments, a gene editor polynucleotide construct is delivered to a cell cytoplasm by an exosome. In some embodiments, the exosome comprises a gene editor protein and associated guide nucleic acids.
[0355] In some embodiments, the prime editor or Gene Writer protein fusion, either of which may have a fused/linked integrase, is incorporated (i.e., packaged) into LNP as protein. Further, associated atgRNA and optional ngRNAs may be co-packaged with gene editor proteins in LNP.
[0356] In some embodiments, the gene editor polynucleotide construct comprises (a) a polynucleotide sequence encoding a prime editor fusion protein or a Gene Writer™ protein, (b) a polynucleotide sequence encoding an attachment site-containing guide RNA (atgRNA), (c) optionally, a polynucleotide sequence encoding a nickase guide RNA (ngRNA), (d) a polynucleotide sequence encoding an integrase, (e) and optionally, a polynucleotide sequence encoding a recombinase.
[0357] In some embodiments, the prime editor or Gene Writer protein fusion, either of which may have a fused/linked integrase, is expressed as a split construct. In typical embodiments, the split construct in reconstituted in a cell. In some embodiments, the split construct can be fused or ligated via intein protein splicing. In some embodiments, the split construct can be reconstituted via protein-protein inter-molecular bonding and/or interactions. In some embodiments, the split construct can be reconstituted via chemical, biological, or environmental induced oligomerization. In certain embodiments, the split construct can be adapted into one or more nucleic acid constructs described herein.
5.10.1. Gene Editor Polynucleotide
[0358] In some embodiments, the systems described include a gene editor polynucleotide that is delivered to a cell using the methods described herein. In some embodiments, the gene editor polynucleotide is delivered as a polynucleotide (e.g., an mRNA). In some embodiments, the gene editor polynucleotide is delivered as a protein. In some embodiments, the gene editor polynucleotide or protein is packaged, and thereby vectorized, within a lipid nanoparticle (LNP). In some embodiments, the gene editor polynucleotide or protein is packaged in a LNP and is codelivered with a template polynucleotide (i.e., nucleic acid “cargo” or nucleic acid “payload”)
packaged into a separate vector (e.g., a viral vector (e.g., an AAV or adenovirus)) or a second lipid nanoparticle (LNP).
[0359] In some embodiments, the gene editor polynucleotide is delivered to the cells as a polynucleotide. For example, the gene editor polynucleotide is delivered to the cells as an mRNA encoding the gene editor polynucleotide (e.g., the gene editor protein or the prime editor system). In some embodiments, the mRNA comprises one or more modified uridines. In some embodiments, the mRNA comprises a sequence where each of the uridines is a modified uridine. In some embodiments, the mRNA is uridine depleted. In some embodiments, the mRNA encoding the nickase comprises one or more modified uridines. In some embodiments, the mRNA encoding the reverse transcriptase comprises one or more modified uridines. In some embodiments, the mRNA encoding the nickase comprises one or more modified uridines, and the mRNA encoding the reverse transcriptase comprises one or more modified uridines. In some embodiments, where the integrase is encoded in an mRNA, the mRNA comprises modified uridines. In some embodiments, a modified uridine is a Nl-Methylpseudouridine-5’ -Triphosphate. In some embodiments, a modified uridine is a pseudouridine. In some embodiments, the mRNA comprises a 5’ cap. In some embodiments, the 5’ cap comprises a molecular formula of C32H43N15O24P4 (free acid).
[0360] In some embodiments, the gene editor polynucleotide (e.g., a gene editor polynucleotide construct) comprises a polynucleotide sequence encoding a primer editor system (e.g., any of the prime editor systems described herein). In some embodiments, the prime editor system comprises a nucleotide sequence encoding a nickase (e.g., any of the Cas proteins or variants thereof (e.g., nickases) and nickases described herein, see Tables 4-8) and a nucleotide sequence encoding a reverse transcriptase (e.g., any of the reverse transcriptases described herein). In some embodiments, the nucleotide sequence encoding the nickase and the nucleotide sequence encoding the reverse transcriptase are positioned in the construct such that when expressed the nickase is linked to the reverse transcriptase. In some embodiments, the nickase is linked to the reverse transcriptase by in-frame fusion. In some embodiments, the nickase is linked to the reverse transcriptase by a linker. In some embodiments, the linker is a peptide fused in-frame between the nickase and reverse transcriptase.
[0361] In some embodiments, the gene editor polynucleotide (e.g., a gene editor polynucleotide construct) further comprises a polynucleotide sequence encoding at least a first integrase (e.g., any of the integrases described herein, e.g., as described in Table 10 and also in Yamall et al., Nat.
BiotechnoL, 2022, doi.org/10.1038/s41587-022-01527-4 and Durrant et al., Nat. BiotechnoL, 2022, doi.org/10.1038/s41587-022-01494-w, each of which are herein incorporated by reference in their entireties). In some embodiments, the linked nickase-reverse transcriptase are further linked to the first integrase.
[0362] In some embodiments, the gene editor polynucleotide construct further comprises a polynucleotide sequence encoding at least a first recombinase (e.g., any of the recombinases described herein).
5.10.2. Split Nickase and Reverse Transcriptase
[0363] Also provided herein are prime editor systems where the nucleotide sequence encoding the nickase and the nucleotide sequence encoding the reverse transcriptase are positioned in a construct such that when expressed the nickase is not linked to the reverse transcriptase. In a nonlimiting example, a sequence encoding a self-cleaving peptide (e.g., a P2A) is positioned between the nucleotide sequence encoding the nickase and the nucleotide sequence encoding the reverse transcriptase such that when expressed the nickase is not linked to the reverse transcriptase (see, e.g., FIG. 34).
[0364] Also provided herein are prime editor systems where the nucleotide sequence encoding the nickase and the nucleotide sequence encoding the reverse transcriptase are on separate polynucleotides. In some embodiments where the prime editor system includes separate polynucleotides encoding the nickase and the reverse transcriptase, the nickase and/or the reverse transcriptase can be engineered such that when expressed they are linked to a binding domain, where the binding domain is part of a binding pair that enables recruitment of the reverse transcriptase to the genomic location of the nickase (e.g., nCas9). A binding pair can be any pair of molecules wherein upon forming the binding pair the binding domains enable recruitment of the reverse transcriptase to the genomic location of the nickase. Once in sufficient proximity with each other the binding domains can dimerize. This dimerization (or other type of interaction) brings the nickase and RT into proximity with each other, thereby enhancing the reverse transcription of the RT template in the atgRNA.
[0365] Non-limiting examples of binding pairs that can aid recruitment of the RT to the genomic location of the nickase include without limitation: dimerizing leucine zippers, an antigen and a corresponding antigen binding domains (e.g., antibody), RNA aptamers and corresponding RNA aptamer binding proteins, affinity tags and corresponding affinity tag binding proteins.
[0366] In some embodiments, dimerizing leucine zippers that can serve as a binding pair includes two binding domains: a Leucine Zip LZ1 domain having a sequence with at least 80%, 85%, 90%, 95%, or 99% sequence identity to a sequence of SEQ ID NO: 568 and a Leucine Zip LZ2 domain having a sequence with at least 80%, 85%, 90%, 95%, or 99% sequence identity to a sequence of SEQ ID NO: 569. In one embodiment, the Leucine Zip LZ1 domain is positioned either N- terminally or C-terminally to the nickase and the Leucine Zip LZ2 is positioned either N-terminally or C-terminally to the RT. In one embodiment, the Leucine Zip LZ1 domain is positioned either N-terminally or C-terminally to the RT and the Leucine Zip LZ2 domain is positioned either N- terminally or C-terminally to the Cas9.

[0367] In one embodiment, the system and methods described herein include a construct that encodes a polypeptide having a sequence with at least 80%, 85%, 90%, 95%, or 99% sequence identity to a sequence of SEQ ID NO: 576 (a Leucine Zip LZ1 domain positioned C-terminally to the nickase), and a construct that encodes a polypeptide having a sequence with at least 80%, 85%, 90%, 95%, or 99% sequence identity to a sequence of SEQ ID NO: 577 (a Leucine Zip LZ2 domain positioned N-terminally to the RT).
[0368] In one embodiment, the system and methods described herein include a construct that encodes a polypeptide having a sequence with at least 80%, 85%, 90%, 95%, or 99% sequence identity to a sequence of SEQ ID NO: 578 (a Leucine Zip LZ1 domain positioned N-terminally to the nickase), and a construct that encodes a polypeptide having a sequence with at least 80%, 85%, 90%, 95%, or 99% sequence identity to a sequence of SEQ ID NO: 577 (a Leucine Zip LZ2 domain positioned N-terminally to the RT).
[0369] In some embodiments, dimerizing leucine zippers that can serve as a binding pair includes two binding domains: a EE1234L peptide having a sequence with at least 80%, 85%, 90%, 95%, or 99% sequence identity to a sequence of SEQ ID NO: 570 and a RR1234L peptide having a sequence with at least 80%, 85%, 90%, 95%, or 99% sequence identity to a sequence of SEQ ID NO: 571.
In one embodiment, the EE1234L peptide is positioned either N-terminally or C-terminally to the nickase and the RR1234L peptide is positioned either N-terminally or C-terminally to the RT. In one embodiment, the EE1234L peptide is positioned either N-terminally or C-terminally to the RT and the RR1234L peptide is positioned either N-terminally or C-terminally to the Cas9.

[0370] In one embodiment, the system and methods described herein include a construct that encodes a polypeptide having a sequence with at least 80%, 85%, 90%, 95%, or 99% sequence identity to a sequence of SEQ ID NO: 579 (a EE1234L peptide positioned N-terminally to the nickase), and a construct that encodes a polypeptide having a sequence with at least 80%, 85%, 90%, 95%, or 99% sequence identity to a sequence of SEQ ID NO: 580 (a RR1234L peptide positioned N-terminally to the RT).
[0371] In some embodiments, an affinity tag is a SpyTag and the corresponding affinity tag binding protein is SpyCatcher, where upon binding of the SpyTag with the SpyCatcher the fusion protein is a covalently stabilized multi-protein complex. In one embodiment, the affinity tag (e.g., SpyTag) is positioned either N-terminally or C-terminally to the nickase and the affinity tag binding protein (e.g,. SpyCatcher) is positioned either N-terminally or C-terminally to the RT. In one embodiment, the affinity tag (e.g., SpyTag) is positioned either N-terminally or C-terminally to the RT and the affinity tag binding protein (e.g,. SpyCatcher) is positioned either N-terminally or C-terminally to the nickase.
[0372] In some embodiments, an antigen and corresponding antigen binding domain includes an antigen such as a peptide present within a tag or a tag itself and an antigen binding domain that binds specifically to the antigen. In one embodiments, an antigen is a GCN4 peptide within a SunTag and the antigen binding domain is an anti-GCN4 scFv. In one embodiment, the SunTag (GCN4 peptide) is positioned either N-terminally or C-terminally to the nickase and the anti-GCN4 antigen binding domain is positioned either N-terminally or C-terminally to the RT. In one embodiment, the SunTag (GCN4 peptide) is positioned either N-terminally or C-terminally to the
RT and the anti-GCN4 antigen binding domain is positioned either N-terminally or C-terminally to the nickase.
[0373] In one embodiment, the system and methods described herein include a construct that encodes a polypeptide having a sequence with at least 80%, 85%, 90%, 95%, or 99% sequence identity to a sequence of SEQ ID NO: 574 (SunTag (GCN4 peptide) positioned C-terminally to the nickase), and a construct that encodes a polypeptide having a sequence with at least 80%, 85%, 90%, 95%, or 99% sequence identity to a sequence of SEQ ID NO: 575 (anti-GCN4 antigen binding domain positioned N-terminally to the RT).
[0374] In some embodiments, provided herein are prime editor systems where a construct includes a nucleotide sequence encoding a self-cleaving peptide (e.g., a P2A) positioned between the nucleotide sequence encoding the nickase and the nucleotide sequence encoding the RT. In such cases, the position of the nucleotide sequence encoding the nickase, the nucleotide sequence encoding the self-cleaving peptide (e.g., a P2A), and the nucleotide sequence encoding the RT is such that when expressed the nickase is not linked to the reverse transcriptase. In one embodiment, the construct includes the nucleotide sequences positioned 5 ’-3’: nucleotide sequence encoding the nickase, nucleotide sequence encoding the self-cleaving peptide (e.g., a P2A), and nucleotide sequence encoding the RT. In one embodiment, the construct includes the nucleotide sequences positioned 5 ’-3’: nucleotide sequence encoding the RT, nucleotide sequence encoding the selfcleaving peptide (e.g., a P2A), and nucleotide sequence encoding the nickase.
[0375] In one embodiment, the construct that includes a nucleotide sequence encoding a selfcleaving peptide (e.g., a P2A) positioned between the nucleotide sequence encoding the nickase and the nucleotide sequence encoding the RT has an amino acid sequence with at least 80%, 85%, 90%, 95%, or 99% sequence identity to a sequence of SEQ ID NO: 581 or 582 (see FIG. 34).







