WO2025049848A2 - Proteases and aldc enzymes for beer haze and diacetyl reduction - Google Patents
Proteases and aldc enzymes for beer haze and diacetyl reduction Download PDFInfo
- Publication number
- WO2025049848A2 WO2025049848A2 PCT/US2024/044586 US2024044586W WO2025049848A2 WO 2025049848 A2 WO2025049848 A2 WO 2025049848A2 US 2024044586 W US2024044586 W US 2024044586W WO 2025049848 A2 WO2025049848 A2 WO 2025049848A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- seq
- polypeptide
- protease
- amino acids
- aldc
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Y—ENZYMES
- C12Y401/00—Carbon-carbon lyases (4.1)
- C12Y401/01—Carboxy-lyases (4.1.1)
- C12Y401/01005—Acetolactate decarboxylase (4.1.1.5)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12C—BEER; PREPARATION OF BEER BY FERMENTATION; PREPARATION OF MALT FOR MAKING BEER; PREPARATION OF HOPS FOR MAKING BEER
- C12C11/00—Fermentation processes for beer
- C12C11/003—Fermentation of beerwort
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12C—BEER; PREPARATION OF BEER BY FERMENTATION; PREPARATION OF MALT FOR MAKING BEER; PREPARATION OF HOPS FOR MAKING BEER
- C12C11/00—Fermentation processes for beer
- C12C11/11—Post fermentation treatments, e.g. carbonation, or concentration
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
- C12N15/52—Genes encoding for enzymes or proenzymes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/0004—Oxidoreductases (1.)
- C12N9/0006—Oxidoreductases (1.) acting on CH-OH groups as donors (1.1)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/14—Hydrolases (3)
- C12N9/48—Hydrolases (3) acting on peptide bonds (3.4)
- C12N9/50—Proteinases, e.g. Endopeptidases (3.4.21-3.4.25)
- C12N9/58—Proteinases, e.g. Endopeptidases (3.4.21-3.4.25) derived from fungi
- C12N9/62—Proteinases, e.g. Endopeptidases (3.4.21-3.4.25) derived from fungi from Aspergillus
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/88—Lyases (4.)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Y—ENZYMES
- C12Y101/00—Oxidoreductases acting on the CH-OH group of donors (1.1)
- C12Y101/01—Oxidoreductases acting on the CH-OH group of donors (1.1) with NAD+ or NADP+ as acceptor (1.1.1)
- C12Y101/01037—Malate dehydrogenase (1.1.1.37)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Y—ENZYMES
- C12Y101/00—Oxidoreductases acting on the CH-OH group of donors (1.1)
- C12Y101/01—Oxidoreductases acting on the CH-OH group of donors (1.1) with NAD+ or NADP+ as acceptor (1.1.1)
- C12Y101/01038—Malate dehydrogenase (oxaloacetate-decarboxylating) (1.1.1.38)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Y—ENZYMES
- C12Y101/00—Oxidoreductases acting on the CH-OH group of donors (1.1)
- C12Y101/01—Oxidoreductases acting on the CH-OH group of donors (1.1) with NAD+ or NADP+ as acceptor (1.1.1)
- C12Y101/01039—Malate dehydrogenase (decarboxylating) (1.1.1.39)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/14—Hydrolases (3)
- C12N9/48—Hydrolases (3) acting on peptide bonds (3.4)
- C12N9/50—Proteinases, e.g. Endopeptidases (3.4.21-3.4.25)
Definitions
- the present invention relates to novel, stable forms of acetolactate decarboxylases (ALDCs) which have improved stability in the presence of proteases. More particularly, the present invention relates to an improved brewing process where stable ALDCs are used in conjunction with proline specific endoproteases during beer fermentation to provide a beer having less off-flavor which is colloidally stable in a shortened time.
- ALDCs acetolactate decarboxylases
- Beer tends to be colloidally unstable in that a haze will form after the beer is bottled and refrigerated prior to sale. This phenomenon is called chill haze.
- chill haze When beer is cooled, polyphenols in the beer interact with proline rich proteins (haze active proteins) and form a precipitate or haze. Chill haze is highly undesirable in a bottled beer.
- a cold stabilization phase prior to beer filtration and bottling helps to eliminate or at least reduce chill haze.
- a typical stabilization phase can be seven days or longer and require chilling to 0 or even -2°C. The energy costs for bringing thousands of hectoliters of beer to this low temperature are substantial.
- Fermentation (converting fermentable sugars in the wort to alcohol) produces what is called “green beer”.
- Green beer contains high levels of undesirable flavor components, notably diketones, such as diacetyl.
- Diacetyl has a strong buttery off-flavor and is considered highly undesirable in many beers.
- the conversion of diacetyl into bland tasting compounds is an important aspect of the subsequent beer maturation phase.
- the reduction of diacetyl into tasteless acetoin is a time-consuming process but of paramount importance.
- Enzymes have been employed to shorten or eliminate the stabilization and/or maturation phase. For example, it is known in the art to use protease to decrease the stabilization phase.
- An appropriate protease can be employed to selectively degrade proteins in the beer that can bind to polyphenols and cause colloidal instability of the beer.
- Acetolactate decarboxylase (ALDC) can be used to convert ⁇ -acetolactate into the flavorless acetoin, shortening the maturation phase.
- ADC Acetolactate decarboxylase
- protease added to a fermenting beer to provide colloidal stability can possibly proteolyze other exogenously added enzymes, including ALDC enzymes. Whether further shortening of the brew process can be accomplished by combining a protease with an ALDC enzyme in the same step is unclear.
- a polypeptide having acetolactate decarboxylase activity which is resistant to protease inactivation is presented, the polypeptide having an amino acid sequence with at least 80, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:3 wherein the polypeptide has at most 250 amino acids.
- the polypeptide is at most 249, 248, 247, 246, 245, 244, 243, 242 or 241 amino acids.
- the polypeptide having acetolactate decarboxylase activity which is resistant to protease inactivation has an amino acid sequence having at least 80, 90, 95, 98, 99 or 100% sequence identity to according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12.
- the polypeptide has an amino acid sequence according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12.
- the protease to which the ALDC is resistant is optionally a proline specific protease.
- the proline specific protease is from Aspergillus niger.
- the protease has at least 80, 85, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:17.
- the protease has an amino acid sequence according to SEQ ID NO:17.
- the polypeptide has an amino acid sequence with at least 80, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:3 wherein the polypeptide has at most 250 amino acids.
- the polypeptide is at most 249, 248, 247, 246, 245, 244, 243, 242 or 241 amino acids.
- the polypeptide having acetolactate decarboxylase activity which is resistant to protease inactivation has an amino acid sequence having at least 80, 90, 95, 98, 99 or 100% sequence identity to according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12.
- the polypeptide has an amino acid sequence according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12.
- the polypeptide having acetolactate decarboxylase activity is added to the wort first.
- the proline specific protease is added to the wort first.
- the polypeptide having acetolactate decarboxylase activity and the proline specific protease are added simultaneously to the wort.
- the proline specific protease is optionally from Aspergillus niger.
- the proline specific protease has at least 80, 85, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:17.
- a Bacillus host cell is presented for producing a heterologous polypeptide of interest, wherein one or more protease genes are inactivated.
- the polypeptide of interest is expressed without a secretion signal peptide.
- the polypeptide of interest is expressed with a secretion signal.
- the polypeptide of interest is an enzyme.
- the enzyme is an oxidoreductase, transferase, hydrolase, lyase, isomerase, ligase, aminopeptidase, amylase, asparaginase, carbohydrase, carboxypeptidase, catalase, cellulase, chitinase, cutinase, cyclodextrin glycosyltransferase, deoxyribonuclease, esterase, alpha-galactosidase, beta- galactosidase, glucoamylase, alpha-glucosidase, beta-glucosidase, hyaluronic acid synthase, invertase, laccase, lipase, mannosidase, mutanase, oxidase, a pectinolytic enzyme, peroxidase, phytase, polyphenoloxidase, protease, ribonucle
- the enzyme is an ALDC enzyme.
- the ALDC enzyme is a polypeptide with an amino acid sequence having at least 80, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:3 wherein the polypeptide has at most 250 amino acids.
- the polypeptide is at most 249, 248, 247, 246, 245, 244, 243, 242 or 241 amino acids.
- the polypeptide having acetolactate decarboxylase activity has an amino acid sequence having at least 80, 90, 95, 98, 99 or 100% sequence identity to according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12.
- the polypeptide has an amino acid sequence according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12.
- the ALDC enzyme is expressed with a secretion signal.
- the heterologous polypeptide of interest is encoded by an exogenous polynucleotide integrated into the chromosome of the host cell in at least one copy.
- the exogenous polynucleotide is a nucleic acid sequence having 80, 85, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:28.
- the exogenous polynucleotide is a nucleic acid sequence according to SEQ ID NO:28.
- the one or more protease gene is inactivated by a non-sense mutation in said one or more gene, a partial deletion of said in the one or more gene or a full deletion of the one or more gene.
- the Bacillus host cell is Bacillus subtilis or Bacillus licheniformis.
- the Bacillus host cell is Bacillus subtilis.
- the one or more protease genes are selected from the group consisting of a nucleic acid sequence having at least 80, 85, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:18, SEQ ID NO:19, SEQ ID NO:20, SEQ ID NO:21, SEQ ID NO:22, SEQ ID NO:23, SEQ ID NO:24, SEQ ID NO:25 and SEQ ID NO:27.
- the one or more protease genes comprise nine inactivated proteases wherein the proteases have nucleic acid sequences with at least 80, 85, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:18, SEQ ID NO:19, SEQ ID NO:20, SEQ ID NO:21, SEQ ID NO:22, SEQ ID NO:23, SEQ ID NO:24, SEQ ID NO:25 and SEQ ID NO:27.
- the nine inactivated proteases genes have nucleic acid sequences according to SEQ ID NO:18, SEQ ID NO:19, SEQ ID NO:20, SEQ ID NO:21, SEQ ID NO:22, SEQ ID NO:23, SEQ ID NO:24, SEQ ID NO:25 and SEQ ID NO:27.
- a method for producing a polypeptide of interest having the steps of: i) providing a Bacillus host cell wherein one or more protease genes are inactivated and wherein said host cell is transformed with a nucleic acid encoding a heterologous polypeptide in operable combination with a promoter; and ii) cultivating said host cell under conditions suitable for the production of said heterologous polypeptide, such that said heterologous polypeptide is produced.
- the method has the further step of recovering the produced polypeptide.
- the polypeptide of interest is expressed with or without a secretion signal peptide.
- the polypeptide of interest is expressed with a secretion signal peptide.
- the polypeptide of interest is an enzyme.
- the enzyme is an oxidoreductase, transferase, hydrolase, lyase, isomerase, ligase, aminopeptidase, amylase, asparaginase, carbohydrase, carboxypeptidase, catalase, cellulase, chitinase, cutinase, cyclodextrin glycosyltransferase, deoxyribonuclease, esterase, alpha-galactosidase, beta- galactosidase, glucoamylase, alpha-glucosidase, beta-glucosidase, hyaluronic acid synthase, invertase, laccase, lipase, mannosidase, mutanase, oxidase, a pectinolytic enzyme, peroxidase, phytase, polyphenoloxida
- the enzyme is an ALDC enzyme.
- the ALDC enzyme is a polypeptide with an amino acid sequence having at least 80, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:3 wherein the polypeptide has at most 250 amino acids.
- the polypeptide is at most 249, 248, 247, 246, 245, 244, 243, 242 or 241 amino acids.
- the polypeptide having acetolactate decarboxylase activity has an amino acid sequence having at least 80, 90, 95, 98, 99 or 100% sequence identity to according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12.
- the polypeptide has an amino acid sequence according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12.
- the ALDC enzyme is expressed with a secretion signal.
- the heterologous polypeptide of interest is encoded by an exogenous polynucleotide integrated into the chromosome of the host cell in at least one copy.
- the exogenous polynucleotide is a nucleic acid sequence having 80, 85, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:28.
- the exogenous polynucleotide is a nucleic acid sequence according to SEQ ID NO:28.
- the one or more protease gene is inactivated by a non-sense mutation in said one or more gene, a partial deletion of said in the one or more gene or a full deletion of the one or more gene.
- the Bacillus host cell is Bacillus subtilis or Bacillus licheniformis.
- the Bacillus host cell is Bacillus subtilis.
- the one or more protease genes are selected from the group consisting of a nucleic acid sequence having at least 80, 85, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:18, SEQ ID NO:19, SEQ ID NO:20, SEQ ID NO:21, SEQ ID NO:22, SEQ ID NO:23, SEQ ID NO:24, SEQ ID NO:25 and SEQ ID NO:27.
- the one or more protease genes comprise nine inactivated proteases wherein the proteases have nucleic acid sequences with at least 80, 85, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:18, SEQ ID NO:19, SEQ ID NO:20, SEQ ID NO:21, SEQ ID NO:22, SEQ ID NO:23, SEQ ID NO:24, SEQ ID NO:25 and SEQ ID NO:27.
- the nine inactivated proteases genes have nucleic acid sequences according to SEQ ID NO:18, SEQ ID NO:19, SEQ ID NO:20, SEQ ID NO:21, SEQ ID NO:22, SEQ ID NO:23, SEQ ID NO:24, SEQ ID NO:25 and SEQ ID NO:27.
- a stable liquid formulation comprising a polypeptide having acetolactate decarboxylase activity having an amino acid sequence with at least 80, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:3 wherein said polypeptide has at most 250 amino acids and a protease.
- the polypeptide is at most 249, 248, 247, 246, 245, 244, 243, 242 or 241 amino acids.
- the polypeptide having acetolactate decarboxylase activity has an amino acid sequence having at least 80, 90, 95, 98, 99 or 100% sequence identity to according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12.
- the polypeptide has an amino acid sequence according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12.
- the protease is optionally a proline specific protease.
- the proline specific protease is from Aspergillus niger.
- the protease has at least 80, 85, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:17.
- the protease has an amino acid sequence according to SEQ ID NO:17.
- the ALDC polypeptide maintains at least 99, 98, 95, 90, 85, 80, 75 or 70% activity when the stable liquid formulation is stored at 10 o C for 30, 60, 90, 120, 150 for 180 days.
- the ALDC polypeptide maintains at least 99, 98, 95, 90, 85, 80, 75 or 70% activity when the stable liquid formulation is stored at 30 o C for 8, 16, 24, 32, 40 or 48 hours.
- a proline specific protease formulation which is substantially depleted of other protease activities comprising the protease wherein when said protease is combined with a polypeptide having acetolactate decarboxylase activity, the polypeptide is stable over time.
- the polypeptide having acetolactate decarboxylase activity has an amino acid sequence having at least 80, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:3 wherein said polypeptide has at most 250 amino acids and a protease.
- the polypeptide is at most 249, 248, 247, 246, 245, 244, 243, 242 or 241 amino acids.
- the polypeptide having acetolactate decarboxylase activity has an amino acid sequence having at least 80, 90, 95, 98, 99 or 100% sequence identity to according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12.
- the polypeptide has an amino acid sequence according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12.
- the protease is optionally a proline specific protease.
- the proline specific protease is from Aspergillus niger.
- the protease has at least 80, 85, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:17.
- the protease has an amino acid sequence according to SEQ ID NO:17.
- the ALDC polypeptide maintains at least 99, 98, 95, 90, 85, 80, 75 or 70% activity when the stable liquid formulation is stored at 10 o C for 30, 60, 90, 120, 150 for 180 days.
- the ALDC polypeptide maintains at least 99, 98, 95, 90, 85, 80, 75 or 70% activity when the stable liquid formulation is stored at 30 o C for 8, 16, 24, 32, 40 or 48 hours.
- SEQ ID NO:1 sets forth the nucleotide sequence of the wild type aldB gene.
- SEQ ID NO:2 sets forth the amino acid sequence of the ALDC aldB precursor protein.
- SEQ ID NO:3 sets forth the predicted amino acid sequence of the mature acetolactate decarboxylase (ALDC) aldB.
- SEQ ID NO:4 sets forth the nucleotide sequence of the mature form of aldB gene in plasmid alrA(CB)RIHI-Bbr.
- SEQ ID NO:5 sets forth the amino acid sequence of the aldB precursor protein expressed from plasmid alrA(CB)RIHI-Bbr.
- SEQ ID NO:6 sets forth the amino acid sequence of aldB_BS truncation variant 1.
- SEQ ID NO:7 sets forth the amino acid sequence of aldB_BS truncation variant 2.
- SEQ ID NO:8 sets forth the amino acid sequence of aldB_BS truncation variant 3.
- SEQ ID NO:9 sets forth the amino acid sequence of aldB_BS truncation variant 4.
- SEQ ID NO:10 sets forth the amino acid sequence of aldB_BS truncation variant 5.
- SEQ ID NO:11 sets forth the amino acid sequence of aldB_BS truncation variant 6.
- SEQ ID NO:12 sets forth the amino acid sequence of aldB_BS truncation variant 7.
- SEQ ID NO:13 sets forth the amino acid sequence of aldB_Bl truncation variant 1.
- SEQ ID NO:14 sets forth the amino acid sequence of aldB_Bl truncation variant 2.
- SEQ ID NO:15 sets forth the amino acid sequence of aldB_Bl truncation variant 3.
- SEQ ID NO:16 sets forth the AniPro_2 precursor protein.
- SEQ ID NO:17 sets forth the AniPro_2 mature protein.
- SEQ ID NO:18 sets forth the Bacillus subtilis aprE gene sequence.
- SEQ ID NO:19 sets forth the Bacillus subtilis nprE gene sequence.
- SEQ ID NO:20 sets forth the Bacillus subtilis nprE gene sequence.
- SEQ ID NO:21 sets forth the Bacillus subtilis ispA gene sequence.
- SEQ ID NO:22 sets forth the Bacillus subtilis bpr gene sequence.
- SEQ ID NO:23 sets forth the Bacillus subtilis wprA gene sequence.
- SEQ ID NO:24 sets forth the Bacillus subtilis vpr gene sequence.
- SEQ ID NO:25 sets forth the Bacillus subtilis Mpr gene sequence.
- SEQ ID NO:26 sets forth the Bacillus subtilis ybfj gene sequence.
- SEQ ID NO:27 sets forth the Bacillus subtilis nprB gene sequence.
- SEQ ID NO:28 sets forth the DNA sequence of aldB gene fused to aprE signal peptide.
- SEQ ID NO:29 sets forth the AL2 primer.
- SEQ ID NO:30 sets forth the AL9 primer.
- SEQ ID NO:31 sets forth the AL3 primer.
- SEQ ID NO:32 sets forth the AL10 primer.
- SEQ ID NO:33 sets forth the AL19, alrA(CB)RIHI Fwd primer.
- SEQ ID NO:34 sets forth the AL20, alrA(CB)RIHI Rev primer.
- SEQ ID NO:35 sets forth the AL21, Bbrev-RIHI Fwd primer.
- SEQ ID NO:36 sets forth the AL22, Bbrev-RIHI Rev primer.
- SEQ ID NO:37 sets forth the ydoC400F primer.
- SEQ ID NO:38 sets forth the alrA-ATG-R primer.
- SEQ ID NO:39 sets forth the yhfO-RI-R primer.
- SEQ ID NO:40 sets forth the Bbrev-R(aprE) primer.
- Figure 1 shows a plasmid map for pCB_alr.
- Figure 2 shows a plasmid map of alrA(CB)RIHI-Bbr for expression of Acetolactate Decarboxylase, aldB.
- Figures 3A-B shows a) the maximum VDK levels reached during all-malt beer fermentation with combination of ALDC and PEP and b) number of hours to decrease VDK to 0.100 mg/L to the right with combination of ALDC and PEP applied as single additions.
- Figures 4A-B shows a) the maximum VDK levels reached during all-malt beer fermentation with combination of ALDC and PEP (added as premixed blend kept 6hours at 30°C) and b) number of hours to decrease VDK to 0.100 mg/L to the right with combination of ALDC and PEP applied as premixed blend kept 6 hours at 30°C.
- Figures 5A-B shows the turbidity (EBC 90°) of beer with and without proline-specific protease, ALDC and combinations hereof added. Forced Haze was measured according to EBC TOHA method and A) Initial Total haze and B) Final Total haze are shown. Standard deviation was determined from 2 determinations. All enzymes dosed at 0.5 or 2.0 g/hL.
- Figures 6A-B shows the turbidity (EBC 25°) of beer with and without proline-specific protease, ALDC and combinations hereof added. Forced Haze was measured according to EBC TOHA method and A) Initial Total haze and B) Final Total haze are shown. Standard deviation was determined from 2 determinations. All enzymes dosed at 0.5 or 2.0 g/hL.
- Figures 7A-E shows SDS-PAGE showing combinations of ALDC (aldB, 29-32 kDa) and PEP (56-62kDa) or individual in samples retained 30°C for up to 24 hours.
- FIG. 8A-B shows Vicinal Di-Ketone (VDK) generation during beer fermentation with the use of aldB_Bl on its own or as blend (bl- 50:50%) with AnPro, as blend (bl) with AniPro_2.
- aldB_Bs on its own or as blend (bl- 50:50%) with AnPro, as blend (bl) with AniPro_2 is applied. Further AniPro_2 on its own is shown as well as control (Ctrl) sample without enzyme addition.
- amino acid sequence is synonymous with the terms “polypeptide,” “protein,” and “peptide,” and are used interchangeably. Where such amino acid sequences exhibit activity, they may be referred to as an “enzyme.”
- the conventional one-letter or three- letter codes for amino acid residues are used, with amino acid sequences being presented in the standard amino-to-carboxy terminal orientation (i.e., N ⁇ C).
- nucleic acid encompasses DNA, RNA, heteroduplexes, and synthetic molecules capable of encoding a polypeptide. Nucleic acids may be single stranded or double stranded.
- nucleic acid and “polynucleotide” are used interchangeably. Because the genetic code is degenerate, more than one codon may be used to encode a particular amino acid, and the present compositions and methods encompass nucleotide sequences that encode a particular amino acid sequence. Unless otherwise indicated, nucleic acid sequences are presented in 5′-to-3′ orientation.
- a “vector” refers to a polynucleotide sequence designed to introduce nucleic acids into one or more cell types. Vectors include cloning vectors, expression vectors, shuttle vectors, plasmids, phage particles, cassettes, and the like.
- an “expression vector” refers to a DNA construct comprising a DNA sequence encoding a polypeptide of interest, which coding sequence is operably linked to a suitable control sequence capable of effecting expression of the DNA in a suitable host.
- control sequences may include a promoter to effect transcription, an optional operator sequence to control transcription, a sequence encoding suitable ribosome binding sites on the mRNA, enhancers and sequences which control termination of transcription and translation.
- the present invention encompasses variants, homologues, derivatives, and fragments thereof.
- variant is used to mean a nucleotide sequence or amino acid sequence which differs from a wild-type sequence.
- a variant may include substitutions, insertions, deletions, truncations, transversions and/or inversions at one or more position(s) relative to a wild-type sequence.
- Variants can be made using methods known in the art for example site scanning mutagenesis, insertional mutagenesis, random mutagenesis, site-directed mutagenesis, and directed-evolution as well as using recombinant methods well known in the art.
- Polynucleotide sequences encoding variant amino acid sequences may readily be synthesized using methods known in the art.
- the variant is a naturally occurring nucleotide sequence or amino acid sequence which differs from a wild-type sequence.
- the variant may be a natural genetic variant.
- the variant is an engineered variant.
- the variant may be engineered by recombinant methods.
- the protein sequences of the instant invention may also have deletions, insertions or substitutions of amino acid residues which produce a silent change and result in a functionally equivalent substance. Deliberate amino acid substitutions may be made based on similarity in polarity, charge, solubility, hydrophobicity, hydrophilicity, and/or the amphipathic nature of the residues as long as the secondary binding activity of the substance is retained.
- negatively charged amino acids include aspartic acid and glutamic acid; positively charged amino acids include lysine and arginine; and amino acids with uncharged polar head groups having similar hydrophilicity values include leucine, isoleucine, valine, glycine, alanine, asparagine, glutamine, serine, threonine, phenylalanine, and tyrosine.
- Conservative substitutions may be made, for example according to the Table below.
- Amino acids in the same block in the second column and preferably in the same line in the third column may be substituted for each other as set forth in Table 1.
- Non-homologous substitution may also occur i.e., from one class of residue to another or alternatively involving the inclusion of unnatural amino acids such as ornithine (hereinafter referred to as Z), diaminobutyric acid ornithine (hereinafter referred to as B), norleucine ornithine (hereinafter referred to as O), pyriylalanine, thienylalanine, naphthylalanine and phenylglycine.
- Replacements may also be made by synthetic amino acids (e.g.
- unnatural amino acids include; alpha* and alpha-disubstituted* amino acids, N-alkyl amino acids*, lactic acid*, halide derivatives of natural amino acids such as trifluorotyrosine*, p-Cl-phenylalanine*, p-Br- phenylalanine*, p-I-phenylalanine*, L-allyl-glycine*, ß-alanine*, L-a-amino butyric acid*, L-g- amino butyric acid*, L-a-amino isobutyric acid*, L-e-amino caproic acid # , 7-amino heptanoic acid*, L-methionine sulfone #* , L-norleucine*, L-norvaline*, p-nitro-L-phenylalanine*, L- hydroxyproline # , L-thioproline*, methyl derivatives of phenyla
- Variant amino acid sequences may include suitable spacer groups that may be inserted between any two amino acid residues of the sequence including alkyl groups such as methyl, ethyl, or propyl groups in addition to amino acid spacers such as glycine or b-alanine residues.
- alkyl groups such as methyl, ethyl, or propyl groups in addition to amino acid spacers such as glycine or b-alanine residues.
- a further form of variation involves the presence of one or more amino acid residues in peptoid form, will be well understood by those skilled in the art.
- the peptoid form is used to refer to variant amino acid residues wherein the a-carbon substituent group is on the residue’s nitrogen atom rather than the a-carbon.
- Processes for preparing peptides in the peptoid form are known in the art, for example Simon RJ et al., PNAS (1992) 89(20), 9367-9371 and Horwell DC, Trends Biotechnol. (1995) 13(4), 132-134.
- the nucleotide sequences for use in the present invention may include within them synthetic or modified nucleotides. Several different types of modification to oligonucleotides are known in the art.
- nucleotide sequences described herein may be modified by any method available in the art. Such modifications may be carried out to enhance the in vivo activity or life span of nucleotide sequences of the present invention.
- the present invention also encompasses the use of nucleotide sequences that are complementary to the sequences presented herein. Other variants of the sequences described herein may be obtained for example by probing DNA libraries made from a range of individuals, for example individuals from different populations.
- homologues may be obtained and such homologues and fragments thereof in general will be capable of selectively hybridizing to the sequences shown in the sequence listing herein.
- sequences may be obtained by probing cDNA libraries or genomic DNA libraries made from other animal species and probing such libraries with probes comprising all or part of any one of the sequences in the attached sequence listings under conditions of medium to high stringency. Similar considerations apply to obtaining species homologues and allelic variants of the polypeptide or nucleotide sequences of the invention.
- Variants and strain/species homologues may also be obtained using degenerate PCR which will use primers designed to target sequences within the variants and homologues encoding conserved amino acid sequences within the sequences of the present invention.
- conserveed sequences can be predicted, for example, by aligning the amino acid sequences from several variants/homologues. Sequence alignments can be performed using computer software known in the art. For example, the GCG Wisconsin PileUp program is widely used. The primers used in degenerate PCR will contain one or more degenerate positions and will be used at stringency conditions lower than those used for cloning sequences with single sequence primers against known sequences. Alternatively, such polynucleotides may be obtained by site directed mutagenesis of characterized sequences. This may be useful where for example silent codon sequence changes are required to optimize codon preferences for a particular host cell in which the polynucleotide sequences are being expressed.
- sequence changes may be desired to introduce restriction enzyme recognition sites, or to alter the property or function of the polypeptides encoded by the polynucleotides.
- the present invention employs, unless otherwise indicated, conventional techniques of biochemistry, molecular biology, microbiology, and recombinant DNA, which are within the capabilities of a person of ordinary skill in the art. Such techniques are explained in the literature. See, for example, J. Sambrook, E. F. Fritsch, and T. Maniatis, 1989, Molecular Cloning: A Laboratory Manual, Second Edition, Books 1-3, Cold Spring Harbor Laboratory Press; Ausubel, F. M. et al. (1995 and periodic supplements; Current Protocols in Molecular Biology, ch.
- percent (%) sequence identity means that a particular sequence has at least a certain percentage of amino acid residues identical to those in a specified reference sequence, when aligned using the CLUSTAL W algorithm with default parameters. See Thompson et al. (1994) Nucleic Acids Res. 22:4673-4680.
- Default parameters for the CLUSTAL W algorithm are: Gap opening penalty: 10.0 Gap extension penalty: 0.05 Protein weight matrix: BLOSUM series DNA weight matrix: IUB Delay divergent sequences %: 40 Gap separation distance: 8 DNA transitions weight: 0.50 List hydrophilic residues: GPSNDQEKR Use negative matrix: OFF Toggle Residue specific penalties: ON Toggle hydrophilic penalties: ON Toggle end gap separation penalty: OFF Deletions are counted as non-identical residues, compared to a reference sequence. Deletions occurring at either terminus are included.
- proteins, including enzymes, of the present invention exist in multiple forms. Proteins of the instant invention may be clipped or trimmed (i.e., removing amino acids) from the N-terminus and/or the C-terminus, resulting in a shorter protein. Proteins of the instant invention can also have internal deletions.
- pre-pro-protein is a protein, including an enzyme, which has an N-terminal signal peptide that targets the protein for secretion.
- a pre-pro- protein is sometimes referred to herein as “full length” or “full length protein”.
- the N-terminal signal peptide is cleaved off in the endoplasmic reticulum to yield a “pro-protein”.
- a pro- protein as used herein, is shorter in length than the full-length protein (it is missing the signal peptide) but longer than the mature protein. In general, a pro-protein is inactive or less active than the mature protein.
- a pro-protein can be activated or converted to a more active mature form by post-translational modification such as N- or C- terminal clipping.
- a pro-protein which is an enzyme may be called a “proenzyme” or a “zymogen.”
- the clipped active protein (derived from the pro-protein) is also referred to herein as the mature protein. It is to be noted that the above terms are used for convenience and are not meant to override or determine the activities of a protein of the instant invention. It is also to be noted that any protein of the instant invention can have more than one variant described by the same term. All references cited in the present specification are hereby incorporated by reference in their entirety. In particular, the teachings of all references herein specifically referred to are incorporated by reference.
- Beer traditionally refers to an alcoholic beverage derived from malt, which is derived from barley, and optionally adjuncts, such as cereal grains, and flavored with hops. Beer can be made from a variety of grains by essentially the same process. All grain starches are glucose homopolymers in which the glucose residues are linked by either alpha-1, 4- or alpha-1,6-bonds, with the former predominating.
- the process of making fermented malt beverages is commonly referred to as brewing.
- the principal raw materials used in making these beverages are water, hops and malt.
- adjuncts such as common corn grits, refined corn grits, brewer's milled yeast, rice, sorghum, refined corn starch, barley, barley starch, dehusked barley, wheat, wheat starch, torrified cereal, cereal flakes, rye, oats, potato, tapioca, and syrups, such as corn syrup, sugar cane syrup, inverted sugar syrup, barley and/or wheat syrups, and the like may be used as a source of starch.
- the starch will eventually be converted into dextrins and fermentable sugars.
- the malt which is produced principally from selected varieties of barley, has the greatest effect on the overall character and quality of the beer.
- the malt is the primary flavoring agent in beer.
- the malt provides the major portion of the fermentable sugar.
- the malt provides the proteins, which will contribute to the body and foam character of the beer.
- the malt provides the necessary enzymatic activity during mashing.
- the term “Hops” refers to it use in contributing significantly to beer quality, including flavoring. Hops (or hop constituents) add desirable bittering substances to the beer.
- the hops act as protein precipitants, establish preservative agents and aid in foam formation and stabilization.
- the “process for making beer” is one that is well known in the art, but briefly, it involves five steps: (a) mashing and/or adjunct cooking (b) wort separation and extraction (c) boiling and hopping of wort (d) cooling, fermentation, and storage, and (e) maturation, processing and packaging.
- first step milled or crushed malt is mixed with water and held for a period under controlled temperatures to permit the enzymes present in the malt to convert the starch present in the malt into fermentable sugars.
- the mash is transferred to a "lauter tun” or mash filter where the liquid is separated from the grain residue. This sweet liquid is called “wort” and the left-over grain residue is called “spent grain”.
- the mash is typically subjected to an extraction, which involves adding water to the mash to recover the residual soluble extract from the spent grain.
- the wort is boiled vigorously. This sterilizes the wort and helps to develop the color, flavor, and odor. Hops are added at some point during the boiling.
- the wort is cooled and transferred to a fermenter, which either contains the yeast or to which yeast is added. After addition of yeast, the liquid is referred to as a fermentate.
- the yeast converts the sugars by fermentation into alcohol and carbon dioxide gas; at the end of fermentation the fermenter is chilled, or the fermenter may be chilled to stop fermentation. The yeast flocculates and is removed.
- the beer is cooled and stored for a period of time, during which the beer clarifies, and its flavor develops, and any material that might impair the appearance, flavor and shelf life of the beer settles out.
- the beer Prior to packaging, the beer is carbonated and, optionally, filtered and pasteurized. After fermentation, a beverage is obtained which usually contains from about 2% to about 10% alcohol by weight.
- the non-fermentable carbohydrates are not converted during fermentation and form most of the dissolved solids in the final beer. This residue remains because of the inability of malt amylases to hydrolyze the alpha-1,6-linkages of the starch.
- the non-fermentable carbohydrates contribute about 50 calories per 12 ounces of beer.
- fermentation means, in the context of brewing, the transformation of sugars in the wort, by enzymes in the brewing yeast, into ethanol and carbon dioxide with the formation of other fermentation by-products.
- a “fermentate” is the liquid solution undergoing a fermentation process leading to chemical change of the food, beer, or beverage by the action of yeast or bacteria, which produce carbon dioxide and turns carbohydrates in it into alcohol:
- malt is understood as any malted cereal grain, such as barley.
- wort refers to the unfermented liquor run-off following extracting the grist during mashing.
- the term “spent grains” refers to the drained solids remaining when the grist has been extracted and the wort separated from the mash.
- the term “beer” refers to fermented wort, e.g., an alcoholic beverage brewed from barley malt, optionally adjunct and hops.
- ALDC in some aspects, the invention provides ALDC enzymes having a better stability and activity, and which further can be recovered from microorganisms in improved yields.
- Acetolactate decarboxylase is an enzyme that belongs to the family of carboxy lyases, which are responsible for cleaving carbon-carbon bonds.
- Acetolactate decarboxylase catalyzes the conversion of 2-acetolactate (also known as 2-hydroxy-2-methyl-3-oxobutanoate) to 2-acetoin and releasing CO2.
- Acetolactate decarboxylase enzymes catalyze the enzymatic reaction belonging to the classification EC 4.l.l.5 (acetolactate decarboxylase activity) and gene ontology (GO) term ID of GO: 0047605.
- the GO term ID specifies that any protein characterized as having this associated GO term encodes an enzyme with catalytic acetolactate decarboxylase activity.
- alsD acetolactate decarboxylase
- Examples of alsD genes include but are not limited to gil3751436271reflYP 005006068.11 Acetolactate decarboxylase [Niastella koreensis OR20-10]; gil361 0576731gb1AEV96664.11 Acetolactate decarboxylase [Niastella koreensis OR20-10]; gi12187634151gb1ACL0588l.11 Acetolactate decarboxylase [Desulfatibacillum alkenivorans AK -01]; gil220909520lreflYP 002484831.11 acetolactate decarboxylase [Cyanothece sp.
- aureus JH9 gil 148268650lreflYP 001247593.11 acetolactate decarboxylase [Staphylococcus aureus subsp. aureus JH9]; gil 1485433721reflYP 001270742.11 acetolactate decarboxylase [Lactobacillus reuteri DSM 20016]; gi13805004881emb1CCG51526.11 acetolactate decarboxylase [Bacillus amyloliquefaciens subsp.
- the invention relates to ALDC enzymes from Lactobaclllus Casei (Godtfredsen 1984), Brevibacterium acetylicum (Oshiro, 1989), Lactococcus lactis (Vincent Phalip 1994), Leuconostoc lactis (O sulivan, 2001), Enterobacter aerogenes (Blomquist, 1993), Bacillus subtilis (Renna, 1993), Bacillus brevis (Svendsen, 1989) and Lactococcus lactis DX (Yuxing, 2014) It is to be understood that any suitable ALDC enzymes, i.e., ALDC produced from any microorganism which activity is dependent on metal ions, can be used according to the invention.
- the ALDC used in the methods and compositions described herein is an ALDC from Bacillus brevis or Bacillus licheniformis.
- the ALDC activity of the enzyme composition according to the invention is measured by the ALDC assays as described herein or any suitable assay known in the art.
- the standard assay is carried out at pH 6.0, and it can be performed at different pH values and temperatures for the additional characterization and specification of enzymes.
- One unit of ALDC activity is defined as the amount of enzyme which produces 1 ⁇ mole acetoin per minute under the conditions of the assay (e.g., pH 6.0 (or as specified) and 30 °C).
- the enzyme has a temperature optimum in the range of 5-80 °C, such as in the range of 5-40°C or 15-80°C, such as in the range 20-80 °C, such as in the range 5- 15°C, 15-20°C, 45-65 °C, 50-65 °C, 55-65 °C or 60-80°C. In some embodiments, the enzyme has a temperature optimum in the range of 45-65 °C. In some embodiments, the enzyme has a temperature optimum of about 60°C.
- the enzyme has a total number of amino acids of less than 350, such as less than 340, such as less than 330, such as less than 320, such as less than 310, such as less than 300 amino acids, such as in the range of 200 to 350, such as in the range of 220 to 345 amino acids.
- the ALDC compositions and methods according to the invention comprises any one or more further enzyme.
- the one or more further enzyme is selected from list consisting of acetolactate reductoisomerases, acetolactate isomerases, amylase, glucoamylase, hemicellulose, cellulase, glucanase, pullulanase, isoamylase, endo-glucanase and related beta-glucan hydrolytic accessory enzymes, xylanase, and xylanase accessory enzymes (for example, arabinofuranosidase, ferulic acid esterase, xylan acetyl esterase), beta-glucosidase and protease.
- acetolactate reductoisomerases acetolactate isomerases
- amylase glucoamylase
- hemicellulose cellulase
- glucanase pullulanase
- isoamylase endo-glucanase and related beta-glucan hydrolytic accessory enzymes
- compositions and methods according to the invention comprises an enzyme exhibiting ALDC activity, wherein the activity of said ALDC enzyme is in the range of 950 to 2500 Units per mg of protein. In some embodiments the compositions and methods according to the invention comprises an enzyme exhibiting ALDC activity, wherein the activity of said ALDC enzyme is in the range of 1000 to 2500 Units per mg of protein.
- Proline Specific Proteases Beer-haze a cloudy appearance in beer, is caused by the aggregation of hydrophobic proteins, e.g., hordeins from barley, and polyphenols, resulting in a beer with an undesirable cloudy appearance or haze.
- proline specific endoproteases are still too broad spectrum.
- prior art proline specific endoproteases can destabilize other enzyme added exogenously to beer, including ALDC enzymes added for maturation.
- proline specific endoproteases have been discovered which are less destabilizing to other exogenously added enzymes, including ALDC enzymes.
- a “polyphenol” is a compound having one or more aromatic rings and substituted by one or more hydroxyl groups. Examples of polyphenols are tannins and flavonoids, including catechins, flavonols and anthocyanins.
- the proline-specific protease and/or ALDC of the present invention may be added at different stages during the preparation of a beer. Addition of the enzyme at the beginning of the fermentation yields the best possible results. However, the enzyme may be added to a mash or to a fermented beer before haze has been formed.
- Enzyme of the present invention including ALDC enzymes and proline specific proteases may be in isolated or purified form.
- isolated or purified means and enzyme removed from its native environment.
- proline specific protease or ALDC expressed in a host cell may be considered isolated for purposes of the invention.
- native, or recombinant polypeptides which have been substantially purified by any suitable technique may be considered isolated or purified.
- Production of enzymes The enzymes of the present invention can be produced in host cells, for example, by secretion or intracellular expression.
- a cultured cell material e.g., a whole-cell broth having an enzyme can be obtained following secretion of the enzyme into the cell medium.
- the enzyme can be isolated from the host cells, or even isolated from the cell broth, depending on the desired purity of the final enzyme.
- Suitable host cells include bacterial, fungal (including yeast and filamentous fungi), and plant cells (including algae). Particularly useful host cells include Aspergillus niger, Aspergillus oryzae or Trichoderma reesei. Other host cells include bacterial cells, e.g., Bacillus subtilis or B. licheniformis, as well as Streptomyces, E. coli.
- Vectors A DNA construct comprising a nucleic acid encoding an enzyme can be constructed to be expressed in a host cell.
- nucleic acids encoding enzymes of the present invention can be incorporated into a vector.
- Vectors can be transferred to a host cell using well-known transformation techniques, such as those disclosed below.
- the vector may be any vector that can be transformed into and replicated within a host cell.
- a vector comprising a nucleic acid encoding an enzyme can be transformed and replicated in a bacterial host cell as a means of propagating and amplifying the vector.
- the vector also may be transformed into an expression host, so that the encoding nucleic acids can be expressed as a functional enzyme.
- Host cells that serve as expression hosts can include filamentous fungi, for example.
- the Fungal Genetics Stock Center (FGSC) Catalogue of Strains lists suitable vectors for expression in fungal host cells. See FGSC, Catalogue of Strains, University of Missouri, at www.fgsc.net (last modified January 17, 2007).
- a representative vector is pJG153, a promoterless Cre expression vector that can be replicated in a bacterial host. See Harrison et al. (June 2011) Applied Environ. Microbiol. 77: 3916-22.