[0376] This disclosure also features systems and methods that include supplementing with reverse transcriptase. In some embodiments, the system and methods include supplementation with a polynucleotide encoding a supplemental reverse transcriptase (i.e., a separate polynucleotide from the gene editor polynucleotide, which includes a polynucleotide encoding a reverse transcriptase). Systems and methods that include supplementation with reverse transcriptase increase integration efficiency of the at least first integration recognition site into the genome of the cell when compared to systems and methods that do not include supplementation with reverse transcriptase. These systems and methods refer to the supplemental or supplemental reverse transcriptase molecule as “supplementing” or “supplementation” with reverse transcriptase.
[0377] In some embodiments, the supplemental reverse transcriptase is the same as the reverse transcriptase that is included in the gene editor polynucleotide. In some embodiments, the supplemental reverse transcriptase is different from the reverse transcriptase that is included in the gene editor polynucleotide.
[0378] In some embodiments, the polynucleotide encoding the supplemental reverse transcriptase is an mRNA.
[0379] In some embodiments, the supplemental reverse transcriptase is delivered concurrently with the one or more LNP, one or more atgRNA, and the template polynucleotide. In some embodiments, the supplemental reverse transcriptase is delivered to the cell prior to delivering the one or more LNP, one or more atgRNA, and the template polynucleotide to the cell. In some embodiments, the supplemental reverse transcriptase is delivered to the cell after delivering the one or more LNP, one or more atgRNA, and the template polynucleotide to the cell.
[0380] Also provided herein are methods of increasing integration efficiency of the at least first integration recognition site into the genome of the cell, where the method comprises delivering to the cell one or more LNPs, one or more vectors, one or more template polynucleotides and supplementing with a polynucleotide encoding a supplemental reverse transcriptase, where the increase in integration efficiency is in comparison to methods that do not include supplementation with a polynucleotide encoding RT. In such embodiments, the method includes a ratio of 1 : 1, 1:2, 1 :3, 1 :4, 1 :5, 1 :6, 1 :7, 1 :8, 1 :9, or 1 : 10 of gene editor polynucleotide to polynucleotide encoding the supplemental reverse transcriptase.
[0381] Also provided herein are methods of increasing integration efficiency of the at least first integration recognition site into the genome of the cell, where the method comprises delivering to the cell one or more LNPs, one or more vectors, one or more template polynucleotides, where the gene editor polynucleotide comprises a ratio of a ratio of 1:1, 1:2, 1:3, 1:4, 1:5, 1:6, 1:7, 1:8, 1:9, 1:10 of the nucleotide sequence encoding the nickase to the nucleotide sequence encoding the reverse transcriptase, and where the increase in integration efficiency is in comparison to methods that do not include supplementation with a polynucleotide encoding RT.
5.10.3.1 Supplementation of RT when using a fused design
[0382] In some embodiments, the system and methods described herein include supplementation with a polynucleotide encoding a reverse transcriptase, where the polynucleotide is in addition to the gene editor polynucleotide that also includes a polynucleotide encoding a reverse transcriptase.
[0383] In one embodiment, a method for delivering a system capable of site-specifically integrating a template polynucleotide into the genome of a cell, includes delivering one or more LNPs and one or more vectors, and also includes delivering a separate polynucleotide encoding a supplemental reverse transcriptase (RT). In a non-limiting example, a method for delivering a system capable of site-specifically integrating a template polynucleotide into the genome of a cell comprises delivering the gene editor polynucleotide (e.g., a polynucleotide encoding a nickase and a reverse transcriptase either linked or not linked), one or more atgRNA, a template polynucleotide, and a polynucleotide encoding a supplemental reverse transcriptase.
[0384] In some embodiments, the method includes delivering into the cell a ratio of gene editor polynucleotide to polynucleotide encoding the supplemental RT of 1:1, 1:2, 1:3, 1:4, 1:5, 1:6, 1:7, 1:8, 1:9, 1:10, 1:11, 1:12, 1:13, 1:14, 1:15, 1:16, 1:17, 1:18, 1:19, 1:20, 1:21, 1:22, 1:23, 1:24, 1:25, 1:26, 1:27, 1:28, 1:29, 1:30, 1:32, 1:34, 1:36, 1:38, or 1:40 or more. In some embodiments, the method includes delivering into the cell a gene editor polynucleotide and a polynucleotide encoding the supplemental RT, where the polynucleotide encoding the supplemental RT is delivered at 1.5 times (1.5X), 2X, 3X, 4X, 5X, 6X, 7X, 8X, 9X, 10X, 20X, 30X, 40X, 50X, 60X, 70X, 80X, 90X, or 100X or more the amount of the gene editor polynucleotide.
[0385] In some embodiments, the method includes delivering into the cell a ratio of polynucleotide encoding the nickase to polynucleotide encoding the RT of 1:1, 1:2, 1:3, 1:4, 1:5, 1:6, 1:7, 1:8, 1:9, 1:10, 1:11, 1:12, 1:13, 1:14, 1:15, 1:16, 1:17, 1:18, 1:19, 1:20, 1:21, 1:22, 1:23, 1:24, 1:25, 1:26, 1:27, 1:28, 1:29, 1:30, 1:32, 1:34, 1:36, 1:38, or 1:40 or more. In some embodiments, the method includes delivering into the cell a polynucleotide encoding the nickase to polynucleotide
encoding the RT, where the polynucleotide encoding the RT is delivered at 1.5 times (1.5X), 2X, 3X, 4X, 5X, 6X, 7X, 8X, 9X, 10X, 20X, 30X, 40X, 50X, 60X, 70X, 80X, 90X, or 100X or more the amount of the nickase polynucleotide.
[0386] In some embodiments, the systems and methods that include delivering of the gene editor polynucleotide supplemented with a polynucleotide encoding a supplemental RT increases integration efficiency of the at least first integration recognition site into the genome of the cell when compared to systems and methods that include delivering to the cell a gene editor polynucleotide not supplemented with a polynucleotide encoding supplemental reverse transcriptase.
5.10.3.2 Split design plus further supplementation
[0387] In some embodiments, the system and methods described herein include supplementation with a polynucleotide encoding a reverse transcriptase, where the system includes a gene editor polynucleotide split over two polynucleotides: a first polynucleotide encoding the nickase and a second polynucleotide encoding the reverse transcriptase. In such cases, the first and second polynucleotides can be delivered to the cell at a ratio of 1 : 1 and supplementation is achieved by adding a polynucleotide encoding a supplemental reverse transcriptase. In such cases, the supplementation comprises adding the polynucleotide encoding the supplemental reverse transcriptase at a ratio of 1 : 1, 1 :2, 1 :3, 1 :4, 1 :5, 1 :6, 1 :7, 1 :8, 1 :9, 1 : 10, 1 : 11, 1 : 12, 1 : 13, 1 : 14, 1 : 15, 1 : 16, 1 : 17, 1 : 18, 1 : 19, 1 :20, 1 :21, 1 :22, 1 :23, 1 :24, 1 :25, 1 :26, 1 :27, 1 :28, 1 :29, 1 :30, 1 :32, 1 :34, 1 :36, 1 :38, or 1 :40 (gene editor polynucleotide (e.g., the first and second polynucleotides delivered to the cell at a ratio of 1 : 1) to the polynucleotide encoding the supplemental reverse transcriptase).
[0388] In some embodiments, a method of delivering to the cell the one or more atgRNA and the gene editor polynucleotide (where the nickase and the RT are encoded on different polynucleotides and delivered at a ratio 1 : 1 (nickase to RT)) and supplementing with a polynucleotide encoding a supplemental RT increases integration efficiency of the at least first integration recognition site into the genome of the cell when compared to methods that do not include supplementing with a polynucleotide encoding a supplemental RT.
5.10.3.3 Supplementation via increase in ratios when using split design
[0389] In some embodiments, the system and methods described herein include supplementation with a polynucleotide encoding a reverse transcriptase, where the system includes a gene editor polynucleotide split over two polynucleotides: a first polynucleotide encoding the nickase and a
second polynucleotide encoding the reverse transcriptase, and supplementation is achieved by increasing the amount of the second polynucleotide encoding the reverse transcriptase.
[0390] In some embodiments, where the gene editor polynucleotide is such that the nucleotide sequence encoding the nickase and the nucleotide sequence encoding the reverse transcriptase are not linked, supplementation of a supplemental reverse transcriptase refers to delivering a greater than 1 : 1 ratio of nickase to reverse transcriptase (e.g., a 1 :2 or greater ratio). In a non-limiting example, the gene editor polynucleotide delivered to the cell in a ratio 1 : 1, 1 :2, 1 :3, 1 :4, 1 :5, 1 :6, 1 :7, 1 :8, 1 :9, 1 : 10, 1 : 11, 1 : 12, 1 : 13, 1 : 14, 1 : 15, 1 : 16, 1 : 17, 1 : 18, 1 : 19, 1 :20, 1 :21, 1 :22, 1 :23, 1 :24, 1 :25, 1 :26, 1 :27, 1 :28, 1 :29, 1 :30, 1 :32, 1 :34, 1 :36, 1 :38, or 1 :40 of polynucleotide sequence encoding the nickase to the polynucleotide sequence encoding the reverse transcriptase.
[0391] In some embodiments, a method of delivering to the cell the one or more atgRNA and the gene editor polynucleotide where the nickase and the RT are encoded on different polynucleotides and delivered at a ratio greater than 1 : 1 (nickase to RT) increases integration efficiency of the at least first integration recognition site into the genome of the cell when compared to methods comprising delivering to the cell a gene editor polynucleotide at a ratio of 1 : 1 (nickase to RT).
5.10.3.4 Supplementation of RT via vector design
[0392] In some embodiments, the system and methods described herein include supplementing with reverse transcriptase, where in supplementing comprises using controllable expression (e.g., inducible promoters, strong promoters) of a polynucleotide encoding a reverse transcriptase.
[0393] In one embodiment, the polynucleotide encoding the nickase and the polynucleotide encoding the RT are on a dual promoter vector, where each polynucleotide is controlled by a different promoter. In a non-limiting example, the polynucleotide encoding the RT is operably linked to a stronger promoter than the polynucleotide encoding the nickase, whereby the RT is expressed at higher levels than the nickase. In another non-limiting example, the polynucleotide encoding the RT is operably linked to an inducible promoter, whereby inducing expression of the RT results in higher levels of the RT compared to the nickase. In another non-limiting example, the polynucleotide encoding the nCas9 is operably linked to an inducible promoter, whereby inducing expressing of the nCas9 results in lower levels of the nCas9 than the RT.
5.10.4. Vector
[0394] In some embodiments, the systems and methods described herein include a vector that is capable of co-delivering a template polynucleotide, one or more attachment site-containing gRNA,
one or more integrases, one or more recombinases, a gene editor polynucleotide, one or more integration recognition sites, one or more recombinase recognition sites, or a combination thereof.
[0395] Non-limiting examples of vectors that can be used in the methods or systems described herein include the vectors described in FIGs. 3-6.
5.10.4.1 AtgRNA and/or ngRNA
[0396] In some embodiments, the vector includes a polynucleotide sequence encoding an attachment site-containing guide RNA (atgRNA). In such embodiments, the polynucleotide sequence encoding the attachment site-containing guide RNA (atgRNA) is operably linked to a regulatory element (e.g., a U6 promoter) that is capable of driving expression of the atgRNA. In such embodiments, the atgRNA comprises (i) a domain that is capable of guiding the prime editor system to a target sequence; and (ii) a reverse transcriptase (RT) template that comprises at least a portion of a first integration recognition site. In some embodiments, where the system, and thereby the vector, include a polynucleotide encoding only a first atgRNA, the RT template comprises the entirety of the first integration recognition site. In such embodiments, the vector or the LNP includes a polynucleotide sequence encoding a nicking gRNA.
[0397] In some embodiments, the vector includes a polynucleotide sequence encoding a first attachment site-containing guide RNA (atgRNA) and a polynucleotide sequence encoding a second attachment site-containing guide RNA (atgRNA). In such embodiments, the first atgRNA and the second atgRNA are an at least first pair of atgRNAs, wherein the at least first pair of atgRNAs have domains that are capable of guiding the prime editor system to a target sequence, the first atgRNA further includes a first RT template that comprises at least a portion of the a first integration recognition site; the second atgRNA further includes a second RT template that comprises at least a portion of the first integration recognition site, and the first atgRNA and the second atgRNAs collectively encode the entirety of the first integration recognition site. In such embodiments, the first atgRNA and second atgRNA include at least a 6bp overlap.
5.10.4.2 Template Polynucleotide
[0398] In typical embodiments, the vector includes a template polynucleotide and a sequence that is an integration cognate of an integration recognition site site-specifically incorporated into the genome of a cell. For example, the vector includes a template polynucleotide and a second integration recognition site that is a cognate pair with the first integration recognition site site- specifically incorporated into the genome of the cell. In such embodiments, the sequence that is an
integration cognate (e.g., a second integration recognition site) enables integration of the template polynucleotide or portion thereof when contacted with an integrase and the site-specifically incorporated first integration recognition site.
[0399] In typical embodiments, the vector comprising a template polynucleotide is a recombinant adenovirus, a helper dependent adenovirus, an AAV, a lentivirus, an HSV, an annelovirus, a retrovirus, a Doggybone™ DNA (dbDNA), a minicircle, a plasmid, a miniDNA, an exosome, a fusosome, or an nanoplasmid. In preferred embodiments, the vector is capable of localizing to the nucleus.
[0400] In certain embodiments, the template polynucleotide is delivered to the cytoplasm and localizes to the nucleus. In certain embodiments, the template polynucleotide is delivered to the cytoplasm by LNP. In certain embodiments, the donor template polynucleotide construct comprises a recognition sequence that is recognized by a DNA binding protein (DNA binding domain) or a transcription factor binding domain. In certain embodiments, the donor template polynucleotide construct is delivered to the nucleus by an integrase or recombinase.
[0401] In certain embodiments, the template polynucleotide is delivered to the mitochondria. In certain embodiments, the donor template polynucleotide construct comprises a mitochondria targeting sequence.
[0402] In certain embodiments, the vector comprising a template polynucleotide is AAV. In some embodiments, the AAV contains a 5’ inverted terminal repeat (ITR). In some embodiments, the AAV contains a 3’ inverted terminal repeat (ITR). In some embodiments, the AAV contains a 5’ and a 3’ ITR. In some embodiments, the 5’ and 3’ ITR are not derived from the same serotype of virus. In some embodiments, the ITRs are derived from adenovirus, AAV2, and/or AAV5.
[0403] In certain embodiments, the vector comprising a template polynucleotide is single stranded AAV (ssAAV). In certain embodiments, the vector comprising a donor template polynucleotide construct is self-complementary AAV (scAAV).
[0404] In some embodiments, a vector comprises an attachment site-containing guideRNA (atgRNA), a nicking-guideRNA (ngRNA), and template polynucleotide. In typical embodiments, the vector comprising an attachment site-containing guideRNA (atgRNA), a nicking-guideRNA (ngRNA), and template polynucleotide is recombinant adenovirus, helper dependent adenovirus, AAV, lentivirus, HSV, annelovirus, retrovirus, Doggybone™ DNA (dbDNA), minicircle, plasmid, miniDNA, exosome, fusosome, or nanoplasmid. In preferred embodiments, the vector is
capable of localizing to the nucleus. In typical embodiments, the attachment site-containing guideRNA (atgRNA) sequence and the nicking-guideRNA (ngRNA) sequence contain a terminal poly dT.
[0405] In some embodiments, a vector comprises an attachment site-containing guideRNA (atgRNA), and donor template. In typical embodiments, the vector comprising an attachment sitecontaining guideRNA (atgRNA) and donor template is recombinant adenovirus, helper dependent adenovirus, AAV, lentivirus, HSV, annelovirus, retrovirus, Doggybone™ DNA (dbDNA), minicircle, plasmid, miniDNA, exosome, fusosome, or nanoplasmid. In preferred embodiments, the vector is capable of localizing to the nucleus. In typical embodiments, the attachment sitecontaining guideRNA (atgRNA) sequence contain a terminal poly dT.
[0406] In typical embodiments, the template polynucleotide is capable of being integrated into a genomic locus that contains an integrase target recognition site or a recombinase target recognition site.
[0407] In certain embodiments, the template polynucleotide comprises at least one of the following: a gene, a gene fragment, an expression cassette, a logic gate system, or any combination thereof. In some embodiments, the template polynucleotide comprises at least one intron or exon.
[0408] In typical embodiments, the template polynucleotide further comprises at least one integrase target recognition site or a recombinase target integrase site. In certain embodiments, at least one integrase target recognition site or a recombinase target integrase site is placed within the donor template vector inverted terminal repeat.
5.10.4.3 Integrase- or recombinase-mediated self-circularization of a subsequence of a vector delivered as part of the codelivery system
[0409] In some embodiments, the delivery system (e.g., co-delivery system) includes a vector having a sub-sequence that is capable of self-circularizing to form a self-circular nucleic acid. In some embodiments, the vector comprises a physical portion or region of the vector that is capable of self-circularizing to form a circular construct. As used herein, the term “sub -sequence” refers to a portion of the vector that is capable of self-circularizing, where the sub-sequence is flanked by integration recognition sites or recombinase recognition sites positioned to enable selfcircularization. As used herein, the term “self-circular nucleic acid” refers to a double-stranded, circular nucleic acid construct produced as a result of recombination of a cognate pair of integrase or recombinase recognition sites present on the vector. Recombination occurs when the vector is
contacted with an integrase or a recombinase under conditions that allow for recombination of the cognate pair of integrase or recombinase recognition sites.
[0410] In some embodiments, the sub-sequence of the vector includes a first recombinase recognition site and a second recombinase recognition site, wherein the first and second recombinase recognition sites are capable of being recombined by a recombinase. In some embodiments, the sub-sequence of the vector includes a first recombinase recognition site, a second recombinase recognition site, and a second integration recognition site (e.g., the second integration recognition site is a cognate pair of the first integration recognition site), where the first and second recombinase recognition sites flank the integration recognition site. In such cases, the first recombinase recognition site, the second recombinase recognition, and a recombinase enable the self-circularizing and formation of the circular construct.
[0411] In some embodiments, the sub-sequence of the vector includes a third integration recognition site and a fourth integration recognition site, wherein the third and fourth integration recognition sites are a cognate pair. In some embodiments, the subsequence of the vector includes the second integration recognition site, the third integration recognition site, the fourth integration recognition site, where the third and fourth integration recognition sites flank the second integration recognition site (where the second integration recognition site is a cognate pair of the first integration recognition site). In such cases, the third integration recognition site, the fourth integration recognition site, and an integrase enable self -circularization and formation of the circular construct. In such cases, the third integration recognition site and/or the fourth integration recognition sites cannot recombine with the first integration recognition site and/or the second integration recognition site due, in part, to having different central dinucleotides than the first and second integration recognition sites.
[0412] In some embodiments where the subsequence includes three or more integration recognition sites, each integration recognition site or each pair of integration recognition is capable of being recognized by a different integrase. In some embodiments where the subsequence includes three or more integration recognition sites, each integration recognition site or each pair of integration recognition comprises a different central dinucleotide.
[0413] In some embodiments, self-circularizing is mediated at the integration recognition sites or recombinase recognition sites. In some embodiments, the self-circularizing is mediated by an integrase or a recombinase.
[0414] In some embodiments, upon introducing the vector into a cell and after selfcircularizing to form the self-circular nucleic acid, the self-circular nucleic acid comprising the second integration recognition site is capable of being integrated into the cell’s genome at the target sequence that contains the first integration recognition site.
[0415] In some embodiments, following self-circularization, the self-circular nucleic acid comprises one or more additional integration recognition sites that enable integration of an additional nucleic acid cargo. In such cases, the additional nucleic acid cargo includes a sequence that is a cognate pair with one or more of the additional integration recognition sites in the selfcircular nucleic acid. For example, integration of the self-circular nucleic acid into the genome of a cell results in integration of the one or more additional integration recognition sites into the genome along with the nucleic acid cargo. The integrated one or more additional integration recognition sites serve as an integration recognition site (beacon) for placing the additional nucleic acid cargo. Upon contacting the cell harboring the integrated nucleic acid cargo and the one or more additional integration recognition sites with an integrase and the second additional nucleic acid cargo that includes a sequence that is an integration cognate to the one or more additional integration recognition sites the additional nucleic acid cargo is integrated into the cell’s genome.
[0416] In typical embodiments, the self-circularized nucleic acid comprises a DNA cargo, embodiments, the DNA cargo is a gene or gene fragment. In some embodiments the DNA cargo is an expression cassette. In some embodiments, the DNA cargo is a logic gate or logic gate system. The logic gate or logic gate system may be DNA based, RNA based, protein based, or a mix of DNA, RNA, and protein. In some embodiments, the nucleic acid cargo is a genetic, protein, or peptide tag and/or barcode.
5.10.4.4 A second vector
[0417] In some embodiments, the system or methods described herein include a second vector. In some embodiments, where the gene editor polynucleotide encodes a prime editor system comprising a nickase (e.g., any of the Cas proteins or variants thereof (e.g., nickases) and nickases described herein, see Tables 4-8) and a reverse transcriptase (e.g., any of the reverse transcriptase described herein), the second vector comprises a polynucleotide sequence encoding an integrase (e.g., any of the integrases described herein, e.g., as described in Table 10 and also in Yamall et al., Nat. Biotechnol., 2022, doi.org/10.1038/s41587-022-01527-4 and Durrant et al., Nat. BiotechnoL, 2022, doi.org/10.1038/s41587-022-01494-w, each of which are herein incorporated by reference in their entireties).
[0418] In some embodiments, where the gene editor polynucleotide encodes a prime editor system comprising a nickase and a reverse transcriptase, the second vector comprises a polynucleotide sequence encoding at least a first recombinase. In some embodiments, where the gene editor polynucleotide encodes a prime editor system comprising a nickase, a reverse transcriptase, and an integrase, the second vector comprises a polynucleotide sequence encoding at least a first recombinase. In some embodiments, where the gene editor polynucleotide encodes a prime editor system comprising a nickase, a reverse transcriptase, and an integrase, the second vector comprises a polynucleotide sequence encoding at least a second integrase.
[0419] In some embodiments, the second vector includes a template polynucleotide and a sequence that is an integration cognate of an integration recognition site site-specifically incorporated into the genome of a cell. For example, the second vector includes a template polynucleotide and a second integration recognition site that is a cognate pair with the first integration recognition site site-specifically incorporated into the genome of the cell. In such embodiments, the sequence that is an integration cognate (e.g., a second integration recognition site) enables integration of the template polynucleotide or portion thereof when contacted with an integrase and the site-specifically incorporated first integration recognition site.
[0420] In some embodiments, the second vector is a vector selected from: adenovirus, AAV, lentivirus, HSV, annelovirus, retrovirus, Doggybone™ DNA (dbDNA), minicircle, plasmid, miniDNA, exosome, fusosome, or nanoplasmid.
[0421] In some embodiments, the polynucleotide sequence encoding the prime editor system is encoded on at least two different vectors. In one embodiment, a first vector comprises a polynucleotide sequence encoding a nickase and a second vector comprises a polynucleotide sequence encoding a reverse transcriptase. In such cases, the first vector and second are delivered concurrently.
[0422] In some embodiments, the polynucleotide sequence(s) encoding the prime editor system is encoded on at least two (non-contiguous) polynucleotide sequences. In one embodiment, a first polynucleotide sequence encodes a nickase and a second polynucleotide sequence encodes a reverse transcriptase. In such cases, the first vector and second are delivered concurrently (e.g., in a first LNP).
5.10.5. Split Lipid Nanoparticles (LNPs)
[0423] Also provided herein are methods of co-delivering a system capable of site-specifically integrating at least a first integration recognition site into the genome of a cell, where the method includes delivering to a cell a mixture of a first LNP and a second LNP (“split LNPs”). In one embodiment, the method includes co-delivering to a cell a first gene editor polynucleotide construct and a first attachment site-containing guide RNA (atgRNA) are packaged, and thereby vectorized, within the first LNP, and a second gene editor polynucleotide construct and a second attachment site containing guide RNR (atgRNA) are packaged, and thereby vectorized, within the second LNP, where the first atgRNA and the second atgRNA are an at least first pair of atgRNA. The at least first pair of atgRNAs comprise domains that are capable of guiding the prime editor system to a target sequence. The first atgRNA further includes a first RT template that comprises at least a portion of a first integration recognition site. The second atgRNA further includes a second RT template that comprises at least a portion of the first integration recognition site. The first atgRNA and the second atgRNAs collectively encode the entirety of the first integration recognition site. In such embodiments, the first atgRNA and second atgRNA include at least a 6bp overlap.
[0424] In some embodiments, where the method includes delivering a first LNP (e.g., a first LNP comprising a first gene editor polynucleotide construct and a first atgRNA) and a second LNP (e.g., a second LNP comprising a second gene editor polynucleotide construct and a second atgRNA), the first LNP and the second LNP are mixed prior to delivering to a cell. In some embodiments, the first LNP and the second LNP are mixed at a ratio of first LNP to second LNP of l:10, 1 :9, 1 :8, 1 :7, 1 :6, 1 :5, 1 :4, 1 :3, 1 :2, 1 : 1, 1 :0.75, 0.75: 1, 2: 1, 3: 1, 4: 1, 5: 1, 6: 1, 7: 1, 8: 1, 9: 1, or 10: 1. In some embodiments, the first LNP and the second LNP are mixed at a ratio of 1 : 1.
[0425] In some embodiments, a first LNP comprising a first gene editor polynucleotide construct and a first attachment site-containing guide RNA (atgRNAl) comprises a ratio of ratio of gene editor polynucleotide construct (e.g., mRNA) to atgRNAl of 1 : 10, 1 :9, 1 :8, 1 :7, 1 :6, 1 :5, 1 :4, 1 :3, 1 :2, 1 : 1, 1 :0.75, 0.75: 1, 2: 1, 3: 1, 4: 1, 5: 1, 6: 1, 7: 1, 8: 1, 9: 1, or 10: 1. In some embodiments, the first LNP comprises a ratio of mRNA to atgRNAl of 2: 1.
[0426] In some embodiments, a second LNP comprising a second gene editor polynucleotide construct and a second attachment site-containing guide RNA (atgRNA2) comprises a ratio of gene editor polynucleotide construct (e.g., mRNA) to atgRNA2 of 1 : 10, 1 :9, 1 :8, 1 :7, 1 :6, 1 :5, 1 :4,
1 :3, 1 :2, 1 : 1, 1 :0.75, 0.75: 1, 2: 1, 3: 1, 4:1, 5: 1, 6: 1, 7: 1, 8: 1, 9: 1, or 10: 1. In some embodiments, the second LNP comprises a ratio of mRNA to atgRNA2 of 2: 1.
[0427] In some embodiments, where the method includes delivering a first LNP (e.g., a first LNP comprising a first gene editor polynucleotide construct and a first atgRNA) and a second LNP (e.g., a second LNP comprising a second gene editor polynucleotide construct and a second atgRNA), the first LNP and the second LNP are mixed such that the ratio of gene editor polynucleotide construct (e.g., mRNA) to first atgRNA (atgRNAl) to second atgRNA (atgRNA2) is 1 :0.25:0.25, l :0.5:0.5, 1 :0.75:0.75, or 1 : 1 : 1.
[0428] In some embodiments, the method of co-delivering to a cell a mixture of LNPs includes co-delivering three or more LNPs, four or more LNPs, five or more LNPs, six or more LNPs, seven or more LNPs, eight or more LNPs, nine or more LNPs, or ten or more LNPs.
[0429] Also provided herein is a system capable of site-specifically integrating at least a first integration recognition site into the genome of a cell, the system comprising: a first gene editor polynucleotide construct and a first attachment site-containing guide RNA (atgRNA) are packaged, and thereby vectorized, within the first LNP, and a second gene editor polynucleotide construct and a second attachment site containing guide RNR (atgRNA) are packaged, and thereby vectorized, within the second LNP, where the first atgRNA and the second atgRNA are an at least first pair of atgRNA. The at least first pair of atgRNAs comprise domains that are capable of guiding the prime editor system to a target sequence. The first atgRNA further includes a first RT template that comprises at least a portion of a first integration recognition site. The second atgRNA further includes a second RT template that comprises at least a portion of the first integration recognition site. The first atgRNA and the second atgRNAs collectively encode the entirety of the first integration recognition site. In such embodiments, the first atgRNA and second atgRNA include at least a 6bp overlap.
[0430] In some embodiments, the system comprises a first LNP (e.g., any of the first LNPs described herein) and a second LNP (e.g., any of the second LNPs described herein) at a ratio of first LNP to second LNP of 1 : 10, 1 :9, 1 :8, 1 :7, 1 :6, 1 :5, 1 :4, 1 :3, 1 :2, 1 : 1, 1 :0.75, 0.75: 1, 2: 1, 3: 1, 4: 1, 5: 1, 6: 1, 7: 1, 8: 1, 9:1, or 10: 1. In some embodiments, the system comprise the first LNP and the second LNP at a ratio of 1 : 1.
[0431] In some embodiments, the system comprises a first LNP having a ratio of a first gene editor polynucleotide construct to a first attachment site-containing guide RNA (atgRNAl) of 1 : 10, 1 :9,
1 :8, 1 :7, 1 :6, 1 :5, 1:4, 1 :3, 1 :2, 1 : 1, 1 :0.75, 0.75: 1, 2: 1, 3: 1, 4: 1, 5: 1, 6: 1, 7: 1, 8: 1, 9: 1, or 10: 1. In some embodiments, the system includes a first LNP having a ratio of mRNA (i.e., mRNA encoding the gene editor protein) to atgRNAl of 2: 1.
[0432] In some embodiments, the system comprise a second LNP having a ratio of a second gene editor polynucleotide construct to a second attachment site-containing guide RNA (atgRNA2) of 1 : 10, 1 :9, 1 :8, 1 :7, 1:6, 1 :5, 1 :4, 1 :3, 1 :2, 1 : 1, 1 :0.75, 0.75: 1, 2: 1, 3: 1, 4: 1, 5: 1, 6: 1, 7: 1, 8: 1, 9: 1, or 10: 1. In some embodiments, the system includes a second LNP having a ratio of mRNA (i.e., mRNA encoding the gene editor protein) to atgRNA2 of 2: 1.
[0433] In some embodiments, the system comprises a ratio of gene editor polynucleotide construct (e.g., mRNA encoding the gene editor protein) to first atgRNA (atgRNAl) to second atgRNA (atgRNA2) of 1 :0.25:0.25, l :0.5:0.5, 1 :0.75:0.75, or 1 : 1 : 1.
[0434] In some embodiments, the system comprises a mixture of LNPs comprising three or more LNPs, four or more LNPs, five or more LNPs, six or more LNPs, seven or more LNPs, eight or more LNPs, nine or more LNPs, or ten or more LNPs.
[0435] In some embodiments, where a split LNP (e.g., a mixture of two LNPs packaged with different cargo) is being used to site-specifically integrate the at least first integration recognition site into the genome, a vector comprising a template polynucleotide and a sequence that is an integration cognate (i.e., cognate to an integration recognition site site-specifically incorporated into the genome of a cell) can be delivered to the cell concurrently with the split LNPs or after delivery of the split LNPs. For example, after delivering the split LNPs to the cell, a vector that includes a template polynucleotide and a second integration recognition site that is a cognate pair with the first integration recognition site is delivered to the cell. In such embodiments, the sequence that is an integration cognate (e.g., a second integration recognition site) enables integration of the template polynucleotide or portion thereof when contacted with an integrase and the site- specifically incorporated first integration recognition site.
5.10.6. Vector Delivery of a template polynucleotide
[0436] In certain aspects the invention involves vectors, e.g. for delivering or introducing in a cell, but also for propagating these components (e.g. in prokaryotic cells). A used herein, a "vector" is a tool that allows or facilitates the transfer of an entity from one environment to another. It is a replicon, such as a plasmid, phage, or cosmid, into which another DNA segment may be inserted so as to bring about the replication of the inserted segment. Generally, a vector is capable of
replication when associated with the proper control elements. In general, the term "vector" refers to a nucleic acid molecule capable of transporting another nucleic acid to which it has been linked. Vectors include, but are not limited to, nucleic acid molecules that are single-stranded, doublestranded, or partially double-stranded; nucleic acid molecules that comprise one or more free ends, no free ends (e.g. circular); nucleic acid molecules that comprise DNA, RNA, or both; and other varieties of polynucleotides known in the art. One type of vector is a ’’plasmid,” which refers to a circular double stranded DNA loop into which additional DNA segments can be inserted, such as by standard molecular cloning techniques. Another type of vector is a viral vector, wherein virally- derived DNA or RNA sequences are present in the vector for packaging into a virus (e.g. retroviruses, replication defective retroviruses, adenoviruses, replication defective adenoviruses, and adeno-associated viruses (AAVs)). Viral vectors also include polynucleotides carried by a virus for transfection into a host cell. Certain vectors are capable of autonomous replication in a host cell into which they are introduced (e.g. bacterial vectors having a bacterial origin of replication and episomal mammalian vectors). Other vectors (e.g., non-episomal mammalian vectors) are integrated into the genome of a host cell upon introduction into the host cell, and thereby are replicated along with the host genome. Moreover, certain vectors are capable of directing the expression of genes to which they are operatively-linked. Such vectors are referred to herein as "expression vectors." Vectors for and that result in expression in a eukaryotic cell can be referred to herein as "eukaryotic expression vectors." Common expression vectors of utility in recombinant DNA techniques are often in the form of plasmids.
[0437] Recombinant expression vectors can comprise a nucleic acid of the invention in a form suitable for expression of the nucleic acid in a host cell, which means that the recombinant expression vectors include one or more regulatory elements, which may be selected on the basis of the host cells to be used for expression, that is operatively-linked to the nucleic acid sequence to be expressed. Within a recombinant expression vector, "operably linked" is intended to mean that the nucleotide sequence of interest is linked to the regulatory element(s) in a manner that allows for expression of the nucleotide sequence (e.g. in an in vitro transcription/translation system or in a host cell when the vector is introduced into the host cell). With regards to recombination and cloning methods, mention is made of U.S. patent application Ser. No. 10/815,730, published Sep. 2, 2004, as US 2004-0171156 Al, the contents of which are herein incorporated by reference in their entirety.
[0438] Vector delivery, e.g., plasmid, viral delivery: The CRISPR enzyme, for instance a Type V protein such as C2cl or C2c3, and/or any of the present RNAs, for instance a guide RNA, can be
delivered using any suitable vector, e.g., plasmid or viral vectors, such as adeno associated virus (AAV), lentivirus, adenovirus or other viral vector types, or combinations thereof. Effector proteins and one or more guide RNAs can be packaged into one or more vectors, e.g., plasmid or viral vectors. In some embodiments, the vector, e.g., plasmid or viral vector is delivered to the tissue of interest by, for example, an intramuscular injection, while other times the delivery is via intravenous, transdermal, intranasal, oral, mucosal, or other delivery methods. Such delivery may be either via a single dose, or multiple doses. One skilled in the art understands that the actual dosage to be delivered herein may vary greatly depending upon a variety of factors, such as the vector choice, the target cell, organism, or tissue, the general condition of the subject to be treated, the degree of transformation/modification sought, the administration route, the administration mode, the type of transformation/modification sought, etc.
[0439] Such a dosage may further contain, for example, a carrier (water, saline, ethanol, glycerol, lactose, sucrose, calcium phosphate, gelatin, dextran, agar, pectin, peanut oil, sesame oil, etc.), a diluent, a pharmaceutically-acceptable carrier (e.g., phosphate-buffered saline), a pharmaceutically-acceptable excipient, and/or other compounds known in the art. The dosage may further contain one or more pharmaceutically acceptable salts such as, for example, a mineral acid salt such as a hydrochloride, a hydrobromide, a phosphate, a sulfate, etc.; and the salts of organic acids such as acetates, propionates, malonates, benzoates, etc. Additionally, auxiliary substances, such as wetting or emulsifying agents, pH buffering substances, gels or gelling materials, flavorings, colorants, microspheres, polymers, suspension agents, etc. may also be present herein. In addition, one or more other conventional pharmaceutical ingredients, such as preservatives, humectants, suspending agents, surfactants, antioxidants, anticaking agents, fillers, chelating agents, coating agents, chemical stabilizers, etc. may also be present, especially if the dosage form is a reconstitutable form. Suitable exemplary ingredients include microcrystalline cellulose, carboxymethylcellulose sodium, polysorbate 80, phenylethyl alcohol, chlorobutanol, potassium sorbate, sorbic acid, sulfur dioxide, propyl gallate, the parabens, ethyl vanillin, glycerin, phenol, parachlorophenol, gelatin, albumin and a combination thereof. A thorough discussion of pharmaceutically acceptable excipients is available in REMINGTON'S PHARMACEUTICAL SCIENCES (Mack Pub. Co., N.J. 1991) which is incorporated by reference herein.
[0440] In an embodiment herein the delivery is via an adenovirus, which may be at a single booster dose containing at least 1 x 105 particles (also referred to as particle units, pu) of adenoviral vector. In an embodiment herein, the dose preferably is at least about 1 x 106 particles (for example, about 1 x 106- 1 x 1011 particles), more preferably at least about 1 x 107 particles, more preferably at least
about 1 x 108 particles (e.g., about 1 x 108-l x 1011 particles or about 1 x 109-l x 1012 particles), and most preferably at least about 1 x IO10 particles (e.g., about 1 x 109- 1 x IO10 particles or about 1 x 109-l x 1012 particles), or even at least about 1 x IO10 particles (e.g., about 1 x 1010-l x 1012 particles) of the adenoviral vector. Alternatively, the dose comprises no more than about 1 x 1014 particles, preferably no more than about 1 x 1013 particles, even more preferably no more than about 1 x 1012 particles, even more preferably no more than about 1 x 1011 particles, and most preferably no more than about 1 x IO10 particles (e.g., no more than about 1 x 109 particles). Thus, the dose may contain a single dose of adenoviral vector with, for example, about 1 x 106 particle units (pu), about 2 x 106 pu, about 4 x 106 pu, about 1 x 107 pu, about 2 x 107 pu, about 4 x 107 pu, about 1 x 108 pu, about 2 x 108 pu, about 4 x 108 pu, about 1 x 109 pu, about 2 x 109 pu, about 4 x 109 pu, about 1 x IO10 pu, about 2 x IO10 pu, about 4 x IO10 pu, about 1 x 1011 pu, about 2 x 1011 pu, about 4 x 1011 pu, about 1 x 1012 pu, about 2 x 1012 pu, or about 4 x 1012 pu of adenoviral vector. See, for example, the adenoviral vectors in U.S. Pat. No. 8,454,972 B2 to Nabel, et. al., granted on Jun. 4, 2013; incorporated by reference herein, and the dosages at col 29, lines 36-58 thereof. In an embodiment herein, the adenovirus is delivered via multiple doses.
[0441] In an embodiment herein, the delivery is via an AAV. A therapeutically effective dosage for in vivo delivery of the AAV to a human is believed to be in the range of from about 20 to about 50 ml of saline solution containing from about 1 x 1010 to about 1 x 1050 functional AAV/ml solution. The dosage may be adjusted to balance the therapeutic benefit against any side effects. In an embodiment herein, the AAV dose is generally in the range of concentrations of from about 1 x 105 to 1 x 1050 genomes AAV (sometimes referred to herein as “vector genomes” or “vg”), from about 1 x 108 to 1 x IO20 genomes AAV, from about 1 x 1010 to about 1 x 1016 genomes, or about 1 x 1011 to about 1 x 1016 genomes AAV. A human dosage may be about 1 x 1013 genomes AAV. Such concentrations may be delivered in from about 0.001 ml to about 100 ml, about 0.05 to about 50 ml, or about 10 to about 25 ml of a carrier solution. Other effective dosages can be readily established by one of ordinary skill in the art through routine trials establishing dose response curves. See, for example, U.S. Pat. No. 8,404,658 B2 to Hajjar, et al., granted on Mar. 26, 2013, at col. 27, lines 45-60.
[0442] The promoter used to drive nucleic acid-targeting effector protein coding nucleic acid molecule expression can include: AAV ITR can serve as a promoter: this is advantageous for eliminating the need for an additional promoter element (which can take up space in the vector). The additional space freed up can be used to drive the expression of additional elements (gRNA, etc.). Also, ITR activity is relatively weaker, so can be used to reduce potential toxicity due to over
expression of nucleic acid-targeting effector protein. For ubiquitous expression, can use promoters: CMV, CAG, CBh, PGK, SV40, Ferritin heavy or light chains, etc. For brain or other CNS expression, can use promoters: SynapsinI for all neurons, CaMKIIalpha for excitatory neurons, GAD67 or GAD65 or VGAT for GABAergic neurons, etc. For liver expression, can use Albumin promoter. For lung expression, can use SP-B. For endothelial cells, can use ICAM. For hematopoietic cells can use IFNbeta or CD45. For Osteoblasts can use OG-2.
[0443] The promoter used to drive guide RNA can include: Pol III promoters such as U6 or Hl Use of Pol II promoter and intronic cassettes to express guide RNA Adeno Associated Virus (AAV).
[0444] Nucleic acid-targeting effector protein and one or more guide RNA can be delivered using adeno associated virus (AAV), lentivirus, adenovirus or other plasmid or viral vector types, in particular, using formulations and doses from, for example, U.S. Pat. No. 8,454,972 (formulations, doses for adenovirus), U.S. Pat. No. 8,404,658 (formulations, doses for AAV) and U.S. Pat. No. 5,846,946 (formulations, doses for DNA plasmids) and from clinical trials and publications regarding the clinical trials involving lentivirus, AAV and adenovirus. For examples, for AAV, the route of administration, formulation and dose can be as in U.S. Pat. No. 8,454,972 and as in clinical trials involving AAV. For Adenovirus, the route of administration, formulation and dose can be as in U.S. Pat. No. 8,404,658 and as in clinical trials involving adenovirus. For plasmid delivery, the route of administration, formulation and dose can be as in U.S. Pat. No. 5,846,946 and as in clinical studies involving plasmids. Doses may be based on or extrapolated to an average 70 kg individual (e.g., a male adult human), and can be adjusted for patients, subjects, mammals of different weight and species. Frequency of administration is within the ambit of the medical or veterinary practitioner (e.g., physician, veterinarian), depending on usual factors including the age, sex, general health, other conditions of the patient or subject and the particular condition or symptoms being addressed. The viral vectors can be injected into the tissue of interest. For celltype specific genome modification, the expression of nucleic acid-targeting effector can be driven by a cell-type specific promoter. For example, liver-specific expression might use the Albumin promoter and neuron-specific expression (e.g., for targeting CNS disorders) might use the Synapsin I promoter.
[0445] In terms of in vivo delivery, AAV is advantageous over other viral vectors for a couple of reasons: Low toxicity (this may be due to the purification method not requiring ultra centrifugation
of cell particles that can activate the immune response) and Low probability of causing insertional mutagenesis because it doesn't integrate into the host genome.
[0446] AAV has a packaging limit of 4.5 or 4.75 Kb. This means that nucleic acid-targeting effector protein (such as a Type V protein such as C2cl or C2c3) as well as a promoter and transcription terminator have to be all fit into the same viral vector. Therefore embodiments of the invention include utilizing homologs of nucleic acid-targeting effector protein (such as a Type V protein such as C2cl or C2c3) that are shorter.
[0447] As to AAV, the AAV can be AAV1, AAV2, AAV5 or any combination thereof. One can select the AAV of the AAV with regard to the cells to be targeted; e.g., one can select AAV serotypes 1, 2, 5 or a hybrid capsid AAV1, AAV2, AAV5 or any combination thereof for targeting brain or neuronal cells; and one can select AAV4 for targeting cardiac tissue. AAV8 is useful for delivery to the liver. The herein promoters and vectors are preferred individually.
[0448] Packaging cells are typically used to form virus particles that are capable of infecting a host cell. Such cells include 293 cells, which package adenovirus, and psi2 cells or PA317 cells, which package retrovirus. Viral vectors used in gene therapy are usually generated by producing a cell line that packages a nucleic acid vector into a viral particle. The vectors typically contain the minimal viral sequences required for packaging and subsequent integration into a host, other viral sequences being replaced by an expression cassette for the polynucleotide(s) to be expressed. The missing viral functions are typically supplied in trans by the packaging cell line. For example, AAV vectors used in gene therapy typically only possess ITR sequences from the AAV genome which are required for packaging and integration into the host genome. Viral DNA is packaged in a cell line, which contains a helper plasmid encoding the other AAV genes, namely rep and cap, but lacking ITR sequences. The cell line may also be infected with adenovirus as a helper. The helper virus promotes replication of the AAV vector and expression of AAV genes from the helper plasmid. The helper plasmid is not packaged in significant amounts due to a lack of ITR sequences. Contamination with adenovirus can be reduced by, e.g., heat treatment to which adenovirus is more sensitive than AAV. Additional methods for the delivery of nucleic acids to cells are known to those skilled in the art. See, for example, US20030087817, incorporated herein by reference.
[0449] Millington-Ward et al. (Molecular Therapy, vol. 19 no. 4, 642-649 April 2011) describes adeno-associated virus (AAV) vectors to deliver an RNA interference (RNAi)-based rhodopsin suppressor and a codon-modified rhodopsin replacement gene resistant to suppression due to nucleotide alterations at degenerate positions over the RNAi target site. An injection of either 6.0
x 108 vp or 1.8 x IO10 vp AAV were subretinally injected into the eyes by Millington-Ward et al. The AAV vectors of Millington-Ward et al. may be applied to the system of the present invention, contemplating a dose of about 2 x 1011 to about 6 x 1011 vp administered to a human.
[0450] Dalkara et al. (Sci Transl Med 5, 189ra76 (2013)) also relates to in vivo directed evolution to fashion an AAV vector that delivers wild-type versions of defective genes throughout the retina after noninjurious injection into the eyes' vitreous humor. Dalkara describes a 7 mer peptide display library and an AAV library constructed by DNA shuffling of cap genes from AAV1, 2, 4, 5, 6, 8, and 9. The rcAAV libraries and rAAV vectors expressing GFP under a CAG or Rho promoter were packaged and deoxyribonuclease-resistant genomic titers were obtained through quantitative PCR. The libraries were pooled, and two rounds of evolution were performed, each consisting of initial library diversification followed by three in vivo selection steps. In each such step, P30 rho-GFP mice were intravitreally injected with 2 ml of iodixanol -purified, phosphate- buffered saline (PBS)-dialyzed library with a genomic titer of about 1. times.10. sup.12 vg/ml. The AAV vectors of Dalkara et al. may be applied to the nucleic acid-targeting system of the present invention, contemplating a dose of about 1 x 1015 to about 1 x 1016 vg/ml administered to a human.
[0451] The tropism of a retrovirus can be altered by incorporating foreign envelope proteins, expanding the potential target population of target cells. Lentiviral vectors are retroviral vectors that are able to transduce or infect non-dividing cells and typically produce high viral titers. Selection of a retroviral gene transfer system would therefore depend on the target tissue. Retroviral vectors are comprised of cis-acting long terminal repeats with packaging capacity for up to 6-10 kb of foreign sequence. The minimum cis-acting LTRs are sufficient for replication and packaging of the vectors, which are then used to integrate the therapeutic gene into the target cell to provide permanent transgene expression. Widely used retroviral vectors include those based upon murine leukemia virus (MuLV), gibbon ape leukemia virus (GaLV), Simian Immuno deficiency virus (SW), human immuno deficiency virus (HIV), and combinations thereof (see, e.g., Buchscher et al., J. Virol. 66:2731-2739 (1992); Johann et al., J. Virol. 66: 1635-1640 (1992); Sommnerfelt et al., Virol. 176:58-59 (1990); Wilson et al., J. Virol. 63:2374-2378 (1989); Miller et al., J. Virol. 65:2220-2224 (1991); PCT/US94/05700). In applications where transient expression is preferred, adenoviral based systems may be used. Adenoviral based vectors are capable of very high transduction efficiency in many cell types and do not require cell division. With such vectors, high titer and levels of expression have been obtained. This vector can be produced in large quantities in a relatively simple system. Adeno-associated virus ("AAV") vectors may also be used to transduce cells with target nucleic acids, e.g., in the in vitro production of
nucleic acids and peptides, and for in vivo and ex vivo gene therapy procedures (see, e.g., West et al., Virology 160:38-47 (1987); U.S. Pat. No. 4,797,368; WO 93/24641; Kotin, Human Gene Therapy 5:793-801 (1994); Muzyczka, J. Clin. Invest. 94: 1351 (1994). Construction of recombinant AAV vectors are described in a number of publications, including U.S. Pat. No. 5,173,414; Tratschin et al., Mol. Cell. Biol. 5:3251-3260 (1985); Tratschin, et al., Mol. Cell. Biol. 4:2072-2081 (1984); Hermonat & Muzyczka, PNAS 81 :6466-6470 (1984); and Samulski et al., J. Virol. 63:03822-3828 (1989).
[0452] Packaging cells are typically used to form virus particles that are capable of infecting a host cell. Such cells include 293 cells, which package adenovirus, and yr2 cells or PA317 cells, which package retrovirus. Viral vectors used in gene therapy are usually generated by producing a cell line that packages a nucleic acid vector into a viral particle. The vectors typically contain the minimal viral sequences required for packaging and subsequent integration into a host, other viral sequences being replaced by an expression cassette for the polynucleotide(s) to be expressed. The missing viral functions are typically supplied in trans by the packaging cell line. For example, AAV vectors used in gene therapy typically only possess ITR sequences from the AAV genome which are required for packaging and integration into the host genome. Viral DNA is packaged in a cell line, which contains a helper plasmid encoding the other AAV genes, namely rep and cap, but lacking ITR sequences. The cell line may also be infected with adenovirus as a helper. The helper virus promotes replication of the AAV vector and expression of AAV genes from the helper plasmid. The helper plasmid is not packaged in significant amounts due to a lack of ITR sequences. Contamination with adenovirus can be reduced by, e.g., heat treatment to which adenovirus is more sensitive than AAV. Additional methods for the delivery of nucleic acids to cells are known to those skilled in the art. See, for example, US20030087817, incorporated herein by reference.
[0453] In some embodiments, a host cell is transiently or non-transiently transfected with one or more vectors described herein. In some embodiments, a cell is transfected as it naturally occurs in a subject. In some embodiments, a cell that is transfected is taken from a subject. Cells taken from a subject include, but are not limited to, hepatocytes or cells isolated from muscle, the CNS, eye or lung. Immunological cells are also contemplated, such as but not limited to T cells, HSCs, B-cells and NK cells.
[0454] Another useful method to deliver proteins, enzymes, and guides comprises transfection of messenger RNA (mRNA). Examples of mRNA delivery methods and compositions that may be utilized in the present disclosure including, for example, PCT/US2014/028330, US8822663B2,
NZ700688A, ES2740248T3, EP2755693A4, EP2755986A4, WO2014152940A1, EP3450553B1, BRI 12016030852A2, and EP3362461A1. Expression of CRISPR systems in particular is described by W02020014577. Each of these publications are incorporated herein by reference in their entireties. Additional disclosure hereby incorporated by reference can be found in Kowalski et al., “Delivering the Messenger: Advances in Technologies for Therapeutic mRNA Delivery,” Mol Therap., 2019; 27(4): 710-728.
[0455] In some embodiments, the cell is derived from cells taken from a subject, such as a cell line. A wide variety of cell lines for tissue culture are known in the art. Examples of cell lines include, but are not limited to, C8161, CCRF-CEM, MOLT, mIMCD-3, NHDF, HeLa-S3, Huhl, Huh4, Huh7, HUVEC, HASMC, HEKn, HEKa, MiaPaCell, Panel, PC-3, TF1, CTLL-2, CIR, Rat6, CV1, RPTE, A10, T24, J82, A375, ARH-77, Calul, SW480, SW620, SKOV3, SK-UT, CaCo2, P388D1, SEM-K2, WEHL231, HB56, TIB55, Jurkat, J45.01, LRMB, Bcl-1, BC-3, IC21, DLD2, Raw264.7, NRK, NRK-52E, MRC5, MEF, Hep G2, HeLa B, HeLa T4, COS, COS-1, COS-6, COS-M6A, BS-C-1 monkey kidney epithelial, BALB/3T3 mouse embryo fibroblast, 3T3 Swiss, 3T3-L1, 132-d5 human fetal fibroblasts; 10.1 mouse fibroblasts, 293-T, 3T3, 721, 9L, A2780, A2780ADR, A2780cis, A172, A20, A253, A431, A-549, ALC, B16, B35, BCP-1 cells, BEAS-2B, bEnd.3, BHK-21, BR 293, BxPC3, C3H-10T1/2, C6/36, Cal-27, CHO, CHO-7, CHOIR, CHO-K1, CHO-K2, CHO-T, CHO Dhfr-/-, COR-L23, COR-L23/CPR, COR-L23/5010, COR- L23/R23, COS-7, COV-434, CML Tl, CMT, CT26, D17, DH82, DU145, DuCaP, EL4, EM2, EM3, EMT6/AR1, EMT6/AR10.0, FM3, H1299, H69, HB54, HB55, HCA2, HEK-293, HeLa, Hepalclc7, HL-60, HMEC, HT-29, Jurkat, JY cells, K562 cells, Ku812, KCL22, KG1, KYO1, LNCap, Ma-Mel 1-48, MC-38, MCF-7, MCF-10A, MDA-MB-231, MDA-MB-468, MDA-MB- 435, MDCK II, MDCK II, MOR/0.2R, MONO-MAC 6, MTD-1A, MyEnd, NCLH69/CPR, NCI- H69/LX10, NCI-H69/LX20, NCI-H69/LX4, NIH-3T3, NALM-1, NW-145, OPCN/OPCT cell lines, Peer, PNT-1A/PNT 2, RenCa, RIN-5F, RMA/RMAS, Saos-2 cells, Sf-9, SkBr3, T2, T-47D, T84, THP1 cell line, U373, U87, U937, VCaP, Vero cells, WM39, WT-49, X63, YAC-1, YAR, and transgenic varieties thereof. Cell lines are available from a variety of sources known to those with skill in the art (see, e.g., the American Type Culture Collection (ATCC) (Manassas, Va.)). In some embodiments, a cell transfected with one or more vectors described herein is used to establish a new cell line comprising one or more vector-derived sequences.
[0456] In some embodiments, one or more vectors described herein are used to produce a nonhuman transgenic animal or transgenic plant. In some embodiments, the transgenic animal is a mammal, such as a mouse, rat, or rabbit. In certain embodiments, the organism or subject is a plant.
In certain embodiments, the organism or subject or plant is algae. Methods for producing transgenic plants and animals are known in the art, and generally begin with a method of cell transfection, such as described herein.
[0457] In one aspect, the invention provides for methods of modifying a target polynucleotide in a prokaryotic or eukaryotic cell, which may be in vivo, ex vivo or in vitro. In some embodiments, the method comprises sampling a cell or population of cells from a human or non-human animal or plant (including micro-algae) and modifying the cell or cells. Culturing may occur at any stage ex vivo. The cell or cells may even be re-introduced into the non-human animal or plant (including micro-algae).
[0458] In plants, pathogens are often host-specific. For example, Fusariumn oxysporum f. sp. lycopersici causes tomato wilt but attacks only tomato, and F. oxysporum f. dianthii Puccinia graminis f. sp. tritici attacks only wheat. Plants have existing and induced defenses to resist most pathogens. Mutations and recombination events across plant generations lead to genetic variability that gives rise to susceptibility, especially as pathogens reproduce with more frequency than plants. In plants there can be non-host resistance, e.g., the host and pathogen are incompatible. There can also be Horizontal Resistance, e.g., partial resistance against all races of a pathogen, typically controlled by many genes and Vertical Resistance, e.g., complete resistance to some races of a pathogen but not to other races, typically controlled by a few genes. In a Gene-for-Gene level, plants and pathogens evolve together, and the genetic changes in one balance changes in other. Accordingly, using Natural Variability, breeders combine most useful genes for Yield. Quality, Uniformity, Hardiness, Resistance. The sources of resistance genes include native or foreign Varieties, Heirloom Varieties, Wild Plant Relatives, and Induced Mutations, e.g., treating plant material with mutagenic agents. Using the present invention, plant breeders are provided with a new tool to induce mutations. Accordingly, one skilled in the art can analyze the genome of sources of resistance genes, and in Varieties having desired characteristics or traits employ the present invention to induce the rise of resistance genes, with more precision than previous mutagenic agents and hence accelerate and improve plant breeding programs.
[0459] Examples of target polynucleotides include a sequence associated with a signaling biochemical pathway, e.g., a signaling biochemical pathway-associated gene or polynucleotide. Examples of target polynucleotides include a disease associated gene or polynucleotide. A “disease-associated” gene or polynucleotide refers to any gene or polynucleotide which is yielding transcription or translation products at an abnormal level or in an abnormal form in cells derived
from a disease-affected tissues compared with tissues or cells of a non disease control. It may be a gene that becomes expressed at an abnormally high level; it may be a gene that becomes expressed at an abnormally low level, where the altered expression correlates with the occurrence and/or progression of the disease. A disease-associated gene also refers to a gene possessing mutation(s) or genetic variation that is directly responsible or is in linkage disequilibrium with a gene(s) that is responsible for the etiology of a disease. The transcribed or translated products may be known or unknown, and may be at a normal or abnormal level.
5.10.7. Lipid Nanoparticle Delivery
[0460] In some embodiments, the delivery system is packaged in one or more LNPs and administered intravenously. In some embodiments, the co-delivery system is packaged in one or more LNPs and administered intrathecally. In some embodiments, the co-delivery system is packaged in one or more LNPs and administered by intracerebral ventricular injection. In some embodiments, the co-delivery system is packaged in one or more LNPs and administered by intraci sternal magna administration. In some embodiments, the co-delivery system is packaged in one or more LNPs and administered by intravitreal injection.
[0461] The preparation of lipidmucleic acid complexes, including targeted liposomes such as immunolipid complexes, is well known to one of skill in the art (see, e.g., Crystal, Science 270:404-410 (1995); Blaese et al., Cancer Gene Ther. 2:291-297 (1995); Behr et al., Bioconjugate Chem. 5:382-389 (1994); Remy et al., Bioconjugate Chem. 5:647-654 (1994); Gao et al., Gene Therapy 2:710-722 (1995); Ahmad et al., Cancer Res. 52:4817-4820 (1992); U.S. Pat. Nos. 4,186,183, 4,217,344, 4,235,871, 4,261,975, 4,485,054, 4,501,728, 4,774,085, 4,837,028, and 4,946,787). In some embodiments, the LNP formulations are selected from LP01 (Cas No. 1799316-64-5), ALC-0315 (Cas No. 2036272-55-4), and cKK-E12 (Cas No. 1432494-65-9). In some embodiments, the LNP formulation is LP01. In some embodiments, the LNP formulation is ALC-0315. In some embodiment, the LNP formulation is cKK-E12.
[0462] In some embodiments, LNP doses range from about 0.1 mg/kg to about 100 mg/kg (or any of the values or subranges therein). In some embodiments, LNP doses is about 0.1 mg/kg, about 0.2 mg/kg, about 0.3 mg/kg, about 0.4 mg/kg, about 0.5 mg/kg, about 0.6 mg/kg, about 0.7 mg/kg, about 0.8 mg/kg, about 0.9 mg/kg, about 1.0 mg/kg, 1.5 mg/kg, about 2 mg/kg, about 2.5 mg/kg, about 3 mg/kg, about 3.5 mg/kg, about 4 mg/kg, about 4.5 mg/kg, about 5 mg/kg, about 6 mg/kg, about7 mg/kg, about 7 mg/kg, about 8 mg/kg, about 9 mg/kg, about 10 mg/kg, about 15
mg/kg, about 20 mg/kg, about 25 mg/kg, about 30 mg/kg, about 35 mg/kg, about 40 mg/kg, about 45 mg/kg, or about 50 mg/kg or more.
[0463] In another embodiment, LNP doses of about 0.01 to about 1 mg per kg of body weight administered intravenously are contemplated. Medications to reduce the risk of infusion-related reactions are contemplated, such as dexamethasone, acetaminophen, diphenhydramine or cetirizine, and ranitidine are contemplated. Multiple doses of about 0.3 mg per kilogram every 4 weeks for five doses are also contemplated.
[0464] The charge of the LNP must be taken into consideration. As cationic lipids combined with negatively charged lipids to induce nonbilayer structures that facilitate intracellular delivery. Because charged LNPs are rapidly cleared from circulation following intravenous injection, ionizable cationic lipids with pKa values below 7 were developed (see, e.g., Rosin et al, Molecular Therapy, vol. 19, no. 12, pages 1286-2200, December 2011). Negatively charged polymers such as RNA may be loaded into LNPs at low pH values (e.g., pH 4) where the ionizable lipids display a positive charge. However, at physiological pH values, the LNPs exhibit a low surface charge compatible with longer circulation times. Four species of ionizable cationic lipids have been focused upon, namely l,2-dilineoyl-3-dimethylammonium-propane (DLinDAP), 1,2- dilinoleyloxy-3-N,N-dimethylaminopropane (DLinDMA), l,2-dilinoleyloxy-keto-N,N-dimethyl- 3 -aminopropane (DLinKDMA), and l,2-dilinoleyl-4-(2-dimethylaminoethyl)-[l,3]-dioxolane (DLinKC2-DMA). It has been shown that LNP siRNA systems containing these lipids exhibit remarkably different gene silencing properties in hepatocytes in vivo, with potencies varying according to the series DLinKC2-DMA>DLinKDMA>DLinDMA»DLinDAP employing a Factor VII gene silencing model (see, e.g., Rosin et al, Molecular Therapy, vol. 19, no. 12, pages 1286-2200, December 2011). A dosage of 1 pg/ml of LNP in or associated with the LNP may be contemplated, especially for a formulation containing DLinKC2-DMA.
[0465] In some embodiments, the LNP composition comprises one or more one or more ionizable lipids. As used herein, the term "ionizable lipid" has its ordinary meaning in the art and may refer to a lipid comprising one or more charged moieties. In some embodiments, an ionizable lipid may be positively charged or negatively charged. In principle, there are no specific limitations concerning the ionizable lipids of the LNP compositions disclosed herein. In some embodiments, the one or more ionizable lipids are selected from the group consisting of 3-(didodecylamino)- N1 ,N1 ,4-tridodecyl- 1 -piperazineethanamine (KL 10), N1 -[2-(didodecylamino)ethyl]-N 1 ,N4,N4- tridodecyl-l,4-piperazinediethanami- ne (KL22), 14,25-ditridecyl-15,18,21,24-tetraaza-
octatriacontane (KL25), l,2-dilinoleyloxy-N,N-dimethylaminopropane (DLin-DMA), 2,2- dilinoleyl-4-dimethylaminomethyl-[l,3]-dioxolane (DLin-K-DMA), heptatriaconta-6,9,28,31- tetraen- 19-yl 4-(dimethylamino)butanoate (DLin-MC3-DMA), 2,2-dilinoleyl-4-(2- dimethylaminoethyl)-[l,3]-di oxolane (DLin-KC2-DMA), 1, 2-di oleyloxy -N,N- dimethylaminopropane (DODMA), 2-({8-[(3)-cholest-5-en-3-yloxy]octyl}oxy)-N,N-dimethyl-3- [(9Z,12Z)-octad- eca-9,12-dien-l-yloxy]propan-l -amine (Octyl-CLinDMA), (2R)-2-({8-[(3)- cholest-5-en-3-yloxy]octyl}oxy)-N,N-dimethyl-3-[(9Z,12Z)— octadeca-9,12-dien-l- yloxy]propan-l -amine (Octyl-CLinDMA (2R)), and (2S)-2-({8-[(3)-cholest-5-en-3- yloxy]octyl}oxy)-N,N-dimethyl-3-[(9Z,12Z)— octadeca-9,12-dien-l-y loxy]propan-l -amine (Octyl-CLinDMA (2S)). In one embodiment, the ionizable lipid may be selected from, but not limited to, an ionizable lipid described in International Publication Nos. WO2013086354 and WO2013116126.
[0466] In some embodiments, the lipid nanoparticle may include one or more (e.g., 1, 2, 3, 4, 5, 6, 7, or 8) cationic and/or ionizable lipids. Such cationic and/or ionizable lipids include, but are not limited to, 3-(didodecylamino)-Nl,Nl,4-tridodecyl-l-piperazineethanamine (KL10), Nl-[2- (didodecylamino)ethyl]-Nl,N4,N4-tridodecyl-l,4-piperazinediethanami- ne (KL22), 14,25- ditridecyl- 15,18,21 ,24-tetraaza-octatriacontane (KL25), 1 ,2-dilinoleyloxy-N,N- dimethylaminopropane (DLin-DMA), 2,2-dilinoleyl-4-dimethylaminomethyl-[l,3]-dioxolane (DLin-K-DMA), heptatriaconta-6,9,28,31-tetraen- 19-yl 4-(dimethylamino)butanoate (DLin- MC3-DMA), 2,2-dilinoleyl-4-(2-dimethylaminoethyl)-[l,3]-dioxolane (DLin-KC2-DMA), 2-({8- [(3.beta.)-cholest-5-en-3-yloxy]octyl}oxy)-N,N-dimethyl-3-[(9Z,12Z)- -octadeca-9,12-dien-l- yloxy]propan-l -amine (Octyl-CLinDMA), (2R)-2-({8-[(3.beta.)-cholest-5-en-3- yloxy]octyl}oxy)-N,N-dimethyl-3-[(9Z- ,12Z)-octadeca-9,12-dien-l-yl oxy]propan-l -amine (Octyl-CLinDMA (2R)), (2S)-2-({8-[(3Pcholest-5-en-3-yloxy]octyl}oxy)-N,N-dimethyl-3-[(9Z- ,12Z)-octadeca-9,12-dien-l-yl oxy]propan-l -amine (Octyl-CLinDMA (2S)).N,N-dioleyl-N,N- dimethylammonium chloride ("DODAC"); N-(2,3 -di oleyloxy)propyl-N,N— N-tri ethylammonium chloride ("DOTMA"); N,N-distearyl-N,N-dimethylammonium bromide ("DDAB"); N-(2,3- dioleoyloxy)propyl)-N,N,N-trimethylammonium chloride ("DOTAP"); l,2-Dioleyloxy-3- trimethylaminopropane chloride salt ("DOTAP. Cl"); 3-.beta.-(N— (N',N'-dimethylaminoethane)- carbamoyl)cholesterol ("DC-Chol"), N-(l-(2,3-dioleyloxy)propyl)-N-2-
(sperminecarboxamido)ethyl)-N,N-dimethyl- -ammonium trifluoracetate ("DOSPA"), dioctadecylamidoglycyl carboxyspermine ("DOGS"), 1, 2-di oleoyl-3 -dimethylammonium propane ("DODAP"), N,N-dimethyl-2,3-dioleyloxy)propylamine ("DODMA"), and N-(l,2- dimyristyloxyprop-3-yl)-N,N-dimethyl-N-hydroxyethyl ammonium bromide ("DMRIE").
Additionally, a number of commercial preparations of cationic and/or ionizable lipids can be used, such as, e.g., LIPOFECTIN.RTM. (including DOTMA and DOPE, available from GIBCO/BRL), and LIPOFECTAMINE.RTM. (including DOSPA and DOPE, available from GIBCO/BRL). KL10, KL22, and KL25 are described, for example, in U.S. Pat. No. 8,691,750.
[0467] In some embodiments, the LNP composition comprises one or more amino lipids. The terms "amino lipid" and "cationic lipid" are used interchangeably herein to include those lipids and salts thereof having one, two, three, or more fatty acid or fatty alkyl chains and a pH-titratable amino head group (e.g., an alkylamino or dialkylamino head group). In principle, there are no specific limitations concerning the amino lipids of the LNP compositions disclosed herein. The cationic lipid is typically protonated (i.e., positively charged) at a pH below the pKa of the cationic lipid and is substantially neutral at a pH above the pKa. The cationic lipids can also be termed titratable cationic lipids. In some embodiments, the one or more cationic lipids include: a protonatable tertiary amine (e.g., pH-titratable) head group; alkyl chains, wherein each alkyl chain independently has 0 to 3 (e.g., 0, 1, 2, or 3) double bonds; and ether, ester, or ketal linkages between the head group and alkyl chains. Such cationic lipids include, but are not limited to, DSDMA, DODMA, DOTMA, DLinDMA, DLenDMA, . gamma. -DLenDMA, DLin-K-DMA, DLin-K-C2- DMA (also known as DLin-C2K-DMA, XTC2, and C2K), DLin-K-C3-DMA, DLin-K-C4-DMA, DLen-C2K-DMA, y-DLen-C2-DMA, C12-200, cKK-E12, cKK-A12, cKK-012, DLin-MC2- DMA (also known as MC2), and DLin-MC3-DMA (also known as MC3).
[0468] Anionic lipids suitable for use in lipid nanoparticles include, but are not limited to, phosphatidylglycerol, cardiolipin, diacylphosphatidylserine, diacylphosphatidic acid, N- dodecanoyl phosphatidylethanoloamine, N-succinyl phosphatidylethanolamine, N-glutaryl phosphatidylethanolamine, lysylphosphatidylglycerol, and other anionic modifying groups joined to neutral lipids.
[0469] Neutral lipids (including both uncharged and zwitterionic lipids) suitable for use in lipid nanoparticles include, but are not limited to, diacylphosphatidylcholine, diacylphosphatidylethanolamine, ceramide, sphingomyelin, dihydrosphingomyelin, cephalin, sterols (e.g., cholesterol) and cerebrosides. In some embodiments, the lipid nanoparticle comprises cholesterol. Lipids having a variety of acyl chain groups of varying chain length and degree of saturation are available or may be isolated or synthesized by well-known techniques. Additionally, lipids having mixtures of saturated and unsaturated fatty acid chains and cyclic regions can be used. In some embodiments, the neutral lipids used in the disclosure are DOPE, DSPC, DPPC,
POPC, or any related phosphatidylcholine. In some embodiments, the neutral lipid may be composed of sphingomyelin, dihydrosphingomyeline, or phospholipids with other head groups, such as serine and inositol.
[0470] In some embodiments, amphipathic lipids are included in nanoparticles. Exemplary amphipathic lipids suitable for use in nanoparticles include, but are not limited to, sphingolipids, phospholipids, fatty acids, and amino lipids.
[0471] The lipid composition of the pharmaceutical composition may comprise one or more phospholipids, for example, one or more saturated or (poly)unsaturated phospholipids or a combination thereof. In general, phospholipids comprise a phospholipid moiety and one or more fatty acid moieties.
[0472] A phospholipid moiety can be selected, for example, from the non-limiting group consisting of phosphatidyl choline, phosphatidyl ethanolamine, phosphatidyl glycerol, phosphatidyl serine, phosphatidic acid, 2-lysophosphatidyl choline, and a sphingomyelin.
[0473] A fatty acid moiety can be selected, for example, from the non-limiting group consisting of lauric acid, myristic acid, myristoleic acid, palmitic acid, palmitoleic acid, stearic acid, oleic acid, linoleic acid, alpha-linolenic acid, erucic acid, phytanoic acid, arachidic acid, arachidonic acid, eicosapentaenoic acid, behenic acid, docosapentaenoic acid, and docosahexaenoic acid.
[0474] Particular amphipathic lipids can facilitate fusion to a membrane. For example, a cationic phospholipid can interact with one or more negatively charged phospholipids of a membrane (e.g., a cellular or intracellular membrane). Fusion of a phospholipid to a membrane can allow one or more elements (e.g., a therapeutic agent) of a lipid-containing composition (e.g., LNPs) to pass through the membrane permitting, e.g., delivery of the one or more elements to a target tissue.
[0475] Non-natural amphipathic lipid species including natural species with modifications and substitutions including branching, oxidation, cyclization, and alkynes are also contemplated. For example, a phospholipid can be functionalized with or cross-linked to one or more alkynes (e.g., an alkenyl group in which one or more double bonds is replaced with a triple bond). Under appropriate reaction conditions, an alkyne group can undergo a copper-catalyzed cycloaddition upon exposure to an azide. Such reactions can be useful in functionalizing a lipid bilayer of a nanoparticle composition to facilitate membrane permeation or cellular recognition or in conjugating a nanoparticle composition to a useful component such as a targeting or imaging moiety (e.g., a dye).
[0476] Phospholipids include, but are not limited to, glycerophospholipids such as phosphatidylcholines, phosphatidylethanolamines, phosphatidylserines, phosphatidylinositols, phosphatidy glycerols, and phosphatidic acids. Phospholipids also include phosphosphingolipid, such as sphingomyelin.
[0477] In some embodiments, the LNP composition comprises one or more phospholipids. In some embodiments, the phospholipid is selected from the group consisting of 1,2-dilinoleoyl-sn- glycero-3 -phosphocholine (DLPC), 1,2-dimyristoyl-sn-glycero-phosphocholine (DMPC), 1,2- dioleoyl-sn-glycero-3 -phosphocholine (DOPC), 1 ,2-dipalmitoyl-sn-glycero-3 -phosphocholine (DPPC), l,2-distearoyl-sn-glycero-3-phosphocholine (DSPC), 1,2-diundecanoyl-sn-glycero- phosphocholine (DUPC), l-palmitoyl-2-oleoyl-sn-glycero-3 -phosphocholine (POPC), 1,2-di-O- octadecenyl-sn-glycero-3-phosphocholine (18:0 Diether PC), l-oleoyl-2- cholesterylhemi succinoyl -sn-gly cero-3 -phosphocholine (OChemsPC), 1 -hexadecyl -sn-gly cero-3 - phosphocholine (Cl 6 Lyso PC), 1,2-dilinolenoyl-sn-gly cero-3 -phosphocholine, 1,2- diarachidonoyl-sn-glycero-3-phosphocholine, l,2-didocosahexaenoyl-sn-glycero-3- phosphocholine, l,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), 1,2-diphytanoyl-sn- gly cero-3 -phosphoethanolamine (ME 16:0 PE), l,2-distearoyl-sn-glycero-3- phosphoethanolamine, 1,2-dilinoleoyl-sn-gly cero-3 -phosphoethanolamine, 1,2-dilinolenoyl-sn- gly cero-3 -phosphoethanolamine, l,2-diarachidonoyl-sn-glycero-3-phosphoethanolaminel,2- didocosahexaenoyl— sn-glycero-3-phosphoethanolamine, 1,2-dioleoyl-sn-gly cero-3 -phospho-rac- (1 -glycerol) sodium salt (DOPG), sphingomyelin, and any mixtures thereof.
[0478] Other phosphorus-lacking compounds, such as sphingolipids, glycosphingolipid families, diacylglycerols, and P-acyloxyacids, may also be used. Additionally, such amphipathic lipids can be readily mixed with other lipids, such as triglycerides and sterols.
[0479] In some embodiments, the LNP composition comprises one or more helper lipids. The term "helper lipid" as used herein refers to lipids that enhance transfection (e.g., transfection of an LNP comprising an mRNA that encodes a site-directed endonuclease, such as a SpCas9 polypeptide). In principle, there are no specific limitations concerning the helper lipids of the LNP compositions disclosed herein. Without being bound to any particular theory, it is believed that the mechanism by which the helper lipid enhances transfection includes enhancing particle stability. In some embodiments, the helper lipid enhances membrane fusogenicity. Generally, the helper lipid of the LNP compositions disclosure herein can be any helper lipid known in the art. Non-limiting examples of helper lipids suitable for the compositions and methods include steroids, sterols, and
alkyl resorcinols. Particularly helper lipids suitable for use in the present disclosure include, but are not limited to, saturated phosphatidylcholine (PC) such as distearoyl-PC (DSPC) and dipalymitoyl-PC (DPPC), di oleoylphosphatidylethanolamine (DOPE), 1,2-dilinoleoyl-sn- glycero-3 -phosphocholine (DLPC), cholesterol, 5-heptadecylresorcinol, and cholesterol hemisuccinate. In some embodiments, the helper lipid of the LNP composition includes cholesterol.
[0480] In some embodiments, the LNP composition comprises one or more structural lipids. As used herein, the term "structural lipid" refers to sterols and also to lipids containing sterol moi eties. Without being bound to any particular theory, it is believed that the incorporation of structural lipids into the LNPs mitigates aggregation of other lipids in the particle. Structural lipids can be selected from the group including but not limited to, cholesterol, fecosterol, sitosterol, ergosterol, campesterol, stigmasterol, brassicasterol, tomatidine, tomatine, ursolic acid, alpha-tocopherol, hopanoids, phytosterols, steroids, and mixtures thereof. In some embodiments, the structural lipid is a sterol. As defined herein, "sterols" are a subgroup of steroids consisting of steroid alcohols. In certain embodiments, the structural lipid is a steroid. In some embodiments, the structural lipid is cholesterol. In certain embodiments, the structural lipid is an analog of cholesterol.
[0481] The lipid component of a lipid nanoparticle composition may include one or more molecules comprising polyethylene glycol, such as PEG or PEG-modified lipids. In some embodiments, the LNP composition disclosed herein comprise one or more polyethylene glycol (PEG) lipid. The term "PEG-lipid" refers to polyethylene glycol (PEG)-modified lipids. Such lipids are also referred to as PEGylated lipids. Non-limiting examples of PEG-lipids include PEG- modified phosphatidylethanolamine and phosphatidic acid, PEG-ceramide conjugates (e.g., PEG- CerC14 or PEG-CerC20), PEG-modified dialkylamines and PEG-modified 1,2-diacyloxypropan- 3-amines For example, a PEG lipid can be PEG-c-DOMG, PEG-DMG, PEG-DLPE, PEG-DMPE, PEG-DPPC, or a PEG-DSPE lipid. In some embodiments, the PEG-lipid includes, but not limited to 1,2-dimyristoyl-sn-glycerol methoxypolyethylene glycol (PEG-DMG), 1,2-distearoyl-sn- glycero-3-phosphoethanolamine-N-[amino(polyethylene glycol)] (PEG-DSPE), PEG-disteryl glycerol (PEG-DSG), PEG-dipalmetoleyl, PEG-dioleyl, PEG-distearyl, PEG-diacylglycamide (PEG-DAG), PEG-dipalmitoyl phosphatidylethanolamine (PEG-DPPE), or PEG- 1,2- dimyristyloxlpropyl-3-amine (PEG-c-DMA). In some embodiments, the PEG-lipid is selected from the group consisting of a PEG-modified phosphatidylethanolamine, a PEG-modified phosphatidic acid, a PEG-modified ceramide, a PEG-modified dialkylamine, a PEG-modified diacylglycerol, a PEG-modified dialkylglycerol, and mixtures thereof. In some embodiments, the
lipid moiety of the PEG-lipids includes those having lengths of from about C. sub.14 to about C. sub.22, preferably from about C. sub.14 to about C. sub.16. In some embodiments, a PEG moiety, for example a mPEG-NH.sub.2, has a size of about 1000, 2000, 5000, 10,000, 15,000 or 20,000 daltons. In some embodiment, the PEG-lipid is PEG2k-DMG. In some embodiments, the one or more PEG lipids of the LNP composition comprises PEG-DMPE. In some embodiments, the one or more PEG lipids of the LNP composition comprises PEG-DMG.
[0482] In some embodiments, the ratio between the lipid components and the nucleic acid molecules of the LNP composition, e.g., the weight ratio, is sufficient for (i) formation of LNPs with desired characteristics, e.g., size, charge, and (ii) delivery of a sufficient dose of nucleic acid at a dose of the lipid component(s) that is tolerable for in vivo administration as readily ascertained by one of skill in the art.
[0483] In certain embodiments, it is desirable to target a nanoparticle, e.g., a lipid nanoparticle, using a targeting moiety that is specific to a cell type and/or tissue type. In some embodiments, a nanoparticle may be targeted to a particular cell, tissue, and/or organ using a targeting moiety. In particular embodiments, a nanoparticle comprises a targeting moiety. Exemplary non-limiting targeting moieties include ligands, cell surface receptors, glycoproteins, vitamins (e.g., riboflavin) and antibodies (e.g., full-length antibodies, antibody fragments (e.g., Fv fragments, single chain Fv (scFv) fragments, Fab' fragments, or F(ab')2 fragments), single domain antibodies, cam elid antibodies and fragments thereof, human antibodies and fragments thereof, monoclonal antibodies, and multispecific antibodies (e.g., bispecific antibodies)). In some embodiments, the targeting moiety may be a polypeptide. The targeting moiety may include the entire polypeptide (e.g., peptide or protein) or fragments thereof. A targeting moiety is typically positioned on the outer surface of the nanoparticle in such a manner that the targeting moiety is available for interaction with the target, for example, a cell surface receptor. A variety of different targeting moieties and methods are known and available in the art, including those described, e.g., in Sapra et al., Prog. Lipid Res. 42(5):439-62, 2003 and Abra et al., J. Liposome Res. 12: 1-3, 2002.
[0484] In some embodiments, a lipid nanoparticle (e.g., a liposome) may include a surface coating of hydrophilic polymer chains, such as polyethylene glycol (PEG) chains (see, e.g., Allen et al., Biochimica et Biophysica Acta 1237: 99-108, 1995; DeFrees et al., Journal of the American Chemistry Society 118: 6101-6104, 1996; Blume et al., Biochimica et Biophysica Acta 1149: ISO- 184, 1993; Klibanov et al., Journal of Liposome Research 2: 321-334, 1992; U.S. Pat. No. 5,013,556; Zalipsky, Bioconjugate Chemistry 4: 296-299, 1993; Zalipsky, FEBS Letters 353: 71-
74, 1994; Zalipsky, in Stealth Liposomes Chapter 9 (Lasic and Martin, Eds) CRC Press, Boca Raton Fla., 1995). In one approach, a targeting moiety for targeting the lipid nanoparticle is linked to the polar head group of lipids forming the nanoparticle. In another approach, the targeting moiety is attached to the distal ends of the PEG chains forming the hydrophilic polymer coating (see, e.g., Klibanov et al., Journal of Liposome Research 2: 321-334, 1992; Kirpotin et al., FEBS Letters 388: 115-118, 1996).
[0485] Standard methods for coupling the targeting moiety or moieties may be used. For example, phosphatidylethanolamine, which can be activated for attachment of targeting moieties, or derivatized lipophilic compounds, such as lipid-derivatized bleomycin, can be used. Antibody- targeted liposomes can be constructed using, for instance, liposomes that incorporate protein A (see, e.g., Renneisen et al., J. Bio. Chem., 265: 16337-16342, 1990 and Leonetti et al., Proc. Natl. Acad. Sci. (USA), 87:2448-2451, 1990). Other examples of antibody conjugation are disclosed in U.S. Pat. No. 6,027,726. Examples of targeting moieties can also include other polypeptides that are specific to cellular components, including antigens associated with neoplasms or tumors. Polypeptides used as targeting moieties can be attached to the liposomes via covalent bonds (see, for example Heath, Covalent Attachment of Proteins to Liposomes, 149 Methods in Enzymology 111-119 (Academic Press, Inc. 1987)). Other targeting methods include the biotin-avidin system.
[0486] In some embodiments, a lipid nanoparticle includes a targeting moiety that targets the lipid nanoparticle to a cell including, but not limited to, hepatocytes, colon cells, epithelial cells, hematopoietic cells, epithelial cells, endothelial cells, lung cells, bone cells, stem cells, mesenchymal cells, neural cells, cardiac cells, adipocytes, vascular smooth muscle cells, cardiomyocytes, skeletal muscle cells, beta cells, pituitary cells, synovial lining cells, ovarian cells, testicular cells, fibroblasts, B cells, T cells, reticulocytes, leukocytes, granulocytes, and tumor cells (including primary tumor cells and metastatic tumor cells). In particular embodiments, the targeting moiety targets the lipid nanoparticle to a hepatocyte.
[0487] The lipid nanoparticles described herein may be lipidoid-based. The synthesis of lipidoids has been extensively described and formulations containing these compounds are particularly suited for delivery of polynucleotides (see Mahon et al., Bioconjug Chem. 2010 21 : 1448-1454; Schroeder et al., J Intern Med. 2010 267:9-21; Akinc et al., Nat. Biotechnol. 2008 26:561-569; Love et al., Proc Natl Acad Sci USA. 2010 107: 1864-1869; Siegwart et al., Proc Natl Acad Sci USA. 2011 108: 12996-3001).
[0488] The characteristics of optimized lipidoid formulations for intramuscular or subcutaneous routes may vary significantly depending on the target cell type and the ability of formulations to diffuse through the extracellular matrix into the blood stream. While a particle size of less than 150 nm may be desired for effective hepatocyte delivery due to the size of the endothelial fenestrae (see e.g., Akinc et al., Mol Ther. 2009 17:872-879), use of lipidoid oligonucleotides to deliver the formulation to other cells types including, but not limited to, endothelial cells, myeloid cells, and muscle cells may not be similarly size-limited.
[0489] In one aspect, effective delivery to myeloid cells, such as monocytes, lipidoid formulations may have a similar component molar ratio. Different ratios of lipidoids and other components including, but not limited to, a neutral lipid (e.g., diacylphosphatidylcholine), cholesterol, a PEGylated lipid (e.g., PEG-DMPE), and a fatty acid (e.g., an omega-3 fatty acid) may be used to optimize the formulation of the mRNA or system for delivery to different cell types including, but not limited to, hepatocytes, myeloid cells, muscle cells, etc. Exemplary lipidoids include, but are not limited to, DLin-DMA, DLin-K-DMA, DLin-KC2-DMA, 98N12-5, C12-200 (including variants and derivatives), DLin-MC3-DMA and analogs thereof. The use of lipidoid formulations for the localized delivery of nucleic acids to cells (such as, but not limited to, adipose cells and muscle cells) via either subcutaneous or intramuscular delivery, may also not require all of the formulation components which may be required for systemic delivery, and as such may comprise the lipidoid and the mRNA or system.
[0490] According to the present disclosure, a system described herein may be formulated by mixing the mRNA or system, or individual components of the system, with the lipidoid at a set ratio prior to addition to cells. In vivo formulations may require the addition of extra ingredients to facilitate circulation throughout the body. After formation of the particle, a system or individual components of a system is added and allowed to integrate with the complex. The encapsulation efficiency is determined using a standard dye exclusion assays.
[0491] In vivo delivery of systems may be affected by many parameters, including, but not limited to, the formulation composition, nature of particle PEGylation, degree of loading, oligonucleotide to lipid ratio, and biophysical parameters such as particle size (Akinc et al., Mol Ther. 2009 17:872-879; herein incorporated by reference in its entirety). As an example, small changes in the anchor chain length of poly(ethylene glycol) (PEG) lipids may result in significant effects on in vivo efficacy. Formulations with the different lipidoids, including, but not limited to penta[3-(l- laurylaminopropionyl)]-triethylenetetramine hydrochloride (TETA-5LAP; aka 98N12-5, see
Murugaiah et al., Analytical Biochemistry, 401 :61 (2010)), C12-200 (including derivatives and variants), MD1, DLin-DMA, DLin-K-DMA, DLin-KC2-DMA and DLin-MC3-DMA can be tested for in vivo activity. The lipidoid referred to herein as "98N12-5" is disclosed by Akinc et al., Mol Ther. 2009 17:872-879). The lipidoid referred to herein as "C12-200" is disclosed by Love et al., Proc Natl Acad Sci USA. 2010 107: 1864-1869 and Liu and Huang, Molecular Therapy. 2010 669-670.
[0492] The LNPs of the present disclosure, in which a nucleic acid is entrapped within the lipid portion of the particle and is protected from degradation, can be formed by any method known in the art including, but not limited to, a continuous mixing method, a direct dilution process, and an in-line dilution process. Additional techniques and methods suitable for the preparation of the LNPs described herein include coacervation, microemulsions, supercritical fluid technologies, phase-inversion temperature (PIT) techniques.
[0493] In some embodiments, the LNPs used herein are produced via a continuous mixing method, e.g., a process that includes providing an aqueous solution a nucleic acid described herein in a first reservoir, providing an organic lipid solution in a second reservoir (wherein the lipids present in the organic lipid solution are solubilized in an organic solvent, e.g., a lower alkanol such as ethanol), and mixing the aqueous solution with the organic lipid solution such that the organic lipid solution mixes with the aqueous solution so as to substantially instantaneously produce a lipid vesicle (e.g., liposome) encapsulating the nucleic acid molecule within the lipid vesicle. This process and the apparatus for carrying out this process are known in the art. More information in this regard can be found in, for example, U.S. Patent Publication No. 20040142025. The action of continuously introducing lipid and buffer solutions into a mixing environment, such as in a mixing chamber, causes a continuous dilution of the lipid solution with the buffer solution, thereby producing a lipid vesicle substantially instantaneously upon mixing. By mixing the aqueous solution comprising a nucleic acid molecule with the organic lipid solution, the organic lipid solution undergoes a continuous stepwise dilution in the presence of the buffer solution (e.g., aqueous solution) to produce a nucleic acid-lipid particle.
[0494] In some embodiments, the LNPs used herein are produced via a direct dilution process that includes forming a lipid vesicle (e.g., liposome) solution and immediately and directly introducing the lipid vesicle solution into a collection vessel containing a controlled amount of dilution buffer. In some embodiments, the collection vessel includes one or more elements configured to stir the contents of the collection vessel to facilitate dilution. In some embodiments, the amount of dilution
buffer present in the collection vessel is substantially equal to the volume of lipid vesicle solution introduced thereto.
[0495] In some embodiments, the LNPs are produced via an in-line dilution process in which a third reservoir containing dilution buffer is fluidly coupled to a second mixing region. In these embodiments, the lipid vesicle (e.g., liposome) solution formed in a first mixing region is immediately and directly mixed with dilution buffer in the second mixing region. These processes and the apparatuses for carrying out direct dilution and in-line dilution processes are known in the art. More information in this regard can be found in, for example, U.S. Patent Publication No. 20070042031.
5.11. Additional delivery modalities
[0496] This disclosure is not limited to systems and methods described herein. Any delivery method that is capable of delivering the systems described herein can be used as long as it is capable of site-specifically integrating a template polynucleotide into the genome of a cell.
5.12. Genes and Targets
[0497] This disclosure provides compositions, systems and methods for correcting or replacing genes or gene fragments (including introns or exons) or inserting genes in new locations. In certain embodiments, such a method comprises recombination or integration into a safe harbor site (SHS). A frequently used human SHS is the AAVS1 site on chromosome 19q, initially identified as a site for recurrent adeno-associated virus insertion. Another locus comprises the human homolog of the murine Rosa26 locus. Yet another SHS comprises the human Hl 1 locus on chromosome 22. In some cases, a complete gene may be prohibitively large and replacement of an entire gene impractical. In certain embodiments, a method of the disclosure comprises recombining corrective gene fragments into a defective locus.
[0498] The methods and compositions can be used to target, without limitation, stem cells for example induced pluripotent stem cells (iPSCs), HSCs, HSPCs, mesenchymal stem cells, or neuronal stem cells and cells at various stages of differentiation. In certain embodiments, methods and compositions of the disclosure are adapted to target organoids, including patient derived organoids.
[0499] In certain embodiments, methods and compositions of the disclosure are adapted to treat muscle cells, not limited to cardiomyocytes for Duchene Muscular Dystrophy (DMD). The dystrophin gene is the largest gene in the human genome, spanning ~2.3 Mb of DNA. DMD is
composed of 79 exons resulting in a 14-kb full-length mRNA. Common mutations include mutations that disrupt the reading frame of generate a premature stop codon. An aspect of DMD that lends it to gene editing as a therapeutic approach is the modular structure of the dystrophin protein. Redundancy in the central rod domain permits the deletion of internal segments of the gene that may harbor loss-of-function mutations, thereby restoring the open reading frame (ORFs). In some embodiments, the methods and systems described herein are used to treat DMD by site- specifically integrating in the genome a polynucleotide template that repairs or replaces all or a portion of the defective DMD gene.
[0500] The following are non-limiting diseases that may be treated utilizing the methods and compositions of the present disclosure:
Inherited Retinal Diseases:
• Stargardt Disease (ABCA4)
• Leber congenital amaurosis 10 (CEP290)
• X linked Retinitis Pigmentosa (RPGR)
• Autosomal Dominant Retinitis Pigmentosa (RHO)
Liver Diseases:
• Wilson’s disease (ATP7B)
• Alpha-1 antitrypsin (SERPINA1)
Intellectual Disabilities:
• Rett Syndrome (MECP2)
• S YNGAP 1 -ID (S YNGAP 1 )
• CDKL5 deficiency disorder (CDKL5)
Peripheral Neuropathies:
• Charcot-Marie-Tooth 2A (MFN2)
Lung Diseases:
• Cystic Fibrosis (CFTR)
• Alpha-1 Antitrypsin (SERPINA1)
Autoimmune diseases:
• IgA Nephropathy (Berger’s disease)
• Anti-Neutrophil Cytoplasmic Antibody (ANCA) Vasculitis
• Systemic Lupus Erythematosus (SLE) / Lupus Nephritis (LN)
• Membranous Nephropathy (MN)
C3 glomerulonephritis (C3GN)
Blood disorders:
• Sickle Cell
• Hemophilia
• Factor VIII or
• Factor IX
• Ornithine transcarbamylase deficiency (OTCD)
• Homocystinuria (HCU)
• Phenylketonuria (PKU)
Cancer
• Prostate cancer
• Renal cell cancer
• Thyroid cancer
[0501] CFTR (cystic fibrosis transmembrane conductance regulator). The most common cystic fibrosis (CF) mutation F508del removes a single amino acid. In some embodiments, recombining human CFTR into an SHS of a cell that expresses CFTR F508del is a corrective treatment path. In some embodiments, the methods and systems described herein are used to CF by site-specifically integrating in the genome a polynucleotide template that corrects the mutation causing CF. Proposed validation is detection of persistent CFTR mRNA and protein expression in transduced cells.
[0502] Sickle cell disease (SCD) is caused by mutation of a specific amino acid — valine to glutamic acid at amino acid position 6. In some embodiments, SCD is corrected by recombination of the HBB gene into a safe harbor site (SHS) and by demonstrating correction in a proportion of target cells that is high enough to produce a substantial benefit. In some embodiments, the methods and systems described herein are used to sickle cell disease by site-specifically integrating in the genome a polynucleotide template that corrects the mutation causing the disease. In some embodiments, validation is detection of persistent HBB mRNA and protein expression in transduced cells.
[0503] DMD — Duchenne Muscular Dystrophy. The dystrophin gene is the largest gene in the human genome, spanning ~2.3 Mb of DNA. DMD is composed of 79 exons resulting in a 14- kb full-length mRNA. Common mutations include mutations that disrupt the reading frame of
generate a premature stop codon. An aspect of DMD that lends it to gene editing as a therapeutic approach is the modular structure of the dystrophin protein. Redundancy in the central rod domain permits the deletion of internal segments of the gene that may harbor loss-of-function mutations, thereby restoring the open reading frame (ORFs).
[0504] In some embodiments, recombination will be into safe harbor sites (SHS). A frequently used human SHS is the A4FS7 site on chromosome 19q, initially identified as a site for recurrent adeno-associated virus insertion. In some embodiments, the site is the human homolog of the e murine Rosa26 locus (pubmed. ncbi.nlm.nih.gov/18037879). In some embodiments, the site is the human Hl 1 locus on chromosome 22. Proposed target cells for recombination include stem cells for example induced pluripotent stem cells (iPSCs) and cells at various stages of differentiation. In some cases, a complete gene may be prohibitively large and replacement of an entire gene impractical. In such instances, rescuing mutants by recombining in corrected gene fragments with the methods and systems described herein is a corrective option.
[0505] In some embodiments, correcting mutations in exon 44 (or 51) by recombining in a corrective coding sequence downstream of exon 43 (or 50), using the methods and systems described herein is a corrective option. Proposed validation is detection of persistent DMD mRNA and protein expression in transduced cells.
[0506] F8 (Factor VIII). A large proportion of severe hemophilia A patients harbor one of two types of chromosomal inversions in the F VIII gene. The recombinase technology and methods described herein are well suited to correcting such inversions (and other mutations) by recombining of the FVIII gene into a SHS.
[0507] In some embodiments, correcting factor VIII deficiency by recombining the FVIII gene into an SHS is a corrective path. In some embodiments, the methods and systems described herein are used to correct factor VIII deficiency by site-specifically integrating in the genome a polynucleotide template that corrects the mutation causing the FIX deficiency. Proposed validation is detection of persistent FVIII mRNA and protein expression in transduced cells.
[0508] Factor 9 (Factor IX) Hemophilia B, also called factor IX (FIX) deficiency is a genetic disorder caused by missing or defective factor IX, a clotting protein.
[0509] In some embodiments, the methods and systems described herein are used to correct factor IX deficiency by site-specifically integrating in the genome a polynucleotide template that
corrects the mutation causing the FIX deficiency. Proposed validation is detection of persistent FiX mRNA and protein expression in transduced cells.
[0510] Ornithine transcarbamylase deficiency (OTCD). Ornithine transcarbamylase deficiency is a rare genetic condition that causes ammonia to build up in the blood. The condition - more commonly called OTC deficiency - is more common in boys than girls and tends to be more severe when symptoms emerge shortly after birth.
[0511] In some embodiments, the methods and systems described herein are used to correct OTC deficiency by site-specifically integrating in the genome a polynucleotide template that corrects the mutation causing the OTC deficiency or integrates a polynucleotide encoding a functional ornithine transcarbamylase enzyme. Proposed validation is detection of persistent OTC mRNA and protein expression in transduced cells.
[0512] Phenylketonuria, also called PKU, is a rare inherited disorder that causes an amino acid called phenylalanine to build up in the body. PKU is caused by a change in the phenylalanine hydroxylase (PAH) gene. This gene helps create the enzyme needed to break down phenylalanine.
[0513] In some embodiments, the methods and systems described herein are used to correct PKU by site-specifically integrating in the genome a polynucleotide template that corrects the mutation causing the PKU deficiency or integrates a polynucleotide encoding a functional phenylalanine hydroxylase (PAH) gene. Proposed validation is detection of persistent PAH mRNA and protein expression in transduced cells.
[0514] Homocystinuria (HCU). Homocystinuria is elevation of the amino acid, homocysteine (protein building block coming from our diet) in the urine or blood. Common causes of HCU include: problems with the enzyme cystathionine beta synthase (CBS), which converts homocysteine to the amino acid cystathionine (which then becomes cysteine) and needs the vitamin B6 (pyridoxine); and problems with converting homocysteine to the amino acid methionine.
[0515] In some embodiments, the methods and systems described herein are used to correct HCU by site-specifically integrating in the genome a polynucleotide template that corrects the mutation causing the HCU or integrates a polynucleotide encoding a functional copy of a gene (e.g., CBS) able to reduce or prevent buildup of homocysteine in the urine. Proposed validation is detection of persistent CBS mRNA and protein expression in transduced cells.
[0516] IgA Nephropathy (Berger’s disease). IgA nephropathy, also known as Berger's disease, is a kidney/autoimmune disease that occurs when an antibody called immunoglobulin A (IgA) builds up in the kidneys.
[0517] In some embodiments, the methods and systems described herein are used to treat Berger’s disease by administering to a patient an iPSC-derived Natural Killer cell that includes a polynucleotide site-specifically integrated in the genome of the cell using the methods described herein. Upon administering the iPSC-NK cell to the patient, the iPSC-NK cell is capable of removing native cells (e.g., B cells) that are responsible, at least in part, for the symptoms of Berger’s disease.
[0518] Anti-Neutrophil Cytoplasmic Antibody (ANCA) Vasculitis. ANCA vasculitis is an autoimmune disease affecting small blood vessels in the body. It is caused by autoantibodies called ANCAs, or Anti-Neutrophilic Cytoplasmic Autoantibodies. ANCAs target and attack a certain kind of white blood cells called neutrophils.
[0519] In some embodiments, the methods and systems described herein are used to treat ANCA vasculitis by administering to a patient an iPSC-derived Natural Killer cell that includes a polynucleotide site-specifically integrated in the genome of the cell using the methods described herein. Upon administering the iPSC-NK cell to the patient, the iPSC-NK cell is capable of removing native cells (e.g., B cells) that are responsible, at least in part, for the symptoms of ANCA vasculitis.
[0520] Systemic Lupus Erythematosus (SLE) / Lupus Nephritis (LN). Lupus is an autoimmune — a disorder in which the body’s immune system attacks the body’s own cells and organs.
[0521] In some embodiments, the methods and systems described herein are used to treat SLE/LN by administering to a patient an iPSC-derived Natural Killer cell that includes a polynucleotide site-specifically integrated in the genome of the cell using the methods described herein. Upon administering the iPSC-NK cell to the patient, the iPSC-NK cell is capable of removing native cells (e.g., B cells) that are responsible, at least in part, for the symptoms of SLE/LN.
[0522] Membranous Nephropathy (MN). MN is a kidney disease that affects the filters (glomeruli) of the kidney and can cause protein in the urine, as well as decreased kidney function
and swelling. It can sometimes be called membranous glomerulopathy as well (these terms can be used interchangeably and mean the same thing).
[0523] In some embodiments, the methods and systems described herein are used to treat MN by administering to a patient an iPSC-derived Natural Killer cell that includes a polynucleotide site-specifically integrated in the genome of the cell using the methods described herein. Upon administering the iPSC-NK cell to the patient, the iPSC-NK cell is capable of removing native cells (e.g., B cells) that are responsible, at least in part, for the symptoms of MN.
[0524] C3 glomerulonephritis (C3GN). C3 glomerulopathy is a group of related conditions that cause the kidneys to malfunction. The major features of C3 glomerulopathy include high levels of protein in the urine (proteinuria), blood in the urine (hematuria), reduced amounts of urine, low levels of protein in the blood, and swelling in many areas of the body. Affected individuals may have particularly low levels of a protein called complement component 3 (or C3) in the blood.
[0525] In some embodiments, the methods and systems described herein are used to treat C3 glomerulopathy by administering to a patient an iPSC-derived Natural Killer cell that includes a polynucleotide site-specifically integrated in the genome of the cell using the methods described herein. Upon administering the iPSC-NK cell to the patient, the iPSC-NK cell is capable of removing native cells (e.g., B cells) that are responsible, at least in part, for the symptoms of C3 glomerulopathy.
5.13. Methods of treatment
[0526] In another aspect, methods of treatment are presented. The method comprises administering an effective amount of the pharmaceutical composition comprising the nucleic acid construct or vectorized nucleic acid construct described above to a patient in need thereof. In some embodiments, the system (e.g., any of the systems described herein) are delivered to a cell ex vivo and the cell is then administered to the subject. In some embodiments, the systems (e.g., any of the systems described herein) are delivered to a patient, thereby delivering to a cell in vivo.
[0527] DNA or RNA viral vectors can be administered directly to patients (in vivo) or they can be used to treat cells in vitro, and the modified cells may optionally be administered to patients (ex vivo). Conventional viral based systems to be used herein could include retroviral, lentivirus, adenoviral, adeno-associated and herpes simplex virus vectors for gene transfer. Integration in the host genome is possible with the retrovirus, lentivirus, and adeno-associated virus gene transfer
methods, often resulting in long term expression of the inserted transgene. Additionally, high transduction efficiencies have been observed in many different cell types and target tissues.
[0528] In some embodiments, the co-delivery system described herein (e.g., a gene editor construct packaged in a LNP and a donor template packaged in a vector) is administered intravenously. In some embodiments, the co-delivery system described herein (e.g., a gene editor construct packaged in a LNP and a donor template packaged in a vector) is administered intrathecally. In some embodiments, the co-delivery system described herein (e.g., a gene editor construct packaged in a LNP and a donor template packaged in a vector) is administered by intracerebral ventricular injection. In some embodiments, the co-delivery system described herein (e.g., a gene editor construct packaged in a LNP and a donor template packaged in a vector) is administered by intraci sternal magna administration. In some embodiments, the co-delivery system described herein (e.g., a gene editor construct packaged in a LNP and a donor template packaged in a vector) is administered by intravitreal injection.
[0529] Methods of non-viral delivery of the donor DNA template described herein include lipofection, nucleofection, microinjection, biolistics, virosomes, liposomes, immunoliposomes, polycation or lipidmucleic acid conjugates, naked DNA, artificial virions, and agent-enhanced uptake of DNA. Lipofection is described in e.g., U.S. Pat. Nos. 5,049,386, 4,946,787; and 4,897,355) and lipofection reagents are sold commercially (e.g., Transfectam™ andLipofectin™). Cationic and neutral lipids that are suitable for efficient receptor-recognition lipofection of polynucleotides include those of Feigner, WO 91/17424; WO 91/16024. Delivery can be to cells (e.g. in vitro or ex vivo administration) or target tissues (e.g. in vivo administration).
5.13.1.1 mRNA delivery
[0530] Another useful method to deliver proteins, enzymes, and guides comprises transfection of messenger RNA (mRNA). Examples of mRNA delivery methods and compositions that may be utilized in the present disclosure including, for example, PCT/US2014/028330, US8822663B2, NZ700688A, ES2740248T3, EP2755693A4, EP2755986A4, WO2014152940A1, EP3450553B1, BRI 12016030852A2, and EP3362461A1. Expression of CRISPR systems in particular is described by W02020014577. Each of these publications are incorporated herein by reference in their entireties. Additional disclosure hereby incorporated by reference can be found in Kowalski et al., “Delivering the Messenger: Advances in Technologies for Therapeutic mRNA Delivery,” Mol Therap., 2019; 27(4): 710-728.
6. EXAMPLES
6.1. Example 1: Delivery of gene editor polynucleotide sequence packaged in LNP and donor template packaged in AAV
[0531] A gene editor polynucleotide construct is packaged into a LNP (FIG. 1), wherein the gene editor polynucleotide sequence comprises a polynucleotide sequence encoding a prime editor protein linked to an integrase via peptide linker a polynucleotide sequence encoding an attachment site-containing guide RNA (atgRNA); a polynucleotide sequence encoding a nickase guide RNA (ngRNA).
[0532] A donor template polynucleotide construct is packaged in an AAV vector (FIG. 2).
[0533] Co-administration of the gene editor construct packaged LNP and the donor template packaged AAV co-delivers the gene editor construct to a cell cytoplasm and the donor template to a cell nucleus. By use of programmable genome editing to place integrase landing site at a desired location in the genome, the direct activity of the associated integrase to the specific genomic site is guided. Gene editor construct expression, with template co-delivery, results in integration of template “cargo” at a precisely defined target location.
6.2. Example 2: Delivery of gene editor polynucleotide sequence packaged in LNP and donor template capable of self-circularization packaged in AAV
[0534] A gene editor polynucleotide construct is packaged into a LNP (FIG. 1), wherein the gene editor polynucleotide sequence comprises a polynucleotide sequence encoding a prime editor protein linked to an integrase via peptide linker a polynucleotide sequence encoding an attachment site-containing guide RNA (atgRNA); a polynucleotide sequence encoding a nickase guide RNA (ngRNA).
[0535] A donor template polynucleotide construct is packaged in an AAV vector (FIG. 2).
[0536] Co-administration of the gene editor construct packaged LNP and the donor template packaged AAV co-delivers the gene editor construct to a cell cytoplasm and the donor template to a cell nucleus. Integrase-mediated self-circularization of donor template occurs at integration target recognition sites within the AAV genome (FIG. 3). By use of programmable genome editing to place an orthogonal integrase landing site (i.e., distinct att site from att sites used for selfcircularization) at a desired location in the genome, the direct activity of the associated integrase to the specific genomic site is guided. Gene editor construct expression, with template co-delivery
and integrase-mediated circularization of template, results in integration of template “cargo” at a precisely defined target location.
6.3. Example 3: Delivery of gene editor polynucleotide sequence packaged in LNP and atgRNA, ngRNA, and donor template co-packaged in AAV
[0537] A gene editor polynucleotide construct is packaged into a LNP (FIG. 4), wherein the gene editor polynucleotide sequence comprises a polynucleotide sequence encoding a prime editor protein linked to an integrase via peptide linker.
[0538] A polynucleotide sequence encoding an attachment site-containing guide RNA (atgRNA), a polynucleotide sequence encoding a nicking guide RNA (ngRNA), and donor template are packaged in an AAV vector (FIG. 4).
[0539] Co-administration of the gene editor construct packaged LNP and the atgRNA, ngRNA, donor template packaged AAV co-delivers the gene editor construct to a cell. Integrase- mediated self-circul arization of donor template occurs at integration target recognition sites within the AAV genome (FIG. 3). By use of programmable genome editing to place an orthogonal integrase landing site (i.e., distinct att site from att sites used for self-circularization) at a desired location in the genome, the direct activity of the associated integrase to the specific genomic site is guided. Gene editor construct expression, with atgRNA, ngRNA, and template co-delivery and integrase-mediated circularization of template, results in integration of template “cargo” at a precisely defined target location.
6.4. Example 4: Delivery of gene editor polynucleotide sequence and ngRNA packaged in LNP and atgRNA and donor template co-packaged in AAV
[0540] A gene editor polynucleotide construct and a nicking guide RNA (ngRNA) are packaged into a LNP (FIG. 5), wherein the gene editor polynucleotide sequence comprises a polynucleotide sequence encoding a prime editor protein linked to an integrase via peptide linker.
[0541] A polynucleotide sequence encoding an attachment site-containing guide RNA (atgRNA) and donor template are packaged in an AAV vector (FIG. 5).
[0542] Co-administration of the gene editor construct and ngRNA packaged LNP and the atgRNA, donor template packaged AAV co-delivers the gene editor construct to a cell. Integrase- mediated self-circul arization of donor template occurs at integration target recognition sites within the AAV genome (FIG. 3). By use of programmable genome editing to place an orthogonal integrase landing site (i.e., distinct att site from att sites used for self-circularization) at a desired
location in the genome, the direct activity of the associated integrase to the specific genomic site is guided. Gene editor construct expression, with atgRNA, ngRNA, and template co-delivery and integrase-mediated circularization of template, results in integration of template “cargo” at a precisely defined target location.
6.5. Example 5: Intramolecular circularization of plasmid and packaged AAV genomes
[0543] Three self-complementary AAV (scAAV) genomes were designed and generated to verify recombinase/integrase-mediated intramolecular circularization of a DNA cargo from within a linear AAV genome (FIGs. 6A-6B). Circularization of a scAAV genome is mediated by one of Cre, FLPe (thermostable mutant), or Bxbl. Further, the scAAV genomes are comprised of a DNA cargo of interest (“payload”) and an attP site (GT central dinucleotide for circularization orthogonality) for gene insertion into a genome placed attB beacon site. Expected recombinase/integrase-mediated intramolecular circularization products are illustrated in FIG. 7. A universal ddPCR probe capable of binding any linear or circularized AAV genome was designed, wherein the universal ddPCR probe is designed to only give signal upon cognate recombinase/integrase mediated circularization (FIGs. 8A-8B). Circularization products are amplified by use of a circle junction PCR primer set that is designed to amplify only circular products due to primer direction constraints. To confirm Bxbl mediated circularization specifically, an attR scar quencher-fluorophore probe was designed. In addition, a template reference primer set was designed and generated to quantify total template DNA (linear or circular confirmation) (FIGs. 8A-8B).
[0544] Intracellular circularization of either plasmid or packaged AAV genomes were screened in HEK293 cells (35K cells per well) (FIG. 9). Plasmids (25 fmol pDNA = IX or 50 frnol pDNA = 2X) encoding one of Cre, FLPe, or Bxbl were transfected by Lipofectamine 3000. Plasmid genome substrates were transfected at a dose of 1E10 copies per well using Lipofectamine 3000 (FIG. 9). Additionally, AAV genomes were packaged in AAV-DJ capsids and delivered at a dose of 3E5 genomes per cell or 1E10 genomes per well. Circularization ddPCR analysis was conducted three days post transfection.
[0545] FIG. 10 demonstrates circularization of AAV pDNA and packaged AAV genomic DNA for both IX Bxbl and 2X Bxbl conditions (confirmed by use of attR ddPCR primer set). Further, replicates that lacked either Bxbl or AAV pDNA substrate demonstrated insignificant circularization. All three of the Cre-, FLPe-, and Bxbl -targeted AAV pDNA substrates demonstrated circularization upon cognate recombinase/integrase introduction, as confirmed by
using the universal ddPCR probe (FIG. 11). Moreover, Cre-, FLPe-, and Bxbl-mediated circularization of packaged AAV DJ genomes substrates were demonstrated and confirmed using the universal ddPCR probe (FIG. 12).
[0546] As shown in FIG. 13, the Bxbl-mediated attR scar probe provided similar percent circularization quantification compared to the universal probe.
6.6. Example 6: In vitro beacon placement in primary mouse hepatocytes and primary human hepatocytes using mRNA and AAV for co-delivery
[0547] This example assessed the efficiency of in vitro beacon placement in primary human hepatocytes using mRNA delivering of a polynucleotide encoding a gene editor polynucleotide construct and AAV to deliver the first and second atgRNA. See FIG. 15 for a non-limiting example of a dual atgRNA-mediated insertion of an integration recognition site.
[0548] In the mouse experiments, the mRNA and AAV were delivered into the primary mouse hepatocytes (PMH) using (i) concurrent delivery (“co-dose”), (ii) AAV delivery followed by a “1- day delay” before delivery of the mRNA, or (iii) AAV delivery followed by a “2-day delay” before delivery of the mRNA. Beacon placement was then assessed using next-generation sequencing of DNA isolated from cells subjected to the delivery conditions mentioned above. The mRNA encoding the gene editor polynucleotide construct was delivered in various amounts per well: 2000 ng, 1000 ng, 500 ng, 250 ng, 125 ng, 62.5 ng, and 31.25 ng. AAV encoding the first and second atgRNA (see Table 12). The primary mouse hepatocyte data is shown in FIG. 16 and the human primary hepatocyte data is shown in FIG. 17.
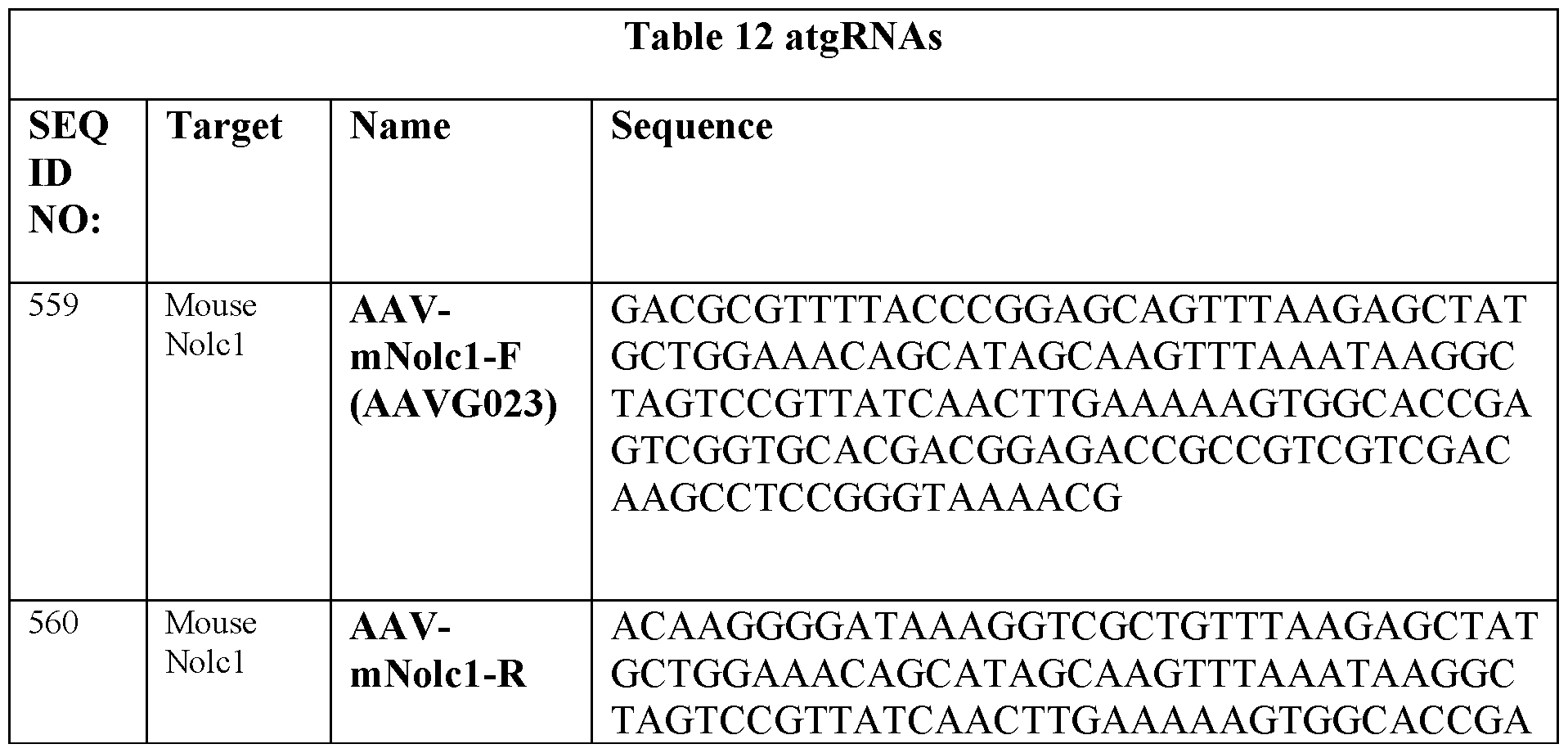