- pJG153 can be modified with routine skill to comprise and express a nucleic acid encoding an enzyme.
- a nucleic acid encoding an enzyme can be operably linked to a suitable promoter, which allows transcription in the host cell.
- the promoter may be any DNA sequence that shows transcriptional activity in the host cell of choice and may be derived from genes encoding proteins either homologous or heterologous to the host cell.
- Exemplary promoters for directing the transcription of the DNA sequence encoding an enzyme, especially in a bacterial host are the promoter of the lac operon of E.
- the Streptomyces coelicolor agarase gene dagA or celA promoters the promoters of the Bacillus licheniformis ⁇ -amylase gene (amyL), the promoters of the Bacillus stearothermophilus maltogenic amylase gene (amyM), the promoters of the Bacillus amyloliquefaciens ⁇ -amylase (amyQ), the promoters of the Bacillus subtilis xylA and xylB genes etc.
- examples of useful promoters are those derived from the gene encoding Aspergillus oryzae TAKA amylase, Rhizomucor miehei aspartic proteinase, Aspergillus niger neutral ⁇ -amylase, A. niger acid stable ⁇ -amylase, A. niger glucoamylase, Rhizomucor miehei lipase, A. oryzae alkaline protease, A. oryzae triose phosphate isomerase, or A. nidulans acetamidase.
- TAKA amylase Rhizomucor miehei aspartic proteinase
- Aspergillus niger neutral ⁇ -amylase A. niger acid stable ⁇ -amylase
- A. niger glucoamylase Rhizomucor miehei lipase
- Rhizomucor miehei lipase Rhizomucor miehe
- a suitable promoter can be selected, for example, from a bacteriophage promoter including a T7 promoter and a phage lambda promoter.
- suitable promoters for the expression in a yeast species include but are not limited to the Gal 1 and Gal 10 promoters of Saccharomyces cerevisiae and the Pichia pastoris AOX1 or AOX2 promoters.
- cbh1 is an endogenous, inducible promoter from Trichoderma reesei. See Liu et al. (2008) “Improved heterologous gene expression in Trichoderma reesei by cellobiohydrolase I gene (cbh1) promoter optimization,” Acta Biochim. Biophys.
- the coding sequence can be operably linked to a signal sequence.
- the DNA encoding the signal sequence may be the DNA sequence naturally associated with the enzyme gene to be expressed or from a different Genus or species.
- a signal sequence and a promoter sequence comprising a DNA construct or vector can be introduced into a fungal host cell and can be derived from the same source.
- the signal sequence is the cbh1 signal sequence that is operably linked to a cbh1 promoter.
- An expression vector may also comprise a suitable transcription terminator and, in eukaryotes, polyadenylation sequences operably linked to the DNA sequence encoding a variant enzyme.
- Termination and polyadenylation sequences may suitably be derived from the same sources as the promoter.
- the vector may further comprise a DNA sequence enabling the vector to replicate in the host cell. Examples of such sequences are the origins of replication of plasmids pUC19, pACYC177, pUB110, pE194, pAMB1, and pIJ702.
- the vector may also comprise a selectable marker, e.g., a gene the product of which complements a defect in the isolated host cell, such as the dal genes from B. subtilis or B.
- the vector may comprise Aspergillus selection markers such as amdS, argB, niaD and xxsC, a marker giving rise to hygromycin resistance, or the selection may be accomplished by co-transformation, such as known in the art. See e.g., International PCT Application WO 91/17243. Intracellular expression may be advantageous in some respects, e.g., when using certain bacteria or fungi as host cells to produce large amounts of enzyme for subsequent enrichment or purification.
- Extracellular secretion of enzyme into the culture medium can also be used to make a cultured cell material comprising the isolated enzyme.
- the expression vector typically includes the components of a cloning vector, such as, for example, an element that permits autonomous replication of the vector in the selected host organism and one or more phenotypically detectable markers for selection purposes.
- the expression vector normally comprises control nucleotide sequences such as a promoter, operator, ribosome binding site, translation initiation signal and optionally, a repressor gene or one or more activator genes.
- the expression vector may comprise a sequence coding for an amino acid sequence capable of targeting the enzyme to a host cell organelle such as a peroxisome, or to a particular host cell compartment.
- Such a targeting sequence includes but is not limited to the sequence, SKL.
- the nucleic acid sequence of the enzyme is operably linked to the control sequences in proper manner with respect to expression.
- the procedures used to ligate the DNA construct encoding an enzyme, the promoter, terminator, and other elements, respectively, and to insert them into suitable vectors containing the information necessary for replication, are well known to persons skilled in the art (see, e.g., Sambrook et al., MOLECULAR CLONING: A LABORATORY MANUAL, 2 nd ed., Cold Spring Harbor, 1989, and 3 rd ed., 2001).
- An isolated cell is advantageously used as a host cell in the recombinant production of an enzyme according to the instant invention.
- the cell may be transformed with the DNA construct encoding the enzyme, conveniently by integrating the DNA construct (in one or more copies) in the host chromosome.
- This integration is generally considered to be an advantage, as the DNA sequence is more likely to be stably maintained in the cell. Integration of the DNA constructs into the host chromosome may be performed according to conventional methods, e.g., by homologous or heterologous recombination.
- the cell may be transformed with an expression vector as described above in connection with the different types of host cells.
- suitable bacterial host organisms are Gram positive bacterial species such as Bacillaceae including Bacillus subtilis, Bacillus licheniformis, Bacillus lentus, Bacillus brevis, Geobacillus (formerly Bacillus) stearothermophilus, Bacillus alkalophilus, Bacillus amyloliquefaciens, Bacillus coagulans, Bacillus lautus, Bacillus megaterium, and Bacillus thuringiensis; Streptomyces species such as Streptomyces murinus; lactic acid bacterial species including Lactococcus sp. such as Lactococcus lactis; Lactobacillus sp.
- Bacillaceae including Bacillus subtilis, Bacillus licheniformis, Bacillus lentus, Bacillus brevis, Geobacillus (formerly Bacillus) stearothermophilus, Bacillus alkalophilus, Bacillus amyloliquefaciens, Bacillus coagulans,
- strains of a Gram-negative bacterial species belonging to Enterobacteriaceae including E. coli, or to Pseudomonadaceae can be selected as the host organism.
- a suitable yeast host organism can be selected from the biotechnologically relevant yeasts species such as but not limited to yeast species such as Pichia sp., Hansenula sp., or Kluyveromyces, Yarrowinia, Schizosaccharomyces species or a species of Saccharomyces, including Saccharomyces cerevisiae or a species belonging to Schizosaccharomyces such as, for example, S. pombe species.
- a strain of the methylotrophic yeast species, Pichia pastoris can be used as the host organism.
- the host organism can be a Hansenula species.
- Suitable host organisms among filamentous fungi include species of Aspergillus, e.g., Aspergillus niger, Aspergillus oryzae, Aspergillus tubigensis, Aspergillus awamori, or Aspergillus nidulans.
- strains of a Fusarium species e.g., Fusarium oxysporum or of a Rhizomucor species such as Rhizomucor miehei can be used as the host organism.
- Other suitable strains include Thermomyces and Mucor species.
- Trichoderma sp. can be used as a host.
- a suitable procedure for transformation of Aspergillus host cells includes, for example, that described in EP 238023.
- An enzyme expressed by a fungal host cell can be glycosylated, i.e., will comprise a glycosyl moiety.
- the glycosylation pattern can be the same or different as present in the wild-type enzyme.
- the type and/or degree of glycosylation may impart changes in enzymatic and/or biochemical properties. It may be advantageous to delete genes from expression hosts, where the gene deficiency can be cured by the transformed expression vector.
- Known methods may be used to obtain a fungal host cell having one or more inactivated genes.
- Gene inactivation may be accomplished by complete or partial deletion, by insertional inactivation or by any other means that renders a gene nonfunctional for its intended purpose, such that the gene is prevented from expression of a functional protein.
- Any gene from a Trichoderma sp. or other filamentous fungal host that has been cloned can be deleted, for example, cbh1, cbh2, egl1, and egl2 genes.
- Gene deletion may be accomplished by inserting a form of the desired gene to be inactivated into a plasmid by methods known in the art.
- Introduction of a DNA construct or vector into a host cell includes techniques such as transformation; electroporation; nuclear microinjection; transduction; transfection, e.g., lipofection mediated and DEAE-Dextrin mediated transfection; incubation with calcium phosphate DNA precipitate; high velocity bombardment with DNA-coated microprojectiles; and protoplast fusion.
- General transformation techniques are known in the art. See, e.g., Sambrook et al. (2001), supra.
- the expression of heterologous protein in Trichoderma is described, for example, in U.S. Patent No. 6,022,725. Reference is also made to Cao et al. (2000) Science 9:991-1001 for transformation of Aspergillus strains.
- Trichoderma sp. for transformation may involve the preparation of protoplasts from fungal mycelia. See Campbell et al. (1989) Curr. Genet. 16: 53- 56.
- the mycelia can be obtained from germinated vegetative spores.
- the mycelia are treated with an enzyme that digests the cell wall, resulting in protoplasts.
- the protoplasts are protected by the presence of an osmotic stabilizer in the suspending medium.
- These stabilizers include sorbitol, mannitol, potassium chloride, magnesium sulfate, and the like. Usually, the concentration of these stabilizers varies between 0.8 M and 1.2 M, e.g., a 1.2 M solution of sorbitol can be used in the suspension medium. Uptake of DNA into the host Trichoderma sp. strain depends upon the calcium ion concentration. Generally, between about 10-50 mM CaCl2 is used in an uptake solution. Additional suitable compounds include a buffering system, such as TE buffer (10 mM Tris, pH 7.4; 1 mM EDTA) or 10 mM MOPS, pH 6.0 and polyethylene glycol.
- TE buffer 10 mM Tris, pH 7.4; 1 mM EDTA
- MOPS polyethylene glycol
- the polyethylene glycol is believed to fuse the cell membranes, thus permitting the contents of the medium to be delivered into the cytoplasm of the Trichoderma sp. strain. This fusion frequently leaves multiple copies of the plasmid DNA integrated into the host chromosome.
- transformation of Trichoderma sp. uses protoplasts or cells that have been subjected to a permeability treatment, typically at a density of 10 5 to 10 7 /mL, particularly 2x10 6 /mL.
- a volume of 100 ⁇ L of these protoplasts or cells in an appropriate solution e.g., 1.2 M sorbitol and 50 mM CaCl2
- an appropriate solution e.g., 1.2 M sorbitol and 50 mM CaCl2
- JGI PID Protein Identification
- a method of producing an enzyme of the instant invention may comprise cultivating a host cell as described above under conditions conducive to the production of the enzyme and recovering the enzyme from the cells and/or culture medium.
- the medium used to cultivate the cells may be any conventional medium suitable for growing the host cell in question and obtaining expression of an enzyme.
- Suitable media and media components are available from commercial suppliers or may be prepared according to published recipes (e.g., as described in catalogues of the American Type Culture Collection).
- An enzyme secreted from the host cells can be used in a whole broth preparation.
- the preparation of a spent whole fermentation broth of a recombinant microorganism can be achieved using any cultivation method known in the art resulting in the expression of an enzyme. Fermentation may, therefore, be understood as comprising shake flask cultivation, small- or large-scale fermentation (including continuous, batch, fed-batch, or solid- state fermentations) in laboratory or industrial fermenters performed in a suitable medium and under conditions allowing the enzyme to be expressed or isolated.
- Culturing can occur from about 12 to about 100 hours or greater (and any hour value there between, e.g., from 24 to 72 hours).
- the culture broth is at a pH of about 4.0 to about 8.0, again depending on the culture conditions needed for the host relative to production of an enzyme.
- Methods for Enriching and Purifying enzymes Fermentation, separation, and concentration techniques are well known in the art and conventional methods can be used to prepare an enzyme polypeptide-containing solution. After fermentation, a fermentation broth is obtained, the microbial cells and various suspended solids, including residual raw fermentation materials, are removed by conventional separation techniques to obtain an enzyme solution.
- Concentration may be performed using, e.g., a precipitation agent, such as a metal halide precipitation agent.
- a precipitation agent such as a metal halide precipitation agent.
- Metal halide precipitation agents include but are not limited to alkali metal chlorides, alkali metal bromides and blends of two or more of these metal halides.
- Exemplary metal halides include sodium chloride, potassium chloride, sodium bromide, potassium bromide and blends of two or more of these metal halides.
- the metal halide precipitation agent, sodium chloride can also be used as a preservative.
- the metal halide precipitation agent is used in an amount effective to precipitate an enzyme.
- the optimal concentration of the metal halide precipitation agent will depend, among others, on the nature of the specific enzyme polypeptide and on its concentration in the concentrated enzyme solution.
- Another alternative way to precipitate the enzyme is to use organic compounds.
- Exemplary organic compound precipitating agents include: 4-hydroxybenzoic acid, alkali metal salts of 4-hydroxybenzoic acid, alkyl esters of 4-hydroxybenzoic acid, and blends of two or more of these organic compounds.
- the addition of the organic compound precipitation agents can take place prior to, simultaneously with or subsequent to the addition of the metal halide precipitation agent, and the addition of both precipitation agents, organic compound, and metal halide, may be carried out sequentially or simultaneously.
- the organic precipitation agents are selected from the group consisting of alkali metal salts of 4-hydroxybenzoic acid, such as sodium or potassium salts, and linear or branched alkyl esters of 4-hydroxybenzoic acid, wherein the alkyl group contains from 1 to 12 carbon atoms, and blends of two or more of these organic compounds.
- the organic compound precipitation agents can be, for example, linear or branched alkyl esters of 4-hydroxybenzoic acid, wherein the alkyl group contains from 1 to 10 carbon atoms, and blends of two or more of these organic compounds.
- Exemplary organic compounds are linear alkyl esters of 4- hydroxybenzoic acid, wherein the alkyl group contains from 1 to 6 carbon atoms, and blends of two or more of these organic compounds.
- Methyl esters of 4-hydroxybenzoic acid, propyl esters of 4-hydroxybenzoic acid, butyl ester of 4-hydroxybenzoic acid, ethyl ester of 4-hydroxybenzoic acid and blends of two or more of these organic compounds can also be used.
- Additional organic compounds also include but are not limited to 4-hydroxybenzoic acid methyl ester (named methyl PARABEN), 4-hydroxybenzoic acid propyl ester (named propyl PARABEN), which also are both preservative agents.
- Addition of the organic compound precipitation agent provides the advantage of high flexibility of the precipitation conditions with respect to pH, temperature, enzyme concentration, precipitation agent concentration, and time of incubation.
- the organic compound precipitation agent is used in an amount effective to improve precipitation of the enzyme by means of the metal halide precipitation agent.
- the selection of at least an effective amount and an optimum amount of organic compound precipitation agent, as well as the conditions of the precipitation for maximum recovery including incubation time, pH, temperature, and concentration of enzyme will be readily apparent to one of ordinary skill in the art, in light of the present disclosure, after routine testing.
- the concentrated polypeptide solution containing the metal halide precipitation agent, and the organic compound precipitation agent, can be adjusted to a pH, which will, of necessity, depend on the enzyme to be enriched or purified.
- the pH is adjusted at a level near the isoelectric point of the enzyme.
- the pH can be adjusted at a pH in a range from about 2.5 pH units below the isoelectric point (pI) up to about 2.5 pH units above the isoelectric point.
- the incubation time necessary to obtain an enriched or purified enzyme precipitate depends on the nature of the specific enzyme, the concentration of enzyme, and the specific precipitation agent(s) and its (their) concentration. Generally, the time effective to precipitate the enzyme is between about 1 to about 30 hours; usually it does not exceed about 25 hours. In the presence of the organic compound precipitation agent, the time of incubation can still be reduced to less about 10 hours and in most cases even about 6 hours. Generally, the temperature during incubation is between about 4°C and about 50°C. Usually, the method is carried out at a temperature between about 10°C and about 45°C (e.g., between about 20°C and about 40°C).
- the optimal temperature for inducing precipitation varies according to the solution conditions and the enzyme or precipitation agent(s) used.
- the overall recovery of enriched or purified enzyme precipitate, and the efficiency with which the process is conducted, is improved by agitating the solution comprising the enzyme, the added metal halide, and the added organic compound.
- the agitation step is done both during addition of the metal halide and the organic compound, and during the subsequent incubation period. Suitable agitation methods include mechanical stirring or shaking, vigorous aeration, or any similar technique.
- the enriched or purified enzyme is then separated from the dissociated pigment and other impurities and collected by conventional separation techniques, such as filtration, centrifugation, microfiltration, rotary vacuum filtration, ultrafiltration, press filtration, cross membrane microfiltration, cross flow membrane microfiltration, or the like. Further enrichment or purification of the enzyme precipitate can be obtained by washing the precipitate with water. For example, the enriched or purified enzyme precipitate is washed with water containing the metal halide precipitation agent, or with water containing the metal halide and the organic compound precipitation agents. During fermentation, an enzyme polypeptide accumulates in the culture broth.
- the culture broth is centrifuged or filtered to eliminate cells, and the resulting cell-free liquid is used for enzyme enrichment or purification.
- the cell-free broth is subjected to salting out using ammonium sulfate at about 70% saturation; the 70% saturation-precipitation fraction is then dissolved in a buffer and applied to a column such as a Sephadex G-100 column and eluted to recover the enzyme-active fraction.
- a conventional procedure such as ion exchange chromatography may be used.
- Enriched or purified enzymes can be made into a final product that is either liquid (solution, slurry) or solid (granular, powder).
- the polypeptide is at most 249, 248, 247, 246, 245, 244, 243, 242 or 241 amino acids.
- the polypeptide having acetolactate decarboxylase activity which is resistant to protease inactivation has an amino acid sequence having at least 80, 90, 95, 98, 99 or 100% sequence identity to according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12. More preferably, the polypeptide has an amino acid sequence according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12.
- the protease to which the ALDC is resistant is preferably a proline specific protease. More preferably, the proline specific protease is from Aspergillus niger. Still more preferably, the protease has at least 80, 85, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:17. Yet more preferably, the protease has an amino acid sequence according to SEQ ID NO:17.
- an improved brewing process is presented having the step of fermenting a wort in the presence of a polypeptide having acetolactate decarboxylase activity which is resistant to protease inactivation and a proline specific protease wherein both enzymes are present in the wort at the same time.
- the polypeptide has an amino acid sequence with at least 80, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:3 wherein the polypeptide has at most 250 amino acids. More preferably, the polypeptide is at most 249, 248, 247, 246, 245, 244, 243, 242 or 241 amino acids. Yet more preferably, the polypeptide having acetolactate decarboxylase activity which is resistant to protease inactivation has an amino acid sequence having at least 80, 90, 95, 98, 99 or 100% sequence identity to according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12.
- the polypeptide has an amino acid sequence according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12.
- the polypeptide having acetolactate decarboxylase activity is added to the wort first.
- the proline specific protease is added to the wort first.
- the polypeptide having acetolactate decarboxylase activity and the proline specific protease are added simultaneously to the wort.
- the proline specific protease is preferably from Aspergillus niger.
- the proline specific protease has at least 80, 85, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:17.
- a Bacillus host cell is presented for producing a heterologous polypeptide of interest, wherein one or more protease genes are inactivated.
- the polypeptide of interest is expressed without a secretion signal peptide.
- the polypeptide of interest is expressed with a secretion signal.
- the polypeptide of interest is an enzyme.
- the enzyme is an oxidoreductase, transferase, hydrolase, lyase, isomerase, ligase, aminopeptidase, amylase, asparaginase, carbohydrase, carboxypeptidase, catalase, cellulase, chitinase, cutinase, cyclodextrin glycosyltransferase, deoxyribonuclease, esterase, alpha-galactosidase, beta- galactosidase, glucoamylase, alpha-glucosidase, beta-glucosidase, hyaluronic acid synthase, invertase, laccase, lipase, mannosidase, mutanase, oxidase, a pectinolytic enzyme, peroxidase, phytase, polyphenoloxidase, protease, ribonu
- the polypeptide having acetolactate decarboxylase activity which is resistant to protease inactivation has an amino acid sequence having at least 80, 90, 95, 98, 99 or 100% sequence identity to according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12. More preferably, the polypeptide has an amino acid sequence according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12.
- the ALDC enzyme is expressed with a secretion signal.
- the heterologous polypeptide of interest is encoded by an exogenous polynucleotide integrated into the chromosome of the host cell in at least one copy.
- the exogenous polynucleotide is a nucleic acid sequence having 80, 85, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:28. More preferably, the exogenous polynucleotide is a nucleic acid sequence according to SEQ ID NO:28.
- the at least one protease gene is inactivated by a non-sense mutation in said at least one gene, a partial deletion of said at least one gene or a full deletion of said at least one gene.
- the Bacillus host cell is Bacillus subtilis or Bacillus licheniformis. More preferably, the Bacillus host cell is Bacillus subtilis.
- the one or more protease genes are selected from the group consisting of a nucleic acid sequence having at least 80, 85, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:18, SEQ ID NO:19, SEQ ID NO:20, SEQ ID NO:21, SEQ ID NO:22, SEQ ID NO:23, SEQ ID NO:24, SEQ ID NO:25 and SEQ ID NO:27.
- the one or more protease genes comprise nine inactivated proteases wherein the proteases have nucleic acid sequences with at least 80, 85, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:18, SEQ ID NO:19, SEQ ID NO:20, SEQ ID NO:21, SEQ ID NO:22, SEQ ID NO:23, SEQ ID NO:24, SEQ ID NO:25 and SEQ ID NO:27.
- the nine inactivated proteases genes have nucleic acid sequences according to SEQ ID NO:18, SEQ ID NO:19, SEQ ID NO:20, SEQ ID NO:21, SEQ ID NO:22, SEQ ID NO:23, SEQ ID NO:24, SEQ ID NO:25 and SEQ ID NO:27.
- a method for producing a polypeptide of interest having the steps of: i) providing a Bacillus host cell wherein one or more protease genes are inactivated and wherein said host cell is transformed with a nucleic acid encoding a heterologous polypeptide in operable combination with a promoter; and ii) cultivating said host cell under conditions suitable to produce said heterologous polypeptide, such that said heterologous polypeptide is produced.
- the method has the further step of recovering the produced polypeptide.
- the polypeptide of interest is expressed with or without a secretion signal peptide. More preferably, the polypeptide of interest is expressed with a secretion signal peptide.
- polypeptide of interest is an enzyme.
- the enzyme is an oxidoreductase, transferase, hydrolase, lyase, isomerase, ligase, aminopeptidase, amylase, asparaginase, carbohydrase, carboxypeptidase, catalase, cellulase, chitinase, cutinase, cyclodextrin glycosyltransferase, deoxyribonuclease, esterase, alpha-galactosidase, beta- galactosidase, glucoamylase, alpha-glucosidase, beta-glucosidase, hyaluronic acid synthase, invertase, laccase, lipase, mannosidase, mutanase, oxidase, a pectinolytic enzyme, peroxidase, phytase, polyphenoloxid
- the enzyme is an ALDC enzyme.
- the ALDC enzyme is a polypeptide with an amino acid sequence having at least 80, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:3 wherein the polypeptide has at most 250 amino acids.
- the polypeptide is at most 249, 248, 247, 246, 245, 244, 243, 242 or 241 amino acids.
- the polypeptide having acetolactate decarboxylase activity has an amino acid sequence having at least 80, 90, 95, 98, 99 or 100% sequence identity to according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12.
- the polypeptide has an amino acid sequence according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12.
- the ALDC enzyme is expressed with a secretion signal.
- the heterologous polypeptide of interest is encoded by an exogenous polynucleotide integrated into the chromosome of the host cell in at least one copy.
- the exogenous polynucleotide is a nucleic acid sequence having 80, 85, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:28.
- the exogenous polynucleotide is a nucleic acid sequence according to SEQ ID NO:28.
- the at least one protease gene is inactivated by a non-sense mutation in said at least one gene, a partial deletion of said at least one gene or a full deletion of said at least one gene.
- the Bacillus host cell is Bacillus subtilis or Bacillus licheniformis. More preferably, the Bacillus host cell is Bacillus subtilis.
- the one or more protease genes are selected from the group consisting of a nucleic acid sequence having at least 80, 85, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:18, SEQ ID NO:19, SEQ ID NO:20, SEQ ID NO:21, SEQ ID NO:22, SEQ ID NO:23, SEQ ID NO:24, SEQ ID NO:25 and SEQ ID NO:27.
- the one or more protease genes comprise nine inactivated proteases wherein the proteases have nucleic acid sequences with at least 80, 85, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:18, SEQ ID NO:19, SEQ ID NO:20, SEQ ID NO:21, SEQ ID NO:22, SEQ ID NO:23, SEQ ID NO:24, SEQ ID NO:25 and SEQ ID NO:27.
- the nine inactivated proteases genes have nucleic acid sequences according to SEQ ID NO:18, SEQ ID NO:19, SEQ ID NO:20, SEQ ID NO:21, SEQ ID NO:22, SEQ ID NO:23, SEQ ID NO:24, SEQ ID NO:25 and SEQ ID NO:27.
- a stable liquid formulation comprising a polypeptide having acetolactate decarboxylase activity having an amino acid sequence with at least 80, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:3 wherein said polypeptide has at most 250 amino acids and a protease.
- the polypeptide is at most 249, 248, 247, 246, 245, 244, 243, 242 or 241 amino acids.
- the polypeptide having acetolactate decarboxylase activity has an amino acid sequence having at least 80, 90, 95, 98, 99 or 100% sequence identity to according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12. More preferably, the polypeptide has an amino acid sequence according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12.
- the protease is preferably a proline specific protease. More preferably, the proline specific protease is from Aspergillus niger. More preferably, the protease has at least 80, 85, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:17. Still more preferably, the protease has an amino acid sequence according to SEQ ID NO:17.
- the ALDC polypeptide maintains at least 99, 98, 95, 90, 85, 80, 75 or 70% activity when the stable liquid formulation is stored at 10 o C for 30, 60, 90, 120, 150 for 180 days.
- the ALDC polypeptide maintains at least 99, 98, 95, 90, 85, 80, 75 or 70% activity when the stable liquid formulation is stored at 30 o C for 8, 16, 24, 32, 40 or 48 hours.
- a proline specific protease formulation is presented which is substantially depleted of other protease activities comprising the protease wherein when said protease is combined with a polypeptide having acetolactate decarboxylase activity, the polypeptide is stable over time.
- the polypeptide having acetolactate decarboxylase activity has an amino acid sequence having at least 80, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:3 wherein said polypeptide has at most 250 amino acids and a protease.
- the polypeptide is at most 249, 248, 247, 246, 245, 244, 243, 242 or 241 amino acids.
- the polypeptide having acetolactate decarboxylase activity has an amino acid sequence having at least 80, 90, 95, 98, 99 or 100% sequence identity to according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12.
- the polypeptide has an amino acid sequence according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12.
- the protease is preferably a proline specific protease. More preferably, the proline specific protease is from Aspergillus niger. More preferably, the protease has at least 80, 85, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:17. Still more preferably, the protease has an amino acid sequence according to SEQ ID NO:17.
- the ALDC polypeptide maintains at least 99, 98, 95, 90, 85, 80, 75 or 70% activity when the stable liquid formulation is stored at 10 o C for 30, 60, 90, 120, 150 for 180 days. In other preferred embodiments, the ALDC polypeptide maintains at least 99, 98, 95, 90, 85, 80, 75 or 70% activity when the stable liquid formulation is stored at 30 o C for 8, 16, 24, 32, 40 or 48 hours.
- the present disclosure is described in further detail in the following examples, which are not in any way intended to limit the scope of the disclosure as claimed.
- the attached figures are meant to be considered as integral parts of the specification and description of the disclosure. The following examples are offered to illustrate, but not to limit the claimed disclosure.
- EXAMPLE 1 As an example of a proline-specific endoprotease (PEP), the proline-specific endoprotease sold as Brewers Clarex® (5 PPU / g product) by DSM from Aspergillus niger (AnPro), was used.
- the defined activity of the proline specific endo-protease (PEP) is based on hydrolysis of the synthetic peptide Z-Gly-Pro-pNA at 37°C in a citrate/disodium phosphate buffer pH 4.6.
- the reaction product is monitored spectro-photometrically at 405 nm and one unit (1PPU) is defined as the quantity of enzyme that liberates 1 mmol of p-nitroanilide per minute under these test conditions.
- aldB As an example of an acetolactate decarboxylase, aldB, from Brevibacillus brevis (which may be referred to as Bacillus brevis) produced in Bacillus licheniformis and sold as Maturex® Pro (2500 ADU-L/ g product) by Novozymes (Bagsv ⁇ rd, Denmark) was used.
- This aldB expressed in Bacillus licheniformis, is referred to herein as aldB_Bl.
- the precursor protein encoded by the AniPro_2 gene is shown in SEQ ID NO:16 (JGI Reference Sequence: Aspni5_52703).
- the protease protein has a signal peptide with a length of 21 amino acids as predicted by SignalP version 4.0 (Nordahl Petersen et al. (2011) Nature Methods, 8:785-786) and derived from a Hypocrea jecorina aspartate protease (uniport id. G0R8T0).
- the presence of a signal sequence ensures that AniPro_2 is a secreted enzyme.
- the predicted mature form of AniPro_2 is shown in SEQ ID NO:17.
- EXAMPLE 3 Expression, fermentation and purification of AniPro_2 DNA sequences encoding AniPro_2, were chemically synthesized and inserted into a Trichoderma reesei expression vector pGXT (the same as the pTTTpyr2 vector as described in published PCT Application WO2015/017256, incorporated by reference herein) by Generay (Shanghai, China). The resulting plasmid was labeled as pGXT-AniPro2. The expression plasmid was then transformed into a suitable Trichoderma reesei strain (described in published PCT application WO 05/001036) using protoplast transformation (Te’o et al. (2002) J. Microbiol.
- Transformants were selected on a medium containing acetamide as a sole source of nitrogen. After 5 days of growth on acetamide plates, transformants were collected and subjected to fermentation via DASGIP (Eppendorf, Juelich, Germany).
- seed culture was grown in 1 L shake flask, each contained 100 mL defined medium (pH5.5 before sterilization) having 50 g/L glucose monohydrate, 6 g/L glycine, 5 g/L (NH4)2SO4, 4.5 g/L KH2PO4, 1 g/L CaCl2 ⁇ 2H2O, 1 g/L MgSO4 ⁇ 7H2O, 2 g/L Mazu 6000K and 2.5 mL 400 ⁇ T.
- defined medium pH5.5 before sterilization
- Trace Metals stock (400 ⁇ Trace Metals stock ( ⁇ pH 1) contains 175 g/L C 6 H 8 O 7 ⁇ H 2 O, 200 g/L FeSO 4 ⁇ 7H 2 O, 16 g/L ZnSO 4 ⁇ 7H 2 O, 3.2 g/L CuSO4 ⁇ 5H2O, 1.4 g/L MnSO4 ⁇ H2O, and 0.8 g/L H3BO3).
- the seed culture was shaken for 48 hours at 250 rpm and 30 °C. Following completion of this incubation, 200 mL of seed culture was transferred to the 2-L bioreactor (DASGIP).
- the medium for fermentation in the 2-L bioreactor contains 60 g/L dextrose, 6 g/L glycine, 1 g/L CaCl 2 ⁇ 2H 2 O, 4.5 g/L KH 2 PO 4 , 4 g/L (NH 4 ) 2 SO 4 , 1 g/L MgSO 4 ⁇ 7H 2 O, 1.2 g/L Mazu 6000K and 2.5 ml 400 ⁇ T. reesei Trace Metals.
- An induction solution containing 250 g glucose/sophorose per kg was prepared and sterilized. Following inoculation, batch fermentation was initiated with working volume of 1 L and was controlled at pH 3.5 and 34 °C.
- Dissolved oxygen level was controlled above 35% by adjusting airflow rate, oxygen supply, and agitation during whole fermentation process.
- the glucose in broth was depleted, and the feed of 250 g (glucose/sophorose)/kg solution was started at this time.
- Stepwise feed rates of 4 mL feed/hr and 6 mL feed/hr were applied in the time intervals of 22-46 hours and 46-72 hours, respectively.
- pH was linearly changed to 4.0, and temperature was adjusted to 28 o C. Fermentation was completed after 72 hours run, and broth was harvested by centrifugation, filtered, and subsequently concentrated.
- the resulting solution was applied to a HiLoad TM Q FF 16/10 column pre-equilibrated with Buffer B.
- the target protein was eluted from the column with 0.3 M NaCl.
- the fractions containing active protein were pooled, concentrated and exchanged buffer into 20 mM NaAc (pH5.0), 150 mM NaCl via the 10K Amicon Ultra devices, then stored in 40% glycerol at -20 o C until usage.
- the proenzyme encoded by the aldB gene is depicted in SEQ ID NO: 2.
- the protein has a signal peptide with a length of 24 amino acids as predicted by SignalP-NN (Emanuelsson et al., Nature Protocols (2007) 2: 953-971).
- the presence of a signal peptide indicates that this acetolactate decarboxylase, aldB is a secreted enzyme.
- the sequence of the predicted, fully processed mature chain (aldB, 261 amino acids) is depicted in SEQ ID NO: 3.
- the aldB gene that encodes an acetolactate decarboxylases enzyme (ALDC) was produced in B.
- subtilis using the synthetic gene inserted into the pCB_alr vector, see Figure 1.
- the aldB gene containing was transcribed using aprE promoter followed by B. subtilis aprE signal sequence.
- the alrA(CB)RIHI-Bbr vector was integrated into a protease deficient B. subtilis strain.
- a map of the pCB_alr vector containing the aldB gene (alrA(CB)RIHI-Bbr) is shown in Figure 2.
- subtilis strain transformant containing the alrA(CB)RIHI-Bbr cassette (strain BRA8014 described below) was cultured in 15-mL Falcon tubes for 16 hours in TSB (broth) with 10 ppm neomycin, and 300 ⁇ L of this pre-culture was added to a 500-mL flask filled with 30 mL of cultivation media (described below) supplemented with 10 ppm neomycin. The flasks were incubated for 24, 48 and 72 hours at 33°C with constant rotational mixing at 180 rpm. Cultures were harvested by centrifugation at 14500 rpm for 20 minutes in conical tubes. The culture supernatants were used for protein determination and assays.
- the cultivation media was an enriched semi-defined media based on MOPs buffer, with urea as major nitrogen source, glucose as the main carbon source, 50 ⁇ M ZnSO4 to ensure high enzyme activity and supplemented with 1% soytone for robust cell growth.
- the aldB expressed in Bacillus subtilis is here within termed, aldB_Bs.
- the nucleotide mature sequence of the aldB gene in plasmid alrA(CB)RIHI-Bbr is depicted in SEQ ID NO:4.
- the amino acid sequence of the aldB precursor protein expressed from plasmid alrA(CB)RIHI-Bbr is depicted in SEQ ID NO:5.
- Protein Determination by Stain Free Imager Criterion Protein was quantified by SDS-PAGE gel and densitometry using Gel DocTM EZ imaging system.
- Reagents used in the assay Concentrated (2x) Laemmli Sample Buffer (Bio-Rad, Catalogue #161-0737); 26-well XT 4-12% Bis-Tris Gel ( Bio-Rad, Catalogue #345-0125); protein markers “Precision Plus Protein Standards” (Bio-Rad, Catalogue #161- 0363); protein standard BSA (Thermo Scientific, Catalogue #23208) and SimplyBlue Safestain (Invitrogen, Catalogue #LC 6060.
- the assay was carried out as follow: In a 96well-PCR plate 50 ⁇ L diluted enzyme sample were mixed with 50 ⁇ L sample buffer containing 2.7 mg DTT. The plate was sealed by Microseal ‘B’ Film from Bio-Rad and was placed into PCR machine to be heated to 70°C for 10 minutes. After that the chamber was filled by running buffer, gel cassette was set. Then 10 ⁇ L of each sample and standard (0.125-1.00 mg/mL BSA) was loaded on the gel and 5 ⁇ L of the markers were loaded. After that the electrophoresis was run at 200 V for 45 minutes. Following electrophoresis, the gel was rinsed 3 times 5 min in water, then stained in Safe-stain overnight and finally destained in water.
- a protein band from an SDS-PAGE gel of aldB ferment sample was subjected to a series of chemical treatments. Between the individual steps the gel pieces were washed and shrunk using Milli-Q water, 50w/w% ethanol and absolute ethanol respectively.
- the protein was reduced/alkylated by DTT/Iodoacetamide treatment.
- a guanidination step was performed to convert lysines to homoarginines to protect lysine side chains from acetylation.
- the acetylation reaction using Sulfo-NHS-Acetate (Sulfosuccinimidyl Acetate) only modifies the protein N-terminal residue.
- the gel pieces were swelled with a buffer containing 40v/v% 18 O water:60v/v% 16 O water and the proteolytic enzymes used for protein digestion (Trypsin and ⁇ -Chymotrypsin).
- the resulting peptides will contain mixtures of 18 O and 16 O, except for the Carboxyl terminus which will retain the native 16 O, as will be apparent from the isotopic pattern of the peptides.
- the peptide, originating from the protein N-terminus, will appear as the only acetylated peptide.
- the peptides were extracted from the gel pieces using 5w/w% formic acid and acetonitrile, then lyophilized and re-dissolved in 0.1w/w% TFA.
- the digestion products were separated (C18 column) and analyzed using a Proxeon nano- LC system followed by LTQ Orbitrap (Thermo Fisher) high resolution mass spectrometer and the amino acid sequence was deduced from the MS/MS fragment spectrum of the peptides, and the isotopic pattern of the peptides (using Xcalibur 2.0 SR2 software). Based on this analysis, the N-terminus of the isolated full-length protein was confirmed to begin with A[25] (according to SEQ ID NO.
- the N-terminus position A[25] at the mature aldB_Bs correspond with the predicted signal peptide cleavage determined by the Signal P 3.0 program (http://www.cbs.dtu.dk/services/SignalP/), set to SignalP-NN system, (Emanuelsson et al., (2007), Nature Protocols, 2: 953-971) of the gene transcript.
- N- and C-terminal truncation variants were further identified and their respective N- and C-terminus position according to SEQ ID NO. 2 are given in table 2.
- Table 2 Identified N- and C-terminus positions of aldB_Bs variants, position according to according to SEQ ID NO. 2. Position N- Position C- SEQ ID NO: terminus terminus
- the longest mature variant of aldB_Bs was found to contain 248 amino-acids and shortest 242 amino acids, all truncations were at N-termini.
- the mature polypeptide sequence of aldB_Bl (aldB produced in Bacillus licheniformis) was analyzed by MS.
- the reaction product acetoin can be quantified colourimetrically.
- Acetoin mixed with ⁇ - naphtol and creatine forms a characteristic red color absorbing at OD522 nm.
- ALDC activity was calculated based on OD 522 nm and an acetoin calibration curve.
- the assay was carried out as follows: 20 mM acetolactate substrate was prepared by mixing 100 ⁇ L ethyl-2-acetoxy-2- methylacetoacetate (Sigma, Catalogue# 220396) with 3.6 ml 0.5 M NaOH at 10°C for 10 min. 20 ml 50 mM MES pH 6.0 was added, pH was adjusted to pH 6.0 and volume adjusted to 25 ml with 50 mM MES pH 6.0.
- 80 ⁇ L 20 mM acetolactate substrate was mixed with 20 ⁇ L enzyme sample diluted in 50 mM MES, pH 6.0, 0.6 M NaCl, 0.05% BRIJ 35 and 0.01% BSA.
- the substrate/enzyme mixture was incubated at 30°C for 10 min.
- 16 ⁇ L substrate/enzyme mixture was transferred to 200 ⁇ L 1 M NaOH, 1.0% ⁇ -naphtol (Sigma, Catalogue# 33420) and 0.1% creatine (Sigma, Catalogue# C3630).
- the substrate/enzyme/color reagent mixture was incubated at 30°C for 20 min and then OD 522 nm was read.
- ALDC activity is defined as the amount of enzyme which produces 1 ⁇ mole acetoin per minute under the conditions of the assay.
- ALDC activity was determined as described in concentrated fermentation samples and is shown in table 4. The activity showed to be highest for aldB_Bs (10396 ADU/g), followed by aldB_Bl (4020 ADU/g), whereas no ALDC activity (0 ADU/g) was found in samples of proline- specific endoproteases AniPro_2 or AnPro, respectively.
- Table 4 ALDC ( ⁇ -Acetolactate Decarboxylase) activity (ADU) of aldB_Bs, aldB_Bl, AniPro_2 and AnPro samples.
- protease cleavage activity was measured using Z-Gly-Pro-pNA, (Z-, benzyloxycarbonyl-; -pNA,-p-nitroanilide) (Bachem, Bubendorf, Germany) (PEPU activity). All pNA substrate were dissolved in 100% DMSO at 100mM and further diluted to 0.4mM in the reaction buffer McIlvaine, pH 4.6. Activity measurements were performed at 30°C for 10 minutes if not stated otherwise. The released pNA was measured by spectrophotometry absorbance at 405nm using a Spectramax plate reader (Molecular Device, UK).
- PEPU activity measurements of AniPro_2, AnPro, aldB_Bs and aldBL were also performed on Z-Gly-Pro-pNA under condition described above and the enzymatic activity of the enzyme solutions obtained is shown in table 5, with one enzyme unit being defined as the activity that liberates 1 mol of pNA from Z-Gly-Pro-pNA in 1 min under the reaction conditions. It is clear that, AniPro_2 and AnPro contained high post-proline protease cleavage activity, with 6.99 and 5.16 PEPU/g respectively, whereas aldB_Bs and aldBL contained very minor post-proline protease cleavage activities 0.11 and 0.01 PEPU/g, respectively.
- a proline-specific endoprotease is used as an alternative to PVPP or silica-gel treatment to prevent chill haze formation.