[0549] As shown in FIG. 16, in primary mouse hepatocytes (PMH) delivering the first atgRNAs (SEQ ID NO: 543) and the second atgRNA (SEQ ID NO: 544) using AAV at day 0 and then delivering the mRNA encoding the gene editing polynucleotide construct at day 2 (“2 day delay”) resulted in greater than 10% beacon placement for each amount of mRNA tested. Surprisingly, a 2 day delay resulted in greater beacon placement than either no delay (“co-dose) or a 1 day delay.
[0550] As shown in FIG. 17, in primary human hepatocytes (PHH), using AAV to deliver the first atgRNA (SEQ ID NO: 545) and the second atgRNA (SEQ ID NO: 546) and mRNA to deliver the gene editing polynucleotide construct resulted in about 17% beacon placement.
[0551] Taken together, this data showed robust ex vivo beacon placement in primary mouse and primary human hepatocytes.
6.7. Example 7: In vivo beacon placement with mRNA + AAV guide
[0552] In vivo beacon placement in mice was assessed using AAV to deliver the first and second atgRNAs and mRNA to delivery the gene editing polynucleotide construct.
[0553] In these experiments, mice were administered AAV containing the first atgRNA (SEQ ID NO: 543; Table 12) and the second atgRNA (SEQ ID NO: 544) targeting the Nolcl locus at 3E11 to 1E12 vector genomes (vg) per animal two 2 weeks prior to administration of the mRNA containing the gene editing polynucleotide construct (see FIG. 18). mRNA was delivered using various LNP formulations (e.g., LNP #F1 , LNP #F2, and LNP #F3) at concentrations ranging from 5 mg/kg to 0.5 mg/kg via intravenous injection (see FIG. 18). After delivery of the mRNA, liver tissue was harvested, genomic DNA was isolated, and beacon efficiency was assessed by NGS. As shown in FIG. 18, three conditions resulted in vivo beacon placement efficiency greater than 10%.
[0554] Taken together, this data provided proof-of-concept for successful in vivo beacon placement using AAV to deliver the first and second atgRNA and LNPs to deliver the mRNA encoding the gene editor polynucleotide construct.
6.8. Example 8: In vivo integration in mice using AAV to deliver the template polynucleotide and Adenovirus to deliver BxBl
[0555] In vivo integration efficiency in AttP mice was assessed using adenovirus to deliver an integrase (e.g., Bxbl) and an AAV to deliver the template polynucleotide.
[0556] For these experiments, the adenovirus (i.e., adenovirus containing polynucleotide encoding the integrase) and the AAV (i.e., AAV containing the template polynucleotide and an attB site) were administered to mice containing dual AttP sites integrated in to the Rosa26 locus (B6.RosaBxb-GT/GA; female, Strain# 036152). The Rosa26 locus included a first AttP site comprising a GT dinucleotide and a second AttP site comprising a GA dinucleotide. The AAV was a scAAV8 containing a vector having a template polynucleotide and a 38 bp GT AttB site. The Adenovirus was an adenovirus-type 5 (Ad5) containing a polynucleotide encoding Bxbl (“Bxbl AdV”) (SEQ ID NO: 563; Table 14). Mice were administered the adenovirus and AAV according to the experimental details in Table 13.




[0557] Ten days after administration of the AdV and AAV viruses, liver punches were collected and genomic DNA was isolated. ddPCR of the genomic DNA was used to assess integration efficiency.
[0558] As shown in FIG. 19, administering the AAV and AdV resulted in in vivo integration of the donor polynucleotide template into the AttP mice. In particular, 3E10 vg/animal BxB 1 AdV resulted in about 7% in vivo integration efficiency (see FIG. 19). Administering increased amounts of BxB 1 AdV, 1E11 vg/animal, resulted in higher integration efficiency, about 11%, in AttP mice than with lower amount of 3E10 vg/animal (see FIG. 19).
[0559] Overall, this data establishes proof-of-concept for in vivo integration using an adenovirus to deliver and drive expression of Bxbl and an AAV to deliver the template polynucleotide to be integrated into a mammalian genome, in this case, the mouse genome.
6.9. Example 9: In vivo beacon placement in neonatal mice using split LNP
[0560] In vivo beacon placement was assessed in neonatal mice following administration of a single dose of a mixture of two LNPs. The first LNP contained mRNA encoding a prime editing system and a first synthetic atgRNA (atgRNAl). The mRNA and atgRNAl were included at 1 : 1 ratio in the first LNP. The second LNP contained mRNA encoding a prime editing system and a second synthetic atgRNA (atgRNA2). The mRNA and atgRNA2 were included at a 1 : 1 ratio in the second LNP. Each of the first and second atgRNAs targeted the mouse Nolcl locus and each encoded a portion of an integration recognition site (a “beacon”). AtgRNAl and atgRNA2 together included a 6bp overlap. The first and second LNPs were combined 1 : 1 as mixture prior to administration. The first atgRNA and second atgRNA are provide in Table 15, where the atgRNA include one or more 2’0-methyl modifications and one or more phosphorothioate linkages.
 [0561] The LNP mixture was administered to the neonatal mice (2-5 day old CD-I mice) according to the experimental details in Table 16.
[0561] The LNP mixture was administered to the neonatal mice (2-5 day old CD-I mice) according to the experimental details in Table 16.