- the enzymes proline-specific endoprotease
- ALDC and PEP enzymes are added to the beer fermentation process to increase the flexibility in the brewery and to save capital costs.
- ⁇ -Acetolactate Decarboxylase activity (ADU/g) were determined throughout stability period after 0, 1, 3, 24 and 48 hours and results are shown in table 6 and 7.
- the ALDC samples aldB_Bs and aldB_Bl showed no loss of ⁇ -Acetolactate Decarboxylase activity at both 5°C and 30°C for up to 48 hours. Variation in Acetolactate Decarboxylase activity is estimated up to 10%.
- No ALDC activity (0 ADU/g) was found in samples of proline-specific endoproteases: AniPro_2 or AnPro, throughout stability test period respectively.
- aldB_Bs blended with either AniPro_2 or AnPro showed no loss of ⁇ - Acetolactate Decarboxylase activity at both 5°C and 30°C for up to 48 hours.
- aldB_Bl blended with AniPro_2 showed 82% and 48% residual activity at 5°C and 30°C after 48 hours.
- aldB_Bl blended with AnPro showed less stability having only 68% and 6% residual activity at 5°C and 30°C after 48 hours.
- aldB_Bl is clearly sensitive towards blending with proline- specific endoproteases as compared with aldB_Bs.
- ALDC ⁇ -Acetolactate Decarboxylase activity (ADU/g) of aldB_Bs, aldB_Bl, AniPro_2, AnPro and combinations samples mixed 50:50 % (w/w) (aldB_Bs + AniPro_2, aldB_Bl + AniPro_2, aldB_Bs + AnPro and aldB_Bl + AnPro), stored at 5°C. Activity of all samples were determined in duplicates up to 48hrs, having residuals calculated against activity at 0 hrs.
- PEPU post-proline, protease cleavage activity
- EXAMPLE 10 Reduction in diacetyl and 2,3-pentanedione during beer fermentation by use of aldB
- the objective of this analysis was to test acetolactate decarboxylase ALDC (aldB) expressed in both B. subtilis and B. licheniformis in combination with proline-specific endoprotease ability to reduce development of diacetyl and 2,3-pentanedione (Vicinal di-ketones, VDK) during a 7-day fermentation at 14°C.
- Pure malt brew analysis 1100 g Malt Extract, Harboe Barlex 7203 Light Malt Extract (Batch 2139121, expiry date 02.12.2024) was dissolved in 6000 ml warm tapwater (45°C).
- Each conical flask was dosed with 0.5% W34/70 (Weihenstephan) freshly produced yeast (1.0g yeast per 200g wort).
- the enzymes were dosed according to table 10, all at the start of the fermentation. Two fermentation trials were performed for each enzyme addition. Table 10 Dosage of ALDC enzymes (aldB_Bs and aldB_Bl), PEP enzyme (AniPro_2) and combinations of ALDC and PEP; AniPro_2 + aldB_Bs, AniPro_2 + aldB_Bl and AnPro + aldB_Bl in strat of beer fermentation trials.
- This slurry was transferred to a headspeace vial and heat-treated at 65°C for 30 minutes before analysis for diacetyl and 2,3-pentanedione were carried out by gas chromatography with mass spectrometric detection (GCMS).
- GCMS gas chromatography with mass spectrometric detection
- Analyses were carried out at an Agilent 6890N/5973N GC with CombiPAL headspace autosampler and MSChemStation acquisition and analysis software.
- the samples were equilibrated at 70°C for 10 minutes before 500 ⁇ l of the gas phase above the sample was injected onto a J&W 122-0763 DB-1701column (60m x 0.25mmID x 1 ⁇ m).
- the injection temperature was 260 o C and the system was operated with a constant helium flow of 2 ml/min.
- the oven temperature was: 50 o C (2 min), 160 o C (20 o C/min), 220°C (40°C/min), hold 2 min.
- MS detection were made with 500 ⁇ L at a split ratio of 5:1at selected ions. All sample were run in duplicates and standards were made using tap water with the addition of diacetyl or 2,3-pentanedione.
- RDF Real Degree of Fermentation
- RDF was measured using an Anton Paar (DMA 5000) following Standard Instruction Brewing, 23.8580-B28 and alcohol by Standard Instruction Brewing, 23.8580-B28.
- RDF Real degree of fermentation
- VDK diacetyl and 2,3-pentanedione
- aldB_Bl on its own or as blend (bl- 50:50%) with AnPro, as blend (bl) with AniPro_2.
- aldB_Bs on its own or as blend (bl- 50:50%) with AnPro, as blend (bl) with AniPro_2 is applied.
- AniPro_2 on its own is shown as well as control (Ctrl) sample without enzyme addition.
- the maximum VDK reached during fermentation and number of fermentation hours required to go below VDK threshold level of 0.1 mg/L, for samples treated with aldB_Bs or aldB_Bl on their own or in combination with one of the two PEP enzymes are shown in Figure 3a and b for ALDC and PEP applied individually.
- aldB_Bl was more negatively impacted than aldB_Bs. And mostly impacted when AnPro was used as PEP enzyme, seen with the requirement of 164 hours fermentation compared to 137 hours with AniPro_2 to reach VDK of 0.1 mg/L. Thus, aldB_Bl is found more sensitive for being used together with PEP enzyme during fermentation, than aldB_Bs. We acknowledge that the relative increase in hours to reach 0.1 mg VDK per L ferment seemed affected for both aldB enzymes. However, these estimates were extrapolated from very few data points. Moreover, aldB_Bl was less impacted when used together with AniPro_2 compared to AnPro and when the ALDC and PEP enzymes were applied separately to the beer fermentation.
- Table 12 Average Diacetyl, 2,3-pentanedione and their sum VDK content throughout all malt beer fermentation from 0 to 164 hours. Control without enzyme, AniPro_2 + aldB_Bs 0.5 g/hL + 0.5 g/hL, AniPro_2 + aldB_Bl 0.5 g/hL + 0.5 g/hL and AnPro + aldB_Bl 0.5 g/hL + 0.5 g/hL.
- aldB_Bs performed good when pre-mixed with AniPro_2 and delivered same VDK reduction as when applied on its own (0.331 versus 0.314 mg/L) and slightly worse when pre-mixed with AnPro (0.354 versus 0.314 mg/L), see also figure 4a and 4b.
- aldB_Bl was much more sensitive for being pre-mixed with PEP enzyme where max VDK increased from 0.462 to 0.771 mg/L when pre-mixed with AniPro_2 and to 1.401 mg/L when premixed with AnPro.
- the turbidity was measured at 20°C, termed Blind Value.
- the samples were then placed in a thermostatic water bath (Julabo, Germany), and the temperature was decreased to 0°C and kept for 24 hours.
- the turbidity was measured at 0°C and was termed Initial Total Haze.
- the beer samples were replaced in the thermostatic water bath and the temperature was increased to 60°C which was kept for 48 hours followed by a decrease in temperature to 0°C which was kept for 24 hours.
- the turbidity was measured at 0°C and was termed Final Total Haze.
- the ALDC enzymes (aldB_Bs and aldB_Bl) showed no significant influence on haze on their own. However, both ALDC enzymes showed minor but significant negative influence on both proline specific endoproteases. This effect was highest at low dosage of proline specific endoproteases (0.5 g/hL), however observed for haze evaluated by EBC 25° and EBC 90° and most significant at Final Total haze. The negative effect was largest for aldB_Bl as compared to aldB_Bs in all cases, this not taking into account that aldB_Bl only contains 4020 ADU/g as compared to aldB_Bs containing 10396 ADU/g.
- the gel was cooled to 20°C rinsed 3 times 5 min in water, then stained in SafeStain (SimplyBlue SafeStain from Invitrogen) overnight and finally destained in water. Then the gel was transferred to Geldoc Go Imaging System from BioRad. Image Lab software was used for calculation of intensity of each band. By knowing the protein amount of the standard sample, the calibration curve can be made. The amount of protein band variants was be determined relative to the band intensity as shown on gel. Similar to samples prepared in example 9, we studied stability of ALDC mixed with proline-specific endoprotease.
- AldB_Bs and AldB_Bl enzyme protein (Mw: 31-35kDa) are stable individually and bands do not seem to change upon incubation for 24 hours at 30°C. The same was observed for PEPs AniPro_2 and AnPro (Mw: 56-62kDa).
- combinations (aldB_Bs + AniPro_2, aldB_Bl + AniPro_2, aldB_Bs + AnPro and aldB_Bl + AnPro) made as 50:50 % (w/w) blend, showed difference in band pattern development upon incubation 24 hours at 30°C.
- aldB_Bl truncation variant (14.5kDa) correspond approximately to the calculated mass (A28-K162) of 14.9kDa and aldB_Bl truncation variant (7.8kDa) correspond approximately to the calculated mass (A28-K109) of 8.7kDa.
- Table 17 Identified N- and C-terminus positions of aldB_Bl variants, position according to according to SEQ ID NO. 2.
- Position N- Position C- EXAMPLE 13 Construction of Strain BRA8014
- the ALDC strain is a descent of B. subtilis subsp. subtilis. In this strain the protease genes were removed by homologous recombination, similar to the way other genes were introduced.
- the epr, isp, bpf,vpr, wprA, mpr-ybfJ, nprB, and alrA genes were deleted, and the comK gene was introduced at amyE locus, yielding the final host strain BG6014. Then the plasmid alrA(CB)RIHI-Bbrev containing the Brevibacillus brevis acetolactate decarboxylase (ALDC) gene, transcribed by the aprE promoter and fused to the alrA gene used as a selectable marker was transformed into strain BG6014. Finally, the competence regulatory gene comK was removed again from the strain, resulting in strain BRA8014.
- ADC Brevibacillus brevis acetolactate decarboxylase
- EXAMPLE 15 Deletion of neutral protease
- the next step in the strain construction was the introduction of a deletion in a second extracellular protease, the neutral protease (nprE) (SEQ ID NO:19).
- nprE neutral protease
- recombinant DNA techniques were used to create a deletion in the nprE gene in a research strain (Yang et al., J. Bacteriology 160, 15-61 (1984)) before introduction of the mutated gene into the aprE deleted strain.
- EXAMPLE 20 Removal of vpr gene An in-vitro created deletion of the wild-type vpr gene (extra cellular serine protease) (SEQ ID NO:24) was introduced into B. subtilis.
- the plasmid vpr/pUCTsKan which carries a deletion of 650 base pairs in the middle of the vpr gene, was transformed into the strain by natural competence. This deletion is carried by a plasmid bearing a kanamycin resistance gene (kan) and a temperature sensitive origin of replication (TsOri).
- this plasmid integrated into the chromosome at the region of homology with the vpr gene at the non-permissive temperature, e.g., 48 °C. After integration, the strain carrying the integrated plasmid, was grown extensively at permissive temperature and in the absence of kanamycin. This allows the excision and loss of the plasmid giving rise either to the wt vpr sequence, or to a deletion mutant, which has lost vpr gene.
- EXAMPLE 21 Removal of mpr-ybfJ genes: The entire mpr gene (extra-cellular serine protease) (SEQ ID NO:25), including the upstream ribosome binding site, and most of the downstream overlapping ybfJ gene (SEQ ID NO:26) were deleted (1.2 kb) using a cre-lox method essentially as described (Yan, X, Hao-Jie, Y, Hong, Q, and Shun-Peng, L, Appl. Env. Microbiol., 74:5556-5562, 2008). The integration of the deletion DNA by a double cross-over was selected using a spectinomycin resistance gene flanked by lox sites.
- the spectinomycin gene was then removed by the introduction of a plasmid with a temperature sensitive origin of replication (TsOri) that expresses the cre recombinase gene under the control of the Pspac promoter.
- the TsOri plasmid can be selected by the phleomycin resistance gene present on the vector. Strains that have lost the spectinomycin resistance gene by recombination at the lox sites were confirmed by sensitive to spectinomycin.
- the TsOri plasmid was eliminated by growth at a non-permissive temperature (e.g., 42 o C) and the loss of the plasmid detected by phleomycin sensitivity. The correct deletion was double-checked by PCR analysis.
- the aldB gene was made synthetically (IDT) with the signal peptide from the B. subtilis aprE gene (Ferrari et al. (1988) J. Bacteriol. 170(1), 289-95) fused to the mature protein coding region of aldB.
- the synthetic aldB gene sequence was the same as reported previously (Diderichsen et al., 1990, surpa), however, two restriction sites (KpnI and BsrGI) were removed by silent mutations.
- Plasmid DNA from pCB-EZ1-alrA was used as a template to amplify the pCB-alrA vector backbone using the primers: AL2 (5’- ACTAGTTACCCTCTCCTTTTAAAAA-3’) (SEQ ID NO:29) and AL9 (5’- ATAAAAGCTTACATAAAAAACCGGCCT-3’) (SEQ ID NO:30).
- AL2 5’- ACTAGTTACCCTCTCCTTTTAAAAA-3’
- AL9 5’- ATAAAAGCTTACATAAAAAACCGGCCT-3’
- this PCR fragment on one end contains approximately 600 bp of the B. subtilis aprE promoter starting at the EcoRI site upstream of the aprE promoter and ending at a SpeI site introduced at the GTG start codon (from the original pBNppt based plasmid).
- subtilisin BPN’
- Bacillus amyloliquefaciens on an approximately 250 bp HindIII BamHI fragment (also from the original pBNppt based plasmid).
- the mature aldB coding sequence including the B. subtilis aprE signal sequence was amplified by PCR using the following primers: AL3 (5’- AACTAGTGAGAAGCAAAAAATTGTG-3’) (SEQ ID NO:31) and AL10 (5’- AGCTTTTATTTTCTTTCTGACTCAGCT-3’) (SEQ ID NO:32).
- the amplicons were purified using the Wizard PCR Clean Up KIT following the manufactures instructions.
- the alrA expression cassette was generated by PCR using plasmid DNA from pUCalrA(CB)RIHI as a template with the primers: AL19, alrA(CB)RIHI Fwd (5’- GGATCCTGACTGCCTGAG-3’) (SEQ ID NO:33) and AL20, alrA(CB)RIHI Rev (5’- GGAGAAAGGCCAAACATG-3’) (SEQ ID NO:34).
- the aldB expression cassette was amplified by PCR using the pCB-alrA-Brev as a template with the following primers: AL21, Bbrev-RIHI Fwd (5’- TGTTTGGCCTTTCTCCGAATTCCTCCATTTTCTTCTG-3’) (SEQ ID NO:35) and AL22, Bbrev-RIHI Rev (5’- AGGCAGTCAGGATCCGATTACGAATGCCGTCTC-3’) (SEQ ID NO:36).
- the amplicons were column cleaned by the wizard PCR clean up KIT following manufactures instructions.
- telomeres were patched onto a fresh 500 ppm CDA plate and grown at 37°C for 1 day. A colony was selected named BRA7997.
- the aldB gene was verified by DNA sequencing.
- PCR was performed using the primers ydoC400F (GGATGTCGCCACAAGCGCAAAGCCTTCC) (SEQ ID NO:37) and alrA-ATG-R (ATCGCGGACAAGTCAATTTCCGCCCAC) (SEQ ID NO:38), which should give a band of about 537 bp if the integration is at the alrA locus and no band if it is not.
- integration at the aprE locus was assessed by PCR using the primers yhfO-RI-R (CGTTGGATAGAGCTGGGTAAAGCCTATG) (SEQ ID NO:39) and Bbrev-R(aprE) (CGTTGAGTATTGAAACAGTA) (SEQ ID NO:40), which should give a band of about 832 bp if the integration is at aprE locus and no band if it is not.
- the site of integration was verified to be at the aprE locus. While preferred embodiments of the present invention have been shown and described herein, it will be obvious to those skilled in the art that such embodiments are provided by way of example only.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Genetics & Genomics (AREA)
- Zoology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Wood Science & Technology (AREA)
- General Engineering & Computer Science (AREA)
- Biochemistry (AREA)
- General Health & Medical Sciences (AREA)
- Biomedical Technology (AREA)
- Molecular Biology (AREA)
- Biotechnology (AREA)
- Microbiology (AREA)
- Medicinal Chemistry (AREA)
- Food Science & Technology (AREA)
- Mycology (AREA)
- Physics & Mathematics (AREA)
- Biophysics (AREA)
- Plant Pathology (AREA)
- Enzymes And Modification Thereof (AREA)
Abstract
The present invention relates to novel, stable forms of acetolactate decarboxylases (ALDCs) which have improved stability in the presence of proteases. More particularly, the present invention relates to an improved brewing process where stable ALDCs are used in conjunction with proline specific endoproteases during beer fermentation to provide a beer having less off-flavor which is colloidally stable in a shortened time.
Description
PROTEASES AND ALDC ENZYMES FOR BEER HAZE AND DIACETYL REDUCTION CROSS-REFERENCE TO RELATED APPLICATIONS This application claims priority to U.S. Application No. 63/580,057, filed September 1, 2023, which is hereby incorporated by reference in its entirety. TECHNICAL FIELD The present invention relates to novel, stable forms of acetolactate decarboxylases (ALDCs) which have improved stability in the presence of proteases. More particularly, the present invention relates to an improved brewing process where stable ALDCs are used in conjunction with proline specific endoproteases during beer fermentation to provide a beer having less off-flavor which is colloidally stable in a shortened time. REFERENCE TO A SEQUENCE LISTING The contents of the electronic submission of the text file Sequence Listing, named “NB42237USPSP_SequenceListing.xml” was created on August 31, 2023, and is 60 KB in size, which is hereby incorporated by reference in its entirety. BACKGROUND In industrial beer brewing, the overall speed of the brewing process is of paramount importance. Holding and chilling large volumes of beer dramatically impacts capital costs in terms of energy and storage space. Before bottling, beer typically must undergo both a maturation and a stabilization phase. The stabilization phase provides colloidal stability to the beer so that it can be stored in cold conditions pending consumer purchase. Maturation is a phase required for ridding the beer of undesirable off-flavors. Beer tends to be colloidally unstable in that a haze will form after the beer is bottled and refrigerated prior to sale. This phenomenon is called chill haze. When beer is cooled, polyphenols in the beer interact with proline rich proteins (haze active proteins) and form a precipitate or haze. Chill haze is highly undesirable in a bottled beer. A cold stabilization phase
prior to beer filtration and bottling helps to eliminate or at least reduce chill haze. However, a typical stabilization phase can be seven days or longer and require chilling to 0 or even -2°C. The energy costs for bringing thousands of hectoliters of beer to this low temperature are substantial. Fermentation (converting fermentable sugars in the wort to alcohol) produces what is called “green beer”. Green beer contains high levels of undesirable flavor components, notably diketones, such as diacetyl. Diacetyl has a strong buttery off-flavor and is considered highly undesirable in many beers. The conversion of diacetyl into bland tasting compounds is an important aspect of the subsequent beer maturation phase. However, the reduction of diacetyl into tasteless acetoin is a time-consuming process but of paramount importance. Enzymes have been employed to shorten or eliminate the stabilization and/or maturation phase. For example, it is known in the art to use protease to decrease the stabilization phase. An appropriate protease can be employed to selectively degrade proteins in the beer that can bind to polyphenols and cause colloidal instability of the beer. Acetolactate decarboxylase (ALDC) can be used to convert α-acetolactate into the flavorless acetoin, shortening the maturation phase. However, protease added to a fermenting beer to provide colloidal stability can possibly proteolyze other exogenously added enzymes, including ALDC enzymes. Whether further shortening of the brew process can be accomplished by combining a protease with an ALDC enzyme in the same step is unclear. There is a continuing need for ALDC enzymes which have lower susceptibility to proteolysis and for proteases that can be used for increasing beer colloidal stability the have greater specificity for haze active proteins. SUMMARY OF THE INVENTION In accordance with an aspect of the present invention, it has been discovered that ALDC enzymes which are used in brewing for shortening the maturation phase are unstable in the presence of proline specific proteases which are used to increase colloidal stability of the beer. The loss of ALDC by degradation can prolong the maturation phase as opposed to shortening it. Accordingly, in an aspect of the present invention, a polypeptide having acetolactate decarboxylase activity which is resistant to protease inactivation is presented, the polypeptide having an amino acid sequence with at least 80, 90, 95, 98, 99 or 100% sequence identity to SEQ
ID NO:3 wherein the polypeptide has at most 250 amino acids. Optionally, the polypeptide is at most 249, 248, 247, 246, 245, 244, 243, 242 or 241 amino acids. Optionally, the polypeptide having acetolactate decarboxylase activity which is resistant to protease inactivation has an amino acid sequence having at least 80, 90, 95, 98, 99 or 100% sequence identity to according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12. Optionally, the polypeptide has an amino acid sequence according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12. The protease to which the ALDC is resistant is optionally a proline specific protease. More preferably, the proline specific protease is from Aspergillus niger. Optionally, the protease has at least 80, 85, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:17. Optionally, the protease has an amino acid sequence according to SEQ ID NO:17. In another aspect of the present invention, an improved brewing process is presented having the step of fermenting a wort in the presence of a polypeptide having acetolactate decarboxylase activity which is resistant to protease inactivation and a proline specific protease wherein both enzymes are present in the wort at the same time. Optionally, the polypeptide has an amino acid sequence with at least 80, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:3 wherein the polypeptide has at most 250 amino acids. Optionally, the polypeptide is at most 249, 248, 247, 246, 245, 244, 243, 242 or 241 amino acids. Optionally, the polypeptide having acetolactate decarboxylase activity which is resistant to protease inactivation has an amino acid sequence having at least 80, 90, 95, 98, 99 or 100% sequence identity to according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12. Optionally, the polypeptide has an amino acid sequence according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12. Optionally, the polypeptide having acetolactate decarboxylase activity is added to the wort first. In other preferred embodiments, the proline specific protease is added to the wort first. Optionally, the polypeptide having acetolactate decarboxylase activity and the proline specific protease are added simultaneously to the wort. The proline specific protease is optionally from Aspergillus niger. Optionally, the proline specific protease has at least 80, 85, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:17.
In another aspect of the present invention, a Bacillus host cell is presented for producing a heterologous polypeptide of interest, wherein one or more protease genes are inactivated. Optionally, the polypeptide of interest is expressed without a secretion signal peptide. Optionally, the polypeptide of interest is expressed with a secretion signal. Optionally, the polypeptide of interest is an enzyme. Optionally, the enzyme is an oxidoreductase, transferase, hydrolase, lyase, isomerase, ligase, aminopeptidase, amylase, asparaginase, carbohydrase, carboxypeptidase, catalase, cellulase, chitinase, cutinase, cyclodextrin glycosyltransferase, deoxyribonuclease, esterase, alpha-galactosidase, beta- galactosidase, glucoamylase, alpha-glucosidase, beta-glucosidase, hyaluronic acid synthase, invertase, laccase, lipase, mannosidase, mutanase, oxidase, a pectinolytic enzyme, peroxidase, phytase, polyphenoloxidase, protease, ribonuclease, transglutaminase, or xylanase. Optionally, the enzyme is an ALDC enzyme. Optionally, the ALDC enzyme is a polypeptide with an amino acid sequence having at least 80, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:3 wherein the polypeptide has at most 250 amino acids. Optionally, the polypeptide is at most 249, 248, 247, 246, 245, 244, 243, 242 or 241 amino acids. Optionally, the polypeptide having acetolactate decarboxylase activity has an amino acid sequence having at least 80, 90, 95, 98, 99 or 100% sequence identity to according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12. Optionally, the polypeptide has an amino acid sequence according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12. Optionally, the ALDC enzyme is expressed with a secretion signal. Optionally, the heterologous polypeptide of interest is encoded by an exogenous polynucleotide integrated into the chromosome of the host cell in at least one copy. Optionally, the exogenous polynucleotide is a nucleic acid sequence having 80, 85, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:28. Optionally, the exogenous polynucleotide is a nucleic acid sequence according to SEQ ID NO:28. Optionally, the one or more protease gene is inactivated by a non-sense mutation in said one or more gene, a partial deletion of said in the one or more gene or a full deletion of the one or more gene. Optionally, the Bacillus host cell is Bacillus subtilis or Bacillus licheniformis. Optionally, the Bacillus host cell is Bacillus subtilis.
Optionally, the one or more protease genes are selected from the group consisting of a nucleic acid sequence having at least 80, 85, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:18, SEQ ID NO:19, SEQ ID NO:20, SEQ ID NO:21, SEQ ID NO:22, SEQ ID NO:23, SEQ ID NO:24, SEQ ID NO:25 and SEQ ID NO:27. Optionally, the one or more protease genes comprise nine inactivated proteases wherein the proteases have nucleic acid sequences with at least 80, 85, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:18, SEQ ID NO:19, SEQ ID NO:20, SEQ ID NO:21, SEQ ID NO:22, SEQ ID NO:23, SEQ ID NO:24, SEQ ID NO:25 and SEQ ID NO:27. Optionally, the nine inactivated proteases genes have nucleic acid sequences according to SEQ ID NO:18, SEQ ID NO:19, SEQ ID NO:20, SEQ ID NO:21, SEQ ID NO:22, SEQ ID NO:23, SEQ ID NO:24, SEQ ID NO:25 and SEQ ID NO:27. In another aspect of the present invention, a method for producing a polypeptide of interest is presented having the steps of: i) providing a Bacillus host cell wherein one or more protease genes are inactivated and wherein said host cell is transformed with a nucleic acid encoding a heterologous polypeptide in operable combination with a promoter; and ii) cultivating said host cell under conditions suitable for the production of said heterologous polypeptide, such that said heterologous polypeptide is produced. Optionally, the method has the further step of recovering the produced polypeptide. Optionally, the polypeptide of interest is expressed with or without a secretion signal peptide. Optionally, the polypeptide of interest is expressed with a secretion signal peptide. Optionally, the polypeptide of interest is an enzyme. Optionally, the enzyme is an oxidoreductase, transferase, hydrolase, lyase, isomerase, ligase, aminopeptidase, amylase, asparaginase, carbohydrase, carboxypeptidase, catalase, cellulase, chitinase, cutinase, cyclodextrin glycosyltransferase, deoxyribonuclease, esterase, alpha-galactosidase, beta- galactosidase, glucoamylase, alpha-glucosidase, beta-glucosidase, hyaluronic acid synthase, invertase, laccase, lipase, mannosidase, mutanase, oxidase, a pectinolytic enzyme, peroxidase, phytase, polyphenoloxidase, protease, ribonuclease, transglutaminase, or xylanase. Optionally, the enzyme is an ALDC enzyme. Optionally, the ALDC enzyme is a polypeptide with an amino acid sequence having at least 80, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:3 wherein the polypeptide has at most 250 amino acids. Optionally, the polypeptide is at most 249, 248, 247, 246, 245, 244, 243, 242 or 241 amino acids. Optionally, the polypeptide having acetolactate decarboxylase activity has an amino acid
sequence having at least 80, 90, 95, 98, 99 or 100% sequence identity to according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12. Optionally, the polypeptide has an amino acid sequence according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12. Optionally, the ALDC enzyme is expressed with a secretion signal. Optionally, the heterologous polypeptide of interest is encoded by an exogenous polynucleotide integrated into the chromosome of the host cell in at least one copy. Optionally, the exogenous polynucleotide is a nucleic acid sequence having 80, 85, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:28. Optionally, the exogenous polynucleotide is a nucleic acid sequence according to SEQ ID NO:28. Optionally, the one or more protease gene is inactivated by a non-sense mutation in said one or more gene, a partial deletion of said in the one or more gene or a full deletion of the one or more gene. Optionally, the Bacillus host cell is Bacillus subtilis or Bacillus licheniformis. Optionally, the Bacillus host cell is Bacillus subtilis. Optionally, the one or more protease genes are selected from the group consisting of a nucleic acid sequence having at least 80, 85, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:18, SEQ ID NO:19, SEQ ID NO:20, SEQ ID NO:21, SEQ ID NO:22, SEQ ID NO:23, SEQ ID NO:24, SEQ ID NO:25 and SEQ ID NO:27. Optionally, the one or more protease genes comprise nine inactivated proteases wherein the proteases have nucleic acid sequences with at least 80, 85, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:18, SEQ ID NO:19, SEQ ID NO:20, SEQ ID NO:21, SEQ ID NO:22, SEQ ID NO:23, SEQ ID NO:24, SEQ ID NO:25 and SEQ ID NO:27. Optionally, the nine inactivated proteases genes have nucleic acid sequences according to SEQ ID NO:18, SEQ ID NO:19, SEQ ID NO:20, SEQ ID NO:21, SEQ ID NO:22, SEQ ID NO:23, SEQ ID NO:24, SEQ ID NO:25 and SEQ ID NO:27. In another aspect of the present invention, a stable liquid formulation comprising a polypeptide having acetolactate decarboxylase activity having an amino acid sequence with at least 80, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:3 wherein said polypeptide has at most 250 amino acids and a protease. Optionally, the polypeptide is at most 249, 248, 247, 246, 245, 244, 243, 242 or 241 amino acids.
Optionally, the polypeptide having acetolactate decarboxylase activity has an amino acid sequence having at least 80, 90, 95, 98, 99 or 100% sequence identity to according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12. Optionally, the polypeptide has an amino acid sequence according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12. The protease is optionally a proline specific protease. Optionally, the proline specific protease is from Aspergillus niger. Optionally, the protease has at least 80, 85, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:17. Optionally, the protease has an amino acid sequence according to SEQ ID NO:17. Optionally, the ALDC polypeptide maintains at least 99, 98, 95, 90, 85, 80, 75 or 70% activity when the stable liquid formulation is stored at 10oC for 30, 60, 90, 120, 150 for 180 days. Optionally, the ALDC polypeptide maintains at least 99, 98, 95, 90, 85, 80, 75 or 70% activity when the stable liquid formulation is stored at 30oC for 8, 16, 24, 32, 40 or 48 hours. In another aspect of the present invention, a proline specific protease formulation is presented which is substantially depleted of other protease activities comprising the protease wherein when said protease is combined with a polypeptide having acetolactate decarboxylase activity, the polypeptide is stable over time. Optionally, the polypeptide having acetolactate decarboxylase activity has an amino acid sequence having at least 80, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:3 wherein said polypeptide has at most 250 amino acids and a protease. Optionally, the polypeptide is at most 249, 248, 247, 246, 245, 244, 243, 242 or 241 amino acids. Optionally, the polypeptide having acetolactate decarboxylase activity has an amino acid sequence having at least 80, 90, 95, 98, 99 or 100% sequence identity to according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12. Optionally, the polypeptide has an amino acid sequence according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12. The protease is optionally a proline specific protease. Optionally, the proline specific protease is from Aspergillus niger. Optionally, the protease has at least 80, 85, 90, 95, 98, 99 or
100% sequence identity to SEQ ID NO:17. Optionally, the protease has an amino acid sequence according to SEQ ID NO:17. Optionally, the ALDC polypeptide maintains at least 99, 98, 95, 90, 85, 80, 75 or 70% activity when the stable liquid formulation is stored at 10oC for 30, 60, 90, 120, 150 for 180 days. Optionally, the ALDC polypeptide maintains at least 99, 98, 95, 90, 85, 80, 75 or 70% activity when the stable liquid formulation is stored at 30oC for 8, 16, 24, 32, 40 or 48 hours. BRIEF DESCRIPTION OF THE BIOLOGICAL SEQUENCES SEQ ID NO:1 sets forth the nucleotide sequence of the wild type aldB gene. SEQ ID NO:2 sets forth the amino acid sequence of the ALDC aldB precursor protein. SEQ ID NO:3 sets forth the predicted amino acid sequence of the mature acetolactate decarboxylase (ALDC) aldB. SEQ ID NO:4 sets forth the nucleotide sequence of the mature form of aldB gene in plasmid alrA(CB)RIHI-Bbr. SEQ ID NO:5 sets forth the amino acid sequence of the aldB precursor protein expressed from plasmid alrA(CB)RIHI-Bbr. SEQ ID NO:6 sets forth the amino acid sequence of aldB_BS truncation variant 1. SEQ ID NO:7 sets forth the amino acid sequence of aldB_BS truncation variant 2. SEQ ID NO:8 sets forth the amino acid sequence of aldB_BS truncation variant 3. SEQ ID NO:9 sets forth the amino acid sequence of aldB_BS truncation variant 4. SEQ ID NO:10 sets forth the amino acid sequence of aldB_BS truncation variant 5. SEQ ID NO:11 sets forth the amino acid sequence of aldB_BS truncation variant 6. SEQ ID NO:12 sets forth the amino acid sequence of aldB_BS truncation variant 7. SEQ ID NO:13 sets forth the amino acid sequence of aldB_Bl truncation variant 1. SEQ ID NO:14 sets forth the amino acid sequence of aldB_Bl truncation variant 2. SEQ ID NO:15 sets forth the amino acid sequence of aldB_Bl truncation variant 3. SEQ ID NO:16 sets forth the AniPro_2 precursor protein. SEQ ID NO:17 sets forth the AniPro_2 mature protein.
SEQ ID NO:18 sets forth the Bacillus subtilis aprE gene sequence. SEQ ID NO:19 sets forth the Bacillus subtilis nprE gene sequence. SEQ ID NO:20 sets forth the Bacillus subtilis nprE gene sequence. SEQ ID NO:21 sets forth the Bacillus subtilis ispA gene sequence. SEQ ID NO:22 sets forth the Bacillus subtilis bpr gene sequence. SEQ ID NO:23 sets forth the Bacillus subtilis wprA gene sequence. SEQ ID NO:24 sets forth the Bacillus subtilis vpr gene sequence. SEQ ID NO:25 sets forth the Bacillus subtilis Mpr gene sequence. SEQ ID NO:26 sets forth the Bacillus subtilis ybfj gene sequence. SEQ ID NO:27 sets forth the Bacillus subtilis nprB gene sequence. SEQ ID NO:28 sets forth the DNA sequence of aldB gene fused to aprE signal peptide. SEQ ID NO:29 sets forth the AL2 primer. SEQ ID NO:30 sets forth the AL9 primer. SEQ ID NO:31 sets forth the AL3 primer. SEQ ID NO:32 sets forth the AL10 primer. SEQ ID NO:33 sets forth the AL19, alrA(CB)RIHI Fwd primer. SEQ ID NO:34 sets forth the AL20, alrA(CB)RIHI Rev primer. SEQ ID NO:35 sets forth the AL21, Bbrev-RIHI Fwd primer. SEQ ID NO:36 sets forth the AL22, Bbrev-RIHI Rev primer. SEQ ID NO:37 sets forth the ydoC400F primer. SEQ ID NO:38 sets forth the alrA-ATG-R primer. SEQ ID NO:39 sets forth the yhfO-RI-R primer. SEQ ID NO:40 sets forth the Bbrev-R(aprE) primer. BRIEF DESCRIPTION OF THE DRAWINGS Figure 1 shows a plasmid map for pCB_alr. Figure 2 shows a plasmid map of alrA(CB)RIHI-Bbr for expression of Acetolactate Decarboxylase, aldB. Figures 3A-B shows a) the maximum VDK levels reached during all-malt beer fermentation with combination of ALDC and PEP and b) number of hours to decrease VDK to 0.100 mg/L to the right with combination of ALDC and PEP applied as single additions.