[0562] Eight days after administration of the LNP mixture in vivo beacon placement was assessed. In particular, at day 8 post administration, liver samples (either whole liver for groups 1-3 or liver punches from each lobe for groups 4-6 (see Table 13)) were collected and genomic DNA was isolated. Beacon placement was detected using ddPCR and NGS.
[0563] As shown in FIG. 20 A, ddPCR revealed about 1% beacon placement (in Nolcl alleles) following administration of a 3mg/kg dose of the LNP mixture. Confirmation of beacon placement using NGS showed about 7% beacon placement (in Nolcl alleles) following administration of a 3 mg/kg dose of the LNP mixture (see FIG. 20B). In order to determine what percentage of the integrated beacons included the expected integration recognition site (“perfect beacon”), an NGS- based assay was used to make this assessment. As shown in FIG. 20C, about 1% of the integrated beacons contained the expected integration recognition site.
[0564] Neonates were also assessed at six weeks after administration of the LNP mixture. Beacon placement was detected using ddPCR and NGS. As shown in FIG. 21 A., at six weeks post administration, ddPCR revealed about 4% beacon placement (in Nolcl alleles) for a 3mg/kg dose
of the LNP mixture. Confirmation of beacon placement using NGS showed about 15% beacon placement (in Nolcl alleles) for a 3 mg/kg dose of the LNP mixture (see FIG. 2 IB). Assessment of the percent of integrated beacons that included the expected integration recognition site (“perfect beacon”) revealed that about 3.5% of beacons were comprised of perfect beacons (see FIG. 21C).
[0565] Overall, this data demonstrated successful in vivo site-specific integration of an integration recognition site. In particular, this data showed that a split LNP approach can be used for site- specifically integrating an integration recognition site in vivo in a mammalian genome, in this case neonatal mice.
6.10. Example 10: In vivo beacon placement in mice using split LNP
[0566] In vivo beacon placement was assessed in adult mice using a single dose mixture of two LNPs. The first LNP contained mRNA encoding a prime editing system and a first synthetic atgRNA (atgRNAl). The mRNA and atgRNAl were included at different ratios (e.g., 1 :0.5, 1: 1, and 1 :2) ratio in the first LNP. The second LNP contained mRNA encoding a prime editing system and a second synthetic atgRNA (atgRNA2). The mRNA and atgRNA2 were included at different ratios (e.g., 1 :0.5, 1 : 1, and 1 :2) ratio in the second LNP. Here, the first and second atgRNAs targeted mouse Factor IX (“mF9”) locus and each encoded a portion of an integration recognition site (“beacon”). Similar to Example 9, atgRNAl and atgRNA2 together included a 6bp overlap and were combined 1 : 1 as mixture prior to administration. The first atgRNA and second atgRNA are provide in Table 17, where the atgRNA include one or more 2’O-methyl modifications and one or more phosphorothioate linkages.


[0567] In particular, the LNP mixture was administered to female CD-I mice 6-8 weeks old according to the experimental details in Table 18.


*1:0.25:0.25 = mRNA:atgRNAl 1:0.5; mRNA:atgRNA21:0.5; LNPs mixed 1:1
**1:0.5:0.5 = mRNA:atgRNAl 1:1; mRNA:atgRNA21:1; LNPs mixed 1:1
***1:1:1= mRNA:atgRNAl 1:2; mRNA:atgRNA21:2; LNPs mixed 1:1
[0568] Eight days after administration of the LNP mixture in vivo beacon placement was assessed. In particular, at day 8 post administration, liver samples (i.e., liver punches of each lobe (see Table 14)) were collected and genomic DNA was isolated. Beacon placement was detected using ddPCR and NGS.
[0569] As shown in FIG. 22A, ddPCR revealed about 0.8% beacon placement (in mF9 alleles) following administration of a 1 :0.25:0.25 ratio of mRNA:atgRNAl :atgRNA2. Confirmation of beacon placement using NGS showed about 14% beacon placement (in mF9 alleles) following administration of the 1 :0.25:0.25 ratio of mRNA:atgRNAl :atgRNA2 (see FIG. 22B). Similar to Example 9, an NGS-based assay was used to determined what percentages of the integrated beacons included the expected integration recognition site (“perfect beacon”). As shown in FIG. 22C, about 0.02% of the beacons placed in the mF9 locus were “perfect” beacons.
[0570] Overall, this data showed successful in vivo site-specific integration of an integration recognition site in adult mice. In particular, this data showed that the ratio of mRNA to atgRNA is an important consideration in determining efficacy of in vivo site-specific integration of an integration recognition site.
6.11. Example 11: Assessment of Engineered Integration Enzymes
[0571] Engineered integrases described in FIG. 23 (see also Table 26) were evaluated for ability to mediate programmable gene insertion, measured as Integration % (see FIG. 24B) or “Beacon Occupancy” (see FIG. 24C). Percent beacon placement, which precedes integration, is illustrated in FIG. 24 A.
[0572] For these experiments, the engineered integrases were introduced into primary human hepatocytes (PHH) at a high dose (750ng) and a low dose (250ng) along with an mRNA encoding a gene editor polypeptide (i.e., a Cas9 nickase fused to a reverse transcriptase (SEQ ID NO: 589)); an AAV expressing an atgRNA (the atgRNA comprising a spacer sequence with sequence complementarity to a sequence in the human factor IX locus, a scaffold, an RT template comprising an integration recognition site, and a primer binding site (See Table 19 below); and a donor polynucleotide comprising a cognate integration recognition site. In these experiments, the AAV used to deliver the atgRNA also included the donor polynucleotide sequence. The AAV sequence is AAVG048 (SEQ ID NO: 592; See Table 20).








[0573] Genomic DNA was harvested 3 days after transduction. Data is shown in FIG. 24A- 24C
6.12. Example 12: Assessment of Integration Enzymes Encoded by Engineered mRNA
[0574] Engineered integrases described in FIG. 25 (see also Table 26) were evaluated for their ability to mediate programmable gene insertion (as measured by “Occupancy %” (FIG. 26)).
[0575] For these experiments, the engineered integrases were introduced into primary human hepatocytes (PHH) at a high dose (1.3 pmol) and a low dose (0.2 pmol) along with a gene editor polypeptide (i.e., mRNA encoding a gene editor polypeptide (i.e., a Cas9 nickase fused to a reverse transcriptase)); an AAV expressing an atgRNA (the atgRNA comprising a spacer sequence with sequence complementarity to a sequence in the human factor IX locus, a scaffold, an RT template comprising an integration recognition site, and a primer binding site (see Table 19 above for atgRNA sequences); and a donor polynucleotide template comprising a cognate integration recognition site. Similar to Example 11, the AAV used to deliver the atgRNA also included the donor polynucleotide template sequence. The AAV sequence is AAVG048 (SEQ ID NO: 592; see Table 20 above). Genomic DNA was harvested 3 days after transduction. Data is shown in
FIG. 26
6.13. Example 13: Assessment of Tag/Domain placement in Engineered Integration Enzymes on PGI
[0576] Engineered integrases described in FIG. 23 (see also Table 26) comprising were evaluated for their ability to mediate programmable gene insertion (as indicated by “Occupancy %” (FIGs. 27A-27D)). In particular, additional experiments were performed to assess engineered BxBl integrases having a c-terminal tag. In particular, these engineered integrases were assessed for their ability to mediate programmable gene insertion (e.g., as indicated by “% Beacon Occupancy”).
[0577] For these experiments, the engineered integrases were introduced into a primary human hepatocyte (PHH) at amounts of 1000 ng, 500 ng, and 250 ng, along with a gene editor polypeptide (i.e., mRNA encoding a gene editor polypeptide (i.e., a Cas9 nickase fused to a reverse transcriptase)); an atgRNA comprising a spacer sequence with sequence complementarity to a sequence in the human factor IX locus, a scaffold, an RT template comprising an integration recognition site, and a primer binding site (see Table 19 above for atgRNA sequences); and a donor polynucleotide template comprising a cognate integration recognition site at 40 fmol, 20 fmol, and 10 fmol. The donor polynucleotide template was introduced into the cells as an AAV vector (PL 753 (SEQ ID NO: 593)). Genomic DNA was harvested 3 days after transduction.
 [0578] Data is shown in FIGs. 27A-27D. Overall, this data showed that a c-terminal tag reduced integration efficiency of BxBl.
[0578] Data is shown in FIGs. 27A-27D. Overall, this data showed that a c-terminal tag reduced integration efficiency of BxBl.
6.14. Example 14: Assessment of Integration Enzymes Engineered to include Stabilization Domains
[0579] The integration enzymes engineered to include stabilization domains were also assessed for their dose dependent impact on PGI. Additional engineered integration enzymes are described in FIG. 28 (see also Table 26).
[0580] For these experiments, the engineered integrases were introduced into a primary human hepatocyte (PHH) at a 250 ng along with a gene editor polypeptide (i.e., mRNA encoding a gene editor polypeptide (i.e., a Cas9 nickase fused to a reverse transcriptase)); an atgRNA comprising a spacer sequence with sequence complementarity to a sequence in the human factor IX locus, a scaffold, an RT template comprising an integration recognition site, and a primer binding site (see Table 19 above in Example 11 for atgRNA sequences); and a donor polynucleotide template comprising a cognate integration recognition site. Genomic DNA was harvested 3 days after transduction. Data for programmable gene insertion (PGI) is shown in FIGs. 29A-29B. This data showed that an engineered BxBl comprising a stabilon domain resulted in increased programmable gene insertion (PGI) compared to the other BxB 1 polypeptides, including other engineered BxBl polypeptides.
[0581] Additional experiments were performed to assess the engineered integrases introduced into a primary human hepatocyte (PHH) at 500 ng, 62.5 ng, and 7.81 ng along with a gene editor polypeptide (i.e., mRNA encoding a gene editor polypeptide (i.e., a Cas9 nickase fused to a reverse transcriptase)); an atgRNA comprising a spacer sequence with sequence complementarity to a sequence in the human factor IX locus, a scaffold, an RT template comprising an integration recognition site, and a primer binding site (see Table 19 above for atgRNA sequences); and a donor polynucleotide template comprising a cognate integration recognition site. Genomic DNA was harvested 3 days after transduction. Data for programmable gene insertion (PGI) is shown in FIGs.
29C-29D
[0582] Overall, similar to the data in FIG. 29A-29B, the data in FIG. 29C-29D showed that an engineered BxBl comprising a stabilon domain has increased programmable gene insertion (PGI) compared to the other BxBl polypeptides, including other engineered BxBl polypeptides.
6.15. Example 15: Assessment of Engineered Integration Enzymes in Pluripotent Stem Cells
[0583] Engineered integration enzymes were assessed for their ability to mediate PGI in pluripotent stem cells. A non-limiting exemplary workflow is as described in FIG. 30A. Engineered integration enzymes that were used in these experiments are as described in FIG. 30B (see also Table 26).
[0584] For these experiments, the engineered integrases were introduced into a pluripotent stem cell clone 52 or clone 17 along with a BxBl mRNA ; and a donor polynucleotide template comprising a cognate integration recognition site (PL1113 (SEQ ID NO: 594; see Table 22 below)).
[0585] Electroporation conditions -




[0586] Genomic DNA was harvested 3 days after transduction. Data for cell viability is shown in FIG. 30C. Data for programmable gene insertion (PGI) is shown in FIG. 30D. As shown in FIG. 30D shows that PL1323 and PL1325 yield up to 30% PGI in Clone 52 and up to 20% in Clone 17.
[0587] The PGI in pluripotent stem cells was also evaluated at day 6 after transduction. FIG. 31A shows ddPCR data for PGI for clone 52 at day 3 and day 6. Flow cytometry was used to assess GFP expression which indicates programmable gene insertion (PGI) (see FIG. 31B). This data shows that PL1323 and PL1325 yield % GFP+ cells by flow -50%.
6.16. Example 16: Assessment of Engineered Integration Enzyme in Hematopoietic Stem Cells
[0588] Engineered integrases were also assessed for their ability to mediate programmable gene insertion in hematopoietic stem cells.
[0589] For these experiments, the engineered integrases were introduced into a hematopoietic stem cell along with a gene editor polypeptide (i.e., mRNA encoding a gene editor polypeptide (i.e., a Cas9 nickase fused to a reverse transcriptase)); an atgRNA comprising a spacer sequence a scaffold, an RT template comprising an integration recognition site, and a primer binding site; and a donor polynucleotide template comprising a cognate integration recognition site. AtgRNA sequences used in this Example are provided in Table 23 below.
 Table 23.
Table 23.


[0590] In these experiments, a dual electroporation approach was used. In one experimental condition, the engineered integrase, the gene editor polypeptide (i.e., mRNA encoding a gene editor polypeptide), and the atgRNA were introduced into a cell using a first electroporation and the donor polynucleotide template was introduced into the cell using a second electroporation. In a second experimental condition, the gene editor polypeptide (i.e., mRNA encoding a gene editor polypeptide), and the atgRNA were introduced into a cell using a first electroporation and the engineered integrase and the donor polynucleotide template (mini circle) were introduced into the cell using a second electroporation. Engineered integrases used in these experiments are as shown in FIG. 32A
[0591] Genomic DNA was harvested 3 days after transduction. Data for programmable gene insertion (PGI) is shown in FIG. 32B. This data shows that a BxB 1 integrase engineered to include a stabilon motif improves PGI by 3-5 fold.
6.17. Example 17: Assessment of Additional Engineered Integration Enzymes
[0592] Additional iterations of engineered integration enzymes were tested for PGI (measured as beacon occupancy). In particular, BxBl was engineered to include a stabilon domain or an Exin21 domain (see, e.g., engineered integration enzymes shown in FIG. 33 (see also Table 26)).
[0593] For these experiments, the engineered integration enzymes described in FIG. 33 were introduced into a HEK293 cell along with a gene editor polypeptide (i.e., mRNA encoding a gene editor polypeptide (i.e., a Cas9 nickase fused to a reverse transcriptase)); an atgRNA comprising a spacer sequence with sequence complementarity to a sequence in the human factor IX locus, a scaffold, an RT template comprising an integration recognition site, and a primer binding site (see Table 19 above for atgRNA sequences); and a donor polynucleotide template comprising a cognate integration recognition site. The data is shown in FIG. 34. The sub-set of engineered BxBl integrases shown in FIG. 35 (see also Table 26) were selected for further study.
6.18. Example 18: Assessment of Engineered Integration Enzymes Comprising an Amino Acid Modification in a Degron Motif
[0594] BxBl integrases were engineered to include an amino acid modification with the aim that the modification would improve stability (e.g., half-life). FIG. 36A shows degron motifs and lysine residues that are candidate for amino acid modifications that may increase the stability of a BxBl integrase.
[0595] In these experiments, a BxBl was engineered to substitute a lysine for an arginine at position 10. Half-life of the integrase was assessed 24 and 48 hours after transduction into the cell using western blot (see FIG. 36B). This data showed that a specific lysine to arginine substitution (K10R) did not alter the stability of BxBl. Further assessment is ongoing to determine whether substituting another lysine residue (see, e.g., FIG. 36A) would be sufficient to increase the stability of the BxB 1 integrase.
6.19. Example 19: Assessment of mRNA Optimization to Increase Stability of mRNA encoding BxBl
[0596] In another set of experiments, mRNAs encoding BxBl were optimized with the aim of improving stability (i.e., half-life of the mRNA) upon being introduced into a cell.
[0597] In these experiments, the mRNA encoding BxBl was optimized using the LinearDesign algorithm described in Zhang et al. (Algorithm for Optimized mRNA Design Improves Stability and Immunogenicity, Nature 2023, doi.org/10.1038/s41586-023-06127-z), which is hereby
incorporated by reference in its entirety. LinearDesign optimizes both structural stability via second structure and codon usage.
[0598] FIGs. 37A-37B show first generations attempts at optimizing BxB 1 RNA structure using the linear design algorithm comparing non-optimized mRNA encoding BxB 1 in FIG. 37A with optimized mRNA encoding BxBl in FIG. 37B.
[0599] FIGs. 38A-38B show first generations attempts at optimizing an RNA structure for RNA encoding nCas9-RT using the LinearDesign algorithm comparing non-optimized mRNA encoding nCas9-RT in FIG. 38A with optimized mRNA encoding nCsa9-RT in FIG. 38B.
[0600] The nCas9-RTs in FIGs. 38A-38B, were tested for their ability to insert integration recognition sites into a genome at the human factor IX locus. In particular, the nCas9-RTs in FIGs. 38A-38B were introduced into a cell along with an atgRNA comprising a spacer sequence with sequence complementarity to a sequence in the human factor IX locus, a scaffold, an RT template comprising an integration recognition site, and a primer binding site (see Table 19 above in Example 11 for atgRNA sequences). Beacon placement data is shown in FIG. 39. The expectation was that the LinearDesign codon optimized mRNA would have a longer half-life due in at least in part to the second structure - predominately folded, which should lead to higher beacon placement. However, the linear design approach and the algorithm predicted codons didn’t perform as anticipated.
6.20. Example 20: Assessment of Fusion Proteins Comprising Engineered Integration Enzymes and a Gene Editor Polypeptide
[0601] Fusion proteins comprising an nCas9-RT are fused with engineered integration enzymes (e.g., BxBl) and tested for their ability to mediate PGI.
[0602] For these experiments, fusion proteins included those as shown in FIG. 40 (see also Table 26). These fusions proteins are introduced into cells along with an atgRNA and a donor polynucleotide template. Genomic DNA is harvested three days after transduction.
6.21. Example 21: Assessment of Split Fusion Proteins
[0603] Fusion proteins comprising a nCas9 and RT are fused with engineered integration enzymes (e.g., BxBl) in various combinations and tested for their ability to mediate PGI.
[0604] For these experiments, fusion proteins included those as shown in FIG. 41 (see also Table 26). These fusions proteins are introduced into cells along with an atgRNA and a donor polynucleotide template. Genomic DNA is harvested three days after transduction.
6.22. Example 22: Additional Assessment of Engineered Integration Enzyme in Induced Pluripotent Stem Cells
[0605] Engineered integration enzymes described and designed herein, including those in FIG. 42 (see also Table 26), were assessed for their ability to mediate PGI in pluripotent stem cells. A non-limiting exemplary workflow is as described in FIG. 30A.
[0606] For these experiments, the engineered integrases were introduced into a pluripotent stem cell clone 52 or clone 17 along and a donor polynucleotide template comprising a cognate integration recognition site (PL1113 (SEQ ID NO: 591; see Table 22) or PL2134. iPSC clones 52 and 17 include beacons site-specifically integrated into their genomes.
[0607] Electroporation conditions -

[0608] Genomic DNA was harvested 3 days after transduction. Digital droplet PCR (ddPCR) data for programmable gene insertion (PGI) with PL1113 donor at day 3 is shown in FIG. 43 for the indicated engineered BxBl polypeptides. Flow cytometry data for PGI with PL1113 donor at day 7 is shown in FIGs. 44A and 44B. FIG. 44A provides a comparison between percent PGI between ddPCR data and flow cytometry data. Flow cytometry was used to assess GFP expression, which indicates programmable gene insertion (PGI). As shown in FIG. 45, PGI with
PL2134 donor was also assessed and compared to PGI with PL1113. This data shows that PGI was higher with PL1113 than PL2134.
6.23. Example 23: Assessment of Engineered Integration Enzymes Fused to a Gene Editor Polypeptide
[0609] Engineered integration enzymes fused to a gene editor polypeptide as described and designed herein, including those in FIG. 46 (see also Table 26), were assessed for their ability to mediate PGI. The engineered integration enzymes fused to a gene editor polypeptide are referred to herein as the all-in-one constructs.
[0610] In a first set of experiments, mRNA encoding the engineered integrases from FIG. 46 were introduced into a primary human hepatocytes (line HU8403) along with a pair of atgRNA (AA089: atgRNAl is SEQ ID NO: 611 and atgRNA2 is SEQ ID NO: 612 (see Table 24)). Electroporation was performed as described elsewhere herein (see Example 22). mRNA was electroporated at either 187 fmol or 374 fmol. Controls included mRNA encoding nCas9-RT with and without a stabilization domain and a mRNA encoding RT-IRES-nCas9.


[0611] Genomic DNA was harvested 4 days after electroporation. Digital droplet PCR (ddPCR) was used to assess beacon placement for each of the constructs tested (see FIG. 46). As shown in FIG. 47, ddPCR data for beacon placement revealed for the all-in-one mRNAs were slightly lower than the control (PL883) but were comparable. Notably, the nCas9-RT-Bxbl orientation produced more efficient beacon placement compared to the Bxbl-nCas9-RT orientation (see FIG. 47).
[0612] In a second set of experiments, mRNA encoding PL1931 ((SEQ ID NO: 631 (amino acid) and SEQ ID NO: 632 (nucleic acid); see FIG. 46) was introduced into a primary human hepatocyte (line HU8403) along with a pair of atgRNA (AA115 (atgRNAl (SEQ ID NO: 613) and atgRNA2 (SEQ ID NO: 614); see Table below) using MessengerMax lipofection. At day 0, 33,000 HU8403 cells were plated per well and MessengerMax, 0.15 pl atgRNA pair, and either 0.15 l or 0.3 pl of various concentrations of Bxbl mRNA (see FIG. 48). PL883 (SEQ ID NO: 617) was used as a control. Genomic DNA was harvested 4 days after lipofection. Digital droplet PCR (ddPCR) was used to assess beacon placement. As shown in FIG. 48, beacon placement was similar between mRNA PL1931 and mRNA PL883 at equimolar input.


[0613] In a third set of experiments, PGI in PHH was compared following lipofection with an mRNA encoding PL1931 (“all-in-one”) (SEQ ID NO: 631 (amino acid) and SEQ ID NO: 632 (nucleic acid)); see FIG. 46) or two mRNAs one encoding nCas9-RT and a second encoding BxB 1. For these experiments, mRNA was introduced into PHH line HU8403 along with a pair of atgRNA (AA115) using MessengerMax lipofection. In particular, for the PL1931 all-in-one, 0.15 pl of atgRNA pair, and either 0.15 pl or 0.3 pl of various concentrations of Bxbl mRNA were transduced into the PHH (see FIG. 49 for concentrations of BxB 1). For the two mRNA condition (i.e., separate mRNA encoding nCas9-RT and BxBl), BxBl was introduced at 210 fmol (or -125 ng) (note specific constructs included: PL1323 (cmyc-NLS-no stabilon), PL1325 (SV40-NLS- Stabilon), and PL 1709 (SV40-exin21 -stabilon). For some conditions, a 3 lipoplex approach was used: guides 0.15pl per well (20 mM) with 0.15 pl MessengerMax (or 30 pl MessengerMax);
BxBl mRNA at 250 ng with 0.15 pl MessengerMax (or 30 pl MessengerMax) per well; and nCas9- RT mRNA PL883 (SEQ ID NO: 617) fixed at 400 ng or 187 fmol with 0.15 pl MesserngerMax (or 30 pl MessengerMax) per well. Genomic DNA was harvested 4 days after lipofection. Digital droplet PCR (ddPCR) was used to assess PGI. As shown in FIG. 49, PGI was around 1% for each of the conditions tested. Looking closer at each PGI event, FIG. 50 and FIG. 51 show data for total edits (AttB + AttL). Analysis of whether MessengerMax volume impacted beacon placement revealed that increased MessengerMax volume decreased beacon placement but increased integration (FIG. 52). Analysis of the 3 lioplex approach in FIG. 53, revealed a 20-30% hit to beacon placement ratio observed with integration samples (3 lipo) compared to beacon only samples (2 lipo). This data shows that mRNAs encoding a BxBl-stabilon result in increased beacon occupancy compared to mRNA encoding BxBl without a stabilon (see PL1323 (SEQ ID NO: 618 (amino acid) and SEQ ID NO: 619 (nucleic acid)).
[0614] Taken together, these experiments show that PL 1931 all-in-one achieves full PGI in PHH; modulating MessengerMax volume decreases overall beacon placement but does increase integration and subsequent occupancy with the 2 lipoplex approach; and integration efficiency is similar from one shot transfections of all-in-one mRNA PL 1931 (2 lipoplex) versus the two mRNA (separate nCas9-RT and BxBl).
6.24. Example 24: Assessment of Additional Engineered Integration Enzymes II
[0615] Additional iterations of engineered integration enzyme constructs were evaluated for ability to mediate programmable gene insertion (PGI) as measured by percent integration which can also be as percent beacon conversion. In particular, BxB 1 was engineered to include a stabilon peptide motif and/or an Exin21 peptide motif. See FIGs. 54, 57, 59, 61, and 63, see also Table 27.
[0616] The engineered integration enzyme constructs described in FIG. 54, 61, and 63 (See also Table 26 and 27) were introduced into HEK293 mediating integration of a donor polynucleotide (cargo) having an integration site cognate to the integration site in the target genome. Recombination site attL was produced and percent integration was measured by ddPCR. The data is provided in FIG. 55 and FIG. 64. The results showed that various engineered bxbl integrases showed significant improved percent integration as compared to others. Percent integration was measured by ddPCR.
[0617] To assess PGI as measured by beacon conversion, first, beacon placement was performed in neonatal mice. Beacon placement was conducted using methods such as those already described in this text. For example, see Example 9. Next, we performed integration of cargo into the genome
of the cell using methods previously described herein this document. The results showed that various Bxbl integrases displayed significant beacon conversion compared to others. See FIG. 56. Beacon conversion was measured using ddPCR.
[0618] Additionally, PGI as measured by beacon conversion mediated by the engineered integration enzymes was assessed in primary human hepatocytes (PHH, line HU8403). Data is shown in FIG. 58 and FIG. 60. See also Table 26 and 27. Results showed that various engineered BxBl constructs resulted in increased integration of the donor polynucleotide compared to the other engineered BxB 1 polypeptides.
[0619] In vivo integration efficiency in mice mediated by the engineered integrases was assessed using adenovirus to deliver the engineered integrases (see Table 26 and 27), and an AAV to deliver the donor polynucleotide. Saline was used as control. For these experiments methods previously recited in this text were employed, i.e., see Example 8. FIG. 62A shows percent integration resulting in recombination site attL. FIG. 62B shows percent integration resulting in recombination site attR.
7. SEQUENCES






























































































