Figures 4A-B shows a) the maximum VDK levels reached during all-malt beer fermentation with combination of ALDC and PEP (added as premixed blend kept 6hours at 30°C) and b) number of hours to decrease VDK to 0.100 mg/L to the right with combination of ALDC and PEP applied as premixed blend kept 6 hours at 30°C. Figures 5A-B shows the turbidity (EBC 90°) of beer with and without proline-specific protease, ALDC and combinations hereof added. Forced Haze was measured according to EBC TOHA method and A) Initial Total haze and B) Final Total haze are shown. Standard deviation was determined from 2 determinations. All enzymes dosed at 0.5 or 2.0 g/hL. Figures 6A-B shows the turbidity (EBC 25°) of beer with and without proline-specific protease, ALDC and combinations hereof added. Forced Haze was measured according to EBC TOHA method and A) Initial Total haze and B) Final Total haze are shown. Standard deviation was determined from 2 determinations. All enzymes dosed at 0.5 or 2.0 g/hL. Figures 7A-E shows SDS-PAGE showing combinations of ALDC (aldB, 29-32 kDa) and PEP (56-62kDa) or individual in samples retained 30°C for up to 24 hours. Truncation variants of aldB in pre-mix with PEP shown at lower molecular weight (aldB truncations < 28 kDa). Figures 8A-B shows Vicinal Di-Ketone (VDK) generation during beer fermentation with the use of aldB_Bl on its own or as blend (bl- 50:50%) with AnPro, as blend (bl) with AniPro_2. aldB_Bs on its own or as blend (bl- 50:50%) with AnPro, as blend (bl) with AniPro_2 is applied. Further AniPro_2 on its own is shown as well as control (Ctrl) sample without enzyme addition. DETAILED DESCRIPTION OF THE INVENTION Definitions The term “amino acid sequence” is synonymous with the terms “polypeptide,” “protein,” and “peptide,” and are used interchangeably. Where such amino acid sequences exhibit activity, they may be referred to as an “enzyme.” The conventional one-letter or three- letter codes for amino acid residues are used, with amino acid sequences being presented in the standard amino-to-carboxy terminal orientation (i.e., N→C). The term “nucleic acid” encompasses DNA, RNA, heteroduplexes, and synthetic molecules capable of encoding a polypeptide. Nucleic acids may be single stranded or double stranded. The terms “nucleic acid” and “polynucleotide” are used interchangeably. Because the
genetic code is degenerate, more than one codon may be used to encode a particular amino acid, and the present compositions and methods encompass nucleotide sequences that encode a particular amino acid sequence. Unless otherwise indicated, nucleic acid sequences are presented in 5′-to-3′ orientation. A “vector” refers to a polynucleotide sequence designed to introduce nucleic acids into one or more cell types. Vectors include cloning vectors, expression vectors, shuttle vectors, plasmids, phage particles, cassettes, and the like. An “expression vector” refers to a DNA construct comprising a DNA sequence encoding a polypeptide of interest, which coding sequence is operably linked to a suitable control sequence capable of effecting expression of the DNA in a suitable host. Such control sequences may include a promoter to effect transcription, an optional operator sequence to control transcription, a sequence encoding suitable ribosome binding sites on the mRNA, enhancers and sequences which control termination of transcription and translation. In addition to the specific amino acid sequences and polynucleotides mentioned herein, the present invention encompasses variants, homologues, derivatives, and fragments thereof. The term "variant" is used to mean a nucleotide sequence or amino acid sequence which differs from a wild-type sequence. For example, a variant may include substitutions, insertions, deletions, truncations, transversions and/or inversions at one or more position(s) relative to a wild-type sequence. Variants can be made using methods known in the art for example site scanning mutagenesis, insertional mutagenesis, random mutagenesis, site-directed mutagenesis, and directed-evolution as well as using recombinant methods well known in the art. Polynucleotide sequences encoding variant amino acid sequences may readily be synthesized using methods known in the art. In some aspects, the variant is a naturally occurring nucleotide sequence or amino acid sequence which differs from a wild-type sequence. For example, the variant may be a natural genetic variant. In some aspects, the variant is an engineered variant. For example, the variant may be engineered by recombinant methods. The protein sequences of the instant invention may also have deletions, insertions or substitutions of amino acid residues which produce a silent change and result in a functionally equivalent substance. Deliberate amino acid substitutions may be made based on similarity in
polarity, charge, solubility, hydrophobicity, hydrophilicity, and/or the amphipathic nature of the residues as long as the secondary binding activity of the substance is retained. For example, negatively charged amino acids include aspartic acid and glutamic acid; positively charged amino acids include lysine and arginine; and amino acids with uncharged polar head groups having similar hydrophilicity values include leucine, isoleucine, valine, glycine, alanine, asparagine, glutamine, serine, threonine, phenylalanine, and tyrosine. Conservative substitutions may be made, for example according to the Table below. Amino acids in the same block in the second column and preferably in the same line in the third column may be substituted for each other as set forth in Table 1. Table 1 ALIPHATIC Non-polar G A P
 e p ese ve o a so e co passes o o ogous su s u o su stitution and replacement are both used herein to mean the interchange of an existing amino acid residue, with an alternative residue) that may occur i.e., like-for-like substitution such as basic for basic, acidic for acidic, polar for polar etc. Non-homologous substitution may also occur i.e., from one class of residue to another or alternatively involving the inclusion of unnatural amino acids such as ornithine (hereinafter referred to as Z), diaminobutyric acid ornithine (hereinafter referred to as B), norleucine ornithine (hereinafter referred to as O), pyriylalanine, thienylalanine, naphthylalanine and phenylglycine. Replacements may also be made by synthetic amino acids (e.g. unnatural amino acids) include; alpha* and alpha-disubstituted* amino acids, N-alkyl amino acids*, lactic acid*, halide derivatives of natural amino acids such as trifluorotyrosine*, p-Cl-phenylalanine*, p-Br-
phenylalanine*, p-I-phenylalanine*, L-allyl-glycine*, ß-alanine*, L-a-amino butyric acid*, L-g- amino butyric acid*, L-a-amino isobutyric acid*, L-e-amino caproic acid#, 7-amino heptanoic acid*, L-methionine sulfone#*, L-norleucine*, L-norvaline*, p-nitro-L-phenylalanine*, L- hydroxyproline#, L-thioproline*, methyl derivatives of phenylalanine (Phe) such as 4-methyl- Phe*, pentamethyl-Phe*, L-Phe (4-amino)#, L-Tyr (methyl)*, L-Phe (4-isopropyl)*, L-Tic (1,2,3,4-tetrahydroisoquinoline-3-carboxyl acid)*, L-diaminopropionic acid # and L-Phe (4- benzyl)*. The notation * has been utilized for the purpose of the discussion above (relating to homologous or non-homologous substitution), to indicate the hydrophobic nature of the derivative whereas # has been utilized to indicate the hydrophilic nature of the derivative, #* indicates amphipathic characteristics. Variant amino acid sequences may include suitable spacer groups that may be inserted between any two amino acid residues of the sequence including alkyl groups such as methyl, ethyl, or propyl groups in addition to amino acid spacers such as glycine or b-alanine residues. A further form of variation, involves the presence of one or more amino acid residues in peptoid form, will be well understood by those skilled in the art. For the avoidance of doubt, “the peptoid form” is used to refer to variant amino acid residues wherein the a-carbon substituent group is on the residue’s nitrogen atom rather than the a-carbon. Processes for preparing peptides in the peptoid form are known in the art, for example Simon RJ et al., PNAS (1992) 89(20), 9367-9371 and Horwell DC, Trends Biotechnol. (1995) 13(4), 132-134. The nucleotide sequences for use in the present invention may include within them synthetic or modified nucleotides. Several different types of modification to oligonucleotides are known in the art. These include methylphosphonate and phosphorothioate backbones and/or the addition of acridine or polylysine chains at the 3' and/or 5' ends of the molecule. For the purposes of the present invention, it is to be understood that the nucleotide sequences described herein may be modified by any method available in the art. Such modifications may be carried out to enhance the in vivo activity or life span of nucleotide sequences of the present invention. The present invention also encompasses the use of nucleotide sequences that are complementary to the sequences presented herein. Other variants of the sequences described herein may be obtained for example by probing DNA libraries made from a range of individuals, for example individuals from different
populations. In addition, other homologues may be obtained and such homologues and fragments thereof in general will be capable of selectively hybridizing to the sequences shown in the sequence listing herein. Such sequences may be obtained by probing cDNA libraries or genomic DNA libraries made from other animal species and probing such libraries with probes comprising all or part of any one of the sequences in the attached sequence listings under conditions of medium to high stringency. Similar considerations apply to obtaining species homologues and allelic variants of the polypeptide or nucleotide sequences of the invention. Variants and strain/species homologues may also be obtained using degenerate PCR which will use primers designed to target sequences within the variants and homologues encoding conserved amino acid sequences within the sequences of the present invention. Conserved sequences can be predicted, for example, by aligning the amino acid sequences from several variants/homologues. Sequence alignments can be performed using computer software known in the art. For example, the GCG Wisconsin PileUp program is widely used. The primers used in degenerate PCR will contain one or more degenerate positions and will be used at stringency conditions lower than those used for cloning sequences with single sequence primers against known sequences. Alternatively, such polynucleotides may be obtained by site directed mutagenesis of characterized sequences. This may be useful where for example silent codon sequence changes are required to optimize codon preferences for a particular host cell in which the polynucleotide sequences are being expressed. Other sequence changes may be desired to introduce restriction enzyme recognition sites, or to alter the property or function of the polypeptides encoded by the polynucleotides. The present invention employs, unless otherwise indicated, conventional techniques of biochemistry, molecular biology, microbiology, and recombinant DNA, which are within the capabilities of a person of ordinary skill in the art. Such techniques are explained in the literature. See, for example, J. Sambrook, E. F. Fritsch, and T. Maniatis, 1989, Molecular Cloning: A Laboratory Manual, Second Edition, Books 1-3, Cold Spring Harbor Laboratory Press; Ausubel, F. M. et al. (1995 and periodic supplements; Current Protocols in Molecular Biology, ch. 9, 13, and 16, John Wiley & Sons, New York, N. Y.); B. Roe, J. Crabtree, and A. Kahn, 1996, DNA Isolation and Sequencing: Essential Techniques, John Wiley & Sons; M. J. Gait (Editor), 1984, Oligonucleotide Synthesis: A Practical Approach, IrI Press; and, D. M. J. Lilley and J. E. Dahlberg,
1992, Methods of Enzymology: DNA Structure Part A: Synthesis and Physical Analysis of DNA Methods in Enzymology, Academic Press. Each of these general texts is herein incorporated by reference. As used herein, “percent (%) sequence identity” means that a particular sequence has at least a certain percentage of amino acid residues identical to those in a specified reference sequence, when aligned using the CLUSTAL W algorithm with default parameters. See Thompson et al. (1994) Nucleic Acids Res. 22:4673-4680. Default parameters for the CLUSTAL W algorithm are: Gap opening penalty: 10.0 Gap extension penalty: 0.05 Protein weight matrix: BLOSUM series DNA weight matrix: IUB Delay divergent sequences %: 40 Gap separation distance: 8 DNA transitions weight: 0.50 List hydrophilic residues: GPSNDQEKR Use negative matrix: OFF Toggle Residue specific penalties: ON Toggle hydrophilic penalties: ON Toggle end gap separation penalty: OFF Deletions are counted as non-identical residues, compared to a reference sequence. Deletions occurring at either terminus are included. For example, a variant with five amino acid deletions of the C-terminus of the mature 617 residue polypeptide would have a percent sequence identity of 99% (612 / 617 identical residues × 100, rounded to the nearest whole number) relative to the mature polypeptide. Such a variant would be encompassed by a variant having “at least 99% sequence identity” to a mature polypeptide. In accordance with the instant invention, proteins, including enzymes, of the present invention exist in multiple forms. Proteins of the instant invention may be clipped or trimmed (i.e., removing amino acids) from the N-terminus and/or the C-terminus, resulting in a shorter protein. Proteins of the instant invention can also have internal deletions. Shorter proteins as described herein can have higher activity or lower activity than longer counterparts. Without
being bound by theory, as used herein the term “pre-pro-protein” is a protein, including an enzyme, which has an N-terminal signal peptide that targets the protein for secretion. A pre-pro- protein is sometimes referred to herein as “full length” or “full length protein”. The N-terminal signal peptide is cleaved off in the endoplasmic reticulum to yield a “pro-protein”. A pro- protein, as used herein, is shorter in length than the full-length protein (it is missing the signal peptide) but longer than the mature protein. In general, a pro-protein is inactive or less active than the mature protein. A pro-protein can be activated or converted to a more active mature form by post-translational modification such as N- or C- terminal clipping. A pro-protein which is an enzyme may be called a “proenzyme” or a “zymogen.” The clipped active protein (derived from the pro-protein) is also referred to herein as the mature protein. It is to be noted that the above terms are used for convenience and are not meant to override or determine the activities of a protein of the instant invention. It is also to be noted that any protein of the instant invention can have more than one variant described by the same term. All references cited in the present specification are hereby incorporated by reference in their entirety. In particular, the teachings of all references herein specifically referred to are incorporated by reference. Unless otherwise defined, all terms used in disclosing the invention, including technical and scientific terms, have the meaning as commonly understood by one of ordinary skill in the art to which this invention belongs. By means of further guidance, term definitions are included to better appreciate the teaching of the present invention. Other definitions are set forth below. Beer As used herein, the term “beer” traditionally refers to an alcoholic beverage derived from malt, which is derived from barley, and optionally adjuncts, such as cereal grains, and flavored with hops. Beer can be made from a variety of grains by essentially the same process. All grain starches are glucose homopolymers in which the glucose residues are linked by either alpha-1, 4- or alpha-1,6-bonds, with the former predominating. The process of making fermented malt beverages is commonly referred to as brewing. The principal raw materials used in making these beverages are water, hops and malt. In addition, adjuncts such as common corn grits, refined corn grits, brewer's milled yeast, rice, sorghum, refined corn starch, barley, barley starch,
dehusked barley, wheat, wheat starch, torrified cereal, cereal flakes, rye, oats, potato, tapioca, and syrups, such as corn syrup, sugar cane syrup, inverted sugar syrup, barley and/or wheat syrups, and the like may be used as a source of starch. The starch will eventually be converted into dextrins and fermentable sugars. For several reasons, the malt, which is produced principally from selected varieties of barley, has the greatest effect on the overall character and quality of the beer. First, the malt is the primary flavoring agent in beer. Second, the malt provides the major portion of the fermentable sugar. Third, the malt provides the proteins, which will contribute to the body and foam character of the beer. Fourth, the malt provides the necessary enzymatic activity during mashing. As used herein, the term “Hops” refers to it use in contributing significantly to beer quality, including flavoring. Hops (or hop constituents) add desirable bittering substances to the beer. In addition, the hops act as protein precipitants, establish preservative agents and aid in foam formation and stabilization. As used herein, the “process for making beer” is one that is well known in the art, but briefly, it involves five steps: (a) mashing and/or adjunct cooking (b) wort separation and extraction (c) boiling and hopping of wort (d) cooling, fermentation, and storage, and (e) maturation, processing and packaging. In the first step, milled or crushed malt is mixed with water and held for a period under controlled temperatures to permit the enzymes present in the malt to convert the starch present in the malt into fermentable sugars. In the second step, the mash is transferred to a "lauter tun" or mash filter where the liquid is separated from the grain residue. This sweet liquid is called "wort" and the left-over grain residue is called “spent grain”. The mash is typically subjected to an extraction, which involves adding water to the mash to recover the residual soluble extract from the spent grain. In the third step, the wort is boiled vigorously. This sterilizes the wort and helps to develop the color, flavor, and odor. Hops are added at some point during the boiling. In the fourth step, the wort is cooled and transferred to a fermenter, which either contains the yeast or to which yeast is added. After addition of yeast, the liquid is referred to as a fermentate. The yeast converts the sugars by fermentation into alcohol and carbon dioxide gas; at the end of fermentation the fermenter is chilled, or the fermenter may be chilled to stop fermentation. The yeast flocculates and is removed. In the last step, the beer is cooled and stored for a period of time, during which the beer clarifies, and its flavor develops, and any material that might impair the appearance, flavor and shelf life of the beer settles out.
Prior to packaging, the beer is carbonated and, optionally, filtered and pasteurized. After fermentation, a beverage is obtained which usually contains from about 2% to about 10% alcohol by weight. The non-fermentable carbohydrates are not converted during fermentation and form most of the dissolved solids in the final beer. This residue remains because of the inability of malt amylases to hydrolyze the alpha-1,6-linkages of the starch. The non-fermentable carbohydrates contribute about 50 calories per 12 ounces of beer. The term "fermentation" means, in the context of brewing, the transformation of sugars in the wort, by enzymes in the brewing yeast, into ethanol and carbon dioxide with the formation of other fermentation by-products. As used herein, a “fermentate” is the liquid solution undergoing a fermentation process leading to chemical change of the food, beer, or beverage by the action of yeast or bacteria, which produce carbon dioxide and turns carbohydrates in it into alcohol: As used herein the term "malt" is understood as any malted cereal grain, such as barley. As used herein, the term "wort" refers to the unfermented liquor run-off following extracting the grist during mashing. As used herein, the term "spent grains" refers to the drained solids remaining when the grist has been extracted and the wort separated from the mash. As used herein, the term "beer" refers to fermented wort, e.g., an alcoholic beverage brewed from barley malt, optionally adjunct and hops. ALDC In some aspects, the invention provides ALDC enzymes having a better stability and activity, and which further can be recovered from microorganisms in improved yields. Acetolactate decarboxylase (ALDC) is an enzyme that belongs to the family of carboxy lyases, which are responsible for cleaving carbon-carbon bonds. Acetolactate decarboxylase catalyzes the conversion of 2-acetolactate (also known as 2-hydroxy-2-methyl-3-oxobutanoate) to 2-acetoin and releasing CO2. Acetolactate decarboxylase enzymes catalyze the enzymatic reaction belonging to the classification EC 4.l.l.5 (acetolactate decarboxylase activity) and gene ontology (GO) term ID of GO: 0047605. The GO term ID specifies that any protein characterized as having this associated GO term encodes an enzyme with catalytic acetolactate decarboxylase activity.
Various acetolactate decarboxylase (alsD) genes, which encode acetolactate decarboxylase enzymes, are known in the art. Examples of alsD genes include but are not limited to gil3751436271reflYP 005006068.11 Acetolactate decarboxylase [Niastella koreensis OR20-10]; gil361 0576731gb1AEV96664.11 Acetolactate decarboxylase [Niastella koreensis OR20-10]; gi12187634151gb1ACL0588l.11 Acetolactate decarboxylase [Desulfatibacillum alkenivorans AK -01]; gil220909520lreflYP 002484831.11 acetolactate decarboxylase [Cyanothece sp. PCC 7425]; gil2187820311reflYP 002433349.11 acetolactate decarboxylase [Desulfatibacillum alkenivorans AK- Ol]; gi12136930901ref1YP 002323676.11 Acetolactate decarboxylase [Bifidobacterium longum subsp. infantis ATCC 15697 = JCM 1222]; gil 1895002971reflYP 001959767.11 Acetolactate decarboxylase [Chlorobium phaeobacteroides BS 1]; gil 1894237871reflYP 001950964.11 acetolactate decarboxylase [Geobacter lovleyi SZ]; gil 1720582711ref1YP 00181473l.11 acetolactate decarboxylase [Exiguobacterium sibiricum 255-15]; gil1639387751reflYP 001643659.11 acetolactate decarboxylase [Bacillus weihenstephanensis KBAB4]; gil 1585223041reflYP 001530174.11 acetolactate decarboxylase [Desulfococcus oleovorans Hxd3]; gil 157371670lreflYP 001479659.11 acetolactate decarboxylase [Serratia proteamaculans 568]; gi11503951111ref1YP 001317786.11 acetolactate decarboxylase [Staphylococcus aureus subsp. aureus JHl]; gil 1503947151reflYP 001317390.11 acetolactate decarboxylase [Staphylococcus aureus subsp. aureus JHl]; gil 1463116791ref1YP 001176753.11 acetolactate decarboxylase [Enterobacter sp.638]; gil 109900061 IreflYP 663316.11 acetolactate decarboxylase [Pseudoalteromonas atlantica T6c]; giI219866131IgbIACL46470.11 Acetolactate decarboxylase [Cyanothece sp. PCC 7425]; giI213524551IgbIACJ53298.11 Acetolactate decarboxylase [Bifidobacterium longum subsp. infantis ATCC 15697 = JCM 1222]; gil 1894200461gb1ACD94444.11 Acetolactate decarboxylase [Geobacter lovleyi SZ]; giI158511130IgbIABW68097.11 Acetolactate decarboxylase [Desulfococcus oleovorans Hxd3]; gil 1573234341gblABV 42531.11 Acetolactate decarboxylase [Serratia proteamaculans 568]; gi11453185551gb1ABP60702.11 Acetolactate decarboxylase [Enterobacter sp.638]; gi11499475631gb1ABR53499.11 Acetolactate decarboxylase [Staphylococcus aureus subsp.aureus JH1]; gi11499471671gb1ABR53103.11 Acetolactate decarboxylase [Staphylococcusaureus subsp. aureus JH1]; gi11638609721gb1ABY42031.11 Acetolactate decarboxylase [Bacillus weihenstephanensis KBAB4]; gill 097023421gb1ABG42262.11 Acetolactate decarboxylase [Pseudoalteromonas atlantica T6c]; gi11894957381gb1ACE04286.11 Acetolactate decarboxylase [Chlorobium phaeobacteroides BS 1]; gi11719907921gb1ACB61714.11 Acetolactate decarboxylase [Exiguobacterium sibiricum 255-15]; gil2239325631reflZP 03624564.11 Acetolactate decarboxylase
[Streptococcus suis 89/1591]; gil 194467531 IreflZP 03073518.11 Acetolactate decarboxylase [Lactobacillus reuteri 100-23]; gi12238988341gb1EEF65194.11 Acetolactate decarboxylase [Streptococcus suis 89/1591]; gil 1944545671gb1EDX43464.11 Acetolactate decarboxylase [Lactobacillus reuteri 100-23]; gi13842671351ref1YP 005422842.11 acetolactate decarboxylase [Bacillus amyloliquefaciens subsp. plantarum YAU B9601-Y2]; gil3753640371reflYP 005132076.11 acetolactate decarboxylase [Bacillus amyloliquefaciens subsp. plantarum CAU B946]; gil3407932311reflYP 004758694.11 acetolactate decarboxylase [Corynebacterium variabile DSM 44702]; gil3363251191reflYP 004605085.11 acetolactate decarboxylase [Corynebacterium resistens DSM 45100]; gil 1482690321reflYP 001247975.11 acetolactate decarboxylase [Staphylococcus aureus subsp. aureus JH9]; gil 148268650lreflYP 001247593.11 acetolactate decarboxylase [Staphylococcus aureus subsp. aureus JH9]; gil 1485433721reflYP 001270742.11 acetolactate decarboxylase [Lactobacillus reuteri DSM 20016]; gi13805004881emb1CCG51526.11 acetolactate decarboxylase [Bacillus amyloliquefaciens subsp. plantarum YAU B9601-Y2]; gi13715700311emb1CCF06881.11 acetolactate decarboxylase [Bacillus amyloliquefaciens subsp. plantarum CAU B946]; gi13405331411gb1AEK35621.11 acetolactate decarboxylase [Corynebacterium variabile DSM 44702]; gi13361011011gb1AEI08921.11 acetolactate decarboxylase [Corynebacterium resistens DSM 45100]; gi11485304061gb1ABQ82405.11 Acetolactate decarboxylase [Lactobacillus reuteri DSM 20016]; gi11477421011gb1ABQ50399.11 Acetolactate decarboxylase [Staphylococcus aureus subsp. aureus JH9]; giI147741719IgbIABQ50017.11 Acetolactate decarboxylase [Staphylococcus aureus subsp. aureus JH9]; gil392529510lreflZP 10276647.11 acetolactate decarboxylase [Carnobacterium maltaromaticum ATCC 35586]; gil3660540741reflZP 09451796.11 acetolactate decarboxylase [Lactobacillus suebicus KCTC 3549]; gil3396241471reflZP 08659936.11 acetolactate decarboxylase [Fructobacillus Jructosus KCTC 3544]; gil3363937271reflZP 08575126.11 acetolactate decarboxylase [Lactobacillus coryniformis subsp. torquens KCTC 3535]. Each sequence associated with the foregoing accession numbers is incorporated herein by reference. In some embodiments, the invention relates to ALDC enzymes from Lactobaclllus Casei (Godtfredsen 1984), Brevibacterium acetylicum (Oshiro, 1989), Lactococcus lactis (Vincent Phalip 1994), Leuconostoc lactis (O sulivan, 2001), Enterobacter aerogenes (Blomquist, 1993), Bacillus subtilis (Renna, 1993), Bacillus brevis (Svendsen, 1989) and Lactococcus lactis DX (Yuxing, 2014) It is to be understood that any suitable ALDC enzymes, i.e., ALDC produced from any microorganism which activity is dependent on metal ions, can be used according to the invention. In some embodiments, the ALDC used in the methods and compositions described herein is an ALDC from Bacillus brevis or Bacillus licheniformis.
The ALDC activity of the enzyme composition according to the invention is measured by the ALDC assays as described herein or any suitable assay known in the art. The standard assay is carried out at pH 6.0, and it can be performed at different pH values and temperatures for the additional characterization and specification of enzymes. One unit of ALDC activity is defined as the amount of enzyme which produces 1 µmole acetoin per minute under the conditions of the assay (e.g., pH 6.0 (or as specified) and 30 °C). In some embodiments, the enzyme has a temperature optimum in the range of 5-80 °C, such as in the range of 5-40°C or 15-80°C, such as in the range 20-80 °C, such as in the range 5- 15°C, 15-20°C, 45-65 °C, 50-65 °C, 55-65 °C or 60-80°C. In some embodiments, the enzyme has a temperature optimum in the range of 45-65 °C. In some embodiments, the enzyme has a temperature optimum of about 60°C. In some embodiments, the enzyme has a total number of amino acids of less than 350, such as less than 340, such as less than 330, such as less than 320, such as less than 310, such as less than 300 amino acids, such as in the range of 200 to 350, such as in the range of 220 to 345 amino acids. In some embodiments the ALDC compositions and methods according to the invention comprises any one or more further enzyme. In some embodiments the one or more further enzyme is selected from list consisting of acetolactate reductoisomerases, acetolactate isomerases, amylase, glucoamylase, hemicellulose, cellulase, glucanase, pullulanase, isoamylase, endo-glucanase and related beta-glucan hydrolytic accessory enzymes, xylanase, and xylanase accessory enzymes (for example, arabinofuranosidase, ferulic acid esterase, xylan acetyl esterase), beta-glucosidase and protease. In some embodiments the compositions and methods according to the invention comprises an enzyme exhibiting ALDC activity, wherein the activity of said ALDC enzyme is in the range of 950 to 2500 Units per mg of protein. In some embodiments the compositions and methods according to the invention comprises an enzyme exhibiting ALDC activity, wherein the activity of said ALDC enzyme is in the range of 1000 to 2500 Units per mg of protein. Proline Specific Proteases Beer-haze, a cloudy appearance in beer, is caused by the aggregation of hydrophobic proteins, e.g., hordeins from barley, and polyphenols, resulting in a beer with an undesirable
cloudy appearance or haze. See, e.g., Asano, K.; Shinagawa, K.; Hashimoto, N. Characterization of haze-forming proteins of beer and their roles in chill haze formation. J. Am. Soc. Brew. Chem. 1982, 40, 147−154. The same phenomenon is also called chill-haze and similar haze formation may also occur in wine and fruit juices. It has been suggested that acid proteases such as papain can be used to degrade beer proteins and hence prevent haze formation. However, broad spectrum proteases such as papain have been found to impair beer foam formation and stability. See, e.g, Posada, J.; Almenar, J.; Garcia Galindo, J. A practical approach on protein stabilizers. Proc. - Eur. Brew. Conv. 1971, 13, 379−391. For this reason, more selective proteases such as proline specific endopeptidases have been employed to reduce beer haze. However, there is a continuing need for proteases that can be used to reduce beer-haze, because present commercial offerings are overly expensive and do not provide complete beer-haze removal. Moreover, there is a concern that even proline specific endoproteases are still too broad spectrum. For example, it has been discovered in accordance with the present invention that prior art proline specific endoproteases can destabilize other enzyme added exogenously to beer, including ALDC enzymes added for maturation. In accordance with an aspect of the present invention, proline specific endoproteases have been discovered which are less destabilizing to other exogenously added enzymes, including ALDC enzymes. A “polyphenol” is a compound having one or more aromatic rings and substituted by one or more hydroxyl groups. Examples of polyphenols are tannins and flavonoids, including catechins, flavonols and anthocyanins. The proline-specific protease and/or ALDC of the present invention may be added at different stages during the preparation of a beer. Addition of the enzyme at the beginning of the fermentation yields the best possible results. However, the enzyme may be added to a mash or to a fermented beer before haze has been formed. Enzyme of the present invention, including ALDC enzymes and proline specific proteases may be in isolated or purified form. By "isolated" or "purified" means and enzyme removed from its native environment. For example, recombinantly produced proline specific protease or ALDC expressed in a host cell may be considered isolated for purposes of the invention. Similarly, native, or recombinant polypeptides which have been substantially purified by any suitable technique may be considered isolated or purified.
Production of enzymes The enzymes of the present invention can be produced in host cells, for example, by secretion or intracellular expression. A cultured cell material (e.g., a whole-cell broth) having an enzyme can be obtained following secretion of the enzyme into the cell medium. Optionally, the enzyme can be isolated from the host cells, or even isolated from the cell broth, depending on the desired purity of the final enzyme. Suitable host cells include bacterial, fungal (including yeast and filamentous fungi), and plant cells (including algae). Particularly useful host cells include Aspergillus niger, Aspergillus oryzae or Trichoderma reesei. Other host cells include bacterial cells, e.g., Bacillus subtilis or B. licheniformis, as well as Streptomyces, E. coli. Vectors A DNA construct comprising a nucleic acid encoding an enzyme can be constructed to be expressed in a host cell. Because of the well-known degeneracy in the genetic code, variant polynucleotides that encode an identical amino acid sequence can be designed and made with routine skill. It is also well-known in the art to optimize codon use for a particular host cell. Nucleic acids encoding enzymes of the present invention can be incorporated into a vector. Vectors can be transferred to a host cell using well-known transformation techniques, such as those disclosed below. The vector may be any vector that can be transformed into and replicated within a host cell. For example, a vector comprising a nucleic acid encoding an enzyme can be transformed and replicated in a bacterial host cell as a means of propagating and amplifying the vector. The vector also may be transformed into an expression host, so that the encoding nucleic acids can be expressed as a functional enzyme. Host cells that serve as expression hosts can include filamentous fungi, for example. The Fungal Genetics Stock Center (FGSC) Catalogue of Strains lists suitable vectors for expression in fungal host cells. See FGSC, Catalogue of Strains, University of Missouri, at www.fgsc.net (last modified January 17, 2007). A representative vector is pJG153, a promoterless Cre expression vector that can be replicated in a bacterial host. See Harrison et al. (June 2011) Applied Environ. Microbiol. 77: 3916-22. pJG153can be modified with routine skill to comprise and express a nucleic acid encoding an enzyme. A nucleic acid encoding an enzyme can be operably linked to a suitable promoter, which
allows transcription in the host cell. The promoter may be any DNA sequence that shows transcriptional activity in the host cell of choice and may be derived from genes encoding proteins either homologous or heterologous to the host cell. Exemplary promoters for directing the transcription of the DNA sequence encoding an enzyme, especially in a bacterial host, are the promoter of the lac operon of E. coli, the Streptomyces coelicolor agarase gene dagA or celA promoters, the promoters of the Bacillus licheniformis α-amylase gene (amyL), the promoters of the Bacillus stearothermophilus maltogenic amylase gene (amyM), the promoters of the Bacillus amyloliquefaciens α-amylase (amyQ), the promoters of the Bacillus subtilis xylA and xylB genes etc. For transcription in a fungal host, examples of useful promoters are those derived from the gene encoding Aspergillus oryzae TAKA amylase, Rhizomucor miehei aspartic proteinase, Aspergillus niger neutral α-amylase, A. niger acid stable α-amylase, A. niger glucoamylase, Rhizomucor miehei lipase, A. oryzae alkaline protease, A. oryzae triose phosphate isomerase, or A. nidulans acetamidase. When a gene encoding an enzyme is expressed in a bacterial species such as E. coli, a suitable promoter can be selected, for example, from a bacteriophage promoter including a T7 promoter and a phage lambda promoter. Examples of suitable promoters for the expression in a yeast species include but are not limited to the Gal 1 and Gal 10 promoters of Saccharomyces cerevisiae and the Pichia pastoris AOX1 or AOX2 promoters. cbh1 is an endogenous, inducible promoter from Trichoderma reesei. See Liu et al. (2008) “Improved heterologous gene expression in Trichoderma reesei by cellobiohydrolase I gene (cbh1) promoter optimization,” Acta Biochim. Biophys. Sin (Shanghai) 40(2): 158-65. The coding sequence can be operably linked to a signal sequence. The DNA encoding the signal sequence may be the DNA sequence naturally associated with the enzyme gene to be expressed or from a different Genus or species. A signal sequence and a promoter sequence comprising a DNA construct or vector can be introduced into a fungal host cell and can be derived from the same source. For example, the signal sequence is the cbh1 signal sequence that is operably linked to a cbh1 promoter. An expression vector may also comprise a suitable transcription terminator and, in eukaryotes, polyadenylation sequences operably linked to the DNA sequence encoding a variant enzyme. Termination and polyadenylation sequences may suitably be derived from the same sources as the promoter. The vector may further comprise a DNA sequence enabling the vector to replicate in the
host cell. Examples of such sequences are the origins of replication of plasmids pUC19, pACYC177, pUB110, pE194, pAMB1, and pIJ702. The vector may also comprise a selectable marker, e.g., a gene the product of which complements a defect in the isolated host cell, such as the dal genes from B. subtilis or B. licheniformis, or a gene that confers antibiotic resistance such as, e.g., ampicillin, kanamycin, chloramphenicol, or tetracycline resistance. Furthermore, the vector may comprise Aspergillus selection markers such as amdS, argB, niaD and xxsC, a marker giving rise to hygromycin resistance, or the selection may be accomplished by co-transformation, such as known in the art. See e.g., International PCT Application WO 91/17243. Intracellular expression may be advantageous in some respects, e.g., when using certain bacteria or fungi as host cells to produce large amounts of enzyme for subsequent enrichment or purification. Extracellular secretion of enzyme into the culture medium can also be used to make a cultured cell material comprising the isolated enzyme. The expression vector typically includes the components of a cloning vector, such as, for example, an element that permits autonomous replication of the vector in the selected host organism and one or more phenotypically detectable markers for selection purposes. The expression vector normally comprises control nucleotide sequences such as a promoter, operator, ribosome binding site, translation initiation signal and optionally, a repressor gene or one or more activator genes. Additionally, the expression vector may comprise a sequence coding for an amino acid sequence capable of targeting the enzyme to a host cell organelle such as a peroxisome, or to a particular host cell compartment. Such a targeting sequence includes but is not limited to the sequence, SKL. For expression under the direction of control sequences, the nucleic acid sequence of the enzyme is operably linked to the control sequences in proper manner with respect to expression. The procedures used to ligate the DNA construct encoding an enzyme, the promoter, terminator, and other elements, respectively, and to insert them into suitable vectors containing the information necessary for replication, are well known to persons skilled in the art (see, e.g., Sambrook et al., MOLECULAR CLONING: A LABORATORY MANUAL, 2nd ed., Cold Spring Harbor, 1989, and 3rd ed., 2001).
Transformation and Culture of Host Cells An isolated cell, either comprising a DNA construct or an expression vector, is advantageously used as a host cell in the recombinant production of an enzyme according to the instant invention. The cell may be transformed with the DNA construct encoding the enzyme, conveniently by integrating the DNA construct (in one or more copies) in the host chromosome. This integration is generally considered to be an advantage, as the DNA sequence is more likely to be stably maintained in the cell. Integration of the DNA constructs into the host chromosome may be performed according to conventional methods, e.g., by homologous or heterologous recombination. Alternatively, the cell may be transformed with an expression vector as described above in connection with the different types of host cells. Examples of suitable bacterial host organisms are Gram positive bacterial species such as Bacillaceae including Bacillus subtilis, Bacillus licheniformis, Bacillus lentus, Bacillus brevis, Geobacillus (formerly Bacillus) stearothermophilus, Bacillus alkalophilus, Bacillus amyloliquefaciens, Bacillus coagulans, Bacillus lautus, Bacillus megaterium, and Bacillus thuringiensis; Streptomyces species such as Streptomyces murinus; lactic acid bacterial species including Lactococcus sp. such as Lactococcus lactis; Lactobacillus sp. including Lactobacillus reuteri; Leuconostoc sp.; Pediococcus sp.; and Streptococcus sp. Alternatively, strains of a Gram-negative bacterial species belonging to Enterobacteriaceae including E. coli, or to Pseudomonadaceae can be selected as the host organism. A suitable yeast host organism can be selected from the biotechnologically relevant yeasts species such as but not limited to yeast species such as Pichia sp., Hansenula sp., or Kluyveromyces, Yarrowinia, Schizosaccharomyces species or a species of Saccharomyces, including Saccharomyces cerevisiae or a species belonging to Schizosaccharomyces such as, for example, S. pombe species. A strain of the methylotrophic yeast species, Pichia pastoris, can be used as the host organism. Alternatively, the host organism can be a Hansenula species. Suitable host organisms among filamentous fungi include species of Aspergillus, e.g., Aspergillus niger, Aspergillus oryzae, Aspergillus tubigensis, Aspergillus awamori, or Aspergillus nidulans. Alternatively, strains of a Fusarium species, e.g., Fusarium oxysporum or of a Rhizomucor species such as Rhizomucor miehei can be used as the host organism. Other suitable strains include Thermomyces and Mucor species. In addition, Trichoderma sp. can be used as a host. A suitable procedure for transformation of Aspergillus host cells includes, for
example, that described in EP 238023. An enzyme expressed by a fungal host cell can be glycosylated, i.e., will comprise a glycosyl moiety. The glycosylation pattern can be the same or different as present in the wild-type enzyme. The type and/or degree of glycosylation may impart changes in enzymatic and/or biochemical properties. It may be advantageous to delete genes from expression hosts, where the gene deficiency can be cured by the transformed expression vector. Known methods may be used to obtain a fungal host cell having one or more inactivated genes. Gene inactivation may be accomplished by complete or partial deletion, by insertional inactivation or by any other means that renders a gene nonfunctional for its intended purpose, such that the gene is prevented from expression of a functional protein. Any gene from a Trichoderma sp. or other filamentous fungal host that has been cloned can be deleted, for example, cbh1, cbh2, egl1, and egl2 genes. Gene deletion may be accomplished by inserting a form of the desired gene to be inactivated into a plasmid by methods known in the art. Introduction of a DNA construct or vector into a host cell includes techniques such as transformation; electroporation; nuclear microinjection; transduction; transfection, e.g., lipofection mediated and DEAE-Dextrin mediated transfection; incubation with calcium phosphate DNA precipitate; high velocity bombardment with DNA-coated microprojectiles; and protoplast fusion. General transformation techniques are known in the art. See, e.g., Sambrook et al. (2001), supra. The expression of heterologous protein in Trichoderma is described, for example, in U.S. Patent No. 6,022,725. Reference is also made to Cao et al. (2000) Science 9:991-1001 for transformation of Aspergillus strains. Genetically stable transformants can be constructed with vector systems whereby the nucleic acid encoding an enzyme is stably integrated into a host cell chromosome. Transformants are then selected and purified by known techniques. The preparation of Trichoderma sp. for transformation, for example, may involve the preparation of protoplasts from fungal mycelia. See Campbell et al. (1989) Curr. Genet. 16: 53- 56. The mycelia can be obtained from germinated vegetative spores. The mycelia are treated with an enzyme that digests the cell wall, resulting in protoplasts. The protoplasts are protected by the presence of an osmotic stabilizer in the suspending medium. These stabilizers include sorbitol, mannitol, potassium chloride, magnesium sulfate, and the like. Usually, the concentration of these stabilizers varies between 0.8 M and 1.2 M, e.g., a 1.2 M solution of
sorbitol can be used in the suspension medium. Uptake of DNA into the host Trichoderma sp. strain depends upon the calcium ion concentration. Generally, between about 10-50 mM CaCl2 is used in an uptake solution. Additional suitable compounds include a buffering system, such as TE buffer (10 mM Tris, pH 7.4; 1 mM EDTA) or 10 mM MOPS, pH 6.0 and polyethylene glycol. The polyethylene glycol is believed to fuse the cell membranes, thus permitting the contents of the medium to be delivered into the cytoplasm of the Trichoderma sp. strain. This fusion frequently leaves multiple copies of the plasmid DNA integrated into the host chromosome. Usually, transformation of Trichoderma sp. uses protoplasts or cells that have been subjected to a permeability treatment, typically at a density of 105 to 107/mL, particularly 2x106/mL. A volume of 100 μL of these protoplasts or cells in an appropriate solution (e.g., 1.2 M sorbitol and 50 mM CaCl2) may be mixed with the desired DNA. Generally, a high concentration of PEG is added to the uptake solution. From 0.1 to 1 volume of 25% PEG 4000 can be added to the protoplast suspension; however, it is useful to add about 0.25 volumes to the protoplast suspension. Additives, such as dimethyl sulfoxide, heparin, spermidine, potassium chloride and the like, may also be added to the uptake solution to facilitate transformation. Similar procedures are available for other fungal host cells. See, e.g., U.S. Patent No. 6,022,725. As used herein, Protein Identification (“JGI PID”) numbers for native Trichoderma genes reference Version 2 of the Trichoderma reesei QM6a genome sequence assembly generated by the Department of Energy Joint Genome Institute (JGI). (The Genome Portal of the Department of Energy Joint Genome Institute, Grigoriev et al., Nucleic Acids Res 2012 Jan;40(Database issue):D26-32. doi: 10.1093/nar/gkr947). The JGI assembled Scaffold sequences and annotated genes have also been deposited in GeneBank (The National Center for Biotechnology) under the nucleotide accession numbers GL985056.1 through GL985132.1. Expression A method of producing an enzyme of the instant invention may comprise cultivating a host cell as described above under conditions conducive to the production of the enzyme and recovering the enzyme from the cells and/or culture medium. The medium used to cultivate the cells may be any conventional medium suitable for growing the host cell in question and obtaining expression of an enzyme. Suitable media and
media components are available from commercial suppliers or may be prepared according to published recipes (e.g., as described in catalogues of the American Type Culture Collection). An enzyme secreted from the host cells can be used in a whole broth preparation. In the present methods, the preparation of a spent whole fermentation broth of a recombinant microorganism can be achieved using any cultivation method known in the art resulting in the expression of an enzyme. Fermentation may, therefore, be understood as comprising shake flask cultivation, small- or large-scale fermentation (including continuous, batch, fed-batch, or solid- state fermentations) in laboratory or industrial fermenters performed in a suitable medium and under conditions allowing the enzyme to be expressed or isolated. The term “spent whole fermentation broth” is defined herein as unfractionated contents of fermentation material that includes culture medium, extracellular proteins (e.g., enzymes), and cellular biomass. It is understood that the term “spent whole fermentation broth” also encompasses cellular biomass that has been lysed or permeabilized using methods well known in the art. An enzyme secreted from the host cells may conveniently be recovered from the culture medium by well-known procedures, including separating the cells from the medium by centrifugation or filtration, and precipitating proteinaceous components of the medium by means of a salt such as ammonium sulfate, followed using chromatographic procedures such as ion exchange chromatography, affinity chromatography, or the like. The polynucleotide encoding an enzyme in a vector can be operably linked to a control sequence that is capable of providing for the expression of the coding sequence by the host cell, i.e., the vector is an expression vector. The control sequences may be modified, for example by the addition of further transcriptional regulatory elements to make the level of transcription directed by the control sequences more responsive to transcriptional modulators. The control sequences may comprise promoters. Host cells may be cultured under suitable conditions that allow expression of an enzyme. Expression of the enzymes may be constitutive such that they are continually produced, or inducible, requiring a stimulus to initiate expression. In the case of inducible expression, protein production can be initiated when required by, for example, addition of an inducer substance to the culture medium, for example dexamethasone or IPTG or Sophorose. Polypeptides can also be produced recombinantly in an in vitro cell-free system, such as the TNT™ (Promega) rabbit reticulocyte system.