[0620] All references cited herein are incorporated by reference to the same extent as if each individual publication, database entry (e.g. Genbank sequences or GenelD entries), patent application, or patent, was specifically and individually indicated incorporated by reference in its entirety, for all purposes. This statement of incorporation by reference is intended by Applicants, pursuant to 37 C.F.R. §1.57(b)(1), to relate to each and every individual publication, database entry (e.g. Genbank sequences or GenelD entries), patent application, or patent, each of which is clearly identified in compliance with 37 C.F.R. § 1.57(b)(2), even if such citation is not immediately adjacent to a dedicated statement of incorporation by reference. The inclusion of dedicated statements of incorporation by reference, if any, within the specification does not in any way weaken this general statement of incorporation by reference. Citation of the references herein is not intended as an admission that the reference is pertinent prior art, nor does it constitute any admission as to the contents or date of these publications or documents.
[0621] It is an object of the invention not to encompass within the invention any previously known product, process of making the product, or method of using the product such that Applicant reserves the right and hereby disclose a disclaimer of any previously known product, process, or method. It is further noted that the invention does not intend to encompass within the scope of the invention any product, process, or making of the product or method of using the product, which does not meet the written description and enablement requirements of the USPTO (35 U.S.C. 112(a)) or the EPO (Article 83 of the EPC), such that Applicant reserves the right and hereby disclose a disclaimer of any previously described product, process of making the product, or method of using the product. It may be advantageous in the practice of the invention to be in compliance with Art. 53(c) EPC and Rule 28(b) and (c) EPC. Nothing herein is to be construed as a promise. It is noted that in this disclosure and particularly in the claims and/or paragraphs, terms such as "comprises", "comprised", "comprising" and the like can have the meaning attributed to it in U.S. Patent law; e.g., they can mean "includes", "included", "including", and the like; and that terms such as "consisting essentially of' and "consists essentially of' have the meaning ascribed to them in U.S. Patent law, e.g., they allow for elements not explicitly recited, but exclude elements that are found in the prior art or that affect a basic or novel characteristic of the invention.
[0622] While the invention has been particularly shown and described with reference to a preferred embodiment and various alternate embodiments, it is understood by persons skilled in the relevant art that various changes in form and details can be made therein without departing from the spirit and scope of the invention.
Claims
1. An engineered integration enzyme, comprising:
(a) an integration enzyme or functional fragment thereof;
(b) an at least first stabilization domain; and
(c) a nuclear localization signal (NLS).
2. The engineered integration enzyme of claim 1, wherein the integration enzyme or fragment thereof comprises integration, recombinase, or transposase activity.
3. The engineered integration enzyme of claim 1 or 2, wherein the integration enzyme is a large serine integrase.
4. The engineered integration enzyme of any one of claims 1-3, wherein the integration enzyme or functional fragment thereof is selected from: a BxBl polypeptide, SsuINT, SssINT, SscINT, Ssc2INT, SsdINT, SmcINT, UhmINT, SacINT, RsaINT, Rsa2INT, Bxbl, Tp9INT, BtlINT, BceINT, BcyINT, and SluINT, or variants thereof.
5. The engineered integration enzyme of claim 4, wherein the integration enzyme or fragment thereof is a BxBl polypeptide.
6. The engineered integration enzyme of any one of claims 1-3, wherein the integration enzyme or fragment thereof comprises an amino acid sequence that is at least 80% identical to an amino acid sequence selected from SEQ ID NOs: 378-393.
7. The engineered integration enzyme of claim 6, wherein the integration enzyme is a BxBl polypeptide or fragment thereof comprising an amino acid sequence having at least 80% sequence identity to a sequence of SEQ ID NO: 388.
8. The engineered integration enzyme of claim 7, wherein the BxBl polypeptide has the amino acid sequence of SEQ ID NO: 388.
9. The engineered integration enzyme of any one of claims 1-8, wherein the engineered integration enzyme is capable of binding to a cognate pair of integration recognition sites.
393 38909/55808/FW/18396397.1
10. The engineered integration enzyme of claim 9, wherein the cognate pair of integration recognition sites is selected from: (i) an attB site and an attP site and (ii) a modified AttB site and a modified AttP site.
11. The engineered integration enzyme of claim 9, wherein at least one of the integration recognition sites of the cognate pair is integrated into a mammalian cell genome at a target DNA sequence by an attachment site containing guide RNA (atgRNA) and a gene editor polypeptide.
12. The engineered integration enzyme of any one of claims 1-11, further comprising a second stabilization domain.
13. The engineered integration enzyme of any one of claims 1-12, wherein the first stabilization domain and/or second stabilization domain are positioned in the engineered integration enzyme such that the engineered integration enzyme and the first and/or second stabilization domains are an in-frame fusion.
14. The engineered integration enzyme of claim 13, wherein the first stabilization domain and/or second stabilization domain are located N-terminus to the integration enzyme or fragment thereof, C-terminus to the integration enzyme of fragment thereof, or between two consecutive amino acid residues in the amino acid sequence of the integration enzyme or fragment thereof.
15. The engineered integration enzyme of claim 14, wherein the first stabilization domain and/or second stabilization domain are located N-terminus to the integration enzyme or fragment thereof.
16. The engineered integration enzyme of claim 14, wherein the first stabilization domain and/or second stabilization domain are located C-terminus to the integration enzyme or fragment thereof.
17. The engineered integration enzyme of claim 14, wherein the first stabilization domain and/or second stabilization domain are located between two consecutive amino acid residues in the amino acid sequence of the integration enzyme or fragment thereof.
18. The engineered integration enzyme of claim 17, wherein the two consecutive amino acid residues in the amino acid sequence of the integration enzyme are located in a N- terminal catalytic domain, a recombinase domain, or one or more zinc ribbon domain.
19. The engineered integration enzyme of claim 18, wherein the two consecutive amino acid residues in the amino acid sequence of the integration enzyme correspond amino acid residues between the N-terminal catalytic domain and the recombinase domain or between the recombinase domain and one of the zinc ribbon domains.
20. The engineered integration enzyme of any one of claims 1-19, wherein the stabilization domain is selected from: a stabilon motif and an exin21 motif.
21. The engineered integration enzyme of claim 20, wherein the stabilization domain is a stabilon motif having at least 80% sequence identity to KDKDKKSDGKDSQKK (SEQ ID NO: 583 or KDKKSDGKDSQKK (SEQ ID NO: 584).
22. The engineered integration enzyme of claim 20, wherein the stabilization domain is an exin21 motif having the sequence QPRFAAA (SEQ ID NO: 585) or QPRFAAA (SEQ ID NO: 586), optionally with one, two or three amino acid substitutions.
23. The engineered integration enzyme of any one of claims 12-21, wherein the second stabilization domain is selected from: a stabilon motif and an exin21 motif.
24. The engineered integration enzyme of any one of claims 1-23, further comprising one or more additional stabilization domains.
25. The engineered integration enzyme of claim 24, wherein the one or more additional stabilization domains is selected from: a stabilon motif and an exin21 motif.
26. The engineered integration enzyme of any one of claims 1-25, further comprising at least a first tag.
27. The engineered integration enzyme of claim 26, further comprising a second tag.
28. The engineered integration enzyme of claim 27, wherein the first tag and/or the second tag are positioned in the engineered integration enzyme such that the engineered integration enzyme and the first and/or second tag are an in-frame fusion.
29. The engineered integration enzyme of any one of claims 26-28, wherein the first tag and/or second tag are selected from: an HA tag, a HiBit tag, a strep tag, and a SUMO tag.
30. The engineered integration enzyme of any one of claims 1-29, further comprising one or more linkers.
31. The engineered integration enzyme of claim 30, wherein the one or more linkers is a glycine serine linker independently selected from: GS, GSG, or GGGS.
32. The engineered integration enzyme of any one of claims 1-31, wherein the engineered integration enzyme comprises, from N-terminus to C-terminus:
N-I-S;
I-S-N;
S-I-N;
N-S-I;
N-Xi-I-S;
I-Xi-S-N;
S-Xi-I-N;
N-Xi-S-I;
N-I-Xi-S;
I-S-Xi-N;
S-I-Xi-N;
N-S-Xi-I;
N-X1-I-X2-S;
I-X1-S-X2-N;
S-X1-I-X2-N; and
N-X1-S-X2-I; wherein
I is the integration enzyme or fragment thereof,
Xi is a first linker,
S is the stabilization domain, X2 is a second linker, and N is the NLS.
33. An engineered integration enzyme, comprising:
(a) an integration enzyme or functional fragment thereof comprising at least a first amino acid modification that increases the stability of the integration enzyme as compared to the same integration enzyme or functional fragment thereof not comprising the at least first amino acid modification; and
(b) a nuclear localization signal (NLS).
34. The engineered integration enzyme of claim 33, wherein the first amino acid modification is located in a degron motif in the integration enzyme.
35. The engineered integration enzyme of claim 34, wherein the first amino acid modification is an amino acid substitution, insertion, deletion, or a combination thereof.
36. The engineered integration enzyme of claim 34, wherein the first amino acid modification is an amino acid substitution of L275V in SEQ ID NO: 388.
37. The engineered integration enzyme of any one of claims 1-36, wherein the engineered integration enzyme is linked to a gene editor polypeptide.
38. The engineered integration enzyme of claim 37, wherein the C-terminus of the engineered integration enzyme is linked to the gene editor polypeptide.
39. The engineered integration enzyme of claim 37, wherein the C-terminus of the gene editor polypeptide is linked to the engineered integration enzyme.
40. The engineered integration enzyme of any one of claims 37-39, wherein the engineered integration enzyme is linked to the gene editor polypeptide by in-frame fusion.
41. The engineered integration enzyme of any one of claims 37-39, wherein the engineered integration enzyme is linked to the gene editor polypeptide by a linker.
42. The engineered integration enzyme of claim 41, wherein the linker is a peptide fused in-frame between the engineered integration enzyme and the gene editor polypeptide.
43. The engineered integration enzyme of claim 41 or 42, wherein the one or more linkers is selected from Table 3.
44. The engineered integration enzyme of any one of claims 37-43, wherein the gene editor polypeptide comprises a DNA binding domain and a reverse transcriptase domain.
45. A polynucleotide comprising a nucleic acid sequence encoding the engineered integration enzyme of any one of claims 1-36 or the engineered integration enzyme linked to the gene editor polypeptide of any one of claims 37-44.
46. The polynucleotide of claim 45, wherein the nucleic acid sequence encoding the engineered integration enzyme or the engineered integration enzyme linked to the gene editor polypeptide is codon optimized.
47. The polynucleotide of claim 46, wherein the codon optimization is performed using an algorithm.
48. The polynucleotide of any one of claims 45-47, wherein the nucleic acid sequence of the engineered integration enzyme is optimized via its secondary structure such that the structure confers increased stability on the polynucleotide.
49. The polynucleotide of claim 48, wherein the nucleic acid sequence of the engineered integration enzyme is optimized based on a linear design algorithm.
50. A vector comprising the polynucleotide of any one of claims 45-49.
51. A host cell comprising the vector of claim 50.
52. A fusion protein, comprising
(a) a DNA binding domain, optionally comprising a nickase activity;
(b) a reverse transcriptase; and
(c) an engineered integration enzyme of any one of claims 1-36, wherein at least any two of elements (a), (b), or (c) are linked via an in-frame linker.
53. The fusion protein of claim 52, wherein the linker is a linker selected from Table 3.
54. A polynucleotide comprising a nucleic acid sequence encoding the fusion protein of claim 52 or 53.
55. A vector comprising the polynucleotide of claim 54.
56. A host cell comprising the vector of claim 55.
57. A system for site-specifically integrating a donor polynucleotide into a mammalian cell genome at a target DNA sequence, comprising: at least a first attachment site-containing guide RNA (atgRNA) comprising at least a portion of an at least first integration recognition site; a gene editor polypeptide comprising a DNA binding nickase domain linked to a reverse transcriptase domain capable of incorporating the integration recognition site into the target DNA sequence, an engineered integration enzyme of any one of claims 1-44; and a donor polynucleotide linked to a sequence that is an integration cognate of the integration recognition site present in the atgRNA, whereby the gene editor polypeptide site-specifically integrates the integration recognition site into the target DNA sequence, whereby the engineered integration enzyme integrates the donor polynucleotide into the target DNA sequence.
58. The system of claim 57, wherein the first atgRNA comprises:
(i) a domain that is capable of guiding the gene editor polypeptide to the target DNA sequence; and
(ii) a reverse transcriptase (RT) template that comprises at least a portion of an at least first integration recognition site, whereby the at least portion of the at least first integration recognition site is integrated into the genome of the cell at the target sequence.
59. The system of claim 57 or 58, further comprising a second atgRNA.
60. The system of claim 59, wherein the first atgRNA and the second atgRNA are an at least first pair of atgRNAs, wherein the at least first pair of atgRNAs have domains that are capable of guiding the gene editor polypeptide to the target DNA sequence; the first atgRNA further includes a first RT template that comprises at least a portion of an at least first integration recognition site; the second atgRNA further includes a second RT template that comprises at least a portion of the first integration recognition site, and the first atgRNA and the second atgRNAs collectively encode the entirety of the first integration recognition site, whereby the least first integration
recognition site is integrated into the genome of the cell at the target sequence.
61. The system of any one of claims 57-60, wherein, upon introducing the system into the cell, the engineered integration enzyme enhances integration efficiency of the donor polynucleotide template at the site-specifically integrated integration recognition site as compared to the integration efficiency of a system using a non-engineered integration enzyme to integrate donor polynucleotide template at the site-specifically integrated integration recognition site.
62. A method for site-specifically integrating a donor polynucleotide template into a mammalian cell genome at a target DNA sequence, comprising: incorporating an integration recognition site into the genome by delivering into the cell: an attachment site containing guide RNA (atgRNA) comprising at least a portion of an at least first integration recognition site; and a gene editor polypeptide or polynucleotide encoding a gene editor polypeptide, wherein the gene editor polypeptide comprises a DNA binding nickase domain linked to a reverse transcriptase domain and is capable of incorporating the integration recognition site into the target DNA sequence; and optionally, a nicking gRNA; and integrating the donor polynucleotide template into the genome by delivering into the cell: an engineered integration enzyme of any one of claims 1-44; and a donor polynucleotide, wherein the donor polynucleotide is linked to a sequence that is an integration cognate of the integration recognition site present in the atgRNA, and wherein the donor polynucleotide template is integrated into the genome at the incorporated genomic integration recognition site by the integration enzyme.
63. The method of claim 61, wherein the atgRNA, the gene editor polypeptide or polynucleotide encoding the gene editor polypeptide, the optional nicking gRNA, the engineered integration enzyme, and the donor polynucleotide template are introduced into the cell concurrently.
64. The method of claim 62, wherein the first atgRNA comprises:
(i) a domain that is capable of guiding the gene editor polypeptide to the target DNA sequence; and
(ii) a reverse transcriptase (RT) template that comprises at least a portion of an at least first integration recognition site, whereby the at least portion of the at least first integration recognition site is integrated into the genome of the cell at the target sequence.
65. The method of any one of claims 62-64, further comprising a second atgRNA.
66. The method of claim 65, wherein the first atgRNA and the second atgRNA are an at least first pair of atgRNAs, wherein the at least first pair of atgRNAs have domains that are capable of guiding the gene editor polypeptide to the target DNA sequence; the first atgRNA further includes a first RT template that comprises at least a portion of an at least first integration recognition site; the second atgRNA further includes a second RT template that comprises at least a portion of the first integration recognition site, and the first atgRNA and the second atgRNAs collectively encode the entirety of the first integration recognition site, whereby the least first integration recognition site is integrated into the genome of the cell at the target sequence.
67. The method of any one of claims 62-66, wherein the method enhances integration efficiency of the donor polynucleotide template at the site-specifically integrated integration recognition stie at the double-stranded target DNA sequence as compared to the integration efficiency of the donor polynucleotide template at the site-specifically integrated recognition site when using a method that does not comprise the engineered integration enzyme.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US202363580293P | 2023-09-01 | 2023-09-01 | |
| US63/580,293 | 2023-09-01 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2025050069A1 true WO2025050069A1 (en) | 2025-03-06 |
Family
ID=92925802
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2024/044901 Pending WO2025050069A1 (en) | 2023-09-01 | 2024-08-30 | Programmable gene insertion using engineered integration enzymes |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2025050069A1 (en) |
Citations (105)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4186183A (en) | 1978-03-29 | 1980-01-29 | The United States Of America As Represented By The Secretary Of The Army | Liposome carriers in chemotherapy of leishmaniasis |
| US4217344A (en) | 1976-06-23 | 1980-08-12 | L'oreal | Compositions containing aqueous dispersions of lipid spheres |
| US4235871A (en) | 1978-02-24 | 1980-11-25 | Papahadjopoulos Demetrios P | Method of encapsulating biologically active materials in lipid vesicles |
| US4261975A (en) | 1979-09-19 | 1981-04-14 | Merck & Co., Inc. | Viral liposome particle |
| US4485054A (en) | 1982-10-04 | 1984-11-27 | Lipoderm Pharmaceuticals Limited | Method of encapsulating biologically active materials in multilamellar lipid vesicles (MLV) |
| US4501728A (en) | 1983-01-06 | 1985-02-26 | Technology Unlimited, Inc. | Masking of liposomes from RES recognition |
| US4774085A (en) | 1985-07-09 | 1988-09-27 | 501 Board of Regents, Univ. of Texas | Pharmaceutical administration systems containing a mixture of immunomodulators |
| US4797368A (en) | 1985-03-15 | 1989-01-10 | The United States Of America As Represented By The Department Of Health And Human Services | Adeno-associated virus as eukaryotic expression vector |
| US4837028A (en) | 1986-12-24 | 1989-06-06 | Liposome Technology, Inc. | Liposomes with enhanced circulation time |
| US4897355A (en) | 1985-01-07 | 1990-01-30 | Syntex (U.S.A.) Inc. | N[ω,(ω-1)-dialkyloxy]- and N-[ω,(ω-1)-dialkenyloxy]-alk-1-yl-N,N,N-tetrasubstituted ammonium lipids and uses therefor |
| US4946787A (en) | 1985-01-07 | 1990-08-07 | Syntex (U.S.A.) Inc. | N-(ω,(ω-1)-dialkyloxy)- and N-(ω,(ω-1)-dialkenyloxy)-alk-1-yl-N,N,N-tetrasubstituted ammonium lipids and uses therefor |
| US5013556A (en) | 1989-10-20 | 1991-05-07 | Liposome Technology, Inc. | Liposomes with enhanced circulation time |
| US5049386A (en) | 1985-01-07 | 1991-09-17 | Syntex (U.S.A.) Inc. | N-ω,(ω-1)-dialkyloxy)- and N-(ω,(ω-1)-dialkenyloxy)Alk-1-YL-N,N,N-tetrasubstituted ammonium lipids and uses therefor |
| WO1991016024A1 (en) | 1990-04-19 | 1991-10-31 | Vical, Inc. | Cationic lipids for intracellular delivery of biologically active molecules |
| WO1991017424A1 (en) | 1990-05-03 | 1991-11-14 | Vical, Inc. | Intracellular delivery of biologically active substances by means of self-assembling lipid complexes |
| US5173414A (en) | 1990-10-30 | 1992-12-22 | Applied Immune Sciences, Inc. | Production of recombinant adeno-associated virus vectors |
| WO1993024641A2 (en) | 1992-06-02 | 1993-12-09 | The United States Of America, As Represented By The Secretary, Department Of Health & Human Services | Adeno-associated virus with inverted terminal repeat sequences as promoter |
| US5846946A (en) | 1996-06-14 | 1998-12-08 | Pasteur Merieux Serums Et Vaccins | Compositions and methods for administering Borrelia DNA |
| US6027726A (en) | 1994-09-30 | 2000-02-22 | Inex Phamaceuticals Corp. | Glycosylated protein-liposome conjugates and methods for their preparation |
| US20030087817A1 (en) | 1999-01-12 | 2003-05-08 | Sangamo Biosciences, Inc. | Regulation of endogenous gene expression in cells using zinc finger proteins |
| US20040142025A1 (en) | 2002-06-28 | 2004-07-22 | Protiva Biotherapeutics Ltd. | Liposomal apparatus and manufacturing methods |
| US20040171156A1 (en) | 1995-06-07 | 2004-09-02 | Invitrogen Corporation | Recombinational cloning using nucleic acids having recombination sites |
| US20070042031A1 (en) | 2005-07-27 | 2007-02-22 | Protiva Biotherapeutics, Inc. | Systems and methods for manufacturing liposomes |
| US8404658B2 (en) | 2007-12-31 | 2013-03-26 | Nanocor Therapeutics, Inc. | RNA interference for the treatment of heart failure |
| US8454972B2 (en) | 2004-07-16 | 2013-06-04 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Method for inducing a multiclade immune response against HIV utilizing a multigene and multiclade immunogen |
| WO2013086354A1 (en) | 2011-12-07 | 2013-06-13 | Alnylam Pharmaceuticals, Inc. | Biodegradable lipids for the delivery of active agents |
| WO2013116126A1 (en) | 2012-02-01 | 2013-08-08 | Merck Sharp & Dohme Corp. | Novel low molecular weight, biodegradable cationic lipids for oligonucleotide delivery |
| US8691750B2 (en) | 2011-05-17 | 2014-04-08 | Axolabs Gmbh | Lipids and compositions for intracellular delivery of biologically active compounds |
| US8697359B1 (en) | 2012-12-12 | 2014-04-15 | The Broad Institute, Inc. | CRISPR-Cas systems and methods for altering expression of gene products |
| EP2755693A2 (en) | 2011-09-12 | 2014-07-23 | Moderna Therapeutics, Inc. | Engineered nucleic acids and methods of use thereof |
| EP2755986A1 (en) | 2011-09-12 | 2014-07-23 | Moderna Therapeutics, Inc. | Engineered nucleic acids and methods of use thereof |
| US8795965B2 (en) | 2012-12-12 | 2014-08-05 | The Broad Institute, Inc. | CRISPR-Cas component systems, methods and compositions for sequence manipulation |
| US8822663B2 (en) | 2010-08-06 | 2014-09-02 | Moderna Therapeutics, Inc. | Engineered nucleic acids and methods of use thereof |
| WO2014152940A1 (en) | 2013-03-14 | 2014-09-25 | Shire Human Genetic Therapies, Inc. | Mrna therapeutic compositions and use to treat diseases and disorders |
| US8865406B2 (en) | 2012-12-12 | 2014-10-21 | The Broad Institute Inc. | Engineering and optimization of improved systems, methods and enzyme compositions for sequence manipulation |
| US8889356B2 (en) | 2012-12-12 | 2014-11-18 | The Broad Institute Inc. | CRISPR-Cas nickase systems, methods and compositions for sequence manipulation in eukaryotes |
| US8906616B2 (en) | 2012-12-12 | 2014-12-09 | The Broad Institute Inc. | Engineering of systems, methods and optimized guide compositions for sequence manipulation |
| US8993233B2 (en) | 2012-12-12 | 2015-03-31 | The Broad Institute Inc. | Engineering and optimization of systems, methods and compositions for sequence manipulation with functional domains |
| US9023649B2 (en) | 2012-12-17 | 2015-05-05 | President And Fellows Of Harvard College | RNA-guided human genome engineering |
| US9068179B1 (en) | 2013-12-12 | 2015-06-30 | President And Fellows Of Harvard College | Methods for correcting presenilin point mutations |
| US9074199B1 (en) | 2013-11-19 | 2015-07-07 | President And Fellows Of Harvard College | Mutant Cas9 proteins |
| US9163284B2 (en) | 2013-08-09 | 2015-10-20 | President And Fellows Of Harvard College | Methods for identifying a target site of a Cas9 nuclease |
| US9228207B2 (en) | 2013-09-06 | 2016-01-05 | President And Fellows Of Harvard College | Switchable gRNAs comprising aptamers |
| US9267135B2 (en) | 2013-06-04 | 2016-02-23 | President And Fellows Of Harvard College | RNA-guided transcriptional regulation |
| NZ700688A (en) | 2009-12-01 | 2016-02-26 | Shire Human Genetic Therapies | Delivery of mrna for the augmentation of proteins and enzymes in human genetic diseases |
| US9322037B2 (en) | 2013-09-06 | 2016-04-26 | President And Fellows Of Harvard College | Cas9-FokI fusion proteins and uses thereof |
| US9322006B2 (en) | 2011-07-22 | 2016-04-26 | President And Fellows Of Harvard College | Evaluation and improvement of nuclease cleavage specificity |
| US9359599B2 (en) | 2013-08-22 | 2016-06-07 | President And Fellows Of Harvard College | Engineered transcription activator-like effector (TALE) domains and uses thereof |
| US9405700B2 (en) | 2010-11-04 | 2016-08-02 | Sonics, Inc. | Methods and apparatus for virtualization in an integrated circuit |
| US9526784B2 (en) | 2013-09-06 | 2016-12-27 | President And Fellows Of Harvard College | Delivery system for functional nucleases |
| US9587252B2 (en) | 2013-07-10 | 2017-03-07 | President And Fellows Of Harvard College | Orthogonal Cas9 proteins for RNA-guided gene regulation and editing |
| US9777262B2 (en) | 2013-03-13 | 2017-10-03 | President And Fellows Of Harvard College | Mutants of Cre recombinase |
| US9790490B2 (en) | 2015-06-18 | 2017-10-17 | The Broad Institute Inc. | CRISPR enzymes and systems |
| BR112016030852A2 (en) | 2014-07-02 | 2018-01-16 | Shire Human Genetic Therapies | rna messenger encapsulation |
| US9914939B2 (en) | 2013-07-26 | 2018-03-13 | President And Fellows Of Harvard College | Genome engineering |
| US10000772B2 (en) | 2012-05-25 | 2018-06-19 | The Regents Of The University Of California | Methods and compositions for RNA-directed target DNA modification and for RNA-directed modulation of transcription |
| EP3362461A1 (en) | 2015-10-16 | 2018-08-22 | Modernatx, Inc. | Mrna cap analogs with modified phosphate linkage |
| US10077453B2 (en) | 2014-07-30 | 2018-09-18 | President And Fellows Of Harvard College | CAS9 proteins including ligand-dependent inteins |
| US10113163B2 (en) | 2016-08-03 | 2018-10-30 | President And Fellows Of Harvard College | Adenosine nucleobase editors and uses thereof |
| US10167457B2 (en) | 2015-10-23 | 2019-01-01 | President And Fellows Of Harvard College | Nucleobase editors and uses thereof |
| US10190137B2 (en) | 2013-11-07 | 2019-01-29 | Editas Medicine, Inc. | CRISPR-related methods and compositions with governing gRNAS |
| US10266887B2 (en) | 2016-12-09 | 2019-04-23 | The Broad Institute, Inc. | CRISPR effector system based diagnostics |
| US10377998B2 (en) | 2013-12-12 | 2019-08-13 | The Broad Institute, Inc. | CRISPR-CAS systems and methods for altering expression of gene products, structural information and inducible modular CAS enzymes |
| US10375938B2 (en) | 2015-10-08 | 2019-08-13 | President And Fellows Of Harvard College | Multiplexed genome editing |
| US10385336B2 (en) | 2014-09-05 | 2019-08-20 | Vilnius University | Programmable RNA shredding by the type III-A CRISPR-Cas system of Streptococcus thermophilus |
| US10494621B2 (en) | 2015-06-18 | 2019-12-03 | The Broad Institute, Inc. | Crispr enzyme mutations reducing off-target effects |
| EP3450553B1 (en) | 2014-03-24 | 2019-12-25 | Translate Bio, Inc. | Mrna therapy for treatment of ocular diseases |
| US10519454B2 (en) | 2014-08-06 | 2019-12-31 | Toolgen Incorporated | Genome editing using Campylobacter jejuni CRISPR/CAS system-derived RGEN |
| WO2020014577A1 (en) | 2018-07-13 | 2020-01-16 | Allele Biotechnology And Pharmaceuticals, Inc. | Methods of achieving high specificity of genome editing |
| US10550372B2 (en) | 2013-12-12 | 2020-02-04 | The Broad Institute, Inc. | Systems, methods and compositions for sequence manipulation with optimized functional CRISPR-Cas systems |
| ES2740248T3 (en) | 2011-06-08 | 2020-02-05 | Translate Bio Inc | Lipid nanoparticle compositions and methods for mRNA administration |
| US10577630B2 (en) | 2013-06-17 | 2020-03-03 | The Broad Institute, Inc. | Delivery and use of the CRISPR-Cas systems, vectors and compositions for hepatic targeting and therapy |
| US10612011B2 (en) | 2015-07-30 | 2020-04-07 | President And Fellows Of Harvard College | Evolution of TALENs |
| US20200109398A1 (en) | 2018-08-28 | 2020-04-09 | Flagship Pioneering, Inc. | Methods and compositions for modulating a genome |
| US10648020B2 (en) | 2015-06-18 | 2020-05-12 | The Broad Institute, Inc. | CRISPR enzymes and systems |
| US10689691B2 (en) | 2014-12-19 | 2020-06-23 | The Broad Institute, Inc. | Unbiased identification of double-strand breaks and genomic rearrangement by genome-wide insert capture sequencing |
| US10711285B2 (en) | 2013-06-17 | 2020-07-14 | The Broad Institute, Inc. | Optimized CRISPR-Cas double nickase systems, methods and compositions for sequence manipulation |
| US10731181B2 (en) | 2012-12-06 | 2020-08-04 | Sigma, Aldrich Co. LLC | CRISPR-based genome modification and regulation |
| US10745677B2 (en) | 2016-12-23 | 2020-08-18 | President And Fellows Of Harvard College | Editing of CCR5 receptor gene to protect against HIV infection |
| US10781444B2 (en) | 2013-06-17 | 2020-09-22 | The Broad Institute, Inc. | Functional genomics using CRISPR-Cas systems, compositions, methods, screens and applications thereof |
| WO2020191248A1 (en) | 2019-03-19 | 2020-09-24 | The Broad Institute, Inc. | Method and compositions for editing nucleotide sequences |
| US10787684B2 (en) | 2013-11-19 | 2020-09-29 | President And Fellows Of Harvard College | Large gene excision and insertion |
| US10851380B2 (en) | 2012-10-23 | 2020-12-01 | Toolgen Incorporated | Methods for cleaving a target DNA using a guide RNA specific for the target DNA and Cas protein-encoding nucleic acid or Cas protein |
| US10851357B2 (en) | 2013-12-12 | 2020-12-01 | The Broad Institute, Inc. | Compositions and methods of use of CRISPR-Cas systems in nucleotide repeat disorders |
| US10851369B2 (en) | 2016-06-21 | 2020-12-01 | President And Fellows Of Harvard College | Frequency-based modulation of diverse species in a nucleic acid library |
| US10930367B2 (en) | 2012-12-12 | 2021-02-23 | The Broad Institute, Inc. | Methods, models, systems, and apparatus for identifying target sequences for Cas enzymes or CRISPR-Cas systems for target sequences and conveying results thereof |
| US10946108B2 (en) | 2013-06-17 | 2021-03-16 | The Broad Institute, Inc. | Delivery, use and therapeutic applications of the CRISPR-Cas systems and compositions for targeting disorders and diseases using viral components |
| US10954514B2 (en) | 2014-12-12 | 2021-03-23 | The Broad Institute, Inc. | Escorted and functionalized guides for CRISPR-Cas systems |
| US10968257B2 (en) | 2018-04-03 | 2021-04-06 | The Broad Institute, Inc. | Target recognition motifs and uses thereof |
| US11001829B2 (en) | 2014-09-25 | 2021-05-11 | The Broad Institute, Inc. | Functional screening with optimized functional CRISPR-Cas systems |
| US11008588B2 (en) | 2013-06-17 | 2021-05-18 | The Broad Institute, Inc. | Delivery, engineering and optimization of tandem guide systems, methods and compositions for sequence manipulation |
| US11021740B2 (en) | 2017-03-15 | 2021-06-01 | The Broad Institute, Inc. | Devices for CRISPR effector system based diagnostics |
| US11041173B2 (en) | 2012-12-12 | 2021-06-22 | The Broad Institute, Inc. | Delivery, engineering and optimization of systems, methods and compositions for sequence manipulation and therapeutic applications |
| US11060115B2 (en) | 2015-06-18 | 2021-07-13 | The Broad Institute, Inc. | CRISPR enzymes and systems |
| US11071790B2 (en) | 2014-10-29 | 2021-07-27 | Massachusetts Eye And Ear Infirmary | Method for efficient delivery of therapeutic molecules in vitro and in vivo |
| US11085072B2 (en) | 2016-08-31 | 2021-08-10 | President And Fellows Of Harvard College | Methods of generating libraries of nucleic acid sequences for detection via fluorescent in situ sequencing |
| US11104937B2 (en) | 2017-03-15 | 2021-08-31 | The Broad Institute, Inc. | CRISPR effector system based diagnostics |
| US11104967B2 (en) | 2015-07-22 | 2021-08-31 | President And Fellows Of Harvard College | Evolution of site-specific recombinases |
| US11111521B2 (en) | 2011-12-22 | 2021-09-07 | President And Fellows Of Harvard College | Compositions and methods for analyte detection |
| US11111472B2 (en) | 2014-10-31 | 2021-09-07 | Massachusetts Institute Of Technology | Delivery of biomolecules to immune cells |
| WO2021226558A1 (en) | 2020-05-08 | 2021-11-11 | The Broad Institute, Inc. | Methods and compositions for simultaneous editing of both strands of a target double-stranded nucleotide sequence |
| WO2022087235A1 (en) | 2020-10-21 | 2022-04-28 | Massachusetts Institute Of Technology | Systems, methods, and compositions for site-specific genetic engineering using programmable addition via site-specific targeting elements (paste) |
| WO2023064871A1 (en) * | 2021-10-14 | 2023-04-20 | Asimov Inc. | Integrases, landing pad architectures, and engineered cells comprising the same |
| WO2023070031A2 (en) | 2021-10-21 | 2023-04-27 | Massachusetts Institute Of Technology | Discovery and engineering of integrases for high-efficiency gene integration |
| WO2023077148A1 (en) | 2021-11-01 | 2023-05-04 | Tome Biosciences, Inc. | Single construct platform for simultaneous delivery of gene editing machinery and nucleic acid cargo |
-
2024
- 2024-08-30 WO PCT/US2024/044901 patent/WO2025050069A1/en active Pending
Patent Citations (208)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4217344A (en) | 1976-06-23 | 1980-08-12 | L'oreal | Compositions containing aqueous dispersions of lipid spheres |
| US4235871A (en) | 1978-02-24 | 1980-11-25 | Papahadjopoulos Demetrios P | Method of encapsulating biologically active materials in lipid vesicles |
| US4186183A (en) | 1978-03-29 | 1980-01-29 | The United States Of America As Represented By The Secretary Of The Army | Liposome carriers in chemotherapy of leishmaniasis |
| US4261975A (en) | 1979-09-19 | 1981-04-14 | Merck & Co., Inc. | Viral liposome particle |
| US4485054A (en) | 1982-10-04 | 1984-11-27 | Lipoderm Pharmaceuticals Limited | Method of encapsulating biologically active materials in multilamellar lipid vesicles (MLV) |
| US4501728A (en) | 1983-01-06 | 1985-02-26 | Technology Unlimited, Inc. | Masking of liposomes from RES recognition |
| US5049386A (en) | 1985-01-07 | 1991-09-17 | Syntex (U.S.A.) Inc. | N-ω,(ω-1)-dialkyloxy)- and N-(ω,(ω-1)-dialkenyloxy)Alk-1-YL-N,N,N-tetrasubstituted ammonium lipids and uses therefor |
| US4897355A (en) | 1985-01-07 | 1990-01-30 | Syntex (U.S.A.) Inc. | N[ω,(ω-1)-dialkyloxy]- and N-[ω,(ω-1)-dialkenyloxy]-alk-1-yl-N,N,N-tetrasubstituted ammonium lipids and uses therefor |
| US4946787A (en) | 1985-01-07 | 1990-08-07 | Syntex (U.S.A.) Inc. | N-(ω,(ω-1)-dialkyloxy)- and N-(ω,(ω-1)-dialkenyloxy)-alk-1-yl-N,N,N-tetrasubstituted ammonium lipids and uses therefor |
| US4797368A (en) | 1985-03-15 | 1989-01-10 | The United States Of America As Represented By The Department Of Health And Human Services | Adeno-associated virus as eukaryotic expression vector |
| US4774085A (en) | 1985-07-09 | 1988-09-27 | 501 Board of Regents, Univ. of Texas | Pharmaceutical administration systems containing a mixture of immunomodulators |
| US4837028A (en) | 1986-12-24 | 1989-06-06 | Liposome Technology, Inc. | Liposomes with enhanced circulation time |
| US5013556A (en) | 1989-10-20 | 1991-05-07 | Liposome Technology, Inc. | Liposomes with enhanced circulation time |
| WO1991016024A1 (en) | 1990-04-19 | 1991-10-31 | Vical, Inc. | Cationic lipids for intracellular delivery of biologically active molecules |
| WO1991017424A1 (en) | 1990-05-03 | 1991-11-14 | Vical, Inc. | Intracellular delivery of biologically active substances by means of self-assembling lipid complexes |
| US5173414A (en) | 1990-10-30 | 1992-12-22 | Applied Immune Sciences, Inc. | Production of recombinant adeno-associated virus vectors |
| WO1993024641A2 (en) | 1992-06-02 | 1993-12-09 | The United States Of America, As Represented By The Secretary, Department Of Health & Human Services | Adeno-associated virus with inverted terminal repeat sequences as promoter |
| US6027726A (en) | 1994-09-30 | 2000-02-22 | Inex Phamaceuticals Corp. | Glycosylated protein-liposome conjugates and methods for their preparation |
| US20040171156A1 (en) | 1995-06-07 | 2004-09-02 | Invitrogen Corporation | Recombinational cloning using nucleic acids having recombination sites |
| US5846946A (en) | 1996-06-14 | 1998-12-08 | Pasteur Merieux Serums Et Vaccins | Compositions and methods for administering Borrelia DNA |
| US20030087817A1 (en) | 1999-01-12 | 2003-05-08 | Sangamo Biosciences, Inc. | Regulation of endogenous gene expression in cells using zinc finger proteins |
| US20040142025A1 (en) | 2002-06-28 | 2004-07-22 | Protiva Biotherapeutics Ltd. | Liposomal apparatus and manufacturing methods |
| US8454972B2 (en) | 2004-07-16 | 2013-06-04 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Method for inducing a multiclade immune response against HIV utilizing a multigene and multiclade immunogen |
| US20070042031A1 (en) | 2005-07-27 | 2007-02-22 | Protiva Biotherapeutics, Inc. | Systems and methods for manufacturing liposomes |
| US8404658B2 (en) | 2007-12-31 | 2013-03-26 | Nanocor Therapeutics, Inc. | RNA interference for the treatment of heart failure |
| NZ700688A (en) | 2009-12-01 | 2016-02-26 | Shire Human Genetic Therapies | Delivery of mrna for the augmentation of proteins and enzymes in human genetic diseases |
| US8822663B2 (en) | 2010-08-06 | 2014-09-02 | Moderna Therapeutics, Inc. | Engineered nucleic acids and methods of use thereof |
| US9405700B2 (en) | 2010-11-04 | 2016-08-02 | Sonics, Inc. | Methods and apparatus for virtualization in an integrated circuit |
| US8691750B2 (en) | 2011-05-17 | 2014-04-08 | Axolabs Gmbh | Lipids and compositions for intracellular delivery of biologically active compounds |
| ES2740248T3 (en) | 2011-06-08 | 2020-02-05 | Translate Bio Inc | Lipid nanoparticle compositions and methods for mRNA administration |
| US10323236B2 (en) | 2011-07-22 | 2019-06-18 | President And Fellows Of Harvard College | Evaluation and improvement of nuclease cleavage specificity |
| US9322006B2 (en) | 2011-07-22 | 2016-04-26 | President And Fellows Of Harvard College | Evaluation and improvement of nuclease cleavage specificity |
| EP2755693A2 (en) | 2011-09-12 | 2014-07-23 | Moderna Therapeutics, Inc. | Engineered nucleic acids and methods of use thereof |
| EP2755986A1 (en) | 2011-09-12 | 2014-07-23 | Moderna Therapeutics, Inc. | Engineered nucleic acids and methods of use thereof |
| WO2013086354A1 (en) | 2011-12-07 | 2013-06-13 | Alnylam Pharmaceuticals, Inc. | Biodegradable lipids for the delivery of active agents |
| US11111521B2 (en) | 2011-12-22 | 2021-09-07 | President And Fellows Of Harvard College | Compositions and methods for analyte detection |
| WO2013116126A1 (en) | 2012-02-01 | 2013-08-08 | Merck Sharp & Dohme Corp. | Novel low molecular weight, biodegradable cationic lipids for oligonucleotide delivery |
| US11008589B2 (en) | 2012-05-25 | 2021-05-18 | The Regents Of The University Of California | Methods and compositions for RNA-directed target DNA modification and for RNA-directed modulation of transcription |
| US10337029B2 (en) | 2012-05-25 | 2019-07-02 | The Regents Of The University Of California | Methods and compositions for RNA-directed target DNA modification and for RNA-directed modulation of transcription |
| US10487341B2 (en) | 2012-05-25 | 2019-11-26 | The Regents Of The University Of California | Methods and compositions for RNA-directed target DNA modification and for RNA-directed modulation of transcription |
| US10519467B2 (en) | 2012-05-25 | 2019-12-31 | The Regents Of The University Of California | Methods and compositions for RNA-directed target DNA modification and for RNA-directed modulation of transcription |
| US10443076B2 (en) | 2012-05-25 | 2019-10-15 | The Regents Of The University Of California | Methods and compositions for RNA-directed target DNA modification and for RNA-directed modulation of transcription |
| US10526619B2 (en) | 2012-05-25 | 2020-01-07 | The Regents Of The University Of California | Methods and compositions for RNA-directed target DNA modification and for RNA-directed modulation of transcription |
| US10428352B2 (en) | 2012-05-25 | 2019-10-01 | The Regents Of The University Of California | Methods and compositions for RNA-directed target DNA modification and for RNA-directed modulation of transcription |
| US10421980B2 (en) | 2012-05-25 | 2019-09-24 | The Regents Of The University Of California | Methods and compositions for RNA-directed target DNA modification and for RNA-directed modulation of transcription |
| US10415061B2 (en) | 2012-05-25 | 2019-09-17 | The Regents Of The University Of California | Methods and compositions for RNA-directed target DNA modification and for RNA-directed modulation of transcription |
| US10407697B2 (en) | 2012-05-25 | 2019-09-10 | The Regents Of The University Of California | Methods and compositions for RNA-directed target DNA modification and for RNA-directed modulation of transcription |
| US10400253B2 (en) | 2012-05-25 | 2019-09-03 | The Regents Of The University Of California | Methods and compositions or RNA-directed target DNA modification and for RNA-directed modulation of transcription |
| US11028412B2 (en) | 2012-05-25 | 2021-06-08 | The Regents Of The University Of California | Methods and compositions for RNA-directed target DNA modification and for RNA-directed modulation of transcription |
| US11008590B2 (en) | 2012-05-25 | 2021-05-18 | The Regents Of The University Of California | Methods and compositions for RNA-directed target DNA modification and for RNA-directed modulation of transcription |
| US10385360B2 (en) | 2012-05-25 | 2019-08-20 | The Regents Of The University Of California | Methods and compositions for RNA-directed target DNA modification and for RNA-directed modulation of transcription |
| US10533190B2 (en) | 2012-05-25 | 2020-01-14 | The Regents Of The University Of California | Methods and compositions for RNA-directed target DNA modification and for RNA-directed modulation of transcription |
| US10550407B2 (en) | 2012-05-25 | 2020-02-04 | The Regents Of The University Of California | Methods and compositions for RNA-directed target DNA modification and for RNA-directed modulation of transcription |
| US10513712B2 (en) | 2012-05-25 | 2019-12-24 | The Regents Of The University Of California | Methods and compositions for RNA-directed target DNA modification and for RNA-directed modulation of transcription |
| US10358659B2 (en) | 2012-05-25 | 2019-07-23 | The Regents Of The University Of California | Methods and compositions for RNA-directed target DNA modification and for RNA-directed modulation of transcription |
| US11001863B2 (en) | 2012-05-25 | 2021-05-11 | The Regents Of The University Of California | Methods and compositions for RNA-directed target DNA modification and for RNA-directed modulation of transcription |
| US10988780B2 (en) | 2012-05-25 | 2021-04-27 | The Regents Of The University Of California | Methods and compositions for RNA-directed target DNA modification and for RNA-directed modulation of transcription |
| US10988782B2 (en) | 2012-05-25 | 2021-04-27 | The Regents Of The University Of California | Methods and compositions for RNA-directed target DNA modification and for RNA-directed modulation of transcription |
| US10982230B2 (en) | 2012-05-25 | 2021-04-20 | The Regents Of The University Of California | Methods and compositions for RNA-directed target DNA modification and for RNA-directed modulation of transcription |
| US10358658B2 (en) | 2012-05-25 | 2019-07-23 | The Regents Of The University Of California | Methods and compositions for RNA-directed target DNA modification and for RNA-directed modulation of transcription |
| US10982231B2 (en) | 2012-05-25 | 2021-04-20 | The Regents Of The University Of California | Methods and compositions for RNA-directed target DNA modification and for RNA-directed modulation of transcription |
| US10351878B2 (en) | 2012-05-25 | 2019-07-16 | The Regents Of The University Of California | Methods and compositions for RNA-directed target DNA modification and for RNA-directed modulation of transcription |
| US10900054B2 (en) | 2012-05-25 | 2021-01-26 | The Regents Of The University Of California | Methods and compositions for RNA-directed target DNA modification and for RNA-directed modulation of transcription |
| US10563227B2 (en) | 2012-05-25 | 2020-02-18 | The Regents Of The University Of California | Methods and compositions for RNA-directed target DNA modification and for RNA-directed modulation of transcription |
| US10308961B2 (en) | 2012-05-25 | 2019-06-04 | The Regents Of The University Of California | Methods and compositions for RNA-directed target DNA modification and for RNA-directed modulation of transcription |
| US10301651B2 (en) | 2012-05-25 | 2019-05-28 | The Regents Of The University Of California | Methods and compositions for RNA-directed target DNA modification and for RNA-directed modulation of transcription |
| US10793878B1 (en) | 2012-05-25 | 2020-10-06 | The Regents Of The University Of California | Methods and compositions for RNA-directed target DNA modification and for RNA-directed modulation of transcription |
| US10570419B2 (en) | 2012-05-25 | 2020-02-25 | The Regents Of The University Of California | Methods and compositions for RNA-directed target DNA modification and for RNA-directed modulation of transcription |
| US10774344B1 (en) | 2012-05-25 | 2020-09-15 | The Regents Of The University Of California | Methods and compositions for RNA-directed target DNA modification and for RNA-directed modulation of transcription |
| US10752920B2 (en) | 2012-05-25 | 2020-08-25 | The Regents Of The University Of California | Methods and compositions for RNA-directed target DNA modification and for RNA-directed modulation of transcription |
| US10577631B2 (en) | 2012-05-25 | 2020-03-03 | The Regents Of The University Of California | Methods and compositions for RNA-directed target DNA modification and for RNA-directed modulation of transcription |
| US10266850B2 (en) | 2012-05-25 | 2019-04-23 | The Regents Of The University Of California | Methods and compositions for RNA-directed target DNA modification and for RNA-directed modulation of transcription |
| US10000772B2 (en) | 2012-05-25 | 2018-06-19 | The Regents Of The University Of California | Methods and compositions for RNA-directed target DNA modification and for RNA-directed modulation of transcription |
| US10597680B2 (en) | 2012-05-25 | 2020-03-24 | The Regents Of The University Of California | Methods and compositions for RNA-directed target DNA modification and for RNA-directed modulation of transcription |
| US10676759B2 (en) | 2012-05-25 | 2020-06-09 | The Regents Of The University Of California | Methods and compositions for RNA-directed target DNA modification and for RNA-directed modulation of transcription |
| US10669560B2 (en) | 2012-05-25 | 2020-06-02 | The Regents Of The University Of California | Methods and compositions for RNA-directed target DNA modification and for RNA-directed modulation of transcription |
| US10640791B2 (en) | 2012-05-25 | 2020-05-05 | The Regents Of The University Of California | Methods and compositions for RNA-directed target DNA modification and for RNA-directed modulation of transcription |
| US10113167B2 (en) | 2012-05-25 | 2018-10-30 | The Regents Of The University Of California | Methods and compositions for RNA-directed target DNA modification and for RNA-directed modulation of transcription |
| US10626419B2 (en) | 2012-05-25 | 2020-04-21 | The Regents Of The University Of California | Methods and compositions for RNA-directed target DNA modification and for RNA-directed modulation of transcription |
| US10612045B2 (en) | 2012-05-25 | 2020-04-07 | The Regents Of The University Of California | Methods and compositions for RNA-directed target DNA modification and for RNA-directed modulation of transcription |
| US10227611B2 (en) | 2012-05-25 | 2019-03-12 | The Regents Of The University Of California | Methods and compositions for RNA-directed target DNA modification and for RNA-directed modulation of transcription |
| US10851380B2 (en) | 2012-10-23 | 2020-12-01 | Toolgen Incorporated | Methods for cleaving a target DNA using a guide RNA specific for the target DNA and Cas protein-encoding nucleic acid or Cas protein |
| US10731181B2 (en) | 2012-12-06 | 2020-08-04 | Sigma, Aldrich Co. LLC | CRISPR-based genome modification and regulation |
| US8889418B2 (en) | 2012-12-12 | 2014-11-18 | The Broad Institute Inc. | Engineering and optimization of improved systems, methods and enzyme compositions for sequence manipulation |
| US8993233B2 (en) | 2012-12-12 | 2015-03-31 | The Broad Institute Inc. | Engineering and optimization of systems, methods and compositions for sequence manipulation with functional domains |
| US8895308B1 (en) | 2012-12-12 | 2014-11-25 | The Broad Institute Inc. | Engineering and optimization of improved systems, methods and enzyme compositions for sequence manipulation |
| US9822372B2 (en) | 2012-12-12 | 2017-11-21 | The Broad Institute Inc. | CRISPR-Cas component systems, methods and compositions for sequence manipulation |
| US8906616B2 (en) | 2012-12-12 | 2014-12-09 | The Broad Institute Inc. | Engineering of systems, methods and optimized guide compositions for sequence manipulation |
| US8771945B1 (en) | 2012-12-12 | 2014-07-08 | The Broad Institute, Inc. | CRISPR-Cas systems and methods for altering expression of gene products |
| US8932814B2 (en) | 2012-12-12 | 2015-01-13 | The Broad Institute Inc. | CRISPR-Cas nickase systems, methods and compositions for sequence manipulation in eukaryotes |
| US8889356B2 (en) | 2012-12-12 | 2014-11-18 | The Broad Institute Inc. | CRISPR-Cas nickase systems, methods and compositions for sequence manipulation in eukaryotes |
| US10930367B2 (en) | 2012-12-12 | 2021-02-23 | The Broad Institute, Inc. | Methods, models, systems, and apparatus for identifying target sequences for Cas enzymes or CRISPR-Cas systems for target sequences and conveying results thereof |
| US8795965B2 (en) | 2012-12-12 | 2014-08-05 | The Broad Institute, Inc. | CRISPR-Cas component systems, methods and compositions for sequence manipulation |
| US8871445B2 (en) | 2012-12-12 | 2014-10-28 | The Broad Institute Inc. | CRISPR-Cas component systems, methods and compositions for sequence manipulation |
| US8697359B1 (en) | 2012-12-12 | 2014-04-15 | The Broad Institute, Inc. | CRISPR-Cas systems and methods for altering expression of gene products |
| US9840713B2 (en) | 2012-12-12 | 2017-12-12 | The Broad Institute Inc. | CRISPR-Cas component systems, methods and compositions for sequence manipulation |
| US8945839B2 (en) | 2012-12-12 | 2015-02-03 | The Broad Institute Inc. | CRISPR-Cas systems and methods for altering expression of gene products |
| US8865406B2 (en) | 2012-12-12 | 2014-10-21 | The Broad Institute Inc. | Engineering and optimization of improved systems, methods and enzyme compositions for sequence manipulation |
| US11041173B2 (en) | 2012-12-12 | 2021-06-22 | The Broad Institute, Inc. | Delivery, engineering and optimization of systems, methods and compositions for sequence manipulation and therapeutic applications |
| US8999641B2 (en) | 2012-12-12 | 2015-04-07 | The Broad Institute Inc. | Engineering and optimization of systems, methods and compositions for sequence manipulation with functional domains |
| US9023649B2 (en) | 2012-12-17 | 2015-05-05 | President And Fellows Of Harvard College | RNA-guided human genome engineering |
| US10717990B2 (en) | 2012-12-17 | 2020-07-21 | President And Fellows Of Harvard College | RNA-guided human genome engineering |
| US9260723B2 (en) | 2012-12-17 | 2016-02-16 | President And Fellows Of Harvard College | RNA-guided human genome engineering |
| US10435708B2 (en) | 2012-12-17 | 2019-10-08 | President And Fellows Of Harvard College | RNA-guided human genome engineering |
| US9970024B2 (en) | 2012-12-17 | 2018-05-15 | President And Fellows Of Harvard College | RNA-guided human genome engineering |
| US10273501B2 (en) | 2012-12-17 | 2019-04-30 | President And Fellows Of Harvard College | RNA-guided human genome engineering |
| US9777262B2 (en) | 2013-03-13 | 2017-10-03 | President And Fellows Of Harvard College | Mutants of Cre recombinase |
| WO2014152940A1 (en) | 2013-03-14 | 2014-09-25 | Shire Human Genetic Therapies, Inc. | Mrna therapeutic compositions and use to treat diseases and disorders |
| US9267135B2 (en) | 2013-06-04 | 2016-02-23 | President And Fellows Of Harvard College | RNA-guided transcriptional regulation |
| US10640789B2 (en) | 2013-06-04 | 2020-05-05 | President And Fellows Of Harvard College | RNA-guided transcriptional regulation |
| US10767194B2 (en) | 2013-06-04 | 2020-09-08 | President And Fellows Of Harvard College | RNA-guided transcriptional regulation |
| US10711285B2 (en) | 2013-06-17 | 2020-07-14 | The Broad Institute, Inc. | Optimized CRISPR-Cas double nickase systems, methods and compositions for sequence manipulation |
| US10577630B2 (en) | 2013-06-17 | 2020-03-03 | The Broad Institute, Inc. | Delivery and use of the CRISPR-Cas systems, vectors and compositions for hepatic targeting and therapy |
| US10946108B2 (en) | 2013-06-17 | 2021-03-16 | The Broad Institute, Inc. | Delivery, use and therapeutic applications of the CRISPR-Cas systems and compositions for targeting disorders and diseases using viral components |
| US11008588B2 (en) | 2013-06-17 | 2021-05-18 | The Broad Institute, Inc. | Delivery, engineering and optimization of tandem guide systems, methods and compositions for sequence manipulation |
| US10781444B2 (en) | 2013-06-17 | 2020-09-22 | The Broad Institute, Inc. | Functional genomics using CRISPR-Cas systems, compositions, methods, screens and applications thereof |
| US10329587B2 (en) | 2013-07-10 | 2019-06-25 | President And Fellows Of Harvard College | Orthogonal Cas9 proteins for RNA-guided gene regulation and editing |
| US9587252B2 (en) | 2013-07-10 | 2017-03-07 | President And Fellows Of Harvard College | Orthogonal Cas9 proteins for RNA-guided gene regulation and editing |
| US9914939B2 (en) | 2013-07-26 | 2018-03-13 | President And Fellows Of Harvard College | Genome engineering |
| US10563225B2 (en) | 2013-07-26 | 2020-02-18 | President And Fellows Of Harvard College | Genome engineering |
| US10508298B2 (en) | 2013-08-09 | 2019-12-17 | President And Fellows Of Harvard College | Methods for identifying a target site of a CAS9 nuclease |
| US9163284B2 (en) | 2013-08-09 | 2015-10-20 | President And Fellows Of Harvard College | Methods for identifying a target site of a Cas9 nuclease |
| US10954548B2 (en) | 2013-08-09 | 2021-03-23 | President And Fellows Of Harvard College | Nuclease profiling system |
| US9359599B2 (en) | 2013-08-22 | 2016-06-07 | President And Fellows Of Harvard College | Engineered transcription activator-like effector (TALE) domains and uses thereof |
| US11046948B2 (en) | 2013-08-22 | 2021-06-29 | President And Fellows Of Harvard College | Engineered transcription activator-like effector (TALE) domains and uses thereof |
| US10227581B2 (en) | 2013-08-22 | 2019-03-12 | President And Fellows Of Harvard College | Engineered transcription activator-like effector (TALE) domains and uses thereof |
| US9340800B2 (en) | 2013-09-06 | 2016-05-17 | President And Fellows Of Harvard College | Extended DNA-sensing GRNAS |
| US9388430B2 (en) | 2013-09-06 | 2016-07-12 | President And Fellows Of Harvard College | Cas9-recombinase fusion proteins and uses thereof |
| US9737604B2 (en) | 2013-09-06 | 2017-08-22 | President And Fellows Of Harvard College | Use of cationic lipids to deliver CAS9 |
| US10597679B2 (en) | 2013-09-06 | 2020-03-24 | President And Fellows Of Harvard College | Switchable Cas9 nucleases and uses thereof |
| US9526784B2 (en) | 2013-09-06 | 2016-12-27 | President And Fellows Of Harvard College | Delivery system for functional nucleases |
| US9322037B2 (en) | 2013-09-06 | 2016-04-26 | President And Fellows Of Harvard College | Cas9-FokI fusion proteins and uses thereof |
| US9228207B2 (en) | 2013-09-06 | 2016-01-05 | President And Fellows Of Harvard College | Switchable gRNAs comprising aptamers |
| US10858639B2 (en) | 2013-09-06 | 2020-12-08 | President And Fellows Of Harvard College | CAS9 variants and uses thereof |
| US9340799B2 (en) | 2013-09-06 | 2016-05-17 | President And Fellows Of Harvard College | MRNA-sensing switchable gRNAs |
| US10912833B2 (en) | 2013-09-06 | 2021-02-09 | President And Fellows Of Harvard College | Delivery of negatively charged proteins using cationic lipids |
| US10682410B2 (en) | 2013-09-06 | 2020-06-16 | President And Fellows Of Harvard College | Delivery system for functional nucleases |
| US9999671B2 (en) | 2013-09-06 | 2018-06-19 | President And Fellows Of Harvard College | Delivery of negatively charged proteins using cationic lipids |
| US10190137B2 (en) | 2013-11-07 | 2019-01-29 | Editas Medicine, Inc. | CRISPR-related methods and compositions with governing gRNAS |
| US10640788B2 (en) | 2013-11-07 | 2020-05-05 | Editas Medicine, Inc. | CRISPR-related methods and compositions with governing gRNAs |
| US10100291B2 (en) | 2013-11-19 | 2018-10-16 | President And Fellows Of Harvard College | Mutant Cas9 proteins |
| US10435679B2 (en) | 2013-11-19 | 2019-10-08 | President And Fellows Of Harvard College | Mutant Cas9 proteins |
| US10787684B2 (en) | 2013-11-19 | 2020-09-29 | President And Fellows Of Harvard College | Large gene excision and insertion |
| US10683490B2 (en) | 2013-11-19 | 2020-06-16 | President And Fellows Of Harvard College | Mutant Cas9 proteins |
| US9074199B1 (en) | 2013-11-19 | 2015-07-07 | President And Fellows Of Harvard College | Mutant Cas9 proteins |
| US10377998B2 (en) | 2013-12-12 | 2019-08-13 | The Broad Institute, Inc. | CRISPR-CAS systems and methods for altering expression of gene products, structural information and inducible modular CAS enzymes |
| US9068179B1 (en) | 2013-12-12 | 2015-06-30 | President And Fellows Of Harvard College | Methods for correcting presenilin point mutations |
| US10550372B2 (en) | 2013-12-12 | 2020-02-04 | The Broad Institute, Inc. | Systems, methods and compositions for sequence manipulation with optimized functional CRISPR-Cas systems |
| US10851357B2 (en) | 2013-12-12 | 2020-12-01 | The Broad Institute, Inc. | Compositions and methods of use of CRISPR-Cas systems in nucleotide repeat disorders |
| US9840699B2 (en) | 2013-12-12 | 2017-12-12 | President And Fellows Of Harvard College | Methods for nucleic acid editing |
| US10465176B2 (en) | 2013-12-12 | 2019-11-05 | President And Fellows Of Harvard College | Cas variants for gene editing |
| US11053481B2 (en) | 2013-12-12 | 2021-07-06 | President And Fellows Of Harvard College | Fusions of Cas9 domains and nucleic acid-editing domains |
| EP3450553B1 (en) | 2014-03-24 | 2019-12-25 | Translate Bio, Inc. | Mrna therapy for treatment of ocular diseases |
| BR112016030852A2 (en) | 2014-07-02 | 2018-01-16 | Shire Human Genetic Therapies | rna messenger encapsulation |
| US10077453B2 (en) | 2014-07-30 | 2018-09-18 | President And Fellows Of Harvard College | CAS9 proteins including ligand-dependent inteins |
| US10704062B2 (en) | 2014-07-30 | 2020-07-07 | President And Fellows Of Harvard College | CAS9 proteins including ligand-dependent inteins |
| US10519454B2 (en) | 2014-08-06 | 2019-12-31 | Toolgen Incorporated | Genome editing using Campylobacter jejuni CRISPR/CAS system-derived RGEN |
| US10385336B2 (en) | 2014-09-05 | 2019-08-20 | Vilnius University | Programmable RNA shredding by the type III-A CRISPR-Cas system of Streptococcus thermophilus |
| US11001829B2 (en) | 2014-09-25 | 2021-05-11 | The Broad Institute, Inc. | Functional screening with optimized functional CRISPR-Cas systems |
| US11071790B2 (en) | 2014-10-29 | 2021-07-27 | Massachusetts Eye And Ear Infirmary | Method for efficient delivery of therapeutic molecules in vitro and in vivo |
| US11111472B2 (en) | 2014-10-31 | 2021-09-07 | Massachusetts Institute Of Technology | Delivery of biomolecules to immune cells |
| US10954514B2 (en) | 2014-12-12 | 2021-03-23 | The Broad Institute, Inc. | Escorted and functionalized guides for CRISPR-Cas systems |
| US10689691B2 (en) | 2014-12-19 | 2020-06-23 | The Broad Institute, Inc. | Unbiased identification of double-strand breaks and genomic rearrangement by genome-wide insert capture sequencing |
| US10648020B2 (en) | 2015-06-18 | 2020-05-12 | The Broad Institute, Inc. | CRISPR enzymes and systems |
| US11060115B2 (en) | 2015-06-18 | 2021-07-13 | The Broad Institute, Inc. | CRISPR enzymes and systems |
| US10494621B2 (en) | 2015-06-18 | 2019-12-03 | The Broad Institute, Inc. | Crispr enzyme mutations reducing off-target effects |
| US9790490B2 (en) | 2015-06-18 | 2017-10-17 | The Broad Institute Inc. | CRISPR enzymes and systems |
| US10876100B2 (en) | 2015-06-18 | 2020-12-29 | The Broad Institute, Inc. | Crispr enzyme mutations reducing off-target effects |
| US11091798B2 (en) | 2015-06-18 | 2021-08-17 | The Broad Institute, Inc. | CRISPR enzymes and systems |
| US11104967B2 (en) | 2015-07-22 | 2021-08-31 | President And Fellows Of Harvard College | Evolution of site-specific recombinases |
| US11078469B2 (en) | 2015-07-30 | 2021-08-03 | President And Fellows Of Harvard College | Evolution of TALENs |
| US10612011B2 (en) | 2015-07-30 | 2020-04-07 | President And Fellows Of Harvard College | Evolution of TALENs |
| US10375938B2 (en) | 2015-10-08 | 2019-08-13 | President And Fellows Of Harvard College | Multiplexed genome editing |
| US10925263B2 (en) | 2015-10-08 | 2021-02-23 | President And Fellows Of Harvard College | Multiplexed genome editing |
| US11064684B2 (en) | 2015-10-08 | 2021-07-20 | President And Fellows Of Harvard College | Multiplexed genome editing |
| US10959413B2 (en) | 2015-10-08 | 2021-03-30 | President And Fellows Of Harvard College | Multiplexed genome editing |
| EP3362461A1 (en) | 2015-10-16 | 2018-08-22 | Modernatx, Inc. | Mrna cap analogs with modified phosphate linkage |
| US10167457B2 (en) | 2015-10-23 | 2019-01-01 | President And Fellows Of Harvard College | Nucleobase editors and uses thereof |
| US10851369B2 (en) | 2016-06-21 | 2020-12-01 | President And Fellows Of Harvard College | Frequency-based modulation of diverse species in a nucleic acid library |
| US10947530B2 (en) | 2016-08-03 | 2021-03-16 | President And Fellows Of Harvard College | Adenosine nucleobase editors and uses thereof |
| US10113163B2 (en) | 2016-08-03 | 2018-10-30 | President And Fellows Of Harvard College | Adenosine nucleobase editors and uses thereof |
| US11085072B2 (en) | 2016-08-31 | 2021-08-10 | President And Fellows Of Harvard College | Methods of generating libraries of nucleic acid sequences for detection via fluorescent in situ sequencing |
| US10266886B2 (en) | 2016-12-09 | 2019-04-23 | The Broad Institute, Inc. | CRISPR effector system based diagnostics |
| US10266887B2 (en) | 2016-12-09 | 2019-04-23 | The Broad Institute, Inc. | CRISPR effector system based diagnostics |
| US10745677B2 (en) | 2016-12-23 | 2020-08-18 | President And Fellows Of Harvard College | Editing of CCR5 receptor gene to protect against HIV infection |
| US11104937B2 (en) | 2017-03-15 | 2021-08-31 | The Broad Institute, Inc. | CRISPR effector system based diagnostics |
| US11021740B2 (en) | 2017-03-15 | 2021-06-01 | The Broad Institute, Inc. | Devices for CRISPR effector system based diagnostics |
| US10968257B2 (en) | 2018-04-03 | 2021-04-06 | The Broad Institute, Inc. | Target recognition motifs and uses thereof |
| WO2020014577A1 (en) | 2018-07-13 | 2020-01-16 | Allele Biotechnology And Pharmaceuticals, Inc. | Methods of achieving high specificity of genome editing |
| US20200109398A1 (en) | 2018-08-28 | 2020-04-09 | Flagship Pioneering, Inc. | Methods and compositions for modulating a genome |
| WO2020191245A1 (en) | 2019-03-19 | 2020-09-24 | The Broad Institute, Inc. | Methods and compositions for editing nucleotide sequences |
| WO2020191234A1 (en) | 2019-03-19 | 2020-09-24 | The Broad Institute, Inc. | Methods and compositions for editing nucleotide sequences |
| WO2020191242A1 (en) | 2019-03-19 | 2020-09-24 | The Broad Institute, Inc. | Methods and compositions for editing nucleotide sequences |
| WO2020191233A1 (en) | 2019-03-19 | 2020-09-24 | The Broad Institute, Inc. | Methods and compositions for editing nucleotide sequences |
| WO2020191153A2 (en) | 2019-03-19 | 2020-09-24 | The Broad Institute, Inc. | Methods and compositions for editing nucleotide sequences |
| WO2020191171A1 (en) | 2019-03-19 | 2020-09-24 | The Broad Institute, Inc. | Methods and compositions for editing nucleotide sequences |
| WO2020191249A1 (en) | 2019-03-19 | 2020-09-24 | The Broad Institute, Inc. | Methods and compositions for editing nucleotide sequences |
| WO2020191241A1 (en) | 2019-03-19 | 2020-09-24 | The Broad Institute, Inc. | Methods and compositions for editing nucleotide sequences |
| WO2020191243A1 (en) | 2019-03-19 | 2020-09-24 | The Broad Institute, Inc. | Methods and compositions for editing nucleotide sequences |
| WO2020191248A1 (en) | 2019-03-19 | 2020-09-24 | The Broad Institute, Inc. | Method and compositions for editing nucleotide sequences |
| WO2020191239A1 (en) | 2019-03-19 | 2020-09-24 | The Broad Institute, Inc. | Methods and compositions for editing nucleotide sequences |
| WO2020191246A1 (en) | 2019-03-19 | 2020-09-24 | The Broad Institute, Inc. | Methods and compositions for editing nucleotide sequences |
| WO2021226558A1 (en) | 2020-05-08 | 2021-11-11 | The Broad Institute, Inc. | Methods and compositions for simultaneous editing of both strands of a target double-stranded nucleotide sequence |
| WO2022087235A1 (en) | 2020-10-21 | 2022-04-28 | Massachusetts Institute Of Technology | Systems, methods, and compositions for site-specific genetic engineering using programmable addition via site-specific targeting elements (paste) |
| US11572556B2 (en) | 2020-10-21 | 2023-02-07 | Massachusetts Institute Of Technology | Systems, methods, and compositions for site-specific genetic engineering using programmable addition via site-specific targeting elements (paste) |
| WO2023064871A1 (en) * | 2021-10-14 | 2023-04-20 | Asimov Inc. | Integrases, landing pad architectures, and engineered cells comprising the same |
| WO2023070031A2 (en) | 2021-10-21 | 2023-04-27 | Massachusetts Institute Of Technology | Discovery and engineering of integrases for high-efficiency gene integration |
| WO2023077148A1 (en) | 2021-11-01 | 2023-05-04 | Tome Biosciences, Inc. | Single construct platform for simultaneous delivery of gene editing machinery and nucleic acid cargo |
Non-Patent Citations (67)
| Title |
|---|
| ABRA ET AL., J. LIPOSOME RES., vol. 12, 2002, pages 1 - 3 |
| AHMAD ET AL., CANCER RES., vol. 52, 1992, pages 4817 - 4820 |
| AKINC ET AL., MOL THER., vol. 17, 2009, pages 872 - 879 |
| AKINC ET AL., NAT. BIOTECHNOL., vol. 26, 2008, pages 561 - 569 |
| ALLEN ET AL., BIOCHIMICA ET BIOPHYSICA ACTA, vol. 1237, 1995, pages 99 - 108 |
| ANZALONE ET AL., BIORXIV, 2 November 2021 (2021-11-02) |
| ANZALONE ET AL., NATURE, vol. 576, 2019, pages 149 |
| BLAESE ET AL., CANCER GENE THER., vol. 2, 1995, pages 291 - 297 |
| BLUME ET AL., BIOCHIMICA ET BIOPHYSICA ACTA, vol. 1149, 1993, pages 180 - 184 |
| BUCHSCHER ET AL., J. VIROL., vol. 66, 1992, pages 1635 - 1640 |
| CHEN ET AL., CELL, vol. 184, 28 October 2021 (2021-10-28), pages 1 - 18 |
| CRYSTAL, SCIENCE, vol. 270, 1995, pages 404 - 410 |
| DALKARA ET AL., SCI TRANSL MED, vol. 5, 2013, pages 189ra76 |
| DEFREES ET AL., JOURNAL OF THE AMERICAN CHEMISTRY SOCIETY, vol. 118, 1996, pages 6101 - 6104 |
| GAO ET AL., GENE THERAPY, vol. 2, 1995, pages 710 - 722 |
| GASIUNAS ET AL.: "A catalogue of biochemically diverse CRISPR-Cas9 orthologs", NATURE COMMUNICATIONS, vol. 11, pages 5512 |
| HEATH: "Methods in Enzymology", vol. 149, 1987, ACADEMIC PRESS, INC., article "Covalent Attachment of Proteins to Liposomes", pages: 111 - 119 |
| HERMONATMUZYCZKA, PNAS, vol. 81, 1984, pages 6466 - 6470 |
| IONNIDI ET AL.: "Drag-and-drop genome insertion without DNA cleavage with CRISPR directed integrases", BIORXIV 2021.11.01.466786, vol. 466786, 1 November 2021 (2021-11-01) |
| JIANG ET AL., NAT. BIOTECHNOLOGY, 14 October 2021 (2021-10-14) |
| KARVELIS ET AL.: "PAM recognition by miniature CRISPR-Casl2f nucleases triggers programmable double-stranded DNA target cleavage", NUCLEIC ACIDS RESEARCH, vol. 48, no. 9, 21 May 2020 (2020-05-21), pages 5016 - 23, XP055920188, DOI: 10.1093/nar/gkaa208 |
| KIRPOTIN ET AL., FEBS LETTERS, vol. 388, 1996, pages 115 - 118 |
| KLIBANOV ET AL., JOURNAL OF LIPOSOME RESEARCH, vol. 2, 1992, pages 321 - 334 |
| KOTIN, HUMAN GENE THERAPY, vol. 5, 1994, pages 793 - 801 |
| KOWALSKI ET AL.: "Delivering the Messenger: Advances in Technologies for Therapeutic mRNA Delivery", MOL THERAP., vol. 27, no. 4, 2019, pages 710 - 728, XP055634628, DOI: 10.1016/j.ymthe.2019.02.012 |
| LEONETTI ET AL., PROC. NATL. ACAD. SCI., vol. 87, 1990, pages 2448 - 2451 |
| LIUHUANG, MOLECULAR THERAPY., 2010, pages 669 - 670 |
| LOVE ET AL., PROC NATL ACAD SCI USA., vol. 107, 2010, pages 1864 - 1869 |
| MAHON ET AL., BIOCONJUG CHEM., vol. 21, 2010, pages 1448 - 1454 |
| MAKAROVA ET AL.: "C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector", SCIENCE, vol. 353, no. 6299, 2016, XP055407082, DOI: 10.1126/science.aaf5573 |
| MAKAROVA ET AL.: "Classification and Nomenclature of CRISPR-Cas Systems: Where from Here?", THE CRISPR JOURNAL, vol. 1, no. 5, 2018, XP055619311, DOI: 10.1089/crispr.2018.0033 |
| MILLER ET AL., J. VIROL., vol. 65, 1991, pages 2220 - 2224 |
| MILLINGTON-WARD ET AL., MOLECULAR THERAPY, vol. 19, no. 4, April 2011 (2011-04-01), pages 642 - 649 |
| MURUGAIAH ET AL., ANALYTICAL BIOCHEMISTRY, vol. 401, 2010, pages 61 |
| MUZYCZKA, J. CLIN. INVEST., vol. 94, 1994, pages 1351 |
| NISHIMASU ET AL.: "Crystal Structure of Cas9 in Complex with Guide RNA and Target DNA", CELL, vol. 156, 27 February 2014 (2014-02-27), pages 935 - 949, XP028667665, DOI: 10.1016/j.cell.2014.02.001 |
| OH, Y. ET AL.: "Expansion of the prime editing modality with Cas9 from Francisella novicida", BIORXIV 2021.05.25.445577, vol. 445577, 25 May 2021 (2021-05-25) |
| RAMAKRISHNAN ET AL.: "Nuclear export signal (NES) of transposases affects the transposition activity of mariner-like elements Ppmarl and Ppmar2 of moso bamboo", MOB DNA., vol. 10, 19 August 2019 (2019-08-19), pages 35 |
| REMY ET AL., BIOCONJUGATE CHEM., vol. 5, 1994, pages 647 - 654 |
| RENNEISEN ET AL., J. BIO. CHEM., vol. 265, 1990, pages 16337 - 16342 |
| RETHI-NAGY ZSUZSANNA ET AL: "STABILON, a Novel Sequence Motif That Enhances the Expression and Accumulation of Intracellular and Secreted Proteins", INTERNATIONAL JOURNAL OF MOLECULAR SCIENCES, vol. 23, no. 15, 25 July 2022 (2022-07-25), Basel, CH, pages 8168, XP093230271, ISSN: 1422-0067, DOI: 10.3390/ijms23158168 * |
| ROSIN ET AL., MOLECULAR THERAPY, vol. 19, no. 1432494-65-9, December 2011 (2011-12-01), pages 1286 - 2200 |
| SAMULSKI ET AL., J. VIROL., vol. 63, 1989, pages 03822 - 3828 |
| SAPRA ET AL., PROG. LIPID RES., vol. 42, no. 5, 2003, pages 439 - 62 |
| SCHMIDT ET AL.: "Improved CRISPR genome editing using small highly active and specific engineered RNA-guided nucleases", NAT COMMUN, vol. 12, 2021, pages 4219 |
| SCHROEDER ET AL., J INTERN MED., vol. 267, 2010, pages 9 - 21 |
| SHAH ET AL.: "Protospacer recognition motifs: mixed identities and functional diversity", RNA BIOLOGY, vol. 10, no. 5, pages 891 - 899 |
| SIEGWART ET AL., PROC NATL ACAD SCI USA., vol. 108, 2011, pages 12996 - 3001 |
| SOMMNERFELT ET AL., VIROL., vol. 176, 1990, pages 58 - 59 |
| SWARTS ET AL.: "Structural Basis for Guide RNA Processing and Seed-Dependent DNA Targeting by CRISPR-Cas12a", MOLECULAR CELL, vol. 66, 20 April 2017 (2017-04-20), pages 221 - 233, XP055569665, DOI: 10.1016/j.molcel.2017.03.016 |
| TRATSCHIN ET AL., MOL. CELL. BIOL., vol. 4, 1984, pages 2072 - 2081 |
| TRATSCHIN ET AL., MOL. CELL. BIOL., vol. 5, 1985, pages 3251 - 3260 |
| WEST ET AL., VIROLOGY, vol. 160, 1987, pages 38 - 47 |
| XU ET AL.: "Accuracy and efficiency define Bxb1 integrase as the best of fifteen candidate serine recombinases for the integration of DNA into the human genome", BMC BIOTECHNOL., vol. 13, 20 October 2013 (2013-10-20), pages 87 |
| XU ET AL.: "Comparison and optimization often phage encoded serine integrases for genome engineering in Saccharomyces cerevisiae", BMC BIOTECHNOL., vol. 16, 9 February 2016 (2016-02-09), pages 13 |
| XU, X. ET AL.: "Engineered Miniature CRISPR-Cas System for Mammalian Genome Regulation and Editing", MOLECULAR CELL, vol. 81, no. 20, 21 October 2021 (2021-10-21), pages 4333 - 45 |
| YAMANO ET AL.: "Crystal structure of Cpfl in complex with guide RNA and target DNA", CELL, no. 165, 2016, pages 949 - 962 |
| YAMANO ET AL.: "Crystal structure of Cpfl in complex with guide RNA and target DNA", CELL, vol. 165, 5 May 2016 (2016-05-05), pages 949 - 962 |
| YARNALL ET AL., NAT. BIOTECHNOL., 2022 |
| YARNALL ET AL., NAT., 2022 |
| YARNALL MATTHEW T. N. ET AL: "Drag-and-drop genome insertion of large sequences without double-strand DNA cleavage using CRISPR-directed integrases", NATURE BIOTECHNOLOGY, 24 November 2022 (2022-11-24), New York, XP093033989, ISSN: 1087-0156, DOI: 10.1038/s41587-022-01527-4 * |
| ZALIPSKY, BIOCONJUGATE CHEMISTRY, vol. 4, 1993, pages 296 - 299 |
| ZALIPSKY, FEBS LETTERS, vol. 353, 1994, pages 71 - 74 |
| ZALIPSKY: "Stealth Liposomes", 1995, CRC PRESS |
| ZHANG ET AL.: "Algorithm for Optimized mRNA Design Improves Stability and Immunogenicity", NATURE, 2023 |
| ZHENGYAO XU ET AL: "Accuracy and efficiency define Bxb1 integrase as the best of fifteen candidate serine recombinases for the integration of DNA into the human genome", BMC BIOTECHNOLOGY, BIOMED CENTRAL LTD, vol. 13, no. 1, 20 October 2013 (2013-10-20), pages 1 - 17, XP021171155, ISSN: 1472-6750, DOI: 10.1186/1472-6750-13-87 * |
| ZHU YUANJUN ET AL: "Protein expression/secretion boost by a novel unique 21-mer cis-regulatory motif (Exin21) via mRNA stabilization", MOLECULAR THERAPY, vol. 31, no. 4, 1 April 2023 (2023-04-01), US, pages 1136 - 1158, XP093098897, ISSN: 1525-0016, DOI: 10.1016/j.ymthe.2023.02.012 * |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20250057980A1 (en) | Co-Delivery of a Gene Editor Construct and a Donor Template | |
| US20260022386A1 (en) | Single Construct Platform for Simultaneous Delivery of Gene Editing Machinery and Nucleic Acid Cargo | |
| JP2024003220A (en) | Gene editing using modified closed-ended DNA (CEDNA) | |
| JP7564102B2 (en) | mRNA encoding CAS9 optimized for use in LNPs | |
| US20240263153A1 (en) | Integrase compositions and methods | |
| WO2023039440A9 (en) | Hbb-modulating compositions and methods | |
| JP2024504611A (en) | Compositions and methods for treating Fabry disease | |
| JP2025537750A (en) | RNA compositions containing lipid nanoparticles or lipid-reconstituted natural messenger packs | |
| US20240110201A1 (en) | Compositions and Methods for Treating Hereditary Angioedema | |
| WO2023205744A1 (en) | Programmable gene insertion compositions | |
| WO2023215831A1 (en) | Guide rna compositions for programmable gene insertion | |
| WO2023225670A2 (en) | Ex vivo programmable gene insertion | |
| WO2024234006A1 (en) | Systems, compositions, and methods for targeting liver sinusodial endothelial cells (lsecs) | |
| WO2024138194A1 (en) | Platforms, compositions, and methods for in vivo programmable gene insertion | |
| WO2025050069A1 (en) | Programmable gene insertion using engineered integration enzymes | |
| WO2025224182A2 (en) | Single construct platform for simultaneous delivery of gene editing machinery and nucleic acid cargo | |
| WO2025224107A1 (en) | Method and compositions for detecting off-target editing | |
| CN118829727A (en) | A single construct platform for simultaneous delivery of gene editing machinery and nucleic acid cargo | |
| WO2025166325A1 (en) | MODIFIED GUIDE RNAs | |
| WO2025235576A1 (en) | Myotis lucifugus transposase engineering | |
| WO2023225471A2 (en) | Helitron compositions and methods | |
| WO2025226730A1 (en) | Revision of genetic material using direct replacement editing | |
| WO2025226912A1 (en) | Cell-type specific delivery of gene editors and methods of use thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 24782375 Country of ref document: EP Kind code of ref document: A1 |