An expression host also can be cultured in the appropriate medium for the host, under aerobic conditions. Shaking or a combination of agitation and aeration can be provided, with production occurring at the appropriate temperature for that host, e.g., from about 25°C to about 75°C (e.g., 30°C to 45°C), depending on the needs of the host and production of the desired enzyme. Culturing can occur from about 12 to about 100 hours or greater (and any hour value there between, e.g., from 24 to 72 hours). Typically, the culture broth is at a pH of about 4.0 to about 8.0, again depending on the culture conditions needed for the host relative to production of an enzyme. Methods for Enriching and Purifying enzymes Fermentation, separation, and concentration techniques are well known in the art and conventional methods can be used to prepare an enzyme polypeptide-containing solution. After fermentation, a fermentation broth is obtained, the microbial cells and various suspended solids, including residual raw fermentation materials, are removed by conventional separation techniques to obtain an enzyme solution. Filtration, centrifugation, microfiltration, rotary vacuum drum filtration, ultrafiltration, centrifugation followed by ultra-filtration, extraction, or chromatography, or the like, are generally used. It is desirable to concentrate an enzyme polypeptide-containing solution to optimize recovery. Use of unconcentrated solutions requires increased incubation time to collect the enriched or purified enzyme precipitate. The enzyme containing solution is concentrated using conventional concentration techniques until the desired enzyme level is obtained. Concentration of the enzyme containing solution may be achieved by any of the techniques discussed herein. Exemplary methods of enrichment and purification include but are not limited to rotary vacuum filtration and/or ultrafiltration. The enzyme solution is concentrated into a concentrated enzyme solution until the enzyme activity of the concentrated enzyme polypeptide-containing solution is at a desired level. Concentration may be performed using, e.g., a precipitation agent, such as a metal halide precipitation agent. Metal halide precipitation agents include but are not limited to alkali metal chlorides, alkali metal bromides and blends of two or more of these metal halides. Exemplary metal halides include sodium chloride, potassium chloride, sodium bromide, potassium bromide and blends of two or more of these metal halides. The metal halide precipitation agent, sodium
chloride, can also be used as a preservative. The metal halide precipitation agent is used in an amount effective to precipitate an enzyme. The selection of at least an effective amount and an optimum amount of metal halide effective to cause precipitation of the enzyme, as well as the conditions of the precipitation for maximum recovery including incubation time, pH, temperature, and concentration of enzyme, will be readily apparent to one of ordinary skill in the art, after routine testing. Generally, at least about 5% w/v (weight/volume) to about 25% w/v of metal halide is added to the concentrated enzyme solution, and usually at least 8% w/v. Generally, no more than about 25% w/v of metal halide is added to the concentrated enzyme solution and usually no more than about 20% w/v. The optimal concentration of the metal halide precipitation agent will depend, among others, on the nature of the specific enzyme polypeptide and on its concentration in the concentrated enzyme solution. Another alternative way to precipitate the enzyme is to use organic compounds. Exemplary organic compound precipitating agents include: 4-hydroxybenzoic acid, alkali metal salts of 4-hydroxybenzoic acid, alkyl esters of 4-hydroxybenzoic acid, and blends of two or more of these organic compounds. The addition of the organic compound precipitation agents can take place prior to, simultaneously with or subsequent to the addition of the metal halide precipitation agent, and the addition of both precipitation agents, organic compound, and metal halide, may be carried out sequentially or simultaneously. Generally, the organic precipitation agents are selected from the group consisting of alkali metal salts of 4-hydroxybenzoic acid, such as sodium or potassium salts, and linear or branched alkyl esters of 4-hydroxybenzoic acid, wherein the alkyl group contains from 1 to 12 carbon atoms, and blends of two or more of these organic compounds. The organic compound precipitation agents can be, for example, linear or branched alkyl esters of 4-hydroxybenzoic acid, wherein the alkyl group contains from 1 to 10 carbon atoms, and blends of two or more of these organic compounds. Exemplary organic compounds are linear alkyl esters of 4- hydroxybenzoic acid, wherein the alkyl group contains from 1 to 6 carbon atoms, and blends of two or more of these organic compounds. Methyl esters of 4-hydroxybenzoic acid, propyl esters of 4-hydroxybenzoic acid, butyl ester of 4-hydroxybenzoic acid, ethyl ester of 4-hydroxybenzoic acid and blends of two or more of these organic compounds can also be used. Additional organic compounds also include but are not limited to 4-hydroxybenzoic acid methyl ester (named
methyl PARABEN), 4-hydroxybenzoic acid propyl ester (named propyl PARABEN), which also are both preservative agents. For further descriptions, see, e.g., U.S. Patent No. 5,281,526. Addition of the organic compound precipitation agent provides the advantage of high flexibility of the precipitation conditions with respect to pH, temperature, enzyme concentration, precipitation agent concentration, and time of incubation. The organic compound precipitation agent is used in an amount effective to improve precipitation of the enzyme by means of the metal halide precipitation agent. The selection of at least an effective amount and an optimum amount of organic compound precipitation agent, as well as the conditions of the precipitation for maximum recovery including incubation time, pH, temperature, and concentration of enzyme, will be readily apparent to one of ordinary skill in the art, in light of the present disclosure, after routine testing. Generally, at least about 0.01% w/v of organic compound precipitation agent is added to the concentrated enzyme solution and usually at least about 0.02% w/v. Generally, no more than about 0.3% w/v of organic compound precipitation agent is added to the concentrated enzyme solution and usually no more than about 0.2% w/v. The concentrated polypeptide solution, containing the metal halide precipitation agent, and the organic compound precipitation agent, can be adjusted to a pH, which will, of necessity, depend on the enzyme to be enriched or purified. Generally, the pH is adjusted at a level near the isoelectric point of the enzyme. The pH can be adjusted at a pH in a range from about 2.5 pH units below the isoelectric point (pI) up to about 2.5 pH units above the isoelectric point. The incubation time necessary to obtain an enriched or purified enzyme precipitate depends on the nature of the specific enzyme, the concentration of enzyme, and the specific precipitation agent(s) and its (their) concentration. Generally, the time effective to precipitate the enzyme is between about 1 to about 30 hours; usually it does not exceed about 25 hours. In the presence of the organic compound precipitation agent, the time of incubation can still be reduced to less about 10 hours and in most cases even about 6 hours. Generally, the temperature during incubation is between about 4°C and about 50°C. Usually, the method is carried out at a temperature between about 10°C and about 45°C (e.g., between about 20°C and about 40°C). The optimal temperature for inducing precipitation varies according to the solution conditions and the enzyme or precipitation agent(s) used. The overall recovery of enriched or purified enzyme precipitate, and the efficiency with
which the process is conducted, is improved by agitating the solution comprising the enzyme, the added metal halide, and the added organic compound. The agitation step is done both during addition of the metal halide and the organic compound, and during the subsequent incubation period. Suitable agitation methods include mechanical stirring or shaking, vigorous aeration, or any similar technique. After the incubation period, the enriched or purified enzyme is then separated from the dissociated pigment and other impurities and collected by conventional separation techniques, such as filtration, centrifugation, microfiltration, rotary vacuum filtration, ultrafiltration, press filtration, cross membrane microfiltration, cross flow membrane microfiltration, or the like. Further enrichment or purification of the enzyme precipitate can be obtained by washing the precipitate with water. For example, the enriched or purified enzyme precipitate is washed with water containing the metal halide precipitation agent, or with water containing the metal halide and the organic compound precipitation agents. During fermentation, an enzyme polypeptide accumulates in the culture broth. For the isolation, enrichment, or purification of the desired enzyme, the culture broth is centrifuged or filtered to eliminate cells, and the resulting cell-free liquid is used for enzyme enrichment or purification. In one embodiment, the cell-free broth is subjected to salting out using ammonium sulfate at about 70% saturation; the 70% saturation-precipitation fraction is then dissolved in a buffer and applied to a column such as a Sephadex G-100 column and eluted to recover the enzyme-active fraction. For further enrichment or purification, a conventional procedure such as ion exchange chromatography may be used. Enriched or purified enzymes can be made into a final product that is either liquid (solution, slurry) or solid (granular, powder). Description of the Preferred Embodiments In accordance with an aspect of the present invention, it has been discovered that ALDC enzymes which are used in brewing for shortening the maturation phase are unstable in the presence of proline specific proteases which are used to increase colloidal stability of the beer. The loss of ALDC by degradation via proteolysis can prolong the maturation phase as opposed to shortening it. Accordingly, in an aspect of the present invention, a polypeptide having acetolactate decarboxylase activity which is resistant to protease inactivation is presented, the
polypeptide having an amino acid sequence with at least 80, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:3 wherein the polypeptide has at most 250 amino acids. Preferably, the polypeptide is at most 249, 248, 247, 246, 245, 244, 243, 242 or 241 amino acids. Preferably, the polypeptide having acetolactate decarboxylase activity which is resistant to protease inactivation has an amino acid sequence having at least 80, 90, 95, 98, 99 or 100% sequence identity to according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12. More preferably, the polypeptide has an amino acid sequence according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12. The protease to which the ALDC is resistant is preferably a proline specific protease. More preferably, the proline specific protease is from Aspergillus niger. Still more preferably, the protease has at least 80, 85, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:17. Yet more preferably, the protease has an amino acid sequence according to SEQ ID NO:17. In another aspect of the present invention, an improved brewing process is presented having the step of fermenting a wort in the presence of a polypeptide having acetolactate decarboxylase activity which is resistant to protease inactivation and a proline specific protease wherein both enzymes are present in the wort at the same time. Preferably, the polypeptide has an amino acid sequence with at least 80, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:3 wherein the polypeptide has at most 250 amino acids. More preferably, the polypeptide is at most 249, 248, 247, 246, 245, 244, 243, 242 or 241 amino acids. Yet more preferably, the polypeptide having acetolactate decarboxylase activity which is resistant to protease inactivation has an amino acid sequence having at least 80, 90, 95, 98, 99 or 100% sequence identity to according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12. Still more preferably, the polypeptide has an amino acid sequence according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12. Preferably, the polypeptide having acetolactate decarboxylase activity is added to the wort first. In other preferred embodiments, the proline specific protease is added to the wort first. In yet other preferred embodiments, the polypeptide having acetolactate decarboxylase activity and the proline specific protease are added simultaneously to the wort.
The proline specific protease is preferably from Aspergillus niger. More preferably, the proline specific protease has at least 80, 85, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:17. In another aspect of the present invention, a Bacillus host cell is presented for producing a heterologous polypeptide of interest, wherein one or more protease genes are inactivated. Preferably, the polypeptide of interest is expressed without a secretion signal peptide. In other preferred embodiments, the polypeptide of interest is expressed with a secretion signal. Preferably, the polypeptide of interest is an enzyme. More preferably, the enzyme is an oxidoreductase, transferase, hydrolase, lyase, isomerase, ligase, aminopeptidase, amylase, asparaginase, carbohydrase, carboxypeptidase, catalase, cellulase, chitinase, cutinase, cyclodextrin glycosyltransferase, deoxyribonuclease, esterase, alpha-galactosidase, beta- galactosidase, glucoamylase, alpha-glucosidase, beta-glucosidase, hyaluronic acid synthase, invertase, laccase, lipase, mannosidase, mutanase, oxidase, a pectinolytic enzyme, peroxidase, phytase, polyphenoloxidase, protease, ribonuclease, transglutaminase, or xylanase. Still more preferably, the enzyme is an ALDC enzyme. Preferably, the ALDC enzyme is a polypeptide with an amino acid sequence having at least 80, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:3 wherein the polypeptide has at most 250 amino acids. Preferably, the polypeptide is at most 249, 248, 247, 246, 245, 244, 243, 242 or 241 amino acids. Preferably, the polypeptide having acetolactate decarboxylase activity which is resistant to protease inactivation has an amino acid sequence having at least 80, 90, 95, 98, 99 or 100% sequence identity to according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12. More preferably, the polypeptide has an amino acid sequence according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12. Preferably, the ALDC enzyme is expressed with a secretion signal. Preferably, the heterologous polypeptide of interest is encoded by an exogenous polynucleotide integrated into the chromosome of the host cell in at least one copy. Preferably, the exogenous polynucleotide is a nucleic acid sequence having 80, 85, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:28. More preferably, the exogenous polynucleotide is a nucleic acid sequence according to SEQ ID NO:28.
Preferably, the at least one protease gene is inactivated by a non-sense mutation in said at least one gene, a partial deletion of said at least one gene or a full deletion of said at least one gene. Preferably, the Bacillus host cell is Bacillus subtilis or Bacillus licheniformis. More preferably, the Bacillus host cell is Bacillus subtilis. Preferably, the one or more protease genes are selected from the group consisting of a nucleic acid sequence having at least 80, 85, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:18, SEQ ID NO:19, SEQ ID NO:20, SEQ ID NO:21, SEQ ID NO:22, SEQ ID NO:23, SEQ ID NO:24, SEQ ID NO:25 and SEQ ID NO:27. More preferably, the one or more protease genes comprise nine inactivated proteases wherein the proteases have nucleic acid sequences with at least 80, 85, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:18, SEQ ID NO:19, SEQ ID NO:20, SEQ ID NO:21, SEQ ID NO:22, SEQ ID NO:23, SEQ ID NO:24, SEQ ID NO:25 and SEQ ID NO:27. Still more preferably, the nine inactivated proteases genes have nucleic acid sequences according to SEQ ID NO:18, SEQ ID NO:19, SEQ ID NO:20, SEQ ID NO:21, SEQ ID NO:22, SEQ ID NO:23, SEQ ID NO:24, SEQ ID NO:25 and SEQ ID NO:27. In another aspect of the present invention, a method for producing a polypeptide of interest is presented having the steps of: i) providing a Bacillus host cell wherein one or more protease genes are inactivated and wherein said host cell is transformed with a nucleic acid encoding a heterologous polypeptide in operable combination with a promoter; and ii) cultivating said host cell under conditions suitable to produce said heterologous polypeptide, such that said heterologous polypeptide is produced. Preferably, the method has the further step of recovering the produced polypeptide. Preferably, the polypeptide of interest is expressed with or without a secretion signal peptide. More preferably, the polypeptide of interest is expressed with a secretion signal peptide. Preferably, polypeptide of interest is an enzyme. Preferably, the enzyme is an oxidoreductase, transferase, hydrolase, lyase, isomerase, ligase, aminopeptidase, amylase, asparaginase, carbohydrase, carboxypeptidase, catalase, cellulase, chitinase, cutinase, cyclodextrin glycosyltransferase, deoxyribonuclease, esterase, alpha-galactosidase, beta- galactosidase, glucoamylase, alpha-glucosidase, beta-glucosidase, hyaluronic acid synthase, invertase, laccase, lipase, mannosidase, mutanase, oxidase, a pectinolytic enzyme, peroxidase, phytase, polyphenoloxidase, protease, ribonuclease, transglutaminase, or xylanase. More
preferably, the enzyme is an ALDC enzyme. Preferably, the ALDC enzyme is a polypeptide with an amino acid sequence having at least 80, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:3 wherein the polypeptide has at most 250 amino acids. Preferably, the polypeptide is at most 249, 248, 247, 246, 245, 244, 243, 242 or 241 amino acids. Preferably, the polypeptide having acetolactate decarboxylase activity has an amino acid sequence having at least 80, 90, 95, 98, 99 or 100% sequence identity to according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12. More preferably, the polypeptide has an amino acid sequence according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12. Preferably, the ALDC enzyme is expressed with a secretion signal. Preferably, the heterologous polypeptide of interest is encoded by an exogenous polynucleotide integrated into the chromosome of the host cell in at least one copy. Preferably, the exogenous polynucleotide is a nucleic acid sequence having 80, 85, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:28. More preferably, the exogenous polynucleotide is a nucleic acid sequence according to SEQ ID NO:28. Preferably, the at least one protease gene is inactivated by a non-sense mutation in said at least one gene, a partial deletion of said at least one gene or a full deletion of said at least one gene. Preferably, the Bacillus host cell is Bacillus subtilis or Bacillus licheniformis. More preferably, the Bacillus host cell is Bacillus subtilis. Preferably, the one or more protease genes are selected from the group consisting of a nucleic acid sequence having at least 80, 85, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:18, SEQ ID NO:19, SEQ ID NO:20, SEQ ID NO:21, SEQ ID NO:22, SEQ ID NO:23, SEQ ID NO:24, SEQ ID NO:25 and SEQ ID NO:27. More preferably, the one or more protease genes comprise nine inactivated proteases wherein the proteases have nucleic acid sequences with at least 80, 85, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:18, SEQ ID NO:19, SEQ ID NO:20, SEQ ID NO:21, SEQ ID NO:22, SEQ ID NO:23, SEQ ID NO:24, SEQ ID NO:25 and SEQ ID NO:27. Still more preferably, the nine inactivated proteases genes have nucleic acid sequences according to SEQ ID NO:18, SEQ ID NO:19, SEQ ID NO:20, SEQ ID NO:21, SEQ ID NO:22, SEQ ID NO:23, SEQ ID NO:24, SEQ ID NO:25 and SEQ ID NO:27.
In another aspect of the present invention, a stable liquid formulation comprising a polypeptide having acetolactate decarboxylase activity having an amino acid sequence with at least 80, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:3 wherein said polypeptide has at most 250 amino acids and a protease. Preferably, the polypeptide is at most 249, 248, 247, 246, 245, 244, 243, 242 or 241 amino acids. Preferably, the polypeptide having acetolactate decarboxylase activity has an amino acid sequence having at least 80, 90, 95, 98, 99 or 100% sequence identity to according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12. More preferably, the polypeptide has an amino acid sequence according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12. The protease is preferably a proline specific protease. More preferably, the proline specific protease is from Aspergillus niger. More preferably, the protease has at least 80, 85, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:17. Still more preferably, the protease has an amino acid sequence according to SEQ ID NO:17. Preferably, the ALDC polypeptide maintains at least 99, 98, 95, 90, 85, 80, 75 or 70% activity when the stable liquid formulation is stored at 10oC for 30, 60, 90, 120, 150 for 180 days. In other preferred embodiments, the ALDC polypeptide maintains at least 99, 98, 95, 90, 85, 80, 75 or 70% activity when the stable liquid formulation is stored at 30oC for 8, 16, 24, 32, 40 or 48 hours. In another aspect of the present invention, a proline specific protease formulation is presented which is substantially depleted of other protease activities comprising the protease wherein when said protease is combined with a polypeptide having acetolactate decarboxylase activity, the polypeptide is stable over time. Preferably, the polypeptide having acetolactate decarboxylase activity has an amino acid sequence having at least 80, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:3 wherein said polypeptide has at most 250 amino acids and a protease. Preferably, the polypeptide is at most 249, 248, 247, 246, 245, 244, 243, 242 or 241 amino acids. Preferably, the polypeptide having acetolactate decarboxylase activity has an amino acid sequence having at least 80, 90, 95, 98, 99 or 100% sequence identity to according to SEQ ID
NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12. More preferably, the polypeptide has an amino acid sequence according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12. The protease is preferably a proline specific protease. More preferably, the proline specific protease is from Aspergillus niger. More preferably, the protease has at least 80, 85, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:17. Still more preferably, the protease has an amino acid sequence according to SEQ ID NO:17. Preferably, the ALDC polypeptide maintains at least 99, 98, 95, 90, 85, 80, 75 or 70% activity when the stable liquid formulation is stored at 10oC for 30, 60, 90, 120, 150 for 180 days. In other preferred embodiments, the ALDC polypeptide maintains at least 99, 98, 95, 90, 85, 80, 75 or 70% activity when the stable liquid formulation is stored at 30oC for 8, 16, 24, 32, 40 or 48 hours. The present disclosure is described in further detail in the following examples, which are not in any way intended to limit the scope of the disclosure as claimed. The attached figures are meant to be considered as integral parts of the specification and description of the disclosure. The following examples are offered to illustrate, but not to limit the claimed disclosure. EXAMPLES EXAMPLE 1 As an example of a proline-specific endoprotease (PEP), the proline-specific endoprotease sold as Brewers Clarex® (5 PPU / g product) by DSM from Aspergillus niger (AnPro), was used. The defined activity of the proline specific endo-protease (PEP) is based on hydrolysis of the synthetic peptide Z-Gly-Pro-pNA at 37°C in a citrate/disodium phosphate buffer pH 4.6. The reaction product is monitored spectro-photometrically at 405 nm and one unit (1PPU) is defined as the quantity of enzyme that liberates 1 mmol of p-nitroanilide per minute under these test conditions. As an example of an acetolactate decarboxylase, aldB, from Brevibacillus brevis (which may be referred to as Bacillus brevis) produced in Bacillus licheniformis and sold as Maturex® Pro (2500 ADU-L/ g product) by Novozymes (Bagsværd, Denmark) was used. This aldB, expressed in Bacillus licheniformis, is referred to herein as aldB_Bl.
EXAMPLE 2 Cloning of Aspergillus niger ATCC 1015 protease AniPro_2 (CRC02753-WT) Aspergillus niger ATCC 1015 was selected as a potential source of enzymes which may be useful in various industrial applications. One of the genes identified in Aspergillus niger ATCC 1015, named AniPro_2 (CRC02753-WT), encodes a protein with homology to a protease as determined from a BLAST search (Altschul et al., J Mol Biol, 215: 403–410, 1990). The precursor protein encoded by the AniPro_2 gene is shown in SEQ ID NO:16 (JGI Reference Sequence: Aspni5_52703). At the N-terminus, the protease protein has a signal peptide with a length of 21 amino acids as predicted by SignalP version 4.0 (Nordahl Petersen et al. (2011) Nature Methods, 8:785-786) and derived from a Hypocrea jecorina aspartate protease (uniport id. G0R8T0). The presence of a signal sequence ensures that AniPro_2 is a secreted enzyme. The predicted mature form of AniPro_2 is shown in SEQ ID NO:17. EXAMPLE 3 Expression, fermentation and purification of AniPro_2 DNA sequences encoding AniPro_2, were chemically synthesized and inserted into a Trichoderma reesei expression vector pGXT (the same as the pTTTpyr2 vector as described in published PCT Application WO2015/017256, incorporated by reference herein) by Generay (Shanghai, China). The resulting plasmid was labeled as pGXT-AniPro2. The expression plasmid was then transformed into a suitable Trichoderma reesei strain (described in published PCT application WO 05/001036) using protoplast transformation (Te’o et al. (2002) J. Microbiol. Methods 51:393-99). Transformants were selected on a medium containing acetamide as a sole source of nitrogen. After 5 days of growth on acetamide plates, transformants were collected and subjected to fermentation via DASGIP (Eppendorf, Juelich, Germany). To initiate the fermentation of AniPro_2, seed culture was grown in 1 L shake flask, each contained 100 mL defined medium (pH5.5 before sterilization) having 50 g/L glucose monohydrate, 6 g/L glycine, 5 g/L (NH4)2SO4, 4.5 g/L KH2PO4, 1 g/L CaCl2·2H2O, 1 g/L MgSO4·7H2O, 2 g/L Mazu 6000K and 2.5 mL 400×T. reesei Trace Metals (400× Trace Metals stock (~pH 1) contains 175 g/L C6H8O7·H2O, 200 g/L FeSO4·7H2O, 16 g/L ZnSO4·7H2O, 3.2 g/L CuSO4·5H2O, 1.4 g/L MnSO4·H2O, and 0.8 g/L H3BO3). The seed culture was shaken for 48 hours
at 250 rpm and 30 °C. Following completion of this incubation, 200 mL of seed culture was transferred to the 2-L bioreactor (DASGIP). The medium for fermentation in the 2-L bioreactor (DASGIP) contains 60 g/L dextrose, 6 g/L glycine, 1 g/L CaCl2·2H2O, 4.5 g/L KH2PO4, 4 g/L (NH4)2SO4, 1 g/L MgSO4·7H2O, 1.2 g/L Mazu 6000K and 2.5 ml 400× T. reesei Trace Metals. An induction solution containing 250 g glucose/sophorose per kg was prepared and sterilized. Following inoculation, batch fermentation was initiated with working volume of 1 L and was controlled at pH 3.5 and 34 °C. Dissolved oxygen level was controlled above 35% by adjusting airflow rate, oxygen supply, and agitation during whole fermentation process. At the elapsed fermentation time of 22 hours, the glucose in broth was depleted, and the feed of 250 g (glucose/sophorose)/kg solution was started at this time. Stepwise feed rates of 4 mL feed/hr and 6 mL feed/hr were applied in the time intervals of 22-46 hours and 46-72 hours, respectively. Accompanying the start of fed-batch phase, pH was linearly changed to 4.0, and temperature was adjusted to 28 oC. Fermentation was completed after 72 hours run, and broth was harvested by centrifugation, filtered, and subsequently concentrated. To purify AniPro_2, the crude from 1 L Dasgip fermenter was concentrated and added ammonium sulfate to the final concentration of 1 M. The solution was loaded onto a HiPrepTM Phenyl FF 16/10 column pre-equilibrated with 20 mM NaAc (pH5.0) supplemented with additional 1 M ammonium sulfate (Buffer A). The target protein was eluted from the column with 0.75 M ammonium sulfate. The corresponding fractions were pooled, concentrated and exchanged buffer into 20 mM NaPi (pH7.0) (Buffer B), using a VivaFlow 200 ultra-filtration device (Sartorius Stedim). The resulting solution was applied to a HiLoadTM Q FF 16/10 column pre-equilibrated with Buffer B. The target protein was eluted from the column with 0.3 M NaCl. The fractions containing active protein were pooled, concentrated and exchanged buffer into 20 mM NaAc (pH5.0), 150 mM NaCl via the 10K Amicon Ultra devices, then stored in 40% glycerol at -20 oC until usage. EXAMPLE 4 HETEROLOGOUS EXPRESSION OF ACETOLACTATE DECARBOXYLASE, ALDB The Brevibacillus brevis (which may be referred to as Bacillus brevis) acetolactate decarboxylases (ALDC) aldB gene was previously identified (Diderichsen et al., J Bacteriol.
(1990) 172(8): 4315), with the sequence set forth as UNIPROT Accession No. P23616.1. The sequence of this gene, aldB, is depicted in SEQ ID NO:1. Nucleotides 1 to 72 encode the signal peptide. The aldB gene and corresponding encoded proenzyme are also referred to as the wildtype (WT). The proenzyme encoded by the aldB gene is depicted in SEQ ID NO: 2. At the N-terminus, the protein has a signal peptide with a length of 24 amino acids as predicted by SignalP-NN (Emanuelsson et al., Nature Protocols (2007) 2: 953-971). The presence of a signal peptide indicates that this acetolactate decarboxylase, aldB is a secreted enzyme. The sequence of the predicted, fully processed mature chain (aldB, 261 amino acids) is depicted in SEQ ID NO: 3. The aldB gene that encodes an acetolactate decarboxylases enzyme (ALDC) was produced in B. subtilis using the synthetic gene inserted into the pCB_alr vector, see Figure 1. The aldB gene containing was transcribed using aprE promoter followed by B. subtilis aprE signal sequence. For expression the alrA(CB)RIHI-Bbr vector was integrated into a protease deficient B. subtilis strain. A map of the pCB_alr vector containing the aldB gene (alrA(CB)RIHI-Bbr) is shown in Figure 2. To produce aldB, a B. subtilis strain transformant containing the alrA(CB)RIHI-Bbr cassette (strain BRA8014 described below) was cultured in 15-mL Falcon tubes for 16 hours in TSB (broth) with 10 ppm neomycin, and 300 µL of this pre-culture was added to a 500-mL flask filled with 30 mL of cultivation media (described below) supplemented with 10 ppm neomycin. The flasks were incubated for 24, 48 and 72 hours at 33°C with constant rotational mixing at 180 rpm. Cultures were harvested by centrifugation at 14500 rpm for 20 minutes in conical tubes. The culture supernatants were used for protein determination and assays. The cultivation media was an enriched semi-defined media based on MOPs buffer, with urea as major nitrogen source, glucose as the main carbon source, 50µM ZnSO4 to ensure high enzyme activity and supplemented with 1% soytone for robust cell growth. The aldB expressed in Bacillus subtilis is here within termed, aldB_Bs. The nucleotide mature sequence of the aldB gene in plasmid alrA(CB)RIHI-Bbr is depicted in SEQ ID NO:4. The amino acid sequence of the aldB precursor protein expressed from plasmid alrA(CB)RIHI-Bbr is depicted in SEQ ID NO:5. EXAMPLE 5 Protein Determination Methods Protein Determination by Stain Free Imager Criterion
Protein was quantified by SDS-PAGE gel and densitometry using Gel Doc™ EZ imaging system. Reagents used in the assay: Concentrated (2x) Laemmli Sample Buffer (Bio-Rad, Catalogue #161-0737); 26-well XT 4-12% Bis-Tris Gel ( Bio-Rad, Catalogue #345-0125); protein markers “Precision Plus Protein Standards” (Bio-Rad, Catalogue #161- 0363); protein standard BSA (Thermo Scientific, Catalogue #23208) and SimplyBlue Safestain (Invitrogen, Catalogue #LC 6060. The assay was carried out as follow: In a 96well-PCR plate 50µL diluted enzyme sample were mixed with 50 µL sample buffer containing 2.7 mg DTT. The plate was sealed by Microseal ‘B’ Film from Bio-Rad and was placed into PCR machine to be heated to 70°C for 10 minutes. After that the chamber was filled by running buffer, gel cassette was set. Then 10 µL of each sample and standard (0.125-1.00 mg/mL BSA) was loaded on the gel and 5 µL of the markers were loaded. After that the electrophoresis was run at 200 V for 45 minutes. Following electrophoresis, the gel was rinsed 3 times 5 min in water, then stained in Safe-stain overnight and finally destained in water. Then the gel was transferred to Imager. Image Lab software was used for calculation of intensity of each band. A calibration curve was made using BSA (Thermo Scientific, Catalogue #23208) and the amount of the target protein was determined by the band intensity and calibration curve. The protein quantification method was employed to prepare enzyme samples of used in subsequent Examples. The protease protein concentration was determined in: A concentrated sample AniPro_2 sample was determined to 55 mg/ml and a sample of AnPro was determined to 39mg/ml. EXAMPLE 6 ALDC enzyme sequence identification by MS including N and C-terminal amino acid determination In preparation for sequence confirmation, an SDS-PAGE gel of isolated aldB_Bs truncation variants were analyzed by LC-MS/MS as described subsequently. In preparation for sequence confirmation, including N- and C-terminal determination, a protein band from an SDS-PAGE gel of aldB ferment sample was subjected to a series of chemical treatments. Between the individual steps the gel pieces were washed and shrunk using Milli-Q water, 50w/w% ethanol and absolute ethanol respectively. The protein was reduced/alkylated by DTT/Iodoacetamide treatment. A guanidination step was performed to convert lysines to homoarginines to protect lysine side chains from acetylation. The acetylation reaction using Sulfo-NHS-Acetate (Sulfosuccinimidyl Acetate) only modifies the protein N-terminal residue. The gel pieces were swelled with a buffer
containing 40v/v% 18O water:60v/v% 16O water and the proteolytic enzymes used for protein digestion (Trypsin and α-Chymotrypsin). The resulting peptides will contain mixtures of 18O and 16O, except for the Carboxyl terminus which will retain the native 16O, as will be apparent from the isotopic pattern of the peptides. The peptide, originating from the protein N-terminus, will appear as the only acetylated peptide. After digestion the peptides were extracted from the gel pieces using 5w/w% formic acid and acetonitrile, then lyophilized and re-dissolved in 0.1w/w% TFA. The digestion products were separated (C18 column) and analyzed using a Proxeon nano- LC system followed by LTQ Orbitrap (Thermo Fisher) high resolution mass spectrometer and the amino acid sequence was deduced from the MS/MS fragment spectrum of the peptides, and the isotopic pattern of the peptides (using Xcalibur 2.0 SR2 software). Based on this analysis, the N-terminus of the isolated full-length protein was confirmed to begin with A[25] (according to SEQ ID NO. 2) and the C-terminus of the isolated full-length protein was confirmed to end at position K[285] (according to SEQ NO. 2) by the acetylation and 18O-label methods as described above (see table 2). The N-terminus position A[25] at the mature aldB_Bs correspond with the predicted signal peptide cleavage determined by the Signal P 3.0 program (http://www.cbs.dtu.dk/services/SignalP/), set to SignalP-NN system, (Emanuelsson et al., (2007), Nature Protocols, 2: 953-971) of the gene transcript. Different N- and C-terminal truncation variants were further identified and their respective N- and C-terminus position according to SEQ ID NO. 2 are given in table 2. [001] Table 2 Identified N- and C-terminus positions of aldB_Bs variants, position according to according to SEQ ID NO. 2. Position N- Position C- SEQ ID NO: terminus terminus
e p ese ve o a so e co passes o o ogous su s u o su stitution and replacement are both used herein to mean the interchange of an existing amino acid residue, with an alternative residue) that may occur i.e., like-for-like substitution such as basic for basic, acidic for acidic, polar for polar etc. Non-homologous substitution may also occur i.e., from one class of residue to another or alternatively involving the inclusion of unnatural amino acids such as ornithine (hereinafter referred to as Z), diaminobutyric acid ornithine (hereinafter referred to as B), norleucine ornithine (hereinafter referred to as O), pyriylalanine, thienylalanine, naphthylalanine and phenylglycine. Replacements may also be made by synthetic amino acids (e.g. unnatural amino acids) include; alpha* and alpha-disubstituted* amino acids, N-alkyl amino acids*, lactic acid*, halide derivatives of natural amino acids such as trifluorotyrosine*, p-Cl-phenylalanine*, p-Br-
phenylalanine*, p-I-phenylalanine*, L-allyl-glycine*, ß-alanine*, L-a-amino butyric acid*, L-g- amino butyric acid*, L-a-amino isobutyric acid*, L-e-amino caproic acid#, 7-amino heptanoic acid*, L-methionine sulfone#*, L-norleucine*, L-norvaline*, p-nitro-L-phenylalanine*, L- hydroxyproline#, L-thioproline*, methyl derivatives of phenylalanine (Phe) such as 4-methyl- Phe*, pentamethyl-Phe*, L-Phe (4-amino)#, L-Tyr (methyl)*, L-Phe (4-isopropyl)*, L-Tic (1,2,3,4-tetrahydroisoquinoline-3-carboxyl acid)*, L-diaminopropionic acid # and L-Phe (4- benzyl)*. The notation * has been utilized for the purpose of the discussion above (relating to homologous or non-homologous substitution), to indicate the hydrophobic nature of the derivative whereas # has been utilized to indicate the hydrophilic nature of the derivative, #* indicates amphipathic characteristics. Variant amino acid sequences may include suitable spacer groups that may be inserted between any two amino acid residues of the sequence including alkyl groups such as methyl, ethyl, or propyl groups in addition to amino acid spacers such as glycine or b-alanine residues. A further form of variation, involves the presence of one or more amino acid residues in peptoid form, will be well understood by those skilled in the art. For the avoidance of doubt, “the peptoid form” is used to refer to variant amino acid residues wherein the a-carbon substituent group is on the residue’s nitrogen atom rather than the a-carbon. Processes for preparing peptides in the peptoid form are known in the art, for example Simon RJ et al., PNAS (1992) 89(20), 9367-9371 and Horwell DC, Trends Biotechnol. (1995) 13(4), 132-134. The nucleotide sequences for use in the present invention may include within them synthetic or modified nucleotides. Several different types of modification to oligonucleotides are known in the art. These include methylphosphonate and phosphorothioate backbones and/or the addition of acridine or polylysine chains at the 3' and/or 5' ends of the molecule. For the purposes of the present invention, it is to be understood that the nucleotide sequences described herein may be modified by any method available in the art. Such modifications may be carried out to enhance the in vivo activity or life span of nucleotide sequences of the present invention. The present invention also encompasses the use of nucleotide sequences that are complementary to the sequences presented herein. Other variants of the sequences described herein may be obtained for example by probing DNA libraries made from a range of individuals, for example individuals from different
populations. In addition, other homologues may be obtained and such homologues and fragments thereof in general will be capable of selectively hybridizing to the sequences shown in the sequence listing herein. Such sequences may be obtained by probing cDNA libraries or genomic DNA libraries made from other animal species and probing such libraries with probes comprising all or part of any one of the sequences in the attached sequence listings under conditions of medium to high stringency. Similar considerations apply to obtaining species homologues and allelic variants of the polypeptide or nucleotide sequences of the invention. Variants and strain/species homologues may also be obtained using degenerate PCR which will use primers designed to target sequences within the variants and homologues encoding conserved amino acid sequences within the sequences of the present invention. Conserved sequences can be predicted, for example, by aligning the amino acid sequences from several variants/homologues. Sequence alignments can be performed using computer software known in the art. For example, the GCG Wisconsin PileUp program is widely used. The primers used in degenerate PCR will contain one or more degenerate positions and will be used at stringency conditions lower than those used for cloning sequences with single sequence primers against known sequences. Alternatively, such polynucleotides may be obtained by site directed mutagenesis of characterized sequences. This may be useful where for example silent codon sequence changes are required to optimize codon preferences for a particular host cell in which the polynucleotide sequences are being expressed. Other sequence changes may be desired to introduce restriction enzyme recognition sites, or to alter the property or function of the polypeptides encoded by the polynucleotides. The present invention employs, unless otherwise indicated, conventional techniques of biochemistry, molecular biology, microbiology, and recombinant DNA, which are within the capabilities of a person of ordinary skill in the art. Such techniques are explained in the literature. See, for example, J. Sambrook, E. F. Fritsch, and T. Maniatis, 1989, Molecular Cloning: A Laboratory Manual, Second Edition, Books 1-3, Cold Spring Harbor Laboratory Press; Ausubel, F. M. et al. (1995 and periodic supplements; Current Protocols in Molecular Biology, ch. 9, 13, and 16, John Wiley & Sons, New York, N. Y.); B. Roe, J. Crabtree, and A. Kahn, 1996, DNA Isolation and Sequencing: Essential Techniques, John Wiley & Sons; M. J. Gait (Editor), 1984, Oligonucleotide Synthesis: A Practical Approach, IrI Press; and, D. M. J. Lilley and J. E. Dahlberg,
1992, Methods of Enzymology: DNA Structure Part A: Synthesis and Physical Analysis of DNA Methods in Enzymology, Academic Press. Each of these general texts is herein incorporated by reference. As used herein, “percent (%) sequence identity” means that a particular sequence has at least a certain percentage of amino acid residues identical to those in a specified reference sequence, when aligned using the CLUSTAL W algorithm with default parameters. See Thompson et al. (1994) Nucleic Acids Res. 22:4673-4680. Default parameters for the CLUSTAL W algorithm are: Gap opening penalty: 10.0 Gap extension penalty: 0.05 Protein weight matrix: BLOSUM series DNA weight matrix: IUB Delay divergent sequences %: 40 Gap separation distance: 8 DNA transitions weight: 0.50 List hydrophilic residues: GPSNDQEKR Use negative matrix: OFF Toggle Residue specific penalties: ON Toggle hydrophilic penalties: ON Toggle end gap separation penalty: OFF Deletions are counted as non-identical residues, compared to a reference sequence. Deletions occurring at either terminus are included. For example, a variant with five amino acid deletions of the C-terminus of the mature 617 residue polypeptide would have a percent sequence identity of 99% (612 / 617 identical residues × 100, rounded to the nearest whole number) relative to the mature polypeptide. Such a variant would be encompassed by a variant having “at least 99% sequence identity” to a mature polypeptide. In accordance with the instant invention, proteins, including enzymes, of the present invention exist in multiple forms. Proteins of the instant invention may be clipped or trimmed (i.e., removing amino acids) from the N-terminus and/or the C-terminus, resulting in a shorter protein. Proteins of the instant invention can also have internal deletions. Shorter proteins as described herein can have higher activity or lower activity than longer counterparts. Without
being bound by theory, as used herein the term “pre-pro-protein” is a protein, including an enzyme, which has an N-terminal signal peptide that targets the protein for secretion. A pre-pro- protein is sometimes referred to herein as “full length” or “full length protein”. The N-terminal signal peptide is cleaved off in the endoplasmic reticulum to yield a “pro-protein”. A pro- protein, as used herein, is shorter in length than the full-length protein (it is missing the signal peptide) but longer than the mature protein. In general, a pro-protein is inactive or less active than the mature protein. A pro-protein can be activated or converted to a more active mature form by post-translational modification such as N- or C- terminal clipping. A pro-protein which is an enzyme may be called a “proenzyme” or a “zymogen.” The clipped active protein (derived from the pro-protein) is also referred to herein as the mature protein. It is to be noted that the above terms are used for convenience and are not meant to override or determine the activities of a protein of the instant invention. It is also to be noted that any protein of the instant invention can have more than one variant described by the same term. All references cited in the present specification are hereby incorporated by reference in their entirety. In particular, the teachings of all references herein specifically referred to are incorporated by reference. Unless otherwise defined, all terms used in disclosing the invention, including technical and scientific terms, have the meaning as commonly understood by one of ordinary skill in the art to which this invention belongs. By means of further guidance, term definitions are included to better appreciate the teaching of the present invention. Other definitions are set forth below. Beer As used herein, the term “beer” traditionally refers to an alcoholic beverage derived from malt, which is derived from barley, and optionally adjuncts, such as cereal grains, and flavored with hops. Beer can be made from a variety of grains by essentially the same process. All grain starches are glucose homopolymers in which the glucose residues are linked by either alpha-1, 4- or alpha-1,6-bonds, with the former predominating. The process of making fermented malt beverages is commonly referred to as brewing. The principal raw materials used in making these beverages are water, hops and malt. In addition, adjuncts such as common corn grits, refined corn grits, brewer's milled yeast, rice, sorghum, refined corn starch, barley, barley starch,
dehusked barley, wheat, wheat starch, torrified cereal, cereal flakes, rye, oats, potato, tapioca, and syrups, such as corn syrup, sugar cane syrup, inverted sugar syrup, barley and/or wheat syrups, and the like may be used as a source of starch. The starch will eventually be converted into dextrins and fermentable sugars. For several reasons, the malt, which is produced principally from selected varieties of barley, has the greatest effect on the overall character and quality of the beer. First, the malt is the primary flavoring agent in beer. Second, the malt provides the major portion of the fermentable sugar. Third, the malt provides the proteins, which will contribute to the body and foam character of the beer. Fourth, the malt provides the necessary enzymatic activity during mashing. As used herein, the term “Hops” refers to it use in contributing significantly to beer quality, including flavoring. Hops (or hop constituents) add desirable bittering substances to the beer. In addition, the hops act as protein precipitants, establish preservative agents and aid in foam formation and stabilization. As used herein, the “process for making beer” is one that is well known in the art, but briefly, it involves five steps: (a) mashing and/or adjunct cooking (b) wort separation and extraction (c) boiling and hopping of wort (d) cooling, fermentation, and storage, and (e) maturation, processing and packaging. In the first step, milled or crushed malt is mixed with water and held for a period under controlled temperatures to permit the enzymes present in the malt to convert the starch present in the malt into fermentable sugars. In the second step, the mash is transferred to a "lauter tun" or mash filter where the liquid is separated from the grain residue. This sweet liquid is called "wort" and the left-over grain residue is called “spent grain”. The mash is typically subjected to an extraction, which involves adding water to the mash to recover the residual soluble extract from the spent grain. In the third step, the wort is boiled vigorously. This sterilizes the wort and helps to develop the color, flavor, and odor. Hops are added at some point during the boiling. In the fourth step, the wort is cooled and transferred to a fermenter, which either contains the yeast or to which yeast is added. After addition of yeast, the liquid is referred to as a fermentate. The yeast converts the sugars by fermentation into alcohol and carbon dioxide gas; at the end of fermentation the fermenter is chilled, or the fermenter may be chilled to stop fermentation. The yeast flocculates and is removed. In the last step, the beer is cooled and stored for a period of time, during which the beer clarifies, and its flavor develops, and any material that might impair the appearance, flavor and shelf life of the beer settles out.
Prior to packaging, the beer is carbonated and, optionally, filtered and pasteurized. After fermentation, a beverage is obtained which usually contains from about 2% to about 10% alcohol by weight. The non-fermentable carbohydrates are not converted during fermentation and form most of the dissolved solids in the final beer. This residue remains because of the inability of malt amylases to hydrolyze the alpha-1,6-linkages of the starch. The non-fermentable carbohydrates contribute about 50 calories per 12 ounces of beer. The term "fermentation" means, in the context of brewing, the transformation of sugars in the wort, by enzymes in the brewing yeast, into ethanol and carbon dioxide with the formation of other fermentation by-products. As used herein, a “fermentate” is the liquid solution undergoing a fermentation process leading to chemical change of the food, beer, or beverage by the action of yeast or bacteria, which produce carbon dioxide and turns carbohydrates in it into alcohol: As used herein the term "malt" is understood as any malted cereal grain, such as barley. As used herein, the term "wort" refers to the unfermented liquor run-off following extracting the grist during mashing. As used herein, the term "spent grains" refers to the drained solids remaining when the grist has been extracted and the wort separated from the mash. As used herein, the term "beer" refers to fermented wort, e.g., an alcoholic beverage brewed from barley malt, optionally adjunct and hops. ALDC In some aspects, the invention provides ALDC enzymes having a better stability and activity, and which further can be recovered from microorganisms in improved yields. Acetolactate decarboxylase (ALDC) is an enzyme that belongs to the family of carboxy lyases, which are responsible for cleaving carbon-carbon bonds. Acetolactate decarboxylase catalyzes the conversion of 2-acetolactate (also known as 2-hydroxy-2-methyl-3-oxobutanoate) to 2-acetoin and releasing CO2. Acetolactate decarboxylase enzymes catalyze the enzymatic reaction belonging to the classification EC 4.l.l.5 (acetolactate decarboxylase activity) and gene ontology (GO) term ID of GO: 0047605. The GO term ID specifies that any protein characterized as having this associated GO term encodes an enzyme with catalytic acetolactate decarboxylase activity.
Various acetolactate decarboxylase (alsD) genes, which encode acetolactate decarboxylase enzymes, are known in the art. Examples of alsD genes include but are not limited to gil3751436271reflYP 005006068.11 Acetolactate decarboxylase [Niastella koreensis OR20-10]; gil361 0576731gb1AEV96664.11 Acetolactate decarboxylase [Niastella koreensis OR20-10]; gi12187634151gb1ACL0588l.11 Acetolactate decarboxylase [Desulfatibacillum alkenivorans AK -01]; gil220909520lreflYP 002484831.11 acetolactate decarboxylase [Cyanothece sp. PCC 7425]; gil2187820311reflYP 002433349.11 acetolactate decarboxylase [Desulfatibacillum alkenivorans AK- Ol]; gi12136930901ref1YP 002323676.11 Acetolactate decarboxylase [Bifidobacterium longum subsp. infantis ATCC 15697 = JCM 1222]; gil 1895002971reflYP 001959767.11 Acetolactate decarboxylase [Chlorobium phaeobacteroides BS 1]; gil 1894237871reflYP 001950964.11 acetolactate decarboxylase [Geobacter lovleyi SZ]; gil 1720582711ref1YP 00181473l.11 acetolactate decarboxylase [Exiguobacterium sibiricum 255-15]; gil1639387751reflYP 001643659.11 acetolactate decarboxylase [Bacillus weihenstephanensis KBAB4]; gil 1585223041reflYP 001530174.11 acetolactate decarboxylase [Desulfococcus oleovorans Hxd3]; gil 157371670lreflYP 001479659.11 acetolactate decarboxylase [Serratia proteamaculans 568]; gi11503951111ref1YP 001317786.11 acetolactate decarboxylase [Staphylococcus aureus subsp. aureus JHl]; gil 1503947151reflYP 001317390.11 acetolactate decarboxylase [Staphylococcus aureus subsp. aureus JHl]; gil 1463116791ref1YP 001176753.11 acetolactate decarboxylase [Enterobacter sp.638]; gil 109900061 IreflYP 663316.11 acetolactate decarboxylase [Pseudoalteromonas atlantica T6c]; giI219866131IgbIACL46470.11 Acetolactate decarboxylase [Cyanothece sp. PCC 7425]; giI213524551IgbIACJ53298.11 Acetolactate decarboxylase [Bifidobacterium longum subsp. infantis ATCC 15697 = JCM 1222]; gil 1894200461gb1ACD94444.11 Acetolactate decarboxylase [Geobacter lovleyi SZ]; giI158511130IgbIABW68097.11 Acetolactate decarboxylase [Desulfococcus oleovorans Hxd3]; gil 1573234341gblABV 42531.11 Acetolactate decarboxylase [Serratia proteamaculans 568]; gi11453185551gb1ABP60702.11 Acetolactate decarboxylase [Enterobacter sp.638]; gi11499475631gb1ABR53499.11 Acetolactate decarboxylase [Staphylococcus aureus subsp.aureus JH1]; gi11499471671gb1ABR53103.11 Acetolactate decarboxylase [Staphylococcusaureus subsp. aureus JH1]; gi11638609721gb1ABY42031.11 Acetolactate decarboxylase [Bacillus weihenstephanensis KBAB4]; gill 097023421gb1ABG42262.11 Acetolactate decarboxylase [Pseudoalteromonas atlantica T6c]; gi11894957381gb1ACE04286.11 Acetolactate decarboxylase [Chlorobium phaeobacteroides BS 1]; gi11719907921gb1ACB61714.11 Acetolactate decarboxylase [Exiguobacterium sibiricum 255-15]; gil2239325631reflZP 03624564.11 Acetolactate decarboxylase
[Streptococcus suis 89/1591]; gil 194467531 IreflZP 03073518.11 Acetolactate decarboxylase [Lactobacillus reuteri 100-23]; gi12238988341gb1EEF65194.11 Acetolactate decarboxylase [Streptococcus suis 89/1591]; gil 1944545671gb1EDX43464.11 Acetolactate decarboxylase [Lactobacillus reuteri 100-23]; gi13842671351ref1YP 005422842.11 acetolactate decarboxylase [Bacillus amyloliquefaciens subsp. plantarum YAU B9601-Y2]; gil3753640371reflYP 005132076.11 acetolactate decarboxylase [Bacillus amyloliquefaciens subsp. plantarum CAU B946]; gil3407932311reflYP 004758694.11 acetolactate decarboxylase [Corynebacterium variabile DSM 44702]; gil3363251191reflYP 004605085.11 acetolactate decarboxylase [Corynebacterium resistens DSM 45100]; gil 1482690321reflYP 001247975.11 acetolactate decarboxylase [Staphylococcus aureus subsp. aureus JH9]; gil 148268650lreflYP 001247593.11 acetolactate decarboxylase [Staphylococcus aureus subsp. aureus JH9]; gil 1485433721reflYP 001270742.11 acetolactate decarboxylase [Lactobacillus reuteri DSM 20016]; gi13805004881emb1CCG51526.11 acetolactate decarboxylase [Bacillus amyloliquefaciens subsp. plantarum YAU B9601-Y2]; gi13715700311emb1CCF06881.11 acetolactate decarboxylase [Bacillus amyloliquefaciens subsp. plantarum CAU B946]; gi13405331411gb1AEK35621.11 acetolactate decarboxylase [Corynebacterium variabile DSM 44702]; gi13361011011gb1AEI08921.11 acetolactate decarboxylase [Corynebacterium resistens DSM 45100]; gi11485304061gb1ABQ82405.11 Acetolactate decarboxylase [Lactobacillus reuteri DSM 20016]; gi11477421011gb1ABQ50399.11 Acetolactate decarboxylase [Staphylococcus aureus subsp. aureus JH9]; giI147741719IgbIABQ50017.11 Acetolactate decarboxylase [Staphylococcus aureus subsp. aureus JH9]; gil392529510lreflZP 10276647.11 acetolactate decarboxylase [Carnobacterium maltaromaticum ATCC 35586]; gil3660540741reflZP 09451796.11 acetolactate decarboxylase [Lactobacillus suebicus KCTC 3549]; gil3396241471reflZP 08659936.11 acetolactate decarboxylase [Fructobacillus Jructosus KCTC 3544]; gil3363937271reflZP 08575126.11 acetolactate decarboxylase [Lactobacillus coryniformis subsp. torquens KCTC 3535]. Each sequence associated with the foregoing accession numbers is incorporated herein by reference. In some embodiments, the invention relates to ALDC enzymes from Lactobaclllus Casei (Godtfredsen 1984), Brevibacterium acetylicum (Oshiro, 1989), Lactococcus lactis (Vincent Phalip 1994), Leuconostoc lactis (O sulivan, 2001), Enterobacter aerogenes (Blomquist, 1993), Bacillus subtilis (Renna, 1993), Bacillus brevis (Svendsen, 1989) and Lactococcus lactis DX (Yuxing, 2014) It is to be understood that any suitable ALDC enzymes, i.e., ALDC produced from any microorganism which activity is dependent on metal ions, can be used according to the invention. In some embodiments, the ALDC used in the methods and compositions described herein is an ALDC from Bacillus brevis or Bacillus licheniformis.
The ALDC activity of the enzyme composition according to the invention is measured by the ALDC assays as described herein or any suitable assay known in the art. The standard assay is carried out at pH 6.0, and it can be performed at different pH values and temperatures for the additional characterization and specification of enzymes. One unit of ALDC activity is defined as the amount of enzyme which produces 1 µmole acetoin per minute under the conditions of the assay (e.g., pH 6.0 (or as specified) and 30 °C). In some embodiments, the enzyme has a temperature optimum in the range of 5-80 °C, such as in the range of 5-40°C or 15-80°C, such as in the range 20-80 °C, such as in the range 5- 15°C, 15-20°C, 45-65 °C, 50-65 °C, 55-65 °C or 60-80°C. In some embodiments, the enzyme has a temperature optimum in the range of 45-65 °C. In some embodiments, the enzyme has a temperature optimum of about 60°C. In some embodiments, the enzyme has a total number of amino acids of less than 350, such as less than 340, such as less than 330, such as less than 320, such as less than 310, such as less than 300 amino acids, such as in the range of 200 to 350, such as in the range of 220 to 345 amino acids. In some embodiments the ALDC compositions and methods according to the invention comprises any one or more further enzyme. In some embodiments the one or more further enzyme is selected from list consisting of acetolactate reductoisomerases, acetolactate isomerases, amylase, glucoamylase, hemicellulose, cellulase, glucanase, pullulanase, isoamylase, endo-glucanase and related beta-glucan hydrolytic accessory enzymes, xylanase, and xylanase accessory enzymes (for example, arabinofuranosidase, ferulic acid esterase, xylan acetyl esterase), beta-glucosidase and protease. In some embodiments the compositions and methods according to the invention comprises an enzyme exhibiting ALDC activity, wherein the activity of said ALDC enzyme is in the range of 950 to 2500 Units per mg of protein. In some embodiments the compositions and methods according to the invention comprises an enzyme exhibiting ALDC activity, wherein the activity of said ALDC enzyme is in the range of 1000 to 2500 Units per mg of protein. Proline Specific Proteases Beer-haze, a cloudy appearance in beer, is caused by the aggregation of hydrophobic proteins, e.g., hordeins from barley, and polyphenols, resulting in a beer with an undesirable
cloudy appearance or haze. See, e.g., Asano, K.; Shinagawa, K.; Hashimoto, N. Characterization of haze-forming proteins of beer and their roles in chill haze formation. J. Am. Soc. Brew. Chem. 1982, 40, 147−154. The same phenomenon is also called chill-haze and similar haze formation may also occur in wine and fruit juices. It has been suggested that acid proteases such as papain can be used to degrade beer proteins and hence prevent haze formation. However, broad spectrum proteases such as papain have been found to impair beer foam formation and stability. See, e.g, Posada, J.; Almenar, J.; Garcia Galindo, J. A practical approach on protein stabilizers. Proc. - Eur. Brew. Conv. 1971, 13, 379−391. For this reason, more selective proteases such as proline specific endopeptidases have been employed to reduce beer haze. However, there is a continuing need for proteases that can be used to reduce beer-haze, because present commercial offerings are overly expensive and do not provide complete beer-haze removal. Moreover, there is a concern that even proline specific endoproteases are still too broad spectrum. For example, it has been discovered in accordance with the present invention that prior art proline specific endoproteases can destabilize other enzyme added exogenously to beer, including ALDC enzymes added for maturation. In accordance with an aspect of the present invention, proline specific endoproteases have been discovered which are less destabilizing to other exogenously added enzymes, including ALDC enzymes. A “polyphenol” is a compound having one or more aromatic rings and substituted by one or more hydroxyl groups. Examples of polyphenols are tannins and flavonoids, including catechins, flavonols and anthocyanins. The proline-specific protease and/or ALDC of the present invention may be added at different stages during the preparation of a beer. Addition of the enzyme at the beginning of the fermentation yields the best possible results. However, the enzyme may be added to a mash or to a fermented beer before haze has been formed. Enzyme of the present invention, including ALDC enzymes and proline specific proteases may be in isolated or purified form. By "isolated" or "purified" means and enzyme removed from its native environment. For example, recombinantly produced proline specific protease or ALDC expressed in a host cell may be considered isolated for purposes of the invention. Similarly, native, or recombinant polypeptides which have been substantially purified by any suitable technique may be considered isolated or purified.
Production of enzymes The enzymes of the present invention can be produced in host cells, for example, by secretion or intracellular expression. A cultured cell material (e.g., a whole-cell broth) having an enzyme can be obtained following secretion of the enzyme into the cell medium. Optionally, the enzyme can be isolated from the host cells, or even isolated from the cell broth, depending on the desired purity of the final enzyme. Suitable host cells include bacterial, fungal (including yeast and filamentous fungi), and plant cells (including algae). Particularly useful host cells include Aspergillus niger, Aspergillus oryzae or Trichoderma reesei. Other host cells include bacterial cells, e.g., Bacillus subtilis or B. licheniformis, as well as Streptomyces, E. coli. Vectors A DNA construct comprising a nucleic acid encoding an enzyme can be constructed to be expressed in a host cell. Because of the well-known degeneracy in the genetic code, variant polynucleotides that encode an identical amino acid sequence can be designed and made with routine skill. It is also well-known in the art to optimize codon use for a particular host cell. Nucleic acids encoding enzymes of the present invention can be incorporated into a vector. Vectors can be transferred to a host cell using well-known transformation techniques, such as those disclosed below. The vector may be any vector that can be transformed into and replicated within a host cell. For example, a vector comprising a nucleic acid encoding an enzyme can be transformed and replicated in a bacterial host cell as a means of propagating and amplifying the vector. The vector also may be transformed into an expression host, so that the encoding nucleic acids can be expressed as a functional enzyme. Host cells that serve as expression hosts can include filamentous fungi, for example. The Fungal Genetics Stock Center (FGSC) Catalogue of Strains lists suitable vectors for expression in fungal host cells. See FGSC, Catalogue of Strains, University of Missouri, at www.fgsc.net (last modified January 17, 2007). A representative vector is pJG153, a promoterless Cre expression vector that can be replicated in a bacterial host. See Harrison et al. (June 2011) Applied Environ. Microbiol. 77: 3916-22. pJG153can be modified with routine skill to comprise and express a nucleic acid encoding an enzyme. A nucleic acid encoding an enzyme can be operably linked to a suitable promoter, which
allows transcription in the host cell. The promoter may be any DNA sequence that shows transcriptional activity in the host cell of choice and may be derived from genes encoding proteins either homologous or heterologous to the host cell. Exemplary promoters for directing the transcription of the DNA sequence encoding an enzyme, especially in a bacterial host, are the promoter of the lac operon of E. coli, the Streptomyces coelicolor agarase gene dagA or celA promoters, the promoters of the Bacillus licheniformis α-amylase gene (amyL), the promoters of the Bacillus stearothermophilus maltogenic amylase gene (amyM), the promoters of the Bacillus amyloliquefaciens α-amylase (amyQ), the promoters of the Bacillus subtilis xylA and xylB genes etc. For transcription in a fungal host, examples of useful promoters are those derived from the gene encoding Aspergillus oryzae TAKA amylase, Rhizomucor miehei aspartic proteinase, Aspergillus niger neutral α-amylase, A. niger acid stable α-amylase, A. niger glucoamylase, Rhizomucor miehei lipase, A. oryzae alkaline protease, A. oryzae triose phosphate isomerase, or A. nidulans acetamidase. When a gene encoding an enzyme is expressed in a bacterial species such as E. coli, a suitable promoter can be selected, for example, from a bacteriophage promoter including a T7 promoter and a phage lambda promoter. Examples of suitable promoters for the expression in a yeast species include but are not limited to the Gal 1 and Gal 10 promoters of Saccharomyces cerevisiae and the Pichia pastoris AOX1 or AOX2 promoters. cbh1 is an endogenous, inducible promoter from Trichoderma reesei. See Liu et al. (2008) “Improved heterologous gene expression in Trichoderma reesei by cellobiohydrolase I gene (cbh1) promoter optimization,” Acta Biochim. Biophys. Sin (Shanghai) 40(2): 158-65. The coding sequence can be operably linked to a signal sequence. The DNA encoding the signal sequence may be the DNA sequence naturally associated with the enzyme gene to be expressed or from a different Genus or species. A signal sequence and a promoter sequence comprising a DNA construct or vector can be introduced into a fungal host cell and can be derived from the same source. For example, the signal sequence is the cbh1 signal sequence that is operably linked to a cbh1 promoter. An expression vector may also comprise a suitable transcription terminator and, in eukaryotes, polyadenylation sequences operably linked to the DNA sequence encoding a variant enzyme. Termination and polyadenylation sequences may suitably be derived from the same sources as the promoter. The vector may further comprise a DNA sequence enabling the vector to replicate in the
host cell. Examples of such sequences are the origins of replication of plasmids pUC19, pACYC177, pUB110, pE194, pAMB1, and pIJ702. The vector may also comprise a selectable marker, e.g., a gene the product of which complements a defect in the isolated host cell, such as the dal genes from B. subtilis or B. licheniformis, or a gene that confers antibiotic resistance such as, e.g., ampicillin, kanamycin, chloramphenicol, or tetracycline resistance. Furthermore, the vector may comprise Aspergillus selection markers such as amdS, argB, niaD and xxsC, a marker giving rise to hygromycin resistance, or the selection may be accomplished by co-transformation, such as known in the art. See e.g., International PCT Application WO 91/17243. Intracellular expression may be advantageous in some respects, e.g., when using certain bacteria or fungi as host cells to produce large amounts of enzyme for subsequent enrichment or purification. Extracellular secretion of enzyme into the culture medium can also be used to make a cultured cell material comprising the isolated enzyme. The expression vector typically includes the components of a cloning vector, such as, for example, an element that permits autonomous replication of the vector in the selected host organism and one or more phenotypically detectable markers for selection purposes. The expression vector normally comprises control nucleotide sequences such as a promoter, operator, ribosome binding site, translation initiation signal and optionally, a repressor gene or one or more activator genes. Additionally, the expression vector may comprise a sequence coding for an amino acid sequence capable of targeting the enzyme to a host cell organelle such as a peroxisome, or to a particular host cell compartment. Such a targeting sequence includes but is not limited to the sequence, SKL. For expression under the direction of control sequences, the nucleic acid sequence of the enzyme is operably linked to the control sequences in proper manner with respect to expression. The procedures used to ligate the DNA construct encoding an enzyme, the promoter, terminator, and other elements, respectively, and to insert them into suitable vectors containing the information necessary for replication, are well known to persons skilled in the art (see, e.g., Sambrook et al., MOLECULAR CLONING: A LABORATORY MANUAL, 2nd ed., Cold Spring Harbor, 1989, and 3rd ed., 2001).
Transformation and Culture of Host Cells An isolated cell, either comprising a DNA construct or an expression vector, is advantageously used as a host cell in the recombinant production of an enzyme according to the instant invention. The cell may be transformed with the DNA construct encoding the enzyme, conveniently by integrating the DNA construct (in one or more copies) in the host chromosome. This integration is generally considered to be an advantage, as the DNA sequence is more likely to be stably maintained in the cell. Integration of the DNA constructs into the host chromosome may be performed according to conventional methods, e.g., by homologous or heterologous recombination. Alternatively, the cell may be transformed with an expression vector as described above in connection with the different types of host cells. Examples of suitable bacterial host organisms are Gram positive bacterial species such as Bacillaceae including Bacillus subtilis, Bacillus licheniformis, Bacillus lentus, Bacillus brevis, Geobacillus (formerly Bacillus) stearothermophilus, Bacillus alkalophilus, Bacillus amyloliquefaciens, Bacillus coagulans, Bacillus lautus, Bacillus megaterium, and Bacillus thuringiensis; Streptomyces species such as Streptomyces murinus; lactic acid bacterial species including Lactococcus sp. such as Lactococcus lactis; Lactobacillus sp. including Lactobacillus reuteri; Leuconostoc sp.; Pediococcus sp.; and Streptococcus sp. Alternatively, strains of a Gram-negative bacterial species belonging to Enterobacteriaceae including E. coli, or to Pseudomonadaceae can be selected as the host organism. A suitable yeast host organism can be selected from the biotechnologically relevant yeasts species such as but not limited to yeast species such as Pichia sp., Hansenula sp., or Kluyveromyces, Yarrowinia, Schizosaccharomyces species or a species of Saccharomyces, including Saccharomyces cerevisiae or a species belonging to Schizosaccharomyces such as, for example, S. pombe species. A strain of the methylotrophic yeast species, Pichia pastoris, can be used as the host organism. Alternatively, the host organism can be a Hansenula species. Suitable host organisms among filamentous fungi include species of Aspergillus, e.g., Aspergillus niger, Aspergillus oryzae, Aspergillus tubigensis, Aspergillus awamori, or Aspergillus nidulans. Alternatively, strains of a Fusarium species, e.g., Fusarium oxysporum or of a Rhizomucor species such as Rhizomucor miehei can be used as the host organism. Other suitable strains include Thermomyces and Mucor species. In addition, Trichoderma sp. can be used as a host. A suitable procedure for transformation of Aspergillus host cells includes, for
example, that described in EP 238023. An enzyme expressed by a fungal host cell can be glycosylated, i.e., will comprise a glycosyl moiety. The glycosylation pattern can be the same or different as present in the wild-type enzyme. The type and/or degree of glycosylation may impart changes in enzymatic and/or biochemical properties. It may be advantageous to delete genes from expression hosts, where the gene deficiency can be cured by the transformed expression vector. Known methods may be used to obtain a fungal host cell having one or more inactivated genes. Gene inactivation may be accomplished by complete or partial deletion, by insertional inactivation or by any other means that renders a gene nonfunctional for its intended purpose, such that the gene is prevented from expression of a functional protein. Any gene from a Trichoderma sp. or other filamentous fungal host that has been cloned can be deleted, for example, cbh1, cbh2, egl1, and egl2 genes. Gene deletion may be accomplished by inserting a form of the desired gene to be inactivated into a plasmid by methods known in the art. Introduction of a DNA construct or vector into a host cell includes techniques such as transformation; electroporation; nuclear microinjection; transduction; transfection, e.g., lipofection mediated and DEAE-Dextrin mediated transfection; incubation with calcium phosphate DNA precipitate; high velocity bombardment with DNA-coated microprojectiles; and protoplast fusion. General transformation techniques are known in the art. See, e.g., Sambrook et al. (2001), supra. The expression of heterologous protein in Trichoderma is described, for example, in U.S. Patent No. 6,022,725. Reference is also made to Cao et al. (2000) Science 9:991-1001 for transformation of Aspergillus strains. Genetically stable transformants can be constructed with vector systems whereby the nucleic acid encoding an enzyme is stably integrated into a host cell chromosome. Transformants are then selected and purified by known techniques. The preparation of Trichoderma sp. for transformation, for example, may involve the preparation of protoplasts from fungal mycelia. See Campbell et al. (1989) Curr. Genet. 16: 53- 56. The mycelia can be obtained from germinated vegetative spores. The mycelia are treated with an enzyme that digests the cell wall, resulting in protoplasts. The protoplasts are protected by the presence of an osmotic stabilizer in the suspending medium. These stabilizers include sorbitol, mannitol, potassium chloride, magnesium sulfate, and the like. Usually, the concentration of these stabilizers varies between 0.8 M and 1.2 M, e.g., a 1.2 M solution of
sorbitol can be used in the suspension medium. Uptake of DNA into the host Trichoderma sp. strain depends upon the calcium ion concentration. Generally, between about 10-50 mM CaCl2 is used in an uptake solution. Additional suitable compounds include a buffering system, such as TE buffer (10 mM Tris, pH 7.4; 1 mM EDTA) or 10 mM MOPS, pH 6.0 and polyethylene glycol. The polyethylene glycol is believed to fuse the cell membranes, thus permitting the contents of the medium to be delivered into the cytoplasm of the Trichoderma sp. strain. This fusion frequently leaves multiple copies of the plasmid DNA integrated into the host chromosome. Usually, transformation of Trichoderma sp. uses protoplasts or cells that have been subjected to a permeability treatment, typically at a density of 105 to 107/mL, particularly 2x106/mL. A volume of 100 μL of these protoplasts or cells in an appropriate solution (e.g., 1.2 M sorbitol and 50 mM CaCl2) may be mixed with the desired DNA. Generally, a high concentration of PEG is added to the uptake solution. From 0.1 to 1 volume of 25% PEG 4000 can be added to the protoplast suspension; however, it is useful to add about 0.25 volumes to the protoplast suspension. Additives, such as dimethyl sulfoxide, heparin, spermidine, potassium chloride and the like, may also be added to the uptake solution to facilitate transformation. Similar procedures are available for other fungal host cells. See, e.g., U.S. Patent No. 6,022,725. As used herein, Protein Identification (“JGI PID”) numbers for native Trichoderma genes reference Version 2 of the Trichoderma reesei QM6a genome sequence assembly generated by the Department of Energy Joint Genome Institute (JGI). (The Genome Portal of the Department of Energy Joint Genome Institute, Grigoriev et al., Nucleic Acids Res 2012 Jan;40(Database issue):D26-32. doi: 10.1093/nar/gkr947). The JGI assembled Scaffold sequences and annotated genes have also been deposited in GeneBank (The National Center for Biotechnology) under the nucleotide accession numbers GL985056.1 through GL985132.1. Expression A method of producing an enzyme of the instant invention may comprise cultivating a host cell as described above under conditions conducive to the production of the enzyme and recovering the enzyme from the cells and/or culture medium. The medium used to cultivate the cells may be any conventional medium suitable for growing the host cell in question and obtaining expression of an enzyme. Suitable media and
media components are available from commercial suppliers or may be prepared according to published recipes (e.g., as described in catalogues of the American Type Culture Collection). An enzyme secreted from the host cells can be used in a whole broth preparation. In the present methods, the preparation of a spent whole fermentation broth of a recombinant microorganism can be achieved using any cultivation method known in the art resulting in the expression of an enzyme. Fermentation may, therefore, be understood as comprising shake flask cultivation, small- or large-scale fermentation (including continuous, batch, fed-batch, or solid- state fermentations) in laboratory or industrial fermenters performed in a suitable medium and under conditions allowing the enzyme to be expressed or isolated. The term “spent whole fermentation broth” is defined herein as unfractionated contents of fermentation material that includes culture medium, extracellular proteins (e.g., enzymes), and cellular biomass. It is understood that the term “spent whole fermentation broth” also encompasses cellular biomass that has been lysed or permeabilized using methods well known in the art. An enzyme secreted from the host cells may conveniently be recovered from the culture medium by well-known procedures, including separating the cells from the medium by centrifugation or filtration, and precipitating proteinaceous components of the medium by means of a salt such as ammonium sulfate, followed using chromatographic procedures such as ion exchange chromatography, affinity chromatography, or the like. The polynucleotide encoding an enzyme in a vector can be operably linked to a control sequence that is capable of providing for the expression of the coding sequence by the host cell, i.e., the vector is an expression vector. The control sequences may be modified, for example by the addition of further transcriptional regulatory elements to make the level of transcription directed by the control sequences more responsive to transcriptional modulators. The control sequences may comprise promoters. Host cells may be cultured under suitable conditions that allow expression of an enzyme. Expression of the enzymes may be constitutive such that they are continually produced, or inducible, requiring a stimulus to initiate expression. In the case of inducible expression, protein production can be initiated when required by, for example, addition of an inducer substance to the culture medium, for example dexamethasone or IPTG or Sophorose. Polypeptides can also be produced recombinantly in an in vitro cell-free system, such as the TNT™ (Promega) rabbit reticulocyte system.
An expression host also can be cultured in the appropriate medium for the host, under aerobic conditions. Shaking or a combination of agitation and aeration can be provided, with production occurring at the appropriate temperature for that host, e.g., from about 25°C to about 75°C (e.g., 30°C to 45°C), depending on the needs of the host and production of the desired enzyme. Culturing can occur from about 12 to about 100 hours or greater (and any hour value there between, e.g., from 24 to 72 hours). Typically, the culture broth is at a pH of about 4.0 to about 8.0, again depending on the culture conditions needed for the host relative to production of an enzyme. Methods for Enriching and Purifying enzymes Fermentation, separation, and concentration techniques are well known in the art and conventional methods can be used to prepare an enzyme polypeptide-containing solution. After fermentation, a fermentation broth is obtained, the microbial cells and various suspended solids, including residual raw fermentation materials, are removed by conventional separation techniques to obtain an enzyme solution. Filtration, centrifugation, microfiltration, rotary vacuum drum filtration, ultrafiltration, centrifugation followed by ultra-filtration, extraction, or chromatography, or the like, are generally used. It is desirable to concentrate an enzyme polypeptide-containing solution to optimize recovery. Use of unconcentrated solutions requires increased incubation time to collect the enriched or purified enzyme precipitate. The enzyme containing solution is concentrated using conventional concentration techniques until the desired enzyme level is obtained. Concentration of the enzyme containing solution may be achieved by any of the techniques discussed herein. Exemplary methods of enrichment and purification include but are not limited to rotary vacuum filtration and/or ultrafiltration. The enzyme solution is concentrated into a concentrated enzyme solution until the enzyme activity of the concentrated enzyme polypeptide-containing solution is at a desired level. Concentration may be performed using, e.g., a precipitation agent, such as a metal halide precipitation agent. Metal halide precipitation agents include but are not limited to alkali metal chlorides, alkali metal bromides and blends of two or more of these metal halides. Exemplary metal halides include sodium chloride, potassium chloride, sodium bromide, potassium bromide and blends of two or more of these metal halides. The metal halide precipitation agent, sodium
chloride, can also be used as a preservative. The metal halide precipitation agent is used in an amount effective to precipitate an enzyme. The selection of at least an effective amount and an optimum amount of metal halide effective to cause precipitation of the enzyme, as well as the conditions of the precipitation for maximum recovery including incubation time, pH, temperature, and concentration of enzyme, will be readily apparent to one of ordinary skill in the art, after routine testing. Generally, at least about 5% w/v (weight/volume) to about 25% w/v of metal halide is added to the concentrated enzyme solution, and usually at least 8% w/v. Generally, no more than about 25% w/v of metal halide is added to the concentrated enzyme solution and usually no more than about 20% w/v. The optimal concentration of the metal halide precipitation agent will depend, among others, on the nature of the specific enzyme polypeptide and on its concentration in the concentrated enzyme solution. Another alternative way to precipitate the enzyme is to use organic compounds. Exemplary organic compound precipitating agents include: 4-hydroxybenzoic acid, alkali metal salts of 4-hydroxybenzoic acid, alkyl esters of 4-hydroxybenzoic acid, and blends of two or more of these organic compounds. The addition of the organic compound precipitation agents can take place prior to, simultaneously with or subsequent to the addition of the metal halide precipitation agent, and the addition of both precipitation agents, organic compound, and metal halide, may be carried out sequentially or simultaneously. Generally, the organic precipitation agents are selected from the group consisting of alkali metal salts of 4-hydroxybenzoic acid, such as sodium or potassium salts, and linear or branched alkyl esters of 4-hydroxybenzoic acid, wherein the alkyl group contains from 1 to 12 carbon atoms, and blends of two or more of these organic compounds. The organic compound precipitation agents can be, for example, linear or branched alkyl esters of 4-hydroxybenzoic acid, wherein the alkyl group contains from 1 to 10 carbon atoms, and blends of two or more of these organic compounds. Exemplary organic compounds are linear alkyl esters of 4- hydroxybenzoic acid, wherein the alkyl group contains from 1 to 6 carbon atoms, and blends of two or more of these organic compounds. Methyl esters of 4-hydroxybenzoic acid, propyl esters of 4-hydroxybenzoic acid, butyl ester of 4-hydroxybenzoic acid, ethyl ester of 4-hydroxybenzoic acid and blends of two or more of these organic compounds can also be used. Additional organic compounds also include but are not limited to 4-hydroxybenzoic acid methyl ester (named
methyl PARABEN), 4-hydroxybenzoic acid propyl ester (named propyl PARABEN), which also are both preservative agents. For further descriptions, see, e.g., U.S. Patent No. 5,281,526. Addition of the organic compound precipitation agent provides the advantage of high flexibility of the precipitation conditions with respect to pH, temperature, enzyme concentration, precipitation agent concentration, and time of incubation. The organic compound precipitation agent is used in an amount effective to improve precipitation of the enzyme by means of the metal halide precipitation agent. The selection of at least an effective amount and an optimum amount of organic compound precipitation agent, as well as the conditions of the precipitation for maximum recovery including incubation time, pH, temperature, and concentration of enzyme, will be readily apparent to one of ordinary skill in the art, in light of the present disclosure, after routine testing. Generally, at least about 0.01% w/v of organic compound precipitation agent is added to the concentrated enzyme solution and usually at least about 0.02% w/v. Generally, no more than about 0.3% w/v of organic compound precipitation agent is added to the concentrated enzyme solution and usually no more than about 0.2% w/v. The concentrated polypeptide solution, containing the metal halide precipitation agent, and the organic compound precipitation agent, can be adjusted to a pH, which will, of necessity, depend on the enzyme to be enriched or purified. Generally, the pH is adjusted at a level near the isoelectric point of the enzyme. The pH can be adjusted at a pH in a range from about 2.5 pH units below the isoelectric point (pI) up to about 2.5 pH units above the isoelectric point. The incubation time necessary to obtain an enriched or purified enzyme precipitate depends on the nature of the specific enzyme, the concentration of enzyme, and the specific precipitation agent(s) and its (their) concentration. Generally, the time effective to precipitate the enzyme is between about 1 to about 30 hours; usually it does not exceed about 25 hours. In the presence of the organic compound precipitation agent, the time of incubation can still be reduced to less about 10 hours and in most cases even about 6 hours. Generally, the temperature during incubation is between about 4°C and about 50°C. Usually, the method is carried out at a temperature between about 10°C and about 45°C (e.g., between about 20°C and about 40°C). The optimal temperature for inducing precipitation varies according to the solution conditions and the enzyme or precipitation agent(s) used. The overall recovery of enriched or purified enzyme precipitate, and the efficiency with
which the process is conducted, is improved by agitating the solution comprising the enzyme, the added metal halide, and the added organic compound. The agitation step is done both during addition of the metal halide and the organic compound, and during the subsequent incubation period. Suitable agitation methods include mechanical stirring or shaking, vigorous aeration, or any similar technique. After the incubation period, the enriched or purified enzyme is then separated from the dissociated pigment and other impurities and collected by conventional separation techniques, such as filtration, centrifugation, microfiltration, rotary vacuum filtration, ultrafiltration, press filtration, cross membrane microfiltration, cross flow membrane microfiltration, or the like. Further enrichment or purification of the enzyme precipitate can be obtained by washing the precipitate with water. For example, the enriched or purified enzyme precipitate is washed with water containing the metal halide precipitation agent, or with water containing the metal halide and the organic compound precipitation agents. During fermentation, an enzyme polypeptide accumulates in the culture broth. For the isolation, enrichment, or purification of the desired enzyme, the culture broth is centrifuged or filtered to eliminate cells, and the resulting cell-free liquid is used for enzyme enrichment or purification. In one embodiment, the cell-free broth is subjected to salting out using ammonium sulfate at about 70% saturation; the 70% saturation-precipitation fraction is then dissolved in a buffer and applied to a column such as a Sephadex G-100 column and eluted to recover the enzyme-active fraction. For further enrichment or purification, a conventional procedure such as ion exchange chromatography may be used. Enriched or purified enzymes can be made into a final product that is either liquid (solution, slurry) or solid (granular, powder). Description of the Preferred Embodiments In accordance with an aspect of the present invention, it has been discovered that ALDC enzymes which are used in brewing for shortening the maturation phase are unstable in the presence of proline specific proteases which are used to increase colloidal stability of the beer. The loss of ALDC by degradation via proteolysis can prolong the maturation phase as opposed to shortening it. Accordingly, in an aspect of the present invention, a polypeptide having acetolactate decarboxylase activity which is resistant to protease inactivation is presented, the
polypeptide having an amino acid sequence with at least 80, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:3 wherein the polypeptide has at most 250 amino acids. Preferably, the polypeptide is at most 249, 248, 247, 246, 245, 244, 243, 242 or 241 amino acids. Preferably, the polypeptide having acetolactate decarboxylase activity which is resistant to protease inactivation has an amino acid sequence having at least 80, 90, 95, 98, 99 or 100% sequence identity to according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12. More preferably, the polypeptide has an amino acid sequence according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12. The protease to which the ALDC is resistant is preferably a proline specific protease. More preferably, the proline specific protease is from Aspergillus niger. Still more preferably, the protease has at least 80, 85, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:17. Yet more preferably, the protease has an amino acid sequence according to SEQ ID NO:17. In another aspect of the present invention, an improved brewing process is presented having the step of fermenting a wort in the presence of a polypeptide having acetolactate decarboxylase activity which is resistant to protease inactivation and a proline specific protease wherein both enzymes are present in the wort at the same time. Preferably, the polypeptide has an amino acid sequence with at least 80, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:3 wherein the polypeptide has at most 250 amino acids. More preferably, the polypeptide is at most 249, 248, 247, 246, 245, 244, 243, 242 or 241 amino acids. Yet more preferably, the polypeptide having acetolactate decarboxylase activity which is resistant to protease inactivation has an amino acid sequence having at least 80, 90, 95, 98, 99 or 100% sequence identity to according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12. Still more preferably, the polypeptide has an amino acid sequence according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12. Preferably, the polypeptide having acetolactate decarboxylase activity is added to the wort first. In other preferred embodiments, the proline specific protease is added to the wort first. In yet other preferred embodiments, the polypeptide having acetolactate decarboxylase activity and the proline specific protease are added simultaneously to the wort.
The proline specific protease is preferably from Aspergillus niger. More preferably, the proline specific protease has at least 80, 85, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:17. In another aspect of the present invention, a Bacillus host cell is presented for producing a heterologous polypeptide of interest, wherein one or more protease genes are inactivated. Preferably, the polypeptide of interest is expressed without a secretion signal peptide. In other preferred embodiments, the polypeptide of interest is expressed with a secretion signal. Preferably, the polypeptide of interest is an enzyme. More preferably, the enzyme is an oxidoreductase, transferase, hydrolase, lyase, isomerase, ligase, aminopeptidase, amylase, asparaginase, carbohydrase, carboxypeptidase, catalase, cellulase, chitinase, cutinase, cyclodextrin glycosyltransferase, deoxyribonuclease, esterase, alpha-galactosidase, beta- galactosidase, glucoamylase, alpha-glucosidase, beta-glucosidase, hyaluronic acid synthase, invertase, laccase, lipase, mannosidase, mutanase, oxidase, a pectinolytic enzyme, peroxidase, phytase, polyphenoloxidase, protease, ribonuclease, transglutaminase, or xylanase. Still more preferably, the enzyme is an ALDC enzyme. Preferably, the ALDC enzyme is a polypeptide with an amino acid sequence having at least 80, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:3 wherein the polypeptide has at most 250 amino acids. Preferably, the polypeptide is at most 249, 248, 247, 246, 245, 244, 243, 242 or 241 amino acids. Preferably, the polypeptide having acetolactate decarboxylase activity which is resistant to protease inactivation has an amino acid sequence having at least 80, 90, 95, 98, 99 or 100% sequence identity to according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12. More preferably, the polypeptide has an amino acid sequence according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12. Preferably, the ALDC enzyme is expressed with a secretion signal. Preferably, the heterologous polypeptide of interest is encoded by an exogenous polynucleotide integrated into the chromosome of the host cell in at least one copy. Preferably, the exogenous polynucleotide is a nucleic acid sequence having 80, 85, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:28. More preferably, the exogenous polynucleotide is a nucleic acid sequence according to SEQ ID NO:28.
Preferably, the at least one protease gene is inactivated by a non-sense mutation in said at least one gene, a partial deletion of said at least one gene or a full deletion of said at least one gene. Preferably, the Bacillus host cell is Bacillus subtilis or Bacillus licheniformis. More preferably, the Bacillus host cell is Bacillus subtilis. Preferably, the one or more protease genes are selected from the group consisting of a nucleic acid sequence having at least 80, 85, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:18, SEQ ID NO:19, SEQ ID NO:20, SEQ ID NO:21, SEQ ID NO:22, SEQ ID NO:23, SEQ ID NO:24, SEQ ID NO:25 and SEQ ID NO:27. More preferably, the one or more protease genes comprise nine inactivated proteases wherein the proteases have nucleic acid sequences with at least 80, 85, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:18, SEQ ID NO:19, SEQ ID NO:20, SEQ ID NO:21, SEQ ID NO:22, SEQ ID NO:23, SEQ ID NO:24, SEQ ID NO:25 and SEQ ID NO:27. Still more preferably, the nine inactivated proteases genes have nucleic acid sequences according to SEQ ID NO:18, SEQ ID NO:19, SEQ ID NO:20, SEQ ID NO:21, SEQ ID NO:22, SEQ ID NO:23, SEQ ID NO:24, SEQ ID NO:25 and SEQ ID NO:27. In another aspect of the present invention, a method for producing a polypeptide of interest is presented having the steps of: i) providing a Bacillus host cell wherein one or more protease genes are inactivated and wherein said host cell is transformed with a nucleic acid encoding a heterologous polypeptide in operable combination with a promoter; and ii) cultivating said host cell under conditions suitable to produce said heterologous polypeptide, such that said heterologous polypeptide is produced. Preferably, the method has the further step of recovering the produced polypeptide. Preferably, the polypeptide of interest is expressed with or without a secretion signal peptide. More preferably, the polypeptide of interest is expressed with a secretion signal peptide. Preferably, polypeptide of interest is an enzyme. Preferably, the enzyme is an oxidoreductase, transferase, hydrolase, lyase, isomerase, ligase, aminopeptidase, amylase, asparaginase, carbohydrase, carboxypeptidase, catalase, cellulase, chitinase, cutinase, cyclodextrin glycosyltransferase, deoxyribonuclease, esterase, alpha-galactosidase, beta- galactosidase, glucoamylase, alpha-glucosidase, beta-glucosidase, hyaluronic acid synthase, invertase, laccase, lipase, mannosidase, mutanase, oxidase, a pectinolytic enzyme, peroxidase, phytase, polyphenoloxidase, protease, ribonuclease, transglutaminase, or xylanase. More
preferably, the enzyme is an ALDC enzyme. Preferably, the ALDC enzyme is a polypeptide with an amino acid sequence having at least 80, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:3 wherein the polypeptide has at most 250 amino acids. Preferably, the polypeptide is at most 249, 248, 247, 246, 245, 244, 243, 242 or 241 amino acids. Preferably, the polypeptide having acetolactate decarboxylase activity has an amino acid sequence having at least 80, 90, 95, 98, 99 or 100% sequence identity to according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12. More preferably, the polypeptide has an amino acid sequence according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12. Preferably, the ALDC enzyme is expressed with a secretion signal. Preferably, the heterologous polypeptide of interest is encoded by an exogenous polynucleotide integrated into the chromosome of the host cell in at least one copy. Preferably, the exogenous polynucleotide is a nucleic acid sequence having 80, 85, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:28. More preferably, the exogenous polynucleotide is a nucleic acid sequence according to SEQ ID NO:28. Preferably, the at least one protease gene is inactivated by a non-sense mutation in said at least one gene, a partial deletion of said at least one gene or a full deletion of said at least one gene. Preferably, the Bacillus host cell is Bacillus subtilis or Bacillus licheniformis. More preferably, the Bacillus host cell is Bacillus subtilis. Preferably, the one or more protease genes are selected from the group consisting of a nucleic acid sequence having at least 80, 85, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:18, SEQ ID NO:19, SEQ ID NO:20, SEQ ID NO:21, SEQ ID NO:22, SEQ ID NO:23, SEQ ID NO:24, SEQ ID NO:25 and SEQ ID NO:27. More preferably, the one or more protease genes comprise nine inactivated proteases wherein the proteases have nucleic acid sequences with at least 80, 85, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:18, SEQ ID NO:19, SEQ ID NO:20, SEQ ID NO:21, SEQ ID NO:22, SEQ ID NO:23, SEQ ID NO:24, SEQ ID NO:25 and SEQ ID NO:27. Still more preferably, the nine inactivated proteases genes have nucleic acid sequences according to SEQ ID NO:18, SEQ ID NO:19, SEQ ID NO:20, SEQ ID NO:21, SEQ ID NO:22, SEQ ID NO:23, SEQ ID NO:24, SEQ ID NO:25 and SEQ ID NO:27.
In another aspect of the present invention, a stable liquid formulation comprising a polypeptide having acetolactate decarboxylase activity having an amino acid sequence with at least 80, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:3 wherein said polypeptide has at most 250 amino acids and a protease. Preferably, the polypeptide is at most 249, 248, 247, 246, 245, 244, 243, 242 or 241 amino acids. Preferably, the polypeptide having acetolactate decarboxylase activity has an amino acid sequence having at least 80, 90, 95, 98, 99 or 100% sequence identity to according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12. More preferably, the polypeptide has an amino acid sequence according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12. The protease is preferably a proline specific protease. More preferably, the proline specific protease is from Aspergillus niger. More preferably, the protease has at least 80, 85, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:17. Still more preferably, the protease has an amino acid sequence according to SEQ ID NO:17. Preferably, the ALDC polypeptide maintains at least 99, 98, 95, 90, 85, 80, 75 or 70% activity when the stable liquid formulation is stored at 10oC for 30, 60, 90, 120, 150 for 180 days. In other preferred embodiments, the ALDC polypeptide maintains at least 99, 98, 95, 90, 85, 80, 75 or 70% activity when the stable liquid formulation is stored at 30oC for 8, 16, 24, 32, 40 or 48 hours. In another aspect of the present invention, a proline specific protease formulation is presented which is substantially depleted of other protease activities comprising the protease wherein when said protease is combined with a polypeptide having acetolactate decarboxylase activity, the polypeptide is stable over time. Preferably, the polypeptide having acetolactate decarboxylase activity has an amino acid sequence having at least 80, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:3 wherein said polypeptide has at most 250 amino acids and a protease. Preferably, the polypeptide is at most 249, 248, 247, 246, 245, 244, 243, 242 or 241 amino acids. Preferably, the polypeptide having acetolactate decarboxylase activity has an amino acid sequence having at least 80, 90, 95, 98, 99 or 100% sequence identity to according to SEQ ID
NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12. More preferably, the polypeptide has an amino acid sequence according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12. The protease is preferably a proline specific protease. More preferably, the proline specific protease is from Aspergillus niger. More preferably, the protease has at least 80, 85, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:17. Still more preferably, the protease has an amino acid sequence according to SEQ ID NO:17. Preferably, the ALDC polypeptide maintains at least 99, 98, 95, 90, 85, 80, 75 or 70% activity when the stable liquid formulation is stored at 10oC for 30, 60, 90, 120, 150 for 180 days. In other preferred embodiments, the ALDC polypeptide maintains at least 99, 98, 95, 90, 85, 80, 75 or 70% activity when the stable liquid formulation is stored at 30oC for 8, 16, 24, 32, 40 or 48 hours. The present disclosure is described in further detail in the following examples, which are not in any way intended to limit the scope of the disclosure as claimed. The attached figures are meant to be considered as integral parts of the specification and description of the disclosure. The following examples are offered to illustrate, but not to limit the claimed disclosure. EXAMPLES EXAMPLE 1 As an example of a proline-specific endoprotease (PEP), the proline-specific endoprotease sold as Brewers Clarex® (5 PPU / g product) by DSM from Aspergillus niger (AnPro), was used. The defined activity of the proline specific endo-protease (PEP) is based on hydrolysis of the synthetic peptide Z-Gly-Pro-pNA at 37°C in a citrate/disodium phosphate buffer pH 4.6. The reaction product is monitored spectro-photometrically at 405 nm and one unit (1PPU) is defined as the quantity of enzyme that liberates 1 mmol of p-nitroanilide per minute under these test conditions. As an example of an acetolactate decarboxylase, aldB, from Brevibacillus brevis (which may be referred to as Bacillus brevis) produced in Bacillus licheniformis and sold as Maturex® Pro (2500 ADU-L/ g product) by Novozymes (Bagsværd, Denmark) was used. This aldB, expressed in Bacillus licheniformis, is referred to herein as aldB_Bl.
EXAMPLE 2 Cloning of Aspergillus niger ATCC 1015 protease AniPro_2 (CRC02753-WT) Aspergillus niger ATCC 1015 was selected as a potential source of enzymes which may be useful in various industrial applications. One of the genes identified in Aspergillus niger ATCC 1015, named AniPro_2 (CRC02753-WT), encodes a protein with homology to a protease as determined from a BLAST search (Altschul et al., J Mol Biol, 215: 403–410, 1990). The precursor protein encoded by the AniPro_2 gene is shown in SEQ ID NO:16 (JGI Reference Sequence: Aspni5_52703). At the N-terminus, the protease protein has a signal peptide with a length of 21 amino acids as predicted by SignalP version 4.0 (Nordahl Petersen et al. (2011) Nature Methods, 8:785-786) and derived from a Hypocrea jecorina aspartate protease (uniport id. G0R8T0). The presence of a signal sequence ensures that AniPro_2 is a secreted enzyme. The predicted mature form of AniPro_2 is shown in SEQ ID NO:17. EXAMPLE 3 Expression, fermentation and purification of AniPro_2 DNA sequences encoding AniPro_2, were chemically synthesized and inserted into a Trichoderma reesei expression vector pGXT (the same as the pTTTpyr2 vector as described in published PCT Application WO2015/017256, incorporated by reference herein) by Generay (Shanghai, China). The resulting plasmid was labeled as pGXT-AniPro2. The expression plasmid was then transformed into a suitable Trichoderma reesei strain (described in published PCT application WO 05/001036) using protoplast transformation (Te’o et al. (2002) J. Microbiol. Methods 51:393-99). Transformants were selected on a medium containing acetamide as a sole source of nitrogen. After 5 days of growth on acetamide plates, transformants were collected and subjected to fermentation via DASGIP (Eppendorf, Juelich, Germany). To initiate the fermentation of AniPro_2, seed culture was grown in 1 L shake flask, each contained 100 mL defined medium (pH5.5 before sterilization) having 50 g/L glucose monohydrate, 6 g/L glycine, 5 g/L (NH4)2SO4, 4.5 g/L KH2PO4, 1 g/L CaCl2·2H2O, 1 g/L MgSO4·7H2O, 2 g/L Mazu 6000K and 2.5 mL 400×T. reesei Trace Metals (400× Trace Metals stock (~pH 1) contains 175 g/L C6H8O7·H2O, 200 g/L FeSO4·7H2O, 16 g/L ZnSO4·7H2O, 3.2 g/L CuSO4·5H2O, 1.4 g/L MnSO4·H2O, and 0.8 g/L H3BO3). The seed culture was shaken for 48 hours
at 250 rpm and 30 °C. Following completion of this incubation, 200 mL of seed culture was transferred to the 2-L bioreactor (DASGIP). The medium for fermentation in the 2-L bioreactor (DASGIP) contains 60 g/L dextrose, 6 g/L glycine, 1 g/L CaCl2·2H2O, 4.5 g/L KH2PO4, 4 g/L (NH4)2SO4, 1 g/L MgSO4·7H2O, 1.2 g/L Mazu 6000K and 2.5 ml 400× T. reesei Trace Metals. An induction solution containing 250 g glucose/sophorose per kg was prepared and sterilized. Following inoculation, batch fermentation was initiated with working volume of 1 L and was controlled at pH 3.5 and 34 °C. Dissolved oxygen level was controlled above 35% by adjusting airflow rate, oxygen supply, and agitation during whole fermentation process. At the elapsed fermentation time of 22 hours, the glucose in broth was depleted, and the feed of 250 g (glucose/sophorose)/kg solution was started at this time. Stepwise feed rates of 4 mL feed/hr and 6 mL feed/hr were applied in the time intervals of 22-46 hours and 46-72 hours, respectively. Accompanying the start of fed-batch phase, pH was linearly changed to 4.0, and temperature was adjusted to 28 oC. Fermentation was completed after 72 hours run, and broth was harvested by centrifugation, filtered, and subsequently concentrated. To purify AniPro_2, the crude from 1 L Dasgip fermenter was concentrated and added ammonium sulfate to the final concentration of 1 M. The solution was loaded onto a HiPrepTM Phenyl FF 16/10 column pre-equilibrated with 20 mM NaAc (pH5.0) supplemented with additional 1 M ammonium sulfate (Buffer A). The target protein was eluted from the column with 0.75 M ammonium sulfate. The corresponding fractions were pooled, concentrated and exchanged buffer into 20 mM NaPi (pH7.0) (Buffer B), using a VivaFlow 200 ultra-filtration device (Sartorius Stedim). The resulting solution was applied to a HiLoadTM Q FF 16/10 column pre-equilibrated with Buffer B. The target protein was eluted from the column with 0.3 M NaCl. The fractions containing active protein were pooled, concentrated and exchanged buffer into 20 mM NaAc (pH5.0), 150 mM NaCl via the 10K Amicon Ultra devices, then stored in 40% glycerol at -20 oC until usage. EXAMPLE 4 HETEROLOGOUS EXPRESSION OF ACETOLACTATE DECARBOXYLASE, ALDB The Brevibacillus brevis (which may be referred to as Bacillus brevis) acetolactate decarboxylases (ALDC) aldB gene was previously identified (Diderichsen et al., J Bacteriol.
(1990) 172(8): 4315), with the sequence set forth as UNIPROT Accession No. P23616.1. The sequence of this gene, aldB, is depicted in SEQ ID NO:1. Nucleotides 1 to 72 encode the signal peptide. The aldB gene and corresponding encoded proenzyme are also referred to as the wildtype (WT). The proenzyme encoded by the aldB gene is depicted in SEQ ID NO: 2. At the N-terminus, the protein has a signal peptide with a length of 24 amino acids as predicted by SignalP-NN (Emanuelsson et al., Nature Protocols (2007) 2: 953-971). The presence of a signal peptide indicates that this acetolactate decarboxylase, aldB is a secreted enzyme. The sequence of the predicted, fully processed mature chain (aldB, 261 amino acids) is depicted in SEQ ID NO: 3. The aldB gene that encodes an acetolactate decarboxylases enzyme (ALDC) was produced in B. subtilis using the synthetic gene inserted into the pCB_alr vector, see Figure 1. The aldB gene containing was transcribed using aprE promoter followed by B. subtilis aprE signal sequence. For expression the alrA(CB)RIHI-Bbr vector was integrated into a protease deficient B. subtilis strain. A map of the pCB_alr vector containing the aldB gene (alrA(CB)RIHI-Bbr) is shown in Figure 2. To produce aldB, a B. subtilis strain transformant containing the alrA(CB)RIHI-Bbr cassette (strain BRA8014 described below) was cultured in 15-mL Falcon tubes for 16 hours in TSB (broth) with 10 ppm neomycin, and 300 µL of this pre-culture was added to a 500-mL flask filled with 30 mL of cultivation media (described below) supplemented with 10 ppm neomycin. The flasks were incubated for 24, 48 and 72 hours at 33°C with constant rotational mixing at 180 rpm. Cultures were harvested by centrifugation at 14500 rpm for 20 minutes in conical tubes. The culture supernatants were used for protein determination and assays. The cultivation media was an enriched semi-defined media based on MOPs buffer, with urea as major nitrogen source, glucose as the main carbon source, 50µM ZnSO4 to ensure high enzyme activity and supplemented with 1% soytone for robust cell growth. The aldB expressed in Bacillus subtilis is here within termed, aldB_Bs. The nucleotide mature sequence of the aldB gene in plasmid alrA(CB)RIHI-Bbr is depicted in SEQ ID NO:4. The amino acid sequence of the aldB precursor protein expressed from plasmid alrA(CB)RIHI-Bbr is depicted in SEQ ID NO:5. EXAMPLE 5 Protein Determination Methods Protein Determination by Stain Free Imager Criterion
Protein was quantified by SDS-PAGE gel and densitometry using Gel Doc™ EZ imaging system. Reagents used in the assay: Concentrated (2x) Laemmli Sample Buffer (Bio-Rad, Catalogue #161-0737); 26-well XT 4-12% Bis-Tris Gel ( Bio-Rad, Catalogue #345-0125); protein markers “Precision Plus Protein Standards” (Bio-Rad, Catalogue #161- 0363); protein standard BSA (Thermo Scientific, Catalogue #23208) and SimplyBlue Safestain (Invitrogen, Catalogue #LC 6060. The assay was carried out as follow: In a 96well-PCR plate 50µL diluted enzyme sample were mixed with 50 µL sample buffer containing 2.7 mg DTT. The plate was sealed by Microseal ‘B’ Film from Bio-Rad and was placed into PCR machine to be heated to 70°C for 10 minutes. After that the chamber was filled by running buffer, gel cassette was set. Then 10 µL of each sample and standard (0.125-1.00 mg/mL BSA) was loaded on the gel and 5 µL of the markers were loaded. After that the electrophoresis was run at 200 V for 45 minutes. Following electrophoresis, the gel was rinsed 3 times 5 min in water, then stained in Safe-stain overnight and finally destained in water. Then the gel was transferred to Imager. Image Lab software was used for calculation of intensity of each band. A calibration curve was made using BSA (Thermo Scientific, Catalogue #23208) and the amount of the target protein was determined by the band intensity and calibration curve. The protein quantification method was employed to prepare enzyme samples of used in subsequent Examples. The protease protein concentration was determined in: A concentrated sample AniPro_2 sample was determined to 55 mg/ml and a sample of AnPro was determined to 39mg/ml. EXAMPLE 6 ALDC enzyme sequence identification by MS including N and C-terminal amino acid determination In preparation for sequence confirmation, an SDS-PAGE gel of isolated aldB_Bs truncation variants were analyzed by LC-MS/MS as described subsequently. In preparation for sequence confirmation, including N- and C-terminal determination, a protein band from an SDS-PAGE gel of aldB ferment sample was subjected to a series of chemical treatments. Between the individual steps the gel pieces were washed and shrunk using Milli-Q water, 50w/w% ethanol and absolute ethanol respectively. The protein was reduced/alkylated by DTT/Iodoacetamide treatment. A guanidination step was performed to convert lysines to homoarginines to protect lysine side chains from acetylation. The acetylation reaction using Sulfo-NHS-Acetate (Sulfosuccinimidyl Acetate) only modifies the protein N-terminal residue. The gel pieces were swelled with a buffer
containing 40v/v% 18O water:60v/v% 16O water and the proteolytic enzymes used for protein digestion (Trypsin and α-Chymotrypsin). The resulting peptides will contain mixtures of 18O and 16O, except for the Carboxyl terminus which will retain the native 16O, as will be apparent from the isotopic pattern of the peptides. The peptide, originating from the protein N-terminus, will appear as the only acetylated peptide. After digestion the peptides were extracted from the gel pieces using 5w/w% formic acid and acetonitrile, then lyophilized and re-dissolved in 0.1w/w% TFA. The digestion products were separated (C18 column) and analyzed using a Proxeon nano- LC system followed by LTQ Orbitrap (Thermo Fisher) high resolution mass spectrometer and the amino acid sequence was deduced from the MS/MS fragment spectrum of the peptides, and the isotopic pattern of the peptides (using Xcalibur 2.0 SR2 software). Based on this analysis, the N-terminus of the isolated full-length protein was confirmed to begin with A[25] (according to SEQ ID NO. 2) and the C-terminus of the isolated full-length protein was confirmed to end at position K[285] (according to SEQ NO. 2) by the acetylation and 18O-label methods as described above (see table 2). The N-terminus position A[25] at the mature aldB_Bs correspond with the predicted signal peptide cleavage determined by the Signal P 3.0 program (http://www.cbs.dtu.dk/services/SignalP/), set to SignalP-NN system, (Emanuelsson et al., (2007), Nature Protocols, 2: 953-971) of the gene transcript. Different N- and C-terminal truncation variants were further identified and their respective N- and C-terminus position according to SEQ ID NO. 2 are given in table 2. [001] Table 2 Identified N- and C-terminus positions of aldB_Bs variants, position according to according to SEQ ID NO. 2. Position N- Position C- SEQ ID NO: terminus terminus
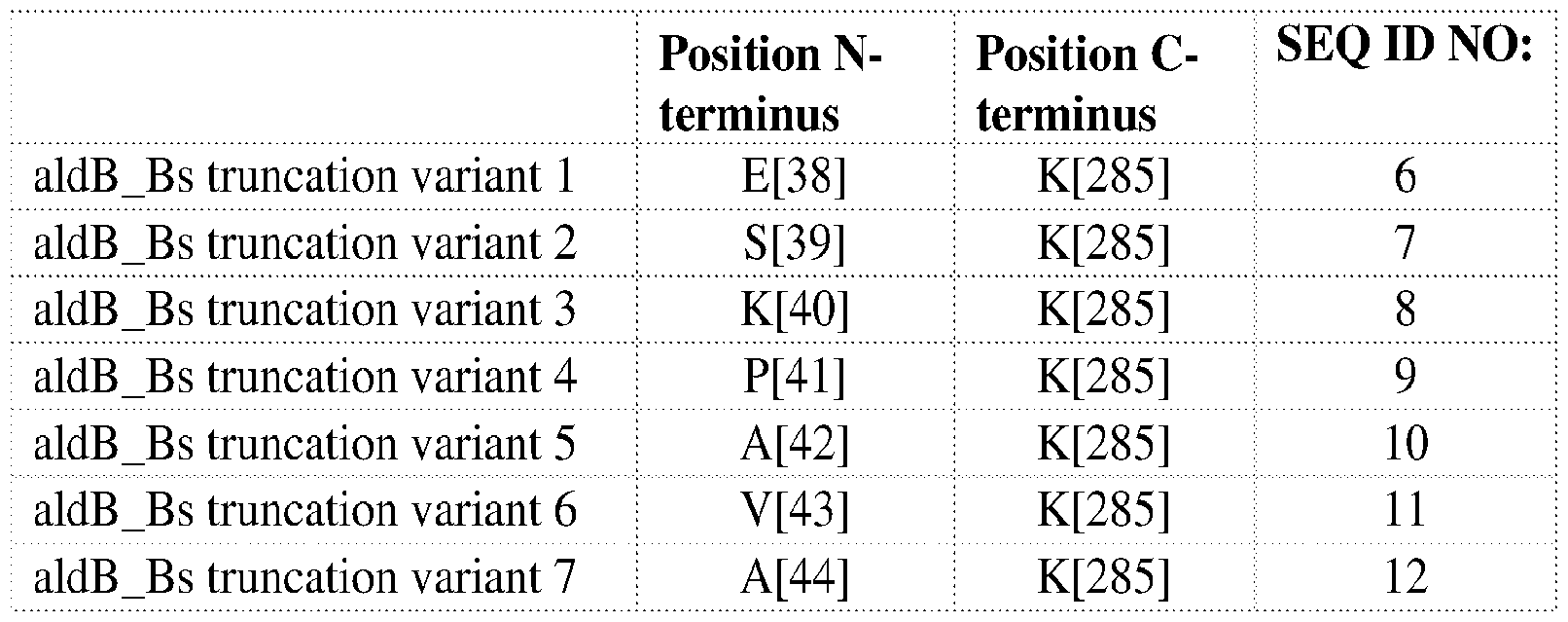 Thus, the longest mature variant of aldB_Bs was found to contain 248 amino-acids and shortest 242 amino acids, all truncations were at N-termini. In addition, the mature polypeptide sequence of aldB_Bl (aldB produced in Bacillus licheniformis) was analyzed by MS. In preparation for sequence confirmation, an SDS-PAGE gel of isolated aldB_Bl truncation variants were analyzed by LC-MS/MS as described subsequently. In preparation for sequence confirmation, including N- and C-terminal determination, a protein band from an SDS-PAGE gel of dB_Bl sample was undertaken. N- and C-terminal truncation variants of aldB_Bl were further identified and their respective N- and C- terminus position according to SEQ ID NO. 2 are given in table 3. [002] Table 3 Identified N- and C-terminus positions of aldB_Bl variants, position according to according to SEQ ID NO. 2. Position N- Position C- SEQ ID NO:
Thus, the longest mature variant of aldB_Bs was found to contain 248 amino-acids and shortest 242 amino acids, all truncations were at N-termini. In addition, the mature polypeptide sequence of aldB_Bl (aldB produced in Bacillus licheniformis) was analyzed by MS. In preparation for sequence confirmation, an SDS-PAGE gel of isolated aldB_Bl truncation variants were analyzed by LC-MS/MS as described subsequently. In preparation for sequence confirmation, including N- and C-terminal determination, a protein band from an SDS-PAGE gel of dB_Bl sample was undertaken. N- and C-terminal truncation variants of aldB_Bl were further identified and their respective N- and C- terminus position according to SEQ ID NO. 2 are given in table 3. [002] Table 3 Identified N- and C-terminus positions of aldB_Bl variants, position according to according to SEQ ID NO. 2. Position N- Position C- SEQ ID NO:
 Thus, the longest mature variant of aldB_Bl was found to contain 258 amino-acids and shortest 253 amino acids, all truncations were at N-termini. Production of aldB in Bacillus subtilis generated shorter aldB enzyme variants as compared to Production of aldB in Bacillus licheniformis. EXAMPLE 7 α-Acetolactate Decarboxylase Activity Assay Method Spectrophotometric assay of α-acetolactate decarboxylase α-Acetolactate decarboxylase (ALDC) catalyzes the decarboxylation of α-acetolactate to acetoin. The reaction product acetoin can be quantified colourimetrically. Acetoin mixed with α- naphtol and creatine forms a characteristic red color absorbing at OD522 nm. ALDC activity was calculated based on OD522 nm and an acetoin calibration curve. The assay was carried out as follows: 20 mM acetolactate substrate was prepared by mixing 100 µL ethyl-2-acetoxy-2- methylacetoacetate (Sigma, Catalogue# 220396) with 3.6 ml 0.5 M NaOH at 10°C for 10 min.
20 ml 50 mM MES pH 6.0 was added, pH was adjusted to pH 6.0 and volume adjusted to 25 ml with 50 mM MES pH 6.0. 80 µL 20 mM acetolactate substrate was mixed with 20 µL enzyme sample diluted in 50 mM MES, pH 6.0, 0.6 M NaCl, 0.05% BRIJ 35 and 0.01% BSA. The substrate/enzyme mixture was incubated at 30°C for 10 min. Then 16 µL substrate/enzyme mixture was transferred to 200 µL 1 M NaOH, 1.0% α-naphtol (Sigma, Catalogue# 33420) and 0.1% creatine (Sigma, Catalogue# C3630). The substrate/enzyme/color reagent mixture was incubated at 30°C for 20 min and then OD522 nm was read. One unit of ALDC activity is defined as the amount of enzyme which produces 1 µmole acetoin per minute under the conditions of the assay. ALDC activity was determined as described in concentrated fermentation samples and is shown in table 4. The activity showed to be highest for aldB_Bs (10396 ADU/g), followed by aldB_Bl (4020 ADU/g), whereas no ALDC activity (0 ADU/g) was found in samples of proline- specific endoproteases AniPro_2 or AnPro, respectively. Table 4 ALDC (α-Acetolactate Decarboxylase) activity (ADU) of aldB_Bs, aldB_Bl, AniPro_2 and AnPro samples. 4 9 0 0
Thus, the longest mature variant of aldB_Bl was found to contain 258 amino-acids and shortest 253 amino acids, all truncations were at N-termini. Production of aldB in Bacillus subtilis generated shorter aldB enzyme variants as compared to Production of aldB in Bacillus licheniformis. EXAMPLE 7 α-Acetolactate Decarboxylase Activity Assay Method Spectrophotometric assay of α-acetolactate decarboxylase α-Acetolactate decarboxylase (ALDC) catalyzes the decarboxylation of α-acetolactate to acetoin. The reaction product acetoin can be quantified colourimetrically. Acetoin mixed with α- naphtol and creatine forms a characteristic red color absorbing at OD522 nm. ALDC activity was calculated based on OD522 nm and an acetoin calibration curve. The assay was carried out as follows: 20 mM acetolactate substrate was prepared by mixing 100 µL ethyl-2-acetoxy-2- methylacetoacetate (Sigma, Catalogue# 220396) with 3.6 ml 0.5 M NaOH at 10°C for 10 min.
20 ml 50 mM MES pH 6.0 was added, pH was adjusted to pH 6.0 and volume adjusted to 25 ml with 50 mM MES pH 6.0. 80 µL 20 mM acetolactate substrate was mixed with 20 µL enzyme sample diluted in 50 mM MES, pH 6.0, 0.6 M NaCl, 0.05% BRIJ 35 and 0.01% BSA. The substrate/enzyme mixture was incubated at 30°C for 10 min. Then 16 µL substrate/enzyme mixture was transferred to 200 µL 1 M NaOH, 1.0% α-naphtol (Sigma, Catalogue# 33420) and 0.1% creatine (Sigma, Catalogue# C3630). The substrate/enzyme/color reagent mixture was incubated at 30°C for 20 min and then OD522 nm was read. One unit of ALDC activity is defined as the amount of enzyme which produces 1 µmole acetoin per minute under the conditions of the assay. ALDC activity was determined as described in concentrated fermentation samples and is shown in table 4. The activity showed to be highest for aldB_Bs (10396 ADU/g), followed by aldB_Bl (4020 ADU/g), whereas no ALDC activity (0 ADU/g) was found in samples of proline- specific endoproteases AniPro_2 or AnPro, respectively. Table 4 ALDC (α-Acetolactate Decarboxylase) activity (ADU) of aldB_Bs, aldB_Bl, AniPro_2 and AnPro samples. 4 9 0 0
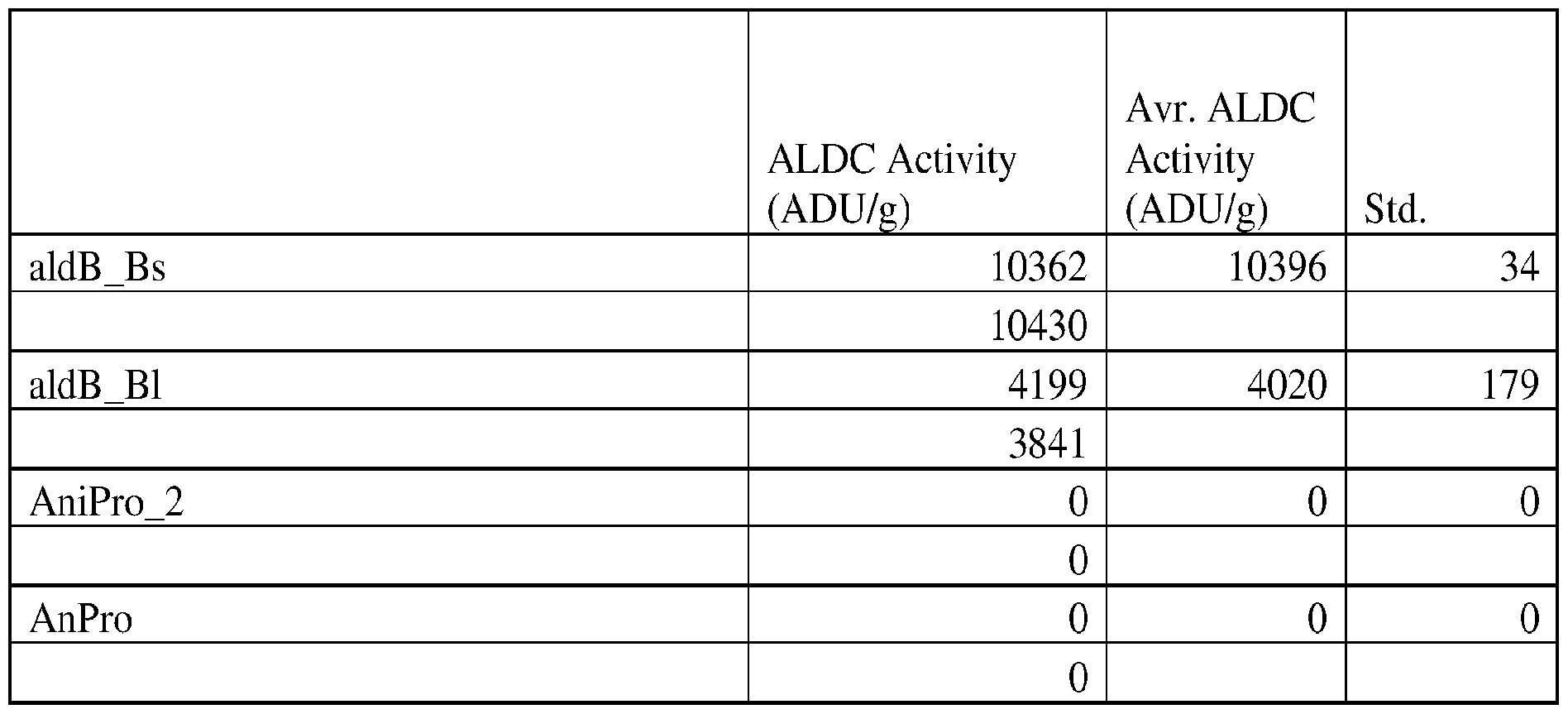 Proline specific Endo-Protease Activity Assay Method Enzyme activity test
Post-proline, protease cleavage activity was measured using Z-Gly-Pro-pNA, (Z-, benzyloxycarbonyl-; -pNA,-p-nitroanilide) (Bachem, Bubendorf, Germany) (PEPU activity). All pNA substrate were dissolved in 100% DMSO at 100mM and further diluted to 0.4mM in the reaction buffer McIlvaine, pH 4.6. Activity measurements were performed at 30°C for 10 minutes if not stated otherwise. The released pNA was measured by spectrophotometry absorbance at 405nm using a Spectramax plate reader (Molecular Device, UK). PEPU activity measurements of AniPro_2, AnPro, aldB_Bs and aldBL were also performed on Z-Gly-Pro-pNA under condition described above and the enzymatic activity of the enzyme solutions obtained is shown in table 5, with one enzyme unit being defined as the activity that liberates 1 mol of pNA from Z-Gly-Pro-pNA in 1 min under the reaction conditions. It is clear that, AniPro_2 and AnPro contained high post-proline protease cleavage activity, with 6.99 and 5.16 PEPU/g respectively, whereas aldB_Bs and aldBL contained very minor post-proline protease cleavage activities 0.11 and 0.01 PEPU/g, respectively. Table 5 Post-proline cleavage activity measured using Z-Gly-Pro-pNA (PEPU activity) of aldB, aldBL, AniPro_2 and AnPro samples. Avr. PEPU 5 4 3 2
Proline specific Endo-Protease Activity Assay Method Enzyme activity test
Post-proline, protease cleavage activity was measured using Z-Gly-Pro-pNA, (Z-, benzyloxycarbonyl-; -pNA,-p-nitroanilide) (Bachem, Bubendorf, Germany) (PEPU activity). All pNA substrate were dissolved in 100% DMSO at 100mM and further diluted to 0.4mM in the reaction buffer McIlvaine, pH 4.6. Activity measurements were performed at 30°C for 10 minutes if not stated otherwise. The released pNA was measured by spectrophotometry absorbance at 405nm using a Spectramax plate reader (Molecular Device, UK). PEPU activity measurements of AniPro_2, AnPro, aldB_Bs and aldBL were also performed on Z-Gly-Pro-pNA under condition described above and the enzymatic activity of the enzyme solutions obtained is shown in table 5, with one enzyme unit being defined as the activity that liberates 1 mol of pNA from Z-Gly-Pro-pNA in 1 min under the reaction conditions. It is clear that, AniPro_2 and AnPro contained high post-proline protease cleavage activity, with 6.99 and 5.16 PEPU/g respectively, whereas aldB_Bs and aldBL contained very minor post-proline protease cleavage activities 0.11 and 0.01 PEPU/g, respectively. Table 5 Post-proline cleavage activity measured using Z-Gly-Pro-pNA (PEPU activity) of aldB, aldBL, AniPro_2 and AnPro samples. Avr. PEPU 5 4 3 2
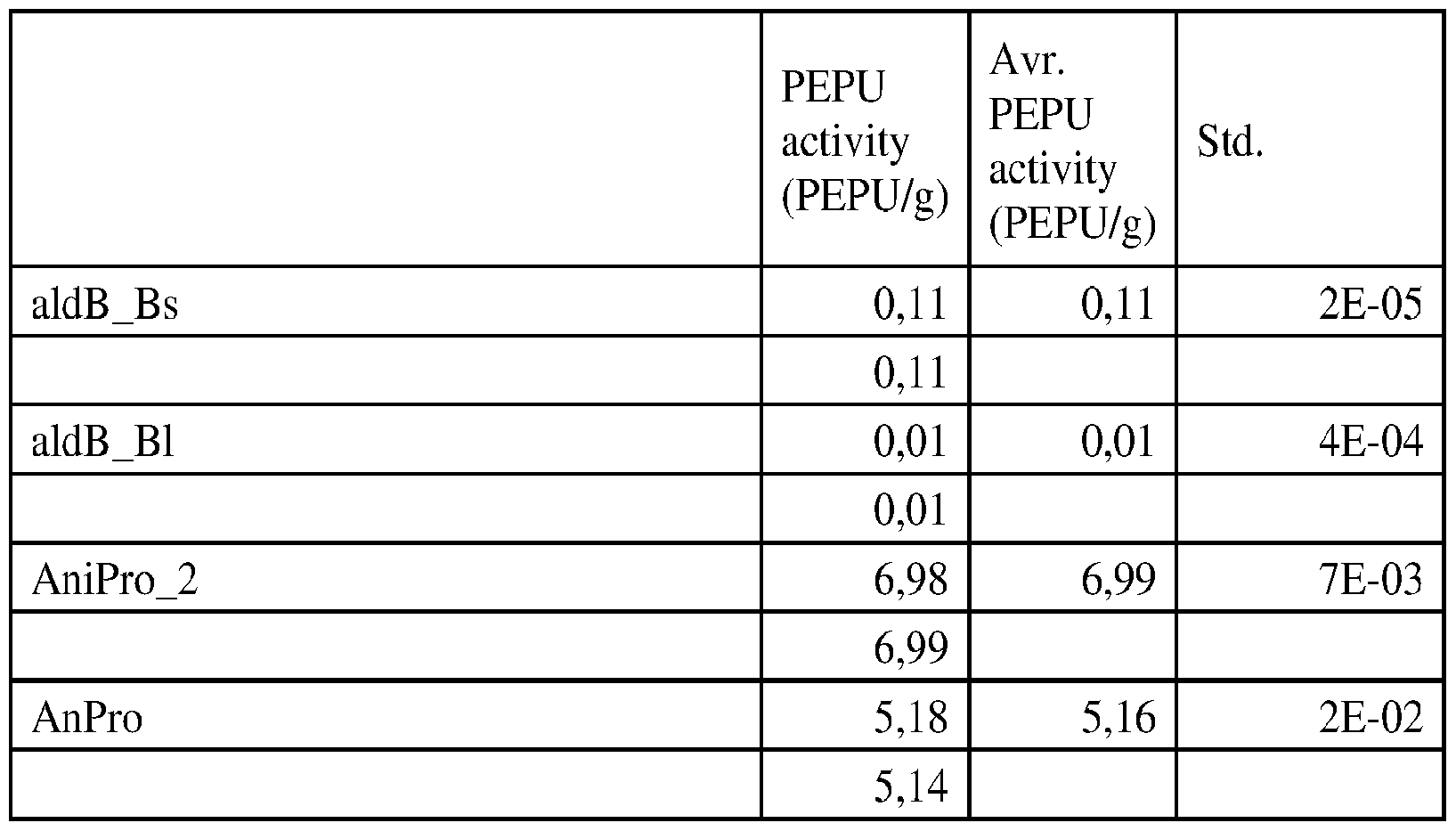 EXAMPLE 9 Enzyme activity stability of samples with combinations of ALDC and PEP enzymes
Acetolactate decarboxylase (ALDC) can also be used as an enzyme to prevent the formation of diacetyl. α-acetolactate can be converted into acetoin by adding an ALDC enzyme during fermentation. In addition, it is known that more selective proteases such as proline specific endopeptidases have been employed to reduce beer haze. In a recent approach, a proline-specific endoprotease is used as an alternative to PVPP or silica-gel treatment to prevent chill haze formation. During beer fermentation, the enzymes (proline-specific endoprotease) are added to selectively hydrolyze the haze-active, proline-rich proteins and may hereby prevent the precipitation of protein-polyphenol complexes. Both ALDC and PEP enzymes are added to the beer fermentation process to increase the flexibility in the brewery and to save capital costs. Over the years, a lot of work has been done to minimize the fermentation, maturation and stabilization phases of the brewing process (Narziss, L. 1990 Ferment, 3, 54-62). In order to dose both enzyme product simultaneous or in conjunction or as blended mixture we studied stability of ALDC mixed with proline-specific endoprotease. Thus, the following combinations made as 50:50 % (w/w) blend: aldB_Bs + AniPro_2, aldB_Bl + AniPro_2, aldB_Bs + AnPro and aldB_Bl + AnPro. These were mixed thoroughly and analyzed together with the individual ALDC samples (aldB_Bs and aldB_Bl) and proline-specific endoprotease samples (AniPro_2 and AnPro) respectively. 10g aliquots of each sample were stored in closed Wheaton vials at either 5°C or 30°C for up to 48 hours. α-Acetolactate Decarboxylase activity (ADU/g) were determined throughout stability period after 0, 1, 3, 24 and 48 hours and results are shown in table 6 and 7. The ALDC samples aldB_Bs and aldB_Bl showed no loss of α-Acetolactate Decarboxylase activity at both 5°C and 30°C for up to 48 hours. Variation in Acetolactate Decarboxylase activity is estimated up to 10%. No ALDC activity (0 ADU/g) was found in samples of proline-specific endoproteases: AniPro_2 or AnPro, throughout stability test period respectively. Moreover, aldB_Bs blended with either AniPro_2 or AnPro showed no loss of α- Acetolactate Decarboxylase activity at both 5°C and 30°C for up to 48 hours. However, aldB_Bl blended with AniPro_2 showed 82% and 48% residual activity at 5°C and 30°C after 48 hours. aldB_Bl blended with AnPro showed less stability having only 68% and 6% residual activity at 5°C and 30°C after 48 hours. Thus, aldB_Bl is clearly sensitive towards blending with proline- specific endoproteases as compared with aldB_Bs.
Table 6 ALDC (α-Acetolactate Decarboxylase) activity (ADU/g) of aldB_Bs, aldB_Bl, AniPro_2, AnPro and combinations samples mixed 50:50 % (w/w) (aldB_Bs + AniPro_2, aldB_Bl + AniPro_2, aldB_Bs + AnPro and aldB_Bl + AnPro), stored at 5°C. Activity of all samples were determined in duplicates up to 48hrs, having residuals calculated against activity at 0 hrs. ADU/
EXAMPLE 9 Enzyme activity stability of samples with combinations of ALDC and PEP enzymes
Acetolactate decarboxylase (ALDC) can also be used as an enzyme to prevent the formation of diacetyl. α-acetolactate can be converted into acetoin by adding an ALDC enzyme during fermentation. In addition, it is known that more selective proteases such as proline specific endopeptidases have been employed to reduce beer haze. In a recent approach, a proline-specific endoprotease is used as an alternative to PVPP or silica-gel treatment to prevent chill haze formation. During beer fermentation, the enzymes (proline-specific endoprotease) are added to selectively hydrolyze the haze-active, proline-rich proteins and may hereby prevent the precipitation of protein-polyphenol complexes. Both ALDC and PEP enzymes are added to the beer fermentation process to increase the flexibility in the brewery and to save capital costs. Over the years, a lot of work has been done to minimize the fermentation, maturation and stabilization phases of the brewing process (Narziss, L. 1990 Ferment, 3, 54-62). In order to dose both enzyme product simultaneous or in conjunction or as blended mixture we studied stability of ALDC mixed with proline-specific endoprotease. Thus, the following combinations made as 50:50 % (w/w) blend: aldB_Bs + AniPro_2, aldB_Bl + AniPro_2, aldB_Bs + AnPro and aldB_Bl + AnPro. These were mixed thoroughly and analyzed together with the individual ALDC samples (aldB_Bs and aldB_Bl) and proline-specific endoprotease samples (AniPro_2 and AnPro) respectively. 10g aliquots of each sample were stored in closed Wheaton vials at either 5°C or 30°C for up to 48 hours. α-Acetolactate Decarboxylase activity (ADU/g) were determined throughout stability period after 0, 1, 3, 24 and 48 hours and results are shown in table 6 and 7. The ALDC samples aldB_Bs and aldB_Bl showed no loss of α-Acetolactate Decarboxylase activity at both 5°C and 30°C for up to 48 hours. Variation in Acetolactate Decarboxylase activity is estimated up to 10%. No ALDC activity (0 ADU/g) was found in samples of proline-specific endoproteases: AniPro_2 or AnPro, throughout stability test period respectively. Moreover, aldB_Bs blended with either AniPro_2 or AnPro showed no loss of α- Acetolactate Decarboxylase activity at both 5°C and 30°C for up to 48 hours. However, aldB_Bl blended with AniPro_2 showed 82% and 48% residual activity at 5°C and 30°C after 48 hours. aldB_Bl blended with AnPro showed less stability having only 68% and 6% residual activity at 5°C and 30°C after 48 hours. Thus, aldB_Bl is clearly sensitive towards blending with proline- specific endoproteases as compared with aldB_Bs.
Table 6 ALDC (α-Acetolactate Decarboxylase) activity (ADU/g) of aldB_Bs, aldB_Bl, AniPro_2, AnPro and combinations samples mixed 50:50 % (w/w) (aldB_Bs + AniPro_2, aldB_Bl + AniPro_2, aldB_Bs + AnPro and aldB_Bl + AnPro), stored at 5°C. Activity of all samples were determined in duplicates up to 48hrs, having residuals calculated against activity at 0 hrs. ADU/
 Table 7 ALDC (α-Acetolactate Decarboxylase) activity (ADU/g) of aldB_Bs, aldB_Bl, AniPro_2, AnPro and combinations samples mixed 50:50 % (w/w) (aldB_Bs + AniPro_2, aldB_Bl + AniPro_2, aldB_Bs + AnPro and aldB_Bl + AnPro), stored at 30°C. Activity of all samples were determined in duplicates up to 48hrs, having residuals calculated against activity at 0 hrs. ADU/g 0 hrs 1 hrs 3 hrs 24 hrs 48 hrs
Table 7 ALDC (α-Acetolactate Decarboxylase) activity (ADU/g) of aldB_Bs, aldB_Bl, AniPro_2, AnPro and combinations samples mixed 50:50 % (w/w) (aldB_Bs + AniPro_2, aldB_Bl + AniPro_2, aldB_Bs + AnPro and aldB_Bl + AnPro), stored at 30°C. Activity of all samples were determined in duplicates up to 48hrs, having residuals calculated against activity at 0 hrs. ADU/g 0 hrs 1 hrs 3 hrs 24 hrs 48 hrs



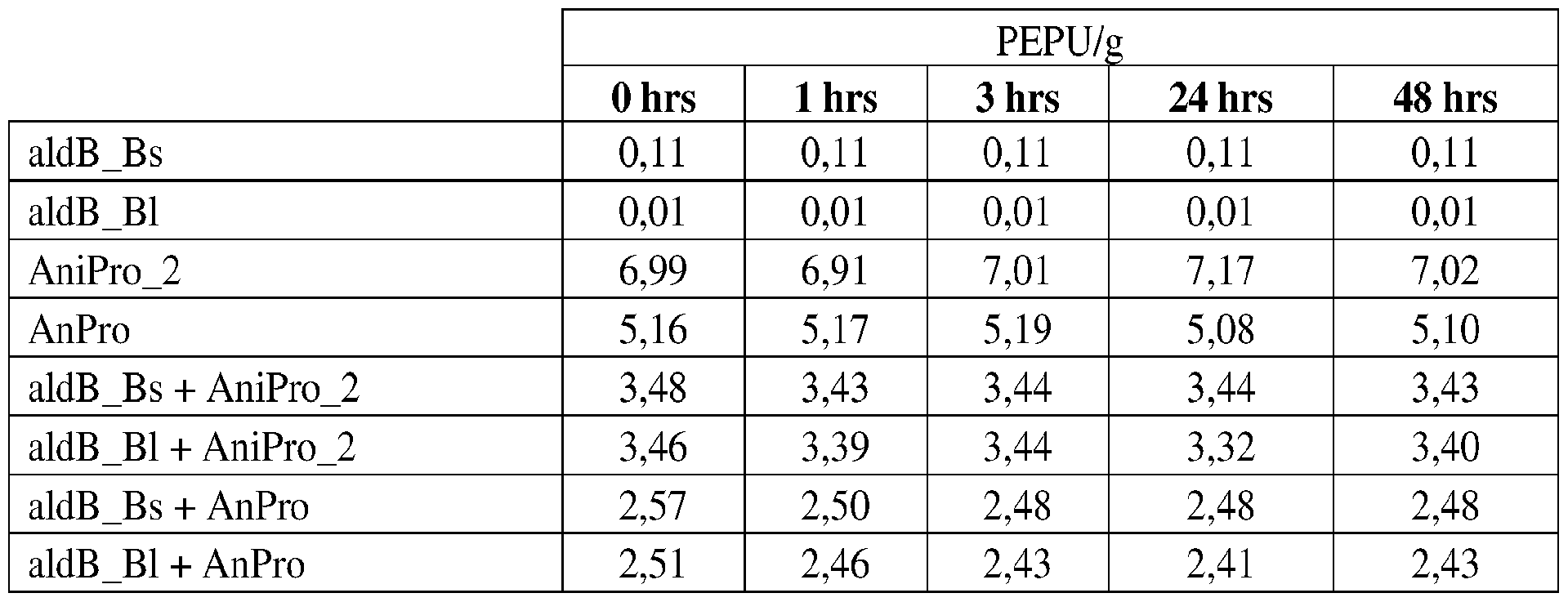






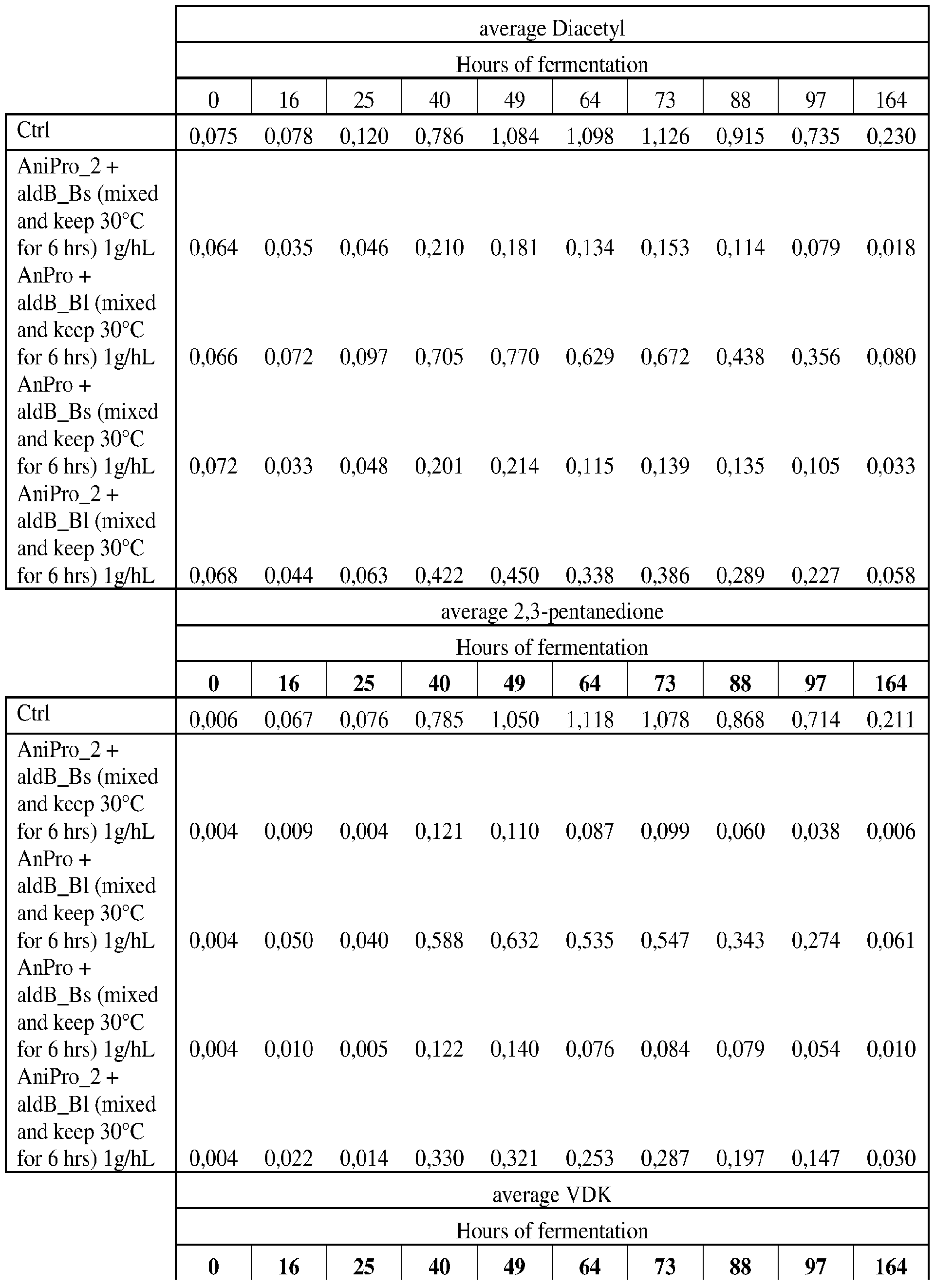
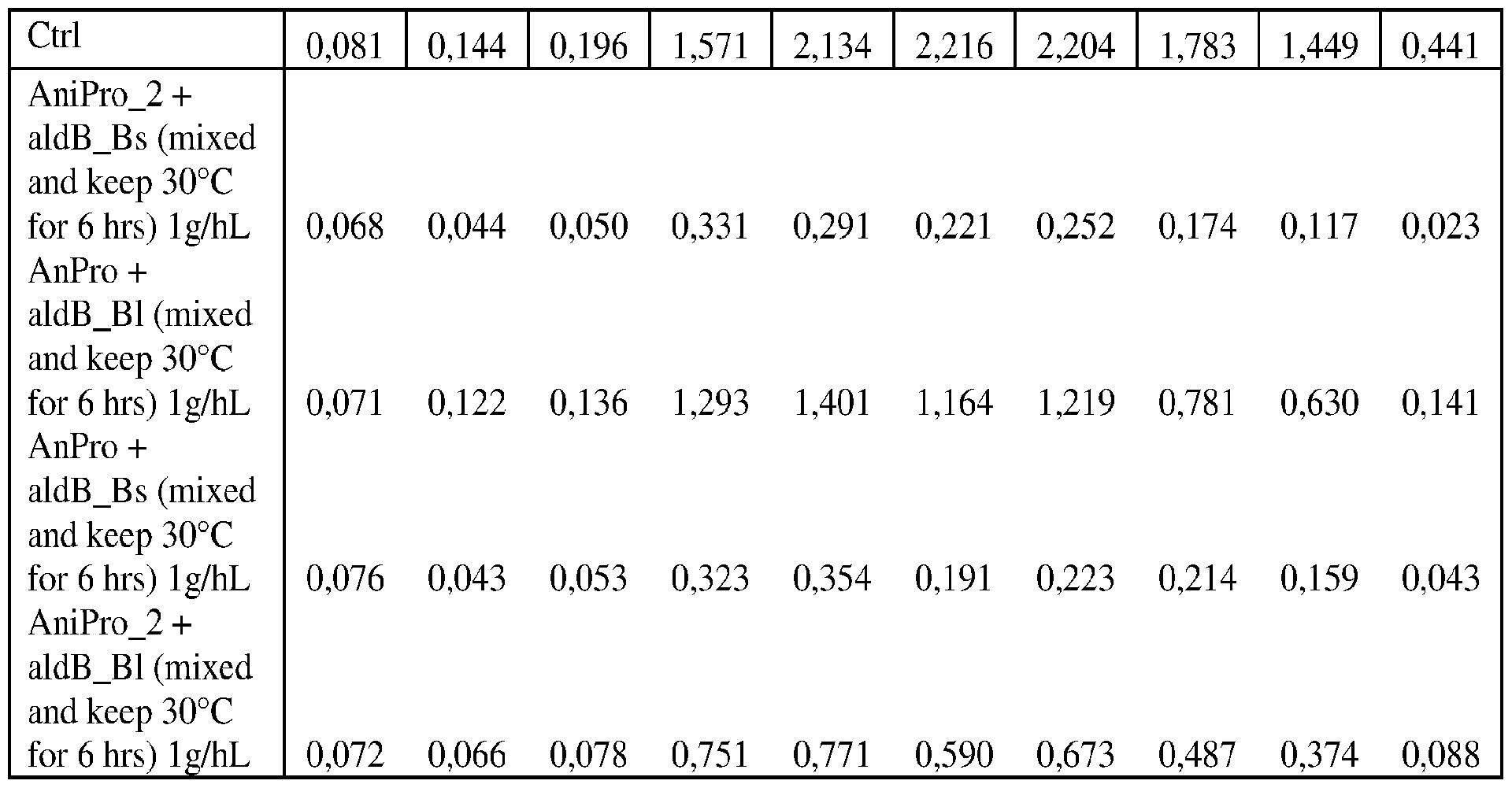

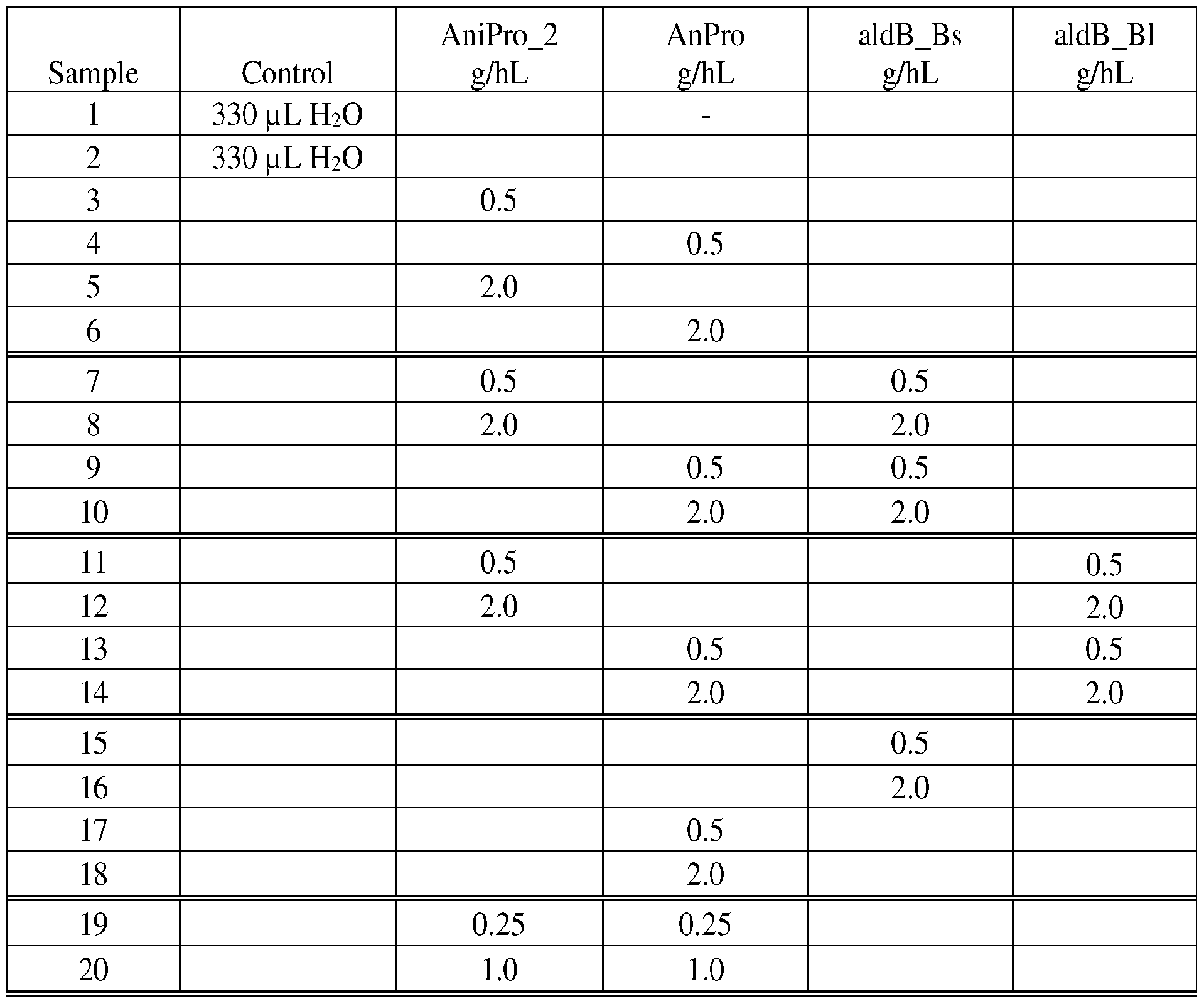





Claims
What is claimed is: 1. A polypeptide having acetolactate decarboxylase activity which is resistant to protease inactivation comprising an amino acid sequence having at least 80, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:3 wherein said polypeptide has at most 250 amino acids.
2. The polypeptide of claim 1 wherein said polypeptide has at most 249 amino acids.
3. The polypeptide of claim 2 wherein said polypeptide has at most 248 amino acids.
4. The polypeptide of claim 3 wherein said polypeptide has at most 247 amino acids.
5. The polypeptide of claim 4 wherein said polypeptide has at most 246 amino acids.
6. The polypeptide of claim 5 wherein said polypeptide has at most 245 amino acids.
7. The polypeptide of claim 6 wherein said polypeptide has at most 244 amino acids.
8. The polypeptide of claim 7 wherein said polypeptide has at most 243 amino acids.
9. The polypeptide of claim 3 wherein said polypeptide has at most 242 amino acids.
10. The polypeptide of claim 3 wherein said polypeptide has at most 241 amino acids.
11. The polypeptide of any of claims 1 to 10 comprising an amino acid sequence having at least 80, 90, 95, 98, 99 or 100% sequence identity to according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12.
12. The polypeptide of claim 11 wherein said polypeptide has an amino acid sequence according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12.
13. The polypeptide of any of the preceding claims wherein said protease is a proline specific protease.
14. The polypeptide of claim 13 wherein said protease is from Aspergillus niger.
15. The polypeptide of claims 13 or 14 wherein said protease has at least 80, 85, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:17.
16. The polypeptide of claim 15 wherein said protease has an amino acid sequence according to SEQ ID NO:17.
17. An improved brewing process comprising fermenting a wort in the presence of a polypeptide having acetolactate decarboxylase activity which is resistant to protease inactivation and a proline specific protease wherein both enzymes are present in the wort at the same time.
18. The improved brewing process of claim 17 wherein the polypeptide having acetolactate decarboxylase activity has an amino acid sequence with at least 80, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:3 wherein the polypeptide has at most 250 amino acids.
19. The improved brewing process of claim 18 wherein said polypeptide has at most 249 amino acids.
20. The improved brewing process of claim 19 wherein said polypeptide has at most 248 amino acids.
21. The improved brewing process of claim 20 wherein said polypeptide has at most 247 amino acids.
22. The improved brewing process of claim 21 wherein said polypeptide has at most 246 amino acids.
23. The improved brewing process of claim 22 wherein said polypeptide has at most 245 amino acids.
24. The improved brewing process of claim 23 wherein said polypeptide has at most 244 amino acids.
25. The improved brewing process of claim 24 wherein said polypeptide has at most 243 amino acids.
26. The improved brewing process of claim 25 wherein said polypeptide has at most 242 amino acids.
27. The improved brewing process of claim 26 wherein said polypeptide has at most 241 amino acids.
28. The improved brewing process of any of claims 17 to 27 wherein said polypeptide has an amino acid sequence with at least 80, 90, 95, 98, 99 or 100% sequence identity to according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12.
29. The improved brewing process of claim 28 wherein said polypeptide has an amino acid sequence according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12.
30. The improved brewing process of any of claims 17 to 29 wherein the polypeptide having acetolactate decarboxylase activity is added to the wort first.
31. The improved brewing process any of claims 17 to 29 wherein the proline specific protease is added to the wort first.
32. The improved brewing process of any of claims 17 to 29 wherein the polypeptide having acetolactate decarboxylase activity and the proline specific protease are added simultaneously to the wort.
33. The improved brewing process of any of claims 17 to 32 wherein said proline specific protease is from Aspergillus niger.
34. The improved brewing process of any of claims 17 to 33 wherein said proline specific protease has at least 80, 85, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:17.
35. A Bacillus host cell producing a heterologous polypeptide of interest, wherein one or more protease genes are inactivated.
36. The host cell of claim 35, wherein the polypeptide of interest is expressed without a secretion signal peptide.
37. The host cell of claim 35, wherein the polypeptide of interest is expressed with a secretion signal.
38. The host cell of any of claims 35 to 37, wherein the polypeptide of interest is an enzyme.
39. The host cell of claim 38, wherein the enzyme is an oxidoreductase, transferase, hydrolase, lyase, isomerase, ligase, aminopeptidase, amylase, asparaginase, carbohydrase, carboxypeptidase, catalase, cellulase, chitinase, cutinase, cyclodextrin glycosyltransferase, deoxyribonuclease, esterase, alpha-galactosidase, beta- galactosidase, glucoamylase, alpha- glucosidase, beta-glucosidase, hyaluronic acid synthase, invertase, laccase, lipase, mannosidase, mutanase, oxidase, a pectinolytic enzyme, peroxidase, phytase, polyphenoloxidase, protease, ribonuclease, transglutaminase, or xylanase.
40. The host cell of claim 39 wherein the enzyme is an ALDC enzyme.
41. The host cell of claim 40 wherein said ALDC enzyme comprises a polypeptide with an amino acid sequence having at least 80, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:3 wherein said polypeptide has at most 250 amino acids.
42. The host cell of claim 41 wherein said polypeptide has at most 249 amino acids.
43. The host cell of claim 42 wherein said polypeptide has at most 248 amino acids.
44. The host cell of claim 43 wherein said polypeptide has at most 247 amino acids.
45. The host cell of claim 44 wherein said polypeptide has at most 246 amino acids.
46. The host cell of claim 45 wherein said polypeptide has at most 245 amino acids.
47. The host cell of claim 46 wherein said polypeptide has at most 244 amino acids.
48. The host cell of claim 47 wherein said polypeptide has at most 243 amino acids.
49. The host cell of claim 48 wherein said polypeptide has at most 242 amino acids.
50. The host cell of claim 49 wherein said polypeptide has at most 241 amino acids.
51. The host cell of any of claims 41 to 50 wherein said polypeptide has an amino acid sequence with at least 80, 90, 95, 98, 99 or 100% sequence identity to according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12.
52. The host cell of claim 51 wherein said polypeptide has an amino acid sequence according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12.
53. The host cell of 40 to 52 wherein the ALDC enzyme is expressed with a secretion signal.
54. The host cell of any of claims 35 to 53, wherein the heterologous polypeptide of interest is encoded by an exogenous polynucleotide integrated into the chromosome of the host cell in at least one copy.
55. The host cell of claim 54 wherein said exogenous polynucleotide comprises a nucleic acid sequence having 80, 85, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:28.
56. The host cell of claim 55 wherein said exogenous polynucleotide comprises a nucleic acid sequence comprising SEQ ID NO:28.
57. The host cell of any of claims 35 to 56, wherein the one or more protease genes is inactivated by a non-sense mutation in said gene, a partial deletion of said gene or a full deletion of said gene.
58. The host cell of any of claims 35 to 57 wherein the Bacillus host is Bacillus subtilis or Bacillus licheniformis.
59. The host cell of claim 58 wherein the Bacillus host is Bacillus subtilis.
60. The host cell of any of claims 35 to 59 wherein the one or more protease genes are selected from the group consisting of a nucleic acid sequence having at least 80, 85, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:18, SEQ ID NO:19, SEQ ID NO:20, SEQ ID NO:21, SEQ ID NO:22, SEQ ID NO:23, SEQ ID NO:24, SEQ ID NO:25 and SEQ ID NO:27.
61. The host cell of claim 60 wherein the one or more protease genes comprise nine inactivated proteases wherein said proteases have nucleic acid sequences with at least 80, 85, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:18, SEQ ID NO:19, SEQ ID NO:20, SEQ ID NO:21, SEQ ID NO:22, SEQ ID NO:23, SEQ ID NO:24, SEQ ID NO:25 and SEQ ID NO:27.
62. The host cell of claim 61 wherein the one or more protease genes have nucleic acid sequences according to SEQ ID NO:18, SEQ ID NO:19, SEQ ID NO:20, SEQ ID NO:21, SEQ ID NO:22, SEQ ID NO:23, SEQ ID NO:24, SEQ ID NO:25 and SEQ ID NO:27.
63. A method for producing a polypeptide of interest comprising: i) providing a Bacillus host cell wherein one or more protease genes are inactivated and wherein said host cell is transformed with a nucleic acid encoding a heterologous polypeptide in operable combination with a promoter; and ii) cultivating said host cell under conditions suitable for the production of said heterologous polypeptide, such that said heterologous polypeptide is produced.
64. The method of claim 63, further comprising recovering said produced polypeptide.
65. The method of claim 63 or 64, wherein the polypeptide of interest is expressed with or without a secretion signal peptide.
66. The method of claim 63 or 64 wherein the polypeptide of interest is expressed with a secretion signal peptide.
67. The method of any of claims 63 to 66, wherein the polypeptide of interest is an enzyme.
68. The method of claim 67, wherein the enzyme is an oxidoreductase, transferase, hydrolase, lyase, isomerase, ligase, aminopeptidase, amylase, asparaginase, carbohydrase, carboxypeptidase, catalase, cellulase, chitinase, cutinase, cyclodextrin glycosyltransferase, deoxyribonuclease, esterase, alpha-galactosidase, beta- galactosidase, glucoamylase, alpha- glucosidase, beta-glucosidase, hyaluronic acid synthase, invertase, laccase, lipase, mannosidase, mutanase, oxidase, a pectinolytic enzyme, peroxidase, phytase, polyphenoloxidase, protease, ribonuclease, transglutaminase, or xylanase.
69. The method of claim 68 wherein the enzyme is an ALDC enzyme.
70. The method of claim 69 wherein said ALDC enzyme comprises a polypeptide with an amino acid sequence having at least 80, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:3 wherein said polypeptide has at most 250 amino acids.
71. The method of claim 70 wherein said polypeptide has at most 249 amino acids.
72. The method of claim 71 wherein said polypeptide has at most 248 amino acids.
73. The method of claim 72 wherein said polypeptide has at most 247 amino acids.
74. The method of claim 73 wherein said polypeptide has at most 246 amino acids.
75. The method of claim 74 wherein said polypeptide has at most 245 amino acids.
76. The method of claim 75 wherein said polypeptide has at most 244 amino acids.
77. The method of claim 76 wherein said polypeptide has at most 243 amino acids.
78. The method of claim 77 wherein said polypeptide has at most 242 amino acids.
79. The method of claim 78 wherein said polypeptide has at most 241 amino acids.
80. The method of any of claims 69 to 79 wherein said polypeptide has an amino acid sequence with at least 80, 90, 95, 98, 99 or 100% sequence identity to according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12.
81. The method of claim 80 wherein said polypeptide has an amino acid sequence according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12.
82. The method any of claims 69 to 81 wherein the ALDC enzyme is secreted.
83. The method of any of claims 63 to 82, wherein the heterologous polypeptide of interest is encoded by an exogenous polynucleotide integrated into the chromosome of the host cell in at least one copy.
84. The method of claim 83 wherein said exogenous polynucleotide comprises a nucleic acid sequence having 80, 85, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:28.
85. The method of claim 84 wherein said exogenous polynucleotide comprises a nucleic acid sequence comprising SEQ ID NO:28.
86. The method of any of claims 63 to 85, wherein the at least one protease gene is inactivated by a non-sense mutation in said at least one gene, a partial deletion of said at least one gene or a full deletion of said at least one gene.
87. The method of any of claims 63 to 86 wherein the Bacillus host is Bacillus subtilis or Bacillus licheniformis.
88. The method of claim 87 wherein the Bacillus host is Bacillus subtilis.
89. The method of any of claims 63 to 88 wherein the one or more protease genes are selected from the group consisting of a nucleic acid sequence having at least 80, 85, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:18, SEQ ID NO:19, SEQ ID NO:20, SEQ ID NO:21, SEQ ID NO:22, SEQ ID NO:23, SEQ ID NO:24, SEQ ID NO:25 and SEQ ID NO:27. 90. The method of claim 89 wherein the one or more protease genes comprises nine inactivated protease genes having nucleic acid sequences with at least 80, 85,
90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:18, SEQ ID NO:19, SEQ ID NO:20, SEQ ID NO:21, SEQ ID NO:22, SEQ ID NO:23, SEQ ID NO:24, SEQ ID NO:25 and SEQ ID NO:27.
91. The method of claim 90 wherein the one or more protease genes have nucleic acid sequences according to SEQ ID NO:18, SEQ ID NO:19, SEQ ID NO:20, SEQ ID NO:21, SEQ ID NO:22, SEQ ID NO:23, SEQ ID NO:24, SEQ ID NO:25 and SEQ ID NO:27.
92. A stable liquid formulation comprising a polypeptide having acetolactate decarboxylase activity comprising an amino acid sequence having at least 80, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:3 wherein said polypeptide has at most 250 amino acids and a protease.
93. The stable liquid formulation of claim 92 wherein said ALDC polypeptide has at most 249 amino acids.
94. The stable liquid formulation of claim 93 wherein said ALDC polypeptide has at most 248 amino acids.
95. The stable liquid formulation of claim 94 wherein said ALDC polypeptide has at most 247 amino acids.
96. The stable liquid formulation of claim 95 wherein said ALDC polypeptide has at most 246 amino acids.
97. The stable liquid formulation of claim 96 wherein said ALDC polypeptide has at most 245 amino acids.
98. The stable liquid formulation of claim 97 wherein said ALDC polypeptide has at most 244 amino acids.
99. The stable liquid formulation of claim 98 wherein said ALDC polypeptide has at most 243 amino acids.
100. The stable liquid formulation of claim 99 wherein said ALDC polypeptide has at most 242 amino acids.
101. The stable liquid formulation of claim 100 wherein said ALDC polypeptide has at most 241 amino acids.
102. The stable liquid formulation of any of claims 92 to 101 wherein said ALDC polypeptide comprises an amino acid sequence having at least 80, 90, 95, 98, 99 or 100% sequence identity to according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12.
103. The stable liquid formulation of claim 102 wherein said ALDC polypeptide comprises an amino acid sequence according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12.
104. The stable liquid formulation of any of claims 92 to 103 wherein said protease is a proline specific protease.
105. The stable liquid formulation of claim 104 wherein said protease is from Aspergillus niger.
106. The stable liquid formulation of claims 104 or 105 wherein said protease has at least 80, 85, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:17.
107. The stable liquid formulation of claim 106 wherein said protease has an amino acid sequence according to SEQ ID NO:17.
108. The stable liquid formulation of any of claims 92 to 107 wherein said ALDC polypeptide maintains at least 99, 98, 95, 90, 85, 80, 75 or 70% activity when the stable liquid formulation is stored at 10oC for 30, 60, 90, 120, 150 or 180 days.
109. The stable liquid formulation of any of claims 92 to 107 wherein said ALDC polypeptide maintains at least 99, 98, 95, 90, 85, 80, 75 or 70% activity when the stable liquid formulation is stored at 30oC for 8, 16, 24, 32, 40 or 48 hours.
110. A proline specific protease formulation which is substantially depleted of other protease activities comprising the protease wherein when said protease is combined with a polypeptide having acetolactate decarboxylase activity, the polypeptide is stable over time.
111. The protease formulation of claim 110 wherein the polypeptide having acetolactate decarboxylase activity comprises an amino acid sequence having at least 80, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:3 wherein said polypeptide has at most 250 amino acids.
112. The protease formulation of claim 111 wherein said ALDC polypeptide has at most 249 amino acids.
113. The protease formulation of claim 112 wherein said ALDC polypeptide has at most 248 amino acids.
114. The protease formulation of claim 113 wherein said ALDC polypeptide has at most 247 amino acids.
115. The protease formulation of claim 114 wherein said ALDC polypeptide has at most 246 amino acids.
116. The protease formulation of claim 115 wherein said ALDC polypeptide has at most 245 amino acids.
117. The protease formulation of claim 116 wherein said ALDC polypeptide has at most 244 amino acids.
118. The protease formulation of claim 117 wherein said ALDC polypeptide has at most 243
amino acids.
119. The protease formulation of claim 118 wherein said ALDC polypeptide has at most 242 amino acids.
120. The protease formulation of claim 119 wherein said ALDC polypeptide has at most 241 amino acids.
121. The protease formulation of any of claims 110 to 120 wherein said ALDC polypeptide comprises an amino acid sequence having at least 80, 90, 95, 98, 99 or 100% sequence identity to according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12.
122. The protease formulation of claim 121 wherein said ALDC polypeptide comprises an amino acid sequence according to SEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 or SEQ ID NO:12.
123. The protease formulation of any of claims 110 to 122 wherein said protease is a proline specific protease.
124. The protease formulation of claim 123 wherein said protease is from Aspergillus niger.
125. The protease formulation of claims 123 or 124 wherein said protease has at least 80, 85, 90, 95, 98, 99 or 100% sequence identity to SEQ ID NO:17.
126. The protease formulation of claim 125 wherein said protease has an amino acid sequence according to SEQ ID NO:17.
127. The protease formulation of any of claims 110 to 126 which is a solid.
128. The protease formulation of any of claims 110 to 126 which is a liquid.
129. The protease formulation of claim 128 wherein said ALDC polypeptide maintains at least 99, 98, 95, 90, 85, 80, 75 or 70% activity when the formulation is stored at 10oC for 30, 60, 90, 120, 150 or 180 days.
130. The protease formulation of claim 128 wherein said ALDC polypeptide maintains at least 99, 98, 95, 90, 85, 80, 75 or 70% activity when the formulation is stored at 30oC for 8, 16, 24, 32, 40 or 48 hours.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US202363580057P | 2023-09-01 | 2023-09-01 | |
| US63/580,057 | 2023-09-01 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2025049848A2 true WO2025049848A2 (en) | 2025-03-06 |
| WO2025049848A3 WO2025049848A3 (en) | 2025-04-10 |
Family
ID=92882485
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2024/044586 Pending WO2025049848A2 (en) | 2023-09-01 | 2024-08-30 | Proteases and aldc enzymes for beer haze and diacetyl reduction |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2025049848A2 (en) |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0238023A2 (en) | 1986-03-17 | 1987-09-23 | Novo Nordisk A/S | Process for the production of protein products in Aspergillus oryzae and a promoter for use in Aspergillus |
| WO1991017243A1 (en) | 1990-05-09 | 1991-11-14 | Novo Nordisk A/S | A cellulase preparation comprising an endoglucanase enzyme |
| US5281526A (en) | 1992-10-20 | 1994-01-25 | Solvay Enzymes, Inc. | Method of purification of amylase by precipitation with a metal halide and 4-hydroxybenzic acid or a derivative thereof |
| US6022725A (en) | 1990-12-10 | 2000-02-08 | Genencor International, Inc. | Cloning and amplification of the β-glucosidase gene of Trichoderma reesei |
| WO2005001036A2 (en) | 2003-05-29 | 2005-01-06 | Genencor International, Inc. | Novel trichoderma genes |
| WO2015017256A1 (en) | 2013-07-29 | 2015-02-05 | Danisco Us Inc. | Variant enzymes |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DK252483D0 (en) * | 1983-06-03 | 1983-06-03 | Novo Industri As | ALPHA-ACETOLACTATE DECARBOXYLASEEMZYM PRODUCT AND PREPARATION thereof |
| DK2402425T3 (en) * | 2006-07-13 | 2021-07-05 | Dsm Ip Assets Bv | IMPROVED BREWING PROCESS |
| PL3298138T3 (en) * | 2015-05-22 | 2022-12-05 | Dupont Nutrition Biosciences Aps | Acetolactate decarboxylase |
| EP4355872A2 (en) * | 2021-06-18 | 2024-04-24 | International N&H Denmark ApS | Proteases for beer haze reduction |
-
2024
- 2024-08-30 WO PCT/US2024/044586 patent/WO2025049848A2/en active Pending
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0238023A2 (en) | 1986-03-17 | 1987-09-23 | Novo Nordisk A/S | Process for the production of protein products in Aspergillus oryzae and a promoter for use in Aspergillus |
| WO1991017243A1 (en) | 1990-05-09 | 1991-11-14 | Novo Nordisk A/S | A cellulase preparation comprising an endoglucanase enzyme |
| US6022725A (en) | 1990-12-10 | 2000-02-08 | Genencor International, Inc. | Cloning and amplification of the β-glucosidase gene of Trichoderma reesei |
| US5281526A (en) | 1992-10-20 | 1994-01-25 | Solvay Enzymes, Inc. | Method of purification of amylase by precipitation with a metal halide and 4-hydroxybenzic acid or a derivative thereof |
| WO2005001036A2 (en) | 2003-05-29 | 2005-01-06 | Genencor International, Inc. | Novel trichoderma genes |
| WO2015017256A1 (en) | 2013-07-29 | 2015-02-05 | Danisco Us Inc. | Variant enzymes |
Non-Patent Citations (24)
| Title |
|---|
| ALTSCHUL ET AL., J MOL BIOL, vol. 215, 1990, pages 403 - 410 |
| ASANO, KSHINAGAWA, KHASHIMOTO, N: "Characterization of haze-forming proteins of beer and their roles in chill haze formation", J. AM. SOC. BREW. CHEM, vol. 40, 1982, pages 147 - 154, XP002795519, DOI: 10.1094/ASBCJ-40-0147 |
| B. ROEJ. CRABTREEA. KAHN: "DNA Isolation and Sequencing: Essential Techniques", 1996, JOHN WILEY & SONS |
| CAMPBELL ET AL., CURR. GENET, vol. 16, 1989, pages 53 - 56 |
| CAO ET AL., SCIENCE, vol. 9, 2000, pages 991 - 1001 |
| D. M. J. LILLEYJ. E. DAHLBERG: "Methods in Enzymology", 1992, ACADEMIC PRESS, article "Methods of Enzymology: DNA Structure Part A: Synthesis and Physical Analysis of DNA" |
| DIDERICHSEN ET AL., J BACTERIOL, vol. 172, no. 8, 1990, pages 4315 |
| DIDERICHSEN ET AL., J. BACTERIOL, vol. 172, no. 8, 1990, pages 4315 |
| EMANUELSSON ET AL., NATURE PROTOCOLS, vol. 2, 2007, pages 953 - 971 |
| FERRARI ET AL., J. BACTERIOL, vol. 170, no. 1, 1988, pages 289 - 95 |
| GRIGORIEV ET AL.: "Nucleic Acids Res", vol. 40, January 2012, article "The Genome Portal of the Department of Energy Joint Genome Institute", pages: D26 - 32 |
| HARRISON ET AL., APPLIED ENVIRON. MICROBIOL, vol. 77, June 2011 (2011-06-01), pages 3916 - 22 |
| HORWELL DC, TRENDS BIOTECHNOL, vol. 13, no. 4, 1995, pages 132 - 134 |
| LIU ET AL.: "Improved heterologous gene expression in Trichoderma reesei by cellobiohydrolase I gene (cbhl) promoter optimization", ACTA BIOCHIM. BIOPHYS. SIN (SHANGHAI), vol. 40, no. 2, 2008, pages 158 - 65, XP002595560, DOI: 10.1111/J.1745-7270.2008.00388.X |
| NARZISS, L, FERMENT, vol. 3, 1990, pages 54 - 62 |
| NORDAHL PETERSEN ET AL., NATURE METHODS, vol. 8, 2011, pages 785 - 786 |
| POSADA, J.ALMENAR, JGARCIA GALINDO, J: "A practical approach on protein stabilizers", PROC. - EUR. BREW. CONV, vol. 13, 1971, pages 379 - 391 |
| SAMBROOK ET AL.: "MOLECULAR CLONING: A LABORATORY MANUAL", 2001, COLD SPRING HARBOR |
| SIMON RJ ET AL., PNAS, vol. 89, no. 20, 1992, pages 9367 - 9371 |
| STAHLFERRARI, J. BACTERIOLOGY, vol. 160, 1984, pages 411 - 418 |
| TE'O ET AL., J. MICROBIOL. METHODS, vol. 51, 2002, pages 393 - 99 |
| THOMPSON ET AL., NUCLEIC ACIDS RES., vol. 22, 1994, pages 4673 - 4680 |
| TRIEU-COET, PP. COURVALIN, GENE, vol. 23, 1983, pages 331 - 341 |
| YAN, XHAO-JIE, YHONG, QSHUN-PENG, L, APPL. ENV. MICROBIOL, vol. 74, 2008, pages 5556 - 5562 |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2025049848A3 (en) | 2025-04-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US12503674B2 (en) | Proteases for beer haze reduction | |
| US20230295601A1 (en) | Aldc production methods | |
| US11873470B2 (en) | Brewing method | |
| WO2017085210A1 (en) | Preparation of a stable beer | |
| WO2014191298A1 (en) | Polypeptides having protease activity for colloidal stability | |
| WO2025049848A2 (en) | Proteases and aldc enzymes for beer haze and diacetyl reduction | |
| US20210315238A1 (en) | Proline specific endopeptidases | |
| US20260042983A1 (en) | Proteases for beer haze reduction | |
| US20200216787A1 (en) | Riboflavinase enzymes and their use to prevent off flavor in brewing | |
| US10450539B2 (en) | Use of M4 metalloprotease in wort production | |
| EP4658746A1 (en) | Improved production of rye-based alcoholic beverages | |
| EP2986701B1 (en) | Polypeptides having protease activity and polynucleotides encoding same | |
| EP3377603A1 (en) | Preparation of a stable beer |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 24776066 Country of ref document: EP Kind code of ref document: A2 |